Drugs, Health Technologies, Health Systems
Reimbursement Review
Aflibercept (Eylea HD)
Sponsor: Bayer Inc.
Therapeutic area: Diabetic Macular Edema
This multi-part report includes:
Clinical_Review
Pharmacoeconomic_Review
Clinical_Review
Abbreviations
APTC
antiplatelet trialists’ collaboration
ANCOVA
analysis of covariance
BCVA
best corrected visual acuity
CCB
Canadian Council of the Blind
CRT
central retinal thickness
CST
central subfield thickness
DME
diabetic macular edema
DRM
dose-regimen modification
DIC
deviance information criterion
ETDRS
Early Treatment Diabetic Retinopathy Study
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development and Evaluation
IFA
International Federation on Ageing
IOP
intraocular pressure
IRF
intraretinal fluid
ITC
indirect treatment comparison
LOCF
last observation carried forward
LSM
least squares mean
MID
minimal important difference
MMRM
mixed model for repeated measures
NMA
network meta-analysis
OCT
optical coherence tomography
PlGF
placental growth factor
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SE
standard error
SRF
subretinal fluid
TEAE
treatment-emergent adverse event
VEGF
vascular endothelial growth factor
VEGF-A
vascular endothelial growth factor A
VFQ
Visual Function Questionnaire
VFQ-25
25-Item Visual Function Questionnaire
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Aflibercept (Eylea HD) 8 mg/0.07 mL, solution for intravitreal injection |
Sponsor | Bayer Inc. |
Indication | For the treatment of diabetic macular edema (DME) |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard Review Aflibercept 8 mg has been submitted for participation in the Aligned Health Canada/Health Technology Assessment Review process |
NOC date | February 2, 2024 |
Recommended dose | Aflibercept 8 mg/0.07 mL is administered by intravitreal injection every month (4 weeks) for the first 3 consecutive doses and, based on the physician’s judgment of visual and/or anatomic outcomes, is followed by 8 mg/0.07 mL via intravitreal injection once up to every 16 weeks in the first year and up to 20 weeks thereafter. |
NOC = Notice of Compliance.
Introduction
Diabetic macular edema (DME) is the principal cause of vision impairment among people with diabetes,1 affecting the central region of the retina and leading to fluid accumulation and macular thickening.2 The multifactorial pathogenesis involves chronic hyperglycemia resulting in oxidative stress, retinal hypoxia, and increased levels of inflammatory cytokines like vascular endothelial growth factor (VEGF), which further compromise the integrity of the blood-retina barrier.3 This condition is prevalent among adults in Canada, with about 60,000 people experiencing DME-related vision loss.4 The highest rates are among people aged older than 60 years and among Indigenous people,5 contributing significantly to morbidity by decreasing quality of life and increasing the risk of mental health issues and social isolation.6
Current diagnostic protocols for DME involve a series of retinal imaging and visual acuity assessments, with optical coherence tomography (OCT) being a cornerstone noninvasive imaging technique for detailed retinal evaluation. The primary therapeutic strategy consists of intravitreal injections of anti-VEGF therapies that directly target the pathophysiological mechanisms underlying DME. These anti-VEGF therapies include aflibercept 2 mg, ranibizumab, brolucizumab, and faricimab. Bevacizumab is also an off-label treatment for this condition. Such therapies, recommended by several international ophthalmology societies, are vital in managing disease progression and improving vision outcomes. However, challenges such as frequent injections contribute to high treatment burden, highlighting the need for therapies that allow for extended treatment intervals. Safety concerns with these therapies include intraocular inflammation, necessitating a balance between efficacy and safety in terms of patient care. The clinical expert consulted by Canada’s Drug Agency (CDA-AMC) for the purpose of this review noted that there are different treatment strategies currently in practice for the management of DME, including a fixed-dosing regimen, an as-needed regimen, and a treat-and-extend regimen where, after initial treatment, the duration between doses is extended for as long as possible while maintaining treatment response goals.
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of aflibercept 8 mg through intravitreal injection in the treatment of adults with DME.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to the call for input and from clinical expert(s) consulted for the purpose of this review.
Patient Input
Input from the Canadian Council of the Blind (CCB); joint patient input from Fighting Blindness Canada, the CCB, Vision Loss Rehabilitation Canada, Diabetes Canada, and the International Federation on Aging (IFA); and a commentary from the IFA were summarized for this report. Overall, patients reported that DME had substantial and life-altering impacts on their daily lives, and they worried about losing vision over time. Patients reported experiencing significant emotional, psychological, and social issues. DME impacted how they completed daily tasks such as reading, using a phone, and driving, and patients described needing help to get to appointments. Although most patients expressed satisfaction with their current treatment options, a significant number described feeling anxiety or fear regarding treatments because of events that occurred after the injections. Some patients experienced notable vision complications such as scratchiness or pain in the eye; others indicated that they were unable to complete at least 1 regular activity such as watching television, reading, or driving and that they required assistance carrying out everyday tasks. Overall, patients across surveys expressed the need for treatments that reduce the impact of injections (e.g., pain) and the burden of repeated appointments as is the case with current treatments. In addition, patients living in rural communities and vulnerable populations experienced greater travel burdens (e.g., increased challenges attending appointments), contributing to missed appointments. Barriers to treatment access can potentially discourage patients from attending their appointments, resulting in vision worsening, and a consequent increase in health care expenditure, according to the patient groups. The patient groups highlighted current issues with the health care system such as surgery backlogs and the inability to overcome the backlog due to the limited number of specialists. Therefore, any treatment that reduces physical, psychological, and logistical strain on patients and the health care system would be preferred, according to the groups.
Clinician Input
Input From Clinical Expert Consulted by CDA-AMC
The clinical expert consulted by CDA-AMC underscored the burden on patients of the frequency of the treatments required to manage DME. The expert noted that the effects of most existing treatments typically do not last beyond 8 weeks, causing significant inconvenience and hindering optimal outcomes. There is a demand for therapies that allow for longer intervals between treatments to reduce treatment burden. Newer anti-VEGF drugs, like faricimab and brolucizumab, could allow intervals between treatments of up to 12 or even 16 weeks, though the safety profiles of these drugs is not as well known as those of the older ones. There remains an unmet need for a longer-lasting treatment that has an acceptable safety profile.
The expert also highlighted the potential for the use of aflibercept 8 mg as a first-line treatment for DME or as an alternative when other treatments fail to control the disease or pose too great a patient burden. Aflibercept 8 mg is suitable for use by a wide range of patients with DME, particularly those who have no experience of treatment or with DME that has responded to prior anti-VEGF treatments but who require a longer-lasting effect.
In clinical practice, visual acuity and OCT measurements, alongside fundus examinations, are critical for monitoring response to treatment. After an initial phase of monthly treatments, intervals may extend to 12 weeks and be adjusted based on treatment response. Treatment discontinuation may be necessary if there is no improvement in or worsening of the condition.
Aflibercept 8 mg should be prescribed in a clinical setting by an ophthalmologist with expertise in retinal diseases.
Clinician Group Input
Input from 6 clinician groups, the Southwestern Ontario Community Ophthalmologists, Toronto Retina Institute, the Canadian Retina Society, Retina Division of the Ottawa Hospital, the Northeastern Ontario Ophthalmology Group, and the Toronto Ophthalmologists were summarized for this review. Treatment goals highlighted were consistent across inputs, that is, to maintain vision (i.e., stabilizing visual acuity and prevent vision worsening) and to improve quality of life, while extending the duration between treatments. The clinician groups highlighted that current treatments are not curative despite targeting the underlying disease mechanism and that the extent and duration of damage to the retina may impact the ability to achieve improvement. Thus, there is an unmet need for efficacious and durable treatments that can reliably extend the treatment interval to minimize treatment burden for patients, caregivers, and the health care system. The clinician groups also highlighted the need for safer treatments because of known safety concerns related to inflammation and occlusive retinal vasculitis observed with treatment with brolucizumab. According to the clinician groups, aflibercept 8 mg may become the drug of choice for patients with no prior treatment experience, and they anticipate that it will replace aflibercept 2 mg formulation, establishing it as a new first-line treatment choice for DME. Response to treatment will be determined by assessing vision stabilization and anatomic outcomes, with eye anatomy measured via OCT scans. According to the clinician groups, factors that will impact any decisions to discontinue treatment with aflibercept 8 mg will be similar to those that apply to the aflibercept 2 mg formulation (e.g., no response or the presence of irreversible macular damage). Treatment with aflibercept 8 mg will be primarily administered in the ophthalmologist’s office, and rarely at hospital outpatient clinics, according to the clinician groups.
Drug Program Input
Input was obtained from the drug programs that participate in the Reimbursement Review process. The following were identified as key factors that could potentially impact the implementation of a recommendation for aflibercept 8 mg:
relevant comparators
considerations for initiation of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
system and economic issues.
Clinical Evidence
Systematic Review
Description of Studies
The PHOTON trial (N = 660) met the inclusion criteria for the systematic review conducted by the sponsor. The PHOTON trial was a phase II/III, active-controlled, noninferiority, multinational (138 sites, including 4 sites in Canada) trial that randomized 660 patients with DME in a 1:2:1 ratio to receive aflibercept 2 mg every 8 weeks, aflibercept 8 mg every 12 weeks, or aflibercept 8 mg every 16 weeks, respectively. The primary outcome was change from baseline in best corrected visual acuity (BCVA) measured using the Early Treatment Diabetic Retinopathy Study (ETDRS) letter score at week 48, and a key secondary outcome was change from baseline in BCVA measured using the ETDRS letter score at week 60. Other secondary and exploratory outcomes relevant to this review included the proportion of participants with no intraretinal fluid (IRF) and no subretinal fluid (SRF) in the foveal centre and the central subfieldat week 48 and week 60, the proportion of participants gaining at least 15 letters in BCVA from baseline at week 48 and 60, and vision-related quality of life at week 48 and 60. Total number of injections, treatment-emergent adverse events (TEAEs), and serious adverse events (SAEs) through week 60 were reported as harms.
The treatment arms were generally well-balanced with respect to baseline disease and demographic characteristics. Patients were similar in age across treatment arms. The mean age of patients in the aflibercept 8 mg every 16 weeks group was 61.9 years (standard deviation [SD] = 9.50 years), which was slightly younger than the mean age of patients in the aflibercept 2 mg every 8 weeks group (63.0 years; SD = 9.78 years). A numerically higher proportion of patients were male in the higher-dosage groups (aflibercept 8 mg every 12 weeks [64.0%] and every 16 weeks [60.7%]) than in the lower-dosage group (aflibercept 2 mg every 8 weeks [55.1%]). The majority of patients were white, with a numerically higher proportion in receiving aflibercept 8 mg every 16 weeks (78.5%) versus 2 mg every 8 weeks (67.1%). The mean duration of diabetes was similar across groups, and the majority of patients had type 2 diabetes. Ocular characteristics like BCVA and central retinal thickness (CRT) were also similar across groups, with marginal variations in BCVA and CRT means between the different dosage groups.
Efficacy Results
Change from Baseline in BCVA at Week 48
The change from baseline in BCVA at week 48 was the primary noninferiority end point in the PHOTON trial. The primary end point was met: treatment with aflibercept 8 mg every 12 weeks and every 16 weeks demonstrated noninferiority to aflibercept 2 mg every 8 weeks using a noninferiority margin of 4 letters. The least squares mean (LSM) changes in BCVA from baseline to week 48 were 8.1 letters (standard error [SE] = 0.61 letters) and 7.2 letters (SE = 0.71 letters) for the aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks arms, respectively, compared with 8.7 letters (SE = 0.73 letters) in the aflibercept 2 mg every 8 weeks arm. Between-group differences in LSM changes from baseline were –0.57 letters (95% confidence interval [CI], –2.26 letters to 1.13 letters; noninferiority P < 0.0001) and –1.44 letters (95% CI, –3.27 letters to 0.39 letters; noninferiority P = 0.0031) for the aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks arms, respectively, compared with the aflibercept 2 mg every 8 weeks arm. The supplementary per-protocol analysis was consistent with the main analysis.
Change from Baseline in BCVA at Week 60
The corresponding key secondary end point of change from baseline in BCVA at week 60 was met: treatment with aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks demonstrated noninferiority to aflibercept 2 mg every 8 weeks using a noninferiority margin of 4 letters, with LSM changes from baseline BCVA to week 60 of 8.5 letters (SE = 0.63 letters) and 7.6 letters (SE = 0.75 letters), respectively, compared with 9.4 letters (SE = 0.77 letters) in the aflibercept 2 mg every 8 weeks arm. Between-group differences in LSM changes from baseline were –0.88 letters (95% CI, –2.67 letters to 0.91 letters; noninferiority P = 0.0003) and –1.76 letters (95% CI, –3.71 letters to 0.19 letters; noninferiority P = 0.0122) letters for the groups receiving aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks, respectively, compared to the aflibercept 2 mg every 8 weeks group.
Proportion of Patients Gaining 15 or More ETDRS Letters at Week 60
At week 60, in the aflibercept 2 mg every 8 weeks group, 43 out of 165 patients (26.1%) gained at least 15 letters in BCVA from baseline. In the aflibercept 8 mg every 12 weeks group, 70 out of 326 patients (21.5%) showed at least a 15-letter gain. In the aflibercept 8 mg every 16 weeks group, 26 out of 163 patients (16.0%) recorded such gains. When compared to the aflibercept 2 mg every 8 weeks group, the differences in proportions of patients achieving at least a 15-letter gain were –5.01% (95% CI, –13.04% to 3.02%) for the aflibercept 8 mg every 12 weeks group and –10.78% (95% CI, –19.27% to –2.29%) for the aflibercept 8 mg every 16 weeks group. This was an exploratory end point.
Proportion of Patients With BCVA of 69 or More ETDRS Letters at Week 60
At week 60, in the aflibercept 2 mg every 8 weeks group, 100 out of 165 patients (60.6%) had a BCVA of 69 or more ETDRS letters. In the aflibercept 8 mg every 12 weeks group, 211 out of 326 patients (64.7%) had a BCVA of 69 or more ETDRS letters. In the aflibercept 8 mg every 16 weeks group, 101 out of 163 patients (62.0%) recorded such scores. When compared to the aflibercept 2 mg every 8 weeks group, the differences in the proportions of patients with BCVA of 69 or more ETDRS letters at week 60 were 4.34% (95% CI, –4.72% to 13.40%) for the aflibercept 8 mg every 12 weeks group and 1.63% (95% CI, –8.91% to 12.17%) for the aflibercept 8 mg every 16 weeks group. This was an exploratory end point.
Proportion of Patients Without Fluid in the Foveal Centre at Week 60
In terms of fluid in the foveal centre (no IRF and no SRF) at week 60, 113 out of 165 patients (68.5%) in the aflibercept 2 mg every 8 weeks group had no fluid. In contrast, in the aflibercept 8 mg every 12 weeks group, 201 out of 325 patients (61.8%) had no fluid in the foveal centre, with a difference of –5.98% (█████ ██████ ██ ████). In the aflibercept 8 mg every 16 weeks group, 94 out of 162 patients (58.0%) had no fluid in the foveal centre, resulting in a difference of –9.88 ██████ ██████ ██ █████ compared with the aflibercept 2 mg every 8 weeks group. This was an exploratory end point.
Frequency of Injections at Week 60
At week 60, 90.3% of 289 patients who completed treatment with aflibercept 8 mg every 12 weeks and 85.5% of 152 patients who completed treatment with aflibercept 8 mg every 16 weeks maintained their randomized treatment interval. This resulted in mean numbers of active injections through week 60 of 7.0 (SD = ████) and 6.0 (SD = ████), respectively, compared with 9.8 (SD = ████) in the aflibercept 2 mg every 8 weeks arm. Comparative differences were not reported.
National Eye Institute 25-Item Visual Function Questionnaire at Week 60
At week 60, the LSM increases in the National Eye Institute (NEI) 25-item Visual Function Questionnaire (VFQ-25) scores were 4.55 (SE = ████) and 3.21 (SE = ████) for patients in the aflibercept 8 mg every 12 weeks and every 16 weeks arms, respectively, compared to 3.05 (SE = ████) for patients in the aflibercept 2 mg every 8 weeks arm. Between-group differences in LSM changes from baseline were 1.50 points (95% CI, █████ ██ ████) and 0.17 points (95% CI, █████ ██ ████) for patients in the aflibercept 8 mg every 12 weeks and every 16 weeks groups, respectively, compared to patients in the aflibercept 2 mg every 8 weeks group.
Harms Results
Ocular TEAEs were reported among less than half of the enrolled patients. Specifically, 73 out of 167 patients (43.7%) receiving aflibercept 2 mg every 8 weeks experienced at least 1 ocular TEAE, as did 147 out of 328 patients (44.8%) receiving aflibercept 8 mg every 12 weeks and 73 out of 163 patients (44.8%) receiving aflibercept 8 mg every 16 weeks. At least 1 ocular treatment-emergent SAE was reported by 1 out of 167 patients (0.6%) receiving aflibercept 2 mg every 8 weeks, 2 out of 328 patients (0.6%) receiving aflibercept 8 mg every 12 weeks, and 2 out of 163 patients (1.2%) receiving aflibercept 8 mg every 16 weeks. Specific events in this category included conditions such as cataract subcapsular (1 patient receiving aflibercept 8 mg every 12 weeks), retinal detachment (1 patient receiving aflibercept 8 mg every 12 weeks), ulcerative keratitis (1 patient receiving aflibercept 2 mg every 8 weeks), and vitreous hemorrhage (1 patient receiving aflibercept 8 mg every 16 weeks).
Non-ocular SAEs were experienced by 32 out of 167 patients (19.2%) receiving aflibercept 2 mg every 8 weeks, 61 out of 328 patients (18.6%) receiving aflibercept 8 mg every 12 weeks, and 27 out of 163 patients (16.6%) receiving aflibercept 8 mg every 16 weeks.
For adverse events of special interest, in the aflibercept 2 mg every 8 weeks group, 1 out of 167 patients (0.6%) experienced intraocular inflammation, while 6 out of 167 (3.6%) experienced increased intraocular pressure (IOP), and 6 out of 167 (3.6%) underwent an antiplatelet trialists’ collaboration (APTC) event. In the aflibercept 8 mg every 12 weeks group, 4 out of 328 patients (1.2%) presented with intraocular inflammation, 7 out of 328 (2.1%) reported increased IOP, and 13 out of 328 (4.0%) experienced an APTC event. Meanwhile, in the aflibercept 8 mg every 16 weeks group, 1 out of 163 patients (0.6%) experienced intraocular inflammation, 1 out of 163 (0.6%) experienced an increase in IOP, and 9 out of 163 (5.5%) experienced an APTC event. No cases of endophthalmitis or retinal vasculitis were reported in any of the treatment groups.
In the aflibercept 2 mg every 8 weeks group, 5 out of 167 patients (3.0%) died. Specific causes of death in this group included cardiac arrest (1.2%), myocardial infarction (0.6%), diabetic metabolic decompensation (0.6%), and acute kidney injury (0.6%). In the aflibercept 8 mg every 12 weeks group, 9 out of 328 patients (2.7%) died. Specific causes of death in this group were cardiac arrest (0.6%), myocardial infarction (0.3%), COVID-19 infection (0.3%), pneumonia (0.3%), endometrial cancer (0.3%), and unknown (0.6%). In the aflibercept 8 mg every 16 weeks group, 4 out of 163 patients (2.5%). Specific causes of death in this group were cardiorespiratory arrest (0.6%), myocardial infarction (0.6%), left ventricular failure (0.6%), and sudden death (0.6%).
Critical Appraisal
The overall design of the PHOTON trial was appropriate for the objectives of the study. Randomization was stratified by baseline BCVA and geographic region, utilizing an interactive response system to maintain allocation concealment. Baseline demographic and disease characteristics and concurrent treatments were mostly evenly distributed across the treatment groups. Notable imbalances in the baseline characteristics included a higher proportion of patients who were male and white in the higher-dosage aflibercept groups than in the aflibercept 2 mg every 8 weeks group. Statistical analytical approaches were similarly appropriate. Statistical analyses, including subgroup analyses, were predefined in the study protocol and the statistical analysis plan. A hierarchical testing procedure was applied to primary and key secondary end points to control for type I error, though no such adjustment was made for week 60 outcomes. The noninferiority margin was set at 4 ETDRS letters, supported by evidence and expert consultation. Both the full analysis set (FAS) and per-protocol set analyses indicated noninferiority of aflibercept 8 mg given at 12-week or 16-week intervals. Missing data in primary and key secondary outcomes was addressed using a mixed model for repeated measures (MMRM) with sensitivity analyses employing last observation carried forward (LOCF) and other models assuming different missing data mechanisms, which corroborated the primary analysis results. In exploratory outcomes, missing data were handled through LOCF with observed cases sensitivity analysis or no sensitivity analysis. This may increase the risk of bias due to missing data in exploratory outcomes. Adjustments for type I error were accounted for in the primary and key secondary end points through a hierarchical testing procedure. However, no such adjustment was made for outcomes at week 60, which are of high clinical value. This increases the possibility of type I error in statistically significant week 60 end points.
The PHOTON trial included 4 sites in Canada. The inclusion and exclusion criteria as well as patients’ baseline characteristics were representative of patients with DME in Canada. In addition, outcomes reported in the trial are clinically important and commonly utilized in clinical practice in Canada. Nonetheless, the dosing regimen of aflibercept 2 mg every 8 weeks does not correspond with the treat-and-extend regimen practised in clinics in Canada. This discrepancy raises questions about the generalizability of the study results, particularly the frequency of injections.
There is no evidence to support the efficacy and safety of switching from other anti-VEGF drugs and no direct evidence to inform the comparative efficacy and safety of aflibercept 8 mg versus other anti-VEGF therapies.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The outcomes were finalized in consultation with expert committee members.
Table 2 presents the GRADE summary of findings for aflibercept 8 mg every 12 weeks and every 16 weeks versus aflibercept 2 mg every 8 weeks as treatment for patients with DME.
Table 2: Summary of Findings for Treatment of Patients With DME With Aflibercept 8 mg Every 12 Weeks and Every 16 Weeks Versus Aflibercept 2 mg Every 8 Weeks
Outcome and follow-up | Intervention: patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Aflibercept 2 mg q.8.w. | Aflibercept 8 mg q.12.w. or q.16.w. | Difference | |||||
Change from baseline in BCVA | |||||||
Change from baseline in BCVA, LSM (SE) Follow-up: 48 weeks (0 [worst] to 100 [best]) | Aflibercept 8 mg q.12.w.: 328 (1 RCT) | NA | 8.7 (0.73) | 8.1 (0.61) | 0.57 fewer letters (2.26 fewer to 1.13 more) | Higha | Aflibercept 8 mg q.12.w. results in little to no clinically important difference in change in BCVA when compared with aflibercept 2 mg q.8.w. |
Aflibercept 8 mg q.16.w.: 163 (1 RCT) | NA | 8.7 (0.73) | 7.2 (0.71) | 1.44 fewer letters (3.27 fewer to 0.39 more) | Higha | Aflibercept 8 mg q.16.w. results in little to no clinically important difference in change in BCVA when compared with aflibercept 2 mg q.8.w. | |
Change from baseline in BCVA, LSM (SE) Follow-up: 60 weeks (0 [worst] to 100 [best]) | Aflibercept 8 mg q.12.w.: 328 (1 RCT) | NA | 9.4 (0.77) | 8.5 (0.63) | 0.88 fewer letters (2.67 fewer to 0.91 more) | Higha | Aflibercept 8 mg q.12.w. results in little to no clinically important difference in the change in BCVA when compared with aflibercept 2 mg q.8.w. |
Aflibercept 8 mg q.16.w.: 163 (1 RCT) | NA | 9.4 (0.77) | 7.6 (0.75) | 1.76 fewer letters (3.71 fewer to 0.19 more) | Higha | Aflibercept 8 mg q.16.w. results in little to no clinically important difference in the change in BCVA when compared with aflibercept 2 mg q.8.w. | |
Proportion of patients without fluid in the foveal centreb | |||||||
Proportion of patients without fluid in the foveal centre Follow-up: 60 weeks | Aflibercept 8 mg q.12.w.: 328 (1 RCT) | 0.90 ██████ █████ | 68.5 per 100 (NR) | 61.8 per 100 (NR) | 5.98 fewer per 100 ██████ █████ ██ ████ ████ ███ ████ | Moderatec | Aflibercept 8 mg q.12.w. likely results in a decrease in the proportion of patients without fluid in the foveal centre when compared with aflibercept 2 mg q.8.w. The clinical importance of the decrease is uncertain. |
Aflibercept 8 mg q.16.w.: 163 (1 RCT) | 0.85 ██████ █████ | 68.5 per 100 (NR) | 58.0 per 100 (NR) | 9.88 fewer per 100 ██████ █████ ██ ████ ████ ███ ████ | Moderatec | Aflibercept 8 mg q.16.w. likely results in a decrease in the proportion of patients without fluid in the foveal centre when compared with aflibercept 2 mg q.8.w. The clinical importance of the decrease is uncertain. | |
Proportion of patients with ETDRS letters gainb | |||||||
Proportion of patients gaining ≥ 15 letters in BCVA from baseline Follow-up: 60 weeks | Aflibercept 8 mg q.12.w.: 328 (1 RCT) | 0.82 █████ ██ █████ | 26.1 per 100 (NR) | 21.5 per 100 (NR) | 5.01 fewer per 100 (13.04 fewer to 3.02 more per 100) | Moderatec | Aflibercept 8 mg q.12.w. likely results in a decrease in the proportion of patients gaining ≥ 15 letters from baseline when compared with aflibercept 2 mg q.8.w. The clinical importance of the decrease is uncertain. |
Aflibercept 8 mg q.16.w.: 163 (1 RCT) | 0.61 █████ ██ █████ | 26.1 per 100 (NR) | 16.0 per 100 (NR) | 10.78 fewer per 100 (19.27 fewer to 2.29 fewer per 100) | Highd | Aflibercept 8 mg q.16.w. results in a decrease in the proportion of patients gaining ≥ 15 letters from baseline when compared with aflibercept 2 mg q.8.w. The clinical importance of the decrease is uncertain. | |
Proportion of patients with BCVA ≥ 69 letters Follow-up: 60 weeks | Aflibercept 8 mg q.12.w.: 328 (1 RCT) | 1.07 █████ ██ █████ | 60.6 per 100 (NR) | 64.7 per 100 (NR) | 4.34 more per 100 (4.27 fewer to 13.40 more per 100) | Moderatec | Aflibercept 8 mg q.12.w. likely results in an increase in the proportion of patients with ≥ 69 letters when compared with aflibercept 2 mg q.8.w. The clinical importance of the increase is uncertain. |
Aflibercept 8 mg q.16.w.: 163 (1 RCT) | 1.02 █████ ██ █████ | 60.6 per 100 (NR) | 62.0 per 100 (NR) | 1.63 more per 100 (8.91 fewer to 12.17 more per 100) | Lowe | Aflibercept 8 mg q.16.w. may result in an increase in the proportion of patients with ≥ 69 letters when compared with aflibercept 2 mg q.8.w. | |
Vision-related QoL (NEI-VFQ-25)b | |||||||
Change from baseline in NEI-VFQ-25 total score, LSM (SE) points Follow-up: 60 weeks (0 [worst] to 100 [best]) | Aflibercept 8 mg q.12.w.: 328 (1 RCT) | NA | 3.05 | 4.55 ██████ | 1.50 more points █████ █████ ██ ████ ████ ███████ | Highf | Aflibercept 8 mg q.12.w. results in little to no clinically important difference in the change from baseline in vision-related QoL when compared with aflibercept 2 mg q.8.w. |
Aflibercept 8 mg q.16.w.: 163 (1 RCT) | NA | 3.05 | 3.21 ██████ | 0.17 more points █████ █████ ██ ████ ████ ███████ | Highf | Aflibercept 8 mg q.16.w. results in little to no clinically important difference in the change from baseline in vision-related QoL when compared with aflibercept 2 mg q.8.w. | |
Number of active injectionsb | |||||||
LSM (95% CI) Follow-up: 60 weeks | Aflibercept 8 mg q.12.w.: 289 (1 RCT) | NA | 9.8 | 7.0 ████ ██ ███ | 2.8 fewer injections (███ ██ █ █ ██ ███████ | Lowg | Aflibercept 8 mg q.12.w. likely results in a decrease in the frequency of injections when compared with aflibercept 2 mg q.8.w. The clinical importance of the decrease is uncertain. |
Aflibercept 8 mg q.16.w.: 152 (1 RCT) | NA | 9.8 | 6.0 ████ ██ ████ | 3.8 fewer injections ████ █████ ███ ███ ███████ | Lowg | Aflibercept 8 mg q.16.w. likely results in a decrease in the frequency of injections when compared with aflibercept 2 mg q.8.w. The clinical importance of the decrease is uncertain. | |
Ocular SAEs | |||||||
Proportion of patients with ocular SAEs Follow-up: 60 weeks | Aflibercept 8 mg q.12.w.: 328 (1 RCT) | NR | 0.6 per 100 (NR) | 0.6 per 100 (NR) | NR | Lowh | Aflibercept 8 mg q.12.w. may have similar proportion of patients with ocular SAEs when compared with aflibercept 2 mg q.8.w. |
Aflibercept 8 mg q.16.w.: 163 (1 RCT) | NR | 0.6 per 100 (NR) | 0.6 per 100 (NR) | NR | Lowh | Aflibercept 8 mg q.16.w. may have similar proportion of patients with ocular SAEs when compared with aflibercept 2 mg q.8.w. | |
BCVA = best corrected visual acuity; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; ETDRS = Early Treatment Diabetic Retinopathy Study; LSM = least squares mean; MID = minimal important difference; NA = not applicable; NEI = National Eye Institute; NR = not reported; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; QoL = quality of life; RCT = randomized controlled trial; SAE = serious adverse event; SE = standard error; VFQ-25 = 25-Item Visual Function Questionnaire.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aDid not rate down for imprecision. The threshold for a clinically important difference was considered to be 4 letters (i.e., the noninferiority margin); the point estimate and entire CI suggest little to no difference.
bNot part of predefined statistical testing in the trial.c No published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Rated down 1 level for serious imprecision as the lower bound of the CI suggests harm and the upper bound of the 95% CI suggests benefit and/or little to no difference.
dNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Did not rate down for imprecision; a between-group difference of less than the null and a CI that excludes the null suggest harm compared to aflibercept 2 mg every 8 weeks.
eNo published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects; therefore, the null was used. Rated down 2 level down for very serious imprecision as the CI is very wide and contains potential for considerable harm in the lower bound of the CI and the upper bound may suggest considerable benefit.
fDid not rate down for imprecision. Based on the literature, a 6.13-point change from the baseline in NEI-VFQ-25 total score was clinically important, the point estimate and entire CI suggest little to no difference.
gRated down 1 level for serious concerns about risk of bias due to missing outcome data. Rated down 1 level for serious indirectness because the number of injections was driven by the protocol and not reflective of how injections would be provided in practice. Did not rate down for imprecision. No published between-group MID was identified, and the clinical experts consulted by CDA-AMC were unable to estimate a threshold for clinically important effects, therefore the null was used. The point estimate, the lower bound, and the upper bound suggest benefit.
hRated down 2 levels for very serious concerns about imprecision due to very small number of events.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
Description of Studies
The sponsor-submitted indirect treatment comparison (ITC) used a Bayesian network meta-analysis (NMA) approach, under fixed-effects and random-effects models, to compare aflibercept 8 mg every 12 weeks and every 16 weeks in the treatment of patients with DME against other anti-VEGF drugs. The following outcome measures are reported here: change in BCVA, gain of 15 or more ETDRS letters, ocular adverse events, and the mean number of injections. The sponsor-submitted NMA identified relevant evidence through a systematic review approach. Depending on the outcome type, different statistical models and links were applied, including normal likelihood with an identity link for BCVA changes, binomial likelihood with a logit link for adverse events, and multinomial likelihood with a probit link for letter gains. Methodological and clinical heterogeneity were evaluated based on study and patient characteristics, with statistical heterogeneity measured using I2 statistics and network inconsistency assessed via node-splitting methods. The mean number of injections was analyzed as an absolute outcome within each intervention node but not comparatively across interventions. Missing data were imputed from external sources, and continuous and binary model inputs were adjusted for SEs derived from various statistical distributions.
Efficacy Results
A total of 17 studies were included in the NMA; 1 assessed aflibercept 8 mg, 11 assessed aflibercept 2 mg, 6 assessed ranibizumab, 2 assessed faricimab, 9 assessed laser therapy as needed, 2 assessed brolucizumab, and 2 assessed bevacizumab. Risk-of-bias assessment of the included studies in the sponsor-submitted ITC determined that 2 studies were high risk, as determined by the Cochrane risk-of-bias tool (version 2.0). The sponsor-submitted ITC did not report any specific actions taken with these studies (e.g., sensitivity analyses).
Results from the majority of comparative outcomes under the random-effects model did not exclude the null in the credible intervals, and the point estimates were, similarly, around the null. ███████ ██████████ include █████████ results in letters gained for patients receiving aflibercept 2 mg every 4 weeks compared to aflibercept 8 mg, and █████████ results for aflibercept 8 mg when compared to laser therapy in the outcome of BCVA. In the outcome of letters gained, patients receiving aflibercept 8 mg every 12 weeks showed an ████████████ response compared with those receiving aflibercept 2 mg every 4 weeks (██ ████ ███████ █ █████ ██████ and a favourable response against laser therapy ███ ████ ███████ █████ ███████ Patients receiving aflibercept 8 mg every 16 weeks showed an ████████████ response compared with those receiving aflibercept 2 mg every 8 weeks ███ ████ ███████ █████ ██████ and those receiving aflibercept 2 mg every 4 weeks ███ ████ ██████ █████ ██████, and showed a favourable odds ratio versus laser therapy ███ ████ ████ █████ ██████.
Based on predetermined injection frequency regimens, certain interventions are expected to have an average number of injections for each treatment regimen and tend to be consistent with the number of injections planned. Interventions administered on a fixed schedule did not show much variability between the planned and the actual number of injections given. Treat-and-extend and as-needed regimens are not predetermined and showed a mean number of injections between ████ ██ ████ across the interventions in the first year and between ████ ██ ████ across the interventions in the second year. Absolute noncomparative results of injection frequency show that treatment with aflibercept 8 mg every 12 weeks has a mean injection frequency of 6.00 in the first year and 3.50 in the second year, while treatment with aflibercept 8 mg every 16 weeks has a mean injection frequency of 5.00 in the first year and 2.80 in the second year.
Harms Results
The relative effect of treatments on the number of ocular adverse events did not exclude the null in any of the credible intervals, except for the serious ocular adverse event comparison with bevacizumab, which shows favourable results safety finding for aflibercept 8 mg.
For other comparisons, the 95% credible intervals were wide, suggesting that either treatment could be favoured. No other safety end point was reported.
Critical Appraisal
The systematic literature review supporting the sponsor-submitted ITC for aflibercept 8 mg in DME followed an acceptable systematic review approach. The review process was adequate for reducing the risk of bias and error in study selection and risk of bias appraisal. Two studies with a high risk of bias were identified; however, the authors did not conduct any analyses (e.g., sensitivity analyses) to investigate the impact of these studies on the results. Clinically relevant outcomes were measured, but the fixed injection regimens in the majority of included studies reduces the applicability of the findings to clinical settings in Canada, which favour treat-and-extend regimens. Despite appropriate Bayesian NMA methods, the clinical heterogeneity observed in the study populations — evidenced by variations in age, baseline visual acuity, glycemic control, and treatment history — raises concerns about the homogeneity assumptions of the NMA models. The sponsor’s statistical testing for heterogeneity identified a number of heterogenous comparisons within the network. In addition, given that many treatment effects were supported by single-trial evidence, study and baseline characteristics variability increased the possibility of bias due to effect modifiers (e.g., disease duration, baseline disease severity, and so on). The absence of comparative data for injection frequency limits the interpretability of the potential benefits of aflibercept 8 mg in reducing injection frequency versus other interventions and regimens.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Conclusions
DME is a progressive condition characterized by central vision loss as a complication of diabetes. There is an unmet need for new treatments to improve visual acuity, reduce frequency of injections, improve vision-related quality of life, and reduce adverse events. According to evidence from the PHOTON trial, aflibercept 8 mg administered every 12 weeks and every 16 weeks demonstrates noninferiority (but not superiority) to aflibercept 2 mg every 8 weeks in terms of the change in BCVA ETDRS letters from baseline at 48- and 60-weeks follow-up.
There is high-certainty evidence that the mean difference in BCVA achieved as a result of treatment with aflibercept 8 mg every 12 weeks and every 16 weeks compared to 2 mg every 8 weeks is of little to no clinical importance. Similarly, there is high-certainty evidence that aflibercept 8 mg every 12 weeks and every 16 weeks result in little to no clinically important difference in vision-related quality of life. There is moderate-certainty evidence that aflibercept 8 mg every 12 weeks and every 16 weeks likely result in little to no difference in the proportion of patients without fluid in the foveal centre compared to aflibercept 2 mg every 8 weeks. There is high-certainty and moderate-certainty evidence that aflibercept 8 mg every 16 weeks and every 12 weeks, respectively, result in a smaller proportion of patients gaining 15 ETDRS letters or more over 60 weeks compared to 2 mg aflibercept every 8 weeks, although the clinical importance of this difference is uncertain. There is moderate-certainty and low-certainty evidence that aflibercept 8 mg every 12 weeks and every 16 weeks, respectively, result in an increase in the proportion of patients with a BCVA of 69 or more letters at week 60 compared to aflibercept 2 mg every 8 weeks. Assessment of the certainty of ocular SAEs was rated as low.
There is low-certainty evidence that treatment with aflibercept 8 mg every 12 weeks and every 16 weeks results in patients receiving fewer injections than when receiving treatment with aflibercept 2 mg every 8 weeks. This is partly due to the limited generalizability of this finding as aflibercept 2 mg is administered according to a treat-and-extend regimen in clinical practice, rather than every 8 weeks, as in the trial.
Comparative efficacy findings in the ITC are insufficient, as standalone evidence, to inform the efficacy and safety of aflibercept 8 mg every 12 weeks and every 16 weeks versus other comparators. This is due to clinical and statistical heterogeneity, the imprecision in the results, as well as the lack of reporting on relevant clinical outcomes such as quality of life. Absolute noncomparative results of injection frequency suggest that aflibercept 8 mg every 12 weeks and every 16 weeks result in a smaller number of injections when compared with other interventions in the network. However, due to the lack of statistical comparison, no inference can be made as to the comparative difference in number of injections.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of aflibercept 8 mg/0.07 mL solution for intravitreal injection for the treatment of DME.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team at CDA-AMC.
DME is a vision-related, microvascular complication of diabetes (type 1 and type 2),1 and the most common cause of vision impairment (about 75% of cases), leading to blindness among working-age adults.3 DME is characterized by the thickening of the centre of the retina (macula) as a result of fluid accumulation (edema).2 The pathophysiology of DME is multifactorial,3 reportedly mediated by both angiogenic VEGF and inflammatory pathways.8-11 Persistent hyperglycemia damages the retinal blood vessels, leading to oxidative damage, retinal hypoxia, and the upregulation of inflammatory cytokines such as VEGF.12 Increased VEGF leads to increased permeability of the retinal blood vessels, causing the breakdown of the blood-retina barrier and subsequent fluid accumulation within the retina and subretinal space.13 People with DME experience blurriness and distortion of central vision, reflected as a reduction in BCVA.14 Other signs and symptoms include retinal hemorrhages, retinal detachment, colours appearing washed out or faded, changes in contrast sensitivity, impaired colour vision, gaps in vision (scotomas), and potential permanent vision loss.15-17
About 60,000 people in Canada have DME-related vision impairment, making DME 1 of the leading causes of vision loss and a significant health concern.4 Approximately 8.9% of people in Canada are diagnosed with type 1 and type 2 diabetes every year, and the reported prevalence rate is 6.6% and 26.8% among patients aged between 18 and 64 years and older than 65 years, respectively.18 An estimated 71,391 patients experience DME-associated vision impairment in Canada.19 A retrospective study conducted in Ontario in 2012 estimated the prevalence of DME in adult patients with diabetes to be 15.7% and DME-associated vision loss at 2.56%.5 Based on this study, the incidence of DME-related vision impairment was 0.37%.5 This study also revealed that more than 50% of patients with DME experiencing vision loss were older than 60 years and more than 22% of patients were Indigenous.5 Vision loss due to DME negatively impacts health-related quality of life of patients and may lead to increased social isolation, depression, anxiety, and restriction of social activities.6
DME is diagnosed based on signs and symptoms presented during eye exams that consist of standard retinal imaging techniques and vision assessments. Common retinal imaging techniques used in practice include colour fundus photography, fluorescein angiography, OCT, and OCT-angiography. OCT is a noninvasive imaging tool that provides detailed cross-sectional images of ocular structures and pathology that are analogous to histologic images.20,21 OCT is usually the first diagnostic test for people with retinal disease. Assessment of visual acuity is a standard part of the eye examination and the visual acuity of patients with DME is routinely evaluated. Visual acuity refers to the acuteness or clearness of vision, which depends on the sharpness of the retinal focus (macula) within the eye.22,23
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team at CDA-AMC.
Intravitreal injections of anti-VEGF therapies represent the current accepted standard of care for DME.23-26 Anti-VEGFs target the underlying pathophysiology of DME, decreasing vascular leakage and neovascularization. Anti-VEGF therapies are recommended for first-line use by guidelines from ophthalmology societies including the American Academy of Ophthalmology, the European Retina Society, and the Royal College of Ophthalmology in the UK, as well as Diabetes Canada.23-26 The maintenance of vision gains during anti-VEGF therapy requires constant monitoring and re-treatment, and the need for such frequent treatment and follow-up visits contributes to poor compliance and suboptimal management.27,28 The anti-VEGF therapies that are publicly reimbursed by at least 1 participating drug plan in Canada or recommended for reimbursement by CDA-AMC for DME include aflibercept 2 mg, ranibizumab, brolucizumab, faricimab, and bevacizumab; bevacizumab is not indicated for ophthalmic use but is used off-label.
The clinical expert consulted by CDA-AMC indicated that anti-VEGF drugs have completely changed DME management paradigms with the ability to improve vision by reducing the exudation, arresting choroidal neovascularization, and converting active choroidal neovascularization to fibrosis. Through several landmark pivotal clinical trials, anti-VEGF drugs have become the standard of care. Current anti-VEGF therapies include aflibercept, ranibizumab, bevacizumab, faricimab, brolucizumab, and bevacizumab (used off-label). The clinical expert noted that different treatment strategies currently in practice for the management of DME include a fixed-dosing regimen, an as-needed regimen, and a treat-and-extend regimen.28
The clinical expert consulted by CDA-AMC indicated that the cost of travelling to appointments and the burden on caregivers or family members for assistance are just some of the obstacles that limit optimal treatment outcomes for patients with DME. Therefore, drugs or treatment programs that allow less frequent appointments are an important consideration for reducing treatment burden. The newer anti-VEGF drugs, faricimab and brolucizumab, can extend the treatment interval to 12 weeks and even up to 16 weeks.27,29 However, according to the clinical expert consulted for this review, brolucizumab has been reported to be associated with a potentially higher frequency of intraocular inflammation than that observed with other anti-VEGF treatments. Although rare, the severe ocular inflammatory reactions, e.g., retinal vasculitis, can cause severe damage to the vision. Therefore, a more durable treatment with good efficacy and no increase in adverse side effects is needed.
Drug Under Review
Mechanism of Action
Aflibercept is an anti-VEGF drug that inhibits predominant signalling pathways responsible for angiogenesis and vascular leakage: vascular endothelial growth factor A (VEGF-A) and placental growth factor (PlGF).30 VEGF-A and PlGF are members of the VEGF family of proangiogenic factors that can act as potent mitogenic, chemotactic, and vascular permeability factors for endothelial cells. VEGF acts via 2 receptor tyrosine kinases, VEGFR-1 and VEGFR-2, present on the surface of endothelial cells. PlGF is a proangiogenic factor that activates VEGFR-1. Excessive activation of these receptors by VEGF-A can result in pathological neovascularization and excessive vascular permeability that is believed to contribute to vision loss in a variety of ocular diseases.
Aflibercept 8 mg/0.07 mL is administered as an intravitreal injection every month (4 weeks) for the first 3 consecutive doses and, based on the physician’s judgment of visual and/or anatomic outcomes, is followed by another 8 mg dose once up to every 16 weeks in the first year and up to 20 weeks thereafter. Treatment intervals of 1 month (4 weeks) for more than 3 consecutive doses have not been studied, and there is limited data for treatment intervals longer than 5 months (20 weeks).30
Aflibercept 8 mg/0.07 mL is indicated for the treatment of DME. The sponsor’s reimbursement request aligns with the proposed Health Canada indication. Aflibercept 8 mg was approved by the FDA on August 18, 2023 for the treatment of neovascular (wet) age-related macular degeneration, DME, and diabetic retinopathy, and is currently under review by the European Medicines Agency. Aflibercept 2 mg has previously been reviewed by CDA-AMC for the treatment of DME and macular edema secondary to central retinal vein occlusion and received a recommendation on May 7, 2015, to reimburse with conditions (i.e., aflibercept 2 mg should be listed in a manner similar to ranibizumab, and it should provide cost savings for drug plans relative to ranibizumab for the treatment of DME).31 On July 27, 2016, another recommendation to reimburse was issued by CDA-AMC for the treatment of branch retinal vein occlusion.32 Aflibercept 2 mg is funded across participating jurisdictions for DME.
Table 3 provides key characteristics of commonly used anti-VEGF treatments for DME.
Table 3: Key Characteristics of Aflibercept 8 mg, Aflibercept 2 mg, Faricimab, Ranibizumab, Bevacizumab, and Brolucizumab
Characteristic | Aflibercept 8 mg33 | Aflibercept 2 mg | Faricimab | Ranibizumab | Bevacizumaba | Brolucizumab |
|---|---|---|---|---|---|---|
Mechanism of action | VEGF inhibitor (soluble decoy receptor targets VEGF-A and PlGF) | VEGF inhibitor (soluble decoy receptor, targets VEGF-A and PlGF) | VEGF inhibitor (mAb, targets ang-2 and VEGF-A) | VEGF inhibitor (mAb, targets VEGF-A isoforms) | VEGF inhibitor (mAb, targets VEGF) | VEGF inhibitor that binds to VEGF-A isoforms (e.g., VEGF110, VEGF121, and VEGF165), thereby preventing binding of VEGF-A to its receptors VEGFR-1 and VEGFR-2. |
Indicationb | For the treatment of DME | For the treatment of DME | For the treatment of DME | For the treatment of DME | None (off-label) | For the treatment of DME |
Route of administration | Intravitreal | Intravitreal | Intravitreal | Intravitreal | Intravitreal | Intravitreal |
Recommended dose | Every 4 weeks for the first 3 doses followed by treatment intervals of up to 16 weeks in the first year and up to 20 weeks thereafter, based on visual and/or anatomic outcomes. | Every 4 weeks for the first 5 doses followed by treatment intervals every 8 weeks. The treatment interval may be maintained at 8 weeks or extended by up to 2-week increments at a time if visual and/or anatomic outcomes remain stable. If visual and/or anatomic outcomes deteriorate, the treatment interval should be shortened. | Recommended to be administered following 1 of 2 dose regimens:
| Monthly until maximum VA is achieved, confirmed by stable VA for 3 consecutive monthly assessments performed while the patient continues to receive ranibizumab treatment. Thereafter patients should be monitored monthly for VA. Treatment with monthly injections is resumed when monitoring indicates a loss of VA due to DME; treatment is continued until stable VA is reached again for 3 consecutive monthly assessments. | Bevacizumab 1.25 mg, used off-label for DME, is administered every 4 weeks for the first 3 doses followed by treatment intervals of every 8 to 12 weeks. | Every 6 weeks for the first 5 doses. Thereafter, treatment intervals may be modified based on VA or anatomic parameters. Treatment intervals of up to every 12 weeks may be considered for patients without disease activity and every 8 weeks for patients with disease activity. The intervals between 2 doses should not be less than 8 weeks. |
Serious adverse effects or safety issues |
|
|
|
|
|
|
ang-2 = angiopoietin-2; ATE = arterial thromboembolic events; DME = diabetic macular edema; IOP = intraocular pressure; mAb = monoclonal antibody; PlGF = placental growth factor; VA = visual acuity; VEGF = vascular endothelial growth factor; VEGF-A = vascular endothelial growth factor A; VEGFR-1 = vascular endothelial growth factor receptor-1; VEGFR-2 = vascular endothelial growth factor receptor-2.
aBevacizumab is used off-label in the treatment of DME.
bHealth Canada–approved indication.
cArterial thromboembolic events include nonfatal stroke, nonfatal myocardial infarction, or vascular death.
Sources: Vabysmo product monograph,34 Eylea product monograph,35 Lucentis product monograph,36 Beovu product monograph,37 Avastin product monograph.38
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups. The full original patient input(s) received by CDA-AMC have been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
Input from the CCB, a joint patient input from Fighting Blindness Canada, the CCB, Vision Loss Rehabilitation Canada, Diabetes Canada, and the IFA, and a commentary from the IFA were summarized for this report. The CCB, Fighting Blindness Canada, Vision Loss Rehabilitation Canada, and the IFA are not-for-profit organizations that cater to research and activities promoting vision health and well-being of people with DME and other vision-related conditions. Diabetes Canada is a national health charity representing millions of people in Canada affected by diabetes.
Vision loss due to DME has substantial and life-altering impacts on patients’ daily life according to the patient group inputs. Information from the joint patient input was sourced from an online survey conducted among people in Canada living with diabetic retinopathy or DME in early 2020. In total, 67 people with DME participated in the survey; most were aged between 61 and 80 years and resided in Alberta, British Columbia, Ontario, and Quebec. Survey respondents worried about vision loss worsening and the inability to carry out daily activities. The survey revealed that respondents had concerns about vision loss as a result of not getting regular injections. Patients described persistent emotional, social, and psychological challenges associated with DME, such as loneliness and feelings of isolation, and required help getting to injection appointments. When patients were asked which activities were most impacted, they emphasized the ability to read, use a phone, and drive.
More than half (56.4%) of respondents in the joint patient input survey indicated that they were currently receiving injections for DME. Common treatments outlined included ranibizumab, aflibercept, bevacizumab, and dexamethasone. There were no reported experiences with the drug dose under review. More than half (54.5%) of the respondents indicated that they were “satisfied” with their injections and that the injections helped respondents avoid losing more of their eyesight. Wait time and travel were ranked high as “difficult aspects” of receiving current treatments. Frequent injections also pose an emotional and/or physical burden for patients. Respondents reported experiencing anxiety or fear associated with the injections, emphasizing that injections into the eye can be emotionally burdensome. Some patients reported experiencing vision complications after injections, such as scratchiness or pain in the eye. A significant majority (81.8%) indicated that their current injections were “slightly painful,” 9.1% indicated that their injections were “not painful at all,” and 9.1% indicated that their injections were “painful.” Other respondents reported experiencing blurred vision that stayed until they fell asleep at night (31.6%), for 1 to 3 hours after their injections (26.3%), or for 4 to 6 hours after their injections (21.1%). Some patients said that after injections they were unable to complete at least 1 regular activity, such as watching television, reading, or driving, and that they required assistance carrying out everyday tasks. Due to these complications, the patients reported frequently requiring assistance in completing everyday tasks.
The inability to access treatments due to missed visits and transportation or logistical issues were other significant concerns, especially for patients residing in rural communities in Canada. Responses to the patient group surveys revealed that a significant number of patients (31.8%) had missed regular eye injections, with the most common reason being the inability to get assistance or someone to accompany them. The IFA highlighted that these barriers can potentially discourage patients from attending their appointments, resulting in vision worsening and increase in health care expenditure. The IFA pointed out that reduced treatment frequency could play a role in reducing logistical demands and decrease dependency on caregivers. The CCB pointed out the surgery backlogs in the health care system and the inability to reduce these because of the limited number of treating ophthalmologists or retinal specialists. According to the patient groups, a new treatment that decreases the number of patients seen by retinal specialists would free up time for surgery and other backlogs, thereby improving vision health for all patients.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
All review teams at CDA-AMC include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of DME.
Unmet Needs
The clinical expert indicated that the need for ongoing treatment at regular intervals poses the greatest challenge for the successful management of DME. The clinical expert noted that current therapies do not reliably resolve macular edema for more than 8 weeks. Patients have to be assessed and, if necessary, treated within this period to prevent macular edema recurrence and vision decline. The expert noted the cost of travelling to appointments and the burden on patients and caregivers limit optimal treatment outcomes for patients with DME. As such, a drug or treatment program that allows for less frequent visits is an important option to reduce the treatment burden. The clinical expert highlighted that the newer anti-VEGF drugs, faricimab and brolucizumab, can extend the treatment interval to 12 weeks and even up to 16 weeks. However, based on the clinical expert’s observations, brolucizumab is associated with higher frequency of intraocular inflammation than other anti-VEGF treatments. Although rare, severe ocular inflammatory reaction can cause severe damage to the vision. Therefore, the clinical expert concluded that a more durable treatment with a high efficacy and without the increase of adverse effects is an unmet need.
Place in Therapy
The clinical expert noted that the introduction of a single use syringe with long therapeutic durability is important as a treatment option to increase safety, improve compliance, and reduce patient burden. The expert indicated that aflibercept 2 mg has had a long history of use and its safety profile is well known. The clinical expert indicated that aflibercept 8 mg could be considered as first-line treatment for DME. Aflibercept 8 mg can also be considered as replacement therapy when the other anti-VEGF treatments are ineffective or optimal control is not achieved due to patient burden. The clinical expert consulted by CDA-AMC highlighted that there is no evidence to support the use of aflibercept 8 mg in combination with other treatments.
Patient Population
The clinical expert indicated that aflibercept 8 mg is suitable for all patients with DME, especially those with no experience of treatment. Aflibercept 8 mg can also be considered for patients with DME that has responded to anti-VEGF treatment, including aflibercept 2 mg, but who wish to extend the treatment interval beyond the 8 weeks that is standard for most currently available anti-VEGF treatments, or for those with DME that has not responded to the other anti-VEGF treatments. Specifically, the clinical expert expected patients with centre-involving DME with minimal macular microstructural damage and minimal ischemia to benefit the most. This is in line with other anti-VEGF treatments. The clinical expert noted that comorbidities such as poor glucose control, poorly controlled hypertension, and incipient renal failure can have a detrimental effect on the odds of successful treatment of DME and diabetic retinopathy with anti-VEGF therapy.
Assessing the Response Treatment
The clinical expert consulted by CDA-AMC noted that visual acuity, OCT assessment of IRF or SRF and CRT measurement, and fundus examination for retinal or subretinal hemorrhage are the usual outcomes measured in clinical practice. The expert indicated that these measurements are taken at each clinical visit for treatment and that assessments are also conducted to determine if treatment needs to be maintained or modified. Following the initial monthly treatment for 3 months, the treatment interval can be extended to every 12 weeks, and subsequently, the interval can be adjusted by increments or reductions of 4 weeks for the next treatment cycle.
Discontinuing Treatment
The clinical expert noted that treatment discontinuation might be considered if there is no sign of response to treatment or if deterioration continues despite ongoing treatment. The expert indicated that the features of treatment failure are decreasing visual acuity, persistent or increased IRF or SRF, or development of new subretinal hemorrhage, despite active treatment.
Prescribing Considerations
The clinical expert noted the treatment with aflibercept 8 mg can occur in a clinic or hospital. The treatment should be provided by an ophthalmologist familiar with the diagnosis and management of retinal diseases including DME.
Clinician Group Input
This section was prepared by the review team at CDA-AMC based on the input provided by clinician groups. The full original clinician group input(s) received by CDA-AMC have been included in the Perspectives of Patients, Clinicians, and Drug Programs section of this report.
Input from 6 clinician groups, the Southwestern Ontario Community Ophthalmologists, Toronto Retina Institute, Northeastern Ontario Ophthalmology Group, Retina Division of the Ottawa Hospital, Toronto Ophthalmologists, and the Canadian Retina Society, were summarized for this review. A total of 17 clinician experts participated in the clinician input submission. Input across groups were generally sourced through telephone conversations, virtual meetings and discussions, emails, literature reviews, conference presentations, systematic reviews, and meta-analyses.
Treatment goals for DME were consistent across inputs, that is, to maintain vision (i.e., stabilizing visual acuity and prevent vision worsening) and to improve quality of life, while extending the duration between treatments. The current treatments were also consistent across groups: listed were anti-VEGFs such as aflibercept 2 mg, ranibizumab, brolucizumab, faricimab, and bevacizumab (off-label use). Bevacizumab, according to the clinician group experts, is poorly accessible for patients aged older than 65 years and brolucizumab is associated with risks of intraocular adverse events. Other treatments noted included laser therapy and corticosteroid injections. The clinician groups highlighted that increased risks of retinal scarring associated with laser therapy and of cataract formation and increased ocular pressure associated with corticosteroids often preclude their use in practice and reinforce the preference for anti-VEGF treatments. In addition, according to the clinician group experts, intravitreal steroid injections are not covered by drug plans, which hinders accessibility and places a financial burden on patients. The clinician groups mentioned that although current treatments target the underlying disease mechanism of DME, they are not curative, and the extent and duration of damage to the retina may impact treatment efficacy. The highlighted unmet needs were consistent across groups. The clinician groups highlighted that treatment of DME is ongoing, requiring repeated visits with trained specialists, which poses a significant burden to patients. Therefore, there is a need for efficacious, durable, and long-lasting treatments that can minimize treatment burden compared to the burden of existing treatments. A treatment formulation designed and studied with an extended dosing interval could help reduce the patient burden and promote compliance. The clinician groups also highlighted the need for safer treatments with minimal ocular complications because of the known safety concerns related to inflammation and occlusive retinal vasculitis observed with treatment with brolucizumab.
According to the clinician groups, aflibercept 8 mg may become the drug of choice for patients with no experience of treatment as it is anticipated that aflibercept 8 mg will replace the aflibercept 2 mg formulation and become established as a new first-line option for DME. This aligns with the input from the clinical expert consulted for the review. According to the clinician groups, all patients who require treatment with anti-VEGFs will be eligible to receive aflibercept 8 mg, although this treatment may be slightly less suitable for patients with monocular disease (disease in only 1 eye) due to the potential risk of infection if the vial is not designed for multiple use. The groups stated that response to treatment will be assessed by measuring vision stabilization and anatomic outcomes. According to the clinician groups, response assessment is highly standardized across clinical trials and clinical practice; thus, the outcomes assessed in the trials are the same as used in clinical practice. The clinician groups noted that the factors that will impact decisions to discontinue aflibercept 8 mg will be similar to those pertaining to the aflibercept 2 mg formulation (e.g., no response or the presence of irreversible macular damage). According to the experts, aflibercept 8 mg will be administered by physicians, primarily in ophthalmologists’ offices and rarely in hospital outpatient clinics.
Drug Program Input
The drug programs provide input on each drug being reviewed through the Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CDA-AMC are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The PHOTON study is a phase II/III, multicentre, randomized, double-masked, active-controlled trial that assessed the effectiveness, safety, and tolerability of a higher dose of aflibercept (8 mg) against the standard aflibercept (Eylea) 2 mg dose. The aim of the study was to evaluate whether 2 extended dosing regimens of aflibercept 8 mg were at least as effective as aflibercept 2 mg. It is important to note that no trials were conducted comparing aflibercept 8 mg with other extended-interval anti-VEGF medications like brolucizumab (Beovu) and faricimab (Vabysmo). | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for initiation of therapy | |
Eligibility for disease diagnosis, scoring, or staging varies across provinces, with most having retinal programs in place. PHOTON trial inclusion criteria specify that patients must have DME with central involvement and central retinal thickness of at least 300 μm — or at least 320 μm on Spectralis — confirmed by a reading centre at the screening visit. In addition, patients must have a BCVA Early Treatment Diabetic Retinopathy Study (ETDRS) letter score from 78 to 24, equivalent to a Snellen visual acuity fraction of 20/32 to 20/320, with vision loss attributed to DME. | This is a comment from the drug plans to inform CDEC deliberations. |
The initiation criteria for treatments in the same category as aflibercept 8 mg have seen changes over time. The 2012 recommendation for ranibizumab (Lucentis) included specific initiation criteria, such as the presence of clinically significant DME where laser photocoagulation is also indicated, and a hemoglobin A1C level of less than 11%. This recommendation, however, might be considered outdated, especially since it mandates an A1C level, a requirement that has since been removed by many jurisdictions. In contrast, the 2014 guidance for aflibercept (Eylea) 2 mg suggested listing it in a manner akin to ranibizumab (Lucentis). Recommendations for brolucizumab (Beovu) and faricimab (Vabysmo) also advised listing them similarly to other anti-VEGF drugs. A question arises for CDEC and clinical experts: Are the 2012 ranibizumab (Lucentis) criteria relevant to the application for aflibercept 8 mg in the treatment of DME? If they are no longer suitable, updated criteria are needed. For instance, could the criteria used for the PHOTON trial be more appropriate for current practice? | The clinical expert noted that the 2012 recommendation for ranibizumab (Lucentis) is based on the eligibility of patients to undergo laser photocoagulation. Current practice and guidelines have changed and patients no longer undergo laser photocoagulation as they did in 2012. Furthermore, the clinical expert noted that glycemic control is important in achieving optimal therapeutic outcomes. However, glycemic control can be achieved in a reasonable period of time. |
Considerations for discontinuation of therapy | |
Should discontinuation criteria be included in the recommendation? | The clinical expert noted that a number of key considerations should be taken into account when considering discontinuation. These would include decreasing visual acuity, persistent or increasing intraretinal or subretinal fluid, or development of new subretinal hemorrhage despite active treatment. Typically, this assessment can take place after at least 3 injections. In such instances, it is important to consider either changing the treatment or stopping it altogether, given the lack of intended effects and the inherent risks associated with each injection. In addition, for patients in the advanced stages of the disease who have substantial scarring, the benefits of anti-VEGF treatments are likely to be minimal, suggesting that treatment discontinuation should be considered. |
Considerations for prescribing of therapy | |
The sponsor notes that aflibercept 8 mg meets an unmet need by having a dosing frequency of every 12 weeks to 16 weeks. The recommended dose of brolucizumab is 6 mg every 6 weeks for the first 5 doses then every 12 weeks. The recommended dose of faricimab is 6 mg every 4 weeks for the first 4 doses and then every 8, 12, or 16 weeks. | This is a comment from the drug plans to inform CDEC deliberations. |
Does aflibercept 8 mg meet an unmet need given there are other products marketed with an extended dosing interval? | The clinical expert noted that aflibercept 8 mg has an established option to extend to 16 weeks and comes with the added advantage of a known safety profile after more than 10 years of clinical experience administering 2 mg aflibercept. |
System and economic issues | |
Aflibercept 8 mg would have significant budget impact on public drug plans. Biosimilars have already been marketed for ranibizumab (Lucentis). Biosimilars are anticipated for aflibercept 2 mg in 2024. There has been a significant increase in drug utilization of aflibercept 2 mg as a result of prescribers switching from ranibizumab to avoid the recently implemented ranibizumab (Lucentis) biosimilar switch initiative. Question for CDEC: Should the pricing recommendation for reimbursement recommend that aflibercept 8 mg be negotiated so that it provides cost savings to drug programs relative to the cost of currently funded anti-VEGF drugs for DME. | — |
Confidential pricing agreements exist for most anti-VEGF drugs. | This is a comment from the drug plans to inform CDEC deliberations. |
Retinal programs or provincial eye centres exist in a number of provinces. Bevacizumab-first policies are in place in a number of provinces. | This is a comment from the drug plans to inform CDEC deliberations. |
BCVA = best corrected visual acuity; CDEC = Canadian Drug Expert Committee; DME = diabetic macular edema; VEGF = vascular endothelial growth factor.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of aflibercept 8 mg administered through intravitreal injection in the treatment of adults with DME. The focus is on comparing aflibercept 8 mg with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of aflibercept 8 mg is presented in 4 sections with the critical appraisal of the evidence from CDA-AMC included at the end of each section. The first section, the systematic review, includes pivotal studies and randomized controlled trials (RCTs) that were selected according to the sponsor’s systematic review protocol. The assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section would include sponsor-submitted long-term extension studies; however, none were submitted. The third section includes indirect evidence submitted by the sponsor. The fourth section would include additional studies that were considered by the sponsor to address important gaps in the systematic review evidence; however, none were submitted.
Included Studies
Clinical evidence from the following studies are included in the review and appraised in this document:
1 pivotal study identified in the systematic review
1 ITC.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team at CDA-AMC.
Description of Studies
Characteristics of the included study are summarized in Table 5.
Table 5: Details of the PHOTON Trial
Detail | PHOTON |
|---|---|
Study design | Phase II/III, multicentre, randomized, double-masked, active-controlled, noninferiority study |
Locations | 138 study sites across Canada, Czech Republic, Germany, Hungary, Japan, the UK, and the US (4 study sites in Canada) |
Patient enrolment dates | Start date: June 29, 2020 End date: June 28, 2021 |
Randomized (N) | N = 660 randomized in a 1:2:1 ratio:
|
Inclusion criteria |
|
Exclusion criteria |
|
Intervention |
|
Comparator(s) |
|
Screening phase | 3 weeks |
Treatment phase | 48 and 60 weeks (primary and select secondary efficacy end points analyzed within double-masked phase); 96 weeks (additional secondary and exploratory end points analyzed within double-masked phase) |
Follow-up phase | NA; patients could consent to continue in an extension period |
Extension phase | Optional open-label treatment extension up to 156 weeks (60 weeks following the double-masked phase) |
Primary end point | Change from baseline in BCVA measured using the ETDRS letter score at week 48 |
Secondary and exploratory end points | Key secondary:
Additional secondary:
Exploratory:
|
Publications | Brown D, Boyer D, Do DV, et al. Intravitreal aflibercept 8 mg in diabetic macular oedema (PHOTON): 48-week results from the randomized, double-masked, noninferiority, phase II/III trial. Unpublished manuscript. Trial registration (ClinicalTrials.gov): NCT04429503 |
ang-2 = angiopoietin-2; BCVA = best corrected visual acuity; CRT = central retinal thickness; DME = diabetic macular edema; DRSS = Diabetic Retinopathy Severity Scale; ETDRS = Early Treatment Diabetic Retinopathy Study; IOP = intraocular pressure; IRF = intraretinal fluid; NA = not applicable; NEI = National Eye Institute; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; SAE = serious adverse event; SRF = subretinal fluid; TEAE = treatment-emergent adverse event; VEGF = vascular endothelial growth factor; VFQ-25 = 25-item Visual Function Questionnaire.
Source: PHOTON Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
The PHOTON trial is an international, multicentre, randomized, double-masked, active-controlled, noninferiority phase II/III study with a primary objective to determine if treatment with aflibercept 8 mg administered with 2 extended dosing intervals (every 12 or 16 weeks) provides noninferior BCVA changes compared to aflibercept 2 mg every 8 weeks in adult patients with DME that involves the centre of the macula. The primary end point, change from baseline in BCVA measured using the ETDRS letter score, was measured at week 48 with 660 patients with DME across 138 study sites in 7 countries, including Canada, where there were 4 sites. Patients were randomized via interactive web response system in a 1:2:1 ratio to 1 of 3 dosing regimens:
aflibercept 2 mg administered every 8 weeks after 5 initial monthly doses (aflibercept 2 mg every 8 weeks)
aflibercept 8 mg administered every 12 weeks after 3 initial monthly doses (aflibercept 8 mg every 12 weeks)
aflibercept 8 mg administered every 16 weeks after 3 initial monthly doses (aflibercept 8 mg every 16 weeks).
Patients in the aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks arms could move to a more frequent dosing regimen based on their visual and anatomic outcomes. From week 52, in addition to shortening of treatment intervals, extension of the dosing intervals for patients in the aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks arms was allowed by 4-week increments based on visual and anatomic criteria. Randomization was stratified based on CRT at baseline (< 400 µm versus ≥ 400 µm), prior DME treatment (yes versus no), and geographic region (rest of the world versus Japan). The study consisted of a screening period of 3 weeks. Thereafter, patients entered a treatment period, where the primary efficacy end point of mean change in BCVA from baseline was measured at week 48.7
At time of submission, data from the 48-week analysis (cut-off date: August 19, 2022) and 60-week analysis (cut-off date: October 7, 2022) are available through the PHOTON Clinical Study Report provided in the submitted dossier. These are the same datasets under review by Health Canada for regulatory approval.
The masked part of the PHOTON trial was completed at week 96 (end of main study visit with a last patient last visit date of April 27, 2023), and therefore only topline results corresponding to this dataset are included in this report (the Clinical Study Report for the 96-week analysis was not available from the sponsor). The optional extension part of the study started immediately after the end of the week 96 study visit, during which the study drug could be administered in an open-label treatment period until week 156.7 The study design and dosing schedule in the PHOTON trial are illustrated in Figure 1.
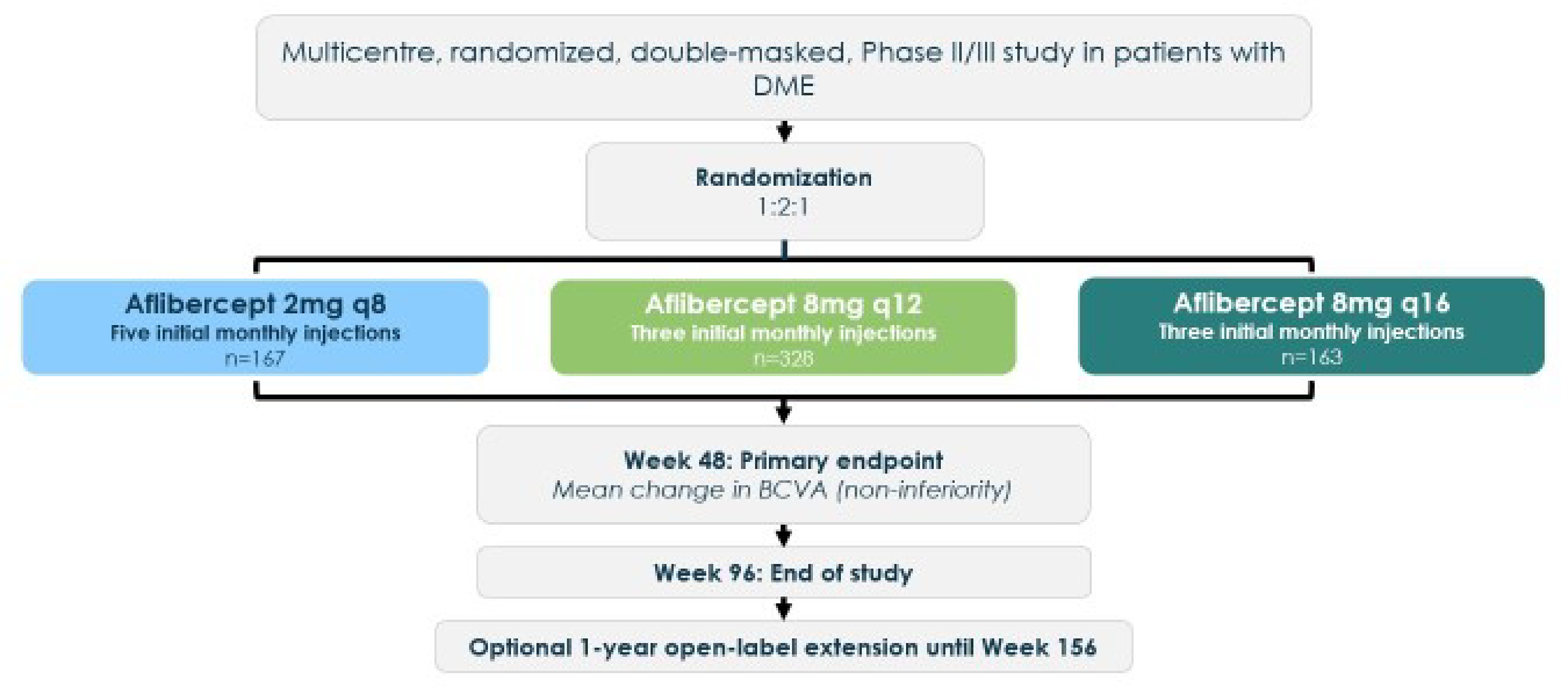
BCVA = best corrected visual acuity; DME = diabetic macular edema; q8 = every 8 weeks; q12 = every 12 weeks; q16 = every 16 weeks.
Source: PHOTON Clinical Study Report.39
Populations
Inclusion and Exclusion Criteria
Eligible patients were aged at least 18 years (or the country’s legal age of adulthood) and lived with type 1 or type 2 diabetes mellitus, DME with central involvement in the study eye with CRT greater than or equal to 300 μm (or ≥ 320 μm on Spectralis), and a BCVA ETDRS letter score of 78 to 24 (equivalent to Snellen visual acuity 20/32 to 20/320) in the study eye with decreased vision determined to be primarily the result of DME. One eye per patient was selected as the study eye. If both eyes were eligible, the study eye was selected based on clinician discretion. Key exclusion criteria included evidence of macular edema due to any cause other than diabetes in either eye, and active proliferative diabetic retinopathy in the study eye, among other criteria.7 Details of the key inclusion and exclusion criteria are shown in Table 5.
Interventions
In the aflibercept 2 mg arm, patients received intravitreal injections every 4 weeks for 5 loading doses, followed by maintenance dosing every 8 weeks to week 92, with a last study visit at week 96. In the aflibercept 8 mg arms, patients received intravitreal injections every 4 weeks for 3 loading doses, followed by maintenance dosing every 12 weeks or every 16 weeks, to week 92, with a last study visit at week 96. A sham procedure was performed on visits when an active injection was not planned for masking purposes. Active injections and sham procedures were administered by study site personnel. During the study, treatment intervals for patients receiving the aflibercept 8 mg every 12 weeks, or aflibercept 8 mg every 16 weeks could be shortened (year 1 and year 2) or extended (year 2) based on prespecified dose-regimen modification (DRM) criteria. Patients in the aflibercept 2 mg group remained on fixed dosing every 8 weeks throughout the study (i.e., their treatment intervals were not modified regardless of the outcomes of the DRM assessments).7
Anatomic measures used as secondary end points in the PHOTON trial included the proportion of patients without fluid in the foveal centre, change from baseline in CRT, and the proportion of patients without leakage on fluorescein angiography. These end points helped determine the timing and degree to which treatment intervals with aflibercept 8 mg could be modified relative to aflibercept 2 mg as described in the “DRM Criteria for Interval Shortening” and “DRM Criteria for Interval Extension” subsections.
All treatments given from the time of informed consent to the end of the study were considered concomitant medications. For the study eye, patients could only receive their assigned treatment for DME, barring any other standard or experimental treatments, while the fellow eye could receive 2 mg aflibercept or another approved indication for DME. Medications or procedures that were considered crucial for the patient's well-being and would not interfere with the study drug's evaluation were permitted.7
DRM Criteria for Interval Shortening
Two criteria were applied to ensure that treatment intervals were shortened only for patients with clinically relevant disease activity or vision deterioration. These criteria were designed in consultation with a steering committee comprising an external, independent panel of thought leaders. These criteria had to be met to shorten treatment intervals and would determine if patients were able to achieve noninferior efficacy end points with increased dosing intervals. Starting at week 16, patients assigned to the aflibercept 8 mg arms were assessed for DRM criteria at every visit (i.e., every 4 weeks). The 2 DRM criteria for shortening the dosing interval were:
greater than 10-letter loss in BCVA from week 12 BCVA in association with persistent or worsening DME
greater than 50 μm increase in CRT from week 12.
If a patient on aflibercept 8 mg every 12 weeks met DRM criteria for shortening at week 16 or week 20, they were dosed with 8 mg at that visit and subsequently continued with dosing every 8 weeks. If a patient on aflibercept 8 mg every 16 weeks met DRM criteria for shortening at week 16 or week 20, they were dosed with 8 mg at that visit and continued on with dosing every 8 weeks. If a patient on aflibercept 8 mg every 16 weeks met DRM criteria for shortening at week 24, they were dosed with 8 mg at that visit and continued on with dosing every 12 weeks. Subsequently, patients who met DRM criteria at any active treatment visit had their intervals shortened by 4 weeks, to a minimum interval of every 8 weeks.
Starting at week 52, all patients randomized to either 8 mg treatment arm were eligible for adjustments of their treatment intervals (shortening or extension) with the dose interval adjustments becoming effective at or after week 60.7
DRM Criteria for Interval Extension
In year 2 (week 52 to week 96), all patients in the 8 mg treatment arms (including patients whose interval was shortened during year 1) were eligible for treatment interval extension (by 4-week increments) if both of the following DRM extension criteria were met at visits with an active injection:
less than 5-letter loss in BCVA from week 12
CRT of less than 300 μm on spectral domain OCT (or < 320 μm on Spectralis spectral domain OCT).7
Outcomes
Patients were examined every 4 weeks throughout the study. Standard examinations were performed at every visit to evaluate safety and efficacy. A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6. This is followed by descriptions of the outcome measures, and a summary of the outcome measures and their measurement properties are in Table 7. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical expert(s) consulted by CDA-AMC and input from patient and clinician groups and public drug plans. Using the same considerations, the review team at CDA-AMC selected end points that were considered to be most relevant to inform CDEC deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing CDEC deliberations were also assessed using GRADE.
Table 6: Summary of Outcomes of the PHOTON Trial
Outcome measure | Time point | PHOTON |
|---|---|---|
Change from baseline in BCVA | At week 48 | Primarya |
Change from baseline in BCVA | At week 60 | Key secondarya |
Patients gaining ≥ 15 ETDRS letters | At week 60 | Exploratory |
Patients achieving an ETDRS score ≥ 69 letters | At week 60 | Exploratory |
Proportion of patients without fluid in the foveal centre | At week 60 | Exploratory |
Change from baseline in vision-related QoL (NEI-VFQ-25) | At week 60 | Exploratory |
Number of injections | Through week 60 | Exploratory |
BCVA = best corrected visual acuity; ETDRS = Early Treatment Diabetic Retinopathy Study; NEI = National Eye Institute; QoL = quality of life; VFQ-25 = 25-item Visual Function Questionnaire.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Source: PHOTON Clinical Study Report.39
Best Corrected Visual Acuity
The objective of the PHOTON study was to assess if aflibercept 8 mg administered at 12-week or 16-week intervals was noninferior to aflibercept 2 mg every 8 weeks for change from baseline in BCVA based on ETDRS letter score, using a noninferiority margin of 4 ETDRS letters. The primary end point was change from baseline in BCVA at week 48 and change in BCVA at week 60 was a key secondary end point. The proportion of patients gaining 15 letters or more at week 60 was an exploratory outcome. The proportion of patients achieving an ETDRS score of greater than or equal to 69 letters (equivalent to Snellen visual acuity fraction of 20/40) at week 60 was also an exploratory outcome. A BCVA Snellen visual acuity fraction equivalent to at least 20/40 is the minimum required for a driver’s licence in most regions of the US. In Canada, the minimum visual acuity required for a driver’s licence is 20/50. Visual acuity examiners must be certified to ensure consistent measurement of BCVA, and they must have remained masked to treatment assignment. Whenever possible, the same examiner must have performed all assessments for each patient.
An ETDRS chart is a standardized visual acuity testing chart with a series of 5 letters of equal difficulty in each row, with standardized spacing between letters and rows, for a total of 14 lines (70 letters in total).40 The letter sizing decreases with each consecutive row down the chart, resulting in increased difficulty reading them. When 20 or more letters are read correctly at 4 m, the visual acuity letter score is equal to the total number of letters read correctly plus 30. If fewer than 20 letters are read correctly at 4 m, the visual acuity letter score is equal to the total number of letters read correctly at 4 m (number of letters recorded on line 1), plus the total number of letters in the first 6 lines read correctly at 1 m. The maximum score could therefore be 100. An increase in letter score corresponds to improvement in visual acuity. No estimates for a minimal important difference (MID) in change from baseline in BCVA were identified in the sponsor’s submission to CDA-AMC. The clinical expert identified 4 to 5 letters as a threshold that could be clinically meaningful. The clinical expert did not provide a threshold of clinical meaningfulness on the difference in the proportion of patients gaining 15 or more ETDRS letters at week 60 or the difference in the proportion of patients achieving 69 or more ETDRS letters at week 60.
Fluid in the Foveal Centre
The proportion of participants without fluid (no IRF and no SRF) in the foveal centre (as assessed by the central reading centre) at week 60 was an exploratory outcome. The accumulation of retinal fluid is an indication of the pathophysiology of DME. An MID has not been estimated for retinal fluid41
The clinical expert did not provide a threshold of clinical importance for the difference in the proportion of patients without fluid in the foveal centre.
Vision-Related Quality of Life
Vision-related quality of life was assessed using the NEI-VFQ-25 administered by a masked interviewer. The change from baseline in NEI-VFQ-25 total score at week 60 was an exploratory end point. The NEI-VFQ-25 has 25 items, including 11 vision-related subscales and a single-item general health scale. The overall composite score is created by averaging the 11 vision-related subscales.42 Each subscale score is the average score of all items in the subscale transformed to a 0 to 100 scale, with 0 indicating the worst possible score and 100 indicating the best possible score. A psychometric validation study of the NEI-VFQ-25 with patients with DME found that the MID ranged from 8.80 (general vision) to 14.40 (role difficulties) and resulted in a composite score MID of 3.33 points to 6.13 points, depending on the approach used for estimating the MID.43 The clinical expert agreed that the estimated MID of 6 from the published literature would be appropriate threshold for the clinical meaningfulness of a between-group difference.
Frequency of Injection
The exploratory end points related to injection frequencies in the PHOTON trial were measured through the mean number of injections given in each group at week 60. In addition, measures of frequency of injection included the proportion of patients randomized to receive aflibercept 8 mg every 16 weeks maintaining a dosing interval of 16 weeks or longer through weeks 48, 60, and 96; and the proportion of patients randomized to receive aflibercept 8 mg every 12 weeks maintaining a dosing interval of 12 weeks or longer through weeks 48, 60 and 96.7
Harms Outcomes
The safety analyses included TEAEs (ocular and non-ocular), SAEs (ocular and non-ocular), withdrawals due to adverse events (WDAEs), and deaths that occurred through week 60. No adverse events of special interest were defined in the study protocol; however, notable harms included intraocular inflammation, endophthalmitis, IOP, retinal vasculitis, and APTC events.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
NEI-VFQ-25 | The NEI-VFQ assesses the impact of vision impairment on the health-related quality of life across a broad range of eye conditions.44 The NEI-VFQ-25 (a shortened version of the original 51-item questionnaire) is administered as an interview and consists of 25 items relevant to 11 subscales, in addition to a single-item general health component.42 Each subscale score is the average score of all items in the subscale transformed to a 0 to 100 scale, with 0 indicating the worst possible score and 100 indicating the best possible score. The composite score is the unweighted average score of all items except for the general health rating, which is considered a standalone item representing overall health status.42 | Validity Lloyd et al.43 conducted a study to evaluate the psychometric properties (construct, convergent, and concurrent validity) of the NEI-VFQ-25 among patients with DME who participated in an RCT (N = 235). Adult patients were randomized to receive either intravitreal pegaptanib injection or sham treatment. Known-groups validity was assessed. The NEI-VFQ-25 distinguishes different visual acuity groups based on number of ETDRS letters. The NEI-VFQ-25 was found to correlate poorly to moderately with the EQ-5D VAS.43 For concurrent validity, the Pearson correlation coefficient for the NEI-VFQ-25 subscale scores and the EQ-5D VAS score ranged from 0.16 to 0.43 for role difficulties and general health, respectively. The Pearson correlation coefficient was 0.38 for the NEI-VFQ-25 composite score with the EQ-5D VAS.43 For construct validity, known-groups validity was evaluated based on subgroups of patients according to their visual acuity. Results showed higher NEI-VFQ-25 subscale scores for patients documented as having better ETDRS-based visual acuity. A higher mean NEI-VFQ-25 composite score was reported in the quartile of patients with the best visual acuity, compared to the quartile of patients with the worst visual acuity; 72.1 (SD = 17.91) vs. 56.1 (SD = 18.00), respectively.43 Convergent validity assessed yielded low to moderate correlations with ETDRS letter scores ranging from 0.10 to 0.41 in the study eye and from 0.01 to 0.51 in the fellow eye. The Pearson correlation with ETDRS total letters in the study eye was reported as 0.35 for the near-vision subscale and 0.34 for the distance-vision subscale. Issues with multidimensionality were observed, rendering composite score questionable.45-47 Reliability Internal consistency was acceptable (Cronbach alpha ≥ 0.748) for 6 of the 8 multi-item subscales. Limitations of internal consistency, due to the presence of single-item domains, were noted.43 Responsiveness All but 2 subscale scores (general health and ocular pain) have been shown to be responsive to changes in visual acuity in the better-seeing eye.45,49 | A 2013 psychometric validation study of the NEI-VFQ-25, specifically among patients with DME, used 2 distribution-based methods to determine an MCID from baseline to week 54.43 Using a half-standard deviation–based approach, the MCID for each VFQ-25 domain ranged from 8.80 (general vision) to 14.40 (role difficulties), resulting in a composite score MCID of 6.13 points. A standard error of measurement approach yielded similar MCID estimates from 8.79 (driving) to 14.04 (role difficulties), with a composite score MCID estimate of 3.33 points.43 |
ETDRS letters | ETDRS charts are used to measure visual acuity. ETDRS charts present a series of 5 letters of equal difficulty of reading in each row, with standardized spacing between letters and rows, to a total of 14 lines and a total of 70 letters. Letters range from 58.18 mm to 2.92 mm in height, corresponding to Snellen visual acuity fractions of 20/200 to 20/10, respectively. Charts are used in a standard light box. The standard testing distance is 4 m. Visual acuity is documented as the smallest line read by each eye in the absence of any errors.50 Scores are based on the number of letters that are correctly read. An ETDRS letter score is calculated when 20 or more letters are read correctly at 4 m, and the visual acuity letter score is equal to the total number of letters read correctly at 4 m plus 30. The maximum score is 100.51 Higher scores indicate better visual acuity. | Validity No relevant evidence of validity found among patients with DME. Reliability Two studies (study 1, n = 40 healthy eyes;52 study 2, n = 265, comprising 53 healthy eyes and 212 eyes with uncorrected refractive error, age-related macular degeneration, diabetic retinopathy, cataract, optic nerve corneauveitis, glaucoma, amblyopia, or other53) reported the test-retest reliability to be moderate to almost perfect agreement (study 1: ICC = 0.580 to 0.866, depending on lighting and constrast;52 study 2: ICC = 0.9953). The test-retest variability, which refers to the difference in visual acuity when a patient is tested and retested in the absence of any true clinical change, helps guide what would be considered a clinically meaningful change. Literature-based estimates of test-retest variability range from ± 0.07 to ± 0.19 logMAR.54 This suggests that any change in score between baseline and follow-up of approximately 4 to 10 letters results in insufficient certainty that the difference in letters is not due to chance alone. Responsiveness No relevant evidence of responsiveness was found among patients with DME. | There has been no estimation of an MID for the ETDRS among patients with DME. |
DME = diabetic macular edema; ETDRS = Early Treatment Diabetic Retinopathy Study; ICC = intraclass correlation coefficient; MCID = minimal clinically important difference; MID = minimal important difference; NEI = National Eye Institute; SD = standard deviation; VAS = visual analogue scale; VFQ = Visual Function Questionnaire; VFQ-25 = 25-Item Visual Function Questionnaire.
Statistical Analysis
Statistical analysis of the outcomes reported in the systematic review are summarized in Table 9 for the PHOTON trial.
Noninferiority Margin
For the primary end point, a noninferiority margin of 4 letters was used. Based on the pooled data analysis of VIVID55 and VISTA56 studies with patients with DME treated with aflibercept 2 mg every 8 weeks, the mean change from baseline in BCVA at week 48 was 10.4 letters (95% CI, 9.41 letters to 11.4 letters). Using the lower limit of this CI, the sponsor asserted that a noninferiority margin of 4 letters would preserve greater than 50% (57.5%) of the treatment effect of aflibercept 2 mg every 8 weeks when compared to a putative placebo. The assumption that mean change in vision for a placebo group would be 0 letters is considered conservative by the sponsor as placebo groups in other studies have shown a higher likelihood of vision loss compared to groups receiving laser therapy.39,57,58
Sample Size and Power Calculation
The sample size calculation was based on the primary end point, change from baseline in BCVA at week 48, assuming a noninferiority margin of 4 ETDRS letters in 2 pairwise comparisons: aflibercept 8 mg every 12 weeks versus aflibercept 2 mg every 8 weeks; and aflibercept 8 mg every 16 weeks versus aflibercept 2 mg every 8 weeks. Under the original testing strategy, assuming an SD of 9.07 letters for each treatment group,59 it was determined that:
129 patients per treatment arm would provide 90% power using a 2-sample t test to demonstrate noninferiority with 1-sided alpha of 0.0125 ( = 0.025 ÷ 2) for each comparison.
The overall family-wise type I error rate of 0.025 (1-sided) would be preserved.
19% of patients would drop out before week 48.
Therefore, 160 patients randomized to each treatment arm would provide 90% power for each pairwise comparison. The sample size in the 8 mg every 12 weeks arm was doubled to meet regulatory requirements for the safety database. This resulted in a total of 640 patients for 3 treatment arms (160, 160, and 320 patients for aflibercept 2 mg every 8 weeks, aflibercept 8 mg every 16 weeks, and aflibercept 8 mg every 12 weeks, respectively). With these sample sizes, the power for the pairwise comparisons were as follows: 90% for aflibercept 8 mg every 16 weeks versus aflibercept 2 mg every 8 weeks, and approximately 97% for aflibercept 8 mg every 12 weeks versus aflibercept 2 mg every 8 weeks. The power to reject each of the primary hypotheses with the proposed multiple testing procedure was at least as high. Under the current hierarchical testing procedure (following protocol amendment 4, which was implemented after the end of enrolment but before database lock or unmasking), using the same assumptions as described, a total sample size of 640 patients for 3 groups provided 98% power for the comparison of aflibercept 8 mg every 12 weeks versus aflibercept 2 mg every 8 weeks, and subsequently 92% power for the comparison of aflibercept 8 mg every 16 weeks versus aflibercept 2 mg every 8 weeks, for the primary end point assessing noninferiority, with a 1-sided t test at significance level of 0.025.7
Statistical Test or Model
The primary analysis was based on the estimand concept. The estimand of primary interest was based mainly on a hypothetical strategy. It describes the change from baseline for all patients who started treatment, assuming all patients continued receiving treatment until week 48.7
The estimand was specified through the following definitions of population, variable, treatment condition, intercurrent events, and population-level summary:
Population: Defined by the inclusion or exclusion criteria. All efficacy analyses were conducted using the FAS.
Variable: Change from baseline to week 48 in BCVA.
Treatment condition: Intention to treat with aflibercept 8 mg administered every 12 weeks after 3 initial monthly injections or every 16 weeks after 3 initial monthly injections each versus aflibercept 2 mg administered every 8 weeks after 5 initial monthly injections; DRMs as detailed in the “DRM Criteria for Interval Extension” subsection do not affect patient's assigned intention-to-treat regimen.
Intercurrent events: Premature discontinuation from treatment (hypothetical strategy).
Population-level summary: Difference in LSM change from baseline to week 48 in BCVA between aflibercept 8 mg every 12 weeks and aflibercept 2 mg every 8 weeks (and aflibercept 8 mg every 16 weeks and aflibercept 2 mg every 8 weeks) resulting from an MMRM.7
The primary efficacy variable (change in BCVA from baseline to week 48) was analyzed based on the FAS and treatment group as randomized using an MMRM. The model included baseline BCVA as a covariate, treatment group, baseline CRT category (from reading centre) (< 400 μm, ≥ 400 μm), prior DME treatment (per electronic data capture) (yes, no), geographic region (rest of world, Japan), and visit-as-fixed effects, as well as interaction terms for treatment by visit and baseline BCVA by visit. A Kenward-Roger approximation was used for the denominator degrees of freedom. Superiority testing for change from baseline in BCVA at week 48 was also performed as part of the hierarchical testing procedure.7
Exploratory end points were subject to descriptive analyses at every scheduled appointment from the initial baseline through either week 48 or week 60, contingent upon the study design. Such analyses potentially encompassed statistical evaluations (yielding nominal P values) and 2-tailed 95% CIs for these efficacy measures, consistent with the methodology applied to the primary and secondary end points.
Missing Data Handling
As a sensitivity analysis for the primary end point, LOCF was conducted for patients who had at least 1 postbaseline value but had any further missing postbaseline BCVA values until week 48, and analysis of covariance (ANCOVA) was applied for the change from baseline in BCVA at week 48. Another approach that assumed that data were missing at random was implemented by using multiple imputation.
Multiple Testing Procedure
The type I error was controlled at 0.025 (1-sided tests) for testing the primary and key secondary end points. This approach allows the confirmatory testing of a hypothesis at 0.025 after successful rejection of the hypotheses that are ranked higher in the hierarchy.7
The testing hierarchy relevant for regulatory review by Health Canada and the European Medicines Agency is presented in Table 8. Appendix 3 describes a testing hierarchy presented for other regulatory agencies requiring a different testing strategy, based on the 48-week analysis only.
Table 8: Order of Hierarchical Testing Procedurea
EP-SAPb |
|---|
Q.12.w. BCVA week 48 noninferiority |
Q.12.w. BCVA week 60 noninferiority |
Q.16.w. BCVA week 48 noninferiority |
Q.16.w. BCVA week 60 noninferiority |
Q.12.w. DRSS week 48 noninferiority |
Q.16.w. DRSS week 48 noninferiority |
Q.12.w. BCVA week 48 superiority |
Q.12.w. BCVA week 60 superiority |
Q.16.w. BCVA week 48 superiority |
Q.16.w. BCVA week 60 superiority |
BCVA = best corrected visual acuity; DRSS = Diabetic Retinopathy Severity Scale; EP-SAP = Statistical Analysis Plan for European Medicines Agency and Pharmaceuticals and Medical Devices Agency; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks ;vs. = versus.
aAccording to EP-SAP.
bComparison vs. aflibercept 2 mg q.8.w.
Source: PHOTON Statistical Analysis Plan.7
Data Imputation Methods
The PHOTON study addressed missing data using 2 principal methods: the MMRM and LOCF. The end points “change in BCVA from baseline to week 48,” “change in BCVA from baseline to week 60,” and “change from baseline in NEI-VFQ-25 total score at week 48” used the MMRM, where it was assumed that data were missing at random. Conversely, the end points “proportion of patients gaining 15 or more letters at week 60,” “proportion of patients with BCVA of 69 or more letters at week 60,” and “proportion of patients without fluid in the foveal centre at week 60” used the LOCF method. In this approach, when data were missing, the most recent observed value for a participant was used to fill the gaps. To ensure the validity of their conclusions, the researchers also carried out sensitivity analyses, particularly where an MMRM was used, to consider potential scenarios wherein missing data might not be entirely random.7
Subgroup Analyses
The following subgroups were considered for prespecified subgroup analyses of the primary and key secondary efficacy end points: age at enrolment (< 55 years, ≥ 55 years to < 65 years, ≥ 65 years to < 75 years, ≥ 75 years), sex (male, female), racial identity (Asian, Black or African American, white), ethnicity (Hispanic or Latino, not Hispanic or Latino), baseline BCVA (≤ 73 letters, > 73 letters), geographic region (Japan, rest of the world), baseline CRT category (< 400 μm, ≥ 400 μm), prior DME treatment (yes, no). Subgroups were analyzed descriptively, that is, no confirmatory tests with adjustments for multiplicity were conducted.7
Sensitivity Analyses
Sensitivity analyses in the PHOTON study were performed to test the robustness and validity of the main analysis outcomes, especially in scenarios where the data might not be missing at random. For the end point “change in BCVA from baseline to week 48,” 3 sensitivity analyses were used: ANCOVA with missing data imputed using the LOCF method; ANCOVA with missing data imputed by multiple imputation assuming missing at random; and a tipping point analysis (if the multiple imputation results under the missing-at-random assumption showed noninferiority of high-dose groups compared to the low-dose group).7 For “change in BCVA from baseline to week 60,” besides LOCF, another approach assuming missing at random was used using multiple imputation. If the multiple imputation data indicated that missing data were not missing at random, a tipping point analysis was executed. The “change from baseline in NEI-VFQ-25 total score at week 48” had ANCOVA with LOCF as its sensitivity analysis.7
Analysis Populations
Results are reported for the FAS, the per-protocol set, and the safety analysis set in the PHOTON trial. The analysis sets are summarized in Table 10.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
PHOTON | ||||
Change from baseline in BCVA at week 48 | MMRM | The model included baseline BCVA as a covariate, treatment group, baseline CRT category (from reading centre) (< 400 μm, ≥ 400 μm), prior DME treatment (per EDC) (yes, no), geographic region (rest of world, Japan), and visit-as-fixed effects, as well as interaction terms for treatment by visit and baseline BCVA by visit. | The MMRM assumes data were MAR for participants who discontinued the study prematurely, i.e., missingness only depended on observed data. Alternative assumptions (not MAR) were included in the sensitivity analyses. |
|
Change from baseline in BCVA at week 60 | MMRM | The model included baseline BCVA as a covariate, treatment group, baseline CRT category (from reading centre) (< 400 μm, ≥ 400 μm), prior DME treatment (per EDC) (yes, no), geographic region (rest of world, Japan), and visit-as-fixed effects, as well as interaction terms for treatment by visit and baseline BCVA by visit. | The MMRM assumes data were MAR for participants who discontinued the study prematurely, i.e., missingness only depended on observed data. Alternative assumptions (not MAR) were included in the sensitivity analyses. | The LOCF method was conducted for participants who had at least 1 postbaseline value but had any further missing postbaseline BCVA values until week 48, and ANCOVA was applied for the change from baseline in BCVA at week 48. Another approach assuming MAR was implemented by using multiple imputation. To check the assumption that the missing data were not MAR, a tipping point analysis was also conducted based on the multiple imputation analysis. |
Proportion of patients gaining ≥ 15 letters at week 60 | CMH | Stratified by baseline CRT, prior DME treatment, and geographic region | LOCF | Observed case analysis |
Proportion of patients with BCVA ≥ 69 letters at week 60 | CMH | Stratified by baseline CRT, prior DME treatment, and geographic region | LOCF | Observed case analysis |
Proportion of patients without fluid in the foveal centre at week 60 | CMH | Stratified by baseline CRT, prior DME treatment, and geographic region | LOCF | Observed case analysis |
Change from baseline in NEI-VFQ-25 total score at week 60 | MMRM | NA | The MMRM assumes MAR for participants who discontinued the study prematurely, i.e., missingness only depended on observed data. | LOCF |
Mean number of injections at week 60 | Descriptive statistics | NA | NA | NA |
Proportion of patients maintaining ≥ q.12.w. dosing intervals through week 48 | Descriptive analysis | NA | NA | NA |
Proportion of patients maintaining ≥ q.12.w. dosing intervals through week 60 | Descriptive analysis | NA | NA | NA |
ANCOVA = analysis of covariance; BCVA = best corrected visual acuity; CMH = Cochran-Mantel-Haenszel; CRT = central retinal thickness; DME = diabetic macular edema; EDC = electronic data capture; LOCF = last observation carried forward; MAR = missing at random; MMRM = mixed model for repeated measures; NA = not applicable; NEI = National Eye Institute; q.12.w. = every 12 weeks; VFQ-25 = 25-Item Visual Function Questionnaire.
Source: PHOTON Statistical Analysis Plan.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Table 10: Analysis Populations of the PHOTON Trial
Study | Population | Definition | Application |
|---|---|---|---|
PHOTON | FAS | The FAS included all randomized patients who received at least 1 dose of the study drug; it was based on the treatment assigned to the participant at baseline (as randomized). | All efficacy analyses |
PPS | The PPS included all patients in the FAS who had a baseline and at least 1 postbaseline assessment of BCVA and did not have any relevant important protocol violations that affect the primary efficacy variable. The final determination of the exclusion of patients from the PPS was based on masked data before the first database lock. | Primary efficacy analysis | |
SAF | The SAF included all randomized patients who received any study treatment; it was based on the treatment received (as treated). | Safety analyses |
BCVA = best corrected visual acuity; FAS = full analysis set; PPS = per-protocol set; SAF = safety analysis set.
Source: PHOTON Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Results
Patient Disposition
In the PHOTON study, 660 patients were randomized to receive aflibercept 2 mg every 8 weeks (n = 167), aflibercept 8 mg every 12 weeks (n = 329), or aflibercept 8 mg every 16 weeks (n = 164). A total of 596 patients completed 60 weeks of the trial. There were no notable differences across the treatment arms with regard to reasons for early discontinuation.7 Important protocol deviations were reported for 5.5% of patients, with the most common important deviation pertaining to reconsenting of an amended informed consent form. A summary of patient disposition in the PHOTON trial is shown in Table 11.
Table 11: Summary of Patient Disposition in the PHOTON Trial
Patient disposition | PHOTON | ||
|---|---|---|---|
Aflibercept 2 mg q.8.w. (N = 167) | Aflibercept 8 mg q.12.w. (N = 329) | Aflibercept 8 mg q.16.w. (N = 164) | |
Screened, N | 970 | ||
Randomized, N (%) | 167 (100) | 329 (100) | 164 (100) |
Completed the study until week 48, n (%) | 157 (94.0) | 300 (91.2) | 156 (95.1) |
Discontinued from study before week 48, n (%) | 10 (6.0) | 29 (8.8) | 8 (4.9) |
Reason for discontinuation, n (%) | |||
Noncompliance with protocol | 1 (0.6) | 0 | 0 |
Adverse event | 0 | 4 (1.2) | 1 (0.6) |
Decision by the investigator or sponsor | 0 | 4 (1.2) | 1 (0.6) |
Withdrawal of consent | 4 (2.4) | 7 (2.1) | 2 (1.2) |
Lost to follow-up | 1 (0.6) | 5 (1.5) | 1 (0.6) |
Death | 4 (2.4) | 9 (2.7) | 3 (1.8) |
COVID-19 | 0 | 0 | 0 |
Completed the study until week 60, n (%) | 155 (92.8) | 289 (87.8) | 152 (92.7) |
Discontinued from study until week 60, n (%) | 12 (7.2) | 40 (12.2) | 12 (7.3) |
Reason for discontinuation, n (%) | |||
Noncompliance with protocol | 1 (0.6) | 1 (0.3) | 0 |
Adverse event | 0 | 4 (1.2) | 2 (1.2) |
Decision by the investigator or sponsor | 0 | 6 (1.8) | 2 (1.2) |
Withdrawal of consent | 4 (2.4) | 12 (3.6) | 2 (1.2) |
Lost to follow-up | 2 (1.2) | 8 (2.4) | 2 (1.2) |
Death | 5 (3.0) | 9 (2.7) | 4 (2.4) |
COVID-19 | 0 | 0 | 0 |
FAS, N | 167 (100) | 328 (99.7) | 163 (99.4) |
PPS, N | 164 (98.2) | 322 (97.9) | 163 (99.4) |
Safety analysis set, N | 167 (100) | 328 (99.7) | 163 (99.4) |
FAS = full analysis set; PPS = per-protocol set; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks.
Source: PHOTON Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Baseline Characteristics
A summary of baseline characteristics of the FAS population in the PHOTON trial is shown in Table 12. The mean age of patients was numerically similar across the groups; at 61.9 years (SD = 9.50 years), patients in the aflibercept 8 mg every 16 weeks group were slightly younger than those in the aflibercept 2 mg every 8 weeks group (63.0 years [SD, 9.78]). There were higher percentages of patients who were male in the aflibercept 8 mg every 12 weeks group (64.0%) and every 16 weeks group (60.7%) compared to the aflibercept 2 mg every 8 weeks group (55.1%). The majority of patients across all groups were white, and the proportions in the aflibercept 8 mg every 16 weeks group (78.5%) and 8 mg every 12 weeks group (70.4%) were higher than in the aflibercept 2 mg every 8 weeks group (67.1%). The duration of diabetes was consistent across groups, with a mean duration ranging from 15.1 years to 15.9 years. The majority of patients had type 2 diabetes, with a similar distribution in each group (> 93%). In terms of ocular characteristics, the mean BCVA was slightly higher in the aflibercept 8 mg every 12 weeks group (63.6 ETDRS letter score [10.10]) compared to 61.5 (SD, 11.22) and 61.4 (SD, 11.76) ETDRS letter score in the aflibercept 2 mg every 8 weeks group and aflibercept 8 mg every 16 weeks group, respectively. The distribution of patients with a BCVA of 73 letters or less was also similar, with a slightly higher proportion in the aflibercept 2 mg every 8 weeks group (88.0%). The means for CRT were consistent across groups, with patients in the aflibercept 8 mg every 16 weeks group having a marginally higher mean (460.3 µm) than those in the aflibercept 2 mg every 8 weeks group (457.2 µm). Mean hemoglobin A1C levels were slightly lower in the aflibercept 8 mg every 16 weeks group (7.84%) than in the aflibercept 2 mg every 8 weeks group (8.14%), with a higher percentage of patients in the aflibercept 8 mg every 16 weeks group having levels equal or less than 8% (65.0%). Prior treatment of DME was reported among approximately 44% of patients across all groups. The distribution of Diabetic Retinopathy Severity Scale scores at baseline was broadly similar, with a small variation in the higher severity levels across the groups.7
Table 12: Summary of Baseline Characteristics in the PHOTON Trial (Full Analysis Set)
Characteristic | PHOTON | ||
|---|---|---|---|
Aflibercept 2 mg q.8.w. (N = 167) | Aflibercept 8 mg q.12.w. (N = 329) | Aflibercept 8 mg q.16.w. (N = 163) | |
Age (years), mean (SD) | 63.0 (9.78) | 62.1 (11.13) | 61.9 (9.50) |
Female, n (%) | 75 (44.9) | 118 (36.0) | 64 (39.3) |
Male, n (%) | 92 (55.1) | 210 (64.0) | 99 (60.7) |
Racial or ethnic identity, n (%) | |||
American Indian or Alaska Native [wording from original source] | 0 | 2 (0.6) | 0 |
Asian | 30 (18.0) | 48 (14.6) | 23 (14.1) |
Black or African American | 18 (10.8) | 35 (10.7) | 9 (5.5) |
Native Hawaiian or other Pacific Islander | 0 | 1 (0.3) | 0 |
White | 112 (67.1) | 231 (70.4) | 128 (78.5) |
Not reported | 4 (2.4) | 6 (1.8) | 1 (0.6) |
Multiracial | 0 | 1 (0.3) | 0 |
Duration of diabetes, years (SD) | 15.9 (10.04) | 15.1 (9.96) | 15.7 (10.67) |
Diabetes type, n (%) | |||
Type 1 | 11 (6.6) | 18 (5.5) | 9 (5.5) |
Type 2 | 156 (93.4) | 310 (94.5) | 154 (94.5) |
BCVA (ETDRS letter score) | |||
Mean (SD) | 61.5 (11.22) | 63.6 (10.10) | 61.4 (11.76) |
≤ 73, n (%) | 147 (88.0) | 269 (82.0) | 140 (85.9) |
> 73, n (%) | 20 (12.0) | 59 (18.0) | 23 (14.1) |
Intraocular pressure (mm Hg) | |||
Mean (SD) | 15.9 (2.99) | 15.3 (3.24) | 14.9 (3.25) |
CRT (µm) | |||
Mean (SD) | 457.2 (144.00) | 449.1 (127.39) | 460.3 (117.84) |
Hemoglobin A1C | |||
Mean (SD) | 8.14 (1.48) | 7.94 (1.55) | 7.84 (1.50) |
≤ 8%, n (%) | 90 (53.9) | 193 (58.8) | 106 (65.0) |
> 8%, n (%) | 76 (45.5) | 133 (40.5) | 55 (33.7) |
Missing | 1 (0.60) | 2 (0.61) | 2 (1.23) |
Prior DME treatmenta | |||
Yes, n (%) | 74 (44.3) | 143 (43.6) | 71 (43.6) |
No, n (%) | 93 (55.7) | 185 (56.4) | 92 (56.4) |
NEI-VFQ-25 total score | |||
Mean (SD) | 76.65 (15.89) | 76.79 (17.32) | 77.86 (15.58) |
DRSS score | |||
10 | 0 | 1 (0.3) | 2 (1.2) |
12 | 0 | 2 (0.6) | 0 |
14 | 1 (0.6) | 1 (0.3) | 1 (0.6) |
15 | 1 (0.6) | 0 | 0 |
20 | 3 (1.8) | 13 (4.0) | 2 (1.2) |
35 | 66 (39.5) | 121 (36.9) | 66 (40.5) |
43 | 34 (20.4) | 59 (18.0) | 36 (22.1) |
47 | 17 (10.2) | 46 (14.0) | 15 (9.2) |
53 | 22 (13.2) | 34 (10.4) | 11 (6.7) |
61 | 9 (5.4) | 20 (6.1) | 9 (5.5) |
65 | 4 (2.4) | 11 (3.4) | 9 (5.5) |
71 | 1 (0.6) | 1 (0.3) | 2 (1.2) |
75 | 0 | 1 (0.3) | 0 |
90 (nongradable) | 9 (5.4) | 18 (5.5) | 10 (6.1) |
BCVA = best corrected visual acuity; CRT = central retinal thickness; DME = diabetic macular edema; DRSS = Diabetic Retinopathy Severity Scale; ETDRS = Early Treatment Diabetic Retinopathy Study; NEI = National Eye Institute; SD = standard deviation; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; VFQ-25 = 25-Item Visual Function Questionnaire; VEGF = vascular endothelial growth factor.
aPrior DME treatment included laser photocoagulation, anti-VEGF treatment, and/or corticosteroids.
Source: PHOTON Clinical Study Report.7 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Exposure to Study Treatments
At 48 weeks, mean duration of exposure to the study treatment was similar between treatment arms, from 45.7 weeks to 47.1 weeks. Adherence to the treatment schedule was high with a mean treatment adherence through week 48 and week 60 of greater than or equal to 94% in all arms.7 A summary of patient exposure is shown in Table 13.
There were no data on subsequent treatment for aflibercept. Treatment with aflibercept 8 mg does not require any concomitant medications for the treatment of DME. A summary of concomitant medication in the PHOTON trial is shown in Table 14.
Table 13: Summary of Patient Exposure in the PHOTON Trial (Safety Analysis Set)
Exposure | PHOTON | ||
|---|---|---|---|
Aflibercept 2 mg q.8.w. (N = 167) | Aflibercept 8 mg q.12.w. (N = 329) | Aflibercept 8 mg q.16.w. (N = 163) | |
Through week 48 | |||
Duration (weeks), mean (SD) | 46.7 (6.89) | 45.7 (9.04) | 47.1 (6.00) |
Duration (weeks), median (IQR) | 48.0 (47.9 to 48.7) | 48.0 (47.7 to 48.7) | 48.0 (48.0 to 48.6) |
Treatment adherence (%), mean (SD) | 95.0 (12.3) | 94.3 (13.1) | 96.4 (8.2) |
Through week 60 | |||
Duration (weeks), mean (SD) | 57.8 (9.73) | 56.5 (12.5) | 58.5 (8.47) |
Duration (weeks), median (IQR) | 60.0 (59.9 to 60.6) | 60.0 (59.9 to 60.7) | 60.1 (60.0 to 60.6) |
Treatment adherence (%), mean (SD) | 94.9 (12.8) | 93.7 (13.5) | 95.9 (8.9) |
IQR = interquartile range; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; SD = standard deviation.
Source: PHOTON Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Table 14: Summary of Concomitant Treatment in the PHOTON Trial (Safety Analysis Set at Week 60)
Exposure | PHOTON | ||
|---|---|---|---|
Aflibercept 2 mg q.8.w. (N = 167) | Aflibercept 8 mg q.12.w. (N = 329) | Aflibercept 8 mg q.16.w. (N = 163) | |
Drugs used in diabetes, n (%) | 167 (100) | 315 (96.0) | 159 (97.5) |
Stomatological preparations, n (%) | 149 (89.2) | 287 (87.5) | 152 (93.3) |
Ophthalmologicals, n (%) | 136 (81.4) | 256 (78.0) | 141 (86.5) |
Agents acting on the renin-angiotensin system, n (%) | 110 (65.9) | 217 (66.2) | 113 (69.3) |
Lipid-modifying agents, n (%) | 114 (68.3) | 209 (63.7) | 115 (70.6) |
Cardiac therapy, n (%) | 94 (56.3) | 181 (55.2) | 97 (59.5) |
Vaccines, n (%) | 77 (46.1) | 137 (41.8) | 74 (45.4) |
Analgesics, n (%) | 58 (34.7) | 110 (33.5) | 61 (37.4) |
Beta-blocking agents, n (%) | 45 (26.9) | 101 (30.8) | 55 (33.7) |
Antibiotics and other chemotherapeutics for dermatological use, n (%) | 54 (32.3) | 93 (28.4) | 61 (37.4) |
Antipruritics (including antihistamines, anesthetics, and others), n (%) | 53 (31.7) | 96 (29.3) | 52 (31.9) |
Diuretics, n (%) | 45 (26.9) | 85 (25.9) | 49 (30.1) |
Drugs for acid-related disorders, n (%) | 46 (27.5) | 78 (23.8) | 45 (27.6) |
Vitamins, n (%) | 54 (32.3) | 74 (22.6) | 38 (23.3) |
Calcium-channel blockers, n (%) | 38 (22.8) | 74 (22.6) | 36 (22.1) |
Antibacterials for systemic use, n (%) | 41 (24.6) | 60 (18.3) | 47 (28.8) |
Antithrombotic agents, n (%) | 35 (21.0) | 64 (19.5) | 28 (17.2) |
Psycholeptics, n (%) | 32 (19.2) | 36 (11.0) | 22 (13.5) |
Antianemic preparations | 23 (13.8) | 41 (12.5) | 15 (9.2) |
Thyroid therapy, n (%) | 17 (10.2) | 39 (11.9) | 17 (10.4) |
Urologicals, n (%) | 16 (9.6) | 35 (10.7) | 15 (9.2) |
Antihypertensives, n (%) | 25 (15.0) | 33 (10.1) | 16 (9.8) |
Antiobesity preparations, excluding diet products, n (%) | 24 (14.4) | 29 (8.8) | 20 (12.3) |
Anti-inflammatory and antirheumatic products, n (%) | 13 (7.8) | 35 (10.7) | 10 (6.1) |
Blood substitutes and perfusion solutions, n (%) | 13 (7.8) | 26 (7.9) | 11 (6.7) |
q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks.
Note: > 10% in any arm.
Source: PHOTON Clinical Study Report.39
Efficacy
Efficacy results from the PHOTON study that are relevant to the current review are shown in Table 15 and detailed in this section.
Change From Baseline in BCVA at Week 48
Change from baseline in BCVA at week 48 was the primary end point in the PHOTON study. The primary end point was met: treatment with aflibercept 8 mg every 12 weeks and every 16 weeks demonstrated noninferiority to aflibercept 2 mg every 8 weeks using a noninferiority margin of 4 letters. The LSM changes in BCVA from baseline to week 48 were 8.1 letters (SE = 0.61 letters) and 7.2 letters (SE = 0.71 letters) for the aflibercept 8 mg every 12 weeks and every 16 weeks arms, respectively, compared with 8.7 letters (SE = 0.73 letters) in the aflibercept 2 mg every 8 weeks arm. Between-group differences in LSM changes from baseline were –0.57 letters (95% CI, –2.26 letters to 1.13 letters; P < 0.0001) and –1.44 letters (95% CI, –3.27 letters to 0.39 letters; P = 0.0031) letters for the aflibercept 8 mg every 12 weeks and every 16 weeks arms, respectively, compared with the aflibercept 2 mg every 8 weeks arm. The analysis of the primary end point was repeated on the per-protocol set from the week 48 analysis as a supplementary analysis and the results were consistent with those in the FAS.7
Change From Baseline in BCVA at Week 60 and Week 96
The corresponding key secondary end point at week 60 was also met: treatment with aflibercept 8 mg every 12 weeks and every 16 weeks demonstrated noninferiority to Eylea 2 mg every 8 weeks using a noninferiority margin of 4 letters, with LSM changes from baseline BCVA to week 60 of 8.5 letters (SE = 0.63 letters) and 7.6 letters (SE = 0.75 letters), respectively, compared with 9.4 letters (SE = 0.77 letters) in the aflibercept 2 mg every 8 weeks arm. Between-group differences in LSM changes from baseline were –0.88 letters (95% CI, –2.67 letters to 0.91 letters) and –1.76 letters (95% CI, –3.71 letters to 0.19 letters) for the aflibercept 8 mg every 12 weeks and every 16 weeks arms, respectively, compared to the aflibercept 2 mg every 8 weeks arm.7 Changes from baseline in BCVA score at week 48 and week 60 for the subgroups and sensitivity analyses were generally consistent with those in the overall population and base-case analysis.
The topline results of change from baseline in BCVA at week 96 indicate that the vision gains achieved with patients receiving aflibercept 8 mg continued to be noninferior to aflibercept 2 mg every 8 weeks, demonstrating lasting vision control. At week 96, the groups receiving aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks achieved LSM change in BCVA from baseline of 8.2 letters and 6.6 letters, respectively, versus 7.7 letters in the groups receiving aflibercept 2 mg every 8 weeks, maintaining noninferiority. Between-group differences in LSM changes from baseline were + 0.5 letters and –1.1 for aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks, respectively, compared to aflibercept 2 mg every 8 weeks.7 At the time of this report, no further results were available.
Proportion of Patients Gaining 15 or More ETDRS Letters at Week 60
At week 60, in the aflibercept 2 mg every 8 weeks group, 43 out of 165 patients (26.1%) gained at least 15 letters in BCVA from baseline. In the aflibercept 8 mg every 12 weeks group, 70 out of 326 patients (21.5%) showed at least a 15-letter gain. In the aflibercept 8 mg every 16 weeks group, 26 out of 163 patients (16.0%) recorded such gains. When compared to the aflibercept 2 mg every 8 weeks group, the differences in proportions of patients achieving at least a 15-letter gain were –5.01% (95% CI, –13.04% to 3.02%) in the aflibercept 8 mg every 12 weeks group and –10.78% (95% CI, –19.27% to 2.29%) in the aflibercept 8 mg every 16 weeks group.7
Proportion of Patients With BCVA of 69 or More ETDRS Letters at Week 60
At week 60, in the aflibercept 2 mg every 8 weeks group, 100 out of 165 patients (60.6%) had a BCVA of 69 or more ETDRS letters. In the aflibercept 8 mg every 12 weeks group, 211 out of 326 patients (64.7%) had a BCVA of 69 or more ETDRS letters. In the aflibercept 8 mg every 16 weeks group, 101 out of 163 patients (62.0%) recorded such scores. When comparing to the aflibercept 2 mg every 8 weeks group, the differences in the proportions of patients with a BCVA of 69 or more ETDRS letters at week 60 were 4.34% (95% CI, –4.72% to 13.40%) for the aflibercept 8 mg every 12 weeks group and 1.63% (95% CI, –8.91% to 12.17%) for the aflibercept 8 mg every 16 weeks group.7
Proportion of Patients Without Fluid in the Foveal Centre at Week 60
At week 60, 113 out of 165 patients (68.5%) in the aflibercept 2 mg every 8 weeks group showed no fluid in the foveal centre (no IRF and no SRF). In contrast, in the aflibercept 8 mg every 12 weeks group, 201 out of 325 patients (61.8%) showed no fluid in the foveal centre, with a difference of –5.98% ██████ ██████ ██ █████ compared with the aflibercept 2 mg every 8 weeks group. In the aflibercept 8 mg every 16 weeks group, 94 out of 162 patients (58.0%) had no fluid in the foveal centre, resulting in a difference of –9.88 ██████ ██████ ██ █████ compared with the aflibercept 2 mg every 8 weeks group.7
Frequency of Injections at Week 60
At week 60, 90.3% of 289 patients who completed treatment with aflibercept 8 mg every 12 weeks and 85.5% of 152 patients who completed treatment with aflibercept 8 mg every 16 weeks maintained their randomized treatment interval. This resulted in mean numbers of active injections through week 60 of 7.0 (SD = ████) and 6.0 (SD = ████) in the aflibercept 8 mg every 12 weeks and every 16 weeks arms, respectively, compared with 9.8 (SD = ████) in the aflibercept 2 mg every 8 weeks arm. Comparative differences were not reported. Results for the frequency of injections in the PHOTON trial are presented in Table 15.
NEI-VFQ-25 at Week 60
At week 60, the LSM increases in NEI-VFQ-25 scores were 4.55 (SE = ████) and 3.21 (SE = ████) for patients in the aflibercept 8 mg every 12 weeks and every 16 weeks arms, respectively, compared to 3.05 (SE = ████) for patients in the aflibercept 2 mg every 8 weeks arm. Between-group differences in LSM changes from baseline were 1.50 points (95% CI, █████ ██ ████) and 0.17 points (95% CI, █████ ██ ████) for patients in the aflibercept 8 mg every 12 weeks and every 16 weeks groups, respectively, compared to those in the aflibercept 2 mg every 8 weeks group.
Table 15: Summary of Key Efficacy Results From the PHOTON Trial (Full Analysis Set)
Variable | PHOTON | ||
|---|---|---|---|
Aflibercept 2 mg q.8.w. (N = 167) | Aflibercept 8 mg q.12.w. (N = 329) | Aflibercept 8 mg q.16.w. (N = 163) | |
Change from baseline in BCVA at week 48 | |||
Number of patients contributing to the analysis, n (%) | 150 (89.8) | 277 (84.2) | 149 (91.4) |
Baseline mean letters (SD) | 61.5 (11.22) | 63.6 (10.10) | 61.4 (11.76) |
Change from baseline LSM (SE) letters | 8.7 (0.73) | 8.1 (0.61) | 7.2 (0.71) |
Treatment group difference vs. control, letters (95% CI) | Reference | –0.57 (–2.26 to 1.13) | –1.44 (–3.27 to 0.39) |
P value (noninferiority) | Reference | < 0.0001 | 0.0031 |
Change from baseline in BCVA at week 60 | |||
Number of patients contributing to the analysis, n (%) | 133 (79.6) | 252 (76.6) | 138 (84.7) |
Baseline, mean (SD) letters | 61.5 (11.22) | 63.6 (10.10) | 61.4 (11.76) |
Change from baseline, LSM (SE) letters | 9.4 (0.77) | 8.5 (0.63) | 7.6 (0.75) |
Treatment group difference vs. control, letters (95% CI) | Reference | –0.88 (–2.67 to 0.91) | –1.76 (–3.71 to 0.19) |
P value (noninferiority) | Reference | 0.0003 | 0.0122 |
Proportion of patients gaining ≥ 15 letters in BCVA from baseline at week 60 | |||
Patients gaining ≥ 15 letters, n/N (%) | 43/165 (26.1) | 70/326 (21.5) | 26/163 (16.0) |
Treatment group difference vs. control, % (95% CI) | Reference | –5.01 (–13.04 to 3.02) | –10.78 (–19.27 to 2.29) |
Proportion of patients without fluid (no IRF and no SRF) in the foveal centre at week 60 | |||
Patients without fluid in the foveal centre, n/N (%) | 113/165 (68.5) | 201/325 (61.8) | 94/162 (58.0) |
Treatment group difference vs. control, % (95% CI) | Reference | –5.98 ███ ██ █████ | –9.88 ████ ██ █████ |
Frequency of active injection outcomes (safety analysis set completing week 60 visit) | |||
Number of active treatment injections | |||
Number of patients contributing to the analysis, N (%) | 155 (92.8) | 289 (88.1) | 152 (93.3) |
Total number of injections at week 60, mean (SD) | 9.8 ██████ | 7.0 ██████ | 6.0 ██████ |
Dosing intervals through week 60 (safety analysis set) | |||
Number of patients contributing to the analysis, N (%) | 167 (100.0) | 328 (99.7) | 163 (100.0) |
Patients maintained with ≥ q.12.w. dosing interval, N (%) | NA | 261 (90.3) | 142 (93.4) |
Patients maintained with ≥ q.16.w. dosing interval, N (%) | NA | NA | 130 (85.5) |
Patients with ≥ q.16.w. as last intended dosing interval, N (%) | NA | 123 (42.6) | 124 (81.6) |
Patients with q.20.w. as last intended dosing interval, N (%) | NA | 0 | 52 (34.2) |
Change from baseline in NEI-VFQ-25 total score at week 60 | |||
Number of patients with week 60 data, (%) | 130 (77.8) | 252 (76.6) | 138 (84.7) |
Baseline mean score, points (SD) | █████ █████ | ████ ██████ | ████ ██████ |
LSM change from baseline, points (SE) | 3.05 ██████ | 4.55 ██████ | 3.21 ██████ |
Treatment group difference vs. control, points (95% CI) | Reference | 1.50 ████ ██ ████ | 0.17 ████ ██ ████ |
Proportion of patients achieving ETDRS letter score ≥ 69 at week 60 | |||
Patients who achieved ≥ 69 letters, n/N (%) | 100/165 (60.6) | 211/326 (64.7) | 101/163 (62.0) |
Treatment group difference vs. control, % (95% CI) | Reference | 4.34 (–4.72 to 13.40) | 1.63 (–8.91 to 12.17) |
P value (superiority) | Reference | ██████ | ██████ |
BCVA = best corrected visual acuity; CI = confidence interval; ETDRS = Early Treatment Diabetic Retinopathy Study; IRF = intraretinal fluid; LSM = least squares mean; NA = not available; NEI = National Eye Institute; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; q.20.w. = every 20 weeks; SD = standard deviation; SE = standard error; SRF = subretinal fluid; VFQ-25 = 25-Item Visual Function Questionnaire; vs. = versus.
Source: PHOTON Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Harms
Harms data reported for the safety analysis set up to 60 weeks are summarized in Table 16.
Ocular and Non-Ocular TEAEs
Ocular TEAEs were reported by less than half of the enrolled patients. Specifically, 73 out of 167 patients (43.7%) in the aflibercept 2 mg every 8 weeks group experienced at least 1 ocular TEAE, as did 147 out of 328 patients (44.8%) in the aflibercept 8 mg every 12 weeks group and 73 out of 163 patients (44.8%) in the aflibercept 8 mg every 16 weeks group.
Non-ocular TEAEs were experienced by 96 out of 167 patients (57.5%) in the aflibercept 2 mg every 8 weeks group, 195 out of 328 patients (59.5%) in the aflibercept 8 mg every 12 weeks group, and 104 out of 163 patients (63.8%) in the aflibercept 8 mg every 16 weeks group.
Ocular and Non-Ocular SAEs
At least 1 ocular treatment-emergent SAE was reported by 1 out of 167 patients (0.6%) in the aflibercept 2 mg every 8 weeks group, 2 out of 328 patients (0.6%) in the aflibercept 8 mg every 12 weeks group, and 2 out of 163 patients (1.2%) in the aflibercept 8 mg every 16 weeks group. Specific events in this category included conditions such as cataract subcapsular (1 event in the 8 mg every 12 weeks group), retinal detachment (1 event in aflibercept 8 mg every 12 weeks group), ulcerative keratitis (1 event in aflibercept 2 mg every 8 weeks group), and vitreous hemorrhage (1 event in aflibercept 8 mg every 16 weeks group).
Non-ocular SAEs were experienced by 32 out of 167 patients (19.2%) in the aflibercept 2 mg every 8 weeks group, 61 out of 328 patients (18.6%) in the 8 mg every 12 weeks group, and 27 out of 163 patients (16.6%) in the aflibercept 8 mg every 16 weeks group.7
Treatment Discontinuation due to Adverse Events
No patients discontinued treatment due to ocular TEAEs in the aflibercept 2 mg every 8 weeks group, and 2 out of 328 patients (0.6%) discontinued treatment in the aflibercept 8 mg every 12 weeks group. No single adverse event took place among more than 2% of patients in any group.
Mortality
Of the 167 patients in the aflibercept 2 mg every 8 weeks group, 5 patients (3.0%) died. Specific causes of death in this group included cardiac arrest (1.2%), myocardial infarction (0.6%), diabetic metabolic decompensation (0.6%), and acute kidney injury (0.6%). Of the 328 patients in the aflibercept 8 mg every 12 weeks group, 9 patients (2.7%) died. Specific causes of death were cardiac arrest (0.6%), myocardial infarction (0.3%), unknown (0.6%), COVID-19 infection (0.3%), pneumonia (0.3%), and acute kidney injury (0.3%). Of the 163 patients in the aflibercept 8 mg every 16 weeks group, 4 patients (2.5%) died. The causes of death in this group were cardiorespiratory arrest (0.6%), left ventricular failure (0.6%), myocardial infarction (0.6%), and sudden death (0.6%).7
Notable Harms
Of the 167 patients in the aflibercept 2 mg every 8 weeks group, 1 (0.6%) experienced intraocular inflammation, 6 (3.6%) experienced increased IOP, and 6 (3.6%) underwent an APTC event. Of the 328 patients in the aflibercept 8 mg every 12 weeks group, 4 (1.2%) presented with intraocular inflammation, 7 (2.1%) experienced increased IOP, and 13 (4.0%) experienced an APTC event. Meanwhile, of the 163 patients in the aflibercept 8 mg every 16 weeks group, 1 (0.6%) experienced intraocular inflammation, 1 (0.6%) experienced an increase in IOP, and 9 (5.5%) experienced an APTC event. No cases of endophthalmitis or retinal vasculitis were reported in any of the treatment groups.7
Table 16: Summary of Harms Results From the PHOTON Trial Through Week 60 (Safety Analysis Set)
Adverse events | PHOTON | ||
|---|---|---|---|
Aflibercept 2 mg q.8.w. (N = 167) | Aflibercept 8 mg q.12.w. (N = 328) | Aflibercept 8 mg q.16.w. (N = 163) | |
Most common events, n (%) | |||
Patients with ≥ 1 ocular TEAEa | 73 (43.7) | 147 (44.8) | 73 (44.8) |
Vitreous floaters | 4 (2.4) | 18 (5.5) | 6 (3.7) |
Conjunctival hemorrhage | 6 (3.6) | 14 (4.3) | 7 (4.3) |
Cataract | 3 (1.8) | 9 (2.7) | 9 (5.5) |
Vitreous detachment | 3 (1.8) | 10 (3.0) | 4 (2.5) |
Punctate keratitis | 1 (0.6) | 5 (1.5) | 6 (3.7) |
Eye pain | 4 (2.4) | 9 (2.7) | 1 (0.6) |
Vision blurred | 3 (1.8) | 4 (1.2) | 2 (1.2) |
Cataract subcapsular | 1 (0.6) | 5 (1.5) | 0 |
Cataract nuclear | 2 (1.2) | 2 (0.6) | 1 (0.6) |
Corneal erosion | 0 | 1 (0.3) | 2 (1.2) |
Corneal abrasion | 2 (1.2) | 3 (0.9) | 1 (0.6) |
IOP increased | 6 (3.6) | 7 (2.1) | 1 (0.6) |
Patients with ≥ 1 non-ocular TEAEb | 96 (57.5) | 195 (59.5) | 104 (63.8) |
Infections and infestations | 47 (28.1) | 72 (22.0) | 40 (24.5) |
COVID-19 | 7 (4.2) | 24 (7.3) | 18 (11.0) |
Investigations | 15 (9.0) | 27 (8.2) | 16 (9.8) |
Hypertension | 18 (10.8) | 30 (9.1) | 25 (15.3) |
Patients with ≥ 1 ocular SAE, n (%) | 1 (0.6) | 2 (0.6) | 2 (1.2) |
Cataract subcapsular | 0 | 1 (0.3) | 0 |
Retinal detachment | 0 | 0 | 1 (0.6) |
Ulcerative keratitis | 1 (0.6) | 0 | 0 |
Vitreous hemorrhage | 0 | 0 | 1 (0.6) |
IOP increased | 0 | 1 (0.3) | 0 |
Patients with ≥ 1 non-ocular SAE, n (%) | 32 (19.2) | 61 (18.6) | 27 (16.6) |
Acute left ventricular failure | 3 (1.8) | 2 (0.6) | 0 |
Acute myocardial infarction | 2 (1.2) | 5 (1.5) | 2 (1.2) |
Myocardial infarction | 2 (1.2) | 4 (1.2) | 3 (1.8) |
Cerebrovascular accident | 0 | 2 (0.6) | 3 (1.8) |
Acute kidney injury | 2 (1.2) | 5 (1.5) | 1 (0.6) |
Patients who stopped treatment due to adverse events, n (%) | 3 (1.8) | 9 (3.7) | 2 (1.2) |
Ocular TEAEs | 0 | 2 (0.6) | 0 |
Non-ocular TEAEs | 3 (1.8) | 7 (2.1) | 2 (1.2) |
Death, n (%) | 5 (3.0) | 9 (2.7) | 4 (2.5) |
Cardiac arrest | 2 (1.2) | 2 (0.6) | 0 |
Cardiorespiratory arrest | 0 | 0 | 1 (0.6) |
Left ventricular failure | 0 | 0 | 1 (0.6) |
Myocardial infarction | 1 (0.6) | 1 (0.3) | 1 (0.6) |
Death (otherwise unknown) | 0 | 2 (0.6) | 0 |
Sudden death | 0 | 0 | 1 (0.6) |
COVID-19 | 0 | 1 (0.3) | 0 |
Pneumonia | 0 | 1 (0.3) | 0 |
Diabetic metabolic decompensation | 1 (0.6) | 0 | 0 |
Endometrial cancer | 0 | 1 (0.3) | 0 |
Acute kidney injury | 1 (0.6) | 1 (0.3) | 0 |
Notable harms, n (%) | |||
Intraocular inflammation | 1 (0.6) | 4 (1.2) | 1 (0.6) |
Endophthalmitis | 0 | 0 | 0 |
IOP increased | 6 (3.6) | 7 (2.1) | 1 (0.6) |
Retinal vasculitis | 0 | 0 | 0 |
APTC event | 6 (3.6) | 13 (4.0) | 9 (5.5) |
APTC = antiplatelet trialists’ collaboration; IOP = intraocular pressure; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Note: TEAEs are defined as adverse events that started in the time frame from first injection to the last injection (active or sham) in the study plus 30 days.
aFrequency ≥ 2% in at least 1 treatment group.
bFrequency ≥ 5% in at least 1 treatment group.
Source: PHOTON Clinical Study Report.39 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Critical Appraisal
Internal Validity
The PHOTON trial was overall well conducted, with appropriate measures for randomization, which involved stratification by baseline BCVA and geographic region, and maintained allocation concealment through the use of an interactive response system. Masking was adequately facilitated for trial participants and personnel. Adverse events were similar across treatment arms, so it is less likely that patients would have been unmasked. As such, there is low risk of bias in the measurement of the outcomes. Baseline characteristics and concomitant treatments were generally well-balanced across treatment arms. Notable imbalances in the baseline characteristics included a higher proportion of patients who were male and white in the higher-dosage aflibercept groups compared to the aflibercept 2 mg every 8 weeks group. Treatment adherence rate was consistent among the groups.
Statistical analyses and subgroups were prespecified in the clinical study protocol or in the statistical analysis plan. Adjustments for type I errors were accounted for in the primary and key secondary end points through a hierarchical testing procedure. However, no such adjustment was made for outcomes at week 60. This increases the possibility of type I error (i.e., false-positive results) in statistically significant week 60 end points. The trial was not specifically powered to identify subgroup differences. Outcomes used in the study had sufficient validity and reliability to address the objectives of the study.
Enrolled sample sizes were sufficient for primary outcome assessment based on the power calculations in the statistical analysis plan. The noninferiority margin of 4 ETDRS letters was evidence-based and deemed reasonable by the clinical expert consulted by CDA-AMC. The analysis conducted on the per-protocol set as a supplementary analysis supported the findings from the FAS.
The handling of missing data was methodologically sound, with the MMRMs assuming missing-at-random data for participants who discontinued the study early. Sensitivity analyses incorporated alternative assumptions for missingness (not missing at random) and an LOCF approach, although it is noted that LOCF is not the most conservative method for noninferiority trials and may contribute to bias.60 Nevertheless, the results of these sensitivity analyses remained consistent with the primary MMRM analysis in the FAS. However, outcomes other than those that were primary or key secondary only utilized LOCF with observed case sensitivity analysis or no sensitivity analysis, increasing the potential risk of bias due to missing data.
Important protocol deviations were reported by 5.5% of participants, with the most common important deviation pertaining to reconsenting of an amended informed consent form. By week 60, there was considerable attrition that was imbalanced across the treatment groups. This may result in attrition bias of unclear direction.
External Validity
The PHOTON trial was conducted in 138 sites, with only 4 in Canada. Nevertheless, the patients’ baseline characteristics were similar to those of patients with DME in Canada, according to the clinical expert engaged by CDA-AMC. The trial inclusion and exclusion criteria reflected those that would be applied in clinical practice when selecting patients for treatment with aflibercept 8 mg every 12 weeks or every 16 weeks. Approximately half the enrolled study population had no experience of treatment, and the remainder had previously received DME treatment. However, due to the study design, the PHOTON trial cannot inform the efficacy and safety of switching from other anti-VEGF drugs, and this remains an evidence gap.
The trial measured outcomes that are important to patients, including visual acuity. The clinical expert also confirmed that the study outcomes, which included visual acuity, injection frequency, and vision-related quality of life, are clinically relevant. The trial did not incorporate quality-of-life measures other than those to do with vision. Moreover, the dosing regimen of aflibercept 2 mg at every 8 weeks does not correspond with the regimen practised in clinics in Canada, which follows a treat-and-extend protocol. This discrepancy raises questions about the generalizability of the study results to clinical practice in Canada.
PHOTON was the only phase II/III trial submitted by the sponsor that provided direct evidence comparing aflibercept 2 mg every 8 weeks versus 8 mg every 12 weeks and every 16 weeks in the treatment of patients with DME. There is no direct evidence comparing aflibercept to the other anti-VEGF drugs currently used in practice in Canada (i.e., brolucizumab or bevacizumab), which represents an evidence gap. As noted, most clinicians use a flexible dosing regimen for anti-VEGF therapies, and the dosing of aflibercept 8 mg in real-world practice in Canada may not replicate the clinical trial setting. Moreover, lack of evidence on the long-term therapeutic effect of aflibercept 8 mg (beyond 2 years) represents a source of uncertainty.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CDEC deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.61,62
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs is rated as being of high certainty and can be rated down due to concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For change from baseline in BCVA letters and NEI-VFQ-25 score, the target of the certainty of evidence assessment was a clinically important effect. In the absence of a known threshold, for all other outcomes the target of the certain of evidence assessment was a non-null effect.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for aflibercept 8 mg administered every 12 weeks and every 16 weeks versus 2 mg every 8 weeks
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team at CDA-AMC.
Objectives for the Summary of Indirect Evidence
The included studies do not provide evidence on the efficacy or safety of aflibercept 8 mg against other interventions other than aflibercept 2 mg. The ITC was conducted to provide estimates of relative efficacy, safety, and number of injections for aflibercept 8 mg relative to standard interventions for the treatment of DME. The aim of this section is to provide an overview of the conduct, the results, and the critical appraisal of the sponsor-submitted ITC.
Description of ITC
A targeted literature search by the sponsor on October 20, 2023, did not identify any published ITC that included aflibercept 8 mg. The sponsor submitted 1 ITC, conducted for them by Broadstreet Health Economics and Outcomes Research, that used a Bayesian NMA approach, under fixed-effects and random-effects models, to compare the effects of aflibercept 8 mg every 12 weeks and every 16 weeks with other anti-VEGF drugs used in the treatment of patients with DME. The following outcome measures are reported: change in BCVA, gain of 15 ETDRS letters or more, ocular adverse events, and the mean number of injections. The sponsor-submitted NMA identified relevant evidence through a systematic review approach. The sponsor submitted to CDA-AMC the prespecified statistical analysis plan that guided the conduct of the systematic review and NMA.63
Table 17: Study Selection Criteria and Methods for the Systematic Review Portion of the Sponsor-Submitted ITC
Characteristics | Indirect comparison |
|---|---|
Population | Patients with no experience of treatment of DME and patients with experience of treatment of DMEa |
Interventionb |
|
Comparator | No restrictions |
Outcome |
Safety:
|
Study designs |
|
Publication characteristics |
|
Exclusion and inclusion criteria |
|
Databases searched | Ovid MEDLINE In-Process and Other Non-Indexed Citations and Ovid MEDLINE (accessed via the Ovid interface) on May 24, 2022 Embase (access via the Ovid interface) on May 24, 2022 Cochrane Central Register of Controlled Trials (CENTRAL) on May 26, 2022 Clinical trials registry: ClinicalTrials.gov on May 26, 2022 Search was repeated on July 24, 2023 |
Selection process | Two reviewers, working independently, screened the list of titles and abstracts according to the defined inclusion and exclusion criteria to select relevant articles pertaining to the topic of interest. The 2 reviewers’ decisions were combined and any differences were resolved through discussion to consensus or by a third reviewer. The full text of any article that met the inclusion criteria based on the review of the abstract was screened. Full texts were evaluated by 2 reviewers working independently to verify if the articles met the inclusion criteria. Differences were resolved through discussion to consensus or by a third reviewer. All finalized articles were checked for any possible links (i.e., if different articles originated from the same study). |
Data extraction process | Data from studies included in the review were extracted using extraction templates created in Excel. One reviewer extracted the data, while another validated the accuracy of the extracted data. |
Risk-of-bias assessment | The quality of the included studies was appraised according to the Cochrane risk-of-bias tool (version 2.0) checklist. This assessment was performed by 1 reviewer, with a second reviewer validating the first reviewer’s assessments. Disagreements were resolved through discussion to consensus or by a third reviewer. The assessment was performed at the study level. |
AE = adverse event; BCVA = best corrected visual acuity; CNV = choroidal neovascularization; CRT = central retinal thickness; CST = central subfield thickness; DME = diabetic macular edema; DRSS = Diabetic Retinopathy Severity Scale; IOP = intraocular pressure; IRF = intraretinal fluid; ITC = indirect treatment comparison; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; RCT = randomized controlled trial; SAE = serious adverse event; SRF = subretinal fluid; VEGF = vascular endothelial growth factor.
aOnly references presenting data for the overall population and/or relevant subgroups (age, baseline BCVA, baseline CNV area/CNV leakage area, CNV type, baseline CRT, retinal fluid presence, Asian population, patients with no previous experience of treatment, and patients with experience of anti-VEGF treatment) were included.
bBoth anti-VEGF monotherapies and anti-VEGFs combined with laser therapy.
cPooled results from pivotal studies to be included and extracted, only if data per each study not available. Pooled data for different drugs or drug doses or treatment regimens were not included/extracted.
dSmall studies excluded to reduce publication bias and uncertainty associated with small sample sizes.
Source: Sponsor-submitted ITC.63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
NMA Design
Objectives
The objective of the NMA was to assess the comparative efficacy and safety of aflibercept 8 mg (administered every 12 weeks and every 16 weeks) with the interventions listed in Table 18.
Methods for the Search, Selection, Data Extraction, and Risk-of-Bias Appraisal
The identification of studies suitable for the ITC was based on a systematic literature review of RCTs of anti-VEGF drugs for patients with DME. MEDLINE and Embase were searched on May 24, 2022, and the Cochrane Central Register of Controlled Trials and clinical trials registry ClinicalTrials.gov were searched on May 26, 2022.63
The population of interest was limited to patients with a diagnosis of DME (those with no experience of treatment and those who had previously received treatment); no specific geographic restrictions were applied. Included publications were limited to articles written in English and studies reporting data on 40 or more participants or eyes. The sponsor indicated that small studies were excluded to reduce publication bias and uncertainty associated with small sample sizes.63
Two reviewers, working independently, screened the retrieved search results. One reviewer conducted the data extraction while another validated the extracted data.63
The quality of the included studies was appraised by 1 reviewer according to the Cochrane risk-of-bias tool (version 2.0) checklist.64 A second reviewer validated the first reviewer’s assessments. Disagreements were resolved through discussion to consensus or by a third reviewer. The assessments were performed at the study level.
NMA Analysis Methods
NMAs were conducted for all the reported outcomes, except the number of injections. There were 3 different types of NMAs to reflect the nature of the outcomes of interest. The analysis of changes in BCVA was conducted with a normal likelihood and an identity link. Analyses of adverse events were conducted with a binomial likelihood and logit link. Analyses of gaining or losing letters were conducted with multinomial likelihood and a probit link. The NMA was based on a Bayesian approach and computed through a Markov chain Monte Carlo simulation. Vague (noninformative) priors were used. Markov chain Monte Carlo simulation was based on 5,000 burn-ins (adaptation period) followed by at least 10,000 iterations with convergence assessed through trace, density, and Bayesian Gelman-Rubin plots. Each NMA was conducted under both fixed-effects and random-effects models. The model with better model fit as determined by a lower value of the deviance information criterion (DIC) was chosen as the primary model. As applicable, median odds ratios and treatment differences (drawn from posterior distributions) and the corresponding 95% credible intervals were reported.63
Clinical and methodological heterogeneity of included studies was assessed through tabulating and contrasting study characteristics and baseline patient characteristics.63
To assess the inconsistency of a network, the node-splitting method was used. In this approach, each treatment effect is estimated separately using direct and indirect evidence; these 2 estimates were then contrasted.63
Analysis for the outcome of mean number of injections was based on a within-node meta-analysis with no comparison between nodes. Specifically, to estimate the number of injections required for each anti-VEGF drug and dosing regimen, meta-analyses were conducted based on the number of injections reported in RCTs during the first year of treatment, during the first 2 years of treatment, and between year 1 and year 2 of treatment. Random-effects meta-analysis models were used to estimate the average numbers of injections for each treatment and dosing regimen examined. The DerSimonian-Laird random-effects method was used to calculate treatment effects as well as the between-trial variability.63 The P value for Cochran Q test and the I2 statistic were provided on forest plots, quantifying the extent of statistical heterogeneity. Clinical and methodological heterogeneity was not described.
To address missing data in clinical study reports, standard formulas were used to calculate missing quantities, and when data were unavailable, values were imputed based on similar studies in the evidence base. Continuous inputs for the model included the difference from baseline for BCVA gain and CRT or central subfield thickness (CST) change, along with the mean number of injections—all with their respective SEs for each study arm. Binary inputs were incorporated for the proportion of patients achieving specified visual acuity thresholds and for safety outcomes, notably incidence of adverse events.
When SEs required for the NMA were not properly reported, they were derived from CIs or interquartile ranges using specific formulas. For instances where precision data (e.g., SD) were absent, values were imputed based on comparable regimens from other studies. In cases of 0 event counts, a continuity correction was applied to mitigate computational issues, ensuring a balanced risk addition across uneven study arms. In addition, for CRT or CST values, due to baseline variability, a percentage change from baseline was computed, with the SEs adjusted accordingly to maintain accuracy in variance estimation. This rigorous methodology ensures consistency and reliability in the meta-analytical outcomes despite inherent data heterogeneity and gaps.
Standard definition of outcomes was used whenever possible. Variability existed in retinal thickness assessments where “CRT” and “CST” were often used interchangeably. Outcome reporting times varied cross studies; however, for consistency, only those results reported at approximately 1 year (48, 52, or 56 weeks) were included without adjusting the clinical outcomes to ensure minimal bias introduction. Nevertheless, the number of injections—an outcome directly affected by the duration of observation—required time point adjustments to a standardized 52-week period to avoid bias in arm-based pooling analyses. This was accomplished using adherence factors for fixed-dosing regimens and proportionate adjustments for flexible regimens, with SEs adjusted accordingly.63
Of the prespecified outcomes, the sponsor-submitted report included the following outcomes: change in BCVA, change in CRT and CST, percentage change from baseline CRT or CST, gain of 10 or more ETDRS letters, gain of 15 or more ETDRS letters, loss of 10 or more ETDRS letters, loss of 15 or more ETDRS letters, ocular adverse events, and the mean number of injections. The reason for not reporting the rest of the preplanned outcomes listed in Table 17 was not clear. However, to adhere to the list of outcomes that was determined through feedback from parties of interest, CDA-AMC has not reported the outcome of change in CRT and CST, gain of 10 ETDRS letters, loss of 10 ETDRS letters, and loss of 15 ETDRS letters.63
Table 18: ITC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Bayesian network meta-analysis (random effects or fixed effects, based on the value of the deviance information criterion) |
Priors | Vague |
Assessment of model fit | Deviance information criterion |
Assessment of consistency | Node-splitting method |
Assessment of convergence | Trace and Bayesian Gelman-Rubin plots |
Outcomes | Change in BCVA, gain of ≥ 10 letters, gain of ≥ 15 letters, loss of ≥ 10 letters, loss of ≥ 15 letters, ocular adverse events |
Follow-up time points | Outcomes reported between 48 and 52 weeks were treated as if reported at 1 year, while outcomes reported between 96 and 104 weeks were treated as if reported at 2 years. |
Construction of nodes | Not reported |
Sensitivity analyses | None |
Subgroup analysis | None |
Methods for pairwise meta-analysis | DerSimonian-Laird random-effects method (number of injections) |
BCVA = best corrected visual acuity; ITC = indirect treatment comparison.
Source: Sponsor-submitted network meta-analysis.63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Results of the Sponsor-Submitted ITC
Summary of Included Studies
A total of 17 studies were included in the NMA: 1 assessed aflibercept 8 mg,65,66 11 assessed aflibercept 2 mg,66-74 6 assessed ranibizumab,75-79 2 assessed faricimab,70,74 9 assessed laser therapy as needed,66,69,72,75-78,80 2 assessed brolucizumab,81,82 and 2 assessed bevacizumab.71,80
The 17 studies included in the analysis were conducted in a variety of ways. Most were multicentre phase III trials enrolling more than 100 patients, though a few enrolled less than 100 patients. The sample sizes of included studies varied (80 to 951 patients). The BOLT study and Chatzirallis 2020 study both had fewer patients than the other studies.68,80 Differences in baseline CRT and CST and duration of DME were also noticeable, but it should be noted that only 7 studies provided data for duration of DME. Baseline CRT ranged from a mean of 111.8 μm to a mean of 540 μm. Mean baseline age across included studies ranged from 55 years to 65 years, representing a source of heterogeneity. Heterogeneity was also observed in baseline BCVA measurements, ranging from a mean of 56.3 ETDRS to 69 ETDRS, as well as variability in hemoglobin A1C levels, with means ranging from 1.72% to 8.0%.7
Treatment patterns varied across the studies, but in most cases the sponsor considered that the studies were conducted similarly enough that treatment nodes were consistently defined. An exception to this is the bevacizumab as-needed node, which consisted of data from the BOLT and Protocol T studies only.71,80 While the scheduled frequency of administration differed between these 2 trials, they were combined into a single node as they were the only studies in the network that examined bevacizumab. The BOLT study began with an injection at baseline, followed by injections at 6 and 12 weeks. Following this initial phase of the treatment, patients were assessed every 6 weeks, and received injections if the macular thickness was not stable. In the Protocol T study, however, patients received an injection at baseline, and were thereafter assessed every 4 weeks, with injections administered if patients’ clinical condition worsened. Both the criteria to determine whether to give injections, and the frequency at which patients were assessed differed. Furthermore, the Protocol T treatment could be labelled “as needed without loading dose.” Despite these differences, the data were combined into a single node because the sponsor considered them to be sufficiently similar.7
The network of evidence was heterogenous in other ways as well. The proportion of patients with no experience of treatment differed across studies (though this was not reported in 11 out of the 17 included trials). The newer studies, on the outside of the network (the RHINE, YOSEMITE, KITE, and KESTREL studies), had high proportions of patients with no experience of treatment. However, the DA VINCI study included a low proportion of patients with no experience of treatment. This is notable, because the DA VINCI study occupies a central place in the network, and many treatment comparisons are made through this trial.7,67,70
The quality assessment process identified 2 of the included studies as having a high risk of overall bias. No specific actions were taken for these studies.
Table 19: Assessment of Homogeneity for ITC
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity |
|
Treatment history | Variation in the proportion of patients experienced with treatment |
Trial eligibility criteria | Overall, similar inclusion and exclusion criteria |
Dosing of comparators | Certain comparators are dosed as per indication. Some comparators are given at a treat-and-extend regimen |
Placebo response | NA |
Definitions of end points | Similar use of ETDRS in most trials to assess BCVA |
Timing of end point evaluation | Outcomes reported between 48 and 52 weeks were treated as if reported at 1 year, while outcomes reported between 96 and 104 weeks were treated as if reported at 2 years. |
Withdrawal frequency | Unclear due to lack of reporting |
Clinical trial setting | Likely similar due to the nature of the injection |
Study design | Some variation in masking of intervention where 1 trial was open label, 3 were single masked, and 1 was unclear. |
BCVA = best corrected visual acuity; CRT = central retinal thickness; ETDRS = Early Treatment Diabetic Retinopathy Study; ITC = indirect treatment comparison; NA = not applicable.
Source: Sponsor-submitted network meta-analysis.63 Details included in the table are from the sponsor’s Summary of Clinical Evidence.7
Change from Baseline BCVA
The changes in BCVA letters from baseline to assessment at 1 year were analyzed using an NMA. The network of evidence is shown in Figure 2. Of note is that the network has some long pathways between treatments, which results in less precise estimates. As an example, comparisons of aflibercept 8 mg to bevacizumab are calculated through at least 2 other nodes aflibercept 2 mg as needed and every 8 weeks. Both random-effects and fixed-effects models were considered, and based on the DIC, the random-effects models were deemed to fit the data better (Table 20).63
Figure 2: Evidence Network of Change From Baseline BCVA
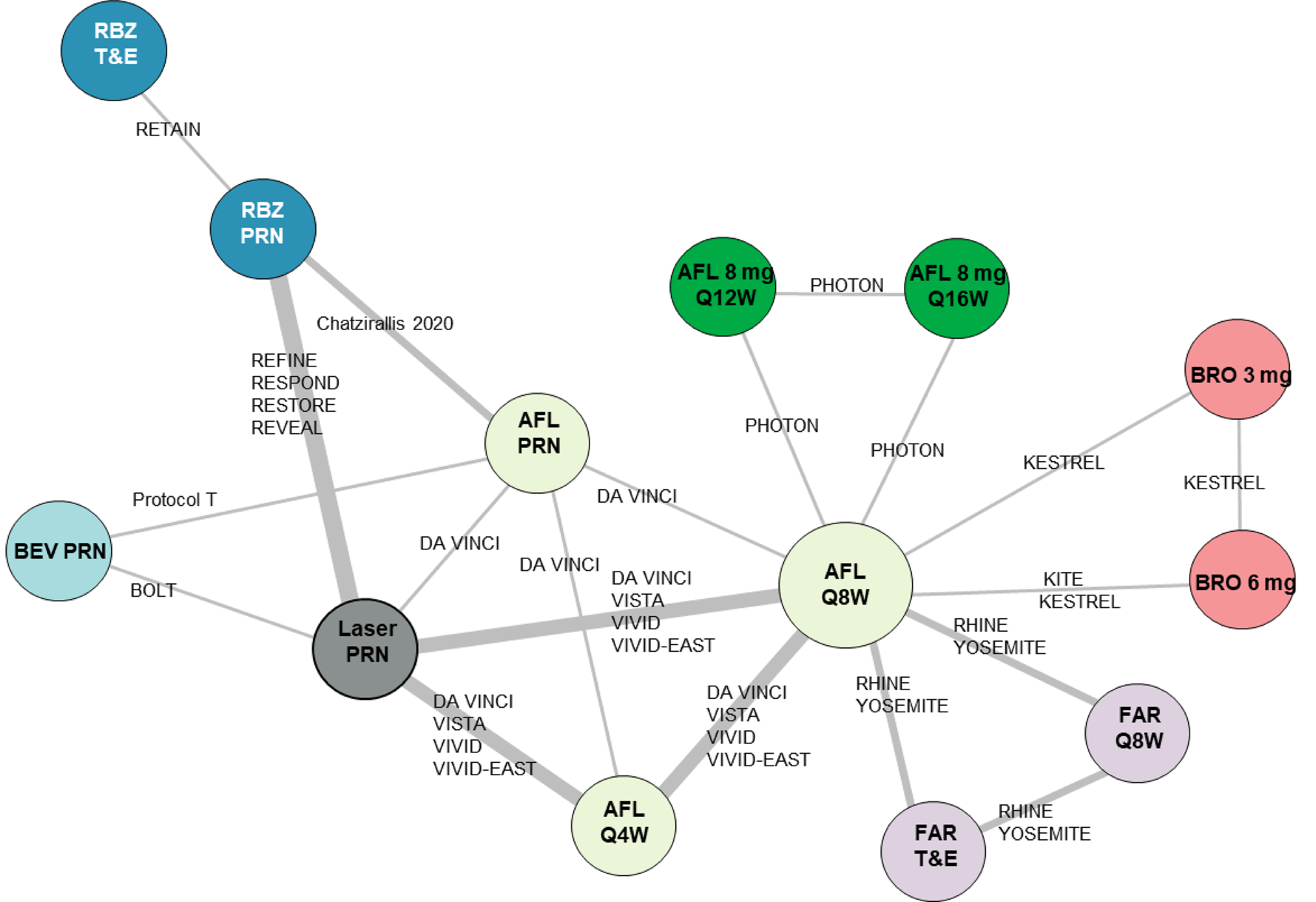
AFL = aflibercept; BCVA = best corrected visual acuity; BEV = bevacizumab; BRO = brolucizumab; FAR = faricimab; PRN = as needed; Q4W = every 4 weeks; Q8W = every 8 weeks; Q12W = every 12 weeks; Q16W = every 16 weeks; RBZ = ranibizumab; T&E = treat and extend.
Source: Sponsor-submitted network meta-analysis.63
Table 20: Model Fit and DIC for Change From Baseline BCVA
Model | Dbar | pD | DIC |
|---|---|---|---|
Fixed effects | 57.97 | 29.02 | 86.99 |
Random effects | 45.00 | 37.06 | 82.06 |
BCVA = best corrected visual acuity; Dbar = posterior mean of the deviance; DIC = deviance information criterion; pD = effective number of parameters.
Source: Sponsor-submitted network meta-analysis.63
In terms of mean change from baseline in BCVA letters, the analysis consistently included the null in the credible interval between aflibercept 8 mg and the other anti-VEGF treatments examined in the NMA, with the differences in numbers of letters being close to zero (null) in almost all cases, and within the 4 letters identified as clinically important, while associated with wide credible intervals. These findings are consistent when dosing every 12 weeks and every 16 weeks. Laser therapy was less effective compared to treatment with aflibercept 8 mg every 12 weeks and every 16 weeks, with the null excluded in the credible interval and the lower bound of the credible interval greater than 4 ETDRS letters.63


Gain or Loss in Number of Letters
Gain and loss of letters was analyzed with a conditional binomial NMA model. Patients were categorized into mutually exclusive groups (those losing ≥ 15 letters, those losing between 10 and 15 letters, those losing < 10 letters to gaining < 10 letters, those gaining between 10 and 15 letters, and those gaining ≥ 15 letters), and the probabilities of falling into each group are modelled simultaneously.63 The network of evidence used in this analysis is presented in Figure 5.
The conditional binomial model is substantially more complicated than the models used for other outcomes. This complication resulted in some difficulties in model fit. The fixed-effects model had a substantially lower DIC than the random-effects model and was selected as the most stable (Table 21).63 The large difference in DICs is due to the random-effects model having a notably larger deviance. This suggests that the random-effects weighting resulted in some trials not fitting the model well.63
Aflibercept 8 mg every 12 weeks showed ██ ████████████ response against aflibercept 2 mg every 4 weeks ███ ████ ███████ █ █████ ██████ ███ █ ██████████ against laser therapy ███ ████ ███████ █████ █████). Aflibercept 8 mg every 16 weeks showed ██ ████████████ response against aflibercept 2 mg every 8 weeks ███ ████ ███████ █████ ██████ ███ against aflibercept 2 mg every 4 weeks ███ ████ ██████ █████ ██████, and showed a █████████ odds ratio versus laser therapy ███ ████ ████ █████ ███████ In all other comparisons, the credible intervals for the odds ratios included the null.
The relative effects of aflibercept 8 mg every 12 weeks and every 16 weeks compared to the other treatments are shown in ██████ | and Figure 7: ██████ |, respectively, for a gain of 15 or more letters.
Figure 5: Evidence Network of Gain or Loss of 10 or 15 Letters
AFL = aflibercept; BEV = bevacizumab; BRO = brolucizumab; FAR = faricimab; PRN = as needed; Q4W = every 4 weeks; Q8W = every 8 weeks; Q12W = every 12 weeks; Q16W = every 16 weeks; RBZ = ranibizumab; T&E = treat and extend.
Source: Sponsor-submitted network meta-analysis.63
Table 21: Model Fit and DIC for Gain or Loss of Letters
Model | Dbar | pD | DIC |
|---|---|---|---|
Fixed effects | 686.18 | 31 | 717.18 |
Random effects | 1,864.1 | 36.86 | 1,900.96 |
DIC = deviance information criterion; Dbar = posterior mean of the deviance? sum of residual deviances; DIC = deviance information criterion; pD = effective number of parameters.
Source: Sponsor-submitted network meta-analysis.63


Harms
Ocular Adverse Events
An NMA on binomial outcomes was used to analyze ocular adverse events. The network of evidence is presented in Figure 8. A total of 13 studies reported the number of ocular adverse events experienced in trials. Based on the DIC (Table 22), fixed-effects models were selected when modelling ocular events.63
Figure 8: Evidence Network of Ocular Adverse Events
AFL = aflibercept; BEV = bevacizumab; BRO = brolucizumab; FAR = faricimab; PRN = as needed; Q4W = every 4 weeks; Q8W = every 8 weeks; Q12W = every 12 weeks; Q16W = every 16 weeks; RBZ = ranibizumab; T&E = treat and extend.
Source: Sponsor-submitted network meta-analysis.63
Table 22: Model Fit and DIC for Ocular Adverse Event
Model | Dbar | pD | DIC |
|---|---|---|---|
Fixed effects | 31.75 | 24.10 | 55.85 |
Random effects | 31.23 | 26.25 | 57.47 |
DIC = deviance information criterion; Dbar = posterior mean of the deviance? sum of residual deviances; DIC = deviance information criterion; pD = effective number of parameters.
Source: Sponsor-submitted network meta-analysis.63
The relative effect of treatments on the number of ocular adverse events do not exclude the null in almost all treatment comparisons. The odds ratios of aflibercept 8 mg regimens are near 1 for almost all comparisons, and credible intervals are wide, such that no comparisons excluded the null in the credible interval. In addition, the extent of the imprecision is uncertain in the absence of absolute effect estimates. While some point estimates show favourable results for aflibercept 8 mg in the comparisons versus faricimab and ranibizumab, none of these comparisons excluded the null in the credible interval. An exception to this consistent finding is the comparison with bevacizumab, which shows a favourable ocular safety finding for aflibercept when compared to bevacizumab.


Non-Ocular Adverse Events
Reporting of non-ocular adverse events was not as comprehensive as the reporting for other outcomes, and there was limited evidence to assess non-ocular adverse events across studies. Therefore, an NMA for non-ocular adverse events was not reported.
Number of Injections
Based on predetermined injections regimens, certain interventions are expected to have an average number of injections observed for each treatment regimen consistent with the number of injections planned. Intervention administered on a fixed schedule did not show much variability between the planned and the actual number of injections given. Treat-and-extend and as-needed regimens are not predetermined and show a mean number of injections between 7.0 and 9.18 across the interventions in the first year.
Estimates of the average number of injections received within the first year of treatment are presented in Table 23 and of the average number of injections received during the second year are presented in Table 24.63
Table 23: Mean Injection Frequency – Meta-Analysis Estimates for 1-Year Results
Regimen | Aflibercept 2 mg | Aflibercept 8 mg | Bevacizumab | Brolucizumab 3 mg | Brolucizumab 6 mg | Ranibizumab | Faricimab |
|---|---|---|---|---|---|---|---|
q.4.w. | ██ ████ | — | — | — | — | — | — |
q.8.w. | ██████ | — | — | — | — | — | ██████ |
q.12.w. | — | 6.00 █ ██ | — | ███ ███ | ██████ | — | — |
q.16.w. | — | 5.00 ████ | — | — | — | — | — |
p.r.n. | ██████ | — | ██████ | — | — | ███ ███ | — |
Treat and extend | — | — | — | — | — | █████ | ███ ███ |
p.r.n. = as needed; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; RBZ = ranibizumab.
Table 24: Mean Injection Frequency – Meta-Analysis Estimates for 1- to 2-Year Results
Regimen | Aflibercept 2 mg | Aflibercept 8 mg | Bevacizumab | Brolucizumab 6 mg | Ranibizumab | Faricimab |
|---|---|---|---|---|---|---|
q.4.w. | ████████ | — | — | — | — | — |
q.8.w. | ███ ████ | — | — | — | — | ████████ |
q.12.w. | — | 3.50 ████ | — | ████ ████ | — | — |
q.16.w. | — | 2.80 ████ | — | — | — | — |
p.r.n. | ████████ | — | ████ ████ | — | ███ █████ | — |
Treat and extend | — | — | — | — | ████ ████ | ███ █████ |
p.r.n. = as needed; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks.
Critical Appraisal of ITC
Studies included in the sponsor-submitted ITC were identified through a systematic literature review approach. The inclusion and exclusion criteria were appropriate with the possible exception of the exclusion of studies with fewer than 40 patients or eyes, which resulted in a total of ███ trials being excluded. The justification for the exclusion of small population studies was to avoid potential biases arising from small sample sizes. However, this approach also increases the potential for publication bias and risk of bias due to missing evidence in the synthesis. The search was originally conducted in 2022 but it was updated in July 2023. The approach where 2 reviewers conducting the screening, with 1 reviewer conducting the risk-of-bias assessment and the other validating the output, minimized the risk of error and bias in the assessments. The risk-of-bias assessment was conducted using the Cochrane risk-of-bias tool (version 2.0) and 2 studies were identified as potentially high-risk, but no clear steps were taken to investigate (e.g., via sensitivity analyses) how studies with high risk of bias may have impacted the results. It is important to note that risk of bias was assessed at the study level, rather than at the level of the reported result. The risk of bias for individual effect estimates reported in each study could differ; as such, the study-level risk-of-bias assessments may not equally apply to all study results.
Outcomes that were included in the ITC were clinically relevant and appropriate; however, some outcomes that are important to patients, such as vision-related quality of life, were not investigated. Furthermore, data related to ocular and non-ocular SAEs could not be analyzed, so no results are available for these outcomes. Results for other outcomes that patients, clinicians, and drug plans considered to be important were included in the protocol but were not reported. It is important to note that studies of interventions that have a preplanned and fixed injection regimen may not reflect clinical practice in Canada where the treat-and-extend approach predominates. As such, it is likely that indirect comparison versus treat-and-extend regimens are the most informative.
The approach by the sponsor ITC to assess the results through a Bayesian NMA was appropriate and transparently communicated.
Clinical heterogeneity across the studies was difficult to assess due to the lack of reporting on several key demographic, baseline, and study characteristics in the studies. Of the available information, several baseline characteristics, including age, baseline BCVA, hemoglobin A1C, and proportion of treatment-experienced patients were variable. It is not clear whether these characteristics would be treatment-effect modifiers. As such, it is uncertain as to whether the assumptions related to homogeneity were met. This is further substantiated by the finding of several pairwise statistical heterogeneity in the network.
The large credible intervals for all estimates suggest that there is substantial variability in the data, which reduces the certainty of the conclusions that can be drawn from these estimates. This may reflect the differences in study populations across the various trials included in the analyses, as the studies differed across many characteristics. In many cases, treatments are informed by only a single trial. Because of this, there is risk that effect modifiers led to biases in the estimates of treatment effect. In indirect comparisons that are informed by only 1 trial at each side, the importance of the overall representation of the population in these trials and the balance of the baseline characteristics between these trials becomes more critical than scenarios where there are multiple trials informing the comparison. This is an assumption that is required in this network given the limitations to the evidence base. When only odds ratios with 95% credible intervals were reported (i.e., in the absence of absolute effect estimates), it was not possible to fully judge the extent of the imprecision.
Assessment of the frequency of injections were based on absolute results and no comparative results were provided. This limits the ability to assess the comparative benefit or harm of various interventions and variability in the comparative results. Descriptive and noncomparative number of injections suggest that treatment with aflibercept 8 mg is likely to result in fewer injections than most other treatments that are administered according to a prescribed standard regimen. However, some regimens that utilize an as-needed or treat-and-extend approach administer a higher but nevertheless relatively similar number of injections as the mean number of injections of aflibercept 8 mg, especially when administered every 12 weeks. Although, considering the lack of comparative assessment in the NMA for injection frequency, no statistical inference can be made regarding the difference in the number of injections. This, combined with the limitations of the NMA, makes any discussion on the comparative number of injections highly speculative and not supported by evidence. Considering the limitations associated with the clinical heterogeneity and the resulting wide credible intervals, the indirect comparative efficacy results of 8 mg aflibercept cannot be used to inform decision-making on their own and in the absence of other type of evidence.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Discussion
Summary of Available Evidence
The evidence included in this review consisted of 1 pivotal phase II/III double-blind RCT and 1 ITC submitted by the sponsor.
One trial, PHOTON (N = 660), met the inclusion criteria for the systematic review conducted by the sponsor. PHOTON was a phase III, active-controlled, noninferiority, multinational (138 sites, including 4 sites in Canada) trial that randomized 660 patients with DME in a 1:2:1 ratio to aflibercept 2 mg every 8 weeks, aflibercept 8 mg every 12 weeks, or aflibercept 8 mg every 16 weeks, respectively. The primary outcome was change from baseline in BCVA measured using the ETDRS letter score at week 48, and a key secondary outcome was change from baseline in BCVA measured using the ETDRS letter score at week 60. Other secondary and exploratory outcomes included the proportion of participants with no IRF and no SRF in the foveal centre and the central subfield, the proportion of participants gaining at least 15 letters in BCVA from baseline, and vision-related quality of life. These outcomes were all assessed at weeks 48 and 60. Total number of injections, TEAEs, and SAEs through week 60 were reported as harms.
The treatment arms were generally well-balanced with a few exceptions. Patients were similar in age across treatment arms, with those receiving aflibercept 8 mg every 16 weeks on average slightly younger (mean age of 61.9 years [SD = 9.50 years]) than the patients receiving aflibercept 2 mg every 8 weeks (mean age of 63.0 years [SD = 9.78 years]). There was a higher proportion of patients who were male in the aflibercept 8 mg every 12 weeks (64.0%) and every 16 weeks (60.7%) groups than in the aflibercept 2 mg every 8 weeks group (55.1%). The majority of patients were white, with a higher proportion who were white in the aflibercept 8 mg every 16 weeks group (78.5%) than in the 2 mg every 8 weeks group (67.1%). The mean duration of diabetes was similar across groups, and the majority of patients had type 2 diabetes. Ocular characteristics like BCVA and CRT were also similar across groups, with marginal variations in BCVA and CRT means between the different dosage groups. Overall, this study was well conducted, but has limitations to do with attrition and the generalizability of the control group.
The sponsor's Bayesian NMA, incorporating 17 studies, compared aflibercept 8 mg every 12 weeks and every 16 weeks with other anti-VEGF therapies for patients with DME, applying fixed-effects and random-effects models across varied outcomes, including BCVA change and ETDRS letter gain or loss. There was considerable clinical and statistical heterogeneity in the included studies and a lack of sufficient reporting to determine potential imbalances in treatment-effect modifiers. Comparative results from the end points show large credible intervals that frequently include the null. Aflibercept was associated with a consistent mean injection frequency that was aligned with the fixed schedule in the first year, that was notably less frequent in the second year, while variable regimens reported a broader range of numbers of injections.
Interpretation of Results
Efficacy
Patient group and clinician input has highlighted the significance of visual acuity, reduced frequency of treatments, and the overall impact on the quality of life for patients as critical considerations. According to clinical inputs received, there is a requirement for durable treatments that minimize adverse effects for individuals with DME. The PHOTON trial included clinically relevant outcomes to assess the efficacy of higher dose of aflibercept given at a longer treatment interval.
The outcomes of the PHOTON trial provided evidence supporting the noninferiority of aflibercept 8 mg given at 12-week or 16-week intervals compared with the standard aflibercept 2 mg at 8-week intervals in terms of mean BCVA change from baseline at week 60 among patients with DME. Topline results at week 96 were in support of the findings at week 60 and week 48. Study limitations included relatively high and imbalanced attrition and the inability to generalize to the context in Canada because of the use of a fixed regimen control group, which does not reflect clinical practice in Canada. The results at the 60-week mark indicated that the aflibercept regimens with longer treatment intervals were likely to have similar changes from baseline in BCVA as aflibercept 2 mg administered every 8 weeks, with little to no clinically important difference.
The percentage of patients who experienced a significant improvement in BCVA (gaining 15 or more ETDRS letters) was higher in the standard dosing-regimen group, albeit with low-certainty evidence due to risk of bias as a result of missing outcome data and imprecision. However, in the absence of a threshold for a clinically important difference, it is not clear whether the observed between-group difference is clinically important. Results from the vision-related quality-of-life measures suggest that there is little to no difference compared with aflibercept 2 mg. Similarly, there is little evidence to suggest that high-dose aflibercept is better or worse than 2 mg aflibercept in increasing the proportion of patients who do not have fluids in the foveal centre.
Aflibercept 8 mg given at 12-week or 16-week intervals showed a lower mean number of injections at week 48 and week 60 than the standard aflibercept 2 mg dosed at 8-week intervals; it is not clear whether the same benefit would be observed in clinical practice, where a treat-and-extend regimen of aflibercept 2 mg is typically used. The frequency of injections is a key outcome for patients and clinicians due to its implications on adverse events and quality of life. Based on this lower injection rate combined with the noninferiority in BCVA and on the primary results from the PHOTON trial, it can be concluded that the longer-interval aflibercept regimens are noninferior and not superior to the 2 mg every 8 weeks dose in terms of effect on the mean difference in BCVA. Nevertheless, that the 2 mg dose led to a higher proportion of patients gaining 15 or more ETDRS letters compared to 16 mg aflibercept is important to consider in the totality of evidence. In addition, the control group regimen, 2 mg aflibercept every 8 weeks, has limited generalizability because the treat-and-extend approach is the clinical practice in Canada.
Due to the high uncertainty in the clinical and statistical heterogeneity in the ITC, the results cannot be used to inform policy decision on their own and must be considered in totality with the available evidence from direct treatment comparisons.
Evidence gaps were identified in the lack of comparison to clinically relevant regimen of aflibercept (i.e., treat and extend), lack of direct evidence to inform the efficacy and safety versus other anti-VEGF therapies, and the lack of appropriate evidence to inform switching from other anti-VEGFs or regimens.
Harms
Ocular TEAEs were reported for less than half of the enrolled patients. Specifically, 43.7% of patients (n = 73) receiving aflibercept 2 mg every 8 weeks, 44.8% of patients (n = 147) receiving aflibercept 8 mg every 12 weeks, and 44.8% of patients (n = 73) receiving aflibercept 8 mg every 16 weeks experienced at least 1 ocular TEAE. At least 1 ocular treatment-emergent SAE was reported by 0.6% of patients (n = 1) receiving 2 mg every 8 weeks, 0.6% of patients (n = 2) receiving 8 mg every 12 weeks, and 1.2% of patients (n = 2) receiving 8 mg every 16 weeks. Specific examples included cataract subcapsular (1 event among patients receiving aflibercept 8 mg every 12 weeks), retinal detachment (1 event among patients receiving aflibercept 8 mg every 16 weeks), ulcerative keratitis (1 event among patients receiving aflibercept 2 mg every 8 weeks), and vitreous hemorrhage (1 event among patients receiving aflibercept 8 mg every 16 weeks). Because of the small number of SAEs, the evidence presented in this review is insufficient to inform the comparative safety of aflibercept 8 mg versus aflibercept 2 mg. Non-ocular SAEs were experienced by 19.2% of the patients receiving 2 mg every 8 weeks, 18.6% of the patients receiving 8 mg every 12 weeks, and 16.6% of the patients receiving 8 mg every 16 weeks.
Conclusion
DME is a progressive condition characterized by central vision loss as a complication of diabetes, and there is an unmet need for new treatments to improve visual acuity, reduce frequency of injections, improve vision-related quality of life, and reduce adverse events. According to evidence from the PHOTON trial, aflibercept 8 mg every 12 weeks and every 16 weeks demonstrates noninferiority (but not superiority) to aflibercept 2 mg every 8 weeks in terms of the change in BCVA from baseline at 48- and 60-weeks follow-up.
High-certainty evidence suggests that the mean difference in BCVA between treatment with aflibercept 8 mg every 12 weeks and every 16 weeks and treatment with aflibercept 2 mg every 8 weeks is of little to no clinical importance. Similarly, there is high-certainty evidence that aflibercept 8 mg every 12 weeks and every 16 weeks results in little to no clinically important difference in vision-related quality of life. Moderate-certainty evidence shows that aflibercept 8 mg every 12 weeks and every 16 weeks likely results in little to no difference in the proportion of patients without fluid in the foveal centre compared to aflibercept 2 mg every 8 weeks. There is high-certainty evidence that aflibercept 8 mg every 16 weeks results in a smaller proportion of patients gaining a BCVA of 15 ETDRS letters or more over 60 weeks compared to aflibercept 2 mg every 8 weeks, while evidence that aflibercept 8 mg every 12 weeks results in a smaller proportion of patients gaining a BCVA of 15 ETDRS letters or more over 60 weeks compared to aflibercept 2 mg every 8 weeks is of moderate certainty; however, the clinical importance of these differences is uncertain. There is moderate-certainty and low-certainty evidence that treatment with aflibercept 8 mg every 12 weeks and every 16 weeks, respectively, results in a higher proportion of patients with a BCVA of 69 letters or more at 60 weeks compared to aflibercept 2 mg every 8 weeks. Assessment of the certainty of ocular SAEs was rated as low.
There is low-certainty evidence that aflibercept 8 mg every 12 weeks or every 16 weeks, versus aflibercept 2 mg every 8 weeks, results in patients receiving fewer injections. This is partly due to the limited generalizability of this finding as aflibercept 2 mg is administered according to a treat-and-extend regimen in clinical practice, as opposed to every 8 weeks regimen in the trial.
Comparative efficacy findings in the ITC are insufficient, as standalone evidence, to inform the efficacy and safety of aflibercept 8 mg every 12 weeks and every 16 weeks versus other comparators. This is due to clinical and statistical heterogeneity, the imprecision in the results, as well as the lack of reporting on relevant clinical outcomes such as quality of life. Absolute noncomparative results of injection frequency suggest that aflibercept 8 mg every 12 weeks and every 16 weeks result in administration of a smaller number of injections compared with other interventions in the network. However, due to the lack of statistical comparison, no inference can be made as to the comparative difference in number of injections.
References
1.Romero-Aroca P. Targeting the pathophysiology of diabetic macular edema. Diabetes Care. 2010;33(11):2484-2485. PubMed
2.International Diabetes Federation. The Diabetic Retinopathy Barometer Report: Global findings [accessed by sponsor]. 2016; https://www.idf.org/our-activities/advocacy-awareness/resources-and-tools/92:diabetic-retinopathy-barometer.html.
3.Bandello F, Battaglia Parodi M, Lanzetta P, et al. Diabetic macular edema. Dev Ophthalmol. 2017;58:102-138. PubMed
4.Bailey I. Designation of visual acuity in logarithmic units [accessed by sponsor]. Optometric Monthly. 1980;71:80-85.
5.Petrella RJ, Blouin J, Davies B, Barbeau M. Prevalence, demographics, and treatment characteristics of visual impairment due to diabetic macular edema in a representative Canadian cohort. J Ophthalmol. 2012;2012:159167. PubMed
6.Kempen GI, Ballemans J, Ranchor AV, van Rens GH, Zijlstra GA. The impact of low vision on activities of daily living, symptoms of depression, feelings of anxiety and social support in community-living older adults seeking vision rehabilitation services. Qual Life Res. 2012;21(8):1405-1411. PubMed
7.Aflibercept 8 mg / 0.07 mL for the treatment of patients with diabetic macular edema (DME) Sponsor Summary of Clinical Evidence [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Aflibercept 8 mg / 0.07 mL, solution for intravitreal injection. Mississauga (ON): Bayer Inc.; 2022 Mar.
8.Drug Reimbursement Review sponsor submission: Ozurdex (dexamethasone intravitreal implant), 0.7 mg [internal sponsor's package]. Markham (ON): Allergan Inc.; 2022 Jun 7.
9.Bandello F, Lattanzio R, Zucchiatti I, Del Turco C. Pathophysiology and treatment of diabetic retinopathy. Acta Diabetol. 2013;50(1):1-20. PubMed
10.Mrugacz M, Bryl A, Zorena K. Retinal vascular endothelial cell dysfunction and neuroretinal degeneration in diabetic patients. J Clin Med. 2021;10(3). PubMed
11.Romero-Aroca P, Baget-Bernaldiz M, Pareja-Rios A, Lopez-Galvez M, Navarro-Gil R, Verges R. Diabetic macular edema pathophysiology: Vasogenic versus inflammatory. J Diabetes Res. 2016;2016:2156273. PubMed
12.Bhagat N, Grigorian RA, Tutela A, Zarbin MA. Diabetic macular edema: Pathogenesis and treatment. Surv Ophthalmol. 2009;54(1):1-32. PubMed
13.Singh A, Stewart JM. Pathophysiology of diabetic macular edema. Int Ophthalmol Clin. 2009;49(2):1-11. PubMed
14.Schmidt-Erfurth U, Garcia-Arumi J, Bandello F, et al. Guidelines for the management of diabetic macular edema by the European Society of Retina Specialists (EURETINA). Ophthalmologica. 2017;237(4):185-222. PubMed
15.Ciulla TA, Amador AG, Zinman B. Diabetic retinopathy and diabetic macular edema: Pathophysiology, screening, and novel therapies. Diabetes Care. 2003;26(9):2653-2664. PubMed
16.Mohamed Q, Gillies MC, Wong TY. Management of diabetic retinopathy: A systematic review. JAMA. 2007;298(8):902-916. PubMed
17.Williams R, Airey M, Baxter H, Forrester J, Kennedy-Martin T, Girach A. Epidemiology of diabetic retinopathy and macular oedema: A systematic review. Eye (Lond). 2004;18(10):963-983. PubMed
18.Public Health Agency of Canada. Framework for diabetes in Canada [accessed by sponsor]. 2022; https://www.canada.ca/en/public-health/services/publications/diseases-conditions/framework-diabetes-canada.html.
19.Advocating for improved treatment and outcomes for diabetic macular edema in Canada: A report based on the Canadian National Multi-Stakeholder Expert Summit for Diabetic Macular Edema convened in Toronto, January 2015 [accessed by sponsor]. Cambridge (MA): Angiogenesis Foundation: https://angio.org/wp-content/uploads/2016/01/Canada-DME-WP.pdf.
20.Bennett TJ, Barry CJ. Ophthalmic imaging today: An ophthalmic photographer's viewpoint - a review. Clin Exp Ophthalmol. 2009;37(1):2-13. PubMed
21.Trichonas G, Kaiser PK. Optical coherence tomography imaging of macular oedema. Br J Ophthalmol. 2014;98 Suppl 2(Suppl 2):ii24-29.
22.Flaxel CJ, Adelman RA, Bailey ST, et al. Age-Related Macular Degeneration Preferred Practice Pattern®. Ophthalmology. 2020;127(1):P1-P65. PubMed
23.Schmidt-Erfurth U, Chong V, Loewenstein A, et al. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA). Br J Ophthalmol. 2014;98(9):1144-1167. PubMed
24.Diabetic retinopathy guidelines. London (GB): The Royal College of Ophthalmologists; 2012: https://www.rcophth.ac.uk/wp-content/uploads/2021/08/2012-SCI-267-Diabetic-Retinopathy-Guidelines-December-2012.pdf. Accessed 2023 Oct 3.
25.Elyasi N, Hemmati HD. Diabetic macular edema: Diagnosis and management. EyeNet Magazine. 2021;May 2021. https://www.aao.org/eyenet/article/diabetic-macular-edema-diagnosis-and-management. Accessed 2023 Oct 3.
26.Diabetes Canada 2018 Clinical Practice Guidelines for the Prevention and Management of Diabetes in Canada. Can J Diabetes. 2018;42:S1-S325. https://www.canadianjournalofdiabetes.com/issue/S1499-2671(17)X0005-1. Accessed 2023 Oct 3.
27.Drug Reimbursement Review clinical guidance report: Faricimab (Vabysmo) For the treatment of diabetic macular edema. Can J Health Technol. 2022;2(10). https://www.cadth.ca/sites/default/files/DRR/2022/SR0729%20Vabysmo%20-%20Confidential%20Final%20CADTH%20Recommendation-KH_BF%20-%20KH-meta.pdf. Accessed 2023 Oct 3.
28.Udaondo P, Parravano M, Vujosevic S, Zur D, Chakravarthy U. Update on current and future management for diabetic maculopathy. Ophthalmol Ther. 2022;11(2):489-502. PubMed
29.CADTH Reimbursement Review: Brolucizumab (Beovu) [accessed by sponsor]. Can J Health Technol. 2023;3(3). https://www.cadth.ca/sites/default/files/DRR/2023/SR0747-Beovu_combined.pdf.
30.Adams BS, Sorhaitz W, Stringham J. Aflibercept [accessed by sponsor]. StatPearls. Treasure Island (FL): StatPearls Publishing; 2023.
31.Drug Reimbursement Review clinical guidance report: Aflibercept (Eylea) for macular edema secondary to central retinal vein occlusion. Ottawa (ON): CADTH; 2015: https://www.cadth.ca/sites/default/files/cdr/complete/cdr_complete_SR0401_Eylea-CRVO_May-11-15.pdf. Accessed 2023 Oct 3.
32.CADTH Canadian Drug Expert Committee final recommendation: Aflibercept (Eylea – Bayer Inc.) new indication: Macular edema secondary to branch retinal vein occlusion. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/cdr/complete/SR0460_Eylea%20BRVO_complete_Jul-29_16.pdf. Accessed 2023 Oct 3.
33.Aflibercept 8mg/ 0.07 mL, solution for intravitreal injection [product monograph]. Mississauga (ON): Bayer Inc.; 2024 Feb 2.
34.Vabysmo (faricimab injection): single use vials, 6mg/0.05mL solution for intravitrial injection [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2022 May 27.
35.Eylea (aflibercept injection): single use pre-filled syringes and single use vials for the treatment of a single eye. 2 mg / 0.05 mL solution for intravitreal injection [product monograph]. Mississauga (ON): Bayer Inc; 2022 Feb 10: https://pdf.hres.ca/dpd_pm/00064757.PDF. Accessed 2022 Apr 26.
36.Lucentis (ranibizumab injection): single use vials. Single use pre-filled syringes. 10 mg/mL solution for injection [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2021 Dec 21: https://pdf.hres.ca/dpd_pm/00064117.PDF. Accessed 2022 Apr 26.
37.Beovu® brolucizumab injectionSingle-use pre-filled syringes, single-use vials, 6 mg / 0.05 mL solution for intravitreal injection [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2022 Apr 12.
38.Avastin (bevacizumab for injection): 100 mg and 400 mg vials (25 mg/mL solution for injection) [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Jan 14: https://pdf.hres.ca/dpd_pm/00059621.PDF.
39.Clinical Study Report: VGFTe-HD-DME-1934. A randomized, double-masked, active-controlled Phase 2/3 study of the efficacy and safety of high-dose aflibercept in patients with diabetic macular edema PHOTON [internal sponsor's report]. Mississauga (ON): Bayer Inc.; 2023 Jan 12.
40.Ferris FL, Kassoff A, Bresnick GH, Bailey I. New visual acuity charts for clinical research. Am J Ophthalmol. 1982;94(1):91-96. PubMed
41.Riedl S, Vogl WD, Waldstein SM, Schmidt-Erfurth U, Bogunovic H. Impact of intra- and subretinal fluid on vision based on volume quantification in the HARBOR Trial. Ophthalmol Retina. 2022;6(4):291-297. PubMed
42.Mangione CM, Lee PP, Gutierrez PR, Spritzer K, Berry S, Hays RD. Development of the 25-item National Eye Institute Visual Function Questionnaire. Arch Ophthalmol. 2001;119(7):1050-1058. PubMed
43.Lloyd AJ, Loftus J, Turner M, Lai G, Pleil A. Psychometric validation of the Visual Function Questionnaire-25 in patients with diabetic macular edema. Health Qual Life Outcomes. 2013;11:10. PubMed
44.Mangione CM, Berry S, Spritzer K, et al. Identifying the content area for the 51-item National Eye Institute Visual Function Questionnaire: Results from focus groups with visually impaired persons. Arch Ophthalmol. 1998;116(2):227-233. PubMed
45.Revicki DA, Rentz AM, Harnam N, Thomas VS, Lanzetta P. Reliability and validity of the National Eye Institute Visual Function Questionnaire-25 in patients with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51(2):712-717. PubMed
46.Marella M, Pesudovs K, Keeffe JE, O'Connor PM, Rees G, Lamoureux EL. The psychometric validity of the NEI VFQ-25 for use in a low-vision population. Invest Ophthalmol Vis Sci. 2010;51(6):2878-2884. PubMed
47.Pesudovs K, Gothwal VK, Wright T, Lamoureux EL. Remediating serious flaws in the National Eye Institute Visual Function Questionnaire. J Cataract Refract Surg. 2010;36(5):718-732. PubMed
48.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
49.Orr P, Rentz AM, Margolis MK, et al. Validation of the National Eye Institute Visual Function Questionnaire-25 (NEI VFQ-25) in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(6):3354-3359. PubMed
50.Kniestedt C, Stamper RL. Visual acuity and its measurement. Ophthalmol Clin North Am. 2003;16(2):155-170, v. PubMed
51.Beck RW, Maguire MG, Bressler NM, Glassman AR, Lindblad AS, Ferris FL. Visual acuity as an outcome measure in clinical trials of retinal diseases. Ophthalmology. 2007;114(10):1804-1809. PubMed
52.Sánchez-González MC, García-Oliver R, Sánchez-González JM, Bautista-Llamas MJ, Jiménez-Rejano JJ, De-Hita-Cantalejo C. Minimum detectable change of visual acuity measurements using ETDRS charts (Early Treatment Diabetic Retinopathy Study). Int J Environ Res Public Health. 2021;18(15). PubMed
53.Beck RW, Moke PS, Turpin AH, et al. A computerized method of visual acuity testing: Adaptation of the early treatment of diabetic retinopathy study testing protocol. Am J Ophthalmol. 2003;135(2):194-205. PubMed
54.Rosser DA, Cousens SN, Murdoch IE, Fitzke FW, Laidlaw DA. How sensitive to clinical change are ETDRS logMAR visual acuity measurements? Invest Ophthalmol Vis Sci. 2003;44(8):3278-3281. PubMed
55.Bayer. NCT01331681: Intravitreal Aflibercept Injection in Vision Impairment Due to DME (VIVID-DME). ClinicalTrials.gov. Bethesda (MD): National Library of Medicine; 2016: https://clinicaltrials.gov/study/NCT01331681?tab=results. Accessed 2023 Oct 3.
56.Regeneron Pharmaceuticals. NCT01363440: Study of Intravitreal Aflibercept Injection (IAI; EYLEA®; BAY86-5321) in Patients With Diabetic Macular Edema (VISTA DME). ClinicalTrials.gov. Bethesda (MD): National Library of Medicine; 2011: https://classic.clinicaltrials.gov/ct2/show/NCT01363440. Accessed 2023 Oct 3.
57.Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Early Treatment Diabetic Retinopathy Study research group. Arch Ophthalmol. 1985;103(12):1796-1806. PubMed
58.Blankenship GW. Diabetic macular edema and argon laser photocoagulation: A prospective randomized study. Ophthalmology. 1979;86(1):69-78. PubMed
59.Brown DM, Schmidt-Erfurth U, Do DV, et al. Intravitreal aflibercept for diabetic macular edema: 100-week results from the VISTA and VIVID studies. Ophthalmology. 2015;122(10):2044-2052. PubMed
60.Mallinckrodt CH, Sanger TM, Dube S, et al. Assessing and interpreting treatment effects in longitudinal clinical trials with missing data. Biol Psychiatry. 2003;53(8):754-760. PubMed
61.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
62.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
63.NMA: Aflibercept 8 mg for the treatment of diabetic macular edema (DME) in Canada [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Aflibercept 8 mg / 0.07 mL, solution for intravitreal administration. Vancouver, BC: Bayer Inc.; 2023 Aug 16.
64.Sterne JAC, Savovic J, Page MJ, et al. RoB 2: A revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898. PubMed
65.Bayer. PHOTON clinical trial: Data on file [sponsor supplied reference]. 2023.
66.Do DV, Schmidt-Erfurth U, Gonzalez VH, et al. The DA VINCI Study: Phase 2 primary results of VEGF Trap-Eye in patients with diabetic macular edema. Ophthalmology. 2011;118(9):1819-1826. PubMed
67.Brown DM, Emanuelli A, Bandello F, et al. KESTREL and KITE: 52-week results from two Phase III pivotal trials of brolucizumab for diabetic macular edema. Am J Ophthalmol. 2022;238:157-172. PubMed
68.Chatzirallis A, Theodossiadis P, Droutsas K, Koutsandrea C, Ladas I, Moschos MM. Ranibizumab versus aflibercept for diabetic macular edema: 18-month results of a comparative, prospective, randomized study and multivariate analysis of visual outcome predictors. Cutan Ocul Toxicol. 2020;39(4):317-322. PubMed
69.Chen YX, Li XX, Yoon YH, et al. Intravitreal aflibercept versus laser photocoagulation in Asian patients with diabetic macular edema: The VIVID-East study. Clin Ophthalmol. 2020;14:741-750. PubMed
70.Eter N, Singh RP, Abreu F, et al. YOSEMITE and RHINE: Phase 3 randomized clinical trials of faricimab for diabetic macular edema: Study design and rationale. Ophthalmol Sci. 2022;2(1):100111. PubMed
71.Glassman AR, Wells JA, 3rd, Josic K, et al. Five-year outcomes after initial aflibercept, bevacizumab, or ranibizumab treatment for diabetic macular edema (Protocol T extension study). Ophthalmology. 2020;127(9):1201-1210. PubMed
72.Heier JS, Korobelnik JF, Brown DM, et al. Intravitreal aflibercept for diabetic macular edema: 148-week results from the VISTA and VIVID studies. Ophthalmology. 2016;123(11):2376-2385. PubMed
73.Regeneron. Two-Year results for aflibercept 8 mg from pivotal PHOTON trial demonstrate durable vision gains at extended dosing intervals in diabetic macular edema [accessed by sponsor]. 2023; https://investor.regeneron.com/news-releases/news-release-details/two-year-results-aflibercept-8-mg-pivotal-photon-trial.
74.Shimura M, Kitano S, Ogata N, et al. Efficacy, durability, and safety of faricimab with extended dosing up to every 16 weeks in Japanese patients with diabetic macular edema: 1-year results from the Japan subgroup of the phase 3 YOSEMITE trial. Jpn J Ophthalmol. 2023;67(3):264-279. PubMed
75.Berger A, Sheidow T, Cruess AF, Arbour JD, Courseau AS, de Takacsy F. Efficacy/safety of ranibizumab monotherapy or with laser versus laser monotherapy in DME. Can J Ophthalmol. 2015;50(3):209-216. PubMed
76.Ishibashi T, Li X, Koh A, et al. The REVEAL Study: Ranibizumab monotherapy or combined with laser versus laser monotherapy in Asian patients with diabetic macular edema. Ophthalmology. 2015;122(7):1402-1415. PubMed
77.Li X, Dai H, Li X, et al. Efficacy and safety of ranibizumab 0.5 mg in Chinese patients with visual impairment due to diabetic macular edema: Results from the 12-month REFINE study. Graefes Arch Clin Exp Ophthalmol. 2019;257(3):529-541. PubMed
78.Mitchell P, Bandello F, Schmidt-Erfurth U, et al. The RESTORE study: Ranibizumab monotherapy or combined with laser versus laser monotherapy for diabetic macular edema. Ophthalmology. 2011;118(4):615-625. PubMed
79.Prünte C, Fajnkuchen F, Mahmood S, et al. Ranibizumab 0.5 mg treat-and-extend regimen for diabetic macular oedema: The RETAIN study. Br J Ophthalmol. 2016;100(6):787-795. PubMed
80.Rajendram R, Fraser-Bell S, Kaines A, et al. A 2-year prospective randomized controlled trial of intravitreal bevacizumab or laser therapy (BOLT) in the management of diabetic macular edema: 24-month data: Report 3. Arch Ophthalmol. 2012;130(8):972-979. PubMed
81.Novartis Pharmaceuticals. NCT03481660: A Study of the Efficacy and Safety of Brolucizumab vs. Aflibercept in Patients With Visual Impairment Due to Diabetic Macular Edema (KITE). ClinicalTrials.gov. Bethesda (MD): National Library of Medicine; 2018: https://clinicaltrials.gov/study/NCT03481660. Accessed 2023 Oct 3.
82.Novartis Pharmaceuticals. NCT03481634: Study of Efficacy and Safety of Brolucizumab vs. Aflibercept in Patients With Visual Impairment Due to Diabetic Macular Edema (KESTREL). ClinicalTrials.gov. Bethesda (MD): National Library of Medicine; 2018: https://classic.clinicaltrials.gov/ct2/show/NCT03481634. Accessed 2023 Oct 3.
Pharmacoeconomic_Review
Abbreviations
AE
adverse event
BCVA
best corrected visual acuity
BIA
budget impact analysis
DME
diabetic macular edema
ITC
indirect treatment comparison
NMA
network meta-analysis
QALY
quality-adjusted life-year
VEGF
vascular endothelial growth factor
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Aflibercept 8 mg (Eylea HD), solution for intravitreal injection |
Submitted price | Aflibercept 8 mg, 30 mg per 0.263 mL, single-use vial: $1,250.00 |
Indication | For the treatment of diabetic macular edema |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | February 2, 2024 |
Reimbursement request | As per indication |
Sponsor | Bayer Inc. |
Submission history | Previously reviewed: In progress Indication: Neovascular (wet) age-related macular degeneration Recommendation: TBD |
NOC = Notice of Compliance; TBD = to be determined.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults with DME |
Treatments | Aflibercept 8 mg, administered every 16 weeks (q.16.w.)a |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (38 years) |
Key data sources |
|
Submitted results | Aflibercept 8 mg q.16.w. was less effective and less costly than faricimab, and was dominant (i.e., more effective and less costly) compared with bevacizumab, ranibizumab, brolucizumab, and aflibercept 2 mg. |
Key limitations |
|
CDA-AMC reanalysis results |
|
BCVA = best corrected visual acuity; CDA-AMC = Canada’s Drug Agency; DME = diabetic macular edema; ITC = indirect treatment comparison; NMA = network meta-analysis; q.16.w. = every 16 weeks; QALY = quality-adjusted life-year; VEGF = vascular endothelial growth factor.
aIn the sponsor’s base case, aflibercept 8 mg was assumed to be administered every 16 weeks. Administration of aflibercept 8 mg every 12 weeks was considered in scenario analysis.
Conclusions
Based on the Canada’s Drug Agency (CDA-AMC) Clinical Review of the PHOTON trial, the available evidence suggests that aflibercept 8 mg is noninferior, but not superior, to aflibercept 2 mg in terms of mean change in best corrected visual acuity (BCVA). Results of the sponsor’s network meta-analysis (NMA) suggest that aflibercept 8 mg is associated with similar changes in BCVA compared to other currently available anti-vascular endothelial growth factors (VEGFs) (i.e., aflibercept 2 mg, bevacizumab, brolucizumab, faricimab, ranibizumab). However, the Clinical Review concluded that the comparative efficacy findings in the sponsor-submitted indirect treatment comparison (ITC) are insufficient as standalone evidence to inform decision-making about the efficacy and safety of aflibercept 8mg versus other anti-VEGF drugs.
It is uncertain as to whether treatment with aflibercept 8 mg will result in fewer injections than other anti-VEGFs in clinical practice due to limitations with the sponsor’s noncomparative results and the individualized approach to administration frequency in clinical practice.
Given the uncertainty in the clinical evidence, there is insufficient evidence to suggest that aflibercept 8 mg should be priced higher than other anti-VEGF treatments for diabetic macular edema (DME).
Patient, Clinician, and Drug Plan Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from the Canadian Council of the Blind, Fighting Blindness Canada, Vision Loss Rehabilitation Canada, Diabetes Canada, and the International Federation on Ageing. These organizations conducted an online survey in 2020 in Canada in which 67 people with diabetic retinopathy or DME participated. The respondents reported that DME has a substantial impact on their daily lives, including physical, psychological, and social impacts. Respondents indicated that vision loss resulting from diabetic retinopathy and DME affects daily activities such as reading, using a phone, and driving. Respondents indicated that they were receiving injections of bevacizumab, ranibizumab, aflibercept 2 mg, and dexamethasone to treat DME; faricimab and aflibercept 8 mg were not available at the time of the survey. The respondents described the injections as being at least somewhat painful and that pain and blurry vision could occur after the injections. The potential benefit associated with extended treatment intervals was highlighted in a statement by the International Federation on Ageing as being very important to patients and caregivers.
Clinical input was received from 6 groups: Southwestern Ontario Community Ophthalmologists, the Northeastern Ontario Ophthalmology Group, the Canadian Retina Society, the Retina Division of the Ottawa Hospital, Toronto Ophthalmologists, and the Toronto Retina Institute. Clinician input noted that current treatment of DME consists of intravitreal injections of drugs that inhibit VEGF and that aflibercept 8 mg is likely to have high uptake due to its longer treatment interval, replacing aflibercept 2 mg as the standard first-line treatment for DME. Clinical input noted that the incidence of DME and thus the demand for these treatments is expected to rise in light of the aging population in Canada, and that treatments with extended intervals will help retinal specialists meet the demands of patients requiring these treatments.
Participating drug plans noted that there have been no trials comparing aflibercept 8 mg with anti-VEGF therapies that can be administered at the same extended dosing interval (i.e., faricimab, brolucizumab). Given the extended dosing intervals of faricimab and brolucizumab, drug plans questioned what unmet need would be addressed by aflibercept 8 mg. Drug plan input noted that biosimilars are available for ranibizumab and are anticipated for aflibercept 2 mg within the next year, and that these are expected to affect the budget impact of aflibercept 8 mg. Finally, the drug plans noted the presence of confidential negotiated prices for comparators.
The potential extended dosing interval of aflibercept 8 mg was raised by patient, clinician, and drug plan input as particularly important to this review; however, CDA-AMC notes that the comparative clinical efficacy and frequency of dosing are areas of uncertainty in the sponsor’s submitted evidence and therefore was unable to address these concerns.
Economic Review
The current review is for aflibercept 8 mg (Eylea HD) for patients with DME.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of aflibercept 8 mg compared with aflibercept 2 mg, brolucizumab, faricimab, ranibizumab (biosimilar), and bevacizumab, in patients with DME.1 The modelled population is aligned with the draft Health Canada indication and was based on patients enrolled in the PHOTON trial.
Aflibercept 8 mg is supplied in single-use vials containing 30 mg aflibercept in 0.263 mL solution (114 mg/mL). The recommended dose of aflibercept 8 mg is 8 mg administered by intravitreal injection every 4 weeks (monthly) for the first 3 doses, followed by 8 mg at a dosing interval of every 8 to 16 weeks (4 months), depending on the physician’s judgment of the individual patient’s visual and anatomic outcomes.2 The schedule for monitoring between dosing visits should be based on the patient’s status and at the physician’s discretion. The sponsor’s submitted price for aflibercept 8 mg is $1,250.00 per vial, which corresponds to an annual per-patient cost of $7,500 in the first year ($4,375 in subsequent years) if administered every 12 weeks (based on 6.0 injections and 3.5 injections in the first and subsequent years, respectively), and an annual per-patient cost of $6,250 in the first year ($3,500 in subsequent years) if administered every 16 weeks (based on 5.0 and 2.8 injections in first and subsequent years, respectively).1
In the model, the sponsor assumed that aflibercept 8 mg would be administered every 16 weeks, aflibercept 2 mg, ranibizumab, and bevacizumab would be administered as needed, faricimab would be administered at intervals extending from 8 to 16 weeks, and brolucizumab would be administered every 8 or 12 weeks.1 The first-year annual per-patient costs for comparators estimated by the sponsor ranged from $4,764 (bevacizumab) to $10,507 (aflibercept 2 mg), while the annual per-patient costs in subsequent years ranged from $2,590 (bevacizumab) to $7,090 (aflibercept 2 mg).1
The clinical outcomes were life-years and quality-adjusted life-years (QALYs), estimated over a lifetime time horizon (38 years; 4-week cycle length) from the perspective of publicly funded health care system in Canada. Costs and QALYs were discounted at a rate of 1.5% per annum, and a half-cycle correction was applied.
Model Structure
The sponsor submitted a Markov model consisting of 65 health states: 64 visual acuity–based health states (defined by Early Treatment for Diabetic Retinopathy Study letter score), and death. Gains in visual acuity in the first year of treatment (informed by change from baseline in BCVA from the sponsor’s NMA) were assumed to be maintained for the duration of treatment (which was limited to a maximum of 5 years). Patients could have DME in 1 or both eyes, with different visual acuity status in each eye accounted for.
Model Inputs
The baseline characteristics and initial distribution of patients across health states in the model were based on the PHOTON trial, which randomized patients with DME (mean age of 62.3 years, 39.1% female) to receive aflibercept 8 mg every 12 weeks, aflibercept 8 mg every 16 weeks, or aflibercept 2 mg every 8 weeks.1 Movement between visual acuity health states in the model was based on the probability that a patient would gain or lose 10 or 15 letters in BCVA as measured using the Early Treatment Diabetic Retinopathy Study (ETDRS) letter score; this was derived using odds ratios for BCVA from the sponsor’s NMA. Severe adverse events (AEs) were also informed by the sponsor’s NMA. The number of injections per year was informed by naive (visual) comparisons across pairwise meta-analyses provided by the sponsor. Discontinuation rates were based on observations from the PHOTON trial (aflibercept 8 mg) and published sources (comparators).3-7 The probability of death was based on age-specific background mortality of people in Canada,8 to which the sponsor applied a hazard ratio of 1.52 to account for higher mortality associated with diabetes9 and 1.36 to account for higher mortality associated with visual impairment in 1 or both eyes.10
Utility values by visual acuity state were estimated using regression coefficients from a published study that simulated visual impairment in healthy volunteers in the UK.11 Disutilities associated with AEs and the duration of events were derived from the literature.12-15 The sponsor assumed that half of patients would experience zero utility on an injection day.16
The economic model included costs related to drugs (acquisition, administration), monitoring, AEs, and blindness. Treatment costs were estimated by using the drug cost for 1 injection, the estimated number of injections per year, and the administration cost per injection. The sponsor assumed that all vials were single-use and that any unused product would be wasted. Costs were derived from Ontario’s schedule of benefits,17 the Ontario Case Costing Initiative,18 the Canadian Institute for Health Information Patient Cost Estimator,19 Saskatchewan’s Payment Schedule for Insured Services Provided by a Physician,20 and the literature.21
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented below. The sponsor’s base-case analysis assumed that aflibercept 8 mg will be administered every 16 weeks; administration every 12 weeks was considered in scenario analysis.
Base-Case Results
In the sponsor’s base case, aflibercept 8 mg every 16 weeks was associated with an estimated cost of $34,277 and 10.60 QALYs over a 38-year horizon (Table 3). In sequential analysis, aflibercept 8 mg every 16 weeks was both less costly and less effective than faricimab (incremental costs: –$17,310; incremental QALYs: –0.14).
The sponsor’s model predicts that aflibercept 8 mg every 16 weeks will be associated with fewer QALYs gained compared with faricimab, but more than with other anti-VEGFs. Of the 10.60 QALYs predicted for aflibercept 8 mg every 16 weeks, approximately 94% were accrued in the extrapolation period of the model (i.e., beyond the 48-week duration of the PHOTON trial). The sponsor’s model predicts a survival benefit for aflibercept 8 mg every 16 weeks relative to most anti-VEGFs (except faricimab), which has not been shown in clinical trials.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total life-years | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Aflibercept 8 mg q.16.w. | 34,277 | 18.46 | 10.60 | Reference |
Faricimab | 51,587 | 18.47 | 10.74 | 116,474 vs. aflibercept 8 mg q.16.w. |
Dominated treatments | ||||
Bevacizumab | 34,729 | 18.43 | 10.42 | Dominated by aflibercept 8 mg q.16.w. |
Ranibizumab | 40,326 | 18.42 | 10.36 | Dominated by aflibercept 8 mg q.16.w. and bevacizumab |
Brolucizumab | 42,617 | 18.44 | 10.56 | Dominated by aflibercept 8 mg q.16.w. |
Aflibercept 2 mg | 59,647 | 18.45 | 10.58 | Dominated by aflibercept 8 mg q.16.w. and faricimab |
ICER = incremental cost-effectiveness ratio; q.16.w. = every 16 weeks; QALY = quality-adjusted life-year; vs. = versus.
aThe sponsor assumed that the cost of bevacizumab was $519.18 per vial based on the cost of branded bevacizumab (Avastin) and that 1 dose would be obtained per vial. The total cost of bevacizumab would be lower if generic bevacizumab is used and/or vial sharing occurs in clinical practice.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted a sequential scenario analysis in which aflibercept 8 mg was assumed to be administered every 12 weeks. In this scenario, aflibercept 8 mg was associated with higher costs and more QALYs versus bevacizumab (incremental costs: $4,137; incremental QALYs: 0.21) over a 38-year horizon, resulting in a sequential incremental cost-effectiveness ratio of $19,941. As with the sponsor’s base case, this analysis assumed that branded bevacizumab would be used, with no vial sharing.
The sponsor also conducted several scenario analyses including multiple dosing assumptions for bevacizumab and ranibizumab, discontinuation assumptions, and treatment efficacy assumptions; however, sequential analyses were not provided (i.e., aflibercept 8 mg every 16 weeks was compared to each of the other treatments in a pair-wise fashion), limiting the interpretation of the findings.
The sponsor conducted a scenario analysis from a societal perspective. This analysis included additional costs associated with productivity loss of caregivers; however, sequential analyses were not provided, limiting the interpretation of the findings.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative clinical efficacy and safety of aflibercept 8 mg versus other anti-VEGFs are uncertain. There is a lack of head-to-head evidence comparing aflibercept 8 mg to anti-VEGF drugs other than aflibercept 2 mg. The results of the PHOTON trial suggest that aflibercept 8 mg is noninferior to aflibercept 2 mg for improvement in visual acuity (i.e., change from baseline in BCVA). In the absence of head-to-head evidence for most comparators, the sponsor conducted NMAs to inform various parameters in the economic model for all anti-VEGF agents, including BCVA and severe ocular AEs. As noted in the Clinical Review, the results of the sponsor’s NMA suggest that there may be no meaningful differences between aflibercept 8 mg and relevant comparators, although the presence of substantial imprecision and unresolved heterogeneity precludes meaningful conclusions.
Given the lack of direct evidence for aflibercept 8 mg relative to anti-VEGF agents other than aflibercept 2 mg and limitations with the sponsor’s NMA, it is uncertain whether aflibercept 8 mg provides a net benefit above any of the currently available treatments for DME.
The relative frequency of anti-VEGF injections is uncertain. In the pharmacoeconomic model, the number of injections per year (and hence drug acquisition and administration costs) for each anti-VEGF drug was informed by sponsor-submitted ITCs. As noted in the Clinical Review, the results of the submitted naive (visual) comparisons across pairwise meta-analyses suggest that aflibercept 8 mg may be associated with numerically fewer injections in year 1 and year 2 when compared to some anti-VEGFs administered on a set injection schedule (e.g., every 4 or 8 weeks). However, clinical expert feedback obtained by CDA-AMC for this review indicated that, in clinical practice, anti-VEGF therapies are typically administered using a treat-and-extend approach, not a set frequency of injections. Results of the sponsor’s noncomparative evidence suggest that there may be minimal differences in the number of injections in year 1 or 2 when compared to some anti-VEGF treatments that are administered as needed or using a treat-and-extend strategy. However, the Clinical Review noted that there is uncertainty in this finding owing to a lack of comparative data and associated measures of variability across treatments.
Owing to limitations with the sponsor-submitted evidence regarding administration frequency and the individualized approach to administration frequency in clinical practice, it is uncertain whether treatment with aflibercept 8 mg will result in fewer injections than treatment with other anti-VEGFs in clinical practice.
Issues for Consideration
Biosimilars for aflibercept 2 mg are currently under review by Health Canada, including for use in the treatment of DME. The introduction of such biosimilars may affect the cost-effectiveness of aflibercept 8 mg versus aflibercept 2 mg, depending on the list price.
The sponsor’s analyses rely on publicly accessible list prices and do not reflect existing confidential prices negotiated by public drug plans. Given that aflibercept 2 mg,22,23 brolucizumab,24,25 faricimab,26 ranibizumab,27,28 and bevacizumab29,30 have successfully undergone price negotiations for the treatment of DME, it is likely that the current unit cost paid by public drug plans for these treatments are lower than the submitted prices. Should the price of these anti-VEGFs be lower than incorporated in the model, the incremental savings predicted with aflibercept 8 mg every 16 weeks may not be realized. In particular, if generic instead of branded bevacizumab is used and if more than 1 dose is obtained per bottle, it is likely that the total treatment cost associated with treatment with bevacizumab would be less than that of aflibercept 8 mg every 16 weeks.
Overall Conclusions
Based on the Clinical Review, data from the PHOTON trial suggest that aflibercept 8 mg is noninferior, but not superior, to aflibercept 2 mg for mean change in BCVA. Results of the sponsor’s NMA suggest that treatment with aflibercept 8 mg is associated with similar changes in BCVA to those associated with other currently available anti-VEGFs (i.e., aflibercept 2 mg, bevacizumab, brolucizumab, faricimab, and ranibizumab). However, the Clinical Review concluded that the comparative efficacy findings from the sponsor’s NMA are insufficient as standalone evidence to inform the efficacy and safety of aflibercept 8 mg versus other anti-VEGF inhibitors owing to substantial imprecision and clinical and statistical heterogeneity. Noncomparative results submitted by the sponsor regarding injection frequency suggest that aflibercept 8 mg may be associated with relatively close but numerically fewer annual injections per year compared with other anti-VEGF inhibitors that utilize a treat-and-extend or an as-needed approach; however, due to the lack of statistical comparison, no inferences can be made as to the comparative differences in number of injections.
Given that the sponsor-submitted indirect evidence suggests that there may be no difference between aflibercept 8 mg and currently available anti-VEGFs in terms of improvements in visual acuity or number of injections per year, there is insufficient evidence to suggest that aflibercept 8 mg should be priced higher than other anti-VEGF treatments for DME. Thus, to ensure cost-effectiveness, aflibercept 8 mg should be priced no more than the lowest cost anti-VEGF that is funded for the treatment of DME.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Eylea HD (aflibercept), 8 mg / 0.07 mL for intravitreal injection. Mississauga (ON): Bayer Inc.; 2023 Aug 18.
2.Eylea HD (aflibercept): 8 mg / 0.07 mL, solution for intravitreal injection in single use vials for the treatment of a single eye [product monograph]. Mississauga (ON): Bayer Inc; 2024 Feb 2.
3.Wykoff CC, Abreu F, Adamis AP, et al. Efficacy, durability, and safety of intravitreal faricimab with extended dosing up to every 16 weeks in patients with diabetic macular oedema (YOSEMITE and RHINE): Two randomised, double-masked, phase 3 trials. Lancet. 2022;399(10326):741-755. PubMed
4.Prünte C, Fajnkuchen F, Mahmood S, et al. ranibizumab 0.5 mg treat-and-extend regimen for diabetic macular oedema: The RETAIN study. Br J Ophthalmol. 2016;100(6):787-795. PubMed
5.Li X, Dai H, Li X, et al. Efficacy and safety of ranibizumab 0.5 mg in Chinese patients with visual impairment due to diabetic macular edema: Results from the 12-month REFINE study. Graefes Arch Clin Exp Ophthalmol. 2019;257(3):529-541. PubMed
6.Lang GE, Berta A, Eldem BM, et al. Two-year safety and efficacy of ranibizumab 0.5 mg in diabetic macular edema: Interim analysis of the RESTORE extension study. Ophthalmology. 2013;120(10):2004-2012. PubMed
7.Brown DM, Emanuelli A, Bandello F, et al. KESTREL and KITE: 52-Week results from two phase III pivotal trials of brolucizumab for diabetic macular edema. Am J Ophthalmol. 2022;238:157-172. PubMed
8.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island [accessed by sponsor]. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401.
9.Canoy D, Tran J, Zottoli M, et al. Association between cardiometabolic disease multimorbidity and all-cause mortality in 2 million women and men registered in UK general practices. BMC Med. 2021;19(1):258. PubMed
10.Zhang T, Jiang W, Song X, Zhang D. The association between visual impairment and the risk of mortality: A meta-analysis of prospective studies. J Epidemiol Community Health. 2016;70(8):836-842. PubMed
11.Czoski-Murray C, Carlton J, Brazier J, Young T, Papo NL, Kang HK. Valuing condition-specific health states using simulation contact lenses. Value Health. 2009;12(5):793-799. PubMed
12.Ziemssen F, Hammer T, Grueb M, et al. Reporting of safety events during anti-VEGF treatment: Pharmacovigilance in a noninterventional trial. J Ophthalmol. 2020;2020:8652370. PubMed
13.Brown GC, Brown MM, Brown HC, Kindermann S, Sharma S. A value-based medicine comparison of interventions for subfoveal neovascular macular degeneration. Ophthalmology. 2007;114(6):1170-1178. PubMed
14.Mok CH, Kwok HHY, Ng CS, Leung GM, Quan J. Health state utility values for type 2 diabetes and related complications in East and Southeast Asia: A systematic review and meta-analysis. Value Health. 2021;24(7):1059-1067. PubMed
15.Gower EW, Cassard SD, Bass EB, Schein OD, Bressler NM. A cost-effectiveness analysis of three treatments for age-related macular degeneration. Retina. 2010;30(2):212-221. PubMed
16.National Institute for Health and Care Excellence. Age-related macular degeneration. (NICE guideline NG82). [accessed by sponsor]. 2018.
17.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 23, 2023)) [accessed by sponsor]. Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf.
18.Ontario Case Costing Initiative (OCCI) [accessed by sponsor]. Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi.
19.Canadian Institute for Health Information. Patient cost estimator [accessed by sponsor]. 2023; https://www.cihi.ca/en/patient-cost-estimator, 2023.
20.eHealth Saskatchewan. Payment schedule for insured services provided by a physician [accessed by sponsor]. 2022; https://www.ehealthsask.ca/services/resources/establish-operate-practice/Documents/Payment%20Schedule%20-%20April%201%2C%202022.pdf, 2023.
21.Cruess AF, Gordon KD, Bellan L, Mitchell S, Pezzullo ML. The cost of vision loss in Canada. 2. Results. Can J Ophthalmol. 2011;46(4):315-318. PubMed
22.pan-Canadian Pharmaceutical Alliance. Eylea (aflibercept). 2017; https://www.pcpacanada.ca/negotiation/20917. Accessed 2023 Sep 25.
23.pan-Canadian Pharmaceutical Alliance. Eylea (aflibercept). 2015; https://www.pcpacanada.ca/negotiation/20741. Accessed 2023 Sep 25.
24.pan-Canadian Pharmaceutical Alliance. Beovu (brolucizumab). 2023; https://www.pcpacanada.ca/negotiation/22193. Accessed 2023 Sep 25.
25.pan-Canadian Pharmaceutical Alliance. Beovu (brolucizumab). 2021; https://www.pcpacanada.ca/negotiation/21223. Accessed 2023 Sep 25.
26.pan-Canadian Pharmaceutical Alliance. Vabysmo (faricimab). 2023; https://www.pcpacanada.ca/negotiation/22056. Accessed 2023 Sep 25.
27.pan-Canadian Pharmaceutical Alliance. Ranopto (ranibuzumab). 2023; https://www.pcpacanada.ca/negotiation/22329. Accessed 2023 Sep 25.
28.pan-Canadian Pharmaceutical Alliance. Byooviz (ranibizumab). 2023; https://www.pcpacanada.ca/negotiation/22008. Accessed 2023 Sep 25.
29.pan-Canadian Pharmaceutical Alliance. Mvasi (bevacizumab). 2022; https://www.pcpacanada.ca/negotiation/21696. Accessed 2023 Sep 25.
30.pan-Canadian Pharmaceutical Alliance. Avastin (bevacizumab). 2027; https://www.pcpacanada.ca/negotiation/20921. Accessed 2023 Sep 25.
31.DeltaPA. [Ottawa (ON)]: IQVIA; 2023: https://www.iqvia.com/. Accessed 2023 Aug 30.
32.CADTH Reimbursement Review: Faricimab (Vabysmo) [accessed by sponsor]. Can J Health Technol. 2023;3(1). https://www.cadth.ca/sites/default/files/DRR/2022/SR0729-Vabysmo_combined.pdf.
33.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Eylea (aflibercept), 8 mg / 0.07 mL for intravitreal injection. Mississauga (ON): Bayer Inc.; 2023 Aug 18.
34.Government of Canada. Census profile: 2021 census of population. 2021; https://www12.statcan.gc.ca/census-recensement/2021/dp-pd/prof/index.cfm?Lang=E. Accessed 2023 Mar 29.
35.Govenment of Canada. Canadian Chronic Disease Surveillance System (CCDSS) [sponsor provided reference]. 2021, Accessed 2023.
36.Petrella RJ, Blouin J, Davies B, Barbeau M. Prevalence, demographics, and treatment characteristics of visual impairment due to diabetic macular edema in a representative Canadian cohort. J Ophthalmol. 2012;2012:159167. PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Table 4: CDA-AMC Cost Comparison Table for DME
Treatment | Strength / concentration | Form | Price ($) | Recommended dosagea | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Aflibercept 8 mg (Eylea HD) | 114.3 mg/mL | 0.07 mL solution for intravitreal injection | 1,250.0000b | 8 mg every 4 weeks for the first 3 doses followed by 8 mg at a dosing interval of every 8 to 16 weeks | Year 1: 17.11 to 27.38 Subsequent: 13.69 to 23.96 | Year 1: 6,250 to 10,000 (5 to 8 injections) Subsequent: 5,000 to 8,750 (4 to 7 injections) |
Anti-VEGF inhibitors | ||||||
Aflibercept (Eylea) | 40 mg/mL | 0.05 mL solution for intravitreal injection | 1,418.0000 | 2 mg every 4 weeks for the first 5 doses followed by 2 mg every 8 weeks in the first year, and up to 16 weeks thereafter | Year 1: 34.94 Subsequent: 15.53 to 27.18 | Year 1: 12,762 (9 injections) Subsequent: 5,672 to 9,926 (4 to 7 injections) |
Bevacizumab (Avastin) | 25 mg/mL | 4 mL 16 mL solution for intravitreal injection | 519.1800c 2,076.7104c | 1.25 mg every 4 weeks for the first 3 doses followed by 1.25 mg every 6 to 8 weeksc | Year 1: 0.38 to 0.47 Subsequent: 0.33 to 0.43 | Year 1: 138 to 173 (8 to 10 injections)d Subsequent: 121 to 156 (7 to 9 injections)d |
Bevacizumab (Mvasi) | 25 mg/mL | 4 mL 16 mL solution for intravitreal injection | 347.0000c 1,388.0000c | 1.25 mg every 4 weeks for the first 3 doses followed by 1.25 mg every 6 to 8 weeksd | Year 1: 0.25 to 0.32 Subsequent: 0.22 to 0.29 | Year 1: 93 to 116 (8 to 10 injections) Subsequent: 81 to 104 (7 to 9 injections) |
Brolucizumab (Beovu) | 120 mg/mL | 0.05 mL solution for intravitreal injection | 1,390.0000 | 6 mg every 6 weeks for the first 5 doses followed by 6 mg every 8 to 12 weeks | Year 1: 26.64 to 30.44 Subsequent: 19.03 to 26.64 | Year 1: 9,730 to 11,120 (7 to 8 injections) Subsequent: 6,950 to 9,730 (5 to 7 injections) |
Faricimab (Vabysmo) | 120 mg/mL | 0.05 mL solution for intravitreal injection | 1,350.0000b | 6 mg every 4 weeks for the first 4 doses followed by 6 mg at a dosing interval of up to every 16 weeks | Year 1: 22.18 to 51.75 to Subsequent: 14.78 to 51.75 | Year 1: 8,100 to 18,900 (6 to 14 injections) Subsequent: 5,400 to 18,900 (4 to 14 injections) |
Ranibizumab (Lucentis) | 10 mg/mL | 0.23 mL solution for intravitreal injection | 1,616.5500 | 0.5 mg monthly until maximum visual acuity is achieved, and resumed when monitoring indicates loss of visual acuity | Year 1: 13.28 to 53.11 Subsequent: 4.43 to 53.11 | Year 1: 4,850 to 19,399 (3 to 12 injections) Subsequent: 1,617 to 19,399 (1 to 12 injections) |
Ranibizumab (biosimilar) | 10 mg/mL | 0.23 mL | 995.0000 | 0.5 mg monthly until maximum visual acuity is achieved, and resumed when monitoring indicates loss of visual acuity | Year 1: 8.17 to 32.69 Subsequent: 2.72 to 32.69 | Year 1: 2,985 to 11,940 (3 to 12 injections) Subsequent: 995 to 11,940 (1 to 12 injections) |
CDA-AMC = Canada’s Drug Agency; DME = diabetic macular edema; VEGF = vascular endothelial growth factor.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed November 2023),31 unless otherwise indicated, and do not include dispensing fees. Annual costs are based on 52 weeks per year. Annual and daily costs for aflibercept 8 mg includes a dose frequency of once every 12, 16, or 20 weeks, as per the PHOTON trial.
aRecommended doses are from the respective product monographs, unless otherwise indicated.
bSponsor-submitted price.1
cPrice obtained from the IQVIA DeltaPA database (accessed November 2023).31
dBevacizumab is used off-label in this population and, as such, does not have a recommended dosage for DME in the product monograph. Dosage and number of administrations per vial (30 per 4 mL vial) were obtained from a previous CDA-AMC review;32 number of doses per year was based on clinical input received by CDA-AMC for the current review.
Appendix 2: Submitted Budget Impact Analysis and Appraisal by CDA-AMC
Please note that this appendix has not been copy-edited.
Table 5: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
BIA = budget impact analysis; DME = diabetic macular edema; VEGF = vascular endothelial growth factor.
Summary of the Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor estimated the incremental budget impact of reimbursing aflibercept 8 mg for the treatment of DME.33 The BIA was undertaken using an epidemiologic approach from the perspective of a public payer in Canada over a 3-year time horizon (January 2024 to December 2026). The number of patients eligible for aflibercept 8 mg was estimated based the population of Canada, prevalence of diabetes (type 1 and 2), the incidence and prevalence of DME, the proportion that have been diagnosed and are being treated for DME, the proportion of patients eligible for public coverage, and the rate of bilateral disease.3 The sponsor’s analysis included drug acquisition costs and excluded dispensing fees and markups.
The reference scenario included aflibercept 2 mg, faricimab, ranibizumab, ranibizumab (biosimilar), brolucizumab, and bevacizumab. The market share estimates for these products were informed by jurisdiction-specific market research and clinical expert consultation conducted by the sponsor. In the new drug scenario, aflibercept 8 mg was assumed to primarily displace aflibercept 2 mg and faricimab, with market share of aflibercept 8 mg based on clinician input solicited by the sponsor. Market share and injection frequency were considered separately for prevalent and incident patients, with the number of injections per year informed by a sponsor-submitted ITC.3 Key inputs to the BIA are documented in Table 7.
Table 6: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (year 1 / year 2 / year 3) | |
|---|---|---|
Target population | ||
Population across Canada (excluding Quebec) | Aged 18 to 64 years | Aged ≥ 65 years |
19,055,22134 | 5,456,04534 | |
Prevalence of diabetes | 6.6%35 | 26.8%35 |
Prevalence of DME | 2.56%36 | |
Incidence of DME | 0.37%36 | |
Proportion of patients whose DME is diagnosed | 80%a | |
Proportion of patients who receive treatment | 70%a | |
Proportion eligible for public coverage | 78%b | |
Rate of bilateral disease | 40%a | |
Number of patients eligible for drug under review | 30,701 / 31,008 / 31,318 | |
Number of eyes eligible for drug under review | 42,982 / 43,412 / 43,846 | |
Market uptake (3 years) | ||
Uptake (reference scenario) | Incident patients | Prevalent patients |
Aflibercept 2 mg | 38% / 34% / 32% | 40% / 38% / 34% |
Ranibizumab | 0% / 0% / 0% | 0% / 0% / 0% |
Ranibizumab biosimilar | 6% / 6% / 5% | 11% / 11% / 11% |
Brolucizumab | 1% / 1% / 1% | 1% / 1% / 1% |
Faricimab | 22% / 25% / 28% | 14% / 17% / 21% |
Bevacizumab | 33% / 33% / 33% | 33% / 33% / 33% |
Uptake (new drug scenario) | ||
Aflibercept 8 mg | 15% / 19% / 22% | 9% / 16% / 20% |
Aflibercept 2 mg | 29% / 25% / 21% | 36% / 30% / 24% |
Ranibizumab | 0% / 0% / 0% | 0% / 0% / 0% |
Ranibizumab biosimilar | 6% / 5% / 5% | 11% / 9% / 9% |
Brolucizumab | 1% / 1% / 1% | 1% / 1% / 1% |
Faricimab | 16% / 17% / 18% | 10% / 11% / 12% |
Bevacizumab | 33% / 33% / 33% | 33% / 33% / 33% |
Annual cost of treatment per patient (induction year / subsequent years)c | ||
Aflibercept 8 mg | $6,250 / $3,500 | |
Aflibercept 2 mg | $10,507 / $7,090 | |
Ranibizumab | $12,108 / $6,127 | |
Ranibizumab biosimilar | $7,453 / $3,771 | |
Brolucizumab | $9,591 / $4,935 | |
Faricimab | $10,260 / $5,616 | |
Bevacizumabd | $4,765 / $2,590 | |
DME = diabetic macular degeneration; NMA = network meta-analysis.
aBased on the sponsor’s assumption.
bBased on assumption and the CADTH review of faricimab (Vabysmo).32
cAnnual cost was calculated by multiplying the cost per dose by the number of administrations per year predicted by the sponsor’s NMA.
dThe sponsor assumed that the cost of bevacizumab was $519.18 per vial based on the cost of branded bevacizumab (Avastin) and that 1 dose would be obtained per vial. The total cost of bevacizumab would be lower if generic bevacizumab is used and/or vial sharing occurs in clinical practice.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the cost savings of funding aflibercept 8 mg for patients with DME will be $4,801,532 in year 1, $16,505,625 in year 2, and $25,097,040 in year 3, for a cumulative cost savings of $46,404,196 over the 3-year time horizon.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable impacts on the results of the BIA:
The frequency of anti-VEGF administration is uncertain. In the BIA, the annual cost of aflibercept 2 mg and comparators was calculated based on the number of injections per year as predicted by the sponsor’s noncomparative pair-wise assessment. As noted in the Clinical Review, the sponsor submitted naive (visual) pairwise comparisons, and no comparative analyses were performed. As such, the uncertainty in the relative injection frequency described in the Appraisal of the Sponsor’s Economic Evaluation also applies to the submitted BIA. The sponsor’s noncomparative assessment suggests that the number of injections of aflibercept 8 mg per year may be similar to that of other anti-VEGFs administered using a treat-and-extend or an as-needed schedule.
CDA-AMC conducted a scenario analysis that assumed equal frequency of injections for all comparators.
Multiple administrations from a single vial may be possible for some comparators. The sponsor assumed that 1 dose would be obtained from each vial of aflibercept 2 mg, ranibizumab, and bevacizumab. Clinical expert opinion obtained from CDA-AMC for this review and previous reviews have indicated that multiple administrations from a single vial may be possible for these anti-VEGFs. Given that the volume within a vial is greater than that required for a single dose, with the proper syringes multiple administrations can be obtained; however, this practice may be jurisdiction-specific. CDA-AMC also notes that, because the sponsor assumed that aflibercept would not displace bevacizumab, the assumption of 1 administration per vial did not impact the incremental results.
As part of a scenario analysis, CDA-AMC assumed that 3 administrations per vial of aflibercept 2 mg and ranibizumab and 30 administrations per vial of bevacizumab were possible.
The displacement of other anti-VEGFs by aflibercept 2 mg is uncertain. The sponsor estimated market shares of aflibercept 8 mg and comparators based on market research conducted by the sponsor and expert opinion solicited by the sponsor, with aflibercept 8 mg assumed to primarily displace aflibercept 2 mg and faricimab. Input received by CDA-AMC for this review indicated that aflibercept 8 mg is likely to predominantly displace aflibercept 2 mg, with a lesser impact on faricimab.
CDA-AMC explored uncertainty in the displacement of anti-VEGF comparators by aflibercept 8 mg in scenario analyses.
The price of drugs paid by public drug plans is uncertain: Both the sponsor’s analysis and the analysis by CDA-AMC are based on publicly available list prices for all comparators. Drug plan feedback received for this review indicated there are confidential negotiated prices for the comparators. Thus, the actual costs paid by the public drug plans for anti-VEGFs are unknown. Depending on the negotiated prices, reimbursing aflibercept 8 mg for the treatment of DME may lead to lower or no cost savings compared to other available anti-VEGFs.
CDA-AMC was unable to incorporate the presence of confidential negotiated prices in the reanalysis.
CDA-AMC Reanalyses of the BIA
In the absence of more reliable estimates to inform the key parameters of the BIA, the sponsor’s submitted base case was maintained. CDA-AMC expects that the budget impact of reimbursing aflibercept 8 mg for the treatment DME will be sensitive to more reliable inputs which may affect the comparator costs (e.g., administration frequency, vial sharing, availability of biosimilars). CDA-AMC conducted scenario analyses to explore the impact of uncertainty in the number of administrations per year of anti-VEGFs, the number of administrations per vial of aflibercept 2 mg and ranibizumab, displacement of anti-VEGFs by aflibercept 8 mg, and the introduction of a biosimilar for aflibercept 2 mg (Table 9).
The results of these scenario analyses suggest that the budgetary impact of reimbursing aflibercept 8 mg for the treatment of DME is sensitive to the administration frequency of each drug and the potential for vial sharing; results of these analyses suggested that the introduction of aflibercept 8 mg would not be cost saving, but would result in $8.0 million and $10.3 million additional costs over 3 years, respectively, indicating that the cost savings associated with the reimbursement of aflibercept 8 mg may have been overestimated in the sponsor’s base case. In addition, the predicted cost savings associated with the introduction of aflibercept 8 mg is highly influenced by assuming a hypothetical price of an aflibercept 2 mg biosimilar with an estimated decline in cost savings of 76% compared to the sponsor’s base-case analysis. Thus, the budget impact of reimbursing aflibercept 8 mg for DME is sensitive to assumptions about comparator prices, and its reimbursement may lead to additional costs to the health care system rather than cost savings.
Table 7: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $242,904,773 | $242,689,160 | $245,232,525 | $246,478,879 | $734,400,565 |
New drug | $242,904,773 | $237,887,629 | $228,726,900 | $221,381,840 | $687,996,369 | |
Budget impact | $0 | –$4,801,532 | –$16,505,625 | –$25,097,040 | –$46,404,196 | |
CDA-AMC scenario 1: Equal administration frequencya | Reference | $147,911,485 | $148,292,138 | $150,344,925 | $152,115,282 | $450,752,345 |
New drug | $147,911,485 | $155,163,773 | $153,015,469 | $150,587,727 | $458,766,969 | |
Budget impact | $0 | $6,871,635 | $2,670,544 | –$1,527,555 | $8,014,624 | |
CDA-AMC scenario 2: Multiple administrations of aflibercept 2 mg, ranibizumab, and bevacizumabb | Reference | $96,102,629 | $101,720,436 | $109,318,225 | $117,701,743 | $328,740,404 |
New drug | $96,102,629 | $109,567,972 | $113,266,308 | $116,246,509 | $339,080,789 | |
Budget impact | $0 | $7,847,536 | $3,948,083 | –$1,455,234 | $10,340,386 | |
CDA-AMC scenario 3: Aflibercept 8 mg primarily displaced aflibercept 2 mgc | Reference | $242,904,773 | $242,689,160 | $245,232,525 | $246,478,879 | $734,400,565 |
New drug | $242,904,773 | $237,212,478 | $227,711,808 | $220,020,074 | $684,944,360 | |
Budget impact | $0 | –$5,476,682 | –$17,520,717 | –$26,458,805 | –$49,456,205 | |
CDA-AMC scenario 4: Biosimilar of aflibercept 2 mg is availabled | Reference | $179,543,780 | $184,443,392 | $190,069,664 | $196,215,817 | $570,728,873 |
New drug | $179,543,780 | $187,439,198 | $186,170,659 | $185,883,993 | $559,493,849 | |
Budget impact | $0 | $2,995,805 | –$3,899,005 | –$10,331,824 | –$11,235,024 | |
CDA-AMC scenario 5: Aflibercept 8 mg q.12.w. dosing frequencye | Reference | $242,904,773 | $242,689,160 | $245,232,525 | $246,478,879 | $734,400,565 |
New drug | $242,904,773 | $243,346,689 | $236,373,975 | $230,166,045 | $709,886,709 | |
Budget impact | $0 | $657,529 | –$8,858,550 | –$16,312,834 | –$24,513,855 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; q.12.w. = every 12 weeks; NMA = network meta-analysis.
aThe frequency of administration was set to be equal to aflibercept 8 mg frequency for all comparators.
bCDA-AMC assumed that 3 administrations per vial of aflibercept 2 mg and ranibizumab and 30 administrations per vial of bevacizumab were possible.
cCDA-AMC assumed that the displacement of faricimab was half that estimated by the sponsor, and the displacement was shifted to aflibercept 2 mg.
dThe cost per injection of aflibercept 2 mg biosimilar ($796 per injection) was obtained from the sponsor’s BIA report. The sponsor assumed hypothetical potential list prices for aflibercept 2 mg biosimilar; however, such a price may not be predictive of any actual price offered by a third party.
eCDA-AMC assumed that aflibercept 8 mg would follow a q.12.w. dosing regimen, and applied the number of injections in the first and subsequent years based on the sponsor-submitted NMA.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.