Drugs, Health Technologies, Health Systems
Reimbursement Review
Trofinetide (Daybue)
Sponsor: Acadia Pharmaceuticals Canada Inc.
Therapeutic area: Rett syndrome
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Clinical Review
Abbreviations
AE
adverse event
CDA-AMC
Canada’s Drug Agency
CGI-I
Clinical Global Impressions-Improvement
CGI-S
Clinical Global Impressions-Severity
CI
confidence interval
CSBS-DP-IT
Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
LSM
least squares mean
MAR
missing at random
MID
minimal important difference
MLPA
multiplex ligation-dependent probe amplification
MMRM
mixed model for repeated measures
MNAR
missing not at random
NGS
next-generation sequencing
OLE
open-label extension
RCT
randomized controlled trial
RSBQ
Rett Syndrome Behaviour Questionnaire
RTT-AMB
Rett Syndrome Clinician Rating of Ambulation and Gross Motor Skills
RTT-CBI
Rett Syndrome Caregiver Burden Inventory
RTT-COMC
Rett Syndrome Clinician Rating of Ability to Communicate Choices
RTT-CSS
Rett Syndrome Clinical Severity Scale
RTT-HF
Rett Syndrome Clinician Rating of Hand Function
RTT-VCOM
Rett Syndrome Clinician Rating of Verbal Communication
SAE
serious adverse event
SD
standard deviation
SE
standard error
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Trofinetide (Daybue), 200 mg/mL, solution, oral or by gastrostomy tube |
Sponsor | Acadia Pharmaceuticals Canada Inc. |
Indication | For the treatment of Rett syndrome in adults and pediatric patients 2 years of age and older and weighing at least 9 kg. |
Reimbursement request | As per the Health Canada–approved indication, with the following requested reimbursement criteria. Initiation:
Patients with any of the following should not be eligible for reimbursement of trofinetide if there is evidence of:
Must be prescribed by clinicians with expertise in the diagnosis and management of Rett syndrome. For renewal following 12 months of therapy, the physician must provide evidence of beneficial clinical effect, defined as stabilization or improvement in sign/symptoms of Rett syndrome from baseline that is considered clinically beneficial by the treating physician. |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | October 11, 2024 |
Recommended dose | Patients aged 2 years and older:a
|
NOC = Notice of Compliance.
aThe Health Canada product monograph recommends that trofinetide be titrated, starting with 50% of the recommended dose taken twice daily, then the dose be increased over 4 to 8 weeks until the recommended dose is reached.
Sources: Sponsor’s submission package for review of trofinetide and trofinetide product monograph.1,2
Introduction
Rett syndrome is a rare, neurodevelopmental disorder characterized by normal early development followed by a progressive loss of speech, purposeful hand use, and motor skills.3 Rett syndrome is most often caused by genetic variants in MECP2, which is located on the X chromosome, and primarily affects females, although it can occur in males in rare cases.3 The disorder progresses through 4 stages, categorized based on speed of development, regression of learned skills, appearance of symptom stabilization, and late motor deterioration.3 With the discovery of MECP2 variants, the diagnostic value of staging now is limited, but it can be used to anticipate potential clinical issues and offer guidance to parents and caregivers.3 In a natural history study of females with genetically confirmed disease who contributed data to the Australian Rett Syndrome Database between 2000 and 2019, Rett Syndrome Behaviour Questionnaire (RSBQ) scores were shown to gradually decline (i.e., suggesting behavioural improvements) with increasing age over a 10-year to 14-year observation period.4 Patients living with Rett syndrome require lifelong care and assistance with daily activities, which has a significant impact on both patients and their caregivers.5 Rett syndrome is clinically diagnosed with criteria that differentiate between classic and atypical disease.6 Genetic testing, particularly the detection of pathogenic MECP2 variants, confirms the diagnosis, although MECP2 variants may not be detected in up to 5% of patients with typical Rett syndrome and approximately 25% of patients with atypical disease.3 The clinical experts consulted for this review emphasized that a clinical diagnosis by a Rett syndrome specialist is required because there are other diseases with a MECP2 variant that appear similar to Rett syndrome.
Rett syndrome is estimated to affect 1 in 10,000 females aged 12 years and younger, with a worldwide prevalence of 1 in 20,000 to 1 in 40,000.7 According to the sponsor, the estimated prevalence is between 600 and 900 cases in Canada, based on the extrapolation of US epidemiological data.8
The panel of experts consulted for this review stated that improving health-related quality of life (HRQoL) is one of the main goals when treating Rett syndrome. Currently there are no Health Canada–approved therapies or disease-modifying treatments indicated for Rett syndrome. Available therapies (drugs and nondrug treatments) are supportive in nature but only partially manage the multisystem symptoms of the disease.9 Clinical management guidance exists, and expert opinion gathered for this review indicated that this guidance is relevant to practice in Canada.7 Pharmacological treatments for symptom management can include drugs for seizures, bone health, contractures, gastrointestinal disturbances, sleep, anxiety, and pain.7,9 Nonpharmacological treatments are used to optimize developmental potential, promote communication, and treat musculoskeletal complications, and can include physical and occupational therapy, speech therapy, and surgery.9 Additionally, specialist care is necessary to address the various comorbidities that patients have.9
Trofinetide is a synthetic analogue of the N-terminal tripeptide of insulin-like growth factor 1 and the mechanism of action is unknown.2 The Health Canada–approved indication for trofinetide is for the treatment of Rett syndrome in adults and pediatric patients aged 2 years and older.2 Trofinetide is administered orally or by gastrostomy tube twice a day, in the morning and evening, with or without food, according to patient weight.2 When tolerability is a concern, administration can be temporarily interrupted or the dose can be reduced to 50% of the recommended dose and then titrated up slowly until the recommended dose is reached.2
Mutations in MECP2 are neither necessary nor sufficient for the diagnosis of Rett syndrome. Nevertheless, a majority of individuals with Rett syndrome have an MECP2 mutation, and thus MECP2 testing is routinely conducted to confirm a Rett syndrome diagnosis. Two testing methods — either next-generation sequencing (NGS) used alone or Sanger sequencing used together with multiplex ligation-dependent probe amplification (MLPA) — can be used to identify an MECP2 mutation in patients with suspected Rett syndrome.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of trofinetide solution, 200 mg/mL, for oral or gastrostomy tube administration in the treatment of Rett syndrome in adults and pediatric patients aged 2 years and older and weighing at least 9 kg.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the call for input and from clinical experts consulted by for the purposes of this review.
Patient Input
Canada's Drug Agency (CDA-AMC) received patient group input from 3 organizations. The Ontario Rett Syndrome Association — with support from Rett syndrome advocacy groups in British Columbia, Alberta, Saskatchewan, Manitoba, and Quebec — provided an overview of the challenges experienced by patients living with Rett syndrome in Canada, and their caregivers, with data gathered from a series of surveys in which caregivers provided their experiences caring for an individual living with Rett syndrome in Canada. Input received from Cure Rett Canada and the International Rett Syndrome Foundation provided information from families and caregivers of patients with Rett syndrome about their experience with trofinetide.
The groups stated that Rett syndrome is a rare and devastating neurodevelopmental disorder that impacts nearly every aspect of an individual’s life, including the ability to speak, walk, eat, and breathe; in addition, approximately 80% to 90% of patients experience epilepsy. Patients often have severe physical and cognitive impairments, communication difficulties, sensory sensitivities, and behavioural issues (such as anxiety, agitation, and mood disorders), as well as respiratory problems, gastrointestinal issues, cardiac abnormalities, and osteoporosis. Caregivers not only face emotional and physical exhaustion, but also financial challenges because they may need to reduce working hours or quit their jobs to provide full-time care. Rett syndrome can disrupt family dynamics, put a strain on relationships, and affect the well-being of siblings and other family members.
The input received from caregivers described how current treatments for Rett syndrome in Canada focus on symptom management, with the use of antiseizure medications and surgical interventions, as well as physical, occupational, and speech therapies. Caregivers reported dissatisfaction with the slow progress of symptom management, particularly with respect to motor skills and communication.
The patient groups stated that any new therapies that result in minor improvements in motor function would result in HRQoL benefits for patients living with Rett syndrome. There is also a need for better treatment options that could lead to improvements in communication abilities and behavioural and emotional stability, as well as reductions in seizures, gastrointestinal issues, and respiratory problems. According to the groups, trofinetide may represent a novel treatment option for patients who have not responded adequately to existing therapies or who are seeking alternatives to current management strategies. Among patients who have had experience with trofinetide, there were reported improvements in patient motor function and hand use, communication abilities, behaviour, and HRQoL, as well as a reduction in seizures. Common side effects included gastrointestinal disturbances, fatigue, and irritability, which may have an impact on drug tolerability and adherence to treatment.
Clinician Input
Input From Clinical Experts Consulted for This Review
According to the panel of experts consulted for this review, one of the main unmet needs of patients with Rett syndrome is that there are no approved disease-modifying treatments in Canada; current supportive therapies do not sufficiently manage the disease. In general, there is a need for treatment that promotes better HRQoL and addresses the individualized needs of patients (which may include treatment for seizures and issues with communication, motor skills, cognition, behaviour, feeding, and sleep). Therapies that better support families and that support patient and caregiver daily activities are also important.
The experts stated that patients would not have to exhaust supportive therapies before accessing trofinetide. Once a diagnosis of Rett syndrome is confirmed, trofinetide would be used as a first-line therapy along with other drugs and nonpharmacological supportive therapies to manage symptoms, as outlined in the care management guidelines.7
The clinical experts stated that, at this time, there are no specific characteristics or markers that would identify the group of patients who would most benefit from trofinetide. Despite available evidence from the LAVENDER study, the experts indicated that patients aged 2 years and older with a confirmed clinical diagnosis of classic Rett syndrome (with or without a disease-causing MECP2 variant) or atypical Rett syndrome (with a disease-causing MECP2 variant) would be suitable candidates for trofinetide. The clinicians also noted that there is a small part of the population that is clinically diagnosed with Rett syndrome but does not have an MECP2 variant; if these patients are diagnosed by Rett syndrome experts, they may also be candidates for trofinetide. However, the experts agreed they would not treat patients with atypical Rett syndrome without a confirmed MECP2 variant. The experts explained that Rett syndrome may be suspected clinically in patients as young as 2 years and, once a patient has a confirmed diagnosis, it would be reasonable to start treatment at that time. One expert also highlighted they would not treat patients with trofinetide who weighed less than 9 kg because there are added concerns with diarrhea, hydration, and nutrition.
Clinicians routinely record developmental and functional history to track changes in a patient and to help determine if there are clinical responses to treatment. The family’s perspective, caregiver reports (including the primary care physicians, specialists, therapists, and educational assistants in the school), and physician symptom assessments were highlighted as being valuable for providing detailed insight into a patient’s day-to-day wellness and needs that lead to changes in care management. The experts stated that they would assess whether a drug provides meaningful benefits and improvements in HRQoL for the patient and caregivers while balancing those benefits against the adverse effects patients can experience. The clinical experts confirmed that there are currently no standard outcomes used in clinics across Canada to measure response to treatment, and that the outcomes used in the LAVENDER study are not used in clinical practice.
When deciding to discontinue treatment, the experts stated that adverse effects (specifically vomiting, diarrhea, dehydration, and weight loss), hospitalization due to adverse effects, and the impact on a patient’s HRQoL are factors to consider and discuss with caregivers. There were different perspectives on deciding when to discontinue treatment, with 1 expert suggesting an adequate trial of trofinetide at the target dose for 3 months (based on the duration of the LAVENDER study) and others suggesting a trial of 6 to 12 months at the target dose to avoid premature discontinuation, should benefits only be observed after at least 3 months of use. The experts also suggested that if there was no improvement or clinical change despite an adequate trial of trofinetide at the target dose, it would be reasonable to conduct a trial off the medication and evaluate whether there is a difference. If there was an abrupt decline in the patient’s health during the trial off the medication, the experts suggested restarting treatment. The clinical experts noted that this approach could also be used for patients outside of the LAVENDER population (i.e., males, patients older than 20 years). Overall, they agreed that most families would not continue treatment with trofinetide if they believed that the patient was not benefiting from the treatment.
The experts noted that they may follow-up with patients more often when starting a new treatment or if the disease is not stable. Additionally, they stated that consultation with the patient’s primary care team, with whom the patient and caregivers have more regular interactions (particularly for monitoring and treating adverse effects), is important. They suggested that patients starting trofinetide may visit Rett syndrome specialists 1 month after initiation, 3 months after initiation, and then less frequently if the disease is stable.
The experts indicated that Rett syndrome specialists, as well as pediatricians and neurologists with expertise in Rett syndrome, would prescribe trofinetide at first because it is a new medication. However, it was noted that as experience with the drug increases, it may be possible for other physicians to prescribe the drug, which would improve access to patients in remote areas and outside major cities, where specialists practice.
Clinician Group Input
The Canadian Rett Syndrome Consortium (including Acadia Pharmaceuticals Inc. advisory board members for trofinetide) provided input for this review. The input included 6 clinicians consisting of pediatric neurologists, developmental pediatricians, and medical geneticists in Canada.
The clinician group stated that, in Canada, there is currently no approved treatment for Rett syndrome (aside from trofinetide), and that existing medications focus on the management of disease symptoms only. The group stated that no medications to date have targeted the underlying biology, the course of the disease, or the deteriorating developmental trajectory characteristic of Rett syndrome. According to the group input, trofinetide is unique in this space and a first-in-class drug, although its exact mechanism of action is unknown. The clinician group input was consistent with the clinical experts consulted for this review, noting that trofinetide would be used as a first-line treatment and that other medications may be added to address associated symptoms tailored to an individual patient's needs.
Outcomes used to determine whether a patient is responding to trofinetide would rely on caregiver reports. A clinically meaningful response to treatment would include improvements in communication, alertness, engagement, and respiratory symptoms; the ability to move independently; and decreases in repetitive movements or stereotypies. The clinician group anticipates that trofinetide will initially be prescribed in specialized medical centres on an outpatient basis and, over time, with education and experience, by community physicians, such as pediatricians or internists. Considerations for discontinuing trofinetide include no improvement in symptoms after 6 months to 12 months of therapy and persistent moderate-to-severe diarrhea or vomiting with weight loss that are not controlled with appropriate medications or lowering the dose of trofinetide.
Drug Program Input
The drug programs identified the following jurisdictional implementation issues: relevant comparators, initiation of therapy, continuation or renewal of therapy, discontinuation of therapy, prescribing of therapy, generalizability, care provision issues, and system and economic issues. Refer to Table 5 for details.
Clinical Evidence
Systematic Review
Description of Studies
One phase III, double-blind, randomized controlled trial (RCT) (LAVENDER; N = 187) of female patients aged 5 years to 20 years, weighing at least 12 kg, who were diagnosed with classic or typical Rett syndrome and had a documented disease-causing MECP2 variant; at 12 weeks, the efficacy and safety of trofinetide 200 mg/mL (n = 93) twice a day was compared to placebo (n = 94).10 Efficacy was measured with the coprimary end points of RSBQ total score and Clinical Global Impressions-Improvement (CGI-I) score; other clinically relevant outcomes included communication (through nonverbal means and symbolic behaviours). Outcomes related to physical function, HRQoL, and caregiver burden were noted as being important to patient and clinician groups and were included as supportive evidence (the Grading of Recommendations Assessment, Development and Evaluation [GRADE] approach was not applied) when data were available from the sponsor’s submission.
The LAVENDER study included only females with Rett syndrome, and the mean age of patients was 11.0 years (standard deviation [SD] = 4.7 years) in the trofinetide group and 10.9 years (SD = 4.6 years) in the placebo group. Clinical characteristics were generally balanced in the 2 groups, and the mean Clinical Global Impressions-Severity (CGI-S) score was 4.9 points (SD = 0.8 points) for both groups, indicating patients were moderately or markedly ill. Disease history was generally similar in the 2 groups.
Efficacy Results
Rett Syndrome Behaviour Questionnaire
The RSBQ is a 45-item, caregiver-completed assessment with 8 subscales that evaluate various neurobehavioural symptoms known to be impaired in patients with Rett syndrome, rated as 0 (not true), 1 (somewhat or sometimes true), or 2 (very true).11 A total score is calculated as the sum of the scores for all 45 items, and ranges from 0 to 90 points, with higher scores indicating symptoms are more frequent. No minimal important difference (MID) was identified from the literature, and clinical expert opinion indicated that a between-group difference of 3 points would be meaningful.
The mean change from baseline in RSBQ score was –4.9 points (standard error [SE] = 0.9 points) and –1.7 points (SE = 0.9 points) in the trofinetide and placebo groups, respectively, resulting in a between-group difference of –3.1 points (95% confidence interval [CI], –5.7 to –0.6 points; P = 0.0175).
Clinical Global Impressions-Improvement Score
The CGI-I is a clinician-rated scale that ranges from 1 (very much improved) to 7 (very much worse) and is used to assess improvement or worsening from baseline.12 Disease-specific anchors used to guide the assessor and anchor descriptions included the characterization of impairment levels across core Rett syndrome signs and symptoms. No MID was identified from the literature, and clinical expert opinion indicated that a between-group difference of 1 would be meaningful.
The mean CGI-I score at week 12 was 3.5 points (SE = 0.1 points) in the trofinetide group and 3.8 points (SE = 0.1 points) in the placebo group, resulting in a between-group difference of –0.3 points (95% CI, –0.5 to –0.1 points; P = 0.0030).
Rett Syndrome Clinician Rating of Ability to Communicate Choices
The Rett Syndrome Clinician Rating of Ability to Communicate Choices (RTT-COMC) is a clinician-completed instrument that assesses a patient’s ability to communicate choices (including by nonverbal means) using a Likert scale that ranges from 0 (normal functioning) to 7 (most severe impairment).12 No MID was identified from the literature and there was no suggested meaningful threshold, based on clinical expert opinion.
The mean change from baseline in RTT-COMC score was –0.4 points (SE = 0.1 points) and 0.0 points (SE = 0.1 points) in the trofinetide and placebo groups, respectively, resulting in a between-group difference of –0.3 points (95% CI, –0.6 to 0.0 points).
Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler Checklist
The Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler (CSBS-DP-IT) Checklist is a caregiver-completed assessment of communication and prelinguistic skills in young children and older children with developmental delay.12 Each item is rated on a scale of 0 (not yet), 1 (sometimes), or 2 (often). The social composite score (emotion and eye gaze, communication rate and function, and gestures) consists of items 1 to 13, and scores range from 0 to 26 points, with higher scores indicating better social communication development. No MID was identified from the literature and, according to clinical expert opinion, there is no suggested meaningful threshold.
The mean change from baseline in CSBS-DP-IT Checklist social composite score was –0.1 points (SE = 0.3 points) in the trofinetide group and –1.1 points (SE = 0.3 points) in the placebo group, resulting in a between-group difference of 1.0 points (95% CI, 0.3 to 1.7 points; P = 0.0064).
Health-Related Quality of Life
There was no information on HRQoL in the LAVENDER study.
Other Efficacy Results Related to Outcomes Important to Patients, Caregivers, and Clinicians
The Rett Syndrome Clinician Rating of Hand Function (RTT-HF) (the ability to use hands for functional purposes), the Rett Syndrome Clinician Rating of Ambulation and Gross Motor Skills (RTT-AMB) (the ability to sit, stand, and ambulate), and the Rett Syndrome Clinician Rating of Verbal Communication (RTT-VCOM) (the ability to communicate verbally) are clinician-completed, disease-specific instruments that rate a patient’s abilities from 0 (normal functioning) to 7 (most severe impairment).12 The between-group difference for the RTT-HF score was –0.1 points (95% CI, –0.3 to 0.1 points), for the RTT-AMB score was –0.1 points (95% CI, –0.3 to 0.1 points), and for the RTT-VCOM score was 0.0 points (95% CI, –0.2 to 0.2 points).
The patient’s overall quality of life was rated from 1 (poor) to 6 (excellent). The between-group difference for the overall quality of life rating was 0.1 points (95% CI, –0.1 to 0.4 points).
The Rett Syndrome Caregiver Burden Inventory (RTT-CBI) is completed by the caregiver to assess the burden of caring for the patient on daily life in 4 areas (physical, emotional, and social burden, and time dependence).12 Caregivers rate how often a statement describes their feelings or experiences, with frequency rated on a Likert scale from 0 (never) to 4 (nearly always). Items 1 to 24 yield a total burden score from 0 to 96 points, and items 25 and 26 make up the optimism index (which was not used in the analyses). Higher scores indicate a greater burden on the caregiver. The between-group difference for the RTT-CBI total score was –0.8 points (95% CI, –3.5 to 2.0 points).
Harms Results
In the LAVENDER study, 92.5% of patients in the trofinetide group and 54.3% of patients in the placebo group experienced at least 1 treatment-emergent adverse event (TEAE). Diarrhea and vomiting were the most common TEAEs and were imbalanced in the 2 treatment groups; 80.6% of patients in trofinetide group and 19.1% of patients in the placebo group reported diarrhea and 26.9% of patients in the trofinetide group and 9.6% of patients in the placebo group reported vomiting. In the trofinetide group, 2.2%, 36.6%, and 41.9% of patients experienced severe, moderate, or mild diarrhea, respectively, whereas 1.1%, 6.5%, and 19.4% of patients experienced severe, moderate, or mild vomiting, respectively. In the trofinetide group, 3 (3.2%) patients reported 5 serious adverse events (SAEs) and in the placebo group, 3 (3.2%) patients reported 3 SAEs. During the study, 17.2% of patients in the trofinetide group and 2.1% of patients in the placebo group stopped treatment due to TEAEs; the most frequently reported TEAE leading to treatment discontinuation was diarrhea (12.9% in the trofinetide group and 0.0% in the placebo group). There were no deaths in the study.
TEAEs considered clinically important by the clinical experts and noted in the product monograph included diarrhea and vomiting, as previously described.
Critical Appraisal
Reports of TEAEs were imbalanced in the 2 treatment groups, particularly for diarrhea and vomiting. This most likely resulted in functional unblinding and may have impacted the ratings of assessors who were aware of a patient’s TEAEs (e.g., caregivers, clinicians). Furthermore, efficacy outcomes were subjective in nature, and there is the potential for overestimation of the treatment effect if assessors suspect the treatment assignment. The RSBQ was originally developed as a diagnostic tool, rather than for measuring treatment effect.11 The RTT-COMC and CSBS-DP-IT Checklist social composite instruments have not been validated in patients with Rett syndrome, and both Health Canada and the FDA have indicated that the CSBS-DP-IT Checklist is not adequate for establishing the efficacy of trofinetide.13,14 No MIDs were identified from the literature for any of the trial outcomes. Moreover, the clinical experts consulted for this review stated that trial outcomes are not commonly used in practice and there are no standardized measures used in clinics in Canada. The greatest number of patients who discontinued study treatment was in the trofinetide group, and the difference between groups was large. This introduces the potential for bias against the null, as the data driving the model are largely from patients who stayed in the study and were likely better responders and had fewer TEAEs. The investigators assumed that data were missing at random (MAR), which is not supported by the differential losses to follow-up and reasons for discontinuations.
The Health Canada indication is broader than the trial population. The LAVENDER study did not enrol patients who were male, younger than 5 years or older than 20 years, did not have classic or typical Rett syndrome, did not have a confirmed disease-causing MECP2 variant, were not at least 6 months after regression, did not have a CGI-S score of at least 4, and did not receive stable standard therapies or have a stable pattern of seizures. The panel of experts consulted for this review indicated that patients would be treated with trofinetide if there was a confirmed clinical diagnosis of classic Rett syndrome (with or without a disease-causing MECP2 variant) or atypical Rett syndrome (with a disease-causing MECP2 variant). Patients without an MECP2 variant would have to have a confirmed clinical diagnosis of classic Rett syndrome to receive trofinetide, but those without an MECP2 variant and with a diagnosis of atypical Rett syndrome would not receive trofinetide because of the lack of evidence in this population. Because none of the LAVENDER outcomes are used in clinical practice, it is a challenge to apply the results to a real-world setting. There was no comprehensive measure of HRQoL and it is uncertain how trofinetide impacts this outcome, which is an important treatment goal in the management of Rett syndrome. The LAVENDER study was 12 weeks long, which is not long enough to assess meaningful, long-term changes in motor skills, communication, or harms in patients with Rett syndrome.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, the GRADE approach was used to assess the certainty of the evidence for outcomes considered most relevant to expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.15,16
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
RSBQ total score
CGI-I score
RTT-COMC score
CSBS-DP-IT Checklist social composite score.
Table 2: Summary of Findings for Trofinetide vs. Placebo in Patients With Rett Syndrome
Outcome and follow-up | Patients (studies), N | Absolute effects | Certainty | What happens | |||
|---|---|---|---|---|---|---|---|
Placebo | Trofinetide | Difference (95% CI) | |||||
RSBQ total score | |||||||
RSBQ total score (0 [symptoms are less frequent] to 90 [symptoms are more frequent]) Follow-up: 12 weeks | 187 (1 RCT) | –1.7 | –4.9 (SE = 0.9) | –3.1 (–5.7 to –0.6) | Very lowa | The evidence is very uncertain about the effect of trofinetide on RSBQ total score when compared with placebo. | |
CGI-I score | |||||||
CGI-I score (1 [very much improved] to 7 [very much worse]) Follow-up: 12 weeks | 187 (1 RCT) | 3.8 | 3.5 (SE = 0.1) | –0.3 (–0.5 to –0.1) | Very lowb | The evidence is very uncertain about the effect of trofinetide on CGI-I score when compared with placebo. | |
RTT-COMC score | |||||||
RTT-COMC score (0 [normal functioning] to 7 [most severe impairment]) Follow-up: 12 weeks | 187 (1 RCT) | 0.0 | –0.4 (SE = 0.1) | –0.3 (–0.6 to 0.0) | Very lowc,d | The evidence is very uncertain about the effect of trofinetide on RTT-COMC score when compared with placebo. | |
CSBS-DP-IT Checklist social composite score | |||||||
CSBS-DP-IT Checklist social composite score (0 [worst] to 26 [best]) Follow-up: 12 weeks | 187 (1 RCT) | –1.1 | –0.1 (SE = 0.3) | 1.0 (0.3 to 1.7) | Lowe | Trofinetide may result in an increase in prelinguistic communication skills, when compared with placebo. The clinical importance of the increase is unclear. | |
HRQoL | |||||||
Not reported | NA | NA | NA | NA | NA | There is no evidence for any effect of trofinetide on HRQoL. | |
CGI-I = Clinical Global Impressions-Improvement; CI = confidence interval; CSBS-DP-IT = Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler; HRQoL = health-related quality of life; NA = not applicable; RCT = randomized controlled trial; RSBQ = Rett Syndrome Behaviour Questionnaire; RTT-COMC = Rett Syndrome Clinician Rating of Ability to Communicate Choices; SE = standard error.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious study limitations (risk of bias due to missing data [results were missing for 16% and 9% of the trofinetide and placebo groups, respectively], potential for functional unblinding due to differences in harms and the instrument not being widely used in clinical practice for measuring treatment effect). Rated down 1 level for serious indirectness (trial population is narrower than the Health Canada indication and the requested reimbursement population; results based on the trial population may not be generalizable to the Health Canada–indicated population). Rated down 1 level for serious imprecision (based on a meaningful threshold of 3 suggested by a clinical expert; CI for difference between groups includes possibility of a difference that is not clinically meaningful).
bRated down 1 level for serious study limitations (risk of bias due to missing data [results missing for 15% and 8% of trofinetide and placebo groups, respectively] and the instrument not being widely used in clinical practice for measuring treatment effect). Rated down 1 level for serious indirectness (trial population is narrower than the Health Canada indication and the requested reimbursement population; results based on the trial population may not be generalizable to the Health Canada–indicated population). Rated down 2 levels for very serious imprecision (based on a meaningful threshold of 1 suggested by a clinical expert; CI for difference between groups suggests no difference).
cRated down 1 level for serious study limitations (risk of bias due to missing data [results missing for 16% and 12% of trofinetide and placebo groups, respectively]; potential for functional unblinding due to differences in harms; lack of evidence supporting the instrument’s validity, reliability, or responsiveness; and the instrument not being widely used in clinical practice to measure treatment effect). Rated down 1 level for serious indirectness (trial population is narrower than the Health Canada indication and the requested reimbursement population; results based on the trial population may not be generalizable to the Health Canada–indicated population). Rated down 2 levels for very serious imprecision (no known minimal important difference [MID]), so the target of certainty appraisal was any effect; CI for the difference between groups includes the possibility of no difference).
dStatistical testing for this outcome was not adjusted for multiplicity. The results are considered to be supportive evidence.
eRated down 1 level for serious study limitations (risk of bias due to missing data [results missing for 20% and 13% of trofinetide and placebo groups, respectively]; potential for functional unblinding due to differences in harms; lack of evidence supporting the instrument’s validity, reliability, or responsiveness; and the instrument not being widely used in clinical practice for measuring treatment effect). Rated down 1 level for serious indirectness (trial population is narrower than the Health Canada indication and the requested reimbursement population; results based on the trial population may not be generalizable to the Health Canada–indicated population). No known MID, so the target of certainty appraisal was any effect.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
Description of Studies
Two open-label extension (OLE) studies — LILAC (N = 154) and LILAC-2 (N = 77) — have been summarized to provide evidence regarding the long-term safety and tolerability of trofinetide in females with Rett syndrome. Patients who completed the LAVENDER study could enrol in the LILAC study, a 40-week OLE study (N = 154) and, upon completion, could continue in the LILAC-2 study (N = 77), an OLE study with up to 32 months of additional treatment time. The inclusion and exclusion criteria for the OLE studies were consistent with those for the LAVENDER study. All patients received trofinetide in these studies. Seventy (45.5%) patients withdrew early from the LILAC study, primarily due to TEAEs, with 84 (54.5%) patients completing the study. In the LILAC-2 study, most patients (79.2%) discontinued due to the study's termination after market approval, 5 (6.5%) patients discontinued due to TEAEs, and 4 (5.2%) patients died.
Demographic characteristics in the LILAC and LILAC-2 studies were similar to those in the LAVENDER study. The mean age of patients was 11.0 years (SD = 4.6 years) in the LILAC study and 12.0 years (SD = 4.4 years) in the LILAC-2 study. The mean baseline CGI-S score was 4.8 (SD = 0.8) in the LILAC study and 4.8 (SD = 0.9) in the LILAC-2 study, and most patients were moderately ill (36.4% and 41.6%, respectively) or markedly ill (41.6% and 31.2%, respectively) in the 2 studies. Dosing in the OLE studies was weight-based, and the weight bands used in the OLE studies were the same as those used in the LAVENDER study.
Outcomes
The primary outcomes in both the LILAC and LILAC-2 studies focused on safety and included TEAEs, SAEs, withdrawals due to TEAEs, and potentially clinically important changes in other safety assessments. Relevant secondary and exploratory efficacy outcomes included RSBQ, CGI-I, RTT-COMC, CSBS-DP-IT Checklist social composite, RTT-HF, RTT-AMB, RTT-VCOM, overall quality-of-life rating, and RTT-CBI scores at various time points in both studies. For both OLE studies, all results were summarized using descriptive statistics and performed using the safety analysis set unless otherwise noted.
Efficacy Results
Patients who received trofinetide in the LAVENDER study showed a decrease in RSBQ total scores from LAVENDER baseline, with a mean change of –7.3 points (SD = 10.7 points) at week 40 in the LILAC study (N = 44) and a mean change of –9.8 points (SD = 11.2 points) at week 104 in the LILAC-2 study (N = 10). Patients who received placebo in the LAVENDER study experienced decreases from LAVENDER baseline after switching to trofinetide in the LILAC study, with a mean change of –7.0 points (SD = 10.7 points) at week 40 in the LILAC study (N = 44) and a mean change of –13.8 points (SD = 12.0 points) at week 104 in the LILAC-2 study (N = 11). Overall, patients who tolerated trofinetide showed at least a 5-point decrease in the RSBQ total score, which persisted throughout the extension studies.
For both OLE studies, changes in the CGI-I score were assessed relative to the patient’s baseline state of illness in the LILAC study. As such, no CGI-I scores were assessed for the baseline visit in the LILAC study. Overall, mean CGI-I scores remained stable over time among patients in the OLE studies.
Other efficacy outcomes, including RTT-COMC, CSBS-DP-IT Checklist social composite, RTT-HF, RTT-AMB, RTT-VCOM, overall quality-of-life rating, and RTT-CBI scores, generally remained stable over time among patients who continued in the OLE studies.
Harms Results
In the LILAC study, 132 patients (85.7%) reported at least 1 TEAE, with diarrhea (59.1%), vomiting (25.3%), and COVID-19 (11.0%) being the most common. SAEs occurred in 19 patients (12.3%), and 48 patients (31.2%) experienced TEAEs leading to drug discontinuation, primarily due to diarrhea (19.5%) and vomiting (5.8%). In the LILAC-2 study, 68 patients (88.3%) reported at least 1 TEAE, with COVID-19 (26.0%), diarrhea (16.9%), pyrexia (16.9%), and urinary tract infection (15.6%) being the most common. SAEs occurred in 23 (29.9%) patients, and 6 (7.8%) patients discontinued the drug. The most common SAEs were seizures (6.5%), followed by vomiting, pneumonia, urinary tract infection, and acute respiratory failure (2.6% each). A total Of 4 deaths were reported, of which 3 (3.9%) were reported as TEAEs (cardiac arrest, aspiration and vomiting, and sudden unexplained death in epilepsy in 1 patient each).
Critical Appraisal
The OLE studies provided efficacy and safety data for trofinetide for up to 104 weeks. The open-label design increases the potential for bias, particularly in the reporting of subjective outcomes and adverse events (AEs). Because completion of the pivotal trial was required for enrolment, patients who discontinued the LAVENDER study were excluded, resulting in a patient population that was more tolerant and responsive to trofinetide and introducing selection bias. Additionally, with a high discontinuation rate in the LILAC study (45.5%), mainly due to TEAEs, the impact of patient dropout on outcomes is unclear; analyses were not conducted to assess how discontinuation affected treatment results.
Indirect Comparisons
No indirect treatment comparisons were submitted for the review of trofinetide.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
Two studies have been summarized to provide additional evidence to the systematic review. The DAFFODIL study is a multicentre, open-label, long-term, phase II/III study conducted in the US that provides evidence on the efficacy and safety of trofinetide in females aged 2 to 5 years with Rett syndrome. The LOTUS study is an ongoing, phase IV, observational, real-world study of patients prescribed trofinetide under routine clinical care in the US for up to 24 months; interim results at 6 months were submitted by the sponsor.
The DAFFODIL study enrolled 15 female patients in the US living with Rett syndrome, an MECP2 variant, and a CGI-S score of 4 or greater at screening and baseline. Eligible patients were aged 2 years to 4 years and had a body mass that ranged from 9 kg to less than 20 kg or were aged 5 years with a body mass that ranged from 9 kg to less than 12 kg. Aside from the age restriction, the inclusion and exclusion criteria were comparable to those used in the pivotal trial. The mean age at the time of diagnosis was 1.9 years (SD = 0.1 years). Trofinetide was dosed by weight and administered orally or by gastrostomy tube.
In total, 154 patients were included in the interim analysis of the LOTUS study. Most patients had classic Rett syndrome (66.7%) and were female (96.1%), and the age of patients in the study ranged from 2 years to 60 years. The mean age at the time of diagnosis was 5.2 years (SD = 5.37 years) and the mean age at the time of trofinetide initiation was 16.5 years (SD = 11.16 years). There were no exclusion criteria in the study.
Outcomes
Relevant exploratory efficacy outcomes in the DAFFODIL study included the CGI-I score from baseline to week 104 and the overall quality-of-life rating. In both the DAFFODIL and LOTUS studies, safety assessments were based on the proportion of patients experiencing TEAEs, SAEs, and withdrawals due to TEAEs. Efficacy and safety data were summarized using descriptive statistics in both studies.
Efficacy Results
In the DAFFODIL study, the mean CGI-I score decreased from week 2 (n = 13) through to week 78 (n = 9) from 3.5 (SD = 0.66) to 2.2 (SD = 0.67). The mean change from baseline in the overall quality-of-life rating at week 12 was 0.3 (SD = 0.72), which continued to increase through to week 78, when mean change was 0.7 (SD = 0.95).
Harms Results
In the DAFFODIL study, safety outcomes were similar to those in the pivotal and OLE studies. In total, 14 (93.3%) patients experienced at least 1 TEAE, the most common of which were diarrhea (73.3%) and vomiting (46.7%). Four (26.7%) patients reported SAEs and 2 (13.3%) patients experienced TEAEs leading to drug and study discontinuation. As of the 9-month LOTUS interim analysis (with data up to June 26, 2024), 22 (11.5%) patients reported 57 TEAEs, with diarrhea, vomiting, constipation, and insomnia being the most common. Six (3.1%) patients reported SAEs (constipation, diarrhea, vomiting, viral gastroenteritis, pneumonia, pneumonia aspiration, and dehydration) and 14 (7.3%) patients withdrew due to a TEAE. No deaths were reported in either study.
Critical Appraisal
The longer-term harms data from both the DAFFODIL and LOTUS studies are generally consistent with those from the pivotal trial, with diarrhea and vomiting being the most common. Although the 2 studies attempted to fill the evidence gaps for patients aged 2 years to 5 years (the DAFFODIL study) or aged 20 years or older who were diagnosed with atypical disease or who were male (the LOTUS study), there remains uncertainty in the study results due to various limitations with the data. Neither study was designed to assess the efficacy and safety of trofinetide in a statistically rigorous manner. Other limitations include potential selection bias, lack of blinding, small study population (the DAFFODIL study), and lack of comparator group, which may have affected the internal validity of the safety and efficacy results. In addition, the lack of blinding could have introduced bias in the reporting of subjective AEs in favour of trofinetide if patients and/or caregivers believed the drug was beneficial.
Conclusions
Rett syndrome is a rare, neurodevelopmental disorder associated with the progressive loss of learned skills, intellectual disability, and various comorbidities. There is a need for safe and effective treatments that address the underlying disease mechanism and improve communication, motor skills, and HRQoL. Based on the evidence from 1 phase III, double-blind RCT (the LAVENDER study), females aged 5 years to 20 years with typical Rett syndrome who have a confirmed, disease-causing MECP2 variant and who receive trofinetide (weight-based dosing) twice a day for 12 weeks are more likely to demonstrate improvements in Rett syndrome–specific neurobehavioural symptoms (measured by the RSBQ). Evidence that trofinetide results in global improvement from a clinician’s perspective or increased communication skills (measured by CGI-I, RTT-COMC, and CSBS-DP-IT Checklist social composite scores) is uncertain because of missing trial data, a lack of MIDs, and the modest treatment effects that may not translate into meaningful changes for patients or caregivers in the real-world setting. It is unclear what impact trofinetide has on HRQoL, or whether changes in motor function or caregiver burden observed in the trial would result in meaningful, long-term improvements for patients or caregivers. Longer-term results (mean time on trofinetide was approximately 2 years) indicated possible further reductions in neurobehavioural symptom frequency, whereas the clinician’s global impression and the patient’s ability to communicate choices remained stable for those who continued in the OLE studies. However, the study design, large number of discontinuations, and relatively short follow-up prevent definitive conclusions from being drawn, as it is likely that those who were receiving perceived benefits remained in the OLE studies longer. Harms, such as diarrhea and vomiting, were a key reason that patients discontinued treatment and are expected to contribute to reduced tolerability, reduced HRQoL, and increased caregiver burden. Furthermore, the Health Canada indication for trofinetide is broader than the pivotal trial population, so there is a lack of rigorous evidence in patients who are male, who are younger than 5 years or older than 20 years, who have an atypical or variant Rett syndrome diagnosis, who have a non-MECP2 variant, and who are in other stages of the disease (i.e., not at least 6 months after regression).
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of trofinetide solution, 200 mg/mL, for oral or gastrostomy tube administration in the treatment of Rett syndrome in adults and pediatric patients aged 2 years and older and weighing at least 9 kg.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Rett syndrome is a rare, neurodevelopmental disorder characterized by normal early development followed by a progressive loss of speech, purposeful hand use, and motor skills.3 Common symptoms include stereotypic hand movements (such as wringing, clapping, or mouthing), gait abnormalities, breathing irregularities (like hyperventilation or breath-holding), seizures, and intellectual disabilities. Rett syndrome is most often caused by genetic variants in MECP2, which is located on the X chromosome, and primarily affects females, although it can occur in males in rare cases.3 A study of 30 males with MECP2 variants (identified through Rett Syndrome Natural History Study databases) showed that the clinical phenotype observed in males is very heterogenous and not always consistent with a classic Rett syndrome diagnosis in females, which may decrease the likelihood of detection in males.17 The study also noted that there appears to be greater clinical severity in a higher proportion of males with MECP2 variants than females with Rett syndrome.17
The disorder progresses through 4 stages: stage I (stagnation, 6 to 18 months), in which developmental progress slows and previously acquired skills are lost; stage II (rapid regression, 1 year to 4 years), which is marked by significant loss of motor and communication skills, worsening microcephaly, and the emergence of seizures and breathing abnormalities; stage III (plateau, 2 years to 10 years), in which symptoms stabilize and some improvement in hand skills or behaviour may occur, although significant cognitive and motor impairments persist; and stage IV (late motor deterioration, 10 years and onward), which is characterized by further loss of mobility, dystonia, and physical disability, leading to wheelchair dependency. With the discovery of MECP2 variants, staging now has limited diagnostic value, but it can be used to anticipate potential clinical issues and offer guidance to parents and caregivers.3
In a natural history study of females with genetically confirmed disease who contributed data to the Australian Rett Syndrome Database between 2000 and 2019, RSBQ scores were shown to gradually decline (i.e., suggesting behavioural improvements) with increasing age.4 The authors noted that although clinical disease severity tends to increase with age, the natural history study results suggest that issues related to behaviour and emotions improved (based on RSBQ scores) over a 10-year to 14-year observation period.
Patients living with Rett syndrome require lifelong care and assistance with daily activities, significantly impacting both patients and their caregivers.5 The condition leads to a high caregiver burden and reduces the quality of life of families.18 It is estimated to affect 1 in 10,000 females aged 12 years and younger, with a worldwide prevalence of 1 in 20,000 to 1 in 40,000.7 According to the sponsor, in Canada, the estimated prevalence is between 600 and 900 cases, based on the extrapolation of US epidemiological data, and around 220 to 240 prevalent cases are diagnosed in Ontario annually, based on assessments of Institute for Clinical Evaluative Sciences datasets.8 The sponsor extrapolated the diagnosed prevalence rate from Ontario to other CDA-AMC participating jurisdictions, resulting in the population estimates by jurisdiction presented in Table 3.8
Table 3: Estimated Prevalence of Rett Syndrome in Each Region
Region | Estimated diagnosed prevalence, na |
|---|---|
Newfoundland and Labrador | 8.74 |
Prince Edward Island | 2.82 |
Nova Scotia | 17.17 |
New Brunswick | 13.54 |
Ontario | 258.00 |
Manitoba | 21.48 |
Saskatchewan | 17.39 |
Alberta | 76.05 |
British Columbia | 91.67 |
Non-Insured Health Benefits | 13.88 |
Canada | 520.74 |
Note: Data are representative of the population aged 2 years and older.
aInstitute for Clinical Evaluative Sciences data for Ontario, extrapolated to all CDA-AMC jurisdictions.
Source: Sponsor’s Summary of Clinical Evidence.8 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Diagnostic Testing Requirements
The diagnosis of Rett syndrome is made on a clinical basis, using the following criteria.6
Main criteria:
partial or complete loss of acquired purposeful hand skills
partial or complete loss of acquired spoken language; note that loss of acquired language is based on best acquired spoken language skill, not strictly on the acquisition of distinct words or higher language skills
gait abnormalities, which include impaired (dyspraxia) or absence of ability (apraxia)
stereotypic hand movements, such as hand wringing or squeezing, clapping or tapping, mouthing, and washing or rubbing automatisms.
Exclusion criteria (for typical Rett syndrome):
brain injury secondary to trauma (perinatal or postnatal), neurometabolic disease, or severe infection that causes neurological problems; note there should be clear evidence (neurological or ophthalmological examination and MRI or CT) that the presumed injury directly resulted in neurological dysfunction
grossly abnormal psychomotor development in first 6 months of life; note that grossly abnormal development indicates that normal milestones (acquiring head control, swallowing, or developing a social smile) are not met; mild generalized hypotonia and other previously reported subtle developmental alterations during the first 6 months of life are common in Rett syndrome and do not constitute exclusionary criteria.
Supportive criteria (for atypical Rett syndrome); note that if an individual has or ever had a clinical feature listed, it is counted as a supportive criterion and that many of these features have an age dependency that manifests or becomes more predominant at certain ages:
breathing disturbances when awake
bruxism when awake
impaired sleep pattern
abnormal muscle tone
peripheral vasomotor disturbances
scoliosis or kyphosis
growth retardation
small, cold hands and feet
inappropriate laughing and/or screaming spells
diminished response to pain
intense eye communication (eye pointing).
There are 2 types of Rett syndrome diagnoses: typical or classic Rett syndrome; and atypical or variant Rett syndrome.19 Typical and atypical Rett syndrome both require a period of regression followed by recovery or stabilization. For typical Rett syndrome, meeting all the main criteria and no exclusionary criteria is required.19 Atypical Rett syndrome requires meeting at least 2 main criteria and 5 supportive criteria.6 Genetic testing, particularly the detection of pathogenic MECP2 variants, confirms the diagnosis, although MECP2 variants may not be detected in up to 5% of patients with typical Rett syndrome cases and approximately 25% of patients with atypical disease.3 The clinical experts consulted for this review emphasized that a clinical diagnosis by a Rett syndrome specialist is required, as diseases similar to Rett syndrome can also involve an MECP2 variant. Information provided by the public drug plans and the experts indicated that testing procedures vary across jurisdictions. Depending on the request, testing can take place locally or tests can be sent out of province or out of country.
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
According to the panel of experts consulted for this review, one of the main goals when treating patients with Rett syndrome is improving HRQoL, which is done mainly through symptom management.
Currently, there are no Health Canada–approved therapies or disease-modifying treatments indicated for Rett syndrome. Available therapies (drugs and nondrug treatments) are supportive in nature but only partially manage the multisystem symptoms of the disease.9 According to the clinical experts consulted for this review, in the absence of approved treatments, care is symptom based and aims to optimize developmental outcomes using rehabilitative therapies. Furthermore, patients with Rett syndrome often have multiple comorbidities and their management requires a multidisciplinary approach.9 Clinical management guidance exists, and expert opinion gathered for this review indicated that this guidance is relevant to practice in Canada.7
Pharmacological treatments for symptom management can include antiepileptic medications for seizures, calcium and vitamin D supplementation to promote bone health, spasticity treatment, laxatives, stool softeners, proton pump inhibitors for gastrointestinal disturbances, and other drugs for sleep, anxiety, and pain.7,9 Careful monitoring of AEs resulting from medications is also important.
According to the experts consulted for this review, nonpharmacological treatments are used to optimize developmental potential, promote communication, and treat musculoskeletal complications. These can include physical and occupational therapy, speech therapy, and surgery (to correct scoliosis).9 Additionally, specialist care is necessary to address nutrition and feeding issues, gastrointestinal dysfunction, possible prolonged QTc intervals, breathing irregularities, seizures, sleep disturbances, movement disorders, bone and joint health, mobility and coordination, communication, and the use of assistive devices.9 An estimated one-third of individuals with Rett syndrome have a gastrostomy tube, which becomes an added consideration when it comes to feeding and administering medications.7
Because the physical, cognitive, and behavioural symptoms begin in early childhood and persist, individuals with Rett syndrome require continuous care throughout their lives, with caregivers and families taking on much of the responsibility, although patients may also live in residential care homes.7 As a result, the burden on families and caregiver support are important considerations.
Drug Under Review
Trofinetide is a synthetic analogue of the N-terminal tripeptide of insulin-like growth factor 1.2 The mechanism by which trofinetide exerts therapeutic effects in patients with Rett syndrome is unknown. The Health Canada–approved indication for trofinetide is for the treatment of Rett syndrome in adults and pediatric patients 2 years and older. According to the Health Canada product monograph, trofinetide is to be administered orally or by gastrostomy tube twice a day, in the morning and evening, with or without food, according to patient weight:2
9 kg to less than 12 kg receive 4 g trofinetide twice daily (20 mL twice daily)
12 kg to less than 20 kg receive 6 g trofinetide twice daily (30 mL twice daily)
20 kg to less than 35 kg receive 8 g trofinetide twice daily (40 mL twice daily)
35 kg to less than 50 kg receive 10 g trofinetide twice daily (50 mL twice daily)
50 kg or greater receive 12 g trofinetide twice daily (60 mL twice daily).
Doses administered by gastrojejunal tubes must be administered through the gastrostomy port.2 If a dose of trofinetide is missed, it should be taken as soon as possible, unless it is nearly time for the next dose. Doses should not be doubled. If vomiting occurs after taking trofinetide, the missed dose should be skipped. If tolerability is a concern, administration should start at a lower dose (i.e., starting at 50% of the recommended dose twice a day and increasing the dose over 4 weeks to 8 weeks until the recommended dose is reached).
The recommended dosage of trofinetide for patients with moderate renal impairment (an estimated glomerular filtration rate of 30 to 59 mL/min/1.73 m2) is half the dosage used in patients without renal impairment.2 Trofinetide is not recommended in patients with severe renal impairment (an estimated glomerular filtration rate 15 to 29 mL/min/1.73 m2).
Testing Procedure Considerations
In the LAVENDER trial,12 only patients with Rett syndrome and a documented disease-causing MECP2 mutation were eligible to be included. According to the sponsor and the clinical experts consulted for this review, a positive MECP2 mutation status is neither necessary nor sufficient for the diagnosis of Rett syndrome because MECP2 mutations may cause the alternative phenotypes found in other neurodevelopmental diseases and patients may be diagnosed with Rett syndrome based on the clinical diagnostic criteria, despite having no MECP2 mutation.20 Nevertheless, MECP2 mutations have been identified in 95% to 97% of people living with typical Rett syndrome and in 50% to 70% of people living with atypical Rett syndrome.21 Given the prevalence, a one-time genetic test for MECP2 mutations is recommended by Rett syndrome specialists to establish or confirm a molecular diagnosis in patients with suspected Rett syndrome.7
Two genetic testing methods can be used to identify MECP2 mutations in patients with suspected Rett syndrome: NGS used alone; or Sanger sequencing used together with MLPA.21,22 According to sponsor-submitted information and supporting literature,21 NGS testing can be used to detect single nucleotide changes and copy number variants in MECP2 associated with Rett syndrome. Single-gene testing using Sanger sequencing can detect point mutations and small insertions or deletions, whereas MLPA can detect deletions or duplications of significant regions (e.g., spanning 1 or more exons).22 NGS, Sanger sequencing, and MLPA testing all use DNA from blood samples for genetic testing analysis, which requires adequate sample collection, storage, and shipping considerations, when needed.22,23 According to the clinical experts consulted for this review and information provided by a targeted jurisdictional survey, single-gene testing using both Sanger sequencing and MLPA together is the testing method used in Canada at the time of this report, which agrees with information from the literature.22
This review considered the potential impacts of genetic testing for MECP2 mutations in patients with Rett syndrome if trofinetide becomes funded, including impacts on health systems, patients (and their families and caregivers), and costs. Key considerations and relevant information available from materials submitted by the sponsor, input from patient groups, input from the clinical experts consulted by the review team, responses from a targeted jurisdictional survey, and sources from jurisdictional molecular testing inventories and the literature were validated by the review team, when possible, and are summarized in Table 4.
Table 4: Considerations Before Testing for MECP2 Mutations in Patients With Suspected Rett Syndrome
Consideration | Criterion | Available Information |
|---|---|---|
Health system-related | Number of individuals in Canada expected to require the test each year | According to sponsor-submitted information, there are approximately 8 to 12 incident cases diagnosed annually in Ontario. The sponsor estimated that, based on the population growth rate for Ontario, the prevalent population diagnosed in Canada, excluding Quebec, will increase from 521 individuals in 2024 (used as the baseline) to 530 at year 1, 539 at year 2, and 549 at year 3, resulting in at least 28 individuals who are expected to receive MECP2 testing in the next 3 years across jurisdictions in Canada, excluding Quebec. A 2014 report by INESSS anticipates that approximately 30 genetic tests for MECP2 would be sent out of province for patients with suspected of Rett syndrome in Quebec annually;22 however, this estimate could not be validated. No other direct incident data were identified. |
Availability and reimbursement status of the testing procedure in jurisdictions across Canada | The availability and reimbursement status of MECP2 testing vary across jurisdictions in Canada. According to the clinical experts consulted for this review and based on a molecular testing inventory scan, MECP2 testing is available and publicly funded for any individual with suspected Rett syndrome in Ontario and Alberta.24 Alberta also provides MECP2 genetic testing to patients in Saskatchewan25 and Quebec.26 Based on responses from a targeted jurisdictional survey:
No information was provided by or identified for Northwest Territories, Nova Scotia, Nunavut, or Prince Edward Island. | |
Testing procedure as part of routine care | According to clinical guidelines on Rett syndrome management, it is recommended that patients with suspected Rett syndrome undergo MECP2 testing to confirm a diagnosis.7 The clinical experts consulted for this review confirmed that patients should undergo MECP2 genetic testing to diagnose Rett syndrome and that this confirmatory testing is the standard of care in all Canadian jurisdictions. | |
Repeat testing requirements | Repeat testing is not required once the MECP2 mutation status is confirmed. | |
Impacts on human and other health care resources of the provision of testing procedures | Based on the relatively low number of individuals estimated to be diagnosed with Rett syndrome each year, and because testing may already be routinely conducted in Canada to confirm the diagnosis of Rett syndrome, if trofinetide becomes funded, the provision of MECP2 testing is not anticipated to have a substantial impact on human or other health care resources. | |
Patient-related | Accessibility of the testing procedure in jurisdictions across Canada | According to the clinical experts consulted for this review and in line with recommendations on Rett syndrome management,7 all patients with suspected Rett syndrome should have access to confirmatory MECP2 genetic testing. However, 1 patient group (Cure Rett Canada) indicated that there are concerns about genetic testing accessibility in patients from remote areas. According to 1 of the clinical experts consulted for this review, MECP2 testing is not limited to Rett syndrome clinics and may be ordered by a development pediatrician, neurologist, or medical geneticist involved in patient care in Ontario. In Manitoba, it is limited to neurologists and medical geneticists, according to the targeted jurisdictional survey, as well as a 2014 publication.27 Test ordering protocols were not identified for other jurisdictions. |
Expected turnaround times for the testing procedure | The turnaround time for MECP2 testing varies and may depend on whether testing is sent for out-of-province processing and the testing method used. For example:
No other relevant information on expected turnaround times in other jurisdictions was identified. | |
Burden associated with the testing procedure for patients, families, and/or caregivers | Patients and caregivers may experience psychological burdens as they await the results of MECP2 genetic testing, which may be exacerbated by longer turnaround times, which vary depending on the jurisdiction. From the targeted jurisdictional survey, British Columbia indicated that in cases of MECP2 testing not approved for provincial reimbursement, out-of-pocket costs may be incurred by patients and their families. | |
Clinical | Clinical utility and validity of the testing procedure | The presence of an MECP2 mutation alone is not sufficient for a Rett syndrome diagnosis. Nevertheless, MECP2 testing can be used to confirm the diagnosis of Rett syndrome, particularly in patients with suspected atypical Rett syndrome, in whom clinical diagnoses may be complicated.21 One clinical expert consulted for this review indicated that the severity of Rett syndrome may be associated with specific MECP2 mutation subtypes; therefore, the results of MECP2 testing may provide clinicians, patients, and caregivers with an indication of the expected Rett syndrome severity. The use of both Sanger sequencing and MLPA together has demonstrated a high diagnostic yield in sample populations with diverse phenotypes that may not meet the criteria for typical Rett syndrome.21 NGS techniques, such as a targeted gene panel or whole exome sequencing, can identify causative variants in MECP2 and other genes in patients with Rett syndrome.21 It has been reported, however, that NGS alone carries the risk of not being able to detect patients with small exonic alterations, like deletions or duplications.21 |
Risks of harm associated with the testing procedure | No risk of harms was reported or identified for NGS, Sanger sequencing, or MLPA for MECP2 testing. | |
Cost | Projected cost of the testing procedure | The cost of MECP2 testing may vary by jurisdiction and depend on the need for out-of-province testing. For example, according to the targeted jurisdictional survey, Saskatchewan provided a cost breakdown of MECP2 testing. The total cost of MECP2 testing is approximately $1,307.45 per sample, which includes:
The anticipated cost of ordering an MECP2 sequence and CNV detection test using NGS is US$990;23 however, this may vary, depending on where NGS testing is being used. |
CNV = copy number variant; INESSS = Institut national d'excellence en santé et en services sociaux; MLPA = multiplex ligation-dependent probe amplification; NGS = next-generation sequencing.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by 3 patient groups.
CDA-AMC received patient group input from 3 organizations. The Ontario Rett Syndrome Association — with support from Rett syndrome advocacy groups in British Columbia, Alberta, Saskatchewan, Manitoba, and Quebec — provided an overview of the challenges experienced by patients living with Rett syndrome, and their caregivers, with data gathered from a series of surveys in which caregivers provided their experiences caring for an individual living with Rett syndrome in Canada. Patient group input received from Cure Rett Canada and the International Rett Syndrome Foundation provided information from families and caregivers of patients with Rett syndrome about their experience with trofinetide.
The groups stated that Rett syndrome is a rare and devastating neurodevelopmental disorder that impacts nearly every aspect of an individual’s life, including the ability to speak, walk, eat, and breathe, and approximately 80% to 90% of patients experience epilepsy. Patients often have severe physical and cognitive impairments, communication difficulties, sensory sensitivities, behavioural issues (such as anxiety, agitation, and mood disorders), as well as respiratory problems, gastrointestinal issues, cardiac abnormalities, and osteoporosis. Caregivers not only face emotional and physical exhaustion, but also financial challenges, as they may need to reduce working hours or quit their jobs to provide full-time care. Rett syndrome can disrupt family dynamics, put a strain on relationships, and affect the well-being of siblings and other family members.
The input received from caregivers described how current treatments for Rett syndrome in Canada focus on symptom management, with the use of antiseizure medications and surgical interventions, as well as physical, occupational, and speech therapies. Caregivers reported dissatisfaction with the slow progress of symptom management, particularly with respect to motor skills and communication.
The patient groups stated that any new therapies that result in minor improvements in motor function would result in HRQoL benefits for patients living with Rett syndrome. There is also a need for better treatment options that could lead to improvements in communication abilities and behavioural and emotional stability, as well as reductions in seizures, gastrointestinal issues, and respiratory problems. According to the groups, trofinetide may represent a novel treatment option for patients who have not responded adequately to existing therapies or who are seeking alternatives to current management strategies. Among patients who have had experience with trofinetide, there were reported improvements in motor function and hand use, communication abilities, behaviour, and HRQoL, as well as a reduction in seizures. Common side effects included gastrointestinal disturbances, fatigue, and irritability, which may have had an impact on tolerability and adherence to treatment.
Clinician Input
Input From the Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the review of trofinetide, a panel of 3 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations in which there are gaps in the evidence that could be addressed with the collection of additional data, promote the early identification of potential implementation challenges, gain insight into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
According to the panel of experts consulted for this review, one of the main unmet needs of patients with Rett syndrome is the lack of disease-modifying treatments in Canada; current supportive therapies do not sufficiently manage the disease. The needs differ for each patient with Rett syndrome (and can change over time); therefore, the goals of treatment are individualized. In general, there is a need for treatments that promote better HRQoL and improve issues with communication, motor skills, cognition, behaviour, feeding, and sleep, as well as address seizures. Therapies that better support families, as well as patient and caregiver daily activities, are also important.
Place in Therapy
The experts stated that patients would not have to exhaust standard treatments before accessing trofinetide, and once a diagnosis of Rett syndrome is confirmed, trofinetide would be used as a first-line therapy, along with other drugs and nonpharmacological supportive therapies to manage symptoms, as outlined in the care management guidelines.7
Patient Population
The clinical experts stated that at this time, there are no specific characteristics or markers that would identify the group of patients who would benefit more from trofinetide. Despite available evidence from the LAVENDER study (which enrolled female patients aged 5 years to 20 years with typical Rett syndrome who have a confirmed, disease-causing MECP2 variant), the experts indicated that patients aged 2 years and older with a confirmed clinical diagnosis of classic Rett syndrome (with or without a disease-causing MECP2 variant) or atypical Rett syndrome (with a disease-causing MECP2 variant) would be suitable candidates for trofinetide. The clinicians also noted that there is a small part of the population that is clinically diagnosed with classic Rett syndrome but does not have an MECP2 variant; if these patients are diagnosed by Rett syndrome experts, they may also be candidates for trofinetide. However, the experts agreed they would not treat patients with an atypical Rett syndrome diagnosis without an MECP2 variant. The experts explained that Rett syndrome may be suspected clinically in patients as young as 2 years, and that once a patient has a confirmed diagnosis, it would be reasonable to start treatment. It is less certain what the treatment effects and harms would be in individuals outside of the LAVENDER population (i.e., males or patients who have atypical Rett syndrome without a disease-causing MECP2 variant). One expert also highlighted that they would not treat patients with trofinetide who weigh less than 9 kg as there are added concerns related to diarrhea, hydration, and nutrition.
One expert suggested that there may be preference for using trofinetide in patients who have retained functions because there could be greater potential for preventing disease progression (and patients who have lost functions may not regain them), while other experts indicated they may treat with trofinetide, regardless of disease severity.
Assessing the Response Treatment
Clinicians routinely record developmental and functional history to track changes in a patient and to help determine if there are clinical responses to treatment. The family’s perspective, caregiver reports (including the primary care physician, specialists, therapists, and educational assistants in the school), and physician symptom assessments were highlighted as being valuable for providing detailed insight into a patient’s day-to-day wellness and needs that lead to changes in care management. The experts stated that they would assess whether a drug was beneficial in a meaningful way to the patient and caregivers and if HRQoL improved while being balanced with the adverse effects patients can experience.
The clinical experts confirmed that there are currently no standard outcomes used in clinics across Canada to measure treatment response in patients with Rett syndrome, and that the outcomes used in the LAVENDER study are not used in clinical practice. One expert has used the RSBQ in practice but noted that this is not common among clinicians at this time.
Discontinuing Treatment
When deciding to discontinue treatment, the experts stated that adverse effects (specifically vomiting, diarrhea, dehydration, and weight loss), hospitalization due to adverse effects, and the impact on a patient’s HRQoL are factors to consider and discuss with caregivers. There were different perspectives on deciding when to discontinue treatment, with 1 expert suggesting an adequate trial of trofinetide at the target dose for 3 months (based on the duration of the LAVENDER study) and others suggesting a trial of 6 months to 12 months at the target dose to avoid premature discontinuation, in case benefits are only observed after at least 3 months of use. The experts also suggested that if there was no improvement or clinical change despite an adequate trial of trofinetide at the target dose, it would be reasonable to have a trial off the medication. If the patient experiences an abrupt decline in the patient’s health during the trial off the medication, the experts suggested restarting treatment. The experts noted this approach could also be used for patients outside of the LAVENDER population (i.e., males, patients older than 20 years). Overall, they agreed that most families would not continue treatment with trofinetide if they believed that the patient was not benefiting from the treatment.
The experts noted that they may follow-up with patients more often when starting a new treatment or if the disease is not stable. Additionally, they noted that consultation with the patient’s primary care team, with whom the patient and caregivers have more regular interactions (particularly for monitoring and treating adverse effects), is important. They suggested that patients starting trofinetide may visit Rett syndrome specialists 1 month after initiation, 3 months after initiation, and then less frequently if the disease is stable.
Prescribing Considerations
The experts indicated that Rett syndrome specialists, as well as pediatricians and neurologists with expertise in Rett syndrome, would prescribe trofinetide at first because it is a new medication. However, it was noted that as experience with the drug increases, it may be possible for other physicians, in consultation with Rett syndrome experts, to prescribe the drug, which would improve access to patients in remote areas.
Clinician Group Input
This section was prepared by the review team based on the input provided by 1 clinician group.
The Canadian Rett Syndrome Consortium (including Acadia Pharmaceuticals Inc. advisory board members for trofinetide) provided input for this review. The input was from 6 clinicians, including pediatric neurologists, developmental pediatricians, and medical geneticists in Canada.
The clinician group stated that, in Canada, there is currently no approved treatment for Rett syndrome (aside from trofinetide), and that existing medications focus on the management of disease symptoms only. The group stated that no medications to date have targeted the underlying biology, the course of the disease, or the deteriorating developmental trajectory that is characteristic of Rett syndrome. According to the clinician group, trofinetide is unique in this space and a first-in-class drug, although its exact mechanism of action is unknown. The clinician group input was consistent with the clinical experts consulted for this review, noting that trofinetide would be used as a first-line treatment and that other medications may be added to address associated symptoms tailored to an individual patient's needs.
Outcomes used to determine whether a patient is responding to trofinetide would be reported by caregivers. A clinically meaningful response to treatment would include improvements in communication, alertness, engagement, and respiratory symptoms; the ability to move independently; and decreases in repetitive movements or stereotypies. The clinician group anticipates that trofinetide will initially be prescribed in specialized medical centres on an outpatient basis and, over time, with education and experience, by community physicians, such as pediatricians or internists. According to the clinician group, considerations for discontinuing trofinetide include no improvement in symptoms after 6 months to 12 months of therapy and persistent moderate-to-severe diarrhea or vomiting with weight loss that are not controlled with appropriate medications or a lower dose of trofinetide.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by for this review are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The sponsor considered a basket of drugs (for symptomatic treatment only) to be relevant comparators:
| Comment from the drug program to inform CDEC deliberations. |
Considerations for initiation of therapy | |
Rett syndrome is an X-linked neurodevelopmental disorder. It is estimated that there are 600 to 900 patients in Canada with Rett syndrome. It mostly affects females and is the most common genetic cause of neurologic disability in girls and women, after Down syndrome. The disease is a multisystem disorder with heterogenous symptoms. The sponsor noted that diagnosis is typically based on clinical evaluation and consensus diagnostic criteria for Rett syndrome, rather than genetic confirmation of the disease. Diagnosis typically occurs in patients around 3 years of age. Loss-of-function mutations in MECP2 are thought to play a role in Rett syndrome. Approximately 90% of patients have an identifiable MECP2 mutation. However, an MECP2 mutation is neither necessary nor sufficient for the diagnosis of Rett syndrome. Is MECP2 testing readily available and reimbursed in all jurisdictions? Should genetic testing to confirm MECP2 mutations be considered part of the initiation criteria? | Information from the public drug plans indicated that the availability of MECP2 testing varies across jurisdictions in Canada (e.g., it is not available in New Brunswick or The Yukon). According to the clinical experts consulted for this review, depending on where a patient lives, testing is conducted locally and may be sent outside the province to be completed. The experts agreed that the diagnosis of Rett syndrome is often made on clinical grounds, although it would be reasonable to include genetic testing to confirm an MECP2 variant as part of the initiation criteria for trofinetide. However, the clinicians stated that if a patient is followed by a Rett syndrome specialist and does not have a confirmed disease-causing MECP2 variant but meets the diagnostic criteria of classic Rett syndrome (not atypical disease), trofinetide should be reimbursed. They indicated that this would be a small number of patients. The experts also noted that patients cared for in Rett syndrome clinics often have already undergone genetic testing. |
The LAVENDER study included only female patients. Rett syndrome is X-linked, and while it is more common in females, it can occur in males. Males tend to have more severe disease and have greater clinical severity than females and may have shortened life expectancy. The indication does not specify treatment for females only. Should treatment be limited to female patients (as per the LAVENDER study) or would male patients also be considered? | The experts expect that reimbursement for treatment with trofinetide for Rett syndrome would be limited to female patients, based on the available RCT evidence (the LAVENDER study), which excluded males with Rett syndrome. |
Considerations for continuation or renewal of therapy | |
Clinical trials used the following coprimary end points:
Are these validated scoring tools for use in Rett syndrome to determine response to drug therapy? Do positive results on these scoring tools correlate with clinically meaningful outcomes? | RSBQ and CGI-I are both validated tools in Rett syndrome; however, RSBQ is a diagnostic tool used to differentiate between Rett syndrome and severe intellectual disability in females and is not validated for use as a measure of treatment effect. The clinical experts stated that neither of these tools is regularly used in Rett syndrome clinics; there are currently no standardized instruments for measuring response to treatment. According to expert opinion, a between-group difference of 3 points on the RSBQ scale and 1 point on the CGI-I scale would be clinically meaningful. |
The sponsor has suggested the following renewal criterion:
Note that this does not reference any specific scoring tool to measure treatment response. What is the most appropriate instrument to measure treatment response in clinical practice to base drug renewal decisions on? | Clinicians routinely record developmental and functional history to track changes in a patient and to help determine if there is a clinical response to treatment. The family’s perspective, caregiver reports (including the primary care physician, specialists, therapists, and educational assistants in the school), and symptom history were highlighted as being valuable for providing detailed insight into a patient’s day-to-day wellness and needs that lead to changes in care management. The clinical experts indicated that because there are no standardized instruments used in clinical practice, clinicians use caregiver and family reports, as well as their own judgment, to assess treatment response and continuation of therapy. The experts stated that they would assess, with families, whether the drug is beneficial in a meaningful way to patients and caregivers and whether HRQoL improved while being balanced with the adverse effects. If use of the drug at the target dose for at least 3 months, and up to 12 months, does not demonstrate a meaningful benefit to patients and caregivers, an off-drug trial would be discussed with the family. |
Considerations for discontinuation of therapy | |
In the LAVENDER study, 19% of patients receiving trofinetide had adverse reactions that led to withdrawal from the study. The most common side effect resulting in discontinuation was diarrhea. Rett syndrome has 4 stages of disease progression:
Considering that Rett syndrome is a progressive disease, is trofinetide effective in later stages of disease? Was there any subgroup analysis based on age group and response? | The clinicians were unable to confirm whether trofinetide is effective in later stages of the disease due to lack of evidence, specifically in patients with late-stage disease. The experts also noted that there is variability in progression as patients age, and that some patients may have stable disease whereas others continue to progress. Subgroup analyses based on age groups (5 years to 11 years, 12 years to 16 years, and 17 years to 20 years) were conducted in the LAVENDER study. In general, results of the subgroup analyses supported those of the primary analyses for the coprimary end points (i.e., mean change from baseline in RSBQ and CGI-I scores was greater in the trofinetide group than the placebo group for all age groups). However, based on the forest plots for the coprimary outcomes, the 95% CI for most age groups appeared to touch or include the null (suggesting the possibility of no difference between trofinetide and placebo): age groups 12 to 16 years and 17 to 20 years for RSBQ and all age groups for CGI-I. Additionally, the subgroup analyses were not controlled for multiplicity and were likely too small and underpowered to provide conclusive evidence to inform reimbursement decisions. |
Considerations for prescribing of therapy | |
Clinical trials dosing:
Trofinetide, 200 mg/mL, is a solution that can be administered orally or via gastrostomy tube. Dosing is weight-based, given twice daily. | Comment from the drug program to inform CDEC deliberations. |
The sponsor suggested that the drug must be prescribed by clinicians with expertise in the diagnosis and management of Rett syndrome. There are 5 Rett syndrome clinics in Canada, located in Vancouver (British Columbia), Calgary (Alberta), London (Ontario), Toronto (Ontario), and Montreal (Quebec). Does this specialty exist outside of the 5 Rett syndrome clinics? | The clinical experts confirmed that there are physicians who manage Rett syndrome outside of the 5 listed clinics. The experts noted that in remote areas, patients have limited access to specialists and may be mostly managed by family doctors, pediatricians, or neurologists, depending on who is available in the local community. |
Generalizability | |
Clinical trials were conducted in patients 2 years of age and older (the DAFFODIL study included patients aged 2 years to 5 years). Are patients diagnosed before the age of 2 years? Would there be interest in initiating treatment before 2 years of age? | The experts confirmed that because genetic testing is available and people are more aware of Rett syndrome, the disease can be diagnosed before a patient is 2 years old. Because of that, the experts expect that there could be interest in the treatment of patients younger than 2 years with a confirmed diagnosis; however, that would be off-label use. |
Care provision issues | |
Trofinetide needs to be refrigerated, but not frozen. The product monograph recommends discarding any unused solution in the bottle after 14 days. This may contribute to drug wastage and, therefore, higher costs. | Comment from the drug program to inform CDEC deliberations. |
A slow titration to target dose is recommended (4 weeks to 8 weeks). It is suggested to reduce, interrupt, or discontinue dosage if diarrhea occurs. | Comment from the drug program to inform CDEC deliberations. |
System and economic issues | |
The submitted price for a 450 mL bottle of 200 mg/mL (90,000 mL) bottle is $13,714.11. Dosing is weight-based so the cost of therapy increases as patients grow. The total budget impact over 3 years is $136,525,194. | Comment from the drug program to inform CDEC deliberations. |
CDEC = Canadian Drug Expert Committee; CGI-I = Clinical Global Impressions-Improvement; CI = confidence interval; HRQoL = health-related quality of life; RCT = randomized controlled trial; RSBQ = Rett Syndrome Behaviour Questionnaire.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of trofinetide, 200 mg/mL, solution for oral or gastrostomy tube administration in the treatment of Rett syndrome in adults and pediatric patients aged 2 years and older and weighing at least 9 kg. The focus will be placed on comparing trofinetide to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of trofinetide is presented in 4 sections, with a critical appraisal of the evidence included at the end of each section. The first section, the Systematic Review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. An assessment of the certainty of the evidence in this first section, which uses the GRADE approach, follows the critical appraisal of the evidence. The second section includes sponsor-submitted OLE studies. The third section includes indirect evidence from the sponsor, of which none was identified or submitted for review. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 pivotal RCT identified in the systematic review
2 OLE studies
2 additional studies addressing gaps in evidence.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
One phase III, double-blind, placebo-controlled RCT was included in the systematic review: LAVENDER (N = 187).11 Characteristics of the LAVENDER study are summarized in Table 6. The study was conducted to evaluate the efficacy and safety of trofinetide in female patients with Rett syndrome. The study consisted of a 3-week screening period and a 12-week treatment period, after which patients could enrol directly into the first of 2 OLE studies (the 40-week LILAC study followed by the 32-month LILAC-2 study); those who did not enrol were followed for safety monitoring for 30 days.28,29
Table 6: Details of the Study Included in the Systematic Review
Detail | LAVENDER |
|---|---|
Designs and populations | |
Study design | Phase III, double-blind, placebo-controlled RCT |
Locations | 21 sites in the US |
Patient enrolment dates | Start date: October 29, 2019 End date: October 28, 2021 |
Randomized (N) | N = 187
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Trofinetide, orally or via gastrostomy tube for 12 weeks, based on body mass:
|
Comparator | Matching placebo |
Study duration | |
Screening phase | Up to 3 weeks |
Treatment phase | 12 weeks |
Follow-up phase | Entry into a 40-week OLE study (the LILAC study), or a 30-day follow-up period for patients who did not continue into the OLE study |
Outcomes | |
Primary end point | Coprimary
|
Secondary and exploratory end points | Secondary
Exploratory
|
Publication status | |
Publications | |
CGI-I = Clinical Global Impressions-Improvement; CGI-S = Clinical Global Impressions-Severity; CSBS-DP-IT = Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler; ECG = electrocardiogram; ICND = Impact of Childhood Neurologic Disability; IGF-1 = insulin-like growth factor 1; OLE = open-label extension; QTcF = corrected QT interval using Fridericia’s correction method; RCT = randomized controlled trial; RSBQ = Rett Syndrome Behaviour Questionnaire; RTT-AMB = Rett Syndrome Clinician Rating of Ambulation and Gross Motor Skills; RTT-CBI = Rett Syndrome Caregiver Burden Inventory; RTT-COMC = Rett Syndrome Clinician Rating of Ability to Communicate Choices; RTT-CSS = Rett Syndrome Clinical Severity Scale; RTT-HF = Rett Syndrome Clinician Rating of Hand Function; RTT-VCOM = Rett Syndrome Clinician Rating of Verbal Communication.
aClassic or typical Rett syndrome diagnostic criteria include the following:6 Required for typical or classic Rett syndrome: a period of regression followed by recovery or stabilization; all main criteria and all exclusion criteria; and supportive criteria are not required, although are often present in typical Rett syndrome. Main criteria include partial or complete loss of acquired purposeful hand skills; partial or complete loss of acquired spoken language (loss of acquired language is based on the best acquired spoken language skill, not strictly on the acquisition of distinct words or higher language skills). Thus, an individual who had learned to babble but then loses this ability is considered to have a loss of acquired language); gait abnormalities, such as impaired (dyspraxic) or absence of ability; and stereotypic hand movements, such as hand wringing or squeezing, clapping or tapping, mouthing, and washing or rubbing automatisms. Exclusion criteria include: brain injury secondary to trauma (perinatally or postnatally), neurometabolic disease, or severe infection that causes neurological problems There should be clear evidence [neurological or ophthalmological examination and MRI or CT] that the presumed injury directly resulted in neurological dysfunction); and grossly abnormal psychomotor development in the first 6 months of life (grossly abnormal to the point that normal milestones [acquiring head control, swallowing, or developing a social smile] are not met. Mild generalized hypotonia or other previously reported subtle developmental alterations during the first 6 months of life are common in patients with Rett syndrome and do not constitute an exclusionary criterion). Supportive criteria include breathing disturbances when awake; bruxism when awake; impaired sleep pattern; abnormal muscle tone; peripheral vasomotor disturbances; scoliosis or kyphosis; growth retardation; small, cold hands and feet; inappropriate laughing or screaming spells; diminished response to pain; and intense eye communication or eye pointing.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Populations
Inclusion and Exclusion Criteria
Eligible patients included females aged 5 years to 20 years, weighing at least 12 kg, who must have been diagnosed with classic or typical Rett syndrome (based on the Rett syndrome diagnostic criteria 20106), have a documented disease-causing MECP2 variant, and symptoms and treatment must have been stable. Individuals were not eligible if they had been treated with growth hormone or insulin in the 12 weeks before baseline or had a history of or current clinically significant medical conditions other than Rett syndrome.
Interventions
Patients were randomized in a 1:1 ratio to receive either trofinetide (n = 93) (weight-based banded dosing) or matching placebo (n = 94) for 12 weeks. Randomization was stratified by age group (5 years to 10 years, 11 years to 15 years, and 16 years to 20 years) and baseline RSBQ severity (< 35 total score and ≥ 35 total score). At least 12 patients were required to be randomized to each age group.
Study medication was administered either orally or by gastrostomy tube, twice a day, with at least 8 hours between doses. Patients who were unable to tolerate the assigned dose were treated with the highest dose they could tolerate, which could not be less than half the assigned dose. After the week 6 visit, the dose could not be changed.
Prohibited medications included insulin-like growth factor 1, growth hormone, and insulin. Psychoactive concomitant medications and nonpharmacological somatic treatments that have an effect on the central nervous system were to remain stable throughout the trial.
Based on consultation with gastroenterologists, recommendations for preventing and managing diarrhea AEs were made to the investigators and were implemented partway through the study. The recommendations included adjusting or discontinuing laxative medications that are commonly used to treat Rett syndrome–associated constipation, starting fibre supplements and antidiarrheal medication, and dose reduction or interruption of trofinetide, if necessary.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, and descriptions of the outcome measures are provided in Table 8. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence, as well as on any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, we selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
The following considerations went into the selection of the outcomes summarized in the report and assessed using GRADE:
The use of a disease-specific instrument (e.g., RSBQ, which is a caregiver-reported measure), coupled with a clinician-rated assessment if the main outcome was not clinician-reported.
Maintaining basic skills (e.g., communication, coordination, and motor skills) and preventing regression were important outcomes to caregivers and clinicians. Expert input noted that many patients do not have verbal communication skills or hand function, making these less clinically relevant for measuring treatment effect.
A patient’s HRQoL and caregiver’s well-being were identified as priorities by all groups who provided input. There was no comprehensive measure of HRQoL used in the study.
Reduction in the severity and/or frequency of symptoms (e.g., seizures, gastrointestinal and breathing issues) were key outcomes noted as being important to caregivers and clinicians but were not measured in the study.
Harms of treatment were also noted as important in the input provided by patient groups, clinician groups, and clinical experts.
Table 7: Outcomes Summarized From the Study Included in the Systematic Review
Outcome measure | Time point | End point |
|---|---|---|
RSBQ total score | Change from baseline to week 12 | Coprimarya |
CGI-I score | At week 12 | Coprimarya |
RTT-COMC | Change from baseline to week 12 | Secondary |
CSBS-DP-IT Checklist social composite score | Change from baseline to week 12 | Key secondarya |
RTT-HF | Change from baseline to week 12 | Secondary |
RTT-AMB | Change from baseline to week 12 | Secondary |
RTT-VCOM | Change from baseline to week 12 | Secondary |
Overall quality-of-life rating | Change from baseline to week 12 | Secondary |
RTT-CBI | Change from baseline to week 12 | Secondary |
CGI-I = Clinical Global Impressions-Improvement; CSBS-DP-IT = Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler; RSBQ = Rett Syndrome Behaviour Questionnaire; RTT-AMB = Rett Syndrome Clinician Rating of Ambulation and Gross Motor Skills; RTT-CBI = Rett Syndrome Caregiver Burden Inventory; RTT-COMC = Rett Syndrome Clinician Rating of Ability to Communicate Choices; RTT-HF = Rett Syndrome Clinician Rating of Hand Function; RTT-VCOM = Rett Syndrome Clinician Rating of Verbal Communication.
aStatistical testing for these end points was adjusted for multiple comparisons.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Rett Syndrome Behaviour Questionnaire
The RSBQ is completed by the caregiver to assess a range of neurobehavioural symptoms that are known to be impaired in patients with Rett syndrome.12 The questionnaire contains 45 items, of which 38 are grouped into 8 domains or subscales (general mood, breathing problems, hand behaviour, face movements, body rocking or expressionless face, nighttime behaviours, fear or anxiety, and walking or standing). The following 7 items are uncategorized but contribute to the total score: spells of laughter for no apparent reason, hand wounds resulting from repetitive hand movements, gaze shift with slow horizontal turn of head, repetitive hand movements apart, appears isolated, teeth grinding, and vacant staring spells.11 Items are rated on a Likert scale for symptom occurrence and frequency, as 0 (not true), 1 (somewhat or sometimes true), or 2 (very true), and a total score is calculated as the sum of the scores for all 45 items, ranging from 0 to 90 points.11 Higher scores indicate that symptoms are more frequent. For patients with Rett syndrome, item 31 (uses eye gaze to convey feelings, needs, and wishes) is reversed when calculating the subscore and total score, as the score of 2 (very true) reflects a better outcome. The RSBQ was assessed at screening, baseline, and visit 3, visit 4, and visit 5. An MID for the RSBQ was not identified in the literature.
Clinical Global Impressions-Improvement Score
The CGI-I is used to rate how the patient’s illness has improved or worsened compared to baseline.12 The scale includes 1 (very much improved), 2 (much improved), 3 (minimally improved), 4 (no change), 5 (minimally worse), 6 (much worse), and 7 (very much worse). The CGI-I was assessed at visit 3, visit 4, and visit 5.
Disease-specific anchors were used during the assessment to guide the assessor.12 The anchor descriptions included characterization of impairment levels across core Rett syndrome signs and symptoms. An MID for the CGI-I specific to patients with Rett syndrome was not identified in the literature.
Rett Syndrome–Specific Clinician Assessments
The study included 4 disease-specific, clinician-completed instruments that assess a patient’s capabilities: RTT-COMC (ability to communicate choices, which includes nonverbal means), RTT-HF (ability to use hands for functional purposes), RTT-AMB (ability to sit, stand, and ambulate), and RTT-VCOM (ability to communicate verbally).12 Each of these tools uses a Likert scale that ranges from 0 (normal functioning) to 7 (most severe impairment) and is a standalone measure (i.e., is not combined for a total score). The 4 tools were assessed at baseline and at visit 3, visit 4, and visit 5. MIDs for any of the Rett syndrome–specific clinician assessments were not identified in the literature.
Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler Checklist
The CSBS-DP-IT Checklist is completed by the caregiver to assess communication and prelinguistic skills in children aged 1 year to 2 years, and can be used for older children with developmental delay.12 The tool has 24-items, and ratings include 0 (not yet), 1 (sometimes), or 2 (often). Three composite scores assess 7 skills areas: social composite (emotion and eye gaze, communication rate and function, and gestures), speech composite (sounds and words), and symbolic composite (understanding and object use).
Previous studies of young patients with Rett syndrome have used the first 16 items of the CSBS-DP-IT Checklist to calculate the social composite score,31,32 and previous studies of developmental disorders have indicated that the score is sensitive to changes in behavioural interventions.33,34 The social composite score consists of items 1 to 13, and scores range from 0 to 26, with higher scores indicating better social communication development. The CSBS-DP-IT Checklist social composite score was assessed at baseline and at visit 3, visit 4, and visit 5. An MID for the CSBS-DP-IT Checklist specific to patients with Rett syndrome was not identified in the literature.
Overall Quality-of-Life Rating
The patient’s overall quality of life was rated from 1 (poor) to 6 (excellent). The quality-of-life rating was assessed at baseline and at visit 5. An MID specific to patients with Rett syndrome was not identified in the literature.
Rett Syndrome Caregiver Burden Inventory
The RTT-CBI is completed by the caregiver to assess the burden of caring for the patient on their daily life in 4 areas: physical, emotional, and social burden, and time dependence.12 Caregivers rate how often a statement describes their feelings or experiences, with frequency rated on a Likert scale of 0 (never), 1 (rarely), 2 (sometimes), 3 (frequently), and 4 (nearly always). Items 1 to 24 yield a total burden score from 0 to 96, and items 25 and 26 make up the optimism index (which were not used in the analyses). Higher scores indicate a greater burden on the caregiver. The RTT-CBI was assessed at baseline and at visit 5. An MID for the RTT-CBI was not identified in the literature.
Safety
Safety outcomes included the proportion of patients who reported TEAEs (coded using the Medical Dictionary for Regulatory Activities, version 24.0), SAEs, withdrawals from treatment due to TEAEs, and death. Based on clinical expert opinion, diarrhea and vomiting were deemed relevant to the current review of trofinetide.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
RSBQ | A 45-item caregiver-completed assessment with 8 subscales that evaluate various neurobehavioural symptoms, rated from 0 (not true) to 2 (very true). A total score is calculated as the sum of the scores for all 45 items, and ranges from 0 to 90 points, with higher scores indicating that symptoms are more frequent.11 | Construct, convergent, and discriminant validity have been demonstrated in studies of patients with Rett syndrome.35,36 Internal consistency is high for the total score and 8 subscales, and interrater and test-retest reliability scores are good in studies of patients with Rett syndrome.37 | No MID was identified from the literature for this disease area. |
CGI-I | A 7-point clinician-rated scale that ranges from 1 to 7 used to assess improvement or worsening compared to baseline. Higher scores indicate worsening disease. | Psychometric assessments for the CGI-I have not been validated in studies of patients with Rett syndrome.38 | No MID was identified from the literature for this disease area. |
Rett syndrome–specific clinician assessments | Four new, clinician-rated instruments assessed a patient’s capabilities: RTT-COMC (ability to communicate choices, which includes nonverbal means), RTT-HF (ability to use hands for functional purposes), RTT-AMB (ability to sit, stand, and ambulate), and RTT-VCOM (ability to communicate verbally). Scales for each instrument range from 0 (normal functioning) to 7 (most severe impairment). Scores were not combined. | Moderate interrater reliability was reported for the RTT-HF.39 Psychometric assessments have not been validated in studies of patients with Rett syndrome. | No MIDs were identified from the literature for this disease area. |
CSBS-DP-IT Checklist social composite score | A 24-item caregiver-rated assessment for communication and prelinguistic skills in young children and older children with developmental delay. Each item is rated on a scale as 0 (not yet), 1 (sometimes), or 2 (often). The social composite consists of items 1 to 13, and scores range from 0 to 26 points, with higher scores indicating better social communication development. | A psychometric assessment has not been validated in studies of patients with Rett syndrome. The instrument has shown concurrent and convergent validity, as well as test-retest reliability, in a population with motor impairments and in various languages.40-42 | No MID was identified from the literature for this disease area. |
Overall quality of life rating | The patient’s overall quality of life is rated from 1 (poor) to 6 (excellent). | A psychometric assessment has not been validated in studies of patients with Rett syndrome. | No MID was identified from the literature for this disease area. |
RTT-CBI | A caregiver-completed questionnaire that assesses 4 different aspects of burden (physical, emotional, and social burden, and time dependence) and uses a 5-point Likert scale to rate the frequency with which each statement describes the caregivers’ feelings or experiences, as 0 (never), 1 (rarely), 2 (sometimes), 3 (frequently), or 4 (nearly always). The 24 items yield a total score from 0 to 96 points, with higher scores indicating a higher frequency of feelings of burden. | Evidence of construct validity, high internal consistency, and good test-retest reliability was reported in a study of caregivers of patients with Rett syndrome.18 | No MID was identified from the literature for this disease area. |
CGI-I = Clinical Global Impressions-Improvement; CSBS-DP-IT = Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler; MID = minimal important difference; RSBQ = Rett Syndrome Behaviour Questionnaire; RTT-AMB = Rett Syndrome Clinician Rating of Ambulation and Gross Motor Skills; RTT-CBI = Rett Syndrome Caregiver Burden Inventory; RTT-COMC = Rett Syndrome Clinician Rating of Ability to Communicate Choices; RTT-HF = Rett Syndrome Clinician Rating of Hand Function; RTT-VCOM = Rett Syndrome Clinician Rating of Verbal Communication.
Statistical Analysis
Statistical analysis methods used in the LAVENDER study are summarized in Table 9.
Table 9: Statistical Analysis of Efficacy End Points in the LAVENDER Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
RSBQ total score | MMRM | Fixed effects: Effects for treatment group, age group (5 years to 10 years, 11 years to 15 years, and 16 years to 20 years), baseline RSBQ severity (total score < 35 and total score ≥ 35), visit, and treatment-by-visit interaction Covariates: Baseline total score, and baseline total score-by-visit interaction Random effect: Patients | MAR | Multiple imputation:
Other:
|
CGI-I score | MMRM | Adjustment factors are the same as those for RSBQ total score | MAR | The same as those for RSBQ total score |
RTT-COMC | MMRM | Effects: Effects for treatment group, age group (5 years to 10 years, 11 years to 15 years, and 16 years to 20 years), baseline RSBQ severity (total score < 35 and total score ≥ 35), visit, treatment-by-visit interaction, baseline score, and baseline score-by-visit interaction | MAR | None |
CSBS-DP-IT Checklist social composite score | MMRM | Adjustment factors are the same as those for RSBQ total score | MAR | None |
RTT-HF | MMRM | Adjustment factors are the same as those for RTT-COMC | MAR | None |
RTT-AMB | MMRM | Adjustment factors are the same as those for RTT-COMC | MAR | None |
RTT-VCOM | MMRM | Adjustment factors are the same as those for RTT-COMC | MAR | None |
Overall quality-of-life rating | ANCOVA | Fixed effects: Treatment group, age group (5 years to 10 years, 11 years to 15 years, and 16 years to 20 years), and baseline RSBQ severity (total score < 35 and total score ≥ 35) Covariates: Baseline value | MAR | None |
RTT-CBI | ANCOVA | Adjustment factors are the same as those for the overall quality-of-life rating | MAR | None |
ANCOVA = analysis of covariance; CGI-I = Clinical Global Impressions-Improvement; CSBS-DP-IT = Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler; MAR = missing at random; MMRM = mixed model for repeated measures; MNAR = missing not at random; RSBQ = Rett Syndrome Behaviour Questionnaire; RTT-AMB = Rett Syndrome Clinician Rating of Ambulation and Gross Motor Skills; RTT-CBI = Rett Syndrome Caregiver Burden Inventory; RTT-COMC = Rett Syndrome Clinician Rating of Ability to Communicate Choices; RTT-HF = Rett Syndrome Clinician Rating of Hand Function; RTT-VCOM = Rett Syndrome Clinician Rating of Verbal Communication.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sample Size and Power Calculation
The sample size was calculated for the coprimary efficacy end points as a family of 2 hypothesis tests at an overall 2-sided significance level of 0.05. A sample size of 174 patients, randomized in a 1:1 ratio to trofinetide or placebo, was estimated to provide at least 90% power to detect a difference for both coprimary end points, assuming the following treatment differences, estimated from phase II data: –4.4 (SD = 8) for the mean change from baseline to week 12 in the RSBQ total score and –0.5 (SD = 0.7) for the CGI-I mean score at week 12. With an estimated discontinuation rate of up to 5%, enrolling approximately 184 patients would result in 174 patients completing the study.
Statistical Testing
The coprimary outcomes — change from baseline to week 12 in the RSBQ total score and the CGI-I score at week 12 — were analyzed using the direct likelihood mixed model for repeated measures (MMRM) method, which included the baseline stratification variables from the randomization process as covariates (i.e., baseline total score and baseline total score-by-visit interaction). An unstructured covariance matrix was used, and the Kenward-Roger approximation was used to adjust the denominator degrees of freedom. If the model failed to converge using the unstructured covariance matrix, the following covariance structures were modelled, in the order given (i.e., from least parsimonious to most parsimonious): heterogeneous Toeplitz, heterogeneous compound symmetry, heterogeneous autoregressive, Toeplitz, compound symmetry, and autoregressive, variance components. The first covariance structure that allowed for convergence was selected for the final model. Summary statistics, least squares means (LSMs), the between-group difference in LSM, corresponding 95% CI, and P value were presented at each postbaseline visit.
Multiple Testing Procedure
The coprimary end points and key secondary end point (CSBS-DP-IT Checklist social composite score) were analyzed in a hierarchical manner, providing control of the family-wise error rate at the nominal alpha of 5%. The coprimary hypotheses formed 1 family and the key secondary hypothesis formed 1 family. The family of the key secondary hypothesis was tested (at a 2-sided alpha of 5%) if, and only if, trofinetide was superior to placebo with respect to both coprimary efficacy end points. The coprimary hypotheses were tested (at a 2-sided alpha of 5%) without multiplicity adjustment because both hypotheses in the family must be rejected to move forward to the next family.
Data Imputation Methods
For the RSBQ total score, if there were 9 items or fewer missing, the total score was calculated based on the mean of nonmissing items multiplied by 45 and rounded to the nearest integer. When there were 10 items or more missing, the total score was considered missing.
The coprimary efficacy analyses were performed assuming MAR data, using the direct likelihood–based MMRMs and missing scores (after any imputation of individual missing items), were not imputed.
Sensitivity analyses assessing the impact of missing data were assessed with the following imputation techniques.
Pattern-mixture models assumed data missing not at random (MNAR). Missing data were imputed based on the distribution of placebo group responses over time to account for treatment discontinuations and missing assessments. The underlying assumption was that patients with missing data due to early withdrawal evolved in the same way as patients in the placebo group who remained in the study. The following steps were taken for imputation:
Nonmonotone (intermediate) missing data at baseline (not applicable for CGI-I score) and at week 2, week 6, and week 12 were multiply imputed using the Markov chain Monte Carlo method to create 50 monotone datasets. The imputation models included the same MMRM model predictors as previously described.
Monotone missing data were imputed using a parametric, sequential linear regression method, in which the missing data were imputed only on data from the placebo group. A single imputation was performed sequentially at each visit for each of the 50 imputed datasets, with the same predictors as for the MMRM model described previously.
Values for the change from baseline to each postbaseline visit were then calculated and analyzed for each of the 50 fully imputed datasets using the analysis of covariance model, with treatment group, baseline RSBQ severity, and age groups as fixed factors, and baseline RSBQ total score or CGI-S score as a covariate.
Missing data due to COVID-19: Missing data after withdrawal due to COVID-19 were assumed to be MAR, whereas missing data after withdrawal not due to COVID-19 were assumed to be MNAR and to evolve in the same way as patients in the placebo group who remained in the study. After the monotone missing datasets were imputed, patients receiving trofinetide who did not have COVID-19–related withdrawal had their treatment group temporarily switched to placebo, then missing data due to COVID-19 withdrawal was imputed using a parametric, sequential linear regression method, based on data from the respective treatment group. Afterward, patients in the trofinetide group had their original treatment restored. The analysis was then performed in the same way as the pattern-mixture models described previously.
Sensitivity Analyses
A sensitivity analysis was performed to assess the impact of COVID-19 on data collection. With this method, the MMRM model described for the coprimary end points had the baseline RSBQ severity strata from interactive response technology replaced with a derived baseline RSBQ severity strata score, in which item 31 (uses eye gaze to convey feelings, needs, and wishes) was reversed. A supportive analysis of the coprimary end points was analyzed, as it was in the primary analysis, using the per-protocol analysis set.
Subgroup Analyses
Based on clinical expert opinion, the prespecified subgroup for age groups (5 years to 11 years, 12 years to 16 years, and 17 years to 20 years) was deemed to be potentially relevant to clinical decision-making. Other subgroups (baseline RSBQ severity and MECP2 variant) were deemed to be not relevant to the review, as they were felt to be exploratory in nature and not based on clear clinical evidence that the characteristic would identify a subpopulation of patients who might benefit more from treatment.
Secondary Outcomes of the LAVENDER Study
For the CSBS-DP-IT Checklist social composite score, if there were 2 items or fewer missing, the social composite score was calculated based on the mean of nonmissing items multiplied by 13 and rounded to the nearest integer. When there were 3 items or more missing, the score was considered missing. Calculations for the RTT-CBI score were handled in a similar way if there were 4 items or fewer missing and the mean was multiplied by 24.
The key secondary end point (CSBS-DP-IT Checklist social composite score) and select other secondary end points (RTT-COMC, RTT-HF, RTT-AMB, and RTT-VCOM) were analyzed using the MMRM model described for the coprimary end points. Overall quality-of-life rating and RTT-CBI were summarized by treatment group and analyzed using the analysis of covariance model. Summary statistics (observed and change from baseline), LSM, between-group treatment difference in LSM, corresponding 95% CI, and P value were presented.
Data imputation methods for the key secondary end point were the same as for the coprimary end points, and analyses for the other secondary efficacy end points were performed using the observed values, without any imputation.
Analysis Populations
Analysis populations included in the LAVENDER study are summarized in Table 10.
Table 10: Analysis Populations in the LAVENDER Study
Population | Definition | Application |
|---|---|---|
Randomized analysis set | All patients who were randomized and classified according to the randomized treatment assignment. | Baseline demographic and disease characteristics |
Safety analysis set | All randomized patients who received ≥ 1 dose of the study drug and analyzed according to the actual treatment received. | Safety evaluation |
Full analysis set | Patients who were randomized, received ≥ 1 dose of study drug, and had both a baseline value and ≥ 1 postbaseline value for the RSBQ total score or had ≥ 1 CGI-I score after taking study medication. Patients were analyzed according to the treatment they were randomized regardless of the actual treatment received. | Efficacy evaluation, demographic characteristics, and baseline characteristics |
Per-protocol analysis set | All patients in the full analysis set who did not have a major protocol violation that would affect interpretation of the efficacy data and who had adequate treatment adherence (≥ 75%). The per-protocol analysis set was analyzed according to randomized treatment assignment. | Primary and key secondary efficacy evaluation |
CGI-I = Clinical Global Impressions-Improvement; RSBQ = Rett Syndrome Behaviour Questionnaire.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Patient disposition in the LAVENDER study is summarized in Table 11. In total, 208 individuals were screened for eligibility and 40 were screened out. The most common reasons for ineligibility were meeting the exclusion criteria (42.5%) — which included having clinically significant comorbid disease (n = 2), having clinically significant laboratory readings (n = 10), having issues related to QT prolongation (n = 4), and in the judgment of the medical monitor (n = 1) — and not meeting the inclusion criteria (20.0%) — which included not having the required Rett Syndrome Clinical Severity Scale (RTT-CSS) or CGI-S severity scores (N = 5), not having stable or having discontinued anticonvulsants or psychoactive treatments (n = 2), and not having a stable pattern of seizures or having no seizures (n = 2). Of the 187 eligible patients, 93 were randomized to receive trofinetide and 94 were randomized to receive placebo. A total of 155 (82.9%) patients completed the study. More patients discontinued from the trofinetide group (24.7%) than from the placebo group (9.6%), with the largest imbalance being for discontinuations due to TEAEs (17.2% and 2.1%, respectively).
Table 11: Summary of Patient Disposition in the LAVENDER Study — Safety Analysis Set
Patient disposition | Trofinetide (N = 93) | Placebo (N = 94) |
|---|---|---|
Screened, N | 208 | |
Reason for screening failure, n (%) | 40 (100.0)a | |
Meeting exclusion criteria | 17 (42.5) | |
Not meeting inclusion criteria | 8 (20.0) | |
Consent withdrawn | 2 (5.0) | |
Other | 13 (32.5) | |
Randomized, N (%) | 93 (100.0) | 94 (100.0) |
Reasons for study discontinuation, n (%) | 23 (24.7) | 9 (9.6) |
TEAEs | 16 (17.2) | 2 (2.1) |
Nonadherence to the study drug | 4 (4.3) | 0 (0.0) |
Consent withdrawn | 1 (1.1) | 1 (1.1) |
Lack of efficacy | 1 (1.1) | 0 (0.0) |
Other (COVID-19 quarantine measures) | 1 (1.1) | 5 (5.3) |
Protocol deviation | 0 (0.0) | 1 (1.1) |
Randomized analysis set, N | 93 | 94 |
Safety analysis set, N | 93 | 94 |
Full analysis set, Nb | 91 | 93 |
Per-protocol analysis set, N | 89 | 90 |
TEAE = treatment-emergent adverse event.
aAlthough there were 208 unique participants screened, some were rescreened, for a total of 227 screenings.
bThe 3 patients missing from the full analysis set who were included in the randomized analysis set had a baseline assessment but no postbaseline efficacy assessments.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
Baseline characteristics are summarized in Table 12 and are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Only females with Rett syndrome were eligible for LAVENDER. The mean age of patients was 11.0 years (SD = 4.7 years) and 10.9 years (SD = 4.6 years) in the trofinetide and placebo groups, respectively. All patients must have had a CGI-S score of 4 or higher to be eligible and the mean score was 4.9 (SD = 0.8) points for both groups indicating that most patients in the study were considered moderately or markedly ill. Disease history was generally similar between the groups.
Table 12: Summary of Baseline Characteristics in the LAVENDER Study — Randomized Analysis Set
Characteristic | Trofinetide (N = 93) | Placebo (N = 94) |
|---|---|---|
Demographic characteristics | ||
Sex, n (%) | ||
Female | 93 (100.0) | 94 (100.0) |
Age (years) | ||
Mean (SD) | 11.0 (4.7) | 10.9 (4.6) |
Median (range) | 10.0 (5 to 20) | 10.0 (5 to 20) |
Age by category, n (%) | ||
5 to < 12 years | 53 (57.0) | 55 (58.5) |
12 to < 17 years | 23 (24.7) | 24 (25.5) |
≥ 17 years | 17 (18.3) | 15 (16.0) |
Race, n (%) | ||
Asian | 5 (5.4) | 1 (1.1) |
Black | 1 (1.1) | 1 (1.1) |
Native Hawaiian or Pacific islander | 1 (1.1) | 0 (0.0) |
Nonwhite | 11 (11.8) | 4 (4.3) |
White | 82 (88.2) | 90 (95.7) |
Other | 4 (4.3) | 2 (2.1) |
Body mass (kg) | ||
Mean (SD) | 30.5 (12.6) | 29.2 (10.4) |
Median (range) | 29.6 (13 to 78) | 27.3 (13 to 57) |
Clinical characteristics | ||
Derived baseline RSBQa | ||
Total score, mean (SD) | 43.8 (11.4) | 44.4 (12.1) |
Total score, median (range) | 42.0 (21 to 74) | 43.0 (14 to 69) |
Baseline RSBQ severity, n (%) | ||
Total score < 35 | 23 (24.7) | 25 (26.6) |
Total score ≥ 35 | 70 (75.3) | 69 (73.4) |
Baseline CGI-S score | ||
Mean (SD) | 4.9 (0.8) | 4.9 (0.8) |
Median (range) | 5 (4 to 6) | 5 (4 to 7) |
Baseline CGI-S category, n (%) | ||
4, moderately ill | 32 (34.4) | 33 (35.1) |
5, markedly ill | 38 (40.9) | 42 (44.7) |
6, severely ill | 23 (24.7) | 18 (19.1) |
7, extremely ill | 0 (0.0) | 1 (1.1) |
Rett Syndrome Clinical Severity Scale score at screening | ||
Mean (SD) | 24.1 (6.4) | 24.2 (6.7) |
Median (range) | 24 (11 to 36) | 23.5 (12 to 40) |
Disease history | ||
Age at diagnosis (years) | ||
Mean (SD) | 2.2 (1.0) | 2.6 (2.0) |
Median (range) | 2.0 (1 to 6) | 2.0 (0 to 12) |
Patients with > 1 MECP2 variant, n (%) | 0 (0.0) | 4 (4.3) |
MECP2 variant severity, n (%) | ||
Mild | 30 (32.3) | 37 (39.4) |
Moderate | 13 (14.0) | 8 (8.5) |
Severe | 46 (49.5) | 46 (48.9) |
Unknown | 4 (4.3) | 3 (3.2) |
MECP2 variant category, n (%) | ||
T158M | 13 (14.0) | 8 (8.5) |
R168X | 9 (9.7) | 10 (10.6) |
R255X | 9 (9.7) | 7 (7.4) |
R270X | 8 (8.6) | 7 (7.4) |
R133C | 6 (6.5) | 7 (7.4) |
R294X | 5 (5.4) | 6 (6.4) |
R306C | 6 (6.5) | 5 (5.3) |
R106W | 1 (1.1) | 3 (3.2) |
C-terminal truncations | 2 (2.2) | 7 (7.4) |
Large deletions | 7 (7.5) | 7 (7.4) |
Other variants | 27 (29.0) | 27 (28.7) |
CGI-S = Clinical Global Impressions-Severity; RSBQ = Rett Syndrome Behaviour Questionnaire; SD = standard deviation.
aThe derived baseline RSBQ score was assessed using the same analysis model as for the baseline RSBQ score, but the score on the Likert scale for item 31 (uses eye gaze to convey feelings, needs, and wishes) was reversed.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure and Adherence to Study Treatments
Treatment exposure and adherence are summarized in Table 13. Mean duration of treatment was shorter in the trofinetide group than in the placebo group, at 73.5 days (SD = 24.1 days) and 81.5 days (SD = 16.1 days), respectively. More patients stopped treatment early in the trofinetide group than the placebo group (28.0% versus 13.8%). Mean adherence was 99.1% (SD = 22.2%) in the trofinetide group and 97.0% (7.0%) in the placebo group, which was calculated based on the duration of exposure (before discontinuation) and accounted for dose adjustments.
Table 13: Summary of Patient Exposure and Adherence in the LAVENDER Study — Safety Analysis Set
Exposure | Trofinetide (N = 93) | Placebo (N = 94) |
|---|---|---|
Duration (days) | ||
Mean (SD) | 73.5 (24.1) | 81.5 (16.1) |
Median (range) | 85.0 (8 to 93) | 85.0 (10 to 100) |
Duration categories, n (%) | ||
< 2 weeks | 6 (6.5) | 1 (1.1) |
2 weeks to < 4 weeks | 1 (1.1) | 2 (2.1) |
4 weeks to < 6 weeks | 7 (7.5) | 1 (1.1) |
6 weeks to < 8 weeks | 5 (5.4) | 4 (4.3) |
8 weeks to < 10 weeks | 0 (0.0) | 0 (0.0) |
10 weeks to < 12 weeks | 7 (7.5) | 5 (5.3) |
≥ 12 weeks | 67 (72.0) | 81 (86.2) |
Adherence, % | ||
Mean (SD) | 99.1 (22.2) | 97.0 (7.0) |
Median (range) | 97.8 (68 to 295) | 98.3 (74 to 125) |
SD = standard deviation.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Concomitant medications are summarized in Table 14. All patients in the trofinetide group and nearly all patients in the placebo group (98.9%) were receiving some concomitant treatment(s) during the LAVENDER study. The most common medications used in the trofinetide group were antiepileptics, antipropulsives, and drugs for constipation. The medications with the largest imbalances between treatment groups were antidepressants, antipropulsives, and intestinal adsorbents.
Table 14: Summary of Concomitant Treatments in the LAVENDER Study — Safety Analysis Set
Concomitant treatmenta | Trofinetide (N = 93) | Placebo (N = 94) |
|---|---|---|
Any concomitant treatments, n (%) | 93 (100.0) | 93 (98.9) |
Adrenergics, inhalants | 15 (16.1) | 12 (12.8) |
Antidepressants | 8 (8.6) | 19 (20.2) |
Antidiarrheal micro-organisms | 10 (10.8) | 7 (7.4) |
Antiepileptics | 60 (64.5) | 68 (72.3) |
Antihistamines for systemic use | 24 (25.8) | 28 (29.8) |
Anti-inflammatory and antirheumatic products, nonsteroids | 30 (32.3) | 27 (28.7) |
Antipropulsives | 47 (50.5) | 3 (3.2) |
Anxiolytics | 13 (14.0) | 15 (16.0) |
Decongestants and other nasal preparations for topical use | 11 (11.8) | 11 (11.7) |
Drugs for constipation | 56 (60.2) | 66 (70.2) |
Drugs for functional gastrointestinal disorders | 16 (17.2) | 14 (14.9) |
Drugs for peptic ulcer and gastroesophageal reflux disease | 28 (30.1) | 33 (35.1) |
Hypnotics and sedatives | 22 (23.7) | 27 (28.7) |
Intestinal adsorbents | 25 (26.9) | 0 (0.0) |
Multivitamins, combinations | 9 (9.7) | 13 (13.8) |
Multivitamins, plain | 29 (31.2) | 28 (29.8) |
Muscle relaxants, centrally acting drugs | 6 (6.5) | 14 (14.9) |
Other alimentary tract and metabolism products | 18 (19.4) | 22 (23.4) |
Other analgesics and antipyretics | 24 (25.8) | 30 (31.9) |
Other mineral supplements | 6 (6.5) | 11 (11.7) |
Other nutrients | 13 (14.0) | 11 (11.7) |
Other plain vitamin preparations | 6 (6.5) | 14 (14.9) |
Psychostimulants, drugs used for ADHD and nootropics | 16 (17.2) | 11 (11.7) |
Vitamins A and D, including combinations | 32 (34.4) | 27 (28.7) |
ADHD = attention-deficit/hyperactivity disorder.
aTreatments reported in at least 10% of patients in either treatment group.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Protocol Deviations
Overall, 11 (11.8%) patients in the trofinetide group and 13 (13.8%) patients in the placebo group experienced a major protocol deviation. More than half of the deviations were related to the COVID-19 public health emergency (6.5% of patients in the trofinetide group and 9.6% of patients in the placebo group). In the trofinetide group, most deviations had to do with study procedures (8.6%), followed by visit-schedule interval (2.2%) and the investigational product (1.1%). In the placebo group, most deviations had to do with study procedures (10.6%), followed by efficacy or safety ratings (1.1%), eligibility and entry (1.1%), and visit-schedule interval (1.1%).
Efficacy
The main efficacy results are summarized in Table 15.
Table 15: Main Efficacy Outcomes in the LAVENDER Study — Full Analysis Set
Variable | Trofinetide (N = 91) | Placebo (N = 93) |
|---|---|---|
RSBQ total score | ||
Number of patients contributing to the analysis, n (%) | 76 (83.5) | 85 (91.4) |
RSBQ total score at baseline, mean (SD) | 43.7 (11.5) | 44.5 (12.2) |
RSBQ total score at week 12, mean (SD) | 39.9 (12.0) | 42.8 (13.1) |
Change from baseline in RSBQ total score, LSM (SE)a | –4.9 (0.9) | –1.7 (0.9) |
Treatment group difference vs. control group, LSM (95% CI)a | –3.1 (–5.7 to –0.6) | |
P valuea,b | 0.0175 | |
CGI-I score | ||
Number of patients contributing to the analysis, n (%) | 77 (84.6) | 86 (92.5) |
CGI-I score at week 12, mean (SD) | 3.5 (0.7) | 3.8 (0.6) |
CGI-I score at week 12, LSM (SE)a | 3.5 (0.1) | 3.8 (0.1) |
Treatment group difference vs. control group, LSM (95% CI)a | –0.3 (–0.5 to –0.1) | |
P valuea,b | 0.0030 | |
RTT-COMC score | ||
Number of patients contributing to the analysis, n (%) | 76 (83.5) | 82 (88.2) |
RTT-COMC score at baseline, mean (SD) | 3.5 (1.5) | 3.3 (1.8) |
RTT-COMC score at week 12, mean (SD) | 3.1 (1.6) | 3.3 (1.7) |
Change from baseline in RTT-COMC score, LSM (SE)a | –0.4 (0.1) | 0.0 (0.1) |
Treatment group difference vs. control group, LSM (95% CI)a | –0.3 (–0.6 to 0.0) | |
P valuea,c | 0.0257 | |
CSBS-DP-IT Checklist social composite score | ||
Number of patients contributing to the analysis, n (%) | 73 (80.2) | 81 (87.1) |
CSBS-DP-IT Checklist social composite score at baseline, mean (SD) | 8.7 (3.3) | 8.8 (3.2) |
CSBS-DP-IT Checklist social composite score at week 12, mean (SD) | 8.9 (3.7) | 7.5 (3.0) |
Change from baseline in CSBS-DP-IT Checklist social composite score, LSM (SE)a | –0.1 (0.3) | –1.1 (0.3) |
Treatment group difference vs. control group, LSM (95% CI)a | 1.0 (0.3 to 1.7) | |
P valuea,b | 0.0064 | |
CGI-I = Clinical Global Impressions-Improvement; CI = confidence interval; CSBS-DP-IT = Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler; LSM = least squares mean; RSBQ = Rett Syndrome Behaviour Questionnaire; RTT-COMC = Rett Syndrome Clinician Rating of Ability to Communicate Choices; SD = standard deviation; SE = standard error.
aThe mixed effects model for repeated measures includes age group, baseline RSBQ severity, planned treatment, study visit, treatment-by-visit interaction, baseline-by-visit interaction, and baseline score as fixed effects. An unstructured covariance matrix was used to model within-patient errors. The Kenward-Roger method was used to calculate the denominator degrees of freedom for tests of fixed effects.
bP value has been adjusted for multiple testing.
cP value has not been adjusted for multiple testing.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Rett Syndrome Behaviour Questionnaire Total Score
The mean total RSBQ score at baseline was 43.7 points (SD = 11.5 points) in the trofinetide group and 44.5 points (SD = 12.2 points) in the placebo group. The mean change from baseline in RSBQ score was –4.9 points (SE = 0.9 points) in the trofinetide group and –1.7 points (SE = 0.9 points) in the placebo group, resulting in a between-group difference of –3.1 points (95% CI, –5.7 to –0.6 points; P = 0.0175).
The results of the sensitivity analysis using pattern-mixture models that assumed MNAR and missing data due to COVID-19 supported the primary analysis.
Subgroup analyses showed that the mean change from baseline in RSBQ score was greater in the trofinetide group than in the placebo group for all age groups (Table 27 and Figure 1).
Clinical Global Impressions-Improvement Score
CGI-I was not measured at baseline. The mean CGI-I score at week 12 was 3.5 points (SE = 0.1 points) in the trofinetide group and 3.8 points (SE = 0.1 points) in the placebo group, resulting in a between-group difference of –0.3 points (95% CI, –0.5 to –0.1 points; P = 0.0030).
The results of the sensitivity analysis using pattern-mixture models that assumed MNAR and missing data due to COVID-19 supported the primary analysis.
Subgroup analyses showed that the mean CGI-I score was greater in the trofinetide group than in the placebo group for all age groups (Table 27 and Figure 1).
Rett Syndrome Clinician Rating of Ability to Communicate Choices
The mean RTT-COMC score at baseline was 3.5 points (SD = 1.5 points) in the trofinetide group and 3.3 points (SD = 1.8 points) in the placebo group. The mean change from baseline in RTT-COMC score was –0.4 points (SE = 0.1 points) in the trofinetide group and 0.0 points (SE = 0.1 points) in the placebo, resulting in a between-group difference of –0.3 points (95% CI, –0.6 to 0.0 points).
Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler Checklist Social Composite Score
The mean CSBS-DP-IT Checklist social composite score at baseline was 8.7 points (SD = 3.3 points) in the trofinetide group and 8.8 points (SD = 3.2 points) in the placebo group. The mean change from baseline in CSBS-DP-IT Checklist social composite score was –0.1 points (SE = 0.3 points) in the trofinetide group and –1.1 points (SE = 0.3 points) in the placebo group, resulting in a between-group difference of 1.0 points (95% CI, 0.3 to 1.7 points; P = 0.0064).
Health-Related Quality of Life
There were no HRQoL outcomes reported in the LAVENDER study.
Other Efficacy Results Related to Outcomes Important to Patients, Caregivers, and Clinicians
Efficacy results for other outcomes are summarized in Table 16. The between-group difference for the RTT-HF score was –0.1 points (95% CI, –0.3 to 0.1 points), for the RTT-AMB score was –0.1 points (95% CI, –0.3 to 0.1 points), for the RTT-VCOM score was 0.0 points (95% CI, –0.2 to 0.2 points), for the overall quality-of-life rating was 0.1 points (95% CI, –0.1 to 0.4 points), and for the RTT-CBI total score was –0.8 points (95% CI, –3.5 to 2.0 points).
Table 16: Other Efficacy Outcomes in the LAVENDER Study — Full Analysis Set
Variable | Trofinetide (N = 91) | Placebo (N = 93) |
|---|---|---|
RTT-HF score | ||
Number of patients contributing to the analysis, n (%) | 76 (83.5) | 83 (89.2) |
Baseline, mean (SD) | 4.9 (1.7) | 4.8 (1.7) |
Change from baseline, LSM (SE)a | –0.1 (0.1) | 0.0 (0.1) |
Treatment group difference vs. control group, LSM (95% CI)a | –0.1 (–0.3 to 0.1) | |
P valuea,b | 0.3649 | |
RTT-AMB score | ||
Number of patients contributing to the analysis, n (%) | 76 (83.5) | 83 (89.2) |
Baseline, mean (SD) | 4.3 (2.1) | 4.4 (2.0) |
Change from baseline, LSM (SE)a | –0.2 (0.1) | 0.0 (0.1) |
Treatment group difference vs. control group, LSM (95% CI)a | –0.1 (–0.3 to 0.1) | |
P valuea,b | 0.2114 | |
RTT-VCOM score | ||
Number of patients contributing to the analysis, n (%) | 76 (83.5) | 83 (89.2) |
Baseline, mean (SD) | 5.5 (1.0) | 5.5 (1.0) |
Change from baseline, LSM (SE)a | 0.0 (0.1) | 0.0 (0.1) |
Treatment group difference vs. control group, LSM (95% CI)a | 0.0 (–0.2 to 0.2) | |
P valuea,b | 0.9799 | |
Overall quality-of-life rating | ||
Number of patients contributing to the analysis, n (%) | 73 (80.2) | 80 (86.0) |
Baseline, mean (SD) | 3.7 (1.1) | 3.6 (1.1) |
Change from baseline, LSM (SE)c | 0.2 (0.1) | 0.1 (0.1) |
Treatment group difference vs. control group, LSM (95% CI)c | 0.1 (–0.1 to 0.4) | |
P valueb,c | 0.2507 | |
RTT-CBI total score (items 1 to 24) | ||
Number of patients contributing to the analysis, n (%) | 73 (80.2) | 81 (87.1) |
Baseline, mean (SD) | 38.4 (15.0) | 39.1 (15.6) |
Change from baseline, LSM (SE)c | –1.1 (1.0) | –0.4 (1.0) |
Treatment group difference vs. control group, LSM (95% CI)c | –0.8 (–3.5 to 2.0) | |
P valueb,c | 0.5855 | |
CI = confidence interval; LSM = least squares mean; RTT-AMB = Rett Syndrome Clinician Rating of Ambulation and Gross Motor Skills; RTT-CBI = Rett Syndrome Caregiver Burden Inventory; RTT-HF = Rett Syndrome Clinician Rating of Hand Function; RTT-VCOM = Rett Syndrome Clinician Rating of Verbal Communication; SD = standard deviation; SE = standard error.
aThe mixed effects model for repeated measures includes age group, baseline Rett Syndrome Behaviour Questionnaire (RSBQ) severity, planned treatment, study visit, treatment-by-visit interaction, baseline-by-visit interaction, and baseline score as fixed effects. An unstructured covariance matrix is used to model within-patient errors. The Kenward-Roger method is used to calculate the denominator degrees of freedom for tests of fixed effects.
bP value has not been adjusted for multiple testing.
cThe analysis of covariance includes age group, baseline RSBQ severity, and planned treatment as factors, and baseline total score as a covariate.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Harms results from the LAVENDER study are summarized in Table 17.
Treatment-Emergent Adverse Events
Overall, 92.5% of patients in the trofinetide group and 54.3% of patients in the placebo group experienced at least 1 TEAE. Diarrhea and vomiting were the most common TEAEs, and both were reported at a greater frequency in the trofinetide group (80.6% and 26.9%, respectively) than in the placebo group (19.1% and 9.6%, respectively). In the trofinetide group, 2.2%, 36.6%, and 41.9% of patients experienced severe, moderate, and mild diarrhea, respectively, whereas in the placebo group, 1.1%, 6.5%, and 19.4% of patients experienced severe, moderate, and mild vomiting, respectively.
Serious Adverse Events
In the trofinetide group, 3 (3.2%) patients reported 5 SAEs (bacteremia, bronchiolitis, COVID-19 pneumonia, seizure, and urinary tract infection), and in the placebo group, 3 (3.2%) patients reported 3 SAEs (constipation, pneumatosis intestinalis, and respiratory distress).
Withdrawals Due to Adverse Events
During the study, 17.2% of patients in the trofinetide group and 2.1% of patients in the placebo group stopped treatment due to TEAEs. The most frequently reported TEAE leading to treatment discontinuation was diarrhea (12.9% in the trofinetide group and 0.0% in the placebo group). All other TEAEs leading to treatment discontinuation were reported in fewer than 4 patients.
Mortality
There were no deaths reported in the study.
Notable Harms
TEAEs considered clinically important by the clinical experts and noted in the Health Canada product monograph included diarrhea and vomiting, which have previously been described.
Table 17: Summary of Harms Results From the LAVENDER Study — Safety Analysis Set
Adverse events | Trofinetide (N = 93) | Placebo (N = 94) |
|---|---|---|
Most common TEAEs, n (%)a | ||
Patients with ≥ 1 TEAE | 86 (92.5) | 51 (54.3) |
Diarrhea | 75 (80.6) | 18 (19.1) |
Vomiting | 25 (26.9) | 9 (9.6) |
Seizure | 8 (8.6) | 5 (5.3) |
Pyrexia | 8 (8.6) | 4 (4.3) |
Irritability | 6 (6.5) | 0 (0.0) |
Decreased appetite | 5 (5.4) | 2 (2.1) |
SAEs, n (%) | ||
Patients with ≥ 1 SAE | 3 (3.2) | 3 (3.2) |
Patients who stopped treatment due to TEAEs, n (%)a | ||
Patients who stopped | 16 (17.2) | 2 (2.1) |
Diarrhea | 12 (12.9) | 0 (0.0) |
Deaths, n (%) | ||
Patients who died | 0 (0.0) | 0 (0.0) |
AEs of special interest, n (%) | ||
Diarrhea | 75 (80.6) | 18 (19.1) |
Vomiting | 25 (26.9) | 9 (9.6) |
AE = adverse event; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aOccurring in at least 5% of patients in either treatment group.
Sources: LAVENDER Clinical Study Report and sponsor’s Summary of Clinical Evidence.8,12 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
In the LAVENDER study, patients were randomized using an interactive response technology system, randomization was stratified by age group and baseline RSBQ severity, and assignment was performed based on a pregenerated permuted-block randomization schedule. Furthermore, patients, caregivers, investigators, outcome assessors, site personnel, and the sponsor were blinded to treatment assignment, and a matching placebo was used for the control group. As such, the risk of bias due to randomization, blinding, and treatment allocation procedures was likely low.
There was a risk of bias in the measurement of study outcomes. When looking at the harms results, reports of TEAEs were imbalanced between the treatment groups (92.5% in the trofinetide group versus 54.3% in the placebo group), particularly for diarrhea and vomiting. Diarrhea and vomiting are known AEs associated with trofinetide and most likely resulted in functional unblinding. This may have also had an impact on the ratings of assessors who were aware of a patient’s TEAEs (e.g., caregivers, clinicians). This is problematic, given that all efficacy outcomes were subjective in nature, and there is the potential for overestimation of the treatment effect if the suspected treatment assignment was revealed to patients, caregivers, and assessors.
The change from baseline in RSBQ score was a coprimary end point in the LAVENDER study. The RSBQ was originally developed and validated as a diagnostic tool to distinguish between Rett syndrome and severe intellectual disability in females,11 and although it has been accepted by the FDA,43 it is unclear how appropriate it is for measuring treatment effect over time, given the lack of information verifying its responsiveness to change.35-37 On the advice of the FDA, the CGI-I was included as a coprimary end point to aid in the interpretation of the clinical meaningfulness of the RSBQ.14 Clinical expert opinion supported the combined use of caregiver-reported and clinician-reported outcomes when assessing patients; however, the evidence is mixed on whether these outcomes are correlated.11,44 The RTT-COMC and CSBS-DP-IT Checklist social composite scores have not been validated in patients with Rett syndrome, and both Health Canada and the FDA have noted limitations in the use of the CSBS-DP-IT Checklist social composite score, indicating that it is not adequate to establish the efficacy of trofinetide.13,14 In general, there was limited evidence or a lack of evidence of validity, reliability, and responsiveness for other outcomes in the LAVENDER study (motor skills, overall quality of life, and caregiver burden) in patients with Rett syndrome. No MIDs for any of the outcomes were identified from the literature, and clinical meaningfulness was based on expert opinion. Moreover, the clinical experts consulted for this review noted that the trial outcomes are not commonly used in practice (and standardized measures of treatment effect are not used in clinics across Canada), making it a challenge to apply the results to real-world practice.
For the coprimary and key secondary outcomes, multiplicity was controlled for. Significance for other secondary outcomes (e.g., RTT-COMC) was nominal. One clinical expert consulted for the review indicated that the subgroup analyses by age group may be relevant for clinical decision-making, based on what is understood about other neurological and neuromuscular conditions; however, it is unknown whether earlier treatment would impact clinical outcomes or whether there would be differences in the treatment effect in different age groups. Regardless, the subgroup analyses were not controlled for multiplicity and were likely too small and underpowered to provide conclusive evidence to inform reimbursement decisions.
There is likely a low risk of bias due to deviations from the assigned intervention. Analyses were based on the full analysis set, not the intention-to-treat population (or randomized analysis set), which is the preferred analysis population. However, the numbers for the groups were similar, and there was likely little impact on the results with the use of the full analysis set. The clinical experts indicated that they had no concerns about the concomitant treatments used in the trial; however, it is possible that imbalances in the treatments used could have had an impact on subjective outcomes, such as symptom ratings and harms reporting.
There was a risk of bias due to nonadherence to study interventions. There was a notable imbalance between the trofinetide and placebo groups in the proportion of patients who discontinued (24.7% in the trofinetide group versus 9.6% in the placebo group), and the 2 most common reasons for discontinuation in the trofinetide group were TEAEs and nonadherence to the study drug. According to a Health Canada report, patient narratives about discontinuations because of withdrawn consent or nonadherence to the study drug indicated that there were issues with medication tolerability (e.g., patient not liking or unwilling to take the medication, difficulty administering the medication, and the caregiver unwilling to resume dosing with a lower dose of concomitant treatment).13 Vomiting was more common in the trofinetide group than in the placebo group, and the product monograph notes that if vomiting occurs after drug administration, additional doses should not be given. As a result, it is possible that adherence rates reported for trofinetide may not have been accurate. Nonadherence to the study treatment could also have underestimated harms reporting in the LAVENDER study.
There is a high risk of bias due to missing data in the study. The greatest number of patients who discontinued the intervention was from the trofinetide group and the difference between groups is large. This introduces the potential for bias against the null (i.e., toward an inflated efficacy of trofinetide), as the data driving the model are largely from patients who stayed in the study (likely better responders with fewer TEAEs). The investigators assumed that data were MAR, which is not supported by the differential losses to follow-up or reasons for discontinuations. Moreover, the MMRM approach (used for the coprimary end points) assumes that data are MAR and that patients with missing data will continue to behave or change in a similar fashion as estimated by those with ongoing data points. This assumption is strong and unverifiable, and may, particularly when patients discontinue therapy because of TEAEs or due to lack of efficacy (as observed in the LAVENDER study), increase the bias in the observed results. Although the investigators used sensitivity analyses to account for missing data (which are only appropriate when data are truly MAR or missing completely at random), such analyses are unlikely to fully account for the bias potentially introduced by the missing data. However, it is worth noting that sensitivity analyses were conducted based on pattern-mixture models assuming data were MNAR and support the primary analyses. The Health Canada report had similar conclusions for the data imputation methods used in the LAVENDER study.13
External Validity
One of the most important limitations of the evidence from the LAVENDER study is that the Health Canada indication is broader than the trial population. The Health Canada indication includes patients with Rett syndrome 2 years and older. The LAVENDER study did not enrol patients who were male, younger than 5 years or older than 20 years, did not have classic or typical Rett syndrome, did not have a confirmed disease-causing MECP2 variant, were not at least 6 months after regression, did not have a CGI-S score of at least 4 (indicating at least moderate disease severity), did not have a stable pattern of seizures, and were not taking stable standard therapies. The rationale provided by the sponsor to support the eligibility criteria noted that few males are diagnosed with Rett syndrome, and their inclusion would make it difficult to stratify by another variable. Additionally, the upper and lower age limits were used to create a more homogenous population, given the difficulty controlling for variability in services for those who were no longer school aged (in the US, services are generally available through school districts until patients are aged 20 years or 21 years), and patients younger than 5 years have variable clinical presentations and developmental regressions early in the disease course that could make it difficult to analyze the results. The panel of experts consulted for this review indicated that patients would be treated with trofinetide if there was a confirmed clinical diagnosis of classic (typical) Rett syndrome (with or without a disease-causing MECP2 variant) or atypical Rett syndrome (with a disease-causing MECP2 variant). Patients without an MECP2 variant would have to have a confirmed clinical diagnosis of classic Rett syndrome to receive trofinetide, but those without an MECP2 variant and with an atypical Rett syndrome diagnosis would not receive trofinetide due to lack of evidence in this population. The sponsor submitted 2 additional studies that may provide some insight into treatment with trofinetide in these populations (the DAFFODIL study included female patients aged 2 years to 5 years with Rett syndrome and the LOTUS study included any patients who were prescribed trofinetide and were under routine clinical care in the US). Based on the baseline characteristics of patients in the LAVENDER study, the experts believed that patients in Canada were likely similar, apart from race; the LAVENDER study had a disproportionately low representation of patients who are nonwhite.
The sponsor has indicated that trofinetide would be used as a first-line treatment in conjunction with supportive therapies for Rett syndrome, which the experts agreed with. In the study, trofinetide was titrated to improve tolerability, and dose reductions or interruptions were permitted to mitigate TEAEs. Similar recommendations are outlined in the product monograph, and the clinical experts stated that they would titrate at a rate that is tolerable to patients (which could be slower than the protocol used in the LAVENDER study). Partway through the study, a diarrhea management plan was implemented that involved stopping or reducing treatments for constipation, increasing fibre intake, using loperamide as needed, and monitoring hydration; the experts indicated that management would be similar in clinical practice.
The outcomes included in the LAVENDER study are reflective of the treatment goals considered to be important to patients and caregivers (behaviour, communication, and motor skills), but because none are used in clinical practice, it is a challenge to apply the results to a real-world setting. The clinical experts highlighted that patients often lack hand function and verbal communication skills, making these outcomes less practical for measuring clinical change. There was no comprehensive measure of HRQoL in the trial, and the clinicians noted that caregiver burden is not consistently measured in practice; however, they agreed that these are both important considerations in this disease area, it is unclear what effect trofinetide has on HRQoL and on patient and caregiver daily living.
The LAVENDER study was a 12-week trial, which the clinical experts noted is likely insufficient to assess meaningful, long-term changes in motor skills, communication, and behaviours in patients with Rett syndrome. Longer-term treatment with trofinetide was assessed in the LILAC and LILAC-2 studies, which are discussed later in this report.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered to be most relevant to expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.15,16
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs starts as high-certainty evidence and can be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for trofinetide versus placebo.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Two extension studies — LILAC (N = 154) and LILAC-2 (N = 77) — have been summarized to provide evidence regarding the long-term safety and tolerability of trofinetide in females living with Rett syndrome. Patients who completed the LAVENDER study were able to enrol in the LILAC study, a 40-week OLE study of trofinetide conducted between January 2020 and August 2022. Patients who completed the LILAC study were able to enrol in the LILAC-2 study, an OLE of trofinetide with a duration of approximately 32 months. The inclusion and exclusion criteria of the LILAC studies were consistent with those of the LAVENDER study.
Populations
Baseline Characteristics
The baseline characteristics of patients in the LILAC and LILAC-2 studies are summarized in Table 18. The demographic characteristics were similar to those in the LAVENDER study. The overall mean age was 11.0 years (SD = 4.6 years) in the LILAC study and 12.0 years (SD = 4.4 years) in the LILAC-2 study, and about half of the patients in each study were aged 5 years to 10 years. The overall mean baseline CGI-S score was 4.8 points (SD = 0.8 points) in the LILAC study and 4.8 points (SD = 0.9 points) in the LILAC-2 study. At baseline, most patients in the LILAC and LILAC-2 studies were moderately ill (36.4% and 41.6%, respectively) or markedly ill (41.6% and 31.2%, respectively).
Table 18: Summary of Baseline Characteristics in the LILAC and LILAC-2 Studies
Characteristic | LILAC | LILAC-2 | ||
|---|---|---|---|---|
LAVENDER PBO to LILAC TRO (N = 85) | LAVENDER TRO to LILAC TRO (N = 69) | LAVENDER PBO to LILAC-2 TRO (N = 36) | LAVENDER TRO to LILAC-2 TRO (N = 41) | |
Demographic characteristics | ||||
Sex, n (%) | ||||
Female | 85 (100.0) | 69 (100.0) | 36 (100.0) | 41 (100.0) |
Age (years) | ||||
Mean (SD) | 11.0 (4.5) | 10.9 (4.6) | 11.8 (4.2) | 12.1 (0.7) |
Range | 5 to 21 | 5 to 21 | 6 to 20 | 6 to 21 |
Age by category, n (%) | ||||
5 years to 10 years | 48 (56.5) | 37 (53.6) | 16 (44.4) | 17 (41.5) |
11 years to 15 years | 20 (23.5) | 17 (24.6) | 12 (33.3) | 14 (34.1) |
16 years to 20 years | 16 (18.8) | 14 (20.3) | 8 (22.2) | 9 (22.0) |
> 20 years | 1 (1.2) | 1 (1.4) | 0 (0.0) | 1 (2.4) |
Race, n (%) | ||||
Asian | 1 (1.2) | 4 (5.8) | 0 (0.0) | 1 (2.4) |
Black | 0 (0.0) | 1 (1.4) | 0 (0.0) | 1 (2.4) |
Native Hawaiian or other Pacific Islander | 0 (0.0) | 1 (1.4) | 0 (0.0) | 0 (0.0) |
White | 82 (96.5) | 61 (88.4) | 34 (94.4) | 37 (90.2) |
Other | 2 (2.4) | 2 (2.9) | 2 (5.6) | 2 (4.9) |
Body mass (kg) | ||||
n (%) | 85 (100.0) | 69 (100.0) | 36 (100.0) | 41 (100.0) |
Mean (SD) | 29.6 (10.6) | 28.9 (11.0) | 32.0 (11.1) | 30.5 (9.6) |
Body mass index (kg/m2) | ||||
n | 84 (98.8) | 67 (97.1) | 36 (100.0) | 39 (95.1) |
Mean (SD) | 17.1 (3.3) | 16.9 (3.8) | 18.1 (3.3) | 17.2 (3.1) |
Clinical characteristics | ||||
RSBQ total score | ||||
n | 85 (100.0) | 68 (98.6) | 36 (100.0) | 40 (97.6) |
Mean (SD) | 42.8 (13.0) | 39.5 (11.9) | 35.6 (11.8) | 37.2 (13.5) |
Range | 16 to 69 | 9 to 69 | 13 to 59 | 7 to 68 |
CGI-S score | ||||
n | 85 (100.0) | 69 (100.0) | 36 (100.0) | 41 (100.0) |
Mean (SD) | 4.8 (0.8) | 4.9 (0.8) | 4.8 (0.9) | 4.7 (0.9) |
Range | 4 to 7 | 3 to 6 | 3 to 7 | 3 to 6 |
CGI-S category, n (%) | ||||
3: Mildly ill | 0 (0.0) | 1 (1.4) | 1 (2.8) | 2 (4.9) |
4: Moderately ill | 32 (37.6) | 24 (34.8) | 14 (38.9) | 18 (43.9) |
5: Markedly ill | 36 (42.4) | 28 (40.6) | 12 (33.3) | 12 (29.3) |
6: Severely ill | 16 (18.8) | 16 (23.2) | 8 (22.2) | 9 (22.0) |
7: The most extremely ill patients | 1 (1.2) | 0 (0.0) | 1 (2.8) | 0 (0.0) |
CGI-S = Clinical Global Impressions-Severity; PBO = placebo; RSBQ = Rett Syndrome Behaviour Questionnaire; SD = standard deviation; TRO = trofinetide.
Note: The LAVENDER PBO to LILAC TRO or to LILAC-2 TRO groups comprise patients who received placebo in the LAVENDER study and trofinetide in the open-label extension (OLE) studies, whereas the LAVENDER TRO to LILAC TRO or to LILAC-2 TRO groups comprise patients who received trofinetide in both the LAVENDER and the OLE studies.
Sources: LILAC and LILAC-2 Clinical Study Reports and sponsor’s Summary of Clinical Evidence.8,45,46 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Interventions
In the OLE studies, trofinetide oral solution was administered twice daily, either orally or via gastrostomy tube. Dosing was weight-based, using the same weight bands as in the LAVENDER study. In the LILAC study, patients received the baseline dose from the LAVENDER study, unless their dose was reduced for tolerability, in which case they remained on that dose with the possibility of an increase if well tolerated. In the LILAC-2 study, patients continued their final LILAC dose, with potential adjustments for weight changes or tolerability, as decided by the investigator.
Outcomes
The primary outcomes in both the LILAC and LILAC-2 studies were focused on safety, and included TEAEs, SAEs, withdrawals due to TEAEs, and potentially clinically important changes in other safety assessments. Relevant secondary and/or exploratory efficacy outcomes in both studies included RSBQ, CGI-I, RTT-COMC, CSBS-DP-IT Checklist social composite score, RTT-HF, RTT-AMB, RTT-VCOM, overall quality-of-life rating, and RTT-CBI at various time points.
Statistical Analysis
For both extension studies, all results were summarized using descriptive statistics and were performed using the safety analysis set unless otherwise noted.
For each continuous measure in the safety and efficacy analyses, change from baseline results were shown using the baseline of the current study and/or using the baseline from the LAVENDER study to report the changes across the time points of the OLE study.
Results
Patient Disposition
Disposition of patients enrolled in the LILAC and LILAC-2 studies is summarized in Table 19. Of the 187 patients enrolled in the LAVENDER study, 161 patients completed it, and 154 patients continued into the LILAC study (85 transitioned from placebo and 69 from trofinetide). Seventy (45.5%) patients withdrew early from the LILAC study, primarily due to TEAEs; 84 (54.5%) patients completed the 40 weeks of treatment.
In the LILAC-2 study, 78 patients consented to roll over from the LILAC study, but 1 was ineligible, resulting in 77 patients in the Safety Analysis Set (36 from the placebo group and 41 from the trofinetide group in the LAVENDER study). Following market approval of trofinetide on March 10, 2023, the study was terminated by the sponsor. Most patients (79.2%) discontinued due to the study’s termination, while 5 (6.5%) discontinued due to TEAEs and 4 (5.2%) patients died.
Table 19: Patient Disposition in the LILAC and LILAC-2 Studies — Safety Analysis Set
Patient disposition | LILAC | LILAC-2 | ||
|---|---|---|---|---|
LAVENDER PBO to LILAC TRO (N = 85) | LAVENDER TRO to LILAC TRO (N = 69) | LAVENDER PBO to LILAC-2 TRO (N = 36) | LAVENDER TRO to LILAC-2 TRO (N = 41) | |
Completed study, n (%) | 39 (45.9) | 45 (65.2) | 0 | 0 |
Discontinued study, n (%) | 46 (54.1) | 24 (34.8) | 36 (100.0) | 41 (100.0) |
Reasons for discontinuation, n (%) | ||||
TEAEs | 36 (42.4) | 19 (27.5) | 4 (11.1) | 1 (2.4) |
Death | 0 | 0 | 2 (5.6) | 2 (4.9) |
Lack of efficacy | 4 (4.7) | 1 (1.4) | 3 (8.3) | 0 |
Nonadherence with study drug | 1 (1.2) | 2 (2.9) | 2 (5.6) | 0 |
Study terminated by sponsor | NA | NA | 24 (66.7) | 37 (90.2) |
Patient withdrew consent | 3 (3.5) | 2 (2.9) | 0 | 0 |
Other | 2 (2.4) | 0 | 1 (2.8) | 1 (2.4) |
Safety analysis set, N | 85 | 69 | 36 | 41 |
NA = not applicable; PBO = placebo; TEAE = treatment-emergent adverse event; TRO = trofinetide.
Sources: LILAC and LILAC-2 Clinical Study Reports and sponsor’s Summary of Clinical Evidence.8,45,46 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Treatment exposure by study and treatment cohort in the LILAC and LILAC-2 studies is presented in Table 20. A total of 92.8% and 97.4% of patients were at least 80% adherent to the study drug in the LILAC and LILAC-2 studies, respectively. The mean duration of exposure to trofinetide in the LILAC study for all 154 patients was 201.6 days (SD = 106.2 days), with a range of 8 to 324 days. In the LILAC-2 study, the mean duration of exposure to trofinetide for all 77 patients was 426.2 days (SD = 174.7 days), with a range of 18 to 813 days.
Concomitant Medications and Cointerventions
Concomitant medications taken by at least 10% of patients in each of the LILAC and LILAC-2 studies are shown in Table 21. All patients in both the LILAC and LILAC-2 studies received at least 1 concomitant medication during the respective study. The most frequently used concomitant medications in the LILAC and LILAC-2 studies were consistent with those in the pivotal LAVENDER trial, and included antiepileptics (72.7% and 77.9%, respectively), antipropulsives (62.3% and 45.5%, respectively), and drugs for constipation (59.7% and 55.8%, respectively).
Table 20: Treatment Exposure and Adherence to the Study Drug in the LILAC and LILAC-2 Studies — Safety Analysis Set
Exposure | LILAC | LILAC-2 | ||
|---|---|---|---|---|
LAVENDER PBO to LILAC TRO (N = 85) | LAVENDER TRO to LILAC TRO (N = 69) | LAVENDER PBO to LILAC-2 TRO (N = 36) | LAVENDER TRO to LILAC-2 TRO (N = 41) | |
Duration (days)a | ||||
Mean (SD) | 189.2 (105.66) | 217.0 (105.55) | 407.0 (201.46) | 443.0 (147.90) |
Range | 8 to 323 | 11 to 324 | 25 to 737 | 18 to 813 |
Adherence ≥ 80%, n (%) | 80 (94.2) | 63 (91.3) | 35 (97.2) | 40 (97.6) |
PBO = placebo; SD = standard deviation; TRO = trofinetide.
aDuration of exposure = (last dose date) – (first open-label dose date) + 1.
Sources: LILAC and LILAC-2 Clinical Study Reports and sponsor’s Summary of Clinical Evidence.8,45,46 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 21: Concomitant Treatments in the LILAC and LILAC-2 Studies — Safety Analysis Set
Concomitant treatment | LILAC | LILAC-2 | ||
|---|---|---|---|---|
LAVENDER PBO to LILAC TRO (N = 85) | LAVENDER TRO to LILAC TRO (N = 69) | LAVENDER PBO to LILAC-2 TRO (N = 36) | LAVENDER TRO to LILAC-2 TRO (N = 41) | |
Any concomitant medications, n (%)a | 85 (100.0) | 69 (100.0) | 36 (100.0) | 41 (100.0) |
Adrenergics, inhalants | 9 (10.6) | 11 (15.9) | 7 (19.4) | 11 (26.8) |
Antibiotics for topical use | 8 (9.4) | 5 (7.2) | 5 (13.9) | 6 (14.6) |
Antidepressants | 15 (17.6) | 6 (8.7) | 4 (11.1) | 5 (12.2) |
Antidiarrheal micro-organisms | 9 (10.6) | 8 (11.6) | 5 (13.9) | 6 (14.6) |
Antiemetics and antinauseants | 16 (18.8) | 7 (10.1) | 8 (22.2) | 5 (12.2) |
Antiepileptics | 63 (74.1) | 49 (71.0) | 26 (72.2) | 34 (82.9) |
Antifungals for topical use | 10 (11.8) | 9 (13.0) | 5 (13.9) | 6 (14.6) |
Antihistamines for systemic use | 26 (30.6) | 21 (30.4) | 11 (30.6) | 16 (39.0) |
Anti-inflammatory and antirheumatic products, nonsteroids | 26 (30.6) | 26 (37.7) | 18 (50.0) | 21 (51.2) |
Antipropulsives | 58 (68.2) | 38 (55.1) | 16 (44.4) | 19 (46.3) |
Anxiolytics | 19 (22.4) | 8 (11.6) | 6 (16.7) | 7 (17.1) |
Beta-lactam antibacterials, penicillins | 4 (4.7) | 6 (8.7) | 6 (16.7) | 11 (26.8) |
Calcium | 10 (11.8) | 6 (8.7) | 1 (2.8) | 5 (12.2) |
Corticosteroids, plain | 8 (9.4) | 6 (8.7) | 4 (11.1) | 6 (14.6) |
Decongestants and other nasal preparations for topical use | 9 (10.6) | 10 (14.5) | 7 (19.4) | 8 (19.5) |
Drugs for constipation | 59 (69.4) | 33 (47.8) | 19 (52.8) | 24 (58.5) |
Drugs for functional gastrointestinal disorders | 15 (17.6) | 15 (21.7) | 10 (27.8) | 13 (31.7) |
Drugs for peptic ulcer and gastroesophageal reflux disease | 29 (34.1) | 22 (31.9) | 16 (44.4) | 12 (29.3) |
Emollients and protectives | 10 (11.8) | 10 (14.5) | 4 (11.1) | 8 (19.5) |
Hypnotics and sedatives | 26 (30.6) | 19 (27.5) | 14 (38.9) | 10 (24.4) |
Intestinal adsorbents | 30 (35.3) | 19 (27.5) | 5 (13.9) | 11 (26.8) |
Multivitamins, plain | 26 (30.6) | 25 (36.2) | 8 (22.2) | 13 (31.7) |
Muscle relaxants, centrally acting drugs | 11 (12.9) | 6 (8.7) | 7 (19.4) | 4 (9.8) |
Muscle relaxants, peripherally acting drugs | 9 (10.6) | 11 (15.9) | 4 (11.1) | 9 (22.0) |
Other alimentary tract and metabolism products | 21 (24.7) | 12 (17.4) | 9 (25.0) | 10 (24.4) |
Other analgesics and antipyretics | 32 (37.6) | 29 (42.0) | 24 (66.7) | 23 (56.1) |
Other beta-lactam antibacterials | 14 (16.5) | 16 (23.2) | 14 (38.9) | 11 (26.8) |
Other nutrients | 11 (12.9) | 9 (13.0) | 3 (8.3) | 6 (14.6) |
Other plain vitamin preparations | 14 (16.5) | 6 (8.7) | 6 (16.7) | 6 (14.6) |
Psychostimulants, drugs used for ADHD and nootropics | 10 (11.8) | 10 (14.5) | 4 (11.1) | 10 (24.4) |
Vitamins A and D, including combinations | 27 (31.8) | 22 (31.9) | 10 (27.8) | 14 (34.1) |
ADHD = attention deficit hyperactive disorder; PBO = placebo; TRO = trofinetide.
aTreatments reported in at least 10% of patients in either treatment group.
Sources: LILAC and LILAC-2 Clinical Study Reports and sponsor’s Summary of Clinical Evidence.8,45,46 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
RSBQ Total Score
Efficacy results for the RSBQ total score in the OLE studies are presented in Table 22. For patients who received trofinetide in the LAVENDER study, decreases were observed in RSBQ total scores from LAVENDER baseline, with a mean change of –7.3 points (SD = 10.7 points) at week 40 in the LILAC study (n = 44) and –9.8 points (SD = 11.2 points) at week 104 in the LILAC-2 study (n = 10). For those who received placebo in the LAVENDER study, decreases were seen starting at week 2 after switching to trofinetide in the LILAC study, with a mean change from LAVENDER baseline of –7.0 points (SD = 10.7 points) at week 40 in the LILAC study (n = 44) and –13.8 points (SD = 12.0 points) at week 104 in the LILAC-2 study (n = 11). Overall, patients who tolerated trofinetide showed at least a 5-point decrease in RSBQ total score, which persisted throughout the extension studies.
Table 22: Summary of RSBQ Total Score and Change From LAVENDER Baseline in the LILAC and LILAC-2 Studies — Safety Analysis Set
Visit | LILAC | LILAC-2 | ||
|---|---|---|---|---|
LAVENDER PBO to LILAC TRO (N = 85) | LAVENDER TRO to LILAC TRO (N = 69) | LAVENDER PBO to LILAC-2 TRO (N = 36) | LAVENDER TRO to LILAC-2 TRO (N = 41) | |
LILAC baseline and LILAC-2 baseline | ||||
n (%) | 85 (100.0) | 68 (98.6) | 36 (100.0) | 40 (97.6) |
Mean (SD) | 42.8 (13.0) | 39.5 (11.9) | 35.6 (11.8) | 37.2 (13.5) |
Change from LAVENDER baseline, mean (SD) | –1.5 (9.1) | –5.2 (9.1) | –7.3 (11.6) | –7.3 (10.2) |
LILAC week 12 and LILAC-2 week 12 | ||||
n (%) | 74 (87.1) | 61 (88.4) | 35 (97.2) | 40 (97.6) |
Mean (SD) | 37.7 (11.9) | 40.7 (13.3) | 34.6 (12.1) | 39.1 (12.8) |
Change from LAVENDER baseline, mean (SD) | –5.6 (9.8) | –4.9 (9.6) | –8.5 (11.8) | –5.7 (8.5) |
LILAC week 26 | ||||
n (%) | 54 (63.5) | 49 (71.0) | NA | NA |
Mean (SD) | 38.4 (13.5) | 37.2 (12.2) | NA | NA |
Change from LAVENDER baseline, mean (SD) | –6.3 (10.0) | –8.0 (8.5) | NA | NA |
LILAC week 40 and LILAC-2 week 52 | ||||
n (%) | 44 (51.8) | 44 (63.8) | 28 (77.8) | 39 (95.1) |
Mean (SD) | 37.1 (12.2) | 38.4 (14.2) | 35.8 (13.3) | 36.8 (13.7) |
Change from LAVENDER baseline, mean (SD) | –7.0 (10.7) | –7.3 (10.7) | –8.6 (11.0) | –7.6 (10.9) |
LILAC-2 week 104 | ||||
n (%) | NA | NA | 11 (30.6) | 10 (24.4) |
Mean (SD) | NA | NA | 36.2 (12.9) | 31.5 (12.2) |
Change from LAVENDER baseline, mean (SD) | NA | NA | –13.8 (12.0) | –9.8 (11.2) |
NA = not applicable; PBO = placebo; RSBQ = Rett Syndrome Behaviour Questionnaire; SD = standard deviation; TRO = trofinetide.
Notes: LAVENDER baseline is the latest nonmissing value before the first dose of the double-blind study drug in the antecedent study. LILAC baseline is the latest nonmissing value before the first dose of the study drug in the LILAC study. LILAC-2 baseline is the latest nonmissing value before the first dose of study drug in the LILAC-2 study.
Sources: LILAC and LILAC-2 Clinical Study Reports and sponsor’s Summary of Clinical Evidence.8,45,46 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
CGI-I Score
Efficacy results for the CGI-I score in the OLE studies are presented in Table 23. For both OLE studies, changes in the CGI-I score were assessed by the investigator relative to the patients’ baseline state of illness in the LILAC study. As such, there were no CGI-I scores for the baseline visit in the LILAC study.
For patients who received trofinetide in the LAVENDER study, the mean CGI-I score was 3.1 points (SD = 0.7 points) at week 40 (n = 47) of the LILAC study and 3.2 points (SD = 0.9 points) at week 12 (n = 39) of the LILAC-2 study. For patients who received placebo in the LAVENDER study, the mean CGI-I score was 3.2 points (SD = 0.9 points) at week 40 (n = 44) of the LILAC study and 3.0 points (SD = 0.9 points) at week 12 (n = 34) of the LILAC-2 study. Overall, CGI-I scores remained stable over time among patients in the OLE studies.
Table 23: Summary of CGI-I Scores by Selected Visits in the LILAC and LILAC-2 Studies —Safety Analysis Set
Visit | LILAC | LILAC-2 | ||
|---|---|---|---|---|
LAVENDER PBO to LILAC TRO (N = 85) | LAVENDER TRO to LILAC TRO (N = 69) | LAVENDER PBO to LILAC-2 TRO (N = 36) | LAVENDER TRO to LILAC-2 TRO (N = 41) | |
LILAC week 2 | ||||
n (%) | 80 (94.1) | 67 (97.1) | 35 (97.2) | 40 (97.6) |
Mean (SD) | 3.7 (0.6) | 3.6 (0.7) | 3.7 (0.5) | 3.6 (0.7) |
LILAC week 12 | ||||
n (%) | 77 (90.6) | 63 (91.3) | 36 (100.0) | 40 (97.6) |
Mean (SD) | 3.5 (0.8) | 3.6 (0.9) | 3.4 (0.6) | 3.5 (0.8) |
LILAC week 40 | ||||
n (%) | 44 (51.8) | 47 (68.1) | 36 (100.0) | 41 (100.0) |
Mean (SD) | 3.2 (0.9) | 3.1 (0.7) | 3.0 (0.8) | 3.0 (0.7) |
LILAC-2 week 12 | ||||
n (%) | NA | NA | 34 (94.4) | 39 (95.1) |
Mean (SD) | NA | NA | 3.0 (0.9) | 3.2 (0.9) |
CGI-I = Clinical Global Impressions-Improvement; NA = not applicable; PBO = placebo; SD = standard deviation; TRO = trofinetide.
Sources: LILAC and LILAC-2 Clinical Study Reports and sponsor’s Summary of Clinical Evidence.8,45,46 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Other Secondary Outcomes
The results of the other secondary efficacy outcomes relative to the LAVENDER baseline are summarized in Table 24. In general, secondary outcomes remained stable over time among patients who remained in the OLE studies.
Table 24: Change From LAVENDER Baseline at Week 40 in the LILAC Study and at Week 52 in the LILAC-2 Study in Other Secondary Outcomes — Safety Analysis Set
Variable | LILAC | LILAC-2 | ||
|---|---|---|---|---|
LAVENDER PBO to LILAC TRO (N = 85) | LAVENDER TRO to LILAC TRO (N = 69) | LAVENDER PBO to LILAC-2 TRO (N = 36) | LAVENDER TRO to LILAC-2 TRO (N = 41) | |
RTT-COMC | ||||
n (%) | 43 (50.6) | 46 (66.7) | 28 (77.8) | 38 (92.7) |
Mean (SD) | –0.7 (1.2) | –0.5 (1.4) | –1.1 (1.4) | –0.7 (1.7) |
CSBS-DP-IT Checklist | ||||
n (%) | 44 (51.8) | 47 (68.1) | 11 (30.6) | 11 (26.8) |
Mean (SD) | 0.1 (3.20) | 0.2 (3.41) | 2.9 (4.11) | 1.6 (3.20) |
RTT-HF | ||||
n (%) | 44 (51.8) | 46 (66.7) | 28 (77.8) | 39 (95.1) |
Mean (SD) | –0.3 (0.7) | –0.2 (1.0) | –0.2 (0.7) | –0.2 (0.8) |
RTT-AMB | ||||
n (%) | 44 (51.8) | 46 (66.7) | 28 (77.8) | 39 (95.1) |
Mean (SD) | –0.2 (0.7) | –0.2 (0.8) | –0.3 (0.8) | –0.2 (0.7) |
RTT-VCOM | ||||
n (%) | 44 (51.8) | 46 (66.7) | 29 (80.6) | 38 (92.7) |
Mean (SD) | –0.2 (0.8) | –0.3 (0.7) | –0.4 (1.0) | –0.3 (0.7) |
Overall quality-of-life rating | ||||
n (%) | 43 (50.6) | 47 (68.1) | 27 (75.0) | 39 (95.1) |
Mean (SD) | 0.6 (1.0) | 0.4 (0.7) | 0.9 (1.0) | 0.2 (0.9) |
RTT-CBI | ||||
n (%) | 44 (51.8) | 47 (68.1) | 28 (77.8) | 39 (95.1) |
Mean (SD) | –5.6 (13.7) | –1.8 (10.2) | –9.1 (10.2) | –1.3 (14.0) |
CSBS-DP-IT = Communication and Symbolic Behaviour Scales Developmental Profile Infant-Toddler; PBO = placebo; RTT-AMB = Rett Syndrome Clinician Rating of Ambulation and Gross Motor Skills; RTT-CBI = Rett Syndrome Caregiver Burden Inventory; RTT-COMC = Rett Syndrome Clinician Rating of Ability to Communicate Choices; RTT-HF = Rett Syndrome Clinician Rating of Hand Function; RTT-VCOM = Rett Syndrome Clinician Rating of Verbal Communication; SD = standard deviation; TRO = trofinetide
Sources: LILAC and LILAC-2 Clinical Study Reports and sponsor’s Summary of Clinical Evidence.8,45,46 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Refer to Table 25 for harms data. Reported TEAEs included those that started on or after the baseline visit of the current study. TEAEs included those that began in the previous study and were still ongoing at the baseline visit of the current study.
In the LILAC study, 132 patients (85.7%) reported at least 1 TEAE. The most common TEAEs were diarrhea (59.1%), vomiting (25.3%), and COVID-19 (11.0%). SAEs occurred in 19 patients (12.3%), and 48 patients (31.2%) had TEAEs leading to drug withdrawal, primarily due to diarrhea (19.5%) and vomiting (5.8%). No deaths were reported in the LILAC study.
In the LILAC-2 study, 68 patients (88.3%) reported at least 1 TEAE. The most common were COVID-19 (26.0%), diarrhea (16.9%), pyrexia (16.9%), and urinary tract infection (15.6%). SAEs occurred in 23 (29.9%) patients, with 6 (7.8%) discontinuing the drug. The most common SAEs were seizures (6.5%), followed by vomiting, pneumonia, urinary tract infection, and acute respiratory failure (2.6% each). A total of 4 deaths were reported in this study, of which 3 (3.9%) were reported as TEAEs (cardiac arrest, aspiration and vomiting, and sudden unexplained death in epilepsy in 1 patient each).
Table 25: Summary of Harms Results From the LILAC and LILAC-2 Studies — Safety Analysis Set
Adverse events | LILAC (N = 154) | LILAC-2 (N = 77) |
|---|---|---|
Most common TEAEs, n (%)a | ||
Patients with ≥ 1 TEAE | 132 (85.7) | 68 (88.3) |
Diarrhea | 91 (59.1) | 13 (16.9) |
Vomiting | 39 (25.3) | 11 (14.3) |
COVID-19 | 17 (11.0) | 20 (26.0) |
Seizure | 13 (8.4) | 11 (14.3) |
Pyrexia | 12 (7.8) | 13 (16.9) |
Upper respiratory tract infection | 12 (7.8) | 9 (11.7) |
Urinary tract infection | 10 (6.5) | 12 (15.6) |
Decreased appetite | 10 (6.5) | 3 (3.9) |
Decreased weight | 9 (5.8) | 4 (5.2) |
Constipation | 5 (3.2) | 9 (11.7) |
Cough | 5 (3.2) | 6 (7.8) |
Pneumonia | 4 (2.6) | 5 (6.5) |
Viral gastroenteritis | 4 (2.6) | 4 (5.2) |
Dehydration | 3 (1.9) | 4 (5.2) |
Lethargy | 3 (1.9) | 4 (5.2) |
Nasal congestion | 2 (1.3) | 4 (5.2) |
Influenza | 1 (0.6) | 7 (9.1) |
Streptococcal pharyngitis | 1 (0.6) | 6 (7.8) |
Viral upper respiratory tract infection | 1 (0.6) | 4 (5.2) |
Prolonged QT on electrocardiogram | 0 (0.0) | 4 (5.2) |
SAEs, n (%) | ||
Patients with ≥ 1 SAE | 19 (12.3) | 23 (29.9) |
Seizure | 5 (3.2) | 5 (6.5) |
Pneumonia | 4 (2.6) | 2 (2.6) |
Dehydration | 3 (1.9) | 0 (0.0) |
Acute respiratory failure | 2 (1.3) | 2 (2.6) |
Pyrexia | 2 (1.3) | 1 (1.3) |
Rhinovirus infection | 2 (1.3) | 1 (1.3) |
Status epilepticus | 2 (1.3) | 1 (1.3) |
Viral infection | 2 (1.3) | 0 (0.0) |
Urinary tract infection | 1 (0.6) | 2 (2.6) |
Vomiting | 0 (0.0) | 2 (2.6) |
Treatment discontinuation due to TEAEs, n (%) | ||
Patients who stopped | 48 (31.2) | 6 (7.8) |
Diarrhea | 30 (19.5) | 0 (0.0) |
Vomiting | 9 (5.8) | 2 (2.6) |
Decreased weight | 3 (1.9) | 0 (0.0) |
Seizure | 2 (1.3) | 1 (1.3) |
Seizure cluster | 2 (1.3) | 0 (0.0) |
COVID-19 | 1 (0.6) | 1 (1.3) |
Aspiration | 1 (0.6) | 1 (1.3) |
Aspiration pneumonia | 1 (0.6) | 1 (1.3) |
Cardiac arrest | 0 (0.0) | 1 (1.3) |
Sudden unexplained death in epilepsy | 0 (0.0) | 1 (1.3) |
Cardiac arrest | 0 (0.0) | 1 (1.3) |
Deaths, n (%) | ||
Patients who died | 0 (0.0) | 3 (3.9) |
Cardiac arrest | 0 (0.0) | 1 (1.3) |
Aspiration and vomiting | 0 (0.0) | 1 (1.3) |
Sudden unexplained death in epilepsy | 0 (0.0) | 1 (1.3) |
SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aOccurring in at least 5% of patients in either treatment group.
Sources: LILAC and LILAC-2 Clinical Study Reports and sponsor’s Summary of Clinical Evidence.8,45,46 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
The OLE studies provided longer-term efficacy and safety data for trofinetide, for up to 104 weeks. The open-label design increases the potential for bias, particularly in subjective outcomes, such as the RSBQ total score, CGI-I, and AEs. Because completion of the pivotal trial was required for enrolment, patients who discontinued the LAVENDER study were excluded, resulting in a patient population that was more responsive and more tolerant to therapy and introducing selection bias. Additionally, with a high discontinuation rate in the LILAC study (45.5%), mainly due to TEAEs, the selection bias for responders and more tolerant patients is further compounded in the LILAC-2 study, and the impact of patient dropout on outcomes is unclear, as analyses were not conducted to assess how discontinuation affected treatment results.
Indirect Evidence
No indirect treatment comparisons were submitted for the review of trofinetide.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Table 26 summarizes 2 sponsor-submitted studies (DAFFODIL and LOTUS) that provide additional evidence to the systematic review. DAFFODIL is a multicentre, open-label, long-term, phase II/III study conducted in the US to investigate the efficacy and safety of trofinetide in females aged 2 years to 5 years living with Rett syndrome.
LOTUS is an ongoing phase IV, observational, real-world evidence study of patients prescribed trofinetide under routine clinical care in the US for up to 24 months. The study aims to assess the efficacy and tolerability of trofinetide, and interim results at 6 months were submitted by the sponsor.
Table 26: Summary of Gaps in the Systematic Review Evidence
Evidence gap | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
Studies to demonstrate the safety and efficacy of trofinetide in females 5 years or younger. | DAFFODIL is an open-label, phase II/III study of trofinetide for the treatment of females aged 2 years to 5 years who have Rett syndrome. | Exploratory efficacy results demonstrated a decrease in CGI-I scores and an increase in overall quality-of-life ratings from baseline to week 78. Trofinetide demonstrated a consistent safety profile, in line with the results of the primary trial. |
Real-world experience with trofinetide, including patients outside the LAVENDER, LILAC, and LILAC-2 patient populations (e.g., patients aged 20 years or older, those diagnosed with atypical Rett syndrome, and males). | LOTUS is an ongoing, phase IV, observational, real-world, prospective study of patients prescribed trofinetide under routine clinical care. | Trofinetide demonstrated a consistent safety profile, in line with the results of the primary trial. |
CGI-I = Clinical Global Impressions-Improvement.
Source: Sponsor’s Summary of Clinical Evidence.8 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Populations
The DAFFODIL study enrolled 15 females with Rett syndrome, an MECP2 variant, and a CGI-S score of 4 or greater at screening and baseline. The sample size for this study was not based on statistical considerations but was believed to be adequate to characterize the pharmacokinetics of trofinetide in this population. Eligible patients were aged 2 years to 4 years with a body mass that ranged from 9 kg to less than 20 kg or aged 5 years with a body mass that ranged from 9 kg to less than 12 kg. Aside from the age restriction, the inclusion and exclusion criteria were comparable to those used in the pivotal LAVENDER trial.
Patients prescribed trofinetide under routine clinical care in the US were eligible to participate in the LOTUS study. There were no exclusion criteria applied to the LOTUS study.
Interventions
In the DAFFODIL study, trofinetide was dosed by weight and administered orally or by gastrostomy tube; the starting dosage was 2 g (10 mL) twice a day. If well tolerated, the dosage was increased over 3 weeks to a highest dosage of 5 g (25 mL) twice a day for patients weighing 9 kg to 12 kg or of 6 g (30 mL) twice a day for patients weighing 12 kg to less than 20 kg. The mean duration of exposure to trofinetide was 433.7 days (SD = 179.4 days), with a mean daily dose of 8.4 g (SD = 2.2 g). The most commonly used concomitant medications were anilides and propionic acid derivatives, antipropulsives, osmotically acting laxatives, melatonin receptor antagonists, other antiepileptics, and vitamin D.
The LOTUS study is a noninterventional, observational study of patients with Rett syndrome treated with trofinetide in a real-world clinical setting in the US.
Outcomes
Relevant exploratory efficacy outcomes in the DAFFODIL study included the CGI-I score from baseline through to week 104 and the overall quality-of-life rating. The assessment of safety was based on the proportion of patients experiencing TEAEs, SAEs, withdrawals due to TEAEs, and potentially clinically important changes in other safety assessments.
Relevant outcomes from the LOTUS study include the proportion of patients experiencing TEAEs, SAEs, and withdrawals due to TEAEs as of the interim analysis.
Statistical Analysis
In both the DAFFODIL and LOTUS studies, efficacy and safety data were summarized using descriptive statistics.
Patient Disposition
Of the 15 patients enrolled into the DAFFODIL study at 7 US sites, 12 (80%) remained in the study at study termination and received trofinetide for at least 12 months. Mean age at the time of diagnosis was 1.9 years (SD = 0.1 years), and mean age at first Rett syndrome symptoms was 0.85 years (SD = 0.1 years).
In total, 154 patients were included in the interim analysis of the LOTUS study. Most patients had classic Rett syndrome (66.7%) and were female (96.1%), with an age range from 2 years to 60 years. Mean age at the time of diagnosis was 5.2 years (SD = 5.37 years), and mean age at the time of trofinetide initiation was 16.5 years (SD = 11.16 years).
Results
Efficacy
In the DAFFODIL study, the mean CGI-I score decreased from 3.5 points (SD = 0.66 points) at week 2 (n = 13) to 2.2 points (SD = 0.67 points) at week 78 (n = 9). The mean change from baseline in overall quality-of-life rating at week 12 was 0.3 points (SD = 0.72 points), which continued to increase through to week 78, with a mean change of 0.7 points (SD = 0.95 points).
Efficacy outcomes relevant to the systematic review were not assessed in the LOTUS study.
Harms
In the DAFFODIL study, safety outcomes aligned with those of the pivotal and extension studies. In total, 14 (93.3%) patients experienced at least 1 TEAE, the most common of which were diarrhea (73.3%) and vomiting (46.7%). Four (26.7%) patients reported SAEs, including dysphagia, gastroenteritis sapovirus, seizure, and altered state of consciousness. Two (13.3%) patients experienced TEAEs leading to drug withdrawal and study discontinuation consisting of vomiting and diarrhea, respectively. No deaths were reported.
As of the interim analysis in the LOTUS study, 20 patients reported 43 TEAEs, with diarrhea, vomiting, and insomnia being the most common. Other reported TEAEs included asthenia, constipation, fatigue, muscle spasms, and gastrointestinal issues. Five SAEs occurred, including 2 cases of recurrent constipation leading to hospitalization, 1 hospitalization due to severe dehydration from diarrhea, and 1 each for pneumonia, and viral gastroenteritis.
A follow-up interim analysis (for data up to June 26, 2024 [N = 192], including 12-month data from the first patient who enrolled in the LOTUS study) reported that 22 (11.5%) patients experienced 57 TEAEs; the most common were diarrhea (5.2%), vomiting (4.2%), constipation (1.6%), and insomnia (1.6%).47 Six (3.1%) patients reported 10 SAEs (constipation, diarrhea, vomiting, viral gastroenteritis, pneumonia, pneumonia aspiration, and dehydration), 14 (7.3%) patients reported a TEAE leading to discontinuation, and there were no deaths.
Critical Appraisal
Evidence from the DAFFODIL study suggests an acceptable tolerability for trofinetide in a younger cohort of patients, aged 2 years to 5 years; however, few patients were enrolled (N = 15). The longer-term harms data from both the DAFFODIL and LOTUS studies are consistent with those from the pivotal LAVENDER trial, with diarrhea and vomiting being the most common. Although the 2 studies attempt to fill the evidence gaps for patients aged 2 years to 5 years (the DAFFODIL study) or 20 years or older, those diagnosed with atypical disease, or those who are male (the LOTUS study), there remains uncertainty in the study results due to various limitations with the data. Neither study was designed to assess the efficacy and safety of trofinetide in a statistically rigorous manner. Other limitations include potential selection bias, lack of blinding, and lack of comparator group, which may have affected the internal validity of the safety and efficacy results.
Discussion
Summary of Available Evidence
The evidence included in this review consisted of 1 pivotal study identified by the sponsor’s systematic review (the LAVENDER study), 2 OLE studies of the pivotal trial (the LILAC and LILAC-2 studies), and 2 studies addressing gaps in the evidence (the DAFFODIL and LOTUS studies). No indirect treatment comparisons were included.
LAVENDER was a phase III, double-blind, placebo-controlled RCT of trofinetide in female patients aged 5 years to 20 years with classic Rett syndrome (N = 187). Patients were randomized in a 1:1 ratio to 12 weeks of treatment with trofinetide 200 mg/mL twice a day (n = 93), or matching placebo (n = 94). To be eligible, patients must have had a documented disease-causing MECP2 variant, must have been 6 months after regression, and must have had at least moderate disease severity, stable background care, and a stable pattern of seizures. The coprimary end points were the change from baseline in RSBQ and CGI-I score at week 12. Secondary end points relevant to the systematic review focused on communication and symbolic behaviours. Mean age of the patients was 11.0 years (SD = 4.7 years) the trofinetide group and 10.9 years (SD = 4.6 years) in the placebo group, and clinical and disease history characteristics were generally balanced between the groups.
The OLE studies, LILAC (N = 154), and LILAC-2 (N = 77), assessed the longer-term safety and efficacy of trofinetide, for up to an additional 3.5 years (after 12 weeks in the LAVENDER study). Eligible patients must have completed the LAVENDER study, and all patients received trofinetide 200 mg/mL twice a day. After US market approval of trofinetide on March 10, 2023, the LILAC-2 study was terminated by the sponsor. Mean duration of exposure to trofinetide (including the LAVENDER, LILAC, and LILAC-2 studies) was 755.6 days (SE = 20.8 days). Outcomes of interest aligned with those in the LAVENDER study.
DAFFODIL (N = 15) was a multicentre, open-label, long-term, phase II/III study investigating the efficacy and safety of trofinetide in females aged 2 years to 5 years living with Rett syndrome and an MECP2 variant. LOTUS is an ongoing, phase IV, observational, real-world evidence study of patients prescribed trofinetide under routine clinical care in the US for up to 24 months. Outcomes of interest included the CGI-I (the DAFFODIL study) and safety (thee DAFFODIL and LOTUS studies).
Interpretation of Results
Efficacy
Rett syndrome is a rare, neurodevelopmental disorder primarily affecting females that results in loss of communication and motor skills, intellectual disability, and various multisystem issues (including seizures, gastrointestinal dysfunction, and breathing problems). Patients require lifelong care, and it often falls to caregivers and families to manage the heavy burdens of the disease and treatment. At the time of this review, there are no approved disease-modifying therapies for Rett syndrome in Canada (aside from trofinetide), and treatment is largely symptom management aimed at improving HRQoL. Input from patient and clinician groups highlighted the importance of improving overall behavioural symptoms and function, thus improving HRQoL, in patients with Rett syndrome.
The LAVENDER study demonstrated that 12 weeks of treatment with trofinetide resulted in a statistically significant decrease in RSBQ score, compared to placebo (–3.1 points; 95% CI, –5.7 to –0.6 points; P = 0.0175; where a negative indicates improvement); however, the results were considered very uncertain. There were no MIDs identified from the literature for this instrument (or any of the study outcomes), and clinical meaningfulness was based on expert opinion. Based on a threshold of 3 points suggested by the clinical expert, the RSBQ results in the LAVENDER study represent a meaningful benefit with trofinetide, compared to placebo; however, the 95% CI includes the possibility of a difference that is not clinically meaningful from placebo. There are some additional caveats to consider with this outcome. Missing data were an issue for all outcomes in the study (16% and 9% of patients did not contribute week 12 data from the trofinetide and placebo groups, respectively), and the RSBQ is not commonly used in practice. The tool was originally developed to distinguish between a diagnosis of Rett syndrome and a diagnosis of severe intellectual disability,11 and although it has been accepted by the FDA for use in clinical trials,43 it is unclear how appropriate it is for measuring treatment effect over time. Health Canada noted that the RSBQ focuses less on core issues of the disease (e.g., verbal language, regression of hand use, atypical gait, and stereotyped hand movements),48 and there is a lack of information verifying its responsiveness to change.35-37 It has also been suggested that the RSBQ would need to show a large effect size in a clinical trial to be a useful tool, but no estimate was provided in the literature.44 Input from the clinician group stated that the instrument is relevant, and some health care providers may elect to track changes in symptoms with the tool, but not all clinics find it practical to perform in routine visits due to time and resource constraints and the addition to caregiver burden. Results from the LILAC and LILAC-2 studies suggested that trofinetide resulted in further decreases in RSBQ scores for patients who remained on treatment; however, the limitations in the LAVENDER study also apply to the OLE studies, with the added risk of selection bias for patients who were able to better tolerate the treatment. RSBQ was not assessed in either the DAFFODIL or LOTUS studies, and it is not clear what effect trofinetide has on neurobehavioural symptoms of patients outside of those eligible for the LAVENDER study (i.e., females aged 2 years to 5 years, males, patients with atypical Rett syndrome, or patients without MECP2 variants).
Regulators have recommended that, along with caregiver-reported outcomes (i.e., RSBQ), clinician ratings be used to aid in the interpretation of clinical changes; expert opinion supported this approach in practice. The LAVENDER study showed that 12 weeks of treatment with trofinetide led to a reduction in CGI-I score, compared to placebo (–0.3 points; 95% CI, –0.5 to –0.1; where a negative indicates improvement); however, the results were very uncertain. No published MID was available and, based on expert opinion that a 1-point change would be clinically meaningful, it is difficult to know if the modest change represents a clinically meaningful improvement. Results from the LILAC and LILAC-2 studies showed that CGI-I scores had a modest decline over time (less than 1 point) for patients who remained in the studies, but selection bias contributes to the uncertainty of the findings. The DAFFODIL study indicated that treatment with trofinetide also resulted in lower CGI-I scores from week 2 to week 78 for females aged 2 years to 5 years; however, this was an uncontrolled study of 15 patients, thus the results were uncertain. Clinician input noted that the CGI-I has disease-specific anchors, which increases the relevance and utility of the tool, but it requires training and additional resources for proper administration and interpretation, which can restrict its use in clinical practice.
Input from patient and clinician groups ranked improvements in communication and motor skills as being some of the most important outcomes hoped for with new treatments. In the LAVENDER study, nonverbal, prelinguistic, and verbal communication were assessed using the RTT-COMC, CSBS-DP-IT Checklist social composite, and RTT-VCOM instruments, respectively, and hand function and ambulation were assessed using the RTT-HF and RTT-AMB instruments, respectively. Although none of these tools are commonly used in practice, expert opinion indicated that clinical assessment was similar to that of the RTT-COMC. Health Canada noted that the CSBS-DP-IT Checklist was developed as a screening tool to assess communication in healthy infants but it was not intended to track communication skills over time, and the FDA report stated that the tool has not been well validated in patients with Rett syndrome.13,14 Clinicians noted that many patients with Rett syndrome do not communicate verbally and have limited hand function and ambulation, making the RTT-VCOM less useful for detecting changes in verbal communication skills and making the RTT-HF and RTT-AMB less clinically relevant to evaluate changes in motor skills, so it is difficult to detect a change in these outcomes (therefore, none were included for GRADE). No published MIDs were identified for any of the communication or motor skills outcomes, expert opinion indicated that the differences were modest, making it a challenge to interpret how clinically meaningful the changes are, and the certainty of the evidence was considered low or very low. The difference in treatment effect between the trofinetide and placebo groups in the LAVENDER study were minimal for the other outcomes (RTT-VCOM, RTT-HF, and RTT-AMB), and changes over time in the LILAC and LILAC-2 studies were small. Although caregivers value improvements in communication and motor function, it is unclear if the study results translate to a meaningful change in patient and caregiver interactions. The clinical experts explained that, in practice, improvements are often due to effective use of alternative communication technology. It has also been reported that assessing communication in individuals with severe disabilities is difficult, and that improvements observed over time might be the result of a caregiver (or an assessor in a trial setting) learning how to interpret a patient’s communicative intent.13,49
Because Rett syndrome is associated with a heavy symptom and treatment burden and because there are no disease-modifying therapies available in Canada, improving HRQoL is a key goal of treatment. However, there were no comprehensive measures of HRQoL used in the studies, and tools commonly used in clinical trials likely would not be suitable for patients who are unable to communicate. Instead, a single rating from 1 to 6 was provided by caregivers based on their impression of the patient’s overall quality of life (outcome not included for GRADE). It is unlikely that this type of tool could be validated in patients with severe developmental disease, and the accuracy of such ratings is unknown. In the LAVENDER study, the difference between groups was minimal, and changes over time in the LILAC and LILAC-2 studies were small, with relatively large variability among patients who remained in the studies. It is unknown if the changes observed in the studies would meet caregivers’ expectations for meaningful, long-term improvements in patient HRQoL.
Given patients’ need for continual support, reducing caregiver burden was noted as an important outcome in the input received from patient and clinician groups and the clinical experts. According to expert input, this is not an outcome that is consistently measured in practice, and the RTT-CBI used in the LAVENDER study is not used in practice (the outcome was not included for GRADE). In the LAVENDER study, the difference between groups was small for the RTT-CBI. Results from the LILAC and LILAC-2 studies suggested a reduction in caregiver burden over time, but the certainty of the results is diminished by study attrition, large SDs, and apparent variability between caregivers of patients who originally received trofinetide and those who originally received placebo, with no clear reason why. It is also unclear if the study results would represent meaningful changes in the long-term relief of caregiver burden in a real-life setting. The sponsor conducted qualitative interviews with a subset of caregivers of patients who completed the OLE studies. In line with the input received from patient groups, these caregivers highlighted improvements in communication, hand use, seizures, and mobility as the most desired outcomes of treatment with trofinetide. Of the 26 caregivers who participated in these interviews, 21 felt that there were improvements in patients with Rett syndrome during the OLE studies, including in engagement with others (11 of 26 caregivers), hand use (10 of 26 caregivers), eye gaze (8 of 26 caregivers), attention or focus or concentration (7 of 26 caregivers), ability to walk (5 of 26 caregivers), and seizures (4 of 26 caregivers).
Treatment duration in the LAVENDER study was only 12 weeks, and although the LILAC and LILAC-2 studies provide longer-term data, early study termination meant that the mean duration of trofinetide was only approximately 2 years, which is relatively short for a drug used to treat a lifelong disease. Another important limitation of the pivotal trial evidence is that the study population is narrower than the Health Canada–approved population. There is a lack of high-quality RCT evidence for patients who are male, who are younger than 5 years or older than 20 years, who have an atypical or variant Rett syndrome diagnosis, or who have a non-MECP2 variant. The DAFFODIL and LOTUS studies attempt to fill these gaps but had few patients (the DAFFODIL study enrolled 15 patients), had limited efficacy results (CGI-I and safety), and lacked the rigour associated with RCTs. Patients enrolled in the LAVENDER study must have been at least 6 months after regression, which may be reasonable for a clinical trial setting (to enrol patients whose symptoms are mostly stable), but there is a lack of evidence (and long-term data) on a treatment effect with trofinetide during the early onset and rapid regression stages, and it is unclear if the drug is effective at preventing disease progression during the late motor deterioration stage.
The sponsor also submitted an externally led, patient-focused drug development meeting report from 2022,50 in which caregivers were surveyed and discussed the symptoms and daily impacts of Rett syndrome, as well as current and future treatments for the disease. In this report, functional improvements in communication, speech, and hand use were rated the most important aspects for new therapeutics to address. The report also highlighted the lack of research and opportunity to participate in studies for males with Rett syndrome and patients with atypical disease. It also emphasized the importance of study end points that measure improvements that are meaningful to patients and caregivers as well as changes in HRQoL, areas in which current studies do not explore. As discussed throughout the CDA-AMC reimbursement review, it is unclear if trofinetide adequately addresses these unmet needs for improvements in communication, speech, and hand function. As previously noted, the LOTUS study included patients who are male and patients with atypical disease; however, there are important limitations with the study that prevent firm conclusions from being made.
Harms
In the LAVENDER study, nearly all patients in the trofinetide group (92.5%) experienced at least 1 TEAE, compared to just more than half of patients in the placebo group (54.3%). This imbalance was also apparent when looking at the most common TEAEs: diarrhea and vomiting. Because these are harms known to be associated with trofinetide, it is likely that functional unblinding occurred. In the LILAC and LILAC-2 studies, diarrhea and vomiting continued to be some of the more common TEAEs reported; reductions in reported frequency with successive studies may be due to discontinuation of patients for whom the harms were intolerable. Diarrhea and vomiting were also the most frequently reported TEAEs in the DAFFODIL and LOTUS studies. Harms were reported as the number and proportion of patients experiencing AEs, and it is unclear what impact the duration of these events had on patients and caregivers (e.g., potentially reducing HRQoL and increasing caregiver burden).
The frequency of SAEs was low in the LAVENDER study, with 3 patients in each group reporting at least 1 event. The proportion of patients experiencing SAEs was greater in the LILAC and LILAC-2 studies, which may be due to the longer duration of the studies. Few SAEs were reported in the DAFFODIL and LOTUS studies. Withdrawals due to TEAEs were imbalanced in the LAVENDER study, with 17.2% of patients stopping trofinetide and 2.1% of patients stopping placebo. Withdrawals due to TEAEs continued in the OLE studies, with 31.2% and 7.8% of patients stopping trofinetide in the LILAC and LILAC-2 studies, respectively. Diarrhea and vomiting were the most common reasons for stopping trofinetide in the LAVENDER, LILAC, LILAC-2, and DAFFODIL studies. There were no deaths in the LAVENDER, LILAC, or DAFFODIL studies, but 3 deaths were reported in the LILAC-2 study.
Part way through the LAVENDER study, the sponsor developed a diarrhea management plan that recommended that caregivers stop or reduce constipation medications and start fibre once trofinetide was started.51,52 If or when diarrhea occurred, caregivers were to start oral loperamide or another antidiarrhea medication, as needed, and to reduce the dosage of trofinetide if diarrhea persisted. It was also recommended that caregivers carefully monitor diet and hydration. Similar recommendations are found in the Health Canada product monograph.2 Input from patient groups stated that it is important that new treatments have minimal harms and demonstrate long-term tolerability, especially because Rett syndrome is a lifelong disease.
To improve tolerability, dosage titration (and interruptions, if diarrhea occurs) is recommended. Expert opinion indicated that clinicians would titrate at a rate that manages TEAEs and still reaches the target dose, which could be slower in clinical practice than in the trial (target dose was to be reached within the first 6 weeks). The clinical experts expected that AEs may still be intolerable for some patients and caregivers, and discontinuations would still be expected. Input from patient groups that assessed families who had experience with trofinetide described the challenges managing diarrhea and vomiting, with 1 family discontinuing treatment due to the latter.
Conclusion
Rett syndrome is a rare, neurodevelopmental disorder associated with the progressive loss of learned skills, intellectual disability, and various comorbidities. There is a need for safe and effective treatments that address the underlying disease mechanism and improve communication, motor skills, and HRQoL. Based on the evidence from 1 phase III, double-blind RCT (the LAVENDER study), females aged 5 years to 20 years with typical Rett syndrome who have a confirmed, disease-causing MECP2 variant and who receive trofinetide (weight-based dosing) twice a day for 12 weeks are more likely to demonstrate improvements in Rett syndrome–specific neurobehavioural symptoms (measured by the RSBQ). Evidence that trofinetide results in global improvement from a clinician’s perspective or increased communication skills (measured by CGI-I, RTT-COMC, and CSBS-DP-IT Checklist social composite scores) is uncertain because of missing trial data, a lack of MIDs, and the modest treatment effects that may not translate to meaningful changes for patients and caregivers in the real-world setting. It is unclear what impact trofinetide has on HRQoL, or whether changes in motor function or caregiver burden observed in the trial would result in meaningful, long-term improvements for patients or caregivers. Longer-term results (mean time on trofinetide was approximately 2 years) indicated possible further reductions in neurobehavioural symptom frequency, whereas the clinician’s global impression and the patient’s ability to communicate choices remained stable for those who continued in the OLE studies. However, the study design, large number of discontinuations, and relatively short follow-up prevent definitive conclusions from being drawn, as it is likely that those who were receiving perceived benefits remained in the OLE studies longer. Harms, such as diarrhea and vomiting, were a key reason that patients discontinued treatment and are expected to contribute to reduced tolerability, reduced HRQoL, and increased caregiver burden. Furthermore, the Health Canada indication for trofinetide is broader than the pivotal trial population, so there is a lack of rigorous evidence in patients who are male, who are younger than 5 years or older than 20 years, who have an atypical or variant Rett syndrome diagnosis, who have a non-MECP2 variant, and who are in other stages of the disease (i.e., not at least 6 months after regression).
References
1.Acadia Pharmaceuticals Canada Inc. Drug Reimbursement Review sponsor submission: Daybue (trofinetide), solution, 200 mg/mL, oral or gastrostomy tube [internal sponsor's package]. October 17, 2024.
2.Acadia Pharmaceuticals Inc. Daybue (trofinetide): solution, 200 mg/mL, oral or gastrostomy tube [product monograph]. October 11, 2024.
3.Schultz R, Suter B. Post TW, ed. Rett syndrome: Genetics, clinical features, and diagnosis. UpToDate; 2023. Accessed December 3, 2024. https://www.uptodate.com/contents/rett-syndrome-genetics-clinical-features-and-diagnosis
4.Downs J, Wong K, Doshi D, et al. Acadia Pharmaceuticals Canada Inc. response to Canada's Drug Agency request for additional information regarding Daybue (trofinetide) review on May 9, 2025: Longitudinal Rett syndrome behaviour questionnaire scores and their associations with genotype and trajectories of mobility, weight and seizure frequency status [internal additional sponsor's information]. 2025.
5.Kirby RS, Lane JB, Childers J, et al. Longevity in Rett syndrome: analysis of the North American Database. J Pediatr. 2010;156(1):135-138 e1. doi:10.1016/j.jpeds.2009.07.015 PubMed
6.Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68(6):944-50. doi:10.1002/ana.22124 PubMed
7.Fu C, Armstrong D, Marsh E, et al. Consensus guidelines on managing Rett syndrome across the lifespan. BMJ Paediatr Open. 2020;4(1):e000717. doi:10.1136/bmjpo-2020-000717 PubMed
8.Acadia Pharmaceuticals Canada Inc. Daybue (trofinetide): Sponsor Summary of Clinical Evidence [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Daybue (trofinetide), solution, 200 mg/mL, oral or gastrostomy tube. October 29, 2024.
9.Gold WA, Krishnarajy R, Ellaway C, et al. Rett syndrome: A genetic update and clinical review focusing on comorbidities. ACS Chem Neurosci. 2018;9(2):167-176. doi:10.1021/acschemneuro.7b00346 PubMed
10.Neul JL, Percy AK, Benke TA, et al. Trofinetide for the treatment of Rett syndrome: a randomized phase 3 study. Nat Med. 2023;29(6):1468-1475. doi:10.1038/s41591-023-02398-1 PubMed
11.Percy AK, Neul JL, Benke TA, et al. A review of the Rett Syndrome Behaviour Questionnaire and its utilization in the assessment of symptoms associated with Rett syndrome. Front Pediatr. 2023;11:1229553. doi:10.3389/fped.2023.1229553 PubMed
12.Acadia Pharmaceuticals Inc. Clinical Study Report: ACP-2566-003. LAVENDER: A randomized, double-blind, placebo-controlled, parallel-group study of trofinetide for the treatment of girls and women with Rett syndrome [internal sponsor's report]. July 1, 2022.
13.Health Canada. Safety and efficacy review report: Daybue, trofinetide solution, 200 mg/mL, oral, Glycine-Proline-Glutamate (GPE) analog [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Daybue (trofinetide), solution, 200 mg/mL, oral or gastrostomy tube. October 3, 2024.
14.Center for Drug Evaluation and Research. Clinical review(s). Daybue (trofinetide) solution, 200 mg/mL, oral. Company: ACADIA Pharmaceuticals Inc. Application No.:217026Orig1s000. Approval date: 03/10/2023 (FDA approval package). 2023. Accessed December 10, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/217026Orig1s000MedR.pdf
15.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
16.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
17.Neul JL, Benke TA, Marsh ED, et al. The array of clinical phenotypes of males with mutations in Methyl-CpG binding protein 2. Am J Med Genet B Neuropsychiatr Genet. 2019;180(1):55-67. doi:10.1002/ajmg.b.32707 PubMed
18.Lane JB, Salter AR, Jones NE, et al. Assessment of Caregiver Inventory for Rett syndrome. J Autism Dev Disord. 2017;47(4):1102-1112. doi:10.1007/s10803-017-3034-3 PubMed
19.Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68(6):944-50. doi:10.1002/ana.22124 PubMed
20.Xiol C, Heredia M, Pascual-Alonso A, et al. Technological improvements in the genetic diagnosis of Rett syndrome spectrum disorders. Int J Mol Sci. 2021;22(19):26. doi:10.3390/ijms221910375 PubMed
21.Kim SY, Woo H, Lim BC, et al. Exploring the clinical utility of targeted MECP2 testing in real-world practice. Pediatr Neurol. 2024;161:28-33. doi:10.1016/j.pediatrneurol.2024.08.001 PubMed
22.Institut national d'excellence en santé et en services sociaux. MECP2 sequencing for Rett Syndrome. 2014. Accessed December 17, 2024. https://www.cda-amc.ca/sites/default/files/pdf/lab-tests/11_MECP2_Sequencing_for_Rett_Syndrome.pdf
23.Prevention Genetics. Rett Syndrome via the MECP2 gene. 2024. Accessed December 19, 2024. https://www.preventiongenetics.com/testInfo?val=Rett-Syndrome-via-the-MECP2-Gene
24.Alberta Precision Laboratories. Rett Syndrome (synonym: MECP2). 2022. Accessed January 2, 2025. https://www.albertahealthservices.ca/webapps/labservices/indexAPL.asp?id=8611&tests=&zoneid=1&details=true
25.Saskatchewan Health Authority. Rett Syndrome - RRPL. 2024. Accessed January 2, 2025. https://www.saskhealthauthority.ca/intranet/departments-programs/provincial-clinical-and-support-services/laboratory-medicine/laboratory-medicine-compendium/laboratory-test/rett-syndrome-rrpl
26.MUHC Clinical Laboratories. List of testing done out of Québec. 2024. Accessed January 2, 2025. https://cusm.ca/sites/default/files/docs/m-Labs/list-testing-done-out-quebec.pdf
27.St. Boniface Hospital. Lab information manual. 2014. Accessed December 19, 2024. https://apps.sbgh.mb.ca/labmanual/test/view?seedId=13241
28.Percy AK, Neul JL, Benke TA, et al. Trofinetide for the treatment of Rett syndrome: Results from the open-label extension LILAC study. Med. 2024;5(9):1178-1189.e3. doi:10.1016/j.medj.2024.05.018 PubMed
29.Percy AK, Neul JL, Benke TA, et al. Trofinetide for the treatment of Rett syndrome: Long-term safety and efficacy results of the 32-month, open-label LILAC-2 study. Med. 2024;5(10):1275-1281.e2. doi:10.1016/j.medj.2024.06.007 PubMed
30.Acadia Pharmaceuticals Inc. NCT04181723: Study of trofinetide for the treatment of girls and women with Rett syndrome (LAVENDER). ClinicalTrials.gov; 2024. Accessed November 20, 2024. https://clinicaltrials.gov/study/NCT04181723
31.Urbanowicz A, Downs J, Girdler S, et al. An exploration of the use of eye gaze and gestures in females with Rett syndrome. J Speech Lang Hear Res. 2016;59(6):1373-1383. doi:10.1044/2015_jslhr-l-14-0185 PubMed
32.O'Leary HM, Kaufmann WE, Barnes KV, et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann Clin Transl Neurol. 2018;5(3):323-332. doi:10.1002/acn3.533 PubMed
33.Wetherby AM, Guthrie W, Woods J, et al. Parent-implemented social intervention for toddlers with autism: an RCT. Pediatrics. 2014;134(6):1084-93. doi:10.1542/peds.2014-0757 PubMed
34.Anagnostou E, Jones N, Huerta M, et al. Measuring social communication behaviors as a treatment endpoint in individuals with autism spectrum disorder. Autism. 2015;19(5):622-36. doi:10.1177/1362361314542955 PubMed
35.Barnes KV, Coughlin FR, O'Leary HM, et al. Anxiety-like behavior in Rett syndrome: characteristics and assessment by anxiety scales. J Neurodev Disord. 2015;7(1):30. doi:10.1186/s11689-015-9127-4 PubMed
36.Romero-Galisteo R, González-Sánchez M, Costa L, et al. Outcome measurement instruments in Rett syndrome: A systematic review. Eur J Paediatr Neurol. 2022;39:79-87. doi:10.1016/j.ejpn.2022.06.003 PubMed
37.Oberman LM, Downs J, Cianfaglione R, et al. Assessment of a clinical trial metric for Rett syndrome: critical analysis of the Rett Syndrome behaviour questionnaire. Pediatr Neurol. 2020;111:4. doi:10.1016/j.pediatrneurol.2020.04.020 PubMed
38.Leonard H, Gold W, Samaco R, et al. Improving clinical trial readiness to accelerate development of new therapeutics for Rett syndrome. Orphanet J Rare Dis. 2022;17(1):108. doi:10.1186/s13023-022-02240-w PubMed
39.Lotan M, Downs J, Stahlhut M, et al. Evaluation tools developed for Rett syndrome. Diagnostics. 2023;13(10):1708. doi:10.3390/diagnostics13101708 PubMed
40.Sobieski M, Wrona S, Flakus M, et al. Reliability and validity of the Polish version of Communication and Symbolic Behaviour Scales-Developmental Profile-Infant-Toddler Checklist. Res Autism Spectr Disord. 2024;117:102454. doi:10.1016/j.rasd.2024.102454
41.Saldaris J, Leonard H, Wong K, et al. Validating the Communication and Symbolic Behavior Scales–Developmental Profile Infant–Toddler Checklist (CSBS–DP ITC) beyond infancy in the CDKL5 deficiency disorder. J Autism Dev Disord. 2024;54(7):2526-2535. doi:10.1007/s10803-023-06002-w PubMed
42.Eadie PA, Ukoumunne O, Skeat J, et al. Assessing early communication behaviours: structure and validity of the Communication and Symbolic Behaviour Scales—Developmental Profile (CSBS‐DP) in 12‐month‐old infants. Int J Lang Commun Disord. 2010;45(5):572-585. doi:10.3109/13682820903277944 PubMed
43.Mount RH, Charman T, Hastings RP, et al. The Rett Syndrome Behaviour Questionnaire (RSBQ): refining the behavioural phenotype of Rett syndrome. J Child Psychol Psychiatry. 2002;43(8):1099-110. doi:10.1111/1469-7610.00236 PubMed
44.Hou W, Bhattacharya U, Pradana WA, et al. Assessment of a clinical trial metric for Rett syndrome: Critical analysis of the Rett Syndrome Behavioural Questionnaire. Pediatr Neurol. 2020;107:48-56. doi:10.1016/j.pediatrneurol.2020.01.009 PubMed
45.Acadia Pharmaceuticals Inc. Clinical Study Report: ACP-2566-004. LILAC: A 40-week, open-label extension study of trofinetide for the treatment of girls and women with Rett syndrome [internal sponsor's report]. August 17, 2023.
46.Acadia Pharmaceuticals Inc. Clinical Study Report: ACP-2566-005. LILAC-2: An open-label extension study of continuing treatment with trofinetide for Rett syndrome [internal sponsor's report] January 5, 2024.
47.Acadia Pharmaceuticals Inc. Clinical Study Protocol: ACP-2566-014. LOTUS: Real-world benefits and tolderability of Daybue (trofinetide): Interim clinical study report. March 7, 2023.
48.Health Canada. Pharmaceutical safety and efficacy assessment – Advanced consideration of request for notice of compliance with conditions and/or priority review: Daybue (trofinetide): 200 mg/mL, solution, oral or via gastrostomy tube [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Daybue (trofinetide), solution, 200 mg/mL, oral or gastrostomy tube. March 7, 2024.
49.Didden R, Korzilius H, Smeets E, et al. Communication in individuals with Rett Syndrome: an assessment of forms and functions. J Dev Phys Disabil. 2010;22(2):105-118. doi:10.1007/s10882-009-9168-2 PubMed
50.The International Rett Syndrome Foundation (IRSF) and the Rett Syndrome Research Trust (RSRT). Voice of the patient report: Rett syndrome externally-led patient-focused drug development meeting. March 11, 2022.
51.Motil KJ, Beisang A, Smith-Hicks C, et al. Recommendations for the management of gastrointestinal comorbidities with or without trofinetide use in Rett syndrome. Expert Rev Gastroenterol Hepatol. 2024;18(6):227-237. doi:10.1080/17474124.2024.2368014 PubMed
52.Marsh ED, Beisang A, Buie T, et al. Recommendations for the management of diarrhea with trofinetide use in Rett syndrome. Expert Opin Orphan Drugs. 2023;11(1):1-8. doi:10.1080/21678707.2023.2217328
53.Acadia Pharmaceuticals Inc. Acadia Pharmaceuticals Canada Inc. response to Canada's Drug Agency request for additional information regarding Daybue (trofinetide) review on May 9, 2025: Rett syndrome behaviour questionnaire reanalyses [internal additional sponsor's information]. 2024.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Subgroup analysis results for the coprimary end points of RSBQ total score change from baseline to week 12 and CGI-I at week 12 for age subgroups 5 years to 11 years, 12 years to 16 years, and 17 years to 20 years are summarized in Table 27 and Figure 1.
Table 27: Subgroup Analysis Results for Coprimary End Points in the LAVENDER Study — Full Analysis Set
Variable | Trofinetide (N = 91) | Placebo (N = 93) |
|---|---|---|
RSBQ total score for patients aged 5 years to 11 years | ||
Number of patients contributing to the analysis, n (%) | 45 (49.5) | 50 (53.8) |
RSBQ total score at baseline, mean (SD) | 44.5 (11.2) | 46.7 (12.7) |
RSBQ total score at week 12, mean (SD) | 41.0 (11.2) | 45.8 (12.8) |
Change from baseline in RSBQ total score, LSM (SE)a | –4.1 (1.17) | –0.3 (1.11) |
Treatment group difference vs. control, LSM (95% CI)a | –3.8 (–7.0 to –0.6) | |
P valuea,b | 0.0216 | |
RSBQ total score for patients aged 12 years to 16 years | ||
Number of patients contributing to the analysis, n (%) | 17 (18.7) | 20 (21.5) |
RSBQ total score at baseline, mean (SD) | 42.2 (13.1) | 39.5 (11.0) |
RSBQ total score at week 12, mean (SD) | 36.7 (13.5) | 37.0 (12.4) |
Change from baseline in RSBQ total score, LSM (SE)a | –6.6 (1.86) | –3.8 (1.72) |
Treatment group difference vs. control, LSM (95% CI)a | –2.8 (–7.9 to 2.4) | |
P valuea,b | 0.2828 | |
RSBQ total score for patients aged 17 years to 20 years | ||
Number of patients contributing to the analysis, n (%) | 14 (15.4) | 15 (16.1) |
RSBQ total score at baseline, mean (SD) | 43.2 (10.9) | 44.0 (10.4) |
RSBQ total score at week 12, mean (SD) | 39.9 (13.1) | 40.6 (12.3) |
Change from baseline in RSBQ total score, LSM (SE)a | –5.1 (2.7) | –3.5 (2.7) |
Treatment group difference vs. control, LSM (95% CI)a | –1.6 (–9.5 to 6.2) | |
P valuea,b | 0.6751 | |
CGI-I score for patients aged 5 years to 11 years | ||
Number of patients contributing to the analysis, n (%) | 46 (50.5) | 51 (54.8) |
CGI-I score at week 12, mean (SD) | 3.5 (0.8) | 3.8 (0.6) |
CGI-I score at week 12, LSM (SE)a | 3.5 (0.1) | 3.8 (0.1) |
Treatment group difference vs. control, LSM (95% CI)a | –0.3 (–0.6 to 0.0) | |
P valuea,b | 0.0283 | |
CGI-I score for patients aged 12 years to 16 years | ||
Number of patients contributing to the analysis, n (%) | 17 (18.7) | 20 (21.5) |
CGI-I score at week 12, mean (SD) | 3.4 (0.7) | 3.8 (0.6) |
CGI-I score at week 12, LSM (SE)a | 3.5 (0.17) | 3.7 (0.15) |
Treatment group difference vs. control, LSM (95% CI)a | –0.3 (–0.7 to 0.2) | |
P valuea,b | 0.2362 | |
CGI-I score for patients aged 17 years to 20 years | ||
Number of patients contributing to the analysis, n (%) | NR | NR |
CGI-I score at week 12, mean (SD) | NR | NR |
CGI-I score at week 12, LSM (SE)a | NR | NR |
Treatment group difference vs. control, LSM (95% CI)a | NR | |
P valuea,b | NR | |
CGI-I = Clinical Global Impressions-Improvement; CI = confidence interval; LSM = least squares mean; MMRM = mixed effects model for repeated measures; NR = not reported; RSBQ = Rett Syndrome Behaviour Questionnaire; SD = standard deviation; SE = standard error.
aThe MMRM includes age group, baseline RSBQ severity, planned treatment, study visit, treatment-by-visit interaction, baseline-by-visit interaction, and baseline score as fixed effects. An unstructured covariance matrix was used to model within-patient errors. The Kenward-Roger method was used for calculating the denominator degrees of freedom for tests of fixed effects.
bP value has not been adjusted for multiple testing.
Source: LAVENDER Clinical Study Report.8,12
Figure 1: Forest Plots of LSM Treatment Difference With 95% CIs for Coprimary End Points by Age Group — Full Analysis Set
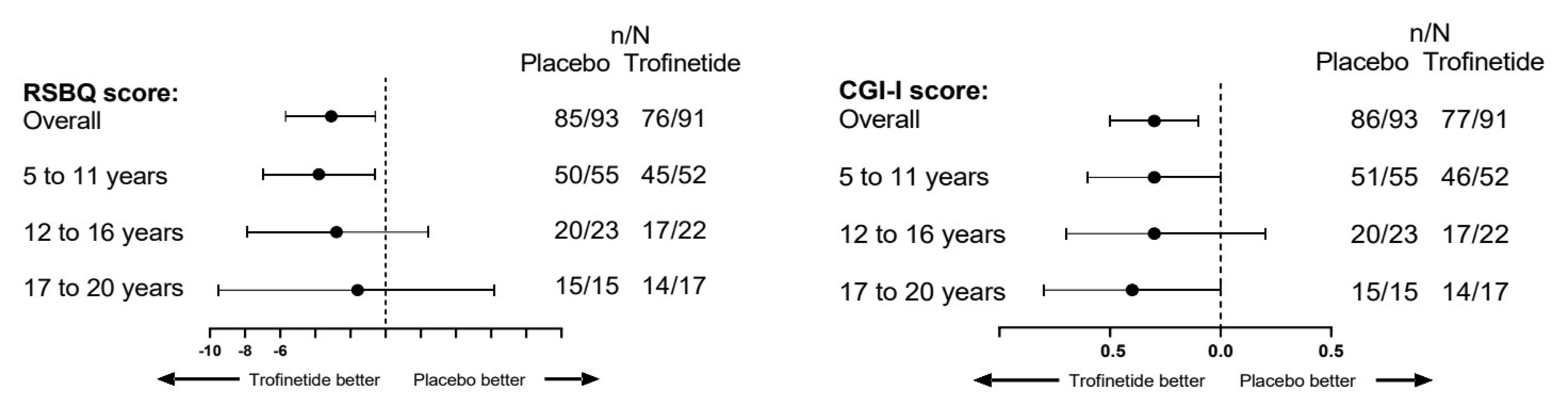
CGI-I = Clinical Global Impressions-Improvement; CI = confidence interval; LSM = least squares mean; RSBQ = Rett Syndrome Behaviour Questionnaire.
RSBQ subscale-level analysis comparing trofinetide and placebo is summarized in Figure 2.
Figure 2: Forest Plot of Treatment Differences in LSM Change From Baseline to Week 12 in RSBQ — Full Analysis Set
CI = confidence interval; LSM = least squares mean; RSBQ = Rett Syndrome Behaviour Questionnaire.
Source: LAVENDER Clinical Study Report.12
Appendix 2: Additional Data Provided to CDA-AMC for the Reconsideration Process
Please note that this appendix has not been copy-edited.
Following the issuance of the draft CDEC recommendation for trofinetide in March 2025, the following additional information was provided to CDA-AMC.
A natural history study by Downs et al. that provides evidence for changes in RSBQ scores in a population of females with genetically confirmed disease who contributed data to the Australian Rett Syndrome Database between 2000 and 2019 (included in the main report);4
An interim analysis from LOTUS for data up to June 26, 2024, with additional harms results (included in the main report);47
An externally led, patient-focused drug development meeting report from 2022 reporting caregiver responses to discussions about the symptoms and daily impacts of Rett syndrome as well as current and future treatments for the disease (included in the main report);50 and
Post hoc item- and domain-level analyses of the RSBQ based on LAVENDER data (included in Appendix 2).53
These references were not included in the original submission to CDA-AMC and were provided by the sponsor to address gaps in the evidence for the systematic review and CDEC initial recommendation decision.


Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BSC
best supportive care
CDA-AMC
Canada’s Drug Agency
CGI-I
Clinical Global Impressions-Improvement
DCE
discrete choice experiment
ICER
incremental cost-effectiveness ratio
QALY
quality-adjusted life-year
RSBQ
Rett Syndrome Behaviour Questionnaire
TEAE
treatment-emergent adverse event
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Trofinetide (Daybue), 450 mL bottle of 200 mg/mL oral solution |
Indication | For the treatment of Rett syndrome in adults and pediatric patients 2 years of age and older and weighing at least 9 kg. |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | October 11, 2024 |
Reimbursement request | As per the Health Canada–approved indication, with the following requested reimbursement criteria. Initiation:
Patients with any of the following should not be eligible for reimbursement of trofinetide if there is evidence of:
Must be prescribed by clinicians with expertise in the diagnosis and management of Rett syndrome For renewal following 12 months of therapy, the physician must provide evidence of beneficial clinical effect, defined as stabilization or improvement in signs/symptoms of Rett syndrome from baseline that is considered clinically beneficial by the treating physician. |
Sponsor | Acadia Pharmaceuticals Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of the Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Rett syndrome in adults and pediatric patients aged 2 years and older and weighing more than 9 kg |
Treatment | Trofinetide plus BSC |
Dose regimen | Patients aged 2 years and older who weigh:a
|
Submitted price | Trofinetide: $13,714.11 per 450 mL bottle of 200 mg/mL oral solution |
Submitted treatment cost | Annual cost (year 1 and year 2 and beyond) for patients who weigh:a
|
Comparator |
|
Perspectives | Publicly funded health care payer Societal perspective |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (79.1 years, until the age of 90 years) |
Key data sources | LAVENDER trial, and long-term extensions LILAC and LILAC-2 |
Submitted results | ICER = $5,864,321 per QALY gained (incremental costs = $3,386,675 and incremental QALYs = 0.578) for both the publicly funded health care payer perspective and societal perspective |
Key limitations |
|
CDA-AMC reanalysis results |
|
AE = adverse event; BSC = best supportive care; CDA-AMC = Canada's Drug Agency; CSR = Clinical Study Report; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; RSBQ = Rett Syndrome Behaviour Questionnaire.
aThe Health Canada product monograph recommends that trofinetide be titrated starting with 50% of the recommended dose taken twice daily, then the dose can be increased over 4 to 8 weeks until the recommended dose is reached.
bAdrenergics, antidepressants, antiepileptics, anxiolytics, laxatives, antipropulsives, drugs for peptic ulcer and gastro-oesophageal reflux disease, muscle relaxants, other alimentary track and metabolism products, and psychostimulants.
Conclusions
The Canada’s Drug Agency (CDA-AMC) Clinical Review noted, that based on evidence from the LAVENDER trial, females aged 5 to 20 years with typical Rett syndrome who have a confirmed, disease-causing MECP2 variant and who received trofinetide (weight-based dosing) twice a day for 12 weeks are more likely to demonstrate improvements in Rett syndrome–specific neurobehavioural symptoms (measured by the Rett Syndrome Behaviour Questionnaire [RSBQ]) than those who did not receive trofinetide. Evidence that trofinetide results in global improvement from a clinician’s perspective or increased communication skills was very uncertain, and there was a lack of information on the impact of trofinetide on patients’ quality of life. Longer-term results from 2 extension studies, LILAC and LILAC-2 (mean duration on trofinetide was approximately 2 years), indicated a possible further reduction in neurobehavioural symptom frequency, and the clinician’s global impression and the patient’s ability to communicate choices remained stable for those who continued in the extension studies. Feedback from clinical experts noted that the RSBQ is not routinely used in practice in Canada. Treatment-emergent adverse events (TEAEs) were more common among patients in the trofinetide group than in the placebo group and were a key reason for discontinuation. TEAEs are expected to contribute to reduced tolerability, decreased quality of life, and increased caregiver burden. The pharmacoeconomic model derived all estimates of treatment efficacy from the LAVENDER trial and the LILAC extension trials. CDA-AMC noted the high discontinuation rate, missing RSBQ data, and the lack of quality-of-life evidence from the trials, which, when considered alongside the limited use of the RSBQ in practice to assess patient health status, results in substantial uncertainty with the clinical information used to inform the cost-effectiveness estimate of trofinetide.
Due to the aforementioned limitations of the clinical evidence, CDA-AMC was not able to generate a robust estimate of cost-effectiveness for trofinetide plus best supportive care (BSC), compared with BSC alone. Based on the sponsor’s submitted results, trofinetide in addition to BSC is more effective (incremental quality-adjusted life-years [QALYs] = 0.578) and associated with greater total costs (incremental costs = $3,386,675) than BSC alone, resulting in an incremental cost-effectiveness ratio (ICER) of $5,864,321 per QALY gained. The sponsor’s results were the same for the public health care payer perspective and societal perspective, as the sponsor assumed that indirect costs associated with caregiver time and productivity were equivalent in both treatment arms and not affected by health state. If a decision-maker is willing to pay $5,864,321 per QALY gained, then trofinetide would be considered cost-effective; if a decision-maker is willing to pay less than $5,864,321 per QALY gained, then a price reduction will be required (Table 5). Due to concerns about some strong assumptions made by the sponsor, CDA-AMC undertook scenario analyses related to the maintenance of treatment effect and discontinuation, which resulted in higher ICERs.
CDA-AMC estimates that approximately 190 patients will receive trofinetide plus BSC over the first 3 years of reimbursement, with an anticipated budget impact of approximately $166 million to the CDA-AMC–participating public drug plans compared to BSC alone. The actual budget impact will depend on the number of eligible patients and the uptake of trofinetide; scenario analyses using alternate assumptions increase the budget impact of reimbursing trofinetide.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from the Ontario Rett Syndrome Association, which provided information gathered from the Canadian Rett Syndrome Registry, as well as Cure Rett Canada and the International Rett Syndrome Foundation, which both included information from US families and the caregivers of patients with Rett syndrome about their experiences with trofinetide. The groups stated that Rett syndrome is a devastating neurodevelopmental disorder that typically becomes apparent at 6 to 18 months of age and impacts nearly every aspect of life; patients can experience a progressive loss of motor skills, cognitive function, and communication abilities, and seizures, behavioural issues, mood disturbances, respiratory problems, gastrointestinal issues, cardiac abnormalities, scoliosis, and osteoporosis. The groups noted that Rett syndrome can be overwhelming, and patients may require constant care, impacting caregivers and families emotionally, physically, and financially. Caregivers may need to reduce or leave employment to provide full-time care, while also facing increased expenses related to medical care, therapies, specialized equipment, home modifications, and travel to specialized centres. Current treatments for Rett syndrome were described as focusing on symptom management, including antiseizure medication; surgical procedures; and physical, occupational, and speech therapies, which do not address the underlying cause of Rett syndrome and, according to the International Rett Syndrome Foundation reporting of an online caregiver poll, typically only control symptoms “somewhat” or “a little.” Some caregivers whose loved ones have experience with trofinetide reported some improvement in communication, motor skills, and seizures; reported side effects mainly included gastrointestinal issues, such as diarrhea and vomiting, and did lead to discontinuation in some patients. The groups emphasized the need for better treatment options and that trofinetide is the only treatment approved for Rett syndrome.
Six members of Canadian Rett Syndrome Consortium, including Acadia Pharmaceuticals Inc. advisory board members for trofinetide, contributed to the clinician group input submitted. The clinicians noted that Rett syndrome is caused by pathogenic variants in the MECP2 gene, leading patients with Rett syndrome to experience a gradual loss of motor function and development, a loss of verbal and social communication skills, seizures, and impairments in behaviour, sleep, breathing, and gastrointestinal motility. Patients with Rett syndrome were noted to require caregiver support and supervision, with management aimed at symptom-based strategies and rehabilitating therapies. The group noted that trofinetide has been shown to impact the mechanism predicted by the genetic mutation associated with Rett syndrome, and that trofinetide would be a first-line treatment for all patients who have Rett syndrome with clinical manifestations consistent with Health Canada–approval specifications. The group stated that patients suspected of having Rett syndrome based on clinical criteria are already tested genetically for pathological variants; therefore, no additional diagnostic testing would be required, and misdiagnosis is unlikely. Outcomes used to determine patient response would most likely rely on caregiver reports of the frequency and severity of symptoms, particularly communication, level of alertness and engagement, respiratory symptoms, decreases in hand and repetitive movements and stereotypies, and improvements in ability to move independently. Although the RSBQ and Clinical Global Impressions-Improvement (CGI-I) scales were noted to be relevant, their application may not be feasible due to clinic time and resource use required, and because of caregiver burden. The discontinuation of trofinetide would be considered when persistent moderate to severe diarrhea cannot be controlled with appropriate medications or dose reductions, persistent vomiting causing weight loss that cannot be likewise controlled, or when there is a lack of improvement in symptoms after 6 to 12 months of therapy. Although the group indicated that trofinetide may facilitate other rehabilitation therapies by increasing alertness and communication, it is not expected to replace any medications or therapies associated with symptom control.
The drug plans noted that there are no therapies approved by Health Canada for the treatment of Rett syndrome and the relevant comparator in the economic model was a basket of drugs that included antiepileptics, antidepressants, adrenergics, gastrointestinal treatments, muscle relaxants, and psychostimulants for symptoms management.
Several of these concerns were addressed in the sponsor’s model:
The comparator in the model was BSC, to reflect that no other indicated options are available.
Quality of life was incorporated in the sponsor’s model through a discrete choice experiment (DCE) study commissioned by an external consulting company.
The societal approach was adopted in the sponsor's model to reflect the economic burden on the caregivers.
In addition, CDA-AMC addressed some of these concerns as follows:
The sponsor assumed sustainable long-term benefits from trofinetide even after discontinuation; however, there is limited evidence to support this assumption. CDA-AMC conducted a scenario analysis to explore the impact of the assumption that patients who discontinued treatments returned to the baseline health state for the duration of the model.
CDA-AMC was unable to address the following concerns raised in patient, clinician, and drug plan input:
The sponsor used RSBQ to measure treatment response in the trial; however, this is not a standard practice, and treatment response is often based on conversations with family or clinical observations.
Economic Review
Economic Evaluation
Summary of the Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing the cost-effectiveness of trofinetide plus BSC compared with BSC alone for the treatment of Rett syndrome in adults and pediatric patients aged 2 years and older and weighing at least 9 kg, which aligns with the proposed reimbursement request. The modelled population is based on the clinical trials (LAVENDER and LILAC), which comprised patients aged 5 to 20 years with Rett syndrome. This is narrower than the Health Canada–proposed indication.
Trofinetide is available as a 450 mL bottle of 200 mg/mL oral solution that can be used for up to 14 days after opening the bottle. The submitted price for trofinetide is $13,714.11.1 Patients start with 50% of the recommended dose of trofinetide and are titrated to a higher dose over 4 to 8 weeks. The recommended dosing for trofinetide is 4 g to 12 g twice daily (morning and evening) depending on patient weight. Trofinetide can be given with or without food and may be administered orally or via a preexisting gastrostomy tube.2 There were no administration costs for trofinetide in the model, because it was assumed to be self-administered. The 12-week (1 model cycle) cost of trofinetide in the model was $84,479 to $211,797 in cycle 1 with titration, and $102,399 to $255,996 in subsequent cycles; annual cost is $455,251 to $1,113,127 (up to 40 kg), assuming zero wastage. BSC comprised concomitant medications observed in the LAVENDER and LILAC trials, the cost per 12-week cycle for BSC in individuals treated with trofinetide was $444 (annual cost = $1,925), and $524 for BSC only (annual cost = $2,271).
The clinical outcomes reported were QALYs and life-years. The analyses were conducted from both the public health care payer and societal perspectives. The time horizon in the base case was a lifetime (79.1 years), and there was a 1.5% annual discount rate for costs and effectiveness.
Model Structure
The sponsor submitted a Markov model with 5 distinct and mutually exclusive health states. Four health states were based on the RSBQ scores: mild to moderate (0 to 25), moderate to severe (26 to 50), severe (51 to 75), and very severe (76 to 90); and dead (Figure 1 in Appendix 3). The RSBQ thresholds to define health states were arbitrarily chosen by the sponsor. The cycle length was 12 weeks, and a half-cycle correction was applied to the calculation of life-years and QALYs. Patients can enter the model in any RSBQ health state and transition between any states or remain in the same state over time. Death is modelled as an absorbing state.
Model Inputs
The baseline population characteristics were derived from the LAVENDER trial (n = 187), a phase III, randomized, double-blind, placebo-controlled trial. The LAVENDER trial evaluated the efficacy and safety of trofinetide in females aged 5 to 20 years with Rett syndrome over 12 weeks, with an initial 4-week to 8-week titration period. The mean age of the patients was 10.9 years, mean body mass index was 17.4 kg/m2, and most patients were moderately ill (34.8%) or markedly ill (42.8%), according to the CGI-I severity score.3,4 After completion of the 12-week, double-blind treatment phase, patients had the option to roll over into the similarly designed open-label extension studies: LILAC (40 weeks; n = 154), and LILAC-2 (104 weeks; n = 78).
The transition probabilities of moving between RSBQ categories were informed by data from the LAVENDER, LILAC, and LILAC-2 trials. Starting RSBQ scores (n = 183) from the LAVENDER trial informed the baseline distribution into health states, and RSBQ category at 12 weeks informed the transition probabilities in the first cycle for both treatment arms. For the trofinetide plus BSC arm of the model, the LILAC RSBQ scores of patients who continued trofinetide informed RSBQ health state transitions in the model over the next 40 weeks, and the LILAC-2 RSBQ scores were used to inform health states for up to 104 weeks. In the event of missing data, the last RSBQ value was carried forward. Beyond 104 weeks, the sponsor assumed that patients in the trofinetide plus BSC arm remained in the same state for the duration of the model, including patients who discontinued trofinetide. It was assumed that patients in the BSC alone arm remained in the same health state as their last state in the LAVENDER trial, based on a published Australian cohort study that indicated that RSBQ scores are relatively stable over time.5 Rett-specific mortality was informed by a study conducted by Kirby et al. (2010),6 which determined longevity in patients with Rett syndrome using a large cohort from a North American database that included patients living in Canada. The sponsor assumed that the treatment would not impact mortality rates, so mortality was equivalent for both treatment arms and irrespective of health state. Treatment discontinuation rates per cycle were calculated using discontinuation data from the LAVENDER and LILAC studies. The proportion of patients who discontinued treatment in the first year was approximately 48%, and discontinuation was assumed to be approximately 10% per year for year 2 and beyond.
Adverse events (AEs) were informed by the LAVENDER, LILAC, and LILAC-2 clinical studies. Only moderate to severe TEAEs related to the study drug observed in at least 5% of patients in either of the studies were included in the model. The most frequently reported TEAEs (≥ 5% of patients in either treatment group) were diarrhea, vomiting, and irritability. None of the gastrointestinal TEAEs were considered serious AEs. AEs were assumed to occur in the first 2 years of treatment and to resolve thereafter, or to be so mild as to not incur significant costs.
A DCE study and caregiver interviews were commissioned to develop utility-based scoring weights for the RSBQ based on the general public in Canada and the UK (N = 561). Disutility weights for each level of each attribute were calculated using the DCE model coefficients and the caregiver valuation task anchors for the best and worst health states, using EQ-5D-5L. Health state utility values for each health state are reported in Table 8. The sponsor assumed AEs did not impact utility values. Furthermore, the sponsor assumed that there were no impacts on quality of life associated with caregivers.
The sponsor included costs related to drug acquisition, health care use, AE management, and indirect costs. Drug acquisition costs for trofinetide were based on the sponsor’s submitted price,1 whereas the weighted average acquisition costs for BSC were obtained from the Ontario Drug Benefit Formulary.7 Health care resource use costs were estimated based on published literature.8,9 It was assumed that health care resource use was the same for each treatment arm and every health state. Costs associated with AEs for gastrointestinal events were estimated by assuming outpatient management and considering medication use in addition to that used in BSC (antipropulsives and antiemetics). Costs for the management of AEs were derived from Alberta’s databases of hospital ambulatory care costs (Comprehensive Ambulatory Care Classification System [CACS] 2019 edition, for code B128 [Disease or Disorder of the Digestive System with or without Minor Intervention]).10 To estimate the indirect costs (caregiver time and productivity loss), caregiving time for spinal muscular atrophy was used as a proxy based on a study conducted by Aranda-Reneo et al.11 of informal caregivers in 4 European countries (France, Germany, Spain, and the UK). An average 8.17 hours per day was assumed to be an approximation of time for a caregiver in Canada, multiplied by the mean hourly wage of $35;12 the indirect cost per cycle was estimated to be $15,613. The sponsor assumed that indirect costs were equivalent in both treatment arms and were not affected by health state due to the lack of data to assume differences in cost. The sponsor commented that symptom improvement may lead to a decrease in caregiver time.
Summary of the Sponsor’s Economic Evaluation Results
The sponsor’s base-case analysis was run probabilistically (8,000 probabilistic iterations, based on model convergence). The deterministic and probabilistic results were similar. The probabilistic findings are presented here. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
In the sponsor’s base-case analysis, trofinetide plus BSC was associated with 0.578 additional QALYs and $3,386,675 additional costs compared to BSC alone over a lifetime horizon (disaggregated results are presented in Appendix 3). The ICER for trofinetide plus BSC was $5,864,321 per QALY gained, compared to BSC alone. Trofinetide plus BSC had a 0% probability of being cost-effective at willingness-to-pay thresholds of $50,000 per QALY gained and of $100,000 per QALY gained.
Almost all the incremental cost ($3,386,315) was attributable to the drug acquisition cost of trofinetide. The key driver of QALY gains for trofinetide plus BSC was the greater proportion of patients in the less severe RSBQ health states (Table 13 in Appendix 3). No additional life-years were gained because the sponsor assumed that trofinetide would not affect survival.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. BSC ($/QALY) |
|---|---|---|---|---|---|
Results from the societal perspective | |||||
BSC alone | 2,093,941 | Reference | 14.144 | Reference | Reference |
Trofinetide plus BSC | 5,480,617 | 3,386,675 | 14.721 | 0.578 | 5,864,321 |
Results from the publicly funded health care payer perspectivea | |||||
BSC alone | 241,414 | Reference | 14.144 | Reference | Reference |
Trofinetide plus BSC | 3,628,090 | 3,386,675 | 14.721 | 0.578 | 5,864,321 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aResults for the public payer perspective were derived by removing the indirect costs ($1,852,527). Deterministic analysis results are reported in the next section.
Source: Sponsor’s pharmacoeconomic submission.
Sensitivity and Scenario Analysis Results
The sponsor conducted several one-way sensitivity analyses and deterministic scenario analyses to test the impact of alternative parameters and assumptions on the modelled results for trofinetide plus BSC, compared with BSC alone. The largest change from the base-case results was seen with variations in the utility value in the moderate to severe state ($3,217,467 per QALY gained to $15,823,786 per QALY gained) and the utility value in the severe state ($3,339,168 per QALY gained to $13,418,556 per QALY gained).
The sponsor conducted deterministic analyses for the societal and public payer perspectives. In these analyses, the ICER was $5,347,601 per QALY gained, compared with BSC alone.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations of the sponsor’s analysis that have notable implications for the economic analysis:
An uncertain relationship between RSBQ and clinically meaningful health-related quality of life status: The sponsor used the caregiver-reported RSBQ score as the outcome measure to determine the health status of patients with Rett syndrome in their economic model. Clinical expert feedback obtained by CDA-AMC for this review indicated that RSBQ scores are not a clinical tool used in current clinical practice in Canada; it is not implemented systematically and is not the standard of care. In Canada, given the broad spectrum of the condition and a general lack of objective markers, the outcomes or health status of individuals with Rett syndrome are usually evaluated with subjective measures, such as conversations with members of the patient’s family, clinical observations, patient history, or biomedical markers. The sponsor acknowledged in its submission that as Rett syndrome “is a very heterogeneous disease, the RSBQ has limitations in providing a comprehensive manifestation of the totality of the disease.”1 Furthermore, the CDA-AMC Clinical Review Report noted that RSBQ focuses less on core issues of the disease (e.g., verbal language, regression of hand use, atypical gait, and stereotyped hand movements), and there is a lack of information verifying its responsiveness to change. The sponsor justified using wide ranges in its arbitrary RSBQ thresholds for the health states to ensure a detectable difference in mean health utility between states. Clinical expert feedback received by CDA-AMC for this review indicated that the arbitrary thresholds used by the sponsor to define health states in the model were not meaningful. As a result of the limitations associated with the sponsor-defined health states, the sponsor’s utility values (collected using a DCE that was adapted by a third-party calculator to determine utility values for the health states) are associated with uncertainty. CDA-AMC noted that there was a notable amount of missing RSBQ data, especially at later time points in the extension studies; only 38 (42%) and 11 (12%) patients reported RSBQ scores at the end of 52 and 104 weeks, respectively. Given the limitations identified with the model pathway and the relevance of the clinical outcomes — that RSBQ is not used in clinical decision-making for patients with Rett syndrome in Canada — the sponsor’s model lacks face validity in terms of its ability to reflect health state occupancy and changes in health-related quality of life with treatment, either in the short term or long term. Any estimated QALYs produced by this model are, therefore, highly uncertain.
CDA-AMC was not able to address this in its reanalysis, as substantial uncertainty exists in absolute and incremental QALYs.
The long-term efficacy of trofinetide is unknown, and the persistence of any benefit after discontinuation is not established: The sponsor assumed that patients who discontinued treatments would remain in the same health state for the duration of the model (i.e., 79 years), indicating that any clinical improvement obtained from trofinetide would be sustained indefinitely, even after discontinuation. CDA-AMC noted that there were missing RSBQ data for a high proportion of individuals in the extension trials; additionally, feedback from the clinical experts consulted by CDA-AMC indicated that treatment benefit is unlikely to be maintained beyond treatment discontinuation. As a result, there is no robust evidence to support the assumption of a treatment effect beyond discontinuation; thus, the benefit associated with trofinetide plus BSC is overestimated.
In the CDA-AMC scenario analysis, transition probabilities were recalculated (refer to Appendix 4 for details) to test the robustness of the results by changing to the assumption that patients returned to the baseline health state after discontinuation for the duration of the model.
The long-term discontinuation rate is uncertain: The sponsor’s model assumed an annual discontinuation rate of 10% for years 2 and beyond, although the extension trials only informed discontinuation rates up to 2 years. This assumption resulted 82% of patients discontinuing trofinetide by year 10 in the model. Clinical expert feedback obtained by CDA-AMC indicated that discontinuation rates are likely to be lower; for example, by mitigating the TEAEs through slower titration. As such, it is plausible that more patients will remain on the drug over time than modelled, with a subsequent greater total drug acquisition cost over time.
In the CDA-AMC reanalysis, the long-term discontinuation rate was set to 0% after 2 years (the end of the LILAC-2 trial), resulting in 55% of patients discontinuing trofinetide by year 10.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
AEs were assumed to occur within the first 2 years of treatment and assumed to resolve thereafter, or to be mild so as to not incur significant costs. | Uncertain. There are no trial data to inform on AEs after 2 years. However, the AE costs were small relative to the drug costs and would not significantly impact the cost-effectiveness results. |
No disutilities were attributed to AEs. | Reasonable. AEs (diarrhea and vomiting) were assumed to be mild and to have resolved. The clinical experts consulted by CDA-AMC also indicated that diarrhea and vomiting can be mitigated. However, these side effects may lead to the discontinuation of treatment in some patients. |
Mortality remained the same across treatment arms and across health states. | Acceptable. There is no robust evidence to indicate that trofinetide will impact survival in patients with Rett syndrome. |
Health care resource use remained the same across treatment arms and across health states. | Not acceptable. The sponsor assumed no difference in health care resource use with treatment. Clinical expert input obtained by CDA-AMC noted that patients may be monitored more frequently if they were receiving a drug for their condition. |
Atypical Rett syndrome and male-specific characteristics were not modelled. | Acceptable. Clinical expert feedback solicited by CDA-AMC suggested that males and patients with atypical Rett syndrome are very different populations from patients with typical Rett syndrome, and that most clinicians would not prescribe trofinetide to those populations without further clinical evidence. |
Due to a lack of data specific to patients with Rett syndrome, costs associated with health care resource use and caregiver burden hours were informed by literature in other disease areas (e.g., genetic diseases and SMA). | Uncertain. Although health care resource use and indirect costs were assumed to be the same in both treatment arms and across health states, and thus did not impact the cost-effectiveness results, it is unclear whether SMA can be considered a proxy for Rett syndrome. The sponsor noted that this condition requires lifelong, around-the-clock care and assistance with all aspects of daily living but stated that treatment with trofinetide was not expected to modify these outcomes. |
AE = adverse event; CDA-AMC = Canada's Drug Agency; SMA = spinal muscular atrophy.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
Given the limitations that CDA-AMC identified in the sponsor’s economic submission, CDA-AMC was unable to use the model to derive robust estimates of the cost-effectiveness of trofinetide or to help quantify the impact of uncertainty. The model was based on health states using RSBQ scores for treatment response that have an unclear relationship with the evaluation used in the assessment of patients with Rett syndrome.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor’s base case (Table 5). A 99% price reduction would be required for trofinetide plus BSC to be considered cost-effective at a willingness-to-pay threshold of $100,000 per QALY gained compared with BSC alone.
CDA-AMC conducted a scenario analysis to explore the impact of the sponsor's assumption that patients who discontinued treatments would remain in the same state for the duration of the model. This change resulted in $3,383,240 of added incremental costs (versus the sponsor’s base-case estimate of $3,386,675 per patient), 0.476 QALYs gained (versus the sponsor’s base-case estimate of 0.578 QALYs), and the ICER increased to $7,102,879 per QALY gained.
An additional scenario analysis was performed to explore the impact of discontinuation rate beyond 2 years. In this scenario, the discontinuation rate was set to 0 after year 2. This change resulted in $9,814,871 in incremental costs, and the ICER increased to $17,053,041 per QALY gained.
Table 5: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost ($) | ICERs for trofinetide plus BSC vs. BSC ($/QALY) |
|---|---|---|
Price reduction | Sponsor base case | |
No price reduction | 13,714.11 | 5,864,321 |
10% | 12,342.70 | 5,211,401 |
20% | 10,791.29 | 4,680,067 |
30% | 9,599.88 | 4,130,749 |
40% | 8,228.47 | 3,505,709 |
50% | 6,857.06 | 2,969,262 |
60% | 5,485.64 | 2,352,342 |
70% | 4,114.23 | 1,744,933 |
80% | 2,742.82 | 1,186,239 |
90% | 1,371.41 | 583,415 |
98% | 274.28 | 111,984 |
99% | 137.14 | 52,622 |
BSC = best supportive care; CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: The results are applicable to both the societal and public payer perspectives.
Issues for Consideration
Genetic testing for MECP2 mutations in patients with Rett syndrome is likely to be needed if trofinetide becomes funded. This might incur extra costs for patients without a documented MECP2 mutation. However, clinical expert feedback indicated that most current patients with suspected Rett syndrome would have a genetic test to confirm a molecular diagnosis of Rett syndrome, as clinical guidelines on Rett syndrome management recommend that patients with suspected Rett syndrome receive MECP2 testing for confirming a Rett syndrome diagnosis.
CDA-AMC undertook a scan of molecular testing inventories and received feedback from the clinical experts and CDA-AMC–participating public drug plans that indicated that there is publicly funded genetic testing for Rett syndrome in some provinces; in other provinces, the test may be paid by patients as out-of-pocket costs. The test may be preapproved for select medical specialties, but physicians in other specialties may have to inquire about getting their patients tested. Jurisdictions may undertake the preparatory work but send the material out of province for testing. In some jurisdictions, it may take up to 6 months to receive test results. Refer to the Testing Procedure Considerations section of the Clinical Review for further information.
Clinical expert feedback noted that there is a new version of the RSBQ currently in development. It is unclear whether the RSBQ version used in the trial will be generalizable to the new version of the RSBQ. Additionally, it is uncertain whether uptake of an updated RSBQ would be different to current practice.
A slow titration to the target dose is recommended (4 to 8 weeks) to mitigate AEs. If this occurs in clinical practice, the treatment cost of trofinetide during the titration phase might be lowered.
The study population in the LAVENDER trial is narrower than the Health Canada–approved population; patients were aged between 5 years and 20 years at initiation in the LAVENDAR trial, whereas the indication is for individuals aged 2 years or older. The clinical experts indicated that there may be some clinicians who would like to access trofinetide for individuals aged less than 2 years. Clinical expert feedback noted that there might be off-label use in individuals with a variant Rett syndrome diagnosis or with a non-MECP2 variant.
Overall Conclusions
The CDA-AMC Clinical Review noted that, based on the evidence from the LAVENDER trial, females aged 5 to 20 years with typical Rett syndrome who have a confirmed, disease-causing MECP2 variant and who received trofinetide (weight-based dosing) twice a day for 12 weeks are more likely to demonstrate improvements in Rett syndrome–specific neurobehavioural symptoms (measured with the RSBQ) than those who did not receive trofinetide. Evidence that trofinetide results in global improvement from a clinician’s perspective or increased communication skills was very uncertain, and there was a lack of information informing how trofinetide impacts patients’ quality of life. Longer-term results from 2 extension studies — LILAC and LILAC-2 (mean duration treated with trofinetide was approximately 2 years) — indicated a possible further reduction in neurobehavioural symptom frequency, and the clinician’s global impression and patient’s ability to communicate choices remained stable for those who continued in the extension studies. Feedback from the clinical experts noted that the RSBQ is not routinely used in practice in Canada. TEAEs were more common among patients in the trofinetide group than in the placebo group, were a key reason for discontinuation, and are expected to contribute to reduced tolerability, decreased quality of life, and increased caregiver burden. The pharmacoeconomic model derived all estimates of treatment efficacy from the LAVENDER trial and LILAC extension trials. CDA-AMC noted the high discontinuation rate, missing RSBQ data, and the lack of quality-of-life evidence from the trials, which, when considered alongside the limited use of the RSBQ in practice to assess patient health status, results in substantial uncertainty with the clinical information used to inform the cost-effectiveness estimate of trofinetide.
CDA-AMC identified limitations in the sponsor’s pharmacoeconomic submission. First, RSBQ is not the standard of care in Canada, and the clinical experts indicated treatment response is usually determined by conversations with family, clinical observation, or biomedical markers. Further, the modelled relationship between RSBQ score changes and clinically important changes in health-related quality of life is highly uncertain. The trial results were extrapolated over a 79-year time horizon, and 93% of incremental QALYs estimated occurred beyond the observation period in the LAVENDER and LILAC trials. The clinical benefits were assumed to be maintained after discontinuation and for the duration of the model. The sponsor also assumed that patients who discontinued treatment would remain in the same health state for the duration of the model, which might be optimistic. The discontinuation rate beyond year 2 was not supported by clinical evidence and might underestimate the treatment costs associated with trofinetide in the long term.
Due to the aforementioned limitations of the clinical evidence, CDA-AMC was not able to generate a robust estimate of cost-effectiveness for trofinetide plus BSC, compared with BSC alone. Based on the sponsor’s submitted results, trofinetide plus BSC is more effective (incremental QALYs = 0.578) and associated with greater total costs (incremental costs = $3,386,675) than BSC alone, resulting in an ICER of $5,864,321 per QALY gained. The sponsor’s results were the same for the public health care payer perspective and societal perspective because the sponsor assumed that indirect costs associated with caregiver time and productivity were equivalent in both treatment arms and were not affected by health state. If a decision-maker is willing to pay $5,864,321 per QALY gained, then trofinetide would be considered cost-effective; if a decision-maker is willing to pay less than $5,864,321 per QALY gained, then a price reduction would be required (Table 5). Due to concerns about some strong assumptions made by the sponsor, CDA-AMC undertook scenario analyses related to the maintenance of treatment effect and discontinuation, which resulted in higher ICERs.
CDA-AMC estimates that approximately 190 patients will receive trofinetide plus BSC over the first 3 years of reimbursement, with an anticipated budget impact of approximately $166 million on the CDA-AMC–participating public drug plans, compared to BSC alone. The actual budget impact will depend on the number of eligible patients and the uptake of trofinetide; scenario analyses using alternate assumptions increase the budget impact of reimbursing trofinetide.
References
1.Acadia Pharmaceuticals Canada Inc. Pharmacoeconomic evaluation [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Daybue (trofinetide), solution, 200 mg/mL, oral or gastrostomy tube. October 28, 2024. URL.
2.Acadia Pharmaceuticals Inc. Daybue (trofinetide): solution, 200 mg/mL, oral or gastrostomy tube [product monograph]. October 11, 2024.
3.Neul JL, Percy AK, Benke TA, et al. Trofinetide for the treatment of Rett syndrome: a randomized phase 3 study. Nat Med. 2023;29(6):1468-1475. doi:10.1038/s41591-023-02398-1 PubMed
4.Acadia Pharmaceuticals Inc. Clinical Study Report: ACP-2566-003. LAVENDER: A randomized, double-blind, placebo-controlled, parallel-group study of trofinetide for the treatment of girls and women with Rett syndrome [internal sponsor's report]. July 1, 2022.
5.Leonard H, Wong K, Doshi D, et al. Rett Syndrome Behaviour Questionnaire (RSBQ): The natural history of RSBQ scores [poster]. presented at: 9th World Rett Congress; 2024;
6.Kirby RS, Lane JB, Childers J, et al. Longevity in Rett syndrome: analysis of the North American Database. J Pediatr. 2010;156(1):135-138 e1. doi:10.1016/j.jpeds.2009.07.015 PubMed
7.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary [sponsor supplied reference].
8.Marshall DA, Benchimol EI, MacKenzie A, et al. Direct health-care costs for children diagnosed with genetic diseases are significantly higher than for children with other chronic diseases. Genet Med. 2019;21(5):1049-1057. doi:10.1038/s41436-018-0289-9 PubMed
9.May DM, Neul JL, Satija A, et al. Real-world clinical management of individuals with Rett syndrome: a physician survey. J Med Econ. 2023;26(1):1570-1580. doi:10.1080/13696998.2023.2286778 PubMed
10.Data from: Hospital Ambulatory Care costs - CACS 2019 edition [sponsor supplied reference]. 2019.
11.Aranda-Reneo I, Peña-Longobardo LM, Oliva-Moreno J, et al. The burden of spinal muscular atrophy on informal caregivers. Int J Environ Res Public Health. 2020;17(23):8989. doi:10.3390/ijerph17238989 PubMed
12.Statistics Canada. Table 14-10-0134-01. Average weekly earnings, average hourly wage rate and average usual weekly hours by union status, annual. Updated 2024-09-17. Accessed by sponsor, no date provided. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1410013401
13.Acadia Pharmaceuticals Inc. Prescribing information: Daybue (trofinetide) oral solution. U.S. Food and Drug Administration,; 2024. Accessed January 7, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/217026s001s003lbl.pdf
14.Acadia Pharmaceuticals Canada Inc. Acadia Pharmaceuticals Canada Inc. response to Canada's Drug Agency request for additional information regarding Daybue (trofinetide) review on December 23, 2024 [internal additional sponsor's information]. January 6, 2025.
15.Acadia Pharmaceuticals Inc. Clinical Study Report: ACP-2566-004. LILAC: A 40-week, open-label extension study of trofinetide for the treatment of girls and women with Rett syndrome [internal sponsor's report]. August 17, 2023.
16.Acadia Pharmaceuticals Inc. Clinical Study Report: ACP-2566-005. LILAC-2: An open-label extension study of continuing treatment with trofinetide for Rett syndrome [internal sponsor's report]. January 5, 2024. .
17.Acadia Pharmaceuticals Canada Inc. Acadia Pharmaceuticals Canada Inc. response to Canada's Drug Agency request for additional information regarding Daybue (trofinetide) review on December 3, 2024 [internal additional sponsor's information]. December 6, 2024.
18.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed January 21, 2025. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
19.Acadia Pharmaceuticals Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Daybue (trofinetide), solution, 200 mg/mL, oral or gastrostomy tube. October 29, 2024.
20.Acadia Pharmaceuticals Inc. Acadia Pharmaceuticals announces Health Canada Approval of Daybue (trofinetide) for the treatment of Rett syndrome. 2024. Accessed January 17, 2025. https://acadia.com/en-ca/media/news-releases/acadia-pharmaceuticals-announces-health-canada-approval-daybuetm
21.Tarquinio DC, Motil KJ, Hou W, et al. Growth failure and outcome in Rett syndrome: specific growth references. Neurology. 2012;79(16):1653-61. doi:10.1212/WNL.0b013e31826e9a70 PubMed
22.Acadia Pharmaceuticals Inc. Data on File. IQVIA rare disease anonymized patient-level data with payer data from November 1, 2016 to October 31, 2019 [sponsor supplied reference].
23.Dyke P, Leonard H, eds. The Australian Rett syndrome study report 2006. Telethon Institute for Child Health Research; 2006. Accessed January 23, 2025. https://rett.thekids.org.au/siteassets/media-docs—rett-syndrome/2006_report.pdf#:~:text=The%20majority%20of%20women%20who%20indicated%20they,level%20secretarial%20government%20job%20prior%20to%20my
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 6: CDA-AMC Cost Comparison for Add-On Therapies for the Treatment of Rett Syndrome in Adult and Pediatric Patients
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($)a |
|---|---|---|---|---|---|---|
Trofinetide (Daybue) | 200 mg/mL | 450 mLa oral solution for oral or gastronomy tube use | 13,714.1100 | Twice daily doses by patient weight: 9 to < 12 kg: 4g 12 to < 20 kg: 6g 20 to < 35 kg: 8 g 35 to < 50 kg: 10 g ≥ 50 kg: 12 g Titration: start with 50% of recommended dose (e.g., 2g twice daily for a 9 kg patient), increasing over 4 to 8 weeks until recommended dose reached | Maintenance 1,219.03 to 3,657.10 | Maintenance 445,251 to 1,335,754 |
Costs assume a 365.25 day year and do not include dispensing fees or markups.
aNote that while the product monograph states that trofinetide is supplied in a 500 mL bottle, the actual volume of trofinetide within each bottle is not specified.2 Upon request, the sponsor clarified that each bottle contains 450 mL of trofinetide 200 mg/mL oral solution, consistent with the description of the product in the Federal Drug Administration product label.13,14
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The study population is narrower than the Health Canada–approved population. There is a lack of high-quality RCT evidence for patients who are male, younger than 5 years or older than 20 years, with an atypical or variant Rett syndrome diagnosis, or with a non-MECP2 variant. |
Model has been adequately programmed and has sufficient face validity | No | Model structure lacks face validity. Refer to CDA-AMC critical appraisal section for limitations. The model lacks face validity due to the uncertain relationship between RSBQ score and changes and health-related quality of life. |
Model structure is adequate for decision problem | No | Using RSBQ and arbitrary threshold may not accurately model quality of life. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
RCT = randomized controlled trial; RSBQ = Rett Syndrome Behaviour Questionnaire.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 8: Sponsor’s Health State Utility Values
Health State | Mild to Moderate RSBQ Score: 0 to 25 | Moderate to Severe RSBQ Score: 26 to 50 | Severe RSBQ Score: 51 to 75 | Very Severe RSBQ Score: 76 to 90 |
|---|---|---|---|---|
Utility Value | 0.6409 | 0.5386 | 0.4158 | 0.2160 |
Detailed Results of the Sponsor’s Base Case
Table 9: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Trofinetide plus BSC | BSC alone |
|---|---|---|
Discounted LYs | ||
Total | 27.417 | 27.417 |
RSBQ Mild to Moderate | 3.455 | 2.823 |
RSBQ Moderate to Severe | 20.633 | 17.099 |
RSBQ Severe | 3.329 | 7.494 |
RSBQ Very Severe | 0.000 | 0.000 |
Discounted QALYs | ||
Total | 14.721 | 14.144 |
RSBQ Mild to Moderate | 2.217 | 1.811 |
RSBQ Moderate to Severe | 11.121 | 9.216 |
RSBQ Severe | 1.383 | 3.116 |
RSBQ Very Severe | 0.000 | 0.000 |
Discounted costs ($) | ||
Total | 5,480,617 | 2,093,941 |
Drug Acquisition Cost | 3,448,671 | 62,356 |
Treatment Health care Resource Use Cost | 179,058 | 179,058 |
Adverse Event Management Cost | 360 | 0 |
Indirect Costsa | 1,852,527 | 1,852,527 |
aIncluded in the sponsor’s analysis from the societal perspective.
BSC = best supportive care; RSBQ = Rett Syndrome Behaviour Questionnaire.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of Scenario Analyses
A series of scenario analyses were performed on the sponsor’s base case to investigate the impact of critical assumptions on the cost-effectiveness of trofinetide plus BSC compared to BSC. These scenario analyses explored the impact of the following model parameters and assumptions on the ICER: assuming patients returned to original health state after discontinuation or missing RSBQ score (refer to the following for new transition probabilities); and assuming 0% discontinuation rate after 2 years (Table 13).
Table 10: New Transition Probabilities Based on the LAVENDER Trial at Week 12
Disease severity | Baseline BSC (N = ██) Trofinetide + BSC (N = 91) | Mild to Moderate RSBQ Score: 0 to 25 n (%) | Moderate to Severe RSBQ Score: 26 to 50 n (%) | Severe RSBQ Score: 51 to 75 n (%) | Very Severe RSBQ Score: 76 to 90 n (%) |
|---|---|---|---|---|---|
Mild to Moderate RSBQ Score: 0 to 25 | BSC (n = ██) | █████ | | █████ | ██ | ██ |
Trofinetide + BSC (n = ██) | █████ | | █████ | ██ | ██ | |
Moderate to Severe RSBQ Score: 26 to 50 | BSC (n = ██) | ███████ | ██ ███████ | | ██████ | ██ |
Trofinetide + BSC (n = ██) | ██████ | ██ ███████ | ██ | ██ | |
Severe RSBQ Score: 51 to 75 | BSC (n = ██) | | ██████ | | ███████ | ██ ███████ | ██ |
Trofinetide + BSC (n = ██) | | ██████ | ██ ███████ | ██ ███████ | ██ | |
Very Severe RSBQ Score: 76 to 90 | BSC (n = ██) | ██ | ██ | ██ | ██ |
Trofinetide + BSC (n = ██) | ██ | ██ | ██ | ██ |
BSC = best supportive care; RSBQ, Rett Syndrome Behaviour Questionnaire.
Source: Sponsor’s pharmacoeconomic submission Tables 26 to 31 and subject disposition.
Table 11: New Transition Probabilities Based on the LILAC Trial at Week 40 (Trofinetide Plus BSC)
Disease severity | Baseline BSC (N = ██) Trofinetide + BSC (N = 91) | Mild to Moderate RSBQ Score: 0 to 25 n (%) | Moderate to Severe RSBQ Score: 26 to 50 n (%) | Severe RSBQ Score: 51 to 75 n (%) | Very Severe RSBQ Score: 76 to 90 n (%) |
|---|---|---|---|---|---|
Mild to Moderate RSBQ Score: 0 to 25 | Trofinetide + BSC (n = ██) | ███████ | | ███████ | ██ | ██ |
Probability per cycle | ██ | ██████ | ██ | ██ | |
Moderate to Severe RSBQ Score: 26 to 50 | Trofinetide + BSC (n = ██) | ██████ | ██ ███████ | ██████ | ██ |
Probability per cycle | ██████ | ██ | ██████ | ██ | |
Severe RSBQ Score: 51 to 75 | Trofinetide + BSC (n = ██) | ██ | ██ | ██ ██████ | ██ |
Probability per cycle | ██ | ██ | ██ | ██ | |
Very Severe RSBQ Score: 76 to 90 | Trofinetide + BSC (n = ██) | ██ | ██ | ██ | ██ |
Probability per cycle | ██ | ██ | ██ | ██ |
BSC = best supportive care; NA = not applicable; RSBQ, Rett Syndrome Behaviour Questionnaire.
Note: Probabilities not required for remaining in the same state.
Source: Sponsor’s pharmacoeconomic submission Tables 26 to 31 and subject disposition.
Table 12: New Transition Probabilities Based on the LILAC-2 Trial at Week 52 (Trofinetide Plus BSC)
Disease severity | Baseline BSC (N = ██) Trofinetide + BSC (N = 91) | Mild to Moderate RSBQ Score: 0 to 25 n (%) | Moderate to Severe RSBQ Score: 26 to 50 n (%) | Severe RSBQ Score: 51 to 75 n (%) | Very Severe RSBQ Score: 76 to 90 n (%) |
|---|---|---|---|---|---|
Mild to Moderate RSBQ Score: 0 to 25 | Trofinetide + BSC (n = ██) | ███████ | | ███████ | ██ | ██ |
Probability per cycle | ██ | ██████ | ██ | ██ | |
Moderate to Severe RSBQ Score: 26 to 50 | Trofinetide + BSC (n = ██) | | ██████ | ██ ███████ | | ██████ | ██ |
Probability per cycle | ██████ | ██ | ██████ | ██ | |
Severe RSBQ Score: 51 to 75 | Trofinetide + BSC (n = ██) | ██ | | ███████ | ██ ███████ | ██ |
Probability per cycle | ██ | ██████ | ██ | ██ | |
Very Severe RSBQ Score: 76 to 90 | Trofinetide + BSC (n = ██) | ██ | ██ | ██ | ██ |
Probability per cycle | ██ | ██ | ██ | ██ |
BSC = best supportive care; NA = not applicable; RSBQ, Rett Syndrome Behaviour Questionnaire.
Note: Probabilities not required for remaining in the same state.
Source: Sponsor’s pharmacoeconomic submission Tables 26 to 31 and subject disposition.1
The transition probabilities were assumed to be the same as BSC (0%) at week 104 and onward due to missing RSBQ scores.
Table 13: Additional Scenario Analysis Results
Analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor's base case | BSC | $2,093,941 | 14.144 | Reference |
Trofinetide + BSC | $5,480,617 | 14.721 | $5,864,321 | |
Scenario 1: Assumption of patients returning to baseline health state | BSC | $2,091,889 | 14.069 | Reference |
Trofinetide + BSC | $5,475,129 | 14.545 | $7,102,879 | |
Scenario 2: 0% discontinuation rate after year 2 | BSC | $2,092,486 | 14.128 | Reference |
Trofinetide + BSC | $11,907,356 | 14.704 | $17,053,041 |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus; [add as required].
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) estimating the expected incremental budget impact of reimbursing trofinetide for the treatment of Rett syndrome in the requested reimbursement population (i.e., patients 2 years or older, weighing 9 kg or more, with a confirmed Rett syndrome diagnosis in accordance with international consensus guidelines as described in Table 1 of this report). The sponsor submitted a revised budget impact report and analysis in January 2025. The following inputs and results are reflective of these revised files. The BIA was conducted from the perspective of the pan-Canadian drug plans over a 3-year time horizon (2025 to 2027). The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets, excluding Quebec, as well as the Non-Insured Health Benefits Program (NIHB). The sponsor’s base case includes drug acquisition costs only. Trofinetide is assumed to not displace any other therapy, but BSC is assumed to be different between those using and not using trofinetide due to between-group background therapies in Study 003. As bottles of trofinetide are stable with refrigeration for 14 days after opening, and treatment with the maintenance dose appropriate for the smallest body weight category would take approximately 11 days to finish a 450 mL 200 mg/mL bottle, wastage was not assumed. Key inputs to the BIA are documented in Table 15.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target Population | |
Prevalent population diagnosed with Rett syndrome | ███ / ███ / ███a |
Proportion patients with public coverage | 72%b |
Number of patients eligible for drug under review | ███ / ███ / ███ |
Market Uptake (3 years, reference scenario)c | |
Trofinetide plus BSC | 0% / 0% / 0% |
BSC | 100% / 100% / 100% |
Market Uptake (3 years, new drug scenario)c | |
Trofinetide plus BSC | ███ / ███ / ███ |
BSC | ███ / ███ / ███ |
Cost of treatment (per patient, per year)d | |
Trofinetide | $851,615 Year 1, $890,503 Years 2 or 3 |
BSC | $1,930 with trofinetide, $2,271 when used alonee |
Cost modifiers | |
Adherence rate | ██%f |
Proportion trofinetide patients remaining on trofinetide at 12 months | |
Proportion of 12-month patients remaining on trofinetide at 24 months | 89.6%16 |
Proportion of 24-month patients remaining on trofinetide at 36 months | 89.6%16 |
Length of treatment for patients discontinuing trofinetide within 12 months |
BSC = best supportive care.
aAs identified within a sponsor-commissioned study identifying patients in Ontario with an ICD-10-CA code F84.2 on a hospital record during the selection period of 2018 to 2022, with cases from 2002 to 2018 accrued if the patient was still alive. Results (n = ███ in 2018 to 2023, inflated to ███ in 2024, the base year of the BIA) were then extrapolated to estimate the number of patients within CDA-AMC-participating jurisdictions (i.e., the 9 provinces, excluding Quebec, and the NIHB).17
bBased on a weighted average of the proportion of the general population eligible for public plans in each jurisdiction.18
cCited as data on file based on US sales data and Canadian market research for year 1 and internal projections for years 2 and 3 (data not provided).
dCosts in this table are based on a mean patient weight of 31.31 kg19 and are reported before adjustments for adherence or discontinuation.
eCosts for BSC when used alone or with trofinetide are based on the proportion of patients using specific concomitant medications in the placebo and trofinetide groups of the LAVENDER study, respectively, and include costs for: salbutamol, trazodone, levetiracetam, clonazepam, lamotrigine, oxcarbazepine, clobazam, diazepam, sennosides, loperamide hydrochloride, lansoprazole, baclofen, levocarnitine, and clonidine.4,19
fCited as data on file, US compliance rate. Upon query by CDA-AMC, the sponsor clarified that the adherence rate was calculated based on the ratio of the number of bottles that were shipped to US patients through the US Patient Support Program relative to the number that should have been shipped if taken as directed (based on their weight).14
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analysis suggest that the reimbursement of trofinetide for the treatment of Rett syndrome in the requested reimbursement population (i.e., patients ≥ 2 years, weighing ≥ 9 kg, with a confirmed Rett syndrome diagnosis in accordance with international consensus guidelines as described in Table 1) would be associated with a three-year incremental cost of $118,401,892 (year 1: $38,238,039, year 2: $38,200,818, year 3 $41,963,035) when added on to BSC alone. Under the sponsor’s base-case assumptions, 95 patients would start publicly reimbursed trofinetide in year 1, 21 in year 2, and 22 in year 3.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of patients with Rett syndrome in Canada is uncertain: The sponsor commissioned a database study to identify the number of patients with Rett syndrome in Ontario, based on patients having ICD-10 code F84.2 within their health care record during the study period. The ███ patients identified within the database study were then extrapolated to the rest of Canada, and adjusted for population differences between the study period and the years of the BIA, resulting in an estimated ███ patients in CDA-AMC-participating jurisdictions in Year 1 of the analysis. According to clinical expert input obtained by CDA-AMC, as well as a news release published by the sponsor,20 there are approximately 600 to 900 patients with Rett syndrome in Canada. The estimate of ███ patients is therefore consistent (i.e., when accounting for the removal of patients who live in Quebec or the territories and who are not clients of NIHB), but at the lower end of this estimate. According to clinical expert input obtained by CDA-AMC, not all patients would be expected to have code F84.2 in their health records, as patients may have instead received a code for more generic genetic abnormalities. Additionally, some patients may have been coded for their primary reason for visiting a health care institution within the study period (e.g., digestive issues, acute illness) without additional coding for their underlying condition. As such, it is unclear what proportion of patients with Rett syndrome in Ontario were captured by the database study.
CDA-AMC conducted a scenario analysis increasing the estimated number of patients with Rett syndrome by 50%, consistent with what would be expected if prevalence is at the higher end of the estimated 600 to 900 patients in Canada range.
The average cost of trofinetide treatment is underestimated: The sponsor’s model assumes a weighted average patient weight of 31 kg, with all patients then assumed to receive 40 mL (8 g) of trofinetide per dose, appropriate for a patient weighing 31 kg. However, in clinical practice, each individual patient would receive the dose appropriate for their body weight, not the dose appropriate for the average patient in the population. Additionally, in calculating the weighted average patient weight, the sponsor included patients weighing less than 9 kg, who are not part of the Health Canada indicated population.
When estimating the per patient cost of a weight-based therapy, using the mean body weight of a population is a better representation of what it would cost to treat the average patient than the median is. Body weight is a skewed parameter, with median typically lower than mean due to floor effects. This appears to be true when considering growth charts from patients with Rett syndrome, where the 98th percentile is further from the median than the second percentile is for any given age, indicating a skew in weight distribution rather than a normal curve.21 In estimating the weight of patients with Rett syndrome, the sponsor’s analysis used median weight at each age, thus underestimating the average dose and thus the average cost of therapy, per person.
Finally, the sponsor used American data22 to estimate the distribution of ages in patients with Rett syndrome, rather than data from Canada.17 While the sponsor noted that the distribution of patient ages was expected to be the same between the US and Canada, this is not consistent with the data submitted as part of the database study conducted in Ontario. For example, in the US data, ████% of patients were less than 20 years of age, while in the Ontario data, ██% of patients were at least 21 years of age. This suggests that patients in Canada may be slightly older than those in the US, which in turn would mean that patients in Canada may weigh more, on average, than those in the US.
In its reanalysis, CDA-AMC included costs consistent with the dosing of trofinetide for each weight category for the proportion of modelled patients within that category. Patients younger than 2 years of age or weighing less than 9 kg were not included, in line with the product monograph. CDA-AMC was unable to adjust for the use of median weight by age rather than mean, nor for the use of US rather than age group data from Canada; it is likely that both outstanding issues lead to underestimates in the average cost of treating patients with Rett syndrome in Canada and thus underestimates in the estimated budget impact of trofinetide.
Proportion of patients who are publicly funded is underestimated: The sponsor’s analysis assumed that a weighted average of 72% of patients with Rett syndrome will be publicly reimbursed for trofinetide, consistent with the proportion of the general population of CDA-AMC-participating jurisdictions who are eligible for public drug funding.18 However, according to clinical expert opinion obtained by CDA-AMC, adult patients with Rett syndrome are unlikely to be employed at the same rate as the general population, and caregivers of children with Rett syndrome are more likely than the general population to be unemployed or underemployed due to their caregiving responsibilities,23 reducing their level of household income and their likelihood of access to private drug insurance. As such, it is likely that the proportion of patients with Rett syndrome who are funded by public plans is substantially higher than that of the general population. In real world evidence provided by the sponsor,14 95% of identified patients with Rett syndrome in Ontario (234 of 246) had at least 1 public drug claim during the 5-year study period.
In its reanalysis, CDA-AMC assumed that in jurisdictions where fewer than 100% of the general population is eligible for public drug plan funding, 95% of patients with Rett syndrome would have public funding. Jurisdictions where 100% of the population is eligible for public funding remained at 100%.
Assumption that trofinetide alters BSC is inappropriate: While the proportion of patients on each permitted background therapy in Study 003 (LAVENDER) differed between the trofinetide and placebo groups,4 it is unclear what proportion of these differences, if any, was due to the addition of trofinetide rather than resulting from variation between individual patients in the 2 groups. As such, unless the assumption is made that between-group differences in background therapies are caused by the use of trofinetide, it is inappropriate to assume differences between background BSC therapies with and without trofinetide in the BIA, an analysis intended to reflect costs within the same group of patients if trofinetide is and is not funded.
In its reanalyses, CDA-AMC assumed that patients receiving only BSC would receive the same BSC medications in the same proportions as those also receiving trofinetide.
Relative dose intensity is uncertain: Based on data from the US Patient Support Program for Daybue, the sponsor estimated that patients within the BIA use trofinetide at a ██% relative dose intensity (RDI).14 This consideration of RDI is uncertain as this parameter can be influenced by several factors. The dose received by a patient may differ from the fully planned dose of the drug due to dose delays, missed doses, dose reductions to manage toxicity, or subsequent dose re-escalation; each of these have differing impacts on drug costs. Additionally, adherence rates derived from a US patient support program may not reflect adherence rates for patients reimbursed by public drug plans in Canada.
In a scenario analysis, the RDI for trofinetide was set to 100%.
The discontinuation rate is uncertain: The sponsor’s analysis assumed that of patients initiating trofinetide therapy, 48% would discontinue treatment after an average of 3.3 months, with a further 10.5% discontinuing between month 12 to 24 (at month 15.3) as well as between month 24 to 36 (at month 27.3). As discussed in the CDA-AMC appraisal of the sponsor’s economic evaluation, discontinuation after month 24 is based on assumption and is thus uncertain. Assuming the same rate in later years as in year 2 of treatment may underestimate the long-term cost of treatment with trofinetide. Additionally, while it may be appropriate to assume that patients discontinuing trofinetide in the first year of treatment do so at an average of 3.3 months, based on discontinuation within Study-003 and Study-004,4,15 it is likely that patients discontinuing in years 2 or 3 of their therapy would be more evenly distributed throughout the year in question. Clinical expert input obtained by CDA-AMC also indicated that in clinical practice, discontinuation of trofinetide may be slower than seen in the clinical studies, occurring at an average of 6 months after initiation.
In a scenario analysis, CDA-AMC assumed a 0% discontinuation rate after 24 months of treatment with trofinetide. As the model was not programmed to allow for discontinuation at different time points depending on year of discontinuation, CDA-AMC conducted an additional scenario where discontinuation occurred 6 months into each year, rather than 3.3.
The estimated uptake of trofinetide is uncertain: The sponsor’s analysis assumed that in its first year of funding, ██% of identified patients with Rett’s syndrome would try trofinetide, with an additional ██% starting treatment in years 2 and 3 (for a total of ██% and ██% by years 2 and 3, respectively). However, according to clinical expert input obtained by CDA-AMC, this is likely an underestimate, and the proportion of patients or their caregivers requesting access to trofinetide might approach 90% to 100%, with an estimated 50% of eligible patients receiving it in its first year of funding, rising to 70 to 80% having trialled it by Year 3. As such, the sponsor’s estimated market share may underestimate the proportion of patients with Rett syndrome who would try trofinetide, should it be funded.
In a scenario analysis, CDA-AMC assumed that the uptake (i.e., market share) of trofinetide would be double the sponsor’s estimate.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analysis by assuming dosing consistent with the distribution of patient weights of modelled patients, by increasing the proportion of patients who will be eligible for public funding, and by assuming that BSC does not change due to the addition of trofinetide therapy. The changes applied to derive the CDA-AMC base case are described in Table 16.
Table 16: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Dosing by weight distribution | 8 g BID: 100% | 4 g BID: 5.2% 6 g BID: 8.4% 8 g BID: 26.8% 10 g BID: 54.7% 12 g BID: 0% Patients less than 2 years old or less than 9 kg were excluded from weighted dosing calculations and from receiving trofinetide. |
2. Increased eligibility for public funding | Weighted average of 72% | Weighted average of 97% |
3. BSC equal between treatments | Proportion of patients using each BSC medication differs between groups. BSC only group: $2,271 per year | Proportion of patients using each BSC medication is the same between groups. BSC only group: $1,930 per year. |
CDA-AMC base case | 1 + 2 + 3 | |
BID = twice daily; BSC = best supportive care.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18. For the treatment of Rett syndrome in the requested reimbursement population (i.e., patients ≥ 2 years, weighing ≥ 9 kg, with a confirmed Rett syndrome diagnosis in accordance with international consensus guidelines as described in Table 1), CDA-AMC reanalyses suggest that the reimbursement of trofinetide will be associated with a 3-year budgetary cost of $166,461,725 (year 1: $53,775,386; year 2: $53,706,466; year 3: $58,979,873). Under the CDA-AMC base-case assumptions, 129 patients would start publicly funded trofinetide in year 1, 29 in year 2, and 30 in year 3. The average annual cost per full year of trofinetide therapy, excluding reductions for adherence, would be $885,774 in Year 1 and $926,221 in Years 2 and 3.
Table 17: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | $118,401,892 |
CDA-AMC reanalysis 1: Dosing by weight distribution | $123,139,064 |
CDA-AMC reanalysis 2: Increased eligibility for public funding | $159,974,867 |
CDA-AMC reanalysis 3: BSC equal between treatments | $118,465,826 |
CDA-AMC base case (1 + 2 + 3) | $166,461,725 |
BIA = budget impact analysis.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 18:
Increasing the estimated number of patients with Rett syndrome by 50% to reflect the upper end of prevalence estimates.
Assuming a 100% RDI.
Assuming that 0% of patients discontinue after 24 months of trofinetide therapy.
Assuming all discontinuations occur 6 months into the year of discontinuation, rather than 3.3 months.
Assuming that the uptake of trofinetide is doubled. Under this scenario, 258 patients would start publicly funded trofinetide in Year 1, 57 in year 2, and 59 in year 3.
Table 18: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $851,075 | $866,316 | $881,836 | $897,638 | $3,496,865 |
New drug | $851,075 | $39,104,356 | $39,082,654 | $42,860,673 | $121,898,757 | |
Budget impact | $0 | $38,238,039 | $38,200,818 | $41,963,035 | $118,401,892 | |
CDA-AMC Base Case | Reference | $977,585 | $994,863 | $1,012,452 | $1,030,359 | $4,015,259 |
New drug | $977,585 | $54,770,249 | $54,718,918 | $60,010,232 | $170,476,984 | |
Budget impact | $0 | $53,775,386 | $53,706,466 | $58,979,873 | $166,461,725 | |
CDA-AMC scenario analysis 1: 50% more patients with Rett syndrome | Reference | $1,466,378 | $1,492,294 | $1,518,678 | $1,545,538 | $6,022,888 |
New drug | $1,466,378 | $82,155,373 | $82,078,377 | $90,015,348 | $255,715,476 | |
Budget impact | $0 | $80,663,079 | $80,559,699 | $88,469,809 | $249,692,587 | |
CDA-AMC scenario analysis 2: 100% RDI | Reference | $977,585 | $994,863 | $1,012,452 | $1,030,359 | $4,015,259 |
New drug | $977,585 | $75,619,811 | $75,544,316 | $82,880,471 | $235,022,183 | |
Budget impact | $0 | $74,624,948 | $74,531,864 | $81,850,112 | $231,006,924 | |
CDA-AMC scenario analysis 3: 0% discontinuation after 2 years treatment | Reference | $977,585 | $994,863 | $1,012,452 | $1,030,359 | $4,015,259 |
New drug | $977,585 | $54,770,249 | $54,718,918 | $63,081,312 | $173,548,064 | |
Budget impact | $0 | $53,775,386 | $53,706,466 | $62,050,953 | $169,532,805 | |
CDA-AMC scenario 4: discontinuation occurs 6 months in all years | Reference | $977,585 | $994,863 | $1,012,452 | $1,030,359 | $4,015,259 |
New drug | $977,585 | $63,645,062 | $57,680,304 | $63,155,443 | $185,458,394 | |
Budget impact | $0 | $62,650,199 | $56,667,852 | $62,125,084 | $181,443,135 | |
CDA-AMC scenario 5: trofinetide uptake doubled | Reference | $977,585 | $994,863 | $1,012,452 | $1,030,359 | $4,015,259 |
New drug | $977,585 | $108,545,635 | $108,425,384 | $118,990,105 | $336,938,709 | |
Budget impact | $0 | $107,550,772 | $107,412,931 | $117,959,746 | $332,923,450 |
BIA = budget impact analysis; RDI = relative dose intensity.
Ethics Review
Abbreviations
CDA-AMC
Canada’s Drug Agency
CGI-I
Clinical Global Impression-Improvement
OLE
open-label extension
RSBQ
Rett Syndrome Behaviour Questionnaire
Ethical Considerations
Rett syndrome is a rare, life limiting, genetic neurodevelopmental disorder, primarily caused by mutations in MECP2. After a brief period of seemingly normal development, people with Rett syndrome progress through a series of 4 stages: developmental stagnation (age 6 to 18 months), rapid regression (age 1 to 4 years), symptom stabilization (age 2 to 10 years), and late motor deterioration (age ≥ 10 years).1 Symptoms are heterogenous and may include stereotypic hand movements, seizures, intellectual disabilities, mobility impairments, and breathing irregularities. Although Rett syndrome can occur in males, it is primarily found in females.
This brief report is informed by the sponsor’s submission and by patient group, clinician group, and drug plan input received by Canada’s Drug Agency (CDA-AMC). Direct consultation with 3 clinical experts (a pediatrician and 2 pediatric neurologists) who have experience treating patients with Rett syndrome also informed this report.
This brief report highlights ethical considerations regarding the use of trofinetide (Daybue) for the treatment of Rett syndrome in adults and pediatric patients aged 2 years and older weighing at least 9 kg. It outlines considerations relevant to decision-making regarding the public reimbursement and implementation of trofinetide in Canada. However, it does not necessarily present an exhaustive list of all ethical considerations associated with Rett syndrome and its treatment.
Diagnosis, Treatment, and Experiences of People Living With Rett Syndrome
Diagnostic Complexity and Equity Concerns
Rett syndrome includes classic and atypical forms, which differ in patterns of regression and severity. Although 95% to 97% of classic cases and 50% to 70% of atypical cases are associated with pathogenic MECP2 mutations,2 some individuals lack identifiable genetic variants, adding complexity to diagnosis and treatment planning. Although not required for the diagnosis of Rett syndrome, clinical experts indicated that genetic confirmation is commonly pursued and generally accessible in Canada. Patient group input noted that families in rural areas may face greater challenges in accessing genetic testing, raising concerns about equity. The phenotypic variability of Rett syndrome, even among those with MECP2 mutations, further complicates efforts to predict disease progression and develop equitable and broadly applicable treatment options. This variability also poses challenges when defining treatment eligibility, as relying solely on a clinical diagnosis or genetic status may inadvertently exclude some individuals who could benefit.
Challenges in Caregiving and the Burden of Illness
Caring for individuals with Rett syndrome is challenging due to the condition’s complexity and unpredictability. Patient group input highlighted acute concerns (e.g., seizures, respiratory distress, feeding difficulties, and mobility issues) that place a heavy emotional, physical, and financial burden on families. The regression after an initial period of typical development is especially distressing, with some caregivers describing it as if “someone pulled the plug on everything.” Many caregivers have reported reducing work hours or leaving employment entirely to provide care, exacerbating financial strain. Patient group input also underscored a lack of societal resources to support families managing these burdens, contributing to feelings of isolation and unmet need.
Lack of Disease-Modifying Treatments and Barriers to Care
Currently, there are no disease-modifying treatments for Rett syndrome, and care is focused on symptom management to improve health-related quality of life. Interventions include pharmacological treatments for seizures, gastrointestinal disturbances, and sleep issues. It also includes nonpharmacological approaches, like physical therapy for motor impairments, speech therapy to address communication challenges, and occupational therapy to support daily activities. This multidisciplinary care often requires coordination across multiple specialists. There are only 5 Rett specialty clinics in Canada — all located in urban centres — and families without access to these clinics are frequently left to organize and manage complex care pathways themselves. Delays in accessing care, particularly for families in rural or remote areas, may limit opportunities to mitigate complications or improve outcomes. Symptom management approaches also fail to address the underlying disease mechanisms, leaving families without the hope of slowing disease progression. This may contribute to emotional distress and a sense of unmet need. Access to supportive services (e.g., speech therapy, physiotherapy) is further complicated by funding structures that often require alternative diagnoses (e.g., autism spectrum disorder) for coverage. This can lead to out-of-pocket costs and/or the need for additional advocacy from families. These barriers highlight significant inequities in accessing timely, comprehensive care for a population already managing substantial burdens.
Therapeutic Needs and Expectations
Input from both clinical experts and patient groups emphasized the urgent need for therapies that address the ability to communicate, motor function, cognition, and seizure management. Caregivers consistently highlighted communication — whether speech, gestures, or assistive technologies — as a top priority, as even small improvements could significantly enhance their ability to understand and respond to their child’s needs. As such, they believe that small improvements would improve their child’s quality of life. Additionally, patient group input suggested that it would also foster greater autonomy for individuals with Rett syndrome by enabling them to better express their needs and preferences.
Clinical Evidence Used in the Evaluation of Trofinetide
Pivotal Trial Evidence and Limitations
The sponsor submitted evidence from the phase III, double-blind, randomized, placebo-controlled LAVENDER trial (N = 187), which evaluated trofinetide in female patients aged 5 to 20 years with typical Rett syndrome and an MECP2 variant. The coprimary outcomes, change from baseline to week 12 in Rett Syndrome Behaviour Questionnaire (RSBQ) and Clinical Global Impression-Improvement (CGI-I) scores at week 12, showed greater decreases (indicating improvement) in the trofinetide group than in the placebo group. However, the absence of validated minimal important differences for these outcomes limits the ability to interpret what constitutes a clinically meaningful change. Similarly, missing data and concerns about the utility of the RSBQ as an outcome assessment further reduced confidence in the findings. Adverse events, particularly diarrhea and vomiting, were frequently reported and contributed to trial dropouts, highlighting the importance of balancing potential benefits against the burdens of treatment.
Long-Term Safety, Efficacy, and Evidence Gaps
The long-term safety and efficacy of trofinetide were assessed in 2 open-label extension (OLE) studies, LILAC (N = 154) and LILAC-2 (N = 77), which were intended to follow patients from the LAVENDER study for up to 3.5 years. The mean duration of exposure to trofinetide across the pivotal trial and 2 OLEs was approximately 2 years. Although this follow-up period offers some insight into long-term use, Rett syndrome is a lifelong condition, so uncertainty remains regarding long-term clinical benefit and safety. Both studies demonstrated continued decreases in RSBQ and CGI-I scores through week 104; however, improvements in CGI-I were modest, at less than a 1-point change during the OLE period. Both studies were also limited by high dropout rates in LILAC due to adverse events and the early termination of the LILAC-2 study after US market approval. Because these studies included only patients who completed the LAVENDER study, individuals who dropped out of the pivotal LAVENDER trial were excluded. This potentially skewed results toward a more favourable representation of the clinical effectiveness of trofinetide by focusing on participants who are more likely to tolerate and benefited from the treatment.
Representation and Generalizability
The pivotal trial and OLE studies excluded males, patients younger than 5 years or older than 20 years, individuals without an MECP2 variant, and those with atypical Rett syndrome. This creates uncertainty regarding the safety and efficacy of trofinetide in these populations. Additionally, clinical experts noted that the predominance of white participants in these studies does not reflect the diversity of patients seen in clinical practice in Canada, raising concerns about the generalizability of the findings. This is particularly important, given that the proposed Health Canada indication includes all individuals aged 2 years and older with Rett syndrome, regardless of sex, clinical diagnosis (i.e., atypical or classic Rett syndrome), or genetic status. To address some of these gaps, the sponsor submitted data from 2 additional studies: DAFFODIL, an open-label study evaluating trofinetide in females aged 2 to 5 years, and LOTUS, an ongoing observational study assessing the real-world use of trofinetide in individuals under routine care in the US. However, both studies have notable limitations, including the absence of control groups, the potential for selection bias, and the open-label designs, which introduce challenges in objectively interpreting results. These factors limit the reliability of the data and contribute to ongoing uncertainty about the safety and efficacy of trofinetide in populations not included in the pivotal trial. These gaps in evidence underscore the importance of robust informed consent processes to ensure that patients and families are aware of the uncertainties and potential risks associated with trofinetide, especially for those whose clinical profiles fall outside the trial population. Providers navigating these complexities will need to engage in shared decision-making and weigh potential benefits and harms on a case-by-case basis. Postmarket data collection will be critical to address these gaps and ensure equitable and evidence-based access to trofinetide in all populations living with Rett syndrome.
Clinical Use of Trofinetide
Balancing Benefits and Risks
Trofinetide represents the first Health Canada–approved therapy targeting the underlying disease course of Rett syndrome. As such, the clinical experts and clinician group input expect it to become the first-line treatment option for patients with Rett syndrome if recommended for public reimbursement. Despite the very low certainty in the Clinical Review Report regarding improvements, assessed by the coprimary outcomes (RSBQ and CGI-I) in the LAVENDER and OLE trials, input from the clinical experts and clinician group indicated a willingness to prescribe trofinetide to eligible patients with Rett syndrome. Although diarrhea and vomiting were the primary side effects leading to trial withdrawal, the clinical experts believed that these could be managed more effectively in clinical practice than in the trial setting.
Caregivers cited in patient group input emphasized that even modest improvements in their child’s communication, cognition, or mobility could have a meaningful impact on quality of life. However, the potential caregiving challenges posed by side effects like diarrhea underscore the need for providers to work closely with families to ensure that expectations are aligned with treatment realities. Given the trade-offs between potential benefits and caregiving burdens, the clinical experts stressed the importance of robust shared decision-making processes. Additionally, there are limited data on the long-term safety and effectiveness of trofinetide. Providers must prioritize patient and caregiver autonomy by presenting clear, comprehensive information about risks, benefits, and uncertainties to enable informed choices.
Eligibility
The heterogeneity of Rett syndrome — including distinctions between the classic and atypical forms and the variability in MECP2 status — complicates questions of eligibility for trofinetide. Clinical evidence from the pivotal LAVENDER trial is limited to females aged 5 to 20 years with typical Rett syndrome and a confirmed MECP2 variant. As a result, there is uncertainty regarding the safety and efficacy of trofinetide for patients with the atypical form of Rett syndrome, males, and those without MECP2 variants. Although the DAFFODIL and LOTUS studies provide some evidence on the use of trofinetide in a broader population, their design limitations leave residual uncertainty regarding the populations excluded from the pivotal trial. Given these uncertainties, the clinical experts suggested limiting eligibility to patients aligned with the trial population, patients with classic Rett without a known MECP2 variant and patients with atypical Rett syndrome and a confirmed MECP2 variant. In contrast, clinician group input supported the broader Health Canada indication, which includes all patients aged 2 and older with Rett syndrome, regardless of sex, clinical diagnosis (i.e., typical or atypical), or genetic status. Caregivers may also advocate for trofinetide access to patients who fall outside the narrower eligibility criteria, creating ethical and emotional challenges for providers navigating these conversations. The clinical experts emphasized the need for clear prescribing guidance beyond the parameters of the pivotal trial, along with robust, shared decision-making processes to facilitate informed decisions. Without such guidance, inconsistencies in prescribing practices may lead to inequitable access across providers and jurisdictions.
Prioritization
The clinical experts suggested that individuals with a lower disease burden, often younger patients, may be more likely to benefit from trofinetide. As a noncurative treatment, they said they expect trofinetide to have a greater impact when introduced earlier in the course of disease before significant progression occurs. However, there is no clinical evidence to support this claim. As such, the experts emphasized that eligibility criteria should not rely solely on age or disease burden, as some older patients or those with more advanced disease may still experience meaningful improvements in communication, mobility, or overall quality of life. Overly rigid prioritization criteria could inadvertently exclude patients who fall outside the typical profile of trial participants but could still derive significant benefit from treatment.
Geographic and Access Disparities
Input from the clinician group and clinical experts recommended that trofinetide initially be prescribed only by specialists with expertise in Rett syndrome, likely based in specialized medical centres. Although this approach may help to ensure appropriate use during early implementation, it could create geographic access barriers for patients living in rural or remote areas who do not have access to Rett specialists or specialty clinics. Over time, broader familiarity with trofinetide may enable the prescribing and management of treatment by a wider range of providers, potentially reducing these inequities. Families with fewer resources or limited access to specialized care may face greater challenges in managing treatment protocols. Addressing these barriers will be crucial to promoting equitable outcomes for all individuals with Rett syndrome.
Health Systems Impact
Impact on Health System Budgets and Equitable Resource Allocation
Expensive drugs for rare diseases, such as trofinetide, raise ethical considerations regarding equity, distributive justice, and the sustainability of health care budgets.3,4 Although there is a significant unmet need for effective, targeted therapies for Rett syndrome, uncertainty surrounding the clinical value of trofinetide, as described previously, complicates decisions about resource allocation. The conclusion of the CDA-AMC Pharmacoeconomic Review Report that trofinetide is not cost-effective intensifies these concerns. As a result, establishing equitable and sustainable eligibility criteria is crucial, ensuring that patients who stand to benefit most have access while maintaining a fair distribution of health resources.
Changes in the Roles and Workloads of Rett Specialists
The introduction of trofinetide would shift the roles and workloads of Rett specialists. Currently, these specialists act primarily as consultants, guiding the treatment of the diverse symptomatic presentations of Rett syndrome. With the approval of trofinetide, specialists would likely take on more direct care responsibilities, requiring additional follow-up visits for monitoring and collaboration with primary care providers. This shift could strain specialist availability and increase administrative costs, potentially creating bottlenecks in care delivery. Implementing clear protocols and investing in education for nonspecialist providers could help mitigate these challenges over time.
References
1.Schultz R, Suter B. Rett syndrome: Genetics, clinical features, and diagnosis. In: Patterson M, Firth H, Dashe J, eds. UpToDate. Waltham (MA): UpToDate; 2023: https://www.uptodate.com/contents/rett-syndrome-genetics-clinical-features-and-diagnosis. Accessed 2024 Dec 03.
2.Kim SY, Woo H, Lim BC, Kim KJ, Chae JH. Exploring the Clinical Utility of Targeted MECP2 Testing in Real-World Practice. Pediatr Neurol. 2024;161:28-33. PubMed
3.Kacetl J, Marešová P, Maskuriy R, Selamat A. Ethical Questions Linked to Rare Diseases and Orphan Drugs - A Systematic Review. Risk Manag Healthc Policy. 2020;13:2125-2148. PubMed
4.Wagner M, Goetghebeur MM, Ganache I, et al. HTA challenges for appraising rare disease interventions viewed through the lens of an institutional multidimensional value framework. Expert Rev Pharmacoecon Outcomes Res. 2023;23(2):143-152. PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.