Drugs, Health Technologies, Health Systems
Reimbursement Review
Lebrikizumab (Ebglyss)
Sponsor: Eli Lilly Canada Inc.
Therapeutic area: Atopic dermatitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AD
atopic dermatitis
AE
adverse event
BSA
body surface area
CDA
Canadian Dermatology Association
CDA-AMC
Canada's Drug Agency
CDLQI
Children’s Dermatology Life Quality Index
CI
confidence interval
CrI
credible interval
CSPA
Canadian Skin Patient Alliance
CSR
clinical study report
DAO
Dermatology Association of Ontario
DLQI
Dermatology Life Quality Index
EASI-50
at least a 50% reduction in EASI score
EASI-75
at least a 75% reduction in EASI score
EASI-90
at least a 90% reduction in EASI score
EASI
Eczema Area and Severity Index
EMA
European Medicines Agency
ESC
Eczema Society of Canada
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
HRQoL
Health-related quality of life
IGA
Investigator Global Assessment
IL
interleukin
ITC
indirect treatment comparison
ITT
intention to treat
JAK
Janus kinase
LS
least squares
MCMC-MI
Markov chain Monte Carlo multiple imputation
MID
minimal important difference
MMRM
mixed model for repeated measures
NMA
network meta-analysis
NRS
Numeric Rating Scale
OR
odds ratio
POEM
Patient-Oriented Eczema Measure
RCT
randomized controlled trial
RD
risk difference
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
SE
standard error
SLR
systematic literature review
TCS
topical corticosteroids
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Lebrikizumab (Ebglyss) injection solution for SC injection, 250 mg/2 mL (prefilled pen or prefilled syringe with needle shield) |
Sponsor | Eli Lilly Canada, Inc. (Eli Lilly) |
Indication | Lebrikizumab injection is indicated for the treatment of moderate-to-severe atopic dermatitis in adults and adolescents 12 years of age and older with a body weight of at least 40 kg, whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Lebrikizumab can be used with or without topical corticosteroids. |
Reimbursement request | As per the indication |
Health Canada approval status | Approved |
Health Canada review pathway | Standard review |
NOC date | June 24, 2024 |
Recommended dose | Initial dose of 500 mg SC at week 0 and week 2, then 250 mg SC every 2 weeks until week 16. Once clinical response is achieved, the recommended maintenance dose is 250 mg every 4 weeks starting at week 16. Continued therapy beyond 16 weeks should be carefully considered in a patient who does not show treatment benefit within this time period. |
NOC = Notice of Compliance; SC = subcutaneous.
Introduction
Atopic dermatitis (AD) is a chronic, relapsing, inflammatory, and noncontagious skin disease that is commonly associated with other atopic expressions, such as asthma, allergic rhinitis, and food allergy.1 The burden of disease and its impact on quality of life may be profound, particularly in the case of moderate-to-severe AD.2 Itch or pruritus; soreness, pain, or tenderness; and skin dryness are the signs and symptoms most frequently cited as having a clinical impact.3 Itch, the major symptom, has a negative impact on quality of life and is associated with mental distress and an increased risk for suicidal thoughts.1 Depression, anxiety, and sleep disturbance are frequently reported comorbidities.2,3 Moreover, AD can result in embarrassment related to appearance and can have a negative impact on a patient’s self-esteem and social life.1 Patients with AD are at increased risk of skin infections because of excessive rubbing or scratching.1 Exacerbations, or flares, are an integral part of the disease course and generally indicate a worsening of AD that requires escalation or intensification of treatment.4
AD has no impact on approximately 15% to 20% of children and approximately 1% to 3% of adults worldwide; in high-income countries, AD affects around 20% of children and up to 10% of adults.1,5 Approximately 50% of adult patients have moderate-to-severe disease based on clinical disease severity scales.6
Initial treatment for most patients with AD is emollients (moisturizers) plus topical anti-inflammatory therapy, including topical corticosteroids (TCS) and topical calcineurin inhibitors.7 For patients with more severe AD or with AD that is refractory to topical therapy, advanced treatments, including phototherapy and systemic treatment, are considered. According to clinical practice guidelines from the American Academy of Dermatology and the American Academy of Allergy, Asthma & Immunology, biologics, and particularly dupilumab, are considered first-line systemic therapy.7,8 Other options include tralokinumab (another biologic) and oral Janus kinase (JAK) inhibitors (upadacitinib, abrocitinib).7,8 According to the clinical expert consulted, off-label immunomodulators (cyclosporine, methotrexate, mycophenolate, and azathioprine) are generally only used when mandated by a medication payer as step-through therapy or when the previously mentioned biologics and JAK inhibitors fail or are contraindicated. These drugs were not listed as first-line systemic therapies in the 2023 American Academy of Dermatology clinical practice guidelines due to the lower certainty of evidence for newer drugs, the potential for serious adverse events (SAEs), the need for stringent laboratory monitoring, and the lack of regulatory approval for use in AD.7
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of lebrikizumab (Ebglyss) 250 mg per 2 mL subcutaneous (SC) injection for the treatment of moderate-to-severe AD in adults and adolescents 12 years and older with a body weight of at least 40 kg, whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.
Perspectives of Patients, Clinicians and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to calls for input from Canada’s Drug Agency (CDA-AMC) and the clinical expert consulted by CDA-AMC for the purpose of this review.
Patient Input
Three patient groups provided input to this submission. Eczema Quebec gathered information through review of scientific literature, informal conversations with patients, The Skin I'm In 2022 Update (a joint report by Eczema Quebec and the Canadian Skin Patient Alliance [CSPA]), expert opinion from the Centre of Excellence for Atopic Dermatitis at the McGill University Health Centre, 9 written patient testimonials, interviews with 14 patients, and feedback from 3 patient-group discussions. The CSPA gathered information from previous submissions to CDA-AMC, data from the Canadian Institute for Health Information on AD-related emergency department visits, hospitalizations from 2016 to 2020 (reported in The Skin I'm In), and guidelines. The Eczema Society of Canada (ESC) gathered information through a survey and through one-on-one interviews from more than 3,000 patients with AD and their caregivers who live in Canada.
According to the input from patient groups, symptoms of patients with AD include inflamed, painful, dry, and itchy skin that cracks, oozes, bleeds, and, in some cases, involves thickening and/or infections of the skin. Conditions associated with AD include asthma, seasonal and environmental allergies, food intolerances, sleep disorders, anxiety, and depression. Patient groups stated that physical manifestations and visibility of the disease contribute to psychological distress through stigmatization, which impacts a patient’s self-esteem, professional commitments, and social engagements.
Based on patient-group input, the burden of AD also extends to caregivers and family members. Caregivers reported feelings of anxiety, depression, helplessness, guilt, frustration, and a lack of control over the situation. Caregivers and family members also shared that their own health and emotional wellness, lifestyle, sleep, intimacy, social activities, and family dynamics were affected by the disease. Further, the cost of treatment and other skincare products can place financial stress not only on the patient, but also on the family.
Important desired outcomes reported by patient groups included the following: better, fast, and long-term control of the disease; reduction of flares; relief from itch; reduction of skin symptoms; pain and discomfort relief; improved psychological status; improved daily and social activities; increased productivity; improved emotional well-being; improved sleep quality; and the ability to maintain intimate relationships. In addition, treatments should be affordable or covered by insurance, and should be easy to use (i.e., not administered by injection or topically).
Access to health care presents another challenge to patients with AD. Canada has a low ratio of dermatologists to the population, making specialized care difficult to obtain, particularly in remote areas. Additionally, 36% of caregivers reported feeling a lack of support from the health care system and 30% reported financial challenges related to managing their child’s disease.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
According to the clinical expert consulted for this review, there is an unmet need for more treatment options for people who are refractory to or do not tolerate current biologic treatments for AD, as well as for people who are concerned about the safety profile of oral JAK inhibitors, particularly people with comorbidities and those who are older.
Patients with moderate-to-severe AD that is refractory to topical therapy are most likely to respond to treatment with lebrikizumab, according to the clinical expert. The clinical expert anticipates that lebrikizumab’s use will be similar to that of other systemic medications that allow the concomitant use of emollients and topical anti-inflammatory treatments (e.g., corticosteroids). Given the clinical experience with dupilumab and the evidence supporting its use, the expert anticipates that lebrikizumab will be considered a second-line biologic after dupilumab and may be chosen for patients for whom dupilumab is contraindicated, ineffective, or not tolerated.
In clinical practice, clinicians generally use a gestalt assessment of improvement in clinical signs and a patient’s history of change in symptoms (e.g., itch) and quality of life, the clinical expert explained. Clinicians only use the tools used in clinical trials (e.g., Eczema Area and Severity Index [EASI] score) if mandated by a medication payer to obtain coverage. According to the clinical expert, a meaningful response to treatment would be an improvement of approximately 50% to 75% in signs and symptoms; the specific proportion likely differs by clinician and by patient. The improvement should include a reduction in the severity and frequency of symptoms and is often accompanied by an improvement in quality of life and the ability to perform household, work, and/or school activities. A reduction in skin infections and disease flares is also important.
The clinical expert indicated that lebrikizumab should be discontinued if it is inadequately effective, if the patient experiences intolerable adverse effects, or if the patient wishes to interrupt or discontinue therapy. The clinical expert noted that in most instances, a specialist (dermatologist, allergist, pediatrician) would be required to treat a patient with AD with a biologic, although in areas where access to specialty care is difficult, some family physicians could gain comfort with biologics for AD.
Clinician-Group Input
CDA-AMC received inputs from 2 clinician groups for this review. The Canadian Dermatology Association (CDA) submitted input from 3 clinicians from its Pharmacy and Therapeutics Advisory Board, and the Dermatology Association of Ontario (DAO) submission included input from 11 clinicians.
Clinician groups and the clinical expert consulted by CDA-AMC agreed that a lack of adequate response to treatment, incomplete effectiveness, adverse effects related to treatments, a lack of feasibility of some treatments, and relapses are unmet needs of patients with AD. One of the clinician groups added that challenges in access to care, multitiered treatment regimens, treatment intolerance or contraindications, and comorbid bacterial skin infections are unmet needs as well.
The CDA and the clinical expert consulted by CDA-AMC agree that the goals of treatment are improving quality of life and maximizing efficacy and safety. Regarding the place of lebrikizumab in therapy, the DAO and the clinical expert consulted by CDA-AMC indicate that lebrikizumab will not cause a shift in the treatment paradigm and would be considered another treatment option. In contrast, the CDA stated that lebrikizumab contributes to an important shift in the current treatment paradigm toward a new era of focus on novel disease mechanisms that target and modify disease and have favourable safety and efficacy profiles.
According to the DAO, adult patients with moderate-to-severe AD who have failed topical therapies and those who have failed or do not have access to phototherapy would be best suited for treatment with lebrikizumab. The CDA stated that patients best suited for treatment with lebrikizumab would be those with uncontrolled moderate-to-severe AD who are candidates for systemic therapy or who meet criteria for biologic therapy. The CDA noted that dupilumab is indicated for patients with other severe forms of atopic or allergic conditions, such as severe asthma or eosinophilic esophagitis; thus, dupilumab may be chosen for these patients instead of the interleukin (IL)-13 inhibitors, such as lebrikizumab, which are not approved for use in patients with these conditions.
The DAO noted that a patient’s response to treatment would be assessed with the Investigator Global Assessment (IGA), EASI, Pruritus Numerical Rating Scale (NRS), and Dermatology Life Quality Index (DLQI) scoring systems at 4 to 6 months and annually thereafter. The CDA stated that the assessment of a patient’s response would be based on a clinical exam, the patient’s history, physician-reported clinical scoring systems (EASI, body surface area [BSA], IGA), and patient-reported outcomes (DLQI, Children’s DLQI [CDLQI], and Pruritus NRS). The CDA added that in clinical practice, due to time limitations, only some of the scoring systems are used.
The clinician groups reported that adverse events (AEs) and a lack of efficacy should be considered when deciding to discontinue the treatment.
Based on clinician-group input, the treatment and monitoring of patients on lebrikizumab should be limited to specialists trained in this area, which would include those from the fields of dermatology, allergy, immunology, or pediatrics.
Drug Program Input
The drug programs identified issues related to relevant comparators; considerations for the initiation, renewal, discontinuation, and prescribing of therapy; and system and economic issues. For more information, refer to Table 4.
Clinical Evidence
Systematic Review
Description of Studies
Three double-blind, randomized controlled trials (RCTs) met the inclusion criteria for the systematic review (ADvocate 1, ADvocate 2, and ADhere).9-11 The objective of the ADvocate 1 (N = 424) and ADvocate 2 (N = 427) studies was to evaluate the safety and efficacy of lebrikizumab as monotherapy in patients with moderate-to-severe AD. Eligible patients were adults or adolescents (aged 12 years to less than 18 years and weighing more than 40 kg) who had a diagnosis of chronic AD that was rated as moderate to severe, based on an EASI score of at least 16, an IGA score of at least 3, and AD covering a BSA of 10% or more. All patients had a history of inadequate response to topical therapies for AD. Both studies included a 16-week induction period (parallel design), followed by a 36-week maintenance period (randomized withdrawal design). The double-blind studies randomized patients in a ratio of 2:1 to receive a lebrikizumab 500 mg SC loading dose at week 0 and week 2 and then 250 mg SC every 2 weeks up to week 16, or placebo for the 16-week induction period. At week 16, patients in the lebrikizumab group who responded to treatment (i.e., an IGA score of 0 or 1 or at least a 75% reduction in EASI score [EASI-75], and who did not receive rescue therapy) were randomly reassigned in a ratio of 2:2:1 to double-blind lebrikizumab 250 mg every 2 weeks until week 36, lebrikizumab 250 mg every 4 weeks until week 36, or placebo for the 36-week maintenance period.
The objective of the ADhere study was to compare the safety and efficacy of lebrikizumab in combination with low-to-midpotency TCS with placebo plus TCS in patients with moderate-to-severe AD. The study was a 16-week randomized, double-blind, parallel-design trial (N = 211). Adults or adolescents (aged 12 years to less than 18 years and weighing more than 40 kg) with moderate-to-severe AD (EASI score of ≥ 16, IGA score of ≥ 3, AD coverage of a BSA of 10% or more) were eligible to enrol. Patients were randomized in a 2:1 ratio to receive a 500 mg lebrikizumab SC loading dose at week 0 and week 2 followed by 250 mg SC once every 2 weeks up to week 16 plus TCS, or placebo plus TCS for the 16-week treatment period.
In all 3 trials, the coprimary outcomes were the proportion of patients with an IGA score of 0 or 1 and at least a 2-point reduction from baseline to week 16, and the proportion of patients with an EASI-75 response at week 16. The IGA measures the investigator’s global assessment of the patient’s overall severity of AD at that visit, based on a static, numeric 5-point scale that ranges from 0 (clear) to 4 (severe). The EASI is a composite index based on the physician’s assessment of 4 clinical signs of the disease (erythema, infiltration and/or papulation, excoriation, and lichenification) and the extent of BSA involved at that visit. It is scored from 0 to 72, with higher scores indicating greater disease severity and/or extent of disease. Other key outcomes reported were the proportion of patients with a Pruritus NRS score of at least 4 points at baseline who reported at least a 4-point reduction from baseline at week 16, and the change from baseline to week 16 in the Patient-Oriented Eczema Measure (POEM) score, the DLQI total score, or the CDLQI total score.
The patients enrolled in the trials had a mean age that ranged from 34.2 years (standard deviation [SD] = 16.4) to 37.5 years (SD = 19.9) per treatment group. In the ADvocate 1, ADvocate 2, and ADhere studies, 13%, 11%, and 22% of patients, respectively, were adolescents. There were roughly equal proportions of females and males in the studies. On average, the patients enrolled in the study had been diagnosed with AD for 20 or more years; most patients (59% to 73%) were classified as having disease of moderate severity based on an IGA score of 3 at baseline, whereas 27% to 41% of patients were classified as having severe AD (i.e., an IGA score of 4). Almost all patients enrolled had previously used TCS (97% to 100%), and 33% to 46% of patients had received topical calcineurin inhibitors. Systemic therapies had been previously received by 43% to 56% of patients, and 12% to 24% of patients had used phototherapy before enrolment in the trials.
Efficacy Results
Induction Period
At week 16, the proportion of patients with an IGA score of 0 or 1 and at least a 2-point reduction from baseline favoured the lebrikizumab groups over the placebo groups in all 3 studies. In the ADvocate 1 study, 43.1% and 12.7% of patients attained an IGA 0 or 1 response in the lebrikizumab and placebo groups, respectively, with a risk difference (RD) of 29.7% (95% confidence interval [CI], 21.6% to 37.8%; P < 0.001). In the ADvocate 2 study, 33.2% and 10.8% of patients attained an IGA 0 or 1 response (RD = 21.9%; 95% CI, 14.2% to 29.6%; P < 0.001) in the lebrikizumab and placebo groups, respectively. The IGA 0 or 1 response also favoured lebrikizumab plus TCS over placebo plus TCS in the ADhere study (41.2% versus 22.1%; RD = 18.3%; 95% CI, 5.1% to 31.5%; P = 0.01).
In all 3 studies, a higher proportion of patients reported an EASI-75 response at week 16 in the lebrikizumab groups than in the placebo groups. An EASI-75 response was attained by 58.8% and 16.2% of patients in the lebrikizumab and placebo groups, respectively, in the ADvocate 1 study (RD = 42.0%; 95% CI, 33.3% to 50.6%; P < 0.001), and by 52.1% and 18.1% of patients, respectively, in the ADvocate 2 study (RD = 33.3%; 95% CI, 24.4% to 42.2%; P < 0.001). In the ADhere study, 69.5% and 42.2% of patients attained an EASI-75 response at week 16 (RD = 26.4%; 95% CI, 12.1% to 40.8%; P < 0.001) in the lebrikizumab plus TCS and placebo plus TCS groups, respectively.
The severity of itch was assessed using the Pruritus NRS, for which patients rated their worst itch symptoms over the previous 24 hours from 0, indicating no itch, to 10, indicating the worst itch imaginable. Among patients who had a Pruritus NRS score of 4 or more at baseline, 45.9% and 13.0% in the lebrikizumab and placebo groups, respectively, reported at least a 4-point reduction at week 16 in the ADvocate 1 study (RD = 32.9%; 95% CI, 24.6% to 41.3%; P < 0.001). The proportion of Pruritus NRS responders was 39.8% and 11.5% in the lebrikizumab and placebo groups, respectively, in the ADvocate 2 study (RD = 28.3%; 95% CI, 20.0% to 36%; P < 0.001), favouring lebrikizumab. In the lebrikizumab plus TCS group in the ADhere study, 50.6% of patients met the Pruritus NRS response criteria, as did 31.9% of patients in the placebo plus TCS group (RD = 19.2%; 95% CI, 4.3% to 34.1%; P = 0.02).
A secondary outcome in the pivotal trials was the change from baseline in the POEM score; the 7-item, self-reported POEM questionnaire was used to assess the frequency of disease symptoms (skin dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping) over the previous week. It is scored from 0 to 28, with a higher score indicating worse disease severity.12 A minimal important difference (MID) of 3.4 points was identified as the threshold for a clinically relevant between-group difference.13 The ADvocate 1 study reported a least squares (LS) mean difference of |||| |||||| |||| ||| |||| || ||||) in the POEM score change from baseline to week 16 for lebrikizumab versus placebo. The LS mean difference was |||| |||||| |||| ||| |||| || ||||| for the lebrikizumab versus placebo groups in the ADvocate 2 study, and the ADhere study reported a LS mean difference of −4.0 points (95% CI, −6.3 to −1.7 points) for lebrikizumab plus TCS versus placebo plus TCS. Of note, this outcome was potentially biased due to the extent of missing data and the analysis methods used to handle missing data. Moreover, the change in POEM score was not part of the graphical testing strategy used to control the family-wise type I error rate, and thus this outcome should be interpreted as supportive evidence only.
In the 3 pivotal trials, the DLQI was used to measure health-related quality of life (HRQoL) in patients 17 years and older, and the CDLQI was used for those who were aged 12 to 16 years. These instruments are scored from 0 to 30, with higher scores indicating poorer HRQoL. MIDs of 4 points for the DLQI and 6 points for the CDLQI were selected as the thresholds for clinically relevant between-group differences.14,15 In the ADvocate 1 study, the LS mean difference in the change from baseline to week 16 in the DLQI total score for lebrikizumab versus placebo was −5.8 points (95% CI, −7.1 to −4.5 points; P < 0.001), and in the ADvocate 2 study, the LS mean difference was −4.9 points (95% CI, −6.3 to −3.5 points; P < 0.001) for lebrikizumab versus placebo. The ADhere study reported a LS mean difference in the change from baseline in DLQI score of −3.3 points (95% CI, −5.3 to −1.3 points; P = 0.001) for the lebrikizumab plus TCS group versus the placebo plus TCS group. These analyses included 75% to 86% of patients randomized to a treatment group who were 17 years or older at the start of the studies.
Among adolescents aged 12 to 16 years, the LS mean difference in the change from baseline in the CDLQI was |||| |||||| |||| ||| ||||| || ||||| in the ADvocate 1 study, |||| |||||| |||| ||| |||| || |||| in the ADvocate 2 study, and −4.6 points (95% CI, −7.2 to −2.0 points) in the ADhere study for the lebrikizumab versus placebo groups at week 16. The change in CDLQI was not controlled for the type I error rate, and thus the outcome should be interpreted as supportive evidence only. Also of note, the number of patients per treatment group was small, ranging from 5 to 11 patients in the placebo groups and from 17 to 26 patients in the lebrikizumab groups.
Maintenance Period
At week 16 of the ADvocate 1 and ADvocate 2 studies, patients in the lebrikizumab group who met the treatment response criteria were rerandomized to placebo or to lebrikizumab every 4 weeks or lebrikizumab every 2 weeks for the maintenance period. This review focuses on the results of the lebrikizumab every-4-weeks groups to be consistent with the Health Canada–recommended maintenance dosing. The ADvocate 1 study reported that 79.2% of patients in the lebrikizumab every-4-weeks group maintained an EASI-75 response at week 52, compared with 61.3% of patients who were switched to placebo (RD = ||||| |||| ||| |||| || |||||). In the ADvocate 2 study, 84.7% and 72.0% of patients maintained an EASI-75 response in the lebrikizumab every 4 weeks and placebo (i.e., lebrikizumab withdrawal) groups, respectively, (RD = ||||| |||| || |||| || |||||||
Harms Results
Induction Period
During the induction period of the trials, the proportion of patients in the ADvocate 1, ADvocate 2, and ADhere studies who experienced 1 or more treatment-emergent adverse events (TEAEs) was 46% versus 52%, 53% versus 66%, and 43% versus 35% in the lebrikizumab and placebo groups, respectively. The most common AEs in the lebrikizumab groups were conjunctivitis, headache, and nasopharyngitis.
The frequency of SAEs was generally low, with 2.1% versus 0.7%, 0.7% versus 2.8%, and 1.4% versus 1.5% reporting an SAE in the lebrikizumab versus placebo groups of the ADvocate 1, ADvocate 2, and ADhere studies, respectively. One patient who received placebo died of a myocardial infarction in the ADvocate 2 study. No other deaths were reported.
During the induction period in the ADvocate 1, ADvocate 2, and ADhere studies, 1.1% versus 0.7%, 3.2% versus 2.8%, and 2.1% versus 0% of patients in the lebrikizumab versus placebo groups, respectively, stopped treatment due to AEs.
Conjunctivitis-related AEs, which was a notable harm, were reported by 4.8% to ||||| of patients in the lebrikizumab groups and by 0.0% to 3.5% of patients in the placebo groups. The RD for conjunctivitis in the lebrikizumab versus placebo groups was |||| |||| ||| ||| || ||||| in the ADvocate 1 study, |||| |||| ||| ||| || ||||| in the ADvocate 2 study, and |||| |||| ||| ||| || |||| in the ADhere study.
Maintenance Period
During the maintenance period, ||| |||||| |||| ||| ||| |||||| ||| of patients experienced a TEAE in the lebrikizumab every-4-weeks group versus the placebo (i.e., lebrikizumab withdrawal) group in the ADvocate 1 and ADvocate 2 trials, respectively. A total of | patients reported an SAE, including |||||||| |||||| in the lebrikizumab every-4-weeks group of the ADvocate 1 study, and ||||||| |||||| in the placebo group and |||||||| |||||| in the lebrikizumab every-2-weeks group of the ADvocate 2 study. No deaths were reported during the maintenance period.
Between week 16 and week 52, 1 patient each in the lebrikizumab every-4-weeks groups of the ADvocate 1 and ADvocate 2 studies stopped treatment due to AEs. No patients in the placebo groups stopped therapy due to AEs during the maintenance period. Overall, conjunctivitis was reported || |||| |||||| ||||| ||| ||||| |||||| ||||| of patients in the lebrikizumab every-4-weeks versus placebo (lebrikizumab withdrawal) groups, respectively, of the ADvocate 1 and ADvocate 2 studies.
Critical Appraisal
No major concerns were identified with the randomization, allocation concealment, blinding, or statistical methods used in the trials included in the systematic review. The key outcomes tested (EASI-75, Pruritus NRS, POEM, and DLQI) were important to patients and had evidence to support their validity and reliability in patients with AD or other dermatologic conditions. The primary estimand for EASI-75, IGA, Pruritus NRS, and DLQI outcomes was used to analyze patients who discontinued due to lack of efficacy or who required rescue therapy as nonresponders, and multiple imputation methods were used to impute data for patients who discontinued for other reasons. These methods should address any potential bias due to the differential use of rescue treatments in the lebrikizumab and placebo groups.
The key limitations of the change in POEM, DLQI, and CDLQI were related to missing data. The analyses of the change in POEM and CDLQI scores were based on the supportive (hypothetical) estimand and the mixed model for repeated measures (MMRM), which assumed that data are missing at random. These outcomes were not based on the true intention-to-treat (ITT) population, as they excluded patients with missing data at baseline. In addition, there were differences between the groups in the frequency of missing outcome data at week 16, and it is unclear if the missing-at-random assumption is valid. Similar issues were noted with regard to missing data for the change in DLQI scores. Due to the missing data imputation methods and the extent and differential rate of missing data, there is potential for bias in the change in POEM and CDLQI scores. The changes in POEM and CDLQI scores were not part of the graphical testing strategy used to control the family-wise type I error rate; therefore, these results should be interpreted as supportive evidence only.
The 52-week data from the ADvocate trials were limited by the enriched population, carry-over effects of lebrikizumab in the placebo group, and the small sample size. At week 16 of the ADvocate studies, patients treated with lebrikizumab who met the response criteria were rerandomized to 1 of 3 groups. This represents an enriched population, and thus the 1-year treatment effects of lebrikizumab may be overestimated compared with what would be observed in an unselected population. Given the long half-life of lebrikizumab (24.5 days16), it is reasonable to assume that there are substantial carry-over effects for patients who switched from lebrikizumab to placebo, which may impact efficacy assessments, as well as the frequency of harms.
The clinical expert consulted for this review did not identify any major limits to the generalizability of the findings of the trials, and the baseline characteristics of patients enrolled were generally consistent with those who may receive systemic treatments for AD in clinical practice. However, the expert noted that the studies excluded some patients with comorbidities who may receive lebrikizumab for AD. Due to these exclusions, the safety and efficacy of lebrikizumab is uncertain for patients with chronic conditions that may require treatment with oral corticosteroids, acute or chronic infections, severe mental or physical illnesses, or a history of immunosuppression. Given that 11% to 22% of patients enrolled were adolescents, the results are mainly reflective of adult patients. The dosing of lebrikizumab during the induction period of the trials was consistent with the Health Canada–recommended dose; however, the clinical expert anticipates that most patients using lebrikizumab will also use TCS as needed. The concurrent use of TCS was prohibited in the ADvocate studies, and thus the magnitude of effects observed in the ADhere study may be more consistent with what may occur in clinical practice. Also, the generalizability of the 52-week efficacy and safety data may be limited, given the enriched population and the carry-over effects of lebrikizumab in patients who switched to placebo. The results at 52 weeks are reflective of the effects of lebrikizumab maintenance therapy, not of lebrikizumab withdrawal among patients who initially tolerate and respond to treatment during the 16-week induction period.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development, and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered to be most relevant to CDA-AMC expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.17,18
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment for the proportion of patients with an IGA 0 or 1 response, EASI-75 response, or at least a 4-point improvement on the Pruritus NRS were based on thresholds informed by the clinical expert consulted for this review. The certainty of evidence assessments for the change in POEM, DLQI, and CDLQI scores were based on thresholds identified in the literature, and the certainty assessments for SAEs and conjunctivitis were based on the presence or absence of any (nonnull) effect.
For the GRADE assessments, findings from the ADvocate 1, ADvocate 2, and ADhere studies were considered together and summarized narratively by outcome, because these studies were similar in population, interventions, design, and outcome measures.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
the proportion of patients with an IGA score of 0 or 1 and at least a 2-point reduction from baseline
the proportion of patients with an EASI-75 response
the proportion of patients who reported at least a 4-point reduction in Pruritus NRS score
the change from baseline in POEM score
the change from baseline in DLQI and CDLQI total scores
SAEs and conjunctivitis AEs.
Table 2: Summary of Findings for Lebrikizumab Versus Placebo for Patients With Moderate-to-Severe Atopic Dermatitis
Outcome and follow-up | Patients (studies), N | Effect | Certainty | What happens |
|---|---|---|---|---|
IGA response | ||||
Proportion of patients with an IGA score of 0 or 1 and a ≥ 2-point improvement from baselinea Follow-up: 16 weeks | 1,062 (3 RCTs) | ADvocate 1
ADvocate 2
ADhere
| High | Lebrikizumab results in an increase in the proportion of patients with an IGA response compared with placebo, with or without concomitant TCS. |
EASI-75 response | ||||
Proportion of patients with an EASI-75 responseb Follow-up: 16 weeks | 1,062 (3 RCTs) | ADvocate 1
ADvocate 2
ADhere
| High | Lebrikizumab results in an increase in the proportion of patients with an EASI-75 response compared with placebo, with or without concomitant TCS. |
Proportion of patients who maintained an EASI-75 response among patients who exhibited an EASI-75 response at week 16 with lebrikizumab 250 mg every 2 weeks induction therapyb Follow-up: 52 weeks | 172 (2 RCTs) | ADvocate 1
ADvocate 2
| Moderatec | Among patients with an EASI-75 response to lebrikizumab induction therapy, lebrikizumab every-4-weeks maintenance therapy likely results in an increase in the proportion of patients who maintain an EASI-75 response compared with patients who switched to placebo. |
Pruritus NRS ≥ 4-point reduction | ||||
Proportion of patients with a ≥ 4-point reduction in Pruritus NRS score from baselined Follow-up: 16 weeks | 964 (3 RCTs) | ADvocate 1
ADvocate 2
ADhere
| High | Lebrikizumab results in an increase in the proportion of patients with at least a 4-point reduction in Pruritus NRS score compared with placebo, with or without concomitant TCS. |
Change in POEM total score | ||||
POEM total score (0 [best] to 28 [worst]) LS mean change from baselinee Follow-up: 16 weeks | 996 (3 RCTs) | ADvocate 1
ADvocate 2
ADhere
| Lowf | Lebrikizumab may result in a reduction in POEM score, compared with placebo, with or without concomitant TCS. |
Change in DLQI score | ||||
DLQI score (0 [best] to 30 [worst]) LS mean change from baselineg Follow-up: 16 weeks | 856 (3 RCTs) | ADvocate 1
ADvocate 2
ADhere
| Lowh | Lebrikizumab may result in a reduction in DLQI score compared with placebo, with or without concomitant TCS. |
Change in CDLQI score | ||||
CDLQI score (0 [best] to 30 [worst]) LS mean change from baselineg Follow-up: 16 weeks | || (3 RCTs) | ADvocate 1
ADvocate 2
ADhere
| Very lowi | The evidence is very uncertain about the effect of lebrikizumab on the change in CDLQI compared with placebo. |
Serious adverse events | ||||
Proportion of patients with SAEs Follow-up: 16 weeks | 1,060 (3 RCTs) | ADvocate 1
ADvocate 2
ADhere
| Very lowj | The evidence is very uncertain about the effect of lebrikizumab on the proportion of patients with 1 or more SAEs compared with placebo, with or without concomitant TCS. |
Proportion of patients with SAEs among patients who met the treatment response criteria at week 16 with lebrikizumab 250 mg every-2-weeks induction therapy Follow-up: 52 weeks | 178 (2 RCTs) | ADvocate 1
ADvocate 2
| Very lowk | Among patients who achieve a response to lebrikizumab induction therapy, the evidence is very uncertain about the effect of lebrikizumab maintenance therapy on the proportion of patients with 1 or more SAEs compared with placebo (i.e., lebrikizumab withdrawal). |
Conjunctivitis | ||||
Proportion of patients with conjunctivitis AEs Follow-up: 16 weeks | 1,060 (3 RCTs) | ADvocate 1
ADvocate 2
ADhere
| Moderatel | Lebrikizumab may result in an increase in the proportion of patients with 1 or more conjunctivitis events compared with placebo, with or without concomitant TCS. The clinical importance of the increase is uncertain. |
Proportion of patients with conjunctivitis AEs among patients who met the treatment response criteria at week 16 with lebrikizumab 250 mg every-2-weeks induction therapy Follow-up: 52 weeks | 178 (2 RCTs) | ADvocate 1
ADvocate 2
| Very lowm | Among patients who achieve a response to lebrikizumab induction therapy, the evidence is very uncertain about the effect of lebrikizumab maintenance therapy on the proportion of patients with 1 or more conjunctivitis events when compared with placebo (lebrikizumab withdrawal). |
AE = adverse event; aRD = adjusted risk difference; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-75 = at least a 75% reduction in EASI score; IGA = Investigator Global Assessment; LEB = lebrikizumab; LS = least squares; NR = not reported; NRS = Numeric Rating Scale; PBO = placebo; POEM = Patient-Oriented Eczema Measure; RCT = randomized controlled trial; RD = risk difference; SAE = serious adverse event; TCS = topical corticosteroids.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThe IGA measures the investigator’s global assessment of the patient’s overall severity of AD at that visit, based on a static, numeric 5-point scale ranging from 0 (clear) to 4 (severe). Based on clinical expert input, the threshold for a clinically important between-group difference was 100 per 1,000 for the proportion of patients with an IGA score of 0 or 1 and at least a 2-point reduction from baseline.
bThe EASI is a composite index, based on the physician’s assessment of 4 clinical signs of the disease (erythema, infiltration and/or papulation, excoriation, and lichenification) and the extent of BSA involved at that visit. It is scored from 0 to 72, with higher scores indicating greater disease severity and/or extent of disease. Based on clinical expert input, the threshold for a clinically important between-group difference was 100 per 1,000 for the proportion of patients with at least an EASI-75 response.
cEASI-75 response at week 52: rated down 1 level for serious imprecision. The CI for differences between groups included the potential for little to no difference (based on the threshold for a clinically important between-group difference of 100 per 1,000 for the proportion of patients who maintained at least an EASI-75 response at week 52).
dThe Pruritus NRS is a patient-reported, single-item, daily, 11-point scale. The scale is used by patients to rate their worst itch severity over the previous 24 hours, with 0 indicating no itch and 10 indicating the worst itch imaginable. Based on clinical expert input, the threshold for a clinically important between-group difference was 100 per 1,000 for the proportion of patients with at least a 4-point reduction from baseline. This outcome was analyzed for the subgroup of patients who had a Pruritus NRS score of 4 or higher at baseline.
eThe POEM is a 7-item, patient-reported questionnaire used to assess the frequency of disease symptoms in adults and children over the previous week. The patients respond to 7 questions on skin dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping. The total score ranges from 0 to 28, with a high score indicating worse disease severity. The MID of 3.4 points was selected as the threshold for a clinically important between-group difference based on the literature and clinical expert input.12,13
fChange in POEM score at week 16: rated down 2 levels for very serious study limitations. The extent of missing data was large and the method for accounting for missing data was potentially biased. Note that there was no control for the type I error rate for this end point, so outcomes should be interpreted as supportive evidence only.
gThe DLQI (for patients 16 years and older) and CDLQI (for those younger than 16 years) are patient-reported, 10-item, HRQoL questionnaires that cover 6 domains (symptoms and feelings, daily activities, leisure, work and school, personal relationships, and treatment) over the previous week. The total score ranges from 0 (no impact of skin disease on quality of life) to 30 (maximum impact on quality of life). MIDs of 4 points for the DLQI and 6 points for the CDLQI were selected as the thresholds for clinically important between-group differences based on the literature and clinical expert input.15,19-21
hChange in DLQI at week 16: rated down 1 level for serious imprecision (the CI for differences between groups included the potential for little to no difference based on an MID of 4 points) and rated down 1 level for serious study limitations (due to missing data). Also considered was the possibility of inconsistency, given that the point estimate for 1 of the 3 trials falls below the MID, although a decision was made not to rate down for inconsistency.
IChange in CDLQI at week 16: rated down 1 level for serious imprecision (the CI for differences between groups included the potential for little to no difference based on a MID of 6 points) and rated down 2 levels for very serious study limitations. The extent of missing data was large and the method for accounting for missing data was potentially biased. Note that there was no control for the type I error rate for this end point, so outcomes should be interpreted as supportive evidence only.
jSAE at week 16: rated down 2 levels for very serious indirectness (follow-up duration limited to 16 weeks, which may be insufficient to detect uncommon SAEs or those that develop over time; the clinical expert noted that worsening AD may be reported as an SAE, whereas this more accurately reflects lack of efficacy) and rated down 1 level for serious imprecision (the CI for differences between groups includes the possibility of no difference, benefit [fewer harms], or increased harms).
kSAE at week 52: rated down 2 levels for very serious indirectness (AEs were reported for an enriched population of patients who had received lebrikizumab 250 mg every-2-weeks induction therapy and met the treatment response criteria at week 16; the AEs reported in the placebo group may be confounded due to the carry-over effects of lebrikizumab before the switch to placebo; follow-up duration and sample size may be insufficient to detect uncommon SAEs or those that develop over time) and rated down 1 level for serious imprecision (the CI for differences between groups includes the possibility of no difference, benefit [fewer harms], or increased harms).
lConjunctivitis at week 16: rated down 1 level for serious indirectness (the clinical expert stated that dermatologists may not have sufficient expertise to distinguish between eye disorders with a similar presentation, so the reported conjunctivitis-related AEs may be flawed).
mConjunctivitis at week 52: rated down 2 levels for very serious indirectness (the clinical expert stated that dermatologists may not have sufficient expertise to distinguish between eye disorders with a similar presentation, so the reported conjunctivitis-related AEs may be flawed; AEs were reported for an enriched population of patients who had received lebrikizumab 250 mg every-2-weeks induction therapy and met the treatment response criteria at week 16; the AEs reported in the placebo group may be confounded due to the carry-over effects of lebrikizumab before the switch to placebo) and rated down 2 levels for very serious imprecision (the CI for differences between groups includes the possibility of no difference, benefit [fewer harms], or increased harms).
Sources: Clinical study report (CSR) for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere,9 additional information supplied by sponsor.22
Long-Term Extension Study
Description of Study
One long-term extension study was summarized to provide evidence on the long-term (100-week) efficacy and safety of lebrikizumab among patients with moderate-to-severe AD who were enrolled in the ADvocate 1, ADvocate 2, ADhere, ADore, and ADopt-VA studies (parent trials).23 This study was conducted at 199 centres that enrolled 999 patients in Australia, Bulgaria, Canada, Estonia, France, Germany, Latvia, Lithuania, Mexico, Poland, Singapore, South Korea, Spain, Taiwan, Ukraine, and the US. This report presents interim safety data from the ADjoin study and limited efficacy data at week 40 for a subset of patients who completed the 16-week ADhere study (i.e., up to 56 weeks of lebrikizumab treatment). |||| |||| |||||||| || |||||| ||| |||||| |||| |||| ||||||||| ||||| || ||||||| ||| |||||||| |||| ||| |||||||| || ||| ||||||| |||||||| ||||| |||||||
Efficacy Results
Efficacy outcomes were assessed up to || ||||| (week 16 to week 104). Evaluation of efficacy in the interim report was conducted on a subset of the main cohort, which included || |||||||||||| who were responders to lebrikizumab plus TCS in the ADhere study.
At week 40, the proportion of patients with an IGA score of 0 or 1 was ||||| in the lebrikizumab 250 mg every-4-weeks group and ||||| in the lebrikizumab 250 mg every-2-weeks group.
At week 40, the mean (standard error [SE]) percent change from baseline in EASI score in the lebrikizumab 250 mg every-4-weeks and lebrikizumab 250 mg every-2-weeks groups were |||||| ||||| ||| |||||| |||||| respectively. The proportion of patients with an EASI-75 response at week 40 in the lebrikizumab 250 mg every-4-weeks and lebrikizumab 250 mg every-2-weeks groups was ||||| ||| |||||| respectively.
Among patients who had a Pruritus NRS score of 4 or more points at baseline, the proportion of patients who reported an improvement of at least 4 points at week 40 in the lebrikizumab 250 mg every-4-weeks and lebrikizumab 250 mg every-2-weeks groups was ||||| ||| |||||| respectively.
The mean (SE) percent change in POEM score from baseline to week 40 in the lebrikizumab 250 mg every-4-weeks and lebrikizumab 250 mg every-2-weeks groups was |||||| |||||| ||| |||||| ||||||, respectively.
Harms Results
Overall, || |||||||| (|| || ||| |||||||| |||||| ||||||||||) discontinued study treatment due to AEs. Discontinuation due to an AE was noted in || |||||| || ||| ||||| |||||||| ||| || | |||||| || ||| |||||||||| ||||||||.
One death due to natural causes occurred in the lebrikizumab 250 mg every-2-weeks group.
The most frequently reported TEAEs were in the infections and infestations system organ class, with COVID-19 (|||| in lebrikizumab 250 mg every-4-weeks group and |||| in the lebrikizumab 250 mg every-2-weeks group) and nasopharyngitis (|| in lebrikizumab 250 mg every-4-weeks group and |||| in the lebrikizumab 250 mg every-2-weeks group) being the most common TEAE. A similar proportion of patients in the lebrikizumab 250 mg every-2-weeks group (||||) and the lebrikizumab 250 mg every-4-weeks group (||||) reported an AE of atopic dermatitis exacerbation. The proportion of patients experiencing 1 or more AEs in the conjunctivitis cluster (narrow terms) was similar in both the lebrikizumab 250 mg every-4-weeks group (||||) and the lebrikizumab 250 mg every-2-weeks group (||||).
Critical Appraisal
Internal Validity
There is no randomized comparison to another treatment or a placebo, which limits the ability to draw inferences on the effects of lebrikizumab in the study population. The patients were aware they were receiving active treatment, so their expectations of treatment may have influenced their reporting of subjective patient-reported outcomes, such as the POEM, and subjective AEs or investigator-reported IGA and EASI responses, which are measures that require subjective judgments. Discontinuation rates were |||| in the lebrikizumab every-4-weeks and ||||| in the lebrikizumab every-2-weeks groups. Among patients from the ADhere study (efficacy assessment), the rates of discontinuation are ||||| in the every-4-weeks group and ||||| in every-2-weeks group. Thus, there is potential bias due to missing data. All analyses were conducted descriptively without statistical comparisons between the cohorts or adjustment for multiple comparisons.
External Validity
Only responders in the ADhere study were included in the efficacy assessment. Patients were excluded if, during their participation in the parent trial, they developed an SAE deemed to be related to lebrikizumab, developed an AE that was deemed to be related to lebrikizumab and led to study treatment discontinuation, or had conditions in the parent trial that led to investigator-initiated or sponsor-initiated withdrawal from the study. This is a select population, so the results apply only to patients who initially tolerate and respond to lebrikizumab. The proportion of patients with concomitant TCS use and systemic rescue therapy was higher in the every-4-weeks group than in the every-2-weeks group. The effect of these differences between groups on the efficacy results remains unclear.
Indirect Comparisons
Description of Studies
The sponsor-submitted indirect treatment comparison (ITC) first conducted a systematic literature review (SLR) to identify evidence for inclusion in a network meta-analysis (NMA). The relative efficacy of lebrikizumab (with or without TCS) from the ADvocate 1, ADvocate 2, J2T-DM-KGAF, ADhere, ADhere-J, ADopt-VA, and ADvantage trials was indirectly compared to alternative treatments for AD using a Bayesian NMA. Comparators of interest for the sponsor-submitted NMA included abrocitinib, dupilumab, and upadacitinib. All networks in the sponsor-submitted NMA also included baricitinib and tralokinumab as comparators.24 However, baricitinib does not have Health Canada approval for the treatment of AD, and tralokinumab is not currently reimbursed by public drug plans in Canada. As such, results comparing lebrikizumab to baricitinib or tralokinumab were not included in this report. Outcomes of interest included EASI response, IGA 0 or 1 response, a reduction of greater than or equal to 4 points in the Pruritus NRS at week 16, and a reduction of greater than or equal to 4 points in Pruritus NRS at week 4.24
Efficacy Results
The SLR identified a total of ||||| citations. A total of || unique studies identified by the SLR were assessed for eligibility to be included in the NMAs. Three studies of lebrikizumab that were not identified as part of the SLR were also assessed for inclusion. In total, || studies were eligible for inclusion in the NMAs: || monotherapy studies and || combination therapy studies.24
Networks were generated for all eligible interventions as monotherapy and combination therapy for the outcomes of EASI response, IGA 0 or 1 response, and Pruritus NRS response at time points of interest. In all cases, the baseline risk-adjusted random-effects model was selected as the favoured model, based on the deviance information criterion and residual deviance.24
Primary Analysis
EASI response (week 16): In the primary analysis for EASI response at week 16 in the monotherapy network, there was insufficient evidence to show a difference between lebrikizumab and dupilumab 300 mg every 2 weeks or abrocitinib 100 mg daily. Abrocitinib 200 mg daily (probit difference, ||||| |||| |||||||| |||||||| |||||| |||||| ||||||), upadacitinib 15 mg daily (probit difference ||||| |||| |||| |||||| ||||||), and upadacitinib 30 mg daily (probit difference ||||| |||| |||| |||||| ||||||) were favoured over lebrikizumab.24
|| ||| ||||||| |||||||| ||| |||| |||||||| || |||| || || ||| ||||||||||| ||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| ||| || ||| |||||||||| |||||||||||||
IGA response of 0 or 1 (week 16): In the primary analysis for an IGA 0 or 1 response at week 16 in the monotherapy network, there was insufficient evidence to show a difference between lebrikizumab and dupilumab 300 mg every 2 weeks, abrocitinib 100 mg daily or 200 mg daily, or upadacitinib 15 mg daily. Upadacitinib 30 mg daily was favoured over lebrikizumab (odds ratio [OR], |||| |||| |||| ||||| ||||||.24
|| ||| ||||||| |||||||| ||| ||| ||| |||||||| || |||| || || ||| ||||||||||| ||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| ||| || ||| |||||||||| ||||||||||| |||||| ||| |||||||||||| || || ||||| |||| |||| ||||| ||| |||||||| |||| |||||||||||| |||| ||| |||| |||| |||| |||| ||||| |||||||||
A reduction of greater than or equal to 4 points in Pruritus NRS (week 16): In the primary analysis for Pruritus NRS response at week 16 in the monotherapy network, there was insufficient evidence to show a difference between lebrikizumab and dupilumab 300 mg every 2 weeks, abrocitinib 100 mg daily or 200 mg daily, or upadacitinib 15 mg daily. Upadacitinib 30 mg daily was favoured over lebrikizumab (OR, |||| |||| |||| ||||| |||||).24
|| ||| ||||||| |||||||| ||| ||||||||||||| ||| |||||||| || |||| || || ||| ||||||||||| ||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| ||||||||||| ||| || ||||| |||| |||| ||| |||||||||||| || || ||||| |||| |||| ||||||||| ||| || ||||| | ||||| |||| ||| |||| |||| |||| |||| ||||| ||||||| ||||||||||| ||| || ||||| |||| ||| |||| |||| |||| |||| ||||| ||||||| ||| |||||||||||| || || ||||| |||| ||| |||| |||| |||| |||| ||||| |||||| |||| |||||||| |||| |||||||||||| |||| ||||||
A reduction of greater than or equal to 4 points in Pruritus NRS (week 4): In the primary analysis for Pruritus NRS response at week 4 in the monotherapy network, there was insufficient evidence to show a difference between lebrikizumab and dupilumab 300 mg every 2 weeks or abrocitinib 100 mg daily. Abrocitinib 200 mg daily (||| |||| |||| |||| ||||| ||||]), upadacitinib 15 mg daily (||| |||| |||| |||| ||||| ||||]), and upadacitinib 30 mg daily (||| |||| |||| |||| ||||| |||||| were favoured over lebrikizumab.24
|| ||| ||||||| |||||||| ||| ||||||||||||| ||| |||||||| || |||| | || ||| ||||||||||| ||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| ||||||||| ||| || ||||| | ||||| |||| |||| ||||||||||| ||| || ||||| |||| |||| ||| ||||||||||| ||| || ||||| |||| |||| |||||||||||| || || ||||| |||| ||| |||| |||| |||| |||| ||||| |||||| ||| |||||||||||| || || ||||| |||| ||| |||| |||| |||| ||| ||||| |||||| |||| |||||||| |||| |||||||||||| |||| ||||||
Secondary Analysis
Phase III studies only in monotherapy networks: ||||||||| |||||||| |||| ||||||||| ||||||||| |||| ||||| ||| ||||||| || ||| ||||||||||| ||||||||| | ||||| || || |||||| |||| |||||||| || ||| |||||||| |||| |||| ||||| ||| |||||||| || ||| |||| |||||||| ||| |||||||||| |||||| ||||||| ||||| ||| |||||||| || ||| |||| ||||| ||| ||| ||||||| |||| |||||||||| |||| ||| ||||||| ||||||||| ||| ||| ||||||||| |||||||||| ||| |||||| ||||||| ||||| |||||||| ||| |||||||| |||| ||| ||| ||||| || ||||||| ||| ||||||| |||| |||||||||| |||| ||| ||||||| ||||||||| || ||| ||| ||| |||||||| |||||||| |||||||||||| || || ||||| ||| |||| |||||||| |||| |||||||||||| |||| |||| |||| |||| |||| || |||||||||
Meta-regression analysis: ||||||||| |||||||| ||||| ||||||||||||||| || |||||| ||| |||||||| |||||||| |||| ||||||||| ||| ||||||||||| ||| ||||||||||| ||||||| |||||||| ||| ||| |||||||| || |||| ||||| || |||| || ||| ||| ||| |||||||| || |||| |||| ||| ||||||||| |||||||| | ||||||| ||||||||||||||| || |||| |||||||| || |||| || |||||||| ||| |||||||| |||| |||| ||||| ||| |||||||||| ||||| ||| || ||||||||||| |||||||||| || ||| ||||||| ||||| ||| |||||| ||||||| ||||||| |||||| |||||| ||||||| |||||| |||| |||||||| ||||| || ||| ||||| |||||||| ||||||||| ||||||| |||| |||||||||| |||| ||| ||||||| ||| ||||||||| ||||||||| |||| ||||||||||| ||| || |||||| |||||||||||| || || |||||| ||| |||||||||||| || || ||||| |||||||| |||| |||||||||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| ||| ||||| ||||||||||| ||| ||| ||| |||||||| || ||| ||||||||||| |||||||| | |||||| ||||||| ||||| |||||||| ||| ||| |||||||||| || |||||||| |||| || ||| ||||| || | || |||| || |||||||| ||| |||||||| || ||| ||||||||| |||||| ||| ||||||| |||| |||||||||| |||| ||| ||||||| ||||||||| |||| |||||||||||| || || ||||| |||||||| |||| |||||||||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| ||| ||||| |||||||||||||||| ||| ||||||||||| ||||||| |||||||| ||| ||||| ||||||| ||||| |||||||| ||| |||||||| |||| ||||| ||| ||| ||||||||| ||||| || ||||| |||| ||| |||| |||||||||| || |||||||| ||| |||||| ||||||| |||||| ||||||||||| ||| ||||||| |||| |||||||||| |||| ||| ||||||| ||| ||||||||| ||||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| |||||||||||| || || ||||| |||| |||||||||| ||| ||||||||| ||| |||||| ||||||| ||||| |||||||| ||| |||||||| ||| |||||||| ||| |||||||| || ||| ||||||||| |||||| ||||||| |||| |||||| |||||||||| |||| ||| ||||||| ||||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| |||||||||||| || || ||||| |||| |||
Harms Results
Harms were not evaluated in the sponsor-submitted NMA.
Critical Appraisal
The sponsor-submitted NMA was informed by an SLR that included comprehensive searches (updated in April 2023) of multiple databases, conference proceedings, clinical trial databases, and health-technology assessment websites. Additionally, the risk-of-bias assessment conducted by the sponsor did not indicate a serious risk of bias in the included studies. However, it should be noted that methods for risk-of-bias appraisals were incompletely reported (i.e., it is not clear how many reviewers were involved and whether they worked independently). As such, the risk for bias and error in the appraisals could not be ascertained. Further, the risk-of-bias appraisal was undertaken at the study level, rather than at the level of the reported effects. Appraisals undertaken at the study level do not account for differences in the risk of bias that can exist across reported results (within and across outcomes) within trials.25 Additionally, there is a risk of bias due to missing results in the networks, because trials of relevant comparator treatments without a placebo control group were excluded. ||| ||| ||||||||| |||||||| ||||||| ||||| | |||||| |||| |||||||| |||| ||| ||||||||| ||| |||| |||||||
A feasibility assessment was conducted, evaluating potential heterogeneity in study design; patient baseline characteristics; interventions; and outcomes, time points, and placebo response. The sponsor noted that some heterogeneity was observed across studies in both the monotherapy and combination therapy networks. Studies for abrocitinib used a 12-week time of assessment, as opposed to the 16 weeks used for other trials. The effect of the difference in time of assessment was not evaluated in the NMA and remains unknown. There were differences in age across studies, with the mean age ranging from |||| ||||| || |||| years. There was also heterogeneity in the proportion of patients |||| ||| |||||||| |||||| || ||||||| ||| |||| |||||||| |||||| || ||||| |||||| ||||||||| || ||| ||||||||||| ||||||| |||||||| ||||||| ||||||| || ||||||||||||| || ||||| |||||| ||| |||||||| ||||||||||||||| |||| |||||||| || ||||||||| |||||||| |||| ||| |||||| |||| || |||| |||||| ||| |||||||||| |||| ||||| || |||||| ||| |||| ||||||||||| ||||| || ||||||| Adjustment for baseline EASI and IGA responses did not bring about improvements in model fit, and conclusions were considered to be comparable to the primary analysis. Other differences were noted by the CDA-AMC review team in weight and ethnicity across studies, although the impact of these differences remains unclear. The sponsor also noted heterogeneity in other features, such as race and time since AD diagnosis, although it is not clear whether these are important treatment-effect modifiers. No formal search for potential treatment-effect modifiers was conducted; instead, the sponsor relied on internal clinical opinion, only including AD severity measured by EASI and IGA, and weight, which the clinical expert consulted by CDA-AMC agreed with, although there was a risk of bias in the selection of treatment-effect modifiers, and it was not clear whether the list was comprehensive. Additionally, the sponsor highlighted differences in the ||||| |||||||| ||| ||||||||| || ||| ||||||||| || ||| ||||||||||| ||||||| ||||||| |||||| |||||||. Differences in TCS treatment may have biased the reported response rates and limited the reliability of comparing responses in patients receiving the active interventions; however, baseline risk-adjusted analysis models were included to mitigate the potential for bias. No scenario analyses were conducted to compare the difference between adjusted and unadjusted results; thus, it is unclear what effect not adjusting for baseline risk had on the results. Overall, the notable heterogeneity in the baseline characteristics raises concern about the plausibility of the transitivity assumption, so the resulting effect estimates may not be valid.
Baricitinib and tralokinumab were included as comparators in the NMAs; however, the use of baricitinib for AD is limited in Canada, given the lack of a specific indication for AD and the availability of more efficacious and tolerable JAK inhibitors (i.e., abrocitinib, upadacitinib). Tralokinumab, although indicated for AD, received a do not reimburse recommendation from CDA-AMC and is not reimbursed in Canada. As such, comparative results for these treatments were not included in this report.
Outcomes included in the NMA were relevant to the treatment of AD in Canada, although the clinical expert consulted by CDA-AMC highlighted the fact that EASI scores are generally not calculated in routine clinical practice. Additionally, outcomes of importance to this review, including harms and HRQoL, were not included in the NMA.
In all random-effects analyses, results were associated with wide 95% credible interval (CrIs), with most estimates crossing the 0 or 1 threshold, suggesting notable imprecision in the results and precluding conclusions to be drawn about which treatment is favoured. For some comparisons in the monotherapy ||| ||||||||||| ||||||| |||||||| |||||| |||||||||||| |||||| ||||||||| ||| || ||||| | ||||| || ||||| ||| ||||||||||| ||| || ||||| || |||||| there was generally insufficient evidence to demonstrate a difference between treatments for most outcomes. Further, abrocitinib 200 mg daily, upadacitinib 15 mg daily, and upadacitinib 30 mg daily (± TCS) were favoured over lebrikizumab (± TCS) for most outcomes but were also associated with wide 95% CrIs. Overall, this imprecision limits the interpretability of the treatment effect of lebrikizumab relative to other comparators. Furthermore, this NMA was primarily restricted to adults, so it is unclear whether the results may be generalized to adolescents with AD.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
The sponsor submitted 4 studies that provided additional data to cover gaps in the systematic review evidence:
ADvantage, a phase III, 52-week (16-week double-blind induction period followed by a 36-week open-label maintenance period), RCT designed to address uncertainty regarding the efficacy and safety of lebrikizumab, specifically in patients whose AD is not adequately controlled with cyclosporine or for whom cyclosporine is not medically advisable (N = 331).
ADopt-VA, a 16-week, phase III, randomized, double-blind, placebo-controlled, parallel-group trial designed to address uncertainty regarding the impact of lebrikizumab on vaccine immune responses. This trial also provides evidence of the efficacy and safety of lebrikizumab (N = 247).
ADhere-J, a 68-week (16-week induction period plus a 52-week maintenance period), phase III, randomized, double-blind, placebo-controlled, parallel-group study designed to address uncertainty regarding the efficacy and safety of lebrikizumab, specifically for patients in Japan (N = 268).
ADore, a 52-week, open-label, single-arm study designed to address uncertainty regarding the efficacy and safety of lebrikizumab, specifically among adolescent patients (N = 206 received treatment, 172 completed the treatment period).
The ADvantage Study
Results
A summary of efficacy results for patients randomized to lebrikizumab plus TCS relative to placebo plus TCS at week 16 is provided here.
EASI-75: 68.4% versus 40.8%, P < 0.0001, || | |||||| ||| || |||||| |||||
IGA 0 or 1 and an improvement of at least 2 points: 42.0% versus 24.5%, ||||||||| || | |||||| ||| || ||||| |||||
Pruritus NRS improvement of at least 4 points: 49.9% versus 29.7%, ||||||||| || | |||||| ||| || ||||||||||
POEM mean (SD) change from baseline: ||||| ||||| |||||| |||| |||||| ||||||||| || |||| |||||||||| | ||||| ||| || |||||| |||||
DLQI mean (SD) change from baseline: |||| ||||| |||||| |||| ||||||||||| || |||| |||||||||| | ||||| ||| || ||||||| ||||||
CDLQI mean (SD) change from baseline: |||| ||||| |||||| |||| |||||| |||||||| || |||| |||||||||| | ||||| ||| ||| ||||||| ||||||
In terms of safety, a summary of harms results for patients randomized to lebrikizumab plus TCS relative to placebo + TCS at week 16 is provided here.
Proportion of patients with at least 1 AE: 61.8% versus 53.2%
Proportion of patients with at least 1 SAE: |||| |||||| ||||
Proportion of patients with at least 1 AE leading to study drug discontinuation: 0.9% versus 1.8%
Proportion of patients with conjunctivitis AE: ||||| |||||| |||||
Up to week 52, harm results for patients randomized to lebrikizumab plus TCS were reported as ||| for patients with at least 1 AE, |||| for patients with at least 1 SAE, and |||| for patients with at least 1 AE leading to study drug discontinuation.
Critical Appraisal
Because few adolescents were enrolled in this study, generalizability to this age group is limited. No control for multiplicity was included for the analyses of the secondary efficacy end points; therefore, the study is at risk of type I error (false-positive results) for all end points except EASI-75 response. Dosage of maintenance therapy was 250 mg every 2 weeks, which is not consistent with the Health Canada product monograph, which recommends 250 mg every 4 weeks after 16 weeks. In the lebrikizumab group versus the placebo group, |||| ||| |||| discontinued the study which might increase risk of bias due to missing outcomes data.
The ADopt-VA Study
Results
The efficacy results reported in the ADopt-VA study that correspond to patients randomized to lebrikizumab versus placebo at week 16 are provided here.
EASI-75: 58.0% versus 32.7%, P < 0.001, || | |||||| ||| || |||||| |||||
IGA 0 or 1 and an improvement of at least 2 points: 40.6% versus 18.9%, P < 0.001, || | |||||| ||| || |||||| |||||
A Pruritus NRS improvement of at least 4 points: ||||| |||||| |||||| |||||||| || | |||||| ||| || ||||| |||||
POEM LS mean change from baseline (SE): −9.4 (0.8) versus −6.6 (0.8), |||||| || |||| |||||||||| | ||||| ||| || |||||| ||||||
In terms of safety, a summary of the harms for patients randomized to lebrikizumab versus placebo at week 16 is provided here.
Proportion of patients with at least 1 AE: 38.4% versus 34.4%
Proportion of patients with at least 1 SAE: 0.8% versus 0.8%
Proportion of patients with at least 1 AE leading to study drug discontinuation: 2.4% versus 4.1%
Proportion of patients with conjunctivitis AE: |||| |||||| |||
Critical Appraisal
There is an increased risk of type I error (false-positive results) for all end points. The results of this study may not be generalizable to adolescent patients. The use of TCS was |||| in lebrikizumab group and ||||| in the placebo group, and its effect on the results is not clear. The discontinuation rate was ||| in the placebo group and |||| in the lebrikizumab group, which might increase the risk of bias due to missing outcomes data.
The ADhere-J Study
A total of ||| |||||||||||| in the ADhere-J study completed the induction period, including patients receiving placebo, patients in the lebrikizumab every-4-weeks group, and patients in the lebrikizumab every-2-weeks group. Responders in the lebrikizumab every-4-weeks group continued on with 250 mg lebrikizumab every 4 weeks. Responders in the lebrikizumab every-2-weeks group were randomly allocated to receive 250 mg lebrikizumab every 2 weeks or 250 mg lebrikizumab every 4 weeks. The nonresponders and those who used rescue therapy in the induction period moved to the escape arm and received 250 mg lebrikizumab every 2 weeks. In the placebo group, responders continued to receive placebo, whereas nonresponders and those who used rescue therapy in the induction period moved to the escape arm and received a loading dose of 500 mg lebrikizumab at week 16 and week 18.
Results
A summary of the efficacy results for the induction period corresponding to patients randomized to placebo plus TCS versus lebrikizumab every 2 weeks plus TCS at week 16 is provided here.
EASI-75: 13.4% versus 51.2%, P < 0.001
IGA 0 or 1 and an improvement of at least 2 points: 6.1% versus 33.4%, P < 0.001
A Pruritus NRS improvement of at least 4 points: 3.3% versus 32.7%, P < 0.001
DLQI LS mean (SE) change from baseline: ||| |||| ||||| |||||| |||| |||||||||||
CDLQI LS mean (SE) change from baseline: ||| |||| ||||| |||||| |||| |||||||||||
POEM LS mean (SE) change from baseline: |||| ||||| |||||| |||| |||||| | |||||||
||| ||| ||||||||||| ||||||| ||||||| ||||| || |||||||| |||||||| |||| || |||||||| || |||| || ||||| ||| |||||||| || |||||||| ||| ||||||||| || |||||||||||| ||||| | ||||| ||||||||| ||||||| ||| |||| |||||||| || |||||||||||| ||||| | ||||| ||||||||||| |||||||.
Harm results for induction period in the placebo versus lebrikizumab every-2-weeks plus TCS groups:
Proportion of patients with at least 1 AE: 63.4% versus 75.6%
Proportion of patients with at least 1 SAE: 2.4% versus 0.8%
Proportion of patients with AEs leading to study drug discontinuation: ||| || |||||| ||||
Proportion of patients with conjunctivitis AE: ||| ||||| |||||| |||||
Harm results for the maintenance blinded period: || |||||||| |||||||| || |||||||||||| ||||| | ||||| ||||||||||| ||||||||
Proportion of patients with at least 1 AE: ||| |||||
Proportion of patients with at least 1 SAE: ||| ||
Proportion of patients with AEs leading to study drug discontinuation: ||| ||
Proportion of patients with conjunctivitis AE: ||| |||||
Critical Appraisal
This study is limited to patients in Japan, and generalizability to patients in Canada is uncertain. Not all patients in the induction phase received the Health Canada–recommended dose. High-potency TCS use was ||||| in the placebo group, |||| in the lebrikizumab every-4-weeks group, and |||| in the lebrikizumab every-2-weeks group; the effect of this difference on the results is unclear. DLQI, CDLQI, and POEM were not included in multiplicity testing and are at risk of type I error. For the maintenance period, discontinuation was |||| in the placebo group versus |||| in the lebrikizumab every-2-weeks responder and/or every-4-weeks plus TCS group. The impact of missing data on the findings is unclear.
The ADore Study
Results
The efficacy results reported in the ADore study at week 52 are summarized here.
EASI-75 (Markov chain Monte Carlo multiple imputation [MCMC-MI] analysis): 81.9%
IGA 0 or 1 and an improvement of at least 2 points (MCMC-MI analysis): 62.6%
DLQI mean (SE) change from baseline (MCMC-MI): −8.9 (0.9), N = 35
CDLQI mean (SE) change from baseline (MCMC-MI): −6.5 (0.5), N = 168.
The harms results reported in the ADore study at week 52 are summarized here.
Proportion of patients with at least 1 AE: 65%
Proportion of patients with at least 1 SAEs: 2.4%
Proportion of patients with at least 1 AE leading to study treatment discontinuation: 2.4%
Proportion of patients with conjunctivitis AE: 6.8%
One death (0.5%), the cause of which was reported as cardiac arrest.
Critical Appraisal
There is a risk of bias in the measurement of the outcomes due to the open-label design and the subjectivity of the outcomes. There is no comparator, which limits the ability to determine causal inferences. Maintenance therapy doses were not consistent with the Health Canada product monograph. There is a ||||| ||||||||||||||| rate, which might contribute to the risk of bias due to missing outcome data.
Key Take Aways for Studies Addressing Gaps in the Evidence
In patients with moderate-to-severe AD who received induction therapy with lebrikizumab 250 mg every 2 weeks (with or without TCS), the results of the supplementary trials (ADvantage, ADhere-J, and ADopt-VA) were generally consistent with the findings of the pivotal trials. The efficacy findings favoured lebrikizumab over placebo for EASI-75, IGA 0 or 1, and a Pruritus NRS score of at least 4 points at 16 weeks in the RCTs addressing gaps in the evidence (ADvantage, ADhere-J, and ADopt-VA).
In terms of harms results at week 16, in the ADvantage study, a higher proportion of patients in the lebrikizumab group than in the placebo group reported TEAEs and serious TEAEs. In the ADopt-VA study, the proportion of patients with TEAEs and the proportion of patients with at least 1 AE leading to study drug discontinuation were higher in the lebrikizumab group than in the placebo group. In the ADhere-J study, the proportion of patients who reported TEAEs and the proportion of patients with 1 or more AEs leading to study drug discontinuation were higher in the lebrikizumab every-2-weeks group than in the placebo group. In the open-label ADore study, 2.4% of patients reported at least 1 AE leading to permanent discontinuation from the study treatment, including 1 death.
Some of the limitations of the ADvantage study include uncertain generalizability to adolescent patients, dosage inconsistency with the Health Canada–recommended dose, lack of control for multiplicity for secondary efficacy end points (increasing the risk of type I errors), and risk of bias due to missing outcomes data. In the ADopt-VA study, there is an increased risk of type I error, uncertain generalizability to adolescent patients, between-group differences in the use of TCS, and risk of bias due to missing outcome data. In the ADhere-J study, there was uncertain generalizability to patients in Canada, the dosage was inconsistent with the Health Canada–recommended dose for the induction period, there were between-group differences in the use of high-potency TCS, there was an increased risk of type I error for DLQI, CDLQI, and POEM scores, and there were between-group differences in discontinuations during the maintenance period.
Conclusions
In patients with moderate-to-severe AD that was not adequately controlled with topical therapies, 3 pivotal RCTs demonstrated that lebrikizumab induction therapy provided a clinically relevant improvement in physician-assessed signs of AD and reduced patient-reported symptoms of itch relative to placebo, measured based on EASI-75 response, IGA 0 or 1 response, or Pruritus NRS response at week 16. The benefits were observed when lebrikizumab was used as monotherapy and in combination with TCS. Lebrikizumab may improve HRQoL and reduce other symptoms of AD at 16 weeks compared with placebo, but the evidence is less certain.
There was no direct evidence comparing lebrikizumab to other biologics or JAK inhibitors used to treat AD in Canada; however, the sponsor submitted indirect evidence from an NMA that assessed the short-term comparative efficacy. The results of the NMA were inconclusive for lebrikizumab compared with dupilumab and abrocitinib, with most estimates affected by serious imprecision. The NMA results suggest that upadacitinib may be favoured over lebrikizumab for the proportion of patients with an EASI or Pruritus NRS response, although differences were not consistently detected, and the clinical relevance of any differences is unclear.
Lebrikizumab may increase the short-term risk of conjunctivitis relative to placebo. The NMA did not assess any safety end points, so the comparative safety of lebrikizumab is unknown. The longer-term safety and efficacy of lebrikizumab derived from the RCTs and extension study is uncertain due to limitations with the data. These limitations include an enriched population, carry-over effects for the 52-week data in the pivotal trials (i.e., effect estimates apply to lebrikizumab maintenance therapy relative to lebrikizumab withdrawal among patients who tolerate the treatment and initially experience a response), and the lack of a comparator group for the extension study.
The supplementary evidence available from the sponsor-submitted trials addressing the gaps was generally consistent with the findings of the pivotal trials, including in patients whose AD was not adequately controlled with cyclosporine or for whom cyclosporine was not medically advisable. No new safety signals were detected in the single-arm study in adolescents.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of lebrikizumab 250 mg per 2 mL SC injection in adult and adolescent patients aged 12 years and older for the treatment of moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.
Disease Background
The contents of this section have been informed by materials submitted by the sponsor and clinical expert input. The following information has been summarized and validated by the CDA-AMC review team.
AD is a chronic, relapsing, inflammatory, and noncontagious skin disease that is commonly associated with other atopic expressions, such as asthma, allergic rhinitis, and food allergy.1 Approximately 90% of patients with AD develop the disease within the first 5 years of life.1 Risk factors for the development of AD are family history of atopy (a major risk factor), and a genetic defect in the filaggrin gene.1,26 Although AD primarily affects the skin, accumulating evidence suggests that it is a systemic disease with both atopic and nonatopic comorbidities.27 Patients with AD are at an increased risk of skin infections because of excessive rubbing or scratching.1 Exacerbations, or flares, are an integral part of the disease course, and generally indicate a worsening of AD that requires the escalation or intensification of treatment.4 Flares can be aggravated or triggered by endogenous factors, including alteration of skin microbiota, skin barrier dysfunction, dysregulation of cytokine production, stressful life events, and hormonal changes, or by environmental factors, such as allergen exposure, hot and humid or dry and cold environments, sweating, sun exposure, or clothing made from irritable fibres.4
AD impacts approximately 15% to 20% of children and approximately 1% to 3% of adults worldwide; in high-income countries, AD affects around 20% of children and up to 10% of adults.1,5 Approximately 50% of adult patients have moderate-to-severe disease, based on clinical disease severity scales.6
AD can adversely impact all aspects of life and the productivity of patients and their families, especially in its severe state.3,28 The burden of disease and its impact on quality of life may be profound, particularly in cases of moderate-to-severe AD.2 An SLR that focused on the burden of AD in adolescents and adults reported that itch or pruritus; soreness, pain, or tenderness; and skin dryness were the signs and symptoms most frequently cited as having a clinical impact.3 Itch, the major symptom, has a negative impact on quality of life and is associated with mental distress and an increased risk for suicidal thoughts.1 Depression, anxiety, and sleep disturbance are frequently reported comorbidities.2,3 Patients may experience significant sleep disturbance leading to lack of concentration, lethargy, and increased absenteeism.2,3 Moreover, AD can result in embarrassment related to appearance and can have a negative impact on self-esteem and a patient’s social life.1 During adolescence, when individuals are developing their identity, body-image and self-image are particularly important, and AD may impact these patients to a greater degree than patients in other age groups.29
A clinical diagnosis of AD is based on the patient’s medical history and an evaluation of morphology, the distribution of skin lesions, and associated signs and symptoms.30 Numerous tools have been developed to assess disease severity in AD, such as the EASI, Pruritus NRS, POEM, SCORing Atopic Dermatitis (SCORAD), and IGA; although not all have been validated.2 These severity measures are primarily used in the research setting and are not generally practical for the measurement of disease severity in routine clinical practice.27
Standards of Therapy
The contents of this section have been informed by materials submitted by the sponsor and clinical expert input. The following information has been summarized and validated by the CDA-AMC review team.
Initial treatment for most patients with AD is emollients (moisturizers) plus topical anti-inflammatory therapy, including TCS and topical calcineurin inhibitors.7 For patients with more severe AD or with AD that is refractory to topical therapy, advanced treatments, including phototherapy and systemic treatment, are considered. Ultraviolet B phototherapy can be tried, but its efficacy is not as well established, and it is often not feasible, requiring 2 or 3 clinic visits per week for about 10 to 14 weeks.7 Additionally, phototherapy cannot be used for the long term, so it cannot be relied upon as a long-term solution for people with more chronic AD.
As discussed in the clinical practice guidelines issued by the American Academy of Dermatology and the American Academy of Allergy, Asthma and Immunology, biologics, and particularly dupilumab, are considered first-line systemic therapy.7,8 Other options include tralokinumab (another biologic) and oral JAK inhibitors (upadacitinib, abrocitinib).7,8 Because of potential safety concerns with the JAK inhibitor drug class, upadacitinib and abrocitinib are generally not considered first-line systemic therapies for AD.7 Regulators have included warnings for all JAK inhibitors based on safety data from other populations with tofacitinib (another JAK inhibitor), which was associated with an increased risk of major cardiovascular AEs, thrombosis, cancer, and death.7 According to the clinical expert consulted, off-label immunomodulators (cyclosporine, methotrexate, mycophenolate, and azathioprine) are generally only used when mandated by a medication payer as step-through therapy or when the previously mentioned biologics and JAK inhibitors fail or are contraindicated. These drugs were not listed a first-line systemic therapies in the 2023 American Academy of Dermatology clinical practice guidelines due to their certainty of evidence being lower than for newer drugs, the potential for SAEs, the need for stringent laboratory monitoring, and lack of regulatory approval for use in AD.7
The clinical expert noted that all therapies treat the underlying inflammation of AD; they do make symptoms better, but that is secondary to treating the inflammation. Goals of treatment include reducing the symptoms of AD, particularly itch; reducing the visible signs of AD; improving quality of life, sleep quality and work productivity. Reducing potential adverse effects is important. Because AD is a chronic disease, maintaining low levels of disease activity and reducing flares is important.
Drug Under Review
The key characteristics of lebrikizumab, dupilumab, abrocitinib, and upadacitinib are summarized in Table 3. Of note, although tralokinumab is approved for the treatment of AD in Canada, it is not currently reimbursed by any publicly funded drug plan and so was not considered a relevant comparator for this review.
Lebrikizumab is approved by Health Canada for the treatment of moderate-to-severe AD in adults and adolescents aged 12 years and older with a body weight of at least 40 kg, whose disease is not adequately controlled with topical prescription therapies or for whom those therapies are inadvisable.16 Lebrikizumab can be used with or without TCS.16 The product monograph also states that lebrikizumab may be used in combination with topical calcineurin inhibitors for problem areas, such as the face, neck, and intertriginous and genital areas.16
Lebrikizumab is available as a 250 mg per 2 mL solution in a prefilled pen or prefilled syringe with needle shield for SC injection.16 The recommended initial dose is 500 mg (two 250 mg injections) at week 0 and week 2, followed by 250 mg (1 injection) every 2 weeks until week 16. Once a clinical response is achieved, the recommended maintenance dose is 250 mg every-4-weeks starting at week 16. The product monograph states that continued therapy beyond 16 weeks should be carefully considered in a patient who does not show a treatment benefit within this time period.16
Lebrikizumab is an immunoglobulin G4 (IgG4) monoclonal antibody that binds with high affinity and a slow off-rate to IL-13, and inhibits IL-13 signalling through the IL-4 receptor alpha (IL-4RAlpha) and IL-13 receptor alpha 1 (IL-13RAlpha1) pathways, thereby blocking the downstream effects of IL-13.16
Lebrikizumab has not been previously reviewed by CDA-AMC. The sponsor’s reimbursement request is the same as the Health Canada indication.31
Lebrikizumab was under review by Health Canada when this report was drafted and received a Notice of Compliance on June 24, 2024. Lebrikizumab was approved in 2024 by the FDA for adults and adolescents with moderate-to-severe AD for the same indication as in Canada. The European Medicines Agency (EMA) authorized lebrikizumab for adults and adolescents with moderate-to-severe AD for whom treatment applied directly to the skin cannot be used or is not sufficient.
Table 3: Key Characteristics of Lebrikizumab, Dupilumab, Abrocitinib, and Upadacitinib
Characteristic | Lebrikizumab | Dupilumab | Abrocitinib | Upadacitinib |
|---|---|---|---|---|
Mechanism of action | Blockade of IL-13 as an immunoglobulin G4 monoclonal antibody. | Inhibits IL-4 and IL-13 signalling. | A selective JAK1 inhibitor that prevents the phosphorylation and activation of the STATs that modulate intracellular activity, including gene expression. | A JAK inhibitor that prevents the phosphorylation and activation of STATs. |
Indicationa | For moderate-to-severe AD in adults and adolescents aged 12 years and older with a body weight of at least 40 kg, whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. | For the treatment of patients aged 6 months and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Can be used with or without TCS. | For the treatment of patients aged 12 years and older with refractory moderate-to-severe AD, including the relief of pruritus, who have had an inadequate response to other systemic drugs (e.g., steroid or biologic) or for whom these treatments are not advisable. Can be used with or without medicated topical therapies for AD. | For the treatment of adults and adolescents aged 12 years and older with refractory moderate-to-severe AD that is not adequately controlled with a systemic treatment (e.g., steroid or biologic) or when the use of those therapies is inadvisable. Can be used with or without TCS. |
Route of administration | SC | SC | Oral | Oral |
Recommended dose | Initial dose of 500 mg (two 250 mg injections) injected subcutaneously at week 0 and week 2, followed by 250 mg (1 injection) every 2 weeks until week 16. Maintenance dose: 250 mg every 4 weeks. | Patients aged 6 months to 5 years:
Patients aged 6 years to 17 years
Adults:
| 100 mg or 200 mg orally once daily for adolescents and adults under 65 years of age, based on an individual’s goals of therapy and potential risk for adverse reactions. For patients using the 200 mg once daily dosage, after symptom control is achieved by week 12, consider a dose reduction to 100 mg once daily. Relative to patients who maintained the 200 mg dose, the risk of occurrence of serious adverse reactions decreased in patients who reduced their dose to 100 mg beyond 12 weeks in clinical studies. If symptom control is lost after dose reduction, the dose can be increased to 200 mg. Exceeding a daily dosage of 200 mg is not recommended. | Adults:
Adolescents (from 12 to 17 years of age):
|
Serious adverse effects or safety issues | Before initiating therapy, complete all age-appropriate immunizations. Avoid the use of live vaccines in patients treated with lebrikizumab. The most frequently reported adverse reactions (> 1%) were injection-site reactions and conjunctivitis. | Hypersensitivity reactions, eosinophilic conditions, conjunctivitis and keratitis, musculoskeletal and connective tissue disorders. Should not be used to treat acute asthma symptoms or acute exacerbations. | Serious infections, malignancy, thrombosis, MACE, hematologic abnormalities, liver enzyme elevation, lipid parameter elevations. Avoid the use of live, attenuated vaccines during or immediately before therapy. | Serious infections, malignancy, thrombosis, MACE, gastrointestinal perforation, anemia, lymphopenia, neutropenia, lipid parameter elevations, hypersensitivity reactions, liver enzyme elevation. |
AD = atopic dermatitis, EASI-75 = at least a 75% reduction in Eczema Area and Severity Index score; IL = interleukin, JAK = Janus kinase, MACE = major adverse cardiovascular events, SC = subcutaneous, STATs = signal transducers and activators of transcription; TCS = topical corticosteroids.
aHealth Canada–approved indication.
Source: Lebrikizumab Product Monograph,16 Dupilumab Product Monograph,32 Abrocitinib Product Monograph,33 Upadacitinib Product Monograph,34 Sponsor’s Summary of Clinical Evidence.35
Perspectives of Patients, Clinicians, and Drug Programs
Patient-Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient inputs received by CDA-AMC have been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
Three patient groups provided input to this submission. Eczema Quebec gathered information through review of scientific literature, informal conversations with patients, The Skin I'm In 2022 Update: A National Report of the Patient and Caregiver Experience With Atopic Dermatitis (a joint report by Eczema Quebec and the CSPA), expert opinion from the Centre of Excellence for Atopic Dermatitis at the McGill University Health Centre, 9 written patient testimonials, interviews with 14 patients, and feedback from 3 patient-group discussions. The CSPA gathered information from previous submissions to CADTH, data from the Canadian Institute for Health Information on AD-related emergency department visits, hospitalizations from 2016 to 2020 (reported in The Skin I'm In), and guidelines. ESC gathered information through a survey, and one-on-one interviews with more than 3,000 patients with AD and their caregivers who live in Canada.
According to the patient-group input, the symptoms experienced by patients with AD include inflamed, painful, dry, and itchy skin that cracks, oozes, bleeds, and, in some cases, involves thickening and/or infections of the skin. ESC noted that 62% of survey respondents with moderate AD and 87% of survey respondents with severe AD reported having scars or marks on their skin from scratching. Conditions associated with AD include asthma, seasonal and environmental allergies, food intolerances, sleep disorders, anxiety, and depression. Often, patients with AD experience flares, which are periods of worsening of the disease and its symptoms, and periods of remissions. Itch is frequently reported as the most burdensome symptom and has been described as uncontrollable, incapacitating, debilitating, and bugs crawling all over, leading to disrupted sleep, fatigue, decreased functionality, and significant impacts on daily life, work, and school.
According to ESC, 72% of adult respondents with moderate AD and 95% of respondents with severe AD reported feeling itchy multiple times each day, whereas 44% of ESC survey respondents with severe AD reported feeling itchy all the time. Furthermore, 71% and 42% of adult survey respondents with moderate or severe AD rated their overall itch as 7 out of 10 and 10 out of 10 (the worst itch imaginable), respectively. Also, 54% of adult survey respondents with severe AD reported rarely being able to control their urge to scratch their skin. According to ESC, 69% of survey respondents with moderate AD and 87% of survey respondents with severe AD reported that itch negatively impacts stress. ESC added that feelings of depression and anxiety, as well as poor self-esteem, low energy, and, in some extreme cases, suicidal thoughts can be common among the patients with more severe AD. Based on the Eczema Quebec and CSPA joint report, 89% of survey respondents acknowledged the significant impact of the emotional and psychological burden of AD on their quality of life. ESC reported that 63% of survey respondents with moderate AD and 86% of survey respondents with severe AD reported that itch negatively impacted their sleep, and 50% of survey respondents with severe AD reported experiencing sleep loss 8 nights per month or more.
Patient groups stated that physical manifestations and the visibility of the disease contribute to psychological distress through stigmatization, which impacts a patient’s self-esteem, professional commitments, and social engagements. ESC reported that 32% of adult survey respondents with moderate or severe AD had missed work events due to their disease, and 30% had to change careers or give up certain activities. Eczema Quebec stated that access to health care presents another challenge to patients with AD; Canada's low ratio of dermatologists to the population makes specialized care difficult to obtain, particularly in remote areas.
Eczema Quebec and CSPA cited data from the Canadian Institute for Health Information that showed the frequency of hospitalization and emergency department visits for patients with AD.
Based on patient-group input, the burden of AD also extends to caregivers and family members. Caregivers reported feelings of anxiety, depression, helplessness, guilt, frustration, and a lack of control over the situation. Caregivers and family members also shared that their own health and emotional wellness, lifestyle, sleep, intimacy, social activities, and family dynamics were affected by the disease. Further, the cost of treatment and other skincare products can place financial stress not only on patients, but also on the family. ESC noted that 55% of caregivers of a teenager with moderate-to-severe AD reported experiencing sleep loss, 69% of caregivers reported experiencing anxiety related to managing a youth with moderate-to-severe AD, and 25% reported experiencing depression related to their child’s moderate-to-severe AD. Additionally, 62% of caregivers reported that time management was a challenge when trying to care for a child with moderate-to-severe AD; 63% reported experiencing physical, mental, or emotional stress; 36% reported feeling a lack of support from the health care system; and 30% reported financial challenges related to managing their child’s disease.
ESC noted that adolescents with AD can suffer significantly with itch and pain; however, the impact goes far beyond those symptoms. ESC reported that the daily life of 52% of families with a patient who has moderate-to-severe disease are negatively impacted by AD, according to survey data. In the same moderate-to-severe disease data, 70% of youth reported loss of sleep, 30% reported difficulty participating in sports or physical activities, and 21% reported avoiding social activities. ESC reported that 30% of teenagers experienced anxiety related to their AD and 20% of adolescents with moderate-to-severe disease missed school days specifically due to their AD, with 23% of those respondents missing 10 or more days of school per year and 12% missing 20 or more days of school per year. The caregiver-reported rate of bullying of children with moderate-to-severe AD was 14%.
In terms of experience with currently available treatments, the patient groups reported that topical treatments that are not eligible for reimbursement and necessary nongeneric products contribute to the financial burden of managing AD. The efficacy level and adverse effects of current treatments, the inconvenience of product use, and the high cost or unavailability of newer therapies were reported as important concerns. According to ESC, 87% of adult respondents with moderate AD reported that their disease is not well controlled; moreover, 74% and 24% of respondents have lived without adequate treatment for “more than a year” and “a decade or longer,” respectively.
Important desired outcomes reported by the patient groups were better, fast, and long-term control of the disease; reduction of flare; relief from itch; reduction of skin symptoms; pain and discomfort relief; improved psychological status; improved daily and social activities; increased productivity; improved emotional well-being; improved sleep quality; and the ability to maintain intimate relationships. In addition, treatments should be affordable or covered by insurance and should be easy to use (i.e., not administered by injection or topically).
Clinician Input
Input From Clinical Expert Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of AD.
Unmet Needs
The clinical expert stated there is an unmet need for more treatment options for people who are refractory to or do not tolerate current biologic treatments and for people who are concerned about the safety profile of oral JAK inhibitors, such as those with comorbidities or who are older.
Place in Therapy
The clinical expert did not expect lebrikizumab to cause a shift in the treatment paradigm; rather, it would be another biologic medication used to treat patients with moderate-to-severe AD whose disease is not adequately controlled with topical therapy. Lebrikizumab is not the first treatment to address the underlying disease process and inflammation; its mechanism of action is similar to other biologics, particularly tralokinumab. The clinical expert anticipates that the use of lebrikizumab will be similar to that of other systemic medications that allow the concomitant use of emollients and topical anti-inflammatory treatments (e.g., corticosteroids). Given the clinical experience with and the evidence supporting the use of dupilumab, the expert anticipated that lebrikizumab would be considered a second-line biologic after dupilumab, and that it may be chosen for patients for whom dupilumab is contraindicated, ineffective, or not tolerated.
The clinical expert stated that it would be appropriate for patients to try topical anti-inflammatory medications (e.g., TCS or topical calcineurin inhibitors) and dupilumab before initiating lebrikizumab. Topical anti-inflammatory treatments are almost always used in the first-line setting and can be effective even in severe cases. However, when patients are refractory, systemic treatment should be considered. According to the clinical expert and clinical practice guidelines,7 dupilumab, is recommended as the first-line systemic treatment option, based on its efficacy and safety profile. Other options, such as tralokinumab and lebrikizumab, are reserved for second-line therapy.
Patient Population
Patients with moderate-severe AD refractory to topical therapy are most likely to respond to treatment with lebrikizumab, according to the clinical expert. Patients who are refractory to treatment with dupilumab are most in need of a new intervention such as lebrikizumab. There are no known disease characteristics among patients with moderate-severe AD refractory to topical therapy that differentiate potential lebrikizumab responders from nonresponders.
Clinician examination and history, including assessment of the impact of AD on quality of life, would establish which patients are suitable for treatment with lebrikizumab. No laboratory tests or other diagnostic tools would be necessary. No companion diagnostic test is required. Most cases of AD are not difficult to diagnose, and misdiagnosis is rare. Occasionally, tests such as skin biopsy or patch testing can help differentiate AD from other mimickers, but these are not necessary in the vast majority of cases.
Assessing the Response Treatment
In clinical practice, clinicians generally use a gestalt assessment of improvement in clinical signs and a patient’s history of change in symptoms (e.g., itch) and quality of life, the clinical expert stated. Clinicians only use the tools used in clinical trials (e.g., EASI score) if mandated by a medication payer to obtain coverage. The outcomes used in clinical trials are often reflective of what would be considered in a clinician’s gestalt response, but they are cumbersome and not meant for use in routine clinical practice.
According to the clinical expert, a meaningful response to treatment would be an approximately 50% to 75% improvement in signs and symptoms; the specific proportion likely differs by clinician and by patient. The improvement should include a reduction in the severity and frequency of symptoms, often accompanied by improvement in quality of life and the ability to perform household, work, and/or school activities. Disease flares should also be reduced (fewer episodes of intense itching and widespread and severe eruptions). For patients who are prone to secondary skin infections, any treatment that reduces the inflammation of AD and improves the skin barrier should result in fewer infections.
Discontinuing Treatment
The clinical expert indicated that lebrikizumab would be discontinued if it is inadequately effective, as judged by the gestalt response to treatment. If patients are satisfied with treatment, even if an arbitrary cut-off like EASI-75 is not met, they might continue with treatment.
Patients may discontinue treatment if AEs are intolerable. Lebrikizumab has been associated with conjunctivitis, but this adverse effect is usually mild and transient; if conjunctivitis is severe and/or refractory to eyedrops, lebrikizumab could be discontinued. Other unanticipated SAEs, like severe allergy, could lead to discontinuation.
The clinical expert stated that some patients do not like the idea of being on a medication indefinitely. In such cases, patients and their physicians can use shared decision-making to decide whether to stop lebrikizumab to see if the dermatitis recurs; if it does, lebrikizumab would likely be restarted.
Prescribing Considerations
The clinical expert indicated that in most instances, a specialist (dermatologist, allergist, pediatrician) would be required to treat a patient with AD with a biologic. This can be done in a community setting, hospital clinic, or specialty clinic. Family physicians are adept at diagnosing AD but are likely not comfortable prescribing biologic therapy. In areas where access to specialty care is difficult, some family physicians could gain comfort with biologics for AD, because minimal clinical monitoring is required.
Clinician-Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full original clinician-group inputs received by CDA-AMC have been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
CDA-AMC received inputs from 2 clinician groups for this review. The CDA submitted input from 3 clinicians from their Pharmacy and Therapeutics Advisory Board, and the DAO submission included input from 11 clinicians.
Clinician groups and the clinical expert consulted by CDA-AMC agreed that a lack of adequate response to treatment, incomplete effectiveness, the adverse effects of treatments, a lack of feasibility of some of treatments, and relapses are unmet needs of patients with AD. One of the clinician groups added that challenges in access to care, multitiered treatment regimens, treatment intolerance or contraindications, and comorbid bacterial skin infections are unmet needs as well.
The CDA and the clinical expert consulted by CDA-AMC agree that the goals of treatment are to improve quality of life and maximize efficacy and safety. Regarding the place of lebrikizumab in therapy, the DAO and the clinical expert consulted by CDA-AMC indicate that lebrikizumab will not cause a shift in the treatment paradigm and would fit into the current paradigm as another treatment option. In contrast, the CDA indicated that lebrikizumab contributes to an important shift in the current treatment paradigm toward a new era of focus on novel disease mechanisms that target and modify disease and have favourable safety and efficacy profiles.
According to the DAO, adult patients with moderate-to-severe AD who have failed topical therapies and those who have failed or do not have access to phototherapy would be best suited to treatment with lebrikizumab. The CDA stated that patients best suited for treatment with lebrikizumab would be those with uncontrolled moderate-to-severe AD who are candidates for systemic therapy or who meet criteria for biologic therapy. The CDA noted that dupilumab is indicated for patients with other severe forms of atopic or allergic conditions, such as severe asthma, or eosinophilic esophagitis; thus, dupilumab may be chosen for these patients instead of the IL-13 inhibitors, which are not indicated for these conditions.
The DAO noted that a patient’s response to treatment would be assessed with the IGA, EASI, Pruritus NRS, and DLQI scoring systems at 4 to 6 months and annually thereafter. The CDA stated that assessment of a patient’s response would be based on a clinical exam, patient history, physician-reported clinical scoring systems (EASI, BSA, IGA) and patient-reported outcomes (DLQI, CDLQI, and Pruritus NRS). The CDA added that in clinical practice, due to time limitations, only some of the scoring systems are used.
Clinician groups reported AEs and poor efficacy of treatment as factors that should be considered when deciding whether to discontinue the treatment.
Based on clinician-group input, the treatment and monitoring of patients on lebrikizumab should be limited to specialists trained in this area, which would include the fields of dermatology, allergy, immunology, or pediatrics.
Drug Program Input
The drug programs provide input on each drug being reviewed through the CDA-AMC reimbursement review process by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 4.
Table 4: Summary of Drug-Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The clinical trials compared lebrikizumab to placebo, and no head-to-head studies were identified that compared lebrikizumab to treatments that are funded in most provinces. What is the place in therapy for lebrikizumab? | The clinical expert indicated that lebrikizumab will likely be used as second-line biologic therapy after dupilumab and stated that it may be chosen for patients for whom dupilumab is contraindicated, ineffective, or not tolerated. |
Considerations for initiation of therapy | |
Should eligibility include an adequate trial of phototherapy, methotrexate, and/or cyclosporine? Should reimbursement be provided for patients who lost response to or never achieved clinical benefit from a trial of dupilumab? What about other drugs in this therapeutic space? | The expert stated that topical therapies are first-line options for AD, and if these fail to control the disease, biologics are recommended as second-line therapies. Of the available biologics, dupilumab is considered a first-line treatment, whereas lebrikizumab, tralokinumab, and JAK inhibitors may be considered third-line treatments. The expert did not endorse a trial of other immunosuppressants, such as methotrexate or cyclosporin, before prescribing a biologic such as dupilumab or lebrikizumab, due to the lower efficacy and risk of toxicity associated with these immunosuppressants. |
Consider alignment with the initiation criteria for dupilumab in AD, as applicable, including definitions regarding moderate-to-severe AD, refractory disease, and adequate trials for different prerequisite therapies. | For consideration by CDEC. |
Considerations for continuation or renewal of therapy | |
The initial eligibility period for dupilumab is 6 months. Would the same initial eligibility period be applicable to lebrikizumab, or would the recommended initial approval be less, due to the 16-week induction period in the clinical studies? Should CDEC consider alignment with the renewal criteria for dupilumab in AD, as applicable? | The expert recommended a 6-month initial eligibility period for lebrikizumab to allow sufficient time for the full treatment response to be achieved. Alignment with the renewal criteria for dupilumab would be reasonable. |
Considerations for discontinuation of therapy | |
Will alignment with dupilumab be considered? | The expert suggested that alignment with the dupilumab discontinuation criteria would be reasonable. Maintenance of an EASI-75 response is an adequate threshold; however, there are some patients who may show a lower change in EASI score but may view their disease as being substantially improved. The EASI score is based on the physician’s assessment of the extent and severity of AD. It is important for the patient perspective to also be considered when making treatment and reimbursement decisions. |
Considerations for prescribing of therapy | |
Per the draft product monograph, the recommended dose of lebrikizumab is 500 mg SC (two 250 mg injections) at week 0 and week 2, followed by 250 mg every 2 weeks until week 16, and 250 mg every 4 weeks thereafter, with some patients maintained on an every-8-week dosing schedule. In what situations would every-8-week dosing be appropriate (or required) based on clinical trial results? | The expert noted that decisions on dosing frequency would be made in consultation with the patient, based on treatment response, goals of therapy, and individual wishes. The CDA-AMC reviewer notes that an every-8-week maintenance dose was suggested in the draft product monograph, but this regimen was removed when the drug was approved by Health Canada. The recommended maintenance dose is 250 mg every 4 weeks. |
Should CDEC consider alignment with the criteria for dupilumab? | The clinical expert indicated that in most instances, a specialist (dermatologist, allergist, pediatrician) would be required to treat AD with a biologic. In areas where access to specialty care is difficult, some family physicians could gain comfort with biologics for AD, because minimal clinical monitoring is required. The expert indicated that lebrikizumab will be used in combination with TCS, but evidence is currently lacking on its use in combination with other biologics, JAK inhibitors, or immunosuppressants. |
System and economic issues | |
Dupilumab has been negotiated by pCPA for AD in adults. It is currently under consideration for negotiation in the pediatric group. Abrocitinib and upadacitinib both concluded pCPA negotiations with a letter of intent. | For consideration by CDEC. |
AD = atopic dermatitis; CDEC = Canadian Drug Expert Committee; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% reduction in Eczema Area and Severity Index score; JAK = Janus kinase; pCPA = pan-Canadian Pharmaceutical Alliance; SC = subcutaneous; TCS = topical corticosteroids.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of lebrikizumab 250 mg per 2 mL solution for SC injection in the treatment of moderate-to-severe AD in adults and adolescents aged 12 years and older with a body weight of at least 40 kg, whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. The focus will be placed on comparing lebrikizumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of lebrikizumab is presented in 4 sections, with the CDA-AMC-conducted critical appraisal of the evidence included at the end of each section. The first section, the Systematic Review, includes pivotal studies and RCTs that were selected in accordance with the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section, using the GRADE approach, follows the critical appraisal of the evidence. The second section includes a sponsor-submitted long-term extension study. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following are included in the CDA-AMC review and appraised in this document:
3 pivotal studies identified in the systematic review
1 long-term extension study
1 ITC
4 additional studies addressing gaps in evidence.
Systematic Review
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
The objective of the ADvocate 1 and ADvocate 2 studies was to evaluate the safety and efficacy of the 250 mg lebrikizumab SC injection as monotherapy in patients with moderate-to-severe AD. Both were randomized, double-blind, placebo-controlled studies that included a 16-week induction period (parallel design), followed by a 36-week maintenance period that used a randomized withdrawal design. Eligible patients were adults or adolescents (aged 12 years to less than 18 years and weighing more than 40 kg) who had a diagnosis of chronic AD that was rated as moderate-to-severe based on an EASI score of at least 16, an IGA score of at least 3, and AD covering a BSA of 10% or more.
Table 5: Details of the Studies Included in the Systematic Review
Detail | ADvocate 1 | ADvocate 2 | ADhere |
|---|---|---|---|
Designs and populations | |||
Study design | Phase III, double-blind, placebo-controlled RCT | Phase III, double-blind, placebo-controlled RCT | Phase III, double-blind, placebo-controlled RCT |
Locations | 89 centres in Australia, Canada, Estonia, France, Latvia, Lithuania, Poland, South Korea, Spain, the US | 82 centres in Bulgaria, Canada, Germany, Mexico, Singapore, Taiwan, Ukraine, the US | 54 centres in Canada, Germany, Poland, the US |
Key dates | Start: September 24, 2019 End: May 3, 2022 | Start: October 29, 2019 End: April 28, 2022 | Start: February 3, 2020 End: September 16, 2021 |
Randomized (N) | N = 424 Placebo = 141 Lebrikizumab 250 mg = 283 | N = 427 Placebo = 146 Lebrikizumab 250 mg = 281 | N = 211 Placebo + TCS = 66 Lebrikizumab 250 mg + TCS = 145 |
Inclusion criteria |
| Same as ADvocate 1 | Same as ADvocate 1 |
Exclusion criteria |
| Same as ADvocate 1 | Same as ADvocate 1, with following exceptions:
Additional exclusion criteria:
|
Drugs: Induction period | |||
Intervention | Lebrikizumab 250 mg every 2 weeks: LD of lebrikizumab 500 mg (4 mL) SC at baseline and week 2, then 250 mg (2 mL) given every 2 weeks through week 14 | Lebrikizumab 250 mg every 2 weeks: LD of lebrikizumab 500 mg SC at baseline and week 2, then 250 mg given every 2 weeks through week 14 | Lebrikizumab 250 mg every 2 weeks + TCS: LD of lebrikizumab 500 mg SC at baseline and week 2, then 250 mg given every 2 weeks through week 14, with TCS treatment initiated at baseline and applied as needed |
Comparator(s) | Placebo every 2 weeks: 4 mL SC administered at baseline and week 2, and 2 mL given every 2 weeks through week 14 | Placebo every 2 weeks: 4 mL administered at baseline and week 2, and 2 mL given every 2 weeks through week 14 | Placebo + TCS: 4 mL SC administered at baseline and week 2, and 2 mL given every 2 weeks through week 14, with TCS treatment initiated at baseline and applied as needed |
Drugs: Maintenance period | |||
Intervention and comparator | Patients who responded to treatment from baseline to week 16 were randomly reassigned to 1 of the following double-blind treatment groups:
| Same as ADvocate 1 | NA |
Study duration | |||
Screening phase | 30 days to 7 days before baseline (day 1) | 30 to 7 days before baseline day 1 | Maximum of 30 days |
Treatment phase | 52 weeks (induction phase:16 weeks, maintenance phase: 36 weeks) | 52 weeks (induction phase:16 weeks, maintenance phase: 36 weeks) | 16 weeks |
Follow-up phase | 12 weeks after last injection | 12 weeks after last injection | 12 weeks after last injection |
Outcomes | |||
Coprimary end pointsa |
| Same as ADvocate 1 | Same as ADvocate 1 |
Secondary and exploratory end points | Major secondary during the induction period (week 16, unless otherwise specified):
Major secondary during the maintenance period:
Other:
| Same as ADvocate 1 | Major secondary (at week 16):
Other:
|
Publication status | |||
Publications | Silverberg et al. (2023)36 Blauvelt et al. (2023)37 NCT04146363 | Silverberg et al. (2023)36 Blauvelt et al. (2023)37 NCT04178967 | Simpson et al. (2023)38 NCT04250337 |
ACQ-5 = Asthma Control Questionnaire; AD = atopic dermatitis; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = at least a 50% reduction in EASI score; EASI-75 = at least a 75% reduction in EASI score; EASI-90 = at least a 90% reduction in EASI score; IGA = Investigator Global Assessment; LD = loading dose; NA = not applicable; NRS = Numeric Rating Scale; POEM = Patient-Oriented Eczema Measure; PROMIS = Patient-Reported Outcomes Measurement Information System; RCT = randomized controlled trial; SC = subcutaneous; SCORAD = SCORing Atopic Dermatitis; TCS = topical corticosteroids.
aPrimary and secondary outcomes are listed according to the statistical analysis plan for the European Medicines Agency.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere,9 Sponsor’s Summary of Clinical Evidence.35
The ADvocate 1 study was conducted in Australia, Canada, Europe, South Korea, and the US, and a total of 424 patients were randomized, including 23 patients from Canada. The ADvocate 2 study randomized 427 patients (including 58 from Canada) from Asia, Europe, and North America. The study schematic for the ADvocate 1 and ADvocate 2 studies is shown in Figure 1. Using an electronic data capture system, the studies randomized patients in a ratio of 2:1 to receive 250 mg lebrikizumab or placebo once every 2 weeks for the 16-week induction period. Randomization was stratified by region (Europe, the US, or the rest of the world), age (adolescent or adult), and disease severity (IGA score of 3 or 4). At week 16, patients who responded to treatment (defined as either an IGA score of 0 or 1 or an EASI-75 response in patients who did not receive rescue therapy) were randomly reassigned in a ratio of 2:2:1 to double-blind lebrikizumab 250 mg every 2 weeks, lebrikizumab 250 mg every 4 weeks, or placebo for the 36-week maintenance period. Separate randomizations were used for patients who responded to lebrikizumab and patients who responded to placebo. Patients who did not meet the response criteria at week 16 or who required rescue therapy during the induction phase were enrolled in the escape arm and received open-label lebrikizumab 250 mg every 2 weeks through to week 52. In addition, rerandomized patients who did not maintain at least a 50% reduction in EASI score from baseline (EASI-50) during the maintenance phase were assigned to the escape arm.
Patients who completed the ADvocate 1 or ADvocate 2 study were eligible to enrol in the extension study (ADjoin).
Figure 1: Study Design of the ADvocate 1 and ADvocate 2 Studies
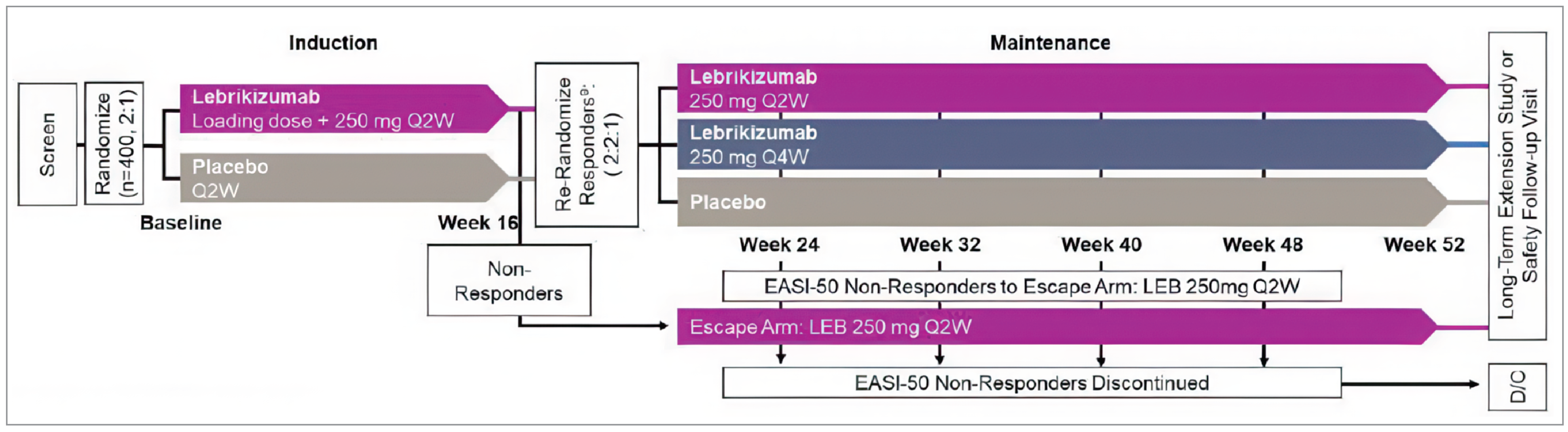
D/C = discontinue; EASI-50 = at least a 50% reduction in Eczema Area and Severity Index score; LEB = lebrikizumab; Q2W = every 2 weeks; Q4W = every 4 weeks.
Source: CSR for ADvocate 1.11
The objective of the ADhere study was to evaluate the safety and efficacy of the lebrikizumab in combination with TCS compared with placebo plus TCS in patients with moderate-to-severe AD. The study was a 16-week randomized, double-blind, placebo-controlled, parallel-design trial. Adults or adolescents (aged 12 years to less than 18 years weighing more than 40 kg) with moderate-to-severe AD (EASI score of ≥ 16, IGA score of ≥ 3, AD covering a BSA of 10% or more) were eligible to enrol. Patients were randomized in a 2:1 ratio to receive 250 mg lebrikizumab SC once every 2 weeks in addition to TCS or placebo plus TCS for the 16-week treatment period. An electronic data capture system was used to allocate patients to treatment, and randomization was stratified by region (Europe, the US, or the rest of the world), age (adolescent or adult), and disease severity (IGA score of 3 or 4). A total of 211 patients were randomized, including 22 patients from Canada. The study was conducted at 54 centres in Canada, Germany, Poland, and the US.
Patients who completed the ADhere study were eligible to enter the extension study (ADjoin).
Populations
Inclusion and Exclusion Criteria
All 3 studies used similar inclusion and exclusion criteria and enrolled adults and adolescents with chronic AD, defined by the American Academy of Dermatology consensus criteria,39 for at least 1 year before the screening visit and with disease that was classified as moderate-to-severe (EASI score of ≥ 16, IGA score of ≥ 3, and AD covering a BSA of 10% or more) (Table 5). Patients were also required to have a history of inadequate response to topical medications or to be in a position in which topical treatments are medically inadvisable.
In the ADvocate 1 and ADvocate 2 studies, patients were excluded if they had previously received dupilumab, tralokinumab, or lebrikizumab, and were required to stop all other AD therapies before randomization. For the ADhere study, patients who had received tralokinumab were not excluded from the trial and those who had received dupilumab were eligible, provided the drug was stopped 8 weeks before study entry. In all trials, patients underwent a 1-week washout period for topical treatments and a 4-week to 6-month washout period for other AD treatments (refer to the Interventions section for details) to be eligible for either study.
Other key exclusion criteria for the 3 trials were uncontrolled chronic disease that might require bursts of oral corticosteroids or may interfere with the study assessments of clinical status, active chronic or acute infection requiring recent systemic treatment, HIV, hepatitis, or cirrhosis, and a history of or suspected immunosuppression.
Additional inclusion and exclusion criteria were applied at week 16 in the ADvocate 1 and ADvocate 2 trials. To quality for rerandomization for the maintenance phase, patients had to meet the treatment response criteria and had to have achieved an IGA score of 0 or 1 or EASI-75 at week 16. Patients who did not meet the response criteria at week 16 or who required rescue therapy during the induction phase were eligible to enter the escape arm of the studies.
Interventions
The ADvocate 1 and ADvocate 2 Studies
In the ADvocate 1 and ADvocate 2 studies, patients were randomized to receive double-blind lebrikizumab 250 mg or placebo SC once every 2 weeks for the 16-week induction period. Patients in the lebrikizumab group received a 500 mg lebrikizumab loading dose at week 0 and week 2, and then 250 mg every 2 weeks until week 14. The lebrikizumab or placebo injections were supplied as 2-mL prefilled syringes with a preassembled needle safety device. The placebo solution was identical in appearance and content to the active solution, except for lebrikizumab.
At week 16, patients who responded to treatment were randomly reassigned in a ratio of 2:2:1 to double-blind lebrikizumab 250 mg every 2 weeks, lebrikizumab 250 mg every 4 weeks, or placebo for the maintenance period. Separate randomizations were used for lebrikizumab responders (maintenance primary population) and placebo responders (maintenance secondary population). Responders who received placebo during the first 16 weeks of the study and who were randomly reassigned to 1 of the lebrikizumab arms received loading doses of lebrikizumab 500 mg at week 16 and week 18, and then received lebrikizumab 250 mg every 2 or 4 weeks, depending on the randomized treatment group. To maintain blinding during the induction and maintenance phases, all patients received the same number of injections through a combination of active and placebo injections. All doses of the study drug were administered at the study centres during the induction period and were self-administered after week 16.
Both studies provided access to escape therapy if the study treatments did not adequately control the AD. Patients who did not meet the response criteria at week 16 or who received topical or systemic rescue therapy during the first 16 weeks were enrolled in the escape arm and received open-label lebrikizumab 250 mg SC every 2 weeks through to week 52 (including a blinded loading dose if the patient had not received lebrikizumab previously). In addition, rerandomized patients who failed to maintain an EASI-50 response during the maintenance phase were assigned to the escape arm and received open-label lebrikizumab 250 mg every 2 weeks. Patients who did not achieve an EASI-50 response after 8 weeks in the escape arm were terminated from the study.
Concomitant medications to treat AD were prohibited during the induction period of the trials unless they were part of rescue therapy. Rescue therapy started with topical therapies first (e.g., midpotency TCS) and, if symptoms were not controlled, systemic treatments could be initiated (e.g., oral corticosteroids, phototherapy, cyclosporine). The study drug was stopped if systemic rescue therapy was required, but these patients continued to attend study visits for assessments of safety and efficacy. During the maintenance period, the intermittent use of topical rescue medications was permitted, including by patients in the escape arm. The short-term use of systemic rescue therapies was determined on a case-by-case basis after consultation with the medical monitor. In both studies, all patients were required to use nonmedicated topical moisturizers daily.
Patients receiving specific AD therapies before the study underwent a washout period before randomization. The washout period was as follows: 1 week for TCS, calcineurin inhibitors, and phosphodiesterase type 4 inhibitors; 4 weeks for systemic immunosuppressive drugs (e.g., corticosteroids, cyclosporine, mycophenolate-mofetil, interferon gamma, JAK inhibitors, azathioprine, and methotrexate) and for phototherapy or photochemotherapy; 6 months for B-cell-depleting biologics (e.g., rituximab); 16 weeks or 5 half-lives for other biologics; and 1 week for prescription moisturizers.
The ADhere Study
In the ADhere study, patients were randomized to receive double-blind lebrikizumab 250 mg or placebo SC once every 2 weeks in combination with TCS for 16 weeks. Loading doses of 500 mg lebrikizumab at week 0 and week 2 were administered to patients randomized to lebrikizumab, followed by 250 mg every 2 weeks thereafter. To maintain blinding, the study drug was supplied as identical-looking prefilled syringes containing 2 mL of either lebrikizumab or placebo (vehicle), and all patients received the same number of injections. All patients were prescribed a midpotency TCS (triamcinolone acetonide 0.1% cream) plus a low-potency TCS (hydrocortisone 1% cream) for use in sensitive areas. The use of topical calcineurin inhibitors was allowed for sensitive areas only. TCS were initiated at baseline, and patients were allowed to taper, stop, or reinitiate TCS as needed. Using an electronic diary, patients recorded the topical AD therapies they used daily. All patients were required to use a nonmedicated topical moisturizer daily.
The ADhere study had the same washout criteria for prior AD treatments as the ADvocate studies, with 1 exception. Patients who had previously received dupilumab were eligible for the ADhere study provided treatment had stopped 8 weeks before the start of the trial. Patients who had received tralokinumab previously were not excluded from the ADhere study.
Rescue therapy for patients who experienced a clinical worsening of symptoms that were intolerable consisted of high-potency TCS or systemic therapy (e.g., oral corticosteroids, phototherapy, and cyclosporine). Patients who required systemic rescue therapy stopped their study drug but continued the scheduled study visits and underwent assessments for safety and efficacy.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6 and is followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence, as well as any outcomes identified as important to this review, according to the clinical expert consulted by CDA-AMC and input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to the CDA-AMC expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. The efficacy outcomes selected for assessment using GRADE are shown in Table 6. Other supportive efficacy outcomes were reported in the Results section only. Select notable harms outcomes considered important for the CDA-AMC expert committee deliberations were also assessed using GRADE.
All the outcomes listed in Table 6 were identified as clinically important measures by the clinical expert consulted by CDA-AMC. The patient-group input also identified AD severity, symptoms (particularly itch), and HRQoL as key outcomes. Moreover, EASI, DLQI and CDLQI, POEM, and Pruritus NRS scores were identified as part of the core outcome set for clinical trials of AD.40 For outcomes in which there were multiple analyses for the same instrument (e.g., change from baseline and proportion of responders), only 1 measure was selected for GRADE. IGA response and EASI-75 response were selected, as they were coprimary end points. The proportion of patients with a Pruritus NRS 4-point response was selected to be consistent with the outcomes reported in the ITC. Conjunctivitis was identified by the clinical expert as a common and potentially troublesome adverse effect of some AD medications, and the risk of SAEs was an important end point, given that most patients will require long-term treatment to manage their disease.
All 3 studies had a separate statistical analysis plan for submission to the EMA and to the FDA. The EMA submission included 2 coprimary end points and 15 major secondary end points for the ADvocate 1 and ADvocate 2 studies, and 2 coprimary end points and 8 major secondary end points for the ADhere trial. The FDA submission considered a single primary end point (IGA 0 or 1 response) and 8 major secondary end points for the ADvocate 1 and ADvocate 2 studies, and a single primary end point and 4 major secondary end points for the ADhere study. The sponsor focused on the results in the EMA submission in its clinical summary, and the same approach was used in the CDA-AMC review.
Evidence for validity, reliability, and responsiveness of the outcome measures of interest is summarized in Table 8.
Investigator Global Assessment
A coprimary end point for all 3 studies was the percentage of patients with an IGA score of 0 or 1 and an improvement of greater than or equal to 2 points from baseline at week 16. The IGA measures the investigator’s global assessment of the patient’s overall severity of AD at that visit, based on a static, numeric 5-point scale that ranges from 0 (clear) to 4 (severe) (Table 7). A score was selected using descriptors that best described the overall appearance of the lesions at a given time point. The CSRs state that assessors were trained and certified by the sponsor before conducting this assessment and that a single assessor was assigned to each patient for as many visits as possible. No MID was identified for adult or adolescent patients with AD.
Based on input from the clinical expert consulted for this review, an absolute difference of at least 10% between the lebrikizumab and placebo groups was considered clinically important with respect to the proportion of patients achieving an IGA score of 0 or 1.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | ADvocate 1a | ADvocate 2 | ADherea |
|---|---|---|---|---|
Assessed using GRADE | ||||
Percentage of patients with an IGA score of 0 or 1 and a reduction of ≥ 2 points from baseline | At week 16 | Coprimaryb | Coprimaryb | Coprimaryb |
Percentage of patients with an EASI-75 response | At week 16 | Coprimaryc | Coprimaryc | Coprimaryc |
Percentage of patients with a Pruritus NRS score of ≥ 4 points at baseline who report a ≥ 4-point reduction from baseline | At week 16 | Major secondaryc | Major secondaryc | Major secondaryc |
Change from baseline in POEM score | At week 16 | Other secondaryd | Other secondaryd | Other secondaryd |
Change from baseline in DLQI total score | At week 16 | Major secondary | Major secondary | Major Secondary |
Change from baseline in CDLQI total score | At week 16 | Otherd | Otherd | Other secondaryd |
Percentage of patients from those randomly reassigned after having achieved EASI-75 at week 16 who continue to exhibit EASI-75 (EASI-75 calculated relative to baseline EASI) | At week 52 | Major secondary | Major secondary | NA |
Percentage of patients with an SAE | At week 16 | Otherd | Otherd | Otherd |
Percentage of patients with conjunctivitis | At week 16 | Otherd | Otherd | Otherd |
Percentage of patients with an SAE | At week 52 | Otherd | Otherd | NA |
Percentage of patients with conjunctivitis | At week 52 | Otherd | Otherd | NA |
Supportive outcomes | ||||
Percentage of patients achieving an EASI-90 | At week 16 | Major secondaryc | Major secondaryc | Major Secondaryc |
Percentage change in EASI score from baseline | At week 16 | Major secondary | Major secondary | Major Secondary |
Percentage change in Pruritus NRS score from baseline | At week 16 | Major secondary | Major secondary | Major Secondary |
CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index score; EASI-75 = at least a 75% reduction in Eczema Area and Severity Index score; EASI-90 = at least a 90% reduction in Eczema Area and Severity Index score; GRADE = Grading of Recommendations Assessment, Development, and Evaluation; IGA = Investigator Global Assessment; NA = not applicable; NRS = Numeric Rating Scale; POEM = Patient-Oriented Eczema Measure; SAE = serious adverse event.
aThe outcomes in the table are described according to the statistical plan for the European Medicines Agency (EMA). Outcomes listed as primary and major secondary were adjusted for multiple comparisons. Other outcomes were not controlled for multiplicity.
bPrimary end point according to the statistical plan for the FDA.
cMajor secondary outcome according to the statistical plan for the FDA, which was adjusted for multiple comparisons.
dOther outcome that was not adjusted for multiple comparisons according to the statistical plan for the EMA or the FDA.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9
Table 7: Investigator Global Assessment Scoring
Score | Grade | Definition |
|---|---|---|
0 | Clear | Minor, residual discoloration; no erythema or induration or papulation; no oozing or crusting; no edema |
1 | Almost clear | Trace, faint pink erythema with barely perceptible induration or papulation and no oozing or crusting; no edema |
2 | Mild | Faint pink erythema with papulation and edema perceptible upon palpation and no oozing or crusting; minimal induration |
3 | Moderate | Pink-red erythema with definite edema of skin papules and plaques; there may be some oozing or crusting; palpable induration |
4 | Severe | Deep or bright red erythema with significant swelling and obvious raised borders of papules and plaques with oozing or crusting; significant induration |
Source: CSR for ADhere.9
Eczema Area and Severity Index
The percentage of patients who attained a reduction greater than or equal to 75% from baseline in EASI score at week 16 was used as the coprimary end point in the 3 trials. The EASI assesses the extent of disease at 4 body regions, and measures 4 clinical signs at the visit, each on a scale of 0 to 3: erythema, edema or papulation, excoriation, and lichenification. The EASI confers a maximum score of 72, with higher values indicating more severe disease and/or more extensive disease. The EASI evaluates 2 dimensions of AD: disease extent and clinical signs. Assessors were trained and certified by the sponsor before conducting this assessment. The overall MID has been reported to be 6.6 based on a study of mainly adults and an unknown number of adolescents with AD.13
The studies also reported the percentage of patients who achieved at least a 90% reduction from baseline in EASI score (EASI 90) and the percent change from baseline in EASI score at week 16.
The clinical expert consulted for this review identified at least a 10% difference between groups in the proportion of patients who achieved an EASI-75 response as a clinically important difference.
Pruritus NRS
The Pruritus NRS is a patient-reported, single-item, daily, 11-point scale. The scale is used by patients to rate their worst itch severity over the previous 24 hours, with 0 indicating no itch and 10 indicating the worst itch imaginable. Patients were asked to record their assessment daily in an electronic diary. In adults, a change from baseline of2 to 4 points may be considered an important within-person change.41,42 According to the CSR, a 4-point change was selected for the responder analysis as a conservative assessment of clinical impact.
At least a 10% difference between groups in the proportion of patients who reported a Pruritus NRS 4-point response was identified as a clinically important difference, according to the clinical expert consulted.
DLQI and CDLQI
In the pivotal trials, patients aged 17 years and older completed the DLQI and those aged 12 to 16 years completed the text version of the CDLQI and continued to complete the CDLQI for the duration of the study.
The DLQI is a patient-reported, 10-item, HRQoL questionnaire for adults that covers 6 domains (symptoms and feelings, daily activities, leisure, work and school, personal relationships, and treatment). The recall period of this scale is the previous week. Response categories include not at all, a little, a lot, and very much, with corresponding scores of 0, 1, 2, and 3; unanswered (or not relevant) responses were scored as 0. The 10 questions are scored from 0 to 3, giving a possible total score range of 0 (no impact of skin disease on quality of life) to 30 (maximum impact on quality of life). A DLQI total score of 0 or 1 is considered to have no effect on a patient’s HRQoL, and a 4-point change from baseline is considered to be the minimal clinically important difference threshold.14,43 Estimates of the MID have ranged from 2.2 to 6.9, but no information about MID was found specifically for adult patients with AD.19-21
The CDLQI questionnaire is based on the adult version (DLQI) and is designed and validated in patients with dermatological conditions who are aged 3 to 16 years. It is available in text and cartoon versions. The questionnaire consists of 10 items addressing the patient’s perception of the impact of their skin disease on various aspects of their quality of life over the previous week, including dermatology-related symptoms and feelings, leisure, school, friendships, sleep, and the impact of treatment. The total score ranges from 0 to 30, with a higher score indicating a poorer HRQoL.15,44,45 In adolescents, a reduction in CDLQI score of 6 to 8 points has been suggested as the clinically relevant threshold for a within-person change in patients with moderate-to-severe AD.15
Based on input from the clinical expert consulted and a review of the literature, a MID of 4 points for the DLQI and 6 points for the CDLQI were selected as the clinically important threshold for GRADE.14,15
Patient-Oriented Eczema Measure
The POEM is a 7-item, self-reported questionnaire used to assess the frequency of disease symptoms in adults and children over the previous week. Patients use an electronic diary to respond to 7 questions on skin dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping. Response categories include no days, 1 to 2 days, 3 to 4 days, 5 to 6 days, and every day, with corresponding scores of 0, 1, 2, 3, and 4.46 The total possible score ranges from 0 to 28, with a high score indicating worse disease severity.12 The MID in AD has been estimated to be 3.4 to 5.0 points in adults and to be 3.0 to 3.9 points in children.12,13,47 Based on expert input, an MID of 3.4 points was selected as the threshold for a clinically relevant difference.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
IGA of AD severity | Investigator-reported assessment instrument used in clinical trials to rate AD severity. Five-point scale, ranging from 0 (clear) to 4 (severe), with distinct, morphological descriptors for each category.48 | Validity: Moderate to strong correlation with EASI score (r = 0.66 to 0.72) in adult patients with AD.48 Reliability: Moderate intrarater (ICC = 0.54, SD = 0.28) and interrater reliability (CV = 33.0, SD = 12.3) in adult patients with AD.48 Responsiveness: No evidence identified. | No MID has been identified in adult or adolescent patients with AD. |
EASI | A physician-administered, composite index that assesses the severity and extent of AD.49 The severity of 4 AD disease characteristics (erythema, induration or papulation, excoriation, and lichenification) on 4 body regions (head and/or neck, trunk, upper extremities, and lower extremities) is assessed by the investigator on a 4-point scale, ranging from 0 (none or absent) to 3 (severe). The EASI score equals the sum of the weighted scores obtained for each body region. Scores range from 0 to 72, with higher values indicating a more severe and/or more extensive condition.49 EASI-50, EASI-75, and EASI-90 represent a ≥ 50%, ≥ 75%, and ≥ 90% reduction from baseline in EASI score, respectively.49 | Validity: In adult patients with AD, a moderate to strong correlation with SCORAD score (r = 0.84 to 0.93) was shown.13,50,51 In pediatric patients with AD, including those older than 12 years, EASI response was correlated strongly with IGA response (r > 0.8 at day 43 and at 6 months).49 Reliability: In adult patients with AD, the internal consistency of EASI is adequate, with Spearman and Cronbach alpha values of 0.86 and 0.94, respectively.51 Test-retest reliability was also adequate (intrarater and interrater reliability kappa = 0.76),51 whereas the reliability of each component of the EASI ranged from 0.38 (ICC, lichenification) to 0.75 (ICC, area), indicating poor to good intrarater reliability.48 No evidence of reliability in adolescent patients with AD was identified. Responsiveness: In a study of adult patients with AD (the MAcAD trial), responsiveness to an improvement or decline in global severity based on IGA over 24 weeks was demonstrated (AUC = 0.67; 95% CI = 0.60 to 0.76).13 In pediatric patients with AD, sensitivity to change was judged to be adequate (P < 0.001; n = 1,068) to detect an improvement in disease status from baseline after 8 days of treatment.49 | In a study of mainly adults and an unknown number of adolescents with AD, the overall MID has been estimated to be 6.6.13 |
Pruritus NRS | Patient-reported worst itch over the previous 24 hours using an 11-point NRS, with 0 indicating no itch and 10 indicating the worst itch possible.42 | Psychometric assessment was performed in adult patients with moderate-to-severe AD from 2 clinical trial populations (SOLO 1 and SOLO 2).42 Validity: Content validity was ensured through concept elicitation during development and with in-depth, one-to-one patient interviews (n = 14). Construct validity with similar constructs (PCS, DLQI itch item, SCORAD itch VAS) was strong (Pearson r = 0.61 to 0.77), whereas for those with dissimilar constructs (EASI, IGA), it was weak to moderate (r = 0.09 to 0.24). Known-group validity has been established; patients with absent or mild itch based on the PCS, no impact on the DLQI, or excellent on the PGADS had a significantly lower score on the NRS (P < 0.0001).42 Reliability: Test-retest reliability over 1 week was adequate (ICC, 0.95 to 0.96).42 Responsiveness: Change from baseline at week 16 on the NRS correlated well with that on the PCS (Pearson r = 0.71), DLQI itch item (r = 0.66), and SCORAD itch VAS (r = 0.77), but less well on the EASI (r = 0.50) and IGA (r = 0.50).42 Psychometric assessment in the adolescent population with AD has not been identified. | Improvement of at least 3 to 4 points from baseline is estimated to be a clinically meaningful change, calculated using anchor-based and distribution-based methods in adults with moderate-to-severe AD.34 In adults with moderate-to-severe AD, MID estimates were 2 to 4 points based on anchors (EASI, IGA, PCS) and 1.0 point based on distribution methods (SD = 0.5).41,42 Evidence of an MID in adolescents was not identified. |
POEM | A patient-reported, AD-specific, symptom questionnaire, with the assessment period being the previous week.47 Consists of 7 items (itching, sleep, bleeding, weeping, cracking, flaking, and dryness), each assessed on a 5-point categorical response scale (0 = no days; 1 = 1 to 2 days; 2 = 3 to 4 days; 3 = 5 to 6 days; 4 = every day). The total score is the sum of the 7 items (ranging from 0 to 28) and reflects disease-related morbidity, with a higher score indicating worse symptoms.47 | Validity: In adult patients, concurrent validity was reported in those with moderate-severe self-reported AD severity (Spearman r = 0.53); however, a weak correlation (r = 0.39) with clear-mild AD was shown. Convergent validity with the DLQI (r = 0.59), correlation with the EASI (r = 0.52), and weaker correlation with the worst itch NRS (r = 0.45) were shown in adult patients with AD.47 Reliability: Internal consistency was acceptable (Cronbach alpha = 0.88), and test-retest reliability was acceptable, with 95% of the scores falling within 2.6 points on repeat testing (mean score difference = 0.04; SD = 1.32) in adult patients with AD.47 Responsiveness: In the Prove trial conducted in adult patients with AD, responsiveness to improvement and decline in global severity, measured by IGA over 18 weeks, was noted.13 | The MID has been estimated to be 3.4 points in adults with AD and from 3.0 to 3.9 points in children with AD.12,13 Another study, which used the global severity of AD as an anchor, estimated 5 points to be the MID for adults.47 |
DLQI | A patient-reported, dermatology-specific, HRQoL instrument for use in adults. Consists of 10 items that address the patient’s perception of the impact of their skin disease on 5 different aspects of HRQoL, each scored on a 4-point Likert scale (0 = not at all; 1 = only a little; 2 = quite a lot; 3 = very much):
The total score is the sum of the 10 items (0 to 30 points), with a higher score indicating a poorer HRQoL (0 to 1 = no effect; 2 to 5 = small effect; 6 to 10 = moderate effect; 11 to 20 = very large effect; 21 to 30 = extremely large effect). Recall period is the previous 1 week.19-21 | Validity: Content validity was ensured with input from adult patients with AD (n = 9; other eczema n = 10) during the development phase.52 Construct validity was demonstrated by a strong correlation with the POEM (r = 0.78) and a moderate correlation with the SCORAD (r = 0.42). Reliability: In patients with stable AD, test-retest reliability was adequate (ICC > 0.7). Among adult patients with mixed skin diseases, including AD, internal consistency was acceptable (Cronbach alpha = 0.75 to 0.92).19,53,54 Responsiveness: In patients older than 16 years with a variety of skin conditions, including AD (n = 192, patients with eczema = 12.5%), improved DLQI scores were observed in those whose disease severity decreased over a 1-to-3-month period (P < 0.0001).19 | Estimates of the MID have ranged from 2.2 to 6.9, but no information about MID was found specifically for adult patients with AD.19-21 |
CDLQI | A patient-reported, dermatology-specific questionnaire based on the adult version of the DLQI and designed for patients aged 3 to 16 years with dermatological conditions. Available in text and cartoon versions. Consists of 10 items that address the patient’s perception of the impact of AD on various aspects of HRQoL over the previous week, including dermatology-related symptoms and feelings, leisure, school, friendships, sleep, and the impact of treatment.15,44,45 Each question is scored on a 4-point Likert scale (0 = not at all; 1 = only a little; 2 = quite a lot; 3 = very much). The total score is the sum of the 10 items (0 to 30 points), with a higher score indicating a worse HRQoL.15,44,45 | Validity: Three studies demonstrated concurrent validity, 2 between CDLQI and the Cardiff Acne Disability Index, and 1 between CDLQI and the Childhood Atopic Dermatitis Impact Scale.45 Convergent construct validity and divergent construct validity of the CDLQI were demonstrated in 45 and 6 studies, respectively.45 Reliability: Good internal consistency of the CDLQI (examined in 6 studies), with Cronbach alpha values ranging from 0.82 to 0.92.44,45 Test-retest reliability is adequate, with Spearman’s rank order correlation coefficient calculated in 4 studies (range, 0.74 to 0.97).44,45 One study showed an ICC of 0.80.44,45 Responsiveness: Examined in 26 studies that demonstrated responsiveness to change of CDLQI.45 | In adolescent patients with moderate-to-severe AD, a reduction of 6.0 to 8.0 points has been suggested as the clinically relevant threshold for a within-person change, corresponding to improvement in anchors.15 |
AD = atopic dermatitis; AUC = area under the curve; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; CV = coefficient of variation; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = at least a 50% reduction in EASI score; EASI-75 = at least a 75% reduction in EASI score; EASI-90 = at least a 90% reduction in EASI score; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; IGA = Investigator Global Assessment; MID = minimal important difference; NRS = Numeric Rating Scale; PCS = Pruritus Categorical Scale; PGADS = Patient Global Assessment of Disease Status; POEM = Patient-Oriented Eczema Measure; SCORAD = SCORing Atopic Dermatitis; SD = standard deviation; VAS = visual analogue scale.
Statistical Analysis
The 3 studies used similar statistical methods to analyze the data for the induction phase (Table 9). The Cochran-Mantel-Haenszel test stratified by geographic region (US versus Europe versus the rest of the world), age group (adolescent versus adult), and disease severity (IGA 3 versus IGA 4) was used to compare treatment groups for categorical end points (e.g., EASI-75 response). The common RD adjusted for stratification factors was reported, with 95% CI calculated using the Mantel-Haenszel-Sato method. The major secondary continuous efficacy end points were analyzed using an analysis of covariance model, with treatment group, baseline value, and stratification factors as covariates (e.g., change from baseline in DLQI score). The LS mean and 95% CI for the difference between groups was reported. Continuous efficacy outcomes with multiple measures (i.e., change from baseline in POEM score) were analyzed using an MMRM that included treatment, baseline value, visit, the interaction of baseline values-by-visit and treatment-by-visit, and the stratification factors.
For the induction phase, all 3 studies used a hybrid estimand for the primary and major secondary end points, and defined a second, supportive estimand for categorical end points (composite) and continuous end points (hypothetical) (Table 10). The estimand defined how missing data and intercurrent events (i.e., the use of rescue therapy or the stoppage of treatment due to AEs or for other reasons) were handled in the analyses (Table 11). Table 9 outlines the primary and/or supportive estimand used for each of the efficacy outcomes of interest to this review. For the change in POEM score, there was no imputation for missing data, and the MMRM used assumed that all missing data were missing at random (i.e., missingness can be explained by associations with observed data). For the IGA 0 or 1, EASI-75, and EASI-90, and at least 4-point Pruritus NRS responder analyses, a tipping point analysis was run as a sensitivity analysis only if the results of the primary analyses were statistically significant. In this analysis, all patients who used rescue medication or discontinued treatment due to lack of efficacy were imputed as nonresponders. Multiple iterations were run in which the probability of response was varied for other intercurrent events or missing data to determine if there was a tipping point, where the results were no longer statistically significant.
For the maintenance phase in the ADvocate 1 and ADvocate 2 studies, the proportion of patients who maintained an EASI-75 response at week 52 was analyzed using a Cochran-Mantel-Haenszel test stratified by region and the maintenance primary estimand (hybrid strategy) (Table 10). Two supportive estimands were defined (hybrid and composite strategies) (Table 12) that used alternate approaches for missing data.
The studies’ protocols stated that subgroup analyses were to be conducted based on age group, sex, race, baseline IGA score, and region. The statistical analysis plans also list the following subgroups to be tested: ethnicity, weight category, body mass index category, duration since AD onset category, baseline Pruritus NRS score (< 4 versus ≥ 4), and prior use of systemic treatments. The sponsor identified 1 subgroup of interest in the protocol for their systematic review: patients using lebrikizumab in combination with TCS.
Table 9: Statistical Analyses of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
ADvocate 1, ADvocate 2, and ADhere studies: induction period (week 16) | ||||
Proportion of patients with the following:
| ||| | Geographic region (Europe, the US, or the rest of the world), age group (adolescent vs. adult), and disease severity (IGA 3 vs. IGA 4) | Primary estimand (hybrid), MCMC-MI Supportive estimand (composite), NRI | Tipping point analysis |
Percent change from baseline in:
Change from baseline in DLQI total score | |||||| | Geographic region, age group, and disease severity | Primary estimand (hybrid), MCMC-MI Supportive estimand (hypothetical), no imputation | Not reported |
Change from baseline in POEM, CDLQI score | |||| | Baseline value, visit, baseline value-by-visit interaction, treatment-by-visit interaction, geographic region, age group, disease severity | Supportive estimand (hypothetical), no imputation | Not reported |
ADvocate 1 and ADvocate 2 studies: maintenance period (week 52) | ||||
Percent of patients who continue to exhibit EASI-75 from baseline to week 52 (among EASI-75 responders re-randomized at week 16) | ||| | Geographic region | Primary maintenance estimand (hybrid), MCMC-MI Maintenance supportive estimand, MCMC-MI, and NRI | Not reported |
CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% reduction in EASI score; EASI-90 = at least a 90% reduction in EASI score; IGA = Investigator Global Assessment; MCMC-MI = Markov chain Monte Carlo multiple imputation; NRI = nonresponder imputation; NRS = Numeric Rating Scale; POEM = Patient-Oriented Eczema Measure; vs. = versus.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9
Table 10: Estimands for the Studies Included in the Systematic Review
Estimand | End point | Accounting for ICE | Population summary | |
|---|---|---|---|---|
ADvocate 1, ADvocate 2, and ADhere studies: induction period | ||||
Primary estimand (hybrid) | Primary and major secondary (categorical and continuous end points) |
| Difference in response proportions or means between treatment conditions | |
Supportive estimand (composite) | Categorical end points |
| Difference in response proportions between treatment conditions | |
Supportive estimand (hypothetical) | Continuous end points |
| Difference in means between treatment conditions | |
ADvocate 1 and ADvocate 2 studies: maintenance period | ||||
Maintenance primary estimand (hybrid) | Major and other secondary end points (categorical and continuous) |
| Difference in response proportions or means between treatment conditions | |
ICE = intercurrent event.
aRescue medications during the ADvocate 1 and ADvocate 2 studies included any topical treatments (e.g., midpotency TCS) or systemic therapies (e.g., oral corticosteroids, phototherapy, and cyclosporine). Rescue therapy in the ADhere study included high-potency TCS or systemic AD therapies (e.g., oral corticosteroids, phototherapy, and cyclosporine). Note that in the ADhere study, all patients were prescribed low-to-midpotency TCS for use as needed during the trial.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9
Table 11: Analysis of Primary and Supportive Estimands — Induction Period
Estimand | ICE: used rescue medication | ICE: discontinued due to lack of efficacy | ICE: discontinued for other reasons | Missing data imputation method |
|---|---|---|---|---|
Primary estimand (hybrid) | Composite: set to baseline | Composite: set to baseline | Hypothetical: set to missing | Primary analysis: MCMC-MI Sensitivity analysis: tipping point analysis |
Supportive estimand for categorical end points (composite) | Composite: set to nonresponder | Composite: set to nonresponder | Composite: set to nonresponder | Nonresponder imputation |
Supportive estimand for continuous end points (hypothetical) | Hypothetical: set to missing | Hypothetical: set to missing | Hypothetical: set to missing | || |||||||||| (MMRM) |
ICE = intercurrent event; MCMC-MI = Markov chain Monte Carlo multiple imputation; MMRM = mixed model repeated measures.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9
Table 12: Analysis of Primary and Supportive Estimands — Maintenance Period
Estimand | ICE: used topical rescue medication | ICE: used systemic rescue medication | ICE: discontinued due to lack of efficacy | ICE: Discontinued for other reasons | Transferred to escape arm | Missing data imputation method |
|---|---|---|---|---|---|---|
Maintenance primary estimand (hybrid) | Hypothetical: set to missing | Composite: set to baseline | Composite: set to baseline | Hypothetical: set to missing | Composite: set to baseline | MCMC-MI |
||||||||||| |||||||||| |||||||| |||||||| | ||||||||| ||||||| || |||||||| | |||||||||| ||| || |||||||| | |||||||||| ||| || |||||||| | ||||||||||||| ||| || ||||||| | |||||||||| ||| || |||||||| | ||||||| |
||||||||||| |||||||||| |||||||| ||| ||||||||||| ||| |||||| ||||||||||| | |||||||||| ||| || ||||||||||||| | |||||||||| ||| || ||||||||||||| | |||||||||| ||| || ||||||||||||| | |||||||||| ||| || ||||||||||||| | |||||||||| ||| || ||||||||||||| | ||||||||||||| |||||||||| |
ICE = intercurrent event; MCMC-MI = Markov chain Monte Carlo multiple imputation.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2.10
Control of Type I Error Rate
According to the statistical analysis plan for the EMA, a graphical approach was used to control the overall type I error rate at a 2-sided alpha of 0.05 for all primary and major secondary end points in the induction period of the ADvocate 1 and ADvocate 2 studies (13 outcomes in total). A hierarchical approach was used for the maintenance phase, in which the major secondary end points were tested in a prespecified order, and for each of the lebrikizumab every-2-weeks and every-4-weeks groups, the outcome of interest to this review (i.e., EASI-75 at week 52) was tested first; subsequent end points were tested only if the results were statistically significant at a 2-sided alpha of 0.05. For the FDA, a graphical approach was also used to control the overall type I error rate at a 2-sided alpha of 0.05 for the primary and 8 major secondary end points for the induction period of the ADvocate 1 and ADvocate 2 studies.
In the ADhere study, a prespecified, multiple testing, gatekeeping approach was used for all primary and major secondary end points to control the overall type I error rate at a 2-sided alpha of 0.05. There were separate gatekeeping approaches for the EMA and the FDA. The primary and major secondary end points were tested in the following sequential order for the EMA:
IGA 0 or 1 with an improvement of at least 2 points at week 16 (coprimary)
EASI-75 at week 16 (coprimary)
EASI-90 at week 16
percent change in EASI score at week 16
an improvement of at least 4 points in Pruritus NRS score at week 16
percent change in Pruritus NRS at week 16
an improvement of at least 4 points in Pruritus NRS score and EASI-75 at week 16
change in DLQI total score at week 16
an improvement of at least 4 points in DLQI total score at week 16
change in Sleep Loss Scale score at week 16.
The sequence for testing for the FDA was as follows:
IGA 0 or 1 with an improvement of at least 2 points at week 16 (primary)
EASI-75 at week 16
an improvement of at least 4 points in Pruritus NRS score at week 16
an improvement of at least 4 points in Pruritus NRS score and EASI-75 at week 16
EASI-90 at week 16.
Sample Size
For the ADvocate studies, a sample size of 96 patients for lebrikizumab and 48 patients for placebo was estimated to have at least 90% power to detect a statistically significant difference between groups for IGA 0 or 1 and EASI-75 responses at week 16, based on a chi-square distribution with a 2-sided significance of 0.05. The CSR stated that the planned sample size was increased to 400 patients to ensure that sufficient safety information was collected and that there was an adequate number of patients for the maintenance phase of the trials. The power estimates were based on data from the phase II study (DRM06-AD01),55 which reported an EASI-75 and IGA 0 or 1 response rate of ||||| ||| ||||| for lebrikizumab and |||| ||| ||||| for placebo, respectively. || ||||| |||||||||||| |||| |||||||| ||| ||| ||||||||||| ||||||
For the ADhere study, a sample size of 225 patients was planned (randomized in a ratio of 2:1 to lebrikizumab and placebo) to have at least 95% power to test the superiority of lebrikizumab for the coprimary end points based on a 2-sided Fisher’s exact test with an alpha of 0.05. These estimates assumed 16-week IGA 0 or 1 and EASI-75 response rates of 38% and 58%, respectively, for lebrikizumab plus TCS and of 13% and 20% for placebo plus TCS. ||| |||||||| ||||| |||| ||||| || | ||||| ||| ||||| ||| |||||||||||| |||||||||||||| | |||||| || |||||||||| ||| |||||||| |||||| ||| || ||| ||||||||| ||||||||||
Analysis Populations
Efficacy analyses for the induction period were conducted in the ITT population of the ADvocate 1 study and the modified ITT population of the ADvocate 2 and ADhere studies (Table 13). In the ADvocate 2 and ADhere trials, the modified ITT population excluded patients from a study site that was closed due to critical audit findings that threatened the validity of the data. The safety analyses were based on all randomized patients who received at least 1 dose of the study drug in the ADvocate 1 study (safety population), and in the modified safety population in the ADvocate 2 and ADhere studies that excluded patients from the closed study site.
The maintenance period efficacy and safety analyses of interest to this review were based on the maintenance primary population or the modified maintenance primary population in the ADvocate studies. These populations included patients who had received lebrikizumab during the induction phase and who were rerandomized and received at least 1 dose of the study drug during the maintenance phase. For patients who entered the escape arm after week 16, only information gathered before escape was included.
Table 13: Analysis Populations of the Studies Included in the Systematic Review
Population | Definition | Application |
|---|---|---|
ADvocate 1 | ||
ITT population | All randomized patients, even those who did not take the assigned treatment, did not receive the correct treatment, or otherwise did not follow the protocol. Patients were analyzed according to the treatment assigned. | All efficacy outcomes during the induction period. |
Safety population | All randomized patients who received at least 1 dose of the study treatment. | All safety analyses during the induction period. |
Maintenance primary population | All patients initially randomized to lebrikizumab who, at week 16, were randomly reassigned and received at least 1 dose of the study treatment during the maintenance period. Patients were analyzed according to the treatment to which they were randomly reassigned. Only information gathered before escape is presented. | Efficacy and safety outcomes during the maintenance period. |
Maintenance secondary population | All patients initially randomized to placebo who, at week 16, were randomly reassigned and received at least 1 dose of the study treatment during the maintenance period. Patients were analyzed according to the treatment to which they were rerandomized. Only information gathered before escape is presented. | Select efficacy outcomes during the maintenance period. |
Escape population | Includes patients not rerandomized at week 16 and who received at least 1 dose of the study treatment during the maintenance period. | Select efficacy outcomes during the maintenance period. |
ADvocate 2 | ||
ITT population | All randomized patients, even those who did not take the assigned treatment, did not receive the correct treatment, or otherwise did not follow the protocol. Patients were analyzed according to the treatment assigned. | Lists of SAEs and primary AEs leading to study treatment discontinuation. |
mITT population | ITT population, excluding all patients from a specific study site with critical audit findings. Patients were analyzed according to the treatment assigned. | Efficacy outcomes for the induction period. |
Modified safety population | All randomized patients who received at least 1 dose of the study treatment and excluding all patients from a specific study site with critical audit findings. | Safety analyses for the induction period. |
Modified maintenance primary population | All patients initially randomized to lebrikizumab who, at week 16, were randomly reassigned and received at least 1 dose of the study treatment during the maintenance period (excluding patients from a specific study site with critical audit findings). Patients were analyzed according to the treatment to which they were rerandomized. Only information gathered before escape is presented. | Efficacy and safety outcomes during the maintenance period. |
Modified maintenance secondary population | All patients initially randomized to placebo who, at week 16, were randomly reassigned and received at least 1 dose of the study treatment during the maintenance period (excluding patients from a specific study site with critical audit findings). Patients were analyzed according to the treatment to which they were rerandomized. Only information before escape is presented. | Select efficacy outcomes during the maintenance period. |
Modified escape population | Includes patients not rerandomized at week 16 and who received at least 1 dose of the study treatment during the maintenance period (excluding patients from a specific study site with critical audit findings). | Select efficacy outcomes during the maintenance period. |
ADhere | ||
ITT population | All randomized patients, even those who did not take the assigned treatment, did not receive the correct treatment, or otherwise did not follow the protocol. Patients were analyzed according to the treatment to which they were assigned. | Lists of SAEs and primary AEs leading to study treatment discontinuation. |
mITT population | ITT population, excluding all patients from a specific study site with critical audit findings. Patients were analyzed according to the treatment to which they were assigned. | Efficacy and health outcomes analyses. |
Modified safety population | All randomized patients who received at least 1 dose of the study treatment and excluding all patients from a specific study site with critical audit findings. | Safety analyses. |
AE = adverse event; ITT = intention to treat; mITT = modified intention to treat; SAE = serious adverse event.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9
Results
Patient Disposition
Patient disposition during the induction phase is shown in Table 14 and during the maintenance phase is shown in Table 15 for the studies included in the systematic review.
In the ADvocate 1 study, 536 patients were enrolled, 112 patients (|||) were excluded at the screening stage, and 424 patients were randomized (|||). Of these patients, ||| ||| || stopped treatment during the induction phase in the placebo and lebrikizumab groups, respectively. Lack of efficacy (||), withdrawal by patient (||), and protocol violations (||) were the most common reasons for discontinuation in the placebo group, whereas protocol violations (||) were the most common reason in the lebrikizumab group. More patients in the placebo group entered the escape arm than the lebrikizumab group (||| |||||| |||) during the first 16 weeks. A total of 157 (|||) patients from the lebrikizumab group met the response criteria at week 16 and were rerandomized. |||| |||||||| || ||| ||| of patients discontinued treatment during the maintenance phase in the placebo group (i.e., lebrikizumab withdrawal), the lebrikizumab every-4-weeks group, and the lebrikizumab every-2-weeks group, respectively.
The ADvocate 2 study screened 606 patients, of which 179 patients (|||) failed to meet the screening criteria or were excluded due to protocol violations at a study site. A total of 427 patients (|||) were randomized and analyzed. In the placebo group, ||| of patients stopped treatment during the induction phase, compared with || of patients in the lebrikizumab group. The most common reasons for stopping treatment included withdrawal by the patient |||), AEs (||), and lack of efficacy (||) in the placebo group, and AEs (||) and protocol violations (||) in the lebrikizumab group. The CSR reported that ||| |||||| ||| of patients in the placebo and lebrikizumab groups, respectively, entered the escape arm during the first 16 weeks. A total of 134 (|||) of patients from the lebrikizumab group met the response criteria at week 16 and were rerandomized. During the maintenance phase, |||| || ||| || of patients stopped treatment in the placebo group (i.e., lebrikizumab withdrawal), the lebrikizumab every-4-weeks group, and the lebrikizumab every-2-weeks group, respectively.
The ADhere study screened 312 patients, of which 211 patients were randomized and analyzed |||||| The proportion of patients who stopped treatment during the first 16 weeks was ||| in the placebo group and || in the lebrikizumab group. The most common reasons for stopping treatment included withdrawal by the patient |||| and protocol violations (|||) in the placebo group, and AEs |||||, lack of efficacy |||||, and withdrawal by the patient |||| in the lebrikizumab group.
In the ADvocate 2 and ADhere studies, a study site was identified |||| ||| ||||| |||||||| |||||||||| |||| |||||||| || |||||||| ||||| ||||||||| ||| |||| |||| |||| |||| ||| |||||| ||||||||||| ||| |||||||| |||| |||| |||| |||| |||||||| |||| ||| |||||||| ||| |||||| |||||||||| ||||| ||||||||| |||| |||||||| ||||| ||| |||||||| |||| || ||||||| ||| |||||||| || ||| |||||||| ||| ||| |||||||| |||||| |||||||||||| ||||| |||||||| ||| |||||||| |||| ||| |||||||| ||||| ||||. In total, 18 patients |||| from the ADvocate 2 study and 17 patients |||| from the ADhere study were excluded.
Table 14: Summary of Patient Disposition From the Studies Included in the Systematic Review — Induction Period
Patient disposition | ADvocate 1 | ADvocate 2 | ADhere | |||
|---|---|---|---|---|---|---|
PBO | LEB every 2 weeks | PBO | LEB every 2 weeks | PBO + TCS | LEB every 2 weeks + TCS | |
Screened, N | 536 | 606 | 312 | |||
Screen failed, n (%) | 112 (|||) | 161(||||) | 84 (||||) | |||
Site excluded, n (%) | NA | 18 (|||) | 17 (|||) | |||
Reason for screening failure, n (%) | ||||||
Inclusion or exclusion criteria | 97 (|||) | 144 (||) | 78 (|||) | |||
Pandemic | 1(|||||) | 1(|||||) | 1(||||) | |||
Lost to follow-up | 1 (||||) | 1(|||||) | 1 (||||) | |||
Withdrawal of consent | 13 (|||) | 15 (||) | 4 (||) | |||
Randomized, N | 141 | 283 | 146a | 281a | 66a | 145a |
Discontinued treatment, n (%) | 21 (|||) | 20(|||) | 16 (||) | 22 (||) | 8(|||) | 11(|||) |
Reason for discontinuation, n (%) | ||||||
Adverse events | 1 (|) | 2 (|) | 4 (|) | 6 (|) | 0 | 3 (|) |
Pandemic | 1 (|) | 2 (||) | 1 (|) | 4 (|) | NR | NR |
Lack of efficacy | 7 (|) | 2 (|) | 4 (|) | 1 (|||) | 1 (||) | 3 (|) |
Lost to follow-up | 1 (|) | 4 (|) | 2 (|) | 0 | NR | NR |
Protocol deviation | 5 (||) | 6 (|) | 0 | 6 (||) | 2 (|) | 2 (|) |
Withdrawal by patient | 6 (||) | 3 (|) | 5 (|) | 4 (|)) | 4 (||) | 3 (||) |
Physician decision | NR | NR | NR | NR | 1 (|) | 0 |
Other | 0 | 1 (0.4) | 0 | 1 (|||) | NR | NR |
Completed week 16, n (%) | 120 (|||) | 263 (||) | 130 (|||) | 259 (||) | 58 (|||) | 134 (|||) |
Enrolled in escape arm at week 16, n (%) | 96 (|||) | 106 (|||) | 108 (||) | 125 ||||) | NR | NR |
Rerandomized to maintenance treatment, n (%) | 24 (|||) | 157 (||) | 22 (||) | 134 (|||) | NA | NA |
ITT, N | 141 | 283 | 150 | 295 | 75 | 153 |
mITT, Na | NA | NA | 146 | 281 | 66 | 145 |
Safety, Nb | 141 | 282 | 145 | 281 | 66 | 145 |
ITT = intention to treat; LEB = lebrikizumab; mITT = modified intention to treat; NA = not applicable; NR = not reported; PBO = placebo; TCS = topical corticosteroids.
aData presented are for the modified ITT population, which excludes data from a study site with critical audit findings.
bData for the ADvocate 2 and ADhere studies are for the modified safety population, which excludes data from a study site with critical audit findings.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9
Table 15: Summary of Patient Disposition From the ADvocate 1 and ADvocate 2 Studies — Maintenance Period Primary Population
Patient disposition | ADvocate 1a | ADvocate 2a | ||||
|---|---|---|---|---|---|---|
PBO (LEB withdrawal) | LEB every 4 weeks | LEB every 2 weeks | PBO (LEB withdrawal) | LEB every 4 weeks | LEB every 2 weeks | |
Randomized, N (%) | 32 | 63 | 62 | 28 | 55 | 51 |
Discontinued treatment, n (%) | 3 (||) | 5 (||) | 8 (||) | 3 (||) | 3 (|||) | 4 (|||) |
Reason for discontinuation, n (%) | ||||||
Adverse events | 0 | 1 (||) | 1 (||) | 1 (||) | 2 (||) | 1 (||) |
Lack of efficacy | 0 | 0 | 1 (|) | 1 (||) | 0 | 0 |
Lost to follow-up | 1 (||) | 1 (||) | 1 (||) | 0 | 0 | 0 |
Withdrawal by patient | 2 (||) | 3 (||) | 5 (||) | 1 (||) | 0 | 3 (||) |
Physician decision | NR | NR | NR | 0 | 1 (||) | 0 |
Enrolled in escape arm, N | 7 (||) | 4 (||) | 6 (||) | 3 (||) | 1 (||) | 6 (||) |
Completed week 52 | 22 (||) | 54 (||) | 48 (||) | 22 (||) | 51 (||) | 41 (||) |
MPP, N | 32 | 63 | 62 | 30 | 59 | 59 |
Modified MPP, Nb | NA | NA | NA | 28 | 55 | 51 |
LEB = lebrikizumab; MPP = maintenance primary population; NA = not applicable; NR = not reported; PBO = placebo.
aData reported are for the maintenance primary population that included all patients randomized to lebrikizumab at baseline who were responders at week 16 and were rerandomized at the start of the maintenance period. All patients received at least 1 dose of the study drug in the maintenance period.
bThe modified MPP population excludes data from the study site with critical audit findings.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2.10
Baseline Characteristics
The characteristics outlined in Table 16, Table 17, and Table 18 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
The baseline characteristics of the patients enrolled were similar between groups within studies, and generally comparable across the trials. The mean age per treatment group ranged from 34.2 years (SD = 16.4) to 37.5 years (SD = 19.9) across the studies. In the ADvocate studies, ||| || ||| of patients were adolescents, compared to ||| || ||| in the ADhere study. There were roughly equal proportions of females and males in the studies. Most patients identified as white (||| || |||), but ||| || ||| of patients identified as Asian and || || ||| identified as Black or African American. People of other races made up a smaller proportion of those enrolled. Of note, there was a higher proportion of patients who identified as Asian in the placebo groups than in the lebrikizumab groups in the ADvocate 1 (||| |||||| |||) and ADhere (||| |||||| |||) studies. On average, patients enrolled in the study had been diagnosed with AD for 20 or more years, with most patients (||| || |||) classified as having disease of moderate severity based on an IGA score of 3 at baseline, whereas ||| || ||| were classified as having severe AD (i.e., IGA score of 4). The mean BSA affected ranged from 38.2% (SD = 20.8) to 47.8% (SD = 23.9).
Table 16: Summary of Baseline Characteristics From the Studies Included in the Systematic Review — Induction Period
Characteristic | ADvocate 1 (ITT) | ADvocate 2 (mITT) | ADhere (mITT) | |||
|---|---|---|---|---|---|---|
PBO (N = 141) | LEB every 2 weeks (N = 283) | PBO (N = 146) | LEB every 2 weeks (N = 281) | PBO + TCS (N = 66) | LEB every 2 weeks + TCS (N = 145) | |
Age, years, mean (SD) | 34.2 (16.4) | 36.1 (17.8) | 35.3 (17.2) | 36.6 (16.8) | 36.7 (17.9) | 37.5 (19.9) |
Adolescents (12 to < 18 years; ≥ 40 kg), n (%) | 18 (12.8) | 37 (13.1) | 17 (11.6) | 30 (10.7) | 14 (21.2) | 32 (22.1) |
Adults (≥ 18 years) n (%) | 123 (87.2) | 246 (86.9) | 129 (88.4) | 251 (89.3) | 52 (78.8) | 113 (77.9) |
Female, n (%) | 73 (51.8) | 141 (49.8) | 75 (51.4) | 136 (48.4) | 33 (50.0) | 70 (48.3) |
Male, n (%) | 68 (48.2) | 142 (50.2) | 71 (48.6) | 145 (51.6) | 33 (50.0) | 75 (51.7) |
Race, n (%) | ||||||
American Indian or Alaska Native | 0 | 7 (2.5) | 2 (1.4) | 3 (1.1) | 2 ||||| | 5 ||||| |
Asian | 31 (22.0) | 39 (13.8) | 44 (30.1) | 78 (27.8) | 13 (19.7) | 18 (12.4) |
Black or African American | 16 (11.3) | 33 (11.7) | 10 (6.8) | 25 (8.9) | 9 (13.6) | 19 (13.1) |
Native Hawaiian or Other Pacific Islander | 0 | 2 (0.7) | 1 (0.7) | 2 (0.7) | 0 | 3 ||||| |
White | 93 (66.0) | 196 (69.3) | 85 (58.2) | 168 (59.8) | 40 (60.6) | 90 (62.1) |
Multiple | 1 (0.7) | 4 (1.4) | 3 (2.1) | 4 (1.4) | 1 ||||| | 8 (||) |
Other | 0 | 1 (0.4) | 1 (0.7) | 1 (0.4) | 1 ||||| | 2 ||||| |
Not reported | |||| | ||||| | || | || | || | || |
Duration since AD onset, years, mean (SD) | 23.8 (15.4) | 22.0 (14.9) | 20.1 (14.1) | 20.8 (15.2) | 21.2 (13.9) | 21.0 (17.4) |
Weight, kg, mean (SD) | 79.0 (22.7) | 77.0 (19.7) | 76 (21.2) | 76.7 (20.5) | 79.8 (24.4) | 74.6 (23.3) |
Weight < 60 kg, n (%) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Weight ≥ 60 to < 100 kg, n (%) | || |||||| | ||| |||||| | ||| |||||| | ||| |||||| | || |||||| | || |||||| |
Weight ≥ 100 kg, n (%) | || |||||| | || |||||| | || ||||| | || |||||| | || |||||| | || |||||| |
Body mass index, kg/m2, mean (SD) | 27.8 (7.2) | 26.6 (5.8) | 26.3 (6.3) | 26.7 (6.6) | 27.9 (7.5) | 26.5 (7.2) |
Baseline disease characteristics | ||||||
IGA score of 3 (moderate), n (%) | 83 (58.9) | 170 (60.1) | 95 (65.1) | 175 (62.3) | 48 (72.7) | 98 (67.6) |
IGA score of 4 (severe), n (%) | 58 (41.1) | 113 (39.9) | 51 (34.9) | 106 (37.7) | 18 (27.3) | 47 (32.4) |
EASI score, mean (SD) | 31.0 (12.9) | 28.8 (11.3) | 29.6 (10.8) | 29.7 (12.0) | 26.4 (10.6) | 27.7 (11.1) |
Pruritus NRS, mean (SD)a | 7.3 (1.7) | 7.2 (1.9) | 7.2 (1.9) | 7.1 (1.9) | 6.8 (2.0) | 7.3(1.8) |
Pruritus NRS score ≥ 4, n (%)a | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | || |||||| | ||| |||||| |
% BSA affected, mean (SD) | 47.8 (23.9) | 45.3 (22.5) | 46.0 (21.1) | 46.1 (22.6) | 38.2 (20.8) | 40.4 (21.9) |
DLQI, mean (SD)b | 15.7 (7.2) | 15.3 (7.4) | 15.9 (7.6) | 15.4 (7.0) | 13.5 (7.5) | 14.9 (7.2) |
CDLQI, mean (SD)c | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| |
POEM, mean (SD)d | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| |
AD = atopic dermatitis; BSA = body surface area; CDLQI = Children Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = Investigator Global Assessment; ITT = intention to treat; LEB = lebrikizumab; mITT = modified intention to treat; NRS = Numeric Rating Scale; PBO = placebo; POEM = Patient-Oriented Eczema Measure; SD = standard deviation; TCS = topical corticosteroids.
aNumber of patients with nonmissing Pruritus NRS scores at baseline was as follows: |||||||| | ||| | ||| |||||||| ||| | ||| |||||||| |||||||| || ||| | ||| |||||||| ||| | ||| |||||||| ||||||| ||| | || |||||||| ||| | ||| ||||||||
bNumber of patients with nonmissing DLQI scores at baseline was as follows: |||||||| | ||| | |||| ||| | |||| |||||||| || ||| | |||| ||| | |||| ||||||| ||| | ||| ||| | ||||
cNumber of patients with nonmissing CDLQI scores at baseline was as follows: |||||||| | ||| | ||| ||| | ||| |||||||| || ||| | ||| ||| | ||| ||||||| ||| | ||| ||| | |||
dNumber of patients with nonmissing POEM scores at baseline was as follows: |||||||| | ||| | ||| |||||||| ||| | ||| |||||||| |||||||| || ||| | ||| |||||||| ||| | ||| |||||||| ||||||| ||| | || |||||||| ||| | ||| ||||||||
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere,9 Sponsor’s Summary of Clinical Evidence.35
Most patients enrolled had previously used TCS |||| || ||||| and ||| || ||| of patients had used topical calcineurin inhibitors. Before enrolment, ||| || ||| of patients had received phototherapy (Table 17). The proportion of patients who had received systemic therapies previously was ||| |||||| |||| ||| |||||| |||| ||| ||| |||||| ||| in the lebrikizumab versus placebo groups of the ADvocate 1, ADvocate 2, and ADhere studies, respectively. In the ADvocate studies, the most common systemic therapies were oral corticosteroids (||| || |||) and cyclosporine (|| || |||). Patients who had previously received dupilumab and tralokinumab were excluded from the ADvocate trials ||| || || || ||| |||||||| ||||| |||||| ||||||||||. In the ADhere study, |||| |||| |||| || ||| || of patients had received oral corticosteroids, cyclosporine, dupilumab, JAK inhibitors, or tralokinumab, respectively. ||| |||||||||| || |||||||| ||| ||| |||||||| ||||| |||||||||| ||| ||||||||| ||||||| ||||||| |||||| |||||| |||||| |||||| ||| |||||||| || ||||| ||||||||| ||||||||||| |||| ||||| ||| |||||||||||| |||| |||||| |||| ||| |||||||| ||||||||||||||| |||| |||||| |||| || ||| |||||||||||| ||| ||||||| ||||||| |||||||||||||
After rerandomization at week 16, the baseline characteristics of patients who continued in the maintenance primary population are shown in Table 18. There was some variation noted between groups in mean age, racial and sex distribution, mean body weight (and body weight categories), baseline IGA score, and baseline EASI score.
Table 17: Summary of AD Treatment History at Baseline for the Studies Included in the Systematic Review
Prior therapy, n (%) | ADvocate 1 (ITT) | ADvocate 2 (mITT) | ADhere (mITT) | |||
|---|---|---|---|---|---|---|
PBO (N = 141) | LEB every 2 weeks (N = 283) | PBO (N = 146) | LEB every 2 weeks (N = 281) | PBO + TCS (N = 66) | LEB every 2 weeks + TCS (N = 145) | |
None | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Topical corticosteroids | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Topical calcineurin inhibitors | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Systemic treatment | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Systemic corticosteroids | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Azathioprine | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Cyclosporine | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
IFN-gamma | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Methotrexate | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Mycophenolate-mofetil | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Janus kinase inhibitors | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Dupilumaba | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Tralokinumaba | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Phototherapy | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Photochemotherapy | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Other biologics (e.g., cell-depleting biologics) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Other nonbiologic medication or treatment | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
AD = atopic dermatitis; IFN = interferon; ITT = intention to treat; LEB = lebrikizumab; mITT = modified intention to treat; NA = not applicable; NR = not reported; PBO = placebo; TCS = topical corticosteroids.
aIn the ADvocate 1 and ADvocate 2 studies, patients were excluded if they had previously received dupilumab or tralokinumab.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere,9 Sponsor’s Summary of Clinical Evidence.35
Table 18: Summary of Baseline Characteristics From the ADvocate 1 and ADvocate 2 Studies — Maintenance Period (Maintenance Primary Population)
Characteristic | ADvocate 1a | ADvocate 2b | ||||
|---|---|---|---|---|---|---|
PBO (LEB withdrawal) (N = 32) | LEB every 4 weeks (N = 63) | LEB every 2 weeks (N = 62) | PBO (LEB withdrawal) (N = 28) | LEB every 4 weeks (N = 55) | LEB every 2 weeks (N = 51) | |
Age, years, mean (SD) | 33.0 (16.6) | 34.4 (17.4) | 37.3 (17.9) | 34.7 (16.8) | 37.5 (17.2) | 34.5 (15.9) |
Adolescents (12 to < 18 years; ≥ 40 kg), n (%) | 3 (9.4) | 10 (15.9) | 7 (11.3) | 5 (17.9) | 7 (12.7) | 6 (11.8) |
Adults (≥ 18 years) n (%) | 29 (90.6) | 53 (84.1) | 55 (88.7) | 23 (82.1) | 48 (87.3) | 45 (88.2) |
Female, n (%) | 21 (65.6) | 38 (60.3) | 28 (45.2) | 15 (53.6) | 31 (56.4) | 25 (49.0) |
Male, n (%) | 11 (34.4) | 25 (39.7) | 34 (54.8) | 13 (46.4) | 24 (43.6) | 26 (51.0) |
Race, n (%) | ||||||
American Indian or Alaska Native | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Asian | 6 (18.8) | 5 (7.9) | 5 (8.1) | 9 (32.1) | 12 (21.8) | 14 (27.5) |
Black or African American | 5 (15.6) | 8 (12.7) | 5 (8.1) | 3 (10.7) | 4 (7.3) | 4 (7.8) |
Native Hawaiian or Other Pacific Islander | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
White | 19 (59.4) | 49 (77.8) | 47 (75.8) | 14 (50.0) | 37 (67.3) | 33 (64.7) |
Multiple | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Other | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Duration since AD onset, years, mean (SD) | 20.7 (12.6) | 22.8 (14.3) | 22.7 (14.8) | 20.1 (17.4) | 22.4 (15.5) | 20.5 (13.6) |
Weight, kg, mean (SD) | 71.5 (16.7) | 73.7 (19.3) | 77.7 (19.8) | 74.1 (18.0) | 75.6 (18.2) | 74.4 (25.0) |
Weight < 60 kg, n (%) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Weight ≥ 60 to < 100 kg, n (%) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Weight ≥ 100 kg, n (%) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Body mass index, kg/m2, mean (SD) | 24.9 (4.2) | 25.8 (6.0) | 26.7 (6.0) | 25.8 (5.4) | 26.6 (5.7) | 25.8 (7.8) |
Disease characteristicsc | ||||||
IGA score of 3 (moderate), n (%) | 20 (62.5) | 40 (63.5) | 38 (61.3) | 17 (60.7) | 38 (69.1) | 32 (62.7) |
IGA score of 4 (severe), n (%) | 12 (37.5) | 23 (36.5) | 24 (38.7) | 11 (39.3) | 17 (30.9) | 19 (37.3) |
EASI, mean (SD) | 27.6 (10.7) | 28.9 (12.7) | 30.0 (11.9) | 30.3 (11.8) | 28.7 (12.6) | 29.0 (9.5) |
Pruritus NRS, mean (SD)d | 7.6 (1.9) | 7.0 (2.1) | 7.4 (1.7) | 7.3 (1.7) | 6.9 (2.1) | 7.0 (1.7) |
Pruritus NRS score ≥ 4, n (%)d | 30 (93.8) | 58 (93.5) | 58 (96.7) | 27 (100.0) | 49 (90.7) | 50 (98.0) |
AD = atopic dermatitis; EASI = Eczema Area and Severity Index; IGA = Investigator Global Assessment; LEB = lebrikizumab; NRS = Numeric Rating Scale; PBO = placebo; SD = standard deviation.
aData for the maintenance primary population of patients who initially received lebrikizumab and met the treatment response criteria at week 16 (EASI-75 or IGA 0 or 1). Patients were rerandomized at week 16 and received at least 1 dose of the study drug during the maintenance phase.
bData presented for the ADvocate 2 study are for the modified maintenance primary population (MPP), which included patients in the MPP as described for the ADvocate 1 study but excluded patients from a study site with critical audit findings.
cDisease characteristics were recorded at the baseline visit for the study (day 1).
dPruritus NRS data in the ADvocate 1 study are based on ||| ||| ||| || ||||||||| ||| ||| |||||||| | ||||| ||||| || ||| || ||| || |||||||| || ||| |||| ||| |||||| ||| ||| |||||| ||||||| ||||||||||||
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 Sponsor’s Summary of Clinical Evidence.35
Exposure to Study Treatments
During the induction period of the trials, the mean duration of exposure ranged from ||||| |||| ||| | ||||| || ||||| |||| ||| | ||||| || ||| ||||||| ||||||| ||| |||| ||||| |||| ||| | ||||| || ||||| |||| ||| | ||||| || ||| |||||||||||| |||||| (Table 19). Patients were considered adherent to therapy if they received at least ||| of the study drug doses while enrolled in the study. During the induction period, ||| || |||| of patients were classified as adherent.
Table 19: Summary of Patient Exposure From the Studies Included in the Systematic Review — Induction Period (Safety Population)
Exposure | ADvocate 1 | ADvocate 2a | ADherea | |||
|---|---|---|---|---|---|---|
PBO (N = 141) | LEB every 2 weeks (N = 282) | PBO (N = 145) | LEB every 2 weeks (N = 281) | PBO + TCS (N = 66) | LEB every 2 weeks + TCS (N = 145) | |
Total, patient-years | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Duration, days, mean (SD) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Duration, days, median (range) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Number of patients who were adherent (received ≥ 75% of the study drug doses), n (%) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
LEB = lebrikizumab; PBO = placebo; SD = standard deviation; TCS = topical corticosteroids.
aData for the ADvocate 2 and ADhere studies were based on the modified safety population, which excludes data from the study site with critical audit findings.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere,9 Sponsor’s Summary of Clinical Evidence.35
Table 20: Summary of Patient Exposure From the ADvocate 1 and ADvocate 2 Studies — Maintenance Period (MPP)
Exposure | ADvocate 1 | ADvocate 2a | ||||
|---|---|---|---|---|---|---|
PBO (LEB withdrawal) (N = 32) | LEB every 4 weeks (N = 63) | LEB every 2 weeks (N = 62) | PBO (LEB withdrawal) (N = 28) | LEB every 4 weeks (N = 55) | LEB every 2 weeks (N = 51) | |
Total, patient-years | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Duration, days, mean (SD) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Duration, days, median (range) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Number of patients who were adherent (received ≥ 75% of the study drug doses), n (%) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
LEB = lebrikizumab; PBO = placebo; SD = standard deviation.
aData presented for the ADvocate 2 study are for the modified maintenance primary population (MPP), which included patients in the MPP except for patients from the study site with critical audit findings.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 Sponsor’s Summary of Clinical Evidence.35
|||||| ||| ||||||||||| |||||| ||| |||| |||||||| || |||||||| ||| ||||| |||| ||| | ||||| ||| ||||| |||| ||| | ||||| || ||| |||||||| | |||||| ||| ||||| |||| ||| | ||||| ||| ||||| |||| ||| | ||||| || ||| |||||||| | |||||| ||| ||| ||||||| ||| |||||||||||| ||||| ||||||| |||||||||||| (Table 20). ||| |||||||| ||| ||| ||||||||| ||||||||| |||||||||
At the start of the ADhere study, all patients were prescribed low- and midpotency corticosteroids for use as needed. ||| ||| | ||||||| |||||||| |||||| |||| ||| ||| (Table 21). || ||||||||| ||| ||| ||| || |||||||| || ||| ||||||| ||| |||||||||||| ||||||| ||||||||||||| |||||||| ||||| ||||||| ||||||||||| |||||||||| |||||| ||| ||||||
Table 21: Concomitant Topical AD Treatments — The ADhere Study (mITT)
AD treatment, n (%) | PBO + TCS (N = 66) | LEB every 2 weeks + TCS (N = 145) |
|---|---|---|
Patients using topical therapy (case report form data) | ||||| | ||||| |
Low- or midpotency TCS | ||||| | ||||| |
Triamcinolone | ||||| | ||||| |
Hydrocortisone | ||||| | ||||| |
Desonide | ||||| | ||||| |
Topical calcineurin inhibitors | ||||| | ||||| |
Tacrolimus | ||||| | ||||| |
Pimecrolimus | ||||| | ||||| |
Patients with ≥ 1 AD treatment (patient diary data) | ||||| | ||||| |
TCS | ||||| | ||||| |
Triamcinolone acetonide cream 0.1% | ||||| | ||||| |
Hydrocortisone 1% cream | ||||| | ||||| |
Other TCS | ||||| | ||||| |
Topical calcineurin inhibitors | ||||| | ||||| |
Pimecrolimus 1% | ||||| | ||||| |
Tacrolimus 0.03% | ||||| | ||||| |
Tacrolimus 0.1% | ||||| | ||||| |
Other topical calcineurin inhibitor | ||||| | ||||| |
AD = atopic dermatitis; LEB = lebrikizumab; PBO = placebo; TCS = topical corticosteroids.
Sources: CSR for ADhere,9 Sponsor’s Summary of Clinical Evidence.35
During the induction phase of the pivotal trials, more patients in the placebo group used rescue therapy than in the lebrikizumab group (Table 22). In the ADvocate 1 study, 33.3% versus 11.0% of patients used rescue treatments in the placebo and lebrikizumab groups, respectively, as did 39.7% versus 18.5% of patients, respectively, in the ADvocate 2 study. Most of these patients used topical treatments; less than 8% of patients used systemic therapies. In the ADhere study, 4.5% versus 1.4% of patients used high-potency TCS in the placebo and lebrikizumab groups, respectively, and 7.6% versus 3.4% of patients used systemic therapies.
During the maintenance period, ||||| |||||| ||||| of patients used rescue treatments in the placebo (lebrikizumab withdrawal) versus lebrikizumab every-4-weeks groups, respectively, in the ADvocate 1 study, as did ||||| |||||| ||||| of patients in the ADvocate 2 study (Table 23).
Table 22: Types of Rescue Therapy in the Studies Included in the Systematic Review — Induction Period
AD treatment, n (%) | ADvocate 1 (ITT) | ADvocate 2 (mITT) | ADhere (mITT) | |||
|---|---|---|---|---|---|---|
PBO (N = 141) | LEB every 2 weeks (N = 283) | PBO (N = 146) | LEB every 2 weeks (N = 281) | PBO + TCS (N = 66) | LEB every 2 weeks + TCS (N = 145) | |
Patients with ≥ 1 AD rescue therapy | 47 (33.3) | 31 (11.0) | 58 (39.7) | 52 (18.5) | 7 (10.6) | 6 (4.1) |
Topical treatment | 44 (31.2) | 27 (9.5) | 54 (37.0) | 48 (17.1) | NAa | NAa |
TCS | 42 (29.8) | 24 (8.5) | 54 (37.0) | 47 (16.7) | NA | NA |
Low-to-mid potency | 38 (27.0) | 20 (7.1) | 24 (16.4) | 27 (9.6) | NA | NA |
High potency | 15 (10.6) | 6 (2.1) | 36 (24.7) | 25 (8.9) | 3 (4.5) | 2 (1.4) |
Topical calcineurin inhibitor | 9 (6.4) | 4 (1.4) | 6 (4.1) | 8 (2.8) | NA | NA |
Systemic therapy | 11 (7.8) | 7 (2.5) | 9 (6.2) | 8 (2.8) | 5 (7.6) | 5 (3.4) |
Systemic corticosteroids | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Immunosuppressant | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Biologics | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Phototherapy | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
AD = atopic dermatitis; ITT = intention to treat; LEB = lebrikizumab; mITT = modified intention to treat; NA = not applicable; PBO = placebo; TCS = topical corticosteroids.
aAll patients in the ADhere study were required to use mild or moderate TCS at the start of the study and were allowed to taper, stop, and restart use as needed throughout the 16-week trial. Low-potency topical calcineurin inhibitors were allowed for sensitive areas. High-potency TCS were designated as rescue therapy.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9
Table 23: Types of Rescue Therapy in the ADvocate 1 and ADvocate 2 Studies — Maintenance Period
AD treatment, n (%) | ADvocate 1 (MPP) | ADvocate 2 (modified MPP) | ||||
|---|---|---|---|---|---|---|
PBO (LEB withdrawal) (N = 32) | LEB every 4 weeks (N = 63) | LEB every 2 weeks (N = 62) | PBO (LEB withdrawal) (N = 28) | LEB every 4 weeks (N = 55) | LEB every 2 weeks (N = 51) | |
Patients with ≥ 1 AD rescue therapy | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Topical treatment | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Topical corticosteroids | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Low-to-mid potency | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
High potency | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Topical calcineurin inhibitor | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Systemic therapy | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Systemic corticosteroids | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Immunosuppressant | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Biologics | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Phototherapy | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
AD = atopic dermatitis; LEB = lebrikizumab; PBO = placebo.
Sources: CSR for J2T-DM-KGAB (Table KGAB.4.11); CSR for J2T-DM-KGAC (Table KGAC.4.11).
Efficacy
Disease Severity
At week 16, the proportion of patients with an IGA score of 0 or 1 and at least a 2-point reduction from baseline favoured the lebrikizumab groups over the placebo groups in all 3 studies (Table 24). For the ADvocate 1 study, 43.1% versus 12.7% of patients attained an IGA 0 or 1 response in the placebo and lebrikizumab groups, respectively, with a RD of 29.7% (95% CI, 21.6 to 37.8) favouring lebrikizumab (P < 0.001). Similar results were reported for the ADvocate 2 study (33.2% versus 10.8%; RD = 21.9%; 95% CI, 14.2 to 29.6; P < 0.001 for the lebrikizumab versus placebo groups, respectively). The IGA 0 or 1 response rate also favoured lebrikizumab plus TCS over placebo plus TCS in the ADhere study (41.2% versus 22.1%; RD = 18.3%; 95% CI, 5.1 to 31.5; P = 0.01).
A higher proportion of patients attained an EASI-75 response at week 16 in the lebrikizumab than in the placebo groups in the ADvocate 1 study (58.8% versus 16.2%; RD = 42.0%; 95% CI, 33.3 to 50.6; P < 0.001) and in the ADvocate 2 study (52.1% versus 18.1%; RD = 33.3%; 95% CI, 24.4 to 42.2; P < 0.001). In the ADhere study, 69.5% versus 42.2% of patients attained an EASI-75 response (RD = 26.4%; 95% CI, 12.1 to 40.8; P < 0.001) in the lebrikizumab plus TCS and placebo plus TCS groups, respectively (Table 24). For the EASI-90 response threshold, the results favoured lebrikizumab over placebo at week 16 in all 3 studies.
From a baseline EASI score of 28.8 (SD = 11.3) and 31.0 (SD = 12.9) in the lebrikizumab and placebo groups, respectively, the ADvocate 1 study reported a LS mean difference in the EASI percent change from baseline to week 16 of ||||| |||| ||| ||||| || ||||||, favouring the lebrikizumab group (P < 0.001) (Table 24). Similar findings were reported for the ADvocate 2 study, with a LS mean difference of |||||| |||| ||| ||||| || |||||; P < 0.001) favouring the lebrikizumab group over the placebo group, and for the ADhere study, with a LS mean difference of −23.6% (95% CI, −33.6% to −13.7%; P < 0.001), favouring the lebrikizumab plus TCS over placebo plus TCS group.
During the maintenance period, the ADvocate 1 study reported that 79.2% of patients in lebrikizumab every-4-weeks group maintained an EASI-75 response at week 52 compared with 61.3% of patients who were switched to placebo ||| || ||||| |||| ||| |||| || ||||||. In the ADvocate 2 study, 84.7% versus 72.0% maintained an EASI-75 response in the lebrikizumab every-4-weeks and placebo (i.e., lebrikizumab withdrawal) groups, respectively, ||| || ||||| |||| || |||| || |||||| (Table 25).
Of note, the every-2-weeks maintenance dose of lebrikizumab is not consistent with the recommended dosing in the product monograph and has been included in the data tables as supplemental information only.
Symptom Severity
Among patients who had a Pruritus NRS score of 4 or more points at baseline, the proportion of patients who reported at least a 4-point reduction in their Pruritus NRS score at week 16 was higher in the lebrikizumab groups than in the placebo groups (Table 26). These results were based on ||| |||||| |||| ||| |||||| |||| ||| ||| |||||| ||| of patients randomized in the lebrikizumab versus placebo groups of the ADvocate 1, ADvocate 2, and ADhere studies, respectively. In the ADvocate 1 study, 45.9% versus 13.0% of patients reported at least a 4-point reduction in their Pruritus NRS score in the lebrikizumab versus placebo groups, respectively, with a RD of 32.9% (95% CI, 24.6% to 41.3%; P < 0.001). The proportion of Pruritus NRS responders was 39.8% versus 11.5% in the ADvocate 2 study (RD = 28.3%; 95% CI, 20.0% to 36.5%; P < 0.001), favouring the lebrikizumab groups over the placebo groups (). In the ADhere study, 50.6% of patients in the lebrikizumab plus TCS group met the response criteria, compared with 31.9% in the placebo plus TCS group (RD = 19.2%; 95% CI, 4.3% to 34.1%; P = 0.02).
Table 24: Summary of Key Disease Severity Results From the Studies Included in the Systematic Review — Induction Period
Variable | ADvocate 1 (ITT) | ADvocate 2 (mITT) | ADhere (mITT) | |||
|---|---|---|---|---|---|---|
PBO (N = 141) | LEB every 2 weeks (N = 283) | PBO (N = 146) | LEB every 2 weeks (N = 281) | PBO +TCS (N = 66) | LEB every 2 weeks +TCS (N = 145) | |
Percentage of patients with an IGA score of 0 or 1 and a ≥ 2-point reduction from baseline to week 16 | ||||||
n (%) | || (12.7) | ||| (43.1) | || (10.8) | || (33.2) | 15 (22.1) | 60 (41.2) |
RD (95% CI) vs. PBOb | Reference | 29.7 (21.6 to 37.8) | Reference | 21.9 (14.2 to 29.6) | Reference | 18.3 (5.1 to 31.5) |
P valueb | Reference | < 0.001 | Reference | < 0.001 | Reference | 0.01 |
Percentage of patients with an EASI-75 response at week 16 | ||||||
n (%) | || (16.2) | ||| (58.8) | || (18.1) | ||| (52.1) | 28 (42.2) | 101 (69.5) |
RD (95% CI) vs. PBOb | Reference | 42.0 (33.3 to 50.6) | Reference | 33.3 (24.4 to 42.2) | Reference | 26.4 (12.1 to 40.8) |
P valueb | Reference | < 0.001 | Reference | < 0.001 | Reference | < 0.001 |
Percentage of patients with an EASI-90 response at week 16 | ||||||
n (%) | || (9.0) | ||| (38.3) | || (9.5) | || (30.7) | 14 (21.7) | 60 (41.2) |
RD (95% CI) vs. PBOb | Reference | 28.8 (21.3 to 36.3) | Reference | 20.7 (13.3 to 28.1) | Reference | 18.9 (6.1 to 31.7) |
P valueb | Reference | < 0.001 | Reference | < 0.001 | Reference | 0.008 |
Change in EASI score from baseline to week 16 | ||||||
Number of patients contributing to the analysis, n (%) | 141 (100.0) | 283 (100.0) | 146 (100.0) | 281 (100.0) | 66 (100.0) | 145 (100.0) |
Baseline EASI total score, mean (SD) | 31.0 (12.9) | 28.8 (11.3) | 29.6 (10.8) | 29.7 (12.0) | 26.4 (10.6) | 27.7 (11.1) |
Absolute change from baseline, LS mean (SE) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
LS mean difference in absolute change (95% CI)a,c | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Absolute change P valuea,c | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Percent change from baseline, LS mean (SE) | ||||| | ||||| | ||||| | ||||| | −53.1 (5.1) | −76.8 (4.1) |
LS mean difference in percent change (95% CI) vs. PBOa | ||||| | ||||| | ||||| | ||||| | Reference | −23.6 (−33.6 to −13.7) |
Percent change P valuea | ||||| | ||||| | ||||| | ||||| | Reference | < 0.001 |
CI = confidence interval; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% reduction in EASI score; EASI-90 = at least a 90% reduction in EASI score; IGA = Investigator Global Assessment; ITT = intention to treat; LEB = lebrikizumab; LS = least squares; mITT = modified intention to treat; PBO = placebo; RD = risk difference; SD = standard deviation; SE = standard error; TCS = topical corticosteroids; vs. = versus.
a|| |||| |||| ||| ||||| || |||||| ||||| |||||||| ||| |||||||||||||| ||||||| |||||||| ||| ||||| ||||||||||| |||||| ||||||| |||||||| ||| ||||| || |||||| ||| ||| |||||||| ||||| || | |||||||||| |||||||| ||| ||||| || ||| ||||||| |||||||| |||||||| ||||||||| ||| |||||||| |||||| ||||||| || |||||||||||| ||| || |||| || |||||||| ||| |||||| ||| || ||||||||| |||| ||||||| ||| ||||| ||||||| |||||||
b|||||| |||| |||||||||| |||| ||| ||||| || ||| ||| |||| |||||||| ||| |||||||||||||| ||||||| |||||||| ||| ||||| ||||||||||| |||||| ||||||| |||||||| ||| ||||| || |||||| ||| ||| ||| ||||||| |||||||| |||||||| ||||||||| ||| |||||||| |||||| ||||||| || |||||||||||| ||| || |||| || |||||||| ||| |||||| ||| || ||||||||| |||| ||||||| ||| ||||| ||||||| ||||||
cP value was not controlled for multiple testing.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9
Table 25: Summary of EASI-75 Response Results From the ADvocate 1 and ADvocate 2 Studies — Maintenance Period
Variable | ADvocate 1 (MPP) | ADvocate 2 (modified MPP) | ||||
|---|---|---|---|---|---|---|
PBO (LEB withdrawal) (N = 32) | LEB every 4 weeks (N = 63) | LEB every 2 weeks (N = 62) | PBO (LEB withdrawal) (N = 28) | LEB every 4 weeks (N = 55) | LEB every 2 weeks (N = 51) | |
Percentage of patients who continued to exhibit an EASI-75 response at week 52a | ||||||
Number of patients contributing to the analysis, N (%) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
n (%) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
RD (95% CI) vs. PBOb | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
P valueb | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
CI = confidence interval; EASI-75 = at least a 75% reduction in Eczema Area and Severity Index score; LEB = lebrikizumab; MPP = maintenance primary population; PBO = placebo; RD = risk difference.
aAmong patients who reported an EASI-75 response at week 16 and were rerandomized to placebo, lebrikizumab 250 mg every 2 weeks, or lebrikizumab 250 mg every 4 weeks.
b|||||| |||| |||||||||| |||| ||| ||||| || ||| ||| |||| |||||||| ||| |||||||||||||| |||||| |||||||| ||| ||| ||||||| ||||||||||| |||||||| |||||||| ||||||||| ||| |||||||| |||||||| |||||| |||||||| |||||||||||| ||| || |||| || |||||||| || ||||||| ||| |||||| ||| ||| |||||| ||| || ||||||||| |||||||| ||| |||||||| ||||||| |||||| ||||||| || |||||||||||| ||| || ||||| ||||||| ||| |||||| ||| || |||||||| ||||||| ||| |||| || |||||| ||||| ||||||| ||||||
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2.10
Table 26: Summary of Symptom Severity Results From the Studies Included in the Systematic Review — Induction Period
Variable | ADvocate 1 (ITT) | ADvocate 2 (mITT) | ADhere (mITT) | |||
|---|---|---|---|---|---|---|
PBO (N = 141) | LEB every 2 weeks (N = 283) | PBO (N = 146) | LEB every 2 weeks (N = 281) | PBO + TCS (N = 66) | LEB every 2 weeks + TCS (N = 145) | |
Percentage of patients with a Pruritus NRS ≥ 4-point reduction at week 16 (in patients with an NRS score ≥ 4 at baseline) | ||||||
Number of patients contributing to the analysis, N (%) | 130 |||| | 263 |||| | 134 |||| | 250 |||| | 57 |||| | 130 |||| |
n (%) | || (13.0) | ||| (45.9) | || (11.5) | ||| (39.8) | || (31.9) | || (50.6) |
RD (95% CI) vs. PBOb | Reference | 32.9 (24.6 to 41.3) | Reference | 28.3 (20.0 to 36.5) | Reference | 19.2 (4.3 to 34.1) |
P valueb | Reference | < 0.001 | Reference | < 0.001 | Reference | 0.02 |
Change in Pruritus NRS score from baseline to week 16 | ||||||
Number of patients contributing to the analysis, N (%) | ||||| | ||||| | ||||| | ||||| | || (95) | ||| (96) |
Baseline Pruritus NRS score, mean (SD) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Absolute change from baseline, LS mean (SE) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
LS mean difference in absolute change (95% CI)a,d | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Absolute change P valuea,d | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Percent change from baseline, LS mean (SE) | ||||| | ||||| | ||||| | ||||| | −35.5 (6.4) | −50.7 (4.5) |
LS mean difference in percent change (95% CI) vs. PBOa | ||||| | ||||| | ||||| | ||||| | Reference | −15.2 (−27.7 to −2.7) |
Percent change P valuea | ||||| | ||||| | ||||| | ||||| | Reference | 0.02 |
Change in POEM total score from baseline to week 16 | ||||||
Number of patients contributing to the analysis, N (%) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Baseline POEM total score, mean (SD) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Absolute change from baseline, LS mean (SE) | ||||| | ||||| | ||||| | ||||| | −6.2 (1.0) | −10.2 (0.7) |
LS mean difference (95% CI) vs. PBOc,d | ||||| | ||||| | ||||| | ||||| | Reference | −4.0 (−6.3 to −1.7) |
P valuec,d | ||||| | ||||| | ||||| | ||||| | Reference | < 0.001 |
CI = confidence interval; ITT = intention to treat; LEB = lebrikizumab; LS = least squares; mITT = modified intention to treat; NRS = Numeric Rating Scale; PBO = placebo; POEM = Patient-Oriented Eczema Measure; SD = standard deviation; SE = standard error; TCS = topical corticosteroids; vs. = versus.
a|| |||| |||| ||| ||||| || |||||| ||||| |||||||| ||| |||||||||||||| ||||||| |||||||| ||| ||||| ||||||||||| |||||| ||||||| |||||||| ||| ||||| || |||||| ||| ||| |||||||| ||||| || | |||||||||| |||||||| ||| ||||| || ||| ||||||| |||||||| ||||||||
bCommon RD (95% CI) based on the Cochran-Mantel-Haenszel test adjusted for stratification factors (region, age group [adolescent vs. adult], baseline IGA score [3 vs. 4]) and the primary (hybrid) estimand (patients who received rescue therapy or discontinued due to lack of efficacy had values set to baseline, with Markov chain Monte Carlo multiple imputation for other missing data).
c|| |||| |||||||||| |||| ||| ||||| || |||| ||||| |||| |||||||||| ||| |||||||||| |||||||| |||||| |||||| |||||||| |||||||||||||| |||||||||||| |||||||||||||||||| |||||||||||| |||||||||| ||||||| ||| |||||| |||||||| ||| |||||| ||| ||||||||||
dP value was not controlled for multiple testing.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere,9 additional information supplied by sponsor.22
The percent change from baseline in Pruritus NRS scores at week 16 favoured the lebrikizumab groups over the placebo groups in the ADvocate 1 study (|| |||| |||||||||| |||||| |||| ||| ||||| || ||||||||||||), the ADvocate 2 study (|||||| |||| ||| ||||| || ||||||| | ||||||), and the ADhere study (−15.2%; 95% CI, −27.7% to −2.7%; P = 0.02) (Table 26).
The POEM symptom frequency scale is scored from 0 to 28, with a high score indicating more severe disease.12 A MID of 3.4 points was selected as the threshold for a clinically relevant difference.13 The analyses at week 16 excluded |||| || |||| || |||||||| ||| |||||| ||| |||| || |||| |||| ||||||| |||| ||||| || ||||| || |||||||| ||| ||||||||| |||||.The change in POEM score was not part of the graphical testing strategy used to control the family-wise type I error rate.
The ADvocate 1 study reported a LS mean difference in the change from baseline to week 16 in POEM scores of |||| |||||| |||| ||| |||| || ||||| | ||||||| for lebrikizumab versus placebo. The LS mean difference was |||| |||||| |||| ||| |||| || ||||| | ||||||| in the ADvocate 2 study for lebrikizumab versus placebo, and the ADhere study reported a LS mean difference of −4.0 points (95% CI, −6.3 to −1.7 points; P < 0.001) for lebrikizumab plus TCS versus placebo plus TCS.
Health-Related Quality of Life
In the pivotal trials, the DLQI was used to measure HRQoL in patents 17 years and older, and the CDLQI was used for those aged 12 to 16 years. The instruments are scored from 0 to 30, with higher scores indicating poorer HRQoL. The change in CDLQI was not part of the graphical statistical testing strategy to control the family-wise type I error rate. MIDs of 4 points for the DLQI and 6 points for the CDLQI were selected as the clinically relevant threshold of change.14,15
In the ADvocate 1 study, the LS mean difference in the change in baseline in the DLQI was −5.8 points (95% CI, −7.1 to −4.5 points; P < 0.001) for lebrikizumab versus placebo at week 16, and in the ADvocate 2 study, it was −4.9 points (95% CI, −6.3 to −3.5 points; P < 0.001). The ADhere study reported a LS mean difference in the change from baseline in the DLQI of −3.3 points (95% CI, −5.3 to −1.3 points; P = 0.001) for lebrikizumab plus TCS versus placebo plus TCS. These analyses included 75% to 86% of patients initially randomized in the trials (Table 27).
The number of patients per treatment group included in the analysis of CDLQI was small, ranging from 5 to 11 patients in the placebo groups and from 17 to 26 patients in the lebrikizumab groups. The LS mean difference in the change from baseline in the CDLQI was −7.0 points (95% CI, −10.1 to −3.9 points; P < 0.001) in the ADvocate 1 study, −4.2 points (95% CI, −9.1 to 0.6 points; P = 0.085) in the ADvocate 2 study, and −4.6 points (95% CI, −7.2 to −2.0 points; P = 0.001) in the ADhere study for the lebrikizumab groups versus the placebo groups at week 16.
Table 27: Summary of HRQoL Results From the Studies Included in the Systematic Review — Induction Period
Variable | ADvocate 1 (ITT) | ADvocate 2 (mITT) | ADhere (mITT) | |||
|---|---|---|---|---|---|---|
PBO (N = 141) | LEB every 2 weeks (N = 283) | PBO (N = 146) | LEB every 2 weeks (N = 281) | PBO + TCS (N = 66) | LEB every 2 weeks + TCS (N = 145) | |
Change in DLQI score from baseline to week 16 | ||||||
Number of patients contributing to the analysis, N (%) | 121 |||| | 239 |||| | 118 |||| | 218 ||| | 51 |||| | 109 |||| |
Baseline DLQI total score, mean (SD) | 15.7 (7.2) | 15.3 (7.4) | 15.9 (7.6) | 15.4 (7.0) | 13.5 (7.5) | 14.9 (7.2) |
Absolute change from baseline, LS mean (SE) | ||||| | ||||| | ||||| | ||||| | −6.5 (1.9) | −9.8 (1.8) |
LS mean difference (95% CI) vs. PBOa | ||||| | ||||| | ||||| | ||||| | Reference | −3.3 (−5.3 to −1.3) |
P valuea | ||||| | ||||| | ||||| | ||||| | Reference | 0.001 |
Change in CDLQI score from baseline to week 16 | ||||||
Number of patients contributing to the analysis, N (%)b | 9 ||| | 26 ||| | 5 ||| | 17 || | 11 ||| | 24 |||| |
Baseline CDLQI total score, mean (SD) | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Absolute change from baseline, LS mean (SE) | ||||| | ||||| | ||||| | ||||| | −4.7 (1.2) | −9.3 (0.9) |
LS mean difference (95% CI) vs. PBOc,d | ||||| | ||||| | ||||| | ||||| | Reference | −4.6 (−7.2 to −2.0) |
P valuec,d | ||||| | ||||| | ||||| | ||||| | Reference | 0.001 |
CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; ITT = intention to treat; LEB = lebrikizumab; LS = least squares; mITT = modified intention to treat; PBO = placebo; SD = standard deviation; SE = standard error; TCS = topical corticosteroids; vs. = versus.
aLS mean (95% CI) based on an analysis of covariance (ANCOVA) model adjusted for stratification factors (region, age group [adolescent vs. adult], baseline IGA score [3 vs. 4]), with the baseline value as a covariate. Analysis was based on the primary (hybrid) estimand (patients who received rescue therapy or discontinued due to lack of efficacy had values set to baseline, with Markov chain Monte Carlo multiple imputation for other missing data).
b|||||| || |||||||| |||| ||||||||||| ||||| |||||| || |||| ||| ||| ||||| |||||||||| ||| |||| || |||||||| || ||||| ||| |||||||| |||||||| ||||| |||||||| || || ||| || |||||||| || |||||||| || || ||| || |||||||| || |||||||| || ||| || |||
cLS mean difference (95% CI) based on the mixed model for repeated measures (MMRM), with covariates for treatment, baseline value, visit, baseline value-by-visit interaction, treatment-by-visit interaction, geographic region, age group, baseline IGA score. The supportive (hypothetical) estimand was used for missing data (i.e., MMRM).
dP value was not controlled for multiple testing.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9
Harms
Refer to Table 28, Table 29, and Table 30 for harms data.
Adverse Events
For the induction period of the trials, the proportion of patients who experienced 1 or more TEAEs was 46% versus 52%, 53% versus 66%, and 43% versus 35% in the lebrikizumab and placebo groups, respectively, of the ADvocate 1, ADvocate 2, and ADhere studies (Table 28). The most common AEs in the lebrikizumab groups were conjunctivitis (5% to 8%), headache (3% to 5%), and nasopharyngitis (2% to 5%). For the placebo groups, conjunctivitis was reported by 0% to 3% of patients, headache was reported by 1% to 4% of patients, and nasopharyngitis was reported by 2% to 6% of patients.
Table 28: Summary of Harms From the Studies Included in the Systematic Review — Induction Period (Safety Population)
Adverse events | ADvocate 1 | ADvocate 2a | ADherea | |||
|---|---|---|---|---|---|---|
PBO (N = 141) | LEB every 2 weeks (N = 282) | PBO (N = 145) | LEB every 2 weeks (N = 281) | PBO + TCS (N = 66) | LEB every 2 weeks + TCS (N = 145) | |
Most common adverse events, n (%)b | ||||||
Patients with ≥ 1 TEAEs | 73 (51.8) | 129 (45.7) | 96 (66.2) | 150 (53.4) | 23 (34.8) | 63 (43.4) |
Conjunctivitis | 4 (2.8) | 21 (7.4) | 3 (2.1) | 21 (7.5) | 0 | 7 (4.8) |
Nasopharyngitis | 4 (2.8) | 11 (3.9) | 3 (2.1) | 14 (5.0) | 4 (6.1) | 3 (2.1) |
Upper respiratory tract infection | 2 (1.4) | 1 (0.4) | 2 (1.4) | 0 | ||||| | ||||| |
Oral herpes | 5 (3.5) | 9 (3.2) | 3 (2.1) | 4 (1.4) | ||||| | ||||| |
Dermatitis atopic | 30 (21.3) | 16 (5.7) | 38 (26.2) | 28 (10.0) | 3 (4.5) | 3 (2.1) |
Pruritus | 6 (4.3) | 3 (1.1) | 1 (0.7) | 5 (1.8) | NR | NR |
Headache | 2 (1.4) | 9 (3.2) | 6 (4.1) | 14 (5.0) | 1 (1.5) | 7 (4.8) |
Serious adverse events, n (%) | ||||||
Patients with ≥ 1 SAEs | 1 (0.7) | 6 (2.1) | 4 (2.8) | 2 (0.7) | 1 (1.5) | 2 (1.4) |
RD (95% CI) for LEB vs. placeboc | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Events reported | Sepsis, cellulitis | Accidental overdose,d carpal tunnel syndrome, myocardial infarction, synovitis, arthralgia, peripheral edema | Uterine leiomyoma, myocardial infarction, fibula fracture, tibia fracture, AD | Cardiac failure, large intestine infection, multiple injuries, cerebellar syndrome, AD | |||||||||||| ||||| | ||||| |||| ||||||||||| |
Patients who stopped treatment due to adverse events, n (%) | ||||||
Patients with ≥ 1 AEs leading to treatment discontinuation | 1 (0.7) | 3 (1.1) | 4 (2.8) | 9 (3.2) | 0 | 3 (2.1) |
Events reported | |||||||||||||| | ||||||||||||||||||||||| || | ||||||||||||||||||||| | ||||||||||||||||||||| | || | ||||||||||||||||||||| |
Deaths, n (%) | ||||||
Patients who died | 0 | 0 | 1 (0.7) | 0 | 0 | 0 |
AD = atopic dermatitis; AE = adverse event; CI = confidence interval; COVID-19 = coronavirus disease 2019; LEB = lebrikizumab; NA = not applicable; NR = not reported; PBO = placebo; q.2.w. = every 2 weeks; RD = risk difference; ref = reference; SAE = serious adverse event; TCS = topical corticosteroids; TEAE = treatment-emergent adverse event; vs. = versus.
aThe ADvocate 2 and ADhere data are based on the modified safety population that excluded patients from a study site with critical protocol violations.
bAEs reported with ≥ 3% frequency in any treatment group.
c|||| ||||||||||| ||| ||||||||| || ||| |||||| ||||| ||| ||| ||| ||| || ||| |||||||||| ||| |||||| ||||||||||||| |||||||
d||||||||||| |||||||| |||||||| || |||||||||||
eAD was the reason for discontinuation in 3 patients in each group in the ADvocate 2 study.
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere.9 Additional information supplied by sponsor,22 Sponsor’s Summary of Clinical Evidence.35
Among patients who received placebo during the induction period, AD was commonly reported as an AE in the ADvocate 1 and ADvocate 2 studies (21% and |||, respectively), but it was reported less often in the placebo plus TCS group in the ADhere study (5%). AD was reported as an AE in 6%, 10%, and 2% of patients who received lebrikizumab in the ADvocate 1, ADvocate 2, and ADhere studies, respectively.
During the maintenance period, ||| |||||| |||| ||| ||| |||||| ||| of patients experienced 1 or more TEAEs in the lebrikizumab every-4-weeks group versus the placebo group (i.e., lebrikizumab withdrawal) in the ADvocate 1 and ADvocate 2 trials, respectively (Table 30).
Serious Adverse Events
The frequency of SAEs during the induction period was generally low, with 2.1% versus 0.7%, 0.7% versus 2.8%, and 1.4% versus 1.5% reporting a SAE in the lebrikizumab versus placebo groups of the ADvocate 1, ADvocate 2, and ADhere studies, respectively. The RD for SAEs is reported in Table 28. All SAEs listed in the table were reported in 1 patient per group.
A total of 5 patients from the MPP reported a SAE during the maintenance period of the trials. In the ADvocate 1 study, 2 patients |||||| in the lebrikizumab every-4-weeks group reported a SAE, and in the ADvocate 2 study, 1 patient |||||| in the and placebo group and 2 patients |||||| in the lebrikizumab every-2-weeks group reported an event. No patients in the other groups experienced a SAE.
Withdrawal Due to Adverse Events
During the induction period, 1.1% versus 0.7%, 3.2% versus 2.8%, and 2.1% versus 0.0% of patients in the lebrikizumab versus placebo groups stopped treatment due to ASs in the ADvocate 1, ADvocate 2, and ADhere studies, respectively. |||||| ||| |||||| |||||||||| ||||||| |||||| || ||| |||||||| | |||||| ||| |||||||| ||||||| |||||| |||| |||||||| || | ||||||| ||| ||||||
In the MPP, ||||||| |||||| || ||| |||||||||||| ||||| ||| ||||| | ||||| |||||| ||||||| ||||||||| || ||| |||||||| | |||||| || ||| |||||||| | |||||| | ||||||| |||||| || ||| |||||||||||| ||||| | ||||| ||||| ||||||| |||||||| ||| |||||||||||||||| |||| ||| || ||||||||||||||. No other patients stopped treatment due to AEs between week 16 and week 52.
Mortality
One patient died of a myocardial infarction during the induction period of the ADvocate 2 study. This patient was randomized to the placebo group. No other deaths were reported during the induction period, or during the maintenance phase in the MPP.
Notable Harms
Conjunctivitis was reported by |||| || ||||| of patients in the lebrikizumab groups and || || |||| of patients in the placebo groups during the induction period. The sponsor-reported RD for conjunctivitis was |||| |||| ||| ||| || ||||| for the ADvocate 1 study, |||| |||| ||| ||| || ||||| for the ADvocate 2 study, and |||| |||| ||| ||| || |||| in the ADhere study for lebrikizumab versus placebo (Table 29). During the maintenance period, |||| |||||| ||||| ||| ||||| |||||| ||||| of patients in the lebrikizumab every-4-weeks group versus the placebo group (lebrikizumab withdrawal) of the ADvocate 1 and ADvocate 2 studies, respectively, reported conjunctivitis (Table 30).
Keratitis AEs were reported in |||| || |||| of patients in the lebrikizumab groups and || || |||| of patients in the placebo groups over the first 16 weeks of the trials. During the maintenance period, only ||||||| |||||| in the lebrikizumab every-4-weeks group of the ADvocate 2 study and ||||||| |||||| in the lebrikizumab every-2-weeks group of the ADvocate 1 study reported keratitis. No keratitis AEs were reported in the other groups.
The sponsor identified herpes virus and parasitic infections as AEs of special interest. The frequency of herpes infections was generally similar in the lebrikizumab and placebo groups during the induction period (2% to 5%) and the maintenance period ||| || |||| No parasitic infections were reported during the induction or maintenance periods of the trials.
Table 29: Summary of Notable Harms From the Studies Included in the Systematic Review — Induction Period (Safety Population)
Adverse events | ADvocate 1 | ADvocate 2a | ADherea | |||
|---|---|---|---|---|---|---|
PBO (N = 141) | LEB every 2 weeks (N = 282) | PBO (N = 145) | LEB every 2 weeks (N = 281) | PBO + TCS (N = 66) | LEB every 2 weeks + TCS (N = 145) | |
Adverse events of special interest, n (%) | ||||||
Conjunctivitis clusterb | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
RD (95% CI) for LEB vs. placeboc | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Conjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Allergic conjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Bacterial conjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Keratitis clusterd | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
RD (95% CI) for LEB vs. placeboc | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Atopic keratoconjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Vernal keratoconjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Keratitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Infection of herpes or zoster | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Parasitic infection | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
CI = confidence interval; LEB = lebrikizumab; NR = not reported; PBO = placebo; RD = risk difference; ref = reference; TCS = topical corticosteroids.
aThe ADvocate 2 and ADhere data are based on the modified safety population |||| |||||||| |||||||| |||| | ||||| |||| |||| |||||||| |||||||| ||||||||||||
bThe conjunctivitis cluster includes the following preferred terms: ||||||||||||||| |||||||||||||| ||||||||| |||||||||||||| |||||||||| |||||||||||||| ||||| ||| ||||| ||||||||| ||||||||||||||||
cRDs are expressed as ||| |||||| ||||| ||| ||| ||| ||| || ||| |||||||||| ||| |||||| ||||||||||||| ||||||||
dThe keratitis cluster includes the following preferred terms: |||||||||| |||||| ||||||||||||||||||||| |||||||| |||||||||| |||||||||| |||||||||| ||| |||||| ||||||||||||||||||||||
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 CSR for ADhere,9 Sponsor’s Summary of Clinical Evidence,35 additional information supplied by the sponsor.22
Critical Appraisal
Internal Validity
Induction Period
The CDA-AMC reviewer identified no major concerns regarding the randomization, allocation concealment, or blinding methods used in the trials included in the systematic review. Randomization was conducted using an electronic data capture system, and was stratified by region, age group, and baseline IGA score. At the start of the trials, the characteristics of the patients appeared to be similar between groups within studies. The 1 exception was the higher proportion of patients who identified as Asian in the placebo groups than in the lebrikizumab groups in the ADvocate 1 and ADhere studies. These differences may be due to chance and were not expected to bias the findings. To maintain blinding, an identical schedule of injections in the placebo and lebrikizumab groups was used, and the placebo injection was indistinguishable from the active treatment. The CDA-AMC reviewer did not identify any substantial imbalances in the frequency of adverse effects that may have led to significant unblinding.
Table 30: Summary of Harms Results From the ADvocate 1 and ADvocate 2 Studies — Maintenance Period (MPP)
Adverse events | ADvocate 1a | ADvocate 2b | ||||
|---|---|---|---|---|---|---|
PBO (LEB withdrawal) (N = 32) | LEB every 4 weeks (N = 63) | LEB every 2 weeks (N = 62) | PBO (LEB withdrawal) (N = 28) | LEB every 4 weeks (N = 55) | LEB every 2 weeks (N = 51) | |
Most common adverse events n (%)c | ||||||
Patients with ≥ 1 TEAEs | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
COVID-19 | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Nasopharyngitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Upper respiratory tract infection | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Conjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Urinary tract infection | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Folliculitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Atopic dermatitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Allergic conjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Headache | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Anxiety | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Serious adverse events, n (%) | ||||||
Patients with ≥ 1 SAEs | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
RD (95% CI) LEB vs. placebod | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Events reported | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Patients who stopped treatment due to adverse events, n (%) | ||||||
Patients with ≥ 1 AEs leading to permanent discontinuation of study treatment | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Event reported | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Deaths, n (%) | ||||||
Patients who died | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Adverse events of special interest, n (%) | ||||||
Conjunctivitis clustere | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
RD (95% CI) LEB vs. placebod | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Conjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Allergic conjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Keratitis clusterf | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
RD (95% CI) LEB vs. placebod | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Vernal keratoconjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Atopic keratoconjunctivitis | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Herpes infection or zoster | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Parasitic infection | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
AE = adverse event; CI = confidence interval; LEB = lebrikizumab; MPP = maintenance primary population; NA = not applicable; NR = not reported; PBO = placebo; RD = risk difference; ref = reference; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
a |||| ||| ||| ||||||||||| ||||||| |||||||||| ||| ||| ||||||||| |||||||| |||||||||||| ||| ||| ||| ||||||||| |||||||| |||||||| || |||| ||| |||||||| |||| ||||||||||||| || |||| || ||| |||||||| || ||||| | |||| || ||||| |||| |||||| ||| ||||||||||
b |||| ||||||||| ||| |||||||| | || ||| ||| |||||||| |||| ||||| |||||||| |||||||| || ||| ||| || ||||||||| ||| |||||||| | ||| |||||||| |||||||| |||| | ||||| |||| |||| ||| |||||||| ||||| ||||||||||
cAEs reported with ≥ 5% frequency in any treatment group.
dRDs are expressed as ||| |||||| ||||| ||| ||| ||| ||| || ||| |||||||||| ||| |||||| ||||||||||||| ||||||||
eConjunctivitis cluster includes the following preferred terms: ||||||||||||||| |||||||||||||| ||||||||| |||||||||||||| |||||||||| |||||||||||||| ||||| ||| ||||| ||||||||| ||||||||||||||||
fKeratitis cluster includes the following preferred terms: |||||||||| |||||| ||||||||||||||||||||| |||||||| |||||||||| |||||||||| |||||||||| ||| |||||| ||||||||||||||||||||||
Sources: CSR for ADvocate 1,11 CSR for ADvocate 2,10 Sponsor’s Summary of Clinical Evidence,35 additional information supplied by sponsor.22
During the induction period, the dosing of lebrikizumab in the trials was consistent with the Health Canada–recommended dose; however, the trials varied in terms of concomitant therapies. The concurrent use of topical AD therapies was not allowed in the ADvocate studies but was permitted in the ADhere trial. According to the clinical expert consulted for this review, the concurrent use of topical anti-inflammatory drugs is a potential effect modifier. A higher proportion of patients in the placebo groups used rescue therapies than in the lebrikizumab groups; however, the use of rescue treatments was accounted for in the primary estimand, in which patients who required rescue therapy were analyzed as nonresponders. In the ADhere study, all patients were prescribed low-to-midpotency TCS to be used as needed. Based on the information available, no important imbalances between groups were noted regarding the use of TCS during the ADhere study.
With regard to the conduct of the trials, there were study sites in the ADvocate 2 and ADhere studies that were found to be noncompliant with good clinical practice guidelines and the clinical trials’ protocols. ||||| || ||| ||||| |||||||| || ||| |||||||||| |||| ||| ||||||| |||| ||||| ||||| |||| |||||||||| ||||| |||||||| | |||||| || |||| ||||||| |||||||| || ||||||| ||||| |||||||| |||| ||| ||||||||. These exclusions involved 18 patients |||| from the ADvocate 2 study and 17 patients |||| from the ADhere study. The CDA-AMC reviewer did not consider their exclusion to be an important source of bias. The COVID-19 pandemic began shortly after the trials were started, and protocol changes were made to allow the trial to continue with accommodations for pandemic-related restrictions. ||| |||| |||| |||| ||| ||| |||||||| |||||||| |||| |||||||| || ||||||| ||| ||||| ||||| |||| ||||||||||||| ||| || |||||||| ||| ||| |||||| || ||||||||||| || |||| ||||| ||||||| ||| |||||| || ||| |||||||| || ||| |||||| ||| ||||||| || || ||||||||
The coprimary end points — IGA and EASI scores — are commonly reported outcomes in clinical trials for AD. Moreover, the EASI scale is recommended as a part of the core outcome set for clinical trials in patients with AD.40 The CSRs stated that assessors were trained and certified by the sponsor before conducting this assessment and that a single assessor was assigned to each patient for as many visits as possible to avoid interassessor variability in scoring. The CDA-AMC review of the reliability of the physician-reported outcomes found that the IGA scale had moderate intrarater and interrater reliability in adult patients with AD.48 Test-retest reliability of the EASI scale was adequate (intrarater and interrater reliability kappa = 0.76),51 whereas the reliability of each component of the EASI scale ranged from 0.38 (intraclass correlation for lichenification) to 0.75 (intraclass correlation for area), indicating poor to good intrarater reliability.48 These data suggest that the IGA may be less reliable than the EASI, and using the same investigator to assess specific patients throughout the trial cannot eliminate interassessor variability across patients, nor does it address intra-assessor variability (e.g., from 1 visit to the next, even within a single patient). The patient-reported outcomes — DLQI and CDLQI, POEM, and Pruritus NRS —reported in the trials are also part of the recommended core outcome set for clinical trials in AD,40 and the patient-group input received by CDA-AMC confirmed their importance. The patient-reported instruments have data to support their validity and reliability in AD or other dermatologic conditions.19,42,44,45,47,52-54
In general, there were no major issues identified by CDA-AMC with the statistical analysis methods or the control of type I errors. The efficacy analyses were conducted using the ITT population, but it should be noted that for the Pruritus NRS, DLQI, CDLQI, and POEM scores, there were missing values for some patients at baseline, so the ITT population was not fully reflected. Moreover, the Pruritus NRS responder analysis was based on patients who had a baseline score of 4. Randomization was not stratified by baseline Pruritus NRS score, so there is a possibility of prognostic imbalance between groups. The proportion of patients with missing Pruritus NRS data at baseline was |||| || ||||| and the proportion excluded due to baseline scores lower than 4 was |||| || |||| per group; thus, overall, the Pruritus NRS responder analyses excluded || || ||| of randomized patients per treatment group. The CDA-AMC reviewer also noted that the proportion of patients who discontinued treatment was numerically higher in the placebo groups than in the lebrikizumab groups during the induction period (|||||||| || ||| |||||| ||| |||||||| ||||| |||||| ||| ||||||| ||| |||||| ||). The primary estimand used a hybrid strategy, in which patients who stopped treatment due to AEs or who required rescue treatment were imputed as nonresponders. Patients who discontinued for other reasons had data imputed using MCMC-MI methods, which assume that patients remaining in the model can predict the response of patients who dropped out. The supportive estimand, which used a conservative assumption (nonresponder imputation), showed similar findings for the proportion of patients with an IGA 0 or 1 response, an EASI-75 response, and a 4-point reduction in Pruritus NRS score. Given that the primary and nonresponder analyses had similar findings, CDA-AMC had no major concerns regarding missing data for these 3 end points at week 16.
The CDA-AMC review team identified potential missing data issues for the change in POEM score in the 3 studies. The analyses were based on the supportive (hypothetical) estimand and the MMRM model, which assumes that data are missing at random (missing data are systematically related to the observed but not the unobserved data). The analyses at week 16 excluded |||| || |||| of patients per group and, therefore, were not based on the ITT population. In addition, there were differences in missing data rates between the groups at week 16, and it is unclear if the missing-at-random assumption is valid. Specifically, week 16 POEM data were missing from |||||| ||||| ||| ||||| of patients in the placebo groups and from |||||| ||||| ||| ||||| of patients in the lebrikizumab groups in the ADvocate 1, ADvocate 2, and ADhere studies, respectively. The sponsor noted that some of these between-group differences may be explained by the |||| |||||||| ||| || |||||| ||||||||| || |||||||| ||| |||||||| |||||||. In the statistical analysis, any follow-up time after the start of rescue therapy was classified as missing. Due to the missing data imputation method and the extent and differential rate of missing data, there is potential for bias in the change in POEM scores. Similar concerns were identified with missing data for the analysis of the change in DLQI and CDLQI total scores and with the potential for bias in the CDLQI data due to the use of the supportive (hypothetical) estimand to impute missing data. In addition, changes in POEM and CDLQI scores were not part of the graphical testing strategy used to control the family-wise type I error rate; therefore, there is an increased risk of false-positive conclusions for statistically significant results. As such, these results should be interpreted as supportive evidence only.
Maintenance Period
The key limitations of the 52-week data from the ADvocate trials are the enriched population, carry-over effects of lebrikizumab in the placebo group, and the small sample size. At week 16 in the ADvocate studies, patients treated with lebrikizumab who met the response criteria were rerandomized to 1 of 3 groups (primary maintenance population). This represents an enriched population, so the 1-year treatment effects of lebrikizumab may be higher than would be observed in an unselected population. Given the long half-life of lebrikizumab (24.5 days16), it is reasonable to assume that there are substantial carry-over effects for patients who switched from lebrikizumab to placebo, which may impact efficacy assessments, as well as the frequency of harms. The observed effects are relevant to the efficacy and harms of lebrikizumab maintenance, compared with lebrikizumab withdrawal, among patients who initially tolerate and respond to lebrikizumab.
The CDA-AMC reviewer also identified some imbalances between groups after rerandomization, which were likely attributable to chance due to the smaller sample sizes (28 to 63 patients per treatment group). The clinical expert consulted for this review did not identify any clinically important differences that would potentially bias the findings. || |||| || ||||| |||| |||||| || ||||||||||||| ||| || |||||||| ||||| |||||||||||| |||||||| |||||||||||||| ||| ||||| || |||||| |||| || |||||||| || |||||||||| ||| |||||||||||||| ||| ||||| |||||||| | ||||| |||||| || |||||||| ||| |||| ||| |||||||||| | ||||||||||| |||||| || ||||. Numerical differences were noted in the frequency of withdrawals in the ADvocate 2 study (placebo: ||| |||||| |||||||||||| ||||| | |||||| ||), but not in the ADvocate 1 study. No major concerns were identified with the statistical methods used to analyze the 52-week EASI-75 response outcome.
External Validity
The pivotal trials enrolled patients with moderate-to-severe AD that had not adequately responded to topical therapies. The average age of the patients was in the mid-30s, and they had been diagnosed with AD for 20 years. In the trials, just 11% to 22% of patients were adolescents, so the results are mainly reflective of adult patients. All 3 trials included patients in Canada; however, the overall study population may not represent the racial diversity in Canada. The clinical expert consulted did not identify any major limits to the generalizability of the findings, but did note that the trials excluded some patients with comorbidities who may have received lebrikizumab in clinical practice. For patients with chronic conditions that may require treatment with oral corticosteroids and for patients with acute or chronic infections, severe mental or physical illnesses, or a history of immunosuppression, the safety and efficacy of lebrikizumab is uncertain.
The dosing of lebrikizumab during the induction period was consistent with the Health Canada–recommended dose; however, the clinical expert noted that most patients using a biologic for AD would also use TCS as needed. The concurrent use of TCS was prohibited in the ADvocate studies, and thus the magnitude of the treatment effects observed in the ADhere study may be more consistent with what would occur in clinical practice. The generalizability of the 52-week efficacy and safety data may be limited, given the enriched population and the carry-over effects of lebrikizumab in patients who switched to placebo. In addition, the every-2-weeks maintenance dosing of lebrikizumab was not consistent with Health Canada recommendations.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to the CDA-AMC expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group:17,18
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word likely for evidence of moderate certainty (e.g., X intervention likely results in Y outcome).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word may for evidence of low certainty (e.g., X intervention may result in Y outcome).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of the effect. We describe evidence of very low certainty as very uncertain.
Following the GRADE approach, evidence from RCTs starts as high-certainty evidence and can be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment for the proportion of patients with an EASI-75 response, IGA 0 or 1 response, or at least a 4-point improvement in Pruritus NRS response was based on thresholds informed by the clinical expert consulted for this review. Changes in the POEM, DLQI, and CDLQI certainty of evidence assessments were based on thresholds identified in the literature; SAE and conjunctivitis assessments were based on the presence or absence of any (nonnull) effect.
For the GRADE assessments, findings from the ADvocate 1, ADvocate 2, and ADhere studies were considered together and summarized narratively by outcome because these studies were similar in population, interventions, design, and outcome measures.56
Results of GRADE Assessments
Lebrikizumab Versus Placebo
Table 2 presents the GRADE summary of findings for lebrikizumab versus placebo.
Long-Term Extension Studies
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Description of Studies
One long-term extension study — ADjoin — was summarized to provide evidence regarding the long-term (100-week) efficacy and safety of lebrikizumab among patients with moderate-to-severe AD who were enrolled in the ADvocate 1, ADvocate 2, ADhere, ADore, and ADopt-VA studies (parent trials).23 This study was conducted at ||| centres, which enrolled ||| patients in Australia, Bulgaria, Canada, Estonia, France, Germany, Latvia, Lithuania, Mexico, Poland, Singapore, South Korea, Spain, Taiwan, Ukraine, and the US. |||| |||||| |||||||| ||||||| |||||| |||| |||| |||||| ||| ||||||| |||||||| |||| || |||| || ||| | |||||| || |||||||| ||| ||||||||| ||| ||||||| |||||| ||||| |||||| || || || ||||| || |||||||||||| ||||||||||| |||| |||| |||||||| || |||||| ||| |||||| |||| |||| ||||||||| ||||| || ||||||| ||| |||||||| |||| ||| |||||||| || ||| ||||||| |||||||| ||||| ||||||.
Populations
Patients from parent trials: Patients were included if they received treatment in 1 of the parent trials and adequately completed the study treatments and last patient visit. Patients were excluded if, during their participation in the parent trial, they developed an SAE deemed to be related to lebrikizumab, developed an AE that was deemed to be related to lebrikizumab and led to study treatment discontinuation, or had conditions in the parent trial that led to investigator-initiated or sponsor-initiated withdrawal from the study.
Direct-entry patients: Inclusion and exclusion criteria for direct entry into the ADjoin study |||||||| ||||||||||||| || | |||||| |||||||||||| |||| |||| |||||||||| |||| ||||| ||| ||| |||||| |||||| |||| ||||||||||| |||||||| || ||||||| ||||||||||||| |||||||| ||||| ||| |||| |||||||| | |||| || |||||||||||| || ||| ||||| |||||||| |||||| || |||||||||| ||||||||| ||||||||| ||||||||| |||| ||| |||||||| |||||||| ||||||||| |||||||||| |||||||||||| || |||||| || |||||| || ||||| ||| ||| |||||||||| ||| ||||||||||| || ||||||||||||||| |||||||||||| ||||| ||||||||| |||||||||
The main cohort in the ADjoin study included all patients who entered through the parent trials or directly, and who received at least 1 dose of lebrikizumab. The responder cohort, a subset of the main cohort, included treated patients from the ADhere study who achieved a response (EASI-75 or IGA 0 or1) at week 16 without receiving rescue therapy. ||| |||||||||||| ||||||||||| | |||||| || ||| |||||| |||| ||||||| |||||||| ||| |||||||| ||| |||||||| || ||||| | |||| || |||||||||||| |||| || |||||||||||| || |||||||| ||||
Interventions
Patients received lebrikizumab for up to 100 weeks. Patients in the ADjoin main cohort were assigned to either blinded lebrikizumab 250 mg SC every 2 weeks or lebrikizumab 250 mg SC every 4 weeks, based on the group to which they were randomly assigned for the maintenance blinded period of the parent trial. |||||||||||| ||||||| ||||| |||| |||||||||||| || |||||||| ||| |||||||| ||||||| || ||| |||||| ||||| || ||| |||| || |||||||||| ||| || |||| || |||||||| || ||| || |||||||||||| ||||| | |||||. Placebo injections were used to maintain the blinding of loading doses and every-4-weeks dosing. Patients who moved to the escape arm in the parent trials continued to receive open-label 250 mg lebrikizumab every 2 weeks.
Direct-entry patients were assigned to ||| || |||||||||||| |||||||||||| || |||||||| ||| |||| | |||||||| ||||| |||||||| || ||| || |||||||||||| ||||| | |||||| Addendum 2.2 patients (enrolled under a modified study protocol) used an autoinjector for 1 full visit interval (starting on or after visit 3; lebrikizumab 250 mg every 2 weeks administered with a 2 mL injection of 125 mg/mL).
To evaluate lebrikizumab maintenance efficacy in combination with TCS, patients from the 16-week ADhere study, who were lebrikizumab plus TCS responders at week 16, |||| |||||||| |||||||| ||| || ||||||| ||| || |||||||||||| ||||| | ||||| || ||||| | |||||
||| ||| || ||||||||||| ||||||||||| ||| ||||| ||||||| |||||||||| |||||| ||||||||||||| ||||||||| ||||| ||||||||||| || ||||||||| |||||| |||| |||||| |||||||||||| ||| || ||||||| |||||| ||||||||||| |||||| |||| ||||| ||| |||| ||||||||||| ||| || || ||||||||| ||| ||| ||||||||| || ||||||| |||||| |||||| ||| ||||||
Outcomes
The primary end point of the ADjoin study was the proportion of patients who discontinued the study treatment due to AEs through the last treatment visit. The secondary end points are the proportion of patients with a response of IGA 0 or 1 at each visit and the proportion of patients achieving a response of EASI-75 from baseline of the parent trial at each visit. Additional secondary end points were the percentage change from baseline in EASI total score (EASI-90, |||| ||), the Pruritus NRS percentage change from baseline, a Pruritus NRS 4-point improvement, ||| |||||| |||| ||||||||| |||||||||| ||||| |||||||||| |||||| |||| ||||||||| |||||||||| ||||||| |||||||||||| ||| |||| |||||||||| |||||| |||| |||||||||
Efficacy end points were evaluated in a subset of patients who were responders in the parent trials. As a result, efficacy outcomes were assessed during 2 periods, depending on which parent trial the patient had been enrolled in. For patients from the ADvocate 1 and ADvocate 2 studies, efficacy outcomes were assessed during the maintenance period of the parent studies (week 16 to week 52) and then for 52 weeks in the ADjoin study (week 52 to week 104). For patients from the ADhere study, efficacy outcomes were assessed for up to 88 weeks in the ADjoin study (week 16 to week 104). The evaluation of efficacy in the interim report was conducted on a subset of the main cohort, which included 86 patients who were responders to lebrikizumab plus TCS in the ADhere study.
Statistical Analysis
Missing data were imputed for the efficacy end points using MCMC-MI and the observation carried forward methods. For the MCMC-MI method, ||| |||| ||||||||| ||||| ||||||||| ||||||||||||||| ||| || |||| || |||||||| |||| ||| || ||| ||||||||| |||||||| |||||| ||| |||| ||||||||| ||||| ||||||||| ||||||||||||||| ||| || ||||| ||||||| |||| ||| || |||||||| ||||||| ||| |||| |||| || |||||| ||| ||||||| |||||| ||| |||| |||||| ||||||||||| |||||||||| ||| |||| ||||||||| ||||| ||||||||| ||||||||||||||| |||| ||| || |||||||||| ||| |||| |||| || |||||| ||| ||||||| ||||| |||||||| |||| ||||| || |||||||||| ||| ||||||| ||||| |||| |||| ||| ||| |||||||| || |||| |||||||| |||||||| |||| ||||||||| ||||||||||||| ||||||| |||||||||| ||| |||||||| |||||||||||.
The statistical methods for analyzing outcomes were consistent with the methods used in the parent trials. The modified ITT analysis set included all patients assigned to treatment, excluding patients from 1 study site who were excluded due to ||| |||||||| || |||||||| |||| ||||||. Patients were analyzed in the groups to which they were assigned. The modified safety analysis set included all patients who received at least 1 dose of lebrikizumab, excluding patients from the 1 study site.
The primary estimand for the ADjoin study was the proportion of modified safety population patients who discontinued the study treatment due to AEs through the last treatment visit of the reporting period. The secondary estimand for the ADjoin study was the proportion of modified ITT (mITT) for patients who met the clinical requirements for response and who did not discontinue due to lack of efficacy.
Results
Patient Disposition
Main Cohort
A total of ||| patients entered the ADjoin study from the ADvocate 1 and ADvocate 2 studies, ||| from the ADhere study, ||| from the ADore study, and || through direct entry. Of ||| randomized patients (ITT), || were excluded due to a critical audit finding at 1 site, resulting in ||| patients in the mITT population (Figure 2). In the main cohort, the rates of discontinuation were |||| in the lebrikizumab every-4-weeks group and ||||| in the lebrikizumab every-2-weeks group. ||| |||| |||||| ||||||| ||| ||||||||||||||| || |||| |||||| ||| ||| || ||| || |||||||||| || ||| ||||||||
ADhere Responder Cohort
A total of || patients from the ADhere study met the treatment response criteria and continued in the ADjoin study. Patients were rerandomized to lebrikizumab every 2 weeks or lebrikizumab every 4 weeks (Figure 3). One patient who was a nonresponder in the ADhere study at week 16 (i.e., did not achieve an IGA 0 or1 or EASI-75 response) was erroneously noted as a responder and rerandomized at baseline of the ADjoin study with the ADhere responder group, and received lebrikizumab 250 mg every 2 weeks. This patient is removed from the analysis.
Among the responder subpopulation from the ADhere study, ||||| of patients in the lebrikizumab every-4-weeks group and ||||| in the lebrikizumab every-2-weeks group discontinued treatment. ||| |||| |||||| ||||||| ||| ||||||||||||||| |||| |||| || |||||| || ||| |||||||||| || ||| ||||||| || ||| |||||||||||| ||||| | ||||| ||||| ||| |||| || |||||||| || ||| |||||||||||| ||||| | ||||| ||||||
Figure 2: Patient Disposition for the ADjoin Main Cohort (Data Cut-Off of July 6, 2022) [Redacted]

ITT = intention to treat; mITT = modified intention to treat; N = number of patients in the group; n = number of patients in the specified category; Q2W = every 2 weeks; Q4W = every 4 weeks. ||||| | ||||||||||| || ||| |||||||||||| ||| ||||| ||| ||||||||| ||||| |||||
Source: CSR for ADjoin.23
Figure 3: Patient Disposition for ADhere Responders (Data Cut-Off of July 6, 2022) [Redacted]

N = number of patients in the group; n = number of patients in the specified category; Q2W = every 2 weeks; Q4W = every 4 weeks.
Source: CSR for ADjoin.23
Baseline Characteristics
Table 31 outlines the baseline disease and demographic characteristics of patients enrolled in the ADjoin study, at the parent trial baseline, based on the interim CSR.
Table 31: Patient Characteristics in the ADjoin Study (mITT Population, Data Cut-Off of July 6, 2022)
Characteristics | ADjoin (main cohort) | ADhere responders who enrolled in ADjoin | ||
|---|---|---|---|---|
LEB 250 mg every 4 weeks (N = 141) | LEB 250 mg every 2 weeks (N = 838) | LEB 250 mg every 4 weeks (N = 29) | LEB 250 mg every 2 weeks (N = 57) | |
Demographic | ||||
Age, years, mean (SD) | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
Adolescents (12 to < 18 years; ≥ 40 kg), n (%) | || |||||| | ||| |||||| | || |||||| | || |||||| |
Adults (≥ 18 years) n (%) | ||| |||||| | ||| |||||| | || |||||| | || |||||| |
Female, n (%) | || |||||| | ||| |||||| | || |||||| | || |||||| |
Male, n (%) | || |||||| | ||| |||||| | || |||||| | || |||||| |
Race, n (%) | ||||
American Indian or Alaska Native | ||||| | ||||| | ||||| | ||||| |
Asian | ||||| | ||||| | ||||| | ||||| |
Black or African American | ||||| | ||||| | ||||| | ||||| |
Native Hawaiian or Other Pacific Islander | ||||| | ||||| | ||||| | ||||| |
White | ||||| | ||||| | ||||| | ||||| |
Multiple | ||||| | ||||| | ||||| | ||||| |
Other | ||||| | ||||| | ||||| | ||||| |
Not reported | ||||| | ||||| | ||||| | ||||| |
Duration since AD onset, years, mean (SD) | ||||| | ||||| | ||||| | ||||| |
Weight, kg, mean (SD) | ||||| | ||||| | ||||| | ||||| |
Weight, kg, n (%) | ||||| | ||||| | ||||| | ||||| |
< 60 kg, n (%) | ||||| | ||||| | ||||| | ||||| |
≥ 60 kg to < 100 kg, n (%) | ||||| | ||||| | ||||| | ||||| |
≥ 100 kg, n (%) | ||||| | ||||| | ||||| | ||||| |
Clinical characteristics | ||||
IGA score of 3 (moderate), n (%) | ||||| | ||||| | ||||| | ||||| |
IGA score of 4 (severe), n (%) | ||||| | ||||| | ||||| | ||||| |
EASI, mean (SD) | ||||| | ||||| | ||||| | ||||| |
BSA, mean (SD) | ||||| | ||||| | ||||| | ||||| |
Pruritus NRS, mean (SD) | ||||| | ||||| | ||||| | ||||| |
Pruritus NRS ≥ 4, n (%) | ||||| | ||||| | ||||| | ||||| |
POEM, mean (SD) | ||||| | ||||| | ||||| | ||||| |
AD = atopic dermatitis; BSA = body surface area; EASI = Eczema Area and Severity Index; IGA = Investigator Global Assessment; LEB = lebrikizumab; mITT = modified intention to treat; NRS = Numeric Rating Scale; POEM = Patient-Oriented Eczema Measure; SD = standard deviation.
Source: CSR for ADjoin.23
Exposure to Study Treatments
In the main cohort (modified safety population), patients were exposed to lebrikizumab every 4 weeks for a mean ||||| |||| |||||| |||| and to lebrikizumab every 2 weeks for a mean of ||||| |||| |||||| days. In the lebrikizumab every-4-weeks group, |||| |||||||||| ||||| ||||||||| ||| ||||| |||||||| patients had ||| ||||| |||| ||||| ||| |||| |||| of exposure, respectively. In the lebrikizumab every-2-weeks group, ||||| |||||||||| ||||| |||||||||| ||| ||||| ||||||||| patients had at least || ||||| |||| ||||| ||| |||| days of exposure, respectively.
In the main cohort, ||| of patients in the lebrikizumab every-4-weeks group and ||| of patients in the lebrikizumab every-2-weeks group adhered to the treatment (defined as receiving ≥ 75% of the expected number of injections). In the ADhere responders cohort, ||| and ||| of patients adhered to the treatment in the lebrikizumab every-4-weeks group and the lebrikizumab every-2-weeks group, respectively.
Concomitant Medications and Cointerventions
Common concomitant AD treatments (used in ≥ 10% of patients) were TCS in the main cohort and ADhere responders cohort. Because the ADjoin study is a long-term extension of the ADhere protocol, a large proportion (||||| in the lebrikizumab 250 mg every-4-weeks group, and ||||| in the lebrikizumab 250 mg every-2-weeks group) of ADhere responder patients recorded concomitant TCS use (Table 32).
Table 32: Summary of Concomitant AD Therapy in the ADjoin Study (mITT Population, Data Cut-Off of July 6, 2022)
Exposure, n (%) | ADjoin (main cohort) | ADjoin (ADhere responders) | ||
|---|---|---|---|---|
LEB 250 mg every 4 weeks (N = 141) | LEB 250 mg every 2 weeks (N = 838) | LEB 250 mg every 4 weeks (N = 29) | LEB 250 mg every 2 weeks (N = 57) | |
Patients with ≥ 1 concomitant AD medications | || |||||| | ||| |||||| | || |||||| | || |||||| |
Topical treatment | || |||||| | ||| |||||| | || |||||| | || |||||| |
Topical corticosteroids | || |||||| | ||| |||||| | || |||||| | || |||||| |
Low mid potency | || |||||| | ||| |||||| | || |||||| | || |||||| |
High potency | ||||| | ||||| | ||||| | ||||| |
Topical calcineurin inhibitors | ||||| | ||||| | ||||| | ||||| |
Systemic treatment | ||||| | ||||| | ||||| | ||||| |
Systemic corticosteroids | ||||| | ||||| | ||||| | ||||| |
Immunosuppressants | ||||| | ||||| | ||||| | ||||| |
Biologics | ||||| | ||||| | ||||| | ||||| |
Phototherapy | ||||| | ||||| | ||||| | ||||| |
AD = atopic dermatitis; LEB = lebrikizumab; mITT = modified intent to treat.
Source: CSR for ADjoin.23
Rescue Therapy Use
||| |||| |||||||||| |||| |||||| ||||||||| ||||| |||||||| || ||| |||||| |||||||||| |||||| ||| |||| ||||||| ||| || ||| |||||||||||| ||| || ||||| | ||||| |||||| ||| |||||||| |||||| ||||||| || ||| |||||||||||| ||||| | ||||| ||||| |||||| ||||
Table 33: Summary of AD Rescue Therapy for ADhere Responders (mITT Population, Data Cut-Off of July 6, 2022)
Rescue therapy, n (%) | LEB 250 mg every 4 weeks (N = 29) | LEB 250 mg every 2 weeks (N = 57) |
|---|---|---|
Any rescue therapya | ||||| | ||||| |
High-potency TCS | ||||| | ||||| |
Systemic rescue therapyb | ||||| | ||||| |
AD = atopic dermatitis; LEB = lebrikizumab; mITT = modified intention to treat; TCS = topical corticosteroids.
a||| |||||| ||||||| |||||||| |||| ||||||| ||| ||| |||||||| |||||| ||||||||
b|||||||| |||||| ||||||| |||||||| |||||||| |||||||||||||||| |||||||||||||||||| |||||||||| ||||||||||||| ||| ||||||||||||||||||
Source: CSR for ADjoin.23
Efficacy
Efficacy outcomes were assessed for up to 88 weeks (week 16 to week 104). |||||||||| || |||||||| || ||| ||||||| |||||| ||| ||||||||| || | |||||| || ||| |||| ||||||| ||||| |||||||| || |||||||||||| ||| |||| |||||||||| || |||||||||||| |||| ||| || ||||||| |||||||| |||| || |||| || ||| ||| |||||| || |||||||||||| ||| ||||||||| ||| ||||||| ||||| ||| ||||||||| || ||||| |||
Harms
|||||||| || ||||||||| || || ||| |||||||| |||||| ||||||||||| |||||||||||| ||||| ||||||||| ||| || |||| ||||||||||||||| ||| || || || ||| ||||| || || |||||| || ||| ||||| |||||||| ||| || | |||||| || ||| |||||||||| |||||
||| ||||| ||| || ||||||| |||||| |||||||| || ||| |||||||||||| ||| || ||||| | ||||| ||||||
The most frequently reported TEAEs |||| || ||| |||||||||| ||| |||||||||||| |||||| ||||| ||||| | ||||||||||||||| ||||||| |||| ||||| || |||||||||||| ||| || ||||| | ||||| ||||| ||| |||| || ||| |||||||||||| ||| || ||||| | ||||| |||||| ||| ||||||||||||||| ||| || |||||||||||| ||| || ||||| | ||||| ||||| ||| |||| || ||| |||||||||||| ||| || ||||| | ||||| |||||| ||||| ||| |||| |||||| |||||||||| |||||||||| || |||||||| || ||| |||||||||||| ||| || ||||| | ||||| ||||| |||||| ||| ||| |||||||||||| ||| || ||||| | ||||| ||||| |||||| |||||||| || || || |||||| |||||||||| ||||||||||||. The proportion of patients experiencing 1 or more AEs in the conjunctivitis cluster (|||||| |||||) was similar in the lebrikizumab 250 mg every-4-weeks group |||||| and the lebrikizumab 250 mg every-2-weeks group |||||||
The proportion of patients reporting 1 or more SAEs was similar in the 2 treatment groups (Table 35).
Table 34: Summary of the Results of Secondary End Points Among ADhere Responders (mITT Population, Data Cut-Off of July 6, 2022)
Efficacy end points at ADjoin week 40 (week 56 from ADhere baseline)a | LEB 250 mg every 4 weeks (N = 29) | LEB 250 mg every 2 weeks (N = 57) |
|---|---|---|
Proportion of patients with a response of IGA 0 or 1 | ||
Response, n (%) | || |||||| | || |||||| |
95% CIb | |||||| ||||| | |||||| ||||| |
Percent change from baseline in EASI score | ||
Mean (SE) | ||||| ||||| | ||||| ||||| |
95% CIb | ||||||| |||||| | ||||||| |||||| |
Proportion of patients achieving a response of EASI-75 | ||
Response, n (%) | || |||||| | || |||||| |
95% CIb | |||||| ||||| | |||||| ||||| |
Percent change from baseline in Pruritus NRS score | ||
Mean (SE) | ||||| ||||| | ||||| ||||| |
95% CIb | ||||||| |||||| | ||||||| |||||| |
Percent of patients with a Pruritus NRS 4-point improvement from baseline and a baseline score of ≥ 4 pointsc | ||
Response, n (%) | || |||||| | || |||||| |
95% CIb | |||||| ||||| | |||||| ||||| |
Percent change from baseline in POEM scores | ||
Mean (SE) | ||||| |||||| | ||||| |||||| |
CI = confidence interval; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% reduction in EASI score; IGA = Investigator Global Assessment; LEB = lebrikizumab; mITT = modified intention to treat; NRS = Numeric Rating Scale; POEM = Patient-Oriented Eczema Measure; SE = standard error.
a||||||||||| |||||||| |||| |||||| ||||||||
b|||||||||| ||||||||| ||| |||||||||| ||||| |||||||||| |||||| ||||||| |||||||||| ||||||||||| |||| || |||||| ||||||||||||| || ||| |||||||| ||||||||||||||||||||| ||| |||||| |||||||||||||| || ||| |||| |||||||||| |||| |||||||| |||||||| ||| ||||| || || ||||| || ||| ||| || |||||| | ||| ||| ||| || |||||| | ||| ||| ||| | || |||||| ||| ||| ||||||||| |||||| ||||| ||||||| |||| |||| ||||||| ||||| ||||||| ||||||| ||| |||| |||||||| |||||| |||| |||||||||||
Source: CSR for ADjoin.23
Table 35: Summary of Harms (Main Cohort Modified Safety Population, Data Cut-Off of July 6, 2022)
Adverse events, n (%) | LEB 250 mg every 4 weeks (N = 141) | LEB 250 mg every 2 weeks (N = 838) |
|---|---|---|
Overview of AEs | ||
All TEAEs | ||||| | ||||| |
Deaths | ||||| | ||||| |
SAEs | ||||| | ||||| |
AEs leading to discontinuation of the study treatment (including death) | ||||| | ||||| |
AEs leading to permanent discontinuation from the study treatment | ||
Patients with ≥ 1 AE leading to permanent discontinuation of the study treatment | ||||| | ||||| |
|||| ||| |||||||||||| |||||| ||||||||| | ||||| | ||||| |
|||||||||| |||||||| | ||||| | ||||| |
|||||||||| |||||| | ||||| | ||||| |
||||||||| | ||||| | ||||| |
|||||||||| ||| |||||||||||| | ||||| | ||||| |
|||||||||||||| | ||||| | ||||| |
||| ||||||||| | ||||| | ||||| |
|||||||||||||| |||||||| | ||||| | ||||| |
|||||| |||||| ||||||||| | ||||| | ||||| |
|||||||||||||||| | ||||| | ||||| |
Most commonly reported (≥ 1% in either group) TEAEs | ||
|||||||||| ||| |||||||||||| | ||||| | ||||| |
|||||||| | ||||| | ||||| |
||||||||||||||| | ||||| | ||||| |
|||||||||||||| | ||||| | ||||| |
||||| ||||||||||| ||||| ||||||||| | ||||| | ||||| |
||||||| ||||| ||||||||| | ||||| | ||||| |
|||| |||||| | ||||| | ||||| |
|||| ||| |||||||||||| |||||| ||||||||| | ||||| | ||||| |
|||||||||| |||||| | ||||| | ||||| |
||| ||||||||| | ||||| | ||||| |
|||||||||||||| |||||||| | ||||| | ||||| |
||||||| |||||| ||||||||| | ||||| | ||||| |
|||||||| | ||||| | ||||| |
||||||| ||||||||| ||| |||||||||||||| |||| |||||||||| | ||||| | ||||| |
||||||||| |||| ||||||||| | ||||| | ||||| |
SAEs | ||
Patients with ≥ 1 SAE | ||||| | ||||| |
AEs of special interest | ||
|||||||||||||| ||||||| |||||| | ||||| | ||||| |
|||||||||||||| | ||||| | ||||| |
|||||||||||||| |||||||| | ||||| | ||||| |
|||||||||||||| ||||||||| | ||||| | ||||| |
||||||||| ||||||| | ||||| | ||||| |
|||||| |||||||||||||||||||| | ||||| | ||||| |
||||||||| | ||||| | ||||| |
|||||||||||| |||| || || ||||||||| ||||||||||||| |||||||||| ||||||| | ||||| | ||||| |
|||||| ||||||| ||||||||| ||||||| ||||| | ||||| | ||||| |
|||||| ||||||| | ||||| | ||||| |
|||||| ||||| ||||||||| | ||||| | ||||| |
|||||| |||||| |||| ||||| | ||||| | ||||| |
AE = adverse event; LEB = lebrikizumab; PT = preferred term; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SAE = serious adverse event; TEAE = treatment-emergent adverse event; TE = treatment emergent.
Sources: CSR for ADjoin,23 Sponsor’s Summary of Clinical Evidence.35
Critical Appraisal
Internal Validity
There is no randomized comparison to another treatment or a placebo, which limits the ability to draw inferences on the effects of lebrikizumab in the study population. The patients were aware they were receiving active treatment, although the dosage received was blinded. Thus, their expectations of treatment may have influenced the reporting of subjective patient-reported outcomes, such as the POEM, and subjective AEs or investigator-reported IGA and EASI scores.
Discontinuation rates were |||| in the lebrikizumab every-4-weeks group and ||||| in the lebrikizumab every-2-weeks group. Among patients from the ADhere study (efficacy assessment), the rates of discontinuation were ||||| in the lebrikizumab every-4-weeks group and ||||| in the lebrikizumab every-2-weeks group. Thus, there is potential bias due to missing data.
All analyses were conducted descriptively, without statistical comparisons between the cohorts.
External Validity
Only responders in the ADhere study were included in the efficacy assessment. Patients were excluded if, during their participation in the parent trial, they developed an SAE deemed to be related to lebrikizumab, developed an AE deemed to be related to lebrikizumab that led to study treatment discontinuation, or had conditions in the parent study that led to investigator-initiated or sponsor-initiated withdrawal from the study. Therefore, the long-term treatment effects apply only to patients who previously responded to lebrikizumab and tolerated it well enough that they did not need to discontinue its use during the parent trial. This is a select population and a generalizability issue. Among ADhere responders, a higher proportion of patients in the lebrikizumab every-4-weeks group than in the lebrikizumab every-2-weeks group recorded using concomitant TCS and systemic rescue therapy. The effect of these differences on the efficacy results remains uncertain.
Indirect Evidence
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
Multiple placebo-controlled, phase III clinical trials have been conducted to determine the safety and efficacy of lebrikizumab in patients with AD. To support health-technology assessment submissions and to inform the cost-effectiveness model, the sponsor conducted an NMA to establish the relative efficacy and safety of lebrikizumab compared to key comparators in the treatment of adults and adolescents with moderate-to-severe AD.24
||||||||||| || ||||||||||||||||| ||| |||| ||||||| ||||||||| || ||| || |||||||| |||||||| ||| ||||||||| || || |||| ||| |||||||| |||||||| || |||||||||||| ||||| || ||||||| |||| |||| ||| |||||||| || |||||||| || |||||||||||| ||||||| ||||||||| ||||||||| ||| ||||||||| |||||| ||| |||||||||| |||||||| || ||||||||||| |||||||||| ||| || ||| |||||||| |||| ||||||||||| || |||||||| ||| ||| ||||||||||||||||| ||| |||||||| |||||||||||| |||||||||| ||| ||||||||||||| ||| |||||||| || ||| ||||||| ||||||||| ||| |||| |||||||| ||||||||||| ||| |||||||||||| || |||||||||||||||||| ||||||||||| |||| ||| |||| |||||| |||||| |||||||| ||| ||| ||||||||| || ||| ||| |||||||||||| |||||||| | |||||||||||||| || ||| ||| |||||||||| |||| ||||| ||| || ||| ||||||||| |||||| || ||||||| || ||||| ||||||| ||||||||| |||||||||||| || ||||||||||| || |||||||||||| |||| ||| |||||||| || |||| ||||||| |||||||| || |||||||| |||||||| |||| ||||||||| ||| ||| ||||||||| ||| ||||||| |||| || ||||| || ||||||| ||||||||| || ||||||||||||| ||| || |||| ||| ||| ||||||| |||| || ||||| || ||||||| ||||||||| || ||||||||||||| ||| || |||| |||||||||||| || ||| ||||||||||||||||| |||||||||||||||||| ||||||||| || ||| ||||||||||||||||| ||| ||| || ||||||| ||||||||| || ||| |||||||| |||||||| || |||||||||||| || |||| ||||||||||| ||| ||||||||||| ||||||| ||| ||| ||||||||| ||||||||| |||||| ||||||||| || ||||| || || |||| |||||||| |||| |||||||| |||||||| || |||||||| |||||||||| || |||||| ||| |||||||||| ||||||||||| |||| |||||||||||||||||| ||| ||||||||| |||||||||| || ||| ||||||||| ||| |||| || |||||| ||| ||||||||||| || ||| ||||||| ||| |||||||||||| || ||||||||||| || ||||||||||| |||||||| || |||| ||||| ||| ||||||| ||| || |||||| ||| ||||||| || ||| |||||||| ||||||||| ||| |||||||| |||||||| || |||| ||| ||||||||||| ||| ||||||||||| ||||||| ||||||||||||||||| ||||||||| ||||||||||| ||||||| ||||||||| ||| ||| |||||||| || || ||| || |||||||| |||||||| |||| ||||||||| || |||||||| |||| |||||||||||||||||| ||| ||| ||||||| ||| ||| ||||||||| ||||||||| || |||||||| ||||| ||| |||||||||||| ||||||| || ||| ||||| |||||||| ||||| ||| ||||| ||||||||||||||| ||| |||||||||||||| || ||||||||| |||||||| |||||||| || ||| |||||||| |||||||| ||||||| ||| ||||||| ||| ||||||| ||||||||||| |||||||||| |||| |||||||||| ||||||| ||||||||| |||||||||| |||||||||||| |||||||| ||||| |||||||||| ||| ||| |||||||| |||| |||| |||||||| || |||||||| |||||||||| |||||||||| ||||||||| || ||||||| |||||||| |||||| ||| ||||||||| |||||||||||| ||||||||| ||||||||| ||||| || |||||||||| |||||||||| ||||||| ||| ||||||||||||| |||| |||| |||||||| ||| ||||||| ||||||| || ||||||||| ||||||||| |||| ||||| |||||||| || |||||| ||| ||||||||| ||| |||| || ||||||||| ||||| |||||||||| |||| ||||| ||||| ||| ||||||||| |||||||| ||||||||| || ||||| || || ||||||||| |||||||||||| ||||||||| |||| |||||||||| ||||||| ||||||||||| |||| | ||||| ||||||||||| |||||||| || || ||||||||| ||| ||| |||||||| |||| ||| ||||||||| |||| ||||||||| |||||||||||| |||||||||| ||| ||||||| |||| ||| |||| |||||| || |||||||| || |||||| ||| ||| |||||||| |||||| ||||||||| |||| || ||||| |||||||||||| || |||||||| |||| |||| |||||||| || |||||||||||| || ||||| |||| ||||| ||| |||| |||| |||| |||||||||||||| |||| ||| ||||||| ||||| |||||||||| ||||||||| ||||||| ||||||| |||||||||| |||||||| |||||||||||| || ||| ||||||||| |||| |||| |||||||| ||||||| || | |||||||||| ||| ||| ||| ||||||| ||| ||||||| |||| ||||||||||| ||| |||| || |||| ||| |||||||| ||| |||| |||||||| ||||| ||||| ||| |||||||| |||| || |||| |||||||||| |||||||||||||| ||| |||||||||| ||| |||| || |||| |||||||||| |||| ||| ||||||||||||||| || |||||||||| ||| ||||||||||| ||||| ||||||||| |||||||| ||| ||| ||| || ||| ||||||||||||||||| |||| ||| ||| ||| ||||||| || ||||| |||| ||| |||| |||||| |||||||||| |||| ||||||| |||| ||||||||| |||||||| || ||||||||||||| || |||||||||| |||| |||| ||| || |||||||| ||| ||| |||| ||| |||||||| ||||||| |||||||||| ||| ||| ||| |||||||| ||||||||||| ||| |||||| |||| ||| ||||||||||||| || |||||||| || ||| ||| |||||||| |||||||||||| ||| || ||||| | |||||| ||||||||||| ||| || ||||| || ||| || |||||| ||||||||||| | || ||||| || | || |||||| ||||||||| ||| || ||||| | |||||| |||||||||||| ||| || ||||| | |||||| ||| |||||||||||| || || ||||| || || || |||||| ||| |||||||| ||| ||| ||| |||| |||| ||| || ||| ||||||||||| || |||| ||||| |||| |||||||| || |||| ||| ||| ||||| || | || | || |||| ||| ||| | ||||||| ||||||||| || ||||||||||||| ||| || |||| | ||| |||| ||| ||| |||||||| ||||| ||| |||| |||||||||||| |||| ||| |||| || |||| ||||||||||| ||| ||||||||||| ||||||| |||||||| || ||| ||||||||||| ||||||| |||||||| ||||| |||||||| ||||| |||| ||||| ||||||||| |||||||||||| ||| ||||| ||||||||| |||||||| ||| ||||||| ||| ||| ||| ||||||||| ||| |||
||||||||| || | ||||||||| |||||||| |
|---|---|
|||||||||| | |||||| ||||||||||| || ||||||||| |||||||| |||| |||||||||||||||||| || |
|||||||||||| | |||||||||||| |
|||||||||| | |||||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||||| |||||||||| |||||||||| |||||| ||||||||||||| ||||||||| |||||||||||| |||||||| ||||||||||||| |||||||||||| ||||||||||||| ||||||||||||| ||||||||||||| ||| || ||||| ||||||||||||| || |||||||| ||||| || || ||||||||||| |
||||||| | ||||||| ||||||||||||||||| || |||||||||||| ||||||||| ||| ||| ||||||||||||||||| || |||||||||||| ||||||||| ||||||| ||| |||||||| |||||||| || |||||||||||| ||||||||| |||| |||| || ||| ||||||||||| || |||||||||||| |||||| |||| |||||||| || |||| ||||| |||||||| || |||||||||||| ||||||||| |||| || ||| || ||||| | ||||||| ||||||||| || ||||||||||||||| || |||||||||||| ||||||||| |||| ||| ||||||||||||||| |||| |||||||| || |||| ||||| |||||||| |||||| |||| |||||||| || ||||||||||||| |||| |||||||| || ||||| ||||||| |||| |||||||| || |||| ||||||| |||| |||||||| || ||||||||||||| || |||||||| ||||||||| |||| |||| ||| ||| ||||||||||| || |||||| ||||||| |||| |||||||| || |||| |||||||| |||||| |||| |||||||| || |||||||| ||| |||||||| || |||||||| ||||||||||| ||||||||| |||| |||||||| || ||||||||||||| ||| |||||||||||||| || |||||||| ||||||||||| ||||||||| |||| |||||||| || |||| |||||||| ||| |||||||||||||||| ||||||| |||| |||||| ||||||||||||||||||| |||| || |||| || |||||||| || ||| || |||| |
||||| |||||| | ||||||||||| ||||||||||| ||||||||||| |||||||| ||||||||||||||||| |||||||| ||||||||| ||||||||||||| |
||||||||||| ||||||||||||||| | || |||| ||||||||||| ||| ||||||||| ||||||||||||||||||| ||||||||| ||||||||| |||| |||||||| ||||||||||||| ||||||||| |
|| | |||||| ||||||||||| || | ||||||| |||||| ||||| | |||||||||| ||||||||||| |||| ||||||| |||||| |||| | ||||||||||| |||| ||||||| |||||| |||| | |||||| |||| ||| |||||||| ||||||||||| |||| | |||||||| ||||||| ||| |||||||||| |||||| ||| | |||||||||||| |||||| ||||||||||| ||| | ||||||| |||||| |||||| |||| | |||||||||||||||| |||||| |||||||| ||| | |||||||||||||||| |||||||| |||||| | ||||||| |||||| ||||||||||| ||||||| | ||||||||| |||||||||||| |||||| |||||||||| ||| |||||| |||||||||||||||||||||||||||||| ||||||| ||| ||||||| || |||||||||| |||||||| ||||||| || ||||||| |||||||||||| ||| |||||||| |||| |||||| |||||||||| ||||||||| | ||||| || || ||||| ||||||||||||| |||| ||| |||||| |||||||||| ||||||||| | ||||| || || ||||| ||| || |||| || ||| ||||||||||| ||| |||||||||||| |||||||| ||| ||||| || ||||||||||||||||| ||||||||||||||||| ||| |||||||||||| ||||||||||||||||||| ||||||||||||||| |||||||||| || ||| |||| ||| ||||||||||| || |||||||||| |||| ||| ||||||||||| || |||||||||||| |||||| ||||||||||||| || |||||||| || ||||||||||| ||| |||||| |||| || ||| |||||||| ||| ||||||||||||| || ||||| ||| |||||||| |||||||||||||||| |||||||||||||| ||||||||||||||| |||||||||||||| |||||||| ||| |||||||||| || ||||||||| ||| ||||||||| ||||||||| |||||| ||||||||| ||||||||| ||||||| ||| |||||||| ||||||||||||||| |||||||||| || || ||||||||| |||||| ||||||||| |||| |||||||||| ||||| || |||||||| |||||||| |||||||| ||||| |||||||||| ||||||||||| |||||||||||| ||||||| |||| |||||||||| || |||||||| ||| ||| |||| |||| |||| ||||||||| |||| |||| || |||| |||| ||| ||||| ||| ||||||||| |||||||| ||| |||||||||| |||| |||| |||| ||||||||| || ||||||| |||||||||||| ||||||| |||| ||| |||||| ||||||||||| || |||||| | ||| |||||| | || || |||| || || ||| || |||| || ||||||||||| ||| |||||||| |||||||||| ||| ||||||| ||||||||||||| ||||||||||| ||||||||||||| ||| ||||| || ||||||||||| |||| ||||||||||||| || |||||| ||||||| ||| ||||| ||||||||| || ||| ||| |||||| ||| ||||||||||| |||||||||||||| ||||||||||| ||||||| ||| ||||||||| || ||||||| |||||||||| |||| ||| ||| || ||| |||| || |||||| || |||||| ||||||| |||| |||||||||| ||| ||||||||| || ||| |||| |||| | |||||||||| ||||||||||| ||||||| || |||||||||||| |||||||| ||| |||||| ||||||| ||||||| | ||||| || || ||||||| |||| |||||||| |||| ||| ||||||||||| ||||||||||| || ||| ||| ||||||||| || |||||||||||| || |||| || ||||||||| | ||||| || ||||| |||||| ||||||||||||||| || ||||||||| ||||||||| | ||||||| ||||||| | ||||||| |||||||| ||| | ||||||| ||| ||||||||| || |||||||| |||||||| || ||||||||||| || |||||| || ||||||| |||| |||||||| ||| ||||||||| || ||||| || ||||||||||| |||||||| ||| || ||||||||||| ||||||| |||||||| ||||| | |||||| ||||||||||| ||| | ||||||| |||||||||||||| ||| | |||||||||| |||||||||| ||||||||||||||| ||||||||||||||||| ||| |||||||||||| ||||||||||| ||| ||||||| ||| ||||||| |||||||||| || ||||| ||| ||| ||||||| |||||||| || ||| ||| ||| ||||||||| || |||||||| ||||||||| ||| |||||||| ||||||| |||| |||||||| || ||||||||||||| |||||||||||| |||||||||||| |||||||||| |||||||||||| ||| ||||||||||||| ||| ||| ||||||||| |||||||||| ||| ||||||||||| ||| ||||||||||| |||||||| ||||||||| |||||||| |||| ||||||||| || ||||||| |||| ||||| ||| ||||||| |||| ||| ||| ||||||||||| |||||||| |||||||||| ||||||||| |||||||| ||||| ||||||| ||||||||||||||| |||||| |||| |||||| |||||||||| || ||| ||||||||||| ||| ||||||||||| ||||||| ||||||| |||||||| |||||||| ||| |||| ||| |||| ||||||||| ||| |||| ||| ||| |||||| || ||||||||| ||| ||| ||||||||| ||||||||| |||||||| |||| |||||||| ||||||||| ||| |||||||| ||| ||| ||||||||||||||| || ||||||||| || |||| ||| ||| |||||| |||| ||||| ||| |||||| ||||||| |||||| |||||| || ||||||||| |||||| || || |||||||||| ||||||||| ||||||||| |||||||| |||||||| |||||||| || |||| |||| ||||| ||| ||| |||||||||| || |||||||| |||| || ||| ||||| || | || |||||||| |||| |||||||| ||| ||| |||||||||||||||| ||||||||| ||| |||||||| ||||||||| || |||| ||| ||| | ||| | |||||||||| ||||||||||||||||||||| |||||||| || ||| ||| |||| |||| ||||| || |||| ||| ||| ||||| || |||| ||| ||| ||||||| || |||| ||||||||| || ||||||||||||| ||| || |||| | ||| |||| ||| ||||||| ||||||||| |||| |||||||| ||||||||| |||||||| ||| ||||||||||| || |||||||| |||| | ||| || |||||||| ||| || |||||||| || ||| || ||||||| |||||| |||| |||||||| || |||| |||||| ||| ||| |||||||| || ||||| ||| |||||||| |||||| || |||||||| |||| ||||||||||| || |||| |||||| |||| |||||||| ||||||| |||||| ||||||| ||||||| ||| ||| || ||||| |||| |||||||||| ||||| |||| |||||||||| ||||||||||||| || ||| |||||| || |||||||| |||||| | ||||| |||| |||||||| ||| |||||||| ||| ||| |||||||||| |||| || ||||| ||| ||||||||||| |||| |||||||||| ||| |||||| || |||||||| |||| ||||||||||| ||| ||||||||||| ||||||||||||| || |||| |||| |||||||| ||| ||||| |||| |||| |||| ||||||||||| |||| | ||||||| |||||| || |||| |||||||||| ||| ||||||||| ||| |||| ||||| || ||| |||||| ||||||||| ||| |||||||| |||||||| ||| ||||||| |||||||| || |||| ||| ||| ||| |||||||| ||||||||| ||| |||||||| |||||| || |||||||| |||| |||||| ||| ||| ||||||||||| |||| |||| || |||||| || ||| |||||| || |||||||| |||| |||||| ||| |||||||| ||| ||| |||||||||| |||| || ||||| ||| ||||||||||| |||| |||||||||| ||| |||||| || |||||||| |||| |||||| ||| ||||||||||| ||||| ||||||| |||||||| |||| |||| |||| || ||||||| ||||||||||||||||| ||||||| |||||||| |||| |||| ||||||| || ||| |||| || |||| |||||||||||||| |||||| ||||||| || ||| ||||||||||||||| ||| ||| ||||||||| || | |||||||| ||||||||| ||| |||| ||||||| ||| ||||||||| |||||||| ||||| |||| ||||| ||| |||||| ||||||| ||||||| ||| ||||| ||| ||| |||||| ||| |||||||| |||| |||||| ||| ||| |||||| ||| ||||||||||| || ||||||| |||||||||| ||| ||| ||||| |||||||| ||| |||||||| ||||| ||| | ||||| ||| |||||| ||||||| |||||| |||| ||| ||| |||||| ||| |||||||| |||| |||| | |||||| |||| |||||||| ||| ||||||||||| |||||||||||| ||| ||||||| |||||||| ||||| |||||||| ||||||| ||| | ||||| |||| |||||||| ||| |||||||| |||||||||||| ||| |||||||| |||||||| |||| ||| |||||||| ||| ||||||||||||| ||| |||||||||| ||| |||||| ||||| ||| |||||| |||||| |||||| |||||||| ||| |||||||| ||||| ||||||||| |||||||||| ||| |||||||||||| || ||| |||||| ||||||| |||||| || ||||||| ||| ||||||||| ||||||||||||| ||||||||| ||| |||||||| |||||||| || |||| ||| ||| ||||| |||||||| |||| ||||| ||| |||||||| |||| ||| |||||||| ||||| || || ||||||||||| ||||||| ||||||| ||| |||||||| |||| ||| ||||||||| ||||||| || ||||||||| || ||| |||||||| |||||||| ||||| ||| ||| ||||||||||| |||||| ||||||||||||||| |||||||||| |||| ||||||||| ||||| | |||||| ||||| ||||| ||||| ||||||| ||| |||| ||||||||| ||||| |||||| |||| ||||| ||| || ||||||| |||||| |||||||||| |||| ||| || | ||||||| |||||| || ||||||| |||||||||||| ||||||| |||||||||| |||| |||||||||| ||| ||| |||||||| ||||||| |||| ||||| || || ||||| | ||||||| |||||| |||||||||| ||| ||||| |||| | |||||||| |||||| || | ||| | ||||| || || ||||| ||||||| ||||||||||| ||||||| |||||||||| |||| ||||||||| || ||||| |||| |||||||| |||| |||||||||||| ||| ||||| ||||| |||||||| |||||| |||| ||||||| || |||||| |||||||||| ||||||||| ||| ||| ||||||| |||||||| || |||| |||||||| ||||| ||| || ||||| |||||||| ||| |||||||| ||||| ||||||||||| ||| |||||||| ||||| ||||||| ||||| |||||| |||||||| |||||| ||||||||| ||||||| ||||| ||| |||||||||||||||||| |||||||||| |||||||||||| |||||||| ||||||||||| ||||||||| |||||| ||||| |||||||| ||||||||| ||| ||||||||| |||||| || |||||||||| |||| ||||||||| ||| ||||||||| ||||| ||| ||||| |||| ||| |||||| ||| ||| ||||||||| || ||||||||||||| ||||| |||||||||| |||| ||||||| |||||| ||| |||| |||||| ||||| |||||||| |||||||| ||||| || ||||||||| ||| ||||||||||||| ||| |||||||||| || ||| |||||| || ||||||||| || |||||||| ||||||||| ||||||| |||||| |||||||| ||| |||| ||||||||| ||||| ||||| ||| || ||||| ||| |||||| ||||||| |||||| |||| ||||||| |||||| | |||||||||| || ||| || |||| |||| ||| |||||| ||||||| |||||| |||| |||||||| ||| |||||||||||||| |||||||| |||||||||| || ||||||||||||| || ||||| ||||||| ||||||| |||||||| ||||||||||||||| ||| ||||||| |||||||| ||| ||||||||| ||| ||| |||||||| ||||||| |||||| ||| ||||||||||| ||||||||||| ||| ||||||||| ||||||||||||| || ||| |||||||| |||| |||||| ||||||| |||||| ||| |||| |||||||| || ||||||| | ||||||| || ||||||||||| |||||||||||||| ||||| |||| ||||||| || |||| ||| |||| ||||||||||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| ||||||||||||| ||| |||||||| ||||||| ||||||||||||| || ||||||| |||||||| ||| ||| |||||||| || ||||| || |||||||| ||| ||| ||||||| ||| ||||||||| ||||||||||||||||| |||||| |||| ||||||||| ||| ||||| ||||| ||||||||||| ||||||||||| |||||| |||| |||| || |||||||| ||| ||||||||||||| || ||| || ||| |||| || ||||||||||| || |||||||||| || |||| ||||||||| |||| |||||||||| | ||||| ||||||||| |||||| ||||| || |||||||||| || ||||||||||||||| ||| ||||| ||||| |||||| |||||||||| |||| ||| |||||||||||| ||||||| || ||| ||| |||| ||||||||| || |||||||| ||||||||| ||||||| |||| ||| |||| ||| ||| |||||||| ||||||||| ||| ||||||||| |||||| || |||||| ||||||||||| ||| ||||| ||| ||| ||||||||| |||||| || ||| ||| |||||||| ||||||||| |||||||| ||||||||| ||||||| ||| ||| |||| ||| ||| |||||||||| |||||||||| |||| ||||||||| || |||||| |||||| ||| |||| |||||||| |||||| ||||| |||| |||| ||||||||| ||| |||| |||||||||| |||||| |||||||| |||||||| |||||||| ||||| ||||| ||||||||| |||||| ||| ||||||||||| || ||||||||| ||| ||| |||||||| ||||||||||||||| ||| ||||||||| ||| ||| |||||||||| |||| ||||| || ||||||||| ||||||||| |||||||| ||||||| |||| |||||||| |||||||| ||||| ||| |||||||| ||| |||||||| ||||||||| |||||||| |||||||| ||||| ||| ||||||| |||| ||||||| ||||| || || ||||||| || ||||||| |||||||| ||||| || ||| |||||||| |||||| ||| ||||| | ||||| ||| ||||||||| || ||| |||||||| ||||||||||| || ||||||||| ||||||| ||| |||||||| || ||| |||| ||| ||| ||||| |||||||||| ||| |||| ||||| |||| ||||||| ||||| || | |||||| ||||||| ||||||||||||| || ||||||| ||||||||| ||| ||||||| ||| |||| ||||||| || ||| |||||| |||||||||||||| ||| ||| |||||||| |||||||
||||||| | ||||||||||||||||| ||| |
|---|---|
||| ||||||| | ||| ||| ||| ||||||||| ||||| | |||||||| ||||||||| ||| |||| |||||||||||| ||| |||||||||||||| |||||| |||| ||||||||||| |
|||||| | ||||| ||| ||||||||||| ||||| ||||||||||||| |||| ||||| |
|||||||||| || ||||| ||| | ||||||||||||||| ||| |||||||| ||||| ||| |||| |||| ||||| |||||| |||||||||| |||||| ||||| |||| |
|||||||||| || ||||||||||| | ||||||||||| ||| |||||||| ||||| || ||| |||||||| ||||||| ||||||||| ||||||||| ||| ||||||||| |
|||||||||| || ||||||||||| | ||||||||||| ||| |||||||| ||||| ||||||| ||||| |||||| |||||||| |||||| ||||||||| ||||||| ||||| ||| |||||||||||||||||| |||||||||| ||||| ||| ||| ||||||| |||||||| || |||| ||||||||| ||||| ||| |||||| ||||||| ||||| |||||||| ||| |||||||| ||||| |
|||||||| | |||| ||| || ||| ||||||||||| || |||| ||||| |||| ||||||||| ||| ||||| || | || || ||| |||||||| ||||||||| || ||||||||||||| ||| |||||||||||||| | |
||||||||| |||||||||| | |||||||| |||| |||||||||||| || |||| ||| |||||| ||| |||| |||| || ||||||||| |||| || |||||||| ||| |||| ||||| |||||||||||| ||| ||||||||||||| ||| |||||||| ||| |||| |||||||||||| || |||| | || | |||||||| |||||||| |
|||||||||||| || ||||| | ||||| |||| ||||||| || |||||||| ||||| ||| |||| |||||||||||| ||||| |||| ||| |||| || |||| ||||||||||| ||| ||||||||||| ||||||| |||||||| || ||| ||||||||||| ||||||| |||||||| ||||| |||||||| ||||| |||| ||||| ||||||||| ||| |||| ||| |||||||| || || |||||||||||| | ||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| |
||||||||||| |||||||| | ||||||| |||||||||||||||| |||| |||| |||||||||| || ||||||||||| |||||||| || |||||| ||| |||||||| |||| ||||| ||| ||| |||||||||| || |||||||| |||| || ||| ||||| || | || ||||||||| ||||| ||||||||||||| |||||| || |||||||| ||||| || ||||||| |||| ||| ||||||||||| |||||||| |||| |||||| |
|||||||| |||||||| | ||| ||||||||||| |
||||||| ||| |||||||| ||||||||||||| | ||||||||||| ||||||| ||| |||||| |||||||| ||| ||||| || ||| |||| || |||| ||||||||||||| ||| |||||| |||||||||||||||||||||||||||| ||||||| ||| ||||||| |||||||| ||| ||||| || ||| |||| || |||||||| |||||||||||||| |||||| |||||| || ||||| ||| |||||| |||||||||||| |
||| | |||||||| ||||||||||| |||||||||| |||| | |||||| |||| ||| |||||||| ||||||||||| ||| | |||||||||||| |||||| ||||||||||| ||| | |||||||| ||||||||| ||||||||||| ||| | ||||||| |||||||||||||| ||| | ||||||| |||||| |||||| || | |||| |||||| ||| | ||||||| |||||||||||||||||||||||| ||||||||||||||||| ||| |||||||||||||||| || ||| ||||||||||||||||| ||||||||||| || |||||||| ||||||| ||| ||||||| || ||| ||||||||||| |||||||||||||| ||| |||||||||| | ||||| || ||||| |||||||||| ||||| ||||||||| ||| |||||||||||| ||| ||||||| |||| |||||||| || ||| |||||||||| ||||||| | ||||| || || |||||| ||||||| |||||||||| || ||| ||| |||| |||||||| ||| ||||||||||| || || |||||||| || ||||| ||||| ||||||| || |||||||||||| |||| ||| |||||||||| || |||| || ||| ||| |||| |||| |||||||| ||| |||||||||| ||||||| |||| ||| ||| |||||| || | |||||||||||| |||| || ||||||||| || || |||||||||||| || |||| ||| |||||||| || ||| ||| |||| ||||||||| ||||||||||||| ||||||| |||| |||| ||| ||||||| ||||||||||| || ||| ||| |||||| |||| || |||||||| || |||||||| |||| ||||||||| || |||||| || ||||||| |||| |||||||| ||| ||||||||| || ||||| || ||||||||||| |||||||| ||| || ||||||||||| ||||||| ||||||||||||||| || |||| |||||||||||| |||| || |||| |||||||||| ||| ||||||||| ||| ||| ||||||| |||||||| || ||| |||| ||| ||||||| |||| |||||||| || |||||| | |||| |||| || |||| || ||| || ||| |||||||||| ||||| |||||||||||||| |||||||| ||||||| |||| |||||||||| || |||||||| |||| ||| |||||| ||| || |||| || ||||||||||| ||| ||| ||| ||| |||| |||| ||||||| || ||| |||||| || |||||||||||||| |||||||||| ||||||||||| ||| ||||||| ||||||||| ||| ||| |||||||| |||| ||||||| |||| |||||||||| || ||| |||| || |||| |||| |||||| || |||||| || ||||||||||||| ||| ||||||||| |||||||||| |||| ||| ||||||| |||| || |||| ||||| ||| ||||||| |||||||| || |||| |||||| ||| |||||||||| |||||||||||||||||||||| |||||||||||||| ||||||||||| || |||||||||| || ||| ||| |||||||| ||| ||||| |||||||||||||||| |||||||| ||| ||||||||||| |||||||||||||| |||||||| |||||||||||||||| ||| ||||||||||||| || ||||||| ||||||||||| ||||||||||||||||||||| || ||||||| |||| |||||||| ||| ||||||||| || |||| ||| ||||||||||| ||||||| ||| || ||||||||||| ||||||| ||||||||| ||| ||||||| |||| |||||| ||||| ||||||| |||||||||| |||| ||| ||||||||| ||||||| |||| ||| ||||| |||||| ||| ||||||||| ||| |||||||| |||||||||||| ||||||| ||||||||| ||||| |||| ||| ||| |||||||||| ||| || ||| ||||||| |||||||| || ||| ||||||||||| ||||||| |||| ||||| || || ||| ||||||| |||||||||| |||||| || |||||||||||| ||||||||| ||||||| ||| |||||||||||||| |||| ||| ||||||||| ||||||| ||||| ||||| ||| |||||||| || ||| ||||||||||| ||||||| |||||||| | ||||| ||| ||||||||| || ||| ||||| || ||||||| |||||||||||||| |||| ||| ||||||||| ||||||| ||||| ||||| ||| |||||||| |||| ||||||||||| ||||||| ||||||| ||||||||||| ||||||||||| |||||| | ||| ||||||| || ||||| |||| ||||||||| || |||||||| |||||||||| ||||||| |||| ||||||||||||| ||||| |||| ||||||| |||||||||| |||| |||| |||||||||| || ||| |||||||| || |||||||| |||||||||| ||||||| |||| ||||||||||||| |||||||||| |||||||||| || || |||||| |||||| ||| ||||| |||| |||||||| || |||||||| ||||||| ||||||||| |||| || ||| ||||||||||||||||| ||||||||| |||||||| || |||| |||| |||||||||| |||| |||| |||||||| ||||||| |||| ||| ||||||| ||| ||| ||||||||||||||||| |||||||| |||||||| || ||| |||||||||| ||||||||||| ||| || |||||||||||| |||||||||| ||||| |||| || ||| ||||||||||||||||| |||||||||| |||| ||| |||||||||| || || || ||||| ||| |||| ||| |||||||||||||||||||||| |||||||| ||| |||||||||| || |||||||| ||| ||| ||| ||||||| |||| ||| ||||||| ||| ||| ||||||||| ||||||||| || ||| |||||||||||| |||||||| |||||| ||| |||||||| ||| |||||||||| |||| ||| || ||| ||||||||||| || |||| ||||| |||| ||||||||| ||| ||||| || | || || ||| |||||||| ||||||||| || ||||||||||||| |||| ||||| |||||||| |||| |||||||||||| || |||| ||| |||||| ||| |||| |||| || ||||||||| |||| || |||||||| ||| |||| ||||| |||||||||||| |||| ||||||| ||||||| |||||||| || ||||||| |||| ||| ||||||||||||| ||| |||||||| ||| |||| |||||||||||| || |||| | || | |||||||| |||||||| ||| ||||||| ||| |||| ||||||||| ||| ||| |||| ||| ||| ||||||||| || ||| ||||||||||| ||| ||||||||||| ||||||| ||||||||| || ||| ||||||||||| ||||||| |||| ||||| ||||||| ||| |||| ||||||||| ||| ||||||||| || ||||||||||||| ||| |||| |||||||| || |||| || ||| ||||| || |||| ||| || ||| ||||||||||| ||||||| |||||||| |||| ||||| ||||||| |||| |||| ||||||||| ||| ||||||||| || ||||||||||||| ||| |||| |||||||| || |||| || ||| ||||| || |||| |||||||||||||||||||| ||||||||||||| || |||||||| || ||| ||| |||| |||||||| |||| ||| |||| |||| ||| |||||||| ||||||| || |||||||| ||||||||| |||||||||||| ||| || ||||| | |||||| ||||||||||| ||| || ||| ||| || |||||| ||||||||||| | || ||| | || |||||| ||||||||| ||| || ||||| | |||||| |||||||||||| ||| || ||||| | |||||| ||| |||||||||||| || || ||| || || |||||| || ||| ||||||||||| ||||||| |||||||| ||||| |||||||| ||||| |||| ||||| ||||||||| |||| ||| ||| |||| || ||||||||||| ||||||||| |||| || |||||||| ||| ||||||||||| ||| |||||||| || |||| ||| ||| ||||||| |||||||| |||| ||| ||||||||||| ||||||||||| ||||||| |||||| ||| ||||| |||||||||||||| |||| |||| || |||||||| ||| ||||||||||||||||| |||||||||||||||||||||||| ||||||||||||||| |||||||||| || || ||||||||| |||||| ||||||||| |||| |||||||||| ||||| || |||||||| |||||||| |||||||| ||||||||| |||||| ||||||||| |||||||||| || ||| ||| |||||||| ||| |||||||| || || ||| |||||||| || |||| |||| ||||| ||| ||| |||||||||| || |||||||| |||| || ||| ||||| || | || ||||||||| ||| ||||||| |||| |||| ||| |||| |||| |||||||||| |||||||||| ||||||| |||||| |||| ||||||||| |||||| |||||||||||||||||||||||||| || ||||||| |||||||| ||||||||||||||| ||| |||||||| ||| ||||||| |||||||| ||| ||| |||| ||| ||||||||||| |||||||| ||| ||||||| ||||| |||| ||||||||||||| || ||| |||| ||| || ||| ||||||| ||||||| | ||| ||||||| || |||||| ||||| |||||||| |||||| || |||||||||| |||||||| |||| |||||| |||| || |||| ||||||| ||| ||||| |||||||||| |||||||| |||||| ||||| ||||||||| ||||| ||| ||||||||| ||||||| |||||||| |||||| ||||| |||||||| ||||||||||| |||||| || || ||||||| ||||| |||| ||| ||| ||||||| ||||||||| || |||||||| |||||| || ||||||||||||| |||||| ||| ||||||||||| || ||| |||| |||||||| || ||| ||||||| |||| ||||||| |||| |||||||||| |||| |||||||| || ||||| || ||||||| ||||| || ||| ||||||||| |||| ||||||| |||| || |||| |||| ||||| ||| |||| ||||||| ||||||||| || ||| |||||||||| || |||| |||||||| |||||| || ||||||| |||||||| |||| |||||| ||| ||| |||||||||| || |||||||| |||| | |||||||| ||| ||||| || | |||| |||| ||||||||| |||| |||| ||||||||| |||||||||| || |||||||| |||| |||||| |||||| |||||| ||||| || |||||| ||| |||||||| |||| || ||| ||||| || | |||||| |||||| ||||| || ||||||||||||| ||||||||||| ||||||| |||||||| ||| ||||||| ||||| |||||||| || ||||||||||||| || |||| |||| ||||| ||| ||||| ||||| ||||| |||||||| |||||| || |||||||||| ||||||||| |||||| |||| ||| ||| |||||||||| |||||||||| || ||| ||||| ||||||| ||||||| |||| || |||| ||||||| ||||| ||| |||| ||||||||||||| || ||||| ||||| ||||| ||||||| ||||||| || |||||| ||| ||| ||||||||| |||||||| |||| ||||| ||||||||| ||| ||||| ||||||| ||||||| || ||||||| || ||||| ||| |||||||||| |||||||| |||||| ||||||||||| ||||| |||||||| ||||||||||| |||||| || || ||||||| ||||| ||| |||| ||||||| ||||||||| || ||| |||||||||| || |||| |||||||| |||||| || |||||| ||| |||||||| |||| |||||| ||||| || |||| |||| |||||||| |||| |||||| ||| ||| |||||||||| || |||||||| |||| | |||||||| ||| ||||| || | |||| |||| ||||||||| |||| |||| ||||||||| |||||||||| || |||||||| |||| |||||| ||||||| ||||| || |||||| ||| |||||||| |||| || ||| ||||| || | ||||||| ||||| || ||||||||||||||| ||||||||||||| |||||| |||| ||||||| |||| |||| |||||||||| ||| ||||||||| |||||||| || | |||||||||| |||| ||||||| |||||||||||| ||| ||| ||||||||||| || ||||||||| ||| ||||||||||||| || ||| ||||||| |||| || |||| |||||||| ||||| ||| ||||||||| ||| ||||||| ||||| |||| || |||| ||||||||||| ||| ||||||||||| ||||||| ||||||||| ||||| ||| ||||||||| || ||||||| |||||||| ||| ||| ||||||| || ||||||| ||||||||| ||||||| |||| ||||||||||||| ||| || ||| || ||| ||||||||||| ||||||| |||| ||| ||||||||||| ||||||| ||||||| |||||| || |||| ||||| ||||||||| || ||||||||| ||||||| ||||||| ||| || |||||||||| ||||||||||||||| |||||||| |||||||||||||||| |||| ||||||||| ||| ||| |||||||| ||||||||||||| || ||||||||||| ||| ||||||||||| ||||||| ||| ||| |||||||| || |||| ||||||||| ||| ||| ||||||||| ||| ||||||||||||| ||| |||||||| || |||||||||| || |||||||| ||||||| | || |||||| ||| || ||| |||||| ||||| || ||| |||| ||||| ||| ||||||| |||||||||| || || || ||||||||| ||||||| |||||| ||| ||| ||||||||| ||| ||| |||||||| ||||||||| ||| |||||||| |||||| ||||||| ||||| ||| |||||||| || ||| |||||||| ||||| ||| ||| ||||||||||||| |||||||| ||||| ||||||| ||||||||||| ||| ||||||||||| ||||||| ||||||| |||||||| ||| ||| |||| |||||||||| |||| |||||||| || |||| || ||| |||||||||| || |||||| || ||||||| || ||||||| ||||||| | |||| |||||||| |||||||||||| ||| ||||||||||| |||||||||||| ||||||||||| ||||||| ||| ||| ||||||||||| ||| ||| ||||||||||| ||||||| ||| |||| ||| ||| ||||||| || |||| |||||||| || |||| ||| || ||| ||||||||||| |||||||| ||| |||||||| ||||||||| || || |||||| |||||||||| || ||||||||||||| ||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||||| |||||||||| ||||||||||||| ||| |||||||| || ||| ||||||||||| ||||||| |||||||| ||| |||||||| ||||||||| || || |||||| |||||||||| || ||||||||||||| ||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||||| |||||||||| ||||||||||||| ||| ||||||| ||| || ||||||||||| |||| |||| | |||||| |||| ||| |||||| ||||||| ||||||||||| ||| ||||||||||| || ||| ||||||||||| ||||||| |||||||| ||||| |||| ||| ||||| |||||| ||||| || |||| ||| ||||||||||| ||| ||||||||||| ||||||| ||||||||| |||||| ||||| |||||||| ||||||||| ||||||| || ||| |||| ||||| |||||| ||| |||| |||||||| ||| |||||||||| ||| || ||||| ||| |||||| |||||||||| || ||||||||||||| | |||||| |||| ||| |||||||| |||||| || | |||||| ||| | ||||| ||| |||||| ||| | ||||||| ||||||||||||||||||||| || ||| ||||||||||| |||||||| |||| ||||||||| ||||||| ||||||| ||||| ||||||||| ||||| ||| ||||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| |||| ||||| ||||||||| ||| |||||| || |||||| ||||||||| | ||||| ||||||||||| |||||| || ||| ||||||||||| ||||||| |||||||| |||| ||||||||| ||||||| ||||||| ||||| ||||||||| ||||| ||| ||||||| |||||| ||||||||| |||| ||| ||||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| |||| ||||| ||||||||| ||| |||||| || |||||| ||||||||| | ||||| ||||||||||||||||||| ||||||||||||||||| ||| |||||||||||||||| ||| ||| |||||||| ||||||||| |||||| || |||||||||||| || |||| |||||||| || |||| || || ||| ||||||||||| ||| ||||||||||| ||||||| |||| ||| |||||||||| || ||||| ||| || ||| ||||||| |||||||| ||| |||| |||||||| || |||| || || ||| ||||||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| ||| ||||||||| ||| || ||||| | ||||| ||| ||||||||||| ||| || |||||| ||||||||||| ||| || ||||| ||||||| ||||||||||| ||||| |||| |||| |||||| |||||||| ||| |||||||||||| || || ||||| ||||||| ||||||||||| ||||| |||| |||| |||||| |||||| ||| || || ||||| ||||||| ||||||||||| ||||| |||| |||| |||||| ||||||| |||| |||||||| |||| |||||||||||||||||| ||| ||||||| |||||||| ||| |||| |||||||| || |||| || || ||| ||||||||||| ||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| |||| || ||| |||||||||| ||||||||||| |||||||| ||||||||||| ||| || ||||| ||||||| ||||||||||| ||||| |||| |||| |||||| ||||||| ||| |||||||||||| || || ||||| ||||||| ||||||||||| ||||| |||| |||| |||||| |||||| |||| |||||||| |||| |||||||||||||| |||||||| ||| ||||| ||||||| ||||||||||| ||| ||||||||||| ||||||| ||||||| |||||||| ||| ||| |||| |||||||||| ||| ||| |||||||| || |||| || ||| |||||||||| || |||||| ||||||||| || ||||||| ||||||| | ||| ||| |||||||| |||||||||||| ||| ||||||||||| |||||||||||| |||||||||| ||||||| ||| ||| ||||||||||| ||| ||| ||||||||||| ||||||| ||| |||| ||| ||| ||||||| || ||| ||| |||||||| || |||| ||| || ||| ||||||||||| |||||||| ||| |||||||| ||||||||| || || |||||| |||||||||| || ||||||||||||| ||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||||| |||||||||| ||||||||||||| ||| |||||||| || ||| ||||||||||| ||||||| |||||||| ||| |||||||| ||||||||| || || |||||| |||||||||| || ||||||||||||| ||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||||| |||||||||| ||||||||||||| ||| ||||||| ||| || ||||||||||| |||| |||| | |||||| |||| ||| |||||| ||||||| ||||||||||| ||| ||||||||||| || ||| ||||||||||| ||||||| |||||||| ||||| |||| ||| ||||| |||||| ||||| || |||| ||| ||||||||||| ||| ||||||||||| ||||||| ||||||||| |||||| ||||| |||||||| ||||||||| ||||||| || ||| |||| ||||| |||||| ||| |||| |||||||| ||| |||||||||| ||| || ||||| ||| |||||| |||||||||| || |||||||||||||| | |||||||||||| |||||| ||||||||||| || | |||||| ||| | ||||| ||| |||||| ||| | ||||||| |||||||||||||||||||||| || ||| ||||||||||| |||||||| |||| ||||||||| ||||||| ||||||| ||||| ||||||||| ||||| ||| ||||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| |||| ||||| ||||||||| ||| |||||| || |||||| ||||||||| | ||||| ||||||||||| |||||| || ||| ||||||||||| ||||||| |||||||| |||| ||||||||| ||||||| ||||||| ||||| ||||||||| ||||| ||| ||||||| |||||| ||||||||| |||| ||| ||||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| |||| ||||| ||||||||| ||| |||||| || |||||| ||||||||| | ||||| ||||||||||||||||||| ||||||||||||||||| ||| |||||||||||||||| ||| ||| |||||||| ||||||||| |||||| || |||||||||||| || ||| ||| |||||||| || |||| || || ||| ||||||||||| ||| ||||||||||| ||||||| |||| ||| |||||||||| || ||||| ||| || ||| ||||||| |||||||| ||| ||| ||| |||||||| || |||| || || ||| ||||||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| ||| ||||||||| ||| || ||||| | |||||| ||||||||||| ||| || ||||| || ||| || |||||| || |||||||||||| || || |||||| |||||||||||| || || ||||| ||| |||||||| |||| |||||||||||| |||| |||| |||| |||| ||||| |||||||||||| ||| ||||||| |||||||| ||| ||| ||| |||||||| || |||| || || ||| ||||||||||| ||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| ||| || ||| |||||||||| ||||||||||| |||||| ||| |||||||||||| || || ||||| |||| |||| ||||| ||| |||||||| |||| |||||||||||| |||| ||| |||| |||| |||| |||| ||||| ||||||||||||| |||| || ||||| || | ||||| ||||||||| || ||||||||||||| ||| ||||| ||||||| ||||||||||| ||| ||||||||||| ||||||| ||||||| |||||||| ||| ||| |||| |||||||||| ||||||||||||| ||| |||||||| || |||| || ||| |||||||||| || |||||| ||||||||| || ||||||| ||||||| | ||||||||||||| ||| |||||||| || |||| || |||||||||||| ||| ||||||||||| |||||||||||| |||||||||| ||||||| ||| ||| ||||||||||| ||| ||| ||||||||||| ||||||| ||| |||| ||| ||| ||||||| || ||||||||||||| ||| |||||||| || |||| ||| || ||| ||||||||||| |||||||| ||| |||||||| ||||||||| || || |||||| |||||||||| || ||||||||||||| ||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||||| |||||||||| ||||||||||||| ||| |||||||| || ||| ||||||||||| ||||||| |||||||| ||| |||||||| ||||||||| || || |||||| |||||||||| || ||||||||||||| ||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||||| |||||||||| ||||||||||||| ||| ||||||| ||| || ||||||||||| |||| |||| ||| |||||| |||| ||| |||||| ||||||| ||||||||||| ||| ||||||||||| || ||| ||||||||||| ||||||| |||||||| ||||| |||| ||| ||||| |||||| ||||| || |||| ||| ||||||||||| ||| ||||||||||| ||||||| ||||||||| |||||| ||||| |||||||| ||||||||| ||||||| || ||| |||| ||||| |||||| ||| |||| |||||||| ||| |||||||||| ||| || ||||| ||| |||||| |||||||||| || ||||||||||||| | ||||||||| |||||| |||||| || | |||||| ||| | ||||| ||| |||||| ||| | ||||||| |||||||||||||||||||||| || ||| ||||||||||| |||||||| |||| ||||||||| ||||||| ||||||| ||||| ||||||||| ||||| ||| ||||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| |||| ||||| ||||||||| ||| |||||| || |||||| ||||||||| | ||||| ||||||||||||||||| || ||| ||||||||||| ||||||| |||||||| |||| ||||||||| ||||||| ||||||| ||||| ||||||||| ||||| ||| ||||||| |||||| ||||||||| |||| ||| ||||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| |||| ||||| ||||||||| ||| |||||| || |||||| ||||||||| | ||||| ||||||||||||||||||| ||||||||||||||||| ||| |||||||||||||||| ||| ||| |||||||| ||||||||| |||||| || |||||||||||| || ||||||||||||| ||| |||||||| || |||| || || ||| ||||||||||| ||| ||||||||||| ||||||| |||| ||| |||||||||| || ||||| ||| || ||| ||||||| |||||||| ||| ||||||||||||| ||| |||||||| || |||| || || ||| ||||||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| ||| ||||||||| ||| || ||||| | |||||| ||||||||||| ||| || ||||| || ||| || |||||| || |||||||||||| || || |||||| |||||||||||| || || ||||| ||| |||||||| |||| |||||||||||| |||| |||| |||| |||| ||||| |||||||||||| ||| ||||||| |||||||| ||| ||||||||||||| ||| |||||||| || |||| || || ||| ||||||||||| ||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| ||||||||||| ||| || ||||| |||| |||| ||| |||||||||||| || || ||||| |||| |||| ||||||||| ||| || ||||| | ||||| |||| ||| |||| |||| |||| |||| ||||| ||||||| ||||||||||| ||| || ||||| |||| ||| |||| |||| |||| |||| ||||| ||||||| ||| |||||||||||| || || ||||| |||| ||| |||| |||| |||| |||| ||||| |||||| |||| |||||||| |||| |||||||||||| |||| ||||||||||| |||| || ||||| || | ||||| ||||||||| || ||||||||||||| ||| ||||| |||||| ||||||||||| ||| ||||||||||| ||||||| ||||||| |||||||| ||| ||| |||| |||||||||| ||||||||||||| ||| |||||||| || |||| | ||| |||||||||| || |||||| ||||||||| || ||||||| ||||||| | ||||||||||||| ||| |||||||| || |||| | |||||||||||| ||| ||||||||||| |||||||||||| |||||||||| ||||||| ||| ||| ||||||||||| ||| ||| ||||||||||| ||||||| ||| |||| ||| ||| ||||||| || ||||||||||||| ||| |||||||| || |||| || || ||| ||||||||||| |||||||| ||| |||||||| ||||||||| || || |||||| |||||||||| || ||||||||||||| ||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||||| |||||||||| ||||||||||||| ||| |||||||| || ||| ||||||||||| ||||||| |||||||| ||| |||||||| ||||||||| || || |||||| |||||||||| || ||||||||||||| ||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||||| |||||||||| ||||||||||||| ||| ||||||| ||| || ||||||||||| |||| |||| ||| |||||| |||| ||| |||||| ||||||| ||||||||||| ||| ||||||||||| || ||| ||||||||||| ||||||| |||||||| ||||| |||| ||| ||||| |||||| ||||| || |||| ||| ||||||||||| ||| ||||||||||| ||||||| ||||||||| |||||| ||||| |||||||| ||||||||| ||||||| || ||| |||| ||||| |||||| ||| |||| |||||||| ||| |||||||||| ||| || ||||| ||| |||||| |||||||||| || ||||||||||||| | ||||||||| |||||| |||||| || | |||||| ||| | ||||| ||| |||||| ||| | ||||||| |||||||||||||||||||||| || ||| ||||||||||| |||||||| |||| ||||||||| ||||||| ||||||| ||||| ||||||||| ||||| ||| ||||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| |||| ||||| ||||||||| ||| |||||| || |||||| ||||||||| | ||||| ||||||||||||||||| || ||| ||||||||||| ||||||| |||||||| |||| ||||||||| ||||||| ||||||| ||||| ||||||||| ||||| ||| ||||||| |||||| ||||||||| |||| ||| ||||||| |||| |||| ||||||||| ||| |||||| || |||||||| ||||||||| |||| ||||||||||||| |||| ||||| ||||||||| ||| |||||| || |||||| ||||||||| | ||||| ||||||||||||||||||| ||||||||||||||||| ||| |||||||||||||||| ||| ||| |||||||| ||||||||| |||||| || |||||||||||| || ||||||||||||| ||| |||||||| || |||| | || ||| ||||||||||| ||| ||||||||||| ||||||| |||| ||| |||||||||| || ||||| ||| || ||| ||||||| |||||||| ||| ||||||||||||| ||| |||||||| || |||| | || ||| ||||||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| ||| ||||||||| ||| || ||||| | |||||| ||| ||||||||||| ||| || |||||| ||||||||||| ||| || ||||| |||| |||| |||| |||| ||||| ||||||| |||||||||||| || || ||||| |||| |||| |||| |||| ||||| ||||||| ||| |||||||||||| || || ||||| |||| |||| |||| |||| ||||| |||||| |||| |||||||| |||| |||||||||||||||||| ||| ||||||| |||||||| ||| ||||||||||||| ||| |||||||| || |||| | || ||| ||||||||||| ||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| ||||||||| ||| || ||||| | ||||| |||| |||| ||||||||||| ||| || ||||| |||| |||| ||| ||||||||||| ||| || ||||| |||| |||| |||||||||||| || || ||||| |||| ||| |||| |||| |||| |||| ||||| |||||| ||| |||||||||||| || || ||||| |||| ||| |||| |||| |||| ||| ||||| |||||| |||| |||||||| |||| |||||||||||| |||| |||||||||||||| ||| ||||||| || ||| ||||||| |||||||| ||||||| ||||||| |||||||| |||||||| ||||||||||||||
||||||||||||||||| | |||||||||||| |||||||||||| ||| || |||||| || ||||||||| ||||||||| | ||||||||||| |||||||| |||||||||||| ||| || |||||| | ||| || ||||||||| |||||| | ||||||
|---|---|---|---|---|---|---|---|---|
|||| |||||||| | | |||| |||||||| | | |||| |||||||| | | |||| |||||||| | | |||| |||||||| | | |||| |||||||| | | |||| |||||||| | | |||| |||||||| | | |
||||||||| ||| || | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | |
||||||||| ||| || | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | |
||||||||| ||| || | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | |
||||||||| ||| || | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | |
||||||||| ||| || | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | |
||||||| | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | | |||| ||||||| | |
|||| | |||||| |||| ||| |||||||| |||||| ||| | |||||||||||| |||||| ||||||||||| ||| | ||||||| |||||||||||||| ||| | ||||||||| |||||| |||||| |||||| | ||||| ||| |||||| ||| | ||||||| |||||||||||||||||||||| ||||| || |||| |||||||| ||| |||| |||| |||||||| ||| |||||||||| ||||||||||| |||| ||||| |||||||| |||||| |||||| |||||||||||| |||||| ||| ||||||||| ||||||||||||| |||| |||| |||||| ||||||| |||| | |||||| |||||||||||| |||||| ||| ||||||||| |||||||||||||||||| ||||||||||||||||| ||| |||||||||||||||||||| |||||||| ||||||| |||||| ||| ||||||| ||||| | ||||||||||| |||||||||||||||||| |||||||| |||| ||||||||| ||||||||| |||| ||||| ||| ||||||| || ||| ||||||||||| ||||||||| | ||||| || || |||||| |||| ||||||||| || ||| |||| |||||||| ||| |||||||||| |||||| ||||||| ||||| ||| |||||||| || ||| |||| |||||| ||||||| |||| |||||||||| |||| ||| ||||||| ||||||||| ||| ||| ||||||||| |||||||||| ||| |||||| ||||||| ||||| |||||||| ||| |||||||| |||| ||| ||| ||||| || ||||||| ||| ||||||| |||| |||||||||| |||| ||| ||||||| ||||||||| || ||| ||| ||| |||||||| |||||||| |||||||||||| || || ||||| ||| |||| |||||||| |||| |||||||||||| |||| |||| |||| |||| |||| || ||||||||||||||||||| |||||||| ||||||| | ||||||||||||||| |||||||||||||||| |||||||| ||||| ||||||||||||||| || |||||| ||| |||||||| |||||||| |||| ||||||||| ||| ||||||||||| ||| ||||||||||| ||||||| |||||||| ||| ||| |||||||| || |||| ||||| || |||| || ||| ||| ||| |||||||| || |||| |||||||| ||| ||||||||||| |||||||| | ||||||| ||||||||||||||| || |||| |||||||| || |||| || |||||||| ||| |||||||| |||| |||| ||||| ||| |||||||||| ||||| ||| || ||||||||||| |||||||||| || ||| ||||||| ||||| ||| |||||| ||||||| ||||||| |||||| |||||| ||||||| |||||| |||| |||||||| ||||| || ||| ||||| |||||||| ||||||||| ||||||| |||| |||||||||| |||| ||| ||||||| ||| ||||||||| ||||||||| |||| ||||||||||| ||| || |||||| |||||||||||| || || |||||| ||| |||||||||||| || || ||||| |||||||| |||| |||||||||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| ||| ||||| ||||||||||| ||| ||| ||| |||||||| || ||| ||||||||||| |||||||| | |||||| ||||||| ||||| |||||||| ||| ||| |||||||||| || |||||||| |||| || ||| ||||| || | || |||| || |||||||| ||| |||||||| || ||| ||||||||| |||||| ||| ||||||| |||| |||||||||| |||| ||| ||||||| ||||||||| |||| |||||||||||| || || ||||| |||||||| |||| |||||||||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| ||| ||||| |||||||||||||||| ||| ||||||||||| ||||||| |||||||| ||| ||||| ||||||| ||||| |||||||| ||| |||||||| |||| ||||| ||| ||| ||||||||| ||||| || ||||| |||| ||| |||| |||||||||| || |||||||| ||| |||||| ||||||| |||||| ||||||||||| ||| ||||||| |||| |||||||||| |||| ||| ||||||| ||| ||||||||| ||||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| |||||||||||| || || ||||| |||| |||||||||| ||| ||||||||| ||| |||||| ||||||| ||||| |||||||| ||| |||||||| ||| |||||||| ||| |||||||| || ||| ||||||||| |||||| ||||||| |||| |||||| |||||||||| |||| ||| ||||||| ||||||||| |||||||| ||||| ||| |||||||||||| |||||||| || |||| | |||||||||| ||||||| |||||||||||| |||| ||| ||| |||||||||||| || || ||||| |||| ||||||||||||||| ||||||||| || ||| ||||||||||||||||| ||||||| ||||||||||||||||| ||| ||| |||||||| || | ||| |||| |||||||| ||||||||||||| |||||||| |||||||| || ||||| ||||| || |||||||| |||||||||| |||||||||| |||||||||||| |||||||| ||||| |||||||||| ||| ||| ||||||||| ||||||||| || ||||||| ||| ||||| || |||||||||||| ||||||||||| ||||||||||||| ||||||||||| ||||||||| ||| ||||| ||||||| ||||||||| ||||| |||| ||||||| ||| ||| ||| |||| ||| ||| |||| ||||||||||| ||||||| |||| ||||||||| |||||||| || ||||||||||||| || |||||||||| |||| |||| ||| || |||||||| ||| ||| |||| |||||| |||||||| ||| ||||||||| |||||||| |||| |||||||||| |||||||| ||| ||||||||||| || ||| |||||||| |||||||| ||||||| ||| ||||||||| ||| |||| |||||||||| |||| |||||||||| || |||||||| ||| |||| || ||||| ||| ||||| ||| |||| || |||| |||||||||| ||||||||| || ||| ||||||| ||| ||| |||||||||| || ||||||| |||| || |||| || ||| |||||||| |||||||| |||||||| || |||||| || ||||| |||| ||||||| ||| |||| || |||| ||||||||| |||| |||||||||||| |||||||| |||||| || || ||| ||||| ||| |||| ||||||||| |||| |||||||| ||| ||||||| |||| |||||| ||||||||||||||| || ||||| ||| |||| ||| |||| ||| ||||| || ||| |||||||||| ||||| ||| || |||||||||||| |||||||| ||| |||| || |||| ||||||||| ||| |||||||||| || ||| ||||| |||||| |||||| |||| || ||| ||||| || ||| |||||||| |||||||| |||||||||| |||||||||| || ||| ||||| ||||| || ||| ||||||| ||| ||||||||||| || ||| |||| || |||| |||| ||| ||||| |||||| |||||||| ||||||| ||||||| ||| |||||| ||||||||| |||||| ||||||||||||||||||| ||||| || | |||| || |||| ||| || ||||||| ||||||| || ||| ||||||||| ||||||| |||||| || |||||||| |||||||||| |||||||||| ||||||| | ||||||| ||||||| ||||| |||| ||||||||| ||| ||| ||||||||| |||||||| ||||||| ||||| | |||||| |||| |||||||| |||| ||| ||||||||| ||| |||| ||||||| || ||||||||||| |||||||||| ||| |||||||||| |||||||||| ||||||||| ||||||||||||| || ||||| ||||||| ||||||| |||||||| |||||||||||||||| |||||||||||||| ||| ||||||||| ||||||||||| ||| ||||||| ||||||||| ||| ||||||| ||||| |||| |||| ||||||||||||| ||| |||||||| |||||| ||||||| || |||| ||| ||||||||||| ||| ||||||||||| ||||||| ||||||||| || ||| ||||||||||| |||||||| |||| || ||| |||| || ||||||| || |||| ||| ||||| ||| ||||||||| ||||||||||||| ||||| ||| |||||||||| || |||||||||| ||||| ||||||||| ||| |||||| || ||| |||||||||| || |||| || |||||||||| ||| ||| ||||||||| || ||| ||| ||| ||||||| |||||||| ||||| |||| |||| ||||||||||| || ||| |||||| |||||||| |||| ||| |||| ||| ||||||| |||| |||| ||||| || |||| |||||| ||||| ||| |||| ||||||||||||| || ||| |||||||||| || |||||||| |||| ||| |||||||| |||||| || ||||||| ||| |||| |||||||| |||||| || ||||| |||||| ||||||||| || ||| ||||||||||| ||||||| |||||||| ||||||| ||||||| || ||||||||||||| || ||||| |||||| ||| |||||||| ||||||||||||||| |||| |||||||| || ||||||||| |||||||| |||| ||| |||||| |||| || |||| |||||| ||| |||||||||| |||| ||||| || |||||| ||| |||| ||||||||||| ||||| || ||||||| |||||||||| ||| |||||||| |||| ||| ||| ||| ||| ||||| ||||| |||||||||||| || ||||| ||| ||| ||||||||||| |||| |||||||||| || || |||||||||| || ||| ||||||| ||||||||| ||||| ||||||||||| |||| ||||| || ||| ||||| |||||| |||| || ||||||| ||| ||||||||| |||||| |||||||| |||||| ||| |||||| || ||||| ||||||||||| ||||||| |||||||| ||| ||||||| |||| ||||| ||||||||||||| || ||||| |||||||| |||| || |||| ||| |||| ||||| || |||||||||| |||||| || || ||| ||||| ||||||| ||||| ||| ||||||||| ||||||||| |||||| |||||||||| ||||||||||||| ||| ||||||| |||| ||||||||||| ||||||||||| || ||| ||||| |||||||| ||| ||||||||| || ||| ||||||||| || ||| ||||||||||| ||||||| ||||||| |||||| |||||||| ||||||||||| || ||| ||||||||| ||| |||| |||||||| |||||||| ||||| ||| ||||| ||| ||||||||||| || ||||||||| ||||||||| || ||| |||||| |||||||||||||| |||||||| |||||||| |||| |||||||| |||||||| |||||| |||| |||||||| || |||||||| ||| ||||||||| ||| ||||| || |||||||| |||||||| |||| ||||||||| || ||||||| ||| |||||||||| ||||||| |||||||| ||| |||||||||| |||||||| ||||| || || ||||||| |||| |||||| ||| ||||||||| ||| |||||||| |||| ||| || ||| |||||||| |||||||| ||| ||||||| ||||||||||||| || ||| |||||||| ||||||||||||||| |||||| ||||||| ||||| ||| |||||||||||| || ||| |||||||||||| ||||||||||| |||||||| ||| ||||||||| |||||| ||||||||| ||| ||| || |||||||||||||||||| ||| |||||||||||| |||| |||||||| || ||||||||||| || ||| ||||| |||||||| ||| ||| || ||||||||||| ||| || || ||||||| || |||||| ||||| ||| |||| || | |||||||| |||||||||| ||| ||| ||| ||| |||||||||||| || |||| ||||||||||| ||| ||||||||| ||| |||||||||| |||||| |||||||||||| |||||||||||||| ||||||||||||| |||||| ||||||||| ||| ||| |||||||| | || ||| ||||||||| |||||||||||||| |||| ||||| ||| || ||| |||||||||| || ||||||||||| ||||| ||||||||||| ||||||| |||||| ||||| |||||||||| |||| ||| |||||||| || |||| |||||||||| |||||| |||||| ||| ||||||||| ||||||||| |||||| ||||||||| ||| |||||||||| |||||||| ||| ||||||| |||||| || |||||||| |||||||| |||||||| |||| ||||||||| || |||||||| |||||||| || |||| ||| |||| ||| ||||||| || ||||| ||||| || |||| || |||| || ||| ||||||||| || ||||||||| |||||| |||||||||| ||| || || ||| ||||| |||| ||| |||| || |||||||||||||| ||| ||||||| ||||||||||| |||| |||||| ||| ||| |||| || || |||||||| ||| |||||| ||| ||| ||||| || |||| || ||||||||| || ||| |||||||| ||||||| || ||||||||||| ||||||||| ||||||||||| ||| |||||||| |||||| ||||||||| || ||||| |||||| |||| ||| |||| || ||||||||| ||||||||| |||||| |||||||||| |||| |||||| |||| |||||| || ||| ||||| || ||||||||||| || || ||||| |||||| ||||| |||| ||||||||| |||||||| |||||| |||||| |||| |||| |||||||| |||||| ||||||| ||||||||| || ||| |||||||| ||||| |||||| ||| ||||||||||||||| |||| |||| ||||||||| |||||| | |||||||| ||||||||| ||| |||| |||| |||||||| |||| ||||||||| ||||| |||||| ||||||| |||||| |||||||| ||| |||||||| ||||| ||||| ||| |||||||||| |||||||||||| ||||| ||| |||||| ||||||||| ||||||| ||| ||| ||| ||||| ||| |||||| ||||||| |||||| ||| |||| ||||||| ||| |||||||| ||||| |||| ||||| ||||||| |||||| ||||||||||||| ||||| |||| |||||||| ||||||||||| || |||||||| |||||||| |||| ||| |||| |||||| || |||||||| |||||| ||||||| |||||| || |||| ||| ||||||||| ||||| ||| ||| |||||| ||||||| ||||||| || |||| |||||| ||||||||||| ||| |||||| ||| ||||||||| ||||||||||||| |||| ||||| || |||||| |||||||| ||| |||||||| |||||| ||||| |||| |||||| || ||| ||||||||||| ||||||||| |||||| |||||| |||| ||| ||||||||| |||||| |||||||| || ||| |||||||| || ||||||||| |||||| ||| ||||||||| |||||||||| ||||||||||| ||||| ||| || ||||||| |||| |||||| || |||||||| || |||||||| ||| |||||| ||||||||| || ||| ||||||||||| ||||||| |||||||| |||||| ||||| |||| |||| |||||| |||| ||| ||||||||| |||||| ||| |||||| ||||||||| ||| |||||||| |||||||| ||||| ||| |||| | |||| || |||||||| ||||||| ||||||||||| ||| || ||||| |||| |||| ||||||||||| ||| || ||||| |||| |||| ||| ||||||||| ||| || ||||| | ||||| |||| ||| ||||| ||| |||| |||||||||| |||||||||||||| ||||||||| |||| ||||||| |||||| |||| |||||| || ||| ||||||||||| ||||||| |||||||| ||| ||| |||||||| || ||| ||| |||||||| || |||| || ||| ||||||||||||| ||| |||||||| || |||| || |||||| || |||||||| || ||||||||||||| ||| |||||||| ||||| || ||| ||| ||| |||||||| |||||||| ||||| |||| ||||||| || ||| ||||||| |||||||| |||||| ||| |||||| |||| ||| ||||||| ||| ||| |||||||| ||||| |||||||| ||| ||||||||||||| ||| |||||||| || |||| |||| ||||| ||||||||||||| ||| |||||| || || ||||||||| ||| ||||||| |||||||||||| ||| ||||| ||||||||| |||||||||||| ||| |||||||||| || |||||||| ||| ||| ||||||||||||| ||||||| ||||||| |||||||| |||||||| |||| || ||| ||||||| ||||||||| || ||| ||||||| |||||| ||| |||||||||||| |||||| |||| |||||||| |||| | ||||| ||||||| |||| || ||||| || | || |||||||| |||| |||||||||| |||| ||||| || ||||||||| |||| ||||||||||||| ||| ||| |||||||||| ||| |||| |||||||| ||||||||| |||||||| || ||| ||| |||| |||||||| || ||| ||||||||| || || || ||||||| |||||| ||| |||||||| |||||| ||||||||| || ||||| ||||||||||| |||| |||| |||||| ||| ||||||||| ||| |||||||||| || ||||||| |||||||| ||||||||| ||||||||||||| |||||||| || |||||||||| || |||| ||||||| ||||||||| ||||| ||| |||||| |||| ||| |||||||| || ||| |||| | ||||||||||| ||||| ||| ||||||||| ||| |||| ||||||| ||||| ||| ||||||||||
Table 39: Summary of Gaps Addressed in the ADvantage Study
Detail | Description |
|---|---|
Evidence gap | There is uncertainty regarding the efficacy and safety of lebrikizumab, specifically in patients whose AD is not adequately controlled with cyclosporine or for whom cyclosporine is not medically advisable. |
Study design |
|
Population |
|
Interventions |
|
Key findings | A summary of the efficacy and safety results for patients randomized to lebrikizumab + TCS vs. placebo + TCS follows. Efficacy at week 16:
Harms at week 16:
Harms up to week 52 (patients treated with lebrikizumab + TCS):
|
Limitations |
|
AD = atopic dermatitis; AE = adverse event; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; diff = difference; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% reduction in EASI score; IGA = Investigator Global Assessment; LSM = least squares mean; NRS = Numeric Rating Scale; q.2.w. = every 2 weeks; RD = risk difference; SAE = serious adverse event; SD = standard deviation; SC = subcutaneous; TCS = topical corticosteroids.
Sources: CSR for ADvantage,60 Sponsor’s Summary of Clinical Evidence.35
Studies Addressing Gaps in the Systematic Review Evidence
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
The sponsor submitted 4 studies that provided additional data on the efficacy and safety of lebrikizumab in patients with AD whose disease was not adequately controlled with cyclosporine or for whom cyclosporine is not medically advisable (ADvantage study), on the short-term treatment effects and vaccine immune response (ADopt-VA study), and on the efficacy and safety of lebrikizumab in patients from Japan (ADhere-J study) and in adolescent patients (ADore study). A brief summary of these studies and their key results are provided in Table 39, Table 40, Table 41, and Table 42.
Table 40: Summary of Gaps Addressed in the ADopt-VA Study
Detail | Description |
|---|---|
Evidence gap | There is uncertainty regarding the impact of lebrikizumab on vaccine immune responses. This trial also provides additional evidence on the efficacy and safety of lebrikizumab. |
Study design |
|
Population |
|
Interventions | 500 mg lebrikizumab SC at baseline and week 2 and 250 mg every 2 weeks thereafter through week 14 or placebo. |
Key findings | A summary of the efficacy and safety results for patients randomized to lebrikizumab vs. placebo at week 16 follows. Efficacy:
Harms:
|
Limitations |
|
AD = atopic dermatitis; AE = adverse event; CI = confidence interval; diff = difference; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% reduction in EASI score; IGA = Investigator Global Assessment; LSM = least squares mean; NRS = Numeric Rating Scale; q.2.w. = every 2 weeks; RD = risk difference; SAE = serious adverse event; SE = standard error; SC = subcutaneous; TCS = topical corticosteroids; vs. = versus.
a||| |||| |||||||| || |||||||| ||| |||||||
b||| |||| |||||||| || |||||||||| |||||| ||| |||||| || |||||| |||| || ||||||| ||| ||||||||||| |||||||||||| || || ||| |||||| |||||| ||| |||||| ||| ||||||| |||||||| |||| | |||||| ||||
c|||||||||| |||||||| |||| ||| |||||||||| |||||||| ||| ||||||||| |||||| |||| |||| ||||||||| ||||||||||
Sources: CSR for ADopt-VA,61 Sponsor’s Summary of Clinical Evidence.35
Table 41: Summary of Gaps Addressed in the ADhere-J Study
Detail | Description |
|---|---|
Evidence gap | There is uncertainty regarding the efficacy and safety of lebrikizumab, specifically in patients in Japan. |
Study design |
|
Population |
|
Interventions | There were 3 treatment groups:
Nonresponders moved to the escape arm and received 250 mg lebrikizumab every 2 weeks. All patients also received concomitant TCS. |
Key findings | Efficacy results for the induction period in the placebo + TCS group vs. the LEB every-4-weeks + TCS group vs. the LEB every-2-weeks + TCS group at week 16:
Efficacy results for the maintenance blinded period (||) in the LEB every-4-weeks responders who switched to LEB every-4-weeks + TCS group vs. the LEB every-2-weeks responders who switched to the every-4-weeks + TCS group vs. the LEB every-2-weeks responders who switched to every 2 weeks + TCS at week 52:
Harm results for the induction period (baseline to week 16) in the placebo vs. LEB every-4-weeks + TCS vs. LEB every-2-weeks + TCS groups:
Harm results for the maintenance blinded period (week 16 to week 68) in the LEB every-4-weeks responders who switched to the every-4-weeks + TCS group vs. the LEB every-2-weeks responders who switched to the every-4-weeks + TCS group vs. the LEB every-2-weeks responders who switched to the every-2-weeks + TCS group:
|
Limitations |
|
AD = atopic dermatitis; AE = adverse event; BSA = body surface area, CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% reduction in EASI score; IGA = Investigator Global Assessment; HC = Health Canada; LEB = lebrikizumab; LS = least squares; NRS = Numeric Rating Scale; q.2.w. = every 2 weeks, q.4.w. = every 4 weeks, RD = risk difference; SAE = serious adverse event; Res = responders; SE = standard error; SC = subcutaneous; TCS = topical corticosteroids.
Sources: CSR for ADhere-J,62 Sponsor’s Summary of Clinical Evidence.35
Table 42: Summary of Gaps Addressed in the ADore Study
Detail | Description |
|---|---|
Evidence gap | There is uncertainty regarding the efficacy and safety of lebrikizumab, specifically among adolescent patients. |
Study design |
|
Population |
|
Interventions | 500 mg lebrikizumab SC at baseline and week 2. From week 4 onward, 250 mg lebrikizumab every 2 weeks through week 52. Concomitant AD therapies were prohibited unless part of rescue therapy. |
Key findings | Efficacy results at week 52:
Harm results:
|
Limitations |
|
AD = atopic dermatitis; AE = adverse event; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; diff = difference; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-75 = at least a 75% reduction in EASI score; IGA = Investigator Global Assessment; HC = Health Canada, LEB = lebrikizumab; LSM = least squares mean; MCMC-MI = Markov chain Monte Carlo multiple imputation; q.2.w. = every 2 weeks, SAE = serious adverse event; SE = standard error; SC = subcutaneous.
Source: CSR for ADore,63 Sponsor’s Summary of Clinical Evidence.35
Discussion
Summary of Available Evidence
The Systematic Review section of this report included 3 pivotal, double-blind, RCTs that evaluated the safety and efficacy of lebrikizumab versus placebo in adults and adolescents (aged 12 years and older) with moderate-to-severe AD who had history of inadequate response to topical AD therapies. The ADvocate 1 (N = 424) and ADvocate 2 (N = 427) studies compared lebrikizumab 250 mg SC every 2 weeks with placebo for the 16-week induction period. At 16 weeks, patients in the lebrikizumab group who responded to therapy were rerandomized to switch to placebo or to continue receiving lebrikizumab every 2 weeks or every 4 weeks up to week 52. The 16-week ADhere study evaluated the safety and efficacy of lebrikizumab 250 mg SC every 2 weeks in combination with low-to-midpotency TCS, compared with placebo plus TCS in adult and adolescent patients with moderate-to-severe AD (N = 211). In all 3 trials, the coprimary outcomes were the proportion of patients with an IGA score of 0 or 1 and at least a 2-point reduction from baseline to week 16, and the proportion of patients with an EASI-75 response at week 16. Other key outcomes reported were the proportion of patients with a Pruritus NRS score of at least 4 points at baseline who reported at least a 4-point reduction from baseline at week 16, and the change from baseline to week 16 in the POEM score, the DLQI total score, or the CDLQI total score.
The patients enrolled in the trials had a mean age that ranged from 34.2 years (SD = 16.4) to 37.5 years (SD = 19.9) per treatment group. On average, the patients enrolled had been diagnosed with AD for 20 or more years, with most patients (59% to 73%) classified as having disease of moderate severity based on an IGA score of 3 at baseline, whereas 27% to 41% were classified as having severe AD (i.e., an IGA score of 4). Almost all patients enrolled had previously used TCS (||| || ||||) and ||| || ||| of patients had received topical calcineurin inhibitors. Systemic therapies were previously received by ||| || ||| of patients, and ||| || ||| of patients had used phototherapy before enrolment in the trials.
This report summarized interim data from 1 extension study (ADjoin) that provided longer-term safety data (N = |||), as well as 40-week efficacy data for a subset of patients (N = ||). The ADjoin study included patients with moderate-to-severe AD who had completed another lebrikizumab clinical trial or who were directly enrolled without prior lebrikizumab exposure. All patients received lebrikizumab 250 mg every 2 weeks or every 4 weeks, alone or in combination with TCS.
The sponsor submitted an NMA to estimate the efficacy of lebrikizumab relative to abrocitinib, dupilumab, and upadacitinib in the short-term treatment of adults and adolescents with moderate-to-severe AD. The outcomes reported were EASI response and IGA 0 or 1 response at week 16, and at least a 4-point reduction in the Pruritus NRS at week 4 and week 16.
Also included in this report is a summary of 4 studies the sponsor submitted to address gaps in the systematic review evidence. The studies provided data on the efficacy and safety of lebrikizumab in patients with moderate-to-severe AD whose disease was not adequately controlled with cyclosporine or for whom cyclosporine is not medically advisable (ADvantage RCT, N = |||), on the short-term treatment effects and vaccine immune response (ADopt-VA RCT, N = 254), and of the efficacy or safety and lebrikizumab in patients from Japan (ADhere-J RCT, N = 286) and in adolescent patients (ADore open-label, single-arm trial, N = 206).
Interpretation of Results
Efficacy
The 3 pivotal placebo-controlled RCTs demonstrated that lebrikizumab 250 mg every 2 weeks as induction therapy (with or without TCS) increased the proportion of patients with an IGA 0 or 1 response, an EASI-75 response, or at least a 4-point improvement in the Pruritus NRS score at 16 weeks, relative to placebo. The treatment effects for all 3 outcomes were clinically relevant, as most analyses showed point estimates and 95% CIs that exceeded the 10% between-group difference that was deemed to be clinically important. The 10% threshold, however, was based on clinical expert input and, therefore, is subject to some uncertainty. The findings of the ADvocate monotherapy trials were similar to those for combination therapy in the ADhere study. However, the magnitude of the RDs in the ADhere study tended to be smaller than were observed in the ADvocate studies, which may be due to the effects of TCS and the higher response rate in placebo plus TCS group of the ADhere study. Even with these potential differences, no serious inconsistency was detected between the monotherapy and combination therapy trials.
Other outcomes that the patient-group input indicated were important to patients were HRQoL and symptoms of AD. The results of the 3 pivotal trials suggest that lebrikizumab (with or without TCS) may result in a reduction in the POEM score and the DLQI score at 16 weeks. The between-group differences were considered clinically relevant, based on an MID of 3.4 points for POEM and 4 points for DLQI, although there were some concerns regarding imprecision of the DLQI. In addition, these outcomes were limited by the extent of missing data, and the analyses of POEM scores were potentially biased due to the imputation methods used. Changes in POEM scores were not controlled for multiplicity in any of the trials, so the potential for an inflated risk of type I error should be considered when interpretating these results. Changes in CDLQI scores were reported for a small proportion of patients aged 12 to 16 years, and these analyses were not controlled for multiplicity. The results were also limited by the potential bias due to missing data and the imputation methods used and had limitations due to serious imprecision. As such, the effects of lebrikizumab on CDLQI scores in adolescents is unclear.
The longer-term efficacy data from the ADvocate trials had a number of limitations. Specifically, the data were reported for an enriched population that had shown a treatment response to lebrikizumab, so may not be representative of the effects observed in an unselected population. Moreover, the patients who switched to placebo showed a high EASI-75 response rate at week 52 (||| and 72%), which may be due to the carry-over effects of lebrikizumab. Because of these issues, the treatment effects are difficult to interpret; the observed effects are relevant to the efficacy of lebrikizumab maintenance, compared with lebrikizumab withdrawal, among patients who initially tolerate and respond to lebrikizumab. In addition, the results were based on a small sample size (28 to 63 patients per treatment group), and the effect estimates were affected by serious imprecision. The longer-term efficacy data from the extension study and the studies addressing gaps also had limitations, which included an enriched population, the lack of a relevant comparator group, maintenance doses that were not consistent with the Health Canada–recommended dose, and potential bias due to missing data. Thus, the longer-term efficacy of lebrikizumab is unclear.
Because there were no controlled trials that compared lebrikizumab to other systemic treatments for AD, the sponsor submitted an NMA that compared the short-term efficacy of lebrikizumab with abrocitinib, dupilumab, and upadacitinib. In the monotherapy and combination therapy (concurrent TCS) networks, the NMA results for EASI response, IGA 0 or 1 response, or at least a 4-point reduction in Pruritus NRS response showed 95% CrIs that overlapped the null for lebrikizumab versus dupilumab and abrocitinib 100 mg daily. The effect estimates were affected by imprecision (i.e., wide CrIs), precluding a conclusion as to which treatment may be favoured. The results for EASI response and Pruritus NRS response for lebrikizumab versus abrocitinib 200 mg daily and lebrikizumab versus upadacitinib 15 mg daily favoured the comparator treatments. Upadacitinib 30 mg daily was favoured over lebrikizumab for all outcomes. The NMAs were limited by heterogeneity in the age of patients enrolled, and the type, potency, and frequency of TCS used in the combination therapy analyses. The EASI response was analyzed using a multinomial model, with treatment effects reported as probit differences, which can be difficult to interpret clinically. Moreover, in all random-effects analyses, the results were associated with wide 95% CrIs, with many estimates crossing the null, precluding conclusions as to which treatment may be favoured.
The sponsor provided 4 studies to address gaps in the systematic review evidence. In patients with moderate-to-severe AD who received induction therapy with lebrikizumab 250 mg every 2 weeks (with or without TCS), the results of the supplementary trials were generally consistent with the findings of the pivotal trials. The efficacy findings favoured lebrikizumab over placebo for EASI-75 response, IGA 0 or 1 response, and at least a 4-point reduction in Pruritus NRS response at 16 weeks in the RCTs addressing gaps (ADvantage, ADhere-J, and ADopt-VA). The proportion of patients with an EASI-75, IGA, and Pruritus NRS response in the single-arm study of adolescents showed response frequencies that were consistent with other lebrikizumab trials; however, the study design precluded causal conclusions.
No major limitations to the external validity of the trials were identified, although the clinical expert consulted did note that the pivotal trials excluded patients with certain comorbidities; thus, the safety and efficacy of lebrikizumab in these patients is uncertain. Moreover, there is less information available from RCTs regarding the efficacy of lebrikizumab versus placebo (with or without TCS) among adolescent patients. The dosing of lebrikizumab during the induction period was consistent with the Health Canada–recommended dose; however, the clinical expert noted that most patients using a biologic for AD will also use TCS as needed. The concurrent use of TCS was prohibited in some studies, so the magnitude of benefit observed in the combination therapy trials (ADhere, ADvantage, ADhere-J studies) may be more consistent with what would occur in clinical practice.
Harms
In the trials included in the systematic review, 43% to 53% of patients in the lebrikizumab groups experienced 1 or more TEAEs during the induction period, compared with 35% to 66% of patients in the placebo groups. In general, the frequency of SAEs was low in all treatment groups (≤ 2.8%), as was the proportion of patients who stopped treatment due to AEs (≤ 3.2%) in the first 16 weeks of therapy. SAEs and conjunctivitis AEs were identified as key harms associated with lebrikizumab. Based on the available evidence, lebrikizumab may increase the short-term frequency of conjunctivitis relative to placebo; however, there was too much uncertainty in the longer-term evidence to draw conclusions. There was too much uncertainty in the evidence to determine if lebrikizumab increased, decreased, or had no effect on the frequency of SAEs in the short term or longer term, compared with placebo.
No new safety signals were detected in the extension study or in the 4 supplemental studies submitted by the sponsor. The frequency of AEs reported in these trials was generally consistent with data reported in the pivotal trials. The longer-term safety data from the ADvocate trials and the ADhere-J study were difficult to interpret because of the enriched patient population (all had achieved a treatment response with lebrikizumab induction therapy), as well as the carry-over effects of lebrikizumab in the patients who switched to placebo. The observed effects are relevant to the harms of lebrikizumab maintenance, compared with lebrikizumab withdrawal, among patients who initially tolerate and respond to lebrikizumab. In the ADjoin, ADvantage, ADhere-J, and ADore studies, some patients received a maintenance therapy dose (i.e., lebrikizumab every 2 weeks) that was not consistent with the Health Canada–recommended regimen. There was no comparator group for the safety data in the ADjoin and ADore studies, nor for the longer-term data in the ADvantage study, so it is difficult to determine what proportion of AEs may be attributable to lebrikizumab and what proportion may be attributable to other factors.
There was no direct or indirect evidence on the safety of lebrikizumab versus other systemic therapies used to treat AD in Canada. All the clinical trials were placebo-controlled, and the sponsor-submitted NMA did not assess any safety end points. Thus, the comparative safety of lebrikizumab is unknown.
Conclusion
In patients with moderate-to-severe AD that was not adequately controlled with topical therapies, 3 pivotal RCTs demonstrated that lebrikizumab induction therapy provided a clinically relevant improvement in physician-assessed signs of AD, and reduced patient-reported symptoms of itch relative to placebo, measured with EASI-75 response, IGA 0 or 1 response, or Pruritus NRS response at week 16. The benefits were observed when lebrikizumab was used as monotherapy and in combination with TCS. Lebrikizumab may improve HRQoL and reduce other symptoms of AD at 16 weeks compared with placebo, but the evidence is less certain.
There was no direct evidence comparing lebrikizumab to other biologics or JAK inhibitors used to treat AD in Canada; however, the sponsor submitted indirect evidence from an NMA that assessed short-term comparative efficacy. The results of the NMA were inconclusive for lebrikizumab compared with dupilumab and abrocitinib, with most estimates affected by serious imprecision. The NMA results suggest that upadacitinib may be favoured over lebrikizumab for the proportion of patients with an EASI or Pruritus NRS response, although differences were not consistently detected and the clinical relevance of any differences is unclear.
Lebrikizumab may increase the short-term risk of conjunctivitis relative to placebo. The NMA did not assess any safety end points, so the comparative safety of lebrikizumab is unknown. The longer-term safety and efficacy of lebrikizumab from the RCTs and extension study is uncertain due to limitations with the data. These limitations included an enriched population and the carry-over effects for the 52-week data in the pivotal trials (i.e., effect estimates apply to lebrikizumab maintenance therapy, relative to lebrikizumab withdrawal, among patients who tolerate the treatment and initially experience a response), and the lack of a comparator group for the extension study.
The supplementary evidence available from the sponsor-submitted trials addressing the gaps was generally consistent with the findings of the pivotal trials, including in patients whose AD was not adequately controlled with cyclosporine or for whom cyclosporine was not medically advisable. No new safety signals were detected in the single-arm study of adolescents.
References
1.Avena-Woods C. Overview of atopic dermatitis. Am J Manag Care. 2017;23(8 Suppl):S115-S123. PubMed
2.Fishbein AB, Silverberg JI, Wilson EJ, Ong PY. Update on atopic dermatitis: diagnosis, severity assessment, and treatment selection. J Allergy Clin Immunol Pract. 2020;8(1):91-101. PubMed
3.Fasseeh AN, Elezbawy B, Korra N, et al. Burden of atopic dermatitis in adults and adolescents: a systematic literature review. Dermatol Ther (Heidelb). 2022;12(12):2653-2668. PubMed
4.Girolomoni G, Busà VM. Flare management in atopic dermatitis: from definition to treatment. Ther Adv Chronic Dis. 2022;13. PubMed
5.Laughter MR, Maymone MBC, Mashayekhi S, et al. The global burden of atopic dermatitis: lessons from the Global Burden of Disease Study 1990–2017*. Br J Dermatol. 2021;184(2):304-309. PubMed
6.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: results from an international survey. Allergy. 2018;73(6):1284-1293. PubMed
7.Davis DMR, Drucker AM, Alikhan A, et al. Guidelines of care for the management of atopic dermatitis in adults with phototherapy and systemic therapies. J Am Acad Dermatol. 2023.
8.Chu DK, Schneider L, Asiniwasis RN, et al. Atopic dermatitis (eczema) guidelines: 2023 American Academy of Allergy, Asthma and Immunology/American College of Allergy, Asthma and Immunology Joint Task Force on Practice Parameters GRADE- and Institute of Medicine-based recommendations. Ann Allergy Asthma Immunol. 2024;132(3):274-312. PubMed
9.Clinical Study Report: 2T-DM-KGAD/DRM06-AD06. A randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of lebrikizumab when used in combination with topical corticosteroid treatment in patients with moderate-to-severe atopic dermatitis [internal sponsor's report]. Menlo Park (CA): Dermira, Inc; 2022 Apr 14.
10.Clinical Study Report: J2T-DM-KGAC/DRM06-AD05. A randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of lebrikizumab in patients with moderate to severe atopic dermatitis [internal sponsor's report]. Menlo Park (CA): Dermira, Inc; 2022 Aug 22.
11.Clinical Study Report: J2T-DM-KGAB/DRM06-AD04, A randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of lebrikizumab in patients with moderate to severe atopic dermatitis [internal sponsor's report]. Menlo Park (CA): Dermira, Inc; 2022 Aug 29.
12.Howells L, Ratib S, Chalmers JR, Bradshaw L, Thomas KS. How should minimally important change scores for the Patient-Oriented Eczema Measure be interpreted? A validation using varied methods. Br J Dermatol. 2018;178(5):1135-1142. PubMed
13.Schram ME, Spuls PI, Leeflang MMG, Lindeboom R, Bos JD, Schmitt J. EASI, (objective) SCORAD and POEM for atopic eczema: responsiveness and minimal clinically important difference. Allergy. 2012;67(1):99-106. PubMed
14.Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27-33. PubMed
15.Simpson EL, de Bruin-Weller M, Eckert L, et al. Responder threshold for Patient-Oriented Eczema Measure (POEM) and Children’s Dermatology Life Quality Index (CDLQI) in adolescents with atopic dermatitis. Dermatol Ther (Heidelb). 2019;9(4):799-805. PubMed
16.Ebglyss (lebrikizumab injection): solution for subcutaneous injection, 250 mg/2 mL single-dose prefilled pen, 250 mg/2 mL single-dose prefilled syringe with needle shield [product monograph]. Toronto (ON): Eli Lilly Canada, Inc; 2024 Jun 24.
17.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
18.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
19.Basra MK, Fenech R, Gatt RM, Salek MS, Finlay AY. The Dermatology Life Quality Index 1994-2007: a comprehensive review of validation data and clinical results. Br J Dermatol. 2008;159(5):997-1035. PubMed
20.Heinl D, Prinsen CA, Deckert S, et al. Measurement properties of adult quality-of-life measurement instruments for eczema: a systematic review. Allergy. 2016;71(3):358-370. PubMed
21.Shikiar R, Harding G, Leahy M, Lennox RD. Minimal important difference (MID) of the Dermatology Life Quality Index (DLQI): results from patients with chronic idiopathic urticaria. Health Qual Life Outcomes. 2005;3:36. PubMed
22.Eli Lilly Inc. response to December 8, 2023 request for additional information regarding lebrikizumab CADTH review: [internal additional sponsor's information]. Toronto (ON): Eli Lilly Inc; 2023 Dec 20.
23.Clinical Study Report: J2T-DM-KGAA/DRM06-AD07. A long-term study to assess the safety and efficacy of lebrikizumab in patients with moderate-to-severe atopic dermatitis [internal sponsor's report]. Menlo Park (CA): Dermira, Inc; 2022 Sep 6.
24.Lebrikizumab in atopic dermatitis network meta-analysis of outcomes at week 16 (and week 4 where applicable) technical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lebrikizumab, 250 mg/ 2 mL solution for subcutaneous injection. Toronto (ON): Eli Lilly Canada, Inc; 2023 Nov 1.
25.Sterne JAC, Savović J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898. PubMed
26.Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396(10247):345-360. PubMed
27.Boguniewicz M, Alexis AF, Beck LA, et al. Expert perspectives on management of moderate-to-severe atopic dermatitis: a multidisciplinary consensus addressing current and emerging therapies. J Allergy Clin Immunol Pract. 2017;5(6):1519-1531. PubMed
28.Global report on atopic dermatitis. London (GB): International League of Dermatological Societies: https://www.eczemacouncil.org/assets/docs/global-report-on-atopic-dermatitis-2022.pdf. Accessed 2024 Aug 7.
29.Ezzedine K, Shourick J, Merhand S, Sampogna F, Taïeb C. Impact of atopic dermatitis in adolescents and their parents: a French study. Acta Derm Venereol. 2020;100(17). PubMed
30.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis. J Am Acad Dermatol. 2014;71(1):116-132. PubMed
31.Drug Reimbursement Review sponsor submission: lebrikizumab, 250 mg/ 2 mL solution for subcutaneous injection [internal sponsor's package]. Toronto (ON): Eli Lilly Canada, Inc; 2023 Nov 1.
32.Dupixent (dupilumab injection): solution for subcutaneous injection, 300 mg single-use syringe (300 mg/2 mL), 300 mg single-use pen (300 mg/2 mL), 200 mg single-use syringe (200 mg/1.14 mL), 200 mg single-use pen (200 mg/1.14 mL), 100 mg single-use syringe (100 mg/0.67 mL) [product monograph]. Laval (QC): Sanofi-aventis Canada, Inc; 2023 Jul 12.
33.Cibinqo (abrocitinib tablets): tablets, 50 mg, 100 mg and 200 mg abrocitinib, oral [product monograph]. Kirkland (QC): Pfizer Canada, U. L. C.; 2023 Jun 30.
34.Rinvoq (upadacitinib): extended-release tablets, 15 mg upadacitinib, oral, 30 mg upadacitinib, oral, 45 mg upadacitinib, oral [product monograph]. St-Laurent (QC): AbbVie Corporation; 2023 Jul 19.
35.Sponsor's summary of clinical evidence [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lebrikizumab, 250 mg/ 2 mL solution for subcutaneous injection. Toronto (ON): Eli Lilly Canada, Inc; 2023 Nov 1.
36.Silverberg JI, Guttman-Yassky E, Thaçi D, et al. Two phase 3 trials of lebrikizumab for moderate-to-severe atopic dermatitis. N Engl J Med. 2023;388(12):1080-1091. PubMed
37.Blauvelt A, Thyssen JP, Guttman-Yassky E, et al. Efficacy and safety of lebrikizumab in moderate-to-severe atopic dermatitis: 52-week results of two randomized double-blinded placebo-controlled phase III trials. Br J Dermatol. 2023;188(6):740-748. PubMed
38.Simpson EL, Gooderham M, Wollenberg A, et al. Efficacy and safety of lebrikizumab in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis. JAMA Dermatol. 2023;159(2):182. PubMed
39.Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338-351. PubMed
40.Williams HC, Schmitt J, Thomas KS, et al. The HOME Core outcome set for clinical trials of atopic dermatitis. J Allergy Clin Immunol. 2022;149(6):1899-1911. PubMed
41.Simpson E, Beck LA, Gadkari A, et al. Defining a responder on the Peak Pruritus Numerical Rating Scale (NRS) in patients with moderate-to-severe atopic dermatitis: detailed analysis from randomized trials of dupilumab. J Am Acad Dermatol. 2017;76(6, Supplement 1):AB93.
42.Yosipovitch G, Reaney M, Mastey V, et al. Peak Pruritus Numerical Rating Scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol. 2019;181(4):761-769. PubMed
43.Hongbo Y, Thomas CL, Harrison MA, Salek MS, Finlay AY. Translating the science of quality of life into practice: What do dermatology life quality index scores mean? J Invest Dermatol. 2005;125(4):659-664. PubMed
44.Lewis-Jones MS, Finlay AY. The Children's Dermatology Life Quality Index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132(6):942-949. PubMed
45.Salek MS, Jung S, Brincat-Ruffini LA, et al. Clinical experience and psychometric properties of the Children's Dermatology Life Quality Index (CDLQI), 1995-2012. Br J Dermatol. 2013;169(4):734-759. PubMed
46.Charman CR, Venn AJ, Williams HC. The patient-oriented eczema measure: development and initial validation of a new tool for measuring atopic eczema severity from the patients' perspective. Arch Dermatol. 2004;140(12):1513-1519. PubMed
47.Silverberg JI, Lei D, Yousaf M, et al. Comparison of Patient-Oriented Eczema Measure and Patient-Oriented Scoring Atopic Dermatitis vs Eczema Area and Severity Index and other measures of atopic dermatitis: a validation study. Ann Allergy Asthma Immunol. 2020;125(1):78-83. PubMed
48.Bożek A, Reich A. Assessment of intra- and inter-rater reliability of three methods for measuring atopic dermatitis severity: EASI, Objective SCORAD, and IGA. Dermatology. 2017;233(1):16-22. PubMed
49.Barbier N, Paul C, Luger T, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150(1):96-102. PubMed
50.Hanifin JM, Thurston M, Omoto M, et al. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. Exp Dermatol. 2001;10(1):11-18. PubMed
51.Schmitt J, Langan S, Deckert S, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation. J Allergy Clin Immunol. 2013;132(6):1337-1347. PubMed
52.Barrett A, Hahn-Pedersen J, Kragh N, Evans E, Gnanasakthy A. Patient-reported outcome measures in atopic dermatitis and chronic hand eczema in adults. Patient. 2019;12(5):445-459. PubMed
53.Badia X, Mascaró JM, Lozano R. Measuring health-related quality of life in patients with mild to moderate eczema and psoriasis: clinical validity, reliability and sensitivity to change of the DLQI. The Cavide Research Group. Br J Dermatol. 1999;141(4):698-702. PubMed
54.Lewis V, Finlay AY. 10 years experience of the Dermatology Life Quality Index (DLQI). J Investig Dermatol Symp Proc. 2004;9(2):169-180. PubMed
55.Guttman-Yassky E, Blauvelt A, Eichenfield LF, et al. Efficacy and safety of lebrikizumab, a high-affinity interleukin 13 inhibitor, in adults with moderate to severe atopic dermatitis: a phase 2b randomized clinical trial. JAMA Dermatol. 2020;156(4):411-420. PubMed
56.Popay J, Roberts HF, Sowden AG, et al. Guidance on the conduct of narrative synthesis in systematic reviews. a product from the ESRC methods programme. Lancaster (GB): Lancaster University; 2026 Apr: https://www.lancaster.ac.uk/media/lancaster-university/content-assets/documents/fhm/dhr/chir/NSsynthesisguidanceVersion1-April2006.pdf. Accessed 2023 Feb 28.
57.Dias S, Sutton AJ, Welton NJ, Ades AE. NICE DSU technical support document 3: heterogeneity: subgroups, meta-regression, bias and bias-adjustment. London (GB): National Institute for Health and Care Excellence (NICE); 2012: https://www.ncbi.nlm.nih.gov/books/NBK395886/pdf/Bookshelf_NBK395886.pdf. Accessed 2024 Jan 4.
58.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU technical support document 2: a generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials London (GB): National Institute for Health and Care Excellence (NICE); 2014: https://www.ncbi.nlm.nih.gov/books/NBK310366/pdf/Bookshelf_NBK310366.pdf. Accessed 2024 Jan 4.
59.CADTH reimbursement recommendation tralokinumab (Adtralza - Leo Pharma Inc). Can J Health Technol. 2022;2(3). https://www.cadth.ca/sites/default/files/DRR/2022/SR0689%20Adtralza%20-%20Confidential%20Final%20CADTH%20Rec-Final.pdf. Accessed 2024 Jan 22.
60.Clinical Study Report: M-17923-30. A randomised, double-blind, placebo-controlled phase 3 clinical trial to assess the efficacy and safety of lebrikizumab in combination with topical corticosteroids in adult and adolescent patients with moderate-to-severe atopic dermatitis that are not adequately controlled with cyclosporine or for whom cyclosporine is not medically advisable [internal sponsor's report]. Barcelona (ES): Almirall, S. A.; 2023 Oct 19.
61.Clinical Study Report: J2T-MC-KGAK/DRM06-AD18. A phase 3, 16-week, randomized, double-blind, placebo-controlled, parallel-group study to assess the impact of lebrikizumab on vaccine responses in adult patients with moderate-to-severe atopic dermatitis [internal sponsor's report]. Indianapolis (IN): Eli Lilly Company; 2023 Mar 27.
62.Clinical Study Report: J2T-JE-KGAL. A randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of lebrikizumab when used in combination with topical corticosteroid treatment in Japanese patients with moderate-to-severe atopic dermatitis [internal sponsor's report]. Kobe Hyogo (JP): Eli Lilly Japan K.K.; 2023 Jan 25.
63.Clinical Study Report:J2T-DM-KGAE/DRM06-AD17. An open-label, single-arm study to assess the safety and efficacy of lebrikizumab in adolescent patients with moderate-to-severe atopic dermatitis [internal sponsor's report]. Menlo Park (CA): Dermira, Inc; 2022 Aug 11.
Pharmacoeconomic Review
Abbreviations
AD
atopic dermatitis
AE
adverse event
BIA
budget impact analysis
BSC
best supportive care
CDA
Canadian Dermatology Association
CDA-AMC
Canada's Drug Agency
DAO
Dermatologist Association of Ontario
EASI
Eczema Area and Severity Index
EASI-75
a reduction of at least 75% in EASI score
EASI-90
a reduction of at least 90% in EASI score
IST
immunosuppressant therapy
JAK
Janus kinase
NMA
network meta-analysis
QALY
quality-adjusted life-year
SC
subcutaneous
TCS
topical corticosteroids
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Lebrikizumab (Ebglyss) injection, solution for SC injection, 250 mg/2 mL (prefilled pen or prefilled syringe with needle shield) |
Indication | For the treatment of moderate-to-severe atopic dermatitis in adults and adolescents 12 years of age and older with a body weight of at least 40 kg whose disease is not adequately controlled with topical prescription therapies, or when those therapies are not advisable |
Health Canada approval status | Approved |
Health Canada review pathway | Standard review |
NOC date | June 24, 2024 |
Reimbursement request | As per indication |
Sponsor | Eli Lilly Canada, Inc. (Eli Lilly) |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance; SC = subcutaneous.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with moderate-to-severe AD |
Treatment | Lebrikizumab plus TCS |
Submitted price | Lebrikizumab, 250 mg/2 mL single-dose prefilled pen: $1,876.71 Lebrikizumab, 250 mg/2 mL single-dose prefilled syringe with needle shield: $1,876.71 |
Submitted treatment cost | First year: $35,657 Subsequent years: $24,397 |
Comparators | Abrocitinib 100 mg plus TCS Abrocitinib 200 mg plus TCS Dupilumab plus TCS Upadacitinib 15 mg plus TCS Upadacitinib 30 mg plus TCS BSC, assumed to be equivalent to placebo |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (70 years) |
Key data source | Clinical efficacy data were informed by sponsor-submitted NMAs |
Submitted results | Lebrikizumab plus TCS was dominated (i.e., associated with fewer total QALYs and greater total costs) by abrocitinib 100 mg plus TCS, upadacitinib 15 mg plus TCS, and abrocitinib 200 mg plus TCS |
Key limitations |
|
CDA-AMC reanalysis results |
|
AD = atopic dermatitis; BSC = best supportive care; CDA-AMC = Canada's Drug Agency; EASI = Eczema Area and Severity Index; JAK = Janus kinase; LY = life-year; NMA = network meta-analysis; QALY = quality-adjusted life-year; TCS = topical corticosteroids.
Conclusions
The CDA-AMC clinical review found that, compared with placebo, lebrikizumab (Ebglyss), whether used as monotherapy or in combination with topical corticosteroids (TCS), provided a clinically relevant improvement in physician-assessed signs of atopic dermatitis (AD) and reduced patient-reported symptoms of itch relative to placebo at week 16, based on 3 pivotal randomized controlled trials. There was no direct evidence comparing lebrikizumab with other biologics or Janus kinase (JAK) inhibitors used to treat AD in Canada; however, the sponsor submitted indirect evidence from a network meta-analysis (NMA) that assessed short-term comparative efficacy. In the combination-therapy NMA for the Eczema Area and Severity Index (EASI) response outcome, the evidence was insufficient to show a difference between lebrikizumab compared with dupilumab, abrocitinib 100 mg, or upadacitinib 15 mg. The NMA results suggested that abrocitinib 200 mg and upadacitinib 30 mg may result in a greater proportion of patients achieving an EASI response. Results were similar (i.e., either insufficient evidence to show a difference between lebrikizumab compared with other comparators, or other comparators led to greater improvements — such as an increased Investigator Global Assessment for AD response or reduced itch — than lebrikizumab) across all other outcomes considered in the NMA in both the combination-therapy and the monotherapy networks. The NMA did not assess any safety end points; thus, the comparative safety of lebrikizumab relative to other biologics or JAK inhibitors is unknown.
CDA-AMC undertook reanalyses to address limitations in the sponsor’s economic model, resulting in a CDA-AMC base case with findings that were generally aligned with those submitted by the sponsor: lebrikizumab is dominated by (i.e., associated with higher total costs and lower quality-adjusted life-years [QALYs]) a number of biologics and JAK inhibitors used to treat AD in Canada (i.e., abrocitinib 100 mg, abrocitinib 200 mg, and upadacitinib 15 mg).
Given the uncertainty in the clinical evidence — the sponsor-submitted NMA suggested that there is either insufficient evidence to show a difference between lebrikizumab and comparators and, in some cases, lebrikizumab may result in less favourable clinical outcomes — there is no clinical evidence to support a price premium for lebrikizumab over other biologics or JAK inhibitors used to treat AD in Canada. Thus, to ensure cost-effectiveness, lebrikizumab should be priced no more than the lowest-cost biologic or JAK inhibitor that is funded in the population to be reimbursed.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process.
Patient input was received from 3 patient groups: Eczema Quebec, The Canadian Skin Patient Alliance; and the Eczema Society of Canada. Eczema Quebec gathered information through a review of scientific literature, informal conversations with patients, 9 written patient testimonials, interviews with 14 patients, and feedback from 3 patient group discussions. The Canadian Skin Patient Alliance gathered information from previous CADTH reviews, guidelines on the management of AD, and data from the Canadian Institute for Health Information on AD-related emergency department visits and hospitalizations. The Eczema Society of Canada gathered information through surveys and interviews with more than 3,000 patients with AD and their caregivers who reside in Canada. Patients reported that topical treatments that are not eligible for reimbursement and other nongeneric products contribute to the financial burden of managing AD. Major concerns regarding current topical treatments include adverse effects such as skin thinning, skin discoloration, increased intraocular pressure, cataracts, and hormonal disturbances. Input also noted that current topical treatments are inconvenient to use and are associated with high costs. Major concerns regarding biologics were injection-site reactions and conjunctivitis. Concerns regarding the use of JAK inhibitors included side effects (e.g., upper respiratory tract infections, headaches, and mild gastrointestinal symptoms). Additionally, as JAK inhibitors are relatively new treatments, patients noted that the potential long-term effects of these treatments remain a concern. No patients had experience with lebrikizumab. Patients noted that desired treatment options would include treatments that do not require injection (including topical treatments), that are covered by insurance or are affordable, that are easy to use, that produce fast results with long-term control of the disease, and that reduce flares, reduce skin redness and inflammation, and improve daily and social activities.
CDA-AMC received input from 2 clinician groups: the Canadian Dermatology Association (CDA); and the Dermatology Association of Ontario (DAO). Input from the DAO noted that the reimbursement of lebrikizumab is not predicted to cause a shift in the treatment paradigm, and that lebrikizumab would fit well as an additional treatment option. The CDA, conversely, noted that lebrikizumab would cause a shift in the current treatment paradigm toward a focus on novel disease mechanisms with favourable safety and efficacy profiles. The CDA noted that lebrikizumab would adopt the same criteria as are used for dupilumab and would fit as a first-line biologic therapy for patients with moderate-to-severe AD. The DAO indicated that adult patients with moderate-to-severe AD who have failed topical therapies and those who have failed or do not have access to phototherapy would be best suited for the treatment with lebrikizumab, whereas the CDA stated that patients best suited for treatment with lebrikizumab would be those with uncontrolled moderate-to-severe AD who are candidates for systemic therapy or who meet the criteria for biologic therapy.
Drug plan input for this review noted that no head-to-head studies were identified for lebrikizumab versus treatments that are funded in most jurisdictions. The public drug plans sought input on the cost-effectiveness of lebrikizumab in comparison to dupilumab. The plans noted that the requested reimbursement indication for lebrikizumab differs from the indication for dupilumab, as it does not include patients who are refractory to or ineligible for systemic immunosuppressant therapies (ISTs).
Several of these concerns were addressed in the sponsor’s model:
Adverse events (AEs) were included in the sponsor’s submission.
Response in the model was based on a reduction of at least 75% in EASI score (EASI-75). The EASI assesses disease extent in the head and neck, trunk, and upper and lower limbs, and clinical signs (erythema, induration or papulation, excoriation, and lichenification) for each body region.
The sponsor included a one-time cost for subcutaneous (SC) injections for lebrikizumab and dupilumab.
The effectiveness of lebrikizumab relative to other treatments was informed by an NMA submitted by the sponsor.
CDA-AMC was unable to address the following concerns raised from stakeholder input:
Disutilities for SC injections were not included in the sponsor-submitted model and could not be included in the CDA-AMC reanalysis.
There is a lack of direct comparative evidence between lebrikizumab and other relevant comparators in the modelled population. There is also a lack of comparative evidence to between lebrikizumab and other relevant comparators in patients who are refractory to or ineligible for systemic ISTs.
AE rates and discontinuation rates are uncertain due to a lack of indirect or direct evidence.
Economic Review
The current review is for lebrikizumab (Ebglyss) for the treatment of adult and adolescent patients 12 years of age and older with moderate-to-severe AD with a body weight of at least 40 kg whose disease is not adequately controlled with topical prescription therapies, or when those therapies are not advisable.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis that compared lebrikizumab plus TCS with abrocitinib plus TCS (100 mg and 200 mg), dupilumab plus TCS, upadacitinib plus TCS (15 mg and 30 mg), and best supportive care (BSC). BSC was not defined in the sponsor’s submission. The sponsor also submitted a scenario analysis of lebrikizumab as monotherapy; in this analysis, all comparators were also examined as monotherapy. The modelled population was based on the characteristics of the ADvocate 1 and ADvocate 2 trial populations.1 The target population is aligned with the Health Canada–indicated population.
Lebrikizumab is available as a single-dose prefilled pen and a single-dose prefilled syringe with needle shield, containing 250 mg/2mL of solution for SC injection at a submitted price of $1,876.7100 per prefilled pen or prefilled syringe with needle shield.2 The recommended dosage is initial doses of 500 mg injected subcutaneously at week 0 and week 2, followed by 250 mg every 2 weeks until week 16, at which time clinical response is assessed.3 Upon clinical response, the recommended maintenance dose is 250 mg every 4 weeks starting at week 16.3 In the sponsor-submitted model, a maintenance dose of 250 mg every 4 weeks was used. The product monograph notes that lebrikizumab can be used with or without TCS or topical calcineurin inhibitors.
Based on sponsor data on file, a total of 19 doses of lebrikizumab is required in the first year of treatment, with a total of 13 doses required every year thereafter.2 As a result, in the sponsor’s model, the total cost of lebrikizumab was $35,741 and $24,481 in the first year and subsequent years of treatment, respectively.2
The economic analysis was undertaken over a lifetime time horizon (assumed to be 70 years) from the perspective of the Canadian publicly funded health care system. Costs and clinical outcomes (life-years and QALYs) were discounted at a rate of 1.5% per annum.
Model Structure
The sponsor submitted a Markov model structure with 4 health states that had 4-week cycle lengths: induction, maintenance, BSC, and death. All patients enter the model in the induction health state, representing the induction phase of treatment, and receive the selected first-line treatment (i.e., lebrikizumab plus TCS or 1 of the other comparators plus TCS). Patients remain in the induction health state for up to 16 weeks, depending on the treatment. At the end of the induction period, response, defined in the sponsor’s base case as the achievement of an EASI-75, is assessed. Patients who respond to the treatment at the end of the induction period then transition to the maintenance health state where they continue to receive their initial treatment until discontinuation, or they transition to the death health state. Patients who do not respond in the induction phase or do not maintain the response in the maintenance phase discontinue treatment and move to the induction health state of the next treatment option. Patients can repeat this process for up to 2 lines of treatments in the model; those who do not respond to first-line and second-line treatments transition to the last line of the BSC health state. In the sponsor’s base case, BSC is the next treatment option if patients do not respond to first-line treatment with lebrikizumab or other biologics or JAK inhibitors (i.e., only 1 line of therapy is assessed, and all patients move to the BSC health state if they do not respond or if they discontinue a drug). The conceptual model is provided in Appendix 3 (Figure 1).
Model Inputs
The model’s baseline population characteristics were derived from the ADvocate 1 and ADvocate 2 trial populations, 2 phase III, randomized, placebo-controlled trials that compared lebrikizumab monotherapy with placebo for the treatment of moderate-to-severe AD (mean age, 35.9 years; 49.9% female, 12% adolescents).1 In the base case, the sponsor compared lebrikizumab in combination with TCS to abrocitinib, dupilumab, and upadacitinib (all in combination with TCS), and to BSC. As such, the pharmacoeconomic model was primarily informed by inputs from the ADhere trial, a phase III, randomized, placebo-controlled trial that assessed the safety and efficacy of lebrikizumab in combination with TCS compared with placebo in combination with TCS in patients with moderate-to-severe AD.4 Subgroup analyses for background therapy (i.e., lebrikizumab monotherapy) and prior cyclosporine exposure were performed (in those who were cyclosporine naïve and in those with prior cyclosporine exposure).2
Clinical efficacy inputs in the sponsor’s submitted base case were derived from the combination-therapy network of the sponsor-conducted NMAs.5 In the sponsor-submitted base case, response was defined based on EASI-75. Transitions from the induction health state to the maintenance health state or to the BSC health state were, therefore, based on the proportion of patients achieving an EASI-75 derived from the NMA.
The sponsor-submitted model allowed for exploration of discontinuation for patients in the maintenance phase. An all-cause discontinuation rate of 6.3% was identified from the CADTH dupilumab review and was assumed to apply to all comparators.2,6 Mortality was based on age-specific and sex-specific all-cause mortality data from Statistics Canada.7 No additional AD-related disease-specific mortality was applied.2 Treatment-emergent AEs were included in the model. AE rates for lebrikizumab were based on an integrated analysis of 8 lebrikizumab clinical trials,8 and rates for all other active comparators were naively derived from a National Institute for Health and Care Excellence (NICE) report.9
Health-state utility values used in the base case were derived from the ADhere study.4 The health-state utility value for the induction and BSC health states were based on the baseline utility value in the ADhere study (██████). The utility in the maintenance health state was based on a weighted average of the proportion of people who achieved an EASI-75 versus a reduction of at least 90% in EASI score (EASI-90) on a given treatment, multiplied by the EASI-75 (██████) and EASI-90 (██████) utility values, resulting in treatment-specific maintenance utility values.2,4 Utilities were age-adjusted based on the general population utilities in Canada.10 AE disutilities were sourced from an Institute for Clinical and Economic Review report and were included in the base case.11
Costs in the model included drug-acquisition and administration costs, disease-management costs, and AE costs. Drug-acquisition costs for lebrikizumab were based on the sponsor’s submitted price.2 Unit costs for comparators were sourced from the IQVIA DeltaPA database.12 Costs of concomitant medications were not included in the base-case analysis, as the sponsor assumed that concomitant medication usage would be similar across treatments.2 Patients receiving SC treatments were assumed to incur a one-time administration cost for their first injection; subsequent doses were assumed to be self-administered.2,13 Disease-management costs, including dermatologist consultation, general practitioner consultation, accident and emergency department visits, and full blood counts, differed between the induction phase and the maintenance phase and between JAK inhibitors and biologic treatment.2 Unit costs for disease management were derived from the Ontario Schedule of Benefits of Physician Services, the Canadian Institute for Health Information, and a report by Hale.14-16 AE costs were derived from Health Data Branch Web Portal in Canada.17
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 1,000 iterations. In the sponsor submission, the deterministic total cost and total QALY results were similar to the probabilistic results; however, incremental results were different. In the deterministic results, dupilumab plus TCS was reported to have fewer total QALYs than lebrikizumab plus TCS, whereas in the probabilistic analysis, dupilumab plus TCS had more total QALYs than lebrikizumab plus TCS. The probabilistic findings follow. Deterministic results are presented in Appendix 3 (Table 10).
Base-Case Results
In the sponsor’s base-case analysis over a 70-year time horizon, lebrikizumab plus TCS was dominated by abrocitinib 100 mg plus TCS, upadacitinib 15 mg plus TCS, and abrocitinib 200 mg plus TCS (Table 3). Apart from BSC, lebrikizumab plus TCS was associated fewer total QALYs than all comparators. Lebrikizumab plus TCS was also more costly than all comparators, apart from dupilumab plus TCS and upadacitinib 30 mg plus TCS, which were both associated with higher total QALYs than lebrikizumab (Table 3). Total life-years across all treatments were reported to be the same, at 39.236. At a willingness-to-pay threshold of $50,000 per QALY, there was a 0% probability of lebrikizumab plus TCS being cost-effective. Additional results from the sponsor’s submitted economic evaluation base case, including deterministic analysis results, are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. BSC ($/QALY) | Sequential ICER ($/QALY) |
|---|---|---|---|---|
BSC | 132,116 | 31.971 | Reference | Reference |
Upadacitinib 15 mg | 271,489 | 32.500 | 263,364 | 263,364 vs. BSC |
Upadacitinib 30 mg | 391,603 | 32.610 | 405,753 | 1,088,819 vs. upadacitinib 15 mg |
Dominated treatments | ||||
Abrocitinib 100 mg | 258,348 | 32.445 | 266,308 | Extendedly dominated by upadacitinib 15 mg |
Abrocitinib 200 mg | 294,457 | 32.518 | 296,463 | Extendedly dominated by upadacitinib 15 mg |
Lebrikizumab | 314,584 | 32.443 | 386,372 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, and abrocitinib 200 mg |
Dupilumab | 330,045 | 32.482 | 387,441 | Dominated by upadacitinib 15 mg and abrocitinib 200 mg |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: All treatments were used in combination with topical corticosteroids.
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analysis Results
The sponsor conducted subgroup analyses that assessed the cost-effectiveness of lebrikizumab as a monotherapy treatment for the population of interest and for patients who had received prior cyclosporin treatment. In the scenario analysis that assessed the cost-effectiveness of lebrikizumab as a monotherapy treatment, lebrikizumab was dominated by upadacitinib 15 mg. In the second scenario analysis, in which the target population had prior cyclosporine exposure, only BSC, lebrikizumab plus TCS, and dupilumab plus TCS were included as comparators. In this scenario, lebrikizumab plus TCS was associated with an incremental cost-effectiveness ratio of $438,737 compared to BSC.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative clinical efficacy of lebrikizumab and other comparators is uncertain. There was no direct evidence comparing lebrikizumab to other biologics or JAK inhibitors used to treat AD in Canada. To inform the pharmacoeconomic analysis, the sponsor conducted NMAs that assessed the short-term efficacy of lebrikizumab compared to biologics and JAK inhibitors. According to the CDA-AMC Clinical Review Report, in the primary analysis for EASI response at week 16 in the combination-therapy network, there was insufficient evidence to show a difference between lebrikizumab plus TCS and dupilumab plus TCS, abrocitinib 100 mg plus TCS, or upadacitinib 15 mg plus TCS. Further, the indirect evidence submitted by the sponsor suggested that abrocitinib 200 mg plus TCS and upadacitinib 30 mg plus TCS were favoured over lebrikizumab plus TCS in the combination-therapy network.
Additionally, in the primary analysis of EASI response at week 16 in the monotherapy network, the CDA-AMC Clinical Review Report noted there was insufficient evidence to show a difference between lebrikizumab, dupilumab, and abrocitinib 100 mg. In the monotherapy network, the CDA-AMC Clinical Review Report noted that abrocitinib 200 mg, upadacitinib 15 mg, and upadacitinib 30 mg were favoured over lebrikizumab. Overall, the results of the NMA for both the combination and monotherapy networks were inconclusive for lebrikizumab compared with dupilumab and abrocitinib, with most estimates crossing the null threshold, which suggested notable imprecision. The results suggested that upadacitinib may be favoured over lebrikizumab; however, differences were not consistently detected, and the clinical relevance of any differences is unclear.
Due to the lack of direct evidence and limitations of the comparative evidence used by the sponsor in the pharmacoeconomic analysis, the cost-effectiveness of lebrikizumab compared with other biologics and JAK inhibitors is highly uncertain. Because the sponsor-submitted NMAs suggested that there may be no difference in efficacy between lebrikizumab plus TCS and other biologics and JAK inhibitors (used in combination with TCS), it is uncertain whether lebrikizumab provides a net benefit relative to currently funded treatments. CDA-AMC was unable to address this limitation in reanalyses.
The relevance of BSC as a comparator is uncertain. In the sponsor-submitted pharmacoeconomic analysis, BSC was included as a comparator to lebrikizumab plus TCS. Details regarding what BSC was assumed to be comprised of were not provided in the sponsor’s submission. Further, in the sponsor’s analysis, no costs were associated with BSC; it was assumed that the use of BSC would not vary between treatments, even if it was used alone. According to clinical expert feedback obtained by CDA-AMC for this review, BSC would consist of treatment with prescription and over-the-counter emollients and anti-inflammatory treatments such as TCS and calcineurin inhibitors. Further, clinical expert feedback emphasized that, given the number of existing biologic and JAK inhibitor treatments available to treat moderate-to-severe AD, the proportion of people in this patient population that would receive BSC rather than the other comparators is very low (approximately 5% to 10%). Clinical expert feedback also highlighted the fact that the proportion of patients switching from BSC to lebrikizumab plus TCS, should it become available, would be negligible (1% to 2%). As such, the relevance of BSC as a comparator to lebrikizumab plus TCS is highly uncertain. Uncertainty regarding the relevance of BSC as a comparator is further highlighted in the sponsor’s budget impact analysis (BIA), which did not include BSC as a comparator. As such, the BIA implies that the entry of lebrikizumab will not impact the proportion of patients receiving BSC, meaning that BSC will not be displaced by lebrikizumab.
Furthermore, comparative efficacy data that could inform a comparison of lebrikizumab plus TCS and BSC were not available. The comparative efficacy of lebrikizumab plus TCS versus BSC in the sponsor’s base case was informed by the sponsor’s submitted NMA results for placebo. As reported in the CDA-AMC Clinical Review Report, the 3 pivotal randomized, placebo-controlled trials included in the sponsor’s submission or the NMA did not include BSC as a comparator for lebrikizumab plus TCS. In the ADhere trial, patients receiving both lebrikizumab and placebo received concomitant TCS, meaning that comparative effect estimates from this trial are for lebrikizumab versus placebo rather than BSC. Further, in the ADvocate trials, patients were prohibited from using other AD treatments, aside from topical emollients, meaning that the comparative efficacy estimates are for lebrikizumab versus placebo rather than BSC. In the sponsor-submitted NMAs, the analyses for both the monotherapy and combination-therapy networks only included placebo; no comparisons were made against BSC. Therefore, the submitted sponsor analysis did not include any data that evaluated the use of lebrikizumab plus TCS in comparison to BSC. This adds a high level of uncertainty to the analysis, as the efficacy of lebrikizumab compared to BSC could not be derived.
Due to uncertainty regarding the relevance of BSC as a comparator in the pharmacoeconomic evaluation and the lack of comparative efficacy data to inform relative effects between BSC and lebrikizumab plus TCS, BSC was removed as a comparator in the CDA-AMC reanalysis. BSC was included as a comparator in a scenario analysis.
AE rates are naively derived. AEs were not included as an outcome in the sponsor’s NMA. As such, in the sponsor’s submitted pharmacoeconomic analysis, 16-week rates of AEs (injection-site reaction, allergic conjunctivitis, infectious conjunctivitis, and oral herpes) were derived from a study by Stein Gold et al.8 for lebrikizumab and from a NICE report for all comparators treatments.9 As such, AE rates were naively derived from each comparator’s respective clinical trials. The resulting rates of AEs were notably higher for dupilumab than for the other biologics, JAK inhibitors, and lebrikizumab. The higher AE rates associated with dupilumab were the driver for the lower total QALYs in the sponsor’s deterministic base-case analysis of dupilumab plus TCS compared with lebrikizumab plus TCS, despite dupilumab plus TCS being associated with a better response rate than lebrikizumab plus TCS.
Because no direct or indirect evidence was available to inform AE rates in the model, it is uncertain if the presented rates are representative of the target population or whether differences in AE rates are due to differences in baseline population characteristics between clinical trials. Clinical expert feedback obtained by CDA-AMC suggested that rates of AEs are expected to be low with all treatment options. Clinical expert feedback noted that although dupilumab likely has the highest risk of conjunctivitis, the treatment is unlikely to be associated with a higher risk for other AEs. The CDA-AMC Clinical Review Report emphasized that lebrikizumab may increase the short-term risk of conjunctivitis relative to placebo; however, the NMA did not assess any safety end points and, therefore, the comparative safety of lebrikizumab is unknown.
CDA-AMC was unable to address the lack of direct or indirect evidence that could inform comparative AE rates. To address the unresolved uncertainty in AE rates, CDA-AMC performed a scenario analysis that removed AEs from the reanalysis.
The use of treatment-specific health-state utility values is inappropriate. In the sponsor’s submitted pharmacoeconomic analysis, the health-state utility value for the maintenance health state was derived by multiplying the distribution of responders (i.e., the proportion of responders on a given treatment who achieved an EASI-75 and the proportion who achieved an EASI-90) by the EASI-75 and EASI-90 utility values.2 Because the distribution of response varied by treatment, the maintenance health utility values were treatment-specific. The use of treatment-specific utility values is contradictory to the CDA-AMC recommendation that utilities should reflect the health states in the economic model.18 Instead, all outcomes associated with treatment, along with their impact on patient utility, should be explicitly modelled, rather than captured using a treatment-specific utility value. Including treatment-specific utilities to capture a difference in consequences between treatments that has not been modelled is, therefore, inappropriate. Given that the response rate for lebrikizumab plus TCS was lower than that for all other biologic and JAK inhibitor treatments, the use of treatment-specific utility values in the sponsor’ base case was conservative.
In the CDA-AMC reanalysis, the maintenance health-state utility value was equal for all comparators and reflected the response outcome (EASI-75) used to define the health state.
Upadacitinib costs are underestimated in the sponsor’s analysis. In their pharmacoeconomic analysis, the sponsor used unit costs for upadacitinib 15 mg and upadacitinib 30 mg from the IQVIA DeltaPA database.19 CDA-AMC noted that pricing for upadacitinib is available from the Ontario Drug Benefit Formulary20 for 15 mg and from the Exceptional Access Program for 30 mg.21
CDA-AMC corrected the cost of upadacitinib based on the formulary prices.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Lebrikizumab was assumed to be used in combination with TCS in the sponsor’s base case. | Likely appropriate. According to clinical expert feedback received for this review, the majority of the use of lebrikizumab is expected to be as combination therapy. To highlight its cost-effectiveness as a monotherapy, CDA-AMC conducted a scenario analysis in the monotherapy population. |
Discontinuation rates among responders for all treatments were assumed to be equal. | Uncertain, but unlikely to have an important effect on the ICER. Clinical expert feedback received by CDA-AMC indicated that discontinuation rates maybe higher among patients who respond to JAK inhibitors than to biologics, due to safety concerns, particularly with long-term use; however, data to inform treatment-specific discontinuation rates were not available. |
A 75% reduction in EASI score from baseline was assumed to represent a treatment response. | Reasonable. Clinical expert feedback received by CDA-AMC indicated that an EASI-75 score would likely represent a clinically meaningful reduction in eczema severity. CDA-AMC noted that EASI-75 has been used in previous submissions in this clinical area. Treatment decisions in practice are not made based on the EASI score, although the EASI score is routinely used due to reimbursement requirements. |
Resource use per year differed when patients were using biologics rather than JAK inhibitors; JAK inhibitors were associated with higher resource use. | Reasonable. Clinical expert feedback received by CDA-AMC emphasized that monitoring lab work is not required for patients receiving biologics; therefore, it is reasonable for JAK inhibitors to be associated with higher resource use. |
No cost was incorporated for BSC, reflecting the assumption that the use of BSC would not vary between treatments. | Uncertain, but unlikely to have an important effect on the ICER. Clinical expert feedback received by CDA-AMC indicated that is it reasonable to assume similar BSC use between treatments; however, patients may be able to reduce the number of topical treatments used based on response. If this is the case, not including BSC costs would favour lebrikizumab, which had the poorest response rate among biologics and JAK inhibitor comparators. |
All patients would receive BSC in the second line if they do not respond to biologics or JAK inhibitors in the first line. | Likely inappropriate. Clinical expert feedback received by CDA-AMC noted that patients who do not respond to a biologic or JAK inhibitor in the first line will likely move onto a second biologic or JAK inhibitor. Clinical expert feedback indicated that BSC is expected to be a third-line or fourth-line therapy. Although the sponsor’s model incorporated the ability to explore alternative second-line therapies, it is unclear whether the data used to inform efficacy in the second line was based on trials conducted in the first line or second line of treatment. CDA-AMC reviewed the impact of including a second biologic or JAK inhibitor as a second-line treatment and found that it did not have impactful results. |
The modelled population, which explored lebrikizumab in combination with TCS, was based on characteristics from the ADvocate 1 and ADvocate 2 trial populations, which were monotherapy trials. | Uncertain but likely reasonable. The ADvocate 1, ADvocate 2 (monotherapy), and ADhere (combination therapy) trials had similar baseline patient characteristics. Changes in characteristics are unlikely to have an important effect on the ICER. |
Abrocitinib monotherapy had an induction period of 12 weeks, whereas all other treatments had a 16-week induction period. | Reasonable. |
BSC = best supportive care; CDA-AMC = Canada's Drug Agency; EASI = Eczema Area and Severity Index; EASI-75 = a reduction of at least 75% in EASI score; ICER = incremental cost-effectiveness ratio; JAK = Janus kinase; TCS = topical corticosteroids.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC undertook reanalyses that addressed limitations of the model, as summarized in Table 5. The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. All CDA-AMC probabilistic reanalyses were based on 1,000 iterations. CDA-AMC was unable to address other key limitations, including the lack of robust comparative clinical effectiveness data, including data to inform the impact of AE rates.
CDA-AMC undertook a stepped analysis incorporating each change proposed in Table 5 to the sponsor’s base case to highlight the impact of each change (Table 11). As in the sponsor’s base case, lebrikizumab plus TCS was associated with the fewest total QALYs compared with all biologic and JAK inhibitor comparators and was more costly than all comparators apart from dupilumab plus TCS and upadacitinib 30 mg plus TCS, which were both associated with higher total QALYs than lebrikizumab plus TCS (Table 6). As a result, findings from the CDA-AMC base case were similar to those of the sponsor’s: lebrikizumab plus TCS was dominated (i.e., more costly and less effective) by abrocitinib 100 mg, abrocitinib 200 mg, and upadacitinib 15 mg (all in combination with TCS) (Table 6). The CDA-AMC base-case results were driven by drug-acquisition costs and small differences in response rates and AE rates between the comparators (Table 12).
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
1. Incorrect comparator pricing | Unit costs for upadacitinib were sourced from the IQVIA DeltaPA database.12 Upadacitinib 15 mg = $48.6800 Upadacitinib 30 mg = $74.0000 | Unit costs for upadacitinib are available in the ODB (15 mg)20 and EAP (30 mg).21 Upadacitinib 15 mg = $51.6810 Upadacitinib 30 mg = $76.9600 |
Changes to derive the CDA-AMC base case | ||
1. BSC included as a comparator | Included | Excluded |
2. Maintenance health-state utility value | Treatment specific | Health-state specifica |
CDA-AMC base case | Reanalysis 1 + 2 | |
BSC = best supportive care; CDA-AMC = Canada's Drug Agency; EAP = Exceptional Access Program; ODB = Ontario Drug Benefit Formulary.
aThis change was derived by aligning the health-state utility value with the response outcome (EASI-75) selected for the model (i.e., EASI-75 utility = ██████).
Scenario Analysis Results
Based on the comparative clinical information submitted by the sponsor, there is insufficient evidence to show a difference in efficacy between lebrikizumab plus TCS and dupilumab plus TCS, abrocitinib 100 mg plus TCS, or upadacitinib 15 mg plus TCS; further, lebrikizumab plus TCS may result in less favourable clinical outcomes than abrocitinib 200 mg plus TCS and upadacitinib 30 mg plus TCS. As such, there is no clinical evidence to support a price premium for lebrikizumab. To ensure cost-effectiveness, lebrikizumab should not be priced higher than other biologics and JAK inhibitors used to treat AD in Canada. CDA-AMC undertook price reduction analyses based on the sponsor’s base case and found that a price reduction of 79% would be required in order for lebrikizumab to be considered cost-effective compared with BSC at a willingness-to-pay threshold of $50,000 per QALY gained (Table 13).
Scenario analyses were conducted using CDA-AMC reanalyses to investigate the impact of:
including BSC as a comparator
removing the impact of AEs from the CDA-AMC base case due to uncertainties resulting from the AE rates being naively derived in the sponsor-submitted pharmacoeconomic model
evaluating lebrikizumab monotherapy.
The results of these scenario analyses are presented in Appendix 4 (Table 14). Including BSC as a comparator did not change results in the sequential analysis for lebrikizumab; it remained dominated by abrocitinib 100 mg, upadacitinib 15 mg, and abrocitinib 200 mg. Removing AEs from the analysis resulted in dupilumab plus TCS being associated with higher total QALYs than lebrikizumab plus TCS. Lebrikizumab plus TCS remained dominated by abrocitinib 100 mg plus TCS, abrocitinib 200 mg plus TCS, and upadacitinib 15 mg plus TCS.
In the scenario analysis that assessed the impact of lebrikizumab and comparators as a monotherapy treatments, lebrikizumab was dominated by upadacitinib 15 mg.
Table 6: Summary of the CDA-AMC Reanalysis Res ults
Drug | Total costs | Total QALYs | ICER vs. reference treatment ($/QALY) | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Sponsor-corrected base case | ||||
BSC | 132,218 | 31.884 | Reference | Reference |
Abrocitinib 100 mg | 259,093 | 32.373 | 259,634 | 259,634 vs. BSC |
Upadacitinib 15 mg | 281,134 | 32.421 | 277,682 | 462,925 vs. abrocitinib 100 mg |
Abrocitinib 200 mg | 294,487 | 32.441 | 291,417 | 649,912 vs. upadacitinib 15 mg |
Upadacitinib 30 mg | 402,750 | 32.531 | 418,268 | 1,203,409 vs. abrocitinib 200 mg |
Dominated treatments | ||||
Lebrikizumab | 313,593 | 32.332 | 405,672 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg |
Dupilumab | 330,136 | 32.282 | 497,983 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg |
CDA-AMC base case | ||||
Abrocitinib 100 mg | 259,355 | 32.337 | Reference | Reference |
Upadacitinib 15 mg | 281,530 | 32.385 | 462,818 | 462,818 vs. abrocitinib 100 mg |
Abrocitinib 200 mg | 294,269 | 32.396 | 588,151 | 1,112,646 vs. upadacitinib 15 mg |
Upadacitinib 30 mg | 402,395 | 32.472 | 1,059,243 | 1,428,775 vs. abrocitinib 100 mg |
Dominated treatments | ||||
Lebrikizumab | 313,860 | 32.328 | Dominated by abrocitinib 100 mg | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg |
Dupilumab | 331,060 | 32.368 | 2,297,200 | Dominated by upadacitinib 15 mg, abrocitinib 200 mg |
BSC = best supportive care; CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: All treatments were used in combination with topical corticosteroids.
Issues for Consideration
Ruxolitinib is undergoing a concurrent reimbursement review by CDA-AMC for the treatment of patients with AD in patients aged 12 years and older whose disease is not adequately controlled with conventional topical prescription therapies (topical calcineurin inhibitors, TCS), or when those therapies are not advisable.22
Tralokinumab underwent a reimbursement review by CADTH for the treatment of AD in patients aged 12 years and older whose disease is not adequately controlled with conventional topical prescription therapies or when those therapies are not advisable. The Canadian Drug Expert Committee recommended that tralokinumab not be reimbursed on November 16, 2023.23
Overall Conclusions
The CDA-AMC clinical review found that, compared with placebo, lebrikizumab, whether used as monotherapy or in combination with TCS, provided a clinically relevant improvement in physician-assessed signs of AD and reduced patient-reported symptoms of itch relative to placebo at week 16, based on 3 pivotal randomized controlled trials. There was no direct evidence comparing lebrikizumab with other biologics or JAK inhibitors used to treat AD in Canada; however, the sponsor submitted indirect evidence from a NMA that assessed short-term comparative efficacy. In the combination-therapy NMA, the evidence for the EASI response outcome was insufficient to show a difference between lebrikizumab and dupilumab, abrocitinib 100 mg, or upadacitinib 15 mg. The NMA results suggested that abrocitinib 200 mg and upadacitinib 30 mg may result in a greater proportion of patients achieving an EASI response. Results were similar (i.e., either insufficient evidence to show a difference between lebrikizumab compared with other comparators, or other comparators led to greater improvements — such as an increased Investigator Global Assessment for AD response or reduced itch — than lebrikizumab) across all other outcomes considered in the NMA in both the combination-therapy and the monotherapy networks. The NMA did not assess any safety end points; thus, the comparative safety of lebrikizumab relative to other biologics or JAK inhibitors is unknown.
CDA-AMC undertook reanalyses to address limitations in the sponsor’s economic model by excluding BSC as a comparator and by removing treatment-specific utilities in the maintenance treatment health state. CDA-AMC base-case findings were generally aligned with those submitted by the sponsor: lebrikizumab is dominated by (i.e., associated with higher total costs and lower QALYs) a number of biologics and JAK inhibitors used to treat AD in Canada (i.e., abrocitinib 100 mg, abrocitinib 200 mg, and upadacitinib 15 mg).
Given the uncertainty in the clinical evidence — the sponsor-submitted NMA suggested that there is either insufficient evidence to show a difference between lebrikizumab and comparators and in some cases, lebrikizumab may result in less favourable clinical outcomes — there is no clinical evidence to support a price premium for lebrikizumab over other biologics or JAK inhibitors used to treat AD in Canada. Thus, to ensure cost-effectiveness, lebrikizumab should be priced no more than the lowest-cost biologic or JAK inhibitor that is funded in the population to be reimbursed.
References
1.Warren RB, Silverberg JI, Guttman-Yassky E, et al. Efficacy and safety of lebrikizumab at 16 weeks: pooled analyses from ADvocate1 and ADvocate2 phase III trials in patients with moderate-to-severe atopic dermatitis. Br J Dermatol. 2023;188(Supplement 4):iv54.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lebrikizumab, 250 mg/ 2 mL solution for subcutaneous injection Toronto (ON): Eli Lilly Canada, Inc; 2023 Oct 31.
3.Ebglyss (lebrikizumab injection): solution for subcutaneous injection, 250 mg/2 mL single-dose prefilled pen, 250 mg/2 mL single-dose prefilled syringe with needle shield [product monograph]. Toronto (ON): Eli Lilly Canada, Inc; 2024 Jun 24.
4.Eli Lilly and Company. Safety and efficacy of lebrikizumab (LY3650150) in combination with topical corticosteroid in moderate-to-severe atopic dermatitis (ADhere). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2023: https://classic.clinicaltrials.gov/ct2/show/NCT04250337?term=adhere&cond=Atopic+Dermatitis&draw=2&rank=1. Accessed 2023 Dec 1.
5.Network Meta Analysis of outcomes at week 16 (and week 4 where applicable) technical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lebrikizumab, 250 mg/ 2 mL solution for subcutaneous injection Toronto (ON): Eli Lilly Canada, Inc; 2023 Nov 1.
6.CADTH reimbursement review. Dupilumab (Dupixent) Ottawa (ON): CADTH; 2023.
7.Statistics Canada. Table: 13-10-0710-01. Mortality rates, by age group. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310071001&pickMembers%5B0%5D=1.1&pickMembers%5B1%5D=3.2&cubeTimeFrame.startYear=2016&cubeTimeFrame.endYear=2020&referencePeriods=20160101%2C20200101. Accessed 2023 Dec 1.
8.Stein Gold L, Thaçi D, Thyssen JP, et al. Safety of lebrikizumab in adults and adolescents with moderate-to-severe atopic dermatitis: an integrated analysis of eight clinical trials. Am J Clin Dermatol. 2023:1-13. PubMed
9.National Institute for Health and Care Excellence. Abrocitinib, tralokinumab or upadacitinib for treating moderate to severe atopic dermatitis. (Technology appraisal guidance TA814) 2022; https://www.nice.org.uk/guidance/ta814. Accessed 2023 Dec 1.
10.Yan J, Xie S, Johnson JA, et al. Canada population norms for the EQ-5D-5L. Eur J Health Econ. 2023:1-9. PubMed
11.Dupilumab and crisaborole for atopic dermatitis: effectiveness and value. Final evidence report. Boston (MA): Institute for Clinical and Economic Review; 2017: https://icer.org/wp-content/uploads/2020/10/MWCEPAC_ATOPIC_FINAL_EVIDENCE_REPORT_060717.pdf. Accessed 2023 Dec 1.
12.Iqvia. IQVIA customer portal. 2023; https://login.customerportal.iqvia.com/EB2/User/CustomerLogin.aspx?TYPE=33554432&REALMOID=06-74dfaa3e-e0b7-466a-8827-11ea74bf42aa&GUID=&SMAUTHREASON=0&METHOD=GET&SMAGENTNAME=$SM$nbDLucHgMqLqNmzcWrvjUmaahxcB4ST2%2blG7WqgOT4LWLGX%2ff3DA%2ffWWcjVsYjde&TARGET=$SM$HTTP%3a%2f%2fbaeappsw%2ecustomerportal%2eiqvia%2ecom%2fIMAC%2fDashboard%2easpx%3fModule%3dDeltaPA. Accessed 2023 Dec 1.
13.Government of Canada. Labour market information. Registered Nurse (R.N.) in Canada. 2023; https://www.jobbank.gc.ca/marketreport/summary-occupation/993/ca. Accessed 2023 Dec 1.
14.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 23, 2023)). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 Dec 1.
15.Canadian Institute for Health Information. Patient cost estimator. 2023; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2023 Dec 1.
16.Hale I. Add to cart? Can Fam Physician. 2015;61(11):937-939. PubMed
17.Ministry of Health Long-term Care. Health data branch web portal. 2023; https://hsim.health.gov.on.ca/hdbportal/. Accessed 2023 Dec 1.
18.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2023 Dec 1.
19.DeltaPA. Ottawa (ON): IQVIA; 2023: https://www.iqvia.com/. Accessed 2023 Dec 1.
20.Ontario Drug Benefit Formulary. Ontario Drug Benefit (ODB) formulary. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Dec 1.
21.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2023: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 Dec 1.
22.CADTH. Ruxolitinib for the treatment of atopic dermatitis. 2024; https://www.cadth.ca/ruxolitinib-1. Accessed 2023 Dec 1.
23.CADTH reimbursement review recommendation: tralokinumab (Adtralza - LEO Pharma Inc) [DRAFT]. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/SR0787%20Adtralza%20-%20Draft%20CADTH%20Recommendation%20for%20posting%20November%2016%2C%202023.pdf. Accessed 2023 Dec 1.
24.CADTH reimbursement review. Upadacitinib (Rinvoq) Ottawa (ON): CADTH; 2022.
25.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lebrikizumab, 250 mg/ 2 mL solution for subcutaneous injection. Toronto (ON): Eli Lilly Canada, Inc; 2023 Nov.
26.CADTH reimbursement review. Abrocitinib (Cibinqo) Ottawa (ON): CADTH; 2023.
27.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: results from an international survey. Allergy. 2018;73(6):1284-1293. PubMed
28.Silverberg JI, Toth D, Bieber T, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial*. Br J Dermatol. 2021;184(3):450-463. PubMed
29.Dupixent. dermatite atopique (adolescents). Montreal (QC): Institut national d'excellence en santé et en services sociaux; 2020.
Appendix 1: Cost Comparison Table
Table 7: CDA-AMC Cost Comparison Table for the Treatment of Moderate-to-Severe Atopic Dermatitis
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Lebrikizumab (TBC) | 250 mg / 2 mL | Prefilled pen or prefilled syringe with needle shield | 1,876.7100a | Initial dose of 500 mg, followed by 250 mg every 2 weeks until Week 16. Once clinical response is achieved (usually observed within 16 weeks), maintenance dose is 250 mg every 4 weeks | Year 1: 97.69 Year 2+: 66.84 | Year 1: 35,657 Year 2+: 24,397 |
Systemic therapies | ||||||
Abrocitinib (Cibinqo) | 50 mg 100 mg 200 mg | Tablet | 48.6667b 48.6667b 54.4667b | 100 mg or 200 mg once daily | 48.67 or 54.47 | 17,763 or 19,880 |
Dupilumab (Dupixent) | 200 mg / 1.14 mL 300 mg / 2 mL | Prefilled syringe or prefilled pen | 978.7000b | Adolescents 30 kg to < 60 kg: 400 mg as an initial dose, followed by 200 mg every 2 weeks Adolescents ≥ 60 kg: 600 mg as an initial dose, followed by 300 mg every 2 weeks Adults: 600 mg as an initial dose, followed by 300 mg every 2 weeks | Year 1: 72.40 Year 2+: 69.72 | Year 1: 26,425 Year 2+: 25,446 |
Upadacitinib (Rinvoq) | 15 mg 30 mg | Tablet | 51.6810c 76.9600b | Adolescents (12 to 17 years) > 40 kg: 15 mg once daily Adults: 15 mg or 30 mg once daily depending on individual patient presentation | Adolescents: 51.68 Adults: 51.68 to 76.96 | Adolescents: 18,864 Adults: 18,864 to 28,090 |
The comparators presented in the above table have been deemed to be appropriate based on feedback from clinical experts and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
aSponsor submitted price.2
bEAP (accessed December 2023).21
cOntario Drug Benefit Formulary (accessed December 2023).20
Note: This table has not been copy-edited.
Table 8: Cost Comparison Table for the Treatment of Moderate-to-Severe Atopic Dermatitis (Off-Label)
Drug/ comparator | Strength / concentration | Dosage form | Price ($) | Recommended dosagea | Daily cost ($) | Annual ($) |
|---|---|---|---|---|---|---|
Immunosuppressants | ||||||
Azathioprine (generic) | 50 mg | Tablet | 0.2405 | Adolescent: 1.0 to 4.0 mg/kg per day Adult: 1.0 to 3.0 mg/kg per day | Adolescent: 0.24 to 0.96 Adult: 0.48 to 1.20 | Adolescent: 88 to 351 Adult: 176 to 439 |
Cyclosporine (generic) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.7115 0.7870 1.5350 3.0720 | Adolescent: 3.0 to 6.0 mg/kg per day Adult: 150 to 300 mg per day | Adolescent: 4.61 to 19.21 Adult: 4.61 to 9.22 | Adolescent: 1,681 to 7,012 Adult: 1,681 to 3,364 |
Methotrexate (generic) | 2.5 mg | Tablet | 0.2513 | Adolescent:0.2 to 0.7 mg/kg per week Adult: 7.5 to 25 mg per week | Adolescent: 0.14 to 0.47 Adult: 0.11 to 0.36 | Adolescent: 52 to 170 Adult: 39 to 131 |
Mycophenolate mofetil | 250 mg 500 mg | Capsule | 0.3712 0.7423 | Adolescent: 30.0 to 50.0 mg/kg per day Adult: 2,000 to 13,000 mg daily | Adolescent: 2.23 to 3.71 Adult: 2.97 to 19.30 | Adolescent: 813 to 1,355 Adult: 1,084 to 7,045 |
Note: Unit prices of medications are taken from the Ontario Drug Benefit Formulary (accessed December 2023), unless otherwise indicated, and do not include dispensing fees.20 Recommended dosage based on the American Atopic Dermatology Guidelines and previous CDA-AMC Pharmacoeconomic Review of upadacitinib.24 Annual costs assumes 52 weeks or 365 days for all comparators.
aMean adolescent weight of 45 kg and mean adult weight of 70 kg used.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The sequential analysis was conducted using lebrikizumab as the reference treatment rather than the least costly comparator. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 10: Deterministic Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. BSC ($/QALY) | Sequential ICER ($/QALY) |
|---|---|---|---|---|
BSC | 132,218 | 31.884 | Ref. | Ref. |
Abrocitinib 100 mg | 259,093 | 32.373 | 259,634 | 259,634 vs. BSC |
Upadacitinib 15 mg | 271,506 | 32.421 | 259,729 | 260,713 vs. abrocitinib 100 mg |
Upadacitinib 30 mg | 391,580 | 32.531 | 400,998 | 1,086,542 vs. upadacitinib 15 mg |
Dominated treatments | ||||
Abrocitinib 200 mg | 294,487 | 32.441 | 291,417 | Extendedly dominated by abrocitinib 100 mg and upadacitinib 15 mg |
Lebrikizumab | 313,593 | 32.332 | 405,672 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg and abrocitinib 200 mg |
Dupilumab | 330,136 | 32.282 | 497,983 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg and lebrikizumab |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: All results are for treatments in combination with TCS.
Source: Sponsor pharmacoeconomic report.2
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 11: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results
Scenario analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case – deterministic | BSC | 132,218 | 31.884 | Ref. |
Abrocitinib 100 mg | 259,093 | 32.373 | 259,634 vs BSC | |
Upadacitinib 15 mg | 271,506 | 32.421 | 260,713 vs abrocitinib 100 mg | |
Upadacitinib 30 mg | 391,580 | 32.531 | 1,086,542 vs upadacitinib 15 mg | |
Dominated treatments | ||||
Abrocitinib 200 mg | 294,487 | 32.441 | Extendedly Dominated by abrocitinib 100 mg and upadacitinib 15 mg | |
Lebrikizumab | 313,593 | 32.332 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, and abrocitinib 200 mg | |
Dupilumab | 330,136 | 32.282 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg and lebrikizumab | |
Sponsor’s base case (corrected) – deterministic | BSC | 132,218 | 31.884 | Ref. |
Abrocitinib 100 mg | 259,093 | 32.373 | 259,634 vs BSC | |
Upadacitinib 15 mg | 281,134 | 32.421 | 462,925 vs abrocitinib 100 mg | |
Abrocitinib 200 mg | 294,487 | 32.441 | 649,912 vs upadacitinib 15 mg | |
Upadacitinib 30 mg | 402,750 | 32.531 | 1,203,409 vs abrocitinib 200 mg | |
Dominated treatments | ||||
Lebrikizumab | 313,593 | 32.332 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
Dupilumab | 330,136 | 32.282 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
CDA-AMC reanalysis 1 – BSC excluded as a comparator | Abrocitinib 100 mg | 259,093 | 32.373 | Ref. |
Upadacitinib 15 mg | 281,134 | 32.421 | 462,925 vs abrocitinib 100 mg | |
Abrocitinib 200 mg | 294,487 | 32.441 | 649,912 vs upadacitinib 15 mg | |
Upadacitinib 30 mg | 402,750 | 32.531 | 1,203,409 vs abrocitinib 200 mg | |
Dominated treatments | ||||
Lebrikizumab | 313,593 | 32.332 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
Dupilumab | 330,136 | 32.282 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
CDA-AMC reanalysis 2 – health-state utility value | BSC | 132,218 | 31.826 | Ref. |
Abrocitinib 100 mg | 259,093 | 32.265 | 288,704 vs BSC | |
Upadacitinib 15 mg | 281,134 | 32.304 | 572,247 vs abrocitinib 100 mg | |
Abrocitinib 200 mg | 294,487 | 32.321 | 773,346 vs upadacitinib 15 mg | |
Upadacitinib 30 mg | 402,750 | 32.393 | 1,499,292 vs abrocitinib 200 mg | |
Dominated treatments | ||||
Lebrikizumab | 313,593 | 32.225 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
Dupilumab | 330,136 | 32.168 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg, and lebrikizumab | |
CDA-AMC base case – deterministic | Abrocitinib 100 mg | 259,093 | 32.265 | Ref. |
Upadacitinib 15 mg | 281,134 | 32.304 | 572,247 vs. abrocitinib 100 mg | |
Abrocitinib 200 mg | 294,487 | 32.321 | 773,346 vs. upadacitinib 15 mg | |
Upadacitinib 30 mg | 402,750 | 32.393 | 1,499,292 vs abrocitinib 100 mg | |
Dominated treatments | ||||
Lebrikizumab | 313,593 | 32.225 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
Dupilumab | 330,136 | 32.168 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg and lebrikizumab | |
CDA-AMC base case – probabilistic | Abrocitinib 100 mg | 259,355 | 32.337 | Ref. |
Upadacitinib 15 mg | 281,530 | 32.385 | 462,818 vs. abrocitinib 100 mg | |
Abrocitinib 200 mg | 294,269 | 32.396 | 1,112,646 vs. upadacitinib 15 mg | |
Upadacitinib 30 mg | 402,395 | 32.472 | 1,428,775 vs abrocitinib 100 mg | |
Dominated treatments | ||||
Lebrikizumab | 313,860 | 32.328 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
Dupilumab | 331,060 | 32.368 | Dominated by upadacitinib 15 mg, abrocitinib 200 mg | |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; Ref = reference.
Note: All treatments were used in combination with TCS.
Table 12: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Detail | Abrocitinib 100 mg | Upadacitinib 15 mg | Abrocitinib 200 mg | Lebrikizumab | Dupilumab | Upadacitinib 30 mg |
|---|---|---|---|---|---|---|
Discounted QALYs | ||||||
1L | 7.013 | 7.690 | 7.890 | 6.909 | 7.470 | 9.015 |
BSC | 25.324 | 24.695 | 24.506 | 25.418 | 24.898 | 23.457 |
Total | 32.337 | 32.385 | 32.396 | 32.328 | 32.368 | 32.472 |
Discounted LYs | ||||||
1L | 8.033 | 8.801 | 9.034 | 7.918 | 8.553 | 10.316 |
BSC | 31.203 | 30.435 | 30.202 | 31.318 | 30.683 | 28.921 |
Total | 39.236 | 39.236 | 39.236 | 39.236 | 39.236 | 39.236 |
Discounted costs ($) | ||||||
1L health-state costs | 11,259 | 12,321 | 12,642 | 6,412 | 6,911 | 14,402 |
BSC health-state costs | 104,679 | 102,100 | 101,312 | 105,059 | 102,928 | 97,000 |
1L drug-acquisition costs | 142,799 | 166,135 | 179,719 | 201,354 | 219,373 | 289,968 |
1L admin costs | 0 | 0 | 0 | 40 | 40 | 0 |
AE costs | 619 | 976 | 596 | 994 | 1,808 | 1,026 |
Total | 259,355 | 281,530 | 294,269 | 313,860 | 331,060 | 402,395 |
1L = first year; AE = adverse event; BSC = best supportive care; LY = life-year; QALY = quality-adjusted life-year.
Note: All treatments were used in combination with TCS.
Scenario Analyses
Table 13: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost per prefilled pen or prefilled syringe | ICERs for lebrikizumab vs. comparators ($/QALY) |
|---|---|---|
Price reduction | $ | Sponsor base case |
No price reduction | 1,876.71 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg |
10% | 1,689.04 | Dominated by abrocitinib 100 mg and upadacitinib 15 mg |
20% | 1,501.37 | Dominated by abrocitinib 100 mg and upadacitinib 15 mg |
30% | 1,313.70 | 257,911 vs. BSC |
40% | 1,126.03 | 215,091 vs. BSC |
50% | 938.36 | 172,270 vs. BSC |
60% | 750.68 | 129,450 vs. BSC |
70% | 563.01 | 86,629 vs. BSC |
80% | 375.34 | 43,809 vs. BSC |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; vs. = versus.
Note: All treatments were used in combination with TCS.
Table 14: Detailed Results of the CDA-AMC Scenario Analyses
Scenario analysis | Drug | Total costs | Total QALYs | ICER vs. abrocitinib 100 mg ($/QALY) | Sequential ICER ($/QALY) |
|---|---|---|---|---|---|
CDA-AMC base case (deterministic) | Abrocitinib 100 mg | 259,093 | 32.265 | Ref. | Ref. |
Upadacitinib 15 mg | 281,134 | 32.304 | 572,247 | 572,247 vs. abrocitinib 100 mg | |
Abrocitinib 200 mg | 294,487 | 32.321 | 634,497 | 773,346 vs. upadacitinib 15 mg | |
Upadacitinib 30 mg | 402,750 | 32.393 | 1,122,386 | 1,499,292 vs abrocitinib 100 mg | |
Dominated treatments | |||||
Lebrikizumab | 313,593 | 32.225 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
Dupilumab | 330,136 | 32.168 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg and lebrikizumab | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg and lebrikizumab | |
Scenario analysis 1: BSC included as a comparator | BSC | 132,218 | 31.826 | Ref. | Ref. |
Abrocitinib 100 mg | 259,093 | 32.265 | 288,704 vs. BSC | 288,704 vs. BSC | |
Upadacitinib 15 mg | 281,134 | 32.304 | 311,552 vs. BSC | 572,247 vs. abrocitinib 100 mg | |
Abrocitinib 200 mg | 294,487 | 32.321 | 327,654 vs. BSC | 773,346 vs. upadacitinib 200 mg | |
Upadacitinib 30 mg | 402,750 | 32.393 | 476,746 vs. BSC | 1,499,292 vs. abrocitinib 200 mg | |
Dominated treatments | |||||
Lebrikizumab | 313,593 | 32.225 | 454,391 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
Dupilumab | 330,136 | 32.168 | 578,002 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg and lebrikizumab | |
Scenario analysis 2: Adverse event removal | Abrocitinib 100 mg | 258,450 | 32.332 | Ref. | Ref. |
Upadacitinib 15 mg | 280,152 | 32.376 | 496,874 | 496,874 vs. abrocitinib 100 mg | |
Abrocitinib 200 mg | 293,865 | 32.391 | 600,609 | 896,995 vs. upadacitinib 15 mg | |
Upadacitinib 30 mg | 401,716 | 32.465 | 1,081,321 | 1,466,843 vs. abrocitinib 200 mg | |
Dominated treatments | |||||
Lebrikizumab | 312,552 | 32.326 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg and abrocitinib 200 mg | Dominated by abrocitinib 100 mg, upadacitinib 15 mg and abrocitinib 200 mg | |
Dupilumab | 328,340 | 32.361 | 2,461,677 | Dominated by abrocitinib 100 mg, upadacitinib 15 mg, abrocitinib 200 mg | |
Scenario analysis 3: Monotherapy | Abrocitinib 100 mg | 246,107 | 29.718 | Ref. | Ref. |
Upadacitinib 15 mg | 287,952 | 30.010 | 143,386 | 143,386 vs. abrocitinib 100 mg | |
Abrocitinib 200 mg | 302,410 | 30.043 | 173,118 | 432,934 vs. upadacitinib 15 mg | |
Upadacitinib 30 mg | 404,827 | 30.229 | 310,430 | 550,453 vs. abrocitinib 200 mg | |
Dominated treatments | |||||
Lebrikizumab | 298,864 | 30.043 | 777,210 | Dominated by upadacitinib 15 mg | |
Dupilumab | 306,760 | 29.792 | 820,053 | Dominated by upadacitinib 15 mg and abrocitinib 200 mg | |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref = reference.
Note: All treatments were used in combination with TCS.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 15: Summary of Key Take Aways
Key take aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA) estimating the expected incremental budgetary impact of reimbursing lebrikizumab for the treatment of adult and adolescent patients 12 years of age and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.25 The BIA was conducted from the perspective of the pan-Canadian public drug plans over a 3-year time horizon (2025 to 2027) with 2024 as the base year. The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits (NIHB) program. Adjustments were made to the provincial populations to remove NIHB patients to estimate the provincial public plan population. The sponsor’s base-case analysis included drug-acquisition costs only. Market shares for comparator treatments were estimated based on internal forecasts conducted by the sponsor and previous CADTH reimbursement reviews of abrocitinib and upadacitinib.24,26 Market uptake for lebrikizumab was estimated based on sponsor internal forecasts. Key inputs to the BIA are documented in Table 16.
The following key assumptions were made by the sponsor:
The sponsor used a blended method to determine drug-acquisition costs for lebrikizumab and relevant comparators. In each year, it was assumed that 1 third of the patients are in the induction year of treatment (year 1), and 2 thirds of patients are in the maintenance years of treatment (Years 2+).
The sponsor assumed no market share would be captured from immunosuppressive therapies (ISTs) if lebrikizumab would be reimbursed.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Adults Proportion diagnosed with AD Proportion with moderate-to-severe AD Proportion treated Proportion whose disease cannot be adequately controlled by topical therapies Proportion eligible for systemic therapy Proportion eligible for enrolment in public drug plans | 3.5%27 52%28 72%24 38%29 40%24 76%24 |
Adolescents Proportion diagnosed with AD Proportion with moderate-to-severe AD Proportion treated Proportion whose disease cannot be adequately controlled by topical therapies Proportion eligible for systemic therapy Proportion eligible for enrolment in public drug plans | 15.8%28 40.2%28 91%29 55%29 40%24 64.5%24 |
Number of patients eligible for drug under review | 57,442 / 58,179 / 58,916 |
Market shares (3 years) | |
Market shares (reference scenario) Abrocitinib Upadacitinib Dupilumab ISTs | 2.5% / 5% / 7% 2.5% / 5% / 7% 17% / 18% / 19% 78% / 72% / 67% |
Market shares (new drug scenario) Lebrikizumab Abrocitinib Upadacitinib Dupilumab ISTs | 1.5% / 4.5% / 6% 2.3% / 4.2% / 5.7% 2.3% / 4.2% / 5.7% 15.8% / 15.1% / 15.5% 78% / 72% / 67% |
Cost of treatment (per patient) | |
Cost of treatment over 1 yeara Lebrikizumab Abrocitinib Upadacitinib Dupilumab ISTs (Adolescents) ISTs (Adults) | $25,336 $13,398 $13,204 $23,195 $845 $1,264 |
AD = atopic dermatitis; ISTs = immunosuppressive therapy.
aCost of treatment was determined using a blended cost that combines year 1 and year 2+ costs in proportion to the number of patients assumed to be in each of the initial year of treatment vs. year 2+. In each year, it was assumed that one-third of patients are in year 1 treatment two-thirds are years 2+.
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement of lebrikizumab for the treatment of adult and adolescent patients 12 years of age and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable would result to an incremental budget impact of $9,602,989 in year 1, $14,854,824 in year 2, and $22,404,206 in year 3. The total 3-year incremental cost of reimbursing lebrikizumab is $46,862,019.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The proportion of patients eligible to receive therapy is uncertain: The sponsor estimated that approximately 38% and 55% of adolescent and adult patients, respectively, who diagnosed with moderate-to-severe AD cannot be adequately controlled with topical prescription therapies, based on a review of dupilumab in adolescents from INESSS.29 Clinical expert feedback obtained by CDA-AMC indicated that the proportion of patients whose AD cannot be adequately controlled with topical therapies is likely switched between the adult and adolescent groups as adults with moderate-to-severe AD are more likely to not be adequately controlled with topical prescription therapies.
Additionally, the sponsor estimated that among those diagnosed with moderate-to-severe AD whose disease cannot be adequately controlled with topical prescription therapies, 40% would be eligible to receive systemic therapy.25 This statistic was sourced from internal sponsor’s report for upadacitinib as reported in the CADTH reimbursement review.24 Clinical expert feedback obtained by CDA-AMC indicated that this proportion is underestimated and that it is unlikely that a patient would be ineligible to receive systemic therapy. The clinical expert highlighted that not all patients that are eligible to receive systemic therapy will choose to, therefore, the clinical expert feedback indicated that the proportion may be closer to 95%.
To address these limitations, CDA-AMC conducted a reanalysis whereby 38% of adolescent and 55% of adult patients diagnosed with moderate-to-severe AD cannot be adequately controlled with topical prescription therapies.
To explore the impact of the proportion of patients eligible to receive systemic therapy, CDA-AMC performed a scenario analysis that assumed that 95% of those whose disease cannot be adequately controlled by topical therapies would be eligible to receive systemic therapy.
The market share estimates in the reference scenario are highly uncertain: In the reference scenario of the sponsor’s submitted BIA, it was assumed that 2.5%, 5.0% and 7.0% of eligible patients would be receiving abrocitinib and upadacitinib in years 1, 2, and 3, respectively, based on previous CADTH reimbursement reviews for upadacitinib and abrocitinib.24-26 This is uncertain because clinical expert feedback obtained by CDA-AMC indicated that approximately twice as many patients will receive upadacitinib compared with abrocitinib. Additionally, the sum of the market shares for abrocitinib and upadacitinib (the 2 JAK inhibitors in the analysis) in the sponsor’s reference scenario in year 1 (2025) were approximately half of what abrocitinib was expected to reach in its third year of reimbursement (i.e., 2025),26 indicating that the market shares for these comparators may be underestimated in the sponsor’s analysis.
Additionally, the sponsor assumed that the majority of the eligible population would remain on ISTs (i.e., 78%, 72% and 67% of the population using ISTs in years 1, 2 and 3, respectively). Clinical expert feedback obtained by CDA-AMC highlighted that the proportion of patients receiving ISTs throughout the 3 years is likely overestimated. Clinical expert feedback indicated that it is expected that the majority of eligible patients would currently be receiving a biologic or JAK inhibitor and that the proportion of patients receiving ISTs would represent all patients that choose not to use biologics or JAK inhibitors. As such, the proportion of patients receiving ISTs in the reference scenario is likely overestimated and the proportion receiving abrocitinib, upadacitinib and dupilumab are likely underestimated.
Finally, market shares in the reference scenario for biologic and JAK inhibitor treatments are increasing year over year, meaning that existing biologic and JAK inhibitors are continuing to capture market share from ISTs. The extent that these treatments will continue to capture market share from ISTs over the next 3 years is highly uncertain given the duration these drugs have been on the market. This uncertainty is exacerbated by the sponsor’s assumption that the entry of lebrikizumab will not capture market share from ISTs, while other biologics and JAK inhibitors will continue to capture IST market share over the same time frame.
To address this limitation, CDA-AMC conducted a reanalysis to double the proportion of patients on upadacitinib based on clinical expert feedback.
CDA-AMC was unable to address the proportion of patients receiving ISTs due to a lack of evidence regarding the impact to the market dynamics of the other treatments in the reference scenario since assuming stability in the IST market across 3 years would mean a decrease in the market shares of 1 of the comparator treatments over time, which is highly uncertain.
Total treatment costs are uncertain: In the sponsor’s submitted BIA, the sponsor used a blended cost method to combine induction and maintenance costs for all treatments to simplify calculations in the model. In the model, it was assumed that one-third of patients are in the induction year of treatment (year 1) and two-thirds of patients are in the maintenance phase (years 2+). The use of blended cost method adds uncertainty to the total treatment costs, as the assumption may not represent clinical practice.
CDA-AMC could not undertake reanalysis to address this limitation as the sponsor’s BIA model lacked flexibility to incorporate treatment costs separately during induction and maintenance phases.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by revising epidemiological estimates and revising reference scenario market share estimates to align with current Canadian clinical practice. The changes applied to derive the CDA-AMC base case and key scenario analysis for both perspectives are described in Table 17.
Table 17: CDA-AMC Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CDA-AMC base case | ||
1. Proportion of patients who disease cannot be adequately controlled with topical therapies is uncertain. | Adults: 38.0% Adolescents: 55.0% | Adults: 55.0% Adolescents: 38.0% |
2. Market shares for upadacitinib are underestimated. | Year 0 / 1 / 2 / 3 Abrocitinib: 1.5% / 2.5% / 5.0% / 7.0% Upadacitinib: 1.5% / 2.5% / 5.0% / 7.0% Dupilumab: 16.0% / 17.0% / 18.0% / 19.0% ISTs: 81.0% / 78.0% / 72.0% / 67.0% | Year 0 / 1 / 2 / 3 Abrocitinib: 1.5% / 2.5% / 5.0% / 7.0% Upadacitinib: 3.0% / 5.0% / 10.0% / 14.0% Dupilumab: 16.0% / 17.0% / 18.0% / 19.0% ISTs: 79.5% / 75.5% / 67.0% / 60.0% |
CDA-AMC base case | Reanalysis 1 + 2 | |
ISTs = immunosuppressive therapies.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19. Based on the CDA-AMC base case, the budget impact associated with the reimbursement of lebrikizumab for the treatment of adult and adolescent patients 12 years of age and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable would be associated with a budgetary increase of $65,018,149 over 3 years (year 1: $12,449,072; year 2: $21,089,890; year 3: $31,419,187).
CDA-AMC conducted the following scenario analyses (Table 19) to highlight uncertainty associated with the potential budget impact.
Increasing the proportion of patients eligible for systemic therapies from 40% to 95%.
Table 18: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $46,862,019 |
CDA-AMC reanalysis 1 | $56,751,513 |
CDA-AMC reanalysis 2 | $53,687,799 |
CDA-AMC base case | $65,018,149 |
BIA = budget impact analysis.
Table 19: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $285,086,244 | $315,454,362 | $367,743,572 | $414,084,712 | $1,097,282,646 |
New drug | $285,086,244 | $325,057,351 | $382,598,396 | $436,488,918 | $1,144,144,665 | |
Budget impact | $0 | $9,602,989 | $14,854,824 | $22,404,206 | $46,862,019 | |
CDA-AMC base case | Reference | $360,507,597 | $405,819,024 | $490,490,480 | $564,287,616 | $1,460,597,120 |
New drug | $360,507,597 | $418,268,095 | $511,580,370 | $595,766,803 | $1,525,615,269 | |
Budget impact | $0 | $12,449,072 | $21,089,890 | $31,479,187 | $65,018,149 | |
CDA-AMC scenario analysis 1: proportion eligible for systemic therapy | Reference | $856,205,544 | $963,820,181 | $1,164,914,890 | $1,340,183,088 | $3,468,918,159 |
New drug | $856,205,544 | $993,386,726 | $1,215,003,379 | $1,414,946,157 | $3,623,336,263 | |
Budget impact | $0 | $29,566,545 | $50,088,490 | $74,763,069 | $154,418,104 |
BIA = budget impact analysis; IST = immunosuppressive therapies.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health-technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health-technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.