Drugs, Health Technologies, Health Systems
Reimbursement Review
Remdesivir (Veklury)
Sponsor: Gilead Sciences Canada, Inc.
Therapeutic area: COVID-19 in hospitalized patients
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
aHR
adjusted hazard ratio
ALT
alanine aminotransferase
AST
aspartate aminotransferase
CI
confidence interval
CRP
c-reactive protein
ECMO
extracorporeal membrane oxygenation
eGFR
estimated glomerular filtration rate
EHR
electronic health record
ESKD
end-stage kidney disease
HFNC
high-flow nasal cannula
HR
hazard ratio
ICD-10-CM
International Classification of Diseases, 10th revision, Clinical Modification
ICU
intensive care unit
IPTW
inverse probability of treatment weighting
IQR
interquartile range
ITT
intention to treat
KM
Kaplan-Meier
NIPPV
noninvasive positive pressure ventilation
OR
odds ratio
PCFS
post–COVID-19 functional status
PS
propensity score
RCT
randomized controlled trial
RNA
ribonucleic acid
RR
relative risk
RRR
risk of recovery ratio
RWE
real-world evidence
SAE
serious adverse event
SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
SD
standard deviation
SOC
standard of care
VOC
variant of concern
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on Application Submitted for Review
Item | Description |
|---|---|
Drug product | Remdesivir (Veklury), 100 mg/vial, IV infusion |
Sponsor | Gilead Sciences Canada, Inc. |
Indication | For the treatment of COVID-19 in hospitalized adults and pediatric patients (at least 4 weeks of age and weighing at least 3 kg) with pneumonia requiring supplemental oxygen |
Reimbursement request | For the treatment of COVID-19 in hospitalized patients ≥ 12 years of age (weighing at least 40 kg) with pneumonia requiring supplemental oxygen |
Health Canada approval status | NOC |
Health Canada review pathway | For use in relation to COVID-19 |
NOC date | July 27, 2020 |
Recommended dose | In patients weighing at least 40 kg, a single loading dose of 200 mg on day 1 followed by 100 mg maintenance doses on day 2 onward, for at least 5 days and not more than 10 days |
NOC = Notice of Compliance.
Introduction
COVID-19 is an illness caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).1 The rapid global spread of the virus led to a pandemic, as declared by WHO on March 11, 2020. Subsequently, the proliferation of COVID-19 has presented significant challenges to health care systems globally, including those in Canada.2,3 As of April 3, 2024, the cumulative reported COVID-19 cases and deaths in Canada were 4,946,090 and 59,034, respectively, and the weekly percentage of positive cases out of the total tests conducted was 5.2%.4 From April 2022 to March 2023 in Canada, according to the Canadian Institute for Health Information, there were 120,524 hospitalizations due to COVID-19;5 the year prior (April 2021 to March 2022) there were 125,986 hospitalizations due to COVID-19.5 In 2022 to 2023, of patients admitted to hospital due to COVID-19, 10% died in the facility and 13% were admitted to the intensive care unit (ICU).5 Of those admitted to the ICU, 39% received ventilation.5 The estimated total cost of COVID-19 hospitalizations in Canada in 2022 to 2023 was approximately $2.9 billion, and costs continue to increase each fiscal year.5
People with symptomatic COVID-19 have a wide range of symptoms, ranging from no or mild symptoms (e.g., fever, cough, headache, malaise, muscle pain, nausea, vomiting, loss of taste and smell) in most cases to severe symptoms, including pneumonia and acute respiratory distress syndrome. Severe cases are also associated with pulmonary embolism, arrhythmia, cardiovascular shock, and heart damage or heart attack. At its worst, COVID-19 can lead to critical illness, where individuals experience respiratory failure, septic shock, and/or organ dysfunction known to be associated with high morbidity and mortality.6-12 Mortality risk estimates reported by WHO for people with nonsevere disease are 0.6% for those at high risk of hospitalization, 0.3% for those at moderate risk of hospitalization, and 0.05% for those at low risk of hospitalization.13
Typical COVID-19 symptoms may appear 2 to 14 days after exposure, and they generally resolve within 14 days, but in severe cases, symptoms can last for over a month.14 Currently, the risk factors for progressing to severe disease are not the same as during the early stages of the pandemic;7 as population immunity has built up over time, the characteristics of patients being hospitalized due to COVID-19 have changed. The common risk factors for progression in COVID-19 now include older age, chronic comorbidities, cerebrovascular and cardiovascular disease, diabetes, hypertension, kidney failure, dementia, neurodevelopmental disorders, cancer, and a history of smoking.6,15
Remdesivir is a nucleotide prodrug of an adenosine triphosphate analogue that competes with the natural adenosine triphosphate substrate for incorporation into nascent ribonucleic acid (RNA) chains by the SARS-CoV-2 RNA-dependent RNA polymerase and inhibits viral replication by terminating RNA transcription prematurely.16
IV remdesivir has been approved by Health Canada (Notice of Compliance) for the treatment of COVID-19 in:
hospitalized adults and pediatric patients (aged 4 weeks or older and weighing at least 3 kg) with pneumonia requiring supplemental oxygen
nonhospitalized adults and pediatric patients (weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing who are at high risk for progression to severe COVID-19, including hospitalization and death.16
For this review, the sponsor has requested reimbursement of remdesivir for the treatment of COVID-19 in hospitalized patients aged 12 years or older (weighing at least 40 kg) with pneumonia requiring supplemental oxygen.
The recommended dosing regimen for remdesivir includes a single loading dose of 200 mg on day 1, then 100 mg once daily from day 2 onward. For hospitalized patients with pneumonia requiring supplemental oxygen, treatment is recommended daily for at least 5 days and not more than 10 days. Remdesivir is administered via IV infusion, and its administration should take place under conditions where the management of severe hypersensitivity reactions (e.g., anaphylaxis) is possible.16
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of remdesivir for the treatment of COVID-19 in hospitalized patients aged 12 years or older (weighing at least 40 kg) with pneumonia requiring supplemental oxygen.
Input From Patients, Clinicians, and Drug Plans
The information in this section is a summary of the input provided by the patient and clinician groups who responded to the Canada’s Drug Agency (CDA-AMC) call for input and from the clinical expert consulted by CDA-AMC for the purpose of this review.
Patient Input
No patient groups provided input for this review.
Clinician Input
Input From Clinical Expert Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of COVID-19 in the inpatient setting.
Unmet Needs
The clinical expert described COVID-19 as no longer being a significant cause of hospitalization and death as a result of the evolution of the virus since the beginning of the pandemic. The expert noted that an ideal intervention for COVID-19 focuses on prevention rather than treatment. For patients who do require treatment, a treatment option that is effective across all disease severities would be ideal, and an oral delivery of the medication would be ideal. A significant treatment goal is also to reduce unnecessary antimicrobial use in COVID-19. However, there is an information gap in clinical data relevant to the currently prevalent variants of the virus, as the majority of clinical evidence was generated with early variants and may no longer apply.
Place in Therapy
The clinical expert noted that remdesivir may have infrequent application, given the lower prevalence of hospitalization and death caused by current COVID-19 variants. Remdesivir would be used in combination with other treatments as a first-line treatment based on WHO’s Living Guidance for Clinical Management of COVID-19. The clinical expert also noted that remdesivir is unique in its antiviral action as, unlike other therapies for COVID-19, it does not target host immune response. The clinical expert stated that remdesivir would not change clinical practice as it has been rarely used since the early stages of the pandemic.
Patient Population
As this review is focused on the inpatient application of remdesivir, outpatient populations will not be discussed.
Among inpatients, those most in need of an intervention are those at risk of death. Diagnosis of COVID-19 is based on polymerase chain reaction testing (more accurate, more expensive, less accessible) or antigen testing (less accurate, less expensive, more accessible). There is a lack of current data to indicate which patients would most benefit from remdesivir, as the available data primarily evaluate early COVID-19 variants and largely unvaccinated patient populations.
However, the clinical expert noted that patients who are sick enough to require oxygen support as a result of COVID-19, but who have not yet progressed to needing ventilation, may be the most likely to benefit from remdesivir. This observation is reflected in trial data, but as these trials were conducted in populations with different variants, there are serious limitations in the generalizability of the results. Nonetheless, speculatively, the clinical expert discussed whether the reason for this observation in the trial data may be related to the pathogenesis of COVID-19 (i.e., earlier stages of the disease are virologically mediated, while later stages of the disease are immune mediated), making the application of an antiviral such as remdesivir less helpful in patients whose medical distress is caused by immune response rather than virological activity.
Assessing the Response Treatment
The clinical expert identified that the key outcomes (among patients already admitted to hospital) are need for oxygen or organ support and rate of mortality. Meaningful response would be a change in the status of oxygen or organ support requirements. The status of these requirements does not vary by physician interpretation; they are objective outcomes. Clinical symptoms and viral load are not relevant clinical outcomes, and they do not correlate with the objective outcomes.
Discontinuing Treatment
The clinical expert stated that remdesivir would generally be given for the entire treatment course (5 days or 10 days), and it would not be stopped due to progression or additional treatments, although it may be stopped as a result of adverse events (AEs) if necessary. Dosing is 200 mg on the first day, followed by 100 mg daily. The expert noted that the shortest effective duration of treatment should be used.
Prescribing Considerations
The clinical expert stated that inpatient treatment with remdesivir would be prescribed in hospital settings, with no need for a specialist to diagnose or treat.
Clinician Group Input
One clinician group, the Ontario Health Infectious Diseases Advisory Committee, responded to the CDA-AMC call for clinician group input. Information was gathered through discussion with 4 clinicians.
According to the clinician group, the treatment regimen for COVID-19 for hospitalized patients includes supplemental oxygen therapy and immunomodulators such as corticosteroid, which is recommended as first-line treatment for hospitalized adults with COVID-19 requiring supplemental oxygen; Janus kinase inhibitors; or anti–interleukin 6 receptor monoclonal antibodies. Remdesivir can be added to other immunomodulatory treatments that work on the hyperinflammatory pathway that tends to drive the disease course in the later stages of illness.
The main treatment goals are to accelerate recovery; reduce the severity of symptoms and the duration of hospitalization; prevent progression to critical COVID-19 disease conditions and long-term sequelae; prevent the need for new high-flow supplemental oxygen, noninvasive ventilation (e.g., BiPAP), mechanical ventilation, or extracorporeal membrane oxygenation (ECMO); and prevent death.
The clinician group indicated that not all patients respond to currently available treatment. The group also noted some limitations with remdesivir, such as the drug formulation (IV) and the lack of randomized controlled trials (RCTs) on the effectiveness of remdesivir on all COVID-19 variants, especially Omicron.
According to the clinician group, hospitalized patients who require supplemental low-flow oxygen are best suited for treatment with remdesivir. Remdesivir should ideally be started early in the disease course, when viral replication predominates.
The clinician group stated that the outcomes used in clinical practice typically align with those used in clinical trials (e.g., duration of hospitalization, ICU admission, length of ICU stay, time to improvement in clinical status, progression to high-flow oxygen or noninvasive ventilation, progression to mechanical ventilation or ECMO, time to receipt of mechanical ventilation, time to clinical improvement, mortality, length of hospital stay, serious AEs [SAEs]) and that improvements in these outcomes would be considered clinically meaningful responses.
According to the clinician group, the factors that should be considered when deciding to discontinue treatment with remdesivir include disease progression to critical COVID-19, severe allergic reaction, adverse drug reaction, and AEs.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may affect their ability to implement a recommendation. For the review of remdesivir, the drug plans provided questions pertaining to relevant comparators, considerations for initiation of therapy, considerations for prescribing of therapy, and system and economic issues (Table 3).
Clinical Evidence
Systematic Review
Description of Studies
The primary sources of evidence for review included 5 studies, of which 3 were RCTs conducted in adults (ACTT-1,17 WHO Solidarity,18 and Wang et al. [2020]19), 1 was an RCT conducted in patients aged 12 years or older (Spinner et al. [2020]20), and 1 was a single-arm, open-label study in pediatric patients (CARAVAN).21 The studies were all conducted in patients hospitalized with COVID-19 requiring inpatient treatment. The ACTT-1 study (N = 1,062) was a double-blind, placebo-controlled, multicentre, international, phase III RCT in adults aged 18 years or older admitted to hospital with confirmed COVID-19. The study by Wang et al. (2020) (N = 237) was a double-blind, placebo-controlled, multicentre RCT conducted in 10 hospitals in China in patients aged 18 years or older admitted to hospital with confirmed COVID-19. The WHO Solidarity trial (N = 8,320 for remdesivir and its control group) was an open-label, standard of care (SOC)-controlled RCT of several putative treatments for COVID-19 across the globe in adults with definite COVID-19, although only the remdesivir group and its associated control group are described in this report. The study by Spinner et al. (2020) (N = 596) was an open-label, multicentre, international RCT evaluating 5 days or 10 days treatment with remdesivir compared with SOC in hospitalized patients aged 12 years or older with moderate COVID-19 pneumonia. The CARAVAN study (N = 53) was a single-arm, open-label, phase II/III, international study in pediatric patients, of which only those in cohort 1 (N = 12) were aged 12 years or older and weighed at least 40 kg. The outcomes of interest from these studies, for the purpose of this review, were mortality, duration of hospitalization, time to recovery or clinical improvement, and initiation of ventilation.
Efficacy Results
Mortality
In the ACTT-1 intention-to-treat (ITT) population, the risk of death by day 15 was lower in the remdesivir group than in the placebo group (hazard ratio [HR] = 0.55; 95% confidence interval [CI], 0.36 to 0.83; P = 0.004). At day 29 the difference between groups was less apparent (HR = 0.73; 95% CI, 0.52 to 1.02; P = 0.066). The median time to death through day 15 or day 29 was not estimable for either treatment group in the ITT or as-treated populations. In ad hoc subgroup analyses of mortality by disease stratum as defined by level of oxygen support required, the greatest differences in percentages of deaths among participants with a known mortality status at day 29 in the remdesivir group compared with the placebo group were observed in the subgroup of patients requiring supplemental oxygen but not high-flow oxygen or ventilation at baseline. The percentage of deaths among patients receiving low-flow oxygen was 4.1% in the remdesivir group (9 of 222 participants) versus 12.8% in the placebo group (25 of 195 participants) (HR = 0.30; 95% CI, 0.14 to 0.64; P < 0.001 [without adjustments for multiplicity]) and among patients in the severe disease stratum was 12.5% in the remdesivir group (57 of 457 participants) versus 16.3% in the placebo group (74 of 453 participants). Patients with a baseline ordinal score of 5 (which represents patients requiring supplemental oxygen but not high-flow oxygen or ventilation) also represent by far the most populous subgroup by ordinal score in the ACTT-1 study.
In the study by Wang et al. (2020), mortality was similar between the treatment groups at day 28 in the ITT population. In the remdesivir group, 22 of 158 patients (14%) died, and in the placebo group, 10 of 78 patients (13%) died, yielding a difference of 1.1% (95% CI, –8.1% to 10.3%; P value not reported). Mortality was similar between treatment groups in the subgroup analyses of patients who used remdesivir “early” (within 10 days of symptom onset) or “late” (more than 10 days after symptom onset), but the numerical results differed in direction: in the early-use subgroup, mortality was numerically higher in the placebo group, but in the late-use subgroup, mortality was numerically higher in the remdesivir group.
In the WHO Solidarity trial, of 8,275 patients in the overall remdesivir analyses, 602 of 4,146 patients (14.5%) assigned to remdesivir and 643 of 4,129 patients (15.6%) assigned to the control group died (relative risk [RR] = 0.91; 95% CI, 0.82 to 1.02; P = 0.12). These analyses of in-hospital mortality include 15 palliative discharges in the remdesivir group and 11 in the control group. Analyses were also subdivided by oxygen support requirements at baseline, and the subgroup of patients who were already on oxygen (low or high flow) but not ventilated at baseline experienced benefit from remdesivir compared with the control group in terms of in-hospital mortality (RR = 0.87; 95% CI, 0.76 to 0.99; P = 0.03).
In the study by Spinner et al. (2020), in the 10-day remdesivir group (N = 193), 5-day remdesivir group (N = 191), and SOC group (N = 200), a total of 3 (2%), 2 (1%), and 4 (2%) patients died from any cause through 28 days of the trial. The Kaplan-Meier (KM) estimates of all-cause mortality at day 28 were 1% (95% CI, 0.0% to 2.6%; P = 0.43) compared with SOC for the 5-day remdesivir group, 2% (95% CI, 0.0% to 3.6%; P = 0.72) compared with SOC for the 10-day remdesivir group, and 2% (95% CI, 0.1% to 4.1%) for the SOC group.
In cohort 1 (N = 12) of the CARAVAN study, there was 1 treatment-emergent death (8.3% of patients).
Duration of Hospitalization
Only the ACTT-1 study reported a benefit of remdesivir on the duration of hospitalization. The median duration of initial hospitalization, including imputations for participants who died, was 12 days (interquartile range [IQR], 6 to 28 days) in the remdesivir group (n = 541) and 17 days (IQR, 8 to 28 days) in the placebo group (n = 521), yielding a median difference of 5 days shorter with remdesivir, and a 95% CI of 2.3 to 7.7 days.
In contrast, the WHO Solidarity trial reported that allocation to remdesivir delayed discharge by about 1 day during the 10-day treatment period, owing to the duration of the treatment regimen itself potentially delaying discharge.
Both Wang et al. (2020) and Spinner et al. (2020) reported that no difference was observed between treatment arms in the duration of hospitalization.
In cohort 1 of the CARAVAN study, the mean duration of hospitalization from day 1 (days from first dose to date discharged alive; n = 9) was 12 days (standard deviation [SD] = 5.5 days) and the median was 12 days (IQR, 8 to 15 days; range, 6 to 24 days).
Time to Recovery or Clinical Improvement
The results from the ACTT-1 study were stratified by disease severity within the ITT population; “mild-moderate” disease was defined as having a blood oxygen saturation of more than 94% and a respiratory rate of fewer than 24 breaths per minute without supplemental oxygen, and “severe” disease was defined as requiring mechanical ventilation, requiring oxygen, having blood oxygen saturation equal to or less than 94% on room air, or experiencing tachypnea (respiratory rate ≥ 24 breaths per minute). In patients in the mild-moderate disease stratum at randomization (remdesivir: n = 82; placebo: n = 77), the median time to recovery was 5 days (95% CI, 4 to 6 days) in the remdesivir group and 7 days (95% CI, 5 to 9 days) in the placebo group (risk of recovery ratio [RRR] = 1.10; 95% CI, 0.80 to 1.53). In patients in the severe disease stratum at randomization, the median time to recovery was 12 days (95% CI, 10 to 14 days) in the remdesivir group versus 19 days (95% CI, 16 to 21 days) in the placebo group (RRR = 1.34; 95% CI, 1.14 to 1.58). In patients with any disease severity, the median time to recovery in the ITT population was 10 days (95% CI, 9 to 11 days) in the remdesivir group (n = 541) and 15 days (95% CI, 13 to 18 days) in the placebo group (n = 521). Subgroup analyses were also conducted according to ordinal score at baseline, which was defined by the level of oxygen support required. Only patients who required supplemental oxygen but not high-flow oxygen or any level of ventilation (i.e., patient at ordinal score level 5) demonstrated a benefit of remdesivir in time to recovery; this was also the most populous subgroup (remdesivir: n = 232; placebo: n = 203).
In the ITT population of the study by Wang et al. (2020), the time to clinical improvement in the remdesivir group (median = 21.0 days; IQR, 13.0 to 28.0 days) was not significantly different from that of the control group (median = 23.0 days; IQR, 15.0 to 28.0 days; HR = 1.23; 95% CI, 0.87 to 1.75).
In the study by Spinner et al. (2020), there were no significant differences between the 10-day remdesivir group and the SOC group for time to 2-point or greater improvement in clinical status (HR = 1.16; 95% CI, 0.93 to 1.43), time to 1-point or greater improvement in clinical status (HR = 1.10; 95% CI, 0.90 to 1.36), time to recovery (HR = 1.11; 95% CI, 0.90 to 1.37), or time to modified recovery (HR = 1.10; 95% CI, 0.90 to 1.36). There were also no significant differences between the 5-day remdesivir group and the SOC group for time to 2-point or greater improvement in clinical status (HR = 1.15; 95% CI, 0.93 to 1.42), time to 1-point or greater improvement in clinical status (HR = 1.19; 95% CI, 0.97 to 1.47), time to recovery (HR = 1.18; 95% CI, 0.96 to 1.45), or time to modified recovery (HR = 1.19; 95% CI, 0.19 to 1.46).
The median time to recovery in cohort 1 of the CARAVAN study was 12 days (IQR, 6 to 24 days).
This outcome was not assessed in the WHO Solidarity trial.
Initiation of Ventilation
Only the ACTT-1 study and the WHO Solidarity trial reported this outcome.
In the ACTT-1 study, the incidence rate of new noninvasive ventilation or high-flow oxygen use among patients who were not already on these supports (nor ventilated) at baseline, was 0.17 (95% CI, 0.13 to 0.22) in the remdesivir group and 0.24 (95% CI, 0.19 to 0.30) in the placebo group. The incidence rate in the remdesivir group was numerically lower, but the 95% CIs of each group overlapped. The incidence rate of new invasive mechanical ventilation or ECMO use among patients not already on these supports at baseline was 0.13 (95% CI, 0.10 to 0.17) in the remdesivir group and 0.23 (95% CI, 0.19 to 0.27) in the placebo group. The incidence rate in the remdesivir group was numerically lower, and the 95% CIs of each group did not overlap.
In the WHO Solidarity trial, assignment to remdesivir was associated with a lower rate of progression to ventilation (event RR = 0.88; 95% CI, 0.77 to 1.00; P = 0.04) and with a lower composite outcome of death or ventilation (event RR = 0.84; 95% CI, 0.75 to 0.93; P = 0.001). For both outcomes, results for the subgroup of patients not receiving oxygen support at entry had an associated 95% CI that crossed the null, whereas results for the subgroup of patients who were receiving low-flow or high-flow oxygen at entry showed a statistically significant benefit of remdesivir. The latter subgroup was also much larger, and so this subgroup (patients already on low-flow or high-flow oxygen at baseline) appears to drive the observed benefit of remdesivir for this outcome. In the Canadian substudy, CATCO,22 among patients not mechanically ventilated at baseline, 8.0% of those assigned to the remdesivir group required mechanical ventilation during the study, compared to 15.0% of those assigned to the SOC group (RR = 0.53; 95% CI, 0.38 to 0.75).
Although duration of oxygen support or ventilation was not selected as a key outcome of interest based on consultation with the clinical expert, related outcomes are reported in Appendix 1, Table 26. The ACTT-1 study reported the median days on oxygen, on noninvasive ventilation or high-flow oxygen, or on invasive mechanical ventilation or ECMO. Although statistical comparisons were not conducted and the IQRs overlapped between groups, the median days on oxygen or on invasive mechanical ventilation or ECMO were lower in the remdesivir group than in the placebo group. The median days on noninvasive ventilation or high-flow oxygen was the same in both groups. The CATCO study reported a significant benefit associated with allocation to remdesivir in terms of mean oxygen-free days and mean ventilator-free days at day 28. Wang et al. (2020)19 reported a lower median number of days of invasive mechanical ventilation and of oxygen support in the remdesivir group than in the placebo group, although again the IQRs overlapped. Spinner et al. (2020)20 reported no significant difference between either of the remdesivir groups and the SOC group in the duration of oxygen support. There is therefore some evidence to suggest a modest benefit of remdesivir on the duration of some forms of oxygen support, but the magnitude is uncertain and there is inconsistency between the studies.
Harms Results
Remdesivir was generally well tolerated in all the included studies. The proportion of patients who experienced at least 1 AE ranged from 51% to 64% across the 4 RCTs and was 91.7% in cohort 1 of the CARAVAN study. The studies differed substantially in which particular AEs were reported, but there was a trend across the trials of focus on biomarkers related to kidney and liver function, hyperglycemia, and some clinical AEs, such as headache, constipation, pyrexia, and diarrhea. Where reported, AEs were generally similar between treatment groups, although in some cases there were numerically more AEs in the placebo or SOC group than the remdesivir group.
In the 4 RCTs, the proportion of patients who experienced at least 1 SAE ranged from 5% in both remdesivir groups of the study by Spinner et al. (2020) to 32% in the placebo group of the ACTT-1 study. In cohort 1 of the CARAVAN study, 5 patients (41.7%) experienced an SAE.
In the ACTT-1 study, a substantially higher proportion of patients experienced SAEs in the placebo arm (32%) than in the remdesivir arm (25%). This was also the case in the study by Wang et al. (2020) (26% in the placebo arm versus 18% in the remdesivir arm). In the study by Spinner et al. (2020), in both remdesivir groups — 10 day and 5 day — 5% of patients experienced at least 1 SAE, while in the SOC group 9% of patients experienced at least 1 SAE. The WHO Solidarity trial did not report this outcome.
The studies were inconsistent with regard to which SAEs they reported. The most common AE reported in the ACTT-1 study and the study by Wang et al. (2020) was respiratory failure, which occurred in 7% and 10% of patients in the remdesivir groups, respectively, and in 11% and 8% of patients in the placebo groups, respectively.
Withdrawals due to AEs were relatively high in the ACTT-1 study, occurring in 11.1% of patients in the remdesivir group and 15% of patients in the placebo group. In the study by Wang et al. (2020), 15% and 13% of patients in the remdesivir and placebo groups, respectively, withdrew due to AEs. In the WHO Solidarity trial, 14.5% and 15.6% in the remdesivir and control groups, respectively, withdrew due to AEs. In the study by Spinner et al. (2020), the rate of withdrawal due to AEs was lower: 2% in the 10-day remdesivir group, 1% in the 5-day remdesivir group, and 2% in the SOC group. In the CARAVAN study, 1 patient (8.3%) withdrew due to AEs.
Mortality is discussed in depth in the Efficacy section of this report.
Critical Appraisal
This review included 5 clinical trials: 4 RCTs and 1 single-arm study. Of the 4 RCTs, 2 were double blind (ACTT-1 and Wang et al. [2020]). The WHO Solidarity trial and the study by Spinner et al. (2020), being open label, have an elevated risk of bias with regard to subjective outcomes and a potential for different treatment decisions by clinicians, which was observed in some instances (i.e., patients in the control arm were more likely to receive other putative treatments for COVID-19 such as in the Spinner et al. [2020] and WHO Solidarity trials).
The authors of the WHO Solidarity trial18 criticized the balance of the treatment groups in the ACTT-1 study and suggested that patients with “good” prognosis (i.e., patients who were unventilated at baseline) were overrepresented in the remdesivir group compared to the placebo group. Patients with an ordinal score of 5, which in the ACTT-1 study represents those hospitalized and requiring supplemental oxygen (but not high-flow oxygen or ventilation), formed the largest subgroup in the ACTT-1 study: 43% of the remdesivir group and 39% of the placebo group fit into this category. However, the clinical expert consulted by CDA-AMC did not feel this was an important difference in terms of risk of bias. Subgroup analysis results for outcomes related to clinical recovery in the ACTT-1 study demonstrated that only the subgroup of patients who were hospitalized and required supplemental oxygen (but not high-flow oxygen or ventilation) showed a benefit of remdesivir over placebo; all other clinical status subgroups (i.e., patients not requiring any oxygen support, patients requiring high-flow oxygen or noninvasive ventilation, and patients requiring invasive ventilation or ECMO) each demonstrated no significant benefit in time to recovery. Similarly, results for mortality in the ACTT-1 study were the strongest in the subgroup of patients who were hospitalized and required supplemental oxygen (but not high-flow oxygen or ventilation). The randomization stratification categories in the ACTT-1 study (“mild-moderate” and “severe”) were broad, the latter hypothetically encompassing patients from all reported ordinal score subgroups or, in other words, all levels of oxygen support requirements.
The results of the WHO Solidarity trial demonstrated a benefit in mortality for the subgroup of patients who were already on oxygen (low or high flow, but not ventilated). As the ACTT-1 study and the WHO Solidarity trial differed in which subgroup contained patients on high-flow oxygen, it is uncertain whether there is a benefit of remdesivir in these patients or whether the apparent benefit is driven entirely by patients on low-flow oxygen. When patients on high-flow oxygen were grouped with those receiving noninvasive ventilation in the ACTT-1 study, there was uncertainty about the benefit of remdesivir on mortality in this subgroup; however, when patients on high-flow oxygen were grouped with those on low-flow oxygen in the WHO Solidarity trial, there was an apparent benefit of remdesivir on mortality in this subgroup. Taken together, the subgroup of patients receiving low-flow oxygen — and perhaps including those receiving high-flow oxygen as well, but this was inconsistent between the studies — was both the largest subgroup and the one most likely to benefit from treatment with remdesivir, at least in time to recovery or clinical improvement (the ACTT-1 study) and mortality (both the ACTT-1 study and the WHO Solidarity trial). As such, the imbalance between groups in the ACTT-1 study may be clinically important and may bias the results in favour of remdesivir; while it is otherwise possible the other subgroups were too small to demonstrate a benefit, the larger WHO Solidarity trial confirms the findings.
The included studies were each conducted in the early stages of the COVID-19 pandemic. There are substantial concerns regarding the external validity and generalizability of every study included in this review because of the fast-evolving nature of the pandemic and the virus itself: prevalent variants, levels of vaccination, and clinical outcomes in today’s world are substantially different than those observed in the early pandemic. The clinical expert consulted by CDA-AMC highlighted that the current need for remdesivir is infrequent as relatively few patients are now presenting with COVID-19 severe enough to warrant hospitalization, and the profile of patients at highest risk for hospitalization and death may have changed. The clinical expert expressed that the differences in variants and levels of vaccination are both critically important and undermine the ability to generalize the results from these trials to a current population.
Additionally, background care and SOC were often sparsely defined in the studies, and it is therefore uncertain whether those care regimens are representative of those experienced by the current patient population of interest.
Long-Term Extension Studies
No long-term extension studies were submitted.
Indirect Comparisons
No indirect comparisons were submitted.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
The sponsor noted the following gaps in the submitted evidence: limited evidence on the efficacy and safety of remdesivir in a real-world setting, for immunocompromised patients, for patients discharged after hospitalization for COVID-19, for post–COVID-19 condition (referred to as “long COVID syndrome” in the study), for patients with renal disease, in combination with dexamethasone among hospitalized patients and vaccinated nonhospitalized patients, and across different COVID-19 variants.
To strengthen the totality of the evidence for remdesivir and address the evidence gaps, the sponsor submitted 9 real-world observational studies: Mozaffari et al. (2023),23 Mozaffari et al. (2024),24 Finn et al. (2022),25 Boglione et al. (2022),26 Kikuchi et al. (2021),27 Seethapathy et al. (2022),28 Seethapathy et al. (2023),29 Mozaffari et al. (2023),30 and Garibaldi et al. (2021).31
The study by Mozaffari et al. (2023)23 was a retrospective cohort study that examined the effect of remdesivir on the outcomes of 14-day and 28-day mortality among in-hospital patients with COVID-19 who required supplemental oxygen, including low-flow oxygen, high-flow oxygen or noninvasive ventilation, and invasive mechanical ventilation or ECMO, across variant of concern (VOC) periods, in a large US health care network. The study by Mozaffari et al. (2024)24 (N = 440 to 601) was a retrospective study evaluating the effect of remdesivir among adult patients discharged after hospitalization for COVID-19 on 30-day COVID-19–related and all-cause readmission across different variants and time periods. The study by Finn et al. (2022)25 (N = 2,062) was a retrospective study evaluating the effect of remdesivir in patients discharged after hospitalization for COVID-19 for the outcomes of length of hospital stay, 30-day readmission, and postdischarge 30-day all-cause mortality. The study by Boglione et al. (2022)26 (N = 449) was a prospective study that aimed to analyze the prevalence of and risk factors for post–COVID-19 condition in patients hospitalized for COVID-19. The study included patients hospitalized at a single hospital in Italy, who were followed for at least 6 months postdischarge. The study by Kikuchi et al. (2021)27 (N = 1,010) was a registry study evaluating risk factors for mortality in patients receiving dialysis who were hospitalized for COVID-19. The study by Seethapathy et al. (2022)28 (N = 62) was a retrospective cohort study that examined the association between remdesivir and AEs in patients hospitalized for COVID-19 with an estimated glomerular filtration rate (eGFR) less than 30 mL/min/1.73 m2 within the Mass General Brigham health care system in the Boston, Massachusetts, region of the US. The study by Seethapathy et al. (2023)29 (N = 350) was a retrospective study evaluating the safety of remdesivir in relation to adverse kidney outcomes in patients hospitalized for COVID-19 with an eGFR between 15 mL/min/1.73 m2 and 60 mL/min/1.73 m2. The study by Mozaffari et al. (2023)30 (N = 28,338) was a retrospective cohort study that examined, across different levels of oxygen requirements and different VOC periods in a large US health care network, the effect of remdesivir on the outcomes of 14-day and 28-day mortality among in-hospital patients with COVID-19 who were immunocompromised. The study by Garibaldi et al. (2021)31 (18,328 pairs of patients treated with and not treated with remdesivir) was a retrospective study that included a sensitivity analysis of remdesivir plus dexamethasone versus dexamethasone alone in patients hospitalized for COVID-19 across different VOC periods for the outcomes of time to improvement and time to death.
Results
Study by Mozaffari et al. (2023)23
In the low-flow oxygen group of the study by Mozaffari et al. (2023), 4,315 patients (6.4%) who received remdesivir and 5,918 matched patients (8.8%) who did not receive remdesivir died within 14 days after hospitalization for COVID-19. By 28 days, 6,641 patients (9.8%) from the remdesivir group and 8,305 patients (12.3%) from the matched nonremdesivir group had died within 28 days after hospitalization for COVID-19 across VOC periods. The in-hospital mortality adjusted hazard ratio (aHR) among patients requiring low-flow oxygen across VOC periods in the remdesivir group compared to the nonremdesivir group was 0.72 (95% CI, 0.66 to 0.79) at 14 days and 0.79 (95% CI, 0.73 to 0.85) at 28 days. Estimates were adjusted for covariates (age, admission month, admission venue [ICU versus general ward], and baseline concomitant COVID-19 treatments [anticoagulants, convalescent plasma, corticosteroids, baricitinib, tocilizumab]).
Among the patients receiving high-flow oxygen or noninvasive ventilation, 5,853 (16.8%) who received remdesivir and 6,770 (19.4%) who did not receive remdesivir died within 14 days. By 28 days, 9,009 such patients (25.8%) from the remdesivir group and 9,853 (28.3%) from the nonremdesivir group had died. After adjustment for covariates, the in-hospital mortality aHR among patients requiring high-flow oxygen or noninvasive ventilation across VOC periods in the remdesivir group compared to the nonremdesivir group was 0.83 (95% CI, 0.77 to 0.89) at 14 days and 0.88 (95% CI, 0.82 to 0.93) at 28 days.
Of the patients receiving invasive mechanical ventilation or ECMO, 1,157 patients (27.8%) who received remdesivir and 1,470 patients (35.3%) who did not receive remdesivir died within 14 days. By 28 days, 1,724 such patients (41.4%) from the remdesivir group and 2,105 (50.6%) from the nonremdesivir group had died. After adjustment for covariates, the in-hospital mortality aHR among patients requiring invasive mechanical ventilation or ECMO across VOC periods in the remdesivir group compared to the nonremdesivir group was 0.73 (95% CI, 0.65 to 0.82) at 14 days and 0.74 (95% CI, 0.67 to 0.82) at 28 days.23
Study by Mozaffari et al. (2024)24
The remdesivir group in the study by Mozaffari et al. (2024) had a 30-day COVID-19–related readmission rate of 3.0% and an all-cause readmission rate of 6.3%, compared with 5.4% and 9.1%, respectively, for the nonremdesivir group. After adjusting for demographics and clinical characteristics, the odds ratio (OR) for 30-day COVID-19–related readmission and all-cause readmission among the remdesivir group was 0.60 (95% CI, 0.58 to 0.62) and 0.73 (95% CI, 0.72 to 0.75), respectively. Similar patterns of OR for 30-day readmission in the remdesivir group were observed across all VOC periods.24
Study by Finn et al. (2022)25
In the study by Finn et al. (2022), remdesivir treatment was associated with a longer hospital stay, with a 3.27-day average increase among patients who were treated with remdesivir relative to patients who were not (95% CI, 2.11 to 4.44 days). This effect was most pronounced in patients with severe COVID-19 symptoms, where the increase in the length of stay was 6.70 days, but the 95% CIs crossed the null (95% CI, 0.47 to 12.92 days); patients with mild or moderate symptoms had only a slight increase in their hospital stay.
Overall, patients treated with remdesivir had a 19% reduced risk of being readmitted to the hospital within 30 days, but the 95% CIs crossed the null (RR = 0.81; 95% CI, 0.59 to 1.13). This reduction in readmission risk was pronounced in patients with mild COVID-19 symptoms, who were 69% less likely to be readmitted if they received remdesivir, with an RR of 0.31 (95% CI, 0.13 to 0.75).
Remdesivir treatment was associated with a 35% decrease in the risk of dying within 30 days of being discharged from hospital, with an HR of 0.65 (95% CI, 0.49 to 0.85).25
Study by Boglione et al. (2022)26
In the study by Boglione et al. (2022), after multivariate adjustment that considered the principal baseline parameters, ICU admission (OR = 2.551; 95% CI, 1.998 to 6.819; P = 0.019), time of hospitalization (OR = 2.255; 95% CI, 1.018 to 6.992; P = 0.016), and treatment with remdesivir (OR = 0.641; 95% CI, 0.413 to 0.782; P < 0.001) were found to be independent predictors of post–COVID-19 condition. Treatment with remdesivir led to a 35.9% reduction in the post–COVID-19 condition rate in the follow-up period.
At visit 1, of the patients who received remdesivir compared with the patients who did not, 123 versus 81 patients were not affected by post–COVID-19 condition, 27 versus 120 patients had a post–COVID-19 functional status (PCFS) score of 2 to 3, and 13 versus 85 patients with a PCFS score greater than 3 . All differences in the 2 groups were statistically significant (P < 0.001).
Survival analysis that compared the patients treated with remdesivir and those not treated with remdesivir according to the diagnosis of post–COVID-19 condition in the follow-up period found significant difference between the 2 groups (χ2 = 14.614; P < 0.001).26
Study by Kikuchi et al. (2021)27
The multivariate analysis in the study by Kikuchi et al. (2021) showed that the HR for mortality risk was 4.92 (95% CI, 3.10 to 7.80) in patients aged 70 years or older and 1.58 (95% CI, 0.90 to 2.77) in patients aged 60 to 69 years. Mortality was increased with a longer duration of dialysis, and the HR among patients with peripheral arterial disease was 1.49 (95% CI, 1.05 to 2.10). Mortality risk was lower in patients who were treated with remdesivir, with an HR of 0.60 (95% CI, 0.37 to 0.98).
In total, 392 patients were analyzed: 98 patients treated with remdesivir, matched with 294 patients not treated with remdesivir. The HR for overall survival was 0.45 (95% CI, 0.26 to 0.80) in the group treated with remdesivir — higher than in the group not treated with remdesivir. The mean duration of hospitalization was 20.9 days (SD = 13.2 days) in the group treated with remdesivir and 16.2 days (SD = 8.1 days) in the group not treated with remdesivir (difference = 4.7 days; 95% CI, 2.2 to 7.4 days).27
Study by Seethapathy et al. (2022)28
In the study by Seethapathy et al. (2022), 1 patient who was not on dialysis before initiating remdesivir developed worsening kidney function (defined as a ≥ 50% increase in creatinine or initiation of kidney replacement therapy), compared with 3 in the historical control group.
There were no significant differences in AEs between the matched groups, with the exception of an increased risk of hyperglycemia (glucose > 200 mg/dL), which occurred in 81% of patients in the remdesivir-treated population and 55% of patients in the control group (P = 0.03). No significant differences were observed between the 2 groups in terms of lowest hemoglobin or peak alanine aminotransferase (ALT) level; only peak glucose level was significantly different. Among patients treated with remdesivir, only 1 met the predefined criteria for worsening kidney function due to initiation of kidney replacement therapy, and among patients in the control group, 3 experienced a greater than 50% increase in serum creatinine.
Early discontinuation of remdesivir occurred in 4 patients (14%) due to safety concerns relating to elevated transaminase levels and low eGFR. The overall mortality rate during the hospital stay was 19% (n = 6) in patients treated with remdesivir and 23% (n = 7) in patients in the control group (P = 0.71).28
Study by Seethapathy et al. (2023)29
In the study by Seethapathy et al. (2023), the mean peak creatinine level was 2.3 mg/dL (95% CI, 1.98 to 2.57 mg/dL) and 2.5 mg/dL (95% CI, 2.13 to 2.89 mg/dL) among patients in the remdesivir-treated group and patients in the non–remdesivir-treated comparator group, respectively. The sensitivity analyses only included patients who received a full course of remdesivir or those with at least 5 posttreatment creatinine measurements.
Eighteen patients treated with remdesivir (10.3%) and 23 patients in the non–remdesivir-treated comparator group (13.1%) experienced doubling of serum creatinine during hospitalization.
Of the patients treated with remdesivir, 8 (4.6%) received kidney replacement therapy during their hospitalization, compared to 11 patients (6.3%)in the non–remdesivir-treated comparator group.
The eGFR of 120 surviving patients was measured, and the average eGFR at day 90 was 54.7 mL/min/1.73 m2 (SD = 20.0 mL/min/1.73 m2) among patients treated with remdesivir (n = 66), compared to 51.7 mL/min/1.73 m2 (SD = 19.5 mL/min/1.73 m2) among patients in the non–remdesivir-treated comparator group (n = 54).29
Study by Mozaffari et al. (2023)30
The study by Mozaffari et al. (2023) found that unadjusted mortality rates were lower among patients treated with remdesivir than in patients not treated with remdesivir across all VOC periods and all levels of baseline supplemental oxygen requirement. In the remdesivir group, 11.1% of patients died within 14 days and 17.7% died within 28 days after hospitalization for COVID-19. In the nonremdesivir group, 15.4% of patients died within 14 days and 22.4% died within 28 days. After adjustment for baseline and clinical covariates, the HR for mortality risk in the remdesivir group on admission was 0.70 (95% CI, 0.62 to 0.78) and 0.75 (95% CI, 0.68 to 0.83) at 14 days and 28 days, respectively. Similar results were seen during each VOC period and were most pronounced during the pre–Delta variant period at the 14-day assessment, with the HRs for the pre–Delta, Delta, and Omicron variant periods being 0.59 (95% CI, 0.48 to 0.71), 0.77 (95% CI, 0.65 to 0.92), and 0.75 (95% CI, 0.63 to 0.90), respectively. At 28 days, the HRs for the pre–Delta, Delta, and Omicron variant periods were 0.65 (95% CI, 0.56 to 0.76), 0.79 (95% CI, 0.68 to 0.91), and 0.84 (95% CI, 0.72 to 0.97), respectively.
For the mortality rate among the subgroup of patients with no supplemental oxygen charges (in hospitals documented to charge for supplemental oxygen) on admission, the HR was 0.71 (95% CI, 0.58 to 0.87) and 0.78 (95% CI, 0.66 to 0.93) at day 14 and day 28, respectively, in favour of the remdesivir group. For those who required low-flow oxygen on admission, the HR was 0.56 (95% CI, 0.46 to 0.68) and 0.62 (95% CI, 0.53 to 0.72) at day 14 and day 28, respectively in favour of the remdesivir group. The HR among those who required high-flow oxygen, noninvasive ventilation, invasive mechanical ventilation, or ECMO on admission was 0.83 (95% CI, 0.70 to 0.99) and 0.86 (95% CI, 0.75 to 0.99) at day 14 and day 28, respectively in favour of the remdesivir group.30
Study by Garibaldi et al. (2021)31
Of the 36,656 matched patients in the study by Garibaldi et al. (2021), 13,569 (74.0%) from the remdesivir group and 12,510 (68.3%) from the nonremdesivir group achieved clinical improvement before 28 days, with a median time to clinical improvement of 7 days (IQR, 5 to 19 days) in the remdesivir group and 9 days (IQR, 5 to 28 days) in the nonremdesivir group. The aHR for clinical improvement at 28 days in the remdesivir group was 1.19 (95% CI, 1.16 to 1.22). The aHR for clinical improvement among the patients treated with remdesivir receiving no oxygen was 1.30 (95% CI, 1.22 to 1.38), with a median of 5 days (IQR, 4 to 13 days) for the remdesivir group compared to 7 days (IQR, 5 to 15 days) for matched patients in the nonremdesivir group.
The aHR for clinical improvement among patients treated with remdesivir receiving low-flow oxygen was 1.23 (95% CI, 1.19 to 1.27), with a median of 6 days (IQR, 4 to 11 days) for the remdesivir group compared to 7 days (IQR, 5 to 15 days) for matched patients in the nonremdesivir group. The aHR for clinical improvement among patients treated with remdesivir receiving high-flow nasal cannula (HFNC) or noninvasive positive pressure ventilation (NIPPV) was 0.95 (95% CI, 0.89 to 1.01), with a median of 15 days (IQR, 7 to 28 days) compared to 17 days (IQR, 8 to 28 days) for matched patients in the nonremdesivir group. The aHR for clinical improvement among patients treated with remdesivir who were receiving invasive mechanical ventilation at the time of initiation was 0.92 (95% CI, 0.81 to 1.04), with a median of 28 days (IQR, 10 to 28 days) in the remdesivir group compared to 28 days (IQR, 9 to 28 days) for matched patients in the nonremdesivir group.
Remdesivir showed no significant impact on mortality overall, with an aHR of 1.02 (95% CI, 0.97 to 1.08) and a 28-day mortality rate of 15.7% (2,879 deaths) for the remdesivir group, compared to 19.6% (3,586 deaths) for the nonremdesivir group.
Among patients on room air, the aHR for mortality was 1.08 (95% CI, 0.92 to 1.27), and the 28-day mortality rate was 11.4% (325 deaths) for the remdesivir group, compared to 13.3% (329 deaths) for matched patients in the nonremdesivir group. The aHR for mortality among patients treated with remdesivir and receiving low-flow oxygen was 0.85 (95% CI, 0.77 to 0.92), and the 28-day mortality rate was 8.4% (865 deaths) for the remdesivir group, compared to 12.5% (1,334 deaths) for matched patients in the nonremdesivir group.
Among the patients treated with remdesivir and receiving HFNC or NIPPV, the aHR was 1.10 (95% CI, 1.01 to 1.20), and the 28-day mortality rate was 28.6% (1,137 deaths) in the remdesivir group, compared to 34.0% (1,237 deaths) for matched patients in the nonremdesivir group. Among the patients treated with remdesivir and receiving invasive mechanical ventilation, the aHR was 1.17 (95% CI, 1.04 to 1.32), and the 28-day mortality rate was 46.7% (552 deaths) in the remdesivir group, compared to 43.9% (686 deaths) for matched patients in the nonremdesivir group.
The aHR for clinical improvement by day 28 in the group that received remdesivir plus dexamethasone versus the group that received dexamethasone alone was 1.21 (95% CI, 1.18 to 1.25). Similarly, for patients on room air and on low-flow oxygen, the aHR was 1.31 (95% CI, 1.23 to 1.41) and 1.24 (95% CI, 1.20 to 1.28), respectively. In terms of survival benefits, the aHR for patients on low-flow oxygen treated with remdesivir plus dexamethasone versus dexamethasone alone was 0.83 (95% CI, 0.76 to 0.91).31
Critical Appraisal
Guidance for Reporting Real-World Evidence forms the foundation for transparent reporting of real-world evidence (RWE) studies in Canada and facilitates the appraisal of RWE by CDA-AMC.32 All applicable sections in the guidance should be reported on when submitting RWE studies as part of a Reimbursement Review.32 Many RWE studies submitted as part of this review were missing important information. Information on the following was missing: why a setting not in Canada was chosen, differences in health systems, access to care, available health care resources during the pandemic, and other factors that may impact the care of patients with COVID-19 and how those factors might affect the applicability of findings to the current context in Canada. A detailed description of data specifications (access, cleaning, and links, where applicable); data sources, including a data dictionary; variables that could not be captured; and the potential impacts on the study results were not provided.32
The pivotal trial data lack information about the effect of remdesivir on mortality in more recent COVID-19 variants. The study by Mozaffari et al. (2023),23 a large observational study, found that remdesivir reduced 14-day and 28-day mortality compared with no remdesivir treatment in patients hospitalized for COVID-19 between December 2020 and April 2022. The study by Mozaffari et al. (2023)23 may address a gap in the pivotal trial data as it describes the comparative effectiveness of remdesivir on the outcomes of 14-day and 28-day mortality in a population of patients across 3 variants (pre-Delta, Delta, and Omicron). The limitations of that study included a lack of information about the time of symptom onset and the treatments or vaccines administered before hospitalization. Although many comorbidities were controlled for, there remains a potential for imbalances in unmeasured confounders and residual confounding. Similarly, the study by Mozaffari et al. (2024),24 another large observational study, found that remdesivir reduced 30-day all-cause and COVID-19–related rehospitalization compared to no remdesivir treatment in patients who were hospitalized between December 2020 and April 2022 across 3 variants (pre-Delta, Delta, and Omicron). The limitations of that study included that the impact of the potential for missing data on the outcome of rehospitalization is not clear. There is also a lack of information about the time since symptom onset and treatments received before hospitalization. There was a lack of matching. Despite the inclusion of numerous variables in the multivariate regression, there is still a potential for unmeasured confounders. For both the study by Mozaffari et al. (2023)23 and the study by Mozaffari et al. (2024),24 it is unclear whether the information from the US cohort is generalizable to Canada. Vaccine uptake and background disease risk, as well as circulating variants, have changed since the studies were conducted, which may limit the generalizability of these findings to the current COVID-19 treatment landscape. Therefore, it is challenging to assess the exact magnitude of the treatment effect of remdesivir on the reduction of in-hospital 14-day and 28-day mortality compared with no remdesivir treatment and to extrapolate the effect to current practice in Canada.
The pivotal trial data lack clear information about the effect of remdesivir on outcomes that occur after hospital discharge. The study by Finn et al. (2022)25 is a small observational study (742 patients who were treated with remdesivir matched to 1,539 patients who were not) that used electronic health record (EHR) data from 3 hospitals in Rhode Island and found that treatment with remdesivir reduced hospital readmission and 30-day all-cause mortality compared to no remdesivir treatment in patients who were discharged after being hospitalized for COVID-19 between April 2020 and December 2020. The study by Finn et al. (2022)25 may address a gap in the pivotal trial data; however, it is subject to numerous limitations. These limitations include a lack of information about the time since symptom onset, the potential for time-related bias in the assessment of hospitalization, the potential for missing data related to postdischarge outcomes, and the potential for unmeasured confounders and residual confounding. Therefore, it is challenging to assess the exact magnitude of the benefit of remdesivir from this study on outcomes that occur after hospital discharge for patients hospitalized with COVID-19 and to extrapolate the effect to current practice in Canada.
The pivotal trial data lack clear information about the effect of remdesivir on post–COVID-19 condition. The study by Boglione et al. (2022)26 is a small observational study (including 163 patients treated with remdesivir) of hospitalized patients with COVID-19 at a single hospital in Italy from March 2020 to January 2021. The study by Boglione et al. (2022)26 likely has significant methodologic limitations, including risks for confounding by indication and lack of matching, uncertainty in the definition of post–COVID-19 condition, and limited generalizability from the Italian setting to Canada. These limitations preclude drawing conclusions about the effect of remdesivir on post–COVID-19 condition from this study.
The pivotal trial data lack clear information about the effect of remdesivir in patients with renal insufficiency. Three RWE studies were submitted to address this: Kikuchi et al. (2021),27 Seethapathy et al. (2021),28 and Seethapathy et al. (2023).29 The study by Kikuchi et al. (2021)27 is a small observational study (98 patients who were treated with remdesivir matched to 294 patients who were not) that used registry data to assess the effect of remdesivir on mortality in patients who were admitted to hospital with COVID-19 and received dialysis in Japan from April 2020 to June 2021. The study by Seethapathy et al. (2021)28 is a small observational study that used EHR data from a single US hospital to examine the relationship between AEs and remdesivir treatment in patients with an eGFR less than 30 mL/min/1.73 m2 from May 2020 to January 2021 (for patients treated with remdesivir) and from March 2020 to April 2020 (for patients not treated with remdesivir). The study by Seethapathy et al. (2023)29 is a small observational study (that used EHR data from a single US hospital to examine the relationship between adverse laboratory-based renal outcomes and remdesivir treatment in patients with an eGFR of 15 mL/min/1.73 m2 to 60 mL/min/1.73 m2 from April 2020 to November 2020 (for patients treated with remdesivir) and from March 2020 to April 2020 (for patients not treated with remdesivir). However, the limitations of all 3 studies and the inability to extrapolate the effects to current practice in Canada preclude conclusions about the effect of remdesivir in patients admitted to hospital with COVID-19 who are receiving dialysis or have reduced renal function.
The pivotal trial data lacks clear information about the effect of remdesivir in patients who are immunocompromised. The study by Mozaffari et al. (2023)30 is a large observational study that used a US dataset and found that treatment with remdesivir reduced 14-day and 28-day mortality, compared with not receiving treatment with remdesivir, in patients who were immunocompromised and hospitalized for COVID-19 between December 2020 and April 2022. The study by Mozaffari et al. (2023)30 may address a gap in the pivotal trial data as it describes the comparative effectiveness of remdesivir on the outcomes of 14-day and 28-day mortality in immunocompromised patients. The limitations of the study included a lack of information about the time of symptom onset and the treatments or vaccines administered before hospitalization. Although many comorbidities were controlled for, there remains a potential for imbalances in unmeasured confounders. It is difficult to extrapolate the magnitude of the effect of treatment with remdesivir for immunocompromised patients in Canada due to uncertainty about the generalizability of the US cohort to Canada.
The study by Garibaldi et al. (2021)31 was submitted by the sponsor to address the effect of remdesivir in patients receiving dexamethasone and is a large observational study (18,328 pairs of patients who were treated with remdesivir and patients who were not) that used a US dataset to examine the relationship between remdesivir treatment and time to improvement in patients who were hospitalized with COVID-19 from February 2020 to February 2021. The study by Garibaldi et al. (2021)31 may address a gap in the pivotal trial data; however, the analysis of treatment with remdesivir plus dexamethasone compared with dexamethasone alone in terms of time to clinical improvement is based on a sensitivity analysis only and therefore has limitations. Additional limitations include the potential for information bias due to the subjective nature of time to improvement (2-point decrease in the WHO severity score or discharged alive without worsening of the WHO severity score within 28 days) and the lack of information about the time since symptom onset or the treatments received before hospitalization. Approximately half the patients treated with remdesivir were unable to be matched, a potential source of bias. The potential for unmeasured confounders and residual confounding are other limitations. It is unclear whether the information from the US cohort is generalizable to Canada. Vaccine uptake and background disease risk as well as the circulating variants have changed, which may limit the generalizability of these findings to the current COVID-19 treatment landscape. Therefore, it is challenging to assess the exact magnitude of the treatment effect of remdesivir on time to clinical improvement compared with no remdesivir treatment and to extrapolate the effect to current practice in Canada.
Conclusions
The findings from the 4 RCTs17-20 suggest that remdesivir may prevent death in hospitalized patients aged 12 years or older with COVID-19 whose disease severity warrants support with low-flow oxygen and perhaps also in patients who require high-flow oxygen, but the latter subgroup was not evaluated in isolation. Remdesivir does not appear to prevent death in hospitalized patients who do not require oxygen support nor in patients who require any level of ventilation (noninvasive or invasive) or ECMO. The same subgroups, but not the overall population, may benefit from treatment with remdesivir for outcomes related to time to recovery or clinical improvement, as defined by the level of oxygen support required by the patient. Whether there is a benefit of remdesivir on duration of hospitalization is inconclusive due to between-study inconsistency: only 1 study (ACTT-1) reported a benefit; all the others either reported a longer duration of hospitalization among patients treated with remdesivir due to the 10-day regimen used in the study or reported no difference between treatment groups (Spinner et al. [2020], Wang et al. [2020]). Based on the findings of the ACTT-1 study and the WHO Solidarity trial, there may be a benefit of remdesivir in reducing the initiation of new ventilation among patients who were not ventilated at baseline. There may also be a modest benefit of remdesivir in reducing the duration of oxygen support, but there is some uncertainty in this finding due to IQRs overlapping between the treatment groups and inconsistency across the included studies.
Remdesivir was well tolerated in all the included studies, and safety outcomes were similar between the remdesivir treatment groups and the placebo or SOC treatment groups in each study. In terms of the duration of therapy, remdesivir can be given for at least 5 days and not more than 10 days; there appeared to be no obvious additional benefit or harm associated with a 10-day course of remdesivir over a 5-day course according to the comparisons to SOC conducted by Spinner et al. (2020).
The results from this assessment are generally aligned with WHO’s Living Guidance for Clinical Management of COVID-19,6 the Canadian treatment practice guidelines by Grant et al. (2024),33 a systematic literature review by CADTH conducted in 2023,34 and a Cochrane systematic review of remdesivir.35 However, there are critical generalizability concerns because all the RCT evidence was gathered during the early pandemic, at which time patients had little to no prior immunity due to the general unavailability of vaccines and the lack of infection history, and COVID-19 infections were caused by variants no longer prevalent in 2024. There was also an information gap regarding some patient subpopulations in the pivotal trial evidence. The sponsor submitted several RWE studies that supported the use of remdesivir in specific populations, including patients with more recent variants (up to Omicron), patients with renal insufficiency, patients who are immunocompromised, and patients also receiving dexamethasone. Additionally, a single-arm pivotal study (CARAVAN) was submitted to inform the safety and efficacy of remdesivir in adolescent patients. However, there are limitations inherent in these studies that preclude concluding with certainty on the benefit of remdesivir as extrapolated to the current context in Canada.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of remdesivir for IV infusion for the treatment of COVID-19 in hospitalized patients aged 12 years or older and weighing at least 40 kg who have pneumonia requiring supplemental oxygen.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
COVID-19 is an illness caused by SARS-CoV-2.1 The rapid global spread of the virus led to a pandemic, as declared by WHO on March 11, 2020. Subsequently, the proliferation of COVID-19 has presented significant challenges to health care systems globally, including those in Canada.2,3 As of April 3, 2024, the cumulative reported COVID-19 cases and deaths in Canada were 4,946,090 and 59,034, respectively, and the weekly percentage of positive cases out of the total tests conducted was 5.2%.4 From April 2022 to March 2023 in Canada, according to the Canadian Institute for Health Information, there were 120,524 hospitalizations due to COVID-19;5 the year prior there were 125,986 hospitalizations due to COVID-19.5 In 2022 to 2023, of patients admitted to hospital, 10% died in the facility and 13% were admitted to the ICU.5 Of those admitted to the ICU, 39% received ventilation.5 The estimated total cost of COVID-19 hospitalizations in Canada in 2022 to 2023 was approximately $2.9 billion, and costs continue to increase each fiscal year.5
Patients with symptomatic COVID-19 have a wide range of symptoms, ranging from no or mild symptoms (e.g., fever, cough, headache, malaise, muscle pain, nausea, vomiting, loss of taste and smell) in most cases to severe symptoms, including pneumonia and acute respiratory distress syndrome. Severe cases are also associated with pulmonary embolism, arrhythmia, cardiovascular shock, and heart damage or heart attack. At its worst, COVID-19 can lead to critical illness, where individuals experience respiratory failure, septic shock, and/or organ dysfunction known to be associated with high morbidity and mortality.6-12 Mortality risk estimates reported by WHO for patients with nonsevere disease are 0.6% for those at high risk of hospitalization, 0.3% for those at moderate risk of hospitalization, and 0.05% for those at low risk of hospitalization.13
The onset and duration of symptoms can vary widely among patients infected with SARS-CoV-2.36 Typical COVID-19 symptoms may appear 2 to 14 days after exposure, and they generally resolve within 14 days, but in severe cases, symptoms can last for over a month.14 Currently, the risk factors for progressing to severe disease are not the same as during the early stages of the pandemic;7 as population immunity has built up over time, the characteristics of patients being hospitalized due to COVID-19 have changed. The common risk factors for progression in COVID-19 now include older age, chronic comorbidities, cerebrovascular and cardiovascular disease, diabetes, hypertension, kidney failure, dementia, neurodevelopmental disorders, cancer, and a history of smoking.6,15
In Canada, it is estimated that 1 in 4 people aged 15 years or older have an underlying health condition, placing them at higher risk of COVID-19 progression and complications.37
SARS-CoV-2 consistently mutated throughout the pandemic, particularly at different locations within the spike protein, resulting in variants different from that of the original virus.38 As of March 2023, 5 VOCs of SARS-CoV-2 had been recognized by WHO: Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529), with the latter surpassing the others in terms of transmissibility and having a shorter incubation period of approximately 3 days and reduced associated morbidity and mortality.39-41
As the initial symptoms of COVID-19 closely resemble those of other respiratory infections, it is crucial to establish a confirmed diagnosis. Antigen tests are valuable and robust in diagnosing SARS-CoV-2 in symptomatic individuals; the tests are typically self-administered at home and provide rapid results (typically within 15 to 30 minutes).42
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Among patients hospitalized with COVID-19, the goal of treatment is to prevent death and reduce the need for life-sustaining treatments such as ventilation. Reducing the duration of the hospital stay and speeding recovery are secondary goals of treatment.
Canadian clinical practice follows WHO’s Living Guidance for Clinical Management of COVID-19,6 according to the clinical expert consulted by CDA-AMC. The WHO guidance categorizes disease severity as critical, severe, or nonsevere COVID-19. Critical COVID-19 is defined by WHO as the patient experiencing acute respiratory distress syndrome, sepsis, septic shock, or other conditions that would normally require life-sustaining therapies such as mechanical ventilation (invasive or noninvasive) or vasopressor therapy. Severe COVID-19 is defined by WHO as any of: oxygen saturation less than 90% on room air; in adults, signs of severe respiratory distress (accessory muscle use, inability to complete full sentences, respiratory rate more than 30 breaths per minute); in children, very severe chest wall indrawing, grunting, central cyanosis, or the presence of any other general danger signs in addition to the signs of pneumonia. Nonsevere COVID-19 is defined by the absence of the criteria described for the other 2 severity levels. The WHO guidance cautions that the threshold of 90% for oxygen saturation is arbitrary and must be considered in the context of the patient’s medical history and trends.
For the purpose of this review, only the recommendations for severe and critical COVID-19 will be discussed, as the goal of treatment for nonsevere COVID-19 is to prevent hospitalization. Among patients with severe or critical COVID-19, there are strong recommendations by WHO in favour of treatment with corticosteroids, interleukin-6 receptor blockers, and/or baricitinib, which may all be combined.6 Among patients with severe but not critical COVID-19, WHO suggests there are weak or conditional recommendations in favour of treatment with remdesivir; in contrast, for critical COVID-19, WHO provides a weak or conditional recommendation against the use of remdesivir.6
Other therapies with weak or conditional recommendations against their use include ruxolitinib and tofacitinib (only to be used if neither baricitinib nor interleukin-6 receptor blockers are available), ivermectin (to be used only in research settings), and convalescent plasma (to be used only in research settings).6 There are strong WHO recommendations against hydroxychloroquine, lopinavir-ritonavir, and casirivimab plus imdevimab.6
WHO’s Living Guidance for Clinical Management of COVID-196 generally aligns with the updated Canadian practice recommendations published by Grant et al. (2024)33 for the treatment of adults with symptomatic COVID-19 in 2023 to 2024. In the Canadian practice recommendations,33 remdesivir is recommended as an antiviral for patients categorized as being in “moderate” condition (receiving low-flow oxygen). Remdesivir is not recommended for patients in “severe” condition (receiving high-flow oxygen) or “critical” condition (receiving invasive ventilation or organ support) but may be considered in patients in “mild” condition who do not need oxygen support, depending on their degree of risk and their trajectory of symptoms.33 Dexamethasone 6 mg is not recommended in patients in mild condition, but it is recommended for immune suppression in patients in moderate, severe, or critical condition. For immune modulation, tocilizumab or baricitinib are recommended for patients in critical condition, may be considered for patients in severe condition (in case of rapid worsening, high inflammatory markers, and evidence of cytokine storm), and are not recommended for patients in mild condition; for patients in moderate condition, tocilizumab is not recommended, but baricitinib may be considered in the same circumstances as for patients in severe condition. For supportive care, neither prophylactic nor therapeutic anticoagulation are recommended for patients in mild condition. For patients in moderate condition, prophylactic anticoagulation is recommended if therapeutics are not given, and therapeutic anticoagulation is recommended. For patients in severe and critical condition, prophylactic (but not therapeutic) anticoagulation is recommended.33
The Canadian practice recommendations published by Grant et al. (2024)33 identify risk by age bracket, with people aged 70 years or older at the highest risk. But the number of comorbid conditions, severe immunosuppression, or other major underlying conditions may place people in higher risk categories despite younger age, these also interact with the expected immunity received from previous infections and vaccinations: patients not previously infected and who have not received the full set of recommended vaccine doses may be at higher risk (given the same age and comorbid factors) than patients with some immunity or patients who are fully up to date with vaccination.
Drug Under Review
Remdesivir is a nucleotide prodrug of an adenosine triphosphate analogue that competes with the natural adenosine triphosphate substrate for incorporation into nascent RNA chains by the SARS-CoV-2 RNA-dependent RNA polymerase and inhibits viral replication by terminating RNA transcription prematurely.16
IV remdesivir has been approved by Health Canada (Notice of Compliance) for the treatment of COVID-19 in:
hospitalized adults and pediatric patients (aged 4 weeks or older and weighing at least 3 kg) with pneumonia requiring supplemental oxygen
nonhospitalized adults and pediatric patients (weighing at least 40 kg) with positive results from direct SARS-CoV-2 viral testing who are at high risk for progression to severe COVID-19, including hospitalization and death.16
For this review, the sponsor has requested reimbursement of remdesivir for the treatment of COVID-19 in hospitalized patients aged 12 years or older (weighing at least 40 kg) with pneumonia requiring supplemental oxygen.
The recommended dosing regimen for remdesivir comprises a single loading dose of 200 mg on day 1, then 100 mg once daily from day 2 onward. For hospitalized patients with pneumonia requiring supplemental oxygen, treatment is recommended daily for at least 5 days and not more than 10 days. Remdesivir is administered via IV infusion, and its administration should take place under conditions where the management of severe hypersensitivity reactions (e.g., anaphylaxis) is possible.16
The key characteristics of remdesivir are summarized in Table 2, along with those of other treatments available for COVID-19.
Table 2: Key Characteristics of Remdesivir, Nirmatrelvir-Ritonavir, Tixagevimab-Cilgavimab, Sotrovimab, and Casirivimab-Imdevimab
Characteristic | Remdesivir | Nirmatrelvir-ritonavir | Tixagevimab-cilgavimab | Sotrovimab | Casirivimab-imdevimab |
|---|---|---|---|---|---|
Mechanism of action | Polymerase inhibitor that inhibits viral RNA synthesis. | Nirmatrelvir is a protease inhibitor that inhibits viral replication. Ritonavir inhibits the CYP3A-mediated metabolism of nirmatrelvir. | Monoclonal antibodies that bind to the SARS-CoV-2 spike protein preventing host cell entry. | ||
Indicationa | For the treatment of COVID-19 in:
| For the treatment of mild-to-moderate COVID-19 in adults with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death. | For the treatment of mild-to-moderate COVID-19 in adults and adolescents (≥ 12 years of age weighing ≥ 40 kg). For the pre-exposure prophylaxis of COVID-19 in adults and adolescents (≥ 12 years of age weighing ≥ 40 kg), who have not had a known recent exposure to an individual infected with SARS-CoV-2 and:
| For the treatment of mild-to-moderate COVID-19, confirmed by direct SARS-CoV-2 viral testing, in adults and adolescents (≥ 12 years of age weighing ≥ 40 kg) who are at high risk for progressing to hospitalization and/or death. | For the treatment of mild-to-moderate COVID-19, confirmed by direct SARS-CoV-2 viral testing, in adults and adolescents (≥ 12 years of age weighing ≥ 40 kg) who are at high risk for progressing to hospitalization and/or death. |
Route of administration | IV | Oral | Intramuscular | IV | IV |
Recommended dose | Adult and pediatric patients (≥ 40 kg): day 1 loading dose of 200 mg; day 2 onward 100 mg once daily:
Pediatric patients aged ≥ 4 weeks (≥ 3 kg and < 40 kg): day 1 loading dose of 5 mg/kg; day 2 onward 2.5 mg/kg:
| 300 mg nirmatrelvir (2 × 150 mg tablets) plus 100 mg ritonavir (1 × 100 mg tablet), with all 3 tablets taken together orally twice daily for 5 days. Treatment should be initiated as soon as possible after a COVID-19 diagnosis and within 5 days of symptom onset. | Treatment: 600 mg of tixagevimab-cilgavimab, administered as 2 separate 3.0 mL sequential injections of 300 mg of tixagevimab and 300 mg of cilgavimab. Treatment should be given as soon as possible after a positive viral test for SARS-CoV-2 and within 7 days after symptom onset. Prophylaxis: 600 mg of tixagevimab-cilgavimab, administered as 2 separate 3.0 mL sequential injections of 300 mg of tixagevimab and 300 mg of cilgavimab. Subsequent doses given once every 6 months. | 500 mg administered as a single IV infusion. Treatment should be given as soon as possible after symptom onset and confirmation of disease by a positive result obtained using a direct SARS-CoV-2 validated testing method. | 1,200 mg of casirivimab and 1,200 mg of imdevimab administered together as a single IV infusion. Treatment should be given as soon as possible after symptom onset and confirmation of COVID-19 by a positive result obtained using a direct SARS-CoV-2 validated testing method. |
Serious adverse effects or safety issues | No dose adjustment is required for patients with mild, moderate, or severe hepatic impairment (Child-Pugh class A, B, or C). However, due to limited data from patients with severe hepatic impairment, monitoring of liver function should be considered. | Contraindicated with drugs that are highly dependent on CYP3A for clearance and drugs that are potent CYP3A inducers; dose adjustment may be required for patients with moderate renal impairment; not recommended in patients with severe renal impairment or severe hepatic impairment; risk of serious adverse reactions with calcineurin inhibitors. | Risk of antiviral resistance. | Risk of antiviral resistance. | Risk of antiviral resistance. |
RNA = ribonucleic acid; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
aHealth Canada–approved indication.
Sources: Product monographs for remdesivir, nirmatrelvir-ritonavir, tixagevimab-cilgavimab, sotrovimab, and casirivimab-imdevimab.16,43-46
Input From Patients, Clinicians, and Drug Plans
Patient Group Input
No patient groups submitted input for this review.
Clinician Input
Input From Clinical Expert Consulted by CDA-AMC
All review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of COVID-19 in the inpatient setting.
Unmet Needs
The clinical expert described COVID-19 as no longer being a significant cause of hospitalization and death owing to the evolution of the virus since the beginning of the pandemic. The expert noted that an ideal intervention for COVID-19 focuses on prevention, not treatment. For patients who do require treatment, a treatment option that is effective across all disease severities would be ideal, and an oral delivery of the medication would be ideal. A significant treatment goal is also to reduce unnecessary antimicrobial use in COVID-19. However, there is an information gap in clinical data relevant to the currently prevalent variants of the virus, as the majority of clinical evidence was generated with early variants and may no longer apply.
Place in Therapy
The clinical expert noted that remdesivir may have infrequent application, given the lower prevalence of hospitalization and death caused by current COVID-19 variants. Remdesivir would be used in combination with other treatments as a first-line agent based on WHO’s Living Guidance for Clinical Management of COVID-19. The clinical expert also noted that remdesivir is unique in its antiviral action as, unlike other therapies for COVID-19, it does not target host immune response. The clinical expert stated that remdesivir would not change clinical practice as it has been rarely used since the early stages of the pandemic.
Patient Population
As this review is focused on the inpatient application of remdesivir, outpatient populations will not be discussed.
Among inpatients, those most in need of an intervention are those at risk of death. Diagnosis of COVID-19 is based on polymerase chain reaction testing (more accurate, more expensive, less accessible) or antigen testing (less accurate, less expensive, more accessible). There is a lack of current data to indicate which patients would most benefit from remdesivir, as the available data primarily evaluate early COVID-19 variants and largely unvaccinated patient populations.
However, the clinical expert noted that patients who are sick enough to require oxygen support as a result of COVID-19, but who have not yet progressed to needing ventilation, may be the most likely to benefit from remdesivir. This is reflected in trial data, but as these trials were conducted in populations with different variants, there are serious limitations in the generalizability of the results. Nonetheless, speculatively, the clinical expert discussed whether the reason for this observation in the trial data may be related to the pathogenesis of COVID-19 (i.e., earlier stages of the disease are virologically mediated, while later stages of the disease are immune mediated), making the application of an antiviral such as remdesivir less helpful in patients whose medical distress is caused by immune response rather than virological activity.
Assessing the Response Treatment
The clinical expert identified that the key outcomes (among patients already admitted to hospital) are need for oxygen or organ support and rate of mortality. Meaningful response would be a change in the status of oxygen or organ support requirements. The status of these requirements does not vary by physician interpretation; they are objective outcomes. Clinical symptoms and viral load are not relevant clinical outcomes, and they do not correlate with the objective outcomes.
Discontinuing Treatment
The clinical expert stated that remdesivir would generally be given for the entire treatment course (5 days or 10 days), and it would not be stopped due to progression or additional treatments, although it may be stopped as a result of AEs if necessary. Dosing is 200 mg on the first day, followed by 100 mg daily. The expert noted that the shortest effective duration of treatment should be used.
Prescribing Considerations
The clinical expert stated that inpatient treatment with remdesivir would be prescribed in hospital settings, with no need for a specialist to diagnose or treat.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups. The full original clinician group input received by CDA-AMC has been included in the Input From Patients, Clinicians, and Drug Plans section of this report.
One clinician group, the Ontario Health Infectious Diseases Advisory Committee, responded to the CDA-AMC call for clinician group input. Information was gathered through discussion with 4 clinicians.
According to the clinician group, the treatment regimen for COVID-19 for hospitalized patients includes supplemental oxygen therapy and immunomodulators such as corticosteroid, which is recommended as first-line treatment for hospitalized adults with COVID-19 requiring supplemental oxygen; Janus kinase inhibitors; or anti–interleukin 6 receptor monoclonal antibody. Remdesivir can be added to other immunomodulatory treatments that work on the hyperinflammatory pathway that tends to drive the disease course in the later stages of illness.
The main treatment goals are to accelerate recovery; reduce the severity of symptoms and the duration of hospitalization; prevent progression to critical COVID-19 disease conditions and long-term sequelae; prevent the need for new high-flow supplemental oxygen, noninvasive ventilation (e.g., BiPAP), mechanical ventilation, or ECMO; and prevent death.
The clinician group indicated that not all patients respond to currently available treatment. The group also noted some limitations with remdesivir, such as the drug formulation (IV) — which is not an easily self-administered therapy — and the lack of RCTs on the effectiveness of remdesivir on all COVID-19 variants, especially Omicron. In addition, the optimal timing for drug initiation from the date of symptom onset is unclear, the potential subset of patients who might benefit from a 10-day course of remdesivir is still uncertain, and the optimal role for remdesivir as part of combination therapy is not well defined.
According to the clinician group, hospitalized patients who require supplemental low-flow oxygen are best suited for treatment with remdesivir. The patients best suited for treatment with remdesivir would be identified based on a positive COVID-19 result via a polymerase chain reaction, rapid molecular, or rapid antigen test. A clinician assessment of a patient’s signs and symptoms would be required to determine COVID-19 severity. Remdesivir should ideally be started early in the disease course, when viral replication predominates. This treatment is least suitable for those with critical COVID-19 who require high-flow supplemental oxygen support or mechanical ventilation.
The clinician group stated that the outcomes used in clinical practice typically align with those used in clinical trials (e.g., duration of hospitalization, ICU admission, length of ICU stay, time to improvement in clinical status, progression to high-flow oxygen or noninvasive ventilation, progression to mechanical ventilation or ECMO, time to receipt of mechanical ventilation, time to clinical improvement, mortality, length of hospital stay, SAEs) and would be considered clinically meaningful responses. Treatment response should be assessed until hospital discharge or death.
The clinician group highlighted that the full course of remdesivir should still be completed for hospitalized patients on low-flow oxygen who progress to requiring HFNC oxygen, noninvasive ventilation, mechanical ventilation, or ECMO. However, it is still unclear whether there is a benefit to completing the full course of remdesivir treatment in such cases.
According to the clinician group, the factors that should be considered when deciding to discontinue treatment with remdesivir include disease progression to critical COVID-19, severe allergic reaction, adverse drug reaction, and AEs (e.g., elevation of ALT, signs or symptoms of liver inflammation, increasing conjugated bilirubin, alkaline phosphatase, or international normalized ratio). The most appropriate setting for treatment with remdesivir is in a hospital.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CDA-AMC are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
To date, COVID-19 therapeutics have been procured, paid for, and distributed to provinces and territories by the federal government. The criteria used to determine coverage may be significantly different across provinces and territories. | Comment from the drug programs to inform CDEC deliberations. The clinical expert confirmed that there are significant differences between jurisdictions in Canada. |
Considerations for initiation of therapy | |
Submitted trials used different inclusion criteria and definitions of severe disease. If remdesivir is recommended for funding, it will be important to clearly define any disease score or stage (e.g., specific severity or maximum duration of symptoms) required for eligibility. What patients benefit the most from treatment with remdesivir? What patients may not experience appreciable benefit? For example, consider:
| The clinical expert noted that the COVID-19 virus changes quickly and that clinical practice in Canada follows WHO’s Living Guidance for Clinical Management of COVID-19. The clinical expert added that, based on data from the early pandemic, it appears that patients receiving supplemental low-flow oxygen benefit from treatment with remdesivir and that patients receiving high-flow oxygen may also benefit (although this is less certain due to conflicting data). In contrast, patients not sick enough to need oxygen or organ support and patients who have already progressed to ventilation do not appear to benefit from treatment with remdesivir, based on current data and the WHO guidelines. The clinical expert described the former (“medium prognosis”) patients as those who may need access to remdesivir. However, as the virus evolves, the expert noted that this could change. |
Considerations for prescribing of therapy | |
Should remdesivir be given for a duration of 5 days or 10 days? Does the ideal duration vary by patient? Does dosing vary? | The clinical expert stated that the duration of treatment can be 5 days or 10 days and that there are no firm guidelines on which to use. The dosage of remdesivir does not otherwise vary (200 mg on the first day followed by 100 mg daily). Because the data do not show a clear benefit or harm of 10 days over 5 days, the clinical expert suggested that the lowest effective duration should be used. However, as most trials used 10-day regimens, there is a lack of data to support a decision between them. |
System and economic issues | |
The indication under consideration is for hospital inpatients. Funding for drugs administered to hospital inpatients generally comes from hospital global budgets and is not provided by public drug programs. | Comment from the drug programs to inform CDEC deliberations. |
CDEC = Canadian Drug Expert Committee.
Clinical Evidence
The objective of the Clinical Review is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of remdesivir infusion in the treatment of COVID-19 in patients aged 12 years or older and weighing at least 40 kg. The focus will be placed on comparing remdesivir to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of remdesivir is presented in 4 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. No sponsor-submitted long-term extension studies (second section) or indirect evidence (third section) were submitted. The fourth section includes additional studies that the sponsor considered to address important gaps in the systematic review evidence.
Clinical evidence from the following are included in the review and appraised in this document:
5 pivotal studies or RCTs identified in the systematic review
9 additional studies addressing gaps in evidence.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 4.
Table 4: Details of Studies Included in the Systematic Review
Detail | ACTT-1 | Wang et al. (2020) | WHO Solidarity | Spinner et al. (2020) | CARAVAN |
|---|---|---|---|---|---|
Designs and populations | |||||
Study design |
|
|
|
|
|
Locations | 60 study sites and 13 subsites in 10 countries:
| 10 hospitals in Hubei, China | 454 hospitals in 35 countries across 5 continents:
| 105 hospitals in the US, Europe, and Asia | 29 study sites in 4 countries:
|
Patient enrolment dates | Start date: February 21, 2020 End date: April 20, 2020 | Start date: February 6, 2020 End date: March 12, 2020 | Start date: March 22, 2020 End date: January 29, 2021 | Start date: March 15, 2020 End date: April 18, 2020 | Start date: July 21, 2020 End date: May 24, 2021 |
Randomized (N) | 1,062
| 237
| 8,320 (RDV or its control group)
Other patients not counted here were randomized to hydroxychloroquine, lopinavir, IFN-Beta1a, or their respective control arms | 596
| 53 (interim analysis)
|
Inclusion criteria |
|
|
|
|
Only cohort 1 is of interest to this review as the other cohorts exclusively include pediatric patients younger than 12 years |
Exclusion criteria |
|
|
|
|
|
Drugs | |||||
Intervention | RDV 200 mg IV on day 1, followed by 100 mg IV once daily, while hospitalized, for up to 10 days total | RDV 200 mg IV on day 1, followed by 100 mg on day 2 to day 10 in single daily infusions | RDV 200 mg IV on day 0 and 100 mg IV on day 1 to day 9, inclusive, plus local SOC | RDV 200 mg IV on day 1, followed by 100 mg per day for either 5 or 10 days | Dosing schedule varies by cohort. For cohort 1, RDV 200 mg on day 1, followed by 100 mg IV once daily for up to 10 days |
Comparators | Matching placebo, given at an equal volume at the same schedule | Placebo IV in the same volume | SOC alone (no placebo) | SOC alone (no placebo) | NA |
Study duration | |||||
Screening phase | 1 to 2 calendar days (from day –1 to day 1) | NR | NR | NR | Within 2 days before day 1 |
Treatment phase | Up to 10 days | Up to 10 days | Up to 10 days | Up to 10 days | Up to 10 days |
Follow-up phase | Up to day 29 | Up to 28 days | Up to day 28 | Up to day 11 | Same as treatment phase |
Outcomes | |||||
Primary end point | Time to recovery (days), where recovery was defined as a clinical status of 1, 2, or 3 on an 8-point ordinal scale | Time to clinical improvement within 28 days after admission, defined as a 2-point reduction in a patient’s admission status on a 6-point ordinal scale, or live discharge from hospital, whichever came first | In-hospital mortality | Distribution of clinical status on a 7-point ordinal scale on study day 11 | Proportion of patients with treatment-emergent AEs Proportion of patients with treatment-emergent graded laboratory abnormalities Pharmacokinetics assessed by plasma concentrations of RDV and metabolites |
Secondary and exploratory end points | Key secondary:
Other secondary:
| Secondary:
| Secondary:
| Secondary:
Exploratory:
| Secondary:
Exploratory:
|
Publication status | |||||
Publications | Study ID: NCT04280705 Beigel et al. (2020a) Beigel et al. (2020b) | Study ID: NCT04257656 Wang et al. (2020) | Study ID: NCT04315948 WHO Solidarity: WHO Solidarity Trial Consortium (2022); WHO Solidarity Trial Consortium (2021) Substudies:
| Study ID: NCT04292730 Spinner et al. (2020) | Study ID: NCT04431453 Munoz et al. (2021) |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; ECMO = extracorporeal membrane oxygenation; eGFR = estimated glomerular filtration rate; Ig = immunoglobin; IFN-Beta1a = interferon beta-1a; NA = not applicable; NEWS = National Early Warning Score; NR = not reported; PCR = polymerase chain reaction; PEWS = Pediatric Early Warning Score; RCT = randomized controlled trial; RDV = remdesivir; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; SBECD = sulfobutylether-beta-cyclodextrin; SOC = standard of care; SpO2 = peripheral oxygen saturation.
Sources: ACTT-1 Clinical Study Report;17 Wang et al. (2020);19 WHO Solidarity Trial Consortium (2022);18 Spinner et al. (2020);20 CARAVAN Clinical Study Report.21
The primary sources of evidence for this review included 5 studies, of which 3 were RCTs conducted in adults (ACTT-1, WHO Solidarity, and Wang et al. [2020]), 1 was an RCT conducted in patients aged 12 years or older (Spinner et al. [2020]), and 1 was a single-arm, open-label study in pediatric patients (CARAVAN). The studies were all conducted in patients hospitalized with COVID-19 requiring inpatient treatment. Each of the studies differed in their design, primary objective, patient eligibility criteria, and primary and secondary outcomes evaluated as well as in how common outcomes (e.g., clinical status) were defined.
The primary objective of the ACTT-1 study (N = 1,062) was to evaluate the clinical efficacy of remdesivir versus placebo in terms of time to recovery among hospitalized adults with COVID-19, based on an 8-point ordinal scale of clinical status. The ordinal scale ranged from 1 (not hospitalized and no limitations on activities) to 8 (death); refer to the Outcomes section for further details. The ACTT-1 study was a randomized (1:1), double-blind, placebo-controlled study with an adaptive design and interim monitoring to allow early stopping for futility, efficacy, or safety. Enrolment occurred from February 21, 2020, to April 20, 2020. Randomization was stratified by site and disease severity (severe versus mild to moderate). Severe disease was defined as the patient requiring mechanical ventilation, requiring oxygen, having a blood oxygen saturation of less than or equal to 94% on room air, or experiencing tachypnea (respiratory rate ≥ 24 breaths per minute). Mild-moderate disease was defined as the patient having a blood oxygen saturation greater than 94% and a respiratory rate of less than 24 breaths per minute without supplemental oxygen. The ACTT-1 study was conducted across 60 study sites and 13 subsites located in the US, Denmark, the UK, Greece, Germany, the Republic of Korea, Mexico, Spain, Japan, and Singapore. No study sites were located in Canada.
The WHO Solidarity trial was an open-label, randomized, adaptive trial initiated by WHO to evaluate the effects of remdesivir, hydroxychloroquine, lopinavir, and interferon beta-1a on in-hospital mortality in adult inpatients with COVID-19. The 3 interventions other than remdesivir were eventually discontinued for futility. Each intervention (in addition to locally available SOC) was compared against a control arm of SOC alone. For this review, only the remdesivir and associated control arm will be discussed further. Overall, 14,221 patients were enrolled between March 22, 2020, and January 29, 2021, of which 8,320 patients were allocated 1:1 to remdesivir (n = 4,169) or its associated control group (n = 4,151). Randomization was not stratified. The study took place in 454 hospitals in 35 countries worldwide, including 52 sites located in Canada (in Alberta, British Columbia, Manitoba, Newfoundland and Labrador, Nova Scotia, Ontario, and Quebec). The Discovery, CATCO, NOR Solidarity, and Solidarity Finland trials were add-on studies or substudies of the WHO Solidarity trial, with patients who completely (NOR Solidarity and Solidarity Finland) or partially (Discovery and CATCO) overlapped with the WHO Solidarity trial. The CATCO study was conducted in Canada and continued randomization for 2 additional months, randomizing an additional 323 patients who were not part of the WHO Solidarity trial (out of a total of 1,282 enrolled in the CATCO study across Canada). For the purpose of this review, the WHO Solidarity trial will be reported as a whole and the substudies will not be independently described in detail, except in special circumstances. Because the CATCO study was conducted in Canada and the majority of patients enrolled in the CATCO study were included in the WHO Solidarity trial, there will be limited discussion of the CATCO study independently from the WHO Solidarity trial.
The study by Spinner et al. (2020) was a randomized, open-label study of hospitalized patients to determine the efficacy of 5 days or 10 days of remdesivir treatment compared with SOC alone on patients’ clinical status on day 11 after initiation of treatment. Clinical status was based on a 7-point ordinal scale, ranging from death (category 1) to discharge (category 7). Enrolled patients were randomized (N = 596) without stratification 1:1:1 to each treatment arm. The study was conducted in 105 hospitals in the US, Europe, and Asia, with no study sites located in Canada.
The study by Wang et al. (2020) was the first randomized, double-blind, placebo-controlled clinical trial assessing the effect of remdesivir versus placebo in adult inpatients with severe COVID-19 and was conducted in 10 hospitals in Hubei, China. The primary end point was time to clinical improvement up to day 28, defined as the time (in days) from randomization to the point of a decline of 2 levels on a 6-point ordinal scale of clinical status (from 1 [discharged] to 6 [death]) or discharged alive from hospital, whichever came first. Between February 6, 2020, and March 12, 2020, a total of 237 patients were enrolled and randomly assigned 2:1 to a treatment group (158 to remdesivir and 79 to placebo). Randomization was stratified by level of respiratory support: no oxygen support or oxygen support with nasal duct or mask; or high-flow oxygen, noninvasive ventilation, invasive ventilation, or ECMO.
The CARAVAN study was a phase II/III, single-arm, open-label study to evaluate the safety, tolerability, and pharmacokinetics of remdesivir in pediatric patients younger than 18 years with COVID-19. The study was conducted across 19 sites in the US, the UK, Italy, and Spain; no study sites were located in Canada. Of the 53 patients enrolled, 12 were categorized into cohort 1, which falls within the reimbursement request relevant for this review (i.e., aged 12 years or older, but younger than 18 years, with a body weight of at least 40 kg). Cohorts 2 through 8 included younger age groups and lower weights. Data from the CARAVAN study used in this review were sourced from an interim Clinical Study Report.
Populations
Inclusion and Exclusion Criteria
All 5 of the studies included patients hospitalized with COVID-19, and 4 of the studies required laboratory-confirmed SARS-CoV-2 (ACTT-1, Spinner et al. [2020], Wang et al. [2020], and CARAVAN). The WHO Solidarity trial did not require laboratory confirmation but required that patients had “definite COVID-19” in the view of the responsible physician. Patients were required to be adults for enrolment in the ACTT-1 study, the WHO Solidarity trial, and the study by Wang et al. (2020). The study protocol of the study by Spinner et al. (2020) initially required a minimum age of 18 years, but the protocol was amended on March 15, 2020 (the same day as patient enrolment began) to lower the minimum age requirement to 12 years, in addition to other changes made on the basis of emerging understanding of COVID-19. The CARAVAN study specifically recruited patients younger than 18 years, including neonatal and infant age ranges, but cohort 1 (aged 12 years or older but younger than 18 years) is the only cohort relevant for the reimbursement request in this review. The studies also varied in the required severity of illness and in how severity was defined. For enrolment in the ACTT-1 study, patients could have illness of any duration but had to have at least 1 of: radiographic infiltrates confirmed by imaging; blood oxygen saturation of less than or equal to 94% on room air; need for supplemental oxygen; or need for mechanical ventilation. The requirements for the studies by Wang et al. (2020) and Spinner et al. (2020) were similar, requiring blood oxygen saturation of less than or equal to 94% on room air and radiologically confirmed pneumonia. The WHO Solidarity trial and the CARAVAN study did not specify similar details, although the WHO Solidarity trial subdivided (but did not stratify the randomization of) patients by disease severity at entry, which was based on ventilation and supplemental oxygen use. The exclusion criteria of the studies were generally similar, excluding patients with signs of serious liver or kidney disease or existing pregnancy or breastfeeding. The CARAVAN study did allow patients with pregnancy discovered after receiving at least 1 dose of the study drug to continue, after discussion with the study investigator.
Interventions
All the included studies evaluated remdesivir given as IV at a dose of 200 mg on the first day of treatment, followed by 100 mg daily on subsequent days of treatment, in an inpatient hospital setting. In all the studies, patients in all treatment arms were also receiving regionally available SOC. The duration of treatment with remdesivir (and, if applicable, matched placebo) was up to 10 days in 4 of the 5 trials (ACTT-1, Wang et al. [2020], WHO Solidarity, and Spinner et al. [2020]) and was either 5 days or 10 days, depending on treatment group assignment, in the study by Spinner et al. (2020). All the studies except the CARAVAN study (a single-arm study) were RCTs, and the ACTT-1 study and the study by Wang et al. (2020) included a matched-volume placebo in the control arm, which was administered in the same manner as remdesivir. The WHO Solidarity trial and the study by Spinner et al. (2020) did not include a placebo and compared remdesivir plus SOC to SOC alone.
Concomitant antivirals with potential anti–COVID-19 activity were disallowed in the ACTT-1 study, the CARAVAN study, and the study by Spinner et al. (2020); details of allowed or disallowed concomitant medications were not available for the other included studies (Wang et al. [2020] and WHO Solidarity).
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 5, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical expert consulted by CDA-AMC and input from the clinician group and the public drug plans. The review team selected the end points considered most relevant to inform the expert committee deliberations and finalized this list of end points in consultation with members of the expert committee.
Table 5: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | ACTT-1 | Wang et al. (2020) | WHO Solidarity | Spinner et al. (2020) | CARAVAN |
|---|---|---|---|---|---|
Mortalitya | Safety outcomes:
| Safety outcome: Number of patients who died by day 28 | Primary outcome: In-hospital mortality (whether before or after day 28), subdivided by disease severity at study entry | Exploratory outcome: All-cause mortality through day 28 | Safety outcome: Number of patients who died |
Duration of hospitalizationa | Secondary outcome: Duration of hospitalization up to 28 days (median) | Secondary outcome: Duration of hospitalization up to day 28 (median) | Secondary outcome: Duration of hospital stay (time from study entry to discharge) | Exploratory outcome: Duration of hospitalization | Secondary outcome: Duration of hospitalization up to day 30 |
Time to recovery or clinical improvementa | Primary outcome: Time to recovery based on 8-point ordinal scale of clinical status up to day 28 (median) | Primary outcome: Time to clinical improvement of at least 2 points (or live discharge, whichever came first) based on a 6-point ordinal scale up to day 28 (median) | NR | Exploratory outcome: Time to recovery based on 7-point ordinal scale of clinical status up to day 11 (median) | Time-to-event outcome: NR |
Initiation of ventilationa,b | Secondary outcomes:
| NR | Secondary outcome: Initiation of ventilation (yes/no) | NR | NR |
ECMO = extracorporeal membrane oxygenation; NR = not reported.
aNo statistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
bSome studies reported duration of ventilation or other oxygen support modalities, but these were not outcomes of interest for the purpose of this review.
Sources: ACTT-1 Clinical Study Report;17 Wang et al. (2020);19 WHO Solidarity Trial Consortium (2022);18 Spinner et al. (2020);20 CARAVAN Clinical Study Report.21
Mortality
ACTT-1 Study
Analyses of mortality were performed on the ITT and as-treated populations in the ACTT-1 study. The number of patients who died by day 15 and day 29, and the 14-day and 28-day crude mortality rate, were presented by treatment arm. The analysis was repeated, treating patients who died after unblinding as alive, and a similar analysis was repeated treating placebo patients who had received placebo who died after crossover treatment with remdesivir as alive. Mortality through day 15 and day 29 was analyzed as a time-to-event end point and presented with median time to event, with 95% CIs, for each treatment group, along with the HR estimate and stratified log-rank P values. Differences in time-to-event end points by treatment were summarized with KM curves. The ITT summaries were repeated, censoring patients who initially received placebo but were re-treated with remdesivir at the time of re-treatment. Similarly, the summaries were repeated censoring patients who were unblinded at the time of unblinding.
Study by Wang et al. (2020)
Mortality was measured at day 28 in the study by Wang et al. (2020).
WHO Solidarity Trial
The primary outcome used to assess the effects of study drugs in the WHO Solidarity trial was in-hospital mortality (whether before or after day 28), subdivided by disease severity at study entry. Palliative discharges were counted as in-hospital deaths, not discharges.
Study by Spinner et al. (2020)
Death was measured as a component of the 7-point clinical status scale on study day 11 in the study by Spinner et al. (2020).
CARAVAN Study
The CARAVAN study defined treatment-emergent deaths as those that occurred between the first dose date and the last dose date plus 30 days, inclusive.
Duration of Hospitalization
ACTT-1 Study
Duration of hospitalization was a secondary outcome in the ACTT-1 study. If the patient was discharged and no further hospitalization data were available, then the patient would be assumed to not have been readmitted. Thus, no imputed days would be added to the number of days recorded for that patient on available assessments. If a patient died while hospitalized, the number of days of hospitalization would be imputed as 28 days.
Study by Wang et al. (2020)
Duration of hospitalization was a secondary outcome in the study by Wang et al. (2020).
WHO Solidarity Trial
Duration of hospital stay (time from study entry to discharge) was a secondary outcome in the WHO Solidarity trial. Palliative discharges were counted as in-hospital deaths, not discharges.
Study by Spinner et al. (2020)
Duration of hospitalization was a secondary outcome in the study by Spinner et al. (2020).
CARAVAN Study
The duration of hospitalization (i.e., duration from hospital admission and duration from day 1) through to the day 30 follow-up visit was summarized in the CARAVAN study.
Recovery or Clinical Improvement
ACTT-1 Study
The primary efficacy end point of the ACTT-1 study was the time to recovery. Recovery was defined as a clinical status of 1, 2, or 3 on an 8-point ordinal scale, censored at day 29. The time to recovery was the elapsed time (in days) from randomization to the earliest day on which a patient reached recovery.
Clinical status was evaluated based on hospitalization, oxygen requirement, noninvasive mechanical ventilation (via mask) requirement, high-flow oxygen requirement, invasive mechanical ventilation (via endotracheal tube or tracheostomy tube) requirement, ECMO requirement, ongoing medical care preventing hospital discharge (COVID-19 related or other medical conditions), limitations of physical activity (self-assessed), and isolation for infection control purposes. While in hospital, elements of clinical status were measured daily until discharge. If patients were still in hospital or were able to visit in person on an outpatient basis, the following assessments were performed on days 15, 22, and 29: clinical data, vital signs, safety laboratory tests, and research laboratory samples, as able. Recognizing that quarantine and other factors could limit the patient’s ability to return to the study site if already discharged, patients could complete the follow-up assessments on days 15, 22, and 29 by phone, in which case the following were assessed: AEs, clinical status (ordinal scale), readmission to a hospital, and mortality.
The 8-point ordinal scale was ranked as follows, from worst to best:
8. Death
7. Hospitalized, on invasive mechanical ventilation or ECMO
6. Hospitalized, on noninvasive ventilation or high-flow oxygen devices
5. Hospitalized, requiring supplemental oxygen
4. Hospitalized, not requiring supplemental oxygen — requiring ongoing medical care (COVID-19 related or otherwise)
3. Hospitalized, not requiring supplemental oxygen — no longer requiring ongoing medical care (this would include those kept in hospital for quarantine or infection control purposes and those awaiting a bed in a rehabilitation facility or homecare or similar)
2. Not hospitalized, limitation on activities and/or requiring home oxygen
1. Not hospitalized, no limitations on activities
The primary analysis used the stratified log-rank test to compare treatment to control through day 29 with respect to time to recovery. Stratification was based on mild to moderate versus severe disease at randomization. The primary analysis was performed on the ITT population. The treatment RRR estimate, 95% CI, and P value from the stratified log-rank test were presented. The median time to event and 95% CI were summarized by treatment arm and disease severity. KM curves for each treatment arm were presented — supplemented by the RRR estimate, the P value, and the number of patients at risk in each arm and severity stratum — at days 1, 3, 5, 7, 11, 15, 22, and 29. Patient listings of the ordinal scale results by day were generated.
Study by Wang et al. (2020)
The primary outcome of the study by Wang et al. (2020) was time to clinical improvement based on a 6-point ordinal scale of clinical status. Clinical improvement was defined as either a 2-point improvement in the scale from baseline or discharge alive, whichever came first. Time to clinical improvement was assessed after all patients had reached day 28; no clinical improvement at day 28 or death before day 28 were considered as right censored at day 28.
The 6-point ordinal scale was ranked as follows, from worst to best:
6. Death
5. Hospital admission for ECMO or mechanical ventilation
4. Hospital admission for noninvasive ventilation or high-flow oxygen therapy
3. Hospital admission for oxygen therapy (but not requiring high-flow oxygen or noninvasive ventilation)
2. Hospital admission but not requiring oxygen therapy
1. Discharged or having reached discharge criteria (defined as clinical recovery: normalization of pyrexia, respiratory rate < 24 breaths per minute, blood oxygen saturation > 94% on room air, and relief of cough, all maintained for at least 72 hours)
WHO Solidarity Trial
This outcome was not reported in the WHO Solidarity trial.
Study by Spinner et al. (2020)
The primary efficacy end point in the study by Spinner et al. (2020) was the distribution of clinical status assessed on the 7-point ordinal scale on study day 11, which will not be reported for the purpose of this review. An exploratory end point was the time to recovery, where recovery was defined as improvement from a baseline score of 2 to 5 to a score of 6 to 7, or improvement from a baseline score of 6 to a score of 7.
The 7-point ordinal scale was ranked as follows, from worst to best:
1. Death
2. Hospitalized, requiring invasive mechanical ventilation or ECMO
3. Hospitalized, requiring noninvasive ventilation or high-flow oxygen
4. Hospitalized, requiring low-flow supplemental oxygen
5. Hospitalized, not requiring supplemental oxygen but requiring ongoing medical care
6. Hospitalized, not requiring supplemental oxygen or medical care
7. Not hospitalized
CARAVAN Study
A secondary end point of the CARAVAN study was clinical status assessed using a 7-point ordinal scale, which was derived by combining the available death, hospital discharge alive, and ordinal scale assessments reported by the site, where death superseded discharge alive and discharge alive superseded the ordinal scale score reported by the site. The proportion of patients for each ordinal scale category end point was summarized by cohort and expressed as a percentage for presentation purposes. However, no time-to-event outcome was calculated, so this outcome in the CARAVAN study will not be discussed further.
Initiation of New Ventilation
ACTT-1 Study
New oxygen use, new noninvasive ventilation or high-flow oxygen, and new mechanical ventilation or ECMO were measured in the ACTT-1 study among patients who were on or not on the oxygen support of interest at baseline. For the purpose of this review, the outcome of interest was new oxygen support (specifically, noninvasive ventilation or high-flow oxygen, and mechanical ventilation or ECMO) among patients who were not receiving these interventions at baseline. The incidence rate and 95% CI were reported.
For the outcome of new noninvasive ventilation or high-flow oxygen use at baseline, the subgroup of patients not already receiving these interventions also excluded patients who were receiving more invasive ventilation or ECMO support.
Study by Wang et al. (2020)
This outcome was not measured in the study by Wang et al. (2020).
WHO Solidarity Trial
In the WHO Solidarity trial, progression to ventilation was reported as a count of patients, subdivided between those receiving no oxygen at entry or receiving oxygen (but not ventilation) at baseline, and as a calculation of the observed minus the expected number of events, with variance based on log-rank statistics. High-flow and low-flow oxygen were not recorded separately at entry into the study. The supplementary material also reports rate ratios for initiation of ventilation among those not already ventilated at entry, stratified by age and respiratory support at entry.
Study by Spinner et al. (2020)
This outcome was not measured in the study by Spinner et al. (2020).
CARAVAN Study
This outcome was not measured in the CARAVAN study.
Statistical Analysis
ACTT-1 Study
For the primary analysis in the ACTT-1 study, the log-rank test was used to compare treatment arms with respect to time to recovery. For the log-rank test, the 2 key determinants of power were the total number of events (i.e., recoveries) and the treatment-to-control ratio of the rate of recovery. For 85% power, approximately 320 recoveries were required to detect a 40% increase in the rate of recovery from remdesivir. A total of 400 recoveries were needed for a recovery ratio of 1.35 with 85% power. The initial sample size was projected to be 572 patients, to achieve 400 patients with a “recovered” status per the primary objective. The key secondary end point related to clinical status on day 15 was analyzed using a proportional odds model. The total sample size of 396 gives approximately 85% power to detect an OR of 1.75 using a 2-tailed test at an alpha level of 0.05.
For the primary and secondary outcomes analyses, stratification was based on mild to moderate versus severe disease at randomization. Cox proportional hazards models were run within each of the disease severity strata to obtain stratum-specific estimates of the treatment HR. There was only 1 primary outcome measure, and there was no planned adjustment for multiple comparisons in any secondary analyses.
Any data point that appeared to be erroneous or inexplicable based on clinical judgment was investigated as a possible outlier. If data points were identified as outliers, sensitivity analyses may have been performed to examine the impact of including or excluding the outliers. Any substantive differences in these analyses were reported. For time-to-event outcomes, patients who were lost to follow-up or who terminated the study before day 29 and before observing or experiencing the event were censored at the time of their last observed assessment. Patients who died before or on day 29 and before observing or experiencing the event were censored at day 29. Patients who completed the study without observing or experiencing the event were censored at the day of their day 29 visit.
For the analysis of the key secondary outcome, patients who were discharged but were subsequently readmitted before day 15 without a reported clinical score, their clinical score was imputed as 7, which is the highest value for a hospitalized patient. If a patient died within 15 days, their clinical score was imputed as 8 (i.e., death). Otherwise, patients who were not discharged or had died by the day 15 visit but had missing ordinal scores on the day 15 visit were excluded from the analysis. For the analyses of the secondary outcomes that involved a clinical score (i.e., the key secondary outcome and time to improvement), if a patient was discharged from the hospital without a previously or concurrently reported clinical score of 1 or 2, their clinical score at the time of discharge was imputed as 2, which is the highest value for a nonhospitalized patient.
For days of hospitalization, if the patient was discharged and no further hospitalization data were available, the patient would be assumed to not have been readmitted. Thus, no imputed days would be added to the number of days recorded on available assessments. If a patient died while hospitalized, the number of days of hospitalization would be imputed as 28 days.
Several subgroup analyses were conducted, aside from those by disease severity already mentioned, including based on geographic region, duration of symptoms before enrolment, race, comorbidities, age group, and sex. The results for these analyses will not be presented in depth for this report.
Sensitivity analyses were performed where patients who were unblinded and retreated with remdesivir were censored at the time of remdesivir treatment initiation. A similar process was used for the unblinding sensitivity analysis. There was a sensitivity analysis of the primary, key secondary, and mortality outcomes to evaluate the effect of concomitant therapy, including experimental treatment and off-label use of marketed medications that were intended as treatment for COVID-19 and were given to patients before and during the study.
Patients who reported use of the following categories of medications of interest were censored at the time of the earliest start date of any of the medications of interest: protease inhibitors, polymerase inhibitors, other drugs used to treat COVID-19 (including off-label and/or experimental use), corticosteroids, other anti-inflammatory drugs (Janus kinase inhibitors, tyrosine kinase inhibitors, tumour necrosis factor inhibitors, interleukin inhibitors, interferon, plasma, immunoglobulin, T-cell therapies, selective T-cell co-stimulation blockers, and B-cell therapies [CD20 monoclonal antibodies]).
Study by Wang et al. (2020)
The power analysis in the study by Wang et al. (2020) determined that 325 events were required across both groups to provide 80% power under a 1-sided type I error of 2.5% if the HR was 1.4, which corresponded to an improvement of 6 days for time to clinical improvement with remdesivir, assuming the time to clinical improvement was 21 days in the placebo group. After taking into account an assumed 80% event rate within 28 days and a dropout rate of 10%, the target for patient recruitment was approximately 453 (151 to be randomized to placebo and 302 to remdesivir).
For the primary analysis, an ITT approach was used, which included all randomly assigned patients. Assessment occurred after all patients had reached day 28; patients with no clinical improvement or who had died on or before day 28 were considered right censored at day 28. Time to clinical improvement was calculated using a Cox proportional hazards model, reported using a KM plot, and compared with a log-rank test.
Subgroup analyses were conducted for:
those receiving treatment for 10 days or less versus more than 10 days after symptom onset
time to clinical deterioration (defined as a 1-category increase on the 6-point ordinal scale or death)
viral RNA load at entry.
WHO Solidarity Trial
The WHO Solidarity trial did not have a predefined recruitment target, as the numbers entered were to depend on how the epidemic developed, given the time frame during which it was conducted. It was speculated during the protocol phase that it “may be possible to enter several thousand hospitalized patients with relatively mild disease, and a few thousand with severe disease, but realistic sample sizes could not be estimated prior to the trial.”
All analyses were conducted according to the randomly assigned treatment, regardless of the actual treatment, excluding patients with a refuted COVID-19 diagnosis or with consent not encrypted in the database. All entry data were recorded irrevocably before unstratified, computerized treatment assignment, yielding 1:1 randomization with no foreknowledge of treatment assignment.
Mortality analyses were reported as RRs or HRs and calculated from log-rank or Cox analyses, split by disease severity, where severity was defined by ventilation and supplemental oxygen use (low or high flow) at study entry. The mortality rates were stratified by age group (younger than 50 years, 50 to 69 years, and 70 years or older) and by respiratory support group (none, oxygen only, and ventilated), yielding 9 strata. Unstratified KM methods were also used to present mortality by time, and the denominators at each time included previously discharged patients.
Study by Spinner et al. (2020)
For the study by Spinner et al. (2020), power analyses calculated that 600 patients (200 in each of 3 treatment groups) would provide greater than 85% power to detect an OR of 1.8 for each remdesivir group versus the SOC group using a 2-sided significance level of 0.05. The OR of 1.8 was calculated based on proposed group sizes at the time of study development, as there were no prior data available to inform a minimum clinically meaningful treatment effect due to the early stage in the pandemic. An OR of greater than 1 would indicate improvement in the remdesivir group versus the SOC group. All patients who were randomized and received at least 1 dose of remdesivir (or day 1 study visit for the SOC group) were included in the assessments of efficacy and AEs. For clinical status, the ordinal score was recorded as 1 on the day of death and all subsequent days; if a patient was discharged, the ordinal score was recorded as 7 on the day of discharge alive and all subsequent days unless the patient was rehospitalized for COVID-19–related reasons; otherwise, the most recent assessment was used for missing values.
Each remdesivir treatment group was compared with the SOC group at a 2-sided alpha of 0.025 for the primary outcome. Proportional odds models were used with treatment as the independent variable, and the assumption of proportional odds was tested using the score test; supporting P values from the Wilcoxon rank sum test were provided if the proportional odds assumption was not met. For the prespecified exploratory end points, death was considered the competing risk in these time-to-event analyses. Patients without the event of interest were censored on the day of the last nonmissing ordinal scale assessment. All-cause mortality was estimated using the KM product limit method with all available data. Each remdesivir group was compared with the SOC group using the log-rank test. Patients who did not die were censored on the last study day. Duration of hospitalization was compared between groups using the Wilcoxon rank sum test.
Several post hoc sensitivity analyses of the primary outcome were conducted, including adjusting for day 1 clinical score; adjusting for duration of symptoms; using day 28 visit data to confirm day 11 clinical status and imputing patients with missing status as dead; and using all randomized patients whether they received treatment or not (ITT population).
The proportions of patients with a 1-point or greater improvement at day 11 was calculated within subgroups based on symptom duration, body mass index, race, baseline oxygen support, region, sex, and age. To understand if the open-label design and assigned duration of treatment had an effect on hospital discharge, rates of discharge were calculated in each group over time.
CARAVAN Study
Twelve patients for each cohort in cohorts 1 through 4 were planned for enrolment in the CARAVAN study, based on power calculations related to pharmacokinetic outcomes (based on the population of all the cohorts combined), which will not be described in depth for the purpose of this review.
No adjustments for multiple comparisons were planned.
Table 6: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
ACTT-1 | ||||
Mortality | NR | NR | NR | NR |
Duration of hospitalization | NR | NR | If the patient was discharged and no further hospitalization data were available, the patient would be assumed to not have been readmitted. Thus, no imputed days would be added to the number of days recorded on available assessments. If a patient died while hospitalized, the number of days of hospitalization would be imputed as 28 days. | NR |
Time to recovery or clinical improvement | Cox proportional hazards model and log-rank test | Stratified by disease severity at study entry (mild to moderate vs. severe) | Patients lost to follow-up or terminated or died before observing the event were censored at the time of their last observed assessment. Patients who completed the study without the event were censored at the day of their day 29 visit. If a patient died within 15 days of admission, their clinical score was imputed as 8 (i.e., death). Patients who were not discharged or had died by the day 15 visit but had missing ordinal scores on day 15 were excluded from the analysis. If a patient was discharged from the hospital without a previously or concurrently reported clinical score of 1 or 2, their clinical score at the time of discharge was imputed as 2, which is the highest value for a nonhospitalized patient. |
|
Initiation of ventilation | Log-rank test | NR | NR | NR |
Wang et al. (2020) | ||||
Mortality | NR | NR | NR | NR |
Duration of hospitalization | NR | NR | NR | NR |
Time to recovery or clinical improvementa | Cox proportional hazards model and log-rank test | NR | Deaths before day 28 were right censored at day 28; number of patients without clinical improvement was still included in number at risk |
|
WHO Solidaritya | ||||
Mortalitya | Log-rank (rate ratios) or Cox analyses (HRs) | Divided by disease severity at study entry | NR | NR |
Duration of hospitalization | NR | NR | NR | NR |
Initiation of ventilation | Log-rank | NR | NR | NR |
Spinner et al. (2020) | ||||
Mortality | Estimated using the KM product limit method with all available data and log-rank test | NR | NR | NR |
Duration of hospitalization | Wilcoxon rank sum test | NR | NR | NR |
Time to recovery or clinical improvement | NR (exploratory) | NR | NR | NR |
CARAVAN | ||||
Mortality | Cox proportional hazards model and log-rank test | Split by disease severity at entry and stratified by age group and level of required respiratory support at entry | NR | NR |
Duration of hospitalization | NR | NR | NR | NR |
HR = hazard ratio; ITT = intention to treat; KM = Kaplan-Meier; NR = not reported; vs. = versus.
aThere was no formal statistical plan for the WHO Solidarity trial.
Sources: ACTT-1 Clinical Study Report;17 Wang et al. (2020);19 WHO Solidarity Trial Consortium (2022);18 Spinner et al. (2020);20 CARAVAN Clinical Study Report.21
Analysis Populations
Table 7: Analysis Populations of Included Studies
Study | Population | Definition | Application |
|---|---|---|---|
ACTT-1 | ITT population | All randomized patients by planned treatment arm | Main analyses of primary and secondary efficacy outcomes |
As-treated population | All randomized patients who received any study treatment infusion even if halted or slowed, by actual treatment arm | AE, safety, and mortality analyses Sensitivity analyses of efficacy outcomes | |
Wang et al. (2020) | ITT population | All randomly assigned patients | All efficacy analyses |
Per-protocol population | Not specifically defined but appears to reflect patients who received at least 5 days of study treatment (either RDV or placebo) based on Figure 1 of the publication by Wang et al. (2020) | Sensitivity analyses of efficacy outcomes | |
WHO Solidarity | ITT population | All randomly assigned patients | Where the ITT population set is applied is not specifically described, but presumably it is applied for all efficacy analyses as no other analysis population was described by the study publication |
Spinner et al. (2020) | ITT population | All randomized patients whether they received treatment or not | Sensitivity analyses of primary efficacy end point |
As-treated population | Patients who started the treatment they were randomized to | All main analyses for all outcomes | |
CARAVAN | All-enrolled analysis set | All patients who were enrolled in the study | Primary analysis set for per-patient listings |
Full analysis set | All patients who were enrolled in the study and received at least 1 dose of the study drug | Primary analysis set for efficacy analyses | |
Safety analysis set | All patients who were enrolled into the study and received at least 1 dose of study drug | Primary analysis set for safety analyses | |
RDV pharmacokinetic analysis set | All patients who were enrolled and received at least 1 dose of RDV and for whom pharmacokinetic concentrations of RDV were available | Pharmacokinetic analyses | |
Metabolites pharmacokinetic analysis set | All patients who were enrolled and received at least 1 dose of RDV and for whom pharmacokinetic concentrations of the metabolite(s) (analytes) were available | Pharmacokinetic analyses |
AE = adverse event; ITT = intention to treat; RDV = remdesivir.
Sources: ACTT-1 Clinical Study Report;17 Wang et al. (2020);19 WHO Solidarity Trial Consortium (2022);18 Spinner et al. (2020);20 CARAVAN Clinical Study Report.21
Results
Patient Disposition
The patient disposition for the ACTT-1 study is reported in Table 8, for the study by Wang et al. (2020) in Table 9, for the WHO Solidarity trial in Table 10, for the study by Spinner et al. (2020) in Table 11, and for the CARAVAN study (cohort 1) in Table 12.
In the ACTT-1 study, of 1,114 screened patients, 52 (5%) did not proceed past screening due to ineligibility (54% of those who did not proceed past screening) or, despite being eligible, due to not being enrolled (46% of those who did not proceed past screening). The randomization of the 1,062 enrolled patients included stratification into mild-moderate and severe disease as described in the Description of Studies section. Of the patients with mild-moderate disease, 82 and 77 were randomized to remdesivir and placebo, respectively, and of the patients with severe disease, the numbers were 459 and 444. Very few patients did not receive treatment. Slightly more patients in the placebo arm received all 10 doses compared to the remdesivir arm, numerically (no statistical comparison). The most common reason for receiving fewer than 10 doses was recovery. In the mild-moderate disease group, 57% and 51% of patients assigned to remdesivir or placebo, respectively, received fewer than 10 doses due to recovery; in the severe disease group, the proportions were 38% and 27%. Very few patients terminated the study early, other than for death or recovery.
In the study by Wang et al. (2020), of 255 screened patients, 18 (7%) did not proceed past screening, of which 78% did not meet the eligibility criteria and 22% withdrew. Of the patients randomized to remdesivir (N = 158) or placebo (N = 79), 98% to 99% started treatment. Some patients (5 [3%] and 2 [3%] in the remdesivir and placebo arms, respectively) were excluded from the ITT population but were included in the safety analysis set due to receiving the study drug for less than 5 days.
The WHO Solidarity trial did not report screening numbers, but 4,169 patients were randomized to remdesivir and 4,151 were randomized to the control group (SOC). Very few of these patients were excluded (less than 1% in each group), with the main reasons for exclusion being lack of COVID-19 or lack of proof of consent. In total, 602 (14%) and 643 (16%) in the remdesivir and control groups, respectively, died in hospital. The Canadian substudy of the WHO Solidarity trial, CATCO,22 included 1,282 patients randomized to remdesivir (n = 634) or SOC (n = 648). Of the 1,282 patients enrolled in the CATCO study, 951 were also described in the global WHO Solidarity trial.
In the study by Spinner et al. (2020), 16 (3%) of the 612 patients screened did not proceed past screening, of which 81% did not meet the eligibility criteria and 19% withdrew. Patients were randomized to receive remdesivir treatment for 10 days (N = 197), remdesivir treatment for 5 days (N = 199), or SOC (N = 200). Disposition data were not reported for the SOC group. In the remdesivir groups, very few patients did not start treatment. Fewer patients in the remdesivir 10-day group completed treatment (73 [37%]) than in the 5-day group (145 [73%]), primarily due to discharge and, less commonly, due to AEs or withdrawal of consent.
In the CARAVAN study, 12 patients were screened, and all were enrolled. Of these, 3 (25%) completed the study drug. Of the 9 patients (75%) who prematurely discontinued the study drug, 2 discontinued due to AEs, 2 discontinued due to hospital discharge, and 5 discontinued at the investigator’s discretion. The study was completed by 11 of the patients (92%). One patient died.
Table 8: Summary of Patient Disposition in the ACTT-1 Study
Patient disposition | RDV (mild-moderate disease) (N = 82) | PBO (mild-moderate disease) (N = 77) | RDV (severe disease) (N = 459) | PBO (severe disease) (N = 444) |
|---|---|---|---|---|
Screened, N | 1,114 | |||
Did not proceed past screening, n (%) | 52 (5) | |||
Did not meet eligibility criteria | 28 (54) | |||
Eligible but not enrolled | 24 (46) | |||
Randomized, N | 82 | 77 | 459 | 444 |
Received treatment, n (%) | 82 (100) | 76 (99) | 449 (89) | 441 (99) |
Did not receive treatment, n (%) | 0 | 1 (< 1) | 10 (2) | 3 (1) |
Received all 10 doses, n (%) | 26 (32) | 29 (38) | 182 (40) | 197 (44) |
Received less than 10 doses, n (%) | 56 (68) | 47 (61) | 267 (58) | 244 (55) |
Recovery | 47 (57) | 39 (51) | 176 (38) | 119 (27) |
Death | 1 (1) | 0 | 14 (3) | 19 (4) |
SAE or AE other than death | 5 (6) | 4 (5) | 47 (10) | 66 (15) |
Withdrawal by investigator | 1 (1) | 0 | 3 (1) | 1 (< 1) |
Withdrawal by patient | 1 (1) | 2 (3) | 5 (1) | 6 (1) |
Withdrawal by patient, transition to comfort care | 1 (1) | 0 | 3 (1) | 6 (1) |
Transferred to another hospital | 0 | 0 | 0 | 1 (< 1) |
Intermittent missed doses | 0 | 2 (3) | 18 (4) | 24 (5) |
Became ineligible after enrolment | 0 | 0 | 0 | 1 (< 1) |
Protocol deviation | 0 | 0 | 0 | 1 (< 1) |
Completed study, including death and recovery, n (%) | 80 (98) | 74 (96) | 437 (95) | 434 (98) |
Terminated study early, excluding for death or recovery, n (%) | 2 (2) | 2 (3) | 12 (3) | 7 (2) |
Voluntary withdrawal by patient | 1 (1) | 2 (3) | 5 (1) | 5 (1) |
Withdrawal by patient, transition to comfort care | 1 (1) | 0 | 2 (< 1) | 2 (< 1) |
SAE or AE other than death | 0 | 0 | 4 | 0 |
Transferred to another hospital | 0 | 0 | 1 (< 1) | 1 (< 1) |
Withdrawal by investigator | 0 | 0 | 0 | 1 (< 1) |
ITT population, N (%) | 82 (100) | 77 (100) | 459 (100) | 444 (100) |
As-treated population, N (%) | 82 (100) | 76 (99) | 450 (98) | 440 (99) |
Excluded from as-treated population (did not receive at least 1 infusion), n (%) | 0 | 1 (1) | 10 (2) | 3 (1) |
Randomized to PBO but received RDV, n (%) | 0 | 0 | 1 (< 1) | 1 (< 1) |
AE = adverse event; ITT = intention to treat; PBO = placebo; RDV = remdesivir; SAE = serious adverse event.
Note: Percentages were not reported and have been calculated by Canada’s Drug Agency.
Source: ACTT-1 Clinical Study Report.17
Table 9: Summary of Patient Disposition in the Study by Wang et al. (2020)
Patient disposition | RDV (N = 158) | PBO (N = 79) |
|---|---|---|
Screened, N | 255 | |
Did not proceed past screening, n (%) | 18 (7) | |
Did not meet eligibility criteria | 14 (78) | |
Withdrew | 4 (22) | |
Randomized, N | 158 | 79 |
Did not receive treatment, n (%) | 3 (2) | 1 (1) |
Received treatment, n (%) | 155 (98) | 78 (99) |
ITT population, N (%) | 158 (100) | 78 (99) |
Per-protocol population, N (%) | 150 (95) | 76 (96) |
SAS, N (%) | 155 (98) | 78 (99) |
Excluded from ITT population but included in SAS due to receiving study drug for less than 5 days, n (%) | 5 (3) | 2 (3) |
ITT = intention to treat; PBO = placebo; RDV = remdesivir; SAS = safety analysis set.
Note: Percentages were not reported and have been calculated by Canada’s Drug Agency.
Source: Wang et al. (2020).19
Table 10: Summary of Patient Disposition in the WHO Solidarity Trial
Patient disposition | RDV (N = 4,169) | Control (N = 4,151) |
|---|---|---|
Screened, N | NR | |
Did not proceed past screening, n (%) | NR | |
Randomized, N | 4,169 | 4,151 |
Excluded, n (%) | 23 (< 1) | 22 (< 1) |
No COVID-19 | 4 (< 1) | 3 (< 1) |
Consent not in database | 19 (< 1) | 19 (< 1) |
Died in hospital, n (%) | 602 (14) | 643 (16) |
Consent to follow-up withdrawn, n (%) | 51 (< 1) | 50 (< 1) |
Transfers | 5 (< 1) | 2 (< 1) |
Not yet reported on (censored at day 28), n (%) | 23 (< 1) | 24 (< 1) |
ITT population, N (%) | 4,169 (100) | 4,151 (100) |
ITT = intention to treat; NR = not reported; RDV = remdesivir.
Note: Percentages were not reported and have been calculated by Canada’s Drug Agency.
Source: WHO Solidarity Trial Consortium (2022).18
Table 11: Summary of Patient Disposition in the Study by Spinner et al. (2020)
Patient disposition | RDV 10 days (N = 197) | RDV 5 days (N = 199) | SOC (N = 200) |
|---|---|---|---|
Screened, N | 612 | ||
Did not proceed past screening, n (%) | 16 (3) | ||
Did not meet eligibility criteria | 13 (81) | ||
Withdrew | 3 (19) | ||
Randomized, N | 197 | 199 | 200 |
Did not receive treatment, n (%) | 4 (2) | 8 (4) | NR |
Withdrew consent | 2 (1) | 6 (3) | NR |
Protocol violation | 2 (1) | 1 (0.5) | NR |
Investigator discretion | 0 | 1 (0.5) | NR |
Completed treatment, n (%) | 73 (37) | 145 (73) | NR |
Stopped treatment early, n (%) | 120 (61) | 46 (23) | NR |
Discharged | 98 (50) | 35 (16) | NR |
AEs | 8 (4) | 4 (2) | NR |
Withdrew consent | 6 (3) | 5 (3) | NR |
Investigator decision | 4 (2) | 1 (< 1) | NR |
Protocol violation | 2 (1) | 0 | NR |
Death | 1 (1) | 0 | NR |
Nonadherence | 1 (1) | 0 | NR |
Lost to follow-up | 0 | 1 (1) | NR |
Included in primary analysis, n (%) | 193 (98) | 191 (96) | 200 (100) |
Excluded from primary analysis due to not starting treatment, n (%) | 4 (2) | 8 (4) | 0 |
AE = adverse event; NR = not reported; RDV = remdesivir; SOC = standard of care.
Note: Percentages were not reported and have been calculated by Canada’s Drug Agency.
Source: Spinner et al. (2020).20
Table 12: Summary of Patient Disposition in the CARAVAN Study (Cohort 1)
Patient disposition | RDV (N = 12) |
|---|---|
Screened, N | 12 |
Did not proceed past screening, n | 0 |
Enrolled, N | 12 |
Completed study drug, n (%) | 3 (25) |
Prematurely discontinued study drug, n (%) | 9 (75) |
AE | 2 (17) |
Hospital discharge | 2 (17) |
Investigator’s discretion | 5 (42) |
Patient decision | 0 |
Parent or guardian decision | 0 |
Still on study up to data cut-off date, n | 0 |
Completed study, n (%) | 11 (92) |
Prematurely discontinued from study, n (%) | 1 (8) |
Death | 1 (8) |
Withdrew consent | 0 |
Lost to follow-up | 0 |
Full analysis set, N (%) | 12 (100) |
Safety analysis set, N (%) | 12 (100) |
AE = adverse event; RDV = remdesivir.
Source: CARAVAN Clinical Study Report.21
Baseline Characteristics
The baseline characteristics outlined in Table 13 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. Oxygen support status or clinical status at baseline are not reported in the table due to the diversity of metrics used to evaluate this kind of characteristic; however, related details are summarized in this section by study, where available.
Vaccination status and COVID-19 variant details were not reported for any of the included studies. However, all the studies were conducted before widespread availability of vaccination and before the variants Omicron and Delta became prevalent. The CATCO study, which was the Canadian substudy of the WHO Solidarity trial and continued additional recruitment, did recruit into Canada’s third COVID-19 wave during the emergence of the Alpha variant; however, those patients are likely those who are accounted for only in the CATCO study and not in the population of the WHO Solidarity trial represented here. As the majority of patients included in the CATCO study were included in the WHO Solidarity trial, they are not presented separately in this report but may be mentioned to supplement the results of the overall WHO Solidarity trial.
The mean age of patients in the ACTT-1 study was 58.6 years (SD = 14.6) in the remdesivir group and 59.2 years (SD = 15.4) in the placebo group; 35.6% were female and 64.4% were male. Overall, 53.3% were white, 21.3% were Black or African American, 12.7% were Asian, and 0.7% were American Indian or Alaska Native (wording from original source). A total of 54.5% of patients had 2 or more coexisting conditions, and 25% had 1 coexisting conditions, including hypertension (50.2%), obesity (44.8%), and type 2 diabetes (30.3%). Baseline characteristics by 8-point ordinal scale score were only reported for scores 4, 5, 6, and 7, presumably because these scores represent hospitalized patients, which was a requirement for enrolment; lower scores represent nonhospitalized patients, and a score of 8 represents death. At baseline, a majority of patients had an ordinal scale score of 5 (i.e., hospitalized, requiring supplemental oxygen): 232 (43%) in the remdesivir group and 203 (39%) in the placebo group. The next most common baseline ordinal score was 7 (hospitalized, on invasive mechanical ventilation or ECMO) at 131 patients (24%) in the remdesivir group and 154 patients (30%) in the placebo group, followed by an ordinal score of 6 (hospitalized, on noninvasive ventilation or high-flow oxygen devices) at 95 patients (18%) in the remdesivir group and 98 (19%) in the placebo group, and finally an ordinal score of 4 (hospitalized, not requiring supplemental oxygen but requiring ongoing medical care) at 75 patients (14%) in the remdesivir group and 63 patients (12%) in the placebo group. In the ACTT-1 study, the study groups were described as similar in terms of demographic and baseline characteristics; however, the WHO Solidarity Consortium conducted a meta-analysis that included the ACTT-1 study and identified in its publication that the proportion of inpatients with good prognosis (those on no oxygen support or on low-flow oxygen) in the ACTT-1 study was “significantly greater” among those allocated to remdesivir than to placebo, and the WHO Solidarity Consortium identified this as contributing to the risk of bias for the outcome of time to recovery in the primary analysis of the ACTT-1 study.
In the study by Wang et al. (2020), the mean age of the patients was 66 years in the remdesivir group and 64 years in the placebo group; although these were reported in the publication alongside an IQR, standard error, or SD, the units of these values are not defined. The most common comorbidity was hypertension, followed by diabetes and coronary heart disease. Lopinavir-ritonavir was co-administered in 42 patients (18%) at baseline. Most patients were in category 3 on the 6-point ordinal scale of clinical status at baseline, which represents hospital admission requiring supplemental oxygen (82% and 83% in the remdesivir and placebo groups, respectively), followed by category 4, which represents hospital admission requiring HFNC or noninvasive mechanical ventilation (18% and 12% in the remdesivir and placebo groups, respectively). Very few patients were in category 2 (hospital admission, not requiring supplemental oxygen: 3 patients [4%] in the placebo group only) and category 5 (hospital admission requiring ECMO or invasive mechanical ventilation: 1 patient [1%] in the placebo group only). At baseline, 1 patient in the remdesivir group (1%) was labelled as category 6 (i.e., death).
In the WHO Solidarity trial, among patients treated with remdesivir or in the control group for remdesivir, the age group with the largest number of patients (46%) was 50 to 69 years, followed by younger than 50 years (32%) and 70 years or older (22%), which was consistent between treatment groups. The most common comorbidities were diabetes (27% in the remdesivir group and in the control group) and heart disease (22% and 23% in the 2 groups, respectively). At study entry, among patients treated with remdesivir, 869 (21%) required no oxygen support, 2,918 (70%) required oxygen, and 359 (9%) were already ventilated. Among patients in the control group, the corresponding values were 861 (21%), 2,921 (74%), and 347 (8%). More patients were male (62% in the remdesivir group and 64% in the control group) than female (37% in the remdesivir group and 36% in the control group). Race was not reported, but the majority of patients (> 50%) were treated for COVID-19 in Asia and Africa, approximately one-third of patients were from Europe or Canada, and approximately 15% were from Latin America. Compared to the global WHO Solidarity trial, the CATCO study22 (the Canadian substudy) had a higher proportion of patients with a background of diabetes (33.6% in the remdesivir group and 38.4% in the SOC group) and a higher proportion of female patients (41% and 39.4% in the 2 groups, respectively). In terms of racial and ethnic identity, the largest percentage of the CATCO study patients were white (42.4% and 39.4% in the remdesivir group and the SOC group, respectively), followed by South Asian (14.2% and 17.0%), and less than 10% of the patients were East Asian, Indigenous, Black, Arab, Latin American, or West Asian or had another racial or ethnic identity. In the CATCO study, 87.2% of patients were using corticosteroids at baseline. In terms of organ support at day 0 in the CATCO study, in the remdesivir and SOC groups, respectively, 11.2% and 8.4% of patients required no organ support, 52.7% and 56.2% of patients were on low-flow oxygen, 23.5% and 23.7% were on high-flow oxygen, 3.5% and 3.6% were on noninvasive ventilation, and 9.1% and 8.3% were on invasive mechanical ventilation.
In the study by Spinner et al. (2020), the mean age of patients was 58 years (IQR, 48 to 66 years) in the 5-day remdesivir group, 56 years (IQR, 45 to 66 years) in the 10-day remdesivir group, and 57 years (IQR, 45 to 66 years) in the SOC group. Although this was the only trial that included both adult patients and patients aged 12 years and older, the IQRs suggest that few patients were younger than 18 years. Overall, 38.9% of patients were female and 61.1% were male. More than half the patients indicated their race as white (57.8%), 18% indicated their race as Asian, and 17.5% indicated their race as Black. The most commonly reported comorbidities included cardiovascular disease (56.3%), hypertension (42.5%), diabetes (39.7%), and asthma (13.9%). Most patients at baseline had a clinical status of 5 on the 7-point scale, corresponding to being hospitalized, not requiring supplemental oxygen, but requiring ongoing medical care (84% in the 10-day remdesivir group, 84% in the 5-day remdesivir group, and 80% in the SOC group). The next most common clinical status was 4, which represents requiring low-flow supplemental oxygen (12%, 15%, and 18% in the 3 groups, respectively), followed by 6 (hospitalized but not requiring supplemental oxygen nor ongoing medical care: 3%, 0%, and 1% in the 3 groups, respectively) and 3 (requiring noninvasive ventilation or high-flow oxygen: 1% in each treatment group).
The mean age in the CARAVAN study was 15 years (SD = 1.71 years). A third of patients were male, and two-thirds were female. Five patients (41.7%) identified as Black and 7 (58.3%) identified as white; no other racial or ethnic identities were reported. History of asthma was reported in 3 patients, hypertension in 2 patients, and cardiomegaly in 1 patient. At baseline, 1 patient (8.3%) was on invasive mechanical ventilation, 6 (50%) were on high-flow oxygen, 2 (16.7%) were on low-flow oxygen, and 3 (25%) were on room air.
Table 13: Summary of Baseline Characteristics From Studies Included in the Systematic Review
Characteristic | ACTT-1 | Wang et al. (2020) | WHO Solidaritya | Spinner et al. (2020) | CARAVAN | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
RDV (mild-medium disease) (N = 82) | PBO (mild-medium disease) (N = 77) | RDV (severe disease) (N = 459) | PBO (severe disease) (N = 444) | RDV (N = 158) | PBO (N = 79) | RDV (N = 4,169) | Control (N = 4,151) | RDV 10 days (N = 197) | RDV 5 days (N = 199) | SOC (N = 200) | Cohort 1 RDV (N = 12) | |
Age (years), median (IQR) | NR | NR | NR | NR | NR | NR | NR | NR | 56 (45 to 66) | 58 (48 to 66) | 57 (45 to 66) | 15.0 (13.5 to 16.5) |
Age (years), mean (SD) | 57.2 (15.2) | 58.6 (14.6) | 58.9 (14.5) | 55.9 (15.9) | 66.0 (NR) | 64.0 (NR) | NR | NR | NR | NR | NR | 15.0 (1.71) |
Aged < 50 years, n (%) | NR | NR | NR | NR | NR | NR | 1,310 (32) | 1,326 (32) | NR | NR | NR | 12 (100) |
Aged 50 to 69 years, n (%) | NR | NR | NR | NR | NR | NR | 1,920 (46) | 1,908 (46) | NR | NR | NR | 0 |
Aged ≥ 70 years, n (%) | NR | NR | NR | NR | NR | NR | 916 (22) | 895 (22) | NR | NR | NR | 0 |
Male, n (%) | 58 (71) | 43 (56) | 294 (64) | 289 (65) | 89 (56) | 51 (65) | 2,601 (62) | 2,639 (64) | 118 (61) | 114 (60) | 125 (63) | 4 (33.3) |
Female, n (%) | 24 (29) | 34 (44) | 165 (36) | 155 (35) | 69 (44) | 27 (35) | 1,545 (37) | 1,490 (36) | 75 (39) | 77 (40) | 75 (38) | 8 (66.7) |
Race, white, n (%) | 39 (48) | 38 (49) | 240 (52) | 249 (56) | NR | NR | NR | NR | 107 of 188 (57) | 109 of 186 (59) | 112 of 193 (58) | 7 (58.3) |
Race, Black, n (%) | 17 (21) | 15 (19) | 92 (20) | 102 (23) | NR | NR | NR | NR | 37 of 188 (20) | 35 of 186 (19) | 27 of 193 (14) | 5 (41.7) |
Race, Asian, n (%) | 14 (17) | 18 (23) | 18 (23) | 38 (9) | NR | NR | NR | NR | 31 of 188 (16) | 34 of 186 (18) | 37 of 193 (19) | 0 |
Race, other or unknown, n (%) | 12 (14) | 6 (8) | 109 (24) | 55 (12) | NR | NR | NR | NR | 13 of 188 (7) | 8 of 186 (4) | 17 of 193 (9) | 0 |
BMI, median (IQR) | NR | NR | NR | NR | NR | NR | NR | NR | 28 (25 to 32) | 27 (24 to 30) | 27 (24 to 31) | 33.8 (21.6 to 46.5) |
BMI, mean (SD) | 27.94 (5.11) | 28.13 (6.74) | 31.15 (7.77) | 30.97 (7.61) | NR | NR | NR | NR | NR | NR | NR | 34.7 (13.38) |
Current smoker, n (%) | NR | NR | NR | NR | NR | NR | 247 (6) | 233 (6) | NR | NR | NR | NR |
History of diabetes, n (%) | 22 (27)b | 20 (26)b | 142 (32)b | 158 (30)b | 40 (25) | 16 (21) | 1,129 (27) | 1,120 (27) | 85 (44) | 71 (37) | 76 (38) | NR |
History of heart disease, n (%) | NR | NR | NR | NR | NR | NR | 929 (22) | 935 (23) | 111 (58)c | 111 (58)c | 107 (54)c | 1 (8.3)d |
History of hypertension, n (%) | 41 (50) | 34 (45) | 228 (51) | 230 (52) | 112 (71) | 30 (38) | NR | NR | 85 (44) | 82 (43) | 81 (41) | 2 (17) |
History of coronary heart disease, n (%) | 8 (10) | 7 (9) | 61 (14) | 50 (11) | 15 (9) | 2 (3) | NR | NR | NR | NR | NR | 0 |
History of chronic lung disease, n (%) | NR | NR | NR | NR | NR | NR | 281 (7) | 63 (2) | NR | NR | NR | 0e |
History of asthma, n (%) | 5 (6) | 5 (7) | 58 (13) | 52 (12) | NR | NR | 242 (6) | 44 (1) | 31 (16) | 22 (12) | 28 (14) | 3 (25) |
History of chronic liver disease, n (%) | NR | NR | NR | NR | NR | NR | 72 (2) | 15 (< 1) | NR | NR | NR | NR |
Days in hospital before study = 0, n (%) | NR | NR | NR | NR | NR | NR | 888 (21) | 892 (21) | NR | NR | NR | NR |
Days in hospital before study = 1, n (%) | NR | NR | NR | NR | NR | NR | 1,462 (35) | 1,459 (35) | NR | NR | NR | NR |
Days in hospital before study ≥ 2, n (%) | NR | NR | NR | NR | NR | NR | 1,796 (43) | 1,778 (43) | NR | NR | NR | NR |
BMI = body mass index; IQR = interquartile range; NR = not reported; PBO = placebo; RDV = remdesivir; SD = standard deviation; SOC = standard of care.
aPercentages were not reported by the WHO Solidarity trial and have been calculated for this review assuming that N represents the denominator for every baseline characteristic reported. Values reported are for the global WHO Solidarity trial; values specific to the CATCO study22 or other substudies are not reported.
bType 2 only.
cCardiovascular disease.
dCardiomegaly.
eChronic respiratory disease (appears to exclude asthma, which was experienced by 3 patient).
Sources: ACTT-1 Clinical Study Report;17 Wang et al. (2020);19 WHO Solidarity Trial Consortium (2022);18 Spinner et al. (2020);20 CARAVAN Clinical Study Report.21
Exposure to Study Treatments
Because the data for most of the included studies (WHO Solidarity, Spinner et al. [2020], and Wang et al. [2020]) came from publicly available data and not Clinical Study Reports, access to exposure-related data was limited for this review, aside from what is reported in the Patient Disposition section.
Exposure to study treatments in the ACTT-1 study and the CARAVAN study, for which Clinical Study Reports were available for this review, is reported in Table 14. In the ACTT-1 study, 98% and 99% of patients allocated to remdesivir and placebo, respectively, received the loading dose, and 38% and 43%, respectively, received all 10 infusions. The reason for receiving fewer than 10 infusions was most commonly due to discharge in both groups. In the CARAVAN study, of the 12 patients in cohort 1, 3 completed all 10 infusions, and the mean number of doses received was 7 (SD = 2.4), while the median was 5 (IQR, 5 to 9). Concomitant medications received in the ACTT-1 study included polymerase or protease inhibitors, corticosteroids, other anti-inflammatory drugs, and hydroxychloroquine or chloroquine (see Table 15); the differences in the percentage of patients taking these medications between the treatment arms appeared numerically small. In cohort 1 of the CARAVAN study, all 12 enrolled patients received at least 1 concomitant nonstudy COVID-19 medication, including experimental antivirals (25%), immune modulators (100%), anti-inflammatories (100%), enoxaparin (75%), ondansetron (33.3%), and enoxaparin sodium (8.3%).
Detailed exposure data from the WHO Solidarity trial were not available. Compliance was briefly summarized: of 4,077 patients allocated to remdesivir with in-hospital outcomes reported, 3,892 (95.5%) were taking remdesivir halfway through the scheduled treatment period, compared with 73 (1.8%) of 4,057 such patients allocated to the control group. In terms of concomitant medications, there was little difference between the treatment groups in use of corticosteroids (2,782 of 4,146 patients [67.1%] for remdesivir versus 2,820 of 4,129 patients [68.3%] in the control group).
Exposure data other than patient disposition were not reported in the publication by Wang et al. (2020). In terms of concomitant medications, 65% and 68% of patients allocated to remdesivir and placebo, respectively, received corticosteroid therapy during the study, and the mean duration of corticosteroid therapy was 9 days (IQR, 5 to 15 days) and 10 days (IQR, 6 to 16 days), respectively. Patients also received lopinavir-ritonavir (28% and 29% in the 2 groups, respectively), interferon alfa-2b (29% and 38%), and antibiotics (90% and 94%).
Co-interventions in the study by Spinner et al. (2020) included steroids, hydroxychloroquine, lopinavir-ritonavir, tocilizumab, and azithromycin. A similar proportion of patients received steroids across the 10-day remdesivir, 5-day remdesivir, and SOC treatment groups (15% to 19%). Patients randomized to the SOC group were more commonly prescribed other drugs with putative activity against COVID-19. Imbalances can be noted between the groups for concomitant use of lopinavir-ritonavir (6% and 5% in the 10-day and 5-day remdesivir groups, respectively, versus 22% in the SOC group), hydroxychloroquine or chloroquine (11% and 8% versus 45%), and azithromycin (21% and 18% versus 31%).
Table 14: Summary of Exposure in the ACTT-1 and CARAVAN Studies
Exposure | ACTT-1 | CARAVAN Cohort 1 | |
|---|---|---|---|
RDV (N = 541) | PBO (N = 521) | RDV (N = 12) | |
Received loading dose, n (%) | 531 (98) | 517 (99) | 12 (100) |
Completed all 10 infusions,a n (%) | 208 (38) | 226 (43) | 3 (25) |
Completed fewer than 10 infusions due to discharge, n (%) | 223 (41) | 158 (30) | NR |
Completed fewer than 10 infusions due to death, n (%) | 15 (3) | 19 (4) | NR |
Had any infusions slowed or halted, n (%) | 13 (2) | 12 (2) | NR |
Missed any maintenance dose,b n (%) | 87 (16) | 112 (21) | NR |
Doses received, mean (SD) | NR | NR | 7 (2.4) |
Doses received, median (IQR) | NR | NR | 5 (5 to 9) |
Doses received, range | NR | NR | 3 to 10 |
IQR = interquartile range; NR = not reported; PBO = placebo; RDV = remdesivir; SD = standard deviation.
aAn infusion is counted as complete even if it was halted or slowed.
bFor patients who died, were discharged, or were terminated from the study during the dosing period, scheduled doses after the time of death, discharge, or termination were not classified as missed.
Sources: ACTT-1 Clinical Study Report;17 CARAVAN Clinical Study Report.21
Table 15: Summary of Concomitant Medications in the ACTT-1 Study (As-Treated Population)
Concomitant medications by number of patients | RDV (N = 532) | PBO (N = 516) | ||
|---|---|---|---|---|
Mild-moderate disease (N = 55) | Severe disease (N = 477) | Mild-moderate disease (N = 49) | Severe disease (N = 467) | |
Antivirals, n (%) | 0 | 10 (2) | 2 (4) | 6 (1) |
Polymerase inhibitors, n (%) | 0 | 2 (< 1) | 0 | 3 (1) |
Protease inhibitors, n (%) | 0 | 8 (2) | 2 (4) | 3 (1) |
Corticosteroids, n (%) | 10 (18) | 105 (22) | 10 (20) | 116 (25) |
Other anti-inflammatory drugs, n (%) | 5 (9) | 37 (8) | 7 (14) | 30 (6) |
Monoclonal antibodies targeting cytokines, n (%) | 1 (2) | 22 (5) | 3 (6) | 23 (5) |
Other biologic therapies, n (%) | 4 (7) | 17 (4) | 4 (8) | 9 (2) |
Potential treatments for COVID-19, n (%) | 15 (27) | 175 (37) | 20 (41) | 181 (39) |
Hydroxychloroquine or chloroquine, n (%) | 15 (27) | 169 (35) | 19 (39) | 170 (36) |
Other, n (%) | 0 | 8 (2) | 1 (2) | 13 (3) |
PBO = placebo; RDV = remdesivir.
Source: ACTT-1 Clinical Study Report.17
Efficacy
Mortality
ACTT-1 Study
In the ITT population of the ACTT-1 study, the risk of death by day 15 was lower in the remdesivir group than in the placebo group (HR = 0.55; 95% CI, 0.36 to 0.83; P = 0.004). At day 29, the difference between groups was less apparent (HR = 0.73; 95% CI, 0.52 to 1.02; P = 0.066). The median time to death through day 15 or day 29 was not estimable for either treatment group in the ITT or as-treated populations. The KM survival curves for the remdesivir and placebo groups separated after approximately 5 days of study treatment, implying a lower incidence of mortality in the remdesivir group than in the placebo group starting from day 5 in the overall study population. Results were similar between the ITT and as-treated populations.
In ad hoc subgroup analyses of mortality by disease stratum or ordinal score, the greatest difference in percentage of deaths among patients with known mortality status at day 29 in the remdesivir group compared with the placebo group was observed in the subgroup with a baseline ordinal score of 5 (4.1% [9 of 222 patients] versus 12.8% [25 of 195 patients] in the remdesivir and placebo groups, respectively; HR = 0.30; 95% CI, 0.14 to 0.64; P < 0.001 [without adjustments for multiplicity]) and in the severe disease stratum (12.5% [57 of 457 patients] versus 16.3% [74 of 453 patients] in the remdesivir and placebo groups, respectively).
Table 16: Mortality in the ACTT-1 Study (ITT and As-Treated Populations)
Time frame | RDV | PBO | RDV vs. PBO HR for risk of death (95% CI) | P value | ||||
|---|---|---|---|---|---|---|---|---|
N | Died, n (%) | Mortality rate by KM (95% CI) | N | Died, n (%) | Mortality rate by KM (95% CI) | |||
ITT population | ||||||||
Day 15 | 541 | 35 (7) | 0.07 (0.05 to 0.09) | 521 | 61 (12) | 0.12 (0.09 to 0.15) | 0.55 (0.36 to 0.83) | 0.004 |
Day 29 | 541 | 59 (11) | 0.11 (0.09 to 0.15) | 521 | 77 (15) | 0.15 (0.12 to 0.19) | 0.73 (0.52 to 1.02) | 0.066 |
As-treated population | ||||||||
Day 15 | 532 | 35 (7) | 0.07 (0.05 to 0.09) | 516 | 61 (12) | 0.12 (0.09 to 0.15) | 0.55 (0.36 to 0.83) | 0.004 |
Day 29 | 532 | 59 (11) | 0.11 (0.09 to 0.14) | 516 | 77 (15) | 0.15 (0.12 to 0.19) | 0.73 (0.52 to 1.02) | 0.065 |
CI = confidence interval; HR = hazard ratio; ITT = intention to treat; KM = Kaplan-Meier; PBO = placebo; RDV = remdesivir; vs. = versus.
Source: ACTT-1 Clinical Study Report.17
Study by Wang et al. (2020)
Mortality was similar between the treatment groups at day 28 in the study by Wang et al. (2020). In the remdesivir group, 22 of 158 patients (14%) died, and in the placebo group 10 of 78 patients (13%) died, yielding a difference of 1.1% (95% CI, –8.1 to 10.3; P value not reported). Mortality was similar between treatment groups in subgroup analyses of patients who used remdesivir “early” (within 10 days of symptom onset) or “late” (more than 10 days after symptom onset), but the numerical results differed in direction: in the early-use subgroup, mortality was numerically higher in the placebo group, while in the late-use subgroup, mortality was numerically higher in the remdesivir group.
Table 17: Mortality in the Study by Wang et al. (2020) (ITT population)
Time frame | RDV | PBO | Rate difference, % (95% CI) | P value | ||
|---|---|---|---|---|---|---|
N | Died, n (%) | N | Died, n (%) | |||
ITT population | ||||||
Day 28 | 158 | 22 (14) | 78 | 10 (13) | 1.1 (–8.1 to 10.3) | NR |
ITT subgroup: Patients who used RDV early (≤ 10 days from symptom onset) | ||||||
Day 28 | 71 | 8 (11) | 47 | 7 (15) | –3.6 (–16.2 to 8.9) | NR |
ITT subgroup: Patients who used RDV late (> 10 days from symptom onset) | ||||||
Day 28 | 84 | 12 (14) | 31 | 3 (10) | 4.6 (–8.2 to 17.4) | NR |
CI = confidence interval; ITT = intention to treat; NR = not reported; PBO = placebo; RDV = remdesivir.
Source: Wang et al. (2020)19
WHO Solidarity Trial
Of 8,275 patients in the overall remdesivir analyses in the WHO Solidarity trial, 602 (14.5%) of the 4,146 patients assigned to remdesivir and 643 (15.6%) of the 4,129 patients assigned to the control group died (RR = 0.91; 95% CI, 0.82 to 1.02; P = 0.12). These analyses of in-hospital mortality included 15 palliative discharges in the remdesivir group and 11 in the control group. Analyses were also subdivided by oxygen support requirements at baseline; of these, the subgroup of patients who were already on oxygen (low or high flow) but not ventilated at baseline demonstrated a benefit of remdesivir over no remdesivir treatment in terms of in-hospital mortality (RR = 0.87; 95% CI, 0.76 to 0.99; P = 0.03).
In the Canadian substudy, CATCO,22 in-hospital mortality was 18.7% among patients assigned to receive remdesivir, compared with 22.6% in the SOC group (RR = 0.83; 95% CI, 0.67 to 1.03). Mortality at 60 days was 24.8% and 28.2% in the 2 groups, respectively (RR = 0.88; 95% CI, 0.72 to 1.07). In terms of in-hospital mortality among prespecified subgroups, the treatment effect did not vary in relation to age, sex, severity of disease (by respiratory support requirement), or duration of symptoms.
Table 18: In-Hospital Mortality (Including Palliative Discharges) in the WHO Solidarity Trial (ITT Population)
Time frame | RDV | Control | Rate ratio (95% CI) | P value | ||
|---|---|---|---|---|---|---|
N | Died, n (%) | N | Died, n (%) | |||
ITT population | ||||||
Day 28 | 4,146 | 602 (14.5) | 4,129 | 643 (15.6) | 0.91 (0.82 to 1.02) | 0.12 |
ITT subgroup: Patients who were not already on oxygen | ||||||
Day 28 | 869 | 25 (2.9) | 861 | 33 (3.8) | 0.76 (0.46 to 1.28) | 0.31 |
ITT subgroup: Patients who were already on oxygen (low or high flow) but not ventilated | ||||||
Day 28 | 2,918 | 426 (14.6) | 2,921 | 476 (16.3) | 0.87 (0.76 to 0.99) | 0.03 |
ITT subgroup: Patients who were already ventilated | ||||||
Day 28 | 359 | 151 (42.1) | 346 | 134 (38.6) | 1.13 (0.89 to 1.42) | 0.32 |
CI = confidence interval; ITT = intention to treat; RDV = remdesivir.
Source: WHO Solidarity Trial Consortium (2022).18
Study by Spinner et al. (2020)
In the 10-day remdesivir group (N = 193), 5-day remdesivir group (N = 191), and SOC group (N = 200) in the study by Spinner et al. (2020), a total of 3 (2%), 2 (1%), and 4 (2%) patients, respectively, died from any cause through 28 days of the trial.
The KM estimates of all-cause mortality at day 28 were 1% (95% CI, 0.0% to 2.6%; P = 0.43) versus SOC for the 5-day remdesivir group, 2% (95% CI, 0.0% to 3.6%; P = 0.72) versus SOC for the 10-day remdesivir group, and 2% (95% CI, 0.1% to 4.1%) for the SOC group.
CARAVAN Study
In cohort 1 (N = 12) of the CARAVAN study, there was 1 treatment-emergent death (8.3% of patients). Treatment-emergent death refers to deaths that occurred between the first dose date and the last dose date plus 30 days, inclusive, due to any cause.
Duration of Hospitalization
ACTT-1 Study
Among the 1,062 participants in the ITT population of the ACTT-1 study who were hospitalized, the median days of initial hospitalization, including imputations for patients who died, was 12 days (IQR, 6 to 28 days) in the remdesivir group (n = 541) and 17 days (IQR, 8 to 28 days) in the placebo group (n = 521), yielding a median difference of 5 days shorter with remdesivir (95% CI, 2.3 to 7.7 days).
Among the patients who did not die, the median days of initial hospitalization was 10 days (IQR, 5 to 21 days) in the remdesivir group (n = 480) and 14 days (IQR, 7 to 27 days) in the placebo group (n = 443).
The percentage of participants who were readmitted was 5% (95% CI, 3% to 7%) in the remdesivir group and 3% (95% CI, 2% to 5%) in the placebo group. Similar results were observed in the as-treated population.
Study by Wang et al. (2020)
The median duration of hospital stay in the study by Wang et al. (2020) was 25.0 days (IQR, 16.0 to 38.0 days) in the remdesivir group (n = 158) and 24.0 days (IQR, 18.0 to 36.0 days) in the placebo group (n = 78); the difference between groups was 0.0 days (IQR, –4.0 to 4.0 days).
WHO Solidarity Trial
Allocation to the remdesivir group (versus the control group) delayed discharge in the WHO Solidary trial by about 1 day during the 10-day treatment period. Data on duration of hospitalization were not otherwise reported.
In the Canadian substudy, CATCO,22 the duration of hospital stay was not different between the remdesivir and SOC groups: median = 10 of days (IQR, 6 to 18 days) in the remdesivir group versus 9 days (IQR, 6 to 17 days) in the SOC group.
Study by Spinner et al. (2020)
There were no significant differences between the remdesivir and SOC groups in duration of hospitalization in the study by Spinner et al. (2020). Data were not reported.
CARAVAN Study
In cohort 1 of the CARAVAN study, the mean duration of hospitalization from day 1 (days from first dose to date discharged alive; n = 9) was 12 days (SD = 5.5 days) and the median was 12 days (IQR, 8 to 15 days; range, 6 to 24 days). For the total duration of hospitalization (number of days from hospital admission to date discharged alive; n = 9), the mean was 14 days (SD = 5.2) and the median was 14 days (IQR, 9 to 16 days; range, 7 to 24 days). Only patients who were discharged alive on or before day 30 were included in the duration of hospitalization descriptive statistics.
Time to Recovery or Clinical Improvement
ACTT-1 Study
Results for the ACTT-1 study’s ITT population and as-treated population are presented in Table 19.
Among the ITT population, in patients in the severe disease stratum at randomization, the median time to recovery was 12 days (95% CI, 10 to 14 days) in the remdesivir group versus 19 days (95% CI, 16 to 21 days) in the placebo group (HR = 1.34; 95% CI, 1.14 to 1.58). In patients with any disease severity, the median time to recovery in the ITT population was 10 days (95% CI, 9 to 11 days) in the remdesivir group (n = 541) and 15 days (95% CI, 13 to 18 days) in the placebo group (n = 521). The KM curves for estimates of cumulative recoveries in patients in the severe disease stratum showed separation between the remdesivir and placebo groups after approximately day 4, suggesting a higher proportion of recoveries in the remdesivir group versus the placebo group starting from day 4.
In the ITT population, for patients in the mild-moderate disease stratum at randomization (remdesivir: n = 82; placebo: n = 77), the median time to recovery was 5 days (95% CI, 4 to 6 days) in the remdesivir group and 7 days (95% CI, 5 to 9 days) in the placebo group (HR = 1.10; 95% CI, 0.80 to 1.53).
The results were similar in the as-treated population.
Several subgroup analyses were conducted for the related outcome of the odds of better (lower) clinical status. Results by different definitions of disease severity at baseline are presented in Table 20, including baseline randomization stratification category (mild-moderate or severe disease) and baseline ordinal scale category (4, 5, 6, or 7, which correspond to levels of oxygen support requirements while in hospital). Between the study-defined categories of mild-moderate and severe disease, a statistically significant result in favour of remdesivir was only identified in the severe disease subgroup, which aligns with the primary analysis of time to recovery.
In contrast, among the subgroup analyses based on ordinal score, the only ordinal score subgroup demonstrating a statistically significant benefit in favour of remdesivir was ordinal score level 5 at baseline, which represents patients requiring supplemental oxygen but not high-flow oxygen or ventilation. This was also the largest subgroup. The subgroup for ordinal score 4 — patients who did not require any supplemental oxygen at baseline — did not demonstrate a statistically significant difference between treatment groups. Similarly, the more severe ordinal score categories, 6 (noninvasive ventilation or high-flow oxygen at baseline) and 7 (invasive mechanical ventilation or ECMO at baseline), did not demonstrate a statistically significant benefit of remdesivir over placebo. As the patients on low-flow oxygen outnumber the other categories by hundreds and demonstrated a benefit of remdesivir, the difference in number of patients in each category may be largely driving the benefit of remdesivir in time to recovery or clinical improvement seen in the severe disease subgroup.
Table 19: Time to Recovery by Treatment Group and Disease Severity in the ACTT-1 Study (ITT and As-Treated Populations)
Analysis population | Disease severity | Treatment group (N)b | nc | Median time to recovery (days) | HRa | P valued | ||
|---|---|---|---|---|---|---|---|---|
Estimate | 95% CI | Estimate | 95% CI | |||||
ITT population | Mild-moderatee | Remdesivir (82) | 75 | 5.0 | 4.0 to 6.0 | 1.10 | 0.80 to 1.53 | NR |
Placebo (77) | 71 | 7.0 | 5.0 to 9.0 | |||||
Severef | Remdesivir (459) | 324 | 12.0 | 10.0 to 14.0 | 1.34 | 1.14 to 1.58 | NR | |
Placebo (444) | 281 | 19.0 | 16.0 to 21.0 | |||||
Any severity | Remdesivir (541) | 399 | 10.0 | 9.0 to 11.0 | 1.29 | 1.12 to 1.49 | < 0.001 | |
Placebo (521) | 352 | 15.0 | 13.0 to 18.0 | |||||
As-treated population | Mild- moderatee | Remdesivir (82) | 75 | 5.0 | 4.0 to 6.0 | 1.10 | 0.80 to 1.53 | NR |
Placebo (76) | 71 | 7.0 | 5.0 to 9.0 | |||||
Severef | Remdesivir (450) | 324 | 12.0 | 10.0 to 14.0 | 1.33 | 1.14 to 1.57 | NR | |
Placebo (440) | 281 | 19.0 | 16.0 to 21.0 | |||||
Any severity | Remdesivir (532) | 399 | 10.0 | 9.0 to 11.0 | 1.29 | 1.11 to 1.48 | < 0.001 | |
Placebo (516) | 352 | 15.0 | 13.0 to 18.0 | |||||
CI = confidence interval; HR = hazard ratio; ITT = intention to treat; NR = not reported.
aThe HR for the “any severity” group is the HR from the stratified Cox proportional hazards model.
bWhere N is the number of patients in the specified population.
cWhere n is the number of recovered patients.
dP value calculated using the stratified log-rank test.
eMild-moderate disease was defined as having blood oxygen saturation greater than 94% and a respiratory rate less than 24 breaths per minute without supplemental oxygen.
fSevere disease was defined as requiring mechanical ventilation, requiring oxygen, having a blood oxygen saturation less than or equal to 94% on room air, or experiencing tachypnea (respiratory rate ≥ 24 breaths per minute).
Source: ACTT-1 Clinical Study Report.17
Table 20: Subgroup Analyses in the ACTT-1 Study (ITT Population): Odds Ratio for Better (Lower) Clinical Status Score at Day 15 Using Proportional Odds Model
Category | Subgroup | Treatment group | OR | P value | |
|---|---|---|---|---|---|
Estimate | 95% CI | ||||
Randomization stratification category at baseline | Mild-moderatea | Remdesivir (N = 82) | 1.2 | 0.7 to 2.2 | 0.475 |
Placebo (N = 77) | |||||
Severeb | Remdesivir (N = 459) | 1.6 | 1.3 to 2.0 | < 0.001 | |
Placebo (N = 444) | |||||
Ordinal scale category at baselinec | 4 (not requiring supplemental oxygen but requiring ongoing medical care) | Remdesivir (N = 75) | 1.5 | 0.8 to 2.7 | 0.234 |
Placebo (N = 63) | |||||
5 (requiring supplemental oxygen) | Remdesivir (N = 232) | 1.6 | 1.2 to 2.3 | 0.004 | |
Placebo (N = 203) | |||||
6 (on noninvasive ventilation or high-flow oxygen devices) | Remdesivir (N = 95) | 1.4 | 0.9 to 2.3 | 0.186 | |
Placebo (N = 98) | |||||
7 (on invasive mechanical ventilation or ECMO) | Remdesivir (N = 131) | 1.2 | 0.8 to 1.9 | 0.369 | |
Placebo (N = 154) | |||||
CI = confidence interval; ECMO = extracorporeal membrane oxygenation; ITT = intention to treat; OR = odds ratio.
aMild-moderate disease was defined as having blood oxygen saturation greater than 94% and a respiratory rate less than 24 breaths per minute without supplemental oxygen.
bSevere disease was defined as requiring mechanical ventilation, requiring oxygen, having a peripheral oxygen saturation less than or equal to 94% on room air, or experiencing tachypnea (respiratory rate ≥ 24 breaths per minute).
cThe scale ranges from 1 to 8, where 1 represents “not hospitalized, no limitations on activities” and 8 represents “death” and higher odds of a lower score is a desired outcome. Only subgroups for scores 4 to 7, inclusive, are reported in the subgroup analysis, which includes hospitalized patients with various levels of oxygen support requirements.
Source: ACTT-1 Clinical Study Report.17
Study by Wang et al. (2020)
In the ITT population of the study by Wang et al. (2020), the time to clinical improvement in the remdesivir group (median = 21.0 days; IQR, 13.0 to 28.0 days) was not significantly different from that of the placebo group (median = 23.0 days; IQR,15.0 to 28.0 days). The HR was 1.23 (95% CI, 0.87 to 1.75).
Results for time to clinical improvement were similar in the per-protocol population, with a median of 21.0 days (IQR, 13.0 to 28.0 days) in the remdesivir group versus 23.0 days (IQR, 15.0 to 28.0 days) in the placebo group. The HR was 1.27 (95% CI, 0.89 to 1.80).
Although not statistically significant, in the ITT population, patients receiving remdesivir within 10 days of symptom onset had a numerically faster time to clinical improvement than those receiving placebo within 10 days of symptom onset (remdesivir: median = 18.0 days; IQR, 12.0 to 28.0 days; placebo: median = 23.0 days; IQR, 15.0 to 28.0 days). The HR was 1.52 (95% CI, 0.95 to 2.43).
If clinical improvement was defined as a 1-category decline, instead of a 2-cateogry decline, the HR was 1.34 (95% CI, 0.96 to 1.86).
WHO Solidarity Trial
This outcome was not assessed in the WHO Solidarity trial.
Study by Spinner et al. (2020)
In the study by Spinner et al. (2020), there were no significant differences between the 10-day remdesivir group and the SOC group for time to a 2-point or greater improvement in clinical status (HR = 1.16; 95% CI, 0.93 to 1.43), time to a 1-point or greater improvement in clinical status (HR = 1.10; 95% CI, 0.90 to 1.36), time to recovery (HR = 1.11; 95% CI, 0.90 to 1.37), or time to modified recovery (HR = 1.10; 95% CI, 0.90 to 1.36).
There were also no significant differences between the 5-day remdesivir group and the SOC group for time to a 2-point or greater improvement in clinical status (HR = 1.15; 95% CI, 0.93 to 1.42), time to a 1-point or greater improvement in clinical status (HR = 1.19; 95% CI, 0.97 to 1.47), time to recovery (HR = 1.18; 95% CI, 0.96 to 1.45), or time to modified recovery (HR = 1.19; 95% CI, 0.19 to 1.46).
CARAVAN Study
The median time to recovery in cohort 1 of the CARAVAN study was 12 days (IQR, 6 to 24 days).
Initiation of Ventilation
ACTT-1 Study
Incidence rates of new oxygen support usage in the ACTT-1 study are reported in Table 21.
The incidence rate of new noninvasive ventilation or high-flow oxygen use among patients who were not already on these supports (nor ventilated) at baseline, was 0.17 (95% CI, 0.13 to 0.22) in the remdesivir group and 0.24 (95% CI, 0.19 to 0.30) in the placebo group. The incidence rate in the remdesivir group was numerically lower, but the 95% CIs of each group overlap.
The incidence rate of new invasive mechanical ventilation or ECMO use among patients not already on these supports at baseline was 0.13 (95% CI, 0.10 to 0.17) in the remdesivir group and 0.23 (95% CI, 0.19 to 0.27) in the placebo group. The incidence rate in the remdesivir group was numerically lower, and the 95% CIs of each group do not overlap.
Similar results were observed in the as-treated population (not displayed).
Table 21: Initiation of Oxygen Support in the ACTT-1 Study (ITT Population)
Subgroup | Statistic | RDV | PBO |
|---|---|---|---|
Initiation of new noninvasive ventilation or high-flow oxygen use | |||
Patients not on noninvasive ventilation, high-flow oxygen, invasive mechanical ventilation, or ECMO at baseline | Patients in subgroup, N | 307 | 266 |
Patients with event, n | 52 | 64 | |
Incidence rate (95% CI) | 0.17 (0.13 to 0.22) | 0.24 (0.19 to 0.30) | |
Initiation of new mechanical ventilation or ECMO | |||
Patients not on invasive mechanical ventilation or ECMO at baseline | Patients in subgroup, N | 402 | 364 |
Patients with event, n | 52 | 82 | |
Incidence rate (95% CI) | 0.13 (0.10 to 0.17) | 0.23 (0.19 to 0.27) | |
CI = confidence interval; ECMO = extracorporeal membrane oxygenation; ITT = intention to treat; PBO = placebo; RDV = remdesivir.
Source: ACTT-1 Clinical Study Report.17
Study by Wang et al. (2020)
This outcome was not assessed in the study by Wang et al. (2020).
WHO Solidarity Trial
In Table 22, progression to ventilation and progression to a composite outcome of in-hospital death or ventilation are each presented for the WHO Solidarity trial. Assignment to remdesivir was associated with a lower rate of progression to ventilation (event RR = 0.88; 95% CI, 0.77 to 1.00; P = 0.04) and with a lower composite outcome of death or ventilation (event RR = 0.84; 95% CI, 0.75 to 0.93; P = 0.001). For both outcomes, results for the subgroup of patients not receiving oxygen support at entry had an associated 95% CI that crossed the null, whereas results for the subgroup of patients who were receiving low-flow or high-flow oxygen at entry showed a statistically significant benefit of remdesivir. The latter subgroup was also much larger, and so this subgroup (patients already on low-flow or high-flow oxygen at baseline) appears to drive the observed benefit of remdesivir for this outcome.
In the Canadian substudy, CATCO,22 among patients not mechanically ventilated at baseline, 8.0% of those assigned remdesivir required mechanical ventilation during the study, compared to 15.0% of those assigned SOC (RR = 0.53; 95% CI, 0.38 to 0.75).
Table 22: Initiation of Oxygen Support in the WHO Solidarity Trial (ITT Population)
Subgroup | Statistic | RDV | Control |
|---|---|---|---|
Progression to ventilation | |||
No oxygen support at study entry | Patients in subgroup, N | 869 | 861 |
Patients with event, n (%) | 39 (4.5) | 40 (4.6) | |
Observed – expected (variance) | –0.4 (18.6) | ||
Event RR (95% CI) | 0.98 (0.62 to 1.54) | ||
Receiving oxygen but not ventilated at study entrya | Patients in subgroup, N | 2,918 | 2,921 |
Patients with event, n (%) | 496 (17.0) | 553 (18.9) | |
Observed – expected (variance) | –30.2 (213.5) | ||
Event RR (95% CI) | 0.87 (0.76 to 0.99) | ||
Total | Patients in subgroups, N | 3,787 | 3,782 |
Patients with event, n (%) | 535 (14.1) | 593 (15.7) | |
Observed – expected (variance) | –30.6 (232.1) | ||
Event RR (95% CI) | 0.88 (0.77 to 1.00)b | ||
Death or progression to ventilation | |||
No oxygen support at study entry | Patients in subgroup, N | 869 | 861 |
Patients with event, n (%) | 52 (6.0) | 58 (6.7) | |
Observed – expected (variance) | –2.8 (26.3) | ||
Event RR (95% CI) | 0.90 (0.61 to 1.32) | ||
Receiving oxygen but not ventilated at study entrya | Patients in subgroup, N | 2,918 | 2,921 |
Patients with event, n (%) | 692 | 793 | |
Observed – expected (variance) | –58.8 (320.3) | ||
Event RR (95% CI) | 0.83 (0.75 to 0.93) | ||
Total | Patients in subgroups, N | 3,787 | 3,782 |
Patients with event, n (%) | 744 (19.6) | 851 (22.5) | |
Observed – expected (variance) | –61.5 (346.6) | ||
Event RR (95% CI) | 0.84 (0.75 to 0.93)c | ||
CI = confidence interval; ITT = intention to treat; RDV = remdesivir; RR = relative risk.
aHigh-flow and low-flow oxygen were not recorded separately at entry into the WHO Solidarity trial.
bP = 0.04.
cP = 0.001.
Source: WHO Solidarity Trial Consortium (2022).18
Study by Spinner et al. (2020)
This outcome was not assessed in the study by Spinner et al. (2020).
CARAVAN Study
This outcome was not assessed in the CARAVAN study.
Duration of Oxygen Support
Although duration of oxygen support or ventilation was not selected as a key outcome of interest based on consultation with the clinical expert, related outcomes are reported in Appendix 1, Table 26, as supplementary data. The ACTT-1 study reported the median days on oxygen, on noninvasive ventilation or high-flow oxygen, or on invasive mechanical ventilation or ECMO. Although statistical comparisons were not conducted and the IQRs overlapped between groups, the median days on oxygen or on invasive mechanical ventilation or ECMO were lower in the remdesivir group than in the placebo group. The median days on noninvasive ventilation or high-flow oxygen was the same in both groups. The CATCO study22 reported a significant benefit associated with allocation to remdesivir in terms of the mean oxygen-free days and mean ventilator-free days at day 28. Wang et al. (2020) reported a lower median number of days of invasive mechanical ventilation and of oxygen support in the remdesivir group than in the placebo group, although again the IQRs overlapped. Spinner et al. (2020) reported no significant difference between either of the remdesivir groups and the SOC group in the duration of oxygen support. There is therefore some evidence to suggest a modest benefit of remdesivir on the duration of some forms of oxygen support, with some uncertainty due to overlapping IQRs and inconsistency between the studies.
Summary of Major Conclusions Reported in Comparative Study Results
A text summary of the direction of results comparing remdesivir to either placebo, control groups, or SOC is presented in Table 23. The CARAVAN study was single-arm trial, so it is not included in this summary as no comparative conclusions can be drawn.
Table 23: Summary of Major Conclusions Reported in Comparative Study Results (Primary Analysis Populations Unless Otherwise Specified)
Outcome | ACTT-1 | Wang et al. (2020) | WHO Solidarity | Spinner et al. (2020) | |||||
|---|---|---|---|---|---|---|---|---|---|
RDV (N = 532) | PBO (N = 516) | RDV (N = 155) | PBO (N = 78) | RDV (N = 4,146) | Control (N = 4,129) | RDV 10 days (N = 193) | RDV 5 days (N = 191) | SOC (N = 200) | |
Mortality | Statistically significant benefit of RDV vs. PBO at day 15, but no significant difference at day 29. | No statistically significant difference. | No significant benefit in ITT population or in subgroups of patients not on oxygen or of patients already ventilated. Significant benefit of RDV for in-hospital mortality in subgroup of patients on oxygen (high or low flow) but not ventilated at baseline. | No statistically significant difference between either of the RDV groups vs. the SOC group. | |||||
Duration of hospitalization | Duration shorter among patients treated with RDV by a median of 5 days. | No statistically significant difference. | Allocation to RDV delayed discharge by about 1 day (unknown if statistically significant); attributed by authors to duration of RDV course requiring inpatient care, not due to lack of pharmacological activity. | No statistically significant difference. | |||||
Time to recovery or clinical improvement | Benefit of RDV in ITT population. Benefit of RDV in severe but not mild-moderate disease subgroup. Benefit of RDV in subgroup with baseline ordinal score of 5 (low-flow oxygen) but not any other subgroup (no oxygen support; high-flow or noninvasive ventilation; invasive ventilation or ECMO). The subgroup with the baseline ordinal score of 5 is the largest subgroup by a substantial margin and may bias other results. | No significant difference. | Not assessed. | No statistically significant difference. | |||||
Incidence of ventilation | For the incidence rate of initiation of new noninvasive ventilation or high-flow oxygen (in patients not already receiving these at baseline), the 95% CIs between the 2 groups overlap slightly, suggesting there may not be a significant difference between RDV and PBO. However, the incidence rate in the RDV group was numerically lower. However, for the incidence rate of initiation of invasive mechanical ventilation or ECMO (in patients not already receiving these at baseline), the 95% CIs do not overlap, and the incidence rate for the RDV group was numerically lower than in the PBO group, suggesting a possible benefit of RDV in preventing this outcome. | Not assessed. | There was a statistically significant benefit of RDV vs. the control group per the relative risk for initiation of ventilation among patients not already ventilated at entry. Additionally, there was a statistically significant benefit of RDV vs. the control group for the composite outcome of “death or progression to ventilation” among patients not already ventilated at entry. Results appear driven by the largest subgroup of patients (patients who were on low-flow or high-flow oxygen at baseline), who also had the most benefit for this outcome. | Not assessed. | |||||
Duration of oxygen support | Although statistical comparisons were not conducted and the IQRs overlapped between groups, the median days on oxygen or on invasive mechanical ventilation or ECMO were lower in the RDV group than in the PBO group. The median days was similar for duration of noninvasive ventilation or high-flow oxygen. | The median days of invasive mechanical ventilation and the median days of oxygen support were lower in the RDV group than in the PBO group, although the IQRs overlapped. | In the Canadian substudy, CATCO, there was a significant benefit associated with allocation to RDV in terms of the mean number of oxygen-free days and of ventilator-free days at day 28. | There were no significant differences between the RDV and SOC groups in duration of oxygen therapy. | |||||
CI = confidence interval; ECMO = extracorporeal membrane oxygenation; IQR = interquartile range; ITT = intention to treat; PBO = placebo; RDV = remdesivir; SOC = standard of care; vs. = versus.
Sources: ACTT-1 Clinical Study Report;17 Wang et al. (2020);19 WHO Solidarity Trial Consortium (2022);18 Spinner et al. (2020).20
Harms
Adverse Events
The proportion of patients who experienced at least 1 AE ranged from 51% to 64% across the 4 RCTs and was 91.7% in cohort 1 of the CARAVAN study. The studies differed substantially in which particular AEs were reported, but there was a trend across the trials of focus biomarkers related to kidney and liver function, hyperglycemia, and some clinical AEs such as headache, constipation, pyrexia, and diarrhea. Where reported, AEs were generally similar between treatment groups, although in some cases there were numerically more AEs in the placebo or SOC group than the remdesivir group.
Serious AEs
Within the 4 RCTs, the proportion of patients who experienced at least 1 SAE ranged from 5% in both remdesivir groups of the study by Spinner et al. (2010) to 32% in the placebo group of the ACTT-1 study. In cohort 1 of the CARAVAN study, 5 patients (41.7%) experienced an SAE.
In the ACTT-1 study, a substantially higher proportion of patients experienced SAEs in the placebo arm (32%) than in the remdesivir arm (25%). This was also the case in the study by Wang et al. (2020): 26% of patients in the placebo arm versus 18% in the remdesivir arm. In the study by Spinner et al. (2020), in both remdesivir groups — 10 day and 5 day — 5% of patients experienced at least 1 SAE; in the SOC group, 9% of patients experienced at least 1 SAE. The WHO Solidarity trial did not report this outcome.
The studies were inconsistent with regard to which SAEs they reported. The most common SAE reported in the ACTT-1 study and the study by Wang et al. (2020) was respiratory failure, which in the remdesivir groups occurred in 7% and 10% of patients in the 2 studies, respectively, and in the placebo groups occurred in 11% and 8% of patients, respectively.
Withdrawals due to AEs
Withdrawals due to AEs were relatively high in the ACTT-1 study, occurring in 11.1% of patients in the remdesivir group and 15% of patients in the placebo group. In the study by Wang et al. (2020), 12% and 5% of patients in the remdesivir and placebo groups, respectively, withdrew due to AEs. In the WHO Solidarity trial, withdrewal due to AEs was not reported. In the study by Spinner et al. (2020), the rate of withdrawal due to AEs was4% in the 10-day remdesivir group, and 2% in the 5-day remdesivir group. In the CARAVAN study, 2 patients (16.7%) withdrew due to AEs.
Mortality
Mortality has been described in depth in the Efficacy section due to the nature of this review. The ACTT-1 study demonstrated a benefit of remdesivir over placebo in mortality, which may have been driven by the patients on low-flow oxygen support at baseline, based on subgroup analyses. Similarly, the WHO Solidarity trial demonstrated a benefit of remdesivir over SOC in the subgroup of patients on low-flow or high-flow oxygen. In both trials, subgroup analyses of patients on noninvasive ventilation, on invasive mechanical ventilation or ECMO, or on no oxygen support at baseline did not demonstrate a benefit of remdesivir.
Table 24: Summary of Harms Results From Studies Included in the Systematic Review
AEs | ACTT-1 (any severity) | Wang et al. (2020) | WHO Solidarity | Spinner et al. (2020) | CARAVAN | |||||
|---|---|---|---|---|---|---|---|---|---|---|
RDV (N = 532) | PBO (N = 516) | RDV (N = 155) | PBO (N = 78) | RDV (N = 4,146) | Control (N = 4,129) | RDV 10 days (N = 193) | RDV 5 days (N = 191) | SOC (N = 200) | Cohort 1 RDV (N = 12) | |
Overall AEs, n (%) | ||||||||||
≥ 1 AE | 305 (57) | 323 (63) | 102 (66) | 50 (64) | NR | NR | 113 (59) | 98 (51) | 93 (47) | 11 (91.7) |
≥ 1 grade 3 or higher AE | 273 (51) | 295 (57) | 13 (8) | 11 (14) | NR | NR | 24 (12) | 20 (10) | 24 (12) | 6 (50.0) |
Study drug–related AE | 41 (8) | 47 (9) | NR | NR | NR | NR | NR | NR | NR | 4 (33.3) |
Study drug–related grade 3 or higher AE | NR | NR | NR | NR | NR | NR | NR | NR | NR | 3 (25.0) |
Most common AEs, n (%) | ||||||||||
Constipation | NR | NR | 21 (14) | 12 (15) | NR | NR | NR | NR | NR | 3 (25.0) |
Acute kidney injury | 28 (5) | 33 (6) | NR | NR | NR | NR | NR | NR | NR | 4 (33.3) |
Hyperglycaemia | 34 (6) | 34 (7) | 11 (7) | 6 (8) | NR | NR | NR | NR | NR | 1 (8.3) |
Pyrexia | 38 (7) | 34 (7) | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Alanine aminotransferase increased | 18 (3) | 33 (6) | NR | NR | NR | NR | 57 of 177 (32) | 61 of 179 (34) | 71 of 182 (39) | 2 (16.7) |
Aspartate aminotransferase increase | NR | NR | 7 (5) | 9 (12) | NR | NR | 56 of 175 (32) | 56 of 177 (32) | 60 of 182 (33) | 1 (8.3) |
Hypertension | NR | NR | NR | NR | NR | NR | NR | NR | NR | 2 (16.7) |
Blood creatinine increased | 32 (6) | 36 (7) | NR | NR | NR | NR | NR | NR | NR | NR |
Glomerular filtration rate decreased | 60 (11) | 76 (15) | NR | NR | NR | NR | NR | NR | NR | NR |
Creatinine clearance decrease | NR | NR | NR | NR | NR | NR | 45 of 176 (26) | 26 of 178 (15) | 55 of 183 (30) | NR |
Hemoglobin decreased | 49 (9) | 62 (12) | NR | NR | NR | NR | NR | NR | NR | NR |
Lymphocyte count decreased | 45 (8) | 54 (10) | NR | NR | NR | NR | NR | NR | NR | NR |
Respiratory failure | 37 (7) | 60 (12) | NR | NR | NR | NR | NR | NR | NR | NR |
Respiratory distress | 18 (3) | 27 (5) | NR | NR | NR | NR | NR | NR | NR | NR |
Lymphopenia | 13 (2) | 30 (6) | NR | NR | NR | NR | NR | NR | NR | NR |
Nausea | NR | NR | 8 (5) | 2 (3) | NR | NR | 18 (9) | 19 (10) | 6 (3) | NR |
Diarrhea | NR | NR | 5 (3) | 2 (3) | NR | NR | 10 (5) | 12 (6) | 14 (7) | NR |
Hypokalemia | NR | NR | 18 (12) | 11 (14) | NR | NR | 13 (7) | 10 (5) | 4 (2) | NR |
Headache | NR | NR | NR | NR | NR | NR | 10 (5) | 10 (5) | 5 (3) | NR |
Anemia | NR | NR | 18 (12) | 12 (15) | NR | NR | NR | NR | NR | NR |
Vomiting | NR | NR | 4 (3) | 2 (3) | NR | NR | NR | NR | NR | NR |
Hypoalbuminemia | NR | NR | 20 (13) | 12 (15) | NR | NR | NR | NR | NR | NR |
Hypokalemia | NR | NR | 18 (12) | 11 (14) | NR | NR | NR | NR | NR | NR |
Rash | NR | NR | 16 (10) | 5 (6) | NR | NR | NR | NR | NR | NR |
Thrombocytopenia | NR | NR | 15 (10) | 7 (9) | NR | NR | NR | NR | NR | NR |
Increased total bilirubin | NR | NR | 15 (10) | 7 (9) | NR | NR | NR | NR | NR | NR |
Increased blood lipids | NR | NR | 10 (6) | 8 (10) | NR | NR | NR | NR | NR | NR |
Increased white blood cell count | NR | NR | 11 (7) | 6 (8) | NR | NR | NR | NR | NR | NR |
Hyperlipidemia | NR | NR | 10 (6) | 8 (10) | NR | NR | NR | NR | NR | NR |
Increased blood urea nitrogen | NR | NR | 10 (6) | 5 (6) | NR | NR | NR | NR | NR | NR |
Increased neutrophil | NR | NR | 10 (6) | 4 (5) | NR | NR | NR | NR | NR | NR |
SAEs, n (%) | ||||||||||
≥ 1 SAE | 131 (25) | 163 (32) | 28 (18) | 20 (26) | NR | NR | 10 (5) | 9 (5) | 18 (9) | 5 (41.7) |
≥ 1 study drug–related SAE | 2 (< 1) | 3 (1) | NR | NR | NR | NR | NR | NR | NR | 0 |
Multiple organ dysfunction syndrome | NR | NR | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Pyrexia | NR | NR | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Respiratory distress | NR | NR | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Septic shock | NR | NR | 1 (1) | 1 (1) | NR | NR | NR | NR | NR | 1 (8.3) |
Thrombosis | NR | NR | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Acute kidney injury | 7 (2) | 12 (2) | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Empyema | NR | NR | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Negative pressure pulmonary edema | NR | NR | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Pulmonary hemorrhage | NR | NR | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Vomiting | NR | NR | NR | NR | NR | NR | NR | NR | NR | 1 (8.3) |
Respiratory failure | 35 (7) | 58 (11) | 16 (10) | 6 (8) | NR | NR | NR | NR | NR | 0 |
Cardiopulmonary failure | NR | NR | 8 (5) | 7 (9) | NR | NR | NR | NR | NR | NR |
Pulmonary embolism | NR | NR | 1 (1) | 1 (1) | NR | NR | NR | NR | NR | NR |
Cardiac arrest | NR | NR | 1 (1) | 0 | NR | NR | NR | NR | NR | NR |
Acute coronary syndrome | NR | NR | 0 | 1 (1) | NR | NR | NR | NR | NR | NR |
Tachycardia | NR | NR | 0 | 1 (1) | NR | NR | NR | NR | NR | NR |
Patients who stopped treatment due to AEs, n (%) | ||||||||||
≥ 1 AE leading to premature study drug discontinuation | 57 (11) | 77 (15) | 18 (12) | 4 (5) | NR | NR | 8 (4) | 4 (2) | NA | 2 (16.7) |
Deaths, n (%) | ||||||||||
Patients who died | 59 (11.1) | 77 (15) | 22 of 150 (15) | 10 of 77 (13) | 602 (14.5) | 643 (15.6) | 3 (2) | 2 (1) | 4 (2) | 1 (8.3) |
AE = adverse event; NA, not applicable; NR = not reported; PBO = placebo; RDV = remdesivir; SAE = serious adverse event; SOC = standard of care.
Sources: ACTT-1 Clinical Study Report,17 Wang et al. (2020),19 WHO Solidarity Trial Consortium,18 Spinner et al. (2020),20 CARAVAN Clinical Study Report.21
Critical Appraisal
Internal Validity
This review included 5 clinical trials, 4 of which were RCTs and 1 of which was a single-arm study. Of the 4 RCTs, 2 were double blind (ACTT-1 and Wang et al. [2020]). The WHO Solidarity trial and the study by Spinner et al. (2020), being open label, have an elevated risk of bias in subjective outcomes as a result of patients knowing their treatment assignment; however, the outcomes of interest assessed for this review were all objective and unlikely to be affected by this. Nonetheless, the open-label design may have also elevated the risk of between-group differences in the concomitant treatment decisions made by clinicians, and indeed this can be observed in that patients randomized to SOC in the study by Spinner et al. (2020) were more likely to receive therapies with putative anti–COVID-19 activity. A similar observation was not made in the WHO Solidarity trial, and the number of patients receiving corticosteroids was similar between treatment groups. However, reporting of data related to other concomitant therapies and the nature of SOC was sparse in the publication of the WHO Solidarity trial, so it is uncertain whether there are unreported differences. In any case, the potential bias caused by a higher likelihood of receiving putative anti–COVID-19 medications in the control arm would be expected to bias against the intervention (i.e., against remdesivir) by hypothetically improving the outcomes of the control group. As expected, the proportions of patients receiving concomitant medications in the double-blind RCTs (ACTT-1 and Wang et al. [2020]), of those reported, were relatively balanced between the treatment arms of the studies.
Most baseline patient characteristics appeared to be balanced between treatment groups in each study, with the exception of some concerns of imbalance in clinical status (i.e., oxygen support requirements) between the treatment arms of some studies. Most notably, the authors of the WHO Solidarity trial publications criticized the balance of the ACTT-1 study treatment groups and suggested that patients with “good” prognosis (i.e., patients who were unventilated at baseline) were overrepresented in the remdesivir group compared to the placebo group. Patients with an ordinal score of 5, which in the ACTT-1 study represented those who were hospitalized and requiring supplemental oxygen (but not high-flow oxygen or ventilation), formed the largest subgroup in the ACTT-1 study overall: 43% of the remdesivir group and 39% of the placebo group fit into this category. The clinical significance of this between-group difference is uncertain: it was the stance of the authors of the WHO Solidarity trial publications that this represents a substantial source of bias, but the clinical expert consulted by CDA-AMC did not feel it was an important difference. Nonetheless, the subgroup results for outcomes related to clinical recovery in the ACTT-1 study demonstrated that only this subgroup showed a benefit of remdesivir over placebo, in contrast to any other clinical status–related subgroup (i.e., patients not requiring any oxygen support, patients requiring high-flow oxygen or noninvasive ventilation, and patients requiring invasive ventilation or ECMO), which all demonstrated no significant benefit. Similarly, this subgroup demonstrated the strongest benefit in mortality associated with remdesivir. The randomization stratification categories in the ACTT-1 study, “mild-moderate” and “severe,” were broad, with the latter hypothetically encompassing patients from all reported ordinal score subgroups (4 to 7, inclusive). Given the smaller population sizes of the groups that did not show benefit, this result could hypothetically be due the sample size being insufficient to demonstrate a benefit.
The results of the WHO Solidarity trial demonstrated a benefit in mortality for a similar subgroup (patients already on oxygen, low flow or high flow, but not ventilated) but not other subgroups (patients not on oxygen support or patients who were already ventilated). As the ACTT-1 study and the WHO Solidarity trial differed in which subgroup contained patients on high-flow oxygen, it is uncertain whether there is a benefit of remdesivir in these patients or whether the apparent benefit is driven entirely by patients on low-flow oxygen. When patients on high-flow oxygen were grouped with those receiving noninvasive ventilation in the ACTT-1 study, there was uncertainty about the benefit of remdesivir on mortality in this subgroup; however, when patients on high-flow oxygen were grouped with those on low-flow oxygen in the WHO Solidarity trial, there was an apparent benefit of remdesivir on mortality in this subgroup. Taken together, the subgroup of patients receiving low-flow oxygen — and perhaps including those receiving high-flow oxygen as well, but this was inconsistent between the studies — was both the largest subgroup and the one most likely to benefit from treatment with remdesivir, at least in clinical status (the ACTT-1 study) and mortality (both the ACTT-1 study and the WHO Solidarity trial). As such, the imbalance between groups in the ACTT-1 study may be clinically important and may bias the results in favour of remdesivir. These observations from the ACTT-1 study and the WHO Solidarity trial align with the recommendations by WHO’s Living Guidance for Clinical Management of COVID-196 and the Canadian practice recommendations by Grant et al. (2024),33 which identify patients on low-flow oxygen as the strongest candidates for treatment with remdesivir and recommend its use (with a caveat for weak or conditional recommendation in the WHO guidance, specifically).
The Canadian substudy of the WHO Solidarity trial, CATCO,22 generally demonstrated consistent results with the global Solidarity study, with the exception that the subgroup analysis by baseline organ support requirements did not demonstrate any particular difference between subgroups in mortality. However, the substudy was much smaller than the global study and, as such, may not have been powered to detect differences in these subgroups.
Mortality, which was highlighted by the clinical expert as the most critical outcome, was reported in every included study. Duration of hospital stay, time to recovery or clinical improvement, and initiation of new ventilation were also noted by the clinical expert to be important outcomes and were commonly reported by at least some of the studies. The ordinal scales used to assess clinical status (as an abstraction of what level of oxygen support patients required and a patient’s hospitalization status) varied between studies and were not standardized, nor was the definition of clinical “recovery” or “improvement.” It was unclear in the Clinical Study Report of the ACTT-1 study whether the primary outcome, time to recovery, was adjusted for multiplicity. No other reported outcomes appeared to be adjusted for multiple comparisons in any included study, which increases the risk of type I error.
The ACTT-1 study included sensitivity analyses for identified outliers. Those results were consistent with the primary analyses.
External Validity
The included studies were each conducted in the early stages of the COVID-19 pandemic. There are substantial concerns regarding external validity and generalizability in every study included in this review because of the fast-evolving nature of the pandemic and the virus itself: prevalent variants, levels of vaccination, and clinical outcomes in today’s world are substantially different than those observed in the early pandemic. The clinical expert consulted by CDA-AMC highlighted that the current need for remdesivir is infrequent as relatively few patients are now presenting with COVID-19 severe enough to warrant hospitalization, and the profile of patients at highest risk for hospitalization and death may have changed. The clinical expert expressed that the differences in variants and levels of vaccination are both critically important and undermine the ability to generalize the results from these trials to a current population.
Additionally, background care and SOC were often sparsely defined in the studies, and it is therefore uncertain whether those care regimens are representative of those experienced by the current patient population of interest.
Long-Term Extension Studies
No long-term extension studies were submitted.
Indirect Evidence
No indirect evidence was submitted.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Real-World Evidence
Certain gaps in the pivotal trial data were not addressed in the RCTs, including the efficacy and safety of remdesivir in a real-world setting, for immunocompromised patients, for patients discharged after hospitalization for COVID-19, for post–COVID-19 condition, for patients with renal disease, in combination with dexamethasone among hospitalized patients and vaccinated nonhospitalized patients, and across different COVID-19 variant periods. The sponsor submitted 9 RWE papers to attempt to address these gaps. A high-level summary of the relevant RWE is presented in Table 25, and more details (including critical appraisals) are provided in this section.
Table 25: Gaps in the Systematic Review Evidence (Identified by the Sponsor) and Studies That Address Them
Gaps in pivotal and RCT evidence | Study description | Summary of key results | Summary of CDA-AMC critical appraisal |
|---|---|---|---|
Effectiveness (mortality) of RDV treatment in patients hospitalized for COVID-19 across different variant periods | Mozaffari et al. (2023)23 was a retrospective study conducted in the US on adults hospitalized for COVID-19 between December 2020 and April 2022. Patients treated with RDV were 1:1 PS-matched to patients who did not receive RDV. The primary outcome was all-cause in-hospital mortality at 14 days and 28 days. Cox proportional hazards models were used to derive aHRs and 95% CIs. Data were stratified according to the supplemental oxygen requirements (LFO, HFO/NIV, IMV/ECMO) and variant period (pre-Delta, Delta, Omicron). | 106,603 patients treated with RDV (67,582 with LFO, 34,857 with HFO/NIV, and 4,164 with IMV/ECMO) were matched to patients not treated with RDV. RDV was associated with a statistically significant reduction in in-hospital mortality at 14 days (LFO: aHR = 0.72; 95% CI, 0.66 to 0.79; HFO/NIV: aHR = 0.83; 95% CI, 0.77 to 0.89; IMV/ECMO: aHR = 0.73; 95% CI, 0.65 to 0.82) and 28 days (LFO: aHR = 0.79; 95% CI, 0.73 to 0.85; HFO/NIV: aHR = 0.88; 95% CI, 0.82 to 0.93; IMV/ECMO: aHR = 0.74; 95% CI, 0.67 to 0.82) compared with no RDV treatment. The lower risk of mortality among patients treated with RDV was observed across all variant periods. Study conclusions: RDV treatment is associated with significantly reduced mortality among patients hospitalized for COVID-19 requiring supplemental oxygen upon admission, including those requiring HFO/NIV or IMV/ECMO with severe or critical disease, across all variant periods. | Limitations included lack of information about time of symptom onset and about treatments or vaccines administered before hospitalization. Although many comorbidities were controlled for, there remains a potential for imbalances in unmeasured confounders and residual confounding. It is unclear whether the information from the US cohort is generalizable to Canada. Vaccine uptake and background disease risk as well as circulating variants have changed, which may limit the generalizability of these findings to the current COVID-19 treatment landscape. |
Effectiveness of RDV treatment on 30-day COVID-19–related and all-cause readmission across different variants and time periods | Mozaffari et al. (2024)24 was a retrospective observational study that explored the effect of RDV during hospitalization for COVID-19 on 30-day COVID-19–related and all-cause readmission across different variants and time periods among adult patients discharged for COVID-19 hospital treatment between May 1, 2020, and April 30, 2022. Hospitalization data were extracted from the US PINC AI Healthcare Database, which records data relating to diagnoses, procedures, and administered medications for approximately 25% of all US hospitalizations. Data included adult patients aged ≥ 18 years admitted to hospital for COVID-19 and discharged alive between May 1, 2020, and April 30, 2022. COVID-19 hospitalizations were based on the presence of a primary discharge diagnosis code of “COVID-19.” Exclusion criteria were discharge documented as “expired” or “transfer to a hospice,” pregnancy, inaccurate or incomplete records, transfer to or from another hospital, admission for elective procedures, and admission to hospitals that did not report charges for supplemental oxygen. | Of the 440,601 patients discharged alive after a COVID-19 hospitalization, 33,217 (7.5%) had an all-cause readmission to the same hospital within 30 days and 248,785 (56.5%) received RDV. The RDV group had a 30-day COVID-19–related readmission rate of 3.0% and an all-cause readmission rate of 6.3%, compared with 5.4% and 9.1%, respectively, for the non-RDV group. After adjusting for demographics and clinical characteristics, the ORs of 30-day COVID-19–related readmission and all-cause readmission among the RDV group were 0.60 (95% CI, 0.58 to 0.62) and 0.73 (95% CI, 0.72 to 0.75), respectively. Study conclusions: RDV treatment is associated with reduced mortality among patients hospitalized for COVID-19 requiring supplemental oxygen upon admission. | Limitations include that the impact of the potential for missing data on the outcome of rehospitalization is not clear. There is also a lack of information about time since symptom onset and treatments received before hospitalization. Despite the inclusion of numerous variables in the multivariate regression, there is still a potential for unmeasured confounders and residual confounding. Changes in vaccine uptake and background disease risk, changing circulating variants, and unclear generalizability from the US to Canada are also limitations. |
Effectiveness (readmission, mortality) of RDV treatment in patients discharged after hospitalization for COVID-19 | Finn et al. (2022)25 was a multicentre retrospective cohort study conducted in the US on adults who were discharged after hospitalization for COVID-19 between April 2020 and December 2020. Patients treated with RDV were compared to patients who did not receive RDV. The main study outcomes were length of hospital stay, 30-day readmission, and postdischarge 30-day all-cause mortality. | 2,062 patients were included in the analytic sample. The RR of being readmitted within 30 days in patients who received RDV relative to those who did not receive RDV was 0.81 (95% CI, 0.59 to 1.13); in patients with mild disease, in particular, treatment with RDV was associated with a lower risk of 30-day readmission (RR = 0.31; 95% CI, 0.13 to 0.75). RDV treatment was also associated with a reduction in all-cause mortality (HR = 0.65; 95% CI, 0.49 to 0.85). Study conclusions: RDV may be an effective strategy for reducing progression to severe COVID-19 disease and limiting morbidity associated with readmission to hospital. | Lack of information about time since symptom onset, potential for time-related bias in assessment of hospitalization, potential for missing data related to postdischarge outcomes, as well as potential for unmeasured confounders and residual confounding are limitations. Changes in vaccine uptake and background disease risk, changing circulating variants, and limited generalizability from 3 US hospitals to Canada are also limitations. |
Effectiveness (post–COVID-19 condition) of RDV treatment during initial hospitalization for COVID-19 | Boglione et al. (2022)26 was a 6-month prospective cohort study conducted in Italy on adults hospitalized with COVID-19 between March 2020 and January 2021. The aim of this study was to examine the prevalence and risk factors of post–COVID-19 condition among this population. | 449 patients were included in the analysis. Post–COVID-19 condition was diagnosed in 322 patients (71.7%) at 1 month and in 206 patients (45.9%) at 6 months. In the multivariate analysis, ICU admission (OR = 2.551; 95% CI, 1.998 to 6.819), duration of hospitalization (OR = 2.255; 95% CI, 1.018 to 6.992), and treatment with RDV (OR = 0.641; 95% CI, 0.413 to 0.782) were independent predictors of post–COVID-19 condition. Study conclusions: Treatment with RDV leads to a 35.9% reduction in post–COVID-19 condition rate in follow-up. | There were significant methodologic limitations, including the potential for missing information and concerns with the validity of the modelling approach leading to potential for confounding by indication. There is potential for uncertainty in the definition of post–COVID-19 condition and a high risk of bias in this outcome due to lack of blinding. There is limited generalizability from the Italian setting early in the pandemic to the current setting in Canada. |
Effectiveness of RDV on mortality for patients on dialysis and hospitalized for COVID-19 | Kikuchi et al. (2021)27 was a registry study conducted in Japan on patients on dialysis and hospitalized for COVID-19. Data from patients registered from April 2020 to June 2021 were extracted. Patients treated with RDV were 1:3 PS-matched to patients who did not receive RDV. Overall survival was estimated by Kaplan-Meier methods and compared by using the log-rank test. | 1,010 patients were included in this analysis. Overall survival was significantly prolonged in patients treated with RDV compared with patients who were not treated with RDV (HR = 0.45; 95% CI, 0.26 to 0.80). Study conclusions: Treatment with RDV of patients on dialysis who are hospitalized with COVID-19 might be effective in shortening the duration of hospitalization and reducing the risk of mortality. | A large proportion of patients were excluded because their outcome or age was unknown. The primary purpose of the analysis was an examination of predictors for mortality, rather than the effect of RDV on mortality. There was a lack of information on time since symptom onset. Potential for time-related bias, incomplete description of PS matching, and unmeasured confounders and residual confounding are other limitations. Changes in vaccine uptake and background disease risk, changing circulating variants, as well as unlikely generalizability from Japan to Canada, are also limitations. |
The safety of RDV in patients with COVID-19 and severe kidney disease | Seethapathy et al. (2022)28 was a retrospective cohort study conducted in the US on patients with severe kidney disease (eGFR < 30 mL/min/1.73 m2) hospitalized for COVID-19 between May 2020 and January 2021. Patients treated with RDV were 1:1 PS-matched to patients who did not receive RDV. Adverse events and hospital outcomes were recorded by manual chart review. | The PS-matched cohort included 31 patients treated with RDV and 31 patients not treated with RDV in the control group. Compared with the matched patients in the control group, the use of RDV in patients with an eGFR < 30 mL/min/1.73 m2 was not associated with a significantly increased risk of cardiac, kidney, liver, or neurologic adverse events. Study conclusions: RDV was well tolerated in patients with an eGFR < 30 mL/min/1.73 m2. | Use of a historic control group with possibly different factors affecting treatment outcomes within a single institution may be a source of bias. Concerns about completeness and accuracy of data collection, potential for unmeasured confounders and residual confounding, and small sample size are limitations. Differences in vaccine uptake and background disease risk, as well as changing circulating variants, may limit generalizability from a single US centre to Canada. |
The effect of RDV on adverse kidney outcomes in hospitalized patients with COVID-19 and impaired kidney function | Seethapathy et al. (2023)29 was a retrospective cohort study conducted in the US on patients with an eGFR between 15 mL/min/1.73 m2 and 60 mL/min/1.73 m2 hospitalized for COVID-19 between April 2020 and November 2020. Patients treated with RDV were 1:1 PS-matched to patients who did not receive RDV. Outcomes included in-hospital peak creatinine, incidence of doubling of creatinine, rate of kidney replacement therapy initiation, and eGFR among surviving patients at day 90. | 175 patients treated with RDV were 1:1 PS-matched to patients not treated with RDV in a historical comparator group. There were no statistically significant differences in peak creatinine during hospitalization (2.3 mg/dL vs. 2.5 mg/dL; P = 0.34), incidence of doubling of creatinine (10.3% vs. 13.1%; P = 0.48), and rate of kidney replacement therapy initiation (4.6% vs. 6.3%; P = 0.49) in patients treated with RDV vs. matched patients in the historical comparator group. Among surviving patients, there was no difference in the average eGFR at day 90 (54.7 mL/min/1.73 m2 for patients treated with RDV vs. 51.7 mL/min/1.73 m2 for patients in the comparator group; P = 0.41). Study conclusions: RDV use in patients with impaired kidney function who present to the hospital with COVID-19 is not associated with increased risk of adverse kidney outcomes. | Use of a historic control group with possibly different factors affecting treatment outcomes, such as use of dexamethasone or other health care resources, within a single institution may be a source of bias. Other limitations include a small sample size, lack of information about time since symptom onset, potential for time-related bias, and potential for unmeasured confounders and residual confounding. Differences in vaccine uptake and background disease risk, as well as changing circulating variants, may limit generalizability from a single US centre to Canada. |
Effectiveness (mortality) of RDV treatment in immunocompromised patients hospitalized for COVID-19 across different variant periods | Mozaffari et al. (2023)30 was a retrospective study conducted in the US on immunocompromised adults hospitalized for COVID-19 between December 2020 and April 2022. Immunocompromised conditions included cancer, solid organ and hematopoietic stem cell transplant, hematologic malignancies, moderate or severe primary immunodeficiencies, use of immunosuppressive medications, asplenia, bone marrow failure or aplastic anemia, HIV, and toxic effects of antineoplastics. Patients treated with RDV were 1:1 PS-matched to patients who did not receive RDV. Cox proportional hazards models were used to examine the effect of RDV on the risk of mortality during different variant periods (pre-Delta, Delta, Omicron). | After matching, 14,169 patients were included in each cohort. Overall, 11.1% and 17.7% of patients treated with RDV died within 14 days and 28 days, respectively, compared with 15.4% and 22.4% of patients not treated with RDV. RDV was associated with a reduction in mortality at 14 days (HR = 0.70; 95% CI, 0.62 to 0.78) and 28 days (HR = 0.75; 95% CI, 0.68 to 0.83). The survival benefit remained significant during the pre-Delta, Delta, and Omicron variant periods. Study conclusions: Prompt initiation of RDV in immunocompromised patients hospitalized for COVID-19 is associated with significant survival benefit across all variant periods. | Limitations included lack of information about time of symptom onset and treatments or vaccines administered before hospitalization. Although many comorbidities were controlled for, there remains a potential for imbalances in unmeasured confounders and residual confounding. It is unclear whether the information from the US cohort is generalizable to Canada. Vaccine uptake and background disease risk as well as circulating variants have changed, which may limit the generalizability of these findings to the current COVID-19 treatment landscape. |
Effectiveness (time to improvement, time to death) of RDV plus dexamethasone vs. dexamethasone alone in patients hospitalized for COVID-19 | Garibaldi et al. (2021)31 was a retrospective, multicentre study conducted in the US on adults hospitalized for COVID-19 between February 2020 and February 2021. Patients treated with RDV were PS-matched to patients not treated with RDV. A sensitivity analysis was conducted to compare patients treated with RDV plus dexamethasone to those treated with dexamethasone alone. The primary outcome was time to improvement, with a secondary outcome of time to death. | For the sensitivity analysis, 39,146 patients received RDV plus dexamethasone, and matches with patients who received dexamethasone alone were found for 15,058 of these patients. Patients who received RDV plus dexamethasone were statistically significantly more likely to achieve clinical improvement by 28 days (aHR = 1.21; 95% CI, 1.18 to 1.25). Patients who received RDV plus dexamethasone on no oxygen (aHR = 1.31; 95% CI, 1.23 to 1.41) or LFO (aHR = 1.24; 95% CI, 1.20 to 1.28) were significantly more likely to achieve clinical improvement by 28 days. Patients who received RDV plus dexamethasone on LFO were significantly less likely to die than matched patients in the control groups by 28 days (aHR = 0.83; 95% CI, 0.76 to 0.91). Study conclusions: These results support the use of RDV plus dexamethasone for patients hospitalized for COVID-19 on no oxygen or LFO. | The analysis of the impact of RDV plus dexamethasone compared with dexamethasone alone on time to improvement is based on a sensitivity analysis only; it is unknown if there were substantial differences such as in baseline disease characteristics between the comparison groups. There is potential for information bias due to the subjective nature of time to improvement (on the WHO scale). There was no information about time since symptom onset or treatments before hospitalization. Despite controlling for numerous variables, there remains a potential for imbalances in unmeasured confounders and residual confounding. It is unclear whether the information from the US cohort is generalizable to Canada. Vaccine uptake and background disease risk as well as circulating variants have changed, which may limit the generalizability of these findings to the current COVID-19 treatment landscape. |
aHR = adjusted hazard ratio; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; ECMO = extracorporeal membrane oxygenation; eGFR = estimated glomerular filtration rate; HFO = high-flow oxygen; HR = hazard ratio; ICU = intensive care unit; IMV = invasive mechanical ventilation; LFO = low-flow oxygen; NIV = noninvasive ventilation; OR = odds ratio; PS = propensity score; RCT = randomized controlled trial; RDV = remdesivir; RR = relative risk; vs. = versus.
Description of the Study by Mozaffari et al. (2023)23
The study by Mozaffari et al. (2023) was a retrospective cohort study that examined the effect of remdesivir on mortality among in-hospital patients with COVID-19 who required supplemental oxygen, including low-flow oxygen, high-flow oxygen or noninvasive ventilation, or invasive mechanical ventilation or ECMO, across VOC periods in a large US health care network from December 2020 to April 2022. The study compared 14-day and 28-day mortality in patients who received remdesivir versus those who did not among those hospitalized for COVID-19 who required supplemental oxygen upon admission across VOC periods: pre-Delta, Delta-predominant, and Omicron-predominant (pre-BA4/5). Data were extracted from the US PINC AI Healthcare Database, a comprehensive hospital administrative database covering approximately 25% of all hospitalizations in the US. It contains information on diagnoses, procedures, and medications, with patient records starting from the first day of hospitalization.
Populations
This study encompassed adult patients who were admitted to hospitals for COVID-19 and required supplemental oxygen between December 1, 2020, and April 30, 2022. Hospitalized patients who received remdesivir upon admission were matched to those who did not.
Patients treated with remdesivir included inpatients who had received at least 1 dose of remdesivir within 2 days of hospitalization for COVID-19. Patients in the nonremdesivir group included inpatients who did not receive remdesivir at any time during their hospitalization. During the study period, patients treated with remdesivir were matched with a 1:1 propensity score (PS) to patients not administered remdesivir during their COVID-19 hospitalization.
Only the first admission for COVID-19 was included in the analyses for patients with multiple admissions. Patients were excluded from the study if they had any of the following criteria: pregnancy, incomplete data, death or hospital discharge within 2 days of admission, transfer from hospice, transfer to or from another hospital, admission for an elective procedure, or initiation of remdesivir after the first 2 days of hospitalization.
Interventions
The intervention of interest was the administration of remdesivir during the study period. Patients treated with remdesivir received at least 1 dose of remdesivir within 2 days of hospitalization, while patients in the nonremdesivir group did not receive remdesivir at any time during their hospitalization.
Outcomes
The baseline was defined as the first 2 days of hospitalization. The primary outcome of the study was all-cause in-hospital mortality at 14 days and 28 days after hospitalization for COVID-19, where in-hospital mortality was defined as discharge status of “expired” or “hospice.”
Statistical Analysis
Patients were stratified by supplemental oxygen requirement upon admission and VOC period. Descriptive statistics were used to describe the treatment group and hospitalized cases at the index date. PS methods were employed to balance patient characteristics and account for indication bias according to remdesivir administration.
Regression models included demographics, comorbidity groups, hospital characteristics, COVID-19 severity, concomitant treatments at baseline, admission month, and source. Covariates were kept in the regression model regardless of their P value. To account for variances in hospital COVID-19 management practices in terms of each VOC, a 1:1 preferential within-hospital matching approach with replacement — with a caliper distance of 0.2 times the SD of the logit of the PS — was employed. All patients included in the analysis had to have at least 3 days of hospital stay after the administration of remdesivir.
The time to 14-day and 28-day in-hospital mortality was assessed using KM curves and compared using log-rank tests. Cox proportional hazards models were used to derive the aHR and 95% CI for 14-day and 28-day mortality.
Results
Patient Disposition
Between December 2020 and April 2022, the database at baseline included 372,468 patients hospitalized for COVID-19, of which 219,028 required low-flow oxygen, 108,171 required high-flow oxygen or noninvasive ventilation, and 45,269 required invasive mechanical ventilation or ECMO. After excluding patients with a secondary discharge diagnosis, the database included 156,132 patients who required low-flow oxygen, 70,863 who required high-flow oxygen or noninvasive ventilation, and 15,772 who required invasive mechanical ventilation or ECMO.
After screening for eligibility, 116,012 patients required low-flow oxygen upon admission, including 81,811 patients (70.5%) who received remdesivir within the first 2 days of hospital admission and 34,201 patients (29.5%) who did not. Another 54,529 patients required high-flow oxygen or noninvasive ventilation: 39,034 (71.6%) received remdesivir, and 15,495 (28.4%) did not. Of the 8,723 patients who required invasive mechanical ventilation or ECMO, 4,961 (56.9%) received remdesivir and 3,762 (43.1%) did not. The most common reasons for exclusion included discharge during the baseline period (n = 26,337), transfer from another hospital or hospice (n = 18,463), transfer to another hospital (n = 6,549), admission for elective procedures (n = 4,252), and death or discharge to hospice during baseline (n = 4,075).
After matching, the low-flow oxygen group included 67,582 patients who received remdesivir and 18,830 who did not, weighted to 67,582 patients after 1:1 matching with replacement. The high-flow oxygen or noninvasive ventilation group included 34,857 patients who received remdesivir and 10,189 who did not, weighted to 34,857 patients. The invasive mechanical ventilation or ECMO group included 4,161 patients who received remdesivir and 1,880 who did not, weighted to 4,161 patients.
Baseline Characteristics
After matching, all covariates were balanced between the remdesivir and nonremdesivir groups. In the low-flow oxygen group, upon admission, about half (47.3%) of the patients who received remdesivir (n = 67,582) were aged 65 years or older, 74.6% were white, and 39.2% were admitted in the pre-Delta period, 42.6% in the Delta period, and 18.2% in the Omicron period. Most of the patients (73.2%) had cardiovascular diseases, 38.6% had obesity, and 36.4% had diabetes. About 22% had immunocompromised conditions, and 14.6% were admitted to the ICU. At baseline, 97.8% of patients had corticosteroids and 18.1% had anticoagulants as other COVID-19 treatments.
Similarly, in the high-flow oxygen or noninvasive ventilation group, upon admission, 46.1% of patients who received remdesivir (n = 34,857) were aged 65 years or older, 73.6% were white, and 34.9% were admitted in the pre-Delta period, 43.5% in the Delta period, and 21.6% in the Omicron period. Most of the patients (82.3%) had cardiovascular diseases, 50.5% had obesity, and 44.1% had diabetes. Also, 25% had immunocompromised conditions and 33.3% were admitted to the ICU. Regarding other COVID-19 treatments at baseline, 98.1% of patients had corticosteroids and 22.9% had anticoagulants.
In the invasive mechanical ventilation or ECMO group, 43.2% of patients who received remdesivir (n = 4,146) were aged 65 years or older, 68.2% were white, and 33.4% were admitted in the pre-Delta period, 42.4% in the Delta period, and 24.2% in the Omicron period. About 90% of the patients had cardiovascular diseases, 53.3% had obesity, and 50.2% had diabetes. Also, 28.2% had immunocompromised conditions and 78.4% were admitted to the ICU. At baseline, 96.4% of patients had corticosteroids and 32.8% had anticoagulants as other COVID-19 treatments. Full details regarding the baseline characteristics are provided in Mozaffari et al. (2023).
Efficacy
In the low-flow oxygen group, 4,315 patients (6.4%) who received remdesivir and 5,918 matched patients (8.8%) who did not receive remdesivir died within 14 days. By 28 days, 6,641 patients (9.8%) from the remdesivir group and 8,305 (12.3%) from the matched nonremdesivir group had died across VOC periods. The 14-day and 28-day in-hospital mortality aHR among patients requiring low-flow oxygen across VOC periods in the remdesivir group compared to the nonremdesivir group was 0.72 (95% CI, 0.66 to 0.79) and 0.79 (95% CI, 0.73 to 0.85), respectively. Estimates were adjusted for covariates (age, admission month, admission venue [ICU versus general ward], and baseline concomitant COVID-19 treatments [anticoagulants, convalescent plasma, corticosteroids, baricitinib, tocilizumab]).
Among the patients receiving high-flow oxygen or noninvasive ventilation, 5,853 (16.8%) who received remdesivir and 6,770 (19.4%) who did not receive remdesivir died within 14 days. By 28 days, 9,009 patients (25.8%) from the remdesivir group and 9,853 (28.3%) from the nonremdesivir group had died. After adjustment for covariates, the 14-day and 28-day in-hospital mortality aHR among patients requiring low-flow oxygen across VOC periods in the remdesivir group compared to the nonremdesivir group was 0.83 (95% CI, 0.77 to 0.89) and 0.88 (95% CI, 0.82 to 0.93), respectively.
In the invasive mechanical ventilation or ECMO group, 1,157 (27.8%) patients who received remdesivir and 1,470 (35.3%) who did not receive remdesivir died within 14 days. By 28 days, 1,724 (41.4%) from the remdesivir group and 2,105 (50.6%) from the nonremdesivir group had died. After adjustment for covariates, the 14-day and 28-day in-hospital mortality aHR among patients requiring invasive mechanical ventilation or ECMO across VOC periods in the remdesivir group compared to the nonremdesivir group was 0.73 (95% CI, 0.65 to 0.82) and 0.74 (95% CI, 0.67 to 0.82), respectively.
Critical Appraisal
Internal Validity
The pivotal trial data lack clear information about the effect of remdesivir on different variants of SARS-CoV-2. The sponsor has indicated that this study fills a gap in the pivotal trial data as it addressed the effectiveness of remdesivir compared to no remdesivir treatment on in-hospital 14-day and 28-day mortality in patients hospitalized with COVID-19 at a large US health care network across 3 variant periods (pre-Delta, Delta, and Omicron) from December 2020 to April 2022.
The study did not include any information about treatments or vaccinations received before hospitalization. Prehospital administration of vaccinations, nirmatrelvir-ritonavir, or monoclonal antibodies for COVID-19 may have impacted the exposure or the outcomes, and this information was not available. The study also lacked information about time since symptom onset; this may have differed between patients who were treated with remdesivir and those who were not.
Although a number of comorbidities were included in the PS match — demographics (age group, gender, race, ethnicity, primary payor), comorbidity groups (obesity, chronic obstructive pulmonary disease, cardiovascular disease, diabetes mellitus, renal disease, cancer, immunosuppressive conditions), hospital characteristics (bed size, teaching, region, urban or rural), COVID-19 severity (hospital ward on admission, admission diagnosis of sepsis, respiratory failure, hypoxemia, pneumonia), baseline treatments (anticoagulants, corticosteroids, convalescent plasma, tocilizumab, baricitinib), admission month, and admission source — and PS were performed separately for different levels of baseline supplemental oxygen, other characteristics may have differed between patients who were treated with remdesivir and those who were not. These variables may not have been captured in comorbidities (obesity, chronic obstructive pulmonary disease, cardiovascular disease, diabetes mellitus, renal disease, cancer, immunosuppressive conditions) that were controlled for in the analysis. The distribution of PS and overlap were not presented.
Despite the covariates that the authors matched for, there is still a possibility of unmeasured confounders and residual confounding. For example, the authors only had lab values for a small proportion of the study population; there was information on serum creatinine for 25% of the study sample. The authors did not describe or justify the rationale for the inclusion of specific variables in the model. The relevant risk factors for progression in COVID-19 have changed over the pandemic. The study authors did not attempt to evaluate the presence or amount of residual confounding.
The primary outcome of the study was all-cause in-hospital mortality at 14 days and 28 days after hospitalization for COVID-19, where in-hospital mortality was defined as a discharge status of “expired” or “hospice.” The validity of this outcome measure for patient mortality status was not described.
External Validity
The data were extracted from the US PINC AI Healthcare Database; this database captures approximately 25% of all hospitalizations in the US. This health care system differs from the Canadian health care system; response to public health measures, levels of vaccination, or prehospital treatments may have been different.
The study occurred during the time period December 1, 2020, to April 20, 2022, and data were analyzed over 3 distinct variant periods (pre-Delta, Delta and Omicron). Vaccine uptake and background disease risk, as well as circulating variants, have since changed, limiting the generalizability of these findings to the current COVID-19 treatment landscape. The comparability of baseline care (e.g., corticosteroids, anticoagulants) may limit generalizability to the setting in Canada.
30-Day Readmission
Description of the Study by Mozaffari et al. (2024)24
The study by Mozaffari et al. (2024) was a retrospective observational study that explored the effect of remdesivir during hospitalization for COVID-19 on 30-day COVID-19–related and all-cause readmission across different variants and time periods among adult patients discharged for COVID-19 hospital treatment in a large US health care network between May 1, 2020, and April 30, 2022. Hospitalization data were extracted from the US PINC AI Healthcare Database, which records data relating to diagnoses, procedures, and administered medications for approximately 25% of all US hospitalizations.
Population
The data included adult patients aged 18 years or older admitted to hospital for COVID-19 and discharged alive between May 1, 2020, and April 30, 2022. COVID-19 hospitalizations were based on the presence of a primary discharge diagnosis code of COVID-19. Exclusion criteria were discharge documented as “expired” or “transfer to a hospice,” pregnancy, inaccurate or incomplete records, transfer to or from another hospital, admission for elective procedures, and admission to hospitals that did not report charges for supplemental oxygen.
Intervention
The remdesivir group received at least a single dose of remdesivir during the index COVID-19 hospitalization, while the nonremdesivir group did not receive remdesivir at any time during the index COVID-19 hospitalization.
Outcomes
The outcome of interest was 30-day readmission.
Statistical Analysis
Analyses were performed by maximal supplemental oxygen requirement during the index COVID-19 hospitalization and by periods defined by the dominant variants: pre-Delta, Delta, and Omicron.
Descriptive analyses of patient characteristics were summarized based on hospital readmission within 30 days after discharge and remdesivir intake during the index COVID-19 hospitalization. The likelihood of 30-day readmission was compared among remdesivir-treated and non–remdesivir-treated groups using multivariable logistic regression models adjusted for age, corticosteroid treatment, Charlson comorbidity index, and ICU stay during the COVID-19 hospitalization. Multicollinearity between the predictors was examined by employing the variance inflation factor and tolerance.
Results
Of the 440,601 patients discharged alive after a COVID-19 hospitalization, 33,217 (7.5%) had an all-cause readmission to the same hospital within 30 days and 248,785 (56.5%) received remdesivir. Compared to those who were not readmitted, patients who were readmitted were older, had more comorbidities, and were more likely to have received corticosteroid monotherapy during the index COVID-19 hospitalization. The proportion of patients who were and were not readmitted who had required high-flow oxygen or noninvasive ventilation or had required invasive mechanical ventilation or ECMO during the index hospitalization was the same, and both groups had comparable lengths of stay. Compared to the nonremdesivir group, patients treated with remdesivir were younger and were more likely to have received some level of supplemental oxygen support.
Efficacy
The remdesivir group had a 30-day COVID-19–related readmission rate of 3.0% and an all-cause readmission rate of 6.3%, compared with 5.4% and 9.1%, respectively, for the nonremdesivir group. After adjusting for demographics and clinical characteristics, the OR of 30-day COVID-19–related readmission and all-cause readmission among the remdesivir group was 0.60 (95% CI, 0.58 to 0.62) and 0.73 (95% CI, 0.72 to 0.75), respectively. Similar patterns of OR of 30-day readmission in patients treated with remdesivir were observed across all variant time periods.
Critical Appraisal
Internal Validity
The pivotal trial data lack clear information about the effect of remdesivir on the outcomes of hospital readmission. The sponsor has suggested that this study fills a gap in the pivotal trial data as it addressed the effectiveness of remdesivir compared to no remdesivir treatment on 30-day all-cause and COVID-19–related rehospitalization in patients who were hospitalized with COVID-19 at a large US health care network from May 1, 2020, to April 30, 2022.
Depending on how the outcome (i.e., 30-day COVID-19–related and all-cause readmission) was collected, recorded, and verified (i.e., the source document), it is difficult to assess any potential bias due to inaccurate or incomplete reporting of readmission in the study. It is unclear if there were any missing data related to hospital readmission. There was no exploration of the extent of missing data for the study outcomes. For example, the authors did not describe that patients were contacted to determine if they were hospitalized at a hospital that was not included in the network. There is a potential that the extent of missing data would be different in those treated with remdesivir versus those not. However, the between-group difference in all-cause hospital readmission was 2.8% (9.1% versus 6.3% for those treated versus not treated with remdesivir), and the difference in COVID-19–related readmission was 2.4% (5.4% versus 3.0%), indicating that the difference in all-cause rehospitalization was nearly all attributed to the difference in COVID-19–related rehospitalization. Therefore, differential missing on rehospitalization seemed unlikely.
The study did not include any information about treatments received before hospitalization or vaccination status. The study also lacked information about time since symptom onset. These factors may have differed between the patients who were treated with remdesivir and those who were not.
Multivariate regression for the effect of remdesivir on 30-day all-cause and COVID-19–related rehospitalization was adjusted for age, corticosteroid use, variant time period, Charlson comorbidity index, maximum supplemental oxygen requirements, and existence of ICU stay. Despite a number of variables that the authors controlled for in the multivariate regression, PS or other matching did not occur. The authors did not justify the rationale for the inclusion of the variables in the model.
This study may be at risk of unmeasured confounders and residual confounding that may have impacted findings related to the disease course, including rehospitalization. The study authors did not attempt to evaluate the presence or amount of residual confounding.
External Validity
The data were extracted from a large private US health care database; this database is described in Mozaffari et al. (2023).23 This health care system differs from the Canadian health care system, and prehospital treatments or response to public health measures may have been different. Minimal details were provided about data suitability (e.g., provenance, relevance, data quality).
The study occurred during the time period of May 1, 2020, to April 30, 2022. However, changes in vaccine uptake and background disease risk, as well as changing circulating variants, may limit the generalizability of these findings to the current COVID-19 treatment landscape. The comparability of baseline care (e.g., corticosteroids, anticoagulants) may limit generalizability to the setting in Canada.
Description of the Study by Finn et al. (2022)25
The study by Finn et al. (2022) was a retrospective multicentre cohort study (3 centres) that explored the effect of remdesivir on length of hospital stay, 30-day readmission, and mortality 30 days postdischarge in adult patients discharged after in-hospital COVID-19 treatment between April 1, 2020, and December 31, 2020, in Rhode Island, US.
Population
Data on sociodemographic and clinical covariates of interest were gathered for 2,062 adult patients aged 18 years or older, who were hospitalized with COVID-19 (2,279 hospital admissions) between April 2020 and December 2020 within the Lifespan network, a large Rhode Island hospital network. Each patient was followed from admission to 30 days after discharge.
Outcomes
The list of outcomes included length of hospital stay, 30-day readmission, and postdischarge 30-day mortality.
Statistical Analysis
The characteristics and outcome events of patients treated or not treated with remdesivir were reported as percentages or as mean and SD. The study used inverse probability of treatment weighting (IPTW) to account for confounding factors and inverse probability of censoring weighting to address selective survival bias. These methods aimed to ensure that the analysis would produce more robust and balanced comparisons between patients who received remdesivir and those who did not.
Logistic regression models were employed to estimate the PS for treatment with remdesivir for each patient compared to not being treated with remdesivir, adjusting for gender, race, ethnicity, language, age, insurance type, smoking status, medical history, laboratory values, and vital signs within 24 hours of index admission.
Logistic regression was used to estimate the PS, which modelled the probability of dying before or within 30 days of discharge compared to survival within the same period of time. This model included the same covariates used in the previous models in addition to the treatment type, high laboratory values during admission, maximum respiratory support, and abnormal vital signs within 24 hours of discharge.
Generalized linear models were employed to estimate the treatment effect of remdesivir on length of hospital stay and 30-day readmission compared to not receiving remdesivir, weighted by the product of IPTW and inverse probability of censoring weighting for each hospitalization, truncated at the 5th and 95th percentiles.
Marginal structural Cox proportional hazards models were used to examine the treatment effect of receiving remdesivir compared to not receiving remdesivir on 30-day survival, weighted by the IPTW. All models accounted for clustering at the patient level.
Patients were stratified according to the supplemental oxygen needed as follows: mild, if no supplemental oxygen was required; moderate, if 0.5 L/minute to 6 L/minute oxygen support was required; severe, if oxygen support of 6.5 L/minute or more was required.
Results
At baseline, 2,279 hospitalized patients were screened; 1,531 received remdesivir, and 748 did not. The mean age of the patients was 63.4 years (SD = 17.9 years), about 52% of patients were white, 46.4% were females, and 53.6% were males. Regarding medical history, 43.8% of patients had cardiac issues, 62.3% had hypertension, 41.3% had diabetes, 38.4% had pulmonary issues, and 14.7% were immunocompromised. About 35.3% did not need maximum respiratory support, and 34.5% needed oxygen support at less than 6 L/minute. In total, 77.9% of patients were neither readmitted to hospital nor deceased within 30 days.
Of the 2,279 hospital admissions, a total of 2,062 hospitalized patients were included in the analysis. Specifically, 742 out of the 752 patients who received remdesivir and 1,369 out of the 1,538 patients who did not receive remdesivir were included in the analysis.
Patients with mild symptoms tended to be younger and were more likely to be Black, Indigenous, or people of colour; those with severe symptoms tended to be older, to be white, and to have more comorbidities. Remdesivir was disproportionally administered to patients who were older, white, and male, who had higher C-reactive protein (CRP) values and admission vitals indicating a respiration rate of more than 30 breaths per minute, and who required some degree of respiratory support during their hospitalization.
Efficacy
Length of Stay
Remdesivir treatment was associated with a longer length of hospital stay, with a 3.27-day average increase relative to not receiving remdesivir (95% CI, 2.11 to 4.44 days). This effect was most pronounced in patients with severe COVID-19 symptoms, where the increase in the length of stay was 6.70 days, but the 95% CIs crossed the null (95% CI, 0.47 to 12.92 days); patients with mild or moderate symptoms had only a slight increase in their hospital stay.
30-Day Readmission
Overall, patients treated with remdesivir had a 19% reduced risk of being readmitted to the hospital within 30 days, but the 95% CIs crossed the null (RR = 0.81; 95% CI, 0.59 to 1.13). This reduction in readmission risk was pronounced in patients with mild COVID-19 symptoms, who were 69% less likely to be readmitted if they received remdesivir, with an RR of 0.31 (95% CI, 0.13 to 0.75).
30-Day All-Cause Mortality
Remdesivir treatment was associated with a 35% decrease in the risk of dying within 30 days of being discharged from hospital, with an HR of 0.65 (95% CI, 0.49 to 0.85).
Critical Appraisal
Internal Validity
The pivotal trial data lack clear evidence about the effect of remdesivir on outcomes that occur after hospital discharge. The sponsor has indicated that this study fills a gap in the pivotal trial data as it assesses the effectiveness of remdesivir compared to not receiving remdesivir in patients who were discharged after hospitalization at a single US hospital network from April 1, 2020, to December 31, 2020. No primary study outcome was indicated, but outcomes included length of hospital stay, 30-day readmission, and postdischarge 30-day mortality.
The study lacked information about time since symptom onset; this may have differed between patients who were treated with remdesivir and those who were not.
This study is at risk for time-related bias. It is not stated when remdesivir was administered during hospitalization. Prior to receiving remdesivir, time should be categorized as unexposed. However, it is not clear how this time was classified; therefore, there is a potential for immortal time bias in the assessment of hospital stay. The longer hospital stay observed in patients treated with remdesivir may be accounted for by the time required to administer the remdesivir course or by the inherent differences in the patients who were selected to receive remdesivir.
PS matching occurred for the following covariates using IPTW: gender, race, ethnicity, language, age, insurance type, smoking status, medical history (yes or no for presence of medical comorbidities including those previously listed), aspartate aminotransferase (AST), ALT, eGFR, hypotension, hypoxia, fever, tachycardia respiratory rate above 30 breaths per minute). Variables were selected for inclusion in the PS model based on potential for confounders or on causes of the outcomes (readmission, extended length of stay, and death that are not in the causal pathway) using directed acyclic graphs. A description of the groups was presented both before and after matching. However, the distribution of PS and overlap was not presented.
This study is at risk for unmeasured confounders and residual confounding. Despite the models for mortality being adjusted for numerous covariates (gender, race, ethnicity language, age, insurance, smoking, comorbidities, laboratory values, vital signs, treatment type, most extreme laboratory values, maximum respiratory support and vital sign abnormalities) using IPTW, there is still a possibility of unmeasured confounders and residual confounding. The study authors did not attempt to evaluate the presence of residual confounding.
It is unclear if there were any missing data related to the postdischarge outcomes of 30-day readmission or death. There was no exploration of the extent of missing data for the study outcomes. Patients were not, for example, contacted to determine if they were readmitted to hospital outside the hospital network of interest. There is a potential that the extent of missing data would be different in those treated with remdesivir versus those not treated with remdesivir — a potential source of bias.
External Validity
The data for this historical cohort were extracted from EHRs from a single US hospital network with 3 hospitals. The health care system differs from the Canadian health care system, and response to public health measures, or prehospital treatments may have been different.
The study occurred during the time period April 1, 2020, to December 31, 2020, before the use of vaccines for COVID-19. However, vaccine uptake and background disease risk, as well as circulating variants, have changed substantively since that time, limiting the generalizability of these findings to the current COVID-19 treatment landscape. The comparability of baseline care (e.g., corticosteroids, anticoagulants) may limit generalizability to the setting in Canada.
Post–COVID-19 Condition
Description of the Study by Boglione et al. (2022)26
The prospective cohort study by Boglione et al. (2022) aimed to analyze the prevalence and risk factors of post–COVID-19 condition in patients hospitalized for COVID-19. The study included patients hospitalized from March 10, 2020, to January 15, 2021, at Saint Andrea Hospital in Vercelli, Italy, where they were followed for at least 6 months postdischarge. Follow-up visits 1 and 2 were performed at approximately 30 days and 180 days after discharge, respectively, and clinical, laboratory, and radiological data were recorded at each visit.
Population
After September 2020, remdesivir was approved for use in patients who met the following inclusion criteria: a confirmed diagnosis of SARS-CoV-2 infection, radiological confirmation of interstitial pneumonia with symptoms that started within 10 days from hospital admission, an eGFR greater than or equal to 30 mL/min/1.73 m2, and a need for low-flow oxygen at the time of admission. Patients were excluded if they did not meet the inclusion criteria.
Outcomes
The end points assessed were the prevalence and severity of post–COVID-19 condition at the 2 follow-up time points. The PCFS scale was used to evaluate the presence and severity of post–COVID-19 condition symptoms: patients were assigned a score based on functional impairment from post–COVID-19 condition. Values greater than 2 refer to significant limitations in daily function.
Statistical Analysis
Descriptive statistics were summarized as medians, frequencies, or percentages, as appropriate. A multivariate logistic regression analysis with stepwise forward selection was used to assess the factors related to the presence of post–COVID-19 condition, with P values of less than 0.05 as the criteria for model inclusion, in a 2-tailed analysis. Survival analysis was carried out comparing the 2 groups (patients treated with remdesivir with those untreated) using the KM plot and the log-rank (Mantel-Cox) test.
Results
Baseline Characteristics
Of the 462 patients discharged after COVID-19 hospital admission, 449 were included in the analysis. At visit 2, a total of 435 patients remained in the study. A total of 163 patients (36.3%) were treated with remdesivir, and 165 patients (36.7%) did not receive any antiviral treatment. Notable characteristics of the patient population included a median age of 65 years and a majority (78%) of male patients. The most frequent comorbidities were cardiovascular diseases (14.2%), diabetes (15.8%), and chronic neurologic conditions (7.3%).
A total of 191 patients (42%) received continuous positive airway pressure or noninvasive ventilation, and 62 (13.8%) needed ICU admission. Regarding COVID-19 treatments, 390 (86.8%) received corticosteroids, 163 (36.3%) received remdesivir, and 165 (36.7%) received any antiviral treatment. In total, 312 patients (69.5%) were discharged to their homes and 137 (30.5%) to long-term care facilities.
Abnormal values of CRP were noticed among 144 patients (32%) at the first visit and in 81 patients (18.6%) at the second visit. High ferritin levels were viewed in 282 patients (62.8%) at visit 1 and in 135 patients (31%) at visit 2; high D dimer levels were detected in 74 patients (16.5%) at visit 1 and 31 patients (7.1%) at visit 2. Persistent abnormal chest X-rays were observed in 224 patients (49.8%) at visit 1 and 151 patients (34.7%) at visit 2.
Post–COVID-19 condition was diagnosed in 322 patients at visit 1 (71.7%) and in 206 at visit 2 (45.9%). At visit 1, according to the PCFS scale, 147 patients had a score of 2 to 3 and 175 patients had a score greater than 3. At visit 2, a total of 133 patients had a score of 2 to 3 and 73 patients had a score greater than 3. Symptoms included fatigue, myalgias or arthralgias, headache, dyspnea, cough, chest pain, anosmia, ageusia or dysgeusia, memory impairment, and dizziness. Psychiatric symptoms, such as sleep disorders, posttraumatic stress disorder, anxiety, depression, psychosis, and behavioural disorders, were also observed. Treatment for various clinical conditions included acetaminophen, analgesics, beta-blockers for tachycardia, anti-hypertensives, and benzodiazepines.
Efficacy
After multivariate adjustment that considered the principal baseline parameters, ICU admission (OR = 2.551; 95% CI, 1.998 to 6.819; P = 0.019), time of hospitalization (OR = 2.255; 95% CI, 1.018 to 6.992; P = 0.016), and treatment with remdesivir (OR = 0.641; 95% CI, 0.413 to 0.782; P < 0.001) were found to be independent predictors of post–COVID-19 condition. Treatment with remdesivir led to a 35.9% reduction in the post–COVID-19 condition rate in the follow-up period.
At visit 1, a total of 123 patients treated with remdesivir versus 81 patients not treated with remdesivir were not affected by post–COVID-19 condition. Twenty-seven patients treated with remdesivir and 120 patients not treated with remdesivir had PCFS scores between 2 and 3. Of the patients treated with remdesivir, 13 had a PCFS score greater than 3, and of the patients not treated with remdesivir, there were 85 with a PCFS score greater than 3. All differences in the 2 groups were statistically significant (P < 0.001).
No results were reported for visit 2.
Survival Analysis
The survival analysis compared the patients treated with remdesivir and those not treated with remdesivir according to the diagnosis of post–COVID-19 condition in the follow-up. Significant difference was found between the 2 groups (χ2 = 14.614; P < 0.001).
Critical Appraisal
Internal Validity
The pivotal trial data lacks clear information about the effect of remdesivir on post–COVID-19 condition. The sponsor has indicated that this study fills a gap in the pivotal trial data as it addressed the impact of remdesivir on symptoms of post–COVID-19 condition in patients hospitalized with COVID-19 at a single hospital in Italy between March 2020 and January 2021, who were followed for 6 months.
The primary study end point was the prevalence and severity of post–COVID-19 condition based on the PCFS scale. The outcome assessment is at high risk of bias due to lack of blinding of the treatment received. The validity, reliability, and minimally important difference of the PCFS scale as a measure of post–COVID-19 condition is unknown. There is a potential for bias due to missing data on the outcome of post–COVID-19 condition at the post–hospital discharge follow-up at approximately 30 days and 189 days, although the extent of the missing data was not quantified.
Remdesivir was included as a covariate in a multivariate, stepwise forward selection, logistic regression analysis of factors on post–COVID-19 condition. Other covariates in the model included baseline disease characteristics, ICU admission, and time of hospitalization. There was significant concern about the validity of these modelling analyses; for example, the stepwise selection of covariates used a P value of less than 0.05 as a criterion. First, confounding by indication may not be appropriately adjusted for in such a multivariate regression model; PS matching to mimic randomization is preferred. Second, confounding variables were included in the model based on statistical significance (P < 0.05). Therefore, the finding of a reduced risk of post–COVID-19 condition from remdesivir is likely biased as it is unknown whether patients treated with remdesivir and patients not treated with remdesivir had comparable prognosis based on baseline disease characteristics, comorbidities, and disease severity.
External Validity
The study was a prospective cohort of all patients who were hospitalized with COVID-19 at a single hospital in Italy from March 2020 to January 2021, who were followed for 6 months. This health care system differs from the Canadian health care system, and response to public health measures, prehospital treatments, and the ability to identify post–COVID-19 condition may have been different.
The study occurred during the time period March 2020 to January 2021, before the widespread use of vaccinations. However, vaccine uptake and population disease exposure as well as circulating variants have changed substantively since this time, limiting the generalizability of these findings to the current COVID-19 treatment landscape.
Renal Safety
Description of the Study by Kikuchi et al. (2021)27
In Japan, the first patient requiring dialysis to also have COVID-19 was reported on March 1, 2020, which sparked the establishment of the COVID-19 Task Force Committee, which included members from the Japanese Association of Dialysis Physicians, the Japanese Society for Dialysis Therapy, and the Japanese Society of Nephrology. This COVID-19 Task Force Committee aimed to create guidance on COVID-19 preventive measures and raise awareness by surveying patients receiving dialysis newly infected with SAR-CoV-2 in Japan in 2020. A total of 1,010 patients receiving dialysis were studied using the Committee’s registry, and predictive factors for mortality and overall survival were investigated by stratification by age group, complication status, and treatment. This prospective cohort study by Kikuchi et al. (2021) reports that the severity of COVID-19 and mortality were higher in patients receiving dialysis than in the general population.
Population
On April 8, 2020, the COVID-19 Task Force Committee began surveillance of new cases of COVID-19 in dialysis facilities in Japan. In this prospective cohort study, 1,948 patients receiving dialysis who also had COVID-19, registered by June 18, 2021, were extracted. Of these, 1,010 patients were included in the analysis; 897 patients were excluded due to unknown outcomes and unknown age. Patient data, including age, gender, primary disease, duration of dialysis, complications, oxygenation, and COVID-19 treatment, were collected. Blood test data at diagnosis or hospitalization were available for patients registered after March 16, 2021 (n = 311 patients).
Outcomes
The investigated outcomes included the overall survival of patients which was assessed after stratification by age, complication status, and treatment — and predictive factors for mortality. The efficacy of remdesivir treatment was assessed among patients matched using a PS for age and oxygenation in a 1:3 ratio of patients treated with remdesivir versus not treated with remdesivir. Length of hospitalization was also investigated in patients who received remdesivir treatment compared to those who did not.
Statistical Analysis
Categorical data were analyzed using the Fisher exact test, with continuous data analyzed with the Welch t test or the Mann–Whitney U test. Survival was measured using KM methods and compared using a log-rank test with Bonferroni correction. HRs and 95% CIs were assessed with the Cox regression hazards model. The univariate and multivariate analyses were performed to identify the risk factors of mortality with the incidence of COVID-19 in facilities and to identify the risk factors of mortality in hospitalized patients with COVID-19 who had blood test data at the time of diagnosis or hospitalization.
Results
Baseline Characteristics
Of the 1,010 patients, 688 (69.2%) recovered and 311 (30.8%) died. Age, duration of dialysis, cardiovascular disease, peripheral arterial disease, oxygenation, or dexamethasone treatment were higher in patients who died. A total of 392 patients were analyzed for predictive factors for mortality: 98 patients treated with remdesivir, matched with 294 patients not treated with remdesivir. Blood test (creatinine, albumin, and CRP) and body mass index data were available for 311 patients.
Efficacy
The overall survival of patients stratified by age (younger than 60 years, 60 to 69 years, or 70 years or older) indicated that mortality risk increased with age. According to the univariate analysis, the HR for mortality risk was 2.02 (95% CI, 1.27 to 3.23) in those aged 60 to 69 years and 3.13 (95% CI, 3.13 to 6.77) among those aged 70 years or older, both higher than in those younger than 60 years.
The multivariate analysis showed that the HR for mortality risk was 4.92 (95% CI, 3.10 to 7.80) for patients aged 70 years or older and 1.58 (95% CI, 0.90 to 2.77) for patients aged 60 to 69 years. Mortality was increased with a longer duration of dialysis, and the HR among patients with peripheral arterial disease was 1.49 (95% CI, 1.05 to 2.10). Mortality was lower in patients who were treated with remdesivir, with an HR of 0.60 (95% CI, 0.37 to 0.98).
In total, 392 patients were analyzed: 98 patients treated with remdesivir, matched with 294 patients not treated with remdesivir. The HR for overall survival was 0.45 (95% CI, 0.26 to 0.80) in the group treated with remdesivir, higher than in those not treated with remdesivir. The mean duration of hospitalization was 20.9 days (SD = 13.2 days) in the patient group treated with remdesivir and 16.2 days (SD = 8.1 days) in the patient group not treated with remdesivir (difference = 4.7 days; 95% CI, 2.2 to 7.4 days).
Among the 311 patients whose blood test data were available and analyzed, the body mass index was lower in the patient group who died.
Critical Appraisal
Internal Validity
The pivotal trial data lack clear information about the effect of remdesivir in patients with renal insufficiency. The sponsor has indicated that this study fills a gap in the pivotal trial data as it includes the effect of remdesivir as a risk factor for mortality in patients admitted to hospital with COVID-19 and receiving dialysis in Japan from April 2020 to June 2021. The overall study examined risk factors for mortality, and the analysis of remdesivir was performed on a smaller group of patients than the full analysis set.
A large number of patients were excluded (897 of 1,948) because their outcome or age was unknown. This leads to significant concern about the overall quality of the registry data on remdesivir treatment and outcomes (including the timing and doses of treatment) and about the completeness of reporting on mortality and whether there were differential missing data on this outcome between the treated and nontreated groups. The analysis of predictive factors for mortality was performed on an even smaller group of 311 patients for whom blood tests were available. The primary purpose of the study was to examine overall risk factors for mortality and not to assess the impact of remdesivir versus no remdesivir on mortality; it was not stated if this analysis was planned a priori. This is a limitation compared to a study where the primary purpose is to compare mortality in patients treated with remdesivir versus patients not treated with remdesivir.
The study data lacked information about time since symptom onset; this may have differed between patients who were treated with remdesivir and those who were not. There was no information about when during the course of hospitalization the patients received remdesivir. Prior to receiving remdesivir, time should be categorized as unexposed. However, it is not clear how time zero was defined, either for starting remdesivir treatment or for the follow-up of survival outcome. Therefore, there is a potential for immortal time bias.
There was no description of the reasons that patients received remdesivir, and it is unclear if it was due to a policy or prescriber choice. The study also lacked information about time since symptom onset; this may have differed between patients who were treated with remdesivir and those who were not. At the time of the study, remdesivir was not recommended for patients with an eGFR less than 30 mL/min/1.73 m2; patient, prescriber, or health care system factors may have influenced the prescribing decision. These factors may have differed between patients who were treated with remdesivir and those who were not.
PS matching occurred for age and oxygenation (with or without oxygen, ventilator, or ECMO). The model included the facility, age, gender, primary renal disease, duration of dialysis, comorbidities (diabetes mellitus, hypertension, cardiovascular disease, peripheral artery disease or malignancy), oxygenation, and treatment with remdesivir or steroids. It was unclear how variables were selected for inclusion in the PS model. No automated approaches for selecting variables were described. The methods used for PS matching were not described. The authors did not describe or justify the rationale for inclusion of the variables in the model. The study authors did not attempt to evaluate the presence or amount of residual confounding. The distribution of PS and overlap was not presented. It is very likely that not all relevant confounders were included in the PS matching.
External Validity
The data for this retrospective cohort were extracted from a registry of patients receiving dialysis who were treated for COVID-19 in hospital in Japan. The health care system differs from the Canadian health care system, and response to public health measures, levels of vaccination, or prehospital treatments may have been different.
The study occurred during the time period April 2020 to June 2021. However, vaccine uptake and background disease risk, as well as circulating variants, have changed substantively since that time, limiting the generalizability of these findings to the current COVID-19 treatment landscape. Reasons for prescribing remdesivir may differ in Japan compared to Canada. The comparability of baseline care (e.g., corticosteroids, anticoagulants) may limit generalizability to the setting in Canada.
Description of the Study by Seethapathy et al. (2022)28
This retrospective cohort study by Seethapathy et al. (2022) examined the association between remdesivir use and AEs in patients hospitalized for COVID-19 with an eGFR less than 30 mL/min/1.73 m2 from May 2020 to January 2021 and in PS-matched patients not treated with remdesivir from March 2020 to April 2020 within the Mass General Brigham health care system located in the Boston, Massachusetts, area in the US.
Population
The inclusion criteria for patients treated with remdesivir comprised remdesivir administration within 72 hours of hospital admission and receipt of kidney replacement therapy or an eGFR less than 30 mL/min/1.73 m2 based on their creatinine level just before the first dose of remdesivir. Patients were excluded if they had taken remdesivir before transfer to the hospital from another hospital.
Patients in the nonremdesivir control group were hospitalized for more than 24 hours and had an eGFR of less than 30 mL/ min/1.73 m2 within the first 72 hours of admission. Inclusion criteria comprised admission to hospital for COVID-19 with an oxygen saturation of less than or equal to 94% on room air or requiring supplemental oxygen within 72 hours of admission and an eGFR less than 30 mL/min/1.73 m2 or receiving kidney replacement therapy within the first 72 hours of admission. Patients were excluded if they had decompensated liver disease within 72 hours of admission, enrolled in a placebo-controlled trial of remdesivir, initiated hospice care before receiving COVID-19–directed therapies, or transferred from another facility.
Outcomes
Outcomes included clinical AEs (cardiac arrhythmia, cardiac arrest, seizure, and altered mental status), and laboratory AEs (transaminitis [more than 5 times the upper limit of normal for AST and ALT], worsening kidney function, anemia [hemoglobin < 8 g/dL], and hyperglycemia [blood glucose > 200 mg/dL]). Worsening kidney function was defined as a rise in serum creatinine greater than or equal to 50% for all patients who were not on kidney replacement therapy before baseline. AEs and hospital outcomes were recorded by manual chart review.
Statistical Analysis
Baseline characteristics were summarized as means and frequencies. Patients treated with remdesivir were matched to patients in a nonremdesivir control group using a PS, which was determined on the basis of a multivariate logistic regression model that estimated the probability of receiving remdesivir. Models were adjusted for age; sex; race/ethnicity; Sequential Organ Failure Assessment score; invasive mechanical ventilation within 72 hours after admission; pretreatment or admission creatinine level; and a history of hypertension, diabetes mellitus, end-stage kidney disease (ESKD), or solid organ transplant.
The patients treated with remdesivir and those not treated with remdesivir were matched using 1:1 nearest neighbour greedy matching without replacement and a caliper of 0.1 SD of the PS. Patient outcomes were examined using the McNemar test (proportion experiencing clinical AEs or 28-day all-cause mortality) for binary outcomes and paired t test or Wilcoxon signed-rank test (lowest hemoglobin and highest AST, ALT, and blood glucose) for continuous outcomes.
Results
Baseline Characteristics
Of the 40 patients with an eGFR less than 30 mL/min/1.73 m2 who received remdesivir between May 10, 2020, and January 31, 2021, four were excluded due to receiving remdesivir more than 72 hours after admission, and 2 were excluded because they received remdesivir in another hospital. Thirty-four patients treated with remdesivir with an eGFR less than 30 mL/min/1.73 m2 met the inclusion criteria.
Of the 34 patients treated with remdesivir, a close match was found for 31 patients. The PS-matched cohort therefore included 31 patients treated with remdesivir and 31 patients not treated with remdesivir in the control group. After matching, patient characteristics were balanced between the remdesivir and nonremdesivir groups, with a standardized difference less than 0.1, with significant imbalance observed in sex and the prevalence of ESKD at baseline. In the patients treated with remdesivir, the mean age was 71.4 years (SD = 16.6 years), 67.7% of the patients were females, and 51.6% of the patients were white. Regarding medical history, 93.5% had a history of hypertension, 83.9% had diabetes mellitus, 41.9% had ESKD, and 6.5% had had a solid organ transplant.
In the nonremdesivir group, the mean age of the patients was 72 years (SD = 16.6 years), 54.8% of the patients were females, 45.2% of the patients were white, and 32.3% of the patients had ESKD. Regarding medical history, 96.8% had a history of hypertension, 83.9% had diabetes mellitus, 32.3% had ESKD, and 6.5% had had a solid organ transplant.
Efficacy
Of the patients who were not on dialysis at baseline, 1 patient in the remdesivir group developed worsening kidney function (defined as ≥ 50% increase in creatinine or the initiation of kidney replacement therapy), as did 3 in the nonremdesivir group.
There were no significant differences in AEs between the matched groups, with the exception of an increased risk of hyperglycemia (glucose > 200 mg/dL), which occurred in 81% of patients in the remdesivir-treated population and 55% of the control population (P = 0.03). No significant differences were observed between the 2 groups in lowest hemoglobin or peak ALT; only peak glucose was significantly different.
Early discontinuation of remdesivir occurred among 4 patients (14%) due to safety concerns of elevated transaminase levels and low eGFR. The overall mortality rate during the hospital stay was 19% (n = 6) among the patients treated with remdesivir and 23% (n = 7) among the patients in the nonremdesivir control group (P = 0.71).
Critical Appraisal
Internal Validity
The pivotal trial data lack clear information about the effect of remdesivir in patients with renal insufficiency. The sponsor has indicated that this study fills a gap in the pivotal trial data as it assesses the effectiveness of remdesivir compared to no remdesivir treatment in patients admitted to a single US hospital with COVID-19 and with an eGFR less than 30 mL/min/1.73 m2 from March 2020 to January 2021. No prehypothesized primary study outcome was specified. The study outcomes included AEs, worsening of kidney function, anemia, and hyperglycemia.
The nonremdesivir control group was drawn from the first 2 months of the pandemic, with clearly different use of concurrent treatments (namely, dexamethasone) for COVID-19 and some postmatching differences in baseline characteristics. This use of a historical control group possibly would have introduced bias to outcome assessment (e.g., adverse effect on liver function). In addition, the completeness and accuracy of the data on outcomes is unknown. For instance, the study reviewed each physician and nursing note to determine AEs for all patients. In the early pandemic, it is unknown if those recordings of AEs were different between patients who received remdesivir and patients who did not. It was noted that patients treated with remdesivir were chart reviewed for their entire remdesivir course plus 48 hours afterward (typically corresponding to 7 days), and patients in the control group were chart reviewed for the first 7 days of hospitalization. The quality assurance applied to the study database, including the amount of missing data, was not reported.
The study data lacked information about time since symptom onset; this may have differed between patients who were treated with remdesivir and those who were not. Remdesivir was administered within 72 hours of hospitalization. Prior to receiving remdesivir, time should be categorized as unexposed. However, it is not clear how this time was classified; therefore, there is a potential for immortal time bias in the assessment of outcomes occurring during the hospitalization.
The study has a small sample size, which is a significant concern when examining AEs and outcomes based on laboratory values at different time points during the course of treatment. With the limited 31 versus 34 patients in the 2 groups, many known confounding factors could not be adjusted for with the regression model. The study may not have sufficient power to find potential differences between the comparison groups. For example, there was variability in outcome measures such as elevated ALT, elevated AST, bradycardia or arrythmia.
External Validity
The data for this retrospective cohort were extracted from EHRs from a single US hospital. The health care system differs from the Canadian health care system, and response to public health measures, levels of vaccination, or prehospital treatments may have been different, thus limiting generalizability to the Canadian health care system. No information was provided to justify the differences in health care systems, access to care, available health care resources during the pandemic, or other factors that may impact the care of patients with COVID-19, and how that might affect the applicability of the findings to the context in Canada.
The study occurred during the time period May 2020 to January 2021 for remdesivir treatment in patients with COVID-19 and an eGFR less than 30 mL/min/1.73 m2; these patients were compared to PS-matched patients not treated with remdesivir from March 2020 to April 2020. Both time periods were before the widespread use of vaccines for COVID-19. Vaccine uptake and background disease risk, as well as circulating variants, have changed substantively since that time, limiting the generalizability of these findings to the current COVID-19 treatment landscape. The comparability of baseline care (e.g., corticosteroids, anticoagulants) may limit generalizability to the setting in Canada.
Description of the Study by Seethapathy et al. (2023)29
Chronic kidney disease is an important risk factor for mortality from COVID-19. However, patients with severe kidney impairment have been excluded from clinical trials with remdesivir due to safety concerns. This retrospective cohort study by Seethapathy et al. (2023) evaluated the safety of remdesivir on hospitalized patients with COVID-19 admitted with an eGFR between 15 mL/min/1.73 m2 and 60 mL/min/1.73 m2. One hundred and seventy-five patients treated with remdesivir were matched in a 1:1 ratio to patients admitted between March 2020 and April 2020, the first wave of COVID-19. Measured dependent outcomes were in-hospital peak creatinine, incidence of doubling creatine, rate of initiation of kidney replacement therapy, and eGFR at day 90.
Population
This retrospective cohort study comprised patients aged 18 years and older who were admitted to the Mass General Brigham health care system due to COVID-19, with an admission creatinine level corresponding to an eGFR between 15 mL/min/1.73 m2 and 60 mL/min/1.73 m2. Patients in the remdesivir-treated group received at least 1 dose of treatment and had at least 1 repeat creatinine measurement after receiving the drug. Patients in the remdesivir-treated group were excluded if they received remdesivir more than 72 hours after admission or if they received remdesivir from another hospital before being transferred to the hospital. PS matching was performed between the patients in the remdesivir-treated cohort and the patients in the untreated historical comparator group, who were adult patients admitted to Mass General Brigham during the first wave of COVID-19 between March 2020 and April 2020. Patients were excluded from the comparator group if they had ESKD or if they were placed into a hospice before receiving COVID-19 treatment.
Outcomes
The primary outcome was peak creatinine level during hospitalization in the remdesivir group compared to the nonremdesivir group. The secondary outcomes included doubling of creatinine from admission, initiation of kidney replacement therapy, and average eGFR among patients alive at day 90.
Statistical Analysis
To control for confounding factors associated with both treatment assignment and outcome, PS matching was used. To compare peak creatinine, duration of hospitalization, and creatinine measurements, a paired t test or Wilcoxon signed-rank test was employed. The McNemar test was used to compare the incidence of doubling creatinine from admission and the rate of initiation of kidney replacement therapy. The average eGFR at day 90 was assessed using an independent sample t test.
Results
Baseline Characteristics
Of the 203 patients with an admission eGFR between 15 mL/min/1.73 m2 and 60 mL/min/1.73 m2 who received remdesivir, 20 were excluded due to either initiation of remdesivir more than 72 hours after admission or due to having received remdesivir from an external hospital before transfer; 183 patients treated with remdesivir were therefore included in the analysis. Of the 556 patients in the historical comparator group, 460 remained after exclusion criteria had been applied. Prior to matching, the historical patients were older and had higher Sequential Organ Failure Assessment scores than the patients treated with remdesivir. A close match was found for 175 of the 184 patients treated with remdesivir (95%). Good balance was achieved in the patient characteristics (standardized mean differences < 0.1). Postmatching, the mean patient age was 74 years, 56.9% of the patients were male, and 59% were white. About 83% had at least 1 comorbidity, 44.3% had pre-existing chronic kidney disease, and 81% received a full course of remdesivir.
Reasons for early discontinuation among those who received a shorter course of remdesivir (< 5 doses) included rapid recovery (n = 19), worsening kidney function (n = 4), elevated transaminases (n = 2), anaphylaxis (n = 1), transition to comfort care (n = 6), and “other” (n = 2).
Efficacy
The mean peak creatinine level was 2.3 mg/dL (95% CI, 1.98 to 2.57 mg/dL) and 2.5 mg/dL (95% CI, 2.13 to 2.89 mg/dL) among patients in the remdesivir-treated group and patients in the non–remdesivir-treated comparator group, respectively.
Eighteen patients treated with remdesivir (10.3%) and 23 patients in the untreated non–remdesivir-treated comparator group (13.1%) experienced doubling of serum creatinine during hospitalization.
Of the patients treated with remdesivir, 8 (4.6%) received kidney replacement therapy during their hospitalization, compared to 11 patients (6.3%) in the non–remdesivir-treated comparator group.
The eGFR of 120 surviving patients was measured, and the average eGFR at day 90 was 54.7 mL/min/1.73 m2 (SD = 20.0 mL/min/1.73 m2) in patients treated with remdesivir (n = 66), compared to 51.7 mL/min/1.73 m2 (SD = 19.5 mL/min/1.73 m2) among patients in the in the non–remdesivir-treated comparator group (n = 54).
Critical Appraisal
Internal Validity
The pivotal trial data lack clear information about the effect of remdesivir in patients with renal insufficiency. The sponsor has indicated that this study addresses a gap in the pivotal trial data as it assesses the effectiveness of remdesivir on adverse laboratory-based renal outcomes compared to no treatment with remdesivir in patients admitted to a single US hospital with COVID-19 and with an eGFR of 15 mL/min/1.73 m2 to 60 mL/min/1.73 m2 from March 2020 to November 2020. The primary study end point was in-hospital peak creatinine.
The study occurred during the time period April 2020 to November 2020 for patients with COVID-19 and an eGFR less than 30 mL/min/1.73 m2 who were treated with remdesivir; these patients were compared to matched patients in a historical non–remdesivir-treated control group from March 2020 to April 2020, before the use of vaccines for COVID-19. The historical control population was drawn from the first 2 months of the pandemic, with possibly different factors affecting treatment outcomes, such as use of dexamethasone or other health care resources within a single institution, which may be a source of bias.
The study data lacked information about time since symptom onset; this may have differed between patients who were treated with remdesivir and those who were not. Remdesivir was administered within 72 hours of hospitalization. Prior to receiving remdesivir, time should be categorized as unexposed. However, it is not clear how this time was classified in this study; therefore, there is a potential for immortal time bias in the assessment of doubling of serum creatinine.
PS matching occurred for a number of covariates (age, sex, race/ethnicity, Sequential Organ Failure Assessment score, invasive ventilation within 72 hours of hospitalization, history of hypertension, diabetes mellitus, eGFR < 60 mL/min/1.73 m2 for 90 days, kidney transplant, pretreatment or admission creatinine, and markers of disease severity). However, the distribution of PS and overlap was not presented. Despite PS matching, this study is at risk of unmeasured confounders and residual confounding. The ability to control for confounders in the model is limited by the study’s small sample size. The study authors did not attempt to evaluate the presence or amount of residual confounding.
The outcome variable of 90-day creatinine had missing data, which leads to the possibility of informative censoring.
External Validity
This study was performed on data from the same hospital as the study by Seethapathy et al. (2022);28 all comments related to the data quality and external validity apply to both studies.
Immunocompromised Patients
Description of the Study by Mozaffari et al. (2023)30
This retrospective cohort study by Mozaffari et al. (2023) investigated the effectiveness of remdesivir treatment on patient mortality among immunocompromised patients hospitalized for COVID-19 across all VOC periods. Data for immunocompromised patients hospitalized for COVID-19 were extracted from the US PINC AI Healthcare Database for the period December 2020 to April 2022. Patients who received remdesivir within 2 days of hospitalization were matched in a ratio of 1:1 using PS matching to patients who did not receive remdesivir. Cox proportional hazards models were used to evaluate the effect of remdesivir on risk of 14-day and 28-day mortality during different VOC periods.
Population
In this retrospective cohort study, patient data were extracted from the US PINC AI Healthcare Database, a dataset that captures data for up to 25% of all hospitalizations in 48 states of the US. The study included patients aged 18 years or older who were hospitalized for COVID-19 and had an International Classification of Diseases, 10th revision, Clinical Modification (ICD-10-CM) code of U7.01 for immunocompromised conditions between December 1, 2020, and April 30, 2022. Immunocompromised conditions were defined as conditions such as cancer, solid organ or hematopoietic stem cell transplant, hematologic malignancies, moderate or severe primary immunodeficiencies, use of immunosuppressive medications, asplenia, bone marrow failure/aplastic anemia, HIV, or toxic effects of antineoplastics, identified through specific ICD-10-CM diagnosis codes. Exclusion criteria were pregnancy, incomplete hospital records, hospitalized for less than 3 days, transferred from hospice, transferred to or from another hospital, admitted for an elective procedure, or received remdesivir as part of a clinical trial. Baseline supplemental oxygen requirements (first 2 days of hospitalization) were categorized as no supplemental oxygen charges in hospitals documented to charge for supplemental oxygen, low-flow oxygen, high-flow oxygen or noninvasive ventilation, and invasive mechanical ventilation or ECMO. Periods for VOCs were defined as pre-Delta (December 2020 to April 2021), Delta (May 2021 to November 2021), and Omicron (December 2021 to April 2022).
Interventions
Patients in the remdesivir group received at least 1 dose of remdesivir within the first 2 days of hospitalization for COVID-19; patients in the nonremdesivir group did not receive remdesivir at any time during hospitalization.
Outcomes
The primary outcome investigated was the effect of remdesivir treatment on inpatient mortality among immunocompromised patients hospitalized for COVID-19 across all VOC periods. All-cause inpatient mortality was assessed at day 14 and day 28 and was defined as a discharge status of either “expired” or “hospice.” Those who were discharged alive were censored at 14 days or 28 days.
Statistical Analysis
PS was used to match patients treated with remdesivir to patients not treated with remdesivir and was estimated separately for each category of baseline supplemental oxygen requirement and each VOC period using logistic regression models. A 1:1 preferential within-hospital matching approach with replacement, with a caliper distance of 0.2 times the SD of the logit of PS, was implemented to account for differences in hospital COVID-19 management practices that may have evolved with each VOC time frame. PS matching was based on age groups (18 to 49 years, 50 to 64 years, > 65 years). All patients included in the analysis had to have at least 3 days of hospital stay since the administration of remdesivir. Time to mortality was assessed using KM curves and compared using log-rank tests. Cox proportional hazards models were used to assess the effect of remdesivir treatment on inpatient mortality. Models were adjusted for hospital-level effects and the following covariates: age, admission month, hospital ward type on admission, and baseline COVID-19 treatments.
Results
Baseline Characteristics
In total, 51,123 immunocompromised patients were included across 819 hospitals. Among them, 30,397 patients met the eligibility criteria: 19,184 (63.1%) who received remdesivir and 11,213 (36.9%) who did not. Following 1:1 matching with replacement, 14,169 patients treated with remdesivir in the first 2 days of the hospitalization were matched to 5,341 unique patients not treated with remdesivir (equivalent to 14,169 patients not treated with remdesivir based on matching with replacement), and 5,015 patients were unmatched.
Following matching, the patients’ baseline characteristics (including the types of immunosuppressive conditions) were well balanced, with each covariate displaying a standardized difference of less than 0.15. In the matched cohort at baseline, 59% of patients were aged 65 years or older, 40% did not require supplemental oxygen, 39% required low-flow oxygen, 19% required high-flow oxygen or noninvasive ventilation, and 2% required invasive mechanical ventilation or ECMO. The median duration of remdesivir therapy was 5 days (IQR, 5.0 to 5.0 days), with 68.2% of patients completing the full 5-day course and 1.8% of patients completing the 10-day course.
Efficacy
Unadjusted mortality rates were lower among patients receiving remdesivir than among patients not receiving remdesivir across all VOC periods and all levels of baseline supplemental oxygen requirement. Among the remdesivir group, 11.1% of patients died within day 14 and 17.7% died within day 28. In the nonremdesivir group, 15.4% of patients died within day 14 and 22.4% died within day 28. After adjusting for baseline and clinical covariates, the HR for mortality risk in the remdesivir group on admission was 0.70 (95% CI, 0.62 to 0.78) and 0.75 (95% CI, 0.68 to 0.83) at 14 days and 28 days, respectively. Similar results were seen during each VOC period and were most pronounced during the pre-Delta period at the 14-day assessment, with the HRs for the pre-Delta, Delta, and Omicron variant periods being 0.59 (95% CI, 0.48 to 0.71), 0.77 (95% CI, 0.65 to 0.92), and 0.75 (95% CI, 0.63 to 0.90), respectively. At 28 days, the HRs for the pre-Delta, Delta, and Omicron variant periods were 0.65 (95% CI, 0.56 to 0.76), 0.79 (95% CI, 0.68 to 0.91), and 0.84 (95% CI, 0.72 to 0.97), respectively.
For the mortality rate among the subgroup of patients with no supplemental oxygen charges on admission, the HR was 0.71 (95% CI, 0.58 to 0.87) and 0.78 (95% CI, 0.66 to 0.93) at day 14 and day 28, respectively, within the remdesivir group. For those who required low-flow oxygen on admission, the HR was 0.56 (95% CI, 0.46 to 0.68) and 0.62 (95% CI, 0.53 to 0.72) at day 14 and day 28, respectively in favour of remdesivir. The HR among those who required high-flow oxygen or noninvasive ventilation and those who required invasive mechanical ventilation or ECMO on admission was 0.83 (95% CI, 0.70 to 0.99) and 0.86 (95% CI, 0.75 to 0.99) at day 14 and day 28, respectively in favour of remdesivir.
Critical Appraisal
Internal Validity
The pivotal trial data lack clear information about the effect of remdesivir in patients who are immunocompromised. The sponsor has indicated that the study by Mozaffari et al. fills a gap in the pivotal trial data as it assesses the effectiveness of treatment with remdesivir compared to no remdesivir treatment on in-hospital 14-day and 28-day mortality in a population who were immunocompromised and hospitalized with COVID-19 at a large US health care network from December 2020 to April 2022.
The primary outcome of the study was all-cause in-hospital mortality at 14 days and 28 days after hospitalization for COVID-19, where in-hospital mortality was defined as a discharge status of “expired” or “hospice.” The validity of this outcome measure of patient mortality status was not described.
The study did not include any information about treatments or vaccinations received before hospitalization. Prehospital administration of vaccinations, nirmatrelvir-ritonavir, or monoclonal antibodies for COVID-19 may have impacted the exposure or the outcomes, and this information was not available. The study also lacked information about time since symptom onset; this may have differed between patients who were treated with remdesivir and those who were not.
Despite the covariates that the authors matched for, and the models being adjusted for age, admission month, hospital ward type, and baseline COVID-19 treatments, there is still a possibility of unmeasured confounders and residual confounding. The authors did not describe or justify the rationale for inclusion of specific variables in the model. The relevant risk factors for progression in COVID-19 has changed over the pandemic. An analysis of the extent of residual confounding was not performed.
External Validity
This study was performed with the same dataset as the study by Mozaffari et al. (2023);23 all comments related to the dataset and external validity apply to both studies.
Effectiveness of Remdesivir in Adults Hospitalized With COVID-19
Description of the Study by Garibaldi et al. (2021)31
The study by Garibaldi et al. (2021) was a retrospective, multicentre, RWE study evaluating the effectiveness of remdesivir in hospitalized patients with COVID-19 in the US. Patients who received remdesivir were matched using time-dependent propensity scoring with patients in a nonremdesivir control group. The primary outcome was time to improvement, and the secondary outcome was time to death.
Population
Data were obtained from HCA Health care, which comprises more than 2,000 care sites including more than 180 acute care facilities. Data included sociodemographics, medical history, ICD-10 codes, laboratory data, vital signs, medications, oxygen support, length of hospitalization, location of discharge, and death. More than 180,000 patients with COVID-19 had been admitted to the HCA facilities by July 2021. The inclusion criteria specified patients hospitalized for COVID-19 for the first time, who were screened through nucleic acid tests and had an FDA emergency use authorization combined with specific ICD-10 codes that indicated symptomatic infection. Discharge and readmission within 24 hours were considered a continuous care episode.
Interventions
At the time of the analysis, HCA guidelines for the use of remdesivir recommended a 5-day treatment course for patients with an oxygen saturation of less than 94% or the need for oxygen.
Outcomes
The primary outcome was time to clinical improvement from the first day of remdesivir treatment or the matched day, defined as a 2-point decrease in the 8-point WHO severity score or discharged alive from the hospital without worsening of the WHO severity score within 28 days. The secondary outcome was time to death from the first day of remdesivir treatment or the matched day. Patients who were discharged to home or self-care alive were censored at 28 days, those who were discharged to another health care facility without a confirmed death date were censored at the last follow-up, and those discharged to a hospice with a recorded death date were included in the group of patients who died.
Statistical Analysis
Only data from facilities using a single EHR system were included, accounting for more than 90% of affiliated facilities. Data included sociodemographics, past medical history, ICD-10 codes, laboratory data, vital signs, medications, oxygen support (HFNC, NIPPV, invasive mechanical ventilation, or ECMO), length of stay, location of discharge, and death.
Patients who received remdesivir were matched to patients in a nonremdesivir control group based on time-dependent propensity scoring calculated using a time-dependent Cox proportional hazards model, with the time from admission to the first dose of remdesivir as the outcome. Dexamethasone was included as a matching variable. To control for changes in the pandemic over time, patients who received remdesivir before October 1, 2020, had to be matched to a patient in the control group who was hospitalized before October 1, 2020.
Cox proportional hazards models were employed to estimate the association between remdesivir treatment and outcomes of interest on the matched sets, demographics, oxygen delivery device, vital signs, key laboratory data, comorbidities, and COVID-19–specific medications (e.g., dexamethasone, tocilizumab) were included as covariates.
Results
Baseline Characteristics
Of 96,859 patients with COVID-19 who were admitted to hospital between February 23, 2020, and February 11, 2021, 42,473 (43.9%) received at least 1 dose of remdesivir; 36,656 patients were matched with patients in a nonremdesivir control group (18,328 in the remdesivir group; 18,328 in the control group). Of those receiving remdesivir, 13,907 patients (32.7%) stopped treatment before day 5, a total of 27,018 (63.6%) received a 5-day course, and 1,548 (3.6%) received treatment of more than 5 days.
The median time from admission to the first dose of remdesivir was 1 day, and the median duration of remdesivir treatment was 5 days. In the remdesivir group, the median age of the patients was 65 years, and 23,701 patients (55.8%) were male.
Of 18,328 successfully matched patients receiving remdesivir (43.2% of eligible patients), 8,207 patients (73.2%) were treated before October 1, 2020, and 10,121 patients (32.4%) were treated after October 1, 2020.
Efficacy
Of the 36,656 matched patients, 13,569 (74.0%) from the remdesivir group and 12,510 (68.3%) from the nonremdesivir group achieved clinical improvement before 28 days, with a median time to clinical improvement of 7 days (IQR, 5 to 19 days) in the remdesivir group and 9 days (IQR, 5 to 28 days) in the nonremdesivir group. The aHR for clinical improvement at 28 days in the remdesivir group was 1.19 (95% CI, 1.16 to 1.22). The aHR for clinical improvement among the patients treated with remdesivir receiving no oxygen was 1.30 (95% CI, 1.22 to 1.38) with a median of 5 days (IQR, 4 to 13 days) for the remdesivir group compared to 7 days (IQR, 5 to 15 days) for matched patients in the nonremdesivir group.
The aHR for clinical improvement among patients treated with remdesivir receiving low-flow oxygen was 1.23 (95% CI, 1.19 to 1.27) with a median of 6 days (IQR, 4 to 11 days) for the remdesivir group compared to 7 days (IQR, 5 to 15 days) for matched patients in the nonremdesivir group. The aHR for clinical improvement among patients treated with remdesivir receiving HFNC or NIPPV was 0.95 (95% CI, 0.89 to 1.01), with a median of 15 days (IQR, 7 to 28 days) compared to 17 days (IQR, 8 to 28 days) for matched patients in the nonremdesivir group. The aHR for clinical improvement among patients treated with remdesivir receiving invasive mechanical ventilation at the time of initiation of remdesivir was 0.92 (95% CI, 0.81 to 1.04), with a median of 28 days (IQR, 10 to 28 days) in the remdesivir group compared to 28 days (IQR, 9 to 28 days) for matched patients in the nonremdesivir group.
Remdesivir showed no significant impact on mortality overall, with an aHR of 1.02 (95% CI, 0.97 to 1.08) and a 28-day mortality rate of 15.7% (2,879 deaths) for the remdesivir group compared to 19.6% (3,586 deaths) for the nonremdesivir group.
Among patients on room air, the aHR for mortality was 1.08 (95% CI, 0.92 to 1.27), and 28-day mortality rate was 11.4% (325 deaths) for the remdesivir group compared to 13.3% (329 deaths) for matched patients in the nonremdesivir group. The aHR for mortality among patients treated with remdesivir receiving low-flow oxygen was 0.85 (95% CI, 0.77 to 0.92), and the 28-day mortality rate was 8.4% (865 deaths) for the remdesivir group compared to 12.5% (1,334 deaths) for matched patients in the nonremdesivir group.
Among the patients treated with remdesivir and receiving HFNC or NIPPV, the aHR was 1.10 (95% CI, 1.01 to 1.20), with a 28-day mortality rate of 28.6% (1,137 deaths) in the remdesivir group compared to 34.0% (1,237 deaths) for matched patients in the nonremdesivir group. Among the patients treated with remdesivir receiving invasive mechanical ventilation, the aHR was 1.17 (95% CI, 1.04 to 1.32), with a 28-day mortality rate of 46.7% (552 deaths) in the remdesivir group compared to 43.9% (686 deaths) for matched patients in the nonremdesivir group.
Critical Appraisal
Internal Validity
The pivotal trial data lack clear information about the effect of remdesivir on time to clinical improvement in patients who are receiving dexamethasone. The sponsor has indicated that this study fills a gap in the pivotal trial data as it addresses the effectiveness of remdesivir plus dexamethasone compared to dexamethasone alone on time to improvement in patients who were hospitalized with COVID-19 at a large US health care network from February 2020 to February 2021.
The sponsor has highlighted the importance of this analysis of patients taking remdesivir plus dexamethasone as compared to dexamethasone alone as addressing a gap in the pivotal trial data. However, that particular analysis was conducted as a sensitivity analysis and not as the primary study analysis. Even though similar results were observed on time to clinical improvement in the comparison groups, it is unknown if there were substantial differences, such as in baseline disease characteristics, between the groups.
There is a potential for information bias due to the subjective nature of time to improvement (2-point decrease in the WHO severity score or discharged alive without worsening of the WHO severity score within 28 days) outside the setting of an RCT, and there is potential for WHO severity to be different between patients who were treated with remdesivir and those who were not. The study did not include any information about treatments received before hospitalization. The study also lacked information about time since symptom onset; this may have differed between patients who were treated with remdesivir and those who were not.
PS matching accounted for a number of time-dependent covariates, including day of hospitalization, comorbidities, and laboratory values. A time-dependent match would have largely reduced the concern of time-related biases. However, the study was unable to match approximately half of the patients treated with remdesivir, largely because many patients received remdesivir after October 1, 2020.
This study is at risk of unmeasured confounders and residual confounding associated with the prognosis of the disease (e.g., the severity of the disease). The analyses were conducted with time-dependent variables that included demographics, oxygen delivery, vital signs, key laboratory values, comorbidities, and specific medications for COVID-19 (acetaminophen, dexamethasone, prednisone, hydrocortisone, tocilizumab). Despite the large number of covariates that the authors matched for, and the inclusion of sensitivity analyses, there is still a possibility of unobserved confounders and residual confounding. The authors did not explicitly justify the rationale for inclusion of the variables in the model.
External Validity
The data were extracted from a large private US health care database; this database captures approximately 6% of all hospitalizations in the US. This health care system differs from the Canadian health care system, and prehospital treatments or response to public health measures may have been different.
The study occurred during the time period February 2020 to February 2021, before the widespread use of vaccinations. However, vaccine uptake and background disease risk, as well as circulating variants, have changed substantively over time, limiting the generalizability of these findings to the current COVID-19 treatment landscape. The comparability of baseline care (e.g., corticosteroids, anticoagulants) may limit generalizability to the setting in Canada.
Discussion
Summary of Available Evidence
Two double-blind RCTs, 2 open-label RCTs, and a cohort from 1 single-arm study were the primary sources of evidence for the efficacy and safety of remdesivir in hospitalized patients with COVID-19 aged 12 years or older, weighing at least 40 kg, and requiring oxygen support due to pneumonia. The ACTT-1 study (N = 1,062) was a double-blind, placebo-controlled, multicentre, international, phase III RCT in adults aged 18 years or older admitted to hospital with confirmed COVID-19. The study by Wang et al. (2020) (N = 237) was a double-blind, placebo-controlled, multicentre RCT conducted in 10 hospitals in China in adult patients aged 18 years or older admitted to hospital with confirmed COVID-19. The WHO Solidarity trial (N = 8,320 for remdesivir and its control group) was an open-label, SOC-controlled RCT of several putative treatments for COVID-19 across the globe in adults with definite COVID-19, although only the remdesivir group and its associated control group are described in this report. The study by Spinner et al. (2020) (N = 596) was an open-label, multicentre, international RCT evaluating 5 days or 10 days of treatment with remdesivir compared with SOC in hospitalized patients aged 12 years or older with moderate COVID-19 pneumonia. The CARAVAN study (N = 53) was a single-arm, open-label, phase II/III, international study in pediatric patients, of which only those in cohort 1 (N = 12) were aged 12 years or older and weighed at least 40 kg. The outcomes of interest from these studies, for the purpose of this review, were mortality, duration of hospitalization, time to recovery or clinical improvement, and initiation of ventilation.
The sponsor also submitted 9 RWE studies as supportive evidence. The study by Mozaffari et al. (2023)23 was a retrospective cohort study that examined the effect of remdesivir on the outcomes of 14-day and 28-day mortality among in-hospital patients with COVID-19 who required supplemental oxygen, including low-flow oxygen, high-flow oxygen or noninvasive ventilation, and invasive mechanical ventilation or ECMO, across VOC periods, in a large US health care network. The study by Mozaffari et al. (2024)24 (N = 440 to 601) was a retrospective study evaluating the effect of remdesivir among adult patients discharged after hospitalization for COVID-19 during hospitalization for COVID-19 on 30-day COVID-19–related and all-cause readmission across different variants and time periods. The study by Finn et al. (2022)25 (N = 2,062) was a retrospective study evaluating the effect of remdesivir in patients discharged after hospitalization for COVID-19 for the outcomes of length of hospital stay, 30-day readmission, and postdischarge 30-day all-cause mortality. The study by Boglione et al. (2022)26 (N = 449) was a prospective study that aimed to analyze the prevalence of and risk factors for post–COVID-19 condition in patients hospitalized for COVID-19. The study included patients hospitalized at a single hospital in Italy, who were followed for at least 6 months postdischarge. The study by Kikuchi et al. (2021)27 (N = 1,010) was a registry study evaluating risk factors for mortality in patients receiving dialysis who were hospitalized for COVID-19. The study by Seethapathy et al. (2022)28 (N = 62) was a retrospective cohort study that examined the association between remdesivir and AEs in patients hospitalized for COVID-19 with an eGFR less than 30 mL/min/1.73 m2 within the Mass General Brigham health care system in the Boston, Massachusetts, region of the US. The study by Seethapathy et al. (2023)29 (N = 350) was a retrospective study evaluating the safety of remdesivir in relation to adverse kidney outcomes in patients hospitalized for COVID-19 with an eGFR between 15 mL/min/1.73 m2 and 60 mL/min/1.73 m2. The study by Mozaffari et al. (2023)30 (N = 28,338) was a retrospective cohort study that examined, across different levels of oxygen requirements and different VOC periods in a large US health care network, the effect of remdesivir on the outcomes of 14-day and 28-day mortality among in-hospital patients with COVID-19 who were immunocompromised. The study by Garibaldi et al. (2021)31 (18,328 pairs of patient treated with and not treated with remdesivir) was a retrospective study that included a sensitivity analysis of remdesivir plus dexamethasone versus dexamethasone alone in patients hospitalized for COVID-19 across different VOC periods for the outcomes of time to improvement and time to death.
Interpretation of Results
Efficacy
Pivotal Trial Data
In the ITT or as-treated populations, of the 4 comparative RCTs, only the ACTT-1 study identified a benefit of remdesivir for mortality (statistically significant at day 15 but not day 29), duration of hospitalization, and time to recovery; the other studies did not demonstrate significant benefits for these outcomes in these populations. There is an important possibility that the positive results of the ACTT-1 study were driven by a subgroup of patients who uniquely benefited from remdesivir, while other subgroups did not benefit. Although the ACTT-1 study stratified randomization by mild-moderate and severe disease and identified that the mild-moderate disease subgroup did not benefit from remdesivir while the severe disease subgroup did, the definition of severe disease was broad and comprised several clinical statuses with distinct prognoses (i.e., patients on low-flow oxygen, high-flow oxygen, noninvasive ventilation, invasive mechanical ventilation, or ECMO). For clinical recovery outcomes, the ACTT-1 study presented subgroup results by oxygen support requirements at baseline and demonstrated that only the subgroup of patients on low-flow oxygen (level 5 on the ordinal scale of clinical status) had a statistically significant benefit associated with allocation to remdesivir; this subgroup was also by far the most populous and was disproportionately larger in the treatment group than in the placebo group. The disproportionate size of this subgroup was criticized as potentially causing bias by the authors of the WHO Solidarity trial; however, the clinical expert consulted by CDA-AMC did not feel this was an important difference in terms of risk of bias. Although the magnitude of potential bias is unknown, these facts considered together suggest that the benefits associated with remdesivir in the overall population or within the subgroup of patients with severe disease in the ACTT-1 study could be biased by or driven by this more specific patient subgroup. Corroborating this are the results of the very large WHO Solidarity trial, which did not demonstrate an overall benefit of remdesivir on mortality in the ITT population but did identify a benefit specifically in the subgroup of patients receiving oxygen support but not ventilation; notably, in the WHO Solidarity trial, low-flow and high-flow oxygen were not recorded separately, so the subgroup definition is not identical to that in the ACTT-1 study. The other RCTs, the studies by Wang et al. (2020) and Spinner et al. (2020), did not demonstrate an overall benefit of remdesivir on mortality, duration of hospitalization, or time to recovery or clinical improvement and did not report on analogous subgroups related to oxygen support requirements. The Canadian substudy of the WHO Solidarity trial, CATCO,22 generally demonstrated consistent results with the global WHO Solidarity study, with the exception that the subgroup analysis by baseline organ support requirements did not demonstrate any particular difference between subgroups in mortality. However, the substudy was much smaller than the global study and, as such, may not have been powered to detect differences in these subgroups.
Taken together, remdesivir appears to be associated with a benefit in mortality in patients who require low-flow oxygen. There is some uncertainty whether patients requiring high-flow oxygen would experience benefit, as these patients were never evaluated in isolation in any study. In the ACTT-1 study, they were grouped with patients on noninvasive ventilation, and there was no identified benefit; in the WHO Solidarity trial, they were grouped with patients on low-flow oxygen, and there was an identified benefit that could possibly have been driven by the positive results in patients on low-flow oxygen.
These observations align with WHO’s Living Guidance for Clinical Management of COVID-196 and the Canadian practice recommendations by Grant et al. (2024),33 which identify patients on low-flow oxygen as the strongest candidates for treatment with remdesivir, and recommend its use (with a caveat for weak or conditional recommendation in the WHO guidance, specifically). The WHO guidance is delineated by severity, where “severe” patients are those with lowered oxygen saturation on room air, signs of pneumonia, and/or signs of severe respiratory distress, and “critical” patients are those requiring life-sustaining treatment. The latter group of patients, who in the Canadian guidance would align with those on the most invasive forms of oxygen or organ support, are not recommended to receive remdesivir by either WHO or the Canadian guidance.
It is uncertain whether there is a benefit from treatment with remdesivir in the duration of hospitalization in the overall target population or in any subgroup, as the results were inconsistent between studies and, critically, the prognosis of patients has substantially changed because of the evolution of the virus and changes in immunity as a result of past infections and vaccinations. Again, only the ACTT-1 study reported a reduction in the duration of hospitalization associated with allocation to treatment with remdesivir versus placebo. In contrast, the WHO Solidarity trial reported a longer hospital stay associated with remdesivir, owing to the duration of therapy keeping patients in hospital to receive treatment who might have otherwise been suitable for discharge earlier. However, patients in the WHO Solidarity trial were receiving a 10-day course of remdesivir; in the real world, patients may be likely to receive a shorter course of treatment as the product monograph recommends a minimum of 5 days of treatment, and the clinical expert consulted by CDA-AMC noted that clinical practice varies widely in using a 5-day or 10-day regimen for remdesivir. The other comparative studies by Wang et al. (2020) and Spinner et al. (2020) reported no significant difference between treatment groups in duration of hospitalization, and the study by Spinner et al. (2020) was unique in including a 10-day remdesivir group as a well as a 5-day remdesivir group, both compared to SOC.
Two studies (ACTT-1 and WHO Solidarity) evaluated the incidence of new ventilation in patients who were not ventilated at baseline. In the ACTT-1 study, new noninvasive ventilation or high-flow oxygen (in patients not on high-flow oxygen or ventilation of any type at baseline) was numerically less common in the remdesivir group, but the difference was not statistically significant compared to placebo. However, the incidence of new invasive mechanical ventilation or ECMO among patients who were not receiving these supports at baseline was statistically significantly lower in the remdesivir group than in the placebo group. In the WHO Solidarity trial, initiation of ventilation was not subdivided by noninvasive versus invasive or ECMO. Among patients who were not ventilated at study entry, remdesivir was associated with a benefit in reducing the initiation of new ventilation and in the composite outcome of “initiation of ventilation or death.” The publication summarizing the Canadian substudy of the WHO Solidarity trial, CATCO,22 concluded that the most important benefit observed in the context in Canada was prevention of the need for new mechanical ventilation, given the limitations on supply for these devices. Additionally, the publication22 reported more ventilator-free and oxygen-free days associated with remdesivir treatment.
Remdesivir for the treatment of adult or adolescent inpatients with COVID-19 was evaluated by CADTH in 2023,34 outside of the reimbursement review pathway. The systematic literature review34 conducted in 2023 included the ACTT-1 study, the study by Spinner et al. (2020), and the WHO Solidarity trial and its add-on studies or substudies (NOR-Solidarity, CATCO, Solidarity Finland, Discovery). The results suggested that remdesivir reduced the need for mechanical ventilation compared to SOC (3 studies) and reduced the need for intubation compared to SOC (1 study). The findings suggested that remdesivir did not have a significant benefit in ICU admissions or length of ICU stay, nor time to ventilation. The impact of treatment with remdesivir on the length of hospitalization, time to clinical improvement, and progression to high-flow oxygen was inconsistent. Pooled meta-analysis results from 6 studies suggest there may have been a benefit of remdesivir on risk of death compared to SOC, but in this systematic literature review34 there was no evaluation of the subgroup effects that may be driving this conclusion.
There is also a publicly available Cochrane review35 of remdesivir for the treatment of COVID-19 in both hospitalized and nonhospitalized patients, although only the in-hospital results will be discussed here. In hospitalized patients with moderate to severe COVID-19, the Cochrane review concluded, with moderate-certainty evidence, that remdesivir probably makes little or no difference in all-cause mortality up to day 28 (4 studies; 7,142 patients) or day 60 (1 study; 1,281 patients) or in in-hospital mortality up to day 150 (1 study; 8,275 patients). The Cochrane review suggested that remdesivir probably increases the chance of clinical improvement up to day 28 slightly and decreases the risk of clinical worsening within 28 days.
This CDA-AMC Reimbursement Review, the clinical practice guidelines by WHO6 and Grant et al. (2024),33 the previous CADTH systematic literature review34 and meta-analysis,34 and the Cochrane review35 are generally aligned in considering that the evidence for the benefit of remdesivir is somewhat inconsistent and that remdesivir is unlikely to have a significant effect on mortality or duration of hospitalization in the overall target population of hospitalized patients with COVID-19 but that (where evaluated) it may have some benefit in reducing the incidence of new ventilation. Moreover, among the guidelines and reviews that considered subgroup effects, there is a consensus that there may be a benefit of treatment with remdesivir in clinical recovery outcomes and mortality, specifically in the subgroup of patients who are receiving low-flow oxygen and, potentially (with less certainty), in patients who are receiving high-flow oxygen, but not in those with severe enough disease to require ventilation or life-saving organ support.
All this evidence has major issues with generalizability given that studies from the early pandemic would be conducted with unvaccinated patients with little to no prior immunity, infected with variants that are no longer prevalent in 2024 and beyond.
Supportive RWE
Several supportive studies were submitted by the sponsor and have been evaluated by CDA-AMC, some of which may address aspects of this generalizability issue, and others which assess outcomes after hospitalization or in specific subpopulations. However, these studies do not represent the totality of RWE available for COVID-19 and were each selected by the sponsor for a specific reason to fill a data gap from the pivotal trials.
Guidance for Reporting Real-World Evidence forms the foundation for transparent reporting of RWE studies in Canada and facilitates the appraisal of RWE by CDA-AMC.32 All applicable sections in the guidance should be reported on when submitting RWE studies as part of a Reimbursement Review.32 Many RWE studies submitted as part of this review were missing important information. Information on the following was missing: why a setting outside of Canada was chosen, differences in health systems, access to care, available health care resources during the pandemic, and other factors that may impact the care of patients with COVID-19 and how those factors might affect the applicability of findings to the current context in Canada. A detailed description of data specifications (access, cleaning, and links, where applicable); data sources, including a data dictionary; variables that could not be captured; and the potential impacts on the study results were not provided.32
The pivotal trial data lack information about the effect of remdesivir on mortality in more recent COVID-19 variants. The study by Mozaffari et al. (2023)23 is a large observational study that found that remdesivir reduced 14-day and 28-day mortality compared with no remdesivir treatment in patients hospitalized for COVID-19 between December 2020 and April 2022. The study by Mozaffari et al. (2023)23 may address a gap in the pivotal trial data as it describes the comparative effectiveness of remdesivir on the outcomes of 14-day and 28-day mortality in a population of patients across 3 variant periods (pre-Delta, Delta, and Omicron). The limitations included a lack of information about the time of symptom onset and the treatments or vaccines administered before hospitalization. Although many comorbidities were controlled for, there remains a potential for imbalances in unmeasured confounders and residual confounding. The study by Mozaffari et al. (2024)24 is another a large observational study that found that remdesivir reduced 30-day all-cause and COVID-19–related rehospitalization compared to no remdesivir treatment in patients who were hospitalized between December 2020 and April 2022 across 3 variant periods (pre-Delta, Delta, and Omicron). The limitations included that the impact of missing data on the outcome of rehospitalization is not clear. There is also a lack of information about the time since symptom onset and treatments received before hospitalization. Despite the inclusion of numerous variables in the multivariate regression, there is still a potential for unmeasured confounders and residual confounding. For the studies by Mozaffari et al. (2023)23 and Mozaffari et al. (2024),24 it is unclear whether the information from the US cohort is generalizable to Canada. Vaccine uptake and background disease risk, as well as circulating variants, have changed since the studies were conducted, which may limit the generalizability of these findings to the current COVID-19 treatment landscape. Therefore, it is challenging to assess the exact magnitude of the treatment effect of remdesivir on the reduction of in-hospital 14-day and 28-day mortality compared with no remdesivir treatment and to extrapolate the effect to current practice in Canada.
The pivotal trial data lack clear information about the effect of remdesivir on outcomes that occur after hospital discharge. The study by Finn et al. (2022)25 is a small observational study (742 patients who were treated with remdesivir matched to 1,539 patients who were not) that used EHR data from 3 hospitals in Rhode Island and found that treatment with remdesivir reduced hospital readmission and 30-day all-cause mortality compared to no remdesivir treatment in patients who were discharged after being hospitalized for COVID-19 between April 2020 and December 2020. The study by Finn et al. (2022)25 may address a gap in the pivotal trial data; however, it is subject to numerous limitations. These limitations include a lack of information about the time since symptom onset, the potential for time-related bias in the assessment of hospitalization, the potential for missing data related to postdischarge outcomes, and the potential for unmeasured confounders and residual confounding. Therefore, it is challenging to assess the exact magnitude of the benefit of remdesivir from this study on outcomes that occur after hospital discharge for patients hospitalized with COVID-19 and to extrapolate the effect to current practice in Canada.
The pivotal trial data lack clear information about the effect of remdesivir on post–COVID-19 condition. The study by Boglione et al. (2022)26 is a small observational study (including 163 patients treated with remdesivir) of hospitalized patients with COVID-19 at a single hospital in Italy from March 2020 to January 2021. The study by Boglione et al. (2022)26 likely has significant methodologic limitations, including risks for confounding by indication and lack of matching, uncertainty in the definition of post–COVID-19 condition, and limited generalizability from the Italian setting to Canada. These limitations preclude drawing conclusions about the effect of remdesivir on post–COVID-19 condition from this study.
The pivotal trial data lack clear information about the effect of remdesivir in patients with renal insufficiency. Three RWE studies were submitted to address this: Kikuchi et al. (2021),27 Seethapathy et al. (2021),28 and Seethapathy et al. (2023).29 The study by Kikuchi et al. (2021)27 is a small observational study (98 patients who were treated with remdesivir matched to 294 patients who were not) that used registry data to assess the effect of remdesivir on mortality in patients who were admitted to hospital with COVID-19 and received dialysis in Japan from April 2020 to June 2021. The study by Seethapathy et al. (2021)28 is a small observational study that used EHR data from a single US hospital to examine the relationship between AEs and remdesivir treatment in patients with an eGFR less than 30 mL/min/1.73 m2 from May 2020 to January 2021 (for patients treated with remdesivir) and from March 2020 to April 2020 (for patients not treated with remdesivir). The study by Seethapathy et al. (2023)29 is a small observational study that used EHR data from a single US hospital to examine the relationship between adverse laboratory-based renal outcomes and remdesivir in patients with an eGFR of 15 mL/min/1.73 m2 to 60 mL/min/1.73 m2 from April 2020 to November 2020 (for patients treated with remdesivir) and from March 2020 to April 2020 (for patients not treated with remdesivir). The limitations of all 3 studies and the inability to extrapolate the effects to current practice in Canada preclude conclusions about the effect of remdesivir in patients admitted to hospital with COVID-19 who are receiving dialysis or have reduced renal function.
The pivotal trial data lack clear information about the effect of remdesivir in patients who are immunocompromised. The study by Mozaffari et al. (2023)30 is a large observational study that used a US dataset and found that treatment with remdesivir reduced 14-day and 28-day mortality, compared with not receiving treatment with remdesivir, in patients who were immunocompromised and hospitalized for COVID-19 between December 2020 and April 2022. The study by Mozaffari et al. (2023)30 may address a gap in the pivotal trial data as it describes the comparative effectiveness of remdesivir on the outcomes of 14-day and 28-day mortality in immunocompromised patients. The limitations included a lack of information about the time of symptom onset and the treatments or vaccines administered before hospitalization. Although many comorbidities were controlled for, there remains a potential for imbalances in unmeasured confounders and residual confounding. It is difficult to extrapolate the magnitude of the effect of treatment with remdesivir for patients who are immunocompromised in Canada due to uncertainty about the generalizability of the US cohort to Canada.
The study by Garibaldi et al. (2021)31 was submitted by the sponsor to address the effect of remdesivir in patients receiving dexamethasone and is a large observational study (18,328 pairs of patients who were treated with remdesivir and patients who were not) that used a US dataset to examine the relationship between remdesivir treatment and time to improvement in patients who were hospitalized with COVID-19 from February 2020 to February 2021. The study by Garibaldi et al. (2021)31 may address a gap in the pivotal trial data; however, the analysis of treatment with remdesivir plus dexamethasone compared with dexamethasone alone in terms of time to improvement is based on a sensitivity analysis only and therefore has limitations. Additional limitations include the potential for information bias due to the subjective nature of time to improvement (2-point decrease in the WHO severity score or discharged alive without worsening of the WHO severity score within 28 days) and the lack of information about the time since symptom onset or the treatments received before hospitalization. Approximately half the patients treated with remdesivir were unable to be matched, a potential source of bias. The potential for unmeasured confounders and residual confounding are other limitations. It is unclear whether the information from the US cohort is generalizable to Canada. Vaccine uptake and background disease risk as well as circulating variants have changed, which may limit the generalizability of these findings to the current COVID-19 treatment landscape. Therefore, it is challenging to assess the exact magnitude of the treatment effect of remdesivir on time to improvement compared with no remdesivir treatment and to extrapolate the effect to current practice in Canada.
Harms
Remdesivir was well tolerated in all the included studies. In the 4 comparative studies, AEs and SAEs were typically similar between remdesivir groups and placebo or SOC groups. There was no substantial difference observed between the 5-day and 10-day remdesivir groups in the study by Spinner et al. (2024).
There is a lack of evidence on the safety of remdesivir for patients older than 70 years and patients with multiple comorbidities who may be at increased risk of negative outcomes.
Conclusion
The findings from the 4 RCTs17-20 suggest that remdesivir may prevent death in hospitalized patients aged 12 years or older with COVID-19 whose disease severity warrants support with low-flow oxygen and perhaps also in patients who require high-flow oxygen, but the latter subgroup was not evaluated in isolation. Remdesivir does not appear to prevent death in hospitalized patients who do not require oxygen support nor in patients who require any level of ventilation (noninvasive or invasive) or ECMO. The same subgroups, but not the overall population, may benefit from treatment with remdesivir for outcomes related to time to recovery or clinical improvement, as defined by the level of oxygen support required by the patient. Whether there is a benefit of remdesivir on duration of hospitalization is inconclusive due to between-study inconsistency: only 1 study (ACTT-1) reported a benefit; all the others either reported a longer duration of hospitalization among patients treated with remdesivir due to the 10-day regimen used in the study (WHO Solidarity) or reported no difference between treatment groups (Spinner et al. [2020], Wang et al. [2020]). Based on the findings of the ACTT-1 study and the WHO Solidarity trial, there may be a benefit of remdesivir in reducing the initiation of new ventilation among patients who were not ventilated at baseline. There may also be a modest benefit of remdesivir in reducing the duration of oxygen support, but there is some uncertainty due to IQRs overlapping between the treatment groups and inconsistency across the included studies.
Remdesivir was well tolerated in all the included studies, and safety outcomes were similar between the remdesivir treatment groups and the placebo or SOC treatment groups in each study. In terms of the duration of therapy, remdesivir can be given for at least 5 days and not more than 10 days; there appeared to be no obvious additional benefit or harm associated with a 10-day course of remdesivir over a 5-day course according to the comparisons to SOC conducted by Spinner et al. (2020).
The results from this assessment are generally aligned with WHO’s Living Guidance for Clinical Management of COVID-19,6 the Canadian treatment practice guidelines by Grant et al. (2024),33 a systematic literature review by CADTH conducted in 2023,34 and a Cochrane systematic review of remdesivir.35 However, there are critical generalizability concerns because all the RCT evidence was gathered during the early pandemic, at which time patients had little to no prior immunity due to the general unavailability of vaccines and the lack of prior infection history, and COVID-19 infections were caused by variants no longer prevalent in 2024. There was also an information gap regarding some patient subpopulations in the pivotal trial evidence. The sponsor submitted several RWE studies that supported the use of remdesivir in specific populations, including patients with more recent variants (up to Omicron), patients with renal insufficiency, patients who are immunocompromised, and patients also receiving dexamethasone. Additionally, a single-arm pivotal study (CARAVAN) was submitted to inform the safety and efficacy of remdesivir in adolescent patients. However, there are limitations inherent to these studies that preclude concluding with certainty on the benefit of remdesivir as extrapolated to the current context in Canada.
References
1.Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int J Antimicrob Agents. 2020;55(3):105924. PubMed
2.Haldane V, De Foo C, Abdalla SM, et al. Health systems resilience in managing the COVID-19 pandemic: lessons from 28 countries. Nat Med. 2021;27(6):964-980. PubMed
3.Canadian Institute for Health Information. Overview: COVID-19’s impact on health care systems. 2021; https://www.cihi.ca/en/covid-19-resources/impact-of-covid-19-on-canadas-health-care-systems/the-big-picture. Accessed 2023 Dec 08.
4.COVID-19 epidemiology update: current situation. Ottawa (ON): Public Health Agency of Canada; 2024: https://health-infobase.canada.ca/covid-19/current-situation.html.
5.Canadian Institute for Health Information. COVID-19 hospitalization and emergency department statistics. 2024; https://www.cihi.ca/en/covid-19-hospitalization-and-emergency-department-statistics. Accessed 2024 May 21.
6.Living guidance for clinical management of COVID-19. Geneva (CH): World Health Organization; 2021: https://www.who.int/publications/i/item/WHO-2019-nCoV-clinical-2021-2. Accessed 2024 Jan 17.
7.Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. 2020;324(8):782-793. PubMed
8.He X, Cheng X, Feng X, Wan H, Chen S, Xiong M. Clinical symptom differences between mild and severe COVID-19 patients in China: a meta-analysis. Front Public Health. 2021;8:561264. PubMed
9.Gandhi RT, Lynch JB, Del Rio C. Mild or moderate Covid-19. N Engl J Med. 2020;383(18):1757-1766. PubMed
10.Hajouli S. A 29-year-old man with COVID-19 pneumonia, heart failure-reduced ejection fraction, and atrial fibrillation with a father and 2 grandparents who were positive for SARS-CoV-2 infection. Am J Case Rep. 2021;22:e933163. PubMed
11.Iqbal QZ, Haider MA, Sattar SBA, Hanif M, Javid I. COVID-19 induced myocarditis: a rare cause of heart failure. Cureus. 2020;12(11):e11690. PubMed
12.Suh YJ, Hong H, Ohana M, et al. Pulmonary embolism and deep vein thrombosis in COVID-19: a systematic review and meta-analysis. Radiology. 2021;298(2):E70-E80. PubMed
13.Therapeutics and COVID-19: living guideline, 10 November 2023. Geneva (CH): World Health Organization; 2023: https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2023.2. Accessed 2024 Apr 03.
14.Gouvernement du Québec. Symptômes, transmission et traitement (COVID-19). 2022; https://www.quebec.ca/sante/problemes-de-sante/a-z/coronavirus-2019/symptomes-transmission-traitement. Accessed 2023 Dec 08.
15.Martono, Fatmawati F, Mulyanti S. Risk factors associated with the severity of COVID-19. Malays J Med Sci. 2023;30(3):84-92. PubMed
16.Veklury (remdesivir): 100 mg/vial powder for solution for infusion [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2024 May 06.
17.Clinical Study Report: CO-US-540-5776, ACTT-1 Final. A multicenter, adaptive, randomized blinded controlled trial of the safety and efficacy of investigational therapeutics for the treatment of COVID-19 in hospitalized adults [internal sponsor's report]. Bethesda (MD): Division of Microbiology and Infectious Diseases (DMID), National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH); 2020 Aug 23.
18.WHO Solidarity Trial Consortium. Remdesivir and three other drugs for hospitalised patients with COVID-19: final results of the WHO Solidarity randomised trial and updated meta-analyses. Lancet. 2022;399(10339):1941-1953. PubMed
19.Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395(10236):1569-1578. PubMed
20.Spinner CD, Gottlieb RL, Criner GJ, et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. JAMA. 2020;324(11):1048-1057. PubMed
21.Clinical Study Report: GS-US-540-5823 (CARAVAN). A phase 2/3 single-arm, open-label study to evaluate the safety, tolerability, pharmacokinetics, and efficacy of remdesivir (GS-5734™) in participants from birth to < 18 years of age with COVID-19 [internal sponsor's report]. Foster City (CA): Gilead Sciences, Inc.; 2021 Oct 04.
22.Ali K, Azher T, Baqi M, et al. Remdesivir for the treatment of patients in hospital with COVID-19 in Canada: a randomized controlled trial. CMAJ. 2022;194(7):E242-E251. PubMed
23.Mozaffari E, Chandak A, Gottlieb RL, et al. Remdesivir is associated with reduced mortality in COVID-19 patients requiring supplemental oxygen including invasive mechanical ventilation across SARS-CoV-2 variants. Open Forum Infect Dis. 2023;10(10):ofad482. PubMed
24.Mozaffari E, Chandak A, Gottlieb RL, et al. Treatment of patients hospitalized for COVID-19 with remdesivir is associated with lower likelihood of 30-day readmission: a retrospective observational study. J Comp Eff Res. 2024;13(4):e230131. PubMed
25.Finn A, Jindal A, Andrea SB, Selvaraj V, Dapaah-Afriyie K. Association of Treatment with Remdesivir and 30-day Hospital Readmissions in Patients Hospitalized with COVID-19. Am J Med Sci. 2022;363(5):403-410. PubMed
26.Boglione L, Meli G, Poletti F, et al. Risk factors and incidence of long-COVID syndrome in hospitalized patients: does remdesivir have a protective effect? QJM. 2022;114(12):865-871. PubMed
27.Kikuchi K, Nangaku M, Ryuzaki M, et al. Survival and predictive factors in dialysis patients with COVID-19 in Japan: a nationwide cohort study. Ren Replace Ther. 2021;7(1):59. PubMed
28.Seethapathy R, Zhao S, Long JD, Strohbehn IA, Sise ME. A propensity score-matched observational study of remdesivir in patients with COVID-19 and severe kidney disease. Kidney360. 2022;3(2):269-278. PubMed
29.Seethapathy R, Wang Q, Zhao S, et al. Effect of remdesivir on adverse kidney outcomes in hospitalized patients with COVID-19 and impaired kidney function. PLoS One. 2023;18(2):e0279765. PubMed
30.Mozaffari E, Chandak A, Gottlieb RL, et al. Remdesivir reduced mortality in immunocompromised patients hospitalized for COVID-19 across variant waves: Findings from routine clinical practice. Clin Infect Dis. 2023;77(12):1626-1634. PubMed
31.Garibaldi BT, Wang K, Robinson ML, et al. Real-world effectiveness of remdesivir in adults hospitalized with coronavirus disease 2019 (COVID-19): a retrospective, multicenter comparative effectiveness study. Clin Infect Dis. 2022;75(1):e516-e524. PubMed
32.Guidance for reporting real-world evidence. (CADTH methods and guidelines). Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/RWE/MG0020/MG0020-RWE-Guidance-Report-Secured.pdf. Accessed 2024 Fe 16.
33.Grant JM, Lam J, Goyal SV, et al. AMMI Canada Practice Point: Updated recommendations for treatment of adults with symptomatic COVID-19 in 2023-2024. J Assoc Med Microbiol Infect Dis Can. 2024;8(4):245-252. PubMed
34.Wang X, Kelly S, Peterson J, et al. Remdesivir (Veklury) for the treatment of COVID-19 in the inpatient setting. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/covid-19/Bundle%20covid%2019/remdesivir_for_the_treatment_of_COVID-19_in_the_inpatient-setting_systematic_review.pdf. Accessed 2024 Apr 30.
35.Grundeis F, Ansems K, Dahms K, et al. Remdesivir for the treatment of COVID-19. Cochrane Database Syst Rev. 2023;1(1):CD014962. PubMed
36.Wu Z, McGoogan JM. Asymptomatic and pre-symptomatic COVID-19 in China. Infect Dis Poverty. 2020;9(1):72. PubMed
37.Ramage-Morin PL, Polsky JY. Health-related concerns and precautions during the COVID-19 pandemic: a comparison of Canadians with and without underlying health conditions. Health Rep. 2020;31(5):3-8. PubMed
38.Ding C, He J, Zhang X, et al. Crucial mutations of spike protein on SARS-CoV-2 evolved to variant strains escaping neutralization of convalescent plasmas and RBD-specific monoclonal antibodies. Front Immunol. 2021:3231. PubMed
39.Dhouib W, Maatoug J, Ayouni I, et al. The incubation period during the pandemic of COVID-19: a systematic review and meta-analysis. Syst Rev. 2021;10(1):101. PubMed
40.Jansen L, Tegomoh B, Lange K, et al. Investigation of a SARS-CoV-2 B.1.1.529 (Omicron) variant cluster - Nebraska, November-December 2021. MMWR Morb Mortal Wkly Rep. 2021;70(5152):1782-1784. PubMed
41.Smith-Jeffcoat SE, Pomeroy MA, Sleweon S, et al. Multistate outbreak of SARS-CoV-2 B.1.1.529 (Omicron) variant infections among persons in a social network attending a convention - New York City, November 18-December 20, 2021. MMWR Morb Mortal Wkly Rep. 2022;71(7):238-242. PubMed
42.Interim guidance on the use of rapid antigen detection tests for the identification of SARS-CoV-2 infection. Ottawa (ON): Public Health Agency of Canada; 2022: https://www.canada.ca/en/public-health/services/diseases/2019-novel-coronavirus-infection/guidance-documents/use-rapid-antigen-detection-tests.html.
43.Paxlovid (nirmatrelvir-ritonavir): 150 mg nirmatrelvir; 100 mg ritonavir, tablets, co-packaged for oral use [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2024 Jan 02: https://webfiles.pfizer.com/file/ffe4e681-59a8-4da8-ad39-bb18703d4271. Accessed 2024 Apr 03.
44.Evusheld (tixagevimab and cilgavimab): 100 mg/mL (tixagevimab) and 100 mg/mL (cilgavimab), solution for intramuscular injection [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2024 Feb 22: https://pdf.hres.ca/dpd_pm/00074797.PDF. Accessed 2024 Apr 03.
45.Sotrovimab: 500 mg/8 mL (62.5 mg/mL) in single use vial, solution for infusion after dilution [product monograph]. Mississauga (ON): GlaxoSmithKline Inc.; 2023: https://pdf.hres.ca/dpd_pm/00069318.PDF. Accessed 2024 Apr 03.
46.Casirivimab and imdevimab: 1332 mg/11.1 mL and 300 mg/2.5 mL (120mg/mL), in single-use vials, solutions for infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2022 Jun 07: https://pdf.hres.ca/dpd_pm/00066206.PDF. Accessed 2024 Apr 03.
Appendix 1: Duration of Supplemental Oxygen or Ventilator Use
Please note that this appendix has not been copy-edited.
Table 26: Duration of Supplemental Oxygen or Ventilator Use
Study | Measure | Population | Value in remdesivir group | Value in placebo or SOC group | Between-group comparison |
|---|---|---|---|---|---|
ACTT-1 | Median days (IQR) on oxygena | ITT; receiving this support at baseline (N = 913) | 13 (5 to 28) | 21 (8 to 21) | NR |
ITT; not receiving this support baseline (N = 138; n with new use = 55) | 4 (2 to 12) | 5.5 (1 to 15) | NR | ||
Median days (IQR) on noninvasive ventilation / high-flow oxygena | ITT; receiving this support at baseline (N = 193) | 6 (3 to 18) | 6 (3 to 16) | NR | |
ITT; not receiving this support at baseline (N = 573; n with new use = 116) | 3 (1 to 10.5) | 3 (4 to 23.5) | NR | ||
Median days (IQR) on invasive mechanical ventilation / ECMOa | ITT; receiving this support at baseline (N = 285) | 17 (9 to 28) | 20 (8 to 28) | NR | |
ITT; not receiving this support at baseline (N = 766; n with new use = 134) | 21.5 (9 to 28) | 23 (12 to 28) | NR | ||
CATCO (Canadian substudy of WHO Solidarity) | Mean oxygen-free days at day 28 (SD) | ITT (N = 634) | 15.9 (10.5) | 14.2 (11) | P = 0.006 |
Mean ventilator-free days at day 28 (SD) | ITT (N = 684) | 21.4 (11.3) | 19.5 (12.3) | P = 0.007 | |
Wang et al. (2020) | Median (IQR) days of invasive mechanical ventilation | ITT (N = 236) | 7.0 (4.0 to 16.0) | 15.5 (6.0 to 21.0) | Difference: –4.0 (95% CI, –14.0 to 2.0) |
Median (IQR) days of oxygen support | ITT (N = 236) | 19.0 (11.0 to 30.0) | 21.0 (14.0 to 30.5) | Difference: –2.0 (95% CI, –6.0 to 1.0) | |
Spinner et al. | Duration of oxygen therapy | NR | NR | NR | “There were no significant differences between the remdesivir and standard care groups in duration of oxygen therapy.” |
CI = confidence interval; IQR = interquartile range; ITT = intention to treat; NR = not reported; SD = standard deviation.
aIncluding imputations for patients who died.
Sources: ACTT-1 Clinical Study Report;17 CATCO;22 Spinner et al. (2020);20 Wang et al. (2020).19
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CIHI
Canadian Institute for Health Information
ECMO
extracorporeal membrane oxygenation
ICER
incremental cost-effectiveness ratio
ICU
intensive care unit
QALY
quality-adjusted life-year
SOC
standard of care
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Remdesivir (Veklury), 100 mg/vial, IV infusion |
Indication | For the treatment of COVID-19 in hospitalized adults and pediatric patients (at least 4 weeks of age and weighing at least 3 kg) with pneumonia requiring supplemental oxygen |
Health Canada approval status | NOC |
Health Canada review pathway | For use in relation to COVID-19 |
NOC date | July 27, 2020 |
Reimbursement request | For the treatment of hospitalized patients ≥ 12 years of age (weighing at least 40 kg) with pneumonia requiring supplemental oxygen |
Sponsor | Gilead Sciences Canada, Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree followed by Markov model |
Target population | Hospitalized patients with COVID-19 requiring supplemental oxygen |
Treatment | Remdesivir |
Dose regimen | Adult and pediatric patients (weighing at least 40 kg): 200 mg on day 1, followed by 100 mg once daily for an additional 4 to 9 days (for a total treatment duration of 5 to 10 days) |
Submitted price | Remdesivir 100 mg vial: $660.53 per vial |
Submitted treatment cost | $3,963.18 per patient, based on a 5-day treatment duration |
Comparator | SOC, in some cases comprising a combination of dexamethasone and therapeutic anticoagulation |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | 6 weeks |
Key data sources | ACTT-1 trial Real-world evidence (Mozaffari et al. [2023]) |
Submitted results | Remdesivir was dominant compared to SOC (incremental costs: –$80; incremental QALYs: 0.0040) |
Key limitations |
|
CDA-AMC reanalysis results |
|
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
Note: Dosing regimen for pediatric patients is as follows: patients aged 4 weeks or older (weighing at least 3 kg but less than 40 kg): 5 mg/kg on day 1, followed by 2.5 mg/kg daily for up to an additional 9 days (for a total treatment duration of up to 10 days).
Conclusions
The Clinical Review reported that remdesivir may prevent death in hospitalized patients aged 12 years or older with COVID-19 whose disease severity warrants support with low-flow oxygen and perhaps also in patients who require high-flow oxygen, but the latter subgroup was not evaluated as a distinct group. There is no evidence to suggest that remdesivir prevents death in hospitalized patients who do not require oxygen support, nor in patients who require any level of ventilation (noninvasive or invasive) or extracorporeal membrane oxygenation (ECMO). The results of 1 study reviewed in the Clinical Review (Wang et al. [2020]) found that mortality was similar between the remdesivir and placebo treatment groups at day 28. The Clinical Review also highlighted that the evaluated trials did not include vaccinated patients or patients who had had COVID-19 in the past and that the trials were performed at a time when the Omicron variant was not yet circulating. There are substantial concerns regarding the external validity and generalizability of every study included in the Clinical Review because of the fast-evolving nature of the pandemic and the virus itself: the prevalent variants, the status of vaccinations, and the clinical outcomes in today’s world are substantially different than those observed in the early pandemic. These differences represent a fundamental challenge in interpreting the results from the sponsor’s submitted evidence dossier and the accompanying pharmacoeconomic model, which are based on the ACTT-1 trial and submitted real-world evidence.
Canada’s Drug Agency (CDA-AMC) conducted several reanalyses that made changes to the sponsor’s submitted model, in consultation with clinical expert feedback. CDA-AMC adjusted the baseline distribution for level of hospital care, adjusted COVID-19 hospitalization costs, and assessed the impact of 4 alternative mortality assumptions that changed the mortality benefit associated with remdesivir and the absolute risk of mortality for all patients hospitalized with COVID-19. In general, the incremental quality-adjusted life-years (QALYs) estimated in the CDA-AMC reanalyses were similar to those in the sponsor’s results (sponsor’s incremental QALYs: 0.0040; the CDA-AMC incremental QALYs: 0.0014 to 0.0009); these benefits equate to an additional 8 to 12 quality-adjusted hours of life in the CDA-AMC reanalyses to an additional 35 quality-adjusted hours of life in the sponsor’s analysis. However, the incremental costs in the CDA-AMC reanalysis were higher (approximately $3,600 in all the CDA-AMC reanalyses). The incremental costs were driven by changes in COVID-19 hospitalization costs and changes in mortality, which had a cost assigned to it.
In the CDA-AMC reanalysis that assumed the mortality benefit applies only to patients requiring low-flow oxygen, remdesivir was associated with an incremental cost-effectiveness ratio (ICER) of $2,542,952 per QALY gained. Similar results were found in CDA-AMC reanalysis that assumed the absolute mortality risk of COVID-19 was lower for all patients than the sponsor assumed. In the CDA-AMC reanalyses that estimated lower incremental QALYs due to assuming no mortality benefit for patients treated with remdesivir, the ICER was $4,208,181. Despite the wide range of ICERs estimated by the CDA-AMC reanalyses, the price reduction analyses were similar across all mortality assumptions. These analyses demonstrated that a price of $317 to $396 per 5-day treatment course (a reduction of approximately 90% to 92%) may be required for remdesivir to be considered cost-effective compared to standard of care (SOC) at a threshold of $50,000 per QALY gained.
The results of these analyses are driven by the efficacy data used and baseline assumptions made about hospitalized patients with COVID-19 infections. Given the differences in patient profile and COVID-19 variants today, the mortality benefit likely to be experienced in the current COVID-19 setting in Canada remains highly uncertain. Notably, the clinical expert opinion solicited by CDA-AMC for this review highlighted that an important distinction exists between patients hospitalized because of their COVID-19 infection and those hospitalized who incidentally have a COVID-19 infection. Patients who are hospitalized for other underlying causes may not benefit from treatment with remdesivir; as such, identifying and treating only those patients hospitalized as a result of COVID-19 is critical to maximizing the clinical benefit and the cost-effectiveness of remdesivir.
Patient, Clinician, and Drug Plan Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the review process.
No patient input was received for this review.
Clinician group input was received from the Ontario Health Infectious Diseases Advisory Committee. The input commented that current drug treatments available for patients requiring supplemental oxygen therapy are remdesivir, dexamethasone, baricitinib, tocilizumab, and sarilumab. The input noted that treatment goals are to reduce the severity of symptoms; prevent progression to critical COVID-19 disease; accelerate symptom recovery and viral clearance; prevent or reduce the need for new high-flow supplemental oxygen, noninvasive ventilation, mechanical ventilation, or ECMO; reduce the duration of hospitalization; prevent long-term sequelae; and prevent death. The clinician group input noted that not all patients respond to currently available treatments and noted specific limitations associated with remdesivir, including the generalizability of the randomized controlled trials, the optimal window for treatment initiation, the optimal duration of treatment, and remdesivir’s role in combination therapy. The input further noted that remdesivir is not expected to cause a shift in the current treatment paradigm as it can be added to other immunomodulatory agents that work on the hyperinflammatory pathway that tends to drive the disease course in the later stages of illness. The clinician group input stated that remdesivir is best suited for use in hospitalized patients who require supplemental low-flow oxygen.
The CDA-AMC–participating drug plans noted that, to date, COVID-19 therapeutics have been procured, paid for, and distributed to provinces and territories by the federal government. As such, the criteria used to determine coverage may be significantly different in provinces and territories. Additionally, the drug plans noted that funding for drugs administered to hospital inpatients generally comes from hospital global budgets and is not provided by public drug programs. Lastly, the drug plans highlighted that the submitted trials used different inclusion criteria, definitions of severe disease, and dosing regimens.
Several of these concerns were addressed in the sponsor’s model:
The submitted model allowed for changes in the need for mechanical ventilation or ECMO and in mortality.
In addition, CDA-AMC addressed some of these concerns, as follows:
CDA-AMC explored the impact of assuming more patients in the model required supplemental low-flow oxygen, the group of patients that clinician input indicated was best suited for treatment with remdesivir.
CDA-AMC was able to adjust the number of patients requiring hospitalization related to COVID-19 in the budget impact analysis (BIA) to better reflect the current COVID-19 landscape.
CDA-AMC was unable to address the following concerns raised by the patient, clinician, and drug plan input:
CDA-AMC was not able to resolve issues relating to the timing of the trials and associated generalizability concerns to patients hospitalized due to COVID-19 in the current setting in Canada.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of remdesivir compared with SOC (a combination of dexamethasone and therapeutic anticoagulation in some cases).1 The modelled population comprised hospitalized patients with COVID-19 who were moderately ill and required conventional oxygen (i.e., low-flow oxygen). The modelled population focuses on hospitalized patients aged 12 and older (weighing at least 40 kg), as per the reimbursement request.
The recommended dose for remdesivir in an inpatient setting for adults and pediatric patients (weighing more than 40 kg) is 200 mg on day 1, followed by 100 mg once daily for an additional 4 to 9 days (for a total treatment duration of 5 to 10 days).2 Remdesivir is administered intravenously and is provided as a powder for solution for infusion (5 mg/mL when reconstituted) at a submitted price of $660.53 per vial. In the submitted model, the sponsor assumed that the cost per patient was $3,963.18, assuming all patients are treated for 5 days. The comparator was SOC. No cost was assumed for SOC as it was assumed to be received by all patients.
The clinical outcomes of interest were QALYs and life-years over a 6-week time horizon. Discounting (1.5% per annum) was applied to both costs and outcomes, and a 2-week cycle length was used. The base-case perspective was that of the Canadian publicly funded health care payer.
Model Structure
The sponsor submitted a short-term acute care decision tree followed by a Markov model. Patients were initially allocated into the inpatient model structure according to the highest level of care received in hospital at baseline using ordinal scale scores, as defined by the level of oxygen support required. The following levels of care received in hospital were modelled: ordinal scale score 1 to 3 (discharged from COVID-19 care), ordinal scale score 4 (no supplemental oxygen, assumed to be in general ward), ordinal scale score 5 (low-flow oxygen, assumed to be in general ward), ordinal scale score 6 (noninvasive ventilation or high-flow oxygen, assumed to be in intensive care unit [ICU]), ordinal scale score 7 (mechanical invasive ventilation or ECMO, assumed to be in ICU). Ordinal scale score 8 represents death. Patients entered the model at the time of hospitalization and could transition to a different level of hospitalized care (or discharge), including death, at week 2 and week 4. Patients who were discharged at week 2 were at risk of rehospitalization at week 4, and all patients were at risk of requiring rehabilitation. A 2-state Markov model (alive or dead) was used to model the postdischarge period.
Model Inputs
Patient baseline characteristics were informed by Canadian population statistics and published literature. The patient starting age in the model was 63 years.3 Patients were allocated to a starting ordinal scale score based on data from the CATCO trial.4
Treatment efficacy was modelled as the transition among ordinal scale scores at week 2 and the reduction of COVID-19–specific mortality at week 4. Treatment efficacy parameters were informed by the ACTT-1 trial (Beigel et al. [2020])5 and an observational study of patients hospitalized for COVID-19 between December 2020 and April 2022 in the US conducted by Mozaffari et al. (2023).6 The sponsor assumed that 17% of patients would require rehabilitation based on published literature7 and that the baseline proportion of people who would require rehospitalization was 10.6%.8 The rehospitalization rate was adjusted for patients treated with remdesivir using a hazard ratio of 0.87, informed by Mozaffari et al. (2023).6 The probability of death for recovered individuals was modelled using general population life tables for people in Canada.9
The sponsor’s model did not include the costs or health outcomes of treatment-related adverse events associated with remdesivir or SOC.
The age-adjusted baseline utility values in the model for the average patient were aligned with the general population in Canada, based on Guertin et al. (2018).10 The sponsor applied utility decrements for hospitalization services (i.e., general ward, ICU, ICU plus mechanical ventilation) that were adjusted using the respective length of hospital stay. The following utility decrements were obtained from published literature:11 0.27 for general ward, 0.36 for ICU, 0.56 for ICU plus mechanical ventilation. These disutilities were derived from a panel of 4 specialist physicians who treated patients with severe acute respiratory syndrome in Toronto in 2003 and did not use standard utility elicitation methods.12 Data on length of hospital stay data for patients receiving SOC were informed by real-world evidence from the US.13 The length of hospital stay for patients in the remdesivir group was estimated by applying rate ratios from the ACTT-1 trial to the length of stay used for patients in the SOC group. Disutilities for rehospitalization (0.003, assumed to be equal to 4 days of general ward stay) and rehabilitation (0.010, based on assumption) were applied as one-off events.
The model included drug acquisition costs, drug administration costs, hospitalization costs, rehabilitation costs, and rehospitalization costs. The drug acquisition costs have been described in the overview. The cost of administration for remdesivir was assumed to be $235.85. The costs of COVID-19–related hospitalizations were obtained from Canadian Institute for Health Information (CIHI) data for time spent in the general ward ($20,097 per stay)14 and from a published economic evaluation for patients with ordinal scale scores 4, 5, and 6 ($44,116, $35,794, and $64,856, respectively).4 The health care costs for ordinal scale score 7 and score 8 (i.e., death) were assumed to be $139,452.4 The 1-time cost of rehabilitation was assumed to be $236 per day, and the sponsor assumed rehabilitation to have a duration of 5 days.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following.
Base-Case Results
In the sponsor’s base-case analysis, treatment with remdesivir was associated with lower costs (–$80) and higher QALYs (0.0040) than SOC (Table 3). Remdesivir was dominant or cost-effective compared to SOC at a $50,000 per QALY gained threshold in 55% of iterations. The sponsor’s analysis estimated that remdesivir was associated with 0.0036 incremental life-years. Full disaggregated results of the sponsor’s economic evaluation are available in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|
SOC | 28,929 | Reference | 0.0780 | Reference | Reference |
Remdesivir | 28,849 | –80 | 0.0820 | 0.0040 | Dominant (lower costs and higher health gains) |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor included scenario analyses involving alternate assumptions for the time horizon and patient starting age. These scenarios had minimal impact on the results; remdesivir remained dominant or cost-effective at a $50,000 per QALY gained threshold.
The sponsor conducted a scenario analysis from a societal perspective in which patients were assumed to miss work due to hospitalization or rehabilitation. The results of this analysis were similar to the sponsor’s base case: remdesivir was associated with lower costs and higher QALYs than SOC. The incremental cost savings in the societal perspective analysis were higher than in the base-case analysis from the payer perspective due to fewer patients missing work in the remdesivir group.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The submitted evidence base does not reflect the current treatment landscape for COVID-19. The primary evidence base for the economic model was the ACTT-1 study, a phase III randomized controlled trial conducted from February 2020 to April 2020. During this time, the circulating variants of COVID-19 were biologically distinct from the variant of COVID-19 circulating at present.15,16 This difference was emphasized by the clinical experts consulted by CDA-AMC, who highlighted that data from the ACTT-1 trial are not externally generalizable to patients infected with the Omicron and later variants of COVID-19 currently circulating in Canada. Furthermore, the ACTT-1 trial was conducted in unvaccinated patients, which is also not reflective of the current state of public health in Canada in which more than 80% of people have received a primary course of vaccines.17 In addition, data from Ontario from February 2021 to April 2023 indicate that COVID-19–related hospitalizations (and deaths) were highest among unvaccinated individuals compared to those that have completed their primary vaccine series with or without additional boosters.18 Thus, remdesivir is not expected to have the same impact on hospitalization in the current vaccinated population in Canada. These differences represent a fundamental challenge in interpreting the results from the sponsor’s submitted evidence dossier and the accompanying pharmacoeconomic model, which are based on the ACTT-1 trial. The clinical experts strongly emphasized that an economic model based on ACTT-1 trial data is unable to meaningfully answer the research question of whether remdesivir is cost-effective for the treatment of moderate-to-severe COVID-19 infections in the current setting in Canada.
In the submitted model, mortality at days 15 and 29 was informed by real-world evidence submitted by the sponsor.6 The Clinical Review highlighted both internal validity concerns and limitations associated with the generalizability of the results of this real-world evidence to Canada in the current COVID-19 landscape. The internal validity concerns included a lack of clear reporting on data sources, data cleaning, and patient characteristics (including time of symptom onset and vaccinations received before hospitalization). Due to the limitations of the real-world evidence, there is uncertainty about the benefit of remdesivir being applied to the current context in Canada. The clinical trials reviewed in the Clinical Review reported mixed findings in relation to mortality outcomes. Some trials appeared to support a benefit in mortality in the subgroup of patients who required low-flow oxygen; however, another trial found that mortality was similar between the remdesivir and SOC treatment groups at day 28. As such, there is outstanding uncertainty about the presence of or magnitude of possible benefits due to when the trials were conducted.
The clinical expert opinion solicited by CDA-AMC also discussed the important distinction between patients hospitalized because of their COVID-19 infection and those hospitalized who incidentally have a COVID-19 infection. Patients who are hospitalized for other underlying causes may not experience a mortality benefit from treatment with remdesivir as it would not address the primary reason that they are hospitalized. As such, identifying and treating only those patients hospitalized as a result of COVID-19 is critical to maximizing the clinical benefit and the cost-effectiveness of remdesivir.
To address the uncertainty specifically with the mortality data in the model, CDA-AMC tested 4 alternative mortality assumptions on the cost-effectiveness of remdesivir compared to SOC:
The first assumed that only patients who entered the hospital requiring low-flow oxygen (ordinal scale score 5) experienced a mortality benefit associated with remdesivir compared to SOC.
The second reduced the overall mortality rates in the model by applying an effect modifier to the mortality estimates. CDA-AMC applied a vaccine effectiveness of 82%, representing the estimated effectiveness against severe outcomes of 2 doses of COVID-19 vaccine received 180 to 239 days prior.19 The vaccine effectiveness estimate was measured in Ontario using a test-negative case-control study using linked provincial administrative databases. Results from the aforementioned study were presented for vaccine effectiveness against infection and against severe outcomes, and considered the timing of the most recent vaccine dose received.19 While this approach assumes that all modelled patients have received 2 doses of vaccine, CDA-AMC notes that as of September 10, 2023, 84% of people living in Canada aged 5 years or older had received at least 2 doses of a COVID-19 vaccine, with that number being even higher in older age groups: 95% of adults aged 60 to 69 years and more than 99% of adults aged 70 years or older.17 Further, CDA-AMC conservatively applied the 2-dose vaccine effectiveness against severe outcomes, despite the high proportion of people living in Canada who have received 3 or more doses of vaccine as of June 2022 (77% of adults aged 60 to 69 years and 85% of adults aged 70 years or older).20 This approach conservatively maintained a benefit for remdesivir for all patients (i.e., patients who entered at all ordinal scale scores). Given the uncertainty in the population immunity against severe outcomes, CDA-AMC also conducted a scenario analysis using an estimate of 50% vaccine effectiveness against severe outcomes.
The third combined the first 2 assumptions and reduced the overall mortality risk for all patients in the model and only maintained a mortality benefit for patients who took remdesivir and entered the hospital at ordinal scale score 5.
The fourth tested the assumption that no mortality benefit was experienced by patients treated with remdesivir.
The baseline distribution of hospital services is not reflective of the levels of care experienced today. The sponsor used an economic evaluation conducted alongside the substudy of the WHO Solidarity trial undertaken in Canada (the CATCO trial; August 14, 2020, to April 1, 2021) for inpatient treatment with remdesivir to inform the baseline distribution of hospital level of care.4 Due to limitations associated with the generalizability of clinical data from that period to the current setting in Canada, this distribution is unlikely to be the same as is experienced today. The clinical expert opinion solicited by CDA-AMC agreed that patients are now entering the hospital in less severe condition than they were at the time of the WHO Solidarity trial due to changes in population immunity and viral pathogenicity.
To address this limitation, CDA-AMC adjusted the baseline distribution of patients upon hospitalization based on evidence from a retrospective analysis conducted using data from Canada.21 CDA-AMC assumed that 4.5% of patients would require mechanical invasive ventilation upon hospital admission, an additional 10% would be admitted to the ICU upon admission, and the remaining 85.5% would start in a general ward, with 20% on supplemental oxygen. CDA-AMC notes that the data informing this distribution are from 2022 and that since then there may have been further changes to population immunity and the severity of the circulating COVID-19 variant that may influence hospital levels of care.
The pharmacoeconomic submission incorporated poor reporting, organization, and modelling practices, which made validation of the submitted evidence difficult. The sponsor’s submission included several discrepancies in reporting within the technical report and the submitted model and lacked detail on how parameter estimates were derived from clinical data sources. Further, the model structure was overly complicated in its reliance on ordinal scale scores at baseline and the transitions between ordinal scale scores. The clinical expert opinion solicited by CDA-AMC indicated that ordinal scale scores are not regularly used in routine clinical practice or research. As a result, applying data from different clinical sources was overly complicated.
CDA-AMC was unable to address this limitation and notes that the sponsor’s model structure made it difficult to assess the impact of alternative data for some influential clinical input parameters, including transitions between ordinal scale scores and mortality.
Hospitalization costs were inaccurately estimated. The sponsor derived the costs of COVID-19–related hospitalizations from CIHI data for time spent in the general ward ($20,097 per stay)14 and from a published economic evaluation for ordinal scale scores 4 to 8.4 The costs from the economic evaluation were derived for hospitalizations that occurred during the early waves of COVID-19 in Canada. As previously described, the clinical outcomes during earlier pandemic waves are not reflective of the current COVID-19 landscape (i.e., a highly vaccinated population and a less severe circulating variant). As such, using hospitalization costs measured at that time introduced uncertainty with regard to the costs that may be experienced in hospitals today. The costs estimated by CIHI for COVID-19 hospitalizations in 2022 to 2023 represent more current and widely applicable cost estimates. The analysis from which the sponsor obtained the cost applied to ordinal scale scores 1 to 3 ($20,097 per stay) also reported that the average cost per hospitalization for patients with COVID-19 requiring ICU admission was $52,774.
In reanalysis, CDA-AMC used hospitalization costs for patients not requiring ICU admission for ordinal scale scores 1 to 5 and the average costs for patients who required ICU admission for ordinal scale scores 6 to 8.
Treatment costs may be underestimated. In the submitted model, the sponsor assumed that the treatment duration of remdesivir is 5 days. The Health Canada–recommended treatment duration is “at least 5 days and not more than 10 days.”2 As noted in the Clinical Review, per Spinner et al. (2020),22 there appears to be no obvious additional benefit or harm of a 10-day course of remdesivir over a 5-day course. Therefore, while the evidence suggests that a 5-day course is sufficient, it is unlikely that all patients will be administered a 5-day course as the Health Canada–recommended dose allows for a longer treatment duration. Therefore, the treatment cost calculated by the sponsor is the minimum cost of reimbursing remdesivir. Should treatment duration exceed 5 days, the cost-effectiveness of remdesivir may change.
CDA-AMC assumed a 10-day treatment course in a scenario analysis.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Adverse events were not included in the model. | Acceptable. The clinical expert opinion solicited by CDA-AMC agreed that remdesivir was not associated with frequent adverse events and that the impact of including the outcomes of adverse events would likely have minimal impact on the results of the model. |
Length of hospital stay differed for patients treated with remdesivir and patients treated with SOC. | Not acceptable. The evidence supporting differences in length of hospital stay for treatment with remdesivir are uncertain, as reported by the Clinical Review. However, this assumption did not have a large impact on the results of the model. |
The modelled time horizon in the sponsor’s submission was 6 weeks. | Acceptable. CDA-AMC agrees, based on the clinical evidence and decision problem, that a 6-week time horizon was appropriate to use in the sponsor’s base-case analysis. Given that the model might predict differences in longer-term outcomes, CDA-AMC conducted a scenario analysis using a 10-year time horizon to explore the impact of a longer time horizon on the cost-effectiveness of remdesivir compared to SOC. |
CDA-AMC = Canada’s Drug Agency; SOC = standard of care.
CDA-AMC Reanalyses of the Economic Evaluation
CDA-AMC Reanalysis Results
The CDA-AMC reanalyses were derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CDA-AMC conducted several reanalyses to assess the impact of alternative assumptions about mortality on the cost-effectiveness of remdesivir versus SOC. All of the CDA-AMC reanalyses incorporated changes to the baseline distribution of level of hospital care and to COVID-19 hospitalization costs. These changes are summarized in Table 5.
CDA-AMC notes that the model structure submitted by the sponsor relies on transitions between ordinal scale scores; however, not all the available clinical data (submitted by the sponsor or identified by the CDA-AMC review team) is easily adaptable to the model structure. As a result, CDA-AMC had to adjust transition probabilities between ordinal scale scores after modifying mortality estimates (i.e., transitions to ordinal scale score 8). The sponsor indicated that it also had to recalibrate transition probabilities following the incorporation of mortality data from Mozaffari et al. (2023) into the model by adjusting the transition probabilities between other ordinal scale scores to ensure that the sum of possible transitions was equal to 1.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC reanalyses | ||
1. Baseline level of care in hospital | Proportion of patients entering hospital by ordinal scale score:
| Proportion of patients entering hospital by ordinal scale score:
|
2. Hospitalization costs |
|
|
3a. Mortality benefit is uncertain | Mortality benefit informed by Mozaffari et al. (2023). | Mortality benefit as implemented by the sponsor is maintained for patients who start at ordinal scale score 5. No mortality benefit is assumed for other patients. |
3b. Mortality benefit is uncertain | Mortality benefit informed by Mozaffari et al. (2023). | Mortality is adjusted for remdesivir and SOC by vaccine effectiveness against severe outcomes from the Omicron variant. |
3c. Mortality benefit is uncertain | Mortality benefit informed by Mozaffari et al. (2023). | Mortality is adjusted for remdesivir and SOC by vaccine effectiveness against severe outcomes from the Omicron variant, and benefit is maintained only for patients who start at ordinal scale score 5. |
3d. Mortality benefit is uncertain | Mortality benefit informed by Mozaffari et al. (2023). | There is no mortality benefit for patients treated with remdesivir. |
CDA-AMC reanalysis A | ― | Reanalysis 1 + 2 + 3a |
CDA-AMC reanalysis B | ― | Reanalysis 1 + 2 + 3b |
CDA-AMC reanalysis C | ― | Reanalysis 1 + 2 + 3c |
CDA-AMC reanalysis D | ― | Reanalysis 1 + 2 + 3d |
CDA-AMC = Canada’s Drug Agency; SOC = standard of care.
The results of the CDA-AMC reanalyses showed remdesivir as having an ICER ranging from $2,542,952 to $4,208,181 per QALY gained, depending on assumptions about the mortality benefit for patients treated with remdesivir (Table 6). A summary of the CDA-AMC stepped analysis can be found in Table 10 (Appendix 4).
The results on cost-effectiveness from CDA-AMC reanalyses A and B were similar, suggesting that assumptions about the magnitude of the mortality benefit of remdesivir and the absolute mortality predicted by the model are both influential parameters. When these changes were combined in CDA-AMC reanalysis C (i.e., reduced overall mortality for all patients and the mortality benefit as implemented by the sponsor is maintained for patients who start at ordinal scale score 5), the combined effect resulted in a higher ICER for remdesivir compared to SOC ($3,748,693 per QALY gained), driven by the reduction in incremental QALYs.
While the incremental QALYs predicted by the CDA-AMC reanalyses are smaller than those predicted by the sponsor’s analysis, the increase in incremental costs in the CDA-AMC reanalyses were an important driver of the high ICERs. In the sponsor’s base-case analysis, the drug acquisition costs associated with remdesivir were offset by the cost savings produced through avoidance of higher ordinal scale score hospital costs (including death, which was associated with a cost of more than $139,000). The changes to ordinal scale score hospitalization costs and mortality assumptions in the CDA-AMC reanalyses minimized the cost offsets, resulting in higher incremental costs associated with remdesivir.
Table 6: Summary of the CDA-AMC Reanalyses for Remdesivir vs. SOC
Key considerations | Reanalysis A | Reanalysis B | Reanalysis C | Reanalysis D |
|---|---|---|---|---|
Mortality assumption | Mortality benefit maintained for patients whose disease severity warrants support with low-flow oxygen (i.e., ordinal scale score 5), but not for other patients, at days 15 and 29. | Mortality for remdesivir and SOC at days 15 and 29 adjusted by vaccine effectiveness against severe outcomes from the Omicron variant (82%). Benefit of remdesivir maintained for all patients. | Mortality benefit maintained for patients whose disease severity warrants support with low-flow oxygen, but not for other patients, at days 15 and 29. Mortality for all patients adjusted by 82% vaccine effectiveness. | No mortality benefit for any patients at days 15 or 29. Mortality assumed to be equal to that associated with SOC regardless of treatment. |
Incremental LYs | 0.0006 | 0.0005 | 0.0000 | 0.0000 |
Incremental costs ($) | 3,631 | 3,639 | 3,683 | 3,668 |
Incremental QALYs | 0.0014 | 0.0014 | 0.0010 | 0.0009 |
ICER vs. SOC ($/QALY) | 2,542,952 | 2,546,227 | 3,748,693 | 4,208,181 |
Price reduction to achieve cost-effectiveness at a threshold of $50,000 per QALY gained (%) | 90 | 90 | 92 | 92 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Note: Full price reduction analyses and stepped analyses are reported in Appendix 4. For reanalyses A, B, and C, differences in incremental LYs and QALYs are observed when results are reported to 6 decimal places; however, we do not have the required precision in estimates to predict to that level.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor base-case analysis and the CDA-AMC reanalyses (Table 11). These analyses demonstrated that remdesivir may require a price reduction of 90% to 92% to be considered cost-effective at a threshold of $50,000 per QALY gained, depending on assumptions about the mortality benefit of remdesivir.
In the scenario analysis that CDA-AMC conducted using 50% vaccine effectiveness against severe outcomes, remdesivir is associated with an ICER of $1,582,044 per QALY gained (incremental costs: $3,569; incremental QALYs: 0.0023). In this scenario, remdesivir may require a price reduction of 87% to be considered cost-effective at a threshold of $50,000 per QALY gained (Table 12).
Additionally, CDA-AMC conducted scenario analyses assuming that patients had a 10-day treatment duration with remdesivir. The results of these analyses are presented in Table 14. These analyses found that the ICERs associated with remdesivir ranged from $4,838,285 to $7,775,426 per QALY gained.
Issues for Consideration
In the inpatient COVID-19 setting, patients may be treated with alternative treatments that were not explicitly included in the economic evaluation or BIA for the current review. The clinician group input indicated that the following treatments may be used in the inpatient COVID-19 population: baricitinib, tocilizumab, and sarilumab. While these treatments may be reserved for patients who are critically ill (as is the case with tocilizumab), the cost-effectiveness of remdesivir compared to these treatments is unknown.
Overall Conclusions
The Clinical Review reported that findings from 4 trials, including the ACTT-1 trial, suggest that remdesivir may prevent death in hospitalized patients aged 12 years or older with COVID-19 whose disease severity warrants support with low-flow oxygen and perhaps also in patients who require high-flow oxygen, but the latter subgroup was not evaluated as a distinct group. There is no evidence to suggest that remdesivir prevents death in hospitalized patients who do not require oxygen support, nor in patients who require any level of ventilation (noninvasive or invasive) or ECMO. The Clinical Review also highlighted that the evaluated trials did not include vaccinated patients or patients who had had COVID-19 in the past and that the trials were performed at a time when the Omicron variant was not yet circulating. There are substantial concerns regarding the external validity and generalizability of every study included in the Clinical Review because of the fast-evolving nature of the pandemic and the virus itself: the prevalent variants, the status of vaccinations, and the clinical outcomes in today’s world are substantially different than those observed in the early pandemic. These differences represent a fundamental challenge in interpreting the results from the sponsor’s submitted evidence dossier and the accompanying pharmacoeconomic model, which are based on the ACTT-1 trial and submitted real-world evidence.
CDA-AMC conducted several reanalyses that made changes to the sponsor’s submitted model, in consultation with clinical experts. CDA-AMC adjusted the baseline distribution for level of hospital care, adjusted COVID-19 hospitalization costs, and assessed the impact of 4 alternative mortality assumptions that changed the mortality benefit associated with remdesivir and the absolute risk of mortality for all patients hospitalized with COVID-19. In general, the incremental QALYs estimated in the CDA-AMC reanalyses were similar to those in the sponsor’s results (sponsor’s incremental QALYs: 0.0040; the CDA-AMC incremental QALYs: 0.0014 to 0.0009); these benefits equate to an additional 8 to 12 quality-adjusted hours of life in the CDA-AMC reanalyses to an additional 35 quality-adjusted hours of life in the sponsor’s analysis. However, the incremental costs in the CDA-AMC reanalysis were higher (approximately $3,600 in all the CDA-AMC reanalyses). The incremental costs were driven by changes in COVID-19 hospitalization costs and changes in mortality, which had a cost assigned to it.
In the CDA-AMC reanalysis that assumed the mortality benefit applies only to patients requiring low-flow oxygen, remdesivir was associated with an ICER of $2,542,952 per QALY gained. Similar results were found in the CDA-AMC reanalysis that assumed the absolute mortality risk of COVID-19 was lower for all patients than the sponsor assumed. In the CDA-AMC reanalyses that estimated lower incremental QALYs due to assuming no mortality benefit for patients treated with remdesivir, the ICER was $4,208,181. Despite the wide range of ICERs estimated by the CDA-AMC reanalyses, the price reduction analyses were similar across all mortality assumptions. These analyses demonstrated that a price of $317 to $396 per 5-day treatment course (a reduction of approximately 90% to 92%) may be required for remdesivir to be considered cost-effective compared to SOC at a threshold of $50,000 per QALY gained.
The results of these analyses are driven by the efficacy data used and the baseline assumptions made about hospitalized patients with COVID-19 infections. Given the differences in patient profile and COVID-19 variants today, the mortality benefit likely to be experienced in the current COVID-19 setting in Canada remains highly uncertain. Notably, the clinical expert opinion solicited by CDA-AMC for this review highlighted that an important distinction exists between patients hospitalized because of their COVID-19 infection and those hospitalized who incidentally have a COVID-19 infection. Patients who are hospitalized for other underlying causes may not benefit from treatment with remdesivir; as such, identifying and treating only those patients hospitalized as a result of COVID-19 is critical to maximizing the clinical benefit and the cost-effectiveness of remdesivir.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Veklury (remdesivir), 100 mg/vial powder for solution for infusion. Mississauga (ON): Gilead Sciences Canada, Inc; 2024 Mar 11.
2.Veklury (remdesivir): 100 mg/vial powder for solution for infusion [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc.; 2024 May 06.
3.Chen C, Haupert SR, Zimmermann L, Shi X, Fritsche LG, Mukherjee B. Global prevalence of post-coronavirus disease 2019 (COVID-19) condition or long COVID: a meta-analysis and systematic review. J Infect Dis. 2022;226(9):1593-1607. PubMed
4.Lau VI, Fowler R, Pinto R, et al. Cost-effectiveness of remdesivir plus usual care versus usual care alone for hospitalized patients with COVID-19: an economic evaluation as part of the Canadian Treatments for COVID-19 (CATCO) randomized clinical trial. CMAJ Open. 2022;10(3):E807-E817. PubMed
5.Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19 - final report. N Engl J Med. 2020;383(19):1813-1826. PubMed
6.Mozaffari E, Chandak A, Gottlieb RL, et al. Remdesivir is associated with reduced mortality in COVID-19 patients requiring supplemental oxygen including invasive mechanical ventilation across SARS-CoV-2 variants. Open Forum Infect Dis. 2023;10(10):ofad482. PubMed
7.Shoucri SM, Purpura L, DeLaurentis C, et al. Characterising the long-term clinical outcomes of 1190 hospitalised patients with COVID-19 in New York City: a retrospective case series. BMJ Open. 2021;11(6):e049488. PubMed
8.McAlister FA, Dong Y, Chu A, et al. The risk of death or unplanned readmission after discharge from a COVID-19 hospitalization in Alberta and Ontario. Can Med Assoc J. 2022;194(19):E666-E673. PubMed
9.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2023: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed by sponsor, no date provided.
10.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
11.Sheinson D, Dang J, Shah A, Meng Y, Elsea D, Kowal S. A Cost-Effectiveness framework for COVID-19 treatments for hospitalized patients in the United States. Adv Ther. 2021;38(4):1811-1831. PubMed
12.Khan K, Muennig P, Gardam M, Zivin JG. Managing febrile respiratory illnesses during a hypothetical SARS outbreak. Emerg Infect Dis. 2005;11(2):191-200. PubMed
13.Barnieh L, Beckerman R, Jeyakumar S, Hsiao A, Jarrett J, Gottlieb RL. Remdesivir for hospitalized COVID-19 patients in the United States: optimization of health care resources. Infect Dis Ther. 2023;12(6):1655-1665. PubMed
14.Canadian Institute For Health Information. COVID-19 hospitalization and emegency department statistics. 2024; https://www.cihi.ca/en/covid-19-hospitalization-and-emergency-department-statistics. Accessed 2024 Apr 23.
15.Centers for Disease Control and Prevention. CDC Museum COVID-19 timeline. 2023; https://www.cdc.gov/museum/timeline/covid19.html. Accessed 2024 Apr 23.
16.Tuite AR, Fisman DN, Odutayo A, et al. COVID-19 hospitalizations, ICU admissions and deaths associated with the new variants of concern. Science Briefs of the Ontario COVID19 Science Advisory Table. 2021;1(18). https://covid19-sciencetable.ca/sciencebrief/covid-19-hospitalizations-icu-admissions-and-deaths-associated-with-the-new-variants-of-concern/. Accessed 2024 Aug 07.
17.COVID-19 vaccination: vaccination coverage. Ottawa (ON): Public Health Agency of Canada; 2023: https://health-infobase.canada.ca/covid-19/vaccination-coverage/archive/2023-05-22/. Accessed 2023 Apr 23.
18.Severe outcomes among confirmed cases of COVID-19 following vaccination in Ontario: December 14 2022 to April 23, 2023. Toronto (ON): Public Health Ontario; 2023: https://www.publichealthontario.ca/-/media/Documents/nCoV/epi/covid-19-epi-confirmed-cases-post-vaccination.pdf?rev=3a3ead21e75640499dc626cc5283d141&sc_lang=en. Accessed 2024 Apr 25.
19.Buchan SA, Chung H, Brown KA, et al. Estimated effectiveness of COVID-19 vaccines against Omicron or Delta symptomatic infection and severe outcomes. JAMA Netw Open. 2022;5(9):e2232760. PubMed
20.Vaccination coverage for COVID-19 additional doses in Canada: results from the 2022 Canadian Community Health Survey (CCHS). Ottawa (ON): Public Health Agency of Canada; 2023: https://www.canada.ca/en/public-health/services/immunization-vaccines/vaccination-coverage/covid-19-additional-doses-results-2022-canadian-community-health-survey.html. Accessed 2024 Apr 23.
21.Lee T, Cheng MP, Vinh DC, et al. Outcomes and characteristics of patients hospitalized for COVID-19 in British Columbia, Ontario and Quebec during the Omicron wave. CMAJ Open. 2023;11(4):E672-E683. PubMed
22.Spinner CD, Gottlieb RL, Criner GJ, et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. JAMA. 2020;324(11):1048-1057. PubMed
23.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Veklury (remdesivir), 100 mg/vial powder for solution for infusion. Mississauga (ON): Gilead Sciences Canada, Inc; 2024 Mar 11.
24.Canadian Institute for Health Information. COVID-19 hospitalization and emergency department statistics, 2022–2023. 2023; https://www.cihi.ca/en/covid-19-hospitalization-and-emergency-department-statistics. Accessed 2023 Nov 06.
25.Lee T, Cheng MP, Vinh DC, et al. Outcomes and characteristics of patients hospitalized for COVID-19 in British Columbia, Ontario and Quebec during the Omicron wave. CMAJ Open. 2023;11(4):E672-E683. PubMed
26.Canadian Institute for Health Information. COVID-19 hospitalization and emergency department statistics, 2023–2024 (Q1 to Q2) — provisional data. Ottawa (ON): CIHI; 2024: https://www.cihi.ca/en/covid-19-hospitalization-and-emergency-department-statistics. Accessed 2024 Apr 11.
27.COVID-19 vaccination: doses administered. Ottawa (ON): Public Health Agency of Canada: https://health-infobase.canada.ca/covid-19/vaccine-administration/. Accessed 2024 Apr 18.
28.BC Centre for Disease Control. Respiratory virus data. http://www.bccdc.ca/health-professionals/data-reports/respiratory-virus-data. Accessed 2024 Apr 11.
29.BC COVID Therapeutics Committee (CTC). Algorithm for treatment of COVID-19 in hospitalized patients. Vancouver (BC): BC Centre for Disease Control; 2023: http://www.bccdc.ca/Health-Professionals-Site/Documents/COVID-treatment/Algorithm_treatment_hospitalized_patient.pdf. Accessed 2024 Apr 22.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CDA-AMC Cost Comparison Table for the Treatment of COVID-19
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Treatment course length | Cost per treatment course ($) |
|---|---|---|---|---|---|---|
Remdesivir (Veklury) | 5 mg/mL | 100 mg vial | 660.5300 | ≥ 40 kg: 200 mg on day 1, followed by 100 mg once daily for an additional 4 to 9 days (for a total treatment duration of 5 to 10 days)a | 5 to 10 days | 3,963 to 7,226 |
CDA-AMC = Canada’s Drug Agency.
Note: The price of remdesivir is based on the sponsor submitted price and does not include dispensing fees.
aThe recommended dose for pediatric patients weighing ≥ 3 kg to ≤ 40 kg is 5 mg/kg on day 1, followed by 2.5 mg/kg daily for up to an additional 9 days (for a total treatment duration of up to 10 days). Patients weighing less than 40 kg were excluded from the sponsor’s reimbursement request.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The population studied in the ACTT-1 trial does not reflect the population eligible for remdesivir at present due to differences in baseline risk, vaccination status, and COVID-19 variant. The population studied in the pivotal trial is not relevant. |
Model has been adequately programmed and has sufficient face validity | No | Refer to key limitation: The pharmacoeconomic submission incorporated poor reporting, organization, and modelling practices, which made validation of the submitted evidence difficult. |
Model structure is adequate for decision problem | No | The model structure was overly complicated and not aligned with the majority of available data. As such, it made addressing the decision problem challenging. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Refer to key limitation: The pharmacoeconomic submission incorporated poor reporting, organization, and modelling practices, which made validation of the submitted evidence difficult. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
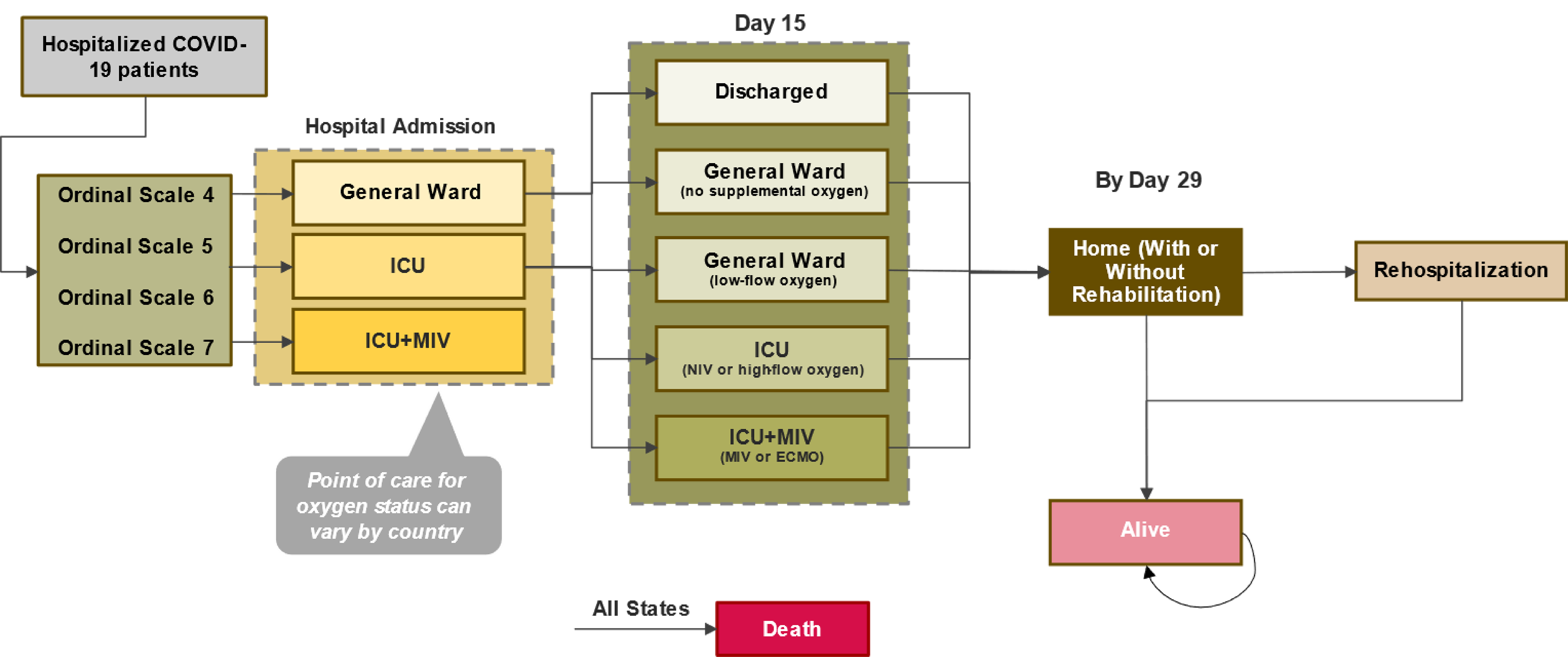
Detailed Results of the Sponsor’s Base Case
Table 9: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Remdesivir | SOC |
|---|---|---|
Discounted LYs | ||
Total | 0.1014 | 0.0978 |
By health state | ||
Ordinal scale score 1 to 3 | 0.0367 | 0.0319 |
Ordinal scale score 4 | 0.0039 | 0.0038 |
Ordinal scale score 5 | 0.0047 | 0.0052 |
Ordinal scale score 6 | 0.0018 | 0.0021 |
Ordinal scale score 7 | 0.0040 | 0.0064 |
Recovered (postdischarge) | 0.0503 | 0.0485 |
Discounted QALYs | ||
Total | 0.0820 | 0.0780 |
By health state | ||
Ordinal scale score 1 to 3 | 0.0300 | 0.0258 |
Ordinal scale score 4 | 0.0031 | 0.0031 |
Ordinal scale score 5 | 0.0038 | 0.0041 |
Ordinal scale score 6 | 0.0014 | 0.0016 |
Ordinal scale score 7 | 0.0023 | 0.0037 |
Recovered (postdischarge) | 0.0424 | 0.0408 |
Utility decrements | ||
Rehabilitation | 0.0008 | 0.0008 |
Rehospitalization | 0.0002 | 0.0002 |
Discounted costs ($) | ||
Total | 28,849 | 28,929 |
Treatment and administration | 4,199 | 0 |
Hospitalization | 24,422 | 28,710 |
Postdischarge | 244 | 238 |
LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
Note: the number of patients who would be needed to treat with remdesivir to prevent 1 rehospitalization is approximately 76 patients, based on the risk of rehospitalization applied in the submitted pharmacoeconomic model.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Reanalyses
Table 10: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | SOC | 28,817 | 0.0781 | Reference |
Remdesivir | 28,798 | 0.0821 | Dominant (lower costs and higher health gains) | |
CDA-AMC reanalysis 1 | SOC | 26,365 | 0.0800 | Reference |
Remdesivir | 26,764 | 0.0835 | 112,853 | |
CDA-AMC reanalysis 2 | SOC | 17,163 | 0.0781 | Reference |
Remdesivir | 20,211 | 0.0821 | 761,686 | |
CDA-AMC reanalysis 3a | SOC | 28,817 | 0.0781 | Reference |
Remdesivir | 29,505 | 0.0817 | 192,116 | |
CDA-AMC reanalysis 3b | SOC | 25,949 | 0.0893 | Reference |
Remdesivir | 25,459 | 0.0829 | Dominated (higher costs and lower health gains) | |
CDA-AMC reanalysis 3c | SOC | 25,459 | 0.0893 | Reference |
Remdesivir | 26,023 | 0.0829 | Dominated (higher costs and lower health gains) | |
CDA-AMC reanalysis 3d | SOC | 28,817 | 0.0781 | Reference |
Remdesivir | 29,755 | 0.0791 | 922,774 | |
CDA-AMC reanalysis A (reanalysis 1 + 2 + 3a) | SOC | 16,434 | 0.0800 | Reference |
Remdesivir | 20,097 | 0.0814 | 2,544,462 | |
CDA-AMC reanalysis B (reanalysis 1 + 2 + 3b) | SOC | 15,817 | 0.0900 | Reference |
Remdesivir | 19,478 | 0.0914 | 2,532,066 | |
CDA-AMC reanalysis C (reanalysis 1 + 2 + 3c) | SOC | 15,817 | 0.0900 | Reference |
Remdesivir | 19,503 | 0.0909 | 3,565,492 | |
CDA-AMC reanalysis D (reanalysis 1 + 2 + 3d) | SOC | 16,434 | 0.0800 | Reference |
Remdesivir | 20,135 | 0.0809 | 4,108,686 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The specific approach to recalibrate ordinal scale score transition probabilities after adjusting mortality estimates used by the sponsor was not provided, and as a result the CDA-AMC approach may differ from that applied by the sponsor.
Scenario Analyses
Table 11: CDA-AMC Price Reduction Analyses
Price reduction | Unit drug cost ($) | ICERs for remdesivir vs. SOC ($/QALY) | ||||
|---|---|---|---|---|---|---|
Sponsor base case | CDA-AMC reanalysis A | CDA-AMC reanalysis B | CDA-AMC reanalysis C | CDA-AMC reanalysis D | ||
No price reduction | 660.53 | Dominant | 2,542,952 | 2,546,226 | 3,748,692 | 4,208,181 |
10% | 594.48 | Dominant | 2,265,414 | 2,268,920 | 3,345,319 | 3,755,891 |
20% | 528.42 | Dominant | 1,987,875 | 1,991,614 | 2,941,946 | 3,303,599 |
30% | 462.37 | Dominant | 1,710,336 | 1,714,308 | 2,538,574 | 2,851,306 |
40% | 396.32 | Dominant | 1,432,798 | 1,437,002 | 2,135,201 | 2,399,014 |
50% | 330.27 | Dominant | 1,155,259 | 1,159,696 | 1,731,829 | 1,946,722 |
60% | 264.21 | Dominant | 877,720 | 882,390 | 1,328,456 | 1,494,429 |
70% | 198.16 | Dominant | 600,182 | 605,084 | 925,083 | 1,042,137 |
80% | 132.11 | Dominant | 322,643 | 327,778 | 521,711 | 589,844 |
90% | 66.05 | Dominant | 45,104 | 50,472 | 118,338 | 137,552 |
100% | 0.00 | Dominant | Dominant | Dominant | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Note: Dominant means that remdesivir was associated with lower costs and higher health gains than SOC.
Table 12: CDA-AMC Scenario Analyses Assuming 50% Vaccine Effectiveness Against Severe Outcomes
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
SOC | 16,015 | 0.0861 | Reference |
Remdesivir | 19,583 | 0.0884 | 1,582,044 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Table 13: CDA-AMC Scenario Analyses Assuming 10-Year Time Horizon
Key considerations | Reanalysis A | Reanalysis B | Reanalysis C | Reanalysis D |
|---|---|---|---|---|
Incremental costs ($) | 3,702 | 3,649 | 3,836 | 3,660 |
Incremental QALYs | 0.0456 | 0.0440 | 0.0091 | 0.0006 |
ICER vs. SOC ($/QALY) | 81,168 | 83,008 | 421,162 | 5,822,793 |
Price reduction to achieve cost-effectiveness at a threshold of $50,000 per QALY gained (%) | 36 | 37 | 85 | 92 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Table 14: CDA-AMC Scenario Analyses Assuming 10-Day Treatment Duration (Deterministic)
Reanalysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CDA-AMC reanalysis A, 10-day treatment duration | SOC | 16,434 | 0.0800 | Reference |
Remdesivir | 23,400 | 0.0814 | 4,838,285 | |
CDA-AMC reanalysis B, 10-day treatment duration | SOC | 15,817 | 0.0900 | Reference |
Remdesivir | 22,780 | 0.0914 | 4,816,586 | |
CDA-AMC reanalysis C, 10-day treatment duration | SOC | 15,817 | 0.0900 | Reference |
Remdesivir | 22,932 | 0.0909 | 7,786,941 | |
CDA-AMC reanalysis D, 10-day treatment duration | SOC | 16,434 | 0.0800 | Reference |
Remdesivir | 22,437 | 0.0809 | 7,775,426 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 15: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Summary of Sponsor’s BIA
In the submitted BIA, the sponsor assessed the introduction of remdesivir for hospitalized COVID-19 patients 12 years and older (at least 40 kg) with pneumonia requiring supplemental oxygen.23 The BIA was undertaken from the perspective of a Canadian public payer over a three-year time horizon (2024 to 2026) using an epidemiological approach. The sponsor compared a reference scenario in which patients were treated with SOC to a new drug scenario in which remdesivir was reimbursed for use in combination with SOC. The sponsor’s submission only considered drug acquisition costs for remdesivir. Data for the model were obtained from various sources including CIHI,24 published literature,25 the sponsor’s internal data, and assumption. Key inputs to the BIA are documented in Table 16.
Key assumptions included:
A flat hospitalization rate due to the challenges with predicting the number of hospitalizations over time.
Remdesivir will capture 90% of the market share in years 1, 2, and 3.
The proportion of hospitalized patients 18 years of age and older is equal to the proportion of hospitalized patients aged 12 years and older.
The proportion of hospitalized COVID-19 patients who received remdesivir during the Omicron wave reflected current clinical practice.
Tocilizumab is not a relevant comparator as it is only recommended for critically ill patients.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Number of hospitalizations with COVID-1924 | 116,043 |
Proportion ≥ 12 years old among COVID-19 hospitalizations24 | 95.28% |
Percentage of hospitalized patients with COVID-19 requiring oxygen therapy25 | 22.6% |
Number of patients eligible for drug under review | 24,987 / 24,987 / 24,987 |
Market uptake (3 years) | |
Uptake (reference scenario) SOC | 100% / 100% / 100% |
Uptake (new drug scenario) Remdesivir + SOC SOC | 90% / 90% / 90% 10% / 10% / 10% |
Cost of treatment (per patient, per 5-day course) | |
Remdesivir + SOC SOC | $3,963.18 $0 |
SOC = Standard of Care.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the 3-year budget impact of reimbursing remdesivir for hospitalized COVID-19 patients 12 years and older (at least 40 kg) with pneumonia requiring supplemental oxygen to cost $265,144,488 ($88,381,496 in each of year 1, year 2, and year 3).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The eligible population size is uncertain: The sponsor used an epidemiologic approach to estimate the number of patients eligible for remdesivir, starting with the number of COVID-19 hospitalizations that occurred from April 2022 to March 2023.24 The sponsor assumed a flat hospitalization rate (i.e., the same number of hospitalizations occur annually as they did from April 2022 to March 2023) due to the challenges with predicting the number of hospitalizations over time but acknowledged hospitalizations will likely decrease over the next years as the prevalence of vaccinations increase.23 CDA-AMC acknowledges the difficulty in predicting the number of hospitalizations over time but notes that hospitalizations have decreased notably since March 202326 and that the number of vaccinations have increased.27 Based on provisional data published by CIHI for April 2023 to September 2023, the number of hospitalizations from April 2022 to September 2023 and from March 2023 to September 2023 have decreased by 75% and 50%, respectively.26 The trends observed in the data are aligned with recent statements made by the BC Centre for Disease Control which noted that within Canada, “COVID-19 activity has continued to slowly decrease or remains at low levels.”28 Given the downward trend of COVID-19 activity and COVID-19 hospitalizations, assuming that the number of COVID-19 hospitalizations that occurred from April 2022 to March 2023 remain constant through to 2026 likely overestimates the eligible population size. Moreover, clinical expert input obtained by CDA-AMC for this review noted that the number of COVID-19 hospitalizations and the number of hospitalizations caused by COVID-19 are vastly different, as a patient may incidentally test positive for COVID-19 and be admitted to hospital, but the patients’ positive COVID-19 test may be completely unrelated to their chief complaint and as such, it is unlikely that they would receive treatment for COVID-19 while in hospital. Thus, using a metric that does not differentiate between hospitalizations with COVID-19 and hospitalizations caused by COVID-19 overestimates the eligible population size. The BC Centre for Disease Control treatment algorithm states that 60% of patients in hospital with severe acute respiratory syndrome coronavirus 2, who do not require consistent oxygen support, exhibit mild to moderate symptoms, and have a positive polymerase chain reaction test, are not hospitalized due to COVID-19.29 Given this estimate, the number of hospitalizations with COVID-19 should be reduced by 60% to more accurately reflect the proportion of hospitalizations caused by COVID-19.
Additionally, the sponsor assumed that 22.2% of patients hospitalized with COVID-19 require oxygen therapy based on a study by Lee et al. that estimated the proportion of patients admitted to hospital during the Omicron wave that were treated with remdesivir.25 Notably, the study excluded patients who were admitted for a reason unrelated to acute COVID-19. As noted by both CDA-AMC and the clinical expert consulted for this review, the sponsor has inherently biased the eligible population size as remdesivir may have been administered correctly, or incorrectly. Clinical expert input stated that the sponsor has likely overestimated the proportion of patients requiring oxygen therapy and stated that if published literature identified the cause of hypoxia in patients admitted to hospital with COVID-19, the majority of the patients would likely be hypoxic for reasons unrelated to COVID-19. At the time of this review, CDA-AMC was unable to identify such literature.
In the CDA-AMC base case, CDA-AMC multiplied the number of COVID-19 hospitalizations that occurred from April 2023 to September 2023 by 2 to estimate the annual number of hospitalizations. CDA-AMC acknowledges the uncertainty associated with this approach but notes that from April 2022 to March 2023, approximately 50% of the cases occurred during the last 6 months thus supporting the multiplicative approach. The annual number of hospitalizations was then multiplied by 0.6 to estimate the proportion of hospitalizations caused by COVID-19. CDA-AMC notes that the number of hospitalizations may still be overestimated as COVID-19 hospitalizations continue to decline.
CDA-AMC was unable to adjust the proportion of hospitalized patients with COVID-19 requiring oxygen therapy owing to a lack of data.
Treatment costs may be underestimated: In the submitted model, the sponsor assumed that the treatment duration of remdesivir is 5 days. The Health Canada–recommended treatment duration is “at least 5 days and not more than 10 days.”2 As noted by the Clinical Review Report, per Spinner et al. (2020)22 there appears to be no obvious additional benefit or harm of a 10-day course of remdesivir over a 5-day course. Therefore, while the evidence suggests that a 5-day course is sufficient, it is unlikely that all patients will be administered a 5-day course as the Health Canada–recommended dose allows for a longer treatment duration. Given this, the sponsor has calculated the minimum cost of reimbursing remdesivir. Should treatment duration exceed 5 days, the budget impact is underestimated.
CDA-AMC assumed a 10-day treatment course in a scenario analysis.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by adjusting the annual number of hospitalizations.
Table 17: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Annual number of hospitalizations | 116,043 hospitalizations (based on annual data from April 2022 to March 2023) | 25,417 hospitalizations (based on provisional data from April 2023 to September 2023 multiplied by 2 to derive the annual number of hospitalizations and then by 0.6 to determine the annual number of hospitalizations caused by COVID-19) |
CDA-AMC base case | Reanalysis 1 | |
CDA-AMC = Canada’s Drug Agency.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19. In the CDA-AMC base case, the 3-year budget impact of reimbursing remdesivir for hospitalized COVID-19 patients 12 years and older (at least 40 kg) with pneumonia requiring supplemental oxygen is estimated to cost $58,058,334 ($19,352,778 in each of year 1, year 2, and year 3). The CDA-AMC base case revised the eligible population size from 24,987 to 5,473.
Table 18: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | 3-year total ($) |
|---|---|
Submitted base case | 265,144,488 |
CDA-AMC reanalysis 1: Annual number of hospitalizations | 58,058,334 |
CDA-AMC base case | 58,058,334 |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 19:
Assuming the treatment duration of remdesivir is 10 days.
Assuming that the price of remdesivir is reduced by 90% (the minimum estimated price reduction from the CDA-AMC cost-utility reanalysis results).
Assuming that the price of remdesivir is reduced by 92% (the maximum estimated price reduction from the CDA-AMC cost-utility reanalysis results).
Table 19: Detailed Breakdown of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | 3-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 88,381,496 | 88,381,496 | 88,381,496 | 265,144,488 | |
Budget impact | 0 | 88,381,496 | 88,381,496 | 88,381,496 | 265,144,488 | |
CDA-AMC base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 19,352,778 | 19,352,778 | 19,352,778 | 58,058,334 | |
Budget impact | 0 | 19,352,778 | 19,352,778 | 19,352,778 | 58,058,334 | |
CDA-AMC scenario analysis 1: 10-day treatment duration | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 35,480,093 | 35,480,093 | 35,480,093 | 106,440,279 | |
Budget impact | 0 | 35,480,093 | 35,480,093 | 35,480,093 | 106,440,279 | |
CDA-AMC scenario analysis 2: 90% price reduction | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 1,935,278 | 1,935,278 | 1,935,278 | 5,805,833 | |
Budget impact | 0 | 1,935,278 | 1,935,278 | 1,935,278 | 5,805,833 | |
CDA-AMC scenario analysis 3: 92% price reduction | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 1,548,222 | 1,548,222 | 1,548,222 | 4,644,667 | |
Budget impact | 0 | 1,548,222 | 1,548,222 | 1,548,222 | 4,644,667 |
CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.