CADTH Reimbursement Review
Infliximab (Remsima SC)
Sponsor: Celltrion Healthcare Co., Ltd.
Therapeutic Area: Crohn Disease and ulcerative colitis
Clinical and Pharmacoeconomic Report
Abbreviations
5-ASA
5-aminosalicylates
AI
autoinjector
AZA
azathioprine
6-MP
mercaptopurine
BIA
budget impact analysis
AE
adverse event
CD
Crohn disease
CDAI
Crohn’s Disease Activity Index
CDAI-70
reduction in CDAI score of at least 70 points from baseline
CDAI-100
reduction in CDAI score of at least 100 points from baseline
CI
confidence interval
CMH
Cochran-Mantel-Haenszel
CPK
creatine phosphokinase
CRP
C-reactive protein
Ctrough
trough concentration calculated from a predose of the next dose observed, if available
Ctrough, week 22
trough concentration calculated from a predose level at week 22, if available
CT-P13
infliximab (Celltrion, Inc.)
DB
double blind
DMARD
disease-modifying antirheumatic drug
EIM
extraintestinal manifestation
EMA
European Medicines Agency
ES
endoscopic subscore
GI
gastrointestinal
HBI
Harvey-Bradshaw Index
HRQoL
health-related quality of life
IBD
inflammatory bowel disease
ITC
indirect treatment comparison
ISR
injection-site reaction
IWRS
interactive web response system
JAK
Janus kinase
LS
least squares
MID
minimal important difference
NSCLC
non–small cell lung cancer
OL
open label
PFS
prefilled syringe
PGA
physician’s global assessment
PK
pharmacokinetic
RA
rheumatoid arthritis
RCT
randomized controlled trial
RHI
Robarts Histopathology Index
S1P
sphingosine-1-phosphate
SAE
serious adverse event
SC
subcutaneous
SEB
subsequent entry biologic
SES-CD
Simplified Endoscopic Activity Score for Crohn’s Disease
SIBDQ
Short Inflammatory Bowel Disease Questionnaire
Study CT-P13 1.6
Study 1.6
TB
tuberculosis
TEAE
treatment-emergent adverse event
TNF
tumour necrosis factor
UC
ulcerative colitis
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on the Application Submitted for Review
Item | Description |
|---|---|
Drug product | Infliximab (Remsima SC), 120 mg/mL solution for subcutaneous injection |
Sponsor | Celltrion Health care Co., Ltd. |
Indication |
|
Reimbursement request | As per indication |
Health Canada approval status | Post-NOC |
Health Canada review pathway | Standard |
NOC date | February 15, 2024 |
Recommended dosage | For patients who have completed an induction regimen with infliximab administered through IV:
There is insufficient information regarding the switching of patients who have received IV infusions of infliximab higher than 5 mg/kg for Crohn disease or ulcerative colitis every 8 weeks to Remsima SC. Information regarding switching patients from the subcutaneous formulation to IV infliximab is not available. |
NOC = Notice of Compliance; SC = subcutaneous.
Introduction
Inflammatory bowel disease (IBD) is an umbrella term describing chronic inflammation of the gastrointestinal (GI) tract caused by 1 of 2 disorders: ulcerative colitis (UC) or Crohn disease (CD). Its etiology is unknown; possible causes include genetics and abnormal immune response to environmental factors, such as pathogens in the GI tract (e.g., viruses, bacteria, fungi, or parasites).1,7,8
Canada has the highest prevalence and incidence of IBD in the world, with an estimated 0.8% of the population (about 322,600 people) living with the disease as of 2023;1 the prevalence of IBD (all types) has been increasing steadily and was estimated at 0.67%,3 0.7%,4 and 0.82%5 of the population in Canada in 2012, 2018, and 2023, respectively. The prevalence of CD and UC in Canada is forecast to increase by 2030 to 493 and 436 per 100,000, respectively, reflecting average annual percentage increases of 2.75% and 2.87%.2 A 2023 report by Crohn and Colitis Canada estimated that 470,000 people will be living with IBD in Canada by 2035.6
CD is caused by inflammation of the GI tract from mouth to rectum, but is mainly observed around the ileum (i.e., small intestine), colon (i.e., beginning of the large intestine), and rectum.9 CD is most common in adolescents and adults aged 20 years to 30 years.10 The estimated prevalence of CD in Canada in 2018 was 368 per 100,000 population, translating to about 135,000 people.11,12 Common symptoms include abdominal pain, rectal bleeding, fatigue, vomiting, diarrhea, perianal disease, weight loss, and bloating.10,13 Patients may experience chronic or intermittent symptoms, and disease activity and severity can vary widely over time. While some patients experience continuous and progressive active disease, about 20% of patients may experience prolonged remission after initial presentation.14
UC, on the other hand, is characterized by inflammation and ulcers in the mucosal layer of the large intestine (colon), typically beginning at the rectum (anus), progressing upward, and in some cases affecting the entire colon.15,1,16 UC has a worldwide annual incidence rate of 1.2 to 20.3 cases per 100,000 people and a prevalence rate of 7.6 to 246.0 cases per 100,000 people.17 UC generally develops in young adulthood18-20 and persists throughout life, marked by periods of spontaneous remission and relapse.21 Symptoms include blood and/or mucus in the stool, frequent diarrhea, loss of appetite, and tenesmus (strong urge to use the bathroom without necessarily having a bowel movement) in addition to abdominal pain, rectal bleeding, and weight loss.2 The disease is characterized as mild, moderate, or severe disease, depending on the specific index score used (Truelove and Witts severity index, Mayo clinic score, or the Montreal classification).22 Although most patients experience a relapsing-remitting disease course, reports show that up to 24% of patients experience continuous UC symptoms.23
UC and CD are diagnosed based on symptoms and clinical tests, such as endoscopic evaluations (endoscopy, biopsy), stool sampling, and histological, radiological, and/or biochemical investigations at initial diagnosis.24 Available treatment options for UC depend on the presence of active disease, the severity and extent of disease, and patient preference. Options for CD depend on location, extent, phenotype, and severity. Treatment options for both diseases are similar. In CD, aminosalicylates, immunosuppressants (e.g., azathioprine [AZA], cyclosporine, methotrexate, and mercaptopurine [6-MP]), corticosteroids (e.g., prednisone), tumour necrosis factor (TNF) alpha antagonists (e.g., infliximab and adalimumab), interleukin inhibitors, and integrin inhibitors (e.g., vedolizumab) are current options.25,26 Conventional therapies for UC include aminosalicylates, corticosteroids, and immunomodulators (such as AZA, 6-MP, and methotrexate); advanced therapies consist of adalimumab, golimumab, infliximab, ustekinumab, tofacitinib, ozanimod, or vedolizumab. Current treatments are unable to meet all patient needs in terms of short- or long-term treatment.
The objective of this report is to review and critically appraise evidence submitted by the sponsor for the beneficial and harmful effects of infliximab 120 mg/mL solution for subcutaneous (SC) injection for the 2 indications highlighted in Table 1. Infliximab (Remsima SC) was approved in 2021 by Health Canada for use in patients with moderately to severely active rheumatoid arthritis (RA). It received a positive conditional CADTH recommendation for the treatment of adult patients with moderately to severely active RA in 2021.27 Infliximab (Remsima SC) has the same main ingredient as the infliximab IV product; however, it is administered at a different dosage and through the SC route.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
One patient input from the GI Society was received and was summarized for this review. The GI Society is a national charitable organization with programs and services that support research, advocate for appropriate patient access to health care, and promote GI and liver health. Information from this input was gathered through questionnaires and interviews. Information was collected from 5 surveys, with a total of 1,633 respondents contributing. Additional data from a 2020 focus group on persons living with IBD and 1-to-1 interviews with patients were also assessed for the input.
The GI Society highlighted that patients with IBD preferred sustained remission and/or treatment response over relieving 1 symptom. Survey respondents expressed various concerns associated with IBD, including fear of running out of medication, uncertainty when trying to determine whether to go to the emergency department based on symptoms, pain, fear of going out due to disease, decreased quality of life, and fear and worry connected to facing mortality at a young age. The patient group highlighted the need for effective treatments that could improve quality of life and eliminate symptoms, pain, frustration, and hardship. The patient advocacy group expressed that inadequate access to treatment causes continual, debilitating disease symptoms; secondary illnesses, such as depression and anxiety disorders; and the loss of family and other social interactions.
According to the patient advocacy group, treatment of CD and UC requires a multifaceted strategy that allows for the management of symptom and disease consequences using therapies that target and reduce the underlying inflammation. The treatment options outlined included 5-aminosalicylates (5-ASAs), corticosteroids, immunosuppressive drugs, and biologics. Newer therapies identified for UC included Janus kinase (JAK) and sphingosine-1-phosphate (S1P) inhibitors. Other therapies included S1P inhibitors, such as ozanimod. The patient advocacy group highlighted that, despite the treatment options available in practice, patients with UC and CD still have trouble achieving remission and adequate symptom relief; thus, there is a need for more treatments that meet their needs. No patients interviewed were currently receiving the treatment under review; however, the majority of patients surveyed had received a biologic. Results from 1 survey showed that 63% of respondents reported symptom reduction after using a biologic, while 23% confirmed remission.
According to the patient advocacy group, patients would like additional effective treatment options with convenient and timely patient access and different administration methods and dosages. The GI Society highlighted that major concerns with available therapies included ensuring adequate supply and continuity of care, especially timely communication between patients and their health care providers. The patient group noted that needing to go to clinics to receive IV treatments — and untimely communications between patients and health care providers — could mean frequently needing to take time off work, which can be difficult and contribute to financial hardship. According to the patient advocacy group, patients desire options that can be administered at home.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
Input from 1 clinical expert with experience treating UC and CD was summarized for this review. The clinical expert highlighted that there is no cure for UC or CD in current practice and that early treatment is crucial because the first medication prescribed has the best chance of improving symptoms and healing. Treatment goals highlighted for patients with UC or CD include symptom resolution (clinical remission), reduced need for surgery, avoidance of the repetitive use of corticosteroids, and improved quality of life (by normalizing bowel movements, normalizing weight and energy levels, and resolving pain, bowel urgency, and rectal bleeding).
According to the expert, treatment selection is complex for patients with UC and CD and depends on disease phenotype and patient preference. Most of the advanced treatments currently available in practice (i.e., anti-TNF alpha therapies, JAK inhibitors, alpha 4 beta 7 integrin inhibition, and interleukin 23 plus interluekin-12 and/or 23 inhibitors) target primary and secondary loss of response in both diseases. However, the expert noted that about half of IBD patients have extraintestinal manifestations (EIMs) of CD, which can be disabling, and only a few treatments address this issue. There is a preference for anti-TNF alpha therapy to treat many of these cases. The expert did not anticipate any shift in treatment paradigm with the use of infliximab SC, apart from the option of switching from IV to SC administration. According to the clinical expert, patients with confirmed moderate to severe CD or UC (based on a pathological and histological diagnosis) are best suited for treatment with infliximab SC. The expert highlighted that misdiagnosis is rarely observed in practice, although delays in diagnosis may occur. The expert noted that not all patients respond well to anti-TNF alpha therapy. Less suitable patients are those who fear self-injection.
The clinical expert consulted noted that patient response to treatment was assessed more frequently in the LIBERTY-UC and LIBERTY-CD trials than it would be in real-world settings. The expert highlighted that colonoscopy is seldom performed every 12 weeks, as it was during the trials, due to logistics and patient preference. C-reactive protein (CRP) and fecal calprotectin are frequently used to monitor patient response to advance treatment in practice, according to the expert, while the Harvey-Bradshaw Index (HBI) (as opposed to the Crohn Disease Activity Index [CDAI] used in the trial) is used to monitor treatment response for patients with CD. Fecal calprotectin is an objective measure to monitor disease activity and treatment response for patients with UC in addition to the partial Mayo score (partial and modified Mayo scores were derived in the LIBERTY-UC trial to evaluate clinical remission), according to the clinical expert consulted by CADTH. The expert noted that the modified Mayo score (which includes an endoscopic assessment) is used in clinical practice for initial patient assessment before treatment initiation, while the partial Mayo score is used routinely for follow-up to assess response. According to the clinical expert consulted by CADTH, factors leading to treatment discontinuation will be consistent with those outlined for current advanced therapies. The expert highlighted that patients are evaluated in practice based on clinical symptoms and an assessment of objective data. The expert mentioned that some patients may present as primary nonresponders during treatment, and some may experience loss of response during treatment (the clinical expert noted that the standard proportion of patients with CD and UC in clinical practice who experience loss of response in the first year of treatment is approximately 10% to 20%). The clinical expert highlighted that UC and CD diagnoses are made by gastroenterologists. However, general internists with a special interest in IBD have sufficient experience to prescribe infliximab for both populations. The expert noted that treatment initiation begins in private infusion centres, where costs are covered by the drug manufacturer or other patient support programs. Patients then transition to self-injection for the SC formulation.
Clinician Group Input
No clinician group input was submitted for this review.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a CADTH recommendation for infliximab SC: relevant comparators, consideration for continuation or renewal of therapy, considerations for discontinuation of therapy, consideration for prescribing of therapy, care provision issues, and system and economic issues. The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug program. Refer to Table 7 for more details.
Clinical Evidence
Systematic Review
Description of Studies
A total of 3 trials supported the clinical efficacy and safety of infliximab SC in patients with IBD. LIBERTY-UC and LIBERTY-CD were 2 identically designed, randomized, double-blind (DB), placebo-controlled, phase III trials designed to assess the superiority of infliximab SC (120 mg) administered every 2 weeks over placebo in adult patients (aged 18 years to 75 years) with moderately to severely active UC and moderately to severely active CD, respectively, who had experienced an inadequate response to conventional therapy. Both trials consisted of an induction phase during which enrolled patients received infliximab (5 mg/kg) through IV; a maintenance phase during which patients who had no safety concerns and had been considered clinical responders before week 10 were randomized in a 2 to 1 ratio to receive infliximab SC or placebo as maintenance treatment for up to 54 weeks; and an extension phase during which patients in both arms who had completed treatment at week 54 were administered open-label (OL) infliximab SC until week 102. The extension phases in both trials are ongoing.
The coprimary objectives of LIBERTY-CD trial were clinical remission (based on CDAI) and endoscopic response. The key secondary end points in LIBERTY-CD were reduction in CDAI score of at least 100 points from baseline (CDAI-100) response, clinical remission based on abdominal pain and stool frequency, endoscopic remission based on central Simplified Endoscopic Activity Score for Crohn Disease (SES-CD), and corticosteroid-free remission at week 54. Health-related quality of life (HRQoL), another secondary outcome, was measured using the Short Inflammatory Bowel Disease Questionnaire (SIBDQ), the patient global scale, and the visual analogue scale (VAS) for local site pain assessment. Baseline characteristics were generally well-balanced between the 2 treatment groups in the trial. The majority of patients were white and male, and the mean age ranged from 32 years to 36 years across the 2 groups.
The primary end point of LIBERTY-UC trial was clinical remission measured using the modified Mayo score. Key secondary end points included clinical response (based on modified Mayo score), endoscopic-histologic mucosal improvement, and corticosteroid-free remission at week 54. HRQoL, another secondary outcome, was measured using the SIBDQ, the patient global scale, and VAS (for local site pain assessment). Baseline characteristics were generally well-balanced between the 2 treatment groups in the trial. The mean age of patients ranged from 38 years to 40 years; most patients were male and white.
Study CT-P13 1.6 (Study 1.6) (n = 131) was an OL, parallel-group, phase I, randomized trial comparing the pharmacokinetic (PK) parameters, efficacy, and safety of infliximab 5 mg/kg IV administered every 8 weeks versus infliximab SC 120 mg or 240 mg administered every 2 weeks in adult patients (18 years to 75 years) with active UC or CD. The study had 2 parts. Part 1 was a PK study designed to find the optimal dose of Remsima SC in patients with active CD and has not been included in this report. Part 2 evaluated PK outcomes as the primary end point and trough concentration calculated from a predose level at week 22, if available (Ctrough, week 22) as well as clinical efficacy end points as secondary outcomes (i.e., reduction in CDAI score of at least 70 points from baseline [CDAI-70], CDAI-100, clinical remission, endoscopic response, clinical response [based on total and partial Mayo score]), mucosal healing, and SIBDQ scores). While the clinical efficacy outcomes are the focus of this review, the PK primary end point is also reported in the Bioequivalence section. Patients in the infliximab IV arm received IV infliximab up to week 22, switched to infliximab SC by week 30, and continued to week 54. Baseline characteristics were generally well-balanced between the 2 treatment groups in the trial; most patients were white and male, and the mean age across the 2 groups was 35 to 36 years.
Efficacy Results: Primary Outcomes
LIBERTY-CD
Clinical Remission: The proportion of patients who achieved clinical remission at week 54 was higher in the infliximab SC group (144 patients [62.3%]) than in the placebo group (36 patients [32.1%]), with an estimated treatment difference of 32.1% (95% confidence interval [CI], 20.9 to 42.1; P < 0.0001).
Endoscopic Response: The proportion of patients who achieved endoscopic response at week 54 was higher in the infliximab SC group (118 patients [51.1%]) than in the placebo group (20 patients [17.9%]), with an estimated treatment difference of 34.7% (95% CI, 24.2 to 43.5; P < 0.0001). Sensitivity and other supportive analyses were consistent with the primary analyses in the LIBERTY-CD trial.
LIBERTY-UC
Clinical Remission: The proportion of patients achieving clinical remission at week 54 was higher in the infliximab SC group (127 patients [43.2%]) than in the placebo group (30 patients [20.8%]), with a 21.1% treatment difference (95% CI, 11.8 to 29.3; P < 0.0001). Sensitivity and other supportive analyses were consistent with the primary analyses in the LIBERTY-UC trial.
Efficacy Results: Key Secondary Outcomes
LIBERTY-CD
Clinical Remission: The proportion of patients who achieved clinical remission at week 54, based on abdominal pain and stool frequency, was greater in the infliximab SC group (131 patients [56.7%]) than in the placebo group (35 patients [31.3%]). The estimated treatment difference was 27.0% (95% CI, 15.8 to 37.1; P < 0.0001).
Endoscopic Remission Based on Central SES-CD: More patients achieved endoscopic remission at week 54, based on central SES-CD score, in the infliximab SC group (80 patients [34.6%]) than in the placebo group (12 patients [10.7%]). The estimated treatment difference was 24.9% (95% CI, 15.4 to 32.8).
Corticosteroid-free Remission: The proportion of patients achieving corticosteroid-free remission at week 54 was higher in the infliximab SC group (39 patients [39.8%]) than in the placebo group (10 patients [22.7%]), with an estimated treatment difference of 17.1% (95% CI, –0.4 to 31.5; P = 0.04).
Maintenance of Clinical Remission: Among patients with clinical remission at week 10, a higher proportion in the infliximab SC group (121 patients [69.5%]) achieved maintenance of clinical remission than in the placebo group (32 patients [35.2%]), with a treatment difference of 34.5% (95% CI, 22.0 to 45.6; P < 0.0001).
Health-Related Quality of Life: Fewer patients completed the SIBDQ for patient-reported outcomes in the LIBERTY-CD trial at week 54 than at baseline in both groups (n = 167 at week 54 versus n = 231 at baseline in the infliximab SC group, and n = 51 at week 54 versus n = 111 at baseline in the placebo group). The least squares (LS) mean was 54.7 (standard error = 1.4), and the LS mean changes from baseline to week 54 in SIBDQ scores were 17.6 in the infliximab group and 15.1 in the placebo group. The estimated treatment difference was 2.6 (95% CI, –2.1 to 7.2; P = 0.28). Of note, many patients in the placebo group required dose adjustments after losing response; therefore, their results were excluded from the descriptive summary of SIBDQ scores from week 30 onward.
LIBERTY-UC Trial
Clinical Response: The proportion of patients who achieved clinical response at week 54 was higher in the infliximab SC group (158 patients [53.7%]) than in the placebo group (45 patients [31.3%]) at week 54, with an estimated treatment difference of 21.1% (95% CI, 11.2 to 30.1; P < 0.0001).
Endoscopic-Histologic Mucosal Improvement: A greater proportion of patients in the infliximab SC group (105 patients [35.7%]) achieved endoscopic-histologic mucosal improvement at week 54 than in the placebo group (24 patients [16.7%]), with an estimated treatment difference of 18.0% (95% CI, 9.1 to 25.7; P < 0.0001).
Corticosteroid-Free Remission: More patients in the infliximab SC group (44 patients [36.7%]) achieved corticosteroid-free remission than in the placebo group (11 patients [18.0%]) at week 54, with an estimated treatment difference of 17.3% (95% CI, 3.1 to 28.9; P = 0.01).
Maintenance of Clinical Remission: Among patients with clinical remission at week 10, a higher proportion of patients in the infliximab SC group (91 patients [63.6%]) achieved maintenance of clinical remission than in the placebo group (18 patients [27.3%]) at week 54, with a treatment difference of 35.5% (95% CI, 21.1 to 47.5; P value < 0.0001).
Total and Partial Clinical Remission: The proportion of patients who achieved total remission at week 54 in the infliximab SC group was 117 (39.8%) compared to 26 (18.1%) in the placebo group (treatment difference = 20.4% [95% CI, 11.3 to 28.3; P < 0.0001]). The proportion of patients who achieved partial clinical remission at week 54 in the infliximab arm was 127 (43.2%) compared to 39 (27.1%) in the placebo group (treatment difference = 14.7% [95% CI, 5.1 to 23.5; P = 0.0017]).
Health-Related Quality of Life: Fewer patients completed patient-reported outcomes using the SIBDQ in the LIBERTY-UC trial at week 54 compared to baseline in both groups (n = 185 patients at week 54 versus n = 294 patients at baseline in the infliximab SC group and n = 61 patients at week 54 versus n = 144 patients at baseline in the placebo group). The LS mean at week 54 for SIBDQ in the infliximab group was 57.7; it was 54.9 in the placebo group. The estimated treatment difference between the 2 groups was 2.9 (95% CI, –0.3 to 6.0; P = 0.08). The LS mean change from baseline at week 54 was 21.9 in the infliximab SC group versus 18.9 in the placebo group; the estimated treatment difference was 3.0 (95% CI, –1.0 to 6.9; P = 0.14).
Study 1.6
The mean (percentage coefficient of variation [CV]) for observed Ctrough, week22 were higher in the infliximab SC group (120 mg or 240 mg) than in the infliximab IV (5 mg/kg) group at week 22 at 21.5 mcg/mL (46.0 mcg/mL) and 2.9 mcg/mL (89.0 mcg/mL), respectively. The ratio of the geometric LS means was 1,154.2, with a lower-bound 90% CI of 786.4%, which was greater than 80%, suggesting that infliximab SC was noninferior to infliximab IV in terms of PK (noninferior margin = 80%). The geometric LS means for observed Ctrough, week22 were 20.9 mcg/mL and 1.8 mcg/mL in the infliximab SC (120 mg or 240 mg) and infliximab IV (5 mg/kg) treatment groups, respectively.
Efficacy Results: Secondary Outcomes
UC Population Within Study 1.6: The proportion of patients achieving a clinical response at week 22 based on total Mayo score was higher among those receiving infliximab SC (n = 24, 63.2%) than among those receiving infliximab IV (n = 17, 43.6%). At week 22, the proportion of patients achieving clinical response, according to partial Mayo score, was 84.2% (n = 32) (in the infliximab SC group versus 76.9% (n = 30) in the infliximab IV group. At week 54, the proportion of patients achieving clinical response was 63.2% (n = 24) in the infliximab SC group versus 61.5% (n = 24) in the infliximab IV group. The proportions of patients achieving partial Mayo scores were as follows infliximab SC 81.6% (n = 31); infliximab IV 71.8% (n = 28).
The proportion of patients achieving clinical remission at week 22 based on total Mayo score was higher in the infliximab SC group (44.7%, n = 17) than in the infliximab IV group (25.6%, n = 10). The proportion of patients achieving clinical remission as measured by partial Mayo scores at week 22 was 60.5% (n = 23) in the infliximab SC group versus 38.5% (n = 15) in the infliximab IV group. At week 54, the proportion of patients achieving clinical remission in the infliximab SC group was 52.6% (n = 20) versus 48.7% (n = 19) in the infliximab IV group. Partial Mayo scores were as follows: infliximab SC (68.4%, n = 26); infliximab IV (61.5%, n = 24).
The proportion of patients achieving mucosal healing at week 22 was higher in the infliximab SC group (47.4%, n = 18) than in the infliximab IV group (30.8%, n = 12). At week 54, the proportion of patients achieving mucosal healing in the infliximab SC group was 55.3% (n = 21) versus 56.4% (n = 22) in the infliximab IV group.
CD Population Within Study 1.6: The proportions of patients achieving clinical remission at week 30 and week 54 in the infliximab SC group were 64.3% (n = 18) and 57.1% (n = 16) respectively, versus 56.0% (n = 14) and 56.0% (n = 14) at week 30 and week 54, respectively, in the infliximab IV group.
Endoscopic remission at week 22 and week 54 was achieved by 5 patients (35.7%) and 6 patients (50%), respectively, in the infliximab SC group versus 1 patient (14.3%) and 5 patients (50.0%), respectively, in the infliximab IV group. Endoscopic response at week 22 and week 54 was achieved by 11 patients (78.6) and 9 patients (75.0), respectively, in the infliximab SC group versus 3 patients (42.9%) and 8 patients (80.0%) in the infliximab IV group.
Harms
LIBERTY-CD Trial
Treatment-emergent adverse events (TEAEs) were numerically higher in the infliximab SC group (72.3%) than in the placebo group (61.9%) in the maintenance phase of LIBERTY-CD. The majority of TEAEs were grade 1 or 2 in intensity. The numbers of patients with at least 1 serious adverse event (SAE) in the maintenance phase were 16 (6.7%) and 8 (7.6%) in the infliximab SC and placebo groups, respectively. The most common SAEs reported were GI disorders (n = 5 [2.1%] in the infliximab SC group and n = 2 [1.9%] in the placebo group) and infections and infestations (n = 6 [2.5%] in the infliximab SC group and n = 1 [1.0%] in the placebo group).
In the LIBERTY-CD trial, the most common grade 3 adverse events (AEs) reported in the infliximab group were decreased neutrophil count (4.6%), increased creatine phosphokinase (CPK) (2.5%), increased blood bilirubin (2.1%), and hypertriglyceridemia (2.1%); the grade 4 events most commonly reported were increased CPK (3.4%) and decreased neutrophil count (0.8%). In the placebo group, decreased lymphocyte count (4.8%), anemia (3.8%), and increased CPK (1.9%) were the most common grade 3 AEs, while increased CPK (1.9%) was the most common grade 4 AE.
AEs of special interest (for the infliximab SC versus placebo groups) included infection (31.1% versus 18.1%), localized injection-site reaction (ISR) (5.9% versus 1.0%), systemic injection reaction (1.3% versus 1.0%), and injection-related reaction (1.3% versus 1.0%).
One death was reported in LIBERTY-CD during the maintenance phase.
LIBERTY-UC Trial
Reported TEAEs were numerically higher in the infliximab SC group (67.6%) compared to the placebo group (59.3%) in the maintenance phase of LIBERTY-UC. The majority of TEAEs were grade 1 or 2 in intensity. The numbers of patients with at least 1 serious AE in the maintenance phase were 19 (6.4%) and 4 (2.9%) in the infliximab SC and placebo groups, respectively. The most common serious AEs (infliximab SC versus placebo) included GI disorders (1.4% versus 1.4%) and infections and infestations (2.4% versus 0.7%).
In the LIBERTY-UC trial, the most common grade 3 AEs reported in the infliximab group were decreased neutrophil count (3.7%), anemia (2.0%), and increased CPK (1.7%); the most commonly reported grade 4 event was increased CPK (1.4%). In the placebo group, increased CPK (2.9%) was the most common grade 3 AE and the most common grade 4 AE (1.4%).
AEs of special interest (infliximab SC versus placebo) included infection (28.0% versus 25.7%), systemic injection reaction (4.1% versus 2.9%), and injection-related reaction (4.1% versus 2.9%).
There were no deaths reported in the LIBERTY-UC trial.
Study 1.6
During the maintenance phase of Study 1.6, a numerically higher proportion of patients reported TEAEs in the infliximab SC group (74.2%) than in the infliximab IV group (58.5%). The most commonly reported AEs during this phase (infliximab SC versus infliximab IV) were localized ISRs (22.7% versus 4.6%), UC (4.5% versus 12.3%), and neutropenia (7.6% versus 4.6%).
The proportions of patients who experienced at least 1 TEAE on or after week 30 were slightly higher in the infliximab SC treatment group (i.e., 31 patients [47.0%] in the infliximab SC group and 21 patients [32.3%] in the infliximab IV treatment group). (The results relating to week 30 and beyond include the pooled safety results of the 2 treatment groups after patients switched to or continued with infliximab SC at week 30.)
The most common AEs of special interest reported during the maintenance phase (infliximab SC versus infliximab IV) included infection (31.8% versus 29.2%), localized ISR (22.7% versus 4.6%), systemic injection reaction (3.0% versus 0%), and malignancy (1.5% versus 0%). An AE of special interest classified as a systemic injection reaction on or after week 30 was reported for 1 patient (1.5%) in the infliximab SC group only.
There were no deaths reported in Study 1.6.
Table 2: Summary of Key Results From the LIBERTY-UC and LIBERTY-CD Pivotal Trials
Category | LIBERTY-UC | LIBERTY-CD | ||
|---|---|---|---|---|
Remsima SC N = 294 | Placebo N = 144 | Remsima SC N = 231 | Placebo N = 112 | |
Primary outcomes | ||||
Clinical remission at week 54, n (%)a | 127 (43.2) | 30 (20.8) | 144 (62.3) | 36 (32.1) |
Difference (95% CI)b | 21.1 (11.8 to 29.3) | 32.1 (20.9 to 42.1) | ||
P valuec | < 0.0001 | < 0.0001 | ||
Endoscopic response at week 54, n (%) | NA | NA | 118 (51.1) | 20 (17.9) |
Difference (95% CI)b | NA | 34.7 (24.2 to 43.5) | ||
P valuec | NA | < 0.0001 | ||
Secondary outcomes | ||||
Clinical response at week 54, n (%)d | 158 (53.7) | 45 (31.3) | NA | NA |
Difference (95% CI)d | 21.1 (11.2 to 30.1) | NA | ||
P value | < 0.0001e | NA | ||
Endoscopic-histologic mucosal improvement at week 54, n (%) | 105 (35.7) | 24 (16.7) | NA | |
Difference (95% CI)d | 18.0 (9.1 to 25.7) | NA | ||
P value | < 0.0001e | NA | ||
Corticosteroid-free remission at week 54, n of N (%) | 44 of 120 (36.7) | 11 of 61 (18.0) | NA | |
Difference (95% CI)d | 17.3 (3.1 to 28.9) | NA | ||
P value | 0.01e | NA | ||
Maintenance of clinical remission at week 54, n of N (%) | 91 of 143 (63.6) | 18 of 66 (27.3) | NA | NA |
Difference (95% CI)d | 35.5 (21.1 to 47.5) | NA | ||
P value | < 0.0001 | NA | ||
Clinical remission at week 54, n (%)f | NA | NA | 131 (56.7) | 35 (31.3) |
Difference (95% CI)i | NA | 27.0 (15.8 to 37.1) | ||
P value | NA | < 0.0001j | ||
Endoscopic remission at week 54 (based on SES-CD), n (%)d | NA | NA | 80 (34.6) | 12 (10.7) |
Difference (95% CI)i | NA | 24.9 (15.4 to 32.8) | ||
P value | NA | < 0.0001j | ||
Corticosteroid-free remission at week 54, n of N (%) | NA | NA | 39 of 98 (39.8) | 10 of 44 (22.7) |
Difference (95% CI)i | NA | 17.1 (–0.4 to 31.5) | ||
P value | NA | 0.04j | ||
Maintenance of clinical remission at week 54, n of N (%) | NA | NA | 121 of 174 (69.5) | 32 of 91 (35.2) |
Difference (95% CI)i | NA | 34.5 (22.0 to 45.6) | ||
P value | NA | < 0.0001 | ||
CD = Crohn disease, CDAI = Crohn Disease Activity Index; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; JAK = Janus kinase; NA = not applicable; SC = subcutaneous; SES-CD = Simplified Endoscopic Activity Score for Crohn Disease; UC = ulcerative colitis.
aClinical remission in LIBERTY-CD was based on CDAI score.
bThe difference in proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CIs with CMH weights were presented (all trials). Analysis was stratified by previous exposure to biologic drugs and/or JAK inhibitors (used or not used), treatment with oral corticosteroids at week 0 (used or not used), and clinical remission at week 10 (remitter or nonremitter, based on modified Mayo score and/or CDAI score) (LIBERTY-UC and LIBERTY-CD). IN LIBERTY-UC and CD, patients with dose adjustment to Remsima SC 240 mg before week 54 were considered nonremitters.
cThe primary and coprimary outcomes in the LIBERTY-UC and LIBERTY-CD trials, respectively, were within the statistical testing hierarchy.
dThe difference in proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CIs with CMH weights were presented (all trials). Analysis was stratified by previous exposure to biologic drugs and/or JAK inhibitors (used or not used), treatment with oral corticosteroids at week 0 (used or not used), and clinical remission at week 10 (remitter or nonremitter, based on modified Mayo score) (LIBERTY-UC). In LIBERTY-UC, patients with dose adjustment to Remsima SC 240 mg before week 54 were considered as nonremitters and/or nonresponders.
eThe P values were part of outcomes within the statistical testing hierarchy.
fClinical remission was based on abdominal pain and stool frequency in LIBERTY-CD, but on CDAI score in Study 1.6.
iThe difference in proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CIs with CMH weights were presented (all trials). Analysis was stratified by previous exposure to biologic drugs and/or JAK inhibitors (used or not used), treatment with oral corticosteroids at week 0 (used or not used), and clinical remission at week 10 (remitter or nonremitter, based on modified CDAI score) (LIBERTY-CD). In LIBERTY-CD, patients with dose adjustment to Remsima SC 240 mg before week 54 were considered as nonremitter/ and/or nonresponder.
jThe P values were part of outcomes within the statistical testing hierarchy.
Source: Sponsor’s submission.28
Table 3: Safety Data in the LIBERTY-UC, LIBERTY-CD, and Study 1.6 Trials
Category | LIBERTY-UC | LIBERTY-CD | Study 1.6 | ||||
|---|---|---|---|---|---|---|---|
Infliximab SC N = 296 | Placebo N = 140 | Infliximab SC N = 238 | Placebo N = 105 | Infliximab SC N = 66 | Infliximab IV N = 65 | After week 30 N = 131a | |
Patients with at least 1 AE, n (%) | |||||||
TEAE | 200 (67.6) | 83 (59.3) | 172 (72.3) | 65 (61.9) | 49 (74.2) | 38 (58.5) | 52 (39.7) |
Patients with at least 1 serious AE, n (%) | |||||||
TESAE | 19 (6.4) | 4 (2.9) | 16 (6.7) | 8 (7.6) | 5 (7.6) | 7 (10.8) | 6 (4.6) |
Most common events (> 1%) by SOC | |||||||
GI disorders | 4 (1.4) | 2 (1.4) | 5 (2.1) | 2 (1.9) | — | — | — |
Infections and infestations | 7 (2.4) | 1 (0.7) | 6 (2.5) | 1 (1.0) | 2 (3.0) | 4 (6.2) | 6 (4.6) |
Patients who stopped treatment due to AEs, n (%) | |||||||
WDAE | 10 (3.4) | 4 (2.9) | 9 (3.8) | 5 (4.8) | 1 (1.5) | 3 (4.6) | 1 (0.8) |
Patients with AEs of special interest, n (%) | |||||||
SIR | 12 (4.1) | 4 (2.9) | 3 (1.3) | 1 (1.0) | 2 (3.0) | 0 | 1 (0.8) |
Localized ISR | 10 (3.4) | 3 (2.1) | 14 (5.9) | 1 (1.0) | 15 (22.7) | 3 (4.6) | 9 (6.9) |
Infection | 83 (28.0) | 36 (25.7) | 74 (31.1) | 19 (18.1) | 21 (31.8) | 19 (29.2) | 21 (16.0) |
Malignancy | 1 (0.3) | 0 | 0 | 1(1.0) | 1 (1.5) | 0 | 1 (0.8) |
IRR | 12 (4.1) | 4 (2.9) | 3 (1.3) | 1 (1.0) | 0 | 2 (3.1) | NR |
AE = adverse event; GI = gastrointestinal; ISR = injection-site reaction; IRR = injection-related reaction; NR = not reported; SC = subcutaneous; SIR = systemic injection reaction; SOC = System Organ Class; TEAE = treatment-emergent adverse event; TESAE = treatment-emergent serious adverse event; WDAE = withdrawal due to adverse event.
aThe “after week 30” group includes pooled safety results for the 2 treatment arms after patients switched to or continued with infliximab SC at week 30.
Source: Sponsor’s submission.28
Critical Appraisal
Internal Validity
LIBERTY-UC and LIBERTY-CD: The LIBERTY-UC and LIBERTY-CD trials were randomized, placebo-controlled, multicentre, phase III studies designed with an OL induction phase, a DB treatment (maintenance) phase, and an OL extension phase. Both trials employed appropriate methods for blinding, treatment allocation, and randomization.
The primary, coprimary, and key secondary outcomes in the LIBERTY-UC and LIBERTY-CD trials, respectively, were considered appropriate and recommended by the FDA and European Medicines Agency (EMA) for assessing treatment effects for patients with UC and CD in the trial settings.29-31 Outcomes assessed in the LIBERTY trials (e.g., CDAI scores, modified Mayo scores, patient-reported outcomes, and safety outcomes) were subjective and potentially prone to assessment bias, which could bias results in both groups in either direction.
There is also a potential bias arising from treatment awareness in both trials due to the frequent dose augmentations observed in both groups from week 22. This may have affected the assessment of subjective outcomes in both populations in the 2 trials. There was also a concern for potential bias due to missing outcome data for the HRQoL results, especially in the placebo group in both trials at week 54, rendering the results inconclusive.
Concomitant drug use in the maintenance phase was similar in both groups for both trials, apart from the use of budesonide, which was numerically higher in the infliximab SC group compared to the placebo group in the LIBERTY-CD trial; this potentially biases the efficacy results in favour of infliximab SC in the population of patients with CD. There was potential for residual drug effect of continued use of corticosteroids in the maintenance phase in both trials, which may have affected disease symptoms in the placebo and infliximab SC groups in the 2 trials.
There were imbalances in study treatment exposures between the 2 groups in both trials, given that there were more dose adjustments observed in the placebo group from week 22 compared to the infliximab SC group (Table 29 and Table 30). Although dose augmentations (up to 2 injections, i.e., 240 mg infliximab SC) were allowed in the trial, frequent dose adjustments in the maintenance phase could have affected treatment awareness within groups as well as the assessment of subjective outcomes. The direction and magnitude of this potential bias are uncertain.
Study 1.6: Study 1.6 study is an OL, randomized, parallel-group, multicentre, phase I study. Appropriate methods for randomization and treatment allocation were implemented. Baseline characteristics were similar between the 2 treatment groups in the trial, suggesting successful randomization.
The key objective of Study 1.6 was to assess the noninferiority of infliximab SC versus infliximab IV in terms of the primary PK outcome; determined by the trough concentration, calculated from the pre-dose level at Week 22 (Ctrough week 22). . Ctrough assessment in the study was considered appropriate and aligned with regulatory guidelines.32 The assessment of plasma concentration of infliximab (Ctrough at week 22) was considered appropriate by the clinical expert consulted by CADTH and aligns with regulatory guideline requirements32 and published literature.33,34 A noninferiority margin of 80%, 1-sided alpha level of 5%, expected ratio of 1.3, dropout rate of 20%, and coefficient of variation (CV) of 100% were assumed for part 1 of the study. The study was powered to detect a statistical difference between the 2 groups of interest for the PK outcome.
Study 1.6 was neither designed nor powered to formally assess comparative efficacy outcomes (i.e., CDAI response, clinical response, clinical remission, endoscopic response and remission, mucosal healing, or HRQoL); this makes it challenging to assess the relative therapeutic efficacy of infliximab SC versus infliximab IV. The sample size of Study 1.6 (i.e., n = 135) was considered relatively small to assess efficacy outcomes in populations of patients with UC and CD. The treatment effect estimates observed may not be replicable in a larger study sample. The protocol did not prespecify a degree of difference from which to formally conclude noninferiority between infliximab SC and infliximab IV in terms of efficacy outcomes. While the evidence from Study 1.6 suggests that infliximab SC is comparable to infliximab IV in terms of PK parameters, the lack of robust evidence on efficacy outcomes (which were presented descriptively, without any statistical comparison) precludes firm conclusions to support switching from infliximab IV to infliximab SC. The clinical expert consulted by CADTH did not anticipate clinically meaningful differences in efficacy between infliximab SC and infliximab IV because the products have the same active ingredient (i.e., infliximab). The clinical expert did not anticipate any clinical concerns from switching patients from IV to SC administration of infliximab as long as the choice to switch was made based on a case-by-case basis after thorough discussion between clinician and patient.
There were concerns related to missing data between the 2 groups for HRQoL data assessed using the SIBDQ and VAS (for local site pain assessment) because fewer patients completed the questionnaires at week 30 and week 54 compared to baseline (Table 21 and Table 22); this may have affected the findings. In addition, no formal statistical tests for significance were conducted for efficacy outcomes, and missing data were not accounted for during the analyses. Therefore, it is uncertain whether switching patients from infliximab IV to infliximab SC at week 30 in Study 1.6 resulted in comparable HRQoL outcomes in the populations of patients with UC and CD.
External Validity
LIBERTY-UC, LIBERTY-CD, and Study 1.6, part 2 were multicentre, international trials that recruited adult patients aged 18 years to 75 years. The inclusion and exclusion criteria of the trials were generally aligned with the selection criteria used in current practice to identify suitable patients for infliximab, according to the clinical expert consulted by CADTH. However, the exclusion of patients with prior experience with 2 or more lines of biologic therapy and or JAK inhibitors was inconsistent with clinical practice, given that patients with prior exposure to other biologic drugs, including JAK inhibitors, are currently considered for treatment with infliximab IV in clinical practice, according to the clinical expert consulted by CADTH. The baseline disease characteristics of the patients in the LIBERTY-UC and LIBERTY-CD trials, such as CDAI scores (for patients with CD), Mayo scores (for patients with UC), the proportions of patients with moderate to severe disease, the types of prior surgeries conducted — and other important objective outcomes (such as CRP and fecal calprotectin) that are important for monitoring patients in practice — were presented. There were no major differences in baseline characteristics between the infliximab SC group and the placebo group in the LIBERTY-UC and LIBERTY-CD trials.
The primary and key secondary outcomes were considered relevant to decision-making and adequately reflected measures of both efficacy and harm, according to the clinical expert consulted by CADTH. Concomitant medications used in the trial were reflective of clinical practice (except for mesalamine, which is seldom used). Corticosteroid tapering was consistent with regulatory guidelines, although the rates differed slightly from clinical practice.
Although the study design (induction and maintenance phases) in the 3 trials is consistent with regulatory guidelines and reflects clinical practice, it generates an enriched population consisting of responders who can better tolerate and respond to infliximab. The induction periods in the 3 trials were also considered short (4 weeks for Study 1.6 and 10 weeks for LIBERTY-UC and LIBERTY-CD trial); such durations fail to accommodate slow responders, which is inconsistent with current practice, according to the clinical expert consulted by CADTH (in practice, dose-loading periods may extend up to 16 weeks). The durations of the maintenance phases were considered adequate to assess treatment effect. The frequencies of endoscopic assessments were considered standard for trials but differed from current practice due to both patient preference and the logistical constraints associated with conducting these (i.e., the practical limitations and invasiveness of the procedure).
The dosing of infliximab IV (5 mg/kg) in the induction phase of the LIBERTY-UC and LIBERTY-CD trials was consistent with the product monograph. The clinical expert consulted by CADTH noted that clinicians may consider higher doses of infliximab IV for patients with more severe disease during the induction and/or dose-loading phase; these doses can then be further adjusted based on patient response, patient preference, and safety profile. The dose of infliximab SC in Study 1.6 differed from the dose recommended by Health Canada for infliximab SC in that weight-based dosing was performed (i.e., 120 mg or 240 mg infliximab SC for patients weighing < 80 kg or ≥ 80 kg, respectively); dose escalation to infliximab SC 240 mg every 2 weeks was allowed from week 30, and patients received only 2 doses during the induction phase rather than the 3 doses recommended by Health Canada. There is some uncertainty as to whether the results of Study 1.6 are generalizable to the use of infliximab SC as per the Health Canda–recommended dosage.
Indirect Comparisons
No indirect treatment comparison (ITC) was submitted for this review.
Cost Information
At the submitted price, the first-year cost of infliximab SC depends on which infliximab IV product is chosen for the induction period. The costs per patient when Inflectra is chosen are $19,357 in the first year and $15,424 in each subsequent year.
The annual costs associated with infliximab SC are less than those associated with the branded IV product (Remicade) and other branded biologic comparators, such as adalimumab (Humira), golimumab SC (Simponi), vedolizumab (Entyvio) IV and SC, and ustekinumab (Stelara). On the other hand, infliximab SC is associated with higher annual costs than other infliximab IV biosimilars (Inflectra, Renflexis, and Avsola) and adalimumab biosimilars, even though it is priced at parity with the least costly biosimilar per mg.
These incremental costs or savings are based on publicly available list prices and may not reflect actual prices paid by Canadian drug plans.
Conclusions
A total of 3 randomized trials supported the clinical efficacy and safety data of infliximab SC for the reimbursement request in patients with UC or CD, which aligns with the Health Canada indication. Infliximab SC demonstrated statistically significant benefits in clinical remission based on the modified Mayo score (UC) and the CDAI scoring system as well as in terms of endoscopic response (CD) in adult patients with moderately to severely active UC and CD, respectively, who have not responded to conventional therapy. The results for key secondary outcomes showed statistically significant benefits in favour of infliximab SC versus placebo. However, the sustainability of the beneficial effect and the potential for recurrence of the disease in the long-term (i.e., beyond 1 year) remain uncertain. A bioequivalence study with a small sample size suggests that infliximab SC may have benefits comparable to the infliximab IV formulation. Due to significant limitations, no conclusion could be drawn as to the benefit of infliximab on improvement in HRQoL.
Overall, infliximab SC treatment was shown to be well-tolerated in patients with UC or CD across 3 trials. Safety data pooled across the trials showed no new or unexpected safety concerns. The safety profile was considered acceptable and comparable to infliximab IV by the clinical expert consulted by CADTH. However, it is unknown if long-term safety among all patients who received infliximab in real-world clinical practice setting will be maintained.
Infliximab (Remsima SC) has the same main ingredient as the infliximab IV product; however, it is administered at a different dosage and through a different route (SC versus IV). It is intended to provide a treatment option in place of infliximab IV that could be self-administered by patients without the need for frequent or lengthy visits to infusion clinics. There was no evidence designed to assess the impact of this more convenient administration on efficacy outcomes.
At the submitted price, and based on the recommended dosing regimen, the annual cost of infliximab SC is $19,357 per patient in the first year and $15,424 every year thereafter. Infliximab is less costly than branded biologic products, but more costly than other biologic disease-modifying antirheumatic drug (DMARD) biosimilars. The submitted price of infliximab SC would have to be reduced by 16% to 40% for its annual cost to be equivalent to that of the least costly subsequent entry biologic (SEB) comparator, depending on the comparator (i.e., infliximab versus noninfliximab products).
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of infliximab (Remsima SC) 120 mg/mL solution for SC injection for the treatment of CD and UC in IBD.
Disease Background
Contents in this section have been informed by materials submitted by the sponsor and by clinical expert input. The following information has been summarized and validated by the CADTH review team.
IBD is an umbrella term describing chronic inflammation of the GI tract caused by 1 of 2 disorders: UC or CD. The etiology of IBD is unknown; possible causes include genetics or abnormal immune response to environmental factors, such as pathogens (viruses, bacteria, fungi, or parasites) in the GI tract.1,7,8 IBD is most prevalent among adolescents and adults aged 40 years to 50 years. Canada has the highest prevalence and incidence of IBD in the world, with estimates of about 0.8%, amounting to about 322,600 people living with the disease as of 2023.1 The prevalence of CD and UC in Canada is forecast to increase by 2030 to 493 and 436 per 100,000, respectively, with average annual percentage increases of 2.75% and 2.87%.2 A 2023 IBD report by Crohn and Colitis Canada estimated that 470,000 people in Canada will be living with IBD by 2035.6 Common risk factors identified are smoking, family history of IBD, infectious gastroenteritis, and frequent use of nonsteroidal anti-inflammatory drugs.24
Crohn Disease
CD can affect any part of the GI tract, from mouth to rectum, but is mainly observed around the ileum (i.e., small intestine), colon (i.e., beginning of the large intestine), and rectum. It is most common in adolescents and adults aged 20 years to 30 years.10 The incidence of CD in Canada ranged from 8.8 to 22.6 per 100,000 from 1990 to 2013.3 The prevalence estimate of CD in Canada in 2018 was 368 per 100,000, according to the Canadian Gastro-Intestinal Epidemiology Consortium. This translates to about 135,000 people living with CD.11,12
CD can manifest in 3 phenotypical forms: inflammatory, stricturing, and penetrating (i.e., fistulas and abscesses).9 Fistulizing disease is characterized by the formation of abnormal, tunnel-like connections between the intestine and skin, usually around the rectum, between loops of intestine, or between the intestine and abdominal wall, especially following surgery.1 Inflammation can also manifest outside the GI tract, affecting the joints (as central or axial arthritis), eyes (as uveitis, iritis, and episcleritis), and skin (as erythema nodosum, pyoderma).14 Patients may present with symptoms such as abdominal pain, rectal bleeding, fatigue, vomiting, diarrhea, perianal disease, weight loss, and bloating.10,13 CD may also cause complications in patients over time, such as malnutrition, weight loss, anemia, bowel obstructions, fistulae, anal fissures, and intra-abdominal and other abscesses and ulcers;10,26 some patients with colonic CD may have an increased risk of developing colon cancer.10 Patients may experience chronic or intermittent symptoms; disease activity and severity can vary widely over time. While some patients experience continuous and progressive active disease, about 20% of patients may experience prolonged remission after initial presentation.14 Relapse rates at 1 year, 2 year, 5 years, and 10 years are estimated at 20%, 40%, 67%, and 76%, respectively, for those presenting with remission.35 Disease severity in CD has been classified using the CDAI, developed by the American College of Gastroenterology (Table 4). For many patients with CD, symptoms are chronic and intermittent, and disease activity and severity can vary widely. Disease severity is measured using the CDAI and Harvey-Bradshaw Index (HBI) which are designed to evaluate bowel-related symptoms including stool frequency, abdominal pain (AP), arthritis/arthralgia, uveitis, skin/mouth lesions, and perianal disease. While the HBI is commonly used in routine gastroenterology practice, the CDAI remains the most common comparable end point across biologics in CD.10 Less precision is expected with the HBI because it is a subset of the CDAI (e.g., the HBI uses single-day readings, includes only 5 of the 8 CDAI variables, and sums variables instead of applying weighted coefficients).10 The correlation coefficients between HBI and CDAI have been reported to be between 0.80 and 0.93.10,12
Diagnosis of CD involves a combination of clinical and endoscopic evaluations as well as histological, radiological, and/or biochemical investigations.24 Ileocolonoscopy with multiple biopsy specimens is usually the first-line procedure for diagnosis.24 The endoscopic hallmark of CD is the patchy distribution of inflammation, with skip lesions, defined as areas of inflammation interposed between normal-appearing mucosa.24 Cross-sectional imaging using MRI, CT enterography, and transabdominal ultrasonography are complementary tests that aid in the detection and staging of inflammatory, obstructive, and fistulizing CD.24
Ulcerative Colitis
UC is characterized by inflammation and ulcers in the mucosal layer of the large intestine (colon), typically beginning at the rectum (anus), progressing upward, and in some cases affecting the entire colon.15,1,16 UC generally develops in young adulthood37 and persists throughout life, with periods of spontaneous remission and relapse.21 UC has a worldwide annual incidence rate of 1.2 to 20.3 cases per 100,000 people and a prevalence of 7.6 to 246.0 cases per 100,000 people.17
Table 4: Classification of Disease Severity in Crohn Disease
Status | CDAI score | Description from ACG guidelines |
|---|---|---|
Remission | < 150 | Asymptomatic or without any symptomatic inflammatory sequelae |
Mild to moderate | 150 to 220 | Ambulatory and able to tolerate oral alimentation without manifestations of dehydration, systemic toxicity, abdominal tenderness, painful mass, intestinal obstruction, or > 10% weight loss |
Moderate to severe | 220 to 450 | Does not respond to treatment for mild to moderate disease, or experiences prominent symptoms of fever, significant weight loss, abdominal pain or tenderness, intermittent nausea or vomiting, or significant anemia |
Severe | > 450 | Experiences persistent symptoms despite the introduction of conventional corticosteroids or biologic drugs as outpatients; or presents with high fevers, persistent vomiting, evidence of intestinal obstruction, or significant peritoneal signs, such as involuntary guarding or rebound tenderness, cachexia, or evidence of an abscess |
ACG = American College of Gastroenterology; CDAI = Crohn Disease Activity Index.
Source: American College of Gastroenterology.36
Patients with UC present with symptoms that include blood and/or mucus in the stool, frequent diarrhea, loss of appetite, and tenesmus (strong urge to use the bathroom without necessarily having a bowel movement) in addition to abdominal pain, rectal bleeding, and weight loss.17,38,39 The most common initial manifestation of UC is bloody diarrhea, with or without mucus. In addition, patients with UC report high rates of fatigue and sleep difficulties.17,38,39 UC is associated with significant morbidity and an increased risk of colorectal cancer, although the incidence of premature mortality does not differ from that of the general population.40,41 Other potentially severe complications associated with UC that may lead to hospitalization include severe blood loss, fulminant colitis, perforated bowel, and toxic megacolon.42 UC substantially reduces patient quality of life and has considerable impacts on many aspects of daily life, such as emotional and psychological functioning, social and physical functioning, and work and academic life.43,44 Chronic, active UC may lead to structural damage of the colon, causing dysmotility, chronic symptoms, reduced quality of life, and risk of colon cancer requiring colectomy.
UC can be further classified in clinical practice based on severity: mild, moderate, or severe disease, depending on the index score used (e.g., the Truelove and Witts severity index, Mayo score, or Montreal classification).22 The Mayo scoring system is described in Table 39, Appendix 1. Most patients experience mild to moderate disease, characterized by active disease at diagnosis, followed by alternating exacerbations and lengthening periods of remission. Overall, 10% to 15% of patients experience aggressive disease, with a cumulative risk of relapse between 70% and 80% at 10 years postdiagnosis.37 Although most patients experience a relapsing-remitting disease course, reports show that up to 24% of patients experience continuous UC symptoms.23
Diagnosis is made based on symptoms at patient presentation and clinical tests using a combination of clinical and endoscopic evaluations, such as endoscopy, biopsy, and stool sampling, to rule out other causes.40
Standards of Therapy
Contents in this section have been informed by materials submitted by the sponsor and clinical expert input. The following information has been summarized and validated by the CADTH review team.
Crohn Disease
Treatment goals for CD highlighted by Canadian45 and American26 published clinical practice guidelines include inducing and maintaining clinical remission and reducing the need for long-term corticosteroid use while minimizing side effects. Long-term goals include endoscopic healing, absence of disability, and normalized HRQoL. Short- to intermediate-term goals include normalizing the biomarkers of disease activity (e.g., CRP and fecal calprotectin).9,46 These goals are consistent with those highlighted by the clinical expert consulted by CADTH for this review.
Treatment selection for CD depends on the location, extent, phenotype, and severity of disease.26 Available treatment options for CD include aminosalicylates, immunosuppressants (e.g., AZA, cyclosporine, methotrexate, and 6-MP), corticosteroids (e.g., prednisone), TNF alpha antagonists (e.g., infliximab and adalimumab), interleukin inhibitors, and integrin inhibitors (e.g., vedolizumab).25,26 The treatment options highlighted are consistent with the clinical expert’s input.
Medical management in practice follows a stepwise approach in which treatments are used sequentially and escalated to newer therapies or higher doses, depending on patients’ responses.47 It is worth noting that most treatments are associated with AEs, depending on short- and long-term use.10,25 Surgery is another treatment option, including total colectomy and ileostomy for patients with serious complications or who do not respond to medical management.26
Ulcerative Colitis
Treatment goals for UC highlighted in the 2015 Canadian guidelines48 include complete remission, defined as symptomatic remission (i.e., normal stool frequency and no blood in the stool) and endoscopic healing (i.e., a Mayo endoscopic subscore [ES] of 0 or 1). The parameters assessed when determining complete remission (stool frequency, rectal bleeding, and findings on endoscopy) are the same 3 that are considered when evaluating the modified Mayo score.49 Another important treatment goal highlighted in the International 2021 STRIDE-II50 initiative document is clinical response, defined as at least a 50% improvement in rectal bleeding and stool frequency as the most immediate target. Another target is clinical remission, defined as Mayo rectal bleeding and stool frequency sub scores of 0 or a partial Mayo score of less than 3, with no Mayo subscore greater than 1. Suggested long-term targets include endoscopic healing and improved quality of life. These goals are consistent with those highlighted by the clinical expert consulted by CADTH for this review.
Treatment options for UC depend on the presence of active disease, the severity and extent of the UC, and patient preference. Treatments are divided into 2 groups: conventional therapies and advanced therapies. Conventional therapies available in Canadian practice include aminosalicylate products, corticosteroids, and immunomodulators (such as AZA, 6-MP and methotrexate). Corticosteroids are recommended as initial or first-line treatments to achieve complete remission for patients with moderate to severe UC and to treat acute flares. Corticosteroids are not recommended for long-term use due to serious side effects and lack of long-term efficacy. Immunomodulators are available as next-line therapy; however, biologic therapy may be administered immediately after steroid failure (or prolonged steroid dependence). Biologics and JAK inhibitors are often grouped together as “advanced therapies.” Available advanced therapies include adalimumab, golimumab, infliximab, ustekinumab, tofacitinib, ozanimod, and vedolizumab. Ustekinumab and ozanimod are not publicly reimbursed for UC in Canada. Tofacitinib is recommended for use only in patients who have not responded to biologics.51 For such patients, a biologic with a different mechanism of action (or tofacitinib) are next options.48,52 Clinicians may switch therapies if patients continue to flare; first, they must confirm adherence, check drug trough levels, and adjust the dose, if subtherapeutic.
When advanced therapies and clinical trials have been exhausted, surgery is an option.41 Although surgery is considered curative,53 it is associated with a high risk of complications.41 Thus, it is usually reserved for patients who cannot be managed medically, patients with acute, severe UC (i.e., toxic megacolon, perforation, and uncontrolled, severe hematochezia), or patients who develop colorectal cancers.41
Drug Under Review
Infliximab is a human-murine chimeric immunoglobulin G1 kappa monoclonal antibody that binds specifically to TNF alpha. By doing so, it prevents TNF alpha receptor activation, thereby neutralizing the biological activity of TNF alpha.
Remsima SC is an SC formulation of infliximab available in a prefilled syringe (PFS) with an automatic needle guard and prefilled pen formats containing 120 mg of active substance. It is recommended for adult patients with moderately to severely active UC or CD. It should be initiated as maintenance therapy 4 weeks after the last administration of 3 IV infusions of infliximab (5 mg/kg) at weeks 0, 2, and 6. The recommended dosage is 120 mg once every 2 weeks.
Health Canada reviewed infliximab SC and the drug received a Notice of Compliance on February 15, 2024 for the following indications:
maintenance treatment of adults with moderately to severely active CD who have had an inadequate response or were intolerant of conventional therapy
maintenance treatment of adults with moderately to severely active UC who have had an inadequate response or were intolerant of conventional therapy.
In both cases, Remsima SC should be used as maintenance therapy only after the completion of an induction period with IV infliximab.
The reimbursement request aligns with the Health Canada indications. Infliximab has also been reviewed by the FDA and received FDA market authorization on October 20, 2023 for CD (i.e., for reducing signs and symptoms and inducing and maintaining clinical remission in adult patients with moderately to severely active disease who have had an inadequate response to conventional therapy) and UC (i.e., for reducing signs and symptoms, inducing and maintaining clinical remission and mucosal healing, and eliminating corticosteroid use in adult patients with moderately to severely active disease who have had an inadequate response to conventional therapy).
It received regulatory authorization by the EMA on June 01, 2020, and by the Medicines and Health care products Regulatory Agency in July 2022.
Infliximab (Remsima SC) was approved in 2021 by Health Canada for use in patients with moderately to severely active RA. It received a positive conditional CADTH recommendation for the treatment of adult patients with moderately to severely active RA in 2021.
Infliximab SB2 (Renflexis) biosimilar for IV infusion has previously been reviewed by CADTH for RA, ankylosing spondylitis, CD (adult and pediatric), fistulizing CD, UC (adult and pediatric), psoriatic arthritis, and plaque psoriasis. It received a conditional positive recommendation from CADTH on February 20, 2018, for UC and CD for the following indications in adult patients:55
reduction of signs and symptoms, induction and maintenance of clinical remission and mucosal healing, and reduction of corticosteroid use in adult patients with moderately to severely active CD who have had an inadequate response to a corticosteroid and/or aminosalicylate (can be used alone or in combination with conventional therapy)
reduction of signs and symptoms, induction and maintenance of clinical remission and mucosal healing, and reduction or elimination of corticosteroid use in adult patients with moderately to severely active UC who have had an inadequate response to conventional therapy (i.e., aminosalicylate and/or corticosteroid and/or an immunosuppressant).
The key characteristics of infliximab SC are summarized in Table 5 and Table 6 along with those of with other treatments available for CD and UC, respectively.
Table 5: Key Characteristics of Treatments Used for Crohn Disease
Characteristic | Mechanism of action | Indicationa | Route of administration | Recommended dosage | Serious adverse effects or safety issues |
|---|---|---|---|---|---|
Risankizumab | Humanized IgG1 monoclonal antibody that binds to the p19 subunit of human IL-23 cytokine and inhibits IL-23 signalling in cell-based assays, including the release of the proinflammatory cytokine, IL-17 | Treatment of patients with moderately to severely active CD who have had an inadequate response to, intolerance of, or demonstrated dependence on corticosteroids; or who have shown an inadequate response to, intolerance of, or loss of response to immunomodulators or biologic therapies | IV (induction) and SC (maintenance) | Adults with moderate to severe CD:
|
|
Ustekinumab | Human IgG1 monoclonal antibody that neutralizes cellular responses mediated by IL-12 and IL-23 | Treatment of patients with moderately to severely active CD who have had an inadequate response to, loss of response to, or intolerance of conventional therapy (i.e., CS or immunomodulators) or 1 or more TNF alpha antagonists, or who were CS-dependent | IV (induction) and SC (maintenance) | Adults with CD:
|
|
Vedolizumab | IgG1 monoclonal antibody that binds to the human alpha 4 beta 7 integrin, acting as a gut-selective, anti-inflammatory biologic | Treatment of patients with moderately to severely active CD who have had an inadequate response to, loss of response to, or intolerance of immunomodulators or a TNF alpha antagonist; or who have had an inadequate response to, intolerance of, or demonstrated dependence on a CS | IV (induction and maintenance) and SC (maintenance) | Adults with moderate to severe CD: IV formulation:
SC formulation:
|
|
Infliximab | Anti-TNF alpha IgG1 kappa monoclonal antibody that neutralizes the biological activity of TNF alpha by binding specifically to its receptors | Reduction of signs and symptoms, induction and maintenance of clinical remission and mucosal healing, and reduction of CS use in adults with moderately to severely active CD who have had an inadequate response to a CS and/or aminosalicylate Adults with fistulizing CD who have not responded to conventional treatment | IV | Adults with moderate to severe CD:
Adults with fistulizing CD:
|
|
Adalimumab | Anti-TNF alpha human IgG1 monoclonal antibody that binds to and blocks TNF alpha and its interaction with p55 and p75 cell-surface TNF alpha receptors | To reduce signs and symptoms and induce and maintain clinical remission in adults with moderately to severely active CD who have had an inadequate response to conventional therapy To reduce signs and symptoms and induce clinical remission in adults with moderately to severely active CD who have stopped responding or are intolerant of infliximab | SC | Adult CD:
|
|
Upadacitinib | Upadacitinib is a selective JAK inhibitor that demonstrates activity against JAK1 JAK2, JAK3, and tyrosine kinase 2 | Treatment of patients with moderately to severely active CD who have not responded to prior treatment (i.e., an inadequate response to, loss of response to, or intolerance of at least 1 conventional and/or biologic therapy) | Oral | Adults with moderate to severe CD:
|
|
CD = Crohn disease; CS = corticosteroid; IgG1 = immunoglobulin G1; IL = interleukin; q.2.w. = every 2 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; SC = subcutaneous; TNF = tumour necrosis factor.
aHealth Canada indication.
Source: Product monographs for risankizumab (Skyrizi),56 vedolizumab (Entyvio),57 infliximab (Remicade and Inflectra),58,59 adalimumab (Humira),60 ustekinumab (Stelara),61 and upadacitinib (Rinvoq).62
Table 6: Key Characteristics of Treatments for UC
Drug | Mechanism of action | Indicationa | Route of administration and recommended dosage | Serious adverse effects or safety issues |
|---|---|---|---|---|
Interleukin-23p19 antagonist | ||||
Mirikizumab | Humanized IgG4 monoclonal antibody, JAK inhibitor | Treatment of adult patients with moderately to severely active UC who have had an inadequate response to, loss of response to, or intolerance of conventional therapy, a biologic treatment, or a JAK inhibitor | Induction: 300 mg IV q.4.w. in weeks 0, 4, and 8 Consider extended induction of 300 mg IV q.4.w. in weeks 12, 16, and 20 in patients who are nonresponders at week 12 Maintenance: 200 mg SC q.4.w. | Upper respiratory tract infection, headache, and injection-site reactions (e.g., rash, maculo-papular rash, popular rash, and pruritic rash) were commonly reported AEs during clinical trials |
S1P receptor modulator | ||||
Ozanimod | S1P receptor modulator that binds to the S1P1 receptors on lymphocytes, preventing egress from lymph nodes The mechanism by which ozanimod and its active metabolites exert their therapeutic effects in MS and UC is unknown, but may involve reduced lymphocyte migration into the CNS and intestine | Treatment of adult patients with moderately to severely active UC who had an inadequate response to, loss of response to, or intolerance of conventional therapy or biologic drug | Dose escalation to 0.92 mg orally once daily Induction (day 1 to day 4): 0.23 mg once daily Dose escalation (day 5 to day 7): 0.46 mg once daily Maintenance (day 8 and onward): 0.92 mg once daily | Malignancies, particularly of the skin, have been reported in patients taking ozanimod in clinical trials Initiation may result in transient reductions in heart rate and atrioventricular delays |
Infliximab | Anti-TNF alpha IgG1k monoclonal antibody that neutralizes the biological activity of TNF alpha by specifically binding to its receptors | Induction and maintenance of clinical remission and mucosal healing, and reduction or elimination of corticosteroid use, in adult patients with moderately to severely active UC who have had an inadequate response to conventional therapy | Induction doses of 5 mg/kg IV at weeks 0, 2, and 6 followed by 5 mg/kg IV every 8 weeks thereafter | Infections and malignancies have been observed |
Golimumab | Anti-TNF alpha human monoclonal antibody that binds with p55 or p75 human TNF alpha receptors | Induction and maintenance of clinical response in adults with moderately to severely active UC who have had an inadequate response to or have medical contraindications to conventional therapy, including corticosteroids, aminosalicylates, azathioprine, or 6-MP | 200 mg administered by SC injection at week 0 followed by 100 mg at week 2 and 50 mg every 4 weeks thereafter | Upper respiratory infections and reactions at the injection site |
Adalimumab | Anti-TNF alpha human IgG1 monoclonal antibody that binds to and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF alpha receptors | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response to conventional therapy, including corticosteroids and/or azathioprine or 6-MP, or who are intolerant of such therapies | 160 mg at week 0 followed by 80 mg at week 2, administered by SC injection | Serious infections (pneumonia), malignancies, and neurologic events have been reported in patients taking adalimumab |
Integrin-blocker | ||||
Vedolizumab | IgG1 monoclonal antibody that binds to the human alpha 4 beta 7 integrin, acting as a gut-selective, anti-inflammatory biologic | Treatment of adult patients with moderately to severely active UC who have had an inadequate response to, loss of response to, or intolerance of conventional therapy or infliximab | 300 mg administered by IV infusion at weeks 0, 2, and 6, then every 8 weeks thereafter SC maintenance dosage: 108 mg every 8 weeks | Infections and malignancies have been reported in patients taking vedolizumab |
Interleukin antagonist | ||||
Ustekinumab | Human IgG1 monoclonal antibody that neutralizes cellular responses mediated by IL-12 and IL-23 | Treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a biologic or have medical contraindications to such therapies. | IV infusion, single-use, weight-based dose (approximately 6 mg/kg): 250 mg for those weighing ≤ 55 kg; 390 for those weighing ≥ 55 kg to ≤ 85 kg; or 520 mg for those weighing ≥ 85 kg for induction therapy, followed by maintenance therapy of 90 mg SC infections every 8 weeks | Immunomodulating drugs have the potential to increase the risk of infections and malignancy. |
JAK inhibitor | ||||
Tofacitinib | Selective JAK inhibitor that blocks several cytokine pathways and lymphocyte activation | For the treatment of adult patients with moderately to severely active UC with an inadequate response to, loss of response to, or intolerance of either conventional UC therapy or a TNF alpha inhibitor | 10 mg orally (as tofacitinib citrate), twice daily | A Health Canada warning indicated an increased risk of thromboses (pulmonary and deep vein thrombosis) and death, and increased risk of serious infection, including herpes zoster infections. Of note, tofacitinib is not recommended in combination with biological UC therapies or with potent immunosuppressants, such as azathioprine or cyclosporine. |
6-MP = mercaptopurine; AE = adverse event; CNS = central nervous system; IgG1 = = immunoglobulin G1; IgG1k = immunoglobulin G1 kappa; IgG4 = immunoglobulin G4; IL = interleukin; IL-12/IL-23p40 = interleukin-12/interleukin-23 p40 subunit; JAK = Janus kinase; MS = multiple sclerosis; q.4.w. = every 4 weeks; S1P = sphingosine-1-phosphate; SIP1 = sphingosine-1-phosphate receptor 1r; S1P5 = = sphingosine-1-phosphate receptor 5; SC = subcutaneous; TNF = tumour necrosis factor; UC = ulcerative colitis.
aHealth Canada–approved indication.
Source: Product monographs for mirikizumab (Omvoh),63 ozanimod (Zeposia),64 ustekinumab (Stelara),65 infliximab (Remicade),66 vedolizumab (Entyvio),67 golimumab (Simponi),68 tofacitinib (Xeljanz),69 and adalimumab (Humira).70
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full patient and clinician group submissions received by CADTH are available in the consolidated patient and clinician group input document for this review on the project website: SR0816 to 000/001 infliximab – Patient and Clinician input.
One patient input from the GI Society was summarized for this review. The GI Society is a national charity with programs and services that support research, advocate for appropriate patient access to health care, and promote GI and liver health.
Information from this input was gathered through questionnaires, interviews, and surveys. There were 5 surveys: a 2015 survey on biologics and biosimilars completed by 423 people in Canada with IBD (CD or UC); a 2018 survey on unmet needs among people in Canada with IBD completed by 432 people; a 2020 survey completed by 579 respondents regarding unmet patient needs in IBD; a 2020 survey on biosimilars with 145 respondents, most of whom had IBD (some had other inflammatory conditions); and a 2022 survey about the IBD patient journey completed by 54 respondents in Canada with IBD. Additional data from a 2020 focus group on persons living with IBD, and 1-to-1 interviews with patients, were also analyzed.
The GI Society expressed that patients with IBD preferred sustained remission and treatment response over relieving any 1 symptom. The group noted that, “it is never just 1 flare that dominates the impact of the disease, but the constant concern that there will be future flares, possibly worse than the last, at unpredictable times, which can disastrously disrupt their lives.” Respondents expressed different concerns associated with IBD. Some of these included fear of running out of medication, anxiety about determining when to go to the emergency department based on symptoms, pain, fear of going out due to disease, decreased quality of life, and fear and worry at being faced with mortality at a young age.
The GI Society highlighted that treatment of CD and UC requires a multifaceted strategy that allows for the management of symptom and disease consequences with therapies that target and reduce the underlying inflammation. Current treatment options identified in the patient advocacy input included 5-ASA, corticosteroids, immunosuppressive drugs, and biologics. The group quoted recent reports published by the Institut national d’excellence en santé et en services sociaux suggesting that clinicians in current practice prescribe more biologics and biosimilars than other drugs in the first-line setting for patients with UC, especially those with known risk of disease progression. The patient advocacy group highlighted that corticosteroids were discouraged for long-term maintenance because these do not prevent flares. Newer therapies for UC identified by the patient advocacy group included JAK and S1P inhibitors. No patients interviewed were receiving the treatment under review; however, most had received a biologic. Results from a survey conducted by the GI Society reported that 63% of respondents indicated they had experienced symptom reduction after using a biologic, while 23% confirmed remission.
The GI Society highlighted that patients would like additional effective treatment options with convenient and timely access and different administration methods and dosages. Patients desire options that can be administered at home and thereby reduce time off work. The GI Society expressed that inadequate access to treatment causes continual, debilitating disease symptoms; secondary illnesses, such as depression and anxiety disorders; and the preventable loss of family and other social interactions. Major concerns associated with access to current therapies noted by the patient group included ensuring adequate supply and continuity of care, especially timely communication between patients and their health care providers. Therefore, there is a need for new, effective treatments for patients that could improve quality of life and eliminate symptoms, pain, frustration, and hardship. The patient advocacy group expressed that while biologic medications are very effective, the ongoing injections or infusions required for a person with a chronic disease can require a lot of work and effort. According to the patient advocacy group, this could mean taking time off work, which can be difficult and contribute to financial hardship for many patients.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of IBD.
Unmet Needs
The clinical expert indicated that there is no cure for UC or CD in current practice, and that early treatment is crucial because the first medication prescribed has the best chance of improving symptoms and mucosal healing. Treatment goals highlighted were similar for patients with UC and CD, and included symptom resolution (clinical remission), improved quality of life (achieved by normalizing bowel movements and weight and energy levels and resolving pain, bowel urgency, and rectal bleeding), reduced need for surgery, and avoidance of the repetitive use of corticosteroids.
The expert highlighted that currently approved treatment options for patients with UC or CD target mucosal healing and histological remission. Therapies listed by the expert were similar across the 2 diseases. These included advanced therapies (anti-TNF alpha biologics, JAK inhibitors, alpha 4 beta 7 integrin inhibitors and interleukin-23 plus interleukin-12 and/or interleukin-23 inhibitors) as well as conventional therapies (immunomodulators and corticosteroids). Most advanced treatments in practice target primary and secondary loss of response in both diseases, according to the expert. However, about half of IBD patients have EIMs of CD, which can be disabling, and only a few treatments address this issue. The expert indicated that currently, there is a preference for anti-TNF alpha therapies to treat many patients who present with concomitant EIMs and or fistulizing perianal disease. The expert also noted that patients desire SC treatment options because these reduce the need to take time off work and give them a greater sense of independence. Infliximab IV is currently available as a treatment option and remains a core component of the treatment paradigm in UC and CD.
Place in Therapy
The clinical expert indicated that treatment selection in practice for patients with UC and CD is complex and depends on disease phenotype and patient preference. Patients with more severe disease are offered a combination therapy with an immunomodulator (e.g., methotrexate, AZA) and advanced therapy for up to a year, after which the immunomodulator can be removed if the patient experiences clinical and endoscopic remission. The expert further noted that anti-TNF alpha drugs are often used as first-line treatments for hospitalized patients with severe UC, a population not included in the LIBERTY-UC trial. The expert highlighted that some clinicians believe IV infusions provide a more rapid response than SC options. The expert added that hospitalized patients with severe UC or CD are more likely to receive the IV formulation of infliximab. The expert highlighted that infliximab SC may be used as first-line treatment in select patients with CD (for example, those with penetrating disease) if approved for funding. The clinical expert did not recommend that patients should have previously tried and not responded to other treatment options before being eligible to receive infliximab SC. Overall, the expert did not anticipate any shift in treatment paradigm with the use of infliximab SC, apart from the option of switching from the IV route to the SC option.
Patient Population
The clinical expert indicated that patients with confirmed, moderate to severe CD or UC (based on pathological and histological diagnosis) are best suited for treatment with infliximab SC. The expert noted that misdiagnosis is rare in practice, but that delays in diagnosis may occur. The expert noted that not all patients respond well to anti-TNF alpha therapy and that the least suitable patients are those who fear self-injection.
Assessing the Response Treatment
The clinical expert consulted noted that the frequencies of treatment response assessment in the LIBERTY-UC and LIBERTY-CD trials are standard for clinical trials but differ from real-world settings due to logistics and patient preference. For patients with CD or UC, the expert highlighted that colonoscopy is seldom performed every 12 weeks because of logistics. The clearest marker of response, according to the expert, is improvement in clinical symptoms (i.e., abdominal pain, stool frequency, energy level, resolution of rectal bleeding [for patients with UC]); however, these do not correlate well with objective markers of disease activity. According to the expert, in clinical practice, among patients with CD, CRP and fecal calprotectin are frequently used to monitor response to advanced treatments, while the HBI is used to monitor treatment response (as opposed to the CDAI, which was used in the trial). Fecal calprotectin is an objective measure used to monitor disease activity and treatment response in patients with UC, alongside the partial Mayo score (which excludes endoscopic assessment), according to the expert. The expert noted that the modified Mayo score (which includes an endoscopic assessment) is used in clinical practice for initial patient assessment, before treatment initiation, while the partial Mayo score is used routinely for follow-up to assess response. The expert highlighted that patients are evaluated in practice based on their presentation of clinical symptoms and an assessment of objective data. The expert mentioned that some patients may present as primary nonresponders during treatment, and some may experience loss of response during treatment (the clinical expert noted that the standard percentage of patients with CD or UC in clinical practice who experience loss of response in the first year of treatment is approximately 10% to 20%). The clinical expert highlighted that treatment response should be assessed after induction, around week 16, and again on maintenance therapy after 1 year of maintenance therapy for reimbursement purposes. The expert added that many clinicians will evaluate fecal calprotectin and CRP every 6 months.
Discontinuing Treatment
The expert noted that the factors leading to treatment discontinuation are consistent with those of other advanced therapies currently used for patients with UC or CD. According to the clinical expert, patients are assessed based on clinical symptoms and objective data, given that some may present as primary nonresponders or experience loss of response following treatment. According to the expert, factors that are considered when evaluating treatment discontinuation include:
persistence or worsening of clinical symptoms (pain and diarrhea for patients with CD, diarrhea; rectal bleeding [except hemorrhoidal] and urgency of defecation for patients with UC)
worsening or persistence of biomarkers (i.e., CRP and fecal calprotectin for patients with CD; fecal calprotectin for patients with UC)
development or worsening of complications of disease (strictures and/or fistulae for patients with CD)
persistence or worsening of endoscopic disease activity (for patients with UC)
inability to taper corticosteroids or requirement of recurrent courses (> 2 full courses in 1 year); some patients will require 1 course of corticosteroid treatment in a year, but this does not lead to automatic discontinuation (for patients with UC or CD)
development of AEs (decision to discontinue based on clinician and patient choice) (for patients with UC or CD).
Prescribing Considerations
The clinical expert noted that UC and CD diagnoses are usually made by gastroenterologists; however, general internists with special interest in IBD have sufficient experience to prescribe infliximab for both populations. The expert indicated that treatment initiation begins at private infusion centres, where costs are covered by the drug manufacturer or other patient support programs. Patients transition to self-injection for the SC formulation if approved for funding.
Clinician Group Input
No clinician group input was submitted for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may have an impact on their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 7.
Table 7: Summary of Drug Plan Questions and Clinical Expert Responses
Drug plan questions | Clinical expert responses |
|---|---|
Relevant comparators | |
There are no direct, phase III, head-to-head trials with other therapies used for the treatment of CD or UC. Question for the clinical expert: Is conducting a head-to-head comparative trial against 1 of the numerous comparative treatments for CD and UC a reasonable expectation in the target population? What could be the rationale for conducting trials against placebo? | The clinical expert recommended conducting a head-to-head comparative trial against currently listed therapies and future therapies for future trials in the UC and CD setting. However, given that the LIBERTY-UC and LIBERTY-CD trials assessed the efficacy of a new mode of administration (i.e., SC) for infliximab that is already approved based on IV administration for use in the indicated populations, the use of a placebo group was considered appropriate. |
Questions for the clinical expert: 1. For what clinical reasons would infliximab IV be selected as therapy for UC or CD rather than the humanized versions of anti-TNF alpha drugs, adalimumab or golimumab? 2. When conventional therapies fail, are anti-TNF alpha drugs the preferred therapy to initiate, or are other biologics with different mechanisms of action being selected due to patient-specific factors? 3. Is there a significant unmet need that infliximab SC fills for the treatment of UC or CD? | 1. According to the clinical expert, infliximab SC will be selected as a treatment of choice according to the same rationale used when selecting any other anti-TNF alpha drug (i.e., the choice of treatment is complex and based on disease phenotype and patient preference; note that golimumab is currently not used to treat patients with CD). The clinical expert highlighted that some clinicians believe IV infusions provoke a more rapid response than SC options. The expert added that hospitalized patients with severe UC or CD are more likely to receive the IV formulation of infliximab. 2. The clinical expert noted that treatment choice in this setting is complex and depends on multiple factors, including patient preference. Anti-TNF alpha drugs are not automatically preferred. According to the expert, conventional therapy should not have to prove ineffective before advanced therapies are considered for patients with moderate to severe UC or CD. In LIBERTY-UC, a total of 432 patients (99.1%) had at least 1 prior medication (292 patients [98.6%] and 140 patients [100%] in the infliximab SC 120 mg and placebo SC groups, respectively); the most reported medications were corticosteroids for systemic use (338 patients [77.5%] in total). In the LIBERTY-CD trial, 325 patients (94.8%) had taken at least 1 prior medication (225 patients [94.5%] in the infliximab SC group and 100 patients [95.2%] in the placebo group). The most common prior medications reported were drugs for constipation (251 patients [73.2%]) in total. 3. According to the clinical expert, infliximab SC provides an SC option for patients already receiving infliximab IV in practice. SC administration of advanced therapies is often desirable for patients because it reduces the need for infusion clinic appointments (e.g., time away from work) and allows them a sense of independence. Many patients find SC administration more convenient. |
Initiation of therapy | |
The LIBERTY-UC and LIBERTY-CD trials assessed the superiority of infliximab SC over placebo in 438 and 343 patients with moderately to severely active UC (i.e., modified Mayo score of 5 to 9, endoscopy subscore ≥ 2) and CD (i.e., CDAI of 220 points to 450 points), respectively. Question for the clinical expert: Some jurisdictions use the HBI in their coverage criteria to determine disease severity. Are there any differences in how the HBI performs against the Mayo score or CDAI score? | The clinical expert noted that although CDAI scores are recommended by regulatory guidelines for the evaluation patients with Crohn disease, these are seldom used in clinical practice, due to the complexity of deriving these scores. According to the clinical expert, the HBI is an easier tool to complete in the clinical setting for patients with CD compared to the CDAI tool (used in clinical trials). Both the CDAI and HBI have limitations, due to the subjective nature of the information being gathered. The clinical expert did not highlight whether CDAI scores are comparable to HBI scores in clinical practice. The HBI tool uses different parameters to derive scores. The HBI is a subset of the CDAI (e.g., the HBI uses single-day readings and only 5 of the 8 CDAI variables, and it sums variables instead of applying weighted coefficients).10 The Mayo score is commonly used for UC in practice and is not comparable to the HBI, according to the expert. |
Infliximab SC is indicated for patients who have had inadequate response or intolerance to conventional therapy. Also, to be started on infliximab SC, patients must be initiated on IV infliximab. Questions for the clinical expert: 1. How many conventional therapies are typically tried before biologic drugs, JAK inhibitors, or S1PRMs are considered for therapy? 2. Is there a standard definition of an inadequate response to conventional (or biologic) therapy for UC and CD? 3. In your opinion, what percentage of patients would choose to switch from IV infliximab every 8 weeks to a biweekly injection of infliximab SC? | 1. The clinical expert highlighted that biologics are now considered as advanced therapies, including S1PRMs and JAK inhibitors. According to the expert, conventional therapy should not have to prove ineffective before advanced therapies are considered for patients with moderate to severe UC or CD. Corticosteroids are not indicated for maintenance of remission in populations with CD or UC. 2. According to the expert, markers to determine inadequate response to conventional therapy include inability to taper off the use of corticosteroids, lack of clinical remission, lack of endomucosal healing, and worsening of objective markers (e.g., fecal calprotectin). 3. According to the expert, it will be difficult to determine the percentage of patients who will switch from IV infliximab to SC treatment. According to the expert, many patients already on stable IV therapy may choose to remain on that treatment plan. However, the expert noted that SC injections often lead to more stable therapeutic drug levels and can be clinically advantageous for some patients. The expert felt that the choice to switch will be made based on a case-by-case approach and after thorough discussion between clinician and patient. |
There is variation in how public drug plans reimburse infliximab across Canada. Question for the clinical expert: If infliximab SC is recommended for reimbursement by CDEC, is it reasonable to use existing initiation criteria for infliximab IV in each jurisdiction? | According to the clinical expert, it would be reasonable to use the existing infliximab IV initiation criteria for infliximab SC in each jurisdiction, although the preference would be not to include the need for a patient to be intolerant of or have an inadequate response to conventional therapies (immunomodulators) as criteria for initiation. |
Continuation or renewal of therapy | |
Question for the clinical expert: If infliximab SC is recommended for reimbursement by CDEC, is it reasonable to use existing renewal criteria for infliximab IV in each jurisdiction? | The clinical expert expressed that it will be reasonable to use the existing renewal criteria for infliximab IV in each jurisdiction for infliximab SC. |
Discontinuation of therapy | |
LIBERTY-UC: Loss of response criteria defined as an increase in modified Mayo score of ≥ 2 points and ≥ 30% from the week 10 modified Mayo score, with actual value of ≥ 5 points and an endoscopic subscore of ≥ 2 points.
LIBERTY-CD: Loss of response criteria defined as an increase in CDAI of ≥ 100 points from the week 10 CDAI score, with a total score ≥ 220.
Question for the clinical expert: 1. Is the loss of response criteria used in the studies consistent with those used in clinical practice? 2. Is a loss of response to infliximab SC 120 mg or 240 mg inevitable for most patients, based on the pathophysiology of CD and UC? 3. In clinical practice, could infliximab SC doses be escalated above 240 mg if patients initially respond to a higher dose but then experience a loss of response? 4. Are the loss of response rates in the LIBERTY studies consistent with loss of response to infliximab IV in your clinical practice? | 1. According to the clinical expert, the definition of loss of response for patients with UC is consistent with clinical practice. The expert noted that the HBI tool is commonly used for patients with CD in clinical practice (whereas the trial used the CDAI). 2. According to the clinical expert, loss of response for infliximab 120 mg or 240 mg SC is not inevitable for patients with UC or CD. The expert noted that many patients will remain on their original advanced therapy for many years. The expert highlighted that they have patients currently in practice that have been on infliximab since starting the medication for their disease. The best chance of achieving remission is commonly observed with the first advanced therapy chosen. 3. The clinical expert noted that there is currently no data on the use of doses of infliximab SC above 240 mg in current practice. According to the expert, given the SC formulation, the likelihood of a patient benefiting from the treatment at a higher dose would be minimal except in specific cases (such as patients with severe perianal disease or other penetrating disease phenotypes). 4. The clinical expert noted that a proportion of patients with CD in clinical practice lose response to advanced therapy over time (10% to 20% in the first year of treatment is the standard expected loss of response, according to the expert). In the trials, 11.9% of patients with UC and 5.5% of patients with CD showed loss of response. |
Question for the clinical expert: If infliximab SC is recommended for reimbursement by CDEC, is it reasonable to use existing discontinuation criteria for infliximab IV in each jurisdiction? | The clinical expert expressed that it will be reasonable to use the existing discontinuation criteria for infliximab IV in each jurisdiction for infliximab SC. |
Prescribing of therapy | |
Question for the clinical expert: If CDEC recommends infliximab SC for reimbursement, is it reasonable to use existing prescribing criteria for infliximab IV in each jurisdiction? | The expert expressed that it will be reasonable to use the existing prescribing criteria for infliximab IV in each jurisdiction for infliximab SC, although they would prefer not to include the need for a patient to have intolerance or an inadequate response to conventional therapies (immunomodulators) as criteria for prescribing. |
Generalizability | |
The LIBERTY studies did not evaluate patients < 18 years of age and did not enrol many patients > 65 years of age. Question for the clinical expert: Is there any desire to use infliximab SC in patients who are outside the age range of 18 years to 65 years, or are there adequate treatment options for these patients? | The expert noted that there may be a need for access to infliximab SC for patients aged less than 18 years. The expert added that anti-TNF alpha drugs are currently used in patients older than 65 years. |
Health system and economic issues | |
Costs of IV infusions are paid by public drug plans (not sponsors), given that these services are intentionally negotiated as part of the total reimbursed price. Question for the clinical expert: Because infliximab SC maintenance therapy does not require IV infusion services, should its reimbursed price be lower than infliximab IV? Would the lowest-priced SC biologic be a reasonable price target? | The clinical expert highlighted that all patients in the trials received IV induction therapy with infliximab, which differed from other currently approved advanced SC therapies. |
CD = Crohn disease; CDAI = Crohn Disease Activity Index; CDEC = Canadian Drug Expert Committee; HBI = Harvey-Bradshaw Index; JAK = Janus kinase; S1PRM = Sphingosine-1-phosphate receptor modulators; SC = subcutaneous; TNF = tumour necrosis factor; UC = ulcerative colitis.
Sponsor’s Summary of the Clinical Evidence
Note that the clinical evidence summarized in this section was prepared by the sponsor in accordance with the CADTH tailored review process and has not been modified by CADTH.
Pivotal Studies
Table 8: Details of Included Studies
Characteristics | CT-P13 3.7 / LIBERTY-UC | CT-P13 3.8 / LIBERTY-CD | CT-P13 1.6 Part 2 |
|---|---|---|---|
Designs and populations | |||
Study design | OL induction, DB maintenance, phase III, RCT | OL induction, DB maintenance, phase III, RCT | OL, phase I, RCT |
Locations | 92 study centers in 14 countries enrolled | 114 study centers in 26 countries enrolled | 50 study centers in 15 countries enrolled |
Patient enrolment dates | 01 September 2020 to 07 July 2022 | 28 October 2019 to 23 August 2022 | 07 May 2018 to 02 October 2019 |
Randomized (N) | 438 | 343 | 131 |
Inclusion criteria |
|
|
|
Exclusion criteria |
|
|
|
Drugs | |||
Intervention | Induction phase: Infliximab IV 5 mg/kg at Weeks 0, 2, and 6 Maintenance phase (among responders): Infliximab SC 120 mg Q2W or 240 mg Q2W (if lost response) via PFS Extension phase: Infliximab SC 120 or 240 mg Q2W via PFS | Induction phase: Infliximab IV 5 mg/kg at Weeks 0, 2, and 6 Maintenance phase (among responders): Infliximab SC 120 mg Q2W or 240 mg Q2W (if lost response) via PFS Extension phase: Infliximab SC 120 or 240 mg Q2W via PFS | Dose-loading phase: Infliximab IV 5 mg/kg at Weeks 0, and 2 Maintenance phase (among completers) SC arm: Infliximab SC 120mg/240mg based on body weight at Week 6, thereafter Q2W via PFS |
Comparator(s) | Maintenance phase (among responders): Placebo SC (matching volume to infliximab SC 120 mg) via PFS | Maintenance phase (among responders): Placebo SC (matching volume to infliximab SC 120 mg) via PFS | Maintenance phase (among completers):
|
Duration | |||
Phase | |||
Screening | Up to 6 weeks | Up to 6 weeks | Up to 6 weeks |
Induction/dose-loading phase (OL) | 6 weeks (Weeks 0, 2, and 6) | 6 weeks (Weeks 0, 2, and 6) | 6 weeks (Week 0 to 6) |
Maintenance phase (DB) | 44 weeks (Week 10 through 54) | 44 weeks (Week 10 through 54) | 48 weeks (Week 6 to 54) |
Extension phase | 46 weeks (Week 56 through Week 102) | 46 weeks (Week 56 through Week 102) | N/A |
Follow-up/End of study | 4 weeks | 4 weeks | 2 weeks |
Outcomes | |||
Primary efficacy end pointc | Clinical remission |
| PK end points assessed as primary end points, no efficacy end points. |
Secondary and exploratory efficacy end pointsc | Key secondary end points:
Other secondary end points:
Exploratory Efficacy End points:
| Key secondary end points:
Other secondary end points:
| Secondary efficacy end points:
|
Notes | |||
Publications | Draft publications are in peer-review. Abstract for key results: Sands B E et al., Subcutaneous infliximab (CT-P13 SC) as maintenance therapy for ulcerative colitis: A Phase 3, randomized, placebo-controlled study: Results of the LIBERTY-UC study. Journal of Crohn and Colitis. 2023;17 (Supplement_1):i623-i624. NCT04205643 | Draft publications are in peer-review. Abstract for key results: Colombel J F et al., Subcutaneous infliximab (CT-P13 SC) as maintenance therapy for ulcerative colitis: A Phase 3, randomized, placebo-controlled study (LIBERTY-CD). Journal of Crohn and Colitis. 2023;17 (Supplement_1):i161-i162 NCT03945019 | Schreiber S. et al. Randomized Controlled Trial: Subcutaneous vs IV Infliximab CT-P13 Maintenance in Inflammatory Bowel Disease. Gastroenterology. 2021 Jun;160(7):2340 to 2353. NCT02883452 |
CD = Crohn Disease; CDAI = Crohn Disease Activity Index; DB = double blind; IV = IV; JAK = Janus kinase; OL = open label; PK = pharmacokinetic; PFS = pre-filled syringe; Q2W = every 2 weeks; RCT = randomized controlled trial; SIBDQ = Short Inflammatory Bowel Disease Questionnaire; TB = tuberculosis; TNFAlpha = Tumor necrosis factor-alpha; UC = ulcerative colitis; VAS = visual analogue scale.
aDefinition of moderate to severely active UC: Modified Mayo score without PGA subscore of 5 to 9 points with endoscopic subscore of ≥ 2 points and had an inadequate response to conventional therapy (Study 3.7) or total Mayo score between 6 and 12 points with endoscopic subscore of ≥ 2 (Study 1.6)
bDefinition of moderate to severely active CD: CDAI of 220 to 450 points with an inadequate response to conventional therapies (Study 3.8 and 1.6).
cDefinition of outcomes provided in the outcome section of this document.
Source: CSRs of LIBERTY-UC, LIBERTY-CD, and Study 1.6.a-c
Description of Studies
Overview
A total of 3 trials supported the clinical efficacy and safety of REMSIMATM SC in patients with inflammatory bowel disease (IBD). LIBERTY-UC (CT-P13 3.7) and LIBERTY-CD (CT-P13 3.8) were 2 identically designed double-blind (DB), parallel-group, placebo controlled, phase III, randomized controlled trials (RCTs) that were designed to assess the superiority of REMSIMATM SC over placebo in patients with moderately to severely active ulcerative colitis (UC) and Crohn disease (CD), respectively, and had an inadequate response to conventional therapy.
Another open label (OL), parallel-group, phase I, RCT, Study CT-P13 1.6 (referred to as Study 1.6 hereafter), compared the pharmacokinetics (PK), efficacy, and safety of IV (IV) infliximab with subcutaneous (SC) infliximab in patients with active UC and CD. The study had 2 parts, part 1 was a PK study designed to find the optimal dose of REMSIMATM SC conducted in patients with active CD and will not be focused on here. Part 2 of the study evaluated both PK and clinical end points, however, this report will be limited to the clinical outcomes only.
All 3 trials comprised of 3 study periods, including screening (up to 6 weeks), treatment phase (54 weeks), and end-of-study (EOS) visit (4 weeks for LIBERTY-UC and LIBERTY-CD, 2 weeks for Study 1.6). Blocked randomization was done using an interactive web response system (IWRS) in all trials, stratified by the following factors:
LIBERTY-UC and CD: Previous exposure to biologic agents and/or JAK inhibitors (used/not used), use of treatment with oral corticosteroids at Week 0 (used/not used), and clinical remission at Week 10 (remitter/non-remitter by modified Mayo score or CDAI score).
Study 1.6: Current use of treatment with azathioprine (AZA) or 6-mercaptourine (6-MP) or methotrexate (MTX) (used or not used), clinical response at Week 6 (responder or non-responder by CDAI-70 for CD or partial Mayo score for UC), body weight at Week 6 (< 80 kg or ≥ 80 kg), and disease (CD or UC).
LIBERTY-UC and CD
The treatment phase of the phase III trials LIBERTY-UC and LIBERTY-CD consisted of a 6-week induction phase during which 548 and 396 patients received OL induction doses of infliximab 5 mg/kg via IV infusion, respectively. The patients who had no safety concerns and were considered clinical responders before Week 10 (438 and 343 in LIBERTY-UC and CD, respectively), defined by a decrease in modified Mayo score from baseline of ≥ 2 points and ≥ 30%, with an accompanying decrease in the rectal bleeding subscore of ≥ 1 point or an absolute rectal bleeding subscore of 0 or 1 point (in LIBERTY-UC) or Crohn disease activity index (CDAI)-100 score (in LIBERTY-CD) were randomized in a 2:1 ratio to receive REMSIMATM SC (CT-P13 SC) or volume matched SC placebo. Patients who completed maintenance treatment up to Week 54 in both trials received OL REMSIMATM SC up to Week 102; however, this extension phase is ongoing, and data will not be presented in this submission. Figure 1 (A) and (B) below provides a schematic diagram of LIBERTY-UC and LIBERTY-CD, respectively.
Study 1.6 Part 2
The phase I Study 1.6 Part 2 had a treatment period, divided into 2 phases: a dose-loading phase from Week 0 to 6, followed by a maintenance phase from Week 6 to 54. During the dose-loading phase, 136 enrolled patients initially received infliximab IV infusion at Weeks 0 and 2. Of the 131 patients who received 2 full doses and had no safety concerns were randomized 1:1 to receive either REMSIMATM SC or infliximab IV starting at Week 6. Patients in the infliximab IV arm received IV infliximab up to week 22, then got switched to receive REMSIMATM SC starting at Week 30 and continued up to Week 54. Patients in the REMSIMATM SC arm started receiving the SC formulation at Week 6 and continued up to Week 54. A schematic diagram of the trial is provided in Figure 2 below.
Figure 1: Schematic Diagram of (A) LIBERTY-UC and (B) LIBERTY-CD
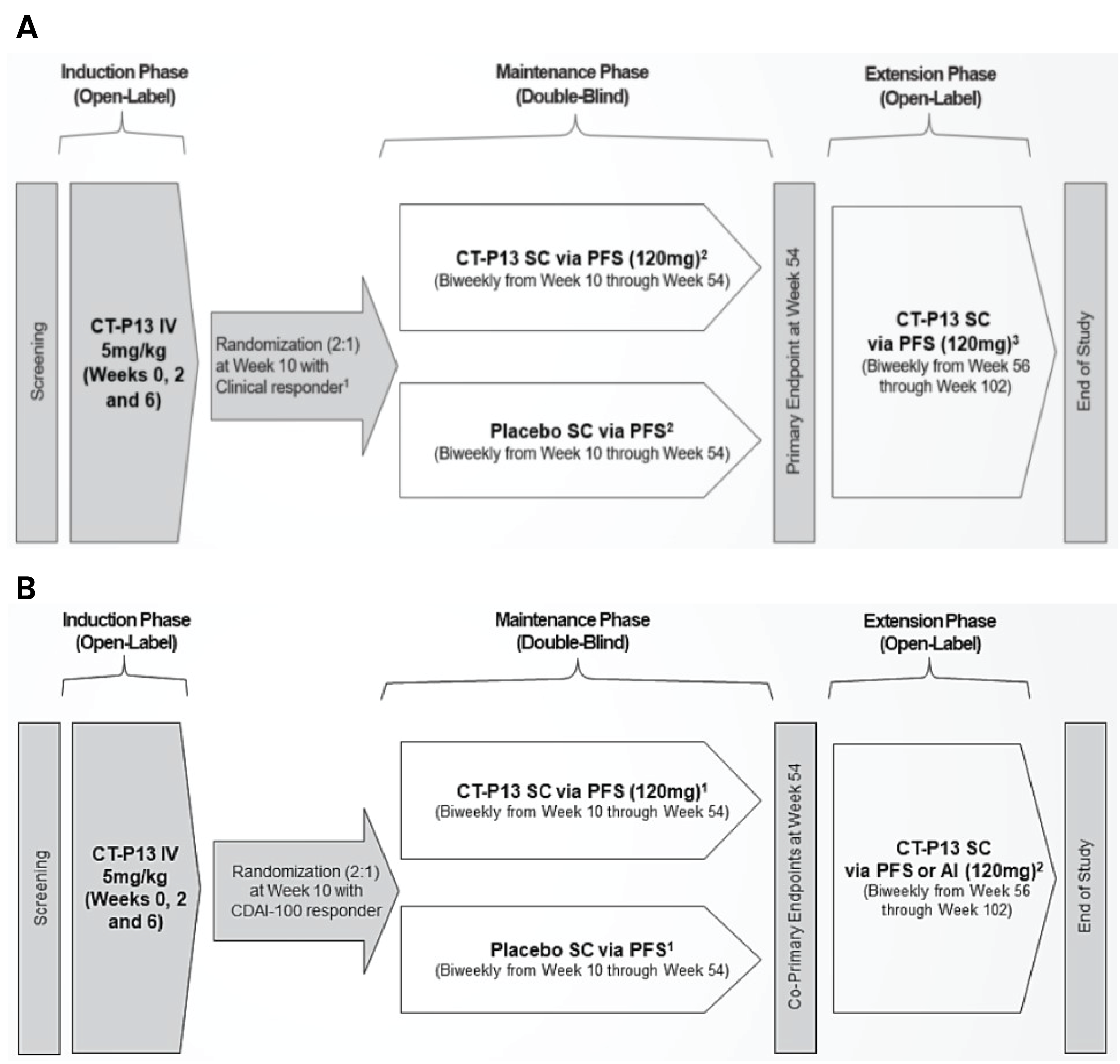
AI = auto injector; CDAI = Crohn Disease Activity Index; CT-P13 = Infliximab; IV = IV; PFS = prefilled syringe; SC = subcutaneous.
1 (A) LIBERTY-UC: Clinical response by modified Mayo score: a decrease in modified Mayo score from baseline of at least 2 points and at least 30%, with an accompanying decrease in the rectal bleeding subscore of at least 1 point or an absolute rectal bleeding subscore of 0 or 1 point.
(B) LIBERTY-CD: From week 22 through week 54, dose adjustment was allowed. The patients who received REMSIMATM SC 120 mg could increase the dose to CT-P13 SC 240 mg every 2 weeks, and the patients who received Placebo SC could receive REMSIMATM SC 240 mg every 2 weeks, if they initially responded but then lost response according to the loss of response criteria.
2 (A) LIBERTY-UC: From week 22 through week 54, dose adjustment was allowed. The patients who received REMSIMATM SC 120 mg could increase the dose to CT-P13 SC 240 mg every 2 weeks, and the patients who received Placebo SC could receive REMSIMATM 240 mg every 2 weeks, if they initially responded but then lost response according to the loss of response criteria.
(B) LIBERTY-CD: In the open-label extension phase, all patients who completed the maintenance phase up to Week 54 and may benefit from continued treatment, in the opinion of the investigator will receive active treatment with REMSIMATM SC 120 mg via PFS or AI from week 56.
3 LIBERTY-UC: In the extension phase, all patients who completed the maintenance phase up to Week 54 and could benefit from continued treatment, in the opinion of the investigator, received active treatment with REMSIMATM SC 120 mg via PFS from Week 56.
Source: CSRs of LIBERTY-UC, and LIBERTY-CD.a-b
Figure 2: Schematic Diagram of Study 1.6 Part 2
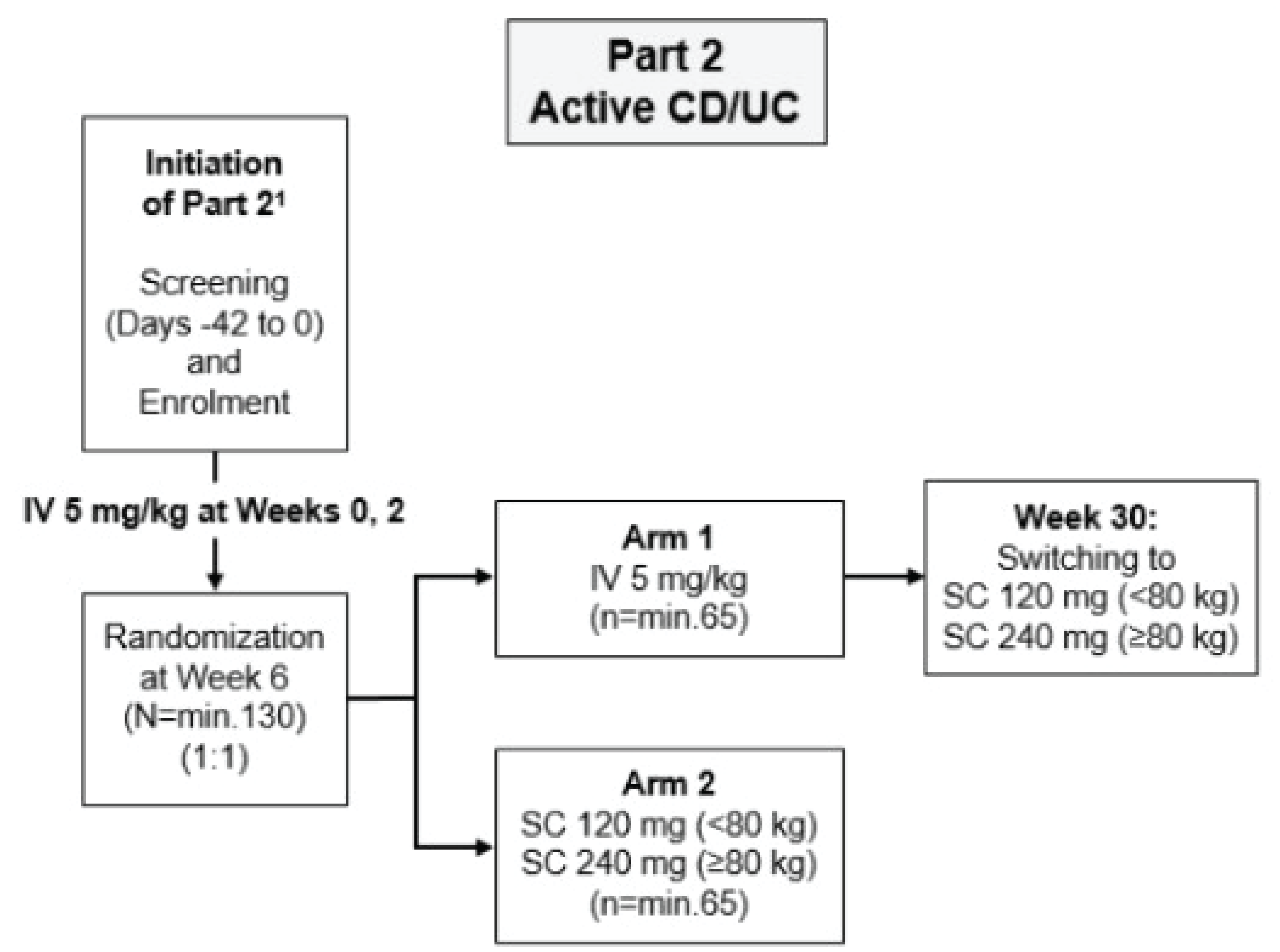
AI = auto injector; CDAI = Crohn Disease Activity Index; CT-P13 = Infliximab; PFS = prefilled syringe; SC = subcutaneous
Source: CSR of Study 1.6.c
Populations
Inclusion and Exclusion Criteria
Inclusion criteria
All 3 trials included male and female patients aged 18 to 75 years (inclusive), and the LIBERTY-UC, LIBERTY-CD, and Study 1.6 Part 2 trials included patients with moderately to severely active UC, CD, and a combination of active CD or UC patients. The definition of moderate to severe UC and CD are in line with clinical trials of UC and CD patients, outlined below. The main inclusion criteria of the trials, all assessed during screening, are provided below.
In LIBERTY-UC, moderately to severely active UC was defined as patients who had a modified Mayo score of 5 to 9 points with endoscopic subscore of ≥ 2 points.
In LIBERTY-CD, moderately to severely active CD was defined as a CDAI score of 220 to 450 points. In addition, the following criteria had to be met:
7-day average daily stool frequency (SF) ≥ 4 points (of type 6 or type 7 on the Bristol stool form scale [BSFS]) and/or a 7-day average worst daily abdominal pain (AP) of ≥ 2 points (using 4-point scale).
Simplified endoscopic activity score for Crohn disease (SES-CD) of ≥ 6 points for ileal-colonic CD or ≥ 4 points including ulcer score from at least 1 segment for ileal CD or colonic CD.
In Study 1.6, patient with active UC had to have a total Mayo score between 6 and 12 points with endoscopic evidence of active colitis as indicated by endoscopic subscore of ≥ 2, and those with active CD had to have a CDAI score of 220 to 450 points in addition to the following:
C-reactive protein (CRP) concentration > 0.5 mg/dL.
Fecal calprotectin (FC) > 100 mcg/g.
SES-CD of ≥ 6 points for ileal-colonic CD or ≥ 4 points including ulcer score from at least 1 segment for ileal CD or colonic CD.
In all trials, diagnosis of UC or CD was confirmed by endoscopic, radiographic, or histological criteria.
All 3 trials included patients who had an inadequate response to conventional therapy or who were intolerant to or had medical contraindications to such therapies. Conventional therapies included corticosteroids alone or in combination with 6-MP or AZA in LIBERTY-UC and Study 1.6 (UC patients); corticosteroids and/or immunosuppressants in LIBERTY-CD and Study 1.6 (CD patients).
Patients receiving stable doses of the following treatments were allowed at the time of the study: AZA, 6-MP, MTX, corticosteroids, budesonide, 5-aminosalicylates (5-ASA), and antibiotics (only in LIBERTY-UC and CD).
Patients with no abnormal clinical hematology, adequate renal and hepatic function based on conventional biomarkers.
Exclusion criteria
A number of general and tuberculosis (TB)-specific exclusion criteria were applied in all trials, and disease-specific criteria were applied in Study 1.6. The main ones are listed below.
Prohibited medications:
Previously received infliximab, or 2 or more biologic agents, 2 or more Janus Kinase (JAK) inhibitors, or 2 or more both biologic agents and JAK inhibitors in LIBERTY-UC and LIBERTY-CD, or any biological agent in Study 1.6.
TNFAlpha inhibitor within 5 half-lives before study treatment in LIBERTY-UC and LIBERTY-CD, and any time in Study 1.6.
Different combinations of the following: parenteral corticosteroids, 5-ASA, antibiotics, alkylating agents, thalidomide, tacrolimus, sirolimus, mycophenolate mofetil, cyclosporine, stem cell therapy, abdominal surgery, apheresis, or colectomy within a pre-specified period before study entry.
Current or previous medical conditions:
Major infections such as hepatitis B or C, HIV (HIV), acute infection requiring antibiotics, recurrent herpes zoster or other chronic or recurrent infections, granulomatous infections or opportunistic infections, cytomegalovirus, or other serious infections.
One or more of the following: UC limited to only the rectum or to < 15 cm of the colon, indeterminate colitis, toxic megacolon, colonic mucosal dysplasia or adenomatous polyps, require surgical intervention, stoma, > 3 small bowel resection procedures, short bowel syndrome, symptomatic stenosis or obstruction of large intestine, BMI ≥ 35 kg/m2, uncontrolled diabetes mellitus, major cardiovascular condition, known malignancy, or any significant medical conditions interfering with the study.
TB exclusion criteria:
Active TB or history of active TB, exposure to persons with active TB, latent TB or previous latent TB.
CD or UC-specific exclusion criteria (Study 1.6 only):
CD: Active entero-vesical, entero-retroperitoneal, entero-cutaneous, and entero-vaginal fistulae, > 3 small-bowel resection procedures within pre-specified periods.
UC: Rectally administered medications containing corticosteroids or 5-ASA pre-specified periods.
Baseline Characteristics
Overall, baseline characteristics were similar between treatment groups across the 3 trials. Details are provided for randomized patients in Error! Reference source not found. since all patients enrolled for the induction/dose-loading phase is not the focus of this review. The mean age across trial ranged between 30 and 40 years, slightly higher proportion of males, and the majority of patients were Caucasian (white). Patients across the trials had their disease for an average of 5 to 6 years. Nearly 10% patients in the LIBERTY-UC and LIBERTY-CD trial used biologics or JAK inhibitors, and nearly 40% used corticosteroids at the start of the trial. The stratification factors were well-balanced across studies. Most common classes of prior and concomitant medications received were generally balanced between treatment groups but varied across trials, and included corticosteroids, constipation drugs, antidiarrheals, intestinal anti-inflammatory/anti-infective agents, and immunosuppressants. Most common type of prior medical condition included gastrointestinal (GI), and surgical and medical procedures in LIBERTY-UC and LIBERTY-CD, and GI, and infections and infestations in Study 1.6.
Table 9: Summary of Baseline Characteristics – All Randomized Population
Characteristics | LIBERTY-UC | LIBERTY-CD | Study 1.6 | |||
|---|---|---|---|---|---|---|
REMSIMATM SC N = 294 | Placebo N = 144 | REMSIMATM SC N = 231 | Placebo N = 112 | REMSIMATM SC N = 66 | Infliximab IV N = 65 | |
Demographic characteristics | ||||||
Age, mean (SD) | 38.2 (12.8) | 40.4 (13.5) | 36.0 (12.5) | 32.3 (11.5) | 37.8 (15.1) | 39.4 (14.0) |
Sex, male, n (%) | 163 (55.4) | 83 (57.6) | 134 (58.0) | 69 (61.6) | 36 (54.5) | 35 (53.8) |
Race, White, n (%) | 288 (98.0) | 140 (97.2) | 211 (91.3) | 101 (90.2) | 62 (93.9) | 60 (92.3) |
Screening BMI, Kg/m2, mean (SD) | 24.16 (4.4) | 25.09(4.2) | 23.27 (4.4) | 22.55 (4.4) | 23.71 (4.1) | 24.18 (4.7) |
Most common class of prior medication, n (%) | ||||||
Corticosteroids | 230 (77.7) | 108 (77.1) | — | — | 35 (53.0) | 34 (52.3) |
Constipation drugs | — | — | 173 (72.7) | 78 (74.3) | — | — |
Antidiarrheals, intestinal anti-inflammatory/ anti-infective agents | — | — | 129 (54.2) | 57 (54.3) | 37 (56.1) | 32 (49.2) |
Disease characteristics | ||||||
Disease, n (%) | 294 (100) | 144 (100) | 231 (100) | 112 (100) | CD: 28 (42.4) | CD: 25 (38.5) |
UC: 38 (57.6) | UC: 40 (61.5) | |||||
Time since diagnosis, Year, mean (SD) | 6.09 (6.0) | 6.80 (6.8) | 4.34 (5.2) | 4.45 (5.8) | 5.7 (6.0) | 5.8 (6.3) |
CD: 4.47 (6.5) | CD: 5.63 (5.6) | |||||
UC: 6.60 (5.5) | UC: 5.98 (6.7) | |||||
Most common class of prior medical conditions, n (%) | ||||||
GI disorders | 146 (49.7%) | 76 (52.8%) | 108 (46.8) | 49 (43.8) | CD: 20 (71.4) UC: 10 (26.3) | CD: 11 (44.0) UC: 12 (30.0) |
Surgical and medical procedures | 60 (20.4%) | 28 (19.4%) | 69 (29.9) | 36 (32.1) | — | — |
Infections and infestations | — | — | — | — | 7 (25.0) | 2 (8.0) |
GI = gastrointestinal; NR = not reported; SC = subcutaneous; SD = standard deviation.
Note: Most common class of prior and concomitant medications, and prior medical conditions are reported as per CSR, not all prior and concomitant medications, and previous medical conditions are listed in this table.
Source: CSRs of LIBERTY-UC, LIBERTY-CD, and Study 1.6.a-c
Interventions
Dosing
LIBERTY-UC and LIBERTY-CD: The 54-week treatment period was divided into 2 phases:
OL induction phase (Weeks 0, 2, and 6): In both trials, all patients received induction doses of 5 mg/kg infliximab IV infusion.
DB maintenance phase (Weeks 10 through 54): Starting at Week 10, patients who were clinical responders received a single injection of 120 mg REMSIMATM SC or volume-matched placebo Q2W in 2:1 ratio via PFS. Patients received the SC injection by study personnel up to Week 12, or until they were properly trained to administer the PFS injection themselves. Dose-adjustment to 240 mg (2 injections of 120 mg) were allowed from Week 22 if patients initially responded but later lost response (defined as an increase in modified Mayo score ≥ 2 points and ≥ 30% from the Week 10 modified Mayo score of ≥ 5 points, and endoscopic subscore of ≥ 2 points in LIBERTY-UC and an increase in CDAI of ≥ 100 points from the Week 10 CDAI score with a total score ≥ 220 in LIBERTY-CD). However, those who had their dose adjusted were considered non-remitter or non-responder for outcome assessment.
It should be noted that patients had the option to self-inject REMSIMATM SC using an auto injector (AI) in the extension phase of LIBERTY-CD; however, this will not be highlighted given this review is limited to the maintenance phase.
Study 1.6: The 54-week treatment period was divided into 2 phases:
OL dose-loading phase (Weeks 0 and 2): All patients received 5 mg/kg infliximab IV infusion.
OL maintenance phase (Weeks 6 through 54): Eligible patients switched to SC formulation of infliximab starting at Week 6, or continued IV treatment up to Week 22, then switched to SC formulation from Week 30 to Week 54. Patients received the SC injection by study personnel, friends or family members, or self-injected the PFS injection if they were adequately trained.
SC arm: Patients received 120 or 240 mg REMSIMATM SC based on body weight (< 80 kg and ≥ 80 kg, respectively) via PFS, starting at Week 6, thereafter Q2W up to Week 54. Dose escalation was only allowed for patients receiving 120 mg REMSIMATM SC, if they initially responded but lost response at or after Week 30. Loss of response was defined as need of the initiation of a new treatment for active CD or UC, or the following: increase in CDAI ≥ 70 points from the lowest CDAI score with a total score ≥ 220 (for CD patients), or increase in rectal bleeding subscore ≥ 1 point from the lowest score with actual value of > 1 point and either increase in partial Mayo score ≥ 2 points from the lowest score with actual value of ≥ 4 points or increase in endoscopic subscore ≥ 1 point from the lowest score with actual value of > 1 point (UC patients).
IV arm: Patients received further 3 doses of 5 mg/kg infliximab IV at Weeks 6, 14, and 22; then switched to receive 120 or 240 mg REMSIMATM SC based on body weight (< 80 kg and ≥ 80 kg, respectively), given Q2W up to Week 54. Dose escalation was only allowed for patients receiving 120 mg REMSIMATM SC, if they initially responded but lost response at or after Week 30. Loss of response was defined as need of the initiation of a new treatment for active CD or UC, or the following: increase in CDAI ≥ 70 points from the lowest CDAI score with a total score ≥ 220 (for CD patients), or increase in rectal bleeding subscore ≥ 1 point from the lowest score with actual value of > 1 point and either increase in partial Mayo score ≥ 2 points from the lowest score with actual value of ≥ 4 points or increase in endoscopic subscore ≥ 1 point from the lowest score with actual value of > 1 point (UC patients).
Concomitant medications (stable doses received for a pre-specified period before study)
Concomitant medications: Stable doses of immunomodulators (such as AZA, 6-MP, or MTX) before study.
Oral corticosteroids < 20 mg/day of prednisone before study, kept at the same dose up to Week 10, then gradually tapered by 2.5 to 5 mg/week.
Oral budesonide < 6 mg/day or < 9 mg/day of prednisone before study, kept at the same dose up to Week 10, then gradually tapered by 3 mg/2 weeks.
5-ASA if stable doses maintained for at least 4 weeks before study.
Antibiotics (such as ciprofloxacin, metronidazole) if stable doses maintained before study.
Prohibited medications (unless specified, applies to all 3 trials)
Any other investigational device or medical product.
Any biological agents or JAK inhibitors for the treatment of UC or CD.
Anakinra, Abatacept, or Tocilizumab (LIBERTY-UC and LIBERTY-CD only).
Cyclosporine, tacrolimus, sirolimus, or mycophenolate mofetil (LIBERTY-UC and LIBERTY-CD only).
Thalidomide, tacrolimus, or cyclosporine (Study 1.6 only).
Parenteral corticosteroids for the treatment of UC or CD.
Rectally administered medications containing corticosteroids or 5-ASA for the treatment of UC.
Any TNFAlpha inhibitor, except for study drug.
Alkylating agents.
Live or live-attenuated vaccine.
Abdominal surgery, including but not limited to, for active gastrointestinal bleeding, peritonitis, intestinal obstruction, gastrointestinal resection, or intra-abdominal or pancreatic abscess requiring surgical drainage.
Nonautologous stem cell therapy (e.g., Prochymal) (LIBERTY-UC and LIBERTY-CD only).
Apheresis (e.g., Adacolumn apheresis) for the treatment of UC.
Use of exclusive enteral or parenteral nutrition.
Antibiotics for the treatment of CD or UC (Study 1.6 only).
Outcomes
The efficacy end points measured in the trials were well-established outcomes assessed commonly in UC, CD, or IBD trials. Below is a description of the clinical measures used in the trials.
Mayo score: Among UC patients in LIBERTY-UC and Study 1.6, the clinical outcomes were based on Mayo score, a disease-specific score that assesses the severity of UC and includes the following components: rectal bleeding, stool frequency, physician global assessment (PGA), and endoscopy findings. The total Mayo score includes all the above components, with each part rated from 0 to 3, yielding a total score of 0 to 12. A score of 3 to 5 points indicates mildly active disease, while a score of 6 to 10 points indicates moderately active disease, and a score of 11 to 12 points indicates severe disease. The full Mayo score was shown to have construct validity, based on strong correlation with patient assessment of disease activity.d Moreover, the Mayo score was found to correlate with patient assessment of change in UC activity, and with improvement in quality of life measures, indicating responsiveness to change.d,e The total Mayo score also showed evidence of reliability, based on high inter-rater agreement.d,f Two abridged versions of the Mayo score have been developed and validated: the modified Mayo score and partial Mayo score that are composed of the 3 components of the total Mayo score excluding PGA subscore and endoscopic subscore, respectively. A strong correlation was found between the partial and total Mayo scores,g with construct validity demonstrated for the partial Mayo score.d Moreover, the FDA guidance (Guidance for Industry. Ulcerative Colitis [2022]), recommended using clinical remission by modified mayo score as the primary end point.h The minimum important difference (MID) for clinical response was reported as a change of 2.5 in total Mayo score from baseline.d The cutpoint for clinical remission varied, ranging from a Mayo score change of 0.6 to 4.5,d,i,j although most commonly a reduction of the baseline total Mayo score of either 2 or 3 points is used.i The FDA defines clinical remission as a Mayo score of 2 or less with no individual subscore greater than 1 (stool frequency subscore of 0 or 1, endoscopy subscore of 0 or 1, and rectal bleeding subscore of 0).k Similarly, clinical response is defined by the FDA as a reduction in the total Mayo score of ≥ 30% or more from baseline with a decrease in rectal bleeding subscore greater ≥ 1 point or absolute rectal bleeding subscore of ≤ 1.k The definitions used in the 3 trials were similar to or in line with the FDA definition, as noted below.
Mucosal improvement or healing: Mucosal improvement or healing was assessed by endoscopic subscore of the Mayo score (both LIBERTY-UC and Study 1.6) and histologic assessment by the Robarts Histopathology Index (RHI), evaluated centrally by an independent reviewer blinded to treatment allocation (LIBERTY-UC only). The construct validity of the endoscopic subscore of the Mayo score was demonstrated by strong correlation with 2 histologic indices, Riley index score and Rubin histologic index score.71,72 g,l The endoscopic subscore also showed a moderate-to-substantial agreement in the inter-rater reliability estimates and a substantial agreement in the intra-rater reliability estimates.m
CDAI score: Among CD patients in LIBERTY-CD and Study 1.6, clinical efficacy was assessed by the evaluation of CDAI score, Simplified Endoscopic Activity Score for Crohn Disease (SES-CD), as well as several other clinical measures based on subscores of CDAI. CDAI is a disease-specific index used to assess the severity of CD, and rated by patients in the previous 7 days. The CDAI consists of 8 items, each of which is independently weighted and weighted differently: stool frequency, abdominal pain, general well-being, sum of 6 findings, antidiarrheal use, hematocrit, and body weight. The overall CDAI score is based on the sum of the weighted value of each item and ranges from 0 to 600, where a score of 150 is defined as the threshold between remission and active disease. Scores ranging between 150 and 219 indicate mild to moderate CD and scores ranging between 220 and 450 indicate moderate to severe CD, whereas scores above 450 indicate very severe CD.n,o The items included in the CDAI were selected by gastroenterologists and are based on accepted features of CD, indicating construct validity.o In addition, evidence of criterion validityo and good to very good test-retest reliability was reported.p While no information regarding MID of CDAI was identified, the FDA and EMA have recently suggested that a change of 100 points in CDAI is considered to be a meaningful response.n In addition to the full CDAI score, the loose (very soft)/watery (liquid) SF and worst daily AP score of the CDAI were measured and reported. While no information regarding the validity, reliability, and MID were found for these subscores, these 2 subscores showed moderate responsiveness.q
Endoscopic response and remission: Endoscopic response or remission was assessed by evaluation of mucosal abnormalities by colonoscopy, reported using the SES-CD score that was evaluated centrally. The SES-CD was designed for the assessment of 4 endoscopic items, including size of ulcers, ulcerated surface, affected surface, and presence of narrowing. Each item is to be scored 0 to 3 with a total score ranging from 0 to 56. Higher scores indicate more severe disease, although no information regarding the MID was identified.r SES-CD was demonstrated to have construct validitys and high inter-rater reliability.t
Short Inflammatory Bowel Disease Questionnaire (SIBDQ): In addition to clinical outcomes, health-related quality of life (HRQoL) was assessed by the SIBDQ in all 3 trials. The SIBDQ is a quality-of-life questionnaire for patients with inflammatory bowel disease that is based on IBDQ, a validated, reliable, and commonly used HRQoL tool in IBD studies. It has 10 questions measuring physical, social, and emotional status. Scores for this questionnaire range from 1 (poorest quality of life) to 7 (best quality of life). A 9-point change in the SIBDQ is considered a minimal important difference (MID) in IBD patients.u
All efficacy results up to Week 10 (LIBERTY-UC and LIBERTY-CD) and Week 6 (Study 1.6) represented the efficacy of the infliximab IV loading dose/induction dose regardless of the randomized treatment group in the maintenance phase. The definition of outcomes measured for patients with UC in the LIBERTY-UC and Study 1.6 trials were largely similar, so were the definition of outcomes measured for patients with CD in the LIBERTY-CD and Study 1.6 trials. Below is a brief description of the outcomes measured in the 3 trials, by type of patients. Unless otherwise specified, all end points were measured at Week 54.
Primary efficacy end points
The primary end point in Study 1.6 were PK end points, which are not listed here.
UC patients in LIBERTY-UC
Clinical remission: Modified Mayo score (stool frequency subscore of 0 or 1 point; rectal bleeding subscore of 0 point; and endoscopic subscore of 0 or 1 point).
CD patients in LIBERTY-CD
Clinical remission (based on CDAI): Absolute CDAI score of < 150 points.
Endoscopic response in LIBERTY-CD: 50% decrease in SES-CD score from baseline.
Secondary efficacy end points
UC patients in LIBERTY-UC and Study 1.6
Clinical response in LIBERTY-UC: Decrease from baseline in modified Mayo score of ≥ 2 points and at least 30%, with accompanying decrease in rectal bleeding subscore of ≥ 1 point or absolute rectal bleeding subscore of 0 or 1 point. Clinical response was assessed at Weeks 10 and 22 in addition to Week 54; however, this data will not be reported here given he focus is on maintenance treatment at Week 54.
Clinical response in Study 1.6
Based on total Mayo score: Decrease from baseline in total Mayo score of ≥ 3 points, with accompanying decrease in rectal bleeding subscore of ≥ 1 point or absolute rectal bleeding subscore of 0 or 1 point.
Based on partial Mayo score: Decrease from baseline in modified Mayo score of ≥ 2 points, with accompanying decrease in rectal bleeding subscore of ≥ 1 point or absolute rectal bleeding subscore of 0 or 1 point. Data for partial Mayo score will not be reported here, unless the results were different compared to total Mayo score.
Clinical remission in LIBERTY-UC and Study 1.6: Stool frequency subscore of 0 or 1 point; rectal bleeding subscore of 0 point; and endoscopic subscore of 0 or 1 point (LIBERTY-UC) or total Mayo score of ≤ 2 points with no individual subscore exceeding 1 point, or partial Mayo score of ≤ 1 point (Study 1.6). Data for partial Mayo score will not be reported here, unless the results were different compared to total Mayo score.
Maintenance of clinical remission in LIBERTY-UC: Maintenance of clinical remission was defined as being in clinical remission by modified Mayo score, among the patients in clinical remission by modified Mayo score at Week 10.
Sustained remission at both week 22 and 54 in LIBERTY-UC: Stool frequency subscore of 0 or 1 point, and rectal bleeding subscore of 0 point at both Week 22 and Week 54.
Endoscopic-histologic mucosal improvement in LIBERTY-UC: Absolute endoscopic subscore of 0 or 1 point from modified Mayo score and absolute RHI score of ≤ 3 points with an accompanying lamina propria neutrophils and neutrophils in epithelium subscore of 0 point.
Corticosteroid-free remission in LIBERTY-UC: Being in clinical remission by modified Mayo score (in UC) in addition to not requiring any treatment with corticosteroid for ≥ 8 weeks at Week 54, among patients who used oral corticosteroids at baseline.
Mucosal healing in Study 1.6: Absolute endoscopic subscore of 0 or 1 from Mayo Scoring System.
SIBDQ score in LIBERTY-UC and Study 1.6: SIBDQ score and change from baseline.
Patient overall satisfaction measured using VAS score in Study 1.6: All patients assessed their overall satisfaction of infliximab IV and SC by using 100-mm visual analogue scale (VAS) after the end of administration of study drug. The patient’s assessment of overall satisfaction about procedure and duration of the study drug administration on that visit day, regardless of other external conditions (e.g., interaction with doctor/nurse, distance from home to hospital, transportation, etc.) was measured by the patient indicating his or her overall satisfaction of the study drug administration by marking a line through the 100-mm line.
Patient’s assessment of local site pain using VAS score in LIBERTY-UC and Study 1.6: All patients assessed local site pain using the 100-mm VAS immediately (not exceeding 1 hour) after the end of administration of study drug, indicating the extent of their pain by marking a line through the 100-mm line. Higher scores of VAS indicated more severe pain.
CD patients in LIBERTY-CD and Study 1.6
Clinical remission (based on abdominal pain and stool frequency) in LIBERTY-CD: Average worst daily abdominal pain score of ≤ 1 (using 4-point scale) and an average loose/watery stool frequency score of ≤ 3 (of Type 6 or Type 7 on Bristol stool form [BSF] scale) with no worsening in either score compared to baseline (based on abdominal pain and stool frequency score).
Maintenance of clinical remission in LIBERTY-CD: Maintenance of clinical remission was defined as being in clinical remission by CDAI score < 150 points, among the patients in clinical remission at Week 10.
Sustained remission at both Week 22 and 54 in LIBERTY-CD: Average worse daily AP score of ≤ 1 (using 4-point scale) and an average loose/watery SF score ≤ 3 (of Type 6 or 7 on BSFS) at both Week 22 and Week 54 with no worsening in either average score compared with the baseline value.
Clinical remission (based on CDAI score) in Study 1.6: Absolute CDAI score of < 150 points.
Corticosteroid-free remission in LIBERTY-CD: Being in clinical remission by an absolute CDAI score of < 150 (in CD) in addition to not requiring any treatment with corticosteroid for ≥ 8 weeks at Week 54, among patients who used oral corticosteroids at baseline.
CDAI-70 response in LIBERTY-CD and Study 1.6: Decrease in CDAI score of ≥ 70 points from baseline.
CDAI-100 response in LIBERTY-CD and Study 1.6: Decrease in CDAI score of ≥ 100 points from baseline.
Maintenance of clinical response based on CDAI-100: Maintenance of CDAI-100 at Week 54, among patients who achieved CDAI-100 response at Week 10.
Endoscopic remission in LIBERTY-CD and Study 1.6: Absolute SES-CD score of ≤ 4 and ≥ 2-point reduction from baseline with no sub-score of > 1 (LIBERTY-CD) or absolute SES-CD score of ≤ 2 points (Study 1.6).
Endoscopic response in Study 1.6: ≥ 50% decrease in Simplified Endoscopic Activity Score for Crohn Disease (SES-CD) score from baseline.
SIBDQ score in LIBERTY-CD and Study 1.6: SIBDQ score and change from baseline.
Patient overall satisfaction measured using VAS score in Study 1.6: Same as above.
Patient’s assessment of local site pain using VAS score in LIBERTY-CD and Study 1.6: Same as above.
Usability assessment in LIBERTY-CD: Usability of patient rating was evaluated using pre and post-self-injection assessment questionnaire (SIAQ), the observer rating of successful self-injections and completion of all instructions using self-injection assessment checklist, and device integrity of used PFS and AI by the observer (using a question that asks clear evidence of damage and/or compromised structural or mechanical integrity based on a visual examination). Results for these outcomes will not be reported here, as these are not efficacy related end points.
Statistical Analysis
Overview
Both LIBERTY-UC and LIBERTY-CD had similar data analysis method for efficacy outcomes; outcomes in Study 1.6 were only reported descriptively. For the safety assessment, only data collected before initiation of dose adjustment was used for the placebo SC arm and all data collected regardless of dose adjustment was used for the REMSIMATM SC arm, unless specified otherwise.
In the LIBERTY-UC and LIBERTY-CD trials, the patients who required dose adjustment to 240 mg before Week 54 were considered non-remitter or non-responder at Week 54 for each efficacy end point. In Study 1.6, if a patient discontinued from the study before a visit or missed endoscopy subscore at a visit, the patient was considered as not to have achieved that clinical outcome at that visit.
Primary Outcome(s) of the Studies
Power Calculation
All 3 trials ensured a minimum of 80% power in detecting the primary end point(s) when estimating the sample size for the randomized maintenance treatment phase. Since the primary end point of Study 1.6 were PK end points, no power calculation was done for the (secondary) efficacy end points. The assumptions made for sample size estimation and other details are provided in the Table 10 below.
Table 10: Overview of Sample Size Estimation
Parameter | LIBERTY-UC | LIBERTY-CD | Study 1.6a |
|---|---|---|---|
Estimated sample size maintenance phase, n | 417 (278 REMSIMATM SC, 139 placebo) | 360 (240 REMSIMATM SC, 120 placebo) | 104 (52 REMSIMATM SC and IV infliximab each), 130 after accounting for dropout rate (65 in each arm) |
Estimated power for primary end point | 80% | 90% | 90% |
Assumptions for primary end point |
|
|
|
Estimated sample size induction phase, nb | 615 | 600 | Not applicable |
Assumptions | 32% non-responder rate of clinical response at Week 10 before randomization | 40% non-responder rate of CDAI-100 at Week 10 before randomization | Not applicable |
Estimated power for key secondary end point | 90% (with 417 patients) | 89% (with 360 patients) | Not applicable |
Assumptions for key secondary end pointc |
|
| Not applicable |
Minimum detectable effect size for other secondary end points with the sample size for primary end point and at least 80% power | Not reported |
| Not applicable |
SC = subcutaneous.
aSample size calculation was based on primary end points, which was PK outcomes in Study 1.6.
bThe number of enrolled patients was adjusted based on the actual number of randomized at week 10.
CKey secondary end point was clinical response in LIBERTY-UC and CDAI-100 response in LIBERTY-CD.
Source: CSRs of LIBERTY-UC, LIBERTY-CD, and Study 1.6.a-c
Statistical Test or Model
LIBERTY-UC and LIBERTY-CD: The statistical analyses plan for the LIBERTY-UC and LIBERTY-CD trials were similar, briefly described below.
The primary/co-primary efficacy end points were analyzed using the Cochrane-Mantel-Haenszel (CMH) test, stratified by previous exposure to biologic agent and/or JAK inhibitors, oral corticosteroid treatment at Week 0, and clinical remission at Week 10. P values for the primary end point(s) was estimated from stratified CMH test, with the difference in proportion between treatment groups estimated using CMH weights and corresponding 95% stratified Newcombe confidence interval (CI) with CMH weights provided. All tests were conducted at the 2-sided significance level of 5%.
Patients in these 2 trials who required dose adjustment to 240 mg before Week 54 were considered non-remitter or non-responder at Week 54 in each efficacy end point. Among patients in the REMSIMATM SC arm, a descriptive comparison of the treatment effect between patients with and without dose adjustment was done for the primary end point(s), without the statistical test. In this analysis, remitter was determined as per remission criteria (described above) regardless of dose adjustment.
For safety assessment, only data collected before initiation of dose adjustment was used for placebo SC arm and all data collected regardless of dose adjustment was used for the REMSIMATM SC arm, unless otherwise specified otherwise.
Data Imputation Methods
Patients who received study drug and discontinued before study completion were not replaced. The impact of missing data on the primary end point(s) was analyzed using the tipping point method on the all-randomized population in LIBERTY-UC and LIBERTY-CD. With this method, patients who underwent dose adjustment before Week 54 or those with missing or incomplete data for the evaluation of the efficacy assessment were considered missing in the analysis. The tipping point analysis was conducted using the same stratified CMH test as the primary analysis, by gradually increasing the number of remitter or responder for each group starting with the scenario where all patients with missing result are non-remitters or non-responders up to the scenario where all patients with missing result were remitters or responders. All P values calculated from the stratified CMH test for the difference between 2 proportions (REMSIMATM SC and Placebo SC) was displayed as a shift table. The results from tipping point analysis were also presented using 2-dimensional plot, although this will not be focused here.
Subgroup Analyses
The following subgroups were analyzed for the primary end point(s), using the same method as above on the all-randomized population: sex, age, and race. The subgroups were pre-specified, without controlling for type I error. For each specific subgroup, if there were not enough (< 5%) patients, the corresponding analyses were not performed.
Sensitivity Analyses
For the primary end point(s), sensitivity analyses were performed using the logistic regression model and Fisher’s exact test. In addition, a sensitivity analyses were performed to evaluate the impact of the war in Ukraine, by excluding war-affected patients in Ukraine and excluding all patients in Ukraine.
Secondary and Exploratory Outcomes of the Studies
The key secondary end points in both LIBERTY-UC and LIBERTY-CD were analyzed in the same manner as the stratified primary analysis, including adjustment for multiplicity, and sensitivity analyses. Of the other secondary and exploratory end points, the binary end points were analyzed following the same procedure as the stratified primary analysis. The continuous end points were analyzed using analysis of covariance (ANCOVA), with the same covariates as the stratification factors used for the primary analysis.
In LIBERTY-UC and LIBERTY-CD, multiplicity adjustment for testing of the primary and key secondary end points was performed using the fixed-sequence testing method. In other words, the key secondary end points were tested in a predefined order only if the primary end point was statistically significant. The other secondary and exploratory end points were not adjusted for multiple comparisons. The fixed sequence was as follows:
Table 11: Fixed Sequence of Key Secondary End points for Multiplicity Testing
LIBERTY-UC | LIBERTY-CD |
|---|---|
Clinical response at Week 54 | CDAI-100 response at Week 54 |
Endoscopic-histologic mucosal improvement at Week 54 | Clinical remission at Week 54 |
Corticosteroid-free remission at Week 54 | Endoscopic remission at Week 54 |
— | Corticosteroid-free remission at Week 54 |
Source: CSRs of LIBERTY-UC and LIBERTY-CD.a,b
The efficacy end points in Study 1.6 were reported descriptively, without any statistical comparison between treatment arms, before or after switching treatment.
Analysis Populations
The analysis sets in LIBERTY-UC and LIBERTY-CD trials were identical. Below are the definitions of the different analysis sets:
Intention-to-treat (ITT) population: All enrolled patients.
All randomized population: All randomly assigned patients at Week 10 (LIBERTY-UC and CD) or at Week 6 (Study 1.6), regardless of the completion of dosing of the study drug. The all-randomized population was used for analysis of primary and key secondary end points in LIBERTY-UC and CD and reported here.
Efficacy population: This was used in Study 1.6 to report efficacy results. This was almost identical to the all-randomized population in the trial, with only 1 UC patient less in the infliximab arm.
The per-protocol (PP) population: All randomly assigned patients who received at least 1 full dose of study drug at Week 10 or thereafter before Week 54 and who have at least 1 efficacy evaluation after Week 10 and who did not have any major protocol violation related to efficacy analysis. The PP population was used for supportive analysis of primary and key secondary end points.
The safety population: All randomly assigned patients who received at least 1 full or partial dose of study drug at Week 10 or thereafter. This population was used to report safety and exposure to study treatment data.
Sponsor’s Summary of the Results
Patient Disposition
Details of patient disposition for the 3 trials are provided in Table 12 below. Screening failures ranged from approximately 30% to 50% across trials, primarily due to not meeting inclusion criteria. Of the patients enrolled in the induction phase (LIBERTY-UC and LIBERTY-CD) or dose-loading phase (Study 1.6) of the trials, approximately 3% to 20% patients discontinued, primarily due to being classified as a non-responder (as per definition above) in the LIBERTY-UC and LIBERTY-CD trials, and adverse events (AE) in Study 1.6. A total of 438, 343, and 131 patients were randomized to the maintenance treatment in LIBERTY-UC, LIBERTY-CD, and Study 1.6, respectively. Overall, 17% to 20% patients discontinued during the maintenance phase of the trials, with no notable difference between the treatment arms. The most common reasons for study discontinuation were withdrawal by patients in LIBERTY-UC, and progressive disease in LIBERTY-CD and Study 1.6. Nearly 80% patients in LIBERTY-UC and LIBERTY-CD were continuing the study at Week 54, and approximately 78% patients completed Study 1.6.
Table 12: Patient Disposition – All-Randomized Population
Characteristics | LIBERTY-UC | LIBERTY-CD | Study 1.6 | |||
|---|---|---|---|---|---|---|
REMSIMATM SC | Placebo | REMSIMATM SC | Placebo | REMSIMATM SC | Infliximab IV | |
Screened, N | 800 | 787 | 195 | |||
Enrolled, N (%) | 548 (68.5) | 396 (50.3) | 136 (69.7) | |||
Discontinued induction/dose-loading phase, N (%) | 110 (20.1) | 53 (13.4) | 5 (3.7) | |||
Reason for discontinuation, N (> 5)a | ||||||
Non-responder | 65 (11.9) | 22 (5.5) | — | |||
Adverse events | 13 (2.4) | 11 (2.8) | 2 (1.5) | |||
Withdrawal by patient | 15 (2.7) | 12 (3.0) | — | |||
Progressive disease | 6 (1.1) | 2 (0.5) | — | |||
Randomized, N | 294 | 144 | 231 | 112 | 66 | 65 |
(38 UC, 28 CD) | (40 UC, 25 CD) | |||||
Discontinued maintenance phase, N (%) | 54 (18.4) | 31 (21.5) | 35 (15.2) | 25 (22.3) | 11 (16.7) | 15 (23.1) |
Reason for discontinuation, N (%) [> 5]a | ||||||
Withdrawal by patient | 23 (7.8) | 12 (8.3) | 6 (2.6) | 8 (7.1) | 2 (3.0) | 5 (7.7) |
Adverse events | 11 (3.7) | 8 (5.5) | 8 (3.5) | 6 (5.4) | 1 (1.5) | 3 (4.6) |
Progressive disease | 13 (4.4) | 9 (6.2) | 12 (5.2) | 6 (5.4) | 4 (6.1) | 5 (7.7) |
Continuing maintenance, N (%) | 240 (81.6) | 113 (78.5) | 196 (84.8) | 87 (77.7) | N/A | N/A |
ITT, N | 548 | 396 | 136 (79 UC, 57 CD) | |||
PP, N | 286 | 135 | 230 | 102 | N/A | |
Efficacy population, N | N/A | N/A | N/A | N/A | 130 (77 UC, 53 CD) | |
Safety, N | 296 | 140 | 238 | 105 | 131 (78 UC, 53 CD) | |
Randomized, N | 294 | 144 | 231 | 112 | 131 (78 UC, 53 CD) | |
CD = Crohn disease; ITT = intention to treat; PP = per protocol; UC = ulcerative colitis.
aDenominator was enrolled and randomized patients for the calculation of study discontinuation.
Source: CSRs of LIBERTY-UC, LIBERTY-CD, and Study 1.6.a-c
Exposure to Study Treatments
Study Treatments
Induction/dose-loading phase
Exposure data for this phase will not be described in detail, given the focus of the review is the maintenance phase. In LIBERTY-UC and LIBERTY-CD, all eligible patients received 3 infusions (Weeks 0, 2, and 6) of infliximab 5 mg/kg, whereas in Study 1.6, patients received 2 infusions (Week 0 and 2). The mean total administered dose was similar across the treatment groups in all trials. None of the patients in LIBERTY-UC and LIBERTY-CD had an overdose of the study drug. One patient in Study 1.6 was overdosed; however, this did not result in any safety issues.
Maintenance phase
In all 3 trials, patients in the REMSIMATM SC arm received an approximate 20 to 22 doses on average, indicating patients in this arm received most of their expected 24 to 25 doses from Week 6/10 to 54. The placebo arms in LIBERTY-UC and LIBERTY-CD trial received less doses compared to the REMSIMATM SC arm, as can be expected given some required dose adjustment. This also explained fewer patients in the REMSIMATM SC arm of these 2 trials requiring dose adjustment compared to placebo. In Study 1.6, the number of patients requiring dose adjustment post Week 30 was similar, as can be expected given all patients received the same SC treatment starting at Week 30. In this study, while the total administered dose was higher in the REMSIMATM SC arm compared to the infliximab IV group over the full maintenance period, the total administered dose post Week 30 was similar between the treatment groups (data not presented). Details are provided in Table 13.
Table 13: Summary of Exposure to Study Treatments during Maintenance Phase
End point | LIBERTY-UC | LIBERTY-CD | Study 1.6 | |||
|---|---|---|---|---|---|---|
REMSIMATM SC N = 296 | Placebo N = 140 | REMSIMATM SC N = 238 | Placebo N = 105 | REMSIMATM SC N = 66 | Infliximab IV N = 65 | |
Proportion of patients achieving Week 54 primary clinical outcomes, n (%) | ||||||
Total number of doses received, Mean (SD) | 20.8 (5.5) | 14.0 (7.9) | 21.32 (4.7) | 14.68 (8.0) | 22.6 (6.0) | 13.2 (5.3) |
Total administered dose, mg, Mean (SD) | 2920.00 (1062.1) | 1685.14 (951.2) | 2799.08 (841.8) | 1762.29 (961.2) | 3541.8 (1511.2) | 2818.0 (1292.9) |
Patients received ≥ 1 adjusted dose, n (%) | 92 (31.1) | 75 (53.6) | 45 (18.9) | 48 (45.1) | 6/44 (13.6)a | 8/38 (21.1)a |
SC = subcutaneous; SD = standard deviation.
Note. Since the patients with dose adjustment in the Placebo SC group in LIBERTY-UC and CD were not included in the analysis after the dose adjustment, the result of the mean total administered dose and number of doses received were lower in the Placebo SC group than in the REMSIMATM SC group.a The denominator in Study 1.6 was patients who were planned to receive REMSIMATM SC 120 mg on or after Week 30, including those switching from infliximab IV. Therefore, the denominator was not the same as the safety population.
Source: CSRs of LIBERTY-UC, LIBERTY-CD, and Study 1.6.a-c
Efficacy
Efficacy results are summarized by diagnosis of patients, i.e., UC patients in LIBERTY-UC and Study 1.6, and CD patients in LIBERTY-CD and Study 1.6. Unless otherwise specified, all end points for LIBERTY-UC and LIBERTY-CD were reported at Week 54. Results for Study 1.6 were presented descriptively without any statistical comparison and provided before, and after, the infliximab IV arm switching to SC treatment. Efficacy analyses were reported at Week 22 (UC patients) or Week 30 (CD patients), and after at Week 54 (all patients). Data for the primary efficacy end points are provided in Table 14, secondary end points in Table 15 through Table 18, and HRQoL end points in Table 21.
Primary efficacy end points
The primary end point in LIBERTY-UC was clinical remission based on modified Mayo score. The 2 co-primary end points in LIBERTY-CD were clinical remission based on CDAI and endoscopic response based on central SES-CD. The primary end point in Study 1.6 were PK outcomes, therefore not discussed here, see below in the “Bioequivalence” section.
UC patients
Clinical remission (LIBERTY-UC): Clinical remission was statistically significantly improved in patients receiving REMSIMATM SC (127 [43.2%] patients) compared to placebo (30 [20.8%] patients), with a 21.1% treatment difference (95% CI, 11.8, 29.3) and p-value of < 0.0001, showing superiority over placebo. Notably, a similar proportion of patients in both groups achieved clinical remission at Week 10 before randomization; however, the REMSIMATM SC arm showed higher remission rate starting at Week 22 after randomization (data not presented, see the CSRs for details). Given the primary outcome was statistically significant, the fixed sequence to assess the key secondary end points was followed.
CD patients
Clinical remission and endoscopic response (LIBERTY-CD): Clinical remission and endoscopic response were statistically significantly improved in patients receiving REMSIMATM SC compared to placebo. Specifically, the proportion of patients who achieved clinical remission at Week 54 was higher in the REMSIMA™ SC group (144 [62.3%]) than in the placebo group (36 [32.1%]) with an estimated treatment difference of 32.1% (95% CI, 20.9, 42.1) and a p-value of < 0.0001. Similarly, the proportion of patients who achieved endoscopic response at Week 54 was higher in the REMSIMA™ SC group (118 [51.1%]) than in the placebo group (20 [17.9%]) with an estimated treatment difference of 34.7% (95% CI, 24.2, 43.5) and a p-value of < 0.0001. Notably, a similar proportion of patients in both groups achieved clinical remission at Week 10 before randomization; however, the REMSIMATM SC arm showed higher remission rate starting at Week 22 after randomization (data not presented, see the CSRs for details). Given the co-primary outcomes were statistically significant, the fixed sequence to assess the key secondary end points was followed.
Table 14: Primary Efficacy Outcomes
End point | LIBERTY-UC | LIBERTY-CD | Study 1.6 | |||
|---|---|---|---|---|---|---|
REMSIMATM SC N = 294 | Placebo N = 144 | REMSIMATM SC N = 231 | Placebo N = 112 | REMSIMATM SC N = 66 | Infliximab IV N = 65 | |
Proportion of patients achieving Week 54 primary clinical outcomes, n (%) | ||||||
Clinical remission, n (%)a | 127 (43.2) | 30 (20.8) | 144 (62.3) | 36 (32.1) | N/A | N/A |
Difference (95% CI)b | 21.1 (11.8, 29.3) | 32.1 (20.9, 42.1) | N/A | |||
P valuec | < 0.0001 | < 0.0001 | N/A | |||
Endoscopic response, n (%) | N/A | N/A | 118 (51.1) | 20 (17.9) | N/A | N/A |
Difference (95% CI)b | N/A | 34.7 (24.2, 43.5) | N/A | |||
P valuec | N/A | < 0.0001 | N/A | |||
CD = Crohn Disease, CDAI = Crohn Disease Activity Index; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; JAK = Janus kinase; N/A = not applicable; SD = standard deviation; SE = standard error; SES-CD = simplified endoscopic activity score for Crohn disease; UC = ulcerative colitis.
aClinical remission in LIBERTY-CD was based on CDAI score.
bThe difference of proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CI with CMH weights were presented (all trials). Analysis was stratified by previous exposure to biologic agent and/or JAK inhibitors (used or not used), use of treatment with oral corticosteroids at Week 0 (used or not used) and clinical remission at week 10 (remitter or non-remitter by modified Mayo score/CDAI score) [LIBERTY-UC and CD]. IN LIBERTY-UC and CD, patients with dose adjustment to REMSIMATM SC SC 240 mg before week 54 were considered as non-remitter.
Specify model, covariates, analysis population and time point for each outcome.
cThe (co)primary outcome(s) in both LIBERTY-UC and CD trials was within the statistical testing hierarchy.
Source: CSRs of LIBERTY-UC, LIBERTY-CD, and Study 1.6.a-c
Table 15: Secondary Efficacy Outcomes in UC patients — Binary Variables
End point | LIBERTY-UC | Study 1.6 | ||
|---|---|---|---|---|
REMSIMATM SC N = 294 | Placebo N = 144 | REMSIMATM SC N = 66 UC = 38 | Infliximab IV N = 65 UC = 39 | |
Proportion of patients achieving Week 54 secondary clinical outcomes, n (%) | ||||
Clinical response, n (%)a | 158 (53.7) | 45 (31.3) | Week 22: 24 (63.2) Week 54: 24 (63.2) | Week 22: 17 (43.6) Week 54: 24 (61.5) |
Difference (95% CI)a | 21.1 (11.2, 30.1) | N/A | ||
P value | < 0.0001b | N/A | ||
Clinical remission, n (%) | N/A | N/A | Week 22: 17 (44.7) Week 54: 20 (52.6) | Week 22: 10 (25.6) Week 54: 19 (48.7) |
Difference (95% CI)a | N/A | N/A | ||
P value | N/A | N/A | ||
Endoscopic-histologic mucosal improvement, n (%) | 105 (35.7) | 24 (16.7) | N/A | N/A |
Difference (95% CI)a | 18.0 (9.1, 25.7) | N/A | ||
P value | < 0.0001b | N/A | ||
Corticosteroid-free remission, n/N (%) | 44/120 (36.7) | 11/61 (18.0) | N/A | N/A |
Difference (95% CI)a | 17.3 (3.1, 28.9) | N/A | ||
P value | 0.01b | N/A | ||
Mucosal healing, n (%) | N/A | N/A | Week 22: 18 (47.4) Week 54: 21 (55.3) | Week 22: 12 (30.8) Week 54: 22 (56.4) |
Difference (95% CI)a | N/A | N/A | ||
P value | N/A | N/A | ||
Maintenance of clinical remission, n/N (%) | 91/143 (63.6) | 18/66 (27.3) | N/A | N/A |
Difference (95% CI)a | 35.5 (21.1, 47.5) | N/A | ||
P value | < 0.0001 | N/A | ||
Sustained clinical remission at Week 22 and 54, n (%) | 142 (48.3) | 35 (24.3) | N/A | N/A |
Difference (95% CI)a | 22.6 (13.0, 31.1) | N/A | ||
P value | < 0.0001 | N/A | ||
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; JAK = Janus kinase; N/A = not applicable; SD = standard deviation; SE = standard error; UC = ulcerative colitis.
aThe difference of proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CI with CMH weights were presented (all trials). Analysis was stratified by previous exposure to biologic agent and/or JAK inhibitors (used or not used), use of treatment with oral corticosteroids at Week 0 (used or not used) and clinical remission at Week 10 (remitter or non-remitter by modified Mayo score) [LIBERTY-UC]. In LIBERTY-UC, patients with dose adjustment to REMSIMATM SC 240 mg before Week 54 were considered as non-remitter/non-responder.
bThe P values were part of outcomes within the statistical testing hierarchy.
Source: CSRs of LIBERTY-UC, and Study 1.6.a,c
Results of supportive, sensitivity, and subgroup analyses
Supportive analyses based on PP analyses; sensitivity analyses utilizing Fisher’s exact test, and logistic regression model – all showed similar results as the primary analysis. Sensitivity analyses excluding war-affected patients in Ukraine, excluding all patients in Ukraine, and using tipping point analysis showed no significant impact of these variables on the results (data not presented). Similarly, the results for the subgroup analysis of primary end point(s) based on sex, age and race showed that patient’s subgroup generally did not have major impact on the results, although no statistical analyses were done to compare the subgroups (data not presented).
Secondary efficacy end points: UC patients
The key secondary end points in LIBERTY-UC were clinical response based on modified Mayo score, endoscopic-histologic mucosal improvement and corticosteroid-free remission at Week 54, the rest were secondary or exploratory. The key secondary end points in LIBERTY-CD were CDAI-100 response, clinical remission based on abdominal pain and stool frequency, endoscopic remission based on central SES-CD, and corticosteroid-free remission at Week 54, the rest were secondary or exploratory.
The secondary efficacy end points measured in patients with UC in Study 1.6 were measured both locally and centrally. Results based on central assessment are provided here, as some local videos either failed to be uploaded onto the central server or was recorded in a condition incapable of central reading. None of the end points in Study 1.6 were compared statistically between treatment arms, and results should be interpreted descriptively.
Clinical response
LIBERTY-UC: The proportion of patients who achieved clinical response was statistically significantly higher in the REMSIMATM SC arm compared to placebo at Week 54, with an estimated treatment difference of 21.1% (95% CI, 11.2, 30.1) and a P value of < 0.0001. This outcome was within the fixed sequence to control the type I error as the primary outcome was statistically significant. Notably, a similar proportion of patients in both groups achieved clinical response at Week 10 before randomization; however, the REMSIMATM SC arm showed higher response rate starting at Week 22 after randomization (data not presented, see the CSRs provided for details).
The actual value for modified Mayo score, total Mayo score, and partial Mayo score – all decreased from baseline to Week 54, with similar decrease between treatment arms up to Week 10 (data not presented, see CSRs for details), but the magnitude of decrease was higher in the REMSIMATM SC arm afterwards, although no statistical comparison was done.
Study 1.6: The proportion of patients who achieved clinical response according to total Mayo score was higher in the REMSIMATM SC arm than in the infliximab IV arm at Week 22 (63.2% vs 43.6%). Of note, 4 (10.5%) and 11 (28.2%) patients in the REMSIMATM SC and infliximab IV group had not had their colonoscopy performed at Week 22, respectively. The impact of greater missing rate in the infliximab IV arm can be apparently seen in relatively lower proportion of patients achieving clinical response in this arm. However, after switching to REMSIMATM SC at Week 30 in the infliximab IV arm, the response rates were similar between the treatment arms at Week 54 (over 60% in both arms).
The mean for total and partial Mayo scores were similar between the treatment arms at baseline. At Week 22, slightly greater improvement of both total and partial Mayo scores was noticed along with greater reduction from the baseline score in the REMSIMATM SC arm. After switching to REMSIMATM SC at Week 30 in the IV treatment arm, the mean actual values and change from the baseline of total and partial Mayo scores were similar between the treatment arms at Week 54.
Clinical remission
Study 1.6: The proportion of patients who achieved clinical remission based on Mayo score was higher in the REMSIMATM SC arm than in the infliximab IV arm at Week 22 (44.7% vs 25.6%). However, after switching to REMSIMATM SC at Week 30 in the infliximab IV arm, the response rates were similar between the treatment arms at Week 54 (approximately 50% in both arms).
Endoscopic-histologic mucosal improvement
LIBERTY-UC: The proportion of patients who achieved Endoscopic-histologic mucosal improvement was statistically significantly higher in the REMSIMATM SC arm compared to placebo at Week 54, with an estimated treatment difference of 18.0% (95% CI, (9.1, 25.7) and a P value of < 0.0001. This outcome was within the fixed sequence to control the type I error as the previous key secondary outcome in the hierarchy was statistically significant. Notably, a similar proportion of patients in both groups achieved endoscopic-histologic mucosal improvement at Week 8; however, the REMSIMATM SC arm showed higher endoscopic-histologic mucosal improvement rate starting at Week 22 after randomization (data not presented, see the CSRs for details).
The actual value for endoscopic and RHI score– all decreased from baseline to Week 54, with similar decrease between treatment arms up to Week 10 (data not presented, see the CSRs for details), but the magnitude of decrease was higher in the REMSIMATM SC arm afterwards, although no statistical comparison was done.
Mucosal healing
Study 1.6: The proportion of patients who achieved mucosal healing was higher in the REMSIMATM SC arm than in the infliximab IV arm at Week 22 (47.4% vs 30.8%). Of note, 4 (10.5%) and 11 (28.2%) patients in the REMSIMATM SC and infliximab IV group had not had their colonoscopy performed at Week 22, respectively. The impact of greater missing rate in the infliximab IV arm can be apparently seen in relatively lower proportion of patients achieving mucosal healing between the treatment arms at Week 54 (over 55% in both arms).
Corticosteroid-free remission
LIBERTY-UC: The proportion of patients who achieved corticosteroid-free remission was statistically significantly higher in the REMSIMATM SC arm compared to placebo at Week 54, with an estimated treatment difference of 17.3% (95% CI, 3.1, 28.9) and a P value of 0.01. This outcome was within the fixed sequence to control the type I error as the previous key secondary outcome in the hierarchy was statistically significant.
Maintenance of clinical remission
LIBERTY-UC: Among the patients with clinical remission at Week 10, a higher proportion of patients in the REMSIMATM SC group achieved maintenance of clinical remission compared to placebo at Week 54, with a treatment difference of 35.5% (95% CI, 21.1, 47.5) and a P value of < 0.0001.
Sustained remission at both Week 22 and 54
LIBERTY-UC: A higher proportion of patients in the REMSIMATM SC group achieved sustained remission at both Week 22 and 54 compared to placebo, with a treatment difference of 22.6% (95% CI, 13.0, 31.1) and a P value of < 0.0001.
Total/partial clinical remission
LIBERTY-UC: Total clinical remission was defined as a total Mayo score (stool frequency, rectal bleeding, endoscopic, and PGA subscores) of ≤ 2 points with no individual subscore exceeding 1 point. Partial clinical remission was defined as a partial Mayo score (stool frequency, rectal bleeding, and PGA subscores) of ≤ 1 point. Both total and partial clinical remission were similar between treatment arms up to Week 10. However, the proportion of patients who achieved these outcomes were statistically higher in the REMSIMATM SC arm up to Week 54 (data not presented, see the CSRs provided for details).
Total/partial clinical response
LIBERTY-UC: Total clinical response was defined as a decrease in total Mayo score from baseline of at least 3 points and at least 30%, with an accompanying decrease in rectal bleeding subscore of at least 1 point or an absolute rectal bleeding subscore of 0 or 1 point. Partial clinical response was defined as a decrease in partial Mayo score from baseline of at least 2 points, with an accompanying decrease in the subscore for rectal bleeding of at least 1 point, or an absolute subscore for rectal bleeding of 0 or 1 point. Both total and partial clinical response were similar between treatment arms up to Week 10. However, the proportion of patients who achieved these outcomes were statistically higher in the REMSIMATM SC arm up to Week 54 (data not presented, see the CSRs provided for details).
Secondary efficacy end points: CD patients
CDAI-100 response
LIBERTY-CD: The proportion of patients who achieved CDAI-100 response was statistically significantly higher in the REMSIMATM SC arm compared to placebo at Week 54, with an estimated treatment difference of 29.0% (95% CI, 17.7, 39.3) and a P value of < 0.0001. This outcome was within the fixed sequence to control the type I error as the primary outcome was statistically significant. Notably, CDAI-100 response rates were similar between treatment arms up to Week 10 before randomization. However, the proportion of patients who achieved CDAI-100 response rates were statistically higher in the REMSIMATM SC arm up to Week 54 (data not presented, see the CSR for details).
The actual value for CDAI score – all decreased from baseline to Week 54, with similar decrease between treatment arms up to Week 10 (data not presented, see the CSRs for details), but the magnitude of decrease was higher in the REMSIMATM SC arm afterwards, although no statistical comparison was done.
Study 1.6: The proportion of patients who achieved CDAI-100 response was similar between the REMSIMATM SC and the infliximab IV arm at Week 30, which remained similar after switching to SC treatment through Week 54 (approximately 65% at both time points).
CDAI-70 response
LIBERTY-CD: The proportion of patients who achieved CDAI-70 response was statistically significantly higher in the REMSIMATM SC arm compared to placebo at Week 54, with an estimated treatment difference of 32.2% (95% CI, 21.0, 42.4) and a P value of < 0.0001. Notably, CDAI-70 response rates were similar between treatment arms up to Week 10 before randomization. However, the proportion of patients who achieved CDAI-70 response rates were statistically higher in the REMSIMATM SC arm up to Week 54 (data not presented, see the CSR for details).
Study 1.6: The proportion of patients who achieved CDAI-70 response was similar between the REMSIMATM SC and the infliximab IV arm at Week 30, which remained similar after switching to SC treatment through Week 54 (under 70% at both time points).
The mean score for CDAI at Week 6 was slightly higher in the REMSIMATM SC treatment arm after the initial IV loading regimen consisting of 2 doses of infliximab IV. However, the mean CDAI score continued to decrease for both treatment arms, and the mean change from baseline of CDAI scores were comparable between the treatment arms up to Week 54.
Clinical remission
LIBERTY-CD: The proportion of patients who achieved clinical remission based on abdominal pain and stool frequency was statistically significantly higher in the REMSIMATM SC arm compared to placebo at Week 54, with an estimated treatment difference of 27.0% (95% CI, 15.8, 37.1) and a P value of < 0.0001. This outcome was within the fixed sequence to control the type I error as the previous key secondary outcome was statistically significant.
Study 1.6: The proportion of patients who achieved clinical remission was higher in the REMSIMATM SC arm than in the infliximab IV arm at Week 22. However, after switching to REMSIMATM SC at Week 30 in the infliximab IV arm, the remission rates were similar between the treatment arms at Week 54.
Endoscopic remission
LIBERTY-CD: The proportion of patients who achieved endoscopic remission based on central SES-CD score was statistically significantly higher in the REMSIMATM SC arm compared to placebo, with an estimated treatment difference of 24.9% (95% CI, 15.4, 32.8). This outcome was within the fixed sequence to control the type I error as the previous key secondary outcome was statistically significant.
Study 1.6: The proportion of patients who achieved endoscopic remission was higher in the REMSIMATM SC arm than in the infliximab IV arm at Week 22 (35.7% vs 14.3%, respectively). However, after switching to REMSIMATM SC at Week 30 in the infliximab IV arm, the remission rates were similar between the treatment arms at Week 54 (50.0% in both arms).
Endoscopic response
Study 1.6: The proportion of patients who achieved endoscopic response was higher in the REMSIMATM SC arm than in the infliximab IV arm at Week 22 (78.6% vs 42.9%, respectively). However, after switching to REMSIMATM SC at Week 30 in the infliximab IV arm, the response rates were similar between the treatment arms at Week 54 (75.0% vs 80.0%, respectively).
At baseline, the mean values of overall SES-CD score (measured centrally) were similar between the treatment arms. They decreased to a similar level at Week 22, with slightly greater magnitude in terms of the change from the baseline in the REMSIMATM SC arm than in the infliximab IV arm (−8.9 vs −4.4, respectively). After switching treatment, the mean SES-CD score at Week 54 was similar between the treatment arms, with slightly greater magnitude in terms of the change from the baseline in the REMSIMATM SC arm than in the infliximab IV arm (−10.2 vs −6.4, respectively).
Corticosteroid-free remission
LIBERTY-CD: The proportion of patients who achieved corticosteroid-free remission was statistically significantly higher in the REMSIMATM SC arm compared to placebo, with an estimated treatment difference of 17.1% (95% CI, −0.4, 31.5) and a P value of 0.04. This outcome was within the fixed sequence to control the type I error as the previous key secondary outcome was statistically significant.
Maintenance of clinical remission
LIBERTY-CD: Among the patients with clinical remission at Week 10, a higher proportion of patients in the REMSIMATM SC group achieved maintenance of clinical remission compared to placebo, with a treatment difference of 34.5% (95% CI, 22.0, 45.6) and a P value of < 0.0001.
Sustained remission at both Week 22 and 54
LIBERTY-CD: A higher proportion of patients in the REMSIMATM SC group achieved sustained remission at both Week 22 and 54 compared to placebo, with a treatment difference of 24.1% (95% CI, 13.0, 34.2) and a P value of < 0.0001.
Maintenance of clinical response
LIBERTY-CD: Among the patients with CDAI-100 response at Week 10, a higher proportion of patients in the REMSIMATM SC group achieved maintenance of clinical response compared to placebo, with a treatment difference of 29.6% (95% CI, 18.4, 39.9) and a P value of < 0.0001.
Sustained remission at both Week 22 and 54
LIBERTY-CD: A higher proportion of patients in the REMSIMATM SC group achieved sustained clinical response based on abdominal pain and stool frequency at both Week 22 and 54 compared to placebo, with a treatment difference of 28.7% (95% CI, s17.5, 39.1) and a P value of < 0.0001.
Table 16: Secondary Efficacy Outcomes in CD Patients — Binary Variables
End point | LIBERTY-CD | Study 1.6 | ||
|---|---|---|---|---|
REMSIMATM SC N = 231 | Placebo N = 112 | REMSIMATM SC N = 66 CD = 28 | Infliximab IV N = 65 CD = 25 | |
CDAI-100 response, n (%) | 152 (65.8) | 43 (38.4) | Week 30: 19 (67.9) Week 54: 18 (64.3) | Week 30: 16 (64.0) Week 54: 16 (64.0) |
Difference (95% CI)a | 29.0 (17.7, 39.3) | N/A | ||
P value | < 0.0001b | N/A | ||
CDAI-70 response, n (%) | 160 (69.3) | 43 (38.4) | Week 30: 19 (67.9) Week 54: 20 (71.4) | Week 30: 17 (68.0) Week 54: 17 (68.0) |
Difference (95% CI)a | 32.2 (21.0, 42.4) | N/A | ||
P value | < 0.0001 | N/A | ||
Clinical remission, n (%)c | 131 (56.7) | 35 (31.3) | Week 30: 18 (64.3) Week 54: 16 (57.1) | Week 30: 14 (56.0) Week 54: 14 (56.0) |
Difference (95% CI)a | 27.0 (15.8, 37.1) | N/A | ||
P value | < 0.0001b | N/A | ||
Endoscopic remission, n (%)d | 80 (34.6) | 12 (10.7) | Week 22: 5/14 (35.7) Week 54: 6/12 (50.0) | Week 22: 1/7 (14.3) Week 54: 5/10 (50.0) |
Difference (95% CI)a | 24.9 (15.4, 32.8) | N/A | ||
P value | < 0.0001b | N/A | ||
Endoscopic response, n/N (%)d | N/A | N/A | Week 22: 11/14 (78.6) Week 54: 9/12 (75.0) | Week 22: 3/7 (42.9) Week 54: 8/10 (80.0) |
Difference (95% CI)a | N/A | N/A | ||
P valueb | N/A | N/A | ||
Corticosteroid-free remission, n/N (%) | 39/98 (39.8) | 10/44 (22.7) | N/A | N/A |
Difference (95% CI)a | 17.1 (−0.4, 31.5) | N/A | ||
P value | 0.04b | N/A | ||
Maintenance of clinical remission, n/N (%) | 121/174 (69.5) | 32/91 (35.2) | N/A | N/A |
Difference (95% CI)a | 34.5 (22.0, 45.6) | N/A | ||
P value | < 0.0001 | N/A | ||
Sustained clinical remission at Week 22 and 54, n (%) | 120 (51.9) | 33 (29.5) | N/A | N/A |
Difference (95% CI)a | 24.1 (13.0, 34.2) | N/A | ||
P value | < 0.0001 | N/A | ||
Maintenance of clinical response, n/N (%) | 152/229 (66.4) | 43/112 (38.4) | N/A | N/A |
Difference (95% CI)a | 29.6 (18.4, 39.9) | N/A | ||
P value | < 0.0001 | N/A | ||
Sustained clinical response at Week 22 and 54, n (%) | 163 (70.6) | 48 (42.9) | N/A | N/A |
Difference (95% CI)a | 28.7 (17.5, 39.1) | N/A | ||
P value | < 0.0001 | |||
CD = Crohn Disease, CDAI = Crohn Disease Activity Index; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; JAK = Janus kinase; N/A = not applicable; SD = standard deviation; SE = standard error; SES-CD = simplified endoscopic activity score for Crohn disease.
aThe difference of proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CI with CMH weights were presented (all trials). Analysis was stratified by previous exposure to biologic agent and/or JAK inhibitors (used or not used), use of treatment with oral corticosteroids at Week 0 (used or not used) and clinical remission at Week 10 (remitter or non-remitter by modified CDAI score) [LIBERTY-CD]. In LIBERTY-CD, patients with dose adjustment to REMSIMATM SC 240 mg before Week 54 were considered as non-remitter/non-responder.
bThe P values were part of outcomes within the statistical testing hierarchy.
cClinical remission in LIBERTY-CD was based on abdominal pain and stool frequency but CDAI score in Study 1.6.
dThe number of patients included in the central analysis in Study 1.6 may have been lower than that included in the adjusted local analysis due to issues involved in a part of local videos which either failed to be uploaded onto the central server or was recorded in a condition incapable of central reading.
Source: CSRs of LIBERTY-CD, and Study 1.6.b,c
Table 17: Secondary Efficacy Outcomes in UC Patients — Continuous Variables
End point | LIBERTY-UC | Study 1.6 | ||
|---|---|---|---|---|
REMSIMATM SC N = 294 | Placebo N = 144 | REMSIMATM SC N = 66 UC = 38 | Infliximab IV N = 65 UC = 39 | |
Modified Mayo Score, mean (SD) | ||||
Baseline, n | 294 | 143 | N/A | N/A |
Baseline value | 6.6 (1.1) | 6.7 (1.2) | N/A | N/A |
Week 54, n | 174 | 53 | N/A | N/A |
Week 54 value | 1.5 (1.8) | 2.2 (2.2) | N/A | N/A |
Change from baseline | −5.1 (2.2) | −4.2 (2.4) | N/A | N/A |
Total Mayo Score, mean (SD) | ||||
Baseline, n | 294 | 143 | 36 | 37 |
Baseline value | 8.8 (1.3) | 8.8 (1.4) | 7.9 (1.6) | 8.2 (1.6) |
Week 22, n | Not relevant | Not relevant | 32 | 26 |
Week 22 value | Not relevant | Not relevant | 2.8 (2.6) | 4.0 (3.0) |
Change from baseline | Not relevant | Not relevant | −4.8 (2.3) | −3.9 (2.8) |
Week 54, n | 174 | 53 | 29 | 31 |
Week 54 value | 2.0 (2.5) | 2.9 (2.7) | 2.2 (2.2) | 2.1 (2.2) |
Change from baseline | −6.7 (2.8) | −5.7 (3.0) | −5.5 (1.9) | −5.7 (2.8) |
Partial Mayo Score, mean (SD) | ||||
Baseline, n | 294 | 143 | 38 | 39 |
Baseline value | 6.3 (1.10) | 6.3 (1.2) | 5.4 (1.3) | 5.9 (1.2) |
Week 22, n | Not relevant | Not relevant | 35 | 36 |
Week 22 value | Not relevant | Not relevant | 1.3 (1.6) | 2.3 (1.9) |
Change from baseline | Not relevant | Not relevant | −4.0 (1.5) | −3.5 (1.9) |
Week 54, n | 184 | 61 | 32 | 32 |
Week 54 value | 1.2 (1.6) | 1.7 (2.1) | 0.9 (1.3) | 1.0 (1.6) |
Change from baseline | −5.1 (2.0) | −4.5 (2.5) | −4.5 (1.27) | −4.7 (2.2) |
Endoscopic subscore, mean (SD) | ||||
Baseline, n | 294 | 144 | N/A | N/A |
Baseline value | 2.5 (0.5) | 2.5 (0.5) | N/A | N/A |
Week 54, n | 175 | 54 | N/A | N/A |
Week 54 value | 0.8 (1.0) | 1.3 (1.0) | N/A | N/A |
Change from baseline | −1.6 (1.1) | −1.2 (1.1) | N/A | N/A |
RHI subscore, mean (SD) | ||||
Baseline, n | 287 | 144 | N/A | N/A |
Baseline value | 17.0 (9.6) | 18.0 (9.2) | N/A | N/A |
Week 54, n | 169 | 54 | N/A | N/A |
Week 54 value | 5.3 (8.7) | 8.9 (10.6) | N/A | N/A |
Change from baseline | −10.9 (11.6) | −9.5 (10.7) | N/A | N/A |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; JAK = Janus kinase; N/A = not applicable; RHI = Robarts Histopathology Index; SD = standard deviation; SE = standard error; UC = ulcerative colitis.
Note: For patients with dose adjustment, data collected before initiating dose adjustment for both treatment groups were included in this table.
Notes: None of the outcomes were controlled for multiplicity. Data for week 22 was not relevant in LIBERTY-UC because there was no switch of treatment done like Study 1.6.
Source: CSRs of LIBERTY-UC, and Study 1.6.a,c
Table 18: Secondary Efficacy Outcomes in CD Patients — Continuous Variables
End point | LIBERTY-CD | Study 1.6 | ||
|---|---|---|---|---|
REMSIMATM SC N = 232 | Placebo N = 112 | REMSIMATM SC N = 66 CD = 28 | Infliximab IV N = 65 CD = 25 | |
CDAI score, mean (SD) | ||||
Baseline, n | N/A | N/A | 28/ | 25/ |
Baseline value | N/A | N/A | 296.4 (59.2) | 294.8 (59.9) |
Week 30, n | N/A | N/A | 24 | 20 |
Week 30 value | N/A | N/A | 103.8 (88.4) | 106.4 (67.7) |
Change from baseline | N/A | N/A | −195.7 (100.7) | 187.1 (93.9) |
Week 54, n | N/A | N/A | 22 | 18 |
Week 54 value | N/A | N/A | 92.0 (77.6) | 79.0 (59.0) |
Change from baseline | N/A | N/A | −210.0 (104.7) | −211.0 (78.4) |
SES-CD Score (central), mean (SD) | ||||
Baseline, n | N/A | N/A | 21 | 16 |
Baseline value | N/A | N/A | 10.9 (8.3) | 8.1 (5.8) |
Week 22, n | N/A | N/A | 19 | 12 |
Week 22 value | N/A | N/A | 3.2 (2.4) | 4.6 (4.0) |
Change from baseline | N/A | N/A | −8.9 (7.6) | −4.4 (4.8) |
Week 54, n | N/A | N/A | 17 | 14 |
Week 54 value | N/A | N/A | 2.2 (2.4) | 2.7 (2.8) |
Change from baseline | N/A | N/A | −10.2 (8.8) | −6.4 (4.9) |
CD = Crohn Disease, CDAI = Crohn Disease Activity Index; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; JAK = Janus kinase; N/A = not applicable; SD = standard deviation; SE = standard error; SES-CD = simplified endoscopic activity score for Crohn disease.
Notes: For patients with dose adjustment, data collected before initiation of dose adjustment for both treatment groups were included in this table. None of the outcomes were controlled for multiplicity.
Source: CSRs of LIBERTY-CD, and Study 1.6.b,c
Results of supportive, sensitivity, and subgroup analyses
These analyses were performed for the key secondary efficacy end points in LIBERTY-UC and LIBERTY-CD. Supportive analyses based on PP analyses; sensitivity analyses utilizing Fisher’s exact test, and logistic regression model – all showed similar results as the primary analysis. Sensitivity analyses excluding war-affected patients in Ukraine, and excluding all patients in Ukraine showed no significant impact of these variables on the results (data not presented). Similarly, the results for the subgroup analysis of primary end point(s) based on sex, age and race showed that patient’s subgroup generally did not have major impact on the results, although no statistical analyses were done to compare the subgroups (data not presented).
Dose adjustment in patients with loss of response to REMSIMATM SC 120 mg
To evaluate the treatment effect in patients with dose adjustment from REMSIMATM SC 120 mg to 240 mg before Week 54, the (co)- primary end points and clinical/endoscopic response were provided for patients with dose adjustment in the REMSIMATM SC group. All patients who met the loss of response criteria adjusted their dose to REMSIMATM SC 240 mg in both LIBERTY-UC and LIBERTY-CD. As presented in Table 19, many patients who received dose adjustment in the REMSIMATM SC group improved in terms of clinical remission and endoscopic response, and also regained clinical response after dose adjustment in LIBERTY-UC and LIBERTY-CD.
Table 19: Analysis of (Co-)Primary End points and Regain of Clinical Response for Patients Who Met the Loss of Response Criteria with Dose Adjustment in LIBERTY-UC and LIBERTY-CD
End point at Week 54 | LIBERTY-UC | LIBERTY-CD | |||
|---|---|---|---|---|---|
Clinical Remission N = 81 | Regained Clinical Response N = 81 | Clinical Remission N = 39 | Endoscopic Response N = 39 | Regained Clinical Response N = 39 | |
Patients with dose adjustment, n (%) | 20 (24.7) | 40 (49.4) | 21 (53.8) | 11 (28.2) | 24 (61.5) |
Note: Patients who adjusted the dose were considered as non-remitter/non-responder for primary efficacy analyses in LIBERTY-UC and LIBERTY-CD, however, for these analyses on the treatment effect in patients with dose adjustment, remitter/responder are determined as per remission/response criteria regardless of dose adjustment.
Source: CSRs of LIBERTY-UC, and LIBERTY-CD.a,b,v
Compared to the first dose adjustment visits, patients who adjusted the dose from REMSIMATM SC 120 mg to 240 mg had almost two-fold, statistically significant, reduction in modified Mayo score in LIBERTY-UC and CDAI score in LIBERTY-CD at Week 54. Although the statistical comparison between the first dose adjustment visits and Week 54 results for SES-CD showed no statistical significance, approximately 20% reduction in mean SES-CD score was observed after dose adjustment in LIBERTY-CD (Table 20).
Table 20: Change in Modified Mayo Score, CDAI Score and SES-CD Score for Patients Who Met Loss of Response Criteria with Dose Adjustment in LIBERTY-UC and LIBERTY-CD
Study | End point | First Visit of Dose Adjustment | Week 54 | Mean Change | P valuea (ANCOVA) |
|---|---|---|---|---|---|
LIBERTY-UC | Modified Mayo | 5.8 (n = 38) | 3.4 (n = 38) | −2.3 | < 0.0001 |
LIBERTY-CD | CDAI | 256.37 (n = 28) | 105.43 (n = 28) | −150.9 | < 0.0001 |
SES-CD | 7.8 (n = 16) | 6.5 (n = 16) | −1.3 | 0.1 |
ANCOVA = analysis of covariance; CDAI = Crohn Disease Activity Index; SES-CD = simplified endoscopic activity score for Crohn disease.
Note: The patients with dose adjustment before Week 54 who had efficacy measurements at both Dose Adjustment Visit and Week 54 were included in this summary.
aP-value for difference in the mean of actual value between Dose Adjustment Visit and Week 54 visit within treatment group was obtained by paired t test.ss
Source: CSRs of LIBERTY-UC, and LIBERTY-CD.a,b,v
HRQoL/PRO end points
SIBDQ score: In LIBERTY-UC and LIBERTY-CD, SIBDQ scores were improved in both treatment groups up to Week 10 (data not presented, see CSRs for details). After randomization, SIBDQ score in the REMSIMATM SC group were well maintained, and higher than placebo up to Week 54, exceeding the MID level in both groups. Data are presented in Table 21 and Table 22. The difference in SIBDQ score between treatment arms were the highest at Week 22 when dose adjustment was allowed for patients who lost response (data not presented, see CSRs for details). Numerous patients in the placebo group in both trials required dose adjustment after losing response and therefore were excluded from descriptive summary from Week 30, explaining this finding.
In Study 1.6, The SIBDQ scores were generally similar between the SC and IV treatment arms up to Week 54, irrespective of UC or CD diagnosis. Similar levels of improvement were observed in both treatment arms at each time point up to Week 54, exceeding the MID levels, illustrating steady improvement in the HRQoL for both treatment arms. Data are presented in Table 21 and Table 22.
Patient Global Scale: In LIBERTY-CD, patients’ position on achieving remission from CD symptoms was evaluated based on PGS. A similar proportion of patients achieved remission based on PGS in both treatment groups in induction phase up to Week 10. After randomization at Week 10, a higher proportion of patients in the REMSIMATM SC arm reported achieving remission compared to those in the placebo arm up to Week 54. Data are presented in Table 22.
Local site pain assessment using VAS: In all 3 trials, the VAS score of local site pain was low at all time points, including the start of SC treatment at Week 6 (Study 1.6) or 10 (LIBERTY-UC and LIBERTY-CD), which was maintained up to Week 54.
Table 21: HRQOL Outcomes in UC Patients
Endpoint | LIBERTY-UC | Study 1.6 | ||
|---|---|---|---|---|
REMSIMATM SC N = 294 | Placebo N = 144 | REMSIMATM SC N = 38 UC | Infliximab IV N = 39 UC | |
SIBDQ score at baseline and change from baseline | ||||
Baseline, n | 294 | 144 | 38 | 39 |
Mean (SD) | 35.9 (11.3) | 36.0 (10.1) | 38.1 (9.9) | 34.8 (10.9) |
Week 30, n | Not relevant | Not relevant | 34 | 36 |
Mean (SD) | Not relevant | Not relevant | 54.4 (9.2) | 49.7 (12.0) |
Change from baseline, mean (SD) | Not relevant | Not relevant | 15.2 (11.9) | 15.0 (13.7) |
Week 54, n | 185 | 61 | 32 | 32 |
Mean (SD)/ LS mean (SE)a | 57.6 (10.1)/ 57.7 (1.4) | 54.5 (12.4)/ 54.9 (1.9) | 57.4 (8.9) | 56.9 (9.3) |
Treatment difference (95% CI)a | 2.9 (−0.3, 6.0) | Not reported | ||
P value | 0.08 | Not reported | ||
Change from baseline, mean (SD)/ LS mean (SE) | 21.4 (13.2)/ 21.9 (1.8) | 18.4 (14.4)/ 18.9 (2.37) | 18.3 (11.1) | 22.3 (12.8) |
Treatment difference (95% CI)a | 3.0 (−1.0, 6.9) | Not reported | ||
P value | 0.14 | Not reported | ||
Local site pain assessment using VAS – Safety population | ||||
Week 10 (LIBERTY-UC)/6 (Study 1.6), n | 296 | 140 | 66 | 65 |
Mean (SD) | 10.4 (13.7) | 6.7 (10.7) | 12.5 (17.4) | 6.7 (14.9) |
Week 54 (LIBERTY-UC, Study 1.6), n | 241 | 53 | 54 | 49 |
Mean (SD) | 8.9 (12.2) | 4.8 (8.8) | 10.3 (20.8) | 6.9 (10.7) |
ANCOVA = analysis of covariance; CI = confidence interval; IV = IV; JAK = Janus kinase; LS mean = least square mean; PGA = Patient Global Scale; SC = subcutaneous; SD = standard deviation; SIBDQ = Short Inflammatory Bowel Disease Questionnaire; SE = standard error; VAS = visual analogue scale.
Notes: The baseline value was considered to be the last non-missing value before the first administration. Local site pain assessment using VAS was reported for the safety population in both trials. Of note, safety population in Study 1.6 was not separated by UC and CD patients, therefore, data for local site pain assessment using VAS includes overall safety population. None of the outcomes were controlled for multiplicity. Week 30 values were not relevant for LIBERTY-UC, since there was no treatment switch at Week 30, which was the case in Study 1.6.
aFor the results after Week 10 randomization in LIBERTY-UC, an ANCOVA comparing the actual value/change from baseline between treatment group was conducted considering the treatment as fixed effect, previous exposure to biologic agent and/or JAK inhibitors (used or not used), use of treatment with oral corticosteroids at Week 0 (used or not used) and clinical remission at Week 10 (remitter or non-remitter by modified Mayo score) as covariates. The LS mean and corresponding SE for each treatment group, estimates of treatment difference, 2-sided 95% CI and p-value obtained from the ANCOVA were displayed. For patients with dose adjustment, data collected before initiation of dose adjustment for both treatment groups were included in this summary.
Source: CSRs of LIBERTY-UC, and Study 1.6.a,c
Patient Overall Satisfaction using VAS: In Study 1.6, most of the patients showed high level of satisfaction about procedure and duration of the study drug administration of both infliximab IV and REMSIMATM SC up to Week 54. The mean overall satisfaction values using the VAS score were above 88 in both the REMSIMATM SC and infliximab IV arm at Week 30, which remained high up to Week 54, over 90 in both treatment arms (data not presented, see CSRs for details).
Harms
Safety Evaluation Plan
The safety of IV infliximab (including the originator Remicade® as well as its biosimilars) has been evaluated in numerous clinical trials, assessed in rheumatoid arthritis (RA), ankylosing spondylitis (AS), psoriatic arthritis (PsA), psoriasis, UC, and CD; including several post-marketing controlled and uncontrolled studies. REMSIMATM SC has the same main ingredient, infliximab, that was a part of a clinical development program to select an optimal SC dose regimen(s) yielding similar efficacy and safety profile as the reference approved IV product. The REMSIMATM SC clinical development program was designed to support the indication of UC and CD and included 2 pivotal phase III studies (LIBERTY-UC and LIBERTY-CD) and 2 phase I dose-finding studies (Studies CT-P13 1.5 and CT-P13 1.6). In addition, the development program included a Phase I bioequivalence (BE) study (Study CT-P13 1.11). A total of 1,768 patients have been treated in 8 controlled, comparative, clinical studies during the development of REMSIMATM SC, listed below:
UC and CD patients:
LIBERTY-UC/Study CT-P13 3.7: Phase III, randomized, double-blind, placebo-controlled, parallel group study in which 436 patients with moderately to severely active UC were treated with Placebo SC or REMSIMATM SC via PFS.
LIBERTY-CD/Study CT-P13 3.8: Phase III, randomized, double-blind, placebo-controlled, parallel group study in which 343 patients with moderately to severely active CD were treated with Placebo SC or REMSIMATM SC via PFS.
Study CT-P13 1.6 Part 1 and 2: Phase I, OL, randomized, multi-dose, parallel-group study in which 175 patients with UC or CD (44 CD patients in Part 1 and 78 UC and 53 CD patients in Part 2) were treated with REMSIMATM SC via PFS or infliximab IV.
Table 22: HRQOL Outcomes in CD Patients
End point | LIBERTY-CD | Study 1.6 | ||
|---|---|---|---|---|
REMSIMATM SC N = 231 | Placebo N = 112 | REMSIMATM SC N = 28 CD | Infliximab IV N = 25 CD | |
SIBDQ score at baseline and change from baseline | ||||
Baseline, n | 231 | 111 | 28 | 25 |
Mean (SD) | 36.7 (11.2) | 37.2 (11.0) | 37.1 (12.7) | 37.8 (11.4) |
Week 30, n | Not relevant | Not relevant | 24 | 18 |
Mean (SD) | Not relevant | Not relevant | 53.6 (9.3) | 53.4 (12.0) |
Change from baseline, mean (SD) | Not relevant | Not relevant | 16.0 (13.8) | 15.3 (13.1) |
Week 54, n | 167 | 51 | 22 | 18 |
Mean (SD)/LS mean (SE)a | 56.0 (10.7)/54.7 (1.4) | 54.3 (14.5)/52.8 (2.0) | 53.6 (9.6) | 53.2 (12.5) |
Treatment difference (95% CI)a | 2.0 (−1.6, 5.6) | Not reported | ||
P value | 0.28 | Not reported | ||
Change from baseline, mean (SD)/ LS mean (SE) | 18.6 (13.9)/ 17.6 (1.9) | 16.0 (17.3)/ 15.1 (2.6) | 15.5 (15.3) | 16.3 (11.8) |
Treatment difference (95% CI)a | 2.6 (−2.1, 7.2) | Not reported | ||
P value | 0.28 | Not reported | ||
Summary of Patient Global Scale | ||||
Week 10, n (%) | ||||
Yes | 172 (74.5) | 90 (80.4) | Not reported | Not reported |
No | 59 (25.5) | 21 (18.8) | Not reported | Not reported |
Week 54, n (%) | ||||
Yes | 139 (60.2) | 40 (35.7) | Not reported | Not reported |
No | 28 (12.1) | 9 (8.0) | Not reported | Not reported |
Difference (95% CI)b | 26.2 (14.9, 36.5) | Not reported | Not reported | |
P value | < 0.0001 | Not reported | Not reported | |
Local site pain assessment using VAS – Safety population | ||||
Week 10 (LIBERTY-CD)/6 (Study 1.6), n | 238 | 104 | 66 | 65 |
Mean (SD) | 13.9 (17.4) | 8.3 (10.3) | 12.5 (17.4) | 6.7 (14.9) |
Week 54 (LIBERTY-CD, Study 1.6), n | 201 | 40 | 54 | 49 |
Mean (SD) | 10.8 (15.1) | 6.3 (7.5) | 10.3 (20.8) | 6.9 (10.7) |
ANCOVA = analysis of covariance; CI = confidence interval; JAK = Janus kinase; LS mean = least square mean; IV = IV; PGA = Patient Global Scale; SC = subcutaneous; SD = standard deviation; SIBDQ = Short Inflammatory Bowel Disease Questionnaire; SE = standard error; VAS = visual analogue scale.
Notes: The baseline value was considered to be the last non-missing value before the first administration. Local site pain assessment using VAS was reported for the safety population in both trials. Of note, safety population in Study 1.6 was not separated by UC and CD patients, therefore, data for local site pain assessment using VAS includes overall safety population. None of the outcomes were controlled for multiplicity. Week 30 values were not relevant for LIBERTY- CD, since there was no treatment switch at Week 30, which was the case in Study 1.6.
aFor the results after Week 10 randomization in LIBERTY-CD, an ANCOVA comparing the actual value/change from baseline between treatment group was conducted considering the treatment as fixed effect, previous exposure to biologic agent and/or JAK inhibitors (used or not used), use of treatment with oral corticosteroids at Week 0 (used or not used) and clinical remission at Week 10 (remitter or non-remitter by modified Mayo score) as covariates. The LS mean and corresponding SE for each treatment group, estimates of treatment difference, 2-sided 95% CI and p-value obtained from the ANCOVA were displayed. For patients with dose adjustment, data collected before initiation of dose adjustment for both treatment groups were included in this summary.
bFor the results after Week randomization, the difference of proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CI with CMH weights were presented with nominal P value (LIBERTY-CD). Analysis was stratified by previous exposure to biologic agent and/or JAK inhibitors (used or not used), use of treatment with oral corticosteroids at Week 0 (used or not used) and clinical remission at Week 10 (remitter or non-remitter by CDAI score) [LIBERTY-CD]. In LIBERTY-CD, data were collected before initiation of dose adjustment for treatment groups.
Source: CSRs of LIBERTY-CD, and Study 1.6.b,c
RA patients:
Study CT-P13 3.5 Part 1 and Part 2: Phase I/III, randomized, multi-dose, parallel-group study in which 391 patients with RA (48 patients in Part 1 and 343 patients in Part 2) were treated with REMSIMATM SC or infliximab IV.
Healthy patients:
Study CT-P13 1.5: Phase I, OL, dose-escalating, single-dose study in which 38 healthy subjects were treated with REMSIMATM SC via PFS or infliximab IV.
Study CT-P13 1.9: Phase I, OL, single-dose PK and safety study in which 215 healthy subjects were treated with REMSIMATM SC via AI or PFS.
Study CT-P13 1.10: Phase I, OL, single-dose PK and safety study in which 24 Japanese healthy subjects were treated with REMSIMATM SC via PFS.
Study CT-P13 1.11: Phase I, OL, single-dose PK and safety study in which 146 healthy subjects were treated with REMSIMATM SC via AI or PFS.
Overview of Safety
Safety data in the 3 trials were provided for the full treatment period (including induction/dose-loading and maintenance phase) as well as the maintenance phase separately. Data for the maintenance phase is primarily focused here, consistent with the focus of this review. Analyses were performed on the observed cases. Adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA) and graded for severity according to the Common Terminology Criteria for Adverse Events (CTCAE). Table 23 provides an overview of the safety results during the maintenance phase across the 3 studies. In addition to safety analyses performed at the individual study level, safety data were integrated across studies to further analyze the safety profile of REMSIMATM SC, summarized in Table 24. The integrated summary of safety is presented below, by 2 subpopulations of pooled dataset:
Pooled REMSIMATM SC dataset (N = 631):
Adverse events (AEs) reported on or after Week 10 from patients in the REMSIMATM 120 mg arms of LIBERTY-UC and LIBERTY-CD, excluding AEs reported after dose adjustment from 120 mg to 240 mg of REMSIMATM.
AEs reported on or after Week 6 from patients in the REMSIMATM 120 mg, 180 mg and 240 mg cohorts of Study CT-P13 1.6 Part 1.
AEs reported on or after Week 6 from patients in the REMSIMATM 120/240 mg arm of Study CT-P13 1.6 Part 2, excluding AEs reported after dose adjustment from 120 mg to 240 mg of REMSIMATM SC.
Pooled Placebo SC dataset (N = 245):
AEs reported on or after Week 10 from patients in the Placebo SC arms in LIBERTY-UC and LIBERTY-CD, excluding AEs reported after dose adjustment from placebo to 240 mg of REMSIMATM SC.
Overall, the safety data support the conclusion that REMSIMATM SC treatment was safe and well-tolerated in UC and CD populations. The safety profile of REMSIMATM SC compared favourably to infliximab IV through 30 weeks. There were no identifiable associations of CT-P13 SC treatment with laboratory, vital sign, or electrocardiogram (ECG) changes.
Table 23: Harms Data in Individual Trials
Adverse events | LIBERTY-UC | LIBERTY-CD | Study 1.6 | ||||
|---|---|---|---|---|---|---|---|
REMSIMATM SC N = 296 | Placebo N = 140 | REMSIMATM SC N = 238 | Placebo N = 105 | REMSIMATM SC N = 66 | Infliximab IV N = 65 | Post Week 30 N = 131a | |
Patients with at least 1 adverse event, n (%) | |||||||
TEAE | 200 (67.6) | 83 (59.3) | 172 (72.3) | 65 (61.9) | 49 (74.2) | 38 (58.5) | 52 (39.7) |
Most common events (≥ 5%) | |||||||
Colitis ulcerative | 20 (6.8) | 14 (10) | — | — | 3 (4.5) | 8 (12.3) | — |
Crohn disease | — | — | 15 (6.3) | 18 (17.1) | — | — | — |
Abdominal pain | — | — | — | — | 5 (7.6) | 1 (1.5) | 4 (3.1) |
Nausea | — | — | — | — | 5 (7.6) | 1 (1.5) | — |
Diarrhea | — | — | — | — | 4 (6.1) | 1 (1.5) | — |
Vomiting | — | — | — | — | 4 (6.1) | 0 | — |
COVID-19 | 30 (10.1) | 9 (6.4) | 27 (11.3) | 5 (4.8) | — | — | — |
Nasopharyngitis | 7 (2.4) | 7 (5) | — | — | 4 (6.1) | 2 (3.1) | — |
Headache | 17 (5.7) | 7 (5) | 18 (7.6) | 5 (4.8) | 4 (6.1) | 3 (4.6) | — |
Localized ISR | — | — | 14 (5.9) | 1 (1.0) | 15 (22.7) | 3 (4.6) | 9 (6.9) |
Anaemia | — | — | 13 (5.5) | 6 (5.7) | — | — | — |
Neutropenia | — | — | — | — | 5 (7.6) | 3 (4.6) | — |
Leukopenia | — | — | — | — | 4 (6.1) | 1 (1.5) | — |
Rash | — | — | — | — | 4 (6.1) | 4 (6.2) | — |
Arthralgia | — | — | — | — | 2 (3.0) | 4 (6.2) | — |
Patients with at least 1 serious adverse event, n (%) | |||||||
TESAE | 19 (6.4) | 4 (2.9) | 16 (6.7) | 8 (7.6) | 5 (7.6) | 7 (10.8) | 6 (4.6) |
Most common events (> 1%) by SOC | |||||||
GI disorders | 4 (1.4) | 2 (1.4) | 5 (2.1) | 2 (1.9) | — | — | — |
Infections and infestations | 7 (2.4) | 1 (0.7) | 6 (2.5) | 1 (1.0) | 2 (3.0) | 4 (6.2) | 6 (4.6) |
Patients who stopped treatment due to adverse events, n (%) | |||||||
WDAE | 10 (3.4) | 4 (2.9) | 9 (3.8) | 5 (4.8) | 1 (1.5) | 3 (4.6) | 1 (0.8) |
Most common events (≥ 1%) by SOC | |||||||
GI disorders | 2 (0.7) | 2 (1.4) | 0 | 1 (1.0) | — | — | — |
Infections and infestation | — | — | 0 | 2 (1.9) | 0 | 1 (1.5) | NR |
Injury, poisoning and procedural complication | — | — | — | — | 0 | 1 (1.5) | NR |
Investigations | — | — | 0 | 1 (1.0) | — | — | — |
Neoplasms benign, malignant and unspecified (including cysts and polyps) | — | — | — | — | 1 (1.5) | 0 | NR |
Skin and subcutaneous tissue disorders | — | — | 5 (2.1) | 1 (1.0) | 0 | 1 (1.5) | NR |
Patients with adverse events of special interest, n (%) | |||||||
SIR | 12 (4.1) | 4 (2.9) | 3 (1.3) | 1 (1.0) | 2 (3.0) | 0 | 1 (0.8) |
Localized ISR | 10 (3.4) | 3 (2.1) | 14 (5.9) | 1 (1.0) | 15 (22.7) | 3 (4.6) | 9 (6.9) |
Infection | 83 (28.0) | 36 (25.7) | 74 (31.1) | 19 (18.1) | 21 (31.8) | 19 (29.2) | 21 (16.0) |
Malignancy | 1 (0.3) | 0 | 0 | 1(1.0) | 1 (1.5) | 0 | 1 (0.8) |
IRR | 12 (4.1) | 4 (2.9) | 3 (1.3) | 1 (1.0) | 0 | 2 (3.1) | NR |
GI = gastrointestinal; ISR = injection site reaction; IRR = injection related reaction; NR = not reported; SC = subcutaneous; SIR = systemic injection reaction; SOC = System Organ Class; TEAE = treatment-emergent adverse event; TESAE = treatment-emergent serious adverse event; WDAE = withdrawal due to adverse event.
Note: The symbol – indicates the adverse event did not occur in frequency of at least 5% (TEAE) or sufficiently high, and should not be interpreted as unavailability of data or 0% incidence.
aPost Week 30 includes pooled safety results of the two treatment arms after switching to or continue with REMSIMATM SC at Week 30.
Source: CSRs of LIBERTY-UC, LIBERTY-CD, Study 1.6.a-c
Adverse Events
Overall, data showed no major or unexpected safety concerns. AEs were reported in higher proportion among patients in the REMSIMATM SC arm compared to placebo during the maintenance phase in LIBERTY-UC and LIBERTY-CD, although this may be a result of receiving placebo. Patients receiving REMSIMATM SC in Study 1.6 also reported AEs at a higher percentage, largely due to injection site reaction (ISR) which is an expected AE with SC injection. The most common AEs included a number of injection or infusion-related AEs, in addition to GI and neurologic AEs.
The majority of AEs were grade 1 or 2 in intensity (AEs data by grade not presented). Grade 4 AEs across trials included increase in blood creatine phosphokinase, COVID-19, duodenal ulcer, hypertriglyceridaemia, neutropenia, increased CD, peritonitis, psychotic disorders, appendicitis – none were considered by the investigators as related to study treatment.
In the pooled datasets, approximately two-thirds of the patients experienced at least 1 AE, slightly higher in patients receiving REMSIMATM SC. Among patients in the pooled REMSIMATM SC group, COVID-19, headache, and localized injection site reaction (ISR) were reported more than 5% of the patients. The proportion of patients with grade 3 or higher AEs were the same in patients receiving REMSIMATM SC and placebo (13.5%). Notably, there were no grade 3 or higher TEAEs that were reported in the pooled REMSIMATM SC group at an incidence rate 1% higher than the pooled Placebo SC group (data not presented for AEs by grade).
Table 24: Harms Data – Integrated Dataset
Adverse events | Pooled safety datasets | |
|---|---|---|
Pooled REMSIMATM SC dataset N = 631 | Pooled Placebo SC dataset N = 245 | |
Patients with at least 1 adverse event, n (%) | ||
TEAE | 422 (66.9) | 148 (60.4) |
Most common events (TEAEs Reported for at least 2% of patients in either group) | ||
Neutropenia | 16 (2.5) | 2 (0.8) |
Abdominal pain | 21 (3.3) | 6 (2.4) |
Diarrhea | 17 (2.7) | 3 (1.2) |
Nausea | 15 (2.4) | 3 (1.2) |
Injection site reaction | 39 (6.2) | 4 (1.6) |
COVID-19 | 49 (7.8) | 14 (5.7) |
Oral herpes | 13 (2.1) | 1 (0.4) |
Pharyngitis | 13 (2.1) | 0 |
Urinary tract infection | 13 (2.1) | 4 (1.6) |
Injection related reaction/infusion related reaction/administration related reaction | 16 (2.5) | 5 (2.0) |
Alanine aminotransferase increased | 19 (3.0) | 3 (1.2) |
Blood creatine phosphokinase increased | 19 (3.0) | 7 (2.9) |
Arthralgia | 23 (3.6) | 5 (2.0) |
Headache | 38 (6.0) | 12 (4.9) |
Hypertension | 16 (2.5) | 2 (0.8) |
Patients with at least 1 serious adverse event, n (%) | ||
TESAE | 40 (6.3) | 12 (4.9) |
Most common events (> 1%) by SOC | ||
Gastrointestinal disorders | 10 (1.6, 2.23) | 4 (1.6, 2.99) |
Infections and infestations | 16 (2.5, 3.56) | 2 (0.8, 1.49) |
Patients who stopped treatment due to adverse events, n (%) | ||
WDAE | 20 (3.2) | 9 (3.7) |
Patients with adverse events of special interest, n (%) | ||
Systemic injection reactions | 16 (2.5) | 5 (2.0) |
Localized injection site reaction | 39 (6.2) | 4 (1.6) |
Infection | 174 (27.6) | 55 (22.4) |
Malignancy | 1 (0.2) | 1 (0.4) |
TEAE = treatment-emergent adverse event; TESAE = treatment-emergent serious adverse event; WDAE = withdrawal due to adverse event.
Source: Submission package for Celltrion.v
Serious Adverse Events
Serious adverse events (SAEs) were reported by less than 10% of patients during the maintenance phase across trials and included primarily GI disorders and infections. The SAEs considered by the investigators as REMSIMATM SC or infliximab IV-related included grade 3 urinary tract infection (UTI) and pneumonia in LIBERTY-UC; grade 2 bacterial arthritis and grade 3 UTI in the same patients in LIBERTY-CD; and grade 3 non–small cell lung cancer (NSCLC), disseminated TB, pneumonia, and spontaneous abortion in Study 1.6. All of the conditions were treated with medications, including the patient with NSCLC whose condition was assigned as possibly related to the drug and received left upper lobectomy as primary treatment.
Only 1 death was reported in LIBERTY-CD during the maintenance phase, which was classified as an accidental death (garage explosion).
In the pooled datasets, a similar proportion of patients experienced SAEs. Aggravation of CD was the most common SAE among patients in the pooled REMSIMATM SC group.
Withdrawal Due to Adverse Events
Less than 5% patients across trials discontinued the study due to AEs, all of which were grade 2 or 3 in intensity. The most frequently reported AEs leading to study discontinuation in patients receiving infliximab (SC or IV) included Colitis ulcerative and increased ALT increased in LIBERTY-UC; hepatitis, hepatotoxicity and a number of dermatological conditions in LIBERTY-CD; disseminated TB, NSCLC (as described above) and psoriasis in Study 1.6.
In the pooled datasets, over 3% patients discontinued the respective studies due to AEs, regardless of treatment received. No AEs leading to study drug discontinuation were reported for > 2 patients in patients receiving REMSIMATM SC.
Adverse Events of Special Interest
Notable AEs included injection and infusion-related AEs, including systemic injection reaction (SIR), injection site reaction (ISR), and injection related reaction (IRR). These AEs were reported in < 5% of patients across trials. The SIR events related to the study treatment (assigned by investigators) were classified as grade 3 or lower but were non-serious. The localized ISRs were grade 1 or 2 in intensity and no serious localized ISRs were reported. The AEs of special interest were generally balanced between the treatment arms, except for ISR, which was reported more commonly in patients receiving SC infliximab compared to IV in Study 16, per expectation. Pooled datasets were consistent with the individual trials. Infections were reported in a similar proportion between treatment arms across trials, and most commonly included Covid-19, nasopharyngitis, UTI, pneumonia, oral herpes, latent tuberculosis, viral respiratory tract infection, bronchitis, and sinusitis.
In terms of malignancy, 1 case in each trial was reported. One patient in LIBERTY-UC experienced non-serious grade 3 prostate cancer, classified as unrelated to study treatment, and withdrew from the study due to AEs of prostate cancer. One patient in LIBERTY-CD in the placebo arm experienced an unrelated grade 3 colon cancer stage III, and although the diagnosis was done after initiating treatment, the biopsy was done before treatment. One patient in Study 1.6 experienced grade 3 NSCLC, which was classified as possibly related to study drug, but was resolved with left upper lobectomy.
Bioequivalence (If Applicable)
The primary objective of Study 1.6 was to demonstrate that REMSIMATM SC was noninferior to infliximab IV in terms of PK, as determined by the observed Ctrough,week22, calculated from the pre-dose level at Week 22. Findings from the PK part of the study are briefly described here.
The mean (CV%) observed Ctrough,week22 was higher in the REMSIMATM SC 120/240 mg treatment arm than the infliximab IV 5 mg/kg treatment arm at Week 22 (21.5 [46.0] and 2.9 [89.0] mcg/mL, respectively). The geometric LS mean of observed Ctrough,week22 was 20.9 and 1.8 mcg/mL in the SC 120/240 mg and IV 5 mg/kg treatment arms, respectively. The ratio of geometric LS means was 1154.2 with lower bound 90% CI of 786.4%, which was greater than 80%, indicating that REMSIMATM SC was indeed noninferior to infliximab IV in terms of PK (noninferior margin 80%). Among the PK parameters, Ctrough has shown to be predictive of therapeutic effect based on a systematic review of available literature where a Ctrough threshold of 5 mcg/mL was considered to be the minimum serum concentration level needed to achieve and maintain improved clinical outcomes, including clinical remission and mucosal healing.w,x The mean predicted Ctrough levels in the SC 120/240 mg arm were substantially greater (above 18 mcg/mL) than the target exposure (5 mcg/mL) throughout the maintenance phase. Even after switching to REMSIMATM SC 120/240 mg at Week 30 in the IV 5 mg/kg treatment arm, the observed Ctrough immediately went above the target threshold within 4 doses of REMSIMATM SC treatment and continued to increase up to level similar to the SC 120/240 mg treatment arm until Week 54, which ensured maintenance of clinical efficacy.
In terms of other PK outcomes, the route of study drug administration impacted to the output of the PK parameters, such as predicted AUCτ, predicted AUCss8w, predicted Cmax,ss, and predicted Tmax,ss. This was mainly because the nature of PK characteristics differed between SC and IV; the absorption of biotherapeutics by SC route is relatively slow due to slow drug transport through the extracellular matrix before reaching the systemic circulation, and mostly incomplete, which results in reduced bioavailability (Richter and Jacobsen, 2014). Furthermore, REMSIMATM SC was administered more frequently (every 2 weeks) at lower dosage per injections compared to infliximab IV (every 8 weeks), which may have led to prolonged and slightly higher drug exposure in REMSIMATM SC.
Overall, the REMSIMATM SC injections at a 2-week interval resulted in the difference in the PK profile compared to the 8 weeks interval of the infliximab IV infusions. The mean infliximab trough level of the REMSIMATM SC 120/240 mg treatment arm was well maintained above the target exposure level of 5 mcg/mL up to Week 54 as well as the IV 5 mg/kg treatment arm after Week 30 up to Week 54 following REMSIMATM SC biweekly injections starting at Week 30.
Sponsor References
a. CELLTRION I. WEEK 54 CLINICAL STUDY REPORT A Randomized, Placebo-Controlled, Double Blind, Phase 3 Study to Evaluate the Efficacy and Safety of the Subcutaneous Injection of CT P13 (CT P13 SC) as Maintenance Therapy in Patients with Moderately to Severely Active Ulcerative Colitis [Confidential internal sponsor's report]. 01 December 2022.
b. CELLTRION I. WEEK 54 CLINICAL STUDY REPORT A Randomized, Placebo-Controlled, Double Blind, Phase 3 Study to Evaluate the Efficacy and Safety of the Subcutaneous Injection of CT P13 (CT P13 SC) as Maintenance Therapy in Patients with Moderately to Severely Active Crohn disease [Confidential internal sponsor's report]. 23 November 2022.
c. CELLTRION I. CLINICAL STUDY REPORT (PART 2) An Open-label, Randomized, Parallel-Group, Phase I Study to Evaluate Pharmacokinetics, Efficacy and Safety between Subcutaneous CT-P13 and IV CT-P13 in Patients with Active Crohn Disease and Active Ulcerative Colitis. 31 March 2020.
d. Lewis JD, Chuai S, Nessel L, Lichtenstein GR, Aberra FN, Ellenberg JH. Use of the noninvasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm Bowel Dis. 2008;14(12):1660 to 1666.
e. Samaan MA, Mosli MH, Sandborn WJ, et al. A systematic review of the measurement of endoscopic healing in ulcerative colitis clinical trials: recommendations and implications for future research. Inflamm Bowel Dis. 2014;20(8):1465 to 1471.
f. Walsh AJ, Ghosh A, Brain AO, et al. Comparing disease activity indices in ulcerative colitis. J Crohns Colitis. 2014;8(4):318 to 325.
g. Dhanda AD, Creed TJ, Greenwood R, Sands BE, Probert CS. Can endoscopy be avoided in the assessment of ulcerative colitis in clinical trials? Inflamm Bowel Dis. 2012;18(11):2056 to 2062.
h. US FDA. Center for Drug Evaluation and Research. Guidance for Industry. Ulcerative Colitis: Developing Drugs for Treatment. 2022.
i. Higgins PD, Schwartz M, Mapili J, Zimmermann EM. Is endoscopy necessary for the measurement of disease activity in ulcerative colitis? Am J Gastroenterol. 2005;100(2):355 to 361.
j. Travis SP, Stange EF, Lémann M, et al. European evidence-based Consensus on the management of ulcerative colitis: Current management. J Crohns Colitis. 2008;2(1):24 to 62.
k. US FDA. Rockville (MD): Center for Drug Evaluation and Research. Ulcerative colitis: clinical trial end points guidance for industry: draft guidance. 2016.
l. Rubin D, Keyashian K, Bunnag A, et al. Correlation Between Clinical, Endoscopic, and Histologic Disease Activity in Ulcerative Colitis: 1712. Official journal of the American College of Gastroenterology | ACG. 2012;107:S694.
m. Mohammed Vashist N, Samaan M, Mosli MH, et al. Endoscopic scoring indices for evaluation of disease activity in ulcerative colitis. Cochrane Database Syst Rev. 2018;1(1):Cd011450.
n. Sandborn WJ, Feagan BG, Hanauer SB, et al. A review of activity indices and efficacy end points for clinical trials of medical therapy in adults with Crohn disease. Gastroenterology. 2002;122(2):512 to 530.
o. Yoshida EM. The Crohn Disease Activity Index, its derivatives and the Inflammatory Bowel Disease Questionnaire: a review of instruments to assess Crohn disease. Can J Gastroenterol. 1999;13(1):65 to 73.
p. Best WR, Becktel JM, Singleton JW, Kern F, Jr. Development of a Crohn disease activity index. National Cooperative Crohn Disease Study. Gastroenterology. 1976;70(3):439 to 444.
q. Khanna R, Zou G, D'Haens G, et al. A retrospective analysis: the development of patient-reported outcome measures for the assessment of Crohn disease activity. Aliment Pharmacol Ther. 2015;41(1):77 to 86.
r. Daperno M, D'Haens G, Van Assche G, et al. Development and validation of a new, simplified endoscopic activity score for Crohn disease: the SES-CD. Gastrointest Endosc. 2004;60(4):505 to 512.
s. Zheng X, Li M, Wu Y, et al. Assessment of pediatric Crohn disease activity: validation of the magnetic resonance enterography global score (MEGS) against endoscopic activity score (SES-CD). Abdom Radiol (NY). 2020;45(11):3653 to 3661.
t. Khanna R, Zou G, D'Haens G, et al. Reliability among central readers in the evaluation of endoscopic findings from patients with Crohn disease. Gut. 2016;65(7):1119 to 1125.
u. Lichtiger S, Binion DG, Wolf DC, et al. The CHOICE trial: adalimumab demonstrates safety, fistula healing, improved quality of life and increased work productivity in patients with Crohn disease who failed prior infliximab therapy. Aliment Pharmacol Ther. 2010;32(10):1228 to 1239.
v. CELLTRION I. Tailored Review submission: Subcutaneous Injection of CT-P13 (CT-P13 SC) Maintenance Therapy in Patients with Moderately to Severely Active Ulcerative Colitis and Crohn disease [CONFIDENTIAL sponsor's submission]. October 2023.
w. Papamichael K, Gils A, Rutgeerts P, et al. Role for therapeutic drug monitoring during induction therapy with TNF antagonists in IBD: evolution in the definition and management of primary nonresponse. Inflamm Bowel Dis. 2015;21(1):182 to 197.
x. Vaughn BP, Sandborn WJ, Cheifetz AS. Biologic concentration testing in inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(6):1435 to 1442.
y. Health Canada. Abbvie Corporation. Product Monograph HUMIRA (adalimumab). 2019 [cited Sept. 20, 2023] Available from https://pdf.hres.ca/dpd_pm/00052133.PDF.
z. Health Canada. Janssen Inc. Product Monograph SIMPONI (golimumab injection). 2019 [cited Sept. 20, 2023] Available from https://pdf.hres.ca/dpd_pm/00052895.PDF.
aa. Health Canada. Celltrion Health care Co. LTD Product Monograph INFLECTRA (infliximab for injection). 2020 [cited Sept. 20, 2023] Available from https://pdf.hres.ca/dpd_pm/00057004.PDF
bb. Health Canada. Takeda Canada Inc. Product Monograph ENTYVIO (vedolizumab) 2022 [cited Sept 20, 2023] Available from https://pdf.hres.ca/dpd_pm/00066565.PDF
cc. Ontario Ministry of Health and Long-term Care. Ontario Drug Benefit Formulary/Comparative Drug Index. 2020 [cited Sept. 20, 2023] Available from https://www.formulary.health.gov.on.ca/formulary/.
dd. Ontario Ministry of Health and Long-term Care. Ontario Drug Benefit EAP Drug Pricing. 2023 [cited Sept. 20, 2023] https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
ee. Health Canada. Janssen Inc. Product Monograph STALARA (ustekinumab) 2018. [cited Sept 20, 2023] Available from https://pdf.hres.ca/dpd_pm/00048246.PDF
ff. CADTH (CADTH). CDR Pharmacoeconomic Review Report for Stelara. 2017 [cited Sept. 20, 2023] Available from https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0501_Stelara_PE_Report.pdf
CADTH’s Critical Appraisal of the Clinical Evidence
CADTH conducted a critical appraisal of the clinical studies for infliximab injection (Remsima SC) based on the summary of the evidence provided by the sponsor.
Internal Validity
LIBERTY-UC and LIBERTY-CD Trials
The LIBERTY-UC and LIBERTY-CD trials were randomized, placebo-controlled, phase III studies designed with an OL induction phase, a DB treatment (maintenance) phase, and an OL extension phase. Both trials employed appropriate methods for blinding, treatment allocation, and randomization. Randomization was implemented in a 2 to 1 ratio using an IWRS and stratified by 3 important treatment modifiers using a permuted block design. Stratification factors in both trials were identical. A concealed treatment allocation was ensured. Baseline characteristics in both trials were generally well-balanced between the placebo and infliximab SC groups, indicating successful randomization.
The outcomes assessed in both trials (CDAI scores, Mayo scores, patient-reported outcomes, and safety outcomes) were subjective and potentially prone to assessment bias. Mayo and CDAI scores are partially derived from accurate reporting by patients of events related to their disease symptoms through the use of electronic diaries and endoscopic assessments. Only endoscopic assessments (i.e., ESs for the Mayo score RHI) were performed centrally by an independent reviewer blinded to treatment assignment; thus, the risk of assessment bias was considered low for endoscopic outcomes. Daily reporting of events related to UC and CD in diaries may be prone to user error. Reporting of events by patients may be further affected if patients (despite double blinding) became aware of treatment allocation — for example, due to developing progressive disease in the maintenance phase — and may influence discontinuation rates. Discontinuation rates in the maintenance phase were greater than 15% across groups in both trials, but slightly higher in the placebo group compared to the infliximab SC group (LIBERTY-CD: 23.3% in placebo versus 15.2% in infliximab SC, respectively; LIBERTY-UC: 21.5% in placebo versus 18.4% in infliximab SC, respectively). The dropout rates in the infliximab SC group were considered high compared to rates for the infliximab IV group in clinical practice setting. The direction and potential magnitude of this bias are uncertain.
The primary and coprimary outcomes in the LIBERTY-UC and LIBERTY-CD trials, respectively, were considered appropriate and are recommended by the FDA29,30 and EMA73,74 for assessing treatment effects for patients with UC or CD in the trial settings. Endoscopic remission, mucosal healing, and endoscopic response were considered objective measures by the clinical expert consulted by CADTH and by regulatory guidelines (FDA29,30 and EMA73,74); these are also considered important in managing moderately to severely active UC or CD. The statistical analysis methods implemented for the primary and secondary outcomes in the 2 trials were considered appropriate by the CADTH review team. Both studies were powered to detect a statistically significant difference between the 2 groups. Methods for the sensitivity analyses (which included excluding war-affected patients in Ukraine, excluding all patients in Ukraine, and using tipping point analysis) were considered appropriate, and results were consistent with the primary analysis in both trials. The methods utilized to assess missing data were valid and appropriate. Subgroup analyses were predefined (sex, age, and race) and conducted for the primary end points in both trials using the all-randomized population; however, the subgroup analyses were likely underpowered to identify subgroup differences and should be considered supportive only.
HRQoL was an important outcome highlighted by the patient advocacy group and clinician expert consulted by CADTH during the review. There was a concern for potential bias due to missing outcome data for the HRQoL results, especially in the placebo groups in both trials at week 54, rendering the results inconclusive. Fewer patients in the placebo group contributed to the analyses at week 54 compared to the infliximab group. (In the LIBERTY-UC trial, 185 of 294 patients [62.9%] in the infliximab SC group reported patient-reported outcomes at week 54 versus 61 of 144 patients [42.3%] in the placebo group. In the LIBERTY-CD trial, 167 of 231 patients [72.3%] in the infliximab SC group reported outcomes for SIBDQ at week 54, while in the placebo group, 51 of 111 patients [45.9%] reported outcomes.)
Concomitant drug use in the maintenance phase was overall comparable between the 2 groups for both trials, with the exception of notable differences in budesonide use in the LIBERTY-CD trial (Table 32 and Table 33). Corticosteroid use was maintained at the same dose during the induction phase in both trials if the patient had received a stable dose for at least 2 weeks before the first administration of the study drug (day 0), and but was tapered in the maintenance phase after week 10 at a rate of 2.5 mg/week if the dose was equal to or less than 10 mg/day (to a maximum rate of 5 mg/week, if the current corticosteroid dose was greater than 10 mg/day). A stable dose of immunomodulators was also allowed throughout week 54, which aligns with clinical practice. All patients, including those who continued to use corticosteroids in the maintenance phase, were included in the primary analyses. The continued use of corticosteroids in the maintenance phase may affect inflammatory symptom improvement and the assessment of subjective outcomes in both groups in both trials due to residual effects of the drug. (For example, patients on placebo in both trials performed well, which could be because of continued corticosteroid use in addition to the use of other immunomodulators allowed during the treatment period.) Of note, the percentage of patients who used budesonide during the maintenance phase was significantly higher in the infliximab SC group (18.5%) than in the placebo group (8.6%) in the LIBERTY-CD trial. This could have biased the efficacy results in favour of infliximab SC in the CD population. The potential magnitude of this bias is uncertain. There were also imbalances in study treatment exposures between the 2 groups in both trials, given that more dose adjustments were observed in the placebo group from week 22 than in the infliximab SC group (Table 34 and Table 35). Although dose augmentations (up to 2 injections; i.e., 240 mg infliximab SC) were allowed in the trial, frequent dose adjustments in the maintenance phase could lead to treatment awareness within groups and affect the assessments of subjective outcomes that depended on the reporting of events in patient diaries. The direction and magnitude of this potential bias are uncertain. (In the LIBERTY-UC trial, the proportions of patients with dose adjustments in the 2 groups in the maintenance phase were n = 92 [31.1%] in the infliximab SC group and n = 75 [53.6%] in the placebo group; in the LIBERTY-CD trial, the proportions of patients with dose adjustments in the 2 groups were n = 45 [18.9%] in the infliximab SC group and n = 48 [45.7%] in the placebo group.)
Study 1.6, Part 2
Study 1.6, part 2 is an OL, randomized, parallel-group, phase I study. Appropriate methods for randomization and treatment allocation were implemented. Randomization was performed in a 1 to 1 ratio using an IWRS and stratified using important blocking factors. Baseline characteristics were similar between the 2 treatment groups in the trial, suggesting successful randomization.
The key objective was to assess the noninferiority of infliximab SC versus infliximab IV for the PK parameter, Ctrough, week22. The assessment of plasma concentration of infliximab (Ctrough at week 22) was considered appropriate by the clinical expert consulted by CADTH and aligns with regulatory guideline requirements32 and published literature.33,34 The clinical expert noted that by week 22, it is expected that infliximab concentrations (IV or SC) should have reached plasma steady state, minimizing the risk of low drug plasma levels when patients move to the SC formulation. A noninferiority margin of 80%, 1-sided alpha level of 5%, expected ratio of 1.3, and coefficient of variation of 100% were assumed for part 1 of the study along with a 20% dropout rate. The study was powered to detect a statistical difference between the 2 groups of interest for the PK outcome. The noninferiority of infliximab SC to infliximab IV was concluded if the lower bound of the 2-sided 90% CI for the ratio of geometric LS means was higher than 80%. The sponsor did not provide any justification for using a noninferiority margin of 80% for the trial. The clinical expert consulted by CADTH agreed that the margin was appropriate (i.e., falling within the range of the biosimilar study conducted to support the reimbursement request for infliximab IV [−15% to 15%])55 and does not affect the interpretability of the findings. Findings from the PK study showed a ratio of geometric LS means of 1,154.2, with a lower-bound 90% CI of 786.4%, which was greater than 80%, indicating that infliximab SC was noninferior to infliximab IV in terms of PK (noninferior margin = 80%). The Ctrough threshold of 5 mcg/mL used in the bioequivalence study was considered the minimum serum concentration level needed to achieve and maintain improved clinical outcomes, according to published literature.33,34 Findings from Study 1.6 showed mean predicted Ctrough levels higher than 18 mcg/mL in the infliximab SC group throughout the maintenance phase, which is beyond the target exposure (5 mcg/mL). Even after the switch to infliximab SC 120 mg or 240 mg at week 30 in the IV 5 mg/kg treatment arm, the observed Ctrough reported was higher than the target threshold within 4 doses of infliximab SC 120 mg or 240 mg of treatment and continued to increase, reaching a level similar to that of the infliximab SC (120 mg or 240 mg) treatment arm until week 54.
Although the evidence from Study 1.6 suggests that infliximab SC is comparable to infliximab IV in terms of PK parameters, the lack of robust evidence on efficacy outcomes (which were presented descriptively, without statistical comparison) precludes firm conclusions to support switching from infliximab IV to infliximab SC. Study 1.6 was not designed or powered to formally assess comparative efficacy outcomes (i.e., CDAI response, clinical response, clinical remission, endoscopic response and remission, mucosal healing, or HRQoL), making assessments of the relative therapeutic efficacy of infliximab SC challenging. The sample size of Study 1.6 (i.e., n = 135) was relatively small. The treatment effect estimates observed in a small study sample may not be replicable in a larger study sample. The protocol did not prespecify a degree of difference from which to formally conclude noninferiority or similarity between infliximab SC and infliximab IV in terms of efficacy outcomes. The results for the key secondary end points assessed in Study 1.6 were presented descriptively, without a statistical comparison provided before or after the infliximab IV group switched to the infliximab SC treatment. The clinical expert consulted by CADTH did not anticipate clinically meaningful differences in efficacy between infliximab SC and infliximab IV due the products’ same active ingredient (i.e., infliximab).
There were concerns related to the risk of assessment bias for key secondary outcomes (CDAI scores, Mayo scores, SES-CD scores, SIBDQ scores, VAS, and safety outcomes) due to the subjective nature of these outcomes and the OL trial design. The impact of assessment bias for subjective outcomes may favour infliximab SC over the IV drug because the SC formulation is more convenient for patients. The potential magnitude of this bias is uncertain. Analyses of the primary outcome (PK) were reviewed and evaluated by an independent data committee, minimizing assessment and performance bias. There were concerns related to missing data between the 2 study groups for HRQoL data assessed using the SIBDQ and VAS (for local site pain assessments). Fewer patients with CD or UC completed questionnaires at week 30 and week 54 compared to baseline (Table 21 and Table 22); this may have affected the analyses presented. In addition, there were no formal statistical tests for significance conducted for efficacy outcomes, and missing data were not accounted for in the analyses. Therefore, it is uncertain whether switching patients from infliximab IV to infliximab SC at week 30 in Study 1.6 resulted in comparable HRQoL outcomes for patient populations with UC and CD, respectively.
External Validity
LIBERTY-UC, LIBERTY-CD, and Study 1.6, part 2 were multicentre, international trials that recruited adult patients aged 18 years to 75 years; Study 1.6 included patients with UC and patients with CD with moderately to severely active disease. The inclusion and exclusion criteria of the 3 trials were generally aligned with the selection criteria for patients in current practice, except for the exclusion of patients who had previously failed 2 or more lines of biologic drugs or JAK inhibitors, which was inconsistent with practice. According to the expert consulted by CADTH, it is not uncommon for patients with prior exposure to other advanced therapies, including JAK inhibitors, to be considered for infliximab. Baseline CDAI scores (for patients with CD), Mayo scores, the proportion of patients with moderate to severe disease, and types of prior surgeries conducted were presented for the 3 trials, including other important objective outcomes (such as CRP and fecal calprotectin) that are recommended for monitoring patients in practice29,30,73,74 (Table 27 and Table 28). There were no major differences in baseline characteristics between the infliximab SC and placebo groups in the LIBERTY-CD and LIBERTY-UC trials.
The use of an induction and maintenance phase across 3 trials is consistent with regulatory guidelines for trials in UC and CD populations.29-31 The design allows for the evaluation of whether response to a loading (induction) dose of 5 mg/kg infliximab IV is maintained in patients in the absence or presence of the continued use of infliximab SC. Although the trial design reflects clinical practice, it generates an enriched population consisting of responders who can better tolerate and respond well to infliximab. The short induction periods of the 3 trials (i.e., 6 weeks for the LIBERTY-UC and LIBERTY-CD trials and 4 weeks for Study 1.6) did not take into consideration delayed responders, and included only patients who were able to achieve a timely response to infliximab, as reflected by the number of nonresponders reported in the induction phases (i.e., n = 65 patients [11.9%] in the LIBERTY-UC trial and n = 22 patients [5.5%] in the LIBERTY-CD trial). The clinical expert consulted by CADTH noted that, in clinical practice, the dose-loading phase for infliximab 5 mg/kg IV may extend up to 16 weeks to accommodate slow responders.
Both trials (LIBERTY-UC and LIBERTY-CD) were placebo-controlled. Placebo-controlled trials are recommended by regulatory guidelines to critically assess the assay sensitivity of new treatments for CD and UC (unless a study aims to demonstrate superiority to an existing treatment);73,74 however, the lack of direct evidence from head-to-head trials of infliximab SC against other available treatments in Canada presents a challenge for assessing comparative effectiveness. Study 1.6 was an active-controlled trial, providing an evaluation of infliximab SC treatment against infliximab IV. The comparator used in Study 1.6 was appropriate, given that infliximab IV is currently used in clinical practice (infliximab SC is composed of the same active ingredient as infliximab IV). Infliximab SC is intended to provide a treatment option that could be self-administered by patients without the need for frequent or lengthy visits to infusion clinics.
Overall, the primary and key secondary outcomes of the LIBERTY trials were consistent with prior trials conducted in the CD and UC setting and were considered relevant to decision-making in clinical practice by the clinical expert consulted by CADTH. The CDAI is recommended by the FDA,29,30 EMA,73,74 and guidelines,26,75 and has been used in historical pivotal trials with infliximab IV in the IBD setting.76 However, the clinical expert consulted by CADTH noted that it is cumbersome to derive and seldom used in clinical practice; the HBI is a more user-friendly tool for clinicians and is used in place of CDAI scores across Canada to assess clinical remission in patients with CD. Endoscopic assessment (which assesses mucosal healing through tissue biopsies) was highlighted by the clinical expert consulted by CADTH as an important objective outcome in assessing treatment efficacy in both patients with UC and patients with CD and is usually used in combination with the modified Mayo score and HBI.
The primary outcome of Study 1.6 was considered appropriate and consistent with PK parameters to show bioequivalence, according to the clinical expert consulted by CADTH. The subgroups (age, sex, and race) predefined in the LIBERTY-UC and LIBERTY-CD trials were considered relevant; however, other important subgroups highlighted by the clinical expert, such as biologic drug–naive patients versus biologic drug–exposed patients, were not investigated. There were no subgroups designed in Study 1.6.
Concomitant medications (such as immunomodulators, oral corticosteroids [prednisone, budesonide], and antibiotics) approved in the 3 studies were consistent with clinical practice, except for a few drugs, such as mesalamine, which is not commonly used in Canada. Corticosteroid tapering was allowed at a rate of 2.5 mg/week for patients up to a maximum rate of 5 mg/week in all 3 trials. This is consistent with regulatory guidelines and current practice; however, according to the expert, tapering rates for corticosteroids may lean toward 5 mg/week as opposed to the schedule implemented in the trials.
The dosing of infliximab IV (5 mg/kg) in the induction phases of the LIBERTY-UC and LIBERTY-CD trials was consistent with the product monograph. The clinical expert by CADTH noted that clinicians may consider higher doses of infliximab IV for patients with more severe disease during the induction or dose-loading phase; these will typically be further adjusted based on patient response, patient preference, and safety profile. A weight-based dosing schedule was implemented in Study 1.6 for patients receiving infliximab SC and infliximab 5 mg/kg IV. The dosing format for infliximab SC for this study did not align with the dosing recommendations in the product monograph and differed from the procedures implemented in the LIBERTY trials (for both UC and CD). Dose escalation was allowed for patients receiving 120 mg infliximab SC if they initially responded, but then stopped responding at or after week 30. In addition, patients received only 2 doses during the induction phase rather than the 3 doses recommended by Health Canada. There is some uncertainty as to whether the results of Study 1.6 are generalizable to the use of infliximab SC as per the Health Canda–recommended dosage.
The durations of the maintenance phases were considered adequate to assess the treatment effects in both populations in the LIBERTY-UC and LIBERTY-CD trials, in addition to patient exposure in the ongoing extension phase (up to week 102). The trial assessments (endoscopy assessments) were considered standard for trials in this setting, according to regulatory guidelines,73,74 but differed from current practice due to logistics and patient preference, according to the clinical expert consulted by CADTH. Endoscopy assessments are typically performed every 6 months to 8 months, due to logistical constraints and the invasiveness of the procedure. For patients with CD or UC, the clinical expert consulted by CADTH noted that treatment response will usually be assessed after induction (at week 16) and during maintenance therapy at 1 year. Clinicians will then assess fecal calprotectin and CRP every 6 months; HBI scores are derived every 3 months to 4 months.
Long-Term Extension Studies
No long-term extension studies were submitted for this review.
Indirect Evidence
No ITC was submitted for this review.
Sponsor-Submitted Cost Information
Infliximab SC (Remsima SC) is a SEB product with a new mode of administration (SC injection). The sponsor submitted a cost comparison of treatments available for CD and UC in which infliximab SC was compared with other infliximab products and with other biologic DMARDs.77 Only drug acquisition costs were considered, under the assumption that the administration costs of IV infusion would be funded by the respective manufacturers rather than by public plans. Costs were reported for both the initial year, including induction with the IV infliximab product, Inflectra, and years thereafter. The submitted price of infliximab SC is $591.60 per PFS or prefilled pen.77
Table 25: Sponsor's Drug Acquisition Cost Comparison for CD and UC
Generic name (brand name) | Strength | Dose form | Price ($) | Recommended dosage regimen | Annual drug cost ($) | Difference in annual cost (savings) ($)a |
|---|---|---|---|---|---|---|
Infliximab (Remsima SC) | 120 mg (in 1 mL) | Prefilled syringe or prefilled pen | 591.6000b | Infliximab-naive: initiated as maintenance therapy 4 weeks after the last administration of 3 IV infusions of infliximab 5 mg/kg given at weeks 0, 2, and 6. The recommended dosage is 120 mg once every 2 weeks. Switching from IV infliximab maintenance: administered 8 weeks after the last administration of the IV infusions of infliximab. | Inflectra induction, year 1: 19,357c Afterward: 15,424d | Reference |
Comparators (biologic DMARDs, infliximab) | ||||||
Infliximab (Remicade) | 100 mg | Vial | 987.5600c | 5 mg/kg at weeks 0, 2, and 6, then every 8 weeks thereafter | Year 1: 34,635 After: 25,747 | (15,278) (9,323) |
Infliximab (Avsola) | 493.0000 | Year 1: 17,290 After: 12,853 | 2,067 2,571 | |||
Infliximab (Inflectra) | 525.0000 | Year 1: 18,413 After: 13,688 | 944 1,736 | |||
Infliximab (Renflexis) | 493.0000 | Year 1: 17,290 After: 12,853 | 2,067 2,571 | |||
Comparators (biologic DMARDs, other anti-TNF alpha drugs) | ||||||
Adalimumab SC (Humira) | 40 mg/0.8 mL | Prefilled syringe or pen | 794.1000 | 160 mg at week 0, 80 mg at week 2, then 40 mg every other week thereafter | Year 1: 23,879 After: 20,703 | (4,522) (5,279) |
Adalimumab (biosimilar) | 471.2700 | Year 1: 14,172 After: 12,287 | 5,185 3,137 | |||
Golimumab SC (Simponi)c | 50 mg/0.5 mL | Prefilled syringe or autoinjector | 1,555.1700c | 200 mg at week 0, 100 mg at week 2, then 50 mg every 4 weeks thereafter | Year 1: 22,735 After: 18,662 | (3,378) |
100 mg/mL | Year 1: 21,062 After: 20,284 | (1,705) (4,860) | ||||
Vedolizumab (Entyvio) IV | 300 mg | Vial with prefilled syringe | 3,401.8600c | 300 mg at weeks 0 and 2, then every 8 weeks thereafter | Year 1: 29,827 After: 22,173 | (10,470) (6,749) |
Vedolizumab (Entyvio) SC | ||||||
Ustekinumab (Stelara)e | 130 mg/26 mL 45 mg/ 0.5mL 90 mg/1 mL | Vial for IV induction Single-use prefilled syringe for SC maintenance | 2,080.0000c 4,593.1400 | 6 mg/kg IV at week 0, then 90 mg SC every 8 weeks thereafter | Year 1: 35,603 After: 29,937 | (16,246) (14,513) |
CD = Crohn disease; DMARD = disease-modifying antirheumatic drug; SC = subcutaneous; UC = ulcerative colitis.
Note: All prices are from the Ontario Drug Benefit Formulary78 (accessed November 2023) unless otherwise indicated and do not include dispensing fees. Costs are based on 365 days per year, using the maintenance dosage where applicable. All weight-based doses assume an average patient weight of 73.2 kg and wastage of excess medication in vials.
aTop row costs represent year 1 Remsima SC costs (i.e., the year 1 comparator cost); bottom row costs represent the subsequent “after” infliximab Remsima SC cost (i.e., the subsequent “after” comparator cost). Paratheses indicate that infliximab SC is less costly than the comparator.
bSponsor-submitted price.
cOntario Exceptional Access Program79 (accessed November 2023).
dGolimumab SC is applicable to UC only.
eUstekinumab is applicable to CD only.
For the treatment of CD and UC, the recommended dosage of infliximab SC in the infliximab-naive setting is induction with 3 infliximab IV infusions of 5 mg/kg at weeks 0, 2, and 6 followed by a maintenance dosage of 120 mg every 2 weeks starting 8 weeks after induction.54 For patients already using infliximab, the recommended maintenance dosage is 120 mg every 2 weeks starting 8 weeks after the last infusion. The sponsor’s analysis indicated that, at a cost of $19,357 in the first year (when inducted with Inflectra) and $15,424 per patient per year thereafter, infliximab SC is priced at parity with the least costly infliximab IV product; however, infliximab SC is more costly in terms of total regimen costs when accounting for dosage, indicating that it is not cost-neutral. Infliximab SC was also less costly than all other relevant biologic DMARD comparators except for adalimumab biosimilars.
Critical Appraisal of Cost Information
CADTH identified 2 key limitations to the sponsor’s analysis that have notable implications on the cost comparison:
The comparative efficacy of infliximab SC with respect to noninfliximab comparators is uncertain. The sponsor’s submitted clinical data compared infliximab SC with infliximab IV only; no direct or indirect evidence for infliximab SC compared with noninfliximab biologic options is available. The clinical review concluded that, based on a bioequivalence study, infliximab SC may have benefits comparable to the infliximab IV formulation. No conclusion could be drawn as to the benefit of infliximab on improvement in HRQoL due to significant limitations in the included studies. The safety profile was considered acceptable by the clinical expert consulted by CADTH and comparable to infliximab IV. However, it is unknown if long-term safety would be maintained beyond the clinical trial setting, in real-world settings.
The sponsor’s submitted pricing for infliximab SC is at parity with infliximab IV on a per mg basis, but does not align with annual costs. The sponsor’s submission noted that infliximab SC is priced at parity with the lowest-priced infliximab IV product on a per mg basis. However, given that infliximab SC is packaged in a 120 mg/mL PFS while infliximab IV is packaged in a 100 mL vial, the unit cost of infliximab SC is higher than that of infliximab IV. Therefore, when accounting for appropriate dosage and total regimen costs, infliximab SC is not cost-neutral compared to the least costly infliximab IV product.
Table 26: CADTH Price Reduction Analyses
Scenario | Submitted price ($) | Reduction needed | Reduced price ($) | Savings relative to submitted pricea ($) |
|---|---|---|---|---|
Price reduction required to equal least costly infliximabb comparators (Renflexis and Avsola) | 591.60 | 16% | 496.94 | Year 1: 2,067.25 After: 2,570.65 |
Price reduction required to equal least costly other biologic DMARD product (adalimumab biosimilar)c | 591.60 | 40% | 354.96 | Year 1: 5,185.70 After: 3,137.18 |
DMARD = disease-modifying antirheumatic drug.
aSavings from the sponsor list price per patient per year.
bAssumes infliximab SC would be inducted with Inflectra.
cThe price reduction required to equal the least costly other biologic DMARD product does not imply that equivalency exists between infliximab and other biologic DMARD products.
Price Reduction Analyses
CADTH conducted price reduction analyses estimating the percentage reduction in the sponsor’s submitted price that would make infliximab SC cost-neutral versus the least costly comparators available. These price reductions assume induction with Inflectra, given that it is the most widely used infliximab biosimilar; this provides a conservative estimate of the total cost of treatment in year 1 because it is priced higher than the other 2 infliximab biosimilars. Based on publicly available list prices, the price of infliximab SC would have to be reduced by 16% for the annual cost of treatment acquisition to be equivalent to that of the least costly infliximab IV treatments (i.e., Renflexis and Avsola). Similarly, the submitted price of infliximab SC would have to be reduced by 40% to be equivalent to the treatment acquisition costs of other biologic DMARDs.
Issues for Consideration
Weight-based dosing: Dosing for some comparators is based on patient weight. Treatment costs relative to infliximab SC for such comparators would differ for patients weighing substantially more or less than 73.2 kg.
Impact of IV administration on total costs: These analyses assume that the costs of IV infusion administration are incurred by product manufacturers rather than by public health care payers. In situations in which the administration of infusions is reimbursed by public payers, the overall cost of all IV products would increase. While infliximab (Remsima SC) is an SC product, it requires induction with another IV infliximab product. Therefore, there is uncertainty regarding the total cost of administration.
Impact on health care resource utilization: Depending on the frequency at which infliximab SC is dispensed, it is possible that small incremental costs or savings related to dispensing fees may be realized related to the fees associated with other biologics.
Analysis based on publicly available list prices: The sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators. The actual costs paid by public drug plans are unknown.
Discussion
Summary of Available Evidence
This report summarizes the evidence of efficacy and safety of infliximab SC in the treatment of CD and UC.
LIBERTY-UC and LIBERTY-CD were identically designed, DB, parallel-group, placebo-controlled, phase III RCTs designed to assess the superiority of infliximab SC (120 mg) administered every 2 weeks over placebo in patients with moderately to severely active UC and moderately to severely active CD, respectively, who had experienced inadequate responses to conventional therapy. Both trials consisted of a 6-week induction phase during which enrolled patients received induction doses of 5 mg/kg of infliximab IV; a maintenance phase during which patients without safety concerns who were considered clinical responders before week 10 were randomized in a 2 to 1 ratio to receive infliximab SC or placebo as maintenance treatment for up to 54 weeks; and an extension phase during which patients in both arms who had completed treatment at week 54 were administered OL infliximab SC until week 102. The extension phases in both trials are ongoing.
The LIBERTY-UC trial (n = 438) included patients aged 18 years to 75 years with moderately to severely active UC who did not respond to conventional therapy, including corticosteroids alone or in combination with 6-MP or AZA, or who were intolerant of or had medical contraindications to these conventional therapies. The primary end point was clinical remission, measured using the modified Mayo score. Key secondary end points were clinical response based on Mayo scores, endoscopic-histologic mucosal improvement, and corticosteroid-free remission at week 54. HRQoL was assessed using the SIBDQ, Patient Global Score, and VAS (for local site pain assessments). Baseline characteristics were generally well-balanced between the 2 treatment groups in the trial; the majority of patients were white and male, with a mean age of 38 years to 40 years across the 2 arms; most had previously received a corticosteroid medication. Concomitant medications received were generally balanced between the treatment groups.
The LIBERTY-CD trial (n = 343) included patients aged 18 years to 75 years with moderately to severely active CD who did not respond to a full and adequate course of therapy with corticosteroids and/or immunosuppressants, or who were intolerant of or had medical contraindications to such therapies. The coprimary objectives were clinical remission (based on CDAI) and endoscopic response. The key secondary end points were CDAI-100 response, clinical remission based on abdominal pain and stool frequency, endoscopic remission based on central SES-CD, corticosteroid-free remission at week 54, and HRQoL using the SIBDQ. Baseline characteristics were generally well-balanced between the 2 treatment groups in the trial. The majority of patients were white and male, with a mean age ranging from 32 years to 36 years across the 2 groups; concomitant medications were generally balanced between the treatment groups (although budesonide use was higher in the infliximab SC group compared to the placebo group).
Study 1.6 (n = 131) was an OL, parallel-group, phase I randomized trial comparing the PK parameters, efficacy, and safety of infliximab 5 mg/kg IV administered every 8 weeks versus infliximab SC (120 mg or 240 mg) administered every 2 weeks in patients with active UC and CD. The study was designed in 2 parts. Part 2, which this review focuses on, evaluated both PK and clinical end points, and was implemented in 2 phases: a dose-loading phase from week 0 to week 6 during which enrolled patients received infliximab IV infusion at week 0 and week 2, followed by a maintenance phase during which a total of 131 patients who had no safety concerns were randomized 1 to 1 to receive infliximab IV or infliximab SC. Patients in the infliximab IV arm received IV infliximab up to week 22, switched to infliximab SC at week 30, and continued with infliximab SC up to week 54. The study enrolled patients with UC and patients with CD aged 18 years to 75 years with moderate to severe active disease. Patients in the infliximab SC arm received the SC formulation at week 6 and continued up to week 54. The primary end point was PK, while key secondary end points were CDAI-70 and CDAI-100 response, clinical remission, endoscopic response, clinical response (based on total and partial Mayo scores), mucosal healing, and SIBDQ scores. Baseline characteristics were generally well-balanced between the 2 treatment groups in the trial; most patients were white and males, with a mean age of 35 years to 36 years across the 2 groups. Concomitant medications were generally balanced between treatment groups.
There were no head-to-head trials submitted comparing infliximab SC to other treatments available to patients in clinical practice. Long-term studies assessing the impact of infliximab SC on in CD and UC is currently ongoing with no data available at the time of submission. No ITC was submitted to assess the efficacy of infliximab SC relative to other treatment options in practice.
Interpretation of Results
Efficacy
Evidence from 2 trials (LIBERTY-UC and LIBERTY-CD) showed the superiority of infliximab SC over placebo for primary and coprimary outcomes, respectively, as maintenance therapy for patients with moderately to severely active UC and CD. After a 10-week induction period with IV infliximab 5 mg/kg, followed by a maintenance period (44 weeks) with infliximab SC or placebo, patients receiving infliximab SC showed improvement in key efficacy outcomes over placebo until week 54.
According to the clinical expert consulted by CADTH, treatment goals for patients with UC or CD include symptom resolution (clinical remission), improved quality of life (through normalized bowel movements; resolution of pain, bowel urgency, and rectal bleeding; and normalization of weight and energy levels), reduced need for surgery, and avoidance of the repetitive use of corticosteroids. These goals were consistent with practice and regulatory guidelines for UC and CD populations.29,30,73,74 Patient respondents in the patient advocacy group input indicated that they preferred sustained remission and treatment response over relieving any 1 symptom, and desired options that could be administered at home to reduce time off work. The design of the LIBERTY-UC and LIBERTY-CD studies and the outcomes assessed were consistent with regulatory guidelines for trials in the UC and CD settings29,30,73,74 and aligned with the desired outcomes expressed by patient respondents and the clinical expert consulted by CADTH. However, other goals, such as reduced need for surgery and normalization of bowel movements, were not assessed in the 3 trials. The clinical expert highlighted that endoscopic response (histological improvement based on colonoscopy and/or tissue biopsies) and clinical remission are valuable objective outcomes that allow for the assessment of treatment benefit and patient recovery (i.e., mucosal healing) for patients with UC or CD. Mucosal healing was assessed in the LIBERTY-UC trial using the ES of the Mayo tool and the RHI. According to the clinical expert consulted by CADTH for the review, Mayo ES is the gold standard for assessing mucosal healing in patients with UC (a Mayo score of 0 or 1 is considered mucosal healing) and aligns with regulatory guidance requirements.29,74 The clinical expert noted that RHI is an additional end point used in current clinical trials, but has not been validated as a definite end point.
UC Population
The LIBERTY-UC trial showed that infliximab SC significantly improved clinical remission in patients in the maintenance phase compared to placebo at week 54. Clinical remission using the modified Mayor score calculation was considered an important clinical outcome as well as a valid and meaningful outcome used in practice by the clinical expert consulted by CADTH. The expert considered the magnitude of the benefit with infliximab SC to be clinically meaningful compared to placebo. The trial met its primary end point and demonstrated statistically significantly higher proportions of patients achieving clinical remission in the infliximab SC group compared to placebo. Sensitivity and other supportive analyses were consistent with the primary analyses in the LIBERTY-UC trial. Subgroup analyses of the primary end point based on sex, age, and race did not show significant differences (Appendix 1).
Results for key secondary outcomes assessed in the LIBERTY-UC trial also showed statistically significant improvements in patients receiving infliximab SC over placebo. The proportion of patients achieving clinical response and endoscopic-histologic mucosal improvement were numerically higher in the infliximab group (53.7% and 35.7%, respectively) compared to placebo, and these findings were considered clinically meaningful by the clinical expert consulted by CADTH. Although the expert considered the results for corticosteroid-free remission in the infliximab SC group versus the placebo group to be clinically meaningful, they also considered the proportion of patients with UC achieving corticosteroid-free remission to be low in the infliximab SC arm, given that the treatment goal in practice is to get as many patients as possible to stay off corticosteroids when using any advanced treatment. However, the finding was considered comparable to findings presented in the initial infliximab IV trials (ACT 1 and ACT 2, respectively)80 for patients with UC, according to the clinical expert. More patients achieved total or partial remission at week 54 in the infliximab group (39.8%) than in the placebo group (18.1%). This finding was considered statistically significant and clinically meaningful by the clinical expert consulted by CADTH.
In the LIBERTY-UC trial, with regard to HRQoL assessments, fewer patients completed questionnaires for patient-reported outcomes (e.g., the SIBDQ patient global scale and local site pain assessment through VAS) at week 54 compared to baseline in both groups. For SIBDQ scores, the numbers of patients completing the questionnaires in both treatment groups were 185 patients at week 54 versus 294 patients at baseline for the infliximab SC group, and 61 patients at week 54 versus 144 patients at baseline for the placebo group. For local site pain assessment using VAS, the numbers of patients completing questionnaires in the infliximab SC group were 241 patients at week 54 and 296 patients at week 10 in the placebo group, 53 patients at week 54 versus 140 patients at week 10. Many patients in the placebo group required dose adjustments after they stopped responding (from week 22); therefore, their results were excluded from the descriptive summary from week 30. Missing data may have significantly affected the patient-reported outcome findings; thus, the impact of infliximab SC on HRQoL outcomes in patients with UC in the trial remains unclear.
CD Population
Evidence from the LIBERTY-CD trial showed improvements in coprimary outcomes at week 54 for patients receiving infliximab SC over placebo. The estimated treatment difference between infliximab SC and placebo for clinical remission (using CDAI score) was 32.1% (95% CI, 20.9 to 42.1); for endoscopic response, it was 34.7% (95% CI, 24.2 to 43.5). Both findings were statistically significant and considered clinically meaningful by the clinical expert consulted by CADTH. Although the CDAI tool is well-established as a measure of clinical remission in trial settings, it is seldom used across jurisdictions in Canada, due to practical challenges involved in deriving the scores. The clinical expert considered the magnitude of the benefit of endoscopic response observed in the infliximab arm (51.1%) clinically important and comparable to (or even slightly better than) outcomes observed for infliximab IV in practice. The sensitivity analyses were consistent with the primary analyses in the LIBERTY-CD trial (Appendix 1). The subgroup analyses for the primary end points based on sex, age, and race showed no significant differences between patient subgroups (Appendix 1).
Significant clinical improvements were also observed in the infliximab SC group over placebo after week 54 for key secondary end points in the trial. Clinical remission, with an estimated difference between the 2 study arms of 21.1% (95% CI, 11.8 to 29.3), was statistically significant in favour of the infliximab SC group compared to the placebo group and considered clinically meaningful by the clinical expert consulted by CADTH. The benefit observed with infliximab SC over placebo for endoscopic remission was also statistically significant and considered clinically meaningful (treatment difference of 24.9%; 95% CI, 15.4 to 32.8). Corticosteroid-free response among patient in the infliximab SC group was statistically significantly higher than among those in the placebo group, with an estimated difference of 17.1% (95% CI, –0.4 to 31.5); this was considered reasonable and meaningful by the clinical expert, given that it is difficult for patients with CD to achieve corticosteroid-free remission in current practice. Clinical remission as measured using the SES-CD was also considered an important outcome. Mucosal healing was assessment-based on central SES-CD score calculations. The proportion of patients who achieved endoscopic remission as measured by central SES-CD scores was statistically significant in the infliximab group compared to placebo.
With regard to HRQoL assessments, as with the LIBERTY-UC trial, fewer patients completed questionnaires for patient-reported outcomes at week 54 compared to baseline in both groups. For SIBDQ scores, 167 patients in the infliximab SC group completed questionnaires at week 54 versus 231 at baseline, and 51 patients in the placebo group completed questions at week 54 versus 111 at baseline. For local site pain assessment using VAS, 201 patients in the infliximab SC group completed questions at week 54 versus 238 patients at week 10; in the placebo group, 40 patients completed questions at week 54 versus 104 at week 10. For the patient global scale questionnaire, 167 patients in the infliximab SC group completed the questionnaire at week 54 versus 231 patients at week 10; in the placebo group, 49 patients completed the questionnaire at week 54 versus 111 at week 10. Many patients in the placebo group required dose adjustments they stopped responding; their results were excluded from the descriptive summary from week 30. Missing data may have significantly affected the patient-reported outcome findings; thus, the impact of infliximab SC on HRQoL outcomes in patients with CD in the trial remains unclear.
Limitations were identified in both trials, such as the potential risk for assessment bias due to the subjective nature of outcome measurements; this may have an impact on the interpretability of the findings in both the UC and CD populations. The potential risk of bias due to the enrichment trial design may have affected the generalizability of the findings, and the potential for residual treatment effects of the use of corticosteroids in the induction and maintenance phases in the 2 arms in both trials may have affected inflammatory symptoms in both populations, biasing the assessment outcomes in either direction. The magnitude of the potential bias on the primary and coprimary outcomes is unknown.
The design in both trials reflected clinical practice. However, the durations of the induction phases in both were short (6 weeks) and included only patients who were able to achieve a timely response to infliximab. This creates an enriched design that may limit the generalizability of the results to current practice. The duration of the induction phase did not take into consideration delayed responders. In practice, induction duration may extend up to 16 weeks to allow patients to benefit from treatment, according to the clinical expert consulted by CADTH. The duration of the maintenance phase in addition to patient exposure in the ongoing extension phase (up to week 102) was considered adequate to assess treatment effect in both populations in the 2 trials.
The concomitant medications (immunomodulators, oral corticosteroids [prednisone, budesonide], and antibiotics) approved in the 3 studies were consistent with clinical practice except for a few drugs not commonly used in Canada, such as mesalamine. The tapering protocol (5 mg/week) for corticosteroids aligned with regulatory requirements but differed slightly from the rates used in current practice.
There were no head-to-head trials submitted comparing infliximab SC to other treatments currently available to patients in clinical practice. Long-term data assessing the impact of infliximab SC on patients with CD or UC is ongoing, with no data available at the time of submission. According to the clinical expert, there is currently no cure for UC or CD, and treatment is usually administered for the long-term. The long-term impact of infliximab SC on key outcomes in patients with UC or CD is uncertain.
Bioequivalence Study 1.6
Results from Study 1.6, part 2 showed that infliximab SC was statistically noninferior to infliximab IV in terms of PK, determined by the observed week 22 Ctrough, calculated from the predose level at week 22. The ratio of geometric LS means was 1,154.2, with a lower-bound 90% CI of 786.4%, which was greater than 80%, indicating that infliximab SC was noninferior to infliximab IV in terms of PK (noninferior margin = 80%). In addition, the mean predicted Ctrough levels in the infliximab SC group throughout the maintenance phase was greater than 18 mcg/mL, higher than the target exposure of 5 mcg/mL, which is considered to be the minimum serum concentration level needed to achieve and maintain improved clinical outcomes, including clinical remission and mucosal healing.33,34The clinical expert consulted by CADTH noted that the PK trough levels achieved by patients on infliximab SC in Study 1.6 were comparable to those receiving the infliximab IV formulation. The clinical expert further agreed that the week 22 Ctrough end point served to support the ability of infliximab SC to maintain therapeutic activity when administered with greater frequency at a lower dose per injection than infliximab IV (i.e., every 2 weeks versus every 8 weeks). When patients with UC or CD were switched from maintenance therapy (infliximab IV) to infliximab SC after achieving steady state (week 22), results suggested similar benefits in the 2 study arms for efficacy outcomes by week 54. According to the clinical expert consulted by CADTH, the PK trough levels for infliximab SC in Study 1.6 were comparable to those for the infliximab IV formulation.
While the evidence from secondary outcomes suggests that infliximab SC (120 mg or 240 mg) is comparable to infliximab IV (5 mg/ kg), the analyses in patients with UC and CD were descriptive, and the trial was not designed or powered to compare secondary outcome results across study groups. Thus, the comparative efficacy of infliximab SC versus infliximab IV remains uncertain. The clinical expert consulted by CADTH did not anticipate clinically meaningful differences in efficacy between infliximab SC and infliximab IV because the products have the same active ingredient (i.e., infliximab). The clinical expert did not anticipate any clinical concerns related to switching from IV to SC administration as long as the choice to switch was made on a case-by-case basis and after a thorough discussion between clinician and patient.
Harms
Overall, no new or unexpected safety concerns were reported across the 3 trials. The safety profile of infliximab SC was considered comparable to that of infliximab IV as currently used in practice, according to the clinical expert consulted by CADTH.
AEs reported in the maintenance phase were numerically higher in the infliximab SC group (i.e., 67.6% and 72.3%) than in the placebo group (59.3% and 61.9%) in the LIBERTY-UC and LIBERTY-CD trials, respectively. The proportion of patients with at least 1 serious AE in the maintenance phase of the 2 trials was reported as follows:
LIBERTY-UC infliximab SC group: n = 19 (6.4%); placebo group: n = 4 (2.9%)
LIBERTY-CD infliximab SC group: n = 16 (6.7%); placebo group: n = 8 (7.6%).
The most common serious AEs reported in the 2 trials were GI disorders:
LIBERTY-UC infliximab SC group: n = 4 (1.4%); placebo group: n = 2 (1.4%)
LIBERTY-CD infliximab SC group: n = 5 (2.1%); placebo group: n = 2 (1.9%).
Results for infections and infestations were as follows:
LIBERTY-UC infliximab SC group: n = 7 (2.4%); placebo group: n = 1 (0.7%)
LIBERTY-CD infliximab SC group: n = 6 (2.5%); placebo group: n = 1 (1.0%).
At least 66.9% of patients (n = 422 patients) in the pooled infliximab dataset and 60.4% of patients (n = 140 patients) in the pooled placebo set reported at least 1 treatment-emergent AE. The most common AEs reported in at least 2% of the population in the infliximab group included headache, ISR, COVID-19, arthralgia, abdominal pain, and alanine aminotransferase increase. The most common AEs reported in the placebo group included COVID-19, headache, blood CPK increase, abdominal pain, and administration-related reaction. There were more AEs reported in the infliximab SC group than in the infliximab IV group in Study 1.6 (74.2% versus 58.5%, respectively).
In the LIBERTY-UC trial, the most common grade 3 AEs reported in the infliximab group were neutrophil count decrease (3.7%), anemia (2.0%), and CPK increase (1.7%) (for comparison, in the placebo group, 2.9% reported increased CPK). The most common grade 4 AEs reported in the infliximab SC group were CPK increase (1.4%) (versus 1.4% in the placebo group), neutrophil count decrease (0.7%), and hypertriglyceridemia (0.7%). In the LIBERTY-CD trial, the most common grade 3 AEs reported in the infliximab SC group were neutrophil count decrease (4.6%), CPK increase (2.5%), blood bilirubin increase (2.1%), and hypertriglyceridemia (2.1%). Grade 4 events commonly reported were CPK increase (3.4%) and neutrophil count decrease (0.8%). In the placebo group, the most commonly reported grade 3 AEs were lymphocyte count decrease (4.8%), anemia (3.8%), and CPK increase (1.9%). The most common grade 4 event was CPK increase (1.9%).
A higher proportion of patients in the infliximab SC group versus the infliximab IV group of Study 1.6 reported AEs (58.7% versus 39.7%, respectively). The most common AEs included localized ISRs (22.7% versus 4.6%), UC (4.5% versus 12.3%), and neutropenia (7.6% versus 4.6%) in the infliximab SC and infliximab IV groups, respectively. The proportions of patients reporting at least 1 serious AE in Study 1.6 were 7.6% and 10.8% for the infliximab SC and infliximab IV groups, respectively. The most common serious AE was infections and infestations (6.2% and 4.6%) in the infliximab SC and infliximab IV groups, respectively. The most common grade 3 AEs reported in the infliximab SC group were neutrophil count decrease (4.5%) and hypertriglyceridemia (3.0%). The most common grade 4 events in the infliximab SC group were CPK increase (3.0%) and neutrophil count decrease (1.5%). In the infliximab IV group, the most common grade 3 events were neutrophil count decrease (6.2%) and white blood cell decrease (3.1%). The most common grade 4 events in the infliximab IV group was neutrophil count decrease (1.5%).
The most common AEs of special interest reported across the 3 trials included systemic injection reaction, ISR, injection-related reaction, and infection. The proportions of patients experiencing AEs of special interest were higher in the pooled infliximab SC dataset compared to the pooled placebo set: systemic injection reactions, 2.5% versus 2%; localized ISR, 6.2% versus 1.6%; and infection, 27.6% versus 22.4%, respectively.
The sponsor did not report associations of infliximab SC treatment with laboratory, vital sign, or electrocardiogram changes in the 3 trials.28
Costs
The first-year cost of infliximab SC will depend on which infliximab IV product is chosen for the induction period. At the submitted price, the cost per patient when Inflectra is chosen is $19,357 for the first year. The cost is $15,424 in each subsequent year.
The annual costs associated with infliximab SC are less than those associated with the branded IV product (Remicade) and with other branded biologic comparators, such as adalimumab (Humira), Golimumab SC (Simponi), Vedolizumab (Entyvio) IV and SC, and ustekinumab (Stelara). On the other hand, infliximab SC is associated with increased annual costs when compared to other infliximab IV biosimilars (Inflectra, Renflexis, and Avsola) and adalimumab biosimilars, even though on a per mg basis, it is priced at parity with the least costly biosimilar. These incremental costs or savings are based on publicly available list prices and may not reflect actual prices paid by Canadian drug plans.
Conclusion
A total of 3 randomized trials supported the clinical efficacy and safety data of infliximab SC for the reimbursement request in patients with UC and CD, which aligns with the Health Canada indication. Infliximab SC demonstrated statistically significant benefits in clinical remission based on modified Mayo score (for UC) and on clinical remission based on CDAI score and endoscopic response (for CD) in adult patients with moderately to severely active UC and CD, respectively, who did not respond to conventional therapies. The results for key secondary outcomes showed statistically significant benefits in favour of infliximab SC versus placebo. However, the sustainability of the beneficial effect and the potential for disease recurrence in the long-term (beyond 1 year) remain uncertain. A bioequivalence study with a small sample size suggests that infliximab SC may have benefits comparable to the infliximab IV formulation. No conclusion could be drawn as to the benefit of infliximab on improvement in HRQoL, due to significant limitations.
Overall, infliximab SC treatment was shown to be well-tolerated among patients with UC or CD across 3 trials. Safety data pooled across trials showed no new or unexpected safety concerns. The safety profile was considered acceptable (and comparable to that of infliximab IV) by the clinical expert consulted by CADTH. However, it is unknown if long-term safety among all patients who receive infliximab in real-world clinical practice settings will be maintained.
Infliximab SC (Remsima SC) has the same main ingredient as the infliximab IV product; however, it is administered at a different dose and frequency and through the SC route rather than through IV. Infliximab SC is intended to provide a treatment option, in place of infliximab IV, that could be self-administered by patients without the need for frequent or lengthy visits to infusion clinics. There was no evidence designed to assess the impact of this more convenient administration on efficacy outcomes.
At the submitted price, and based on the recommended dosage regimen, the annual cost of infliximab SC is $19,357 per patient in the first year and $15,424 every year thereafter. Infliximab is less costly than branded biologic products, but more costly than other biologic DMARD biosimilars. The submitted price of infliximab SC would have to be reduced by 16% to 40% for its annual cost to be equivalent to that of the least costly SEB comparators, depending on the comparator (i.e., infliximab versus noninfliximab products).
References
1.GI Society: Canadian Society of Intestinal Research. Inflammatory bowel disease. 2023; https://badgut.org/information-centre/a-z-digestive-topics/inflammatory-bowel-disease/. Accessed 2023 Oct 23.
2.Coward S, Clement F, Benchimol EI, et al. Past and future burden of inflammatory bowel diseases based on modeling of population-based data. Gastroenterology. 2019;156(5):1345-1353 e1344. PubMed
3.Rocchi A, Benchimol EI, Bernstein CN, et al. Inflammatory bowel disease: A Canadian burden of illness review. Can J Gastroenterol. 2012;26(11):811-817. PubMed
4.Kaplan GG, Bernstein CN, Coward S, et al. The impact of inflammatory bowel disease in Canada 2018: Epidemiology. J Can Assoc Gastroenterol. 2019;2(Suppl 1):S6-S16. PubMed
5.Windsor JW, Kuenzig ME, Murthy SK, et al. The 2023 impact of inflammatory bowel disease in Canada: Executive summary. J Can Assoc Gastroenterol. 2023;6(Suppl 2):S1-S8. PubMed
6.2023: Impact of inflammatory bowel disease in Canada. Toronto (ON): Crohn’s and Colitis Canada; 2023: https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2023-IBD-Report-English-LR.pdf. Accessed 2023 Dec 4.
7.Centers for Disease Control and Prevention. Inflammatory bowel disease (IBD). 2022; https://www.cdc.gov/ibd/what-is-IBD.htm. Accessed 2022 Apr 13.
8.Public Health Agency of Canada. Inflammatory bowel disease (IBD). 2021; https://www.canada.ca/en/public-health/services/chronic-diseases/inflammatory-bowel-disease.html. Accessed 2023 Oct 23.
9.Feuerstein JD, Ho EY, Shmidt E, et al. AGA clinical practice guidelines on the medical management of moderate to severe luminal and perianal fistulizing Crohn's disease. Gastroenterology. 2021;160(7):2496-2508. PubMed
10.2018: Impact of inflammatory bowel disease in Canada. Toronto (ON): Crohn's and Colitis Canada; 2018: https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2018-Impact-Report-LR.pdf?ext=.pdf. Accessed 2020 Nov 30.
11.Benchimol EI, Bernstein CN, Bitton A, et al. The impact of inflammatory bowel disease in Canada 2018: A scientific report from the Canadian Gastro-Intestinal Epidemiology Consortium to Crohn's and Colitis Canada. J Can Assoc Gastroenterol. 2019;2(Suppl 1):S1-S5. PubMed
12.Coward S, Clement F, Benchimol EI, et al. A29 The rising prevalence of inflammatory bowel disease in Canada: Analyzing the past to predict the future. J Can Assoc Gastroenterol. 2018;1(Suppl 2):47-48. https://academic.oup.com/jcag/article/1/suppl_2/47/4916529. Accessed 2023 Oct 23.
13.The impact of inflammatory bowel disease in Canada: 2012 final report and recommendations Toronto (ON): Crohn's and Colitis Foundation of Canada; 2012: http://www.crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/ccfc-ibd-impact-report-2012.pdf. Accessed 2020 Nov 30.
14.Peppercorn MA, Kane SV. Clinical manifestations, diagnosis, and prognosis of Crohn disease in adults. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2022: https://www.uptodate.com/. Accessed 2023 Oct 23.
15.Schroeder KW, Tremaine WJ, Ilstrup DM. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N Engl J Med. 1987;317(26):1625-1629. PubMed
16.Stenson W. Inflammatory bowel diseases. In: Goldman I BJ, ed. Cecil Textbook of Medicine, 21st ed. Philadephia (PA): WB Saunders Co; 2000:722-729.
17.Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med. 2011;365(18):1713-1725. PubMed
18.Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet. 2017;390(10114):2769-2778. PubMed
19.Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46-54 e42; quiz e30. PubMed
20.Benchimol EI, Fortinsky KJ, Gozdyra P, Van den Heuvel M, Van Limbergen J, Griffiths AM. Epidemiology of pediatric inflammatory bowel disease: A systematic review of international trends. Inflamm Bowel Dis. 2011;17(1):423-439. PubMed
21.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785-1794. PubMed
22.Al Hashash J, Regueiro M. Medical management of low-risk adult patients with mild to moderate ulcerative colitis. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2023: https://www.uptodate.com/. Accessed 2023 Mar 22.
23.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785-1794. PubMed
24.Gomollon F, Dignass A, Annese V, et al. 3rd European Evidence-based Consensus on the diagnosis and management of Crohn's disease 2016: Part 1: Diagnosis and medical management. J Crohns Colitis. 2017;11(1):3-25. PubMed
25.Hashash J, Regueiro M. Overview of medical management of high-risk, adult patients with moderate to severe Crohn disease. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2020: www.uptodate.com. Accessed 2020 Oct 27.
26.Lichtenstein GR, Loftus EV, Isaacs KL, Regueiro MD, Gerson LB, Sands BE. ACG clinical guideline: Management of Crohn's disease in adults. Am J Gastroenterol. 2018;113(4):481-517. PubMed
27.CADTH Drug Reimbursement Expert Review Committee final recommendation: Infliximab SC (Remsima SC - Celltrion Healthcare Canada). Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/cdr/complete/SR0659%20Remsima%20SC%20-%20CDEC%20Final%20Recommendation%20April%2026%2C%202021_for%20posting.pdf. Accessed 2024 Jan 11.
28.Drug Reimbursement Review sponsor submission: Infliximab (REMSIMA™ SC), 120 mg/ml, solution for subcutaneous injection [internal sponsor's package]. Toronto (ON): Celltrion Healthcare Canada Limited; 2023 Oct 23.
29.U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for industry: Ulcerative colitis - Developing drugs for treatment (draft guidance). 2022; https://www.fda.gov/media/158016/download. Accessed 2023 Dec 7.
30.Food and Drug Administration: Center for Drug Evaluation and Research Center for Biologics Evaluation and Research. Crohn’s disease: Developing drugs for treatment. 2022; https://www.fda.gov/media/158001/download. Accessed 2023 Oct 23.
31.Guideline on the development of new medicinal products for the treatment of ulcerative colitis (CHMP/EWP/18463/2006 Rev.1). Amsterdam (NL): European Medicines Agency; 2023: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-development-new-medicinal-products-treatment-ulcerative-colitis-revision-1_en.pdf. Accessed 2023 Oct 23.
32.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry: Bioavailability and bioequivalence studies submitted in NDAs or INDs - General considerations (draft guidance). 2014; https://www.fda.gov/files/drugs/published/Bioavailability-and-Bioequivalence-Studies-Submitted-in-NDAs-or-INDs-%E2%80%94-General-Considerations.pdf. Accessed 2024 Jan 8.
33.Papamichael K, Gils A, Rutgeerts P, et al. Role for therapeutic drug monitoring during induction therapy with TNF antagonists in IBD: evolution in the definition and management of primary nonresponse. Inflamm Bowel Dis. 2015;21(1):182-197. PubMed
34.Vaughn BP, Sandborn WJ, Cheifetz AS. Biologic concentration testing in inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(6):1435-1442. PubMed
35.Lapidus A, Bernell O, Hellers G, Lofberg R. Clinical course of colorectal Crohn's disease: A 35-year follow-up study of 507 patients. Gastroenterology. 1998;114(6):1151-1160. PubMed
36.Peyrin-Biroulet L, Panes J, Sandborn WJ, et al. Defining disease severity in inflammatory bowel diseases: Current and future directions. Clin Gastroenterol Hepatol. 2016;14(3):348-354 e317. PubMed
37.Fumery M, Singh S, Dulai PS, Gower-Rousseau C, Peyrin-Biroulet L, Sandborn WJ. Natural history of adult ulcerative colitis in population-based cohorts: A systematic review. Clin Gastroenterol Hepatol. 2018;16(3):343-356 e343. PubMed
38.Travis SP, Stange EF, Lemann M, et al. European evidence-based consensus on the management of ulcerative colitis: Current management. J Crohns Colitis. 2008;2(1):24-62. PubMed
39.Rubin DT, Sninsky C, Siegmund B, et al. International perspectives on management of inflammatory bowel disease: Opinion differences and similarities between patients and physicians from the IBD GAPPS survey. Inflamm Bowel Dis. 2021;27(12):1942-1953. PubMed
40.Lynch WD, Hsu R. Ulcerative colitis. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022: https://www.ncbi.nlm.nih.gov/books/NBK459282/. Accessed 2022 Dec 29.
41.Rubin DT, Ananthakrishnan AN, Siegel CA, Sauer BG, Long MD. ACG clinical guideline: Ulcerative colitis in adults. Am J Gastroenterol. 2019;114(3):384-413. PubMed
42.Pola S, Patel D, Ramamoorthy S, et al. Strategies for the care of adults hospitalized for active ulcerative colitis. Clin Gastroenterol Hepatol. 2012;10(12):1315-1325 e1314. PubMed
43.Dubinsky MC, Watanabe K, Molander P, et al. Ulcerative Colitis Narrative global survey findings: The impact of living with ulcerative colitis - Patients' and physicians' view. Inflamm Bowel Dis. 2021;27(11):1747-1755. PubMed
44.Lopez-Sanroman A, Carpio D, Calvet X, et al. Perceived emotional and psychological impact of ulcerative colitis on outpatients in Spain: UC-LIFE survey. Dig Dis Sci. 2017;62(1):207-216. PubMed
45.Panaccione R, Steinhart AH, Bressler B, et al. Canadian Association of Gastroenterology clinical practice guideline for the management of luminal Crohn's disease. J Can Assoc Gastroenterol. 2019;2(3):e1-e34. PubMed
46.Loftus EV, Reinisch W, Panaccione R, et al. Adalimumab effectiveness up to six years in adalimumab-naive patients with Crohn's disease: Results of the PYRAMID registry. Inflamm Bowel Dis. 2019;25(9):1522-1531. PubMed
47.Panaccione R, Steinhart AH, Bressler B, et al. Canadian Association of Gastroenterology clinical practice guideline for the management of luminal Crohn's disease. Clin Gastroenterol Hepatol. 2019;17(9):1680-1713. PubMed
48.Bressler B, Marshall JK, Bernstein CN, et al. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: The Toronto consensus. Gastroenterology. 2015;148(5):1035-1058 e1033. PubMed
49.Raine T, Bonovas S, Burisch J, et al. ECCO guidelines on therapeutics in ulcerative colitis: Medical treatment. J Crohns Colitis. 2022;16(1):2-17. PubMed
50.Turner D, Ricciuto A, Lewis A, et al. STRIDE-II: An update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) initiative of the International Organization for the Study of IBD (IOIBD): Determining therapeutic goals for treat-to-target strategies in IBD. Gastroenterology. 2021;160(5):1570-1583. PubMed
51.Newton L, Randall JA, Hunter T, et al. A qualitative study exploring the health-related quality of life and symptomatic experiences of adults and adolescents with ulcerative colitis. J Patient Rep Outcomes. 2019;3(1):66. PubMed
52.Feuerstein JD, Isaacs KL, Schneider Y, et al. AGA clinical practice guidelines on the management of moderate to severe ulcerative colitis. Gastroenterology. 2020;158(5):1450-1461. PubMed
53.Kuhn F, Klar E. Surgical principles in the treatment of ulcerative colitis. Viszeralmedizin. 2015;31(4):246-250. PubMed
54.Remsima SC (infliximab injection): 120 mg / ml, solution for subcutaneous injection [product monograph]. Toronto (ON): Celltrion Healthcare Canada Limited; 2023 Oct 23.
55.CADTH Common Drug Review Biosimilar Submission: Infliximab (Renflexis - Merck Canada). Ottawa (ON): CADTH; 2018: https://www.cadth.ca/sites/default/files/cdr/relatedinfo/SE0532_Renflexis_Combined_Report.pdf. Accessed 2023 Oct 23.
56.Skyrizi (risankizumab): 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection; 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection; 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection; 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion [product monograph]. St-Laurent (QC): AbbVie; 2022 Oct 19.
57.Entyvio (vedolizumab): 108 mg / 0.68 mL solution for subcutaneous injection in a pre-filled syringe or pen [product monograph]. Toronto (ON): Takeda Canada Inc.; 2020 Nov 19.
58.Remicade (infliximab): powder for solution, sterile, lyophilized, 100 mg/vial [product monograph]. Toronto (ON): Janssen Inc; 2017 Aug 4: https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/REMICADE-MONOGRAPH.PDF. Accessed 2020 Nov 27.
59.Inflectra (infliximab for injection): powder for solution, sterile, lyophilized, 100 mg/vial [product monograph]. Kirkland (QC): Pfizer Canada Inc; 2018: https://www.pfizer.ca/sites/default/files/201809/INFLECTRA_PM_E_218146_24Aug2018.pdf. Accessed 2020 Nov 30.
60.Humira (adalimumab): 40 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection, 10 mg in 0.1 mL sterile solution (100 mg/mL) subcutaneous injection, 20 mg in 0.2 mL sterile solution (100 mg/mL) subcutaneous injection, 40 mg in 0.4 mL sterile solution (100 mg/mL) subcutaneous injection, 80 mg in 0.8 mL sterile solution (100 mg/mL) subcutaneous injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2019 Jun 25: https://pdf.hres.ca/dpd_pm/00052133.PDF. Accessed 2020 Nov 27.
61.Stelara (ustekinumab): 45 mg/0.5 mL, 90 mg/1.0 mL solution for subcutaneous injection, 130 mg/26 mL (5 mg/mL) solution for intravenous infusion[product monograph]. Toronto (ON): Janssen Inc; 2017 Aug 18: https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/STELARA_MONOGRAPH.PDF. Accessed 2020 Nov 27.
62.AbbVie. Rinvoq (upadacitinib): Extended-release tablets, 15 mg upadacitinib, oral extended-release tablets, 30 mg upadacitinib, oral extended-release tablets, 45 mg upadacitinib, oral [product monograph]. Montreal (QC): AbbVie; 2023 May 10.
63.Omvoh (mirikizumab): 100 mg/mL solutions for subcutaneous injection; 20 mg/mL solution for intravenous infusion doses and administration [draft product monograph]. Toronto (ON): Eli Lilly Canada, Inc.; 2023 Jul 20.
64.Zeposia (ozanimod): 0.23 mg, 0.46 mg and 0.92 mg capsules [product monograph]. Saint-Laurent (QC): Celgene Inc; 2022 Apr 7: https://pdf.hres.ca/dpd_pm/00065373.PDF. Accessed 2022 Apr 29.
65.Stelara (ustekinumab): 45 mg/0.5 mL, 90 mg/1.0 mL solution for subcutaneous injection; 130 mg/26 mL (5mg/ML) solution for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc; 2020 Jan 23: https://pdf.hres.ca/dpd_pm/00054738.PDF. Accessed 2022 Mar 29.
66.Remicade (infliximab): 100 mg/vial powder for solution, sterile, lyophilized [product monograph]. Toronto (ON): Janssen Inc; 2019 Jun 06: https://pdf.hres.ca/dpd_pm/00051606.PDF. Accessed 2022 Mar 29.
67.Entyvio (vedolizumab): 300 mg / vial powder for concentrate for solution for intravenous injection; 108 mg/0.68 mL prefilled syringe or pen solution for subcutaneous injection [product monograph]. Toronto (ON): Takeda Canada Inc; 2015 Jan 29: https://pdf.hres.ca/dpd_pm/00058905.PDF. Accessed 2022 Mar 29.
68.Simponi (golimumab): 50 mg/0.5 mL, 100 mg/1.0 mL solution for subcutaneous injection; 50 50 mg/4.0 mL solution for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc; 2019 Jun 20: https://pdf.hres.ca/dpd_pm/00052895.PDF. Accessed 2022 Mar 29.
69.Xeljanz (tofacitinib): 5 mg, 10 mg oral tablets; 11 mg extended-release oral tablets [product monograph]. Kirkland (QC) Pfizer Canada ULC; 2019 Oct 24: https://pdf.hres.ca/dpd_pm/00053735.PDF. Accessed 2022 Mar 29.
70.Humira (adalimumab injection): 40 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection, 10 mg in 0.1 mL sterile solution (100 mg/mL) subcutaneous injection, 20 mg in 0.2 mL sterile solution (100 mg/mL) subcutaneous injection, 40 mg in 0.4 mL sterile solution (100 mg/mL) subcutaneous injection, 80 mg in 0.8 mL sterile solution (100 mg/mL) subcutaneous injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2021 Apr 21: https://pdf.hres.ca/dpd_pm/00061690.PDF. Accessed 2022 May 2.
71.Rubin D, Keyashian K, Bunnag A, et al. Correlation between clinical, endoscopic, and histologic disease activity in ulcerative colitis: 1712. Official journal of the American College of Gastroenterology | ACG. 2012;107:S694. https://journals.lww.com/ajg/fulltext/2012/10001/correlation_between_clinical,_endoscopic,_and.1712.aspx. Accessed 2023 Oct 23.
72.Dhanda AD, Creed TJ, Greenwood R, Sands BE, Probert CS. Can endoscopy be avoided in the assessment of ulcerative colitis in clinical trials? Inflamm Bowel Dis. 2012;18(11):2056-2062. PubMed
73.Guideline on the development of new medicinal products for the treatment of Crohn’s Disease. Amsterdam (NL): European Medicine Agency; 2018: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-development-new-medicinal-products-treatment-crohns-disease-revision-2_en.pdf. Accessed 2023 Dec 7.
74.Guideline on the development of new medicinal products for the treatment of ulcerative colitis Amsterdam (NL): European Medicines Agency; 2019: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-development-new-medicinal-products-treatment-ulcerative-colitis-revision-1_en.pdf. Accessed 2023 Dec 7.
75.Torres J, Bonovas S, Doherty G, et al. ECCO guidelines on therapeutics in Crohn's disease: Medical treatment. J Crohns Colitis. 2020;14(1):4-22. PubMed
76.Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn's disease. N Engl J Med. 2004;350(9):876-885. PubMed
77.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Remsima SC (infliximab), 120 mg/mL pre-filled syringe or pre-filled pen. Toronto (ON): Celltrion Healthcare Canada Ltd; 2023 Oct 04.
78.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Oct 23.
79.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2023: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 Oct 23.
80.Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462-2476. PubMed
81.PharmaStat. [Ottawa (ON)]: IQVIA; 2023: https://www.iqvia.com/. Accessed 2023 Oct 23.
82.Tadrous M, McCormack D, Martins D, Kitchen S, Singh S, Gomes T. Current and prospective utilization of innovator biologics and biosimilars in Ontario. Toronto (ON): Ontario Drug Policy Research Network; 2020: https://odprn.ca/wp-content/uploads/2020/01/Utilization-of-Innovator-Biologics-and-Biosimilars-in-Ontario.pdf. Accessed 2023 Oct 23.
Appendix 1: Detailed Outcome Data
Note this appendix has not been copy-edited.
Additional baseline characteristics in the LIBERTY-UC and LIBERTY-CD trials
Table 27: Additional Baseline Characteristics Reported in the LIBERTY-UC Trial
Baseline characteristic | Infliximab SC (N = 294) | Placebo (N = 144) |
|---|---|---|
Moderate UC at baseline, n (%) | 131 (44.6) | 64 (44.4) |
Severe UC at baseline, n (%) | 163 (55.4) | 79 (54.9) |
Modified Mayo score at baseline | ||
n | 294 | 143 |
Mean (SD) | 6.6 (1.09) | 6.7 (1.17) |
Total Mayo Score at baseline | ||
n | 294 | 143 |
Mean (SD) | 8.8 (1.30) | 8.8 (1.42) |
Partial Mayo Score at baseline | ||
n | 294 | 143 |
Mean (SD) | 6.3 (1.10) | 6.3 (1.23) |
C-reactive protein (nmol/L) at baseline | ||
n | 292 | 144 |
Mean (SD) | 82.09 (168.721) | 79.18 (178.361) |
Median (min, max) | 27.15 (1.9, 1712.4) | 25.75 (1.9, 1132.4) |
Fecal Calprotectin (mg/kg), at baseline | ||
n | 292 | 144 |
Mean (SD) | 2,681.0 (3,467.64) | 2,441.2 (3,142.89) |
Median (min, max) | 1,481.5 (10, 32,184) | 1,428.5 (10, 20,340) |
Surgical and medical procedures, n (%) | 60 (20.4) | 28 (19.4) |
Appendicectomy | 3 (1.0) | 2 (1.4) |
Caesarean section | 7 (2.4) | 1 (0.7) |
Cholecystectomy | 3 (1.0) | 3 (2.1) |
Inguinal hernia repair | 6 (2.0) | 3 (2.1) |
Large intestinal polypectomy | 5 (1.7) | 1 (0.7) |
Tonsillectomy | 5 (1.7) | 4 (2.8) |
Note: Moderate UC includes patients with baseline modified Mayo score of 5 or 6, and severe UC includes patients with baseline modified Mayo score of 7, 8, or 9.
Source: Sponsor’s submission.28
Table 28: Additional Baseline Characteristics Reported in the LIBERTY-CD Trial
Baseline characteristic | Infliximab SC (N = 231) | Placebo (N = 112) |
|---|---|---|
Baseline CDAI score | ||
Asymptomatic remission (CDAI < 150) | 0 | 0 |
Mild to moderate CD (150 ≤ CDAI < 220): MILD | 0 | 0 |
Moderate to severe CD (220 ≤ CDAI ≤ 450): MODERATE | 229 (99.1) | 112 (100) |
Severe-fulminant disease (CDAI > 450): SEVERE | 0 | 0 |
Missing | 2 (0.9) | 0 |
C-reactive protein (nmol/L) at baseline | ||
n | 231 | 111 |
Mean (SD) | 179.9 (351.9) | 202.8 (314.13) |
Median (min, max) | 46.7 (1.9, 2392.4) | 59 (1.9, 1976.2) |
Fecal Calprotectin (mg/kg) at baseline | ||
n | 230 | 110 |
Mean (SD) | 2,585.2 (7,612.4) | 1951.6 (2,554.9) |
Median (min, max) | 802.5 (10, 76,800) | 1,019 (33, 13,351) |
Surgical and medical procedures, n (%) | 69 (29.9) | 36 (32.1) |
Abscess drainage | 8 (3.5) | 8 (7.1) |
Appendicectomy | 16 (6.9) | 6 (5.4) |
Caesarean section | 5 (2.2) | 2 (1.8) |
Colectomy | 7 (3.0) | 4 (3.6) |
Ileocolectomy | 6 (2.6) | 3 (2.7) |
Ovarian cystectomy | 4 (1.7) | 1 (0.9) |
Note: Inclusion criteria for 3.8 had a condition of 220 ≤ CDAI ≤ 450, so most patients were moderate. Missing patients were classified as having moderate disease based on the screening results; however, due to the inclusion of invalid data, the patients were treated as having a missing baseline during the analysis.
Source: Sponsor’s submission.28
Sensitivity Analyses Results for Coprimary Outcomes in the LIBERTY-CD Trial
Clinical Remission
Fisher exact test: (infliximab SC: 144 (62.3%); placebo: 36 (32.1%); difference- 30.2 (95% CI, 18.8 to 40.6), P value < 0.0001)
Logistic regression: (infliximab SC:144 (62.3%); placebo: 36 (32.1%); difference- 33.6 (95% CI, 22.9 to 44.3); P value < 0.0001)
Excluding war-affected patients in Ukraine: (infliximab SC: 136/214 (63.6%); placebo: 34/104 (32.7%); difference- 32.4 (95% CI, 20.7 to 42.7); P value < 0.0001)
Excluding all patients in Ukraine:(infliximab SC:125/202 (61.9%); placebo: 3//99 (32.3%); difference- 32.1 (95% CI, 20.1 to 43.8); P value < 0.0001).
Endoscopic Response
Fisher exact test: (infliximab SC:118 (51.1%); placebo:20 (17.9%); difference- 33.2 (95% CI, 22.2 to 42.5); P value < 0.0001).
Logistic regression: (infliximab SC:118 (51.1%); placebo: 20 (17.9%); difference- 34.5 (95% CI, 25 to 44); P value < 0.0001).
Excluding war-affected patients in Ukraine: (infliximab SC: 114/214 (53.3%); placebo: 19/104 (18.3%); difference- 36.2 (95% CI, 25.3 to 45.4); P value < 0.0001).
Excluding all patients in Ukraine: (infliximab SC: 107/202 (53.0%); placebo: 18/99 (18.2%); difference- 36.8 (95% CI, 25.5 to 46.2); P value < 0.0001).
Sensitivity Analyses for the Primary Outcome in the LIBERTY-UC Trial
Clinical Remission
Fisher’s Exact Test:(infliximab SC: 127 (43.2%); placebo: 30 (20.8%); difference- 22.4 (95% CI, 12.7 to 30.8); P value < 0.0001).
Logistic Regression: (infliximab SC:127 (43.2%); placebo: 30 (20.8%); difference- 23.2 (95% CI,14.3 to 32.1); P value < 0.0001).
Excluding War-Affected Patients in Ukraine: (infliximab SC: 21/267 (45.3%); placebo: 27/124 (21.8%); difference- 21.4 (95% CI,11.3 to 30.3); P value < 0.0001).
Excluding All Patients in Ukraine (infliximab SC: 119/254 (46.9%); placebo: 25/118 (21.2%); difference- 23.3 (95% CI, 13.0 to 32.3); P value < 0.0001).
Subgroup Analyses
LIBERTY-UC Trial
Table 29: Subgroup Analyses for Clinical Remission at Week 54 (All-Randomized Population) in the LIBERTY-UC Trial
Category | Infliximab SC (N = 294) n/N’ (%) of patients | Placebo (N = 144) n/N’ (%) of patients | Difference (95% CI)a | P-valueb |
|---|---|---|---|---|
Subgroup by Sex | ||||
Male Proportion of patients achieving clinical remission at week 54 | 70/163 (42.9) | 12/83 (14.5) | 26.1 (14.2, 36.2) | < 0.0001 |
Female Proportion of patients achieving clinical remission at week 54 | 57/131 (43.5) | 18/61 (29.5) | 14.3 (−0.7, 27.5) | 0.0500 |
Subgroup by Age | ||||
< 35 years Proportion of patients achieving clinical remission at week 54 | 58/129 (45.0) | 11/54 (20.4) | 23.7 (8.4, 36.0) | 0.0017 |
≥ 35 years Proportion of patients achieving clinical remission at week 54 | 69/165 (41.8) | 19/90 (21.1) | 19.1 (7.0, 29.7) | 0.0014 |
Subgroup by Race | ||||
White Proportion of patients achieving clinical remission at week 54 | 124/288 (43.1) | 30/140 (21.4) | 20.5 (11.0, 28.9) | < 0.0001 |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; JAK = Janus kinase; SC = subcutaneous.
Note: Clinical remission was defined as modified Mayo score with a stool frequency subscore of 0 or 1 point, rectal bleeding subscore of 0 point, and endoscopic subscore of 0 or 1 point. Analysis was stratified by previous exposure to biologic drug and/or JAK inhibitors (used or not used), use of treatment with oral corticosteroids at week 0 (used or not used) and clinical remission at week 10 (remitter or nonremitter by modified Mayo score). Patients with dose adjustment to CT-P13 SC 240 mg before week 54 were considered as nonremitter.
N’ = number of patients in the subgroup.
aThe difference of proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CI with CMH weights were presented.
bThe nominal P value from stratified CMH test is presented in descriptive purpose.
Source: Sponsor’s submission28
LIBERTY-CD Trial
Table 30: Subgroup Analysis for Coprimary End Point Endoscopic Activity Score for CD at Week 54 (All-Randomized Population) in the LIBERTY-CD Trial
Category | Infliximab SC (N = 231) n/N’ (%) of patients | Placebo (N = 112) n/N’ (%) of patients | Difference (95% CI)a | P valueb |
|---|---|---|---|---|
Subgroup by sex | ||||
Male Proportion of patients achieving endoscopic response based on central SES-CD at week 54 | 68/134 (50.7) | 13/69 (18.8) | 32.6 (18.8, 44.0) | < 0.0001 |
Female Proportion of patients achieving endoscopic response based on central SES-CD at week 54 | 50/97 (51.5) | 11/77 (14.3) | 38.2 (20.8, 51.1) | < 0.0001 |
Subgroup by age | ||||
< 35 years Proportion of patients achieving endoscopic response based on central SES-CD at week 54 | 58/108 (53.7) | 11/77 (14.3) | 41.9 (28.2, 53.0) | < 0.0001 |
≥ 35 years Proportion of patients achieving endoscopic response based on central SES-CD at week 54 | 60/123 (48.8) | 9/35 (25.7) | 28.0 (9.1, 42.7) | 0.0034 |
Subgroup by race | ||||
White Proportion of patients achieving endoscopic response based on central SES-CD at week 54 | 105/211 (49.8) | 17/101 (16.8) | 34.5 (23.6, 43.7) | < 0.0001 |
CDAI = Crohn Disease Activity Index; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; SC = subcutaneous; SES-CD = Simplified Endoscopic Activity Score for Crohn Disease.
Note: Endoscopic response was defined as a 50% decrease in SES-CD score from baseline value. Analysis was stratified by previous exposure to biologic drug and/or JAK inhibitors (used or not used), use of treatment with oral corticosteroids at week 0 (used or not used) and clinical remission at week 10 (remitter or nonremitter by modified Mayo score). Patients with dose adjustment to CT-P13 SC 240 mg before week 54 were considered as nonremitter.
N’ = number of patients in the subgroup
aThe difference of proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CI with CMH weights were presented.
bThe nominal P value from stratified CMH test is presented in descriptive purpose.
Source: Sponsor’s submission.28
Table 31: Subgroup Analysis for Coprimary End Point Clinical Remission (Based on CDAI) at Week 54 (All-Randomized Population) in the LIBERTY-CD Trial
Category | Infliximab SC (N = 231) n/N’ (%) of patients | Placebo (N = 112) n/N’ (%) of patients | Difference (95% CI)a | P-valueb |
|---|---|---|---|---|
Subgroup by Sex | ||||
Male Proportion of patients achieving clinical remission based on CDAI at week 54 | 87/134 (64.9) | 24/69 (34.8) | 30.7 (16.1, 43.5) | < 0.0001 |
Female Proportion of patients achieving clinical remission based on CDAI at week 54 | 57/97 (58.8) | 12/43 (27.9) | 35.0 (16.9, 49.6) | < 0.0001 |
Subgroup by Age | ||||
< 35 years Proportion of patients achieving clinical remission based on CDAI at week 54 | 72/108 (66.7) | 25/77 (32.5) | 36.5 (21.8, 49.1) | < 0.0001 |
≥ 35 years Proportion of patients achieving clinical remission based on CDAI at week 54 | 72/123 (58.5) | 11/35 (31.4) | 30.1 (11.2, 45.7) | 0.0009 |
Subgroup by Race | ||||
White Proportion of patients achieving clinical remission based on CDAI at Week 54 | 131/211 (62.1) | 32/101 (31.7) | 32.2 (20.4, 42.6) | < 0.0001 |
CDAI = Crohn Disease Activity Index; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; SC = subcutaneous.
Note: Clinical remission was defined as an absolute CDAI score of < 150 points. Analysis was stratified by previous exposure to biologic drug and/or JAK inhibitors (used or not used), use of treatment with oral corticosteroids at week 0 (used or not used) and clinical remission at week 10 (remitter or nonremitter by modified Mayo score). Patients with dose adjustment to CT-P13 SC 240 mg before week 54 were considered as nonremitter.
N’ = number of patients in the subgroup.
aThe difference of proportions between 2 treatment groups was estimated using CMH weights, and the 95% stratified Newcombe CI with CMH weights were presented.
bThe nominal P value from stratified CMH test is presented in descriptive purpose.
Source: Sponsor’s submission.28
Table 32: Concomitant Medication Use in the Maintenance Phase of the LIBERTY-UC Trial
Category | Infliximab SC 120 mg (N = 296) | Placebo (N = 140) |
|---|---|---|
Number of patients with at least 1 concomitant medication | 295 (99.7%) | 138 (98.6%) |
Mesalazine | 254 (85.8%) | 127 (90.7%) |
Propofol | 131 (44.3%) | 55 (39.3%) |
Fentanyl | 77 (26.0%) | 31 (22.1%) |
Azathioprine | 63 (21.3%) | 27 (19.3%) |
Prednisone | 61 (20.6%) | 30 (21.4%) |
Macrogol 4000; potassium chloride; sodium bicarbonate; sodium chloride; Sodium Sulphate Anhydrous | 60 (20.3%) | 27 (19.3%) |
Midazolam Hydrochloride | 52 (17.6%) | 22 (15.7%) |
Citric Acid; Magnesium Oxide; Sodium Picosulfate | 49 (16.6%) | 26 (18.6%) |
Paracetamol | 48 (16.2%) | 21 (15%) |
Ascorbic Acid; Macrogol3350; potassium chloride; Sodium Ascorbate; sodium chloride; Sodium Sulphate | 38 (12.8%) | 14 (10%) |
Methylprednisolone | 37 (12.5%) | 17 (12.1%) |
Tozinameran | 31 (10.5%) | 17 (12.1%) |
Magnesium Sulphate; Potassium Sulphate; Sodium Sulphate | 27 (9.1%) | 18 (12.9%) |
Midazolam | 24 (8.1%) | 10 (7.1%) |
Budesonide | 23 (7.8%) | 13 (9.3%) |
Cholecalciferol | 21 (7.1%) | 9 (6.4%) |
Sulfasalazine | 18 (6.1%) | 4 (2.9%) |
Cetirizine | 14 (4.7%) | 8 (5.7%) |
Hydrocortisone | 14 (4.7%) | 6 (4.3%) |
Pantoprazole Sodium Sesquihydrate | 14 (4.7%) | 5 (3.6%) |
Folic Acid | 13 (4.4%) | 2 (1.4%) |
Prednisolone | 13 (4.4%) | 3 (2.1%) |
Ascorbic Acid; Ferrous Sulphate | 12 (4.1%) | 9 (6.4%) |
Electrolytes Nos | 11 (3.7%) | 5 (3.6%) |
Pantoprazole | 11 (3.7%) | 5 (3.6%) |
Sodium Chloride | 10 (3.4%) | 3 (2.1%) |
Lidocaine Hydrochloride | 9 (3.0%) | 2 (1.4%) |
Ciprofloxacin | 9 (3.0%) | 1 (0.7%) |
Ibuprofen | 9 (3.0%) | 5 (3.6%) |
Metamizole Sodium | 8 (2.7%) | 4 (2.9%) |
Ferrous Sulphate; Folic Acid | 8 (2.7%) | 4 (2.9%) |
Metronidazole | 8 (2.7%) | 3 (2.1%) |
Ramipril | 7 (2.4%) | 7 (5%) |
Pethidine Hydrochloride | 7 (2.4%) | 3 (2.1%) |
Sufentanil | 7 (2.4%) | 4 (2.9%) |
Cefuroxime axetil | 7 (2.4%) | 1 (0.7%) |
Cetirizine Hydrochloride | 7 (2.4%) | 3 (2.1%) |
Isoniazid | 7 (2.4%) | 7 (5%) |
Acetylsalicylic Acid | 7 (2.4%) | 3 (2.1%) |
Bisoprolol Fumarate | 7 (2.4%) | 7 (5%) |
Potassium Chloride | 7 (2.4%) | 2 (1.4%) |
Bisoprolol | 6 (2.0%) | 3 (2.1%) |
Loperamide Hydrochloride | 5 (1.7%) | 2 (1.4%) |
Source: Sponsor’s submission.28
Table 33: Concomitant Medication Use in the Maintenance Phase of the LIBERTY-CD Trial
Category | Infliximab SC 120 mg (N = 296) | Placebo (N = 105) |
|---|---|---|
Number of patients with at least 1 concomitant medication, n (%) | 235 (98.7%) | 104 (99.0%) |
Mesalazine | 142 (59.7%) | 57 (54.3%) |
Propofol | 126 (52.9%) | 48 (45.7%) |
Fentanyl citrate | 66 (27.7%) | 25 (23.8%) |
Azathioprine | 64 (26.9%) | 33 (31.4%) |
Paracetamol | 55 (23.1%) | 13 (12.4%) |
Midazolam | 49 (20.6%) | 18 (17.1%) |
Budesonide | 44 (18.5%) | 9 (8.6%) |
Ascorbic acid; Macrogol 3350; potassium chloride; Sodium Ascorbate; sodium chloride; Sodium Sulphate | 42 (17.6%) | 16 (15.2%) |
Midazolam hydrochloride | 40 (16.8%) | 22 (21.0%) |
Citric acid; magnesium oxide; sodium picosulfate | 37 (15.5%) | 20 (19.0%) |
Prednisone | 35 (14.7%) | 16 (15.2%) |
Macrogol 4000; potassium chloride; sodium bicarbonate; sodium chloride; Sodium Sulphate Anhydrous | 34 (14.3%) | 18 (17.1%) |
Tozinameran | 34 (14.3%) | 8 (7.6%) |
Macrogol | 22 (9.2%) | 5 (4.8%) |
Cetirizine | 19 (8.0%) | 3 (2.9%) |
Methylprednisolone | 18 (7.6%) | 12 (11.4%) |
Magnesium sulphate; potassium sulphate; sodium sulphate | 18 (7.6%) | 1 (1.0%) |
Pantoprazole sodium sesquihydrate | 15 (6.3%) | 10 (9.5%) |
Folic acid | 14 (5.9%) | 4 (3.8%) |
Metronidazole | 14 (5.9%) | 9 (8.6%) |
Loperamide hydrochloride | 14 (5.9%) | 12 (11.4%) |
Ibuprofen | 14 (5.9%) | 0 |
Lidocaine hydrochloride | 12 (5.0%) | 3 (2.9%) |
Ascorbic acid; ferrous sulphate | 11 (4.6%) | 7 (6.7%) |
Cetirizine hydrochloride | 11 (4.6%) | 1 (1.0%) |
Isoniazid | 11 (4.6%) | 5 (4.8%) |
Prednisolone | 11 (4.6%) | 7 (6.7%) |
Omeprazole | 11 (4.6%) | 8 (7.6%) |
Citric acid; macrogol 4,000; potassium chloride; simeticone; sodium chloride; sodium citrate; sodium sulphate | 11 (4.6%) | 5 (4.8%) |
Pethidine hydrochloride | 10 (4.2%) | 4 (3.8%) |
Sulfasalazine | 10 (4.2%) | 3 (2.9%) |
Hydrocortisone | 10 (4.2%) | 1 (1.0%) |
Metamizole sodium | 9 (3.8%) | 7 (6.7%) |
Colecalciferol | 9 (3.8%) | 4 (3.8%) |
Sufentanil | 8 (3.4%) | 5 (4.8%) |
Ciprofloxacin | 8 (3.4%) | 2 (1.9%) |
Hyoscine butylbromide | 8 (3.4%) | 4 (3.8%) |
Fentanyl | 7 (2.9%) | 5 (4.8%) |
Ketamine hydrochloride | 7 (2.9%) | 2 (1.9%) |
Amoxicillin trihydrate; clavulanate potassium | 7 (2.9%) | 2 (1.9%) |
Acyclovir | 6 (2.5%) | 0 |
Levothyroxine sodium | 6 (2.5%) | 1 (1.0%) |
Azithromycin | 5 (2.1%) | 1 (1.0%) |
Desloratadine | 5 (2.1%) | 0 |
Mercaptopurine | 5 (2.1%) | 2 (1.9%) |
Pantoprazole | 5 (2.1%) | 4 (3.8%) |
Ondansetron | 4 (1.7%) | 2 (1.9%) |
Clotrimazole | 4 (1.7%) | 0 |
Enoxaparin sodium | 4 (1.7%) | 2 (1.9%) |
Nadroparin calcium | 4 (1.7%) | 2 (1.9%) |
Sodium chloride | 4 (1.7%) | 2 (1.9%) |
Metoclopramide hydrochloride | 4 (1.7%) | 0 |
Methotrexate sodium | 4 (1.7%) | 1 (1.0%) |
Potassium chloride | 4 (1.7%) | 3 (2.9%) |
Acetylsalicylic acid | 3 (1.3%) | 1 (1.0%) |
Atorvastatin calcium | 3 (1.3%) | 0 |
Dexpanthenol | 3 (1.3%) | 0 |
Methotrexate | 2 (0.8%) | 1 (1.0%) |
Source: Sponsor’s submission.28
Table 34: Summary of Dose Adjustment in the Safety Population, LIBERTY-UC Trial
Category | Infliximab SC 120 mg (N = 296) | Placebo (N = 140) |
|---|---|---|
Number of patients who received at least 1 adjusted dose | ||
In the treatment period (except for extension phase), n (%) | 92 (31.1%) | 75 (53.6%) |
In the maintenance phase | 92 (31.1%) | 75 (53.6%) |
Number of patients who received adjusted dose- maintenance phase, n (%) | ||
Week 22 | 60 (20.3%) | 49 (35%) |
Week 24 | 61 (20.6%) | 53 (37.9%) |
Week 26 | 63 (21.3%) | 54 (38.6%) |
Week 28 | 60 (20.3%) | 54 (38.6%) |
Week 30 | 64 (21.6%) | 60 (42.9%) |
Week 32 | 62 (20.9%) | 60 (42.9%) |
Week 34 | 62 (20.9%) | 60 (42.9%) |
Week 36 | 62 (20.9%) | 60 (42.9%) |
Week 38 | 62 (20.9%) | 60 (42.9%) |
Week 40 | 60 (20.3%) | 59 (42.1%) |
Week 42 | 63 (21.3%) | 56 (40%) |
Week 44 | 63 (21.3%) | 57 (40.7%) |
Week 46 | 61 (20.6%) | 60 (42.9%) |
Week 48 | 61 (20.6%) | 57 (40.7%) |
Week 50 | 61 (20.6%) | 57 (40.7%) |
Week 52 | 61 (20.6%) | 56 (40%) |
Week 54 | 67 (22.6%) | 56 (40%) |
Source: Sponsor’s submission.28
Table 35: Summary of Dose Adjustment in the Safety Population, LIBERTY-CD Trial
Category | Infliximab SC 120 mg (N = 296) | Placebo (N = 140) |
|---|---|---|
In the treatment period (except for extension phase) | 45 (18.9%) | 48 (45.7%) |
In the maintenance phase | 45 (18.9%) | 48 (45.7%) |
Number of patients who received adjusted dose- maintenance phase | ||
Week 22 | 21 (8.8%) | 29 (27.6%) |
Week 24 | 21 (8.8%) | 33 (31.4%) |
Week 26 | 22 (9.2%) | 35 (33.3%) |
Week 28 | 21 (8.8%) | 35 (33.3%) |
Week 30 | 27 (11.3%) | 38 (36.2%) |
Week 32 | 27 (11.3%) | 39 (37.1%) |
Week 34 | 23 (9.7%) | 38 (36.2%) |
Week 36 | 24 (10.1%) | 39 (37.1%) |
Week 38 | 30 (12.6%) | 39 (37.1%) |
Week 40 | 31 (13.0%) | 39 (37.1%) |
Week 42 | 32 (13.4%) | 39 (37.1%) |
Week 44 | 33 (13.9%) | 39 (37.1%) |
Week 46 | 33 (13.9%) | 40 (38.1%) |
Week 48 | 32 (13.4%) | 39 (37.1%) |
Week 50 | 32 (13.4%) | 40 (38.1%) |
Week 52 | 32 (13.4%) | 40 (38.1%) |
Week 54 | 35 (14.7%) | 41 (39.0%) |
Note: Patient 3504 to 0001 who received infliximab SC 180 mg actual administration at week 52 despite dose adjustment after week 38 is considered as received adjusted dose. Patient 2827 to 0001 who received placebo SC 120 mg actual administration from week 46 to week 52 despite dose adjustment after week 22 is not considered as received adjusted dose. Patient 2874 to 0006 who received infliximab SC 120 mg actual administration. At week 28 despite dose adjustment after week 22 is not considered as received adjusted dose. Patient 2901 to 0007 who received placebo SC 240 mg actual administration at week 30 despite dose adjustment after week 22 is not considered as received adjusted dose.
Source: Sponsor’s submission.28
Table 36: Summary of the Most Severe Adverse Events (Grade 3 or Higher) in the Maintenance Phase of the LIBERTY-UC Trial — Safety Population
Laboratory category CTCAE term CTCAE grade | Infliximab 120 mg (N = 296) | Placebo (N = 140) |
|---|---|---|
Clinical Chemistry, n (%) of patients | ||
Alanine aminotransferase increased | ||
Grade 3 (Severe) | 1 (0.3) | 0 |
Aspartate aminotransferase increased | ||
Grade 3 (Severe) | 2 (0.7) | 0 |
CPK increased | ||
Grade 3 (Severe) | 5 (1.7) | 4 (2.9) |
Grade 4 (Life-Threatening) | 4 (1.4) | 2 (1.4) |
Cholesterol high | ||
Grade 3 (Severe) | 0 | 1 (0.7) |
GGT increased | ||
Grade 3 (Severe) | 0 | 1 (0.7) |
Hyperkalemia | ||
Grade 3 (Severe) | 1 (0.3) | 0 |
Hypertriglyceridemia | ||
Grade 3 (Severe) | 4 (1.4) | 1 (0.7) |
Grade 4 (Life-Threatening) | 0 | 1 (0.7) |
Hyponatremia | ||
Grade 4 (Life-threatening) | 1 (0.3) | 0 |
Hematology | ||
Anemia Grade 3 (Severe) | 6 (2.0) | 1 (0.7) |
Lymphocyte count decreased Grade 3 (Severe) | 1 (0.3) | 1 (0.7) |
Neutrophil count decreased Grade 3 (Severe) | 11 (3.7) | 1 (0.7) |
Grade 4 (Life-Threatening) | 0 | 1 (0.7) |
White blood cell decreased Grade 3 (Severe) | 0 | 1 (0.7) |
CTCAE = Common Terminology Criteria for Adverse Events; CP K = creatine phosphokinase; GGT = gamma glutamyl transferase; SC = subcutaneous.
Note: If a patient’s value fails to satisfy any CTCAE criteria, the result is classified as no grade. For patients with dose adjustment, all data collected regardless of dose adjustment for CT-P13 SC group and data collected before initiation of dose adjustment for placebo SC group are included in this summary.
Source: Sponsor’s submission28
Table 37: Summary of the Most Severe Adverse Events (Grade 3 or Higher) in the Maintenance Phase of the LIBERTY-CD Trial — Safety Population
Laboratory category CTCAE term CTCAE grade | Infliximab SC 120 mg (N = 238) | Placebo SC (N = 105) |
|---|---|---|
Clinical chemistry, n (%) of patients | ||
Alanine aminotransferase increased Grade 3 (Severe) | 2 (0.8) | 0 |
Aspartate aminotransferase increased Grade 3 (Severe) | 1 (0.4) | 0 |
Blood bilirubin increased Grade 3 (Severe) | 5 (2.1) | 1 (1.0) |
CPK increased Grade 3 (Severe) | 6 (2.5) | 1 (1.0) |
Grade 4 (Life-Threatening) | 8 (3.4) | 2 (1.9) |
GGT increased Grade 3 (Severe) | 1 (0.4) | 1 (1.0) |
Hypertriglyceridemia Grade 3 (Severe) | 5 (2.1) | 0 |
Grade 4 (life-threatening) | 1 (0.4) | 0 |
Hypocalcemia Grade 3 (Severe) | 1 (0.4) | 0 |
Hypokalemia Grade 3 (Severe) | 1 (0.4) | 0 |
Hematology | ||
Anemia Grade 3 (Severe) | 5 (2.1) | 4 (3.8) |
Lymphocyte count decreased Grade 3 (Severe) | 2 (0.8) | 5 (4.8) |
Neutrophil count decreased Grade 3 (Severe) | 11 (4.6) | 0 |
Grade 4 (Life-Threatening) | 2 (0.8) | 0 |
CPK = creatine phosphokinase; CTCAE = Common Terminology Criteria for Adverse Events; GGT = gamma-glutamyltransferase; SC = subcutaneous.
Note: At each level of summarization, only the most severe case (before dose adjustment for placebo SC group) was counted. If a patient’s most extreme postbaseline value failed to satisfy any CTCA criteria, the result was classified as “no grade.”
Source: Sponsor’s submission.28
Table 38: Summary of the Most Severe Adverse Events (Grade 3 or Higher) in the Maintenance Phase of Study 1.6 Part 2 — Safety Population
Laboratory category CTCAE term CTCAE grade | Infliximab SC 120/240 mg (N = 66) | Infliximab IV 5 mg/kg (N = 65) |
|---|---|---|
Clinical Chemistry, n (%) of patients | ||
Alanine aminotransferase increased Grade 3 (Severe) | 0 | 1 (1.5) |
CPK increased Grade 4 (Life-Threatening) | 2 (3.0) | 0 |
GGT increased Grade 3 (Severe) | 1 (1.5) | 0 |
Hypertriglyceridemia Grade 3 (Severe) | 2 (3.0) | 0 |
Hematology | ||
Anemia Grade 3 (Severe) | 1 (1.5) | 0 |
Lymphocyte count decreased Grade 3 (Severe) | 2 (3.0) | 0 |
Neutrophil count decreased Grade 3 (Severe) | 3 (4.5) | 4 (6.2) |
Grade 4 (Life-Threatening) | 1 (1.5) | 1 (1.5) |
Platelet count decreased Grade 3 (Severe) | 1 (1.5) | 0 |
White blood cell decreased Grade 3 (Severe) | 0 | 2 (3.1) |
CPK = creatine phosphokinase; CTCAE = Common Terminology Criteria for Adverse Events; GGT = gamma-glutamyltransferase SC = subcutaneous.
Note: The summary included only the most severe case during scheduled visit, unscheduled visit, or end-of-study visit. Percentages were calculated by using the number of patients in the safety population as the denominator. At each level of summarization, only the most severe case was counted. The results after study drug administration at week 6 were included.
Source: Sponsor’s submission28
Mayo Score Assessment
Clinical response and remission were assessed by the Mayo score. The Mayo score composed of the patient’s Mayo score diary entries and assessments performed by the site investigator including PGA and flexible proctosigmoidoscopy. The total Mayo score was summed up of the stool frequency, rectal bleeding, endoscopic, and PGA subscores. The modified Mayo score was summed up of the 3 components of the total Mayo score excluding PGA subscore, and the partial Mayo score was summed up of the 3 components of the total Mayo score excluding ES. The components of the Mayo Scoring System are presented in Table 39.
No. | Items | Score |
|---|---|---|
1 | Stool frequencya Normal no. of stools for this patient | 0 |
1 to 2 stools more than normal | 1 | |
3 to 4 stools more than normal | 2 | |
5 or more stools more than normal | 3 | |
2 | Rectal bleedingb No blood seen | 0 |
Streaks of blood with stool less than half the time | 1 | |
Obvious blood (more than just streaks) or streaks of blood with stool most of the time | 2 | |
Blood alone passes | 3 | |
3 | Findings of flexible proctosigmoidoscopyc Normal or inactive disease | 0 |
Mild disease (erythema, decreased vascular pattern) | 1 | |
Moderate disease (marked erythema, absent vascular pattern, friability, erosions) | 2 | |
Severe disease (spontaneous bleeding, ulceration) | 3 | |
4 | Physician’s global assessmentd | |
Normal | 0 | |
Mild disease | 1 | |
Moderate disease | 2 | |
Severe disease | 3 |
PGA = physician’s global assessment.
Note: Total Mayo score ranged from 0 to 12, with higher scores indicating more severe disease.
Modified Mayo scores ranged from 0 to 9, excluding PGA, with higher scores indicating more severe disease.
aEach patient served as their own control to establish the degree of abnormality of the stool frequency.
bThe daily bleeding score represented the most severe bleeding of the day. Rectal bleeding subscore of the Mayo Score was modified in accordance with FDA guidance so that a value of 2 consisted of obvious blood (more than just streaks) and streaks of blood with stool most of the time.
cThe endoscopic subscore of the Mayo Score was modified in accordance with FDA guidance so that a value of 1 did not included friability.
dThe PGA acknowledged the 3 other criteria: the patient’s recollection of abdominal discomfort and general sense of well-being, and other observations, such as physical findings and the patient’s performance status.
Source: Sponsor’s submission.28
Appendix 2: Submitted Budget Impact Analysis and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 40: Summary of Key Takeaways
Key takeaways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the expected budget impact of reimbursing infliximab SC for reduction of signs and symptoms, and maintenance of clinical remission and mucosal healing, and reduction of corticosteroid use in adult patients with moderately to severely active CD or UC who have had an inadequate response to a corticosteroid and/or aminosalicylate and/or an immunosuppressant.77 The sponsor submitted separate budget impact models and reports for CD and UC, however there was significant overlap and similarity in the models.
A claims-based approach was taken to estimate the number of claims that may be displaced by the introduction of infliximab SC based upon historical drug purchasing behaviour. The sponsor used the IQVIA Pharmastat database81 to estimate the number of claims of infliximab-based products and biologic DMARD products marketed in Canada, which were then inflated by a 2% annual growth rate to estimate the number of claims forecasted in 2024 to 2026. The proportion of these biological claims reimbursed for the indications of interested were estimated based on an Ontario Drug Policy Research Network report82 which reported that, of the patients using the identified comparators in 2019, 35.5% were using them for IBD. Among the IBD claims, half were for CD, and the other half were for patients with UC. 30% of patients were assumed to require induction in any given year, with the exception of infliximab SC, where 100% of patients were assumed to be in the induction phase in year 1, followed by 30% in each year thereafter. Key inputs to the BIA are documented in Table 41.
The sponsor made the following key assumptions:
The availability of infliximab SC is not expected to grow the total market for infliximab or biologic DMARD products, so the total number of claims is constant in both the reference and new drug scenarios.
Table 41: Summary of Key Model Parameters
Crohn disease | Ulcerative colitis | ||
|---|---|---|---|
Parameter | Sponsor’s estimate (Year 1 / Year 2 / Year 3) | Parameter | Sponsor’s estimate (Year 1 / Year 2 / Year 3) |
Proportion of comparator market applied to each indication | |||
IBD CD UC | 35.5%82 18% 18% | IBD CD UC | 35.5%82 18% 18% |
Number of included claims | 87,016 / 88,756 / 90,531 | Number of included claims | 96,638 / 98,571 / 100,542 |
Market Uptake (reference scenario) | |||
Adalimumab (Humira) Adalimumab (B/S) Golimumab (Simponi) Infliximab (Remicade) Infliximab (Avsola) Infliximab (Inflectra) Infliximab (Renflexis) Ustekinumab (Stelara) Vedolizumab (Entyvio) | 33.3% 20.5% 0% 10.9% 0.4% 11.1% 4.1% 5.3% 14.4% | Adalimumab (Humira) Adalimumab (B/S) Golimumab (Simponi) Infliximab (Remicade) Infliximab (Avsola) Infliximab (Inflectra) Infliximab (Renflexis) Ustekinumab (Stelara) Vedolizumab (Entyvio) | 30.0% 18.5% 14.7% 9.8% 0.3% 10.0% 3.7% 0% 13.0% |
Market Uptake (new drug scenario) | |||
Infliximab (Remsima) Adalimumab (Humira) Adalimumab (B/S) Golimumab (Simponi) Infliximab (Remicade) Infliximab (Avsola) Infliximab (Inflectra) Infliximab (Renflexis) Ustekinumab (Stelara) Vedolizumab (Entyvio) | 0.29% / 0.58% / 0.86% 33.23% / 33.13% / 33.04% 20.48% / 20.42% / 20.37% 0% / 0% / 0% 10.84% / 10.81% / 10.78% 0.38% / 0.38% / 0.38% 11.11% / 11.08% / 11.05% 4.07% / 4.06% / 4.05% 5.24% / 5.23% / 5.21% 14.35% / 14.31% / 14.27% | Infliximab (Remsima) Adalimumab (Humira) Adalimumab (B/S) Golimumab (Simponi) Infliximab (Remicade) Infliximab (Avsola) Infliximab (Inflectra) Infliximab (Renflexis) Ustekinumab (Stelara) Vedolizumab (Entyvio) | 0.29% / 0.58% / 0.86% 29.92% / 29.83% / 29.75% 18.44% / 18.39% / 18.34% 14.65% / 14.61% / 14.57% 9.76% / 9.74% / 9.71% 0.35% / 0.34% / 0.34% 10.01% / 9.98% / 9.95% 3.66% / 3.65% / 3.64% 0% / 0% / 0% 12.92% / 12.88% / 12.84% |
Cost of treatment (per 8-week claim, in maintenance year) | |||
Cost per claim Infliximab (Remsima) Adalimumab (Humira) Adalimumab (B/S) Golimumab (Simponi) Infliximab (Remicade) Infliximab (Avsola) Infliximab (Inflectra) Infliximab (Renflexis) Ustekinumab (Stelara) Vedolizumab (Entyvio) | $2,366 $3,176 $1,885 — $3,950 $1,972 $2,100 $1,972 $4,593 $3,402 — | Cost per claim Infliximab (Remsima) Adalimumab (Humira) Adalimumab (B/S) Golimumab (Simponi) Infliximab (Remicade) Infliximab (Avsola) Infliximab (Inflectra) Infliximab (Renflexis) Ustekinumab (Stelara) Vedolizumab (Entyvio) | $2,366 $3,176 $1,885 $2,863 $3,950 $1,972 $2,100 $1,972 — $3,402 — |
CD = Crohn disease; IBD = irritable bowel disease; UC = ulcerative colitis.
Summary of the Sponsor’s BIA Results
For the CD analysis, the sponsor concluded that the reimbursement of infliximab SC would be associated with a budgetary increase of $22,032 in year 1, and budgetary savings of $171,030 and $261,676 in Years 2 and 3, respectively, for a total savings of $410,674 over the three-year time horizon.
For the UC analysis, the sponsor concluded that the reimbursement of infliximab SC would be associated with budgetary savings of $20,416 in year 1, $281,507 in year 2, $430,705 in year 3, for a total savings of $732,628 over the three-year time horizon.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Use of a claims-based approach to estimate market size introduces uncertainty with the anticipated budget impact of infliximab: The sponsor estimated market size based on public claims data for the relevant comparators. The sponsor assumed that approximately 35.5% of claims attributed to patients who were likely using the identified comparators were for treating CD or UC. Given the claims database does not specify indication and the proportion of claims pertaining to use for other indications is unknown, using a claims-based approach to estimate market size introduces significant uncertainty in the estimated market size. Furthermore, the sponsor did not concert the claims data into the number of users; instead, the sponsor assumed unit to unit and claim to claim displacement between infliximab and comparators.
CADTH could not undertake a reanalysis to address this limitation.
Characteristics of patient population used in the model may not align with clinical expectations: In the sponsor-submitted model, a mean weight of 73.20 kg was used to represent adult patients with moderately to severely active CD or UC who have had an inadequate response to a corticosteroid and/or aminosalicylate and/or an immunosuppressant. CADTH obtained feedback from the clinical expert that highlighted that the target population in Canada is likely overweight or obese and the average used in the model may not be representative of the pan-Canadian population. The clinical expert highlighted that the average weight of the target may be closer to 85 kg.
To address this limitation, CADTH undertook a scenario analysis to assess the impact of a higher average target population weight.
Actual price for drugs paid by public drug plans is uncertain: The sponsor’s and CADTH’s analyses are both based on publicly available list prices for all comparators. Actual prices paid by public drug plans are unknown.
This limitation could not be addressed by CADTH. It is possible that the budget impacts estimated by both CADTH and the sponsor for reimbursing infliximab SC at its submitted price are optimistic than would be actualized due to confidential pricing agreements for the comparator products.
CADTH Reanalyses of the BIA
CADTH did not undertake a base-case reanalysis. Instead, CADTH explored the potential impact of several scenario analyses which included:
Increasing the average population weight to 85 kg, due to feedback obtained from the clinical expert consulted by CADTH.
16% reduction in the price of infliximab SC to align with annual costs of the least costly infliximab IV product.
40% reduction in the price of infliximab SC to align with annual costs of the least costly noninfliximab product.
The results are presented in Table 42. The reimbursement of infliximab SC was associated with a three-year incremental budgetary savings of $732,628 and incremental budgetary savings of $410,674 in the UC and CD populations, respectively, for a total incremental savings of $1,143,302 over 3 years.
All of the CADTH scenario reanalyses resulted in incremental cost savings when both diseases were considered separately or together. Increasing the average patient weight led to an increase in product use, which in turn, increased the incremental cost savings reported in both analyses. A 16% price reduction led to doubling the incremental cost savings when both analyses were considered, and a 40% price reduction led to just under four-times the incremental cost savings. Notably, all analyses were conducted assuming publicly available list prices for all comparators, which likely overestimates the budgetary benefit of reimbursing infliximab SC at the submitted price.
Table 42: Summary of the CADTH Exploratory Analysis of the BIA, Reporting 3-Year Totals
Stepped analysis | Crohn disease | Ulcerative colitis | Sum of both analyses |
|---|---|---|---|
CADTH base case | (410,674) | (732,628) | (1,143,302) |
CADTH scenario A: target population characteristics | (576,552) | (881,441) | (1,457,993) |
CADTH scenario B: 16% price reduction to equal annual cost of least costly infliximab product | (958,182) | (1,340,680) | (2,298,862) |
CADTH scenario C: 40% price reduction to equal annual cost of least costly noninfliximab product | (1,779,445) | (2,252,758) | (4,032,203) |
BIA = budget impact analysis. Paratheses indicate budgetary cost savings.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third-party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for noncommercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.