Drugs, Health Technologies, Health Systems
Reimbursement Review
Semaglutide (Wegovy)
Sponsor: Novo Nordisk Canada Inc.
Therapeutic area: Weight management
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ACE
angiotensin-converting enzyme
AE
adverse event
ASA
acetylsalicylic acid
ASCVD
atherosclerotic cardiovascular disease
BMI
body mass index
CABPS
Canadian Association for Bariatric Physicians and Surgeons
CDA-AMC
Canada’s Drug Agency
CDEC
Canadian Drug Expert Committee
CI
confidence interval
CRSP
Cardiac Rehabilitation and Secondary Prevention Program
CV
cardiovascular
EAC
event adjudication committee
eGFR
estimated glomerular filtration rate
FAS
full analysis set
GERD
gastroesophageal reflux disease
GLP-1
glucagon-like peptide-1
GLP-1 RA
glucagon-like peptide-1 receptor agonist
HDL
high-density lipoprotein
HF
heart failure
HR
hazard ratio
LDL
low-density lipoprotein
MACE
major adverse cardiovascular event
MASH
metabolic dysfunction–associated steatohepatitis
MASLD
metabolic dysfunction–associated steatotic liver disease
MI
myocardial infarction
PAD
peripheral arterial disease
RAAS
renin-angiotensin-aldosterone system
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SGLT2
sodium-glucose cotransporter-2
TCR
TotalCardiology Rehabilitation
TIA
transient ischemic attack
UACR
urinary albumin-to-creatinine ratio
VAS
visual analogue scale
WRSSM
weight-related sign and symptom measure
Executive Summary
An overview of the resubmission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Semaglutide injection (Wegovy); 0.25 mg, 0.5 mg, 1 mg, 1.7 mg, or 2.4 mg; solution for subcutaneous injection in a prefilled pen |
Sponsor | Novo Nordisk Canada Inc. |
Indication | As an adjunct to a reduced calorie diet and increased physical activity for chronic weight management in adult patients with an initial body mass index of:
|
Reimbursement request | As an adjunct to a reduced calorie diet and increased physical activity for chronic weight management in adult patients with a BMI of 27 kg/m2 or greater and established cardiovascular disease (MI, stroke, or PAD). |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC datea | November 23, 2021 |
Recommended dose | The therapeutic and maintenance dose of 2.4 mg semaglutide once weekly is reached by starting with a dose of 0.25 mg and then following a dose-escalation regimen, with dose increases every 4 weeks (to doses of 0.5 mg, 1 mg, 1.7 mg, and 2.4 mg per week) until the therapeutic-maintenance dose of 2.4 mg once weekly is reached after 16 weeks. Follow the dose escalation to reduce the likelihood of gastrointestinal symptoms. If patients do not tolerate a dose during dose escalation, consider delaying dose escalation for 4 weeks. If patients (adults aged 18 years and older) do not tolerate the therapeutic-maintenance 2.4 mg dose, the dose can be temporarily decreased to 1.7 mg weekly, for a maximum of 4 weeks. Patients should re-escalate to the therapeutic-maintenance 2.4 mg dose. |
BMI = body mass index; MI = myocardial infarction; NOC = Notice of Compliance; PAD = peripheral arterial disease.
aThe NOC date is for the indication under review. Of note, this is distinct from the NOC issued in 2024 for Wegovy to reduce the risk of nonfatal MI in adults with established cardiovascular disease and BMI of 27 kg/m2 or greater.
Sources: Product monograph for semaglutide injection (Wegovy).1 Details included in the table are from the sponsor’s summary of clinical evidence.2
Submission History
Initial Submission
Semaglutide 2.4 mg was first reviewed by the Canadian Drug Expert Committee (CDEC) for weight management in 2022. CDEC issued a recommendation that semaglutide 2.4 mg not be reimbursed as an adjunct to a reduced calorie diet and increased physical activity for chronic weight management in adult patients with an initial body mass index (BMI) of 30 kg/m2 or greater (obesity) or 27 kg/m2 or greater (overweight) in the presence of at least 1 weight-related comorbidity such as hypertension, type 2 diabetes mellitus, dyslipidemia, or obstructive sleep apnea.3
The 2022 reimbursement recommendation for semaglutide (Wegovy) and the Clinical Review report on semaglutide (Wegovy) that was used to inform the recommendation are both available on the Canada’s Drug Agency (CDA-AMC) project website.
Basis of Resubmission
The rationale for the 2022 recommendation included evidence from 4 placebo-controlled, double-blind, randomized controlled trials (RCTs) (STEP 1, STEP 2, STEP 3, and STEP 4). These trials demonstrated that treatment with semaglutide 2.4 mg injection reduced body weight in individuals with an initial BMI of 30 kg/m2 or greater (obesity) or 27 kg/m2 or greater (overweight) in the presence of at least 1 weight-related comorbidity but did not demonstrate improvement in or prevention of weight-related comorbidities. Comorbidities such as a major adverse cardiovascular event (MACE), osteoarthritis, and obstructive sleep apnea were not outcomes assessed in the STEP trials.3
CDEC noted there was an ongoing trial, the SELECT study,4 comparing semaglutide 2.4 mg injection with placebo for the prevention of MACE occurrences in patients with overweight or obesity who have established cardiovascular (CV) disease but not diabetes mellitus. CDEC concluded in 2022 that the results of that study, once completed, would address the evidence gap regarding the effects of semaglutide 2.4 mg once weekly on CV outcomes in the indicated population.3
Therefore, the objective of this report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of semaglutide 2.4 mg solution for subcutaneous injection in chronic weight management in patients with a BMI of 27 kg/m2 or greater and established CV disease (myocardial infarction [MI], stroke, or peripheral arterial disease [PAD]).
Sponsor’s Clarifying Note on Resubmission
In 2024, Health Canada issued an indication for semaglutide injection (Wegovy) to reduce the risk of nonfatal MI in adults with established CV disease and a BMI of 27 kg/m2 or greater. This indication was based on the evidence from the SELECT trial.
The sponsor indicated that the population for the revised reimbursement request, namely, adults with established CV disease and a BMI of 27 kg/m2 or greater, is fully encompassed in the weight management indication and is the same population for which the CV indication was granted.
Introduction
Disease Background
Obesity is a chronic disease where excess body fat impairs health and increases the risk of long-term health complications.5 The presence of overweight (BMI of greater than 25 kg/m2) and obesity (BMI of greater than 30 kg/m2) are considered major contributors to CV disease progression through direct and indirect mechanisms.6-15 In 2022, the estimated prevalence of obesity was 30% of individuals living in Canada aged 18 years or older.16,17 In a study of patients with atherosclerotic cardiovascular disease (ASCVD), obesity was among the 30 most common comorbid conditions, with a prevalence of 38%.18 Worldwide, CV disease is among the leading causes of morbidity and mortality and is reported to be one of the leading causes of hospitalization in Canada, alongside stroke.19,20
Standards of Therapy
The Canadian obesity guideline states there are 4 medications indicated for long-term obesity management as adjuncts to health-behaviour changes: liraglutide, naltrexone-bupropion in a combination tablet, orlistat, and semaglutide. The Canadian guideline recognizes all 4 medications as effective in producing clinically significant weight loss and health benefits relative to placebo over a period of at least 1 year.21 The clinical experts indicated that another medication, tirzepatide, is currently approved only for adjunct glycemic control in type 2 diabetes but is increasingly being prescribed off label for chronic weight management in Canada. Further, patient groups have indicated that semaglutide (Ozempic) has been prescribed off label for weight management as well. The clinical experts indicated that in clinical practice in Canada, semaglutide is being used in patients with diabetes for weight loss.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups that responded to the call for input and from the clinical experts consulted for the purpose of this review.
Patient Input
Input for this review was submitted by 6 patient groups: GI Society, Obesity Canada, Obesity Matters, Fatty Liver Alliance, HeartLife Foundation, and Diabetes Canada. Input from the GI Society, Obesity Canada, and Diabetes Canada was based on surveys of individuals with obesity or diabetes conducted between 2021 and 2024. The GI Society also included input from an in-person focus group of individuals with obesity held in 2023. Input from the HeartLife Foundation was gathered through interviews and discussions with individuals with CV diseases and their health care providers. The Fatty Liver Alliance gathered physician insights on metabolic dysfunction–associated steatotic liver disease (MASLD) and metabolic dysfunction–associated steatohepatitis (MASH), supplemented by a 2022 to 2023 US survey of patients with nonalcoholic fatty liver disease or nonalcoholic steatohepatitis. Obesity Matters also provided feedback on the CDEC reimbursement recommendation issued for the initial 2022 review of semaglutide for weight management.
Patient groups highlighted the significant physical and mental health impact of obesity and related conditions, including CV diseases, diabetes, MASLD, and MASH. Common symptoms included mobility limitations, chronic pain, fatigue, swelling, shortness of breath, dizziness, and nausea, affecting daily life. Comorbidities were prevalent: in the GI Society’s 2021 survey, 91% of those who responded reported comorbidities such as arthritis (51%), hypertension (33%), sleep apnea (30%), gastroesophageal reflux disease (GERD) (29%), irritable bowel syndrome (29%), high cholesterol (25%), and diabetes (24%). Input from Diabetes Canada also noted high comorbidity rates for patients with diabetes, including high blood pressure, foot or eye problems, nerve damage, heart disease, and kidney-related comorbidities. Obesity not only contributes to these conditions but also complicates disease management and reduces quality of life. Mental health challenges, including anxiety, mood disorders, and social isolation due to stigma, especially in health care, were widely reported.
The patient groups highlighted weight loss as a key treatment outcome, along with reduced risk of comorbidities (e.g., CV disease), improvements in quality of life, and increased ability to perform everyday tasks. When choosing a new therapy, most patients primarily value efficacy (i.e., significant and sustained weight loss) and affordability.
Patient groups emphasized that traditional lifestyle modifications, such as diet and exercise, are often ineffective for long-term weight management. Few medication options exist, with semaglutide, liraglutide, and naltrexone-bupropion being commonly prescribed, but none have public or full private coverage, making cost the biggest barrier. Those who responded to the survey and who reported benefits of these treatments in managing obesity described them as “life-changing,” with most (94%) considering the side effects to be manageable. However, it was also noted that these medications lack long-term effectiveness, with many individuals gaining back the weight that was lost within 5 years. Bariatric surgery, while currently considered the gold standard for obesity treatment, carries risks of severe side effects, postsurgery weight gain, and long wait times, making it a last-resort option.
Patients with experience receiving semaglutide described several benefits, including substantial weight loss. It was highlighted that semaglutide also improved the management of comorbidities, such as CV disease, and improved quality of life. Thus, it was identified that semaglutide has the ability to address multiple health conditions simultaneously. Patients with experience with semaglutide also highlighted that its side effects — most commonly nausea, vomiting, and constipation — were typically temporary and manageable, and the once-weekly administration reduced treatment burden.
Obesity Matters provided additional feedback on the 2022 draft CDEC reimbursement recommendation for semaglutide (Wegovy). The feedback indicated that while the initial draft recommendation acknowledged the effect of semaglutide on body-weight reduction and the increased risk of comorbidities observed with obesity, it did not conclude that weight reduction can directly reduce comorbidities. The feedback also critiqued the rationale for a negative reimbursement recommendation, noting this overlooks the agency of individuals actively working with specialists to make lifestyle changes. The feedback also indicated that the previous recommendation did not adequately consider cost as a barrier to treatment, patient perspectives, the direct impact of treatment costs on quality of life, or how public reimbursement would reduce the long-term costs associated with obesity-related comorbidities and conditions, nor did it offer actionable solutions for integrating semaglutide into existing health care systems.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts identified the following as limitations with weight management medications: barriers to public access, that not all patients experience weight loss with treatment, and that some patients experience adverse effects that necessitate stopping the drug. Specific to bariatric surgery, the clinical experts noted that in addition to barriers to access, not all patients are interested in pursuing this option because of the potential risks associated with surgery.
According to the clinical experts consulted for this review, semaglutide 2.4 mg has been used for weight management in Canada since it was approved by Health Canada in 2021. The clinical experts indicated that the anticipated place of semaglutide with regard to pharmacotherapy for chronic weight management is as a first-line treatment. The clinical experts advised that semaglutide would be combined with therapies that reduce cardiorenal risk, including combination treatment with renin-angiotensin-aldosterone system (RAAS) inhibitors (such as angiotensin-converting enzyme [ACE] inhibitors and angiotensin 2 receptor blockers) and sodium-glucose cotransporter-2 (SGLT2) inhibitor therapies as well as other standards of care to improve CV outcomes. The clinical experts indicated there is currently no high-level evidence to support combination treatment with other pharmacotherapies for weight management; however, they noted there is relevant evidence from small studies (e.g., a retrospective cohort study on the effect of a combined glucagon-like peptide-1 [GLP-1] receptor agonist [GLP-1 RA] and bupropion-naltrexone in weight loss22). The clinical experts indicated that, in practice, specialists in obesity treat patients with combination therapy when needed and appropriate and when cost and insurance coverage are not limiting factors. The clinical experts indicated that the most common combination is a GLP-1 RA combined with bupropion-naltrexone.
The clinical experts deferred to the baseline characteristics of the SELECT trial population to identify patients who would most likely respond to treatment with semaglutide, where response is defined as reduced risk of future CV outcomes and weight loss. Of note, the clinical experts highlighted that the trial population included patients with a pre-existing history of CV disease, and 64.5% of trial participants had prediabetes (hemoglobin A1C > 5.7% per US criteria). Overall, the clinical experts advised that it would be appropriate to treat most patients with a BMI of 27 kg/m2 or greater for the chronic weight management indication, and that patients with established CV disease (including heart failure [HF]), type 2 diabetes, multiple risk factors, severe disease, or a high BMI are most in need of an intervention for the secondary prevention of future events and to improve symptom burden.
When considering which patients would be least suitable for treatment with semaglutide, the clinical experts deferred to the product monograph for absolute contraindications and patients with a potential increased risk of adverse effects (e.g., history of pancreatitis, gallbladder disease, diabetic retinopathy, severe renal impairment, gastrointestinal conditions).
Although weight loss is an outcome that can be easily monitored in practice, the clinical experts advised that monitoring the recurrence of CV events and symptoms is equally as important. The clinical experts further noted that patient-reported outcome tools are used to measure response to treatment.
The clinical experts indicated that treatment with semaglutide would be long term, unless the following occurred: excessive weight loss or nutritional deficiencies (may also be considered for dose reduction), renal failure (however, treatment may be continued, depending on overall nutritional status and if tolerating otherwise), side effects (e.g., persistent severe nausea, vomiting, or diarrhea), development of a contraindication, pregnant or planning to become pregnant, and cost and/or loss of coverage. The experts indicated that the occurrence of a repeat CV event while receiving treatment with semaglutide would not be considered treatment failure, noting there would be too much uncertainty to determine whether treatment was associated with the delayed onset of a second CV event in an individual. Additionally, the clinical experts advised that pausing treatment for an elective surgery may be considered, given reports that semaglutide is associated with slow gastrointestinal transit, which can increase the risk of aspiration with endotracheal intubation.
The clinical experts advised that a specialist is not required to prescribe semaglutide because family physicians and primary care providers have been prescribing semaglutide (Ozempic) for diabetes and obesity and are familiar with its side effect profile and monitoring.
Clinician Group Input
A total of 4 clinician groups provided input for this resubmission: TotalCardiology Rehabilitation (TCR) (5 clinicians) and Cardiac Rehabilitation and Secondary Prevention Program (CRSP) (Western University Division of Cardiology, 5 clinicians), plus a joint input from Obesity Canada and the Canadian Association for Bariatric Physicians and Surgeons (CABPS) (total of 18 clinicians). TCR, a multidisciplinary group of family physicians, internists, and cardiologists, is the sole provider of cardiac rehabilitation in the city of Calgary and the surrounding area. The Western University CRSP program delivers comprehensive, multidisciplinary rehabilitation care, including secondary prevention through lifestyle and pharmacotherapeutic interventions. The CABPS is a group of specialists in Canada with experience treating obesity. Obesity Canada is a national registered charity that assisted with the coordination of the group clinician response from CABPS.
The clinician group input was largely consistent with that of the clinical experts. Both the clinician groups and experts highlighted the limitations of the currently available weight management treatment options and the barriers to patients accessing them. The CABPS noted that obesity management practice currently follows the 2020 Canadian Adult Obesity Clinical Practice Guidelines, which recommend lifestyle modification supported by psychological or behavioural therapy, pharmacotherapy, and bariatric surgery. The clinician groups further highlighted that lifestyle modifications alone are often insufficient to experience the weight loss needed to improve obesity-related complications such as type 2 diabetes and MASLD. The clinician groups noted that liraglutide, semaglutide, and bupropion-naltrexone are the most effective long-term pharmacologic treatments due to their impact on the neuroendocrine pathways associated with obesity. The clinician groups also outlined the role of medical bariatric centres in providing multidisciplinary care and differentiated them from nonmedical weight loss clinics. The CABPS expressed that obesity treatment should extend beyond weight loss to address dysfunctional adipose tissue driving adverse health outcomes. The clinician groups’ overview of therapies that reduce cardiorenal risk in patients with comorbid CV disease was consistent with the expert input. The clinician groups also added that patients with ASCVD become eligible for cardiac rehabilitation, which combines lifestyle and pharmacotherapeutic interventions.
Both the clinicians and experts agreed that a primary goal of CV disease treatment is to reduce the incidence of CV events, but the clinician groups also suggested additional goals for weight management and improved glycemic control, when applicable. Both the clinicians and experts highlighted the access challenges and potential risks associated with bariatric surgery. Both noted a need for additional treatment options that address both CV risk and weight reduction, noting that existing treatments for weight loss and CV disease can lack long-term effectiveness and fail to directly target weight management. The clinician groups noted this as being particularly true for patients with obesity who do not have diabetes, a group for which there are currently no targeted treatment options. The clinician groups noted that these significant treatment gaps contribute to a persisting high risk of morbidity and mortality in these populations.
In contrast to the clinical expert input, which noted that semaglutide would be combined with other therapies that reduce CV risk (i.e., RAAS inhibitors, SGLT2 inhibitors), the clinician groups indicated that semaglutide would be used for first-line treatment alongside lifestyle modifications. The clinician groups noted it is unnecessary to first try other treatments before semaglutide, given its effectiveness in achieving substantial weight loss and improving obesity-related health outcomes. The clinician groups anticipate that semaglutide could shift current treatment paradigms, noting its efficacy approaches that of bariatric surgery and could reduce the number of patients requiring surgery. The patient type that the clinician groups indicated would benefit most from semaglutide aligned exactly with the type indicated by the clinical experts. The clinician groups added that semaglutide might also benefit patients with diabetes who require additional CV protection, or those without diabetes who are at high risk of CV disease and have difficulty experiencing weight loss through lifestyle modifications alone. The CRSP clinicians estimated that 78% of their patients receiving cardiac rehabilitation do not have diabetes and that 45% of patients who complete the rehabilitation program could potentially benefit from an additional therapy such as semaglutide. The patients least suitable for treatment, identified by the clinician groups and experts, also aligned. According to the clinician groups, patients with obesity are identified through self-identification or by health care providers using diagnostic tools such as BMI, waist circumference, and staging systems such as the Edmonton Obesity Staging System, which also classifies the degree of associated conditions (e.g., type 2 diabetes, impaired mobility). However, the CABPS noted there are currently no methods for identifying which patients are likely to respond to semaglutide.
The physician group input was consistent with the expert insights on the use of weight loss to assess treatment response but emphasized additional markers of treatment benefit. The clinician groups highlighted improvements in prediabetes, lipids, blood pressure, mobility, and quality of life as equally important. Conversely, TCR indicated there is no definitive evidence that the absence of weight loss precludes other treatment benefits. The CABPS provided specific criteria for a meaningful treatment response, including a 5% reduction in total body weight after 3 months of treatment (aligning with the expert input) and improved laboratory markers, reduced osteoarthritis pain, and the ability to proceed with procedures like hip replacement. The clinician groups also suggested initial evaluations every 4 to 6 weeks, then every 3 months, recognizing variability based on clinician and patient preference.
Both the clinician groups and experts agreed that semaglutide treatment should be continued indefinitely, with the clinician groups likening the regimen to statin therapy or treatment with acetylsalicylic acid (ASA) after an MI. The clinician groups suggested the discontinuation of semaglutide be considered in the following scenarios: presence of treatment intolerance or intolerable side effects that do not improve over time with appropriate countermeasures, pancreatitis, pregnancy or planning pregnancy, lack of meaningful treatment response, lack of affordability, or a more effective treatment becomes available in the future that requires semaglutide discontinuation. The clinician groups noted that semaglutide treatment could be delivered in family medicine and primary care clinics, community-based obesity management or metabolic medicine clinics, and hospital-based medical and surgical centres. Like the experts, the clinician groups indicated that any health care provider with experience managing obesity could prescribe semaglutide, noting this role should not be restricted to specialists alone.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. The following items were identified as key factors that could potentially impact the implementation of a recommendation to reimburse semaglutide 2.4 mg: relevant comparators, considerations for the continuation or renewal of therapy, considerations for the prescribing of therapy, and systemic and economic issues. The clinical experts consulted for this review provided advice on the potential implementation issues raised by the drug programs and are presented in Table 3.
Clinical Evidence
Systematic Review
Description of Study
The SELECT trial (N = 17,604) was a phase IIIb, multinational, multicentre, randomized, double-blind, parallel-group, placebo-controlled trial that evaluated the effect of semaglutide 2.4 mg subcutaneous injection compared with placebo in reducing the risk of MACE occurrences (CV death, nonfatal MI, or nonfatal stroke) in patients with established CV disease and overweight or obesity and without diabetes. The key secondary objective of the trial was to evaluate the effect of semaglutide in mortality compared with placebo. Patients were randomized in a 1:1 ratio to receive once-weekly treatment with either semaglutide 2.4 mg or placebo, both as an adjunct to the standard of care for CV disease.
A total of 8,803 patients were randomized to receive semaglutide 2.4 mg and 8,801 patients were randomized to receive placebo. The mean age of patients in the semaglutide 2.4 mg group was 61.6 years (standard deviation [SD] = 8.9 years) and 61.6 years (SD = 8.8 years) in the placebo group. The mean BMI of patients in the semaglutide 2.4 mg group was 33.30 kg/m2 (SD = 5.03 kg/m2) and 33.37 kg/m2 (SD = 5.04 kg/m2) in the placebo group. Most patients in the full analysis set (FAS) had a history of MI: 5,962 patients (67.7%) in the semaglutide 2.4 mg group and 5,944 patients (67.5%) in the placebo group. The mean duration of follow-up was 39.9 months (SD = 9.3 months) in the semaglutide 2.4 mg group and 39.7 months (SD = 9.5 months) in the placebo group. The mean duration of exposure to semaglutide 2.4 mg was 33.3 months (SD = 14.4 months) and to placebo was 35.1 months (SD = 13.0 months).
Efficacy Results
Key efficacy results are based on the in-trial observation period with a data cut-off date of July 18, 2023, and are presented in Table 2.
Major Adverse CV Event (CV Death, Nonfatal MI, or Nonfatal Stroke)
At the data cut-off date, the percentages of patients (in the FAS) with their first event adjudication committee (EAC)–confirmed MACE (consisting of CV death, nonfatal MI, or nonfatal stroke) were 6.5% of 8,803 patients in the semaglutide 2.4 mg group and 8.0% of 8,801 patients in the placebo group. Semaglutide 2.4 mg was favoured over placebo (adjusted hazard ratio [HR] = 0.80; 95% confidence interval [CI], 0.72 to 0.90). A cumulative incidence plot of time from randomization to first EAC-confirmed MACE is presented in Figure 3 in Appendix 1. The absolute risk difference in time to first EAC-confirmed MACE between semaglutide 2.4 mg and placebo at week 156 was −1.1% (95% CI, −1.9% to −0.4%).
Consultation with the clinical experts did not identify any relevant potential treatment-effect modifiers to examine for this review. However, BMI was identified as the most relevant subgroup for the purpose of this review to inform expert committee deliberations and, as such, a summary of the subgroup analysis results by BMI for the primary end point is presented in Table 28 in Appendix 1.
CV Death
At the data cut-off date, the percentages of patients (in the FAS) with an EAC-confirmed CV death (including undetermined cause of death) were 2.5% in the semaglutide 2.4 mg group and 3.0% in the placebo group (HR = 0.85; 95% CI, 0.71 to 1.01). A cumulative incidence plot of time from randomization to EAC-confirmed CV death is presented in Figure 4 in Appendix 1. The absolute risk difference in time to EAC-confirmed CV death between semaglutide 2.4 mg and placebo at week 156 was 0% (95% CI, −0.5% to 0.4%).
HF Composite (CV Death or Hospitalization for HF or Urgent HF Visit)
At the data cut-off date, the percentages of patients (in the FAS) with their first EAC-confirmed composite HF outcome (comprising CV death, HF requiring hospitalization or urgent HF visit) were 3.4% in the semaglutide 2.4 mg group and 4.1% in the placebo group (HR = 0.82; 95% CI, 0.71 to 0.96). A cumulative incidence plot of time from randomization to first EAC-confirmed composite HF outcome is presented in Figure 5 in Appendix 1. The absolute risk difference in time to EAC-confirmed composite HF outcome between semaglutide 2.4 mg and placebo at week 156 was −0.2% (95% CI, −0.8% to 0.3%).
All-Cause Death
At the data cut-off date, the percentages of patients (in the FAS) with an EAC-confirmed all-cause death were 4.3% in the semaglutide 2.4 mg group and 5.2% in the placebo group (HR = 0.81; 95% CI, 0.71 to 0.93). A cumulative incidence plot of time from randomization to EAC-confirmed all-cause death is presented in Figure 6 in Appendix 1. The absolute risk difference in time to EAC-confirmed all-cause death between semaglutide 2.4 mg and placebo at week 156 was −0.5% (95% CI, −1.1% to 0.1%).
Nonfatal MI
At the data cut-off date, the percentages of patients (in the FAS) with their first EAC-confirmed nonfatal MI were 2.7% in the semaglutide 2.4 mg group and 3.7% in the placebo group (HR = 0.72; 95% CI, 0.61 to 0.85).
Nonfatal Stroke
At the data cut-off date, the percentages of patients (in the FAS) with their first EAC-confirmed nonfatal stroke were 1.7% in the semaglutide 2.4 mg group and 1.9% in the placebo group (HR = 0.93; 95% CI, 0.74 to 1.15).
Hemoglobin A1C of 6.5% or Greater
Classification of glycemic status was according to the American Diabetes Association Standards of Medical Care in Diabetes, published in 2018.23 According to these standards, a patient with a hemoglobin A1C of 6.5% or greater has diabetes.23
At the data cut-off date, 306 patients (3.5% of the FAS) in the semaglutide 2.4 mg group and 1,059 patients (12.0% of the FAS) in the placebo group experienced a first occurrence of a hemoglobin A1C of 6.5% or greater (HR = 0.27; 95% CI, 0.24 to 0.31).
Composite Nephropathy Event
The 5-component composite nephropathy end point consisted of onset of persistent macroalbuminuria (urinary albumin-to-creatinine ratio [UACR] > 300 mg/g), persistent 50% reduction in estimated glomerular filtration rate (eGFR) compared with baseline, onset of a persistent eGFR of less than 15 mL/min/1.73 m2, initiation of chronic renal replacement therapy (i.e., dialysis or transplant), or renal death.
At the data cut-off date, 155 patients (1.8% of the FAS) in the semaglutide 2.4 mg group and 198 patients (2.2% of the FAS) in the placebo group had experienced a first composite nephropathy event (HR = 0.78; 95% CI, 0.63 to 0.96).
Body Weight
The mean treatment difference in the percent change from baseline in body weight at week 104 between semaglutide 2.4 mg and placebo was −8.51% (95% CI, −8.75% to −8.27%).
The analyses at weeks 156 and 208 were not performed as planned because more than 10% of patients in the FAS missed the relevant annual study visit due to trial termination. However, an analysis of the percent change from baseline in body weight was performed at week 208 and the estimated treatment difference (based on the in-trial period) between semaglutide 2.4 mg and placebo was −8.7% (95% CI, −9.42% to −7.88%). The on-treatment analysis, which defined treatment exposure as the observation period until first time off treatment for greater than 35 days, estimated a treatment difference of −10.2% (95% CI, −11.0% to −9.42%), which was similar to the result from the main analysis for change in body weight at week 208.24
EQ-5D Index Score and EQ Visual Analogue Scale Score
The EQ-5D-5L questionnaire is a patient-reported outcome tool used to estimate health-related quality of life. The tool includes a descriptive system that provides a description of problems experienced by the patient who responded according to dimensions, a visual analogue scale (VAS) that provides a score indicating overall self-rated health (score ranges from 0 to 100), and an index score that ranges from 0 to 1. A higher score indicates better self-reported health status.
The treatment difference in the change from baseline in the EQ-5D index score at week 104 between semaglutide 2.4 mg and placebo was 0.01 (95% CI, 0.01 to 0.02).
The treatment difference in the change from baseline in the EQ VAS score at week 104 between semaglutide 2.4 mg and placebo was 1.60 (95% CI, 1.16 to 2.04).
Weight-Related Sign and Symptom Measure
The weight-related sign and symptom measure (WRSSM) is a patient-reported outcome tool (a self-rated VAS) used to assess the presence and bother associated with weight-related symptoms. Specifically, it is used to assess the impact of multifaceted aspects of obesity on symptom experience in individuals with overweight or obesity. The total score ranges from 0 to 4, with higher scores indicating worse symptomatology.
The mean change from baseline in WRSSM total score at week 104 was −0.26 (SD = 0.71) in the semaglutide 2.4 mg group and −0.12 (SD = 0.68) in the placebo group. The between-group difference was not estimated.
Cardiometabolic Risk Factors
Efficacy results on cardiometabolic risk factors (change from baseline in systolic blood pressure, change from baseline in total cholesterol, and change from baseline in high-density lipoprotein [HDL] cholesterol at week 104) were used to inform the accompanying pharmacoeconomic analysis and are presented in Table 29 in Appendix 1.
Harms Results
Harms results are based on the in-trial observation period with a data cut-off date of July 18, 2023, and are presented in Table 2.
Adverse Events
As described in the Clinical Study Report, nonserious adverse events (AEs) not fulfilling any of the prespecified criteria were not systematically collected. The focus of the safety evaluation per the Clinical Study Report for the SELECT trial was based on the reporting of serious adverse events (SAEs) and other systematically collected events (i.e., AEs of special interest).
Serious AEs
The proportion of patients with an SAE was 33.41% (2,941 of 8,803 patients) in the semaglutide 2.4 mg group and 36.40% (3,204 of 8,801 patients) in the placebo group. The most frequently reported SAEs (by preferred term) (frequency ≥ 2.0%) were coronary arterial stent insertion, acute MI, and unstable angina.
Withdrawals Due to AEs
The proportion of patients who permanently stopped treatment due to AEs was 16.60% (1,461 patients) in the semaglutide 2.4 mg group and 8.16% (718 patients) in the placebo group. The most frequently reported AEs (by preferred term) that led to permanent treatment discontinuation (frequency ≥ 2.0%) were nausea and diarrhea.
The proportion of patients who had their treatment interrupted or dose withdrawn due to AEs was 30.32% (2,669 patients) in the semaglutide 2.4 mg group and 16.00% (1,408 patients) in the placebo group. The most frequently reported AEs (by preferred term) that led to treatment interruption or dose withdrawal (frequency ≥ 2.0%) were nausea, diarrhea, vomiting, constipation, and decreased appetite.
Mortality
There were 375 all-cause deaths (4.26%) in the semaglutide 2.4 mg group and 458 all-cause deaths (5.20%) in the placebo group. There were 371 investigator-reported SAEs with fatal outcome (4.21%) in the semaglutide 2.4 mg group and 460 investigator-reported SAEs with fatal outcome (5.23%) in the placebo group.
Table 2: Summary of Key Results From the In-Trial Observation Period in the SELECT Trial (FAS)
Outcome | Semaglutide 2.4 mg | Placebo |
|---|---|---|
CV outcomes | ||
Time to first EAC-confirmed MACEa | ||
Observation time (patient-years) | 28,655 | 28,297 |
Number of patients with event, n (%) | 569 (6.5) | 701 (8.0) |
CV death and undetermined cause of death | 191 (2.2) | 221 (2.5) |
CV death | 128 (1.5) | 141 (1.6) |
Undetermined cause of death | 63 (0.7) | 80 (0.9) |
Nonfatal acute MI | 230 (2.6) | 321 (3.6) |
Nonfatal stroke | 148 (1.7) | 159 (1.8) |
Non-CV, nonrenal death as competing event, n (%) | 141 (1.6) | 176 (2.0) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.80 (0.72 to 0.89) | |
One-sided P value | < 0.0001 | |
Two-sided P value | < 0.0001 | |
Adjusted HR (95% CI) | 0.80 (0.72 to 0.90) | |
Adjusted 1-sided P value | < 0.0001 | |
Adjusted 2-sided P value | < 0.0001 | |
Time to EAC-confirmed CV deathb | ||
Observation time (patient-years) | 29,283 | 29,112 |
Number of patients with event, n (%) | 223 (2.5) | 262 (3.0) |
CV death | 146 (1.7) | 172 (2.0) |
Undetermined cause of death | 77 (0.9) | 90 (1.0) |
Non-CV, nonrenal death as competing event, n (%) | 152 (1.7) | 196 (2.2) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.85 (0.71 to 1.01) | |
One-sided P valuec | 0.0327 | |
Two-sided P value | 0.0653 | |
Time to first EAC-confirmed composite HF outcomeb | ||
Observation time (patient-years) | 29,165 | 28,944 |
Number of patients with event, n (%) | 300 (3.4) | 361 (4.1) |
CV death and undetermined cause of death | 203 (2.3) | 240 (2.7) |
CV death | 133 (1.5) | 154 (1.7) |
Undetermined cause of death | 70 (0.8) | 86 (1.0) |
HF requiring hospitalization or urgent HF visit | 97 (1.1) | 121 (1.4) |
HF hospitalization | 95 (1.1) | 113 (1.3) |
Urgent HF visit | 2 (< 0.1) | 8 (< 0.1) |
Non-CV, nonrenal death as competing event, n (%) | 144 (1.6) | 190 (2.2) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.82 (0.71 to 0.96) | |
One-sided P valued,e | 0.0066 | |
Two-sided P value | 0.0132 | |
Time to EAC-confirmed all-cause deathb | ||
Observation time (patient-years) | 29,283 | 29,112 |
Number of patients with event, n (%) | 375 (4.3) | 458 (5.2) |
CV death | 146 (1.7) | 172 (2.0) |
Undetermined cause of death | 77 (0.9) | 90 (1.0) |
Non-CV, nonrenal death | 152 (1.7) | 196 (2.2) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.81 (0.71 to 0.93) | |
One-sided P valued,f | 0.0015 | |
Two-sided P value | 0.0029 | |
Time to first EAC-confirmed nonfatal MIb | ||
Observation time (patient-years) | 28,890 | 28,565 |
Number of patients with event, n (%) | 234 (2.7) | 322 (3.7) |
STEMI | 42 (0.5) | 52 (0.6) |
NSTEMI | 139 (1.6) | 192 (2.2) |
Undetermined | 53 (0.6) | 78 (0.9) |
Competing events, n (%) | ||
CV death | 139 (1.6) | 150 (1.7) |
Non-CV, nonrenal death | 146 (1.7) | 178 (2.0) |
Undetermined cause of death | 70 (0.8) | 86 (1.0) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.72 (0.61 to 0.85) | |
Two-sided P valueg | 0.0001 | |
Time to first EAC-confirmed nonfatal strokeb | ||
Observation time (patient-years) | 29,036 | 28,839 |
Number of patients with event, n (%) | 154 (1.7) | 165 (1.9) |
Ischemic | 141 (1.6) | 147 (1.7) |
Hemorrhagic | 10 (0.1) | 14 (0.2) |
Undetermined | 3 (< 0.1) | 4 (< 0.1) |
Competing events, n (%) | ||
CV death | 135 (1.5) | 160 (1.8) |
Non-CV, nonrenal death | 147 (1.7) | 193 (2.2) |
Undetermined cause of death | 69 (0.8) | 84 (1.0) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.93 (0.74 to 1.15) | |
Two-sided P valueg | 0.4985 | |
Glucose metabolism | ||
Time to first occurrence of hemoglobin A1C ≥ 48 mmol/mol (6.5%)h | ||
Number of patients contributing to the analysis, n (%) | 8,800 (99.97) | 8,797 (99.95) |
Observation time (patient-years) | 28,914 | 27,386 |
Number of patients with event, n (%) | 306 (3.5) | 1,059 (12.0) |
Competing events, n (%) | ||
CV death | 146 (1.7) | 154 (1.7) |
Non-CV, nonrenal death | 149 (1.7) | 176 (2.0) |
Undetermined cause of death | 77 (0.9) | 80 (0.9) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.27 (0.24 to 0.31) | |
Two-sided P valueg | < 0.0001 | |
Renal outcomes | ||
Time to first composite nephropathy eventb | ||
Observation time (patient-years) | 28,930 | 28,656 |
Number of patients with event, n (%) | 155 (1.8) | 198 (2.2) |
Persistent macroalbuminuria | 144 (1.6) | 179 (2.0) |
Onset of persistent ≥ 50% reduction in eGFR | 9 (0.1) | 15 (0.2) |
Onset of persistent eGFR < 15 mL/min/1.73 m2 | 1 (< 0.1) | 1 (< 0.1) |
EAC-confirmed initiation of chronic renal replacement therapy | 1 (< 0.1) | 3 (< 0.1) |
EAC-confirmed renal death | Not reported | Not reported |
Competing events, n (%) | ||
CV death | 140 (1.6) | 164 (1.9) |
Non-CV, nonrenal death | 151 (1.7) | 192 (2.2) |
Undetermined cause of death | 73 (0.8) | 84 (1.0) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.78 (0.63 to 0.96) | |
Two-sided P valueg | 0.0191 | |
Anthropometric outcomes | ||
Change from baseline in body weight at week 104i | ||
Body weight at baseline (kg), mean (SD) | 96.53 (17.52) | 96.82 (17.80) |
Body weight at week 104 (kg), mean (SE) | 87.56 (0.08) | 95.79 (0.08) |
Missing, n (%) | ████████ | ████████ |
Number of patients contributing to the analysis, n (%) | 8,605 (97.8) | 8,574 (97.4) |
Change from baseline (%), mean (SE) | −9.39 (0.09) | −0.88 (0.08) |
Treatment group difference vs. placebo (95% CI) | −8.51 (−8.75 to −8.27) | |
Two-sided P valueg | < 0.0001 | |
Health-related quality of life | ||
Change from baseline in EQ-5D index score at week 104i | ||
EQ-5D index score at baseline, mean (SD) | 0.88 (0.15) | 0.88 (0.15) |
EQ-5D index score at week 104, mean (SE) | 0.89 (0) | 0.87 (0) |
Missing, n (%) | ███████████ | ███████████ |
Number of patients contributing to the analysis, n (%) | 8,323 (94.5) | 8,309 (94.4) |
Change from baseline, mean (SE) | 0.01 (0) | −0.01 (0) |
Treatment group difference vs. placebo (95% CI) | 0.01 (0.01 to 0.02) | |
Two-sided P valueg | < 0.0001 | |
Change from baseline in EQ VAS score at week 104i | ||
EQ VAS score at baseline, mean (SD) | 77.15 (15.63) | 77.15 (15.63) |
EQ VAS score at week 104, mean (SE) | 79.83 (0.16) | 78.23 (0.16) |
Missing, n (%) | ███████████ | ███████████ |
Number of patients contributing to the analysis, n (%) | 8,323 (94.5) | 8,309 (94.4) |
Change from baseline, mean (SE) | 2.52 (0.16) | 0.92 (0.16) |
Treatment group difference vs. placebo (95% CI) | 1.60 (1.16 to 2.04) | |
Two-sided P valueg | < 0.0001 | |
Symptom burden | ||
Change from baseline in WRSSM total score at week 104j | ||
WRSSM total score at baseline, mean (SD) | 1.12 (0.77) | 1.13 (0.78) |
WRSSM total score at week 104, mean (SD) | 0.84 (0.73) | 0.98 (0.77) |
Missing, n (%) | ███████████ | ███████████ |
Number of patients contributing to the summary statistic, n (%) | 6,725 (76.4) | 6,635 (75.4) |
Change from baseline, mean (SD) | −0.26 (0.71) | −0.12 (0.68) |
Harmsk | ||
Patients with ≥ 1 SAE, n (%) | 2,941 (33.41) | 3,204 (36.40) |
Cardiac disorders | 1,008 (11.45) | 1,184 (13.45) |
Patients who permanently stopped treatment, n (%) | 1,461 (16.60) | 718 (8.16) |
Gastrointestinal disorders | 880 (10.00) | 172 (1.95) |
Patients with dose interruption or dose withdrawn, n (%) | 2,669 (30.32) | 1,408 (16.00) |
Gastrointestinal disorders | 1,613 (18.32) | 323 (3.67) |
All-cause death, n (%) | 375 (4.26) | 458 (5.20) |
Investigator-reported SAE with fatal outcome, n (%) | 371 (4.21) | 460 (5.23) |
Patients with AEs of special interest, n (%) | ||
SAEs of cardiac disordersl | 1,008 (11.45) | 1,184 (13.45) |
SAEs of gallbladder disorders | ||
Cholelithiasis | 44 (0.50) | 31 (0.35) |
Gallbladder-related disorders (predefined MedDRA search)m | ||
Cholelithiasis | 123 (1.40) | 100 (1.14) |
SAEs of gastrointestinal disorders (predefined MedDRA search)n | ||
Vomiting | 18 (0.20) | 12 (0.14) |
Nausea | 12 (0.14) | 8 (0.09) |
Gastroesophageal reflux disease | 12 (0.14) | 13 (0.15) |
Constipation | 8 (0.09) | 6 (0.07) |
SAEs of exocrine pancreas conditions | ||
Acute pancreatitis | 9 (0.10) | 14 (0.16) |
Pancreatitis | 5 (0.06) | 3 (0.03) |
Obstructive pancreatitis | 4 (0.05) | 7 (0.08) |
Edematous pancreatitis | 1 (0.01) | 1 (0.01) |
Relapsing pancreatitis | 0 | 1 (0.01) |
Pancreatitis (predefined MedDRA search)o | ||
Acute pancreatitis | 12 (0.14) | 15 (0.17) |
Pancreatitis | 7 (0.08) | 6 (0.07) |
Obstructive pancreatitis | 5 (0.06) | 7 (0.08) |
Chronic pancreatitis | 3 (0.03) | 1 (0.01) |
Edematous pancreatitis | 1 (0.01) | 1 (0.01) |
Relapsing pancreatitis | 0 | 1 (0.01) |
EAC-confirmed events of acute pancreatitisp | 17 (0.19) | 24 (0.27) |
AE = adverse event; ANCOVA = analysis of covariance; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; CV = cardiovascular; EAC = event adjudication committee; eGFR = estimated glomerular filtration rate; FAS = full analysis set; HF = heart failure; HR = hazard ratio; MACE = major adverse cardiovascular event; MedDRA = Medical Dictionary for Regulatory Activities; MI = myocardial infarction; NSTEMI = non–ST elevation myocardial infarction; SAE = serious adverse event; SD = standard deviation; SE = standard error; STEMI = ST elevation myocardial infarction; VAS = visual analogue scale; vs. = versus; WRSSM = weight-related sign and symptom measure.
Note: Data cut-off date was July 18, 2023.
aAnalyzed using a Cox proportional hazards model with treatment as categorical fixed factor. The assumption of proportional hazards was evaluated by the standardized score process and was considered supported, per investigator. Patients without events of interest were censored at the end of their in-trial period. Based on the available number of events for analysis, the nominal significance level was updated to 0.02281 using the Lan-DeMets alpha spending function. Adjustment for group sequential design was done using likelihood ratio ordering.
bAnalyzed using a Cox proportional hazards model with treatment as categorical fixed factor. Patients without events of interest were censored at the end of their in-trial observation period.
cBased on the available number of events for analysis, the nominal significance level was updated to 0.01148 using the alpha spending function described in the statistical analysis plan.
dThe superiority of semaglutide 2.4 mg vs. placebo was confirmed for the primary end point of time to first EAC-confirmed MACE comprising CV death, nonfatal MI, and nonfatal stroke. Superiority of semaglutide 2.4 mg vs. placebo was not confirmed for the confirmatory secondary end point of time to EAC-confirmed CV death. As such, the superiority of semaglutide 2.4 mg vs. placebo was not tested for the confirmatory secondary end points that followed the order of the prespecified testing hierarchy (time to first EAC-confirmed composite HF outcome and time to EAC-confirmed all-cause death).
eBased on the available number of events for analysis, the nominal significance level was updated to 0.01149 using the alpha spending function described in the statistical analysis plan.
fBased on the available number of events for analysis, the nominal significance level was updated to 0.01213 using the alpha spending function described in the statistical analysis plan.
gNot controlled for multiplicity.
hAnalyzed using a Cox proportional hazards model with treatment as categorical fixed factor. Patients without events of interest were censored at the end of their in-trial observation period. Patients randomized in error with a baseline hemoglobin A1C of ≥ 48 mmol/mol (6.5%) were excluded from this analysis.
iAnalyzed using an ANCOVA with treatment as fixed factor and baseline value as covariate. Before analysis, missing data were multiple imputed. The imputation model (linear regression) was done separately for each treatment group and included baseline value as a covariate and was fitted to all patients with a measurement regardless of treatment adherence at week 104. The fitted model was used to impute values for patients without a measurement at week 104. Mean estimates were adjusted according to observed baseline distribution.
jPresented descriptively only.
kThe most frequently reported AEs by system organ class are summarized in the table, except for all-cause death and AEs of special interest.
lCV AEs were collected systematically when serious. In addition, potential events of acute coronary syndrome, stroke, coronary artery revascularization, and HF for adjudication by the EAC were reported, irrespective of seriousness. A broad evaluation of CV safety, based on SAEs in the system organ class of cardiac disorders was performed. The evaluation of CV safety was supplemented by the assessment of blood pressure and heart rate.
mAll AEs of gallbladder disease were collected systematically, irrespective of seriousness. Gallbladder-related disorders were evaluated based on a predefined MedDRA search on all AEs, supplemented with additional information collected from specific forms for recording events of gallbladder disease.
nGastrointestinal AEs were collected systematically when serious. Gastrointestinal safety was evaluated based on SAEs in the system organ class of gastrointestinal disorders.
oAll AEs of pancreatitis were collected systematically, irrespective of seriousness. The evaluation of pancreatitis was based on a predefined MedDRA search for pancreatitis among all AEs, which provided a broad evaluation of all types of investigator-reported pancreatitis, including search terms indicative of acute or chronic pancreatitis; additional information collected from a specific form for recording events of pancreatitis; and the outcome of the adjudication by the EAC, where the evaluation focused on events of acute pancreatitis.
pA total of 69 patients (35 in the semaglutide 2.4 mg group and 34 in the placebo group) had a medical history of acute pancreatitis at screening. None of the patients with a history of acute pancreatitis had EAC-confirmed events of acute pancreatitis during the in-trial period.
Sources: Clinical Study Report version 1.0 for the SELECT trial25 and the Novo Nordisk Canada Inc. responses to the January 15, 2025,26 and January 23, 2025,27 requests by CDA-AMC for additional information for the CDA-AMC review of Wegovy. Details included in the table are from the sponsor’s summary of clinical evidence.2
Notable Harms
The AEs of special interest identified for this review included CV AEs, cholelithiasis, nausea, vomiting, constipation, GERD, and pancreatitis. SAEs of cardiac disorders were reported in 11.45% (1,008 patients) in the semaglutide 2.4 mg group and 13.45% (1,184 patients) in the placebo group. All other AEs of special interest were reported in less than 2% of patients in each group.
Critical Appraisal
Internal Validity
The SELECT trial had a group sequential design, a type of adaptive clinical trial design that plans for interim analyses at predetermined points during follow-up and as data are collected in the trial.28 Unlike traditional, fixed-period RCTs, this design offers structured opportunities to make earlier decisions when the intervention shows a significant benefit and improves efficiency in research by allowing adjustments to be made in the trial design. However, this design requires strong and appropriately conservative statistical plans due to the potential for unblinding, investigator influence, overestimation of treatment effect, and an increased chance of inflated type I error.28-30 As will be described, the SELECT trial’s design, conduct, and analysis mitigate some of these potential limitations.
Overall, the baseline characteristics were similar between groups, suggesting randomization successfully distributed potential confounders. In consultation with the clinical experts, the baseline characteristics did not reflect any notable imbalances in the known prognostic factors for CV events and treatment-effect modifiers. As such, the review team concluded that the methods used to achieve allocation and allocation concealment for randomization were appropriate and that the risk of bias arising from the randomization process was low.
The percentage of patients who discontinued the study was relatively small and similar between the groups (approximately 3%). The reasons for discontinuing the study (patient withdrawal and lost to follow-up) were also similar between the groups and, therefore, the risk of attrition bias was likely low. The overall treatment discontinuation rates were similar between groups (approximately 30%). An imbalance in the discontinuation of treatment due to AEs was observed (16.3% in the semaglutide 2.4 mg group and 7.9% in the placebo group). This imbalance could have contributed to differential unblinding because those participants with treatment-emergent AEs — many of which were gastrointestinal in nature — might have inferred their assignment to semaglutide. Similarly, investigators might have formed expectations based on these observations, thereby introducing the potential for performance and reporting bias. While the observed difference between groups in treatment discontinuation due to lack of effect was relatively smaller, it was still differential (0.7% in the semaglutide 2.4 mg group and 2.8% in the placebo group) and, therefore, does not completely negate the potential impact on reporting and performance biases (i.e., relatively less concerning for biases). However, appropriate methods for adjudicating events in a robust and standardized way were employed and safeguards to prevent operational bias (in the context of a group sequential design) were reportedly in place; therefore, the review team concluded the probability for introducing bias into the results was low.
Supplementary analyses of absolute risk difference were conducted at week 156 of the trial to assess the robustness of various end points. A total of 27.3% of patients who were randomized in the trial did not have a visit at week 156 due to trial closure. The trial was event driven and would not be completed until the target number of primary end point events was reached. While the general assumption for censored observations was that the risk of experiencing an event was not changed by censoring, the potential for bias in the estimated cumulative incidence at week 156 would arise only if those patients who were censored due to what was termed “trial closure” in the Clinical Study Report had a different risk of MACE compared with those who remained in the study. However, the definition of trial closure in the Clinical Study Report is not entirely clear. Administrative censoring included trial completion, lost to follow-up, and patient withdrawal. The implications of administrative censoring for the robustness of absolute risk difference in MACE occurrences are difficult to determine based on the available information. The review team noted that the between-group distribution in administrative censoring, including “trial closure,” was similar and, therefore, if bias exists, it is not likely an important factor in the cumulative incidence estimates. Further, a total of 79.4% of patients who were randomized in the trial did not have a visit at week 208 due to trial closure. The review team considered this large percentage of missing data to seriously undermine the validity and reliability of the percent change from baseline in body weight at week 208 reported by Ryan et al.24 There is a high chance that missing information was due to informative censoring and, in the absence of additional analyses evaluating the missing data patterns, it is not possible to draw firm conclusions on the results from the analyses on body weight at week 208.
The use of a single interim analysis in the SELECT trial reduces the risk of bias compared with multiple interim tests in a trial that uses a group sequential design. For the primary end point, the final analysis was adjusted for the group sequential design by using likelihood ratio ordering. The similarity between the unadjusted and adjusted estimates suggested that the single interim analysis had little influence on the final effect size and the reported treatment effect was unlikely influenced by type I error, which is associated with the group sequential design. Further, given the trial continued to its final analysis without stopping for efficacy, the concern for treatment-effect inflation due to early termination does not apply.
The proportional hazards assumption for the primary end point, which the investigators reported as being met, was evaluated graphically using Schoenfeld residuals and the standardized score process. However, an early deviation in the slope suggested a violation of the proportional hazards assumption in the first 12 months, although the stabilization afterward indicated the issue was not persistent over the full duration of the study. The statistical analysis plan for the SELECT trial did not describe methods for assessing the potential impact of this violation on the results. While a visual inspection of the score process suggested a time-dependent effect on the proportional hazards, there was no clear evidence that this influenced the confirmatory results in a meaningful way.
The superiority hypothesis was tested for each confirmatory secondary end point under multiplicity control using a stagewise hierarchical testing scheme according to the prespecified order. The statistical testing strategy for the confirmatory secondary end points used a separate alpha spending function to control the type I error rate at a 1-sided level of 2.5%, which aligned with the P value adjustment for the group sequential design. Importantly, the end points — the composite HF outcome and all-cause death — could not be interpreted formally for superiority because the prespecified hierarchical testing procedure failed to reach statistical significance with the analysis of CV deaths, meaning they should be interpreted as exploratory rather than confirmatory. The SELECT trial performed statistical comparisons for the nonconfirmatory secondary end points and some exploratory end points. However, the statistical comparisons were not included in the approach to adjust for multiple comparisons and, therefore, they increase the risk of type I error. The absence of a prespecified multiplicity control strategy for these end points limits the strength of the conclusions that can be drawn from the observed differences between groups.
The number of events contributing to the subgroup analyses of patients with a BMI of 40 kg/m2 and greater was considered few (< 50 events in each of the 2 treatment groups), and the sample sizes for these 2 subgroups were considered small (< 10% of patients randomized in each group), thereby lowering the certainty in the consistency of the treatment effect for the primary end point in these subgroups.
The main analysis of the time-to-event end points assumed independent censoring of patients who had withdrawn from the trial or were lost to follow-up, while deaths from causes not included in the end point were handled as censored observations but not part of the independent censoring assumption. Sensitivity analyses (addressing the independent censoring assumption and assessing the potential impact of missing data for patients who had withdrawn from the trial or were lost to follow-up) and a supplementary analysis (assessing the influence of the competing risk of death from causes not included in the end point) were performed for the confirmatory end points. The results of the sensitivity analyses were consistent with the main analyses; the review team judged that the risk of bias due to the handling of these intercurrent events and missing outcomes data for the confirmatory end points was low. Of note, the tipping-point sensitivity analysis was conducted only for the primary analysis and not for the key secondary confirmatory analyses. The review team had concerns about the 2 additional sensitivity analyses that were described as applying to patients who withdrew consent or were lost to follow-up; however, these sensitivity analyses yielded results similar to those of the primary analysis (which was further supported by the tipping-point sensitivity analysis) and other confirmatory analyses, which suggests that the assumption of independent censoring was reasonable and that the trial’s conclusions are likely robust.
For all objectives, the primary estimand (intention to treat) was used to evaluate the treatment effect irrespective of adherence to treatment or changes to background medication. Although this approach to the handling of intercurrent events may be reflective of clinical practice, it can be a limitation for the interpretation of efficacy results, given that these intercurrent events also have an effect on CV risk. Further, the proportion of patients who discontinued treatment was relatively high (approximately 30%). A supplementary analysis based on the first on-treatment period was performed for the confirmatory end points to evaluate the effect in all patients who had remained on their randomized treatment. Because the results were consistent with the main analysis, the review team judged that the risk of bias due to treatment discontinuation was low for the confirmatory end points. The proportion of patients initiating a CV-related medication, an SGLT2 inhibitor, or metformin after randomization was, overall, slightly higher with placebo compared with semaglutide 2.4 mg. In consultation with the clinical experts, it was concluded that there is a potential for bias against semaglutide 2.4 mg in the assessment of CV outcomes due to this slight imbalance in the prescribing of these medications.
Continuous supportive secondary end points at week 104 were analyzed, with multiple imputation used for missing values under a missing-at-random assumption, which was likely not plausible because there were notable imbalances in the reasons for treatment discontinuation between groups. A sensitivity analysis that was planned to address this assumption for missing data were performed for the change from baseline in body weight at week 104. Because the results were consistent with the main analysis, the review team judged that the risk of bias due to missing outcomes data for this end point was low. No sensitivity analysis was planned for change from baseline in the EQ-5D, where missing outcome data were relatively high (approximately 16%) at week 104. As such, the review team concluded there is a potential for risk of bias due to missing outcomes data for these HRQoL end points.
WRSSM was an exploratory end point because the measure was not sensitive to weight loss in a psychometric evaluation, per the Clinical Study Report of the SELECT trial. The sponsor indicated that the WRSSM tool was under development at the time of the trial and, as such, no further evidence of its measurement properties in the population of interest was submitted by the sponsor.
External Validity
Evidence from the SELECT trial addressed the evidence gap on the effects of semaglutide 2.4 mg on CV outcomes, which was raised in the previous submission for the weight management indication, but only for a subset of patients in the indicated population.
There was overlap between the SELECT trial population and the population criteria specified by the sponsor in its reimbursement request: patients with a BMI of 27 kg/m2 or greater and established CV disease (MI, stroke, and/or PAD). The clinical experts consulted for this review agreed that the inclusion criteria captured the target population with established CV disease (including HF) that is seen in practice and is in need of an intervention for the secondary prevention of future CV events. Notably, most patients in the trial had prediabetes, according to the hemoglobin A1C range defined in the American Diabetes Association Standards of Medical Care in Diabetes published in 2018 (i.e., 5.7% to < 6.5%).23 However, it is important to note that the Diabetes Canada Clinical Practice Guidelines31 specify a slightly different hemoglobin A1C range (6.0% to < 6.5%) for diagnosing prediabetes. In consultation with the clinical experts, no major concern with the generalizability of the results due to this difference was identified.
Because treatment with semaglutide 2.4 mg in practice is not expected to be limited to patients who do not have diabetes, the review team found the trial’s exclusion of patients with a hemoglobin A1C level of 6.5% or greater to be a concern for the generalizability of the results to the target population. The investigator indicated that the trial population did not include patients with type 1 or type 2 diabetes, to remove any confounding that a diagnosis of diabetes could have on future CV risk. For more information on this evidence gap and how evidence from the SUSTAIN-6 trial addressed that gap, refer to the study addressing the gap in the systematic review evidence section in this document.
In consultation with the clinical experts, it was concluded that the comparison with placebo added to the standard of care for CV disease (i.e., antihypertensives, lipid-lowering drugs, anticoagulants, and ASA and other antiplatelet drugs) was appropriate, given that none of the current and accessible standard-of-care therapies to treat or prevent CV disease directly target weight loss. Regarding the standard of care for CV disease used in the trial, the clinical experts advised that, in practice, semaglutide would be combined with therapies that reduce cardiorenal risk, including combination treatment with ACE inhibitors, angiotensin 2 receptor blockers, statins, and SGLT2 inhibitors.
Long-Term Extension Study
The sponsor did not submit a long-term extension study for this review.
Indirect Evidence
The sponsor did not submit indirect evidence for this review.
Study Addressing a Gap in the Evidence From the Systematic Review
Study Description
Data from the SUSTAIN-6 trial (N = 3,297) was submitted to address the effects of semaglutide on CV outcomes in the target reimbursement population (patients with a BMI of 27 kg/m2 or greater and established CV disease) who also have type 2 diabetes. SUSTAIN-6 (N = 3,297) was a long-term, multicentre, multinational, randomized, double-blind, parallel-group, controlled trial that evaluated the CV safety and long-term outcomes of semaglutide compared with placebo, when added to the standard of care, in patients with type 2 diabetes at high risk of CV events. Patients were randomized 1:1:1:1 to receive a once-weekly subcutaneous injection of either semaglutide 0.5 mg, semaglutide 1.0 mg, or volume-matched placebo. Treatment duration spanned 104 weeks and consisted of a 4-week to 8-week dose-escalation period followed by a 96-week to 100-week treatment maintenance period, and then a 5-week follow-up period.
From the SUSTAIN-6 trial data, a post hoc subgroup analysis was conducted that included patients with a BMI of 27 kg/m2 or greater and established CV disease and type 2 diabetes. Post hoc subgroup analyses were conducted to determine whether the efficacy observed in this patient population was consistent with the SELECT trial and, therefore, the enrolment criteria for this subgroup replicated that of the SELECT study (i.e., limiting the subpopulation to patients with prior MI, stroke [excluding transient ischemic attack; TIA], and/or PAD and a BMI of ≥ 27 kg/m2). Consistent with the exclusion criteria for all SUSTAIN-6 trial participants, patients in the post hoc subgroup analyses did not have type 1 diabetes and had not used a GLP-1 RA or pramlintide within 90 days before screening. Although there were 2 maintenance doses used in the overall SUSTAIN-6 study (0.5 mg and 1.0 mg), only patients who had received 1.0 mg were included in the post hoc subgroup analyses because this dose was considered most comparable to that of the SELECT trial. However, the SUSTAIN-6 trial’s post hoc subgroup had still received a lower dose of semaglutide (1.0 mg) compared with participants in the SELECT trial (2.4 mg). Because the post hoc subgroup analyses were the most relevant for the purposes of this review, the results summarized in this section include only those pertaining to this subgroup.
The CV-related efficacy outcomes investigated in the SUSTAIN-6 study were consistent with those of the SELECT trial. The primary end point was time from randomization to the first occurrence of MACE, defined as CV death, nonfatal MI, or nonfatal stroke. The secondary end points identified by the clinical experts as most relevant to this review were time from randomization to first occurrence of all-cause death, nonfatal MI, or nonfatal stroke.
Demographics and Baseline Characteristics
Patients in the SUSTAIN-6 trial’s post hoc subgroup analyses had a mean age of ███████ (SD = ███), a mean type 2 diabetes disease duration of ██ years (SD = ███), and a mean BMI of 34 kg/m2 (SD = ███). Most patients across both groups had triglyceride levels of 150 mg/dL or greater (███), with a mean triglyceride level of █████ mg/dL (SD = █████). The eGFR of patients across both groups were primarily between 60 mL/min/1.73 m2 to 90 mL/min/1.73 m2 (███), with a mean eGFR of 77.8 mL/min/1.73 m2 (SD = ████). Most patients were taking antihypertensive medication (█████) and/or lipid-lowering drugs (█████). For CV history, only data for prior HF were provided, and ██ (█████) of patients in the semaglutide group had prior HF compared with ███ (███) of the placebo group. Data for other CV inclusion criteria (MI, stroke, and PAD) were not provided.
Results
The outcomes reported for the post hoc subgroup efficacy analyses align with the key outcomes identified for this review. Of all efficacy outcomes included in the post hoc subgroup analysis, only the key secondary outcome of time from randomization to first occurrence of an expanded composite CV outcome, defined as either MACE, revascularization (coronary or peripheral), unstable angina requiring hospitalization, or hospitalization for HF, demonstrated nominal statistical significance; however, this outcome was not among those of interest for the purposes of this review. The remaining CV-related outcomes demonstrated numerical improvement in the semaglutide group compared with the placebo group; however, they did not achieve statistical significance. The overall safety profile of semaglutide observed in the post hoc subgroup of the SUSTAIN-6 trial was consistent with that of the SELECT trial, with fewer CV SAEs with semaglutide 1.0 mg versus placebo.
Critical Appraisal
The post hoc nature of the subgroup analysis in the SUSTAIN-6 trial introduces important internal validity concerns and risk of bias. Because the subgroup was not prespecified, the analysis is exploratory rather than confirmatory, increasing the risk of spurious findings due to multiple comparisons. Additionally, patients in the SUSTAIN-6 study were not randomized within the subgroup of interest, meaning that unmeasured confounders could bias the treatment-effect estimate. Among all CV-related end points, only expanded MACE reached nominal significance, which may have been due to inadequate power. As a result, it is challenging to interpret the efficacy of semaglutide 1.0 mg in this patient subgroup.
The small sample size of the post hoc subgroup limits the generalizability of the findings, given that 80% to 90% of individuals in Canada with type 2 diabetes with overweight or obesity and 32% of people worldwide have CV disease. The high prevalence of potentially eligible individuals in Canada, combined with significant heterogeneity in CV disease presentation, driven by factors like age, diabetes duration, glycemic control, and comorbidities, limits the generalizability of the findings. A major limitation of the SUSTAIN-6 trial in the context of the current resubmission is that the dosages evaluated in the trial, specifically, semaglutide 0.5 mg and 1.0 mg once weekly, do not align with the proposed Health Canada–recommended therapeutic and maintenance dosage of semaglutide (Wegovy), which is 2.4 mg once weekly. The focus of the post hoc subgroup of the SUSTAIN-6 study was on the 1.0 mg dose because, according to the sponsor, it was most comparable to the dose in the SELECT trial; however, this remains a significant limitation to the generalizability of the evidence.
Conclusion
The results from the SELECT trial were submitted as new evidence for this resubmission to address the evidence gap on the effects of semaglutide 2.4 mg once weekly on weight-related comorbidities, such as CV outcomes, in the indicated weight management population. The SELECT trial demonstrated that semaglutide 2.4 mg once weekly, as an adjunct to the standard of care for CV disease, reduced the occurrence of MACE compared with placebo over a mean follow-up period of nearly 40 months in patients with established CV disease and overweight (BMI ≥ 27 kg/m2) or obesity (BMI ≥ 30 kg/m2) but without diabetes. The point estimates of the observed treatment effects for the individual MACE components — CV death, nonfatal MI, or nonfatal stroke — were directionally aligned with the overall reduction in MACE occurrences, although the certainty of the estimated effects varied across components. More specifically, the CIs for CV death and, notably, nonfatal stroke, included the possibility of no benefit with respect to these efficacy outcomes. Additionally, the trial suggested a reduction in the composite HF events and all-cause mortality with semaglutide 2.4 mg compared with placebo; however, these end points could not be interpreted formally for superiority because the prespecified hierarchical testing procedure failed to reach statistical significance.
No new harms with semaglutide 2.4 mg were identified within the SELECT trial. The harms that were observed were consistent with what has been observed clinically, as per the input from the clinical experts consulted for this review regarding experience with semaglutide in clinical practice, as well as what has been observed in previous studies. The AEs leading to the permanent or temporary discontinuation of treatment were notable and reported more frequently with semaglutide 2.4 mg compared with placebo. Throughout the trial and, in particular, during the dose-escalation phase, the most frequently reported AEs were gastrointestinal-related, suggesting that tolerability at the recommended maintenance dose may be an issue. The AEs of special interest for this review (cholelithiasis, nausea, vomiting, constipation, GERD, and pancreatitis) were reported infrequently.
Introduction
The objective of this report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of semaglutide 2.4 mg solution for subcutaneous injection in chronic weight management in patients with a BMI of 27 kg/m2 or greater and established CV disease (MI, stroke, or PAD).
Submission History
Semaglutide 2.4 mg was first reviewed by CDEC for weight management in 2022. CDEC issued a recommendation that semaglutide 2.4 mg not be listed as an adjunct to a reduced calorie diet and increased physical activity for chronic weight management in adult patients with an initial BMI of 30 kg/m2 or greater (obesity) or 27 kg/m2 or greater (overweight) in the presence of at least 1 weight-related comorbidity such as hypertension, type 2 diabetes mellitus, dyslipidemia, or obstructive sleep apnea.3
The 2022 reimbursement recommendation for semaglutide (Wegovy) and the Clinical Review report on semaglutide (Wegovy) that was used to inform that recommendation are both available on the CDA-AMC project website.
Basis of Resubmission
Rationale for the 2022 recommendation included evidence from 4 placebo-controlled, double-blind RCTs (STEP 1, STEP 2, STEP 3, and STEP 4) that demonstrated that treatment with semaglutide 2.4 mg injection reduced body weight in individuals with an initial BMI of 30 kg/m2 or greater (obesity) or 27 kg/m2 or greater (overweight) in the presence of at least 1 weight-related comorbidity but did not demonstrate improvement in or prevention of weight-related comorbidities. Comorbidities such as MACE, osteoarthritis, and obstructive sleep apnea were not outcomes assessed in the STEP trials.3
CDEC noted there was an ongoing trial, the SELECT study,4 comparing semaglutide 2.4 mg injection with placebo for the prevention of MACE occurrences in patients with overweight or obesity who have established CV disease but not diabetes mellitus. CDEC concluded in 2022 that the results of that study would address the evidence gap regarding the effects of semaglutide 2.4 mg once weekly on CV outcomes in the indicated population.3
Hence the focus of this resubmission for the weight management indication was on the sponsor’s revised reimbursement request: as an adjunct to a reduced calorie diet and increased physical activity for chronic weight management in adult patients with a BMI of 27 kg/m2 or greater and established CV disease (MI, stroke, or PAD).
Sponsor’s Clarifying Note on Resubmission
In 2024, Health Canada issued an indication for semaglutide injection (Wegovy) to reduce the risk of nonfatal MI in adults with established CV disease and a BMI of 27 kg/m2 or greater. This indication was based on the evidence from the SELECT trial.
The sponsor indicated that the revised reimbursement request population, namely, adults with established CV disease and a BMI of 27 kg/m2 or greater, is fully encompassed in the weight management indication and is the same population for which the CV indication was granted.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Obesity is a complex chronic disease where adiposity, or excess body fat, impairs health and increases the risk of long-term health complications and comorbidities.5 The presence of overweight (a BMI of greater than 25 kg/m2) and obesity (a BMI of greater than 30 kg/m2) are considered major contributors to CV disease through both direct and indirect mechanisms. Obesity can directly result in changes to cardiac loading, volume status, energy substrate use, tissue metabolism, and systemic inflammation, which are believed to promote CV disease progression.6 Additionally, obesity may contribute to CV disease indirectly through the development of exacerbating conditions that carry high CV mortality risk (e.g., sleep apnea, thromboembolic disease, or MI) and cardiometabolic diseases (e.g., hypertension, dyslipidemia, type 2 diabetes, nonalcoholic fatty liver disease, or steatohepatitis).7-15 Obesity is also associated with an increased risk of a wide range of illnesses and long-term conditions, including type 2 diabetes, hypertension, gallstones, GERD, and cancer, as well as psychological and psychiatric morbidities.32 Individuals with obesity have been shown to have a higher risk of anxiety, psychosis spectrum disorders, eating disorders, and mood disorders, such as major depressive disorder, bipolar disorder, and dysthymia.33-36 Psychiatric morbidities can be compounded by social stigma and discrimination toward individuals with obesity, which can result in social isolation and avoidance of seeking medical care.37 Quality of life is further impacted by difficulty performing everyday activities, mobility challenges, physical discomfort, and pain.38
BMI is the primary measure used to identify patients who may benefit from obesity management and is often employed alongside other indicators, such as waist circumference and presence of cardiometabolic complications.39 Additional diagnostic testing, including blood pressure, blood lipid, liver and kidney panels, electrocardiogram and/or echocardiogram, angiography, Doppler ultrasound, or Holter and event monitoring, are broadly available and can be arranged by primary care providers to help identify metabolic problems and tailor therapy.39 Based on epidemiological studies, overweight is currently classified as a BMI of 25 kg/m2 to less than 30 kg/m2 and obesity is classified as a BMI of 30 kg/m2 or greater.5 Obesity can be further subclassified into class I (BMI of 30 kg/m2 to 34.9 kg/m2), class II (BMI of 35 kg/m2 to 39.9 kg/m2), class III (BMI of 40 kg/m2 to 49.9 kg/m2), class IV (BMI of 50 kg/m2 to 59.9 kg/m2), and class V (BMI of greater than 60 kg/m2).40
In 2022, the Canadian Community Health Survey estimated that, in Canada, 30% of individuals aged 18 years or older were living with obesity.16,17 Across Canada, the prevalence was higher than the national average in New Brunswick (43%), Newfoundland and Labrador (42%), Saskatchewan (38%), Prince Edward Island (36%), Nova Scotia (36%), and Manitoba (34%); lower than the national average in British Columbia (26%) and Quebec (29%); and similar in Ontario (30%) and Alberta (31%).16,17 There are limited data on patients in Canada with overweight or obesity and established CV disease. Across CV diseases, the proportion of people with CV disease who are living with obesity tends to be higher than among those with normal weight, and CV disease rates appear to proportionally increase with BMI.41 For example, hypertension prevalence among those with a normal BMI is 45% compared with 67% for those with overweight, 79% for those with obesity class I and II, and up to 87% among those with obesity class III.41 In a study of patients with ASCVD, obesity was among the 30 most common comorbid conditions, with a prevalence of 38%.18 CV disease is among the leading causes of morbidity and mortality worldwide and is reported to be 1 of the leading causes of hospitalization in Canada, alongside stroke.19,20
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Based on clinical expert input, current chronic weight management in patients with a BMI of 27 kg/m2 or greater and established CV disease is informed in part by the Canadian Adult Obesity Clinical Practice Guidelines: Pharmacotherapy for Obesity Management, updated in 2022;21 the Canadian Cardiovascular Society Guideline for Use of GLP-1 Receptor Agonists and SGLT2 Inhibitors for Cardiorenal Risk Reduction in Adults, published in 2022;42 and the European Society of Cardiology Guidelines for the Management of Chronic Coronary Syndromes, published in 2024.43
The Canadian obesity guideline states that there are 4 medications indicated for long-term obesity management as adjuncts to health-behaviour changes: liraglutide, naltrexone-bupropion in a combination tablet, orlistat, and semaglutide. The Canadian guideline recognizes all 4 medications as effective in producing clinically significant weight loss and health benefits relative to placebo over a period of at least 1 year.21 The clinical experts indicated that another medication, tirzepatide, is currently approved only for adjunct glycemic control in type 2 diabetes but is increasingly being prescribed off label for chronic weight management in Canada. Further, patient groups have indicated that semaglutide (Ozempic) has been prescribed off label for weight management as well. The clinical experts indicated that in clinical practice in Canada, semaglutide is being used in patients with diabetes for weight loss.
Based on clinical expert input, the most important goals of therapy for chronic weight management in patients with a BMI of 27 kg/m2 or greater and established CV disease is treatment and prevention of ASCV and HF outcomes (fatal and nonfatal events). Other important goals of therapy include weight loss and improvement in quality of life and symptom burden. Additional goals of therapy include improvement in health parameters such as CV risk factors (e.g., blood pressure, lipid profile, and glycemic status) and mental health.
Drug Under Review
Semaglutide is a selective long-acting GLP-1 RA that is 94% similar to human GLP-1 that binds to and activates GLP-1 receptors in the same manner as the endogenous hormone incretin. GLP-1 is a physiological regulator of appetite and caloric intake and is present in several brain areas involved in appetite regulation. Compared with native GLP-1, semaglutide has a prolonged half-life of approximately 1 week due to albumin binding, which results in decreased renal clearance and protection from metabolic degradation.
Semaglutide is administered subcutaneously in the abdomen, thigh, or upper arm. The therapeutic and maintenance dose of 2.4 mg of semaglutide once weekly is reached by starting with a dose of 0.25 mg and then following a dose-escalation regimen, with dose increases every 4 weeks (to doses of 0.5 mg, 1 mg, 1.7 mg, and 2.4 mg per week) until the therapeutic-maintenance dose of 2.4 mg once weekly is reached after 16 weeks. This dose-escalation regimen is suggested to decrease the likelihood of gastrointestinal symptoms. If patients do not tolerate a dose during dose escalation, prescribers should consider delaying dose escalation for 4 weeks.
Semaglutide (2.4 mg) was approved by Health Canada in November 2021 as an adjunct to a reduced calorie diet and increased physical activity for chronic weight management in adult patients with an initial BMI of 30 kg/m2 or greater (obesity) or 27 kg/m2 or greater (overweight) in the presence of at least 1 weight-related comorbidity such as hypertension, type 2 diabetes mellitus, dyslipidemia, or obstructive sleep apnea. Semaglutide (2.4 mg), which was previously reviewed by CDA-AMC in 2022, received a “do not reimburse” recommendation for the sponsor’s request that semaglutide be reimbursed as an adjunct to a reduced calorie diet and increased physical activity for chronic weight management in adult patients with an initial BMI of 35 kg/m2 or greater and prediabetes.
For the current resubmission review, the sponsor has requested that semaglutide (2.4 mg) be reimbursed as an adjunct to a reduced calorie diet and increased physical activity for chronic weight management in adult patients with an initial BMI of 27 kg/m2 or greater and established CV disease (MI, stroke, or PAD).
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available on the CDA-AMC project website for this review in the consolidated patient and clinician group input document.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
Input for this review was submitted by 6 patient groups: GI Society, Obesity Canada, Obesity Matters, Fatty Liver Alliance, HeartLife Foundation, and Diabetes Canada. GI Society input was based on 2 international surveys for individuals with obesity from 2021 (1,550 individuals who responded, 62% from Canada) and 2024 (1,050 individuals who responded, 96% from Canada), as well as insights from an in-person focus group of individuals with obesity held in 2023. Obesity Canada input was based on 1 Canadian survey (170 individuals who responded) conducted between September and October 2024 of individuals with obesity. Survey responses were primarily from Ontario (42%), British Columbia (17%), and Alberta (13%), with additional representation from Saskatchewan, New Brunswick, Nova Scotia, and Prince Edward Island; most individuals who responded were female (92%) and between the ages of 45 and 64 years (58%). Input from the HeartLife Foundation was gathered through interviews and discussions with individuals with CV disease and their health care providers. The Fatty Liver Alliance gathered physician insights on MASLD and MASH, supplemented by a 2022 to 2023 US survey of 1,518 patients with nonalcoholic fatty liver disease or nonalcoholic steatohepatitis. Input from Diabetes Canada was based on a Canadian survey (October to November 2024) with 170 individuals who responded, including individuals with diabetes (59% with type 2 and 22% with type 1 diabetes, 12% with prediabetes) and their caregivers. Obesity Matters collected input in October 2024 through a Canadian obesity survey (186 individuals who responded) and a patient testimonial. Obesity Matters also provided feedback on the CDEC reimbursement recommendation issued for the initial 2022 review of semaglutide for weight management.
The patient groups highlighted the significant physical and mental health impact of obesity and related conditions, including CV diseases, diabetes, MASLD, and MASH. Common symptoms included mobility limitations, chronic pain, fatigue, swelling, shortness of breath, dizziness, and nausea, affecting daily life. Comorbidities were prevalent: in the GI Society’s 2021 survey, 91% of individuals who responded reported comorbidities such as arthritis (51%), hypertension (33%), sleep apnea (30%), GERD (29%), irritable bowel syndrome (29%), high cholesterol (25%), and diabetes (24%). Input from Diabetes Canada also noted high comorbidity rates for patients with diabetes, including high blood pressure, foot or eye problems, nerve damage, heart disease, and kidney-related comorbidities. Obesity not only contributes to these conditions but also complicates disease management and reduces quality of life. Mental health challenges, including anxiety, mood disorders, and social isolation due to stigma, especially in health care, were widely reported.
The patient groups highlighted weight loss as a key treatment outcome, along with a reduced risk of comorbidities (e.g., CV disease), improvements in quality of life, and increased ability to perform everyday tasks. When choosing a new therapy, most patients primarily value efficacy (i.e., significant and sustained weight loss) and affordability.
The patient groups emphasized that traditional lifestyle modifications, such as diet and exercise, are often ineffective for long-term weight management. Few medication options exist, with semaglutide, liraglutide, and naltrexone-bupropion being commonly prescribed, but none have public or full private coverage, making cost the biggest barrier. Those who responded to the survey and who reported the benefits of these treatments in managing obesity described them as “life-changing,” with most (94%) considering the side effects to be manageable. However, it was also noted that these medications lack long-term effectiveness, with many individuals gaining back the weight that was lost within 5 years. Bariatric surgery, while currently considered the gold standard for obesity treatment, carries risks of severe side effects, postsurgery weight gain, and long wait times, making it a last-resort option.
Patients with experience taking semaglutide described several benefits, including substantial weight loss. It was highlighted that semaglutide also improved the management of comorbidities, such as CV disease, and improved quality of life. Thus, it was identified that semaglutide has the ability to address multiple health conditions simultaneously. Patients with experience with semaglutide also highlighted that its side effects — most commonly nausea, vomiting, and constipation — were typically temporary and manageable, and the once-weekly administration reduced treatment burden.
Obesity Matters provided additional feedback on the 2022 draft of the CDEC reimbursement recommendation for semaglutide (Wegovy). The feedback indicated that while the initial draft recommendation acknowledged the effect of semaglutide on body-weight reduction and the increased risk of comorbidities observed with obesity, it did not conclude that weight reduction can directly reduce comorbidities. The feedback also critiqued the inclusion of the lack of structured weight programs available in Canada as part of the rationale for a negative reimbursement recommendation, noting this overlooks the agency of individuals actively working with specialists to make lifestyle changes. Obesity Matters indicated that the previous recommendation did not adequately consider cost as a barrier to treatment, patient perspectives, or the direct impact of treatment costs on quality of life. The feedback indicated that certain access barriers and implementation issues did not provide necessary guidance, required further elaboration on relevance, and were not specific to semaglutide, noting these challenges are routinely addressed for other treatments. Obesity Matters felt that the recommendation did not adequately consider how public reimbursement would reduce long-term costs associated with obesity-related comorbidities and conditions, nor did it offer actionable solutions for integrating semaglutide into existing health care systems.
Clinician Input
Input From the Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of overweight and obesity and CV disease.
Unmet Needs
The clinical experts identified the following as limitations with weight management medications: barriers to public access, that not all patients experience weight loss with treatment, and that some patients experience adverse effects that necessitate stopping the drug. Specific to bariatric surgery, the clinical experts noted that in addition to barriers to access, not all patients are interested in pursuing this option because of the potential risks associated with surgery.
Place in Therapy
According to the clinical experts consulted for this review, semaglutide 2.4 mg has been used for weight management in Canada since it was approved by Health Canada in 2021. The clinical experts indicated that the anticipated place of semaglutide with regard to pharmacotherapy for chronic weight management is as a first-line treatment. Of note, the clinical experts anticipated that clinicians will increasingly prescribe medications with public reimbursement that specifically target obesity and have evidence for reduced risk of future CV events.
The clinical experts advised that semaglutide would be combined with therapies that reduce cardiorenal risk, including combination treatment with RAAS inhibitors (such as ACE inhibitors and angiotensin 2 receptor blockers) and SGLT2 inhibitor therapies as well as other standards of care to improve CV outcomes. The clinical experts indicated there is currently no high-level evidence to support combination treatment with other pharmacotherapies for weight management; however, they noted there is relevant evidence from small studies (e.g., a retrospective cohort study on the effect of combining a GLP-1 RA with bupropion-naltrexone in weight loss22). The clinical experts indicated that, in practice, specialists in obesity treat patients with combination therapy when needed and appropriate and when cost and insurance coverage are not limiting factors. The clinical experts indicated that the most common combination is a GLP-1 RA combined with bupropion-naltrexone.
The clinical experts advised that semaglutide would not be combined with another GLP-1 RA (such as tirzepatide, which is increasingly being prescribed off label for weight management).
Patient Population
To identify patients who would most likely respond to treatment with semaglutide, the clinical experts deferred to the baseline characteristics of the SELECT trial population in which response was defined as a reduced risk of future CV outcomes and weight loss. Of note, the clinical experts highlighted that the trial population included patients with a pre-existing history of CV disease, and 64.5% of trial participants had prediabetes (hemoglobin A1C > 5.7% per US criteria). The clinical experts advised caution when prescribing semaglutide for patients with nutritional deficiencies or very low appetite at baseline.
The clinical experts noted that the benefits of semaglutide for primary prevention of CV disease are less established and less clear in patients without established CV disease or type 2 diabetes.
Overall, the clinical experts advised that it would be appropriate to treat most patients with a BMI of 27 kg/m2 or greater for the chronic weight management indication, and that patients with established CV disease (including HF), type 2 diabetes, multiple risk factors, severe disease, or a high BMI are most in need of an intervention for the secondary prevention of future events and to improve symptom burden.
The clinical experts indicated that patients with a history of CV disease best suited for treatment with semaglutide can be identified through their medical records, with supporting documentation on their admission and/or procedure, or results from a diagnostic test such as echocardiogram. BMI is typically measured with a scale and self-reported height per clinical expert input. Of note, the clinical experts indicated that CV disease in the presence of overweight and obesity are underdiagnosed in practice if the clinician does not gather a standard history, perform a physical, and document the BMI in the chart.
When considering which patients would be least suitable for treatment with semaglutide, the clinical experts deferred to the product monograph for absolute contraindications and patients with a potential increased risk of adverse effects (e.g., history of pancreatitis, gallbladder disease, diabetic retinopathy, severe renal impairment, or gastrointestinal conditions).
Assessing Response to Treatment
Although weight loss is an outcome that can be easily monitored in practice, monitoring the recurrence of CV events and symptoms is equally as important.
When considering a clinically meaningful response to treatment in a patient-reported outcome, the clinical experts advised on the following:
A weight loss of 5% or more is often considered clinically meaningful.
A change of 5 points or more in the Kansas City Cardiomyopathy Questionnaire is considered clinically meaningful; some studies have estimated a change of 20 points or more to be clinically meaningful for HF.
An increase of at least 30 m in the 6-minute walk distance is commonly considered clinically meaningful for HF.
Discontinuing Treatment
The clinical experts indicated that treatment with semaglutide would be long term, unless the following occurred (factors for consideration in discontinuing treatment with semaglutide):
excessive weight loss or nutritional deficiencies (may also be considered for dose reduction)
renal failure (however, treatment may be continued, depending on overall nutritional status and if tolerating otherwise)
side effects, such as persistent severe nausea, vomiting, or diarrhea
development of a contraindication
pregnant or planning to become pregnant
cost and/or loss of coverage.
The experts indicated that the occurrence of a repeat CV event while receiving treatment with semaglutide would not be considered treatment failure, noting there would be too much uncertainty to determine whether treatment was associated with the delayed onset of a second CV event in an individual. Additionally, the clinical experts advised that pausing treatment for an elective surgery may be considered, given reports that semaglutide is associated with slow gastrointestinal transit, which can increase the risk of aspiration with endotracheal intubation.
Prescribing Considerations
The clinical experts advised that a specialist is not required to prescribe semaglutide because family physicians and primary care providers have been prescribing semaglutide (Ozempic) for diabetes and obesity and are familiar with its side effect profile and monitoring.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
A total of 4 clinician groups provided input for this resubmission: TCR (5 clinicians) and the CRSP at the Western University Division of Cardiology (5 clinicians), plus a joint input from Obesity Canada and the CABPS (total of 18 clinicians). TCR, a multidisciplinary group of family physicians, internists, and cardiologists, is the sole provider of cardiac rehabilitation in the city of Calgary and surrounding area. The Western University CRSP program delivers comprehensive, multidisciplinary rehabilitation care, including secondary prevention through lifestyle and pharmacotherapeutic interventions. The CABPS is a group of specialists in Canada with experience treating obesity. Obesity Canada is a national registered charity that assisted with the coordination of the group clinician response from CABPS.
The clinician group input was largely consistent with that of the clinical experts. Both the clinician groups and experts highlighted the limitations of the currently available weight management treatment options and the barriers to patients accessing them. The CABPS noted that obesity management practice currently follows the 2020 Canadian Adult Obesity Clinical Practice Guidelines, which recommend lifestyle modification supported by psychological or behavioural therapy, pharmacotherapy, and bariatric surgery. The clinician groups further highlighted that lifestyle modifications alone are often insufficient to experience the weight loss needed to improve obesity-related complications such as type 2 diabetes and MASLD. The clinician groups noted that liraglutide, semaglutide, and bupropion-naltrexone are the most effective long-term pharmacologic treatments due to their impact on the neuroendocrine pathways associated with obesity. The clinician groups also outlined the role of medical bariatric centres in providing multidisciplinary care and differentiated them from nonmedical weight loss clinics. The CABPS expressed that obesity treatment should extend beyond weight loss to address dysfunctional adipose tissue driving adverse health outcomes. The clinician groups’ overview of therapies that reduce cardiorenal risk in patients with comorbid CV disease was consistent with the expert input. The clinician groups also added that patients with ASCVD become eligible for cardiac rehabilitation, which combines lifestyle and pharmacotherapeutic interventions.
Both the clinicians and experts agreed that a primary goal of CV disease treatment is to reduce the incidence of CV events, but the clinician groups also suggested additional goals for weight management and improved glycemic control, when applicable. Both the clinicians and experts highlighted the access challenges and potential risks associated with bariatric surgery. Both noted a need for additional treatment options that address both CV risk and weight reduction, noting that existing treatments for weight loss and CV disease can lack long-term effectiveness and fail to directly target weight management. The clinician groups noted this as being particularly true for patients with obesity who do not have diabetes, a group for which there are currently no targeted treatment options. The clinician groups noted that these significant treatment gaps contribute to a persisting high risk of morbidity and mortality in these populations.
In contrast to the clinical expert input, which noted that semaglutide would be combined with other therapies that reduce CV risk (i.e., RAAS inhibitors, SGLT2 inhibitors), the clinician groups indicated that semaglutide would be used for first-line treatment alongside lifestyle modifications. The clinician groups noted it is unnecessary to first try other treatments before semaglutide, given its effectiveness in achieving substantial weight loss and improving obesity-related health outcomes. The clinician groups anticipate that semaglutide could shift current treatment paradigms, noting its efficacy approaches that of bariatric surgery and could reduce the number of patients requiring surgery. The patient type that clinician groups indicated would benefit most from semaglutide aligned exactly with the type indicated by the clinical experts. The clinician groups added that semaglutide might also benefit patients with diabetes who require additional CV protection, or those without diabetes who are at high risk of CV disease and have difficulty experiencing weight loss through lifestyle modifications alone. The CRSP clinicians estimated that 78% of their patients receiving cardiac rehabilitation do not have diabetes and that 45% of patients who complete the rehabilitation program could potentially benefit from an additional therapy such as semaglutide. The patients least suitable for treatment, identified by the clinician groups and experts, also aligned. According to the clinician groups, patients with obesity are identified through self-identification or by health care providers using diagnostic tools such as BMI, waist circumference, and staging systems such as the Edmonton Obesity Staging System, which also classifies the degree of associated conditions (e.g., type 2 diabetes, impaired mobility). However, the CABPS noted there are currently no methods for identifying which patients are likely to respond to semaglutide.
The physician group input was consistent with the expert insights on the use of weight loss to assess treatment response but emphasized additional markers of treatment benefit. The clinician groups highlighted improvements in prediabetes, lipids, blood pressure, mobility, and quality of life as equally important. Conversely, TCR indicated there is no definitive evidence that the absence of weight loss precludes other treatment benefits. The CABPS provided specific criteria for a meaningful treatment response, including a 5% reduction in total body weight after 3 months of treatment (aligning with the expert input) and improved laboratory markers, reduced osteoarthritis pain, and the ability to proceed with procedures like hip replacement. The clinician groups also suggested initial evaluations every 4 to 6 weeks, then every 3 months, recognizing variability based on clinician and patient preference.
Both the clinician groups and experts agreed that semaglutide treatment should be continued indefinitely, with the clinician groups likening the regimen to statin therapy or treatment with ASA after an MI. The clinician groups suggested the discontinuation of semaglutide be considered in the following scenarios: presence of treatment intolerance or intolerable side effects that do not improve over time with appropriate countermeasures, pancreatitis, pregnancy or planning pregnancy, lack of meaningful treatment response, lack of affordability, or a more effective treatment becomes available in the future that requires semaglutide discontinuation. The clinician groups noted that semaglutide treatment could be delivered in family medicine and primary care clinics, community-based obesity management or metabolic medicine clinics, and hospital-based medical and surgical centres. Like the experts, the clinician groups indicated that any health care provider with experience managing obesity could prescribe semaglutide, noting this role should not be restricted to specialists alone.
Drug Program Input
The drug programs provide input on each drug being reviewed through the CDA-AMC reimbursement review processes by identifying issues that may impact their ability to implement a reimbursement recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 3.
Semaglutide 2.4 mg was first reviewed by CDEC for the weight management indication in 2022. The implementation questions and corresponding responses from the clinical experts consulted for the past review were summarized in the 2022 Clinical Review report on semaglutide (Wegovy) used to inform the reimbursement recommendation for semaglutide (Wegovy). Both the Clinical Review report and the recommendation document are available on the project website.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Treatments for chronic weight management (naltrexone-bupropion, liraglutide, and orlistat) are not a benefit under the participating public drug programs. As such, the drug plans did not identify any issue with the comparator (placebo) in the SELECT trial. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
If a patient is unable to tolerate the target maintenance dose at week 16 after initiation of therapy (in reference to the dose-escalation regimen of 16 weeks per the recommended dose in the product monograph), should they be considered for renewal? | The clinical experts advised that treatment with semaglutide should be continued for the potential benefits in CV outcomes despite a patient not tolerating the target maintenance dose. The clinical experts noted that many patients are unable to tolerate the 2.4 mg semaglutide dose but can still derive benefit from the medication at a lower dose. |
Considerations for prescribing of therapy | |
If a patient is unable to tolerate the target maintenance dose, should treatment with semaglutide be discontinued? In other words, would patients receiving a lower maintenance dose equally benefit from semaglutide and be considered for reimbursement? | As previously mentioned, the clinical experts advised that treatment with semaglutide should be continued for the potential benefits in CV outcomes despite not tolerating the target maintenance dose of 2.4 mg. Based on their experience in clinical practice, the clinical experts indicated that patients taking semaglutide 0.25 mg, 0.5 mg, or 1.0 mg weekly have experienced weight loss. Therefore, the clinical experts advised to continue treatment at the maximum tolerated dose, particularly when the patient is deriving benefit in terms of weight loss, appetite reduction, and improved quality of life. The CDA-AMC review team notes that the recommended dose and dosage adjustment outlined in the product monograph for semaglutide (Wegovy) advised that, “If patients do not tolerate the therapeutic/maintenance 2.4 mg dose, the dose can be temporarily decreased to 1.7 mg weekly, for a maximum of 4 weeks. Patients should re-escalate to the therapeutic/maintenance 2.4 mg dose.”1 |
System and economic issues | |
Cost of therapy per year: $5,069.67. Pan-Canadian net budget impact:
3-year total: $380,761,195. | This is a comment from the drug plans to inform CDEC deliberations. |
CDA-AMC = Canada’s Drug Agency; CDEC = Canadian Drug Expert Committee; CV = cardiovascular.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of semaglutide 2.4 mg solution for subcutaneous injection in chronic weight management in patients with a BMI of 27 kg/m2 or greater and established CV disease (MI, stroke, or PAD). The focus will be placed on comparing semaglutide 2.4 mg with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of semaglutide injection is presented in 2 sections, with the review team’s critical appraisal of the evidence included at the end of the section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The second section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 RCT identified in the systematic review
1 additional study addressing a sponsor-identified gap in the evidence.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of the Study
Characteristics of the included study, the SELECT trial, are summarized in Table 4.
The SELECT trial was a phase IIIb, multinational, multicentre, randomized, double-blind, parallel-group, placebo-controlled trial that evaluated the effect of semaglutide 2.4 mg subcutaneous injection administered once weekly compared with placebo, both of which were added to the standard of care, in reducing the risk of MACE occurrences (CV death, nonfatal MI, or nonfatal stroke) in patients with established CV disease and overweight or obesity and without diabetes (N = 17,604). The key secondary objective of the trial was to evaluate the effect of semaglutide in mortality compared with placebo.
Table 4: Details of the Study Included in the Systematic Review
Detail | SELECT study |
|---|---|
Design and population | |
Study design | Phase IIIb, multinational, multicentre, randomized, double-blind, parallel-group, placebo-controlled trial |
Locations | 804 sites in 41 countries and regions (Algeria, Argentina, Australia, Austria, Belgium, Brazil, Bulgaria, Canada, Columbia, Croatia, Czech Republic, Denmark, Finland, France, Germany, Greece, Hungary, India, Ireland, Israel, Italy, Japan, Latvia, Malaysia, Mexico, Netherlands, Norway, Poland, Portugal, Romania, Russian Federation, Serbia, South Africa, Spain, Sweden, Taiwan, Thailand, Türkiye, UK, Ukraine, US) |
Trial initiation and completion dates | Start date: October 24, 2018 End date: June 29, 2023 |
Randomized (N) | N = 17,604:
|
Key inclusion criteria |
|
Key exclusion criteria | CV-related
Glycemia-related
General safety
|
Drugs | |
Intervention | Semaglutide 2.4 mg subcutaneous injection once weekly as adjunct to standard of care for CV disease. Dose escalation was every fourth week until a maintenance dose of 2.4 mg was reached; dose escalations were 0.24 mg, 0.5 mg, 1.0 mg, and 1.7 mg. |
Comparator | Placebo subcutaneous injection once weekly as adjunct to standard of care for CV disease. Dose escalation was every fourth week (same dosing schedule as the intervention). |
Study duration | |
Screening phase | Up to 3 weeks |
Treatment phase | A 16-week dose-escalation period followed by a maintenance period of up to 53 months. This was an event-driven trial; trial closure was planned when 1,225 EAC-confirmed MACEs were reached. |
Follow-up phase | 5 weeks |
Outcomes | |
Primary end point | Time from randomization to first occurrence of MACE,a a composite end point consisting of CV death, nonfatal MI, or nonfatal stroke |
Secondary and exploratory end points | Confirmatory secondary end points
Supportive secondary end points
Exploratory end points
|
Publication status | |
Clinicaltrials.gov entry | NCT03574597 |
Publications | Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N Engl J Med. 2023;389(24):2221 to 2232.4 Ryan DH, Lingvay I, Deanfield J, et al. Long-term weight loss effects of semaglutide in obesity without diabetes in the SELECT trial. Nat Med. 2024;30(7):2049 to 2057.24 Lingvay I, Deanfield J, Kahn SE, et al. Semaglutide and Cardiovascular Outcomes by Baseline HbA1c and Change in HbA1c in People With Overweight or Obesity but Without Diabetes in SELECT. Diabetes Care. 2024;47(8):1360 to 1369.44 Deanfield J, Verma S, Scirica BM, et al. Semaglutide and cardiovascular outcomes in patients with obesity and prevalent heart failure: A prespecified analysis of the SELECT trial. Lancet. 2024;404(10454):773 to 786.45 |
ABI = ankle-brachial index; BMI = body mass index; BP = blood pressure; CV = cardiovascular; EAC = event adjudication committee; eGFR = estimated glomerular filtration rate; GLP-1 RA = glucagon-like peptide-1 receptor agonist; HDL = high-density lipoprotein; HF = heart failure; hsCRP = high-sensitivity C-reactive protein; LDL = low-density lipoprotein; MACE = major adverse cardiovascular event; MI = myocardial infarction; NYHA = New York Heart Association; PAD = peripheral arterial disease; T1D = type 1 diabetes; T2D = type 2 diabetes; UACR = urinary albumin-to-creatinine ratio; UAP = unstable angina pectoris; VAS = visual analogue scale; WRSSM = weight-related sign and symptom measure.
aEAC-confirmed event.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
The trial design is presented in Figure 1. The trial included a 16-week dose-escalation period, a maintenance period of up to 231 weeks (up to 53 months), and a 5-week follow-up period. The trial was conducted in 41 countries and regions, and patients were randomized at a total of 804 sites, 30 of which were in Canada. Patients were randomized in a 1:1 ratio, using an interactive web response system, to receive once-weekly treatment with either semaglutide 2.4 mg or placebo, both as an adjunct to the standard of care for CV disease. The trial was event driven; trial closure was planned when 1,225 primary end point events (EAC-confirmed MACEs) were reached. The trial used a group sequential design with 1 interim testing planned for superiority.
Based on its evaluation of the results from the interim analysis, on July 19, 2022, the data-monitoring committee advised to continue the trial. The final results presented in this document are based on a data cut-off date of July 18, 2023.
Population
Inclusion and Exclusion Criteria
The trial population was patients aged 45 years and older with established CV disease (prior MI, prior stroke, and/or symptomatic PAD) and a BMI of 27 kg/m2 or greater (overweight or obesity).
The trial population did not include patients with any of the following in the past 60 days before screening: MI, stroke, hospitalization for unstable angina pectoris, or TIA. The trial population also did not include patients with planned coronary, carotid, or peripheral artery revascularization known on the day of screening. Patients with New York Heart Association class IV HF were also excluded from the trial.
The trial population did not include patients with type 1 and type 2 diabetes at randomization; however, patients with a history of gestational diabetes were eligible for enrolment in the trial. The trial population also did not include patients with a hemoglobin A1C of 6.5% or greater at screening. Patients who were treated with any glucose-lowering drug and/or any GLP-1 RA in the past 90 days before screening were also excluded from the trial.
Figure 1: The SELECT Trial Design
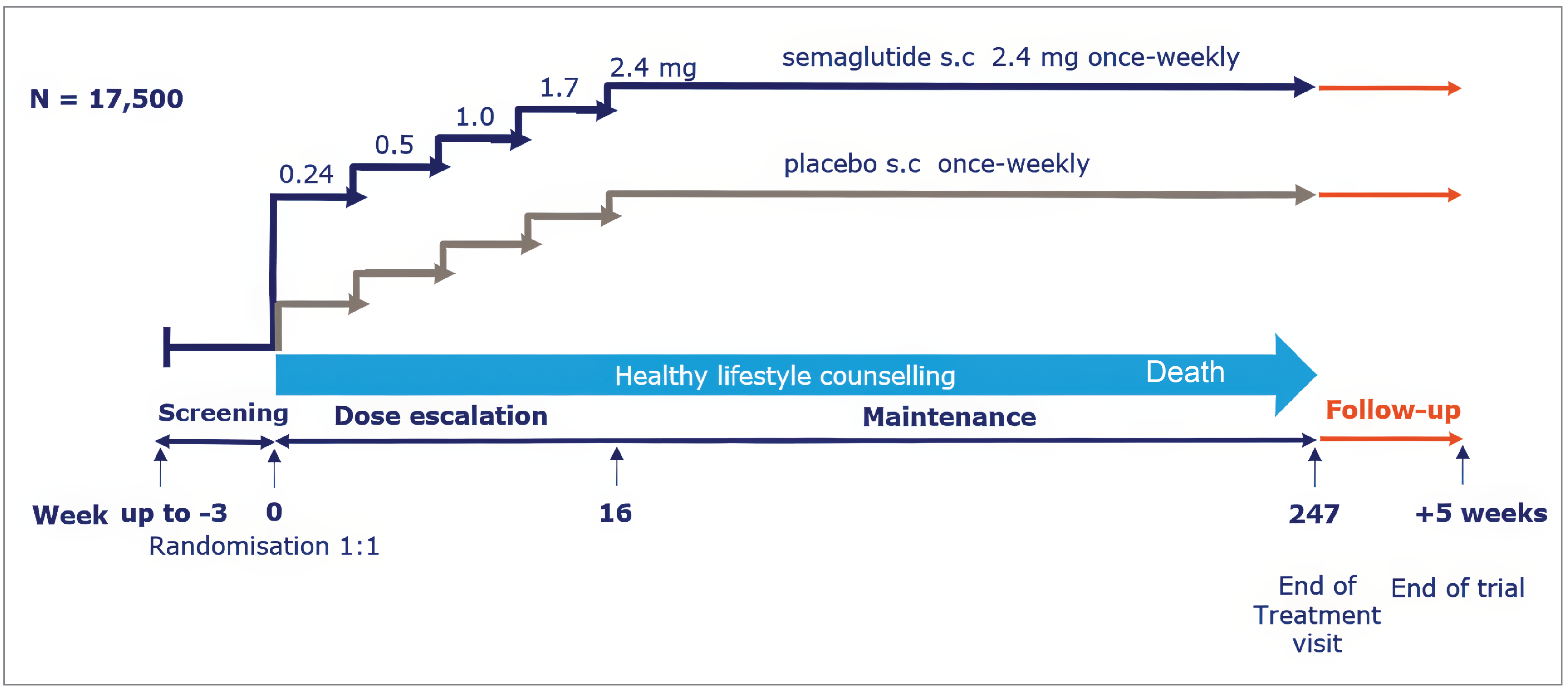
s.c = subcutaneous.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Interventions
The intervention was semaglutide 2.4 mg subcutaneous injection once weekly as an adjunct to the standard of care for CV disease. Dose escalation was every fourth week until a maintenance dose of 2.4 mg was reached; the dose escalations were 0.24 mg, 0.5 mg, 1.0 mg, and 1.7 mg.
The comparator was placebo subcutaneous injection once weekly as an adjunct to the standard of care for CV disease. Dose escalation was every fourth week (same dosing schedule as the intervention).
An extension of dose-escalation intervals and treatment pauses could be considered if treatment was associated with unacceptable AEs or due to other circumstances. If a patient did not tolerate the target maintenance dose, then they may have stayed at the lower dose, preferably 1.7 mg once weekly. Re-escalating to the target dose or restarting treatment after treatment discontinuation was advised if it was considered safe to continue the intervention, per the investigator’s discretion.
Semaglutide 2.4 mg and placebo were supplied as prefilled pen injectors for self-administration in the thigh, abdomen, or upper arm at any time of the day, irrespective of meals. Training in the use of prefilled pen injectors was provided at randomization and at weeks 4, 8, 12, and 16, and at annual visits thereafter, as needed.
Concomitant Therapy
The standard of care (medical treatment) for comorbidities and CV risk factors could be initiated or adjusted (optimized) throughout the trial based on treatment guidelines and local clinical practice, and at the investigator’s discretion. Individualized healthy lifestyle counselling, including diet and physical activity, was offered at every study visit.
Only drugs received at screening or received during the trial for the following indications were recorded:
to treat or prevent CV disease (e.g., antihypertensives, anticoagulants, lipid-lowering drugs, and ASA and other antiplatelet drugs)
to treat overweight or obesity
to treat diabetes and diabetes complications in patients who developed diabetes during the trial
to treat an SAE or a drug that provided further information on the SAE (i.e., alternative etiology)
in relation to a clinical trial for COVID-19 prevention or treatment
in relation to an approved COVID-19 vaccine.
Note that simultaneous participation in a trial with the primary objective of evaluating an approved or nonapproved investigational medicinal product for the prevention or treatment of COVID-19 disease or postinfectious conditions was generally permitted at the investigator’s discretion, without discontinuing trial treatment.
Prohibited Therapy
Starting another GLP-1 RA was prohibited during the trial.
Outcomes
A list of efficacy end points assessed in this Clinical Review report is provided in Table 5, followed by descriptions of the outcome measures. The summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and the input from the patient and clinician groups and public drug plans. Using the same considerations, the review team selected end points that were considered to be the most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee.
Table 5: Outcomes Summarized From the SELECT Trial
Outcome measure | Time point | SELECT trial |
|---|---|---|
CV outcomes | ||
3-component MACE (CV death, nonfatal MI, or nonfatal stroke)a | Results were based on a data cut-off date of July 18, 2023 | Primary end point |
CV deatha | Confirmatory secondary end pointb | |
HF composite (CV death, or HHF or urgent HF visit)a | Confirmatory secondary end pointb | |
All-cause deatha | Confirmatory secondary end pointb | |
Nonfatal MIa | Supportive secondary end point | |
Nonfatal strokea | Supportive secondary end point | |
Glucose metabolism | ||
Hemoglobin A1C ≥ 48 mmol/mol (6.5%)a | Results were based on a data cut-off date of July 18, 2023 | Supportive secondary end point |
Renal outcomes | ||
Five-component nephropathy: onset of persistent macroalbuminuria (UACR > 300 mg/g), persistent 50% reduction in eGFR compared with baseline, onset of persistent eGFR < 15 mL/min/1.73 m2, initiation of chronic renal replacement therapy (dialysis or transplant), or renal death)a | Results were based on a data cut-off date of July 18, 2023 | Supportive secondary end point |
Cardiometabolic risk factorsc | ||
Systolic blood pressure | At week 104d | Supportive secondary end points |
Lipids (total cholesterol, HDL cholesterol) | ||
Anthropometric outcome | ||
Body weight | At week 104d | Supportive secondary end point |
Health-related quality of life | ||
EQ-5D-5L index score | At week 104d | Supportive secondary end points |
EQ VAS score | ||
Symptom burden | ||
WRSSM total score | At week 104d | Exploratory end point |
Notable harms | ||
SAEs, WDAEs, CV AEs, cholelithiasis, nausea, vomiting, constipation, GERD, pancreatitis | Results were based on a data cut-off date of July 18, 2023 | Safety assessments |
AE = adverse event; CV = cardiovascular; eGFR = estimated glomerular filtration rate; GERD = gastroesophageal reflux disease; HDL = high-density lipoprotein; HF = heart failure; HHF = hospitalization for heart failure; MACE = major adverse cardiovascular event; MI = myocardial infarction; SAE = serious adverse event; UACR = urinary albumin-to-creatinine ratio; VAS = visual analogue scale; WDAE = withdrawal due to adverse event; WRSSM = weight-related sign and symptom measure.
aAll time-to-event end points were assessed within the interval from time of randomization to the follow-up visit (up to 59 months or longer) and reported in months. If the event rate was lower than anticipated, then visits and related assessments were repeated every 13 weeks beyond week 234 (the visit scheduled before the end-of-treatment visit) until the necessary number of primary end point events were accrued.
bStatistical testing for these end points was adjusted for multiple comparisons (hierarchal testing).
cCardiometabolic risk factors (systolic blood pressure, total cholesterol, and HDL cholesterol) were included because they were used to inform the accompanying pharmacoeconomic analysis.
dFor analyses of binary and continuous end points at visits after week 52, an analysis was performed only if less than 10% of patients from the full analysis set missed the relevant visit due to trial termination earlier than planned.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
CV Outcomes
The primary end point was time from randomization to the first occurrence of MACE, a composite end point consisting of CV death, nonfatal MI, or nonfatal stroke. Fatal MI was defined as an MI that occurred in 30 days before a CV death that was classified with MI as the cause of death. All other MIs were defined as nonfatal MIs. Similar definitions were applied to fatal and nonfatal stroke. Undetermined cause of death was presumed to be CV death, which was consistent with guidelines, according to the investigator.46,47
The confirmatory secondary end points were time from randomization to CV death, time from randomization to the first occurrence of a composite HF end point consisting of CV death or HF hospitalization or urgent HF visit, and time from randomization to all-cause death.
Supportive secondary end points included time from randomization to the first occurrence of nonfatal MI and time from randomization to the first occurrence of nonfatal stroke.
The evaluation of CV end points comprised the events summarized in Table 6. The descriptions of events were used to guide the investigator in the reporting of AEs, while definitions of events for adjudication were outlined in the charter for the EAC.
Table 6: Events for Adjudication in the SELECT Trial
Type of event | Description of events for reporting AEs | Adjudication outcome |
|---|---|---|
Death | All-cause death |
|
Acute coronary syndrome | All types of acute MI and hospitalization for UAP |
|
Stroke and transient ischemic attack | An episode of focal or global neurologic dysfunction that could be caused by a brain, spinal cord, or retinal vascular injury as a result of hemorrhage or ischemia, with or without infarction | Stroke |
Coronary artery revascularization | A catheter-based (PCI) or surgical procedure (CABG) designed to improve myocardial blood flow | Coronary revascularization procedure |
HF hospitalization or urgent HF visit | Patient presents for an urgent, unscheduled hospital admission or clinic, office, or emergency department visit with a primary diagnosis of HF, including a new episode or worsening of existing HF |
|
Pancreatitis | Any event of pancreatitis should be reported | Acute pancreatitis |
Nephropathy (events leading to renal replacement therapy) | Initiation of dialysis treatment (hemodialysis or peritoneal dialysis) or kidney transplant | Chronic renal replacement therapy |
AE = adverse event; CABG = coronary artery bypass surgery; CV = cardiovascular; EAC = event adjudication committee; HF = heart failure; MI = myocardial infarction; PCI = percutaneous coronary intervention; UAP = unstable angina pectoris.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
An independent external EAC was established to perform ongoing adjudication, based on blinded review, of predefined event types according to definitions and guidelines outlined in the committee’s charter. All relevant events reported from the day of randomization until database lock were adjudicated. Events for adjudication were either directly reported by the investigator, identified through a standardized screening for potential events for adjudication, or identified by the EAC while reviewing source documents for another event for adjudication. The assessments made by both the EAC and the investigator were included in the clinical trial report.
All time-to-event end points were assessed within the interval from time of randomization to the follow-up visit (up to 59 months or longer) and reported in months.
Glucose Metabolism
Supportive secondary end points included time from randomization to the first occurrence of a hemoglobin A1C of 48 mmol/mol (6.5%) or greater.
Classification of glycemic status was according to the American Diabetes Association Standards of Medical Care in Diabetes published in 201823 and summarized in Table 7. Evaluation of glycemic status could involve any method (i.e., fast plasma glucose, plasma glucose, or hemoglobin A1C reflecting current health status) in addition to the investigator’s evaluation of all available relevant information, including prescribed glucose-lowering medications.
Table 7: Classification of Glycemic Status From the SELECT Trial
Glycemic status | FPG | 2-hour PG during oral glucose tolerance test | Hemoglobin A1C |
|---|---|---|---|
Normoglycemia | 3.9 mmol/L to 5.5 mmol/L (70 to 99 mg/dL) | < 7.8 mmol/L (< 140 mg/dL) | < 39 mmol/mol (< 5.6%) |
Prediabetes | 5.6 mmol/L to 6.9 mmol/L (100 to 125 mg/dL) | 7.8 mmol/L to 11.0 mmol/L (140 mmol/L to 199 mg/dL) | 39 mmol/L to 47 mmol/mol (5.7% to 6.4%) |
Diabetes | ≥ 7.0 mmol/L (≥ 126 mg/dL) | ≥ 11.1 mmol/L (≥ 200 mg/dL) | ≥ 48 mmol/mol (≥ 6.5%) |
FPG = fasting plasma glucose; PG = plasma glucose.
Note: Glycemic status classifications are based on the 2018 American Diabetes Association Standards of Medical Care in Diabetes.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Central laboratory assessments of glucose metabolism (hemoglobin A1C parameter) were performed at screening; weeks 20, 52, 104, 156, and 208; and the end-of-treatment visit.
Renal Outcomes
Supportive secondary end points included time from randomization to the first occurrence of a 5-component composite nephropathy end point consisting of the onset of persistent macroalbuminuria (UACR > 300 mg/g), persistent 50% reduction in eGFR compared with baseline, onset of a persistent eGFR of less than 15 mL/min/1.73 m2, initiation of chronic renal replacement therapy (i.e., dialysis or transplant), or renal death.
A persistent change in eGFR was defined as having 2 consecutive samples (at least 4 weeks apart) meeting the criteria. A persistent change in macroalbuminuria was defined as having 2 out of 3 consecutive postbaseline samples (at least 4 weeks apart) above the limit for macroalbuminuria.
The evaluation of kidney function comprised central laboratory assessments of UACR and eGFR, and outcomes from event adjudication by the EAC (events of renal death and nephropathy, per Table 6).
Central laboratory assessments of urine samples to measure UACR (urinalysis) and blood samples to measure creatinine (eGFR was calculated based on the creatinine value using the Chronic Kidney Disease Epidemiology Collaboration equation) were performed at randomization; weeks 20, 52, 104, 156, and 208; and the end-of-treatment visit.
Cardiometabolic Risk Factors
Supportive secondary end points included change from randomization in systolic blood pressure and change from randomization in lipids (total cholesterol, HDL cholesterol, low-density lipoprotein [LDL] cholesterol, triglycerides) at week 104.
Blood pressure measurements were assessed at every study visit with an automated device; manual techniques were used only if an automated device was not available.
Central laboratory assessments of nonfasting lipids (total cholesterol, HDL cholesterol, LDL cholesterol, and triglycerides) were performed at randomization; weeks 20, 52, 104, 156, and 208; and the end-of-treatment visit.
Anthropometric Outcome
Supportive secondary end points included change from randomization in body weight at week 104.
Body weight was measured on a calibrated digital scale and recorded in kilograms or pounds at every visit during the trial.
Health-Related Quality of Life
Supportive secondary end points included change from baseline in the EQ-5D index score and EQ VAS at week 104.
The EQ-5D-5L questionnaire is a patient-reported outcome tool used to estimate health-related quality of life. The tool includes a descriptive system that provides a description of problems experienced by the patient who responded according to dimensions, a VAS that provides a score indicating overall self-rated health (score ranges from 0 to 100), and an index score that ranges from 0 to 1. A higher score indicates better self-reported health status.
The EQ-5D-5L questionnaire was administered at randomization; weeks 20, 52, 104, 156, and 208; and the end-of-treatment visit.
Symptom Burden
Exploratory end points included change from baseline in WRSSM total score at week 104.
The sponsor indicated the WRSSM tool was under development at the time of the trial. The WRSSM is a patient-reported outcome tool (a self-rated VAS) used to assess the presence and bother associated with weight-related symptoms. Specifically, it is used to assess the impact of multifaceted aspects of obesity on symptom experience in individuals with overweight or obesity. The total score ranges from 0 to 4, with higher scores indicating worse symptomatology.
The WRSSM questionnaire was administered at randomization; at weeks 20, 52, 104, 156, and 208; and the end-of-treatment visit.
Safety Assessments
Only the following types of AEs were recorded:
all SAEs
AEs leading to discontinuation of trial product, irrespective of seriousness
AEs of COVID-19, irrespective of seriousness
AEs requiring additional data collection (e.g., pancreatitis, acute renal failure, gallbladder disease, malignant neoplasm) and events for adjudication (e.g., events to evaluate CV end points), irrespective of seriousness.
Nonserious AEs not fulfilling any of the prespecified criteria were not systematically collected. The focus of the safety evaluation per the Clinical Study Report for the SELECT trial was based on the reporting of SAEs and other systematically collected events (i.e., AEs of special interest).
The prespecified AEs were collected at every study visit from the day of randomization until the end-of-trial visit. AEs were coded and reported in the Medical Dictionary for Regulatory Activities Version 26.0.
In consultation with the clinical experts, the notable harms for this review were identified to be CV AEs, cholelithiasis, nausea, vomiting, constipation, GERD, and pancreatitis.
The sponsor established an internal safety committee to endorse appropriate actions in case of a safety signal by conducting integrated blinded assessments of safety data. If the internal safety committee advised to unblind any data for further analyses, then an independent ad hoc group was established to maintain blinding of trial personnel.
Potential withdrawal or rebound effects were investigated for semaglutide 2.4 mg for weight management in the Wegovy phase III program. Except for increased appetite and hunger, no withdrawal effects were observed upon discontinuation of semaglutide treatment, per the investigator. The investigator concluded that withdrawal and rebound effects have therefore not been further investigated in the SELECT trial.
Statistical Analysis
An overview of the statistical analyses of efficacy end points from the SELECT trial is presented in Table 10.
Sample Size and Power Calculation
The trial was designed with 90% power to confirm superiority of the primary end point (i.e., reject the null hypothesis of an HR ≥ 1.0 against the 1-sided alternative of HR < 1.0, where HR was the HR of semaglutide 2.4 mg versus placebo).
The assumed true HR of 0.83 was based on a conservative assessment of the point estimate for the HR in the SUSTAIN-6 trial,48 which was 0.74 (95% CI, 0.58 to 0.95) for a similar definition of MACE. Based on a randomization ratio of 1:1 and under the assumed true HR of 0.83, a total of 1,225 primary end point events were required.
The following assumptions were made to calculate the number of patients to be randomized:
uniform recruitment in 28 months (2,500 patients every 4 months based on the DEVOTE trial49)
annual primary end point rate of 1.8% in the semaglutide 2.4 mg group and 2.2% in the placebo group, based on the SUSTAIN-6, LEADER, FOURIER, SCOUT, and IRIS trials48,50-53 and adjusted to the inclusion and exclusion criteria of the SELECT trial
annual lost to follow-up rate of 1% in both treatment groups.
With a sample size of 17,500 patients and under the alternative hypothesis, the 1,225 primary end point events were expected to be accrued between months 58 and 59 (approximately 5 years after randomization of the first patient), with a mean observation time of 44 months.
Interim Analysis (Group Sequential Design)
One interim test for superiority of the primary end point was introduced through a protocol amendment dated March 7, 2019. The interim test was performed when two-thirds of the planned total number of primary end point events was accrued (817 of 1,225 events). Patients without an EAC-confirmed primary end point event before the interim analysis data cut-off date were considered censored. The censoring date was defined as the end date of the first in-trial observation period or interim analysis data cut-off date. Under the design assumptions and sample size calculation, the timing of the interim test was expected to be at month 44, with a mean observation time of 30 months.
The Lan-DeMets alpha spending function, approximating the O’Brien-Fleming stopping boundaries, was used to test superiority at a studywise 1-sided type I error rate of 2.5%. Testing for futility was not included. The same Cox model described for the primary analysis of the primary end point was used for the interim analysis, addressing the primary estimand and using the fixed-sample 1-sided lower P value from the score test. Only a fixed-sample P value below the boundary specified by the error spending function enabled the data-monitoring committee to advise early trial termination for superiority. The actual stopping boundaries were based on the exact number of events available at the interim analysis.
An independent external data-monitoring committee was established to assess the benefit-risk balance by evaluating accumulated unblinded data at predefined time points and ad hoc. The data-monitoring committee advised on the continuation, modification, or early termination of the trial based on its evaluation of the result from the interim test for superiority of the primary end point. The interim analysis was performed by an external independent statistical service provider. Throughout the trial, the data-monitoring committee and the statistician were the unblinded parties, while the sponsor, investigators, event adjudication vendor, and the EAC were the blinded parties. Unblinded data were discussed at closed session meetings at which only the unblinded parties were present, while blinded data were discussed at open session meetings where all parties were present.
Based on its evaluation of the results from the interim analysis, on July 19, 2022, the data-monitoring committee advised to continue the trial. Per the Clinical Study Report, the interim analysis was found to have no impact on the conduct of the trial or on the interpretation of results; hence, the results from the interim analysis were not submitted by the sponsor.
Definitions of Estimands and Observation Periods
For all objectives, the primary estimand (intention to treat) was used to evaluate the treatment effect irrespective of adherence to treatment or changes to background medication. This estimand was addressed using the FAS and the in-trial observation period, defined in Table 8.
For the confirmatory end points, a secondary estimand was used to evaluate the treatment effect in all patients who were randomized who had remained on their randomized treatment for the entire trial. This estimand was addressed using the FAS and the first on-treatment observation period, defined in Table 8.
Table 8: Observation Periods in the SELECT Trial
Observation period | Definition |
|---|---|
In-trial observation period | The in-trial observation period was defined as the time from randomization to the last day of the trial, irrespective of treatment adherence. For an individual patient, the in-trial observation period was defined as the period from date of randomization to any of the following dates, whichever occurred first, inclusive:
|
On-treatment observation period | The on-treatment observation period was defined as the sum of all time points in the in-trial period during which a patient had received the trial product within the previous 5 weeks. For a patient, the on-treatment observation period could consist of several time intervals with gaps between if treatment had been paused for > 35 consecutive days. |
First on-treatment observation period | The first on-treatment observation period was defined as the on-treatment observation period until first time being off treatment for > 35 days (the first of the time intervals in the on-treatment period). |
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
General Considerations for Time-to-Event End Points
Vital status was systematically ascertained throughout the trial until database lock, but other types of events could not be systematically collected after withdrawal, lost to follow-up, or after the end-of-trial visit. As such, any event occurring after the in-trial observation period was not included in the analyses, unless otherwise stated.
In general, time-to-event end points were time-to-first-event end points. If a patient experienced an event of interest during the in-trial observation period, then the observation of the time-to-event end point was the time from randomization to the date of the event.
The observation of the time-to-event end point was censored if the event of interest did not occur during the in-trial observation period and if the patient was alive at the end of the observation period. The general assumption for censored observations was that the risk of experiencing an event was not changed by censoring (i.e., an assumption of independent censoring).
The observation of the time-to-event end point was terminated if the event of interest did not occur before death, unless death was part of the end point. Terminating events (competing risks) were potentially present for all time-to-event end points except for all-cause death. For the primary end point, non-CV death was a competing risk that could terminate the observation of the event of interest (MACE). Terminated observations due to competing risks from non-CV death were handled as censored observations in the Cox analysis but were not part of the independent censoring assumption.
Statistical Test
The primary analysis addressed the primary estimand (intention to treat). The HR (and the 2-sided 95% CI and 1-sided fixed design P value for hypothesis testing) for comparing semaglutide 2.4 mg versus placebo was estimated from a Cox proportional hazards model with treatment group as a fixed factor under the assumption of independent censoring. The score test from the Cox model was used for testing. The assumption of proportional hazards was investigated by residuals. Tied event times were handled using the exact method and CIs were based on the profile likelihood.
The population-level summary measure for time-to-event end points was the HR for semaglutide 2.4 mg versus placebo. Cumulative incidence functions for time-to-event end points were estimated with the Aalen-Johansen estimator, which accounted for competing risks.
The nominal significance level was calculated using the alpha spending function and based on the actual observed number of events at the analysis. Final inference on termination was adjusted for the group sequential design by using likelihood ratio ordering.
The handling of intercurrent events for the primary estimand and primary end point is summarized in Table 9. Intercurrent events were reported using descriptive statistics.
Table 9: Handling of Intercurrent Events for the Primary Estimand and Primary End Point in the SELECT Trial
Intercurrent event | Approach for estimand | MACE events available after intercurrent event | Statistical approach |
|---|---|---|---|
| Collect events irrespective of the intercurrent event | Yes | Use all available events irrespective of intercurrent event |
Trial discontinuation (withdrawal or lost to follow-up) | Attempts should be made to collect vital events through indirect sources | Vital events potentially available | Censoring at time of discontinuation |
Non-CV death (renal death and non-CV, nonrenal death) | While alive perspective | No | Competing risk |
CV = cardiovascular; MACE = major adverse cardiovascular event.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Missing Data
Unobserved data pertaining to patients who withdrew consent or were lost to follow-up and were not censored at the time point in question were considered missing data, irrespective of any vital status data collected at the end of the trial.
Sensitivity Analyses
Because the primary analysis assumed independent censoring for patients who withdrew consent or were lost to follow-up, a 2-way tipping-point analysis based on the approach described by Zhao et al.54 was performed. In this sensitivity analysis, imputation of event times for these patients was based on the conditional event distribution with a penalty (i.e., the risk of MACE changed after censoring compared with while under observation). Multiple imputed datasets were analyzed with separate Cox regressions and results were combined using the Rubin rule. The tipping points were defined as the combination of changes in penalties in each treatment group needed to turn around the superiority conclusion.
Two additional sensitivity analyses were performed by multiple imputation of event times for patients who withdrew consent or were lost to follow-up:
The first sensitivity analysis used an estimated annual event rate from patients who permanently discontinued treatment but remained in the trial (i.e., the event rate was based on events and time while these patients were permanently off treatment; this period was defined as > 5 weeks since any dose of trial product was administered, and the patient remained off treatment for the remainder of the trial).
The second sensitivity analysis used an estimated annual event rate from patients who discontinued treatment at any point in the trial, which avoids conditioning on the future (i.e., the event rate was based on events that occurred from the first time they were off treatment and until the end of the in-trial observation period; therefore, this can include time periods when patients went back on trial treatment).
For each sensitivity analysis, the off-treatment event time data were fitted within treatment groups to an exponential distribution using Bayesian analysis and accounting for censoring. A noninformative prior distribution was used for the rate parameter in each treatment group. A total of 500 replicates of the 2 off-treatment event rates were randomly sampled from the posterior distribution. A total of 500 copies of the original dataset were created and linked to the corresponding replicate of the off-treatment event rates. For each patient who was censored due to withdrawal or lost to follow-up, the event time was imputed by adding a random variable to the original censoring date. The random variables were generated from an exponential distribution using the off-treatment event rate for the corresponding replicate and treatment group, rounded up to whole days. If the imputed event time occurred after the patient’s planned end-of-trial time, then the patient was censored at the planned end-of-trial time. Each complete dataset was analyzed using the same Cox regression used in the primary analysis to provide the estimated log HR and associated standard error, which were pooled using the Rubin rule to obtain a single point estimate, CI, and P value.
Supplementary Analyses
The following supplementary analyses were performed:
absolute risk difference estimate and 95% CI between semaglutide 2.4 mg and placebo at week 156 based on the cumulative incidence function for each treatment group
an analysis using a Cox proportional hazards model with the first on-treatment period to address the secondary estimand
an analysis of non-CV death using a Cox proportional hazards model to assess the influence of the competing risk of non-CV death on the results from the primary analysis
various supplementary analyses to assess the impact of the COVID-19 pandemic as well as the impact of coparticipation in COVID-19 trials:
Supplementary analyses were done to assess the impact of the COVID-19 pandemic on the primary end point. The analyses addressed 2 scenarios: 1 in which MACEs have been impacted by an increased MACE rate and potentially a different treatment effect for events occurring concurrently with COVID-19 infection, and a second scenario in which there was a reduced MACE rate due to concurrent COVID-19 infection leading to fewer CV deaths because the patient died (prematurely) due to COVID-19 infection and not their underlying atherosclerotic disease.
Subgroup Analyses
To explore the consistency in the treatment effect for the primary end point, subgroup analyses based on the following baseline characteristics were performed:
sex (female, male)
age (< 55 years, 55 to 65 years, 65 to 75 years, ≥ 75 years)
race (white, Black or African American, Asian, other)
ethnicity (Hispanic or Latino, not Hispanic or Latino)
region (Europe, North America, Asia, other)
hemoglobin A1C less than 5.7% (yes, no)
BMI (< 30 kg/m2, 30 kg/m2 to < 35 kg/m2, 35 kg/m2 to < 40 kg/m2, 40 kg/m2 to < 45 kg/m2, ≥ 45 kg/m2)
CV disease (MI alone, stroke alone, PAD alone, any combination)
eGFR of 60 mL/min/1.73 m2 or less per the Chronic Kidney Disease Epidemiology Collaboration equation (yes, no)
chronic HF (yes, no).
The subgroup analyses used Cox proportional hazards models with an interaction between the treatment groups and the specific subgroup as factor.
Of the preceding subgroups listed, BMI was identified as the most relevant for the purpose of this review to inform expert committee deliberations. Consultation with the clinical experts did not identify any relevant potential treatment-effect modifiers to examine for this review.
Testing Hierarchy
One major (classification per investigator) change to the statistical analyses before trial unblinding was implemented through a protocol amendment dated February 9, 2022. The composite HF end point, consisting of EAC-confirmed CV death or HF hospitalization or urgent HF visit, was moved from being a supportive secondary end point to a confirmatory secondary end point. This change was reportedly introduced to add to the observed overall data indicating a beneficial effect of GLP-1 RA in preventing HF events.
A second major (classification per investigator) change to the statistical analyses before trial unblinding was implemented through a protocol amendment dated January 4, 2021. The statistical testing strategy for the confirmatory secondary end points was updated with a separate alpha spending function, as described by Glimm et al.,55 to control the type I error rate at a 1-sided level of 2.5%.
If superiority of the primary end point was confirmed, then the superiority hypothesis was tested for each confirmatory secondary end point under multiplicity control using a stagewise hierarchical testing scheme according to the following order:
time from randomization to EAC-confirmed CV death
time from randomization to first occurrence of a composite HF end point consisting of EAC-confirmed CV death or HF requiring hospitalization or urgent HF visit
time from randomization to EAC-confirmed all-cause death.
The testing procedure was stopped the first time an analysis failed to confirm superiority of the end point in question using the nominal significance level. No adjustment to the results for the group sequential design was made for the confirmatory secondary end points.
The nominal significance levels for the confirmatory secondary end points were derived from the alpha spending function and were based on the actual observed number of events at the analysis.
Secondary End Points
For confirmatory end points controlled for multiplicity, estimated treatment effects were presented together with 2-sided 95% CIs and 1-sided P values for superiority tests. For the reporting of results, the HR and 95% CI were presented with the 2-sided P value.
For nonconfirmatory end points, estimated treatment effects were reported with 2-sided 95% CI and 2-sided P values.
Time-to-Event Confirmatory Secondary End Points
The confirmatory secondary end points were analyzed using a Cox proportional hazards model as described for the primary end point and addressed the primary estimand.
The 2 additional sensitivity analyses with imputation from patients off treatment were also performed for the confirmatory secondary end points. Supplementary analyses (absolute risk difference at week 156 and first on-treatment observation period) described for the primary end point were also performed for the confirmatory secondary end points.
For the composite HF end point, an additional supplementary analysis was performed by replacing the CV death component with all-cause death.
For the end point of all-cause death, an additional supplementary analysis was performed using the FAS and an extended in-trial observation period that included the follow-up for vital status for patients who withdrew consent or were lost to follow-up.
Time-to-Event Supportive Secondary End Points
The supportive secondary end points were analyzed using a Cox proportional hazards model as described for the primary end point and addressed the primary estimand.
For nonfatal MI and nonfatal stroke, supplementary analyses were performed for end points defined as time from randomization to first MI (fatal or nonfatal) and time from randomization to first stroke (fatal or nonfatal).
Continuous Supportive Secondary End Points
The population-level summary measure for continuous end points was the mean difference for semaglutide 2.4 mg versus placebo. End points on lipid parameters were logarithmic transformed and the mean difference on the logarithmic scale was back transformed to the original scale and reported as geometric mean ratios.
The continuous supportive secondary end points (change from baseline at week 104) were analyzed by an analysis of covariance (ANCOVA) with multiple imputation for missing values under a missing-at-random assumption. The imputation model (linear regression) was performed separately for each treatment group and included baseline value as a covariate and fitted to patients with an observed data point, irrespective of adherence to randomized treatment. The fitted model was used to impute values for all patients with missing data at week 104 to create complete datasets. Patients without a baseline measurement were not part of the model. The Rubin rule was used to combine the results. The complete datasets were analyzed by ANCOVA with treatment as fixed factor and baseline value as covariate.
Analyses for body weight were carried out at weeks 52, 156, and 208 if less than 10% of patients from the FAS missed the corresponding visit due to trial termination.
Table 10: Overview of Statistical Analyses of Efficacy End Points From the SELECT Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Primary end point | ||||
Time from randomization to first occurrence of MACE, a composite end point consisting of CV death, nonfatal MI, or nonfatal strokea | Cox proportional hazards model | Treatment group as fixed factor | Unobserved data pertaining to patients who were lost to follow-up or withdrawn and were not censored at the time point in question were considered missing data irrespective of vital status collected at end of trial. The statistical approach for the intercurrent event of trial discontinuation (withdrawal or lost to follow-up) was censoring at time of discontinuation. |
|
Confirmatory secondary end points | ||||
Time from randomization to CV death | Cox proportional hazards model | Treatment group as fixed factor | Unobserved data pertaining to patients who were lost to follow-up or withdrawn and were not censored at the time point in question were considered missing data irrespective of vital status collected at end of trial. The statistical approach for the intercurrent event of trial discontinuation (withdrawal or lost to follow-up) was censoring at time of discontinuation. |
|
Time from randomization to first occurrence of a composite HF end point consisting of CV death, HHF, or urgent HF visita | Cox proportional hazards model | Treatment group as fixed factor | Unobserved data pertaining to patients who were lost to follow-up or withdrawn and were not censored at the time point in question were considered missing data irrespective of vital status collected at end of trial. The statistical approach for the intercurrent event of trial discontinuation (withdrawal or lost to follow-up) was censoring at time of discontinuation. |
|
Time from randomization to all-cause death | Cox proportional hazards model | Treatment group as fixed factor | Unobserved data pertaining to patients who were lost to follow-up or withdrawn and were not censored at the time point in question were considered missing data irrespective of vital status collected at end of trial. The statistical approach for the intercurrent event of trial discontinuation (withdrawal or lost to follow-up) was censoring at time of discontinuation. |
|
Supportive secondary end points | ||||
Time from randomization to nonfatal MI | Cox proportional hazards model | Treatment group as fixed factor | Unobserved data pertaining to patients who were lost to follow-up or withdrawn and were not censored at the time point in question were considered missing data irrespective of vital status collected at end of trial. The statistical approach for the intercurrent event of trial discontinuation (withdrawal or lost to follow-up) was censoring at time of discontinuation. | None prespecified |
Time from randomization to nonfatal stroke | Cox proportional hazards model | Treatment group as fixed factor | Unobserved data pertaining to patients who were lost to follow-up or withdrawn and were not censored at the time point in question were considered missing data irrespective of vital status collected at end of trial. The statistical approach for the intercurrent event of trial discontinuation (withdrawal or lost to follow-up) was censoring at time of discontinuation. | None prespecified |
Time from randomization to first occurrence of hemoglobin A1C ≥ 48 mmol/mol (6.5%) | Cox proportional hazards model | Treatment group as fixed factor | Unobserved data pertaining to patients who were lost to follow-up or withdrawn and were not censored at the time point in question were considered missing data irrespective of vital status collected at end of trial. The statistical approach for the intercurrent event of trial discontinuation (withdrawal or lost to follow-up) was censoring at time of discontinuation. | None prespecified |
Time from randomization to first occurrence of a 5-component composite nephropathy end point consisting of renal death, initiation of chronic renal replacement therapy (dialysis or transplant), onset of persistent eGFR < 15 mL/min/1.73 m2, persistent ≥ 50% reduction in eGFR compared with randomization, or onset of persistent macroalbuminuria (UACR > 300 mg/g)a | Cox proportional hazards model | Treatment group as fixed factor | Unobserved data pertaining to patients who were lost to follow-up or withdrawn and were not censored at the time point in question were considered missing data irrespective of vital status collected at end of trial. The statistical approach for the intercurrent event of trial discontinuation (withdrawal or lost to follow-up) was censoring at time of discontinuation. | None prespecified |
Change from baseline in systolic BP at week 104 | ANCOVA | Treatment group as a fixed factor and baseline value as covariate | Multiple imputation for missing values under a missing-at-random assumption | None prespecified |
Change from baseline in lipids (total cholesterol, HDL cholesterol) at week 104 | ANCOVA on logarithmic transformed assessments | Treatment group as a fixed factor and baseline value as covariate | Multiple imputation for missing values under a missing-at-random assumption | None prespecified |
Change from baseline in body weight at week 104. | ANCOVA | Treatment group as a fixed factor and baseline value as covariate | Multiple imputation for missing values under a missing-at-random assumption | Jump to reference multiple imputation |
Change from baseline in EQ-5D-5L index score at week 104. | ANCOVA | Treatment group as a fixed factor and baseline value as covariate | Multiple imputation for missing values under a missing-at-random assumption | None prespecified |
Change from baseline in EQ VAS score at week 104 | ANCOVA | Treatment group as a fixed factor and baseline value as covariate | Multiple imputation for missing values under a missing-at-random assumption | None prespecified |
Exploratory end point | ||||
Change from baseline in WRSSM total score at week 104 | Descriptive | Not applicable | Not applicable | Not applicable |
ANCOVA = analysis of covariance; BP = blood pressure; CV = cardiovascular; eGFR = estimated glomerular filtration rate; HDL = high-density lipoprotein; HF = heart failure; HHF = hospitalization for heart failure; MACE = major adverse cardiovascular event; MI = myocardial infarction; UACR = urinary albumin-to-creatinine ratio; VAS = visual analogue scale; WRSSM = weight-related sign and symptom measure.
aFor composite end points, the ordering defined the hierarchy of components when reporting events contributing to a composite end point when there was a tie in the event dates. Tied event times were handled using the exact method.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
A sensitivity analysis using a jump to reference multiple imputation approach was performed for change from baseline in body weight at week 104 (and weeks 52, 156, and 208 if less than 10% of patients from the FAS missed the corresponding visit due to trial termination). This analysis used a multiple imputation approach in which missing values from both treatment groups were imputed using an imputation model for the placebo group only, irrespective of adherence to randomized treatment at week 104.
Exploratory End Point
Change from baseline in WRSSM total score at week 104 was presented descriptively.
Analysis Population
The FAS was defined as all patients who were randomized; all efficacy and safety analyses were performed according to the treatment assigned at randomization.
For patients who were randomized more than once, only the patient identification number and treatment corresponding to the first randomization were included in the FAS. Any additional identification numbers of patients who were randomized were excluded from the FAS.
Results
Patient Disposition
A summary of patient disposition from the SELECT trial is presented in Table 11.
A total of 21,089 patients were screened, of which 3,480 patients (16.5%) were not eligible for randomization because they did not meet all eligibility criteria, did not return for randomization, or withdrew consent before randomization. The most frequently reported reason for screening failure was due to a hemoglobin A1C level of 6.5% or greater in 1,724 patients (49.5%).
Initially, a total of 8,805 patients were randomized to receive semaglutide 2.4 mg and 8,804 patients were randomized to receive placebo. However, 5 patient identifiers were removed because they were randomized more than once. Thus, the FAS comprised 8,803 patients in the semaglutide 2.4 mg group and 8,801 patients in the placebo group.
Of the patients who were randomized and included in the FAS, a total of 259 patients (2.9%) in the semaglutide 2.4 mg group and 284 patients (3.2%) in the placebo group discontinued from the study due to either withdrawal by patient or lost to follow-up. A total of █████ patients (█████) in the semaglutide 2.4 mg group and █████ patients (█████) in the placebo group discontinued treatment. Notably, 1,434 patients (16.3%) in the semaglutide 2.4 mg group and 696 patients (7.9%) in the placebo group discontinued treatment due to AEs, while 64 patients (0.7%) in the semaglutide 2.4 mg group and 244 patients (2.8%) in the placebo group discontinued treatment due to lack of effect.
Of the patients who were randomized and included in the FAS, a total of 4 patients (< 0.1%) did not have a study visit at week 104 due to trial closure. Notably, 4,814 patients (27.3%) did not have a study visit at week 156 and 13,977 patients (79.4%) did not have a study visit at week 208 due to trial closure.
Table 11: Summary of Patient Disposition From the SELECT Trial
Patient disposition | Semaglutide 2.4 mg | Placebo |
|---|---|---|
Screened, N | 21,089 | |
Screening unsuccessful, N (%) | 3,480 (16.5) | |
Reason for unsuccessful screening, n (%) | ||
Did not meet all eligibility criteria | 2,740 (78.7) | |
Exclusion criterion: Hemoglobin A1C ≥ 48 mmol/mol (6.5%) as measured by the central laboratory at screeninga | 1,724 (49.5) | |
Did not return for randomization | 356 (10.2) | |
Withdrawal of consent by patient before randomization | 384 (11.0) | |
Missing reason | 5 (0.1) | |
Randomized, N | 8,805 | 8,804 |
Randomizations removed due to patient randomized more than once,b n | 2 | 3 |
Completed study, n (%) | 8,544 (97.1) | 8,517 (96.8) |
Attended follow-up visit | 8,169 (92.8) | 8,059 (91.6) |
Died during trial | 375 (4.3) | 458 (5.2) |
Discontinued from study, n (%) | 259 (2.9) | 284 (3.2) |
Last known vital status, n (%) | ||
Alive | 205 (2.3) | 223 (2.5) |
Deceased | 0 | 6 (< 0.1) |
Unknown | 54 (0.6) | 55 (0.6) |
Reason for study discontinuation, n (%) | ||
Withdrawal by patient | 67 (0.8) | 96 (1.1) |
Lost to follow-up | 192 (2.2) | 188 (2.1) |
Discontinued treatment, n (%) | ███████████ | ███████████ |
Reason for treatment discontinuation, n (%) | ||
Adverse events | 1,434 (16.3) | 696 (7.9) |
Lack of effect | 64 (0.7) | 244 (2.8) |
Unintentional treatment discontinuation | 252 (2.9) | 327 (3.7) |
Currently no contact with patient | 71 (0.8) | 101 (1.1) |
Participation in another clinical trial anytime during the trial | 3 (< 0.1) | 4 (< 0.1) |
Simultaneous use of prohibited medication | 5 (< 0.1) | 29 (0.3) |
COVID-19 pandemic | 39 (0.4) | 43 (0.5) |
Other | 511 (5.8) | 647 (7.4) |
Missing | 306 (3.5) | 265 (3.0) |
FAS, N | 8,803 | 8,801 |
Patients with no visit due to trial closure, n/N (%)c | ||
Visit at week 52 | 0/17,604 (0) | |
Visit at week 104 | 4/17,604 (< 0.1) | |
Visit at week 156 | 4,814/17,604 (27.3) | |
Visit at week 208 | 13,977/17,604 (79.4) | |
FAS = full analysis set.
aFrequency ≥ 5.0%.
bRescreening was permitted for all inclusion and exclusion criteria; a new patient identification number was assigned in the interactive web response system. For patients randomized more than once, only the patient identification number and treatment corresponding to the first randomization were included in the FAS. The additional identification numbers of patients who were randomized were excluded from the FAS.
cProportion of patients in the FAS who missed the annual visit due to trial termination. If 10% or more of patients from the FAS missed the annual visit due to trial termination, then the analysis for the supportive secondary end points, body weight and hemoglobin A1C, was not performed for the corresponding week.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
Protocol Deviations and Serious Breaches
A summary of important protocol deviations from the SELECT trial is presented in Table 12. No patient in the FAS was excluded from the analyses due to protocol deviations.
Several site-level and patient-level important protocol deviations regarding quality and good clinical practice issues were reported at 2 sites, resulting in serious breaches. A total of 11 serious breaches of good clinical practice were reported in the trial. An assessment of the potential impact on patient safety and rights and data integrity was performed by the investigator; only those breaches that were considered to have a potential impact on data integrity are summarized in Table 13.
There were 3 cases of unintentional unblinding before database lock involving unintentional unblinding of clinical study group, treatment, and trial product batch numbers.
There was a total of 724 cases of intentional unblinding before database lock to handle suspected trial product–related unexpected serious adverse reactions as part of the regulatory reporting requirements for SAEs. Breaking the randomization code for a patient before database lock in the case of a suspected unexpected serious adverse reaction was prespecified. It was reported that these cases of intentional unblinding were handled according to internal procedures, and it was concluded that they did not affect trial data integrity.
Table 12: Summary of Important Protocol Deviations From the SELECT Trial (Patient Level)
Protocol deviation | Semaglutide 2.4 mg | Placebo |
|---|---|---|
Informed consent, N | 143 | 135 |
Missing and late reconsent of informed consent, n (%) | 68 (47.6) | 48 (35.6) |
Otherwise incorrect informed consent process, n (%) | 20 (14.0) | 27 (20.0) |
Wrong version of informed consent signed, n (%) | 9 (6.3) | 18 (13.3) |
Other, n (%) | 38 (26.6) | 34 (25.2) |
Inclusion and exclusion criteria, N | 56 | 60 |
Eligibility criteria violated, n (%) | 53 (94.6) | 48 (80.0) |
Discontinuation and withdrawal criteria violated, n (%) | 3 (5.4) | 7 (11.7) |
Treatment administration, N | 350 | 257 |
Wrong dispensing unit number and expired trial product, n (%) | 242 (69.1) | 169 (65.8) |
Treatment nonadherence, n (%) | 82 (23.4) | 77 (30.0) |
Study procedures and assessments, N | 49 | 67 |
Documentation and delegation of tasks, n (%) | 17 (34.7) | 28 (41.8) |
Safety focus assessment, n (%) | 4 (8.2) | 7 (10.4) |
Other assessments, n (%) | 9 (18.4) | 9 (13.4) |
Other, n (%) | 16 (32.7) | 17 (25.4) |
AE and other safety procedures, N | 352 | 420 |
SAE and other AEs requiring additional reporting, n (%) | 311 (88.4) | 367 (87.4) |
Concomitant medication and medical intervention, N | 32 | 94 |
Prohibited medication and medical intervention, n (%) | 31 (96.9) | 93 (98.9) |
Visit schedule, N | 7 | 15 |
Other, n (%) | 7 (100) | 15 (100) |
Privacy and data protection, N | 60 | 64 |
Patient privacy and data protection, n (%) | 47 (78.3) | 53 (82.8) |
Other, n (%) | 13 (21.7) | 11 (17.2) |
AE = adverse event; SAE = serious adverse event.
Notes: The data cut-off date was July 18, 2023.
Protocol deviations deemed important were defined as deviations that could significantly impact the completeness, accuracy, and/or reliability of the trial data or that could significantly affect the patient’s rights, safety, or well-being.
Percentages were of the total number of patient-level protocol deviations within each category.
Only a frequency of ≥ 10.0% by subcategory in any treatment group was summarized in the preceding table.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Table 13: Summary of Serious Breaches With Potential Impact on Data Integrity From the SELECT Trial
Serious breach (date of occurrence) | Description | Investigator assessment of potential impact on trial data |
|---|---|---|
EDC system shutdown (July 13, 2021). | The EDC system was unavailable due to disk-related hardware failure. The time gap between the EDC system becoming unavailable and the restore point resulted in the loss of 436 transactions belonging to patients across 25 countries, including Canada. | Potentially could have had an impact on data integrity but not considered to have had an impact on patient safety or rights. |
Out-of-stability HDL results used for LDL and VLDL calculations (March 16, 2018). | For LDL and VLDL calculations, the central laboratory had been using serum samples with a 5-day stability rating, but samples with a 3-day stability rating received in ambient conditions should have been used. A total of 2,048 samples were affected. | Not considered to have an impact on data integrity or on patient safety or rights. |
Multiple quality issues at 1 study site (March 23, 2020). | The sponsor was informed by the principal investigator of multiple quality issues identified during a review by their quality assurance team:
| Potentially could have had an impact on data integrity (i.e., the reliability and robustness of trial data). |
Persistent GCP-related issues at 1 study site (March 1, 2020). | The site was closed in March 2020 with sponsor oversight conducted by remote monitoring of data in electronic systems and phone and email discussions with site staff. Recruitment was completed in March 2021, and the first onsite monitoring visit took place in May 2021. Numerous GCP-related issues at the site were reported and little improvement was observed after the sponsor’s communication with the site:
| Not considered to have a major impact on data integrity or patient safety. |
Patient duplicated at 2 study sites (May 2, 2019). | One patient was enrolled at 2 sites until death, resulting in approximately 184 double doses taken. | Potentially could have had an impact on patient safety but not considered to have had an impact on data integrity or patient rights. |
Flooding at 1 study site (April 11, 2022). | Severe flooding resulted in damages to 1 study site, including all equipment being destroyed and all research files being submerged in water. | Potentially could have had an impact on data integrity but not considered to have had an impact on patient safety or rights. |
Potential external access to study data, results, and patient identities due to hacking of computers at 1 study site (August 20, 2022). | A hacking occurred on a study site and hacked data (including patient identifiers, access to trial systems, and study documentation) was later confirmed to have been distributed on the dark web. Source data for study were fully recorded on paper. | Potentially could have had an impact on data integrity (the reliability and robustness of trial data) and patient rights. |
Lack of principal investigator oversight at 1 study site (February 1, 2021). |
| Potentially could have had an impact on data integrity (the validity of trial data) and patient safety and rights. |
AE = adverse event; EDC = electronic data capture; GCP = good clinical practice; HDL = high-density lipoprotein; LDL = low-density lipoprotein; SAE = serious adverse event; VLDL = very-low-density lipoprotein.
Note: A total of 11 serious breaches of GCP were reported in the trial. An assessment of the potential impact on patient safety and rights and data integrity was performed by the investigator; only those breaches that were considered for potential impact on data integrity were summarized in the preceding table.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Intercurrent Events
A summary of intercurrent events from the SELECT trial is presented in Table 14.
Notably, █████ of 8,803 patients (█████) in the semaglutide 2.4 mg group and █████ of 8,801 patients (█████) in the placebo group discontinued treatment, and █████ patients (█████) in the semaglutide 2.4 mg group and █████ patients (█████) in the placebo group started or changed medication that modifies CV risk. Medication modifying CV risk comprised glucose-lowering medication, weight-lowering medication, and CV-related medication.
Intercurrent event | Semaglutide 2.4 mg | Placebo |
|---|---|---|
Treatment discontinuation | █████████ | ███████ |
Initiation of medication modifying CV riska | █████████ | ███████ |
Bariatric surgery reported as an AEb | ██████ | ██████ |
Bariatric surgery reported as non-SAE hospitalization (gastrointestinal system) | ███████ | ███████ |
Initiation of chronic renal replacement therapy | ██████ | ██████ |
Trial discontinuation | ████████ | ████████ |
Non-CV death | ████████ | ████████ |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; CV = cardiovascular; SAE = serious adverse event.
aMore than 99% of patients were on medications modifying CV risk. An initiation may be a change in medication and not a reflection of a true modification. Medication modifying CV risk comprised glucose-lowering medication, weight-lowering medication, and CV-related medication.
bOne of the AEs was reported as an SAE. Because bariatric surgery is a surgical procedure, only those procedures related to SAEs were systematically collected, per protocol. Non-SAE hospitalizations were collected, but with less detailed information.
Source: Novo Nordisk Canada Inc. response to the January 15, 2025, CDA-AMC request for additional information regarding the CDA-AMC review of Wegovy.26
Baseline Characteristics
The baseline characteristics outlined in Table 15 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
The mean age of patients in the semaglutide 2.4 mg group was 61.6 years (SD = 8.9 years) and 61.6 years (SD = 8.8 years) in the placebo group. The mean BMI of patients in the semaglutide 2.4 mg group was 33.30 kg/m2 (SD = 5.03 kg/m2) and 33.37 kg/m2 (SD = 5.04 kg/m2) in the placebo group. Most patients in the FAS had a BMI between 30 kg/m2 and 35 kg/m2, 3,693 of 8,803 patients (42.0%) in the semaglutide 2.4 mg group and 3,781 of 8,801 patients (43.0%) in the placebo group.
The mean hemoglobin A1C was 5.78% (SD = 0.34%) in the semaglutide 2.4 mg group and 5.78% (SD = 0.33%) in the placebo group. Most patients in the FAS had prediabetes, 5,701 patients (64.8%) in the semaglutide 2.4 mg group and 5,661 patients (64.3%) in the placebo group, according to the classification of glycemic status published in 2018 in the American Diabetes Association Standards of Medical Care in Diabetes.23
Most patients in the FAS had a history of MI, 5,962 patients (67.7%) in the semaglutide 2.4 mg group and 5,944 patients (67.5%) in the placebo group. A total of 2,155 patients (24.5%) in the semaglutide 2.4 mg group and 2,131 patients (24.2%) in the placebo group had chronic HF, of which 1,174 patients (13.3%) in the semaglutide 2.4 mg group and 1,099 patients (12.5%) in the placebo group had HF with preserved ejection fraction.
Table 15: Summary of Baseline Characteristics From the SELECT Trial (FAS)
Characteristic | Semaglutide 2.4 mg | Placebo |
|---|---|---|
Patient demographics | ||
Age (years), mean (SD) | 61.6 (8.9) | 61.6 (8.8) |
Age (years), median (range) | 61 (45 to 89) | 61 (45 to 93) |
Sex, n (%) | ||
Female | 2,448 (27.8) | 2,424 (27.5) |
Male | 6,355 (72.2) | 6,377 (72.5) |
Race,a n (%) | ||
American Indian or Alaska Native | 23 (0.3) | 21 (0.2) |
Asian | 720 (8.2) | 727 (8.3) |
Black or African American | 348 (4.0) | 323 (3.7) |
Native Hawaiian or other Pacific Islander | 3 (< 0.1) | 5 (< 0.1) |
White | 7,387 (83.9) | 7,404 (84.1) |
Other | 227 (2.6) | 247 (2.8) |
Not reported | 95 (1.1) | 74 (0.8) |
Body measurements | ||
Body weight (kg), mean (SD) | 96.53 (17.52) | 96.82 (17.80) |
Body weight (kg), median (range) | 94.2 (53.0 to 240.0) | 94.3 (52.9 to 233.8) |
BMI (kg/m2), mean (SD) | 33.30 (5.03) | 33.37 (5.04) |
BMI (kg/m2), median (range) | 32.12 (25.51 to 75.92) | 32.14 (24.80 to 71.08) |
BMI, n (%) | ||
< 30 kg/m2 | 2,555 (29.0) | 2,469 (28.1) |
30 kg/m2 to < 35 kg/m2 | 3,693 (42.0) | 3,781 (43.0) |
35 kg/m2 to < 40 kg/m2 | 1,687 (19.2) | 1,659 (18.9) |
40 kg/m2 to < 45 kg/m2 | 579 (6.6) | 595 (6.8) |
≥ 45 kg/m2 | 289 (3.3) | 297 (3.4) |
N | 8,759 | 8,756 |
Waist circumference (cm), mean (SD) | 111.3 (13.1) | 111.4 (13.1) |
Waist circumference (cm), median (range) | 110 (34 to 213) | 110 (36 to 182) |
Glycemic status | ||
N | 8,802 | 8,799 |
Hemoglobin A1C (%), mean (SD) | 5.78 (0.34) | 5.78 (0.33) |
Hemoglobin A1C (%), median (range)b | 5.8 (3.8 to 14.6) | 5.8 (4.0 to 8.4) |
N | 8,802 | 8,800 |
Prediabetes, n (%) | 5,701 (64.8) | 5,661 (64.3) |
Normoglycemic, n (%) | 3,101 (35.2) | 3,139 (35.7) |
Renal function | ||
N | 8,724 | 8,742 |
eGFR (mL/min/1.73 m2), mean (SD) | 82.44 (17.48) | 82.48 (17.32) |
eGFR (mL/min/1.73 m2), median (range) | 85.0 (16.0 to 162.0) | 85.0 (7.0 to 146.0) |
Normal renal function (eGFR ≥ 90 mL/min/1.73 m2),c n (%) | 3,454 (39.2) | 3,534 (40.2) |
Mild renal impairment (60 mL/min/1.73 m2 to < 90 mL/min/1.73 m2), n (%) | 4,307 (48.9) | 4,273 (48.6) |
Moderate renal impairment (30 mL/min/1.73 m2 to 60 mL/min/1.73 m2), n (%) | 922 (10.5) | 905 (10.3) |
Severe renal impairment (15 mL/min/1.73 m2 to 30 mL/min/1.73 m2), n (%) | 41 (0.5) | 28 (0.3) |
End-stage renal disease (< 15 mL/min/1.73 m2), n (%) | 0 | 2 (< 0.1) |
CV disease | ||
CV inclusion criteria, n (%) | ||
Only MI | 5,962 (67.7) | 5,944 (67.5) |
Only stroke | 1,578 (17.9) | 1,556 (17.7) |
Only PAD | 376 (4.3) | 401 (4.6) |
≥ 2 CV disease | 718 (8.2) | 719 (8.2) |
Otherd | 169 (1.9) | 181 (2.1) |
N | 8,802 | 8,798 |
Chronic HF, n (%) | 2,155 (24.5) | 2,131 (24.2) |
Subclass, n (%) | ||
HF with preserved ejection fraction | 1,174 (13.3) | 1,099 (12.5) |
HF with reduced ejection fraction | 654 (7.4) | 693 (7.9) |
Unknown | 326 (3.7) | 337 (3.8) |
NYHA class, n (%) | ||
Class I | 715 (8.1) | 656 (7.5) |
Class II | 1,277 (14.5) | 1,263 (14.4) |
Class III | 156 (1.8) | 208 (2.4) |
Laboratory assessment | ||
N | 8,732 | 8,753 |
hsCRP ≥ 2 mg/L,e n (%) | 4,155 (47.2) | 4,091 (46.5) |
Blood pressure | ||
N | 8,800 | 8,794 |
SBP (mm Hg), mean (SD) | 131.0 (15.6) | 130.9 (15.3) |
DBP (mm Hg), mean (SD) | 79.4 (10.0) | 79.2 (9.9) |
Tobacco use | ||
N | 8,803 | 8,799 |
Current smoker, n (%) | 1,486 (16.9) | 1,464 (16.6) |
Never smoked, n (%) | 3,096 (35.2) | 3,024 (34.4) |
Previous smoker, n (%) | 4,221 (47.9) | 4,311 (49.0) |
BMI = body mass index; CV = cardiovascular; DBP = diastolic blood pressure; eGFR = estimated glomerular filtration rate; FAS = full analysis set; HF = heart failure; hsCRP = high-sensitivity C-reactive protein; MI = myocardial infarction; NYHA = New York Heart Association; PAD = peripheral arterial disease; SBP = systolic blood pressure; SD = standard deviation.
aCategories are as reported in the study.
bA total of 7 patients (3 in the semaglutide 2.4 mg group and 4 in the placebo group) had hemoglobin A1C ≥ 6.5% at screening and were randomized in error (captured in protocol deviations). These 7 patients were categorized by the investigator as being either normoglycemic or having prediabetes at baseline.
cRenal function category was based on the eGFR calculated by the central laboratory based on the creatinine value using the Chronic Kidney Disease Epidemiology Collaboration equation.
dIncluded patients who were not known to have fulfilled only 1 or more than 1 CV inclusion criteria (i.e., a criterion was selected in the electronic case report form but the other criteria were left blank, and it was not assumed that missing responses meant criteria not met), and patients who were randomized in error and did not fulfill any criteria (these cases were categorized as protocol deviations).
eThreshold indicative of inflammation, per investigator.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
Exposure to Study Treatments
A summary of patient exposure from the SELECT trial is presented in Table 16.
After the initial 16 weeks of dose escalation, approximately 75% of patients who were receiving treatment at that time point in the semaglutide group were receiving the planned maintenance dose of 2.4 mg. The sponsor noted that 77.12% of patients were receiving the semaglutide 2.4 mg dose at any point in the study, indicating that the patients not receiving the target dose at week 16 were not just extending the dose-escalation phase. For patients who did not reach the semaglutide 2.4 mg target dose at any time in the study, the maximum semaglutide dose was 1.7 mg for 7.97%, 1.0 mg for 7.22%, 0.5 mg for 4.79%, and 0.24 mg for 2.78% of patients.56
A total of 9 patients (0.1%) in the semaglutide 2.4 mg group and 19 patients (0.2%) in the placebo group in the FAS were not exposed to the trial product; no patient in the FAS was excluded from the analyses.
The mean duration of follow-up was 39.9 months (SD = 9.3 months) in the semaglutide 2.4 mg group and 39.7 months (SD = 9.5 months) in the placebo group. The mean duration of exposure to semaglutide 2.4 mg was 33.3 months (SD = 14.4 months) and to placebo was 35.1 months (SD = 13.0 months).
Patients in the semaglutide 2.4 mg group and the placebo group received their assigned treatment for 82.5% (SD = 30.8%) and 87.7% (SD = 25.2%) of the potential treatment time, respectively.
Table 16: Summary of Patient Exposure From the SELECT Trial (FAS)
Exposure | Semaglutide 2.4 mg | Placebo |
|---|---|---|
Observation time by observation period | ||
In-trial period (months) | ||
Mean (SD) | 39.9 (9.3) | 39.7 (9.5) |
Median (range) | 41.9 (0 to 55.2) | 41.7 (0 to 55.2) |
On-treatment period (months)a | ||
N | 8,794 | 8,782 |
Mean (SD) | 33.3 (14.4) | 35.1 (13.0) |
Median (range) | 37.3 (0.3 to 55.0) | 38.6 (0.5 to 55.0) |
First on-treatment period (months)b | ||
N | 8,794 | 8,782 |
Mean (SD) | 30.7 (16.0) | 33.6 (14.1) |
Median (range) | 33.8 (0.3 to 55.0) | 37.5 (0.5 to 55.0) |
Adherence | ||
Adherence on-treatment period (months)c | ||
N | 8,794 | 8,782 |
Mean (SD) | 32.2 (14.5) | 34.1 (13.0) |
Median (range) | 35.9 (0.2 to 54.0) | 37.5 (0.2 to 54.0) |
Planned on-treatment period (months)d | ||
N | 8,803 | 8,801 |
Mean (SD) | 39.1 (9.2) | 38.9 (9.4) |
Median (range) | 41.0 (0 to 55.0) | 40.8 (0 to 55.2) |
Percent of adherence on-treatment period of the planned on-treatment period | ||
N | 8,794 | 8,782 |
Mean (SD) | 82.5 (30.8) | 87.7 (25.2) |
FAS = full analysis set; SD = standard deviation.
aThe on-treatment period was defined as all days from the date of the first dose to the end of the in-trial period during which a dose was administered within 5 weeks (35 days). The period could include nonconsecutive time intervals.
bThe first on-treatment period was defined as the date of the first dose until the first time that no dose was administered within 5 weeks (35 days) or the end of the in-trial period, whichever occurred first.
cThe adherence on-treatment period was defined as all days from the date of the first dose to the end of the in-trial period during which a dose was administered within 1 week (7 days). The period could include nonconsecutive time intervals. The dose did not have to be the target dose.
dThe planned on-treatment period was defined as the date of randomization until the end-of-treatment visit plus 6 days or the end of the in-trial period, whichever occurred first.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
Concomitant Treatments
A summary of concomitant CV-management and weight management–related treatments ongoing at randomization from the SELECT trial is presented in Table 17. Patients receiving glucose-lowering medication or any GLP-1 RA in the past 90 days before screening were not eligible to enrol in the trial. Notably, the majority of patients (> 85% of patients in the FAS) were receiving CV drugs, lipid-lowering drugs, and platelet aggregation inhibitors at randomization, while few patients (< 0.1% of patients in the FAS) were receiving weight management–related medications.
A summary of concomitant CV- and weight management–related and glucose-lowering treatments initiated in the in-trial observation period is presented in Table 18. Glucose-lowering medications (except GLP-1 RA) could be initiated during the trial if the patient developed diabetes. The initiation of a medication could be due to adding a new drug or switching from 1 product to another, which was perceived as a change in medication by the investigator. Notably, the initiation of any CV-related, weight management–related, or glucose-lowering medication after randomization was slightly higher in the placebo group than observed in the semaglutide 2.4 mg group.
Table 17: Summary of Concomitant CV-Management and Weight Management–Related Treatments Ongoing at Randomization From the SELECT Trial (FAS)
Treatment | Semaglutide 2.4 mg | Placebo |
|---|---|---|
CV-related medications | ||
CV drugs, n (%) | 8,122 (92.3) | 8,074 (91.7) |
Beta blockers | 6,182 (70.2) | 6,175 (70.2) |
ACE inhibitors | 3,963 (45.0) | 3,966 (45.1) |
Angiotensin receptor blockers | 2,618 (29.7) | 2,569 (29.2) |
Calcium channel blockers | 2,407 (27.3) | 2,331 (26.5) |
ARNI | 121 (1.4) | 144 (1.6) |
Lipid-lowering drugs, n (%) | 7,928 (90.1) | 7,929 (90.1) |
Statins | 7,716 (87.7) | 7,709 (87.6) |
Ezetimibe | 1,188 (13.5) | 1,144 (13.0) |
Fibrates | 213 (2.4) | 266 (3.0) |
PCSK-9 inhibitors | 177 (2.0) | 162 (1.8) |
Other omega-3 triglycerides | 89 (1.0) | 90 (1.0) |
Eicosapentaenoic acid ethyl ester | 38 (0.4) | 59 (0.7) |
Omega-3-acid ethyl ester | 19 (0.2) | 25 (0.3) |
Bile acid sequestrants | 10 (0.1) | 15 (0.2) |
Bempedoic acid | 1 (< 0.1) | 1 (< 0.1) |
Platelet aggregation inhibitors, n (%) | 7,612 (86.5) | 7,569 (86.0) |
ASA | 6,909 (78.5) | 6,860 (77.9) |
P2Y12 inhibitors | 2,925 (33.2) | 2,998 (34.1) |
Other | 77 (0.9) | 104 (1.2) |
Diuretics, n (%) | 2,922 (33.2) | 2,978 (33.8) |
Loop diuretics | 1,075 (12.2) | 1,134 (12.9) |
Thiazides | 1,020 (11.6) | 1,007 (11.4) |
Aldosterone antagonists | 893 (10.1) | 927 (10.5) |
Thiazide-like diuretics | 517 (5.9) | 519 (5.9) |
Other potassium sparring diuretics | 35 (0.4) | 34 (0.4) |
Antiangina drugs, n (%) | 1,707 (19.4) | 1,770 (20.1) |
Antithrombotic drugs, n (%) | 1,086 (12.3) | 1,150 (13.1) |
DOAC | 738 (8.4) | 784 (8.9) |
Vitamin K antagonists | 336 (3.8) | 340 (3.9) |
Heparin | 17 (0.2) | 30 (0.3) |
Antiarrhythmic drugs, n (%) | 266 (3.0) | 329 (3.7) |
Weight management–related medications | ||
Phentermine, n (%) | 5 (< 0.1) | 7 (< 0.1) |
Phentermine and topiramate, n (%) | 4 (< 0.1) | 3 (< 0.1) |
Lorcaserin, n (%) | 3 (< 0.1) | 0 |
Orlistat, n (%) | 2 (< 0.1) | 4 (< 0.1) |
Naltrexone-bupropion, n (%) | 2 (< 0.1) | 4 (< 0.1) |
Other, n (%) | 2 (< 0.1) | 2 (< 0.1) |
ACE = angiotensin-converting enzyme; ARNI = angiotensin receptor–neprilysin inhibitor; ASA = acetylsalicylic acid; CV = cardiovascular; DOAC = direct oral anticoagulant; FAS = full analysis set.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
Table 18: Summary of Concomitant CV-Management and Weight Management–Related and Glucose-Lowering Treatments Initiated in the In-Trial Observation Period From the SELECT Trial (FAS)
Treatment | Semaglutide 2.4 mg | Placebo |
|---|---|---|
CV-related medications | ||
CV drugs, n (%) | 2,735 (31.1) | 2,963 (33.7) |
Beta blockers | 1,307 (14.8) | 1,312 (14.9) |
Calcium channel blockers | 924 (10.5) | 1,132 (12.9) |
Angiotensin receptor blockers | 886 (10.1) | 1,021 (11.6) |
ACE inhibitors | 834 (9.5) | 904 (10.3) |
ARNI | 151 (1.7) | 195 (2.2) |
Lipid-lowering drugs, n (%) | 2,046 (23.2) | 2,475 (28.1) |
Statins | 1,493 (17.0) | 1,778 (20.2) |
Ezetimibe | 724 (8.2) | 983 (11.2) |
PCSK-9 inhibitors | 148 (1.7) | 195 (2.2) |
Fibrates | 87 (1.0) | 136 (1.5) |
Eicosapentaenoic acid ethyl ester | 17 (0.2) | 41 (0.5) |
Bempedoic acid | 21 (0.2) | 39 (0.4) |
Other omega-3 triglycerides | 16 (0.2) | 18 (0.2) |
Bile acid sequestrants | 10 (0.1) | 6 (< 0.1) |
Omega-3-acid ethyl ester | 2 (< 0.1) | 11 (0.1) |
Platelet aggregation inhibitors, n (%) | 1,172 (13.3) | 1,296 (14.7) |
P2Y12 inhibitors | 764 (8.7) | 875 (9.9) |
ASA | 557 (6.3) | 620 (7.0) |
Other | 52 (0.6) | 62 (0.7) |
Diuretics, n (%) | 1,139 (12.9) | 1,585 (18.0) |
Loop diuretics | 539 (6.1) | 789 (9.0) |
Aldosterone antagonists | 392 (4.5) | 495 (5.6) |
Thiazides | 332 (3.8) | 443 (5.0) |
Thiazide-like diuretics | 192 (2.2) | 284 (3.2) |
Other potassium sparring diuretics | 11 (0.1) | 12 (0.1) |
Antithrombotic drugs, n (%) | 1,114 (12.7) | 1,204 (13.7) |
Heparin | 656 (7.5) | 702 (8.0) |
DOAC | 564 (6.4) | 616 (7.0) |
Vitamin K antagonists | 89 (1.0) | 108 (1.2) |
Antiangina drugs, n (%) | 735 (8.3) | 783 (8.9) |
Antiarrhythmic drugs, n (%) | 344 (3.9) | 366 (4.2) |
Weight management–related medications | ||
Naltrexone-bupropion, n (%) | 4 (< 0.1) | 21 (0.2) |
Phentermine, n (%) | 4 (< 0.1) | 9 (0.1) |
Phentermine and topiramate, n (%) | 4 (< 0.1) | 3 (< 0.1) |
Orlistat, n (%) | 1 (< 0.1) | 10 (0.1) |
Lorcaserin, n (%) | 1 (< 0.1) | 4 (< 0.1) |
Other, n (%) | 34 (0.4) | 119 (1.4) |
Glucose-lowering medicationsa | ||
SGLT2 inhibitors | 213 (2.4) | 332 (3.8) |
Biguanides and metformin | 110 (1.2) | 504 (5.7) |
Insulins | 39 (0.4) | 69 (0.8) |
GLP-1 RAs (including tirzepatide)b | 30 (0.3) | 100 (1.1) |
DPP-4 inhibitors | 6 (< 0.1) | 23 (0.3) |
Sulfonylureas | 4 (< 0.1) | 31 (0.4) |
Thiazolidinediones | 1 (< 0.1) | 3 (< 0.1) |
Alpha glucosidase inhibitors | 0 | 1 (< 0.1) |
Others | 2 (< 0.1) | 8 (< 0.1) |
ACE = angiotensin-converting enzyme; ARNI = angiotensin receptor–neprilysin inhibitor; ASA = acetylsalicylic acid; CV = cardiovascular; DOAC = direct oral anticoagulant; DPP-4 = dipeptidyl peptidase-4; FAS = full analysis set; GLP-1 RA = glucagon-like peptide-1 receptor agonist; SGLT2 = sodium-glucose cotransporter-2.
Note: Initiation of a medication could be due to adding a new medication or switching from one product to another which was perceived as a change in medication by the investigator.
aGlucose-lowering medications (except GLP-1 RA) could be initiated during the trial if the patient developed diabetes.
bInitiating another GLP-1 RA was not permitted during the entire trial, otherwise, treatment with the trial product was discontinued.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
Efficacy
A summary of key efficacy results based on the in-trial observation period and a data cut-off date of July 18, 2023, is presented in Table 19.
CV Outcomes
MACE (CV Death, Nonfatal MI, or Nonfatal Stroke)
At the data cut-off date, the percentages of patients (in the FAS) with their first EAC-confirmed MACE (consisting of CV death, nonfatal MI, or nonfatal stroke) were 6.5% of 8,803 patients in the semaglutide 2.4 mg group and 8.0% of 8,801 patients in the placebo group. Semaglutide 2.4 mg was favoured over placebo (adjusted HR = 0.80; 95% CI, 0.72 to 0.90). A cumulative incidence plot of time from randomization to first EAC-confirmed MACE is presented in Figure 3 in Appendix 1. A summary of absolute risk difference from the supplementary analysis of confirmatory end points at week 156 is presented in Table 27 in Appendix 1. The absolute risk difference in time to first EAC-confirmed MACE between semaglutide 2.4 mg and placebo at week 156 was −0.011 (95% CI, −0.019 to −0.004) (for interpretation of results, transformed to −1.1%; 95% CI, −1.9% to −0.4%).
The 2-way tipping-point sensitivity analysis suggested that the results from the primary analysis were robust (i.e., the observed treatment effect of semaglutide 2.4 mg compared with placebo was unlikely affected by missing data).
The 2 additional sensitivity analyses, which used multiple imputation to impute events for patients who withdrew consent or were lost to follow-up, estimated the HRs for MACE, which were the same as the result from the primary analysis, and estimated the associated 95% CIs, which were consistent with the result from the primary analysis. For the sensitivity analysis using imputation of event times based on patients who permanently discontinued trial treatment, the HR was 0.80 (95% CI, 0.72 to 0.90). For the sensitivity analysis using imputation of event times based on patients not receiving treatment at some point during the trial, the HR was 0.80 (95% CI, 0.72 to 0.89).
The on-treatment supplementary analysis — which defined treatment exposure as the period from the date of the first dose until the first time during which no dose had been administered in the past 5 weeks or the end of the in-trial period, whichever occurred first — estimated the HR and associated 95% CI for MACE (HR = 0.76; 95% CI, 0.67 to 0.87), and the result was consistent with the result from the primary analysis.
The competing risk supplementary analysis on EAC-confirmed non-CV deaths (141 in the semaglutide 2.4 mg group and 176 in the placebo group) estimated the HR and associated 95% CI, which were similar to the result from the primary analysis (EAC-confirmed non-CV death HR of 0.77; 95% CI, 0.62 to 0.95).
Consultation with the clinical experts did not identify any relevant potential treatment-effect modifiers to examine for this review. However, BMI was identified as the most relevant subgroup for the purpose of this review to inform expert committee deliberations and, as such, a summary of the subgroup analysis results by BMI for the primary end point is presented in Table 28 in Appendix 1.
EAC-confirmed events of MACE with onset after the in-trial observation period comprised 6 events of undetermined cause of death in the placebo group: 1 event of non-CV, nonrenal death in the placebo group; 3 events of acute MI (1 event in the semaglutide 2.4 mg group and 2 events in the placebo group); and 2 events of stroke in the placebo group. Note that events of MI and stroke were not systematically collected outside the in-trial period. For patients who did not complete the study, vital status was collected at the end of trial to the extent possible.
CV Death
At the data cut-off date, the percentages of patients (in the FAS) with an EAC-confirmed CV death (including undetermined cause of death) were 2.5% in the semaglutide 2.4 mg group and 3.0% in the placebo group (HR = 0.85; 95% CI, 0.71 to 1.01). A cumulative incidence plot of time from randomization to EAC-confirmed CV death is presented in Figure 4 in Appendix 1. The absolute risk difference in time to EAC-confirmed CV death between semaglutide 2.4 mg and placebo at week 156 was 0% (95% CI, −0.5% to 0.4%).
As with the primary analysis, the 2 sensitivity analyses using multiple imputation to impute events for those who were lost to follow-up or withdrew consent estimated HRs for CV death and associated 95% CIs (for both sensitivity analyses: HR = 0.84; 95% CI, 0.70 to 1.00) that were consistent with the result from the confirmatory secondary analysis.
The on-treatment supplementary analysis estimated the HR for CV death and associated 95% CI (HR = 0.85; 95% CI, 0.67 to 1.07) that were consistent with the result from the confirmatory secondary analysis.
HF Composite (CV Death or Hospitalization for HF or Urgent HF Visit)
At the data cut-off date, the percentages of patients (in the FAS) with their first EAC-confirmed composite HF outcome (comprising CV death or HF requiring hospitalization or urgent HF visit) were 3.4% in the semaglutide 2.4 mg group and 4.1% in the placebo group (HR = 0.82; 95% CI, 0.71 to 0.96). A cumulative incidence plot of time from randomization to first EAC-confirmed composite HF outcome is presented in Figure 5 in Appendix 1. The absolute risk difference in time to EAC-confirmed composite HF outcome between semaglutide 2.4 mg and placebo at week 156 was −0.2% (95% CI, −0.8% to 0.3%).
The 2 sensitivity analyses using multiple imputation to impute events for those who were lost to follow-up or withdrew consent estimated HRs for the HF composite outcome (and associated 95% CIs) that were consistent with the result from the confirmatory secondary analysis. For the sensitivity analysis using imputation of event times based on patients who permanently discontinued trial treatment, the HR was 0.82 (95% CI, 0.71 to 0.96). For the sensitivity analysis using imputation of event times based on patients not receiving treatment at some point during the trial, the HR was 0.82 (95% CI, 0.70 to 0.96).
The on-treatment supplementary analysis estimated the HR for the HF composite outcome and associated 95% CI (HR = 0.76; 95% CI, 0.63 to 0.93) that were consistent with the result from the confirmatory secondary analysis.
The supplementary analysis replacing the CV death component with all-cause death in the HF composite outcome estimated the HR and associated 95% CI (HR = 0.80; 95% CI, 0.70 to 0.91), which were consistent with the result from the confirmatory secondary analysis.
All-Cause Death
At the data cut-off date, the percentages of patients (in the FAS) with an EAC-confirmed all-cause death were 4.3% in the semaglutide 2.4 mg group and 5.2% in the placebo group (HR = 0.81; 95% CI, 0.71 to 0.93). A cumulative incidence plot of time from randomization to EAC-confirmed all-cause death is presented in Figure 6 in Appendix 1. The absolute risk difference in time to EAC-confirmed all-cause death between semaglutide 2.4 mg and placebo at week 156 was −0.5% (95% CI, −1.1% to 0.1%).
The 2 sensitivity analyses using multiple imputation to impute events for those who were lost to follow-up or withdrew consent estimated HRs for all-cause death and associated 95% CIs that were consistent with the result from the confirmatory secondary analysis. For the sensitivity analysis using imputation of event times based on patients who permanently discontinued trial treatment, the HR was 0.81 (95% CI, 0.71 to 0.93). For the sensitivity analysis using imputation of event times based on patients not receiving treatment at some point during the trial, the HR was 0.81 (95% CI, 0.70 to 0.93).
The on-treatment supplementary analysis estimated the HR for all-cause death and associated 95% CI (HR = 0.74; 95% CI, 0.61 to 0.89) that were consistent with the result from the confirmatory secondary analysis.
The supplementary analysis based on an extended in-trial observation period that included follow-up for vital status in patients who withdrew consent or were lost to follow-up estimated the HR for all-cause death and associated 95% CI (HR = 0.81; 95% CI, 0.71 to 0.92) that were consistent with the result from the confirmatory secondary analysis.
Nonfatal MI
At the data cut-off date, the percentages of patients (in the FAS) with their first EAC-confirmed nonfatal MI were 2.7% in the semaglutide 2.4 mg group and 3.7% in the placebo group (HR = 0.72; 95% CI, 0.61 to 0.85).
The supplementary analysis on first EAC-confirmed fatal or nonfatal MI estimated the HR and associated 95% CI (HR = 0.72; 95% CI, 0.61 to 0.85), which were consistent with the result from the supportive secondary analysis.
Nonfatal Stroke
At the data cut-off date, the percentages of patients (in the FAS) with their first EAC-confirmed nonfatal stroke were 1.7% in the semaglutide 2.4 mg group and 1.9% in the placebo group (HR = 0.93; 95% CI, 0.74 to 1.15).
The supplementary analysis on first EAC-confirmed fatal or nonfatal stroke estimated the HR and associated 95% CI (HR = 0.89; 95% CI, 0.72 to 1.11), which were consistent with the result from the supportive secondary analysis.
Glucose Metabolism
At the data cut-off date, 306 patients (3.5% of the FAS) in the semaglutide 2.4 mg group and 1,059 patients (12.0% of the FAS) in the placebo group experienced a first occurrence of a hemoglobin A1C of 6.5% or greater (HR = 0.27; 95% CI, 0.24 to 0.31).
Renal Outcomes
At the data cut-off date, 155 patients (1.8% of the FAS) in the semaglutide 2.4 mg group and 198 patients (2.2% of the FAS) in the placebo group had experienced a first composite nephropathy event (HR = 0.78; 95% CI, 0.63 to 0.96).
Anthropometric Outcomes
The mean treatment difference in the percent change from baseline in body weight at week 104 between semaglutide 2.4 mg and placebo was −8.51% (95% CI, −8.75% to −8.27%).
The sensitivity analysis that imputed missing values in both groups using an imputation model for the placebo group only, irrespective of adherence to randomized treatment at week 104, estimated the treatment difference (mean treatment difference of −7.39%; 95% CI, −7.63% to −7.16%), which was similar to the result from the main analysis for change in body weight.
The analyses at weeks 156 and 208 were not performed as planned because greater than 10% of patients in the FAS missed the relevant annual study visit due to trial termination. However, an analysis of the percent change from baseline in body weight was performed at week 208 and the estimated treatment difference (based on the in-trial period) between semaglutide 2.4 mg and placebo was −8.7% (95% CI, −9.42% to −7.88%). The on-treatment analysis, which defined treatment exposure as the observation period until first time off treatment for greater than 35 days, estimated the treatment difference (−10.2%; 95% CI, −11.0% to −9.42%), which was similar to the result from the main analysis for change in body weight at week 208.24
Health-Related Quality of Life
The treatment difference in the change from baseline in EQ-5D index score at week 104 between semaglutide 2.4 mg and placebo was 0.01 (95% CI, 0.01 to 0.02).
The treatment difference in the change from baseline in EQ VAS score at week 104 between semaglutide 2.4 mg and placebo was 1.60 (95% CI, 1.16 to 2.04).
Table 19: Summary of Key Efficacy Results From the In-Trial Observation Period in the SELECT Trial (FAS)
Outcome | Semaglutide 2.4 mg (N = 8,803) | Placebo (N = 8,801) |
|---|---|---|
CV outcomes | ||
Time to first EAC-confirmed MACEa | ||
Observation time (patient-years) | 28,655 | 28,297 |
Number of patients with event, n (%) | 569 (6.5) | 701 (8.0) |
CV and undetermined cause of death | 191 (2.2) | 221 (2.5) |
CV death | 128 (1.5) | 141 (1.6) |
Undetermined cause of death | 63 (0.7) | 80 (0.9) |
Nonfatal acute MI | 230 (2.6) | 321 (3.6) |
Nonfatal stroke | 148 (1.7) | 159 (1.8) |
Non-CV, nonrenal death as competing event, n (%) | 141 (1.6) | 176 (2.0) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.80 (0.72 to 0.89) | |
One-sided P value | < 0.0001 | |
Two-sided P value | < 0.0001 | |
Adjusted HR (95 CI%) | 0.80 (0.72 to 0.90) | |
Adjusted 1-sided P value | < 0.0001 | |
Adjusted 2-sided P value | < 0.0001 | |
Time to EAC-confirmed CV deathb | ||
Observation time (patient-years) | 29,283 | 29,112 |
Number of patients with event, n (%) | 223 (2.5) | 262 (3.0) |
CV death | 146 (1.7) | 172 (2.0) |
Undetermined cause of death | 77 (0.9) | 90 (1.0) |
Non-CV, nonrenal death as competing event, n (%) | 152 (1.7) | 196 (2.2) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.85 (0.71 to 1.01) | |
One-sided P valuec | 0.0327 | |
Two-sided P value | 0.0653 | |
Time to first EAC-confirmed composite HF outcomeb | ||
Observation time (patient-years) | 29,165 | 28,944 |
Number of patients with event, n (%) | 300 (3.4) | 361 (4.1) |
CV and undetermined cause of death | 203 (2.3) | 240 (2.7) |
CV death | 133 (1.5) | 154 (1.7) |
Undetermined cause of death | 70 (0.8) | 86 (1.0) |
HF requiring hospitalization or urgent HF visit | 97 (1.1) | 121 (1.4) |
HF hospitalization | 95 (1.1) | 113 (1.3) |
Urgent HF visit | 2 (< 0.1) | 8 (< 0.1) |
Non-CV, nonrenal death as competing event, n (%) | 144 (1.6) | 190 (2.2) |
Administrative censoring, n (%) | ||
Completed trial | █████ ██████ | █████ ██████ |
Lost to follow-up | ███ █████ | ███ █████ |
Withdrawal by patient | ██ █████ | ██ █████ |
HR (95% CI) | 0.82 (0.71 to 0.96) | |
One-sided P valued,e | 0.0066 | |
Two-sided P value | 0.0132 | |
Time to EAC-confirmed all-cause deathb | ||
Observation time (patient-years) | 29,283 | 29,112 |
Number of patients with event, n (%) | 375 (4.3) | 458 (5.2) |
CV death | 146 (1.7) | 172 (2.0) |
Undetermined cause of death | 77 (0.9) | 90 (1.0) |
Non-CV, nonrenal death | 152 (1.7) | 196 (2.2) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.81 (0.71 to 0.93) | |
One-sided P valued,f | 0.0015 | |
Two-sided P value | 0.0029 | |
Time to first EAC-confirmed nonfatal MIb | ||
Observation time (patient-years) | 28,890 | 28,565 |
Number of patients with event, n (%) | 234 (2.7) | 322 (3.7) |
STEMI | 42 (0.5) | 52 (0.6) |
NSTEMI | 139 (1.6) | 192 (2.2) |
Undetermined | 53 (0.6) | 78 (0.9) |
Competing events, n (%) | ||
CV death | 139 (1.6) | 150 (1.7) |
Non-CV, nonrenal death | 146 (1.7) | 178 (2.0) |
Undetermined cause of death | 70 (0.8) | 86 (1.0) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.72 (0.61 to 0.85) | |
Two-sided P valueg | 0.0001 | |
Time to first EAC-confirmed nonfatal strokeb | ||
Observation time (patient-years) | 29,036 | 28,839 |
Number of patients with event, n (%) | 154 (1.7) | 165 (1.9) |
Ischemic | 141 (1.6) | 147 (1.7) |
Hemorrhagic | 10 (0.1) | 14 (0.2) |
Undetermined | 3 (< 0.1) | 4 (< 0.1) |
Competing events, n (%) | ||
CV death | 135 (1.5) | 160 (1.8) |
Non-CV, nonrenal death | 147 (1.7) | 193 (2.2) |
Undetermined cause of death | 69 (0.8) | 84 (1.0) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.93 (0.74 to 1.15) | |
Two-sided P valueg | 0.4985 | |
Glucose metabolism | ||
Time to first occurrence of hemoglobin A1C ≥ 48 mmol/mol (6.5%)h | ||
Number of patients contributing to the analysis, n (%) | 8,800 (99.97) | 8,797 (99.95) |
Observation time (patient-years) | 28,914 | 27,386 |
Number of patients with event, n (%) | 306 (3.5) | 1,059 (12.0) |
Competing events, n (%) | ||
CV death | 146 (1.7) | 154 (1.7) |
Non-CV, nonrenal death | 149 (1.7) | 176 (2.0) |
Undetermined cause of death | 77 (0.9) | 80 (0.9) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.27 (0.24 to 0.31) | |
Two-sided P valueg | < 0.0001 | |
Renal outcomes | ||
Time to first composite nephropathy eventb | ||
Observation time (patient-years) | 28,930 | 28,656 |
Number of patients with event, n (%) | 155 (1.8) | 198 (2.2) |
Persistent macroalbuminuria | 144 (1.6) | 179 (2.0) |
Onset of persistent ≥ 50% reduction in eGFR | 9 (0.1) | 15 (0.2) |
Onset of persistent eGFR < 15 mL/min/1.73 m2 | 1 (< 0.1) | 1 (< 0.1) |
EAC-confirmed initiation of chronic renal replacement therapy | 1 (< 0.1) | 3 (< 0.1) |
EAC-confirmed renal death | Not reported | Not reported |
Competing events, n (%) | ||
CV death | 140 (1.6) | 164 (1.9) |
Non-CV, nonrenal death | 151 (1.7) | 192 (2.2) |
Undetermined cause of death | 73 (0.8) | 84 (1.0) |
Administrative censoring, n (%) | ||
Completed trial | ███████████ | ███████████ |
Lost to follow-up | ████████ | ████████ |
Withdrawal by patient | ███████ | ███████ |
HR (95% CI) | 0.78 (0.63 to 0.96) | |
Two-sided P valueg | 0.0191 | |
Anthropometric outcomes | ||
Change from baseline in body weight at week 104i | ||
Body weight at baseline (kg), mean (SD) | 96.53 (17.52) | 96.82 (17.80) |
Body weight at week 104 (kg), mean (SE) | 87.56 (0.08) | 95.79 (0.08) |
Missing, n (%) | ████████ | ████████ |
Number of patients contributing to the analysis, n (%) | 8,605 (97.8) | 8,574 (97.4) |
Change from baseline (%), mean (SE) | −9.39 (0.09) | −0.88 (0.08) |
Treatment group difference vs. placebo (95% CI) | −8.51 (−8.75 to −8.27) | |
Two-sided P valueg | < 0.0001 | |
Health-related quality of life | ||
Change from baseline in EQ-5D index score at week 104i | ||
EQ-5D index score at baseline, mean (SD) | 0.88 (0.15) | 0.88 (0.15) |
EQ-5D index score at week 104, mean (SE) | 0.89 (0) | 0.87 (0) |
Missing, n (%) | ███████████ | ███████████ |
Number of patients contributing to the analysis, n (%) | 8,323 (94.5) | 8,309 (94.4) |
Change from baseline, mean (SE) | 0.01 (0) | −0.01 (0) |
Treatment group difference vs. placebo (95% CI) | 0.01 (0.01 to 0.02) | |
Two-sided P valueg | < 0.0001 | |
Change from baseline in EQ VAS score at week 104i | ||
EQ VAS score at baseline, mean (SD) | 77.15 (15.63) | 77.15 (15.63) |
EQ VAS score at week 104, mean (SE) | 79.83 (0.16) | 78.23 (0.16) |
Missing, n (%) | ███████████ | ███████████ |
Number of patients contributing to the analysis, n (%) | 8,323 (94.5) | 8,309 (94.4) |
Change from baseline, mean (SE) | 2.52 (0.16) | 0.92 (0.16) |
Treatment group difference vs. placebo (95% CI) | 1.60 (1.16 to 2.04) | |
Two-sided P valueg | < 0.0001 | |
Symptom burden | ||
Change from baseline in WRSSM total score at week 104j | ||
WRSSM total score at baseline, mean (SD) | 1.12 (0.77) | 1.13 (0.78) |
WRSSM total score at week 104, mean (SD) | 0.84 (0.73) | 0.98 (0.77) |
Missing, n (%) | ███████████ | ███████████ |
Number of patients contributing to the summary statistic, n (%) | 6,725 (76.4) | 6,635 (75.4) |
Change from baseline, mean (SD) | −0.26 (0.71) | −0.12 (0.68) |
ANCOVA = analysis of covariance; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; CV = cardiovascular; EAC = event adjudication committee; eGFR = estimated glomerular filtration rate; FAS = full analysis set; HF = heart failure; HR = hazard ratio; MACE = major adverse cardiovascular event; MI = myocardial infarction; NSTEMI = non–ST elevation myocardial infarction; SD = standard deviation; SE = standard error; STEMI = ST elevation myocardial infarction; VAS = visual analogue scale; vs. = versus; WRSSM = weight-related sign and symptom measure.
Note: The data cut-off date was July 18, 2023.
aAnalyzed using a Cox proportional hazards model with treatment as categorical fixed factor. The assumption of proportional hazards was evaluated by the standardized score process and was considered supported, per investigator. Patients without events of interest were censored at the end of their in-trial period. Based on the available number of events for analysis, the nominal significance level was updated to 0.02281 using the Lan-DeMets alpha spending function. Adjustment for group sequential design was done using likelihood ratio ordering.
bAnalyzed using a Cox proportional hazards model with treatment as categorical fixed factor. Patients without events of interest were censored at the end of their in-trial observation period.
cBased on the available number of events for analysis, the nominal significance level was updated to 0.01148 using the alpha spending function described in the statistical analysis plan.
dThe superiority of semaglutide 2.4 mg versus placebo was confirmed for the primary end point of time to first EAC-confirmed MACE comprising CV death, nonfatal MI, and nonfatal stroke. Superiority of semaglutide 2.4 mg versus placebo was not confirmed for the confirmatory secondary end point of time to EAC-confirmed CV death. As such, superiority of semaglutide 2.4 mg versus placebo was not tested for the confirmatory secondary end points that followed the order of the prespecified testing hierarchy (time to first EAC-confirmed composite HF outcome and time to EAC-confirmed all-cause death).
eBased on the available number of events for analysis, the nominal significance level was updated to 0.01149 using the alpha spending function described in the statistical analysis plan.
fBased on the available number of events for analysis, the nominal significance level was updated to 0.01213 using the alpha spending function described in the statistical analysis plan.
gNot controlled for multiplicity.
hAnalyzed using a Cox proportional hazards model with treatment as categorical fixed factor. Patients without events of interest were censored at the end of their in-trial observation period. Patients randomized in error with a baseline hemoglobin A1C ≥ 48 mmol/mol (6.5%) were excluded from this analysis.
IAnalyzed using an ANCOVA with treatment as fixed factor and baseline value as covariate. Before analysis, missing data were multiple imputed. The imputation model (linear regression) was done separately for each treatment group and included baseline value as a covariate and was fitted to all patients with a measurement regardless of treatment adherence at week 104. The fitted model was used to impute values for patients without a measurement at week 104. Mean estimates were adjusted according to observed baseline distribution.
jPresented descriptively only.
Sources: Clinical Study Report version 1.0 for the SELECT trial25 and the Novo Nordisk Canada Inc. responses to the January 15, 2025,26 and January 23, 2025,27 requests by CDA-AMC for additional information for the CDA-AMC review of Wegovy. Details included in the table are from the sponsor’s summary of clinical evidence.2
Symptom Burden
The mean change from baseline in WRSSM total score at week 104 was −0.26 (SD = 0.71) in the semaglutide 2.4 mg group and −0.12 (SD = 0.68) in the placebo group. The between-group difference was not estimated.
Cardiometabolic Risk Factors
Efficacy results on cardiometabolic risk factors (change from baseline in systolic blood pressure, change from baseline in total cholesterol, and change from baseline in HDL cholesterol at week 104) were used to inform the accompanying pharmacoeconomic analysis and are presented in Table 29 in Appendix 1.
Harms
A summary of harms results based on the in-trial observation period is presented in Table 20.
Adverse Events
As described in the Clinical Study Report, nonserious AEs not fulfilling any of the prespecified criteria were not systematically collected. The focus of the safety evaluation per the Clinical Study Report for the SELECT trial was based on the reporting of SAEs and other systematically collected events (i.e., AEs of special interest).
Serious AEs
The proportion of patients with an SAE was 33.41% (2,941 of 8,803 patients) in the semaglutide 2.4 mg group and 36.40% (3,204 of 8,801 patients) in the placebo group. The most frequently reported SAEs (by preferred term) (frequency ≥ 2.0%) were:
coronary arterial stent insertion, reported in 2.78% (245 patients) in the semaglutide 2.4 mg group and 3.17% (279 patients) in the placebo group
acute MI, reported in 2.37% (209 patients) in the semaglutide 2.4 mg group and 3.33% (293 patients) in the placebo group
unstable angina, reported in 2.07% (182 patients) in the semaglutide 2.4 mg group and 2.37% (209 patients) in the placebo group.
Withdrawals Due to AEs
The proportion of patients who permanently stopped treatment due to AEs was 16.60% (1,461 patients) in the semaglutide 2.4 mg group and 8.16% (718 patients) in the placebo group. The most frequently reported AEs (by preferred term) that led to permanent treatment discontinuation (frequency ≥ 2.0%) were:
nausea, reported in 4.29% (378 patients) in the semaglutide 2.4 mg group and 0.40% (35 patients) in the placebo group
diarrhea, reported in 2.35% (207 patients) in the semaglutide 2.4 mg group and 0.36% (32 patients) in the placebo group.
The proportion of patients who had their treatment interrupted or withdrawn due to AEs was 30.32% (2,669 patients) in the semaglutide 2.4 mg group and 16.00% (1,408 patients) in the placebo group. The most frequently reported AEs (by preferred term) that led to dose interruption or withdrawal (frequency ≥ 2.0%) were:
nausea, reported in 8.07% (710 patients) in the semaglutide 2.4 mg group and 0.75% (66 patients) in the placebo group
diarrhea, reported in 5.12% (451 patients) in the semaglutide 2.4 mg group and 0.75% (66 patients) in the placebo group
vomiting, reported in 3.62% (319 patients) in the semaglutide 2.4 mg group and 0.20% (18 patients) in the placebo group
constipation, reported in 2.20% (194 patients) in the semaglutide 2.4 mg group and 0.37% (33 patients) in the placebo group
decreased appetite, reported in 2.15% (189 patients) in the semaglutide 2.4 mg group and 0.22% (19 patients) in the placebo group.
Mortality
There were 375 all-cause deaths (4.26%) in the semaglutide 2.4 mg group and 458 all-cause deaths (5.20%) in the placebo group. There were 371 investigator-reported SAEs with fatal outcome (4.21%) in the semaglutide 2.4 mg group and 460 investigator-reported SAEs with fatal outcome (5.23%) in the placebo group.
Notable Harms
The AEs of special interest identified for this review included CV AEs, cholelithiasis, nausea, vomiting, constipation, GERD, and pancreatitis. SAEs of cardiac disorders were reported in 11.45% (1,008 patients) in the semaglutide 2.4 mg group and 13.45% (1,184 patients) in the placebo group. All other AEs of special interest were reported in less than 2% of patients in each group.
Table 20: Summary of Safety Results From the In-Trial Observation Period in the SELECT Trial (FAS)
AEs | Semaglutide 2.4 mg (N = 8,803) | Placebo (N = 8,801) |
|---|---|---|
SAEs, n (%) | ||
Patients with ≥ 1 SAE | 2,941 (33.41) | 3,204 (36.40) |
Cardiac disorders | 1,008 (11.45) | 1,184 (13.45) |
Coronary artery disorders | 645 (7.33) | 800 (9.09) |
Acute MIs | 209 (2.37) | 293 (3.33) |
Unstable angina | 182 (2.07) | 209 (2.37) |
Cardiac arrhythmias | 287 (3.26) | 319 (3.62) |
Heart failures | 176 (2.00) | 215 (2.44) |
Infections and infestations | 624 (7.09) | 738 (8.39) |
Infections, pathogen unspecified | 323 (3.67) | 388 (4.41) |
Viral infectious disorders | 263 (2.99) | 304 (3.45) |
Nervous system disorders | 444 (5.04) | 496 (5.64) |
Central nervous system vascular disorders | 254 (2.89) | 311 (3.53) |
Surgical and medical procedures | 433 (4.92) | 548 (6.23) |
Vascular therapeutic procedures | 420 (4.77) | 533 (6.06) |
Coronary arterial stent insertion | 245 (2.78) | 279 (3.17) |
Neoplasms — benign, malignant, and unspecified (including cysts and polyps) | 405 (4.60) | 402 (4.57) |
Gastrointestinal disorders | 342 (3.89) | 323 (3.67) |
Injury, poisoning, and procedural complications | 305 (3.46) | 313 (3.56) |
General disorders and administration site conditions | 273 (3.10) | 316 (3.59) |
General system disorders NEC | 163 (1.85) | 189 (2.15) |
Musculoskeletal and connective tissue disorders | 236 (2.68) | 254 (2.89) |
Vascular disorders | 231 (2.62) | 259 (2.94) |
Renal and urinary disorders | 192 (2.18) | 198 (2.25) |
Respiratory, thoracic, and mediastinal disorders | 180 (2.04) | 276 (3.14) |
Patients who permanently stopped treatment due to AEs, n (%) | ||
Patients who permanently stopped treatment | 1,461 (16.60) | 718 (8.16) |
Gastrointestinal disorders | 880 (10.00) | 172 (1.95) |
Gastrointestinal signs and symptoms | 625 (7.10) | 86 (0.98) |
Nausea | 378 (4.29) | 35 (0.40) |
Gastrointestinal motility and defecation conditions | 321 (3.65) | 52 (0.59) |
Diarrhea | 207 (2.35) | 32 (0.36) |
Patients who had their treatment interrupted or treatment withdrawn due to AEs, n (%) | ||
Patients with dose interruption or dose withdrawn | 2,669 (30.32) | 1,408 (16.00) |
Gastrointestinal disorders | 1,613 (18.32) | 323 (3.67) |
Gastrointestinal signs and symptoms | 1,162 (13.20) | 160 (1.82) |
Nausea | 710 (8.07) | 66 (0.75) |
Vomiting | 319 (3.62) | 18 (0.20) |
Gastrointestinal motility and defecation conditions | 687 (7.80) | 107 (1.22) |
Diarrhea | 451 (5.12) | 66 (0.75) |
Constipation | 194 (2.20) | 33 (0.37) |
Infections and infestations | 301 (3.42) | 308 (3.50) |
Viral infectious disorders | 170 (1.93) | 177 (2.01) |
Nervous system disorders | 274 (3.11) | 185 (2.10) |
Metabolism and nutrition disorders | 226 (2.57) | 49 (0.56) |
Appetite and general nutritional disorders | 194 (2.20) | 24 (0.27) |
Decreased appetite | 189 (2.15) | 19 (0.22) |
General disorders and administration site conditions | 214 (2.43) | 94 (1.07) |
General system disorders NEC | 187 (2.12) | 63 (0.72) |
Deaths, n (%) | ||
All-cause deaths | 375 (4.26) | 458 (5.20) |
CV deaths | 146 (1.66) | 172 (1.95) |
Undetermined cause of death | 77 (0.87) | 90 (1.02) |
Non-CV, nonrenal deaths | 152 (1.73) | 196 (2.23) |
Investigator-reported SAEs with fatal outcome | 371 (4.21) | 460 (5.23) |
AEs of special interest, n (%) | ||
SAEs of cardiac disordersa | 1,008 (11.45) | 1,184 (13.45) |
Coronary artery disorders | 645 (7.33) | 800 (9.09) |
Cardiac arrhythmias | 287 (3.26) | 319 (3.62) |
Heart failures | 176 (2.00) | 215 (2.44) |
Myocardial disorders | 18 (0.20) | 23 (0.26) |
Cardiac valve disorders | 12 (0.14) | 21 (0.24) |
Pericardial disorders | 9 (0.10) | 10 (0.11) |
Cardiac disorders, signs and symptoms NEC | 8 (0.09) | 18 (0.20) |
SAEs of gallbladder disorders | ||
Cholelithiasis | 44 (0.50) | 31 (0.35) |
Gallbladder-related disorders (predefined MedDRA search)b | ||
Cholelithiasis | 123 (1.40) | 100 (1.14) |
SAEs of gastrointestinal disorders (predefined MedDRA search)c | ||
Vomiting | 18 (0.20) | 12 (0.14) |
Nausea | 12 (0.14) | 8 (0.09) |
Gastroesophageal reflux disease | 12 (0.14) | 13 (0.15) |
Constipation | 8 (0.09) | 6 (0.07) |
SAEs of exocrine pancreas conditions | ||
Acute pancreatitis | 9 (0.10) | 14 (0.16) |
Pancreatitis | 5 (0.06) | 3 (0.03) |
Obstructive pancreatitis | 4 (0.05) | 7 (0.08) |
Edematous pancreatitis | 1 (0.01) | 1 (0.01) |
Relapsing pancreatitis | 0 | 1 (0.01) |
Pancreatitis (predefined MedDRA search)d | ||
Acute pancreatitis | 12 (0.14) | 15 (0.17) |
Pancreatitis | 7 (0.08) | 6 (0.07) |
Obstructive pancreatitis | 5 (0.06) | 7 (0.08) |
Chronic pancreatitis | 3 (0.03) | 1 (0.01) |
Edematous pancreatitis | 1 (0.01) | 1 (0.01) |
Relapsing pancreatitis | 0 | 1 (0.01) |
EAC-confirmed events of acute pancreatitise | 17 (0.19) | 24 (0.27) |
AE = adverse event; CV = cardiovascular; EAC = event adjudication committee; FAS = full analysis set; MedDRA = Medical Dictionary for Regulatory Activities; MI = myocardial infarction; NEC = not elsewhere classified; SAE = serious adverse event.
Note: The data cut-off date was July 18, 2023.
The preceding table was sorted in descending order by system organ class, high-level group term, and preferred term based on the proportion of patients with at least 1 event in either group. Only frequencies of ≥ 2.0% are summarized in the table, except for all-cause deaths and AEs of special interest.
aCV AEs were collected systematically when serious. In addition, potential events of acute coronary syndrome, stroke, coronary artery revascularization, and heart failure for adjudication by the EAC were reported, irrespective of seriousness. A broad evaluation of CV safety, based on SAEs in the system organ class of cardiac disorders was performed. The evaluation of CV safety was supplemented by the assessment of blood pressure and heart rate.
bAll AEs of gallbladder disease were collected systematically, irrespective of seriousness. Gallbladder-related disorders were evaluated based on a predefined MedDRA search on all AEs, supplemented with additional information collected from specific event forms for events of gallbladder disease.
cGastrointestinal AEs were collected systematically when serious. Gastrointestinal safety was evaluated based on SAEs in the system organ class of gastrointestinal disorders.
dAll AEs of pancreatitis were collected systematically, irrespective of seriousness. The evaluation of pancreatitis was based on a predefined MedDRA search for pancreatitis among all AEs, which provided a broad evaluation of all types of investigator-reported pancreatitis, including search terms indicative of acute or chronic pancreatitis; additional information was collected from a specific event form for pancreatitis, and the outcome of the adjudication by the EAC, the evaluation of which focused on events of acute pancreatitis.
eA total of 69 patients (35 in the semaglutide 2.4 mg group and 34 in the placebo group) had a medical history of acute pancreatitis at screening. None of the patients with a history of acute pancreatitis had EAC-confirmed events of acute pancreatitis during the in-trial period.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
Critical Appraisal
Internal Validity
The SELECT trial had a group sequential design, a type of adaptive clinical trial design that plans for interim analyses at predetermined points during follow-up and as data are collected throughout the trial.28 This trial design offers structured opportunities for making decisions earlier when the intervention shows a significant benefit (i.e., allows early stopping for efficacy, futility, or safety concerns). It can also improve efficiency in research by allowing adjustments to be made in the trial design, such as reducing the required sample size and duration, compared with traditional fixed-period RCTs. However, this trial design requires strong and appropriately conservative statistical plans because of the potential for unblinding, investigator influence, overestimation of treatment effect, and an increased chance of inflated type I error.28-30 These points, combined with the need for a follow-up time that is sufficiently long to observe deaths and CV events, are especially important for evaluating the treatment effect of semaglutide 2.4 mg in preventing further CV morbidity and mortality. As will be described, the SELECT trial’s design, conduct, and analysis mitigate some of the potential limitations.
Randomization used appropriate methods (a 1:1 ratio using an interactive web response system) to achieve allocation and allocation concealment. Overall, the baseline characteristics were similar between groups, suggesting randomization successfully distributed potential confounders. In consultation with the clinical experts, the baseline characteristics did not reflect any notable imbalances in the known prognostic factors for CV events and treatment-effect modifiers. As such, the review team concluded that the methods used for randomization were appropriate and that the risk of bias arising from the randomization process was low.
The percentage of patients who discontinued the study was relatively small (approximately 3%) and similar between the groups. The reasons for discontinuing the study (patient withdrawal and lost to follow-up) were also similar between the groups and, therefore, the risk of attrition bias was likely low. The overall treatment discontinuation rates were similar between groups (approximately 30%). An imbalance in the discontinuation of treatment due to AEs was observed (16.3% in the semaglutide 2.4 mg group and 7.9% in the placebo group). This imbalance could have contributed to differential unblinding because those participants with treatment-emergent AEs — many of which were gastrointestinal in nature — might have inferred their assignment to semaglutide. Similarly, investigators might have formed expectations based on these observations, thereby introducing the potential for performance and reporting bias. While the observed difference between groups in treatment discontinuation due to lack of effect was relatively small, it was still differential (0.7% in the semaglutide 2.4 mg group and 2.8% in the placebo group) and, therefore, does not completely negate the potential impact on reporting and performance biases (i.e., relatively less concerning for biases). In consideration of the group sequential design, an additional key risk of bias is operational bias, in which the knowledge of treatment allocation potentially influences subsequent trial conduct, such as changes in how end points are assessed or reported near interim time points and recruitment patterns. However, an independent external EAC performed ongoing adjudication, based on a blinded review, of predefined event types according to definitions and guidelines outlined in its charter. In addition to reports by the investigator, events for adjudication were identified by the EAC and through standardized screening. Further, it was reported that the 2008 FDA guidance for evaluating CV safety in glucose-lowering therapies was adhered to for best practice on event adjudication and safety evaluation.57 Unblinded data were reviewed exclusively in closed sessions, attended only by the independent external data-monitoring committee and the independent statistical analysts responsible for interim analyses, while blinded data were discussed in open sessions that included all relevant trial members, including sponsor representatives. Overall, the review team concluded the probability for introducing bias into the results was low.
Supplementary analyses of absolute risk difference were conducted at week 156 of the trial to assess the robustness of various end points. A total of 27.3% of patients who were randomized in the trial did not have a visit at week 156 due to trial closure; however, the distribution by treatment group was not reported. The trial was event driven and would not be completed until the target number of primary end point events was reached. While the general assumption for censored observations was that the risk of experiencing an event was not changed by censoring, the potential for bias in the estimated cumulative incidence at week 156 would arise only if those patients who were censored due to what was termed “trial closure” in the Clinical Study Report had a different risk of MACE compared with those who remained in the study. However, the definition of trial closure in the Clinical Study Report is not entirely clear. Administrative censoring included trial completion, lost to follow-up, and patient withdrawal. The implications of administrative censoring for the robustness of absolute risk difference in MACE occurrences are difficult to determine based on the available information. The review team noted that the between-group distribution in administrative censoring, including “trial closure,” was similar and, therefore, if bias exists, it is not likely an important factor in the cumulative incidence estimates. Further, a total of 79.4% of patients who were randomized in the trial did not have a visit at week 208 due to trial closure. The review team considered this large percentage of missing data to seriously undermine the validity and reliability of the percent change from baseline in body weight at week 208 reported by Ryan et al.24 There is a high chance that missing information was due to informative censoring and, in the absence of additional analyses evaluating the missing data patterns, it is not possible to draw firm conclusions on the results from the analyses on body weight at week 208.
Several important site-level and patient-level protocol deviations in quality and good clinical practice were reported at 2 sites that resulted in serious breaches. Other serious breaches (electronic data capture system shutdown, flooding at 1 study site that resulted in the loss of trial data, potential external access to trial data due to hacking at 1 study site, and lack of principal investigator oversight at 1 study site) were concluded by the investigator to have a potential impact on data integrity (validity, reliability, and robustness of the trial data). Therefore, the review team considered these to be serious breaches potentially undermining the validity and reliability of the trial data, particularly when fraudulent data were entered for patient questionnaires (i.e., patient-reported outcomes) by a study nurse for missed visits to enable the questionnaire for the upcoming visit to become activated in the system. However, the overall impact on the trial data is likely low, given the small proportion of patients potentially impacted by these serious breaches.
Important potential limitations of the group sequential design include early stopping rules that may overestimate treatment effect and multiple interim statistical testing points that can introduce bias if the stopping threshold is met at a random peak in treatment effect. While the use of a single interim analysis in the SELECT trial reduces the risk of bias compared with multiple interim tests, the absence of a futility boundary means that the trial was not designed to stop early if semaglutide 2.4 mg showed little or no benefit versus placebo. However, given the trial continued to its final analysis without stopping for efficacy, and a reasonable approach to controlling for multiplicity was used (Lan-DeMets alpha spending function approximating O’Brien-Fleming boundaries) for the primary end point, the concern of treatment-effect inflation due to early termination does not apply.
For the primary end point, the final analysis was adjusted for the group sequential design by using likelihood ratio ordering. This method accounts for the adaptive nature of interim analyses by incorporating the sequential monitoring plan into the estimation of treatment effects, aiming to control the type I error and avoid overestimation of efficacy due to early stopping. In this case, the unadjusted HR was 0.80 (95% CI, 0.72 to 0.89) and the adjusted HR was 0.80 (95% CI, 0.72 to 0.90), indicating minimal impact of the adjustment. The similarity between the unadjusted and adjusted estimates suggested that the single interim analysis conducted in the trial had little influence on the final effect size, likely because the trial was not stopped early for efficacy. This additional information supports the robustness of the reported treatment effect in terms of it being unlikely that the estimated treatment effect was influenced by type I error potentially associated with the group sequential design.
The proportional hazards assumption for the primary end point, which the study investigators reported as being met, was evaluated graphically using Schoenfeld residuals and the standardized score process. However, the standardized score plot showed a strong downward slope from 0 in the first 12 months, returning to 0 at approximately month 21. Thereafter, it fluctuated near 0 until the end of the follow-up period. The early deviation suggested a violation of the proportional hazards assumption in the first 12 months, although the stabilization afterward indicated the issue was not persistent over the full duration of the study. The statistical analysis plan and Clinical Study Report for the SELECT trial did not describe any methods for assessing the potential impact of this violation on the results, such as adding time-dependent covariates to the Cox models or conducting sensitivity analyses with models that do not rely on the proportional hazards assumption. While visual inspection of the score process suggested a time-dependent effect on the proportional hazards, there was no clear evidence that this influenced the confirmatory results in a meaningful way.
The superiority hypothesis was tested for each confirmatory secondary end point under multiplicity control using a stagewise hierarchical testing scheme according to the prespecified order. The statistical testing strategy for the confirmatory secondary end points used a separate alpha spending function to control the type I error rate at a 1-sided level of 2.5%, which aligned with the P value adjustment for the group sequential design. Importantly, the end points — the composite HF outcome and all-cause death — could not be interpreted formally for superiority because the prespecified hierarchical testing procedure failed to reach statistical significance with the analysis of CV deaths, meaning they should be interpreted as exploratory rather than confirmatory.
The SELECT trial performed statistical comparisons for the nonconfirmatory secondary end points and some exploratory end points. However, the statistical comparisons were not included in the approach to adjust for multiple comparisons and, therefore, they increase the risk of type I error (false-positive findings). The absence of a prespecified multiplicity control strategy for these end points limits the strength of the conclusions that can be drawn from the observed differences between groups.
The number of events contributing to the subgroup analyses of patients with a BMI of 40 kg/m2 and greater was considered few (< 50 events in each of the 2 treatment groups), and the sample sizes for these 2 subgroups were considered small (< 10% of patients randomized in each group), thereby lowering the certainty in the consistency of the treatment effect for the primary end point in these subgroups.
The main analysis of the time-to-event end points assumed independent censoring of patients who had withdrawn from the trial or were lost to follow-up, while deaths from causes not included in the end point were handled as censored observations but not part of the independent censoring assumption. Sensitivity analyses (which were planned to address the independent censoring assumption and assess the potential impact of missing data for patients who had withdrawn from the trial or were lost to follow-up) and a supplementary analysis (which was planned to assess the influence of the competing risk of death from causes not included in the end point) were performed for the confirmatory end points. The results of the sensitivity analyses were consistent with the main analyses; the review team judged that the risk of bias due to the handling of these intercurrent events and missing outcomes data for the confirmatory end points was low. Of note, the tipping-point sensitivity analysis was conducted only for the primary analysis and not for the key secondary confirmatory analyses. The review team had concerns about the 2 additional sensitivity analyses that were described as applying to patients who withdrew consent or were lost to follow-up (i.e., permanently discontinued the trial). Both analyses derived event rates from patients who stopped treatment but remained in the trial. As a result, the imputed events for those who permanently discontinued the study may not fully reflect their true risk. These patients might have different characteristics (e.g., worse health status, higher event risk), which could lead to the potential underestimation or overestimation of the true event rate. If the assumption underlying these sensitivity analyses — that off-treatment event rates from retained participants apply equally to those who permanently discontinued the study — does not hold, then the imputed results could introduce bias rather than correct for it. Nonetheless, these sensitivity analyses yielded results similar to those of the primary analysis (which was further supported by the tipping-point sensitivity analysis) and other confirmatory analyses, which suggests that the assumption of independent censoring was reasonable and that the trial’s conclusions are likely robust.
For all objectives, the primary estimand (intention to treat) was used to evaluate the treatment effect irrespective of adherence to treatment or changes to background medication. Although this approach to the handling of intercurrent events may be reflective of clinical practice, it can be a limitation for the interpretation of efficacy results, given that these intercurrent events also have an effect on CV risk. Further, the proportion of patients who discontinued treatment was relatively high (approximately 30%). A supplementary analysis based on the first on-treatment period, which was planned to address the secondary estimand (used to evaluate the effect in all patients who were randomized who had remained on their randomized treatment), was performed for the confirmatory end points. Because the results were consistent with the main analysis, the review team judged that the risk of bias due to treatment discontinuation was low for the confirmatory end points. The proportion of patients initiating a CV-related medication, an SGLT2 inhibitor, or metformin after randomization was, overall, slightly higher with placebo compared with semaglutide 2.4 mg. In consultation with the clinical experts, it was concluded there is a potential for bias against semaglutide 2.4 mg in the assessment of CV outcomes due to this slight imbalance in the prescribing of these medications.
Continuous supportive secondary end points at week 104 were analyzed, with multiple imputation used for missing values under a missing-at-random assumption, which was likely not plausible because there were notable imbalances in the reasons for treatment discontinuation between groups. A sensitivity analysis that was planned to address this assumption for missing data were performed for the change from baseline in body weight at week 104. Because the results were consistent with the main analysis, the review team judged that the risk of bias due to missing outcomes data for this end point was low. In contrast, no sensitivity analysis was planned for change from baseline in the EQ-5D, where missing outcome data were relatively high (approximately 16%) at week 104. As such, the review team concluded there is a potential for risk of bias due to missing outcomes data for these HRQoL end points.
In the protocol amendment dated January 4, 2021, the WRSSM assessment was removed from the supportive secondary end points and included in the exploratory end points because the measure was not sensitive to weight loss in a psychometric evaluation. According to the sponsor, the WRSSM tool was under development at the time of the trial and was used to gather preliminary insights as an exploratory outcome. In this context, no evidence of its measurement properties, including validity, in patients with established CV disease and obesity or overweight was submitted by the sponsor.
External Validity
Evidence from the SELECT trial addressed the evidence gap on the effects of semaglutide 2.4 mg on CV outcomes, which was raised in the previous submission for the weight management indication, but only for a subset of patients in the indicated population.
There was overlap between the SELECT trial population and the population criteria specified by the sponsor in its reimbursement request: patients with a BMI of 27 kg/m2 or greater and established CV disease (MI, stroke, and/or PAD). The clinical experts consulted for this review agreed that the inclusion criteria captured the target population with established CV disease (including HF) that is seen in practice and is in need of an intervention for the secondary prevention of future CV events. Notably, most patients in the trial had prediabetes according to the hemoglobin A1C range defined in the American Diabetes Association Standards of Medical Care in Diabetes published in 2018 (i.e., 5.7% to < 6.5%).23 However, it is important to note that the Diabetes Canada Clinical Practice Guidelines31 specify a slightly different hemoglobin A1C range (6.0% to < 6.5%) for diagnosing prediabetes. In consultation with the clinical experts, no major concern with the generalizability of the results due to this difference was identified.
Because treatment with semaglutide 2.4 mg in practice is not expected to be limited to patients who do not have diabetes, the review team found the trial’s exclusion of patients with a hemoglobin A1C level of 6.5% or greater to be a concern for the generalizability of the results to the target population. The investigator indicated that the trial population did not include patients with type 1 or type 2 diabetes, to remove any confounding that a diagnosis of diabetes could have on future CV risk. For more information on this evidence gap and how evidence from the SUSTAIN-6 trial addressed that gap, refer to the study addressing the gap in the systematic review evidence section in this document.
One exclusion criterion for the SELECT trial was patients who had experienced an MI, stroke, hospitalization for unstable angina pectoris, or TIA in the past 60 days before screening. In clinical practice, the clinical experts indicated that the typical treatment paradigm is to start drugs known to reduce the risk of future CV events at the time of the triggering CV event and continue these drugs long-term.
In consultation with the clinical experts, it was concluded that the comparison with placebo added to the standard of care for CV disease (antihypertensives, lipid-lowering drugs, anticoagulants, and ASA and other antiplatelet drugs) was appropriate, given that none of the current and accessible standard-of-care therapies to treat or prevent CV disease directly target weight loss. Regarding the standard of care for CV disease used in the trial, the clinical experts advised that, in practice, semaglutide would be combined with therapies that reduce cardiorenal risk, including combination treatment with ACE inhibitors, angiotensin 2 receptor blockers, statins, and SGLT2 inhibitors.
Long-Term Extension Study
The sponsor did not submit a long-term extension study for this review.
Indirect Evidence
The sponsor did not submit indirect evidence for this review.
Study Addressing the Gap in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor that addresses a gap in the systematic review evidence (Table 21). The following has been summarized and validated by the review team.
Table 21: Summary of Gap in the Systematic Review Evidence
Evidence gap | Study that addresses the gap | |
|---|---|---|
Study description | Summary of key results | |
SELECT trial excluded patients with T2D | A long-term, randomized, double-blind, placebo-controlled, multinational, multicentre trial to evaluate CV and other long-term outcomes with semaglutide in patients with T2D. Analyses of subgroups of patients with T2D, a BMI of ≥ 27 kg/m2, and established CVD are provided subsequently. | These subgroup analyses in patients with T2D and established CVD demonstrated improvements in CV measures, consistent with the full population of the SUSTAIN-6 study as well as the SELECT trial. |
BMI = body mass index; CV = cardiovascular; CVD = cardiovascular disease; T2D = type 2 diabetes.
Sources: SUSTAIN-6 post hoc subgroup analysis Clinical Study Report.58 Details included in the table are from the sponsor’s summary of clinical evidence.2
Description of Study
SUSTAIN-6 was a long-term, multicentre, multinational, randomized, double-blind, parallel-group, controlled trial that evaluated the CV safety and long-term outcomes of semaglutide compared with placebo, when added to the standard of care, in patients with type 2 diabetes at high risk of CV events. A total of 3,297 patients were randomized (1:1:1:1) to receive a once-weekly subcutaneous injection of either semaglutide 0.5 mg (826 patients), semaglutide 1.0 mg (822 patients), placebo 0.5 mg (824 patients), or placebo 1.0 mg (825 patients). Refer to Table 22 for a summary of the SUSTAIN-6 trial.
From the SUSTAIN-6 trial data, a post hoc subgroup analysis was conducted that included patients with a BMI of 27 kg/m2 or greater and established CV disease and type 2 diabetes. Post hoc subgroup analyses were conducted to determine whether the efficacy observed in this patient population was consistent with the SELECT trial and, therefore, the enrolment criteria for this subgroup replicated that of the SELECT study (i.e., limiting the subpopulation to patients with prior MI, stroke [excluding TIA], and/or PAD) and a BMI of ≥ 27 kg/m2). Additionally, although there were 2 maintenance doses used in the overall SUSTAIN-6 study (0.5 mg and 1.0 mg), only patients who had received 1.0 mg were included in the post hoc subgroup analyses because this dose was considered most comparable with that of the SELECT trial. However, the SUSTAIN-6 post hoc subgroup had still received a lower dose of semaglutide (1.0 mg) compared with participants in the SELECT trial (2.4 mg). Because the post hoc subgroup analyses were the most relevant for the purposes of this review, the results summarized in this section include only those pertaining to this subgroup.
Refer to Figure 2 for an overview of the details of the SUSTAIN-6 study.
Figure 2: Overview of the SUSTAIN-6 Trial Design
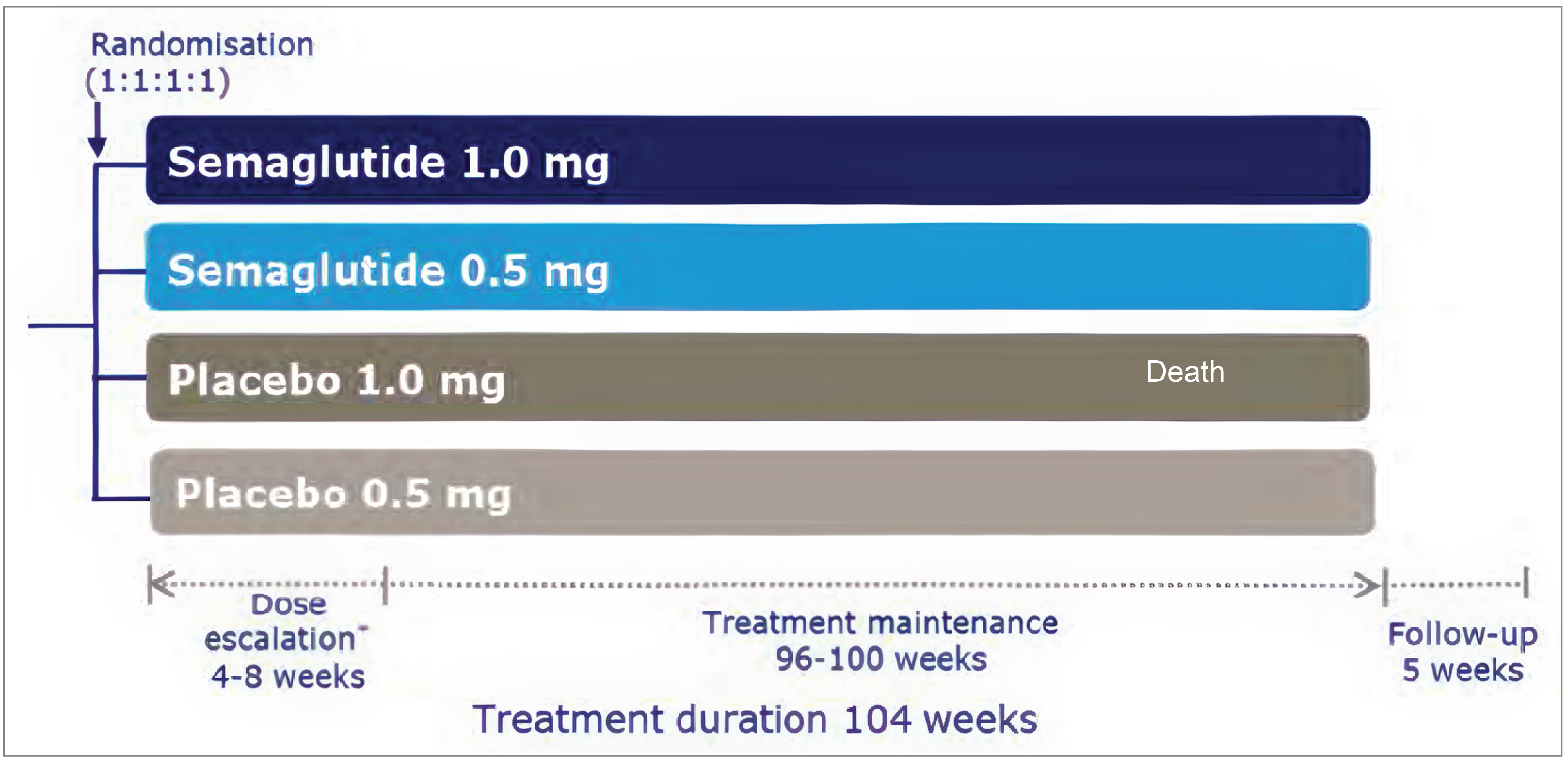
Sources: SUSTAIN-6 post hoc subgroup analysis Clinical Study Report.58 Details included in the table are from the sponsor’s summary of clinical evidence.2
Table 22: Details of the SUSTAIN-6 Study
Detail | SUSTAIN-6 |
|---|---|
Study design | Long-term, multicentre, multinational, randomized, double-blind, parallel-group, controlled trial |
Enrolled, N | 3,297 |
Key inclusion criteria for the SUSTAIN-6 trial |
Inclusion criteria for post hoc subgroup analysis:
|
Key exclusion criteria |
|
Intervention |
Post hoc subgroup analysis:
|
Comparator | Placebo subcutaneous injection (0.5 mg or 1.0 mg) |
Primary end point | Time from randomization to first occurrence of MACE (defined as CV death, nonfatal MI, or nonfatal stroke) |
Secondary end points |
|
Publications | Marso SP, Bain SC, Consoli A, et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016; 375:1834 to 1844.48 |
BMI = body mass index; CHD = coronary heart disease; CV = cardiovascular; HF = heart failure; GLP-1 RA = glucagon-like peptide-1 receptor agonist; MACE = major adverse cardiovascular events; MI = myocardial infarction; NYHA = New York Heart Association; T1D = type 1 diabetes; T2D = type 2 diabetes; TIA = transient ischemic attack.
aBoth dosages in the SUSTAIN-6 study were lower than the recommended dose for the indication under review, aligning more closely with Ozempic than Wegovy.
Sources: SUSTAIN-6 post hoc subgroup analysis Clinical Study Report.58 Details included in the table are from the sponsor’s summary of clinical evidence.2
Population
The post hoc analysis of patients with established CV disease and type 2 diabetes was designed to replicate the enrolment criteria of the SELECT trial, comparing semaglutide 1.0 mg versus placebo 1.0 mg. Patients in the post hoc subgroup analyses were aged 50 years or older with a BMI of 27 kg/m2 or greater, type 2 diabetes, and clinical evidence of CV disease defined by prior MI, stroke (excluding TIA), and/or PAD. Consistent with the exclusion criteria for all SUSTAIN-6 participants, patients in the post hoc subgroup analyses did not have type 1 diabetes and had not used a GLP-1 RA within 90 days before screening.
Interventions
In the post hoc subgroup analyses, patients received either semaglutide 1.0 mg or volume-matched placebo. Although there were 2 maintenance doses in the overall SUSTAIN-6 study, 0.5 mg and 1.0 mg, only patients who had received the 1.0 mg dose were included in the subgroup analyses because this dose was considered by the sponsor as most comparable to that of the SELECT trial. The maintenance dose of 1.0 mg was reached after 4 doses (4 weeks) of 0.25 mg, followed by 4 doses (4 weeks) of 0.5 mg. After the maintenance dose was reached, the dose was not to be changed during the remainder of the trial. The total treatment duration for each patient was 104 weeks.
Outcomes
Refer to Table 22 for a summary of all outcomes included in the SUSTAIN-6 study, which were consistent with the SELECT trial’s CV-related end points. The primary end point was time from randomization to first occurrence of MACE, defined as CV death, nonfatal MI, or nonfatal stroke. The secondary end points identified by the clinical experts as most relevant to this review were time from randomization to first occurrence of all-cause death, nonfatal MI, or nonfatal stroke.
While the expanded composite CV end points (defined as either MACE, revascularization [coronary or peripheral], unstable angina requiring hospitalization, or hospitalization for HF) were also included as secondary outcomes in the SUSTAIN-6 trial, the clinical experts indicated that the 3-component MACE end points were most important to this review because these are the typical CV outcome benchmarks used in clinical trials for CV outcomes and the most clinically important for patients.
Statistical Analysis
An unplanned post hoc analysis of patients with established CV disease and type 2 diabetes replicated the enrolment criteria of the SELECT trial, comparing semaglutide 1.0 mg versus placebo 1.0 mg. Efficacy analyses were conducted using a stratified Cox proportional hazards model.
Results
Patient Disposition
In the post hoc subgroup, approximately ███ of patients in the semaglutide 1.0 mg group and ███ of patients in the placebo group discontinued treatment prematurely. The sponsor did not provide the reasons for discontinuation in this subgroup, but in the full SUSTAIN-6 study population, discontinuation was largely due to gastrointestinal tolerability issues in the semaglutide group (0.5 mg, ████; 1.0 mg, ████) compared with placebo (0.5 mg, ████; 1.0 mg, ████). Treatment discontinuations were reported for 2 different study time frames with different numbers of total participants (N): from 0 to 6 months (patients who were randomized) and from 6 to 24 months (patients who remained and who did not discontinue). In the post hoc subgroup, the highest proportion of premature discontinuations among patients in the semaglutide group took place at between 0 to 6 months (█████ of patients who were randomized) compared with 6 to 24 months (███ of the patients who remained), while placebo-group discontinuations were greatest between 6 and 24 months (█████ of the patients who remained) compared with 0 to 6 months (████ of patients who were randomized) (refer to Table 23 for details). In the larger SUSTAIN-6 study population, common reasons for premature discontinuation in the placebo group were trial fatigue, suspicion of placebo, and introduction of disallowed medication.
Table 23: Summary of Treatment Discontinuations in the SUSTAIN-6 Trial Post Hoc Subgroup
Time to treatment discontinuation | Semaglutide (N = 340) | Placebo (N = 347) | ||
|---|---|---|---|---|
N | Discontinuations, | N | Discontinuations, | |
0 to 6 months | ███ | ████████ | ███ | ███████ |
6 months to 24 months | ███ | ████████ | ███ | ████████ |
Source: SUSTAIN-6 post hoc subgroup analysis Clinical Study Report.58
Baseline Characteristics
For a summary of baseline characteristics and demographics of the post hoc subgroup, refer to Table 24. Overall, the characteristics of the semaglutide 1.0 mg and placebo groups were comparable; patients had a mean age of ██ years (SD = ███), a mean type 2 diabetes disease duration of ██ years (SD = ███), and a mean BMI of ██ kg/m2 (SD = ███). Most patients across both groups had triglyceride levels of ███ mg/dL or greater (███), with a mean triglyceride level of █████ mg/dL (SD = █████). The eGFR levels of patients across both groups were primarily between ██ and ██ mL/min/1.73 m2 (███), with a mean eGFR of ████ mL/min/1.73 m2 (SD = ████). Most patients were taking antihypertensive medication (██████ and/or lipid-lowering drugs ███████. For CV history, only data for prior HF were provided, and ██ (██████) of patients in the semaglutide group had prior HF compared with ███ █████ of the placebo group. Data for other CV inclusion criteria (MI, stroke, and PAD) were not provided.
Table 24: Summary of Baseline Characteristics of the SUSTAIN-6 Trial Post Hoc Subgroup
Characteristic | Semaglutide 1.0 mg | Placebo 1.0 mg |
|---|---|---|
Patient demographics | ||
Age (years), mean (SD) | █████████ | █████████ |
Sex (female), n (%) | █████████ | █████████ |
Type 2 diabetes duration (years), mean (SD) | █████████ | █████████ |
Currently smokes | ████████ | ████████ |
Has previously smoked | █████████ | █████████ |
Has never smoked | █████████ | █████████ |
Body measurements | ||
Body weight (kg), mean (SD) | ██████████ | ██████████ |
BMI (kg/m2), mean (SD) | █████████ | █████████ |
Cardiometabolic health markers | ||
Hemoglobin A1C (%), mean (SD) | ████████ | ████████ |
SBP (mm Hg), mean (SD) | ███████████ | ███████████ |
Total cholesterol (mg/dL), mean (SD) | ███████████ | ███████████ |
HDL cholesterol (mg/dL), mean (SD) | ██████████ | ██████████ |
Triglycerides (mg/dL) | ████████████ | ████████████ |
Triglycerides ≥ 150 mg/dL, n (%) | █████████ | █████████ |
Renal function | ||
eGFR (CKD-EPI; mL/min/1.73 m2), mean (SD) | ██████████ | ██████████ |
eGFR ≥ 90, n (%) | █████████ | █████████ |
60 ≤ eGFR < 90, n (%) | █████████ | █████████ |
eGFR < 60, n (%) | ████████ | ████████ |
Cardiovascular disease | ||
Prior HF, n (%) | ████████ | █████████ |
Antihypertensive medication, n (%) | █████████ | █████████ |
Lipid-lowering drugs, n (%) | █████████ | █████████ |
BMI = body mass index; CKD-EPI = Chronic Kidney Disease Epidemiology Collaboration; eGFR = estimated glomerular filtration rate; HDL = high-density lipoprotein; HF = heart failure; SBP = systolic blood pressure; SD = standard deviation.
Sources: SUSTAIN-6 post hoc subgroup analysis Clinical Study Report.58 Details included in the table are from the sponsor’s summary of clinical evidence.2
Exposure to Study Treatments
No information on exposure to study treatments was provided in the post hoc subgroup analyses.
Efficacy
The outcomes reported for the post hoc subgroup efficacy analyses align with the key outcomes identified for this review. Refer to Table 25 for a summary of efficacy results for the SUSTAIN-6 trial post hoc subgroup, which included patients with a BMI of 27 kg/m2 or greater, established CV disease, and type 2 diabetes. Of all efficacy outcomes included in the post hoc subgroup analysis, only the key secondary outcome of time from randomization to first occurrence of expanded MACE demonstrated nominal statistical significance; however, this outcome was not among those of interest for the purposes of this review for the reasons described earlier (i.e., the 3-component MACE end points were considered the most important to this review because these are the typical CV outcome benchmarks and the most clinically important for patients). The remaining CV-related outcomes demonstrated numerical improvement in the semaglutide group compared with the placebo group; however, they did not achieve statistical significance.
Table 25: Analysis of First Events of Primary and Secondary CV-Related Outcomes in the SUSTAIN-6 Trial Post Hoc Subgroup
Outcome | Semaglutide 1.0 mg | Placebo 1.0 mg |
|---|---|---|
Time from randomization to first MACE | ||
Number of patients with event, n (%) | ███████ | ████████ |
Hazard ratio (95% CI)a | ████████████████ | |
Hazard ratio P value | ██████ | |
Absolute risk ratio (95% CI) | █████████████████ | |
██████████████████████████████████ | ||
Number of patients with event, n (%) | ███████ | ███████ |
Hazard ratio (95% CI)a | ████████████████ | |
Hazard ratio P value | ██████ | |
Absolute risk ratio (95% CI) | █████████████████ | |
██████████████████████████████████ | ||
Number of patients with event, n (%) | █████ | █████ |
Hazard ratio (95% CI)a | ████████████████ | |
Hazard ratio P value | ██████ | |
Absolute risk ratio (95% CI) | █████████████████ | |
██████████████████████████████████ | ||
Number of patients with event, n (%) | █████ | █████ |
Hazard ratio (95% CI)a | ████████████████ | |
Hazard ratio P value | ██████ | |
Absolute risk ratio (95% CI) | █████████████████ | |
███████████████████████████████████████ | ||
Number of patients with event, n (%) | █████ | ███████ |
Hazard ratio (95% CI)a | ████████████████ | |
Hazard ratio P value | ██████ | |
Absolute risk ratio (95% CI) | █████████████████ | |
███████████████████████████████████████████████ | ||
Number of patients with event, n (%) | ███████ | ████████ |
Hazard ratio (95% CI)a | ████████████████ | |
Hazard ratio P value | ██████ | |
Absolute risk ratio (95% CI) | █████████████████ | |
██████████████████████████████████████████████ | ||
Number of patients with event, n (%) | ███████ | ███████ |
Hazard ratio (95% CI)a | ████████████████ | |
Hazard ratio P value | ██████ | |
Absolute risk ratio (95% CI) | █████████████████ | |
██████████████████████████████████████████████████████ | ||
Number of patients with event, n (%) | ███████ | ██████ |
Hazard ratio (95% CI)a | ████████████████ | |
Hazard ratio P value | ██████ | |
Absolute risk ratio (95% CI) | █████████████████ | |
CI = confidence interval; CV = cardiovascular; MACE = major cardiovascular event; MI = myocardial infarction; TIA = transient ischemic attack.
aCox analysis with treatment as a factor, stratified by stratum.
bThe main coronary outcome was either MI or coronary revascularization.
Sources: SUSTAIN-6 post hoc subgroup analysis Clinical Study Report.58 Details included in the table are from the sponsor’s summary of clinical evidence.2
Harms
AEs reported in the post hoc subgroup were limited to SAEs only, from randomization to week 104 of the SUSTAIN-6 trial. The frequencies of SAEs in the post hoc subgroup among patients with established CV disease and type 2 diabetes are reported in Table 26.
Table 26: Summary of Serious Adverse Events Reported by 2% or More of Patients in the SUSTAIN-6 Trial Post Hoc Subgroup [Redacted]
Outcome | Semaglutide 1.0 mg | Placebo 1.0 mg | ||
|---|---|---|---|---|
n (%) | Number of events | n (%) | Number of events | |
Randomization to week 30 visita | ||||
████████████████ | ███ | ███ | ███ | ███ |
████████████████ | ███ | ███ | ███ | ███ |
████████████████ | ███ | ███ | ███ | ███ |
████████████████ | ███ | ███ | ███ | ███ |
████████████████ | ███ | ███ | ███ | ███ |
███████████ | ███ | ███ | ███ | ███ |
████████████████████████ | ||||
██████████████████ | ███ | ███ | ███ | ███ |
████████████████ | ███ | ███ | ███ | ███ |
██████████████████ | ███ | ███ | ███ | ███ |
██████████████████ | ███ | ███ | ███ | ███ |
████████████████████████████████ | ||||
██████████████████ | ███ | ███ | ███ | ███ |
████████████████ | ███ | ███ | ███ | ███ |
██████████████████ | ███ | ███ | ███ | ███ |
██████████████████ | ███ | ███ | ███ | ███ |
██████████████████ | ███ | ███ | ███ | ███ |
██████████████████ | ███ | ███ | ███ | ███ |
███████████ | ███ | ███ | ███ | ███ |
██████████████████ | ███ | ███ | ███ | ███ |
██████████████████ | ███ | ███ | ███ | ███ |
██████████████████ | ███ | ███ | ███ | ███ |
FAS = full analysis set; GI = gastrointestinal.
Note: N is the number of patients experiencing at least 1 event, % is the percentage of patients experiencing at least 1 event, E is the number of events, R is the event rate per 100 years of observation time.
Events with onset between randomization and the week 30 visit. Missing visits are set to the expected number of weeks since randomization, provided the expected date of visit is before the end of the study and consistent with the other dates present. Missing visits are set to the expected number of weeks since randomization, provided the expected date of the visit is before the end of the study and consistent with the other dates present.
aEvents with a missing start date are included in this period.
Source: SUSTAIN-6 post hoc subgroup analysis Clinical Study Report.58
Critical Appraisal
Internal Validity
The post hoc nature of the subgroup analysis in the SUSTAIN-6 trial introduces important internal validity concerns and a risk of bias. Because the subgroup was not prespecified, the analysis is exploratory rather than confirmatory, increasing the risk of spurious findings due to multiple comparisons. Additionally, patients in the SUSTAIN-6 trial were not randomized within the subgroup of interest, meaning that unmeasured confounders could bias the treatment-effect estimate. It is notable that the reported baseline characteristics were generally similar between the groups. The Cox proportional hazards model assumes proportional hazards, but no evidence was provided to confirm that this assumption held within the subgroup, which could affect the validity of the estimated HRs. In the post hoc subgroup analyses, the number of participants was substantially lower than the estimated sample required for adequate power in the SUSTAIN-6 trial. Among all CV-related end points, only expanded MACE reached nominal significance, which may have been due to inadequate power. As such, it is challenging to draw conclusions based on any of the other CV-related outcomes beyond the expanded MACE because they are subject to significant uncertainty and imprecision.
External Validity
A major limitation of the SUSTAIN-6 trial in the context of the current resubmission is that the dosages evaluated in the trial, specifically, semaglutide 0.5 mg and 1.0 mg, do not align with the proposed Health Canada–recommended therapeutic-maintenance dosage of semaglutide (Wegovy), which is 2.4 mg once weekly. The focus of the post hoc subgroup of the SUSTAIN-6 trial was on the 1.0 mg dose because, according to the sponsor, it was most comparable to the dose in the SELECT trial; however, this remains a significant limitation to the generalizability of the evidence. The small sample size of the post hoc subgroup may limit the generalizability of the findings to the broader population of patients with a BMI of 27 kg/m2 or greater, established CV disease, and type 2 diabetes. While the post hoc subgroup included 673 patients, an estimated 80% to 90% of patients in Canada with overweight or obesity have type 2 diabetes, and 32% worldwide have some form of CV disease.59,60 Given the significant heterogeneity in CV disease presentation in this population, driven by factors such as age, duration of diabetes, glycemic control, coexisting conditions (e.g., hypertension, kidney disease), and genetic predisposition,61-64 and the high prevalence of potentially eligible individuals in Canada, the comparatively small sample size somewhat limits the generalizability of the findings. Further, the data describing the baseline characteristics was limited, including a lack of specific data about the history of prior MI, stroke, and/or PAD in the population included in the analysis.
Discussion
Summary of Available Evidence
Evidence from the SELECT trial (N = 17,604) was submitted to address the evidence gap regarding the effects of semaglutide 2.4 mg once weekly on CV outcomes in the indicated population, which was raised in the previous submission for the weight management indication.
The SELECT trial was a phase IIIb, multinational, multicentre, randomized, double-blind, parallel-group, placebo-controlled trial that evaluated the effect of semaglutide 2.4 mg subcutaneous injection compared with placebo in reducing the risk of MACE occurrences (CV death, nonfatal MI, or nonfatal stroke) in patients with established CV disease and overweight or obesity and without diabetes. The key secondary objective of the trial was to evaluate the effect of semaglutide in mortality compared with placebo. Patients were randomized in a 1:1 ratio to receive once-weekly treatment with either semaglutide 2.4 mg or placebo, both as an adjunct to the standard of care for CV disease. A total of 8,803 patients were randomized to receive semaglutide 2.4 mg and 8,801 patients were randomized to receive placebo. The mean age of patients in the semaglutide 2.4 mg group was 61.6 years (SD = 8.9 years) and 61.6 years (SD = 8.8 years) in the placebo group. The mean BMI of patients in the semaglutide 2.4 mg group was 33.30 kg/m2 (SD = 5.03 kg/m2) and 33.37 kg/m2 (SD = 5.04 kg/m2) in the placebo group. Most patients in the FAS had a history of MI — 5,962 patients (67.7%) in the semaglutide 2.4 mg group and 5,944 patients (67.5%) in the placebo group. The mean duration of follow-up was 39.9 months (SD = 9.3 months) in the semaglutide 2.4 mg group and 39.7 months (SD = 9.5 months) in the placebo group. The mean duration of exposure to semaglutide 2.4 mg was 33.3 months (SD = 14.4 months) and to placebo was 35.1 months (SD = 13.0 months).
To supplement this evidence, data from the SUSTAIN-6 trial (N = 3,297) were submitted to address the effects of semaglutide on CV outcomes in the target reimbursement population (patients with a BMI of 27 kg/m2 or greater and established CV disease) who also have type 2 diabetes.
A post hoc subgroup analysis was conducted on patients who received semaglutide 1.0 mg or placebo. Although the CV outcomes investigated in the SUSTAIN-6 trial were consistent with those of the SELECT trial, no efficacy end points achieved statistical significance compared with placebo. However, CV SAEs were fewer with semaglutide 1.0 mg versus placebo, and the harms profile was consistent with that of the SELECT trial.
Interpretation of Results
Efficacy
When considering improved outcomes, patients have emphasized the importance of access to effective treatments that can address both overweight and obesity and CV risks, as well as improve mobility, mental health, and overall well-being. Clinicians have indicated that outcomes that are considered in clinical practice to define treatment response are generally aligned with the outcomes used in clinical trials, namely, percentage and categorical weight loss, cardiometabolic parameters, and patient-reported outcomes.
The SELECT trial demonstrated that semaglutide 2.4 mg once weekly, as an adjunct to the standard of care for CV disease, was associated with a reduction in the estimated HR for MACE (adjusted HR for the composite of CV death, nonfatal MI, or nonfatal stroke = 0.80; 95% CI, 0.72 to 0.90) at any given time during the study period compared with placebo. This finding was observed over a mean follow-up of 39.8 months (SD = 9.4 months) in patients with established CV disease and a BMI of 27 kg/m2 or greater but without diabetes. The absolute risk difference in the cumulative incidence of MACE at week 154 was −0.011 (95% CI, −0.019 to −0.004) (for the interpretation of the results, this was transformed to –1.1%; 95% CI, –1.9% to –0.4%). In the absence of a validated minimal important difference to contextualize the clinical significance of this result, input from the clinical experts consulted for this review suggested that this reduction in MACE occurrences was clinically meaningful to both patients and clinicians. The clinical experts emphasized that even seemingly small absolute reductions in event rates can have substantial implications for patient outcomes, especially in the context of an add-on therapy such as the semaglutide that was used in the trial. Preventing a single fatal or nonfatal MACE is important because it has significant health and economic benefits, including reductions in morbidity and health care resource use and associated costs. However, the 95% CI (–1.9% to –0.4%) indicates some uncertainty in the precise magnitude of the treatment effect. The lower bound of –1.9% suggests the potential for an even greater reduction in MACE occurrences, while the upper bound of –0.4% represents lower certainty in the benefit with semaglutide 2.4 mg, although it does not cross zero (i.e., it does not indicate the absence of treatment effect or possible harm). The reduced precision in the between-group difference may also reflect that 27.3% of patients did not have a week 156 visit due to trial closure. Nonetheless, given the burden of CV events, especially as a leading cause of morbidity and mortality, the clinical experts suggested that this degree of improvement in event rates could lead to significant benefits when applied across large populations and is likely to be clinically relevant for both patients and health care systems.
The point estimates of the analysis of the individual components of MACE were directionally consistent with the treatment effect of the composite MACE outcome favouring semaglutide 2.4 mg, although the CIs varied in their degree of certainty. The reduction in nonfatal MI events was more precisely estimated (HR = 0.72; 95% CI, 0.61 to 0.85), suggesting a benefit with semaglutide 2.4 mg relative to placebo; however, the comparison was not included in the prespecified hierarchical testing procedure and reduces the certainty in the result. The CIs for nonfatal stroke (HR = 0.93; 95% CI, 0.74 to 1.15) and CV death (HR = 0.85; 95% CI, 0.71 to 1.01) were wider and included values beyond the null, indicating greater uncertainty on the magnitude of treatment effect. Specifically, the CIs for CV death and nonfatal stroke included the possibility of no benefit with respect to these efficacy outcomes. The broader CIs for CV death and nonfatal stroke may reflect lower event rates compared with nonfatal MI, resulting in less statistical power to detect a difference. Additionally, because the primary MACE end point was analyzed using a time-to-first-event approach, patients who experienced 1 type of MACE were no longer at risk for another type within that analysis, potentially contributing to differences in the event rates observed between the composite end point and the individual component analyses.
For the confirmatory secondary end points, the estimated HR for the composite HF outcome was 0.82 (95% CI, 0.71 to 0.96), and for all-cause death, it was 0.81 (95% CI, 0.71 to 0.93). While these CIs suggest a potential benefit with semaglutide 2.4 mg, they also indicate varying degrees of precision. The upper bound of the CI for the HF composite approaches 1.0, reflecting some uncertainty about the magnitude of effect. Similarly, while the CI for all-cause death suggests a reduction in events, the range of plausible effects remains relatively broad. Importantly, these end points could not be interpreted formally for superiority because the prespecified hierarchical testing procedure failed to reach statistical significance with the analysis of CV deaths, meaning they should be interpreted as exploratory rather than confirmatory.
The SUSTAIN-6 study post hoc subgroup analyses of patients with type 2 diabetes, a BMI of 27 kg/m2 or greater, and established CV disease included a lower number of participants than the estimated sample required for adequate power in the SUSTAIN-6 study, making it challenging to interpret the efficacy of semaglutide 1.0 mg in this patient subgroup. Of all CV-related efficacy outcomes investigated, only time from randomization to first occurrence of expanded MACE reached statistical significance. However, this end point was not identified by the clinical experts as one of the clinically important outcomes for this review. The wide CIs for other end points, including nonfatal MI (HR = ████; 95% CI, ██████████) and CV death (HR = ████; ███████████████), indicate substantial uncertainty in the estimated treatment effects. Additionally, the post hoc nature of the analysis introduces a higher risk of bias because the subgroup was not prespecified and randomization was not stratified within it, meaning that unmeasured confounders could have influenced the results. The semaglutide dose studied in the post hoc subgroup of the SUSTAIN-6 trial (1.0 mg) was below the Health Canada–approved therapeutic dose of 2.4 mg based on the SELECT trial. However, the post hoc subgroup analysis of the SUSTAIN-6 trial was not designed to assess a dose-dependent cardioprotective effect of semaglutide, and it does not provide conclusive evidence on whether lower doses confer similar CV benefits. While semaglutide 1 mg has already demonstrated a clinically meaningful and sustained reduction in CV risk in patients with type 2 diabetes, regardless of BMI,48 there is no clear biological rationale to assume that a higher dose would necessarily lead to greater CV benefit, nor that it would be insufficient at reducing CV risk in individuals with overweight or obesity. Additionally, the post hoc analysis is not necessarily comparable with the SELECT trial because it was conducted in a different population — patients with type 2 diabetes — whereas the SELECT study specifically evaluated semaglutide 2.4 mg in individuals without diabetes. Furthermore, no formal comparison of the similarity of the populations was made to indicate that the results from the SUSTAIN-6 trial subgroup could be generalized in a similar manner to the SELECT trial population. Given these actual and potential differences in study design, population characteristics, and dosing, the results from the post hoc subgroup analysis of the SUSTAIN-6 trial do not directly address the question of whether a lower dose of semaglutide provides the same level of CV protection as observed in the SELECT trial. However, given that semaglutide 1 mg is already available and publicly reimbursed for patients with type 2 diabetes independent of body weight and BMI, the clinical experts emphasized that current diabetes guidelines do not recommend titration to the maximally tolerated dose (2.4 mg) solely for CV benefit. Instead, the dose may be increased from 1.0 mg in some cases to enhance the effects of semaglutide on improving glycemic control and reducing body weight. Neither sponsor-provided study adequately addresses these issues.
In its December 2022 Clinical Review report on semaglutide (Wegovy), available on the project website, CDA-AMC concluded that evidence from the STEP 1 (N = 1,961), STEP 2 (N = 1,210), STEP 3 (N = 611), and STEP 4 (N = 803) trials demonstrated a reduction in body weight after 68 weeks of treatment with semaglutide 2.4 mg in patients with obesity (BMI ≥ 30 kg/m2) or overweight (BMI ≥ 27 kg/m2) with at least 1 weight-related comorbidity (i.e., hypertension, dyslipidemia, obstructive sleep apnea, or CV disease), including patients with type 2 diabetes, compared with placebo.65 Evidence from the SELECT trial suggested that semaglutide 2.4 mg was associated with a reduction in body weight when compared with placebo at week 104. Consistent results were reported by Ryan et al.24 based on a prespecified analysis of the SELECT trial that evaluated weight loss at week 208; however, 79.4% of patients in the trial did not have a study visit at week 208 due to trial closure and, therefore, the results at this time point may not be valid or reliable. Input from the clinical experts consulted for this review suggested that the magnitude of weight loss observed in the SELECT trial for semaglutide 2.4 mg compared with placebo appeared smaller than they would have expected based on their experience and the magnitude of the weight loss observed in the STEP trials. The clinical experts speculated that this observation may be due in part to the high proportion of patients with prediabetes in the trial, which may mean this was a population similar to the patients with type 2 diabetes in the STEP 2 trial who experienced relatively less weight loss with semaglutide.65 Additionally, the clinical experts noted that the SELECT trial population included patients with likely more complex medical histories compared with the trial populations in the STEP trials, including experience receiving drugs that promote weight gain (e.g., beta blockers).
The Canadian Adult Obesity Clinical Practice Guidelines: Pharmacotherapy in Obesity Management, updated in 2022, indicates that regulatory agencies have traditionally suggested discontinuing pharmacotherapy for weight management if a weight loss of at least 5% (from baseline) has not been attained after 3 months on the recommended therapeutic dose. However, the Canadian guideline advises that potential substantial health improvements may be attained without a weight loss of at least 5%.21 The Canadian guideline also indicates that clinical trials of pharmacotherapy for weight management have consistently demonstrated weight regain after treatment discontinuation. As such, the Canadian guideline advises that medications for weight management are intended as part of a long-term treatment strategy.21 There was no long-term extension of the SELECT trial to inform durability of treatment effect after treatment discontinuation (i.e., no evidence for the duration of treatment benefit, outside of changes in weight following discontinuation, was available for this review), but there was an extension of the STEP 1 trial.66 The STEP 1 trial was reviewed by CDA-AMC in the December 2022 Clinical Review report on semaglutide (Wegovy), available on the project website. In the extension analysis (off-treatment period starting at week 68) of the STEP 1 trial (N = 327),67 the mean change from baseline in body weight at week 120 was 11.6% (SD = 7.7%) in the semaglutide 2.4 mg group and 1.9% (SD = 4.8%) in the placebo group.27 CDA-AMC concluded that evidence from the extension of the STEP 1 trial was suggestive of weight regain after discontinuing semaglutide 2.4 mg in patients with obesity or overweight with at least 1 weight-related comorbidity but without diabetes.65
The clinical experts consulted for this review advised that the potential benefits of semaglutide 2.4 mg beyond weight loss (i.e., improved CV outcomes mediated through biomarkers of CV risk, such as glucose metabolism and kidney function) should be considered when assessing its clinical value relative to existing treatments. Evidence from the SELECT trial suggested that semaglutide 2.4 mg was associated with a lower likelihood of developing or progressing to type 2 diabetes while receiving treatment, with 306 patients (3.5%) in the semaglutide 2.4 mg group versus 1,059 (12.0%) in the placebo group experiencing an event (HR = 0.27; 95% CI, 0.24 to 0.31). This is perhaps not surprising because it aligns with the broader evidence base for the metabolic effects of semaglutide and GLP-1 RA in general. Similarly, a lower frequency of composite nephropathy events was observed with semaglutide 2.4 mg (1.8% of patients) compared with placebo (2.2% of patients) (HR = 0.78; 95% CI, 0.63 to 0.96). Although these end points were evaluated in the SELECT trial outside of the hierarchical analysis plan, the results were consistent with results from previous studies (e.g., the FLOW trial68) of semaglutide and what the clinical experts have observed in practice settings.
Evidence from the SELECT trial suggested that semaglutide 2.4 mg was associated with little to no difference in health-related quality of life as assessed by the generic EQ-5D index and EQ VAS tools. Additionally, the trial did not estimate the between-group difference in weight-related symptom burden, as assessed by the change from baseline in the condition-specific WRSSM total score, leaving uncertainty about whether semaglutide 2.4 mg provided meaningful improvements in condition-specific symptoms related to excess weight.
Harms
A targeted approach for the collection of safety data were applied in the SELECT trial (i.e., only prespecified SAEs were systematically collected) based on the rationale that the safety profile of semaglutide is already characterized. No new harms or unexpectedly high frequencies of known harms with semaglutide 2.4 mg were identified. The clinical experts consulted for this review agreed that the distribution and frequency of AEs were as expected based on previous studies and their experience in practice.
In the SELECT trial, AEs leading to permanent discontinuation of treatment were reported more frequently with semaglutide 2.4 mg compared with placebo (16.60% of patients compared with 8.16% of patients, respectively). Similarly, AEs leading to temporary discontinuation of treatment were reported more frequently with semaglutide 2.4 mg compared with placebo (30.32% of patients compared with 16.00% of patients, respectively). Throughout the trial and, in particular, during the dose-escalation phase, the most frequently reported AEs were gastrointestinal-related, suggesting that tolerability at the recommended maintenance dose may be an issue. Of note, the product monograph for semaglutide (Wegovy) advises following a dose-escalation regimen to reduce the likelihood of gastrointestinal symptoms.1 Notably, approximately 75% of patients who were receiving treatment in the semaglutide 2.4 mg group received the planned maintenance dose of 2.4 mg after the initial 16 weeks of dose escalation. Among those who were receiving treatment, the distribution of patients receiving each dose (semaglutide 0.24 mg, 0.5 mg, 1.0 mg, 1.7 mg, and 2.4 mg) remained consistent throughout the trial. Based on their experience in clinical practice, the clinical experts indicated that patients receiving semaglutide 0.25 mg, 0.5 mg, or 1.0 mg weekly have experienced weight loss. Therefore, the clinical experts advised to continue treatment at the maximum tolerated dose for the potential benefits in CV outcomes and, in particular, when the patient is deriving benefit in terms of weight loss, appetite reduction, and improved quality of life. The Health Canada reviewer’s report noted that nearly all patients in the trial (97%) received a potentially therapeutic dose of semaglutide but only approximately 85% were on a recommended dose level for obesity or overweight (1.7 mg to 2.4 mg).56
In the post hoc subgroup analysis of the SUSTAIN-6 trial, which included patients receiving semaglutide 1.0 mg or volume-matched placebo who had a BMI of 27 kg/m2 or greater, established CV disease, and type 2 diabetes, there were fewer CV SAEs with semaglutide 1.0 mg versus placebo, and the overall safety profile of semaglutide was consistent with that of the SELECT trial.
The AEs of special interest identified for this review included CV AEs, cholelithiasis, nausea, vomiting, constipation, GERD, and pancreatitis. Discussion on CV AEs as well as mortality is presented in the preceding efficacy section. All other AEs of special interest were reported infrequently (< 2% of the trial population) during the in-trial period in the SELECT trial.
Conclusion
The results from the SELECT trial were submitted as new evidence for this resubmission to address the evidence gap on the effects of semaglutide 2.4 mg once weekly on weight-related comorbidities, such as CV outcomes, in the indicated weight management population. The SELECT trial demonstrated that semaglutide 2.4 mg once weekly, as an adjunct to the standard of care for CV disease, reduced MACE occurrences compared with placebo over a mean follow-up period of nearly 40 months in patients with established CV disease and with overweight (BMI ≥ 27 kg/m2) or obesity (BMI ≥ 30 kg/m2) but without diabetes. The point estimates of the observed treatment effects for the individual MACE components — CV death, nonfatal MI, or nonfatal stroke — were directionally aligned with the overall reduction in MACE occurrences, although the certainty of the estimated effects varied across components. More specifically, the CIs for CV death and, notably, nonfatal stroke included the possibility of no benefit with respect to these efficacy outcomes. Additionally, the trial suggested a reduction in composite HF events and all-cause mortality with semaglutide 2.4 mg compared with placebo; however, these end points could not be interpreted formally for superiority because the prespecified hierarchical testing procedure failed to reach statistical significance.
No new harms with semaglutide 2.4 mg were identified within the SELECT trial. The harms that were observed were consistent with what has been observed clinically, as per the input from the clinical experts consulted for this review regarding their experience with semaglutide in clinical practice, as well as what has been observed in previous studies. The AEs leading to the permanent or temporary discontinuation of treatment were notable and reported more frequently with semaglutide 2.4 mg compared with placebo. Throughout the trial and, in particular, during the dose-escalation phase, the most frequently reported AEs were gastrointestinal-related, suggesting that tolerability at the recommended maintenance dose may be an issue. The AEs of special interest for this review (cholelithiasis, nausea, vomiting, constipation, GERD, and pancreatitis) were reported infrequently.
References
1.Wegovy (semaglutide): 0.25 mg, 0.5 mg, 1 mg, 1.7 mg or 2.4 mg; solution for subcutaneous injection in a prefilled pen [product monograph]. Mississauga (ON): Novo Nordisk Canada Inc.; 2024 March 22. .
2.Sponsor summary of clinical evidence template: Wegovy semaglutide (injection) 2.4 mg [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Semaglutide; 0.25 mg, 0.5 mg, 1 mg, 1.7 mg or 2.4 mg; solution for subcutaneous injection in a prefilled pen. Mississauga (ON): Novo Nordisk Canada Inc.; 2024 November 27.
3.CADTH Drug Reimbursement Expert Review Committee final recommendation: semaglutide (Wegovy - Novo Nordisk Canada Inc). Ottawa (ON): CADTH; 2022 Oct: https://www.cda-amc.ca/sites/default/files/DRR/2022/SR0725%20Wegovy%20-%20CADTH%20Final%20Recommendation-meta.pdf. Accessed 2025 February 7.
4.Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N Engl J Med. 2023;389(24):2221-2232. PubMed
5.Wharton S, Lau DCW, Vallis M, et al. Obesity in adults: a clinical practice guideline. CMAJ. 2020;192(31):E875-E891. PubMed
6.Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure With Preserved Ejection Fraction. Circulation. 2017;136(1):6-19. PubMed
7.Lopez-Jimenez F, Almahmeed W, Bays H, et al. Obesity and cardiovascular disease: mechanistic insights and management strategies. A joint position paper by the World Heart Federation and World Obesity Federation. Eur J Prev Cardiol. 2022;29(17):2218-2237. PubMed
8.Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. 1999;282(16):1523-1529. PubMed
9.Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol. 2013;28 Suppl 1:68-76. PubMed
10.Renehan AG, Soerjomataram I, Tyson M, et al. Incident cancer burden attributable to excess body mass index in 30 European countries. Int J Cancer. 2010;126(3):692-702. PubMed
11.Perk J, De Backer G, Gohlke H, et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J. 2012;33(13):1635-1701. PubMed
12.Nordestgaard BG, Palmer TM, Benn M, et al. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med. 2012;9(5):e1001212. PubMed
13.Li TY, Rana JS, Manson JE, et al. Obesity as compared with physical activity in predicting risk of coronary heart disease in women. Circulation. 2006;113(4):99-506. PubMed
14.Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health. 2009;9:88. PubMed
15.Bombelli M, Facchetti R, Sega R, et al. Impact of body mass index and waist circumference on the long-term risk of diabetes mellitus, hypertension, and cardiac organ damage. Hypertension. 2011;58(6):1029-1035. PubMed
16.Statistics C. An overview of weight and height measurements on World Obesity Day. 2024; https://www.statcan.gc.ca/o1/en/plus/5742-overview-weight-and-height-measurements-world-obesity-day. Accessed 19 Aug 2024.
17.Statistics C. Health characteristics, annual estimates. Table: 13-10-0096-01 (formerly CANSIM 105-0508). [Internet]. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310009601. Accessed 19 Aug 2024.
18.Dai D, Fernandes J, Sun X, Lupton L, Payne VW, Berk A. Multimorbidity in Atherosclerotic Cardiovascular Disease and Its Associations With Adverse Cardiovascular Events and Healthcare Costs: A Real-World Evidence Study. J Health Econ Outcomes Res. 2024;11(1):75-85. PubMed
19.University of Ottawa Heart I. Heart Health Education. [Internet]. 2024; https://pwc.ottawaheart.ca/education/heart-health-education/general-information. Accessed 18 Aug 2024.
20.Heart, Stroke. Transforming Recovery. [Internet]. 2024; https://www.heartandstroke.ca/what-we-do/our-impact/transforming-recovery. Accessed 18 Aug 2024.
21.Pedersen SD, Manjoo P, Wharton S. Canadian Adult Obesity Clinical Practice Guidelines: Pharmacotherapy for Obesity Management. Available from: https://obesitycanada.ca/guidelines/pharmacotherapy. Accessed February 7, 2025.
22.Naude J, Zentner A, Suresh P, Bittman J, Khan NA. Effect of combined GLP-1 analogue and bupropion/naltrexone on weight loss: a retrospective cohort study. Int J Obes (Lond). 2024;48(8):1118-1125. PubMed
23.2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care. 2018;41(Suppl 1):S13-s27. PubMed
24.Ryan DH, Lingvay I, Deanfield J, et al. Long-term weight loss effects of semaglutide in obesity without diabetes in the SELECT trial. Nat Med. 2024;30(7):2049-2057. PubMed
25.Clinical Study Report: EX9536-4388. SELECT − Semaglutide effects on cardiovascular outcomes in people with overweight or obesity [internal sponsor's report]. Bagsvaerd, Denmark: Novo Nordisk; 2023 September 01.
26.Novo Nordisk Canada Inc. response to January 15, 2025 CDA-AMC request for additional information regarding Wegovy CDA-AMC review [internal additional sponsor's information]. Mississauga (ON): Novo Nordisk Canada Inc.; January 22, 2025. .
27.Novo Nordisk Canada Inc. response to January 23, 2025 CDA-AMC request for additional information regarding Wegovy CDA-AMC review [internal additional sponsor's information]. Mississauga (ON): Novo Nordisk Canada Inc.; January 30, 2025. .
28.Neumann K, Grittner U, Piper SK, et al. Increasing efficiency of preclinical research by group sequential designs. PLoS Biol. 2017;15(3):e2001307. PubMed
29.Grayling MJ, Wason JM. Point estimation following a two-stage group sequential trial. Stat Methods Med Res. 2023;32(2):287-304. PubMed
30.Zhang J, Saju C. A systematic review of randomised controlled trials with adaptive and traditional group sequential designs - applications in cardiovascular clinical trials. BMC Med Res Methodol. 2023;23(1):200. PubMed
31.Diabetes Canada Clinical Practice Guidelines Expert Committee. Diabetes Canada 2018 Clinical Practice Guidelines for the Prevention and Management of Diabetes in Canada. Can J Diabetes. 2018;42(Suppl 1):S1-S325.
32.Kalra S, Sahay R. A Review on Semaglutide: An Oral Glucagon-Like Peptide 1 Receptor Agonist in Management of Type 2 Diabetes Mellitus. Diabetes Ther. 2020;11(9):1965-1982. PubMed
33.Leutner M, Dervic E, Bellach L, Klimek P, Thurner S, Kautzky A. Obesity as pleiotropic risk state for metabolic and mental health throughout life. Translational Psychiatry. 2023;13(1):175. PubMed
34.Luppino FS, de Wit LM, Bouvy PF, et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry. 2010;67(3):220-229. PubMed
35.Petry NM, Barry D, Pietrzak RH, Wagner JA. Overweight and obesity are associated with psychiatric disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Psychosom Med. 2008;70(3):288-297. PubMed
36.Lin HY, Huang CK, Tai CM, et al. Psychiatric disorders of patients seeking obesity treatment. BMC Psychiatry. 2013;13:1. PubMed
37.Federation WO. Weight Stigma. n.d.: https://www.worldobesity.org/what-we-do/our-policy-priorities/weight-stigma#:~:text=It%20has%20been%20proven%20to,through%20fear%20of%20being%20stigmatised. Accessed 9 Jan 2025.
38.Dyer MT, Goldsmith KA, Sharples LS, Buxton MJ. A review of health utilities using the EQ-5D in studies of cardiovascular disease. Health Qual Life Outcomes. 2010;8:13. PubMed
39.Obesity Canada-Obésité C. Canadian Adult Obesity Clinical Practice Guidelines. 2020: https://obesitycanada.ca/wp-content/uploads/2020/08/6-Obesity-Assessment-v4-with-links.pdf. Accessed 9Jan2025.
40.Rueda-Clausen CF, Poddar M, Lear SA, Poirier P, Sharma AM. Canadian Adult Obesity Clinical Practice Guidelines: Assessment of People Living with Obesity. [Internet]. 2020; https://obesitycanada.ca/guidelines/assessment/. Accessed 18 Aug 2024.
41.Landi F, Calvani R, Picca A, et al. Body Mass Index is Strongly Associated with Hypertension: Results from the Longevity Check-up 7+ Study. Nutrients. 2018;10(12). PubMed
42.Mancini GBJ, O'Meara E, Zieroth S, et al. 2022 Canadian Cardiovascular Society Guideline for Use of GLP-1 Receptor Agonists and SGLT2 Inhibitors for Cardiorenal Risk Reduction in Adults. Can J Cardiol. 2022;38(8):1153-1167. PubMed
43.Vrints C, Andreotti F, Koskinas KC, et al. 2024 ESC Guidelines for the management of chronic coronary syndromes: Developed by the task force for the management of chronic coronary syndromes of the European Society of Cardiology (ESC) Endorsed by the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J. 2024;45(36):3415-3537. PubMed
44.Lingvay I, Deanfield J, Kahn SE, et al. Semaglutide and Cardiovascular Outcomes by Baseline HbA1c and Change in HbA1c in People With Overweight or Obesity but Without Diabetes in SELECT. Diabetes Care. 2024;47(8):1360-1369. PubMed
45.Deanfield J, Verma S, Scirica BM, et al. Semaglutide and cardiovascular outcomes in patients with obesity and prevalent heart failure: a prespecified analysis of the SELECT trial. Lancet. 2024;404(10454):773-786. PubMed
46.Food and Drug Administration, CDER. Standardized Definitions for Cardiovascular Outcomes Trials: Draft Recommendations. Division of Metabolism and Endocrinology Products. 20 Aug 2014. [Accessed by the sponsor].
47.Food and Drug Administration, CDER. Guidance for Industry. Endpoints and Standardized Data Collection for Cardiovascular Outcomes Trials: Draft Recommendations. 22 Jul 2009. [Accessed by the sponsor].
48.Marso SP, Bain SC, Consoli A, et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016;375(19):1834-1844. PubMed
49.Marso SP, McGuire DK, Zinman B, et al. Efficacy and Safety of Degludec versus Glargine in Type 2 Diabetes. N Engl J Med. 2017;377(8):723-732. PubMed
50.Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med. 2016;375(4):311-322. PubMed
51.Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. PubMed
52.James WP, Caterson ID, Coutinho W, et al. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med. 2010;363(10):905-917. PubMed
53.Young LH, Viscoli CM, Schwartz GG, et al. Heart Failure After Ischemic Stroke or Transient Ischemic Attack in Insulin-Resistant Patients Without Diabetes Mellitus Treated With Pioglitazone. Circulation. 2018;138(12):1210-1220. PubMed
54.Zhao Y, Herring AH, Zhou H, Ali MW, Koch GG. A multiple imputation method for sensitivity analyses of time-to-event data with possibly informative censoring. J Biopharm Stat. 2014;24(2):229-253. PubMed
55.Glimm E, Maurer W, Bretz F. Hierarchical testing of multiple endpoints in group-sequential trials. Stat Med. 2010;29(2):219-228. PubMed
56.Health Canada reviewer's report: Wegovy (semaglutide) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: semaglutide; 0.25 mg, 0.5 mg, 1 mg, 1.7 mg or 2.4 mg; solution for subcutaneous injection. Novo Nordisk; 2024 November 5. .
57.U.S. Food and Drug Administration. Guidance for Industry. Diabetes Mellitus - Evaluating Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes. 2008. [Accessed by the sponsor].
58.Novo Nordisk Canada I. NN9535-EXPLORATORY Clinical Trial Report. SUSTAIN-6 Post-Hoc Subgroup Analyses [internal sponsor's report]. 2024.
59.Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovasc Diabetol. 2018;17(1):83. PubMed
60.Wharton S, Pedersen SD, Lau DCW, Sharma AM. Weight Management in Diabetes. Can J Diabetes. 2018;42 Suppl 1:S124-s129. PubMed
61.Dal Canto E, Ceriello A, Rydén L, et al. Diabetes as a cardiovascular risk factor: An overview of global trends of macro and micro vascular complications. Eur J Prev Cardiol. 2019;26(2_suppl):25-32. PubMed
62.De Rosa S, Arcidiacono B, Chiefari E, Brunetti A, Indolfi C, Foti DP. Type 2 Diabetes Mellitus and Cardiovascular Disease: Genetic and Epigenetic Links. Front Endocrinol (Lausanne). 2018;9:2. PubMed
63.Goodarzi MO, Rotter JI. Genetics Insights in the Relationship Between Type 2 Diabetes and Coronary Heart Disease. Circ Res. 2020;126(11):1526-1548. PubMed
64.Halter JB, Musi N, McFarland Horne F, et al. Diabetes and cardiovascular disease in older adults: current status and future directions. Diabetes. 2014;63(8):2578-2589. PubMed
65.Drug Reimbursement Review clinical guidance report: SEMAGLUTIDE (WEGOVY) for WEIGHT MANAGEMENT. Ottawa (ON): CADTH; 2022 December: https://www.cda-amc.ca/sites/default/files/DRR/2022/SR0725-Wegovy_combined.pdf. Accessed 2025 February 7.
66.Wilding JPH, Batterham RL, Calanna S, et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N Engl J Med. 2021;384(11):989-1002. PubMed
67.Wilding JPH, Batterham RL, Davies M, et al. Weight regain and cardiometabolic effects after withdrawal of semaglutide: The STEP 1 trial extension. Diabetes Obes Metab. 2022;24(8):1553-1564. PubMed
68.Perkovic V, Tuttle KR, Rossing P, et al. Effects of Semaglutide on Chronic Kidney Disease in Patients with Type 2 Diabetes. N Engl J Med. 2024;391(2):109-121. PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Figure 3: Cumulative Incidence Plot of Time From Randomization to First EAC-Confirmed MACE From the In-Trial Observation Period in the SELECT Trial (FAS)
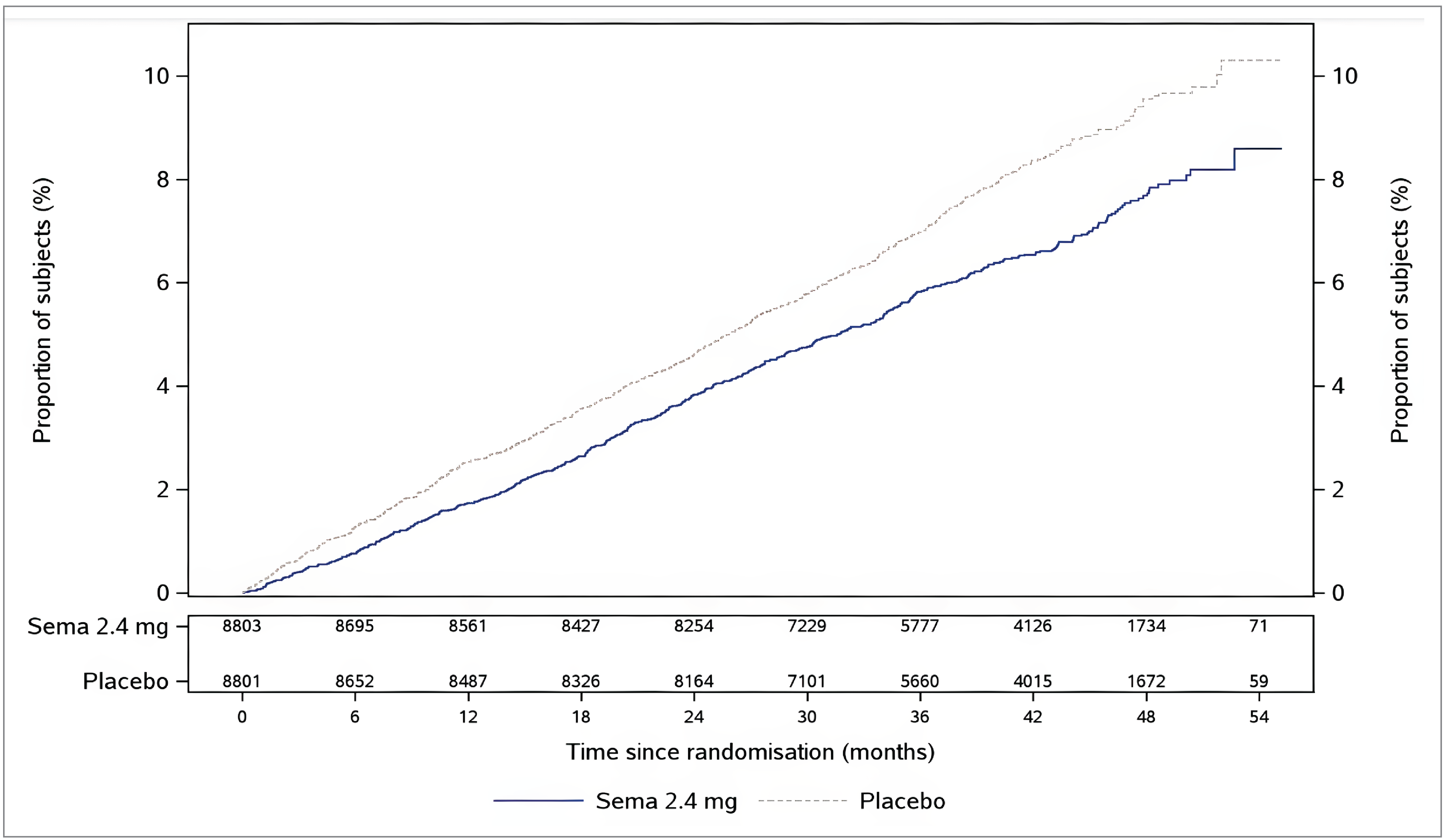
CV = cardiovascular; EAC = event adjudication committee; FAS = full analysis set; MACE = major adverse cardiovascular event.
Note: Cumulative incidence estimates were based on time from randomization to first EAC-confirmed MACE with non-CV death modelled as competing risk using the Aalen-Johansen estimator. Patients without events of interest were censored at the end of their in-trial observation period.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Figure 4: Cumulative Incidence Plot of Time From Randomization to EAC-Confirmed CV Death from the In-Trial Observation Period in the SELECT Trial (FAS)
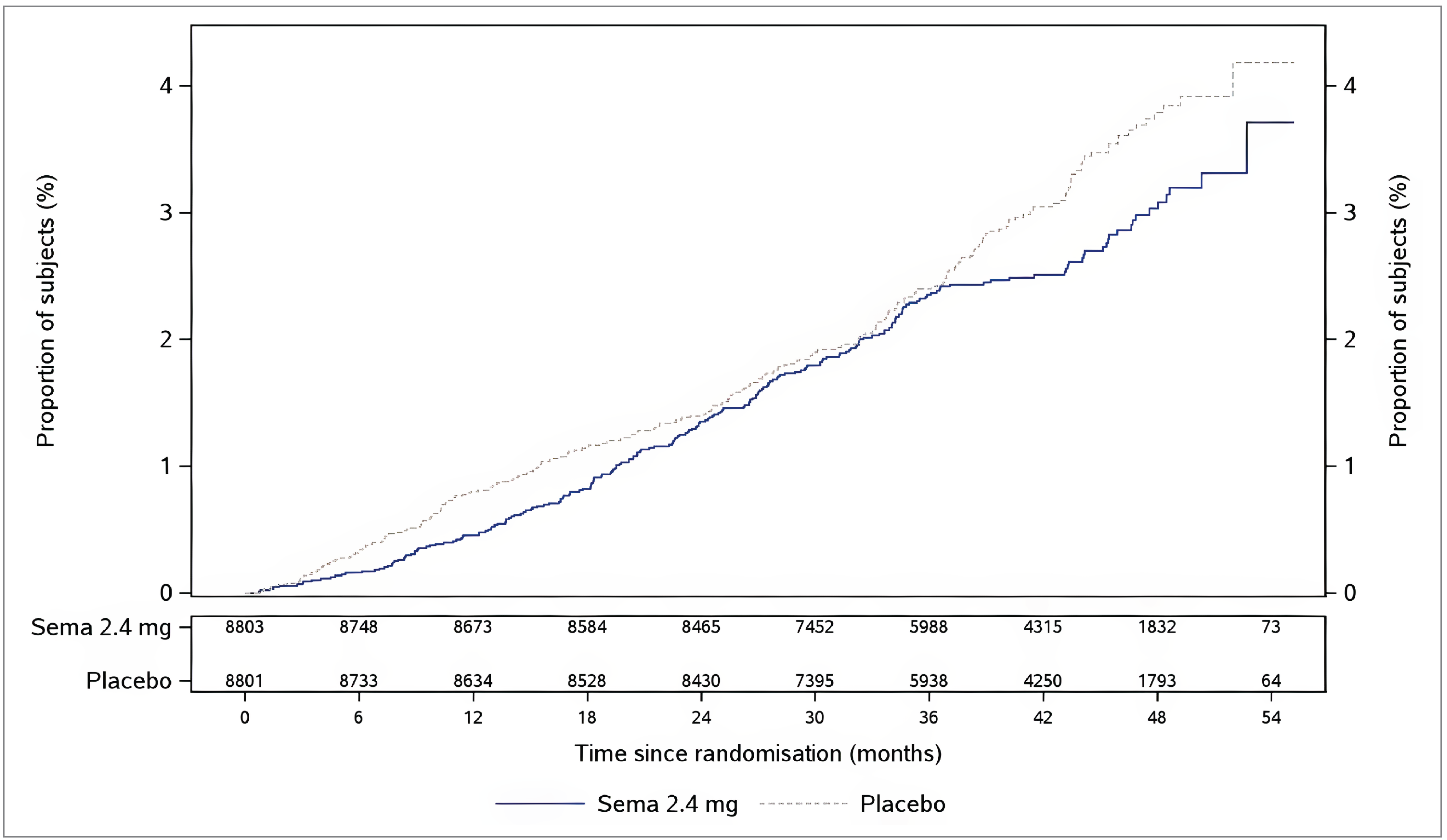
CV = cardiovascular; EAC = event adjudication committee; FAS = full analysis set.
Note: Cumulative incidence estimates were based on time from randomization to EAC-confirmed CV death with non-CV death modelled as competing risk using the Aalen-Johansen estimator. Patients without events of interest were censored at the end of their in-trial observation period. EAC-confirmed CV death included both EAC-confirmed CV death and EAC-confirmed undetermined cause of death.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Figure 5: Cumulative Incidence Plot of Time From Randomization to EAC-Confirmed Composite Heart Failure Outcome From the In-Trial Observation Period in the SELECT Trial (FAS)
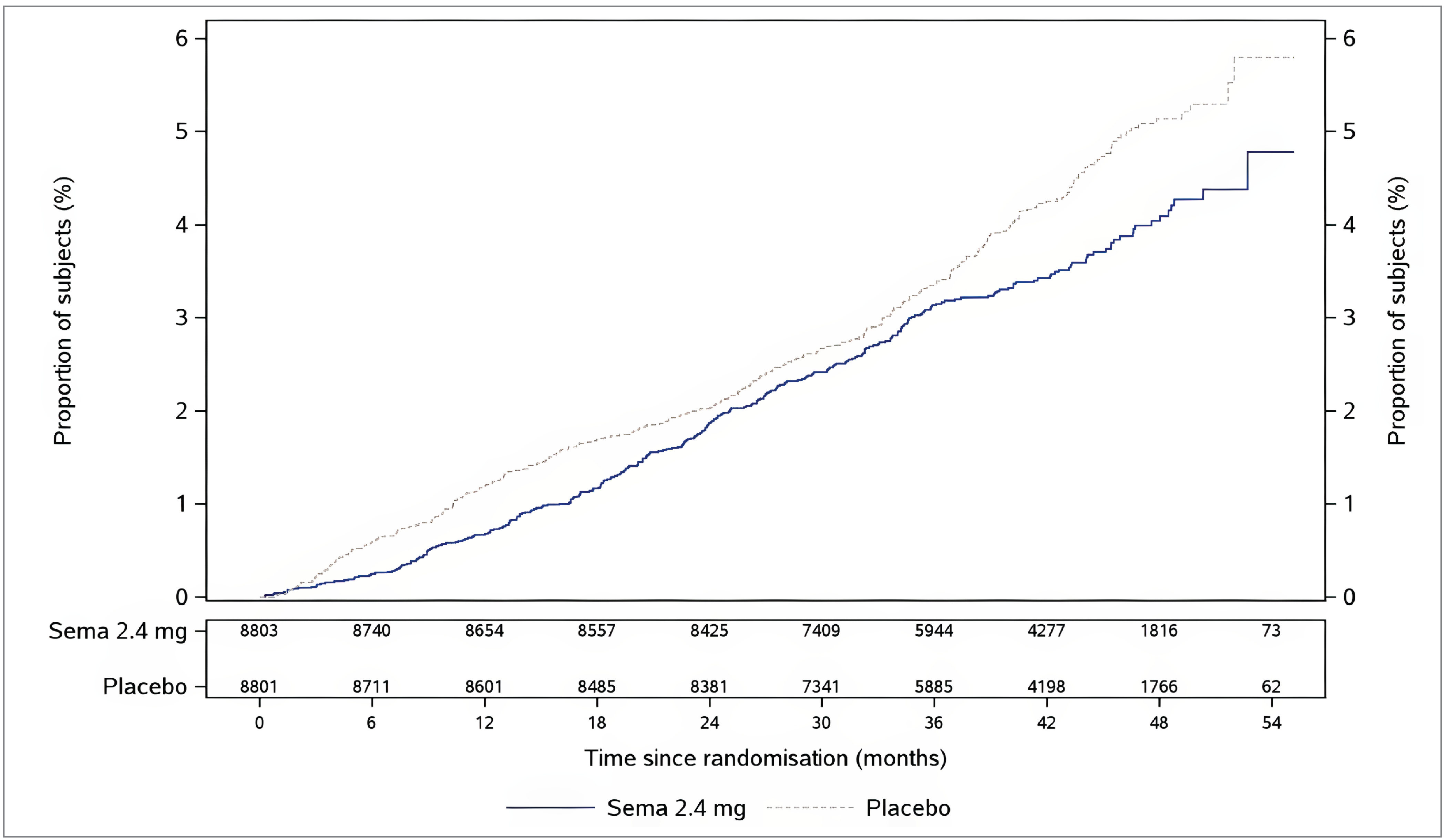
CV = cardiovascular; EAC = event adjudication committee; FAS = full analysis set.
Note: Cumulative incidence estimates were based on time from randomization to first EAC-confirmed composite heart failure outcome with non-CV death modelled as competing risk using the Aalen-Johansen estimator. Patients without events of interest were censored at the end of their in-trial observation period.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Figure 6: Cumulative Incidence Plot of Time From Randomization to EAC-Confirmed All-Cause Death From the In-Trial Observation Period in the SELECT Trial (FAS)
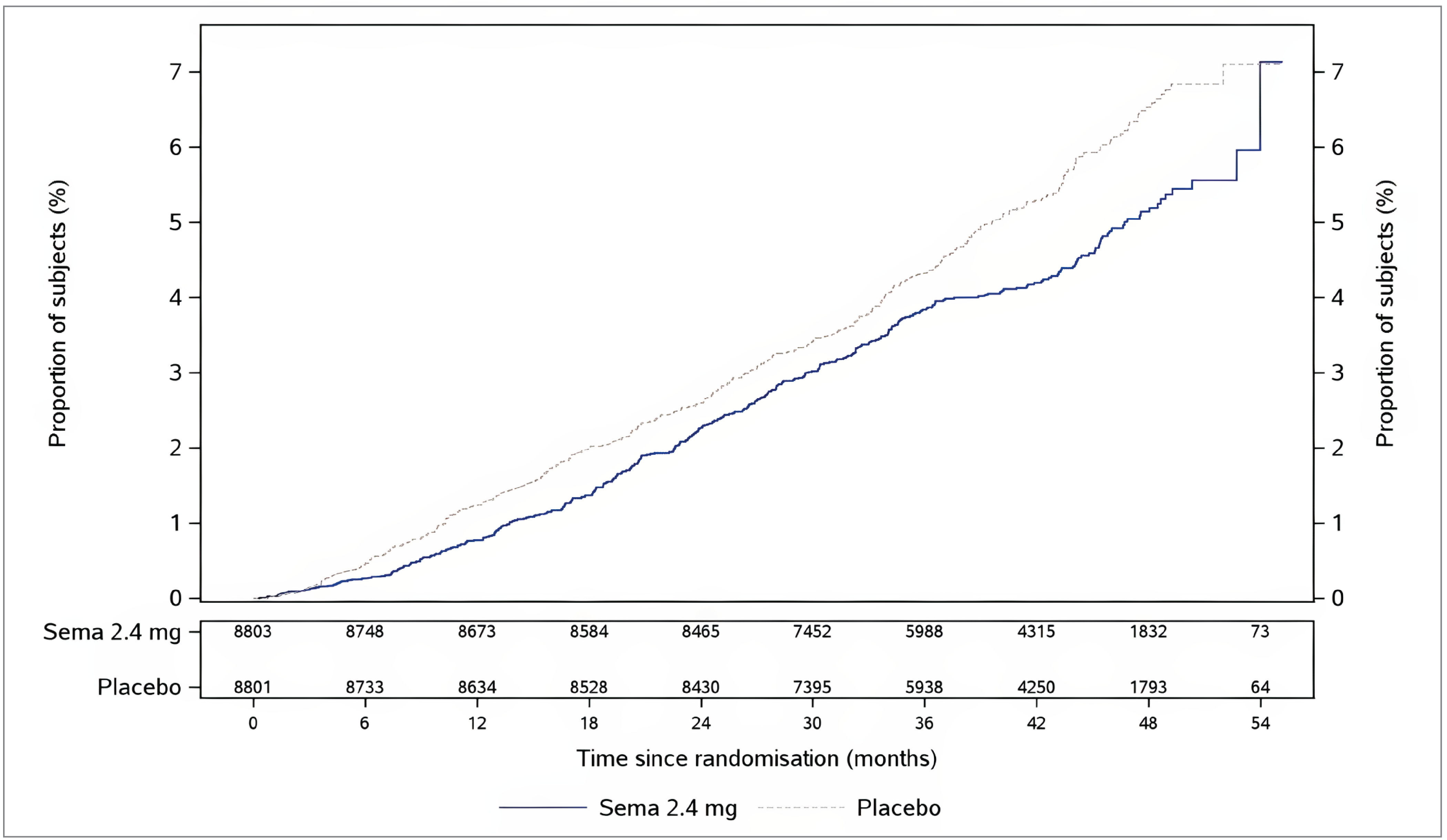
EAC = event adjudication committee; FAS = full analysis set.
Note: Cumulative incidence estimates were based on time from randomization to EAC-confirmed all-cause death using the Aalen-Johansen estimator. Patients without events of interest were censored at the end of their in-trial observation period.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Table 27: Summary of Absolute Risk Difference From the Supplementary Analysis of Key Cardiovascular Outcomes at Week 156 From the SELECT Trial (FAS)
Variable | Semaglutide 2.4 mg | Placebo |
|---|---|---|
Time to first EAC-confirmed MACE (primary end point) | ||
Events, N | 485 | 582 |
Cumulative incidence estimate | 0.058 | 0.070 |
Absolute risk difference vs. placebo at week 156 (95% CI) | −0.011 (−0.019 to −0.004) | |
Two-sided P valuea | 0.0029 | |
Time to EAC-confirmed CV death (confirmatory secondary end point) | ||
Events, N | 193 | 198 |
Cumulative incidence estimate | 0.024 | 0.024 |
Absolute risk difference vs. placebo at week 156 (95% CI) | 0 (−0.005 to 0.004) | |
Two-sided P valuea | 0.8432 | |
Time to first EAC-confirmed composite HF outcome (confirmatory secondary end point) | ||
Events, N | 258 | 277 |
Cumulative incidence estimate | 0.031 | 0.033 |
Absolute risk difference vs. placebo at week 156 (95% CI) | −0.002 (−0.008 to 0.003) | |
Two-sided P valuea | 0.4462 | |
Time to EAC-confirmed all-cause death (confirmatory secondary end point) | ||
Events, N | 317 | 358 |
Cumulative incidence estimate | 0.038 | 0.043 |
Absolute risk difference vs. placebo at week 156 (95% CI) | −0.005 (−0.011 to 0.001) | |
Two-sided P valuea | 0.1146 | |
CI = confidence interval; CV = cardiovascular; EAC = event adjudication committee; FAS = full analysis set; HF = heart failure; MACE = major adverse cardiovascular event; vs. = versus.
Note: The estimate of the absolute risk difference was calculated as the difference between the cumulative incidence estimates for each treatment group at week 156. The cumulative incidence was estimated using the Aalen-Johansen estimator.
aNot controlled for multiplicity.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Table 28: Summary of Subgroup Analysis Results by BMI for Time From Randomization to First EAC-Confirmed MACE From the In-Trial Observation Period in the SELECT Trial (FAS)
Variable | Semaglutide 2.4 mg | Placebo |
|---|---|---|
P value for test of no interaction effect | 0.6109 | |
Subgroup of patients with a BMI of < 30 kg/m2 | ||
Patients contributing to the analysis, N | 2,555 | 2,469 |
Events, N | 155 | 200 |
HR (95% CI) | 0.74 (0.60 to 0.91) | |
Subgroup of patients with a BMI of 30 kg/m2 to < 35 kg/m2 | ||
Patients contributing to the analysis, N | 3,693 | 3,781 |
Events, N | 217 | 286 |
HR (95% CI) | 0.76 (0.64 to 0.91) | |
Subgroup of patients with a BMI of 35 kg/m2 to < 40 kg/m2 | ||
Patients contributing to the analysis, N | 1,687 | 1,659 |
Events, N | 135 | 142 |
HR (95% CI) | 0.93 (0.74 to 1.18) | |
Subgroup of patients with a BMI of 40 kg/m2 to < 45 kg/m2 | ||
Patients contributing to the analysis, N | 579 | 595 |
Events, N | 40 | 49 |
HR (95% CI) | 0.83 (0.55 to 1.26) | |
Subgroup of patients a with BMI of ≥ 45 kg/m2 | ||
Patients contributing to the analysis, N | 289 | 297 |
Events, N | 22 | 24 |
HR (95% CI) | 0.92 (0.51 to 1.65) | |
BMI = body mass index; CI = confidence interval; EAC = event adjudication committee; FAS = full analysis set; HR = hazard ratio; MACE = major adverse cardiovascular event.
Note: The data cut-off date was July 18, 2023.
For the subgroup analyses, estimated HRs and corresponding CIs were calculated using a Cox proportional hazards model with interaction between treatment group and the relevant subgroup as fixed factor.
Source: Clinical Study Report version 1.0 for the SELECT trial.25
Table 29: Summary of Efficacy Results on Cardiometabolic Risk Factors From the SELECT Trial (FAS)
Variable | Semaglutide 2.4 mg (N = 8,803) | Placebo (N = 8,801) |
|---|---|---|
Change from baseline in systolic blood pressure at week 104a | ||
Systolic blood pressure at baseline (mm Hg), mean (SD) | 131.0 (15.6) | 130.9 (15.3) |
Systolic blood pressure at week 104 (mm Hg), mean (SE) | 127.18 (0.16) | 130.49 (0.16) |
Number of patients contributing to the analysis, n (%) | 8,602 (97.7) | 8,567 (97.3) |
Change from baseline (mm Hg), mean (SE) | −3.82 (0.16) | −0.51 (0.16) |
Treatment group difference vs. placebo (95% CI) | −3.31 (−3.75 to −2.88) | |
Two-sided P valueb | < 0.0001 | |
Change from baseline in total cholesterol at week 104c | ||
Total cholesterol at baseline (mg/dL), geometric mean (CEV) | 155.5 (25.8) | 156.0 (25.4) |
Total cholesterol at week 104 (mg/dL), geometric mean (CEV) | 148.39 (NR) | 152.61 (NR) |
Number of patients contributing to the analysis, n (%) | 8,530 (96.9) | 8,517 (96.8) |
Ratio to baseline (CEV) | 0.95 (NR) | 0.98 (NR) |
Treatment ratio to placebo (95% CI) | 0.97 (0.97 to 0.98) | |
Two-sided P valueb | < 0.0001 | |
Change from baseline (%), mean (SD) | −4.63 (NR) | −1.92 (NR) |
Treatment group difference vs. placebo (95% CI) | −2.77 (−3.37 to −2.16) | |
Change from baseline in HDL cholesterol at week 104c | ||
HDL cholesterol at baseline (mg/dL), geometric mean (CEV) | 44.1 (25.5) | 44.2 (25.0) |
HDL cholesterol at week 104 (mg/dL), geometric mean (CEV) | 46.33 (NR) | 44.44 (NR) |
Number of patients contributing to the analysis, n (%) | 8,365 | 8,341 |
Ratio to baseline (CEV) | 1.05 (NR) | 1.01 (NR) |
Treatment ratio to placebo (95% CI) | 1.04 (1.04 to 1.05) | |
Two-sided P valueb | < 0.0001 | |
Change from baseline (%), mean (SD) | 4.86 (NR) | 0.59 (NR) |
Treatment group difference vs. placebo (95% CI) | 4.24 (3.70 to 4.79) | |
ANCOVA = analysis of covariance; CEV = coefficient of variation; CI = confidence interval; FAS = full analysis set; NR = not reported; SD = standard deviation; SE = standard error; vs. = versus.
aAnalyzed using an ANCOVA with treatment as fixed factor and baseline value as covariate. Before analysis, missing data were multiple imputed. The imputation model (linear regression) was done separately for each treatment group and included baseline value as a covariate and was fitted to all patients with a measurement regardless of treatment adherence at week 104. The fitted model was used to impute values for patients without a measurement at week 104. Mean estimates were adjusted according to observed baseline distribution.
bNot controlled for multiplicity.
cAnalyzed using an ANCOVA with treatment as fixed factor and baseline value as covariate. Before analysis, missing data were multiple imputed. The imputation model (linear regression) was done separately for each treatment arm and included baseline value as a covariate and was fitted to all patients with a measurement regardless of treatment status at week 104. The fitted model was used to impute values for patients without a measurement at week 104. The ratio to baseline and the corresponding baseline value were log-transformed before analysis. Mean estimates were adjusted according to observed baseline distribution. The approximate relative changes and differences were derived from estimated ratios by subtracting 1 and multiplying by 100.
Sources: Clinical Study Report version 1.0 for the SELECT trial.25 Details included in the table are from the sponsor’s summary of clinical evidence.2
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
BMI
body mass index
CDA-AMC
Canada’s Drug Agency
CV
cardiovascular
CVD
cardiovascular disease
ICER
incremental cost-effectiveness ratio
MACE
major adverse cardiac event
MASH
metabolic dysfunction–associated steatohepatitis
MASLD
metabolic dysfunction–associated steatotic liver disease
MI
myocardial infarction
NIHB
Non-Insured Health Benefit
PAD
peripheral arterial disease
QALY
quality-adjusted life-year
SGLT2
sodium-glucose cotransporter-2
T2DM
type 2 diabetes mellitus
TIA
transient ischemic attack
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Semaglutide injection (Wegovy) 0.25 mg, 0.5 mg, 1 mg, 1.7 mg, or 2.4 mg solution for subcutaneous injection in a prefilled pen. |
Indication | As an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in adult patients with an initial body mass index of:
|
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC datea | November 23, 2021 |
Reimbursement request | As an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in adult patients with a BMI of 27 kg/m2 or greater and established cardiovascular disease (MI, stroke, or PAD). |
Sponsor | Novo Nordisk Canada Inc. |
Submission history | Previously reviewed: Yes Indication: As an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in adult patients with an initial BMI of:
Recommendation date: October 2022 Recommendation: Do not reimburse |
BMI = body mass index; MI = myocardial infarction; NOC = Notice of Compliance; PAD = peripheral arterial disease.
aThe NOC date is for the indication under review. Of note, this is distinct from the NOC issued in 2024 for Wegovy to reduce the risk of nonfatal MI in adults with established cardiovascular disease and BMI of 27 kg/m2 or greater.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with a BMI ≥ 27 kg/m2 and established CV disease |
Treatment | Semaglutide (Wegovy) |
Dose regimen | 2.4 mg once weekly for no more than 3 years as an adjunct to a reduced-calorie diet and increased physical activity |
Submitted price | Semaglutide (Wegovy): $388.64 per multiuse, prefilled, single-dose pen, regardless of strength (0.25 mg, 0.5 mg, 1 mg, 1.7 mg, or 2.4 mg); each pen contains 4 doses |
Submitted treatment cost | Annual cost of semaglutide, assuming 82.5% compliance, is $4,182 per patient |
Comparator | Standard of care (reduced-calorie diet and increased physical activity) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | 40 years |
Key data sources | SELECT and SUSTAIN-6 clinical trials |
Submitted results | ICER = $39,619 per QALY gained (incremental costs: $9,389; incremental QALYs: 0.24) |
Key limitations |
|
CDA-AMC reanalysis results |
|
BMI = body mass index; CDA-AMC = Canada’s Drug Agency; CV = cardiovascular; ICER = incremental cost-effectiveness ratio; LY = life-year; SBP = systolic blood pressure; QALY = quality-adjusted life-year; T2DM = type 2 diabetes mellitus.
Conclusions
Results from the SELECT trial were submitted as new evidence for this resubmission to address the evidence gap on the effects of semaglutide 2.4 mg once weekly on weight-related comorbidities, such as cardiovascular (CV) outcomes, in the indicated weight management population. The SELECT trial demonstrated that semaglutide 2.4 mg once weekly, as an adjunct to the standard of care for cardiovascular disease (CVD), reduced the occurrence of major adverse cardiac events (MACEs) compared with placebo over a mean follow-up period of nearly 40 months in patients with established CVD and with overweight (body mass index [BMI] ≥ 27 kg/m2) or obesity (BMI ≥ 30 kg/m2) but without diabetes. The point estimates of the observed treatment effects for the individual MACE components — CV death, nonfatal myocardial infarction (MI), and nonfatal stroke — were directionally aligned with the overall reduction in MACE, though the certainty of the estimated effects varied across components. More specifically, the confidence intervals for CV death and, notably, nonfatal stroke, included the potential for no benefit. Additionally, the trial suggested a reduction in composite heart failure events and all-cause mortality with semaglutide 2.4 mg compared with placebo; however, these end points could not be formally interpreted for superiority because the prespecified hierarchical testing procedure failed to reach statistical significance. No new harms with semaglutide 2.4 mg were identified. Adverse events leading to permanent or temporary discontinuation of treatment were notable and reported more frequently with semaglutide 2.4 mg compared with placebo.
In the Canada’s Drug Agency (CDA-AMC) base case, the incremental cost-effectiveness ratio (ICER) for semaglutide (given for a maximum 3 years) compared with the standard of care is $185,646 per quality-adjusted life-year (QALY) gained (incremental costs: $9,595; incremental QALYs: 0.05). The majority of incremental costs is from the cost of semaglutide ($10,402), with small cost savings from delay of type 2 diabetes mellitus (T2DM) onset ($416), less time spent with prediabetes ($150), and reduced costs of CV events ($161). The impact of BMI on patient utility had the most impact on the QALY gains (48%), with smaller gains attributed to reduction in CV events (5%), increased life expectancy (31%), and reduced time with prediabetes and T2DM (16%). To achieve cost-effectiveness at a $50,000 per-QALY threshold, the price of semaglutide would need to decrease by 67%. There is an extremely high degree of uncertainty around the base-case results, given the concerns over the model structure, the uncertainty over the long-term benefits associated with short-term weight loss, and the inability to vary the length of treatment with semaglutide.
The CDA-AMC and sponsor base cases produce similar results when analyzing outcomes during time on treatment (3 years); however, the main difference is that the sponsor assumes long-term benefit after treatment discontinuation; no evidence was provided to support this benefit. For sustained treatment benefit, patients will likely need to remain on treatment. The cost-effectiveness of semaglutide use beyond 3 years is highly uncertain as the sponsor’s model precluded an appropriate assessment of this. If long-term sustained semaglutide use does not translate into sustained weight loss and continued prevention of CV events, then long-term use would likely be less cost-effective than the CDA-AMC base case. If continued use of semaglutide translates into continued reduction in CV events and sustained weight loss, then semaglutide use may be more cost-effective than the CDA-AMC base case.
Finally, the CDA-AMC base case applies only to a restricted population of patients with established CVD, a BMI greater than 27 kg/m2, and who have either normal glucose tolerance or prediabetes. The cost-effectiveness of using semaglutide (Wegovy) versus semaglutide (Ozempic) for patients with T2DM is unknown. At the recommended maintenance dose for weight management (2.4 mg weekly), the annual per-patient cost of Wegovy is $5,066, whereas at the recommended dose for T2DM management (1 mg to 2 mg weekly), the annual per-patient cost of Ozempic is $2,844 to $5,688. Therefore, if patients with T2DM were to receive Wegovy rather than Ozempic, this could increase costs by more than $2,000 per patient per year. The additional benefit of this approach, in terms of health outcomes for patients, could not be established in this submission.
Input Relevant to the Economic Review
This section is a summary of the feedback relevant to the economic review that was received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Input for this review was submitted by 6 patient groups: GI Society, Obesity Canada, Obesity Matters, Fatty Liver Alliance, HeartLife Foundation, and Diabetes Canada. Input from these groups was informed by international surveys of individuals with obesity, surveys of individuals in Canada with obesity and individuals with diabetes or prediabetes and their caregivers, a survey of patients in the US with nonalcoholic fatty liver disease or nonalcoholic steatohepatitis, and interviews, focus groups, and/or testimonials from individuals with obesity, individuals with CVD and their health care providers, and physicians with experience managing patients with metabolic dysfunction–associated steatotic liver disease (MASLD) or metabolic dysfunction–associated steatohepatitis (MASH). Of note, much of the feedback from patient groups was not specific to the reimbursement request (i.e., adult patients with a BMI ≥ 27 kg/m2 and established CVD) but was focused more generally on obesity and weight management. The patient groups emphasized the physical and mental health impacts of obesity and related conditions, including CVD, diabetes, and MASLD or MASH. Respondents reported physical effects such as severe mobility limitations, chronic pain, fatigue, discomfort, shortness of breath, swelling, dizziness, and nausea, which impacted daily functioning; comorbidities such as arthritis, hypertension, sleep apnea, gastroesophageal reflux disease, irritable bowel syndrome, high cholesterol, and diabetes; and mental health impacts such as anxiety disorders, mood disorders, and social isolation from stigma and discrimination, including from health care providers. The patient groups reported that obesity is not only associated with other conditions but also complicates comorbid disease management. Traditional lifestyle modifications involving diet and exercise were noted as a key current treatment for obesity and CVD, but these were typically unsuccessful, especially in the long term. Some patient groups noted that comorbidities and chronic illnesses make adherence to lifestyle modification particularly difficult. The medication options reported by patient groups included semaglutide, liraglutide, and naltrexone-bupropion; however, it was noted that these were not publicly or fully privately funded, with cost noted as the greatest barrier to access. Bariatric surgeries were noted to be the gold standard of obesity treatment; however, they are considered to be a last resort due to the potential serious or fatal side effects and long wait times. Patients with experience with semaglutide (Ozempic or Wegovy) described its benefits as life changing, including substantial weight loss, improved quality of life, normalized relationships with food, and improved management of comorbidities such as CVD. Side effects were generally described as manageable and temporary, although some patients did report gastrointestinal distress–related symptoms that were difficult to manage.
Four clinician groups provided input for this review: TotalCardiology Rehabilitation, the Cardiac Rehabilitation and Secondary Prevention Program, and a joint input from Obesity Canada and the Canadian Association for Bariatric Physicians and Surgeons. The primary goal noted in the clinician group input in treating patients with CVD was reducing the incidence of MACE, with additional goals of weight management and improved glycemic control, where applicable. Current treatment standards for reducing the risk of CV events in patients with CVD include statins, antihypertensives, antiplatelets, sodium-glucose cotransporter-2 (SGLT2) inhibitors, glucagon-like peptide-1 receptor agonists, and lifestyle modifications. Evidence-based management of obesity was noted to include psychological and behavioural therapy, pharmacotherapy, and metabolic bariatric surgery. The clinician groups highlighted that lifestyle modification alone has limited success in achieving or sustaining the weight loss required to improve obesity-related medical complications. The clinician groups also noted that semaglutide is the first Health Canada–approved treatment to support both chronic weight management and to reduce the risk of MI, and that while orlistat, liraglutide, and naltrexone-buproprion are also approved for the treatment of obesity in Canada, access is lacking due to a lack of public and private funding. Successful weight loss was seen as an indication of initial treatment adherence and response, while the resolution of prediabetes, improvements in lipids and blood pressure, and improvements in patient-reported outcomes such as control of eating, mobility, and quality of life, were also seen as equally important. Other outcomes listed as clinically meaningful were a 5% reduction in total body weight within 3 months; improvements in laboratory markers such as fasting glucose, hemoglobin A1C, and triglycerides; improvements in quality of life, mobility, and weight stability; reductions in osteoarthritis pain scores; and reductions in obesity-associated conditions such as obstructive sleep apnea, hypertension, and MASLD and MASH. The groups noted that semaglutide should be continued indefinitely, likening the treatment to statin therapy or the use of acetylsalicylic acid after MI.
The drug plan program input questioned whether patients who cannot tolerate the target maintenance dose should be discontinued from treatment, or whether they would similarly benefit compared with those who can. The plans also noted concern with the anticipated budgetary impact of reimbursing semaglutide for the indication under review.
Several of these concerns were addressed in the sponsor’s model:
The model addresses the primary objective of treating patients with CVD by focusing on reductions in the incidence of major adverse CV events.
The model also addresses additional outcomes relating to weight management and improved glycemic control.
The model encompasses quality-of-life gains from weight loss, independent of the reduction in clinical events.
In addition, CDA-AMC addressed some of these concerns, as follows:
CDA-AMC conducted an array of scenario analyses exploring uncertainty in the estimated budgetary impact of reimbursing semaglutide.
CDA-AMC was unable to address the following concerns raised in the input from the drug plans:
Whether patients who cannot tolerate the target maintenance dose should be discontinued from treatment, given the lack of clinical information.
Economic Review
The current review is for semaglutide (Wegovy) as an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in adult patients with a BMI of 27 kg/m2 or greater and established CVD (MI, stroke, or peripheral arterial disease [PAD]).
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted an economic model1 that estimates outcomes in terms of long-term costs and QALYs, assessing the cost-effectiveness of semaglutide as an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in adult patients. The analysis relates to the following population:
Reimbursement request: For adult patients with a BMI of 27 kg/m2 or greater and established CVD (MI, stroke, or PAD).
This differs from the original 2021 Health Canada indication:
Adult patients with an initial BMI of 30 kg/m2 or greater (obesity), or 27 kg/m2 or greater (overweight) in the presence of at least 1 weight-related comorbidity such as hypertension, T2DM, dyslipidemia, or obstructive sleep apnea.
Semaglutide is delivered weekly as a 2.4 mg subcutaneous injection. Semaglutide is initiated at a dose of 0.25 mg weekly, which is increased every 4 weeks to doses of 0.5 mg (weeks 5 to 8), 1.0 mg (weeks 9 to 12), 1.7 mg (weeks 13 to 16) and, finally, 2.4 mg weekly thereafter. Semaglutide (Wegovy) is provided as multiuse prefilled pens that contain 4 doses. The unit cost is $388.64 per pen (or $97.16 per dose). The 28-day cost is $388.64 because the cost of pens is the same for all doses.
In the economic evaluation, semaglutide as an adjunct to diet and exercise is compared with diet and exercise alone (standard of care). Any comparison with other weight loss medications was considered inappropriate due to the lack of publicly funded weight loss pharmacotherapies. Because semaglutide is an adjunct to diet and exercise, no treatment costs for the comparator therapy are included.
The analysis was conducted from the perspective of a provincial ministry of health with a time horizon of 40 years. A discount rate of 1.5% per annum was applied.
Model Structure
A cohort multistate Markov model was developed in Microsoft Excel to simulate the progression of adult patients either receiving semaglutide as an adjunct therapy to diet and exercise versus diet and exercise alone (standard of care).
To do this, the model assesses the impact that changes in risk factors (e.g., BMI, glycemic status, and cardiometabolic risk factors) have on weight-related complications and events (acute coronary syndrome, stroke, cancer, sleep apnea, and knee replacement).2,3 The probability of patients developing these complications and events was derived from risk prediction models other than the probability of temporary reversal of prediabetes, which was derived from the SELECT and SUSTAIN-6 trials. The risk of death throughout the model is related to the risk of fatal events, the increased risk after events, and underlying age-specific and sex-specific population mortality.2,4,5 The model also applies an additional mortality multiplier to every mortality probability in the model, which accounts for an individual’s BMI. Treatment effectiveness is modelled indirectly through changes in BMI and cardiometabolic risk factors and directly through the effect on the temporary reversal of prediabetes. The model does not directly incorporate the rates of CV events (other than heart failure hospitalizations) or mortality from the SELECT trial.
For the first year, the model cycle length is 3 months, with a subsequent cycle length of 1 year. Although discontinuations from treatment with semaglutide are included, this only impacts the costs of treatment because the model assumes the same effectiveness for patients regardless of whether they remain on treatment. Thus, changes to discontinuations within the model do not affect the expected values for life expectancy and QALYs. Treatment is assumed to be discontinued for all patients at 3 years.
Within the sponsor’s report, after treatment discontinuation at 3 years, patients’ systolic blood pressure, lipids, and hemoglobin A1C are stated as immediately returning to the same value as patients on standard of care. However, within the model, it takes 3 years for values to return to those of the standard of care. As per the sponsor’s report, both patients’ weight and the proportion of patients with prediabetes return to those of the standard of care after 3 years.
Model Inputs
For the reimbursement request, the baseline population comprises 3 distinct subgroups of patients with established CVD: patients with normal glycemic control, patients with prediabetes (defined as a hemoglobin A1C of 5.7% to 6.4% [39 mmol/mol to 46 mmol/mol]), and patients with T2DM. Baseline patient characteristics are derived from all patients within the SELECT trial (patients with normal glucose tolerance and prediabetes) and from a subgroup of the SUSTAIN-6 trial (patients with T2DM who had established CVD). The proportion of patients falling into each of the 3 categories was based on prevalence in the population in Canada.
Treatment effectiveness for semaglutide was derived from the SELECT and SUSTAIN-6 trials and relates to temporary prediabetes reversal and changes in BMI, hemoglobin A1C, systolic blood pressure, total cholesterol, and high-density lipoprotein cholesterol.2,3
Risk equations are used to estimate transition probabilities for complications (prediabetes, T2DM) and events (stroke, MI, angina, transient ischemic attack [TIA]). Mortality is based on literature-based estimates of event-related and complication-related mortality and general population mortality sourced from Statistics Canada Life Tables.2,5 An additional mortality multiplier is also applied to non–disease-specific mortality probabilities based on a patient's BMI.4
Health state utilities in the model were based on a regression analysis that mapped Short Form (36) Health Survey responses from the STEP 1 clinical study to the EQ-5D instrument.6,7 The analysis allowed estimation of utility values by age, presence of heart or circulatory disease, presence of hypertension, smoking status, prediabetes status, and BMI (linear, quadratic, and cubic effects). This allowed the estimation of the utility scores by BMI. Additional decrements in utility were sourced from the literature and related to diabetes, acute coronary syndrome, sleep apnea, cancer, stroke, bariatric surgery knee replacement, and TIAs.8-14 Health state utility values for combined states were based on an additive model. EQ-5D responses are available from the SELECT trial but were not submitted by the sponsor.
Costs included in the model related to obesity treatment costs, the long-term costs of the management of obesity-related complications, and acute costs of events related to obesity complications. Treatment cost related to the costs of semaglutide only, are described earlier. Other costs included within the model relate to the incidence of diabetes complications or acute events related to complications and the long-term management of complications. These costs were sourced from the literature.15-23 The sponsor did not include the cost of the management of patients who always had a normal glucose level or who had a temporary reversal of prediabetes.
Summary of Sponsor’s Economic Evaluation Results
The sponsor submitted results for the reimbursement request based on probabilistic analyses with 1,000 iterations. Several scenario analyses were presented. The results of the CDA-AMC reanalysis are based on the deterministic analysis because CDA-AMC was uncertain as to whether the probabilistic analysis incorporated the many changes to the sponsor’s original model. The results of the sponsor’s deterministic analysis are consistent with the probabilistic analysis.
Base-Case Results
For the reimbursement population, semaglutide was associated with increased treatment and monitoring costs of $10,187 ($10,610 versus $423) but reduced costs associated with health states (−$522: $69,753 versus $70,276) and events (-$276: $12,057 versus $12,333) leading to total incremental costs of $9,389 ($92,421 versus $83,032). The reduced costs associated with health states were primarily due to reduced time with T2DM. Semaglutide was associated with increased QALYs of 0.24 (12.82 versus 12.58). Thus, the estimated ICER was $39,619 per QALY gained (Table 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Treatment | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. Standard of care ($/QALY) |
|---|---|---|---|---|---|
Standard of care | 83,032 | Reference | 12.58 | Reference | Reference |
Semaglutide + standard care | 92,421 | 9,389 | 12.82 | 0.24 | 39,619 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.
Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Various scenario analyses were conducted, although the ICER remained below $50,000 per QALY for all analyses except when an alternative risk equation for the incidence of T2DM was applied. For this analysis, the cost per QALY gained was $54,145.
A scenario analysis relating to the SELECT trial population only (no patients with T2DM), found semaglutide was associated with higher costs ($8,926; $81,129 versus $72,203) and higher QALYs (0.28; 13.85 versus 13.57), resulting in an incremental cost per QALY gained of $31,992.
The sponsor conducted a scenario analysis from a societal perspective. This analysis included additional costs to a private payer and to the patient as well as costs associated with lost productivity. In this analysis, relative to standard of care, the ICER was $36,264 per QALY gained. This was similar to the sponsor’s base-case analysis using a health care payer perspective.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Inclusion of patients with T2DM. The sponsor included 3 subgroups of adult patients with a BMI of 27 kg/m2 or greater and established CVD, normal glucose tolerance, prediabetes, and T2DM. Currently, patients with T2DM are eligible for treatment with semaglutide (Ozempic) at a lower recommended dose (1 mg to 2 mg weekly versus 2.4 mg weekly for weight management). Therefore, a more relevant comparison for individuals with T2DM would be semaglutide using a T2DM management dose (Ozempic) versus semaglutide using a weight management dose (Wegovy). Patients with T2DM within the submitted budget impact analysis (BIA) who were using Ozempic were assumed to switch to Wegovy, indicating that the sponsor implicitly recognizes that Ozempic is a relevant comparator for Wegovy in a population with T2DM. Currently, there is no clinical trial with direct evidence to facilitate a comparison of Wegovy with Ozempic in people with T2DM.
At a maintenance dose of 1 mg, the annual per-patient cost of semaglutide for the management of T2DM is $2,844. For weight management, the annual cost is $5,066 per patient regardless of what maintenance dose is used, as each prefilled pen costs the same regardless of dose. This means for patients with T2DM, switching from Ozempic to Wegovy may generate an additional $2,000 in treatment costs per year. No evidence was presented in this review that allows the cost-effectiveness of Wegovy to be assessed relative to Ozempic in a population with T2DM. The STEP 2 trial does assess the differences in weight loss between semaglutide 2.4 mg (Wegovy) versus semaglutide 1 mg (Ozempic) in a population with T2DM;24 however, this was not considered by the sponsor in the submitted economic analysis.
Because semaglutide at a T2DM management dose was not included as a comparator, the cost-effectiveness in patients with T2DM could not be assessed. The CDA-AMC reanalysis used the functionality of the model provided by the sponsor to conduct an analysis for the patient population within the SELECT trial, i.e., adult patients with a BMI of 27 kg/m2 or greater and established CVD with a normal glucose tolerance or prediabetes while removing the diabetes subgroup, which would be treated with Ozempic.
The modelling of treatment effect with semaglutide creates uncertainty. For patients with a normal glucose tolerance or prediabetes, recurrent CV events were not modelled based on the rates observed within the SELECT trials; rather, they were modelled based on risk equations using BMI and cardiometabolic outputs from the SELECT trial. It is unclear why the sponsor did not directly use the rate of CV events for patients with a normal glucose tolerance and prediabetes directly from the trial, as this would provide greater validity. As a result, the model does not accurately replicate results from the trial (refer to Table 11 for a comparison). For example, in the SELECT trial, death from CV causes made up more than 50% of deaths in both arms of the trial. In the model, death from CV causes is responsible for only 14% of deaths in the analysis. This questions the validity of the risk equations for predicting obesity-related comorbidities. Furthermore, the risk equations assume all impacts on CV events can be predicted through BMI and cardiometabolic factors. There may be a direct treatment effect associated with semaglutide or an unknown indirect effect that impacts CV events. Not using direct output from the SELECT trial leads to a less accurate analysis.
In addition to CV events (i.e., stroke, including TIAs; angina; MI; and heart failure hospitalization), the sponsor’s model incorporated the following comorbidities associated with weight loss: glucose tolerance, sleep apnea, knee replacement, colon cancer, endometrial cancer, and breast cancer. In the SELECT trial, no evidence is presented that shows a direct impact on any of these comorbidities, except T2DM onset. Likewise, the sponsor’s model does not allow an appropriate assessment of the effects of semaglutide beyond 3 years of treatment duration. In the previous submission, CDA-AMC considered each comorbidity in turn and assessed whether short-term weight loss, followed by rapid weight regain, would impact any of these comorbidities. The general conclusion was that weight loss needs to be sustained for impacts on weight-related comorbidities, such as cancer, to be realized.25
Finally, the model does not account for heterogeneity in the patient population. Given the population is broad, the population is likely heterogenous, with some patients having higher risks than others. The model treats all patients as homogenous, meaning they all experience the same risk factors in any given health state. A preferred approach would be to either model more homogeneous stratified populations within the overall population (i.e., distinct subpopulations with average values appropriate to the subpopulation) or to conduct an individual patient simulation. By not accounting for heterogeneity, and using average effects over a diverse population, the analysis may produce biased estimates, the direction of which is unknown.
Given that the risk equations do not directly replicate data from the SELECT trial, any extrapolation beyond 3 years is highly uncertain.
In the CDA-AMC reanalysis, impacts relating to glucose tolerance and recurrent CV events were included. Impacts on sleep apnea, cancer, and knee replacements were excluded, in the absence of evidence.
Treatment effects after treatment discontinuation. In the sponsor’s report, the sponsor indicates that after treatment discontinuation, patients’ systolic blood pressure, lipids, and hemoglobin A1C immediately return to the same value as patients receiving standard of care. However, upon examination of the sponsor’s model, in the base-case analysis, it takes 3 years for these values to reflect those of standard of care. In addition, the prevalence of prediabetes almost returns to the level of the standard of care after 3 years — although there is a slight bias in favour of semaglutide. The proportion of patients with T2DM who received semaglutide do not return to the proportion receiving standard of care.
No data were provided by the sponsor in the pharmacoeconomic submission that explored the impacts of semaglutide on treatment discontinuation. The sponsor assumes that short-term semaglutide use will have an impact for decades, despite BMI and cardiometabolic factors returning to baseline upon discontinuation. Likewise, as previously discussed, the sponsor does not account for heterogeneity in the patient cohort that discontinues treatment. This assumes the patient population is homogenous and that risk factors are equal among all patients. This is unlikely to be the case. Therefore, any extrapolation of treatment benefit postdiscontinuation, without reference to any data from the trial, is highly uncertain and speculative.
In response to a request by CDA-AMC, the sponsor provided functionality within the model to allow for an analysis that assumes that all treatment effects would dissipate by 1 year after treatment discontinuation. This modification did not fully address the request made to the sponsor to provide a model that reflects what was described in the report (i.e., that effects immediately return to the same value as patients receiving standard of care). Of note, there is still a slight bias beyond 1 year in the proportion of patients with prediabetes and T2DM favouring semaglutide. CDA-AMC used the functionality in this revised model to assume that treatment effects persist for 1 year after treatment discontinuation.
CDA-AMC assumed posttreatment benefit would last for 4 years and thus a 7-year time horizon was adopted for the analysis. In the absence of any data, posttreatment benefit is highly uncertain, and this assumption may overestimate or underestimate the cost-effectiveness of semaglutide.
Utility associated with weight loss. To model the direct impact of weight loss on utility, the sponsor mapped Short Form (36) Health Survey responses from the STEP 1 trial to the EQ-5D using UK values. This introduces uncertainty into the analysis because EQ-5D data were not directly gathered from the STEP 1 trial. As noted in the CDA-AMC economic guidelines, mapping as a means of deriving health utilities is not recommended unless there are no alternative approaches.26 In this submission, mapping is used to derive a relationship between BMI and utility. An alternative relationship between BMI and utility was provided by the sponsor using data from Søltoft et al. that explored the relationship between BMI and health-related quality of life in adults (n = 14,416) who were aged 18 years or older using EQ-5D with a UK derived population set of values.13 Given the larger sample size and the absence of mapping, the study by Søltoft likely provides a more robust estimate of the relationship between BMI and EQ-5D. As noted subsequently, CDA-AMC conducted a scenario analysis based on the Søltoft et al. study and found, however, that it made little difference to the CDA-AMC base-case results.
CDA-AMC notes that the EQ-5D instrument was assessed within the SELECT trial.2 At week 104 in the SELECT trial, mean improvement assessed using the EQ-5D-5L was better for patients who received semaglutide versus those who did not (0.01 difference).2 This supports the model assessment that semaglutide will improve utility. Using the EQ-5D-5L values directly measured in the SELECT trial and applying a Canadian population value set would be the most suitable approach. Given the small differences in utility measured by both the trial and the model, it is unlikely that this method would drastically influence the cost-effectiveness results because the base-case analysis only allows semaglutide to be given for 3 years. If the base-case analysis considered continued semaglutide use beyond 3 years, then the impact of not using EQ-5D estimates from the SELECT trial would be more considerable.
In a scenario analysis, CDA-AMC used estimates from Søltoft et al., as provided by the sponsor, to determine the utility benefits associated with weight loss.
Mortality associated with short-term weight loss. The sponsor assumed that every 1-point decrease in BMI is associated with a reduction in mortality risk, starting 3 months after treatment initiation. This mortality risk reduction is on top of the mortality risk reduction associated with preventing weight-related comorbidities. In the previous submission, CDA-AMC noted many issues with respect to the assumption that weight loss would have an immediate impact on mortality and that impact could be estimated from population-level data.25 The review team for the 2022 submission concluded there was insufficient evidence to support this approach.
When mortality associated with short-term weight loss was excluded from the analysis, it led to estimates of mortality that were 3 years lower than the estimate in the SELECT trial.2 This is due to the underestimation of CV mortality. So, while the model underestimates the mortality benefit associated with CV events, it overestimates the mortality benefit associated with weight loss.
CDA-AMC adjusted the model to directly replicate mortality data from the SELECT trial. Because the model assumes all patients discontinue semaglutide at 3 years, only the first 3 years of data could be taken from the semaglutide arm of the trial:
Overall survival for patients who received semaglutide: Year 1, 99.2%; year 2, 97.7%; year 3, 96.2%.
Overall survival for patients who received placebo: Year 1, 98.74%; year 2, 97.42%; year 3, 95.72%; year 4, 93.39%.
CDA-AMC assumed that for patients who received standard of care, the mortality rate during year 4 would continue for years 5, 6, and 7.
CDA-AMC adopted mortality rates for years 4 to 7 for semaglutide such that survival after 7 years would be equivalent to that of standard of care. Given the equivalence of the results for overall survival and other health states at 7 years, the CDA-AMC base case adopted a 7-year time horizon.
Time on treatment. The sponsor assumes patients will be treated with semaglutide for only 3 years, at which point 100% of patients discontinue. No such discontinuation criterion is stated in the product monograph. Likewise, in line with the previous submission, the clinical expert consulted by CDA-AMC noted there would be no desire to stop treatment if the patient wished to remain on the therapy, given the known weight regain associated with treatment discontinuation.
The only option to explore a treatment duration longer than 3 years is by assuming patients do not discontinue treatment beyond 3 years until a specified time point of up to 20 years. For example, if 10 years is selected, then no patients would discontinue after 3 years and then, at 10 years, 100% of patients would discontinue. This was considered inappropriate and an unrealistic interpretation of how semaglutide discontinuation would occur over time. There were also concerns with regard to how treatment efficacy was modelled during this extrapolated period because the impact of treatment discontinuation may not be fully reflected in the trial data, for example. Best modelling practices would explicitly incorporate treatment effects related to treatment and discontinuation. Not accounting for the heterogeneity in outcomes between those who discontinue versus those who remain on treatment leads to inaccurate modelling estimations.27
Due to concerns over the approaches adopted within the sponsor’s model, CDA-AMC found the model is unable to provide an appropriate assessment of a scenario in which patients remain on therapy for longer than 3 years. If patients remain on therapy for longer than 3 years, then weight loss may be sustained. In the absence of long-term evidence, it is unknown whether there would be a treatment-waning effect over time whereby the patient remains on therapy but starts to regain weight.
Given the inappropriate assumptions used to inform longer treatment duration, the CDA-AMC reanalysis is restricted to the assessment of a 3-year treatment duration on semaglutide. This approach will underestimate treatment costs and treatment benefit. If there is a treatment-waning effect over time, then the cost-effectiveness of semaglutide will be overestimated. That is because additional treatment costs will be incurred but additional QALYs will be fewer. If there is no treatment waning and benefit is sustained, then the cost-effectiveness of semaglutide may be underestimated because, although there will be additional treatment costs, there will be greater additional benefit derived from sustained weight loss and continued reduction in CV events.
Prediabetes reversal cost. In the sponsor’s analysis, an individual with prediabetes, defined as having a hemoglobin A1C of less or equal to 5.8%, will incur $117 in health care costs each year. This estimate was calculated by comparing health care costs between patients with T2DM whose condition is insulin resistant versus those with no T2DM and whose condition is not insulin resistant. Although this analysis may show that those with prediabetes have higher costs than those who do not, it does not mean that, upon achieving normal glycemic control, care for these patients will instantly shift. For the previous submission, according to the clinical expert consulted by CDA-AMC, a patient who has prediabetes and then reverts to having normal glycemic control will still receive frequent follow-up and will likely not have an instant shift in care. In the analysis, those with prediabetes have an elevated risk of developing T2DM; therefore, additional costs are incurred for those with prediabetes who then develop T2DM.
CDA-AMC notes there is no functionality within the sponsor’s model to apply a cost to those who revert to having “normal” glycemic control. A scenario analysis was conducted that removed all cost savings associated with prediabetes.
Errors within the modelling approach: CDA-AMC identified minor errors within the sponsor’s analysis that did not impact the sponsor’s submitted results. The sponsor provided a revised analysis that fixed all errors.
CDA-AMC used the revised model in the base-case analysis.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Additive model for combining utility effects within hybrid states. | Likely inappropriate. The analysis adopts an additive assumption for utility values for hybrid states. This approach leads to the largest assumed utility effect of avoiding complications and is therefore likely to overestimate QALY benefits and underestimate the ICER per QALY gained. However, given the small utility benefits associated with treatment, this is unlikely to have a substantial impact. |
Treatment cost is hard coded. | Inappropriate practice. The economic model provided by the sponsor provides disaggregated outcomes. However, the costs of treatment with semaglutide reference a cell where the outcome is hard coded, i.e., not linked to model calculations. Thus, it is not possible to verify the cost estimate. |
Cost of treatment with semaglutide (Wegovy) calculated based on 82.5% compliance, as observed in the SELECT trial. | Uncertain. The actual compliance in clinical practice is unknown. The assumption was deemed appropriate to align with the clinical results from the SELECT trial. |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with the clinical experts.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Analysis adopted the revised model received in January 2025. | — | — |
Changes to derive the CDA-AMC base case | ||
1. Patient population | The sponsor included 3 subgroups of adult patients with a BMI ≥ 27 kg/m2 and established CVD:
| Because patients with T2DM may be eligible for treatment with lower-dose semaglutide (Ozempic), an appropriate analysis would compare Wegovy with Ozempic in this population. The reanalysis used the functionality of the model provided by the sponsor to conduct an analysis for the patient population within the SELECT trial, i.e., adult patients with a BMI ≥ 27 kg/m2 and established CVD with:
|
2. Inclusion of additional complications related to weight | The sponsor included the following comorbidities:
The impact continues for the remainder of the patient’s life even after full weight regain. | As per the previous submission, CDA-AMC excluded those comorbidities that will be impacted by semaglutide use. |
3. Posttreatment time to return for treatment effects | Within the sponsor’s report, after treatment discontinuation at 3 years, patients’ SBP, lipids, and hemoglobin A1C are stated as immediately returning to the same value as patients receiving standard of care. However, within the model, it takes 3 years for values to return to those of the standard of care. As per the sponsor’s report, patients’ weight with semaglutide returns to the level of the standard of care after 3 years. At 3 years, the hemoglobin A1C, SBP, and lipids values among the proportion of patients with prediabetes who are receiving semaglutide return to values similar to those of the standard of care. The hemoglobin A1C, SBP, and lipids values among the proportion of patients with T2DM who are receiving semaglutide do not return to those of the standard of care. | CDA-AMC used the functionality provided in the revised model to assume that all treatment effects dissipate 1 year after treatment discontinuation. Of note, there is still a slight bias in the proportion of patients with prediabetes and T2DM, favouring semaglutide. |
4. Mortality | The sponsor assumed that patients who lose weight will have a lower risk of dying relative to those who do not lose weight starting 3 months after treatment initiation. Estimates of CV death within the submitted model are significantly lower than those found in the SELECT trial. | CDA-AMC revised the model inputs to allow replication of the mortality data from the SELECT trial for semaglutide and standard of care for years 1 to 3. The estimated mortality rate for year 4 with standard of care was applied for years 4 and 5. For semaglutide, a mortality rate was adopted that led to survival being equivalent to the standard of care at 7 years. Given the equivalence of survival at 7 years, a revised time horizon of 7 years was adopted. |
CDA-AMC base case | 1 + 2 + 3 + 4 | |
BMI = body mass index; CDA-AMC = Canada’s Drug Agency; CV = cardiovascular; CVD = cardiovascular disease; SBP = systolic blood pressure; T2DM = type 2 diabetes mellitus.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s submitted base case (reimbursement request) | Standard care | 83,032 | 12.58 | Reference |
Semaglutide | 92,421 | 12.82 | 39,619 | |
Sponsor’s submitted scenario analysis: non-T2DM population | Standard care | 72,203 | 13.57 | Reference |
Semaglutide | 81,129 | 13.85 | 31,922 | |
CDA-AMC reanalysis 2 | Standard care | 68,724 | 13.52 | Reference |
Semaglutide | 77,719 | 13.79 | 32,578 | |
CDA-AMC reanalysis 3 | Standard care | 72,422 | 13.55 | Reference |
Semaglutide | 82,047 | 13.72 | 57,299 | |
CDA-AMC reanalysis 4 | Standard care | 23,711 | 5.49 | Reference |
Semaglutide | 32,807 | 5.55 | 131,152 | |
CDA-AMC base case (1 + 2 + 3 + 4) | Standard care | 22,787 | 5.51 | Reference |
Semaglutide | 32,386 | 5.56 | 185,646 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; T2DM = type 2 diabetes mellitus.
Note Reference product is the least costly alternative.
In the CDA-AMC base-case analysis, semaglutide was associated with more QALYs (5.56 versus 5.51) and higher costs ($32,386 versus $22,787) than the standard of care, leading to an ICER of $185,646 per QALY gained. The incremental treatment costs associated with semaglutide of $10,402 were partially offset by a reduction in the costs of events ($161) and reduced time with prediabetes and T2DM ($642).
Scenario Analysis Results
In the scenario analysis that excluded the costs of management of prediabetes, the ICER increased to $187,120 per QALY gained. In the scenario analysis using the utility values from Søltoft et al., the ICER decreased to $182,193 per QALY gained.
In the sponsor’s submitted analysis, semaglutide is cost-effective at a $50,000 per-QALY threshold for the reimbursement request without any price reductions. In the CDA-AMC base-case analysis, the price reduction necessary for semaglutide to be cost-effective at a threshold of $50,000 per QALY is 67%.
Table 7: CDA-AMC Price Reduction Analyses
Price reduction analysis | Unit drug cost ($) | ICERs for semaglutide vs. standard of care ($/QALY) | |
|---|---|---|---|
Sponsor base case ($) | CDA-AMC reanalysis ($) | ||
No price reduction | 388.64 | 39,619 | 185,646 |
10% | 349.78 | 35,321 | 165,546 |
20% | 310.91 | 31,024 | 145,446 |
30% | 272.05 | 26,726 | 125,345 |
40% | 233.18 | 22,428 | 105,245 |
50% | 194.32 | 18,131 | 85,145 |
60% | 155.46 | 13,833 | 65,045 |
67% | 128.25 | 10,825 | 50,000 |
70% | 116.59 | 9,535 | 44,945 |
80% | 77.73 | 5,237 | 24,845 |
90% | 38.86 | 940 | 4,745 |
100% | 0.00 | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Issues for Consideration
The sponsor assumed that treatment duration with semaglutide for this population would only be for a maximum of 3 years. The justification for this assumption is not supported by the recommended dosing or based on the experience of the clinical experts consulted for this review. It was not possible to conduct an appropriate analysis revising this assumption. The cost-effectiveness of semaglutide when used for longer durations is unknown.
Overall Conclusions
Results from the SELECT trial were submitted as new evidence for this resubmission to address the evidence gap on the effects of semaglutide 2.4 mg once weekly on weight-related comorbidities, such as CV outcomes, in the indicated population for weight management. The SELECT trial demonstrated that semaglutide 2.4 mg once weekly, as an adjunct to the standard of care for CVD, reduced the occurrence of MACE compared with placebo over a mean follow-up period of nearly 40 months in patients with established CVD and with overweight (BMI ≥ 27 kg/m2) or obesity (BMI ≥ 30 kg/m2) but without diabetes. The point estimates of the observed treatment effects for the individual MACE components — CV death, nonfatal MI, or nonfatal stroke — were directionally aligned with the overall reduction in MACE, though the certainty of the estimated effects varied across components. More specifically, the confidence intervals for CV death and, notably, nonfatal stroke, included the potential for no benefit. Additionally, the trial suggested a reduction in composite heart failure events and all-cause mortality with semaglutide 2.4 mg compared with placebo; however, these end points could not be formally interpreted for superiority because the prespecified hierarchical testing procedure failed to reach statistical significance. No new harms with semaglutide 2.4 mg were identified. Adverse events leading to permanent or temporary discontinuation of treatment were notable and reported more frequently with semaglutide 2.4 mg compared with placebo.
In the CDA-AMC base case, the ICER for semaglutide (given for a maximum 3 years) compared with the standard of care is $185,646 per QALY gained (incremental costs: $9,595; incremental QALYs: 0.05). The majority of incremental costs is from the cost of semaglutide ($10,402), with small cost savings from delay of T2DM onset ($416), less time spent with prediabetes ($150), and reduced costs of CV events ($161). The impact of BMI on patient utility had the most impact on the QALY gains (48%), with smaller gains attributed to reduction in CV events (5%), increased life expectancy (31%), and reduced time with prediabetes and T2DM (16%). To achieve cost-effectiveness at a $50,000 per-QALY threshold, the price of semaglutide would need to decrease by 67%. There is an extremely high degree of uncertainty around the base-case results, given the concerns over the model structure, the uncertainty over the long-term benefits associated with short-term weight loss, and the inability to vary the length of treatment with semaglutide.
The CDA-AMC and sponsor base cases produce similar results when analyzing outcomes during time on treatment (3 years); however, the main difference is that the sponsor assumes long-term benefit after treatment discontinuation; no evidence was provided to support this benefit. For sustained treatment benefit, patients will likely need to remain on treatment. The cost-effectiveness of semaglutide use beyond 3 years is highly uncertain because the sponsor’s model precluded an appropriate assessment of this. If long-term sustained semaglutide use does not translate into sustained weight loss and continued prevention of CV events, then long-term use would likely be less cost-effective than the CDA-AMC base case. If continued use of semaglutide translates into continued reduction in CV events and sustained weight loss, then semaglutide use may be more cost-effective than the CDA-AMC base case.
Finally, the CDA-AMC base case applies only to a restricted population of patients with established CVD, a BMI greater than 27 kg/m2, and either normal glucose tolerance or prediabetes. The cost-effectiveness of using semaglutide (Wegovy) versus semaglutide (Ozempic) in T2DM is unknown. At the recommended maintenance dose for weight management (2.4 mg weekly), the annual per-patient cost of Wegovy is $5,066, whereas at the recommended dose for T2DM management (1 mg to 2 mg weekly), the annual per-patient cost of Ozempic is $2,844 to $5,688. Therefore, if patients with T2DM were to receive Wegovy rather than Ozempic, this could increase costs by more than $2,000 per patient, per year. The additional benefit of this approach, in terms of health outcomes for patients, could not be established in this submission.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Wegovy (semaglutide injection): solution for subcutaneous injection in a pre-filled pen, single-use pre-filled pen delivering doses of 0.25 mg, 0.5 mg, 1 mg, 1.7 mg or 2.4 mg and multi-use pre-filled pen (FlexTouch®) delivering doses of 0.25 mg, 0.5 mg, 1 mg, 1.7 mg or 2.4 mg. Mississauga (ON): Novo Nordisk Canada Inc.; November 27, 2024.
2.Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N Engl J Med. 2023;389(24):2221-2232. doi: 10.1056/NEJMoa2307563 PubMed
3.Marso SP, Bain SC, Consoli A, et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016;375(19):1834-1844. doi: 10.1056/NEJMoa1607141 PubMed
4.Bhaskaran K, Dos-Santos-Silva I, Leon DA, Douglas IJ, Smeeth L. Association of BMI with overall and cause-specific mortality: a population-based cohort study of 3.6 million adults in the UK. Lancet Diabetes Endocrinol. 2018;6(12):944-953. doi: 10.1016/S2213-8587(18)30288-2 PubMed
5.Statistics Canada. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. 2022 (Table 13-10-0114-01) [sponsor submitted reference]. Accessed 2023. https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310011401
6.Kral P. Effect and safety of semaglutide 2.4 mg once-weekly in subjects with overweight or obesity. Health Economics and Outcomes Research Study Report on file at Novo Nordisk [sponsor submitted reference]. 2021.
7.Rowen D, Brazier J, Roberts J. Mapping SF-36 onto the EQ-5D index: how reliable is the relationship? Health Qual Life Outcomes. 2009;7:27. doi: 10.1186/1477-7525-7-27 PubMed
8.Campbell J, McGarry LA, Shikora SA, Hale BC, Lee JT, Weinstein MC. Cost-effectiveness of laparoscopic gastric banding and bypass for morbid obesity. Am J Manag Care. 2010;16(7):e174-87. PubMed
9.Clarke P, Gray A, Holman R. Estimating utility values for health states of type 2 diabetic patients using the EQ-5D (UKPDS 62). Med Decis Making. 2002;22(4):340-349. doi: 10.1177/0272989X0202200412 PubMed
10.Foos V, McEwan P. Conversion of Hypoglycemia Utility Decrements from Categorical Units Reflecting Event History into Event Specific Disutility Scores Applicable to Diabetes Decision Models. Value Health. 2018;21:S223. doi: 10.1016/j.jval.2018.04.1506
11.Gough SC, Kragh N, Ploug UJ, Hammer M. Impact of obesity and type 2 diabetes on health-related quality of life in the general population in England. Diabetes Metab Syndr Obes. 2009;2:179-84. doi: 10.2147/dmsott.s7088 PubMed
12.National Institute for Health Care Excellence. Naltrexone–bupropion for managing overweight and obesity (ID757), single technology appraisal [sponsor submitted reference]. 2017. Accessed August 11, 2024. https://www.nice.org.uk/guidance/ta494/documents/committee-papers-2
13.Søltoft F, Hammer M, Kragh N. The association of body mass index and health-related quality of life in the general population: data from the 2003 Health Survey of England. Qual Life Res. 2009;18(10):1293-9. doi: 10.1007/s11136-009-9541-8 PubMed
14.Sullivan PW, Slejko JF, Sculpher MJ, Ghushchyan V. Catalogue of EQ-5D scores for the United Kingdom. Med Decis Making. 2011;31(6):800-804. doi: 10.1177/0272989X11401031 PubMed
15.CADTH. New drugs for type 2 diabetes: second-line therapy – science report [sponsor submitted reference]. 2017.
16.Cohen DJ, Osnabrugge RL, Magnuson EA, et al. Cost-effectiveness of percutaneous coronary intervention with drug-eluting stents versus bypass surgery for patients with 3-vessel or left main coronary artery disease: final results from the Synergy Between Percutaneous Coronary Intervention With TAXUS and Cardiac Surgery (SYNTAX) trial. Circulation. 2014;130(14):1146-57. doi: 10.1161/circulationaha.114.009985 PubMed
17.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. doi: 10.1186/s12885-016-2835-7 PubMed
18.Goeree R, Blackhouse G, Petrovic R, Salama S. Cost of stroke in Canada: A 1-year prospective study. J Med Econ. 2008;8:147-167. doi: 10.3111/200508147167
19.Novo Nordisk. Comparative cost analysis of adult obesity in Alberta, Canada, 2022 [sponsor submitted reference].
20.Paszat L, Sutradhar R, Luo J, Rabeneck L, Tinmouth J, Baxter NN. Overall Health Care Cost During the Year Following Diagnosis of Colorectal Cancer Stratified by History of Colorectal Evaluative Procedures. J Can Assoc Gastroenterol. 2021;4(6):274-283. doi: 10.1093/jcag/gwab001 PubMed
21.Sheppard CE, Lester EL, Chuck AW, Birch DW, Karmali S, de Gara CJ. The economic impact of weight regain. Gastroenterol Res Pract. 2013;2013:379564. doi: 10.1155/2013/379564 PubMed
22.Tan MC, Ayas NT, Mulgrew A, et al. Cost-effectiveness of continuous positive airway pressure therapy in patients with obstructive sleep apnea-hypopnea in British Columbia. Can Respir J. 2008;15(3):159-65. doi: 10.1155/2008/719231 PubMed
23.Tarride JE, Doumouras AG, Hong D, et al. Association of Roux-en-Y Gastric Bypass With Postoperative Health Care Use and Expenditures in Canada (supplementary online content). JAMA surgery. 2020;155(9):e201985. doi: 10.1001/jamasurg.2020.1985 PubMed
24.Davies M, Faerch L, Jeppesen OK, et al. Semaglutide 2.4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet. 2021;397(10278):971-984. doi: 10.1016/S0140-6736(21)00213-0 PubMed
25.Drug Reimbursement Review Combined Clinical and Pharmacoeconomic Review: Semaglutide (Wegovy) for Weight Management. CADTH; 2022. Accessed February 20, 2025. https://www.cda-amc.ca/sites/default/files/DRR/2022/SR0725-Wegovy_combined.pdf
26.CADTH. Guidelines for the Economic Evaluation of Health Technologies: Canada. 4th ed [sponsor submitted reference]. March 2017. https://www.cadth.ca/sites/default/files/pdf/guidelines_for_the_economic_evaluation_of_health_technologies_canada_4th_ed.pdf
27.Zaric GS. The impact of ignoring population heterogeneity when Markov models are used in cost-effectiveness analysis. Med Decis Making. 2003;23(5):379-96. doi: 10.1177/0272989X03256883 PubMed
28.Wegovy (semaglutide injection): solution for subcutaneous injection in a pre-filled pen, single-use pre-filled pen delivering doses of 0.25 mg, 0.5 mg, 1 mg, 1.7 mg or 2.4 mg and multi-use pre-filled pen (FlexTouch®) delivering doses of 0.25 mg, 0.5 mg, 1 mg, 1.7 mg or 2.4 mg [product monograph]. Novo Nordisk Canada Inc.; March 22, 2024.
29.Exceptional Access Program (EAP). Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2024. Accessed January 31, 2025. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
30.Ozempic (semaglutide injection): 2 mg/pen (0.68 mg/mL or 1.34 mg/mL), 4 mg/pen (1.34 mg/mL), 8 mg/pen (2.68 mg/mL) Prefilled pen delivering doses of 0.25 or 0.5 mg, 1 mg, 2 mg [product monograph]. Novo Nordisk Canada Inc.; 2025. Accessed January 31, 2025. https://pdf.hres.ca/dpd_pm/00078459.PDF
31.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Wegovy (semaglutide injection): solution for subcutaneous injection in a pre-filled pen, single-use pre-filled pen delivering doses of 0.25 mg, 0.5 mg, 1 mg, 1.7 mg or 2.4 mg and multi-use pre-filled pen (FlexTouch®) delivering doses of 0.25 mg, 0.5 mg, 1 mg, 1.7 mg or 2.4 mg. Mississauga (ON): Novo Nordisk Canada Inc.; November 27, 2024.
32.Statistics Canada. Table 13-10-0096-01. Health characteristics, annual estimates. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310009601. Accessed 1 May, 2024.
33.Janssen I. The public health burden of obesity in Canada. Can J Diabetes. 2013;37(2):90-6. doi: 10.1016/j.jcjd.2013.02.059 PubMed
34.Novo Nordisk. Data on file: SELECT CTR, Table 10-13 [sponsor submitted reference].
35.Iqvia. Exploratory analysis to understand the feasibility of segmenting individuals with obesity by clinical characteristics (IQVIA#3031365). 2024 [sponsor submitted reference].
36.IQVIA, LRx data, April 2024 MAT Patient Counts. [sponsor submitted reference].
37.Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). Statistics Canada; 2022. Accessed February 12, 2025. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701
38.Non-Insured Health Benefits Program. First Nations and Inuit Health Branch: Annual report 2022 to 2023. https://www.sac-isc.gc.ca/eng/1713194236054/1713194280612 Accessed July 22, 2024.
39.Iqvia. Exploratory analysis to understand the feasibility of segmenting individuals with obesity by clinical characteristics (IQVIA#3031365). [sponsor submitted reference]. 2024.
40.Sutherland G, Dinh T. Understanding the Gap: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage. 2017. Accessed August 14, 2024. https://www.conferenceboard.ca/temp/405c1a8c-733b-4893-9d20-a2b97c091f80/9326_Understanding-the-Gap__RPT.pdf
41.Dellplain M. From diabetes medication to weight-loss wonder drug: Ozempic’s popularity raises big questions. Healthy Debate. 2023. Accessed February 12, 2025. https://healthydebate.ca/2023/07/topic/diabetes-medication-weight-loss-ozempic/
42.Edwards S. Canada has an Ozempic shortage as demand surges. What to know about the drug being used for weight loss and its availability. The Globe and Mail. 2024. Accessed February 12, 2025. https://www.theglobeandmail.com/canada/article-ozempic-shortage-obesity-side-effects/
43.Vanek Smith S. 'You forget to eat': How Ozempic went from diabetes medicine to blockbuster diet drug. NPR. 2023. Accessed February 12, 2025. https://www.npr.org/2023/04/01/1166781510/ozempic-weight-loss-drug-big-business
44.Weikle B. Drugs like Ozempic are popular for weight loss. That's because there's been little other help: obesity doctors. CBC. 2023. Accessed February 12, 2025. https://www.cbc.ca/radio/whitecoat/ozempic-weight-loss-obesity-1.7029033
45.Public Health Agency of Canada. Infographic: Inequalities in obesity in Canada. 2018. Accessed February 12, 2025. https://www.canada.ca/en/public-health/services/publications/science-research-data/inequalities-obesity-infographic.html
46.Batal M, Decelles S. A Scoping Review of Obesity among Indigenous Peoples in Canada. J Obes. 2019;2019(2090-0716 (Electronic)):9741090. doi: 10.1155/2019/9741090
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Chronic Weight Management in Adult Patients With a BMI of 27 kg/m2 or Greater and Established Cardiovascular Disease
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Semaglutide (Wegovy) | 1 mg (0.68 mg/mL) 2 mg (1.34 mg/mL) 4 mg (1.34 mg/mL) 6.8 mg (2.27 mg/mL) 9.6 mg (3.2 mg/mL) | Multiuse prefilled pen | 388.6400a | Week 1 to 4: 0.25 mg once weekly Week 5 to 8: 0.5 mg once weekly Week 9 to 12: 1 mg once weekly Week 13 to 16: 1.7 mg once weekly Week 17+: 2.4 mg once weekly If a 2.4 mg weekly maintenance dose cannot be tolerated, patients can be maintained on a 1.7 mg weekly dose. Otherwise, semaglutide should be discontinued. | 13.88 | 5,066 |
Other forms of injectable semaglutide (indicated for the treatment of type 2 diabetes) | ||||||
Semaglutide (Ozempic) | 2 mg (0.68 mg/mL) 2 mg (1.34 mg/mL) 4 mg (1.34 mg/mL) | Multiuse prefilled pen | 218.0300b | 0.25 mg once weekly for 4 weeks, then increase to 0.5 mg once weekly. If additional glycemic control is needed after a further 4 weeks, the dose may be increased to 1 mg once weekly, and then again to 2 mg once weekly if needed after an additional 4 weeks. | 7.79b or 15.57 | 2,844 or 5,688 |
BMI = body mass index; CDA-AMC = Canada’s Drug Agency.
aSponsor’s submitted price.1 Each pen is calibrated to deliver 4 doses and comes with 4 disposable needles; thus, the choice of pen is dependent on the desired dose. Of note, while the product monograph specifies that patients should be discontinued if they cannot tolerate a 1.7 mg weekly dose, clinical expert opinion obtained by CDA-AMC indicated that clinicians would keep a patient on a lower dose if necessary and if benefit was seen.28
bOntario Drug Benefit Formulary Exceptional Access Program list price.29 The 2 mg pens can deliver doses of 0.25 mg or 0.5 mg and come with 6 disposable needles, while the 4 mg pens can deliver 1 mg doses and come with 4 needles. The daily cost of Ozempic-brand semaglutide is therefore $5.19 for the first 6 weeks of therapy and $7.79 to $15.57 thereafter. A weekly dose of 2 mg would require 2 injections from the 4 mg pen. An 8 mg pen intended to deliver 2 mg per dose has been approved by Health Canada but is neither marketed nor publicly funded.30
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | Yes | No comment |
Model structure is adequate for decision problem | No | The model structure is consistent with previous submission relating to weight management and obesity. SELECT is a cardiovascular outcomes trial, however, and the model could be more reflective of the outcomes measured within the trial. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | The model does not replicate CV mortality from the SELECT trial thus limiting its validity. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | The inability to assign a cost to the temporary reversal of prediabetes is a limitation. The model does not allow an appropriate consideration of treatment continuation beyond 3 years. The model does not allow for the prevalence of T2DM and prediabetes to return to the values of standard of care. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
CV = cardiovascular; T2DM = type 2 diabetes mellitus.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
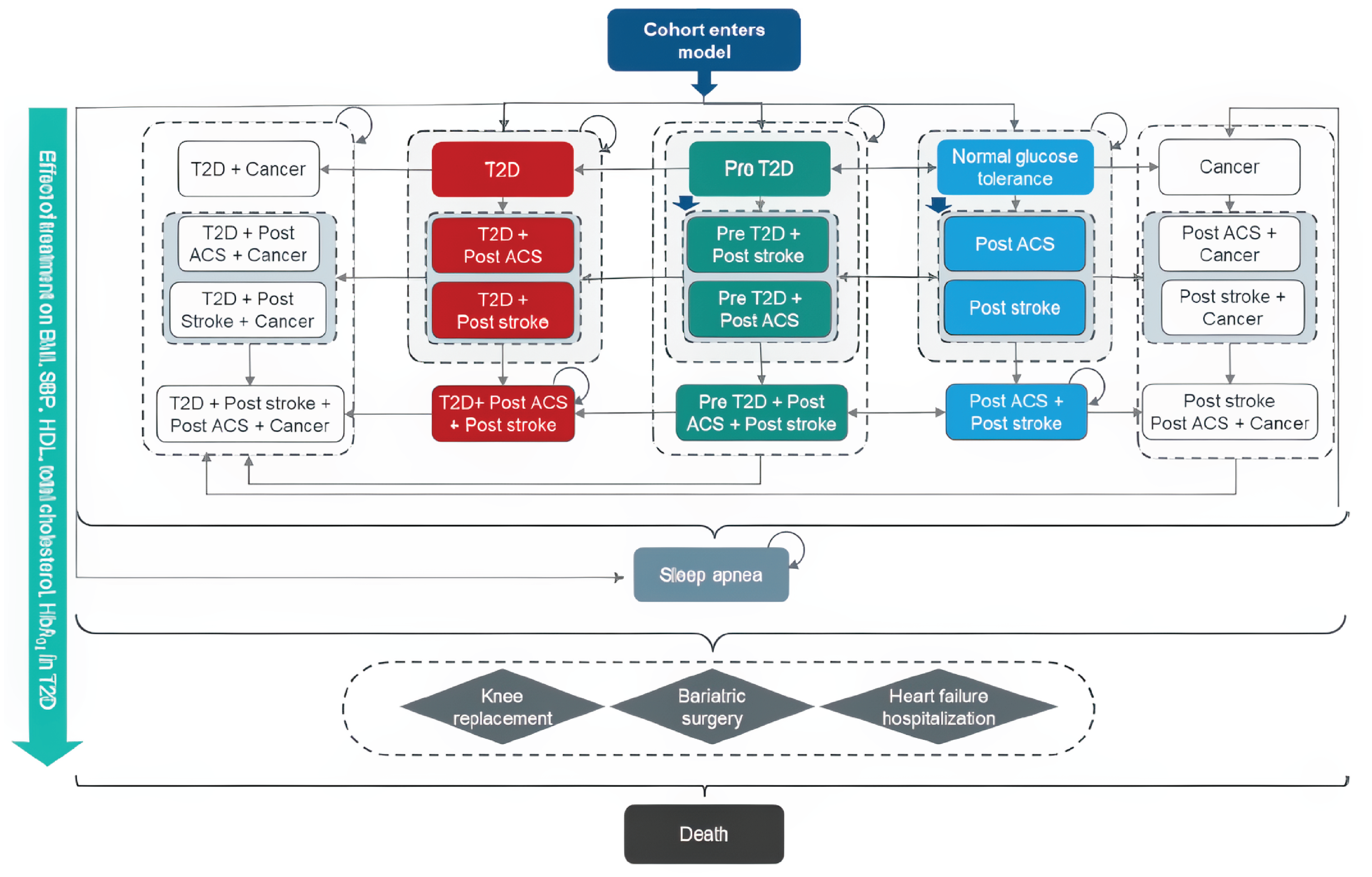
ACS = acute coronary syndrome; BMI = body mass index; HbA1c = glycated hemoglobin; HDL = high-density lipoprotein; SBP = systolic blood pressure; T2D = type 2 diabetes.
Note: Blue colour-coding denotes patients with normal glucose tolerance, green colour-coding denotes patients with prediabetes, and red colour-coding denotes patients with T2D.
ACS includes myocardial infarction and angina; stroke includes transient ischemic attack; cancer includes colorectal, postmenopausal breast, and postmenopausal endometrial cancer.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Item | Semaglutide | Standard care |
|---|---|---|
Total costs ($) | 92,421 | 83,032 |
Treatment costs ($) | 10,181 | 0 |
Blood pressure treatment cost ($) | 429 | 423 |
Complications: State costs ($) | 69,753 | 70,276 |
Normal glucose tolerance after prediabetes with history of CVD | 2,279 | 709 |
Prediabetes with history of CVD | 14,469 | 15,372 |
Normal glucose tolerance with history of CVD | 9,112 | 9,016 |
T2DM with history of CVD | 41,429 | 42,651 |
Normal glucose tolerance with history of cancer and CVD | 403 | 339 |
T2DM with history of cancer and CVD | 1,409 | 1,506 |
Sleep apnea | 652 | 682 |
Complications: Event costs ($) | 12,057 | 12,333 |
Stroke (nonfatal) | 3,270 | 3,270 |
Fatal stroke | 475 | 483 |
TIA | 284 | 289 |
MI (nonfatal) | 2,220 | 2,268 |
Fatal MI | 715 | 739 |
Unstable angina (nonfatal) | 1,074 | 1,102 |
Fatal unstable angina | 393 | 406 |
HF hospitalization (nonfatal) | 2,347 | 2,456 |
Knee replacement (nonfatal) | 1,277 | 1,317 |
Fatal knee replacement | 4 | 4 |
Total QALYS (undiscounted) | 14.85 | 14.57 |
TOTAL QALYs | 12.821 | 12.584 |
QALYs per state | ||
Normal glucose tolerance after prediabetes with history of CVD | 0.747 | 0.230 |
Prediabetes with history of CVD | 3.710 | 3.903 |
Normal glucose tolerance with history of CVD | 2.453 | 2.400 |
T2DM with history of CVD | 6.005 | 6.151 |
Normal glucose tolerance with history of cancer and CVD | 0.028 | 0.023 |
T2DM with history of cancer and CVD | 0.075 | 0.080 |
Disutilities per event | ||
Stroke (nonfatal) | −0.029 | −0.029 |
Fatal stroke | −0.006 | −0.006 |
TIA | −0.002 | −0.002 |
MI (nonfatal) | −0.028 | −0.029 |
Fatal MI | −0.018 | −0.018 |
Unstable angina (nonfatal) | −0.021 | −0.022 |
Fatal unstable angina | −0.015 | −0.015 |
HF hospitalization (nonfatal) | −0.020 | −0.021 |
Fatal HF hospitalization | 0.000 | 0.000 |
Bariatric surgery (nonfatal) | 0.000 | 0.000 |
Knee replacement (nonfatal) | −0.057 | −0.059 |
Total LYs (undiscounted) | 17.49 | 17.24 |
Total LYs | 15.09 | 14.88 |
CVD = cardiovascular disease; HF = heart failure; LY = life-year; MI = myocardial infarction; QALY = quality-adjusted life-year; T2DM = type 2 diabetes mellitus; TIA = transient ischemic attack.
Source: Sponsor submission.1
Table 11: Comparison of Outcomes — Sponsor's Model Based on SELECT Trial Population Versus SELECT Trial
Outcome | Sponsor model | SELECT trial | ||
|---|---|---|---|---|
Semaglutide | Standard of care | Semaglutide | Standard of care | |
Stroke + TIAa | 0.0085 | 0.0104 | — | — |
Stroke | 0.0067 | 0.0076 | 0.0061 | 0.0066 |
TIAa | 0.0018 | 0.0029 | — | — |
Myocardial infarction | 0.0066 | 0.0087 | 0.0089 | 0.0132 |
Angina | 0.0035 | 0.0046 | 0.0041 | 0.0047 |
Heart failure hospitalization | 0.0031 | 0.0039 | 0.0048 | 0.0057 |
CV death | 0.0011 | 0.0014 | 0.0076 | 0.0090 |
CV = cardiovascular; TIA = transient ischemic attack.
aOnly stroke is reported in the SELECT trial.
Source: Sponsor model based on the SELECT trial population and 3-year outcomes and SELECT trial.1,2
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 12: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Item | Semaglutide | Standard care |
|---|---|---|
Total costs ($) | 32,386 | 22,787 |
Treatment costs ($) | 10,402 | 0 |
Blood pressure treatment cost ($) | 175 | 175 |
Complications: State costs ($) | 19,725 | 20,367 |
Normal glucose tolerance after prediabetes with history of CVD | 2,858 | 891 |
Prediabetes with history of CVD | 8,150 | 10,327 |
Normal glucose tolerance with history of CVD | 6,278 | 6,294 |
T2DM with history of CVD | 2,440 | 2,856 |
Complications: Event costs ($) | 2,083 | 2,244 |
Stroke (nonfatal) | 908 | 949 |
Fatal stroke | 80 | 85 |
TIA | 81 | 93 |
MI (nonfatal) | 471 | 518 |
Fatal MI | 49 | 56 |
Unstable angina (nonfatal) | 170 | 189 |
Fatal unstable angina | 23 | 27 |
HF hospitalization (nonfatal) | 302 | 327 |
Total QALYS (undiscounted) | 5.80 | 5.75 |
Total QALYs | 5.558 | 5.507 |
QALYs per state | ||
Normal glucose tolerance after prediabetes with history of CVD | 0.941 | 0.291 |
Prediabetes with history of CVD | 2.358 | 2.924 |
Normal glucose tolerance with history of CVD | 1.908 | 1.882 |
T2DM with history of CVD | 0.377 | 0.439 |
Disutilities per event | ||
Stroke (nonfatal) | −0.009 | −0.009 |
Fatal stroke | −0.001 | −0.002 |
TIA | −0.001 | −0.001 |
MI (nonfatal) | −0.006 | −0.007 |
Fatal MI | −0.002 | −0.002 |
Unstable angina (nonfatal) | −0.003 | −0.004 |
Fatal unstable angina | −0.001 | −0.002 |
HF hospitalization (nonfatal) | −0.003 | −0.003 |
Total LYs (undiscounted) | 6.61 | 6.59 |
Total LYs | 6.33 | 6.31 |
CDA-AMC = Canada’s Drug Agency; CVD = cardiovascular disease; HF = heart failure; LY = life-year; MI = myocardial infarction; QALY = quality-adjusted life-year; T2DM = type 2 diabetes mellitus; TIA = transient ischemic attack.
Scenario Analyses
Table 13: Summary of the CDA-AMC Scenario Analyses
Analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CDA-AMC base case | Standard care | 22,787 | 5.51 | Reference |
Semaglutide | 32,386 | 5.56 | $185,646 | |
Exclude costs of prediabetes | Standard care | 22,399 | 5.51 | Reference |
Semaglutide | 32,074 | 5.56 | $187,120 | |
Utility values from Søltoft et al.13 | Standard care | 22,787 | 5.48 | Reference |
Semaglutide | 32,386 | 5.53 | $182,193 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
BMI = body mass index; CDA-AMC = Canada’s Drug Agency; CVD = cardiovascular disease; MI = myocardial infarction; NIHB = Non-Insured Health Benefits Program; PAD = peripheral artery disease; T2DM = type 2 diabetes mellitus.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a BIA estimating the expected incremental budget impact of reimbursing semaglutide for adult patients with a BMI of 27 kg/m2 or greater and established CVD (MI, stroke, or PAD).31 The BIA was conducted from the perspective of the pan-Canadian drug plans over a 3-year time horizon (2026 to 2028, with 2025 as the base year). The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets, excluding Quebec, as well as the Non-Insured Health Benefits Program (NIHB). The sponsor’s base case included drug acquisition costs only, although a cost offset was assumed for patients who also had T2DM and were already using semaglutide (Ozempic) under the assumption that they would switch to Wegovy if eligible for it. Market uptake of semaglutide was based on the increased uptake of SGLT2 inhibitors when the indication for them expanded from patients with T2DM to also include patients with heart failure.
Key inputs to the BIA are documented in Table 15.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate, base year |
|---|---|
Pan-Canadian population ≥ 18 years | 26,343,563a |
Proportion by sex, varies by jurisdiction (male / female) | 49.5% / 50.5%a |
Prevalence of relevant classes of obesity by sex (male or female),b varies by jurisdiction32,33 | |
Overweight (BMI 27 kg/m2 to 29.9 kg/m2) | 23.3% / 17.1%32,c |
Obesity (BMI ≥ 30.0 kg/m2) | 31.7% / 34.0%32 |
Proportion of patients with pre-existing CVD (overweight or obesity) | ███% / ███%d |
Proportion of eligible patients enrolled in public drug plans | 17.9% to 100%, depending on jurisdictione |
Adherence | 82.5%34 |
Market uptake (reference scenario, 3 years) | |
Semaglutide + SOC | 0% / 0% / 0% |
SOC | 100% / 100% / 100% |
Market uptake (new-drug scenario, 3 years) | |
Semaglutide + SOC | 4.8% / 9.6% / 14.4% |
SOC | 95.2% / 90.4% / 85.6% |
Cost of treatment (per patient) | |
SOC | $0 |
Semaglutide | $5,070 ($4,182 including adherence) |
Cost offset for patients who would have received semaglutide (Ozempic) for diabetes, but instead receive Wegovy | |
Proportion of eligible patients using semaglutide (Wegovy) who also have T2DM | ████%35 |
Proportion of patients with T2DM using semaglutide (Ozempic) | ████%36 |
Annual cost of offset semaglutide (Ozempic), per patient | $2,844 (or $2,346, assuming an 82.5% adherence rate and the switching of 100% of Wegovy-eligible Ozempic users)f |
BIA = budget impact analysis; BMI = body mass index; CVD = cardiovascular disease; NIHB = Non-Insured Health Benefit; SOC = standard of care; T2DM = type 2 diabetes mellitus.
Note: The sponsor’s analysis assumes a year has 365.25 days.
aStatistics Canada medium-growth population projection M637 and NIHB annual report.38 Includes the adult populations of the provinces (excluding Quebec) and the NIHB.
bThe sponsor’s analysis does not consider patients who do not identify as or who are not reported under male or female categories. The prevalence of obesity and CVD in this population is not known. Due to relatively small numbers, the impact of excluding or not separately considering patients who were nonbinary on the pan-Canadian budget impact results is expected to be minimal.
cThe overweight category reported by Statistics Canada includes adults who self-reported a BMI between 25 and ≤ 30 kg/m2. The sponsor multiplied these numbers by three-fifths to approximate the proportion of adults who have a BMI between 27 and ≤ 30 kg/m2.
dBased on the proportion of patients with overweight (27 ≤ BMI < 30kg/m2) or with obesity (BMI ≥ 30 kg/m2) who also have prior CVD as reported in a sponsor-submitted IQVIA analysis of electronic medical records.39
eBased on the Understanding the Gap report from the Conference Board of Canada, with the proportion of patients aged ≥ 25 years used as a proxy for the entire adult population.40
SOC represents the management of CV risk factors including medication for the treatment and prevention of CVD and health lifestyle counselling. As SOC is not expected to change when semaglutide is added, costs for SOC were not included within the BIA.
fThe sponsor’s cost offset for Ozempic-brand semaglutide assumes that ████% of eligible patients using Wegovy also have diabetes, and that ████% would receive funded Ozempic for T2DM if Wegovy was not funded.
Summary of the Sponsor’s BIA Results
Results from the sponsor’s analysis suggested that the reimbursement of semaglutide for adults with a BMI ≤ 27 kg/m2 and pre-existing CVD would be associated with an incremental cost of $61,256,584 in year 1, $125,470,998 in year 2, and $194,033,613 in year 3, for a 3-year incremental budget impact of $380,761,195.
This estimate includes a cost offset for patients who would have received semaglutide (Ozempic) for the treatment of T2DM but would instead receive semaglutide (Wegovy) if funded. The cost offset of Ozempic avoided was: $2,825,531 in year 1, $5,786,765 in year 2, and $8,947,380 in year 3, for a 3-year total of $17,559,676.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Proportion of patients enrolled in public drug plans is underestimated: The sponsor’s analysis cited the Understanding the Gap report when estimating the proportion of the population in each jurisdiction who are enrolled in public drug plans.40 As the indication is limited to the adult population, the sponsor assumed that a proportion of the population would be publicly funded consistent with the proportion of the population aged 25 years or older who are enrolled in public drug plans. However, the sponsor’s reimbursement request is limited to patients who have had a previous CVD event (defined as a previous MI, previous stroke, or symptomatic PAD). Patients with a previous CVD event are older than the general population of Canada. As reported within the IQVIA data used by the sponsor to estimate the proportion of patients with previous CVD, ████% of those with CVD only or heart failure and CVD and a BMI of 27 kg/m2 or greater were aged 65 to 79 years or 80 years or older.35 Patients aged 65 years or older have a much higher rate of public drug funding in Canada than the younger population40 and, thus, the sponsor’s analysis underestimates the proportion of patients eligible for semaglutide who would be publicly reimbursed. Of note, neither the sponsor’s nor the CDA-AMC methods account for additional patients enrolling in public plans specifically to access Wegovy. If a substantial number do so, particularly if they are under 65 years of age, then the budget impact of reimbursing Wegovy would be further increased.
In reanalysis, CDA-AMC assumed that ████% of patients who would be eligible for semaglutide were aged 65 years and older and thus enrolled in public drug plans at rates consistent with that age group for each jurisdiction. The remaining ████% of patients were assumed to be enrolled at rates consistent with those aged 25 to 64 years.
Proportion of patients with prior CVD is uncertain: The sponsor’s analysis assumes that ███% of individuals with overweight (27 ≤ BMI < 30 kg/m2) and ███% of individuals with obesity (BMI ≥ 30 kg/m2) have previous CVD (defined as having had an MI, stroke, or symptomatic PAD), based on a sponsor-submitted IQVIA feasibility study on the clinical characteristics of patients with obesity.35 This study appears to include patients with at least 1 BMI measure in electronic medical records between September 2020 and August 2022, subcategorizing patients by BMI category and diagnosis of diabetes, CVD events, and heart failure. It is unclear which jurisdictions in Canada were included, and detailed methodology was not provided. When queried why patients with obesity had a reportedly lower risk of prior CVD than those classified as being overweight without obesity, the sponsor clarified an age-related effect where those older than 65 years were both more likely to have had a prior CVD event, and less likely to be with obesity than younger patients, which was supported by data from Statistics Canada.32 However, clinical expert opinion obtained by CDA-AMC noted that a 5% to 6% estimate may be too low for the overall population of patients with overweight or obesity who have had a prior CVD event. As such, the proportion of patients with overweight or obesity who have had a prior CVD event is uncertain and potentially underestimated.
CDA-AMC conducted a series of scenarios assuming multiples of 0.5, 1, 1.5, and 2 times the sponsor’s base-case estimate of the proportion of patients with being overweight or obesity who would have had a qualifying CVD event. These values approximate the range of inputs deemed plausible in clinical expert input obtained by CDA-AMC.
The market uptake of semaglutide is highly uncertain: The sponsor’s analysis assumes a 14.4% uptake of semaglutide by year 3 and a linear uptake in years 1 and 2, based on the increase in use of SGLT2 inhibitors after their indication expanded from T2DM to also include patients with heart failure, as reported in the sponsor-submitted electronic medical record analysis by IQVIA.39 Clinical expert input obtained by CDA-AMC indicated that an assumed uptake of 14% over 3 years in a population of patients with overweight and have previously experienced a serious CVD event is very uncertain, with differences of opinion in whether the sponsor’s estimate was slightly too high due to the stigma of requesting a product known as a weight loss medication, or substantially too low due to clinician and patient desire to access such a product. CDA-AMC notes that while SGLT2 inhibitor uptake in patients with heart failure would not otherwise be an unreasonable proxy for the potential uptake of semaglutide in patients with prior CVD, semaglutide in the form of Ozempic is very well known as a weight loss medication by the general population,41-44 and is generally not funded for weight loss by public or private plans in Canada. As such, the ability to access publicly funded semaglutide may be seen as desirable by a larger proportion of eligible patients and their prescribing clinicians than the ability to access SGLT2 inhibitors would have been.
CDA-AMC conducted a series of scenarios assuming multiples of 0.5, 1, 2, and 3 times the sponsor’s base-case estimate of the market uptake of semaglutide in the eligible population. These values approximate the range of inputs deemed plausible in clinical expert input obtained by CDA-AMC.
The NIHB population was inappropriately estimated: The sponsor reportedly calculated the total population of CDA-AMC-participating plans by adding the size of the adult population obtained from Statistics Canada for each jurisdiction, and excluding the proportion covered under the NIHB program. Because the sponsor hard coded the final population size for each jurisdiction without providing their calculations from the source data, it is unclear whether this was derived appropriately. Additionally, NIHB clients residing within Ontario who are aged 25 to 65 years or older are eligible for reimbursement by the Ontario Drug Benefit and thus should be counted as Ontario Drug Benefit clients (and included in the Ontario population estimates) rather than as NIHB clients for the purposes of modelling the budget impact of reimbursing semaglutide; it was unclear whether this was considered.
Additionally, the sponsor’s analysis used obesity prevalence rates for Canada as a proxy to estimate the NIHB population potentially eligible for semaglutide, however higher prevalence rates of obesity in adults have been reported for First Nations (1.6 times higher), Inuit (1.6 times higher), and Métis (1.4 times higher) Peoples compared to the adult population who were not First Nations, Inuit, and Métis Peoples.45,46 By assuming the same prevalences rate for the NIHB client population as for Canada as a whole, the sponsor likely underestimates the budgetary impact of reimbursing semaglutide in the NIHB and pan-Canadian analyses.
CDA-AMC did not adjust for this limitation in reanalysis. The impact on pan-Canadian model results is expected to be relatively minor, though the impact on the Ontario and especially the NIHB jurisdictional analyses is expected to be greater.
The cost offset associated with switching semaglutide brands was inappropriately implemented: The sponsor’s analysis included a cost offset for patients with a BMI of 27 kg/m2 or greater, prior CVD, and T2DM who are currently receiving semaglutide in the form of Ozempic. All such patients were assumed to switch from Ozempic to Wegovy, with the cost of Ozempic subtracted from the budget impact of reimbursing Wegovy. The sponsor assumed that ████% of patients who would be eligible for Wegovy also have T2DM, based on an IQVIA analysis39 of the clinical characteristics of individuals with obesity submitted by the sponsor, and that of these, ████% would use Ozempic in the absence of Wegovy, based on the proportion of patients with T2DM estimated to be receiving Ozempic as derived by the sponsor using IQVIA patient count data from April 2024.36 While Ozempic is not an indicated treatment for reducing CVD events or weight loss, the sponsor’s analysis would be more transparent if Ozempic was considered a comparator to Wegovy, given that it is anticipated to be displaced should Wegovy be funded and that, according to clinician input obtained by CDA-AMC, is often chosen for its weight loss and CVD benefits for patients with T2DM. If Ozempic-brand semaglutide had been included as a comparator with market share in the reference scenario of the BIA, with some of its market captured by Wegovy in the new-drug scenario, results would have been clearer and scenarios incorporating differences in these assumptions could have been explored.
Further, CDA-AMC also notes that in applying the proportion of patients who also have T2DM and then applying the proportion of patients with T2DM who are treated with Ozempic to the population of patients who are eligible for Wegovy, the sponsor implies that these probabilities are independent of each other. However, it is likely that clinicians prescribe Ozempic more often for patients with T2DM who have a BMI of 27 kg/m2 or greater and/or CVD than they would for the general population of patients with T2DM. The sponsor also assumes that all eligible patients who would otherwise receive Ozempic will switch to Wegovy; the clinical expert input obtained by CDA-AMC indicated that a patient who was doing well on Ozempic may not be switched to Wegovy if the patient’s target dose of semaglutide would be less expensive to achieve with Ozempic.
CDA-AMC did not adjust for this limitation due to the structure of the model. The impact on the estimated budget impact is unclear because the sponsor likely underestimated the proportion of patients who have T2DM, a BMI of 27 kg/m2 or greater, and prior CVD who are receiving Ozempic, and overestimated the proportion of such patients who would switch to Wegovy. As the average annual cost of Ozempic when taken at the usual doses used by patients with T2DM ($2,384 in the BIA, including adherence) is less than that of Wegovy when used for weight loss ($4,182, including adherence), the fewer patients using Ozempic for T2DM who are assumed to switch to Wegovy, the lower the estimated budget impact of reimbursing Wegovy.
Hardcoding of analysis inputs made validation uncertain: The sponsor’s analysis included multiple instances of parameters being hardcoded after substantial calculations had been conducted, rather than including such calculations and their source data within the Excel file or detailing their methods in the submitted budget impact report. For example, the starting population size for each jurisdiction for each year was entered after adjustment for the NIHB population had been conducted. This calculation is frequently conducted inappropriately in reimbursement review submissions to CDA-AMC. Similarly, the sponsor hardcoded the proportion of the population within each jurisdiction who are enrolled in public drug plans without providing their derivation assumptions to CDA-AMC until queried. As such, validation of the submitted BIA was time consuming and may not have been adequately completed within the time frame of this review, leading to increased uncertainty in results.
CDA-AMC could not adjust for this limitation in reanalysis.
CDA-AMC Reanalyses of the BIA
Due to high uncertainty in several inputs used to derive the sponsor’s submitted based case and the resulting large impact on results, CDA-AMC did not undertake a base-case reanalysis. Instead, CDA-AMC revised the proportion of the population enrolled in public drug plans to reflect the proportion of patients within each funding-relevant age category (Table 16, Table 17), and then used this corrected version as the basis to explore uncertainty in parameters (i.e., proportion on the population who have pre-existing CVD and estimated uptake of semaglutide) by conducting a series of scenario analyses.
The results of the CDA-AMC underlying analysis altering the proportion of eligible patients in each funding-relevant age category are presented Table 17. For the patients with overweight or obesity who have had a prior CVD event (MI, stroke, symptomatic PAD), the underlying analysis by CDA-AMC suggests that the reimbursement of semaglutide will be associated with a 3-year incremental budgetary impact of $597,965,020 (year 1, $96,284,813; year 2, $197,094,448; year 3, $304,585,759). This estimate includes a cost offset for patients who would have received semaglutide (Ozempic) for the treatment of T2DM but would instead receive semaglutide (Wegovy), if funded. The cost offset of Ozempic avoided was $4,460,728 in year 1, $9,129,689 in year 2, and $14,106,609 in year 3, for a 3-year total of $27,697,026.
Table 16: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC underlying analysis | ||
1. Proportion of patients enrolled in public drug plans | Proportion of the overall adult population enrolled in public plansa | Proportion of the adult population enrolled in public plans, weighted by the proportion of eligible patients who are aged ≥ 65 yearsb |
CDA-AMC underlying analysis | Reanalysis 1 | |
CDA-AMC = Canada’s Drug Agency.
aProportion of the adult population estimated to be enrolled in public funding: British Columbia, 73%; Alberta, 23%; Saskatchewan, 43%; Manitoba, 65%; Ontario, 32%; New Brunswick, 18%; Nova Scotia, 24%; Prince Edward Island, 39%; Newfoundland and Labrador, 22%; Non-Insured Health Benefits, 100%.
bProportion of the adult population estimated to be enrolled in public funding: British Columbia, 73%; Alberta, 61%; Saskatchewan, 63%; Manitoba, 76%; Ontario, 62%; New Brunswick, 28%; Nova Scotia, 42%; Prince Edward Island, 65%; Newfoundland and Labrador, 34%; Non-Insured Health Benefits, 100%.
Table 17: Results of the CDA-AMC Underlying Reanalysis of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Budget impact | $0 | $61,256,584 | $125,470,998 | $194,033,613 | $380,761,195 |
CDA-AMC underlying analysis | Budget impact | $0 | $96,284,813 | $197,094,448 | $304,585,759 | $597,965,020 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: As standard of care is assumed to have a cost of $0, the cost of the reference scenario is $0 across all years for every analysis, with the new-drug scenario and incremental budget impact being identical. Therefore, only the estimated budget impact is presented in this table.
However, CDA-AMC notes that the estimated proportion of patients with overweight or obesity who have also had a CVD event as well as the potential uptake of semaglutide are highly uncertain. Changes in assumptions for both these parameters have large impacts on the estimated budgetary impact of reimbursing semaglutide. CDA-AMC therefore conducted a series of scenario analyses on its underlying analysis varying these inputs Table 18.
The total 3-year budgetary impact of reimbursing semaglutide in its eligible population is highly uncertain and could reach well into the billions of dollars if both the proportion of patients with prior CVD and the uptake of semaglutide were substantially underestimated by the sponsor (Table 18).
Table 18: Summary of the CDA-AMC Scenario Analyses of the BIA
Three-year total ($) | ||||
|---|---|---|---|---|
Uptake of semaglutide (Y1 / Y2 / Y3)a | Proportion of the population with pre-existing CVD (overweight or obesity)b | |||
███% / ███% | ███% / ███%c | ███% / ███% | ████% / ████% | |
2.4% / 4.8% / 7.2% | $149,491,255 | $298,982,510 | $448,473,765 | $597,965,020 |
4.8% / 9.6% / 14.4%c | $298,982,510 | $597,965,020d | $896,947,530 | $1,195,930,041 |
9.6% / 19.2% / 28.8% | $597,965,020 | $1,195,930,041 | $1,793,895,061 | $2,391,860,081 |
14.4% / 28.8% / 43.3% | $896,947,530 | $1,793,895,061 | $2,690,842,591 | $3,587,790,122 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; CVD = cardiovascular disease, defined as having had a myocardial infarction, a stroke, or symptomatic peripheral artery disease; Y = year.
aSponsor’s base-case inputs were varied by multiplying them by 0.5, 1, 2, and 3.
bSponsor’s base-case inputs were varied by multiplying them by 0.5, 1, 1.5, and 2.
cSponsor’s base-case input.
dCDA-AMC underlying analysis.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.