Drugs, Health Technologies, Health Systems
Reimbursement Review
Eplontersen (Wainua)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Polyneuropathy in hereditary transthyretin-mediated amyloidosis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AIC
Akaike information criterion
ANCOVA
analysis of covariance
ASO
antisense oligonucleotide
ALT
alanine aminotransferase
AST
aspartate aminotransferase
CDA-AMC
Canada’s Drug Agency
CDEC
Canadian Drug Expert Committee
CI
confidence interval
CIR
copy increments from reference
CKD-EPI
Chronic Kidney Disease Epidemiology Collaboration
CM
cardiomyopathy
COMPASS
Composite Autonomic Symptom Score
DCO
data cut-off
eGFRcreat-cys
estimated glomerular filtration rate calculated from both serum creatinine and serum cystatin C
ESS
effective sample size
FAP
familial amyloid polyneuropathy
FAS
full analysis set
GalNAc
N-Acetylgalactosamine
GRADE
Grading of Recommendations Assessment, Development and Evaluation
hATTR
hereditary transthyretin-mediated amyloidosis
hATTR-PN
hereditary transthyretin-mediated amyloidosis polyneuropathy
HRdb
heart rate while deep breathing
HRQoL
health-related quality of life
IPD
individual patient data
ITC
indirect treatment comparison
J2R
jump to reference
LOCF
last observation carried forward
LSM
least squares mean
MAIC
matching-adjusted indirect comparison
MAR
missing at random
mBMI
modified body mass index
MedDRA
Medical Dictionary for Regulatory Activities
MMRM
mixed model for repeated measures
mNIS + 7
modified Neuropathy Impairment Score + 7
mRNA
messenger ribonucleic acid
mTCNS
modified Toronto Clinical Neuropathy Score
NCS
nerve conduction study
NIS
Neuropathy Impairment Score
NMA
network meta-analysis
NMD4C
Neuromuscular Disease Network for Canada
Norfolk QoL-DN
Norfolk Quality of Life Questionnaire–Diabetic Neuropathy
NSC
Neuropathy Symptom and Change
ONLS
Overall Neuropathy Limitation Scale
PCS
physical component summary
PN
polyneuropathy
PND
polyneuropathy disability
PPS
per-protocol set
QoL
quality of life
R-ODS
Rasch-built Overall Disability Scale
RCT
randomized controlled trial
SAE
serious adverse event
SAP
statistical analysis plan
SC
subcutaneous
SD
standard deviation
SF-36
Short Form (36) Health Survey
SLR
systematic literature review
STC
simulated treatment comparison
TAC
Transthyretin Amyloidosis Canada
TEAE
treatment-emergent adverse event
TTR
transthyretin
ULN
upper limit of normal
UPCR
urine protein-to-creatinine ratio
wtATTR
wild-type transthyretin-mediated amyloidosis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Eplontersen, 56 mg/mL, 45 mg single-dose prefilled pen for SC injection |
Sponsor | AstraZeneca Canada |
Indication | For the treatment of polyneuropathy associated with stage 1 or stage 2 hATTR in adults |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | June 19, 2024 |
Recommended dose | 45 mg eplontersen SC once per month |
hATTR = hereditary transthyretin-mediated amyloidosis; NOC = Notice of Compliance; SC = subcutaneous.
Introduction
Transthyretin (TTR) amyloidosis is a rare, systemic, life-threatening disease resulting from the deposition of amyloid in multiple tissues. TTR amyloidosis has 2 main forms: hereditary transthyretin-mediated amyloidosis (hATTR), and wild-type transthyretin-mediated amyloidosis (wtATTR). Both conditions are characterized by the abnormal deposition of TTR protein in various organs, leading to organ dysfunction. The hereditary condition (hATTR) is caused by an autosomal dominant mutation in the TTR gene that leads to the production of unstable TTR proteins. Produced primarily in the liver, these are prone to misfolding and amyloid deposition. Normally, the TTR protein exists as a tetramer; however, in hATTR, a mutation destabilizes the tetrameric protein structure, causing it to break down into unstable monomers and TTR fragments. Accumulation of misfolded amyloid fragments in a range of organ systems causes a variety of motor, sensory, and autonomic neuropathies leading to progressive muscle weakness and disability, pain, wasting, gastrointestinal dysfunction, and other autonomic symptoms, such as orthostatic hypotension.1 The peripheral nervous system and cardiac system are heavily affected, leading to 2 of the primary manifestations of the disease: polyneuropathy (PN) and cardiomyopathy (CM).2-4
Clinically, hATTR often progresses rapidly and leads to worsening sensorimotor neuropathy, a condition that damages the patient's sensory and motor nerves, escalating disability over time. Beyond sensorimotor neuropathy, the disease can also instigate a progressive autonomic neuropathy that affects the nerves controlling the body's automatic functions, including digestion, leading to gastrointestinal impairment, weight loss, and cachexia.3 In the clinical setting, hereditary transthyretin amyloidosis polyneuropathy (hATTR-PN) is assessed and classified using 2 key staging systems: the polyneuropathy disability (PND) score and the familial amyloid polyneuropathy (FAP) staging system (developed by Coutinho).3 hATTR-PN can be characterized as early onset (i.e., in patients aged less than 50 years) or late onset (i.e., in patients aged 50 years or older); there is significant worldwide variability regarding age of onset.2 The life expectancy of patients with hATTR-PN ranges from 10 years to 15 years following the development of symptoms.4 The median survival from the time of diagnosis is 4.7 years.5
hATTR-PN is an ultra-rare disease affecting approximately 10,000 individuals worldwide, though the condition may be underdiagnosed.1,3,4 The clinical experts consulted for this review noted that misdiagnosis is common because neuropathy can be attributed to many other diseases. The highest prevalences of hATTR-PN have been observed in northern Portugal and northern Sweden (where it has been observed to be as high as 50 cases per 100,000 inhabitants).4,6 There is a lack of published Canadian prevalence estimates.
Diagnosis of hATTR-PN should include gene sequencing to identify TTR variants and amyloid detection with tissue biopsy or bone scintigraphy scans.7 According to the 2019 consensus recommendation, the minimum criteria to establish the diagnosis of symptomatic hATTR include: “at least one quantified or objective symptom or sign definitively related to the onset of symptomatic hATTR; or at least one probably related symptom plus one abnormal definitive or confirmed test result; or 2 abnormal definitive or confirmed test results in the absence of clinical symptoms.”8 The list of tests and investigations for the follow-up of TTR mutation carriers includes clinical evaluation, neurophysiology assessment, biomarker measurement, and cardiac evaluation.
Two primary treatments have been authorized for market use in Canada for managing hATTR-PN: patisiran and inotersen.9,10 The 2022 Canadian guidelines recommend the use of patisiran and inotersen as first-line treatments. Recently, vutrisiran also received a recommendation for reimbursement with conditions by Canada’s Drug Agency (CDA-AMC) for the treatment of stage 1 or stage 2 PN in adult patients with hATTR.11
Eplontersen is administered at a dose of 45 mg (56 mg/mL) through subcutaneous (SC) injection using a prefilled pen. Eplontersen is an N-Acetylgalactosamine (GalNAc) conjugated antisense oligonucleotide (ASO) that selectively binds eplontersen to the TTR messenger ribonucleic acid (mRNA) in hepatocytes, causing the degradation of TTR mRNA. This prevents the synthesis of TTR protein in the liver, resulting in significant reductions in the levels of TTR protein in circulation.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of eplontersen 45 mg SC injection for the treatment of adult patients with hATTR-PN.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the call for input for this submission and from clinical experts consulted for the purpose of this review.
Patient Input
One patient group provided input for this submission: Transthyretin Amyloidosis Canada (TAC), a not-for-profit organization that supports individuals living with all forms of transthyretin amyloidosis, including hATTR and wtATTR, through community support, research, and education. Qualitative and quantitative information was gathered from a 23-question online survey of 30 patients as well as from 12 one-on-one interviews and a round table discussion (sample size not reported). Across sources, input was gathered from a total of 51 patients and caregivers across Canada. All respondents were older than 65 years of age.
All therapies approved by Health Canada are publicly reimbursed to varying degrees in different provinces. Additionally, TAC noted that all therapies have undesirable side effects as well as prohibitive costs and administration schedules. As such, the patient group input highlighted the need for additional treatment options as well as treatments that offer more convenient modes of administration or dosing schedules. Additionally, treatments that improve quality of life (QoL) were important for patients with hATTR.
The patient group input highlighted that currently available treatments have benefits and side effects. TAC noted that not every therapy has equal efficacy in all patients. As such, allowing patients and physicians access to different treatment options, particularly in a rare, multisystem disease such as hATTR, is paramount in ensuring that no patient is left behind. Among 30 patient and caregiver survey respondents, 83% cited travel for appointments and/or infusions as highly or somewhat intrusive. Other challenges reported by both patients and caregivers included costs associated with travel and parking, costs of medications, and time taken away from work and other activities. It was also noted that decreased hospital admission is an important outcome of treatment to patients, given that many patients are older and have frail immune systems. As a result, 80% felt that at-home administration was an important attribute for a new therapy because it could result in greater freedom, less reliance on infusion networks and clinic visits, and fewer missed workdays.
Patients and caregivers highlighted that losses of autonomy and independence have the greatest impacts on QoL. Of the 30 survey respondents, 67% emphasized that hATTR affected their ability to maintain a career, forcing them to stop work, retire early, or scale back to fewer than 15 hours of work per week. Additionally, 80% of respondents felt that hATTR had a significant or somewhat significant impact on their ability to travel. In all qualitative interviews, patients expressed that hATTR had a significant impact on their ability to maintain their social lives, indicating that their identity is entwined with their disease, partially due to the need to plan their lives around medical and infusion appointments.
None of the patients had experience with eplontersen. However, during the qualitative interviews and in the feedback obtained during the 2-hour round table meeting, all respondents mentioned the void that eplontersen can fill as the only therapy that may be self-administered.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The clinical experts stated that currently, there are no therapies available that can reverse neuropathy in hATTR. The primary aim of existing treatments in hATTR-PN is to decelerate the progression of the disease, which ultimately leads to loss of physical functioning and reduced QoL. Currently available disease-modifying therapies have some important limitations and carry risks of significant morbidity and mortality, including the potential for severe adverse events (AEs) with inotersen and a burdensome infusion schedule with patisiran. With all therapies, there is also a risk of vitamin A deficiency, leading to the need for supplementation to prevent vision loss. As such, the clinical experts highlighted the need for additional therapies with better efficacy, safety profiles, and convenience. They also mentioned the potential to switch to more tolerable therapies in patients with hATTR-PN. Even though some treatments are administered subcutaneously, offering convenience, current treatments still require health care assistance during administration; thus, the clinical experts noted that an ideal treatment could be self-administered or administered by caregivers, whether orally or with the use of an autoinjector.
Patients with hATTR-PN in Canada receive 1 of inotersen, patisiran, or vutrisiran (which recently received a recommendation to reimburse with conditions from the Canadian Drug Expert Committee [CDEC]). These are considered the standard of care in Canada. According to the clinical experts consulted by for this review, eplontersen would be used similarly to other first-line treatment options (i.e., inotersen, patisiran, or vutrisiran), and the clinical decision between eplontersen and other available disease-modifying therapies would be based on AE profile and convenience. The clinical experts also noted that patients should be able to switch between approved treatments based on tolerance and/or convenience, and that failure of other disease-modifying drugs would not be a prerequisite. The clinical experts noted that there is no evidence to support combining disease-modifying treatments; combining TTR silencers is unlikely to provide much additional clinical benefit, though the theoretical rationale of combining TTR silencers and TTR stabilizers could be explored. The clinical experts highlighted that existing patients are likely to have tried pharmacotherapies such as inotersen, patisiran, or tafamidis, or to have undergone liver transplant. They noted that eplontersen may be favoured over the newly available vutrisiran, given its less frequent dosing requirements. Overall, the experts highlighted that, based on the mechanism of action, eplontersen may offer improvements in efficacy, safety, and patient convenience that could position it favourably against existing treatments.
According to the clinical experts, the patients most suitable for eplontersen treatment are those with confirmed neuropathy and a pathogenic mutation in the TTR gene, confirmed by genetic testing. The clinical experts noted that accessibility of genetic testing in Canada has improved significantly, with initiatives by pharmaceutical companies and provincial labs now offering free testing that includes methods such as saliva or cheek swabs in addition to traditional blood tests. (However, awareness of genetic testing is inconsistent across Canada.) The experts noted that the ideal patients for treatment with eplontersen are those who mirror the participants in the relevant clinical trials: specifically, adults with confirmed neuropathy determined through diagnostic assessments, such as nerve conduction studies or evaluations of small fibres to accurately ascertain the presence and extent of neuropathy. Patients with confirmed neuropathy and TTR mutation who experience rapid progression of neuropathy are generally most in need of intervention and should be offered treatment as soon as possible. The clinical experts noted that these patients are most likely to experience a noticeable benefit from eplontersen treatment because they typically exhibit more pronounced symptoms. Additionally, the clinical experts noted that eplontersen may prove particularly advantageous for patients who have not adequately responded to other treatments, or for those seeking a therapy with a less burdensome administration schedule, which can significantly impact QoL. Conversely, patients with very advanced neuropathy (e.g., those who are bedbound) may not have much clinical benefit from a neuropathy perspective and are traditionally excluded from trials.
The overarching goal of hATTR-PN treatment is to stabilize disease progression. Any improvement in symptoms would also be considered a sign of successful treatment. Other critical outcomes of hATTR-PN treatment include improvements in mortality and serious complications requiring hospitalization.
The outcome measures used in hATTR trials are generally not feasible for use in clinical practice. Clinically, patients are assessed through neurologic examination. Some centres may use patient-reported outcomes to follow patients, but this is not standardized across Canada. Nerve conduction studies and small-fibre assessments to evaluate large and small-fibre function may be conducted. Other nonstandardized assessments include autonomic function tests (such as the Composite Autonomic Symptom Score [COMPASS] scale) to gauge patient experiences and neuropathy measures (such as the Toronto Clinical Neuropathy Scale, the Overall Neuropathy Limitation Scale [ONLS], and the Rasch-built Overall Disability Scale [R-ODS]) to track neuropathy severity. Beyond these clinical and diagnostic measures, emphasis is placed on the patient's overall functioning and QoL; these are gauged through comprehensive clinical evaluation, clinical history, and discussions about daily activities.
According to the clinical experts, the primary reasons for discontinuation of treatment are severe AEs. Objective disease progression (e.g., upper and lower extremity functional deterioration) at a rate similar to the natural history of hATTR, despite treatment, may also be considered a reason for discontinuation.
According to the clinical experts consulted for this review, PN, complex and advanced large- and small-fibre neuropathies, and autonomic neuropathies are diagnosed by a neurologist with training in neuromuscular medicine and experience with similar biologic therapies used in other neuromuscular disorders. Given the rarity of hATTR, neuromuscular neurologists or neurologists with experience treating hATTR would be required to prescribe and monitor treatment and follow-up. The clinical experts highlighted that ideally, specialized care would be administered in a hospital or clinic equipped with the necessary capabilities and resources to comprehensively manage all facets of advanced neuropathy, including associated cardiac and autonomic symptoms.
Clinician Group Input
One clinician group provided input for this review: the Neuromuscular Disease Network for Canada (NMD4C). The NMD4C is a pan-Canadian network that brings together the country’s leading clinical, scientific, technical, and patient expertise to improve care, research, and collaboration in neuromuscular disease. In total, 7 clinicians with experience in treating hATTR-PN who have also contributed to published Canadian guidelines for hATTR-PN provided input for this submission.
The treatment goals highlighted by the NMD4C were consistent with those noted by the clinical experts consulted for this review and included the prevention of disease progression, decreased morbidity and mortality, fewer hospital visits, and enhanced QoL. However, the clinician group and the clinical experts consulted for this review emphasized the need for options that offer improved efficacy and tolerability as well as greater convenience and adherence.
Given that the clinical manifestations of hATTR occur after significant build-up of amyloid has occurred in the body, the NMD4C emphasized that the earlier a therapy is initiated, the better outcomes are. Patients who present with several different disease manifestations, mainly PN and CM, are most in need of intervention, given the significant morbidity and mortality and reduced QoL. The NMD4C stated that patients in the early stages of disease (i.e., stage 1 or 2 PN) will demonstrate a better response to treatment. The NMD4C and the clinical experts consulted for this review agreed that it is unknown whether the failure of 1 treatment indicates that a different treatment would also fail; thus, having multiple treatment options available is important. Although eplontersen is not the first drug to be approved, the NMD4C noted that, in its opinion, eplontersen may have advantages over inotersen, not only regarding side effects but also in terms of effectiveness.
In line with the clinical experts consulted for this review, the NMD4C highlighted that the outcome measures often used in trials for hATTR are not feasible for use in clinical practice due to the extensive testing and training required for their use. To determine whether a patient is responding to treatment in clinical practice, the NMD4C and the clinical experts consulted for this review follow the recommendations described in Canadian treatment guidelines and use neurologic history, neurologic examination, and nerve conduction studies. The NMD4C noted that clinically meaningful responses to treatment would be stability or slower progression of symptoms, greater preservation of functional abilities, and improved survival.
Both the NMD4C and the clinical experts consulted for this review agreed that disease progression may be a reason for discontinuation. In clinical practice, outcomes such as loss of walking ability despite intensive treatment could help identify those who are not responding to treatment.
Both the clinician group and the clinical experts consulted for this review stated that neurology specialists and neuromuscular specialists would be required to prescribe and monitor treatment and follow-up. The most appropriate settings include specialized and multidisciplinary tertiary centres with lines of referral to cardiologists. The NMD4C also highlighted that hospital outpatient neuromuscular clinics, community-based neurologic clinics, and referral lines to neuromuscular expertise may be appropriate.
Drug Program Input
The drug programs identified the following jurisdictional implementation issues: relevant comparators, considerations for initiation of therapy, considerations for continuation or renewal of therapy, considerations for discontinuation of therapy, considerations for prescribing of therapy, generalizability, care provision issues, and system and economic issues. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
Only 1 study was included in this review. The NEURO-TTRansform study was an 85-week, phase III, multicentre, randomized, open-label study evaluating the efficacy and safety of eplontersen in patients with hATTR. In total, 168 patients were randomized 6 to 1 to receive 45 mg eplontersen SC once every 4 weeks (n = 144) or 300 mg inotersen SC once per week for up to 34 weeks and were then switched to eplontersen SC once every 4 weeks from week 37 to week 81 (n = 24); the latter group is hereafter referred to as the inotersen-eplontersen group. The NEURO-TTRansform study was conducted at 40 sites in 15 countries including North America, Europe, South America, Australasia, and Asia; 2 sites in Canada (British Columbia and Ontario) enrolled a total of 3 patients. There were 3 analysis time points in the NEURO-TTRansform study: week 35 (interim analysis), week 65 or 66 (final analysis), and week 85 (end-of-treatment analysis). Three coprimary end points were used at the final analysis: the percentage change from baseline in serum TTR, the change from baseline in modified Neuropathy Impairment Score + 7 (mNIS + 7), and the change from baseline in Norfolk Quality of Life Questionnaire–Diabetic Neuropathy (Norfolk QoL-DN).12
The NEURO-TTRansform study also included an external control group using the placebo arm (n = 60) from the NEURO-TTR study. The NEURO-TTR study was a phase III, double-blind, placebo-controlled study that compared the efficacy and safety of inotersen 300 mg SC injection weekly with placebo in patients with stage 1 or 2 hATTR-PN. Eligibility criteria for the NEURO-TTR and NEURO-TTRansform studies were identical. Analyses were adjusted for select baseline covariates using propensity scores.12
Baseline characteristics of the eplontersen group in the NEURO-TTRansform study and the external placebo group in the NEURO-TTR study were generally well-balanced. In the NEURO-TTRansform study, the mean age was 53.0 years (standard deviation [SD] = 15.0 years) in the eplontersen group. The mean age in the placebo group of the NEURO-TTR study was 59.5 years (SD = 14.1 years). In the NEURO-TTRansform study, most patients had stage 1 hATTR-PN (115 patients [79.9%]) and Val30Met (V30M) mutations (85 patients [59.0%]), while in the NEURO-TTR study placebo group, 42 patients (70.0%) had stage 1 hATTR-PN, and 33 patients (55.0%) had V30M mutations. In the NEURO-TTRansform study eplontersen group at baseline, the mean serum TTR was 0.2 g/L (SD = 0.1 g/L); the mean mNIS + 7 composite score was 81.2 (SD = 43.4); and the mean Norfolk QoL-DN total score was 44.1 (SD = 26.6). In the NEURO-TTR study placebo group, the mean serum TTR was 0.2 g/L (SD = 0.04 g/L). Patients in the NEURO-TTR study may have had less severe disease than patients enrolled in the NEURO-TTRansform study, given that the mean mNIS + 7 composite score in that study was 74.8 (SD = 39.0) and the mean Norfolk QoL-DN total score was 48.7 (SD = 26.8).12
Efficacy Results
Change From Baseline in mNIS + 7 Composite Score
The change from baseline in mNIS + 7 composite score was a coprimary end point of the NEURO-TTRansform study at the interim and final analyses. At week 35, the least squares mean (LSM) changes from baseline were 0.22 points (95% CI, –3.46 points to 3.90 points) for eplontersen and 9.23 points (95% CI, 5.54 points to 12.91 points) for external placebo, representing an LSM difference of –9.01 points (95% CI, –13.48 points to –4.54 points) in favour of eplontersen.13 At week 66, the LSM changes from baseline were 0.30 points (95% CI, –4.46 points to 5.06 points) for eplontersen and 25.06 points (95% CI, 20.23 points to 29.88 points) for external placebo, representing an LSM difference of –24.76 points (95% CI, –30.96 points to –18.56 points) in favour of eplontersen.12 At both time points, the reduction in mNIS + 7 scores corresponded to an improvement in the severity of neuropathy when treated with eplontersen.
Results for the prespecified sensitivity analyses were consistent at the interim and final analyses and with the primary analysis, with point estimates for the LSM differences ranging from █████ ██████ ██ █████ ██████ at week 35 and ██████ ██████ ██ ██████ points at week 65 ██ ██████ ██ ███████████ across sensitivity analyses.12,13
In general, the results of the subgroup analyses were consistent with those of the primary analysis at both analyses.12,13
For the exploratory analysis of the eplontersen and inotersen-eplontersen groups at week 35, the mean changes from baseline in mNIS + 7 composite score were –0.03 points (SD = 16.28 points) for eplontersen-treated patients (n = 137) and 4.06 points (SD = 13.39 points) following treatment with inotersen (n = 19).12
Change From Baseline in Norfolk QoL-DN Total Score
The change from baseline in Norfolk QoL-DN total score was a key secondary end point of the NEURO-TTRansform study at the week 35 interim analysis and a coprimary end point at the week 66 final analysis. At week 35, the LSM changes from baseline were –3.12 points (95% CI, –7.19 points to 0.96 points) for eplontersen and 8.67 points (95% CI, 4.53 points to 12.81 points) for external placebo, representing an LSM difference of –11.79 points (95% CI, –16.82 points to –6.76 points) in favour of eplontersen.13 At week 66, the LSM changes from baseline were –5.50 points (95% CI, –10.03 points to –0.96 points) for eplontersen and 14.24 points (95% CI, 9.51 points to 18.97 points) for external placebo, representing an LSM difference of –19.74 points (95% CI, –25.63 points to –13.84 points) in favour of eplontersen.12 At both time points, the reduction in Norfolk QoL-DN scores corresponded to an improvement in health-related quality of life (HRQoL) with eplontersen treatment.
Results for the sensitivity analyses were consistent with those of the primary analysis, with point estimates for the LSM differences ranging from █████ ██████ ██ ██████ ██████ at week 35 and –█████ ██████ ██ ██████ ██████ at week 65 ██ ██████ ██ ███████████ across sensitivity analyses.12,13
In general, the results of the subgroup analyses were consistent with those of the primary analysis at both analysis time points.12,13
For the exploratory analysis of the eplontersen and inotersen-eplontersen groups at week 35, the mean changes from baseline in Norfolk QoL-DN total scores were –4.79 points (SD = 16.51 points) for patients treated with eplontersen (n = 130) and –2.97 points (SD = 12.10 points) for patients treated with inotersen (n = 20).12
Change From Baseline in COMPASS-31
Change from baseline in COMPASS-31 at weeks 37 and 81 was an exploratory outcome of the NEURO-TTRansform study at the final analysis. COMPASS-31 was not assessed in the external placebo group. At baseline, the mean COMPASS-31 score in the eplontersen group was 19.4 points (SD = 11.26 points). The mean changes from baseline at weeks 37 and 81 were ████ ██████ ███ █ █████ and –2.6 points (SD = 7.52 points), respectively.12
Change From Baseline of R-ODS
Change from baseline in R-ODS at weeks 37 and 81 was an exploratory outcome of the NEURO-TTRansform study at the final analysis. ███ █████ ███ ███ ████████ ██ ███ ████████ ███████ ██████ At baseline, the mean R-ODS score in the eplontersen group was ████ ██████ ███ █ ███████ The mean changes from baseline at weeks 37 and 81 were ███ ██████ ███ █ █████ ███ ████ ██████ ███ █ ██████ respectively.12
Change From Baseline in Serum TTR
Change from baseline in serum TTR at week 35 and week 65 was a coprimary end point of the NEURO-TTRansform study. At the week 35 interim analysis, the LSM percentage change from baseline in serum TTR was –81.20% (95% confidence interval [CI], – 84.55% to –77.84%) with eplontersen compared to –14.76% (95% CI, –18.73% to –10.80%) for the external placebo group, representing an LSM difference of –66.43% (95% CI, –71.59%, –61.71%) in favour of eplontersen.13 At week 65, the LSM percentage changes from baseline in serum TTR concentration were –81.65% ████ ███ ███████ ██ ████████ in the eplontersen group and –11.24% ████ ███ ███████ ██ ███████ in the external placebo group, representing an LSM difference of –70.42% ████ ███ ██████ ██ ███████ in favour of eplontersen.12 At both time points, the LSM difference corresponded to a reduction in serum TTR (or improvement in TTR levels) for patients receiving eplontersen.
Results for all prespecified sensitivity analyses of change from baseline in serum TTR at week 65 were consistent with those of the primary analysis, as well as with week 35, with point estimates for the LSM differences in percentage reduction in serum TTR between eplontersen and external placebo ranging from ███████ ██ ███████.12,13
In general, results of the subgroup analyses were consistent at both analysis time points and with the primary analysis. The subgroup of █████████ ████████ ███ █ ███████ █████ LSM difference in percent reduction in serum TTR at week 35 ████████ ████ ███ ██████ ██ ████████ and week 65 ████████ ████ ███ ██████ ██ █████████ ██████ ███ ███████ █████ ████████ ███████████.
For the exploratory analysis of the groups receiving eplontersen and inotersen-eplontersen at week 35, the mean percentage changes from baseline in serum TTR were ███████ ███ █ ██████ for patients treated with eplontersen ██ █ ████ and ███████ ███ █ ██████ for patients treated with inotersen ██ █ ████.12
Change From Baseline in Neuropathy Symptom and Change Total Score
The change from baseline in Neuropathy Symptom and Change (NSC) total score was a secondary outcome of the NEURO-TTRansform study at the final analysis. At week 66, the LSM changes from baseline were –0.03 points (95% CI, –1.92 points to 1.86 points) in the eplontersen group and 8.2 points (95% CI, 6.24 points to 10.12 points) in the external placebo group, representing an LSM difference of –8.2 points (95% CI, –10.65 points to –5.76 points) in favour of eplontersen, corresponding to an improvement in neuropathy symptoms.12
Change From Baseline in PCS Score of the Short Form (36) Health Survey
The change from baseline in the physical component summary (PCS) score of the Short Form (36) Health Survey (SF-36) was a secondary outcome of the NEURO-TTRansform study at the final analysis. At week 65, the LSM changes from baseline were 0.85 points (95% CI, –0.711 points to 2.412 points) in the eplontersen group and –4.46 points (95% CI, –6.139 points to –2.770 points) in the external placebo group, representing an LSM difference of 5.31 points (95% CI, 3.195 points to 7.416 points) in favour of eplontersen, corresponding to an improvement in HRQoL with eplontersen.12
Change From Baseline in PND Score
The change from baseline in PND score was a secondary outcome of the NEURO-TTRansform study at the final analysis. At week 65, the LSM changes from baseline were ███ ██████ ████ ███ ████ ██ ████ in the eplontersen group and ███ ██████ ████ ███ ████ ████ in the external placebo group, representing an LSM difference of ████ ██████ ████ ███ ████ ██ █████
Hospitalizations
███ █████████ ██ ███ █████ ████████████████ ██ ███ ████████ ███ ██ ████████ ████ ██ ███ ██ ███████████ ███████ ██ ███ ████████████████ ██████ █ █████ ██ ██ ████████ ██ ███ ███████████ █████ ███ ██ ████████ ██ ███ ████████ ███████ █████ ████ ████████ ██ ███ ██ █████████ ██ ██████ █ ████████ ██████ ████ ████████████ ██ ███ ███████████ ██████ ███ ███ █ ███████ ██████ ███ ████████████ ██ ███ ████████ ███████ ██████ ███ ████ ███ ██ ████████
Harms Results
At the week 66 final analysis, at least 1 treatment-emergent adverse event (TEAE) was reported by 140 patients (97.2%) in the eplontersen group and by 60 patients (100%) in the external placebo group of the NEURO-TTR study. The most frequently reported TEAEs in the eplontersen group were COVID-19 (35 patients [24.3%]), urinary tract infection (24 patients [16.7%]), diarrhea (24 patients [16.7%]), vitamin A deficiency (17 patients [11.8%]), and nausea (16 patients [11.1%]). The most frequently reported TEAEs in the external placebo group were fall (13 patients [21.7%]), fatigue (12 patients [20.0%]), diarrhea (11 patients [18.3%]), urinary tract infection (10 patients [16.7%]), neuralgia (9 patients [15.0%]), pain in extremity, cough, asthenia, pain (8 patients [13.3%] each), nausea, headache (7 patients [11.7%] each), and nasopharyngitis, dizziness, constipation, thermal burn, hypoesthesia, and muscular weakness (6 patients [10.0%] each).12
Serious adverse events (SAEs) were reported in 21 patients (14.6%) in the eplontersen group and 12 patients (20.0%) in the external placebo group. The most-reported individual SAEs in the eplontersen group were ████████ ██ ███████ ████████ ██████████ ███████ ████████ ███ ███████ █████ █████████ ██ ██████ ██████ The most-reported SAEs in the placebo group included █████████ ███ █████ ████████ ██ ██████ ████
TEAEs leading to discontinuation of treatment occurred in 6 patients (4.2%) in the eplontersen group and 2 patients (3.3%) in the external placebo group.12
Up to week 66, 2 patients in the eplontersen group had died due to arrhythmia and cerebral hemorrhage. At week 85, 1 additional patient in the eplontersen group had died due to myocardial infarction. At week 66, no patients in the external placebo group had died.12
Notable harms included in this review consisted of thrombocytopenia and ocular AEs potentially related to vitamin A deficiency. In the eplontersen group, ██ ████████ ███████ ███ ██████ ███ ███████████ ███████ ██ ███████ █ ███████████ ███ ██████████ ██ ████████ ████ ██████ ███ ██ ███ █████████ █████ ███ █ █████; however, given that investigators in the NEURO-TTR study were blinded to vitamin A levels (so as not to inadvertently become unblinded to treatment allocation), no vitamin A-related AEs were reported. The most common ocular AEs potentially related to ███████ █ ██████████ ████████ ███████ █ ██████████ ███ ████████ ███ ███████ ██████ ██ ███████. For thrombocytopenia, a total of 3 AEs (2.1%) were reported in the eplontersen group, and 1 AE (1.7%) was reported in the external placebo group at week 66.12
Critical Appraisal
The NEURO-TTRansform study was a randomized, open-label study that utilized an external placebo control from the NEURO-TTR study of inotersen compared to placebo. The choice to conduct a study using external control has implications for the overall strength and interpretability of the results. The design, study sites, eligibility criteria, and assessments of disease progression of the NEURO-TTRansform study and NEURO-TTR study were aligned for the purposes of this comparison. Because the NEURO-TTRansform study was an open-label study, there were increased risks of detection bias and performance bias, particularly for subjective outcomes, although the magnitude and direction of these biases remain unclear. Despite randomization, no statistical comparisons were conducted comparing eplontersen with the concurrent inotersen arm for randomized patients as the sponsor considered the large sample size that would have been required to be infeasible for this rare indication. As such, this comparison was only used for safety considerations. The baseline characteristics were generally well-balanced, with the exception of some baseline scores (i.e., the mNIS + 7 and Neuropathy Impairment Score [NIS] composite scores and PND scores), which were generally higher in the eplontersen group, suggesting a population with more severe neuropathy impairment compared to those in the inotersen-eplontersen group. In comparison to the external placebo group from the NEURO-TTR study, the NEURO-TTRansform study included younger patients, had a greater proportion of Asian and Black patients, included more patients with FAP and/or Coutinho stage 1 disease (but fewer with stage 2), had a longer period of time from diagnosis to enrolment, included fewer patients with CM, and included more patients who had previous experience with tafamidis or diflunisal. The impact of these differences on the results remains unknown, but results of the subgroup analyses for these variables were generally consistent with those of the primary results. The NEURO-TTRansform study met its coprimary and key secondary end points at the interim analysis; therefore, further statistical testing was not conducted on these end points at the final analysis. Results at the final analysis were consistent with those at the interim analysis across all study end points, despite the switch from analysis of covariance (ANCOVA) at the interim analysis to a mixed model for repeated measures (MMRM) at the final analysis for the end points of change from baseline in mNIS + 7 and change from baseline in Norfolk QoL-DN. Given the use of the external placebo control, the MMRM for each end point was adjusted by propensity score weights for each patient. It was unclear how the covariates for adjustment were selected or whether all relevant prognostic factors and effect modifiers were considered. Additionally, it was not possible to account for differences in known unmeasured or unknown confounders. As such, there is a risk of bias due to residual baseline confounding of unknown magnitude and direction.
The NEURO-TTRansform trial was an international trial conducted in 15 countries, including Canada (which enrolled 3 patients). Patients eligible for the NEURO-TTRansform study were similar to those eligible for the NEURO-TTR study, given the identical eligibility criteria. Similar to other trials of treatments for hATTR-PN, the NEURO-TTRansform study enrolled adult patients with stage 1 or stage 2 PN with hATTR. Patients receiving current or prior TTR-lowering treatment were excluded from the NEURO-TTRansform trial. As such, the efficacy of eplontersen in patients who have previously received patisiran or vutrisiran is unknown.
The clinical experts consulted for this review also highlighted that there is limited overlap in outcomes between clinical trials for hATTR-PN and clinical practice, emphasizing that many of the measures included as outcomes in the NEURO-TTRansform trial are not routinely used in Canadian clinical practice. The primary end point of the NEURO-TTRansform study was the percentage change from baseline in serum TTR. The abnormal aggregation of TTR is a fundamental manifestation of hATTR-PN; however, the clinical experts noted that serum TTR levels are not measured routinely in clinical practice. Although demonstrative of treatment effect and biological plausibility, serum TTR reduction has not been identified as a validated surrogate outcome for efficacy in hATTR-PN; thus, its use as a primary end point may have been inappropriate. The mNIS + 7 and Norfolk QoL-DN measures have limited application in clinical practice in Canada, given the complexity and time-consuming nature of these tools. The clinical experts noted that COMPASS-31, R-ODS, and other tools (ONLS and the modified Toronto Clinical Neuropathy Score [mTCNS]) are more frequently used, given their simplicity; however, this is not standardized across Canada. COMPASS-31 and R-ODS were exploratory outcomes of the NEURO-TTRansform study, but did not include a comparison to placebo, and should be considered supportive only. The ONLS and mTCNS were not reported in the NEURO-TTRansform study.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development and Evaluations (GRADE) was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the deliberations of the CDA-AMC expert committee, and a final certainty rating was determined, as outlined by the GRADE Working Group. For the comparison of eplontersen to placebo, which leveraged an external placebo group from the NEURO-TTR study, the certainty of evidence started at low, acknowledging the nonrandomized design, risk of selection bias, and residual baseline confounding. The clinical review team assessed the submitted evidence for study limitations (i.e., internal validity or risk of bias), indirectness, imprecision of effects, and publication bias. In the absence of a comparator (i.e., single-arm design), appraisals of the results for COMPASS-31 and R-ODS started at very low certainty, with no opportunity for rating up.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: serum TTR levels, mNIS + 7 composite score, Norfolk QoL-DN total score, COMPASS-31, R-ODS, ocular AEs potentially related to vitamin A deficiency, and thrombocytopenia.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and its location relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of any effect for serum TTR, mNIS + 7 composite score, and Norfolk QoL-DN total score.
Table 2 presents the GRADE summary of findings for eplontersen versus placebo for outcomes in the pivotal NEURO-TTRansform trial.
Table 2: Summary of Findings for Eplontersen Versus Placebo (NEURO-TTR Study) for Patients With hATTR-PN in the NEURO-TTRansform Trial
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certaintya | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo group (NEURO-TTR study) | Eplontersen group (NEURO-TTRansform trial) | Difference | |||||
Neuropathy symptoms and neurologic function | |||||||
mNIS + 7 composite score (–22.3 points [best] to 346.3 points [worst]) LSM change from baseline Follow-up: week 35 | 199 (1 nonrandomized study) | NA | 9.23 | 0.22 (–3.46 to 3.90) | –9.01 (–13.48 to –4.54) | Lowb,c,d | Eplontersen may result in lesser neurologic impairment based on the change from baseline in mNIS + 7 composite score compared to placebo. |
mNIS + 7 composite score (–22.3 points [best] to 346.3 points [worst]) LSM change from baseline Follow-up: week 66 | 180 (1 nonrandomized study) | NA | 25.06 | 0.30 (–4.46 to 5.06) | –24.76 (–30.96 to –18.56) | Lowb,d | Eplontersen may result in lesser neurologic impairment based on the change from baseline in mNIS + 7 composite score compared to placebo. |
COMPASS-31 score (0 points [best] to 100 points [worst]), mean (SD) change from baseline Follow-up: weeks 37 and 81 | 141 (1 single-arm study) | NA | NA | ████ ███ | NA | Very lowa,e | The evidence is uncertain about the effect of eplontersen on COMPASS-31 scores vs. any comparator. |
R-ODS score (0 points [worst] to 48 points [best]), mean (SD) change from baseline Follow-up: weeks 37 and 81 | 141 (1 single-arm study) | NA | NA | ████ ███ | NA | Very lowa,e | The evidence is uncertain about the effect of eplontersen on R-ODS scores vs. any comparator. |
Health-related quality of life | |||||||
Norfolk QoL-DN total score (–4 points [best] to 136 points [worst]), LSM change from baseline Follow-up: week 35 | 191 (1 nonrandomized study) | NA | 8.67 | –3.12 (–7.19 to 0.96) | –11.79 (–16.82 to –6.76) | Lowb,c,f | Eplontersen may result in better HRQoL, based on the change from baseline in Norfolk QoL-DN total score compared to placebo. |
Norfolk QoL-DN total score (–4 points [best] to 136 points [worst]), LSM change from baseline Follow-up: week 66 | 180 (1 nonrandomized study) | NA | 14.24 | –5.50 (–10.03 to –0.96) | –19.74 (–25.63 to –13.84) | Lowb,f | Eplontersen may result in better HRQoL, based on the change from baseline in Norfolk QoL-DN total score compared to placebo. |
Serum TTR | |||||||
Percentage change from baseline in serum TTR, LSM Follow-up: week 35 | 193 (1 nonrandomized study) | NA | –14.76 | –81.20 (–84.55 to –77.84) | –66.43 (–71.39 to –1.47) | Moderatec,g | Eplontersen likely results in an increase (improvement) in serum TTR levels compared with placebo. |
Percentage change from baseline in serum TTR, LSM Follow-up: week 65 | 186 (1 nonrandomized study) | NA | –11.24 | –81.65 (–84.82 to –78.48) | –70.42 (–75.17 to –65.66) | Moderateg | Eplontersen likely results in an increase (improvement) in serum TTR levels compared with placebo. |
Notable harms | |||||||
Ocular AEs potentially related to vitamin A deficiency Follow-up: week 66 | 204 (1 nonrandomized study) | NR | 150 per 1,000 | 271 per 1,000 (NR) | NR | Very lowb,h,i | The evidence is uncertain about the effect of eplontersen on ocular AEs compared with placebo. |
Thrombocytopenia Follow-up: week 66 | 204 (1 nonrandomized study) | NR | 17 per 1,000 | 21 per 1,000 (NR) | NR | Very lowb,h | The evidence is uncertain about the effect of eplontersen on thrombocytopenia compared with placebo. |
AE = adverse event; CI = confidence interval; COMPASS-31 = Composite Autonomic Symptom Score-31; hATTR-PN = hereditary transthyretin amyloidosis polyneuropathy; HRQoL = health-related quality of life; LSM = least squares mean; mNIS + 7 = modified Neuropathy Impairment Score + 7; NA = not applicable; Norfolk QoL-DN = Norfolk Quality of Life–Diabetic Neuropathy; NR = not reported; R-ODS = Rasch-built Overall Disability Score; SD = standard deviation; TTR = transthyretin; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThe NEURO-TTRansform trial used an external control (placebo group in the NEURO-TTR trial) for comparison with the eplontersen group. This observational comparison introduced potential for bias (resulting from confounding and selection bias), and the certainty of evidence was started at low. The clinical review team noted that the external placebo control was collected from a randomized, double-blind, placebo-controlled study (the NEURO-TTR study) that was conducted using the same eligibility criteria and disease assessments as the NEURO-TTRansform trial. However, risk of bias due to residual baseline confounding could not be ruled out. Results for the change from baseline in COMPASS-31 and R-ODS scores lacked a comparator and were started at very low certainty without the opportunity to be rated up.
bAlthough there is a risk of bias arising from the open-label nature of the study and the subjective nature of the outcome, the certainty of evidence was not rated down. Starting the certainty of evidence at low already acknowledges the serious risk of study limitations.
cPotential to be rated down 1 level for serious internal validity limitations, given that results at week 35 are based on an interim analysis. The review team assessed the interim analyses for the potential to overestimate treatment effects; however, it was concluded that the risk of serious study limitations is not a concern because the results of the interim analysis were aligned with the results of the final analysis. As such, the certainty was not rated down.
dImprecision was not rated down. Per the clinical experts consulted for this review, any stabilization or decrease in mNIS + 7 score from baseline is viewed positively; however, the clinical experts consulted for this review were unable to provide a threshold of clinically meaningful improvement. As such, the clinical review team used the null as the threshold. The lower bound of the 95% CIs excluded the null.
eEnd point was an exploratory outcome without statistical testing. The findings should be considered as supportive evidence.
fImprecision was not rated down. No threshold of clinical importance was provided by the clinical experts consulted for this review. As such, the null was used as a threshold. The lower bound of the 95% CIs exceeded the null.
gThe certainty of evidence was starting at low, acknowledging the potential for residual baseline confounding and selection bias as a result of the nonrandomized study design. The certainty of evidence was rated up by 1 level to account for the large effect size, which is biologically plausible and aligned with the mechanism of action of eplontersen. Note that serum TTR is considered a biomarker for efficacy of treatment in patients with hATTR-PN; however, the validity of its relationship as a surrogate for clinical outcomes has not been established.
hRated down 1 level for serious imprecision due to the low number of events and small sample size.
iThis outcome was not measured the same way in both trials. In the NEURO-TTR study, investigators were blinded to vitamin A levels so as not to inadvertently become unblinded to study drug allocation. As such, vitamin A-related AEs were not reported in this study.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023).12,13
Long-Term Extension Studies
One open-label extension study of patients with hATTR-PN who are continuing to receive eplontersen after week 85 in the NEURO-TTRansform trial is currently ongoing. No data were available at the time of this review.
Indirect Comparisons
Description of Studies
Given the lack of head-to-head studies comparing the efficacy and/or safety of eplontersen to other treatments available in Canada (i.e., vutrisiran, patisiran, and inotersen) for hATTR-PN, the sponsor submitted an indirect treatment comparison (ITC) to evaluate the comparative efficacy of eplontersen versus other medical therapies used for the treatment of patients with hATTR-PN.14
The sponsor conducted unanchored matching-adjusted indirect comparisons (MAIC) and simulated treatment comparisons (STCs) comparing eplontersen from the NEURO-TTRansform study to inotersen from the NEURO-TTR trial, patisiran from the APOLLO and HELIOS-A trials, and vutrisiran from the HELIOS-A trial for the outcomes of change from baseline in mNIS + 7, change from baseline in Norfolk QoL-DN, and percentage change from baseline in serum TTR.14
Efficacy Results
For change from baseline in mNIS + 7, there were no statistically significant differences detected between eplontersen and vutrisiran in the HELIOS-A trial, patisiran in the HELIOS-A trial, or inotersen in the NEURO-TTR trial. ███████ ███ ███ ██████████ ██ ███████████ ███ █████████ ████ ███ ██████ █████ ████ █████████████ ███████████ ██ ██████ ██ █████████ █████ ██████████ █████ █████ ████ ███ ████ ██ ████████ ██████████ ████ █████████ ███████ ██ █ ███████ ███████████ ██ ███████████ In the alternative models, there were no statistically significant differences detected between eplontersen and vutrisiran in the HELIOS-A trial or patisiran in the HELIOS-A trial, but there was a statistically significant improvement in the mNIS + 7 composite score compared to inotersen ████ █████ ████ ███ ██████ ██ ████████ ██ ████ ███ █████████ ██████ █████████ ████ ███ ██████ █████ ███ ████████ ████ ███████████ ████ ████ ████ ███ ████ ██ ███████
For the change from baseline in Norfolk QoL-DN, comparisons of eplontersen to vutrisiran in the HELIOS-A trial and to inotersen in the NEURO-TTR trial demonstrated a statistically significant improvement in Norfolk QoL-DN total score ████ ████████ ████ █████ ████ ███ ██████ ██ ██████ ███ ███ █████ ████ ███ ██████ ██ ███████ ██████████████ ██ ██████ ██ ████████████ ██████████ ███████ ████████████ ██ █████ ████ ████████████ ███████████ ██ ███████████ ██ █████████ ████ ███ ██████ ██ ████████ ██████ ███ ███ ███████████ █ █████████████ ███████████ ██████████ ██ ██████ ████ ████████ ██ ███████ ██████ █████ ██████ ███████ ███ ███ ███████████ ██████ ████ ██████████ ████ ███ █████████ ███████ ██████ ███ ███ ██████████ ██ ███████████ ███ █████████ ████ ███ ████████ ██████ ███████████ ███ ████████ ████ █████████ ████ █████ ████ ███ ██████ ██ ██████
For the change from baseline in serum TTR concentration, ███ ██████████ ██ ███████████ ███ █████████ ████ ███ ████████ █████ and eplontersen and inotersen in the NEURO-TTR trial demonstrated statistically significant reductions in serum TTR concentration ████ █████ ████ ███ █████ ██ ███████ ███ ███ █████ ████ ███ ██████ ██ ███████ ██████████████ in favour of eplontersen, which suggested that eplontersen results in greater reductions in serum TTR levels. However, there was no statistically significant difference detected between eplontersen and vutrisiran. Results for the alternative model were generally consistent with the reference model; however ██ █████████████ ███████████ ██████████ ███ ████████ ███ ███ ██████████ ██ ███████████ ███ █████████ ████ ███ ████████ █████ ████ █████ ████ ███ █████ ██ ███████. Percentage change from baseline in serum TTR concentration was not evaluated in the comparison of eplontersen and patisiran in the APOLLO trial.14
Critical Appraisal
The sponsor-submitted MAIC and STCs were informed by an adequately conducted systematic literature review (SLR) that included planned searches of multiple databases and standard screening and extraction methods. Risk-of-bias assessments of the included studies were conducted per the University of York Centre for Reviews and Dissemination criteria; however, the results of this quality assessment were not provided. Thus, the potential impact of study-level biases on the results of the MAICs and STCs could not be comprehensively judged.
In total, 4 trials evaluating eplontersen, vutrisiran, patisiran, and inotersen were identified for inclusion during the sponsor’s feasibility assessment. Given the heterogeneity observed, the lack of a common comparator across the included trials, and the unique design of 2 studies that included randomized reference arms (the NEURO-TTRansform study [inotersen arm] and HELIOS-A study [patisiran arm]), the sponsor concluded that MAIC and STC methods were most appropriate for comparing eplontersen and relevant comparators. Other sources of heterogeneity in the included studies were the baseline characteristics of age, proportion of patients who identify as white, proportion of patients with V30M mutation, proportion of patients with hATTR with CM, proportion of patients previously treated with tafamidis or diflunisal, proportion of patients with stage 1 and stage 2 disease, as well as differences in various outcome scores.
In the base-case (reference) models, comparisons of eplontersen to other treatments resulted in sample-size decreases of █████ ██ █████ across outcomes and treatments. These decreases were generally smaller in the alternative models, given that fewer variables were included in the adjustment. Given the reduction in effective sample size (ESS), there was likely considerable heterogeneity between studies among the variables included in the weighting process. Despite the substantial reduction in ESS for nearly all comparisons following the matching and adjustment, the populations in all MAIC and STC analyses were relatively balanced. Substantial reductions in ESS have implications for generalizability and the precision of effect estimates. A comprehensive list of prognostic factors and treatment-effect modifiers was included and — based on discussions with the clinical experts consulted for this review —considered relevant. However, it was noted that the exclusion of region as a factor may bias the results because there may be regional variation in health care access and treatment approaches that are unrelated to V30M.
Two versions of the mNIS + 7 were utilized in the analyses: mNIS + 7Ionis and mNIS + 7Alnylam composite scores. The mNIS + 7Ionis composite score from the NEURO-TTRansform study was rescored; however, the rescored versions are not validated for use and may not be appropriate, given that certain domains are not captured in the rescored version. Thus, any interpretation of the comparative results of the mNIS + 7 composite score should consider this limitation. Additionally, outcomes for the mNIS + 7 composite score and Norfolk QoL-DN total score were extrapolated to match the time points reported in the comparator trial, which may introduce uncertainty into the magnitude of any estimates of treatment effect. In the reference model, for the outcome of mNIS + 7, there was generally insufficient evidence to determine whether eplontersen or the comparator treatments were favoured, given the wide 95% CIs, which included the potential for stabilization of disease or disease progression. For the Norfolk QoL-DN and change from baseline in serum TTR outcomes, eplontersen was often favoured over other treatments; however, imprecision and uncertainty remained, given the wide 95% CIs.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Conclusions
hATTR-PN is a rare disease with a need for new, more convenient, safer treatments that slow disease progression and improve neurologic symptoms and HRQoL. One study was included in this review: the NEURO-TTRansform study, a phase III study evaluating the efficacy of eplontersen after 65 weeks compared to an external placebo in adult patients with genetic confirmation of hATTR-PN. Important features of the NEURO-TTRansform study design limit the certainty of evidence, notably the inclusion of an external placebo control and the open-label design, which increases the risk of baseline confounding, selection bias, and performance bias.
Key outcomes evaluated in the NEURO-TTRansform study were of limited applicability to clinical practice in Canada; the clinical experts consulted for this review highlighted that these are not used to evaluate treatment effect in routine clinical practice. The NEURO-TTRansform study demonstrated that eplontersen likely resulted in a clinically meaningful decrease in serum TTR levels; however, the clinical importance of this biomarker remains unknown. According to the clinical experts consulted for this review, disease stabilization is among the most important outcomes of treatment. In the NEURO-TTRansform study, neuropathy symptoms and neurologic function were measured using the mNIS + 7; the findings suggested that patients treated with eplontersen experience stabilization of disease, whereas those in the placebo group experience deterioration. Lastly, improved HRQoL was an outcome important to patients. In the NEURO-TTRansform study, the results suggested that patients treated with eplontersen may have clinically meaningful improvements in HRQoL (per the Norfolk QoL-DN) compared to placebo. Outcome measures that are clinically relevant to practice in Canada, including the COMPASS-31 and R-ODS, were consistent with the suggested stabilization from the coprimary end points, but were generally considered supportive of the overall effect of eplontersen because the results were noncomparative. There were few safety concerns with eplontersen relative to other treatments for hATTR-PN, including a lower frequency of thrombocytopenia AEs compared to the randomized inotersen group; however, there were more frequent ocular AEs related to vitamin A deficiency. The clinical experts noted that this was manageable in clinical practice.
There were important limitations in the conduct of the ITCs: the included studies varied in design, including outcome definitions and time of assessment. Additionally, there were notable differences in patient characteristics before adjustment, and the removal of patients in the weighting process substantially reduced the precision of treatment-effect estimates. Overall, the ITCs suggested that there was insufficient evidence to detect a difference between eplontersen and other treatments or to suggest that eplontersen may be favoured over some treatments. However, conclusions could not be drawn with any certainty, owing to methodological limitations and imprecision, as evidenced by wide 95% CIs.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of eplontersen 45 mg single-dose prefilled pen (56 mg/ML solution) for SC injection in the treatment of adult patients with PN associated with hATTR.
Disease Background
The contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CDA-AMC review team.
TTR amyloidosis is a rare, systemic, life-threatening disease resulting from the deposition of amyloid in multiple tissues. TTR amyloidosis has 2 main forms: hATTR and wtATTR. Both are characterized by the abnormal deposition of TTR protein in various organs, leading to organ dysfunction. Hereditary TTR-mediated amyloidosis is a genetic condition caused by an autosomal dominant mutation in the TTR gene. This mutation leads to the production of unstable TTR proteins — primarily in the liver — that are more prone to misfolding and amyloid deposition. In contrast, wtATTR occurs in the absence of TTR gene mutations.15,16 Normally, the TTR protein exists as a tetramer; however, in hATTR, a mutation destabilizes the tetrameric protein structure, causing it to break down into unstable monomers and TTR fragments. Accumulation of misfolded amyloid fragments in a range of organ systems causes a variety of motor, sensory, and autonomic neuropathies leading to progressive muscle weakness and disability, pain, wasting, gastrointestinal dysfunction, and other autonomic symptoms, such as orthostatic hypotension.1 The peripheral nervous system and cardiac system are heavily affected, leading to PN and CM, respectively. These are 2 of the primary manifestations of the disease.2-4
Clinically, hATTR often progresses rapidly and leads to worsening sensorimotor neuropathy, a condition that damages the patient's sensory and motor nerves, escalating their disability over time. Beyond sensorimotor neuropathy, the disease can also instigate a progressive autonomic neuropathy, which affects the nerves controlling the body's automatic functions, including digestion, leading to gastrointestinal impairment, weight loss, and cachexia.3 In the clinical setting, hATTR-PN is assessed and classified using 2 key staging systems: the PND score and FAP staging system (developed by Coutinho).3 Both systems classify disease progression on a categorical scale, ranging from symptom-free (PND 0 and FAP stage 0) to a complete lack of ambulation, where patients may require a wheelchair or be bedridden (PND IV and FAP stage 3). Hereditary TTR-mediated amyloidosis PN can be characterized as early onset (i.e., in patients aged less than 50 years) or late onset (i.e., in patients aged 50 years or older); there is significant worldwide variability regarding age of onset.2 The life expectancy of patients with hATTR-PN ranges from 10 years to 15 years after symptom development.4 The median survival from the time of diagnosis in hATTR-PN is 4.7 years.5
hATTR-PN is an ultra-rare disease. Although it affects approximately 10,000 individuals worldwide,3,4 there are endemic regions where prevalence is noticeably higher (i.e., Europe). The highest prevalences of hATTR-PN have been observed in northern Portugal and northern Sweden (where it is as high as 50 cases per 100,000 inhabitants);4,6 even so, the condition may be underdiagnosed.1,4 The clinical experts consulted for this review noted that misdiagnosis is common because neuropathy can be attributed to many other diseases. There is a lack of published Canadian prevalence estimates.
Diagnosis of hATTR-PN should include gene sequencing to identify TTR variants and amyloid detection with tissue biopsy or bone scintigraphy scans.7 According to the 2019 consensus recommendation, the minimum criteria to establish the diagnosis of symptomatic hATTR include: “at least one quantified or objective symptom or sign definitively related to the onset of symptomatic hATTR; or at least one probably related symptom plus one abnormal definitive or confirmed test result; or 2 abnormal definitive or confirmed test results in the absence of clinical symptoms.”8 The list of tests and investigations for the follow-up of TTR mutation carriers includes clinical evaluation, neurophysiology assessment, biomarker measurement, and cardiac evaluation. In some patients, hATTR manifests in the form of CM, which is characterized by the infiltration of TTR amyloid fibrils in myocardium, leading to CM and heart failure. Cardiac involvement manifestations include diastolic or — later in the disease course — systolic dysfunction, heart failure, palpitations, syncope, arrhythmia, heart block, and angina or infarction.17 Autonomic dysfunction and peripheral neuropathy are the main determinants of QoL, but cardiac involvement is the most important determinant of prognosis, with a median survival of 4 years to 5 years when cardiac amyloidosis is present.18
Standards of Therapy
The contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CDA-AMC review team.
The current treatment landscape for patients with hATTR in Canada is guided by accurate diagnosis and distinction of the disease manifestation (i.e., whether the symptomatic presentation is neuropathy, cardiac disease, or both). Two primary treatments have been authorized for market use in Canada for managing hATTR-PN: patisiran and inotersen.9,10 Recently, vutrisiran also received a recommendation for reimbursement with conditions by CDA-AMC for the treatment of stage 1 or stage 2 PN in adult patients with hATTR.11 Tafamidis (Vyndaqel) is indicated for use in patients with transthyretin-mediated amyloidosis who present primarily with CM.19
Historically, orthotopic liver transplant was employed as a therapeutic option, especially for a selective cohort of patients in the early stages of hATTR-PN. This procedure was essential because it eliminated variant TTR from circulating within the liver by replacing the native liver (responsible for the genetic defect leading to variant TTR production) with 1 free from the defect.20 However, due to the complications and the need for immunosuppression regimens as a result of organ transplant, combined with the evolution of therapies like patisiran and inotersen, there has been a marked decline in resorting to liver transplant over the past 2 decades, making orthotopic liver transplant increasingly obsolete.21
The 2022 Canadian guidelines recommend the use of both patisiran and inotersen as first-line treatments for managing hATTR-PN. The guidelines further emphasize a shift away from liver transplant as a primary intervention, citing potential perioperative complications and the ensuing need for continuous immunosuppression.21
For patients diagnosed with neuropathy, small-interfering ribonucleic acid or oligonucleoside therapies emerge as potent disease-modifying strategies. These treatments stabilize the otherwise persistent disease progression once neuropathy commences. The clinician group input highlighted the importance of having treatment options with diverse mechanisms of action to provide extra scope for the patient to respond to treatment. Furthermore, symptomatic treatments are commonly prescribed; these include cardiac medications, interventions for neuropathic pain, surgical solutions (such as for severe symptomatic compressive neuropathy; e.g., carpal tunnel syndrome), and management techniques for autonomic dysfunction, which often manifests prominently in patients with hATTR.
Drug Under Review
Eplontersen is administered at a dose of 45 mg (56 mg/mL) through SC injection using a prefilled pen. Before initiation, patients and/or caregivers are required to receive training on proper preparation and administration of eplontersen. The first injection administered by the patient or caregiver should be performed under the guidance of an appropriately qualified health care professional.
Eplontersen is a GalNAc conjugated 2′-O-2-methoxyethyl modified with a mixed backbone of phosphorothioate and phosphodiester internucleotide linkages. GalNAc conjugation enables targeted delivery of the ASO to hepatocytes. The selective binding of eplontersen to the TTR mRNA causes degradation of both mutant and normal TTR mRNA. This prevents the synthesis of TTR protein in the liver, resulting in significant reductions in the levels of mutated and wild-type TTR protein secreted by the liver into the circulation. TTR is a carrier protein for retinol binding protein 4, which is the principal carrier of vitamin A (retinol). Therefore, reduction in plasma TTR is expected to result in a reduction of plasma retinol levels to below the lower limit of normal.
The Health Canada indication and reimbursement request are for the treatment PN associated with stage 1 or stage 2 hATTR in adult patients. Eplontersen has not previously been reviewed by CDA-AMC. The key characteristics of eplontersen are summarized in Table 3 along with other treatments available for hATTR-PN.
Table 3: Key Characteristics of Eplontersen and Other Treatments for hATTR-PN
Characteristic | Eplontersen | Vutrisiran | Patisiran | Inotersen |
|---|---|---|---|---|
Mechanism of action | Antisense oligonucleotide silencer of TTR mRNA in the liver | siRNA-mediated degradation of TTR mRNA in the liver | siRNA-mediated degradation of TTR mRNA in the liver | Selective binding of inotersen to the TTR mRNA, causing the degradation of both mutant and wild-type TTR mRNA |
Indicationa | For the treatment of PN associated with stage 1 or stage 2 hATTR in adults | For the treatment of stage 1 or stage 2 PN in adult patients with hATTR | Treatment of PN in adult patients with hATTR | Treatment of stage 1 or 2 PN in adult patients with hATTR |
Route of administration | Subcutaneous | Subcutaneous | IV | Subcutaneous |
Recommended dose | 45 mg monthly | 25 mg every 3 months | 0.3 mg/kg, to a maximum dose of 30 mg once every 3 weeks | 284 mg inotersen (300 mg inotersen sodium) once weekly |
Serious adverse effects or safety issues | Reduced vitamin A levels Contraindications: severe hypersensitivity to the product | Reduced vitamin A levels Contraindications: severe hypersensitivity to the product | IRRs, reduced vitamin A levels Contraindications: severe hypersensitivity to the product | Thrombocytopenia, glomerulonephritis, reduced vitamin A levels Contraindicated in patients with hypersensitivity to the product, platelet count < 100 × 109/L, urine protein-to-creatinine ratio ≥ 113 mg/mmol, eGFR < 45 mL/min/1.73 m2, or severe liver impairment |
Other | Vitamin A supplementation is recommended | Must be administered by a health care professional Vitamin A supplementation is recommended | Must be administered by a health care professional in a supervised setting. Premedications (i.e., oral acetaminophen, IV corticosteroid, IV H1 blocker, and IV H2 blocker) are required to minimize the risk of IRRs. Vitamin A supplementation is recommended | Monitoring of platelet count is required every 2 weeks for platelet levels > 100 × 109/L Increased monitoring and dose adjustments are required for platelet levels < 100 × 109/L; drug discontinuation is required for platelet levels < 25 × 109/L Vitamin A supplementation is recommended |
eGFR = estimated glomerular filtration rate; H1 blocker = histamine-1 receptor antagonist; H2 blocker = histamine-2 receptor antagonist; hATTR = hereditary transthyretin-mediated amyloidosis; hATTR-PN = hereditary transthyretin amyloidosis polyneuropathy; IRR = infusion-related reaction; mRNA = messenger ribonucleic acid; PN = polyneuropathy; siRNA = small-interfering ribonucleic acid; TTR = transthyretin.
aHealth Canada–approved indication.
Sources: Product monographs for eplontersen,22 vutrisiran,23 patisiran,24 and inotersen.25
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient input received by CDA-AMC has been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
One patient group submitted input for this review: TAC, a not-for-profit organization that supports individuals living with all forms of TTR amyloidosis — including hATTR and wtATTR — through community support, research, and education. Qualitative and quantitative information was gathered from a 23-question online survey of 30 patients as well as from 12 one-on-one interviews and a round table discussion (sample size not reported). Across sources, input was gathered from a total of 51 patients and caregivers across Canada. All respondents were older than 65 years of age.
All therapies approved by Health Canada are publicly reimbursed to varying degrees in different provinces. Additionally, TAC noted that all therapies have undesirable side effects as well as prohibitive costs and administration schedules. As such, the patient group input highlighted the need for additional treatment options as well as treatments that offer more convenient modes of administration or dosing schedules. Additionally, treatments that improve QoL were important for patients with hATTR.
The patient group input highlighted that currently available treatments have benefits and side effects. TAC noted that not every therapy has equal efficacy in all patients. As such, allowing patients and physicians access to different treatment options, particularly in a rare, multisystem diseases such as hATTR, is paramount in ensuring that no patient is left behind. Among 30 patient and caregiver survey respondents, 83% cited travel for appointments and/or infusions as highly or somewhat invasive, with some of the reported challenges reported by both patients and caregivers including costs associated with travel and parking, costs of medications, and time taken away from work and other activities. It was also noted that decreased hospital admission is an important outcome of treatment to patients, given that many patients are older and have frail immune systems. As a result, 80% felt that at-home administration was an important attribute for a new therapy because it could result in greater freedom, less reliance on infusion networks and clinic visits, and fewer missed workdays.
The patient group highlighted that according to patients and caregivers, losses of autonomy and independence have the greatest impacts on QoL. Of the 30 survey respondents, 67% emphasized that hATTR affected their ability to maintain a career, forcing them to stop work, retire early, or scale back to fewer than 15 hours of work per week. Additionally, 80% of respondents felt that hATTR had a significant or somewhat significant impact on their ability to travel. In all qualitative interviews, patients expressed that hATTR had a significant impact on their ability to maintain their social lives, indicating that their identity is entwined with their disease, partially due to the need to plan their lives around medical and infusion appointments.
None of the patients had experience with eplontersen. However, during the qualitative interviews and in the feedback obtained during the 2-hour round table meeting, all respondents mentioned the void that eplontersen can fill as the only therapy that may be self-administered.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of hATTR.
Unmet Needs
The clinical experts consulted for this review stated that currently, there are no therapies available that can reverse neuropathy in hATTR. The primary aim of existing treatments is to decelerate the progression of the disease, which ultimately leads to loss of physical functioning and reduced QoL.
Currently available disease-modifying therapies have some important limitations and carry risks of significant morbidity and mortality. Inotersen requires frequent patient monitoring because it is associated with thrombocytopenia and glomerulonephritis, with the potential for severe AEs. Patisiran is generally better tolerated, but requires IV infusion every 3 weeks, which can be burdensome to patients. The clinical experts highlighted that once funding has become available for vutrisiran, it will likely replace patisiran due to its more favourable administration schedule, requiring SC injection only every 3 months (although currently, this must be done by a health care provider). The clinical experts also noted that the therapeutic efficacy of treatments varies for cardiac and neurologic symptoms. Furthermore, there is limited evidence regarding the impact of these therapies on functional outcomes and overall QoL, which are of critical importance to patients with hATTR-PN. With all therapies, there is also a risk of vitamin A deficiency and the need for supplementation to prevent vision loss. One clinical expert also noted that available disease-modifying treatments are ineffective in preventing leptomeningeal amyloidosis, which they considered a gap in therapy.
As such, there is a need for additional therapies with better efficacy and safety profiles as well as formulations that improve convenience. The potential for patients with hATTR-PN to switch to more tolerable therapies is also of interest. Despite some currently available treatments being administered subcutaneously, offering convenience, these still require health care assistance for administration; thus, the clinical experts noted that an ideal treatment would be 1 that patients could self-administer or that caregivers could administer, whether orally or with an autoinjector.
Place in Therapy
The contemporary approach to managing hATTR involves an accurate diagnosis and determining whether the disease manifests as neuropathy, cardiac issues, or both. Since the approval of disease-modifying therapy, patients with hATTR-PN in Canada receive 1 of either inotersen, patisiran, or vutrisiran (which recently received a recommendation to reimburse with conditions from CDEC). These are considered the standard of care in Canada, provided that patients have confirmed neuropathy, no previous liver transplant, and no severe neurologic impairment.
Other therapeutic options include diflunisal and tafamidis. Diflunisal is not widely used because it has only mild TTR-stabilizing properties with minimal clinical efficacy. Tafamidis is approved only for patients diagnosed with hATTR with cardiac dysfunction (i.e., CM) without PN. Patients may also require symptomatic treatment for neuropathic pain (e.g., gabapentin, pregabalin, duloxetine, tramadol, nortriptyline; many of these are used off-label) or surgical management for carpal tunnel syndrome or severe spinal stenosis. The clinical experts noted that liver transplant is also a treatment option in patients with severe, symptomatic hATTR-PN to decrease the production of mutated TTR protein. However, its use is becoming scarcer due to the availability of disease-modifying therapies that are associated with fewer surgical and immunosuppressive challenges and less morbidity overall.
According to the clinical experts consulted by CDA-AMC, eplontersen would be used similarly to other first-line treatment options (i.e., inotersen, patisiran, and vutrisiran). The clinical decision between eplontersen and other available disease-modifying therapies would be based on AE profile and convenience. The clinical experts also noted that patients should be able to switch between approved treatments based on tolerance and/or convenience, and that failure of other disease-modifying drugs would not be a prerequisite.
The clinical experts noted that there is no evidence to support combining disease-modifying treatments for hATTR because the mechanism of action is similar across therapies; combining TTR silencers is unlikely to provide much additional clinical benefit, though the theoretical rationale of combining TTR silencers and TTR stabilizers could be explored.
The clinical experts highlighted that existing patients are likely to have tried pharmacotherapies such as inotersen, patisiran, or tafamidis, or to have undergone liver transplant. The clinical experts noted that eplontersen may be favoured over the newly available vutrisiran, given its less frequent dosing requirements. Overall, the experts highlighted that, based on the mechanism of action, eplontersen may offer improvements in efficacy, safety, and patient convenience that could position it favourably against existing treatments.
Patient Population
According to the clinical experts, the patients most suitable for eplontersen treatment are those with confirmed neuropathy and a pathogenic mutation in the TTR gene (i.e., confirmed diagnosis of hATTR) confirmed by genetic testing. The clinical experts noted that accessibility of genetic testing in Canada has improved significantly, with initiatives by pharmaceutical companies and provincial labs now offering free testing that includes methods such as saliva or cheek swabs in addition to traditional blood tests. (However, awareness of genetic testing is inconsistent across Canada.) Enhancing the reliability and accessibility of these diagnostic assessments is essential to accurately identify candidates for eplontersen and prevent both underdiagnosis and overdiagnosis in the clinical setting.
The ideal patients for treatment with eplontersen are those who mirror the participants in the relevant clinical trials: specifically, adults with confirmed neuropathy determined through reliable and objective assessments. These assessments should ideally involve diagnostic tools, such as nerve conduction studies or evaluations of small fibres, to accurately ascertain the presence and extent of neuropathy. Patients with confirmed neuropathy and TTR mutation who experience rapid progression of neuropathy are generally most in need of intervention and should be offered treatment as soon as possible. The clinical experts noted that these patients are most likely to experience a noticeable benefit from eplontersen treatment because they typically exhibit more pronounced symptoms. Additionally, the clinical experts noted that eplontersen may prove particularly advantageous for patients who have not adequately responded to other treatments, or for those seeking a therapy with a less burdensome administration schedule, which can significantly impact QoL.
Patients with very advanced neuropathy (e.g., those who are bedbound) may not derive much clinical benefit (from a neuropathy perspective) and are traditionally excluded from trials.
Assessing the Response Treatment
The overarching goal of hATTR-PN treatment is to stabilize disease progression. Improvements in neuropathy outcomes and autonomic symptoms provide essential insights into the disease's progression from a neuromuscular perspective; thus, any improvement in symptoms (e.g., dizziness, bladder and/or bowel dysfunction, or abnormalities in sweating) would be considered a sign of successful treatment. Regular assessment and monitoring of autonomic symptoms are essential to determine the overall impact of therapy and guide clinical decisions about its continuation or modification. Other critical outcomes of hATTR-PN treatment include improvements in mortality and serious complications requiring hospitalization.
One clinical expert highlighted that the outcome measures used in hATTR trials are generally not feasible for use in clinical practice. Clinically, patients are assessed through neurologic examination. Some centres may use patient-reported outcomes to follow patients, but this is not standardized across Canada. In some Canadian centres, the progression of hATTR is monitored using both objective measures and patient-reported outcomes. Nerve conduction studies and small-fibre assessments, utilizing tools like laser Doppler imaging and quantitative sensory thresholds to evaluate large- and small-fibre function, may be conducted. Other nonstandardized assessments include autonomic function tests (such as the COMPASS scale) to gauge patient experiences and neuropathy measures (such as the mTCNS, the ONLS, and the R-ODS) to track neuropathy severity. Beyond these clinical and diagnostic measures, emphasis is placed on the patient's overall functioning and QoL; these are gauged through comprehensive clinical evaluation (including gait observation), clinical history, and discussions about daily activities.
Discontinuing Treatment
According to the clinical experts, the primary reasons for discontinuation of treatment are severe AEs, which can include injection-site reactions, liver and/or renal function abnormalities, hematological abnormalities (anemia or thrombocytopenia), infusion-related reactions, or fatigue. Objective disease progression (e.g., upper and lower extremity functional deterioration [patient loses the ability to walk independently]) at a rate similar to the natural history of hATTR, despite treatment, may also be considered a reason for discontinuation. The clinical experts also noted that additional factors, such as the patient's tolerance for and willingness to continue with the therapy, are critical in this decision-making process.
Prescribing Considerations
According to the clinical experts consulted for this review, PN, complex and advanced large- and small-fibre neuropathies, and autonomic neuropathies are diagnosed by a neurologist, often with training in neuromuscular medicine and experience with similar biologic therapies used in other neuromuscular disorders.
Given the rarity of hATTR, neuromuscular neurologists, or neurologists with experience treating hATTR would be required to prescribe and monitor treatment and follow-up. The clinical experts highlighted that specialized care would ideally be administered in a hospital or clinic setting equipped with the necessary capabilities and resources to comprehensively manage all facets of advanced neuropathy, including associated cardiac and autonomic symptoms.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full original clinician group input received by CDA-AMC has been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
One clinician group, the NMD4C, provided input for this review. The NMD4C is a pan-Canadian network that brings together the country’s leading clinical, scientific, technical, and patient expertise to improve care, research, and collaboration in neuromuscular disease. In total, 7 clinicians with experience treating hATTR-PN who have also contributed to published Canadian guidelines for hATTR-PN provided input for this submission.
The treatment goals highlighted by the NMD4C were consistent with those noted by the clinical experts consulted for this review and included the prevention of disease progression, decreased morbidity and mortality, fewer hospital visits, and enhanced QoL. However, the clinician group and the clinical experts consulted for this review also emphasized the need for options that offer improved efficacy and tolerability as well as greater convenience and adherence.
Given that the clinical manifestations of hATTR occur after significant build-up of amyloid has occurred in the body, the NMD4C emphasized that the earlier a therapy is initiated, the better the outcomes are. Patients who present with several different disease manifestations, mainly PN and CM, are most in need of intervention, given the significant morbidity and mortality and reduced QoL. The NMD4C stated that patients in the early stages of disease (i.e., stage 1 or 2 PN) will demonstrate a better response to treatment. The NMD4C and the clinical experts consulted for this review agreed that it is unknown whether the failure of 1 treatment indicates that a different treatment would also fail; thus, having multiple treatment options available is important. Although eplontersen is not the first drug to be approved, the NMD4C noted that, in its opinion, eplontersen may have advantages over inotersen, not only regarding side effects, but also in terms of effectiveness.
In line with the clinical experts consulted for this review, the NMD4C highlighted that the outcome measures often used in trials for hATTR are not feasible for use in clinical practice due to the extensive testing and training required for use. To determine whether a patient is responding to treatment in clinical practice, the NMD4C and the clinical experts consulted for this review follow the recommendations described in Canadian treatment guidelines and use neurologic history, neurologic examination, and nerve conduction studies. The NMD4C noted that clinically meaningful responses to treatment would be stability or slower progression of symptoms, greater preservation of functional abilities, and improved survival.
Both the NMD4C and the clinical experts consulted for this review agreed that disease progression may be considered a reason for discontinuation. In clinical practice, outcomes such as loss of walking ability and deterioration of cardiac measures despite intensive treatment could help identify those who are not responding to treatment. The NMD4C highlighted that there is still a long way to go when it comes to understanding all the effects (including side effects) of new hepatically targeted treatments.
Both the clinician group and the clinical experts consulted for this review stated that neurology and neuromuscular specialists would be required to prescribe and monitor treatment and follow-up. The most appropriate settings include specialized and multidisciplinary tertiary centres with lines of referral to cardiologists, given that hATTR-PN is a multidisciplinary disease. The NMD4C also highlighted that hospital outpatient neuromuscular clinics, community-based neurologic clinics, and referral lines to neuromuscular expertise may be appropriate.
Drug Program Input
The drug programs provide input on each drug being reviewed through the CDA-AMC reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The submitted trial, NEURO-TTRansform, was a phase III, multicentre, open-label RCT. There was a treatment arm and a reference arm (inotersen-eplontersen), with effects compared to an external placebo arm from the NEURO-TTR trial. Given the rare and life-threatening nature of hATTR-PN and the existence of approved therapies, an active placebo arm was considered unethical. The sponsor also conducted ITCs with currently available treatment options. Inotersen and patisiran are reimbursed in most but not all federal, provincial, and territorial jurisdictions. Vutrisiran, another relevant comparator, only recently received a CDEC recommendation to reimburse. | This was a comment from the drug programs to inform CDEC deliberations. |
Considerations for initiation of therapy | |
If recommended for reimbursement, would it be appropriate to consider aligning with the initiation criteria for vutrisiran for adult patients with genetically confirmed, stage 1 or stage 2 hATTR-PN who are symptomatic with early-stage neuropathy? For this question, early-stage neuropathy is defined as:
| The clinical experts noted that there has been little change in the treatment landscape for hATTR-PN; thus, aligning the initiation criteria for eplontersen with the most recently recommended therapy, vutrisiran, is reasonable. |
Considerations for continuation or renewal of therapy | |
The primary end points of the NEURO-TTRansform trial included change from baseline in serum TTR concentration, change from baseline in neuropathy using the mNIS + 7 score, and change from baseline in HRQoL using the Norfolk QoL-DN scoring scale. Although the pivotal study used various tools to assess response to treatment, CDEC noted in a previous review for this indication that monitoring is conducted using continuous clinical assessments, with timing depending on disease severity, ranging from every 3 months to every 6 months or more. With no clearly defined renewal criteria for patisiran, inotersen, or vutrisiran, consistency with currently used approaches for monitoring in clinical practice settings would be reasonable. As such, if recommended for reimbursement, would it be appropriate to consider aligning with the renewal criteria for vutrisiran:
| The clinical experts noted that these criteria are in line with clinical practice, but they highlighted that patients may be seen more frequently initially if there are safety concerns or concerns related to cardiac disease, given that these are contraindications for treatment. |
Considerations for discontinuation of therapy | |
If recommended for reimbursement, would it be appropriate to consider aligning with the following discontinuation criteria for vutrisiran? Treatment should be discontinued for patients who are:
| The clinical experts were in agreement with the discontinuation criteria for eplontersen being aligned with those of vutrisiran. |
Considerations for prescribing of therapy | |
Is there any scenario in which a combination of TTR silencers (i.e., RNA-targeted treatments, such as inotersen, patisiran, or vutrisiran) or TTR stabilizers (such as tafamidis) would be used to treat a patient with hATTR-PN? | For neuropathy, there is no evidence showing that combining a silencer and stabilizer provides additional clinical benefit. The experts hypothesized that the role of stabilizers in the neurologic indication is likely minimal, considering the efficacy of silencers in reducing TTR. |
If recommended for reimbursement, would it be appropriate to consider aligning with the following prescribing criteria for vutrisiran?
| The clinical experts agreed with the alignment of prescribing conditions for eplontersen and vutrisiran, citing that it may be difficult for a general neurologist to diagnose this rare disease. Most provinces have neuromuscular physicians or community neurologists with expertise in treating neuromuscular disease; thus, given that hATTR-PN is a rare disease, most patients would be under the care of these specialists. |
Generalizability | |
The sponsor noted that eplontersen is expected to displace inotersen. Is there a time frame or other factor to indicate when this switch may occur? Under what conditions would it be appropriate to switch from inotersen, patisiran, or vutrisiran to eplontersen? | The clinical experts highlighted that they were not aware of any patients currently using inotersen; thus, the switch would likely occur as soon as eplontersen became available to patients. The experts noted that patients may choose to switch treatments for various reasons (e.g., AEs, dosing schedule, patient preference), but noted that there is currently no evidence supporting a switch from inotersen, patisiran, or vutrisiran to eplontersen. |
Care provision issues | |
Patients in the NEURO-TTRansform trial required genetic testing for diagnostic confirmation and documentation of TTR gene mutations to be eligible for treatment. Is diagnostic confirmation by genetic testing required to initiate treatment in patients with hATTR-PN in Canada? How readily available is genetic testing for hATTR? | The clinical experts highlighted that patients are often misdiagnosed with other types of neuropathies, which limits the use of genetic testing. However, genetic testing for hATTR is readily available through commercial or in-province services. |
System and economic issues | |
The sponsor noted that clinicians expect eplontersen to displace mainly inotersen, and perhaps patisiran in rare occasions in which a switch is deemed appropriate. Given the rarity of the condition, the sponsor did not expect the total market size to change with the introduction of eplontersen. | As previously noted, the clinical experts indicated that in their experience, no patients are currently using inotersen; therefore, they did not expect the market size to shift. |
There are confidential negotiated prices for patisiran and inotersen. There was a recent CDEC recommendation for reimbursement of vutrisiran, which has not yet been negotiated by the pCPA. | This was a comment from the drug programs to inform CDEC deliberations. |
AE = adverse event; CDEC = Canadian Drug Expert Committee; FAP = familial amyloid polyneuropathy; hATTR = hereditary transthyretin amyloidosis; hATTR-PN = hereditary transthyretin amyloidosis polyneuropathy; HRQoL = health-related quality of life; ITC = indirect treatment comparison; mNIS + 7 = modified Neuropathy Impairment Score plus 7; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; NYHA = New York Heart Association; pCPA = pan-Canadian Pharmaceutical Alliance; PND = polyneuropathy disability; RCT = randomized controlled trial; RNA = ribonucleic acid; TTR = transthyretin.
Clinical Evidence
The objective of the CDA-AMC Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of eplontersen 56 mg/mL solution (45 mg/0.8 mL prefilled syringe) for SC injection in the treatment of PN associated with hATTR. The focus will be on comparing eplontersen to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of eplontersen is presented in 2 main sections, with CDA-AMC’s critical appraisal of the evidence included at the end of each. The first main section, the systematic review, includes 1 pivotal study that was selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second main section includes indirect evidence from the sponsor. No long-term extension studies or additional studies that were considered to address important gaps in the systematic review evidence were submitted by the sponsor.
Included Studies
Clinical evidence from the following are included in the CDA-AMC review and appraised in this document:
1 pivotal study identified in the systematic review (the NEURO-TTRansform trial)12,13
2 ITCs (a MAIC and an STC).14
Systematic Review
The contents within this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included study are summarized in Table 5.
One study was included in the review. The NEURO-TTRansform study was an 85-week, phase III, multicentre, randomized, open-label study evaluating the efficacy and safety of eplontersen in patients with hATTR. In total, 168 patients were randomized 6 to 1 using an interactive voice or web response system, stratified by V30M TTR mutation, previous treatment, and disease stage to receive 45 mg eplontersen SC once every 4 weeks (n = 144) or 300 mg inotersen SC once per week for up to 34 weeks; these patients were then switched to eplontersen SC once every 4 weeks from week 37 to week 81 (n = 24). All patients continued dosing with eplontersen until week 81, with end-of-treatment assessments occurring at week 85. The NEURO-TTRansform study also included 2 external control groups (placebo [n = 60] and inotersen [n = 112]) from the NEURO-TTR study (described later). (Results comparing eplontersen to the historical inotersen comparison are not described in this section of the report, but are included in the indirect evidence.) The NEURO-TTRansform study was conducted at 40 sites in 15 countries in North America, Europe, South America, Australasia, and Asia, including 2 sites in Canada (British Columbia and Ontario) that enrolled 3 patients. There were 3 analysis time points in the NEURO-TTRansform study: week 35 (interim analysis), week 65 or 66 (final analysis), and week 85 (end-of-treatment analysis).12
Table 5: Details of Study Included in the Systematic Review
Detail | NEURO-TTRansform |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, randomized, open-label study with 2 arms (eplontersen and a concurrent reference arm [inotersen-eplontersen]) plus an external placebo group |
Locations | 40 trial sites in 15 countries: Argentina, Australia, Brazil, Canada, Cyprus, France, German, Italy, New Zealand, Portugal, Spain, Sweden, Taiwan, Turkey, and the US |
Patient enrolment dates | Start date: December 11, 2019 End date: April 11, 2023 |
Randomized (N) | N = 168 Eplontersen: n = 144 Inotersen-eplontersen: n = 24 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Eplontersen administered as an SC injection at a dose of 45 mg in a single 0.3 mL injection once every 4 weeks from week 1 through week 81 |
Comparator(s) |
|
Study duration | |
Screening phase | ≤ 10 weeks |
Treatment phase | 84 weeks (EOT assessment at week 85) |
Follow-up phase | 20 weeks (i.e., posttreatment evaluation period for patients not enrolling in the OLE study) |
Outcomes | |
Primary end points |
|
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publication status | |
Publications | Coelho T, Ando Y, Benson MD, et al. Design and Rationale of the Global Phase 3 NEURO-TTRansform Study of Antisense Oligonucleotide AKCEA-TTR-LRx (ION-682884-CS3) in Hereditary Transthyretin-Mediated Amyloid Polyneuropathy. Neurol Ther. 2021;10(1):375-389. doi: 10.1007/s40120-021-00235-6. Coelho T, Waddington Cruz M, Chao CC, et al. Characteristics of Patients with Hereditary Transthyretin Amyloidosis-Polyneuropathy (ATTRv-PN) in NEURO-TTRansform, an Open-label Phase 3 Study of Eplontersen. Neurol Ther. 2023;12(1):267-287. doi: 10.1007/s40120-022-00414-z. Coelho T, Marques W Jr, Dasgupta NR, et al. Eplontersen for Hereditary Transthyretin Amyloidosis With Polyneuropathy. JAMA. 2023;330(15):1448-1458. doi: 10.1001/jama.2023.18688. |
Clinical trial record number | NCT04136184 |
10MWT = 10-metre walk test; AL = amyloid light chain; ALT = alanine aminotransferase; AST = aspartate aminotransferase; ATTR-PN = transthyretin amyloidosis with polyneuropathy; eGFR = estimated glomerular filtration rate; ECHO = echocardiogram; EOT = end of treatment; COMPASS-31 = Composite Autonomic Symptom Score-31; FAP = familial amyloid polyneuropathy; GLS = global longitudinal strain; hATTR = hereditary transthyretin-mediated amyloidosis; hATTR-PN = hereditary transthyretin amyloidosis polyneuropathy; HCV = hepatitis C virus; Ig = immunoglobulin; IVS = interventricular septum; LV = left ventricular; mBMI = modified body mass index; mNIS + 7 = modified Neuropathy Impairment Score + 7; mRNA = messenger ribonucleic acid; NIS = Neuropathy Impairment Score; NSC = Neuropathy Symptom and Change; NIS = Neuropathy Impairment Score; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; NT-proBNP = N-terminal pro–B-type natriuretic peptide; NYHA = New York Heart Association; OLE = open-label extension; PCS = physical component summary; PGIC = Patient Global Impression of Change; PGIS = Patient Global Impression of Severity; PND = polyneuropathy disability; RBC = red blood cell; RNA = ribonucleic acid; R-ODS = Rasch-built Overall Disability Score; SC = subcutaneous; SF-36 = Short Form (36) Health Survey; TTR = transthyretin; TUDCA = tauroursodeoxycholic acid; ULN = upper limit of normal.
aIncludes tafamidis, inotersen, patisiran, off-label use of diflunisal or doxycycline, and TUDCA. If a patient was previously treated with tafamidis, diflunisal or doxycycline, and TUDCA, the treatment must have been discontinued for at least 2 weeks before day 1 of the study.
bBased on the ClinicalTrial.gov record for the NEURO-TTR study. No further details on the placebo treatment regimen were provided in the NEURO-TTRansform Clinical Study Report.
Source: NEURO-TTRansform Clinical Study Report.12
Two data cut-offs (DCOs) were submitted for the NEURO-TTRansform study. The first interim analysis was conducted at week 35, based on a DCO date of April 18, 2022, comparing eplontersen with the placebo arm of the NEURO-TTR study.13 A post hoc additional data cut was made on July 19, 2022, based on FDA feedback at the pre–new drug application meeting to include at least 100 patients treated with eplontersen for at least 1 year. This data cut was used to provide an expanded safety database at the first interim analysis.
The second interim analysis describes the protocol-specified final analyses, including week 66 analyses versus external controls from the NEURO-TTR study and exploratory analyses conducted from week 85 onward, based on a DCO of April 7, 2023.12
All interim analyses of the NEURO-TTRansform study were conducted in accordance with the statistical analysis plan (SAP) (Version 2.1, dated June 3, 2022, and a Version 2.1 addendum, dated September 22, 2022).12
Figure 1: Study Design of NEURO-TTRansform

ATTRv-PN = transthyretin-mediated amyloidosis variation with polyneuropathy.
Source: NEURO-TTRansform Clinical Study Report.12
The NEURO-TTR Study (ISIS 420915-CS2)
The placebo arm of the NEURO-TTR study was used as an external control for the NEURO-TTRansform study. The NEURO-TTR study was a phase II and III, double-blind, placebo-controlled study that compared the efficacy and safety of an SC injection of inotersen 300 mg weekly with placebo in patients with stage 1 or 2 hATTR-PN. Eligibility criteria for the NEURO-TTR and NEURO-TTRansform studies were identical. A propensity score adjustment for baseline characteristics from the NEURO-TTR study was conducted for disease stage, previous treatment, and V30M mutation.12
Furthermore, measures were taken to maximize the validity of the comparison of the primary end points. The primary end points of mNIS + 7 and Norfolk QoL-DN were blinded to the sponsor team responsible for oversight of the study. Similar to the NEURO-TTR study, the mNIS + 7 central reader was blinded to treatment. All mNIS + 7 assessors were blinded to the rest of the study conduct. Also, the evaluator was insulated from the patients’ general study procedures and knowledge of the patients’ AEs. For mNIS + 7 evaluations, the conduct of the primary end point assessment was similar to that of the NEURO-TTR study. The mNIS + 7 assessors were trained and certified by the staff from the Peripheral Nerve Research Laboratory who had developed the composite score. The Norfolk QoL-DN was required to be administered before any other study procedures. Any team members who became unblinded at the time of the interim analysis were replaced with blinded team members.12
Populations
Inclusion and Exclusion Criteria
The inclusion and exclusion criteria for the NEURO-TTRansform trial are summarized in Table 5. The eligibility criteria were designed to mirror those of the NEURO-TTR study in defining hATTR-PN disease stage, age range, gender, exclusions of certain comorbidities, restriction of concomitant medications for hATTR, and functional status. Briefly, eligible patients in the NEURO-TTRansform study included adults (aged 18 years to 82 years) who had hATTR-PN defined as FAP or Coutinho stage 1 or 2 disease and documented mutations in the TTR gene. Inclusion also required a NIS ranging from 10 to 130. Patients were excluded if they had clinically significant cardiac abnormalities (e.g., previous acute coronary syndrome) or severe heart failure symptoms (i.e., a New York Heart Association functional classification of ≥ 3). Patients with prior or anticipated liver transplant within 1 year of screening, and those who had previous treatment with inotersen, patisiran, or other oligonucleotide or ribonucleic acid therapeutic, were excluded from the NEURO-TTRansform study.12
Interventions
In the NEURO-TTRansform study, eplontersen was administered through SC injection by study centre personnel or at home by patients or caregivers at a dose of 45 mg in a single 0.3 mL injection once every 4 weeks from week 1 through week 81.12
Inotersen was administered through SC injection at a dose of 300 mg in a single 1.5-mL prefilled syringe injection once weekly from week 1 through week 34. Patients were then switched to eplontersen at week 37. The study drug could be administered by study centre personnel or at home by the patient or caregiver.12
If a study drug (i.e., eplontersen or inotersen) was going to be administered at home by a patient or caregiver, dosing instructions and training were provided by the study centre personnel. If a study drug was going to be administered during a clinic or nonclinic visit, all blood samples were required to be drawn before administration. It was not necessary for the study drug to be administered during clinic visits.12
Dose Modifications, Reductions, or Interruptions
Other than for safety reasons, no dose adjustments were planned. The study drug (eplontersen or inotersen) could be temporarily discontinued or withheld (i.e., dose paused or held) for safety reasons, such as AEs.12
Treatment Discontinuation and Withdrawal From Study
Patients could discontinue from the study by request or withdraw their consent to participate. They could also be required to permanently discontinue the study drug for any of the following reasons:12
protocol-specific requirements (e.g., pregnancy, laboratory test abnormalities [refer to stopping rules in subsequent text], or major organ transplant)
investigator decision (e.g., AE, noncompliance, protocol deviation)
sponsor decision.
All efforts were to be made to complete and report observations up to the date of withdrawal. The reason for withdrawal from study was required to be recorded. Any patient who withdrew consent for study participation was removed from further treatment with the study drug and from study observations immediately at the date of request. Patients who withdrew from the study were not replaced.12
Protocol defined stopping criteria for study drug included the following:12
Dosing of the study drug was stopped permanently for the following liver chemistry evaluations:
alanine aminotransferase (ALT) or aspartate aminotransferase (AST) greater than 8 times the upper limit of normal (ULN)
ALT or AST greater than 5 times the ULN and persisting for greater than or equal to 2 weeks
ALT or AST greater than 3 times the ULN (or 2 times the baseline value, if the baseline value was > ULN), and total bilirubin greater than 2 times the ULN or international normalized ratio greater than 1.5
ALT or AST greater than 3 times the ULN (or 2 times the baseline value, if the baseline value was > ULN) with the new appearance (i.e., onset coincides with changes in hepatic enzymes) of fatigue, nausea, vomiting, right upper quadrant pain or tenderness, fever, rash, and/or concomitant eosinophilia (> ULN).
The stopping rules for renal function test results and the temporary stopping rules for renal function test results were as follows:
Dosing should be stopped temporarily in the event of a persistent elevation observed over 2 consecutive weeks for any of the following 4 criteria:
a decrease in estimated glomerular filtration rate calculated from both serum creatinine and serum cystatin C (eGFRcreat-cys) greater than 50% (eGFRcreat-cys is calculated using the Chronic Kidney Disease Epidemiology Collaboration [CKD-EPI] creatinine-cystatin C equation)
eGFRcreat-cys less than 45 mL/min/1.73 m2 (if baseline eGFRcreat-cys > 60 mL/min/1.73 m2)
eGFRcreat-cys less than 30 mL/min/1.73 m2 (if baseline eGFRcreat-cys ≤ 60 mL/min/1.73 m2)
random urine protein-to-creatinine ratio (UPCR) greater than 5 times baseline and greater than 1,500 mg/g; or absolute UPCR value greater than or equal to 2,000 mg/g (confirmed by a repeat random spot UPCR ≥ 2,000 mg/g or a 24-hour UPCR ≥ 2,000 mg/24 hour)
If a dose is held, dosing may be reinitiated once eGFRcreat-cys increases to greater than or equal to 45 mL/minute/1.73 m2, UPCR decreases to below 1,000 mg/g, or the underlying cause of the decline in renal function is corrected. In the case of a UPCR of 2,000 mg/g or higher, further evaluation for renal disease should be performed. If acute glomerulonephritis is confirmed, treatment should be permanently discontinued.
Dosing is stopped permanently if urinalysis or renal blood tests confirm any of the following values in the absence of an alternative explanation agreed to by a consulting nephrologist:
urine protein greater than 2 g (in 24-hour urine)
creatinine clearance less than 45 mL/min/1.73 m2 (if baseline CKD-EPI > 60 mL/min/1.73 m2)
creatinine clearance less than 30 mL/min/1.73 m2 (if baseline CKD-EPI ≤ 60 mL/min/1.73 m2).
The stopping rules for platelet count results are as follows:
For any platelet count of less than 25 × 109/L, dosing of the study drug was permanently stopped, and platelet count was monitored until 2 successive values higher than 25 × 109/L.
In the event of a platelet count of less than 100 × 109/L and greater than or equal to 25 × 109/L, and in the absence of major bleeding or clinically relevant nonmajor bleeding, dosing with the study drug was suspended temporarily until the platelet count recovered to greater than or equal to 100 × 109/L.
The stopping rule for ocular effects was as follows:
Patients were permanently discontinued from study drug if they had clear signs of vitamin A deficiency confirmed by an ophthalmologist.
Concomitant Medications
Patients were required to take supplemental doses of vitamin A during the study at the level of the recommended dietary allowance. Concomitant therapy with tafamidis, inotersen, patisiran, or off-label use of diflunisal was not permitted. Short-term use (i.e., < 15 days) of doxycycline to treat infections was allowed, as were any other medications deemed necessary by the investigator.12
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review, according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform the CDA-AMC expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing the CDA-AMC expert committee deliberations were also assessed using GRADE.
According to all patient, clinician, and drug program inputs, current treatments options are associated with burdensome administration schedules and safety concerns. As such, clinicians and patients highlight the need for treatments that improve HRQoL and offer a more tolerable safety profile. The clinician group input and clinical experts highlighted that the outcome measures used in amyloidosis drug trials are not typically used in clinical practice in Canada. Therefore, the outcomes related to HRQoL and symptoms were considered the most clinically relevant for assessment using GRADE. In the included trial, these consisted of change from baseline in mNIS + 7 composite score, change from baseline in Norfolk QoL-DN total score, change from baseline in COMPASS-31 score, and change from baseline in R-ODS. Additional outcomes included in the GRADE assessments were related to the reduction of circulating TTR protein (i.e., the percentage change from baseline in serum TTR concentration) and select notable harms related to eplontersen (i.e., thrombocytopenia and ocular AEs due to vitamin A deficiency).
Outcomes from the NEURO-TTRansform study that were summarized but not included in the GRADE assessment included the change from baseline in NSC, change from baseline in PND scores, and change from baseline in the PCS of the SF-36, given that the domains that these measures evaluate were captured within the selected outcomes. Additionally, the frequency of all-cause hospitalizations was included only in text, given the exploratory nature of the outcome. A complete list of the primary and secondary end points of the NEURO-TTRansform trial is summarized in Table 5. A summary of the outcomes included in this review and their measurement properties and validity are provided in Table 7.
Table 6: Summary of Outcomes From the NEURO-TTRansform Study
Outcome measure | Time point | NEURO-TTRansform study |
|---|---|---|
Change from baseline in mNIS + 7 composite score | Week 35 (interim analysis) | Primary |
Week 66 (final analysis) | Primarya | |
Change from baseline in Norfolk QoL-DN total score | Week 35 (interim analysis) | Secondarya |
Week 66 (final analysis) | Primarya | |
Change from baseline in COMPASS-31 | Week 37 and week 81 | Exploratory |
Change from baseline in R-ODS | Week 37 and week 81 | Exploratory |
Percent change from baseline in serum TTR | Week 35 (interim analysis) | Primary |
Week 65 (final analysis) | Primarya | |
Week 85 (EOT analysis) | Exploratory | |
Change from baseline in NSC score | Week 35 (interim analysis) | Secondary |
Week 66 (final analysis) | Secondary | |
Change from baseline in PCS score of the SF-36 | Week 66 (final analysis) | Secondary |
Change from baseline in PND score | Week 66 (final analysis) | Secondary |
Frequency of all-cause hospitalizations | Week 66 (final analysis) | Exploratory |
COMPASS-31 = Composite Autonomic Symptom Score-31; EOT = end of treatment; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; NSC = Neuropathy Symptom and Change; PCS = physical component summary; PND = polyneuropathy disability; R-ODS = Rasch-built Overall Disability Score; SF-36 = Short Form (36) Health Survey; TTR = transthyretin.
aStatistical testing for these end points was adjusted for multiple comparisons.
Source: NEURO-TTRansform Clinical Study Report.12,13
Efficacy Outcomes
Interim Analysis
At the first interim analysis (DCO of April 18, 2022), the coprimary efficacy end points were:13
percentage change from baseline in serum TTR concentration at week 35
change from baseline in mNIS + 7 at week 35.
The key secondary end point was the change from baseline in Norfolk QoL-DN at week 35.13
The exploratory efficacy end point in the first interim analysis was the change from baseline in SF-36 components at week 35.
Final Analysis
At the prespecified final analysis (DCO of April 7, 2023), there were 3 coprimary efficacy end points:12
percentage change from baseline in serum TTR concentration at week 65
change from baseline in mNIS + 7 at week 66
change from baseline in Norfolk QoL-DN at week 66.
Secondary end points of interest to this review at the final analysis included the change from baseline in NSC at weeks 35 and 66, the change from baseline in the PCS score of the SF-36 at week 65, and the change from baseline in PND score at week 65.12
Exploratory efficacy end points of interest to this review at the final analysis included the change from baseline in mNIS + 7 and Norfolk QoL-DN at week 85, the change from baseline in COMPASS-31 and R-ODS at weeks 37 and 81, and the frequency of all-cause hospitalizations in all patients and patients with cardiac involvement at week 66.12
Serum TTR: The percentage change from baseline in serum TTR at weeks 35 and 65 was the primary end point of the NEURO-TTRansform study.12 hATTR is characterized by abnormal, misfolded TTR proteins that aggregate in major organs, including the peripheral nervous system, heart, and gastrointestinal tract. Therefore, effective TTR suppression is an important biomarker of treatment effectiveness. While sustained serum TTR reduction appears indicative of the drugs’ biological activity, no formal assessment of the validity of TTR reduction as a surrogate for clinical efficacy outcomes in patients with hATTR-PN who receive eplontersen was available at the time of this review.
Modified NIS + 7: The mNIS + 7 is a modified version of the NIS. It is considered a relatively objective measure of neuropathy, and has been used in 2 clinical trials involving patients with hATTR-PN.26,27 The mNIS + 7 is a combination of:12
NIS (cranial nerves, muscle weakness, reflexes, and sensation)
modified + 7 (heart rate while deep breathing [HRdb], nerve conduction tests, and standardized quantitative sensory testing [heat pain and touch pressure at multiple body sites]).
The version of mNIS + 7 used in the NEURO-TTRansform study (also called the mNIS + 7Ionis) has a range of –22.3 points to 346.3 points, with higher scores representing greater severity of disease.12
For each individual patient, the study protocol recommended using the same mNIS + 7 evaluator for all mNIS + 7 assessments throughout the study. In addition, evaluators were required to have no involvement in the patients’ general study procedures and no knowledge of their AEs to maintain objectivity with respect to this clinical outcome measure.12
Norfolk QoL-DN: The Norfolk QoL-DN is a neuropathy-specific, 5-domain questionnaire used to assess disease-specific changes in patients’ perceived HRQoL. The questionnaire has been validated in hATTR-PN patients and used in multiple pivotal studies for novel therapies for hATTR.28 Although the typical recall period for the Norfolk QoL-DN is 1 month, the NEURO-TTRansform study used a 1-week recall period; this period was validated and used in the NEURO-TTR study that supported the marketing authorization for inotersen.29 The Norfolk QoL-DN was administered before any other study procedures. At baseline, the Norfolk QoL-DN could be administered on the same day as the first mNIS + 7 assessment. The version of the Norfolk QoL-DN used in this study has a range of –4 to 136 points, with higher scores representing greater impairment.12
NSC Score: The NSC is a questionnaire used to assess neuropathy progression and patients’ neuropathy experiences. The NSC is composed of 38 questions that assess the presence and severity of these neuropathy symptoms, and it consists of 1 total score and 5 domains (muscle weakness, loss of sensation or hyposensation sensory symptoms, paresthesia or hypersensation sensory symptoms, gastrointestinal and urinary incontinence autonomic symptoms, and nongastrointestinal and/or nonurinary incontinence autonomic symptoms). The muscle weakness domain is divided into 4 subdomains (head and neck, chest, upper limbs, and lower limbs). The NSC total score has a range of 0 points to 108 points for males and 0 points to 114 points for females, with higher scores indicating more severe symptoms.12
PND Score: The PND score is a scoring system to assess PN severity in 5 stages, with higher scores indicating greater functional impairment. The 5 stages of hATTR-PN using the PND scale are:12
stage 0 (no impairment)
stage 1: sensory disturbances, but preserved walking capabilities
stage 2: impaired walking capacity, but ability to walk without a stick or crutches
stage 3A or B: walking with help of 1 or 2 sticks or crutches
stage 4: confined to wheelchair or bedridden.
Short Form (36) Health Survey: The SF-36 is a non–disease-specific tool to assess HRQoL. It comprises 8 multi-item scales (35 items) assessing physical function (10 items), role limitations due to physical health problems (4 items), bodily pain (2 items), general health (5 items), vitality (4 items), social functioning (2 items), role limitations due to emotional problems (3 items), and emotional well-being (5 items). These 8 scales can be aggregated into 2 summary measures: PCS scores and mental component summary scores. The 36th item, which asks about health change, is not included in the scale or summary scores. The SF-36 is scored on a scale from 0 to 100, with higher scores indicating better HRQoL.12
Composite Autonomic Symptom Score-31: The COMPASS-31 is a 31-item questionnaire that evaluates 6 autonomic domains (orthostatic intolerance, vasomotor, secretomotor, gastrointestinal, bladder, and pupillomotor). Scores range from 0 to 100, with higher scores representing more severe symptoms. Scores greater than 20 are generally considered to represent moderate to severe symptoms.12,30
Rasch-Built Overall Disability Score: The R-ODS is a 24-item, patient-reported outcome measure validated to measure activities of daily life in patients with inflammatory neuropathies and hATTR-PN. Scores range from 0 to 48, with lower scores indicating greater disability.12,26,31
All-Cause Hospitalization: Additional analyses of all-cause hospitalizations in all patients — and all-cause hospitalizations in patients with cardiac involvement — at week 66 were conducted as exploratory analyses.12
Harms Outcomes
The safety objectives of the NEURO-TTRansform study were to evaluate safety and tolerability in patients with hATTR-PN who were treated with eplontersen. Safety assessments included TEAEs, clinical laboratory tests, vital signs, weight, physical examinations, electrocardiograms, an ocular questionnaire, and concomitant medications.12
Notable harms of interest to this review and included in the GRADE assessment included thrombocytopenia as well as ocular AEs related to vitamin A deficiency, in line with the profile of other ASO drugs. There is no Medical Dictionary for Regulatory Activities (MedDRA) query specific to ocular toxicities potentially related to vitamin A deficiency; therefore, the assessment used a nonspecific, concatenated MedDRA search that included preferred terms for either vitamin deficiency or ocular AEs.12
The ocular questionnaire was designed to screen for vitamin A deficiency and was administered every 2 months to 3 months during the treatment period. Patients who presented with new and persistent ocular symptoms compatible with vitamin A deficiency (e.g., night blindness or dry eyes) were to be referred for an ophthalmology examination. If the ophthalmologist confirmed that the patient’s symptoms were consistent with vitamin A deficiency and/or the examination revealed physical findings consistent with vitamin A deficiency (but that did not fulfill stopping rule criteria), then additional work-up with a corneal specialist was to be considered.12
Thrombocytopenia was an AE of special interest to eplontersen because reductions in platelet count were observed in the phase III studies for inotersen. Consequently, thrombocytopenia was included as an AE of special interest in the current study. Thrombocytopenia AEs were defined as any AE with the preferred terms of thrombocytopenia or platelet count decreased.12
Table 7: Summary of Outcome Measures and Their Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
mNIS + 7 | The mNIS + 7 is a score developed specifically for polyneuropathy in patients with hATTR. It quantifies decreased muscle weakness, muscle stretch reflexes, sensory loss, and autonomic impairment.16 The mNIS + 7Ionis differs from the mNIS + 7 in that it includes NIS sensation and assesses autonomic dysfunction with heart rate decrease with deep breathing rather than postural blood pressure. This was the version used in the NEURO-TTRansform study. The mNIS + 7 score ranges from −22.32 points to 346.32 points, with higher scores indicating greater neuropathic impairment.29 | The clinometric performance of the mNIS + 7Ionis was evaluated by Dyck et al.32 Baseline assessments of neuropathy signs (NIS), NIS + 7, mNIS + 7Ionis, PND score, Norfolk QoL-DN, Dyck/Rankin score, NSC score, and the SF-36 Version 2 were evaluated in the first 100 patients with FAP enrolled in the NEURO-TTR trial (inotersen vs. placebo). Validity: The mNIS + 7Ionis was correlated with Norfolk QoL-DN, PND stage, Dyck/Rankin score, and NSC score (Spearman rank correlation r ≥ 0.5 or r ≤ –0.5). The correlation between the mNIS + 7Ionis and the SF-36 Version 2 was also evaluated with Spearman rank correlation (r ≥ 0.25 or r ≤ –0.25).32 Reliability: The reproducibility level for mNIS + 7Ionis is not available, but test-retest reproducibility for the NIS, the sum of 5 nerve conduction studies, and heart rate with deep breathing was high (i.e., Krippendorff alphas = 0.97, 0.98, and 0.93, respectively). Test-retest reproducibility for QST was lower (Krippendorff alpha = 0.57; 0.44 for touch pressure and 0.65 for heat pain). The repeat tests were conducted within a day or a few days of the first test by the same examiners; therefore, the results may have been influenced by recall.32 | No MID reported |
Norfolk QoL-DN | The Norfolk QoL-DN score assesses 35 measures of symptoms and functional impairment related to nerve function in patients with diabetic neuropathy, with higher scores indicating worse QoL. The 5 domains of the Norfolk QoL-DN are symptoms, activities of daily living, physical functioning and large-fibre neuropathy, small-fibre neuropathy, and autonomic neuropathy.28 The version of the Norfolk QoL-DN used in this study has a range of –4 points to 136 points, with higher scores representing greater impairment. | The Norfolk QoL-DN was validated in 61 patients with hATTR with a V30M mutation and stage 1 to 3 disease and in 16 healthy volunteers from a single study centre in Portugal. The questionnaire was translated into Portuguese and validated linguistically.28 Validity: The Norfolk QoL-DN was correlated with objective measures of neurologic function, which included the modified form of NIS, NIS lower limbs, and QST. Reliability: There were no statistically significant differences between the baseline and week 4 assessments in patients with stage 2 or stage 3 disease. Aside from small-fibre neuropathy in stage 2 patients, there were also no statistically significant differences in the individual domains at baseline and week 4.28 | No between-group MID estimated |
COMPASS-31 | Patient-reported measure for assessing symptoms of dysautonomia. Consists of 31 questions. Scores range from 0 to 100, with higher scores representing more severe symptoms.33 | The COMPASS-31 was validated in 66 patients with and without small-fibre polyneuropathy; COMPASS-31 total scores had excellent internal validity and test-retest reliability, and good convergent validity, as defined by the study investigators. COMPASS-31 scores differed across patients with or without small-fibre polyneuropathy and demonstrated fair diagnostic accuracy.33 No studies were identified that examined the validity or reliability of the COMPASS-31 in patients with hATTR. | No MID reported |
R-ODS | The R-ODS is a 24-item scale used to assess the ability to perform everyday activities and social participation in patients with Guillain-Barré syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. Scores range from 0 to 48. A lower score indicates worsening disability.31 | The validity and reliability of the R-ODS was examined in 294 patients with Guillain-Barré syndrome, chronic inflammatory demyelinating polyradiculoneuropathy, and gammopathy-related polyneuropathy. Validity: The intraclass correlation of the R-ODS with the overall disability sum score was evaluated. The intraclass correlation coefficient was 0.85, which demonstrated good external construct validity.31 Reliability: The Person Separation Index was determined to measure internal reliability; an index > 0.7 was considered acceptable. The resulting index was 0.97, which demonstrated acceptable internal reliability.31 No studies were identified that examined the validity or reliability of the R-ODS in patients with hATTR. | No MID reported |
PND | The PND score provides a measure of the impact of neuropathy on ambulation. PND assesses polyneuropathy severity in 5 stages, with higher scores indicating greater functional impairment. During monitoring, a change in score indicates increased functional impairment.15 | The validity, reliability, and responsiveness to change have not been investigated in patients with hATTR. | No MID reported |
NSC | The NSC is a patient questionnaire consisting of 38 questions about the presence, severity, and change in neurologic symptoms.34 The NSC total score has a range of 0 to 108 points for males and 0 to 114 points for females, with higher scores indicating more severe symptoms. | Validity: Baseline assessments of the NSC score, as well as neuropathy signs (NIS), NIS + 7, mNIS + 7Ionis, PND score, Norfolk QoL-DN, Dyck/Rankin score, and the SF-36 Version 2 were evaluated in the first 100 patients enrolled in an oligonucleotide FAP trial (the NEURO-TTR trial [inotersen vs. placebo] by Dyck et al.)32 The following correlations between the NSC score and other scales were found:34
No information was identified in the literature on the reliability or responsiveness of the NSC score in patients with hATTR or other neurologic conditions. | No MID reported |
COMPASS-31 = Composite Autonomic Symptom Scale-31; FAP = familial amyloid polyneuropathy; hATTR = hereditary transthyretin-mediated amyloidosis; MID = minimum important difference; mNIS + 7 = modified Neuropathy Impairment Score + 7; NIS = neurologic impairment score; Norfolk QoL-DN = Norfolk Quality of Life–Diabetic Neuropathy; NSC = Neuropathy Symptoms and Change; PND = polyneuropathy disability; QST = quantitative sensory testing; R-ODS = Rasch-built Overall Disability Score; SF-36 = Short Form (36) Health Survey; V30M = Val30Met; VDT = vibration-detection threshold; vs. = versus.
Statistical Analysis
Sample Size and Power Calculation
The sample size for the NEURO-TTRansform study was estimated based on the data from the NEURO-TTR clinical trial that were relevant to the coprimary end points of the NEURO-TTRansform study. In the NEURO-TTR trial, 52 evaluable patients received placebo. Assuming a 10% dropout rate, total of 140 patients (120 patients treated with eplontersen) were required to achieve a sample size of 108 eplontersen-treated patients.12
In the placebo group of the NEURO-TTR study, patients had a 23.8-point increase in mNIS + 7 score from baseline to week 66. It was estimated that patients treated with eplontersen will have a 4.2-point increase in mNIS + 7, with a 20-point SD in the change from baseline, providing at least 90% power to detect a 19.6-point difference between patients treated with eplontersen and patients treated with placebo in the NEURO-TTR study, with a 2-sided alpha level of 0.025.12
For the Norfolk QoL-DN, patients in the placebo group of the NEURO-TTR study had a 10.7-point change from baseline to week 66. It was estimated that the eplontersen-treated group would have a 0-point change from baseline, with a 20-point SD, providing at least 80% power to detect a 10.7-point difference between eplontersen-treated patients and the NEURO-TTR study placebo patients, with a 2-sided alpha level of 0.025.12
For the percentage change from baseline in TTR, patients in the placebo group of the NEURO-TTR study had a 9.7% reduction from baseline to week 65. An 80% reduction in serum TTR, with a 13% SD for eplontersen-treated patients, was estimated to provide at least 95% power to detect a 70.3% difference between the group treated with eplontersen and the NEURO-TTR-placebo group, with a 2-sided alpha level of 0.025.12
Interim and Final Analyses
When all ongoing patients had reached at least week 35, an interim analysis was conducted to assess the efficacy and safety of eplontersen compared with the placebo arm of the NEURO-TTR study. For the week 35 interim efficacy analysis (DCO of April 18, 2022), there were 2 coprimary end points: percentage change in TTR from baseline to week 35 and change in mNIS + 7 from baseline to week 35. There was 1 key secondary end point: the change in Norfolk QoL-DN from baseline to week 35, which was tested only if both primary end points were significant. If the coprimary end points were nonsignificant, these were retested; the coprimary end point Norfolk QoL-DN was to be tested at the week 66 final analysis, when the alpha level of the final analysis for each end point was determined by a resampling procedure. However, given that both coprimary end points and the key secondary end point of the interim analysis were significant at the 2-sided alpha level of 0.025, as per the SAP, no further hypothesis testing of TTR, mNIS + 7, or Norfolk QoL-DN was performed at the week 66 final analysis. Regardless of the interim analysis results, the study was to proceed as planned.13
For the week 66 final analysis, there were 3 coprimary end points: the percentage change from baseline in serum TTR to week 65, the change from baseline in mNIS + 7 score to week 66, and the change from baseline in Norfolk QoL-DN score to week 66. The final analysis also included 5 secondary end points that were tested hierarchically at a 2-sided alpha level of 0.05 as follows: change from baseline in NSC to week 66, change from baseline in NSC to week 35, change from baseline in PCS score of SF-36 to week 65, change from baseline in PND score to week 65, and change from baseline in modified body mass index (mBMI) to week 65.12
Statistical and Analytical Plans
A summary of statistical analyses conducted in the NEURO-TTRansform trial is included in Table 8.
Efficacy Analyses
Efficacy analyses for the interim and final analysis are summarized by the patients randomized to eplontersen in the NEURO-TTRansform study and patients randomized to placebo in the NEURO-TTR study.12
At the interim analysis, the coprimary end point of percentage change in TTR from baseline to week 35 was analyzed using the MMRM model adjusted by propensity score weights. The propensity score was calculated for each patient using a logistic regression model with baseline covariates, including disease stage (stage 1 or stage 2), V30M mutation (yes or no), and previous treatment (yes or no). The average treatment effect for treated approach was used, where patients in the eplontersen-treated group served as the reference population. The MMRM model also included the effects of treatment (eplontersen or placebo), time (categorical), disease stage (stage 1 or stage 2), V30M mutation (yes or no), previous treatment (yes or no), treatment-by-time interaction, baseline TTR value, and baseline-by-time interaction. The normality assumptions for the MMRM model were formally tested using a Shapiro-Wilks test at the 0.01 significance level and assessed by an inspection of plots. If the Shapiro-Wilks test was statistically significant at the 0.01 level, a stratified Wilcoxon rank sum test was planned. For patients with missing mNIS + 7 values at week 35, multiple imputation was conducted using an imputation model (based on a missing at random [MAR] assumption) that contained the following variables: disease stage (stage 1 or stage 2), V30M mutation (yes or no), previous treatment with tafamidis or diflunisal (yes or no), and whether the baseline value of the end point and the multiple imputation were stratified by treatment group. Of note, multiple imputation was treated as a primary analysis, which was based on on-treatment data for the full analysis set (FAS). In addition, ANCOVA based on observed data (with missing value) was treated as a sensitivity analysis. The same analysis as described for mNIS + 7 was applied for the Norfolk QoL-DN total score.13
At the final analysis, there were 3 coprimary end points: percentage change in TTR from baseline to week 65, change in mNIS + 7 from baseline to week 66, and change in Norfolk QoL-DN from baseline to week 66. These were analyzed using the MMRM model adjusted by propensity score weights, as conducted for the interim analysis. In this model, missing data were not explicitly imputed. Instead, all available postbaseline assessments up to the week 65 (or week 66) end points during the treatment period (which fell within the visit windows) for patients in the FAS were utilized; through modelling of the within-patient correlation structure, the end point treatment differences (which were adjusted to take account of missing data) were derived.12
Sensitivity analyses: Sensitivity analyses were identical between the interim and final analyses. At the interim analysis, sensitivity analyses were conducted for the coprimary end points and key secondary end point at week 35, while at the final analysis, sensitivity analyses were conducted for the 3 coprimary end points at week 66. The following sensitivity analyses were conducted on the FAS or safety set for each end point, except where noted:12
Sensitivity analysis 1: Nonparametric stratified Wilcoxon rank sum test adjusted by stratified propensity score weights. Hodges-Lehmann estimates of the differences between the eplontersen group and NEURO-TTR study placebo group based on the Wilcoxon rank sum test were provided. The propensity score was calculated using the same logistic regression model in the primary analysis. This sensitivity analysis was applied for each analysis (TTR, mNIS + 7, Norfolk QoL-DN) performed in the week 35 interim analysis and for the week 66 final analyses.
Sensitivity analysis 2: Multiple imputation assuming MAR using the safety set. This sensitivity analysis was applied for serum TTR in the week 35 interim analysis and serum TTR, mNIS + 7, and Norfolk QoL-DN in the week 66 final analyses.
Sensitivity analysis 3: Multiple imputation assuming copy increments from reference (CIR) using the safety set. This sensitivity analysis was applied for serum TTR in the week 35 interim analysis and serum TTR, mNIS + 7, and Norfolk QoL-DN in the week 66 final analyses.
Sensitivity analysis 4: Multiple imputation assuming jump to reference (J2R) using the safety set. This sensitivity analysis was applied for serum TTR in the week 35 interim analysis and serum TTR, mNIS + 7, and Norfolk QoL-DN in the week 66 final analyses.
Sensitivity analysis 5: Repeat of the primary efficacy analysis using the per-protocol set (PPS). This sensitivity analysis was applied for each analysis (TTR, mNIS + 7, Norfolk QoL-DN) performed in the week 35 interim analysis and the week 66 final analyses.
Sensitivity analysis 6: A responder analysis based on change in mNIS + 7 was conducted to examine whether improvement in response is consistent over a range of response thresholds. A responder was defined as a patient whose mNIS + 7 change from baseline to week 35 was smaller than or equal to the threshold value. Thresholds that were tested included –2 points, 0 points, 2 points, 4 points, 6 points, 8 points, and 10 points higher than the baseline value. For each of these specific response thresholds, the response rates at week 35 for both the eplontersen-treated group and the NEURO-TTR study placebo group were calculated and plotted against the response threshold. Patients who terminated treatment early (regardless of reason) or had missing week 35 data were considered nonresponders. This sensitivity analysis was applied for mNIS + 7 performed in the week 35 interim analysis using change from baseline to week 35. The same responder analysis was also presented for Norfolk QoL-DN for both the week 35 interim analysis and the week 66 final analysis.
Sensitivity analysis 7: In addition to the 3 factors (disease stage, V30M, and previous treatment) used in the logistic model for propensity score in the primary analysis, this sensitivity analysis included 3 additional covariates: gender (male or female), mBMI (continuous value), and region (Europe, North America, South America, Australasia, Asia) in the ANCOVA or MMRM model performed for the primary end points.
Sensitivity analysis 8: The same ANCOVA used in the primary analysis was performed based on observed data for mNIS + 7 and Norfolk QoL-DN for the week 35 interim analysis.
Subgroup analyses: Subgroup analyses were identical between the interim and final analyses. At the interim analysis, subgroup analyses were conducted for the coprimary end points and key secondary end point at week 35, while at the final analysis, subgroup analyses were conducted for the 3 coprimary end points at week 66. Subgroup analyses included V30M mutation (yes or no), age (< 65 years old, 65 years to 74 years, or ≥ 75 years), race (white or nonwhite), sex (male or female), region (North America, Europe, South America, Australia, or Asia), previous treatment with tafamidis or diflunisal (yes or no), disease stage (stage 1 or stage 2), diagnosis of familial amyloid cardiomyopathy (yes or no), and CM subgroup (yes or no). The CM subgroup included patients with 1) a diagnosis of TTR CM at study entry, or 2) baseline interventricular septum thickness greater than or equal to 13 mm on echocardiogram and no hypertension or ventricular hypertrophy in past medical history, or who had been diagnosed on study and did not have 2 consecutive systolic blood pressure readings greater than or equal to 150 mm Hg at any time during the study (including during screening and baseline visits). The treatment-by-subgroup interaction at each time point was tested at the significance level of 0.10. Treatment-group differences were evaluated within each category of the subgroup, regardless of whether the interaction was statistically significant.12
For TTR at week 35 and week 65, mNIS + 7 at week 66, and Norfolk QoL-DN at week 66, the MMRM adjusted by propensity score for the change from baseline included fixed categorical effects for treatment, time, disease stage, V30M mutation, previous treatment, treatment-by-time interaction, treatment-by-subgroup interaction, and treatment-by-time-by-subgroup interaction. The baseline value of the end point and the baseline-by-time interaction were included as covariates in the model. The treatment-by-subgroup interaction at each time point was tested at the significance level of 0.10. Treatment-group differences were evaluated within each category of the subgroup, regardless of whether the interaction was statistically significant. The propensity score was calculated using the same logistic regression model in the primary analysis.12
For the mNIS + 7 and Norfolk QoL-DN at week 35, the ANCOVA (adjusted by propensity score for the change from baseline) included fixed categorical effects for treatment, disease stage, V30M mutation, and previous treatment with tafamidis or diflunisal. The baseline value of the end point was included as a covariate in the model. The treatment-by-subgroup interaction was tested at the significance level of 0.10. Treatment-group differences were evaluated within each category of the subgroup, regardless of whether the interaction was statistically significant. The propensity score was calculated using the same logistic regression model in the primary analysis.12
Secondary End Point Analysis
Other than the key secondary end point evaluated at the interim analysis (described previously), no other secondary end points were considered at week 35. Secondary end point analyses were conducted on both the FAS and the PPS populations. At the final analysis, if all the coprimary end points were significant, the following secondary end points (which were not tested in the interim analysis) were tested sequentially using the ranking strategy at a 2-sided alpha level of 0.05:12
comparison of change from baseline to week 66 in the NSC between eplontersen and external placebo in the FAS
comparison of change from baseline to week 35 in the NSC between eplontersen and external placebo in the FAS
comparison of change from baseline to week 65 in the PCS score of SF-36 between eplontersen and external placebo in the FAS
comparison of change from baseline to week 65 in the PND between eplontersen and external placebo in the FAS
comparison of change from baseline to week 65 in mBMI between eplontersen and external placebo in the FAS.
For the secondary end points, treatment-group differences were evaluated using the same method as the primary efficacy analysis (i.e., MMRM). The normality assumptions for the MMRM were assessed for each of the secondary end points by inspecting the following plots:12
histogram of marginal studentized residuals derived from the MMRM model
normal probability plot.
No sensitivity analyses were conducted for the secondary end points at the final analysis. The nonparametric stratified Wilcoxon rank sum test was performed as a sensitivity analysis, if deemed necessary. Hodges-Lehmann estimates of the differences between eplontersen and external placebo as well as distribution-free CIs based on the Wilcoxon rank sum test were also provided.12
Exploratory End Point Analysis
For the exploratory end points of interest to this review, statistical analyses were descriptive. For the mNIS + 7 and Norfolk QoL-DN end points, descriptive statistics were provided for absolute values and change from baseline at week 85. For R-ODS and COMPASS-31, descriptive statistics were provided for absolute values and change from baseline at week 37 and week 81. For all-cause hospitalizations, frequency tables and chi-square tests were used for analysis. The change from baseline was analyzed using an ANCOVA model with fixed effects of treatment, disease stage (stage 1 or stage 2), V30M mutation (yes or no), previous treatment with tafamidis or diflunisal (yes or no), and baseline value as covariate based on FAS adjusted by propensity score. The propensity score was calculated using the same logistic regression model in the primary analysis.12
Multiple Imputation
A repeated-measures Gaussian model was fitted to the data using a Bayesian approach, with noninformative priors for the mean and a variance-covariance matrix to provide a joint posterior for the parameters in this model. The repeated-measures Gaussian model included separate mean profiles for each treatment group and the same covariates as those in the primary MMRM analysis for final analysis and TTR at the interim analysis based on on-study data for the safety set.12
For mNIS + 7 and Norfolk QoL-DN at the interim analysis, the same ANCOVA model used in the primary analysis was used in multiple imputation based on on-treatment data for the FAS.12
Independent samples were drawn from the posterior distributions for the mean and variance-covariance matrix to provide inputs into an imputation model. For each patient with missing data, these sampled values of the parameters for mean vectors and the variance-covariance matrices specified a joint distribution for their observed and unobserved outcome data. The postwithdrawal part of each pattern-specific distribution was modelled using 3 different approaches: MAR, CIR, and J2R. The imputation model had the same covariates as those in the primary MMRM analysis. Based on this imputation model, a single set of data was sampled for the missing data based on the distribution for the patients’ missing data conditional upon their observed data.12
MAR approach: The means and variance-covariances following withdrawal are chosen to reflect the patient’s treatment group. This approach should provide similar numerical results to the primary analysis. For the final analysis and TTR at the week 35 interim analysis, all assessments that are within the analysis windows are included in this analysis, even if conducted more than 52 days after the last dose (on study). For the mNIS + 7 and Norfolk QoL-DN at the week 35 interim analysis, only data within 52 days after the last dose (on treatment) are included.
CIR approach: The CIR approach addresses a potential pattern of informative missingness where the assumption is that the active study drug halts or slows disease progression, and the disease progresses after treatment is discontinued. In CIR, missing data in the NEURO-TTR study placebo group were imputed under a within-treatment-arm MAR assumption. For patients in the eplontersen treatment group, their mean profile (i.e., mean increments) will track that of the NEURO-TTR stud placebo group, but starting from the benefit obtained from the previous visit. All assessments that are within the analysis windows are included in this analysis, even if conducted more than 52 days after the last dose.
J2R approach: The J2R approach is an extremely conservative imputation approach that assumes that a patient receiving active study drug does not sustain benefit after discontinuation of the drug. In J2R, missing data in the NEURO-TTR study placebo group were imputed under a within-treatment-arm MAR assumption. For patients with missing data in the eplontersen treatment group, their mean response distribution is set to equal that of the NEURO-TTR study placebo group. All assessments that are within the analysis windows are included in this analysis, even if conducted more than 52 days after the last dose.
For each imputation method used, at least 500 imputed datasets were generated. The imputed observations in each dataset were checked to ensure these were within the possible change-from-baseline range for the patient to whom these belonged. If a dataset contained out-of-range values, it was discarded, and a new dataset was generated until there were 500 datasets containing no out-of-range values. Each of the 500 imputed datasets was then analyzed using a simple ANCOVA model at week 35 and week 66, and the resulting treatment differences and their standard errors were combined using Rubin’s rules. The ANCOVA model adjusted by propensity score included the effects of treatment (eplontersen or NEURO-TTR study placebo), disease stage (stage 1 or stage 2), V30M mutation (yes or no), previous treatment with tafamidis or diflunisal (yes or no), and the baseline value of the end point. Note that in these analyses, efficacy assessments that were within the analysis window but more than 52 days after last dose were included. This differs from the primary analysis, in which data after 52 days from the last dose were excluded. The number of imputed datasets may have been increased after reviewing results if the simulation error was considered large.12
A random seed to be used by a random number generator with value of 2,855 was used to initiate data imputation for all 3 methods.12
Harms Analysis
Unless otherwise specified, all safety analyses were performed on the safety population, and all analyses were summarized descriptively by treatment by system organ class, preferred term, and severity. All AEs were coded using the MedDRA (Version 25.0 or later). For the inotersen-eplontersen group, TEAEs that first occurred or worsened on or after dosing at week 37 are defined as TEAEs after the switch to eplontersen. Otherwise, these are defined as TEAEs before the switch to eplontersen.12
Table 8: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Primary end points | ||||
% change from baseline in serum TTR at weeks 35 and 65a |
|
| Missing data not explicitly imputed (treatment differences were estimated based on the assumption that missing data followed a MAR mechanism) |
|
Change from baseline in mNIS + 7 composite score at week 35 |
|
|
|
|
Change from baseline in mNIS + 7 composite score at week 66 |
|
| Missing data were not explicitly imputed (treatment differences were estimated based on the assumption that missing data followed a MAR mechanism) |
|
Change from baseline in Norfolk QoL-DN total score at week 66 |
|
| Missing data were not explicitly imputed (treatment differences were estimated based on the assumption that missing data followed a MAR mechanism) |
|
Secondary end points | ||||
Change from baseline in Norfolk QoL-DN total score at week 35 |
|
|
|
|
Change from baseline in NSC total score, PCS score of the SF-36, PND score, and mBMI at week 35c |
|
| Missing data not explicitly imputed (treatment differences were estimated based on the assumption that missing data follow a MAR mechanism) | None |
ANCOVA = analysis of covariance; CIR = copy increments from reference; J2R = jump to reference; MAR = missing at random; mBMI = modified body mass index; MMRM = mixed model for repeated measures; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; NSC = Neuropathy Symptom and Change; PCS = physical component summary; PND = polyneuropathy disability; PPS = per-protocol set; SA = sensitivity analysis; SF-36 = Short Form (36) Health Survey; TTR = transthyretin; V30M = Val30Met.
aThe end points for percentage change from baseline in serum TTR at weeks 35 and 65 were combined into a single row because the statistical analysis summaries (i.e., statistical model, adjustment factors, handling of missing data, and sensitivity analyses) were the same for both outcomes.
bBaseline covariates in the logistic regression model included Coutinho stage (1 or 2), V30M mutations (yes or no), and previous treatment with tafamidis or diflunisal (yes or no).
cEnd points for change from baseline in NSC total score, PCS score of the SF-36, PND score, and mBMI at week 35 were combined into a single row because the statistical analysis summaries (i.e., statistical model, adjustment factors, handling of missing data, and sensitivity analyses) were the same for all outcomes.
Source: NEURO-TTRansform Clinical Study Report12 and sponsor’s Clinical Evidence Summary.35
Analysis Populations
A description of the analysis populations in the NEURO-TTRansform study can be found in Table 9.
Table 9: Analysis Populations of the NEURO-TTRansform Study
Population | Definition | Application |
|---|---|---|
FAS | All randomized patients who received at least 1 injection of eplontersen or inotersen and who had a baseline and at least 1 postbaseline efficacy assessment for the mNIS + 7 or Norfolk QoL-DN total score (for the NEURO-TTR study, the FAS included all randomized patients who received at least 1 injection of the study drug and who had a baseline and at least 1 postbaseline efficacy assessment for the mNIS + 7 score or Norfolk QoL-DN total score) | Efficacy and pharmacodynamic analyses |
Per-protocol set | Subset of the FAS who received at least 80% of the prescribed doses of eplontersen or inotersen and had no significant protocol deviations that would be expected to affect efficacy assessments | Efficacy and pharmacodynamic analyses |
Safety set | All patients who were randomized and received at least 1 injection of eplontersen or inotersen | All safety analyses |
FAS = full analysis set; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy.
Source: NEURO-TTRansform Clinical Study Report.12,13
Protocol Amendments and Deviations
The original study protocol was dated June 28, 2019. The final, global study protocol was dated August 12, 2021 (amendment 5). There were 18 protocol amendments in total across the global and country-specific protocols. Key amendments to the global protocol included the addition of exploratory efficacy assessments for EQ-5D-5L and COMPASS-31 (as of September 18, 2019) and Patient Global Impression of Change and Patient Global Impression of Severity (as of January 30, 2020) and amendments to improve study safety, decrease patient burden, and support recruitment during the COVID-19 pandemic by allowing selected visits and procedures to be conducted remotely, lengthening selected time windows for visits and procedures, and adding the option for providing informed consent remotely (as of December 11, 2020). One country-specific addendum for Japan included a change to the age inclusion criterion; however, the impact of this change was unknown.12 None of these protocol amendments was considered to have affected the conduct or integrity of the study.
In the NEURO-TTRansform trial, there were 137 protocol deviations in the eplontersen-treated patients and 23 deviations in the concurrent inotersen group (Table 10). The most frequent major protocol deviations were procedural errors (75.0% for eplontersen and 79.2% for concurrent inotersen), drug errors (42.4% for eplontersen and 58.3% for concurrent inotersen), and improper informed consent procedures (22.9% for eplontersen and 25.0% for concurrent inotersen). Most study procedure deviations were due to assessments that had been performed outside the protocol-defined windows or to missed assessments. Most drug error deviations were due to missed doses or to dosing with more than 14 days between platelet result reviews.12
There was 1 protocol deviation in the eplontersen group due to eligibility criteria not being met at screening. This patient was participating in another trial with eplontersen and did not inform site personnel at screening.12
Table 10: Summary of Major Protocol Deviations (Data Cut-Off of April 7, 2023)
Deviation category | NEURO-TTR study | NEURO-TTRansform study | |
|---|---|---|---|
External placebo group (N = 60) | Eplontersen group (N = 144) | Concurrent inotersen group (N = 24) | |
Any protocol deviations, n (%) | 60 (100) | 137 (95.1) | 23 (95.8) |
Major protocol deviation, n (%) | 49 (81.7) | 120 (83.3) | 21 (87.5) |
Study procedure | 40 (66.7) | 108 (75.0) | 19 (79.2) |
Drug error | 21 (35.0) | 61 (42.4) | 14 (58.3) |
Improperly informed consent procedures | 4 (6.7) | 33 (22.9) | 6 (25.0) |
Visit out of window | 8 (13.3) | 9 (6.3) | 2 (8.3) |
Eligibility criteria | 1 (1.7) | 1 (0.7) | 0 |
Missed visit | 0 | 1 (0.7) | 2 (8.3) |
Restricted concomitant medications | 3 (5.0) | 1 (0.7) | 0 |
Other | 11 (18.3) | 0 | 0 |
Minor protocol deviation, n (%) | 60 (100) | 125 (86.8) | 21 (87.5) |
Note: A patient could have more than 1 protocol deviation category.
Source: NEURO-TTRansform Clinical Study Report.12,13
Changes to Planned Analyses
The original SAP was dated June 17, 2021, and subsequently updated 5 times. The final version of the SAP (Version 2.3) was dated May 31, 2023. Version 2.1 of the SAP (June 3, 2022) was in effect at the time of the primary efficacy analysis for the interim analysis (April 18, 2022); however, it was finalized before the mNIS + 7 and Norfolk QoL-DN data had been made available.12 Other changes to the planned analyses were not considered to have affected the conduct or integrity of the study.
Results
Patient Disposition
Table 11 summarizes the disposition of patients enrolled in the NEURO-TTRansform trial and the external placebo arm of the NEURO-TTR trial. In the NEURO-TTRansform trial, a total of 217 patients were screened for eligibility, and 168 were randomized 6 to 1 to receive eplontersen 45 mg every 4 weeks (n = 144) or inotersen-eplontersen (concurrent inotersen 300 mg once weekly followed by eplontersen 45 mg every 4 weeks at week 37) (n = 24). In the NEURO-TTR study, 278 patients were screened, and a total of 173 patients were randomized: 60 to the placebo group and 113 to the inotersen group. In total, 49 patients (22.6%) and 105 patients (37.8%) in the NEURO-TTRansform and NEURO-TTR studies were screened out, primarily due to not meeting the eligibility criteria (92.0% and 92.4%, respectively).12
In the NEURO-TTRansform study, most randomized patients completed study treatment through week 66 (93.8% in the eplontersen group and 83.3% in the inotersen-eplontersen group), with 90.3% of patients randomized to the eplontersen group completing 85 weeks of treatment. A greater proportion of patients discontinued treatment in the inotersen-eplontersen group compared to the eplontersen group (20.8% and 9.7%, respectively), mainly due to AEs (12.5% for the inotersen-eplontersen group and 3.5% for eplontersen group).12
The mean duration of treatment before last dose for discontinued patients was longest in the eplontersen group (347.1 days). It was 222.6 days for the external placebo group and156.6 days for the inotersen-eplontersen group.12
From the NEURO-TTRansform study, 75.0% of patients in the eplontersen group and 83.3% of patients in the inotersen-eplontersen group discontinued during the posttreatment follow-up due to voluntary withdrawal, including enrolment in the ongoing open-label extension study (74 patients [51.4%] in the eplontersen group and 15 patients [62.5%] in the inotersen-eplontersen group).12
Table 11: Summary of Patient Disposition in the NEURO-TTRansform Study and the External Control
Patient disposition | NEURO-TTRansform study | NEURO-TTR study | |
|---|---|---|---|
Eplontersen group (N = 144) | Inotersen-eplontersen group (N = 24) | External placebo group (N = 60) | |
Screened, N | 217 | 278a | |
Screened out, n (%) | 49 (22.6) | 105 (37.8)a | |
Reason for being screened out, n (%) | |||
Inclusion or exclusion criteria | 46 (92.0) | 97 (92.4)a | |
Withdrawal of consent | 2 (4.0) | 2 (1.9)a | |
Investigator decision | 0 | 0a | |
Sponsor decision | 1 (2.0) | 0a | |
Other | 1 (2.0) | 6 (5.7)a | |
Randomized, N | 144 | 24 | 60 |
Completed study treatment up to week 66 | 135 (93.8) | 20 (83.3) | 52 (86.7) |
Completed study treatment up to week 85 | 130 (90.3) | 19 (79.2) | NA |
Treatment discontinuation up to week 85 | 14 (9.7) | 5 (20.8) | 8 (13.3)b |
Enrolled in OLE study, n (%) | 74 (51.4) | 15 (62.5) | NA |
Reason for discontinuation up to treatment period,c n (%) | |||
AE or SAE | 5 (3.5) | 3 (12.5) | 1 (1.7) |
Stopping rule met | 0 | 0 | 1 (1.7) |
Investigator judgment | 1 (0.7) | 1 (4.2) | 0 |
Voluntary withdrawal | 4 (2.8) | 1 (4.2) | 3 (5.0) |
Pregnancy | 0 | 0 | 0 |
Ineligibility | 1 (0.7) | 0 | 0 |
Significant protocol deviation | 0 | 0 | 0 |
Liver transplant | 0 | 0 | 0 |
Disease progression | 0 | 0 | 3 (5.0) |
Other | 0 | 0 | 0 |
FAS, N | 141 | 21 | 59 |
PPS, N | 137 | 18 | 52 |
Safety, N | 144 | 24 | 60 |
AE = adverse event; FAS = full analysis set; NA = not applicable; OLE = open-label extension; PPS = per-protocol set; SAE = serious adverse event.
aThe numbers of patients screened (or screened out) refer to the entire NEURO-TTR study (i.e., not specifically to the placebo arm), given that patients were randomized to their respective treatment arms after screening was completed.
bTreatment discontinuation up to week 66.
cThe treatment period for the eplontersen arm and inotersen-eplontersen arm was up to week 85, while the treatment period for the external placebo arm was up to week 66.
Source: NEURO-TTRansform Clinical Study Report.12
Baseline Characteristics
Baseline characteristics for the NEURO-TTRansform study and NEURO-TTR study placebo group are summarized in Table 12. In the NEURO-TTRansform study, the mean ages were 53.0 years (SD = 15.0 years) in the eplontersen group and 51.1 years (SD = 12.53 years) in the inotersen-eplontersen group, and there were more males than females enrolled (69.5% and 66.7%versus 30.6% and 33.3% for the eplontersen and inotersen-eplontersen groups, respectively). Most patients had stage 1 hATTR-PN (79.9% and 75.0%, respectively) as well as V30M mutations (59.0% and 66.7%, respectively). The median times from onset of symptoms to enrolment in the study were 54.0 months and 42.5 months, while the median durations of disease from diagnosis until enrolment in the study were 30.0 months and 28.5 months in the eplontersen and inotersen-eplontersen groups, respectively. A clinical diagnosis of hATTR with CM was reported in 27.1% and 29.2% of patients in the eplontersen and inotersen-eplontersen groups, respectively. The mean serum TTR was 0.2 g/L in both treatment groups. The mean mNIS + 7 composite scores and Norfolk QoL-DN total scores at baseline were 81.2 (SD = 43.4) and 44.1 (SD = 26.6) and 65.1 (SD = 33.5) and 40.1 (SD = 21.5) in the eplontersen and inotersen-eplontersen groups, respectively.12
In the NEURO-TTR study placebo group, the mean age was 59.5 years (SD = 14.1), and there were more males than females enrolled (68.3% and 31.7%, respectively). A total of 42 patients (70.0%) had stage 1 hATTR-PN, and 33 patients (55.0%) had V30M mutations. The median time from onset of symptoms to enrolment in the study was 48.0 months (range, 8.0 months to 277.0 months), while the median duration of disease from diagnosis until enrolment in the study was 24.0 months (range, 1.0 month to 159.0 months). A clinical diagnosis of hATTR with CM was reported in 36.7% of patients. The mean serum TTR was 0.2 g/L (SD = 0.04 g/L). Patients in the NEURO-TTR study may have had less severe disease than patients enrolled in the NEURO-TTRansform study, given that the mean mNIS + 7 composite score was 74.8 (SD = 39.0) and the mean Norfolk QoL-DN total score was 48.7 (SD = 26.8).12
Table 12: Summary of Baseline Characteristics in Studies Included in the Systematic Review
Characteristic | NEURO-TTRansform study (SS) | NEURO-TTR study | |
|---|---|---|---|
Eplontersen group (N = 144) | Inotersen-eplontersen group (N = 24) | External placebo group (N = 60) | |
Demographic and patient characteristics | |||
Age, years | |||
Mean (SD) | 53.0 (15.0) | 51.1 (14.4) | 59.5 (14.1) |
Median (range) | 51.5 (24, 82) | 49.5 (30, 77) | 63.0 (28, 81) |
< 65, n (%) | 100 (69.4) | 16 (66.7) | 34 (56.7) |
65 to 74, n (%) | 36 (25.0) | 7 (29.2) | 17 (28.3) |
≥ 75, n (%) | 8 (5.6) | 1 (4.2) | 9 (15.0) |
Sex, n (%) | |||
Male | 100 (69.4) | 16 (66.7) | 41 (68.3) |
Female | 44 (30.6) | 8 (33.3) | 19 (31.7) |
Race, n (%) | |||
Asian | 22 (15.4) | 2 (8.7) | 3 (5.0) |
Black or African American | 5 (3.5) | 0 | 1 (1.7) |
White | 112 (78.3) | 19 (82.6) | 53 (88.3) |
Other | 3 (2.1) | 2 (8.7) | 2 (3.3) |
Multiple | 1 (0.7) | 0 | 1 (1.7) |
Weight (kg) | |||
Mean (SD) | 70.3 (15.8) | 79.2 (19.3) | 71.1 (18.1) |
Median (range) | 68.5 (40.0 to 113.7) | 75.9 (46.3 to 127.0) | 69.9 (38.2 to 126.0) |
mBMI (kg/m2 × g/L)a | |||
Mean (SD) | 1,025.8 (235.12) | 1,101.7 (246.5) | 1,049.9 (228.4) |
Median (range) | 1,003.1 (615.7 to 1,714.0) | 1,103.9 (544.7 to 1,545.8) | 1,027.6 (668.7 to 1,710.0) |
Clinical and disease characteristics | |||
Disease stage (FAP or Coutinho), n (%) | |||
Stage 1 | 115 (79.9) | 18 (75.0) | 42 (70.0) |
Stage 2 | 29 (20.1) | 6 (25.0) | 18 (30.0) |
TTR genotype, n (%) | |||
Val30Met | 85 (59.0) | 16 (66.7) | 33 (55.0) |
NYHA classification, n (%) | |||
Ib | 105 (72.9) | 17 (70.8) | 40 (66.7) |
Duration of disease from hATTR-PN diagnosis, months | |||
Mean (SD) | 46.8 (58.1) | 45.7 (54.1) | 39.3 (40.3) |
Median (range) | 30.0 (0.0 to 379.0) | 28.5 (2.0 to 232.0) | 24.0 (1.0 to 159.0) |
Duration from onset of hATTR-PN symptoms, months | |||
Mean (SD) | 67.7 (50.9) | 72.5 (111.0) | 64.0 (52.3) |
Median (range) | 54.0 (5.0 to 354.0) | 42.5 (8.0 to 567.0) | 48.0 (8.0 to 277.0) |
hATTR-CM clinical diagnosis from case report form | |||
Yes | 39 (27.1) | 7 (29.2) | 22 (36.7) |
Previous treatment (with tafamidis or diflunisal), n (%) | |||
Yes | 100 (69.4) | 15 (62.5) | 36 (60.0) |
Serum TTR (g/L) | |||
Mean (SD) | 0.2 (0.1) | 0.2 (0.01) | 0.2 (0.04) |
Median (range) | 0.2 (0.1 to 0.4) | 0.2 (0.1 to 0.3) | 0.2 (0.1 to 0.2) |
mNIS + 7 composite scores | |||
Mean (SD) | 81.3 (43.4) | 65.1 (33.5) | 74.8 (39.0) |
Median (range) | 78.1 (7.9 to 205.6) | 58.4 (17.0 to 130.4) | 74.9 (13.2 to 156.7) |
NIS composite scores | |||
Mean (SD) | 46.3 (29.9) | 37.6 (24.39) | 43.8 (24.6) |
Median (range) | 41.6 (4.0 to 127.8) | 29.3 (11.5 to 86.5) | 39.3 (3.5 to 88.4) |
Modified + 7 component scores | |||
Mean (SD) | 35.0 (19.84) | 27.5 (15.1) | 31.0 (18.1) |
Median (range) | 31.5 (–4.1 to 88.4) | 32.8 (–0.2 to 59.6) | 31.8 (2.7 to 85.6) |
Norfolk QoL-DN total scores | |||
Mean (SD) | 44.1 (26.6) | 40.1 (21.5) | 48.7 (26.8) |
Median (range) | 42.0 (1.0 to 106.0) | 44.0 (1.0 to 75.0) | 48.1 (–1.0 to 111.0) |
SF-36 scores | |||
PCS scores | |||
Mean (SD) | 39.7 (9.3) | 39.7 (9.6) | 37.2 (9.8) |
Median (range) | 39.12 (16.3 to 62.2) | 38.6 (19.3 to 58.8) | 36.0 (21.0 to 71.4) |
MCS scores | |||
Mean (SD) | 47.1 (10.0) | 46.1 (14.0) | 50.4 (10.8) |
Median (range) | 48.1 (12.4 to 65.6) | 49.7 (9.6 to 65.7) | 50.3 (17.7 to 67.8) |
NSC total scores | |||
Mean (SD) | 23.1 (12.4) | 20.6 (10.5) | 23.0 (12.6) |
Median (range) | 22.0 (1.5 to 60.0) | 20.5 (0.0 to 40.0) | 22.0 (1.0 to 50.5) |
PND score, n (%) | |||
I | 56 (39.2) | 12 (50.0) | 23 (38.3) |
II | 61 (42.7) | 8 (33.3) | 19 (31.7) |
IIIa | 16 (11.2) | 3 (12.5) | 15 (25.0) |
IIIb | 10 (7.0) | 1 (4.2) | 3 (5.0) |
IV | 0 | 0 | 0 |
BMI = body mass index; CM = cardiomyopathy; FAP = familial amyloid polyneuropathy; hATTR-CM = hereditary transthyretin amyloidosis cardiomyopathy; hATTR-PN = hereditary transthyretin amyloidosis polyneuropathy; mBMI = modified body mass index; MCS = mental component summary; mNIS + 7 = modified Neuropathy Impairment Score + 7; NIS = Neuropathy Impairment Score; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; NSC = Neuropathy Symptom and Change; NYHA = New York Heart Association; PCS = physical component summary; PND = polyneuropathy disability; SD = standard deviation; SF-36 = Short Form (36) Health Survey; TTR = transthyretin.
aDefined as BMI in kg/m2 multiplied by albumin level in g/L; higher scores indicate better nutritional status.36
bAll other patients were in NYHA class II. Patients in class III or greater were excluded from the trial.
Source: NEURO-TTRansform Clinical Study Report.12
Concomitant Medications and Cointerventions
In the NEURO-TTRansform study, 27.1% and 9.0% of patients in the eplontersen group received prior treatment with tafamidis and diflunisal, respectively, compared to 15.0% and 25.0% in the external placebo group. Per the protocol, all patients were required to take vitamin A supplements.12
Aside from vitamin A supplements, the most frequently reported concomitant medications (taken by 10% or more patients in the eplontersen group) were tozinameran (33.3%); paracetamol (33.3%); COVID-19 vaccines (31.3%); gabapentin (22.2%); pregabalin (20.8%); furosemide (14.6%); ibuprofen (13.9%); elasomeran (13.2%); pantoprazole (13.2%); domperidone, clonazepam, loperamide, and COVID-19 vaccine nonreplicating viral vector adenovirus (12.5% each); calcium phosphate, cholecalciferol, retinol, and vitamins and minerals not otherwise specified (11.8% each); ciprofloxacin (11.1%); omeprazole (10.4%); and ascorbic acid with other vitamins (10.4%).12
Table 13: Summary of Relevant Concomitant Medications Used During the NEURO-TTRansform Study (Safety Set) Over 85 Weeks
Concomitant medication | Eplontersen group (N = 144) | External placebo group (N = 60) | Inotersen-eplontersen group (N = 24) |
|---|---|---|---|
Patients with ≥ 1 concomitant medication | 144 (100) | 60 (100) | 24 (100) |
Vitamin A Nos | 93 (64.6) | 0 | 17 (70.8) |
Paracetamol | 48 (33.3) | 29 (48.3) | 16 (66.7) |
Tozinameran | 48 (33.3) | 0 | 5 (20.8) |
Gabapentin | 32 (22.2) | 18 (30.0) | 7 (29.2) |
Pregabalin | 30 (20.8) | 15 (25.0) | 6 (25.0) |
Furosemide | 21 (14.6) | 13 (21.7) | 1 (4.2) |
Ibuprofen | 20 (13.9) | 11 (18.3) | 4 (16.7) |
Covid-19 vaccine | 20 (13.9) | 0 | 1 (4.2) |
Elasomeran | 19 (13.2) | 0 | 10 (41.7) |
Pantoprazole | 19 (13.2) | 2 (3.3) | 2 (8.3) |
Clonazepam | 18 (12.5) | 6 (10.0) | 2 (8.3) |
Domperidone | 18 (12.5) | 3 (5.0) | 2 (8.3) |
Loperamide | 18 (12.5) | 7 (11.7) | 6 (25.0) |
Covid-19 vaccine Nrvv Ad (Chadox1 Ncov-19) | 18 (12.5) | 0 | 4 (16.7) |
Calcium Phosphate; cholecalciferol; retinol | 17 (11.8) | 0 | 2 (8.3) |
Minerals Nos; vitamins Nos | 17 (11.8) | 20 (33.3) | 5 (20.8) |
Ciprofloxacin | 16 (11.1) | 11 (18.3) | 2 (8.3) |
Other vitamins and mineralsa | 15 (10.4) | 0 | 2 (8.3) |
Omeprazole | 15 (10.4) | 14 (23.3) | 4 (16.7) |
Nrvv Ad = nonreplicating viral vector adenovirus.
aIncludes ascorbic acid, beta-carotene, cholecalciferol, cyanocobalamin, ferrous fumarate, folic acid, pyridoxine hydrochloride, riboflavin, thiamine mononitrate, tocopherol acetate, and zinc oxide.
Source: NEURO-TTRansform Clinical Study Report (2023).12
Exposure to Study Treatments
Patient exposure to study treatments in the NEURO-TTRansform study up to week 85 is summarized in Table 14. Most patients were on treatment for greater than 12 months but fewer than 24 months (137 patients [95.1%] for eplontersen and 52 patients [86.7%] for external placebo). The median duration of exposure to eplontersen was 561.0 days (range, 57 days to 582 days), while the median duration of exposure to inotersen was 227.5 days (range, 51 days to 236 days), followed by a median exposure of 166 days (range, 78 days to 323 day) in patients who switched to eplontersen at week 37. In the NEURO-TTR study, the median exposure to placebo was 449.0 days (range, 36 days to 463 days).12
Across treatment groups, the mean adherence ranged from 92.3% (in the external placebo group) to 99.2% (in the inotersen-eplontersen group). A total of 38 patients (26.4%) in the eplontersen group had dose pauses or missed doses during treatment compared to 23 patients (38.3%) and 19 patients (79.2%) in the external placebo group and concurrent inotersen group (up to week 37).12
Table 14: Summary of Patient Exposure in the NEURO-TTRansform Study (Up to Week 85 for the NEURO-TTRansform Study and Up to Week 66 for the NEURO-TTR Study External Placebo Group) (Safety Set)
Characteristic | Eplontersen groupa (N = 144) | External placebo groupb (N = 60) | Inotersen-eplontersen group (N = 24) | |
|---|---|---|---|---|
Inotersen (week 1 to week 37) (N = 24) | Eplontersen (week 37 to week 85) (N = 20) | |||
Duration of study drug exposure (days) | ||||
n | 144 | 60 | 24 | 20 |
Mean (SD) | 540.8 (82.2) | 418.6 (87.1) | 202.4 (59.1) | 187.8 (86.0) |
Median (range) | 561.0 (57 to 582) | 449.0 (36 to 463) | 227.5 (51 to 236) | 166.0 (78 to 323) |
< 6 months, n (%) | 3 (2.1) | 3 (5.0) | 4 (16.7) | 11 (55.0) |
≥ 6 months to < 12 months, n (%) | 4 (2.8) | 5 (8.3) | 20 (83.3) | 9 (45.0) |
≥ 12 months to < 24 months, n (%) | 137 (95.1) | 52 (86.7) | 0 | 0 |
Overall adherence (%)c | ||||
n | 144 | 60 | 24 | 20 |
Mean (SD) | 97.9 (6.1) | 92.3 (18.6) | 99.2 (3.7) | 99.2 (3.7) |
Median (range) | 100.0 (60.0 to 112.8) | 99.9 (10.4 to 101.5) | 100.0 (83.8 to 100.0) | 100.0 (83.8 to 100.0) |
EOT = end of treatment; SD = standard deviation.
aThe eplontersen arm refers to patients who received eplontersen beginning in week 1 (i.e., it does not include patients who switched from inotersen to eplontersen).
bPatients from the external placebo group were evaluated up to only 66 weeks.
cAdherence was defined as (actual dose infused [mg] in each treatment group divided by the expected total dose [mg] to be administered in each treatment group) multiplied by 100.
Note: The duration of study treatment exposure is calculated as the date of the last dose of study drug minus the date of the first dose plus 1. For eplontersen, this was the dose administered between the first dose of the study drug on day 1 and the EOT date or date of premature termination; for inotersen before week 35, this was the dose administered between the first dose of the study drug on day 1 and the last dose date in week 34 or date of premature termination; for those who received inotersen and then switched to eplontersen, this was the dose administered between the first dose of the study drug on day 1 of week 37 and the EOT date or date of premature termination; for external placebo, this was the dose administered between the first dose of the study drug on day 1 and the EOT date or date of premature termination.
Source: NEURO-TTRansform Clinical Study Report.12
Efficacy
Efficacy results of interest to this review from the NEURO-TTRansform trial are summarized in Table 15 except for the outcomes of COMPASS-31, R-ODS, and hospitalization.
Change From Baseline in mNIS + 7 Composite Score
The change from baseline in mNIS + 7 composite score was a coprimary end point of the NEURO-TTRansform study at the week 35 interim analysis and week 66 final analysis.12,13
Week 35 (Interim Analysis)
At baseline, the mean mNIS + 7 composite scores in the eplontersen group (N = 140) and external placebo group (N = 59) were 79.59 points (SD = 42.25 points) and 74.12 points (SD = 39.03 points), respectively. At week 35, the LSM changes from baseline were 0.22 points (95% CI, –3.46 points to 3.90 points) for the eplontersen group and 9.23 points (95% CI, 5.54 points to 12.91 points) for the external placebo group, representing an LSM difference of –9.01 points (95% CI, –13.48 points to –4.54 points; P = 0.00007889) in favour of eplontersen and corresponding to an improvement in the severity of neuropathy with eplontersen treatment.13
Week 66 (Final Analysis)
At week 66, the LSM changes from baseline were 0.30 points (95% CI, –4.46 points to 5.06 points) for eplontersen and 25.06 points (95% CI, 20.23 points to 29.88 points) for external placebo, representing an LSM difference of –24.76 points (95% CI, –30.96 points to –18.56 points; P < 0.00000001) in favour of eplontersen and corresponding to an improvement in the severity of neuropathy with eplontersen treatment.12
A summary of the LSM difference between eplontersen and external placebo for the mNIS + 7 composite and component scores (i.e., cranial nerves, muscle weakness, reflexes, sensation, HRdb, nerve conduction, touch pressure sensation, and heat pain sensation) at week 66 is presented in Figure 5 in Appendix 1. Aside from the cranial nerve score, the individual components were directionally consistent with the composite score.12
Sensitivity analyses: Per the SAP, sensitivity analyses 2, 3, and 4 were not performed for week 35. The results for all sensitivity analyses conducted for the mNIS + 7 are summarized in Table 26 to Table 33. Results for the sensitivity analyses were consistent with those of the primary analysis, with point estimates for the LSM differences ranging from █████ ██████ ██ █████ ██████ at week 35 and ██████ ██████ ██ ██████ ██████ at week 65 ██ ██████ ██ ███████████ across the sensitivity analyses.12,13
In sensitivity analysis 6 (Table 31), which evaluated the difference in response between treatment groups, the proportion of patients considered responders was ███████████ ██████ ██ ███ ███████████ █████ ██ ███ ██████████ ████████ ██ ████████ ████████ ██ ██████ ██ ███████ ███████████████████ ████████ ███ ██ ███████ ███████████████ ████████ ████████ ███████ ██ ████ ████ █ ██████ at week 35 compared to baseline, █████ ██ ███████ ███████████████████ ████████ ███ ██ ███████ ███████████████ ████████ ████████ ███████ ██ ████ ████ █ ██████ at week 66 compared to baseline.12,13
Subgroup analyses: Results of all prespecified subgroup analyses at weeks 35 and 65 are shown in Figure 3 in Appendix 1. In general, the results of the subgroup analyses were consistent with those of the primary analysis at both analysis time points.12,13
Exploratory Analysis: Eplontersen Versus Inotersen-Eplontersen
An exploratory analysis of mNIS + 7 composite score for the eplontersen and inotersen-eplontersen groups up to week 85 was conducted. At week 35, the mean change from baseline in mNIS + 7 composite score was –0.03 points (SD = 16.28 points) for patients treated with eplontersen (n = 137) and 4.06 points (SD = 13.39 points) following treatment with inotersen (n = 19) (i.e., before switching to eplontersen). At week 66, the mean change from baseline in mNIS + 7 composite scores were –0.21 points (SD = 17.62) for patients treated with eplontersen (n = 128) and 3.22 points (SD = 15.44 points) for patients who received inotersen followed by eplontersen at week 37 (n = 19). Results were consistent at week 85 for the eplontersen group (n = 122; –2.86 points [SD = 20.54 points]) and the inotersen-eplontersen group (n = 18; 4.07 points [SD = 20.63 points]). No between-group differences were reported at any follow-up time.12
Change From Baseline in Norfolk QoL-DN Total Score
The change from baseline in Norfolk QoL-DN total score was a key secondary end point of the NEURO-TTRansform study at the week 35 interim analysis and a coprimary end point at the week 66 final analysis.12,13
Week 35 (Interim Analysis)
At baseline, the mean Norfolk QoL-DN total scores in the eplontersen group (N = 134) and external placebo group (N = 58) were 43.33 points (SD = 26.21 points) and 48.60 points (SD = 26.97 points), respectively. At week 35, the LSM changes from baseline were –3.12 points (95% CI, –7.19 points to 0.96 points) for eplontersen and 8.67 points (95% CI, 4.53 points to 12.81 points) for external placebo, representing an LSM difference of –11.79 points (95% CI, –16.82 points to –6.76 points; P = 0.00000430) in favour of eplontersen and corresponding to an improvement in HRQoL with eplontersen treatment.13
Week 66 (Final Analysis)
At week 66, the LSM changes from baseline were –5.50 points (95% CI, –10.03 points to –0.96 points) for eplontersen and 14.24 points (95% CI, 9.51 points to 18.97 points) for external placebo, representing an LSM difference of –19.74 points (95% CI, –25.63 points to –13.84 points; P < 0.00000001) in favour of eplontersen and corresponding to an improvement in HRQoL with eplontersen treatment.12
A summary of the LSM difference between eplontersen and external placebo for the Norfolk QoL-DN domain scores (physical functioning and/or large-fibre neuropathy, symptoms, activities of daily living, small-fibre neuropathy, autonomic neuropathy) at week 66 is presented in Figure 6 in Appendix 1. All individual domains were directionally consistent with the total score.12
Exploratory Analysis: Eplontersen Versus Inotersen-Eplontersen
An exploratory analysis of Norfolk QoL-DN total score for the eplontersen and inotersen-eplontersen groups up to week 85 was conducted. At week 35, the mean changes from baseline in Norfolk QoL-DN total score were –4.79 points (SD = 16.51 points) for patients treated with eplontersen (n = 130) and –2.97 points (SD = 12.10 points) for patients treated with inotersen (n = 20) (i.e., before switching to eplontersen). At week 66, the mean changes from baseline in Norfolk QoL-DN total score were –7.24 points (SD = 18.51 points) for patients treated with eplontersen (n = 128), and –2.37 points (SD = 11.70 points) for patients who received inotersen followed by eplontersen at week 37 (n = 20). Results were consistent at week 85 for eplontersen (n = 119; –6.23 points [SD = 18.01 points]) and inotersen-eplontersen (n = 18; 1.21 points [SD = 14.03 points]). No between-group differences were reported at any follow-up time.12
Sensitivity analyses: Per the SAP, sensitivity analyses 2, 3, and 4 were not performed for week 35. The results for all sensitivity analyses conducted for the Norfolk QoL-DN are summarized in Table 26 to Table 33. Results for sensitivity analyses were consistent with those of the primary analysis, with point estimates for the LSM differences ranging from █████ ██████ ██ ██████ ██████ at week 35 and █████ ██████ ██ ██████ ██████ at week 65 ██ ██████ ██ ███████████ across sensitivity analyses.12,13
In sensitivity analysis 6 (Table 31), which evaluated the difference in response between treatment groups, the proportion of patients considered ██████████ ███ ███████████ ██████ ██ ███ ███████████ █████ ██ ███ ██████████ ████████ ██ ████████ ████████ ██ ██████ ██ ███████ ███████████████████ ████████ ███ ██ ███████ ███████████████ ████████ ████████ ███████ ██ ████ ████ █ ██████ ██ ████ ██ ████████ ██ █████████ █████ ██ ███████ ███████████████████ ████████ ███ ██ ███████ ███████████████ ████████ ████████ ███████ ██ ████ ████ █ ██████ ██ ████ ██ ████████ ██ ████████
Subgroup Analyses: Results of all prespecified subgroup analyses at weeks 35 and 65 are displayed in Figure 4 in Appendix 1. In general, the results of the subgroup analyses were consistent with those of the primary analysis at both analysis time points.12,13
Change From Baseline in COMPASS-31
Change from baseline in COMPASS-31 at weeks 37 and 81 was an exploratory outcome of the NEURO-TTRansform study at the final analysis. COMPASS-31 was not assessed in the external placebo group. At baseline, the mean COMPASS-31 score in the eplontersen group was 19.4 points (SD = 11.26 points). The mean changes from baseline at weeks 37 and 81 were ████ ██████ ███ █ █████ and –2.6 points (SD = 7.52 points), respectively.12
Change From Baseline of R-ODS
Change from baseline in R-ODS at weeks 37 and 81 was an exploratory outcome of the NEURO-TTRansform study at the final analysis. ███ █████ ███ ███ ████████ ██ ███ ████████ ███████ ██████ At baseline, the mean R-ODS score in the eplontersen group was ████ ██████ ███ █ ███████ The mean changes from baseline at weeks 37 and 81 were ███ ██████ ███ █ █████ ███ ████ ██████ ███ █ ██████ respectively.12
Change From Baseline in Serum TTR
Week 35 (Interim Analysis)
Change from baseline in serum TTR at week 35 was a coprimary end point of the NEURO-TTRansform study. Because the prespecified Shapiro-Wilks test rejected normality in the MMRM, the nonparametric Wilcoxon rank sum test (sensitivity analysis 1) was used as the primary analysis. At week 35, the mean percentage changes from baseline for patients in the eplontersen and external placebo groups were –81.98% (SD = 11.70%) and –11.13% (SD = 19.60%), respectively. Based on the MMRM, the LSM percentage change from baseline in serum TTR at week 35 was –1.20% (95% CI, –84.55% to –77.84%) for the eplontersen group compared to –4.76% (95% CI, –18.73% to –10.80%) for the external placebo group, representing an LSM difference of 66.43% (95% CI, –71.59% to –61.71%; P < 0.00000001) in favour of eplontersen and corresponding to a reduction in serum TTR levels for patients receiving eplontersen.13
Using the nonparametric sensitivity analysis, the mean percentage changes from baseline for the eplontersen and external placebo groups were –84.17% (SD = not reported) and –9.45% (SD = not reported), respectively. The Hodges-Lehmann difference between eplontersen and external placebo at week 35 was –67.09% (95% CI, –72.88% to –61.00%).13
Week 65 (Final Analysis)
The percentage change from baseline in serum TTR was also a coprimary end point of the week 65 final analysis. Because the results of the coprimary and key secondary efficacy end points were statistically significant at the week 35 interim analysis, no formal statistical tests were conducted at week 65. However, treatment-group differences for all coprimary end points tested at the 0.05 significance level were evaluated at week 65.12
At week 65, the LSM percentage changes from baseline in serum TTR concentration were –81.65% ████ ███ ███████ ██ ████████ in the eplontersen group and –11.24% ████ ███ ███████ ██ ███████ in the external placebo group, representing an LSM difference of –70.42% ████ ███ ██████ ██ ██████████████████ in favour of eplontersen and corresponding to a reduction in serum TTR levels for patients receiving eplontersen. The corresponding nonparametric LSM difference at week 65 using Hodges-Lehman (sensitivity analysis 1) was –██████ ████ ███ ██████ ██ ███████.
Sensitivity analyses: Results for the prespecified sensitivity analyses of percentage change from baseline in serum TTR at week 35 and week 65 are summarized in Table 26 to Table 30, Table 32, and Table 33. Results for all prespecified sensitivity analyses of change from baseline in serum TTR at week 35 were consistent with the primary analysis, with point estimates for the LSM differences in percentage reduction in serum TTR between eplontersen and external placebo ranging from ███████ ██ ███████. For week 65, results were also consistent with the primary analysis, as well as with week 35 results, with point estimates for the LSM differences in percentage reduction in serum TTR between eplontersen and external placebo ranging from ██████ ██ ███████.12,13
Subgroup analyses: Results of all prespecified subgroup analyses at weeks 35 and 65 are displayed in Figure 2 in Appendix 1. In general, results of the subgroup analyses were consistent at both analysis time points and with the primary analysis. The subgroup of █████████ ████████ ███ █ ███████ █████ LSM difference in percent reduction in serum TTR at week 35 ████████ ████ ███ ██████ ██ ████████ and week 65 ████████ ████ ███ ██████ ██ █████████ ██████ ███ ███████ █████ ████████ ████████████ ████████ █████ ███ █ █████ ██████ ██ ████████ ████████ ██ ███ ████████ ██ █ ██ ██ ████ ██ ████ ██ ████ ███ ███ █████ ████ ██ █████ ███ ████████ ███████████.
Week 85
The percentage change from baseline in serum TTR at week 85 was an exploratory end point of the final analysis. At week 85, the mean percentage change from baseline in the eplontersen group was –81.8% (SD = 13.38%). Week 85 data were not available for the external placebo group because the NEURO-TTR study was only 66 weeks in duration.12
Exploratory Analysis: Eplontersen Versus Inotersen-Eplontersen
An exploratory analysis of TTR for the eplontersen and inotersen-eplontersen groups up to week 85 was conducted. At week 35, the mean percentage changes from baseline in serum TTR were ███████ ███ █ ██████ for patients treated with eplontersen ██ █ ████ and ███████ ███ █ ██████ for patients treated with inotersen ██ █ ███ (i.e., before switching to eplontersen). ██ ████ ███ ███ ████ ███████ ██████ ████ ████████ ██ █████ ███ ███ ███████ ███ █ ██████ ███ ███████████████████ ████████ ██ █ █████ ███ ███████ ███ █ ██████ ███ ████████ ███ ████████ █████████ ████████ ██ ███████████ ██ ████ ██ ██ █ ████ ███████ ████ ██████████ ██ ████ ██ ███ ███████████ ██ █ ████ ███████ ███ █ ███████ ███ █████████████████████ ██ █ ███ ███████ ███ █ █████████ ██ █████████████ ███████████ ████ ████████ ██ ███ █████████ ███
Change From Baseline in NSC Total Score
Change from baseline in NSC total score was a secondary outcome of the NEURO-TTRansform study at the final analysis. At week 66, the LSM changes from baseline were –0.03 points (95% CI, –1.92 points to 1.86 points) in the eplontersen group and 8.2 points (95% CI, 6.24 points to 10.12 points) in the external placebo group, representing an LSM difference of –8.2 points (95% CI, –10.65 points to –5.76 points) in favour of eplontersen.12
Change From Baseline in PCS Score of the SF-36
The change from baseline in the PCS score of the SF-36 was a secondary outcome of the NEURO-TTRansform study at the final analysis. At week 65, the LSM change from baseline was 0.85 points (95% CI, –0.711 points to 2.412 points) in the eplontersen group and –4.46 points (95% CI, –6.139 points to –2.770 points) in the external placebo group, representing an LSM difference of 5.31 points (95% CI, 3.195 points to 7.416 points) in favour of eplontersen.12
Change From Baseline in PND Score
Change from baseline in PND score was a secondary outcome of the NEURO-TTRansform study at the final analysis. At week 65, the LSM changes from baseline were ███ ██████ ████ ███ ████ ██ ████ in the eplontersen group and ███ ██████ ████ ███ ████ ████ in the external placebo group, representing an LSM difference of ████ ██████ ████ ███ ████ ██ ██████.12
Hospitalizations
███ █████████ ██ ███ █████ ████████████████ ██ ███ ████████ ███ ██ ████████ ████ ██████████████ ███ ██ ███████████ ███████ ██ ███ ████████████████ ██████ █ █████ ██ ██ ████████ ██ ███ ███████████ █████ ███ ██ ████████ ██ ███ ████████ ███████ █████ ████ ████████ ██ ███ ██████████████ █████████ ██ ██████ █ ████████ ██████ ████ ████████████ ██ ███ ███████████ ██████ ███ ███ █ ███████ ██████ ███ ████████████ ██ ███ ████████ ███████ ██████ ███ ████ ███ ██████████████ ███████
Table 15: Summary of Key Efficacy Results in the NEURO-TTRansform Study
Variable | Eplontersen group N = 144 | External placebo group N = 60 | Inotersen-eplontersen group N = 24 |
|---|---|---|---|
Change from baseline in mNIS + 7 composite score | |||
Baseline | |||
N | 141 | 59 | 21 |
mNIS + 7 composite score at baseline, mean (SD) | 79.81 (42.25) | 74.12 (39.03) | 65.41 (35.86) |
Week 35 | |||
N | 137 | 55 | 19 |
CFB in mNIS + 7 composite score, mean (SD) | –0.03 (16.28) | 9.76 (14.20) | 4.06 (13.39) |
Week 66 | |||
N | 128 | 52 | 19 |
CFB in mNIS + 7 composite score, mean (SD) | –0.21 (17.62) | 23.89 (24.19) | ████ █ |
Statistical analysis of CFB | |||
CFB in mNIS + 7 composite score at week 35, LSM (95% CI)a | 0.22 (–3.46 to 3.90) | 9.23 (5.54 to 12.91) | NA |
Difference in LSM (eplontersen vs. placebo) at week 35 (95% CI) | –9.01 (–13.48 to –4.54) | NA | |
P value | 0.00007889 | NA | |
CFB in mNIS + 7 composite score at week 66, LSM (SE)b | 0.30 (–4.46 to 5.06) | 25.06 (20.23 to 29.88) | NA |
Difference in LSM (eplontersen vs. placebo) at week 66 (95% CI) | –24.76 (–30.96 to –18.56) | NA | |
P value | < 0.00000001 | NA | |
Change from baseline in Norfolk QoL-DN total score | |||
Baseline | |||
N | 134 | 58 | 21 |
Norfolk QoL-DN total score, mean (SD) | 43.33 (26.21) | 48.60 (26.97) | 37.97 (21.51) |
Week 35 | |||
N | 130 | 57 | 20 |
CFB in mNIS + 7 composite score, mean (SD) | –4.79 (16.51) | 5.51 (20.18) | –2.97 (12.10) |
Week 66 | |||
N | 128 | 52 | 20 |
CFB in mNIS + 7 composite score, mean (SD) | –7.24 (18.51) | 10.77 (21.13) | ████ █ |
Statistical analysis of CFB | |||
CFB in Norfolk QoL-DN total score at week 35, LSM (95% CI)a | –3.12 (–7.19 to 0.96) | 8.67 (4.53 to 12.81) | NA |
Difference in LSM at week 35 (95% CI) | –11.79 (–16.82 to –6.76) | NA | |
P value | 0.00000430 | NA | |
CFB in Norfolk QoL-DN total score at week 66, LSM (95% CI)b | –5.50 (–10.03 to –0.96) | 14.24 (9.51 to 18.97) | NA |
Difference in LSM at week 66 (95% CI) | –19.74 (–25.63 to –13.84) | NA | |
P value | 0.00000001 | NA | |
Percentage change from baseline in serum TTR concentration (g/L)b | |||
Baseline | |||
N | 140 | 59 | 21 |
Serum TTR, mean (SD) | 0.2269 (0.0755) | 0.1541 (0.0375) | 0.21 (0.07) |
Week 35 | |||
N | 136 | 57 | 20 |
Serum TTR, mean (SD) | 0.0396 (0.0266) | 0.1347 (0.0385) | NR |
Absolute CFB in serum TTR, mean (SD) | –0.1894 (0.0737) | –0.0193 (0.0315) | NR |
Percent CFB in serum TTR, mean (SD) | –81.98 (11.70) | –11.13 (19.60) | –74.26 (23.28) |
Week 65 | |||
N | 135 | 51 | 20 |
Serum TTR, mean (SD) | 0.0366 (0.0221) | 0.1427 (0.0410) | NR |
Absolute CFB in serum TTR, mean (SD) | –0.1919 (0.0750) | –0.0122 (0.0333) | NR |
Percent CFB in serum TTR, mean (SD) | –82.96 (10.37) | –5.95 (21.42) | ████ █ |
Statistical analysis of CFB | |||
Percent CFB in serum TTR at week 35, LSM (95% CI) | –81.20 (–84.55 to –77.84) | –14.76 (–18.73 to –10.80) | NA |
Difference in LSM at week 35 (95% CI) | –66.43 (–71.39 to –61.47) | NA | |
P value | < 0.00000001 | NA | |
Percent CFB in serum TTR at week 65, LSM (95% CI) | –81.65 (–84.82 to –78.48) | –11.24 (–15.06 to –7.41) | NA |
Difference in LSM at week 65 (95% CI) | –70.42 (–75.17 to –65.66) | NA | |
P value | < 0.00000001 | NA | |
Change from baseline in NSC score | |||
Baseline | |||
N | 141 | 59 | NA |
NSC at baseline, mean (SD) | 22.70 (12.05) | 22.92 (12.73) | NA |
Week 66 | |||
N | 132 | 52 | NA |
CFB in NSC, LSM (95% CI) | –0.03 (–1.92 to 1.86) | 8.18 (6.24 to 10.12) | NA |
Difference in LSM (95% CI)b | –8.21 (–10.65 to –5.76) | NA | |
P value | 0.00000001 | NA | |
Change from baseline in PCS of the SF-36 | |||
Baseline | |||
N | 141 | 59 | NA |
PCS at baseline, mean (SD) | 39.886 (9.20) | 37.191 (9.93) | NA |
Week 66 | |||
N | 136 | 50 | NA |
CFB in PCS of SF-36, LSM (95% CI) | 0.85 (–0.71 to 2.41) | –4.5 (–6.14 to –2.77) | NA |
Difference in LSM (95% CI)b | 5.31 (3.20 to 7.42) | NA | |
P value | 0.00000558 | NA | |
Change from baseline in PND score | |||
Baseline | |||
N | 140 | 59 | NA |
PND score at baseline, mean (SD) | 1.9 (0.88) | 1.9 (0.92) | NA |
Week 66 | |||
N | 134 | 51 | NA |
CFB in PND score, LSM (95% CI) | 0.3 (0.2 to 0.4) | 0.1 (–0.0 to 0.2) | NA |
Difference in LSM (95% CI)b | –0.21 (–0.4 to –0.0) | NA | |
P value | 0.02407897 | NA | |
ANCOVA = analysis of covariance; CFB = change from baseline; CI = confidence interval; LSM = least squares mean; MMRM = mixed model for repeated measures; mNIS + 7 = modified Neuropathy Impairment Score + 7; NA = not applicable; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; NR = not reported; NSC = Neuropathy Symptom and Change; PCS = physical component summary; PND = polyneuropathy disability; SD = standard deviation; SF-36 = Short Form (36) Health Survey; TTR = transthyretin; V30M = Val30Met; vs. = versus.
aBased on an ANCOVA model adjusted by propensity score with the effects of treatment, disease stage, V30M mutation, previous treatment, and baseline value. Data up to week 35 only are included in the week 35 interim analysis. Patients with a missing value at week 35 had values multiply imputed using an imputation model. All 500 imputed datasets were analyzed using a simple ANCOVA model, and the 500 ANCOVA model results were combined using Rubin's rules.
bBased on an MMRM adjusted by propensity score weights with fixed categorical effects for treatment, time, treatment-by-time interaction, disease stage, V30M mutation, previous treatment, and fixed covariates for the baseline value and the baseline-by-time interaction. Data up to week 35 only are included in the week 35 interim analysis, and data up to week 65 only are included in the modelling for end points at week 65 or 66.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023)12,13
Harms
AEs, SAEs, withdrawals due to AEs, and notable harms of interest to this review are summarized in Table 16.
Adverse Events
At least 1 TEAE was reported by 140 patients (97.2%) in the eplontersen group at week 66 and by 60 patients (100%) in the external placebo group in the NEURO-TTR study. In the inotersen-eplontersen group, 24 patients (100%) reported at least 1 TEAE during treatment with inotersen, and 17 patients (85%) reported 1 TEAE following the switch to eplontersen at week 37 to week 66.12
The most frequently reported TEAEs in the eplontersen group were COVID-19 (35 patients [24.3%]); urinary tract infection (24 patients [16.7%]); diarrhea (24 patients [16.7%]); vitamin A deficiency (17 patients [11.8%]); and nausea (16 patients [11.1%]). The most frequently reported TEAEs in the external placebo group were fall (13 patients [21.7%]); fatigue (12 patients [20.0%]); diarrhea (11 patients [18.3%]); urinary tract infection (10 patients [16.7%]); neuralgia (9 patients [15.0%]); pain in extremity, cough, asthenia, and pain (8 patients [13.3%] each); nausea, headache (7 patients [11.7%] each); and nasopharyngitis, dizziness, constipation, thermal burn, hypoesthesia, and muscular weakness (6 patients [10.0%] each). In patients randomized to inotersen, the most frequently reported TEAEs included injection-site erythema (8 patients [34.8%]); pyrexia (7 patients [30.4%]); fatigue (6 patients [26.1%]); nausea, myalgia, injection-site bruising, decreased appetite, chills, and headache (5 patients [21.7%] each); and vomiting, decreased glomerular filtration rate, and arthralgia (4 patients [16.7%] each).12
TEAEs that occurred more frequently in the eplontersen group (3% or more) than in the external placebo group included COVID-19 (24.3% versus 0%), vitamin A deficiency (11.8% versus. 0%), immunization reaction (8.3% versus 0%), vomiting (8.3% versus 5.0%), proteinuria (8.3% versus 3.3%), and blurred vision (5.6% versus 1.7%).12
The majority of TEAEs in the NEURO-TTRansform study were mild or moderate in severity. In the eplontersen group, 64 AEs (44.4%) were mild and 57 AEs (39.6%) were moderate. Only 20 AEs (13.9%) were severe. In the external placebo group, 7 AEs (11.7%) were mild, 39 AEs (65.0%) were moderate, and 14 AEs (23.3%) were severe. In the inotersen-eplontersen group, during weeks 1 to 37 (inotersen treatment), 8 AEs (33.3%) were considered mild, 13 AEs (54.2%) were moderate, and 3 AEs (12.5%) were severe. Following the switch to eplontersen at week 35, 8 AEs (40.0%) were mild, 7 AEs (35.0%) were moderate, and 4 AEs (20.0%) were severe.12
Serious Adverse Events
SAEs were reported in 21 patients (14.6%) in the eplontersen group and 12 patients (20.0%) in the external placebo group. ███ ████ ████████ ██████████ ████ ██ ███ ███████████ █████ ████ ████████ ██ ███████ ████████ ██████████ ███████ ████████ ███ ███████ █████ █████████ ██ ██████ ██████ ███ ████ ████████ ████ ██ ███ ███████ █████ ████████ █████████ ███ █████ ████████ ██ ██████ ██████ Only 3 patients (12.5%) in the inotersen-eplontersen group had SAEs, and ████ █ ███████ ███████████ ██ ███ █████ █████████ ██ ███████████ ████ ██████████ In the inotersen-eplontersen arm, 3 patients (12.5%) receiving inotersen before week 37 experienced at least 1 serious TEAE. ███████ ███ ██ ████ ██████ ██████████ ███████ ██████████████ ███████████████ █████████████████████ ███ █████ ████████ ████ ████████ ██ █ ██████ ███████ ████ ██ ███ █████████ █████ ██ ██ ████ ██
Withdrawals Due to AEs
Treatment-emergent AEs leading to discontinuation of treatment occurred in 6 patients (4.2%) in the eplontersen group and 2 patients (3.3%) in the external placebo group. The TEAEs that led to discontinuation of eplontersen included fatal AEs of arrhythmia and cerebral hemorrhage as well as TEAEs of urosepsis, proteinuria, renal impairment, and abnormal transaminases. In the placebo group, TEAEs that led to treatment discontinuation included proteinuria, arthralgia, pain, and increased weight in 1 patient (1.7%) each. Serious TEAEs leading to discontinuation from treatment were reported in 4 patients (2.8%) in the eplontersen group compared to 0 patients in the external placebo group.12
Three patients (12.5%) discontinued treatment in the inotersen-eplontersen group during treatment with inotersen due to TEAEs of hyperthyroidism, nephroangiosclerosis, and drug eruption (1 patient [4.2%] each). No patients experienced a TEAE leading to treatment discontinuation after switching to eplontersen.12
Mortality
Up to week 66, 2 patients in the eplontersen group had died due to arrhythmia and cerebral hemorrhage. At week 85, 1 additional patient in the eplontersen group had died due to myocardial infarction. At week 66, no patients in the external placebo group or inotersen-eplontersen group had died.12
Notable Harms
Notable harms included in this review consisted of thrombocytopenia and ocular AEs potentially related to vitamin A deficiency.
In the eplontersen group, ██ ████████ ███████ ███ ██████ ███ ███████████ ███████ ██ ███████ █ ██████████ ████████ ██ █ ████████ ███████ ██ ███ █████████████████████ █████ ███ ████████ ██████████ ███ █ ████████ ███████ ███ ████████ ██ ███████████ after week 37. ███ ██████████ ██ ████████ ████ ██████ ███ in the external placebo group from the NEURO-TTR study was █████; however, because investigators in the NEURO-TTR study were blinded to vitamin A levels so as not to inadvertently become unblinded to treatment allocation, no vitamin A-related AEs were reported. The most common ocular AEs reported in the NEURO-TTR study potentially related to ███████ █ ██████████ ████████ ███████ █ ██████████ ███ ████████ ███ ███████ ██████ ██ ████████. Of the 17 vitamin A deficiency AEs, 16 were mild in severity, and 1 was moderate in severity. All TEAEs of vision blurred were mild, and for the TEAE of decreased vitamin A, 2 of the 6 events were mild and 4 were moderate in severity.12 For thrombocytopenia, a total of 3 AEs (2.1%) were reported in the eplontersen group, and 1 AE (1.7%) was reported in the external placebo group at week 66. Four patients (16.7%) who received inotersen during weeks 1 to 37 had AEs of thrombocytopenia; however, no patients had thrombocytopenia AEs following the switch to eplontersen from weeks 37 to 66. None of the thrombocytopenia AEs of special interest in the eplontersen or external placebo groups led to discontinuation of the study drug.12
Table 16: Summary of Harms Results in the NEURO-TTRansform Study at Week 66 (Safety Set)
Adverse events | Eplontersen group (N = 144) | External placebo group (N = 60) | Inotersen-eplontersen group (N = 24) | |
|---|---|---|---|---|
Inotersen (week 1 to week 37) (N = 24) | Eplontersen (week 37 to week 66) (N = 20) | |||
Most common AEs, n (%)a | ||||
Patients with ≥ 1 TEAE | 140 (97.2) | 60 (100) | 24 (100) | 17 (85.0) |
Vitamin A deficiency | 17 (11.8) | 0 | 1 (4.2) | 2 (10.0) |
Urinary tract infection | 24 (16.7) | 10 (16.7) | 2 (8.3) | 3 (15.0) |
COVID-19 | 35 (24.3) | 0 | 1 (4.2) | 2 (10.0) |
Diarrhea | 24 (16.7) | 11 (18.3) | 1 (4.2) | 1 (5.0) |
Vomiting | 12 (8.3) | 3 (5.0) | 4 (16.7) | 1 (5.0) |
Nausea | 16 (11.1) | 7 (11.7) | 5 (21.7) | 0 |
Edema (peripheral) | 12 (8.3) | 5 (8.3) | 0 | 2 (10.0) |
Dizziness | 10 (6.9) | 6 (10.0) | 0 | 1 (5.0) |
Headache | 9 (6.3) | 7 (11.7) | 5 (21.7) | 1 (5.0) |
Pain in extremity | 9 (6.3) | 8 (13.3) | 3 (13.0) | 1 (5.0) |
Arthralgia | 9 (6.3) | 5 (8.3) | 4 (16.7) | 1 (5.0) |
Nasopharyngitis | 8 (5.6) | 6 (10.0) | ||
Fall | 8 (5.6) | 13 (21.7) | 2 (8.7) | 1 (5.0) |
Fatigue | 7 (4.9) | 12 (20.0) | 6 (26.1) | 2 (10.0) |
Injection-site pain | 5 (3.5) | 5 (8.3) | 3 (13.0) | 0 |
Myalgia | 6 (4.2) | 1 (1.7) | 5 (21.7) | 0 |
Anemia | 6 (4.2) | 1 (1.7) | 0 | 0 |
Cough | 7 (4.9) | 8 (13.3) | 3 (13.0) | 0 |
Injection-site erythema | 5 (3.5) | 0 | 8 (34.8) | 0 |
Syncope | 7 (4.9) | 2 (3.3) | 0 | 0 |
Asthenia | 3 (2.1) | 8 (13.3) | 0 | 0 |
Constipation | 4 (2.8) | 6 (10.0) | 0 | 0 |
Decreased GFR | 3 (2.1) | 2 (3.3) | 4 (16.7) | 2 (10.0) |
Neuralgia | 4 (2.8) | 9 (15.0) | 0 | 1 (5.0) |
Injection-site bruising | 1 (0.7) | 2 (3.3) | 5 (21.7) | 0 |
Thrombocytopenia | 1 (0.7) | 1 (1.7) | 0 | 0 |
Chills | 1 (0.7) | 2 (3.3) | 5 (21.7) | 0 |
Decreased appetite | 1 (0.7) | 0 | 5 (21.7) | 0 |
Pain | 1 (0.7) | 8 (13.3) | 2 (8.7) | 0 |
Pyrexia | 2 (1.4) | 5 (8.3) | 7 (30.4) | 0 |
Hypoesthesia | 2 (1.4) | 6 (10.0) | 0 | 0 |
Muscular weakness | 1 (0.7) | 6 (10.0) | 0 | 0 |
Serious adverse events, n (%) | ||||
Patients with ≥ 1 serious TEAE | 21 (14.6) | 12 (20.0) | 3 (12.5) | 1 (5.0) |
Vomiting | 5 (3.5) | 1 (1.7) | 0 | 0 |
COVID-19 pneumonia | 2 (1.4) | 0 | 0 | 0 |
Nausea | 2 (1.4) | 0 | 0 | 0 |
Syncope | 2 (1.4) | 0 | 0 | 0 |
Urinary tract infection | 2 (1.4) | 1 (1.7) | 0 | 0 |
Dehydration | 1 (0.7) | 0 | 0 | 0 |
Pneumonia | 1 (0.7) | 2 (3.3) | 0 | 0 |
Ankle fracture | 0 | 2 (3.3) | 0 | 0 |
Atrial flutter | 0 | 0 | 0 | 1 (5.0) |
Atrioventricular block (complete) | 0 | 0 | 0 | 1 (5.0) |
Cardiac failure | 0 | 1 (1.7) | 0 | 0 |
Soft tissue infection | 0 | 0 | 1 (4.2) | 0 |
Osteomyelitis (chronic) | 0 | 0 | 1 (4.2) | 0 |
Osteoarthritis | 0 | 0 | 1 (4.2) | 0 |
Nephroangiosclerosis | 0 | 0 | 1 (4.2) | 0 |
Tibia fracture | 0 | 0 | 1 (4.2) | 0 |
Patients who stopped treatment due to AEs, n (%) | ||||
Patients with TEAEs leading to treatment discontinuation | 6 (4.2) | 2 (3.3) | 3 (12.5) | 0 |
Abnormal transaminases | 1 (0.7) | 0 | 0 | 0 |
Arrhythmia | 1 (0.7) | 0 | 0 | 0 |
Cerebral hemorrhage | 1 (0.7) | 0 | 0 | 0 |
Proteinuria | 1 (0.7) | 1 (1.7) | 0 | 0 |
Renal impairment | 1 (0.7) | 0 | 0 | 0 |
Urosepsis | 1 (0.7) | 0 | 0 | 0 |
Arthralgia | 0 | 1 (1.7) | 0 | 0 |
Pain | 0 | 1 (1.7) | 0 | 0 |
Weight (increased) | 0 | 1 (1.7) | 0 | 0 |
Hyperthyroidism | 0 | 0 | 1 (4.2) | 0 |
Nephroangiosclerosis | 0 | 0 | 1 (4.2) | 0 |
Drug eruption | 0 | 0 | 1 (4.2) | 0 |
Deaths, n (%) | ||||
Patients who died | 2 (1.4) | 0 | 0 | 0 |
Adverse events of special interest, n (%)b | ||||
Patients with ≥ 1 ocular TEAE potentially related to vitamin A deficiency | ██ ██████ | 9 (15.0) | 4 (16.7) | 3 (15.0) |
Vitamin A deficiency | 17 (11.8) | NA | 1 (4.2) | 2 (10.0) |
Blurred vision | 8 (5.6) | 1 (1.7) | 2 (8.3) | 0 |
Dry eye | 6 (4.2) | 2 (3.3) | 1 (4.2) | 0 |
Decreased vitamin A levels | 6 (4.2) | 0 | 0 | 0 |
Visual impairment | 3 (2.1) | 0 | 0 | 0 |
Patients with ≥ 1 thrombocytopenia TEAE | 3 (2.1) | 1 (1.7) | 6 (25.0) | 0 |
Decreased platelet count | 2 (1.4) | 0 | 2 (8.3) | 0 |
Thrombocytopenia | 1 (0.7) | 1 (1.7) | 4 (16.7) | 0 |
AE = adverse event; GFR = glomerular filtration rate; NA = not applicable; TEAE = treatment-emergent adverse event.
aDefined as TEAEs occurring in greater than or equal to 10% of patients in any treatment arm.
bFrequency greater than 2% in any treatment arm.
Source: NEURO-TTRansform Clinical Study Report (2023).12
Critical Appraisal
Internal Validity
The NEURO-TTRansform study was a randomized, open-label study that utilized an external placebo control from the NEURO-TTR study of inotersen compared to placebo. The choice to conduct a study using external control has implications for the overall strength and interpretability of the results. However, the design, study sites, eligibility criteria, and assessments of disease progression in the NEURO-TTRansform study and NEURO-TTR study were aligned for the purpose of this comparison. The US FDA has endorsed the use of the external placebo control provided that the effect size is large enough to overcome possible bias inherent in the open-label, nonrandomized design. Given that the NEURO-TTRansform study was an open-label study, there was an increased risk of detection bias and performance bias, particularly for subjective outcomes (i.e., patient-reported outcomes or harms); however, the magnitude and direction of this bias with regard to the NEURO-TTRansform study remains unclear.
Adequate randomization methods were employed in the NEURO-TTRansform study, which used an interactive voice or web response system in which patients were randomized 6 to 1 to either eplontersen or concurrent inotersen, followed by a switch to eplontersen at week 37. However, no statistical comparisons of eplontersen versus the concurrent inotersen arm were conducted for randomized patients because the sponsor considered such comparisons infeasible for this rare indication, given the large sample size that would have been required; a comparison was conducted to assess safety only. All statistical comparisons of efficacy were made versus the external placebo control from the NEURO-TTR study. Although the overall sample of randomized patients was small (a situation that can challenge the achievement of prognostic balance across treatment groups), the baseline characteristics were generally well-balanced, with the exception of some baseline scores (i.e., mNIS + 7 and NIS composite scores as well as PND scores), which were generally higher in the eplontersen group, suggesting a population with more severe neuropathy impairment compared to those enrolled in the inotersen-eplontersen group.
Demographics and patient characteristics (i.e., age, race, disease stage, duration of disease from diagnosis, diagnosis of CM associated with hATTR, and previous treatment with tafamidis or diflunisal) differed somewhat between the NEURO-TTR external placebo group and the population of patients in the NEURO-TTRansform study. The NEURO-TTRansform study included younger patients (mean age = 53.0 years versus 59.5 years), a greater proportion of Asian patients (15.%% versus 5.0%) and Black patients (3.5% versus 1.7%), fewer white patients (78.3% versus 88.3%), more patients with FAP and/or Coutinho stage 1 disease (79.9% versus 70.0%), and fewer patients with stage 2 disease (20.1% versus 30.0%). Patients had a longer period of time from diagnosis to enrolment in the NEURO-TTRansform study (46.8 months versus 39.3 months), and the study included fewer patients with hATTR-associated CM (27.1% versus 36.7%) and a greater number of patients with previous tafamidis or diflunisal treatment (69.4% versus 60.0%). The impact of these differences on the results remains unknown; however, the subgroup analyses for these variables were generally consistent with the primary results.
The NEURO-TTRansform study included a week 35 interim analysis and week 66 final analysis, in line with the NEURO-TTR study. The interim analysis included 2 coprimary end points (change from baseline in serum TTR and change from baseline in mNIS + 7 composite score) and 1 key secondary end point (change from baseline in Norfolk QoL-DN); all were coprimary end points at the final analysis. The NEURO-TTRansform study met its coprimary and key secondary end points at the interim analysis, though no further statistical testing was conducted at the final analysis. Results at the final analysis were consistent with those of the interim analysis across all study end points; however, the end points of change from baseline in mNIS + 7 and change from baseline in Norfolk QoL-DN were switched from ANCOVA at the interim analysis to MMRM at the final analysis. Subgroup analyses generally appeared consistent across prespecified subgroups; however, the trial likely not powered to detect subgroup differences.
Given the use of the external placebo control, the MMRM for the coprimary end points was adjusted by propensity score weights for each patient. The propensity score was calculated for each patient using a logistic regression model with baseline covariates, including disease stage, V30M mutation, and previous treatment; however, it was unclear how these covariates were selected or whether all relevant prognostic factors and effect modifiers were considered. Additionally, this method cannot account for differences in known unmeasured or unknown confounders. As such, there is a risk of bias due to residual baseline confounding of unknown magnitude and direction. Additionally, it was unclear whether harms results were adjusted using propensity scoring; given the differences in collection and in the definitions of some AEs, the certainty in conclusions is reduced.
At the interim analysis, given that both coprimary end points and the key secondary end point were significant, no further hypothesis testing of serum TTR, mNIS + 7, or Norfolk QoL-DN was performed at the week 66 final analysis. Statistical models for the coprimary end points at the interim analysis were based on ANCOVA models, while the week 66 final analysis of the coprimary end points was conducted using an MMRM. Although hypothesis testing was not conducted at the final analysis, the rationale for switching models was unclear, and the impact remains unknown. There were some missing data reported for each outcome: upward of 12% of patients treated with eplontersen and 15% of patients treated with placebo (in the NEURO-TTR study) were missing data at the final analysis. In the primary analyses, missing data were imputed using the MAR assumption, but the impact on the direction of treatment effect over time remains unclear. In the final analysis of MMRM models for coprimary end points, missing data were not explicitly imputed; however, the end point treatment differences were adjusted to account for missing data. No information on this process was provided, thus, it remains unclear how missing data were accounted for in the final analysis. Multiple prespecified sensitivity analyses, including the CIR and J2R approaches, were also incorporated to evaluate the pattern of missingness, and the results were consistent with the primary analysis at both analyses; thus, the missing data potentially had minimal impact on the results.
External Validity
The NEURO-TTRansform trial was an international trial conducted in 15 countries, including Canada, which enrolled 3 patients. Patients enrolled were similar to those enrolled in the NEURO-TTR study, given the identical eligibility criteria. The clinical experts consulted for this review indicated that the inclusion and exclusion criteria of the NEURO-TTRansform study were appropriate. Similar to other studies of treatments for hATTR-PN, the NEURO-TTRansform study enrolled adult patients with stage 1 or stage 2 PN with hATTR. Patients receiving current or prior treatment with TTR-lowering treatments were excluded from the NEURO-TTRansform trial. As such, the efficacy of eplontersen in patients who previously received patisiran or vutrisiran is unknown. The clinical experts consulted for this review highlighted that this would not exclude patients from receiving eplontersen, despite the lack of evidence to support switching from other treatments.
In the NEURO-TTRansform study, eplontersen was administered through SC injection by study centre personnel or at home by patients or caregivers. The sponsor noted that at-home self-administration of eplontersen using a prefilled syringe is a potential feature of this treatment in the real world; however, no evidence on the use of eplontersen administered in this manner was available in the NEURO-TTRansform study.
The clinical experts consulted for this review also highlighted that there is limited overlap in outcomes between clinical trials for hATTR-PN in clinical practice, emphasizing that many of the measures included as outcomes in the NEURO-TTRansform trial are not routinely used in Canadian clinical practice. The primary end point of the NEURO-TTRansform study was the percentage change from baseline in serum TTR. Abnormal aggregation of TTR is a fundamental manifestation of hATTR-PN, but the clinical experts noted that serum TTR levels are not measured routinely in clinical practice. Despite being demonstrative of treatment effect and biological plausibility, serum TTR reduction has not been identified as a validated surrogate outcome for clinical outcomes in hATTR-PN. As such, change in serum TTR was not used as a surrogate for important clinical outcomes in this review. However, clinical outcomes that are important to patients were tested, including neurologic impairment and HRQoL. The mNIS + 7 and Norfolk QoL-DN measures have limited application in clinical practice in Canada, given the complexity and time-consuming nature of these tools. The clinical experts noted that COMPASS-31, R-ODS, and other tools (i.e., ONLS and mTCNS) are more frequently used, given their simplicity (however, the use of these tools is not standardized across Canada). However, COMPASS-31 and R-ODS were exploratory outcomes of the NEURO-TTRansform study and did not include comparisons to placebo, precluding conclusions regarding the efficacy of eplontersen relative to any comparator with respect to either of these end points. ONLS and mTCNS were not reported in the NEURO-TTRansform study.
Hospitalizations, an additional clinically relevant outcome, were evaluated in the NEURO-TTRansform study; however, between-group effect estimates with measures of precision were not reported. As such, the precision of the between-group differences could not be judged. Mortality was reported as part of the safety assessment of eplontersen; however, the duration of the NEURO-TTRansform trial (85 weeks) was considered insufficient to capture the impact of treatment on patients’ survival.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform CDA-AMC expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:37,38
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
For the comparison of eplontersen to placebo, which leveraged an external placebo group from the NEURO-TTR study, the certainty of evidence started at low, acknowledging the nonrandomized design, risk for selection bias, and residual baseline confounding. The clinical review team assessed the evidence for study limitations (i.e., internal validity or risk of bias), indirectness, imprecision of effects, and publication bias. In the absence of a comparator (i.e., the study used a single-arm design), appraisals of results for COMPASS-31 and R-ODS started at very low certainty, with no opportunity for rating up. Appraisals of the previously mentioned considerations were included for transparency.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of any effect for serum TTR, mNIS + 7 composite score, and Norfolk QoL-DN total score.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for eplontersen compared to placebo from the NEURO-TTRansform study in the treatment of adult patients with hATTR-PN.
Long-Term Extension Studies
The contents of this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CDA-AMC review team.
One open-label extension study of patients with hATTR-PN from the NEURO-TTRansform study who continued to receive eplontersen after week 85 is currently ongoing. No data were available at the time of this review.
Indirect Evidence
The contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
Given the lack of head-to-head studies comparing the efficacy and/or safety of eplontersen to other treatments for hATTR-PN available in Canada (i.e., vutrisiran, patisiran, and inotersen), the sponsor submitted ITCs to evaluate the comparative efficacy of eplontersen versus other medical therapies used for the treatment of patients with hATTR-PN.14
Description of Indirect Comparisons
The sponsor-submitted ITCs began with an SLR to identify relevant published studies for the treatment of patients with hATTR-PN. A feasibility assessment was performed to determine the most appropriate ITC methods for the included studies based on cross-trial differences in study design, baseline patient characteristics, treatment regimens evaluated, outcome definitions and availability, and time points of outcome assessment. The results of the feasibility assessment concluded that comparisons of eplontersen to relevant comparators of inotersen, partisan, and vutrisiran using MAIC and STC methods were feasible, based on the design of the NEURO-TTRansform study and the overall heterogeneity in design and patient characteristics of the studies included in the ITC (precluding network meta-analysis [NMA]).14
ITC Designs
Objectives
The overall objective of the sponsor-submitted ITCs (i.e., STC and MAIC) was to compare eplontersen to other treatments available in Canada for patients with hATTR-PN (i.e., vutrisiran, patisiran, and inotersen). The specific objectives of the ITC were to:14
conduct feasibility assessments of pairwise ITC methods and NMAs for the comparison of eplontersen and comparators for efficacy end points of interest
if deemed feasible, conduct pairwise ITCs of eplontersen and comparators using methods deemed appropriate by the feasibility assessment and conduct an NMA of medical therapies for the treatment of patients with hATTR-PN.
Study Selection Methods
The sponsor-submitted ITC was informed by an SLR (search date: July 2022) to identify clinical, humanistic, and economic literature on patients with hATTR-PN.14 Details describing the SLR methods and selection criteria are summarized in Table 17. The authors noted that hATTR-PN is a heterogeneous disease; thus, outcomes were broadly defined in the search strategy. As such, some outcomes of interest, such as serum TTR, were not explicitly defined in the search criteria because it was known that these outcomes were not reported in relevant comparator studies (e.g., serum TTR was not reported in the APOLLO trial for patisiran). Comparators of tafamidis and diflunisal are not currently approved in Canada for hATTR-PN and, although included in the search, were not included in the submitted ITC.14
Table 17: Study Selection Criteria and Methods for the Systematic Review Submitted by the Sponsor
Characteristics | Inclusion criteria |
|---|---|
Population | Adults (≥ 18 years) with a diagnosis of ATTR-PN |
Intervention | Eplontersen |
Comparators |
|
Outcome |
|
Study designs | Phase III RCTs |
Publication characteristics |
|
Databases searched |
|
Selection process | Titles, abstracts, and full-text publications were reviewed against the eligibility criteria by 2 independent reviewers in 2 stages (titles and abstracts, then full texts). Disagreements were resolved by discussion or by consulting a third reviewer. |
Data extraction process | Data extraction was performed by a single individual for each included study and verified for accuracy and completeness by a second reviewer. Discrepancies or missing information were discussed until a consensus was reached. If necessary, a third individual was enlisted to arbitrate the final decision. |
Risk-of-bias assessment | A risk-of-bias assessment of each study was conducted using an appropriate tool, dependent on the study design:
The risk-of-bias assessment was completed by 1 individual and verified by a second independent reviewer. |
ATTR-PN = transthyretin amyloidosis with polyneuropathy; CDSR = Cochrane Database of Systematic Reviews; CENTRAL = Cochrane Central Register of Controlled Trials; CRD = Centre for Reviews and Dissemination; HRQoL = health-related quality of life; HTAD = Health Technology Assessment Database; INAHTA = International Network of Agencies for Health Technology Assessment; ITC = indirect treatment comparison; KCCQ-OS = Kansas City Cardiomyopathy Questionnaire–Overall Summary Score; NHSEED = National Health Service Economic Evaluations Database; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; RCT = randomized controlled trial; ROBINS-I = Risk Of Bias In Non-randomized Studies of Interventions; SF-36 = Short Form (36) Health Survey; WPAI = Work Productivity and Activity Impairment.
Source: Sponsor-submitted ITC.14
ITC Analysis Methods
A feasibility assessment was performed to determine the most appropriate ITC methods for the included studies by evaluating the cross-trial differences in study design, baseline patient characteristics, treatment regimens evaluated, outcome definitions and availability, and time points of outcome assessment. The methods considered included Bucher analyses, NMAs, and population-adjusted methods. Based on the results of the feasibility assessment, population adjustment methods (MAIC and STC) were considered most suitable for comparing eplontersen to relevant treatments.14 A summary of the methods used for the unanchored MAICs and STCs is presented in Table 18.
Matching-Adjusted Indirect Comparison
Based on the results of the SLR and subsequent feasibility assessment (described in the following), the sponsor determined that an unanchored MAIC was the most appropriate approach for comparing eplontersen and relevant comparators in patients with hATTR-PN. Comparators of interest included vutrisiran, patisiran, and inotersen.14
The NEURO-TTRansform study was used as the “index trial” in the MAIC. Patients who did not meet the inclusion criteria for the comparator trials were excluded from the NEURO-TTRansform study dataset before the calculation of patient weights. The number of patients excluded was quantified, given that this may contribute to limitations in the analysis. The distribution of adjustment variables was compared between the 2 trials in the MAIC to ensure overlap between the populations. If there was too much variation in the adjustment variables available, a MAIC was deemed not feasible, and an STC was favoured. To perform the matching adjustment of the index trial onto the population of the comparator trial, a logistic propensity score model was fitted such that the reported statistics of the treatment-modifying patient characteristics were equal to the equivalent derived statistics from the index trial when weighted by the log-odds of entering the comparator trial.14
Table 18: Summary of Unanchored MAIC and STC Analysis Methods
Methods | Description |
|---|---|
Analysis methods |
|
Outcomes |
|
Identification and validation of prognostic factors and treatment-effect modifiers |
|
Assessment of variable selection |
|
Weighting assessment |
|
Missing data imputation |
|
Follow-up time point adjustment |
|
AIC = Akaike information criterion; ESS = effective sample size; IPD = individual patient data; LOCF = last observation carried forward; MAIC = matching-adjusted indirect comparison; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; PF = prognostic factor; PND = polyneuropathy disability; SAP = statistical analysis plan; STC = simulated treatment comparison; TEM = treatment-effect modifier; TTR = transthyretin.
aBoth PND score and disease stage (Coutinho stage) are considered PFs and TEMs, but because the 2 are strongly correlated, these cannot be included in the models simultaneously. Coutinho stage is used because it was available in all studies and avoids the issue of very few patients in a category.
Source: Sponsor’s Summary of Clinical Evidence.14
The patient weights were used to calculate the weighted outcome to compare the index and comparator trials. For continuous variables, a weighted linear model was used; for binary outcomes, including responder analyses, a weighted binomial model was used. Weighted distributions of the index baseline variables were also compared to the comparator trial to confirm that the populations were more closely matched after weighting. The resultant ESS was estimated using the methods of Signorovich et al. The distribution of patient weights was assessed through a histogram along with the frequency of extreme patient weights within the matching set. In the reference model, all reported prognostic factors and treatment-effect modifiers were then adjusted for.14
Simulated Treatment Comparison
When an unanchored MAIC could not be performed, an STC was conducted for the same studies in the MAIC analysis. The unanchored STCs used a regression model on the individual patient data (IPD) from the index trial to evaluate the relationship between the trial population characteristics and outcomes. The model was then used to estimate the outcome for the comparator trial population. A linear model was used for continuous variables, and a binomial model was used for binary outcomes, including responder analyses. The reference model adjusted for all prognostic factors and treatment-effect modifiers identified by clinicians.14
Models
Two models were conducted for the ITCs: a reference model and an alternative model for each method and outcome. The reference model adjusted for all prognostic factors and treatment-effect modifiers (which were identified by literature review and validated by 2 clinicians; these are described subsequently). The adjustment variables included in the reference model varied between comparators, depending on the reporting of baseline characteristics in trial publications. Only the HELIOS-A and APOLLO studies reported all the adjustment variables identified by clinicians. The NEURO-TTR study did not report FAP stage; therefore, this covariate could not be included in the reference model case for this comparison.14
The alternative model adjusted for a smaller subset of prognostic factors and treatment-effect modifiers, formed through stepwise selection based on the Akaike information criterion (AIC). For unanchored comparisons, data from the eplontersen arm of the NEURO-TTRansform study were used in the stepwise selection process to derive the alternative model, which was subsequently used to adjust the eplontersen data from the NEURO-TTRansform study to the aggregate data for comparators. In the alternative model for anchored comparisons, eplontersen data from the NEURO-TTRansform study and external placebo data (i.e., from NEURO-TTR) were used in the stepwise process to derive the models with the lowest AIC; these were then combined to form a model that was the sum of the lowest AIC models. The variables selected in the combined model were used to adjust the eplontersen data from the NEURO-TTRansform study to the population characteristics of the comparator trial treatment group (e.g., vutrisiran, patisiran or inotersen) and the placebo arm to the comparator trial placebo group characteristics.14
For the mNIS + 7 composite score and Norfolk QoL-DN total score, both the mean change-from-baseline outcome and the response outcome were analyzed. For these 2 measures, the alternative model was estimated separately.14
For comparisons versus vutrisiran and patisiran, the definition of cardiac involvement from the NEURO-TTRansform study was altered to match that of the HELIOS-A and APOLLO trials. The definition for the cardiac group for these comparisons was a diagnosis of TTR CM at study entry (defined as familial amyloid CM), baseline interventricular septum thickness greater than or equal to 13 mm on echocardiogram, no medical history of hypertension (i.e., no 2 consecutive systolic blood pressure readings ≥ 150 mm Hg at either of these 2 visits), and no medical history of aortic valve disease. However, for comparisons versus inotersen, the original definition of cardiac involvement from the NEURO-TTRansform study was retained. This was defined as a diagnosis of TTR CM at study entry (defined as familial amyloid CM) or baseline interventricular septum thickness greater than or equal to 13 mm on echocardiogram and no medical history of hypertension (i.e., no 2 consecutive systolic blood pressure readings ≥ 150 mm Hg at any time during the study).14
Outcomes
The priority end points for the ITC were those that formed the coprimary end point at the week 66 final efficacy analysis in the NEURO-TTRansform study and included change from baseline in mNIS + 7 composite score, change from baseline in Norfolk QoL-DN total score, and percentage change from baseline in serum TTR concentration at steady state.14
Modified NIS + 7
For all trials, mNIS + 7 was recorded at baseline and again at multiple time points. The version of mNIS + 7 used in the NEURO-TTRansform and NEURO-TTR trials (i.e., mNIS + 7Ionis) differed from that used in the HELIOS-A and APOLLO trials (i.e., mNIS + 7Alnylam). The differences between the 2 instruments meant it was not possible to conduct a fair comparison without rescoring the mNIS + 7 in 1 set of trials to allow for a better comparison between the versions.39
The mNIS + 7Ionis was rescored for the nerve conduction study (NCS) and HRdb (postural blood pressure in mNIS + 7Alnylam) components, given that the NIS motor strength and weakness, NIS reflexes, and quantitative sensory testing domains were the same in the mNIS + 7Ionis and mNIS + 7Alnylam scores. The sensation component of the mNIS + 7Ionis was excluded because it does not form part of the mNIS + 7Alnylam score. Postural blood pressure and HRdb are both measures of autonomic impairment. Rescorings of HRdb and NCS were conducted similarly, where HRdb and NCS normal components from mNIS + 7Ionis were replaced with NCS percentile-based points, as used in mNIS + 7Alnylam, with each component of the NCS scored as 0, 1, or 2, where 0 = ‘No impairment,’ 1 = ‘Mild impairment,’ 2 = ‘Severe impairment,’ based on the 95th and 99th percentiles. All the components, including the rescored components, were then summed to create the closest approximation of the mNIS + 7Alnylam score possible based on the data available from the NEURO-TTRansform study.14
Norfolk QoL-DN
The Norfolk QoL-DN scale was described previously. The Norfolk QoL-DN total scores from the included studies were considered suitable for ITC because these were consistent across studies. The NEURO-TTRansform and HELIOS-A trials also reported all Norfolk QoL-DN individual domain scores. ITCs of the individual domains were performed based on data availability.14
Serum TTR
Based on discussion with the clinical experts consulted by the authors, serum TTR was included as an outcome of clinical interest. Mean serum TTR levels (mg/L) were extracted for vutrisiran and patisiran from the HELIOS-A study at baseline and steady state levels (mg/L) from month 6 to month 18. For the patisiran arm of the APOLLO study, mean serum TTR levels (mg/L) were extracted at baseline. However, mean serum TTR steady state levels at month 6 to month 18 values from APOLLO were not reported and could not be estimated from other reported values with adequate precision. For inotersen, mean serum TTR was calculated from the IPD because these values were not available in the public domain. Thus, the comparison versus inotersen conflicts with the previous decision not to use IPD from the NEURO-TTR study in the ITC. To match the definition of steady state serum TTR used in the HELIOS-A study, the steady state serum TTR values for eplontersen and inotersen were also calculated using predose serum TTR measurements available from month 6 to month 18. In the NEURO-TTR and NEURO-TTRansform studies, the first predose serum TTR measurements between months 6 and 18 were at week 47 and week 49, respectively.14
The following limitations were identified for the outcome of percentage change from baseline in serum TTR by the authors:14
Percentage change from baseline was not normally distributed and could not be treated as a continuous variable in the analysis.
The included trials collected data at different time points relative to the treatments received. It is not appropriate to compare a time point that is shortly after dosing of eplontersen with a time point that is shortly before dosing of patisiran or vutrisiran due to the relationship between dosing schedule and serum TTR.
Consultation with clinical experts validated observations from major clinical trials that show an initial drop in serum TTR concentrations followed by a plateau. Due to this nonlinear behaviour and the relationship between dosing schedule and serum TTR, extrapolation of serum TTR over time was subject to a high degree of uncertainty and was not recommended.
In the APOLLO study, very few time points were measured, and these were not equivalent to those of the NEURO-TTRansform study. Additionally, the number of patients included in the analysis at each time point was not reported, and the measure of error is uncertain.
As a result of these limitations, alternative methods were explored and used for the ITC of serum TTR. Percentage change from baseline in serum TTR levels at each of the serum TTR measurement time points was calculated by dividing the change from baseline in serum TTR at each time point by the baseline serum TTR concentrations and multiplying by 100. The mean percentage change from baseline in serum TTR was then calculated by averaging the percentage change values across the measurement time points. This was done for each patient in the eplontersen arm of the NEURO-TTRansform study. Aggregate mean serum TTR levels at baseline and aggregate mean serum TTR levels for vutrisiran and patisiran from month 6 to month 18 were used to approximate the percentage change from baseline in serum TTR at steady state. For inotersen, patient-level calculations were performed by averaging the serum TTR value across the predose measurements with data from weeks 47, 59, and 65.14
The MAIC and STC analyses for percentage change from baseline in serum TTR made the following assumptions:14
The mean change from baseline in serum TTR at steady state is approximated by the mean absolute serum TTR level at steady state minus the mean serum TTR level at baseline.
The mean percentage change from baseline in serum TTR at steady state is approximated by the mean change in serum TTR at steady state divided by the mean serum TTR level at baseline and multiplied by 100.
Missing Data Imputation for End Points in the ITCs
For serum TTR, imputation was performed using multiple imputation through chained equations of the absolute serum TTR level at each of the trough serum TTR measurement time points (i.e., weeks 47, 57 and 65 for inotersen, and weeks 49, 57, 65, 73, 81, and 85 for eplontersen). Variables included in the imputation were V30M mutation, sex, prior treatment, FAP stage, and cardiac involvement. Each missing value was imputed 100 times and checked for validity (i.e., no negative values or spurious estimates within each set of 100 imputations). Imputed values were then used to calculate the mean percentage change in serum TTR from baseline at steady state.14
For the Norfolk QoL-DN and mNIS + 7, the base-case imputation method was matched to that of the comparator trial: placebo group mean imputation (in the NEURO-TTR study for mNIS + 7), last observation carried forward (LOCF) (in the NEURO-TTR study for Norfolk QoL-DN), and multiple imputation of mean difference (in the HELIOS-A and APOLLO studies).14
Placebo group mean imputation: If at least half of the subcomponents in a mNIS + 7 component were nonmissing, missing subcomponents were imputed as the mean of the patient's scores for other subcomponents of that component. For baseline values, if more than half of the subcomponents in an mNIS + 7 component were missing, missing subcomponent scores were set to the mean of that subcomponent score for patients in the same treatment arm of that study. For postbaseline values, if more than half of the subcomponents in an mNIS + 7 component were missing, then the missing subcomponent score was set to the mean subcomponent score in the placebo arm. Subsequently, component scores were derived from subcomponent scores. Finally, if only 1 component in the mNIS + 7 still had a missing value, then the missing component score was set to the mean component score of the patients in that treatment arm. For the Norfolk QoL-DN at baseline, if at least half of the items in a domain were non-missing (or if 1 out of 3 items in the autonomic neuropathy domain), then the missing items were imputed as mean value from that patient's study (i.e., group-level, but only ever across 2 arms). For postbaseline, missing items were imputed using LOCF (including both observed and imputed values). For the symptom domain, if an impossible combination was selected (i.e., “no symptom X” but also “symptom X present in Y”), then the item was set to missing and imputed using the method described previously.14
LOCF: If a patient had missing data postbaseline, the last observed value was carried forward. If a patient had no recorded values for a component of score, baseline values were mean imputed (using the method described previously), and baseline value carried forward.14
Multiple imputation of mean difference: Missing values at baseline and postbaseline were imputed. Multiple imputations (replacement values) for multivariate missing data were created. The method is based on fully conditional specification, in which each incomplete variable is imputed by a separate model. For a given end point, missing end point values were multiply imputed separately for each treatment group using a regression procedure, with baseline information including baseline score as a covariate and genotype, prior tetramer stabilizer use, FAP stage (I versus II or III), cardiac subpopulation, and sex as factors. For NIS-related end points, the categorical baseline NIS score (e.g., NIS < 50 versus NIS ≥ 50) was not included in the regression procedure.14
Time of Assessment
All included trials reported end points at an interim time and final time point. The NEURO-TTR and NEURO-TTRansform studies reported times of assessment in weeks (interim at week 35 and final at week 65 or 66), while the other trials reported time points in months. (The HELIOS-A and APOLLO studies’ outcomes at 9 months were measured at weeks 36 to 39, and outcomes at 18 months were measured at weeks 78 to 80). Where a range was provided, the upper range was used as the target extrapolation time point in the base case.14
A linear model was fitted to data for eplontersen from baseline and week 66 to predict the value at 80 weeks (in the HELIOS-A and APOLLO studies). Data at baseline and week 66 were used rather than limiting the linear model to data between baseline and week 35 or week 35 to week 66 to estimate the change over the whole period and attain the most accurate determination of the change per week. Given the importance of baseline measurements, it was expected that ignoring baseline values would lead to less accurate predictions. Due to the small number of data points available to fit the models, the authors noted a high level of uncertainty in the predicted values.14
Prognostic Factors and Treatment-Effect Modifiers
Potentially relevant prognostic factors and treatment-effect modifiers were identified by literature review and validated by clinical expert opinion from 2 experts: 1 based in the US and 1 in the UK. Potential prognostic factors and treatment-effect modifiers included age, sex, race, region, disease stage and/or PND score, V30M mutation, prior treatment, cardiac involvement, and outcome (i.e., as measured using mNIS + 7 or Norfolk QoL-DN) at baseline. However, region was excluded as a prognostic factor on the basis that evidence for its inclusion was not available, and that the prognostic factors that might be the reason for inclusion of region in the models have been included as variables (i.e., region may be a surrogate variable for race, V30M mutation, or previous prescription of medication, per the sponsor).14
Additionally, IPD from the placebo arm of the NEURO-TTR study and the eplontersen arm of the NEURO-TTRansform study were used to support the inclusion of the identified prognostic factors and treatment-effect modifiers into the models. Univariate analysis of each of the variables was used to confirm which of the variables may be prognostic factors. Results of the univariate analysis suggested that cardiac involvement, prior treatment, and age did not meet the criteria for confirmation as prognostic factors. The clinicians consulted by the authors noted that prior treatment would be considered a treatment-effect modifier. Cardiac involvement and age were also known effect modifiers, despite the results of the univariate analysis; thus, these were retained in the model.14
Results of the Sponsor-Submitted ITCs
Summary of Included Studies
The sponsor-submitted SLR included a total of 141 articles representing 92 studies (9 RCTs, 13 single-arm trials, 60 observational studies, 8 economic studies, and 2 studies identified through other sources). Six relevant phase III RCTs were identified for inclusion in the SLR and subsequent ITCs, including studies of inotersen (the NEURO-TTR study), patisiran (the APOLLO study), vutrisiran (the HELIOS-A study), tafamidis (the Fx-005 study), diflunisal (Study NCT00294671), and eplontersen (the NEURO-TTRansform study).14
Feasibility Assessment
The feasibility assessment identified several important sources of heterogeneity in study design, baseline patient characteristics, treatment regimens evaluated, outcome definitions and availability, and time points of outcome assessment (Table 19).14
With regards to study design, both the NEURO-TTRansform (eplontersen) and HELIOS-A (vutrisiran) trials had external placebo control arms; thus, analyses anchoring vutrisiran and eplontersen through the external placebo controls would be biased, given that the analyses assume there are no baseline differences between the placebo and active arms within a study. Additionally, both studies included a reference treatment arm (inotersen in the NEURO-TTRansform study and partisan in the HELIOS-A study); however, the sample size in the NEURO-TTRansform study was small and included a crossover design by which patients in the inotersen group switched to eplontersen after 37 weeks. The authors also noted differences in premedication across trials: in the APOLLO and HELIOS-A studies, patients were required to receive premedication (i.e., IV corticosteroid, oral paracetamol, IV histamine 1 receptor, and histamine 2 receptor blockers) before each administration of patisiran to reduce the risk of infusion-related reactions, whereas premedication was not required for the vutrisiran arm of HELIOS-A or in either arm of the NEURO-TTR and NEURO-TTRansform studies.14
Definitions and time points of assessment for outcomes varied across studies, particularly for the mNIS + 7, which was comparable between APOLLO and HELIOS-A studies and between NEURO-TTR and NEURO-TTRansform studies, but not between these pairs of studies. There was misalignment of the time points at which results were reported: in the NEURO-TTRansform and NEURO-TTR studies, interim analyses were conducted at 35 weeks, and final analyses were conducted at 65 or 66 weeks, whereas in the HELIOS-A and APOLLO studies, interim end points were reported at 9 months (range, 36 weeks to 39 weeks), and final end points were reported at 18 months (range, 79 weeks to 80 weeks).14
Differences in baseline patient characteristics were noted across studies. These included differences in mBMI, disease stage, PND score, mNIS + 7 score at baseline, Norfolk QoL-DN score at baseline, and race (refer to Table 20).14
Due to differences in the underlying patient characteristics of each trial, the authors deemed population adjustment comparisons (i.e., MAIC, STC) to be the most suitable methods of comparing treatments between trials. Given the key limitations identified with regard to the reference arms (not designed for statistical analysis and premedication difference) and placebo arms (premedication differences and heterogeneity in baseline population) across all trials, unanchored comparisons were conducted.14
Table 19: Summary of Study Design Characteristics Across Studies Included in the ITC
Characteristics | NEURO-TTRansform study | NEURO-TTR study | HELIOS-A study | APOLLO study | ||||
|---|---|---|---|---|---|---|---|---|
Eplontersen group (n = 144) | Inotersen group (n = 24) | Inotersen group (n = 112) | Placebo group (n = 60) | Vutrisiran group (n = 122) | Patisiran group (n = 62) | Patisiran group (n = 148) | Placebo group (n = 77) | |
Study design characteristics | ||||||||
Sample size | 168 | 172 | 164 | 225 | ||||
Outcome definitions | ||||||||
Serum TTR | PD biomarker measured in g/L. Measured predose as such considered as trough. | PD biomarker measured in mg/dL. Measured predose as such considered as trough. | PD biomarker measured in mg/L. Steady state peak and steady state trough. | PD biomarker measured in mg/L. Measured predose as such considered as trough. | ||||
mNIS + 7 | Composite measure of 5 components: motor strength, reflexes, sensation, nerve conduction, and HRdb. Scoring: –22.3 to 346.3; higher scores indicate more impairment | Same as NEURO-TTRansform study | Composite measure of 5 components: weakness, reflexes, nerve conduction, sensory testing, postural BP; total of 0 points to 304 points; higher score indicates more impairment | Same as HELIOS-A study | ||||
Norfolk QoL-DN | 35-item composite measure with 5 domains;a scores range from –4 to 136; higher scores indicate worse HRQoL | Same as NEURO-TTRansform study | Same as NEURO-TTRansform study | Same as NEURO-TTRansform study | ||||
Time points reported | Serum TTR: % CFB at 5, 9, 13, 25, 35, 49, 57, 65, 73, 81, and 85 weeks mNIS + 7 and Norfolk QoL-DN: CFB at 35, 66, and 85 weeks | Serum TTR: % CFB at 3, 5, 8, 13, 18, 23, 29, 35, 41, 47, 53, 59, and 65 weeks mNIS + 7: CFB and proportion of patients with increase from baseline at 35 weeks and 66 weeks Norfolk QoL-DN: CFB reported at 35 and 66 weeks. | Serum TTR: % CFB at 3, 6, 12, 18, 24, 30, 36, 39, 42, 48, 54, 60, 66, 72, 78, 81, and 84 weeks mNIS + 7: CFB at 36 to 39 weeks and 79 to 80 weeks Norfolk QoL-DN: CFB at 36 to 39 weeks and 79 to 80 weeks | Serum TTR: % CFB at weeks 3, 18, 36 to 39, 57, and 78 to 81a mNIS + 7: CFB and absolute patient improvement % at 36 to 39 weeks and 79 to 80 weeks Norfolk QoL-DN: CFB at 36 to 39 weeks and 79 to 80 weeks | ||||
BP = blood pressure; CFB = change from baseline; HRdb = heart rate response to deep breathing; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; PD = pharmacodynamic; TTR = transthyretin.
aDomains included large-fibre neuropathy, small-fibre neuropathy, autonomic neuropathy, symptoms, and activities of daily living.
Source: Sponsor-submitted ITC.14
Table 20: Summary of Baseline Characteristics Across Studies Included in the ITC (Before Matching and Adjustment)
Characteristics | NEURO-TTRansform study | NEURO-TTR study | HELIOS-A study | APOLLO study | ||||
|---|---|---|---|---|---|---|---|---|
Eplontersen group (n = 144) | Inotersen group (n = 24) | Inotersen group (n = 112) | Placebo group (n = 60) | Vutrisiran group (n = 122) | Patisiran group (n = 62) | Patisiran group (n = 148) | Placebo group (n = 77) | |
Age (years) | ||||||||
Mean (SD) | 53.0 (15.0) | ███ | ███ | ███ | 57.8 (13.2) | ███ | ███ | ███ |
Sex, n (%) | ||||||||
Male | 100 (69.4) | ███ | ███ | ███ | 79 (64.8) | ███ | ███ | ███ |
Race, n (%) | ||||||||
Asian | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
Black or African American | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
Other | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
White | 112 (77.8) | ███ | ███ | ███ | 86 (70.5) | ███ | ███ | ███ |
Missing | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
TTR variant, n (%) | ||||||||
V30M | 85 (59.0) | ███ | ███ | ███ | 54 (44.3) | ███ | ███ | ███ |
hATTR-CM subpopulation, n (%) | ||||||||
Yes | 24 (16.7) | ███ | ███ | ███ | 40 (32.8) | ███ | ███ | ███ |
mBMI (kg/m2 × g/L) | ||||||||
Mean (SD) | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
Missing, n (%) | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
Previous treatment, n (%) | ||||||||
Tafamidis or diflunisal | 100 (69.4) | ███ | ███ | ███ | 75 (61.5) | ███ | ███ | ███ |
Disease stage, n (%) | ||||||||
I | 115 (79.9) | ███ | ███ | ███ | 85 (70) | ██ | ███ | ███ |
II | 29 (20.1) | ███ | ███ | ███ | 37 (3%) | ██ | ███ | ███ |
PND score, n (%) | ||||||||
I | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
II | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
IIIa | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
IIIb | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
Missing | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
mNIS + 7 score, mean (SD) | ||||||||
mNIS + 7Ionis | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
mNIS + 7Alnylam | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
Norfolk QoL-DN total score | ||||||||
Mean (SD) | 44.10 (26.60) | ███ | ███ | ███ | 47.10 (26.30) | ███ | ███ | ███ |
Missing, n (%) | ███ | ███ | ███ | ███ | ███ | ███ | ███ | ███ |
FAP = familial amyloid polyneuropathy; hATTR-CM = hereditary transthyretin amyloidosis cardiomyopathy; mBMI = modified body mass index; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; NR = not reported; PND = polyneuropathy disability; SD = standard deviation; TTR = transthyretin.
Source: Sponsor-submitted ITC.14
Results
To ensure that the eplontersen patient cohort used for the analyses was as homogeneous as possible, a filtering procedure applying the comparator trial eligibility criteria was applied to patients from the NEURO-TTRansform study. Following the filtering of patients by eligibility criteria, 141 patients from the NEURO-TTRansform study were included in the analyses (except for the comparison to inotersen for the Norfolk QoL-DN outcome).14
Baseline characteristics included in the adjustments comparing eplontersen from the NEURO-TTRansform study to vutrisiran from the HELIOS-A study, patisiran from the APOLLO and HELIOS-A studies, and inotersen from the NEURO-TTR study for outcomes included in the MAIC and STC are summarized in Table 21, Table 22, Table 23, and Table 24, respectively. Variables included in the reference models for each comparison were age, sex, race, V30M mutation, previous treatment with tafamidis or diflunisal, disease stage, cardiac involvement, and baseline measurement of the outcome of interest. Disease stage was not included in the reference model for the comparison of eplontersen and inotersen because FAP stage was not reported in the NEURO-TTR study. A smaller subset of the prognostic factors and treatment-effect modifiers was included in the alternative models and is described later in the report.14
For the comparison of eplontersen to vutrisiran (Table 21), following adjustment in the reference model, the ESSs for eplontersen were 77 patients for the mNIS + 7 composite score outcome, 95 patients for the Norfolk QoL-DN outcome, and 96 patients for the serum TTR concentration outcome. The ESSs were slightly greater for all outcomes in the alternative models ██ █ ██ █████████ ███ █████████ ███ ██ ████████ ███ ███ ████████ ██ ██████ █████████ ██████ ███████ ███████ ███ █████ ███ ██████████████ █████████████, based on the adjustment factors included. The reported baseline characteristics were well-balanced following adjustment in the reference model. Following adjustment in the alternative model, differences remained for some variables not included in the adjustment; notable differences remained in V30M mutation and cardiac involvement for the outcomes of mNIS + 7 composite score and Norfolk QoL-DN. For the alternative model of serum TTR concentration, there were differences in age, race, and prior treatment, given that these were the covariates not included in the alternative model.14
███ ███ ██████████ ██ ███████████ ██ █████████ Table 22 ███ Table 23 ██ █████████ ██████████ ██ ███ █████████ ██████ ██████ █████████ ████ ███ ██████ ███ ████████ ███████ ███ ███ ███ ███████████ ███ ██ ███ ██ ████████ ███ ███ ██████ █████████ █████ ████████ ███ ██ ███ ██ ████████ ███ ███ ███████ ██████ ████████ ███ ███ ██████████ ██ ███████████ ██ █████████ ████ ███ ████████ ██████ ███ ███ ███ ███████████ ███ ██ ████████ ███ ███ ███████ ██ █████ ███ ██████████████ ███ ███ ███ ████████ ███████ ███ ███ ████████ ██ ███ ███████████ ██████ ██ █ ██ ███ ██ ████████ ███ ███████ ██ ███ ███ ████████ ███ ███████ ███████ ███ ███ ████████ ███ █████ ███ █████████████ █████ █████████ ██ ███ ██████████ ██ ███ ████████ ████████ █████ ██ ███ ██████████ ███████ █████████ ███ ████████ ████████ ███████████████ ████ ████ ████████ █████████ ██████████ ██ ███ █████████ ██████ ███ ███████ ███ ███████████ ██████ ███ ███ ███████████ ██ ███████████ ███ █████████ ████ ███ ██████ ███ ████████ ██████ ███ ███████████ ██ ████ ████ ███ █████ ████ ██████ ███████████ ██ ████ ███████████ ███ ██████████ ██ ████████ ████ ███████ ████████████ ███ ███████ ███████ ███ ███████████ █████ █████████ ███████████ ██ █████████ ████ ███ ██████ █████ ███ ███████████ ██ ████ ███ ███ ████ ███ █ ██████ ██████████ ██ ███████ ███████████ █████ ███████████ ████ █████████ ███████████ ██ █████████ ████ ███ ████████ ██████ ███ ███████████ █████ ███ ███████████ ██ ████ ███ ██████ ███████████ ██ ██████████ ██ ████ █████████ ██████████ ██ █████ ██████████ ██████████ ██ ███ ██ ███ ██████████ ██ ███████████ ████ ███████ ███████████ █████ ███████████ ███ █████ ███ ██████████████ ███ ███████████ █████ ████████████ ███████████ ██ ████ █████ ███ █████ ██████████ ███ ██████████ ██ ███████████ ██ █████████ Table 24 ██ █████████ ██████████ ██ ███ █████████ ██████ ███ ███ ███ ███████████ ███ ██ ████████ ███ ███ ██████ █████████ █████ ████████ ██ ████████ ███ ███ ███████ ██████ ████████ ███ ██ ████████ ███ ███ █████ ███ █████████████ ████████ ███ ███ ███ ███████ ███ ███ ████████ ██ ███ ███████████ ██████ ██ █ ███ █████████ ██ █████████ ███ ██ ████████ ███ ███ ████████ ██ ██████ █████████ ██████ ███████ ███████ ███ █████ ███ ██████████████ ██████████████ █████ ██ ███ ██████████ ███████ █████████ ███ ██████████ ██████ ███ █████ ████████████ ██████ █████████ █████ ███ ███ ████ ██ ██ ████████ ███ ███ ██████████ ██ ████████ ██ ███ ███████ ███████████ █████ ██ █████████ ██ ███ ███████ ██████████ ██████████ ███ ███████ ██ ███ ██████████ ██ ███ ██████████ ██████ ███ ████████ ████████ ███████████████ ████ ████ ████████ █████████ ██████████ ██ ███ █████████ ███████ ██ ███ ███████████ █████ ███ ███████ █████ ████ ███████ ███████████ ██ ███ █████████ ██████ ██████████ ██ ████ ███████████ ███ ████████ ██████ █████████ ██████ ███ ███ ███████ ██████ ████████ █████ ████ ███████████ ██ ██████████ ██ █████ ████████████ ███ ██████████ ██ ███████████ ████ █████ █████████ █████ ███████████ ███ █████ ███ ██████████████ ███ ███████████ █████ ███ ███████████ ██ █████ ████ ██████ ███████████ ████████ ███ █████ ████████
Table 21: Trial Population Adjustments for Eplontersen (NEURO-TTRansform Study) and Vutrisiran (HELIOS-A Study)
Variable | Vutrisiran (n = 122) | mNIS + 7 composite score | Norfolk QoL-DN total score | Serum TTR concentration at steady state | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
Eplontersen unadjusted (n = 141) | Eplontersen adjusted | Eplontersen unadjusted (n = 141) | Eplontersen adjusted | Eplontersen unadjusted (n = 141) | Eplontersen adjusted | |||||
Reference (ESS = 77) | Alternative (ESS = 88)a | Reference (ESS = 95) | Alternative (ESS = 114)b | Reference (ESS = 96) | Alternative (ESS = 98)c | |||||
Age, mean (SD) | 57.80 (13.20) | 52.25 (15.01) | 57.80 (14.32) | 54.38 (15.28) | 52.48 (14.98) | 57.80 (14.47) | 57.80 (14.66) | 52.48 (14.98) | 57.80 (14.55) | 55.92 (14.91) |
Sex (% male) | 65 | 69 | 65 | 68 | 70 | 65 | 65 | 70 | 65 | 65 |
Race (% white) | 71 | 77 | 71 | 74 | 77 | 71 | 77 | 77 | 70 | 74 |
V30M mutation (%) | 44 | 6 | 44 | 56 | 60 | 44 | 56 | 60 | 44 | 44 |
Prior treatment (%) | 62 | 72 | 62 | 62 | 70 | 62 | 70 | 70 | 62 | 64 |
FAP stage 1 (%) | 70 | 82 | 70 | 70 | 80 | 70 | 74 | 80 | 70 | 70 |
Cardiac involvement (%) | 33 | 16 | 33 | 14 | 17 | 33 | 19 | 17 | 33 | 33 |
Baseline mNIS + 7, mean (SD) | 60.55 (35.99) | 66.32 (35.38) | 60.55 (33.67) | 60.55 (33.19) | — | — | — | — | — | — |
Baseline Norfolk QoL-DN, mean (SD) | 47.10 (26.30) | — | — | — | 43.01 (25.66) | 47.10 (26.62) | 47.10 (26.09) | — | — | — |
Baseline serum TTR, mean | ███ | — | — | — | — | — | — | ██████ | ██████ | ██████ |
ESS = effective sample size; FAP = familial amyloidotic polyneuropathy; ITC = indirect treatment comparison; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk quality of life–diabetic neuropathy; SD = standard deviation; TTR = transthyretin; V30M = Val30Met.
Note: Multiple imputation of mean difference was conducted for mNIS + 7 composite score and Norfolk QoL-DN total score, and for steady state mean serum TTR concentration. Linear extrapolation to week 80 was conducted for mNIS + 7 composite score and Norfolk QoL-DN total score. No time adjustment was conducted for serum TTR concentration at steady state.
aThe alternative model adjusted for previous treatment, FAP stage, and baseline mNIS + 7 total score.
bThe alternative model adjusted for age, sex, and baseline Norfolk QoL-DN total score.
cThe alternative model adjusted for sex, V30M mutation, FAP stage, cardiac involvement, and serum TTR at baseline.
Source: Sponsor-submitted ITC.14
Table 22: Trial Population Adjustments for Eplontersen (NEURO-TTRansform Study) and Patisiran (APOLLO Study)
Variable | Patisiran ██ █ | mNIS + 7 composite score | Norfolk QoL-DN total score | ||||
|---|---|---|---|---|---|---|---|
Eplontersen unadjusted ██ █ | Eplontersen adjusted | Eplontersen unadjusted ██ █ | Eplontersen adjusted | ||||
Reference ██ █ | Alternative ██ █ | Reference ██ █ | Alternative ██ █ | ||||
Age, mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Sex (% male) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Race (% white) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
V30M mutation (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Prior treatment (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
FAP stage 1 (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Cardiac involvement (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Baseline mNIS + 7, mean (SD) | ████ | ████ | ████ | ████ | — | — | — |
Baseline Norfolk QoL-DN, mean (SD) | ████ | — | — | — | ████ | ████ | ████ |
ESS = effective sample size; FAP = familial amyloidotic polyneuropathy; ITC = indirect treatment comparison; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk quality of life–diabetic neuropathy; SD = standard deviation; V30M = Val30Met.
Note: Multiple imputation of mean difference was conducted for mNIS + 7 composite score and Norfolk QoL-DN total score. Linear extrapolation to week 80 was conducted for mNIS + 7 composite score and Norfolk QoL-DN total score.
aThe alternative model adjusted for previous treatment, FAP stage, and baseline mNIS + 7 total score.
bThe alternative model adjusted for age, sex, and baseline Norfolk QoL-DN total score.
Source: Sponsor-submitted ITC.14
Table 23: Trial Population Adjustments for Eplontersen (NEURO-TTRansform Study) and Patisiran (HELIOS-A Study)
Variable | Patisiran ██ █ | mNIS + 7 composite score | Norfolk QoL-DN total score | Serum TTR concentration at steady state | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
Eplontersen unadjusted ██ █ | Eplontersen adjusted | Eplontersen unadjusted ██ █ | Eplontersen adjusted | Eplontersen unadjusted ██ █ | Eplontersen adjusted | |||||
Reference ██ █ | Alternative ██ █ | Reference ██ █ | Alternative ██ █ | Reference ██ █ | Alternative ██ █ | |||||
Age, mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Sex (% male) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Race (% white) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
V30M mutation (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Prior treatment (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
FAP stage 1 (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Cardiac involvement (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Baseline mNIS + 7, mean (SD) | ████ | ████ | ████ | ████ | — | — | — | — | — | — |
Baseline Norfolk QoL-DN, mean (SD) | ████ | — | — | — | ████ | ████ | ████ | — | — | — |
Baseline serum TTR, mean | ████ | — | — | — | — | — | — | ████ | ████ | ████ |
ESS = effective sample size; FAP = familial amyloidotic polyneuropathy; ITC = indirect treatment comparison; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk quality of life–diabetic neuropathy; SD = standard deviation; TTR = transthyretin; V30M = Val30Met.
Note: Multiple imputation of mean difference was conducted for mNIS + 7 composite score and Norfolk QoL-DN total score, and for steady state mean serum TTR concentration. Linear extrapolation to week 80 was conducted for mNIS + 7 composite score and Norfolk QoL-DN total score. No time adjustment was conducted for serum TTR concentration at steady state.
aThe alternative model adjusted for previous treatment, FAP stage, and baseline mNIS + 7 total score.
bThe alternative model adjusted for age, sex, and baseline Norfolk QoL-DN total score.
cThe alternative model adjusted for sex, V30M mutation, FAP stage, cardiac involvement, and serum TTR at baseline.
Source: Sponsor-submitted ITC.14
Table 24: Trial Population Adjustments for Eplontersen (NEURO-TTRansform Study) and Inotersen (NEURO-TTR Study)
Variable | Inotersen ██ █ | mNIS + 7 composite score | Norfolk QoL-DN total score | Serum TTR concentration at steady state | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
Eplontersen unadjusted ██ █ | Eplontersen adjusted | Eplontersen unadjusted ██ █ | Eplontersen adjusted | Eplontersen unadjusted ██ █ | Eplontersen adjusted | |||||
Reference ██ █ | Alternative ██ █ | Reference ██ █ | Alternative ██ █ | Reference ██ █ | Alternative ██ █ | |||||
Age, mean (SD) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Sex (% male) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Race (% white) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
V30M mutation (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Prior treatment (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Cardiac involvement (%) | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ | ████ |
Baseline mNIS + 7, mean (SD) | ████ | ████ | ████ | ████ | — | — | — | — | — | — |
Baseline Norfolk QoL-DN, mean (SD) | ████ | — | — | — | ████ | ████ | ████ | — | — | — |
Baseline serum TTR, mean | ████ | — | — | — | — | — | — | ████ | ████ | ████ |
ESS = effective sample size; FAP = familial amyloidotic polyneuropathy; ITC = indirect treatment comparison; LOCF = last observation carried forward; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk quality of life–diabetic neuropathy; SD = standard deviation; TTR = transthyretin; V30M = Val30Met.
Note: Multiple imputation of mean difference was conducted for mNIS + 7 composite score and for steady state mean serum TTR concentration. The LOCF method was used for imputation of the Norfolk QoL-DN total score. No time adjustment was conducted for mNIS + 7 composite score, Norfolk QoL-DN total score, or serum TTR concentration at steady state.
aThe alternative model adjusted for sex and baseline mNIS + 7 total score.
bThe alternative model adjusted for age, sex, cardiac involvement, and baseline Norfolk QoL-DN total score.
cThe alternative model adjusted for sex, V30M mutation, cardiac involvement, and serum TTR at baseline.
Source: Sponsor-submitted ITC.14
Results of the MAIC and STC analyses using the reference and alternative models for the outcomes of change from baseline in mNIS + 7, change from baseline in Norfolk QoL-DN, and percentage change from baseline in serum TTR concentration are summarized in Table 25.
MAIC Results
██ ███ █████████ █████ ███ ███ ██████ ████ ████████ ██ ███████ █████ ████ ██ █████████████ ███████████ ███████████ ████████ ███████ ███████████ ███ ██████████ ████ ███ ████████ ██████ █████████ ████ ███ ████████ ██████ ██ █████████ ████ ███ █████████ ██████ ███████ ███ ███ ██████████ ██ ███████████ ███ █████████ ████ ███ ██████ █████ ████ █████████████ ███████████ ██ ██████ ██ █████████ ████ █████ ████ ███ ████ ██ ████████. In the alternative models, there were no statistically significant differences detected between eplontersen and vutrisiran from the HELIOS-A trial, ██ █████████ ████ ███ ████████ ██████ ███ ████████ ██ █ █████████████ ███████████ ███████████ ██ ██████ █████████ █████ ████████ ██ █████████ ████ █████ ████ ███ ██████ ██ ████████ ██ ████ ███ █████████ ██████ █████████ ████ ███ ██████ █████ ███ ████████ ████ ███████████ ████ ████ ████ ███ ████ ██ ███████
In the reference model for the change from baseline in Norfolk QoL-DN, comparisons of eplontersen to vutrisiran from the HELIOS-A trial ███ █████████ ████ ███ █████████ █████ ████████████ █ █████████████ ███████████ ███████████ ██ ███████ ██████ █████ █████ ████ ████████ ████ █████ ████ ███ ██████ ██ ██████ ███ ███ █████ ████ ███ ██████ ██ ███████ ██████████████ ███████████ ██ ███████████ ██ █████████ ████ ███ ██████ ██ ████████ ██████ ███ ███ ███████████ █ █████████████ ███████████ ██████████ ██ ██████ ████ ████████ ██ ███████ ██████ █████ ██████ ███████ ███ ███ ███████████ ██████ ████ ██████████ ████ ███ █████████ ███████ ██████ ███ ███ ██████████ ██ ███████████ ███ █████████ ████ ███ ████████ ██████ ███████████ ███ ████████ ████ █████████ ████ █████ ████ ███ ██████ ██ ███████
██ ███ █████████ █████ ███ ███ ██████ ████ ████████ ██ █████ ███ ██████████████ ███ ██████████ ██ ███████████ ███ █████████ ████ ███ ████████ █████ ███ ███████████ ███ █████████ ████ ███ █████████ █████ ████████████ █████████████ ███████████ ██████████ ██ █████ ███ █████████████ ████ █████ ████ ███ █████ ██ ███████ ███ ███ █████ ████ ███ ██████ ██ ███████ ██████████████ However, there was no statistically significant difference detected between eplontersen and vutrisiran. ███████ ███ ███ ███████████ █████ ████ █████████ ██████████ ████ ███ █████████ ██████ ████████ ██ █████████████ ███████████ ██████████ ███ ████████ ███ ███ ██████████ ██ ███████████ ███ █████████ ████ ███ ████████ █████ ████ █████ ████ ███ █████ ██ ███████ ███████ ██████ ████ ████████ ██ █████ ███ █████████████ ███ ███ █████████ ██ ███ ██████████ ███████ ███████████ ███ █████████ ████ ███ ██████ █████
STC Results
Results of the reference and alternative models in the STC were consistent with the MAIC for the outcome of change from baseline in mNIS + 7 composite score, with statistically significant reductions for the comparison of eplontersen and inotersen from the NEURO-TTR trial ████ █████ ████ ███ ██████ ██ ███████ in favour of eplontersen in the alternative model, and for the comparison of ███████████ ███ █████████ ████ ███ ██████ █████ ██ ████ ███ █████████ ████ ████ ████ ███ ████ ██ ███████ ███ ███████████ ██████ ████ ████ ████ ███ ████ ██ ████████ ██ ██████ ██ █████████
███ ██████ ████ ████████ ██ ███████ ███████ ███████ ████ █████████ ██████████ ████ ███ █████ █████ ███████████ ████████████ █████████████ ███████████ █████████ ██ ███████ ██████ █████ █████ ██ ████ ███ █████████ ███ ███████████ ██████ ███ ███ ███████████ ████ ███████████ █████████ ████ ███ ████████ ██████ ███ ██████████ ████████ ██ █████████████ ███████████ ██████████ ███ ████████ ███ ███████████ ███ █████████ ████ ███ ██████ ████
For the change from baseline in serum TTR, results for the STC suggested that eplontersen resulted in statistically significant reductions in serum TTR concentration compared to inotersen in the NEURO-TTR study in both the reference ████ █████ ████ ███ ██████ ██ ███████ ███ ███████████ ████ █████ ████ ███ ██████ ██ ███████ ███████ ██ █████████████ ███████████ ███████████ ████ ████████ ███ ███ ███████████ ██ ███████████ ███ ██████████ ██████████ ██ █████████ ███████████ ██ ████ ███ █████ ███████ ██████ ████ ████████ ██ █████ ███ █████████████ ███ ███ █████████ ██ ███ ███ ███████ ███████████ ███ █████████ ████ ███ ██████ █████
Table 25: Summary of Unanchored MAIC and STC Results for Outcomes of Interest
Comparator (eplontersen vs.) | Model | CFB in mNIS + 7 composite score (MD [95% CI]) | CFB in Norfolk QoL-DN total score (MD [95% CI]) | Percentage CFB in serum TTR concentration (MD [95% CI]) |
|---|---|---|---|---|
MAIC | ||||
Vutrisiran (HELIOS-A study)a | Reference | ████ ████ | ████ ████ | ████ ████ |
Alternative | ████ ████ | ████ ████ | ████ ████ | |
Patisiran (APOLLO study)a | Reference | ████ ████ | ████ ████ | — |
Alternative | ████ ████ | ████ ████ | — | |
Patisiran (HELIOS-A study)a | Reference | ████ ████ | ████ ████ | ████ ████ |
Alternative | ████ ████ | ████ ████ | ████ ████ | |
Inotersen (NEURO-TTR study)b | Reference | ████ ████ | ████ ████ | ████ ████ |
Alternative | ████ ████ | ████ ████ | ████ ████ | |
STC | ||||
Vutrisiran (HELIOS-A study)a | Reference | ████ ████ | ████ ████ | ████ ████ |
Alternative | ████ ████ | ████ ████ | ████ ████ | |
Patisiran (APOLLO study)a | Reference | ████ ████ | ████ ████ | — |
Alternative | ████ ████ | ████ ████ | — | |
Patisiran (HELIOS-A study)a | Reference | ████ ████ | ████ ████ | ████ ████ |
Alternative | ████ ████ | ████ ████ | ████ ████ | |
Inotersen (NEURO-TTR study)b | Reference | ████ ████ | ████ ████ | ████ ████ |
Alternative | ████ ████ | ████ ████ | ████ ████ | |
CFB = change from baseline; CI = confidence interval; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; MD = mean difference; mNIS + 7 = modified Neuropathy Impairment Score + 7; Norfolk QoL-DN = Norfolk Quality of Life–Diabetic Neuropathy; STC = simulated treatment comparison; TTR = transthyretin; vs. = versus.
Note: Point estimates and 95% CIs less than 0 favour eplontersen.
aResults for comparisons to vutrisiran and patisiran were extrapolated to week 80.
bResults for comparisons to inotersen were conducted at week 66.
Source: Sponsor-submitted ITC.14
Critical Appraisal of Sponsor-Submitted ITCs
The sponsor-submitted MAIC and STCs were informed by an adequately conducted SLR that included planned searches of multiple databases. Screening was conducted based on standard methods, with studies selected independently in duplicate, according to prespecified criteria. Risk-of-bias assessments of the included studies were conducted per University of York Centre for Reviews and Dissemination criteria, which contain all domains with empirical evidence of potential to introduce bias. However, the results of this quality assessment were not included in the submitted report. As a result, the potential impact of study-level biases on the results of the MAICs and STCs could not be comprehensively judged.
In total, 4 trials were identified for inclusion in the sponsor’s feasibility assessment. These evaluated eplontersen in the NEURO-TTRansform study, vutrisiran in the HELIOS-A study, patisiran in the HELIOS-A and APOLLO studies, and inotersen in the NEURO-TTR study. Given the heterogeneity observed, the sponsor’s feasibility assessment determined that MAIC and STC methods were most appropriate for comparing eplontersen and relevant comparators. This choice was justified by the lack of a common comparator across the included trials, which precluded the formation of connected networks, and the unique design ██ ███ ███████ ████ ████████ ██████████ █████████ ████ █████████████████ ██████████ ████ ███ ████████ ██████████ ████ █████████ █████ ███████ ██ █████████████ ██ ███ ████████ ███████ ████ ████████ ███████████████ ██ ███ ████████ ████ ████ ██ ████ █████ ██████ █████████ ████████ ██████████ ██ ████████ ███ ████████ ██ █████ ████████ ████ █████ ██ ███████ ██████████ ██ ████████ ████ ████ ████████ ████████ ████ █████ ██ ███████ ██████████ ██ ████████ ████ █████ ████ ██ ████████ ████ █████ ██ ███████ ██████████ ██ ████████ ████ ████████ █████████ ████ █████████ ██ ██████████ ████████ ████ █████ ██ ███████ ██████████ ██ ████████ ████ █████ █ ████████ ████ █████ ██ ██████ ███ █████ ██ ████████ ████ █████ ██ ██████ ████████ ██ ████ ██ ███████████ ██ ███████ ███████ ██████ ████ Table 19.
Given that there were multiple comparator trials, separate ITCs were conducted for comparisons of eplontersen to each of the relevant comparators. When conducting a MAIC or STC, the inclusion criteria for the index study should be the same as or broader than the comparator study to allow for matching on inclusion and exclusion criteria. After applying the exclusion criteria of the comparator trials to the NEURO-TTRansform trial, the original sample size of 144 patients from the eplontersen arm was reduced to ███ ████████ for most comparisons and outcomes, and to ███ ████████ for 1 outcome in the comparison to inotersen. It was unclear which criteria from the comparator studies were applied to the NEURO-TTRansform study; as such, the implications for the generalizability of the results are uncertain. A reference and alternative matching model were then conducted to optimize the ESS for each comparison. In the reference models, comparisons of eplontersen to vutrisiran resulted in further sample-size decreases of 31.9% to 45.4% across outcomes. Comparisons of eplontersen to patisiran from the APOLLO and HELIOS-A trials resulted in sample-size decreases of █████ ██ █████ ███ █████ ██ ██████ █████████████ . Comparisons of eplontersen to inotersen resulted in sample-size decreases of █████ ██ ██████ Sample-size decreases were generally smaller in the alternative models, given that fewer variables were included in the adjustment. Interestingly, there was a large reduction in sample size for the reference model for the comparison to inotersen, despite the uniform eligibility criteria. Given the reduction in ESS, there was likely considerable heterogeneity between studies among the variables included in the weighting process. Despite the substantial reduction in ESS for nearly all comparisons following the matching and adjustment, the populations in all MAIC and STC analyses were relatively balanced. Substantial reductions in ESS have implications for generalizability and the precision of effect estimates.
The key limitation of the sponsor-submitted MAIC and STC, which is a limitation inherent to all unanchored MAICs and STCs, is that it assumes that all treatment-effect modifiers and prognostic factors are accounted for in the model. This assumption is largely considered impossible to meet, according to the National Institute for Health and Care Excellence (NICE) Decision Support Unit Technical Guidance report on methods for population-adjusted ITCs. A list of prognostic factors and treatment-effect modifiers identified through appropriate channels was included in the report, and based on discussions with the clinical experts consulted for this review, were considered relevant; however, it was noted by the clinical experts that the exclusion of region as a factor may bias the results because there may be regional variation in health care access and treatment approaches that are not related to the V30M mutation. Additionally, the clinical experts noted the heterogeneity in the number and type of mutations in hATTR, each potentially influencing disease progression and treatment response; however, the impact of this remains uncertain. Further, the treatment-effect modifiers and prognostic factors accounted for in the models included only those that were reported within the included trials; unknown (measured or unmeasured) and known, unmeasured treatment-effect modifiers and/or prognostic factors could not be accounted for using the MAIC or STC methods.
Methodological or design differences across trials — such as in blinding, outcome measurement, and/or time point or end point assessment — also could not be adjusted for through these methods, resulting in additional uncertainty. The indirect evidence focused on 3 key outcomes: change from baseline in mNIS + 7, change from baseline in Norfolk QoL-DN, and change from baseline in serum TTR. There were differences in the definitions of some outcomes across trials as well as the times of assessment, particularly the application of the mNIS + 7. Two versions of the mNIS + 7 were utilized in the analyses (mNIS + 7Ionis and mNIS + 7Alnylam composite scores), and naive comparison of these 2 composite scores would have been inappropriate. As such, the mNIS + 7Ionis composite score from NEURO-TTRansform study was rescored; however, the rescored versions are not validated for use and may not be appropriate, given that certain domains are not captured in the rescored version. Any interpretation of the comparative results for the mNIS + 7 composite score should consider this limitation. Multiple missing data imputation methods were described and used for the outcomes in the ITC, including placebo group mean imputation (in the NEURO-TTR study for mNIS + 7), LOCF (in the NEURO-TTR study for Norfolk QoL-DN), and multiple imputation of mean difference (in the HELIOS-A and APOLLO studies). However, the amount of missing data for each outcome was unclear; thus, no conclusions could be drawn as to how this may have affected the results. Additionally, outcomes for the mNIS + 7 composite score and Norfolk QoL-DN total score were extrapolated to match the time points reported in the comparator trial, which may introduce uncertainty into the magnitude of any estimates of treatment effect.
In the reference model, for the outcome of mNIS + 7, there was generally insufficient evidence to determine whether eplontersen or the comparator treatments was favoured given the wide 95% CIs that included the potential for no difference or that either treatment could be favoured. For the Norfolk QoL-DN and change from baseline in serum TTR outcomes, eplontersen was often favoured statistically over other treatments, though uncertainty remained, given the wide 95% CIs. For example, the upper bounds of the 95% CIs for between-group differences in Norfolk QoL-DN scores often included effects that appeared small. In the absence of known MIDs, it is uncertain whether these would be considered clinically important differences.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Discussion
Summary of Available Evidence
The evidence included in this review consisted of 1 pivotal study (the NEURO-TTRansform study) and 1 sponsor-submitted ITC.
The NEURO-TTRansform study was an 85-week, phase III, multicentre, randomized, open-label study to evaluate the efficacy of eplontersen after 65 weeks compared to placebo in adult patients with genetic confirmation of hATTR-PN. Patients were randomized 6 to 1 to eplontersen 45 mg SC once every 4 weeks (n = 144) or to 300 mg inotersen SC once per week for up to 34 weeks before switching to eplontersen SC once every 4 weeks from week 37 to week 81 (n = 24). The NEURO-TTRansform study also included an external control group using the placebo arm (n = 60) from the NEURO-TTR study, with adjustment for a select set of baseline covariates using propensity scores. The NEURO-TTR trial was a phase II and III, double-blind, placebo-controlled study that compared the efficacy and safety of inotersen 300 mg SC injection weekly with placebo in patients with stage 1 or 2 hATTR-PN. The eligibility criteria for the NEURO-TTR and NEURO-TTRansform studies were identical. The coprimary end point of the NEURO-TTRansform study was the percentage change from baseline in serum TTR and change from baseline in mNIS + 7 at weeks 35 and 65. The change from baseline in Norfolk QoL-DN was a key secondary end point of the study at week 35, but was included as a third coprimary end point at the week 66 final analysis.
Baseline characteristics of the eplontersen group in the NEURO-TTRansform study and the external placebo group in the NEURO-TTR study were generally well-balanced. In the NEURO-TTRansform study, the mean age was 53.0 years (SD = 15.0 years) in the eplontersen group. The mean age in the placebo group of the NEURO-TTR study was 59.5 years (SD = 14.1 years). In the NEURO-TTRansform study, most patients had stage 1 hATTR-PN (115 patients [79.9%]) and V30M mutations (85 patients [59.0%]), while in the NEURO-TTR study placebo group, 42 patients (70.0%) patients had stage 1 hATTR-PN and 33 patients (55.0%) had V30M mutations. In the NEURO-TTRansform study eplontersen group at baseline, the mean serum TTR was 0.2 g/L (SD = 0.1 g/L); the mean mNIS + 7 composite score was 81.3 (SD = 43.4); and the mean Norfolk QoL-DN total score was 44.1 (SD = 26.6). In the NEURO-TTR study placebo group, the mean serum TTR was 0.2 g/L (SD = 0.04 g/L); the mean mNIS + 7 composite score was 74.8 (SD = 39.0); and mean Norfolk QoL-DN total score was 48.7 (SD = 26.8). Based on the lower mNIS + 7 scores and higher Norfolk QoL-DN scores, patients in the NEURO-TTR study may have had less severe disease and better HRQoL at baseline than those in the NEURO-TTRansform study.
One sponsor-submitted ITC was summarized and critically appraised, consisting of a MAIC and STC that compared eplontersen to vutrisiran, patisiran, and inotersen for the outcomes of change from baseline in mNIS + 7, change from baseline in Norfolk QoL-DN, and percentage change from baseline in serum TTR.
One open-label extension study of patients with hATTR-PN who are continuing to receive eplontersen after week 85 in the NEURO-TTRansform study is currently ongoing. No data were available at the time of this review.
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Interpretation of Results
Efficacy
The efficacy of eplontersen in adult patients with hATTR-PN was informed by the NEURO-TTRansform trial. The NEURO-TTRansform study featured a design that incorporated an external placebo control. Per the sponsor, a concurrent placebo group was considered unethical, given the existence of multiple approved therapies for hATTR-PN. Additionally, the sponsor noted that an active-controlled study design would have required a larger sample size than what was considered feasible for this rare disease. Additionally, the US FDA was open to using an external placebo control, based on the expectation that the effect size would be large enough to overcome possible bias inherent in the nonrandomized design. However, despite the acceptability of the study design, inherent limitations arise from the lack of randomization, including a greater risk of bias from imbalances in baseline prognostic factors; these decrease the certainty of the evidence. To mitigate some of the uncertainty, the sponsor’s external control was the NEURO-TTR study, which included identical eligibility criteria to minimize the differences between populations.
Because the treatments being studied pertain to a rare disease, the sample size of the included study was small; however, it was adequately powered for the coprimary end points. A larger sample size was likely infeasible due to the rare nature of the condition. The clinical experts consulted for this review also highlighted that the characteristics of the enrolled patients were generally similar to patients in real-world clinical practice despite the small sample sizes.
The primary outcomes of the NEURO-TTRansform study were consistent with the outcomes used in trials for hATTR-PN; however, the clinical experts consulted for this review highlighted that the use of serum TTR, mNIS + 7, and Norfolk QoL-DN as outcome measures in clinical practice is very limited. Additionally, considering the nonrandomized study design utilizing an external control arm, the potential for bias resulting from confounding, and the potential for selection bias, the certainty of evidence for the GRADE assessment started at low.
The overarching aim of treatment in hATTR-PN is stabilization of the disease, such that functional outcomes (e.g., mobility and lower extremity function) do not progressively deteriorate. For the coprimary end point of percentage change from baseline in serum TTR, there was a substantial improvement in circulating TTR levels compared to placebo, indicating that there is some drug activity with eplontersen. The clinical experts consulted for this review noted that a 50% reduction in serum TTR is generally considered clinically meaningful; this was achieved in the eplontersen group, as well as compared to placebo. However, the relationship between circulating TTR and functional outcomes remains unknown because change in TTR is not a validated surrogate for clinical outcomes. Results for the within-group change from baseline in mNIS + 7 composite score at the interim and final analyses were generally unchanged for patients treated with eplontersen, while those treated with placebo experienced deterioration, suggesting stabilization of disease and lesser neurologic impairment relative to placebo. As noted by the clinical experts, clinician groups, and patient groups, stabilization is important because improvements or reversal of disease are unlikely for these patients. For the Norfolk QoL-DN, results were consistent with the findings for the mNIS + 7 in that the difference compared to placebo favoured eplontersen at both time points, suggesting improvements in daily living, physical functioning, neuropathy symptoms, and HRQoL with eplontersen. The clinical experts consulted for this review indicated that a 5- to 10-point decrease in Norfolk QoL-DN score typically signifies clinically important improvement; thus, the results for the Norfolk QoL-DN using these thresholds were considered clinically meaningful. Utilizing the null to determine clinically relevant stabilization or improvement, eplontersen may result in lesser neurologic impairment and better HRQoL compared to placebo. Previous literature has cited correlation between the mNIS + 7Ionis and the Norfolk QoL-DN in hATTR;32 however, the absolute impact that these outcome measures have on each other remains unknown. Overall, the results of the neurologic function, neuropathy, and HRQoL measures suggest disease stability, while the results for placebo suggest disease progression.
As noted, many of the clinical trial outcomes are too burdensome to apply in clinical practice. However, these outcomes adequately address neurologic and functional impairment and change. The clinical experts consulted for this review highlighted that other outcome measures, including COMPASS-31, R-ODS, and hospitalizations, may be more clinically relevant to real-world practice. Although these were evaluated in the NEURO-TTRansform trial, these were exploratory, and the results for COMPASS-31 and R-ODS were noncomparative. Overall, the results for these outcomes were stable, with limited changes from baseline. Despite the lack of a randomized comparison group, the clinical experts highlighted that stability in symptoms and disability would be unlikely without treatment because the natural course of hATTR is progressive.
Given the lack of direct comparative evidence for eplontersen, the sponsor submitted an ITC to compare eplontersen to relevant treatments for hATTR-PN. There were inherent differences in the study design, eligibility criteria, and baseline characteristics, including potential prognostic factors and treatment-effect modifiers. Using the MAIC methodology, following the adjustment of select characteristics, the populations were comparable; however, the substantial reductions in the sample size of the eplontersen groups limit the precision of the findings because these suggest considerable heterogeneity between the included studies among the variables included in the weighting process. The results of the ITCs generally suggested that there was insufficient evidence to detect a difference between eplontersen and other available treatments with respect to the mNIS + 7, with patisiran only mildly favoured over eplontersen. For the Norfolk QoL-DN and serum TTR outcomes, eplontersen was mostly favoured over other treatments, although there was insufficient evidence to detect a difference with vutrisiran. However, all results were associated with wide 95% CIs, often including the potential for no difference, or for either of the treatments being compared to be favoured. Overall, given the limitations outlined previously, it remains uncertain whether eplontersen provided additional benefits versus currently available therapies.
Harms
The safety analysis for eplontersen was based on the safety set, which included 144 patients treated with eplontersen, the 24 patients treated with inotersen from week 1 to week 37, and the 20 patients who switched to eplontersen from week 37 until the end of treatment. Harms for the 60 patients included from the external placebo group were also summarized. Harms occurred at similar frequencies across treatment groups. Overall, the clinical experts consulted for this review noted that the reported harms were in line with other treatments for hATTR and generally manageable.
There was a higher frequency (> 10%) of some TEAEs in the placebo group compared to the eplontersen group, including falls (5.6% versus 21.7%), fatigue (4.9% versus 20.0%) neuralgia (2.8% versus 15.0%), asthenia (2.1% versus 13.3%), and pain (0.7% versus 13.3%). Although the reason for this is uncertain, it may be due to disease progression in the placebo group, resulting in lack of motor function and control.
Vitamin A deficiency and thrombocytopenia were outcomes of special interest to eplontersen as well as this review, given that these are known safety concerns associated with inotersen treatment. As previously noted, no vitamin A-related AEs were reported in the NEURO-TTR trial because the study investigators were blinded to vitamin A levels so as not to inadvertently become unblinded to treatment allocation. As such, more vitamin A deficiency-related AEs were reported in the eplontersen group compared to the external placebo group, and interpretations of this outcome are limited, considering the issues. Although the certainty of evidence of the notable harms was considered very low, the product monograph for eplontersen states that patients should take vitamin A supplements at the recommended daily dose to reduce the potential risk of ocular symptoms due to deficiency.
Although no formal comparisons were made, there was a higher frequency of thrombocytopenia events in the inotersen group compared to the eplontersen group, and there were no thrombocytopenia AEs following the switch to eplontersen. Inotersen use comes with serious warnings about thrombocytopenia.
No safety outcomes were evaluated in the submitted ITC; thus, the comparative safety of eplontersen, vutrisiran, and patisiran remains unknown.
Conclusion
hATTR-PN is a rare disease, and there is a need for new, more convenient, safer treatments that slow disease progression and improve neurologic symptoms and HRQoL. One study was included in this review: the NEURO-TTRansform study, a phase III study evaluating the efficacy of eplontersen after 65 weeks of treatment compared to an external placebo in adult patients with genetic confirmation of hATTR-PN. Important features of the NEURO-TTRansform study design limit the certainty of evidence, notably the inclusion of an external placebo control and an open-label design that increases the risk of baseline confounding, selection bias, and performance bias.
The key outcomes evaluated in the NEURO-TTRansform study were of limited applicability to clinical practice in Canada; the clinical experts consulted for this review highlighted that these outcomes are not used to evaluate treatment effect in routine clinical practice. The NEURO-TTRansform study demonstrated that eplontersen likely resulted in a clinically meaningful decrease in serum TTR levels, but the clinical importance of this biomarker remains unknown. According to the clinical experts consulted for this review, disease stabilization is among the most important outcomes of treatment. In the NEURO-TTRansform study, neuropathy symptoms and neurologic function were measured using the mNIS + 7; these results suggested that patients treated with eplontersen experienced stabilization of disease, whereas those in the placebo group experienced deterioration. Lastly, improved HRQoL was an outcome important to patients. The results of the NEURO-TTRansform study suggest that patients treated with eplontersen may have clinically meaningful improvements in HRQoL (per the Norfolk QoL-DN) compared to placebo. Outcomes that are clinically relevant to practice in Canada, including the COMPASS-31 and R-ODS, were consistent with the suggested stabilization from the coprimary end points, but were generally considered supportive of the overall effect of eplontersen, given that the results were noncomparative. There were few safety concerns with eplontersen relative to other treatments for hATTR-PN. The frequency of thrombocytopenia AEs was lower versus the randomized inotersen group. There were more frequent ocular AEs related to vitamin A deficiency; however, the clinical experts noted that this was manageable in clinical practice.
There were important limitations in the conduct of the ITCs: the included studies varied in design, including outcome definitions and time of assessment. Additionally, there were notable differences in patient characteristics before adjustment, and the removal of patients in the weighting process substantially reduced the precision of treatment-effect estimates. Overall, the ITCs suggest that there was insufficient evidence to detect a difference between eplontersen and other treatments, or that eplontersen may be favoured over some treatments; however, conclusions could not be drawn with any certainty owing to methodological limitations and imprecision, as evidenced by wide 95% CIs.
References
1.Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8(1):31. PubMed
2.Conceicao I, Gonzalez-Duarte A, Obici L, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5-9. PubMed
3.Hawkins PN, Ando Y, Dispenzeri A, Gonzalez-Duarte A, Adams D, Suhr OB. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625-638. PubMed
4.Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829-837. PubMed
5.Swiecicki PL, Zhen DB, Mauermann ML, et al. Hereditary ATTR amyloidosis: A single-institution experience with 266 patients. Amyloid. 2015;22(2):123-131. PubMed
6.Committee For Medicinal Products For Human Use. Assessment Report: Amvuttra. (European public assessment report). Amsterdam (Netherlands): European Medicines Agency; 2022: https://www.ema.europa.eu/en/documents/assessment-report/amvuttra-epar-public-assessment-report_en.pdf. Accessed 2024 May 1.
7.Adams D, Ando Y, Beirao JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109-2122. PubMed
8.Conceição I, Damy T, Romero M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers ofTTRgene mutations*. Amyloid. 2019;26(1):3-9. PubMed
9.CADTH Canadian Drug Expert Committee Recommendation: Patisiran (Onpattro). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/cdr/complete/SR0598%20Onpattro%20-%20CDEC%20Final%20Recommendation%20July%2029%2C%202019%20for%20posting.pdf. Accessed 2024 May 1.
10.CADTH Canadian Drug Expert Committee Recommendation: Inotersen (Tegsedi). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/cdr/complete/SR0603%20Tegsedi%20-%20Final%20CDEC%20Recommendation%20December%2020%2C%202019%20for%20posting.pdf. Accessed 2024 May 1.
11.CADTH Reimbursement Recommendation: Vutrisiran (Amvuttra). Can J Health Technol. 2024;4(2). https://www.cadth.ca/sites/default/files/DRR/2024/SR0801REC-Amvuttra%20Final.pdf. Accessed 2024 Feb 21.
12.Clinical Study Report: ION-682884-CS3. A Phase 3 Global, Open-Label, Randomized Study to Evaluate the Efficacy and Safety of ION-682884 in Patients with Hereditary Transthyretin-Mediated Amyloid Polyneuropathy. [internal sponsor's report]. Carlsbad (CA): Ionis Pharmaceuticals, Inc.; 2023 Sep 7.
13.Clinical Study Report: ION-682884-CS3 (interim study report). A Phase 3 Global, Open-Label, Randomized Study to Evaluate the Efficacy and Safety of ION-682884 in Patients with Hereditary Transthyretin-Mediated Amyloid Polyneuropathy [internal sponsor's report]. Carlsbad (CA): Ionis Pharmaceuticals, Inc.; 2022 Nov 22.
14.AstraZeneca. Data on file. Indirect treatment comparison between eplontersen, vutrisiran, patisiran, and inotersen for the treatment of hereditary transthyretin amyloidosis with polyneuropathy. Prority technical report. Version 7.0. [sponsor supplied reference]. 2024 Mar 27.
15.Ando Y, Adams D, Benson MD, et al. Guidelines and new directions in the therapy and monitoring of ATTRv amyloidosis. Amyloid. 2022;29(3):143-155. PubMed
16.Dyck PJB, Gonzalez-Duarte A, Obici L, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: From NIS to mNIS + 7. J Neurol Sci. 2019;405:116424. PubMed
17.Gorevic PD. Overview of amyloidosis. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate: https://www.uptodate.com. Accessed 2024 May 1.
18.Fontana M. Cardiac amyloidosis: Treatment and prognosis. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate: https://www.uptodate.com. Accessed 2024 May 1.
19.CADTH Reimbursement Recommendation: Tafamidis Meglumine (Vyndaqel). Can J Health Technol. 2022;2(7). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/SF0722/SF0722. Accessed 2024 May 1.
20.Carvalho A, Rocha A, Lobato L. Liver transplantation in transthyretin amyloidosis: Issues and challenges. Liver Transpl. 2015;21(3):282-292. PubMed
21.Alcantara M, Mezei MM, Baker SK, et al. Canadian Guidelines for Hereditary Transthyretin Amyloidosis Polyneuropathy Management. Can J Neurol Sci. 2022;49(1):7-18. PubMed
22.Wainua (eplontersen): 56 mg/mL eplontersen (59 mg/mL as eplontersen sodium), subcutaneous use [product monograph]. Carlsbad (CA): Ionis Pharmaceuticals, Inc.; 2024 Mar 1.
23.Amvuttra (vutrisiran): injection solution; 25 mg/0.5 mL vutrisiran (as vutrisiran sodium), subcutaneous injection [product monograph]. Amsterdam (Netherlands): Alnylam Netherlands B.V.; 2023 Oct 17: https://pdf.hres.ca/dpd_pm/00072887.PDF.
24.Onpattro (patisiran): lipid complex solution; 2 mg/mL patisiran (as patisiran sodium), intravenous [product monograph]. Amsterdam (Netherlands): Alnylam Netherlands B.V.; 2023 Jun 12: https://pdf.hres.ca/dpd_pm/00071220.PDF.
25.Tegsedi (inotersen): injection solution, 284 mg inotersen / 1.5 mL per syringe [189 mg inotersen / mL (as inotersen sodium) [product monograph]. Boston (MA): Akcea Therapeutics, Inc.; 2018 Oct 2: https://pdf.hres.ca/dpd_pm/00047651.PDF. Accessed 2024 May 1.
26.Adams D, Gonzalez-Duarte A, O'Riordan WD, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):11-21. PubMed
27.Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22-31. PubMed
28.Vinik EJ, Vinik AI, Paulson JF, Merkies ISJ, Packman J, et al. Norfolk QOL-DN: validation of a patient reported outcome measure in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2014;19(2):104-114. PubMed
29.Yarlas A, Lovley A, Brown D, Kosinski M, Vera-Llonch M. Responder analysis for neuropathic impairment and quality-of-life assessment in patients with hereditary transthyretin amyloidosis with polyneuropathy in the NEURO-TTR study. J Neurol. 2022;269(1):323-335. PubMed
30.Sletten DM, Suarez GA, Low PA, Mandrekar J, Singer W. COMPASS 31: A refined and abbreviated Composite Autonomic Symptom Score. Mayo Clin Proc. 2012;87(12):1196-1201. PubMed
31.van Nes SI, Vanhoutte EK, van Doorn PA, et al. Rasch-built Overall Disability Scale (R-ODS) for immune-mediated peripheral neuropathies. Neurology. 2011;76(4):337-345. PubMed
32.Dyck PJ, Kincaid JC, Dyck PJB, et al. Assessing mNIS+7(Ionis) and international neurologists' proficiency in a familial amyloidotic polyneuropathy trial. Muscle Nerve. 2017;56(5):901-911. PubMed
33.Treister R, O'Neil K, Downs HM, Oaklander AL. Validation of the composite autonomic symptom scale 31 (COMPASS-31) in patients with and without small fiber polyneuropathy. Eur J Neurol. 2015;22(7):1124-1130. PubMed
34.Drug Reimbursement Review clinical review report: Inotersen (Tegsedi). Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/clinical/sr0603-tegsedi-clinical-review-report.pdf. Accessed 2023 Nov 14.
35.Eplontersen for the treatment of hereditary transthyretin-mediated amyloidosis (hATTR) with polyneuropathy (PN) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Eplontersen, 56 mg/mL single-dose pre-filled syringe. Mississauga (ON): AstraZeneca Canada; 2024 Apr 9.
36.Suhr O, Danielsson A, Holmgren G, Steen L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J Intern Med. 1994;235(5):479-485. PubMed
37.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
38.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: Informative statements to communicate the findings of systematic reviews of interventions. Journal of clinical epidemiology. 2020;119:126-135. PubMed
39.Gorevic P, Franklin J, Chen J, Sajeev G, Wang JCH, Lin H. Indirect treatment comparison of the efficacy of patisiran and inotersen for hereditary transthyretin-mediated amyloidosis with polyneuropathy. Expert Opin Pharmacother. 2021;22(1):121-129. PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 26: Sensitivity Analysis 1 — Nonparametric Analysis (FAS)
Variable | Serum TTR (%) | mNIS + 7 Composite Score | Norfolk QoL-DN Total Score | |||
|---|---|---|---|---|---|---|
Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | |
Week 35 | ||||||
N | ███ | ██ | ███ | ██ | ███ | ██ |
Hodges-Lehmann Estimate of Difference in LSM (95% CI)a | ███████████ | ██████████ | ██████████ | |||
P value | ███████████ | ██████████ | ██████████ | |||
Week 65 or 66 | ||||||
N | ███ | ██ | ███ | ██ | ███ | ██ |
Hodges-Lehmann Estimate of Difference in LSM (95% CI)a | ███████████ | ██████████ | ██████████ | |||
P value | ███████████ | ██████████ | ██████████ | |||
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LSM = least squares mean; TTR = transthyretin
aHodges-Lehmann estimates of the differences between the eplontersen group and the external placebo group as well as distribution-free CIs are based on the stratified Wilcoxon rank sum test. The stratum consists of 4 quartiles levels of the propensity scores (≤ Q1, > Q1 and ≤ Median, > Median and ≤ Q3, > Q3). The propensity score is calculated for each placebo or eplontersen patient using a logistic regression model with baseline value of the end point and covariates including disease stage, V30M mutation, and previous treatment.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023).12,13
Table 27: Sensitivity Analysis 2 — Multiple Imputation Assuming MAR (Safety Set)a
Variable | Serum TTR (%) | mNIS + 7 Composite Score | Norfolk QoL-DN Total Score | |||
|---|---|---|---|---|---|---|
Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | |
Week 35 | ||||||
N | ███ | ██ | ██ | ██ | ██ | ██ |
CFB, LSM (95% CI) | ███ | ██ | ██ | ██ | ██ | ██ |
Difference in LSM (95% CI) | ███ | ██ | ██ | |||
P value | ███ | ██ | ██ | |||
Week 65 or 66 | ||||||
N | ███ | ██ | ██ | ██ | ██ | ██ |
CFB, LSM (95% CI) | ███ | ██ | ██ | ██ | ██ | ██ |
Difference in LSM (95% CI) | ███ | ██ | ██ | |||
P value | ███ | ██ | ██ | |||
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LSM = least squares mean; TTR = transthyretin
aEach of the 500 imputed datasets is analyzed using simple ANCOVA and the 500 ANCOVA model results are combined using Rubin’s rules. The ANCOVA model adjusted by propensity score will include the effects of treatment, disease stage, V30M mutation, and previous treatment, and the baseline values of the end point.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023)12,13
Table 28: Sensitivity Analysis 3 — Multiple Imputation Assuming CIR (Safety Set)a,b
Variable | Serum TTR (%) | mNIS + 7 Composite Score | Norfolk QoL-DN Total Score | |||
|---|---|---|---|---|---|---|
Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | |
Week 35 | ||||||
N | ███ | ███ | ███ | ███ | ███ | ███ |
CFB, LSM (95% CI) | ███ | ███ | ███ | ███ | ███ | ███ |
Difference in LSM (95% CI) | ███ | ███ | ███ | |||
P value | ███ | ███ | ███ | |||
Week 65 or 66 | ||||||
N | ███ | ███ | ███ | ███ | ███ | ███ |
CFB, LSM (95% CI) | ███ | ███ | ███ | ███ | ███ | ███ |
Difference in LSM (95% CI) | ███ | ███ | ███ | |||
P value | ███ | ███ | ███ | |||
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LSM = least squares mean; MAR = missing at random; TTR = transthyretin
aEach of the 500 imputed datasets is analyzed using simple ANCOVA and the 500 ANCOVA model results are combined using Rubin’s rules. The ANCOVA model adjusted by propensity score will include the effects of treatment, disease stage, V30M mutation, and previous treatment, and the baseline values of the end point.
bIn the copy increment from reference approach, missing data in the NEURO-TTR trial placebo group will be imputed under a within-treatment-arm MAR assumption. For a patient in the eplontersen treatment group, their mean profile (i.e., mean increments) will track that of NEURO-TTR trial placebo group but starting from the benefit obtained.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023).12,13
Table 29: Sensitivity Analysis 4 — Multiple Imputation Assuming Jump to Reference (Safety Set)a,b
Variable | Serum TTR (%) | mNIS + 7 Composite Score | Norfolk QoL-DN Total Score | |||
|---|---|---|---|---|---|---|
Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | |
Week 35 | ||||||
N | ███ | ███ | ███ | ███ | ███ | ███ |
CFB, LSM (95% CI) | ███ | ███ | ███ | ███ | ███ | ███ |
Difference in LSM (95% CI) | ███ | ███ | ███ | |||
P value | ███ | ███ | ███ | |||
Week 65 or 66 | ||||||
N | ███ | ███ | ███ | ███ | ███ | ███ |
CFB, LSM (95% CI) | ███ | ███ | ███ | ███ | ███ | ███ |
Difference in LSM (95% CI) | ███ | ███ | ███ | |||
P value | ███ | ███ | ███ | |||
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LSM = least squares mean; MAR = missing at random; TTR = transthyretin
aEach of the 500 imputed datasets is analyzed using simple ANCOVA and the 500 ANCOVA model results are combined using Rubin’s rules. The ANCOVA model adjusted by propensity score will include the effects of treatment, disease stage, V30M mutation, and previous treatment, and the baseline values of the end point.
bIn Jump to Reference approach, missing data in the NEURO-TTR placebo group will be imputed under a within-treatment-arm MAR assumption. For a patient in the eplontersen treatment group, their mean response distribution is set to equal that of the NEURO-TTR placebo group.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023)12,13
Table 30: Sensitivity Analysis 5 — Primary Analysis Using PPSa
Variable | Serum TTR (%) | mNIS + 7 Composite Score | Norfolk QoL-DN Total Score | |||
|---|---|---|---|---|---|---|
Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | |
Week 35 | ||||||
N | ███ | ███ | ███ | ███ | ███ | ███ |
CFB, LSM (95% CI) | ███ | ███ | ███ | ███ | ███ | ███ |
Difference in LSM (95% CI) | ███ | ███ | ███ | |||
P value | ███ | ███ | ███ | |||
Week 65 or 66 | ||||||
N | ███ | ███ | ███ | ███ | ███ | ███ |
CFB, LSM (95% CI) | ███ | ███ | ███ | ███ | ███ | ███ |
Difference in LSM (95% CI) | ███ | ███ | ███ | |||
P value | ███ | ███ | ███ | |||
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LSM = least squares mean; mNIS + 7 = Modified Neuropathy Impairment Score + 7; TTR = transthyretin
aBased on a MMRM adjusted by propensity score weights with fixed categorical effects for treatment, time, treatment-by-time interaction, and disease stage, V30M mutation, previous treatment, and fixed covariates for the baseline value and the baseline-by-time interaction.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023)12,13
Table 31: Sensitivity Analysis 6 — Responder Analysis (Safety Set)
Variable | mNIS + 7a Composite Score | Norfolk QoL-DNb Total Score | ||
|---|---|---|---|---|
Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | |
Week 35 | ||||
With Nonmissing Score, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
With Missing Score, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ −2 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 0 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 2 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 4 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 6 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 8 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 10 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
Week 66 | ||||
With Nonmissing Score, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
With Missing Score, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ −2 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 0 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 2 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 4 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 6 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 8 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
≤ 10 Points Increase, n (%) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
aA responder was defined as a patient whose mNIS + 7 score changes from baseline to week 35/66 ≤ the threshold value. Patients that terminate treatment early irrespective of the reason or have missing week 35 or week 66 data (at each time of assessment) were considered nonresponders.
bA responder is defined as a patient whose Norfolk QoL-DN score changes from baseline to week 35/66 ≤ the threshold value. Patients that terminate early irrespective of the reason or have missing week 35 or week 66 data are considered nonresponders.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023)12,13
Table 32: Sensitivity Analysis 7 — Propensity Analysis Using 6 Covariates (FAS)a
Variable | Serum TTR (%) | mNIS + 7 Composite Score | Norfolk QoL-DN Total Score | |||
|---|---|---|---|---|---|---|
Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | |
Week 35 | ||||||
N | ███ ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
CFB, LSM (95% CI) | ███ ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Difference in LSM (95% CI) | ███ ███ | ███ ███ | ███ ███ | |||
P value | ███ ███ | ███ ███ | ███ ███ | |||
Week 65 or 66 | ||||||
N | ███ ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
CFB, LSM (95% CI) | ███ ███ | ███ ███ | ███ ███ | ███ | ███ ███ | ███ ███ |
Difference in LSM (95% CI) | ███ ███ | ███ ███ | ███ ███ | |||
P value | ███ ███ | ███ ███ | ███ ███ | |||
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LSM = least squares mean; TTR = transthyretin
aBased on a MMRM adjusted by propensity score weights with fixed categorical effects for treatment, time, treatment-by-time interaction, and disease stage, V30M mutation, previous treatment, and fixed covariates for the baseline value and the baseline-by-time interaction. In addition to 3 factors (disease stage, V30M mutation, previous treatment) used in the logistic model for propensity score in the primary analysis, this sensitivity analysis included additional 3 covariates (gender, modified BMI, region).
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023)12,13
Table 33: Sensitivity Analysis 8 — Observed Data, ANCOVA (FAS)
Variable | mNIS + 7 Composite Score | Norfolk QoL-DN Total Score | ||
|---|---|---|---|---|
Eplontersen ███ | External Placebo ███ | Eplontersen ███ | External Placebo ███ | |
Week 35 | ||||
N | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
CFB, LSM (95% CI) | ███ ███ | ███ ███ | ███ ███ | ███ ███ |
Difference in LSM (95% CI) | ███ ███ | ███ ███ | ||
P value | ███ ███ | ███ ███ | ||
CFB = change from baseline; CI = confidence interval; FAS = full analysis set; LSM = least squares mean.
Based on an ANCOVA model adjusted by propensity score with the effects of treatment, disease stage, V30M mutation, previous treatment, and the baseline value. Only data up to week 35 are included in the week 35 interim analysis.
Source: NEURO-TTRansform Clinical Study Report Interim Analyses (2023)13
Figure 2: Subgroup Analysis Forest Plot of Difference in LSM of Percentage Change From Baseline in Serum TTR to Week 35 and Week 65 (FAS) [Redacted]

CI = confidence interval; CM = cardiomyopathy; FAC = familial amyloid cardiomyopathy; LSM = least squares mean; SA = South America; TTR = transthyretin; V30M = Val30Met.
Note: Difference in LSMs, CIs, and p-values are based on an MMRM adjusted by propensity score weights with fixed categorical effects for treatment, time, subgroup factors, treatment-by-time interaction, treatment-by-subgroup interaction, and treatment-by-time-by-subgroup interaction, and disease stage, V30M mutation, previous treatment, and fixed covariates for the baseline value of the end point and the baseline-by-time interaction. n1 represents the eplontersen group and n2 represents the external placebo group.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023)12,13
Figure 3: Subgroup Analysis Forest Plot of Difference in LSM Change From Baseline in mNIS + 7 to Week 35 and Week 65 (FAS) [Redacted]

CI = confidence interval; CM = cardiomyopathy; FAC = familial amyloid cardiomyopathy; LSM = least squares mean; SA = South America; TTR = transthyretin; V30M = Val30Met.
Note: Difference in LSMs, CIs, and p-values are based on an MMRM adjusted by propensity score weights with fixed categorical effects for treatment, time, subgroup factors, treatment-by-time interaction, treatment-by-subgroup interaction, and treatment-by-time-by-subgroup interaction, and disease stage, V30M mutation, previous treatment, and fixed covariates for the baseline value of the end point and the baseline-by-time interaction. n1 represents the eplontersen group and n2 represents the external placebo group.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023).12,13
Figure 4: Subgroup Analysis Forest Plot of Difference in LSM Change From Baseline in Norfolk QoL-DN Total Score to Week 35 and Week [Redacted]

CI = confidence interval; CM = cardiomyopathy; FAC = familial amyloid cardiomyopathy; LSM = least squares mean; SA = South America; TTR = transthyretin; V30M = Val30Met.
Note: Difference in LSMs, CIs, and p-values are based on an MMRM adjusted by propensity score weights with fixed categorical effects for treatment, time, subgroup factors, treatment-by-time interaction, treatment-by-subgroup interaction, and treatment-by-time-by-subgroup interaction, and disease stage, V30M mutation, previous treatment, and fixed covariates for the baseline value of the end point and the baseline-by-time interaction. n1 represents the eplontersen group and n2 represents the external placebo group.
Source: NEURO-TTRansform Clinical Study Report Interim and Final Analyses (2023).12,13
Figure 5: mNIS + 7 Composite and Component Scores for Difference in LSM at Week 66 Final Analysis (FAS) [Redacted]

CI = confidence interval; LSM = least squares mean; mNIS + 7 = modified Neuropathy Impairment Score + 7; NIS = Neuropathy Impairment Score.
Difference in LSMs, CIs, and p-values are based on an MMRM adjusted by propensity score weights with fixed categorical effects for treatment, time, treatment-by-time interaction, and disease stage, V30M mutation, previous treatment, and fixed covariates for the baseline value and the baseline-by-time interaction. Only data up to week 66 are included in the modelling.
Source: NEURO-TTRansform Clinical Study Report12
Figure 6: Forest Plot of Norfolk QoL — DN Total and Domain Scores for Difference in LSM at Week 66 Final Analysis (FAS) [Redacted]

CI = confidence interval; LSM = least squares mean; Norfolk QoL-DN, Norfolk Quality of Life – Diabetic Neuropathy.
Difference in LSMs, CIs, and p-values are based on a MMRM adjusted by propensity score weights with fixed categorical effects for treatment, time, treatment-by-time interaction, and disease stage, V30M mutation, previous treatment, and fixed covariates for the baseline value and the baseline-by-time interaction. Only data up to week 66 are included in the modelling.
Source: NEURO-TTRansform Clinical Study Report12
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CDA-AMC
Canada's Drug Agency
hATTR
hereditary transthyretin-mediated amyloidosis
hATTR-PN
hereditary transthyretin-mediated amyloidosis polyneuropathy
ICER
incremental cost-effectiveness ratio
IPD
individual patient data
ITC
indirect treatment comparison
NICE
National Institute for Health and Care Excellence
Norfolk QoL-DN
Norfolk Quality of Life Questionnaire–Diabetic Neuropathy
OLT
orthotopic liver transplant
pCPA
pan-Canadian Pharmaceutical Alliance
QALY
quality-adjusted life-year
TTR
transthyretin
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Eplontersen (Wainua), 45 mgprefilled pen (56 mg/mL) |
Indication | Treatment of polyneuropathy associated with stage 1 or stage 2 hereditary transthyretin-mediated amyloidosis in adults |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | June 19, 2024 |
Reimbursement request | As per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults with hATTR-PN |
Treatment | Eplontersen |
Dose regimen | 45 mg once per month |
Submitted price | $47,680.33 per prefilled, single-dose pen |
Submitted treatment cost | $572,164 per patient per year |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (47 years) |
Key data source | Efficacy of eplontersen informed by the NEURO-TTRansform study Efficacy of comparators informed by unanchored matching-adjusted indirect comparisons |
Submitted results |
|
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; hATTR-PN = hereditary transthyretin-mediated amyloidosis polyneuropathy; ICER = incremental cost-effectiveness ratio; LY = life-year; Norfolk QoL-DN = Norfolk Quality of Life Questionnaire–Diabetic Neuropathy; QALY = quality-adjusted life-year; SAE = serious adverse event; vs. = versus.
Conclusions
Based on the clinical review by Canada’s Drug Agency (CDA-AMC) and data from the NEURO-TTRansform study, eplontersen likely results in a clinically meaningful decrease in serum transthyretin (TTR) levels among patients with hereditary transthyretin-mediated amyloidosis polyneuropathy (hATTR-PN) and may stabilize neuropathy symptoms and neurologic function. However, the use of an external placebo-control, open-label design limits the certainty of the evidence, and these outcomes were not included in the submitted economic model. There have been no head-to-head trials of eplontersen versus inotersen, vutrisiran, or patisiran for the indicated population, and indirect evidence submitted by the sponsor was insufficient to determine whether eplontersen would be associated with different clinical outcomes relative to vutrisiran, patisiran, or inotersen, owing to methodological limitations and imprecision in the effect estimates.
CDA-AMC identified important limitations with the sponsor’s economic evaluation that precluded reanalysis. Notably, the sponsor’s modelling approach was deemed inappropriate in that it relied on Norfolk Quality of Life Questionnaire–Diabetic Neuropathy (Norfolk QoL-DN) scores to estimate disease progression, utilized overly complex methodology, lacked transparency, and did not adequately characterize parameter uncertainty.
The sponsor’s base case indicates that eplontersen is associated with higher costs than inotersen and patisiran (incremental costs = $1,563,984 and $510,198, respectively) as well as higher quality-adjusted life-years (QALYs) (incremental QALYs = 0.17 and 0.23, respectively), resulting in incremental cost-effectiveness ratios (ICERs) of $9,502,073 and $2,264,909, respectively, per QALY gained. When compared to vutrisiran, the sponsor’s model suggests that eplontersen will be similarly effective (incremental QALYs = 0.09) and less costly (incremental cost = –$41,987). However, limitations with both the comparative clinical data and the sponsor’s modelling approach make the results highly uncertain and prone to bias in an unknown direction. Given the limitations in the indirect clinical evidence and uncertainties with the submitted economic model, there is insufficient evidence to suggest that eplontersen should be priced higher than other currently reimbursed treatments for hATTR-PN. To ensure cost-effectiveness, eplontersen should be priced no more than the lowest cost treatment option that is funded in the population to be reimbursed.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from Transthyretin Amyloidosis Canada. Information was collected in an online survey of patients with hereditary transthyretin-mediated amyloidosis (hATTR) (i.e., not specific to hATTR-PN) and caregivers residing in Ontario, Quebec, British Columbia, Alberta, Manitoba, and Nova Scotia, as well as from patient interviews and a round table discussion with patients and caregivers. Patients noted that hATTR is a multisystem disease that can have a variety of symptoms, including neurologic, cardiac, gastrointestinal, ophthalmologic, fatigue, urologic, and muscular symptoms, with variability in how symptoms are experienced. However, patients reported that hATTR affects all aspects of their lives, including their ability to work, and that polyneuropathy can result in mobility problems and falls. Specific details regarding patient history with existing treatment options or eplontersen were not reported. Eplontersen was identified as a new treatment option that is available at a lower dose and administered less frequently than existing alternatives.
Clinician input was received from the Neuromuscular Disease Network for Canada. The goals of hATTR-PN therapy were described as being to prevent morbidity, reduce mortality, minimize hospital visits, and enhance patient quality of life. Canadian treatment guidelines for hATTR-PN recommend the use of novel TTR gene-silencing therapies, where available. The input noted that eplontersen requires a lower dose and frequency of administration than existing TTR gene-silencing therapies and that this may lead to greater treatment compliance.
Drug plan input noted that genetic testing is required to confirm a patient’s diagnosis and eligibility for treatment. The plans indicated that eplontersen is expected to displace inotersen and raised concerns about budget impact and sustainability. It was noted that confidential negotiated prices are available for patisiran and inotersen. The plans also noted that, although the Canadian Drug Expert Committee recently recommended the reimbursement of vutrisiran, it is currently under consideration for negotiation with the pan-Canadian Pharmaceutical Alliance (pCPA).
Several of these concerns were addressed in the sponsor’s model:
Quality of life was incorporated in the sponsor’s model using EQ-5D data collected during the NEURO-TTRansform trial.
Loss of productivity was included in a scenario analysis.
CDA-AMC was unable to address the following concerns raised from the input relevant to the economic review:
CDA-AMC was unable to consider falls in the economic model, owing to a lack of clinical data and the model structure.
CDA-AMC was unable to consider confidential negotiated prices for patisiran or inotersen.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Eplontersen is indicated for the treatment of polyneuropathy with stage 1 or stage 2 hATTR in adults. The sponsor submitted a cost-utility analysis of eplontersen compared with vutrisiran, patisiran, and inotersen.1 The target population, comprising adults with hATTR-PN, was aligned with the Health Canada indication and the reimbursement request.
Eplontersen is available as a prefilled pen (45 mg/0.8 mL) for subcutaneous injection, with a recommended dosage of 45 mg once every month. The submitted price of eplontersen is $47,780 per prefilled pen, which corresponds to an annual per-patient cost of $570,060.1 The annual per-patient cost of vutrisiran was calculated by the sponsor to be $570,206 per patient per year,1 while the annual per-patient costs of patisiran and inotersen were $546,125 and $8,403, respectively.1,2
The model outcomes were life-years and QALYs. Costs were estimated from the perspective of the Canadian public health care payer over a lifetime horizon (47 years; 4-week cycle length). Costs and outcomes were discounted at 1.5% per annum.1
Model Structure
The sponsor submitted a Markov model with 3 health states based on Coutinho stage (stage 1, stage 2, and stage 3), 2 states related to orthotopic liver transplant (OLT or post-OLT), and death (Figure 1). Patients entered the model in Coutinho stage 1 or 2. In each cycle, patients could remain in their initiate state or transition to another Coutinho-based health state or death.1 Health states related to OLT were not utilized in the sponsor’s base case or scenario analyses.1
Coutinho-based health states were defined by the sponsor using Norfolk QoL-ND total scores, with Coutinho stage 1, stage 2, and stage 3 corresponding to Norfolk QoL-ND scores of greater than or equal to 2.6 to less than 54, greater than or equal to 54 to less than 91, and greater than or equal to 91, respectively.1,3 In the base case, it was assumed that patients would receive eplontersen or 1 of the comparators if they were in Coutinho stage 1 or 2.
Model Inputs
Baseline population characteristics to inform the model were obtained from the NEURO-TTRansform trial, which included patients with a confirmed diagnosis of hATTR-PN who were in Coutinho stage 1 or 2 (69% of patients were male and 31% were female; the mean age was 52.7 years; the mean weight was 71.6 kg).1,4 The NEURO-TTRansform trial randomized participants to receive either eplontersen or inotersen followed by eplontersen.
Clinical efficacy inputs for the model for vutrisiran, patisiran, and inotersen were obtained from a sponsor-submitted indirect treatment comparison (ITC). Due to concerns regarding between-study heterogeneity, the sponsor utilized unanchored, independent matching-adjusted indirect treatment comparisons (MAIC) and simulated treatment comparisons. For the purposes of the economic evaluation, the ITC results of interest were those pertaining to change from baseline in Norfolk QoL-DN score.
The sponsor obtained treatment-specific transition probabilities from contingency tables that summarized the number of patients who transitioned between Coutinho stages within 2 periods: baseline to week 35, and week 35 to week 66.1 While this necessitated patient-level data for each treatment, the NEURO-TTRansform trial reported only Coutinho stages at baseline. The sponsor obtained the necessary patient-level Coutinho staging data indirectly, using cut-off values from Faria et al. to link ranges of the Norfolk QoL-DN to specific Coutinho stages.1,3 For eplontersen, the contingency tables were calculated by converting Norfolk QoL-DN scores for each patient at each time point (i.e., baseline, week 35, week 66), then counting the transitions between each stage within each period. The same approach was used to obtain the contingency tables for the remaining treatments (inotersen, vutrisiran, and patisiran). In the absence of actual individual patient data (IPD), the sponsor used relative effect estimates from the ITC (mean change from baseline) to create pseudo IPD for the Norfolk QoL-DN scores by adjusting actual IPD for eplontersen at each follow-up point. Additional steps were involved in the creation of the pseudo IPD for vutrisiran and patisiran. Due to differences in the timing of the efficacy assessments between the NEURO-TTRansform trial and the trials used in the ITC, a linear model was used to predict eplontersen Norfolk QoL-DN scores at week 39 (from week 35) and week 80 (from week 66). After adjusting the predicted values using the relative effect estimate from the ITC, the sponsor interpolated the pseudo IPD back to weeks 35 and 66 using a separate linear model. Following the estimation of the contingency tables, the count data were converted to probabilities by dividing the number of patients by the total sample size. Additional adjustments were made to match the 4-week cycle length by assuming constant risks over each interval.1
In each cycle of the model, patients were at risk of all-cause death, which increased with age, calculated as the predicted survival probability from a parametric survival model (Weibull) fitted to natural history data. Time-to-event data were obtained from a prospective cohort study.1,5 To account for the younger population in the trial data compared with the prospective cohort study, the sponsor adjusted the predicted survival curve using a hazard ratio calculated from the general population mortality risk at 63 years and 53 years. The sponsor further adjusted mortality risk to ensure that the predicted overall survival values would not fall below the general population mortality risk. Age- and gender-specific general population mortality risks were obtained from Canadian Life Tables published by Statistics Canada.1,6
Treatment-specific adverse events (AEs) were incorporated in the model for serious AEs reported for at least 2% of patients in the NEURO-TTRansform trial for eplontersen or the respective pivotal trials for comparators.1,4,7-10 The probability of each AE was adjusted by the sponsor to reflect the 4-week cycle length, and it was assumed that the risk of AEs was constant over the model time horizon.1
Health state utility values were calculated from EQ-5D-5L questionnaire scores collected at baseline, week 37, and week 81 of the NEURO-TTRansform trial1 and valued using a Canadian value set.1,11 Utility values were calculated for the range of Norfolk QoL-DN scores specific to each Coutinho stage, and a mixed-effects model was fitted to capture variations at distinct time points of the trial.1 Disutilities were included for AEs, with the duration of each AE and corresponding disutility value obtained from the literature.12-16
Costs included in the model were those related to drug acquisition, administration, monitoring, disease progression, disease management, and AEs. Drug acquisition costs for eplontersen were based on the sponsor’s submitted price, while acquisition costs were based on the Ontario Drug Benefit formulary price for patisiran and inotersen and on the price of vutrisiran in the corresponding CDA-AMC submission.1,17,18 Monograph-recommended dosages were used for each drug.1 Premedication costs were incorporated for patisiran to reduce the risk of infusion-related reactions.1,2 Subsequent treatment costs were assumed to be equal to the total AE management costs.1 Administration costs were included for vutrisiran and patisiran.1,19,20 Given that eplontersen and inotersen would be injected by the patient (or a caregiver) at home, the sponsor assumed these treatments would have no associated administration costs.1 Monitoring costs included regular laboratory testing for platelet count as well as liver and kidney function, with unit costs obtained from the Ontario Schedule of Benefits for Laboratory Services.1,21 The sponsor included a 1-time disease progression cost for patients who progress to Coutinho stage 2 or 3.1 Disease management costs were derived through a micro-costing approach that included specialist consultation and laboratory testing associated with polyneuropathy, gastrointestinal disorders, cardiac arrhythmias, bladder dysfunction, ocular problems, primary care visits, mobility aids, home care, and hospitalizations.1 Finally, costs associated with AEs were calculated as the product of unit prices to treat each AE and the predicted incidence rates. Age-specific cost data for treating AEs were obtained from the Canadian Institute for Health Information Patient Cost Estimator and inflated to 2024 Canadian dollars.1,22
Summary of Sponsor’s Economic Evaluation Results
In the sponsor’s base case, costs and benefits were estimated probabilistically using a Monte Carlo simulation of 500 iterations. The deterministic and probabilistic results of the sponsor’s analyses were similar. The probabilistic findings are presented here. The submitted analysis was based on the publicly available prices of the comparator treatments. Additional results from the sponsor’s submitted base case are presented in Appendix 3.
CDA-AMC approved a deviation request by the sponsor to submit pairwise comparisons (i.e., instead of a sequential assessment of cost-effectiveness of all treatments).
Base-Case Results
In the sponsor’s assessment, eplontersen was more effective and more expensive than inotersen and patisiran, but less costly than vutrisiran, with an estimated cost of $5,341,196 and 7.34 QALYs gained over the 47-year horizon (Table 3). In pairwise analyses, eplontersen was associated with an ICER of $9,502,073 per QALY gained compared with inotersen and an ICER of $2,264,909 per QALY gained compared with patisiran. Compared with vutrisiran, eplontersen was predicted by the sponsor’s model to be associated with higher QALYs (incremental QALYs = 0.09) at a lower cost (incremental cost = –$41,987). At a willingness-to-pay threshold of $50,000 per QALY gained, eplontersen was cost-effective in 0% of iterations compared with inotersen or patisiran, and in 100% of iterations compared to vutrisiran.
Results of the sponsor’s analyses were driven by drug acquisition costs (approximately 98% of total costs in each analysis) and predicted differences in life-years and QALYs. The sponsor’s model estimated that eplontersen would generate 7.34 QALYs over a lifetime horizon. Of the QALYs gained, more than 90% were accrued after the first year of treatment on the basis of extrapolation (i.e., NEURO-TTRansform trial = 35 weeks).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
Comparison 1: eplontersen vs. inotersen | |||||
Inotersen | 3,777,212 | Reference | 7.18 | Reference | Reference |
Eplontersen | 5,341,196 | 1,563,984 | 7.34 | 0.17 | 9,502,073 |
Comparison 2: eplontersen vs. patisiran | |||||
Patisiran | 4,830,998 | Reference | 7.12 | Reference | Reference |
Eplontersen | 5,341,196 | 510,198 | 7.34 | 0.23 | 2,264,909 |
Comparison 3: eplontersen vs. vutrisiran | |||||
Eplontersen | 5,341,196 | Reference | 7.34 | Reference | Reference |
Vutrisiran | 5,383,183 | 41,987 | 7.25 | –0.09 | Dominated by eplontersen |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor considered 11 scenario analyses. These explored alternate inputs or assumptions related to the discount rate, time horizon, perspective, AEs, approach used to define Coutinho stage occupancy, treatment discontinuation, inclusion of carer disutility, source of relative effectiveness data, mortality risk calculation, drug wastage, and utility values for Coutinho stage 3. None of the scenarios had a meaningful effect on the cost-effectiveness of eplontersen.
The sponsor conducted a scenario analysis from a societal perspective. This analysis included costs associated with productivity losses in each Coutinho stage health state. Consistent with the sponsor’s base case, eplontersen was more expensive and more effective than patisiran and inotersen and less costly and more effective than vutrisiran.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative efficacy of eplontersen is highly uncertain: The NEURO-TTRansform trial utilized an external placebo-control, open-label design, which limits the certainty of the evidence (e.g., it increases the risk of baseline confounding, selection bias, and performance bias). There have been no head-to-head trials of eplontersen versus vutrisiran, patisiran, or inotersen. To inform the economic evaluation, the sponsor obtained relative effect estimates for changes in Norfolk QoL-DN score from an unanchored MAIC using independent pairwise comparisons with eplontersen.
The CDA-AMC clinical review noted several limitations related to the sponsor’s ITCs, including variations in design, outcome definition, and timing of assessment among the included studies; heterogeneity in patient characteristics before sample size adjustments; and imprecision in the treatment-effect estimates. Overall, the clinical review stated that conclusions could not be drawn with any certainty, owing to methodological limitations and imprecision.
Given the lack of direct evidence for eplontersen to any comparator and to limitations with the sponsor’s ITC, it remains highly uncertain whether eplontersen provides a net clinical benefit relative to vutrisiran, patisiran, or inotersen.
Uncertainty in the long term effectiveness of eplontersen: Evidence of the effectiveness of eplontersen beyond 85 weeks is not available. In the economic model, the sponsor assumed that patients who remain on eplontersen maintain the efficacy of eplontersen estimated from the NEURO-TTRansform trial for the duration of treatment, without consideration of potential waning of treatment effect. Given that the majority of the incremental QALYs (90%) predicted by the sponsor’s model were derived on the basis of extrapolated findings rather than on the basis of observed benefit, the lack of long-term data and the lack of consideration of potential waning of effectiveness introduces considerable uncertainty into the analysis. The open-label extension study of patients with hATTR-PN who continued to receive eplontersen after week 85 in the NEURO-TTRansform trial is ongoing; however, no data were provided to CDA-AMC for review.
CDA-AMC was unable to address this limitation due to a lack of clinical data. The direction and magnitude of the impact of this limitation is unknown, given that the comparative rate of potential effectiveness waning between eplontersen and comparators is unknown.
The validity of using Norfolk QoL-DN scores to estimate disease progression is highly uncertain: The sponsor submitted a Markov model with health states based on Coutinho stage (Figure 1). Given that this outcome was measured only at baseline in the NEURO-TTRansform trial, the sponsor estimated Coutinho stages indirectly from the Norfolk QoL-DN scores. Consistent with a 2012 National Institute for Health and Care Excellence (NICE) appraisal of tafamidis,3 the sponsor assumed that patients with a Norfolk QoL-DN score from 2.7 to 53 are in Coutinho stage 1; patients with a score from 54 to 90 are in stage 2; and patients with a score of 91 or greater are in stage 3.3 However, in its appraisal, NICE noted that, “it is uncertain… whether the Coutinho stages can be defined by TQoL [Total Quality of Life; Norfolk QoL-DN] scores; …whether the TQoL cut-offs for disease stages used in the model are appropriate; …whether TQoL rate of change estimated from a cross-sectional study appropriately captures disease progression…”3 The clinical experts consulted by CDA-AMC expressed similar concerns and noted that the Norfolk QoL-DN and Coutinho stages measure different constructs. That is, the Coutinho staging system categorizes disease severity based on a patient’s ability to walk and degree of neuropathy, while the Norfolk QoL-DN measures a patient’s perceived, disease-specific, health-related quality of life across 5 domains (i.e., activities of daily living, autonomic neuropathy, large-fibre neuropathy and physical functioning, small-fibre neuropathy, and neuropathic symptoms). In practice, the clinical experts indicated that decision-making is not guided by Norfolk QoL-DN score; instead, disease progression is assessed using clinical judgment, which includes a general neurology exam, assessment of mobility, and nerve conduction studies.
CDA-AMC notes that, although patients in the NEURO-TTRansform trial were required to be in Coutinho stage 1 or 2 at enrolment, the reported baseline Norfolk QoL-DN scores (range, 1 to 106) indicate that some patients would have been classified as being in stage 3, using the cut-offs applied in the economic evaluation.4 This poor reliability may also be attributable to the fact that Coutinho stage data were not used in the creation of the referenced cut-offs, which instead relied on assumptions about comparability between modified Polyneuropathy Disability Scale scores and Coutinho stages.3 The aforementioned NICE appraisal raised concerns about the lack of evidence supporting this proxy relationship as well as the absence of any justification for the original methods used to create the cut-off scores,3 as did CDA-AMC in its review of inotersen.23
CDA-AMC was unable to address this limitation. Given the identified limitations, it is highly uncertain whether the sponsor’s model accurately reflects disease progression for patients with hATTR-PN.
The methods used to estimate health state transitions were overly complex and introduced uncertainty: As described in the previous limitation, the sponsor used Norfolk QoL-DN scores to determine disease progression. For vutrisiran and patisiran, the transition probabilities were obtained through a complex procedure that relied on the creation of pseudo IPD. Due to differences in the timing of the efficacy assessments between the NEURO-TTRansform trial and the corresponding trials for the ITC, the sponsor used a linear model to predict eplontersen scores at week 39 (from week 35 data) and week 80 (from week 66 data). After adjusting the predicted values using the relative effect estimate from the ITC, the sponsor interpolated these data back to weeks 35 and 66 using separate linear models.1 This approach assumed a linear effect of treatment on Norfolk QoL-DN over time, which may not be reflective of the true treatment effect. Use of independent regression models in this manner risks distorting the trajectory of the predicted values away from what would have been anticipated from combining the eplontersen data with the relative effect estimates obtained in the ITC. Furthermore, this approach relied on assumptions that are not verifiable using other sources of evidence.
CDA-AMC was unable to address this limitation. The methods used by the sponsor to align the timing of efficacy assessments may introduce uncertainty beyond the imprecision in the estimates used to create the pseudo IPD.
The methods used to track changes in Coutinho stage were not verifiable: Contingency tables summarizing changes in Coutinho stage were created from a complex procedure that predicted each stage from actual or pseudo IPD of Norfolk QoL-DN scores. While the submitted report provided a detailed explanation of the underlying methods, these were not captured within the submitted spreadsheet. This is important because the procedure in question is the primary mechanism by which differences in state membership — and by extension, costs and QALYs — will be observed. Therefore, CDA-AMC was unable to verify that the methods were implemented (without error) in a manner consistent with the sponsor’s description.
CDA-AMC was unable to address this limitation.
Parameter uncertainty was improperly characterized in the model: Consistent with CDA-AMC guidelines, the sponsor’s base case used a Monte Carlo simulation to characterize the uncertainty of the relevant input parameters.24 The purpose of this requirement is to ensure that the uncertainty in the input parameters is reflected in the full range of possible costs and QALYs for each alternative considered in the model. To characterize the uncertainty in state membership, beta and Dirichlet distributions were used to randomly sample possible values for the treatment-specific contingency tables. This approach was problematic because it failed to characterize the uncertainty in the evidence identified by the sponsor. Given the sponsor’s choice to measure Coutinho staging indirectly from the Norfolk QoL-DN scores, the actual eplontersen IPD and relative effectiveness estimates obtained from the ITC should have been treated as random values. For each iteration, this would involve recalculating the contingency tables using a bootstrapped dataset of eplontersen Norfolk QoL-DN scores and randomly drawn values of each relative effect estimate from an appropriate distribution. In other words, the model failed to characterize the uncertainty in the Norfolk QoL-DN scores used to predict the Coutinho stages. Therefore, the expected costs and effects from the submitted model do not reflect the uncertainty in the evidence identified by the sponsor.
CDA-AMC was unable to address this limitation.
The impact of AEs was not adequately considered: In the economic model, the sponsor included serious AEs that occurred in at least 2% of patients in the respective pivotal trials. These included complete cardiovascular block, cardiac failure, congestive heart failure, pneumonia, orthostatic hypotension, vomiting, diarrhea, syncope, and dehydration. Data to inform the rates of these AEs were incorporated in the model through naive comparison, without adjusting or accounting for differences in patient characteristics. Owing to the direct use of clinical trial data, it is not possible to determine if any observed differences between the therapies are solely due to the treatment or rather to bias or confounding factors. The clinical experts consulted by CDA-AMC for this review identified AEs of clinical interest, such as those related to vitamin A deficiency and thrombocytopenia; however, given that these SAEs occurred in less than 2% of patients in all included treatments, these were not included in the model by the sponsor. As noted in the CDA-AMC clinical review, █████████████ ███ ██ ████████ ███ ████████ ███████████ ██ ███ ████████████████ █████ ████ ████████ ██ ████ ███ █ █████████████████ ██ ███████████ ███████ ██ ███████ █ ███████████ ███ ████ ███ █ █████████████████ ██ ███████ ██ ████████████████. Approximately 97% of patients who received eplontersen in the NEURO-TTRansform trial experienced at least 1 AE, including falls (5.6% of patients), which were noted to be important to patients and clinicians in the input received by CDA-AMC for this review.
CDA-AMC was unable to address this limitation. Results of the sponsor’s analysis suggest that eplontersen will be associated with fewer health care costs related to AE management. Whether these savings will be realized in clinical practice is highly uncertain.
Additional limitations were identified, but were not considered to be key limitations:
Health state costs may not be representative of costs to Canadian public payers: The sponsor’s model included costs related to disease management and progression, which included some items unlikely to be covered by public health care payers, such as mobility aids (e.g., wheeled walker, wheelchair).
CDA-AMC was unable to address this limitation.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Costs related to vitamin A supplementation were excluded. | Reasonable. Vitamin A supplementation is recommended for all comparators in the economic evaluation. However, vitamin A is not covered by any of the public drug plans in Canada. |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC was unable to address several key limitations in the sponsor’s submission, including uncertainty in the comparative clinical data, methodological and conceptual limitations related to the model structure and movement of patients through the model, and a lack of transparency in the submitted model (which prevented thorough validation by CDA-AMC). These limitations prevented CDA-AMC from specifying a reanalysis of the economic evaluation. As such, the cost-effectiveness of eplontersen for the treatment of polyneuropathy in stage 1 or stage 2 hATTR in adults is unknown.
Issues for Consideration
Patisiran and inotersen have successfully completed negotiations at the pCPA for hATTR and are listed on public formularies. Therefore, it is likely that these treatments are reimbursed by jurisdictional drug plans at confidential prices that are less than publicly available list prices. Additionally, vutrisiran is currently under consideration for negotiation with the pCPA. Should negotiations conclude with a letter of intent, the price of vutrisiran paid by the drug plans may be lower than that incorporated in the sponsor’s model, which was based on the submitted price in the CADTH review of vutrisiran.18
Overall Conclusions
The CDA-AMC clinical review found that eplontersen likely results in a clinically meaningful decrease in serum TTR levels among patients with hATTR-PN and may stabilize neuropathy symptoms and neurologic function, based on data from the NEURO-TTRansform trial. However, the use of an external placebo-control, open-label design limits the certainty of the evidence; and these outcomes were not included in the submitted economic model. There have been no head-to-head trials of eplontersen versus inotersen, vutrisiran, or patisiran for the indicated population, and indirect evidence submitted by the sponsor was insufficient to determine whether eplontersen would be associated with different clinical outcomes relative to vutrisiran, patisiran, or inotersen, owing to methodological limitations and imprecision in the effect estimates.
CDA-AMC identified important limitations with the sponsor’s economic evaluation that precluded reanalysis. Notably, the sponsor’s modelling approach relied on Norfolk QoL-DN scores to estimate disease progression; the validity of this approach is highly uncertain. Although the sponsor’s model structure was based on Coutinho stage, this outcome was monitored at baseline only, and the sponsor assumed that Norfolk QoL-DN scores could be used to predict changes in Coutinho stage. Norfolk QoL-DN and Coutinho stage measure different constructs, and there is insufficient evidence that 1 is a reliable predictor of the other. Further, the sponsor utilized complex methodology that CDA-AMC could not verify.
The sponsor’s base case indicates that eplontersen is associated with higher costs compared to inotersen and patisiran (incremental costs = $1,563,984 and $510,198, respectively) as well as higher QALYs (incremental QALYs = 0.17 and 0.23, respectively), resulting in ICERs of $9,502,073 and $2,264,909 per QALY gained, respectively. When compared to vutrisiran, the sponsor’s model suggests that eplontersen will be similarly effective (incremental QALYs = 0.09) and less costly (incremental cost = –$41,987). However, limitations with both the comparative clinical data and the sponsor’s modelling approach make the results highly uncertain and prone to bias in an unknown direction.
At the sponsor’s submitted price, eplontersen is expected to cost $572,164 annually, which is more costly than patisiran and inotersen and the same as vutrisiran (based on publicly available prices for patisiran and inotersen and the price of vutrisiran previously submitted to CADTH). Although the sponsor’s model estimates that eplontersen will be similarly effective (incremental QALYs = 0.09) and less costly than vutrisiran (incremental cost = –$41,987), the identified limitations with the sponsor’s modelling approach make it unknown whether these benefits will be realized in clinical practice. Further, vutrisiran is currently under consideration for negotiation with the pCPA; should an agreement be reached, it is likely that the negotiated price would be lower than the price included in the sponsor’s model.
Given the limitations in the indirect evidence and the methodological limitations identified in the sponsor’s economic model, there is insufficient evidence to suggest that eplontersen should be priced higher than other currently reimbursed treatments for hATTR-PN. Thus, eplontersen should be priced no more than the lowest-cost treatment option that is funded in the population to be reimbursed.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Wainua (eplontersen): 56 mg/mL eplontersen (59 mg/mL as eplontersen sodium), 45 mg single-dose pre-filled pen, subcutaneous use. Mississauga (ON): AstraZeneca Canada Inc.; 2024 Apr 9.
2.Onpattro (patisiran): lipid complex for injection, 2 mg/mL intravenous [product monograph]. Amsterdam (Netherlands): Alnylam Netherlands, BV; 2023 Jun 12: https://pdf.hres.ca/dpd_pm/00071220.PDF.
3.Faria R, Walker S, Palmer S, Corbett M, Stirk L, et al. Tafamidis for Transthyretin Familial Polyneuropathy (TTR-FAP) Evidence Review Group assessment of manufacturer submission [accessed by sponsor]. 2012; https://www.york.ac.uk/media/crd/Tafamidis%20ERG%20Report_CRDCHE%20September%204%202013.pdf. Accessed 2024 Mar 27.
4.AstraZeneca. Data on file. NEURO-TTRansform CSR (Final Analysis) [sponsor supplied reference]. 2023.
5.Sattianayagam PT, Hahn AF, Whelan CJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33(9):1120-1127. PubMed
6.Statistics Canada. Table: 13-10-0837-01. Life expectancy and other elements of the complete life table, single-year estimates, Canada, all provinces except Prince Edward Island. Release date: 2023-11-27. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310083701. Accessed 2024 Apr 5.
7.Ionis Pharmaceuticals, Inc. NCT01737398: Efficacy and Safety of Inotersen in Familial Amyloid Polyneuropathy. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine: https://www.clinicaltrials.gov/study/NCT01737398. Accessed 2024 Jan 24.
8.Alnylam Pharmaceuticals. NCT03759379: HELIOS-A: A Study of Vutrisiran (ALN-TTRSC02) in Patients With Hereditary Transthyretin Amyloidosis (hATTR Amyloidosis). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine: https://clinicaltrials.gov/study/NCT03759379. Accessed 2023 Nov 14.
9.Adams D, Gonzalez-Duarte A, O'Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11-21. PubMed
10.Alnylam Pharmaceuticals. NCT01960348: APOLLO: The Study of an Investigational Drug, Patisiran (ALN-TTR02), for the Treatment of Transthyretin (TTR)-Mediated Amyloidosis. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine: https://clinicaltrials.gov/study/NCT01960348?tab=results. Accessed 2023 Nov 14.
11.Bansback N, Tsuchiya A, Brazier J, Anis A. Canadian valuation of EQ-5D health states: Preliminary value set and considerations for future valuation studies. PLoS One. 2012;7(2):e31115. PubMed
12.National Institute for Health and Care Excellence. Alemtuzumab for treating highly active relapsing–remitting multiple sclerosis (NICE Technology appraisal guidance TA312). 2020; https://www.nice.org.uk/guidance/ta312/resources/alemtuzumab-for-treating-highly-active-relapsingremitting-multiple-sclerosis-pdf-82602423084229. Accessed 2024 Mar 28.
13.Briggs AH, Parfrey PS, Khan N, et al. Analyzing health-related quality of life in the EVOLVE Trial: The joint impact of treatment and clinical events. Med Decis Making. 2016;36(8):965-972. PubMed
14.National Institute for Health and Care Excellence. Ponesimod for treating relapsing multiple sclerosis (NICE single technology appraisal ID1393) committee papers. 2021; https://www.nice.org.uk/guidance/ta767/documents/committee-papers. Accessed 2024 Mar 28.
15.Jowett S, Kodabuckus S, Ford GA, et al. Cost-effectiveness of antihypertensive deprescribing in primary care: A Markov Modelling Study using data from the OPTiMISE trial. Hypertension. 2022;79(5):1122-1131. PubMed
16.McEwan P, Darlington O, McMurray JJV, Jhund PS, Docherty KF, et al. Cost-effectiveness of dapagliflozin as a treatment for heart failure with reduced ejection fraction: A multinational health-economic analysis of DAPA-HF. Eur J Heart Fail. 2020;22(11):2147-2156. PubMed
17.Government of Ontario. Ontario Drug Benefit Formulary/Comparative Drug Index. https://www.formulary.health.gov.on.ca/formulary/. Accessed 2024 Apr 5.
18.CADTH Reimbursement Recommendation: Vutrisiran (Amvuttra). Can J Health Technol. 2024;4(2). https://www.cadth.ca/sites/default/files/DRR/2024/SR0801REC-Amvuttra%20Final.pdf. Accessed 2024 Feb 21.
19.Government of Ontario. Schedule of Benefits. Physician Services Under the Health Insurance Act (February 19, 2024 (effective September 25, 2023)) [accessed by sponsor]. 2024; https://www.ontario.ca/files/2024-03/moh-ohip-schedule-of-benefits-2024-03-28.pdf. Accessed 2024 Apr 4.
20.CADTH Common Drug Review pharmacoeconomic review report: Patisiran (Onpattro). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0598-onpattro-pharmacoeconomic-review-report.pdf. Accessed 2024 Mar 18.
21.Government of Ontario. Schedule of Benefits for Laboratory Services. July 5, 2023 (Effective July 24, 2023) [accessed by sponsor]. 2023; https://www.ontario.ca/files/2024-01/moh-ohip-schedule-of-benefits-laboratory-services-2024-01-24.pdf. Accessed 2024 Apr 5.
22.CIHI. Patient Cost Estimator [accessed by sponsor]. 2022; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2023 Oct 4.
23.CADTH Common Drug Review pharmacoeconomic review report: Inotersen (Tegsedi). Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0603-tegsedi-pharmacoeconomic-review-report.pdf. Accessed 2023 Nov 14.
24.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2024 Jun 17.
25.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Wainua (eplontersen): 56 mg/mL eplontersen (59 mg/mL as eplontersen sodium), 45 mg single-dose pre-filled pen, subcutaneous use. Mississauga (ON): AstraZeneca Canada Inc.; 2024 Apr 9.
26.Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829-837. PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CDA-AMC Cost Comparison Table for hATTR-PN
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost | Annual cost ($) |
|---|---|---|---|---|---|---|
Eplontersen | 45 mg / 0.8 mL | Single-dose prefilled pen for SC injection | 47,680.3333a | 45 mg once every month | 1,566.50 | 572,164 |
Comparators | ||||||
Inotersen | 284 mg per 1.5 mL | Prefilled syringe for SC injection | 8,043.4874 | 284 mg weekly | 1,145.14 | 418,262 |
Patisiran | 2 mg/mL in a 5 mL vial | Solution for injection | 2,100.4813 per mL | 0.3 mg / kg (to a maximum of 30 mg) every 3 weeks | 1,552.72 | 567,130b |
Vutrisiran | 25 mg | Prefilled syringe for SC injection | 143,041.0000c | 25 mg every 3 months | 1,566.50 | 572,164 |
SC = subcutaneous.
Note: All prices are from the Ontario Exceptional Access Program (accessed April 2024), unless otherwise indicated, and do not include dispensing fees. Where relevant, a weight of 75 kg was used to calculate the required dose. Costs include wastage for single-use vials.
aSponsor-submitted price.1
bBased on 18 doses per year.
cPrice obtained from CADTH review of vutisiran.18
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | No | Refer to limitations regarding model structure and health state transitions |
Model structure is adequate for decision problem | No | Refer to limitations regarding model structure and health state transitions |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to limitation regarding methods used to track changes in Coutinho stage |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to limitation regarding improper characterization of parameter uncertainty |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | CDA-AMC was unable to verify that the methods to calculate changes in Coutinho stage were implemented without error |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
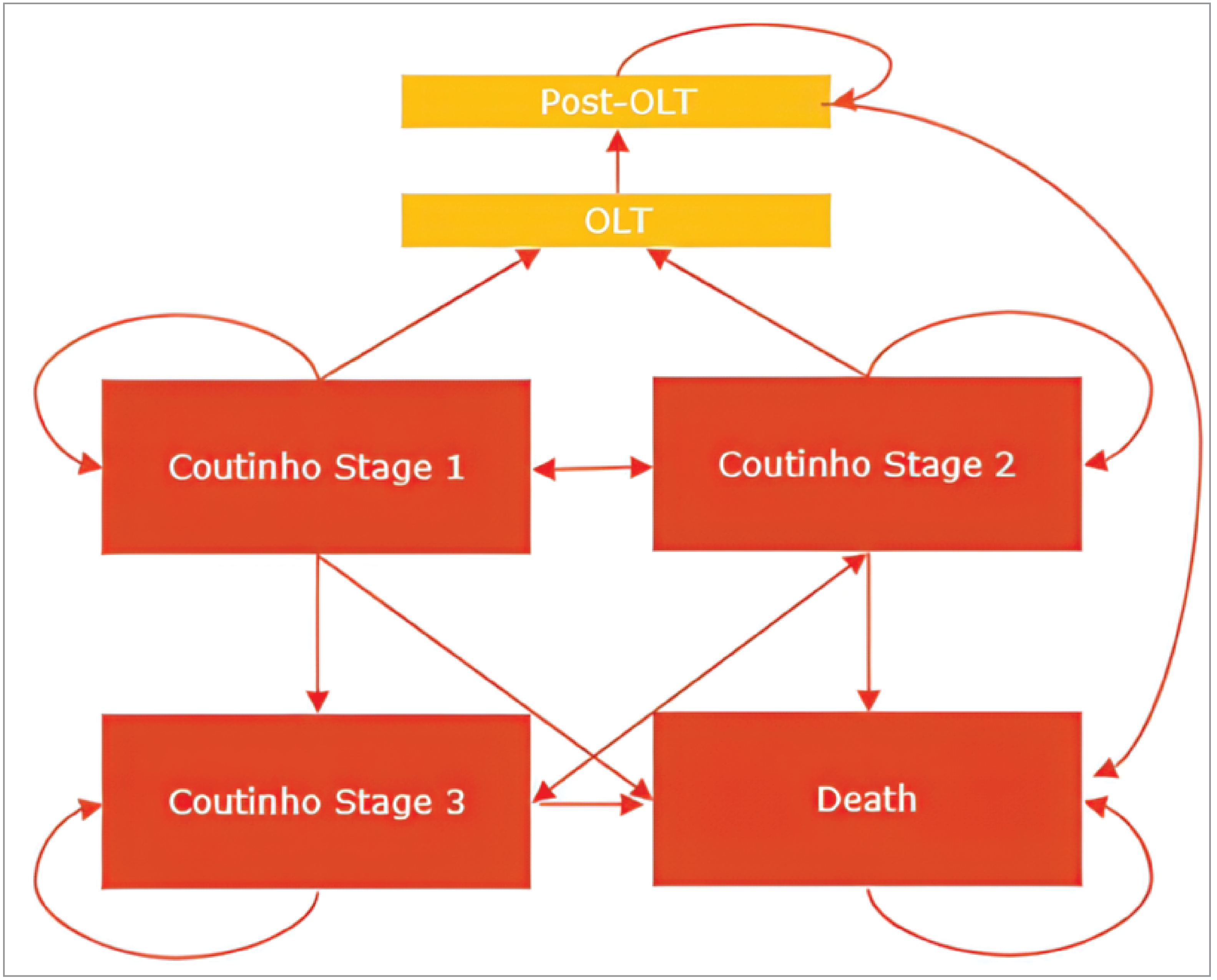
OLT = orthotopic liver transplant.
Note: Coutinho stages were defined as follows. Stage 1: Patients do not require assistance (unimpaired ambulation) and have mostly mild sensory, motor, and autonomic neuropathy in the lower limbs; Coutinho stage 2: Patients require assistance with ambulation and have disease progression in lower limbs with symptoms developing in the hands (weakness and wasting of muscles); Coutinho stage 3: Patients are wheelchair-bound or bedridden and have severe sensory, motor, and autonomic neuropathy of all limbs.1
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 7: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Eplontersen | Inotersen | Patisiran | Vutrisiran |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 10.26 | 10.26 | 10.26 | 10.26 |
Discounted QALYs | ||||
Total | 7.34 | 7.18 | 7.12 | 7.25 |
By health state | ||||
Coutinho stage 1 | 6.32 | 5.72 | 5.52 | 5.87 |
Coutinho stage 2 | 0.90 | 1.29 | 1.39 | 1.27 |
Coutinho stage 3 | 0.12 | 0.17 | 0.20 | 0.12 |
Discounted costs ($) | ||||
Total | 5,341,196 | 3,777,212 | 4,830,998 | 5,383,183 |
Acquisition | 5,302,357 | 3,726,980 | 4,752,415 | 5,340,886 |
Administration | 0 | 0 | 24,555 | 146 |
Disease management | 34,314 | 40,756 | 44,175 | 34,727 |
Subsequent treatmenta | 415 | 582 | 672 | 386 |
Premedication | 0 | 0 | 961 | 0 |
Monitoring | 0 | 1,436 | 0 | 0 |
Adverse events | 4,110 | 7,459 | 8,221 | 7,037 |
aThe sponsor assumed that patients would discontinue their initial treatment upon progressing to Coutinho stage 3 and would subsequently receive best supportive care (i.e., symptom management).
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Given the identified limitations within the submitted pharmacoeconomic model, CDA-AMC was unable to conduct any additional analyses to assess the relative cost-effectiveness of eplontersen for the treatment of adults with hATTR-PN.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 8: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) estimating the budget impact of reimbursing eplontersen for patients with hATTR-PN.25 The BIA was undertaken from the perspective of Canadian public drug plans (excluding Quebec) over a three-year time horizon (2026 to 2028). An epidemiological approach was used to estimate the eligible number of patients in each year of the analysis (Figure 2). The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits (NIHB) Program. Data to inform the model were obtained from various sources, including the published literature, the sponsor’s internal data, and assumptions. Key inputs to the BIA are documented in Table 9.
The sponsor compared a reference scenario, in which patients with hATTR-PN receive a current treatment option for hATTR-PN (i.e., vutrisiran, patisiran, and inotersen). In the new drug scenario, the sponsor assumed that eplontersen would displace market share from the existing alternatives.
Key assumptions:
The sponsor that 0.000752% of adults in Canada have hATTR-PN. This value represented the high prevalence estimate (270 individuals in a population of 35.9 million) for Canada from a study by Schmidt et al.25,26
The proportion of patients diagnosed with hATTR-PN (75%) and those in Coutinho stage 1 or 2 (77.5%) were elicited from clinical experts consulted by the sponsor.25
The sponsor assumed that 94% of patients in Coutinho stage 1 or 2 would be eligible for treatment based on clinical expert opinion obtained by the sponsor.
The sponsor assumed that 90% of patients would be eligible for public drug plan coverage based on clinical expert opinion and that 100% of patients in the NIHB program would have public drug coverage.25
Market share distributions were informed by clinical expert opinion.25
In the new drug scenario, the sponsor assumed that eplontersen would obtain most of its market share from newly diagnosed patients receiving their first silencer treatment, as well as a small proportion of patients switching from patisiran or inotersen.
Figure 2: Sponsor’s Estimation of the Size of the Eligible Population

hATTR = hereditary transthyretin-mediated amyloidosis; NIHB = noninsured health benefits; PN = polyneuropathy.
Source: Sponsor-submitted budget impact analysis.25
Table 9: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Total adult population (Canada, Excluding Quebec) | 26,150,257 / 27,146,783 / 27,549,442 |
Number of patients eligible for drug under review | 97 / 101 / 103 |
Market uptake (3 years) | |
Uptake (reference scenario) Inotersen Patisiran Vutrisiran | 4.5% / 4.0% / 3.0% 43.0% / 14.5% / 9.0% 52.5% / 81.5% / 88.0% |
Uptake (new drug scenario) Eplontersen Inotersen Patisiran Vutrisiran | 7.5% / 20.0% / 22.5% 4.5% / 2.0% / 0.5% 40.5% / 10.5% / 7.0% 47.5% / 67.5% / 70.0% |
Cost of treatment (per patient, per year) | |
Eplontersen Inotersen Patisiran Vutrisiran | $572,164 $419,698 $548,001a $572,164 |
hATTR = hereditary transthyretin-mediated amyloidosis; PN = polyneuropathy.
Note: treatment wastage was not considered for eplontersen, inotersen, or vutrisiran.
aThe recommended dose of patisiran is 0.3 mg / kg. The sponsor’s analysis assumed costs were obtained for patients weighing 71.57 kg, resulted in an average dose of 21.47 mg per administration. Treatment wastage was included in the base case, resulting in the consumption of 3 vials of patisiran for each administration.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analysis suggest that the reimbursement of eplontersen for the treatment of hATTR-PN is expected to be $871,579 over the first 3 years (Year 1: $51,177; Year 2: $390,203; Year 3: $430,199). CDA-AMC notes that the sponsor’s results are predicated on the assumption that no patients with hATTR-CM will receive eplontersen.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of patients with hATTR-PN is uncertain: In the BIA, the sponsor adopted an estimate of 0.000752% for the prevalence of hATTR-PN in Canada (equating to 270 people). This estimate reflects the high end of the range for Canada from a systematic review of the prevalence global prevalence of hATTR-PN; however, this rate was based on data extrapolated from other countries and was not based on Canadian data.25,26 Clinical experts consulted by CDA-AMC noted that an estimate of 270 patients may underestimate the true prevalence in Canada. Further, CDA-AMC notes that true prevalence may be higher than estimated, owing to, for example, the emergence of survival-extending treatments and improved diagnostic processes and clinician knowledge over time.26
CDA-AMC could not address this limitation owing to a lack of alternative estimates.
The prices paid by public drug plans is uncertain: The prices for patisiran and inotersen were based on publicly available list prices and may not reflect the actual prices paid by public drug plans. Patisiran and inotersen have previously gone through negotiations at pCPA, and any potential confidential rebates are not reflected in this analysis. The sponsor adopted the price of vutrisiran submitted to CADTH for review.18 Vutrisiran is currently under consideration for negotiations with pCPA. Should negotiations conclude with a letter of intent, the price paid by public drug plans is likely to be lower than considered in the sponsor’s analysis.
CDA-AMC was unable to address this limitation.
CDA-AMC Reanalyses of the BIA
In the absence of more reliable estimates to inform the key parameters of the BIA, the sponsor’s base case was maintained. CDA-AMC expects that the budget impact of eplontersen will be sensitive to more reliable inputs which may affect the market size calculation. The disaggregated results of the submitted BIA are provided in Table 10.
Table 10: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 52,417,853 | 53,949,117 | 56,684,665 | 57,799,154 | 168,432,936 |
New drug | 52,417,853 | 54,000,294 | 57,074,868 | 58,229,353 | 169,304,515 | |
Budget impact | 0 | 51,177 | 390,203 | 430,199 | 871,579 |
BIA = budget impact analysis.
Note: The submitted analysis was based on publicly available prices for the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.