Drugs, Health Technologies, Health Systems
Reimbursement Review
Sotatercept (Winrevair)
Sponsor: Merck Canada Inc.
Therapeutic area: Pulmonary arterial hypertension (WHO group 1)
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
6MWD
6-minute walk distance
AE
adverse event
AESI
adverse event of special interest
ANCOVA
analysis of covariance
CAMPHOR
Cambridge Pulmonary Hypertension Outcome Review
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
COE
centre of excellence
DBPC
double-blind placebo-controlled
ERA
endothelial receptor antagonist
ERS
European Respiratory Society
ESC
European Society of Cardiology
FAS
full analysis set
FC
functional class
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
LTDB
long-term double-blind
LTFU
long-term follow-up
mPAP
mean pulmonary arterial pressure
OLE
open-label extension
PAH
pulmonary arterial hypertension
PAH-SYMPACT
Pulmonary Arterial Hypertension – Symptoms and Impact
PDE5
phosphodiesterase type 5
PHA Canada
Pulmonary Hypertension Association of Canada
PVR
pulmonary vascular resistance
RHC
right-heart catheterization
SAE
serious adverse event
SF-36
Short Form (36) Health Survey
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Winrevair (sotatercept), 45 mg or 60 mg, powder and solvent for injectable solution |
Sponsor | Merck Canada Inc. |
Indication | In combination with standard pulmonary arterial hypertension (PAH) therapy, for the treatment of adults with WHO [WHO] Group 1 PAH and Functional Class (FC) II or III |
Reimbursement request | As an add-on to optimal background therapy for the treatment of adult patients with PAH who are not at low risk. Low risk is defined as meeting all the following criteria:
Optimal background therapy is defined as the following:
This medication should be prescribed under the direction of a specialist in the area of PAH. Diagnosis must be confirmed by right-heart catheterization. |
Health Canada approval status | Approved |
Health Canada review pathway | Priority review |
NOC date | August 28, 2024 |
Recommended dose | Weight-based dosing, with a starting dose of 0.3 mg/kg at cycle 1 and a target dose of 0.7 mg/kg from cycle 2 |
6MWD = 6-minute walk distance; FC = functional class; NOC = Notice of Compliance; PAH = pulmonary arterial hypertension.
Introduction
Pulmonary arterial hypertension (PAH), classified as WHO Group 1 pulmonary hypertension, is a rare, highly progressive, and disabling chronic disease. It is characterized by the uncontrolled proliferation of endothelial and smooth muscle cells in the pulmonary arteries, leading to vascular remodelling, increased pulmonary arterial pressure, and right-heart dysfunction. This results in progressive symptoms like dyspnea, fatigue, dizziness, and chest pain, ultimately leading to right-heart failure and reduced quality of life and survival. The disease has a complex pathophysiology involving the transforming growth factor beta superfamily and is more prevalent in females, with a median diagnosis age of 62.5 years. Despite advances in treatment, PAH has a poor prognosis, with a 5-year survival rate of about 56% in Canada. The prevalence of PAH in Canada is estimated at 78 per million population, based on registry data, with considerable variation in global estimates due to different study methodologies.
PAH has nonspecific signs and symptoms. The diagnostic process includes cardiac biomarkers like BNP and NT-proBNP. Echocardiograms can reveal abnormalities in the right ventricular chamber and interventricular septum. The gold standard for diagnosing PAH is right-heart catheterization (RHC), an invasive procedure that directly measures pulmonary artery pressure and flow. The current definition of PAH, based on RHC, is a mean pulmonary arterial pressure (mPAP) greater than 20 mm Hg, a pulmonary arterial wedge pressure less than or equal to 15 mm Hg, and a pulmonary vascular resistance (PVR) greater than 2.0 WU (160 dyn·sec·cm-5). Risk status in PAH can be assessed using methods like COMPERA 2.0 or the Simplified French Risk Score. Both methods evaluate 3 noninvasive parameters: WHO functional class (FC), 6-minute walk distance (6MWD), and BNP or NT-proBNP levels, using the same cut-off values. COMPERA 2.0 assigns grades 1 through 4 to each parameter and calculates the risk status based on the average score. In contrast, the Simplified French Risk Score requires meeting all low-risk criteria to achieve a low-risk status. These parameters are clinically relevant and correlate with long-term survival in patients with PAH.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of sotatercept (45 mg or 60 mg, powder and solvent for subcutaneous injection solution) in the treatment of patients with PAH. Sotatercept received a Notice of Compliance from Health Canada on August 28, 2024. It has not been previously evaluated by Canada’s Drug Agency (CDA-AMC).
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to the CDA-AMC call for input and from clinical expert(s) consulted for the purpose of this review.
Patient Input
CDA-AMC received a submission from the Pulmonary Hypertension Association of Canada (PHA Canada), which included patient and caregiver survey data and insights. PHA Canada, a charity focused on supporting the pulmonary hypertension community, collaborated with several organizations to gather information on patients’ experiences and expectations for sotatercept. The survey found that most respondents were adults with PAH; idiopathic PAH and scleroderma-associated PAH were the most common subtypes. Diagnosing PAH often takes more than 2 years due to its nonspecific symptoms, leading to advanced disease at diagnosis. The disease significantly impacts people’s daily lives and work productivity, with many people with PAH experiencing severe limitations in physical activities and requiring caregiver assistance.
The socioeconomic burden of PAH is considerable, with many people with PAH underemployed or dependent on assistance. The survey highlighted that current therapies are only somewhat effective in managing symptoms, particularly the psychological and emotional impacts. Adverse effects of medications are common, and patients face barriers to accessing treatments. Patients and caregivers expressed a willingness to tolerate serious adverse effects for benefits such as slowed disease progression, improved quality of life, and better symptom management. Only a few patients reported experience using sotatercept, likely through clinical trials, and 1 patient from the US shared positive outcomes from the drug’s use.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The following input was provided by 3 clinical specialists with expertise in the diagnosis and management of PAH.
The clinical experts highlighted significant unmet needs in treating PAH due to its rarity and lack of curative treatments. Key issues included the heterogeneous drug response among patients, limited options for right-heart failure, and scarce guidelines for mixed phenotype PAH or PAH with comorbidities. Additionally, the experts mentioned that there is insufficient data on when to switch or stop treatments based on patient response and that treatment side effects hinder adherence and tolerance.
Sotatercept, targeting a novel pathway in PAH biology, may show promise in improving outcomes when added to existing therapies, especially for patients whose disease is not well controlled. The experts mentioned that, because of the current scarcity of evidence about treatment with sotatercept, the drug should be used as an addition to established treatments rather than as a first-line therapy. Sotatercept’s novel mechanism could significantly benefit patients for whom existing therapies are not meeting treatment goals and offer an alternative to chronic infusions. The experts suggested that for new patients with PAH who are not at high risk, dual therapy with phosphodiesterase type 5 (PDE5) inhibitors and endothelial receptor antagonists (ERAs) remains the initial recommendation before considering sotatercept.
Ideal candidates for sotatercept are patients with PAH, especially those with WHO Group 1 pulmonary hypertension, who are on 2 to 3 background therapies and for whom those therapies have not achieved treatment goals. It may also be considered for patients on a single therapy if they cannot tolerate other drugs. Patient selection should involve thorough evaluation by PAH specialists, including RHC, and consideration for those not reaching low-risk status after standard therapies. Treatment efficacy should be assessed through 6-minute walk tests, BNP/NT-proBNP levels, and clinical assessments. Response to treatment should be assessed through WHO FC improvements, 6-minute walk test results, right ventricular function, hemodynamic measurements, and overall quality of life. The clinical experts also considered disease stabilization or a slowed disease progression rate to be meaningful responses. Initial treatment response is typically evaluated within 4 weeks, with a full assessment at 3 months. If deterioration occurs, earlier reassessment and treatment adjustment may be necessary.
According to the experts, discontinuation should be considered for patients experiencing significant adverse events (AEs), like bleeding or telangiectasias, if these significantly impact their quality of life. The decision to discontinue, especially in cases of telangiectasias, should be made on a case-by-case basis, weighing the benefits of continued therapy against the side effects. Treatment should also be stopped if the patient has had a lung transplant.
The clinical experts emphasized that sotatercept should be managed within specialized PAH centres staffed by trained cardiologists or respirologists. These specialists are essential for diagnosing, treating, and monitoring patients with PAH, ensuring comprehensive and high-quality care.
Clinician Group Input
The Canadian Pulmonary Hypertension Health-Care Providers, a nonaffiliated group of physicians and nurse practitioners from specialized pulmonary hypertension centres, provided input on the current state and challenges in PAH treatment. Their insights are based on the 2022 European Society of Cardiology and European Respiratory Society (ESC/ERS) guidelines, the 2020 Canadian Cardiovascular Society and Canadian Thoracic Society position statement, recent multicentre Canadian PAH studies, and the clinicians’ collective clinical experience. They emphasized that while current treatment options, ranging from nonpharmacologic management to combination therapy and lung transplant, offer symptomatic benefits and stability, these benefits are often short-lived. Current PAH medications act mainly as vasodilators and have minimal impact on blocked vessels or the underlying cellular proliferation, leading to disease progression. Despite optimal medical therapy, few patients reach low-risk status, with a 5-year survival rate of only about 60% in Canada. Key end points for patients living in Canada include improvements in symptoms, quality of life, and survival and reductions in clinical deterioration.
The clinician group highlighted significant unmet needs in PAH treatment, such as the inability of current therapies to halt or reverse cell proliferation and vessel remodelling. They also noted the intolerable side effects and complications associated with parenteral therapies. They view sotatercept, the first approved PAH therapy targeting growth factor signalling to control aberrant cell proliferation, as a promising add-on therapy rather than as a first-line treatment. Sotatercept is expected to benefit patients across common PAH types and the major WHO FCs (II and III). The clinician group emphasized the importance of early aggressive treatment in PAH. The group recommends that prescribing of sotatercept be restricted to provincially designated pulmonary hypertension centres.
Drug Program Input
The drug programs noted that the sponsor states that, unlike other PAH-specific medications in Canada, sotatercept is not a vasodilator but a novel activin signalling inhibitor potentially capable of reversing pulmonary vascular remodelling in PAH. However, it is unclear how many current plans allow triple therapy for patients with PAH and how many have defined objective renewal criteria for existing PAH treatments.
The drug programs emphasized that PAH diagnosis requires an invasive RHC by specialists in pulmonary hypertension centres of excellence (COEs). Patients in the pivotal trial were on stable doses of background PAH therapy for at least 3 months, and the sponsor seeks reimbursement for sotatercept as an add-on to optimal background therapy. A clear definition of “optimal background therapy” is essential if sotatercept is recommended for funding. The clinical experts agreed with this notion and emphasized that the definition may vary in clinical practice but is overall known among clinicians and stated in current clinical guidelines. The clinical experts also recognized that although optimal background therapy is defined by guideline recommendations, patient tolerance is important, and with the significant side effect profile of some of these drugs, “maximally tolerated optimal medical therapy” can be different than optimal background therapy.
Decisions regarding therapy continuation may be guided by noninvasive risk status assessments, focusing on exercise capacity, 6MWD, WHO FC, and cardiac biomarkers. Reaching or maintaining a low-risk status predicts better long-term survival, raising the question of whether a minimum response to therapy should justify continuation and how this response should be defined at the first and subsequent renewal assessments. The clinical experts acknowledged that reaching or maintaining low-risk status is 1 of the main goals of treatment. They considered that patients experiencing a maintained or improved risk status would be enough reason for continuing therapy. An increase in a patient’s risk status would be a reason to consider discontinuation of the drug. The experts acknowledged that the evidence that would be used to establish the best continuation or discontinuation criteria is unclear.
In relation to prescribing, the drug programs noted that it is recommended that hemoglobin and platelet count be reviewed before each dose until the disease is stable and that they then be reviewed periodically to determine if dose adjustments are required; the drug programs also noted that sotatercept should be prescribed by PAH specialists. Patients with PAH associated with HIV, portal hypertension, schistosomiasis, and pulmonary veno-occlusive disease were excluded from the pivotal trials, prompting the question of whether these patients should also be ineligible for treatment with sotatercept. The clinical experts responded that patients with these conditions should not be excluded from consideration for treatment with sotatercept but, rather, should be considered on a case-by-case basis, with individual decisions made.
Clinical Evidence
Systematic Review
Description of Studies
The systematic review included 1 pivotal study. The STELLAR trial (NCT04576988), a phase III, randomized, multicentre, double-blind placebo-controlled (DBPC) study, evaluated the efficacy and safety of sotatercept versus placebo on stable background PAH therapy in adults with PAH (WHO Group 1 pulmonary hypertension). The trial was conducted across 21 countries, including 3 sites in Canada, from January 2021 to December 2022, and it enrolled 323 participants with age and sex distributions reflecting typical PAH demographics. The study had 2 treatment periods: a 24-week DBPC phase and a long-term double-blind (LTDB) phase lasting up to 72 weeks or until unblinding. Participants completing the DBPC phase could join a long-term follow-up (LTFU) study, the SOTERIA study (NCT04796337). Participants were randomized 1:1 to receive sotatercept or placebo subcutaneously every 21 days, with the dose starting at 0.3 mg/kg and increasing to 0.7 mg/kg, with adjustments as needed. The trial included 163 participants in the sotatercept group and 160 in the placebo group, with analyses conducted on the full analysis set (FAS) and safety set.
Overall, the patients in the STELLAR trial had a mean age of 47.9 years. Almost half of them had PAH classified as WHO FC III (166 of the 323 participants randomized [51.4%]), with equal distribution between placebo and sotatercept groups. The rest had PAH classified as WHO FC II. The mean time since diagnosis was 8.8 years for all patients. There were no significant differences in the baseline characteristics between study arms.
Efficacy Results
Mortality
In the STELLAR trial, mortality (i.e., the number of patients who died during the follow-up of the study) was assessed as part of a multicomponent end point (also described Multicomponent Improvement section) at the final cut-off date of December 6, 2022. The number of patients who died was relatively low: less than 4%. More deaths were observed in the placebo arm (6 patients died [3.8%]) than in the sotatercept arm (2 patients died [1.2%])
Change From Baseline in 6MWD
Sotatercept significantly improved the 6MWD in adults with PAH on background therapy, with a median treatment difference between the sotatercept and placebo groups of 40.8 m (95% confidence interval [CI], 27.5 m to 54.1 m) at 24 weeks. The improvement was greater in patients with PAH classified in the WHO FC III stratum (61.7 m; 95% CI, 40.9 m to 82.6 m) than in the WHO FC II stratum (21.7 m; 95% CI, 6.6 m to 36.7 m).
Multicomponent Improvement
At week 24, a higher proportion of patients in the sotatercept group (38.9%) met all criteria for improvement in the multicomponent end point (6MWD, NT-proBNP level, and WHO FC) than in the placebo group (10.1%). The risk difference between groups was ████ █ ██████ ████ ██ █████.
Time to Clinical Worsening or Death
By the December 2022 data cut-off, fewer participants in the sotatercept group (11 [6.7%]) than in the placebo group (42 [26.3%]) had died or had at least 1 clinical worsening event. The risk difference between groups was ██████ ███████ █████ ██ ██████; this is a reduction in the risk of the composite end point in favour of sotatercept. Evaluating this composite end point as a time-to-event outcome, the risk of death or a first clinical worsening event was 82% lower in the sotatercept group than in the placebo group (hazard ratio = 0.18; 95% CI, 0.09 to 0.38).
When evaluating the individual components of the composite end point, more patients in the placebo arm (17 patients [10.6%]) required rescue therapy or an increase in the dose of infusion prostacyclin than in the sotatercept arm (2 patients [1.2%]). PAH-related hospitalization was observed in 7 patients in the placebo arm and 1 patient in the sotatercept arm (4.4% versus 0.6%). As mentioned in the Mortality section, 2 patients in the sotatercept arm died, compared to 6 patients from the placebo arm.
Change From Baseline in PVR
Patients in the sotatercept arm demonstrated a reduction in PVR from baseline to week 24 of –165.1 dyn·sec·cm-5 (95% CI, –184.0 dyn·sec·cm-5 to –152.0 dyn·sec·cm-5), whereas the PVR increased in the placebo arm by 32.8 dyn·sec·cm-5 (95% CI, 24.0 dyn·sec·cm-5 to 40.0 dyn·sec·cm-5). The median treatment difference between the sotatercept and placebo groups was –234.6 dyn·sec·cm-5 (95% CI, –288.4 dyn·sec·cm-5 to –180.8 dyn·sec·cm-5). Results from the supportive analysis using the analysis of covariance (ANCOVA) model were consistent with those from the primary analysis. The treatment effect of sotatercept on PVR at week 24 was consistent across the prespecified subgroups and remained consistent in the post hoc subgroups stratified by baseline risk status.
Change From Baseline in NT-proBNP
The median treatment difference between the sotatercept and placebo groups in mean change from baseline was –441.6 pg/mL (95% CI, –573.5 pg/mL to –309.6 pg/mL). Results from the supportive analysis using the ANCOVA model were consistent with those from the primary analysis. The treatment effect of sotatercept on NT-proBNP at week 24 was consistent across the prespecified subgroups and remained consistent in the post hoc subgroups stratified by baseline risk status.
WHO FC Improvement
More patients in the sotatercept group (29.4%) experienced improvement in WHO FC at week 24 than in the placebo group (13.8%). The risk difference was █████ ███████ ███ ██ █████. Specifically, more patients in the sotatercept group than in the placebo group experienced improvement from WHO FC II to FC I (5.0% versus 2.0%, respectively) and from WHO FC III to FC II (24.5% versus 12.2%, respectively) at week 24. The treatment effect of sotatercept on WHO FC improvement at week 24 was consistent across the post hoc subgroups stratified by baseline risk status.
Change From Baseline in PAH-SYMPACT Domain Scores
Patients in the sotatercept group reported greater improvements in both the physical impacts and the cardiopulmonary symptoms domain scores, based on the Pulmonary Arterial Hypertension – Symptoms and Impact (PAH-SYMPACT) questionnaire, than those in the placebo group from baseline to week 24. For the physical impacts domain, the difference between arms was –0.26 points (95% CI, –0.49 to –0.04 points) in favour of sotatercept. For the cardiopulmonary symptoms’ domain, the values of a difference between arms were–0.13 points (95% CI, –0.26 to –0.01 points) in favour of sotatercept. In both cases, negative values indicate improvement.
EQ-5D-5L
Patients treated with sotatercept experienced a greater increase in the EQ visual analogue scale (VAS) than those receiving placebo. Specifically, there was an increase (improvement) in the VAS from baseline that was greater in the sotatercept group ███ ██████ ██████ ███████ ████ ██ █████ than in the placebo group █████ ██████ ████ ███ █████ ██ ██████. The difference between groups was ███ ██████ ███████ ████ ██ █████ in favour of sotatercept.
PAH-Specific Hospitalization
Fewer hospitalizations were observed in the sotatercept group than in the placebo group. This outcome was obtained from the composite end point of time to clinical worsening or death. Overall, 7 patients (4.4%) in the placebo group and 1 patient (0.6%) in the sotatercept group were hospitalized, with a risk difference of █████ ███████ ████ ██ █████ in favour of sotatercept
Harms Results
Through week 24, the most common AEs associated with sotatercept, versus placebo, included epistaxis (12.3% versus 1.9%), telangiectasia (10.4% versus 3.1%), and dizziness (10.4% versus 1.9%). These events were mostly mild to moderate in severity. Serious AEs (SAEs) occurred in 14.1% of participants in the sotatercept group and 22.5% in the placebo group, with no significant patterns emerging. The sotatercept group had isolated instances of atrial flutter, falls, and hemoptysis, with only 2 SAEs (1 fall and 1 hemoptysis) deemed related to the study intervention. In contrast, the placebo group reported multiple cases of PAH, cardiac arrest, right ventricular failure, and dyspnea. No deaths were reported in the sotatercept group during the initial 24 weeks, compared to 6 deaths in the placebo group. By the final data cut-off, 2 deaths had occurred in the sotatercept group.
The sotatercept group had a lower rate of discontinuation due to AEs than the placebo group. Notably, telangiectasia incidents were higher in the sotatercept group but were neither serious nor severe, with only 1 case leading to treatment discontinuation. The sponsor identified several AEs of special interest (AESIs), including increased hemoglobin, thrombocytopenia, and various bleeding events. Epistaxis was the most reported bleeding event in the sotatercept group (12.3% of patients), followed by gingival bleeding (3.1% of patients). None of these bleeding events were serious or severe, though 2 participants discontinued due to bleeding events. Increased hemoglobin levels were observed in 4.3% of participants in the sotatercept group, leading to study intervention interruption in 3 cases, but none were serious or severe. Thrombocytopenia was more common in the sotatercept group than the placebo group (6.1% versus 2.5% of patients).
Critical Appraisal
Internal Validity
The STELLAR trial was a well-designed, phase III, multicentre, randomized DBPC study assessing the efficacy and safety of sotatercept versus placebo over 24 weeks in adult patients with PAH on stable background therapy. The trial used a robust 1:1 random allocation process, generated by a computer algorithm and centrally managed to maintain allocation concealment. Although blinding was effective initially, patients might have inferred their treatment group due to there being more frequent AEs, like telangiectasia and nosebleeds, in the sotatercept group. Adherence was meticulously monitored, with rates exceeding 98%, and deviations were well documented. Missing data were handled appropriately through sensitivity analyses, whose results were in agreement with the primary analysis outcomes for key measures like 6MWD, NT-proBNP, PVR, and PAH-SYMPACT. Outcome measurement methods were validated and reliable, and the reported outcomes and analysis plan adhered to the study protocol.
External Validity
The reimbursement criteria for sotatercept target patients with PAH on background therapy whose disease does not meet low-risk status, defined as having PAH in WHO FC I or II, a 6MWD over 440 m, and specific NT-proBNP or BNP levels.
Overall, the 323 patients in the STELLAR study were deemed representative of the PAH population in Canada, though certain subgroups (e.g., patient with HIV or patients with portal hypertension) and demographics may not be properly represented. The STELLAR study enrolled 53 patients (16.4%) classified as low risk, which is an excluded patient population in the suggested reimbursement criteria, and 157 patients (48.6%) with PAH within the WHO FC II stratum. However, the clinical experts consulted by CDA-AMC considered that the impact of including these patients on the generalizability of results is low and that the effect estimates are still applicable to the target population for reimbursement.
The trial’s restriction to patients with a baseline PVR of at least 400 dyn·sec·cm-5 may not fully represent the broader PAH population. Additionally, the 24-week median treatment duration and study design limit the ability to determine long-term mortality outcomes and extended safety profiles, although the long-term data suggest that efficacy and harm outcomes remain similar to the results of the STELLAR trial.
GRADE Summary of Findings and Certainty of the Evidence
The Grading of Recommendations Assessment, Development and Evaluation (GRADE) tool was used to assess the certainty of the evidence for the outcomes considered most relevant to inform CDA-AMC expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.1,2
Following the GRADE approach, evidence from randomized controlled trials started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effect estimates, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
The GRADE assessments included an evaluation of the main outcomes considered important by clinicians, patient groups, and committee members. The comparison evaluated in the GRADE assessments of this report was that of sotatercept versus placebo. Table 2 presents the GRADE summary of findings.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
mortality or deaths
6MWD
multicomponent improvement
time to first occurrence of clinical worsening event or death
PVR
NT-proBNP
change in WHO FC
health-related quality of life (HRQoL) — PAH-SYMPACT physical impacts and cardiopulmonary symptoms domain scores and EQ-5D-5L
hospitalization (PAH specific)
harms (AEs, SAEs, and AESIs).
Table 2: Summary of Findings for Sotatercept Versus Placebo for Patients With Pulmonary Arterial Hypertension
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Sotatercept | Difference (95% CI) | |||||
Clinical efficacy | |||||||
Mortality Follow-up: median, 24 weeks | 323 | NA | 6 of 160 patients (3.8%) | 2 of 163 patients (1.2%) | ██ █████ ████████ ███ █████████ █████ ██ ██ █████ | Lowa | Sotatercept may reduce the number of deaths compared with placebo. The clinical magnitude of the effect is unclear. |
6MWD, change from baseline | 323 | NA | 1.0 m (range, –1.0 to 5.0 m) | 34.4 m (range, 32.5 to 35.5 m) | 40.8 more metres (27.5 more to 54.1 more) | Highb | Sotatercept results in a clinically important increase in 6MWD compared with placebo. |
Multicomponent improvement (6MWD, NT-proBNP level, and WHO FC) | 321 | NA | 16 of 160 patients (10.1%) | 63 of 163 patients (38.9%) | ███ ████ ███ ██████████ ████ ██ ███ █████ | Highc | Sotatercept results in an important increase in the proportion of patients with multicomponent improvement compared with placebo. |
Composite: time to clinical worsening or death Follow-up: median, 24 weeks | 323 | NA | 42 of 160 patients (26.3%) | 9 of 163 patients (5.5%) | ███ █████ ███ ██████████ █████ ██ ███ ██████ | Highc | Sotatercept results in an important reduction in the proportion of patients with the composite end point compared with placebo. |
Pulmonary vascular resistance, median change from baseline Follow-up: median, 24 weeks | 323 | NA | 32.8 dyn·sec·cm-5 | –165.1 dyn·sec·cm-5 (95% CI, –184.0 to –152.0 dyn·sec·cm-5) | –234.6 dyn·sec·cm-5 (–288.4 to –180.8 dyn·sec·cm-5) | Highd | Sotatercept results in a decrease in pulmonary vascular resistance compared with placebo. The clinical magnitude of the effect is unclear.d |
NT-proBNP, change from baseline Follow-up: median, 24 weeks | 323 | NA | 58.6 pg/mL | –230.3 pg/mL (range, –236.0 to –233.0 pg/mL) | –441.6 pg/mL | Highd | Sotatercept results in a decrease in NT-proBNP compared with placebo. The clinical magnitude of the effect is unclear.d |
Change in WHO FC Follow-up: median, 24 weeks | 322 | NA | 22 of 159 patients (13.8%) | 48 of 163 patients (29.4%) | ███ █████ ███ ███████████ ███ █████ ██ ██ ██████ | Highc | Sotatercept results in an important increase in the proportion of patients who experience improvement in WHO FC compared with placebo. |
PAH-specific hospitalization Follow-up: median, 24 weeks | 323 (1 RCT) | NA | 7 of 160 patients (4.4%) | 1 of 163 patients (0.6%) | ██ █████ ███ ███████████ ██ █████ ██ ██ ██████ | High | Sotatercept results in an important decrease in the proportion of patients hospitalized due to PAH compared to placebo. |
Health-related quality of life | |||||||
Health-related quality of life (PAH-SYMPACT and EQ-5D-5L) | 323 (1 RCT) | The MD in change from baseline between sotatercept and placebo was –0.26 points (95% CI, –0.49 to –0.04 points) in the PAH-SYMPACT physical impacts domain and –0.13 points (–0.26 to –0.01 points) in the cardiopulmonary symptoms domain (negative values mean improvement). For the EQ VAS, the MD was ███ points more in sotatercept ████ ███ █████ ████ (higher values mean improvement) in favour of sotatercept.e | Moderatee | Sotatercept likely results in an important improvement in health-related quality of life measurements (PAH-SYMPACT and EQ-5D-5L) compared with placebo. | |||
Harms | |||||||
Adverse events Follow-up: range, 42 weeks to 72 weeks | 323 | NA | 149 of 160 patients (93.1%) | 151 of 163 patients (92.6%) | | █████ ███ ███████████ ██ █████ ██ ██ █████ | Lowf | Sotatercept may result in little to no clinically meaningful difference in adverse events compared with placebo. |
Serious adverse events Follow-up: range, 24 weeks to 72 weeks | 323 | NA | 47 of 160 patients (29.4%) | 40 of 163 patients (24.5%) | ██ █████ ███ ███████████ ███ █████ ██ ██ █████ | Lowf | Sotatercept may result in little to no clinically meaningful difference in serious adverse events compared with placebo. |
Adverse events of special interest (telangiectasia and epistaxis) Follow-up: range, 24 weeks to 72 weeks | 323 | NA | 7 of 160 patients (4.4%) | 27 of 163 patients (16.6%) | ███ ████ ███ ███████████ ██ ████ ██ ███ █████ | High | Sotatercept results in an increase in the proportion of patients with events of telangiectasia or epistaxis compared with placebo. |
6MWD = 6-minute walk distance; CI = confidence interval; FC = functional class; MD = mean difference; MID = minimally important difference; NA = not applicable; PAH = pulmonary arterial hypertension; PAH-SYMPACT = Pulmonary Arterial Hypertension – Symptoms and Impact; RCT = randomized controlled trial; VAS = visual analogue scale.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
Details included in the table are from the sponsor’s summary of clinical evidence.
aRated down 2 levels for imprecision. The 95% CI is wide and may include important effects of benefit but also the possibility of trivial effects. The study presents a low number of events, and it was not powered to detect a difference for this outcome. No MID or threshold of clinical significance was obtained; hence, the null effect was used to determine the target of the rating of certainty.
bAlthough the lower limit of the CI (27.5 m) was below the MID of 33 m, it was deemed that patients and clinicians would consider this an important effect of benefit. Hence, no rating down for imprecision was performed.
cThe clinical experts considered that if 5 to 10 patients per 1,000 treated with sotatercept vs. placebo improved (or got worse) it would be a meaningful beneficial (or harmful) effect. Hence, no rating down for imprecision was performed.
dNo MID was obtained for this end point. The clinical experts considered the change observed to be clinically meaningful.
eMIDs for the physical impacts and cardiopulmonary symptoms domains were estimated to be –0.3 and –0.2, respectively. Hence, values were rated down 1 level for imprecision because they included the threshold of the MID. No MID was obtained for the EQ VAS.
fMay be little to no difference between groups, but the 95% CI is wide and includes a possible important reduction in total AEs as well as an increase in AEs, using a threshold of benefit/harm of 20 patients per 1,000 treated.
Long-Term Extension Studies
Description of Studies
The evaluation of long-term outcomes is supported by 3 key reports. First, the long-term assessment of AEs and efficacy outcomes of sotatercept were addressed during the LTDB treatment period of the STELLAR trial. Additionally, efficacy and safety end points were evaluated in the open-label extension (OLE) phase of the PULSAR phase II study. Lastly, the ongoing open-label SOTERIA trial provides primary evidence, though current information is based on interim analyses. Subsequent subsections will provide detailed descriptions of each study.
The primary objective of the STELLAR trial extension period was to evaluate the long-term incidence of AEs in patients treated with sotatercept. After completing the 24-week DBPC treatment period, patients entered the LTDB treatment period, which lasted up to 72 weeks, eventually transitioning to the LTFU study (the SOTERIA study) upon unblinding. Due to varying enrolment times in the STELLAR trial, some participants had more visits beyond the initial 24 weeks. The PULSAR study, conducted from June 2018 to March 2022, also evaluated sotatercept’s safety and efficacy over a 24-week DBPC period followed by an 18-month OLE. The SOTERIA trial, initiated in May 2021 and ongoing across 196 sites in 21 countries, aims to assess the long-term efficacy, safety, and tolerability of sotatercept over up to 7 years.
Efficacy Results
STELLAR LTDB Study
The STELLAR LTDB study extended the evaluation of the long-term safety and efficacy of sotatercept beyond the initial 24-week DBPC phase. As patients transitioned into the SOTERIA study (those who completed the DBPC treatment period and were on treatment in the LTDB treatment period were eligible to participate in the open-label LTFU study), the efficacy outcomes during the LTDB period remained descriptive. Sotatercept continued to show superior improvements compared to placebo in 6MWD, PVR, NT-proBNP levels, WHO FC, and the proportion of participants with a low-risk score.
PULSAR Study
The PULSAR study, a phase II trial, assessed the long-term efficacy and safety of sotatercept over a 24-week DBPC period followed by an 18-month OLE phase. Reductions in PVR were maintained from baseline to months 18 to 24 in both the continued-sotatercept group and the placebo-crossed group. Improvements in 6MWD and NT-proBNP levels were also sustained in both groups. The WHO FC improvements were notable, with a high percentage of patients experiencing their PAH improving to or maintaining WHO FC II status and, for some patients, WHO FC I. Time to clinical worsening events was low, and mortality risk scores reflected sustained low-risk status.
SOTERIA Study
The ongoing SOTERIA study, initiated in May 2021, focuses on the long-term efficacy, safety, and tolerability of sotatercept over up to 7 years. At 1 year, patients maintained the improvements in 6MWD, NT-proBNP levels, WHO FC, and low-risk scores consistent with the STELLAR trial results. Clinical worsening events remained low, with only 6.2% of participants in the continued-sotatercept arm experiencing such events, and even fewer in the placebo-crossed arm. Detailed results will become available as the study progresses.
Harms Results
STELLAR LTDB Study
The STELLAR LTDB study showed a consistent profile for the harm outcomes in the sotatercept arm compared to the initial 24-week analysis. Common AEs in the sotatercept group included epistaxis, telangiectasia, dizziness, nasal congestion, thrombocytopenia, and increased hemoglobin levels, primarily mild to moderate. The sotatercept group reported 2 deaths due to AEs, compared to 7 deaths in the placebo group. Discontinuation due to AEs was lower in the sotatercept group (3.7%) than in the placebo group (6.9%).
PULSAR OLE Phase
In the PULSAR OLE phase, all participants in the sotatercept 0.7 mg/kg group reported AEs, similar to those in the STELLAR trial. SAEs related to the study drug were reported in 4.8% of participants and included conditions like fever, increased red blood cells, and systemic lupus erythematosus. Discontinuation due to AEs occurred in 19% of participants in the continued-sotatercept 0.7 mg/kg arm, with 3 deaths reported, including 1 due to a brain abscess. AESIs included leukopenia, neutropenia, and thrombocytopenia, occurring in 17.3% of participants. Hemoglobin increases and telangiectasia were noted, with the latter developing after approximately 1.5 years of treatment.
SOTERIA Study
In the SOTERIA study, 90.8% of participants experienced 1 or more AEs, with 3.5% discontinuing treatment and 2.8% dying due to AEs. SAEs occurred in 30.3% of patients, with telangiectasia reported in 17.4% of participants, no cases deemed serious. Epistaxis was the most common bleeding event (22.1%), with serious bleeding events occurring in 5.2% of participants. Increased hemoglobin levels (nonserious) were observed in 14.3% of participants, and thrombocytopenia occurred in 6.1% of participants, with 3 cases being serious and treatment related.
Critical Appraisal
The LTDB phase of the STELLAR study presented efficacy and harm end points descriptively due to patient attrition, as participants could transition to the SOTERIA trial. Blinding and randomization were maintained, though unblinding was possible due to AEs associated with sotatercept. The open-label PULSAR study, lacking a comparator, posed a higher risk of bias, potentially affecting patient expectations and reporting of preventive measures. The ongoing SOTERIA study, also open-label and without a comparator, faces similar biases, with potential influences on patient-reported outcomes and the inclusion of patients who have experienced good drug performance.
The LTDB phase shared limitations with the pivotal STELLAR trial, particularly the exclusion of patients with certain types of PAH and the inclusion of patients with PAH classified as both WHO FC II and III. Extended observation beyond 24 weeks helped confirm AEs, aligning with the pivotal trial results. The PULSAR study included only patients with PAH classified as WHO FC II and III, presenting similar limitations. Conducted at 43 centres in 8 countries, the PULSAR study lacked Canadian representation, but the clinical experts did not express concerns about the generalizability of the international evidence.
Indirect Comparisons
No indirect treatment comparison was submitted.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the evidence were identified.
Conclusions
The evidence on the effects of sotatercept in patients with PAH (WHO Group 1 pulmonary hypertension) comprises 1 pivotal randomized controlled trial comparing sotatercept to placebo and 3 reports on the long-term effects of sotatercept. Sotatercept was not compared to other interventions due to the nature of the reimbursement request and the drug’s suggested place in therapy as an add-on intervention to optimal background therapy or for the treatment of patients who are already on double or triple therapy, depending on contraindications or the tolerability of available PAH therapies.
The outcomes considered critical for decision-making by clinical experts and patient groups include the impact of sotatercept on mortality, exercise capacity (measured by the 6MWD), risk status assessed as a multicomponent improvement (6MWD, NT-proBNP level, and WHO FC), a composite of time to clinical worsening or death, PVR, NT-proBNP levels, HRQoL, hospitalization rates, and AEs.
The pivotal STELLAR study evaluated all these outcomes (including death as part of the composite end point), providing evidence that sotatercept, when compared to placebo, significantly improves 6MWD and increases the proportion of patients experiencing improvement in the assessment of clinical worsening, in the multicomponent composite end point, and in risk status, according to the Simplified French Risk Score. Additionally, sotatercept demonstrates meaningful benefits in PVR and NT-proBNP levels, as noted by the clinical experts. Patients receiving sotatercept had a lower risk of hospitalization due to PAH, and their PAH showed greater improvement in WHO FC stratum, than patients receiving placebo. Sotatercept also likely leads to improvements in HRQoL measurements (PAH-SYMPACT and EQ-5D-5L) compared to placebo, although the magnitude of these HRQoL effects remains uncertain due to the imprecision of the results.
The frequency of AEs and SAEs was similar between sotatercept and placebo, though bleeding events (especially epistaxis), telangiectasia, and dizziness occurred more frequently with sotatercept. These events were mostly reported as mild or moderate, and the clinical experts identified these events as manageable in practice and likely of lower significance for patients when weighed against the desirable outcomes associated with PAH.
Overall, sotatercept improves exercise capacity, WHO FC, and risk status and reduces time to clinical worsening events compared with placebo and has a similar profile in short-term harms. However, the effects of sotatercept on mortality alone and on longer-term outcomes remain uncertain.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of sotatercept 45 mg and 60 mg for subcutaneous injection in the treatment of adults with PAH (WHO Group 1 pulmonary hypertension).
Disease Background
The contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
Pulmonary hypertension consists of 5 distinct groups of disease defined by WHO. PAH, a rare, highly progressive, and disabling chronic disease, represents WHO Group 1 pulmonary hypertension. It is characterized by uncontrolled proliferation of endothelial and smooth muscle cells of the pulmonary arteries, resulting in extensive vascular remodelling.5 With the arteries narrowing and stiffening, blood flow through the pulmonary vessels becomes difficult, leading to high blood pressure in the arteries that carry blood to the lungs. Consequently, the right side of the heart needs to work harder to pump blood to the lungs, leading to right-heart dysfunction. Ultimately, there is a progressive deterioration in symptoms, functional capacity, HRQoL, and the resulting right-heart failure that negatively impacts patients’ survival.
PAH has a complex pathophysiology. While other pathways are involved in PAH, its initiation and progression are mediated by the transforming growth factor beta super family, including ACTR2A and its ligands, such as activin A, as well as BMPR2.6 The imbalance in the pro-proliferative ACTR2A and the anti-proliferative BMPR2 signalling in PAH results in vascular cell hyperproliferation, pathological remodelling of the pulmonary arterial wall, metabolic reprogramming, and inflammation in the lung.5,7
The common presenting symptoms of PAH include dyspnea, fatigue, dizziness, and chest pain.8 As the disease progresses, the symptoms become increasingly debilitating and may include additional symptoms such as repeated syncope. These symptoms may present at any point or over time. Unfortunately, these symptoms are not specific to PAH. As such, there is often a substantial delay in diagnosing this condition. Confirming a PAH diagnosis can take more than 2 years in Canada,9 further contributing to the continued poor prognosis and high mortality of this rare disease.
PAH can affect anyone at any age or background, but females (particularly those who are premenopausal) are about twice as likely to be diagnosed with PAH than their male counterparts.10 Based on the Canadian Pulmonary Hypertension Registry, the median age of patients at diagnosis is 62.5 years,11 similar to recent reports from registries in the US and Europe.12-15 The prognosis of PAH is generally poor and largely depends on patients’ clinical presentation, including exercise capacity, symptom severity, and level of heart failure biomarkers. The median survival following diagnosis ranges from about 6 years to about 7 years, despite advances in treatments.8,16 In Canada, the 5-year survival rate is about 56% regardless of initial treatment strategy.17
Estimated Disease Prevalence
The sponsor identified PAH as a rare disease in Canada (i.e., having a prevalence less than 500 per million).18,19 The estimates of prevalence of PAH vary for several reasons, including the study method used. Two recent systematic reviews reported the estimated prevalence of PAH from registries, hospital databases, and claims databases globally.20,21 In adults, the estimated incidence ranges from 12.4 to 268 patients per million per year and the prevalence (based on data from the years 2000 to 2010) is 1.5 to 32 patients per million population.20 Using recent data from national registries, the prevalence and incidence of PAH in adults were estimated at 47.6 to 54.7 per million and 5.8 per million, respectively.20
A 2018 article based on a population-based cohort study conducted among Ontario residents identified patients with PAH using International Classification of Diseases codes from hospitalization or emergency department visits.22 It estimated that the prevalence of PAH was 291 per million population in 2012, which corresponds to the upper limit of the global estimates described in the previous paragraph.20,22 However, a major methodological limitation of that study is that the International Classification of Diseases codes were not linked with confirmatory diagnosis using RHC; therefore, the study likely overestimated the prevalence of PAH in Canada.
The Canadian Pulmonary Hypertension Registry was initiated in 2017. As of June 30, 2023, 10 out of the 16 pulmonary hypertension COEs that treat adult patients with pulmonary hypertension have entered data in the registry, and the data show 1,247 patients with PAH.11 Through an extrapolation exercise to account for patients in all COEs, the sponsor estimated that there are approximately 2,497 patients with PAH diagnoses in Canada. Based on the population in Canada (from Statistics Canada),23 this resulted in a prevalence of 78 patients per million population at that time.
Diagnosis
PAH has nonspecific signs and symptoms; hence, the diagnosis of PAH requires extensive investigations. These include medical history, physical examination, and testing for the cardiac biomarkers BNP and/or NT-proBNP.24 These biomarkers are usually elevated in patients with PAH due to cardiac stress as the pressure and volume increase in the heart.25 Echocardiographs may show an enlarged right ventricular chamber, increased thickness of the interventricular septum in comparison to the left ventricular wall, and reduced right ventricular systolic function. While there are no Canadian guidelines for the diagnosis and management of pulmonary hypertension, the Canadian Thoracic Society and Canadian Cardiovascular Society joint position statement in 2020 recommends prompt referral for confirmatory diagnosis.24
The best standard to make a diagnosis of PAH is through RHC,24 an invasive hemodynamic procedure performed by specialists. RHC directly measures the pulmonary artery pressure and flow. Patients undergoing RHC are monitored for a few hours and are generally discharged on the same day.26 The frequency with which RHC is repeated during follow-ups (every 3 to 6 months) is dependent on the treating physicians and on patients if they are experiencing disease worsening. Noninvasive risk assessments are also recommended by treatment guidelines and are used by physicians in Canada for treatment decision-making at diagnosis and during follow-ups.
The current definition of PAH is based on an RHC with an mPAP of more than 20 mm Hg, a pulmonary arterial wedge pressure of less than or equal to 15 mm Hg, and a PVR of more than 2.0 WU.8 The mPAP indicative of PAH was historically defined at greater than or equal to 25 mm Hg, based on expert opinion; however, it has been reassessed and was reduced to more than 20 mm Hg in 2019.27
A comprehensive evaluation also includes testing exercise capacity using a 6-minute walk test and assigning a WHO FC based on a patient’s symptoms. The 6-minute walk test measures the distance an individual can walk during 6 minutes on a hard, flat, indoor surface, and the 6MWD is the most-used measure of exercise capacity in PAH and acts as an indicator of clinical worsening or improvement during follow-ups. The WHO FC system divides patients into 4 groups based on the severity of their PAH symptoms, ranging from asymptomatic (FC I) to having severe symptoms (FC IV); patients with PAH classified as WHO FC II and III are largely symptom-free at rest but experience symptoms upon exertion.8
Risk status was introduced in the 2015 ESC/ERS guidelines for the diagnosis and treatment of PAH,28 which were subsequently updated in 2022.8 While WHO FC has historically been used to guide disease management decisions, several studies have suggested that WHO FC alone is not sufficient to assess patients and their outcomes in response to treatments.8,28 As a result, the guidelines have shifted from WHO FC alone to risk status, suggesting that a comprehensive assessment of patients with PAH is required.
At the time of diagnosis, patients are classified into 3 strata: low risk, intermediate risk, and high risk. The stratification criteria are shown in Table 3.8 These strata are based on hemodynamic parameters and clinical observations including the 6MWD, WHO FC assessment, and NT-proBNP or BNP levels.8 Since the updated 2022 ESC/ERS guidelines, patients at intermediate risk are further divided into intermediate-low risk and intermediate-high risk categories at follow-up (Table 3), which provides more refined risk stratification and can further guide treatment decision-making.8 Patients considered low risk are defined as those with PAH classified as WHO FC I or II and who have a 6MWD greater than 440 m and either an NT-proBNP level less than 300 ng/L or a BNP level less than 100 ng/L.
Risk status can be assessed using the COMPERA 2.0 method, as outlined in the 2022 ESC/ERS guidelines,8,29 or the Simplified French Risk Score, among others. Both take into consideration 3 key noninvasive parameters — WHO FC, 6MWD, and BNP or NT-proBNP — and use the same value cut-offs, as presented in Table 3. The ESC/ERS guideline (or COMPERA 2.0 method) assigns a grade of 1 through 4 to each parameter and calculates a patient’s risk status based on the average score.8 To be classified as low-risk status using the Simplified French Risk Score, each of the low-risk criteria must be met.30 These 3 risk assessment parameters are all clinically relevant and are associated with long-term survival in patients.30
Table 3: Risk Stratification Criteria for COMPERA 2.0 or Simplified French Risk Score
Parameter | Low riska | Intermediate-low risk | Intermediate-high risk | High risk |
|---|---|---|---|---|
WHO FC | I or II | NA | III | IV |
6MWD (m) | > 440 | 320 to 440 | 165 to 319 | < 165 |
BNP (ng/L) or NT-proBNP (ng/L) | < 50 < 300 | 50 to 199 300 to 649 | 200 to 800 650 to 1,100 | > 800 > 1,100 |
6MWD = 6-minute walk distance; FC = functional class; NA = not applicable.
aThe 1-year mortality risk was 0% to 3%, 6%, 5% to 20%, and more than 20% for the low-risk, intermediate-low–risk, intermediate-high–risk, and high-risk groups, respectively.
Sources: Humbert 2022;8 Boucly 2017;30 Boucly 2022.31
Confirmative diagnostic testing for PAH by RHC is available at pulmonary hypertension COEs, which have been established in all provinces except for Prince Edward Island, which is served by the COE in Nova Scotia. The Vancouver pulmonary hypertension COE serves patients in Yukon, while patients from the Northwest Territories and Nunavut are served by COEs in Alberta, Ontario, and Quebec. The sponsor is not undertaking any initiative to increase the availability of the diagnostic tests.
Standards of Therapy
The contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
All the currently approved PAH-specific treatments aim to promote vasodilation by targeting 1 of the 3 pathways linked to PAH pathology: inhibiting the endothelin pathway through ERAs or promoting the nitric oxide pathway through PDE5 inhibitors and the prostacyclin pathway through prostacyclin analogues or a prostacyclin receptor agonist.24
As per the 2015 CADTH recommendation report for drugs in PAH, the initial treatment for patients is PDE5 inhibitor monotherapy, and patients are required to experience inadequate disease control (e.g., disease not classified as WHO FC I or II or a 6MWD > 440 m) before receiving additional treatments.32
The most recent Canadian consensus statement for pulmonary hypertension treatment was published in 2020 by the Canadian Cardiovascular Society and the Canadian Thoracic Society.24 It is aligned with the 2015 ESC/ERS guidelines,28 which were updated in 2022.8 In patients without significant cardiovascular comorbidities, the 2015 ESC/ERS guidelines recommended that patients in low-risk to intermediate-risk categories begin treatment with upfront double oral combination therapy, such as with a PDE5 inhibitor and an ERA.28 Patients in high-risk categories are recommended to commence on a triple combination therapy, such as a combination of an ERA, a PDE5 inhibitor, and a parenteral prostacyclin.28 The guidelines also recommend follow-up risk assessment every 3 to 6 months, with a goal of reaching low-risk status, regardless of the assessment methods.28 Ultimately, patients may need to be listed for lung or heart-lung transplant as the last treatment option, although this is uncommon.
The sponsor provided estimated percentages of patients with PAH receiving the different treatments available in Canada. According to the 2021 PAH impact survey conducted by PHA Canada, almost 2 in 3 patients with PAH are on combination therapy, either dual (33%) or triple (27%).9 As of September 2023, based on the Socioeconomic Burden of PAH in Canada study of 217 patients, also conducted by PHA Canada, 188 respondents (86.6%) were on PAH therapy, of which 30 (13.8%) were on monotherapy, 77 (35.5%) were on dual therapy, and 81 (37.3%) were on triple therapy.33 Specifically, among those on dual therapy, about 75% were treated with ERAs and PDE5 inhibitors, 10% with PDE5 inhibitors and prostanoids, and about 9% with ERAs and prostanoids. And among those taking triple therapy, 64% were adding a prostacyclin receptor agonist and 36% were on prostacyclin analogue infusions.
Drug Under Review
The key characteristics of sotatercept are summarized in Table 4 for the treatment of adults with PAH (WHO Group 1 pulmonary hypertension).
Sotatercept is an activin-signalling inhibitor for activin A. Activin A binding to ACTR2A promotes proliferative signalling and a decrease in BMPR2 signalling. The imbalance of ACTR2A-BMPR2 signalling results in vascular cell hyperproliferation, causing pathological remodelling of the pulmonary arterial wall, narrowing the arterial lumen, and increasing PVR, which lead to increased pulmonary artery pressure and right ventricular dysfunction.
The Health Canada indication states that sotatercept is indicated in combination with standard PAH therapy, for the treatment of adults with PAH (WHO Group 1 pulmonary hypertension) classified as WHO FC II or III. Sotatercept is administered once every 3 weeks by subcutaneous injection according to patient weight. The starting dose is 0.3 mg/kg, with a target dose of 0.7 mg/kg.
Sotatercept has not been previously reviewed by CDA-AMC for any indication. The sponsor’s reimbursement request is as an add-on to optimal background therapy for the treatment of adult patients with PAH who are not at low risk. Low risk is defined as PAH classified as WHO FC I or II and 6MWD greater than 440 m and NT-proBNP less than 300 ng/L or BNP less than 100 ng/L. Optimal background therapy is defined by the sponsor as patients receiving an optimal number and optimal doses of therapies according to clinical guidelines; patients may be on double or triple therapy depending on contraindications and/or the tolerability of available PAH therapies.
Sotatercept received a Notice of Compliance from Health Canada on August 28, 2024. The drug has been approved by the FDA for the treatment of adults with PAH (WHO Group 1 pulmonary hypertension) to increase exercise capacity, improve WHO FC, and reduce the risk of clinical worsening events. Sotatercept is under review by the European Medicines Agency.
Table 4: Key Characteristics of Sotatercept
Characteristic | Sotatercept |
|---|---|
Mechanism of action | Activin-signalling inhibitor for activin A |
Indicationa | In combination with standard pulmonary arterial hypertension (PAH) therapy, for the treatment of adults with WHO [WHO] Group 1 PAH and Functional Class (FC) II or III |
Route of administration | Subcutaneous |
Recommended dosage | 0.3 mg/kg, with a target of 0.7 mg/kg, every 3 weeks |
Serious adverse effects or safety issues | Increased hemoglobin, severe thrombocytopenia |
Other | Intended for use under the guidance of a health care professional |
aHealth Canada–approved indication.
Source: Product monograph from the sponsor.34
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient input(s) received have been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
CDA-AMC received 1 submission from PHA Canada, which is a joint input from PHA Canada, the Pulmonary Arterial Hypertension Foundation of Quebec, Scleroderma Canada, and Scleroderma Quebec.
PHA Canada is a federally registered and accredited charity whose mission is to empower the Canadian pulmonary hypertension community through support, education, advocacy, awareness, and research. The Pulmonary Arterial Hypertension Foundation of Quebec is a provincially registered nonprofit organization that aims to improve the quality of life of people with PAH and their loved ones. Scleroderma Canada is a federally registered charity and national advocate that has worked collaboratively with regional and international organizations to bring health care research, education, and clinical care together to ensure those affected by scleroderma have access to the latest advances in care. Scleroderma Quebec is a federally registered charity that provides medical and moral support to patients, provides information resources for the public and the medical community, and raises funds for scleroderma research.
PHA Canada stated that information for this submission was gathered primarily from 2 sources:
Sotatercept Patient Evidence Submission Survey: an online survey of patients with PAH and their caregivers in Canada, conducted in English and French by PHA Canada from March 13, 2024, to April 1, 2024. This survey aimed to gather feedback from patients with PAH and their caregivers in Canada about their current treatment experiences and expectations for sotatercept. Out of 216 respondents, 82% were adults with PAH, 4% were parents or guardians of children with PAH, and 14% were caregivers of adults (aged > 18 years old) with PAH. Of the patients surveyed, 3 indicated experience taking sotatercept. Half the total responses were from Ontario, 18% were from Quebec, 10% were from British Columbia, and 10% were from Alberta, with the rest from other jurisdictions. Nearly half (46%) of the patient respondents (including those aged < 18 years represented by their parent or guardian) had been diagnosed with PAH less than 5 years ago. The most common PAH subtype reported by patients was idiopathic PAH (46%), followed by scleroderma-associated PAH (26%).
Socioeconomic Burden of PAH in Canada: an online survey of adult patients with PAH in Canada, conducted in English and French by PHA Canada and the University of Alberta from August 15, 2023, to September 10, 2023. This study aimed to evaluate the socioeconomic burden of PAH, with an emphasis on workplace and activity-related limitations, assessed using the Work Productivity and Activity Impairment questionnaire. Most patients who responded to this survey were female (84%), white (84%) with a mean age of 57 years. Just over 40% of the responses were from Ontario; 17% were from British Columbia and 14% were from Alberta. Most patients (84%) self-reported as having PAH classified as WHO FC II or III.
PHA Canada added findings from the 2021 Canadian Pulmonary Hypertension Community Survey, the 2013 Burden of Illness Survey, information from the PHA Canada and Scleroderma Canada’s joint submission to CADTH in April 2016 for selexipag, and personal stories and insights from patients and their families.
According to the patient group input, the most common symptoms of PAH can also be signs of more common medical problems such as asthma, chronic obstructive pulmonary disease, or heart disease, making diagnosing PAH difficult. In Canada, it is common for it to take more than 2 years for patients to be accurately diagnosed with PAH, leading to significant delays in access to appropriate care and treatment. Late diagnosis is associated with more advanced disease and poorer prognosis for patients. PHA Canada noted that the physical symptoms associated with PAH are difficulty breathing upon little or no exertion, fatigue/loss of energy, dizziness upon activity, edema, syncope, bluish lips/hands/feet, chest pain, fainting, light-headedness, heart palpitations, and coughing. PHA Canada reported that the effects of these symptoms included difficulty with climbing stairs (as reported by 86% of patients), doing household chores (79% of patients), walking a short distance (55% of patients), and being intimate with a partner (39% of patients). Many respondents reported limitations to recreation (88% of patients) and travel (74% of patients).
PHA Canada noted that findings from the 2023 Socioeconomic Burden of Illness study demonstrated that patients with PAH are frequently underemployed and dependent on financial and daily living assistance. Only 61 patients (28.1%) surveyed were employed, while 151 (69.6%) were not working, and 5 (2.3%) did not specify their work status. As a result of PAH, 61.3% of the patients had lowered their hours at work, with 44.5% converting from full-time to part-time work. Patients younger than 65 years experienced more frequent changes to work patterns than patients older than 65 years; conversely, the older population more commonly resigned from work or opted for early retirement. Among working patients, diminished workplace productivity and activity were frequently reported. The mean percentage of work missed due to PAH was 12%, impairment while working due to PAH was 42%, overall work impairment due to PAH was 46%, and activity impairment due to PAH was 54%.
PHA Canada clarified that as self-reported WHO FC worsened, the percentage of patients requiring caregiver assistance with daily activities increased (ranging from 12% for FC I to 85% for FC IV), as did the number of hours caregivers lost to caregiving activities (ranging from 6 hours for FC I to 16 hours for FC IV).
Based on PHA Canada’s input, in 2021, 73% of patients reported a lack of understanding of pulmonary hypertension among friends and colleagues and half (53%) felt isolated and excluded from society because pulmonary hypertension is not a “visible” disease. A third of patients felt that pulmonary hypertension has a bigger negative impact on their lives than it did when they were first diagnosed, compared to 45% of caregivers. Furthermore, 64% of patients and 68% of caregivers reported that pulmonary hypertension negatively impacts their daily lives. PHA Canada added that based on the Socioeconomic Burden of Illness study, HRQoL on the EQ VAS was similar regardless of age or sex; however, it decreased with increasing WHO FC (I to IV), with mean values of 82, 66, 52, and 42 for the 4 FCs, respectively. As expected, patients with PAH on triple therapy also reported lower HRQoL.
In terms of experience with currently available treatments, PHA Canada noted that in 2023, 21% of the patients reported being on infusion-based therapies, while 24% reported being on triple oral therapy that included selexipag. By comparison, in 2024’s survey, 16% of the patients reported being on an oral prostacyclin therapy (selexipag) and 16% reported being on an infusion-based therapy (epoprostenol or treprostinil). PHA Canada reported that 58% of the patient respondents of the 2024 survey (including parents or guardians of pediatric patients) found their current therapy only “somewhat effective” at controlling shortness of breath upon exertion; 17% found it to be “highly effective.” Patients were most likely to report that their current therapy was “highly effective” at controlling the following: shortness of breath at rest (50% of patients), chest pain at rest (46%), chest pain upon exertion (31%), fainting (31%), and coughing (25%). They were most likely to report that their current therapy was “not effective” at controlling the psychological and emotional impacts of the disease, such as depression (35% of patients); fatigue or tiredness (28%); and limitations on day-to-day activities (21%). PHA Canada added that the adverse effects of currently approved medications reported by patients in 2024 were headaches and body pain (56% of patients), sleep difficulties (49%), flushing of skin (48%), digestive problems (47%), stuffy or runny nose (45%), and pain or infection at the infusion site (14%). PHA Canada noted the barriers to accessing treatments reported by patients as reliance on a drug manufacturer’s compassionate access program (29% of respondents), intolerance due to AEs (20%), paying out of pocket for treatment (12%), and paying out of pocket for supplies (6%).
PHA Canada mentioned that based on the Socioeconomic Burden of PAH in Canada survey conducted in 2023, the PAH-related expenses paid by patients per year for travel to a doctor or a hospital and for health visits range from $375 for patients with PAH classified as WHO FC I to $747 for patients with PAH classified as WHO FC IV. The parking fees at pharmacies or hospitals or for health-related visits, per patient, range from $60 for patients with PAH classified as WHO FC I to $86 for patients with PAH classified as WHO FC IV.
PHA Canada stated that the top 3 benefits patients were willing to tolerate serious adverse effects for were slowed disease progression (82%), increased quality of life for patients (79%), and improved symptom management (62%). This was very similar to the responses of caregivers of both pediatric and adult patients. A small minority of respondents (7) expressed no willingness to tolerate adverse effects.
Only 3 patients who responded to the Sotatercept Patient Evidence Submission Survey indicated experience taking sotatercept. Since these patients could only have accessed sotatercept through the clinical trial in Canada, it cannot be verified if they received sotatercept or placebo. PHA Canada added that a patient from the US participating in the OLE trial for sotatercept who had been taking the drug at home shared publicly the following: “It is once every 21 days by subcutaneous injection. The side effects are nosebleeds and high hemoglobin. It has given me exceptionally great results and has been so hope-filling.”
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 3 clinical specialists with expertise in the diagnosis and management of PAH.
Unmet Needs
The clinical experts identified several unmet needs in the treatment of patients with PAH. These unmet needs arise from the condition’s rarity and the lack of treatments capable of effectively reversing or curing the disease. According to clinical experts, a key characteristic of PAH is the heterogeneous response from patients to some drugs in their treatment regimens. This variability underscores the need for precision-based regimens targeting the underlying cause.
Experts also highlighted the limited treatment options for right-heart failure, a major cause of death and morbidity in patients with PAH, as well as the scarcity of recommendations for patients with mixed phenotype PAH or PAH with comorbidities. Additionally, there is a lack of data to guide the switching or discontinuation of drugs based on clinical response. The cumulative side effects of treatments further complicate adherence and tolerance, emphasizing the urgent need for better therapeutic strategies that provide better efficacy end points, tolerability, and HRQoL.
Place in Therapy
The experts mentioned that the selection of patients for sotatercept should be guided by a thorough evaluation, including RHC, and should only be performed by PAH specialists.
The experts considered that sotatercept fits into the current treatment paradigm for PAH by affecting a pathway related to the disease’s underlying biology that other available medications do not target.
According to clinical experts, given its mechanism of action and the context of its testing in current trials, sotatercept would be best used as add-on therapy for patients not experiencing disease control (clinical worsening or not reaching the low-risk stratification) with established dual background therapy (PDE5 inhibitors and ERAs) or triple background therapy (dual background therapy plus prostaglandin or prostanoid). The clinical experts also noted that patients who have an absolute contraindication for or who have not tolerated treatment with the components of triple combination therapy would be considered for treatment with sotatercept in clinical practice. Moreover, in their opinion, patients with a contraindication or who cannot tolerate other drug classes while on a single therapy or on dual therapy should also be considered for treatment with sotatercept because this scenario occurs frequently, leaving patients with few alternatives. Thus, it was agreed among the clinical experts that sotatercept would be reserved as an add-on treatment option based on current evidence. The clinical experts also noted that for patients on dual therapy that is not meeting treatment targets, sotatercept would provide an alternative to selexipag when considering escalating to triple therapy. The choice of add-on therapy would be guided by key clinical factors, including patients’ risk stratum and rate of clinical worsening.
Assessing the Treatment Response
According to clinical experts, assessment of treatment efficacy for patients with PAH should involve a comprehensive approach, including 6-minute walk tests, BNP or NT-proBNP levels, and clinical assessments such as improvements in WHO FC, right ventricular function (via echocardiography), hemodynamic measurements, and HRQoL. Additionally, they emphasize that even stabilization or a slowing of disease progression can be considered a clinically meaningful response. The initial assessment of treatment response is typically conducted within 4 weeks of starting therapy for a new diagnosis. This period allows for the full effects of the therapy to manifest. However, if there is evidence of deterioration at any point, earlier assessment and adjustment of treatment may be necessary.
Discontinuing Treatment
When considering discontinuation of sotatercept in the treatment of PAH, several key factors were considered by the clinical experts. Typically, medications are not stopped for lack of effectiveness because this is difficult to judge in a progressive disease like PAH. The primary reason for discontinuation is intolerance or the occurrence of clinically important AEs. Specifically, for sotatercept, if the patients experience SAEs such as bleeding or telangiectasias that require medical attention and/or indicate serious harm to the patient, then treatment would be stopped. Moreover, the experts suggest that if the patient shows progression of the disease to a stage requiring lung transplant, sotatercept should be discontinued posttransplant.
Prescribing Considerations
The appropriate settings for treatment with sotatercept will primarily revolve around the context of PAH. The clinical experts mention that current care for patients with PAH is centralized in specialized centres — often referred to as pulmonary hypertension COEs — a practice that should continue with the introduction of sotatercept. Typically, these centres are staffed by cardiologists or respirologists who have undergone additional training in pulmonary hypertension. It is essential that the prescription and oversight of sotatercept remain within the purview of the specialists working in these centres.
Given the complexity of managing PAH, the clinical experts underscored the critical role of PAH specialists in diagnosing, treating, and monitoring patients who are candidates for sotatercept therapy. These specialists possess the necessary expertise to navigate the intricacies of PAH management and are equipped to tailor treatment plans to individual patient needs. The experts mentioned that this ensures comprehensive quality of care for patients with PAH.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full original clinician group input received has been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
CDA-AMC received input from 1 clinician group, the Canadian Pulmonary Hypertension Health-Care Providers, which is a nonaffiliated group of physicians and nurse practitioners from provincial specialized pulmonary hypertension centres. Fourteen clinicians contributed to this input.
According to the clinician group input, the information in this submission is primarily derived from the recent 2022 ESC/ERS pulmonary hypertension guidelines, the 2020 Canadian Cardiovascular Society and Canadian Thoracic Society position statement on pulmonary hypertension, published evidence from recent multicentre PAH studies in Canada, and the collective perspectives of the authors of these documents based on clinical experience and knowledge of patient outcomes and access to therapies in the Canadian context.
The clinician group stated that the current treatment options range from nonpharmacologic management options to oral or parenteral combination therapy and even lung transplant. The current PAH medications provide some symptomatic benefit and stability to the patients, but it is often short-lived. The medications function mainly as vasodilators of less diseased blood vessels but have minimal effect on the blocked vessels or on the cellular proliferation that leads to disease progression. The existing PAH therapies have not been shown to have disease-reversing or disease-modifying effects. Even with optimal medical therapy, only a minority of patients with PAH experience having or maintaining a low-risk status. According to the clinician group, a recent analysis showed that 5-year survival for PAH was only approximately 60% in the patient population living in Canada. The clinician group mentioned that the most important end points for patients in a Canadian study were to improve symptoms and quality of life, prolong survival, and reduce the risk of clinical deterioration, including delaying or preventing hospitalizations or transplant.
Intolerable side effects (e.g., significant headache, flushing, rashes, nasal congestion, bone and jaw pain, diarrhea), as well as difficulties and complications of parenteral therapies, are among the unmet needs and treatment gaps to be addressed.
Regarding place in therapy, the clinician group believes that sotatercept is the first approved PAH therapy that acts by altering growth factor signalling, which controls the aberrant cell proliferation in PAH. Sotatercept has been in clinical trials as an add-on therapy and is not expected to be used as a first-line treatment.
According to the clinician group input, the outcomes used to determine whether a patient is responding to treatment in clinical practice are changes in hemodynamics, functional capacity, symptoms, clinical deterioration, and measures of right-heart stress, such as NT-proBNP and echocardiogram. In terms of deciding to discontinue treatment, none of the clinicians has had to discontinue the treatment for anyone so far; the clinician group input suggested restriction of prescribing to provincially designated pulmonary hypertension centres only.
Drug Program Input
The drug programs provide input on each drug being reviewed through the CDA-AMC Reimbursement Review process by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The sponsor proposes that there are no comparators for sotatercept and notes that all currently available PAH-specific medications in Canada are vasodilators (e.g., phosphodiesterase type 5 inhibitors, endothelin receptor antagonists, prostanoids), whereas sotatercept is a novel activin -signalling inhibitor that “may reverse the characteristic pulmonary vascular remodeling in PAH.” Comments:
| For CDEC deliberations. |
Considerations for initiation of therapy | |
The sponsor notes that a confirmative diagnosis of PAH requires an invasive right-heart catheterization performed by specialists within the pulmonary hypertension centres of excellence located across Canada. | For CDEC deliberations. |
Patients in the pivotal trial were required to be on stable doses of their respective background PAH therapy for at least 3 months before enrolment, and the sponsor is requesting reimbursement as add-on to “optimal background therapy.” Comment: If recommended for funding, it will be important to clearly define optimal background therapy. | The clinical experts noted that optimal background therapy is well known and defined among experts in PAH and is based on current clinical guidelines. They acknowledged that the optimal number and combination of drugs is used according to the patient’s risk status and with the goal to reach or maintain a low-risk status. The clinical experts emphasized that although optimal background therapy is defined by guideline recommendations, patient tolerance is important, and with the significant side effect profile of some of these drugs, “maximally tolerated optimal medical therapy” can be different than “optimal background therapy.” |
Considerations for continuation or renewal of therapy | |
The sponsor notes that:
Question: Should there be a minimum response to therapy to justify continuation of the intervention? If so, how should it be defined, both at the first renewal assessment and afterwards? | The clinical experts acknowledged that reaching or maintaining low-risk status is 1 of the main goals of treatment and considered that maintaining or improving the patient’s risk status would be enough for continuing therapy. The experts recognized that there are no targeted markers to define if and how a patient is responding and that observing deterioration (an increase) in the patient’s risk status would suggest that discontinuation or escalation of therapy should be considered. The experts acknowledged that the evidence that could be used to establish the best continuation or discontinuation criteria is unclear. |
Considerations for prescribing of therapy | |
Comments:
| For CDEC deliberations. |
Generalizability | |
Those with a diagnosis of PAH associated with HIV, portal hypertension, schistosomiasis, or pulmonary veno-occlusive disease were excluded from the pivotal trial. Question: Should these patients be excluded from treatment eligibility? | The clinical experts considered that patients with these conditions should not be excluded from consideration for treatment with sotatercept but, rather, should be considered on a case-by-case basis, with individualized decisions made. |
System and economic issues | |
Comment: Compared to background therapy alone, the budget impact analysis predicts that funding of sotatercept in adults with PAH who are not at low risk and who are on optimal background therapy would result in incremental total costs of $38,782,232 in year 1, $82,425,708 in year 2, and $115,650,464 in year 3, for total incremental costs of $236,858,404 over the 3-year projection period. | For CDEC deliberations. |
CDEC = Canadian Drug Expert Committee; NA = not applicable; NR = not reported; PAH = pulmonary arterial hypertension.
Clinical Evidence
The objective of the CDA-AMC Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of sotatercept 45 mg and 60 mg, for subcutaneous injection, in the treatment of adult patients with PAH. The focus will be on comparing sotatercept to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of sotatercept is presented in 2 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes 1 pivotal study, which was selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section, using the GRADE approach, follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies.
Included Studies
Information from the following bodies of evidence submitted by the sponsor are included in the CDA-AMC review and appraised in this document:
1 pivotal study included in the sponsor’s systematic review
3 long-term extension studies.
Systematic Review
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CDA-AMC review team.
Description of Studies
One pivotal trial (the STELLAR trial, NCT04576988) was included in the systematic review. The main objectives of the STELLAR trial were to evaluate the efficacy and safety of sotatercept versus placebo on top of stable background PAH therapy in adults with PAH.
The STELLAR trial was a phase III, randomized, multicentre, DBPC, parallel-group intervention study in adult patients with PAH (WHO Group 1 pulmonary hypertension). The trial was conducted across 21 countries, including 3 sites in Canada, between January 2021 and December 2022, enrolling an adult patient population with age and gender distributions that closely reflected the demographics of the patient population with PAH in Canadian clinical practice.
The study design is presented in Figure 1. This study consisted of 2 treatment periods: the DBPC phase and the LTDB phase. The duration of the DBPC period was 24 weeks. Participants who completed the DBPC period entered the LTDB phase, which lasted up to 72 weeks or until the last randomized participant completed the DBPC period and the study was unblinded. Participants who completed the DBPC period and were on treatment (sotatercept or placebo) in the LTDB period were eligible to participate in a separate, open-label LTFU ongoing study, the SOTERIA study (NCT04796337). The STELLAR trial had an end-of-treatment and an end-of-study follow-up period (nontreatment period) of at least 8 weeks, which included at least 2 visits, for participants who prematurely discontinued study intervention during the DBPC or LTDB period or who did not transition to the LTFU study. Participants who met the entry criteria in the STELLAR trial were randomized in a 1:1 ratio to receive either sotatercept or placebo. Randomization was generated through a computerized system, provided by an interactive response technology, and patients were stratified by baseline WHO FC (II or III) and background PAH therapy (monotherapy or double therapy, or triple therapy). A total of 323 participants were randomized (163 in the sotatercept group; 160 in the placebo group). For analyses, 323 participants were included in the FAS and the safety set.
Characteristics of the included study are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Characteristic | Details |
|---|---|
Designs and populations | |
Study design | Phase III, multinational, randomized, placebo-controlled, double-blind, multicentre study |
Locations | 91 sites in 21 countries (Argentina, Australia, Belgium, Brazil, Canada, Czech Republic, France, Germany, Israel, Italy, Mexico, Netherlands, New Zealand, Poland, Serbia, South Korea, Spain, Sweden, Switzerland, UK, and the US) including 3 sites in Canada (Alberta, Ontario, and Quebec) |
Patient enrolment dates | Start date: January 25, 2021 End date: December 6, 2022 |
Randomized (N) | Sotatercept: 163 Placebo: 160 Total: 323) |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Sotatercept (at a starting dose of 0.3 mg/kg administered subcutaneously, with a target dose of 0.7 mg/kg administered subcutaneously, every 21 days) + background PAH therapya for 24 weeks |
Comparator(s) | Placebo (every 21 days) + background PAH therapya for 24 weeks |
Study duration | |
Screening phase | ≤ 4 weeks before randomization |
Run-in phase | NR |
Treatment phase | Phase I: DBPC phase of 24 weeks for the primary outcomes Phase II: LTDB phase up to 72 weeks (i.e., until the last participant randomized completes the DBPC treatment period) |
Follow-up phase | At least 8 weeks Participants who completed the DBPC treatment period and were on treatment in the LTDB treatment period were eligible to participate in a separate, open-label, long-term follow-up study (SOTERIA) |
Outcomes | |
Primary end points |
|
Secondary and exploratory end points | Secondary
Exploratory (baseline to week 24)
|
Publication status | |
Publications | Hoeper et al. (2023) Souza et al. (2023) |
6MWD = 6-minute walk distance; AE = adverse event; DBPC = double-blind placebo-controlled; eGFR = estimated glomerular filtration rate; FC = functional class; LTDB = long-term double-blind; MDRD = modification of diet in renal disease; NR = not reported; PAH = pulmonary arterial hypertension; PAH-SYMPACT = Pulmonary Arterial Hypertension – Symptoms and Impact; PVR = pulmonary vascular resistance; RHC = right-heart catheterization.
aBackground PAH therapy refers to approved PAH-specific medications and consists of monotherapy or combination therapy with endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, soluble guanylate cyclase stimulators, and/or prostacyclin analogues or receptor agonists every 21 days for 24 weeks.
Details included in the table are from the sponsor’s summary of clinical evidence.
Sources: Clinical Study Report;3 ClinicalTrials.gov (NCT04576988).35
Figure 1: STELLAR Study Design
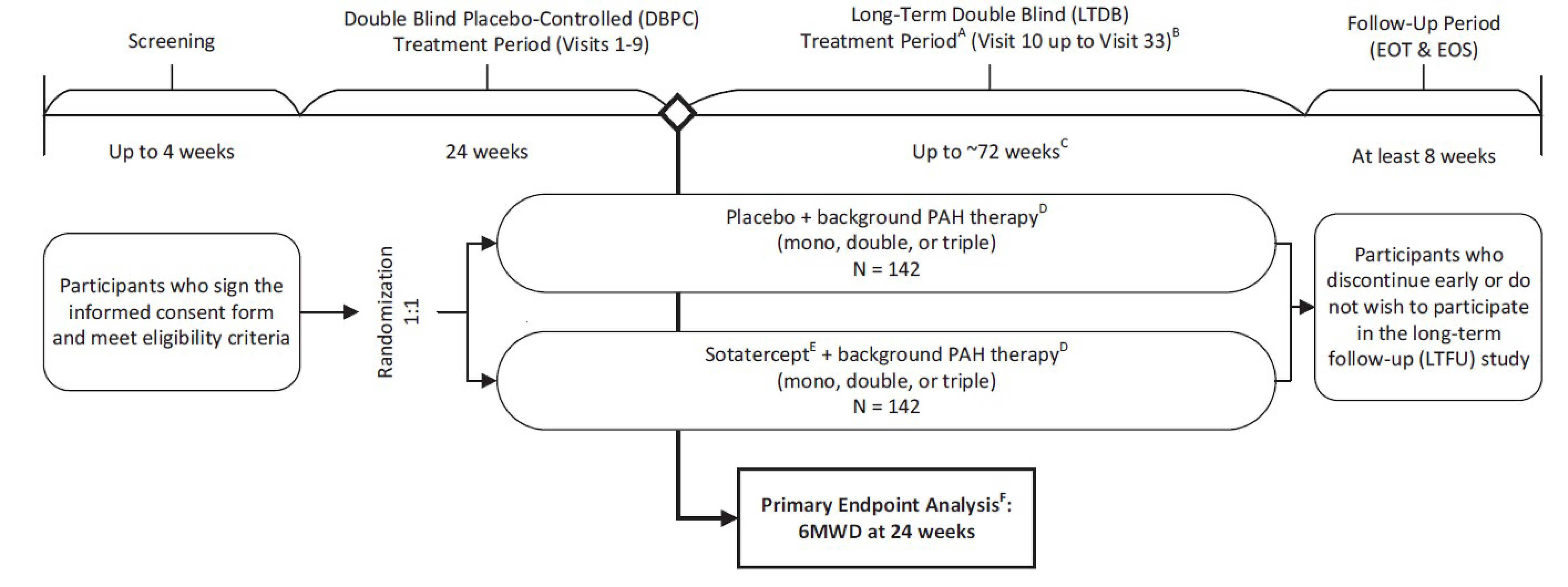
6MWD = 6-minute walk distance; DBPC = double-blind placebo-controlled; EOS = end of study; EOT = end of treatment; LTDB = long-term double-blind; LTFU = long-term follow-up; PAH = pulmonary arterial hypertension.
a During the LTDB treatment period, select study visits may be performed as home health care visit.
b LTDB treatment period will last until the last participant randomized completes the DBPC treatment period, at which point the study will be unblinded and participants may roll over into the LTFU study.
c LTDB treatment period duration is estimated based on projected enrolment duration and time required for the last participant to complete the DBPC treatment period.
d Background PAH therapy refers to approved PAH-specific medications and may consist of monotherapy or combination therapy with endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, soluble guanylate cyclase stimulators, and/or prostacyclin analogues or receptor agonists.
e Sotatercept at a starting dose of 0.3 mg/kg subcutaneous, with a target dose of 0.7 mg/kg subcutaneous.
f Primary end point analysis will be completed after the last participant randomized completes the DBPC treatment period.
Source: Clinical Study Report.3
Populations
Inclusion and Exclusion Criteria
Eligible participants for the STELLAR trial included adults with a documented diagnosis of PAH (WHO Group 1 pulmonary hypertension) via RHC. Patients also had symptomatic PAH in WHO FC II or III and had had stable doses of their respective background PAH therapy for at least 3 months before enrolment. However, the patients required additional therapy beyond standard of care to meet their treatment goals. Detailed inclusion and exclusion criteria are presented in Table 6.
Interventions
Patients were randomized 1:1 to receive sotatercept or its matching placebo every 21 days. The placebo contained all ingredients other than sotatercept. Both the clinical drug product containing sotatercept and its matching placebo were supplied as a lyophilized powder in labelled, rubber-stoppered, type I glass vials. Both the investigator and the participant were blinded, and treatments were administered by study personnel.
Both arms also included background PAH therapy, which was approved PAH-specific medications and which consisted of monotherapy or combination therapy with ERAs, PDE5 inhibitors, soluble guanylate cyclase stimulators, and/or prostacyclin analogues or receptor agonists. Background PAH therapy was not provided as study medication during the study; however, patients continued receiving stable doses of their individual background therapy throughout the trial.
All participants began sotatercept treatment at a starting dose of 0.3 mg/kg at visit 1. At visit 2, the dose was escalated to the target dose of 0.7 mg/kg and remain at 0.7 mg/kg for the duration of the treatment period, unless dose reduction criteria were met. The dose reduction criteria included an increase in hemoglobin greater than 2.0 g/dL since the last dose and hemoglobin above the gender-specific upper normal limit of normal, as well as low platelets count.
Outcomes
A list of efficacy end points assessed in this Clinical Review report is provided in Table 7, followed by descriptions of the outcome measures. The measurement properties of the outcomes of 6MWD, the PAH-SYMPACT, and EQ-5D-5L are depicted in Table 8. These end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by CDA-AMC and input from patient and clinician groups and public drug plans. Using the same considerations, the review team selected the end points that were most relevant to inform the CDA-AMC expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing the CDA-AMC expert committee deliberations were also assessed using GRADE. Outcomes submitted by the sponsor that were not included in the GRADE assessment of this review are presented in Appendix 1.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | Evaluation in STELLAR trial |
|---|---|---|
Mortality | Week 24 | Secondary end point (part of multicomponent) |
6MWD | Week 24 | Primary end pointa |
Multicomponent improvement | Week 24 | Secondary end pointa |
Time to first occurrence of clinical worsening event or death | When the last patient completed the week 24 visit | Secondary end pointa |
PVR | Week 24 | Secondary end pointa |
NT-proBNP | Week 24 | Secondary end pointa |
Change in WHO FC | Week 24 | Secondary end pointa |
Simplified French Risk Score | Week 24 | Secondary end pointa |
PAH-SYMPACT physical impacts domain score | Week 24 | Secondary end pointa |
PAH-SYMPACT cardiopulmonary symptoms domain score | Week 24 | Secondary end pointa |
EQ-5D-5L | Week 24 | Exploratory end point |
Hospitalization (PAH specific) | When the last patient completed the week 24 visit | Secondary end pointa |
Patients experiencing AEs | Week 24 | Primary end point |
Patients discontinuing due to AEs | Week 24 | Primary end point |
6MWD = 6-minute walk distance; AE = adverse event; FC = functional class; PAH = pulmonary arterial hypertension; PAH-SYMPACT = Pulmonary Arterial Hypertension – Symptoms and Impact; PVR = pulmonary vascular resistance.
aA gatekeeping method was used to control the type I error rate in the primary and secondary efficacy end points by starting testing with the primary efficacy end point and then proceeding in the order of the secondary efficacy end points as listed. Secondary end point testing was performed using a 2-sided alpha at the 0.05 level and by proceeding successively in the order of the secondary end points only after each of the preceding end points was found to be statistically significant.
Details included in the table are from the sponsor’s summary of clinical evidence.
Sources: Clinical Study Report;3 ClinicalTrials.gov (NCT04576988).35
Six-Minute Walk Distance
The primary efficacy end point of the STELLAR trial was the change from baseline in 6MWD. The 6MWD is the distance an individual can walk for more than 6 minutes on a hard, flat, indoor surface. It is a validated, clinically relevant measure of exercise and functional capacity that is often severely restricted in patients with PAH and indicative of their disease burden.36 The 6MWD has historically been used as a primary efficacy end point in pivotal trials for PAH therapy approved across various regulatory agencies. The consensus of the point estimates for the minimal clinically important difference in 6MWD is approximately 33 m for patients with PAH, using anchor and distributional methods.37,38
The 6-minute walk test was performed during the screening period; at visits 2, 5, and 9; and during the follow-up visits. From visit 1 onward, the 6-minute walk test was performed on the study visit day or within 10 days before study drug administration. During the study, the 6-minute walk test was performed at approximately the same time of day under the same conditions between screening and visit 9 and included chronic oxygen therapy and the use of walking aids or face coverings (as the STELLAR trial was conducted during the COVID-19 pandemic). The baseline 6MWD is derived using the data from the 6-minute walk test performed at screening. The screening 6-minute walk test is performed twice, at least 4 hours apart, but no longer than 1 week apart. The corresponding 6MWDs are to be within 15% of each other. If the difference between the first and second tests is more than 15%, the test may be repeated once more, provided the repeat test is within 1 week of the previous test. If no test was repeated when the difference was more than 15%, the average of the 2 screening 6MWD measurements was used as the baseline.
Multicomponent Improvement
Multicomponent improvement was the first secondary efficacy end point in hierarchical testing. This was a composite measure that required that all the following criteria be met at week 24 relative to baseline:
Increase in 6MWD greater than or equal to 30 m
Decrease in NT-proBNP greater than or equal to 30% or maintaining or reaching NT-proBNP level less than 300 ng/L
Improvement in WHO FC from III to II or I, or from WHO FC II to I, or maintenance of WHO FC II
The components of this end point include functional assessment (6MWD and WHO FC) and a prognostic biomarker indicating cardiac stress (NT-proBNP). They align with the variables used for the Simplified French Risk Score end point in this study. NT-proBNP and WHO FC were also evaluated independently as 2 secondary end points.
Samples for NT-proBNP analysis were collected during the screening period; at visits 1, 2, 3, 4, 5, and 9; and during the follow-up visits (visit 1 and 2). The baseline NT-proBNP was defined as the last measurement taken before the first dose of the study medication. This could be at the screening or visit 1 assessment.
The WHO FC is categorized from I through IV, representing the symptom severity of PAH. A worsening in WHO FC is an indicator of disease progression. The WHO FC was assessed by the investigator during the screening period; at visits 2, 5, and 9; and during the follow-up visits (visit 1 and 2).
Time to First Occurrence of Clinical Worsening Event or Death
Death or nonfatal clinical worsening was a key secondary end point assessed and recorded by the investigator at each dosing visit to assess disease progression associated with PAH. The components of this composite end point were:
death
worsening-related listing for lung or heart-lung transplant
need to initiate rescue therapy with an approved background PAH therapy or to increase the dose of infusion prostacyclin by 10% or more
need for atrial septostomy
hospitalization for worsening of PAH (≥ 24 hours)
deterioration of PAH defined by both of the following events occurring at any time, even if they began at different times, as compared to their baseline values:
worsening of WHO FC
decrease in 6MWD by 15% or more, confirmed by 2 tests at least 4 hours apart, but no more than 1 week apart.
An independent, blinded adjudication committee adjudicated all clinical worsening events, including death, up to the end of the study to determine whether these events were due to PAH. All other clinically significant abnormal findings that did not meet the above criteria were reported as AEs.
Pulmonary Vascular Resistance
PVR was the second secondary efficacy end point in hierarchical testing. It is a parameter for the evaluation of pulmonary circulation hemodynamics. However, a minimal clinically important difference has not been established for changes in PVR. PVR was calculated as the difference between the mPAP and the pulmonary arterial wedge pressure, divided by the volume of blood pumped by the heart per minute (i.e., cardiac output). Components of PVR were measured via RHC at baseline and at week 24.
Simplified French Risk Score
In the STELLAR study, the noninvasive Simplified French Risk Score was used. “Low risk” was defined as attaining or maintaining all 3 low-risk criteria: WHO FC I or II, 6MWD greater than 440 m, and NT-proBNP less than 300 ng/L. Change from baseline in the percentage of participants who maintained or reached a low-risk score at week 24, using the Simplified French Risk Score calculator, was reported for the DBPC period.
PAH-SYMPACT Questionnaire
The PAH-SYMPACT is a 23-item questionnaire measuring PAH-related symptoms and the impact of PAH on daily life. In the STELLAR study, 3 domains (physical impacts, cardiopulmonary symptoms, and cognitive/emotional impact) were measured as key secondary outcomes, and 1 domain (cardiovascular symptoms) was measured as an exploratory outcome. This instrument has been validated39 by correlations with other patient-reported outcomes.40
The physical impacts domain consists of walking slowly on a flat surface, walking quickly on a flat surface, walking uphill, carrying things, doing light indoor household chores, washing or dressing oneself, and needing help from others. The cardiopulmonary symptoms domain consists of shortness of breath, fatigue, lack of energy, swelling in the ankles or legs, swelling in the stomach area, and cough. The cognitive/emotional impact domain consists of thinking clearly, feeling sad, feeling worried, and feeling frustrated. The cardiovascular symptoms domain consists of heart palpitation, rapid heartbeat, chest pain, chest tightness, and light-headedness. Patients reported, at home, responses to the impact questions on day 7 with a 1-week recall period, and responses to the symptom questions daily for 7 days before the study visit. The score for each item ranges from 0 (not difficult at all) to 4 (extremely difficult). A domain score was calculated by summing the individual responses for each item and dividing by the number of impact items (0 = no physical impact, no cardiopulmonary symptoms, or no cognitive/emotional impact; 4 = severe physical impact, severe cardiopulmonary symptoms, or severe cognitive/emotional impact). A higher score indicated a more severe impact and worse quality of life. Change from baseline (visit 1) in each of the domain scores (i.e., physical impacts, cardiopulmonary symptoms, cognitive/emotional impact, and cardiovascular symptoms) at week 24 was reported for the DBPC period. Minimal clinically important differences for the physical impacts and cardiopulmonary symptoms domains were estimated to be –0.3 and –0.2, respectively, in both anchor-based and distribution-based methods, using data combined across the sotatercept and placebo groups.41
EQ-5D-5L
The EQ-5D-5L was an exploratory end point on HRQoL. It is a standardized measure of health status developed by the EuroQol Group to provide a simple, generic measure of health for clinical and economic appraisal. The EQ-5D-5L descriptive system comprises the following 5 dimensions, each describing a different aspect of health: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 5 response levels of severity: no problems (level 1), slight problems (level 2), moderate problems (level 3), severe problems (level 4), and extreme problems (level 5). The EQ VAS records the patient’s self-rated health on a vertical VAS, where the end points are labelled “the best health you can imagine” and “the worst health you can imagine” on a scale of 0 to 100. The EQ-5D-5L was to be completed before performing other study assessments (6-minute walk test, blood draws, AE discussions, and RHC) and before study drug administration. The EQ-5D-5L index score was reported for each dimension. The baseline for the EQ-5D-5L index score was the measurement from visit 1.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
6MWD | Generic measure of the distance an individual can walk in 6 minutes on a hard, flat, indoor surface. Single score, where higher values represent improvement. | Correlates well with functional class, hemodynamics, and other markers. Validity as surrogate for long-term outcomes is uncertain.36,42 It has good reliability and responsiveness.43 | Estimated as approximately 33 m more than at baseline.37 In previous CADTH reviews, a range from 25.1 m to 38.6 m was used.44 |
PAH-SYMPACT questionnaire | 23-item, patient-reported outcome instrument. Used to measure HRQoL; symptom and impact domain scores range from 0 to 4, where higher scores indicate more severe symptoms or impact. | Good content validity has been shown qualitatively.39 Construct validity supported by correlations with other patient-reported outcomes.40 | MIDs for the physical impacts and cardiopulmonary symptoms domains were estimated to be –0.3 and –0.2, respectively, in both anchor-based and distribution-based methods, using data combined across the sotatercept and placebo groups.41 |
EQ-5D-5L | Generic measure of HRQoL. | No information in patients with PAH. In other populations, there is adequate reliability, with ICCs reported from 0.71 to 0.87. Moderate correlations with clinical scales (0.21 to 0.59). Low responsiveness.45 | No direct MID in patients with PAH. Indirect measures can be found from patients with COVID-19 (7.5 in the VAS46), or interstitial lung disease (ranging from 0.5 to 9.7 in the VAS47). |
6MWD = 6-minute walk distance; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; MID = minimal important difference; PAH = pulmonary arterial hypertension; PAH-SYMPACT = Pulmonary Arterial Hypertension – Symptoms and Impact; VAS = visual analogue scale.
Source: Clinical Study Report.3
Statistical Analysis
The summary of statistical analysis for all efficacy end points is presented in Table 9. All efficacy end points were analyzed using the FAS population.
Table 9: Statistical Analysis of Efficacy End Points for the STELLAR Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Primary end point | ||||
6MWD at week 24 | ARSW | Adjusted from baseline and by randomization stratification factors (baseline WHO FC and treatment utilization) |
|
|
Secondary end points | ||||
Multicomponent improvement, WHO FC improvement, Simplified French Risk Score | Stratified CMH | Adjusted from baseline and by randomization stratification factors (baseline WHO FC and treatment utilization) |
| NA |
Change from baseline in PVR; change from baseline in NT-proBNP; change from baseline in physical impacts domain score, cognitive/emotional impact domain score, and cardiovascular symptoms domain score of PAH-SYMPACT | ARSW (primary) ANCOVA (supportive) | Adjusted from baseline and by randomization stratification factors (baseline WHO FC and treatment utilization) |
| NA |
Time to first clinical worsening or death | Cox regression | Adjusted from baseline and by treatment and randomization stratification factors (baseline WHO FC and treatment utilization) | If no clinical worsening event or death, patient was censored at last visit on treatment through data cut-off at the end of the double-blind, placebo-controlled treatment period | NA |
6MWD = 6-minute walk distance; ANCOVA = analysis of covariance; ARSW = aligned rank–stratified Wilcoxon; CMH = Cochran-Mantel-Haenszel; FC = functional class; NA = not applicable; PAH-SYMPACT = Pulmonary Arterial Hypertension – Symptoms and Impact; PVR = pulmonary vascular resistance.
Sources: Clinical Study Report;3 ClinicalTrials.gov (NCT04576988).35
Sample Size and Power Calculation
The sample size determination was based on the primary efficacy end point and the secondary end point of improvement in WHO FC. Assumptions for the desired treatment effect and estimate of variability (primary end point) are based on data from the PULSAR study (phase II, NCT03496207)48 and from a published clinical trial in patients with PAH.49 For 6MWD, assuming a 1:1 randomization, a 2-sided 0.05 type I error rate, a 25 m improvement in sotatercept treatment compared to placebo, a common standard deviation of 50 m, and 121 patients per arm, the statistical power is approximately 96% under the Wilcoxon rank sum test using N-Query. For the secondary end point — proportion of patients who experience improvement in WHO FC at week 24 — assuming the true proportions among patients treated with placebo and sotatercept are 0.11 and 0.25 respectively, 1:1 randomization, a 2-sided 0.05 type I error rate, and 121 patients per arm, the statistical power is approximately 80% based on the 2-sample chi-square test. Assuming a 15% drop out rate, the total sample size is estimated at 284 (n = 142 patients per treatment group).
Statistical Testing
The change in 6MWD at week 24 from baseline was analyzed using the aligned rank–stratified Wilcoxon test, with the randomization stratification factors as strata.50,51 In this test, the end point values are first aligned across the randomization strata using the stratum-level Hodges-Lehmann location shift estimates, and the aligned values are then analyzed using a Wilcoxon rank sum test. The output from this analysis was used to provide a 2-sided P value and corresponding Hodges-Lehmann location shift estimate of the overall treatment difference with a 95% CI.
A gatekeeping method was used to control the type I error rate in the primary and secondary efficacy end points by starting to test with the primary efficacy end point and then proceeding in the order of the secondary efficacy end points (multicomponent improvement, PVR, NT-proBNP, WHO FC improvement, composite for time to clinical worsening or death). Secondary end point testing was performed using a 2-sided alpha at the 0.05 level and by proceeding successively in the order of the secondary end points only after each of the preceding end points was found to be statistically significant.
The Shapiro-Wilk test of the residuals (P < 0.001) was used to test the normality assumption for statistical tests where an ANCOVA was planned. If the Shapiro-Wilk test revealed that the normality assumption is violated, then the nonparametric analysis was conducted.
Subgroup Analyses
In the STELLAR study, prespecified subgroup analyses were performed on the primary efficacy end point, as well as for 2 secondary end points, PVR and NT-proBNP, if the sample size in each level of the subgroup category was at least 10 patients and based on the analyses for the full population. The subgroups included:
sex (male and female)
PAH subgroups (idiopathic PAH, heritable PAH, drug-induced or toxin-induced PAH, connective tissue disease, congenital heart disease with shunt repair)
monotherapy versus double therapy versus triple combination therapy at baseline
prostacyclin infusion therapy versus non-prostacyclin infusion at baseline
baseline WHO FC (II or III)
baseline PVR (≤ 800 or > 800 dyn·sec·cm-5).
Additionally, a post hoc analysis was performed on 6MWD, PVR, NT-proBNP, WHO FC, and risk of fatal and nonfatal clinical worsening events in patient subgroups based on risk strata at baseline (low risk, intermediate-low risk, or intermediate-high risk). This post hoc analysis was not adjusted for multiplicity.
Analysis Populations
The STELLAR study provides 2 populations of interest. The first is the FAS, consisting of all patients who underwent randomization. The FAS population was assessed in the efficacy analysis. The second is the safety population (safety set), which includes all patients who were randomly assigned to receive at least 1 dose of sotatercept or placebo; this population was assessed in the safety analysis.
Results
Patient Disposition
Of the 434 patients screened for inclusion into the study, 111 patients (25.5%) were excepted from entering for different reasons, described in Table 10. The rest of the patients (323 [74.5%]) were eligible and were randomized 1:1 to the sotatercept (n = 163) or placebo (n = 160) arms.
More patients in the placebo arm than in the sotatercept arm discontinued from the study (16 [10%] versus 8 [4.9%]), and most discontinuations were due to clinical worsening or death or at the patient’s own request (Table 10). Despite the discontinuations, investigators considered all patients in the analyses (FAS and safety set) with the imputations described in the Statistical Analysis section when needed.
Baseline Characteristics
The baseline characteristics of the patients in the STELLAR study outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Table 10: Patient Disposition in the STELLAR Study (Data Cut-Off: August 26, 2022)
Characteristic | Sotatercept | Placebo |
|---|---|---|
Screened, N | 434 | |
Did not meet screening criteria, n | 111 | |
Reason did not meet screening criteria,a n (%) | ||
Inclusion or exclusion criteria | 131 (30.2) | |
Death | 1 (0.2) | |
Disease progression | 1 (0.2) | |
eGFR < 30 mL/min/1.73 m2 | 1 (0.2) | |
Investigator decision | 2 (0.5) | |
Participant request | 3 (0.7) | |
LVEF < 45% at screening | 1 (0.2) | |
QTcF not in range | 1 (0.2) | |
Developed a central line infection | 1 (0.2) | |
Missing | 1 (0.2) | |
Randomized, N | 163 | 160 |
Discontinued from study, n (%) | 8 (4.9) | 16 (10.0) |
Reason for discontinuation, n (%) | ||
Adverse events | 3 (1.8) | 1 (0.6) |
Participant request (withdrawal of consent) | 2 (1.2) | 5 (3.1) |
Participant’s unwillingness or inability to comply with the protocol | 1 (0.6) | 1 (0.6) |
Clinical worsening event | 0 (0.0) | 2 (1.3) |
Death | 2 (1.2) | 6 (3.8) |
Other | 0 (0.0) | 1 (0.6) |
Full analysis set, N | 163 | 160 |
Safety set, N | 163 | 160 |
eGFR = estimated glomerular filtration rate; LVEF = left ventricular ejection fraction; QTcF = Fridericia corrected QT interval.
aPatients could have more than 1 reason for not meeting the screening criteria and may be counted more than once in the categories.
Source: Clinical Study Report.3
Table 11: Summary of Baseline Patient Characteristics in STELLAR Study (FAS)
Characteristic | Sotatercept (N = 163) | Placebo (N = 160) | All treatments (N = 323) |
|---|---|---|---|
Sex female, n (%) | 129 (79.1) | 127 (79.4) | 256 (79.3) |
Sex male, n (%) | 34 (20.9) | 33 (20.6) | 67 (20.7) |
Age (years), mean (SD) | 47.6 (14.1) | 48.3 (15.5) | 47.9 (14.8) |
Geographic region, n (%) | |||
North America | 49 (30.1) | 56 (35.0) | 105 (32.5) |
South America | 13 (8.0) | 15 (9.4) | 28 (8.7) |
Europe | 91 (55.8) | 77 (48.1) | 168 (52.0) |
Asia-Pacific | 10 (6.1) | 12 (7.5) | 22 (6.8) |
Race, n (%) | |||
Asian | 1 (0.6) | 6 (3.8) | 7 (2.2) |
Black | 2 (1.2) | 5 (3.1) | 7 (2.2) |
White | 147 (90.2) | 141 (88.1) | 288 (89.2) |
Other | 7 (4.3) | 6 (3.8) | 13 (4.0) |
Missing | 6 (3.7) | 2 (1.2) | 8 (2.5) |
Time since diagnosis of PAH (years), mean (SD) | 9.2 (7.3) | 8.3 (6.7) | 8.8 (7.0) |
PAH subtype, n (%) | |||
Idiopathic | 83 (50.9) | 106 (66.3) | 189 (58.5) |
Heritable | 35 (21.5) | 24 (15.0) | 59 (18.3) |
Associated with CTD | 29 (17.8) | 19 (11.9) | 48 (14.9) |
Drug induced or toxin induced | 7 (4.3) | 4 (2.5) | 11 (3.4) |
Associated with corrected congenital shunt | 9 (5.5) | 7 (4.4) | 16 (5.0) |
WHO FC, n (%) | |||
FC II | 79 (48.5) | 78 (48.8) | 157 (48.6) |
FC III | 84 (51.5) | 82 (51.2) | 166 (51.4) |
6MWD (m), mean (SD) | 398.5 (83.5) | 407.0 (78.2) | 402.7 (80.9) |
mPAP (mm Hg), mean (SD) | 53 (14.6) | 52.2 (13) | 52.6 (13.8) |
NT-proBNP (pg/mL), mean (SD) | 1,037.5 (2,498.6) | 1,207.8 (2,694.4) | 1,121.1 (2,593) |
PVR (dyn·sec·cm−5), mean (SD) | 781.3 (398.5) | 745.8 (313.5) | 763.7 (358.8) |
6MWD = 6-minute walk distance; CTD = connective tissue disorder; FAS = full analysis set; FC = functional class; mPAP = mean pulmonary arterial pressure; PAH = pulmonary arterial hypertension; PVR = pulmonary vascular resistance; SD = standard deviation.
Sources: Clinical Study Report;3 ClinicalTrials.gov (NCT04576988).35
No significant differences were detected in the baseline characteristics between the arms of the study. Close to 80% of patients were female, and the mean patient age was 47.9 years. Just over half of the patients had PAH classified as WHO FC III (166 of the 323 randomized [51.4%]), with equal distribution between the placebo and sotatercept groups. The rest of the patients had PAH classified as WHO FC II. The mean time since diagnosis was 8.8 years.
Exposure to Study Treatments
Study Treatments
Exposure to randomized study treatments is summarized in Table 12. Compliance with the study intervention was high (mean > 98% in each group) during the DPBC treatment period and during the cumulative DBPC and LTDB treatment periods up to the data cut-off in August 2022.
Table 12: Patient Exposure in the DBPC Treatment Phase of STELLAR Study
Exposure | Sotatercept (N = 163) | Placebo (N = 160) |
|---|---|---|
Duration (days), mean (SD) | 166.3 (12.5) | 162.2 (25.8) |
Duration (days), median (range) | 168.0 (61 to 193) | 168.0 (21 to 196) |
Adherencea (%), mean (SD) | 98.4 (4.7) | 99.0 (4.2) |
DBPC = double-blind placebo-controlled; SD = standard deviation.
aTreatment adherence for each patient (%) = (number of visits where study medication was administered / number of visits in the treatment period where study medication should have been administered) × 100%.
Source: Clinical Study Report.3
Concomitant Medications and Co-Interventions
During screening and throughout the study, participants could take stable doses of medications for chronic preexisting conditions. If there was an immediate clinical need during the study to prescribe a new medication or a new dosage of an existing medication for either a new or worsening preexisting condition, concurrent therapy could be administered at the discretion of the investigator. The investigator could consult the medical monitor regarding what constituted a stable dose or a chronic condition.
Table 13 summarizes the concomitant background PAH therapy taken by participants during the study. Use of background PAH medications were comparable in the sotatercept and placebo groups. Almost all patients (98.5%) were on concomitant medications, the most common being proton pump inhibitors (49.8%), sulfonamides (33.1%), and potassium (31.3%).
Table 13: Summary of Concomitant Medications in STELLAR Study
Concomitant medication | Sotatercept (N = 163) | Placebo (N = 160) | All treatment (N = 323) |
|---|---|---|---|
Prostacyclin infusion therapy,a n (%) | 65 (39.9) | 64 (40.0) | 129 (39.9) |
Monotherapy, n (%) | 9 (5.5) | 4 (2.5) | 13 (4.0) |
Double therapy, n (%) | 56 (34.4) | 55 (34.4) | 111 (34.4) |
Triple therapy, n (%) | 98 (60.1) | 101 (63.1) | 199 (61.6) |
aProstacyclin infusion therapy includes IV epoprostenol and IV or subcutaneous treprostinil.
Source: Clinical Study Report.3
Efficacy
A summary of all key efficacy outcomes considered in this review and obtained from the pivotal study is represented in Table 14.
Table 14: Summary of Key Efficacy Results From STELLAR Study
End point | Sotatercept (N = 163) | Placebo (N = 160) |
|---|---|---|
Mortality (FAS) | ||
Deaths, n (%) | 2 (1.2) | 6 (3.8) |
Risk difference, % (95% CI)a | ████ ████ ███ | NA |
Change from baseline in 6MWD at week 24 | ||
Patients contributing to the analysis (standard multiple imputation),b,c n | 163 | 160 |
Median change estimate from baseline, m (range)c | 34.4 (32.5 to 35.5) | 1.0 (–1.0 to 5.0) |
Hodges-Lehmann location shift from placebo estimate, m (95% CI)d | 40.8 (27.5 to 54.1) | NA |
P valuee | < 0.001 | NA |
Multicomponent improvement at week 24 | ||
Patients contributing to the analysis, n | 162 | 159 |
Patients who met all 3 criteria for improvement in 6MWD, NT-proBNP level, and WHO FC, n (%) | 63 (38.9) | 16 (10.1) |
Risk difference, % (95% CI)f | ███ ████ █████ | NA |
P valueg | < 0.001 | NA |
Time to clinical worsening or death (cut-off December 6, 2022) | ||
Patients in FAS who experienced at least 1 clinical worsening event or death, n (%) | 9 (5.5) | 42 (26.3) |
Risk difference in the FAS, % (95% CI) | ███ ████ ██████ | NA |
Patients in overall study population (including the double-blind extension phase) who experienced at least 1 clinical worsening event or death, n (%) | 11 (6.7) | 42 (26.3) |
Total events, n | 11 | 45 |
Hazard ratio (95% CI)h | 0.18 (0.09 to 0.38) | NA |
Log-rank test P valuei | < 0.001 | NA |
Individual assessments of the composite end point, n (%) | ||
Death | 2 (1.2) | 6 (3.8) |
Worsening-related listing for lung or heart-lung transplant | 1 (0.6) | 1 (0.6) |
Initiation of rescue therapy or increase in the dose of infusion of prostacyclin | 2 (1.2) | 17 (10.6) |
Atrial septostomy | 0 (0.0) | 0 (0.0) |
PAH-specific hospitalization | 1 (0.6) | 7 (4.4) |
Risk difference in the FAS, % (95% CI) | ████ ████ ████ | NA |
Deterioration of PAHj | 5 (3.1) | 15 (9.4) |
PVR at week 24 | ||
Patients contributing to the analysis (standard multiple imputation),k n | 163 | 160 |
Median change estimate from baseline, dyn·sec·cm-5 (95% CI)l | –165.1 (–184.0 to –152.0) | 32.8 (24.0 to 40.0) |
Hodges-Lehmann location shift from placebo estimate, dyn·sec·cm-5 (95% CI)d | –234.6 (–288.4 to –180.8) | NA |
P valuee | < 0.001 | NA |
NT-proBNP at week 24 | ||
Patients contributing to the analysis,m n | 163 | 160 |
Median change estimate from baseline, pg/mL (range) | –230.3 (–236.0 to –233.0) | 58.6 (44.0 to 73.0) |
Hodges-Lehmann location shift from placebo estimate, pg/mL (95% CI)d | –441.6 (–573.5 to –309.6) | NA |
P valuee | < 0.001 | NA |
WHO FC improvement at week 24 | ||
Patients contributing to the analysis, n | 163 | 159 |
WHO FC improved, n (%) | 48 (29.4) | 22 (13.8) |
Risk difference, % (95% CI) | ██ ████ █████ | NA |
P valueg | < 0.001 | NA |
Simplified French Risk Score at week 24 | ||
Patients contributing to the analysis, n | 162 | 159 |
Patients who maintain or reach a low risk score using the Simplified French Risk Score calculator at week 24 vs. baseline, n (%) | 64 (39.5) | 29 (18.2) |
P valueg | < 0.001 | NA |
PAH-SYMPACT physical impacts domain score at week 24 | ||
Patients contributing to the analysis (standard multiple imputation),n n | 163 | 160 |
Median change estimate from baseline (range) | –0.13 (–0.15 to 0.00) | 0.01 (0.00 to 0.14) |
Hodges-Lehmann location shift from placebo estimate (95% CI)d | –0.26 (–0.49 to –0.04) | NA |
P valueg | 0.01 | NA |
PAH-SYMPACT cardiopulmonary symptoms domain score at week 24 | ||
Patients contributing to the analysis (standard multiple imputation),n n | 163 | 160 |
Median change estimate from baseline (range)b | –0.12 (–0.14 to –0.06) | –0.01 (–0.03 to 0.02) |
Hodges-Lehmann location shift from placebo estimate (95% CI)c | –0.13 (–0.26 to –0.01) | NA |
P valueg | 0.03 | NA |
EQ VAS at week 24 | ||
Patients contributing to the analysis, n | 90 | 89 |
LS mean change from baseline (95% CI) | ███ ████ ████ | ███ █████ ████ |
LS mean difference from placebo (95% CI) | ███ ████ ████ | NA |
P valueo | 0.002 | NA |
6MWD = 6-minute walk distance; CI = confidence interval; FAS = full analysis set; FC = functional class; LS = least squares; NA = not applicable; PAH = pulmonary arterial hypertension; PAH-SYMPACT = Pulmonary Arterial Hypertension – Symptoms and Impact; PVR = pulmonary vascular resistance; VAS = visual analogue scale.
aThe risk treatment arm proportions are based on raw percentages; risk differences are computed accounting for the randomization variables (WHO FC [II vs. III] and background PAH therapy [monotherapy or double therapy vs. triple therapy]) as stratification factors.
bA gatekeeping method was used to control the type I error rate in the primary and secondary efficacy end points by starting testing with the primary efficacy end point and then proceeding in the order of the secondary efficacy end points. Secondary end point testing was performed using a 2-sided alpha at the 0.05 level and by proceeding successively in the order of the secondary end points only after each of the preceding end points was found to be statistically significant.
cChange from baseline in 6MWD at week 24 for patients who died was assigned a value of –2,000 m so as to assign the worst rank. Change from baseline in 6MWD at week 24 for patients who had missing data due to a nonfatal clinical worsening event was imputed to –1,000 m so as to assign the next worst rank. Shown is the average of the medians across the imputed datasets (with range) if missing data were imputed.
dThe Hodges-Lehmann location shift from placebo estimate is the median of all paired differences.
eWilcoxon P value, which refers to the P value from the aligned rank–stratified Wilcoxon test with randomization factors as strata.
fBased on Miettinen and Nurminen method stratified by WHO FC (II vs. III) and background PAH therapy (monotherapy or double therapy vs. triple therapy).
gComparison with placebo uses Cochran-Mantel-Haenszel method stratified by randomization factors.
hThe hazard ratio (sotatercept vs. placebo) is derived from a Cox proportional hazard model, with treatment group as the covariate, stratified by the randomization factors.
iLog-rank test comparison, with placebo stratified by the randomization factors.
jDefined by both of the following events occurring at any time, even if they began at different times, as compared to their baseline values: Worsening of WHO FC (II to III, III to IV, II to IV, and so forth) or decrease in 6MWD by at least 15% (confirmed by two 6-minute walk tests at least 4 hours apart but no more than 1 week apart).
kChange from baseline in PVR at week 24 for patients who died was assigned as 20,000 so as to assign the worst rank. Change from baseline in PVR at week 24 for patients who had missing data due to a nonfatal clinical worsening event was imputed as 15,000 so as to assign the next worst rank.
lShown is the average of the medians across the imputed datasets (with range) if missing data were imputed.
mChange from baseline in NT-proBNP at week 24 for patients who died was assigned as 200,000 so as to assign the worst rank. Change from baseline in NT-proBNP at week 24 for patients who had missing data due to a nonfatal clinical worsening event was imputed as 150,000 so as to assign the next worst rank.
nChange from baseline in PAH-SYMPACT scores at week 24 for patients who died was assigned as 200 so as to assign the worst rank. Change from baseline in PAH-SYMPACT scores at week 24 for patients who had missing data due to a nonfatal clinical worsening event was imputed as 150 so as to assign the next worst rank.
oP value is from Shapiro-Wilk test on the residuals from the analysis of covariance model. If the normality assumption is violated (P < 0.001), results from the analysis of covariance are not presented.
Details included in the table are from the sponsor’s summary of clinical evidence.
Sources: Hoeper (2023);52 Souza (2023);53 Clinical Study Reports.3,4
Mortality
The end point of mortality (i.e., the number of patients who died during the follow-up of the study) was assessed as part of a multicomponent end point (also described in the Multicomponent Improvement section) at the final cut-off date of December 6, 2022. More deaths were observed in the placebo arm (6 patients [3.8%]) than in the sotatercept arm (2 patients [1.2%]).
Change From Baseline in 6MWD
In adults with PAH on background PAH therapy, the addition of sotatercept, compared with the addition of placebo, led to higher values (improvement) from baseline in 6MWD at week 24. The median treatment difference (Hodges-Lehmann location shift) between the sotatercept and placebo groups was 40.8 m (95% CI, 27.5 m to 54.1 m). The results from all sensitivity analyses using alternative methods of imputation were consistent with the results from the primary analysis.
In the subgroup analysis based on the WHO FC (Figure 2), the treatment effect of sotatercept on 6MWD at week 24 was greater in patients with PAH classified as WHO FC III (61.7 m; 95% CI, 40.9 m to 82.6 m) than in those with PAH classified as WHO FC II (21.7 m; 95% CI, 6.6 m to 36.7 m). The rest of the subgroups were consistent with the overall effect estimate.
Multicomponent Improvement
The results for the multicomponent improvement end point, which includes the proportion of patients who met all the individual criteria (6MWD, NT-proBNP level, and WHO FC), are presented in Table 14. The proportion of patients who met all 3 criteria of the multicomponent improvement end point was greater in the sotatercept group (38.9%) than in the placebo group (10.1%) (P < 0.001) at week 24. The risk difference between groups was ████ █ ██████ ████ ██ ██████. The specific components of this end point are described as separate outcomes in this section.
Figure 2: Forest Plot: Change From Baseline in 6MWD (m) at Week 24 in Subgroups (FAS)
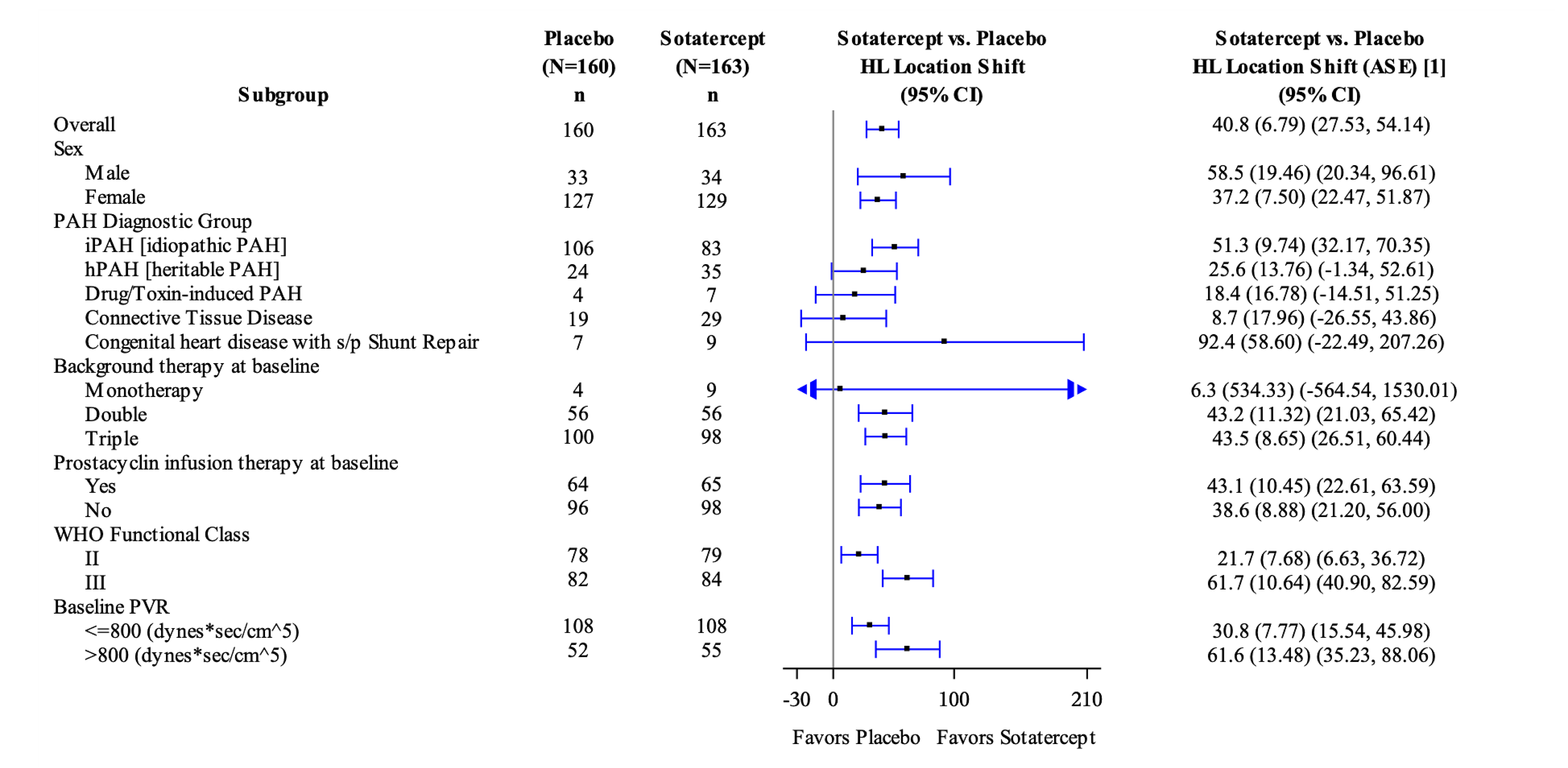
6MWD = 6-minute walk distance; ASE = asymptotic standard error; CI = confidence interval; FAS = full analysis set; FC = functional class; HL = Hodges-Lehmann location shift from placebo estimate (median of all paired differences); PAH = pulmonary arterial hypertension; PVR = pulmonary vascular resistance; s/P = status post; vs. = versus.
Note: Imputation method: Change from baseline in 6MWD at week 24 for patients who died was assigned a value of to –2,000 m so as to assign the worst rank. Change from baseline in 6MWD at week 24 for patients who have missing data due to a nonfatal clinical worsening event was imputed to –1,000 m so as to assign the next worst rank.
Time to Clinical Worsening or Death
At the December 2022 data cut-off, when all patients had completed their week 24 visit, fewer participants in the sotatercept group (11 [6.7%]) than in the placebo group (42 [26.3%]) had died or had experienced at least 1 clinical worsening event. The absolute effect (i.e., the risk difference between groups) was ██████ ███████ █████ ██ ██████ in favour of sotatercept. When evaluating this composite end point as a time-to-event outcome, the hazard ratio for a first clinical worsening event was 0.18 (95% CI, 0.09 to 0.38), favouring the sotatercept group compared with the placebo group.
When evaluating the individual components of the composite end point, it was found that more patients in the placebo arm (17 patients [10.6%]) required rescue therapy or an increase in the dose of infusion prostacyclin than in the sotatercept arm (2 patients [1.2%]; see Table 14). PAH-related hospitalization was observed in 7 patients in the placebo arm and 1 in the sotatercept arm (4.4% versus 0.6%). As mentioned in the Mortality Outcome section, 2 patients in the sotatercept arm died, compared to 6 from the placebo arm.
Change From Baseline in PVR
Patients in the sotatercept arm demonstrated a reduction in PVR from baseline to week 24, whereas the PVR increased in the placebo arm. The median treatment difference between the sotatercept and placebo groups was –234.6 dyn·sec·cm-5 (–288.4 dyn·sec·cm-5 to –180.8 dyn·sec·cm-5). The results from the supportive analysis using the ANCOVA model were consistent with those from the primary analysis. The treatment effect of sotatercept on PVR at week 24 was consistent across the prespecified subgroup and remained consistent in the post hoc subgroups stratified by baseline risk status.
Change From Baseline in NT-proBNP
The results for the NT-proBNP end point are presented in Table 14. As for PVR, only the participants in the sotatercept arm demonstrated a reduction in NT-proBNP levels from baseline to week 24. The median treatment difference between the sotatercept and placebo groups was –441.6 pg/mL (95% CI, –573.5 pg/mL to –309.6 pg/mL). The results from the supportive analysis using the ANCOVA model were consistent with those from the primary analysis. The treatment effect of sotatercept on NT-proBNP at week 24 was consistent across the prespecified subgroups and remained consistent in the post hoc subgroups stratified by baseline risk status.
WHO FC Improvement
The proportion of participants who experienced improvement from baseline in WHO FC at week 24 was greater in the sotatercept group (29.4%) than in the placebo group (13.8%). The risk difference was █████ ███████ ███ ██ ██████. Specifically, more patients in the sotatercept group than in the placebo group experienced improvement from WHO FC II to FC I (5.0% versus 2.0%) and from WHO FC III to FC II (24.5% versus 12.2%) at week 24. The treatment effect of sotatercept on improvement in WHO FC at week 24 was consistent across the post hoc subgroups stratified by baseline risk status.
Simplified French Risk Score
As mentioned in the Multicomponent Improvement section, more patients in the sotatercept arm than the placebo arm experienced improvement in 6MWD, NT-proBNP levels, and WHO FC. As such, the proportion of participants who maintained or reached a low risk score relative to baseline at week 24 was greater in the sotatercept group (39.5%) than in the placebo group (18.2%).
Change From Baseline in PAH-SYMPACT Domain Scores
The results for the PAH-SYMPACT end points are presented in Table 14. Patients in the sotatercept group reported greater improvements in both the physical impacts and the cardiopulmonary symptoms domains from baseline to week 24 than patients in the placebo group. For the physical impacts domain, the difference between arms was –0.26 points (95% CI, –0.49 to –0.04 points) in favour of sotatercept. For the cardiopulmonary symptoms domain, the between-group difference was –0.13 points (95% CI, –0.26 to –0.01 points) in favour of sotatercept. For both values a reduction in the score represents an improvement.
The results from the supportive analysis using the ANCOVA model were consistent with those from the primary analysis.
EQ-5D-5L
The results for the EQ VAS end points are presented in Table 14. The increase (improvement) in the VAS from baseline was greater in the sotatercept group (███ ██████ ██████ ███████ ████ ██ ████) than in the placebo group █████ ██████ ████ ███ █████ ██ █████). The difference between groups was ███ ██████ ███████ ████ ██ ████) in favour of sotatercept.
PAH-Specific Hospitalization
This outcome was considered important by the clinical experts and was obtained from the composite end point of time to clinical worsening or death (Table 14). Overall, the placebo group presented more hospitalizations (7 patients [4.4%]) than the sotatercept group (1 patient [0.6%]), with a risk difference of █████ ███████ ████ ██ █████ in favour of sotatercept.
Harms
To evaluate the safety of sotatercept treatment, the STELLAR study used end points including AEs, laboratory tests, vital signs, immunogenicity (antidrug antibody) and electrocardiograms. The predefined AESI was telangiectasia; however, the clinical experts consulted by CDA-AMC identified bleeding as an additional AESI.
All patients randomized (N = 323) were evaluated in the safety analysis. Key harms data are presented in Table 15.
Adverse Events
The overall proportion of patients reporting at least 1 AE was similar in both groups (84.7% and 87.5% in the sotatercept and placebo groups, respectively). The most common AEs in the sotatercept group through week 24 included epistaxis (nosebleed), experienced by 12.3% of patients; telangiectasia (spider veins), experienced by 10.4% of patients; and dizziness, experienced by 10.4% of patients. The majority of AEs reported for participants in each treatment group were mild or moderate in severity.
Serious AEs
SAEs were reported for 23 patients (14.1%) in the sotatercept group and 36 patients (22.5%) in the placebo group. No notable pattern of SAEs was observed in the sotatercept group relative to the placebo group. Atrial flutter, fall, and hemoptysis (i.e., coughing blood) were each reported for 2 participants in the sotatercept group; no other SAEs were reported for more than 1 participant in this group. One event of fall and 1 event of hemoptysis were considered related to the study intervention by the investigator. In the placebo group, SAEs of PAH, cardiac arrest, right ventricular failure, and dyspnea were reported for at least 2 participants; no other SAEs were reported more than 1 participant in this group.
Through week 24, no deaths were reported as AEs in the sotatercept group, compared with 6 in the placebo group (these were events that did not qualify under the definition of time to first clinical worsening or death in the efficacy outcome). However, at the data cut-off of December 6, 2022, 2 patients in the sotatercept group and 6 in the placebo group had died. None of the deaths in the placebo group were considered by the investigator to be related to the study intervention.
Withdrawals Due to AEs
The proportion of participants who discontinued the study intervention due to an AE was lower in the sotatercept group (1.8%) than the placebo group (6.3%). No notable pattern of AEs leading to discontinuation of study intervention was observed in the sotatercept group; no AE leading to discontinuation of the study intervention was reported in more than 1 participant in this group.
AEs of Special Interest
The incidences of telangiectasia were higher in the sotatercept group than in the placebo group. In the sotatercept group, none of these events were serious or severe, and only 1 led to discontinuation of the treatment.
Several AEs were considered of interest by the sponsor, including increased hemoglobin, thrombocytopenia, and epistaxis and other bleeding events. The most reported bleeding events in the sotatercept group were epistaxis (12.3%), followed by gingival bleeding (3.1%). Participants with epistaxis accounted, almost entirely, for the imbalance between sotatercept and placebo groups in bleeding events. However, none of the epistaxis or gingival bleeding events were serious or severe. One participant in the sotatercept group discontinued due to epistaxis, while another withdrew due to gingival bleeding. Seven patients (4.3%) experienced increased hemoglobin, and they were all in the sotatercept group. None of these events were serious or severe. Increase in hemoglobin led to the interruption of study intervention in 3 participants. Lastly, thrombocytopenia was more commonly reported in the sotatercept group than in the placebo group (6.1% versus 2.5%). In the sotatercept group, 1 event of thrombocytopenia was serious and another was severe; while both events led to the interruption of the study intervention, neither led to the discontinuation of the study intervention.
Table 15: Summary of Key Harms Data in STELLAR Study (Safety Set)
AEs | Sotatercept (N = 163) | Placebo (N = 160) |
|---|---|---|
AEs | ||
Patients reporting ≥ 1 AE, n (%) | 138 (84.7) | 140 (87.5) |
AEs reported in ≥ 10% of patients in either group, n (%) | ||
Headache | 33 (20.2) | 24 (15.0) |
COVID-19 | 24 (14.7) | 21 (13.1) |
Epistaxis | 20 (12.3) | 3 (1.9) |
Diarrhea | 20 (12.3) | 12 (7.5) |
Fatigue | 17 (10.4) | 12 (7.5) |
Telangiectasia | 17 (10.4) | 5 (3.1) |
Dizziness | 17 (10.4) | 3 (1.9) |
Nausea | 16 (9.8) | 18 (11.2) |
SAEs | ||
Patients reporting ≥ 1 SAE, n (%) | 23 (14.1) | 36 (22.5) |
SAEs reported in > 1% of patients in either group, n (%) | ||
Atrial flutter | 2 (1.2) | 0 (0.0) |
Cardiac arrest | 0 (0.0) | 2 (1.3) |
Right ventricular failure | 0 (0.0) | 2 (1.3) |
COVID-19 | 0 (0.0) | 2 (1.3) |
Fall | 2 (1.2) | 0 (0.0) |
Pulmonary arterial hypertension | 1 (0.6) | 4 (2.5) |
Dyspnea | 1 (0.6) | 2 (1.3) |
Hemoptysis | 2 (1.2) | 0 (0.0) |
Patients who stopped treatment due to AEs, n (%) | ||
Total | 3 (1.8) | 10 (6.3) |
Cardiac arrest | 0 (0.0) | 2 (1.3) |
Right ventricular failure | 0 (0.0) | 1 (0.6) |
Sepsis | 0 (0.0) | 1 (0.6) |
Malnutrition | 0 (0.0) | 1 (0.6) |
Arthralgia | 1 (0.6) | 0 (0.0) |
Abortion | 0 (0.0) | 1 (0.6) |
Pulmonary arterial hypertension | 0 (0.0) | 3 (1.9) |
Epistaxis | 1 (0.6) | 0 (0.0) |
Hemoptysis | 1 (0.6) | 0 (0.0) |
Respiratory failure | 0 (0.0) | 1 (0.6) |
Telangiectasia | 1 (0.6) | 0 (0.0) |
Deaths, n (%) | ||
Total | 0 (0.0) | 6 (3.8) |
Cardiac arrest | 0 (0.0) | 2 (1.3) |
Cardiogenic shock | 0 (0.0) | 1 (0.6) |
Right ventricular failure | 0 (0.0) | 1 (0.6) |
Sepsis | 0 (0.0) | 1 (0.6) |
Pulmonary arterial hypertension | 0 (0.0) | 1 (0.6) |
AEs of special interest, n (%) | ||
Telangiectasia | 17 (10.4) | 5 (3.1) |
Bleeding events | 35 (21.5) | 20 (12.5) |
AE = adverse event; SAE = serious adverse event.
Note: Details included in the table are from the sponsor’s summary of clinical evidence.
Source: Clinical Study Report.3
Critical Appraisal
Internal Validity
The STELLAR trial is a phase III, multicentre, randomized DBPC trial assessing the efficacy and safety of sotatercept versus placebo at 24 weeks on top of stable background PAH therapy in adult patients with PAH.
Overall, the STELLAR study is well designed, featuring an appropriate 1:1 random allocation of patients to either the sotatercept or placebo group. The random sequence was generated using a computer algorithm, and the allocation process was centralized, ensuring that the sequence remained concealed from both patients and investigators.
Patients and personnel involved in the STELLAR trial were likely unaware of the assigned intervention during the initial stages of the study. However, as the study progressed, patients might have deduced their treatment due to the higher frequency of AEs, such as telangiectasis or nosebleeds, associated with sotatercept compared to the placebo.
Adherence to the interventions was well monitored through regular follow-ups, showing adherence rates above 98%. Deviations from intended interventions were well documented, and handling of missing data was properly assessed through sensitivity analyses using alternative methods of imputation that were consistent with the results from the primary analysis for the main outcomes (i.e., 6MWD, NT-proBNP, PVR, and PAH-SYMPACT).
The outcomes reported and the analysis plan were consistent with those prespecified in the study protocol. The outcome measurement methods were deemed appropriate and used validated clinical criteria and tools with proper validity and reliability measurements. Specifically, maintaining or reaching an NT-proBNP level less than 300 ng/L aligns with the cut-off used in the low risk for 1-year mortality category of the ESC/ERS guidelines.8 Additionally, previous studies of PAH therapies have shown that decreases of NT-proBNP levels with treatments could be associated with a more than 90% reduction in the risk of morbidity and mortality54,55 and that decreases in PVR with treatments have been associated with long-term transplant-free survival.56
External Validity
The STELLAR study included 323 patients with PAH; however, patients with certain subtypes of PAH — such as PAH associated with HIV, portal hypertension, and pulmonary veno-occlusive disease — were excluded from the pivotal trial, and hence were not represented, as were some demographic groups and individuals outside North America and Europe.
The reimbursement criteria for this submission apply to patients with PAH undergoing background therapy who do not fall into the low-risk category. Low risk is defined as patients with PAH classified as WHO FC I or II, with a 6MWD exceeding 440 m, and with either NT-proBNP levels below 300 ng/L or BNP levels below 100 ng/L. The STELLAR study enrolled 157 of 323 patients (48.6%) with PAH classified as WHO FC II, but only 53 of the 323 patients (16.4%) were classified as having low-risk status, as defined in the trial. The impact of these numbers on the generalizability of the trial results to the reimbursement criteria was deemed low by the clinical experts consulted by CDA-AMC, who considered that the results from the STELLAR study are applicable to the target population for the reimbursement of sotatercept, first because the distinction between the 2 WHO FC levels is not always clinically apparent and, second, because when analyzed by subgroups (see Figure 2), even though patients with PAH classified as WHO FC III had higher and more clinically meaningful effect estimates (in the 6MWD) than those with PAH classified as WHO FC II, both subgroups had meaningful effects of benefit.
Enrolment was restricted to patients with baseline PVR values of at least 400 dyn·sec·cm−5, which may not be fully reflective of the broader hemodynamic definition of PAH according to recent guidelines.8
The pivotal study was not powered to measure the outcome of mortality and serious morbidity from PAH; hence, the median treatment duration of 24 weeks and the design and power calculations of the study limited its capacity to ascertain these end points, as well as long-term treatment response, along with the safety profiles over extended administration periods.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal study identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the CDA-AMC expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.1,2
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from randomized controlled trials started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effect estimates, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
The GRADE assessments included an evaluation of the main outcomes considered important by clinicians, patient groups, and committee members. The comparison evaluated in the GRADE assessments of this report was that of sotatercept versus placebo. Table 2 presents the GRADE summary of findings.
The clinical significance of the improvements noted in the PVR and NT-proBNP numbers remains uncertain due to the absence of established minimal clinically important differences. This limits the possibility of assessing imprecision properly, and the clinical relevance of those effects remains uncertain.
Long-Term Extension Studies
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CDA-AMC review team.
The evaluation of longer-term outcomes is informed by 3 reports. The long-term assessment of AEs and the long-term assessment of AEs and efficacy outcomes of sotatercept were addressed during the LTDB treatment period of the STELLAR trial. Efficacy and safety end points were evaluated in the OLE phase of the PULSAR phase II study. The ongoing open-label SOTERIA trial provides preliminary evidence based on interim analyses. Subsequent subsections will provide detailed descriptions of each study.
STELLAR Study LTDB Treatment Period
Study Design and Objectives
The primary objective of the extension period was to evaluate the longer-term frequency of AEs associated with sotatercept. As described in the study design of the STELLAR trial in the Systematic Review section, patients who completed the 24-week DBPC treatment period of the STELLAR trial entered the LTDB treatment period, which was up to 72 weeks. Once the last randomized participant completed the last visit in the DBPC treatment period, the study was unblinded and eligible participants rolled over to the LTFU study, the SOTERIA study, which will be presented later in this section. Since the timing of enrolment in the STELLAR trial varied, participants who were randomized early in the study would have more visits beyond week 24 of the DBPC phase. After the initial DBPC phase, the number of participants with efficacy assessments decreased over time as participants rolled over to the SOTERIA study.
Eligibility Criteria
In addition to the eligibility criteria of the STELLAR trial described in the Description of Studies section in the systematic review, only patients who completed the DBPC treatment period could enter the LTDB treatment period.
Interventions
The interventions used in the STELLAR trial are described in the Interventions section of the systematic review. Patients who entered the LTDB period remained on the same interventions that were taken during the DBPC phase.
Outcomes
The safety outcomes during the LTDB phase included the frequency of AEs, severe AEs, AEs leading to discontinuation, and AESIs. The cumulative results of the efficacy outcomes from the DBPC and LTDB periods were then reported.
Statistical Analysis
The statistical analyses for all outcomes in the LTDB phase are descriptive in nature.
Population
Since no new participants were randomized in the STELLAR trial after the DBPC phase, the patient baseline characteristics can be found in Table 11.
Patient disposition for the STELLAR trial (cumulative from the DBPC and LTDB phases) are presented in Table 16.
Table 16: Patient Disposition for the STELLAR Trial — Cumulative From DBPC and LTDB Phases
Patient disposition | Sotatercept (N = 163) | Placebo (N = 160) |
|---|---|---|
Randomized, N | 163 | 160 |
Discontinued from study, n (%) | 8 (4.9) | 18 (11.3) |
Reason for discontinuation, n (%) | ||
AE | 3 (1.8) | 1 (0.6) |
Withdrew consent | 2 (1.2) | 6 (3.8) |
Unwillingness/ inability to comply with protocol | 1 (0.6) | 2 (1.3) |
Clinical worsening | 0 (0.0) | 2 (1.3) |
Death | 2 (1.2) | 6 (3.8) |
Other | 0 (0.0) | 1 (0.6) |
Full analysis set, N | 163 | 160 |
Per-protocol set, N | NA | NA |
Safety set, N | 163 | 160 |
AE = adverse event; DBPC = double-blind placebo-controlled; LTDB = long-term double-blind; NA = not applicable.
Sources: Clinical Study Reports;4,57 Preston (2024).58
Study Treatments
Exposure to randomized study treatments, cumulative for the DBPC and LTDB phases, is summarized in Table 17. By the end of the LTDB period in the STELLAR trial, the median time of exposure was higher in the sotatercept group (313.0 days [approximately 45 weeks]) than in the placebo group (273.0 days [39 weeks]). The mean treatment adherence remained similar between the 2 groups.
Table 17: Patient Exposure in the STELLAR Trial — Cumulative From DBPC and LTDB Phases
Exposure | Sotatercept (N = 163) | Placebo (N = 160) |
|---|---|---|
Total, patient-years | NR | NR |
Duration (days), mean (SD) | █████ ███████ | █████ ███████ |
Duration (days), median (range) | █████ ███ ██ ████ | █████ ███ ██ ████ |
Total dose administered (mg), mean (SD) | █████ ████████ | ███ |
Adherence (%), mean (SD) | ████ ██████ | ████ ██████ |
DBPC = double-blind placebo-controlled; LTDB = long-term double-blind; NR = not reported; SD = standard deviation.
Sources: Clinical Study Reports;4,57 Preston (2024).58
Summary of Outcomes in the STELLAR Trial LTDB Phase
Efficacy
As patients enrolled in the STELLAR trial progressively enter the SOTERIA study after the initial 24-week DBPC phase, the efficacy outcomes in the LTDB period are descriptive in nature and thus not presented here. Overall, consistent with the analysis at week 24, the observed mean change from baseline in 6MWD, PVR, NT-proBNP, WHO FC, and the proportion of participants who maintained or reached low-risk status remained higher in the sotatercept group than in the placebo group until the end of study.
Harms
The frequency of AEs (i.e., the overall safety profile) in the LTDB phase remained consistent with the primary analysis at week 24, as shown in Table 18. AEs with a higher frequency in the sotatercept group than in the placebo group were epistaxis, telangiectasia, dizziness, nasal congestion, thrombocytopenia, and increased hemoglobin, as identified at week 24. Most AEs reported for participants in each treatment group were mild or moderate in severity. Two deaths due to AEs were reported in the sotatercept group, compared with 7 in the placebo group in the cumulative DBPC and LTDB analysis. None of the deaths were considered by the investigator to be related to the study intervention. Discontinuation due to AEs was lower in the sotatercept group (3.7%) than in the placebo group (6.9%).
Table 18: Summary of Harms in the STELLAR Trial — Cumulative From DBPC and LTDB Phases (Safety Set)
AEs | Sotatercept (N = 163) | Placebo (N = 160) |
|---|---|---|
AEs | ||
Patients reporting ≥ 1 AE, n (%) | 151 (92.6) | 149 (93.1) |
AEs reported in ≥ 10% of patients in either group, n (%) | ||
COVID-19 | 48 (29.4) | 42 (26.3) |
Headache | 40 (24.5) | 28 (17.5) |
Nausea | 23 (14.1) | 19 (11.9) |
Diarrhea | 25 (15.3) | 16 (10.0) |
Epistaxis | 36 (22.1) | 3 (1.9) |
Fatigue | 23 (14.1) | 16 (10.0) |
Dizziness | 24 (14.7) | 10 (6.3) |
COVID-19–related dyspnea | 5 (3.1) | 17 (10.6) |
Telangiectasia | NA | NA |
SAEs | ||
Patients with ≥ 1 SAE, n (%) | 40 (24.5) | 47 (29.4) |
SAEs reported in ≥ 4 patients in either group by system organ class, n (%) | ||
Cardiac disorders | 6 (3.7) | 11 (6.9) |
Gastrointestinal disorders | 8 (4.9) | 6 (3.8) |
General disorders and administration site conditions | 3 (1.8) | 2 (1.3) |
Infections and infestations | 14 (8.6) | 8 (5.0) |
Investigations | 1 (0.6) | 3 (1.9) |
Metabolism and nutrition disorders | 0 (0.0) | 6 (3.8) |
Musculoskeletal and connective tissue disorders | 4 (2.5) | 1 (0.6) |
Neoplasms, benign, malignant, and unspecified (including cysts and polyps) | 0 (0.0) | 4 (2.5) |
Respiratory, thoracic, and mediastinal disorders | 7 (4.3) | 11 (6.9) |
Stopping treatment due to AEs | ||
Patients who stopped treatment due to AEs, n (%) | 6 (3.7) | 11 (6.9) |
Deaths,a n (%) | ||
Total | 2 (1.2) | 7 (4.4) |
Cardiac arrest | 0 (0.0) | 2 (1.3) |
Acute myocardial infarction | 1 (0.6) | 0 (0.0) |
Cardiogenic shock | 0 (0.0) | 1 (0.6) |
Right ventricular failure | 0 (0.0) | 1 (0.6) |
COVID-19 pneumonia | 0 (0.0) | 1 (0.6) |
Sepsis | 0 (0.0) | 1 (0.6) |
Intracranial hemorrhage | 1 (0.6) | 0 (0.0) |
Pulmonary arterial hypertension | 0 (0.0) | 1 (0.6) |
Brain abscess | 0 (0.0) | 0 (0.0) |
AEs of special interest, n (%) | ||
Telangiectasia | 27 (16.6) | 7 (4.4) |
Bleeding | 36 (22.1) | 3 (1.9) |
AE = adverse event; DBPC = double-blind placebo-controlled; LTDB = long-term double-blind; NA = not applicable; SAE = serious adverse event.
aDefined as treatment-emergent adverse events leading to death.
Sources: Clinical Study Report;3 Humbert (2023);59 Preston (2024).58
PULSAR and SOTERIA Studies
Description of Studies
In this section, 2 long-term extension studies, the PULSAR and SOTERIA studies, have been summarized to provide evidence regarding the longer-term efficacy and safety of sotatercept.
The PULSAR study (NCT03496207) was a phase II, multicentre, randomized, DBPC, parallel-group study evaluating the safety and efficacy of sotatercept on top of background PAH therapy in participants with PAH (WHO Group 1 pulmonary hypertension; WHO FC II or III), including a 24-week placebo-controlled period followed by an 18-month OLE period (up to a maximum of 24 months). This study was conducted between June 2018 and March 2022 at 43 centres in 8 countries (Australia, Brazil, France, Germany, Israel, Spain, UK, and US). The main objective of the extension period was to evaluate the longer-term safety of sotatercept; however, efficacy outcomes were also analyzed.
The SOTERIA study (NCT04796337) is an ongoing, phase III, open-label, LTFU study initiated in May 2021. Currently, there are 196 sites in 21 countries, including 4 sites in Canada (Alberta, Manitoba, Ontario, and Quebec). It aims to assess the efficacy, safety, and tolerability of sotatercept treatment as an add-on to background PAH therapy for up to 7 years.
PULSAR OLE Study
Populations
Eligible patients were adults aged 18 years or older with symptomatic PAH (WHO Group 1 pulmonary hypertension; WHO FC II or III). WHO Group 1 pulmonary hypertension (i.e., PAH) consists of idiopathic PAH, heritable PAH, drug-induced PAH, PAH associated with connective tissue disease, and post–shunt correction PAH. Patients were required to have a PVR of at least 400 dyn·sec·cm-5; a total lung capacity greater than 70% predicted (or, if between 60% and 70%, a confirmatory high-resolution CT indicating no more than mild interstitial lung disease); forced expiratory volume in the first second or forced vital capacity greater than 70% predicted; a 6MWD between 150 m and 450 m (inclusive); a normal ventilation-perfusion scan; stable PAH therapy for at least 90 days before enrolment; and the ability to adhere to the study visits.
Interventions
At the double-blind baseline, participants were randomized in a 3:3:4 ratio to 1 of 3 arms: placebo, 0.3 mg/kg sotatercept, or 0.7 mg/kg sotatercept, all administered subcutaneously every 21 days in addition to stable doses of background therapy. Randomization was stratified based on baseline WHO FC (II or III). Those who completed the placebo-controlled period and completed a week 24 PVR assessment could continue to the extension period. In the OLE, patients originally in the placebo group were rerandomized 1:1 to 0.3 mg/kg sotatercept or 0.7 mg/kg sotatercept. Those initially randomized to sotatercept treatment remained on the same dose in a blinded manner. During this extension period, investigators were allowed to substitute, remove, or adjust the dose of concomitant medications for any chronic conditions or PAH worsening that were not specifically excluded. Although both 0.3 mg/kg and 0.7 mg/kg doses were evaluated in this trial, results from the 0.7 mg/kg group will be presented in this report wherever possible, as this is the therapeutic dose for approval from Health Canda. After completion of the primary end point (i.e., up to 18 to 24 months), the study was unblinded and investigators were provided with the treatment assignment of their respective site participants, allowing investigators the flexibility to up-titrate to the sotatercept 0.7 mg/kg dose.
Outcomes
The primary efficacy end point of the extension period was change in PVR from baseline to months 18 to 24. The key secondary end point was change in 6MWD from baseline to months 18 to 24. Other secondary end points included change in WHO FC and NT-proBNP from baseline to months 18 to 24, time to clinical worsening, mortality risk assessment (Simplified French Risk Score), change in quality of life (Cambridge Pulmonary Hypertension Outcome Review [CAMPHOR], Short Form [36] Health Survey [SF-36]), and change in echocardiogram parameters.
Safety outcomes during the extension period included the frequency of patients with AEs, severe AEs, AEs leading to discontinuation, and AESIs (including leukopenia, neutropenia, and thrombocytopenia).
Statistical Analysis
In the extension period, efficacy end points were analyzed using the FAS-E and PPS-E. These populations are summarized as follows:
FAS-E includes all the FAS participants who transitioned to the extension period. The primary analysis was based on this population.
PPS-E includes all the participants in FAS-E who completed their extension period PVR assessment and completed key procedures with no relevant major protocol deviations.
The extension period safety population includes all randomized participants who received at least 1 dose of the study intervention in the extension period. Participants were analyzed according to the treatment they received in the extension period.
Efficacy was assessed through 2 analyses, regardless of dose. The first analysis involved an analysis of efficacy end points from months 18 to 24 compared to baseline within the patients that crossed to the placebo group. The second analysis, a delayed-start efficacy assessment, compared the change from baseline to months 18 to 24 for efficacy end points between the continued-sotatercept and placebo-crossed groups. Both analyses were conducted on the FAS for the extension period, comprising all participants transitioning to the extension period. The sponsor reported only the statistics of the group of patients that crossed into the placebo group.
Safety end points were summarized based on the safety population and categorized by participants’ randomized doses. The safety population comprised all randomized participants who received at least 1 dose of sotatercept, encompassing data from the initial administration of sotatercept until the data cut-off. The overall safety profile was presented as the number and percentage of all participants treated with sotatercept who reported treatment-emergent AEs (TEAEs), related TEAEs, AESIs, serious TEAEs, related serious TEAEs, and TEAEs leading to treatment and study discontinuation. Also summarized were the number and percentage of participants treated with sotatercept who reported a TEAE that was experienced by more than 10% of all treated participants.
The type I error rate was set at 2-sided 0.05, using the recycle method to control the overall type I error rate. The placebo-crossed efficacy analyses were initially tested at the 2-sided 0.025 level. The gatekeeping method was used to sequentially test each efficacy end point. If all 3 placebo-crossed analyses were statistically significant, the type I error rate of 0.025 was recycled, and each end point in the delayed-start efficacy analysis underwent similar testing.
The primary efficacy end point, PVR, was evaluated using ANCOVA, with a term for treatment group and with baseline value as a covariate. Normality was assessed with the Shapiro-Wilk test and, if violated, a nonparametric Wilcoxon signed-rank test was employed. Missing data were addressed through multiple imputation. Secondary efficacy end points underwent similar analysis with ANCOVA, incorporating respective average baseline values as covariates. Various methods, including excluding missing participants for binary end points, were employed to handle missing data resulting from COVID-19.
SOTERIA Study (Ongoing)
All the information and data for the SOTERIA study are exclusively from the sponsor’s summary of clinical evidence.
Populations
Eligible patients are adults with PAH on stable background PAH therapy who completed a prior sotatercept study (SPECTRA, PULSAR, ZENITH, HYPERION, or STELLAR) without early discontinuation. The SPECTRA study was a phase IIa, single-arm, open-label, multicentre, exploratory study that aimed to assess the resting and exercise hemodynamics and peak oxygen uptake in patients treated with sotatercept; the ZENITH study is an ongoing phase III, randomized DBPC study evaluating the effect of sotatercept as an add-on to maximum tolerated background PAH therapy in patients who are at high risk of mortality; and the HYPERION study is another ongoing phase III trial in patients newly diagnosed with PAH who are at intermediate or high risk of disease progression.
Interventions
Participants rolling over from a blinded study start with sotatercept 0.3 mg/kg plus background PAH therapy. Those rolling over from an unblinded study continue at their current sotatercept dose with background PAH therapy. Those starting at less than 0.7 mg/kg sotatercept who have stable hemoglobin and platelet levels can be titrated up to 0.7 mg/kg of sotatercept at visit 2. Sotatercept is administered subcutaneously once every 21 days. PAH background therapy was not provided as a study medication during the study; however, patients are to continue taking their background therapy according to local practice.
Outcomes
The safety and tolerability of sotatercept are measured through reports of AEs, antidrug antibody levels, clinical laboratory assessments, levels of vital signs, and 12-lead electrocardiogram.
The efficacy outcomes were 6MWD, NT-proBNP, PVR, time to death or nonfatal clinical worsening, and Simplified French Risk Score. The 6MWD, NT-proBNP, PVR, and Simplified French Risk Score are reported as change from the SOTERIA study baseline. The SOTERIA study also included an end point on overall survival, which will be evaluated every 12 months.
Statistical Analysis
Statistical analyses will be conducted with all participants enrolled in the SOTERIA study who received at least 1 dose of sotatercept (i.e., the FAS). The change in 6MWD and NT-proBNP at 1 year in the study will be analyzed using an ANCOVA model, with the parent study the participant belonged to before enrolment in this study and baseline level as covariates. Point estimates of the least squares mean change at 1 year, with the corresponding 95% CI, will be provided for each parent study and across all enrolled participants. The proportion of participants with PAH that reaches or maintains WHO FC II status at year 1 in the study will be analyzed using the binomial test with the normal approximation. The proportion of participants who maintain or reach low-risk status using the Simplified French Risk Score calculator at 1 year in the study will be analyzed using the binomial test with the normal approximation. The change in PVR will be measured at 4 years. A Kaplan-Meier curve will be used to represent the OS, which is currently not available.
Results
Patient Disposition
Patient disposition for the PULSAR OLE study is presented here; patient disposition for the SOTERIA study is not available yet.
Of the 106 patients randomized at the start of PULSAR, 97 patients (92%) who completed the placebo-controlled period and a week 24 PVR assessment could continue in the extension period. Of these, 31 continued receiving sotatercept 0.3 mg/kg and 36 continued receiving sotatercept 0.7 mg/kg. Thirty participants who were initially randomized to placebo continued to the extension period and were rerandomized 1:1 to sotatercept 0.3 mg/kg or 0.7 mg/kg (15 patients in each). In the continued-sotatercept 0.7 mg/kg group, 13.9% of the patients discontinued the study, compared to 0% in the placebo to sotatercept 0.7 mg/kg group. In the continued-sotatercept 0.7 mg/kg group, 2 deaths (5.6% of patients) were reported, compared to 0 in the placebo to sotatercept 0.7 mg/kg group.
Table 19: Patient Disposition in PULSAR OLE and SOTERIA Studies
Patient disposition | PULSAR OLE | SOTERIA | |
|---|---|---|---|
Placebo to sotatercept 0.7 mg/kg (N = 15) | Continued-sotatercept 0.7 mg/kg (N = 36) | Total (N = 426) | |
Randomized, N | 15 | 36 | NA |
Discontinued from study, n (%) | 0 (0) | 5 (13.9) | 15 (3.5) |
Reason for discontinuation, n (%) | |||
AE | 0 (0) | 1 (2.8) | 15 (3.5) |
Withdrawal of consent | 0 (0) | 1 (2.8) | NR |
Unwillingness or inability to comply with protocol | NR | NR | NR |
Clinical worsening | NR | NR | NR |
Death | 0 (0) | 2 (5.6) | NR |
Other | 0 (0) | 1 (2.8) | NR |
Full analysis set, N | 15 | 36 | 426 |
Per-protocol set, N | 12 | 29 | NA |
Safety set, N | 15 | 36 | 426 |
AE = adverse event; NA = not applicable; NR = not reported; OLE = open-label extension.
Sources: Humbert (2023);59 Preston (2024).58
Table 20: Summary of Baseline Characteristics in PULSAR OLE and SOTERIA Studies (FAS)
Characteristic | PULSAR OLE | SOTERIA | |
|---|---|---|---|
Placebo-crossed (N = 30) | Continued-sotatercept (N = 67) | Total (N = 426) | |
Sex female, n (%) | 24 (80.0) | 62 (92.5) | 348 (81.7) |
Sex male, n (%) | 6 (20.0) | 5 (7.5) | 78 (18.3) |
Age (years), mean (SD) | 45.3 (13.6) | 48.6 (14.4) | NA |
Age (years), median (range) | 46.0 (21 to 71) | 48.0 (19 to 80) | 49.0 (18 to 83) |
Race, n (%) | |||
Asian | 0 (0.0) | 0 (0.0) | 9 (2.1) |
White | 28 (93.3) | 61 (91.0) | 380 (89.2) |
Black or African American | 0 (0.0) | 4 (6.0) | 9 (2.1) |
Other or multiple | 2 (6.7) | 2 (3.0) | 20 (4.7) |
Time since PAH diagnosis (years), median (range) | 6.8 (0.3 to 21.9) | 7.4 (0.7 to 26.2) | 7.88 (0.5 to 39.0) |
PAH subtype, n (%) | |||
Drug-induced or toxin-induced PAH | 0 (0.0) | 6 (9.0) | 15 (3.5) |
Heritable PAH | 6 (20.0) | 10 (14.9) | 69 (16.2) |
Idiopathic PAH | 18 (60.0) | 36(53.7) | 247 (58.0) |
PAH associated with CTD | 4 (13.3) | 14 (20.9) | 75 (17.6) |
PAH associated with simple, congenital systemic-to-pulmonary shunts ≥ 1 year following repair | 2 (6.7) | 1 (1.5) | 17 (4.0) |
WHO FC, n (%) | |||
FC I | NA | NA | 41 (9.6) |
FC II | 17 (56.7) | 38 (56.7) | 242 (56.8) |
FC III | 13 (43.3) | 29 (43.3) | 118 (27.7) |
FC IV | NA | NA | 25 (5.9) |
6MWD (m), mean (SD) | 409.2 (65.9) | 397.9 (86.2) | 420.4 (116.40) |
mPAP, (mm Hg), mean (SD) | 53.9 (13.7) | 52.0 (12.9) | NR |
NT-proBNP (pg/mL), mean (SD) | 840.1 (1,246.8) | 777.4 (1,051.0) | 917.5 (2,579.07) |
PVR (dyn·sec·cm−5), mean (SD) | 802.0 (331.1) | 783.7 (371.6) | NR |
6MWD = 6-minute walk distance; CTD = connective tissue disease; FAS = full analysis set; FC = functional class; mPAP = mean pulmonary arterial pressure; NA = not applicable; NR = not reported; OLE = open-label extension; PAH = pulmonary arterial hypertension; PVR = pulmonary vascular resistance; SD = standard deviation.
Sources: Humbert (2023);59 Preston (2024).58
Exposure to Study Treatments
The mean treatment compliance remained similar in the PULSAR OLE study between the placebo-crossed and continued-sotatercept groups. At the current data cut-off on November 8, 2023, for the SOTERIA study the overall median exposure on sotatercept was 922.2 patient-years, which included the patients’ time on the parent studies (e.g., the STELLAR and PULSAR studies). Adherence to the study treatment has not been reported.
Concomitant Medications and Co-Interventions
During the extension period of the PULSAR study, adjustments were made to concomitant PAH medications for 6 participants. Among them, 3 individuals who were initially on triple background PAH therapy involving an ERA, a PDE5 inhibitor, and prostacyclin had their IV (1 participant) or subcutaneous (2 participants) prostacyclin switched to an oral form. Another participant, initially on triple background PAH therapy, had their inhaled prostacyclin changed to an oral formulation. Additionally, a participant receiving triple background therapy with an ERA, a PDE5 inhibitor, and oral prostacyclin underwent a switch from the ERA to a soluble guanylate cyclase. Finally, 1 participant necessitated an escalation of PAH therapy, transitioning from monotherapy with a PDE5 inhibitor to dual therapy with a PDE5 inhibitor and the addition of an ERA.
Table 21: Summary of Concomitant Medications in PULSAR OLE and SOTERIA Studies
Concomitant medication taken by patients | PULSAR OLE (N = 97) | SOTERIA (N = 426) |
|---|---|---|
Prostacyclin infusion therapy, n (%) | 35 (36.1) | 163 (38.3) |
Monotherapy, n (%) | 7 (7.2) | 16 (3.8) |
Double therapy, n (%) | 35 (36.1) | 170 (39.9) |
Triple therapy, n (%) | 55 (56.7) | 238 (55.9) |
OLE = open-label extension.
Sources: Humbert (2023);59 Preston (2024).58
Efficacy
The observed mean (standard deviation) changes from baseline to months 18 to 24 among the placebo-crossed group in the PULSAR OLE study were reported as follows: –223.2 (57.5) dyn·sec·cm−5 in PVR, 60.5 (13.2) m in 6MWD, –506.2 (1,190.0) pg/mL in NT-proBNP, –0.6 (0.74) in WHO FC, and –10.7 (11.5) mm Hg in mPAP. The number of patients with clinical worsening events or death was 5 (16.7%) in the placebo-crossed group. In the FAS-E, at months 18 to 24 relative to baseline, there were no meaningful differences in the CAMPHOR scores (symptom, activity, or quality of life subscales and overall scores) and SF-36 scores.
Table 22: Key Efficacy Outcomes in the PULSAR OLE Study
Outcome | Placebo-crossed (N = 30) | Continued-sotatercept (N = 67) |
|---|---|---|
PVR | ||
Baseline | ||
Patients contributing to the analysis, n | 30 | 67 |
PVR (dyn·sec·cm−5), mean (SD) | 802 (331) | 784 (372) |
Months 18 to 24 | ||
Patients contributing to the analysis, n | 25 | 57 |
PVR (dyn·sec·cm−5), mean (SD) | 583 (310) | 538 (199) |
Change from baseline to months 18 to 24 | ||
Patients contributing to the analysis (multiple imputation), n | 30 | 57 |
Change from baseline (dyn·sec·cm−5), mean (SD) | –223.2 (57.5)a,b | –212.6 (254.24) |
P valuec | < 0.0001b | < 0.0001 |
Change from baseline in 6MWD | ||
Baseline | ||
Patients contributing to the analysis, n | 30 | 67 |
6MWD (m), mean (SD) | 409 (66) | 398 (86) |
Months 18 to 24 | ||
Patients contributing to the analysis, n | 25 | 62 |
6MWD (m), mean (SD) | 480 (73) | 458 (110) |
Change from baseline to months 18 to 24 | ||
Patients contributing to the analysis (multiple imputation), n | 30 | 62 |
Change from baseline (m), mean (SD) | 60.5 (13.2)a,b | 59.8 (80.9) |
P valuec | < 0.0001b | < 0.0001 |
NT-proBNP | ||
Baseline | ||
Patients contributing to the analysis, n | 30 | 66 |
NT-proBNP (pg/mL), mean (SD) | 840 (1,247) | 777.4 (1,051.03) |
Months 18 to 24 | ||
Patients contributing to the analysis, n | 28 | 64 |
NT-proBNP (pg/mL), mean (SD) | 363 (702) | 268 (457) |
Change from baseline to months 18 to 24 | ||
Patients contributing to the analysis, n | 28 | 63 |
Change from baseline (pg/mL), mean (SD) | –506.2 (1,190.0) | –470.5 (910.4) |
P valuec | 0.0004d | < 0.0001 |
WHO FC | ||
Baseline | ||
Patients contributing to the analysis, n | 30 | 67 |
Mean score (SD) | 2.4 (0.5) | 2.4 (0.5) |
Months 18 to 24 | ||
Patients contributing to the analysis, n | 28 | 63 |
Mean score (SD) | 1.9 (0.6) | 1.9 (0.5) |
Change from baseline to months 18 to 24 | ||
Patients contributing to the analysis, n | 28 | 63 |
Change from baseline, mean (SD) | –0.6 (0.74) | –0.4 (0.59) |
P valuec | < 0.0001 | < 0.0001 |
Time to clinical worsening | ||
Patients contributing to the analysis, n | 30 | 67 |
Patients with clinical worsening events or death, n (%) | 5 (16.7) | 9 (13.4) |
Assessment of first event of clinical worsening, n (%) | ||
Death | 1 (3.3) | 3 (4.5) |
Functional deterioration | 4 (13.3) | 5 (7.5) |
PAH-specific hospitalization | 1 (3.3) | 3 (4.5) |
Worsening-related listing for transplant | 0 (0) | 2 (3.0) |
Low Simplified French risk score | ||
Patients contributing to the analysis, n | 30 | 67 |
Patients who met all 3 low-risk criteria (6MWD > 440 m, maintaining WHO FC II or improving, NT-proBNP < 300 pg/mL), n (%) | 16 (53.3) | 32 (47.8) |
mPAP | ||
Baseline | ||
Patients contributing to the analysis, n | 30 | 67 |
mPAP (mm Hg), mean (SD) | 53.9 (13.72) | 52.0 (12.86) |
Months 18 to 24 | ||
Patients contributing to the analysis, n | 25 | 57 |
mPAP (mm Hg), mean (SD) | 44.1 (11.98) | 39.1 (10.48) |
Change from baseline to months 18 to 24 | ||
Patients contributing to the analysis, n | 25 | 57 |
Change from baseline (mm Hg), mean (SD) | –10.7 (11.5) | –12.5 (9.8) |
P valuec | < 0.0001d | < 0.0001d |
6MWD = 6-minute walk distance; FC = functional class; mPAP = mean pulmonary arterial pressure; OLE = open-label extension; PAH = pulmonary arterial hypertension; PVR = pulmonary vascular resistance; SD = standard deviation.
aValues are mean (standard error) instead of mean (SD).
bMultiple imputation was used to handle missing data.
cP values corresponding to change from baseline to months 18 to 24 within each group.
dNominal P value.
Source: Humbert (2023).59
Table 23: Key Efficacy Outcomes in the SOTERIA Study (Up to 1 Year Follow-Up)
Time to clinical worsening | Placebo-crossed (N = 143) | Continued-sotatercept (N = 259) |
|---|---|---|
Patients contributing to the analysis, n | 143 | 259 |
Patients who experienced at least 1 clinical worsening event, n | 4 | 16 |
Time to first event (weeks), median (range) | 33.4 (0.1 to 36.3) | 38.6 (6.7 to 88.3) |
Patients that experienced clinical worsening or death, n | 11 | 45 |
Assessment of first occurrence of clinical worsening events, n (%)a | ||
Death | 2 (1.4) | 3 (1.2) |
Worsening-related listing for lung and/or heart transplant | 2 (1.4) | NA |
Initiate rescue therapy or increase the dose of infusion of prostacyclin by ≥ 10% | NA | 2 (0.8) |
PAH-specific hospitalization (≥ 24 hours) | 1 (0.7) | 6 (2.3) |
Deterioration of PAH | NA | 3 (1.2) |
NA = not applicable; PAH = pulmonary arterial hypertension.
aA patient can have more than 1 assessment recorded for their first event of clinical worsening.
Source: Preston (2024).58
Harms
PULSAR OLE Study
TEAEs were reported in all patients in the PULSAR OLE phase who were assigned to the 0.7 mg/kg continued sotatercept group. TEAEs leading to the discontinuation of the study were experienced in 8 patients (19.0%) in the continued-sotatercept 0.7 mg/kg arm by the end of the OLE period. Three deaths (7.1%) were reported, 2 of which occurred in the OLE period (1 in the placebo-crossed treatment group [0.3 mg/kg] due to worsening PAH, and 1 in the continued-sotatercept treatment group [0.7 mg/kg] as a result of a brain abscess). The AESIs for the PULSAR OLE study were leukopenia, which occurred in 1 patient (2.4%) from the continued-sotatercept 0.7 mg/kg arm; neutropenia, which occurred in 1 patient (2.4%) from the continued-sotatercept 0.7 mg/kg arm; and thrombocytopenia, which occurred in 1 patient (6.7%) the from placebo crossed to sotatercept 0.7 mg/kg arm and 6 patients (14.3%) from the continued-sotatercept 0.7 mg/kg arm.
SOTERIA Study
Most of the participants (90.8%) in the SOTERIA study experienced 1 or more TEAEs, 3.5% discontinued treatment due to a TEAE, and 2.8% died due to a TEAE. SAEs occurred in 30.3% of patients. The AESI of telangiectasia occurred in 71 patients upon or after rolling over into the SOTERIA study. No telangiectasia event was deemed serious, but 1 patient discontinued the study due to telangiectasia. Among the other AEs of interest, epistaxis was the most common bleeding event, occurring in 92 patients (22.1%). Serious bleeding events (including gastrointestinal hemorrhage [1.9% of patients], hemoptysis [1.2%], and heavy menstrual bleeding [0.5%]) occurred in 22 patients (5.2%), but only 2 patients (0.5%) discontinued. Sixty-one patients (14.3%) experienced increased hemoglobin; however, none of the cases were serious. Thrombocytopenia events were experienced by 6.1% of participants, with 3 participants having serious, treatment-related thrombocytopenia.
Table 24: Summary of Harms Results From PULSAR OLE and SOTERIA Studies
Adverse events | PULSAR OLE | SOTERIA | ||
|---|---|---|---|---|
Placebo to sotatercept 0.7 mg/kg (N = 15) | Continued-sotatercept 0.7 mg/kg (N = 42) | Placebo-crossed (N = 143) | Continued- sotatercept (N = 259) | |
TEAEs | ||||
Patients reporting ≥ 1 adverse event, n (%) | 15 (100.0) | 42 (100) | 131 (91.6) | 240 (92.7) |
TEAEs reported in ≥ 10% of patients in either group, n (%) | ||||
COVID-19 | 1 (6.7) | 5 (11.9) | — | — |
Headache | 7 (46.7) | 12 (28.6) | — | — |
Nausea | 4 (26.7) | 8 (19.0) | — | — |
Diarrhea | 5 (33.3) | 11 (26.2) | — | — |
Epistaxis | 4 (26.7) | 8 (19.0) | — | — |
Fatigue | 4 (26.7) | 8 (19.0) | — | — |
Dizziness | 2 (13.3) | 8 (19.0) | — | — |
COVID-19–related dyspnea | 3 (20.0) | 3 (7.1) | — | — |
Nasopharyngitis | 6 (40.0) | 5 (11.9) | — | — |
Peripheral edema | 6 (40.0) | 10 (23.8) | — | — |
Arthralgia | 5 (33.3) | 6 (14.3) | — | — |
Pain in extremity | 3 (20.0) | 6 (14.3) | — | — |
Upper respiratory tract infection | 1 (6.7) | 7 (16.7) | — | — |
Back pain | 2 (13.3) | 3 (7.1) | — | — |
Increased hemoglobin | 5 (33.3) | 9 (21.4) | — | — |
Myalgia | 2 (13.3) | 6 (14.3) | — | — |
Urinary tract infection | 1 (6.7) | 6 (14.3) | — | — |
Hypokalemia | 0 (0.0) | 6 (14.3) | — | — |
Vomiting | 3 (20.0) | 5 (11.9) | — | — |
Gastroenteritis | 2 (13.3) | 6 (14.3) | — | — |
Thrombocytopenia | 1 (6.7) | 6 (14.3) | — | — |
Iron deficiency | 2 (13.3) | 7 (16.7) | — | — |
Pyrexia | 0 (0.0) | 5 (11.9) | — | — |
Anemia | 2 (13.3) | 3 (7.1) | — | — |
Cough | 3 (20.0) | 1 (2.4) | — | — |
Muscle spasms | 2 (13.3) | 5 (11.9) | — | — |
Sinusitis | 3 (20.0) | 3 (7.1) | — | — |
Bronchitis | 2 (13.3) | 4 (9.5) | — | — |
Telangiectasia | 5 (33.3) | 7 (16.7) | — | — |
SAEs | ||||
Patients reporting ≥ 1 SAE, n (%) | 4 (26.7) | 19 (45.2) | 41 (28.7) | 80 (30.9) |
SAEs reported in ≥ 4 patients in either group by system organ class, n (%) | ||||
Cardiac disorders | 0 (0.0) | 5 (11.9) | — | — |
General disorders and administration site conditions | 0 (0.0) | 4 (9.5) | — | — |
Infections and infestations | 1 (6.7) | 10 (23.8) | — | — |
Respiratory, thoracic, and mediastinal disorders | 0 (0.0) | 4 (9.5) | — | — |
Patients who stopped treatment due to adverse events, n (%) | ||||
Total | 0 (0.0) | 8 (19.0) | 2 (1.4) | 12 (4.6) |
Leukopenia | 0 (0.0) | 1 (2.4) | — | — |
Neutropenia | 0 (0.0) | 1 (2.4) | — | — |
Thrombocytopenia | 0 (0.0) | 1 (2.4) | — | — |
Cardiac arrest | 0 (0.0) | 1 (2.4) | — | — |
Sepsis | 0 (0.0) | 1 (2.4) | — | — |
Pleural effusion | 0 (0.0) | 1 (2.4) | — | — |
Respiratory failure | 0 (0.0) | 2 (4.8) | — | — |
Brain abscess | 0 (0.0) | 1 (2.4) | — | — |
Increased hemoglobin | 0 (0.0) | 1 (2.4) | — | — |
Increased red blood cell count | 0 (0.0) | 1 (2.4) | — | — |
Deaths, n (%) | ||||
Total | 0 (0.0) | 3 (7.1) | 4 (2.8) | 6 (2.3) |
Cardiac arrest | 0 (0.0) | 2 (4.8) | — | — |
Brain abscess | 0 (0.0) | 1 (2.4) | — | — |
Adverse events of special interest, n (%) | ||||
Telangiectasia | NA | NA | 37 (25.9) | 34 (13.1) |
Leukopenia | 0 (0.0) | 1 (2.4) | NA | NA |
Neutropenia | 0 (0.0) | 1 (2.4) | NA | NA |
Thrombocytopenia | 1 (6.7) | 6 (14.3) | NA | NA |
NA = not applicable; OLE = open-label extension; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Details included in the table are from the sponsor’s summary of clinical evidence.
Sources: Humbert (2023);59 Preston (2024).58
Critical Appraisal
Internal Validity
Due to the nature of the LTDB phase of the STELLAR study, the efficacy and harm end points are presented in a descriptive manner. Patients who completed the 24-week treatment period and continued to the LTDB phase could move into the SOTERIA trial. The median total follow-up time on treatment was 45 weeks and 39 weeks in the sotatercept and placebo groups, respectively, considerably shorter than the planned follow-up to a possible 72 weeks after the DBPC phase. Hence, there are attritional losses that impact the interpretability of the LTDB phase data, even though the investigators analyzed all patients up to the point they were observed and then moved to the SOTERIA trial or stopped. The blinding and randomization were maintained in the LTDB phase, although with the possibility of unblinding due to the nature of the AEs with sotatercept, as described in the Critical Appraisal section of the systematic review.
The open-label nature of the PULSAR study and lack of comparator may increase the risk of bias in determining the magnitude of the safety outcomes and efficacy end points that include more subjective assessments (i.e., clinical worsening and WHO FC status). The lack of blinding may affect patients’ expectations of treatment and influence reporting of subjective measures such as acceptability or AEs. The direction and magnitude of these potential biases remain unclear. Patients were selected from placebo-controlled study, which puts the results at risk of selection bias.
The SOTERIA study is still ongoing, and limited data have been reported so far. The open-label nature of the study and lack of comparator may increase the risk of bias in determining the magnitude of the efficacy and safety outcomes. The lack of blinding may affect patients’ expectations of treatment and influence reporting of subjective measures such as acceptability or AEs. The direction and magnitude of these potential biases remain unclear. The study included most of the patients who completed the parent trial (including 143 of the 160 patients in the placebo group of the STELLAR trial). It is possible that the patients who continued and remained on sotatercept were also those who experienced good performance from the drug; however, with the high number of patients crossing over from placebo to sotatercept included in the SOTERIA study, it is unclear whether the good performance of the drug was a driving factor in this population.
External Validity
The LTDB part of the study has the same limitations as the pivotal trial, including the lack of information on patients excluded from the trial, like those with PAH associated with HIV, portal hypertension, and pulmonary veno-occlusive disease. The observation of efficacy and harm outcomes beyond 24 weeks helps ascertain AEs over an extended period, which is overall in agreement with the pivotal trial results.
Like the STELLAR trial, the PULSAR study only included patients with PAH classified as WHO FC II and III, and the results may have the same limitations already mentioned. This study was conducted at 43 centres in 8 countries (Australia, Brazil, France, Germany, Israel, Spain, UK, and US), but no centres were in in Canada, although no concerns were raised by the clinical experts in terms of the generalizability of the evidence from other countries.
Indirect Evidence
An indirect treatment comparison was not included in this submission. The reason provided by the sponsor was that the place in therapy of sotatercept will be as an add-on to optimal background PAH therapy in adult patients who are not at low risk. Optimal background therapy is defined as patients receiving an optimal number and optimal doses of therapies according to clinical guidelines, which recommend an upfront double therapy for patients considered low to intermediate risk, or triple therapy for patients considered high risk. Standard of care with background PAH therapy was considered by the sponsor as the most appropriate comparator; hence, no indirect comparison is included in this application.
Discussion
Summary of Available Evidence
The body of evidence informing this submission consists of 1 study assessing sotatercept on top of stable background therapy versus placebo in patients with PAH. The STELLAR trial was a phase III, randomized, multicentre, DBPC trial in adult patients (N = 323) with PAH (WHO Group 1 pulmonary hypertension) who randomly received sotatercept (n = 163) or placebo (n = 160). The trial was conducted across 21 countries, including 3 sites in Canada, enrolling a population with demographic characteristics, including age and gender distributions, that closely reflected those of patients currently seen in clinical practice in Canada and relevant to this submission.
A supplemental body of evidence was submitted in the form of 3 reports — the LTDB treatment period of the STELLAR trial, the PULSAR study, and the SOTERIA study — to provide information on the long-term clinical efficacy and harms associated with sotatercept in adults with PAH.
No evidence from indirect treatment comparisons nor studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Interpretation of Results
Efficacy
For patients with PAH, the end points assessed in this body of evidence (i.e., mortality, improvement in exercise capacity, and hemodynamic values) were highly valued outcomes, as were those related to HRQoL. The clinical experts agreed with these considerations and added that other hemodynamic variables and composite end points, commonly used in clinical studies and in practice, were also key factors that inform decisions.
However, the current information on mortality is based on the single trial (the STELLAR study) with its LTFU phase, which was not designed and powered to detect significant between-group differences in the event of death; hence, the current effect estimates are too imprecise to ascertain a conclusion of a clinically meaningful difference.
Exercise capacity, as assessed by the 6-minute walk test, was determined to be important for decisions in clinical practice by the experts consulted by CDA-AMC and by patients. Overall, the use of sotatercept provides a clinically meaningful mean increase from baseline of 40.8 m more than placebo (95% CI, 27.5 m to 54 m). Although the lower value of the CI for this end point included the minimally important difference published in the literature of 33 m, the clinical experts considered that most patients would value a minimum benefit of 27 m as significant.
Similarly, the effect estimates in the composite end point (which includes the 6MWD, NT-proBNP level and WHO FC), as well as the composite of time to clinical worsening or death, were considered clinically meaningful when comparing sotatercept against placebo.
Hemodynamic variables (PVR and NT-proBNP) were considered relevant for clinical practice by clinical experts as they represent measures of improvement linked to clinical outcomes in patients. These measurements were both improved with the use of sotatercept as compared to placebo. Although a specific minimally important difference was not identified in the literature for patients with PAH, the clinical experts considered the extent of the observed improvements clinically meaningful. Whether the magnitude of the effects is important to patients and linked to better outcomes is still unclear and needs more clarification.
HRQoL was considered an important and valued outcome, which was measured with the PAH-SYMPACT and the EQ VAS. Both measurements showed statistically significant improvements. However, when considering the minimally important difference, both domains (physical impacts and cardiopulmonary symptoms) of the PAH-SYMPACT assessed showed values and 95% CIs that included the threshold of a minimally important difference, denoting some uncertainty (imprecision) about whether the effects are clinically meaningful.
Overall, the clinical experts consulted by CDA-AMC highlighted the observed improvements in the pivotal trials on important outcomes when sotatercept was added to existing therapies, especially in patients whose disease was not controlled by their current regimen, rather than as a first-line treatment. Patients in the STELLAR study, who had significant disease duration, showed notable improvement with sotatercept despite the perceived stability of their disease, indicating the potential impact of sotatercept on those already on multiple PAH agents.
For all efficacy and HRQoL outcomes, long-term extension studies suggest that the effects remain consistent over time. However, due to the inherent design of these studies (open label), there is still some level of uncertainty in the estimates.
Harms
Based on this body of evidence, sotatercept was generally well tolerated. The most common AEs observed in the sotatercept arm were epistaxis (12.3% of patients), telangiectasia (10.4%), and dizziness (10.4%). None of these events were severe, but they were more common in the sotatercept arm than in the placebo arms. Most AEs reported for participants in each treatment group were mild or moderate in severity. According to the clinical experts consulted by CDA-AMC, these AEs are not a critical concern and typically do not necessitate additional assessment beyond routine checks.
The number of SAEs was also similar between the sotatercept group (40 patients; 24.5%) and the placebo group (47 patients; 29.4%), with no specific difference in the individual AEs.
The information comparing sotatercept and placebo does not suggest a large difference in the number of either AEs or SAEs, although it still shows wide CIs, which were considered too imprecise for definite conclusions to be drawn.
Although AESI (epistaxis and telangiectasia) occurred significantly more often in the sotatercept group than in the placebo group, the experts consulted by CDA-AMC considered that patients would deem these harms acceptable weighed against the possible desirable effects of sotatercept.
Conclusion
The evidence on the effects of sotatercept in patients with PAH (WHO Group 1 pulmonary hypertension) comprises 1 pivotal randomized controlled trial comparing sotatercept to placebo and 3 reports on sotatercept’s long-term effects. Sotatercept was not compared to other interventions due to the nature of the reimbursement request and the drug’s suggested place in therapy as an add-on intervention to optimal background therapy or in patients who are already on double or triple therapy, depending on contraindications or the tolerability of available PAH therapies.
The outcomes considered critical for decision-making by clinical experts, patient groups, and stakeholders include the impact of sotatercept on mortality, exercise capacity (measured by the 6MWD), risk status assessed as a multicomponent improvement (6MWD, NT-proBNP level, and WHO FC), a composite of time to clinical worsening or death, PVR, NT-proBNP levels, HRQoL, hospitalization rates, and AEs.
The pivotal STELLAR study evaluated all these outcomes (including death as part of the composite end point), providing evidence that sotatercept, when compared to placebo, significantly improves 6MWD and increases the proportion of patients experiencing improvement in the assessment of clinical worsening, in the multicomponent composite end point, and in risk status, according to the Simplified French Risk Score. Additionally, sotatercept demonstrates meaningful benefits in PVR and NT-proBNP levels, as noted by the clinical experts. Patients receiving sotatercept had a lower risk of hospitalization due to PAH, and their PAH showed greater improvement in WHO FC stratum, than patients receiving placebo. Sotatercept also likely leads to improvements in HRQoL measurements (PAH-SYMPACT and EQ-5D-5L) compared to placebo, although the magnitude of these HRQoL effects remains uncertain due to imprecision of the results.
The frequency of AEs and SAEs was similar between the sotatercept and placebo groups, though bleeding events (especially epistaxis), telangiectasia, and dizziness occurred more frequently with sotatercept. These events were mostly reported as mild or moderate, and the clinical experts identified these as manageable in practice and likely of lower significance for patients when weighed against the desirable outcomes associated with PAH.
Overall, sotatercept improved exercise capacity, WHO FC, and risk status and reduced time to clinical worsening events compared with placebo and had a similar profile in short-term harms. However, the effects of sotatercept on mortality alone and on longer-term outcomes remain uncertain.
References
1.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
2.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
3.Clinical Study Report: P003V01MK7962. A Phase 3, Randomized, Double-Blind, Placebo Controlled Study to Compare the Efficacy and Safety of Sotatercept Versus Placebo When Added to Background Pulmonary Arterial Hypertension (PAH) Therapy for the Treatment of PAH. Rahway (NJ): Merck Sharp & Dohme Corp; 2022 Dec 16.
4.Clinical Study Report: P003MK7962. A Phase 3, Randomized, Double-Blind, Placebo Controlled Study to Compare the Efficacy and Safety of Sotatercept Versus Placebo When Added to Background Pulmonary Arterial Hypertension (PAH) Therapy for the Treatment of PAH. Rahway (NJ): Merck Sharp & Dohme Corp; 2023 May 9.
5.Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492. PubMed
6.International P. P. H. Consortium, Lane KB, Machado RD, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26(1):81-84. PubMed
7.Cuthbertson I, Morrell NW, Caruso P. BMPR2 Mutation and Metabolic Reprogramming in Pulmonary Arterial Hypertension. Circ Res. 2023;132(1):109-126. PubMed
8.Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023;61(1).
9.Pulmonary Hypertension Association of Canada (PHA Canada). The Impact of Pulmonary Hypertension on Canadians [sponsor-supplied reference]. 2022; https://phacanada.ca/Research/Survey.
10.Cheron C, McBride SA, Antigny F, et al. Sex and gender in pulmonary arterial hypertension. Eur Respir Rev. 2021;30(162). PubMed
11.Canadian Pulmonary Hypertension Registry Annual Report [sponsor-supplied reference]. [2022]: https://phacanada.ca/PHA/media/assets/Connections-Magazine-Winter-2022.pdf. Accessed 2023 Oct 24.
12.Hoeper MM, Pausch C, Grunig E, et al. Temporal trends in pulmonary arterial hypertension: results from the COMPERA registry. Eur Respir J. 2022;59(6). PubMed
13.Chang KY, Duval S, Badesch DB, et al. Mortality in Pulmonary Arterial Hypertension in the Modern Era: Early Insights From the Pulmonary Hypertension Association Registry. J Am Heart Assoc. 2022;11(9):e024969. PubMed
14.Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med. 2012;186(8):790-796. PubMed
15.Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376-387. PubMed
16.Hoeper MM, Kramer T, Pan Z, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J. 2017;50(2). PubMed
17.Zelt JGE, Sugarman J, Weatherald J, et al. Mortality trends in pulmonary arterial hypertension in Canada: a temporal analysis of survival per ESC/ERS guideline era. Eur Respir J. 2022;59(6). PubMed
18.PHA Canada. About Pulmonary Hypertension [sponsor-supplied reference]. https://phacanada.ca/For-Physicians/About-Pulmonary-Hypertension.
19.Canadian Organization for Rare Diseases. About CORD Key Facts [sponsor-supplied reference]. https://www.raredisorders.ca/about-cord/.
20.Leber L, Beaudet A, Muller A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: identification of the most accurate estimates from a systematic literature review. Pulm Circ. 2021;11(1):2045894020977300. PubMed
21.Emmons-Bell S, Johnson C, Boon-Dooley A, et al. Prevalence, incidence, and survival of pulmonary arterial hypertension: A systematic review for the global burden of disease 2020 study. Pulm Circ. 2022;12(1):e12020. PubMed
22.Wijeratne DT, Lajkosz K, Brogly SB, et al. Increasing Incidence and Prevalence of World Health Organization Groups 1 to 4 Pulmonary Hypertension: A Population-Based Cohort Study in Ontario, Canada. Circ Cardiovasc Qual Outcomes. 2018;11(2):e003973. PubMed
23.Statistics Canada. Table 17-10-0005-01: Population estimates on July 1, by age and gender [sponsor-supplied reference]. 2024.
24.Hirani N, Brunner NW, Kapasi A, et al. Canadian Cardiovascular Society/Canadian Thoracic Society Position Statement on Pulmonary Hypertension. Can J Cardiol. 2020;36(7):977-992. PubMed
25.Lewis RA, Durrington C, Condliffe R, Kiely DG. BNP/NT-proBNP in pulmonary arterial hypertension: time for point-of-care testing? Eur Respir Rev. 2020;29(156). PubMed
26.Healthwise Staff. Right Heart Catheterization: What to Expect at Home. 2022; https://myhealth.alberta.ca/health/AfterCareInformation/pages/conditions.aspx?hwid=acd9435. Accessed 2023 Oct 30.
27.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. PubMed
28.Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46(4):903-975. PubMed
29.Hoeper MM, Pausch C, Olsson KM, et al. COMPERA 2.0: a refined four-stratum risk assessment model for pulmonary arterial hypertension. Eur Respir J. 2022;60(1):2102311. PubMed
30.Boucly A, Weatherald J, Savale L, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J. 2017;50(2). PubMed
31.Boucly A, Weatherald J, Savale L, et al. External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. Eur Respir J. 2022;59(6). PubMed
32.Drugs for Pulmonary Arterial Hypertension: Comparative Efficacy, Safety, and Cost-Effectiveness — Recommendations Report [sponsor-supplied reference]. (CADTH Therapeutic review report). Ottawa (ON): CADTH; 2015: https://www.cadth.ca/sites/default/files/pdf/TR0006_PAH_Recs_Report.pdf.
33.PHA Canada, Canadian VIGOUR Centre at the University of Alberta. The Socioeconomic Burden of Pulmonary Arterial Hypertension in Canada [sponsor-supplied reference]. 2023.
34.Winrevair (sotatercept): 45 mg for subcutaneous injection and 60 mg for subcutaneous injection [product monograph]. Kirkland (QC): Merck Canada Inc.; 2024 Aug 28.
35.Acceleron Pharma, Inc. NCT04576988: A Study of Sotatercept for the Treatment of Pulmonary Arterial Hypertension (MK-7962-003/A011-11)(STELLAR) [sponsor-supplied reference]. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2023: https://clinicaltrials.gov/ct2/show/NCT04576988.
36.Gabler NB, French B, Strom BL, et al. Validation of 6-minute walk distance as a surrogate end point in pulmonary arterial hypertension trials. Circulation. 2012;126(3):349-356. PubMed
37.Mathai SC, Puhan MA, Lam D, Wise RA. The minimal important difference in the 6-minute walk test for patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(5):428-433. PubMed
38.Moutchia J, McClelland RL, Al-Naamani N, et al. Minimal Clinically Important Difference in the 6-minute-walk Distance for Patients with Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2023;207(8):1070-1079. PubMed
39.McCollister D, Shaffer S, Badesch DB, et al. Development of the Pulmonary Arterial Hypertension-Symptoms and Impact (PAH-SYMPACT(R)) questionnaire: a new patient-reported outcome instrument for PAH. Respir Res. 2016;17(1):72. PubMed
40.Chin KM, Gomberg-Maitland M, Channick RN, et al. Psychometric Validation of the Pulmonary Arterial Hypertension-Symptoms and Impact (PAH-SYMPACT) Questionnaire: Results of the SYMPHONY Trial. Chest. 2018;154(4):848-861. PubMed
41.Addendum to Common Technical Document section 2.7.3: summary of clinical efficacy [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Winrevair (sotatercept), 45 mg for subcutaneous injection and 60 mg for subcutaneous injection. Kirkland (QC): Merck Canada Inc.; 2024 Apr 8.
42.Gaine S, Simonneau G. The need to move from 6-minute walk distance to outcome trials in pulmonary arterial hypertension. Eur Respir Rev. 2013;22(130):487-494. PubMed
43.Bonner N, Abetz L, Meunier J, Sikirica M, Mathai SC. Development and validation of the living with pulmonary hypertension questionnaire in pulmonary arterial hypertension patients. Health Qual Life Outcomes. 2013;11:161. PubMed
44.CADTH. Drug Reimbursement Review: selexipag (Uptravi) for pulmonary arterial hypertension (WHO class II and III). 2016; https://www.cadth.ca/selexipag-reimbursement-review. Accessed 2024 Jul 2.
45.Dams J, Rimane E, Steil R, Renneberg B, Rosner R, Konig HH. Reliability, Validity and Responsiveness of the EQ-5D-5L in Assessing and Valuing Health Status in Adolescents and Young Adults with Posttraumatic Stress Disorder: a Randomized Controlled Trail. Psychiatr Q. 2021;92(2):459-471. PubMed
46.Del Corral T, Fabero-Garrido R, Plaza-Manzano G, Navarro-Santana MJ, Fernandez-de-Las-Penas C, Lopez-de-Uralde-Villanueva I. Minimal Clinically Important Differences in EQ-5D-5L Index and VAS after a Respiratory Muscle Training Program in Individuals Experiencing Long-Term Post-COVID-19 Symptoms. Biomedicines. 2023;11(9). PubMed
47.Tsai APY, Hur SA, Wong A, et al. Minimum important difference of the EQ-5D-5L and EQ-VAS in fibrotic interstitial lung disease. Thorax. 2021;76(1):37-43. PubMed
48.Humbert M, McLaughlin V, Gibbs JSR, et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2021;384(13):1204-1215. PubMed
49.Sitbon O, Channick R, Chin KM, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2015;373(26):2522-2533. PubMed
50.Hodges JL, Lehmann EL. Rank Methods for Combination of Independent Experiments in Analysis of Variance. In: Rojo J, ed. Selected Works of E. L. Lehmann. Boston, MA: Springer US; 2012:403-418.
51.Mehrotra DV, Lu X, Li X. Rank-Based Analyses of Stratified Experiments: Alternatives to the van Elteren Test. The American Statistician. 2010;64(2):121-130.
52.Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N Engl J Med. 2023;388(16):1478-1490. PubMed
53.Souza R, Badesch DB, Ghofrani HA, et al. Effects of sotatercept on haemodynamics and right heart function: analysis of the STELLAR trial. Eur Respir J. 2023;62(3). PubMed
54.Galie N, Jansa P, Pulido T, et al. SERAPHIN haemodynamic substudy: the effect of the dual endothelin receptor antagonist macitentan on haemodynamic parameters and NT-proBNP levels and their association with disease progression in patients with pulmonary arterial hypertension. Eur Heart J. 2017;38(15):1147-1155. PubMed
55.Chin KM, Rubin LJ, Channick R, et al. Association of N-Terminal Pro Brain Natriuretic Peptide and Long-Term Outcome in Patients With Pulmonary Arterial Hypertension. Circulation. 2019;139(21):2440-2450. PubMed
56.Tiede H, Sommer N, Milger K, et al. Short-term improvement in pulmonary hemodynamics is strongly predictive of long-term survival in patients with pulmonary arterial hypertension. Pulm Circ. 2013;3(3):523-532. PubMed
57.Clinical Study Report: P001MK7962. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study to Compare the Efficacy and Safety of Sotatercept (ACE-011) Versus Placebo When Added to Standard of Care for the Treatment of Pulmonary Arterial Hypertension (PAH). Rahway (NJ): Merck Sharp & Dohme Corp; 2023 Mar 28.
58.Preston IR, Badesch D, Ghofrani H-A, et al. SOTERIA: A long-term follow-up study of sotatercept for pulmonary arterial hypertension. Eur Respir J. 2023;62(suppl 67):OA739.
59.Humbert M, McLaughlin V, Gibbs JSR, et al. Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur Respir J. 2023;61(1). PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
These tables represent data from the sponsor that were not included in the main report. Some end points may repeat throughout.
Table 25: Key Efficacy Outcomes
Variable | STELLAR | |
|---|---|---|
Sotatercept N = 163 | Placebo N = 160 | |
Primary end pointa | ||
Change from baseline in 6MWD (m) at week 24 | ||
Number of patients contributing to the analysis (standard multiple imputation)b | 163 | 160 |
Median change estimate from baseline (range)b | 34.4 (32.5 to 35.5) | 1.0 (–1.0 to 5.0) |
Hodges-Lehmann location shift from placebo estimate (95% CI)c | 40.8 (27.5 to 54.1) | NA |
P valued | < 0.001 | NA |
Secondary end pointsa | ||
PAH-SYMPACT cognitive and emotional impacts domain score at week 24 | ||
Number of patients contributing to the analysis (standard multiple imputation)j | 163 | 160 |
Median change estimate from baseline (range)b | 0.00 (0.00 to 0.00) | 0.00 (0.00 to 0.00) |
Hodges-Lehmann location shift from placebo estimate (95% CI)c | –0.16 (–0.40 to –0.08) | NA |
P valued | 0.16 | NA |
Exploratory end points | ||
PAH-SYMPACT cardiovascular symptoms domain score at week 24 | ||
Number of patients contributing to the analysis (standard multiple imputation)j | 163 | 160 |
Median change estimate from baseline (range)b | –0.09 (–0.14 to –0.04) | 0.00 (0.00 to 0.00) |
Hodges-Lehmann location shift from placebo estimate (95% CI)c | –0.14 (–0.25 to –0.02) | NA |
P valued | 0.02 | NA |
mPAP (mm Hg) at week 24 | ||
Number of patients contributing to the analysis | 154 | 144 |
LS mean change from baseline (95% CI) | –13.6 (–15.1 to –12.1) | 0.3 (–1.2 to 1.9) |
LS mean difference from placebo (95% CI) | –13.9 (–16.0 to –11.8) | NA |
P valuek | < 0.001 | NA |
Right ventricular work (g.m) at week 24 | ||
Number of patients contributing to the analysis | 154 | 144 |
LS mean change from baseline (95% CI) | –1.0 (–1.1 to –0.8) | –0.1 (–0.3 to 0.02) |
LS mean difference from placebo (95% CI) | –0.9 (–1.1 to –0.6) | NA |
P value | < 0.0001 | NA |
Right ventricular power (mm Hg.L.min-1) at week 24 | ||
Number of patients contributing to the analysis | 154 | 144 |
LS mean change from baseline (95% CI) | –38.5 (–44.2 to –32.8) | –5.8 (–11.8 to 0.2) |
LS mean difference from placebo (95% CI) | –32.7 (–40.9 to –24.5) | NA |
P value | < 0.0001 | NA |
TAPSE/sPAP (mm Hg−1) at week 24 | ||
Number of patients contributing to the analysis | 76 | 111 |
LS mean change from baseline (95% CI) | 0.11 (0.08 to 0.14) | –0.01 (–0.04 to 0.01) |
LS mean difference from placebo (95% CI) | 0.12 (0.09 to 0.16) | NA |
P value | < 0.0001 | NA |
EQ-5D-5L mobility at week 24 | ||
Number of patients contributing to the analysis | 90 | 89 |
LS mean change from baseline (95% CI) | –0.1 (–0.3 to 0.0) | 0.0 (–0.2 to 0.2) |
LS mean difference from placebo (95% CI) | –0.1 (–0.4 to 0.1) | NA |
P valuek | < 0.001 | NA |
EQ-5D-5L self-care at week 24 | ||
Number of patients contributing to the analysis | 90 | 89 |
LS mean change from baseline (95% CI) | –0.1 (–0.2 to 0.0) | 0.0 (–0.1 to 0.2) |
LS mean difference from placebo (95% CI) | –0.1 (–0.3 to 0.0) | NA |
P valuek | < 0.001 | NA |
EQ-5D-5L usual activities at week 24 | ||
Number of patients contributing to the analysis | 90 | 89 |
LS mean change from baseline (95% CI) | –0.2 (–0.4 to –0.0) | –0.0 (–0.2 to 0.1) |
LS mean difference from placebo (95% CI) | –0.2 (–0.4 to 0.1) | NA |
P valuek | < 0.001 | NA |
EQ-5D-5L pain or discomfort at week 24 | ||
Number of patients contributing to the analysis | 90 | 89 |
LS mean change from baseline (95% CI) | 0.1 (–0.1 to 0.2) | –0.0 (–0.2 to 0.1) |
LS mean difference from placebo (95% CI) | 0.1 (–0.1 to 0.3) | NA |
P valuek | < 0.001 | NA |
EQ-5D-5L anxiety or depression at week 24 | ||
Number of patients contributing to the analysis | 90 | 89 |
LS mean change from baseline (95% CI) | –0.1 (–0.2 to 0.1) | 0.1 (–0.02 to 0.3) |
LS mean difference from placebo (95% CI) | –0.2 (–0.4 to –0.01) | NA |
P valuek | < 0.001 | NA |
6MWD = 6-minute walk distance; CI = confidence interval; FC = functional class; mPAP = mean arterial pressure; PAH-SYMPACT = physical impacts domain score of pulmonary arterial hypertension; PVR = pulmonary vascular resistance; sPAP = systolic pulmonary artery pressure; TAPSE = tricuspid annular plane systole excursion; VAS = visual analogue scale.
Sources: Hoeper (2023);52 Souza (2023);53 Clinical Study Reports.3,4
Pharmacoeconomic Review
Abbreviations
6MWD
6-minute walk distance
BGT
background therapy
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
FC
functional class
ICER
incremental cost-effectiveness ratio
PAH
pulmonary arterial hypertension
PCA
prostacyclin analogue
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Sotatercept (Winrevair), 45 mg or 60 mg, powder, and solvent for injectable solution 45 mg vial kit, 60 mg vial kit, 2 × 45 mg vial kit, 2 × 60 mg vial kit |
Indication | In combination with standard pulmonary arterial hypertension (PAH) therapy, for the treatment of adults with WHO [WHO] Group 1 PAH and Functional Class (FC) II or III |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | August 28, 2024 |
Reimbursement request | As an add-on to optimal background therapy for the treatment of adult patients with PAH who are not at low risk. Low risk is defined as:
Optimal background therapy is defined as:
This medication should be prescribed under the direction of a specialist in the area of PAH. Diagnosis must be confirmed by right-heart catheterization. |
Sponsor | Merck Canada Inc. |
Submission history | Previously reviewed: No |
6MWD = 6-minute walk distance; FC = functional class; NOC = Notice of Compliance; PAH = pulmonary arterial hypertension.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with PAH who are not at low risk Low risk is defined as:
|
Treatment | Sotatercept as an add-on to optimal BGT Optimal BGT is defined as the following:
|
Dosing regimen | 0.3 mg/kg for first dose, followed by 0.7 mg/kg every 3 weeks |
Submitted price | Sotatercept 45 mg vial, $8,717.15 Sotatercept 60 mg vial, $11,622.87 Sotatercept 2 × 45 mg vials, $17,434.30 Sotatercept 2 × 60 mg vials, $23,245.73 |
Submitted treatment costa | First year, per patient annual cost: $152,344 Subsequent years, per patient annual cost: $186,523 |
Comparator | Optimal BGT includes any combination of the following drugs:
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (30 years) |
Key data sources | STELLAR trial COMPERA registry data |
Submitted results | ICER = $339,613 per QALY gained (incremental costs = $4,095,212; incremental QALYs = 12.06) |
Key limitations |
|
CDA-AMC reanalysis results |
|
6MWD = 6-minute walk distance; BGT = background therapy; CDA-AMC = Canada’s Drug Agency; FC = functional class; ICER = incremental cost-effectiveness ratio; PAH = pulmonary arterial hypertension; QALY = quality-adjusted life-year.
aThe weighted average annual cost of sotatercept assumed by the sponsor was calculated based on the patients’ weight distribution derived from the STELLAR trial to determine the utilization rates for each kit (45 mg, 60 mg, 2 × 45 mg, or 2 × 60 mg). This distribution indicated that the utilization rates for the 45 mg, 60 mg, 2 × 45 mg and 2 × 60 mg vial kits are 44.73%, 48.38%, 6.88%, and 0.01%, respectively.
Conclusions
The Canada’s Drug Agency (CDA-AMC) Clinical Review reported that, based on results of the STELLAR trial, sotatercept significantly improves 6-minute walk distance (6MWD) and increases the proportion of patients with pulmonary arterial hypertension (PAH) experiencing improvements in the multicomponent composite end point (i.e., 6MWD, NT-proBNP level, and WHO functional class [FC], which are used to define risk status in the economic model) compared to placebo at 24 weeks. The clinical reviewers also noted that sotatercept likely leads to improvements in health-related quality of life measurements compared to placebo; however, the magnitude of these effects remains uncertain due to imprecision of the results.
CDA-AMC was able to address some limitations associated with the sponsor’s analysis, including the way in which the treatment effect of sotatercept was included in the model and the population from which all-cause mortality was extrapolated. Based on the CDA-AMC base-case analysis, sotatercept plus optimal background therapy (BGT) was associated with 4.09 incremental quality-adjusted life-years (QALYs) at an additional cost of $1,786,879, resulting in an incremental cost-effectiveness ratio (ICER) of $436,796 per QALY gained compared to optimal BGT alone.
The incremental cost is driven by direct treatment costs (sotatercept and optimal BGT), which make up 96% of the total costs associated with treatment with sotatercept plus optimal BGT. The drug acquisition cost of sotatercept is $199,249 per patient in the first year and $202,155 per patient in subsequent years (based on a weight of 67.5 kg to 88.9 kg), resulting in a lifetime cost of $1,561,624 per patient. Relative to those who do not receive sotatercept, patients who receive sotatercept plus optimal BGT are expected to incur $207,243 in additional drug costs. The incremental cost of optimal BGT was driven by the 4.31 additional life-years for those treated with sotatercept plus optimal BGT because these patients are expected to remain on treatment for longer. If the prices of all treatments included in optimal BGT were 50% lower than their public list prices, a price reduction for sotatercept of approximately 95% would be necessary for it to be considered cost-effective at a threshold of $50,000 per QALY gained (reflecting an annual drug cost of $7,970 and $8,086, in the first and subsequent years of treatment with sotatercept, respectively).
CDA-AMC was unable to resolve the uncertainty in the duration of the treatment effect for sotatercept; as such, the CDA-AMC analysis is predicated on a persistent treatment benefit observed from week 12 to week 24 in the STELLAR trial over the 30-year time horizon. The potential value of sotatercept is dependent on patients realizing the uncertain survival benefit compared to optimal BGT alone (4.31 life-years). If these health outcomes are not realized, the ICER for sotatercept plus optimal BGT compared to optimal BGT alone will likely be higher than predicted in these analyses.
Patient, Clinician, and Drug Plan Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from the Pulmonary Arterial Hypertension Association of Canada. Information for this input was gathered from 2 surveys conducted by the association: the Sotatercept Patient Evidence Submission Survey (an online survey of patients with PAH and their caregivers in Canada conducted in English and French between March 13, 2024, and April 1, 2024, that aimed to gather feedback about their current treatment experiences and expectations for sotatercept) and the Socioeconomic Burden of PAH in Canada (an online survey of adult patients with PAH in Canada conducted in English and French from August 15, 2023, to September 10, 2023, which aimed to evaluate the socioeconomic burden of PAH with an emphasis on workplace-related and activity-related limitations, assessed using the Work Productivity and Activity Impairment questionnaire). Both studies included both patients and caregivers. Of the patients surveyed, 3 indicated experience taking sotatercept. As these patients could only have accessed sotatercept through the clinical trial in Canada, CDA-AMC could not determine whether they received the drug or the placebo. Generally, these patients reported high variability in their experience of sotatercept. All 3 reported no changes to their shortness of breath at rest (the only experience shared by all 3 respondents). Shortness of breath upon exertion was unchanged for 1 patient and worsened for 1 patient, and 1 patient was unsure whether there had been a change. Results relating to tiredness and fatigue also varied, with 1 patient reporting an improvement, 1 reporting no change, and 1 reporting worsening symptoms. There were also single reports of the following symptoms worsening: swelling of the abdomen, chest pain upon exertion, light-headedness, and heart palpitations. None of the patients reported any pain or swelling at their injection site or any bleeding, which is a more severe side effect that can be associated with sotatercept. Two of the 3 patients were unsure if sotatercept was more effective than current therapies in slowing disease progression, and the third reported the same effectiveness as current treatments. Two of the 3 also agreed that sotatercept offered the same efficacy in addressing the physical and social limitations of the disease. All 3 patients reported that sotatercept was easy to use.
Clinicians indicated that sotatercept stands out for its focus on inhibiting cell proliferation, a departure from conventional treatments that mainly aim at widening blood vessels. Therefore, sotatercept should be used to complement existing vasodilators rather than to replace them as the initial treatment. Clinicians also noted that sotatercept could be beneficial for patients with various types of PAH, particularly those in WHO FC II and III, suggesting that starting sotatercept treatment as soon as possible after diagnosis is essential.
Participating drug plans sought clarification on the definition of optimal BGT used alongside sotatercept for patients with PAH. They also expressed concerns about how many plans currently allow triple or quadruple therapy regimens for patients with PAH and questioned how many plans have clearly defined, objective criteria for renewing current PAH therapies. Additionally, the drug plans identified challenges in initiation criteria (e.g., diagnosing, scoring, or staging for eligibility), as well as the difficulties in assessing and monitoring therapeutic responses. Lastly, the drug plans asked about the applicability of sotatercept to the broader indicated population, specifically including those diagnosed with PAH related to HIV, portal hypertension, schistosomiasis, and pulmonary veno-occlusive disease.
One of these concerns was addressed in the sponsor’s model:
The submitted model accounted for the preferred administration type for sotatercept.
CDA-AMC was unable to address the following concern raised from patient, clinician, and drug plan input:
CDA-AMC was not able to resolve issues around the definition of optimal BGT.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of adding sotatercept to optimal BGT for PAH management compared with the use of optimal BGT only.1 Optimal BGT was defined as patients receiving the optimal number and doses of therapies according to clinical guidelines, where patients may be on double or triple therapy depending on contraindications and/or the tolerability of available PAH therapies.
The modelled population consisted of adult patients diagnosed with PAH (as defined within WHO Group 1) who were not at low risk and who were on their optimal BGT. Low risk was defined as meeting all the following criteria: WHO FC I or II, and 6MWD greater than 440 m, and NT-proBNP less than 300 ng/L or B-type natriuretic peptide less than 100 ng/L. The modelled population aligned with the reimbursement request criteria.
The recommended dose of sotatercept is 0.3 mg/kg for the initial dose, followed by 0.7 mg/kg once every 3 weeks by subcutaneous injection.2 Sotatercept is provided in 45 mg or 60 mg vials, individually or in kits of 2 vials of the same sizes. The sponsor-submitted cost for sotatercept was $8,717.15 per 45 mg vial and $11,622.87 per 60 mg vial. Based on the distribution of patient weight from the STELLAR trial, the sponsor assumed the annual per patient cost of sotatercept was $152,344 in the first year and $186,523 in subsequent years, accounting for wastage of excess medication.1 There were no administration costs for sotatercept, which was assumed to be self-administered.
The sponsor assumed that an optimal BGT regimen consists of a combination of various vasodilators, including sildenafil citrate and tadalafil (phosphodiesterase type 5 inhibitors); bosentan, ambrisentan, and macitentan (endothelin receptor antagonists); as well as epoprostenol, treprostinil, and selexipag, which are prostacyclin analogues (PCAs). The utilization of BGT was based on the distribution of therapies observed in a Canadian study and on expert opinion,3 and it varied across different health states and over time in the sponsor’s submitted model.1 The sponsor assumed that the annual cost of optimal BGT ranged from $47,471 to $93,099 per patient, depending on health state and time, in the model.
The clinical outcomes of interest reported in the analysis were QALYs and life-years over a 30-year time horizon. Discounting of 1.5% per year was applied to both costs and health-related outcomes. The base-case perspective was that of the Canadian publicly funded health care payer.
Model Structure
The sponsor presented a Markov cohort-level model consisting of 6 mutually exclusive health states.1 Four of the states were based on the 4-strata risk assessment for PAH: low risk, intermediate-low risk, intermediate-high risk, and high risk. In addition, the model included a separate health state for patients who underwent lung or heart transplant and a separate state for patients who left the modelling cohort due to death. The sponsor’s model structure is presented in Figure 1.
The patient flow was described as follows: Upon entering the model, the patients were allocated to 1 of the 4 risk strata. During each model cycle, patients could remain in their current risk state, transition to other risk states, undergo a lung or heart transplant, or die.
The entire model dynamic was split into 2 distinct time periods: the short-term period based on the STELLAR trial period (≤ 24 weeks) and the long-term period following the time starting from the ending of the STELLAR trial and continuing until the end of the model time horizon. Consequently, the model cycle length was adjusted to be in line with the schedule of the STELLAR trial period and follow-up visits; thus, the first model cycle spanned 3 weeks (from baseline to visit 1 in the STELLAR trial), the second cycle was 9 weeks (from visit 1 to visit 2), and the third cycle was 12 weeks (from visit 2 to visit 3). All cycles thereafter were set to a length of 12 weeks. Model transition rates were also adjusted to the reflect the change in cycle length.
Model Inputs
The baseline characteristics and the risk stratification for the model’s initial cohort were derived from the intent-to-treat population of the STELLAR trial, which included 321 patients diagnosed with PAH in WHO FC II or III, 70% of whom were not classified as low risk at baseline. Each participant in the trial had been on a stable dose of their optimal BGT for a minimum of 3 months before enrolment, with 4% on monotherapy, 34% on double therapy, and 62% on triple therapy. The average age of the patients was 47.9 years, and the average body weight was 70.1 kg.
Treatment efficacy was modelled in 2 periods. Initially, for the short-term (up to 24 weeks), transition probabilities were based on data directly observed in the STELLAR trial. For the long-term phase (beyond week 24), transition probabilities extrapolated from weeks 12 to 24 of the STELLAR data were carried forward under the assumption that these would remain constant throughout the model’s duration.1 The sponsor used data from the SOTERIA trial to support this assumption.
Risk stratum–specific mortalities were derived from parametric regression models fitted to long-term Kaplan-Meier survival curves sourced from a subgroup of patients with no comorbidities from Rosenkranz et al. (2023).4 Age-based and sex-based life tables for the population living in Canada were also used to adjust mortality rates.5
Hospitalization probabilities for each nonfatal health state were obtained from the COMPERA registry. The sponsor assumed patients who were considered high risk and intermediate-high risk were eligible for lung or heart transplants. The probability for lung and heart transplant was also obtained from the COMPERA registry; the posttransplant mortality was sourced from the retrospective study by Bernstein (2018).6
Utility score estimates were based on the EQ-5D-5L data collected in the STELLAR trial using a Canadian-specific mapping value set.7 Utilities for PAH risk states were 0.89 for low risk, 0.80 for intermediate-low risk, 0.73 for intermediate-high risk, and 0.64 for high risk, with the utility for the post–lung transplant or post–heart transplant state assumed to be the same as for the low-risk state.
Adverse event data were sourced from the STELLAR trial. The sponsor’s model included all incidence of moderate and severe adverse events reported in at least 1% of participants. The adverse events included were thrombocytopenia, diarrhea, nausea, vomiting, fatigue, headache, epistaxis, flushing, telangiectasia, and increased hemoglobin. Disutilities were associated with drug administration (via continuous IV and subcutaneous infusion, estimated from a sample of the UK general population),8 PAH hospitalization, lung or heart transplant, and adverse events. Disutility values for each were derived from various sources, including Di Tanna (2021),9 Sullivan (2006),10 and Hagiwara (2018).11
The sponsor included costs related to drug acquisition, PAH-related routine care, PAH hospitalization, lung or heart transplant, and terminal care. The details of drug acquisition costs were outlined in the Overview section. Administration costs were considered only for drugs administered via IV and subcutaneous infusion and were estimated at $129.31 and $127.05, respectively. PAH-related routine care costs, sourced from a retrospective observational study by Dufour (2017),12 were specified as follows based on patient risk status: $1,151 for low risk, $1,456 for intermediate-low risk, $1,644 for intermediate-high risk, and $1,648 for high risk. The cost of PAH hospitalization, obtained from the CADTH Therapeutic Review on PAH drugs, was $17,637.13 Expenses related to lung or heart transplants, totalling $171,566, were sourced from the Alberta Interactive Health Data Application.14 Costs for routine care for patients post–lung or heart transplant were assumed to align with those of patients considered low risk. Terminal care costs, amounting to $38,440, were derived from a study by Kendzerska (2019) of people living in Ontario with lung cancer and chronic obstructive pulmonary disease.15 Costs associated with adverse events were included only for thrombocytopenia, where a 6-week course of prednisone was prescribed, with a daily dose of 100 mg.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
Sotatercept plus optimal BGT was associated with an incremental QALY gain of 12.06 and an incremental cost of $4,095,212, resulting in an ICER of $339,613 per QALY gained (Table 3).
The results showed that incorporating sotatercept alongside BGT resulted in substantial increases in both life-years (incremental life-years = 13.20) and QALYs. These increases were mainly attributed to the use of sotatercept resulting in a greater proportion of patients improving to lower-risk health states, which the sponsor’s model predicted would result in improved overall survival. The increased costs in patients treated with sotatercept were primarily due to the increase in the drug acquisition costs for both sotatercept and BGT. The results showed that nearly 99% of the total incremental QALYs and approximately 95% of the total incremental costs were accrued during the posttrial period.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. optimal BGT alone ($/QALY) |
|---|---|---|---|---|---|
Optimal BGT | 479,749 | Reference | 4.19 | Reference | Reference |
Sotatercept plus optimal BGT | 4,574,961 | 4,095,212 | 16.25 | 12.06 | 339,613 |
BGT = background therapy; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor evaluated several scenarios, altering the discount rates for costs and QALYs, and investigated an alternative method for deriving transition probabilities using a relative risk derived from the STELLAR trial. These alternate approaches had a limited impact on the results (the ICER fluctuating between a decrease of 26.31% and an increase of 6.67%).
The sponsor also conducted a scenario analysis from a societal perspective, considering indirect costs associated with work impairment and productivity losses for both patients and their caregivers. In this analysis, relative to optimal BGT alone, the ICER for sotatercept plus optimal BGT was $300,272 per QALY gained.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The treatment effect of sotatercept on clinical outcomes is overestimated. The sponsor’s model demonstrated the clinical effectiveness of sotatercept through the improved risk status (e.g., transitioning from intermediate-high risk to intermediate-low risk) based on results of the STELLAR trial. The risk-based health states were based on the 4–risk strata assessment for PAH that is recommended in the 2022 European Society of Cardiology–European Respiratory Society treatment guidelines for PAH.16 This risk assessment includes assessment of the WHO FC, 6MWD, and BNP or NT-proBNP to classify patients into a risk status.16 According to the clinical guidelines, these measures are the strongest prognostic predictors of death within 12 months.16 In the model there is a reduction in risk-related outcomes for hospitalization, mortality, and the need for escalated PCA treatment associated with the risk status–based health states. However, the sponsor also independently accounts for the effects of sotatercept on these events using hazard ratios (0.08 for hospitalization, 0.25 for mortality, and 0.33 for escalated PCA treatment), which double counts the effect of sotatercept on those outcomes and leads to an overestimation of the clinical benefit of sotatercept. The clinical experts consulted by CDA-AMC during this review indicated that they agreed that lower patient risk status would be associated with reduced mortality, hospitalization, and treatment with PCA infusions, in line with the European Society of Cardiology–European Respiratory Society guidelines, but indicated that there is insufficient evidence to know if treatment with sotatercept is associated with additional risk reductions not captured by the risk status–based health states.
In reanalysis, CDA-AMC removed the hazard ratios relating to mortality, hospitalization, and PCA treatment escalation associated with treatment with sotatercept while maintaining the risk status–associated risk reductions for these clinical outcomes.
The long-term treatment effect of sotatercept is unknown. The sponsor assumed that the treatment effect of sotatercept from week 12 to week 24 of the STELLAR trial would persist indefinitely, such that the same treatment effect is applied every 12-week cycle for up to 30 years. As a result, the probability of patients transitioning to a lower risk health state and the probability of remaining in lower-risk health states (i.e., not experiencing disease progression) is highly uncertain, and likely overestimated, beyond the time period studied in the STELLAR trial. For example, under this assumption, the sponsor’s base-case analysis estimates that treatment with sotatercept will result in an average gain of 13.20 life-years (12.06 QALYs) compared with treatment with only optimal BGT over the lifetime time horizon. PAH is a highly progressive, chronic disease16 with an estimated 5-year survival rate in Canada of 56%;17 however, in the sponsor’s base-case analysis, 96% of patients treated with sotatercept plus optimal BGT remained alive at 5 years. This long-term survival benefit was largely driven by the improved clinical risk status and slower progression for patients being treated with sotatercept. However, the current clinical evidence assesses the benefit of sotatercept added to optimal BGT over 24 weeks. It remains highly uncertain whether the treatment benefit observed for sotatercept over the 24-week trial period will persist or whether the magnitude of benefit observed will change over time.
It was not possible to assess the impact of treatment effect waning in the submitted model due to the model structure and lack of alternative data. As such, CDA-AMC maintained the sponsor’s assumption regarding the treatment effects persisting over a lifetime time horizon, but as a result clinical outcomes (including QALYs and life-years) for sotatercept may be overestimated in both the sponsor and the CDA-AMC base-case analyses.
Mortality for people with PAH is uncertain. The sponsor based mortality on a parametric regression fitted to reconstructed Kaplan-Meier data from the COMPERA registry, using a subgroup of patients with no comorbidities (n = 208).4 The results of the COMPERA analysis by number of comorbidities (groups were stratified based on 0, 1 to 2, or 3 to 4 comorbidities) demonstrated that mortality outcomes at 1, 3, and 5 years differed across these groups; patients with no comorbidities were estimated to have transplant-free survival rates at 1, 3, and 5 years of 99%, 75%, and 73%, respectively, compared to 96%, 67%, and 46% for those with 1 to 2 comorbidities, and 95%, 67%, and 46% for those with 3 to 4 comorbidities.4 By using data only from the group of patients with no comorbidities, the sponsor has likely underestimated mortality in the submitted model. While the median age of this subgroup of patients from the COMPERA registry (51 years) was similar to that of the STELLAR trial (48 years), the exclusion of patients with specific comorbidities (arterial hypertension, diabetes mellitus, coronary heart disease, and obesity) is not reflective of the population of patients with PAH in Canada. From a cohort of patients with PAH (n = 392) identified at 3 major PAH centres in Canada, 28% had diabetes, 18% had coronary artery disease, and 47% had hypertension.17
The sponsor’s model also included the option to inform mortality using data from the French pulmonary hypertension registry (n = 2,879).18 While the mean age of patients in the French registry (61 years) is not as similar to the STELLAR population, the clinical experts consulted by CDA-AMC for this review agreed that the patient population in Canada who may be eligible for treatment with sotatercept is likely more comparable to the full French registry population than the subgroup of patients with no comorbidities from the COMPERA registry.
In reanalysis, CDA-AMC applied the mortality curves estimated using the French pulmonary hypertension registry,18 maintaining the sponsor’s selected parametric model (i.e., Gompertz).
The submitted model included patients classified as low risk at baseline. In the sponsor’s submitted model, 29.6% of patients started in the low-risk health state and were able to receive treatment with sotatercept. However, the target population in this review excluded patients classified as low risk. Further, transition probabilities between health states were based on the full STELLAR trial population and, as such, include patients who were classified as low risk at baseline. As a result, the transition probability associated with staying in the low-risk health state (i.e., not progressing to a higher risk status once you have achieved low-risk status) is uncertain. The clinical experts consulted by CDA-AMC indicated that it is likely appropriate to generalize results for patients who were classified as low risk at baseline to patients who started in a higher risk status and became classified as low risk following treatment.
In consultation with the sponsor, CDA-AMC removed patients starting in the low-risk health state as a correction to the sponsor’s base-case analysis. The transition probabilities maintain the use of data from patients who were classified as low risk at baseline in the STELLAR trial.
An additional limitation was identified but was not considered to be a key limitation. This limitation is described subsequently.
Costs of background therapies were underestimated. In the submitted model, the sponsor applied discounts to the drug acquisition costs for macitentan (65% price reduction), selexipag (42% price reduction), and tadalafil (32% price reduction) based on recommended price reductions from past CADTH Reimbursement Reviews (macitentan and selexipag)19,20 and the potential for more generics entering the market for tadalafil. Although the public list prices may not account for confidentially negotiated prices, actual potential price reductions are uncertain and the public list prices represent the most appropriate drug costs to apply in the model.21
CDA-AMC removed the price reductions applied to macitentan, tadalafil, and selexipag as a correction of the sponsor’s base case.
The cost of treprostinil was incorrectly estimated. In the submitted model, the sponsor incorrectly used the cost per 1 mg of treprostinil ($45 per mg) to estimate the cost of a 200 mg vial resulting in a cost per vial of $9,000. The public list price of a 20 mL vial (which contains a 200 mg dose) is $450 per vial.22
CDA-AMC changed the price of treprostinil to $450 per 20 mL vial (10 mg/mL strength) as a correction of the sponsor’s base case.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Data for both treatment arms (i.e., sotatercept plus optimal BGT and optimal BGT alone) were pooled together and applied to both treatment arms in the model, assuming there would be no difference in transition probabilities from the high-risk health state between the treatment arms. | Acceptable. This assumption is likely conservative in that it does not suggest a treatment benefit for patients receiving treatment with sotatercept from the high-risk health state. |
Patients receiving treatment with sotatercept were assumed to stay on treatment over their entire lives. | Acceptable. The clinical experts consulted as part of this review confirmed that sotatercept would typically not be discontinued unless a patient is intolerant to sotatercept, is experiencing clinically important AEs, or has experience disease progression to the point of requiring lung or heart transplant. |
Drug acquisition costs for treatments included in optimal BGT were the public list prices from Saskatchewan. | Acceptable. While the drug acquisition costs for treatments included in optimal BGT differ across jurisdictions, given that sotatercept is an add-on therapy and all patients alive in the model are receiving treatment with optimal BGT, the impact on the results of the cost-effectiveness analysis are minimal. |
The sponsor assumed that there would be utility decrements depending on the mode of administration of PAH treatments. | Uncertain. The sponsor assumed that there would be a disutility associated with IV and subcutaneous infusions, but not for oral treatments or for IV or subcutaneous injections. The values used in the sponsor’s analysis were estimated in a sample of 150 people from the general population in the UK that compared PAH treatments delivered by continuous infusion, orally, and by inhalation. As a result of this study population and design, there is outstanding uncertainty about the administration-related disutility; however, the impact on the results of the cost-effectiveness analysis are minimal. |
AE = adverse event; BGT = background therapy; CDA-AMC = Canada’s Drug Agency; PAH = pulmonary arterial hypertension.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. These changes, summarized in Table 5, included modifying the risk of hospitalization, PCA infusion escalation, and mortality.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Low-risk patients | Included in the modelled population | Removed from the modelled population |
2. Background therapy costs | Included price reductions for macitentan, tadalafil, and selexipag | Removed price reductions from background therapies |
3. Cost of treprostinil per vial | $9,000 | $450 |
Changes to derive the CDA-AMC base case | ||
1. Treatment effect of sotatercept | Included hazard ratios for mortality, hospitalization, and PCA escalation | Removed hazard ratios for mortality, hospitalization, and PCA escalation |
2. Mortality estimates | Based on the COMPERA registry, using a subgroup of patients with no comorbidities | Based on the French pulmonary hypertension registry |
CDA-AMC base case | ― | 1 plus 2 |
CDA-AMC = Canada’s Drug Agency; PCA = prostacyclin analogue.
The CDA-AMC base-case analysis found that sotatercept was associated with 4.09 incremental QALYs (and 4.31 incremental life-years) at an additional cost of $1,786,879. Therefore, the ICER of sotatercept plus optimal BGT was $436,796 per QALY gained compared to optimal BGT alone. A summary of the stepped analysis and base-case analysis results can be found in Table 6. Approximately 95% of the incremental QALYs were accrued beyond the time for which trial data are available (i.e., 24 weeks).
Sotatercept had a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. The CDA-AMC reanalysis was driven by the treatment acquisition costs — both the cost of sotatercept and the cost of optimal BGT — for patients treated with sotatercept plus optimal BGT. Combined, these costs made up 96% of the total costs associated with treatment with sotatercept plus optimal BGT (73% for sotatercept; 23% for optimal BGT).
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | Optimal BGT | 467,622 | 3.95 | Reference |
Sotatercept plus optimal BGT | 4,812,016 | 16.94 | 334,390 | |
Sponsor’s corrected base case | Optimal BGT | 442,061 | 3.65 | Reference |
Sotatercept plus optimal BGT | 4,548,211 | 16.85 | 311,088 | |
CDA-AMC reanalysis 1 | Optimal BGT | 442,061 | 3.62 | Reference |
Sotatercept plus optimal BGT | 3,221,259 | 10.79 | 387,367 | |
CDA-AMC reanalysis 2 | Optimal BGT | 343,212 | 2.89 | Reference |
Sotatercept plus optimal BGT | 4,014,896 | 13.71 | 339,277 | |
CDA-AMC base case (1 plus 2) | Optimal BGT | 343,212 | 2.87 | Reference |
Sotatercept plus optimal BGT | 2,230,362 | 7.41 | 415,782 | |
CDA-AMC base case (1 plus 2) (probabilistic) | Optimal BGT | 351,248 | 3.01 | Reference |
Sotatercept plus optimal BGT | 2,138,127 | 7.10 | 436,796 |
BGT = background therapy; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor base case and the CDA-AMC base-case analysis (Table 7). The results of both analyses suggest that with a 100% price reduction for sotatercept (i.e., $0 cost), the ICER for sotatercept would be $61,002 per QALY gained (based on the sponsor’s analysis) or $55,062 per QALY gained (based on the CDA-AMC analysis) compared to optimal BGT alone. This is because of the drug acquisition cost of optimal BGT and the extended life expectancy (i.e., longer duration of treatment) for those treated with sotatercept. As such, when reducing the price of sotatercept by 100%, the drug acquisition cost of optimal BGT in the sotatercept plus optimal BGT arm of the model remains higher than the BGT cost for those taking optimal BGT alone.
Table 7: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost per mg ($) | ICERs for sotatercept plus optimal BGT vs. optimal BGT ($/QALY) | |
|---|---|---|---|
Price reduction | Sponsor base casea | CDA-AMC reanalysis | |
No price reduction | 194 | 348,810 | 436,796 |
10% | 174 | 320,029 | 398,623 |
20% | 155 | 291,248 | 360,449 |
30% | 136 | 262,467 | 322,276 |
40% | 116 | 233,687 | 284,102 |
50% | 97 | 204,906 | 245,929 |
60% | 77 | 176,125 | 207,756 |
70% | 58 | 147,344 | 169,582 |
80% | 39 | 118,564 | 131,409 |
90% | 19 | 89,783 | 93,235 |
100% | 0 | 61,002 | 55,062 |
BGT = background therapy; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aCalculated using the probabilistic results of the sponsor’s corrected base-case analysis.
Although based on the CDA-AMC base case there is no price reduction upon which sotatercept would be considered cost-effective at a threshold of $50,000 per QALY gained, it is acknowledged that a 100% price reduction could not be expected to be practically implemented by decision-makers. With a 95% price reduction for sotatercept, the ICER for sotatercept plus optimal BGT compared to optimal BGT alone would be $75,393 per QALY gained (in the sponsor’s analysis) and $74,149 per QALY gained (in the CDA-AMC reanalysis).
CDA-AMC explored a scenario in which costs of treatments included in optimal BGT were lower than their public list prices to estimate the price reduction for which sotatercept would be considered cost-effective at a threshold of $50,000 per QALY gained. In a hypothetical scenario, assuming that all treatments included in optimal BGT were reduced by 50%, a price reduction for sotatercept of approximately 95% would be necessary for it to be considered cost-effective at a threshold of $50,000 per QALY gained.
Issues for Consideration
Input received from drug plans and clinicians as part of this review noted that a clearly defined definition of optimal BGT will be necessary to support the potential implementation of sotatercept in jurisdictions in Canada. Drug plans specifically noted that it is unclear how many public drug plans currently permit triple therapy for patients with PAH and that the proposed reimbursement criteria for sotatercept position the drug as part of a triple or quadruple therapy regimen. The STELLAR trial population included patients on monotherapy, double therapy, and triple therapy at study entry; however, the submitted model and efficacy data do not differentiate by number of background PAH therapies. As such, the impact of varying definitions of optimal BGT on the cost-effectiveness analysis and budget impact analysis (BIA) is uncertain.
In the submitted economic model, the sponsor assumed there was no direct comparator treatment for sotatercept. In current clinical practice, based on the 2022 European Society of Cardiology–European Respiratory Society guidelines, selexipag is considered as a potential add-on therapy for patients receiving dual therapy with endothelin receptor antagonists or phosphodiesterase type 5 inhibitors who present at intermediate-low risk of death.16 However, the clinical experts consulted for this review indicated that for patients who are not meeting treatment targets on dual therapy, sotatercept may provide an alternative to selexipag when considering escalating to triple therapy. In this case, selexipag may be considered a comparator to sotatercept. The cost-effectiveness of sotatercept compared to selexipag for patients requiring escalation to triple therapy has not been assessed.
Overall Conclusions
The CDA-AMC clinical review reported that, based on the results of the STELLAR trial, sotatercept significantly improves 6MWD and increases the proportion of patients with PAH showing improvements in the multicomponent composite end point (i.e., 6MWD, NT-proBNP level, and WHO FC, which are used to define risk status in the economic model), compared to placebo, at 24 weeks. The clinical reviewers also noted that sotatercept likely leads to improvements in health-related quality of life measurements compared to placebo; however, the magnitude of these effects remains uncertain due to imprecision of the results.
CDA-AMC was able to address some limitations associated with the sponsor’s analysis, including the way in which the treatment effect of sotatercept was included in the model and the population from which all-cause mortality was extrapolated. Based on the CDA-AMC base-case analysis, sotatercept plus optimal BGT was associated with 4.09 incremental QALYs at an additional cost of $1,786,879, resulting in an ICER of $436,796 per QALY gained compared to optimal BGT alone.
The incremental cost is driven by direct treatment costs (sotatercept and optimal BGT), which make up 96% of the total costs associated with treatment with sotatercept plus optimal BGT. The drug acquisition cost of sotatercept is $199,249 per patient in the first year and $202,155 per patient in subsequent years (based on a weight of 67.5 kg to 88.9 kg), resulting in a lifetime cost of $1,561,624 per patient. The incremental drug acquisition cost of optimal BGT was estimated to be $207,243 per patient compared to those treated with optimal BGT alone. Relative to those who do not receive sotatercept, patients who receive sotatercept plus optimal BGT are expected to incur $207,243 in additional drug costs. The incremental cost of optimal BGT was driven by the 4.31 additional life-years for those treated with sotatercept plus optimal BGT because these patients are expected to remain on treatment for longer.
If the prices of all treatments included in optimal BGT were 50% lower than their public list prices, a price reduction for sotatercept of approximately 95% would be necessary for it to be considered cost-effective at a threshold of $50,000 per QALY gained (reflecting an annual drug cost of $7,970 and $8,086 in the first and subsequent years of treatment with sotatercept, respectively). With a 95% price reduction for sotatercept (and no change to the cost of BGT), the ICER for sotatercept plus optimal BGT compared to optimal BGT alone would be $75,393 per QALY gained (in the sponsor’s analysis) and $74,149 per QALY gained (in the CDA-AMC reanalysis).
CDA-AMC was unable to resolve uncertainty in the duration of treatment effect for sotatercept; as such, the CDA-AMC analysis is predicated on a persistent treatment benefit observed from week 12 to week 24 in the STELLAR trial over the 30-year time horizon. The potential value of sotatercept is dependent on patients realizing the uncertain survival benefit compared to optimal BGT alone (4.31 life-years). If these health outcomes are not realized, the ICER for sotatercept plus optimal BGT compared to optimal BGT alone will likely be higher than predicted by the CDA-AMC analysis.
References
1.Pharmacoeconomic evaluation [internal sponsor’s report]. In: sotatercept for injection, 45 mg for subcutaneous injection, 60 mg for subcutaneous injection. Kirkland (QC): Merck Canada Inc; 2024 Apr 8. 2024.
2.Sotatercept for injection; 45 mg for subcutaneous injection, 60 mg for subcutaneous injection [product monograph]. Kirkland (QC): Merck Canada Inc.; 2024.
3.The Socioeconomic Burden of Pulmonary Arterial Hypertension in Canada [sponsor supplied reference]. Vancouver (BC): Pulmonary Hypertension Association of Canada; 2023.
4.Rosenkranz S, Pausch C, Coghlan JG, et al. Risk stratification and response to therapy in patients with pulmonary arterial hypertension and comorbidities: A COMPERA analysis. J Heart Lung Transplant. 2023;42(1):102-114. PubMed
5.Statistics Canada. Table 13-10-0114-01: Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island [sponsor supplied reference]. Ottawa (ON): Government of Canada: https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310011401. .
6.Bernstein EJ, Bathon JM, Lederer DJ. Survival of adults with systemic autoimmune rheumatic diseases and pulmonary arterial hypertension after lung transplantation. Rheumatology (United Kingdom). 2018;57(5):831-834. PubMed
7.Xie F, Pullenayegum E, Gaebel K, et al. A Time Trade-off-derived Value Set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. PubMed
8.Davies EW, Llewellyn S, Beaudet A, Kosmas CE, Gin-Sing W, Doll HA. Elicitation of health state utilities associated with the mode of administration of drugs acting on the prostacyclin pathway in pulmonary arterial hypertension. Patient Prefer Adherence. 2018;12:1079-1088. PubMed
9.Di Tanna GL, Urbich M, Wirtz HS, et al. Health State Utilities of Patients with Heart Failure: A Systematic Literature Review. PharmacoEconomics. 2021;39(2):211-229. PubMed
10.Sullivan PW, Ghushchyan V. Preference-Based EQ-5D index scores for chronic conditions in the United States. Med Decis Making. 2006;26(4):410-420. PubMed
11.Hagiwara Y, Shiroiwa T, Shimozuma K, et al. Impact of Adverse Events on Health Utility and Health-Related Quality of Life in Patients Receiving First-Line Chemotherapy for Metastatic Breast Cancer: Results from the SELECT BC Study. Pharmacoeconomics. 2018;36(2):215-223. PubMed
12.Dufour R, Pruett J, Hu N, et al. Healthcare resource utilization and costs for patients with pulmonary arterial hypertension: real-world documentation of functional class. J Med Econ. 2017;20(11):1178-1186. PubMed
13.Drugs for Pulmonary Arterial Hypertension: Comparative Efficacy, Safety, and Cost-Effectiveness — Recommendations Report [sponsor supplied reference]. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2015. CADTH Therapeutic Review Report.
14.Alberta Interactive Health Databranch Grouper CMG+ 160 (Heart or Lung Transplant)- 2019 - Typical cases [sponsor supplied reference]. 2019; http://www.ahw.gov.ab.ca/IHDA_Retrieval/redirectToURL.do?cat=201&subCat=487. Accessed by sponsor.
15.Kendzerska T, Nickerson JW, Hsu AT, et al. End-of-life care in individuals with respiratory diseases: a population study comparing the dying experience between those with chronic obstructive pulmonary disease and lung cancer. Int J Chron Obstruct Pulmon Dis. 2019;14:1691-1701. PubMed
16.Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023;61(1).
17.Zelt JGE, Sugarman J, Weatherald J, et al. Mortality trends in pulmonary arterial hypertension in Canada: a temporal analysis of survival per ESC/ERS guideline era. Eur Respir J. 2022;59(6). PubMed
18.Boucly A, Weatherald J, Savale L, et al. External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. European Respiratory Journal. 2022;59(6). PubMed
19.Common Drug Review Pharmacoeconomic Review Report: macitentan (Opsumit - Actelion Pharmaceuticals Canada Inc.) [sponsor supplied reference]. Ottawa (ON): CADTH; 2015: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0364_Opsumit_PE_Report_e.pdf. Accessed 17 June 2024.
20.Common Drug Review Pharmacoeconomic Review Report: selexipeg (Uptravi - Actelion Pharmaceuticals Canada Inc.) [sponsor supplied reference]. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0482_Uptravi_PE_Report.pdf.
21.Guidance document for the costing of health care resources in the Canadian setting: second edition. Ottawa (ON): Canada’s Drug Agency; 2016: https://www.cadth.ca/guidance-document-costing-health-care-resources-canadian-setting. Accessed 17 June 2024.
22.Saskatchewan Drug Plan: search formulary [sponsor supplied reference]. Regina (SK): Government of Saskatchewan; 2024: https://formulary.drugplan.ehealthsask.ca/SearchFormulary.
23.Budget Impact Analysis [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: sotatercept for injection, 45 mg for subcutaneous injection, 60 mg for subcutaneous inection. Kirkland (QC)): Merck Canada Inc; 2024 Apr 8. 2024.
24.Canadian Pulmonary Hypertension Registry. Canadian Pulmonary Hypertension Registry Annual Report [sponsor supplied reference]. Vancouver (BC): Pulmonary Hypertension Association of Canada; 2023.
25.Understanding the Gap: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage [sponsor supplied reference]. Ottawa (ON): The Conference Board of Canada; 2017.
26.Understanding the Gap 2.0: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage [sponsor supplied reference]. Ottawa (ON): The Conference Board of Canada; 2022.
27.Vaid HM, Camacho X, Granton JT, et al. The Characteristics of Treated Pulmonary Arterial Hypertension Patients in Ontario. Can Respir J. 2016;2016:6279250. PubMed
28.Statistics Canada. Projected population, by projection scenario, age and sex, as of July 1 (x 1,000); Table: 17-10-0057-01. Projection scenario M3: medium-growth [sponsor supplied reference]. Ottawa (ON): Government of Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501.
29.Indigenous Services Canada. First Nations and Inuit Health Branch Non-Insured Health Benefits Program: Annual Report 2021/2022 [sponsor supplied reference]. Ottawa (ON): Government of Canada; 2023: https://sac-isc.gc.ca/eng/1683039690813/1683039973755.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Pulmonary Arterial Hypertension
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Sotatercept (Winrevair) | 50 mg/mL | 45 mg single-use vial for subcutaneous injection (1 or 2 vial kits) 60 mg single-use vial for subcutaneous injection (1 or 2 vial kits) | 8,717.1503a 11,622.8670a | Loading dose of 0.3 mg/kg, followed by 0.7 mg/kg administered every 3 weeks | First year: 505.51 Subsequent years: 553.47 | First year: 199,249b Subsequent years: 202,155b |
CDA-AMC = Canada’s Drug Agency.
Note: As sotatercept is an add-on therapy, the cost of background therapies is the same regardless of treatment with sotatercept. Background treatments include phosphodiesterase type 5 inhibitors (sildenafil citrate and tadalafil), endothelin receptor antagonists (bosentan, ambrisentan, and macitentan), and prostacyclin analogues (epoprostenol, treprostinil, and selexipag). These treatments may be taken in combination, and will vary by patient. Annual costs of these treatments may range from $3,246 (annual cost of sildenafil citrate) to $57,051 (annual cost of epoprostenol IV infusion).22
aSponsor’s submitted price.
bThe annual cost of sotatercept assumes a mean patient weight of 70 kg and assumes wastage of excess medication.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Sotatercept plus optimal BGT | Optimal BGT |
|---|---|---|
Discounted LYs | ||
Total | 18.83 | 5.63 |
By health state or data source | ||
Low risk | 1.50 | 1.27 |
Intermediate-low risk | 0.43 | 0.92 |
Intermediate-high risk | 0.34 | 1.61 |
High risk | 0.01 | 0.05 |
Post lung or heart transplant | 18.83 | 5.63 |
Discounted QALYs | ||
Total | 16.25 | 4.19 |
By health state or data source | ||
Low risk | 14.74 | 1.60 |
Intermediate-low risk | 1.20 | 1.01 |
Intermediate-high risk | 0.31 | 0.67 |
High risk | 0.22 | 1.03 |
Post lung or heart transplant | 0.01 | 0.04 |
QALY gain or loss due to administration | 0.10 | 0.02 |
QALY loss due to hospitalization | –0.02 | –0.09 |
QALY loss due to adverse events | –0.30 | –0.08 |
Discounted costs ($) | ||
Total | 4,574,961 | 479,749 |
Acquisition, sotatercept | 3,507,754 | 0 |
Acquisition, optimal BGT | 946,745 | 392,094 |
Administration | 1,679 | 823 |
Routine care costs | 97,685 | 35,342 |
Hospitalization costs | 4,049 | 15,327 |
Transplant, terminal, and AE costs | 17,049 | 36,164 |
AE = adverse event; BGT = background therapy; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 11: Disaggregated Summary of CDA-AMC Economic Evaluation Results
Parameter | Sotatercept plus optimal BGT | Optimal BGT |
|---|---|---|
Discounted LYs | ||
Total | 8.39 | 4.08 |
By health state or data source | ||
Low risk | 6.60 | 1.14 |
Intermediate-low risk | 1.48 | 1.08 |
Intermediate-high risk | 0.28 | 0.81 |
High risk | 0.02 | 1.01 |
Post lung or heart transplant | 0.00 | 0.03 |
Discounted QALYs | ||
Total | 7.10 | 3.01 |
By health state or data source | ||
Low risk | 5.87 | 1.01 |
Intermediate-low risk | 1.18 | 0.86 |
Intermediate-high risk | 0.21 | 0.60 |
High risk | 0.01 | 0.65 |
Post lung or heart transplant | 0.00 | 0.03 |
QALY gain or loss due to administration | 0.04 | –0.01 |
QALY loss due to hospitalization | –0.08 | –0.07 |
QALY loss due to adverse events | –0.14 | –0.06 |
Discounted costs ($) | ||
Total | 2,138,127 | 351,248 |
Acquisition, sotatercept | 1,561,624 | 0 |
Acquisition, optimal BGT | 484,329 | 277,086 |
Administration | 792 | 589 |
Routine care costs | 44,448 | 25,746 |
Hospitalization costs | 13,512 | 11,086 |
Transplant, terminal, and AE costs | 33,422 | 36,740 |
AE = adverse event; BGT = background therapy; CDA-AMC = Canada’s Drug Agency; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 12: Results of CDA-AMC Scenario Assuming a 50% Lower Price of Optimal BGT
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. Optimal BGT alone ($/QALY) |
|---|---|---|---|---|---|
Optimal BGT | 212,589 | Reference | 3.00 | Reference | Reference |
Sotatercept plus optimal BGT | 1,893,131 | 1,680,542 | 7.09 | 4.09 | 410,907 |
BGT = background therapy; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: In this scenario, assuming that the public list prices of all treatments included in optimal BGT are reduced by 50%, a price reduction of approximately 95% for sotatercept would be required for it to be considered cost-effective at a threshold of $50,000 per QALY gained.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA to estimate the incremental 3-year budget impact of adding sotatercept to optimal BGT for PAH management in patients who are not at low risk.23 The BIA was undertaken from the perspective of a Canadian public payer over a 3-year time horizon using an epidemiological approach. The sponsor compared a reference scenario in which patients were treated with the current standard of care (i.e., optimal BGT) to a new drug scenario in which sotatercept was reimbursed for use in combination with BGT.
Data for the model were obtained from various sources including published literature,3,17,24-27 the sponsor’s internal data, and assumption. Key inputs to the BIA are documented in Table 14.
Key assumptions included:
The sponsor used prevalence-based approach to estimate the eligible population size.
To estimate the total PAH prevalence rate, the sponsor relied on data extracted from the Canadian Pulmonary Hypertension Registry annual report of 2022 to 2023.24 Furthermore, the sponsor hypothesized that 1,250 patients with PAH were absent from the registry. Consequently, the model considered a total of 2,497 patients with PAH, leading to an estimated PAH prevalence rate of 7.8 per 100,000 population.
Patients surveyed in the 2023 study on the socioeconomic burden of PAH in Canada were used to estimate the proportion of patients on PAH therapy (86.6%).3
The model uses the assumption that 80% of patients would be receiving treatment with optimal BGT.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
25,806,100 / 26,224,114 / 26,631,153 | |
PAH prevalence24 | 7.80 per 100,000 |
Percentage of patients with PAH not at low-risk status17 | 73.6% |
Proportion of patients who are treated3 | 86.6% |
Proportion on optimal BGTa | 80.0% |
Proportion of patients with PAH < 65 years old27 | 50.61% |
62.82% | |
Proportion of patients with PAH ≥ 65 years old27 | 49.39% |
98.56% | |
Number of patients eligible for drug under review | 827 / 841 / 854 |
Market uptake (3 years) | |
Uptake (reference scenario) Optimal BGT alone | 100% / 100% / 100% |
Uptake (new drug scenario) Sotatercept Optimal BGT | 40% / 60% / 80% 60% / 40% / 20% |
Cost of treatment (per patient, per annum) | |
Sotatercept Optimal BGT: Bosentan (Oral) Ambrisentan (Oral) Macitentan (Oral) Sildenafil citrate (Oral) Tadalafil (Oral) Selexipag (Oral) Epoprostenol (IV) Treprostinil (IV) | $186,523.24 $11,720.65 $11,422.54 $48,233.09 $3,245.61 $7,394.71 $51,518.15 $57,051.18 $195,669.64 |
BGT = background therapy; PAH = pulmonary arterial hypertension.
aEstimate is based on sponsor assumption.
Summary of the Sponsor’s BIA Results
The sponsor estimated that reimbursing sotatercept for the treatment of PAH would result in incremental costs of $38,782,232 in year 1; $82,425,708 in year 2; and $115,650,464 in year 3, for total costs of $236,858,404 over the 3-year projection period.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The eligible population is uncertain. Drug plan input and feedback received from clinical experts consulted by CDA-AMC noted that in clinical practice in Canada the definition of optimal BGT is uncertain and may vary across clinical practice. The clinical experts consulted for this review indicated that patients who have an absolute contraindication to or who have not tolerated treatment with the components of triple combination therapy would be considered for sotatercept in clinical practice. Additionally, patients with a contraindication or who cannot tolerate other drug classes while on a single therapy or on dual therapy should also be considered for receiving sotatercept. In the absence of a clear definition of optimal BGT, there is uncertainty with regards to how many patients may be deemed eligible for treatment with sotatercept.
In the sponsor’s estimate of the eligible population, they assumed that 86.6% of patients with PAH who are not low risk were being treated and that 80% of those patients’ treatment would be considered optimal BGT. These assumptions resulted in an estimate of 854 eligible patients in year 3 of the analysis. Clinical experts consulted by CDA-AMC indicated that of patients with PAH who are not low risk, it is likely that a higher proportion would be on some treatment, and that there remains a challenge in estimating the proportion of those whose treatment would be considered optimal. As a result of these uncertainties, there may be a range of number of eligible patients for treatment with sotatercept depending on how optimal BGT is defined.
We performed a scenario analysis assuming that 90% of patients with PAH who are not low risk and are being treated are on optimal BGT, thus increasing the eligible population size. This analysis estimated that there would be 960 eligible patients in year 3 of the analysis.
The budget impact of patients starting treatment with sotatercept is not fully captured. The sponsor used a prevalence-based model to estimate the budget impact of sotatercept. In their model, the sponsor assumed that patients would gradually initiate treatment with sotatercept in the new drug scenario such that a quarter of patients started treatment every 3 months. With regards to treatment uptake, the sponsor assumed that 40%, 60%, and 80% of eligible patients would receive treatment with sotatercept in year 1, year 2, and year 3, respectively. In a prevalence-based model, the market shares represent the proportion of patients who are receiving treatment at any point throughout the year, and as such, already accounts for changes in treatment over the course of a year. Additionally, clinician input received by CDA-AMC for this review noted that while reaching a market uptake of 80% in year 3 is likely reasonable, that they expect that uptake will be faster than assumed by the sponsor. Given that sotatercept would be recommended for a prevalent population (i.e., not newly diagnosed patients, given that patients need to be receiving optimal BGT before initiating treatment with sotatercept) and the patient need for additional therapies, it is likely that patients will start sotatercept soon after its public listing in the new drug scenario. As a result of the sponsor’s gradual entry of patients, the treatment costs of sotatercept are underestimated.
In reanalysis, we assumed that there was no gradual entry for patients to initiate treatment with sotatercept.
In a scenario analysis, we assumed that the market uptake of sotatercept would be 70%, 75%, and 80% in years 1, 2, and 3, respectively.
The cost of treprostinil was incorrectly estimated. In the submitted model, the sponsor incorrectly used the cost per 1 mg of treprostinil ($45 per mg) to estimate the cost of a 200 mg vial, resulting in a cost per vial of $9,000. The public list price of a 20 mL vial (that contains a 200 mg dose) is $450 per vial.22
We changed the price of treprostinil to $450 per 20mL vial (10 mg / mL strength) as a correction of the sponsor’s base case.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analysis by removing the gradual entry of patients in the budget impact model.
Table 15: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
1. Cost of treprostinil per vial | $9,000 | $450 |
Changes to derive the CDA-AMC base case | ||
1. Gradual entry of patients in the new drug scenario. | Gradual entry of patients in the new drug scenario. | No gradual entry of patients in the new drug scenario. |
CDA-AMC base case | Reanalysis 1 | |
CDA-AMC = Canada’s Drug Agency.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17. The CDA-AMC reanalysis suggests that reimbursing sotatercept would be associated with an incremental cost of $62,051,571 in year 1; $94,650,189 in year 2; and $128,250,629 in year 3, for a 3-year budgetary impact of $284,952,390. In the CDA-AMC reanalysis, there were a total of 859 patients eligible for sotatercept in year 3, of whom 688 were estimated to receive treatment with sotatercept.
Table 16: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 236,858,404 |
CDA-AMC reanalysis 1 and base case | 284,952,390 |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 17):
Assuming that 90% of patients with PAH who are not low risk and are receiving treatment are on optimal BGT.
Assuming that the market uptake of sotatercept is 70%, 75%, and 80% in years 1, 2, and 3, respectively.
Assuming that the price of sotatercept is reduced by 95%, and the price of treatments included in optimal BGT are all reduced by 50%.
Results of the CDA-AMC scenario analyses demonstrate that the estimated budget impact is sensitive to changes in the eligible population and the market uptake of sotatercept. The scenario analysis assuming more patients were being treated with optimal BGT estimated a 3-year budget impact of $320,571,438, a 12% increase from the CDA-AMC base case. The scenario that assumed a faster uptake of sotatercept estimated that the 3-year budget impact of sotatercept of $355,153,616, a 22% increase from the CDA-AMC base case.
Table 17: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 68,751,085 | 69,931,354 | 71,113,063 | 72,268,437 | 213,312,854 |
New drug | 68,751,085 | 108,713,586 | 153,538,770 | 187,918,901 | 450,171,257 | |
Budget impact | 0 | 38,782,232 | 82,425,708 | 115,650,464 | 236,858,404 | |
Submitted base case, corrected | Reference | 48,536,438 | 49,369,677 | 50,203,932 | 51,019,595 | 150,593,204 |
New drug | 48,536,438 | 88,151,909 | 132,629,639 | 166,670,059 | 387,451,607 | |
Budget impact | 0 | 38,782,232 | 82,425,708 | 115,650,464 | 236,858,404 | |
CDA-AMC base case | Reference | 48,536,438 | 49,369,677 | 50,203,932 | 51,019,595 | 150,593,204 |
New drug | 48,536,438 | 111,421,248 | 144,854,121 | 179,270,224 | 435,545,593 | |
Budget impact | 0 | 62,051,571 | 94,650,189 | 128,250,629 | 284,952,390 | |
CDA-AMC scenario analysis 1: 90% patients on optimal BGT | Reference | 54,603,493 | 55,540,886 | 56,479,423 | 57,397,045 | 169,417,354 |
New drug | 54,603,493 | 125,348,904 | 162,960,886 | 201,679,002 | 489,988,792 | |
Budget impact | 0 | 69,808,018 | 106,481,463 | 144,281,958 | 320,571,438 | |
CDA-AMC scenario analysis 2: Market uptake of sotatercept | Reference | 48,536,438 | 49,369,677 | 50,203,932 | 51,019,595 | 150,593,204 |
New drug | 48,536,438 | 157,959,926 | 168,516,668 | 179,270,224 | 505,746,819 | |
Budget impact | 0 | 108,590,250 | 118,312,737 | 128,250,629 | 355,153,616 | |
CDA-AMC scenario analysis 3: 95% price reduction of sotatercept, and 50% price reduction of optimal BGT | Reference | 24,268,219 | 24,684,838 | 25,101,966 | 25,509,798 | 75,296,602 |
New drug | 24,268,219 | 27,787,417 | 29,834,475 | 31,922,329 | 89,544,221 | |
Budget impact | 0 | 3,102,579 | 4,732,509 | 6,412,531 | 14,247,619 |
BIA = budget impact analysis; BGT = background therapy; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.