Drugs, Health Technologies, Health Systems
Reimbursement Review
Danicopan (Voydeya)
Sponsor: Alexion Pharma GmbH
Therapeutic area: Paroxysmal nocturnal hemoglobinuria
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
BTH
breakthrough hemolysis
C3
complement component 3
C3i
complement component 3 inhibitor
C5
complement component 5
C5i
complement component 5 inhibitor
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EQ VAS
EQ visual analogue scale
ESS
effective sample size
EVH
extravascular hemolysis
FA
full analysis
FACIT-F
Functional Assessment of Chronic Illness Therapy–Fatigue
GRADE
Grading of Recommendations, Assessment, Development, and Evaluations
Hb
hemoglobin
HRQoL
health-related quality of life
IA
interim analysis
IEAS
interim efficacy analysis set
ITT
intention to treat
IVH
intravascular hemolysis
LDH
lactate dehydrogenase
LS
least squares
LTE
long-term extension
MAC
membrane attack complex
MAIC
multiple-adjusted indirect comparison
MID
minimal important difference
PNH
paroxysmal nocturnal hemoglobinuria
QoL
quality of life
RBC
red blood cell
RCT
randomized controlled trial
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
SLR
systematic literature review
TD
treatment difference
TEAE
treatment-emergent adverse event
TP1
treatment period 1
TP2
treatment period 2
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Danicopan (Voydeya), 50 mg and 100 mg film-coated tablets, oral administration |
Sponsor | Alexion Pharma GmbH |
Indication | As an add-on to ravulizumab or eculizumab for the treatment of adult patients with PNH who have residual hemolytic anemia due to EVH |
Reimbursement request | As per Health Canada indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | July 19, 2024 |
Recommended dose | The recommended starting dose is 150 mg t.i.d. administered orally, approximately 8 hours apart (± 2 hours). The dose can be increased to 200 mg t.i.d. if a patient’s hemoglobin level has not increased by at least 2 g/dL after 4 weeks of therapy, if a patient required a transfusion within the previous 4 weeks, or to achieve an appropriate hemoglobin response based on clinical judgment. |
EVH = extravascular hemolysis; NOC = Notice of Compliance; PNH = paroxysmal nocturnal hemoglobinuria; t.i.d. = 3 times a day.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, chronic, and potentially life-threatening blood condition caused by an acquired genetic defect in hematopoietic stem cells.1,2 This defect leads to the production of blood cells that lack 2 glycosylphosphatidylinositol-anchored complement regulatory proteins, CD55 and CD59, at their surface, causing the complement system to recognize red blood cells (RBCs) as damaged. The uncontrolled activation of the complement cascade prematurely attacks these cells resulting in hemolysis. Symptoms of PNH can vary significantly among individuals, and the disease can affect any race, ethnicity, or sex. It may manifest at any age,3,4 although it typically emerges in young adults, with the median age of diagnosis being around 30 years.1,2
Intravascular hemolysis (IVH) occurs in both terminal and proximal pathways when RBCs are directly lyzed because of the activation of the alternative complement pathway.5 Patients with PNH are susceptible to an increased risk of thrombosis, pain, organ damage (e.g., impaired renal function), underlying bone marrow dysfunction,1,3,6-8 and increased risk of morbidity and mortality.1,9 They also have an increased need for transfusions, which can impair health-related quality of life (HRQoL).1,3,6,10-14
In Canada, ravulizumab and eculizumab are complement component 5 inhibitors (C5is) used as first-line therapy to treat hemolytic PNH. This treatment regimen addresses uncontrolled complement activation through a complete complement component 5 (C5) inhibition in the terminal complement cascade and helps reduce symptoms and complications, resulting in improved survival for patients with PNH.15,16 However, some patients receiving C5i treatment remain anemic and transfusion-dependent. Possible causes of this include breakthrough hemolysis (BTH), extravascular hemolysis (EVH), nutritional deficiencies, and bone marrow failure.17
EVH is a mechanistic consequence believed to be caused by ongoing complement component 3 (C3) deposition on surviving yet defective RBCs, which makes them vulnerable to phagocytosis in the liver or spleen.18-20 While symptoms of EVH are not life-threatening, its manifestation is heterogeneous. For some patients, EVH may consist of having normal hemoglobin (Hb) levels21 and being asymptomatic,22,23 while others may develop severe clinical symptoms and may require blood transfusions to manage ongoing anemia.22,24,25 Clinical trial and real-world data show that approximately 20% of patients with PNH who were clinically stable on C5i treatment develop clinically significant EVH.26
The historical approach to managing anemia due to EVH in patients in Canada with PNH has been supportive care (e.g., RBC transfusions, corticosteroids, splenectomy, danazol, and epoetin alfa) and continuing C5i treatment to prevent the life-threatening consequences of IVH.20 Pegcetacoplan, a subcutaneous (SC) proximal C3 inhibitor (C3i) is an approved therapy indicated for patients with inadequate response to, or intolerant of, a C5i.27 Per the clinical experts consulted by Canada’s Drug Agency (CDA-AMC), this option would currently be offered as a second-line pharmacologic option to patients diagnosed with EVH.
Because of the rarity of the disease, the prevalence and incidence of PNH have been poorly reported, and published prevalence and incidence estimates of PNH and EVH are not available for the population of people living in Canada. A study in the US estimated the prevalence of PNH at 1.2 to 1.3 per 100,000 persons between 2016 and 2017. The incidence rate over the study period was 0.57 per 100,000 person-years.28
Danicopan selectively inhibits complement alternative pathway factor D,29 which plays a key role in amplifying complement system response. Danicopan is thought to mediate the deposition of C3 fragments on PNH blood cells, which is a key cause of EVH in patients receiving ravulizumab or eculizumab for PNH. Inhibition of factor D activity specifically targets the control point of the complement cascade amplification loop, blocking C3 convertase formation and thereby reducing the production of C3 fragments and downstream membrane attack complex (MAC) formation.30 Although danicopan blocks the alternative pathway–mediated amplification of the complement classical pathway and lectin pathway, these 2 pathways remain active to provide residual complement-dependent protection against infectious pathogens.31 When co-administered with ravulizumab or eculizumab, danicopan is anticipated to maintain control over C5 and MAC-mediated IVH.
Danicopan has a Health Canada indication as an add-on to ravulizumab or eculizumab for the treatment of adult patients with PNH who have residual hemolytic anemia due to EVH.29 The sponsor reimbursement request is as per the indication. The recommended starting dose of danicopan is 150 mg 3 times a day administered orally, approximately 8 hours apart (± 2 hours). The dose can be increased to 200 mg 3 times a day if a patient’s Hb level has not increased by at least 2 g/dL after 4 weeks of therapy, if a patient required transfusion within the previous 4 weeks, or to achieve an appropriate Hb response based on clinical judgment.29 Danicopan should not be administered as monotherapy and should only be prescribed as an add-on to ravulizumab or eculizumab.29
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of danicopan 50 mg and 100 mg film-coated oral tablets as an add-on to ravulizumab or eculizumab, to treat adult patients with PNH who are experiencing signs and symptoms of EVH.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to the CDA-AMC call for input and from clinical expert(s) consulted by CDA-AMC for the purpose of this review.
Patient Input
The Canadian Association of PNH Patients and the Aplastic Anemia & Myelodysplasia Association of Canada submitted a joint input for this review. A clinical summary of PNH was provided and information was gathered through the personal experiences of 1 patient living in Canada who received danicopan.
The patient group input expressed that PNH significantly impacts the quality of life for both patients and their caregivers. Beyond the persistent fatigue and weakness caused by chronic anemia from hemolysis, patients deal with other symptoms such as abdominal pain and dysphagia which influence their dietary habits and social interactions. Managing symptoms requires ongoing medical interventions, medication adjustments, and lifestyle changes. The input noted that even though currently available treatments for PNH, such as C5is (ravulizumab and eculizumab) and a C3i (pegcetacoplan), effectively inhibit IVH, thrombosis, and EVH, approximately 20% of patients continue to experience EVH and persistent anemia and require frequent blood transfusions. The financial costs associated with treatment exacerbate stress, creating a significant economic strain on patients and families. This wide-ranging impact underscores the importance of holistic management approaches to effectively support both patients and their caregivers in managing PNH.
The input stated that patients, caregivers, and families affected by PNH desire tolerable treatment options that reduce treatment burden, decrease hemolysis symptoms, decrease dependency on blood transfusions, slow disease progression, and improve long-term outcomes and quality of life. The input indicated that the 1 patient with experience with danicopan noticed a remarkable improvement in her symptoms.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
CDA-AMC consulted 2 clinical experts with experience treating PNH for this review. Per the clinical experts, PNH is a complicated disease. The initial goals of therapy are to reduce mortality, reduce complications and morbidities associated with IVH, as well as reduce transfusion needs, improve HRQoL with better Hb support and avoidance of iron overload, and help patients attain better functional status and return to prediagnosis activities and employment. The initial treatment of choice for PNH is a C5i, which controls IVH and thus the major mortality and morbidity of the disease, as most deaths in patients with PNH are because of thrombotic complications.
C5is can provide incomplete control of PNH in some circumstances. Possible causes include rare genetic mutations (in people of Japanese ethnicity), inadequate dosing of the C5i, response to vaccination, or infections leading to BTH or symptomatic EVH related to C5 inhibition. The experts estimated that approximately 40% of patients with PNH will continue to have low Hb despite therapy, approximately 30% will require transfusions, and EVH will contribute to poor HRQoL in 20% to 30% of patients.
Per the experts, there is no standard definition for EVH and a diagnosis of EVH generally requires ruling out other possible causes of anemia, which may be challenging as patients often have other comorbidities and it may not be evident that anemia is due to 1 cause. Clinical diagnosis for EVH typically requires anemia along with normal or minimally elevated lactate dehydrogenase (LDH), as well as elevated bilirubin and reticulocyte counts. Alternative explanations for anemia which the experts noted would have to be ruled out include bone marrow failure, hematinic deficiencies (such as vitamin B12 or ferritin), renal insufficiency, or blood loss.
Treatment goals for patients with PNH and EVH remain to reduce mortality, inhibit IVH, and improve HRQoL by providing better Hb support that does not require transfusion, avoids iron overload, and leads to better functional status for patients. The main nonpharmacologic treatment for EVH and persistent anemia in PNH while on C5i treatment is transfusion support, which is associated with several drawbacks such as lengthy hospital visits and risks with transfusion including infection, antibody development, or iron overload. In addition, most patients receiving transfusions will have significantly reduced HRQoL and be unable to maintain regular employment.
Pegcetacoplan is the primary pharmacologic option offered to patients with clinically significant EVH. Pegcetacoplan is a subcutaneous (SC) infusion with twice-weekly dosing and specific transportation requirements. If BTH occurs, the experts noted that the frequency of pegcetacoplan will usually be increased to 3 times weekly.
The experts noted that danicopan would be an alternative to pegcetacoplan as a second-line drug and would be used as an add-on therapy for patients already on a C5i. Some patients already on pegcetacoplan may wish to switch to danicopan plus C5i if they were having ongoing BTH or issues with SC infusions.
Response to therapy would typically be an improvement in Hb and a reduction in transfusion requirements relative to the baseline for a given patient. The experts noted that ongoing anemia and transfusion needs may or may not be a treatment failure, as it is possible that other concurrent diseases such as bone marrow failure, aplastic anemia, other cancers, or comorbidities could be contributing factors. Intolerance or allergy to danicopan would be reason to discontinue therapy, as would a lack of improvement in Hb levels and transfusion needs. The experts noted that an episode of BTH or transfusion requirement in another setting would not be considered a treatment failure, nor would a required stoppage of therapy because of pregnancy or breastfeeding. Stopping danicopan therapy should be considered independent of the C5i as that treatment controls IVH.
Clinician Group Input
One clinician group, the Canadian PNH Network, submitted input for this review based on contributions from 9 clinicians. Information was gathered through publicly available documents, congress abstracts, and published literature.
The clinician group agreed with the clinical experts that the current standard of care for PNH is a C5i (i.e., eculizumab and ravulizumab), which acts via terminal complement blockade, and that there are still some unmet therapeutic needs within the available PNH treatment regimen. The clinician group input agreed with the clinical experts that some patients remain anemic due to EVH, and some remain transfusion-dependent with C5i.
The clinician group agreed with the experts that a subset of patients would benefit from proximal complement inhibition given the development of clinically significant EVH, but for whom pegcetacoplan is less than ideal. Dual complement blockade (i.e., C5i plus danicopan) would provide these patients with the same benefits of improved Hb but with a lower risk of complications.
The clinician group and the clinical experts were also aligned on the patients most likely to benefit from danicopan — those who have persistent anemia despite stable-dose C5i, in whom EVH is suspected. Patients who may receive proximal inhibition monotherapy (e.g., pegcetacoplan), who may not tolerate it, or have repeated BTH or other concerns could also benefit from the therapy. The input further noted that treatment is least suitable for those who are not anemic, or who meet exclusion criteria in clinical trials such as pregnancy.
The clinician input noted that clinically meaningful response to treatment would be sustained control of LDH but with further Hb increases and improvement in anemia-related symptoms. A lack of improvement in the first few months of therapy would be a prompt to increase the dose. Danicopan discontinuation should be considered in patients who develop adverse events (AEs) that preclude ongoing therapy, including poor treatment compliance and intolerable side effects. The most important feature to monitor for would be evidence of BTH.
The clinical experts and clinician group input agreed that patients with PNH should be followed by clinicians who specialize in the condition.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for danicopan:
considerations for relevant comparators
considerations for initiation or renewal of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
generalizability
care provision issues.
The clinical expert consulted by CDA-AMC provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
The ALPHA trial is an ongoing phase III, double-blind, randomized, placebo-controlled trial which enrolled a total of 86 patients with PNH who had clinically significant EVH and were receiving treatment with ravulizumab or eculizumab. The study used a 45-day screening period and randomization was stratified by transfusion history (> 2 transfusions or ≤ 2 transfusions in the 6 months before screening), Hb at screening (< 8.5 g/dL or ≥ 8.5 g/dL), and Japanese patient (yes or no). Stochastic dynamic allocation rules were used to randomize patients 2:1 through an interactive response technology to either receive danicopan 3 times a day added onto their C5i or a placebo 3 times a day added onto their C5i monotherapy, respectively. The study design consisted of a 12-week treatment period 1 (TP1) which was randomized, double-blind, and placebo-controlled, followed by a 12-week treatment period 2 (TP2) where patients initially randomized to placebo switched to receive danicopan and patients initially randomized to danicopan continued to receive danicopan. Patients completing TP2 were eligible to continue onto a total of 2 long-term extensions (LTE1 or LTE2); results from patients who have completed LTE1 to date were included in the submission.
The prespecified interim analysis (IA) submitted for this reimbursement review was planned for when approximately 75% (N = 63 patients) of the total planned sample had been randomized and completed the TP1; the purpose of this analysis, per the submission, was to assess stopping early for efficacy. The data cut-off for the TP1 IA was conducted on June 28, 2022, and a second interim data analysis for TP2 results was conducted with a data cut-off of September 20, 2022. A total of 63 patients formed the interim efficacy analysis set (IEAS) and a total of 86 patients (the entire randomized study sample) formed the interim safety analysis set.
Patients eligible to participate in the study were required to be aged 18 years or older, have a diagnosis of PNH, and have clinically significant EVH defined as patients presenting with anemia (Hb ≤ 9.5 g/dL) and increased reticulocyte count (≥ 120 × 109/L), with or without the need for transfusion, had to be receiving an approved C5i (ravulizumab or eculizumab) with no change in dose or interval for at least 6 months, as well as meet a platelet count threshold of 30,000 or more per µL and a neutrophil count of 500 or more per µL. Patients were eligible regardless of transfusion status. Patients were excluded if they had a history or presence of any clinically significant medical condition or comorbidity, including any conditions leading to anemia that are not primarily because of PNH; if they had any procedures and/or laboratory anomalies which would put them at undue risk to receive danicopan; or patients who were, or who had partners who were pregnant, nursing, or planning to become pregnant during the study or within 90 days of study intervention.
All patients received either danicopan or placebo in the form of 50 mg or 100 mg film-coated oral tablets. To assess adherence, adherence was calculated as a percentage of danicopan doses taken divided by the doses scheduled to be taken. The dosage administered started at 150 mg 3 times a day; dosing could be escalated up to a maximum of 200 mg at specific time points and specific clinical circumstances in the study.
The primary outcome was change in Hb levels from baseline to week 12. Key secondary outcomes were the proportion of patients with Hb increase of 2 g/dL or greater in the absence of transfusion at week 12, transfusion avoidance (transfusion-free and not requiring transfusion) at week 12, change in absolute reticulocyte count from baseline to week 12, and change in Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F) scores from baseline to week 12. The primary and key secondary outcomes were controlled for multiple comparisons and an alpha-spending procedure was applied to account for the fact that a smaller sample size than was required by the power calculations was used for this analysis. The alpha-spending procedure and hierarchical testing structure controlled the family-wise type I error rate for these end points. Secondary outcomes were the proportion of patients with Hb normalization (defined as patients with Hb values > lower limit of the normal reference range [110 g/L for female patients and 125 g/L for male patients]);32 transfusion burden, defined as the number of RBC units transfused and the number of transfusion instances; and change in LDH from baseline. Exploratory outcomes were change from baseline in the EQ visual analogue scale (EQ VAS) scores and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) global health status/quality of life score. All primary, key secondary, secondary, and exploratory outcomes were measured at weeks 12 and 24; Hb, absolute reticulocyte count, LDH, FACIT-F, EQ-5D-3L, and EORTC QLQ-C30 were also measured at week 72 among patients with data at that time point, and reported as LTE1 results.
Most baseline characteristics were broadly similar between study arms. There was a numeric difference in the proportion of patients (66.7% female in the placebo plus C5i arm, 54.8% female in the danicopan plus C5i arm; 33.3% male in the placebo plus C5i arm, 45.2% male in the danicopan plus C5i arm), and the proportion of patients of Asian descent (33.3% in the placebo plus C5i arm, 42.9% in the danicopan plus C5i arm). There were also numeric differences in the proportion of patients treated with each C5i (64.3% of patients in the danicopan plus C5i arm and 47.6% of patients in the placebo plus C5i arm were treated with ravulizumab). There was a numerically higher LDH in the danicopan plus C5i arm (298.73 U/L) relative to the placebo plus C5i arm (278.25 U/L), and a numerically higher proportion of patients in the danicopan plus C5i arm had received a transfusion within 24 weeks of receiving the study drug (90.5% in the danicopan plus C5i arm, 81.0% in the placebo plus C5i arm).
Efficacy Results
Change in Hb Levels
The least squares (LS) mean change from baseline in Hb level to 12 weeks was the primary outcome. At TP1, the LS mean difference for the change in Hb from baseline between the danicopan plus C5i and the placebo plus C5i arms was 24.44 g/L (98.2% confidence interval [CI], 15.25 g/L to 33.63 g/L; P ≤ 0.0001). At TP2, the LS mean change from baseline to week 24 in the danicopan-emergent arm (patients who received danicopan plus C5i from weeks 0 to 12 and continued to receive danicopan plus C5i from weeks 12 to 24) was 31.67 g/L (95% CI, 25.61 g/L to 27.74 g/L). In the placebo-emergent arm (patients who received placebo plus C5i from weeks 0 to 12 and who subsequently switched to receive danicopan plus C5i from weeks 12 to 24), the LS mean change from baseline to week 24 was 22.58 g/L (95% CI, 15.72 g/L to 29.44 g/L). At LTE1, the observed mean change from baseline in Hb levels was 32.00 g/L (standard deviation [SD] = 11.81 g/L) in the danicopan-emergent arm and 31.50 g/L (SD = 10.61 g/L) in the placebo-emergent arm.
Proportion of Patients With Hb Level Increase of 2 g/dL or Greater in the Absence of Transfusion
The proportion of patients with Hb level increases of 2 g/dL or greater was a key secondary outcome in the analysis. At TP1, the LS mean difference for the proportion of patients with Hb level increase of 2 g/dL or greater between the danicopan plus C5i and the placebo plus C5i arms was 45.90% (95.8% CI, 27.40% to 64.42%; P ≤ 0.0001). At TP2, the proportion of patients with this outcome in the danicopan-emergent arm was 46.3% (95% CI, 30.66% to 62.58%); results were not reported for the placebo-emergent arm. This outcome was not reported at LTE1 in either arm.
Proportion of Patients With Hb Normalization
The proportion of patients with Hb normalization was a secondary outcome. At TP1, the LS mean difference for the change in the proportion of patients with Hb normalization between the danicopan plus C5i and the placebo plus C5i arms was 18.40% (95% CI, –0.84% to 37.71%; P = 0.0080). At TP2, the proportion of patients with this outcome in the danicopan-emergent arm was 19.50% (95% CI, 8.82% to 34.87%). This outcome was not reported for the placebo-emergent arm at TP2 and was not reported at LTE1 for either arm.
Transfusion Avoidance
Transfusion avoidance at TP1 was a key secondary outcome in the analysis. At TP1, the LS mean treatment difference (TD) for the proportion of patients with transfusion avoidance between the danicopan plus C5i and the placebo plus C5i arms was 40.80% (95.8% CI, 21.08% to 60.58%; P = 0.0004). At TP2, the proportion of patients with this outcome in the danicopan-emergent arm was 78.00% (95% CI, 62.39% to 89.44%), and was 90.00% (95% CI, 68.30% to 98.77%) in the placebo-emergent arm. This outcome was not reported at LTE1 in either arm.
Transfusion Burden
Transfusion burden was measured by the number of RBC units transfused and the number of transfusion instances; both were secondary outcomes. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in the number of RBC units transfused between the 12 weeks before study drug initiation and the 12 weeks after study drug initiation was –1.31 (95.8% CI, –2.24 to –0.37; P = 0.0072). At TP2, the change in the number of RBC units transfused in the 24 weeks after treatment initiation relative to the 24 weeks before treatment initiation in the danicopan-emergent arm was –2.80 (95% CI, –4.55 to –1.11). This outcome was not reported in the placebo-emergent arm or at LTE1 in either arm.
At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in the number of transfusion instances between the 12 weeks before study drug initiation and the 12 weeks after study drug initiation was –0.72 (95% CI, –1.32 to –0.11; P = 0.0207). At TP2, the change in the number of transfusion instances between the 24 weeks before study drug initiation and the 24 weeks after study drug initiation in the danicopan-emergent arm was –1.50 (95% CI, –2.36 to –0.67). This outcome was not reported in the placebo-emergent arm or at LTE1 in either arm.
Absolute Reticulocyte Count
Change in absolute reticulocyte count from baseline to week 12 was a key secondary outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in absolute reticulocyte count from baseline was –0.087 × 1012/L (95.8% CI, –0.119 × 1012/L to –0.056 × 1012/L; P ≤ 0.0001). At TP2, the change from baseline in absolute reticulocyte counts in the danicopan-emergent arm was –0.080 × 1012/L (SD = 0.073 × 1012/L), and in the placebo-emergent arm was –0.084 × 1012/L (SD = 0.110 × 1012/L). At LTE1, the observed mean change from baseline in absolute reticulocyte counts in the danicopan-emergent arm was –0.041 × 1012/L (SD = 0.029 × 1012/L), and in the placebo-emergent arm was –0.106 × 1012/L (SD = not applicable; n = 1 patient).
Lactate Dehydrogenase
Change in LDH from baseline was a secondary outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in LDH from baseline was –20.57 U/L (95% CI, –49.28 U/L to 8.15 U/L; P = 0.1569). At TP2, the mean change from baseline in LDH in the danicopan-emergent arm was –23.46 U/L (SD = 105.40 U/L), and in the placebo-emergent arm was 0.21 U/L (SD = 84.89 U/L). At LTE1, the mean change from baseline in LDH in the danicopan-emergent arm was –20.83 U/L (SD = 67.00 U/L), and in the placebo-emergent arm was 5.00 U/L (SD = 111.89 U/L).
Functional Assessment of Chronic Illness Therapy–Fatigue
The change in FACIT-F (ranging from 0 [extreme fatigue] to 52 [no fatigue] with higher scores indicating less fatigue)33 scores from baseline was a key secondary outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in FACIT-F scores from baseline was 6.12 (95.8% CI, 2.18 to 10.06; P = 0.0021). At TP2, the LS mean change from baseline in FACIT-F scores in the danicopan-emergent arm was 6.12 (95% CI, 3.41 to 8.82), and in the placebo-emergent arm was 6.44 (95% CI, 1.23 to 11.64). At LTE1, the mean change from baseline in the danicopan-emergent arm was 3.86 (SD = 7.15) and –4.33 (SD = 9.07) in the placebo-emergent arm.
EQ Visual Analogue Scale
The change in EQ VAS (health rating on a scale from 0 to 100, with 0 representing the worst imaginable health state and 100 the best)34,35 scores from baseline was an exploratory outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change from baseline in EQ VAS scores was 6.27 (95% CI, –2.85 to 15.40; P = 0.1738). At TP2, the mean change from baseline in EQ VAS scores was 13.70 (SD = 20.12) in the danicopan-emergent arm and 9.70 (SD = 21.93) in the placebo-emergent arm. At LTE1, the mean change from baseline in the danicopan-emergent arm was 12.30 (SD = 18.70) and –11.00 (SD = 12.73) in the placebo-emergent arm.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
The change in EORTC QLQ-C30 global health (standardized score ranging from 0 to 100, higher score represents higher HRQoL)36 scores from baseline was an exploratory outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change from baseline in EORTC QLQ-C30 global health scores was 6.62 (95% CI, –1.17 to 14.41; P = 0.0941). At TP2, the mean change from baseline in EORTC QLQ-C30 global health scores was 8.56 (SD = 16.96) in the danicopan-emergent arm and 10.53 (SD = 14.92) in the placebo-emergent arm. At LTE1, the mean change from baseline in the danicopan-emergent arm was 1.19 (SD = 26.97) and 8.33 (SD = 22.05) in the placebo-emergent arm.
Harms Results
Harms were reported separately for TP1, TP2, and LTE1 cut-offs, as well as overall during the entire time patients were exposed to danicopan (total danicopan treatment). Overall, a total of 93.0% of patients in the danicopan-emergent arm and 82.6% of patients in the placebo-emergent arm experienced treatment-emergent AEs (TEAEs) during treatment with danicopan.
During TP1, there were numeric differences in the proportion of patients experiencing TEAEs for anemia (1.8% danicopan plus C5i, 13.8% placebo plus C5i), vomiting (5.3% danicopan plus C5i, 0% placebo plus C5i), upper abdominal pain (1.8% danicopan plus C5i, 6.9% placebo plus C5i), pyrexia (5.3% danicopan plus C5i, 0% placebo plus C5i), asthenia (0% danicopan plus C5i, 13.8% placebo plus C5i), ear infection (0% danicopan plus C5i, 6.9% placebo plus C5i), contusion (1.8% danicopan plus C5i, 10.3% placebo plus C5i), increased aspartate aminotransferase (3.5% danicopan plus C5i, 10.3% placebo plus C5i), pain in extremity (5.3% danicopan plus C5i, 0% placebo plus C5i), dizziness (1.8% danicopan plus C5i, 6.9% placebo plus C5i), and insomnia (1.8% danicopan plus C5i, 10.3% placebo plus C5i). A total of 57 patients in the danicopan plus C5i arm and 29 patients in the placebo plus C5i arm contributed data. There were numeric differences in the proportion of patients experiencing TEAEs for nausea (2.1% danicopan-emergent, 13.0% placebo-emergent), and pyrexia (10.4% danicopan-emergent, 0% placebo-emergent). A total of 48 patients in the danicopan-emergent arm and 23 patients in the placebo-emergent arm contributed data. During the LTE there were numeric differences in the proportion of patients experiencing TEAEs for diarrhea (2.5% danicopan-emergent, 10.0% placebo-emergent), asthenia (2.5% danicopan-emergent, 15.0% placebo-emergent), and back pain (2.5% danicopan-emergent, 10.0% placebo-emergent). A total of 40 patients in the danicopan-emergent arm and 20 patients in the placebo-emergent arm contributed data.
Overall, a total of 12.3% of patients in the danicopan-emergent arm and 26.1% of patients in the placebo-emergent arm experienced any serious AE (SAE) while being treated with danicopan. During TP1, 5.3% of patients in the danicopan plus C5i arm experienced any SAE; the SAEs were pancreatitis, cholecystitis, COVID-19, and blood bilirubin increase (1 report of each). A total of 6.9% of patients in the placebo plus C5i arm experienced any SAE; the SAEs were anemia, abdominal pain, and headache (1 report of each). During TP2, 6.3% of patients in the danicopan-emergent arm experienced any SAE; the SAEs were Dieulafoy vascular malformation, pyrexia, COVID-19 pneumonia, and staphylococcus sepsis (1 report of each). In the placebo-emergent arm, 13.0% of patients experienced any SAE; the SAEs were hemolysis, vertigo, and headache (1 report of each). During LTE, 7.5% of patients in the danicopan-emergent arm experienced any SAE; the SAEs were stent-grant endoleak, decreased Hb, invasive ductal breast carcinoma, pulmonary embolism, and pulmonary hemorrhage (1 report of each). In the placebo-emergent arm, 20.0% of patients experienced any SAE; the SAEs were pericardial effusion, diarrhea, disease progression, COVID-19, and body temperature increased (1 report of each).
During TP1, TEAEs led to withdrawal of the study drug for 5.3% of patients in the danicopan plus C5i arm and 3.4% of patients in the placebo plus C5i arm. SAEs led to withdrawal of the study drug for 1.8% of patients in the danicopan plus C5i arm, and 0% of patients in the placebo plus C5i arm. During TP2, there were no TEAEs or SAEs leading to withdrawal of the study drug in either treatment arm. During LTE, TEAEs led to withdrawal of the study drug in 5.0% of patients in the placebo-emergent arm; there were no TEAEs leading to withdrawal of the study drug in the danicopan-emergent arm. There were no SAEs leading to withdrawal of the study drug in either treatment arm. There were no deaths reported in either study arm, at any time point during the trial to date.
Meningococcal infections and liver enzyme elevations were prespecified AEs of special interest during the ALPHA study. Throughout TP1, TP2, and LTE, there were no reported AEs of meningococcal infections in either study arm. During TP1, liver enzyme elevations occurred in 14.0% of patients in the danicopan plus C5i arm and 10.3% of patients in the placebo plus C5i arm. During TP2, liver enzyme elevations occurred in 6.3% of patients in the danicopan-emergent arm and 13.0% of patients in the placebo-emergent arm. During LTE, liver enzyme elevations occurred in 2.5% of patients in the danicopan-emergent arm and 5.0% of patients in the placebo-emergent arm. There was a total of 8 TEAEs of hemolysis reported in 7 patients during the study to date, 4 which were hemolysis and 4 of which were BTH based on investigator judgment. All patients were stable on their C5i. No case-specific details were provided in the submission on the management of the hemolysis or BTH events. Per the submission, no events led to treatment discontinuation, and none were associated with an LDH level greater than 2.2 × upper limit of normal (ULN).
Critical Appraisal
There are some limitations pertaining to patient disposition and patient characteristics to note. A total of 18.9% of patients failed to meet the inclusion or exclusion criteria, but it is not specified which inclusion or exclusion criteria were not met during screening; therefore, it is not known whether excluded patients were systematically different from included ones. In addition, while baseline characteristics were broadly balanced between study arms, the differences in the proportion of patients treated with each C5i (64.3% of patients in the danicopan plus C5i arm and 47.6% of patients in the placebo plus C5i arm were treated with ravulizumab) may bias the harms results as according to the clinical experts and literature, ravulizumab is the preferred C5i drug.37 In addition, TP1 and TP2 time points had numerically low patient dropout; however, the small number of patients who have completed LTE1 to date make long-term results for efficacy and safety highly uncertain. There are also some potential limitations associated with the study design. The ALPHA trial IA used a prespecified interim stopping criteria at 75% of patients, as well as an alpha-spending procedure for the primary and key secondary end points. However, given the IA was conducted based on 75% of the originally targeted sample size, there is an increased risk that the true effect of danicopan on these end points is overestimated by the IA. In addition, while the primary and key secondary end points were controlled for multiple comparisons, the secondary and exploratory outcomes were not controlled for this or for the smaller sample size, and there is a risk of inflated type I error when interpreting results from these comparisons. Furthermore, there are possible limitations pertaining to the numbers of complete cases in the danicopan plus C5i and subsequent danicopan-emergent arm; without further information on the patients who were missing, the degree to which the missingness may be informative to the results is not known. In addition, there was no placebo comparator after the end of TP1, therefore, observed results in TP2 and LTE may not all be attributable to treatment. Lastly, there are some potential limitations associated with outcome ascertainment. While laboratory outcomes such as Hb or LDH are likely at low risk of bias because of being centrally measured, the open-label design of TP2 and the LTE mean that knowledge of the treatment being received may impact reporting of subjective quality of life outcomes at those time points (impacting FACIT-F, EORTC QLQ-C30, and EQ-5D-3L outcomes). Similarly, while a measure of treatment adherence was reported in the study, this was based on tablet counts and there is a possibility of reporting bias.
There are some limitations regarding the study population to note. Per the clinical experts, most of the inclusion criteria were reasonable for patients with PNH in a Canadian context; however, the minimum thresholds for platelet and neutrophil counts, as well as the exclusion criteria ruling out patients with other causes of anemia or other clinical comorbidities may exclude patients who could be candidates for treatment in a real-world setting. The clinical experts also noted that while there are certain clinical characteristics alongside persistent anemia whose presence indicate that EVH is the likely cause, there is no standard diagnostic definition of the condition. The cut-off used in the ALPHA study to define anemia was a level at which the clinical experts speculated patients would likely feel symptoms and could require intervention, but was not based on a known standard. In addition, the clinical experts noted that transfusion practices vary greatly and are partially dependent on patient factors such as lifestyle or comorbidities. Therefore, the study population included in the ALPHA study may not represent all patients with PNH with EVH. There are also some limitations regarding the generalizability of the results to clinical situations. The frequency of visits used in the trial setting may not exactly reflect daily clinical practice in Canada and therefore the efficacy and safety profile during the trial may not be extrapolatable to the general patient population. During the trial, the approved C5i dose was not permitted to be increased, nor the interval shortened, which also may not reflect clinical practice. FACIT-F and EORTC QLQ-C30 are validated tools in patients with PNH, but the EQ-5D-3L is not validated in PNH specifically; therefore, changes in health status reflected in that score may not translate perfectly to changes in health status in PNH. Furthermore, there were no minimal important differences (MIDs) provided by the sponsor or the clinical experts for all but 1 of the outcomes in patients with PNH; therefore, information on clinically meaningful change for the majority of outcomes remains lacking.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations, Assessment, Development, and Evaluations (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform the CDA-AMC expert committee’s deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.38,39
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). The target of the certainty of evidence assessment was based on thresholds informed by the sponsor submission, input from the clinical experts, and/or thresholds identified in the literature. In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members.
Clinical outcomes — change from baseline to week 12 in the following:
Hb levels
proportion of patients with Hb increase of 2 g/dL or more in the absence of transfusion
transfusion avoidance
absolute reticulocyte count
transfusion burden (number of RBC units transfused; number of transfusion instances)
LDH
proportion of patients with Hb normalization.
Fatigue and HRQoL outcomes — change from baseline to week 12 in the following:
FACIT-F
EQ-5D-3L
EORTC QLQ-C30.
Mortality — proportion of patients who died
Harms — proportion of patients with meningococcal infections, proportion of patients with liver enzyme elevation
Table 2: Summary of Findings for Danicopan Plus C5i Versus Placebo Plus C5i for Patients With PNH Experiencing EVHa
Outcome and follow-up | Patients (studies), Nb | Absolute effects (CI)c | Certainty | What happens | ||
|---|---|---|---|---|---|---|
Placebo plus C5i | Danicopan plus C5i | Difference | ||||
Hematologic outcomes | ||||||
LS mean change in Hb from baseline (g/L) Follow-up: 12 weeks | 63 (1 RCT) | 4.96 (98.2% CI, –2.70 to 12.61) | 29.40 (98.2% CI, 24.23 to 34.57) | 24.44 (98.2% CI, 15.25 to 33.63) | Moderated | Treatment with danicopan plus C5i therapy likely results in an increase in Hb levels when compared to placebo plus C5i therapy. |
Proportion of patients with Hb increase of ≥ 2 g/dL (20 g/L) in the absence of transfusion (%) Follow-up: 12 weeks | 63 (1 RCT) | 0 (95.8% CI, 0.00 to 16.80) | 59.50 (95.8% CI, 42.73 to 74.84) | 45.90 (95.8% CI, 27.40 to 64.42) | Moderated | Treatment with danicopan plus C5i therapy likely results in an increase in the proportion of patients with a Hb increase of ≥ 2 g/dL (20 g/L) in the absence of transfusion when compared to placebo plus C5i therapy. The clinical importance of the increase is unclear. |
Proportion of patients achieving transfusion avoidance (transfusion-free and do not require a transfusion) (%) Follow-up: 12 weeks | 63 (1 RCT) | 38.10 (95.8% CI, 17.56 to 62.32) | 83.30 (95.8% CI, 68.08 to 93.27) | 40.80 (95.8% CI, 21.08 to 60.58) | Moderated | Treatment with danicopan plus C5i therapy likely results in an increase in the proportion of patients achieving transfusion avoidance (i.e., transfusion-free and do not require a transfusion) when compared to placebo plus C5i therapy. The clinical importance of the increase is unclear. |
LS mean change from baseline in absolute reticulocyte counts (1012/L) Follow-up: 12 weeks | 63 (1 RCT) | 0.004 (95.8% CI, –0.023 to 0.030) | –0.084 (95.8% CI, –0.102 to –0.065) | –0.087 (95.8% CI, –0.119 to –0.056) | Moderated | Treatment with danicopan plus C5i therapy likely results in an increase in the LS mean change from baseline in absolute reticulocyte counts when compared to placebo plus C5i therapy. The clinical importance of the increase is unclear. |
LS mean change from baseline in transfusion burden | ||||||
Number of RBC units transfusede Follow-up: 12 weeks pretrial to 12 weeks posttreatment | 63 (1 RCT) | –0.18 (95% CI, –0.94 to 0.59) | –1.48 (95% CI, –2.02 to –0.94) | –1.31 (95% CI, –2.24 to –0.37) | Moderated | Treatment with danicopan plus C5i therapy likely results in a decrease in the number of RBC units transfused when compared to placebo plus C5i therapy. The clinical importance of the decrease is unclear. |
Number of transfusion instancese Follow-up: 12 weeks pretrial to 12 weeks posttreatment | 63 (1 RCT) | –0.21 (95% CI, –0.70 to 0.29) | –0.92 (95% CI, –1.27 to –0.57) | –0.72 (95% CI, –1.32 to –0.11) | Moderated | Treatment with danicopan plus C5i therapy likely results in a decrease in the number of transfusion instances when compared to placebo plus C5i therapy. The clinical importance of the decrease is unclear. |
Proportion of patients with Hb normalization (Hb greater than the LLN for reference range)e Follow-up: 12 weeks | 63 (1 RCT) | 0 (95% CI, 0.00 to 16.11) | 28.6 (95% CI, 15.72 to 44.58) | 18.40 (95% CI, –0.84 to 37.71) | Lowd,f | Treatment with danicopan plus C5i therapy may result in an increase in the proportion of patients with Hb normalization when compared to placebo plus C5i therapy. |
LS mean change from baseline in LDHe Follow-up: 12 weeks | 63 (1 RCT) | –2.92 (95% CI, –26.76 to 20.93) | –23.49 (95% CI, –40.08 to –6.90) | –20.57 (95% CI, –49.28 to 8.15) | Lowd,g | Treatment with danicopan plus C5i therapy may result in a decrease in LDH when compared to placebo plus C5i therapy. The clinical importance of the decrease is unclear. |
Fatigue and HRQoL | ||||||
LS mean change from baseline in FACIT-F scores Follow-up: 12 weeks | 63 (1 RCT) | 1.85 (95.8% CI, –1.31 to 5.02) | 7.97 (95.8% CI, 5.72 to 10.23) | 6.12 (95.8% CI, 2.33 to 9.91) | Lowd,h | Treatment with danicopan plus C5i therapy may result in an increase in FACIT-F scores when compared to placebo plus C5i therapy. |
LS mean change from baseline in EQ VAS scorese Follow-up: 12 weeks | 63 (1 RCT) | 5.25 (95% CI, –2.46 to 12.96) | 11.53 (95% CI, 6.25 to 16.81) | 6.27 (95% CI, –2.85 to 15.40) | Lowd,g | Treatment with danicopan plus C5i therapy may result in little to no change in EQ VAS scores when compared to placebo plus C5i therapy. |
LS mean change from baseline in EORTC QLQ-C30 global health status/QoL scorese Follow-up: 12 weeks | 63 (1 RCT) | 3.80 (95% CI, –2.78 to 10.38) | 10.42 (95% CI, 5.87 to 14.97) | 6.62 (95% CI, –1.17 to 14.41) | Lowd,g | Treatment with danicopan plus C5i therapy may result in little to no change in EORTC QLQ-C30 global health status/QoL scores when compared to placebo plus C5i therapy. |
Harms | ||||||
Number of patients with meningococcal infections, n Follow-up: 72 weeks | 63 (1 RCT) | 0 (NR) | 0 (NR) | NR (NR) | Very lowd,i,j | The evidence is very uncertain about the effect of danicopan plus C5i therapy on the number of patients with meningococcal infections when compared to placebo plus C5i therapy. |
Number of patients with liver enzyme elevations, n Follow-up: 72 weeks | 63 (1 RCT) | 10 (NR) | 4 (NR) | NR (NR) | Very lowd,i,j | The evidence is very uncertain about the effect of danicopan plus C5i therapy on the number of patients with liver enzyme elevations when compared to placebo plus C5i therapy. |
Mortality | ||||||
Proportion of patients who died Follow-up: 72 weeks | 63 (1 RCT) | 0 (NR) | 0 (NR) | NR (NR) | Very lowd,i,j | The evidence is very uncertain about the effect of danicopan plus C5i therapy on the number of patients who died when compared to placebo plus C5i therapy. |
C5i = complement component 5 inhibitor; CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = EQ visual analogue scale; EVH = extravascular hemolysis; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; HRQoL = health-related quality of life; LDH = lactate dehydrogenase; LLN = lower limit of normal; LS = least squares; MID = minimal important difference; NR = not reported; PNH = paroxysmal nocturnal hemoglobinuria; QoL = quality of life; RBC = red blood cell; RCT = randomized controlled trial.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aClinically significant EVH was defined in ALPHA as anemia (Hb ≤ 9.5 g/dL) and absolute reticulocyte count ≥ 120 × 109/L.
bResults are from the interim efficacy analysis of ALPHA (N = 63 patients; 42 patients randomized to receive danicopan add-on therapy and 21 patients randomized to receive placebo add-on therapy).
cCIs for the primary outcome (change in Hb from baseline) are 98.2% and for the key secondary outcomes (proportion of patients with Hb increase of ≥ 2 g/dL in the absence of transfusion, proportion of patients achieving transfusion avoidance, change from baseline in absolute reticulocyte counts, change from baseline in FACIT-F scores) CIs are 95.8%, per the interim analysis alpha-spending procedure. For all other outcomes, CIs are 95%.
dRated down 1 level for serious indirectness. Per the clinical experts, there is no standard definition for EVH, the exclusion criteria do not provide a specific list of comorbidities or laboratory values used in screening, and the minimum requirements for platelet and neutrophil counts may exclude patients with comorbidities who could be considered for treatment with danicopan.
eStatistical testing for this outcome was not adjusted for multiple comparisons in the trial.
fRated down 1 level for serious imprecision. The clinical experts specified that the target for the certainty of evidence would be the presence of a non-null effect. The CI includes the possibility of a decrease in the outcome, no effect on the outcome, and an increase in the outcome.
gRated down 1 level for serious imprecision. The target of the certainty assessment is the presence of a non-null effect. The CI includes the possibility of potential benefit as well as potential harm.
hRated down 1 level for serious imprecision. The MID provided in the submission was a change in scores from baseline of 5 points. The CI includes the possibility of clinically meaningful benefit as well as the possibility of benefit that is not clinically meaningful.
iRated down 1 level for serious study limitations. The evidence submitted for the ALPHA study was an interim analysis, and as the study is still ongoing the reporting of harms information is incomplete and may bias the reported results.
jRated down 2 levels for very serious imprecision. There are a very small number of events captured.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence, the ALPHA Clinical Study Report,40 and additional information provided by the sponsor.41,42
LTE Studies
Results of the LTE of the ALPHA study are summarized in the systematic review section.
Indirect Comparisons
Description of Studies
Indirect evidence was required to be considered as part of the submission because the ALPHA trial compared danicopan plus C5i therapy with placebo plus C5i therapy; however, comparative data against pegcetacoplan, the other second-line therapeutic option for PNH, remains lacking. The submission included a systematic literature review (SLR) and feasibility assessment to undertake a matching-adjusted indirect comparison (MAIC) with the PEGASUS trial, which compared pegcetacoplan with eculizumab in adult patients with PNH. A naive comparison of these 2 trials was also submitted but was not appraised because of considerable methodological limitations with this method.
The feasibility assessment consisted of a comparison of the between-trial heterogeneity in trial design, trial end points, patient eligibility criteria, and baseline patient characteristics.
The MAIC analysis compared a subset of the ALPHA study population which was trimmed to meet the additional inclusion criteria which were a part of the PEGASUS study but not the ALPHA study:
body mass index less than 40 kg/m2
platelet count greater than 50,000/µL
The MAIC used a weighting approach per the methodology reported by Signorovitch et al.43 and qualitatively reported on the 2 methods in terms of balancing characteristics. The weight model included baseline Hb and baseline reticulocyte count. Efficacy results were reported in the anchored MAICs as differences of TDs for each trial (danicopan plus C5i minus placebo plus C5i; or pegcetacoplan minus eculizumab). The unanchored MAICs reported efficacy results as TDs between the danicopan plus C5i arm and the pegcetacoplan arm.
The distribution of calculated weights from both methods was reported, as well as the baseline characteristics after adjustment by both methods. After weighting, anchored and unanchored MAICs were undertaken for the following efficacy outcomes: change in Hb from baseline, change in absolute reticulocyte count from baseline, change in LDH from baseline, change in FACIT-F scores from baseline, and transfusion avoidance. The following safety outcomes were also reported from the MAICs: time-to-hemolysis AE and probability of BTH during extended follow-up (48 weeks for pegcetacoplan and 34.5 weeks for the ALPHA study). Time to discontinuation because of BTH was also reported, but in an unweighted population and therefore was not appraised. All analyses compared results from the ALPHA study at 12 weeks to results from the PEGASUS study at 20 weeks (the study design consisted of a 4-week run-in with C5i monotherapy coadministration, followed by a 16-week randomized period).
Efficacy Results
In the feasibility assessment, the sponsor detailed differences in trial design, inclusion criteria, baseline characteristics, and treatment duration between the ALPHA trial and the PEGASUS trial. Differences in the mean baseline Hb were also highlighted by the sponsor in the baseline characteristics between the trimmed ALPHA study population (7.7 g/dL in the danicopan plus C5i arm, 7.8 g/dL in the placebo plus C5i arm) and the PEGASUS study population (8.69 g/dL in the pegcetacoplan arm, 8.68 g/dL in the eculizumab arm). In addition, there were numeric differences between the trimmed ALPHA study population and the PEGASUS study population in the proportion of patients of Asian descent (47.4% danicopan plus C5i arm, 31.6% placebo plus C5i arm of the ALPHA study, versus 12% in the pegcetacoplan arm, 18% in the eculizumab arm of the PEGASUS study), proportion of white patients (42.1% danicopan plus C5i and 47.4% placebo plus C5i in the ALPHA study, versus 59% pegcetacoplan and 64% eculizumab in the PEGASUS study), absolute reticulocyte count (238.8 × 109 danicopan plus C5i and 242.9 × 109 placebo plus C5i in the ALPHA study, versus 217.5 × 109 pegcetacoplan and 216.2 × 109 eculizumab in the PEGASUS study), and total bilirubin (33.2 µmol/L danicopan plus C5i and 34.8 µmol/L placebo plus C5i in the ALPHA study, versus 42.5 µmol/L pegcetacoplan and 40.5 µmol/L eculizumab in the PEGASUS study). There was no information on the potential clinical importance of these differences in the submission.
The conclusions for the anchored and unanchored MAICs were numerically similar for most efficacy outcomes, with 2 exceptions: transfusion avoidance, where the unanchored MAIC showed that danicopan was favoured for transfusion avoidance, but the anchored MAIC did not (anchored TD = –0.32; 95% CI, –2.70 to 2.06; unanchored TD = 1.64; 95% CI, 0.06 to 3.22), and absolute reticulocyte count, where the reduction reported favoured pegcetacoplan with a greater reduction than danicopan plus C5i (anchored TD = 53.70; 95% CI, 16.90 to 90.50; unanchored TD = 32.80; 95% CI, 13.60 to 51.90). Neither danicopan plus C5i nor pegcetacoplan were favoured for the outcomes of Hb change from baseline, LDH change from baseline, change in FACIT-F scores from baseline, or transfusion avoidance (anchored MAIC only).
Harms Results
Based on a time-to-event analysis of BTH, there was no significant difference between the time to BTH AE for patients in the trimmed ALPHA study sample or in the PEGASUS study. Based on the extended follow-up from the PEGASUS study (48 weeks) and a median follow-up of 34.6 weeks from patients in the danicopan-emergent arm of the ALPHA study, the results from the weighted, unanchored MAIC found that there was no difference in the probability of BTH between the 2 trials.
Critical Appraisal
The indirect evidence assessment is subject to several major limitations that make drawing firm conclusions about the comparative results challenging. With regards to the SLR and feasibility assessment, the submission did not provide a preregistered protocol for the SLR and so it is not known whether the search criteria, study selection, or subgroups of interest were prespecified before the search. It is also not known whether statistical testing was undertaken during the feasibility assessment to determine differences in study population or whether there was a prespecified threshold to determine the meaningfulness of differences between populations. Per the clinical experts consulted by CDA-AMC, the differences highlighted in the feasibility assessment for inclusion criteria and baseline characteristics did not represent clinically meaningful differences. They noted that the anemia and platelet cut-offs being different was not hugely meaningful from a clinical perspective as the mean values for both in the baseline characteristics were similar; they also noted that patient-specific factors such as lifestyle and important symptoms are often a driver of treatment choices. As this information was not included in the submission, the impact of these factors on patient differences is unknown. Ravulizumab is the suggested C5i therapy over eculizumab when both are available; however, the 2 therapies have similar efficacy results.37 Therefore, there is enough overlap between the study populations to suggest that the reported characteristics do not represent enough of a source of heterogeneity to rule out a MAIC.
The MAICs themselves are also subject to considerable limitations. The anchored MAICs provided control on 2 treatment effect modifiers and the sponsor noted that these were the only effect modifiers able to be adjusted on; however, the clinical experts noted that the modifiers used in weighting were not a comprehensive list of possible modifiers or prognostic factors. Therefore, the anchored MAICs would not be able to account for all possible sources of heterogeneity between the study populations. In addition, key differences in the comparator arms for the ALPHA and PEGASUS trials were noted including which C5i therapies were used in the placebo arm and the duration of follow-up, which suggests that the comparators in these 2 trials may not be an appropriate anchor for the MAIC. This increases the uncertainty in the results, and thus, drawing firm conclusions based on these results about the comparative effectiveness of danicopan add-on and pegcetacoplan is not recommended. Unanchored MAICs were also undertaken for all efficacy and safety outcomes. This method requires the assumption that all prognostic factors and treatment effect modifiers are accounted for, which is a strong assumption largely considered impossible to meet — failure of this assumption leads to an unknown amount of bias in the effect estimate.
In addition, the ALPHA and PEGASUS trials differ in other ways which may impact the risk of bias in the results and the generalizability of the results. Patients in the PEGASUS study were exposed to pegcetacoplan monotherapy for 4 weeks longer than patients were exposed to danicopan in the ALPHA study, which may bias the efficacy results to favour pegcetacoplan. Furthermore, the trial design for pegcetacoplan was an open-label trial, which may bias the reporting of FACIT-F, a subjective outcome. The results from the MAICs are subject to the same concerns about generalizability to the PNH population as the ALPHA study, and without detailed information from the PEGASUS study, the generalizability of that study population to the wider PNH population is not known. In addition, results were only reported for efficacy outcomes at week 20 for the PEGASUS study and week 12 for the ALPHA study, and so any information on efficacy past this time is not known. For BTH events, these were reported only up to 48 weeks in the PEGASUS study and 34.6 weeks for the ALPHA study; therefore, longer-term data on safety and information on other harms is unknown.
Studies Addressing Gaps in the Evidence From the Systematic Review
A phase II dose-finding study was submitted providing information on lower doses of danicopan add-on therapy; as these doses either overlapped with the dosing from the ALPHA study or were outside of the approved indication, the study was not appraised.
Conclusions
PNH is a rare disease with significant morbidity and mortality — mortality is predominantly because of thrombosis related to IVH and is treated by C5i therapies (ravulizumab or eculizumab). Approximately 20% of patients with PNH who were clinically stable on C5i treatment develop clinically significant EVH.26 Evidence from the IA of the ALPHA study, a phase III RCT with a 12-week placebo-controlled, double-blind portion plus a 12-week single-arm, open-label extension and a LTE for an additional 52 weeks was appraised to assess the impact of danicopan added on to C5i therapy versus placebo plus C5i therapy. The results demonstrated that over 12 weeks, when compared with placebo plus C5i therapy, danicopan plus C5i therapy likely increased Hb levels, the proportion of patients with Hb increase of 2 g/dL or more in the absence of transfusion, and the proportion of patients with transfusion avoidance. In addition, danicopan plus C5i therapy likely decreased markers of transfusion burden and absolute reticulocyte counts, and may increase the proportion of patients attaining Hb normalization. Results from week 24, the open-label, single-arm treatment period of the ALPHA study where all patients were receiving danicopan therapy, suggested this trend was maintained for most hematologic outcomes. Danicopan plus C5i therapy may result in an increase in FACIT-F scores; however, danicopan plus C5i therapy may result in little to no difference in EQ VAS scores or EORTC QLQ-C30 global health status/quality of life scores at week 12 when compared to placebo plus C5i therapy. Results from week 24 suggest that score increases were maintained for FACIT-F in both treatment arms and suggest a trend toward increased scores in both treatment arms for EQ-5D-3L and EORTC QLQ-C30 scores. Results from the LTE portion were only available from a fraction of patients for all outcomes and therefore remain highly uncertain. With regards to safety, the majority of patients in both trial arms experienced any TEAE, and there was a numerically higher proportion of patients in the placebo-emergent arm who experienced SAEs while being treated with danicopan; there were also imbalances between the treatment arms in the proportion of patients with some TEAEs. However, a numerically low proportion of SAEs led to withdrawal of the study drug across treatment arms. Study limitations include that the ALPHA study is an IA and some missing data were reported for efficacy outcomes; it is unknown whether the missing data are informative or not. There is also no standard clinical definition for danicopan’s indication of EVH, and the study definition as well as the inclusion and exclusion criteria may leave out patients who would be treatment candidates in a clinical context. The safety results are particularly limited by the fact that the ALPHA study is an ongoing trial, therefore potential additional safety signals are possible which would not be captured by this review, particularly since the data from the full sample of patients are not available for the TP2 and LTE phases of the trial. The limitations associated with the indirect evidence submitted did not allow for firm conclusions on the effectiveness of danicopan plus C5i therapy relative to pegcetacoplan, and therefore conclusive information on the comparative effectiveness between danicopan and pegcetacoplan remains lacking.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of danicopan 50 mg and 100 mg film-coated oral tablets as an add-on to ravulizumab or eculizumab, to treat signs and symptoms of EVH in adult patients with PNH.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CDA-AMC review team.
The complement system is a powerful mechanism of the innate immune response, responsible for immune surveillance and host defence. There are 3 distinct pathways through which the complement cascade can be activated on different molecules for their initiation: classical, lectin, and alternative.44,45 The first classical and lectin pathways are activated when specific triggers are recognized by host pattern-recognition receptors, while the alternative pathway is continuously active. When activated after a trigger, the 3 pathways involve a series of reactions that form a C3 convertase, leading to the activation of and a cascade down through C5 to generate a host immune defensive effect.44,45
PNH is a rare, chronic, and potentially life-threatening blood condition caused by an acquired genetic defect in hematopoietic stem cells.1,2 This defect leads to the production of blood cells that lack 2 glycosylphosphatidylinositol-anchored complement regulatory proteins, CD55 and CD59, at their surface, causing the complement system to recognize RBCs as damaged. The uncontrolled activation of the complement cascade prematurely attacks these cells resulting in hemolysis.2
Hemolysis occurs through 2 mechanisms in PNH. IVH occurs in both terminal and proximal pathways when RBCs are directly lyzed because of the activation of the alternative complement pathway5 involving the formation of complexed complement proteins such as C3 convertase, C5 convertase, and the formation of the MAC. EVH occurs in the proximal pathway when RBCs are opsonized by fragments of the complement protein C3, which targets RBCs by macrophages in the spleen and liver.3,18,46 Constant IVH results in hemoglobinuria, mainly characterized by dark-coloured urine, particularly noticeable in the morning because of overnight urine concentration.1,2 In addition to hemolytic anemia and its related symptoms (e.g., fatigue, dyspnea), patients with PNH are susceptible to an increased risk of thrombosis, pain, organ damage (e.g., impaired renal function), and underlying bone marrow dysfunction.1,3,6-8 These symptoms and the IVH association with an increased need for transfusions have a significant effect on patients’ daily living, impair their HRQoL,1,3,6,10-14 and increase the risk of morbidity and mortality with 10-year mortality rates of 29%.1,9 Symptoms of PNH can vary significantly among individuals, and the disease can affect any race, ethnicity, or sex. It may manifest at any age,3,4 although it typically emerges in young adults, with the median age of diagnosis being around 30 years.1,2
In Canada, ravulizumab or eculizumab are C5is used as first-line therapy to treat hemolytic PNH. This treatment regimen addresses uncontrolled complement activation through a complete C5 inhibition in the terminal complement cascade and helps reduce symptoms and complications, resulting in improved survival for patients with PNH.15,16
However, some patients receiving C5i treatment remain anemic and transfusion-dependent. Possible causes of Hb less than 10 g/dL include BTH, EVH, nutritional deficiencies, and bone marrow failure.17 Approximately 11% to 27% of patients may experience BTH on approved doses of eculizumab, and fewer patients experience BTH with ravulizumab. BTH is characterized by the return of IVH and the reappearance of PNH symptoms, and it may occur because of suboptimal C5 inhibition.18,47 EVH is a mechanistic consequence believed to be caused by ongoing C3 deposition on surviving yet defective RBCs, which makes them vulnerable to phagocytosis in the liver or spleen.18-20 While symptoms of EVH are not life-threatening, its manifestation is heterogeneous. For some patients EVH may consist of having normal Hb levels21 and being asymptomatic,22,23 while others may develop severe clinical symptoms and may require blood transfusions to manage ongoing anemia.22,24,25
Because of the rarity of the disease, the prevalence and incidence of PNH have been poorly reported, and published prevalence and incidence estimates of PNH and EVH are not available for the population of people living in Canada. A study in the UK estimated the 15-year prevalence of PNH at 1.59 per 100,000 and the annual incidence of approximately 0.13 per 100,000 persons.48 Another study in the UK reported an overall prevalence of 3.81 per 100,000 and an overall annual incidence rate of 0.35 per 100,000 persons.49 A study in the US estimated the prevalence of PNH at 1.2 to 1.3 per 100,000 persons between 2016 and 2017. The incidence rate over the study period was 0.57 per 100,000 person-years.28 Clinical trial and real-world data showed that approximately 20% of patients with PNH who were clinically stable on C5i treatment develop clinically significant EVH.26
Once suspected by clinical and laboratory data (e.g., low Hb levels, abdominal pain, persistent fatigue dyspnea, cytopenias, iron deficiency, hemolysis), the diagnosis of PNH is established in an appropriate clinical setting by flow cytometry, which demonstrates a deficiency of glycosylphosphatidylinositol-anchored proteins (e.g., CD55, CD59) on RBCs.1,2 Regular clinical workups to identify clinically significant EVH (e.g., lowered Hb levels and elevated reticulocyte counts) are available and conducted as part of the routine monitoring for patients receiving treatment for PNH.
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CDA-AMC review team.
The clinical course of PNH is unpredictable, and symptoms can arise at any age.3,4 As the primary cause of mortality and morbidity in PNH is IVH and complications of thrombosis, in Canada patients receive ravulizumab or eculizumab as standard first-line therapy, which reduces the uncontrolled complement activation and its complications through C5 inhibition in the terminal complement cascade.16,50 The 2 agents have comparable efficacy and toxicity but ravulizumab is dosed less frequently, is dosed by weight, and is associated with fewer episodes of BTH.3 The main C5i are both IV infusions; ravulizumab is administered every 8 weeks and eculizumab is administered every 2 weeks.
EVH is an iatrogenic effect of C5 blockade and is not life-threatening. Per the clinical experts consulted by CDA-AMC, the diagnosis of EVH is complex as it is important to rule out other possible underlying causes of anemia. Once diagnosed, treatment focuses on addressing residual anemia22,24,25 and continued terminal complement blockade remains important to prevent the life-threatening consequences of IVH.2,16,51 EVH can become clinically relevant for patients with persistent symptoms of anemia and patients who become dependent on transfusions.18,22
According to the Canadian PNH Network and clinical experts, the historical approach to managing anemia due to EVH in patients living in Canada with PNH has been supportive care (e.g., RBC transfusions, corticosteroids, splenectomy, danazol, and epoetin alfa) and continuing C5i treatment.20 Per the clinical experts consulted by CDA-AMC, the main nonpharmacologic treatment for EVH and persistent anemia in PNH while on C5i treatment is transfusion support. Folic acid and vitamin B12 support are also supportive options. Hematopoetic stem cell transplant is considered curative, but transplant-related mortality and morbidity are significant and it is reserved for patients with PNH with specific additional comorbidities.52
Pegcetacoplan, a proximal C3i, is an approved therapy indicated for patients with inadequate response to, or intolerant of, a C5i.27 The product monograph recommends pegcetacoplan 1,080 mg SC infusion be given twice weekly with a syringe system infusion pump either by a health care professional, the patient, or caregiver.27 Dosage increase to 1,080 mg every third day may be considered if the LDH level is at least 2 times greater than the ULN on twice-weekly dosing.27 Per the clinical experts consulted by CDA-AMC, this option would currently be offered as a second-line pharmacologic option to patients diagnosed with EVH. Pegcetacoplan is intended to be used as a monotherapy which patients will switch to after an initial period of co-treatment with the C5i and pegcetacoplan. Pegcetacoplan has been previously reviewed by CDA-AMC with the recommendation to reimburse with conditions.16
Drug Under Review
Danicopan selectively inhibits complement alternative pathway factor D,29 which plays a key role in amplifying complement system response. Danicopan is thought to mediate the deposition of C3 fragments on PNH blood cells, which is a key cause of EVH in patients receiving ravulizumab or eculizumab for PNH. Inhibition of factor D activity specifically targets the control point of the complement cascade amplification loop, blocking C3 convertase formation and thereby reducing the production of C3 fragments and downstream MAC formation.30 Although danicopan blocks the alternative pathway-mediated amplification of the complement classical pathway and lectin pathway, these 2 pathways remain active to provide residual complement-dependent protection against infectious pathogens.31 When coadministered with ravulizumab or eculizumab, danicopan is anticipated to maintain control over C5 and MAC-mediated IVH.
Danicopan has a Health Canada indication as an add-on to ravulizumab or eculizumab for the treatment of adult patients with PNH who have residual hemolytic anemia due to EVH.29 The sponsor reimbursement request is as per the indication.
The recommended starting dose of danicopan is 150 mg 3 times a day, administered orally, approximately 8 hours apart (± 2 hours). The dose can be increased to 200 mg 3 times a day if a patient’s Hb level has not increased by at least 2 g/dL after 4 weeks of therapy, if a patient required transfusion within the previous 4 weeks, or to achieve an appropriate Hb response based on clinical judgment.29 Danicopan should not be administered as monotherapy and should only be prescribed as an add-on to ravulizumab or eculizumab.29
If danicopan is discontinued, the dose should be tapered over a 6-day period until complete cessation as follows:29
150 mg regimen — 100 mg 3 times a day for 3 days, followed by 50 mg 3 times a day for 3 days
200 mg regimen — 100 mg 3 times a day for 3 days, followed by 100 mg twice a day for 3 days
Key characteristics of danicopan are summarized in Table 3 with other treatments available for PNH.
Table 3: Key Characteristics of Danicopan, Pegcetacoplan, Eculizumab, and Ravulizumab
Characteristic | Danicopan | Pegcetacoplan | Eculizumab | Ravulizumab |
|---|---|---|---|---|
Mechanism of action | Factor D inhibitor | C3 inhibitor; proximal complement inhibition | C5 inhibitor; terminal complement inhibition | C5 inhibitor; terminal complement inhibition |
Indicationa | As an add-on to eculizumab or ravulizumab for the treatment of adult patients with PNH who have residual hemolytic anemia due to EVH. | For the treatment of adult patients with PNH who have an inadequate response to, or are intolerant of, a C5 inhibitor. | For the treatment of patients with PNH to reduce hemolysis. | For the treatment of adult patients with PNH |
Route of administration | Oral | SC | IV | IV |
Recommended dose | Administered orally at 150 mg 3 times a day, approximately 8 hours apart (± 2 hours). The dose can be increased to 200 mg 3 times a day if a patient’s hemoglobin level has not increased by at least 2 g/dL after 4 weeks of therapy, if a patient required transfusion within the previous 4 weeks, or to achieve an appropriate hemoglobin response based on clinical judgment. | 1,080 mg twice weekly Dose adjustment:
Pegcetacoplan should be administered in addition to the patient’s current dose of C5 inhibitor treatment for the first 4 weeks of treatment to minimize the risk of hemolysis with abrupt treatment discontinuation. | 600 mg every 7 days for the first 4 weeks, followed by 900 mg for the fifth dose 1 week later, then 900 mg every 2 weeks thereafter.b | One loading dose, then 2 weeks later start maintenance dose once every 8 weeks thereafter. Weight-based dosing Loading:
Maintenance:
|
Serious adverse effects or safety issues | Serious infections caused by encapsulated bacteria | Meningococcal infections | Meningococcal infections | Meningococcal infections/sepsis |
Other |
|
|
|
|
C3 = complement component 3; C5 = complement component 5; EVH = extravascular hemolysis; LDH = lactate dehydrogenase; PNH = paroxysmal nocturnal hemoglobinuria; SC = subcutaneous; ULN = upper limit of normal.
aHealth Canada–approved indication.
bDose escalation of eculizumab to 1,200 mg every 14 weeks or reduction of dosing interval to 900 mg every 12 days is considered in patients with PNH experiencing breakthrough hemolysis in clinical practice, as per the Canadian PNH Network.53
Source: Details included in the table are from the product monographs of danicopan,29 pegcetacoplan,27 eculizumab,53 and ravulizumab.54
Perspectives of Patients, Clinicians, and Drug Plans
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website for danicopan.
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups.
The Canadian Association of PNH Patients and the Aplastic Anemia and Myelodysplasia Association of Canada submitted a joint input for this review. A clinical summary of PNH was provided and information was gathered through the personal experiences of 1 patient living in Canada who received danicopan.
The patient group input expressed that PNH significantly impacts the quality of life for both patients and their caregivers. Beyond the persistent fatigue and weakness caused by chronic anemia from hemolysis, patients deal with other symptoms that demand continuous management, such as abdominal pain and dysphagia, influencing their dietary habits and social interactions. Managing symptoms requires ongoing medical interventions, medication adjustments, and lifestyle changes. The disease's nature often leads to social isolation as fatigue and frequent medical appointments hinder participation in social activities, exacerbating feelings of loneliness. Additionally, these symptoms can severely impact work productivity and cause significant emotional and psychological strain not only for patients but also for their caregivers.
According to the input, even though currently available treatments for PNH, such as C5is (e.g., eculizumab, ravulizumab) and a C3i (e.g., pegcetacoplan) effectively inhibit IVH, thrombosis, and EVH, approximately 20% of patients continue to experience EVH and persistent anemia and require frequent blood transfusions. Also, these therapies can introduce side effects, disrupt daily routines, and demand substantial time and resources. The financial costs associated with treatment exacerbate stress, creating a significant economic strain on patients and families. This wide-ranging impact underscores the importance of holistic management approaches to effectively support both patients and their caregivers in managing PNH. Danicopan, an oral factor D inhibitor, has shown promise as an add-on treatment to C5is, targeting residual hemolysis and improving overall disease control in patients with PNH with EVH.
The input stated that patients, caregivers, and families affected by PNH desire tolerable treatment options that reduce treatment burden, decrease hemolysis symptoms, decrease dependency on blood transfusions, slow disease progression, and improve long-term outcomes and quality of life. These improvements could lead to reduced symptom burden, increased independence, and enhanced emotional well-being, significantly impacting daily life and overall well-being for those affected by PNH.
The input indicated that the 1 patient with experience with danicopan noticed a remarkable improvement in her symptoms.
Clinician Input
Input was received from 2 clinical experts and 1 clinician group for this review.
Input From Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of PNH.
Unmet Needs
Per the clinical experts, PNH is a complicated disease and the initial goals of therapy are to reduce mortality, reduce complications and morbidities associated with IVH, as well as reduce transfusion needs and improve HRQoL with better Hb support and avoidance of iron overload, helping patients to attain better functional status and returning to prediagnosis activities and employment.
The clinical experts noted that C5is can provide incomplete control of PNH in some circumstances: rare genetic mutations (in people of Japanese ethnicity), inadequate dosing, response to vaccination, or infections leading to BTH or symptomatic EVH related to C5 inhibition. Per the experts, approximately one-third of patients require higher doses of C5i, although this may be less likely with ravulizumab since it is dosed by weight. Patients may also develop BTH toward the end of their treatment cycles if they would benefit from more frequent perfusion; per the clinical experts, this last situation is not generally considered a treatment failure. The experts estimated that approximately 40% of patients with PNH will continue to have low Hb despite therapy, approximately 30% will require transfusions, and, in 20% to 30% of patients, EVH will contribute to their poor HRQoL. Treatment goals for patients with PNH and EVH remain to reduce mortality, inhibit IVH, and improve HRQoL with better Hb support that does not require transfusion and avoids iron overload, leading to better functional status for patients.
Treatment strategies for clinically relevant EVH include splenectomy, erythropoietin administration, and steroids which, per the experts, have questionable efficacy; the main nonpharmacologic treatment for EVH and persistent anemia in PNH while on C5i treatment is transfusion support. Transfusion is associated with several drawbacks, according to the clinical experts: hospital visits of 2 to 4 hours are required and may be longer if blood typing is not done in advance, and there are risks with transfusion including infection, antibody development, or iron overload which can lead to heart and liver failure or endocrine disorders including diabetes, as well as liver cancer if untreated. In addition, most patients on transfusion will have significantly reduced HRQoL and be unable to maintain regular employment.
Pegcetacoplan, a C3i, is a second-line SC therapy which, per the clinical experts, would be the primary pharmacologic option offered for patients with clinically significant EVH. They noted that pegcetacoplan is a SC infusion with twice-weekly dosing and specific transportation requirements. If BTH occurs, the experts noted that the frequency of pegcetacoplan will usually be increased to 3 times weekly. If BTH is severe, doses of ravulizumab or eculizumab would also be added and the experts noted that these may not be on formulary in all hospitals.
Place in Therapy
If approved, the experts noted that danicopan would be an alternative drug to pegcetacoplan, as a second-line drug, and would be used as an add-on therapy for patients already on C5i.
Patient Population
Per the experts, most patients with clinically significant EVH as the cause of their persistent anemia, with optimized control of other causes of anemia, would be suitable candidates for danicopan. They estimated this to be approximately 30% of patients with PNH. They also highlighted that patients who are potentially undertreated because of not wanting transfusions, whose anemia is not severe enough for transfusion, or for whom SC therapy is unacceptable or unfeasible would likely benefit from danicopan as an oral therapeutic option. The experts noted that some patients already on pegcetacoplan may wish to switch to danicopan plus C5i if they were having ongoing BTH or issues with SC infusions.
The indication and reimbursement criteria for danicopan would require a diagnosis of EVH. Per the experts, a diagnosis of EVH generally required ruling out other possible causes of anemia, including incomplete C5 inhibition. This may be challenging as patients often have other comorbidities and it may not be evident that anemia is because of 1 cause, though they noted that EVH remains an important iatrogenic effect of C5 inhibition. Per the clinical experts, clinical diagnosis for EVH typically requires anemia along with normal or minimally elevated LDH, as well as elevated bilirubin and reticulocyte counts. Alternative explanations for anemia which the experts noted would have to be ruled out include bone marrow failure, hematinic deficiencies (such as vitamin B12 or ferritin), renal insufficiency, or blood loss.
The experts noted that in certain circumstances, a trial with add-on danicopan therapy may be needed to assess for efficacy in certain complicated patients or to avoid the potential confounding effect of recent transfusion as the cause of improvement.
Assessing the Response to Treatment
The clinical experts noted that response to therapy is typically an improvement in Hb and a reduction in transfusion requirements relative to the baseline for a given patient. They noted that ongoing anemia and transfusion needs may or may not be a treatment failure, as it is possible that other concurrent diseases such as bone marrow failure, aplastic anemia, other cancers, or comorbidities could be contributing factors. The experts note that failures or suboptimal responses emphasize the need for full evaluation of the cause of anemia.
Discontinuing Treatment
Per the clinical experts, intolerance or allergy to danicopan would be reason to discontinue therapy, as would a lack of improvement in Hb levels and transfusion needs. The experts noted that an episode of BTH or transfusion requirement in another setting would not be considered a treatment failure, nor would a required stoppage of therapy because of pregnancy or breastfeeding. The experts highlighted that stopping danicopan therapy should be considered independent of the C5i as the purpose of that medication is to manage IVH.
Prescribing Considerations
Per the clinical experts, treatment with danicopan would need to be initiated by a hematologist, preferably with expertise in PNH, and that at the least a consultation with a PNH expert would be warranted if a patient with PNH was being followed in a shared-care model (i.e., a hematologist with expertise in PNH along with a local hematologist).
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups.
One clinician group, the Canadian PNH Network, submitted input for this review based on contributions from 9 clinicians. Information was gathered through publicly available documents, congress abstracts, and published literature.
The group noted that the current standard of care for PNH is C5is (i.e., eculizumab and ravulizumab), which act via terminal complement blockade. Although not curative, these treatments have been shown to be effective in controlling IVH, leading to significant improvement in fatigue, HRQoL, transfusion dependence, thrombosis, and overall survival. In addition, pegcetacoplan (proximal complement inhibitor) has been recently approved for patients with persistent anemia despite C5i therapy or those who are intolerant to C5is. The only curative treatment for PNH is allogeneic hematopoietic stem cell transplant, reserved for patients with predominant or progressive bone marrow failure that can coincide with a diagnosis of PNH.
According to the clinician group, there are still some unmet therapeutic needs within the available PNH treatment regimen. Some patients remain anemic due to EVH, and some remain transfusion-dependent with C5i therapy. Moreover, the delivery mechanism of pegcetacoplan could be unfeasible and time-consuming and can be challenging for some patients with needle phobia, vision problems, poor skin integrity, and/or issues with manual dexterity.
The clinician group indicated that a subset of patients would benefit from proximal complement inhibition given the development of clinically significant EVH but for whom pegcetacoplan is less than ideal. Dual complement blockade (i.e., anti-C5 plus a proximal inhibitor) would provide these patients with the same benefits of improved Hb but with a lower risk of complications.
The group stated that danicopan, an oral factor D (proximal complement) inhibitor taken 3 times a day, could potentially improve Hb in patients with suboptimal response to C5is alone when used as an add-on to C5is for patients with PNH and EVH.
According to the clinician group, the patients most likely to benefit from danicopan are those who have persistent anemia despite stable-dose C5i, in whom EVH is suspected. Patients who may receive proximal inhibition monotherapy (e.g., pegcetacoplan) who may not tolerate it or have repeated BTH or other concerns could also benefit from the therapy. This treatment is least suitable for those who are not anemic or who meet exclusion criteria in clinical trials, such as pregnancy.
The input stated that outcomes used in clinical practice to determine treatment response include an increase in Hb and a reduction in LDH (LDH ratio < 1.5 × ULN), which should be accompanied by decreased fatigue, transfusion requirements, improved HRQoL, and improved overall survival. A clinically meaningful response to treatment would be sustained control of LDH but with further Hb increases and improvement in anemia-related symptoms. Efficacy outcomes would typically be followed every 2 to 4 weeks initially, but follow-up would be required less often (e.g., every 3 to 6 months) as a patient becomes established on the drug and does not show evidence of side effects or other concerns. A lack of improvement in the first few months of therapy would be a prompt to dose increase.
Danicopan discontinuation should be considered in patients who develop AEs that preclude ongoing therapy, including poor treatment compliance and intolerable side effects. The most important feature to monitor for is evidence of BTH. Treatment is contraindicated during pregnancy or breastfeeding.
Patients with PNH should be followed by clinicians who specialize in the area. However, treatment with danicopan plus C5 inhibition could be done entirely at the patient’s home (or the C5i infusions could be given at an infusion clinic or hospital based on local or provincial practices).
Drug Program Input
The drug programs provide input on each drug being reviewed through the CDA-AMC reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The comparator in the ALPHA trial was placebo, which is appropriate for an add-in therapy; however, pegcetacoplan is approved for patients who have had an inadequate response to C5i therapy. Could danicopan be used as an add-on therapy to pegcetacoplan as well? | The clinical experts noted that as there are no studies on the use of danicopan in combination with pegcetacoplan, and that such combination would not be used for the time being. |
Considerations for initiation of therapy | |
The specific requirements in the ALPHA trial for a definition of clinically significant EVH were:
Patients also need to have C5i treatment for at least 6 months and a platelet count ≥ 30,000/µL. Are these measurements typical or standard to define EVH? Do these criteria represent a typical patient? Are these criteria readily measurable? | The clinical experts indicated that there are no specific definitions or standards to define EVH; broadly speaking it consists of signs of hemolysis that are not intravascular, plus suggestive changes in laboratory markers including reticulocytes, bilirubin, or Coombs test. Patients do have to have anemia; however, the cut-off of 9.5 g/dL did not pertain to a specific standard. The experts commented that at 9.5 g/dL they would likely not consider transfusion unless other patient factors suggested it should be done. They noted that the criteria defined in the ALPHA trial represent a typical patient; however, they also noted that the platelet count threshold does not represent an indication or contraindication to therapy. It may be a criteria in the trial to ensure that there are not too many patients with bone marrow failure, which they noted is standard for research practice. All criteria would be measurable with standard laboratory testing. |
Could clinically significant EVH be seen with pegcetacoplan, the current second-line therapy? Could danicopan be added on to pegcetacoplan therapy? | The experts noted that clinically significant EVH could be observed with pegcetacoplan, bearing in mind the caveats about the lack of a specific clinical definition for EVH. Per the experts, because of a lack of studies combining pegcetacoplan and danicopan, they would not use the combination at this time. |
Considerations for continuation or renewal of therapy | |
Frequent monitoring of bloodwork is required. Can this be defined as to what is needed and when, to assess response? | The experts emphasized that their patients are frequently complex and so the type and frequency of bloodwork or transfusions is patient-dependent; they may meet with patients at frequencies varying from weekly to every 6 months, although their baseline visit frequency was usually every 3 months. They highlighted that measures for blood count, creatinine, electrolytes, bilirubin, lactate dehydrogenase, and haptoglobin were regular laboratory tests, with the possibility of adding on measures such as reticulocyte counts, vitamin levels, or other biomarkers to identify the source of patient concerns or symptoms. |
Considerations for discontinuation of therapy | |
Can loss of response or a lack of response to danicopan therapy be defined? | The experts emphasized that an important concern in PNH therapy was defining whether a patient was experiencing a loss of response because of poor adherence or inadequate dosing, which would be considered a loss of response, as opposed to a treatment failure. They indicated that if a patient were to become anemic and transfusion-dependent again, they would consider that a loss of response. However, if a patient did not improve in any measures after starting a new therapy, they considered it a lack of response. |
Is danicopan therapy intended to be indefinite? | The experts indicated that danicopan would be considered to be indefinite, apart from specific situations such as palliative care or bone marrow grafts. |
Considerations for prescribing of therapy | |
Are there concerns about combining danicopan as an add-on to pegcetacoplan? | The experts indicated that as there are no studies on this combination, it is not 1 they would envision using at this time. They specified that danicopan combined with a C5i was preferable as it would control both IVH and EVH, therefore pegcetacoplan would not be necessary. |
Generalizability | |
Should patients have to be on a C5i for at least 6 months before adding on danicopan? It may be desired to add on sooner, and in the previous CADTH review of pegcetacoplan, 3 months was needed before initiating. | The experts noted that the 3-month duration was a requirement for the clinical trial in pegcetacoplan, but in clinical practice they noted that changes to therapy are rarely made before the patient has been on a medication for 6 months. These changes exclude dose adjustments. |
Could patients currently on pegcetacoplan want to be switched back to a C5i with danicopan add-on? | The experts noted that there would likely be some patients who are either suboptimally controlled with pegcetacoplan or who prefer not to use it because of the requirement for infusions, or whose quality of life was otherwise impacted by the medication administration. They noted it would likely not be the majority of patients as, in their experience, patients are often hesitant to switch medications. |
Care provision issues | |
Will Neisseria meningitidis vaccinations and/or antibiotics be required before initiation? | The experts noted that all patients are usually vaccinated for meningitis Group B, C, and D strains every 3 to 5 years but there is inconsistent access for other vaccines which might be required such as pneumococcal vaccines. They noted that access and required vaccines per province is unequal. They did not have issues with vaccine access specifically. |
C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency; EVH = extravascular hemolysis; IVH = intravascular hemolysis; Hb = hemoglobin; PNH = paroxysmal nocturnal hemoglobinuria.
Clinical Evidence
The objective of the CDA-AMC Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of danicopan (Voydeya) 50 mg and 100 mg film-coated oral tablets in the treatment of signs and symptoms of EVH in adults with PNH. The focus will be placed on comparing danicopan to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of danicopan is presented in 4 sections with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted LTE studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following are included in the CDA-AMC review and appraised in this document:
One RCT identified in systematic review
One LTE study (extension of the RCT and therefore presented in the systematic review section)
One indirect treatment comparison.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | ALPHA (ALXN2040-PNH-301) |
|---|---|
Designs and populations | |
Study design | Phase III, randomized, double-blind, placebo-controlled study |
Locations | 80 centres in 18 countries in Europe, North America, South America, and Asia |
Patient enrolment dates | Start date: January 6, 2021 Interim analysis data cut-off: September 20, 2022 |
Randomized (N) | A total of 86 patients were randomized 2:1 to receive danicopan (n = 57) or placebo (n = 29); a total of 75% of randomized patients (n = 63) form the prespecified interim efficacy analysis set. |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Danicopan 150 mg orally t.i.d.; dose escalations of up to a maximum of 200 mg orally t.i.d. were made based on efficacy and safety assessments. All patients were receiving concurrent treatment with ravulizumab or eculizumab. |
Comparator(s) | Placebo 150 mg orally t.i.d. with dose escalation up to a maximum of 200 orally t.i.d. based on efficacy and safety assessments (dose escalation was performed in a similar manner for the placebo group to preserve blinding). All patients were receiving concurrent treatment with ravulizumab or eculizumab. |
Study duration | |
Screening phase | 45 days |
Treatment phase |
|
Follow-up phase | LTE1: 52 weeks Optional LTE2: 52 weeks Patients who discontinued danicopan were dose tapered over 6 days with a follow-up visit approximately 30 days after the last dose |
Outcomes | |
Primary end point | Change in Hb from baseline to week 12 |
Secondary and exploratory end points | Key secondary:
Secondary:
Exploratory:
|
Publication status | |
Publications | Sponsor-provided Clinical Study Report40 Lee et al. (2023)55 ClinicalTrials.gov identifier: NCT0446946556 |
C5 = complement component 5; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EVH = extravascular hemolysis; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; LDH = lactate dehydrogenase; LTE = long-term extension; PNH = paroxysmal nocturnal hemoglobinuria; RBC = red blood cell; t.i.d. = 3 times a day.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The ALPHA trial is an ongoing phase III, double-blind, randomized placebo-controlled trial which enrolled a total of 86 patients with PNH who had clinically significant EVH, on treatment with ravulizumab or eculizumab. The study is ongoing; data presented in the current submission come from a prespecified IA consisting of 75% of the total sample (N = 63 patients) and was based on data from enrolment until database lock on September 20, 2022. The study design consisted of a 12-week TP1 which was randomized, double-blind, and placebo-controlled, followed by a 12-week TP2 where patients initially randomized to placebo switched to receive danicopan and patients initially randomized to danicopan continued to receive danicopan. Patients completing TP2 were eligible to continue onto to a total of 2 LTEs (LTE1 or LTE2); results from LTE1 to date are presented in the submission. Treatment assignment in TP1 was not unblinded until after the first interim database lock occurred in August 2022.
The study sites were located in 18 countries in Europe, Asia, South America, and North America, including Canada. The study used a 45-day screening period and randomization was stratified by transfusion history (> 2 transfusions or ≤ 2 transfusions in the 6 months before screening), Hb at screening (< 8.5 g/dL or ≥ 8.5 g/dL), and Japanese patient (yes or no). Stochastic dynamic allocation rules were used to randomize patients 2:1 through an interactive response technology to either receive danicopan 3 times a day added onto their C5i or a placebo 3 times a day added onto their C5i, respectively. Full study design is in Figure 1.
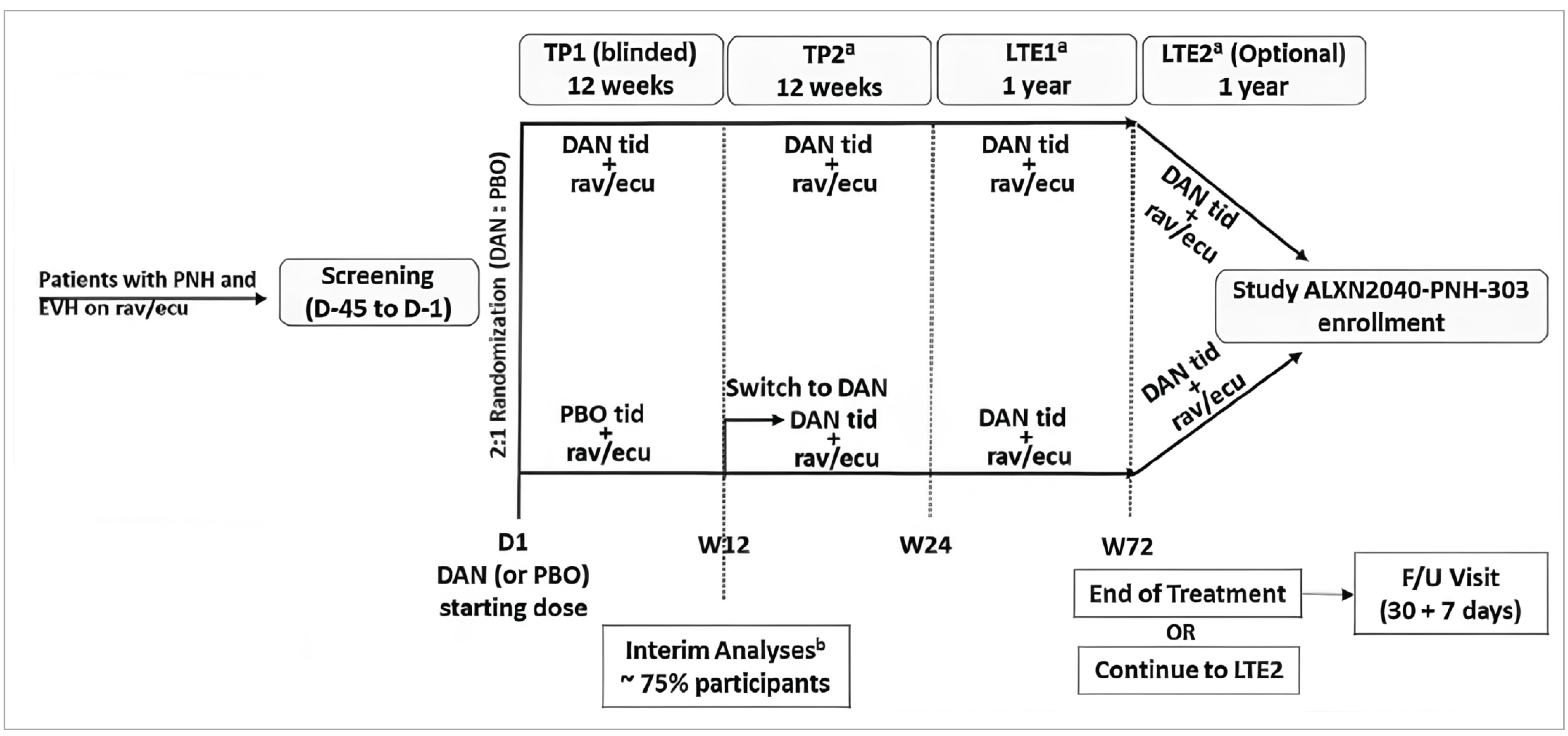
D = day; DAN = danicopan; ecu = eculizumab; EVH = extravascular hemolysis; F/U = follow-up; LTE1 = long-term extension year 1; LTE2 = long-term extension year 2; PBO = placebo; PNH = paroxysmal nocturnal hemoglobinuria; rav = ravulizumab; tid = 3 times a day; TP1 = treatment period 1; TP2 = treatment period 2; W = week.
aAfter TP1, all placebo participants were switched to DAN and remained on DAN throughout the study.
bAs of the interim analysis cut-off date (September 20, 2022), 86 participants were randomized, and 63 participants were included for interim efficacy analysis.
Source: Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Populations
Inclusion and Exclusion Criteria
Patients eligible to participate in the study were required to be aged 18 years or older, have a diagnosis of PNH, and have clinically significant EVH defined as patients presenting with anemia (Hb ≤ 9.5 g/dL) and increased reticulocyte count (≥ 120 × 109/L), with or without the need for transfusion, had to be receiving an approved C5i (ravulizumab or eculizumab) with no change in dose or interval for at least 6 months, as well as meet a platelet count threshold of 30,000 or more per µL and a neutrophil count of 500 or more per µL. Patients were eligible regardless of transfusion status. Patients were excluded if they had a history or presence of any clinically significant medical condition or comorbidity, including any conditions leading to anemia that are not primarily because of PNH; if they had any procedures and/or laboratory anomalies which would put them at undue risk to receive danicopan; or patients who were, or who had partners who were pregnant, nursing, or planning to become pregnant during the study or within 90 days of study intervention. The submission provided some specifics on the medical conditions, laboratory values, or procedures which would meet the criteria for study exclusion in the study protocol.57
Interventions
C5i Therapy
All participants were to be treated with danicopan or placebo in combination with a C5i therapy (i.e., ravulizumab or eculizumab) at stable doses. In countries where ravulizumab was not approved, local amendments were issued to provide it as an investigational medical product. Participants could not switch between ravulizumab or eculizumab during the first 24 weeks but were permitted to do so during the LTE period. The only C5i switch permitted was from eculizumab to ravulizumab. With the exception of ravulizumab’s weight-based dosing changes in response to changes in weight, the approved C5i dose was not permitted to be increased, nor the interval shortened, during the study. The dose was permitted to be decreased if indicated, with a dose re-escalation to the prior dose if the reduction was not tolerated. Changes in C5i administration frequency because of patient convenience or logistical reasons but which did not result in a change of prescribed dose or frequency, were considered stable and were to be discussed with the medical monitor before randomization.
Danicopan or Placebo Add-On
All patients received either danicopan or placebo in the form of 50 mg or 100 mg film-coated oral tablets in white high-density polyethylene bottles with white child-resistant polypropylene screw caps, fitted with induction-sealed aluminum faced liners. The majority of doses were taken at home, with patients supplied enough study drug to last until their next study visit. To assess adherence, adherence was calculated as a percentage of danicopan doses taken divided by the doses scheduled to be taken.
The dosage administered started at 150 mg 3 times a day; an option to start dosing at 100 mg 3 times a day for patients with alanine transferase or direct bilirubin screening values greater than 1.5 × ULN was initially specified and then removed in a protocol amendment; any patients on 100 mg 3 times a day at the time were escalated to 150 mg 3 times a day. Dosing could be escalated up to a maximum of 200 mg. During TP1, dosing could be escalated at week 6 under the following circumstances:
if the participant’s Hb level at week 4 had not increased by 2 g/dL (20 g/L) or more from their baseline value, or
if the participant received a transfusion in the past 4 weeks.
Dose escalation could be performed in a similar manner for both treatment arms to maintain blinding. In TP2, dosing could be escalated at weeks 12 or 18 if, at weeks 10 or 16, respectively, 1 of the following circumstances occurred:
the participant’s Hb had not been normalized to at least the midpoint of the normal range for their sex relative to the baseline value, or
the participant received a transfusion during the previous 4 weeks.
Dose escalation could be done up to a maximum of 200 mg 3 times a day during the LTE phases if they had been on their previous dose for at least 4 weeks and if the investigator, after discussion with the sponsor, believed that additional efficacy could be achieved.
Patients discontinuing the study had to undergo a dose tapering over 6 days, with 2 taper visits and a follow-up visit approximately 30 days after the last dose during the tapering period. Participants continued to receive their background C5i therapy at the same dose and interval they were receiving during the taper and follow-up visits.
Concomitant Medications and Procedures
Concomitant treatments with folic acid, erythropoiesis-stimulating agents, steroids, or other immunosuppressants were permitted if patients were on stable doses for a prespecified period before study initiation and remained on stable doses through week 24 of the trial. Patients taking iron, folic acid, and vitamin B12 supplements were eligible if their dose was stable for at least 30 days. Hormonal therapies were permitted for contraception or hormonal replacement therapy. Prophylactic antibodies for treatment with a complement inhibitor were permitted if deemed appropriate by local clinical practice and/or guidelines.
Transfusions were administered to patients with Hb less than 7 g/dL regardless of symptoms; they were also administered to patients who had a Hb less than 9 g/dL in the presence of signs or symptoms to warrant a transfusion (i.e., angina, change in mental status, syncope, light-headedness, confusion, shortness of breath, and fatigue). The study investigator determined the appropriate number of packed RBCs to administer.
Outcomes
A list of efficacy and safety end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by CDA-AMC and input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform the CDA-AMC expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points reported at week 12 of the ALPHA study were assessed using GRADE. Select notable harms outcomes considered important for informing the expert committee deliberations of the CDA-AMC were also assessed using GRADE.
Table 6: Outcomes Summarized From the ALPHA Study
Outcome measure | ALPHA | End points measured in ALPHA |
|---|---|---|
Hb level | Primary outcome at week 12a | Change in Hb from baseline at weeks 12, 24 |
Key secondary outcome at week 12a | Proportion of patients with Hb increase of ≥ 2 g/dL in the absence of transfusion at weeks 12 and 24 | |
Secondary outcome at week 12 | Proportion of patients with Hb normalization at weeks 12 and 24 | |
Transfusions | Key secondary outcome at week 12a | Proportion of patients achieving transfusion avoidance (transfusion-free and do not require a transfusion) at week 12 and 24 |
Secondary outcome at week 12 | Change in transfusion burden (number of RBC units transfused and transfusion instances) at week 12 and 24 | |
Absolute reticulocyte count | Key secondary outcome at week 12a | Change from baseline in absolute reticulocyte count at weeks 12, 24, and 52 |
LDH | Secondary outcome at week 12 | Change in LDH from baseline at weeks 12, 24, and 52 |
Fatigue | Key secondary outcome at week 12a | Change from baseline in FACIT-F scores from baseline at weeks 12, 24, and 52 |
HRQoL | Exploratory outcome at week 12 | Change from baseline in EQ-5D-3L scores at weeks 12, 24, and 52 |
Change from baseline in EORTC QLQ-C30 scores at weeks 12, 24, and 52 | ||
Survival | Not measured as an efficacy end point | Safety end point |
Total and direct bilirubin | Not appraised as an efficacy end point | Safety end point |
Liver enzyme elevations | Not measured as an efficacy end point | Safety end point |
Meningococcal infections | Not measured as an efficacy end point | Safety end point |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; HRQoL = health-related quality of life; LDH = lactate dehydrogenase; RBC = red blood cell.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Hb Levels
Change in Hb Levels
Hb is used in the diagnosis and monitoring of disease activity of PNH.58 The primary outcome of the ALPHA study was change in Hb levels from baseline to week 12. All blood tests were performed by a central laboratory. A MID has not been established in the PNH population for Hb level.
For the change in Hb at week 12, the submission noted that an increase in Hb of 2 g/dL or greater was considered to be clinically meaningful in anemic patients with cancer.59 The clinical experts consulted by CDA-AMC noted that any change in Hb would likely be meaningful in a real-world setting.
Proportion of Patients With Hb Increases of 2 g/dL or Greater in the Absence of Transfusion
The proportion of patients with Hb increase of 2 g/dL or greater in the absence of transfusion at week 12 was a key secondary end point. All blood tests were performed by a central laboratory. An MID for this outcome has not been established in patients with PNH. Patients with an improvement in Hb of at least 2 g/dL have previously been shown to experience significantly greater increases in FACIT-F subscale scores relative to those who did not achieve this level of Hb response.59 The submission noted that Canadian clinician feedback received is aligned with this MID.42 The clinical experts consulted by CDA-AMC noted that any change in this measure would likely be clinically meaningful in their context.
Proportion of Patients With Hb Normalization
The proportion of patients with Hb normalization, defined as patients with Hb values greater than the lower limit of the normal reference range (110 g/L for females and 125 g/L for males),32 was a secondary outcome. All blood tests were performed by a central laboratory. A MID for this outcome has not been established in patients with PNH. The clinical experts consulted by CDA-AMC noted that any change in this measure would likely be clinically meaningful in their context.
Transfusions
Transfusion Avoidance
Transfusion avoidance was a key secondary outcome of the ALPHA study and was defined as the proportion of patients who were transfusion-free and did not require a transfusion. Patients with severe anemia have high transfusion burden, and there is potential for complications such as iron overload with chronic transfusions.31,60 Transfusions were administered to patients who had a Hb less than 7 g/dL regardless of symptoms and to those who had a Hb less than 9 g/dL with signs or symptoms to warrant a transfusion. A MID has not been established for transfusion avoidance. Canadian clinician feedback received by the sponsor noted that a 50% reduction in transfusion burden over 6 months would represent a clinically meaningful improvement.42 The clinical experts consulted by CDA-AMC noted that the 50% reduction would be an important change, but any change in this measure would likely be clinically meaningful.
Number of RBCs and Number of Transfusion Instances
Transfusion burden, defined as the number of RBC units transfused and the number of transfusion instances, was a secondary outcome. An MID has not been established for transfusion burden. Canadian clinician feedback received by the sponsor noted that a 50% reduction in transfusion burden over 6 months would represent a clinically meaningful improvement.42 The clinical experts consulted by CDA-AMC noted that any change in this measure would likely be clinically meaningful in their context.
Absolute Reticulocyte Count
Change in absolute reticulocyte count from baseline to week 12 was a key secondary outcome. Reticulocyte count is an indicator of hemolytic anemia. Elevated reticulocyte count may indicate overcompensation of marrow production in response to hemolysis.61 For EVH, reticulocyte count correlates better than LDH with increased C3 on PNH erythrocytes, raised bilirubin, and increased transfusion dependence.62 All blood tests were performed by a central laboratory.
An MID has not been established for change in absolute reticulocyte count in patients with PNH. Canadian clinician feedback received by the sponsor noted that normalization of absolute reticulocyte count is considered to be less than 100 × 109/L.42 The clinical experts consulted by CDA-AMC noted that any change in this measure would likely be clinically meaningful.
Lactate Dehydrogenase
LDH is a marker of IVH61 and is used in the diagnosis and monitoring of disease activity of PNH.58 It was reported that an LDH level of at least 1.5 × greater than the ULN was associated with an increased risk of thromboembolism based on data from a national South Korean PNH registry including patients who were eculizumab-naive.63
An MID has not been established for LDH values. The submission noted that participants in the ALPHA study were on stable C5i therapy before entry, and the mean values for LDH from the trial (mean = 292.12 U/L; SD = 95.19 U/L) were within the reference range (135 U/L to 330 U/L) at baseline.42 The clinical experts consulted by CDA-AMC noted that any change in this measure would likely be clinically meaningful in their context; however, LDH measures that are already in normal range should not be expected to change.
Fatigue and HRQoL
Descriptions of the type of outcomes, conclusions about their measurement properties, and relevant information about MIDs in the change in scores are detailed in Table 7, followed by further detail on these outcomes.
Table 7: Summary of Fatigue and HRQoL Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
FACIT-F | 13-item, patient-reported, fatigue-specific, quality of life questionnaire using a 5-point Likert scale. It assesses tiredness, weakness, and difficulty conducting usual activities as a result of fatigue over the past week.33 The 13-item scale ranges from 0 (extreme fatigue) to 52 (no fatigue). Higher scores indicate less fatigue.33 | Patients with PNH: The content validity was confirmed as an appropriate tool to be used in patients with PNH.8,64 Convergent validity between FACIT-F and Hb, ARC, and indirect bilirubin (post hoc analysis using data from the PEGASUS study) were r = 0.47, r = −0.37, and r = −0.25, respectively.65 Responsiveness: Patients with improvements in Hb, indirect bilirubin, and ARC showed improvements in FACIT-F scores (P < 0.0001, P = 0.0002, and P = 0.0002, respectively)65 Patients with cancer or psoriatic arthritis:66 Internal consistency by Cronbach alpha was 0.9533 and test-retest by intraclass correlation coefficient was 0.95.67 No evidence was identified for reliability in patients with PNH. | Per the submission, a change in score of 5 points is considered clinically important in the PNH population. Canadian clinician feedback received by the sponsor align with this MID.68 |
EQ-5D-3L | The EQ-5D is a widely applicable generic HRQoL tool with 2 main components. The first part involves a descriptive system comprising 5 dimensions (mobility, self-care, usual activities, pain/discomfort, and anxiety/depression) The EQ-5D-3L has 3 possible levels (1, 2, or 3) for each domain representing “no problems,” “some problems,” and “extreme problems,” respectively.35 Respondents select the level that reflects their health state for each dimension, resulting in 243 potential health states. These states are then assigned an index score using a scoring function based on population preferences. The second part consists of the EQ VAS where respondents rate their health on a scale from 0 to 100, with 0 representing the worst imaginable health state and 100 the best.34,35 | Patients with cancer: A study69 conducted among patients (N = 184) with breast, colorectal, or lung cancer found the following. Construct validity: Validity was assessed using Pearson correlation coefficient (r) where r between 0 and 0.3 demonstrated weak correlation, between 0.3 and 0.49 was moderate, and > 0.5 was considered strong. The same study69 found the following, between the EORTC QLQ-C30 and EQ-5D, r = 0.43; comparing the EORTC QLQ-C30 and EQ VAS, r = 0.73; and between EQ-5D and EQ VAS, r = 0.43. External validity: The EQ-5D was able to discriminate populations based on self-reported health status (excellent, good vs. fair, very poor; ES = 0.90), and somewhat based on the ECOG PS score (0 vs. 1 to 3; ES = 0.31), but not for stage of cancer (stages 1 and 2 vs. stages 3 and 4; ES = 0.06) Reliability: Evidence of acceptable reliability for 5 functioning scales and global health status in patients with cancer. Responsiveness: The study found this instrument to be unresponsive when compared to other disease-specific instruments. It is worth noting that the EQ-5D was based on a population not living in Canada. No evidence was identified for reliability in patients with PNH. | No MID was identified in patients with PNH. MID 0.033 to 0.074 estimated for the general population. MID 0.07 to 0.11 for UK-index scores and 0.05 to 0.08 for US-index scores for patients with cancer.70 |
EORTC QLQ-C30 | 30-item, patient-reported, cancer-specific, quality of life questionnaire using 4- and 7-point Likert scales. It consists of 5 multi-item functional scales (physical, role, emotional, cognitive, and social), 3 multi-item symptom scales (fatigue, nausea and vomiting, and pain), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial impact), and a 2-item GHS/QoL scale. A 1-week recall period is used to assess the items.36 Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. A higher score on the functional scales represents better functioning, a higher score on the symptom scales represents a higher level of symptomatology, and a higher score on the global health status/HRQoL scale represents a higher HRQoL.36 | Patients with PNH: The content validity was confirmed in patients with PNH.8 Convergent validity between EORTC QLQ-C30 scales and Hb, ARC, and indirect bilirubin were:
Responsiveness: Patients with improvements in Hb, indirect bilirubin, and ARC showed improvements in physical functioning (P = 0.0103, P = 0.0050, and P = 0.0072, respectively) and fatigue scores (P = 0.0093, P = 0.0073, and P = 0.0162, respectively)65 Patients with cancer: Reliability of the EORTC QLQ-C30 in patients with HL or DLBCL undergoing chemotherapy measured by Cronbach alpha was 0.79 for GHS/QoL, 0.51 to 0.85 for functional scales, and 0.82 to 0.86 for symptom scales/items.36 No evidence was identified for reliability in patients with PNH. | Patients with cancer:71
No MID was identified in patients with PNH. |
ARC = absolute reticulocyte count; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = EQ visual analogue scale; ES = effect size; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GHS = global health status; Hb = hemoglobin; HL = Hodgkin lymphoma; HRQoL = health-related quality of life; MID = minimum important difference; PNH = paroxysmal nocturnal hemoglobinuria; QoL = quality of life.
Functional Assessment of Chronic Illness Therapy–Fatigue
FACIT-F is a validated and reliable patient-reported outcome instrument used for evaluation of fatigue associated with anemia in different patient populations, including patients with PNH.68 A change in score of 5 points is considered the MID in the PNH population based on distribution-based estimations using real-world data from the International PNH Registry.68 The clinical experts consulted by CDA-AMC noted that this tool is not commonly used in clinical practice.
EQ-5D-3L
MID values have not been established for this measurement tool in the PNH population. The clinical experts consulted by CDA-AMC noted that this tool is not commonly used in clinical practice.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
The EORTC QLQ-C30 tool has been validated in patients with PNH; however, MID values have not been established for this measurement tool in the PNH population. However, an increase of 10 or more points in EORTC QLQ-C30 score for patients with cancer is considered moderately large and represents a clinically important improvement.72 The clinical experts consulted by CDA-AMC noted that this tool is not commonly used in clinical practice.
Statistical Analysis
Analysis Populations
Planned Interim Analyses
The prespecified IA was planned for when approximately 75% of the total planned sample (N = 63 patients) had been randomized and completed the TP1; the purpose of this analysis, per the submission, was to assess stopping early for efficacy. The data cut-off for the TP1 IA was conducted on June 28, 2022, and a second interim data analysis for TP2 results was conducted with a data cut-off of September 20, 2022. A total of 63 patients formed the IEAS and a total of 86 patients (the entire randomized study sample) formed the interim safety analysis set.
Analyses were based on the intention-to-treat (ITT) principle. Full details of the analysis populations are provided in Table 8.
Table 8: Analysis Populations for the ALPHA Study
Population | Definition | Application |
|---|---|---|
Interim efficacy analysis set | The full analysis set for efficacy analyses at the interim analysis. Per the prespecified plan for IA, the first 75% of randomized participants (N = 63 out of 84 enrolment target) formed the IA set for efficacy analysis. The first IA (data cut-off of June 28, 2022) was performed when all 63 participants reached the end of TP1. The second IA (data cut-off of September 20, 2022) was performed when all 63 participants in the IA set reached the end of TP2 (either completed or discontinued). | Population for primary end point and key secondary end point analyses, and all other efficacy end points analyses in the ALPHA CSR 2.0 (IA). All analyses were to follow the ITT principle. |
Interim safety analysis set | All participants (N = 86) who received at least 1 dose of study intervention by the interim database cut-off date. | All safety analyses on data collected up to the database cut-off date of the interim CSR. |
CSR = Clinical Study Report; IA = interim analysis; ITT = intention to treat; TP1 = treatment period 1; TP2 = treatment period 2.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sample Size and Power Calculations
The submission noted that a minimum difference in the primary outcome of 2 g/dL between danicopan and placebo treatments at 12 weeks would be considered clinically meaningful, and also noted that the mean baseline Hb levels of patients included in the study (≤ 9.5 g/dL) is lower than the average Hb values reported in the literature for patients who are receiving an approved C5i but are still anemic (average Hb is 10.5 g/dL). The submission also anticipated that approximately 10% of all patients would discontinue therapy before the primary end point measurement at week 12.
With a full target sample of 84 patients, for the primary end point of change from baseline to week 12 in Hb level, the statistical power using a 2-sample t test was 99% to detect the difference in mean change from baseline of 2 g/dL (alternative hypothesis), assuming the 2-sided statistical significance level of 0.05 and an SD of 1.6 g/dL, which was estimated from results of Study ACH471-101, a dose-finding study included in the submission.73 For the key secondary end point of patients with a Hb increase or 2 g/dL or more at week 12 with the absence of transfusion, the full study had more than 95% power for detecting a significant difference between treatment groups with a 2-sided alpha of 0.05, assuming at least 35% of patients in the danicopan arm and 5% of patients in the placebo arm met the threshold. For the key secondary end point of patients with transfusion avoidance, the study had 70% power for detecting a significant difference between treatment groups, assuming 90% of patients in the danicopan arm and 64% of patients in the placebo arm had transfusion avoidance. For the key secondary end point of change from baseline to week 12 in FACIT-F scores, the study had 91% power with a 2-sample t test to detect a 9-point difference between treatment arms in mean change from baseline, which per the submission was considered clinically meaningful. The submission did not provide any reference for this assumption. The power calculation for FACIT-F was based on the assumption of an SD of 11, which was observed in Study ALXN1210-PNH-301 in patients with PNH (not included in the submission). The power would be 80% based on the SD assumption of 13, which was observed in Study ACH471-101, the dose-finding study included in the submission.73
IA Sample Adjustment
For the current submission sample size of 63 patients, analyses for the primary and key secondary end points were evaluated using an alpha-spending method to control the family-wise error rate. The evaluation of the primary end point used a 2-sided alpha level of 0.018, and the evaluation of the key secondary end points used a 2-sided alpha of 0.042. The overall family-wise error rate was controlled at a 2-sided 0.05 level across the primary and key secondary end points among the interim and full final analyses.
Efficacy Analysis
Full details of the efficacy analysis methods, adjustments, handling of missing data and sensitivity analyses by end point can be found in Table 9, followed by a detailed description of certain methods.
Primary End Point Analysis
The primary end point was based on the ITT population and analyzed using a mixed model for repeated measures method which included the fixed, categorical effects of treatment group, study visit, and study visit-by-treatment group interaction, as well as the fixed, continuous covariate of a baseline Hb value and the randomization stratification factor of transfusion history. The alpha-spending method described previously was applied. As transfusions could impact the primary outcome, Hb values collected within 4 weeks after a transfusion were not included in the mixed model for repeated measures. An unstructured covariance matrix was used to model the within-patient errors. If the analysis model failed to converge, the covariance matrix structures was evaluated in the following order until model convergence was met: Toeplitz, first-order autoregressive, and compound symmetry. The order was specified according to decreasing number of covariance parameters in the structure. The Kenward-Roger approximation was used to estimate the denominator degrees of freedom.
A sensitivity analysis was conducted exploring TDs using a rerandomization test; rerandomized treatment assignments were simulated for all randomized patients for 1,500 iterations using the same randomization algorithm as the original randomization, keeping patient stratification factors and entry.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Change in Hb from baseline | MMRM | The MMRM model included the randomization stratification factors of transfusion history and screening Hb level. | Missing data were not imputed. Missing-at-random assumed for the missing data mechanism. |
|
Proportion of patients with Hb increase of ≥ 2 g/dL in the absence of transfusion | CMH test; the 95% CI was produced using the Miettinen and Nurminen method | No adjustment factors were planned. | Patients who withdrew from the study or had a missing Hb value at week 12 were considered to have not met the end point. | None |
Proportion of patients achieving transfusion avoidance (transfusion-free and do not require a transfusion) | CMH test; the 95% CI was produced using the Miettinen and Nurminen method | CMH test was stratified by transfusion history (> 2 or ≤ 2 transfusions in the last 6 months) and baseline Hb levels (< 8.5 and ≥ 8.5 g/dL). | Patients who withdrew from the study or had a missing transfusion occurrence assessment at week 12 were considered to have not achieved TA. | None |
Change from baseline in FACIT-F scores | MMRM | The MMRM model included the randomization stratification factors of transfusion history and screening Hb level. | Missing data were not imputed. Missing-at-random assumed for the missing data mechanism. | None |
Change from baseline in absolute reticulocyte count | MMRM | The MMRM model included the randomization stratification factors of transfusion history and screening Hb level. | Missing data were not imputed. Missing-at-random assumed for the missing data mechanism. | None |
Change in transfusion burden (number of RBC units transfused and transfusion instances) | ANCOVA model | No adjustment factors were planned. | Missing data were not imputed. Missing-completely-at-random assumed for the missing data mechanism. | None |
Change from baseline in LDH values | MMRM | No adjustment factors were planned. | Missing data were not imputed. Missing-at-random assumed for the missing data mechanism. | None |
Proportion of patients with Hb normalization | MMRM | No adjustment factors were planned. | Missing data were not imputed. Missing-at-random assumed for the missing data mechanism. | None |
Change from baseline in EQ-5D-3L scores | MMRM | No adjustment factors were planned. | Missing data were not imputed. Missing-at-random assumed for the missing data mechanism. | None |
Change from baseline in EORTC QLQ-C30 scores | MMRM | No adjustment factors were planned. | Missing data were not imputed. Missing-at-random assumed for the missing data mechanism. | None |
ANCOVA = analysis of covariance; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; LDH = lactate dehydrogenase; MMRM = mixed model for repeated measures; RBC = red blood cell; TA = transfusion avoidance.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Key Secondary End Point Analyses
The key secondary end points were analyzed on the ITT population using a hierarchical fixed sequence test procedure, provided that statistical significance was declared for the primary end point. The prespecified alpha-spending method described previously was applied. In order of importance, the sequential testing process for key secondary outcomes was:
difference in proportion of patients with Hb increase of 2 g/dL or greater at week 12 in the absence of transfusions
difference in proportion of patients with RBC transfusion avoidance between danicopan and placebo groups during the first 12 weeks of treatment
difference in changes from baseline in FACIT-F scores between danicopan and placebo groups at week 12
difference in changes from baseline in absolute reticulocyte counts between danicopan and placebo groups at week 12.
Subgroup Analysis
Subgroup analyses were descriptive in nature and did not take multiple comparisons into account. The primary end point was summarized by the following prespecified subgroups (not appraised by CDA-AMC):
transfusion history (≤ 2 or > 2 transfusions in the last 6 months)
Hb levels (< 8.5 g/dL or ≥ 8.5 g/dL)
Japanese ethnicity (yes or no).
The primary and key secondary end points were summarized by subgroups on the basis of the following (not appraised by CDA-AMC):
sex
race
region
age (aged < 65 years and ≥ 65 years)
background C5i (ravulizumab or eculizumab)
Results
Patient Disposition
Complete details of patient disposition for the IEAS are available in Table 10. Briefly, 111 patients were screened for inclusion in the trial and 25 (22.5%) failed screening. A total of 21 (18.9%) of patients did so because of not meeting inclusion or exclusion criteria; the submission did not provide additional detail on which criterion was the most common reason for exclusion. A total of 1 patient discontinued from each treatment arm during TP1, and 1 patient from the danicopan plus C5i arm discontinued during TP2. A total of 6 patients from the danicopan plus C5i arm and 3 patients from the placebo plus C5i arm had completed year 1 of the LTE at the time of study submission.
Table 10: Summary of Patient Disposition From the ALPHA Study — Interim Efficacy Analysis
Patient disposition | ALPHA | |
|---|---|---|
Danicopan plus C5i n = 42 | Placebo plus C5i n = 21 | |
Screened, N | 111 | |
Screening failures, N (%) | 25 (22.5) | |
Reason for screening failure, N (%) | ||
Failure to meet the inclusion or exclusion criteria | 21 (18.9) | |
Other | 4 (3.6) | |
Randomized, N (%) | 57 (100.0) | 29 (100.0) |
ISAS, N | 57 | 29 |
IEAS, N | 42 | 21 |
Treatment period 1 (weeks 0 to 12) — IEAS | ||
Completed | 41 (97.6) | 20 (95.2) |
Discontinued from study, n (%) | 1 (2.4) | 1 (4.8) |
Reason for discontinuation, n (%) | ||
Adverse events | 1 (2.4) | 1 (4.8) |
Treatment period 2 (weeks 12 to 24) — IEAS | ||
Entered treatment period 2 | 41 (97.6) | 20 (95.2) |
Completed | 40 (95.2) | 20 (95.2) |
Discontinued from study, n (%) | 1 (2.4) | 0 |
Reason for discontinuation, n (%) | ||
Adverse events | 1 (2.4) | 0 |
LTE (LTE1 and LTE2) — IEAS | ||
Entered LTEs | 40 (95.2) | 20 (95.2) |
Ongoing | 36 (85.7) | 19 (90.5) |
Completed LTE1 | 6 (14.3) | 3 (14.3) |
Discontinued from study, n (%) | 4 (9.5) | 1 (4.8) |
Reason for discontinuation, n (%) | ||
Adverse events | 0 | 1 (4.8) |
Nonadherence with study intervention | 1 (2.4) | 0 |
Physician decision | 1 (2.4) | 0 |
Withdrawal of consent | 2 (4.8) | 1 (4.8) |
C5i = complement component 5 inhibitor; IEAS = interim efficacy analysis set; ISAS = interim safety analysis set; LTE = long-term extension.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. Briefly, most baseline characteristics were broadly similar between study arms. There was a numeric difference in the proportion of patients (66.7% female in the placebo plus C5i arm, 54.8% female in the danicopan plus C5i arm; 33.3% male in the placebo plus C5i arm, 45.2% male in the danicopan plus C5i arm), and the proportion of patients of Asian descent (33.3% in the placebo plus C5i arm, 42.9% in the danicopan plus C5i arm). There were also numeric differences in the proportion of patients treated with each C5i (64.3% of patients in the danicopan plus C5i arm and 47.6% of patients in the placebo plus C5i arm were treated with ravulizumab). There was a numerically higher LDH in the danicopan plus C5i arm (298.73 U/L) relative to the placebo plus C5i arm (278.25 U/L), and a numerically higher proportion of patients in the danicopan plus C5i arm had received a transfusion within 24 weeks of receiving the study drug (90.5% in the danicopan plus C5i arm, 81.0% in the placebo plus C5i arm).
In addition, a total of 41 (97.6%) patients in the danicopan plus C5i arm and 21 (100%) patients in the placebo plus C5i arm reported experiencing PNH symptoms at any time before consenting to the study. The most commonly reported (> 30% of patients) symptoms in the danicopan plus C5i arm were fatigue or asthenia (90.5%), red or dark urine (71.4%), shortness of breath (59.5%), jaundice (38.1%), or central nervous system symptoms such as headache (31.0%). The most commonly reported (> 30% of patients) in the placebo plus C5i arm were fatigue or asthenia (90.5%), abdominal pain (57.1%), shortness of breath (42.9%), red or dark urine (42.9%), or jaundice (33.3%).
Table 11: Summary of Baseline Characteristics From the ALPHA Study — IEAS
Characteristic | ALPHA | |
|---|---|---|
Danicopan plus C5i n = 42 | Placebo plus C5i n = 21 | |
Age at informed consent (years), mean (SD) | 55.0 (15.64) | 53.1 (14.27) |
Sex | ||
Female | 23 (54.80) | 14 (66.70) |
Male | 19 (45.20) | 7 (33.30) |
Race | ||
American Indian or Alaska Native | 1 (2.40) | 0 |
Asian | 18 (42.90) | 7 (33.30) |
Black or African American | 1 (2.40) | 0 |
White | 19 (45.20) | 9 (42.90) |
Other | 1 (2.40) | 0 |
Not reported | 2 (4.80) | 4 (19.00) |
Unknown | 0 | 1 (4.80) |
Japanese ancestry, n (%) | ||
Yes | 5 (11.90) | 2 (9.50) |
No | 37 (88.10) | 19 (90.50) |
BMI (kg/m2), mean (SD) | 26.74 (5.38) | 24.77 (4.87) |
PNH-related characteristics | ||
Age at PNH diagnosis (years), mean (SD) | 44.20 (16.59) | 40.79 (16.30) |
Years since diagnosis (years), mean (SD) | 11.28 (10.59) | 12.78 (10.42) |
Age at first C5i infusion (years), mean (SD) | 50.05 (15.32) | 47.05 (14.57) |
Duration of C5i therapy, mean (SD) | 5.53 (3.89) | 6.66 (4.62) |
Current C5i, n (%) | ||
Ravulizumab | 27 (64.30) | 10 (47.60) |
Eculizumab | 15 (35.70) | 11 (52.40) |
Hb at baseline (g/dL), mean (SD) | 7.66 (0.94) | 7.74 (1.04) |
FACIT-F score, mean (SD) | 33.46 (11.09) | 33.86 (10.78) |
Absolute reticulocyte count (109/L), mean (SD) | 236.37 (91.38) | 240.64 (120.28) |
LDH (U/L), mean (SD) | 298.73 (105.71) | 278.25 (68.40) |
Transfusion history | ||
Participants with pRBC transfusions 6 months before screening, n (%) | 42 (100) | 21 (100) |
Participants with pRBC transfusions within 24 weeks before first dose, n (%) | 38 (90.50) | 17 (81.00) |
Transfusion instances, mean (SD) | 2.5 (2.16) | 2.6 (2.11) |
Units transfused, mean (SD) | 4.3 (4.66) | 4.4 (3.79) |
Participants with pRBC transfusions within 12 weeks before receiving study drug, n (%) | 29 (69.00) | 15 (71.40) |
Transfusion instances, mean (SD) | 1.4 (1.41) | 1.5 (1.36) |
Units transfused, mean (SD) | 2.1 (2.46) | 2.4 (2.25) |
BMI = body mass index; C5i = complement component 5 inhibitor; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; LDH = lactate dehydrogenase; PNH = paroxysmal nocturnal hemoglobinuria; pRBC = packed red blood cells; SD = standard deviation.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Details of patient exposure to danicopan are summarized in Table 12. Briefly, mean and median exposure durations were similar for TP1 and TP2; the median duration of exposure during LTE1 was numerically larger in the placebo plus C5i arm (251 days) than the danicopan plus C5i arm (179.5 days). Patients in the danicopan plus C5i arm had numerically different mean durations of exposure than the placebo plus C5i arm at 150 mg danicopan dosing (63.5 days in the danicopan plus C5i arm, 55.5 days in the placebo plus C5i arm during TP2; 153.2 days in the placebo plus C5i arm, 143.8 days in the danicopan plus C5i arm in LTE1). This was also reported for exposure to 200 mg danicopan dosing (71.9 days in the danicopan plus C5i arm, 40.5 days in the placebo plus C5i arm during TP2; 200.3 days in the danicopan plus C5i arm, 213 days in the placebo plus C5i arm during LTE1). Overall adherence based on pill counts was greater than 95% for all treatment periods and in all treatment arms.
Efficacy
Detailed outcomes from baseline to week 12 (TP1) are presented in Table 13, and results from baseline to week 24 (TP2) and from baseline to week 72 (LTE1) are presented in Table 14.
Hb Levels
Change in Hb Levels
The LS mean change from baseline in Hb level to 12 weeks (at TP1) was the primary outcome. At TP1, the LS mean difference for the change in Hb from baseline between the danicopan plus C5i and the placebo plus C5i arms was 24.44 g/L (98.2% CI, 15.25 g/L to 33.63 g/L; P ≤ 0.0001). At TP2, the LS mean change from baseline to week 24 in the danicopan-emergent arm (patients who received danicopan plus C5i from weeks 0 to 12 and continued to receive danicopan plus C5i from weeks 12 to 24) was 31.67 g/L (95% CI, 25.61 g/L to 27.74 g/L). In the placebo-emergent arm (patients who received placebo plus C5i from weeks 0 to 12 and who subsequently switched to receive danicopan plus C5i from weeks 12 to 24), the LS mean change from baseline to week 24 was 22.58 g/L (95% CI, 15.72 g/L to 29.44 g/L). At LTE1, the observed mean change from baseline in Hb levels was 32.00 g/L (SD = 11.81 g/L) in the danicopan-emergent arm and 31.50 (SD = 10.61) in the placebo-emergent arm.
Proportion of Patients With Hb Level Increase of 2 g/dL or Greater in the Absence of Transfusion
The proportion of patients with Hb level increases of 2 g/dL or greater was a key secondary outcome in the analysis. At TP1, the LS mean difference for the proportion of patients with Hb level increase of 2 g/dL or greater between the danicopan plus C5i and the placebo plus C5i arms was 45.90% (95.8% CI, 27.40% to 64.42%; P ≤ 0.0001). At TP2, the proportion of patients with this outcome in the danicopan-emergent arm was 46.3% (95% CI, 30.66% to 62.58%); results were not reported for the placebo-emergent arm. This outcome was not measured at LTE1.
Proportion of Patients With Hb Normalization
The proportion of patients with Hb normalization, defined as patients with Hb values greater than the lower limit of the normal reference range, was a secondary outcome. At TP1, the LS mean difference for the change in the proportion of patients with Hb normalization between the danicopan plus C5i and the placebo plus C5i arms was 18.40% (95% CI, –0.84% to 37.71%; P = 0.0080). At TP2, the proportion of patients with this outcome in the danicopan-emergent arm was 19.50% (95% CI, 8.82% to 34.87%). This outcome was not reported for the placebo-emergent arm at TP2 and was not reported at LTE1 for either arm.
Transfusions
Transfusion Avoidance
Transfusion avoidance at TP1 was a key secondary outcome in the analysis. At TP1, the LS mean TD for the proportion of patients with transfusion avoidance between the danicopan plus C5i and the placebo plus C5i arms was 40.80% (95.8% CI, 21.08% to 60.58%; P = 0.0004). At TP2, the proportion of patients with this outcome in the danicopan-emergent arm was 78.00% (95% CI, 62.39% to 89.44%), and was 90.00% (95% CI, 68.30% to 98.77%) in the placebo-emergent arm. This outcome was not reported at LTE1.
Table 12: Summary of Patient Exposure During the ALPHA Study — ISAS
Exposure | TP1 | TP2 | LTE1 | |||
|---|---|---|---|---|---|---|
DAN plus C5i n = 57 | Placebo plus C5i n = 29 | Week 0 to 12: DAN plus C5i Week 12 to 24: DAN plus C5i n = 48 | Week 0 to 12: placebo plus C5i Week 12 to 24: DAN plus C5i n = 23 | Week 0 to 12: DAN plus C5i Week 12 to 24: DAN plus C5i n = 40 | Week 0 to 12: placebo plus C5i Week 12 to 24: DAN plus C5i n = 20 | |
Duration (days), mean (SD) | 79.2 (13.47) | 76.7 (17.16) | 78.2 (17.03) | 78.4 (17.64) | 192.6 (120.25) | 207.9 (133.80) |
Duration (days), median (range) | 84.0 (23.0 to 85.0) | 84.0 (28.0 to 86.0) | 84.0 (12.0 to 94.0) | 84.0 (5.0 to 85.0) | 179.5 (1.0 to 455.0) | 251.0 (7.0 to 419.0) |
Adherence, % mean (SD)a | 97.9 (4.55) | 96.1 (10.21) | 98.7 (9.98) | 98.3 (3.71) | 95.3 (12.57) | 98.1 (3.36) |
Exposure at specific doses | ||||||
DAN 100 mg | ||||||
N patients | 3 | NA | 1 | 0 | 0 | 0 |
Duration (days), mean (SD) | 56.0 (37.04) | NA | 28.0 (NA) | NA | NA | NA |
Duration (days), median (range) | 70.0 (14.0 to 84.0) | NA | 28.0 (28.0 to 28.0) | NA | NA | NA |
DAN 150 mg | ||||||
N patients | 56 | NA | 21 | 23 | 9 | 9 |
Duration (days), mean (SD) | 69.1 (21.35) | NA | 63.5 (24.22) | 55.5 (22.82) | 143.8 (91.08) | 153.2 (146.98) |
Duration (days), median (range) | 84.0 (14.0 to 97.0) | NA | 83.0 (15.0 to 94.0) | 42.0 (5.0 to 85.0) | 112.0 (36.0 to 309.0) | 92.0 (4.0 to 407.0) |
DAN 200 mg | ||||||
N patients | 14 | NA | 33 | 13 | 32 | 13 |
Duration (days), mean (SD) | 35.4 (13.08) | NA | 71.9 (19.76) | 40.5 (6.16) | 200.3 (118.39) | 213.8 (134.76) |
Duration (days), median (range) | 42.0 (6.0 to 48.0) | NA | 84.0 (27.0 to 85.0) | 42.0 (20.0 to 43.0) | 196.0 (1.0 to 413.0) | 251.0 (7.0 to 474.0) |
C5i = complement component 5 inhibitor; DAN = danicopan; ISAS = interim safety analysis set; LTE = long-term extension; NA = not applicable; SD = standard deviation; TP = treatment period.
aAdherence based on tablet counts.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Transfusion Burden
Transfusion burden was measured by the number of RBC units transfused and the number of transfusion instances; both were secondary outcomes. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in the number of RBC units transfused between the 12 weeks before study drug initiation and the 12 weeks after study drug initiation was –1.31 (95.8% CI, –2.24 to –0.37; P = 0.0072). At TP2, the change in the number of RBC units transfused in the 24 weeks after treatment initiation relative to the 24 weeks before treatment initiation in the danicopan-emergent arm was –2.80 (95% CI, –4.55 to –1.11). This outcome was not reported in the placebo-emergent arm or at LTE1 in either arm.
Table 13: Summary of Key Efficacy Results From the ALPHA Study During TP1 — IEAS
Variable | TP1 | |
|---|---|---|
Danicopan plus C5i n = 42 | Placebo plus C5i n = 21 | |
Change in Hb from baseline to week 12 | ||
Complete cases, n | 36 | 20 |
Baseline (g/L), mean (SD) | 76.6 (9.39) | 77.4 (10.35) |
LS mean (98.2% CI)a change from baseline (g/L) | 29.40 (24.23 to 34.57) | 4.96 (–2.70 to 12.61) |
LS mean difference (98.2% CI)a | 24.44 (15.25 to 33.63) | |
P valueb (2-sided alpha = 0.018) | < 0.0001 | |
Proportion of patients with Hb increase ≥ 2 g/dL (20 g/L) in the absence of transfusion | ||
Complete cases, n | NR | NR |
N patients | 25 | 0 |
Proportion of patients (95.8% CI)a | 59.50 (42.73 to 74.84) | 0 (0.00 to 16.80) |
Treatment difference (95.8% CI)a | 45.90 (27.40 to 64.42) | |
Stratified CMH P valueb (2-sided alpha = 0.042) | < 0.0001 | |
Proportion of patients with transfusion avoidance at week 12 | ||
Complete cases, n | NR | NR |
N patients | 35 | 8 |
Proportion of patients (95.8% CI)a | 83.30 (68.08 to 93.27) | 38.10 (17.56 to 62.32) |
Treatment difference (95.8% CI)a | 40.80 (21.08 to 60.58) | |
Stratified CMH P valueb (2-sided alpha = 0.042) | 0.0004 | |
Change in FACIT-F scores from baseline to week 12 | ||
Complete cases, n | 39 | 21 |
Baseline, mean (SD)c | 33.46 (11.09) | 33.86 (10.78) |
LS mean (95.8% CI)a change from baseline | 7.97 (5.63 to 10.32) | 1.85 (–1.44 to 5.14) |
LS mean difference (95.8% CI)a | 6.12 (2.18 to 10.06) | |
P valueb (2-sided alpha = 0.042) | 0.0021 | |
Change in absolute reticulocyte count (1012/L) from baseline to week 12 | ||
Complete cases, n | 35 | 18 |
Baseline, mean (SD)c | 0.236 (0.091) | 0.241 (0.120) |
LS mean change from baseline (95.8% CI)a | –0.084 (–0.102 to –0.065) | 0.004 (–0.023 to 0.030) |
LS mean difference (95.8% CI)a | –0.087 (–0.119 to –0.056) | |
P valueb (2-sided alpha = 0.042) | < 0.0001 | |
Change in the number of RBC units transfused from 12 weeks before treatment initiation to week 12 postinitiation | ||
Complete cases, n | NR | NR |
LS mean (95% CI) change from baseline | –1.48 (–2.02 to –0.94) | –0.18 (–0.94 to 0.59) |
LS mean difference (95% CI) | –1.31 (–2.24 to –0.37) | |
P value | 0.0072 | |
Change in the number of transfusion instances from 12 weeks before treatment initiation to week 12 postinitiation | ||
Complete cases, n | NR | NR |
LS mean (95% CI) change from baseline | –0.92 (–1.27 to –0.57) | –0.21 (–0.70 to 0.29) |
LS mean difference (95% CI) | –0.72 (–1.32 to –0.11) | |
P value | 0.0207 | |
Change in LDH values (U/L) from baseline to week 12 | ||
Complete cases, n | 41 | 20 |
Baseline (U/L), mean (SD)c | 298.73 (105.71) | 278.25 (68.40) |
Observed, mean (SD) | 268.24 (61.38) | 328.38 (224.31) |
LS mean (95% CI) change from baseline | –23.49 (–40.08 to –6.90) | –2.92 (–26.78 to 20.93) |
LS mean difference (95% CI) | –20.57 (–49.28 to 8.15) | |
P value | 0.1569 | |
Proportion of patients with Hb normalization at week 12 | ||
N patients | 12 | 0 |
Proportion of patients (95% CI) | 28.60 (15.72 to 44.58) | 0 (0.00 to 16.11) |
Treatment difference (95% CI) | 18.40 (–0.84 to 37.71) | |
Stratified CMH P value | 0.0080 | |
Change in EQ VAS from baseline to week 12 | ||
Complete cases, n | 39 | 20 |
Baseline, mean (SD)c | 57.40 (19.90) | 62.60 (20.14) |
LS mean (95% CI) change from baseline | 11.53 (6.25 to 16.81) | 5.25 (–2.46 to 12.96) |
LS mean difference (95% CI) | 6.27 (–2.85 to 15.40) | |
P value | 0.1738 | |
Change in EORTC QLQ-C30 global health status/QoL scores from baseline to week 12 | ||
Complete cases, n | 39 | 20 |
Baseline, mean (SD)c | 57.14 (19.09) | 59.17 (15.51) |
LS mean (95% CI) change from baseline | 10.42 (5.87 to 14.97) | 3.80 (–2.78 to 10.38) |
LS mean difference (95% CI) | 6.62 (–1.17 to 14.41) | |
P value | 0.0941 | |
C5i = complement component 5 inhibitor; CI = confidence interval; CMH = Cochran Mantel-Haenszel; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = EQ visual analogue scale; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; IESA = interim efficacy analysis set; LDH = lactate dehydrogenase; LS = least squares; NR = not reported; QoL = quality of life; RBC = red blood cell; SD = standard deviation; TP = treatment period.
aThe CI level which matches the alpha level specified in the multiple testing structure is reported.
bP value adjusted for multiple comparisons.
cBaseline measurement for the full study sample, obtained from the ALPHA Clinical Study Report.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence, additional information provided by the sponsor,41 and the ALPHA Clinical Study Report.40
Table 14: Summary of TP2 and LTE Efficacy Results From the ALPHA Study — IEAS
Variable | TP2 | LTE1 | ||
|---|---|---|---|---|
Weeks 0 to 12: Danicopan plus C5i Weeks 12 to 24: Danicopan plus C5i n = 42 | Weeks 0 to 12: Placebo plus C5i Weeks 12 to 24: Danicopan plus C5i n = 21 | Weeks 0 to 12: Danicopan plus C5i Weeks 12 to 24: Danicopan plus C5i n = 42 | Weeks 0 to 12: Placebo plus C5i Weeks 12 to 24: Danicopan plus C5i n = 21 | |
Change in Hb from baseline | ||||
Complete cases, n | 35 | 16 | 5 | 2 |
Baseline (g/L), mean (SD)a | 76.6 (9.39) | 77.4 (10.35) | 76.6 (9.39) | 77.4 (10.35) |
Observed (g/L), mean (SD) | 107.80 (17.30) | 104.60 (17.01) | 10.380 (10.18) | 115.50 (9.19) |
Change from baseline (g/L), mean (SD) | 31.20 (17.13) | 27.40 (14.14) | 32.00 (11.81) | 31.50 (10.61) |
LS mean change (g/L) from baseline (95% CI) | 31.67 (25.61 to 37.74) | 22.58 (15.72 to 29.44) | NR | NR |
Proportion of patients with Hb increase ≥ 2 g/dL (20 g/L) in the absence of transfusion | ||||
Complete cases, n | NR | NR | NR | NR |
N patients | 19 | NR | NR | NR |
Proportion of patients (95% CI) | 46.30 (30.66 to 62.58) | NR | NR | NR |
Proportion of patients with transfusion avoidance | ||||
Complete cases, n | NR | NR | NR | NR |
N patients | 32 | 18 | NR | NR |
Proportion of patients (95% CI) | 78.00 (62.39 to 89.44) | 90.00 (68.30 to 98.77) | NR | NR |
Change in FACIT-F scores | ||||
Complete cases, n | 37 | 20 | 7 | 3 |
Baseline, mean (SD)a | 33.46 (11.09) | 33.86 (10.78) | 33.46 (11.09) | 33.86 (10.78) |
Observed, mean (SD) | 40.32 (10.54) | 40.55 (10.88) | 41.43 (13.82) | 23.67 (17.16) |
Change from baseline, mean (SD) | 6.48 (9.03) | 5.60 (10.23) | 3.86 (7.15) | –4.33 (9.07) |
LS mean (95% CI) change from baseline | 6.12 (3.41 to 8.82) | 6.44 (1.23 to 11.64) | NR | NR |
Change in absolute reticulocyte count (1012/L) from baseline | ||||
Complete cases, n | 31 | 12 | 5 | 1 |
Baseline, mean (SD)a | 0.236 (0.091) | 0.241 (0.120) | 0.236 (0.091) | 0.241 (0.120) |
Observed count, mean (SD) | 0.148 (0.059) | 0.167 (0.054) | 0.237 (0.097) | 0.138 (NA) |
Change from baseline, mean (SD) | –0.080 (0.073) | –0.084 (0.110) | –0.041 (0.029) | –0.106 (NA) |
Change in the number of RBC units transfused from 24 weeks before treatment initiation to week 24 post initiation | ||||
Complete cases, n | 41 | NR | NR | NR |
Mean (95% CI) change | –2.80 (–4.55 to –1.11) | NR | NR | NR |
Change in the number of transfusion instances from 24 weeks before treatment initiation to week 24 post initiation | ||||
Complete cases, n | 41 | NR | NR | NR |
Mean (95% CI) change | –1.50 (–2.36 to –0.67) | NR | NR | NR |
Change in LDH values (U/L) from baseline | ||||
Complete cases, n | 38 | 19 | 6 | 3 |
Baseline, mean (SD)a | 298.73 (105.71) | 278.25 (68.40) | 298.73 (105.71) | 278.25 (68.40) |
Observed, mean (SD) | 279.21 (88.64) | 277.55 (64.78) | 244.17 (55.53) | 268.33 (36.69) |
Mean (SD) change from baseline | –23.46 (105.40) | 0.21 (84.89) | –20.83 (67.00) | 5.00 (111.89) |
Proportion of patients with Hb normalization | ||||
N patients | 8 | NR | NR | NR |
Proportion of patients (95% CI) | 19.50 (8.82 to 34.87) | NR | NR | NR |
Change in EQ VAS scores from baseline | ||||
Complete cases, n | 37 | 18 | 7 | 2 |
Baseline, mean (SD)a | 57.40 (19.90) | 62.60 (20.14) | 57.40 (19.90) | 62.60 (20.14) |
Observed, mean (SD) | 72.50 (17.57) | 72.50 (15.52) | 74.90 (25.77) | 51.50 (30.41) |
Mean (SD) change from baseline | 13.70 (20.12) | 9.70 (21.93) | 12.30 (18.70) | –11.00 (12.73) |
Change in EORTC QLQ-C30 global health status/QoL scores from baseline | ||||
Complete cases, n | 36 | 19 | 7 | 3 |
Baseline, mean (SD)a | 57.14 (19.09) | 59.17 (15.51) | 57.14 (19.01) | 59.17 (15.51) |
Observed, mean (SD) | 67.82 (19.84) | 70.42 (15.17) | 67.86 (20.65) | 61.11 (25.46) |
Mean (SD) change from baseline | 8.56 (16.96) | 10.53 (14.92) | 1.19 (26.97) | 8.33 (22.05) |
C5i = complement component 5 inhibitor; CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = EQ visual analogue scale; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; IEAS = interim efficacy analysis set; LDH = lactate dehydrogenase; LS = least squares; LTE = long-term extension; NA = not available; NR = not reported; QoL = quality of life; RBC = red blood cell; SD = standard deviation; TP = treatment period.
aBaseline measurement for the full study sample, obtained from the ALPHA Clinical Study Report.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the ALPHA Clinical Study Report.40
At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in the number of transfusion instances between the 12 weeks before study drug initiation and the 12 weeks after study drug initiation was –0.72 (95% CI, –1.32 to –0.11; P = 0.0207). At TP2, the change in the number of transfusion instances between the 24 weeks before study drug initiation and the 24 weeks after study drug initiation in the danicopan-emergent arm was –1.50 (95% CI, –2.36 to –0.67). This outcome was not reported in the placebo-emergent arm or at LTE1 in either arm.
Absolute Reticulocyte Count
Change in absolute reticulocyte count from baseline to week 12 was a key secondary outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in absolute reticulocyte count from baseline was –0.087 × 1012/L (95.8% CI, –0.119 × 1012/L to –0.056 × 1012/L; P ≤ 0.0001). At TP2, the change from baseline in absolute reticulocyte counts in the danicopan-emergent arm was –0.080 × 1012/L (SD = 0.073 × 1012/L), and in the placebo-emergent arm was –0.084 × 1012/L (SD = 0.110 × 1012/L). At LTE1, the observed mean change from baseline in absolute reticulocyte counts in the danicopan-emergent arm was –0.041 × 1012/L (SD = 0.029 × 1012/L), and in the placebo-emergent arm was –0.106 × 1012/L (SD = not applicable; n = 1 patient).
Lactate Dehydrogenase
Change in LDH from baseline was a secondary outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in LDH from baseline was –20.57 U/L (95% CI, –49.28 U/L to 8.15 U/L; P = 0.1569). At TP2, the mean change from baseline in LDH in the danicopan-emergent arm was –23.46 U/L (SD = 105.40 U/L), and in the placebo-emergent arm was 0.21 U/L (SD = 84.89 U/L). At LTE1, the mean change from baseline in LDH in the danicopan-emergent arm was –20.83 U/L (SD = 67.00 U/L), and in the placebo-emergent arm was 5.00 U/L (SD = 111.89 U/L).
Fatigue and HRQoL
Functional Assessment of Chronic Illness Therapy–Fatigue
The change in FACIT-F scores from baseline was a key secondary outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change in FACIT-F scores from baseline was 6.12 (95.8% CI, 2.18 to 10.06; P = 0.0021). At TP2, the LS mean change from baseline in FACIT-F scores in the danicopan-emergent arm was 6.12 (95% CI, 3.41 to 8.82), and in the placebo-emergent arm was 6.44 (95% CI, 1.23 to 11.64). At LTE1, the mean change from baseline in the danicopan-emergent arm was 3.86 (SD = 7.15) and –4.33 (SD = 9.07) in the placebo-emergent arm.
EQ Visual Analogue Scale
The change in EQ VAS scores from baseline was an exploratory outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change from baseline in EQ VAS scores was 6.27 (95% CI, –2.85 to 15.40; P = 0.1738). At TP2, the mean change from baseline in EQ VAS scores was 13.70 (SD = 20.12) in the danicopan-emergent arm and 9.70 (SD = 21.93) in the placebo-emergent arm. At LTE1, the mean change from baseline in the danicopan-emergent arm was 12.30 (SD = 18.70) and –11.00 (SD = 12.73) in the placebo-emergent arm.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
The change in EORTC QLQ-C30 global health scores from baseline was an exploratory outcome. At TP1, the LS mean TD between the danicopan plus C5i arm and the placebo plus C5i arm for the change from baseline in EORTC QLQ-C30 global health scores was 6.62 (95% CI, –1.17 to 14.41; P = 0.0941). At TP2, the mean change from baseline in EORTC QLQ-C30 global health scores was 8.56 (SD = 16.96) in the danicopan-emergent arm and 10.53 (SD = 14.92) in the placebo-emergent arm. At LTE1, the mean change from baseline in the danicopan-emergent arm was 1.19 (SD = 26.97) and 8.33 (SD = 22.05) in the placebo-emergent arm.
Sensitivity Analyses
For each primary and key secondary outcome from baseline to week 12, per-protocol analysis was done as a sensitivity, all of which had similar results to the ITT analysis. Change from Hb at 12 weeks additionally included an analysis using Hb values collected less than 4 weeks from transfusion, with similar results. Transfusion avoidance had an analysis with alternate handling of patients who withdrew from treatment, wherein those who discontinued the study because of lack of efficacy during TP1 were excluded. This analysis also yielded similar results.
Harms
Harms reporting was done on the interim safety analysis set, using the full sample size recruited and including all patients who had received at least 1 dose of study drug as of the data cut-off. As of the data cut-off date, a total of 4 patients were still receiving placebo in TP1 and 2 patients discontinued before the switch to danicopan in TP2. Harms were reported separately for TP1, TP2, and LTE1 cut-offs, as well as overall during the entire time patients were exposed to danicopan (total danicopan treatment). Table 15 contains reporting of the most common harms data.
During TP1, in the danicopan plus C5i arm, 17.5% patients had TEAEs that were grade 3, and 1.8% had TEAEs that were grade 4 (no grade 5 events). In the placebo plus C5i arm, 13.8% of patients had TEAEs that were grade 3 (no grade 4 or 5 events). During TP2, in the danicopan-emergent arm, 12.5% of patients had TEAEs that were grade 3 (no grade 4 or 5 events); in the placebo-emergent arm, 13.0% of patients had TEAEs that were grade 3 and 4.3% had TEAEs that were grade 4. During LTE, 7.5% of patients in the danicopan-emergent arm and 15.0% of patients in the placebo-emergent arm experienced grade 3 TEAEs, 2.5% of patients in the danicopan-emergent arm and 5.0% of patients in the placebo-emergent arm experienced grade 4 TEAEs, and there were no grade 5 TEAEs reported during the LTE.
Adverse Events
Overall, a total of 93.0% of patients in the danicopan-emergent arm and 82.6% of patients in the placebo-emergent arm experienced TEAEs during treatment with danicopan. The most common TEAEs during treatment with danicopan were COVID-19 (21.1% danicopan-emergent and 21.7% placebo-emergent), diarrhea (14.0% danicopan-emergent and 17.4% placebo-emergent), headache (17.5% danicopan-emergent and 8.7% placebo-emergent), pyrexia (15.8% danicopan-emergent and 8.7% placebo-emergent), nausea (12.3% danicopan-emergent and 13.0% placebo-emergent), and fatigue (10.5% danicopan-emergent and 8.7% placebo-emergent).
During TP1, 73.7% of patients in the danicopan plus C5i arm and 62.1% of patients in the placebo plus C5i arm experienced any TEAEs. There were numeric differences in the proportion of patients experiencing TEAEs for anemia (1.8% danicopan plus C5i, 13.8% placebo plus C5i), vomiting (5.3% danicopan plus C5i, 0% placebo plus C5i), upper abdominal pain (1.8% danicopan plus C5i, 6.9% placebo plus C5i), pyrexia (5.3% danicopan plus C5i, 0% placebo plus C5i), asthenia (0% danicopan plus C5i, 13.8% placebo plus C5i), ear infection (0% danicopan plus C5i, 6.9% placebo plus C5i), contusion (1.8% danicopan plus C5i, 10.3% placebo plus C5i), increased aspartate aminotransferase (3.5% danicopan plus C5i, 10.3% placebo plus C5i), pain in extremity (5.3% danicopan plus C5i, 0% placebo plus C5i), dizziness (1.8% danicopan plus C5i, 6.9% placebo plus C5i), and insomnia (1.8% danicopan plus C5i, 10.3% placebo plus C5i). A total of 57 patients in the danicopan plus C5i arm and 29 patients in the placebo plus C5i arm contributed data.
During TP2, when both arms were receiving danicopan, a total of 64.6% of patients in the danicopan-emergent arm and 56.5% of patients in the placebo-emergent arm reported TEAEs. There were numeric differences in the proportion of patients experiencing TEAEs for nausea (2.1% danicopan-emergent, 13.0% placebo-emergent), and pyrexia (10.4% danicopan-emergent, 0% placebo-emergent). A total of 48 patients in the danicopan-emergent arm and 23 patients in the placebo-emergent arm contributed data.
During LTE (up until the data cut-off of September 20, 2022), a total of 62.5% of patients in the danicopan-emergent arm and 80.0% of patients in the placebo-emergent arm reported TEAEs. There were numeric differences in the proportion of patients experiencing TEAEs for diarrhea (2.5% danicopan-emergent, 10.0% placebo-emergent), asthenia (2.5% danicopan-emergent, 15.0% placebo-emergent), and back pain (2.5% danicopan-emergent, 10.0% placebo-emergent). A total of 40 patients in the danicopan-emergent arm and 20 patients in the placebo-emergent arm contributed data.
Serious AEs
Overall, a total of 12.3% of patients in the danicopan-emergent arm and 26.1% of patients in the placebo-emergent arm experienced any SAE while being treated with danicopan.
During TP1, 5.3% of patients in the danicopan plus C5i arm experienced any SAE; the SAEs were pancreatitis, cholecystitis, COVID-19, and blood bilirubin increase (1 report of each). A total of 6.9% of patients in the placebo plus C5i arm experienced any SAE; the SAEs were anemia, abdominal pain, and headache (1 report of each).
During TP2, 6.3% of patients in the danicopan-emergent arm experienced any SAE; the SAEs were Dieulafoy vascular malformation, pyrexia, COVID-19 pneumonia, and staphylococcus sepsis (1 report of each). In the placebo-emergent arm, 13.0% of patients experienced any SAE; the SAEs were hemolysis, vertigo, and headache (1 report of each).
During LTE (up until the data cut-off on September 20, 2022), 7.5% of patients in the danicopan-emergent arm experienced any SAE; the SAEs were stent-grant endoleak, decreased Hb, invasive ductal breast carcinoma, pulmonary embolism, and pulmonary hemorrhage (1 report of each). In the placebo-emergent arm, 20.0% of patients experienced any SAE; the SAEs were pericardial effusion, diarrhea, disease progression, COVID-19, and body temperature increased (1 report of each).
Withdrawals Due to AEs
During TP1, TEAEs led to withdrawal of the study drug for 5.3% of patients in the danicopan plus C5i arm and 3.4% of patients in the placebo plus C5i arm. SAEs led to withdrawal of the study drug for 1.8% of patients in the danicopan plus C5i arm, and 0% of patients in the placebo plus C5i arm. During TP2, there were no TEAEs or SAEs leading to withdrawal of the study drug in either treatment arm. During LTE, TEAEs led to withdrawal of the study drug in 5.0% of patients in the placebo-emergent arm; there were no TEAEs leading to withdrawal of the study drug in the danicopan-emergent arm. There were no SAEs leading to withdrawal of the study drug in either treatment arm.
The submission did not provide further details on whether the TEAEs or SAEs that led to withdrawal of the study drug also led to patient discontinuation from the study; per the submission, there was 1 patient withdrawal in each arm due to AEs during TP1, 1 patient withdrawal due to AEs in the danicopan-emergent arm during TP2 (0 in the placebo-emergent arm), and 1 patient withdrawal in the placebo-emergent arm due to AEs in the LTE (0 in the danicopan-emergent arm).
Mortality
There were no deaths reported in either study arm, at any time point during the trial to date.
Notable Harms
AEs of Special Interest
Meningococcal infections and liver enzyme elevations were prespecified AEs of special interest during the ALPHA study. Throughout TP1, TP2, and LTE until September 20, 2022, there were no reported AEs of meningococcal infections in either study arm.
Liver enzyme elevations were defined using the MedDRA preferred terms for drug-related hepatic disorders – severe events only [narrow] [20000007] and liver-related investigations, signs and symptoms [narrow] [20000008]. During TP1, liver enzyme elevations occurred in 14.0% of patients in the danicopan plus C5i arm and 10.3% of patients in the placebo plus C5i arm. During TP2, liver enzyme elevations occurred in 6.3% of patients in the danicopan-emergent arm and 13.0% of patients in the placebo-emergent arm. During LTE, liver enzyme elevations occurred in 2.5% of patients in the danicopan-emergent arm and 5.0% of patients in the placebo-emergent arm.
Hemolysis and BTH Events
There was a total of 8 TEAEs of hemolysis reported in 7 patients during the study to date, 4 which were hemolysis and 4 of which were BTH based on investigator judgment. All patients were stable on their C5i. No case-specific details were provided in the submission on the management of the hemolysis or BTH events. Per the submission, no events led to treatment discontinuation, and none were associated with an LDH level greater than 2.2 × ULN.
In TP1, 1 patient in the danicopan plus C5i arm had a TEAE of hemolysis (LDH 1.2 × ULN; reference range 135 to 281 U/L) which was graded as nonserious; this event resolved. Another patient in the danicopan plus C5i arm had a nonserious hemolysis TEAE during TP1 (LDH 1.2 × ULN) and a nonserious BTH TEAE during TP2 (LDH 1.6 × ULN); they received a transfusion around the time of the second event.
In TP2, 1 patient in the danicopan-emergent arm had a nonserious BTH TEAE (LDH 1.9 × ULN), which resolved in 15 days. Another patient in the placebo-emergent arm had a serious hemolysis TEAE (normal LDH); they received a transfusion around the time of the event. It resolved after 1 day.
During LTE, 2 patients in the danicopan-emergent arm had nonserious BTH TEAEs (1 with LDH 2.2 × ULN with pyrexia and COVID-19 concurrently, and 1 with LDH 1.5 × ULN). Another patient in the placebo-emergent group had a nonserious TEAE of hemolysis (LDH 1.3 × ULN), which resolved with sequelae after 32 days.
Table 15: Summary of Harms Results From the ALPHA Study — ISAS
Adverse events | ALPHA | |
|---|---|---|
Weeks 0 to 12: Danicopan plus C5i Weeks 12 to 24: Danicopan plus C5i n = 57 | Weeks 0 to 12: Placebo plus C5i Weeks 12 to 24: Danicopan plus C5i n = 29 | |
Most common TEAEs, n (%)a | ||
Treatment period 1 | n = 57 | n = 29 |
Any AE | 42 (73.7) | 18 (62.1) |
Anemia | 1 (1.8) | 4 (13.8) |
Nausea | 5 (8.8) | 3 (10.3) |
Diarrhea | 4 (7.0) | 3 (10.3) |
Vomiting | 3 (5.3) | 0 |
Abdominal pain upper | 1 (1.8) | 2 (6.9) |
Abdominal pain | 0 | 2 (6.9) |
Pyrexia | 3 (5.3) | 0 |
Asthenia | 0 | 4 (13.8) |
Ear infection | 0 | 2 (6.9) |
Contusion | 1 (1.8) | 3 (10.3) |
Alanine aminotransferase increased | 3 (5.3) | 1 (3.4) |
Aspartate aminotransferase increased | 2 (3.5) | 3 (10.3) |
Arthralgia | 4 (7.0) | 2 (6.9) |
Pain in extremity | 3 (5.3) | 0 |
Headache | 6 (10.5) | 3 (10.3) |
Dizziness | 1 (1.8) | 2 (6.9) |
Insomnia | 1 (1.8) | 3 (10.3) |
Hypertension | 3 (5.3) | 1 (3.4) |
Treatment period 2 | n = 48 | n = 23 |
Any AE | 31 (64.6) | 13 (56.5) |
Diarrhea | 6 (12.5) | 2 (8.7) |
Nausea | 1 (2.1) | 3 (13.0) |
Pyrexia | 5 (10.4) | 0 |
Asthenia | 2 (4.2) | 2 (8.7) |
Fatigue | 3 (6.3) | 1 (4.3) |
Headache | 5 (10.4) | 2 (8.7) |
LTE (up to data cut-off on September 20, 2022) | n = 40 | n = 20 |
Any AE | 25 (62.5) | 16 (80.0) |
Diarrhea | 1 (2.5) | 2 (10.0) |
Pyrexia | 3 (7.5) | 2 (10.0) |
Asthenia | 1 (2.5) | 3 (15.0) |
Fatigue | 2 (5.0) | 1 (5.0) |
COVID-19 | 9 (22.5) | 5 (25.0) |
Back pain | 1 (2.5) | 2 (10.0) |
Pain in extremity | 2 (5.0) | 1 (5.0) |
Overall danicopan treatment (up to data cut-off on September 20, 2022) | n = 57 | n = 23 |
Any AE | 53 (93.0) | 19 (82.6) |
COVID-19 | 12 (21.1) | 5 (21.7) |
Diarrhea | 8 (14.0) | 4 (17.4) |
Headache | 10 (17.5) | 2 (8.7) |
Pyrexia | 9 (15.8) | 2 (8.7) |
Nausea | 7 (12.3) | 3 (13.0) |
Fatigue | 6 (10.5) | 2 (8.7) |
Asthenia | 3 (5.3) | 4 (17.4) |
Anemia | 5 (8.8) | 1 (4.3) |
Arthralgia | 4 (7.0) | 2 (8.7) |
Back pain | 3 (5.3) | 3 (13.0) |
Urinary tract infection | 5 (8.8) | 1 (4.3) |
Pain in extremity | 4 (7.0) | 1 (4.3) |
Vomiting | 4 (7.0) | 0 |
Abdominal pain | 1 (1.8) | 3 (13.0) |
Aspartate aminotransferase increased | 3 (5.3) | 1 (4.3) |
Breakthrough hemolysis | 4 (7.0) | 0 |
Chromaturia | 3 (5.3) | 1 (4.3) |
Dizziness | 4 (7.0) | 0 |
Hemolysis | 2 (3.5) | 2 (8.7) |
Most common SAEs, n (%) | ||
Treatment period 1 | n = 57 | n = 29 |
Any SAE | 3 (5.3) | 2 (6.9) |
Anemia | 0 | 1 (3.4) |
Pancreatitis | 1 (1.8) | 0 |
Abdominal pain | 0 | 1 (3.4) |
Cholecystitis | 1 (1.8) | 0 |
COVID-19 | 1 (1.8) | 0 |
Blood bilirubin increased | 1 (1.8) | 0 |
Headache | 0 | 1 (3.4) |
Treatment period 2 | n = 48 | n = 23 |
Any SAE | 3 (6.3) | 3 (13.0) |
Hemolysis | 0 | 1 (4.3) |
Vertigo | 0 | 1 (4.3) |
Dieulafoy vascular malformation | 1 (2.1) | 0 |
Pyrexia | 1 (2.1) | 0 |
COVID-19 pneumonia | 1 (2.1) | 0 |
Staphylococcal sepsis | 1 (2.1) | 0 |
Headache | 0 | 1 (4.3) |
LTE (up to data cut-off on September 20, 2022) | n = 40 | n = 20 |
Any SAE | 3 (7.5) | 4 (20.0) |
Pericardial effusion | 0 | 1 (5.0) |
Diarrhea | 0 | 1 (5.0) |
Disease progression | 0 | 1 (5.0) |
Stent-graft endoleak | 1 (2.5) | 0 |
COVID-19 | 0 | 1 (5.0) |
Body temperature increased | 0 | 1 (5.0) |
Hemoglobin decreased | 1 (2.5) | 0 |
Invasive ductal breast carcinoma | 1 (2.5) | 0 |
Pulmonary embolism | 1 (2.5) | 0 |
Pulmonary hemorrhage | 1 (2.5) | 0 |
Overall danicopan treatment (up to data cut-off on September 20, 2022) | n = 57 | n = 23 |
Any SAE | 7 (12.3) | 6 (26.1) |
Hemolysis | 0 | 1 (4.3) |
Pericardial effusion | 0 | 1 (4.3) |
Vertigo | 0 | 1 (4.3) |
Diarrhea | 0 | 1 (4.3) |
Dieulafoy vascular malformation | 1 (1.8) | 0 |
Pancreatitis | 1 (1.8) | 0 |
Disease progression | 0 | 1 (4.3) |
Pyrexia | 1 (1.8) | 0 |
Stent-graft endoleak | 1 (1.8) | 0 |
Cholecystitis | 1 (1.8) | 0 |
COVID-19 | 1 (1.8) | 1 (4.3) |
COVID-19 pneumonia | 1 (1.8) | 0 |
Staphylococcal sepsis | 1 (1.8) | 0 |
Blood bilirubin increased | 1 (1.8) | 0 |
Body temperature increased | 0 | 1 (4.3) |
Hemoglobin decreased | 1 (1.8) | 0 |
Invasive ductal breast carcinoma | 1 (1.8) | 0 |
Headache | 0 | 1 (4.3) |
Pulmonary embolism | 1 (1.8) | 0 |
Pulmonary hemorrhage | 1 (1.8) | 0 |
Patients who stopped treatment due to AEs, n (%) | ||
Treatment period 1 | n = 57 | n = 29 |
Patients who stopped treatment | 3 (5.3) | 1 (3.4) |
Liver enzyme abnormality | 2 (3.5) | 1 (3.4) |
Blood bilirubin increase and pancreatitis | 1(1.8) | 0 |
Treatment period 2 | n = 48 | n = 23 |
Patients who stopped treatment | 0 | 0 |
LTE (up to data cut-off on September 20, 2022) | n = 40 | n = 20 |
Patients who stopped treatment | 0 | 1 (5.0) |
Hepatic function abnormality | 0 | 1 (5.0) |
Overall danicopan treatment (up to data cut-off on September 20, 2022) | n = 57 | n = 23 |
Patients who stopped treatment | 3 (5.3) | 1 (4.3) |
Pancreatitis | 1(1.8) | 0 |
Hepatic function abnormal | NR | 1 (4.3) |
Alanine aminotransferase increased | 1(1.8) | 0 |
Aspartate aminotransferase increased | 1(1.8) | 0 |
Blood bilirubin increased | 1(1.8) | 0 |
Hepatic enzyme increased | 1(1.8) | 0 |
Deaths, n (%) | ||
Treatment period 1 | n = 57 | n = 29 |
Patients who died | 0 | 0 |
Treatment period 2 | n = 48 | n = 23 |
Patients who died | 0 | 0 |
LTE (up to data cut-off on September 20, 2022) | n = 40 | n = 20 |
Patients who died | 0 | 0 |
Overall danicopan treatment (up to data cut-off on September 20, 2022) | n = 57 | n = 23 |
Patients who died | 0 | 0 |
AEs of special interest, n (%) | ||
Treatment period 1 | n = 57 | n = 29 |
Meningococcal infections | 0 | 0 |
Liver enzyme elevations | 8 (14.0) | 3 (10.3) |
Treatment period 2 | n = 48 | n = 23 |
Meningococcal infections | 0 | 0 |
Liver enzyme elevations | 3 (6.3) | 3 (13.0) |
LTE (up to data cut-off on September 20, 2022) | n = 40 | n = 20 |
Meningococcal infections | 0 | 0 |
Liver enzyme elevations | 1 (2.5) | 1 (5.0) |
Overall danicopan treatment (up to data cut-off on September 20, 2022) | n = 57 | n = 23 |
Meningococcal infections | 0 | 0 |
Liver enzyme elevations | 10 (17.5) | 4 (17.4) |
AE = adverse event; C5i = complement component 5 inhibitor; ISAS = interim safety analysis set; LTE = long-term extension; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aThe cut-off for most common TEAEs was ≥ 5% patients in any treatment arm during treatment period 1, treatment period 2, and LTE, or ≥ 5% in both arms for overall danicopan treatment.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
The IA of the ongoing ALPHA study is a phase III, double-blind RCT with a 12-week placebo-controlled period where patients stable on a C5i were randomized either to danicopan add-on therapy or placebo add-on therapy, with an additional 12-week single-arm portion where both arms received danicopan add-on therapy, and an LTE to 72 weeks and ongoing. There appeared to be a low risk of bias from the treatment blinding process and randomization and treatment allocation processes, as randomization was stratified and done via interactive response technology, and treatment allocation was not unblinded until after a database lock. Per the clinical experts, the criteria for permitted concomitant medications, danicopan or placebo dosing escalations, timing of escalations, and criteria for transfusions were all reasonable in their experience. They also noted that 12 weeks would be a sufficient length of time to note the impacts of treatment, and requiring patients to be stable on C5i therapy for 6 months would also align with clinical practice, as they would usually trial a new therapy for 6 months before considering changes in dose or therapeutic drug.
There are some limitations pertaining to patient disposition and patient characteristics to note. A total of 18.9% of patients failed to meet the inclusion or exclusion criteria, but it is not specified which inclusion/exclusion criteria were not met during screening; therefore, it is not known whether excluded patients were systematically different from included ones. In addition, while baseline characteristics were broadly balanced between study arms, the differences in the proportion of patients treated with each C5i (64.3% of patients in the danicopan plus C5i arm and 47.6% of patients in the placebo plus C5i arm were treated with ravulizumab) may bias the harms results as ravulizumab is suggested as a first-line choice where both agents are available.37 Per the submission, 40 patients in the danicopan-emergent arm and 20 patients in the placebo-emergent arm (95.2% of patients in each arm) completed TP1 and TP2, representing numerically low patient dropout in these phases. However, the LTE phase of this trial is ongoing, and to date, 6 (14.3%) patients in the danicopan-emergent arm and 3 (14.3%) patients in the placebo-emergent arm have completed LTE1; furthermore, 4 (9.5%) patients in the danicopan-emergent arm and 1 (4.8%) patient in the placebo-emergent arm discontinued during the LTE. The small number of patients who have completed the LTE to date make long-term results for efficacy and safety highly uncertain.
There are some potential limitations associated with the study design. The ALPHA trial IA used a prespecified interim stopping criteria of 75% of patients, as well as an alpha-spending procedure for the primary and key secondary end points to account for the fact that a smaller sample size than was required by the power calculations was used for this analysis. The alpha-spending procedure and hierarchical testing structure controlled the family-wise type I error rate for these end points. However, given the IA was conducted based on 75% of the originally targeted sample size, there is an increased risk that the true effect of danicopan on these end points is overestimated by the IA. In addition, while the primary and key secondary outcomes were controlled for multiple comparisons, the secondary and exploratory outcomes were not controlled for this or for the smaller sample size, and there is a risk of inflated type I error when interpreting results from these comparisons. Furthermore, there are possible limitations pertaining to the numbers of complete cases (i.e., patients observed at each time point of the analysis) in the danicopan plus C5i/danicopan-emergent arm. At TP1, in the danicopan plus C5i arm (n = 42), there were 36 complete cases for the change in Hb, 35 cases for absolute reticulocyte count, and the complete cases were missing for the proportion of patients with Hb increase of 2 g/dL (20 g/L) or more in the absence of transfusion and the proportion of patients with transfusion avoidance. The models used in the analysis assumed that missing data are missing-at-random (mixed model for repeated measures) or missing-completely-at-random (analysis of covariance, transfusion burden outcomes only). Without further information on the patients who were missing, the degree to which the missingness may be informative to the results is not known. Lastly, there was no placebo comparator after the end of TP1; therefore, observed results in TP2 and LTE may not all be attributable to treatment.
There are some potential limitations associated with outcome ascertainment. While laboratory outcomes such as Hb or LDH are likely at low risk of bias because of being centrally measured, the open-label design of TP2 and LTE mean that knowledge of the treatment being received may impact reporting of subjective quality of life outcomes at those time points (impacting FACIT-F, EORTC QLQ-C30, and EQ-5D-3L outcomes). Similarly, while a measure of treatment adherence was reported in the study, this was based on tablet counts and there is a possibility of reporting bias.
External Validity
There are some limitations regarding the study population to note. Per the clinical expert, most of the inclusion criteria were reasonable for patients with PNH in a Canadian context; however, the minimum thresholds for platelet and neutrophil counts, and the exclusion criteria which excluded patients with other causes of anemia or other clinical comorbidities, excluded patients who may be treatment candidates in a real-world setting. The clinical expert noted that while there are certain clinical characteristics alongside persistent anemia whose presence indicate that EVH is the likely cause, there is no standard diagnostic definition of the condition. The cut-off used in the ALPHA study to define anemia was a level at which the clinical experts speculated patients would likely feel symptoms and could require intervention but was not based on a known standard. In addition, the clinical experts noted that transfusion practices vary greatly and are partially dependent on patient factors such as lifestyle or comorbidities. Therefore, the study population included in the ALPHA study may not represent all patients with PNH who have EVH.
There are also some limitations regarding the generalizability of the results to clinical situations. The frequency of visits used in the trial setting may not exactly reflect daily clinical practice in Canada and therefore the efficacy and safety profile during the trial may not be extrapolatable to the general patient population. In addition, the additional dosing information in the product monograph includes the phrase “or to achieve an appropriate hemoglobin response based on clinical judgment” for dose increases; this was not in the dosing criteria for the trial, and it is not clear what impact it might have in clinical practice. During the trial, the approved C5i dose was not permitted to be increased, nor the interval shortened, which also may not reflect clinical practice. FACIT-F and EORTC QLQ-C30 are validated tools in patients with PNH, but the EQ-5D-3L is not validated in PNH specifically, therefore changes in health status reflected in this score may not translate perfectly to changes in health status in PNH. Furthermore, there were no MIDs provided by the sponsor or the clinical experts for all but 1 of the outcomes in patients with PNH, therefore information on clinically meaningful change for the majority of outcomes remains lacking.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform the CDA-AMC expert committee’s deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.38,39
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). The target of the certainty of evidence assessment was based on thresholds informed by the sponsor submission, input from the clinical experts, and/or thresholds identified in the literature. In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for danicopan plus C5i therapy versus placebo plus C5i therapy.
Long-Term Extension
Information available to date from the LTE1 phase of the ALPHA study has been reported and appraised in the Systematic Review section.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
Indirect evidence was required to be considered as part of the submission because the ALPHA trial compared danicopan plus C5i therapy with placebo plus C5i therapy, however comparative data against pegcetacoplan, the other second-line therapeutic option for PNH, remained lacking.
Description of Indirect Comparison(s)
The submission included an SLR and feasibility assessment to undertake a MAIC with the PEGASUS trial, which compared pegcetacoplan with eculizumab in adult patients with PNH.
Table 16: Study Selection Criteria and Methods for the SLR Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Individuals with PNH who experience EVH |
Intervention | Any |
Comparator | Any |
Outcome | Efficacy:
Safety:
|
Study designs | Prospective or retrospective observational studies Cross-sectional studies RCTs and single-arm trials |
Publication characteristics | Published articles and conference abstracts |
Exclusion criteria |
|
Databases searched | Bibliographic databases:
Grey literature sources:
|
Selection process | Articles were screened independently by 2 researchers. A third researcher provided arbitration for any discrepancies that occurred between the studies selected for inclusion by the 2 researchers. |
Data extraction process | Double data extraction was performed by 2 data reviewers. Any discrepancies between the data extracted were resolved through discussion to achieve consensus. Data were stored and managed in Microsoft Excel. |
Quality assessment | The PRISMA guidelines for designing, performing, and reporting the systematic review were followed.59 The quality of reporting of publications included in this review was assessed using the Critical Appraisal Skills Programme checklist for observational studies,74 the Cochrane Risk of Bias assessment tool for RCTs and nonrandomized trials (v2.0),75 and the Drummond checklist for economic evaluations.76 Quality assessments were conducted by a single reviewer with auditing by a second, independent reviewer. Discrepancies in the assessments were resolved through discussion to achieve consensus. |
AE = adverse event; BTH = breakthrough hemolysis; EVH = extravascular hemolysis; LDH = lactate dehydrogenase; PNH = paroxysmal nocturnal hemoglobinuria; PRISMA = Preferred Reporting Items for Systematic reviews and Meta-Analyses; RBC = red blood cell; RCT = randomized controlled trial; SAE = serious adverse event; SLR = systematic literature review.
aPer the submission, the focus of the systematic review was on patients with PNH and EVH; however, studies describing investigational treatments of interest in patients not treated with complement component inhibitors- (i.e., those who have not experienced EVH) were also considered for inclusion as part of the full evidence base for these treatments.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence and SLR Report.77
Indirect Treatment Comparison Design
Objectives
The objective of the SLR was to characterize the efficacy, safety, and humanistic impact of currently available and future (in phase III) treatments for EVH in patients with PNH.
Study Selection Methods
Full details of the SLR search strategy, study inclusion or exclusion, data extraction, and quality assessment can be found in Table 16. Briefly, a systematic search of the literature was undertaken on November 1, 2022, and re-run on June 12, 2023. The evidence base for EVH was expected to be small, therefore the search strategy was designed not to include the comparators, outcomes, or study designs; these were instead screened for at the abstract screening and full-text review stages. A supplemental search of the grey literature was also undertaken.
Clinical outcomes were selected according to their relevance in assessing treatment benefit for patients with PNH experiencing EVH and sought to capture outcomes related to IVH, BTH, thrombosis hematological response, and anemia in the evidence base. The submission did not provide any information on whether a specific definition of EVH was used to identify relevant studies during screening, or whether the list of clinical outcomes was prespecified before screening or compiled during screening.
During the data extraction, data which were unavailable was considered missing and was not reported for that outcome. For studies with multiple time points, only the data from the last assessment were summarized.
Indirect Comparison Analysis Methods
Feasibility Study
The submission included a feasibility assessment on the possibility of comparing patient-level data from the ALPHA trial to aggregate data from the PEGASUS study, a phase III trial RCT comparing pegcetacoplan with eculizumab in patients with PNH being treated with a C5i. The submission assessed the feasibility of comparing the 2 studies via a trimmed MAIC (anchored or unanchored) and a trimmed naive comparison. The feasibility assessment consisting of a comparison of the between-trial heterogeneity was conducted for trial design, trial end points, patient eligibility criteria, and baseline patient characteristics.
The submission did not provide further details on whether the list of factors considered in the feasibility assessment were prespecified or compiled based on the studies, and whether there were statistical tests or systematic margins used to determine differences between the ALPHA and PEGASUS studies for individual characteristics.
MAIC Analysis
The MAIC analysis compared a subset of the ALPHA study population which was trimmed to meet the additional inclusion criteria which were a part of the PEGASUS study but not the ALPHA study:
body mass index less than 40 kg/m2
platelet count greater than 50,000/µL.
The MAIC used a weighting approach which approximates a propensity score approach as per the methodology reported by Signorovitch et al.43 as well as an alternate weighting approach proposed by Jackson et al.,78 which was proposed by the submission to maximize effective sample size (ESS), and qualitatively reported on the 2 methods in terms of balancing characteristics. The model used to produce weights included baseline Hb and baseline reticulocyte count. The submission did not provide further details on why these 2 characteristics were selected but noted that overall that the ALPHA and PEGASUS trials contained several differences which would make indirect comparison challenging. Weights were calculated at the level of the trial.
The distribution of calculated weights from both methods was reported, as well as the baseline characteristics after adjustment by both methods. After weighting, anchored and unanchored MAICs were undertaken for the following efficacy outcomes:
change in Hb from baseline
change in absolute reticulocyte count from baseline
change in LDH from baseline
change in FACIT-F scores from baseline
transfusion avoidance.
Efficacy results were reported in the anchored MAICs as differences of TD for each trial (danicopan plus C5i minus placebo plus C5i, or pegcetacoplan minus eculizumab). The unanchored MAICs reported efficacy results as TDs between the danicopan plus C5i arm and the pegcetacoplan arm. The submission did not provide additional information on the definition used for transfusion avoidance.
Anchored and/or unanchored MAICs were also undertaken for the following safety outcomes:
BTH AEs and BTH over extended follow-up (safety outcome).
Time to discontinuation due to BTH, BTH AEs, and BTH AEs over extended follow-up were also reported in an unweighted population and were not appraised. All analyses compared results from the ALPHA study at 12 weeks to results from the PEGASUS study at 20 weeks (the study design consisted of a 4-week run-in with C5i monotherapy followed by a 16-week randomized period).
Results
Summary of Included Studies
The original search identified 35 articles for inclusion in the SLR, 31 of which described clinical, humanistic, and/or economic outcomes, and 4 of which were cost-effectiveness analyses. The search was re-run on June 12, 2023, yielding 385 abstracts from the databases and 25 from grey literature. Following this additional screening and full-text review, 15 articles were retained for descriptive synthesis and combined with the original 35, resulting in 50 studies ultimately being included. Of these articles, 32 described a PNH population that was previously treated with a C5i; these included the phase III trial comparing pegcetacoplan to eculizumab (PEGASUS), and the ALPHA trial. Both the PEGASUS trial and the ALPHA trial were assessed in the SLR to have high risk of bias in the quality assessment; the PEGASUS study because of the open-label nature of the study and the ALPHA study because the only available reference was a conference abstract which did not provide detail on the methods. The IA of the ALPHA study has been appraised by CDA-AMC in the systematic review section.
After excluding conference abstracts, 13 manuscripts were retained. Of these, 2 were MAICs, and their references were screened for additional studies (none were found).
Results
Feasibility Study Results
Full details of the feasibility study conducted by the sponsor are included in Table 17. Briefly, the sponsor detailed differences in trial design, inclusion criteria, baseline characteristics, and treatment duration between the ALPHA trial and the PEGASUS trial.
Table 17: Results of Feasibility Assessment for the MAIC — Study Differences
Characteristic | ALPHA | PEGASUS |
|---|---|---|
Trial design | Double-blind, placebo-controlled RCT for first 12 weeks | Open-label trial for 16 weeks (plus 4 weeks run-in) |
Inclusion criteria | C5i (ravulizumab or eculizumab) therapy for 6 months at randomization | Eculizumab therapy at stable dose for 3 months at randomization |
At least 1 blood transfusion in the 12 months before randomization (removed in a protocol amendment) | No inclusion criteria related to transfusion history | |
Baseline Hb ≤ 9.5 g/dL | Baseline Hb ≤ 10.5 g/dL | |
Platelets ≥ 30,000/L | Platelets ≥ 50,000/L | |
Baseline characteristics | All patients had had a transfusion in the previous 12 months | Approximately 25% of patients had no transfusions within the previous 12 months |
Mean baseline Hb = 7.7 g/dL in both treatment arms (SD = 0.9 g/dL [danicopan arm]; SD = 1.0 g/dL [placebo arm]) | Mean baseline Hb = 8.69 g/dL (SD = 1.08 g/dL) in pegcetacoplan arm; mean baseline Hb = 8.68 g/dL (SD = 0.89 g/dL) in placebo arm | |
Treatment duration | Randomized treatment for initial 12 weeks, followed by switch from placebo to danicopan in the placebo arm for 12 weeks | Both eculizumab and pegcetacoplan for initial 4-week run-in period, followed by randomized treatment |
C5i = complement component 5 inhibitor; Hb = hemoglobin; MAIC = multiple-adjusted indirect comparison; RCT = randomized controlled trial; SD = standard deviation.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the MAIC Feasibility Assessment.79
A comparison of the full baseline characteristics from the ALPHA study, trimmed ALPHA study, and the PEGASUS study are included in Table 18. A difference in the mean baseline Hb was highlighted by the sponsor in the baseline characteristics between the trimmed ALPHA study population (7.7 g/dL in the danicopan plus C5i arm, 7.8 g/dL in the placebo plus C5i arm) and the PEGASUS study population (8.69 g/dL in the pegcetacoplan arm, 8.68 g/dL in the eculizumab arm). Apart from this, there were numeric differences between the trimmed ALPHA study population and the PEGASUS study population in the proportion of Asian patients (47.4% in the danicopan plus C5i arm and 31.6% in the placebo plus C5i arm of the ALPHA study, versus 12% in the pegcetacoplan arm and 18% in the eculizumab arm of PEGASUS study), proportion of white patients (42.1% in the danicopan plus C5i arm and 47.4% in the placebo plus C5i arm in the ALPHA study, versus 59% in the pegcetacoplan arm and 64% in the eculizumab arm in the PEGASUS study), absolute reticulocyte count (238.8 × 109/L in the danicopan plus C5i arm and 242.9 × 109/L in the placebo plus C5i arm in the ALPHA study, versus 217.5 × 109/L in the pegcetacoplan arm and 216.2 × 109/L in the eculizumab arm in the PEGASUS study), and total bilirubin (33.2 µmol/L in the danicopan plus C5i arm and 34.8 µmol/L in the placebo plus C5i arm in the ALPHA study, and 42.5 µmol/L in the pegcetacoplan arm and 40.5 µmol/L in the eculizumab arm in the PEGASUS study). There was no information on the potential clinical importance of these differences in the submission.
Table 18: Comparison of Patient Characteristics Between the ALPHA and PEGASUS Studies
Characteristic | ALPHA — IEAS population | ALPHA – Subset Meeting PEGASUS Criteria (N = 57) | PEGASUS | |||
|---|---|---|---|---|---|---|
Danicopan plus C5i n = 42 | Placebo plus C5i n = 21 | Danicopan plus C5i n = 38 | Placebo plus C5i n = 19 | Pegcetacoplan n = 41 | Eculizumab n = 39 | |
Age (years), mean (range) | 55.0 (25 to 80) | 53.1 (29 to 75) | 56.7 (28 to 80) | 53.6 (29 to 75) | 50.2 (19 to 81) | 47.3 (23 to 78) |
Age > 65 years, n (%) | 12 (28.6) | 4 (19.0) | 12 (31.6) | 4 (21.1) | 10 (24) | 7 (18) |
Sex (female), n (%) | 23 (54.8) | 14 (66.7) | 20 (52.6) | 13 (68.4) | 27 (66) | 22 (56) |
Race, n (%) | ||||||
Asian | 18 (42.9) | 7 (33.3) | 18 (47.4) | 6 (31.6) | 5 (12) | 7 (18) |
Black | 1 (2.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (5) | 0 |
White | 19 (45.2) | 9 (42.9) | 16 (42.1) | 9 (47.4) | 24 (59) | 25 (64) |
Other | 2 (4.8) | 1 (4.8) | 2 (5.3) | 1 (5.3) | 0 | 1 (3) |
Not reported | 2 (4.8) | 4 (19) | 2 (5.3) | 3 (15.8) | 10 (24) | 6 (15) |
BMI (kg/m2), mean (SD) | 26.7 (5.4) | 24.8 (4.9) | 23.9 (2.8) | 25.3 (3.0) | 26.7 (4.3) | 25.9 (4.3) |
No transfusions within previous 12 months, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 10 (24) | 10 (26) |
Time since PNH diagnosis (years), median (range) | 7.3 (0.9 to 49.6) | 10.8 (1.2 to 39.6) | 7.3 (0.9 to 49.6) | 10.5 (1.2 to 39.6) | 6.0 (1 to 31) | 9.7 (1 to 38) |
Duration of prior treatment with eculizumab or C5i (years), median (range) | 3.6 (0.5 to 14.2) | 3.7 (0.7 to 16.8) | 3.7 (0.5 to 14.2) | 3.7 (0.7 to 16.8) | 4.4 (0.4 to 17.1) | 3.4 (0.3 to 13.8) |
Platelets ( × 109/L), mean (SD) | 131.5 (64.1) | 138.0 (76.8) | 137.3 (61.5) | 147.9 (74.0) | 166.6 (98.3) | 146.9 (68.8) |
≥ 4 transfusions in previous 12 months, n (%) | 22 (52.4) | 9 (42.9) | 19 (50.0) | 7 (36.8) | 21 (51) | 23 (59) |
Hb (g/dL), mean (SD) | 7.7 (0.9) | 7.7 (1.0) | 7.7 (0.9) | 7.8 (0.9) | 8.69 (1.08) | 8.68 (0.89) |
Reticulocyte count ( × 10−9/L), mean (SD) | 236.4 (91.4) | 240.6 (120.3) (n = 20) | 238.8 (93.4) | 242.9 (125.9) (n = 18) | 217.5 (75.0) (normal reference range, 30 to 120) | 216.2 (69.1) (normal reference range, 30 to 120) |
LDH (U/L), mean (SD) | 298.7 (105.7) | 278.2 (68.4) | 302.0 (110.4) | 279.6 (71.2) | 257.5 (97.6) (normal reference range, 113 to 226) | 308.6 (284.8) (normal reference range, 113 to 226) |
Total bilirubin (µmol/L), mean (SD) | 32.5 (21.8) | 34.2 (21.0) | 33.2 (22.5) | 34.8 (21.7) | 42.5 (31.5) (normal reference range, 1.7 to 18.8) | 40.5 (26.6) (normal reference range, 1.7 to 18.8) |
Indirect bilirubin (µmol/L), mean (SD) | 23.7 (19.0) | 25.4 (19.6) | 24.4 (19.6) | 26.0 (20.3) | 34.7 (28.5) | 32.9 (23.0) |
FACIT-F score, mean (SD) | 33.5 (11.1) | 33.9 (10.8) | 34.4 (11.1) | 33.4 (10.3) | 32.2 (11.4) | 31.6 (12.5) |
BMI = body mass index; C5i = complement component 5 inhibitor; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; IEAS = interim efficacy safety analysis set; LDH = lactate dehydrogenase; MAIC = multiple-adjusted indirect comparison; PNH = paroxysmal nocturnal hemoglobinuria; SD = standard deviation.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the MAIC Feasibility Assessment.79
MAIC Efficacy Results
Results for the MAICs weighted by the Signorovitch et al.43 methods were reported, as this method is referenced in the NICE Decision Support Unit Technical Support documents and results of the MAICs weighted using Jackson et al.78 were very similar. The ESS reported for the Signorovitch weighting method was 20.276, and the ESS reported for the Jackson weighting method was 22.610.
Table 19 describes the trials included and key efficacy results for the anchored and unanchored MAICs is in Table 20. Briefly, the conclusions for the anchored and unanchored MAICs were numerically similar for most efficacy outcomes, with 2 exceptions: transfusion avoidance, where the unanchored MAIC showed that danicopan was favoured for transfusion avoidance, but the anchored MAIC did not; and absolute reticulocyte count, where pegcetacoplan was favoured over danicopan in both MAICs. The overall conclusions of the MAICs were that neither danicopan add-on therapy nor pegcetacoplan were favoured for the outcomes of Hb change from baseline, LDH change from baseline, change in FACIT-F scores from baseline, or transfusion avoidance (anchored MAIC only).
Table 19: Summary of Trial Arms Included in the MAIC
Study | Study design | N | Interventions | Control |
|---|---|---|---|---|
ALPHA (ongoing) | Double-blind; placebo-controlled to 12 weeks; open-label, single-arm at 24 weeks, long-term extension to week 52 onwards | 63 (57 trimmed) | Danicopan orally t.i.d. plus C5i IV | Placebo orally t.i.d. plus C5i IV until week 12 |
PEGASUS | Open-label RCT with 4-week eculizumab plus study drug run-in followed by 16 weeks on randomized therapy | 80 | Pegcetacoplan SC | Eculizumab IV |
C5i = complement component 5 inhibitor; MAIC = multiple-adjusted indirect comparison; RCT = randomized controlled trial; SC = subcutaneous; t.i.d. = 3 times a day.
Source: Information included in the table is from the sponsor’s MAIC presentation.80
MAIC Safety Results
Weighted MAICs were reported for the time to BTH AEs and the probability of a BTH event; an unweighted MAIC was reported for the time to discontinuation because of BTH and the probability of BTH over extended follow-up (not appraised). The time-to-event curve for the time-to-hemolysis AE is reported in Figure 2. Briefly, based on the information provided, there was no significant difference between the time to BTH AE for patients in the trimmed ALPHA study sample or in the PEGASUS study.
Based on the extended follow-up from the PEGASUS study (48 weeks) and a median follow-up of 34.6 weeks from patients in the danicopan-emergent arm of the ALPHA study, the probability of experiencing a BTH event was calculated and reported in Table 21. Briefly, the results from the weighted, unanchored MAIC did not favour either treatment in the probability of BTH between the 2 trials.
Table 20: Anchored and Unanchored MAIC Efficacy Results
Variable | Anchored MAIC | Unanchored MAIC | ||
|---|---|---|---|---|
ALPHA (danicopan plus C5i) – (placebo plus C5i) | PEGASUS Pegcetacoplan – eculizumab | ALPHA Danicopan plus C5i | PEGASUS Pegcetacoplan | |
Change in Hb from baseline | ||||
Single-arm estimate (95% CI) | 2.57 (1.85 to 3.28) | 3.80 (2.30 to 5.30) | 2.75 (2.35 to 3.15) | 2.40 (1.62 to 3.18) |
Difference between ALPHA and PEGASUS (95% CI) | –1.23 (–2.90 to 0.43) | 0.35 (–0.53 to 1.23) | ||
Change in absolute reticulocyte count from baseline | ||||
Single-arm estimate (95% CI) | –110.30 (–136.00 to –84.60) | –164.00 (–190.30 to –137.70) | –103.20 (–117.60 to –88.90) | –136.00 (–148.70 to –123.30) |
Difference between ALPHA and PEGASUS (95% CI) | 53.70 (16.90 to 90.50) | 32.80 (13.60 to 51.90) | ||
Change in LDH from baseline | ||||
Single-arm estimate (95% CI) | 3.40 (–48.20 to 55.10) | –5.00 (–181.70 to 171.70) | –2.40 (–18.10 to 13.40) | –15.00 (–98.70 to 68.70) |
Difference between ALPHA and PEGASUS (95% CI) | 8.40 (–175.60 to 192.50) | 12.60 (–72.50 to 97.80) | ||
Change in FACIT-F scores from baseline | ||||
Single-arm estimate (95% CI) | 7.70 (1.24 to 14.17) | 11.90 (5.50 to 18.30) | 7.92 (3.52 to 12.32) | 9.20 (6.06 to 12.34) |
Difference between ALPHA and PEGASUS (95% CI) | –4.20 (–13.29 to 4.90) | –12.8 (–6.66 to 4.10) | ||
Transfusion avoidance | ||||
Single-arm estimate (95% CI) | 3.15 (1.11 to 5.19) | 3.47 (2.24 to 4.70) | 3.37 (2.04 to 4.70) | 1.74 (0.88 to 2.59) |
Difference between ALPHA and PEGASUS (95% CI) | –0.32 (–2.70 to 2.06) | 1.64 (0.06 to 3.22) | ||
C5i = complement component 5 inhibitor; CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; LDH = lactate dehydrogenase; MAIC = multiple-adjusted indirect comparison.
Source: Details included in the table are from the sponsor’s MAIC presentation.80
Table 21: BTH Adverse Events During Extended Follow-up — MAIC-Adjusted
Variable | Unanchored MAIC | |
|---|---|---|
ALPHA Weeks 0 to 12: danicopan plus C5i Weeks 12 onwards: danicopan plus C5i | PEGASUS | |
Hemolysis event | ||
Single-arm estimate (95% CI) | 0.079 (0.023 to 0.199) | 0.263 (0.180 to 0.366) |
Difference between ALPHA and PEGASUS (95% CI) | –0.184 (–0.308 to 0.050) | |
AE = adverse event; BTH = breakthrough hemolysis; C5i = complement component 5 inhibitor; CI = confidence interval; MAIC = multiple-adjusted indirect comparison.
Source: Details included in the table are from the sponsor’s MAIC presentation.80
Figure 2: Time to BTH Adverse Event — MAIC-Adjusted
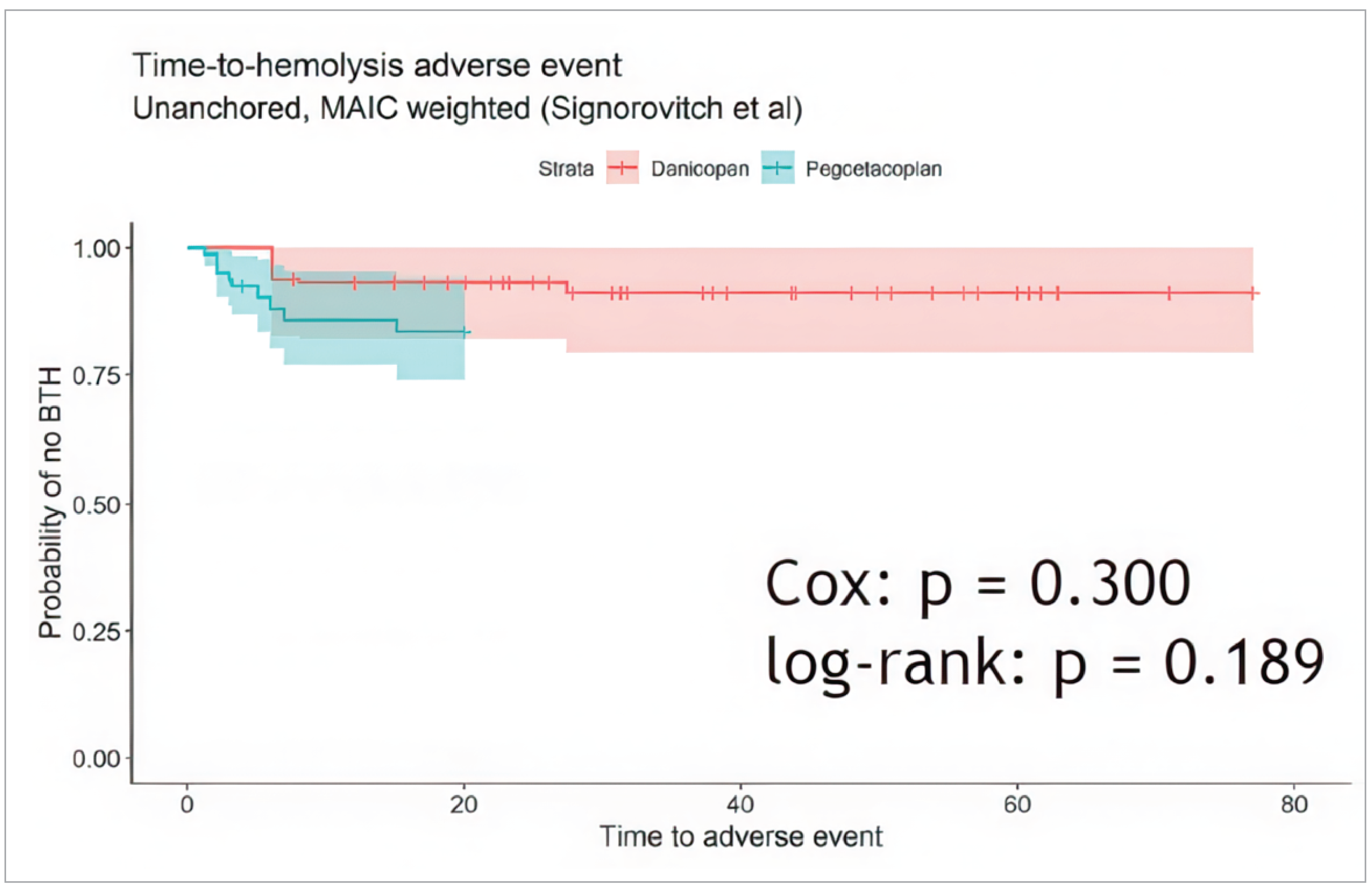
BTH = breakthrough hemolysis; MAIC = multiple-adjusted indirect comparison.
Source: Details included in the figure are from the sponsor’s MAIC presentation.80
Critical Appraisal
The submission provided an SLR, feasibility assessment for a MAIC, naive comparison, and both anchored and unanchored MAICs for select efficacy and safety outcomes. The SLR searched multiple databases and grey literature sources using search terms that were provided in the submission, carried out study selection and data extraction using accepted methods, and provided a Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) diagram of the study selection. There are some limitations of note. The SLR did not provide a preregistered protocol, and so it is not known whether the search criteria, study selection, or subgroups of interest were prespecified before the search.
The results of the SLR informed the feasibility assessment for conducting a MAIC comparing danicopan add-on to pegcetacoplan monotherapy. The feasibility assessment provided a comprehensive description for the justification of a MAIC over other methods such as reweighting and network meta-analyses. Based on the feasibility assessment, the sponsor concluded that a MAIC was infeasible for comparing danicopan add-on to pegcetacoplan monotherapy. However, it is not known whether statistical testing was undertaken to determine differences in study population or whether there was a prespecified threshold to determine the meaningfulness of differences between populations. Since the clinical experts noted that the differences identified between the study populations were not clinically meaningful, the rationale for the sponsor’s conclusion would have provided additional context. Furthermore, other differences across the trials such as race and total bilirubin were not discussed in the feasibility assessment and the submission provided little discussion of whether these or other patient characteristics have prognostic or meaningful impact to study outcomes. Per the clinical experts consulted by CDA-AMC, the differences highlighted in the feasibility assessment for inclusion criteria and baseline characteristics did not represent clinically meaningful differences. They noted that the anemia and platelet cut-offs being different was not overly meaningful from a clinical perspective as the mean values for both in the baseline characteristics were similar; they also noted that patient-specific factors such as lifestyle and important symptoms are often a driver of treatment choices. As this information was not included in the submission, the impact of these factors on patient differences is unknown. Ravulizumab is the suggested C5i therapy over eculizumab when both are available; however, the 2 therapies have similar efficacy results.37 Therefore, there is enough overlap between the study populations to suggest that the reported characteristics do not represent enough of a source of heterogeneity to rule out a MAIC. Furthermore, infeasibility of a MAIC would not be appropriate justification for relying on a naive comparison for drawing conclusions. A naive comparison would be subject to significant limitations: it would break randomization by using a subset of the ALPHA study patient population, provide a narrative comparison with no statistical testing for differences, and would not address any of the concerns highlighted by the feasibility assessment. Thus, drawing conclusions based on a naive comparison would not be more appropriate in this setting.
MAICs would be justified over alternative indirect comparisons such as network meta-analysis due primarily to the size of the network and the number of comparator treatments for EVH. An anchored MAIC was possible as there is a common C5i monotherapy arm used in both trials. However, the sponsor noted key differences in the comparator arms of each trial which could undermine the internal validity of the anchored comparison. The ALPHA study C5i monotherapy arm included patients assigned to both ravulizumab and eculizumab whereas the PEGASUS study C5i monotherapy arm only included patients assigned to eculizumab. Thus, for an anchored comparison to be appropriate, the efficacy of these 2 C5i therapies would need to be the same; the clinical experts consulted by CDA-AMC and the literature suggested that this assumption may be appropriate for efficacy end points.37,47,50 In addition, since the ALPHA study reported comparative results at 12 weeks from baseline whereas the PEGASUS trial reported comparative results at 20 weeks, the anchored comparison compared differences in end points at these time points. This would only be appropriate if the efficacies of the drugs were considered stable after 12 weeks of treatment, which may not be an appropriate assumption. The anchored MAICs provided control on 2 treatment effect modifiers and the sponsor noted that these were the only effect modifiers able to be adjusted on, citing limitations in the study population reporting. In addition, they carried out MAIC weighting using 2 methods, 1 of which would maximize the ESS; results were overall very similar between the 2 methods for the outcomes assessed. However, the clinical experts noted that the modifiers used in weighting were not a comprehensive list of possible modifiers or prognostic factors. Therefore, the anchored MAICs likely did not account for all possible sources of heterogeneity between the study populations. This increases the uncertainty in the results, and thus, drawing firm conclusions about the comparative effectiveness of danicopan add-on and pegcetacoplan therapy based on the results of the anchored MAIC would not be recommended.
Unanchored MAICs were also undertaken for all efficacy and safety outcomes. This method requires the assumption that all prognostic factors and treatment effect modifiers are accounted for, which is a strong assumption largely considered impossible to meet — failure of this assumption leads to an unknown amount of bias in the effect estimate.81 The unanchored comparison should ideally provide sufficient evidence on the likely extent of error because of unaccounted for covariates,81 which this analysis did not do. Cross-validation methods or other sensitivity analyses are suggested methods to explore the impact of the lack of anchoring.81 However, these were not reported in the current submission, which imparts additional uncertainty in the results. The weighted model for the unanchored MAIC included the same covariates as were included for the anchored MAIC. As noted previously, the 2 included covariates were not considered sufficient to account for all effect modifiers and prognostic factors in this setting. Thus, drawing conclusions based on the results of the unanchored MAIC would not be recommended either.
Lastly, the ALPHA and PEGASUS trials differ in other ways that were not accounted for in any of the indirect comparison approaches and which may impact the risk of bias and the generalizability of the results. Patients in the PEGASUS study were exposed to pegcetacoplan monotherapy for 4 weeks longer than patients were exposed to danicopan in the ALPHA study, which may bias the efficacy results to favour pegcetacoplan. Furthermore, the trial design for pegcetacoplan was an open-label trial, which may bias the reporting of FACIT-F, a subjective outcome, and would not provide an appropriate contrast to the ALPHA study which used a double-blinded design. No information is available on the ascertainment of outcomes in the PEGASUS study; however, the ascertainment of the other efficacy outcomes in the ALPHA study was likely at low risk of bias because of the use of a central laboratory. The results from the MAICs are also subject to the same concerns about generalizability to the PNH population as the ALPHA study population, and without detailed information from the PEGASUS study, the generalizability of that study population to the wider PNH population is not known. Results were only reported for efficacy outcomes at week 20 for the PEGASUS study and week 12 for the ALPHA study, and so any information on efficacy past this time is not known. For BTH events, these were reported up to 48 weeks in the PEGASUS study and 34.5 weeks for the ALPHA study, therefore longer-term data on safety and information on other harms is unknown. Of note, the MAICs also did not include comparative information on several outcomes included in the pharmacoeconomic model such as iron overload, proportion of patients with Hb greater or less than 9.5 g/dL, or BTH by severity, which also limits the generalizability of this indirect comparison to the pharmacoeconomic analysis.
Summary
The body of evidence submitted for the indirect comparison consisted of an SLR, feasibility assessment for conducting an indirect comparison of danicopan plus a C5i to pegcetacoplan, a naive comparison of the trimmed ALPHA study population with the full sample from the PEGASUS study, as well as anchored and unanchored MAICs on select efficacy and safety outcomes. Briefly, the sponsor submission trimmed the patient sample in the ALPHA trial to meet 2 additional inclusion criteria from the PEGASUS study and compared the characteristics of the studies and the patient populations to ascertain whether a MAIC would be feasible to undertake. The sponsor concluded that the assumptions required for a MAIC would not be satisfied and provided a naive comparison of the trimmed ALPHA study sample with the PEGASUS study sample by study arm. However, feedback from the clinical experts noted that the differences highlighted by the sponsor were not clinically meaningful as reported, although the CDA-AMC appraisal noted that the trial designs still differed in ways that risk biasing the results. Furthermore, the infeasiblity of a MAIC would not be an appropriate justification for relying on results of a naive comparison. The CDA-AMC appraisal concluded that a naive comparison would be largely uninformative and the information too uncertain to make firm conclusions on efficacy or safety and therefore did not include it in the indirect evidence appraisal although it makes up the base case in the pharmacoeconomic analysis. As it would be possible with a MAIC to control for treatment effect modifiers in the 2 studies, the CDA-AMC team appraised the unanchored and anchored MAICs undertaken on the trimmed ALPHA study sample and the PEGASUS study sample after weighting by the Signorovitch method.43 The efficacy outcomes of interest were change from baseline in Hb levels, absolute reticulocyte count, LDH, FACIT-F, and transfusion avoidance. The safety outcomes of interest analyzed with the MAIC were BTH AEs and BTH AEs during extended follow-up (48 weeks for the PEGASUS study, median follow-up 34.6 weeks for the ALPHA study). The MAICs concluded that neither danicopan plus C5i nor pegcetacoplan were favoured for any of the efficacy or safety outcomes, with the exception of transfusion avoidance in an unanchored MAIC. However, the limitations associated with the indirect evidence overall did not allow for firm conclusions on the relative effectiveness or safety of danicopan plus C5i therapy relative to pegcetacoplan, and information on the comparative effectiveness remains lacking. The MAICs also did not include comparative information on several outcomes included in the pharmacoeconomic model such as iron overload, proportion of patients with Hb greater or less than 9.5 g/dL, or BTH by severity, which also limits the generalizability of this indirect comparison to the pharmacoeconomic analysis.
Discussion
Summary of Available Evidence
This report summarizes the evidence for danicopan as an add-on therapy to ravulizumab or eculizumab for the treatment of signs and symptoms of EVH in patients with PNH. The evidence appraisal was based on 1 IA from an ongoing phase III double-blind, placebo-controlled RCT with single-arm extensions and 1 body of indirect evidence consisting of a series of naive comparisons, using anchored and unanchored MAICs.
The pivotal trial, the ALPHA study, was a phase III RCT with a 12-week placebo-controlled, double-blind portion plus a 12-week single-arm extension and an additional LTE. Data from an interim efficacy analysis containing 75% of the total sample of patients was submitted (42 patients randomized to receive danicopan add-on therapy and 21 patients randomized to receive placebo add-on). An interim safety analysis provided data from the full patient sample enrolled in the trial (57 patients randomized to receive danicopan add-on and 29 patients randomized to receive placebo add-on) and their information collected up until interim database lock on September 20, 2022. The primary outcome of the study was the change in Hb levels from baseline to 12 weeks. Key secondary outcomes were the change from baseline to week 12 in the proportion of patients with Hb increase of 2 g/dL or more in the absence of transfusion, the proportion of patients achieving transfusion avoidance, FACIT-F scores, and absolute reticulocyte counts. Secondary outcomes were the change from baseline in transfusion burden (number of RBC units transfused and the number of transfusion instances), the percentage of patients with Hb normalization (defined as achieving the lower limit of normal reference range [110 g/L for female patients and 125 g/L for male patients]), and the change from baseline in LDH. Exploratory outcomes were the change from baseline to weeks 12 and 24 in EORTC QLQ-C30 scores and EQ-5D-3L scores. The sponsor also submitted interim results from the ALPHA study LTE for those patients in the ALPHA study who had completed an additional 52 weeks of follow-up in a single-arm, open-label format where all patients were receiving danicopan add-on therapy. The change from baseline in Hb, absolute reticulocyte count, LDH, FACIT-F, EQ-5D-3L, and EORTC QLQ-C30 were the outcomes reported. Lastly, the sponsor-submitted indirect evidence consisting of MAICs comparing a sample of the ALPHA study population trimmed to meet the inclusion criteria of the PEGASUS study, a phase III, open-label RCT that assessed the efficacy and safety of pegcetacoplan compared with eculizumab. Change from baseline to week 12 (ALPHA) or week 20 (PEGASUS) in Hb, absolute reticulocyte count, FACIT-F scores, transfusion avoidance, and LDH were efficacy outcomes, while time-to-hemolysis AEs and probability of BTH during extended follow-up were safety outcomes.
Baseline characteristics were generally well balanced between the study arms in the ALPHA study, with the exception of some imbalances in the proportion of patients receiving each C5i (a greater proportion of patients in the danicopan plus C5i arm were receiving ravulizumab as their C5i), sex (more males and fewer females in the danicopan plus C5i arm), race (more Asian patients in the danicopan plus C5i arm and more patients whose race was unknown in the placebo plus C5i arm), and transfusion history (more patients in the danicopan plus C5i arm had received a transfusion within 24 weeks of receiving the study drug, and more patients in the placebo plus C5i arm had received a transfusion within 12 weeks of starting the study drug). The majority of patients (90.5% in the danicopan plus C5i arm and 81.0% in the placebo plus C5i arm) had received a transfusion within 24 weeks of receiving the study drug. In addition, the majority of patients (69.0% of patients in the danicopan plus C5i arm and 71.4% of patients in the placebo plus C5i arm) had received a transfusion in the 12 weeks before receiving the study drug. The inclusion criteria was generally applicable to a Canadian context, although the clinical experts consulted by CDA-AMC noted that the inclusion and exclusion criteria may exclude certain patients with PNH in their practice; specifically, patients in the ALPHA study were required to have a platelet count greater than 30,000/µL and a neutrophil count greater than 500/L, and the exclusion criteria did not specify a list of comorbidities or laboratory values for exclusion. The experts noted that this may exclude patients with PNH with bone marrow suppression or other complex cases who may be candidates for danicopan therapy, although they also noted this was standard practice for clinical research. In addition, there is no accepted standardized definition for EVH according to the clinical experts, therefore the inclusion criteria for EVH may not encompass all patients with the condition.
Interpretation of Results
Efficacy
PNH is a rare disease and both the clinical experts and clinician group inputs consulted by CDA-AMC noted that patients with PNH are frequently complex, their PNH symptoms can be variable, and control can change due to complement-activating situations such as infection or suboptimal dosing, which would require clinical intervention but which might not be considered treatment failure. Improvement in Hb relative to the baseline for a given patient could be used to assess effectiveness of therapy. Overall, results from the ALPHA study for hematologic outcomes suggested that for patients with PNH meeting the study definition for EVH, treatment with danicopan plus C5i likely resulted in greater positive impact on these outcomes when compared to placebo plus C5i after 12 weeks of treatment. Danicopan therapy likely increased Hb levels from baseline to week 12 relative to treatment with placebo plus C5i therapy, with a trend toward further numeric increases at 24 weeks. At week 12, danicopan add-on therapy also likely increased the proportion of patients with a Hb increase of 2 g/dL or greater without transfusion, and may also result in an increase in the proportion of patients attaining Hb normalization (the lower limit of the sex-specific normal Hb range), when compared to placebo plus C5i. Results from 24 weeks suggested a trend toward maintained improvements in these Hb outcomes. The change in Hb from baseline surpassed the MID provided by the sponsor for clinically meaningful change. While no MIDs were available for the other Hb outcomes, the clinical experts noted that any improvement to hematologic outcomes would be clinically meaningful to them and on this basis, the improvements in Hb-related outcomes at 12 weeks appear to meet this criterion. Comparative results at later time points remain uncertain, however, predominantly due to the lack of a comparator arm after 12 weeks and the fact that not all patients had completed the LTE at the time that the ALPHA study was submitted for appraisal.
Reduction in transfusion dependence was highlighted by the clinical experts, the clinician input, and the patient input as important for treatment. Overall, danicopan add-on therapy likely decreased measures of transfusion burden (number of transfusion instances and number of RBCs transfused) and absolute reticulocyte count at 12 weeks. Results from transfusion outcomes suggest that the proportion of patients with transfusion avoidance was maintained at 24 weeks in the danicopan-emergent arm (not measured in the placebo-emergent arm), and a numerically small but non-null reduction in the number of transfusion instances and the number of RBC units transfused was also maintained at 24 weeks, along with an observed reduction in absolute reticulocyte counts. Similar to Hb-related outcomes, the clinical experts noted that any change in transfusion needs would be clinically meaningful to them, and the results at 12 and 24 weeks appear to meet this criterion. Transfusion outcomes at later time points remain uncertain due to the same limitations (lack of comparator arm after 12 weeks, very low number of patients completing LTE1 to date).
Changes in LDH levels was a biomarker outcome of interest in the ALPHA study; the clinical experts noted that if LDH was normal it would not be expected to change during therapy, and the clinician group input highlighted reduction in LDH (LDH ratio < 1.5 × ULN) as a marker for improvement. The results from 12 weeks suggested that treatment with danicopan plus C5i therapy may result in little to no difference in LDH levels when compared with placebo plus C5i therapy. This would align with the expectations noted by clinical experts. Observed changes in LDH from baseline to 24 weeks showed variable changes in the danicopan-emergent and placebo-emergent arms; a numeric decrease in LDH was observed in the danicopan-emergent arm and the placebo-emergent arm had little to no change in LDH from baseline to 24 weeks. Results from the LTE were reported for only a fraction of patients and were too uncertain to conclude the impact of ongoing danicopan therapy. In general, results are uncertain at later time points because of the study-level limitations noted for Hb and transfusion outcomes.
The clinician group input noted that PNH improvements should be accompanied by decreased fatigue, improved HRQoL, and improved overall survival. The patient input also highlighted that slowing disease progression and improving long-term outcomes and HRQoL were desired. Two measures of HRQoL (EQ-5D-3L and EORTC QLQ-C30) and 1 measure of fatigue (FACIT-F) were assessed in the ALPHA study. With regards to patient-reported outcomes of fatigue at 12 weeks, treatment with danicopan plus C5i therapy may result in an increase in FACIT-F scores when compared to placebo plus stable C5i therapy, based on an MID of 5 points provided in the submission, with results of a similar magnitude observed at 24 weeks. Evidence from the EQ-5D-3L and EORTC QLQ-C30 measures suggested danicopan add-on treatment may also result in little to no difference in scores from baseline to 12 weeks. Results from 24 weeks suggested a trend toward an increase in both scores, with variable results at LTE1, which may suggest a lagging improvement in HRQoL. Based on MIDs for patients with cancer, the score changes at 12 weeks in both arms in the EORTC QLQ-C30 measures were within the margin considered to be a small change. At 24 weeks, the change in scores in the placebo-emergent arm marginally passed the MID to be considered a medium change; the danicopan-emergent arm score at 24 weeks was considered a small change. However, the clinical meaningfulness of the EQ-5D-3L and EORTC QLQ-C30 score changes in patients with PNH is unclear as there was no MID available. In general, because of study limitations such as the small number of patients who have completed the LTE phases to date, the impact of danicopan add-on therapy on long-term HRQoL and fatigue outcomes remains unclear.
The body of evidence submitted for the indirect comparison consisted of an SLR, feasibility assessment for conducting an indirect comparison of danicopan plus a C5i to pegcetacoplan, a naive comparison of the trimmed ALPHA study population with the full sample from the PEGASUS study, as well as anchored and unanchored MAICs on select efficacy and safety outcomes. Briefly, the sponsor submission trimmed the patient sample in the ALPHA trial to meet 2 additional inclusion criteria from the PEGASUS study and compared the characteristics of the studies and the patient populations to ascertain whether a MAIC would be feasible to undertake. The sponsor concluded that the assumptions required for a MAIC would not be satisfied and provided a naive comparison of the trimmed ALPHA study sample with the PEGASUS study sample by study arm. However, feedback from the clinical experts noted that the differences highlighted by the sponsor were not clinically meaningful as reported, although the CDA-AMC appraisal noted that the trial designs still differed in ways that risk biasing the results. Furthermore, the infeasibility of a MAIC would not be an appropriate justification for relying on results of a naive comparison. The CDA-AMC appraisal concluded that a naive comparison would be largely uninformative and the information too uncertain to make firm conclusions on efficacy or safety and therefore did not review it or include it in the indirect evidence appraisal, although it makes up the base case in the pharmacoeconomic analysis. As it would be possible with a MAIC to control for treatment effect modifiers in the 2 studies, the CDA-AMC team appraised the unanchored and anchored MAICs undertaken on the trimmed ALPHA study sample and the PEGASUS study sample after weighting by the Signorovitch method.43 The efficacy outcomes of interest were change from baseline in Hb levels, absolute reticulocyte count, LDH, FACIT-F, and transfusion avoidance. The safety outcomes of interest analyzed with the MAIC were BTH AEs and BTH AEs during extended follow-up (48 weeks for the PEGASUS study, median follow-up 34.6 weeks for the ALPHA study). The MAICs concluded that neither danicopan plus C5i nor pegcetacoplan were favoured for any of the efficacy or safety outcomes, with the exception of transfusion avoidance in an unanchored MAIC. However, the limitations associated with the indirect evidence overall precluded firm conclusions on the relative effectiveness or safety of danicopan plus stable C5i therapy relative to pegcetacoplan, and information on the comparative effectiveness remains lacking.
Pegcetacoplan is the main comparator for danicopan, and the clinical experts as well as clinician input referenced the potential for treatment burden associated with SC administration and the possibility of severe BTH because of the nature of the proximal complement blockade when discussing unmet needs. The clinical experts noted that under the approved indication for danicopan, patients would be required to have residual hemolytic anemia due to EVH, while the indication for pegcetacoplan does not specifically require EVH. However, there was no evidence provided in the submission about the comparability of these treatments with respect to BTH severity or adherence, therefore it is not known whether danicopan would address these concerns. The indirect comparison also did not include comparative information on several other outcomes included in the pharmacoeconomic model such as iron overload, proportion of patients with Hb greater or less than 9.5 g/dL, or BTH by severity, which also limits the generalizability of this indirect comparison to the pharmacoeconomic analysis.
Efficacy results from the full analysis (FA) of the ALPHA study are in Appendix 1 and initial findings are presented here. Of note, the patient disposition reported that a total of 70 (81.4%) patients (46 [80.7%] patients initially randomized to danicopan plus C5i and 24 [82.8%] patients initially randomized to placebo plus C5i) completed the study, including years 1 and 2 of the LTE phase (study design available in Figure 1). Results at TP1 were either numerically similar for the FA compared to the IA, or the numeric changes observed did not materially impact the interpretation of the evidence, with some exceptions. A numeric increase in the LS mean TD for the proportion of patients with transfusion avoidance was reported in the FA relative to the IA (IA result = 40.80%; 95% CI, 21.08% to 60.58% and FA result = 48.40%; 95% CI, 31.79% to 64.94%). A slight numeric increase, sufficient to attain statistical significance, was reported for the TD in the proportion of patients with Hb normalization (IA result = 18.40; 95% CI, –0.84 to 37.71; P = 0.008 and FA result = 19.20; 95% CI, 3.34 to 35.10; P = 0.0023).
Results at TP2 and LTE were available in the FA which provided insight into the longer-term impacts of danicopan plus C5i on efficacy outcomes. Results for the proportion of patient with Hb normalization and the proportion of patients with Hb increase of 2 g/dL or greater were numerically similar between TP2 and LTE, suggesting a maintained effect. There was slight numeric reduction in the observed change in Hb from baseline between TP2 and LTE in the placebo-emergent arm (FA observed mean change from baseline at TP2 = 25.00; SD = 14.46 and FA observed mean change from baseline at LTE = 22.70; SD = 18.27) which was not observed in the danicopan-emergent arm. There was a notable reduction in the proportion of patients with transfusion avoidance at LTE in the danicopan-emergent arm relative to the result at TP2 (FA result at TP2 = 69.10; 95% CI, 55.19 to 80.86 and FA result at LTE = 59.30; 95% CI, 45.03 to 72.43). Both results at TP2 and LTE represented a numeric decrease from TP1 in the danicopan-emergent arm. Results for this outcome were not reported from TP2 to LTE for the placebo-emergent arm. LTE results were not reported for the FA for LDH. In terms of the measures of HRQoL, there were observed numeric decreases in both treatment arms between TP2 and LTE for EQ-5D-3L and EORTC QLQ-C30 scores. Overall, the efficacy results are still subject to the limitations (except for those inherent to interim analyses) which were highlighted in the critical appraisal. The clinical experts noted that any improvement to hematologic outcomes would be clinically meaningful to them and on this basis, the additional data from FA demonstrate that the results from the majority of outcomes still meet this criterion, although decreases in some Hb markers and the patient-reported outcomes are reported in the longer term and remain important to note.
Harms
Harms reporting was submitted as part of the IA in the ALPHA study; as of the data cut-off date, a total of 4 patients were still receiving placebo in TP1 and 2 patients discontinued before the switch to danicopan in TP2. Overall, a majority of patients reported any TEAE, with a numerically greater proportion in the danicopan-emergent arm (93.0% of patients) than the placebo-emergent arm (82.6% of patients). However, the proportion of patients with severe AEs or greater was numerically lower. Grade 3 TEAEs was generally balanced between treatment arms during TP1 (17.5% in the danicopan plus C5i arm, 13.8% in the placebo plus C5i arm), and TP2 (12.5% in the danicopan-emergent arm, 13.0% in the placebo-emergent arm). Grade 4 and 5 events during these times were rare (1.8% of patients in the danicopan plus C5i arm had a grade 4 TEAE during TP1, and 4.3% of patients in the placebo-emergent arm had grade 4 TEAEs during TP2); no other grade 4 or 5 TEAEs were reported. During the LTE, there was a numeric difference in the danicopan-emergent arm and the placebo-emergent arm for grade 3 TEAEs (7.5% of patients in the danicopan-emergent arm, 15.0% of patients in the placebo-emergent arm). Grade 4 events were generally balanced between treatment arms for grade 4 TEAEs (2.5% of patients in the danicopan-emergent arm, 5.0% of patients in the placebo-emergent arm). There were no grade 5 TEAEs reported during the LTE. A numerically greater proportion of patients in the placebo-emergent arm (26.1%) relative to the danicopan-emergent arm (12.3%) experienced SAEs while being treated with danicopan. A total of 4 patients (2 per arm) withdrew due to AEs and AEs led to discontinuation of the study drug in some patients during the trial, however those were evenly distributed between treatment arms. No deaths were reported.
Meningococcal infections and liver enzyme abnormalities were the 2 AEs of special interest prespecified in the ALPHA study. The product monograph of danicopan includes a serious warning and precaution about serious infections caused by encapsulated bacteria and elaborates that this may include organisms such as Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B; only meningococcal infections were reported as AEs of special interest in the ALPHA study. During the trial there were no reports of meningococcal infections; susceptibility to meningococcal infection with complement inhibitor therapy is known and was declared on the product monograph. The clinical experts also noted that it is common practice for patients with PNH to be routinely vaccinated for several meningitis strains although access to other vaccines may vary by province. Liver enzyme elevations occurred to a similar extent between treatment arms in patients during TP1; a numerically greater proportion of patients in the placebo arm experienced liver enzyme elevations during TP2 and the same number of patients in the LTE experienced liver enzyme elevations. Of note, during TP1, 2 (3.5%) patients stopped treatment due to liver enzyme abnormalities in the danicopan plus C5i arm, and 1 (3.4%) patient in the placebo plus C5i arm; during TP2, 1 (5.0%) patient in the placebo-emergent arm stopped treatment due to hepatic function abnormality. During LTE, 1 (4.8%) patient in the placebo-emergent arm stopped treatment due to hepatic function abnormality, and 1 (1.8%) patient each stopped treatment due to alanine transaminase and aspartate transaminase increases, respectively, in the danicopan arm. These numbers are overall numerically low, but these discontinuations may be important in the context of the small sample size. An additional AE of special interest identified by CDA-AMC was BTH; overall 4 BTH events were reported in the danicopan-emergent arm during the ALPHA study to date. No BTH events were reported to be serious; 1 hemolysis event was reported as serious, and the patient received a transfusion.
BTH events were also the safety outcome assessed as part of the indirect evidence; no other safety events were assessed. Of note, similar to the efficacy analysis, the limitations in the indirect evidence preclude firm conclusions about the comparative safety of danicopan plus C5i therapy relative to pegcetacoplan monotherapy.
The reporting of harms in the main body of the clinical report is subject to an important limitation in that the ALPHA trial is still ongoing, therefore potential additional safety signals are possible which would not be captured by this review, and the data from the full sample of patients are not available for TP2 (n = 48 patients in the danicopan-emergent arm and n = 23 patients in the placebo-emergent arm) and LTE (n = 40 patients in the danicopan-emergent arm and n = 20 patients in the placebo-emergent arm). A summary of safety data from the FA of the ALPHA study are presented in Appendix 1 and initial findings summarized here. Briefly, the most common AEs during the entire study as per the FA were similar to the IA, with the most common being COVID-19 (26.3% patients in the danicopan-emergent arm, 40.7% patients in the placebo-emergent arm), pyrexia (33.3% patients in the danicopan-emergent arm, 11.1% patients in the placebo-emergent arm), headache (26.3% patients in the danicopan-emergent arm, 11.1% in the placebo-emergent arm), nausea (17.5% in the danicopan-emergent arm, 11.1% in the placebo-emergent arm), and asthenia (10.5% patients in the danicopan-emergent arm, 18.5% patients in the placebo-emergent arm).
The proportion of patients with TEAEs during TP1 did not change notably; during TP1 there was 1 additional SAE reported (cholelithiasis) in the danicopan arm. The proportion of patients with any TEAE during TP2 increased from 64.6% to 74.5% of patients in the danicopan-emergent arm and from 56.5% to 66.7% of patients in the placebo-emergent arm; there were additional SAEs reported of hemolysis, cholecystitis, and femur fracture (1 report of each, placebo-emergent arm). The proportion of patients with any TEAE during LTE (entire study) increased from 62.5% to 88.9% in the danicopan-emergent arm and from 80.0% to 92.3% in the placebo-emergent arm. There were additional SAE reports in the danicopan-emergent arm of anemia, abdominal pain, nausea, vomiting, noncardiac chest pain, pyrexia, COVID-19, and decreased platelet count (1 report of each). There were additional SAE reports in the placebo-emergent arm of hemorrhagic diathesis, upper abdominal pain, COVID-19, pneumonia, cystitis, neutropenic sepsis, arthralgia, and PNH (1 report of each). Relative to the IA, there were additional increases in the proportion of patients who withdrew from the study drug due to AEs or SAEs during all treatment periods; information in the FA was split into withdrawals due to AEs and SAEs instead of because of specific events. During TP1, in the danicopan plus C5i arm 3 (5.3%) patients discontinued due to AEs and 1 (1.8%) discontinued because of SAEs (overall increase of 1 patient who withdrew from the study drug relative to IA); 1 (3.4%) patient withdrew from the study drug due to AEs in the placebo-emergent arm (unchanged from IA). During TP2, 1 (3.7%) patient withdrew from the study drug due to AEs in the placebo-emergent arm (overall increase of 1 patient relative to the IA). During LTE, 1 (1.9%) patient in the danicopan-emergent arm withdrew from the study drug due to AEs, and 1 (3.8%) patient in the placebo-emergent arm withdrew due to AEs (increase of 1 patient in the danicopan-emergent arm relative to IA). There was 1 death reported in the placebo-emergent arm during the study in the FA, which took place in the LTE; the patient had an SAE of pneumonia (increase of 1 patient relative to the IA). The FA did not report any additional AEs of meningococcal infections but reported 1 additional patient in the danicopan-emergent arm with liver enzyme elevations in the LTE. Overall, the safety results from the FA provided additional safety signals including 1 death; however, the overall proportion of patients with SAEs and the proportion of patients who withdrew from the study drug due to AEs or SAEs remained numerically low and broadly similar between study arms, similar to results from the IA.
Conclusion
PNH is a rare disease with significant morbidity and mortality — mortality is predominantly due to thrombosis related to IVH and is treated by C5i therapies (ravulizumab or eculizumab). Approximately 20% of patients with PNH who were clinically stable on C5i treatment develop clinically significant EVH.26 Evidence from the IA of the ALPHA study, a phase III RCT with a 12-week placebo-controlled, double-blind portion plus a 12-week single-arm, open-label extension and a LTE for an additional 52 weeks, was appraised to assess the impact of danicopan added on to C5i therapy versus placebo plus C5i therapy. The results demonstrated that over 12 weeks, when compared with placebo plus C5i therapy, danicopan plus C5i therapy likely increased Hb levels, the proportion of patients with Hb increase of 2 g/dL or more in the absence of transfusion, and the proportion of patients with transfusion avoidance. In addition, danicopan plus C5i therapy likely decreased markers of transfusion burden and absolute reticulocyte counts, and may increase the proportion of patients attaining Hb normalization. Results from week 24, the open-label, single-arm treatment period of the ALPHA study where all patients were receiving danicopan therapy, suggested this trend was maintained for most hematologic outcomes. Danicopan plus C5i therapy may result in an increase in FACIT-F scores, however may result in little to no difference in EQ VAS scores or EORTC QLQ-C30 global health status/quality of life scores at week 12 when compared to placebo plus C5i therapy. Results from week 24 suggest that score increases were maintained for FACIT-F in both treatment arms and suggest a trend toward increased scores in both treatment arms for EQ-5D-3L and EORTC QLQ-C30 scores. Results from the LTE portion were only available from a fraction of patients for all outcomes and therefore remain highly uncertain. With regards to safety, the majority of patients in both trial arms experienced any TEAE, and there was a numerically higher proportion of patients in the placebo-emergent arm who experienced SAEs while being treated with danicopan; there were also imbalances between the treatment arms in the proportion of patients with some TEAEs. However, a numerically low proportion of SAEs led to withdrawal of the study drug across treatment arms. Study limitations include that the ALPHA study is an IA and some missing data were reported for efficacy outcomes; it is unknown whether the missing data are informative or not. There is also no standard clinical definition for danicopan’s indication of EVH, and the study definition as well as the inclusion and exclusion criteria may leave out patients who would be treatment candidates in a clinical context. The safety results are particularly limited by the fact that the ALPHA study is an ongoing trial, therefore potential additional safety signals are possible which would not be captured by this review, particularly since the data from the full sample of patients are not available for the TP2 and LTE phases of the trial. The limitations associated with the indirect evidence submitted did not allow for firm conclusions on the effectiveness of danicopan plus C5i therapy relative to pegcetacoplan, and therefore conclusive information on the comparative effectiveness between danicopan and pegcetacoplan remains lacking.
References
1.National Organization for Rare Disorders. Paroxysmal nocturnal hemoglobinuria. 2024.
2.Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124(18):2804-2811. PubMed
3.Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers. 2017;3(1). PubMed
4.Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253-1258. PubMed
5.de Latour RP RA. Hemolytic processes in paroxysmal nocturnal hemoglobinuria and its treatment: intravascular and extravascular hemolysis, introduction. Nature: Bone Marrow Transplantation. 2020.
6.Schrezenmeier H, Muus P, Socie G, et al. Baseline characteristics and disease burden in patients in the International Paroxysmal Nocturnal Hemoglobinuria Registry. Haematologica. 2014;99(5):922-929. PubMed
7.Brodsky RA, Young NS, Antonioli E, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840-1847. PubMed
8.Weitz I, Meyers G, Lamy T, et al. Cross-sectional validation study of patient-reported outcomes in patients with paroxysmal nocturnal haemoglobinuria. Intern Med J. 2013;43(3):298-307. PubMed
9.Bektas M, Copley-Merriman C, Khan S, Sarda SP, Shammo JM. Paroxysmal nocturnal hemoglobinuria: patient journey and burden of disease. J Manag Care Spec Pharm. 2020;26(12-b Suppl):S8-S14.
10.Schrezenmeier H, Röth A, Araten DJ, et al. Baseline clinical characteristics and disease burden in patients with paroxysmal nocturnal hemoglobinuria (PNH): updated analysis from the International PNH Registry. Ann Hematol. 2020;99(7):1505-1514. PubMed
11.Ueda Y, Obara N, Yonemura Y, et al. Effects of eculizumab treatment on quality of life in patients with paroxysmal nocturnal hemoglobinuria in Japan. Int J Hematol. 2018;107(6):656-665. PubMed
12.Kelly RJ, Hill A, Arnold LM, et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117(25):6786-6792. PubMed
13.Hillmen P, Elebute M, Kelly R, et al. Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2010;85(8):553-559. PubMed
14.Meyers G, Weitz I, Lamy T, et al. Disease-related symptoms reported across a broad population of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(11):3683-3683.
15.Kulasekararaj A, Glasmacher A, Liu P, et al. Composite endpoint to evaluate complement inhibition therapy in patients with paroxysmal nocturnal hemoglobinuria. Eur J Haematol. 2022;108(5):391-402. PubMed
16.CADTH reimbursement reccomendation: pegcetacoplan (Empaveli -Sobi Canada Inc.). Can J Health Technol. 2023;3(5). https://www.cadth.ca/sites/default/files/DRR/2023/SR0748-Empaveli_combined.pdf. Accessed 2023 Sep 14.
17.Shammo J, Gajra A, Patel Y, et al. Low rate of clinically evident extravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria treated with a complement C5 inhibitor: results from a large, multicenter, US real-world study. J Blood Med. 2022;13:425-437. PubMed
18.Notaro R, Sica M. C3-mediated extravascular hemolysis in PNH on eculizumab: mechanism and clinical implications. Semin Hematol. 2018;55(3):130-135. PubMed
19.Risitano AM, Peffault de Latour R, Marano L, Frieri C. Discovering C3 targeting therapies for paroxysmal nocturnal hemoglobinuria: achievements and pitfalls. Semin Immunol. 2022;59. PubMed
20.Patriquin CJ, Kiss T, Caplan S, et al. How we treat paroxysmal nocturnal hemoglobinuria: A consensus statement of the Canadian PNH Network and review of the national registry. Eur J Haematol. 2018;102(1):36-52. PubMed
21.Risitano AM, Marotta S, Ricci P, et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front Immunol. 2019;10. PubMed
22.DeZern AE, Dorr D, Brodsky RA. Predictors of hemoglobin response to eculizumab therapy in paroxysmal nocturnal hemoglobinuria. Eur J Haematol. 2013;90(1):16-24. PubMed
23.Subías Hidalgo M, Martin Merinero H, López A, et al. Extravascular hemolysis and complement consumption in paroxysmal nocturnal hemoglobinuria patients undergoing eculizumab treatment. Immunobiology. 2017;222(2):363-371. PubMed
24.Anliker M, Schmidt CQ, Harder MJ, et al. Complement activation by human red blood cell antibodies: hemolytic potential of antibodies and efficacy of complement inhibitors assessed by a sensitive flow cytometric assay. Transfusion. 2018;58(12):2992-3002. PubMed
25.Kulasekararaj AG, Brodsky RA, Nishimura J-i, Patriquin CJ, Schrezenmeier H. The importance of terminal complement inhibition in paroxysmal nocturnal hemoglobinuria. Ther Adv Hematol. 2022;13. PubMed
26.Kulasekararaj A, Mellor J, Earl L, et al. Pb2056: prevalence of clinically significant extravascular hemolysis in stable C5 inhibitor-treated patients with Pnh and its association with disease control, quality of life and treatment satisfaction. HemaSphere. 2023;7(S3).
27.Empaveli (pegcetacoplan):solution, 1080 mg / 20 mL (54 mg/mL) for subcutaneous infusion use [product monograph]. Stockholm (SE): Swedish Orphan Biovitrum AB; 2022 Dec 7.
28.Jalbert JJ, Chaudhari U, Zhang H, Weyne J, Shammo JM. Epidemiology of PNH and real-world treatment patterns following an incident PNH diagnosis in the US. Blood. 2019;134(Supplement_1):3407-3407.
29.Voydeya (danicopan): 50 mg and 100 mg oral film-coated tablet [product monograph]. Zürich (CH): Alexion Pharma GmbH; 2024 Jul 19.
30.Risitano AM, Kulasekararaj AG, Lee JW, et al. Danicopan: an oral complement factor D inhibitor for paroxysmal nocturnal hemoglobinuria. Haematologica. 2020;106(12):3188-3197. PubMed
31.Kulasekararaj AG, Brodsky RA, Hill A. Monitoring of patients with paroxysmal nocturnal hemoglobinuria on a complement inhibitor. Am J Hematol. 2021;96(7):E232-E235. PubMed
32.Alexion Pharma GmbH response to June 5, 2024 CADTH request for additional information regarding danicopan CADTH review: [internal sponsor's report]. Mississauga (ON): Alexion Pharma GmbH; 2024 Jun 11.
33.Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) Scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19. PubMed
34.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
35.EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
36.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
37.Brodsky R. Treatment and prognosis of paroxysmal nocturnal hemoglobinuria. In: Post TW, ed. UpToDate UpToDate Waltham (MA); 2023: http://www.uptodate.com. Accessed 2024 Jul 2.
38.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
39.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
40.Clinical Study Report 2.0 (Interim Analysis Report): ALXN2040-PNH-301. A phase 3 study of danicopan (ALXN2040) as add-on therapy to a C5 inhibitor (eculizumab or ravulizumab) in patients with paroxysmal nocturnal hemoglobinuria who have clinically evident extravascular hemolysis (EVH) [internal sponsor's report]. Boston (MA): Alexion Pharmaceuticals Inc; 2023 Mar 23.
41.Alexion Pharma GmbH response to April 29, 2024 CADTH request for additional information regarding danicopan CADTH review: [internal sponsor's report]. Mississauga (ON): Alexion Pharma GmBH; 2024 May 6.
42.Alexion Pharma GmbH response to April 30, 2024 CADTH request for additional information regarding danicopan CADTH review: [internal sponsor's report]. Mississauga (ON): Alexion Pharma GmbH; 2024 May 7.
43.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials. Pharmacoeconomics. 2010;28(10):935-945. PubMed
44.Janeway C, Travers, P., Walport, M., & Shlomchik, M. Immunobiology: the immune system in health and disease. Vol 2. New York (NY): Garland Science; 2001.
45.Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20(1):34-50. PubMed
46.Berentsen S, Hill A, Hill QA, Tvedt THA, Michel M. Novel insights into the treatment of complement-mediated hemolytic anemias. Ther Adv Hematol. 2019;10:2040620719873321. PubMed
47.Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530-539. PubMed
48.Hill A, Platts PJ, Smith A, et al. The incidence and prevalence of paroxysmal nocturnal hemoglobinuria (PNH) and survival of patients in Yorkshire. Blood. 2006;108(11):985-985.
49.Richards SJ, Painter D, Dickinson AJ, et al. The incidence and prevalence of patients with paroxysmal nocturnal haemoglobinuria and aplastic anaemia PNH syndrome: a retrospective analysis of the UK's population-based haematological malignancy research network 2004-2018. Eur J Haematol. 2021;107(2):211-218. PubMed
50.Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor–experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540-549. PubMed
51.Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(25):4985-4996. PubMed
52.Oliver M, Patriquin C. Paroxysmal nocturnal hemoglobinuria: Current management, unmet needs, and recommendations J Blood Med. 2023;14:613-628. PubMed
53.Soliris (eculizumab for injection): 30 mL parenteral solution (10 mg/mL) [product monograph]. Baar (SE): Alexion Pharma GmbH; 2021 Mar 25.
54.Ultomiris (ravulizumab for injection): 10 mg/mL & 100 mg/mL concentrate for solution for intravenous infusion [product monograph]. Baar (SE): Alexion Pharma GmbH 2023 Oct 30.
55.Lee JW, Griffin M, Kim JS, et al. Addition of danicopan to ravulizumab or eculizumab in patients with paroxysmal nocturnal haemoglobinuria and clinically significant extravascular haemolysis (ALPHA): a double-blind, randomised, phase 3 trial. Lancet Haematol. 2023;10(12):e955-e965. PubMed
56.Alexion Pharmaceuticals Inc. NCT04469465: Danicopan as add-on therapy to a C5 Inhibitor in paroxysmal nocturnal hemoglobinuria (PNH) participants who have clinically evident extravascular hemolysis (EVH)(ALPHA) ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2024: https://clinicaltrials.gov/study/NCT04469465. Accessed 2024 Feb 12.
57.Clinical Study Report Protocol: ALXN2040-PNH-301. A phase 3 study of danicopan (ALXN2040) as add-on therapy to a C5 inhibitor (eculizumab or ravulizumab) in patients with paroxysmal nocturnal hemoglobinuria who have clinically evident extravascular hemolysis (EVH) [internal sponsor's report]. Boston (MA): Alexion Pharmaceuticals Inc; 2022 Feb 25.
58.Cançado RD, Araújo AdS, Sandes AF, et al. Consensus statement for diagnosis and treatment of paroxysmal nocturnal haemoglobinuria. Hematol Transfus Cell Ther. 2021;43(3):341-348. PubMed
59.Cella D, Kallich J, McDermott A, Xu X. The longitudinal relationship of hemoglobin, fatigue and quality of life in anemic cancer patients: results from five randomized clinical trials. Ann Oncol. 2004;15(6):979-986. PubMed
60.Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699-3709. PubMed
61.Barcellini W, Fattizzo B. Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia. Dis Markers. 2015;2015:635670. PubMed
62.McKinley CE, Richards SJ, Munir T, et al. Extravascular hemolysis due to C3-loading in patients with PNH treated with eculizumab: defining the clinical syndrome. Blood. 2017;130:3471.
63.Lee JW, Jang JH, Kim JS, et al. Clinical signs and symptoms associated with increased risk for thrombosis in patients with paroxysmal nocturnal hemoglobinuria from a Korean Registry. Int J Hematol. 2013;97(6):749-757. PubMed
64.Cella D, Nowinski CJ. Measuring quality of life in chronic illness: the functional assessment of chronic illness therapy measurement system. Arch Phys Med Rehabil. 2002;83(12 Suppl 2):S10-17. PubMed
65.Cella D, Sarda SP, Hsieh R, et al. Changes in hemoglobin and clinical outcomes drive improvements in fatigue, quality of life, and physical function in patients with paroxysmal nocturnal hemoglobinuria: post hoc analyses from the phase III PEGASUS study. Ann Hematol. 2022;101(9):1905-1914. PubMed
66.Hillmen P, Szer J, Weitz I, et al. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2021;384(11):1028-1037. PubMed
67.Chandran V, Bhella S, Schentag C, Gladman DD. Functional assessment of chronic illness therapy-fatigue scale is valid in patients with psoriatic arthritis. Ann Rheum Dis. 2007;66(7):936-939. PubMed
68.Cella D, Johansson P, Ueda Y, et al. Clinically important change for the FACIT-Fatigue scale in paroxysmal nocturnal hemoglobinuria: a derivation from international PNH registry patient data. J Patient Rep Outcomes. 2023;7(1):63. PubMed
69.Teckle P, Peacock S, McTaggart-Cowan H, et al. The ability of cancer-specific and generic preference-based instruments to discriminate across clinical and self-reported measures of cancer severities. Health Qual Life Outcomes. 2011;9:106. PubMed
70.Davda J, Kibet H, Achieng E, Atundo L, Komen T. Assessing the acceptability, reliability, and validity of the EORTC Quality of Life Questionnaire (QLQ-C30) in Kenyan cancer patients: a cross-sectional study. J Patient Rep Outcomes. 2021;5(1):4. PubMed
71.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
72.Cocks K, King MT, Velikova G, et al. Evidence-based guidelines for interpreting change scores for the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Eur J Cancer. 2012;48(11):1713-1721. PubMed
73.Kulasekararaj AG, Risitano AM, Maciejewski JP, et al. Phase 2 study of danicopan in patients with paroxysmal nocturnal hemoglobinuria with an inadequate response to eculizumab. Blood. 2021;138(20):1928-1938. PubMed
74.Critical Appraisal Skills Programme. CASP checklists. Cohort study. 2023; https://casp-uk.net/casp-tools-checklists/. Accessed 2023 April 4.
75.Cochrane. Cochrane risk of bias tool RoB v2.0. 2018; https://methods.cochrane.org/bias/resources/cochrane-risk-bias-tool. Accessed 2024 Apr 5.
76.Drummond MF, Sculpher MJ, Claxton K, Stoddart GL, Torrance G. Methods for the economic evaluation of health care programmes. Oxford (GB): Oxford University Press; 2015.
77.Extravascular hemolysis (EVH) in paroxysmal nocturnal hemoglobinuria (PNH): a systematic literature review (version 3.0) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Voydeya (danicopan), 50 mg and 100 mg oral film-coated tablets. Mississauga (ON): Alexion Pharma GmbH; 2023 Aug 10.
78.Jackson D, Rhodes K, Ouwens M. Alternative weighting schemes when performing matching‐adjusted indirect comparisons. Research Synthesis Methods. 2021;12(3):333-346. PubMed
79.Feasibility of conducting an indirect comparison in paroxysmal nocturnal haemoglobinuria between danicopan + C5 inhibitor and pegcetacoplan [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Voydeya (danicopan), 50 mg and 100 mg oral film-coated tablets. Mississauga (ON): Alexion Pharma GmbH; 2023 Jan 27.
80.Danicopan vs pegcetacoplan: indirect treatment comparison [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Voydeya (danicopan), 50 mg and 100 mg oral film-coated tablets. Mississauga (ON): Alexion Pharma GmbH; 2022 Dec 6.
81.Phillippo D, Ades T, Dias S, Palmer S, Abrams KR, Welton N. NICE DSU technical support document 18: methods for population-adjusted indirect comparisons in submissions to NICE. Sheffield (GB) Decision Support Unit, University of Sheffield: https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf. Accessed 2024 Jul 2.
82.Alexion GmbH response to July 30, 2024 CADTH request for additional information regarding danicopan CADTH review [internal sponsor's report]. Mississauga (ON): Alexion GmbH; 2024 Aug 1.
Appendix 1: Additional Outcome Data
Please note that this appendix has not been copy-edited.
The following additional data from the FA of the ALPHA trial.
Efficacy
Table 22: Summary of Key Efficacy Results From the ALPHA Study During TP1 — FAS
Variable | TP1 | |
|---|---|---|
Danicopan + C5i N = 57 | Placebo + C5i N = 28 | |
Change in Hb from Baseline to Week 12 | ||
LS mean (95% CI) change from baseline (g/L) | 28.08 (24.17, 31.98) | 4.62 (–1.39, 10.64) |
LS mean difference (95% CI) | 23.46 (16.31, 30.61) | |
P valuea | < 0.0001 | |
Proportion of Patients with Hb Increase ≥ 2 g/dL (20 g/L) in the Absence of Transfusion | ||
N patients | 31 | 0 |
Proportion of patients (95% CI) | 54.4 (40.66, 67.64) | 0 (0.00, 11.94) |
Treatment difference (95% CI) | 47.5 (32.63, 62.39) | |
Stratified CMH P valuea | < 0.0001 | |
Proportion of Patients with Transfusion Avoidance at Week 12 | ||
N patients | 45 | 8 |
Proportion of patients (95% CI) | 78.9 (66.11, 88.62) | 27.6 (12.73, 47.24) |
Treatment difference (95% CI) | 48.4 (31.79, 64.94) | |
Stratified CMH P valuea | < 0.0001 | |
Change in FACIT-F Scores from Baseline to Week 12 | ||
LS mean (95% CI) change from baseline | 8.13 (6.30, 9.96)b | 2.35 (–0.22, 4.91) |
LS mean difference (95% CI) | 5.79 (2.68, 8.89) | |
P valuea | 0.0004 | |
Change in Absolute Reticulocyte Count (1012/L) from Baseline to Week 12 | ||
LS mean change from baseline (95% CI) | –0.093 (–0.109, –0.076) | –0.001 (–0.024, 0.023)c |
LS mean difference (95% CI) | –0.092 (–0.120, –0.063) | |
P valuea | < 0.0001 | |
Change in the Number of RBC Units Transfused from 12 Weeks | ||
LS mean (95% CI) change from baseline | –1.44 (–1.86, –1.02) | –0.14 (–0.73, 0.45) |
LS mean difference (95% CI) | –1.29 (–2.02, –0.57) | |
P value | 0.0007 | |
Change in the Number of Transfusion Instances from 12 Weeks | ||
LS mean (95% CI) change from baseline | –0.91 (–1.18, –0.63) | –0.11 (–0.49, 0.27) |
LS mean difference (95% CI) | –0.80 (–1.27, –0.33) | |
P value | 0.0012 | |
Change in LDH Values (U/L) from Baseline to Week 12 | ||
LS mean (95% CI) change from baseline | –25.60 (–41.48, –9.73) | –16.92 (–39.69, 5.85) |
LS mean difference (95% CI) | –8.69 (–36.25, 18.88) | |
P value | 0.5306 | |
Proportion of Patients with Hb Normalization at Week 12 | ||
N patients | 15 | 0 |
Proportion of patients (95% CI) | 26.3 (15.54, 39.66) | 0 (0.00, 11.94) |
Treatment difference (95% CI) | 19.2 (3.34, 35.10) | |
Stratified CMH P value | 0.0023 | |
Change in EQ VAS from Baseline to Week 12 | ||
LS mean (95% CI) change from baseline | 13.35 (9.10, 17.59) | 5.97 (–0.18, 12.12) |
LS mean difference (95% CI) | 7.38 (0.06, 14.70) | |
P value | 0.0483 | |
Change in EORTC QLQ–C30 global health status/QoL Scores from Baseline to Week 12 | ||
LS mean (95% CI) change from baseline | 11.62 (7.39, 15.84) | 7.36 (1.22, 13.49) |
LS mean difference (95% CI) | 4.26 (–3.07, 11.58) | |
P value | 0.2503 | |
C5i = complement component 5 inhibitor; CI = confidence interval; CMH = Cochran Mantel-Haenszel; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = EQ visual analogue scale; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; LDH = lactate dehydrogenase; LS = least squares; NR = not reported; RBC = red blood cell; SD = standard deviation; TP = treatment period.
aP value adjusted for multiple comparisons.
bResults reported for 56 patients in the danicopan + C5i arm.
cResults reported for 26 patients in the placebo + C5i arm.
Source: details included in the table are from additional information provided by the sponsor.82
Table 23: Summary of TP2 and LTE Efficacy Results From the ALPHA Study — FAS
Variable | TP2 | LTE1 | ||
|---|---|---|---|---|
Weeks 0 to 12: Danicopan + C5i Weeks 12 to 24: Danicopan + C5i N = 57 | Weeks 0 to 12: Placebo + C5i Weeks 12 to 24: Danicopan + C5i N = 29 | Weeks 0 to 12: Danicopan + C5i Weeks 12 to 24: Danicopan + C5i N = 57 | Weeks 0 to 12: Placebo + C5i Weeks 12 to 24: Danicopan + C5i N = 29 | |
Change in Hb from Baseline | ||||
Complete cases, n | 47 | 20 | 38 | 20 |
Observed value (g/L) | 105.9 (18.18) | 102.9 (17.20) | 104.00 (15.38) | 100.40 (20.63) |
Change from baseline (g/L), mean (SD) | 28.90 (18.61) | 25.00 (14.46) | 28.10 (14.00) | 22.70 (18.27) |
LS mean change (g/L) from baseline (95% CI) | 29.49 (24.19, 34.79) | 22.51 (17.03, 27.99) | NR | NR |
Proportion of Patients with Hb Increase ≥ 2g/dL (20 g/L) in the Absence of Transfusion | ||||
Number of patients included in analysis, n | 55 | NR | NR | NR |
N patients | 23 | NR | 29 | 12 |
Proportion of patients (95% CI) | 41.80 (28.55, 55.89) | NR | 53.70 (39.61, 67.38) | 46.20 (26.59, 66.63) |
Proportion of Patients with Transfusion Avoidancea | ||||
N patients | 38 | NR | 32 | NR |
Proportion of patients (95% CI) | 69.10 (55.19, 80.86) | NR | 59.30 (45.03, 72.43) | NR |
Change in FACIT-F Scores | ||||
Complete cases, n | NR | NR | 48 | 24 |
Observed, mean (SD) | 40.94 (9.73) | 37.74 (11.54) | 40.29 (10.26) | 36.67 (11.17) |
Change from baseline, mean (SD) | 6.81 (8.77) | 5.33 (9.26) | 4.93 (8.80) | 4.08 (9.43) |
LS mean (95% CI) change from baseline | 6.21 (4.11, 8.31) | 5.64 (1.66, 9.62) | NR | NR |
Change in Absolute Reticulocyte Count (1012/L) from Baseline | ||||
Complete cases, n | 42 | 15 | 36 | 16 |
Observed count, mean (SD) | 0.148 (0.060) | 0.172 (0.052) | 0.172 (0.070) | 0.189 (0.081) |
Change from baseline, mean (SD) | –0.084 (0.078) | –0.065 (0.107) | –0.080 (0.086) | –0.055 (0.070) |
Change in the Number of RBC Units Transfused from 24 Weeks Before Treatment Initiation to Week 24 Post-Initiation | ||||
Complete cases, n | 55 | NR | NR | NR |
Mean (95% CI) change | –2.70 (–4.04, –1.41) | NR | NR | NR |
Change in the Number of Transfusion Instances from 24 Weeks Before Treatment Initiation to Week 24 Post-Initiation | ||||
Complete cases, n | 55 | NR | NR | NR |
Mean (95% CI) change | –1.50 (–2.20, –0.89) | NR | NR | NR |
Change in LDH Values (U/L) from Baseline | ||||
Observed, mean (SD) | NR | NR | NR | NR |
Mean (SD) change from baseline | NR | NR | NR | NR |
Proportion of Patients with Hb Normalization | ||||
N patients | 11 | NR | 7 | 6 |
Proportion of patients (95% CI) | 20.00 (10.43, 32.97) | NR | 13.0 (5.37, 24.90) | 23.10 (8.97, 43.65) |
Change in EQ VAS Scores from Baseline | ||||
Complete cases, n | 50 | 25 | 46 | 21 |
Observed, mean (SD) | 73.80 (16.70) | 69.60 (18.19) | 68.60 (23.17) | 67.00 (19.21) |
Mean (SD) change from baseline | 14.0 (19.08) | 10.70 (21.10) | 7.40 (25.72) | 7.50 (29.28) |
Change in EORTC QLQ–C30 Global Health Status/QoL Scores from Baseline | ||||
Complete cases, n | 49 | 26 | 47 | 23 |
Observed, mean (SD) | 68.88 (19.38) | 64.51 (19.14) | 68.26 (17.86) | 62.50 (19.35) |
Mean (SD) change from baseline | 10.03 (17.26) | 10.90 (16.12) | 6.91 (20.06) | 6.88 (23.93) |
C5i = complement component 5 inhibitor; CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; Hb = hemoglobin; LDH = lactate dehydrogenase; LS = least squares; NR = not reported; RBC = red blood cell; SD = standard deviation; TP = treatment period.
aProportion of patients with transfusion avoidance from baseline to the end of TP2 and LTE, respectively.
Source: Details included in the table are from additional information provided by the sponsor.82
Harms
Table 24: Summary of Harms Results From the ALPHA Study — SAS
Adverse events | ALPHA | |
|---|---|---|
Weeks 0 to 12: Danicopan + C5i Weeks 12 to 24: Danicopan + C5i (N = 57) | Weeks 0 to 12: Placebo + C5i Weeks 12 to 24: Danicopan + C5i (N = 29) | |
Most common (≥ 5% patients) treatment-emergent adverse events, n (%)a | ||
Treatment Period 1 | N = 57 | N = 29 |
Any AE | 43 (75.4) | 18 (62.1) |
Blood and lymphatic system disorders | ||
Anemia | 1 (1.8) | 4 (13.8) |
Gastrointestinal disorders | ||
Nausea | 5 (8.8) | 3 (10.3) |
Diarrhea | 4 (7.0) | 3 (10.3) |
Vomiting | 3 (5.3) | 0 |
Abdominal pain upper | 1 (1.8) | 2 (6.9) |
Abdominal pain | 0 | 2 (6.9) |
General disorders and administration site conditions | ||
Pyrexia | 3 (5.3) | 0 |
Asthenia | 0 | 4 (13.8) |
Infections and infestations | ||
Urinary tract infection | 3 (5.3) | 1 (3.4) |
Ear infection | 0 | 2 (6.9) |
Injury, poisoning, and procedural complications | ||
Contusion | 2 (3.5) | 3 (10.3) |
Investigations | ||
Alanine aminotransferase increased | 3 (5.3) | 1 (3.4) |
Aspartate aminotransferase increased | 2 (3.5) | 3 (10.3) |
Musculoskeletal and connective tissue disorders | ||
Arthralgia | 4 (7.0) | 2 (6.9) |
Pain in extremity | 3 (5.3) | 0 |
Nervous system disorders | ||
Headache | 6 (10.5) | 3 (10.3) |
Dizziness | 1 (1.8) | 2 (6.9) |
Psychiatric disorders | ||
Insomnia | 1 (1.8) | 3 (10.3) |
Vascular disorders | ||
Hypertension | 3 (5.3) | 1 (3.4) |
Treatment Period 2 | N = 55 | N = 27 |
Any AE | 41 (74.5) | 18 (66.7) |
Gastrointestinal disorders | ||
Diarrhea | 6 (10.9) | 2 (7.4) |
General disorders and administration site conditions | ||
Pyrexia | 7 (12.7) | 0 |
Nervous system disorders | ||
Headache | 6 (10.9) | 2 (7.4) |
LTE | N = 54 | N = 26 |
Any AE | 48 (88.9) | 24 (92.3) |
Blood and lymphatic system disorders | ||
Thrombocytopenia | 2 (3.7) | 3 (11.5) |
Anemia | 3 (5.6) | 1 (3.8) |
Breakthrough hemolysis | 4 (7.4) | 0 |
Hemolysis | 2 (3.7) | 2 (7.7) |
Gastrointestinal disorders | ||
Nausea | 4 (7.4) | 1 (3.8) |
Abdominal pain | 2 (3.7) | 2 (7.7) |
Constipation | 0 | 4 (15.4) |
General disorders and administration site conditions | ||
Pyrexia | 14 (25.9) | 3 (11.5) |
Asthenia | 4 (7.4) | 4 (15.4) |
Fatigue | 4 (7.4) | 1 (3.8) |
Infections and infestations | ||
COVID-19 | 12 (22.2) | 9 (34.6) |
Nasopharyngitis | 7 (13.0) | 1 (3.8) |
Urinary tract infection | 4 (7.4) | 2 (7.7) |
Musculoskeletal and connective tissue disorders | ||
Arthralgia | 3 (5.6) | 2 (7.7) |
Back pain | 2 (3.7) | 2 (7.7) |
Pain in extremity | 4 (7.4) | 0 |
Nervous system disorders | ||
Headache | 8 (14.8) | 1 (3.8) |
Psychiatric disorders | ||
Insomnia | 1 (1.9) | 3 (11.5) |
Respiratory, thoracic, and mediastinal disorders | ||
Cough | 3 (5.6) | 1 (3.8) |
Most common serious adverse events, n (%) | ||
Treatment Period 1 | N = 57 | N = 29 |
Any SAE | 3 (5.3) | 2 (6.9) |
Blood and lymphatic system disorders | 0 | 1 (3.4) |
Anemia | 0 | 1 (3.4) |
Gastrointestinal disorders | 1 (1.8) | 1 (3.4) |
Pancreatitis | 1 (1.8) | 0 |
Abdominal pain | 0 | 1 (3.4) |
Hepatobiliary disorders | 2 (3.5) | 0 |
Cholecystitis | 1 (1.8) | 0 |
Cholelithiasis | 1 (1.8) | 0 |
Infections and infestations | 1 (1.8) | 0 |
COVID-19 | 1 (1.8) | 0 |
Investigations | 1 (1.8) | 0 |
Blood bilirubin increased | 1 (1.8) | 0 |
Nervous system disorders | 0 | 1 (3.4) |
Headache | 0 | 1 (3.4) |
Treatment Period 2 | N = 55 | N = 27 |
Any SAE | 3 (5.5) | 6 (22.2) |
Blood and lymphatic system disorders | 0 | 2 (7.4) |
Hemolysis | 0 | 2 (7.4) |
Ear and labyrinth disorders | 0 | 1 (3.7) |
Vertigo | 0 | 1 (3.7) |
Gastrointestinal disorders | 1 (1.8) | 0 |
Dieulafoy vascular malformation | 1 (1.8) | 0 |
General disorders and administration site conditions | 1 (1.8) | 0 |
Pyrexia | 1 (1.8) | 0 |
Hepatobiliary disorders | 0 | 1 (3.7) |
Cholecystitis | 0 | 1 (3.7) |
Infections and infestations | 2 (3.6) | 0 |
COVID-19 pneumonia | 1 (1.8) | 0 |
Staphylococcal sepsis | 1 (1.8) | 0 |
Injury, poisoning, and procedural complications | 0 | 1 (3.7) |
Femur fracture | 0 | 1 (3.7) |
Nervous system disorders | 0 | 1 (3.7) |
Headache | 0 | 1 (3.7) |
LTE | N = 54 | N = 26 |
Any SAE | 7 (13.0) | 6 (23.1) |
Blood and lymphatic system disorders | 1 (1.9) | 1 (3.8) |
Anemia | 1 (1.9) | 0 |
Hemorrhagic diathesis | 0 | 1 (3.8) |
Cardiac disorders | 0 | 1 (3.8) |
Pericardial effusion | 0 | 1 (3.8) |
Gastrointestinal disorders | 1 (1.9) | 1 (3.8) |
Abdominal pain | 1 (1.9) | 0 |
Abdominal pain upper | 0 | 1 (3.8) |
Diarrhea | 0 | 1 (3.8) |
Nausea | 1 (1.9) | 0 |
Vomiting | 1 (1.9) | 0 |
General disorders and administration site conditions | 3 (5.6) | 0 |
Noncardiac chest pain | 1 (1.9) | 0 |
Pyrexia | 1 (1.9) | 0 |
Stent-graft endoleak | 1 (1.9) | 0 |
Infections and infestations | 1 (1.9) | 4 (15.4) |
COVID-19 | 1 (1.9) | 2 (7.7) |
Pneumonia | 0 | 1 (3.8) |
Cystitis | 0 | 1 (3.8) |
Neutropenic sepsis | 0 | 1 (3.8) |
Investigations | 2 (3.7) | 1 (3.8) |
Body temperature increased | 0 | 1 (3.8) |
Hemoglobin decreased | 1 (1.9) | 0 |
Platelet count decreased | 1 (1.9) | 0 |
Musculoskeletal and connective tissue disorders | 0 | 1 (3.8) |
Arthralgia | 0 | 1 (3.8) |
Neoplasms benign, malignant and unspecified (incl cysts and polyps) | 1 (1.9) | 0 |
Invasive ductal breast carcinoma | 1 (1.9) | 0 |
Renal and urinary disorders | 0 | 1 (3.8) |
Paroxysmal nocturnal hemoglobinuria | 0 | 1 (3.8) |
Respiratory, thoracic, and mediastinal disorders | 1 (1.9) | 0 |
Pulmonary embolism | 1 (1.9) | 0 |
Pulmonary hemorrhage | 1 (1.9) | 0 |
Overall danicopan treatment (up to data cut-off on September 20, 2022) | N = 57 | N = 29 |
Any SAE | 11 (19.3) | 9 (33.3) |
Patients who stopped treatment due to adverse events, n (%) | ||
Treatment Period 1 | N = 57 | N = 29 |
AE leading to withdrawal of study intervention | 3 (5.3) | 1 (3.4) |
SAE leading to withdrawal of study intervention | 1 (1.8) | 0 |
Treatment Period 2 | N = 55 | N = 27 |
AE leading to withdrawal of study intervention | 0 | 1 (3.7) |
SAE leading to withdrawal of study intervention | 0 | 0 |
LTE | N = 54 | N = 26 |
AE leading to withdrawal of study intervention | 1 (1.9) | 1 (3.8) |
SAE leading to withdrawal of study intervention | 0 | 0 |
Overall danicopan treatment | N = 57 | N = 29 |
AE leading to withdrawal of study intervention | 4 (7.0) | 2 (7.4) |
SAE leading to withdrawal of study intervention | 1 (1.8) | 0 |
Deaths, n (%) | ||
Treatment Period 1 | N = 57 | N = 29 |
Patients who died | 0 | 0 |
Treatment Period 2 | N = 55 | N = 27 |
Patients who died | 0 | 0 |
LTE | N = 54 | N = 26 |
Patients who died | 0 | 1 (3.8) |
Overall danicopan treatment | N = 57 | N = 29 |
Patients who died | 0 | 1 (3.7) |
Adverse events of special interest, n (%) | ||
Treatment Period 1 | N = 57 | N = 29 |
Meningococcal infections | 0 | 0 |
Liver enzyme elevations | 8 (14.0) | 3 (10.3) |
Treatment Period 2 | N = 55 | N = 27 |
Meningococcal infections | 0 | 0 |
Liver enzyme elevations | 3 (5.5) | 3 (11.1) |
LTE | N = 54 | N = 26 |
Meningococcal infections | 0 | 0 |
Liver enzyme elevations | 2 (3.7) | 1 (3.8) |
Overall danicopan treatment | N = 57 | N = 29 |
Meningococcal infections | 0 | 0 |
Liver enzyme elevations | 11 (19.3) | 4 (14.8) |
AE = adverse event; C5i = complement component 5 inhibitor; EQ VAS = EQ visual analogue scale; LTE = long-term extension; SAE = severe adverse event; TEAE = treatment-emergent adverse event.
Note: In summarizing n (%), if a participant had multiple events for a particular Preferred Term, they were counted only once for that Preferred Term. A TEAE having a starting date during a certain treatment period was regarded as a TEAE in that treatment period. Any TEAE lasting across treatment periods was only counted once in the treatment period the event started.
Source: Details included in the table are from additional information provided by the sponsor.82
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ALT
alanine aminotransferase
BIA
budget impact analysis
BTH
breakthrough hemolysis
C3i
complement component 3 inhibitor
C5i
complement component 5 inhibitor
CDA-AMC
Canada’s Drug Agency
ESS
effective sample size
EVH
extravascular hemolysis
FACIT
Functional Assessment of Chronic Illness Therapy
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
Hb
hemoglobin
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
LDH
lactate dehydrogenase
LTE
long-term extension
MAIC
matching-adjusted indirect comparison
PNH
paroxysmal nocturnal hemoglobinuria
QALY
quality-adjusted life-year
TP1
treatment period 1
TP2
treatment period 2
ULN
upper limit of normal
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Danicopan (Voydeya), 50 mg and 100 mg film-coated tablets, oral administration |
Indication | Proposed: As an add-on to eculizumab or ravulizumab for the treatment of adult patients with PNH who have residual hemolytic anemia due to EVH |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | July 19, 2024 |
Reimbursement request | As per the approved Health Canada indication |
Sponsor | Alexion Pharma GmbH |
Submission history | Previously reviewed: No |
EVH = extravascular hemolysis; NOC = Notice of Compliance; PNH = paroxysmal nocturnal hemoglobinuria.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with PNH with signs or symptoms of EVH (i.e., clinically significant EVH; signs/symptoms of anemia that cannot be explained by other causes of anemia) |
Treatment | Danicopan as an add-on to ravulizumab or eculizumab |
Dose regimen | Recommended starting dose of danicopan is 150 mg t.i.d. (in addition to ravulizumab or eculizumab). Depending on clinical response,a the danicopan dose can be increased to 200 mg t.i.d. |
Submitted price | Danicopan 50 mg: $22.97 per tablet 100 mg: $45.95 per tablet |
Submitted annual treatment cost | Danicopan as an add-on to C5i: $618,485 per patient per year Note: the danicopan treatment cost is $85,282 and the C5i treatment cost is $533,203 |
Comparator(s) |
|
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs |
Time horizon | Lifetime (45.7 years) |
Key data source | The ALPHA trial informed efficacy and safety of danicopan plus C5i and C5i monotherapy; and utility values for health states for all treatment arms The PEGASUS trial (and its analysis by Hakimi et al. [2022]) informed efficacy and safety of pegcetacoplan |
Submitted results | The ICER for danicopan plus C5i compared to C5i monotherapy was $1,232,033 per QALY gained (incremental costs: $1,736,855; incremental QALYs: 1.41) Danicopan plus C5i was dominant compared to pegcetacoplan (i.e., higher QALYs and lower costs). |
Key limitations |
|
CDA-AMC reanalysis results |
|
BTH = breakthrough hemolysis; C5i = complement component 5 inhibitor; CDA-AMC = Canada’s Drug Agency; EVH = extravascular hemolysis; ICER = incremental cost-effectiveness ratio; MAIC = matching-adjusted indirect comparison; PNH = paroxysmal nocturnal hemoglobinuria; QALY = quality-adjusted life-year; t.i.d. = 3 times daily; WTP = willingness to pay.
aClinical response to prompt a dose increase to 200 mg 3 times daily is defined as being required if a patient’s hemoglobin level has not increased by at least 2 g/dL after 4 weeks of therapy, if a patient required a transfusion within the previous 4 weeks, or to achieve an appropriate hemoglobin response based on clinical judgment.
Conclusions
Based on the Canda’s Drug Agency (CDA-AMC) clinical appraisal of the ALPHA trial, at 12 weeks of treatment, danicopan add-on therapy likely increased hemoglobin (Hb) levels from baseline, the proportion of patients with transfusion avoidance and may result in an increase in FACIT-F scores; however, danicopan add-on therapy may have little to no difference on health-related quality of life (HRQoL), and has very uncertain evidence regarding mortality and adverse events (AEs) of special interest, relative to complement component 5 inhibitor (C5i) monotherapy. However, there is an absence of head-to-head clinical evidence comparing danicopan to pegcetacoplan for the treatment of paroxysmal nocturnal hemoglobinuria (PNH). The limitations associated with the matching-adjusted indirect comparisons (MAICs) overall did not allow for firm conclusions on the relative effectiveness or safety of danicopan plus C5i therapy relative to pegcetacoplan for the included outcomes (change from baseline in Hb levels, absolute reticulocyte count, lactate dehydrogenase [LDH], the Functional Assessment of Chronic Illness Therapy [FACIT]–Fatigue, breakthrough hemolysis [BTH], AEs, and transfusion avoidance). Additionally, the MAICs did not assess several outcomes included in the pharmacoeconomic model such as iron overload, proportion of patients with Hb greater or less than 9.5 g/dL, or BTH by severity, which also limits the generalizability of this indirect comparison to the pharmacoeconomic analysis. The infeasibility of an MAIC would not be an appropriate justification for relying on results of a naive comparison, which would be largely uninformative and subject to substantial limitations, and therefore, was not included in the CDA-AMC clinical appraisal. As such, the indirect evidence submitted by the sponsor was insufficient to draw conclusions about the comparative efficacy or safety of danicopan plus C5i relative to pegcetacoplan.
The sponsor’s base case suggested that danicopan add-on is more costly and more effective than C5i monotherapy (associated with an incremental cost-effectiveness ratio [ICER] of $1.2 million per quality-adjusted life-year [QALY]) and dominated pegcetacoplan (i.e., less costly and more effective). In the CDA-AMC base case, pegcetacoplan was no longer dominated by danicopan plus C5i when assuming equal efficacy and safety across treatment regimens for some key parameters. Pegcetacoplan was associated with an ICER of $113,166 per QALY compared to C5i monotherapy. Danicopan was the next best treatment option in the efficiency frontier, and compared to pegcetacoplan, associated with an ICER of $7,056,575 per QALY (incremental QALYs gains = 0.23; incremental cost of $1,606,562; 0% probability of being cost-effective at a willingness-to-pay [WTP] threshold of $50,000 per QALY). However, CDA-AMC was unable to address differences in time to discontinuation within the sponsor’s model structure, which does not allow the QALY estimates for danicopan and pegcetacoplan to be equal.
There is uncertainty associated with the AE profile of the treatments. In the model, BTH events are associated with utility decrements and pegcetacoplan dose increases, which alone can change the relative cost-effectiveness of danicopan. When considering alternative assumptions around the risk of severe BTH events with pegcetacoplan (10-fold higher, as assumed by the sponsor from the naive comparison) the results suggest similar results to the sponsor (danicopan dominates pegcetacoplan and results in an ICER of $1.2 million per QALY compared to C5i monotherapy). At the listed prices, a danicopan price reduction between 90.1% and 90.4% would be needed for danicopan when used in addition to a C5i to be cost-effective compared to C5i monotherapy at a WTP threshold of $50,000 per QALY. There is no robust clinical evidence to justify a price premium for danicopan plus C5i compared to pegcetacoplan in adult patients with PNH who have signs or symptoms of extravascular hemolysis (EVH). Further uncertainty remains regarding the model structure and the confidential discounts negotiated by public plans. The cost-effectiveness of danicopan add-on in third-line treatment remains unknown.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
CDA-AMC received patient input from the Canadian Association of PNH Patients and Aplastic Anemia, a nonprofit patient advocacy group in Canada dedicated to serving individuals affected by PNH. The Canadian clinical trial for danicopan had a limited enrolment size. Personal experiences were gathered from 1 patient living in Canada who was diagnosed with PNH and suffered EVH soon after starting ravulizumab. This patient suffered a near-life-threatening anemia. When this patient was enrolled in the clinical trial and treated with danicopan, fatigue, brain fog, and the patient’s physical ability to care for her infant improved. The patient felt as she did before being diagnosed with PNH. The Canadian Association of PNH Patients and Aplastic Anemia reports that despite the advancements in C5is, approximately 20% of patients with PNH continue to experience EVH, persistent anemia (manifested as fatigue or extreme fatigue) and require frequent blood transfusions while on ravulizumab or eculizumab therapy. Furthermore, the association highlights the gap and need for add-on therapies to address residual hemolysis when on currently approved C5i therapy and reports that danicopan is the first oral complement inhibitor treatment demonstrating efficacy and safety in patients with PNH as an add-on therapy. Potential side effects of danicopan include gastrointestinal symptoms and liver enzyme elevations that could require treatment regimen adjustments or additional medical management. The Canadian Association of PNH and Aplastic Anemia acknowledged pegcetacoplan as a Health Canada–approved treatment for adults with PNH but did not address how the availability of pegcetacoplan would affect the need for add-on therapies to address residual hemolysis.
Clinician input was received from the Canadian PNH network, a group of hematologists in Canada with a special interest and expertise in patients with PNH. Information was obtained via publicly available documents, congress abstracts, published literature (including the ALPHA trial), and input from members of the Canadian PNH Network. The current standard of care in Canada for patients with PNH is C5i monotherapy (eculizumab or ravulizumab). To be approved for either treatment, patients must have PNH clone equal or greater than 10%, LDH greater than 1.5 × upper limit of normal (ULN), and at least 1 significant clinical manifestation such as thrombosis, anemia, transfusion dependence, renal or respiratory failure without other explanation, or smooth muscle dystonic symptoms requiring either hospitalization or opioid analgesia. Allogeneic hematopoietic stem cell transplant is the only curative treatment for PNH, which is reserved for patients with predominant or progressive bone marrow failure (i.e., aplastic anemia), which can coincide with, precede, or follow a diagnosis of PNH. Treatment with a C5i may be recommended over transplant (due to transplant-related risk of complication and mortality) and is associated with control of intravascular hemolysis, which leads to significant improvements in quality of life, fatigue, transfusion dependence, thrombosis, and overall survival. Approximately one-third of patients with PNH treated with a C5i remain anemic and possibly still transfusion-dependent due to increased complement component 3 (C3) split products that drive EVH, mostly via receptors in the liver. Blocking complement at the proximal level with a C3 inhibitor (C3i) such as pegcetacoplan can block EVH and allow Hb to increase. Pegcetacoplan was recently approved in Canada and is available in several provinces. It requires twice-weekly subcutaneous infusion (typically self-administered) for patients with persistent anemia despite at least 6 months of C5i therapy or intolerant to C5i therapy. However, approximately 30% of patients treated with pegcetacoplan have BTH due to incomplete proximal complement blockade. In such cases, intensified dosing of pegcetacoplan is needed temporarily to address the BTH. A subset of patients would benefit from danicopan as a proximal inhibitor added on to C5i to protect patients from repeated BTH and complications of intravascular hemolysis. Danicopan could also provide another option for patients who do not tolerate proximal inhibition monotherapy (i.e., pegcetacoplan) or have repeated BTH while on pegcetacoplan. Pegcetacoplan is a subcutaneous infusion, typically self-administered, which requires refrigeration. Patient treatment burden can also be reduced due to the convenience of danicopan taken as an oral therapy 3 times daily. Response to therapy for patients with PNH should first focus on controlling intravascular hemolysis. This is measured by targeting an LDH of less than 1.5 × UL. Associated with this, improvements in Hb, transfusion dependence, and reduced risk of thrombosis. An increase in Hb is an important clinical outcome for patients on danicopan as an add-on therapy, particularly for patients with suboptimal Hb response to C5i monotherapy because most patients would already have a good control of their LDH levels. Other clinical outcomes of interest include decreased fatigue, transfusion requirements, improved quality of life, and improved overall survival. Danicopan discontinuation should be considered in patients with poor compliance, elevations in liver enzymes that are severe or do not resolve, evidence of BTH, and any patients who become pregnant and/or are breastfeeding.
The drug programs provided input on the drug being reviewed through the CDA-AMC reimbursement review process by identifying issues that may affect their ability to implement a recommendation. Drug plans noted several concerns. First, whether considerations for initiation of danicopan are the standard to define EVH, and whether these are readily measurable (i.e., Hb levels, absolute reticulocyte count, and platelet count while on C5i for at least 6 months). Second, could these considerations be similar to those required to initiate therapy with pegcetacoplan and could danicopan be prescribed sooner (e.g., patients are required to be on a C5i for only 3 months before pegcetacoplan initiation). Third, could danicopan be added to pegcetacoplan and would there be concerns with this combination. Fourth, continuation requirements (i.e., the scheduling and bloodwork specifics) and discontinuation considerations (i.e., the definition of lack of response, and the definition of fixed duration) need clarification. Fifth, could patients on pegcetacoplan be switched back to a C5i with danicopan added on (instead of switched back to C5i monotherapy). Last, special implementation issues of concern include additional costs associated with vaccinations and antibiotic requirements before initiation of danicopan.
Several concerns were addressed in the sponsor’s model:
Clinical effectiveness was based on improvement of Hb levels, need for transfusions, BTH events, AEs, and dose adjustments.
Cost of vaccinations and antibiotics required before initiating danicopan were incorporated into the model.
CDA-AMC was unable to address the following concerns raised from input:
The efficacy of danicopan given in combination with pegcetacoplan, or as add-on therapy to C5i posttreatment with pegcetacoplan could not be evaluated directly due to the structure of the model not allowing sequential treatment, and the lack of data for this scenario.
The cost-effectiveness of initiating danicopan sooner than 6 months of being on a C5i could not be addressed.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of danicopan in combination with a C5i (ravulizumab or eculizumab), compared with C5i monotherapy, and pegcetacoplan (coadministered with C5i therapy during a 4-week run-in period). The modelled population was based on the enrolment criteria in the ALPHA trial, which comprised adult patients with PNH who have signs or symptoms of EVH (i.e., clinically significant EVH; signs/symptoms of anemia that cannot be explained by other causes of anemia). The modelled population aligns with the reimbursement request and trial evidence (i.e., use in the second line) but the Health Canada indication is line agnostic.1
Danicopan inhibits factor D activity, which blocks C3 convertase formation, thereby reducing the production of C3 fragments that lead to the opsonization of PNH cells and subsequent EVH. Danicopan in combination with a C5i demonstrated terminal and proximal inhibition of the complement pathway to improve Hb concentrations and treat EVH in adult patients with PNH.1 Danicopan is available as 50 mg and 100 mg oral tablets. The recommended dosage of danicopan when taken in combination with a C5i is 150 mg 3 times a day, taken orally, approximately 8 hours apart (± 2 hours). Depending on clinical response (i.e., if a patient’s Hb level has not increased by at least 2 g/dL after 4 weeks of therapy, if a patient required a transfusion within the previous 4 weeks, or to achieve an appropriate Hb response based on clinical judgment), the dose can be increased to 200 mg 3 times a day, as per the product monograph.2 As estimated by the sponsor, at the submitted prices of $22.97 per 50 mg tablet and $45.95 per 100 mg tablet, the annual cost of danicopan is $85,282 per year per patient. In combination with a C5i, treatment with danicopan is estimated to cost $618,485 per patient per year. The weighted annual cost of C5i monotherapy is $533,203 per patient (62% ravulizumab, 38% eculizumab, not account for up-dosing). Pegcetacoplan is available as a 1,080 mg/20 mL single-dose vial for subcutaneous infusion. The recommended dose of pegcetacoplan is 1,080 mg twice weekly,3 but the dosage can be increased to 1,080 mg every third day if the patient’s LDH levels rise to greater than 2 × ULN (i.e., a sign of BTH). Pegcetacoplan is given to patients for the first 4 weeks in addition to their current dose of C5i treatment. At the public prices, the annual cost of pegcetacoplan is $518,655. When accounting for run-in period costs of C5i treatment, up-dosing of pegcetacoplan, and discontinuation due to BTH, the weighted annual cost of pegcetacoplan was estimated by the sponsor as $505,523 per patient.4
The main clinical outcomes modelled were Hb levels and transfusion. Secondary outcomes were BTH events, AEs, the average time spent in the “transfusion” health state, and transfusion-related iron overload. The economic outcome of interest was QALYs over a lifetime horizon (i.e., 45.7 years) from the perspective of the Canadian public health care payer. Costs and QALYs were discounted at 1.5% annually.1
Model Structure
The sponsor submitted a Markov model with 4 states: “low Hb (no transfusion),” “moderate Hb (no transfusion),” “transfusion,” and “death” (Figure 1). The low Hb (no transfusion) state was defined as patients with a Hb level less than 9.5 g/dL and not currently receiving a transfusion. The moderate Hb (no transfusion) state was defined as patients with a Hb level of 9.5 g/dL or greater and not currently receiving a transfusion. The transfusion state was defined as patients currently receiving a transfusion.1 The model used a 4-week cycle length and patients could die from any health state at any cycle. All patients entered the model in the low Hb state where they could remain or transition between any other health state except the death state (absorbing state). Patients were assumed to receive a PNH-EVH treatment (danicopan plus C5i, pegcetacoplan, or C5i monotherapy) throughout the entire model time horizon.1
Patients may experience a BTH event while in any health state, assumed to occur at the midpoint of each cycle. For patients treated with pegcetacoplan, BTH events are associated with increasing the pegcetacoplan dose, increasing the frequency of drug administration, or discontinuation of pegcetacoplan and return to C5i monotherapy (Figure 2). However, in the model, if a BTH event occurred for those patients already on the maximum pegcetacoplan dose, patients remained on the maximum pegcetacoplan dose for the rest of the time horizon (unless a background treatment change occurred). Patients receiving danicopan plus C5i did not experience dose changes due to BTH events. During danicopan or pegcetacoplan treatment, patients could experience a background treatment change unrelated to BTH while in any health state (i.e., danicopan dose increase, or discontinue danicopan or pegcetacoplan and return to C5i monotherapy). An increase in the drug dose or frequency only impacts the cost of treatments, but not the probabilities of transition between states, or any other outcomes (i.e., probabilities of BTH event, AEs, utility values, and so forth).1 Patients receiving C5i monotherapy did not experience up-dosing or discontinuation in the model (due to BTH or as a background dose change).
Patients switching to C5i monotherapy after danicopan or pegcetacoplan remained on C5i monotherapy until death or the end of the model horizon and assumed the same transition and event probabilities as those who started treatment with C5i monotherapy.1
In each cycle, patients are at risk of serious AEs irrespective of their health states. Additionally, in the “transfusion” health state, patients are at risk of iron overload which may require treatment with iron chelation or other therapy.1
Model Inputs
The baseline patient characteristics in the sponsor’s model were informed by the ALPHA trial and considered representative of the patients with PNH in Canadian clinical practice (mean age = 54.30 years; 58.73% female, 41.27% male).1
The clinical efficacy and safety of danicopan plus C5i and C5i monotherapy were informed by the ALPHA trial data (an ongoing phase III, randomized, double-blind, placebo-controlled trial). The study design consisted of treatment period 1 (TP1; 12 weeks), treatment period 2 (TP2; 12 weeks), and a long-term extension period (LTE; 1 year and optional second year). At the September 2022 data cut-off, a subset of patients completed 48 weeks of the ALPHA trial.5 The clinical efficacy and safety of pegcetacoplan were informed by the PEGASUS trial (a phase III, open-label, controlled trial that compared pegcetacoplan to eculizumab monotherapy). The trial included a 4-week run-in period where all patients received pegcetacoplan plus eculizumab, the randomized control period (weeks 4 to 16), and an open-label period (weeks 17 to 48).6 The sponsor’s base case used a naive comparison. An option to use estimates derived from an MAIC analysis was available; however, the MAIC was not considered suitable by the sponsor due to the adjustable heterogeneity between the trial designs and the patient characteristics.
The health state transition probabilities for patients treated with danicopan plus C5i or C5i monotherapy (moving between low Hb, moderated Hb, and transfusion states) were calculated using a multinominal logistic regression model applied to the ALPHA trial data, divided by 3 distinct treatment periods: weeks 1 to 12, weeks 13 to 24, and week 25 to 52. The probability of being in the current health state was calculated based on the previous health state (4 weeks earlier), as well as covariates for treatment (i.e., danicopan plus C5i and placebo), treatment period, and age.1 Transition probabilities for patients treated with pegcetacoplan were directly derived from the analysis in the Hakimi et al. (2022) study which employed a similar multinomial logistic regression model to the PEGASUS trial data,7 except it included an interaction variable between treatment and visit (i.e., treatment period), which was not included in the regression model applied to the ALPHA trial data due to collinearity.7
The probability of BTH events in the model was applied in 2 periods and defined differently between the treatment arms. For patients treated with danicopan plus C5i who experienced BTH, period 1 was defined as weeks 1 to 24 (probabilities derived from 24-week data from the ALPHA trial TP1 and TP2), and period 2 was defined as weeks 25 and later (probabilities derived from the ALPHA study LTE data up to 2 years, and extrapolated to the model lifetime). For patients treated with pegcetacoplan who experienced BTH, period 1 was defined as weeks 1 to 16 (probabilities derived from weeks 4 to 16 of the PEGASUS study randomized controlled period) and period 2 was defined as weeks 17 and later (probabilities derived from the PEGASUS study open-label period [week 17 to 48] and extrapolated to the model lifetime). Patients treated with C5i monotherapy were assumed to have the same BTH probabilities as those treated with danicopan plus C5i. As per the respective monographs, BTH events in patients treated with danicopan, ravulizumab, or eculizumab were not associated with dosing or frequency changes, while for those treated with pegcetacoplan these events would trigger an increase in dosing and frequency based on the pegcetacoplan product monograph and clinical expert opinion.1
Additionally, background treatment changes (i.e., independent of BTH events) were implemented in the model. For patients treated with danicopan plus C5i, 2 sets of probabilities were derived from the ALPHA trial: the probability of discontinuing danicopan to return to C5i monotherapy, and the probability of a dose increase (from 150 mg to 200 mg). These were divided into 4 specific periods: from week 1 to 12 (TP1), week 13 to 24 (TP2, crossover period), and week 25 to 52 (open-label 24-week LTE), and from week 53 and beyond the sponsor assumed no further danicopan dose escalation or discontinuation occurred. A proportion of patients treated with pegcetacoplan discontinued treatment to return to C5i only during weeks 17 to 52 and did not experience non-BTH-related dose escalation, informed by the corresponding periods from the PEGASUS trial.7 Patients receiving C5i monotherapy were assumed to not experience background treatment changes in the model.
All patients in the transfusion health state were assumed to have a treatment-dependent probability of experiencing transfusion-related iron overload. The probability of iron overload while treated with pegcetacoplan was derived from the Hakimi et al. study,7 while treated with danicopan plus C5i arm was informed by the ALPHA trial, and while treated with C5i monotherapy was assumed to be the same as for those treated with danicopan plus C5i.5 Patients who experienced iron overload while treated with C5i monotherapy were assumed to be treated with iron chelation while those treated with danicopan or pegcetacoplan were assumed to be treated with phlebotomy. Additionally, the probability of alanine aminotransferase (ALT) increase was modelled for patients treated with danicopan plus C5i and informed by AEs of grade 3 or more in the initial treatment period of the ALPHA trial. AE data were collected for both treatment arms, but ALT increase only occurred in more than 5% of patients in the danicopan plus C5i arm.5 In the PEGASUS trial, there were no grade 3 or greater AEs reported in more than 5% of the patients receiving pegcetacoplan other than hemolysis, which was accounted for with BTH events. Therefore, ALT increase was not modelled for those treated with pegcetacoplan.6
Health state utilities were estimated based on the EQ-5D-3L data directly obtained from patients treated with danicopan plus C5i and C5i monotherapy in the ALPHA trial. EQ-5D-3L scores were collected during TP1 (weeks 0 to 12), TP2 (weeks 13 to 24), and the LTE (weeks 25 to 52).5 A generalized linear model (a beta distribution with a logit link function) was utilized to incorporate data for all randomized subjects for each time period and extrapolate to the model time horizon, then further adjusted to avoid bounds of 0 and 1. These health state utility values (low Hb = 0.8181, moderate Hb = 0.8644, and transfusion = 0.7018) were applied for all treatment arms of the model.1 Additionally, the utility values predicted by the generalized linear model were adjusted for age- and sex-specific Canadian population utilities.8 Disutilities were sourced from published literature and applied in the model associated with BTH events (–0.40, from a published economic analysis with no further information on how this value was elicited),9 iron overload requiring IV infusion chelation therapy (–0.0724),10,11 and administration of eculizumab and pegcetacoplan (–0.02)12 to account for the increased frequency of IV administration. ALT increase and the administration of ravulizumab and danicopan were not assumed to incur any disutilities.
The risk of death followed the general mortality rates for the Canadian general population.13 EVH does not have a clear impact on survival. In addition, long-term data on eculizumab showed the same survival rates as the age- and gender-matched general population.9
Costs captured in the model included those associated with treatment acquisition, administration, clinical events (i.e., BTH, blood transfusion, iron overload), AEs, follow-up and monitoring, and costs related to vaccination and antibiotic therapy. Drug acquisition for danicopan, ravulizumab, and eculizumab was based on the sponsor’s submitted price and treatment costs were calculated based on the regimens reported in the ALPHA and PEGASUS trials, product monographs, and dose change assumptions.2,3,5,6,14,15 The cost of prophylactic antibiotics was sourced from the Ontario Drug Benefit Formulary and calculated based on the dose from the product monograph.16 The cost of a BTH event included the costs of stay in the general ward and intensive care units, and dialysis treatment and were sourced from the Canadian Institute for Health Information and published literature (Tomazos et al. [2020], Ferguson et al. [2021]).17-20 The blood transfusion costs and their frequency per cycle were sourced from the Coyle et al. study (2014).19,21 Iron chelation utilization was sourced from the Murray et al. (2016) study, TA778 Committee Papers, and clinical experts in Canada.11,22 The cost of chelation drugs (deferasirox and deferoxamine mesylate) was sourced from the Ontario Drug Benefit Formulary and the Association Québécoise des Pharmaciens Propriétaires.16,23 The costs of phlebotomy were sourced from Alberta Health, Job Bank Canada, and the Pettigrew et al. (2016) study.24-26 The sponsor assumed 3 phlebotomies occurred per year and annual costs were adjusted for cycle length. Follow-up and monitoring costs included general practitioner visits, hematologist services, and blood tests.27-29 The inpatient cost of managing ALT increase was sourced from the Alberta Interactive Health Data Application.30 Drug administration and vaccination costs were assumed to be covered by the manufacturer and were not included in the sponsor’s base case.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base case).4 In the sponsor submission, the deterministic total costs and total QALY results were similar in comparison to the probabilistic results (< 2% difference); however, the incremental ICER versus C5i monotherapy was higher by 18%. Pegcetacoplan was dominated in the probabilistic and deterministic results. The probabilistic findings are presented in the following.
Base-Case Results
In the sponsor’s base case (Table 3), C5i monotherapy and danicopan plus C5i remained on the cost-effectiveness efficiency frontier. Danicopan plus C5i was associated with higher costs (incremental cost = $1,736,855) and higher QALYs (incremental QALY = 1.410) compared to C5i monotherapy, resulting in a sequential ICER of $1,232,033 per QALY gained. Based on a WTP threshold of $50,000, there is a 0% probability of danicopan plus C5i being cost-effective compared to C5i monotherapy. Pegcetacoplan was more costly and less effective than danicopan plus C5i, resulting in it being dominated.
The majority of the QALY gains for danicopan plus C5i (> 91%) and pegcetacoplan (98%) accrued beyond the duration of the trials (48 weeks to 2 years open-label or LTE data, and 16 to 24 weeks of randomized data from the ALPHA and PEGASUS studies, respectively). Most of the QALYs in the model were acquired in the health state of Hb of 9.5 g/dL or greater and were based on the sponsor’s extrapolations of the trial data over the time horizon of the model (47.5 years). Key drivers of cost-effectiveness results were drug acquisition costs (> 99% of total costs). More than 86% of the danicopan plus C5i drug acquisition cost was associated with the cost of the C5i.
The sponsor’s submitted analysis is based on the publicly available prices for all drug treatments.1 Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
C5i monotherapy | 12,354,309 | 17.74 | Reference |
Danicopan plus C5i | 14,091,164 | 19.15 | 1,232,033 |
Pegcetacoplan | 14,374,559 | 18.81 | Dominated by danicopan plus C5i |
C5i = complement component 5 inhibitor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor assessed several model parameters and assumptions in deterministic scenario analyses which included: danicopan dose escalation to 200 mg for all patients, sustained discontinuation for danicopan plus C5i and pegcetacoplan, pegcetacoplan discontinuation due to BTH management, C5i monotherapy patients received phlebotomies for iron overload, using different sets of health state transition probabilities based on 10.5 g/dL cut-off for low Hb or moderate Hb health states (MAIC-derived), using different sets of utility values, eculizumab treatment dosing distributions to include 1,200 mg and 1,550 mg, including IV administration costs, including vaccination costs, reducing the time horizon, and changing the discount rate.1 In all scenario analyses, the ICERs were higher than the sponsor’s base-case analysis and pegcetacoplan continued to be dominated by danicopan plus C5i, except when assumptions were changed for discontinuation, BTH management, and treatment distribution for eculizumab. In these 3 scenarios, the danicopan plus C5i ICER when compared to pegcetacoplan ranged from $195,622 to $303,716 per QALY gained.1 If discontinuation was assumed to occur after 1 year in the scenario analysis, the pegcetacoplan arm had a higher probability of discontinuing to C5i compared to danicopan plus C5i. In the BTH management scenario, patients on the maximum pegcetacoplan dose due to BTH events discontinued to C5i (the danicopan plus C5i arm maintained the same treatment doses from the base case and did not discontinue). In the eculizumab dose distribution scenario analysis, higher doses of eculizumab were given to a proportion of patients, increasing the C5i cost in combined therapy and monotherapy drug acquisition. These scenarios led to the increased drug acquisition cost of danicopan plus C5i and removed the dominance of danicopan plus C5i over pegcetacoplan.1
The sponsor conducted a scenario analysis from a societal perspective which included additional costs associated with productivity loss (i.e., indirect costs associated with the working hours lost due to PNH symptoms and treatment by including age-specific workforce participation, and average daily wage loss). In this analysis, the ICER was $1,446,664 per QALY gained relative to C5i monotherapy. This is higher than the sponsor’s base-case analysis using a public health care payer perspective.1
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
Comparative clinical efficacy and safety versus pegcetacoplan is highly uncertain. There is an absence of head-to-head clinical evidence comparing danicopan to pegcetacoplan for the treatment of PNH. The sponsor adopted a naive comparison as the basis for the economic evaluation (deriving inputs from the ALPHA trial for danicopan plus C5i and C5i monotherapy, and from the PEGASUS trial for pegcetacoplan). The sponsor conducted indirect treatment comparisons in the form of MAICs in an attempt to provide comparative clinical effectiveness data and concluded that the assumptions required for an MAIC would not be satisfied. However, feedback from the clinical experts noted that the differences highlighted by the sponsor were not clinically meaningful as reported, although the CDA-AMC clinical appraisal noted that the trial designs still differed in ways that risked biasing the results. The clinical appraisal concluded that the infeasibility of an MAIC would not be an appropriate justification for relying on the results of a naive comparison, since a naive comparison would be largely uninformative and the information too uncertain to make firm conclusions on comparative efficacy or safety. As it would be possible with an MAIC to control for treatment effect modifiers in the 2 studies, the CDA-AMC clinical team appraised the unanchored and anchored MAICs. The MAICs concluded that there was no evidence of difference between danicopan plus C5i and pegcetacoplan for any of the included efficacy or safety outcomes (i.e., change from baseline in Hb levels, absolute reticulocyte count, LDH, Functional Assessment of Chronic Illness Therapy–Fatigue, BTH, and AEs), with the exception of transfusion avoidance in an unanchored MAIC only. However, unanchored MAICs require the assumption that all prognostic factors and treatment effect modifiers are accounted for, which is a strong assumption largely considered impossible to meet, leading to an unknown amount of bias in the effect estimate. There were also no MAICs done on several outcomes included in the pharmacoeconomic model such as iron overload, proportion of patients with Hb greater or less than 9.5 g/dL, or BTH by severity, which also limits the generalizability of this indirect comparison to the pharmacoeconomic analysis. As such, the indirect evidence submitted by the sponsor does not allow firm conclusions about the relative safety and efficacy of danicopan compared to pegcetacoplan. The implications of the sponsor’s base case using a naive comparison for some key parameters are further discussed in the following limitations.
In CDA-AMC reanalyses, danicopan and pegcetacoplan were assumed to produce equivalent results as no evidence was presented that suggests danicopan is superior to pegcetacoplan. This was conducted by assuming danicopan and pegcetacoplan have equivalent probabilities of transitioning between the health states, BTH events, and iron overload, acknowledging that any cost-effectiveness estimates remain highly uncertain (due to further limitations discussed in the following).
Limited validity in the transition probabilities between health states for different treatment arms. The sponsor’s base case used naive efficacy values to inform the transition probabilities. The danicopan plus C5i and C5i transition probabilities were generated by applying a multinomial logistic regression model to the patient-level data from the ALPHA trial. The danicopan plus C5i and C5i monotherapy probability of being in the current health state was calculated based on the previous health state, and covariates for treatment and age. The selected model partly matched the structure and covariates of the multinomial logistic regression model generated for a cost-effectiveness study of pegcetacoplan (by Hakimi et al. [2022])7 except for an interaction variable between treatment and visit, not included in the model used to derive the danicopan plus C5i and C5i monotherapy transition probabilities. The objective of including covariates in the regression equation is to adjust for the independent effect that baseline patient characteristics (as factors explaining variability in the hazard function) may have on the probability of transitioning between Hb levels and transfusion health states. Each covariate should be evaluated in a step-wise sequence to assess the effect on the transitions between health states based on ranking the resulting R2 and P values. Decisions made by the sponsor to include (and exclude) variables in the specification of the risk equation effectively impact the estimation of the rate of transition between health states. Therefore, it is unclear whether relevant variables were omitted from the risk equation, as the sponsor did not select covariates specific to the ALPHA trial. Instead, they replicated the structure and covariates from the model used for the PEGASUS trial. Consequently, the validity of the calculated transition probabilities for danicopan plus C5i and C5i monotherapy remains uncertain and potentially inappropriate. Upon the request of CDA-AMC, the sponsor provided the Akaike I information criterion (AIC), Bayesian information criterion (BIC), log-likelihood, and deviance to justify the multinomial logistic regression model for the naive comparison, MAIC, and maximized effective sample size (ESS) MAIC data. The provided statistics compare the fit between each model but do not provide reasoning for the inclusion/exclusion of covariates. In addition, the results of the fit statistics indicate that, among the 3 options available within the submitted model, the model applied to the maximized ESS MAIC data had the best fit, and the naive comparison had the most inappropriate fit. Furthermore, a naive comparison would not be recommended to compare danicopan plus C5i and pegcetacoplan transition probabilities in the absence of a head-to-head trial, as the naive comparison would have significant limitations and would not address any of the concerns raised by the sponsor in the feasibility assessment. The danicopan plus C5i and C5i monotherapy transition probabilities used a 9.5 g/dL cut-off to differentiate between low and moderate Hb levels to be in line with the ALPHA trial entry limit. The pegcetacoplan transition probabilities used a 10.5 g/dL cut-off to differentiate between low and moderate Hb levels to be in line with the PEGASUS trial entry limit. Therefore, there is a high degree of uncertainty associated with the resulting calculated transition probabilities between the ALPHA and PEGASUS trials when derived from the unweighted raw ALPHA trial efficacy data (i.e., lacking the weights to modify variables to match effect-modifying variables to the comparator trial). On the other hand, choosing the transition probabilities calculated from the adjusted data (from the MAIC or maximized ESS MAIC) would introduce further uncertainty to the economic assessment as it would have distorted the comparison between danicopan plus C5i to C5i monotherapy due to the applied weights. Based on the CDA-AMC clinical appraisal, at 12 weeks of treatment, danicopan add-on therapy likely increased Hb levels from baseline, increased the proportion of patients with transfusion avoidance, and may result in an increase in FACIT-C scores; however, danicopan therapy may have little to no difference in HRQoL and the evidence is very uncertain regarding mortality and AEs of special interest. Therefore, the naive comparison (i.e., using the unweighted raw ALPHA trial data) remained the preferred source of evidence to compare danicopan plus C5i versus C5i monotherapy for the purposes of the pharmacoeconomic analysis. Clinical experts consulted by CDA-AMC did not expect significant differences in efficacy and safety between danicopan plus C5i and pegcetacoplan based on the available data, but support both treatment options being superior to C5i monotherapy in certain patients.
In the CDA-AMC reanalyses, danicopan and pegcetacoplan were assumed to have equivalent probabilities of transitioning between the health states (“low Hb,” “moderate Hb,” and “transfusion”) based on the transition probabilities the sponsor estimated for danicopan plus C5i in the naive comparison. This was chosen to preserve the relative efficacy of danicopan plus C5i compared to C5i monotherapy observed in the ALPHA trial.
CDA-AMC explored the sponsor’s transition probabilities derived from the MAICs in a scenario analysis.
BTH event probabilities for the different treatment arms are uncertain. In the sponsor’s base case using a naive comparison, patients treated with pegcetacoplan had an approximately 10 times greater BTH event probability compared to the danicopan plus C5i arm; the clinical report did not appraise BTH probability as it was a naive comparison. This resulted in greater disutility and costs for patients treated with pegcetacoplan due to costs associated with BTH treatment and pegcetacoplan up-dosing following BTH events. The sponsor noted that the definition of BTH was different between the ALPHA and PEGASUS trials (i.e., the LDH value × ULN) and both trials had very small numbers of patients and BTH events. The CDA-AMC clinical review of the MAICs concluded that there was no evidence of difference between danicopan plus C5i and pegcetacoplan treatment for any included safety outcomes. Further, the MAICs also did not include comparative information on BTH by severity. Therefore, of the evidence appraised, there is no evidence from the clinical review report submission to support a 10-fold increase in the risk of BTH events of any severity. The CDA-AMC clinical review did not appraise the naive comparisons as these were subject to substantial limitations. The sponsor argues that danicopan is less likely to cause BTH events compared to pegcetacoplan because danicopan is used in combination with a C5i, maintaining proximal and terminal inhibition. Clinical experts consulted by CDA-AMC noted that they do not expect a difference in the risk of BTH events between patients treated with danicopan plus C5i and pegcetacoplan in clinical practice. However, when a BTH event occurs, it is plausible to be far more severe with pegcetacoplan as this drug is a proximal complement inhibitor monotherapy, which sometimes may require higher transfusion volumes. The submitted model was not designed to reflect the different severity of BTH events and associated effects on transfusion requirements. Therefore, the naive comparison of BTH probabilities likely overestimated the total cost of pegcetacoplan and underestimated the total QALYs used to calculate the ICER, which contributed to pegcetacoplan being dominated in the sponsor’s base-case results.
In the CDA-AMC reanalysis, the BTH event probability for pegcetacoplan was assumed to be the same as danicopan plus C5i based on the ALPHA trial data, as no evidence was presented that suggests danicopan is superior to pegcetacoplan.
CDA-AMC explored the sponsor’s BTH event probabilities derived from the naive comparison in a scenario analysis.
The probability of patients developing iron overload in the transfusion health state is uncertain. The CDA-AMC clinical review noted that the MAICs concluded that there was no evidence of difference between danicopan plus C5i and pegcetacoplan in transfusion avoidance, and did not include comparative information on iron overload. As such, due to the naive comparison approach to inform the sponsor’s base-case values for iron overload, results are highly uncertain. Clinical experts consulted by CDA-AMC note that the risk of iron overload during transfusion is not inherently affected by the treatment, but instead, more closely related to the volume of the transfusions. Furthermore, clinical experts note that once the patient is transfused, the risk of iron overload should reasonably be the same between treatments, unless the model had accounted for the different volumes of transfusion between treatment arms, which is not included in the submitted model. Therefore, the use of a naive comparison may lead to an overestimation of the benefit of danicopan compared to pegcetacoplan related to iron overload.
In the CDA-AMC reanalysis, the probability of developing iron overload with pegcetacoplan was assumed to be equal to danicopan plus C5i, as no evidence was presented that suggests danicopan is superior to pegcetacoplan.
CDA-AMC explored the sponsor’s iron overload probabilities derived from the naive comparison in a scenario analysis.
The model structure does not allow for a complete assumption of equivalent efficacy and safety between danicopan and pegcetacoplan. In the model, the treatment with pegcetacoplan was programmed to use a different set of health state transition probabilities for the run-in period. Additionally, the model has a different structure, including different time points, for background treatment changes between pegcetacoplan and danicopan leading to treatment discontinuation and return to C5i monotherapy. These values for time points for discontinuation and their probabilities are derived from a naive comparison. Altogether these data and model structure issues result in differences in health state membership between danicopan and pegcetacoplan. The model structure does not allow revisions to the model to consider equal QALY estimates for danicopan and pegcetacoplan, potentially biasing the cost-effectiveness in favour of danicopan.
CDA-AMC is unable to address these limitations within the submitted model.
Assumptions of the management of patients who experience iron overload are not aligned with Canadian clinical practice. The sponsor assumed that transfusion-related iron overload is managed with phlebotomy in all patients treated with danicopan plus C5i and pegcetacoplan and managed with chelation therapy in patients treated with C5i monotherapy. Clinical experts consulted by CDA-AMC note that it is highly unlikely for patients with PNH to be treated with phlebotomy because patients must have Hb levels greater than 130 g/dL and robust bone marrow activity to tolerate 500 mL of blood removal. Patients with PNH are baseline anemic and would not qualify to receive phlebotomy. Furthermore, clinicians note that once patients develop iron overload, all patients with PNH are equally likely to be treated with iron chelation independent of their PNH treatment. In the submitted model, chelation therapy is more costly than phlebotomy and associated with disutilities. Therefore, the total cost of pegcetacoplan and danicopan plus C5i may have been underestimated, and the total QALYs overestimated. Additionally, the sponsor assumed that 54.8% of patients treated with chelation therapy receive deferasirox and 45.2% receive deferoxamine mesylate based on the NICE submission for pegcetacoplan (2021) and the Cherry et al. (2012) study.10,31 Clinical experts consulted by CDA-AMC note that deferasirox is more likely to be used in Canadian settings.
In the CDA-AMC reanalysis, based on clinical expert input, all patients regardless of their PNH treatment (danicopan plus C5i, pegcetacoplan, or C5i monotherapy) who are treated for iron overload were assumed to receive iron chelation therapy, with the majority receiving deferasirox (80%).
The submitted model does not align with the indicated population or capture all aspects of the condition and its management. The approved indication for danicopan is as an add-on to ravulizumab or eculizumab for the treatment of residual hemolytic anemia due to EVH in adult patients with PNH, and therefore line agnostic. Clinical experts consulted by CDA-AMC indicated that danicopan may be used in subsequent treatment lines. The ALPHA trial was restricted to patients receiving their second line of treatment. As a result, there is no direct comparative evidence for the use of danicopan in third-line settings. The submitted model does not allow for exploring the impact of patients who had a suboptimal response on pegcetacoplan receiving danicopan plus C5i as a subsequent therapy. Pegcetacoplan is a proximal complement inhibitor, provided as a monotherapy. Clinical experts consulted by CDA-AMC noted that in clinical practice, if danicopan is publicly reimbursed, patients who have a suboptimal response to pegcetacoplan and continue to experience severe BTH events may switch to danicopan plus C5i to provide terminal and proximal inhibition of the complement pathway (instead of remaining on maximum doses of pegcetacoplan or switching back to C5i monotherapy). The total costs of danicopan treatment may have been underestimated and the impact of using either drug (pegcetacoplan or danicopan) as subsequent lines of therapy is unknown. Additionally, the sponsor’s submitted model did not explicitly account for cost and HRQoL associated with thrombosis. Input from clinician experts consulted by CDA-AMC and patients recognized thrombosis as the most devastating consequence of PNH, yet the impact of thrombosis on the overall cost-effectiveness of treatments is unknown. Further, the sponsor did not assume dosing up of danicopan due to continuous BTH events, or discontinuation of danicopan due to liver toxicity. Clinical experts consulted by CDA-AMC noted that both management strategies could occur in clinical practice and could affect the total treatment costs for danicopan in the model in different directions: underestimate costs when not accounting for up-dosing, overestimate costs when not accounting for discontinuation, and overestimate the utilities (patients switching to monotherapy would accrue lower utilities due to more transfusions and longer stay in the low Hb states).
CDA-AMC is unable to address these limitations within the submitted model and the influence of not including all aspects of the condition in the model on the cost-effectiveness results remains unknown.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Background mortality was assumed to be equal to that of the general population and there was no excess mortality risk associated with health states or complications. | Inappropriate. Clinical experts consulted by CDA-AMC noted that there is a mortality risk associated with PNH-related complications (such as BTH, thrombotic events, anemia, organ dysfunction) although the magnitude of the risk is uncertain. |
Pegcetacoplan discontinuation due to BTH events is underestimated. | Inappropriate. The sponsor states in the PEGASUS trial and real-world studies, patients may need to discontinue pegcetacoplan and return to C5i. However, in the sponsor’s base case, all patients treated with pegcetacoplan who experience BTH events have a 2-step dose escalation of pegcetacoplan and stay at the maximum dose (i.e., none of the patients discontinue pegcetacoplan due to BTH). Clinical experts consulted by CDA-AMC note that patients experiencing persistent BTH while on pegcetacoplan for more than 157 weeks will likely stay on the maximum dose of pegcetacoplan, but a portion of patients who have been on pegcetacoplan for less than 157 weeks may discontinue pegcetacoplan, possibly because patients do not want to maintain the subcutaneous injections. As a result, the drug acquisition cost for pegcetacoplan may have been overestimated. |
Cost of vaccines is assumed to be paid by the manufacturer. | Inappropriate. Clinical experts consulted by CDA-AMC noted that as the second-line treatment option for patients with PNH, manufacturers are unlikely to pay for vaccination as patients’ needs for vaccination are likely to have been covered by the Canadian publicly funded vaccination programs or other private options with the first-line treatment. This likely has a minimal effect on the overall results. |
In the ALPHA trial, the dose escalation to 200 mg to achieve an appropriate hemoglobin response based on clinical judgment was not an included criterion. | Uncertain. In the updated product monograph, the danicopan dose can be increased to 200 mg if a patient’s hemoglobin level has not increased by at least 2 g/dL after 4 weeks of therapy, if a patient required a transfusion within the previous 4 weeks, or to achieve an appropriate hemoglobin response based on clinical judgment.2 CDA-AMC notes that in the ALPHA trial, the dose escalation to 200 mg to achieve an appropriate hemoglobin response based on clinical judgment was not an included criterion. The proportion of patients who escalate their danicopan dose in the submitted model is based on data from the ALPHA trial. As such, there is uncertainty on whether the proportion of patients with an escalated danicopan dose in the model will reflect clinical practice to include clinical judgment. |
BTH = breakthrough hemolysis; C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency; PNH = paroxysmal nocturnal hemoglobinuria.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CDA-AMC reanalysis addressed several limitations within the economic model, as summarized in Table 5, which included: assuming equal transition probabilities between health states, equal BTH event probabilities, and equal probability of experiencing iron overload between danicopan plus C5i and pegcetacoplan. Also, the CDA-AMC reanalysis assumed all patients treated for iron overload receive chelation therapy regardless of their PNH treatment, with an increased proportion of patients receiving deferasirox as their chelation therapy.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Pegcetacoplan transition probability between health states equal to danicopan plus C5i | Naive comparison | Assumed equal transition probabilities used for danicopan plus C5i |
2. Pegcetacoplan BTH event probability | Naive comparison | Assumed equal to the BTH event probability used for danicopan plus C5i |
3. Pegcetacoplan iron overload probability | Naive comparison: 0.65% | Assumed equal to the iron overload probability for danicopan plus C5i: 0.47% |
4. Percent of patients receiving chelation therapy for iron overload | Danicopan plus C5i: 0% Pegcetacoplan: 0% C5i monotherapy: 100% | Danicopan plus C5i: 100% Pegcetacoplan: 100% C5i monotherapy: 100% |
5. Proportion of patients receiving deferasirox and deferoxamine mesylate as chelation therapy | Deferasirox: 54.8% Deferoxamine mesylate: 45.2% | Deferasirox: 80% Deferoxamine mesylate: 20% |
CDA-AMC base case | — | 1 + 2 + 3 + 4 + 5 |
BTH = breakthrough hemolysis; C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency.
The results of the CDA-AMC base case are presented in Table 6. The results of the CDA-AMC stepped analysis are presented in Table 14. In the CDA-AMC base case, pegcetacoplan is no longer dominated by danicopan plus C5i. Pegcetacoplan treatment resulted in an ICER of $113,166 per QALY compared to C5i monotherapy. Danicopan was the next best treatment option in the efficiency frontier, and compared to pegcetacoplan, the ICER of danicopan plus C5i was $7,056,575 per QALY (incremental QALYs gains = 0.23; incremental costs = $1,606,562). The probability that danicopan plus C5i is cost-effective at a WTP threshold of $50,000 per QALY is 0% compared to pegcetacoplan. CDA-AMC notes that even when assuming an equivalent effect on Hb levels, transfusion, and BTH events, due to remaining issues with the model structure that CDA-AMC was unable to address, it was not possible to ensure QALYs were exactly equivalent between the danicopan add-on and pegcetacoplan treatment arms. CDA-AMC advises caution on the applicability of these results as they continue to be biased in favour of danicopan.
Consistent with the sponsor’s results, the majority (approximately 91%) of the QALYs for danicopan plus C5i and pegcetacoplan were accrued beyond the duration of the trials (24 weeks of randomized data and 2 years of LTE data from the ALPHA study, and 16 weeks of randomized data and 48 weeks of open-label data from the PEGASUS study). Most of the QALYs in the model were acquired in the health state with a Hb of 9.5 g/dL or greater and were based on the sponsor’s extrapolations of the trial data over the time horizon of the model (47.5 years). Costs and QALYs are most sensitive to the assumptions concerning the risk of severe BTH events. When assuming equivalent probabilities of severe BTH events, pegcetacoplan is less costly than danicopan plus C5i (by approximately $1.6 million), and the incremental QALYs of danicopan plus C5i compared to pegcetacoplan are reduced (from 0.34 in the sponsor’s base case to 0.16 in the CDA-AMC reanalysis #2).
Table 6: Summary of the CDA-AMC Reanalysis Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY)a |
|---|---|---|---|
Sponsor base case (probabilistic) | |||
C5i monotherapy | 12,354,309 | 17.74 | Reference |
Danicopan plus C5i | 14,091,164 | 19.15 | 1,232,033 |
Pegcetacoplan | 14,374,559 | 18.81 | Dominated by danicopan plus C5i |
CDA-AMC base case (probabilistic) | |||
C5i monotherapy | 12,362,803 | 17.75 | Reference |
Pegcetacoplan | 12,496,858 | 18.94 | 113,166 |
Danicopan plus C5i | 14,103,420 | 19.17 | 7,056,575 |
C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aReference product is the least costly alternative.
Source: Sponsor’s pharmacoeconomic submission.1
Scenario Analysis Results
CDA-AMC undertook price reduction analyses based on the sponsor’s and the CDA-AMC base-case results (Table 7). At a WTP threshold of $50,000 per QALY, the CDA-AMC base case suggests that a 90.4% price reduction for danicopan would be required for the danicopan plus C5i regimen to be considered cost-effective relative to C5i monotherapy (similar to the sponsor’s results). There is no robust clinical evidence to justify a price premium for danicopan plus C5i versus pegcetacoplan. At the listed prices, a minimum price reduction of 86.8% would be required for danicopan to be similar to pegcetacoplan in terms of total costs. However, there are remaining issues with the model structure and the nature of the naive comparison, that CDA-AMC was unable to address, to fully reproduce equivalent efficacy and safety between danicopan and pegcetacoplan. Therefore, these price reduction estimates remain uncertain and may require further price reductions to ensure similar total costs between regimens.
Table 7: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost ($) | Sequential ICERs for danicopan plus C5i ($/QALY) | |
|---|---|---|---|
Price reduction | Sponsor base case | CDA-AMC reanalysis | |
No price reduction | 23 | 1,232,033 vs. C5i | 7,056,575 vs. Peg |
10% | 21 | 1,100,755 vs. C5i | 6,263,308 vs. Peg |
20% | 18 | 969,697 vs. C5i | 5,449,251 vs. Peg |
30% | 16 | 838,640 vs. C5i | 4,635,194 vs. Peg |
40% | 14 | 707,582 vs. C5i | 3,821,137 vs. Peg |
50% | 11 | 576,525 vs. C5i | 3,007,079 vs. Peg |
60% | 9 | 445,467 vs. C5i | 2,193,022 vs. Peg |
70% | 7 | 314,410 vs. C5i | 1,378,965 vs. Peg |
80% | 5 | 183,352 vs. C5i | 564,907 vs. Peg |
90% | 2 | 52,295 vs. C5i | 54,885 vs. C5i |
C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; Peg = pegcetacoplan; QALY = quality-adjusted life-year; vs. = versus.
Note: All analyses were performed probabilistically.
Additionally, CDA-AMC conducted a series of scenario analyses to explore the impact of alternative assumptions on the cost-effectiveness of danicopan plus C5i (Table 16). These scenarios analyses explored the impact of the following model parameters and assumptions on the ICER:
Set transition probabilities from the cut-off Hb of 10.5 g/dL MAIC weights
Set transition probabilities from the cut-off Hb of 10.5 g/dL MAIC maximized ESS weights
Revert the pegcetacoplan’s BTH probability to the sponsor’s original estimate (approximately 40% higher, as per sponsor original assumptions)
Revert the pegcetacoplan’s iron overload probability to the sponsor’s original estimate (10-fold higher, as per sponsor’s original assumptions)
The results ranged from being similar to the CDA-AMC base case (i.e., pegcetacoplan is no longer dominated by danicopan and the ICERs of danicopan versus pegcetacoplan ranged from $6.5 to $6.9 million per QALY gained) to similar to the sponsor’s base case (danicopan dominates pegcetacoplan and results in a ICER of $1.2 million per QALY gained compared to C5i monotherapy, with a 0% probability of being cost-effective at a WTP threshold of $50,000). This confirms that the cost-effectiveness estimates are most sensitive to the assumptions concerning the risk of severe BTH events for which evidence of comparative effectiveness remains insufficient to conclude any difference between danicopan and pegcetacoplan. In the scenario in which it was assumed a 10-fold higher probability of BTH for pegcetacoplan (scenario #4), a price reduction of 90.1% would be necessary for danicopan to be cost-effective compared to C5i monotherapy at WTP threshold of $50,000 per QALY gained.
Issues for Consideration
The modelled prices of all other comparators (e.g., danicopan, pegcetacoplan, eculizumab, and ravulizumab) are based on publicly accessible list prices and do not reflect existing confidential pricing that has been negotiated by public plans. When existing confidential discounts are considered, greater price reductions than those referenced in this report may be required to achieve cost-effectiveness.
Clinical experts noted that pegcetacoplan is the most relevant comparator to danicopan. Furthermore, clinicians note patients with a suboptimal response on pegcetacoplan are likely to receive danicopan plus C5i as a subsequent therapy. Yet, direct comparative evidence comparing danicopan to pegcetacoplan as second or third-line treatment remains lacking.
The same sponsor manufacturers danicopan and both C5i monotherapies (eculizumab and ravulizumab). Additionally, C5i is the first line of treatment, and danicopan add-on allows patients to continue on C5i therapy. The sponsor has control of all 3 drugs, potentially creating logistical and negotiation advantages for danicopan.
Overall Conclusions
Based on the CDA-AMC clinical appraisal of the ALPHA trials, at 12 weeks of treatment and relative to C5i monotherapy, danicopan add-on therapy likely increased Hb levels from baseline, increased the proportion of patients with transfusion avoidance, and may result in an increase in FACIT-F scores; however, danicopan add-on therapy may have little to no difference on HRQoL, and the evidence is very uncertain regarding mortality and AEs of special interest. There is an absence of head-to-head clinical evidence comparing danicopan to pegcetacoplan for the treatment of PNH. As it would be possible with an MAIC to control for treatment effect modifiers in the 2 studies assessing danicopan plus C5i and its relevant comparator, pegcetacoplan, the CDA-AMC clinical team appraised the unanchored and anchored MAICs. The limitations associated with the MAICs overall did not allow for firm conclusions on the relative effectiveness or safety of danicopan plus C5i therapy relative to pegcetacoplan for the included outcomes (change from baseline in Hb levels, absolute reticulocyte count, LDH, Functional Assessment of Chronic Illness Therapy–Fatigue, BTH, AEs, and transfusion avoidance). Additionally, the MAICs did not assess several outcomes included in the pharmacoeconomic model such as iron overload, proportion of patients with Hb greater or less than 9.5 g/dL, or BTH by severity, which also limits the generalizability of this indirect comparison to the pharmacoeconomic analysis. The infeasibility of an MAIC would not be an appropriate justification for relying on results of a naive comparison, which would be largely uninformative and subject to substantial limitations, and therefore, the naive comparison was not included in the CDA-AMC clinical appraisal despite forming the base case for the pharmacoeconomic model. As such, the indirect evidence submitted by the sponsor was insufficient to determine whether danicopan would be associated with different clinical outcomes relative to pegcetacoplan.
In addition to the aforementioned limitations with the clinical evidence, CDA-AMC identified several limitations with the sponsor’s economic submission that were addressed in reanalysis: CDA-AMC assumed equal health state transition probabilities, equal BTH event probabilities, and equal probability of experiencing iron overload for danicopan plus C5i and pegcetacoplan, based on the estimates derived from the ALPHA trial for patients treated with danicopan plus C5i. The rationale is that the use of MAIC-derived estimates would have distorted the comparison between danicopan plus C5i to C5i monotherapy due to the applied weights for which the trial is the preferred source of evidence. The CDA-AMC reanalysis attempts to preserve the comparison in efficacy between danicopan plus C5i versus C5i monotherapy by maintaining the data derived from the ALPHA trial data, but assumed similar efficacy and safety of danicopan plus C5i and pegcetacoplan to estimate incremental costs and QALYs, as the clinical evidence did not allow for firm conclusions on the comparative safety or efficacy. Additionally, the CDA-AMC base case assumed all patients treated for iron overload receive chelation therapy; with an increased proportion of patients receiving deferasirox as chelation therapy, according to input from clinical experts consulted by CDA-AMC. CDA-AMC was unable to address the limited validity of the health state transition probabilities for danicopan add-on and C5i monotherapy; the omission of important aspects of the condition and its management, and the model structure does not allow revisions to the model to consider equal QALY estimates for danicopan and pegcetacoplan.
The sponsor’s base case suggested that danicopan add-on is more costly and more effective than C5i monotherapy (associated with an ICER of $1.2 million per QALY) and dominated pegcetacoplan (i.e., less costly and more effective). In the CDA-AMC base case, all treatment options remained on the cost-effectiveness frontier which differed from the sponsor’s results. Pegcetacoplan was no longer dominated when assuming equal efficacy and safety across treatment regimens for some key parameters. Pegcetacoplan was associated with an ICER of $113,166 per QALY gained compared to C5i monotherapy. Danicopan was the next best treatment option in the efficiency frontier, and compared to pegcetacoplan, the ICER of danicopan plus C5i was $7,056,575 per QALY (incremental QALY gains = 0.23; incremental cost = $1,606,562; 0% probability of being cost-effective at a WTP threshold of $50,000 per QALY). At the listed prices, danicopan would require a price reduction of 90.4% to achieve cost-effectiveness compared to C5i monotherapy. However, these results remain uncertain as CDA-AMC was unable to address differences in time to discontinuation within the sponsor’s model structure, which does not allow the QALY estimates for danicopan and pegcetacoplan to be equal, and there were concerns with the validity of the health state transition probabilities derived from the ALPHA trial data.
There is uncertainty associated with the AE profile of the treatments. Scenario analyses were conducted to explore the use of the adjusted data to derive health state transition probabilities (from the MAICs), and the effects of reverting pegcetacoplan’s severe BTH probabilities and iron overload probabilities to the sponsor’s original estimates (from the naive comparison). The use of MAICs or higher iron overload (40% higher) assumptions resulted in similar results to the CDA-AMC base case (i.e., pegcetacoplan is not dominated by danicopan and the ICERs of danicopan versus pegcetacoplan ranged from $6.5 to 6.9 million per QALY gained). Considering alternative assumptions around the risk of severe BTH events with pegcetacoplan (10-fold higher, as assumed by the sponsor from the naive comparison) results in pegcetacoplan being dominated by danicopan plus C5i (similar to the sponsor’s submitted base case). The results are largely driven by utility decrements (–0.40 per BTH event) and, in the pegcetacoplan arm of the model, associated with pegcetacoplan dose increases, where patients stay on the maximum pegcetacoplan dose for the remainder of the modelled time horizon. This single parameter can change the relative cost-effectiveness of danicopan. In this scenario, at the listed prices, a price reduction of 90.1% would be needed for danicopan when used in addition to a C5i to be cost-effective compared to C5i monotherapy at a WTP threshold of $50,000 per QALY. There is no robust clinical evidence to justify a price premium for danicopan plus C5i compared to pegcetacoplan in adult patients with PNH who have signs or symptoms of EVH.
Further uncertainty remains regarding the model structure (i.e., important aspects of the condition and treatment pathways were not captured) and the confidential discounts negotiated by public plans. Moreover, when comparing the duration of follow-up in the ALPHA trials to the model’s time horizon (2 years open label versus 47.5 years), it is important to note that the majority (91%) of the QALY benefit realized by patients in the CDA-AMC base case was accrued in the posttrial period of the model based on extrapolation. Finally, the cost-effectiveness of danicopan add-on in third-line treatment remains unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Voydeya (danicopan), 50 mg and 100 mg oral film-coated tablets. Mississauga (ON): Alexion Pharma GmbH; 2024 Apr 4.
2.Voydeya (danicopan): 50 mg and 100 mg oral film-coated tablet [product monograph]. Zürich (CH): Alexion Pharma GmbH; 2024 Jul 19.
3.Empaveli (pegcetacoplan):solution, 1080 mg / 20 mL (54 mg/mL) for subcutaneous infusion use [product monograph]. Stockholm (SE): Swedish Orphan Biovitrum AB; 2022 Dec 7.
4.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Voydeya (danicopan), 50 mg and 100 mg oral film-coated tablets. Mississauga (ON): Alexion Pharma GmbH; 2024 Apr 4.
5.Clinical Study Report: ALXN2040-PNH-301. A phase 3 study of danicopan (ALXN2040) as add-on therapy to a C5 inhibitor (eculizumab or ravulizumab) in patients with paroxysmal nocturnal hemoglobinuria who have clinically evident extravascular hemolysis (EVH) [internal sponsor's report]. Boston (MA): Alexion Pharmaceuticals Inc; 2023.
6.de Latour RP, Szer J, Weitz IC, et al. Pegcetacoplan versus eculizumab in patients with paroxysmal nocturnal haemoglobinuria (PEGASUS): 48-week follow-up of a randomised, open-label, phase 3, active-comparator, controlled trial. Lancet Haematol. 2022;9(9):e648-e659. PubMed
7.Hakimi Z, Wilson K, McAughey E, et al. The cost-effectiveness, of pegcetacoplan compared with ravulizumab for the treatment of paroxysmal nocturnal hemoglobinuria, in a UK setting. J Comp Eff Res. 2022;11(13):969-985. PubMed
8.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
9.O'Connell T, Buessing M, Johnson S, Tu L, Thomas SK, Tomazos I. Cost-utility analysis of ravulizumab compared with eculizumab in adult patients with paroxysmal nocturnal hemoglobinuria. Pharmacoeconomics. 2020;38(9):981-994. PubMed
10.Cherry MG, Greenhalgh J, Osipenko L, et al. The clinical effectiveness and cost-effectiveness of primary stroke prevention in children with sickle cell disease: a systematic review and economic evaluation. Health Technol Assess. 2012;16(43):1-129. PubMed
11.National Institute for Health and Care Excellence. Pegcetacoplan for treating paroxysmal nocturnal haemoglobinuria. (Technology appraisal guidance TA778) 2022; https://www.nice.org.uk/guidance/ta778. Accessed 2023 Jul 20.
12.Ludt S, Wensing M, Szecsenyi J, et al. Predictors of health-related quality of life in patients at risk for cardiovascular disease in European primary care. PLoS One. 2011;6(12):e29334. PubMed
13.Statistics Canada. Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2024 May 9.
14.Ultomiris (ravulizumab for injection): 10 mg/mL & 100 mg/mL concentrate for solution for intravenous infusion [product monograph]. Baar (SE): Alexion Pharma Gmb, H; 2023 Oct 30.
15.Soliris (eculizumab for injection): 30 mL parenteral solution (10 mg/mL) [product monograph]. Baar (SE): Alexion Pharma GmbH; 2021 Mar 25.
16.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Aug 2.
17.Canadian Institute for Health Information. Cost of a standard hospital stay. 2023; https://www.cihi.ca/en/indicators/cost-of-a-standard-hospital-stay. Accessed 2024 Feb.
18.Care in Canadian ICUs. Ottawa (ON): Canadian Institute for Health Information; 2016: https://secure.cihi.ca/free_products/ICU_Report_EN.pdf. Accessed by sponsor, no date provided.
19.Tomazos I, Sierra JR, Johnston KM, Cheung A, Brodsky RA, Weitz IC. Cost burden of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria receiving ravulizumab versus eculizumab. Hematology. 2020;25(1):327-334. PubMed
20.Ferguson TW, Whitlock RH, Bamforth RJ, et al. Cost-utility of dialysis in Canada: hemodialysis, peritoneal dialysis, and nondialysis treatment of kidney failure. Kidney Med. 2021;3(1):20-30 e21. PubMed
21.Coyle D, Cheung MC, Evans GA. Opportunity cost of funding drugs for rare diseases: the cost-effectiveness of eculizumab in paroxysmal nocturnal hemoglobinuria. Med Decis Making. 2014;34(8):1016-1029. PubMed
22.Murray C, De Gelder T, Pringle N, Johnson JC, Doherty M. Management of iron overload in the Canadian hematology/oncology population: Implications for nursing practice. Can Oncol Nurs J. 2016;26(1):19-28. PubMed
23.Association québécoise des pharmaciens propriétaires. Price list. [Last accessed by sponsor 2024 Jan 12]. 2024.
24.My Health Alberta. Phlebotomy for too much iron. 2023; https://myhealth.alberta.ca/Health/Pages/conditions.aspx?hwid=uh1552&lang=en-ca#:~:text=Health%20professionals%20perform%20phlebotomy%20in,procedure%20takes%20about%2030%20minutes. Accessed 2024 Feb.
25.Government of Canada. Job Bank Canada - phlebotomist hourly wage (NOC33101). 2022; https://www.jobbank.gc.ca/wagereport/occupation/4220. Accessed by sponsor, no date provided.
26.Pettigrew M KPSL, Lim HJ. Comparative net cost impact of the utilization of panitumumab versus cetuximab for the treatment of patients with metastatic colorectal cancer in Canada. J Med Econ. 2016;19(2):135-147. PubMed
27.Fishman J, Wilson K, Drzewiecka A, Pochopien M, Dingli D. The cost-effectiveness of pegcetacoplan in complement treatment-naive adults with paroxysmal nocturnal hemoglobinuria in the USA. J Comp Eff Res. 2023;12(10):e230055. PubMed
28.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Toronto (ON): Ontario Ministry of Health; 2023. Accessed 2024 Jan.
29.Schedule of benefits for laboratory services. (effective July 24, 2023). Toronto (ON): Ontario Ministry of Health; 2023: https://www.ontario.ca/files/2024-01/moh-ohip-schedule-of-benefits-laboratory-services-2024-01-24.pdf. Accessed 2024 Jan.
30.Alberta Interactive Health Data Application. Health costing. 2019; http://www.ahw.gov.ab.ca/IHDA_Retrieval/selectSubCategory.do. Accessed 2024 Feb.
31.Government of Alberta. Interactive drug benefit list. 2024; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2024 May 9.
32.Hill A, Platts PJ, Smith A, et al. The incidence and prevalence of paroxysmal nocturnal hemoglobinuria (PNH) and survival of patients in Yorkshire. Blood. 2006;108(11):985.
33.CADTH reimbursement reccomendation: pegcetacoplan (Empaveli -Sobi Canada Inc.). Can J Health Technol. 2023;3(5).
34.Kulasekararaj A, Mellor J, Earl L, et al. Pb2056: Prevalence of clinically significant extravascular hemolysis in stable C5 inhibitor-treated patients with Pnh and its association with disease control, quality of life and treatment satisfaction. HemaSphere. 2023;7(S3).
35.Pan-Canadian prescription drug data landscape. Ottawa (ON): Canadian Institute for Health Information; 2024: https://www.cihi.ca/sites/default/files/document/pan-canadian-prescription-drug-data-landscape-report-en.pdf. Accessed 2024 Aug 2.
Appendix 1: Cost Comparison Table
Table 8: CDA-AMC Cost Comparison Table for the Treatment of EVH in Adult Patients With PNH
Treatment | Strength / concentration | Form (Vial size if single use/ If multidose pen state # doses) | Price ($) | Recommended dosage | Daily cost ($)a | Annual cost ($)a |
|---|---|---|---|---|---|---|
Complement Component 3 Inhibitor Therapy | ||||||
Danicopan (TBC) | 50 mg 100 mg | tablet | 22.9750b 45.9500 | The recommended starting dose is 150 mg 3 times daily. Depending on clinical response, dose can be increased to 200 mg 3 times dailyc | 206.78 to 275.70 | 75,525 to 100,699 |
Danicopan (150 mg – 200 mg) + Ravulizumab (2,400 mg – 3,600 mg) | First yeard: 1,616.81 to 1,975.72 Subsequent yearsd: 1,507.16 to 1,836.16 | First yeard: 590,542 to 721,633 Subsequent yearsd: 550,490 to 670,658 | ||||
Danicopan (150 mg – 200 mg) + Eculizumab | First yeare: 1,701.16 to 1,770.09 Subsequent yearse: 1,637.20 to 1,706.12 | First yeare: 621,350 to 646,524 Subsequent yearse: 597,986 to 623,161 | ||||
Pegcetacoplan (Empaveli) | 54 mg/mL 1,080 mg/20 mL | 20 mL vial single-dose vial for subcutaneous infusion | 4,970.0000 | First 4 weeks: 1,080 mg twice weekly in addition to patient’s current dose of C5i Subsequent weeks: 1,080 mg twice weekly as monotherapy | 1,420.00f | 518,655f |
Pegcetacoplan + Ravulizumab (4 weeks run-in period) (3,000 mg to 3,600 mg) | 1,420.00g | 518,655 g | ||||
Pegcetacoplan + Eculizumab (4 weeks run-in period) | First yearh: 1,529.66 Subsequent weeks: 1,420.00 | First yearh: 558,707 Subsequent years: 518,655 | ||||
Complement Component 5 Inhibitor Monotherapies | ||||||
Ravulizumab (Ultomiris) | 10 mg / mL | 300 mg single-dose vial for IV infusion | 7,282.1500 | Loading dosei: 2,400 to 3,000 mg then maintenance dose starting 2 weeks after loading dose Maintenance dose: 3,000 mg to 3,600 mg once every 8 weeks thereafter | First yearj: 1,410.04 to 1,700.02 Subsequent yearsd: 1,300.38 to 1,560.46 | First yearj: 515,017 to 620,933 Subsequent yearsd: 474,965 to 569,958 |
Eculizumab (Soliris) | 10 mg / mL | 300 mg single-use vial for IV infusion | 6,675.3000 | Loading: 600 mg every 7 days for the first 4 weeks, then 900 mg for the fifth dose 1 week later Maintenance: 900 mg every 2 weeks thereafter | First yeark: 1,494.39 Subsequent yearse: 1,430.42 | First yeark: 545,825 Subsequent yearse: 522,461 |
CDA-AMC = Canada's Drug Agency.
Notes: The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
All prices are from IQVIA Delta PA (accessed May 2024), unless otherwise indicated, and do not include dispensing fees.
This table has not been copy-edited.
aAnnual and daily costs assumed 365.25 days in a year.
bSponsor’s submitted price and recommended dosage.
cThe recommended starting dose is 150 mg (one 50 mg tablet and one 100 mg tablet) 3 times daily. Depending on clinical response (if a patient’s hemoglobin level has not increased by at least 2 g/dL after 4 weeks of therapy, if a patient required a transfusion within the previous 4 weeks, or to achieve an appropriate hemoglobin response based on clinical judgment), dose can be increased to 200 mg 3 times daily.
dAssume 6.5 maintenance doses of ravulizumab.
eSubsequent year costs assume 26.1 administrations per year of eculizumab.
fCosts assumed 104.4 1080mg doses in 1 year of pegcetacoplan.
gYear 1 assume 104.4 1080mg doses of pegcetacoplan only (assumed patient would receive pegcetacoplan treatment during the last 4 weeks of the 8-week ravulizumab treatment cycle).
hYear 1 assume 2 900mg doses of eculizumab and 104.4 1080mg doses of pegcetacoplan.
iLoading dose and maintenance doses are weight based (refer to the product monograph for more options) – ranges were calculated based on doses for patients ≥ 40 kg to < 60 kg (lower range) and ≥ 100 Kg (upper range).
jYear 1 assume 1 loading dose and 6.3 maintenance doses of ravulizumab.
kYear 1 costs assume four 600 mg doses and 24.6 900 mg doses of eculizumab.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Commentsa |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Clinical experts consulted by CDA-AMC noted that thrombosis should be included as a relevant outcome |
Model has been adequately programmed and has sufficient face validity | No | The model failed to run probabilistically for the CDA-AMC Scenario Analysis 2 |
Model structure is adequate for decision problem | No | Refer to CDA-AMC limitations on model structure not allowing full assumption of equal efficacy and safety |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
CDA-AMC = Canada's Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
BTH = breakthrough hemolysis; Hb = hemoglobin; Rx = Dose; Tr = transfusion
Source: Sponsor’s pharmacoeconomic submission.1
Figure 2: Implementation of Treatment Change Probabilities Within a Cycle
C5i = complement component 5 inhibitor; BTH = breakthrough hemolysis
*Includes an additional 3 doses of pegcetacoplan in 3 days during the cycle in which the BTH event occurred.
Source: Sponsor’s pharmacoeconomic submission.1
Table 10: Danicopan Plus C5i Health State Transition Probabilities in Base Case
Beginning health state | Hb < 9.5 g/dL | Hb ≥ 9.5 g/dL | Transfusion |
|---|---|---|---|
Hb < 9.5 g/dL | 0.4299 | 0.5401 | 0.0301 |
Hb ≥ 9.5 g/dL | 0.0634 | 0.9217 | 0.0149 |
Transfusion | 0.1744 | 0.7573 | 0.0683 |
Hb = hemoglobin
Source: Sponsor’s pharmacoeconomic submission. Derived from the ALPHA trial by applying a multinomial logistic regression model1
Table 11: Pegcetacoplan Health State Transition Probabilities in Base Case
Beginning health state | Hb < 9.5 g/dL | Hb ≥ 9.5 g/dL | Transfusion |
|---|---|---|---|
Hb < 9.5 g/dL | 0.4370 | 0.4900 | 0.0730 |
Hb ≥ 9.5 g/dL | 0.0310 | 0.9660 | 0.0030 |
Transfusion | 0.2660 | 0.6120 | 0.1220 |
Hb = hemoglobin
Source: Sponsor’s pharmacoeconomic submission. Obtained from Hakimi (2022),6 which derived them from the PEGASUS trial by applying a multinomial logistic regression model1
Table 12: Per Model Cycle Probability of BTH Events in Base Case
Drug | Period 1 | Period 2 | Period description | Source |
|---|---|---|---|---|
Danicopan + C5i | 0% | 0.24% | Week 1 to 24 / Week 25+ | ALPHA trial: TP1 and TP2 (Week 0 to 24) LTE (Week 25 to 52) |
Pegcetacoplan | 2.53% | 2.67% | Week 1 to 16 / Week 17+ | PEGASUS trial Randomized controlled period (Week 4 to 16) Open-label period (Week 17 to 48) |
C5i Monotherapy | 0% | 0.24% | Week 1 to 24 / Week 25+ | Assumed same as danicopan + C5i |
C5i = complement component 5 inhibitor; TP1 = time period 1; TP2 = time period 2; LTE = long-term extension
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 13: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Danicopan + C5i | Pegcetacoplan | C5i Monotherapy |
|---|---|---|---|
Discounted LYs | |||
Total | 23.63 | 23.63 | 23.63 |
Hb < 9.5 g/dL | 3.18 | 2.35 | 9.24 |
Hb ≥ 9.5 g/dL | 18.84 | 20.14 | 5.92 |
Transfusion | 1.62 | 1.15 | 8.47 |
Discounted QALYs | |||
Total | 19.15 | 18.81 | 17.74 |
Hb < 9.5 g/dL | 2.49 | 1.80 | 7.22 |
Hb ≥ 9.5 g/dL | 15.59 | 16.27 | 4.90 |
Transfusion | 1.08 | 0.75 | 5.63 |
Discounted costs ($) | |||
Total | 14,091,164 | 14,374,559 | 12,354,309 |
Acquisition | 14,050,332 | 14,339,093 | 12,201,942 |
Administration | 0 | 0 | 0 |
Adverse events | 48 | 0 | 0 |
BTH costs | 632 | 2,998 | 167 |
Iron overload costs | 73 | 61 | 537 |
Health state-specific costs | 40,079 | 32,407 | 151,662 |
One-off costs | 0 | 0 | 0 |
BTH = breakthrough hemolysis; C5i = complement component 5 inhibitor; Hb = hemoglobin; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission, probabilistic results.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 14: Summary of the Stepped Analysis of the CDA-AMC Base-Case Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY)a |
|---|---|---|---|---|
Sponsor base case | C5i monotherapy | 12,320,000 | 17.66 | Reference |
Danicopan + C5i | 14,078,898 | 18.87 | 1,459,446 | |
Pegcetacoplan | 14,366,580 | 18.51 | Dominated by danicopan + C5i | |
1. CDA-AMC reanalysis 1: pegcetacoplan health state transition probabilities equal to danicopan + C5i | C5i monotherapy | 12,320,000 | 17.66 | Reference |
Danicopan + C5i | 14,078,898 | 18.87 | 1,459,446 | |
Pegcetacoplan | 14,369,705 | 18.43 | Dominated by danicopan + C5i | |
2. CDA-AMC reanalysis 2: pegcetacoplan BTH event probability equal to danicopan + C5i | C5i monotherapy | 12,320,000 | 17.66 | Reference |
Pegcetacoplan | 12,471,984 | 18.71 | 145,334 | |
Danicopan + C5i | 14,078,898 | 18.87 | 10,079,212 | |
3. CDA-AMC reanalysis 3: pegcetacoplan iron overload probability equal to danicopan + C5ib | C5i monotherapy | 12,320,000 | 17.66 | Reference |
Danicopan + C5ia | 14,078,898 | 18.87 | 1,459,446 | |
Pegcetacoplan | 14,366,580 | 18.51 | Dominated by danicopan + C5i | |
4. CDA-AMC reanalysis 4: % patients receiving chelation therapy for iron equal across treatmentsc | C5i monotherapy | 12,320,000 | 17.66 | Reference |
Danicopan + C5i | 14,078,920 | 18.87 | 1,459,612 | |
Pegcetacoplan | 14,366,597 | 18.51 | Dominated by danicopan + C5i | |
5. CDA-AMC reanalysis 5: increased % patients to use deferasirox as chelation therapy | C5i monotherapy | 12,320,047 | 17.66 | Reference |
Danicopan + C5i | 14,078,904 | 18.87 | 1,459,331 | |
Pegcetacoplan | 14,366,585 | 18.51 | Dominated by danicopan + C5i | |
CDA-AMC base case (deterministic) 1 + 2 + 3 + 4 + 5 | C5i monotherapya | 12,320,047 | 17.66 | Reference |
Pegcetacoplan | 12,475,142 | 18.64 | 159,416 | |
Danicopan + C5i | 14,078,929 | 18.87 | 6,903,946 | |
CDA-AMC base case (probabilistic) 1 + 2 + 3 + 4 + 5 | C5i monotherapy | 12,362,803 | 17.75 | Reference |
Pegcetacoplan | 12,496,858 | 18.93 | 113,166 | |
Danicopan + C5i | 14,103,420 | 19.17 | 7,056,575 |
C5i = complement component 5 inhibitor; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is presented both deterministically and probabilistically.
aReference product is least costly alternative.
bCDA-AMC analyses 3, 4, and 5 need to be made together in the model order to produce changes in costs (multivariate analysis).
cCDA-AMC analysis 4 results in slightly lower QALYs in danicopan + C5i and pegcetacoplan in the third and fourth decimal places, respectively, and not captured in QALYs reported in this table.
Table 15: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Parameter | Danicopan + C5i | Pegcetacoplan | C5i Monotherapy |
|---|---|---|---|
Discounted LYs | |||
Total | 23.66 | 23.66 | 23.66 |
Hb < 9.5 g/dL | 3.19 | 3.04 | 9.27 |
Hb ≥ 9.5 g/dL | 18.854 | 19.15 | 5.92 |
Transfusion | 1.61 | 1.47 | 8.47 |
Discounted QALYs | |||
Total | 19.17 | 18.94 | 17.75 |
Hb < 9.5 g/dL | 2.49 | 2.35 | 7.24 |
Hb ≥ 9.5 g/dL | 15.60 | 15.63 | 4.89 |
Transfusion | 1.07 | 0.96 | 5.62 |
Discounted costs ($) | |||
Total | 14,103,420 | 12,496,858 | 12,362,803 |
Acquisition | 14,062,659 | 12,458,739 | 12,210,640 |
Administration | 0 | 0 | 0 |
Adverse events | 48 | 0 | 0 |
BTH costs | 638 | 501 | 168 |
Iron overload costs | 113 | 103 | 591 |
Health state-specific costs | 39,963 | 37,516 | 151,404 |
One-off costs | 0 | 0 | 0 |
BTH = breakthrough hemolysis; C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency; Hb = hemoglobin; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 16: Scenario Analyses Conducted on the CDA-AMC Base-Case Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY)a |
|---|---|---|---|---|
Sponsor base case (probabilistic) | C5i monotherapy | 12,354,309 | 17.74 | Reference |
Danicopan + C5i | 14,091,164 | 19.15 | 1,232,033 | |
Pegcetacoplan | 14,374,559 | 18.81 | Dominated by danicopan + C5i | |
CDA-AMC base case (probabilistic) | C5i monotherapy | 12,335,150 | 17.72 | Reference |
Pegcetacoplana | 12,464,785 | 18.90 | 109,560 | |
Danicopan + C5i | 14,069,880 | 19.13 | 7,057,987 | |
CDA-AMC Scenario 1: 10.5 Hb MAIC: MAIC weightsc | C5i monotherapy | 12,283,058 | 18.23 | Reference |
Pegcetacoplan | 12,470,697 | 18.83 | 314,356 | |
Danicopan + C5i | 14,073,570 | 19.07 | 6,569,654 | |
CDA-AMC Scenario 2: 10.5 Hb MAIC: Maximized ESS weightsb | C5i monotherapy | 12,244,406 | 18.15 | Reference |
Pegcetacoplan | 12,464,784 | 18.58 | 506,758 | |
Danicopan + C5i | 14,066,921 | 18.83 | 6,531,222 | |
CDA-AMC Scenario 3: Sponsor’s pegcetacoplan BTH probabilityc | C5i monotherapy | 12,338,826 | 17.73 | Reference |
Danicopan + C5i | 14,074,026 | 19.13 | 1,236,283 | |
Pegcetacoplan | 14,359,823 | 18.70 | Dominated by danicopan + C5i | |
CDA-AMC Scenario 4: Sponsor’s pegcetacoplan iron overload probability probabilisticc | C5i monotherapy | 12,341,701 | 17.72 | Reference |
Pegcetacoplan | 12,475,656 | 18.90 | 113,424 | |
Danicopan + C5i | 14,078,242 | 19.13 | 6,936,496 |
BTH = breakthrough hemolysis; C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency; ESS = effective sample size; Hb = hemoglobin; ICER = incremental cost-effectiveness ratio; LY = life-year; MAIC = match adjusted indirect comparison; QALY = quality-adjusted life-year.
aReference product is least costly alternative.
bResults shown deterministically as the probabilistic model fails to execute. For comparison, the CDA-AMC base case’s deterministic ICER for danicopan vs. pegcetacoplan is $6,903,946 per QALY gained.
cBased on probabilistic analysis with 1,000 iterations.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 17: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
BIA = budget impact analysis; C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency; CUA = cost-utility analysis; EVH = extravascular hemolysis; PNH = paroxysmal nocturnal hemoglobinuria.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) estimating the incremental 3-year budget impact of reimbursing danicopan + C5i for the treatment of adult patients who have signs or symptoms of EVH (i.e., clinically significant EVH; signs/symptoms of anemia that cannot be explained by other causes of anemia) compared with C5i monotherapy and pegcetacoplan. The base case of the BIA reflects the full Health Canada indication and the reimbursement requested population. The analysis was undertaken from a Canadian public drug plan payer perspective over a 3-year time horizon using an epidemiological approach. The sponsor’s base-case analysis included drug acquisition costs of danicopan + C5i and 2 weeks of prophylactic antibiotic costs.4 Data inputs informing the BIA were obtained from literature and assumptions. Key inputs to the BIA are documented in Table 18.
Key assumptions made by the sponsor include:
60% of patients with PNH-EVH would have public plan coverage.
The weighted annual cost for the C5i monotherapy was based on the C5i monotherapy treatment distribution in the ALPHA trial (62% ravulizumab, 38% eculizumab). The BIA considered patients experiencing EVH after stable treatment with C5 inhibition. Therefore, the BIA included maintenance doses and not the initial loading doses of C5i monotherapy. The maintenance doses of C5i drugs were considered as per product monographs: ravulizumab 3,000 mg to 3,600 mg once every 8 weeks depending on patient weight (average of 6.5 doses per year) and eculizumab 900 mg every 2 weeks (average of 26.1 doses per year).
The total annual cost of pegcetacoplan was calculated assuming 70% of patients receiving pegcetacoplan were on the initial dose (twice weekly), 15% were on their first dose escalation due to BTH (every 3 days), and 15% were on their second escalation due to BTH (3 times a week), based on estimates from the PEGASUS trial. Additionally, 10% were assumed to be new patients (i.e., would receive C5i during the initial 4-week run-in period, assumed as 0.5 doses of ravulizumab or 2 doses of eculizumab), 88% were on maintenance dose, and 2% were on intensive dosing (1,080 mg dose every 24 hours for 3 doses administered in addition to increased maintenance dosing).
The weighted annual cost for danicopan was calculated assuming 29% of patients were on the 150 mg dose and 71% on the 200 mg dose, 3 times a day.
Pre-treatment vaccination costs and drug administration costs were assumed to be covered by the manufacturer (explored in a scenario analysis).
Market shares were assumptions based on input from clinicians in Canada.
Table 18: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Prevalence of PNH (per 100,000 in general population) | 1.5932 |
Proportion of PNH patients treated with C5i | 39.90%33 |
Proportion of PNH patients clinically stable on C5i and develop clinically significant EVH | 20.00%34 |
Proportion of patients with public plan coverage | 60.00% |
Number of patients eligible for drug under review | 24 / 24 / 25 |
Market uptake (3 years) | |
Uptake (reference scenario) C5i monotherapy Pegcetacoplan | 30% / 20% / 10% 70% / 80% / 90% |
Uptake (new drug scenario) Danicopan + C5i C5i Monotherapy Pegcetacoplan | 35% / 40% / 45% 30% / 20% / 10% 35% / 40% / 45% |
Cost of treatment (per patient, per year duration) | |
Danicopan + C5i C5i Monotherapy Pegcetacoplan | $609,052a $515,609b $573,828c |
C5i = Complement component 5 inhibitor; EVH = extravascular hemolysis; PNH = paroxysmal nocturnal hemoglobinuria.
aWeighted total annual cost danicopan + C5i assuming 29% of patients are on the 150mg starting dose, 71% on the 200mg maximum dose; 38% as an add-on to eculizumab, 62% as an add-on to ravulizumab (weight-based dose: assumed 30% of patients’ weigh ≥ 40 to < 60 kg, 63% weigh ≥ 60 to < 100 kg, 7% weight ≥ 100 kg).
bWeighted total annual cost of C5i monotherapy assuming 38% of patients taking eculizumab, and 62% ravulizumab (weight-based dose: assumed 30% of patients’ weigh ≥ 40 to < 60 kg, 63% weigh ≥ 60 to < 100 kg, 7% weight ≥ 100 kg).
cWeighted total annual cost of pegcetacoplan assuming that 70% of patients would remain on the initial dose (twice weekly), 15% would remain on the dose after one escalation due to their first BTH event (every 3 days), and 15% would remain on the dose after second escalation due to their second BTH event (3 times a week); 10% assumed to be new patients (i.e., would receive C5i during the initial 4-week run-in period), 88% are on maintenance dose, and 2% are on intensive dosing).
Source: Sponsor’s budget impact submission.4
Summary of the Sponsor’s BIA Results
The sponsor estimated that the reimbursement of danicopan + C5i for the treatment of adult patients who have signs or symptoms of EVH (i.e., clinically significant EVH; signs/symptoms of anemia that cannot be explained by other causes of anemia) will be associated with an incremental cost of $296,737 in year 1, $343,214 in year 2, $390,713 in year 3, for a 3-year cumulative total incremental costs of $1,030,665.4
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Drug acquisition costs are uncertain and misaligned with the pharmacoeconomic model. The sponsor calculated the danicopan + C5i total drug acquisition costs based on the assumption that 71% of patients were on the escalated dose (200 mg) in the BIA. Clinical experts consulted by CDA-AMC noted that this is likely an overestimation. On the other hand, the BIA did not account for danicopan + C5i discontinuation. This is not aligned with the assumptions in the cost-effectiveness analysis which accounts for dose escalation and discontinuation, and likely overestimated the annual cost of danicopan + C5i in the BIA. In addition, the sponsor referenced the PEGASUS trial to estimate that 30% of patients would experience BTH events while on pegcetacoplan and require dose escalation in the BIA (from a naive comparison) and did not account for non-BTH related discontinuation. The CDA-AMC clinical review concluded that the evidence was insufficient to determine whether danicopan would be associated with different clinical outcomes relative to pegcetacoplan. This may have overestimated drug acquisition cost in the BIA.
In reanalysis, CDA-AMC updated the annual drug acquisition cost to reflect the average costs from the pharmacoeconomic model from the CDA-AMC base case.
CDA-AMC conducted a scenario analysis exploring the budget impact to reflect the average costs from the pharmacoeconomic model from the scenario in which pegcetacoplan is assumed to have a higher (10-fold) BTH probability, to assess the uncertainty associated with the relative safety of the treatments.
Coverage rates are uncertain. The sponsor assumed a coverage rate of 60%. However, PNH is a rare disease and the PNH-EVH available treatments are extremely costly. Many jurisdictions also have support programs available to help cover the cost of expensive drugs for rare diseases.35 The sponsor’s approach of estimating the public coverage based on an assumption may underestimate public coverage, especially for a drug for a rare disease because it fails to account for the role public support programs play in supporting patients with expensive and rare diseases across Canada.
CDA-AMC conducted a scenario analysis exploring a 100% coverage rate.
Anticipated market share is uncertain. Clinical experts consulted by CDA-AMC deemed it reasonable to assume that the reimbursement of danicopan in the new drug scenario would displace market shares from pegcetacoplan but not from C5i monotherapy. In their clinical experience, there would be a proportion of patients contraindicated to pegcetacoplan and expect to switch to danicopan or would choose to stay on C5i monotherapy for a variety of reasons (e.g., pregnancy, breastfeeding). Also, clinical experts consulted by CDA-AMC deemed it reasonable for an equal split of the market share between pegcetacoplan and danicopan. However, the same sponsor of danicopan also controls the market for both C5i monotherapies currently available (eculizumab and ravulizumab), which may create logistical and negotiation advantages for danicopan. Additionally, since C5i is the first line of treatment, and danicopan add-on allows patients to continue on C5i therapy to maintain the terminal inhibition, it is possible that some clinicians and patients may favour the add-on therapy instead of switching to pegcetacoplan (and discontinuation of C5i monotherapy).
CDA-AMC conducted a scenario analysis exploring higher market shares for danicopan + C5i.
Price of drugs paid by public drug plans is uncertain: Both the sponsor’s and the CDA-AMC analyses are based on publicly available list prices for all comparators. Actual costs paid by public drug plans are unknown.
CDA-AMC could not address this limitation in reanalysis.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s base case by applying the estimated drug acquisition cost to patients in the BIA. Table 19 notes the assumptions used by the sponsor in comparison to those used by CDA-AMC in the reanalysis.
Table 19: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Annual treatment costs | Danicopan + C5i: $609,052 C5i monotherapy: $515,609 Pegcetacoplan: $573,828 | Danicopan + C5i: $599,404a C5i monotherapy: $519,366a Pegcetacoplan: $537,854a |
CDA-AMC base case | Reanalysis 1 | |
C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency.
aCosts were calculated as the average annual drug costs from the pharmacoeconomic model (CDA-AMC base case), run for a 3-year time horizon without discount, added of antibiotics costs.
Table 20: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 1,030,665 |
CDA-AMC base case | 1,800,996 |
CDA-AMC = Canada's Drug Agency.
The results of the CDA-AMC reanalyses are presented in summary format in Table 20 and a more detailed breakdown is presented in Table 21. In the CDA-AMC base case, the 3-year budget impact is expected to be $1,800,996 (year 1: $518,523; year 2: $599,737; year 3: $682,737) should danicopan be reimbursed.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 21). Consistent with the sponsor’s base case, the results are based on publicly available prices of the comparator treatments.
Assuming the average total annual treatment cost for pegcetacoplan aligned with the pharmacoeconomic model scenario analysis with a higher (10-fold) probability of BTH events while treated with pegcetacoplan ($580,119)
Assuming a 100% coverage rate.
Assuming a higher market share for danicopan + C5i in the new drug scenario than pegcetacoplan (2:1 ratio)
danicopan + C5i market shares for year 1: 47%; year 2: 53%; year 3: 60%;
pegcetacoplan market shares for year 1: 23%; year 2: 27%; year 3: 30%.
Table 21: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 13,091,820 | 13,391,612 | 13,694,781 | 14,001,327 | 41,087,720 |
New drug | 13,091,820 | 13,688,350 | 14,037,996 | 14,392,040 | 42,118,385 | |
Budget impact | — | 296,737 | 343,214 | 390,713 | 1,030,665 | |
CDA-AMC base case | Reference | 12,614,072 | 12,812,407 | 13,011,815 | 13,212,295 | 39,036,517 |
New drug | 12,614,072 | 13,330,930 | 13,611,552 | 13,895,031 | 40,837,513 | |
Budget impact | — | 518,523 | 599,737 | 682,737 | 1,800,996 | |
CDA-AMC scenario analysis 1: Pegcetacoplan annual treatment cost with a higher BTH probability in the CUA | Reference | 13,217,105 | 13,524,526 | 13,835,470 | 14,149,939 | 41,509,934 |
New drug | 13,217,105 | 13,686,989 | 14,023,380 | 14,363,853 | 42,074,222 | |
Budget impact | — | 162,463 | 187,909 | 213,915 | 564,287 | |
CDA-AMC scenario analysis 2: Higher coverage rates | Reference | 21,023,453 | 21,354,012 | 21,686,358 | 22,020,491 | 65,060,861 |
New drug | 21,023,453 | 22,218,216 | 22,685,920 | 23,158,386 | 68,062,521 | |
Budget impact | — | 864,204 | 999,562 | 1,137,895 | 3,001,660 | |
CDA-AMC scenario analysis 3: Higher market shares for danicopan | Reference | 12,614,072 | 12,812,407 | 13,011,815 | 13,212,295 | 39,036,517 |
New drug | 12,614,072 | 13,503,770 | 13,811,464 | 14,122,610 | 41,437,845 | |
Budget impact | — | 691,363 | 799,649 | 910,316 | 2,401,328 |
BTH = breakthrough hemolysis; C5i = complement component 5 inhibitor; CDA-AMC = Canada's Drug Agency; CUA = cost-utility analysis.
Results of the CDA-AMC scenario analyses demonstrate that the budget impact is sensitive to assumptions regarding the increased coverage rate and increased market share for danicopan + C5i. CDA-AMC reanalysis suggests that the potential budget impact may lie between $564,287 (if the pegcetacoplan BTH probability in the cost-utility analysis is assumed to be the sponsor’s estimated value) and $3,001,660 (if the coverage rate is 100%).
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.