Drugs, Health Technologies, Health Systems
Reimbursement Review
Remdesivir (Veklury)
Sponsor: Gilead Sciences Canada, Inc.
Therapeutic area: COVID-19 in nonhospitalized patients
This multi-part report includes:
Clinical_Review
Pharmacoeconomic_Review
Clinical_Review
Abbreviations
AE
adverse event
AMMI
Association of Medical Microbiology and Infectious Disease Canada
CI
confidence interval
DB
double blind
FLU-PRO
InFLUenza Patient-Reported Outcome
FLU-PRO Plus
COVID-19–adapted InFLUenza Patient-Reported Outcome
HR
hazard ratio
HRQoL
health-related quality of life
ICU
intensive care unit
KM
Kaplan-Meier
MAV
medically attended visit
MID
minimal important difference
PCR
polymerase chain reaction
RAT
rapid antigen test
RCT
randomized controlled trial
RNA
ribonucleic acid
RWE
real-world evidence
SAE
serious adverse event
SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
SD
standard deviation
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on the Application Submitted for Review
Item | Description |
|---|---|
Drug product | Remdesivir (Veklury), 100 mg/vial, IV infusion |
Sponsor | Gilead Sciences Canada, Inc. |
Indication | For the treatment of COVID-19 in nonhospitalized adults and pediatric patients (weighing ≥ 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization and death |
Reimbursement request | For the treatment of COVID-19 in nonhospitalized patients aged ≥ 12 years (weighing ≥ 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progression to severe COVID-19, including hospitalization and death |
Health Canada approval status | NOC |
Health Canada review pathway | Expedited: for use in relation to COVID-19 |
NOC date | July 27, 2020 |
Recommended dose | A single loading dose of 200 mg on day 1, then 100 mg once daily on days 2 and 3. For nonhospitalized patients who are at high risk of progressing to severe COVID-19, treatment is recommended daily for 3 days, starting as soon as possible after diagnosis of COVID-19 and within 7 days of the onset of symptoms. |
NOC = Notice of Compliance; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Source: Sponsor’s submission package for review of remdesivir; remdesivir product monograph.1,2
Introduction
COVID-19 is an illness caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in which patients present variably, from having absent or mild symptoms (for most current cases of infection) to severe symptoms.3,4 WHO has estimated the mortality risks for patients with nonsevere disease to be 0.6% for those at high risk of hospitalization, 0.3% for patients at moderate risk of hospitalization, and 0.05% for patients at low risk of hospitalization.5 According to the literature and the clinical expert we consulted, the most important risk factors for progression to severe disease include lack of SARS-CoV-2 immunity (through prior infection or vaccination), severe immune suppression, multiple chronic comorbidities, and older age (e.g., 80 years and older).4 Compared to previous variants, the omicron variant of concern is more transmissible, has a shorter incubation period, and is associated with reduced morbidity and mortality.6-8
Since the approval of the first COVID-19 vaccine by Health Canada in December 2020, 83.7% of the population has received at least 1 dose.9 Vaccination remains the first line of defence to prevent SARS-CoV-2 infection.9 Based on serology, it is estimated that nearly everyone in Canada (> 99% of the population) has some form of infection-acquired or vaccine-induced immunity to the virus.10 Most patients with COVID-19 can be managed at home with symptomatic care, monitoring for clinical deterioration, and isolation to prevent transmission.11 The need for additional treatment is based on a patient’s severity of illness and risk of progressing to severe disease, the assessments of which vary across jurisdictions. According to updated recommendations from the Association of Medical Microbiology and Infectious Disease Canada (AMMI), nirmatrelvir-ritonavir and remdesivir may be considered in patients with mild disease (i.e., not requiring oxygen supplementation) based on the patient’s risk, severity, and trajectory of symptoms.12 WHO guidelines for COVID-19 therapeutics provide a conditional recommendation for the use of remdesivir to treat patients with nonsevere disease who are at high risk of hospitalization, acknowledging the challenges associated with IV administration.5 Other factors to consider when deciding which treatment to use include local availability of therapies, duration of symptoms, feasibility of administration, and potential drug interactions.11
Remdesivir is a nucleotide prodrug that incorporates into nascent viral ribonucleic acid (RNA) and inhibits viral replication by prematurely terminating RNA transcription.2 The recommended dosage of remdesivir is a single loading dose of 200 mg on day 1 and a 100 mg dose on day 2 and day 3 for nonhospitalized patients (Table 1).2 For this review, the sponsor has requested reimbursement of remdesivir for the treatment of COVID-19 in nonhospitalized patients who are aged 12 years or older (weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and at high risk of progression to severe COVID-19, including hospitalization and death. Remdesivir also has a Health Canada indication for the treatment of hospitalized adult and pediatric patients (at least 4 weeks of age and weighing at least 3 kg) with pneumonia requiring supplemental oxygen.2 It is undergoing review for the treatment of COVID-19 in hospitalized patients aged 12 years or older (weighing at least 40 kg) with pneumonia requiring supplemental oxygen.2
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of remdesivir, 100 mg/vial, IV infusion for the treatment of COVID-19 in nonhospitalized patients who are aged 12 years or older, weigh at least 40 kg with positive results of direct SARS-CoV-2 viral testing, and are at high risk of progression to severe COVID-19, including hospitalization and death.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to our call for input and from the clinical expert we consulted for the purpose of this review.
Patient Input
One group, the Gastrointestinal Society, responded to our call for patient group input by gathering information through meetings and discussions with health care professionals, researchers, academics, and staff with first-hand experience with COVID-19. According to the patient group, SARS-CoV-2 infection can damage the intestinal lining, leading to irritating and sometimes severe symptoms, and can modify the microbiome in the gastrointestinal tract, leaving the patient more susceptible to opportunistic infections. The patient group stated that, due to possible reinfection and recurrent illness, the availability of effective vaccines and treatments remains paramount. It was emphasized that despite the available options to protect patients from severe disease or death, treatments are difficult to access. There are also concerns related to contraindications with current medications.
Clinician Input
Input From Clinical Experts Consulted
The clinical expert we consulted stated that the aim of treatment for nonhospitalized patients with SARS-CoV-2 infection is to avoid hospitalization, death, and disability. While most patients will improve without intervention, the expert indicated that the 2 key criteria for identifying patients who could receive remdesivir for COVID-19 are having a high-risk underlying medical condition and showing evidence of substantial symptoms. Although there are fewer administrative challenges with nirmatrelvir-ritonavir compared to remdesivir, the latter would be an alternative option for patients who cannot receive nirmatrelvir-ritonavir due to intolerance and/or drug-drug interactions. According to the clinical expert, the evidence supporting the claim that either drug reduces symptom severity, viral transmission, or long COVID is inconclusive, especially in immune populations; this topic is subject to ongoing research. The expert indicated that between the 2 options, nirmatrelvir-ritonavir is generally preferred for patients who do not have a contraindication.
According to the expert, few patients are likely to benefit from treatment for COVID-19; only those with severe immune suppression are likely to benefit. Moreover, these patients tend to be receiving specialty care already and can be identified as high-risk patients based on their underlying illnesses. The expert emphasized that specific viral testing is necessary to differentiate SARS-CoV-2 infection from other influenza-like illnesses and to allow access to treatment.
More recently, because of the milder disease trajectory of COVID-19, fewer patients are being admitted to hospital because of the virus, and intensive care unit (ICU) admissions are rare. However, patients who are severely immune-compromised (e.g., from B-cell depletion) are unable to clear viral infections effectively and may develop chronic infection. The expert identified these patients as being of great concern for progression to severe disease.
There are no clinical markers for assessing patients with mild disease; however, medical intervention should be pursued in those who continue to worsen. As per the expert, commonly used clinical trial outcomes (e.g., hospitalization, ICU admission, and death) are also important in practice. Remdesivir for nonhospitalized patients is indicated for a set 3-day duration, and it is expected that most patients will complete treatment.
Given that remdesivir is an IV medication, a team of health care providers who can insert, care for, and remove an IV is necessary for administration. The care team is also required to monitor the patient during infusion and assess potential infection at the IV site. The expert stated that treatment typically takes place at hospitals or infusion clinics, although at-home nursing care may be an option for some.
Clinician Group Input
Two clinician groups, BC Transplant Clinicians (7 authors) and the Ontario Health Infectious Diseases Advisory Committee (4 authors), responded to our call for clinician group input. Both groups indicated that the goals of treatment include reducing COVID-related mortality, hospitalizations, ICU admissions, and symptom severity; preventing progression to severe disease and long-term sequelae; and accelerating recovery. The groups highlighted the ongoing need for treatments and vaccines with evidence of long-term safety and efficacy as well as drug options that improve the convenience of administration. Timely access to testing and COVID-19 treatments (considering the narrow time frames for initiating therapy) were noted as ongoing issues.
The groups noted that nirmatrelvir-ritonavir is not often used in patients in whom interactions between the drug and their current medications cannot be safely managed and that, in such cases, remdesivir may be used where contraindications to nirmatrelvir-ritonavir exist or in patients who are outside the 5-day initiation window for nirmatrelvir-ritonavir. British Columbia currently recommends and has prioritized remdesivir for nonhospitalized, symptomatic patients with solid organ transplants, regardless of vaccine status or previous infection, while the Ontario clinician group noted that older age, immunocompromised status, nonvaccinated status, presence of multiple and/or uncontrolled comorbidities, and specific medical or social vulnerabilities are considerations when assessing the need for intervention. Those who are asymptomatic or beyond the 7-day onset would be least suited to receive remdesivir.
According to the clinician group in British Columbia, people with solid organ transplant receiving remdesivir as outpatients are monitored by infusion clinic nurses during the 3-day treatment course and followed up by their respective transplant centres. The clinician group in Ontario indicated that, in general, a specialist is not required for managing patients receiving remdesivir and that the drug can be administered in a community setting (e.g., nursing and long-term care homes), hospital outpatient clinic, or hospital emergency department. Both groups noted that a clinically meaningful response would be indicated by a significant reduction in hospitalizations, ICU admissions, and deaths; no need for new or increased supplemental oxygen; and improved or resolved symptoms. Discontinuation of therapy should be considered in patients experiencing adverse events (AEs) — such as increased serum creatinine, increased serum liver enzymes, or hypersensitivity or infusion reactions — or if new data indicate that viral variants are no longer susceptible to remdesivir.
Drug Program Input
The public drug plans asked about appropriate reimbursement criteria for remdesivir, how SARS-CoV-2 infection should be confirmed, and how high-risk patients aged less than 12 years and weighing less than 40 kg are managed. The plans also asked if reimbursement of remdesivir should account for nirmatrelvir-ritonavir and if criteria for the 2 drugs should be aligned, where appropriate. Lastly, the drug plans asked if there were situations where patients would receive longer or additional remdesivir treatment and if there is evidence to support the combination use of antiviral therapies.
The clinical expert we consulted stated that the eligibility criteria used in the PINETREE trial would not be appropriate for use as reimbursement criteria for remdesivir due to changes in disease and care management since the trial was conducted — that is, population immunity has increased, viral pathogenicity has changed, and hospitalization rates have decreased. It was emphasized that SARS-CoV-2 infection must be confirmed before receiving remdesivir and that any approved test would be acceptable. For patients aged less than 12 years or weighing less than 40 kg, the possibility of antiviral treatment should be discussed on a case-by-case basis with specialists who have knowledge of infectious diseases and experience managing pediatric patients. The expert agreed that nirmatrelvir-ritonavir should be considered before remdesivir, for practical reasons. Furthermore, it would be reasonable to align the reimbursement criteria for the 2 antivirals, where appropriate. According to the clinical expert, most nonhospitalized patients receive remdesivir for 3 days, and it would be rare to require longer and/or additional courses of the drug in clinical practice as prophylaxis for hospitalization. Lastly, the expert noted that there is a lack of evidence to support the combination use of antiviral therapies, including remdesivir.
Clinical Evidence
Systematic Review
Description of Studies
One multicentre, phase III, double-blind (DB), randomized controlled trial (RCT) (the PINETREE trial, N = 584) assessed whether remdesivir reduced COVID-19–related hospitalizations (defined as at least 24 hours of acute care) or all-cause death by day 28 compared to placebo in nonhospitalized patients with confirmed SARS-CoV-2 infection weighing at least 40 kg who were at high risk of progression to severe COVID-19 (based on prespecified patient characteristics at the time the study was conducted). Secondary and exploratory end points included COVID-19–related medically attended visits (MAVs), all-cause mortality, a new requirement for oxygen supplementation, ICU admission, mechanical ventilation, and patient-reported symptom alleviation.
The mean age of patients was 50 years (standard deviation [SD] = 15.1); demographic characteristics were generally balanced between treatment groups. Patients aged less than 60 years must have had at least 1 pre-existing risk factor. Across treatment groups, the most frequently reported baseline risk factors were diabetes (62%), obesity (56%), hypertension (48%), and chronic lung disease (24%), while other comorbidities were less common (i.e., fewer than 10% of patients had cardiovascular or cerebrovascular disease, chronic mild or moderate kidney disease, chronic liver disease, current cancer, sickle cell disease, or were in an immunocompromised state). Individuals were excluded if they had received any other antiviral treatment or vaccination against SARS-CoV-2.
Efficacy Results
COVID-19–Related Hospitalization or All-Cause Death
Two patients (0.7%) who received remdesivir and 15 patients (5.3%) who received placebo met the primary efficacy end point (hazard ratio [HR] = 0.13; 95% confidence interval [CI], 0.03 to 0.59; P value = 0.0076) of COVID-19–related hospitalization (defined as at least 24 hours of acute care) or all-cause death by day 28. We calculated the number needed to treat was 22. A sensitivity analysis using baseline stratification factors as strata supported the results of the primary analysis. The primary composite outcome was driven by COVID-19–related hospitalizations, given that there were no deaths in either group by day 28. Furthermore, all COVID-19–related hospitalizations occurred by day 14, and the number of events by day 14 was the same as that by day 28.
Two subgroups were considered relevant to the review. Of the 15 patients who were residing in a skilled nursing facility and the 23 patients who had a baseline risk factor of being immunocompromised, none experienced a COVID-19–related hospitalization or death from any cause by day 28.
COVID-19–Related MAV or All-Cause Death
By day 28, 4 patients (1.6%) in the remdesivir group and 21 patients (8.3%) in the placebo group met the composite end point of COVID-19–related MAV (defined as a medical visit attended in person by the patient and a health care professional) or all-cause death (HR = 0.19; 95% CI, 0.07 to 0.56). By day 14, 2 patients (0.8%) in the remdesivir group and 20 patients (7.9%) in the placebo group met the composite end point (HR = 0.10; 95% CI, 0.02 to 0.43).
Patients Progressing to Requiring Oxygen Supplementation
Of the 6 patients who progressed to requiring oxygen supplementation by day 28, 1 patient (0.4%) was in the remdesivir group and 5 patients (1.8%) were in the placebo group.
Patients Admitted to the ICU
Of the 6 patients who were admitted to the ICU by day 28, 3 patients (1.1%) were in the remdesivir group and 3 patients (1.1%) were in the placebo group.
Patients Started on Mechanical Ventilation
One patient (0.4%) in the trial (randomized to the placebo group) required mechanical ventilation by day 28.
InFLUenza Patient-Reported Outcome Plus
The InFLUenza Patient-Reported Outcome (FLU-PRO) is a standardized questionnaire for reporting influenza symptoms in clinical trials that categorizes 32 items or symptoms into 6 domains: nose, throat, eyes, chest or respiratory, gastrointestinal, and body or systemic.13-15 The COVID-19–adapted InFLUenza Patient-Reported Outcome (FLU-PRO Plus) includes a seventh domain for senses, with 2 items: loss of taste and smell.14,15 Patients rate the extent of their symptoms from 0 (not at all) to 4 (very much) over the preceding 24 hours.14,15 The mean score of symptoms for each domain is used to determine a total score.14,15
Baseline data captured before the first dose were gathered for 66 patients in the remdesivir group and 60 patients in the placebo group. For this subset of patients, 23 patients in the remdesivir group and 15 patients in the placebo group reported alleviation of baseline COVID-19 symptoms (to mild or absent) (HR = 1.41; 95% CI, 0.73 to 2.69) at day 14.
Harms Results
In the PINETREE trial, 42.3% of patients in the remdesivir group and 46.3% of patients in the placebo group reported at least 1 treatment-emergent adverse event (TEAE). The most common TEAEs in the trial included nausea (10.8% for remdesivir and 7.4% for placebo), headache (5.7% for remdesivir and 6.0% for placebo), and cough (3.6% for remdesivir and 6.4% for placebo). In general, the incidence of harm was similar between treatment groups.
Overall, 1.8% of patients in the remdesivir group and 6.7% of patients in the placebo group reported at least 1 serious adverse event (SAE). The most common SAEs were related to COVID-19 and pneumonia.
In total, 0.7% of patients in the remdesivir group and 1.8% of patients in the placebo group stopped treatment due to a TEAE. TEAEs included COVID-19, pneumonia, respiratory failure, hypoxia, and dyspnea.
There were no patient deaths during the trial.
Elevated transaminases and hypersensitivity reactions were considered notable harms for the review. In the trial, 1 patient in the remdesivir group and 3 patients in the placebo group reported elevated alanine aminotransferase levels, while 1 patient in each of the treatment groups reported elevated aspartate aminotransferase levels. In terms of hypersensitivity, there were 6 patients in the remdesivir group and 4 patients in the placebo group who reported infusion-site reactions. Anaphylactic reactions were not captured in the trial.
Table 2: Summary of Key Results From the PINETREE Trial
Key results | Remdesivir (N = 279) | Placebo (N = 283) |
|---|---|---|
Composite end point of COVID-19–related hospitalization or all-cause death by day 28 (full analysis set) | ||
Patients, n (%)a | 2 (0.7) | 15 (5.3) |
HR (95% CI)b | 0.13 (0.03 to 0.59) | |
P valuec | 0.0076 | |
NNT (patients)d | 22 | |
Composite end point of COVID-19–related MAV or all-cause death by day 28 (full analysis set) | ||
Patients, n (%)e | 4 (1.6) | 21 (8.3) |
HR (95% CI)b | 0.19 (0.07 to 0.56) | |
P valuec | 0.0024 | |
Composite end point of COVID-19–related MAV or all-cause death by day 14 (full analysis set) | ||
Patients, n (%)e | 2 (0.8) | 20 (7.9) |
HR (95% CI)b | 0.10 (0.02 to 0.43) | |
P valuec | 0.0019 | |
Patients requiring oxygen supplementation by day 28 (full analysis set) | ||
Patients, n (%) | 1 (0.4) | 5 (1.8) |
Patients admitted to the ICU by day 28 (full analysis set) | ||
Patients, n (%) | 3 (1.1) | 3 (1.1) |
Patients started on mechanical ventilation by day 28 (full analysis set) | ||
Patients, n (%) | 0 (0) | 1 (0.4) |
Time to alleviation of baseline COVID-19 symptoms (to mild or absent), as reported on the FLU-PRO Plus at day 14 (full analysis set) | ||
Patients in analysis, n | 66 | 60 |
Patients with event, n | 23 | 15 |
HR (95% CI)f | 1.41 (0.73 to 2.69) | |
P valuec | 0.2987 | |
Harms, n (%) (safety analysis set) | ||
TEAEs | 118 (42.3) | 131 (46.3) |
SAEs | 5 (1.8) | 19 (6.7) |
Treatment stopped due to TEAEs | 2 (0.7) | 5 (1.8) |
Deaths | 0 (0) | 0 (0) |
Notable harms, n (%) | ||
Transaminase elevations | ||
Alanine aminotransferase | 1 (0.4) | 3 (1.1) |
Aspartate aminotransferase | 1 (0.4) | 1 (0.4) |
Hypersensitivity reactions | ||
Infusion-site reactions | 6 (2.2) | 4 (1.4) |
Anaphylactic reactions | NR | NR |
CI = confidence interval; FLU-PRO Plus = COVID-19–adapted InFLUenza Patient-Reported Outcome; HR = hazard ratio; MAV = medically attended visit; NNT = number needed to treat; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aProportion of COVID-19–related hospitalization or all-cause death based on Kaplan-Meier estimates.
bHR, 2-sided 95% CI, and P value were estimated using Cox regression, with baseline stratification factors as covariates (region, age, and resident of skilled nursing facility or not).
cP value has not been adjusted for multiple testing.
dWe approximated the NNT by dividing 1 by the reported absolute risk difference between treatment groups.
eProportion of COVID-19–related MAV or all-cause death based on Kaplan-Meier estimates.
fHR and 2-sided 95% CIs were estimated using Cox regression, with baseline stratification factors as covariates (region, age, and resident of skilled nursing facility or not). P value was based on the stratified log-rank test, with baseline stratification factors as strata.
Source: Clinical Study Report for the PINETREE trial.16
Critical Appraisal
The overall risk of bias in the PINETREE trial was low for randomization, deviations from intended interventions, missing outcomes data, and reported results. Study enrolment was stopped early, and the final trial population was less than half (46.2%) of the planned sample size; however, this did not have an impact on the statistical analysis of results, given that the primary efficacy end point was met. However, event rates were low during the trial, limiting the assessment of key clinical end points for this disease (e.g., requirements for supplemental oxygen, ICU admissions, and mechanical ventilation). No deaths were reported. The efficacy, as observed, was driven by hospitalization only. It was not specified how the need for hospitalization or MAV was determined; this may have been subjective. The trial also did not report what the causes of hospitalization or MAV were, aside from being COVID-19–related, which itself was not further defined. No minimal important difference (MID) for the FLU-PRO Plus was identified from the literature, and the clinical expert confirmed that it is not an instrument used in clinical practice. The trial was relatively short in duration, and there is a lack of long-term safety evidence for remdesivir.
The intervention and treatment setting in the trial were considered generalizable to clinical practice in Canada. Considering when the trial took place (September 2020 to May 2021) and the significant changes in population immunity, viral pathogenesis, and disease management since then, the clinical expert we consulted indicated that the eligibility and baseline characteristics are no longer relevant to how remdesivir would be used in practice today. There were 8 adolescent patients in the trial, and there are limited data to inform the use of remdesivir in patients aged 12 to 18 years. The trial excluded individuals who had received COVID-19 vaccinations, which greatly limits the generalizability of the results to current practice; it is expected that the magnitude of the treatment effect would be smaller than that reported in the trial. The definition of “high risk of progression to severe COVID-19” has narrowed over time to focus on vaccination status, immunocompromised status (severe), and age (older). Infection with current SARS-CoV-2 variants no longer carries a high risk of hospitalization. Relevant subgroup analyses were not available and/or not informative for these populations of interest. According to the expert, most of the comorbidities listed for enrolment eligibility alone are no longer considered to significantly increase the risk of worse disease outcomes. Evidence for the treatment effect of remdesivir was based largely on hospitalization (with no conclusions on the impact of remdesivir on death), which varies among different clinical practices, regions (no trial sites were in Canada), and the availability of health care resources. The PINETREE trial took place before the omicron variant was the predominant circulating variant. Therefore, the estimate of the treatment effect from the trial may not be applicable in the context of the current COVID treatment landscape.
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Comparisons
No indirect treatment comparisons were submitted for this review.
Studies Addressing Gaps in the Evidence from the Systematic Review
Based on the information provided from the pivotal PINETREE trial, the sponsor identified an evidence gap for the effectiveness of remdesivir on patients with COVID-19 who were nonhospitalized, vaccinated, immunosuppressed and at high risk of disease progression.
Description of Studies
From December 1, 2021, to April 30, 2022 (i.e., early in the period when omicron became the dominant circulating variant), a total of 196 high-risk patients in Mexico were diagnosed with COVID-19, 126 of whom were included in the prospective, real-world evidence (RWE) study; 43% received remdesivir and 57% did not. Baseline clinical characteristics were similar between the groups; autoimmune diseases (31%), solid organ transplant (25%), and malignant neoplasms (19%) were the most common immunocompromising conditions. Most patients were vaccinated (79%) and immunosuppressed (94%). The primary efficacy composite outcome was all-cause hospitalization or death at 28 days after symptom onset.
Efficacy Results
Treatment with remdesivir significantly reduced the likelihood of hospitalization or death (adjusted HR = 0.16; 95% CI, 0.06 to 0.44; P < 0.01). The results were largely driven by all-cause hospitalization events (in 5 patients and 22 patients in the remdesivir and control groups, respectively) compared to all-cause deaths (in 0 patients and 9 patients in the remdesivir and control groups, respectively). There were 20 COVID-19–related hospitalizations; all were among patients in the control group. Diabetes mellitus was strongly associated with the primary outcome in both groups. Prior SARS-CoV-2 infection or vaccination were not independently associated with COVID-19 progression.
Harms Results
Harms results were not reported in the study.
Critical Appraisal
Overall, this was a small observational study (with 54 remdesivir users and 72 nonusers) that relied on data from a single tertiary referral centre in Mexico City for a highly selected group of patients with immunosuppression to examine the relationship between remdesivir exposure and 28-day hospitalization or mortality from December 2021 to April 2022. There were minimal details provided about data suitability (such as provenance, relevance, and data quality).
The study was submitted to address gaps in the RCT evidence; however, it is at risk of bias, residual confounding, and potential for unmeasured confounders. Population disease exposure as well as circulating variants have changed substantively since the time of this study, limiting the generalizability of these findings to the current COVID-19 treatment landscape. Due to these limitations, the comparative effectiveness estimates may be biased, and it is not possible to quantify or identify the direction of the bias. The results were susceptible to bias due to potential imbalances in unmeasured confounders. Therefore, it is challenging to draw any conclusions from this study.
Conclusions
Evidence from the PINETREE trial indicated that patients aged 12 years or older weighing 40 kg or more and at high risk of progression to severe COVID-19 were less likely to experience COVID-19–related hospitalization if they received remdesivir IV for 3 days in an outpatient setting. However, evidence that remdesivir reduces mortality, MAVs, need for oxygen supplementation, ICU admission, need for mechanical ventilation, and symptoms was lacking due to significant limitations of the study. It is important to note that the patient population in the trial (which was conducted early in the pandemic) was significantly different from the current patient population (e.g., given the milder viral variants now circulating and the greater proportion of people in Canada who have been vaccinated). While the results of the RWE study are aligned with those of the RCT in terms of remdesivir reducing hospitalization, there remains some uncertainty in the magnitude of the drug’s effectiveness in a highly vaccinated patient population with significant immunosuppression, given that the study was small and likely subject to significant concerns related to biases. As a result, the evidence included in this review is of uncertain or low clinical relevance in terms of how remdesivir would be used in clinical practice today. Based on the limited trial results, remdesivir appears to be a well-tolerated treatment, and both the literature and expert opinion indicate that it could be preferred for high-risk patients who cannot receive nirmatrelvir-ritonavir, although the IV administration of remdesivir is a limiting factor for patient access. There remains a lack of long-term efficacy and safety data for the use of remdesivir in relevant populations (i.e., those who are considered high risk today and are more likely to receive the IV drug in practice). No direct or indirect comparisons between remdesivir and other COVID-19 treatments were appraised in this review.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of remdesivir, 100 mg/vial, IV infusion for the treatment of COVID-19 in nonhospitalized patients weighing at least 40 kg with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progression to severe COVID-19, including hospitalization and death.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
COVID-19 is an illness caused by SARS-CoV-2 and the rapid global spread of the virus led to a pandemic, as declared by WHO on March 11, 2020.3 Subsequently, the proliferation of COVID-19 has presented significant challenges to health care systems globally, including in Canada.17-19 As of April 3, 2024, the cumulative numbers of reported COVID-19 cases and deaths in Canada were 4,946,090 and 59,034, respectively; the weekly percentage of positive cases out of the total tests conducted was 5.2%.20,21
Patients with COVID-19 present variably, with most current cases showing absent or mild symptoms.4 Mild symptoms can include sinus congestion, headache, sore throat, diarrhea, cough, fever, loss of taste and smell.4 Severe symptoms can include dyspnea, pneumonia, and acute respiratory distress syndrome.4 Mortality risk estimates reported by WHO for patients with nonsevere disease are 0.6% for those at high risk of hospitalization, 0.3% for those at moderate risk of hospitalization, and 0.05% for those at low risk of hospitalization.5 According to the clinical expert we consulted and the literature, the current omicron variants are associated with low morbidity, and patients tend to have self-limiting disease.6 Overall mortality and hospitalization risks were estimated to be 0.06% and 0.24%, respectively, based on first-ever infection with omicron variants in Canada during the second half of 2022.22
Currently, the relevance of risk factors for progressing to severe disease is not the same as it was early in the pandemic; over time, as viral pathogenicity has changed and population immunity has increased, the characteristics of patients being hospitalized due to COVID-19 have changed.6 According to the literature and the clinical expert we consulted, the most important risk factors for progression to severe disease now include lack of SARS-CoV-2 immunity (through prior infection, vaccination, or both), severe immune suppression, multiple chronic comorbidities, and older age (e.g., 80 years and older).4
SARS-CoV-2 has continually mutated throughout the pandemic, particularly at different locations within the spike protein, resulting in variants of the original virus.5 As of March 2023, 5 SARS-CoV-2 variants of concern had been recognized by WHO: alpha (B.1.1.7), beta (B.1.351), gamma (P.1), delta (B.1.617.2), and omicron (B.1.1.529), with omicron being more transmissible, having a shorter incubation period (approximately 3 days), and being associated with reduced morbidity and mortality, compared to the other variants.6-8
Given that the initial symptoms of COVID-19 closely resemble those of other respiratory infections, it is necessary to confirm diagnosis before treatment.12 Viral testing for COVID-19 has included molecular diagnostic tests, such as reverse-transcription polymerase chain reaction (PCR) tests, and rapid antigen tests (RATs).23
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by our review team.
Since the approval of the first COVID-19 vaccine by Health Canada in December 2020, 83.7% of the population has received at least 1 dose of vaccine (initial vaccination series).9 Vaccination remains the first line of defence to prevent SARS-CoV-2 infection.9 Based on serology, it is estimated that nearly everyone in Canada (> 99% of the population) has some form of infection-acquired or vaccine-induced immunity to the virus.10
Most patients with COVID-19 can be managed at home, which typically includes symptomatic care, monitoring for clinical deterioration, and isolation to prevent transmission.11 Patients with conditions that increase the risk of progression to severe disease (e.g., older age, immunocompromised status, chronic comorbidities, or inadequate vaccination) may need additional intervention.4,11 Currently, each province and territory adapts and implements treatments authorized for adults with mild to moderate COVID-19 within their own health care systems considering local context and resources. Treatment approaches are based on a patient’s severity of illness and risk of progressing to severe disease; the assessments of these vary across jurisdictions.
For individuals who are at high risk of progression to severe COVID-19 (i.e., leading to hospitalization or death), nirmatrelvir-ritonavir is generally the recommended first-line treatment; however, when it is not available or is contraindicated, remdesivir is an option.5,11 It has been suggested that, due to its IV administration, remdesivir may be suitable for patients living in institutional settings (e.g., skilled nursing facilities) or who are hospitalized with incidental nonsevere COVID-19, and that access can be improved if the drug is administered at infusion centres.11 According to the AMMI updated recommendations, nirmatrelvir-ritonavir and remdesivir may be considered in patients with mild disease (i.e., not requiring oxygen supplementation) based on the patient’s risk, severity, and trajectory of symptoms.12 Risk of hospitalization or death due to COVID-19 was based on vaccination status, age, and comorbidities.12 WHO guidelines for COVID-19 therapeutics provides a conditional recommendation, acknowledging the challenges associated with IV administration, for the use of remdesivir to treat patients with nonsevere disease who are at high risk of hospitalization.5 Other factors to consider when deciding which treatment to use include local availability of therapies, duration of symptoms (treatment should be started as soon as possible after confirming SARS-CoV-2 infection and within 5 days to 7 days of symptom onset), feasibility of administration (oral versus IV), and potential drug interactions (particularly with nirmatrelvir-ritonavir).11 Combination treatment is not recommended for outpatient management.11
Earlier in the pandemic, monoclonal antibodies were possible options for treating mild to moderate COVID-19; however, these are no longer recommended due to evidence showing limited benefit against omicron subvariants, which are now the prevailing circulating variants.11,12 There is also uncertain benefit for the use of other antivirals (e.g., simnotrelvir-ritonavir, ensitrelvir, molnupiravir), inhaled glucocorticoids, and pegylated interferon lambda in an outpatient setting.11
Drug Under Review
Remdesivir is a nucleotide prodrug of an adenosine triphosphate analogue that competes with the natural adenosine triphosphate substrate for incorporation into nascent RNA chains by the SARS-CoV-2 RNA-dependent RNA polymerase.2 It inhibits viral replication by terminating RNA transcription prematurely.2
Remdesivir has received a Notice of Compliance from Health Canada for the treatment of COVID-19 in nonhospitalized adult and pediatric patients (weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk of progression to severe COVID-19, including hospitalization and death.2 For this review, the sponsor has requested reimbursement of remdesivir for the treatment of COVID-19 in nonhospitalized patients aged 12 years or older (weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk of progression to severe COVID-19, including hospitalization and death. The recommended dose of remdesivir for nonhospitalized patients is a single loading dose of 200 mg on day 1 and a 100 mg dose on days 2 and 3.2 Treatment is to be started as soon as possible after diagnosis and within 7 days of symptom onset. Remdesivir is administered through IV infusion under conditions where severe hypersensitivity reactions (e.g., anaphylaxis) can be managed.
Remdesivir also has a Health Canada indication for the treatment of hospitalized adult and pediatric patients (aged at least 4 weeks and weighing at least 3 kg) with pneumonia requiring supplemental oxygen.2 It is also undergoing a review for the treatment of COVID-19 in hospitalized patients aged 12 years or older (weighing at least 40 kg) with pneumonia requiring supplemental oxygen.
Key characteristics of remdesivir and nirmatrelvir-ritonavir are summarized in Table 3.
Table 3: Key Characteristics of Remdesivir and Nirmatrelvir-Ritonavir
Characteristic | Remdesivir | Nirmatrelvir-ritonavir |
|---|---|---|
Mechanism of action | Remdesivir is a polymerase inhibitor that inhibits viral RNA synthesis. | Nirmatrelvir is a protease inhibitor that inhibits viral replication. Ritonavir inhibits the CYP3A-mediated metabolism of nirmatrelvir. |
Health Canada–approved indication | For the treatment of COVID-19 in:
| For the treatment of mild to moderate COVID-19 in adults with positive results of direct SARS-CoV-2 viral testing, and who are at high risk of progression to severe COVID-19, including hospitalization or death |
Route of administration | IV | Oral |
Recommended dose | Adult and pediatric patients (≥ 40 kg): loading dose of 200 mg on day 1; 100 mg once daily on days 2 and 3. Nonhospitalized patients are treated for 3 days starting as soon as possible after diagnosis and within 7 days of symptom onset. Hospitalized patients with pneumonia requiring supplemental oxygen are treated daily for at least 5 days and no more than 10 days. | 300 mg nirmatrelvir (2 × 150 mg tablets) with 100 mg ritonavir (1 × 100 mg tablet), with all 3 tablets taken together orally twice daily for 5 days. Treatment should be initiated as soon as possible after a COVID-19 diagnosis and within 5 days of symptom onset. |
Serious adverse effects or safety issues | Not to be used in patients with hepatic impairment. | Contraindicated with drugs that are highly dependent on CYP3A for clearance and drugs that are potent CYP3A inducers; dose adjustment required for patients with moderate renal impairment; not recommended in patients with severe renal or hepatic impairment; risk of serious adverse reactions with calcineurin inhibitors. |
CYP3A = cytochrome P450, family 3, subfamily A; RNA = ribonucleic acid; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Source: Product monographs for remdesivir and nirmatrelvir-ritonavir.2,24
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by our review team based on the input provided by 1 patient group.
One group, the Gastrointestinal Society, responded to our call for patient group input. The Gastrointestinal Society is a national charity committed to improving the lives of people living with gastrointestinal and liver conditions, supporting research, advocating for appropriate patient access to health care, and promoting gastrointestinal and liver health. Information was gathered primarily through meetings and discussions with health care professionals, researchers, academics, and staff with first-hand experience with COVID-19. The input highlights the impact of COVID-19 on the digestive tract.
According to the patient group, symptoms of COVID-19 can range from mild to severe and can include fever, fatigue, dry cough, difficulty breathing, aches and pains, nasal congestion, and sore throat. COVID-19 can affect the digestive tract in 2 key ways: first, by causing damage and affecting the intestinal lining, leading to diarrhea, stomach upset, vomiting, and inflammation (severe cases may even lead to obstructions, co-infections, intestinal necrosis, and organ failure); and second, by modifying the microbiome in the gastrointestinal tract, which can lead to opportunistic infections, severe gastrointestinal symptoms (pain, nausea, and diarrhea), and even anxiety and depression.
The patient input stated that, due to possible reinfection with the virus and recurrent illness, the availability of effective vaccines and treatments remains paramount. The group emphasized that, while a few options do exist to protect patients from severe COVID-19 infection or death — such as cilgavimab and tixagevimab, bamlanivimab, casirivimab and imdevimab, and sotrovimab — these treatments are difficult to access. According to the group, nirmatrelvir-ritonavir is the only oral medication available in Canada, and access to it is limited. The patient group stated that remdesivir is an option for individuals who have contraindications to nirmatrelvir-ritonavir; however, issues with access exist. The group indicated that individuals who are at increased risk of severe COVID-19 need access to treatments that are effective against the newer viral variants and that do not present contraindications with their other medicines and therapies.
Clinician Input
Input From Clinical Experts Consulted
All of our review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of COVID-19 in nonhospitalized patients.
Unmet Needs
The clinical expert we consulted stated that the aim of treatment for nonhospitalized patients with SARS-CoV-2 infection is to avoid hospitalization, death, and disability. While most patients will improve without intervention, the expert indicated that the 2 key criteria for identifying patients who could receive remdesivir for COVID-19 are the presence of a high-risk underlying medical condition and evidence of substantial symptoms (with no improvement within 5 days to 7 days). Although there are fewer administrative challenges with nirmatrelvir-ritonavir (an oral drug with no risk of infection from IV administration) compared to remdesivir, the latter would be an alternative option for patients who cannot receive nirmatrelvir-ritonavir due to intolerance and/or drug-drug interactions. According to the clinical expert, the evidence supporting the claim that either drug reduces symptom severity, viral transmission, or long COVID is inconclusive, especially in immune populations; this topic is subject to ongoing research.
Place in Therapy
The clinical expert indicated that between remdesivir and nirmatrelvir-ritonavir, nirmatrelvir-ritonavir is generally preferred for patients who do not have a contraindication. For patients who have an existing IV line, remdesivir may be a reasonable alternative.
Patient Population
According to the expert, in general, few patients are likely to benefit from treatment for COVID-19; it is only those with severe immune suppression who are likely to benefit. Moreover, patients with severe immune suppression tend to be receiving specialty care already (e.g., from a rheumatologist, transplant team, and/or hematologist), and can be identified as high risk based on their underlying illness. The expert emphasized that specific viral testing is necessary to differentiate SARS-CoV-2 infection from other influenza-like illnesses and for access to treatment. Additionally, disease trajectory is important because many high-risk patients will improve without intervention.
More recently, due to mild disease trajectory, fewer patients are being admitted to the hospital for COVID-19, and ICU admissions are rare. The expert also noted that patients with previously mild COVID-19 are likely to have less severe disease upon reinfection, except in cases where their medical condition has substantially changed. Patients who are severely immune-compromised (e.g., with B-cell depletion) are unable to effectively clear viral infections and may develop chronic infection. The expert identified these patients as being of great concern for progression to severe disease.
Assessing Treatment Response
There are no clinical markers for assessing patients with mild disease, although medical intervention should be pursued in those who continue to worsen. As per the expert, commonly used clinical trial outcomes (e.g., hospitalization, ICU admissions, and death) are important to practice.
Discontinuing Treatment
Remdesivir for nonhospitalized patients is indicated for a set 3-day duration and it is expected that most patients will complete treatment.
Prescribing Considerations
Given that remdesivir is an IV medication, a team of health care providers who can insert, care for, and remove an IV is necessary for administration. The care team is also required to monitor the patient during infusion and assess potential infection at the IV site. The expert stated that treatment typically takes place at hospitals or infusion clinics, although at-home nursing care may be an option for some.
Clinician Group Input
This section was prepared by our review team based on the input provided by 2 clinician groups.
Two clinician groups, BC Transplant Clinicians (7 authors) and the Ontario Health Infectious Diseases Advisory Committee (4 authors), responded to our call for clinician group input. The group from British Columbia gathered information through consultation with several key transplant clinicians involved in COVID-19 research and from clinical practice in patients with solid organ transplants. The group from Ontario jointly discussed the information for the submission through email.
Both clinician groups stated that the goals of treatment include reducing COVID-19–related mortality, hospitalization, ICU admissions, and symptom severity; preventing progression to severe disease and long-term sequelae; and accelerating recovery. The groups highlighted the ongoing need for treatments and vaccines with evidence of long-term safety and efficacy as well as drug options that improve the convenience of administration (i.e., oral versus IV infusion and fewer drug-drug interactions). Timely access to testing and COVID-19 treatments (considering the narrow time frames for initiating therapy) were noted as ongoing issues.
According to the clinician group from British Columbia, several monoclonal antibody treatments were previously approved as COVID-19 treatments; however, most were later found to be ineffective against newer viral variants, such as omicron BA.2. The groups noted that nirmatrelvir-ritonavir is not often used in patients in whom interactions between the drug and their current medications cannot be safely managed (e.g., those on antiarrhythmic medications, antiepileptic medications, or immunosuppressants to prevent graft rejection). Remdesivir may be used in such cases where contraindications to nirmatrelvir-ritonavir exist or in those who are outside the 5-day initiation window for nirmatrelvir-ritonavir.
British Columbia currently recommends and has prioritized the use of remdesivir in nonhospitalized patients with solid organ transplants with mild to moderate COVID-19 symptoms within 7 days of symptom onset, regardless of vaccine status or previous infection, because of its improved safety profile compared to that of nirmatrelvir-ritonavir. However, due to a lack of research including patients with solid organ transplants, the clinician group from British Columbia acknowledged that there is some uncertainty as to whether remdesivir is the best COVID-19 therapy for these patients. The group from Ontario noted that older age, immunocompromised status, nonvaccinated status, the presence of multiple and/or uncontrolled comorbidities, and specific medical or social vulnerabilities (e.g., cognitive disabilities or racialized status) are considerations when assessing greater need for intervention. Those who are asymptomatic or beyond the 7-day onset would be least suited to receive remdesivir.
According to the group from British Columbia, people with solid organ transplant receiving remdesivir as outpatients are monitored by infusion clinic nurses during the 3-day treatment course and followed up by their respective transplant centres. The group from Ontario indicated that, in general, a specialist is not required for managing patients receiving remdesivir, and that the drug can be administered in a community setting (e.g., nursing and long-term care homes), hospital outpatient clinic, or hospital emergency department. Both groups noted that a clinically meaningful response would be indicated by a significant reduction in hospitalizations, ICU admissions, and deaths; no need for new or increased supplemental oxygen; and improved or resolved symptoms. Discontinuation of therapy should be considered in patients with AEs — such as increased serum creatinine, increased serum liver enzymes, or hypersensitivity or infusion-site reactions — or if new data indicate that viral variants are no longer susceptible to remdesivir.
Drug Program Input
The drug programs provide input on each drug being reviewed through our Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts we consulted are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
Eligibility criteria for the pivotal trial included:
Are the eligibility criteria from the pivotal trial appropriate as reimbursement criteria for this indication? How should “confirmed SARS-CoV-2 infection” be determined? How would “pre-existing risk factor for progression to hospitalization” be defined? Are the pre-existing risk factors outlined in the eligibility criteria for the pivotal trial still relevant? | The clinical expert we consulted stated that, due to changes in population immunity and virus pathogenicity, the eligibility criteria for the PINETREE trial would not be appropriate as reimbursement criteria for remdesivir. According to the expert, it is essential that symptomatic patients who are to receive remdesivir have confirmed SARS-CoV-2 infection. The expert indicated that any approved test is acceptable (although a PCR test is preferred), and that patients who attend an infusion clinic may be able to better access PCR testing. The clinical expert noted that the pre-existing risk factors included in the PINETREE trial are no longer relevant for defining those who are at high risk of progression to severe disease. This is because most of the population of Canada has some natural or induced SARS-CoV-2 immunity, and hospitalization rates among the general and high-risk populations have decreased over time. However, some patients at high risk of progression, such as those who are severely immune suppressed and not able to produce a sufficient immune response (to vaccination or infection), may benefit from antiviral treatments. |
The Health Canada indication and sponsor’s reimbursement request specify “nonhospitalized adults and pediatric patients (weighing ≥ 40 kg).” How are nonhospitalized pediatric patients < 12 years of age and/or weighing < 40 kg who are at high risk of progression to severe COVID-19 managed? | The clinical expert suggested that the possibility of antiviral treatment for these patients (aged < 12 years and/or weighing < 40 kg) should be discussed on a case-by-case basis with specialists who have knowledge of infectious diseases and experience managing pediatric patients. |
Should reimbursement criteria for remdesivir include consideration of nirmatrelvir-ritonavir before remdesivir (where appropriate), given the ease of availability and access? | The clinical expert agreed that nirmatrelvir-ritonavir should be considered before remdesivir, and that due to its IV administration, remdesivir has a significant impact on health care resources (to administer) compared to oral nirmatrelvir-ritonavir. |
Is it possible to align the reimbursement criteria for remdesivir with those for nirmatrelvir-ritonavir, where appropriate? | As per the clinical expert consulted for this review, it would be reasonable to align the reimbursement criteria of the 2 drugs, where appropriate. |
Considerations for prescribing of therapy | |
For nonhospitalized patients, the recommended total treatment duration for remdesivir is 3 days. However, NIH has suggested that longer and/or additional courses of remdesivir may be used in immunocompromised patients with prolonged, symptomatic COVID-19 and evidence of ongoing viral replication. How often do patients require longer and/or additional courses of remdesivir in clinical practice? | The clinical expert indicated that most patients in an outpatient setting receive remdesivir for 3 days, and that it would be rare to require longer and/or additional courses of the drug as prophylaxis for hospitalization in clinical practice. |
Is there any evidence to support the combination use of antiviral therapies? | According to the expert, there is a lack of evidence to support the combination use of antiviral therapies, including remdesivir. |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; eGFR = estimated glomerular filtration rate; NIH = National Institutes of Health; PCR = polymerase chain reaction; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; ULN = upper limit of normal.
Clinical Evidence
The objective of our Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of remdesivir, 100 mg/vial, IV infusion for the treatment of COVID-19 in nonhospitalized patients weighing at least 40 kg with positive results of direct SARS-CoV-2 viral testing and who are at high risk of progression to severe COVID-19, including hospitalization and death. The focus is on comparing remdesivir to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of remdesivir is presented in 2 sections, with our critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes 1 RCT selected according to the sponsor’s systematic review protocol. The second section includes 1 additional RWE study considered by the sponsor to address important gaps in the systematic review evidence. No long-term extension studies or indirect comparisons were submitted by the sponsor.
Included Studies
Clinical evidence from the following 2 studies were included in the review and are appraised in this document:
1 pivotal RCT identified in the systematic review
1 RWE study addressing gaps in the evidence.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the our review team.
Description of Studies
The characteristics of the PINETREE study are summarized in Table 5. One multicentre, phase III, DB, placebo-controlled RCT was included in the sponsor’s systematic review of the efficacy and safety of remdesivir. The PINETREE trial (N = 584) assessed the superiority of remdesivir compared with placebo for the treatment of COVID-19 in nonhospitalized patients aged 12 years or older (weighing at least 40 kg) with SARS-CoV-2 infection confirmed through direct viral testing who were at high risk of progression to severe COVID-19, including hospitalization and death (with risk based on prespecified patient characteristics at the time the study was performed). Patients were randomized 1:1 to either remdesivir or matching placebo for 3 days. Randomization was stratified by patients who resided in a skilled nursing facility (yes versus no), age (less than 60 years versus at least 60 years), and region (US versus non-US). The primary outcome of the trial was a combined outcome of the proportion of patients with COVID-19–related hospitalization (defined as at least 24 hours of acute care) or all-cause death by day 28.
Table 5: Details of the Studies Included in the Systematic Review
Detail | PINETREE trial |
|---|---|
Designs and populations | |
Study design | Phase III, DB, placebo-controlled RCT |
Locations | 64 sites in US, Denmark, Spain, and UK |
Patient enrolment dates | First patient screened: September 18, 2020 Last patient last visit for primary end point: May 6, 2021 |
Randomized (N) | N = 584, randomized 1:1 to the following groups:
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Remdesivir 100 mg IV for 3 days: 200 mg on day 1, and 100 mg on each of days 2 and 3 |
Comparator | Matching placebo administered in a manner similar to remdesivir |
Study duration | |
Screening phase | Within 2 days before randomization |
Treatment phase | 3 days |
Follow-up phase | Day 4 through day 28 |
Outcomes | |
Primary end points |
|
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publication status | |
Publications | |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; DB = double blind; eGFR = estimated glomerular filtration rate; FLU-PRO Plus = COVID-19–adapted InFLUenza Patient-Reported Outcome; ICU = intensive care unit; MAV = medically attended visit; PCR = polymerase chain reaction; PK = pharmacokinetic; RCT = randomized controlled trial; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; TEAE = treatment-emergent adverse event; ULN = upper limit of normal.
aPre-existing risk factors included chronic lung disease (e.g., chronic obstructive pulmonary disease, moderate to severe asthma, cystic fibrosis, pulmonary fibrosis); hypertension (e.g., systemic or pulmonary); cardiovascular or cerebrovascular disease (e.g., coronary artery disease, congenital heart disease, heart failure, cardiomyopathy, history of stroke, atrial fibrillation, or hyperlipidemia); diabetes mellitus (i.e., type 1, type 2, or gestational); obesity (i.e., body mass index ≥ 30 kg/m2); immunocompromised state (e.g., having a solid organ, blood, or bone marrow transplant, immune deficiencies, HIV with low cluster of differentiation 4 cell count or not receiving HIV treatment, prolonged use of corticosteroids, or use of other immunity-weakening medicines); chronic mild or moderate kidney disease; chronic liver disease; current cancer; or sickle cell disease.
bHospitalization was defined as receiving at least 24 hours of acute care.
cMAV was defined as a medical visit attended in person by the patient and a health care professional.
Source: Clinical Study Report for the PINETREE trial.16
Populations
Inclusion and Exclusion Criteria
Patients must have been aged at least 12 years and weighed at least 40 kg. If aged less than 60 years, they must have had at least 1 pre-existing risk factor for progression to hospitalization. Pre-existing risk factors included chronic lung disease (e.g., chronic obstructive pulmonary disease, moderate to severe asthma, cystic fibrosis, pulmonary fibrosis); hypertension (e.g., systemic or pulmonary); cardiovascular or cerebrovascular disease (e.g., coronary artery disease, congenital heart disease, heart failure, cardiomyopathy, history of stroke, atrial fibrillation, or hyperlipidemia); diabetes mellitus (i.e., type 1, type 2, or gestational); obesity (i.e., body mass index of at least 30 kg/m2); immunocompromised state (e.g., having a solid organ, blood, or bone marrow transplant; immune deficiencies; HIV with a low cluster of differentiation 4 cell count or not receiving HIV treatment, prolonged use of corticosteroids, or use of other immune weakening medicines); chronic mild or moderate kidney disease; chronic liver disease; current cancer; and sickle cell disease. Patients who were at least 60 years old were permitted trial entry regardless of pre-existing risk factors. SARS-CoV-2 infection was confirmed by PCR test or RAT within 4 days of screening, and patients must have been symptomatic. Patients were excluded if they were receiving or required supplemental oxygen or required hospitalization during the trial. Prior COVID-19–related hospitalization, experimental or preventive treatment for COVID-19 (including vaccination), and low kidney function (estimated glomerular filtration rate of less than 30 mL/min/1.73 m2) were also reasons for exclusion.
Interventions
Patients receiving active treatment were administered a single 200 mg dose of remdesivir IV infusion over 30 minutes to 120 minutes on day 1 followed by remdesivir 100 mg IV on days 2 and 3. Patients not receiving remdesivir were given a matching placebo for the same duration. Study treatments were administered at the study centre or at home (93 patients received at least 1 infusion at home; 16 patients received at least 1 infusion in a skilled nursing facility; and 1 patient received treatment in both settings).
Prohibited concomitant medications included investigational or approved drugs for treating SARS-CoV-2 (including, but not limited to, HIV protease inhibitors, such as lopinavir-ritonavir and interferon, although their use was permitted for approved indications other than SARS-CoV-2 infection), hydroxychloroquine, chloroquine, and strong inducers of P-glycoprotein (e.g., rifampin, herbal medications).
Remdesivir was discontinued for any of the following reasons: intercurrent illness significantly affecting assessments, unacceptable toxicity, elevated liver function test results (ALT, AST, total bilirubin), lack of efficacy, patient choice, nonadherence, sponsor or regulatory request, drug-related grade 3 or grade 4 abnormal laboratory results, grade 2 or higher infusion-related systemic reaction, or grade 3 or higher infusion-related localized reaction.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review, according to the clinical expert we consulted and input from patient and clinician groups and public drug plans. Using the same considerations, the review team selected end points that were considered most relevant to inform our expert committee deliberations and finalized this list of end points in consultation with members of the expert committee.
Table 6: Outcomes Summarized From the PINETREE Trial
Outcome measure | Time point | Study 1 |
|---|---|---|
Composite end point of COVID-19–related hospitalizationa or all-cause death by day 28 | Through 28 days | Primary |
Proportion of patients with safety events (e.g., TEAEs) | Through 28 days | Primary |
Composite end point of COVID-19–related hospitalizationa or all-cause death by day 14 | Through 14 days | Secondary |
Composite end point of COVID-19–related MAVb or all-cause death by day 28 | Through 28 days | Secondary |
Composite end point of COVID-19–related MAVb or all-cause death by day 14 | Through 14 days | Secondary |
All-cause mortality at day 28 | Through 28 days | Secondary |
Proportion of patients hospitalized by day 28 | Through 28 days | Secondary |
Proportion of patients requiring oxygen supplementation by day 28 | Through 28 days | Secondary |
Proportion of patients admitted to the ICU by day 28 | Through 28 days | Exploratory |
Proportion of patients started on mechanical ventilation by day 28 | Through 28 days | Exploratory |
Time to alleviation of baseline COVID-19 symptoms (to mild or absent), as reported on the FLU-PRO Plus | Through 14 days | Secondary |
FLU-PRO Plus = COVID-19–adapted InFLUenza Patient-Reported Outcome; ICU = intensive care unit; MAV = medically attended visit; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; TEAE = treatment-emergent adverse event.
Note: Outcomes were not adjusted for multiple comparisons (e.g., hierarchal testing).
aHospitalization was defined as receiving at least 24 hours of acute care.
bMAV was defined as a medical visit attended in person by the patient and a health care professional.
Source: Clinical Study Report for the PINETREE trial.16
FLU-PRO Plus Questionnaire
The FLU-PRO questionnaire that was adapted for assessing COVID-19 symptoms is referred to as the FLU-PRO Plus.
The FLU-PRO is a standardized questionnaire for reporting influenza symptoms in clinical trials.13 According to the literature, the FLU-PRO categorizes 32 items or symptoms into 6 domains: nose, throat, eyes, chest or respiratory, gastrointestinal, and body or systemic.14,15 The FLU-PRO Plus includes a seventh domain for senses, with 2 items: loss of taste and smell.14,15 Patients rate the extent of their symptoms from 0 (not at all) to 4 (very much) over the preceding 24 hours, and the mean score of symptoms for each domain is used to determine a total score.14,15 Where available, patients in the PINETREE trial completed the questionnaire daily from day 1 to day 14; this was to be done before drug dosing.16
A 2020 study of US Military Health System adult beneficiaries (N = 226) suggested that the FLU-PRO Plus demonstrates construct validity (i.e., moderate to high Spearman correlations with the patient global assessment of disease severity and the domain and total scores) and known-groups validity (i.e., larger differences between the mild and moderate groups and mild and severe groups, but smaller differences between the moderate and severe groups).14 A second study using data from the COMET-ICE trial (N = 845) suggested a similar finding for construct validity (i.e., moderate correlation between the total score and the Short Form [12] Health Survey as well as the Work Productivity and Activity Impairment questionnaire: General Health Survey) and known-groups validity (compared to the Work Productivity and Activity Impairment questionnaire: General Health Survey).15 Two-day test-retest reliability was measured using intraclass correlation coefficients (range, 0.86 to 0.93) and indicated good internal consistency for all domains in the US beneficiaries study.14 Similar internal consistency reliability results were found in the data from the COMET-ICE trial (i.e., Cronbach coefficient alpha of 0.94 for total score, ranging from 0.71 to 0.90 for domain scores).15 In terms of responsiveness, the instrument was sensitive to changes that differentiated responders from nonresponders at day 6 or day 7 of the questionnaire in the US beneficiaries survey and from day 1 to day 29 in the COMET-ICE trial data.14,15 The time between symptom onset and questionnaire responses varied in the US beneficiaries survey, with a median of 6 days between the time points.14 No MID has been identified from the literature.
Safety Events
Safety was assessed based on the proportion of patients experiencing TEAEs (coded using Version 24.0 of the Medical Dictionary for Regulatory Activities), SAEs, withdrawal from treatment due to AEs, and death during the trial. An AE was any untoward medical occurrence in a patient who received the study drug, and included pretreatment or posttreatment complications, lack of efficacy, overdose, drug misuse, occupational exposure, and changes in or increased severity of pre-existing events. An SAE was an event that resulted in a life-threatening situation, inpatient hospitalization (or prolongation of existing hospitalization), persistent significant disability or incapacity, congenital anomaly or birth defect, death, or other medically significant event requiring medical or surgical intervention.
Based on the Health Canada product monograph, transaminase elevations and hypersensitivity reactions (e.g., infusion-related and anaphylactic reactions) were notable harms included in the review.2
Statistical Analysis
The statistical analysis methods are summarized in Table 7.
Table 7: Statistical Analysis of Efficacy End Points in the PINETREE Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Composite end point of COVID-19–related hospitalizationa or all-cause death by day 28 (primary end point) | Cox model | Region, patient age, and resident of a skilled nursing facility (yes or no) | Premature study discontinuations, missing hospitalization, and death status were censored at last date of contact | CMH test using baseline stratification factors as strata Premature study discontinuations were considered as no hospitalization or death |
Composite end point of COVID-19–related hospitalizationa or all-cause death by day 14 | Same as primary end point | Not reported | Not reported | Not reported |
Composite end point of COVID-19–related MAVb or all-cause death by day 28 | Same as primary end point | Not reported | Not reported | Not reported |
Composite end point of COVID-19–related MAVb or all-cause death by day 14 | Same as primary end point | Not reported | Not reported | Not reported |
All-cause mortality at day 28 | Fisher exact test | Not reported | Premature study discontinuations and missing death status were not included in the analysis | Not reported |
Proportion of patients hospitalized by day 28 | KM estimate, log-rank test, Cox regression | Region, patient age, and resident of a skilled nursing facility (yes or no) | Not reported | Not reported |
Proportion of patients requiring oxygen supplementation by day 28 | Fisher exact test | Not reported | Premature study discontinuations were considered not to require oxygen supplementation | Not reported |
Proportion of patients admitted to the ICU by day 28 | Fisher exact test | Not reported | Not reported | Not reported |
Proportion of patients started on mechanical ventilation by day 28 | Not reported | Not reported | Not reported | Not reported |
Time to alleviation of baseline COVID-19 symptoms (to mild or absent) as reported on the FLU-PRO Plus | KM estimate, log-rank test, Cox regression | Region, patient age, and resident of a skilled nursing facility (yes or no) | Not reported | Not reported |
CMH = Cochran-Mantel-Haenszel; FLU-PRO Plus = COVID-19–adapted InFLUenza Patient-Reported Outcome; ICU = intensive care unit; KM = Kaplan-Meier; MAV = medically attended visit.
aHospitalization was defined as receiving at least 24 hours of acute care.
bMAV was defined as a medical visit attended in person by the patient and a health care professional.
Source: Clinical Study Report for the PINETREE trial.16
Sample Size and Power Calculation
Initially, a sample size of 1,264 patients (632 patients in each treatment group) was planned to provide more than 90% power to detect a ratio of 0.55 (remdesivir to placebo) for the primary outcome (i.e., the proportions of COVID-19–related hospitalization or all-cause death; equivalent HR of 0.53). The analyses used a 2-sided 0.05 significance level, and it was assumed that the overall COVID-19–related hospitalization or all-cause death rate was 9.3% (6.6% in the remdesivir group and 12% in the placebo group), with a 5% dropout rate. The sample size would also provide approximately 80% power to detect a smaller treatment effect size with a ratio of 0.60 (remdesivir to placebo) based on the assumption that the overall COVID-19–related hospitalization or all-cause death rate was 9.6% (7.2% in the remdesivir group and 12% in the placebo group). The placebo group rate was based on the 13.5% of high-risk patients (aged at least 65 years or with a body mass index of at least 35 kg/m2) experiencing COVID-19–related hospitalization or emergency department visits and the recent declining hospitalization rate.29
Statistical Testing
The primary efficacy end point was evaluated in a stratified analysis based on 3 randomization stratification variables: region (US versus non-US), age (less than 60 years versus at least 60 years), and whether they were a resident of a skilled nursing facility (yes versus no). Observed imbalances between treatment groups in baseline characteristics may have been considered as covariates for the sensitivity analyses.
Efficacy was assessed based on the primary efficacy end point at the 0.05 significance level. The HR, 95% CI, P value from the Cox model, and proportion of COVID-19–related hospitalization or all-cause death at day 28 (based on Kaplan-Meier [KM] estimates) were provided. No multiplicity adjustment was made in the final analysis, and efficacy end points beyond the primary outcome were exploratory in nature.
Categorical data were compared between treatment groups using the Cochran-Mantel-Haenszel test. Continuous data were analyzed using the 2-sided Wilcoxon rank sum test.
Data Imputation Methods
Patient with missing data for the primary efficacy end point due to premature study discontinuation or missing hospitalization or death status were censored at the last study date. Missing data were not imputed.
Subgroup Analyses
Prespecified subgroup analyses were conducted for the primary efficacy outcome and were not adjusted for multiplicity. Of the subgroups assessed in the trial, patients who resided in a skilled nursing facility and had a baseline risk factor of being in an immunocompromised state were identified as relevant to the review.
Sensitivity Analyses
A sensitivity analysis to test the robustness of the primary efficacy end point was conducted using the Cochran-Mantel-Haenszel test, with baseline stratification factors as strata. Patients who discontinued the study prematurely were considered as having no hospitalization or death.
Analysis of Secondary and Exploratory Outcomes
Secondary end points of COVID-19-related hospitalization or all-cause death by day 14, COVID-19–related MAV or death by day 28, and COVID-19–related MAV or death by day 14 were analyzed in the same manner as the primary end point.
The proportion of patients with COVID-19–related hospitalization by day 28 was estimated using the KM method and compared between treatment groups using a log-rank test. The HR and 2-sided 95% CI were estimated using Cox regression, with baseline stratification factors as covariates.
All-cause mortality by day 28 was compared between treatment groups using the Fisher exact test. Patients who prematurely discontinued the study or had a missing death status were not included in the analysis.
The number and proportion of patients requiring oxygen supplementation by day 28 were summarized and compared using the Fisher exact test. Patients who prematurely discontinued the study before requiring oxygen supplementation were considered as not requiring oxygen supplementation.
The time to alleviation of baseline COVID-19 symptoms (for 2 consecutive days), as reported on the FLU-PRO Plus, used the KM product limit method and log-rank test to estimate and compare treatment groups. The HR and 2-sided 95% CI were estimated using the Cox regression, with baseline stratification factors as covariates. Patients who had not achieved symptom alleviation by the last FLU-PRO Plus assessment date or who prematurely discontinued were censored at the last questionnaire date.
Analysis Populations
The analysis populations of the PINETREE trial are summarized in Table 8.
Table 8: Analysis Populations of the PINETREE Trial
Study | Population | Definition | Application |
|---|---|---|---|
PINETREE trial | All-randomized analysis set | All patients randomized in the study. | Number and percentage of patients randomized at each investigator site |
Full analysis set | All patients who were randomized in the study and received ≥ 1 dose of study treatment. Patients were grouped according to the treatment to which they were randomized. | Primary analysis set for efficacy analysis | |
Modified full analysis set | All patients who were randomized in the study, received ≥ 1 dose of study treatment, and enrolled under protocol amendment 2 or later. Patients were grouped according to the treatment to which they were randomized. | For secondary end points of the composite end point of COVID-19–related MAV or deatha | |
Safety analysis set | All patients who were randomized in the study and received ≥ 1 dose of study treatment. Patients were grouped according to the treatment they received. | Primary analysis set for safety analyses For demographic and baseline characteristic summaries | |
Virology analysis set | All patients who were randomized in the study, received ≥ 1 dose of study treatment, and had positive SARS-CoV-2 viral load at baseline.b | Analyses for SARS-CoV-2 end points |
MAV = medically attended visit; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
aThe modified full analysis set includes patients who were enrolled under protocol amendment 2 and excludes patients for whom MAV data were not collected.
bA “no SARS-CoV-2 detected” result was considered negative, while “inconclusive,” “< 2,228 cp/mL SARSCoV2 detected,” and numerical results were considered positive.
Source: Clinical Study Report for the PINETREE trial.16
Results
Patient Disposition
Patient disposition is summarized in Table 9. Overall, 630 individuals were screened, 46 of whom were screened out. Of those screened out, 24 did not meet the eligibility criteria, and 22 met all the criteria but were not randomized in the trial. Of the 584 individuals randomized to treatment, 292 were assigned to each of the remdesivir and placebo groups. However, 13 patients and 9 patients from the respective groups were never treated.
Of the 20 patients who discontinued the study drug, 6 patients (2.2%) were from the remdesivir group and 14 patients (4.9%) were from the placebo group. The most common reasons for discontinuing were patient decision (1.1%) in the remdesivir group and AEs (2.1%) in the placebo group. Of the 24 patients who discontinued the study, 13 patients (4.7%) were from the remdesivir group and 11 patients (3.9%) were from the placebo group. The most common reasons for discontinuing were loss to follow-up (2.5%) in the remdesivir group and withdrawn consent (1.4%) in the placebo group. Reasons for discontinuing treatment and the study were generally balanced between treatment groups.
Study enrolment was stopped on April 8, 2021, at which time less than half the planned sample size had been randomized. Reasons for stopping enrolment included declining COVID-19 case rates, increasing availability of single-infusion monoclonal antibodies as an alternative to placebo, and increasing vaccination rates among high-risk patients during the trial.
Table 9: Summary of Patient Disposition in the PINETREE Trial
Patient disposition | PINETREE trial | |
|---|---|---|
Remdesivir | Placebo | |
Screened, N | 630 | |
Reason for being screening out, n | 22 | |
Withdrawn consent | 14 | |
Outside of visit window | 3 | |
Lost to follow-up | 2 | |
Investigator's discretion | 1 | |
Other | 2 | |
Randomized, N | 292 | 292 |
Received ≥ 1 dose of study drug, N (%) | 279 (100) | 283 (100) |
Completed study drug, n (%) | 273 (97.8) | 269 (95.1) |
Discontinued study drug, n (%) | 6 (2.2) | 14 (4.9) |
Reason for discontinuation, n (%) | ||
Patient decision | 3 (1.1) | 5 (1.8) |
Adverse event | 1 (0.4) | 6 (2.1) |
Investigator discretion | 1 (0.4) | 1 (0.4) |
Protocol violation | 1 (0.4) | 1 (0.4) |
Noncompliance with study drug | 0 (0) | 1 (0.4) |
Completed study, n (%) | 266 (95.3) | 272 (96.1) |
Discontinued study, n (%) | 13 (4.7) | 11 (3.9) |
Reason for discontinuation, n (%) | ||
Lost to follow-up | 7 (2.5) | 2 (0.7) |
Withdrawn consent | 5 (1.8) | 4 (1.4) |
Protocol violation | 1 (0.4) | 1 (0.4) |
Adverse event | 0 (0) | 3 (1.1) |
Investigator’s discretion | 0 (0) | 1 (0.4) |
All-randomized analysis set, N (%) | 292 (100) | 292 (100) |
Full analysis set, n (%) | 279 (95.5) | 283 (96.9) |
Modified full analysis set, n (%) | 246 (84.2) | 252 (86.3) |
Safety analysis set, n (%) | 279 (95.5) | 283 (96.9) |
Virology analysis set, n (%) | 217 (74.3) | 214 (73.3) |
Source: Clinical Study Report for the PINETREE trial.16
Baseline Characteristics
The baseline characteristics outlined in Table 10 are limited to those that are most relevant to this review or were considered to affect the outcomes or interpretation of the study results. The mean ages of patients were 50 years (SD = 15.3 years) in the remdesivir group and 51 years (SD = 14.8 years) in the placebo group. There were 3 patients (1.1%) and 5 patients (1.8%) in the remdesivir and placebo groups, respectively, who were aged 12 years to 18 years, and there were 83 patients (29.7%) and 87 patients (30.7%) in the remdesivir and placebo groups, respectively, who were aged older than 60 years. Other baseline demographic and clinical characteristics were generally balanced between the groups.
Table 10: Summary of Baseline Characteristics in the PINETREE Trial, Safety Analysis Set
Characteristic | PINETREE trial | |
|---|---|---|
Remdesivir (N = 279) | Placebo (N = 283) | |
Demographic characteristics | ||
Age (years), mean (SD) | 50 (15.3) | 51 (14.8) |
Age (years), median (range) | 51 (13 to 89) | 52 (14 to 98) |
Age category, n (%) | ||
< 18 years | 3 (1.1) | 5 (1.8) |
≥ 18 years to < 60 years | 193 (69.2) | 191 (67.5) |
≥ 60 years | 83 (29.7) | 87 (30.7) |
Sex, n (%) | ||
Male | 148 (53.0) | 145 (51.2) |
Female | 131 (47.0) | 138 (48.8) |
Race, n (%) | ||
White | 228 (81.7) | 224 (79.2) |
Black | 20 (7.2) | 22 (7.8) |
Asian | 6 (2.2) | 7 (2.5) |
Other | 25 (9.0) | 30 (10.6) |
Clinical characteristics | ||
Baseline risk factor present, n (%) | ||
Diabetes mellitus | 173 (62.0) | 173 (61.1) |
Obesity | 154 (55.2) | 156 (55.1) |
Hypertension | 138 (49.5) | 130 (45.9) |
Chronic lung disease | 67 (24.0) | 68 (24.0) |
Cardiovascular or cerebrovascular disease | 20 (7.2) | 24 (8.5) |
Immunocompromised state | 14 (5.0) | 9 (3.2) |
Current cancer | 12 (4.3) | 18 (6.4) |
Chronic mild or moderate kidney disease | 7 (2.5) | 11 (3.9) |
Chronic liver disease | 1 (0.4) | 1 (0.4) |
Sickle cell disease | 0 (0) | 0 (0) |
Duration of symptoms before first dose of study drug (days) | ||
Mean (SD) | 5 (1.9) | 5 (1.9) |
Median (range) | 5 (0 to 18) | 5 (0 to 13) |
Duration from SARS-CoV-2 confirmation to first dose of study drug (days) | ||
Mean (SD) | 2 (1.5) | 3 (1.5) |
Median (range) | 2 (0 to 6) | 3 (0 to 7) |
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; SD = standard deviation.
Source: Clinical Study Report for the PINETREE trial.16
Protocol Deviations
During the trial, 23 patients (11 patients in the remdesivir group and 12 patients in the placebo group) reported at least 1 protocol deviation, for a total of 26 protocol deviations. Reasons for protocol deviations included eligibility criteria (8 patients), informed consent (5 patients), wrong treatment or incorrect dose (2 patients), use of excluded concomitant medication (1 patient), missing data (1 patient), and other reasons (7 patients). Numbers were similar between groups.
Exposure to Study Treatments
Patient exposure to study treatments is summarized in Table 11. Most patients (97.8% of patients in the remdesivir group and 95.4% of patients in the placebo group) received all 3 doses of the study drug.
Nearly all patients (93% to 94% of patients in the groups) reported using at least 1 concomitant medication during the trial. The use of any specific medication was similar between groups (i.e., < 5% difference). The most frequently used medications (i.e., used by > 15% of patients in any group) were paracetamol, ascorbic acid, acetylsalicylic acid, and salbutamol.
Table 11: Summary of Patient Exposure From the PINETREE Trial, Safety Analysis Set
Exposure | PINETREE trial | |
|---|---|---|
Remdesivir (N = 279) | Placebo (N = 283) | |
Mean (SD) | 3 (0.3) | 3 (0.3) |
1 dose, n (%) | 4 (1.4) | 5 (1.8) |
2 doses, n (%) | 2 (0.7) | 8 (2.8) |
3 doses, n (%) | 273 (97.8) | 270 (95.4) |
SD = standard deviation.
Source: Clinical Study Report for the PINETREE trial.16
Efficacy
COVID-19–Related Hospitalization or All-Cause Death
Results for COVID-19–related hospitalization and all-cause death by day 28 are summarized in Table 12. Two patients (0.7%) who received remdesivir and 15 patients (5.3%) who received placebo met the primary efficacy end point (HR = 0.13; 95% CI, 0.03 to 0.59; P value = 0.0076). We calculated the number needed to treat was 22. A sensitivity analysis using baseline stratification factors as strata supported the results of the primary analysis.
Two subgroups were considered relevant to the our review. Among the 15 patients who were residing in a skilled nursing facility and the 23 patients who had a baseline risk factor of being immunocompromised, none experienced a COVID-19–related hospitalization or death from any cause by day 28.
The primary composite outcome was driven by COVID-19–related hospitalizations because there were no deaths in either group by day 28. Furthermore, all COVID-19–related hospitalizations occurred by day 14, and the number of events by day 14 was the same as that by day 28.
COVID-19–Related MAV or All-Cause Death
COVID-19–related MAV or all-cause death by day 28 and day 14 are summarized in Table 13. By day 28, 4 patients (1.6%) in the remdesivir group and 21 patients (8.3%) in the placebo group met the composite end point of COVID-19–related MAV or all-cause death (HR = 0.19; 95% CI, 0.07 to 0.56). By day 14, 2 patients (0.8%) in the remdesivir group and 20 patients (7.9%) in the placebo group met the composite end point (HR = 0.10; 95% CI, 0.02 to 0.43).
Table 12: Summary of COVID-19–Related Hospitalization or All-Cause Death From the PINETREE Trial by Day 28, Full Analysis Set
Outcome | PINETREE trial | |
|---|---|---|
Remdesivir (N = 279) | Placebo (N = 283) | |
Composite end point of COVID-19–related hospitalization or all-cause death by day 28 | ||
Patients, n (%)a | 2 (0.7) | 15 (5.3) |
HR (95% CI)b | 0.13 (0.03 to 0.59) | |
P valuec | 0.0076 | |
NNT (patients)d | 21.7 | |
CI = confidence interval; HR = hazard ratio; NNT = number needed to treat.
aProportion of COVID-19–related hospitalization or all-cause death based on KM estimates.
bHR, 2-sided 95% CI, and P value were estimated using Cox regression, with baseline stratification factors as covariates (region, age, and resident of skilled nursing facility or not).
cP value has not been adjusted for multiple testing.
dWe approximated the NNT by dividing 1 by the reported absolute risk difference between treatment groups.
Source: Clinical Study Report for the PINETREE trial.16
Table 13: Summary of COVID-19–Related MAV or All-Cause Death From the PINETREE Trial, Modified Full Analysis Set
Outcome | PINETREE trial | |
|---|---|---|
Remdesivir (N = 246) | Placebo (N = 252) | |
Composite end point of COVID-19–related MAV or all-cause death by day 28 | ||
Patients, n (%)a | 4 (1.6) | 21 (8.3) |
HR (95% CI)b | 0.19 (0.07 to 0.56) | |
P valuec | 0.0024 | |
Composite end point of COVID-19–related MAV or all-cause death by day 14 | ||
Patients, n (%)a | 2 (0.8) | 20 (7.9) |
HR (95% CI)b | 0.10 (0.02 to 0.43) | |
P valuec | 0.0019 | |
CI = confidence interval; HR = hazard ratio; MAV = medically attended visit.
aProportion of COVID-19–related MAV or all-cause death based on KM estimates.
bHR, 2-sided 95% CI, and P value were estimated using Cox regression, with baseline stratification factors as covariates (region, age, and resident of skilled nursing facility or not).
cP value has not been adjusted for multiple testing.
Source: Clinical Study Report for the PINETREE trial.16
Patients Progressing to Requiring Oxygen Supplementation
Of the 6 patients who progressed to requiring oxygen supplementation by day 28, 1 patient (0.4%) was in the remdesivir group and 5 patients (1.8%) were in the placebo group.
Patients Admitted to the ICU
Of the 6 patients who were admitted to the ICU by day 28, 3 patients (1.1%) were in the remdesivir group and 3 patients (1.1%) were in the placebo group.
Patients Started on Mechanical Ventilation
One (0.4%) patient in the trial (randomized to the placebo group) required mechanical ventilation by day 28.
FLU-PRO Plus Results
FLU-PRO Plus results from the PINETREE trial are summarized in Table 14. Baseline data captured before the first dose were gathered for 66 patients in the remdesivir group and 60 patients in the placebo group. For this subset of patients, 23 patients in the remdesivir group and 15 patients in the placebo group reported alleviation of baseline COVID-19 symptoms (to mild or absent) at day 14 (HR = 1.41; 95% CI, 0.73 to 2.69).
In a post hoc analysis that included questionnaire results collected before or on the first dosing date, 61 patients of the 169 patients in the remdesivir group and 33 patients of the 165 patients in the placebo group reported alleviation of baseline COVID-19 symptoms at day 14 (HR = 1.92; 95% CI, 1.26 to 2.94).
Table 14: Summary of FLU-PRO Plus Results From the PINETREE Trial, Full Analysis Set
Outcome | PINETREE trial | |
|---|---|---|
Remdesivir (N = 279) | Placebo (N = 283) | |
Time to alleviation of baseline COVID-19 symptoms (to mild or absent) as reported on the FLU-PRO Plus at day 14 – prespecified outcome (data collected before first dosing) | ||
Patients in analysis, n | 66 | 60 |
Patients with events, n | 23 | 15 |
HR (95% CI)a | 1.41 (0.73 to 2.69) | |
P valueb | 0.2987 | |
CI = confidence interval; FLU-PRO Plus = COVID-19–adapted InFLUenza Patient-Reported Outcome; HR = hazard ratio.
aHR and 2-sided 95% CI were estimated using Cox regression, with baseline stratification factors as covariates (region, age, and resident of skilled nursing facility or not). P value was based on the stratified log-rank test, with baseline stratification factors as strata.
bP value has not been adjusted for multiple testing.
Source: Clinical Study Report for the PINETREE trial.16
Harms
Harms are summarized in Table 15.
Adverse Events
In the PINETREE trial, 42.3% of patients in the remdesivir group and 46.3% of patients in the placebo group reported at least 1 TEAE. The most common events occurring in the trial included nausea (10.8% for remdesivir and 7.4% for placebo), headache (5.7% for remdesivir and 6.0% for placebo), and cough (3.6% for remdesivir and 6.4% for placebo). In general, the incidences of harms were similar between treatment groups.
Serious Adverse Events
Overall, 1.8% of patients in the remdesivir group and 6.7% of patients in the placebo group reported at least 1 SAE. The most common SAEs were related to COVID-19 and pneumonia.
Withdrawal Due to AEs
In total, 0.7% of patients in the remdesivir group and 1.8% of patients in the placebo group stopped treatment due to TEAEs, including COVID-19, pneumonia, respiratory failure, hypoxia, and dyspnea.
Mortality
There were no patient deaths during the trial.
Notable Harms
Elevated transaminases and hypersensitivity reactions were considered notable harms for the review. In the trial, 1 patient in the remdesivir group and 3 patients in the placebo group reported elevated alanine aminotransferase levels, while 1 patient in each of the treatment groups reported elevated aspartate aminotransferase levels. In terms of hypersensitivity reactions, there were 6 patients in the remdesivir group and 4 patients in the placebo group who reported infusion-site reactions. Anaphylactic reactions were not captured in the trial.
Table 15: Summary of Harms Results From the PINETREE Trial, Safety Analysis Set
Harms | PINETREE trial | |
|---|---|---|
Remdesivir (N = 279) | Placebo (N = 283) | |
Most common adverse events, n (%)a | ||
Patients with ≥ 1 TEAE | 118 (42.3) | 131 (46.3) |
Nausea | 30 (10.8) | 21 (7.4) |
Headache | 16 (5.7) | 17 (6.0) |
Cough | 10 (3.6) | 18 (6.4) |
Serious adverse events, n (%)b | ||
Patients with ≥ 1 SAE | 5 (1.8) | 19 (6.7) |
Pneumonia | 2 (0.7) | 3 (1.1) |
COVID-19 | 1 (0.4) | 2 (0.7) |
COVID-19 pneumonia | 0 (0) | 7 (2.5) |
Hypoxia | 0 (0) | 3 (1.1) |
Patients who stopped treatment due to adverse events, n (%)b | ||
Patients who stopped treatment | 2 (0.7) | 5 (1.8) |
Deaths, n (%)c | ||
Patients who died | 0 (0) | 0 (0) |
Adverse events of special interest, n (%) | ||
Transaminase elevations | ||
Alanine aminotransferase | 1 (0.4) | 3 (1.1) |
Aspartate aminotransferase | 1 (0.4) | 1 (0.4) |
Hypersensitivity reactions | ||
Infusion-site reactions | 6 (2.2) | 4 (1.4) |
Anaphylactic reactions | NR | NR |
NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aOccurring in at least 5% of patients overall.
bOccurring in at least 3 patients overall.
cDeaths that occurred between the first dose date and the last dose date plus 30 days (inclusive).
Source: Clinical Study Report for the PINETREE trial.16
Critical Appraisal
Internal Validity
The overall risk of bias in the PINETREE trial was low for the randomization process, deviations from intended interventions, missing outcomes data, and reported results. Patients were randomized and assigned to treatment groups using a stratified randomization schedule. Study personnel and patients were blinded to treatment assignment, and a matching placebo was used. Additionally, baseline characteristics were generally balanced between the groups. The modified full analysis set included patients enrolled under protocol amendment 2 for whom MAV data were collected; the proportions of patients were similar between groups for this analysis set. In general, discontinuations were low overall and unlikely to bias the results.
There may be some issues with how outcomes were measured; however, the direction of bias is unclear. Study enrolment was stopped on April 8, 2021, due to declining COVID-19 case rates, increasing availability of single-infusion monoclonal antibodies as a treatment, and increasing vaccination rates among high-risk patients during the trial. The final trial population was less than half (46.2%) of the planned sample size. This did not have an impact on the statistical analysis of results because the primary efficacy end point was met. However, event rates were low during the trial, limiting the assessment of key clinical end points for this disease (e.g., requirements for supplemental oxygen, ICU admissions, and mechanical ventilation); no death events were observed. The efficacy, as observed, was driven by hospitalization only. It was not specified how the need for hospitalization or MAV was determined; this may have been subjective (e.g., dependent on the severity of a patient’s symptoms). The trial also did not report what the causes of hospitalization or MAV were, aside from being COVID-19–related, which itself was not further defined. There is evidence of validity, reliability, and responsiveness of the FLU-PRO Plus in populations with COVID-19; however, no MID was identified from the literature, and the clinical expert confirmed that it is not a questionnaire used in clinical practice. The trial was relatively short in duration, and there is a lack of long-term safety evidence for remdesivir.
External Validity
The intervention and treatment setting in the trial were considered generalizable to clinical practice in Canada.
Considering when the trial took place (September 2020 to May 2021) and the significant changes in population immunity, viral pathogenesis, and disease management since then, the clinical expert we consulted indicated that the eligibility and baseline characteristics are no longer relevant to how remdesivir would be used in practice. There were 8 adolescent patients (aged 12 years to 18 years) in the PINETREE trial, and there are limited data to inform the use of remdesivir in patients aged 12 to 18 years. The clinical expert we consulted indicated that it would be rare for a pediatric patient to receive remdesivir and that the decision to use the drug would be made in consultation with specialists who have knowledge of infectious diseases and experience managing pediatric patients. The trial excluded individuals who had received COVID-19 vaccinations, which greatly limits the generalizability of the results to current practice, given that most people in Canada have received at least 1 vaccine dose.9 Now that many people in Canada have immunity through vaccination and/or infection, the magnitude of the treatment effect is expected to be smaller than that reported in the trial.30
The clinical expert noted that, contrary to earlier in the pandemic, infection with current SARS-CoV-2 variants no longer carries a high risk of hospitalization. The definition of being at high risk of progression to severe COVID-19 has narrowed over time to focus on vaccination status, severity of immunocompromised status, and age; the results of the trial are no longer broadly generalizable to patients who would be considered high risk today. Relevant subgroup analyses were not available for these categories or included few patients (i.e., 5% immunocompromised in the remdesivir group), and results were not informative. Less than a third of patients were older than 60 years of age, and subgroup analyses for older adults (e.g., 80 years and older) were not available, limiting the conclusions that can be drawn for this population. According to the expert, most of the comorbidities listed for enrolment eligibility alone are no longer considered to significantly increase the risk of worse disease outcomes. Moreover, many comorbidities had low representation in the trial (i.e., less than 10%), making it difficult to apply the results to clinical decision-making.
Evidence for the treatment effect of remdesivir was based largely on hospitalization (with no conclusions concerning the impact of remdesivir on death), which varies among different clinical practices, regions (no trial sites were in Canada), and availability of health care resources. The PINETREE trial took place before the omicron variant was the predominant circulating variant; therefore, the estimate of treatment effect from the trial may not be applicable in the context of the current COVID treatment landscape.31
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Evidence
No indirect treatment comparisons were submitted for this review.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by our review team.
Description of Studies
One prospective cohort study has been summarized to provide evidence regarding the efficacy of remdesivir in adults with high-risk conditions and confirmed, mild to moderate COVID-19 who attended the emergency department of a tertiary referral centre in Mexico City from December 1, 2021, to April 30, 2022.32 High-risk conditions included being at least 60 years of age; having a body mass index greater than 35 kg/m2, uncontrolled diabetes, uncontrolled arterial hypertension, cerebrovascular disease, ischemic heart disease, chronic renal disease on renal replacement therapy, or liver cirrhosis; being pregnant; having no vaccine history against SARS-CoV-2 or an incomplete vaccine schedule (i.e., only 1 dose from a 2-dose primary series); and immunosuppression. The investigators defined immunosuppression as the presence or experience of at least 1 of the following: primary immunodeficiency, receiving chemotherapy and/or immunotherapy for active cancer, having HIV with a cluster of differentiation 4-positive (T-cell) count of less than 200 cells per mL, receiving chronic steroid therapy (i.e., more than 2 weeks of at least 15 mg/d prednisone or equivalent); or any of the following: solid organ transplant, hematopoietic stem cell transplant, active autoimmune disease, immunosuppressive treatment, or recent use of anti–cluster of differentiation 20 drugs or antimetabolite drugs.
Based on the information provided by the pivotal PINETREE trial, the sponsor identified an evidence gap for the effectiveness of remdesivir on nonhospitalized, vaccinated, immunosuppressed patients with COVID-19 who are at high risk of disease progression and submitted the Rajme-Lopez et al. (2022) study to address this gap (Table 16).32
Table 16: Summary of Gaps in the Systematic Review Evidence
Evidence gap | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
Effectiveness (reduced hospitalization and mortality) of remdesivir treatment in nonhospitalized, vaccinated, immunosuppressed patients with COVID-19 at high risk of progression. | Rajme-Lopez et al. (2022) was a prospective cohort study conducted in Mexico of nonhospitalized adults with mild to moderate COVID-19 at high risk of progression during the omicron period from December 2021 to April 2022.32 Most patients were vaccinated (79%) and immunosuppressed (94%). The primary efficacy composite outcome was hospitalization or death at 28 days after symptom onset. A Cox proportional hazards regression model was used to identify associations with the primary outcome. | 126 patients were included in this study (54 received remdesivir and 72 did not receive remdesivir). Remdesivir was associated with an 84% reduction in the risk of hospitalization or all-cause death at day 28 after symptom onset compared to those did not receive remdesivir (aHR = 0.16; 95% CI, 0.06 to 0.44). Study conclusions: Early outpatient treatment with remdesivir significantly reduces hospitalization or death by 84% in a high-risk, majority-immunosuppressed population of patients with omicron variant COVID-19. |
aHR = adjusted hazard ratio; CI = confidence interval.
Source: Sponsor’s Summary of Clinical Evidence.33
Populations
Eligible patients must have been adults with mild to moderate COVID-19 confirmed through a PCR test or RAT and more than 1 high-risk condition for COVID-19 progression. Patients who received remdesivir must have started experiencing symptoms within the past 7 days and have had an oxygen saturation of 90% or higher. Patients who did not receive remdesivir were diagnosed before the policy on remdesivir for outpatients was implemented on January 17, 2022, to include the use of remdesivir in high-risk patients with early COVID-19. There was consecutive sampling of all patients who met the inclusion criteria.
Interventions
Patients either received 200 mg of remdesivir on day 1 and 100 mg of remdesivir on days 2 and 3, or did not receive any remdesivir. This dosage of remdesivir is consistent with that used in the PINETREE trial.
Outcomes
The composite primary end points were all-cause hospitalization (defined as hospitalization for more than 24 hours) or death at day 28 after symptom onset.
Statistical Analysis
For the omicron variant period, the investigators assumed prevalences of 21% versus 0.07% for hospitalization or death among patients who were immunosuppressed versus high-risk patients treated with remdesivir, respectively. The investigators also considered a 0.05% probability of type I error and a statistical power of 90% to determine a sample size of at least 94 patients (47 per treatment group) for the study. Factors associated with the primary outcome were identified using bivariate analysis, and HRs and 95% CIs were calculated. A multivariate Cox proportional hazards regression model with clinically and biologically relevant variables (with P < 0.2) was used to identify an independent association between remdesivir and the primary efficacy outcome, and adjusted HRs and 95% CIs were calculated. Variables used for the multivariate regression model included sex, age, diabetes mellitus, cirrhosis, malignant hematologic disorders, autoimmune disorders, solid organ transplant, previous SARS-CoV-2 vaccination, previous SARS-CoV-2 infection, and treatment with remdesivir. Missing data were not imputed.
Results
Patient Disposition
In total, 2,588 individuals were screened, among whom 196 patients were considered high risk. Of these, 126 had mild to moderate COVID-19 and were enrolled in the study. Individuals were screened out for not having high-risk conditions (n = 2,392), having severe or critical COVID-19 (n = 55), and having incomplete data (n = 15). Of those included in the study, 54 patients (42.9%) received remdesivir and 72 patients (57.1%) did not.
Baseline Characteristics
Overall, there were more female patients (57.1%) than male patients (42.9%) in the study. Among the patients who did not receive remdesivir, there were more female patients (63.9%) than male patients (36.1%). The median age of the study patients was 49 years (interquartile range, 35 years to 63 years), and the median age was younger among patients who received remdesivir than among those who did not (43 years versus 51.5 years, respectively). All but 1 patient (99.2%) reported comorbidities; the 1 patient reporting none was considered high risk for being older than 65 years and having received no SARS-CoV-2 vaccines. Most patients (93.7%) were in an immunosuppressed state, and most patients (88.1%) had at least 2 factors considered high risk for progression of COVID-19.
Comorbidities included arterial hypertension (31.8%), autoimmune disorders (31.0%), solid organ transplant recipient (24.6%), type 2 diabetes mellitus (21.4%), obesity (19.1%), malignant hematologic disorders (11.9%), advanced chronic kidney disease (7.1%), solid organ malignant neoplasm (7.1%), hematopoietic stem cell transplant (5.6%), chronic lung disease (4.8%), liver cirrhosis (4.8%), ischemic heart disease (4.0%), cerebrovascular disease (3.2%), and uncontrolled HIV infection (1.6%). Comorbidities were generally balanced between treatment groups, aside from arterial hypertension, which was lower in the remdesivir group (25.9%) than in the group that did not receive remdesivir (36.1%). Most patients (79.4%) had received a complete vaccine series, and few (9.3%) had self-reported prior SARS-CoV-2 infection. Diagnosis by PCR testing (92.8%) was more common than diagnosis by antigen testing, and omicron was identified as the predominant variant.
Efficacy
Of the 36 patients who met the primary end point, 5 patients (9.3%) received remdesivir and 31 patients (43.1%) did not (bivariate analysis HR = 0.18; 95% CI, 0.07 to 0.45; P < 0.001; multivariate analysis adjusted HR = 0.16; 95% CI, 0.06 to 0.44; P < 0.001). The results were largely driven by all-cause hospitalization events (5 patients and 22 patients in the remdesivir and control groups, respectively) compared to all-cause deaths (0 patients and 9 patients in the remdesivir and control groups, respectively). There were 20 COVID-19–related hospitalizations, all from the control group.
Results of bivariate analysis indicated that age (60 years or older) (HR = 2.57; 95% CI, 1.31 to 3.80), diabetes mellitus (HR = 3.40; 95% CI, 1.76 to 6.59), and cirrhosis (HR = 3.38; 95% CI, 1.19 to 9.58) were associated with higher rates of hospitalization or death, whereas prior vaccination (HR = 0.48; 95% CI, 0.24 to 0.96) was associated with lower rates of hospitalization or death. Diabetes mellitus (adjusted HR = 3.35; 95% CI, 1.58 to 7.07) was also associated with the primary outcome using multivariate analysis.
Harms
Harms results were not reported in the study.
Critical Appraisal
The pivotal trial data lack clear information about the effects of remdesivir on outpatients who are immunosuppressed, vaccinated, and at high risk of COVID-19 progression (using a current definition); this was noted in the critical appraisal. The sponsor indicated that this RWE study fills a gap in the pivotal trial data because it assesses the effectiveness of remdesivir by comparing outcomes (i.e., hospitalization or mortality by day 28) for vaccinated, immunosuppressed outpatients who received remdesivir versus those who did not. These were patients who sought treatment at a single tertiary referral centre in Mexico City from December 2021 to April 2022 (early in the period when omicron became the dominant circulating variant).
Guidance for Reporting Real-World Evidence forms the foundation for the transparent reporting of RWE studies in Canada and facilitates our appraisal of RWE.34 All applicable sections of the guidance should be reported when submitting RWE studies as part of a reimbursement review.34 Some of the most important missing information in the submitted RWE study, organized by the 12 sections from the guidance, includes:34
Setting and context: No information was provided about why a setting outside of Canada was chosen or about differences in health systems, access to care, available health care resources during the pandemic, and other factors that can affect the care of patients with COVID-19. In addition, there was no description of how these factors might affect the applicability of the study’s findings to the context of Canada.
Data specifications: No information was provided about access, cleaning, and linkage.
Data sources, data dictionary, and variables: No detailed descriptions of data sources, data dictionary, or variables were provided. Information on important variables that should have been considered, but were not captured in the study, were also not included. The potential impact of these omissions on the study results was not discussed.
Statistical methods: A detailed explanation and justification of the model(s) and all variables was not provided.
Internal Validity
Because of how the outcome data were collected, recorded, and verified (i.e., the source document), it is difficult to assess any potential bias due to inaccurate or incomplete reporting of hospitalization or death occurring during the time period from symptom onset to day 28 in the study.
The study authors used a bivariate analysis to identify factors associated with the primary outcome. Multivariate regression for the effect of remdesivir on 28-day all-cause and COVID-19–related rehospitalizations was adjusted for a number of clinical and biologic factors as well as those that were identified through the bivariate analysis (i.e., sex, age, diabetes mellitus, cirrhosis, malignant hematologic disorders, autoimmune disorders, solid organ transplant, previous SARS-CoV-2 vaccination, previous SARS-CoV-2 infection, and treatment with remdesivir). The selection of variables for the model may have been a source of bias, given that a rationale for including specific variables in the model was not provided. The authors did not provide model fit statistics to justify the inclusion of the many variables in the model, with only 54 remdesivir recipients and 72 nonrecipients. The study authors did not attempt to evaluate the presence or amount of residual confounding through bias analysis.
Historic controls may affect time-related bias because treatments beyond remdesivir, providers, availability of hospital resources, health care systems, and other factors affecting treatment or access to health care may have differed between the time periods, potentially introducing bias.
It is unclear if there were any missing data related to hospitalization, and there was no exploration of the extent of missing data for the study outcomes. For example, the authors did not describe if or how patients were contacted to determine if they were hospitalized at a different hospital from the 1 where they first sought care. It is possible that the extent of missing data would differ in patients who were treated with remdesivir versus those who were not.
External Validity
The data were extracted from patients who attended the emergency department of a single, tertiary, national referral centre in Mexico City for patients who have rheumatologic, oncologic, hematologic, or renal diseases or have received a solid organ or hematopoietic stem cell transplant. This health care system differs from the health care system in Canada; therefore, response to public health measures, access to care, and outpatient treatments for COVID-19 may have differed.
The study occurred from December 2021 to April 2022. Since then, population disease exposure as well as circulating variants have changed substantively, limiting the generalizability of these findings to the current COVID-19 treatment landscape. The patient population that entered the cohort was a complex group of patients who accessed a specific, single emergency department and were highly selected based on the inclusion criteria. It is unknown how this patient population would be generalizable to a those living in Canada. The comparability of best supportive care (e.g., corticosteroids, anticoagulants) may limit its generalizability to a clinical setting in Canada.
Discussion
Summary of Available Evidence
One DB, placebo-controlled RCT (the PINETREE trial, N = 584) of remdesivir for the treatment of COVID-19 in nonhospitalized patients aged 12 years or older (weighing at least 40 kg) with confirmed SARS-CoV-2 infection, and who are at high risk of progression to severe COVID-19 was submitted by the sponsor for the systematic review.16 Patients were randomized 1:1 to 3 days of treatment with remdesivir or matching placebo. The primary efficacy end point of the trial was the composite of COVID-19–related hospitalization (defined as receiving 24 hours of acute care) or all-cause death by day 28. Secondary and exploratory end points relevant to our review included COVID-19–related MAVs, mortality, new requirement for oxygen supplementation, ICU admission, mechanical ventilation, and patient-reported symptom alleviation. The mean age of patients was 50 years (SD = 15.1 years), and demographic characteristics were generally balanced between the treatment groups. Patients aged less than 60 years must have had at least 1 pre-existing risk factor. Across treatment groups, the most frequently reported baseline risk factors were diabetes (62%), obesity (56%), hypertension (48%), and chronic lung disease (24%), while other comorbidities were less common (i.e., less than 10% each for cardiovascular or cerebrovascular disease, immunocompromised state, chronic mild or moderate kidney disease, chronic liver disease, current cancer, and sickle cell disease). Individuals were excluded if they had received any other antiviral treatment for or vaccination against SARS-CoV-2.
One prospective cohort RWE study (N = 126) has been summarized to provide evidence regarding the efficacy of remdesivir in adults with high-risk conditions and confirmed, mild to moderate COVID-19 who attended the emergency department of a tertiary referral centre in Mexico City from December 1, 2021, to April 30, 2022.32 Patients received remdesivir (n = 54) for 3 days as indicated; those who did not (n = 72) had been diagnosed before the policy on remdesivir for outpatients was implemented on January 17, 2022. The primary composite end point was all-cause hospitalization (i.e., more than 24 hours in hospital) or death at day 28 after symptom onset. The median age of patients was 49 years (interquartile range, 35 years to 63 years). All but 1 patient (99%) reported comorbidities; the 1 patient reporting none was considered high risk for being older than 65 years and having received no SARS-CoV-2 vaccines. In the study, 94% of patients were in an immune-suppressed state, 88% had at least 2 factors considered to be high risk for progression of COVID-19, 79% had received a complete vaccine series, and 9% had self-reported prior COVID-19 infection. Omicron was identified as the predominant variant.
Interpretation of Results
Efficacy
Hospitalizations were noted as an important outcome to the patient and clinician groups who provided input for this review, and clinical expert we consulted. Results from the PINETREE trial indicate that remdesivir reduces COVID-19–related hospitalizations at 28 days compared to placebo in the context of the RCT. The primary end point of the trial was a composite end point of COVID-19–related hospitalizations or all-cause death, but was entirely driven by hospitalizations, given that there were no deaths during the trial. Furthermore, all events took place early in the trial, and day 14 and day 28 results were the same. The clinical expert indicated that patients residing in a skilled nursing facility and patients with an immunocompromised status were relevant populations; however, no patients from either subgroup experienced a COVID-19–related hospitalization or death from any cause by day 28, and results were not informative for clinical decision-making in these groups. Older age groups were of interest (e.g., stratification by decade older than 60 years), but these data were not available for review. It is necessary to emphasize that the PINETREE trial took place before omicron became the main circulating variant, when patients were unvaccinated, and patients were enrolled based on a definition of high risk that the clinical expert considered no longer relevant to today’s clinical practice. The omicron variant is associated with less severe disease, and the expert confirmed that COVID-19–related hospitalizations have decreased.35 Because of the evolving disease landscape, it would be reasonable to expect that the estimate of treatment effect would be smaller today than what was observed in the trial. Therefore, the results may be of limited clinical relevance for current disease management and the use of remdesivir in practice now.
The pivotal trial data lacked clear information about the effect of remdesivir on outpatients who are immunosuppressed, vaccinated, and at high risk of COVID-19 progression (using a current definition of high risk). Results from the Rajme-Lopez et al. (2022) RWE study indicated that receiving remdesivir resulted in fewer all-cause hospitalizations (and fewer COVID-19–related hospitalizations) compared to not receiving remdesivir. The sponsor attempted to address a gap in the pivotal trial data with the RWE study, but the study had numerous limitations. Overall, it was a very small, observational study using data from a single tertiary referral centre in Mexico City in a highly selected group of immunosuppressed patients. The potential for inaccurate or incomplete reporting of outcomes, use of historic controls, and limitations to the model are potential sources of bias. Population background disease risk as well as circulating variants have changed substantively since the time of this study, limiting the generalizability of these findings to the current COVID-19 treatment landscape. Although the results of the RWE study were in agreement with the RCT, it is challenging to assess the magnitude of the effect of receiving treatment with remdesivir on reduction in hospitalization or mortality at day 28 compared with not receiving it for outpatients who are immunosuppressed, vaccinated, and at high risk of COVID-19 progression and to extrapolate the effect to current practice in Canada.
COVID-19–related MAVs were defined as medical visits attended in person by the patient and a health care professional. Results from the PINETREE trial showed that MAVs were numerically fewer in the remdesivir group than in the placebo group and that results by treatment group were similar between day 14 and day 28 (outcomes not controlled for multiplicity). It is expected that there is some subjectivity around how MAVs were determined to be COVID-19–related or not, which adds uncertainty to the interpretation of the outcome. Furthermore, the clinical expert suggested that treatment with remdesivir may not reduce overall MAVs (or the associated use of health care resources) compared to treatments like oral nirmatrelvir-ritonavir because each day of remdesivir IV infusion could be considered a MAV. MAVs were not an outcome in the RWE study.
Other clinical outcomes from the PINETREE trial for oxygen supplementation, ICU admissions, and mechanical ventilation were noted as being important to both the clinician groups who submitted input for the review and the clinical expert we consulted. The clinical expert also indicated that there would be greater concern about more severe outcomes (such as ICU admissions and mechanical ventilation versus supplemental oxygen). Overall, there were few events for each of the outcomes and similar proportions between treatment groups, limiting the conclusions that can be drawn from the data. These outcomes were not assessed in the RWE study.
Both the patient and clinician groups noted that preventing or reducing symptoms is an important outcome. In the PINETREE trial, symptom alleviation was measured using the FLU-PRO Plus questionnaire, and a greater number of patients in the remdesivir group reported symptom alleviation by day 14 (outcome not controlled for multiplicity). Not all patients in the trial completed the questionnaire; of those who did, not all completed it before drug dosing, as was intended. Overall, the number of patients who reported symptom alleviation was small. While there is evidence of instrument validity, no MID was identified from the literature, making it difficult to interpret what a clinically meaningful improvement would be based on changes in questionnaire scores. Moreover, the FLU-PRO Plus is not a measure of overall health-related quality of life (HRQoL) and how remdesivir affected patients’ HRQoL in the trial is unknown. The clinical expert stated that the questionnaire is not used in clinics; thus, the results are not particularly relevant to clinical practice. No patient-reported outcomes or HRQoL outcomes were captured in the RWE study.
The results of the PINETREE trial and the Rajme-Lopez et al. (2022) study must be interpreted in light of the current disease landscape — including changes in population immunity, viral pathogenicity, and treatment recommendations — versus when the 2 studies were conducted. Population immunity (both infection-acquired and vaccine-induced) has increased since the beginning of the pandemic. Based on serology, it is estimated that nearly everyone in Canada (> 99% of the population) has some form of infection-acquired or vaccine-induced immunity to the virus.10 Additionally, the clinical expert stated that most individuals who are infected experience self-limiting disease and recover without specific interventions. As a result, the definition of high risk for progression to severe disease has changed over time. According to the literature and the clinical expert, the most important risk factors for progression include lack of SARS-CoV-2 immunity, severe immune suppression, multiple chronic comorbidities, and older age (e.g., 80 years and older).4 The patients of greatest concern include those who cannot produce a sufficient immune response to clear the infection; as a result, these patients may be more likely to benefit from an antiviral drug. It should also be noted that those who are at high risk of severe disease are not necessarily the same as those who derive the most benefit from treatment. It will be important for future research to assess whether these 2 populations overlap. Remdesivir has a Health Canada–approved indication for pediatric patients weighing more than 40 kg, whereas nirmatrelvir-ritonavir is indicated for adults. Because there were few adolescent patients in the PINETREE trial, there remains a lack of evidence to support a treatment benefit for young patients. In addition, although the Health Canada indication includes pediatric patients, the clinical expert stated that it would be rare to treat young individuals with remdesivir (unless they were severely immunocompromised and had contraindications to nirmatrelvir-ritonavir), and that the decision to treat pediatric patients would require more specialized consultation. The limitations of the included studies prevent generalization of the results to clinical practice today.
Molecular tests (RATs or PCR tests) are necessary for confirming viral infection and accessing antiviral treatment. However, access to tests is limited and variable across jurisdictions, and getting results in a timely manner presents another hurdle because remdesivir and nirmatrelvir-ritonavir must be initiated within 7 days and 5 days of symptom onset, respectively. Access to remdesivir can be challenging for patients living in remote areas because they must be able to travel to an appropriate care setting for a health care professional to administer the IV drug and monitor for postinfusion reactions. Moreover, the clinical expert stated that the need for additional health care resources to administer remdesivir is a factor that restricts widespread use of the drug. Both the literature and expert opinion indicated that there is a general preference for oral nirmatrelvir-ritonavir, except in cases where patients have potential drug-drug interactions or other contraindications. Both the patient and clinician groups who provided input and the clinical expert noted the importance of having access to treatment that limits drug-drug interactions.
WHO recommendations suggest that nirmatrelvir-ritonavir would likely be used before remdesivir due to the former having an oral route of administration versus the practical challenges related to the IV administration of remdesivir.5 However, remdesivir may be an option for patients who have contraindications to nirmatrelvir-ritonavir, who are already receiving IV therapy, or who are past the 5-day window (after symptom onset and confirmed diagnosis) for accessing nirmatrelvir-ritonavir but still within the 7-day window for remdesivir. The AMMI updated recommendations suggest that remdesivir can be considered for patients with mild disease (i.e., who do not need oxygen supplementation) based on their degree of risk and trajectory of symptoms.12 The recommendations describe the highest-risk patients as those who have no previous infection or immunization and who have severe immune suppression or other major underlying conditions.12 Such conditions can include solid organ transplant, hematological malignancy in the last year, bone marrow transplant in the past 2 years, anti–cluster of differentiation 20 or B-cell depleting drugs in the past 2 years, chemotherapy for cancer in the last 6 months, radiation therapy for cancer in the last 3 months, immune suppression medications, including corticosteroids (equivalent to 20 mg/d of prednisone) and/or immunosuppressive biologics, severe structural lung disease, and severe or moderate primary immune deficiencies.12
Harms
Overall, fewer than half of the patients in the PINETREE trial reported at least 1 TEAE, and the types and frequencies of events were similar between the treatment groups. Reports of SAEs and patients stopping treatment due to a TEAE were low in the trial overall, and lower in the remdesivir group than in the placebo group. There were no deaths during the trial. Notable harms identified from the Health Canada product monograph (i.e., elevated transaminase levels and hypersensitivity reactions) were also rare. Safety results reported in the RWE study were limited to deaths, of which there were 9 in total, all in the group that did not receive remdesivir. Input from the patient and clinician groups broadly noted safety concerns with treatments for COVID-19, and the clinical expert indicated that remdesivir is a well-tolerated drug. Because of the known contraindications associated with nirmatrelvir-ritonavir, it is generally accepted that remdesivir is an alternative for patients who cannot receive nirmatrelvir-ritonavir.5 There was a lack of long-term safety evidence for remdesivir from the trial.
Conclusion
Evidence from the PINETREE trial indicated that patients who were aged 12 years or older, weighing 40 kg or more, and at high risk of progression to severe COVID-19 were less likely to experience COVID-19–related hospitalization if they received remdesivir IV for 3 days in an outpatient setting. However, evidence that remdesivir reduces mortality, MAVs, need for oxygen supplementation, ICU admission, need for mechanical ventilation, and symptoms was lacking due to significant limitations of the study. It is important to note that the patient population in the trial, which was conducted early in the pandemic, was significantly different from the current patient population because immunization protection status has evolved since then and different viral variants are now circulating. Although the results of the RWE study aligned with those of the RCT in terms of remdesivir being associated with reduced hospitalization, there remains some uncertainty in the magnitude of the drug’s effectiveness in a highly vaccinated patient population with significant immunosuppression because the RWE study was small and likely subject to significant bias concerns. As a result, the evidence included in this review is of uncertain or low clinical relevance in terms of its ability to determine how remdesivir would be used in clinical practice today. Based on the limited trial results, remdesivir appears to be a well-tolerated treatment, and both the literature and expert opinion indicate that it could be preferred for high-risk patients who cannot receive nirmatrelvir-ritonavir (although its IV route of administration limits who is likely to access the drug). There remains a lack of long-term efficacy and safety data for the use of remdesivir in relevant populations (i.e., those who are considered high-risk today and are more likely to receive the drug in practice). No direct or indirect comparisons between remdesivir and other COVID-19 treatments were appraised in this review.
References
1.Drug Reimbursement Review sponsor submission: Veklury (remdesivir), 100 mg/vial powder for solution for infusion [internal sponsor's package]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2024 Mar 11.
2.Veklury (remdesivir): 100 mg/vial powder for solution for infusion [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2024 May 06.
3.Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int J Antimicrob Agents. 2020;55(3):105924. PubMed
4.McIntosh K, Gandhi RT. COVID-19: clinical features. In: Post TW, ed. UpToDate. Waltham (MA0: UpToDate; 2024: http://www.uptodate.com. Accessed 2024 Feb 22.
5.Therapeutics and COVID-19: living guideline, 10 November 2023. Geneva (CH): World Health Organization; 2023: https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2023.2. Accessed 2024 Mar 28.
6.McIntosh K. COVID-19: epidemiology, virology, and prevention. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2024: http://www.uptodate.com. Accessed 2024 Feb 22.
7.Wang C, Liu B, Zhang S, et al. Differences in incidence and fatality of COVID-19 by SARS-CoV-2 Omicron variant versus Delta variant in relation to vaccine coverage: a world-wide review. J Med Virol. 2023;95(1):e28118. PubMed
8.Xu X, Wu Y, Kummer AG, et al. Assessing changes in incubation period, serial interval, and generation time of SARS-CoV-2 variants of concern: a systematic review and meta-analysis. BMC Med. 2023;21(1):374. PubMed
9.COVID-19 vaccination: vaccination coverage. Ottawa (ON): Public Health Agency of Canada; 2024: https://health-infobase.canada.ca/covid-19/vaccination-coverage/. Accessed 2024 Mar 14.
10.The COVID-19 Immunity Task Force. SARS-CoV-2 seroprevalence in Canada (April 13, 2020 to December 31, 2023). 2024; https://www.covid19immunitytaskforce.ca/seroprevalence-in-canada/. Accessed 2024 Apr 18.
11.Cohen P, Gebo K. COVID-19: management of adults with acute illness in the outpatient setting. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2024: http://www.uptodate.com. Accessed 2024 Mar 08.
12.Grant JM, Lam J, Goyal SV, et al. AMMI Canada Practice Point: updated recommendations for treatment of adults with symptomatic COVID-19 in 2023-2024. J Assoc Med Microbiol Infect Dis Can. 2024;8(4):245-252. PubMed
13.Powers JH, 3rd, Bacci ED, Leidy NK, et al. Performance of the inFLUenza Patient-Reported Outcome (FLU-PRO) diary in patients with influenza-like illness (ILI). PLoS One. 2018;13(3):e0194180. PubMed
14.Richard SA, Epsi NJ, Pollett S, et al. Performance of the inFLUenza Patient-Reported Outcome Plus (FLU-PRO Plus) instrument in patients with coronavirus disease 2019. Open Forum Infect Dis. 2021;8(12):ofab517. PubMed
15.Keeley TJH, Satram S, Ghafoori P, et al. Content validity and psychometric properties of the inFLUenza Patient-Reported Outcome Plus (FLU-PRO Plus©) instrument in patients with COVID-19. Qual Life Res. 2023;32(6):1645-1657. PubMed
16.Clinical Study Report: GS-US-540-9012, PINETREE Final. A phase 3, randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of remdesivir (GS-5734™) treatment of COVID-19 in an outpatient setting [internal sponsor's report]. Foster City (CA): Gilead Sciences, Inc.; 2021 Oct 01.
17.Filip R, Gheorghita Puscaselu R, Anchidin-Norocel L, Dimian M, Savage WK. Global challenges to public health care systems during the COVID-19 pandemic: a review of pandemic measures and problems. J Pers Med. 2022;12(8):1295. PubMed
18.Zeitouny S, Cheung DC, Bremner KE, et al. The impact of the early COVID-19 pandemic on healthcare system resource use and costs in two provinces in Canada: an interrupted time series analysis. PLoS One. 2023;18(9):e0290646. PubMed
19.Experiences of health care workers during the COVID-19 pandemic, September to November 2021. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/n1/daily-quotidien/220603/dq220603a-eng.htm. Accessed 2024 Apr 02.
20.COVID-19 epidemiology update: summary. Ottawa (ON): Public Health Agency of Canada; 2024: https://health-infobase.canada.ca/covid-19/. Accessed 2024 Mar 14.
21.COVID-19 epidemiology update: testing and variants. Ottawa (ON): Public Health Agency of Canada; 2024: https://health-infobase.canada.ca/covid-19/testing-variants.html. Accessed 2024 Mar 14.
22.Skowronski DM, Kaweski SE, Irvine MA, et al. Risk of hospital admission and death from first-ever SARS-CoV-2 infection by age group during the Delta and Omicron periods in British Columbia, Canada. CMAJ. 2023;195(42):E1427-E1439. PubMed
23.Caliendo AM, Hanson KE. COVID-19: diagnosis. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2022 Sep 01: http://www.uptodate.com. Accessed 2024 Feb 22.
24.Paxlovid (nirmatrelvir-ritonavir): 150 mg nirmatrelvir; 100 mg ritonavir, tablets, co-packaged for oral use [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2024 Jan 02: https://webfiles.pfizer.com/file/ffe4e681-59a8-4da8-ad39-bb18703d4271. Accessed 2024 Mar 14.
25.Stokes EK, Zambrano LD, Anderson KN, et al. Coronavirus disease 2019 case surveillance - United States, January 22-May 30, 2020. MMWR Morb Mortal Wkly Rep. 2020;69(24):759-765. PubMed
26.Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19). Geneva (CH): World Health Organization; 2020: https://www.who.int/publications/i/item/report-of-the-who-china-joint-mission-on-coronavirus-disease-2019-(covid-19). Accessed 2024 Mar 14.
27.Gottlieb RL, Vaca CE, Paredes R, et al. Early remdesivir to prevent progression to severe Covid-19 in outpatients. N Engl J Med. 2022;386(4):305-315. PubMed
28.Gilead Sciences. NCT04501952: study to evaluate the efficacy and safety of remdesivir (GS-5734™) treatment of coronavirus disease 2019 (COVID-19) in an outpatient setting. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/study/NCT04501952. Accessed 2024 Mar 14.
29.Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA. 2021;325(7):632-644. PubMed
30.COVID-19 epidemiology update: cases following vaccination. Ottawa (ON): Public Health Agency of Canada; 2024: https://health-infobase.canada.ca/covid-19/cases-following-vaccination.html. Accessed 2024 Mar 28.
31.Multiple Technology Appraisal: remdesivir and tixagevimab plus cilgavimab for treating COVID-19 [ID6261]. Supporting evidence: committee papers. London (UK): National Institute for Health and Care Excellence (NICE); 2024: https://www.nice.org.uk/guidance/gid-ta11297/documents/committee-papers-3. Accessed 2024 Apr 01.
32.Rajme-López S, Martinez-Guerra BA, Zalapa-Soto J, et al. Early outpatient treatment with remdesivir in patients at high risk for severe COVID-19: a prospective cohort study. Open Forum Infect Dis. 2022;9(10):ofac502. PubMed
33.Veklury (remdesivir) for the treatment of COVID-19 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Veklury (remdesivir), 100 mg/vial powder for solution for infusion. Mississauga (ON): Gilead Sciences Canada, Inc.; 2024.
34.Guidance for reporting real-world evidence. (CADTH methods and guidelines). Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/RWE/MG0020/MG0020-RWE-Guidance-Report-Secured.pdf. Accessed 2024 Feb 16.
35.Clinical management of COVID-19: living guideline, 18 August 2023. Geneva (CH): World Health Organization; 2023: https://iris.who.int/bitstream/handle/10665/372288/WHO-2019-nCoV-clinical-2023.2-eng.pdf?sequence=1. Accessed 2024 Apr 04.
Pharmacoeconomic_Review
Abbreviations
BIA
budget impact analysis
CIHI
Canadian Institute for Health Information
ICER
incremental cost-effectiveness ratio
ICU
intensive care unit
NNT
number needed to treat
QALY
quality-adjusted life-year
SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
SoC
standard of care
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Remdesivir (Veklury), 100 mg/vial, IV infusion |
Indication | For the treatment of COVID-19 in nonhospitalized adults and pediatric patients (weighing ≥ 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high-risk for progression to severe COVID-19, including hospitalization and death |
Health Canada approval status | NOC |
Health Canada review pathway | Expedited: For use in relation to COVID-19 |
NOC date | July 27, 2020 |
Reimbursement request | As per indication |
Sponsor | Gilead Sciences Canada, Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Table 2: Summary of the Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree followed by Markov model |
Target population | Nonhospitalized patients with COVID-19 at high risk of progression to severe disease |
Treatment | Remdesivir |
Dose regimen | 200 mg on day 1 followed by 100 mg once daily for an additional 2 days (for a total treatment duration of 3 days) |
Submitted price | Remdesivir 100 mg vial: $660.53 per vial |
Submitted treatment cost | $2,642.12 per patient, based on a 3-day treatment duration |
Comparator | SoC, comprising over-the-counter and off-label steroid medications |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | 12 weeks |
Key data sources | PINETREE trial ACTT-1 trial and real-world evidence (Mozaffari et al., 2023) to inform inpatient clinical efficacy |
Submitted results | ICER = $30,362 per QALY gained (incremental costs = $201; incremental QALYs = 0.007) |
Key limitations |
|
CDA-AMC reanalysis results |
|
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SoC = standard of care.
Conclusions
In the PINETREE pivotal trial, remdesivir reduced the incidence of COVID-19–related hospitalization or death from any cause through day 28 compared to placebo. As noted in the clinical review, PINETREE did not include vaccinated patients or patients who had had COVID-19 in the past, and it was conducted at a time when the omicron variant had not yet begun circulating. Furthermore, the trial was conducted in a population that is not considered to be at high risk of progressing to severe disease, as defined in clinical practice at the time of this review. These differences represent a fundamental challenge in interpreting the results from the sponsor’s submitted evidence dossier and accompanying pharmacoeconomic model, which are based on the PINETREE trial.
CDA-AMC made several changes to the sponsor’s submitted model, in consultation with clinical experts, to derive a reanalysis. The CDA-AMC reanalysis found that remdesivir was associated with 0.006 additional quality-adjusted life-years (QALYs) at an additional cost of $2,372 and an incremental cost-effectiveness ratio (ICER) of $390,996 per QALY gained compared to standard of care (SoC). A price of $486 per 3-day treatment course (i.e., a price reduction of approximately 82%) would be required for remdesivir to be considered cost-effective at a threshold of $50,000 per QALY gained. The findings of CDA-AMC’s reanalysis differ from those of the sponsor due to the choice of clinical data sources and hospitalization costs; the alternate choices better reflect the current understanding and experience with COVID-19 in Canada. However, CDA-AMC notes that the incremental QALYs estimated by the sponsor (incremental QALYs = 0.007) are similar to the results of the CDA-AMC reanalysis; these benefits equate to an additional 2 quality-adjusted days of life.
CDA-AMC included an alternative approach considering the number needed to treat (NNT) to avoid 1 severe case of COVID-19 (i.e., a case resulting in hospitalization or death), as reported by the CDA-AMC clinical review. Based on the PINETREE trial, 22 high-risk individuals would need to be treated to avoid 1 case of severe COVID-19. Based on CDA-AMC’s reduction to the risk of hospitalization in the economic model using vaccine effectiveness against severe outcomes, 122 high-risk individuals would need to be treated. When comparing the drug acquisition costs of remdesivir for 22 individuals and 122 individuals (approximately $63,000 and $351,000, respectively) with the cost of a general ward admission to treat COVID-19 ($20,000), a price reduction of approximately 68% to 94% would be required to ensure minimal financial impact on the health care system.
The results of this analysis are driven by assumptions about high-risk patients and COVID-19 risk informed by the clinical evidence that could not be assessed, given the lack of comparative clinical evidence that more accurately reflects the current COVID-19 setting in Canada. Given that the majority of patients (> 90% in both the sponsor’s and CDA-AMC’s analyses) will not require hospitalization, even on SoC, the effectiveness of remdesivir is limited to patients who would potentially require hospitalization. Notably, the description of patients who are considered to be at high risk of hospitalization in the current COVID-19 context in Canada has narrowed, and COVID-19 variants have changed such that infection no longer carries as high a risk of hospitalization. Therefore, identifying and treating only those patients at highest risk of hospitalization is critical to maximizing the clinical benefit and cost-effectiveness of remdesivir.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from the Gastrointestinal Society based on meetings and discussions with health care professionals, researchers, academics, and first-hand experiences among staff who were affected by COVID-19. Input was focused on the impacts of COVID-19 on the digestive tract. No patients had experience with remdesivir. Patients reported experience with tixagevimab-cilgavimab, bamlanivimab, casirivimab-imdevimab, sotrovimab, and nirmatrelvir-ritonavir. The group input noted that while individuals have a few options to protect them from severe COVID-19 or death, treatments are difficult to access and may be limited in their effectiveness against newer variants. The input also noted a desire among patients for access to treatments that are effective against newer variants and do not present contraindications to their current medicines and therapies.
Clinician group input was received from BC Transplant Clinicians and the Ontario Health Infectious Diseases Advisory Committee. The British Columbia group gathered information based on member experiences with patients who had solid organ transplants and developed COVID-19. The Ontario group compiled its information through email discussions. The groups noted that current treatment options for mild to moderate COVID-19 included remdesivir and nirmatrelvir-ritonavir, and that other, previously approved treatments have been shown to be ineffective against the newer COVID-19 variants. However, the input noted that nirmatrelvir-ritonavir is not commonly prescribed for patients who may face drug-drug interactions that cannot be safely managed. As such, remdesivir is commonly regarded as an alternative option, and is usually the only option for patients at very high risk of severe disease (e.g., those with solid organ transplants). The input suggested that the reimbursement of remdesivir would not cause a shift in the current treatment paradigm unless the eligibility criteria for reimbursement were to change.
CDA-AMC participating drug plans noted that remdesivir and nirmatrelvir-ritonavir are currently used in clinical practice and that most jurisdictions have implemented their own criteria and/or guidelines to outline their place in therapy; thus, coverage criteria and access differ across jurisdictions. Drug plans commented that remdesivir is generally being used in patients who are ineligible for or have a contraindication to nirmatrelvir-ritonavir. The input highlighted that the pivotal trial excluded patients who had been vaccinated and that only approximately 5% of patients in the trial population were immunocompromised. It was further noted that while the recommended total treatment duration for remdesivir in nonhospitalized patients is 3 days, longer and/or additional courses of remdesivir may be used in patients who are immunocompromised and have prolonged, symptomatic COVID-19 with evidence of ongoing viral replication. Additionally, because remdesivir is administered by IV infusion once daily (over 30 minutes to 120 minutes) for 3 consecutive days, nursing resources and travel requirements should be considered. Drug plans noted that access to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) testing may be an issue in some jurisdictions.
CDA-AMC was unable to address the following concerns raised from stakeholder input:
Nirmatrelvir-ritonavir was not considered a relevant comparator by the sponsor for outpatient treatment of COVID-19 in this economic evaluation because it is assumed that remdesivir will be provided only to patients who are not eligible for nirmatrelvir-ritonavir. As a result, there is no information on the relative cost-effectiveness of remdesivir versus nirmatrelvir-ritonavir.
Alternative treatment durations and re-treatment were not considered in the submitted model due to a lack of evidence to inform comparative clinical efficacy.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of remdesivir compared with SoC (comprising over-the-counter and off-label steroid medications). The modelled population comprised nonhospitalized patients with COVID-19 at high risk of progression to severe disease.1,2 The modelled population is consistent with the Health Canada indication and reimbursement request and aligned with the PINETREE trial.
The recommended dose of remdesivir in an outpatient setting for adults and pediatric patients weighing at least 40 kg is 200 mg on day 1 followed by 100 mg once daily for an additional 2 days (for a total treatment duration of 3 days), starting as soon as possible after diagnosis and within 7 days of the onset of symptoms. Remdesivir is administered through IV and provided as a powder for infusion (5 mg/mL when reconstituted) at a submitted price of $660.53 per vial. In the submitted model, the sponsor assumed that the cost per patient was $2,642.12 for 3 days of treatment. The comparator was SoC. No cost was assumed for SoC because it was assumed to be received by all patients.
The clinical outcomes of interest were QALYs and life-years over a 12-week time horizon. Discounting (1.5% per annum) was applied to both costs and outcomes, and a 2-week cycle length was used. The base-case perspective was that of the Canadian publicly funded health care payer.
Model Structure
The sponsor submitted a short-term, acute-care decision tree followed by a postdischarge Markov model.2 All patients entered the model in the outpatient decision tree (acute care). After 2 weeks, patients could remain symptomatic and not hospitalized or become hospitalized (Figure 1). If patients remained outpatients, after 2 weeks they could recover from their acute infection with or without long COVID. Individuals who were hospitalized at 2 weeks were allocated into the inpatient model structure (Figure 2).
Patients who were hospitalized were initially allocated into the inpatient model structure according to the highest level of care they received in the hospital at baseline using ordinal scale scores, defined by the level of oxygen support required. The following levels of care received in hospital were modelled: ordinal scale score 1 to 3 (discharged from COVID-19 care), ordinal scale score 4 (general ward with no supplemental oxygen), ordinal scale score 5 (general ward with low-flow oxygen), ordinal scale score 6 (intensive care unit [ICU] with noninvasive ventilation or high-flow oxygen), and ordinal scale score 7 (ICU with mechanical invasive ventilation or extracorporeal membrane oxygenation). Ordinal scale score 8 represents death. Patients could transition to an alternate level of hospital care, be discharged, or die. A 2-state Markov model (alive or death) was used to model the postdischarge period.
Model Inputs
Patient baseline characteristics were informed by Canadian population statistics and published literature. The starting age in the model was 63 years.3 The proportion of high-risk patients (37.8%) was informed by Statistics Canada.4
Treatment efficacy in the outpatient setting was modelled using the following clinical outcomes from the PINETREE trial: the proportion hospitalized at 2 weeks, the proportion requiring medically attended visits, and the proportion recovered at 2 weeks. The sponsor assumed that a proportion of all alive patients at week 4 experienced long COVID, which was modelled as a one-off event. For the proportion of patients who were subsequently admitted to the hospital after 2 weeks, patients’ starting ordinal scale score (as described in the model overview section) was based on data from the CATCO trial.5 Transitions between ordinal scale scores in hospital were informed by the ACTT-1 trial (Beigel et al., 2020)6 and an observational study conducted by Mozaffari and colleagues (2023),7 and were assumed to be the same for patients treated with remdesivir and SoC in the outpatient setting. The sponsor assumed that, based on published literature, 17% of patients would require rehabilitation,8 and the baseline proportion of people who would require rehospitalization would be 10.6%.9 The rehospitalization rate was adjusted for patients treated with remdesivir using a hazard ratio of 0.87, informed by Mozaffari et al.7 The probability of death for recovered individuals was modelled using general Canadian population life tables.10
The sponsor’s model did not include costs or health outcomes of treatment-related adverse events associated with remdesivir or SoC.
The age-adjusted baseline utility values in the model for the average patient aligned with those of the general population in Canada, based on Guertin et al. (2018).11 The sponsor applied the following disutilities in the outpatient setting, sourced from published literature: 0.32 disutility for symptomatic patients12 and 0.46 disutility for those who experienced long COVID.13 The sponsor applied hospitalization-related utility decrements for hospitalization services (i.e., general ward, ICU, ICU with mechanical ventilation) that were adjusted using the respective lengths of stay. The following utility decrements were obtained from published literature: 0.27 for general ward, 0.36 for ICU, and 0.56 for ICU with mechanical ventilation.14 These disutilities were derived originally from a panel of 4 specialist physicians who had treated severe acute respiratory syndrome patients in Toronto in 2003 and did not use standard utility elicitation methods.15 Disutilities for rehospitalization (0.003, assumed equal to 4 days of general ward stay) and rehabilitation (0.010, based on assumption) were applied as one-off events.
The model included drug acquisition costs, drug administration costs, outpatient medical appointment costs (including those related to emergency departments, physician services, and outpatient clinics), hospitalization costs, rehabilitation costs, and long COVID–related costs. Drug acquisition costs have already been described. The cost of administration for remdesivir was assumed to be $235.85. Outpatient costs were sourced from published literature.16 The costs of COVID-19–related hospitalizations were obtained from Canadian Institute for Health Information (CIHI) data for time spent in the general ward ($20,097 per stay)17 and from a published economic evaluation for ordinal scale scores 4, 5, and 6 ($44,116, $35,794, and $64,856, respectively).5 The health care costs for ordinal scale scores 7 and 8 (i.e., death) were assumed to be $139,452, based on the same economic evaluation.5 Data on hospital lengths of stay for patients receiving SoC were informed by published literature from an observational US study.18 The hospital length of stay for the remdesivir arm was estimated by applying rate ratios from the ACTT-1 trial to the length of stay used for the SoC group. The 1-time cost of rehabilitation was assumed to be $236 per day; the sponsor assumed a duration of 5 days.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar.
CDA-AMC identified an error in the sponsor’s pharmacoeconomic model that affected the results. The sponsor’s model incorrectly estimated the proportion of patients requiring hospitalization. The sponsor acknowledged this error and corrected it upon request. The probabilistic findings of the corrected sponsor’s model are presented here.
Base-Case Results
In the sponsor’s base-case analysis, treatment with remdesivir was associated with higher costs ($201) and higher QALYs (0.007) than SoC (Table 3). Remdesivir had an ICER of $30,362 compared to SoC and was cost-effective at a threshold of $50,000 per QALY gained in 54% of iterations. Full disaggregated results of the sponsor’s economic evaluation are available in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SoC ($/QALY) |
|---|---|---|---|---|---|
SoC | 2,988 | Reference | 0.166 | Reference | Reference |
Remdesivir | 3,189 | 201 | 0.173 | 0.007 | 30,362 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care; vs. = versus.
Source: Sponsor’s revised pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor included scenario analyses involving alternate assumptions for the time horizon and patient starting age. These scenarios had minimal impact on the results; remdesivir remained cost-effective at a threshold of $50,000 per QALY gained. The sponsor also conducted a scenario analysis including nirmatrelvir-ritonavir as a comparator. In this scenario, remdesivir was associated with higher costs and fewer health gains than nirmatrelvir-ritonavir; as such, remdesivir was dominated by nirmatrelvir-ritonavir.
The sponsor conducted a scenario analysis from a societal perspective in which patients were assumed to miss work due to hospitalization or rehabilitation. Results in this analysis were similar to those in the sponsor’s base case, given that remdesivir was cost-effective at a threshold of $50,000 per QALY gained.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The submitted evidence base does not reflect the current treatment landscape for COVID-19. The primary evidence base for the economic model was the PINETREE study, a phase III, randomized controlled trial conducted from September 2020 to May 2021. During this time, the circulating variants of COVID-19 were biologically distinct from the variant of COVID-19 circulating at present.19,20 The difference in COVID-19 variants was emphasized by the clinical experts consulted by CDA-AMC, who highlighted that data from the PINETREE trial are not externally generalizable to patients infected with the omicron and later variants of COVID-19 now circulating in Canada. Furthermore, the PINETREE trial was conducted in unvaccinated patients, which is also not reflective of the current state of public health in Canada, given that more than 80% of people have now received a primary course of vaccines.21 In addition, data from Ontario from February 2021 to April 2023 indicate that COVID-19–related hospitalizations (and deaths) were higher among unvaccinated individuals than among those who had completed their primary vaccine series, with or without additional boosters.22 Thus, remdesivir is not expected to have the same impact on hospitalization in the current vaccinated population in Canada. These differences represent a fundamental challenge in interpreting the results from the sponsor’s submitted evidence dossier and accompanying pharmacoeconomic model, which are based on the PINETREE trial. The clinical experts strongly emphasized that an economic model based on PINETREE trial data is unable to meaningfully answer the research question of whether remdesivir is cost-effective for the treatment of mild to moderate COVID-19 infections in the current setting in Canada.
Upon hospitalization, the sponsor used placebo arm data from the ACTT-1 trial and sponsor-submitted real-world evidence study (Mozaffari et al., 2023)6,7 to inform transitions between ordinal scale scores. CDA-AMC’s clinical review highlighted that there are substantial concerns regarding the external validity and generalizability of ACTT-1 and the real-world evidence used to inform inpatient mortality as a result of the fast-evolving nature of the pandemic and the virus itself, given that prevalent variants, vaccination status, and clinical outcomes in today’s world are substantially different than those observed early in the pandemic. Clinical expert opinion solicited by CDA-AMC also discussed the important distinction between patients hospitalized because of their COVID-19 infection and those hospitalized who incidentally have a COVID-19 infection. Patients who are hospitalized for other underlying causes may not experience a mortality benefit from treatment with remdesivir if their primary reason for hospitalization is not related to their COVID-19 infection.
CDA-AMC was unable to address this limitation due to the lack of alternative comparative data that could reasonably be applied within the submitted model structure. CDA-AMC notes that the clinical outcomes in the model are highly uncertain.
The proportion of patients requiring hospitalization is uncertain. The sponsor used the proportion of patients requiring hospitalization following outpatient treatment with remdesivir or SoC from the PINETREE trial. As described in the prior limitation, the results of the PINETREE trial are not externally generalizable to patients infected with the omicron and later variants of COVID-19 or to vaccinated patients. In the PINETREE trial, 5.3% of patients treated with SoC in the outpatient setting required hospitalization, which likely overestimates the number of patients at risk of hospitalization today, given the reduced severity of the omicron variant,23-25 high vaccine coverage,21 and history of prior infection. While the results of the PINETREE trial suggest a reduction in hospitalization with remdesivir treatment, the magnitude of that benefit is unknown for the circulating variant of COVID-19 and patient characteristics today (i.e., prior immunity from vaccination or history of infection). CDA-AMC’s clinical review reported that because of the evolving disease landscape, it would be reasonable to expect that the estimate of treatment effect would be smaller today than what was observed in the trial and that the results may be of limited clinical relevance for current disease management and how remdesivir would be used in practice now.
To address this limitation, CDA-AMC applied an effect modifier to the proportion of patients requiring hospitalization. CDA-AMC applied a vaccine effectiveness rate of 82%. This rate represented the estimated effectiveness (in preventing severe outcomes) of 2 doses of COVID-19 vaccine received 180 days to 239 days prior.26 The vaccine effectiveness estimate was measured in Ontario using a test-negative case-control study using linked provincial administrative databases. Vaccine effectiveness results were presented for effectiveness against both infection and severe outcomes; these also considered the timing of the most recent vaccine dose received.26 This approach assumes that all modelled patients received 2 doses of vaccine. CDA-AMC notes that as of September 10, 2023, 84% of Canadians aged 5 years and older had received at least 2 doses of a COVID-19 vaccine, with that number being higher in older age groups: 95% for adults aged 60 years to 69 years, and greater than 99% for adults aged 70 years and older.21 Further, CDA-AMC conservatively applied the 2-dose vaccine effect modifier against severe outcomes, despite the high proportion of Canadians who had received 3 or more doses of vaccine as of June 2022 (i.e., 77% of adults aged 60 years to 69 years and 85% of adults aged 70 years or older).27 CDA-AMC also conservatively maintained the sponsor’s magnitude of benefit in favour of treatment with remdesivir, and did not account for the reduction in severity of the omicron virus compared to past COVID-19 variants.24 CDA-AMC notes that the inpatient mortality risk was not adjusted by vaccine effectiveness, and mortality likely remains overestimated, as described in the subsequent limitation.
Given the uncertainty about population immunity against severe outcomes, CDA-AMC conducted a scenario analysis using an estimate of 50% vaccine effectiveness against severe outcomes.
The baseline distribution of hospital services is not reflective of the levels of care experienced today. The sponsor informed the baseline distribution of hospital levels of care from an economic evaluation conducted alongside the WHO Solidarity Trial in Canada (August 14, 2020 to April 1, 2021) for inpatient treatment with remdesivir.5 Due to limitations associated with the generalizability of clinical data from that period to the current setting in Canada, this distribution is unlikely to be the same as what is experienced today. Clinical expert opinion solicited by CDA-AMC agreed that patients are entering the hospital in less severe conditions than they were at the time of the WHO Solidarity Trial, due to changes in population immunity and viral pathogenicity.
To address this limitation, CDA-AMC adjusted the baseline distribution of patients upon hospitalization based on evidence from a retrospective analysis of several provinces in Canada.28 CDA-AMC assumed that 4.5% of patients would require mechanical invasive ventilation upon hospital admission, that an additional 10% would be admitted to the ICU upon admission, and that the remaining 85.5% would start in the ward, with 20% on supplemental oxygen. CDA-AMC notes that the data informing this distribution are from 2022, and since then, further changes to population immunity and the severity of the circulating COVID-19 variant may have influenced hospital levels of care. Clinical expert opinions solicited by CDA-AMC highlighted that hospitalization rates have continued to decline since 2022; CIHI hospital and emergency department statistics also support this information.17
Although this change addressed the baseline distribution, the movement between ordinal scale scores, as informed by the placebo arm of the ACTT-1 trial, continues to lack generalizability to the current COVID-19 landscape. After adjusting the baseline distribution of hospital care, the model predicts that 14% of patients who require hospitalization will die, which is nearly 3 times the all-cause mortality for patients in hospital observed in the omicron wave from March 20, 2022 to May 28, 2022, as reported by the Canadian Nosocomial Infection Surveillance Program.24 The sponsor’s base-case analysis estimated that 16% of patients who require hospitalization died.
The pharmacoeconomic submission incorporated poor reporting, organization, and modelling practices, which made validation of the submitted evidence difficult. The sponsor’s submission included several discrepancies in reporting within the technical report and submitted model and lacked detail on how parameter estimates were derived from clinical data sources. Further, the model structure was overly complicated in its reliance on ordinal scale scores at baseline and the transitions between ordinal scale scores. Clinical expert opinion solicited by CDA-AMC indicated that ordinal scale scores are not regularly used in routine clinical practice or research. As a result, the application of data from different clinical sources was overly complicated.
CDA-AMC was unable to address this limitation and notes that the sponsor’s model structure made it difficult to assess the impact of alternative data for some influential clinical input parameters, including transitions between ordinal scale scores and mortality.
Hospitalization costs were inaccurately estimated. The sponsor derived costs for COVID-19–related hospitalizations from CIHI data for time spent in the general ward ($20,097 per stay)17 and from a published economic evaluation for ordinal scale scores 4 to 8.5 The costs from the economic evaluation were derived for hospitalizations that occurred during the early waves of COVID-19 in Canada. As previously described, clinical outcomes during earlier pandemic waves are not reflective of the current COVID-19 landscape (which is characterized by a highly vaccinated population and less severe circulating variants). As such, using hospitalization costs measured at that time introduced uncertainty with regards to the costs that may be experienced in hospitals today. The costs estimated by CIHI for COVID-19 hospitalizations in fiscal year 2022 to 2023 represent more current and widely applicable cost estimates. The same analysis from which the sponsor obtained the cost applied to ordinal scale scores 1 to 3 ($20,097 per stay) also reported that the average cost per hospitalization for patients with COVID-19 requiring ICU admission was $52,774.
In its reanalysis, for ordinal scale scores 1 to 5 (patients not requiring ICU admission), CDA-AMC used average hospitalization costs; for ordinal scale scores 6 to 8, it used the cost for patients requiring ICU admission.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Medically attended visits were informed by results of the PINETREE trial. | Not acceptable. The number of medically attended visits measured in the PINETREE trial is subject to the aforementioned generalizability concerns related to circulating COVID-19 variants, vaccination rates, and patients’ histories of prior infections. As such, it is uncertain whether outpatient treatment with remdesivir is associated with fewer medically attended visits than SoC treatment. In fact, clinical expert opinion solicited by CDA-AMC indicated that treatment with remdesivir may result in an increase in medical visits, given that it requires at least 3 visits to an infusion centre for treatment. The impact of the number of medically attended visits on the results of the model is minimal. However, there are likely to be resource implications that are not fully captured. |
Symptom alleviation in the model was informed by results of the PINETREE trial, measured by the FLU-PRO Plus questionnaire. | Not acceptable. The CDA-AMC clinical review noted limitations associated with the use of the FLU-PRO Plus questionnaire to measure symptom alleviation and noted that its use to infer clinically meaningful improvement may be challenging. However, the impact of symptom alleviation on the results of the model was minimal. |
Adverse events were not included in the model. | Acceptable. Clinical expert opinion solicited by CDA-AMC agreed that remdesivir was not associated with frequent adverse events, and that the impact of including the outcomes of adverse events would likely have a minimal impact on the results of the model. |
The modelled time horizon in the sponsor’s submission was 12 weeks. | Acceptable. CDA-AMC agrees that, based on the clinical evidence and decision problem, a 12-week time horizon was appropriate to use in the sponsor’s base-case analysis. Given that the model may predict differences in longer-term outcomes, CDA-AMC conducted a scenario analysis using a 10-year time horizon to explore the impact of a longer time horizon on the cost-effectiveness of remdesivir compared to SoC. |
FLU-PRO Plus = COVID-19–adapted InFLUenza Patient-Reported Outcome; SoC = standard of care.
CDA-AMC Reanalyses of the Economic Evaluation
Reanalysis Results
The CDA-AMC reanalysis was derived by making changes in model parameter values and assumptions in consultation with clinical experts. These changes, summarized in Table 5, included modifying the risk of hospitalization, the distribution of baseline levels of care during hospitalization, and hospitalization costs.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC reanalysis | ||
1. Risk of hospitalization | Informed directly by the PINETREE trial | Applied omicron-specific vaccine effectiveness against severe outcomes to hospitalization rate |
2. Baseline level of care in hospital | Proportion of patients entering hospital by ordinal scale score:
| Proportion of patients entering hospital by ordinal scale score:
|
3. Hospitalization costs | Hospitalization costs:
| Hospitalization costs:
|
CDA-AMC reanalysis | ― | Reanalysis 1 + 2 + 3 |
The CDA-AMC reanalysis found that remdesivir was associated with 0.006 additional QALYs at an additional cost of $2,372. Therefore, the ICER of remdesivir was $390,996 per QALY gained compared to SoC. A summary of the CDA-AMC stepped analysis and reanalysis results can be found in Table 6. Remdesivir had a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
The number of incremental QALYs predicted by the CDA-AMC reanalysis was similar to that predicted in the sponsor’s submitted analysis; however, adjustments to inpatient care led CDA-AMC’s reanalysis to estimate a higher incremental cost for remdesivir compared to SoC. This was driven by the reduction in costs in the SoC arm of the model, which was a combined effect of lowering the absolute number of patients requiring hospitalization and revising the COVID-19 hospitalization costs.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | SoC | 2,990 | 0.166 | Reference |
Remdesivir | 3,204 | 0.173 | 29,767 | |
CDA-AMC reanalysis 1 | SoC | 714 | 0.166 | Reference |
Remdesivir | 2,894 | 0.173 | 316,813 | |
CDA-AMC reanalysis 2 | SoC | 2,729 | 0.166 | Reference |
Remdesivir | 3,169 | 0.173 | 62,573 | |
CDA-AMC reanalysis 3 | SoC | 1,222 | 0.166 | Reference |
Remdesivir | 2,964 | 0.173 | 242,429 | |
CDA-AMC reanalysis (reanalysis 1 + 2 + 3) | SoC | 372 | 0.166 | Reference |
Remdesivir | 2,848 | 0.173 | 361,319 | |
CDA-AMC reanalysis (reanalysis 1 + 2 + 3) (probabilistic) | SoC | 480 | 0.167 | Reference |
Remdesivir | 2,851 | 0.173 | 390,996 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor’s base case and CDA-AMC reanalysis (Table 7). This analysis demonstrated that a price reduction of 82% would be necessary to achieve cost-effectiveness at a threshold of $50,000 per QALY gained in the CDA-AMC reanalysis.
Table 7: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost ($) | ICERs for remdesivir vs. SoC ($/QALY) | |
|---|---|---|---|
Price reduction | $ | Sponsor base case | CDA-AMC reanalysis |
No price reduction | 660.53 | 30,362 | 390,996 |
10% | 594.48 | Dominant | 349,230 |
20% | 528.42 | Dominant | 307,464 |
30% | 462.37 | Dominant | 265,697 |
40% | 396.32 | Dominant | 223,931 |
50% | 330.27 | Dominant | 182,165 |
60% | 264.21 | Dominant | 140,399 |
70% | 198.16 | Dominant | 98,632 |
80% | 132.11 | Dominant | 56,866 |
90% | 66.05 | Dominant | 15,100 |
100% | 0.00 | Dominant | Dominant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care; vs. = versus.
Note: Dominant means that remdesivir was associated with lower costs and higher health gains than SoC.
Additionally, the CDA-AMC clinical review indicated that, based on the PINETREE trial results, an NNT of 22 would be needed to prevent 1 case of severe COVID-19. When the NNT is multiplied by the cost of remdesivir (i.e., $2,878 per 3-day treatment course), a cost of $63,316 is obtained; this is the cost to prevent 1 case of severe COVID-19. If this is compared to the cost of a general ward hospital stay ($20,097),17 a price reduction of approximately 68% is required to achieve cost neutrality.
Given that the proportion of patients hospitalized is highly uncertain (refer to the key limitation described earlier about the risk of hospitalization), CDA-AMC also estimated the NNT using the adjusted hospitalization rates in the CDA-AMC reanalysis. The NNT using the adjusted proportion of patients requiring hospitalization is 122 to prevent 1 case of severe COVID-19. When this NNT is multiplied by the cost of remdesivir, a cost of $351,116 is obtained. If this is compared to the cost of a general ward hospital stay, a price reduction of approximately 94% is required to achieve cost neutrality.
In the scenario analysis that CDA-AMC conducted using 50% vaccine effectiveness, while maintaining the other changes to derive the CDA-AMC reanalysis, remdesivir is associated with an ICER of $300,855 per QALY gained (incremental cost = $1,952; incremental QALYs = 0.006) (Table 13).
CDA-AMC also conducted a scenario analysis using an extended time horizon of 10 years while maintaining CDA-AMC’s other changes to derive the CDA-AMC reanalysis. The results of this scenario estimate that remdesivir is associated with an ICER of $166,036 per QALY gained (incremental cost = $2,365; incremental QALYs = 0.014) compared to SoC (Table 13).
Issues for Consideration
The clinical expert opinion solicited by CDA-AMC and the input received from drug plans indicated that there may be issues with patient access to remdesivir due to its mode of administration (i.e., patients may face challenges accessing infusion centres). This may disproportionately affect people living in remote parts of Canada who are unable to travel long distances to infusion centres. Further, the health system impacts of IV treatments, including the need for nursing staff and capacity at infusion centres, are not captured in the cost-utility analysis or budget impact analysis (BIA).
At the time the remdesivir review was initiated, nirmatrelvir-ritonavir was not reimbursed by provincial drug plans for outpatient treatment of COVID-19. However, on April 11, 2024, CDA-AMC published a CDA-AMC Canadian Drug Expert Committee recommendation recommending nirmatrelvir-ritonavir for reimbursement after a diagnosis of COVID-19 has been made and within 5 days of symptom onset for adult patients who are severely immunocompromised with specific conditions (e.g., solid organ transplant recipients, malignant hematologic conditions, severe primary immunodeficiencies), and moderately immunosuppressed due to 1 or more specific conditions (e.g., treatment for cancer including solid tumours, advanced untreated HIV infection or treated HIV).29 Although the sponsor submitted a scenario analysis including nirmatrelvir-ritonavir, the cost-effectiveness of nirmatrelvir-ritonavir versus remdesivir in the CDEC-recommended population is unknown.
Overall Conclusions
In the PINETREE pivotal trial, remdesivir reduced the incidence of COVID-19–related hospitalization or death from any cause through day 28 compared to placebo. As noted in the clinical review, the PINETREE trial did not include vaccinated patients or patients who had had COVID-19 in the past, and it was performed at a time when the omicron variant was not yet circulating. Furthermore, the trial was performed in a population not considered to be at high risk of progressing to severe disease, as defined in clinical practice at the time of this review. These differences represent a fundamental challenge in interpreting the results from the sponsor’s submitted evidence dossier and accompanying pharmacoeconomic model, which are based on the PINETREE trial.
CDA-AMC made several changes to the sponsor’s submitted model, in consultation with clinical experts, to derive a reanalysis. The CDA-AMC reanalysis found that remdesivir was associated with 0.006 additional QALYs at an additional cost of $2,372 and an ICER of $390,996 per QALY gained compared to SoC. A price of $486 per 3-day treatment course (i.e., a price reduction of approximately 82%) would be required for remdesivir to be considered cost-effective at a threshold of $50,000 per QALY gained. The findings of CDA-AMC’s reanalysis differ from the sponsor’s due to the choice of clinical data sources and hospitalization costs; the alternate choices better reflect the current understanding and experience with COVID-19 in Canada. However, CDA-AMC notes that the incremental QALYs estimated by the sponsor (incremental QALYs = 0.007) are similar to the results of the CDA-AMC reanalysis; these benefits equate to an additional 2 quality-adjusted days of life.
CDA-AMC included an alternative approach considering the NNT to avoid 1 severe case of COVID-19 (i.e., a case resulting in hospitalization or death), as reported by the CDA-AMC clinical review. Based on the PINETREE trial, 22 high-risk individuals would need to be treated to avoid 1 case of severe COVID-19. Based on CDA-AMC’s reduction to the risk of hospitalization in the economic model using vaccine effectiveness against severe outcomes, 122 high-risk individuals would need to be treated. When comparing the drug acquisition costs of remdesivir for 22 individuals and 122 individuals (approximately $63,000 and $351,000, respectively) with the cost of a general ward admission to treat COVID-19 ($20,000), a price reduction of approximately 68% to 94% would be required to ensure minimal financial impact on the health care system.
The results of this analysis are driven by assumptions about high-risk patients and COVID-19 risk informed by clinical evidence that could not be assessed, given the lack of comparative clinical evidence that more accurately reflects the current COVID-19 setting in Canada. Given that the majority of patients (> 90% in both the sponsor’s and CDA-AMC’s analysis) will not require hospitalization, even on SoC, the effectiveness of remdesivir is limited to patients who would potentially require hospitalization. Notably, the description of patients who are considered to be at high risk of hospitalization in the current COVID-19 context in Canada has narrowed, and COVID-19 variants have changed such that infection no longer carries as high a risk of hospitalization. Therefore, identifying and treating only those patients at highest risk of hospitalization is critical to maximizing the clinical benefit and cost-effectiveness of remdesivir.
References
1.Drug Reimbursement Review sponsor submission: Veklury (remdesivir), 100 mg/vial powder for solution for infusion [internal sponsor's package]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2024 Mar 11.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Veklury (remdesivir), 100 mg/vial powder for solution for infusion. Mississauga (ON): Gilead Sciences Canada, Inc.; 2024 Mar 11.
3.Chen C, Haupert SR, Zimmermann L, Shi X, Fritsche LG, Mukherjee B. Global prevalence of post-coronavirus disease 2019 (COVID-19) condition or long COVID: a meta-analysis and systematic review. J Infect Dis. 2022;226(9):1593-1607. PubMed
4.The prevalence of underlying health conditions that increase the risk of severe health outcomes related to COVID-19. The Daily. Ottawa (ON): Statistics Canada; 2020: https://www150.statcan.gc.ca/n1/en/daily-quotidien/200703/dq200703a-eng.pdf?st=4KOzoYHi. Accessed 2023 Dec 12.
5.Lau VI, Fowler R, Pinto R, et al. Cost-effectiveness of remdesivir plus usual care versus usual care alone for hospitalized patients with COVID-19: an economic evaluation as part of the Canadian Treatments for COVID-19 (CATCO) randomized clinical trial. CMAJ Open. 2022;10(3):E807-E817. PubMed
6.Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19 - final report. N Engl J Med. 2020;383(19):1813-1826. PubMed
7.Mozaffari E, Chandak A, Gottlieb RL, et al. Remdesivir is associated with reduced mortality in COVID-19 patients requiring supplemental oxygen including invasive mechanical ventilation across SARS-CoV-2 variants. Open Forum Infect Dis. 2023;10(10):ofad482. PubMed
8.Shoucri SM, Purpura L, DeLaurentis C, et al. Characterising the long-term clinical outcomes of 1190 hospitalised patients with COVID-19 in New York City: a retrospective case series. BMJ Open. 2021;11(6):e049488. PubMed
9.McAlister FA, Dong Y, Chu A, et al. The risk of death or unplanned readmission after discharge from a COVID-19 hospitalization in Alberta and Ontario. CMAJ. 2022;194(19):E666-E673. PubMed
10.Table 13-10-0114-01 Life expectancy and other elements of the complete life table, three-year estimates (2019-2021), Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2023: https://open.canada.ca/data/en/dataset/d0958223-ba07-4803-aeb5-1e7086f2cb92. Accessed 2023 Dec 12.
11.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
12.Poteet S, Craig BM. QALYs for COVID-19: a comparison of US EQ-5D-5L value sets. Patient. 2021;14(3):339-345. PubMed
13.Goswami H, Alsumali A, Jiang Y, et al. Cost-effectiveness analysis of molnupiravir versus best supportive care for the treatment of outpatient COVID-19 in adults in the US. Pharmacoeconomics. 2022;40(7):699-714. PubMed
14.Sheinson D, Dang J, Shah A, Meng Y, Elsea D, Kowal S. A cost-effectiveness framework for COVID-19 treatments for hospitalized patients in the United States. Adv Ther. 2021;38(4):1811-1831. PubMed
15.Khan K, Muennig P, Gardam M, Zivin JG. Managing febrile respiratory illnesses during a hypothetical SARS outbreak. Emerg Infect Dis. 2005;11(2):191-200. PubMed
16.Tsui TCO, Zeitouny S, Bremner KE, et al. Initial health care costs for COVID-19 in British Columbia and Ontario, Canada: an interprovincial population-based cohort study. CMAJ Open. 2022;10(3):E818-E830. PubMed
17.Canadian Institute for Health Information. COVID-19 hospitalization and emegency department statistics. 2024; https://www.cihi.ca/en/covid-19-hospitalization-and-emergency-department-statistics. Accessed 2024 Apr 23.
18.Barnieh L, Beckerman R, Jeyakumar S, Hsiao A, Jarrett J, Gottlieb RL. Remdesivir for hospitalized COVID-19 patients in the United States: optimization of health care resources. Infect Dis Ther. 2023;12(6):1655-1665. PubMed
19.Centers for Disease Control and Prevention. CDC Museum COVID-19 timeline. 2023; https://www.cdc.gov/museum/timeline/covid19.html. Accessed 2024 Apr 23.
20.Tuite AR, Fisman DN, Odutayo A, et al. COVID-19 Hospitalizations, ICU Admissions and Deaths Associated with the New Variants of Concern. Science Briefs of the Ontario COVID19 Science Advisory Table. 2021;1:8. https://covid19-sciencetable.ca/sciencebrief/covid-19-hospitalizations-icu-admissions-and-deaths-associated-with-the-new-variants-of-concern/. Accessed 2024 Aug 07.
21.COVID-19 vaccination: vaccination coverage. Ottawa (ON): Public Health Agency of Canada; 2023: https://health-infobase.canada.ca/covid-19/vaccination-coverage/archive/2023-05-22/. Accessed 2023 Apr 23.
22.Severe outcomes among confirmed cases of COVID-19 following vaccination in Ontario: December 14 2022 to April 23, 2023. Toronto (ON): Public Health Ontario; 2023: https://www.publichealthontario.ca/-/media/Documents/nCoV/epi/covid-19-epi-confirmed-cases-post-vaccination.pdf. Accessed 2024 Apr 23.
23.Ulloa AC, Buchan SA, Daneman N, Brown KA. Estimates of SARS-CoV-2 Omicron variant severity in Ontario, Canada. JAMA. 2022;327(13):1286-1288. PubMed
24.Mitchell R, Cayen J, Thampi N, et al. Trends in severe outcomes among adult and pediatric patients hospitalized with COVID-19 in the Canadian Nosocomial Infection Surveillance Program, March 2020 to May 2022. JAMA Netw Open. 2023;6(4):e239050. PubMed
25.Harrigan SP, Wilton J, Chong M, et al. Clinical severity of severe acute respiratory syndrome coronavirus 2 Omicron variant relative to Delta in British Columbia, Canada: a retrospective analysis of whole-genome sequenced cases. Clin Infect Dis. 2023;76(3):e18-e25. PubMed
26.Buchan SA, Chung H, Brown KA, et al. Estimated effectiveness of COVID-19 vaccines against Omicron or Delta symptomatic infection and severe outcomes. JAMA Netw Open. 2022;5(9):e2232760. PubMed
27.Vaccination coverage for COVID-19 additional doses in Canada: results from the 2022 Canadian Community Health Survey (CCHS). Ottawa (ON): Public Health Agency of Canada; 2023: https://www.canada.ca/en/public-health/services/immunization-vaccines/vaccination-coverage/covid-19-additional-doses-results-2022-canadian-community-health-survey.html. Accessed 2024 Apr 23.
28.Lee T, Cheng MP, Vinh DC, et al. Outcomes and characteristics of patients hospitalized for COVID-19 in British Columbia, Ontario and Quebec during the Omicron wave. CMAJ Open. 2023;11(4):E672-E683. PubMed
29.Drug Reimbursement Expert Review Committee final recommendation: nirmatrelvir-ritonavir (Paxlovid - Pfizer Canada ULC). Can J Health Technol. 2024;4(4). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/SR0808/SR0808. Accessed 2024 Apr 22.
30.DeltaPA. Ottawa (ON): IQVIA; 2023: https://www.iqvia.com/. Accessed 2024 Mar 27.
31.COVID-19 related procurement: Committee of the Whole—May 19, 2022. Ottawa (ON): Public Services and Procurement Canada; 2022: https://www.canada.ca/en/public-services-procurement/corporate/transparency/briefing-materials/committee-of-the-whole/2022-05-19/covid-19-related-procurement.html. Accessed 2024 Apr 22.
32.Lefebvre C. Paxlovid to no longer be provided to provinces for free. Winnipeg (MB): CTV Winnipeg; 2024: https://winnipeg.ctvnews.ca/paxlovid-to-no-longer-be-provided-to-provinces-for-free-1.6829335. Accessed 2024 Apr 22.
33.pan-Canadian Pharmaceutical Alliance (pCPA). Paxlovid (nirmatrelvir/ritonavir). 2024; https://www.pcpacanada.ca/negotiation/22592. Accessed 2024 Apr 22.
34.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Veklury (remdesivir), 100 mg/vial powder for solution for infusion. Mississauga (ON): Gilead Sciences Canada, Inc.; 2024 Mar 11.
35.COVID-19 epidemiology update: summary. Ottawa (ON): Public Health Agency of Canada: https://health-infobase.canada.ca/covid-19/. Accessed 2023 Nov 21.
36.Epidemiological summary of COVID-19 cases in First Nations communities. Ottawa (ON): Indigenous Services Canada; 2023: https://www.sac-isc.gc.ca/eng/1589895506010/1589895527965#cmtv. Accessed 2023 Nov 29.
37.Centers for Disease Control and Prevention. Estimated COVID-19 burden. 2022; https://archive.cdc.gov/#/details?q=Centers%20for%20Disease%20Control%20and%20Prevention%20(CDC).%20Estimated%20COVID-19%20Burden%202022&start=0&rows=10&url=https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/burden.html. Accessed 2023 Nov 06.
38.Living guidance for clinical management of COVID-19. Geneva (CH): World Health Organization; 2021: https://apps.who.int/iris/bitstream/handle/10665/349321/WHO-2019-nCoV-clinical-2021.2-eng.pdf. Accessed 2023 Nov 06.
39.Table 13-10-0777-01 Number and percentage of adults (aged 18 years and older) in the household population with underlying health conditions, by age and sex (two-year period). Ottawa (ON): Statistics Canada; 2020: https://doi.org/10.25318/1310077701-eng. Accessed 2024 Apr 22.
40.Sun N, Hua CL, Qiu X, Brown JS. Determinants of COVID-19 testing among late middle-aged and older adults: Applying the health belief model. Aging Health Res. 2022;2(2):100066. PubMed
41.Kojima N, Klausner JD. Usage and awareness of antiviral medications for coronavirus disease 2019 (COVID-19) among individuals at risk for severe COVID-19, March 2021 to 1 August 2022. Clin Infect Dis. 2023;76(4):775-776. PubMed
42.COVID-19 epidemiology update: current situation. Ottawa (ON): Public Health Agency of Canada: https://health-infobase.canada.ca/covid-19/current-situation.html. Accessed 2024 Apr 11.
43.Oksuz E, Malhan S, Gonen MS, et al. Cost-effectiveness analysis of remdesivir treatment in COVID-19 patients requiring low-flow oxygen therapy: payer perspective in Turkey. Adv Ther. 2021;38(9):4935-4948. PubMed
44.Clinical Study Report: GS-US-540-9012, PINETREE Final. A phase 3, randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of remdesivir (GS-5734™) treatment of COVID-19 in an outpatient setting [internal sponsor's report]. Foster City (CA): Gilead Sciences, Inc.; 2021 Oct 01.
45.Ontario COVID-19 Drugs and Biologics Clinical Practice Guidelines Working Group. Clinical practice guideline summary: recommended drugs and biologics in adult patients with COVID-19. Toronto (ON): Ontario COVID-19 Science Advisory Table; 2022: https://dx.doi.org/10.47326/ocsat.cpg.2022.11.0. Accessed by sponsor, no date provided.
46.British Columbia COVID-19 Therapeutics Committee (CTC) and COVID-19 Therapeutics Review and Advisory Working Group (CTRAWG). Clinical practice guidelines for antimicrobial and immunomodulatory therapy in adult patients with COVID-19. Vancouver (BC): BC Centre for Disease Control; 2023: http://www.bccdc.ca/Health-Professionals-Site/Documents/Antimicrobial-Immunomodulatory-Therapy-adults.pdf. Accessed 2023 Nov 06.
Appendix 1: Cost Comparison Table
Please note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for the Treatment of COVID-19
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosage | Treatment course length | Cost per treatment course ($) |
|---|---|---|---|---|---|---|
Remdesivir (Veklury) | 5 mg/mL | 100 mg vial | 660.5300 | 200 mg on Day 1 and 100 mg on Days 2 and 3 | 3 days | 2,642 |
Note: The price of remdesivir is based on the sponsor-submitted price and does not include dispensing fees.
Table 9: CDA-AMC Cost Comparison Table for Other COVID-19 Treatments
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosage | Treatment course length | Cost per treatment course ($) |
|---|---|---|---|---|---|---|
Nirmatrelvir-ritonavir (Paxlovid) | 150 mg / 100 mg | 10 or 20 tablets nirmatrelvir (depending on patient’s renal status) and 10 tablets ritonavir One carton per treatment course containing 5 daily-dose blister cards (2 or 4 tablets nirmatrelvir and 2 tablets ritonavir each) | 1,288.8848a | 300 mg nirmatrelvir with 100 mg ritonavir twice daily for 5 days | 5 days | 1,289 |
aPrice retrieved from Draft CDA-AMC Recommendation for nirmatrelvir-ritonavir.29 DeltaPA has a list price of $0.01 per 30 tablet blister pack30 as the Public Health Agency of Canada was providing nirmatrelvir-ritonavir to provinces.31 Per a recent news release, provinces will now be responsible for the procurement of nirmatrelvir-ritonavir.32 At the time of this review, nirmatrelvir-ritonavir is in active negotiations with the pCPA.33
Appendix 2: Submission Quality
Please note this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The population studied in the PINETREE trial does not reflect the population eligible for remdesivir at present due to differences in baseline risk, vaccination status, and COVID-19 variant. The population studied in the pivotal trial is not relevant. |
Model has been adequately programmed and has sufficient face validity | No | Refer to key limitation: The pharmacoeconomic submission incorporated poor reporting, organization, and modelling practices, which made validation of the submitted evidence difficult. |
Model structure is adequate for decision problem | No | The model structure was overly complicated and not aligned with the majority of available data. As such, it made addressing the decision problem challenging. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Refer to key limitation: The pharmacoeconomic submission incorporated poor reporting, organization, and modelling practices, which made validation of the submitted evidence difficult. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note this appendix has not been copy-edited.
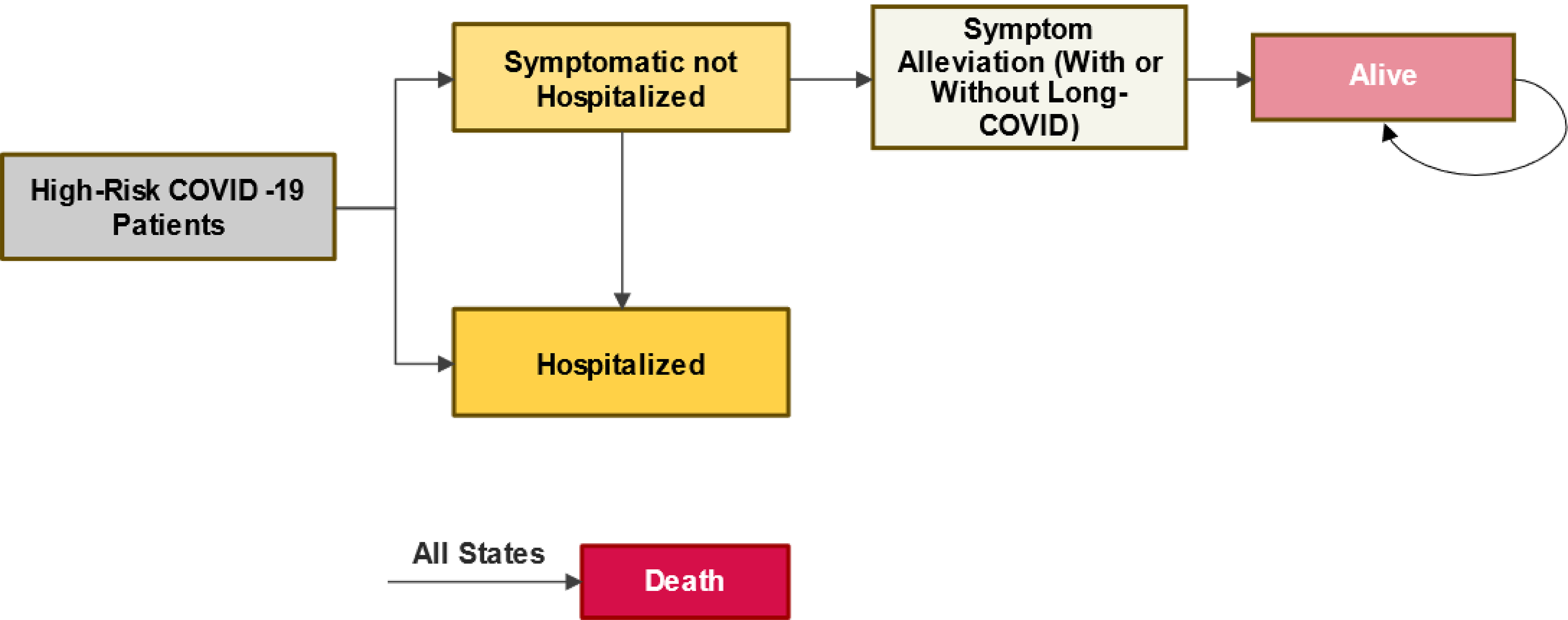
Figure 2: Model Structure, Inpatient
ICU = intensive care unit; MIV = mechanical invasive ventilation.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Remdesivir | SoC |
|---|---|---|
Discounted LYs | ||
Total | 0.219 | 0.217 |
Outpatient, symptomatic | 0.033 | 0.045 |
Outpatient, recovered | 0.181 | 0.147 |
Outpatient, long COVID | 0.003 | 0.003 |
Ordinal scale score 1 to 3 | 0.001 | 0.005 |
Ordinal scale score 4 | 0.000 | 0.001 |
Ordinal scale score 5 | 0.000 | 0.003 |
Ordinal scale score 6 | 0.000 | 0.001 |
Ordinal scale score 7 | 0.000 | 0.001 |
Recovered (postdischarge) | 0.001 | 0.010 |
Discounted QALYs | ||
Total | 0.173 | 0.166 |
Outpatient, symptomatic | 0.017 | 0.023 |
Outpatient, recovered | 0.153 | 0.124 |
Outpatient, long COVID | 0.001 | 0.001 |
Ordinal scale score 1 to 3 | 0.000 | 0.004 |
Ordinal scale score 4 | 0.000 | 0.001 |
Ordinal scale score 5 | 0.000 | 0.002 |
Ordinal scale score 6 | 0.000 | 0.001 |
Ordinal scale score 7 | 0.000 | 0.001 |
Recovered (postdischarge) | 0.001 | 0.008 |
Utility decrements | ||
Rehabilitation | 0.000 | 0.000 |
Rehospitalization | 0.000 | 0.000 |
Discounted costs ($) | ||
Total | 3,550 | 6,506 |
Treatment and administration | 2,758 | 0 |
Long COVID | 42 | 57 |
Medically attended visit | 12 | 134 |
Hospitalization | 735 | 6,287 |
Postdischarge | 3 | 27 |
LY = life-year; QALY = quality-adjusted life-year; SoC = standard of care
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note this appendix has not been copy-edited.
Detailed Results of CDA-AMC Reanalysis
Table 12: Disaggregated Summary of CDA-AMC’s Economic Evaluation Results
Parameter | Remdesivir | SoC |
|---|---|---|
Discounted LYs | ||
Total | 0.220 | 0.219 |
Outpatient, symptomatic | 0.034 | 0.051 |
Outpatient, recovered | 0.183 | 0.163 |
Outpatient, long COVID | 0.003 | 0.004 |
Ordinal scale score 1 to 3 | 0.000 | 0.000 |
Ordinal scale score 4 | 0.000 | 0.000 |
Ordinal scale score 5 | 0.000 | 0.000 |
Ordinal scale score 6 | 0.000 | 0.000 |
Ordinal scale score 7 | 0.000 | 0.000 |
Recovered (postdischarge) | 0.000 | 0.001 |
Discounted QALYs | ||
Total | 0.173 | 0.167 |
Outpatient, symptomatic | 0.018 | 0.027 |
Outpatient, recovered | 0.154 | 0.137 |
Outpatient, long COVID | 0.001 | 0.001 |
Ordinal scale score 1 to 3 | 0.000 | 0.000 |
Ordinal scale score 4 | 0.000 | 0.000 |
Ordinal scale score 5 | 0.000 | 0.000 |
Ordinal scale score 6 | 0.000 | 0.000 |
Ordinal scale score 7 | 0.000 | 0.000 |
Recovered (postdischarge) | 0.000 | 0.001 |
Utility decrements | ||
Rehabilitation | 0.000 | 0.000 |
Rehospitalization | 0.000 | 0.000 |
Discounted costs ($) | ||
Total | 2,851 | 480 |
Treatment and administration | 2,760 | 0 |
Long COVID | 43 | 64 |
Medically attended visit | 12 | 143 |
Hospitalization | 37 | 270 |
Postdischarge | 0 | 2 |
LY = life-year; QALY = quality-adjusted life-year; SoC = standard of care
Scenario Analyses
Table 13: Results of the CDA-AMC Scenario Analyses
Drug | Total costs ($) | Total QALYs | ICER vs. SoC ($/QALY) |
|---|---|---|---|
Scenario assuming 50% vaccine effectiveness against severe outcomes | |||
SoC | 964 | 0.167 | Reference |
Remdesivir | 2,915 | 0.173 | 300,855 |
Scenario assuming a 10-year time horizon | |||
SoC | 484 | 7.074 | Reference |
Remdesivir | 2,849 | 7.088 | 166,036 |
QALY = quality-adjusted life-year; SoC = standard of care; vs. = versus.
Note: The scenario analysis assuming a 10-year time horizon demonstrated that a price reduction of 65% would be necessary to achieve cost-effectiveness at a threshold of $50,000 per QALY gained.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 14: Summary of Key Take Aways
Key take aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s BIA
In the submitted BIA, the sponsor assessed the introduction of remdesivir for nonhospitalized COVID-19 patients (weighing at least 40 kg) with positive SARS-CoV-2 test results who are at high risk of progression to severe COVID-19, including hospitalization and death.34 The BIA was undertaken from the perspective of a Canadian public payer over a three-year time horizon (2024 to 2026) using an epidemiological approach (Figure 3). The sponsor compared a reference scenario in which patients were treated with SoC to a new drug scenario in which remdesivir was reimbursed for use in combination with SoC. The sponsor’s submission only considered drug acquisition costs for remdesivir. Data for the model were obtained from various sources including published literature,35-41 the sponsor’s internal data, and assumption. Key inputs to the BIA are documented in Table 14.
Key assumptions included:
5% of patients are not eligible to oral antiviral therapy (nirmatrelvir-ritonavir).
A flat number of COVID-19 cases due to challenges with predicting infection trends over time.
The ratio of reported to unreported COVID-19 infections (1:4) from February 2020 to September 2021 in the US were reflective of current reporting patterns in Canada.
Only patients of at least 12 years of age were included in the eligible population as remdesivir is indicated for patients weighing at least 40 kg.
A sample of community-dwelling adults aged 51 and older residing in the US who were asked “Have you been tested for the coronavirus?” from June 2020 to November 202040 is equivalent to the proportion of high-risk COVID-19 cases getting tested in Canada.
The proportion of adults 65 years of age or older residing in the US who sought treatment from March 2021 to August 2022 were reflective of patterns in Canada.
All eligible patients will receive remdesivir in years 1, 2, and 3 (i.e., 100% market share).
Figure 3: Sponsor’s Estimation of the Size of the Eligible Population
Source: Sponsor’s budget impact analysis submission.34
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/ year 2/year 3, if appropriate) |
|---|---|
Target population | |
COVID-19 cases with mild to moderate symptoms | |
906,236 | |
Number of COVID-19 cases not reported37 | 679,677 (assumed an additional 75% of the reported cases are unreported) |
Percentage reporting mild to moderate symptoms38 | 80% |
Patients < 60 years old with mild-moderate COVID-19 at high risk | |
Proportion < 60 years old among COVID-19 cases35 | 67.3% |
Proportion immunocompromised or with a chronic condition39 | 27.8% |
Patients ≥ 60 years old with mild-moderate COVID-19 at high risk | |
Proportion ≥ 60 years old among COVID-19 cases35 | 22.75% |
High-risk COVID-19 cases getting tested and seeking treatment | |
Proportion of cases getting tested40 | 21% |
Proportion of cases seeking treatment41 | 36.3% |
Proportion of cases not eligible to oral antiviral therapya | 5% |
Number of patients eligible for drug under review | 2,010 / 2,010 / 2,010 |
Market Uptake (3 years) | |
Uptake (reference scenario) | |
SoC | 100% / 100% / 100% |
Uptake (new drug scenario) | |
Remdesivir + SoC | 100% / 100% / 100% |
SoC | 0% / 0% / 0% |
Cost of treatment (per patient, per 3-day treatment) | |
Remdesivir + SoC | $2,642.12 |
SoC | $0 |
SoC = standard of care.
aEstimate is based on sponsor assumption.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the 3-year budget impact of reimbursing remdesivir for nonhospitalized COVID-19 patients (weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk of progression to severe COVID-19, including hospitalization and death, would be $15,935,256 ($5,311,752 in each of year 1, year 2, and year 3).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The eligible population size is highly uncertain: The sponsor used an epidemiologic approach to estimate the number of patients eligible for remdesivir, starting with the number of COVID-19 cases reported from March 2022 to February 2023.35,36 The number of reported cases across Canada was retrieved from the Government of Canada COVID-19 dataset35 and the number for cases for First Nations individuals residing on reserve36 were assumed to represent the entire noninsured health benefits (NIHB) population and were removed from their respective province. Cases reported in Quebec were excluded. CDA-AMC notes that the number of reported cases is inclusive of hospitalized cases with COVID-19 which are captured by the concurrent review for remdesivir use in hospitalized COVID-19 patients.
First, the sponsor assumed that the number of cases that occurred from March 2022 to February 2023 would remain constant until 2026. The number of reported cases is published weekly and cases have declined significantly since February 2023.42 For example, from the last week in February 2023 to the last week in March 2024, the number of cases reported decreased by approximately 570%.42 The trends observed in the data are aligned with recent statements made by the BC Centre for Disease Control which noted that within Canada, “COVID-19 activity has continued to slowly decrease or remains at low levels.”43 Given the downward trend of COVID-19 activity, assuming that the number of COVID-19 cases remains constant through to 2026 likely overestimates the eligible population size. CDA-AMC notes that more recent data on the number of reported cases in First Nations communities is unavailable and thus while it is likely that the number of cases among the NIHB population have also decreased, no new data are available to substantiate this information.
Second, the sponsor assumed that the number of cases underrepresented the proportion of patients who would get tested and assumed that for every 1 case reported, 4 cases were unreported. This assumption was based on the estimated rate of reported to unreported COVID-19 infections from February 2020 to September 2021 in the US.37 Presumably, all reported cases tested positive for COVID-19. As such, to estimate the number of unreported cases, in addition to the number of reported cases, and then assume that 21% of all cases (reported and unreported) get tested is unnecessary as the individuals getting tested are captured by the reported cases. Furthermore, the sponsor’s estimate of the number of cases getting tested was obtained from a sample of community-dwelling adults aged 51 and older residing in the US who were asked “Have you been tested for the coronavirus?” from June 2020 to November 2020.40 It is unlikely that the methodology and time period captured by Sun et al. accurately reflects current testing practices in Canada considering the decreased availability of COVID-19 tests and the change in testing practices.
Third, the population considered at high risk of progression was divided into 2 groups according to the PINETREE trial: 1) patients less than 60 years of age with at least 1 underlying disease or immunocompromised and 2) patients aged 60 or older.44 All individuals 60 years of age or older were considered high-risk. To estimate the proportion of patients less than 60 with at least 1 underlying disease or immunocompromised, the sponsor assumed that this population was equivalent to the proportion of adults 18 to 59 years old with either obesity, chronic obstructive pulmonary disease, heart disease, high blood pressure, diabetes, dementia, suffer from the effects of a stroke, or current cancer.39 CDA-AMC notes that while the chronic and immunocompromising conditions are fairly consistent with current guidelines,45,46 clinical expert input received for this review noted that patients that are considered at high risk of progression primarily include those with severe immune suppression, specifically those who do not have functional B-cells, as these are the patient’s being hospitalized because of COVID-19. As such, clinical expert input commented that a much narrower definition of high-risk is employed in practice and thus the sponsor’s estimate of 27.8% likely overestimates the proportion of patients that would be considered at risk. Additionally, clinical expert input noted that assuming all individuals 60 years and older are at high risk of progression is not reflective of current practice. Input noted that age 70 is more reflective of the population eligible for treatment, as noted by current guidelines.45,46 Furthermore, the sponsor used all COVID-19 data available at the time of their submission to calculate the age distribution of patients. It is more appropriate to use data from the same period used for the number of cases reported.
Fourth, the sponsor estimated that 36.5% of patients seek treatment based on a study from the US that estimated the proportion of adults 65 years of age or older who sought treatment from March 2021 to August 2022.41 Clinical expert input commented that this was likely an overestimate further stating that based on the sponsor’s definition of patients at high risk of progression, 1% would be more reflective of the proportion of patients seeking treatment. However, CDA-AMC notes that this parameter is highly uncertain as the proportion of patients seeking treatment will likely be linked to disease severity, which fluctuates over time based on vaccination rates, prior exposure, and the COVID-19 variant.
Lastly, the sponsor assumed that 5% of patients are not eligible for oral antiviral therapy (nirmatrelvir-ritonavir). Clinical expert input noted that the proportion of patients who cannot take nirmatrelvir-ritonavir may be as high as 25%.
In the CDA-AMC base case, CDA-AMC revised the number of reported cases and the age distribution of cases based on data from April 2023 to March 2024, adjusted the age threshold for high-risk patients to 70 years and older and 12 to 69 with an immunocompromising or chronic condition, removed the number of COVID-19 cases not reported and assumed that all reported cases were tested for COVID-19. CDA-AMC notes that these base-case changes decreased the eligible population size from 2,010 to 1,761.
CDA-AMC explored uncertainty in the proportion of patients not eligible for oral antiviral therapy and the proportion of patients seeking treatment in scenario analyses.
CDA-AMC was unable to address uncertainty in the proportion of patients with an immunocompromising or chronic condition.
The uptake of remdesivir is uncertain: The sponsor’s submitted base case assumed that 100% of eligible patients would receive remdesivir in years 1, 2, and 3. Clinician input received by CDA-AMC for this review suggested that approximately 25% of the patients eligible for treatment receive treatment as patients are often feeling better by the time a practitioner is available to assess them. Additionally, clinician input noted that 100% uptake does not meet face validity as there are rural and Northern communities in which patients would be unable to access remdesivir. Notably, in a study cited by the sponsor,41 only 1.7% of adults who tested positive for COVID-19 from March 2021 to August 2022 were treated with an antiviral therapy. CDA-AMC acknowledges that while these estimates may not be reflective of current treatment practices in Canada, it provides evidentiary support for the clinical expert statements that uptake of remdesivir is significantly less than 100%. Moreover, CDA-AMC notes that public coverage rates were not incorporated in the sponsor’s model and as such, the sponsor’s estimate implicitly assumes that 100% of all eligible patients will be covered by public drug plans.
In the CDA-AMC base case, CDA-AMC assumed 25% uptake of remdesivir in years 1, 2, and 3 and explored uncertainty in scenario analyses.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by revising the number of COVID-19 cases reported, adjusting the age distribution of COVID-19 cases, removing the number of COVID-19 cases not reported, assuming that all reported cases were tested for COVID-19, and adjusting the uptake of remdesivir.
Table 16: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Number of COVID-19 cases reported | 906,236 (based on data from March 2022 to February 2023) | 180,358 (based on data from April 2023 to March 2024) |
2. Age distribution of COVID-19 cases | Aged 12 to 59: 68% Aged 60+: 23% (date range not specified by sponsor – assumed to be based on data from 2020 to 2023) | Aged 12 to 69: 42% Aged 70+: 53% (based on data from April 2023 to March 2024) |
3a. Number of COVID-19 cases not reported | 679,677 (assumed an additional 75% of the reported cases are unreported) | None |
3b. Proportion of cases getting tested | 21% | 100% |
4. Uptake of remdesivir | 100% | 25% |
CDA-AMC base case | reanalysis 1 + 2 + 3a + 3b + 4 | |
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17. In the CDA-AMC base case, the 3-year budget impact of reimbursing remdesivir for nonhospitalized COVID-19 patients (weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk of progression to severe COVID-19, including hospitalization and death, is estimated to be $3,489,179 ($1,163,060 in each of year 1, year 2, and year 3). The CDA-AMC base case revised the eligible population size from 2,010 to 1,761.
Table 17: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | 3-year total ($) |
|---|---|
Submitted base case | 15,935,256 |
CDA-AMC reanalysis 1: Number of COVID-19 cases reported | 3,296,104 |
CDA-AMC reanalysis 2: Distribution of COVID-19 cases | 24,796,982 |
CDA-AMC reanalysis 3a: Number of COVID-19 cases not reported | 9,105,861 |
CDA-AMC reanalysis 3b: Proportion of cases getting tested | 75,882,171 |
CDA-AMC reanalysis 4: Uptake of remdesivir | 3,983,814 |
CDA-AMC base case | 3,489,179 |
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 17:
Assuming 25% of patients are not eligible for oral antiviral therapy (nirmatrelvir-ritonavir).
Assuming the proportion of cases seeking treatment is 1.7% based on a study by Kojima et al. which found that 1.7% of patients received an antiviral treatment for COVID-19.41 As this assumption intrinsically considers uptake, remdesivir uptake was set to 100%.
Assuming 75% uptake of remdesivir in years 1, 2, and 3.
Assuming that the price of remdesivir is reduced by 82% (CDA-AMC’s estimated price reduction from the cost-utility analysis).
Table 18: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 5,311,752 | 5,311,752 | 5,311,752 | 15,935,256 | |
Budget impact | 0 | 5,311,752 | 5,311,752 | 5,311,752 | 15,935,256 | |
CDA-AMC base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 1,163,060 | 1,163,060 | 1,163,060 | 3,489,179 | |
Budget impact | 0 | 1,163,060 | 1,163,060 | 1,163,060 | 3,489,179 | |
CDA-AMC scenario analysis 2: 25% not eligible for oral antiviral therapy | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 5,815,299 | 5,815,299 | 5,815,299 | 14,445,897 | |
Budget impact | 0 | 5,815,299 | 5,815,299 | 5,815,299 | 14,445,897 | |
CDA-AMC scenario analysis 1: 1.7% seeking treatment and 100% uptake | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 217,893 | 217,893 | 217,893 | 653,620 | |
Budget impact | 0 | 217,893 | 217,893 | 217,893 | 653,620 | |
CDA-AMC scenario analysis 1: 75% uptake | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 3,489,179 | 3,489,179 | 3,489,179 | 10,467,538 | |
Budget impact | 0 | 3,489,179 | 3,489,179 | 3,489,179 | 10,467,538 | |
CDA-AMC scenario analysis 3: 82% price reduction | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 209,351 | 209,351 | 209,351 | 628,052 | |
Budget impact | 0 | 209,351 | 209,351 | 209,351 | 628,052 |
BIA = budget impact analysis.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.