Drugs, Health Technologies, Health Systems
Reimbursement Review
Aflibercept 8 mg (0.07 mL) (Eylea HD)
Sponsor: Bayer Inc.
Therapeutic area: Macular degeneration, age-related
This multi-part report includes:
Clinical_Review
Pharmacoeconomic_Review
Clinical_Review
Abbreviations
AE
adverse event
AMD
age-related macular degeneration
APTC
Antiplatelet Trialists' Collaboration
BCVA
best corrected visual acuity
CCB
Canadian Council of the Blind
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CNV
choroidal neovascularization
CRT
central retinal thickness
CST
central subfield retinal thickness
DRM
dose regimen modification
ETDRS
Early Treatment of Diabetic Retinopathy Study
FAS
full analysis set
FBC
Fighting Blindness Canada
GRADE
Grading of Recommendations Assessment, Development and Evaluation
IFA
International Federation on Ageing
IRF
intraretinal fluid
ITC
indirect treatment comparison
LOCF
last observation carried forward
LS
least squares
MAR
missing at random
MID
minimal important difference
MMRM
mixed model for repeated measures
nAMD
neovascular (wet) age-related macular degeneration
NEI VFQ-25
National Eye Institute Visual Function Questionnaire-25
NMA
network meta-analysis
OCT
optical coherence tomography
PIGF
placental growth factor
PCV
polypoidal choroidal vascularization
PPS
per-protocol set
PRN
pro re nata
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SE
standard error
SLR
systematic literature review
SRF
subretinal fluid
TEAE
treatment-emergent adverse event
VEGF
vascular endothelial growth factor
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Aflibercept (Eylea HD) 8 mg (0.07 mL), solution for intravitreal injection |
Sponsor | Bayer Inc. |
Indication | For the treatment of nAMD |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard Review |
NOC date | February 2, 2024 |
Recommended dose | Aflibercept 8 mg (0.07 mL) is administered by intravitreal injection every month (4 weeks ± 1 week) for the first 3 consecutive doses, followed by 8 mg (0.07 mL) every 8 to 16 weeks (± 1 week) based on the physician’s judgment of visual and anatomic outcomes |
HD = high dose; nAMD = neovascular (wet) age-related macular degeneration; NOC = Notice of Compliance.
Introduction
Age-related macular degeneration (AMD) is a progressive condition characterized by central vision loss due to aging.1,2 Neovascular (wet) age-related macular degeneration (nAMD) is a late-stage version of AMD, affecting about 10% of patients and accounting for 90% of cases of severe vision loss in Canada.3 The overall prevalence of any AMD in Canada is estimated at 9% among adults aged 45 years and older, with about 10% of patients reportedly presenting with the neovascular form.4,5 Age-related macular degeneration affects more than 2.5 million patients in Canada, with about 180,000 experiencing vision loss.6,7 Patients experience rapid vision loss with worsening of central vision (caused by scotoma) and/or distortion of straight lines.8 If left untreated, nAMD produces scarring and irreversible vision loss.9 Prompt treatment is imperative, given that patients who experience treatment delay have lower chances of an improvement in visual outcomes.10 Patients with impaired visual acuity caused by progressive disease will experience difficulties with activities associated with daily living and an increased risk of falls, and are at higher risk of social dependence and premature admission to nursing homes.11,12
A clinical expert consulted by Canada’s Drug Agency (CDA-AMC) for this review indicated that intravitreal injections of drugs that inhibit vascular endothelial growth factor (VEGF), including aflibercept 2 mg, ranibizumab, brolucizumab, and faricimab, have become the current standard of care for nAMD. Bevacizumab is an off-label treatment for this condition. Anti-VEGF therapies are recommended as the first line of treatment by guidelines from international ophthalmology societies, including the Canadian Retina Society, American Academy of Ophthalmology, European Retina Society, and British Royal College of Ophthalmology.3,13,14 The clinical expert we consulted noted that different treatment strategies are currently in practice to manage nAMD, including a fixed dosing regimen; an as-needed, or pro re nata (PRN), regimen; and a treat-and-extend regimen.
The objective of the CDA-AMC Clinical Review is to critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of aflibercept 8 mg (0.07mL) through intravitreal injection in the treatment of adults with nAMD.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from a clinical expert we consulted for the purpose of this review.
Patient Input
Input was received from the Canadian Council of the Blind (CCB) and a joint input from Fighting Blindness Canada (FBC), the Canadian Council of the Blind, Vision Loss Rehabilitation Canada, and the International Federation on Ageing (IFA). They surveyed patients living with nAMD including 337 people in Canada.
Vision loss due to AMD has substantial and life-altering impacts on patients’ daily life, manifesting as physical, psychological, and social impacts, according to the patient groups. Patients expressed they often relied on assistance from others to attend appointments, and felt isolated or lonely. Patients worried about their condition worsening due to missed injection appointments. The patient groups noted that burdens associated with injection appointments increased with appointment frequency.
None of the patients surveyed reported having experience with the drug under review. Respondents indicated they were satisfied with their current therapies and noted that it helped them avoid losing more eyesight. According to the patient groups, an efficacious treatment that reduces the number of visits to the ophthalmologist (i.e., a treatment that requires fewer injections) will undoubtedly lead to fewer missed appointments and improved outcomes.
Clinician Input
Input From Clinical Experts We Consulted
The clinical expert indicated that the cost of travelling to medical appointments and the burden placed on family members to provide assistance are some of the obstacles to an optimal treatment outcome that older adult patients with nAMD face. A drug or treatment program that allows for less-frequent visits would therefore improve patient compliance and fill this treatment gap. The clinical expert emphasized that the newer emerging anti-VEGF drugs, faricimab and brolucizumab, can extend the treatment interval to 12 or even 16 weeks. However, the expert noted that brolucizumab is associated with intraocular inflammation. The clinical expert concluded that a more durable and efficacious treatment that does not involve an increase in adverse side effects is an unmet need.
The clinical expert noted that the introduction of longer-acting therapy represents a shift in the treatment paradigm. The expert indicated that aflibercept 2 mg has been used for more than 10 years and has a known safety profile. The clinical expert noted that aflibercept 8 mg can be considered a first-line treatment for nAMD and a replacement therapy when other anti-VEGF treatments are ineffective, or as a treatment for those who do not respond to other anti-VEGF treatments.
The clinical expert we consulted noted that the outcome measures used in clinical practice align with those in the trial: visual acuity, optical coherence tomography (OCT) to assess intraretinal fluid (IRF) or subretinal fluid (SRF), central retinal thickness (CRT) measurement, and fundus examination for retinal or subretinal hemorrhages. Following the initial monthly treatment of aflibercept 8 mg for 3 months, the treatment interval can be extended to every 12 weeks, and the interval can be adjusted by increases or reductions of 4 weeks for the subsequent treatment cycles. The clinical expert described the features of treatment failure as decreasing visual acuity, persistent or increasing volumes of IRF or SRF, and the development of a new subretinal hemorrhage despite active treatment. The clinical expert noted that aflibercept 8 mg can be administered in a clinic or hospital by an ophthalmologist familiar with the diagnosis and management of retinal diseases, including nAMD.
Clinician Group Input
Southwestern Ontario Community Ophthalmologists, the Toronto Retina Institute, the Canadian Retina Society, the Retina Division of the Ottawa Hospital, the Northeastern Ontario Ophthalmology Group, and Toronto Ophthalmologists provided input to this review.
Treatment goals highlighted for AMD (i.e., to maintain vision while extending the duration between treatments to reduce the treatment burden) were consistent across the clinician groups. The clinician groups highlighted that, although current treatments (anti-VEGFs) target the underlying disease mechanism, they are not curative, and the extent and duration of damage to the retina may affect patients’ abilities to achieve improvement. There is therefore a need for new treatments that are efficacious and durable, improve long-term visual outcomes, and maintain a favourable safety profile that minimizes the risk of ocular complications. The clinician groups agreed that a treatment formulation designed and studied with an extended dosing interval would help address the high burden on patients, caregivers, ophthalmologists posed by repeated injections, and reduce backlogs in the health care system. One group added that a treatment that promotes a fluid-free retina for longer durations may help patients avoid declines in quality-of-life metrics associated with vision loss secondary to nAMD. The clinician groups anticipated that aflibercept 8 mg will replace the aflibercept 2 mg formulation, establishing it as a new first-line treatment choice for AMD. The clinician groups inputs aligned with the input submitted by the clinical expert consulted for this review.
Drug Program Input
Input was obtained from the drug programs that participated in the CDA-AMC reimbursement review process. The following key factors could potentially impact the implementation of a CDA-AMC recommendation for aflibercept 8 mg (0.07 mL):
relevant comparators
consideration for initiation of therapy
consideration for discontinuation of therapy
consideration for prescribing of therapy
system and economic issues.
Clinical Evidence
Systematic Review
Description of Study
The PULSAR study (N = 1,009) was a phase III, multicentre (3 sites in Canada), double-blind, randomized, active-controlled noninferiority trial of the efficacy and safety of aflibercept 8 mg administered either every 12 weeks or every 16 weeks compared to aflibercept 2 mg every 8 weeks in treatment-naive adults with nAMD. The study included a screening period (up to 3 weeks) followed by a treatment period. Outcomes were assessed at the 48-week and 60-week time points of the treatment period. The primary outcome of the PULSAR study was the change from baseline in best corrected visual acuity (BCVA) at 48 weeks. Secondary outcomes relevant to the review included the proportion of patients with no IRF or SRF at week 48, proportion of participants gaining at least 15 letters in BCVA from baseline at week 48, frequency of injection through week 48, change from baseline in National Eye Institute Visual Function Questionnaire-25 (NEI VFQ-25) total score at week 48, treatment-emergent adverse events (TEAEs), and serious adverse events (SAEs) through week 60.
The overall proportions of male and female participants were 45.5% and 54.5%, respectively. The median age was 75 years, ranging from 50 to 96 years, and the majority of participants were white (75.8%) or Asian (23.2%). Most patients (86.2%) had a baseline BCVA of 73 or fewer letters on Early Treatment of Diabetic Retinopathy Study (ETDRS) charts.
Efficacy Results
Change From Baseline in Best Corrected Visual Acuity at Week 48
The difference in least squares (LS) mean change from baseline to week 48 was −0.97 letters (95% confidence interval [CI], −2.87 to 0.92) for 8 mg every 12 weeks versus 2 mg every 8 weeks (noninferiority P = 0.0009; superiority P = ██████) and −1.14 letters (95% CI, −2.97 to 0.69) for 8 mg every 16 weeks versus 2 mg every 8 weeks (noninferiority P = 0.0011, superiority P = ██████). Results of analysis of the per-protocol set (PPS) and sensitivity analyses using different approaches to imputing missing data were consistent with those of the full analysis set (FAS). The differences in LS mean change from baseline to week 60 were −0.86 letters (95% CI, −2.57 to 0.84) (noninferiority P = 0.0002; superiority P = ██████) and −0.92 letters (95% CI, −2.51, 0.66; noninferiority P < 0.0001; superiority P = ██████) for the 8 mg every 12 weeks and 8 mg every 16 weeks arms, respectively, compared to the 2 mg every 8 weeks arm. The results from the PPS for week 60 were consistent with those from the FAS.
Proportion of Patients Gaining 15 or More ETDRS Letters at Week 48
The between-group differences in the proportion of patients gaining 15 or more letters in BCVA from baseline to week 48 were −1.75% (95% CI, −7.78 to 4.29%; P = ██████) for aflibercept 8 mg every 12 weeks versus 2 mg every 8 weeks and −0.94% (95% CI, −7.00 to 5.12%; P = ██████) for aflibercept 8 mg every 16 weeks versus 2 mg every 8 weeks, based on the last observation carried forward (LOCF) in the FAS. The observed findings were maintained at week 60.
Presence of Intraretinal or Subretinal Fluid at Week 48
At the week 48 time point, 71.1% of patients in the aflibercept 8 mg every 12 weeks arm and 66.8% of those in the aflibercept 8 mg every 16 weeks arm had no IRF or SRF compared with 59.4% in the aflibercept 2 mg every 8 weeks arm. This resulted in differences in the proportion of patients with no IRF or SRF in the central subfield of 11.73% (95% CI, 4.52% to 18.92%, superiority P = ██████) for 8 mg every 12 weeks versus 2 mg every 8 weeks and 7.45% (95% CI, 0.14% to 14.76%, superiority P = ██████) for 8 mg every 16 weeks versus 2 mg every 8 weeks, based on the LOCF in the FAS. The observed findings were maintained at week 60.
Frequency of Injections
At week 48, a total of 251 of completers (79.4%) in the aflibercept 8 mg every 12 weeks and 239 (76.6%) of those in the 8 mg every 16 weeks arms maintained their randomized treatment interval. This resulted in mean numbers of active injections through week 48 of 6.1 and 5.2 in the aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks arms, respectively, compared to 6.9 in the aflibercept 2 mg every 8 weeks arm. The treatment-group difference between aflibercept either 2 mg every 8 weeks or 8 mg every 12 weeks and treatment with aflibercept 2 mg every 8 weeks was −0.9 █████ ██ █████ injections and the difference between treatment with aflibercept 2 mg every 8 weeks or 8 mg every 16 weeks and treatment with aflibercept 2 mg every 8 week was −1.8 █████ ██ █████ injections. At week 60, the mean numbers of injections were 8.8 (standard deviation [SD] = ███), 7.1 (SD = ███), and 6.2 (SD = ███) for the aflibercept 2 mg every 8 weeks, 8 mg every 12 weeks, and 8 mg every 16 weeks, respectively.
National Eye Institute Visual Function Questionnaire-25
Changes in the LS mean from baseline were observed in all arms at week 48, ranging from 3.35 (standard error [SE] = ████) in the aflibercept 8 mg every 16 weeks arm to 4.22 (SE = ████) in the aflibercept 2 mg every 8 weeks arm. The differences in the LS mean change from baseline using the mixed model for repeated measures (MMRM) in the FAS, were −0.72 ████ ███ █████ ██ █████ | | ███████ for 8 mg every 12 weeks versus 2 mg every 8 weeks and −0.87 ████ ███ █████ ██ █████ | | ███████ for 8 mg every 16 weeks versus 2 mg every 8 weeks. The results were consistent at week 60.
Harms Results
The proportions of patients in the trial who reported at least 1 ocular TEAE were similar across the treatment arms (45% in the aflibercept 2 mg every 8 weeks arm, 42.4% in the aflibercept 8 mg every 12 weeks, and 42.3% in the aflibercept 8 mg every 16 weeks). The most common ocular TEAEs in all treatment arms were reduced visual acuity (6.3% in the aflibercept 2 mg every 8 weeks arm, 3.9% in the aflibercept 8 mg every 12 weeks, and 5.9% in the aflibercept 8 mg every 16 weeks arm), cataracts (3.9%, 4.8%, and 4.4%, respectively), and retinal hemorrhaging (4.5%, 3.6%, and 3.8%, respectively). The proportions of patients with a nonocular TEAE were 59.8%, 59.4%, and 61.2% in the aflibercept 2 mg every 8 weeks, 8 mg every 12 weeks, and 8 mg every 16 weeks arms, respectively. At least 1 treatment-emergent SAE was reported in 1.2% of patients in the aflibercept 2 mg every 8 weeks arm, and 2.1% of patients in each of the 8 mg every 12 weeks and every 16 weeks arms. Retinal hemorrhaging and retinal detachment were the most common SAEs, with similar percentages of 0.3%, 0.6%, and 0.6% for the former and 0.3%, 0.6%, and 0.3% for the latter reported in the aflibercept 2 mg every 8 weeks, aflibercept 8 mg every 12 weeks, and aflibercept 8 mg every 16 weeks arm, respectively.
The proportions of patients who discontinued treatment due to an ocular TEAE were 0.6% in the aflibercept 2 mg every 8 weeks arm and 1.2% in both the 8 mg every 12 weeks and every 16 weeks arms. In the 2 mg every 8 weeks arm, 1.5% of patients died. In the aflibercept 8 mg every 12 weeks arm, deaths were reported for 0.9% and 0.6% of patients in the 8 mg every 12 weeks arm and 8 mg every 16 weeks arm respectively. In terms of notable harms, cataracts occurred in 3.9% of patients treated with aflibercept 2 mg every 8 weeks and 4.8% of patients treated with aflibercept 8 mg every 12 weeks, and 4.4% patients treated with the aflibercept 8 mg every 16 weeks. The incidences of increased intraocular pressure were 2.7% in the 2 mg every 8 weeks arm, 3.3% in the 8 mg every 12 weeks arm, and 3.0% in the 8 mg every 16 weeks arm. The percentages of patients experiencing a retinal pigment epithelium tear were 0.9% in the 2 mg every 8 weeks arm, 1.8% in the 8 mg every 12 weeks arm, and 0.9% in the 8 mg every 16 weeks arm.
Critical Appraisal
The PULSAR study was a randomized, double-blind, active-controlled, noninferiority, phase III trial comparing aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks to aflibercept 2 mg every 8 weeks. No particular concern with the methods of randomization, allocation concealment, or blinding were identified. Baseline characteristics were well balanced among treatment arms. There was agreement between both the FAS and PPS analyses, with both showing noninferiority of high-dose aflibercept (8 mg) compared with aflibercept 2 mg. Adjustment for multiple comparisons in the primary and key secondary end points was made with a hierarchical testing procedure. Aside from change from baseline in BCVA, the remaining outcomes in this report were not subject to hypothesis testing (i.e., they were presented descriptively with 95% CIs), and should be considered supportive evidence only. The MMRM imputation strategy used for the primary analysis assumes data were missing at random (MAR) for participants who discontinued the study prematurely (i.e., missingness depended only on observed data); this appeared to be the case for approximately ██ ██ ███ of participants. Multiple sensitivity analyses employing different techniques for imputing missing data were supportive of the findings. There was some concern regarding missing outcome data across the remaining efficacy outcomes, mainly because it is not clear whether the imputation approaches would result in unbiased estimates. Data for numbers of injections used observed cases only.
In terms of generalizability, only 3 of 251 study sites included in the PULSAR trial were in Canada. The study enrolled only treatment-naive patients who were excluded if they had any prior or concomitant anti-VEGF treatment. The clinical expert we consulted noted that aflibercept may be used in patients after failing another anti-VEGF treatment. This created a gap in the evidence with respect to the efficacy of aflibercept in patients with treatment experience. In terms of clinical relevance of the outcomes assessed in the studies, the most important outcomes of interest to clinicians and patients were measured in the PULSAR trial. The dosing regimen of aflibercept 8 mg in the trial does not align completely with the recommended dosing in the draft product monograph for aflibercept 8 mg, in which aflibercept 8 mg can be administered for up to every 16 weeks in the first year and up to 20 weeks thereafter.15 The clinical expert described aflibercept 2 mg every 8 weeks as an appropriate comparator; however, the dosing regimen in the trial is not aligned with the treat-and-extend approach used by most clinicians for the treatment of nAMD in Canada. The use of a fixed interval (every 8 weeks) for aflibercept 2 mg is a more rigid approach than would be expected in practice, according to the clinical expert, and the inability to modify the dosing schedule raises questions about the generalizability of the injection-frequency outcome. The PULSAR study was the only phase III trial submitted by the sponsor that provided direct evidence comparing aflibercept 8 mg every 12 weeks and every 16 weeks to a relevant comparator in patients with nAMD. No direct evidence comparing aflibercept to the other anti-VEGF drugs currently used in Canadian practice (brolucizumab, faricimab, and bevacizumab) was submitted by the sponsor, creating an evidence gap in the systematic review. The lack of evidence on the long-term (beyond week 60) therapeutic effect of aflibercept 8 mg may also represent a source of uncertainty. Another external validity issue is that the younger patients were underrepresented in this study as the mean age in the study is 74.5 years.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to informing our expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.16,17
In the GRADE approach, evidence from RCTs is initially treated as offering a high degree of certainty and can be rated downward for concerns related to study limitations (internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance was unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The selection of outcomes for GRADE assessments was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
Efficacy:
change from baseline in BCVA
proportion of patients with no IRF and SRF
proportion of patients gaining 15 or more ETDRS letters in BCVA
number of injections
Health-related QoL outcome:
NEI VFQ-25 total score
Harms:
ocular SAEs.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for aflibercept 8 mg every 12 weeks and every 16 weeks versus aflibercept 2 mg every 8 weeks.
Table 2: Summary of Findings for Aflibercept 8 mg Every 12 Weeks and Every 16 Weeks Versus Aflibercept 2 mg Every 8 Weeks for Patients With Treatment-Naive nAMD
Outcome and follow-up | Intervention: patients (studies, N) | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Aflibercept 2 mg q.8.w. | Aflibercept 8 mg q.12.w. or q.16.w | Difference (95% CI) | |||||
Visual acuity | |||||||
Change from baseline in BCVA, LS mean (SE) letters Follow-up: 48 weeks | Aflibercept 8 mg q.12.w.: 671 (1 RCT) | NA | 7.03 (0.74) | 6.06 (0.77) | 0.97 fewer (2.87 fewer to 0.92 more) | High | Aflibercept 8 mg q.12.w. results in little to no clinically important difference in the change in BCVA compared with aflibercept 2 mg q.8.w. |
Aflibercept 8 mg q.16.w.: 674 (1 RCT) | NA | 7.03 (0.74) | 5.89 (0.72) | 1.14 fewer (2.97 fewer to 0.69 more) | High | Aflibercept 8 mg q.16.w. results in little to no clinically important difference in the change in BCVA compared with aflibercept 2 mg q.8.w. | |
Proportion of patients gaining ≥ 15 letters in BCVA from baseline Follow-up: 48 weeks | Aflibercept 8 mg q.12.w.: 671 (1 RCT) | NA | 22.1 per 100 | 20.7 per 100 | 1.8 fewer per 100 (7.8 fewer to 4.3 more per 100) | Moderatea,b | Aflibercept 8 mg q.12.w. likely results in little to no clinically important difference in the proportion of patients gaining ≥ 15 letters from baseline compared with aflibercept 2 mg q.8.w. |
Aflibercept 8 mg q.16.w.: 674 (1 RCT) | NA | 22.1 per 100 | 21.7 per 100 | 0.9 fewer per 100 (7.0 fewer to 5.1 more per 100) | Moderatea,b | Aflibercept 8 mg q.16.w. likely results in little to no clinically important difference in the proportion of patients gaining ≥ 15 letters from baseline compared with aflibercept 2 mg q.8.w. | |
Proportion of patients with no IRF and no SRF | |||||||
Proportion of patients with no IRF and no SRF Follow-up: 48 weeks | Aflibercept 8 mg q.12.w.: 671 (1 RCT) | NR | 59.4 per 100 | 71.1 per 100 | 11.7 more per 100 (4.5 to 18.9 more per 100) | Moderateb,c | Aflibercept 8 mg q.12.w. likely results in little to no clinically important difference in the proportion of patients without IRF and SRF compared with aflibercept 2 mg q.8.w. |
Aflibercept 8 mg q.16.w.: 674 (1 RCT) | NR | 59.4 per 100 | 66.8 per 100 | 7.5 more per 100 (0.1 to 14.8 more per 100) | Moderateb,c | Aflibercept 8 mg q.16.w. likely results in little to no clinically important difference in the proportion of patients without IRF and SRF compared with aflibercept 2 mg q.8.w. | |
Vision-related QoL (NEI VFQ-25) | |||||||
Change from baseline in NEI VFQ-25 total score, LS mean (SE) Follow-up: 48 weeks | Aflibercept 8 mg q.12.w.: 671 (1 RCT) | NA | 4.22 ██████ | 3.50 ██████ | 0.72 less █████ ████ ██ ████ █████ | Moderatea,d | Aflibercept 8 mg q.12.w. likely results in little to no clinically important difference in the change from baseline in vision-related QoL compared with aflibercept 2 mg q.8.w. |
Aflibercept 8 mg q.16.w.: 674 (1 RCT) | NA | 4.22 ██████ | 3.35 ██████ | 0.87 fewer █████ ████ ██ ████ █████ | Moderatea,d | Aflibercept 8 mg q.16.w. likely results in little to no clinically important difference in the change from baseline in vision-related QoL compared with aflibercept 2 mg q.8.w. | |
Number of injections | |||||||
Number of injections, LS mean (95% CI) Follow-up: 48 weeks | Aflibercept 8 mg q.12.w.: 625 (1 RCT) | NA | 7.0 (███ ██ ██ | 6.1 ████ ██ ████ | 0.9 fewer ████ ██ ███ █████ |g | Lowa,e | Aflibercept 8 mg q.12.w. may result in little to no clinically important difference in the frequency of injections when compared with aflibercept 2 mg q.8.w. |
Aflibercept 8 mg q.16.w.: 621 (1 RCT) | NA | 7.0 ████ ██ ██ | 5.2 ████ ██ ████ | 1.8 fewer ████ ██ ███ █████ |g | Lowa,e | Aflibercept 8 mg q.16.w. may result in a reduction in the frequency of injections when compared with aflibercept 2 mg q.8.w. | |
Ocular SAEs | |||||||
Proportion of patients with ocular SAEs Follow-up: 60 weeks | Aflibercept 8 mg q.12.w.: 671 (1 RCT) | NR | 1.2 per 100 | 2.1 per 100 | 0.9 more per 100 (NR) | Moderatea,f | Aflibercept 8 mg q.12.w. likely results in little to no difference in the proportion of patients with ocular SAEs when compared with aflibercept 2 mg q.8.w.; there may be some uncertainty about the clinical importance of the effect |
Aflibercept 8 mg q.16.w.: 674 (1 RCT) | NR | 1.2 per 100 | 2.1 per 100 | 0.9 more per 100 (NR) | Moderatea,f | Aflibercept 8 mg q.16.w. likely results in little to no difference in the proportion of patients with ocular SAEs when compared with aflibercept 2 mg q.8.w.; there may be some uncertainty about the clinical importance of the effect | |
BCVA = best corrected visual acuity; CI = confidence interval; IRF = intraretinal fluid; LS = least squares; NEI VFQ-25 = National Eye Institute Visual Functioning Questionnaire-25; NA = not applicable; NR = not reported; SAE = serious adverse event; QoL = quality of life; RCT = randomized controlled trial; SE = standard error; SRF = subretinal fluid; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks.
Note: Study limitations (internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThere was no hypothesis test for this outcome in the trial; the results can be considered supportive evidence.
bRated down 1 level for serious concerns about risk of bias due to missing outcome data. Did not rate down for imprecision; a between-group difference of greater than 20% was clinically significant according to the clinical experts; the entire CI is compatible with little to no difference.
cThere is no multiplicity adjustment; the result can be considered supportive evidence.
dRated down 1 level for serious concerns about risk of bias due to missing outcome data. Did not rate down for imprecision. Based on the literature, a 6-point change from the baseline in NEI VFQ-25 total score was clinically important; the point estimate and entire CI suggest little to no difference.
eRated down 1 level for serious concerns about risk of bias due to missing outcome data. Did not rate down for imprecision; the clinical expert considered a difference of 2 injections in this time frame to be clinically important; the sample size was adequately large. Rated down 1 level for serious indirectness because the number of injections was driven by the protocol and not reflective of how injections would be provided in practice.
fBecause the clinical expert we consulted was unable to estimate a threshold for clinically important effects, the null was used. Rated down 1 level for serious imprecision due to the small number of events.
gThe information is based on the sponsor's calculation.
Source: PULSAR Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence and the sponsor’s response to requested additional information.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
Description of Studies
The sponsor-submitted an indirect treatment comparison (ITC) that used a Bayesian network meta-analysis (NMA) approach and fixed-effect and random-effects models to compare aflibercept 8 mg every 12 weeks and every 16 weeks in patients with nAMD against other anti-VEGF drugs used for this condition. The following outcome measures are reported: change in BCVA, gain of 15 ETDRS letters, ocular adverse events (AEs), and the mean number of injections. The NMA identified relevant evidence through a systematic review. Depending on the outcome type, different statistical models and links were applied, including a normal likelihood with an identity link for BCVA changes, a binomial likelihood with a logit link for AEs, and a multinomial likelihood with a probit link for letter gains or losses. Methodological and clinical heterogeneity was evaluated using study and patient characteristics, with heterogeneity measured by I2 statistics, and network inconsistency assessed via node-splitting. The mean number of injections was analyzed as an absolute outcome within each intervention node but not comparatively across interventions. Missing data were imputed from external sources, and continuous and binary model inputs were adjusted for SEs derived from various statistical distributions.
Efficacy Results
A total of 34 studies were included in the NMA: 1 assessed aflibercept 8 mg in nAMD, 13 assessed aflibercept 2 mg, 20 assessed ranibizumab, 3 assessed faricimab, 3 assessed brolucizumab, and 12 assessed bevacizumab. Risk-of-bias assessment of the included studies in the sponsor ITC determined that 9 studies were of “low risk,” 14 as of “some concern,” and 11 as of “high risk” as determined by the Cochrane risk-of-bias tool. The sponsor’s ITC did not report any specific actions taken with the studies that were determined as having a high risk of bias.
Results from all comparative outcomes under the random-effects model did not exclude the null in the credible intervals, and the point estimates were often similarly around the null. This applied to the difference in BCVA, 15-letter gain in ETDRS score, and ocular AEs.
Based on predetermined injection regimens, certain interventions are expected to have an average number of injections observed for each treatment regimen and tend to be consistent with the number of injections planned. Intervention administered on a fixed schedule showed little variability between the planned and the actual number of injections given. Treat-and-extend and PRN regimens are not predetermined and, in the first year, showed a mean number of injections of ████ in aflibercept 2 mg PRN (█████ ████ ██ ████), ████ ██████ █████ █████) in bevacizumab PRN, ████ ██████ █████ █████ in ranibizumab PRN, ████ ██████ █████ ██████ PRN nonlinear, ████ ██████ █████ ██████ in ranibizumab PRN nonlinear, ████ ██████ █████ █████ in aflibercept 2 mg treat-and-extend, ████ ██████ █████ █████ in bevacizumab treat-and-extend, ████ ██████ █████ █████ in ranibizumab treat-and-extend, and ████ ██████ █████ █████ for aflibercept 2 mg on a 4 week interval mg treat-and-extend ██████ ████ ██ █████. In the second year, the mean number of injections for treat-and-extend and PRN regimens were ████ ██████ █████ █████ for aflibercept 2 mg PRN, ████ ██████ █████ █████ for bevacizumab, ████ ██████ █████ █████ for ranibizumab PRN, ████ ██████ █████ █████ for aflibercept 2 mg treat-and-extend, ████ ██████ █████ for bevacizumab treat-and-extend, ████ ██████ █████ █████ for ranibizumab treat-and-extend, and ████ ██████ █████ █████ for aflibercept 2 mg treat-and-extend. Noncomparative results of an analysis of injection frequency suggest that aflibercept 8 mg every 12 weeks has a mean injection frequency of 6.10 in the first year, and 3.60 in the second year, while every 16 weeks has a mean injection frequency of 5.20 in the first year, and 3.00 in the second year.
Harms Results
Ocular AEs were reported. The odds ratios of aflibercept 8 mg regimens were near 1 for almost all comparisons, and credible intervals were wide, such that no comparisons excluded the null in the credible interval. While some numerical differences existed in the comparisons versus faricimab and ranibizumab, none excluded the null. Nonocular AEs were not analyzed due to a lack of comprehensive reporting in the included studies.
Critical Appraisal
The systematic literature review supporting the sponsor-submitted ITC for aflibercept 8 mg in nAMD lacked a pre-established protocol and excluded studies with fewer than 40 participants, which may introduce publication bias. The review process, involving dual reviewers for screening but a single reviewer for quality assessment, identified high-risk studies without a clear strategy for mitigating the introduced biases. Clinically relevant outcomes were measured (including BCVA and 15-letter gains, but not IRF or SRF or health-related quality-of-life measures), but the use of fixed injection regimens in the majority of included studies reduces the applicability of the findings to clinical settings in Canada, which favours treat-and-extend regimens. Despite appropriate Bayesian NMA methods, the clinical heterogeneity observed in the study populations — in age, baseline visual acuity, and retinal thickness — raises concerns about the homogeneity assumptions of the NMA models. The sponsor did not use node-splitting to identify inconsistencies. However, the analysis revealed a number of large statistical heterogeneities in direct comparisons as measured through the I2 measure. In addition, given that many treatment effects were supported by single-trial evidence, study and baseline characteristics variability increase the possibility of bias due to effect modifiers. The absence of comparative data for injection frequency limits the interpretability of the potential benefits of aflibercept 8 mg in terms of reduction of injection frequency versus other interventions and regimens.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Conclusions
Based on the PULSAR trial, there is evidence of high certainty that aflibercept 8 mg every 12 or 16 weeks is noninferior (but not superior) to aflibercept 2 mg every 8 weeks as measured by change in BCVA from baseline over 48 weeks of treatment among treatment-naive adults with nAMD. The evidence for other outcomes was considered supportive. Moderate-certainty evidence showed that aflibercept 8 mg every 12 or 16 weeks likely results in little to no difference in important outcomes, such as the proportion of patients gaining 15 or more letters in BCVA and vision-related quality of life when compared with aflibercept 2 mg every 8 weeks. The evidence from the PULSAR trial revealed that the higher dose of aflibercept, if administered every 16 weeks, may (with low certainty) reduce the frequency of injections compared to low-dose aflibercept, but the generalizability of findings is limited as the number of injections were driven by the trial protocol, which is not aligned with clinical practice, where a treat-and-extend approach is commonly used. Moderate-certainty evidence revealed aflibercept 8 mg every 12 weeks and every 16 weeks likely results in little to no difference in the risk of ocular SAEs when compared with aflibercept 2 mg every 8 weeks at 60 weeks. The safety profile of aflibercept 8 mg over 60 weeks was similar to that of aflibercept 2 mg in terms of ocular and nonocular TEAEs, deaths, and notable harms.
Comparative efficacy findings in the ITC are insufficient, as standalone evidence, to inform on the efficacy and safety of aflibercept 8 mg every 12 weeks and every 16 weeks versus other comparators. In general, between-group differences for efficacy outcomes (visual acuity) showed point estimates versus other relevant comparators (including PRN and treat-and-extend strategies) that were close to the null, with wide credible intervals suggesting uncertainty about what treatment might be favoured. This is due to clinical variability among studies and broad credible intervals indicating limited data strength. Noncomparative results of an analysis of injection frequency suggest that aflibercept 8 mg every 12 weeks and every 16 weeks may have a smaller number of injections when numerically contrasted against other interventions with a fixed frequency as well as treat-and-extend regimens; however, there is uncertainty in this finding due to a lack of comparative data and associated measures of variability.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of an aflibercept 8 mg (0.07 mL) solution for intravitreal injection in the treatment of nAMD.
Disease Background
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CDA-AMC review team.
Age-related macular degeneration is a progressive condition characterized by central vision loss due to aging.1,2 AMD is classified into dry or wet forms. The overall prevalence of any AMD in Canada is estimated at 9% among adults aged 45 years and older, with about 10% of patients reportedly presenting with the neovascular form.4,5 The late-stage version of the disease, nAMD, accounts for 90% of severe vision loss in Canada.3 The risk of developing nAMD increases with age, particularly beyond 75 years.19 Neovascular AMD occurs when abnormal new blood vessels under the retina propagate and begin to grow under the macula in a process known as choroidal neovascularization (CNV). These new blood vessels are thin and fragile, and can leak blood, lipids, and fluid into the retina, causing the macula to swell.2,20 The development of CNV is determined by the interplay of stimulators and inhibitors of angiogenesis.8,21,22 VEGF is among the most important angiogenic factors, and its inhibition is known to have a strong antiangiogenic effect.23
More than 2.5 million Canadians have been diagnosed with AMD, with about 180,000 patients experiencing vision loss.6,7 The annual incidence rate of late-stage AMD (encompassing geographic atrophy and the neovascular form of AMD) according to pooled data from 4 incidence studies reported in a meta-analysis study by Li and colleagues, was 1.4 per 1,000 individuals above the age of 60 years.24 Patients experience rapid vision loss with worsening of central vision (caused by scotoma) and/or distortion of straight lines.8 If left untreated, nAMD produces scarring and irreversible vision loss.9 Prompt treatment is imperative, given that patients who experience treatment delay have lower chances of an improvement in visual outcomes.10 Patient mobility and independence depend heavily on vision. Patients with impaired visual acuity caused by progressive disease will experience difficulties with daily living, an increased risk of falls, and are at higher risk of social dependence and premature admission to nursing homes.11,12
A diagnosis of nAMD is based on the presence of characteristic findings (e.g., the presence of IRF and/or SRF, retinal and subretinal hemorrhage, and retinal thickening) in an eye examination using standard retinal imaging techniques and visual assessments. Retinal imaging techniques include colour fundus photography, fluorescein angiography, OCT, and OCT angiography. OCT is a noninvasive imaging tool routinely used across ophthalmology settings as the first diagnostic test for patients with retinal disease.25,26
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by our review team.
Intravitreal injections with anti-VEGF therapies have become the current standard of care for nAMD, according to the clinical expert we consulted. Anti-VEGFs target the pathology of nAMD, decreasing vascular leakage and neovascularization. Guidelines from international ophthalmology societies, including the Canadian Retina Society, American Academy of Ophthalmology, the European Retina Society and the British Royal College of Ophthalmology3,13,14 recommend anti-VEGF therapies for first-line use. Anti-VEGF therapies, which are publicly reimbursed by at least 1 participating drug plan, reimbursed on an off-label basis, or positively recommended by CADTH for nAMD, include aflibercept 2 mg, ranibizumab, brolucizumab, faricimab, and bevacizumab. Bevacizumab is not indicated for ophthalmic use (i.e., its use in nAMD is off-label).
The clinical expert we consulted by also indicated that anti-VEGF drugs have changed the nAMD management paradigms, introducing an opportunity to improve vision by reducing exudation, arresting the growth of the CNV, and converting active CNV to fibrosis. The clinical expert noted that different treatment strategies involving anti-VEGF injections are currently in use for the management of nAMD, including a fixed dosing regimen, a PRN regimen, and a treat-and-extend regimen. The treat-and-extend approach is used by most clinicians to treat nAMD in Canada.
The clinical expert we consulted by indicated that the cost of travelling and the burden of family members posed by providing assistance are some of the obstacles that prevent older adult patients with nAMD from achieving an optimal treatment outcome. A drug or treatment program that allows for less-frequent visits is therefore likely to improve patient compliance. The clinical expert emphasized that the risk of severe ocular inflammatory reactions (e.g., retinal vasculitis) with brolucizumab, although rare, can be devastating to vision. A more durable and efficacious treatment that does not result in an increase in adverse side effects is therefore needed, according to the clinical expert.
Drug Under Review
Aflibercept is an anti-VEGF drug that inhibits the predominant signalling pathways responsible for angiogenesis and vascular leakage:27 VEGF-A and placental growth factor (PIGF), which are members of the VEGF family of proangiogenic factors, can act as potent mitogenic, chemotactic, and vascular permeability factors for endothelial cells. VEGF acts via 2 receptor tyrosine kinases, VEGFR-1 and VEGFR-2, that are present on the surface of endothelial cells. PlGF is a proangiogenic factor that activates VEGFR-1. Excessive activation of these receptors by VEGF-A can result in pathological neovascularization and excessive vascular permeability, which is believed to contribute to vision loss in a variety of ocular diseases.15 Aflibercept inhibits PlGF in addition to all isoforms of VEGF-A.28 Aflibercept 8 mg (0.07 mL) has a different pharmacokinetic profile compared to aflibercept 2 mg, which leads to longer inhibition of VEGF.29
Aflibercept 8 mg (0.07 mL) is administered by intravitreal injection every month (4 weeks ± 1 week) for the first 3 consecutive doses, followed by 8 mg (0.07 mL) every 8 to 16 weeks (± 1 week) based on the physician’s judgment of visual and anatomic outcomes. Treatment intervals of 1 month (4 weeks) for more than 3 consecutive doses have not been studied, and there are limited data on treatment intervals longer than 5 months (20 weeks).15
Aflibercept 8 mg (0.07 mL) is indicated for the treatment of nAMD. The sponsor’s reimbursement request aligns with the proposed Health Canada indication. Aflibercept 8 mg (0.07 mL) was approved by the FDA on August 18, 2023, for nAMD, diabetic macular edema, and diabetic retinopathy, and is currently under review by the European Medicines Agency. Aflibercept 2 mg was previously reviewed by CADTH for nAMD and received a recommendation to reimburse with conditions on October 20, 2014.30 Aflibercept 2 mg is funded for the treatment of nAMD across CDA-AMC–participating jurisdictions.
Table 3 provides key characteristics of commonly used anti-VEGF treatments for nAMD.
Table 3: Key Characteristics of Aflibercept 8 mg, Aflibercept 2 mg, Faricimab, Ranibizumab, Bevacizumab, and Brolucizumab
Characteristic | Aflibercept 8 mg | Aflibercept 2 mg | Faricimab | Ranibizumab | Bevacizumaba | Brolucizumab |
|---|---|---|---|---|---|---|
Mechanism of action | VEGF inhibitor (soluble decoy receptor, targets VEGF-A and PIGF) | VEGF inhibitor (soluble decoy receptor, targets VEGF-A and PIGF) | VEGF inhibitor (mAb, targets Ang-2 and VEGF-A) | VEGF inhibitor (mAb, targets VEGF-A isoforms) | VEGF inhibitor (mAb, targets VEGF) | VEGF inhibitor that binds to VEGF-A isoforms (VEGF110, VEGF121, and VEGF165), preventing binding of VEGF-A to its receptors, VEGFR-1 and VEGFR-2 |
Indicationb | For treatment of nAMD | For treatment of nAMD | For treatment of nAMD | For treatment of nAMD | None for nAMD (off-label use) | For treatment of nAMD |
Route of administration | IVT | IVT | IVT | IVT | IVT | IVT |
Recommended dosing | Every month (4 weeks ± 1 week) for the first 3 consecutive doses, followed by 8 mg (0.07 mL) every 8 to 16 weeks (± 1 week) based on physician’s judgment of visual and anatomic outcomes | Every 4 weeks for the first 3 doses followed by treatment intervals every 8 weeks; treatment may be maintained every 8 weeks or extended in 2-week increments based on visual and anatomic outcomes | Every 4 weeks for the first 4 doses, followed by treatment intervals of every 8, 12 or 16 weeks, based on anatomic and visual acuity evaluations at week 20 and 24 | Once a month; treatment may be reduced to 1 injection every 3 months after the first 3 injections if monthly dosing is not feasible | Every 4 weeks for the first 3 doses followed by treatment intervals of every 8 to 12 weeks | Every 4 weeks for the first 3 doses, followed by treatment intervals of 8 or 12 weeks based on visual and anatomic outcomes assessed 16 weeks after treatment start and regularly after that |
Serious adverse effects or safety issues |
|
|
|
|
|
|
Ang-2 = angiopoietin-2; ATE = arterial thromboembolic events (includes nonfatal stroke, nonfatal myocardial infarction, or vascular death); IOP = intraocular pressure; IVT = intravitreal; mAb = monoclonal antibody; nAMD = neovascular (wet) age-related macular degeneration; PIGF = placental growth factor; VA = visual acuity; VEGF = vascular endothelial growth factor.
aBevacizumab is used off-label in the treatment of age-related macular degeneration.
bHealth Canada–approved indication.
Sources: Product monographs for Vabysmo,31 aflibercept,15 Lucentis,32 and Avastin.33
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient input(s) we received have been included in the Patient and Clinician Group Input.
Input from the CCB and a joint patient input from FBC, the Canadian Council of the Blind, Vision Loss Rehabilitation Canada, and the IFA were summarized for this report. The CCB, FBC, and IFA are not-for-profit patient groups that promote research in vision loss and provide support and advocacy for patients with living with vision loss and other related vision issues.
Information from the CCB input was sourced from a joint survey conducted by FBC in early 2020 in patients with AMD and diabetic macular edema, including 2 other surveys conducted in April 2020 and June to July 2022. Information from the joint patient input was gathered through an online survey made available to Canadians living with wet or dry AMD during the first months of 2020. The survey gathered lived experiences, particularly perceptions of the disease, treatments, and the specific burdens associated with living with wet and dry AMD. The survey presented in the joint patient input had a total of 337 patients in Canada with AMD participating. About half of the respondents (47.1%) reported they had wet AMD, 37.7% reported dry AMD, 12.8% indicated either wet in 1 eye and dry in the other, and 2.4% were unsure of the type. The number of patients who participated in the CCB surveys were not presented.
Vision loss due to AMD has substantial and life-altering impacts on patients’ daily life, manifesting as physical, psychological, and social challenges, according to the patient group inputs. Patients reported that vision loss resulting from AMD significantly affected daily activities, such as personal care and hygiene, interactions with electronic devices (phones and tablets), and reading books and newspapers. Patients indicated they frequently relied on assistance from others to attend injection appointments, and often felt isolated or lonely. Patients worried that their condition could worsen due to missed injection appointments. Survey results from the CCB and FBC revealed that a significant number of patients were missing their regular eye injections, the most common reason being travel logistics and payment. Both groups noted that the burden associated with injection appointments increased when appointments were frequent. Challenges related to treatment access were notably exacerbated for respondents living in rural areas and remote communities, where access to specialized care is often limited. Ophthalmologists interviewed by CCB also reported that patients that had missed injection visits presented with significant vision loss.
The majority of respondents (75.4%) in the joint patient input indicated they were currently receiving injections for AMD. The most common treatments listed included bevacizumab, ranibizumab, aflibercept 2 mg, and dexamethasone. No patients reported receiving aflibercept 8 mg since the surveys were conducted in early 2020. Almost half (46%) of the respondents indicated they were satisfied with their injections and added that it helped them avoid losing more eyesight. Wait times and travel ranked high on lists of difficult aspects of receiving treatment. Respondents also expressed anxiety or fear associated with the injections they received. Some patients experienced visual complications such as scratchiness or pain in the eye following injections while others reported blurry vision for 1 to 3 hours after injections (48.2%), followed by 4 to 6 hours (25.9%). Other respondents indicated that they were unable to complete at least 1 regular activity postinjection.
According to both patient advocacy groups, a treatment with a less-demanding injection regimen would ease the burden associated with AMD as patients would prefer a treatment that is taken less frequently. According to the patient advocacy groups, a new medication that decreases the number of patients being seen by specialists could free up ophthalmologists’ time for surgery and other backlogs, consequently improving vision health for all patients.
Clinician Input
Input From Clinical Experts We Consulted
All of our review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of nAMD.
Unmet Needs
The clinical expert indicated that the need for ongoing treatment at regular intervals poses the greatest challenge for the successful management of nAMD. The expert noted the cost of travelling and the burden on family members who supply assistance can keep some older adults with nAMD from achieving an optimal treatment outcome. A drug or treatment program that allows for less-frequent visits would therefore likely improve patient adherence and fill this treatment gap. The clinical expert emphasized that the newer emerging anti-VEGF drugs, faricimab and brolucizumab, can extend the treatment interval to 12 weeks or even 16 weeks. However, brolucizumab is reportedly associated with a higher frequency of intraocular inflammation. The risk of severe, although rare, ocular inflammatory reactions, such as retinal vasculitis, can be devastating to vision. The clinical expert therefore concluded that a more durable and efficacious treatment that does not increased the risk of adverse side effects is an unmet need.
Place in Therapy
The clinical expert noted that, after more than a decade of anti-VEGF treatments for nAMD patients requiring frequent intravitreal injections, the introduction of a longer-acting therapy represents a shift in the treatment paradigm. The expert indicated that aflibercept 2 mg has been used for more than 10 years and has a known safety profile. The clinical expert indicated that aflibercept 8 mg could be considered a first-line treatment for nAMD and a replacement therapy when other anti-VEGF treatments are ineffective. The clinical expert we consulted emphasized that there is no evidence to support aflibercept 8 mg use in combination with another treatment.
Patient Population
The clinical expert indicated that aflibercept 8 mg (0.07 mL) is suited for all nAMD patients, particularly those who are treatment-naive. It can also be considered for patients who responded to an anti-VEGF treatment, including aflibercept 2 mg, but were unable to tolerate it extend beyond 8 weeks, or those who do not respond with the other anti-VEGF treatments.
Assessing the Response Treatment
The clinical expert we consulted noted that visual acuity, OCT, assessment of IRF or SRF and CRT measurement and fundus examination for retinal and/or subretinal hemorrhaging are the usual outcome measurements used in clinical practice and are the same as in the clinical trial. The expert indicated that these measurements are taken at each clinical visit for treatment, while the assessment is also performed to determine if treatment needs to be maintained or modified. Following the initial monthly treatment for 3 months, the treatment interval can be extended to every 12 weeks, and subsequently, the interval can be adjusted up or down by increments of 4 weeks for the next treatment cycle.
Discontinuing Treatment
The clinical expert noted that discontinuation of the treatment could be considered when there is no sign of treatment response or there is deterioration of the condition despite ongoing treatment. The expert indicated that treatment failure is characterized by decreasing visual acuity, persistent or increased IRF or SRF, or development of new subretinal hemorrhaging despite active treatment.
Prescribing Considerations
The clinical expert noted that aflibercept 8 mg (0.07 mL) can be administered in a clinic or hospital. The treatment should be provided by an ophthalmologist who is familiar with the diagnosis and management of retinal diseases, including nAMD.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full original clinician group inputs we receive are included in the Patient and Clinician Group Input.
Inputs from 6 clinician groups were summarized for this review: Southwestern Ontario Community Ophthalmologists, the Toronto Retina Institute, the Canadian Retina Society, Retina Division of the Ottawa Hospital, the Northeastern Ontario Ophthalmology Group, and Toronto Ophthalmologists. In total, 17 clinician experts contributed to the clinician group submission. Inputs across the 6 clinician groups were sourced from telephone conversations, virtual meetings and discussions, emails, literature reviews, conference presentations, systematic reviews, and meta-analyses.
Treatment goals highlighted for AMD were consistent across groups. Maintaining vision (improving retinal anatomy and achieving stabilization or improvement in visual acuity) while extending the duration between treatments to reduce the treatment burden was highlighted. All groups highlighted similar treatments currently in use in practice, including anti-VEGFs such as aflibercept 2 mg, ranibizumab, brolucizumab, faricimab, and bevacizumab (off-label use). Bevacizumab, according to the clinician groups, is difficult for patients aged over 65 years to access, and brolucizumab is associated with risks of intraocular side effects. The clinician groups noted that, although current treatments target the underlying disease mechanism, they are not curative, and the extent and duration of damage to the retina may affect patients’ abilities to achieve improvement. There is therefore a need for efficacious, durable, and long-lasting treatments that current therapies do not provide. The groups also emphasized the need for treatments that improve outcomes in the long term for this population. Treatment for AMD is also ongoing, requiring repeated visits (at least every 7 to 8 weeks). The clinician groups noted that treatment formulations designed with an extended dosing interval (such as the case of aflibercept 8 mg) would help address unmet needs and promote treatment compliance for this patient population. The Canadian Retinal Society added that a fluid-free retina for a longer duration allows for maintenance of maximal vision gains over the patient’s lifetime, which may translate into improved quality of life, increased independence, reduction in the risk of falls, and reduced depression. All groups emphasized the need for safer treatments to minimize the risk of ocular complications.
The clinician group inputs aligned with the input submitted by the clinical expert consulted for this review. The clinician groups anticipated that aflibercept 8 mg would replace the aflibercept 2 mg formulation, establishing it as a new first-line treatment choice for AMD. The groups indicated it may also be considered as an alternative to ranibizumab’s biosimilar formulation. According to the clinician groups, all patients requiring treatment with an anti-VEGF will be eligible to receive aflibercept 8 mg; however, monocular patients (those with disease in only 1 eye), may be slightly less suitable due to the potential risk of infection if the vial is not designed for multi-use. The clinician groups reported that response to treatment will be assessed by examining vision stabilization, anatomic outcomes, and clinical exams for hemorrhaging. Eye anatomy will be measured via OCT. Response assessment, according to the clinician groups, is highly standardized across trials and clinical practice; the same outcomes assessed in the trials will be used in clinical practice. The clinical groups noted that factors that will affect decisions to discontinue will include end-stage disease with significant atrophying and/or fibrosis and no improvement in vision despite regular treatments. The clinician groups also noted that aflibercept 8 mg will be administered only by physicians who specialize in ophthalmology and who are equipped to assess and manage the disease and address AEs. The groups added that aflibercept 8 mg will be primarily administered in an ophthalmologist’s office, and may in rare cases be given at hospital outpatient clinics.
Drug Program Input
The drug programs provide input on each drug being reviewed through the CDA-AMC reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts we consulted are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The PULSAR study is a phase III, multicentre, randomized, double-masked, active-controlled study that compared aflibercept high-dose (8 mg) to aflibercept 2 mg for efficacy, safety, and tolerability and to determine if aflibercept 8 mg administered in 2 extended dosing regimens was noninferior to aflibercept 2 mg. There were no trials comparing aflibercept 8 mg with other anti-VEGF drugs (brolucizumab and faricimab) that can be administered at the same extended dosing interval. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for initiation of therapy | |
Most provinces have retinal programs and therefore no published criteria or criteria is not adjudicated against. Some provinces have initiation criteria that were developed by a working group and may be outdated. Inclusion criteria for the PULSAR study are not consistent with existing drug plan criteria for nAMD. Ranibizumab recommendation is from 2008 with no initiation or discontinuation criteria. Aflibercept 2 mg recommendation is from 2014 and also did not include initiation or discontinuation criteria. More recently, the brolucizumab recommendation included wording from existing drug plan (discontinuation) criteria, and faricimab was to be listed in a manner similar to other anti-VEGF drugs. | The clinical experts consulted by CADTH advised that the initiation of treatment for patients diagnosed with nAMD, defined by the presence of retinal fluid (either intraretinal or subretinal) or hemorrhages, is warranted. In terms of discontinuing treatment, the clinical expert we consulted noted that, several factors should be considered, such as the absence of a positive response in a patient after receiving the treatment for at least 3 interval injections, as well as a lack of improvement in retinal fluid or visual acuity. In such cases, it is advisable to contemplate switching or discontinuing the medication, as it may not be delivering the intended benefits, while acknowledging that each injection carries inherent risks. Conversely, the clinical expert noted that patients at the end stages of the disease with extensive scarring are unlikely to derive significant benefits from anti-VEGF treatment. This also warrants consideration in terms of treatment cessation. |
Considerations for discontinuation of therapy | |
Consistency with discontinuation criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for prescribing of therapy | |
The manufacturer notes that aflibercept 8 mg meets an unmet need by having a dosing frequency of every 12 to 16 weeks. Recommended dose of brolucizumab is 6 mg every 6 weeks for the first 5 doses then every 12 weeks. Recommended dose of faricimab is 6 mg every 4 weeks for the first 4 doses then every 8, 12 or 16 weeks. | This is a comment from the drug plans to inform CDEC deliberations. |
Does aflibercept 8 mg meet an unmet need given there are other products marketed with an extended dosing interval? | The clinical expert consulted by CADTH indicated that currently, 3 medications that offer extended dosing intervals are available: aflibercept 8 mg, faricimab, and brolucizumab. The clinical expert indicated that it is essential to note that brolucizumab has been associated with a higher frequency of intraocular inflammations and severe cases of hemorrhagic retinal vasculitis. These severe effects have the potential to cause significant vision loss, to the extent that some patients may even experience complete blindness as a result of complications arising from the treatment. The clinical expert noted that faricimab represents a relatively new medication that can extend treatment intervals up to 12 weeks. While it is not clear if the intention is to extend treatment to 16 weeks, this 16-week extension is the optimal treatment goal. This is noteworthy because even aflibercept 2 mg allows for extension up to 12 weeks in some cases when using a treat-and-extend protocol. The clinical expert noted that aflibercept 2 mg, with a history of over a decade in clinical use, demonstrated an appropriate safety profile. The clinical expert highlighted that the primary objective, as dictated by the unmet need, is to extend treatment intervals and alleviate the treatment burden on both patients and clinicians. |
System and economic issues | |
Aflibercept 8 mg would have a significant budget impact on public drug plans. Biosimilars have already been marketed for Lucentis. Biosimilars are anticipated for aflibercept 2 mg next year. Should the pricing recommendation for reimbursement recommend that aflibercept 8 mg be negotiated so that it provides cost savings to drug programs relative to the cost of currently funded anti-VEGF drugs for AMD? | This is a question for CDEC. |
Confidential pricing agreements exist for most anti-VEGF drugs. Based on current list price, aflibercept 8 mg is not a cost-effective treatment option. | This is a comment from the drug plans to inform CDEC deliberations. |
Retinal programs/provincials eye centres exist in a number of provinces. Bevacizumab first policies in place in a number of provinces. | This is a comment from the drug plans to inform CDEC deliberations. |
AMD = age-related macular degeneration; CDA-AMC = Canada’s Drug Agency; CDEC = Canadian Drug Expert Committee; nAMD = neovascular (wet) age-related macular degeneration; VEGF = vascular endothelial growth factor.
Clinical Evidence
The objective of the CDA-AMC Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of aflibercept 8 mg (0.07 mL) through intravitreal injection in the treatment of adults with nAMD. The focus will be placed on comparing aflibercept 8 mg (0.07 mL) to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of aflibercept 8 mg (0.07 mL) is presented in 2 sections, with our critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor. No long-term extensions studies or studies addressing gaps in the pivotal and RCT evidence were submitted by the sponsor.
Included Studies
Clinical evidence from 1 pivotal RCT identified in the systematic review and 1 ITC are included in our review and appraised in this document:
Systematic Review
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CDA-AMC review team.
Description of Study
Characteristics of the included study are summarized in Table 5.
Detail | PULSAR |
|---|---|
Study design | Phase III, multicentre, randomized, double-masked, active-controlled, noninferiority study |
Locations | 251 study sites in 27 countries/regions in Europe, North America, Latin America, Australia, and Asia Pacific (3 study sites in Canada) |
Patient enrolment dates | Start date: August 11, 2020 End date: July 30, 2021 |
Randomized (N) | N = 1,009 randomized in a 1:1:1 ratio:
|
Inclusion criteria |
|
Exclusion criteria | Causes of CNV other than nAMD in the study eye Prior or concomitant conditions in the study eye:
|
Intervention |
|
Comparator | Aflibercept 2 mg intravitreal injection administered every 8 weeks after 3 initial injections at 4-week intervals |
Screening phase | 3 weeks |
Treatment phase | 48 and 60 weeks (primary and select secondary efficacy end points analyzed within double-masked phase) 96 weeks (additional secondary and exploratory end points analyzed within double-masked phase) |
Follow-up phase | NA; patients could consent to continue in an extension period |
Primary end point | Change from baseline in BCVA measured by the ETDRS letter score at week 48 |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publications | Lanzetta P, Korobelnik J, Heier JS, et al. Intravitreal aflibercept 8 mg vs. 2 mg in neovascular age-related macular degeneration at 48 weeks (PULSAR): a randomized, double-masked, noninferiority, phase III trial; unpublished manuscript. Trial registration: https://clinicaltrials.gov/study/NCT04429503 |
BCVA = best corrected visual acuity; CNV = choroidal neovascularization; CRT = central retinal thickness; ETDRS = Early Treatment Diabetic Retinopathy Study; IOP = intraocular pressure; IRF = intraretinal fluid; nAMD = neovascular (wet) age-related macular degeneration; NEI VFQ-25 = National Eye Institute Visual Function Questionnaire-25; OCT = optical coherence tomography; SAE = serious adverse event; SRF = subretinal fluid; TEAE = treatment-emergent adverse event; VEGF = vascular endothelial growth factor.
Sources: PULSAR Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The PULSAR study is multi-centre, randomized, double-masked, active-controlled phase III trial with a primary objective of determining if treatment with aflibercept 8 mg administered with 2 extended dosing intervals (every 12 or 16 weeks) provides noninferior BCVA change compared to aflibercept 2 mg every 8 weeks in adult patients with treatment-naive nAMD. Patients (N = 1,009) were enrolled across 27 countries, including 3 sites in Canada, and were randomized in a 1:1:1 ratio to 1 of 3 dosing regimens:
aflibercept 2 mg administered every 8 weeks after 3 initial monthly doses (2 mg every 8 weeks)
aflibercept 8 mg administered every 12 weeks after 3 initial monthly doses (8 mg every 12 weeks)
aflibercept 8 mg administered every 16 weeks after 3 initial monthly doses (8 mg every 16 weeks).
The randomization was stratified by baseline BCVA and geographical region. The study included a screening period of 3 weeks. Thereafter, patients entered a treatment period, where the primary efficacy end point of mean change in BCVA from baseline was measured at week 48. Additional end points were tested at 60 and 96 weeks. In this report, we used a data cut-off at 48 weeks for efficacy outcomes and 60 weeks for safety outcomes. Evidence at the 60-week data cut-off had been considered for efficacy outcomes. The masked phase of PULSAR was completed at week 96 (end of main study visit with a last patient last visit date of June 29, 2023); the sponsor therefore presented only selected results corresponding to this dataset in their submission (the clinical study report for the 96-week analysis is not available at time of submission). The extension phase of the study starts immediately after at the end of the week 96 study visit, during which the study drug will be administered in an open-label treatment period until week 156. However, the open-label extension is ongoing. The study design is illustrated in Figure 1.
Figure 1: Study Design of PULSAR
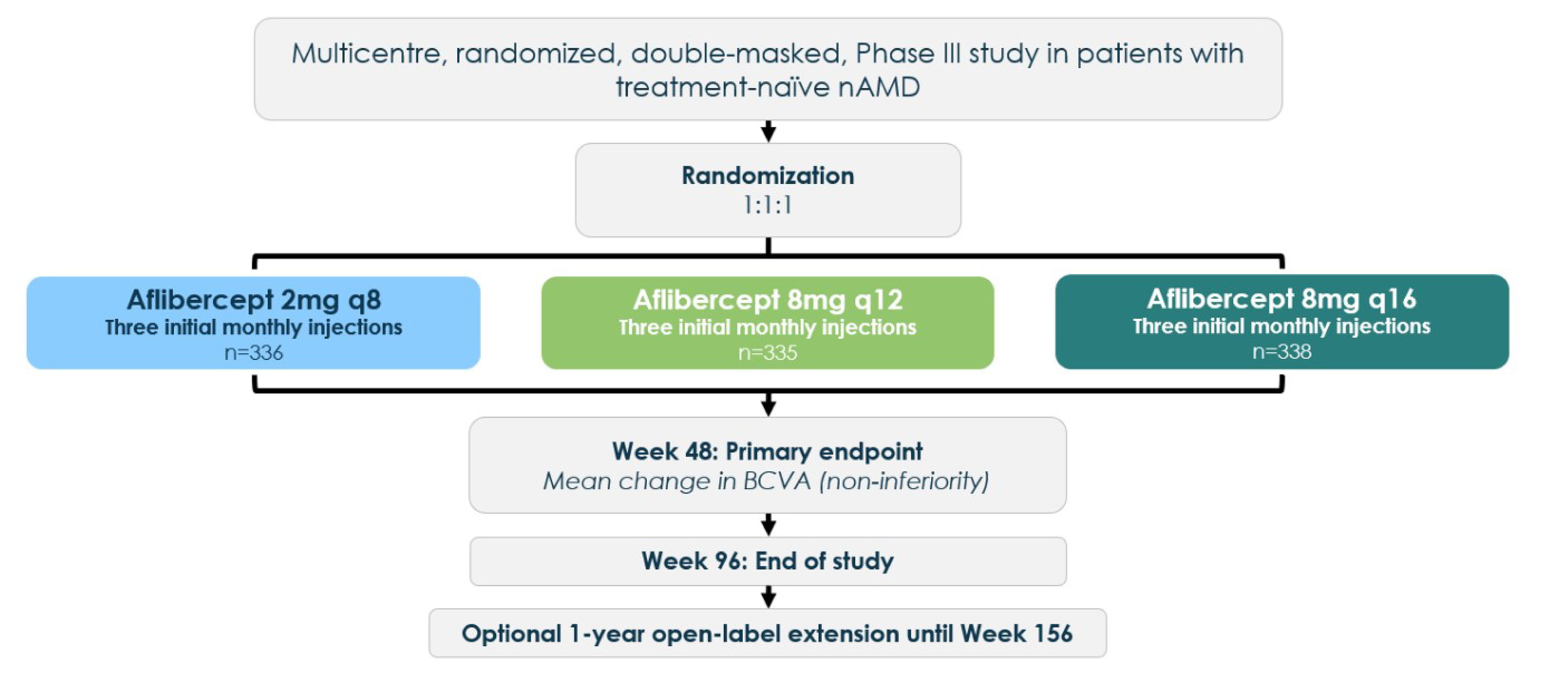
BCVA = best corrected visual acuity; n = number of participants per group; nAMD = neovascular (wet) age-related macular degeneration; q8 = every 8 weeks; q12 = every 12 weeks; q16 = every 16 weeks.
Source: PULSAR Clinical Study Report.18
Populations
A detailed description of the inclusion and exclusion criteria for the PULSAR trial is provided in Table 5. Eligible patients were at least 50 years of age with treatment-naive active CNV lesions secondary to nAMD (> 50% of the total lesion area), BCVA ETDRS letter scores of 78 to 24 (Snellen equivalent of 20/32 to 20/320), and with IRF and/or SRF affecting the central subfield on OCT. Only treatment-naive nAMD patients were enrolled in PULSAR. Patients were excluded if they received prior or concomitant ocular (in the study eye) or systemic treatment with an investigational or approved, anti-VEGF, or other drug. They also were excluded if the cause of CNV was something other than nAMD and if they had some concomitant conditions, including but not limited to subretinal hemorrhage, scar or fibrosis, and presence of retinal pigment epithelial tears.
Interventions
In the PULSAR trial, eligible patients were randomized in a 1:1:1 ratio to 3 parallel treatment arms: aflibercept 2 mg every 8 weeks, aflibercept 8 mg every 12 weeks, or aflibercept 8 mg every 16 weeks. In the aflibercept 2 mg arm, patients received intravitreal injections every 4 weeks for 3 loading doses, followed by maintenance dosing every 8 weeks to week 92, with a final study visit at week 96. In the aflibercept 8 mg arms, patients received intravitreal injections every 4 weeks for 3 loading doses, followed by maintenance dosing every 12 weeks or every 16 weeks, respectively. A sham procedure was performed on visits when an active injection was not planned for masking purposes. Active injections and sham procedures were administered by study-site personnel. During the study, treatment intervals could be shortened or extended in the 8 mg arms based on prespecified dose regimen modification (DRM) criteria. According to protocol, no adjustments to the treatment interval were allowed in the 2 mg arm. Participants in the aflibercept 2 mg group remained on a fixed dosing regimen of every 8 weeks until the end-of-masked-study visit at week 96 (i.e., no modifications of treatment intervals were allowed regardless of the outcomes of the DRM assessments).
DRM Criteria for Interval Shortening
Starting at week 16, patients assigned to the aflibercept 8 mg arms were assessed for DRM criteria at every visit. DRM criteria for shortening the dosing interval were:
greater than a 5-letter loss in BCVA from week 12 BCVA
greater than a 25 µm increase in CRT from week 12 or new onset foveal neovascularization or foveal hemorrhage.
If a patient in the 8 mg every 12 weeks arm met the DRM criteria for shortening at week 16 or week 20, they were dosed with 8 mg at that visit and subsequently continued with every 8 weeks dosing. If a patient on 8 mg every 16 weeks met the DRM criteria for shortening at week 16 or week 20, they were dosed with 8 mg at that visit and continued with every 8 weeks dosing. If a patient on 8 mg every 16 weeks met the DRM criteria for shortening at week 24, they were dosed with 8 mg at that visit and continued with every 12 weeks dosing. Subsequently, patients who met the DRM criteria at any active treatment visit had their intervals shortened by 4 weeks, to a minimum interval of 8 weeks.
Starting at week 52, all patients randomized to either 8 mg treatment arm were eligible for adjustments of their treatment intervals (shortening or extension) with the dose interval adjustments becoming effective at or after week 60.
Dose Regimen Modification Criteria for Interval Extension
In year 2, all patients in the aflibercept 8 mg treatment arms (including patients whose interval was shortened during year 1) were eligible for an extension of the treatment interval (by 4-week increments) if the following DRM extension criteria were met at visits with active injection:
greater than a 5-letter loss in BCVA from week 12
no fluid at the central subfield on OCT
no new onset foveal neovascularization or foveal hemorrhage.
Concomitant Therapy
Participants were not eligible to receive any standard or investigational drugs for treatment of nAMD in the study eye until they had completed the end-of-study or early-termination-visit assessments. This includes medications administered locally (e.g., intravitreal, by juxta scleral or periorbital routes), as well as those administered systemically with the intent of treating the fellow eye. If a pretreatment concomitant medication (e.g., antibiotic or topical anesthetic) was administered in the study eye before injection, it was administered for sham procedures as well.
Only 1 eye per participant could be enrolled in the study. If a participant’s fellow (nonstudy) eye required anti-VEGF treatment during the study, the fellow eye was to be treated with aflibercept 2 mg according to the approved treatment regimen in the respective country, irrespective of the randomization assignment of the participant.
Outcomes
A list of efficacy end points assessed in this Clinical Review is provided in Table 6, followed by descriptions of the outcome measures. Patients were examined every 4 weeks throughout the study. Standard evaluations of safety and efficacy were performed at every visit. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical expert we consulted and input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform the expert committee deliberations and then finalized this list of end points in consultation with the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing the expert committee deliberations were also assessed using GRADE. Specifically, the following were considered:
The clinical expert as well as clinician and patient groups noted that visual acuity (change from baseline and proportion of patients gaining at least 15 EDTRS letters in BCVA) was the foremost clinical outcome of interest. The clinical expert suggested that a change of 15 letters would represent a large improvement.
The clinical expert noted that the presence of IRF and SRF are criteria to start treatment, and a persistent increase would be indicative of treatment failure.
Vision-related quality of life was an important outcome to patients.
Patient and clinician groups as well as the clinical expert noted a desire for less-burdensome treatments (i.e., fewer injections).
Table 6: Outcomes Summarized — PULSAR
Outcome measure | Time point | PULSAR |
|---|---|---|
Change from baseline in BCVA | At week 48 | Primarya |
IRF and SRF | At week 48 | Secondary |
Patients gaining ≥ 15 ETDRS letters in BCVA | At week 48 | Secondary |
NEI VFQ-25 | At week 48 | Secondary |
Frequency of injection | Through week 48 | Exploratory |
BCVA = best corrected visual acuity; EDTRS = Early Treatment Diabetic Retinopathy Study; NEI VFQ-25 = National Eye Institute Visual Functioning Questionnaire-25.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Sources: PULSAR Clinical Study Report18 and the sponsor’s Summary of Clinical Evidence.
Best Corrected Visual Acuity
The change from baseline in BCVA as measured by the ETDRS letter score at week 48 was the primary outcome in PULSAR to determine if treatment with aflibercept 8 mg at intervals of 12 or 16 weeks was associated with a noninferior BCVA change compared to aflibercept 2 mg every 8 weeks. Visual acuity examiners needed to be certified to ensure consistent measurements of BCVA, and they needed to remain masked to treatment assignment. For each participant, the same examiner was required to perform all assessments whenever possible. An ETDRS chart is a standardized visual acuity testing chart with a series of 5 letters of equal difficulty in each row, with standardized spacing between letters and rows, for a total of 14 lines and 70 letters.34 The letter size decreases with each consecutive row, resulting in increased difficulty. An increase in letter score corresponds to an improvement in visual acuity. No minimal important difference (MID) in change from baseline in BCVA has been identified; however, according to FDA guidance, an improvement of 15 or more letters on the ETDRS chart is clinically significant.35 The change from baseline in BCVA at week 60 is a key secondary end point. The proportion of patients gaining 15 or more letters in BCVA from baseline at week 48 was a secondary end point, and BCVA was assessed using the ETDRS protocol starting at 4 m by an examiner masked to treatment.
Intraretinal and Subretinal Fluid
The proportion of patients in each treatment arm with no IRF or SRF in the central subfield at week 48 was a secondary end point. The accumulation of retinal fluid is a hallmark symptom in patients with nAMD.36
Vision-Related Quality of Life
Vision-related quality of life was self-reported by patients, assessed using the NEI VFQ-25, and administered by a masked interviewer at baseline and weeks 48 and 60. The change from baseline in NEI VFQ-25 total score at week 48 was a secondary end point. The NEI VFQ-25 includes 25 items relevant to 11 vision-related constructs, in addition to a single-item general-health component. The composite score is the unweighted average score of all items except for the general-health rating.37 Responses for each item are converted to a 0-to-100 scale, with 0 representing the worst and 100 the best vision-related quality of life.38 In patients with nAMD, the MID is estimated to be 4 to 6 points on the composite score, because this corresponds to a 15-letter change in BCVA.39 The change in NEI VFQ-25 total score at week 60 was measured as an exploratory end point.
Frequency of Injections
The exploratory end points related to injection frequencies in the PULSAR trial were the proportion of patients randomized to 8 mg every 16 weeks who maintained a 16-week dosing interval or longer through weeks 48, 60, and 96, and the proportion of patients randomized to 8 mg every 12 weeks who maintained a 12-week dosing interval or longer through weeks 48, 60, and 96.
Harms Outcomes
An AE was defined as any untoward medical occurrence in a patient or clinical study participant who was associated with the use of study intervention, whether or not it was considered related to the study intervention itself. TEAEs were AEs that started in the time frame from first injection to the last injection (active or sham) in the study plus 30 days. SAEs were defined as an untoward medical occurrence that resulted in death; was life-threatening; required inpatient hospitalization or prolongation of existing hospitalization, featured persistent disability or incapacity; involved a congenital anomaly or birth defect, or was medically important according to medical and scientific judgment. Important ocular medical events were defined as an AE that required either surgical or medical intervention to prevent permanent loss of vision and substantial, unexplained vision loss or an AE that caused substantial vision loss.
The collection of safety information included reporting of ocular and nonocular TEAEs, SAEs (ocular and nonocular), discontinuation due to TEAEs, and death that occurred through week 60. AEs were to be reported by the patient in response to open-ended, nonleading verbal questions, with the investigator responsible for detecting and documenting events meeting the definition of an AE or SAE. No AEs of special interest were defined in the study protocol; however, notable harms included intraocular inflammation, endophthalmitis, intraocular pressure, retinal vasculitis, and Antiplatelet Trialists' Collaboration (APTC) events.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
NEI VFQ-25 | The NEI VFQ is used to assess the self-reported impact of visual impairment on the health-related quality of life across a broad range of eye conditions.40 The NEI VFQ-25 (a shortened version of the original 51-item questionnaire) is administered as an interview and consists of 25 items relevant to 11 subscales, in addition to a single-item general health component.38 Each subscale score is the average score of all items in the subscale transformed to a 0 to 100 scale, with 0 indicating the worst possible score and 100 indicating the best possible score. The composite score is the unweighted average score of all items except for the general health rating, which is considered a standalone item representing overall health status.37 | Validity: Content validity confirmed as the original 51-item VFQ was developed based on focus groups composed of people with common eye conditions (including AMD).37 Convergent validity of the VFQ-25 has been demonstrated in patients with nAMD (N = 1,13441 and N = 9242) using correlations with visual acuity38,41,42 and the SF-36 physical and mental component summary scores.41
Reliability: Acceptable internal consistency (Cronbach alpha of ≥ 0.746) for all of the NEI VFQ-25 subscale scores (for subscales with more than 1 item) and the composite score in a mixed population of patients with eye diseases,38 as well as for the composite score in patients with nAMD was observed.41 Internal consistency was acceptable for most subscale scores in patients with nAMD, with Cronbach alpha values ranging from 0.62 to 0.92.41,42 Test-retest reliability was not assessed in the above studies. Responsiveness: A change in 9.61 to 10.57 points corresponded to a medium effect size.39 | A 3-line (15-letter) change in visual acuity has been used as the outcome of interest in clinical trials, and corresponding changes in the NEI VFQ-25 are suggested as clinically meaningful end points. Evidence from 2 studies in patients with nAMD (N = 716 and N = 423) suggested that a 15-letter change in visual acuity in the study eye (typically the worse-seeing eye) corresponded to a change in 3.90 to 4.34 points in the composite score.39 |
BCVA using ETDRS charts | ETDRS charts measure visual acuity in clinical trials by presenting a series of 5 letters of equal difficulty in reading on each line, for a total of 14 lines (70 letters). A greater number of letters means better visual acuity. Charts are used in a standard light box. The standard testing distance is 4 m. Visual acuity is documented as the smallest line read by each eye in the absence of any errors. | Validity: No data were identified in patients with nAMD. Reliability: 2 studies including patients with nAMD47) reported test-retest reliability to be moderate to almost perfect (study 1, ICC = 0.580 to 0.866, depending on lighting and constrast;47 study 2, ICC = 0.9948). | No MID identified. A loss or gain of 2 lines (15 letters) is considered a moderate degree of change and is commonly used as an outcome in clinical trials.49 |
AMD = age-related macular degeneration; BCVA = best correct visual acuity; ETDRS = Early Treatment of Diabetic Retinopathy Study; ICC = intraclass correlation coefficient; MID = minimal important difference; nAMD = neovascular (wet) age-related macular degeneration; NEI VFQ-25 = National Eye Institute Visual Function Questionnaire-25; SF-36 = Short Form (36) Health Survey.
Statistical Analysis
Statistical analyses of the outcomes reported in the systematic review of the PULSAR study are summarized in Table 8.
The primary and key secondary efficacy variables were evaluated on both the FAS and the PPS, and all other efficacy variables were evaluated on the FAS only. Safety variables were analyzed using the safety analysis set. The exploratory efficacy end points were analyzed descriptively for the FAS.
Sample Size and Power Calculation
The sample size calculation was based on the primary end point, change from baseline in BCVA at week 48, assuming a noninferiority margin of 4 ETDRS letters in 2 pairwise comparisons: 8 mg every 12 weeks versus 2 mg every 8 weeks; and 8 mg every 16 weeks versus 2 mg every 8 weeks. It was determined that 288 evaluable patients per treatment arm would provide 94% power to reject the initial null hypothesis (8 mg every 12 weeks versus 2 mg every 8 week) for the primary end point to assess noninferiority with a 1-sided t test at a significance level of 0.025. The power to reject both primary null hypotheses (8 mg every 12 weeks versus 2 mg every 8 weeks and 8 mg every 16 weeks versus 2 mg every 8 weeks) would be 88%. Assuming that 10% of patients drop out before week 48, approximately 320 patients would be required to be randomized to each treatment arm to demonstrate noninferiority.
Noninferiority Margin
The noninferiority margin for the primary outcome (change from baseline in BCVA at 48 weeks) was defined as 4 ETDRS letters. The justification was that recent controlled phase III clinical trials studying nAMD (HARBOR, HAWK and HARRIER)50 used this same margin.51,52
Multiplicity Control Procedure
Overall familywise type I error for the primary and key secondary end points was controlled at a 1-sided alpha of 0.025 using a hierarchical testing procedure. Testing of hypotheses later in the hierarchy required rejection of earlier hypotheses, as described in the -statistical analysis plan (submitted to Health Canada):
noninferiority of aflibercept 8 mg every 12 weeks versus 2 mg every 8 weeks for change from baseline in BCVA at week 48
noninferiority of aflibercept 8 mg every 12 weeks versus 2 mg every 8 weeks for change from baseline in BCVA at week 60
noninferiority of aflibercept 8 mg every 16 weeks versus 2 mg every 8 weeks for change from baseline in BCVA at week 48
noninferiority of aflibercept 8 mg every 16 weeks versus 2 mg every 8 weeks for change from baseline in BCVA at week 60
superiority of pooled aflibercept 8 mg arms versus 2 mg every 8 weeks in proportion of patients with no IRF or SRF at week 16
superiority of aflibercept 8 mg every 12 weeks versus 2 mg every 8 weeks in change from baseline in BCVA at week 48
superiority of aflibercept 8 mg every 12 weeks versus 2 mg every 8 weeks in change from baseline in BCVA at week 60
superiority of aflibercept 8 mg every 16 weeks versus 2 mg every 8 weeks in change from baseline in BCVA at week 48
superiority of aflibercept 8 mg every 16 weeks versus 2 mg every 8 weeks in change from baseline in BCVA at week 60.
Analysis of the Primary End Point
The estimand of primary interest was mainly based on a hypothetical strategy. It describes the change from baseline for all participants who started treatment assuming all participants had stayed on treatment until week 48. The estimand is specified as follows:
Target population: Defined by the inclusion and exclusion criteria; used the FAS.
Variable: Absolute change from baseline to week 48 in BCVA.
Treatment condition: Aflibercept 8 mg administered every 12 weeks after 3 initial monthly injections with the option for DRM/rescue regimen, or aflibercept 8 mg administered every 16 weeks after 3 initial monthly injections with the option for DRM/rescue regimen, versus aflibercept 2 mg administered every 8 weeks after 3 initial monthly injections.
Intercurrent events: Premature discontinuation from treatment (handled by hypothetical strategy). Shortening or extension of the dosing interval (DRM or rescue regimen) was not considered an intercurrent event, but part of the randomized treatment regimen.
Population-level summary: Difference in LS mean change from baseline to week 48 in BCVA between aflibercept 8 mg every 12 weeks and aflibercept 2 mg every 8 weeks and aflibercept 8 mg every 16 weeks and aflibercept 2 mg every 8 week.
For the analysis of the primary end point, change from baseline in BCVA at week 48, an MMRM was used with baseline BCVA measurement as a covariate, and treatment arm, visit, and stratification variables (geographic region [Japan versus the rest of the world] and baseline BCVA [< 60 versus ≥ 60]), as fixed factors as well as terms for the interaction between baseline BCVA and visit and for the interaction between treatment and visit. The MMRM assumes MAR data for participants who discontinued the study prematurely. Sensitivity analyses were performed using different data-imputation methods.
Analysis of Secondary End Points
The proportion of participants gaining at least 15 letters in BCVA from baseline at week 48 was summarized descriptively. A Cochran-Mantel-Haenszel test stratified by geographic region and baseline BCVA was applied with 2-sided 95% CIs provided for descriptive purposes, using observed cases.
The proportion of participants with no IRF or SRF in the central subfield at week 48 was analyzed by a Cochran-Mantel-Haenszel test stratified by geographic region (Japan versus the rest of world) and baseline (day 1) BCVA (< 60 versus ≥ 60) LOCF to impute missing data. Two-sided 95% CIs were provided for descriptive purposes.
Change from baseline in the NEI VFQ-25 total score at week 48 was analyzed using an MMRM with the baseline score as a covariate and treatment group, visit, and stratification variables as fixed factors, as well as terms for the interaction between baseline score and visit, and treatment and visit. Two-sided 95% CIs for LS means were provided for descriptive purposes. Sensitivity analyses were performed using analysis of covariance and LOCF to impute missing values.
The frequency of injections was presented descriptively, using LS means and 95% CIs.
Subgroup Analyses
The following prespecified subgroups were considered for primary and key secondary efficacy end points: age at enrolment (< 65, ≥ 65 to < 75, ≥ 75 to < 80, ≥ 80 to < 85, ≥ 85), sex (male, female), geographic region (Japan, rest of the world), ethnicity (not Hispanic or Latino, Hispanic or Latino), race (white, Asian), baseline BCVA baseline (≤ 73, > 73), baseline polypoidal choroidal vascularization (PCV) (yes, no), medical history of cerebrovascular disease (no, yes), medical history of ischemic heart disease (no, yes), medical history of renal impairment (normal, mild, moderate, severe), and medical history of hepatic impairment (no, yes). Statistical analyses were conducted for the FAS population and analyzed descriptively. For the subgroup analyses by geographic region and baseline BCVA the corresponding variables were removed as stratification variables from the statistical models. Subgroups for continuous end points were determined using the MMRM without imputation of missing values and subgroups for categorical end points were determined using the Cochran-Mantel-Haenszel test with imputation by LOCF.
Analysis Populations
The analysis sets are summarized in Table 9.
Table 8: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
PULSAR | ||||
Change from baseline in BCVA at week 48 | MMRM | Treatment group (8 mg every 16 week vs. 2 mg every 8 weeks and 8 mg every 12 weeks vs. 2 mg every 8 week), visit, and the stratification variables baseline BCVA ([< 60 vs. ≥ 60]) and (geographic region [Japan vs. rest of world]) as fixed factors as well as terms for the interaction between baseline BCVA and visit and for the interaction between treatment and visit | The MMRM assumes data MAR for participants who discontinued the study prematurely, i.e., missingness only depended on observed data. | LOCF was conducted for participants who had at least 1 postbaseline value but had any further missing postbaseline BCVA values until week 48 or 60 and ANCOVA was applied for the change from baseline in BCVA at week 48 or 60. Another approach assuming MAR was implemented by using MI; to check the assumption that the missing data were not MAR; a tipping-point analysis was conducted based on the MI analysis |
Proportion of patients gaining ≥ 15 letters in BCVA from baseline at week 48 | CMH | Stratified by baseline BCVA (< 60 vs. ≥ 60) and geographic region (Japan vs. rest of world) | LOCF | Observed cases |
Proportion of patients with no IRF and no SRF in the central subfield at week 48 | CMH | Stratified by baseline BCVA (< 60 vs. ≥ 60) and geographic region (Japan vs. rest of world) | LOCF imputation for participants with missing SD-OCT | Observed cases |
Change from baseline in NEI VFQ-25 total score at week 48 | MMRM | Treatment group (8 mg every 16 weeks vs. 2 mg every 8 weeks and 8 mg every 12 weeks vs. 2 mg every 8 week), visit, and the stratification variables (geographic region [Japan vs. rest of world] and baseline BCVA [< 60 vs. ≥ 60]) as fixed factors as well as terms for the interaction between baseline CRT and the visit and for the interaction between treatment and visit | The MMRM assumes data were MAR for patients who discontinued the study prematurely, i.e., missingness only depended on observed data. Alternative assumptions (not MAR) were included in the sensitivity analysis | An ANCOVA was calculated using the LOCF method for imputation of missing values for participants discontinuing |
Treatment intervals | Descriptive statistics | NA | NA | NA |
ANCOVA = analysis of covariance; BCVA = best corrected visual acuity; CMH = Cochran-Mantel-Haenszel; LOCF = last observation carried forward; MAR = missing at random; MI = multiple imputation; MMRM = mixed model for repeated measures; NA = not applicable; NEI VFQ-25 = National Eye Institute Visual Function Questionnaire-25; SD-OCT = spectral domain optical coherence tomography; vs. = versus.
Source: PULSAR Clinical Study Report18 and Statistical Analysis Plan. Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 9: Analysis Populations of PULSAR
Study | Population | Definition | Application |
|---|---|---|---|
PULSAR | FAS | The FAS included all patients randomly assigned to study treatment and who received at least 1 dose of study treatment. Patients were analyzed within their original randomized group (as randomized). | All efficacy analyses |
PPS | The PPS included all patients in the FAS who did not have an important deviation from the protocol affecting the primary efficacy variable or a validity findings as listed below. The PPS included all patients in the FAS who:
Other relevant deviations from the protocol affecting efficacy were considered as intercurrent events in the context of the estimands strategy. Analysis of the PPS was performed according to the treatment the patient actually received (as treated). | Primary and key secondary efficacy analyses | |
SAF | The SAF included all patients who were randomly assigned to study treatment and who received at least 1 dose of study treatment. Analysis of the SAF was performed according to the treatment the patient actually received (as treated). | Safety analyses |
BCVA = best corrected visual acuity; FAS = full analysis set; IRF = intraretinal fluid; PPS = per-protocol set; SAF = safety analysis set; SRF = subretinal fluid.
Source: PULSAR Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Of the 1,395 patients screened in the PULSAR study, 383 did not complete screening and 1,009 were randomized to aflibercept 2 mg every 8 weeks (n = 336), aflibercept 8 mg every 12 weeks (n = 335), or aflibercept 8 mg every 16 weeks (n = 338). A total of 937 patients completed treatment through week 48, and 923 patients completed treatment through week 60. There were no notable differences in the reasons for early discontinuation across the treatment arms. Table 10 summarizes patient disposition in the PULSAR study.
Table 10: Summary of Patient Disposition in the PULSAR Study
Patient disposition | Aflibercept 2 mg q.8.w. (N = 336) | Aflibercept 8 mg q.12.w. (N = 335) | Aflibercept 8 mg q.16.w. (N = 338) |
|---|---|---|---|
Screened, N | 1,395 | ||
Randomized, N (%) | 337 (100) | 336 (100) | 338 (100) |
Completed the study until week 48, n (%) | 309 (91.7) | 316 (94.0) | 312 (92.3) |
Discontinued from study by week 48, n (%) | 25 (7.4) | 18 (5.4) | 25 (7.4) |
Reason for discontinuation, n (%) | |||
Adverse events | 5 (1.5) | 1 (0.3) | 5 (1.5) |
Lost to follow-up | 1 (0.3) | 1 (0.3) | 0 |
Physician decision | 1 (0.3) | 3 (0.9) | 2 (0.6) |
Withdrawal by patient | 5 (1.5) | 5 (1.5) | 12 (3.6) |
Death | 5 (1.5) | 3 (0.9) | 1 (0.3) |
COVID-19, patient decision | 2 (0.6) | 2 (0.6) | 2 (0.6) |
Lack of efficacy | 2 (0.6) | 0 | 0 |
Other | 4 (1.2) | 2 (0.6) | 2 (0.6) |
Completed the study until week 60, n (%) | 305 (90.5) | 310 (92.3) | 308 (91.1) |
Discontinued from study by week 60, n (%) | 29 (8.6) | 23 (6.8) | 29 (8.6) |
Adverse event | 6 (1.8) | 2 (0.6) | 5 (1.5) |
Physician decision | 1 (0.3) | 4 (1.2) | 1(0.3) |
Protocol deviation | 0 | 1 (0.3) | 1 (0.3) |
Lost to follow-up | 1 (0.3) | 1 (0.3) | 2 (0.6) |
Lack of efficacy | 2 (0.6) | 0 | 0 |
Withdrawal by patient | 6 (1.8) | 8 (2.4) | 14 (4.1) |
Death | 5 (1.5) | 3 (0.9) | 1 (0.6) |
COVID-19 pandemic:patient decision | 2 (0.6) | 2 (0.6) | 2 (0.6) |
Other | 6 (1.8) | 2 (0.6) | 2 (0.6) |
FAS, N | 336 (99.7) | 335 (99.7) | 338 (100) |
PPS, N | 320 (95.0) | 325 (96.7) | 325 (96.2) |
Safety, N | 336 (99.7) | 335 (99.7) | 338 (100) |
FAS = full analysis set; PPS = per-protocol set; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks.
Source: PULSAR Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. The FAS (and safety analysis set) consisted of 459 male patients (45.5%) and 550 female patients (54.5%) with a mean age of 75 years (SD = 8.4). The majority of patients were white (75.8%) or Asian (23.2%). The mean BCVA at baseline was 59.6 letter (SD = 13.3). All lesion types (i.e., occult, minimally classic, and predominantly classic) were represented. Overall, the 3 treatment arms were balanced with respect to demographic and disease characteristics.
Table 11: Summary of Baseline Characteristics in the PULSAR Study
Characteristic | Aflibercept 2 mg q.8.w. (N = 336) | Aflibercept 8 mg q.12.w. (N = 335) | Aflibercept 8 mg q.16.w. (N = 338) | Total (N = 1,009) |
|---|---|---|---|---|
Age (years), mean (SD) | 74.2 (8.8) | 74.7 (7.9) | 74.5 (8.5) | 74.5 (8.4) |
Female, n (%) | 188 (56.0) | 182 (54.3) | 180 (53.3) | 550 (54.5) |
Male, n (%) | 148 (44.0) | 153 (45.7) | 158 (46.7) | 459 (45.5) |
Race, n (%) | ||||
White | 249 (74.1) | 256 (76.4) | 260 (76.9) | 765 (75.8) |
Asian | 83 (24.7) | 74 (22.1) | 77 (22.8) | 234 (23.2) |
Black or African American | 2 (0.6) | 2 (0.6) | 0 | 4 (0.4) |
Not reported | 2 (0.6) | 2 (0.6) | 1 (0.3) | 5 (0.5) |
Multiple | 0 | 1 (0.3) | 0 | 1 (0.1) |
Native Hawaiian or other Pacific Islander | 0 | 0 | 0 | 0 |
American Indian or Alaska Native | 0 | 0 | 0 | 0 |
Fellow eye with history of nAMD, n (%) | ||||
No | 321 (95.5) | 324 (96.7) | 326 (96.4) | 971 (96.2) |
Yes | 15 (4.5) | 11 (3.3) | 12 (3.6) | 38 (3.8) |
BCVA, ETDRS letter score (SD) | ||||
Mean | 58.9 (14.0) | 59.9 (13.4) | 60.0 (12.4) | 59.6 (13.3) |
≤ 73 | 287 (85.4) | 293 (87.5) | 290 (85.8) | 870 (86.2) |
> 73 | 49 (14.6) | 42 (12.5) | 48 (14.2) | 139 (13.8) |
< 60 | 136 (40.5) | 141 (42.1) | 144 (42.6) | 421 (41.7) |
≥ 60 | 200 (59.5) | 194 (57.9) | 194 (57.4) | 588 (58.3) |
Intraocular pressure, mm Hg | ||||
Mean | 14.8 (3.0) | 14.9 (3.2) | 14.9 (3.2) | 14.9 (3.2) |
Geographic atrophy, n (%) | ||||
No | 328 (97.6) | 326 (97.3) | 326 (96.4) | 980 (97.1) |
Yes | 3 (0.9) | 3 (0.9) | 6 (1.8) | 12 (1.2) |
Not available | 5 (1.5) | 6 (1.8) | 6 (1.8) | 17 (1.7) |
CRT, µm | ||||
Mean (SD) | 367.1 (133.6) | 370.3 (123.7) | 370.7 (132.7) | 369.3 (130.0) |
CNV size, mm2 | ||||
Mean (SD) | 6.36 (5.04) | 5.98 (4.83) | 6.55 (5.53) | 6.29 (5.14) |
CNV classification, n (%) | ||||
Predominantly classic | 71 (21.1) | 71 (21.2) | 67 (19.8) | 209 (20.7) |
Minimally classic | 61 (18.2) | 56 (16.7) | 68 (20.1) | 185 (18.3) |
Occult only | 192 (57.1) | 197 (58.8) | 186 (55.0) | 575 (57.0) |
RAP | 5 (1.5) | 4 (1.2) | 5 (1.5) | 14 (1.4) |
PVC | 2 (0.6) | 1 (0.3) | 3 (0.9) | 6 (0.6) |
Cannot grade | 0 | 0 | 1 (0.3) | 1 (0.1) |
NA | 5 (1.5) | 6 (1.8) | 6 (1.8) | 17 (1.7) |
Hypertension, n (%) | ||||
No | 132 (39.3) | 113 (33.7) | 119 (35.2) | 364 (36.1) |
Yes | 204 (60.7) | 222 (66.3) | 219 (64.8) | 645 (63.9) |
NEI VFQ-25 total score | ||||
Mean (SD) | 77.81 (14.42) | 76.36 (15.12) | 77.67 (15.40) | 77.27 (14.98) |
BCVA = best corrected visual acuity; CNV = choroidal neovascularization; CRT = central subfield retinal thickness; EDTRS = Early Treatment Diabetic Retinopathy Study; nAMD = neovascular (wet) age-related macular degeneration; NEI VFQ-25 = National Eye Institute Visual Functioning Questionnaire-25; PCV = polypoidal choroidal vascularization; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; RAP = retinal angiomatous proliferation; SD = standard deviation.
Source: PULSAR Clinical Study Report18 and the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
At 48 weeks, the mean duration of exposure was similar between treatment arms, ranging from 46.2 weeks to 46.5 weeks. At 60 weeks, the mean duration of exposure remained similar between treatment arms, ranging from 57.2 weeks to 57.7 weeks. Adherence to the treatment schedule was high in all groups with a mean treatment adherence through week 48 and through week 60 of more than 97% in all arms. A summary of patient exposure is available in Table 12.
Table 12: Summary of Patient Exposure in the PULSAR Study
Exposure | Aflibercept 2 mg q.8.w. (N = 336) | Aflibercept 8 mg q.12.w. (N = 335) | Aflibercept 8 mg q.16.w. (N = 338) |
|---|---|---|---|
Through week 48 | |||
Duration (weeks), mean (SD) | 46.3 (6.6) | 46.5 (6.6) | 46.2 (7.0) |
Duration (weeks), median (IQR) | 48.0 (47.9 to 48.3) | 48.0 (47.9 to 48.3) | 48.0 (47.7 to 48.3) |
Treatment adherence (%), mean (SD) | 97.7 (5.8) | 98.0 (5.5) | 98.0 (5.2) |
Through week 60 | |||
Duration (weeks), mean (SD) | 57.2 (9.6) | 57.7 (9.1) | 57.4 (9.8) |
Duration (weeks), median (IQR) | 60.0 (59.7 to 60.3) | 60.0 (59.9 to 60.3) | 60.0 (59.9 to 60.3) |
Treatment adherence (%), mean (SD) | 97.2 (6.0) | 97.6 (5.8) | 97.9 (4.9) |
IQR = interquartile range; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; SD = standard deviation.
Source: PULSAR Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
There were no important imbalances in concomitant treatments across the treatment groups and any imbalances that were observed did not appear to affect assessments of efficacy. Subgroup analyses for the primary and key secondary end points, which were performed on a descriptive level by age, sex, geographic region, ethnicity, race, baseline BCVA letters, and baseline PCV, appeared to be consistent with those of the main analysis.
Efficacy
In this report, efficacy outcomes were reported based on a data cut-off at 48 weeks. Additionally, evidence at the 60-week data cut-off had been considered. All relevant efficacy results are presented in Table 13.
Change From Baseline in BCVA at Week 48
The difference in LS mean change from baseline to week 48 was −0.97 letters (95% CI, −2.87 to 0.92) for 8 mg every 12 weeks versus 2 mg every 8 weeks (noninferiority P = 0.0009; superiority P = ██████) and −1.14 letters (95% CI, −2.97 to 0.69) for 8 mg every 16 weeks versus 2 mg every 8 weeks (noninferiority P = 0.0011, superiority P = ██████). Results of analysis of the PPS for week 48 were consistent with those in the FAS. The observed findings were maintained at week 60. The trajectory of BCVA up to week 60 is presented in Figure 2. The results of sensitivity analyses were consistent with those of the primary analysis using MMRM in the FAS. Subgroup analyses for changes from baseline in BCVA at week 48 by age, sex, geographic region, ethnicity, race, baseline BCVA letters, and baseline PCV were performed using MMRM in the FAS. In all of the subgroups considered, changes from baseline in BCVA at week 48 were consistent with those in the overall population. Overall, no clinically meaningful differences between the subgroup population and the total population were observed.
Change From Baseline in BCVA at Week 60
The differences in LS mean changes from baseline to week 60 were −0.86 (95% CI, −2.57 to 0.84) letters (noninferiority P = 0.0002; superiority P = ██████) and −0.92 (95% CI, −2.51 to 0.66) letters (noninferiority P < 0.0001; superiority P = ██████) for the 8 mg every 12 weeks and 8 mg every 16 weeks arms, respectively, compared to the 2 mg every 8 weeks arm. The results for the PPS for week 60 were consistent with those from the FAS. The results of these sensitivity analyses were consistent with those of the primary analysis using a MMRM in the FAS. Subgroup analyses for change from baseline in BCVA at week 60 by age, sex, geographic region, ethnicity, race, baseline BCVA letters, and baseline PCV were performed using an MMRM in the FAS. In all of the subgroups considered, changes from baseline in BCVA at week 60 were consistent with those in the overall population. Overall, no clinically meaningful differences between the subgroup population and the total population were observed.
Proportion of Patients Gaining 15 or More ETDRS Letters at Week 48
The between-group difference in the proportion of patients gaining 15 or more letters in BCVA from baseline to week 48 was −1.75% (95% CI, −7.78 to 4.29%; P = ██████) for aflibercept 8 mg every12 weeks versus 2 mg every 8 weeks and −0.94% (95% CI, −7.00 to 5.12%; P = ██████) for aflibercept 8 mg every 16 weeks versus 2 mg every 8 weeks, based on LOCF in the FAS. The observed findings were maintained at week 60. The results of these sensitivity analyses were consistent with those of the primary analysis in the FAS.
Figure 2: Least Squares Mean Change in BCVA as Measured by ETDRS Letter Score From Baseline Through Week 60 (Full Analysis Set)
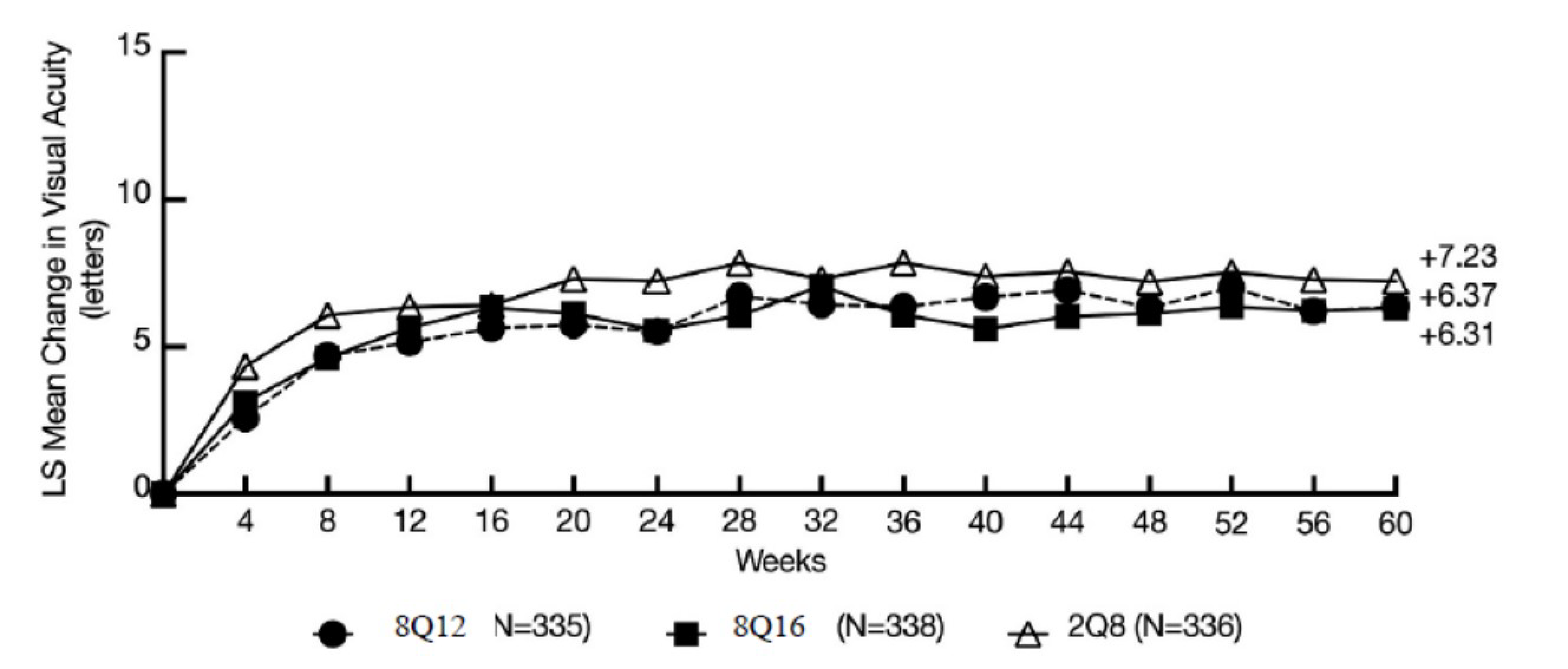
LS = least squares; 8Q12 = aflibercept 8 mg every 12 weeks; 8Q16 = aflibercept 8 mg every 16 weeks; 2Q8 = aflibercept 2 mg every 8 weeks.
Source: PULSAR Clinical Study Report.18
Presence of IRF or SRF at Week 48
The proportion of patients with no IRF and no SRF in the central subfield at week 48 was a secondary end point in the PULSAR study. At week 48, 71.1% and 66.8% of patients in the aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks arms, respectively, had no IRF or SRF compared with 59.4% in the aflibercept 2 mg every 8 weeks arm. This resulted in a difference in the proportion of patients with no IRF or SRF in the central subfield of 11.73% (95% CI, 4.52% to 18.92%, superiority P = ██████) for 8 mg every 12 weeks versus 2 mg every 8 weeks and 7.45% (95% CI, 0.14% to 14.76%, superiority P = ██████) for 8 mg every 16 weeks, based on the LOCF in the FAS. The observed findings were maintained at week 60. The results of these sensitivity analyses were consistent with those of the primary analysis using the LOCF in the FAS.
Frequency of Injections
At week 48, totals of 251 (79.4%) and 239 (76.6%) of completers in the aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks arms maintained their randomized treatment interval. This resulted in mean numbers of active injections through week 48 of 6.1 and 5.2 in the aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks arms, respectively, compared to 6.9 in the aflibercept 2 mg every 8 weeks arm. The treatment-group difference between 2 mg every 8 weeks aflibercept 8 mg every 12 weeks and aflibercept 2 mg every 8 weeks was −0.9 █████ ██ █████ injections and the difference between aflibercept 2 mg every 8 weeks and aflibercept 8 mg every 16 weeks and aflibercept 2 mg every 8 week was −1.8 █████ ██ █████ injections. At week 60, the mean numbers of injections were 8.8 █████, 7.1 █████, and 6.2 █████ for the aflibercept 2 mg every 8 weeks, 8 mg every 12 weeks and 8 mg every 16 week, respectively.
National Eye Institute Visual Function Questionnaire-25
Least squares mean changes from baseline were observed in all arms at week 48, ranging from 3.35 (SE = ████) in the aflibercept 8 mg every 16 weeks arm to 4.22 ██████ in the aflibercept 2 mg every 8 weeks arm. The differences in the LS mean change from baseline using the MMRM in the FAS, were −0.72 ████ ███ █████ ██ █████ | | ███████ for 8 mg every 12 weeks versus 2 mg every 8 weeks and −0.87 ████ ███ █████ ██ █████ | | ███████ for 8 mg every 16 weeks versus 2 mg every 8 weeks. The results were consistent at week 60. The results of these sensitivity analyses were consistent with those of the primary analysis based on MMRM in the FAS.
Table 13: Summary of Key Efficacy Results From PULSAR (Full Analysis Set)
Variable | Aflibercept 2 mg q.8.w. (N = 336) | Aflibercept 8 mg q.12.w. (N = 335) | Aflibercept 8 mg q.16.w. (N = 338) |
|---|---|---|---|
Change from baseline in BCVA at week 48 (primary end point) | |||
Number of patients contributing to the analysis, n (%) | 285 (84.82) | 299 (89.25) | 298 (88.16) |
Baseline, mean (SD)a | 58.9 (14.0) | 59.9 (13.4) | 60.0 (12.4) |
Change from baseline, LS mean (SE) | 7.0 (0.74) | 6.1 (0.77) | 5.9 (0.72) |
Treatment group difference vs. control (95% CI)b | Reference | −0.97 (−2.87 to 0.92) | −1.14 (−2.97 to 0.69) |
P value (noninferiority) | Reference | 0.0009 | 0.0011 |
P value (superiority)c | Reference | ██████ | ██████ |
Proportion of patients gaining ≥ 15 letters in BCVA from baseline at week 48 (secondary end point) | |||
Patients gaining ≥ 15 letters, n/N (%) | 74/335 (22.1) | 69/334 (20.7) | 73/337 (21.7) |
Difference vs. aflibercept 2 mg q.8.w. (95% CI) | Reference | −1.75 (−7.78 to 4.29) | −0.94 (−7.00 to 5.12) |
P value (superiority)c | Reference | 0.5704 | 0.7611 |
Proportion of patients with no IRF and no SRF at week 48 (secondary end point) | |||
n/N (%) | 199/335 (59.4) | 236/332 (71.1) | 223/334 (66.8) |
Treatment-group difference vs. aflibercept 2 mg q.8.w. (95% CI) | Reference | 11.725 (4.52 to 18.92) | 7.451 (0.14 to 14.76) |
P value (superiority)c | Reference | ██████ | ██████ |
Frequency of injection outcomes (safety analysis set) | |||
Number of treatment injections | |||
Week 48, mean (SD)d | 6.9 █████ | 6.1 █████ | 5.2 █████ |
LS mean (95% CI) | 7.0 ███ ██ ████ | 6.1 ████ ██ ████ | 5.2 ████ ██ ████ |
LS mean difference vs. aflibercept 2 mg q.8.w. (95% CI) | Reference | −0.9 █████ ██ █████ | −1.8 █████ ██ █████ |
Week 60, mean (SD)d | 8.8 █████ | 7.1 █████ | 6.2 █████ |
Dosing intervals through week 48 | |||
Patients with ≥ q.12.w. dosing interval, n (%) | NA | 251 (79.4) | 272 (87.2) |
Patients with q.16.w. dosing interval, n (%) | NA | NA | 239 (76.6) |
Change from baseline in NEI VFQ-25 total score over time | |||
Change from baseline in NEI VFQ-25 total score at week 48 | |||
Number of patients with week 48 data | 266 | 285 | 266 |
Baseline mean | ████ | ████ | ████ |
LS mean change from baseline (SE) | 4.22 ████ | 3.50 ██████ | 3.35 ██████ |
LS mean difference vs. aflibercept 2 mg every 8 weeks (95% CI) | Reference | −0.72 ███████ █████ | −0.87 ███████ █████ |
P value (superiority)c | Reference | ██████ | ██████ |
BCVA = best corrected visual acuity; CI = confidence interval; IRF = intraretinal fluid; LS = least squares; NA = not applicable; NEI VFQ-25 = National Eye Institute Visual Functioning Questionnaire-25; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; SD = standard deviation; SE = standard error; SRF = subretinal fluid; vs. = versus.
aBased on observed assessments.
bEstimate based on the MMRM model, was computed for the differences of high-dose q.12.w. minus 2 mg every 8 weeks and high-dose q.16.w. minus 2 mg every 8 week, respectively, with 2-sided 95% CIs.
cP value is not adjusted for multiplicity.
dSafety analysis set, only participants considered as completers.
Source: PULSAR Clinical Study Report.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Harms data reported for the safety analysis set up to 60 weeks are included in Table 14.
Adverse Events
Patients in the study reported at least 1 ocular TEAE at almost the same proportions (45.2% in the aflibercept 2 mg every 8 weeks arm, 42.4% in the aflibercept 8 mg every 12 weeks, and 42.3% in the aflibercept 8 mg every 16 week). The most common ocular TEAEs in all treatment arms were reduced visual acuity (6.3% in the aflibercept 2 mg every 8 weeks arm, 3.9% in the aflibercept 8 mg every 12 weeks, and 5.9% in the aflibercept 8 mg every 16 weeks arm), cataracts (3.9%, 4.8%, and 4.4%, respectively), and retinal hemorrhaging (4.5%, 3.6%, and 3.8%, respectively). The proportions of patients with a nonocular TEAE were 59.8%, 59.4%, and 61.2% in the aflibercept 2 mg every 8 weeks, the aflibercept 8 mg every 12 weeks, and the aflibercept 8 mg every 16 weeks arms, respectively. The most common nonocular TEAEs in all treatment arms were COVID-19 (4.8%, 5.7%, and 9.2%), nasopharyngitis (4.8%, 4.2%, and 6.2%), nasopharyngitis (5.4%, 4.5%, and 5.0%), and hypertension (3.6%, 5.7%, and 5.3%) in the aflibercept 2 mg every 8 weeks, the aflibercept 8 mg every 12 weeks, and the aflibercept 8 mg every 16 weeks groups, respectively.
Serious Adverse Events
At least 1 treatment-emergent SAE was reported in 1.2% of patients in the aflibercept 2 mg every 8 weeks arm, and in 2.1% of patients in each of the aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks arms. Retinal hemorrhaging and retinal detachment were the most common SAE in the treatment groups, occurring in 0.3%, 0.6%, and 0.6% of patients in the aflibercept 2 mg every 8 weeks, 8 mg every 12 weeks, and 8 mg every 16 weeks arms, respectively. Nonocular SAEs were reported in 15.8% of patients in the aflibercept 2 mg every 8 weeks arm and 12.2% of patients in the aflibercept 8 mg every 12 weeks arm and 12.1% of patients in the aflibercept 8 mg every 16 weeks arm.
The proportions of patients who discontinued treatment due to an ocular TEAE were 0.6% in the aflibercept 2 mg every 8 weeks arm, and 1.2% in both the aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks arms. The proportions of patients who discontinued treatment due to nonocular TEAEs were 1.8%, 0.3% and 0.6% in the aflibercept 2 mg every 8 weeks arm, 8 mg every 12 weeks arm, and 8 mg every 16 weeks arm respectively.
Mortality
In the aflibercept 2 mg every 8 weeks arm, death events were reported for 1.5% of patients. Death events were reported for 0.9% and 0.6% of the aflibercept 8 mg every 12 weeks arm and aflibercept 8 mg every 16 weeks arm respectively.
Notable Harms
Notable harms were selected based on serious warnings and precautions in the Health Canada product monograph for aflibercept 8 mg.15 Cataracts occurred in 3.9% of patients treated with aflibercept 2 mg every 8 weeks, 4.8% of patients treated with aflibercept 8 mg every 12 week, and 4.4% of patients treated with aflibercept 8 mg every 16 week. The incidences of increased intraocular pressure were 2.7% in the aflibercept 2 mg every 8 weeks arm, 3.3% in the aflibercept 8 mg every 12 weeks arm, and 3.0% in the aflibercept every 16 weeks arm. The percentages of patients experiencing retinal pigment epithelium tear were 0.9% in the aflibercept 2 mg every 8 weeks arm, 1.8% in the aflibercept 8 mg every 12 weeks arm, and 0.9% in the aflibercept 8 mg every 16 weeks arm. Other notable harms reported at a lower frequency in the treatment arms were intraocular inflammation, retinal detachments, APTC event, thromboembolic events, intraocular pressure.
Table 14: Summary of Harms Results Through Week 60 — PULSAR Study Safety Analysis Set
Adverse events | Aflibercept 2 mg q.8.w. (N = 336) | Aflibercept 8 mg q.12.w. (N = 335) | Aflibercept 8 mg q.16.w. (N = 338) |
|---|---|---|---|
Most common events, n (%) | |||
Patients with ≥ 1 ocular TEAEa | 152 (45.2) | 142 (42.4) | 143 (42.3) |
Visual acuity reduced | 21 (6.3) | 13 (3.9) | 20 (5.9) |
Cataract | 13 (3.9) | 16 (4.8) | 15 (4.4) |
Retinal hemorrhage | 15 (4.5) | 12 (3.6) | 13 (3.8) |
Vitreous floaters | 13 (3.9) | 4 (1.2) | 14 (4.1) |
Vitreous detachment | 5 (1.5) | 7 (2.1) | 10 (3.0) |
Subretinal fluid | 12 (3.6) | 11 (3.3) | 8 (2.4) |
Macular thickening | 3 (0.9) | 8 (2.4) | 7 (2.1) |
Patients with ≥ 1 nonocular TEAEb | 201 (59.8) | 199 (59.4) | 207 (61.2) |
COVID-19 | 16 (4.8) | 19 (5.7) | 31 (9.2) |
Nasopharyngitis | 16 (4.8) | 14 (4.2) | 21 (6.2) |
Neoplasms benign, malignant, and unspecified (including cysts and polyps) | 18 (5.4) | 15 (4.5) | 17 (5.0) |
Hypertension | 12 (3.6) | 19 (5.7) | 18 (5.3) |
Back pain | 18 (5.4) | 15 (4.5) | 14 (4.1) |
Patients with ≥ 1 ocular SAE, n (%) | 4 (1.2) | 7 (2.1) | 7 (2.1) |
Retinal hemorrhage | 1 (0.3) | 2 (0.6) | 2 (0.6) |
Retinal detachment | 1 (0.3) | 2 (0.6) | 1 (0.3) |
Angle closure glaucoma | 1 (0.3) | 0 | 1 (0.3) |
Dry age-related macular degeneration | 0 | 1 (0.3) | 0 |
Vitreous hemorrhage | 0 | 0 | 1 (0.3) |
Cataract | 0 | 0 | 1 (0.3) |
Patients with ≥ 1 nonocular SAE, n (%) | 53 (15.8) | 41 (12.2) | 41 (12.1) |
Pneumonia | 1 (0.3) | 4 (1.2) | 2 (0.6) |
Urinary tract infection | 4 (1.2) | 1 (0.3) | 0 |
Pyelonephritis acute | 0 | 0 | 2 (0.6) |
Patients who stopped treatment due to AEs, n (%) | |||
Ocular TEAEs | 2 (0.6) | 4 (1.2) | 4 (1.2) |
Nonocular TEAEs | 6 (1.8) | 1 (0.3) | 2 (0.6) |
Death, n (%) | 5 (1.5) | 3 (0.9) | 2 (0.6) |
COVID-19 pneumonia | 0 | 1 (0.3) | 0 |
Sepsis | 0 | 0 | 1 (0.3) |
Metastatic neoplasm | 0 | 1 (0.3) | 0 |
Notable harms, n (%) | |||
Cataracts | 13 (3.9) | 16 (4.8) | 15 (4.4) |
Increased intraocular pressure | 9 (2.7) | 11 (3.3) | 10 (3.0) |
Retinal pigment epithelium tear | 3 (0.9) | 6 (1.8) | 3 (0.9) |
Intraocular inflammation | 4 (1.2) | 4 (1.2) | 1 (0.3) |
Retinal detachments | 1 (0.3) | 2 (0.6) | 1 (0.3) |
Antiplatelet Trialists’ Collaboration event | 8 (2.4) | 1 (0.3) | 2 (0.6) |
Vitreous hemorrhage | 0 | 1 (0.3) | 0 |
Endophthalmitis | 2 (0.6) | 0 | 0 |
AE = adverse event; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; SAE = serious adverse event; TEAE = Treatment-emergent adverse event
Note: TEAEs are defined as AEs that started in the time frame from first injection to the last injection (active or sham) in the study plus 30 days.
aFrequency of ≥ 2% in at least 1 treatment group.
bFrequency of ≥ 5% in at least 1 treatment group.
Sources: PULSAR Clinical Study Report18 and the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
The PULSAR study was a randomized, double-blind, active-controlled, noninferiority, phase III trial comparing aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks to aflibercept 2 mg every 8 weeks. No concern was raised by the chosen method of randomization, which involved stratification by baseline BCVA and geographic region. Adequate methods were used to maintain allocation concealment (an interactive response system). The investigators also took adequate measures (e.g., sham injections) to facilitate blinding of participants and personnel involved in the trial. Baseline characteristics were well balanced among treatment arms. The treatment adherence rates in the treatment groups were almost the same.
All statistical analyses and subgroups were prespecified in the clinical study protocol or in the statistical analysis plan. The analysis was repeated on PPS as a supplementary analysis, and there was agreement between both the FAS and PPS (which is generally considered more conservative for noninferiority testing) analysis, with both showing that high-dose aflibercept was noninferior to aflibercept 2 mg. The noninferiority margin of 4 ETDRS letters, which was based on the similar trials for nAMD, was considered reasonable by the clinical expert we consulted. The enrolled sample sizes were adequate to assess the primary outcome. Adjustment for multiple comparisons in the primary and key secondary end points was adequate, using a hierarchical testing procedure. Aside from change from baseline in BCVA, the remaining outcomes in this report were not subject to hypothesis testing (i.e., they were presented descriptively with 95% CIs), and should be considered supportive evidence. The validity, reliability, and MID of the NEI VFQ-25 are established in the literature. The validity of BCVA using ETDRS charts was not identified. The reliability and MID were reported in the literature.
The MMRM imputation strategy used for the primary analysis assumes data were MAR for participants who discontinued the study prematurely (i.e., missingness only depended on observed data); this appeared to be approximately ███ ██ ███ of participants. The validity of the MAR assumption is difficult to ascertain. Alternative assumptions, including LOCF, multiple imputation, and tipping-point analysis, were included in the sensitivity analyses. The results of these sensitivity analyses were consistent with those of the primary analysis using an MMRM in the FAS. There was some concern regarding missing outcome data across the remaining efficacy outcomes, mainly because it is not clear whether the imputation approaches would result in unbiased estimates. For health-related quality of life, the proportion of missing data at week 48 was large (approximately ███), with these being imputed via an MMRM and a sensitivity analysis via LOCF. The analysis of the proportion of patients gaining 15 or more letters and the proportion of patients without IRF or SRF in the central subfield used LOCF. It is not clear whether the LOCF imputation strategy would represent the true trajectory of the outcomes. Additionally, sensitivity analyses using observed cases may be subject to bias. Data for number of injections used observed cases only.
Overall, █████ participants reported important protocol deviations. The most frequent (≥ 5%) important protocol deviations were related to the categories “procedure deviations,” “treatment deviations,” “time schedule deviations,” and “informed consent” ██████. The most frequent important deviation of the category, “inclusion/exclusion criteria not met but subject entered treatment,” was related to exclusion criterion 4 (participant had uncontrolled blood pressure [defined as systolic > 160 mm Hg or diastolic > 95 mm Hg]), which was reported for ████ of participants. Those deviations may have biased the results of study, but to what extent is difficult to ascertain.
External Validity
Only 3 of 251 study sites included in the PULSAR trial were in Canada. The clinical expert we consulted commented that the baseline characteristics of the study populations were similar to those of patients with nAMD in Canada. The clinical expert considered the inclusion and exclusion criteria reflective of the eligibility criteria used to offer treatment in clinical practice. The trial included only treatment-naive patients who were excluded if they had any prior or concomitant anti-VEGF treatment. The clinical expert noted that aflibercept 8 mg may be used in patients after failing another anti-VEGF treatment. This is therefore a potential gap in the evidence where the efficacy of aflibercept 8 mg in patients with treatment experience was not explored in the PULSAR trial so the results may not be generalizable to patients with treatment experience.
In terms of the clinical relevance of the outcomes assessed in the trial, the most important outcomes of interest to clinicians and patients were vision acuity, frequency of injections, and vision-related quality of life, which were measured in the PULSAR trial. The dosing regimens of aflibercept 8 mg in the trial (i.e., aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks) do not fully align with the dosing specified in the product monograph. In the product monograph, aflibercept 8 mg (0.07 mL) can be dosed up to every 16 weeks in the first year and up to 20 weeks thereafter.15 According to the clinical expert, aflibercept 2 mg every 8 weeks is an appropriate comparator. Although the expert acknowledged that the fixed-dose interval may have contributed to maintaining the internal validity by ensuring consistent dosing intervals, this is not aligned with the treat-and-extend approach used by most clinicians to treat nAMD in Canada. In practice, that interval is usually extended to every 10 weeks or 12 weeks. The clinical expert noted that more than half of patients would be on a 12-week regimen. The use of fixed intervals is more rigid than would be expected in practice, according to the clinical expert, and the absence of the ability to modify the dosing schedule raises questions about the generalizability of the injection-frequency outcome. Additionally, it would not have been possible within the trial to understand the impact of a reduced number of injections on overall health-related quality of life or treatment burden from the patient’s perspective, because blinding required the use of sham injections.
The PULSAR trial was the only phase III trial submitted by sponsor that provides direct evidence comparing aflibercept 2 mg every 8 weeks versus aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks in patients with nAMD. No direct evidence comparing aflibercept to the other anti-VEGF drugs currently used in Canadian practice (i.e., brolucizumab, faricimab, or bevacizumab), and which may be used on extended dosing regimens (e.g., 12 or 16 weeks), was submitted by the sponsor, creating an evidence gap. Moreover, the lack of evidence regarding the long-term therapeutic effect of aflibercept 8 mg (beyond 60 weeks) may represent a source of uncertainty.
In the trial, the mean age was 74.5 years, and younger patients may be underrepresented in this study. Other races are also underrepresented as the majority of enrolled patients were white or Asian.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to informing the expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group16,17
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for aflibercept 8 mg every 12 weeks and every 16 weeks versus aflibercept 2 mg every 8 weeks.
Long-Term Extension Studies
No long-term extension studies with available results were submitted by the sponsor. The long-term extension phase of PULSAR, which is ongoing, consists of a 12-week transition period after the completion of PULSAR, during which study intervention will still be administered in a blinded fashion, followed by an open-label treatment period of 48 weeks. During the blinded transition period of 12 weeks, participants who received aflibercept 2 every 8 weeks will switch to 8 mg every 12 weeks, while participants on 8 mg every 12 weeks and 8 mg every 16 weeks will continue on their individual dosing schedules. After 12 weeks, all participants who remain in the extension period of the study will receive aflibercept 8 mg in an open-label fashion according to their individual dosing schedule. The long-term extension phase will assess safety outcomes. However, no results were available at the time of this review.
Indirect Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
The included studies do not provide evidence regarding the efficacy or safety of aflibercept 8 mg compared with other interventions beyond aflibercept 2 mg. The ITCs were conducted to provide estimates of relative efficacy, safety, and number of injections for aflibercept 8 mg relative to standard interventions for the treatment of nAMD. Specifically, 2 treatment regimens of aflibercept 8 mg (1 regimen of every 12 weeks and every 16 weeks and 1 of every 12 weeks and every 16 weeks) were evaluated against relevant comparators. This section provides an overview of the conduct, the results, and the critical appraisal of the sponsor submitted ITC.
Description of Indirect Comparison
One ITC, conducted by Broadstreet Health Economics and Outcomes Research, was submitted by the sponsor. A targeted literature search by the sponsor on October 2, 2023, did not identify any published ITC that included aflibercept 8 mg. The sponsor-submitted ITC used a Bayesian NMA approach with fixed-effect and random-effects models to compare aflibercept 8 mg every 12 weeks and every 16 weeks in patients with nAMD against other anti-VEGF drugs used for this condition. The following outcome measures are reported here: change in BCVA, gain of 15 ETDRS letters, ocular AEs, and mean number of injections. The sponsor-submitted NMA identified relevant evidence through a systematic literature review (SLR).53 No published protocol was available at the time of this report.
Table 15: Study Selection Criteria and Methods Within the NMA Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Treatment-naive patients with nAMDa |
Intervention |
|
Comparator | No restrictions |
Outcome |
|
Study designs | RCTsd |
Publication characteristics | Published studies and an unpublished pivotal study of aflibercept 8 mg in nAMD (PULSAR) |
Exclusion criteria |
|
Databases searched |
|
Selection process | The list of titles and abstracts was screened by 2 independent reviewers according to the defined inclusion and exclusion criteria, to select relevant articles pertaining to the topic of interest. The decisions from the 2 reviewers were combined and discrepancies were resolved by consensus or by a third reviewer. For any article that met the inclusion criteria or could not be excluded based on the abstract review, the full text was screened to decide on inclusion or exclusion. Full texts were evaluated by 2 independent reviewers to verify if they met the inclusion criteria. Differences were resolved by consensus or by a third reviewer. All finalized references were checked for any possible linking (i.e., to check if different articles originate from the same study). |
Data-extraction process | Data from studies included in the review were extracted using extraction templates created in Excel. One reviewer extracted the data, while another validated the accuracy of the extracted data. |
Quality assessment | The quality of the included studies was appraised according to the Cochrane risk-of-bias tool checklist (version 2.0). This assessment was performed by 1 reviewer, with a second reviewer to validate the first reviewer’s assessment. Disagreements were resolved by consensus or by a third reviewer. The assessment was performed at the study level. |
AE = adverse event; BCVA = best corrected visual acuity; CNV = choroidal neovascularization; CRT = central retinal thickness; CST = central subfield thickness; IRF = intraretinal fluid; ITC = indirect treatment comparison; nAMD = neovascular (wet) age-related macular degeneration; PDS = port delivery system; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; RCT = randomized controlled trial; SAE = serious adverse event; SRF = subretinal fluid; VEGF = vascular endothelial growth factor.
aOnly references presenting data for overall population and/or relevant subgroups (age, baseline BCVA, baseline CNV area and/or leakage area, CNV type, baseline CRT, retinal fluid presence, Asian population, previous treatment-naive, and pretreated patients with anti-VEGF) were included.
bRanibizumab PDS is not presented in the results because it is not a publicly reimbursed treatment and therefore not a relevant comparator.
cThe clinical trials of brolucizumab assessed brolucizumab at 6 mg and 3 mg strengths. While brolucizumab 3 mg is presented in the ITC network and results for completeness, brolucizumab 3 mg is not approved for use in Canada and it is not a relevant comparator.
dPooled results from pivotal studies to be included and extracted, only if data from each study were not available. Pooled data for different drugs or drug doses or treatment regimens were not included and/or extracted.
eRelevant subgroups included those grouped by age, baseline BCVA, baseline CRT, CNV type, baseline CNV area and/or leakage area, retinal fluid presence, Asian patients, previous treatment with any regimen (including pro re nata and treat-and-extend).
Sources: Sponsor-submitted ITC53 and the sponsor’s Summary of Clinical Evidence.
Network Meta-Analysis Design
Objectives
The objective of the NMA was to assess the comparative efficacy and safety of aflibercept 8 mg (every 12 weeks and every 16 weeks) with the interventions listed in Table 15.
Methods for the Search, Selection, Data Extraction, and Risk-of-Bias Appraisal
Studies suitable for the ITC were identified using an SLR of RCTs of anti-VEGF drugs in patients with nAMD. MEDLINE and Embase were searched on May 24, 2022, and the Cochrane Library and ClinicalTrials.gov were searched on May 26, 2022.53
The population of interest was limited to patients with a diagnosis of nAMD who were treatment-naive; no specific geographical restrictions were applied. The list of publications was narrowed down to articles written in English. With regard to additional limitations, only studies reporting data on more than 40 patients or eyes were included. The sponsor excluded small studies with an aim to reduce publication bias and uncertainty associated with small sample sizes.53
Two independent reviewers screened the results retrieved from the search. One reviewer extracted the data while another validated the extracted data.53
The risk of bias of the included studies was appraised according to the Cochrane risk-of-bias tool (version 2.0). This assessment was performed by 1 reviewer, with a second reviewer validating the first reviewer’s assessment. Disagreements were resolved by consensus or by a third reviewer. The assessment was performed at the study level.
Network Meta-Analysis Methods
For all of the reported outcomes, with the exception of the number of injections, NMAs consisting of 3 different types reflecting the nature of the outcomes of interest were conducted. The analysis of change from baseline in BCVA was conducted with a normal likelihood and an identity link. Analyses of AEs were conducted with a binomial likelihood and logit link. The analyses of gaining or losing letters were conducted with multinomial likelihood and a probit link. The NMA was based on a Bayesian approach and computed through a Markov chain Monte Carlo simulation. Vague (noninformative) priors were used. The simulation was based on 5,000 burn-ins (adaptation period) followed by at least 10,000 iterations, with convergence assessed through trace, density, and Brooks-Gelman-Rubin plots. Each NMA was conducted under both a fixed-effect and random-effects models. The model with superior model fit as determined by a lower value of the deviance information criterion was chosen as the primary model. As applicable, median odds ratios and treatment differences (drawn from posterior distributions) and the corresponding 95% credible intervals were reported.53
Clinical and methodological heterogeneity of included studies was assessed by tabulating and contrasting study characteristics and baseline patient characteristics. These included baseline visual acuity, disease duration, and demographic characteristics. For treatment comparisons that included multiple studies, the Cochran Q statistic accompanied by its associated P value, and the I2 statistic were used to quantify the extent of statistical heterogeneity.
To assess the inconsistency of a network, the node-splitting method was used. In this approach, each treatment effect is estimated separately using direct and indirect evidence; these 2 estimates are then contrasted.
The outcome of mean number of injections was analyzed as an absolute-effect (as opposed to a relative-effect) measure based on a within-node (each intervention) meta-analysis; no comparison between interventions was conducted for this outcome. To estimate the number of injections required for each anti-VEGF drug and dosing regimen, meta-analyses were conducted based on the number of injections reported in RCT: over the first year of treatment, over the first 2 years of treatment, or between year 1 and year 2 of treatment. The DerSimonian-Laird random-effects method was used to calculate treatment effects as well as the between-trial variability.53 The P value for Cochran Q and I2 values were provided on forest plots to quantify the extent of statistical heterogeneity. Clinical and methodological heterogeneity was not described.
Imputation of missing data was based on external data from comparable studies when direct information was not available. Continuous model inputs included BCVA gain and mean injection count, each with their associated SEs. Binary inputs involved counts of patients reaching specific visual-acuity thresholds and safety-event occurrences. SE estimates were derived from published data using transformations of CIs, P values, and imputed SDs. The formula for SE from a 95% CI relied on the z score at the 2.5th percentile of the standard normal distribution. The SE for efficacy outcomes related to CRT and/or central subfield retinal thickness (CST) was based on the Student’s t distribution, and the SE of the injection count was computed using the normal distribution with a pooled variance assumption. Counts of zero were adjusted via a continuity correction to facilitate the NMA, using an additive approach that maintained comparability across treatment arms. This correction was defined algebraically for both treatment and control groups. When precise SD data were unavailable, an average SD from comparable arms was used, assuming shared variance among similar treatment regimens.
Of the prespecified outcomes, the sponsor-submitted report included the following outcomes: change in BCVA, gain of 15 ETDRS letters, ocular AEs, and the mean number of injections. The reason for not reporting the rest of the preplanned outcomes per in Table 15 was not clear. However, to adhere to the list of outcomes that was determined through patient and clinician feedback, we will not be reporting the outcome of change in CRT and CFT.
Outcomes reported between 48 to 52 weeks were treated as if they were reported at 1 year, while outcomes reported between 96 to 104 weeks were treated as if they were reported at 2 years.
The analyses and data manipulations were conducted primarily in R version 4.2.2. The Bayesian NMA analyses used JAGS (version 4.3.1) for the Markov chain Monte Carlo sampling, either by using the GeMTC package for binomial or continuous outcomes, or by calling JAGS code directly through the rjags package (for multinomial models). Pairwise meta-analyses were conducted with the metafor package.53
Table 16: Indirect Treatment Comparison Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Bayesian network meta-analysis (random or fixed effects based on the value of the deviance information criterion) |
Priors | Vague |
Assessment of model fit | Deviance information criterion |
Assessment of consistency | Node-splitting method |
Assessment of convergence | Trace and Gelman-Rubin plots |
Outcomes | Change in BCVA, gain of ≥ 10 letters, gain of ≥ 15 letters, loss of ≥ 10 letters, loss of ≥ 15 letters, ocular adverse events |
Follow-up time points | Outcomes reported between 48 to 52 weeks were treated as if reported at 1 year, while outcomes reported between 96 to 104 weeks were treated as if reported at 2 years |
Construction of nodes | Not reported |
Sensitivity analyses | None |
Subgroup analysis | None |
Methods for pairwise meta-analysis | DerSimonian-Laird random-effects method (number of injections) |
BCVA = best corrected visual acuity.
Sources: Sponsor-submitted network meta-analysis53 and the sponsor’s Summary of Clinical Evidence.
Results of the Sponsor-Submitted ITC
Summary of Included Studies
A total of 34 studies were included in the NMA: 1 assessed aflibercept 8 mg in nAMD,54,55 13 assessed aflibercept 2 mg,51,55-62 20 assessed ranibizumab,61,63-80 3 assessed faricimab,56,70 3 assessed brolucizumab,51,62 and 12 assessed bevacizumab.63,64,66,71,72,75,76,81-84
All included trials studied patients with nAMD, but differences in study conduct, definitions, inclusion and exclusion criteria, and patient characteristics were evident, which resulted in variations in the populations of patients across the trials. In several cases, it was possible to identify differences across trials, but in other cases differences in reporting precluded proper assessments of heterogeneity by the sponsor (e.g., there was limited reporting on disease duration as well as on the presence of IRF or SRF). Differences were observed between the included studies that could potentially lead to heterogeneity in the analyses.
The mean baseline age in the included studies ranged from 66 to 79 years, representing a source of heterogeneity. Baseline visual acuity showed some heterogeneity between studies, ranging from 51 ETDRS letters (assessing ranibizumab PRN versus ranibizumab PRN-nonlinear in ARTIS) to 67 ETDRS letters (assessing aflibercept 2 mg PRN versus aflibercept 2 mg every 8 weeks in Mori [2017]).60 The mean baseline BCVA for the PULSAR study was 59.6 ETDRS letters, which is close to the median (59.4) for all studies. Some variation in the mean baseline CRT and/or CST measurements, which varied from 249 to 503.7 μm, was seen across the included studies.
Examination of the studies revealed some anomalous results in 1 trial, RIVAL.68 While both treatments were supposed to be administered in accordance with a treat-and-extend regimen, the mean injection frequency in the aflibercept arm was extraordinarily high (9.7 and 17.0) at 1 and 2 years, respectively. The sponsor determined that the higher injection frequency was likely associated with the lack of European Union approval of the posology and the relatively strict re-treatment criteria compared with other studies.
Risk-of-bias assessment of the included studies in the sponsor-submitted ITC determined that ███ studies were of “low risk,” ███ had “some concerns,” and ███ were of “high risk” as determined by the Cochrane risk-of-bias quality-assessment tool. The sponsor’s ITC did not report on any specific actions taken with the studies that were determined as having “high risk” of bias.
Table 17: Assessment of Homogeneity for Indirect Treatment Comparison
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity |
|
Treatment history | Likely similar across trial as patients are all treatment-naive |
Trial eligibility criteria | In many instances the eligibility criteria were similar; however, some variations exist |
Dosing of comparators | Certain comparators are dosed according to the indication; some comparators given as a treat-and-extend regimen |
Placebo response | NA |
Definitions of end points | Similar use of ETDRS in most trials to assess BCVA |
Timing of end-point evaluation | Outcomes reported between 48 to 52 weeks were treated as if reported at 1 year, while outcomes reported between 96 to 104 weeks were treated as if reported at 2 years |
Withdrawal frequency | Withdrawal frequencies for the studies included in the network meta-analysis were not specifically assessed for homogeneity |
Clinical trial setting | Likely similar due to the nature of the injection |
Study design | Some variation in masking of intervention where several trials were open-label |
BCVA = best corrected visual acuity, CST = central subfield thickness, CRT = central retinal thickness, ETDRS = Early Treatment Diabetic Retinopathy Study, NA = not applicable.
Source: Sponsor-submitted network meta-analysis.53 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Change From Baseline BCVA
An NMA was conducted on change from baseline BCVA at 1 year based on the network of evidence (Figure 3) identified by the SLR. Both fixed-effects and random-effects models were considered; an examination of the deviance information criterion (Table 18) suggested that the random-effects model was a better fit to the data. Pairwise assessments of statistics suggested relatively high heterogeneity between aflibercept 2 mg every 4 weeks and each of ranibizumab 0.5 mg every 4 weeks (|| | ██████) and aflibercept 2 mg every 8 weeks (|| | ██████). Additionally, high statistical heterogeneity was evident between ranibizumab 0.5 mg every 4 weeks and ranbizumab 0.5 mg treat-and-extend (|| | ██████).53
Figure 3: Evidence Network of Change From Baseline BCVA
AFL = aflibercept; BEV = bevacizumab; BRO = brolucizumab; FAR = faricimab; PRN = as needed; Q4 = every 4 weeks; Q8 = every 8 weeks; Q12 = every 12 weeks; Q16 = every 16 weeks; PRNnL = pro re nata no loading; RBZ = ranibizumab; T&E = treat-and-extend.
Source: Sponsor-submitted network meta-analysis.53
Table 18: Model Fit and DIC for Change From Baseline BCVA
Model | Dbar | pD | DIC |
|---|---|---|---|
Fixed effects | 100.19 | 56.90 | 157.09 |
Random effects | 79.39 | 67.59 | 146.98 |
Dbar = posterior mean of the deviance; DIC = deviance information criterion; pD = number of parameters.
Source: Sponsor-submitted network meta-analysis.53
With respect to the BCVA measurement, the credible intervals of the comparative results for aflibercept 8 mg and other anti-VEGF drugs in the NMA consistently included the null. Figure 4 and Figure 5 show the relative effect of aflibercept 8 mg every 12 weeks and every 16 weeks, respectively, compared to the other treatments. As noted in the figures, the effect compared to most treatments is centred around zero (the null), with corresponding wide credible intervals. Additionally, all but 1 of the point estimates ███████ ███████████ ████ ██ █ ███ were smaller than 4 letters. All of the credible intervals included the null and almost all included the clinically meaningful threshold of 4 to 5 letters on either or both the benefits and the harms sides.


Gain or Loss in Number of Letters
Gain and loss of letters was analyzed with a conditional binomial NMA model. Patients were categorized into mutually exclusive groups (those losing ≥ 15 letters, losing between 10 and 15 letters, losing < 10 letters to gaining < 10 letters, gaining between 10 and 15 letters, and gaining ≥ 15 letters), and the probabilities of falling into each group were modelled simultaneously. Figure 6 shows the structure of the network of trials that report this outcome. The conditional binomial model was substantially more complicated than the models employed for other outcomes. The fixed-effects model had a substantially lower DIC compared with the random-effects model and was selected as the most appropriate (Table 19). Pairwise statistical heterogeneity was not reported for this outcome.
Table 19: Model Fit and DIC for Gain or Loss of Letters
Model | Dbar | pD | DIC |
|---|---|---|---|
Fixed effects | 686.18 | 31 | 717.18 |
Random effects | 1,864.1 | 36.86 | 1,900.96 |
Dbar = posterior mean of the deviance; DIC = deviance information criterion; pD = number of parameters.
Source: Sponsor-submitted network meta-analysis.53
Figure 6: Evidence Network of Gain or Loss of 10 and 15 letters
AFL = aflibercept; BEV = bevacizumab; BRO = brolucizumab; FAR = faricimab; PRN = as needed; Q4 = every 4 weeks; q.8.w. = every 8 weeks; Q12 = every 12 weeks; Q16 = every 16 weeks; PRNnL = pro re nata no loading; RBZ = ranibizumab; T&E = treat-and-extend.
Source: Sponsor-submitted network meta-analysis.53
On the basis of this NMA, the results for aflibercept 8 mg and most other treatments with respect to the gain and loss of letters did not exclude the null. Reporting of the number of letters gained and lost was focused on 4 cut-offs: gaining 15 or more letters, gaining 10 or more letters, losing 10 or more letters, and losing 15 or more letters. Only gains of 15 or more letters will be reported here. The relative effects of aflibercept 8 mg every 12 weeks and every 16 weeks compared to the other treatments are shown in Figure 7 and Figure 8, respectively, for a gain of 15 or more letters. For aflibercept 8 mg every 12 weeks and every 16 weeks, all the comparisons (except versus ████████████ | ██ █████ under a fixed-effects model) had credible intervals that overlapped 1 (the null), indicating uncertainty about which treatment might be favoured. Wide credible intervals were noted in all comparisons; It is difficult to judge the clinical relevance of the upper and lower bounds of the credible interval as no absolute effects were provided.


Harms
Ocular Adverse Events
To assess the harms related to treatments, an NMA was conducted on the reported ocular AEs. The network of evidence (Figure 9) identified by the SLR was somewhat smaller than those for the efficacy outcomes, as collection of AE data was not as rigorous as the reporting of efficacy outcomes. A total of 14 studies reported the number of ocular AEs experienced in trials. Both fixed-effects and random-effects models were considered; examination of the deviance information criterion (Table 20) suggested that both models fit the data equally well and the sponsor presented the results for the fixed-effects model. Pairwise assessments showed increased statistical heterogeneity for ranibizumab 0.5 mg PRN versus 0.5 mg every 8 weeks (|| | ████ and ranibizumab 0.5 mg every 4 weeks versus a treat-and-extend regimen ██| | ████.
Figure 9: Evidence Network of Ocular Adverse Events
AFL = aflibercept; BEV = bevacizumab; BRO = brolucizumab; FAR = faricimab; PRNnL = as needed; Q4 = every 4 weeks; Q8 = every 8 weeks; Q12 = every 12 weeks; Q16 = every 16 weeks; PRNnL = pro re nata no loading; RBZ = ranibizumab; T&E = treat-and-extend.
Source: Sponsor-submitted network meta-analysis.53
Table 20: Model Fit and DIC for Ocular Adverse Events
Model | Dbar | pD | DIC |
|---|---|---|---|
Fixed effects | 40.23 | 29.09 | 69.32 |
Random effects | 38.91 | 31.79 | 70.71 |
Dbar = posterior mean of the deviance; DIC = deviance information criterion; pD = number of parameters.
Source: Sponsor-submitted network meta-analysis.53
The relative effect of treatments on the number of ocular AEs are similar across all treatments. The odds ratios for aflibercept 8 mg regimens are near 1 for almost all comparisons, and credible intervals are wide, such that no comparisons excluded the null in the credible interval. It is difficult to judge the clinical relevance of the upper and lower bounds of the credible interval as no absolute effects are provided. While some numerical differences are evident in the comparisons versus faricimab and ranibizumab, none excluded the null (Figure 10 and Figure 11). Pairwise statistical heterogeneity was available for 7 of the total 24 pairwise comparisons. Of these 7, ranibizumab 0.5 PRN versus ranibizumab 0.5 mg every 8 weeks had high pairwise heterogeneity ██| | ██████.



Nonocular Adverse Events
Because reporting of nonocular AEs was not as comprehensive as it was for other outcomes, there was limited evidence to assess nonocular AEs across studies, and an NMA for nonocular AEs was not reported.
Number of Injections
Treat-and-extend and PRN regimens are not predetermined and, in the first year, show a mean number of injections of ████ in aflibercept 2 mg PRN (█████ ████ ██ █████, ████ ██████ █████ ██████ in bevacizumab PRN, ████ ██████ █████ █████ in ranibizumab PRN, ████ ██████ █████ ██████ PRN nonlinear, ████ ██████ █████ ██████ in ranibizumab PRN nonlinear, ████ ██████ █████ █████ in aflibercept 2 mg treat-and-extend, ████ ██████ █████ █████ in bevacizumab treat-and-extend, ████ ██████ █████ █████ in ranibizumab treat-and-extend, and ████ ██████ █████ █████ for aflibercept 2 mg on a 4-week interval treat-and-extend ██████ ████ ██ █████. In the second year, the mean number of injections for treat-and-extend and PRN regimens were ████ ██████ █████ █████ for aflibercept 2 mg PRN, ████ ██████ █████ █████ for bevacizumab, ████ ██████ █████ █████ for ranibizumab PRN, ████ ██████ █████ █████ for aflibercept 2 mg treat-and-extend, ████ ██████ █████ for bevacizumab treat-and-extend, ████ ██████ █████ █████ for ranibizumab treat-and-extend, and ████ ██████ █████ █████ for aflibercept 2 mg treat-and-extend). Estimates of the average number of injections received within the first year of treatment are presented in Table 21. The average number of injections received between the first and second year are presented in Table 22.
Table 21: Mean Injection — Meta-Analysis Estimates for 1-Year Results
Regimen | Aflibercept 2 mg | Aflibercept 8 mg | Bevacizumab | Brolucizumab 6 mg | Ranibizumab | Faricimab |
|---|---|---|---|---|---|---|
q.4.w | ███████ | ██ | ███████ | ██ | ███ ███ | ██ |
q.6.w | ██ | ██ | ███████ | ██ | ██ | ██ |
q.8.w. | ███████ | ██ | ██ | ████ ███ | ████ ███ | ██ |
q.12.w. | ██ | 6.10 ████████ | ███████ | ██ | ██ | ████ ███ |
q.16.w. | ██ | 5.20 ██ █████ | ██ | ██ | ██ | ████ ███ |
q.8.w., q.12.w., q.16.w. | ██ | ██ | ██ | ██ | ██ | ███████ |
PRN | ███████ | ██ | ███████ | ██ | ███████ | ██ |
PRN, nonlinear | ██ | ██ | ███████ | ██ | ████ ██ | ██ |
Treat-and-extend | ██ ██ ███ | ██ | ███ ███ | ██ | ████ ███ | ██ |
4-week interval treat-and-extend | ████████ | ██ | ██ | ██ | ██ | ██ |
NA = not applicable; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; q.8.w., q.12.w., q.16.w. = various intervals including every 8, 12, or 16 weeks; PRN = pro re nata.
Source: Sponsor-submitted network meta-analysis.53
Table 22: Mean Injections — Meta-Analysis Estimates for 1- to 2-Year Results
Regimen | Aflibercept 2 mg | Aflibercept 8 mg | Bevacizumab | Brolucizumab 6 mg | Ranibizumab | Faricimab |
|---|---|---|---|---|---|---|
q.4.w. | ██ | ██ | ███ ████ | ██ | ███ ████ | ██ |
q.8.w. | ████████ | ██ | ██ | ████████ | ██ | ██ |
q.12.w. | ██ | 3.60 ███████ | ██ | ██ | ██ | ██ |
q.16.w. | ██ | 3.00 ███ ████ | ██ | ██ | ██ | ██ |
q.8.w., q.12.w., q.16.w. | ██ | ██ | ██ | ██ | ██ | ████ ███ |
PRN | ████ ███ | ██ | ████ ███ | ██ | ██ █████ | ██ |
Treat-and-extend | ████████ | ██ | ██ ██ ███ | ██ | ████ ███ | ██ |
4-week interval treat-and-extend | ██ ██ ███ | ██ | ██ | ██ | ██ | ██ |
NA = not applicable; q.4.w. = every 4 weeks; Q6 = every 6 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.16.w. = every 16 weeks; q.8.w., q.12.w., q.16.w. = various intervals including every 8, 12, or 16 weeks; PRN = pro re nata.
Source: Sponsor submitted network meta-analysis.53
Critical Appraisal of Indirect Treatment Comparison
Studies included in the sponsor-submitted ITC were identified through an SLR, which was transparently reported but did not include an established protocol before the conduct of the systematic review. The search strategy was conducted more than a year ago, potentially missing evidence published within the past year. The inclusion and exclusion criteria were appropriate, with the possible exception of the exclusion of studies with 40 or fewer patients. The justification of the exclusion of small population studies was to avoid potential biases arising from a small sample size. However, this approach also increases the potential for publication bias. The approach of conducting the SLR was appropriate, with 2 reviewers conducting the screening. Only 1 reviewer conducted the quality assessment, with another validating the output. The quality assessment was done using the Cochrane risk-of-bias assessment tool (version 2.0). A number of studies were identified as potentially “high-risk,” but no clear steps were taken to mitigate potential biases arising from studies with a high risk of bias (e.g., sensitivity analyses). The risk-of-bias appraisal was done at the study level, which fails to recognize that various outcomes are commonly affected by difference sources of bias. The ITC provided a list of excluded studies as well as a list of per-study risk-of-bias assessments.
Outcomes that were included in the ITC were clinically relevant and appropriate. However, a few clinically relevant outcomes were not reported, including quality-of-life measures and serious ocular AEs. Certain outcomes that were predefined in the initial inclusion and exclusion table were not reported and lacked justification for the lack of reporting. It is important to note that interventions of studies that had a preplanned and fixed injection regimen are not reflective of clinical practices. As such, it is likely that indirect comparison versus treat-and-extend regimens are the most informative.
The statistical approach by the sponsor-submitted ITC to assessing the results through a Bayesian NMA was appropriate and transparently communicated.
Clinical heterogeneity among the studies was difficult to assess due to the lack of reporting on several key demographic, baseline, and study characteristics. Mean baseline age in the included studies ranged from 66 to 79 years, representing a potential source of heterogeneity. Baseline visual acuity showed some heterogeneity between studies, ranging from 51 ETDRS letters (assessing ranibizumab PRN versus ranibizumab PRN-no-loading in ARTIS79) to 67 ETDRS letters (assessing aflibercept 2 mg PRN versus aflibercept 2 mg every 8 weeks in Mori [2017]60). The mean baseline BCVA for the PULSAR study was 59.6 ETDRS letters, which is close to the median (59.4) for all studies. There was some variation in the mean baseline CRT and/or CST measurements, which varied from 249 to 503.7 μm across the included studies. As such, whether the assumptions related to homogeneity were met is uncertain. However, statistical assessment of the NMA models showed no evidence of assumption violations. No statistical evidence of inconsistency was observed. When direct treatment comparisons were informed by multiple trials, parameter estimates were consistent, showing that statistical heterogeneity did not lead to poor model fit. While the statistical models were valid, they may still be affected by the differences observed across the included studies.
The credible intervals appeared large for all estimates, indicating substantial variability in the data. This may be reflective of the differences in study populations across the various trials included in the analyses, as the studies differed across many characteristics, such as phase of trial and baseline participant characteristics. In many cases, treatments are informed by only a single trial. Because of this, there is a risk that effect modifiers lead to biases in the estimates of treatment effect. In the network evaluated, the PULSAR study is the only trial that examines aflibercept 8 mg, and only the HAWK and HARRIER51 studies examine brolucizumab. Additionally, 2 bevacizumab regimens are informed by single trials, and 2 aflibercept 2 mg regimens are informed by single trials. Many other direct comparisons are informed by only 2 studies. The ranibizumab PRN versus bevacizumab PRN nodes is the only direct treatment comparison informed by many trials. Should 1 of the direct comparisons be informed by only 1 trial, it is important that the trial be representative of the population overall, and not be affected by effect modification. This is an assumption that is required in this network given the limitations to the evidence base. In addition, the assessment of outcomes in binary end points lacked measures of absolute effects, making it difficult to assess the clinical relevance of these end points.
Assessment of the frequency of injections were based on naive (visual) comparisons across pairwise meta-analyses, and no comparative analyses were performed. This limits the ability to understand the comparative benefit or harm of various interventions and to properly assess potential variability in the comparative results. Descriptive and noncomparative numbers of injections data suggest that aflibercept 8 mg is likely to have fewer injections than most other treatments that were administered according to a prescribed standard regimen. However, some regimens that use a PRN or treat-and-extend approach show a number of injections that can be contrasted with similarity to that of 8 mg. Considering the limitations associated with the clinical heterogeneity and the resulting wide credible intervals suggesting a lack of robust data, the comparative efficacy results of 8 mg aflibercept cannot be used to inform decision-making on their own and must be used within the context of results derived from other evidence.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were submitted by the sponsor.
Discussion
Summary of Available Evidence
One trial, PULSAR (N = 1,009) met the inclusion criteria for the systematic review conducted by the sponsor. The PULSAR study was a phase III, active-controlled, noninferiority, multinational (251 sites, including 3 in Canada) trial that randomized 1,009 patients with treatment-naive nAMD, 1:1:1, to aflibercept 2 mg every 8 weeks, aflibercept 8 mg every 12 weeks, or aflibercept 8 mg every 16 weeks. The primary outcome was change from baseline in BCVA measured by the ETDRS letter score at week 48. The proportion of participants with no IRF or SRF in the central subfield at week 48, proportion of participants gaining at least 15 letters in BCVA from baseline at week 48, vision-related quality of life at week 48, and number of injections through 48 weeks were reported as secondary outcomes. Ocular and nonocular TEAEs and SAEs, deaths, and discontinuations due to TEAEs through week 60 were reported as harms. The median age of the patients in PULSAR was 75 years, ranging from 50 to 96 years overall. The proportion of female patients was 53.3%. The majority of patients were white (75.8%) or Asian (23.2%).
There were no direct comparisons of aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks to other anti-VEGF therapies. The sponsor submitted a Bayesian NMA of 34 studies to compare aflibercept 8 mg every 12 weeks and every 16 weeks against other anti-VEGF therapies in nAMD patients. The NMA applied fixed-effect and random-effects models across varied outcomes, including BCVA change, ETDRS letter gain or loss, and ocular AEs with methodological rigour and diverse statistical links. No indirect comparisons of injection frequency, aside from a naive (visual) comparison of pairwise meta-analyses for each regimen, were provided.
Interpretation of Results
Efficacy
It is clear from inputs to CDA-AMC that visual acuity, less-frequent injections, and patient quality of life were highly important to patients and clinicians. Clinician group input submitted to us also indicated that a long-lasting treatment with reduced adverse effects is a need for patients with nAMD. The PULSAR trial assessed outcomes that correspond to patient needs, including change from baseline in BCVA, proportion of participants gaining at least 15 letters in BCVA as outcomes, vision-related quality of life, and frequency of injections.
The results of the PULSAR trial support the noninferiority of aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks to aflibercept 2 mg every 8 weeks for the mean change from baseline in BCVA at week 48 in treatment-naive patients with nAMD when using a noninferiority margin of 4 letters in the FAS (high certainty). A supplementary per-protocol analysis (often considered more conservative for noninferiority hypotheses) supported the conclusion of noninferiority in the FAS population, as did a number of sensitivity analyses using different imputation approaches to address missing data. However, the results did not show the superiority of aflibercept 8 mg to aflibercept 2 mg for this outcome. Furthermore, other outcomes in this report were not part of a statistical hierarchy and were not subject to hypothesis tests.
Evidence from the PULSAR trial showed (with moderate certainty) that aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks likely results in little to no clinically important difference in the proportion of patients gaining 15 EDTRS letter and the proportion with no IRF or SRF at 48 weeks. The only concern noted for these analyses was related to study limitations, because there was uncertainty about whether the imputation techniques for missing data would result in unbiased findings.
The pivotal trial measured the proportions of patients with no IRF or SRF as a secondary outcome. IRF and SRF are indicators of active disease measured in clinical practice to evaluate clinical response, according to the clinical expert we consulted. The proportions of patients with no IRF or SRF at weeks 48 were approximately 72% in the aflibercept 8 mg every 12 weeks arm and 68% in the aflibercept 8 mg every 16 weeks arm; these results were not considered clinically important by the clinical expert compared to 56% in the aflibercept 2 mg every 8 weeks arm. The clinical expert we consulted indicated that a difference of approximately 20% between groups may be a clinical meaningful difference. We determined there was moderate certainty of little to no difference in the proportion of patients with no IRF and SRF when comparing aflibercept 2 mg every 8 weeks with aflibercept 8 mg every 12 weeks and aflibercept 8 mg every 16 weeks, respectively.
Vision-related quality of life measured by the NEI VFQ-25 at week 48 was a secondary outcome in the PULSAR trial. While improvements in the composite score were observed in the high-dose aflibercept arms, the magnitude of change did not meet the MID of 6 points established in the literature. The difference between treatment arms showed (with moderate certainty) that aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks likely results in little to no clinically important difference in the change from baseline in vision-related quality of life when compared with aflibercept 2 mg every 8 weeks. There was a concern about a potential risk of bias due to missing outcome data.
Frequency of injection was a key outcome of interest for patients and clinicians as it has implications for the frequency of AEs and vision-related quality of life and some challenges patients face, including travel logistics and payment, especially for those living in remote communities. The PULSAR trial determined (with low certainty) that aflibercept 8 mg every 12 weeks may result in little to no clinically important difference, and that aflibercept 8 mg every 16 weeks may reduce the frequency of injections when compared with aflibercept 2 mg every 8 weeks at 48 weeks; however, these results are associated with uncertainty. The difference in number of injections between the 2 treatment groups was lower than the clinically important threshold of 2 suggested by the clinical experts we consulted, although the difference between aflibercept 2 mg every 8 weeks and aflibercept 8 mg every 16 weeks was near the threshold (i.e., 1.8). Furthermore, the generalizability of the results is limited as the protocol-specified dosing interval for the aflibercept 2 mg every 8 weeks arm was not aligned with the treat-and-extend protocol commonly used with aflibercept 2 mg in clinical practice, according to the clinical expert. The rigid protocol-driven injections in comparator arm potentially caused patients in this arm to receive more frequent injections compared with those in the intervention arms. While the CDA-AMC review team acknowledges that protocol-driven injections may be necessary to demonstrate noninferiority between arms, the injections in both arms were not provided as they would be in practice, according to the clinical expert we consulted. The applicability of the results to the real world is therefore limited.
An evidence gap arises from the exclusive enrolment of treatment-naive patients in the PULSAR trial. The clinical expert noted that patients for whom other anti-VEGF drugs fail could be eligible to receive aflibercept 8 mg. Moreover, in the product monograph for aflibercept 8 mg, no criteria are provided for administering the drug to treatment-naive patients. The absence of patients with treatment experience in the trial arms raises uncertainty about the findings from the PULSAR trial to this patient group. Moreover, the absence of head-to-head trials comparing aflibercept 8 mg with other anti-VEGF treatments and the lack of evidence about the long-term therapeutic effect of aflibercept 8 mg may represent gaps in the evidence. Patients and clinicians are in search of a new drug that can offer reduced injection frequencies, enhance vision, and positively affect health-related quality of life compared to currently available treatments. The evidence presented indicates that the higher dose of aflibercept is noninferior to the 2 mg dose of aflibercept, with it being likely that there is no discernible difference in clinical benefits improvements in vision-related quality of life compared to aflibercept 2 mg, which has been available in the market for almost 10 years.
The sponsor submitted an ITC to provide comparative evidence comparing aflibercept 8 mg against other anti-VEGF drugs. Despite methodical evaluation for heterogeneity and inconsistency, including I2 and node-splitting techniques, the random-effects model revealed that none of the comparative outcomes — ranging from BCVA changes to ocular AEs — were subject to credible intervals that excluded the null. The results from the sponsor-submitted ITC showed wide credible intervals in all comparative outcomes of interest. These wide credible intervals frequently included the null and a point estimate that was around the null. The number of injections, while descriptive and not comparative in nature, implies that aflibercept 8 mg may require fewer injections compared to other treatments that involve a fixed regimen. Certain regimens that adopt an as-needed (PRN) or treat-and-extend strategy could have an injection frequency similar to that of aflibercept 8 mg. However, due to clinical variability among studies and broad credible intervals indicating limited data strength, the comparative efficacy findings for 8 mg aflibercept are insufficient as standalone evidence for decision-making. Overall, aflibercept showed a consistent mean injection frequency aligned with the fixed schedule, which appeared to be lower in the second year, while variable regimens reported a broader range of injections.
Harms
The clinician group emphasized the importance of a new drug for nAMD with reduced side effects when compared to existing treatments. In the PULSAR trial, the overall rates of ocular and nonocular TEAEs through week 60 appeared to be similar among the treatment groups. The safety profile of the drug under review demonstrated that no additional AEs were observed in patients who received a higher dose of aflibercept. Almost the same proportion (around 43%) of patients in each arm of the PULSAR study experienced at least 1 ocular TEAE, with the most frequent being reduced visual acuity, cataracts, and retinal hemorrhaging. Similarly, the proportion of nonocular TEAEs was almost the same (around 60%) in the treatment arms. The most common nonocular TEAEs were COVID-19, nasopharyngitis, and benign and malignant neoplasms.
The percentage of patients who experienced at least 1 ocular SAE was similar in the aflibercept 8 mg arms (2.1% for both the aflibercept 8 mg every 12 weeks and the aflibercept 8 mg every 16 weeks arms) compared to aflibercept 2 mg arm (1.2%). However, an assessment of the evidence revealed (with moderate certainty) that aflibercept 8 mg every 12 weeks and 8 mg every 16 weeks likely results in little to no difference in the proportion of patients with ocular SAEs when compared with aflibercept 2 mg every 8 weeks due to imprecision. Moreover, the clinical expert we consulted could not determine a threshold for a clinically important difference between arms for these harms. The proportion of patients who stopped treatment due to ocular AEs in the aflibercept 8 mg arms was numerically higher than the aflibercept 2 mg arm. The frequency of deaths was nearly 1.5% in the aflibercept 2 mg every 8 weeks arm and less than 1% in both aflibercept 8 mg arms. Higher numbers of patients in aflibercept 8 mg arms reported more common notable harms, such as cataracts and increased intraocular pressure, compared to the aflibercept 2 mg arm; however, these differences were trivial. The frequencies of some notable harms, including APTC events and thromboembolic events, were lower in the aflibercept 2 mg arm compared to the aflibercept 8 mg arms.
The NMA indicated odds ratios for ocular AEs that were often near the null, with wide credible intervals suggesting uncertainty about which treatment could be favoured. Due to insufficient data, nonocular AE were not thoroughly assessed.
Conclusion
Based on the PULSAR trial, there is evidence of high certainty that aflibercept 8 mg every 12 and 16 weeks demonstrates noninferiority (but not superiority) to aflibercept 2 mg every 8 weeks in terms of the change in BCVA from baseline over 48 weeks of treatment among treatment-naive adults with nAMD. The evidence for other outcomes was considered supportive. Moderate-certainty evidence showed that aflibercept 8 mg every 12 and 16 weeks likely results in little to no difference in important outcomes, such as the proportion of patients gaining 15 or more letters in BCVA and vision-related quality of life, when compared with aflibercept 2 mg every 8 weeks. The evidence from the PULSAR trial revealed (with low certainty) that the higher dose of aflibercept, if administered every 16 weeks, may reduce the frequency of injections compared to low-dose aflibercept, but the generalizability of these findings is limited as the number of injections was driven by trial protocol and is not aligned with clinical practice, in which the treat-and-extend approach is commonly used. Moderate-certainty evidence revealed aflibercept 8 mg every 12 weeks and every 16 weeks likely results in little to no difference in the risk of ocular SAEs when compared with aflibercept 2 mg every 8 weeks at 60 weeks. The safety profile of aflibercept 8 mg over 60 weeks showed similarities to that of aflibercept 2 mg in terms of ocular and nonocular TEAEs, deaths, and notable harms.
Comparative efficacy findings in the ITC are insufficient, as standalone evidence, to inform on the efficacy and safety of aflibercept 8 mg every 12 weeks and every 16 weeks versus other comparators. In general, between-group differences for efficacy outcomes (visual acuity) showed point estimates versus other relevant comparators (including PRN and treat-and-extend strategies) that were near the null, with wide credible intervals suggesting uncertainty about which treatment might be favoured. This is due to clinical variability among studies, indicating limited data strength. Noncomparative analysis of injection frequency suggests that aflibercept 8 mg every 12 weeks and every 16 weeks may have a smaller number of injections when numerically contrasted against other interventions with fixed frequency as well as treat-and-extend regimens; however, there is uncertainty in this finding due to a lack of comparative data and associated measures of variability.
References
1.de Jong PT. Age-related macular degeneration. N Engl J Med. 2006;355(14):1474-1485. PubMed
2.National Eye Institute. At a glance: AMD. 2021; https://www.nei.nih.gov/learn-about-eye-health/eye-conditions-and-diseases/age-related-macular-degeneration. Accessed 2023 Feb.
3.Flaxel CJ, Adelman RA, Bailey ST, et al. Age-Related Macular Degeneration Preferred Practice Pattern®. Ophthalmology. 2020;127(1):P1-P65. PubMed
4.Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106-116. PubMed
5.Yonekawa Y, Kim IK. Clinical characteristics and current treatment of age-related macular degeneration. Cold Spring Harb Perspect Med. 2014;5(1):a017178. PubMed
6.Flighting Blindness Canada. Age-related macular degeneration. 2022; https://www.fightingblindness.ca/eyehealth/eye-diseases/age-related-macular-degeneration/. Accessed by sponsor, no date provided.
7.The cost of vision loss and blindness in Canada: Canadian Council of the Blind. Sydney (Au): Deloitte Access Economics Pty Ltd; 2021: https://www.fightingblindness.ca/wp-content/uploads/2021/12/Deloitte-Cost-of-vision-loss-and-blindness-in-Canada-report-May-2021.pdf. Accessed by sponsor, no date provided.
8.Nowak JZ. Age-related macular degeneration (AMD): pathogenesis and therapy. Pharmacol Rep. 2006;58(3):353-363. PubMed
9.Hageman G, Gehrs K, Johnson LV, Anderson D. Age-related macular degeneration (AMD). In: Kolb H, Nelson R, Fernandez E, Jones B, eds. Webvision: the organization of the retina and visual system. Salt Lake City (UT): University of Utah Health Sciences Center; 2008: https://webvision.med.utah.edu/book/part-xii-cell-biology-of-retinaldegenerations/age-related-macular-degeneration-amd. Accessed 2023 Feb.
10.Lim JH, Wickremasinghe SS, Xie J, et al. Delay to treatment and visual outcomes in patients treated with anti-vascular endothelial growth factor for age-related macular degeneration. Am J Ophthalmol. 2012;153(4):678-686, 686.e671-672.
11.Mitchell P, Liew G, Gopinath B, Wong TY. Age-related macular degeneration. Lancet. 2018;392(10153):1147-1159. PubMed
12.Mathew RS, Delbaere K, Lord SR, Beaumont P, Vaegan, Madigan MC. Depressive symptoms and quality of life in people with age- related macular degeneration. Ophthalmic Physiol Opt. 2011;31(4):375-380. PubMed
13.Schmidt-Erfurth U, Chong V, Loewenstein A, et al. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA). Br J Ophthalmol. 2014;98(9):1144-1167. PubMed
14.Chandra S, McKibbin M, Mahmood S, et al. The Royal College of Ophthalmologists Commissioning guidelines on age macular degeneration: executive summary. Eye (Lond). 2022;36(11):2078-2083. PubMed
15.Eylea HD (aflibercept injection): 8 mg/0.07 mL, solution for intravitreal injection in single use vials for the treatment of a single eye [product monograph]. Mississauga (ON): Bayer Inc.; 2024 Feb 02.
16.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
17.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
18.Clinical Study Report: 20968 (Week 60). Randomized, double-masked, active-controlled, phase III study of the efficacy and safety of high dose aflibercept in patients with neovascular age-related macular degeneration [internal sponsor's report]. Leverkusen (DE): Bayer AG; 2023 Jan 11.
19.Owen CG, Jarrar Z, Wormald R, Cook DG, Fletcher AE, Rudnicka AR. The estimated prevalence and incidence of late stage age related macular degeneration in the UK. Br J Ophthalmol. 2012;96(5):752-756. PubMed
20.Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. Lancet. 2012;379(9827):1728-1738. PubMed
21.Liu X, Lin Z, Zhou T, et al. Anti-angiogenic and anti-inflammatory effects of SERPINA3K on corneal injury. PLoS One. 2011;6(1):e16712. PubMed
22.Brown DM. Age-related macular degeneration: underlying causes and treatment. Newark (NJ): Medscape; 2005: https://www.medscape.org/viewarticle/506716_3. Accessed 2023 March.
23.Ishikawa M, Jin D, Sawada Y, Abe S, Yoshitomi T. Future therapies of wet age-related macular degeneration. J Ophthalmol. 2015;2015:138070. PubMed
24.Li JQ, Welchowski T, Schmid M, Mauschitz MM, Holz FG, Finger RP. Prevalence and incidence of age-related macular degeneration in Europe: a systematic review and meta-analysis. Br J Ophthalmol. 2020;104(8):1077-1084. PubMed
25.Trichonas G, Kaiser PK. Optical coherence tomography imaging of macular oedema. Br J Ophthalmol. 2014;98 Suppl 2(Suppl 2):ii24-29.
26.Bennett TJ, Barry CJ. Ophthalmic imaging today: an ophthalmic photographer's viewpoint - a review. Clin Exp Ophthalmol. 2009;37(1):2-13. PubMed
27.Adams BS, Sorhaitz W, Stringham J. Aflibercept. StatPearls. Treasure Island (FL): StatPearls Publishing; 2023: https://www.ncbi.nlm.nih.gov/books/NBK582136/. Accessed by sponsor, no date provided.
28.Holash J, Davis S, Papadopoulos N, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A. 2002;99(17):11393-11398. PubMed
29.Brown DM. Preclinical pharmacology data support longer treatment intervals with increased aflibercept dose. Virtual Meeting: 19th Angiogenesis, Exudation, and Degeneration, 2022 Annual Meeting; 2022 Feb 11-12.
30.Drug Reimbursement Expert Review Committee final recommendation: aflibercept (Eylea — Bayer Inc.). Ottawa (ON): CADTH; 2014 Oct 20: https://www.cadth.ca/sites/default/files/cdr/complete/cdr_complete_SR0361−000_eylea_october_22_2014.pdf. Accessed 2024 May 28.
31.Vabysmo (faricimab injection): 6mg/0.05mL solution for intravitrial injection, in single use vials [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2022 May 27.
32.Lucentis (ranibizumab injection): 10 mg/mL solution for injection, in single use vials or single use pre-filled syringes [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2021 Dec 21: https://pdf.hres.ca/dpd_pm/00064117.PDF. Accessed 2022 Apr 26.
33.Avastin (bevacizumab for injection): 100 mg and 400 mg vials (25 mg/mL) solution for injection [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Jan 14: https://pdf.hres.ca/dpd_pm/00059621.PDF. Accessed 2023 Sep 11.
34.Ferris FL, 3rd, Kassoff A, Bresnick GH, Bailey I. New visual acuity charts for clinical research. Am J Ophthalmol. 1982;94(1):91-96. PubMed
35.Neovascular age-related macular degeneration: developing drugs for treatment: guidance for industry: draft guidance. 2023; https://www.fda.gov/media/165606/download. Accessed by sponsor, no date provided.
36.Riedl S, Vogl WD, Waldstein SM, Schmidt-Erfurth U, Bogunovic H. Impact of intra- and subretinal fluid on vision based on volume quantification in the HARBOR trial. Ophthalmol Retina. 2022;6(4):291-297. PubMed
37.Dougherty BE, Bullimore MA. Comparison of scoring approaches for the NEI VFQ-25 in low vision. Optom Vis Sci. 2010;87(8):543-548. PubMed
38.Mangione CM, Lee PP, Gutierrez PR, Spritzer K, Berry S, Hays RD. Development of the 25-item National Eye Institute Visual Function Questionnaire. Arch Ophthalmol. 2001;119(7):1050-1058. PubMed
39.Suner IJ, Kokame GT, Yu E, Ward J, Dolan C, Bressler NM. Responsiveness of NEI VFQ-25 to changes in visual acuity in neovascular AMD: validation studies from two phase III clinical trials. Invest Ophthalmol Vis Sci. 2009;50(8):3629-3635. PubMed
40.Mangione CM, Berry S, Spritzer K, et al. Identifying the content area for the 51-item National Eye Institute Visual Function Questionnaire: results from focus groups with visually impaired persons. Arch Ophthalmol. 1998;116(2):227-233. PubMed
41.Revicki DA, Rentz AM, Harnam N, Thomas VS, Lanzetta P. Reliability and validity of the National Eye Institute Visual Function Questionnaire-25 in patients with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51(2):712-717. PubMed
42.Orr P, Rentz AM, Margolis MK, et al. Validation of the National Eye Institute Visual Function Questionnaire-25 (NEI VFQ-25) in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(6):3354-3359. PubMed
43.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
44.Pesudovs K, Gothwal VK, Wright T, Lamoureux EL. Remediating serious flaws in the National Eye Institute Visual Function Questionnaire. J Cataract Refract Surg. 2010;36(5):718-732. PubMed
45.Wilkinson CP, Ferris FL, 3rd, Klein RE, et al. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology. 2003;110(9):1677-1682. PubMed
46.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centreed outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
47.Sánchez-González MC, García-Oliver R, Sánchez-González JM, Bautista-Llamas MJ, Jiménez-Rejano JJ, De-Hita-Cantalejo C. Minimum detectable change of visual acuity measurements using ETDRS charts (Early Treatment Diabetic Retinopathy Study). Int J Environ Res Public Health. 2021;18(15):7876. PubMed
48.Beck RW, Moke PS, Turpin AH, et al. A computerized method of visual acuity testing: adaptation of the early treatment of diabetic retinopathy study testing protocol. Am J Ophthalmol. 2003;135(2):194-205. PubMed
49.Joussen AM, Lehmacher W, Hilgers RD, Kirchhof B. Is significant relevant? Validity and patient benefit of randomized controlled clinical trials on age-related macular degeneration. Surv Ophthalmol. 2007;52(3):266-278. PubMed
50.Ho AC, Busbee BG, Regillo CD, et al. Twenty-four-month efficacy and safety of 0.5 mg or 2.0 mg ranibizumab in patients with subfoveal neovascular age-related macular degeneration. Ophthalmology. 2014;121(11):2181-2192. PubMed
51.Dugel PU, Koh A, Ogura Y, et al. HAWK and HARRIER: Phase III, Multicentre, Randomized, Double-Masked Trials of Brolucizumab for Neovascular Age-Related Macular Degeneration. Ophthalmology. 2020;127(1):72-84. PubMed
52.Drug Reimbursement Review clinical report, pharmacoeconomic report, and stakeholder input: Faricimab (Vabysmo) for age-related macular degeneration. Can J Health Technol. 2022;2(11). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/SR0719r/SR0719r. Accessed by sponsor, no date provided.
53.Aflibercept 8 mg / 0.07 mL for the treatment of neovascular (wet) age-related macular degeneration (nAMD) in Canada NMA [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Eylea HD (aflibercept injection), 8 mg / 0.07 mL for intravitreal injection. Mississauga (ON): Bayer Inc.; 2023 Aug 18. 2023.
54.Bayer. PULSAR clinical trial: Data on file. 2023 [sponsor provided reference].
55.Regeneron. Two-year PULSAR trial results for aflibercept 8 mg demonstrate durable vision gains at extended dosing intervals in wet age-related macular degeneration. 2023; https://investor.regeneron.com/news-releases/news-release-details/two-year-pulsar-trial-results-aflibercept-8-mg-demonstrate. Accessed by sponsor, no date provided.
56.Heier JS, Khanani AM, Quezada Ruiz C, et al. Efficacy, durability, and safety of intravitreal faricimab up to every 16 weeks for neovascular age-related macular degeneration (TENAYA and LUCERNE): two randomised, double-masked, phase III, noninferiority trials. Lancet. 2022;399(10326):729-740. PubMed
57.Mitchell P, Holz FG, Hykin P, et al. Efficacy and safety of intravitreal aflibercept using a treat-and-extend regimen for neovascular age-related macular degeneration: the ARIES study: a randomized clinical trial. Retina. 2021;41(9):1911-1920. PubMed
58.Ohji M, Takahashi K, Okada AA, Kobayashi M, Matsuda Y, Terano Y. Efficacy and safety of intravitreal aflibercept treat-and-extend regimens in exudative age-related macular degeneration: 52- and 96-week findings from ALTAIR: a randomized controlled trial. Adv Ther. 2020;37(3):1173-1187. PubMed
59.Haga A, Kawaji T, Ideta R, Inomata Y, Tanihara H. Treat-and-extend versus every-other-month regimens with aflibercept in age-related macular degeneration. Acta Ophthalmol. 2018;96(3):e393-e398. PubMed
60.Mori R, Tanaka K, Haruyama M, Kawamura A, Furuya K, Yuzawa M. Comparison of pro re nata versus bimonthly injection of intravitreal aflibercept for typical neovascular age-related macular degeneration. Ophthalmologica. 2017;238(1-2):17-22. PubMed
61.Heier JS, Brown DM, Chong V, et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology. 2012;119(12):2537-2548. PubMed
62.Dugel PU, Jaffe GJ, Sallstig P, et al. Brolucizumab versus aflibercept in participants with neovascular age-related macular degeneration: a randomized trial. Ophthalmology. 2017;124(9):1296-1304. PubMed
63.Berg K, Pedersen TR, Sandvik L, Bragadóttir R. Comparison of ranibizumab and bevacizumab for neovascular age-related macular degeneration according to LUCAS treat-and-extend protocol. Ophthalmology. 2015;122(1):146-152. PubMed
64.Biswas P, Sengupta S, Choudhary R, Home S, Paul A, Sinha S. Comparing ranibizumab with bevacizumab. Ophthalmology. 2011;118(3):600-600.e602. PubMed
65.Busbee BG, Ho AC, Brown DM, et al. Twelve-month efficacy and safety of 0.5 mg or 2.0 mg ranibizumab in patients with subfoveal neovascular age-related macular degeneration. Ophthalmology. 2013;120(5):1046-1056. PubMed
66.Chakravarthy U, Harding SP, Rogers CA, et al. Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: one-year findings from the IVAN randomized trial. Ophthalmology. 2012;119(7):1399-1411. PubMed
67.Feltgen N, Bertelmann T, Bretag M, et al. Efficacy and safety of a fixed bimonthly ranibizumab treatment regimen in eyes with neovascular age-related macular degeneration: results from the RABIMO trial. Graefes Arch Clin Exp Ophthalmol. 2017;255(5):923-934. PubMed
68.Gillies MC, Hunyor AP, Arnold JJ, et al. Macular atrophy in neovascular age-related macular degeneration: a randomized clinical trial comparing ranibizumab and aflibercept (RIVAL study). Ophthalmology. 2020;127(2):198-210. PubMed
69.Kertes PJ, Galic IJ, Greve M, et al. Efficacy of a treat-and-extend regimen with ranibizumab in patients with neovascular age-related macular disease: a randomized clinical trial. JAMA Ophthalmol. 2020;138(3):244-250. PubMed
70.Khanani AM, Patel SS, Ferrone PJ, et al. Efficacy of every four monthly and quarterly dosing of faricimab vs ranibizumab in neovascular age-related macular degeneration: the STAIRWAY phase 2 randomized clinical trial. JAMA Ophthalmol. 2020;138(9):964-972. PubMed
71.Kodjikian L, Souied EH, Mimoun G, et al. Ranibizumab versus bevacizumab for neovascular age-related macular degeneration: results from the GEFAL noninferiority randomized trial. Ophthalmology. 2013;120(11):2300-2309. PubMed
72.Krebs I, Schmetterer L, Boltz A, et al. A randomised double-masked trial comparing the visual outcome after treatment with ranibizumab or bevacizumab in patients with neovascular age-related macular degeneration. Br J Ophthalmol. 2013;97(3):266-271. PubMed
73.Li X, Zhu Q, Egger A, et al. Two different treatment regimens of ranibizumab 0.5 mg for neovascular age-related macular degeneration with or without polypoidal choroidal vasculopathy in Chinese patients: results from the Phase IV, randomized, DRAGON study. Acta Ophthalmol. 2021;99(3):e336-e345. PubMed
74.López Gálvez MI, Arias Barquet L, M SF, García-Layana A, Ruiz Moreno JM. Bimonthly, treat-and-extend and as-needed ranibizumab in naive neovascular age-related macular degeneration patients: 12-month outcomes of a randomized study. Acta Ophthalmol. 2020;98(7):e820-e829. PubMed
75.CATT Research Group, Martin DF, Maguire MG, et al. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364(20):1897-1908. PubMed
76.Nunes RP, Hirai FE, Barroso LF, et al. Effectiveness of monthly and fortnightly anti-VEGF treatments for age-related macular degeneration. Arq Bras Oftalmol. 2019;82(3):225-232. PubMed
77.Scholler A, Richter-Mueksch S, Weingessel B, Vécsei-Marlovits PV. Differences of frequency in administration of ranibizumab and bevacizumab in patients with neovascular AMD. Wien Klin Wochenschr. 2014;126(11-12):355-359. PubMed
78.Silva R, Berta A, Larsen M, et al. Treat-and-Extend versus monthly regimen in neovascular age-related macular degeneration: results with ranibizumab from the TREND study. Ophthalmology. 2018;125(1):57-65. PubMed
79.Wang F, Yuan Y, Wang L, et al. One-year outcomes of 1 dose versus 3 loading doses followed by pro re nata regimen using ranibizumab for neovascular age-related macular degeneration: the ARTIS trial. J Ophthalmol. 2019;2019:7530458. PubMed
80.Wykoff CC, Ou WC, Brown DM, et al. Randomized trial of treat-and-extend versus monthly dosing for neovascular age-related macular degeneration: 2-year results of the TREX-AMD study. Ophthalmol Retina. 2017;1(4):314-321. PubMed
81.Barikian A, Mahfoud Z, Abdulaal M, Safar A, Bashshur ZF. Induction with intravitreal bevacizumab every two weeks in the management of neovascular age-related macular degeneration. Am J Ophthalmol. 2015;159(1):131-137. PubMed
82.El-Mollayess GM, Mahfoud Z, Schakal AR, Salti HI, Jaafar D, Bashshur ZF. Fixed-interval versus OCT-guided variable dosing of intravitreal bevacizumab in the management of neovascular age-related macular degeneration: a 12-month randomized prospective study. Am J Ophthalmol. 2012;153(3):481-489.e481. PubMed
83.Li X, Hu Y, Sun X, Zhang J, Zhang M. Bevacizumab for neovascular age-related macular degeneration in China. Ophthalmology. 2012;119(10):2087-2093. PubMed
84.Lushchyk T, Amarakoon S, Martinez-Ciriano JP, van den Born LI, Baarsma GS, Missotten T. Bevacizumab in age-related macular degeneration: a randomized controlled trial on the effect of injections every 4 weeks, 6 weeks and 8 weeks. Acta Ophthalmol. 2013;91(6):e456-461. PubMed
Pharmacoeconomic_Review
Abbreviations
AE
adverse event
AMD
age-related macular degeneration
BCVA
best corrected visual acuity
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
ITC
indirect treatment comparison
nAMD
neovascular (wet) age-related macular degeneration
NMA
network meta-analysis
QALY
quality-adjusted life-year
VEGF
vascular endothelial growth factor
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Aflibercept 8 mg (Eylea HD), solution for intravitreal injection |
Submitted price | Aflibercept 8 mg, 30 mg per 0.263 mL, single-use vial: $1,250.00 |
Indication | For the treatment of neovascular (wet) age-related macular degeneration |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | February 2, 2024 |
Reimbursement request | As per indication |
Sponsor | Bayer Inc. |
Submission history | Previously reviewed: in progress Indication: diabetic macular edema Recommendation: to be determined |
HD = high dose; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults with nAMD |
Treatment | Aflibercept 8 mg, administered every 16 weeks (q.16.w.)a |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (25 years) |
Key data sources |
|
Submitted results | The ICER for aflibercept 8 mg q.16.w. vs. bevacizumab = $51,463 per QALY gained (incremental costs: $9,515; incremental QALYs: 0.18). |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; BCVA = best corrected visual acuity; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; nAMD = neovascular age-related macular degeneration; QALY = quality-adjusted life-year; q.16.w. = every 16 weeks; VEGF = vascular endothelial growth factor; vs. = versus.
aIn the sponsor’s base case, aflibercept 8 mg was assumed to be administered every 16 weeks. Administration of aflibercept 8 mg every 12 weeks was considered in scenario analysis.
Conclusions
Based on the Canada’s Drug Agency (CDA-AMC) Clinical Review of the PULSAR trial, the available evidence suggests that aflibercept 8 mg is noninferior, but not superior, to aflibercept 2 mg for mean change in best corrected visual acuity (BCVA). Results of the sponsor’s network meta-analysis (NMA) suggest that aflibercept 8 mg is associated with similar changes in BCVA compared to other currently available inhibitors of vascular endothelial growth factor (VEGF) (i.e., aflibercept 2 mg, bevacizumab, brolucizumab, faricimab, ranibizumab). However, the CDA-AMC Clinical Review concluded that the comparative efficacy findings from the sponsor-submitted indirect treatment comparison (ITC) are insufficient as standalone evidence to inform decision-making about the efficacy and safety of aflibercept 8 mg versus other anti-VEGF drugs.
Noncomparative results submitted by the sponsor suggest that the number of annual injections of aflibercept 8 mg may be similar to the number of injections for comparator regimens that use a treat-and-extend or “as-needed” approach.
Given uncertainty in the clinical evidence, there is insufficient evidence to suggest that aflibercept 8 mg should be priced higher than other anti-VEGF treatments for nAMD.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process.
CDA-AMC received patient input from the Canadian Council of the Blind, Fighting Blindness Canada, Vision Loss Rehabilitation Canada, and the International Federation on Ageing that was collected via an online survey of people in Canada with age-related macular degeneration (AMD), 47% of whom reported having neovascular (wet) age-related macular degeneration (nAMD). Patients reported that AMD has a major impact on their daily lives, including physical, psychological, and social impacts. Respondents indicated that vision loss from AMD affects their daily activities, including personal care and hygiene and leisure. Respondents indicated that they were receiving injections to treat AMD including bevacizumab, ranibizumab, aflibercept 2 mg, and dexamethasone (faricimab and aflibercept 8 mg were not available at the time of the survey). Patients described injections as being at least somewhat painful and that pain and blurry vision may occur after injection. Prior patient engagement efforts by these groups found that most patients would prefer a treatment or medication that could be taken less frequently. Additional surveys conducted by the Canadian Council of the Blind in 2020 and 2022 found that many patients experienced increased difficulty attending appointments during the COVID-19 pandemic.
Clinician input was received from 6 groups: Southwestern Ontario Community Ophthalmologists, the Northeastern Ontario Ophthalmology Group, the Canadian Retina Society, the Retina Division of the Ottawa Hospital, Toronto Ophthalmologists, and the Toronto Retina Institute. Clinician input noted that current treatment for nAMD consists of intravitreal injections of drugs that inhibit VEGF and that aflibercept 8 mg is likely to result in high uptake due to its longer treatment interval, replacing aflibercept 2 mg as the standard first-line treatment for nAMD.
Drug plans participating in the CDA-AMC review noted that there have been no trials comparing aflibercept 8 mg with anti-VEGF therapies that can be administered at the same extended dosing interval (i.e., faricimab and brolucizumab). Given the extended dosing intervals of faricimab and brolucizumab, drug plans questioned what unmet need would be addressed by aflibercept 8 mg. Drug plan input noted that biosimilars are available for ranibizumab and are anticipated for aflibercept 2 mg within the next year, and that this would be expected to influence the budget impact of aflibercept 8 mg. Finally, the plans noted the presence of confidentially negotiated prices for comparators.
The potential extended dosing interval for aflibercept 8 mg was described as being particularly important by patients, clinicians, and drug plans; however, the comparative clinical efficacy and frequency of dosing are areas of uncertainty in the sponsor’s submitted evidence and CDA-AMC was unable to address these concerns.
Economic Review
The current review is for aflibercept 8 mg (Eylea HD) for patients with nAMD.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of aflibercept 8 mg compared to other VEGF inhibitors, including aflibercept 2 mg, brolucizumab, faricimab, ranibizumab (a biosimilar), and bevacizumab, in patients with nAMD.1 The modelled population is aligned with the draft Health Canada indication and patients enrolled in the PULSAR trial.
Aflibercept 8 mg is supplied in single-use vials containing 30 mg of aflibercept in a 0.263 mL solution (114 mg/mL). The recommended dosage is 8 mg administered by intravitreal injection every 4 weeks (monthly) for the first 3 doses, followed by 8 mg at a dosing interval of every 8 to 16 weeks (4 months), depending on the physician’s judgment of the individual patient’s visual and anatomic outcomes.2 Monitoring between dosing visits should be based on the patient’s status and at the physician’s discretion. The sponsor’s submitted price for aflibercept 8 mg is $1,250.00 per vial, which corresponds to an annual per-patient cost of $7,625 in the first year ($4,500 in subsequent years) if administered every 12 weeks (based on 6.1 injections and 3.6 injections in the first and subsequent years, respectively) and an annual per-patient cost of $6,500 in the first year ($3,750 in subsequent years) if administered every 16 weeks (based on 5.2 and 3.0 injections in the first and subsequent years, respectively).1
In the model, the sponsor assumed that aflibercept 8 mg would be administered every 16 weeks, aflibercept 2 mg and ranibizumab would be administered using a treat-and-extend approach, faricimab would be administered at intervals extending from 8 to 12 and 16 weeks, brolucizumab would be administered every 8 or 12 weeks, and bevacizumab would be administered “as needed.”1 The first-year per-patient costs for comparators estimated by the sponsor ranged from $4,246 (bevacizumab) to $11,131 (aflibercept 2 mg), while the annual per-patient costs in subsequent years ranged from $3,011 (bevacizumab) to $8,328 (ranibizumab).1
The clinical outcomes were life-years and quality-adjusted life-years (QALYs), estimated over a lifetime time horizon (25 years; 4-week cycle length) from the perspective of Canada’s publicly funded health care system. Costs and QALYs were discounted at a rate of 1.5% per annum, and a half-cycle correction was applied.
Model Structure
The sponsor submitted a Markov model that consisted of 65 health states: 64 states based on visual acuity (defined by Early Treatment for Diabetic Retinopathy Study letter score) and death. Gains in visual acuity in the first year of treatment (informed by change from baseline in BCVA from the sponsor’s NMA) were assumed to be maintained for the duration of treatment. Patients could have nAMD in 1 or both eyes, with a different visual acuity in each eye accounted for.
Model Inputs
The baseline characteristics and initial distribution of patients across visual acuity health states in the model were based on the PULSAR trial, which randomized patients with nAMD (mean age of 74.5 years, 54.4% female) to receive aflibercept 8 mg every 12 weeks, aflibercept 8 mg every 16 weeks, or aflibercept 2 mg every 8 weeks.3 Movement between visual acuity health states in the model was based on the probability that a patient would gain or lose 10 or 15 letters, which was derived using odds ratios for BCVA from the sponsor’s NMA. Severe adverse events (AEs) were also informed by the sponsor’s NMA. The number of injections per year was informed by naive (visual) comparisons across pairwise meta-analyses provided by the sponsor. Discontinuation rates were based on observations from the PULSAR trial (aflibercept 8 mg)3 and from the literature (for comparators).4-12 The probability of death was based on age-specific Canadian background mortality,13 to which the sponsor applied a hazard ratio of 1.36 to account for higher mortality among nAMD patients who are blind in 1 or both eyes.14
Utility values for each visual acuity state were estimated using regression coefficients from a published study that simulated visual impairment in healthy volunteers from the UK.15 Disutilities associated with AEs and the duration of AEs were obtained from the literature.16,17 The sponsor assumed that half of patients would experience zero utility on an injection day.18
The economic model included costs related to drugs (acquisition and administration), monitoring, AEs, and blindness. Treatment costs were estimated by using the drug cost for a single injection, the estimated number of injections per year, and the administration cost per injection. The sponsor assumed that all vials were single-use and that any unused product would be wasted. Costs were derived from Ontario’s Schedule of Benefits,19 the Ontario Case Costing Initiative,20 and the literature.21-23
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following section. The sponsor’s base-case analysis assumed that aflibercept 8 mg will be administered every 16 weeks; administration every 12 weeks was considered in scenario analysis.
Base-Case Results
In the sponsor’s base case, aflibercept 8 mg every 16 weeks was associated with an estimated cost of $53,358 and 8.28 QALYs over a 25-year horizon (Table 3). In sequential analysis, aflibercept 8 mg every 16 weeks was associated with an incremental cost-effectiveness ratio (ICER) of $51,463 versus bevacizumab ($9,515 in incremental costs: 0.18 QALYs), with a 45% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
Results were driven by increased drug acquisition costs associated with aflibercept 8 mg every 16 weeks (incremental costs of $9,515 and 0.18 QALYs) compared with bevacizumab. The sponsor’s model estimated 0.002 incremental QALYs with aflibercept 8 mg every 16 weeks treatment compared with bevacizumab in the first year of treatment (the approximate duration of the PULSAR trial), indicating that approximately 99% of the incremental benefit are accrued during the extrapolated portion of the model. At the end of the 25-year time horizon, the percentage of patients estimated to remain alive was approximately 4% for all treatments.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total life-years | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|---|
Bevacizumab | 43,844a | 13.979 | 8.097 | Reference |
Aflibercept 8 mg q.16.w. | 53,358 | 14.035 | 8.282 | 51,463 vs. bevacizumab |
Faricimab | 94,634 | 14.067 | 8.402 | 344,555 vs. aflibercept 8 mg q.16.w. |
Dominated treatments | ||||
Brolucizumab | 84,308 | 14.033 | 8.277 | Dominated by aflibercept 8 mg q.16.w. |
Aflibercept 2 mg | 85,387 | 13.999 | 8.159 | Dominated by aflibercept 8 mg q.16.w. |
Ranibizumab | 103,444 | 14.046 | 8.328 | Dominated by faricimab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; q.16.w. = every 16 weeks; vs. = versus.
aSponsor assumed that the cost of bevacizumab was $519.18 per vial based on the cost of branded bevacizumab (Avastin) and that 1 dose would be obtained per vial. The total cost of bevacizumab would be lower if generic bevacizumab is used and/or if vial sharing occurs in clinical practice.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted a sequential scenario analysis in which aflibercept 8 mg was assumed to be administered every 12 weeks. In this scenario, aflibercept 8 mg was associated with an estimated cost of $72,898 and 8.36 QALYs over a 25-year horizon, with a sequential ICER of $111,466 versus bevacizumab (incremental costs: $29,054; incremental QALYs: 0.261).
The sponsor conducted several scenario analyses including multiple dosing assumptions for bevacizumab and ranibizumab, discontinuation assumptions, and treatment efficacy assumptions; however, sequential analyses were not provided (i.e., aflibercept 8 mg every 16 weeks was compared to each other treatment in a pairwise fashion), limiting the interpretation of the findings.
The sponsor conducted a scenario analysis from a societal perspective. This analysis included additional costs associated with productivity loss of caregivers; however, sequential analyses were not provided, limiting the interpretation of the findings.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative clinical efficacy and safety of aflibercept 8 mg versus other anti-VEGFs are uncertain. There is a lack of head-to-head evidence comparing aflibercept 8 mg to anti-VEGF drugs other than aflibercept 2 mg. The results of the PULSAR trial suggest that aflibercept 8 mg is noninferior to aflibercept 2 mg for improvement in visual acuity (i.e., change from baseline in BCVA). In the absence of head-to-head evidence for most comparators, the sponsor conducted NMAs to inform various parameters in the economic model for all anti-VEGF agents, including BCVA and severe AEs. As noted in the CDA-AMC Clinical Review, results of the sponsor’s NMA suggests that there may be no meaningful differences in BCVA between aflibercept 8 mg and relevant comparators. However, the CDA-AMC Clinical Review concluded that the comparative findings of the sponsor’s NMAs were insufficient as standalone evidence to inform on the efficacy and safety of aflibercept versus comparators due to the presence of substantial imprecision and unresolved heterogeneity.
Given the lack of direct evidence for aflibercept 8 mg relative to anti-VEGF drugs other than aflibercept 2 mg and limitations with the sponsor’s NMA, it is uncertain whether aflibercept 8 mg provides a net benefit above any of the currently available treatments for nAMD.
The relative frequency of anti-VEGF injections is uncertain. In the pharmacoeconomic model, the number of annual injections (and consequently drug acquisition and administration costs) for each anti-VEGF drug was informed by sponsor-submitted ITCs. As noted in the CDA-AMC Clinical Review, the results of the submitted naive (visual) comparisons across pairwise meta-analyses suggest that aflibercept 8 mg may be associated with numerically fewer injections in year 1 and year 2 when compared to treatments administered on a set injection schedule (e.g., every 4, 8, or 12 weeks). However, clinical expert feedback obtained by CDA-AMC for this review indicated that, in clinical practice, anti-VEGF therapies are typically administered using a treat-and-extend approach, not a set injection frequency. A visual comparison of the sponsor’s results suggests there may be a similar number of injections in year 1 or year 2 with aflibercept 8 mg when compared to anti-VEGF treatments that followed an as-needed (pro re nata) or treat-and-extend strategy. However, the CDA-AMC Clinical Review noted that there is uncertainty in this finding due to a lack of comparative data and associated measures of variability across treatments.
Whether aflibercept 8 mg will result in fewer injections in clinical practice compared with other anti-VEGFs is uncertain due to limitations with the sponsor’s submitted evidence for administration frequency and the individualized approach to administration frequency in clinical practice.
Issues for Consideration
Biosimilars for aflibercept are currently under review by Health Canada. The introduction of such biosimilars may affect the cost-effectiveness of aflibercept 8 mg versus aflibercept 2 mg depending on the list price.
The sponsor’s analyses rely on publicly accessible list prices and do not reflect existing confidential prices negotiated by public plans. Given that aflibercept 2 mg,24 faricimab,25 ranibizumab,26,27 and brolucizumab28 have successfully undergone price negotiations for the treatment of nAMD, it is likely that the current unit costs paid by public drug plans for these treatments are lower than the submitted prices.
Overall Conclusions
Based on the CDA-AMC Clinical Review, data from the PULSAR trial suggest that aflibercept 8 mg is noninferior, but not superior, to aflibercept 2 mg with respect to mean change in BCVA. Evidence from the PULSAR trial suggests that aflibercept 8 mg may result in little to no difference in the frequency of injections compared to aflibercept 2 mg; however, the number of injections was driven by the trial protocol and may not be aligned with clinical practice when a treat-and-extend approach is adopted. Results of the sponsor’s NMA suggest that aflibercept 8 mg is associated with similar changes in BCVA compared to other currently available anti-VEGFs (i.e., aflibercept 2 mg, bevacizumab, brolucizumab, faricimab, and ranibizumab). However, the CDA-AMC Clinical Review concluded that the comparative findings from the sponsor-submitted ITC are insufficient as standalone evidence to inform decision-making about the efficacy and safety of aflibercept 8 mg versus other anti-VEGF drugs due to clinical heterogeneity and imprecision. Noncomparative results of injection frequency submitted by the sponsor suggest that the number of annual injections may be similar among anti-VEGFs administered using a treat-and-extend or pro re nata (as-needed) approach. However, there is uncertainty in this finding due to a lack of comparative data and associated measures of variability across treatments.
Given that the indirect evidence submitted by the sponsor suggests that there may be no difference between aflibercept 8 mg and currently available anti-VEGFs in terms of improvements in VA or number of annual injections, there is insufficient evidence to suggest that aflibercept 8 mg should be priced higher than other anti-VEGF treatments for nAMD. To ensure cost-effectiveness, aflibercept 8 mg should be priced no more than the lowest cost anti-VEGF that is funded for the treatment of nAMD.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Eylea HD (aflibercept injection), 8 mg / 0.07 mL for intravitreal injection. Mississauga (ON): Bayer Inc.; 2023 Aug 18.
2.Eylea HD (aflibercept injection): 8 mg / 0.07 mL, solution for intravitreal injection in single use vials for the treatment of a single eye [product monograph]. Mississauga (ON): Bayer Inc.; 2024 Feb 02.
3.Clinical Study Report: 20968 (Week 60), PULSAR. Randomized, double-masked, active-controlled, phase 3 study of the efficacy and safety of high dose aflibercept in patients with neovascular age-related macular degeneration [internal sponsor's report]. Leverkusen (DE): Bayer AG; 2023 Jan 11.
4.Ferro Desideri L, Traverso CE, Nicolò M, Munk MR. Faricimab for the treatment of diabetic macular edema and neovascular age-related macular degeneration. Pharmaceutics. 2023;15(5):1413. PubMed
5.Kertes PJ, Galic IJ, Greve M, et al. Canadian treat-and-extend analysis trial with ranibizumab in patients with neovascular age-related macular disease: one-year results of the randomized Canadian treat-and-extend analysis trial with ranibizumab study. Ophthalmology. 2019;126(6):841-848. PubMed
6.Silva R, Berta A, Larsen M, et al. Treat-and-extend versus monthly regimen in neovascular age-related macular degeneration: results with ranibizumab from the TREND study. Ophthalmology. 2018;125(1):57-65. PubMed
7.Berg K, Pedersen TR, Sandvik L, Bragadottir R. Comparison of ranibizumab and bevacizumab for neovascular age-related macular degeneration according to LUCAS treat-and-extend protocol. Ophthalmology. 2015;122(1):146-152. PubMed
8.Gillies MC, Hunyor AP, Arnold JJ, et al. Effect of ranibizumab and aflibercept on best-corrected visual acuity in treat-and-extend for neovascular age-related macular degeneration: a randomized clinical trial. JAMA Ophthalmol. 2019;137(4):372-379. PubMed
9.Kodjikian L, Souied EH, Mimoun G, et al. Ranibizumab versus bevacizumab for neovascular age-related macular degeneration: results from the GEFAL noninferiority randomized trial. Ophthalmology. 2013;120(11):2300-2309. PubMed
10.Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364(20):1897-1908. PubMed
11.Dugel PU, Jaffe GJ, Sallstig P, et al. Brolucizumab versus aflibercept in participants with neovascular age-related macular degeneration: a randomized trial. Ophthalmology. 2017;124(9):1296-1304. PubMed
12.Dugel PU, Koh A, Ogura Y, et al. HAWK and HARRIER: phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology. 2020;127(1):72-84. PubMed
13.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed by sponsor, no date provided.
14.Zhang T, Jiang W, Song X, Zhang D. The association between visual impairment and the risk of mortality: a meta-analysis of prospective studies. J Epidemiol Community Health. 2016;70(8):836-842. PubMed
15.Czoski-Murray C, Carlton J, Brazier J, Young T, Papo NL, Kang HK. Valuing condition-specific health states using simulation contact lenses. Value Health. 2009;12(5):793-799. PubMed
16.Ziemssen F, Hammer T, Grueb M, et al. Reporting of safety events during anti-VEGF Treatment: pharmacovigilance in a noninterventional trial. J Ophthalmol. 2020;2020:8652370. PubMed
17.Brown GC, Brown MM, Brown HC, Kindermann S, Sharma S. A value-based medicine comparison of interventions for subfoveal neovascular macular degeneration. Ophthalmology. 2007;114(6):1170-1178. PubMed
18.National Institute for Health Care Excellence. Age-related macular degeneration. (NICE guideline NG82) 2018; https://www.nice.org.uk/guidance/ng82/resources/agerelated-macular-degeneration-pdf-1837691334853. Accessed by sponsor, no date provided.
19.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 23, 2023)). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed by sponsor, no date provided.
20.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed by sponsor, no date provided.
21.Iskedjian M, Walker J, Vicente C, et al. Cost of glaucoma in Canada: analyses based on visual field and physician's assessment. J Glaucoma. 2003;12(6):456-462. PubMed
22.Cruess AF, Gordon KD, Bellan L, Mitchell S, Pezzullo ML. The cost of vision loss in Canada. 2. Results. Can J Ophthalmol. 2011;46(4):315-318. PubMed
23.Canadian Institute for Health Information. The cost of hospital stays: why costs vary. Ottawa (ON): CIHI; 2008: https://secure.cihi.ca/free_products/2008hospcosts_report_e.pdf. Accessed by sponsor, no date provided.
24.pan-Canadian Pharmaceutical Alliance. Eylea (aflibercept). 2015; https://www.pcpacanada.ca/negotiation/20741. Accessed 2023 Sep 25.
25.pan-Canadian Pharmaceutical Alliance. Vabysmo (faricimab). 2023; https://www.pcpacanada.ca/negotiation/21977. Accessed 2023 Sep 25.
26.pan-Canadian Pharmaceutical Alliance. Ranopto (ranibuzumab). 2023; https://www.pcpacanada.ca/negotiation/22329. Accessed 2023 Sep 25.
27.pan-Canadian Pharmaceutical Alliance. Byooviz (ranibizumab). 2023; https://www.pcpacanada.ca/negotiation/22008. Accessed 2023 Sep 25.
28.pan-Canadian Pharmaceutical Alliance. Beovu (brolucizumab). 2021; https://www.pcpacanada.ca/negotiation/21223. Accessed 2023 Sep 25.
29.DeltaPA. Ottawa (ON): IQVIA; 2023: https://www.iqvia.com/. Accessed 2023 Nov.
30.Drug Reimbursement Review clinical report, pharmacoeconomic report, and stakeholder input: Faricimab (Vabysmo) for age-related macular degeneration. Can J Health Technol. 2022;2(11). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/SR0719r/SR0719r. Accessed by sponsor, no date provided.
31.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Eylea HD (aflibercept injection), 8 mg / 0.07 mL for intravitreal injection. Mississauga (ON): Bayer Inc.; 2023 Aug 18.
32.Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106-116. PubMed
33.Li JQ, Welchowski T, Schmid M, Mauschitz MM, Holz FG, Finger RP. Prevalence and incidence of age-related macular degeneration in Europe: a systematic review and meta-analysis. Br J Ophthalmol. 2020;104(8):1077-1084. PubMed
34.Yonekawa Y, Kim IK. Clinical characteristics and current treatment of age-related macular degeneration. Cold Spring Harb Perspect Med. 2014;5(1):a017178. PubMed
35.Mann SS, Rutishauser-Arnold Y, Peto T, et al. The symmetry of phenotype between eyes of patients with early and late bilateral age-related macular degeneration (AMD). Graefes Arch Clin Exp Ophthalmol. 2011;249(2):209-214. PubMed
Appendix 1: Cost-Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing product listing agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: CDA-AMC Cost-Comparison Table for Neovascular Age-Related Macular Degeneration
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosagea | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Aflibercept 8 mg (Eylea HD) | 114.3 mg / mL | 0.07 mL Solution for intravitreal injection | 1,250.0000b | 8 mg every 4 weeks for first 3 doses followed by 8 mg at a dosing interval of every 8 to 16 weeks | Year 1: 17.11 to 27.38 Subsequent: 13.69 to 23.96 | Year 1: 6,250 to 10,000 (5 to 8 inj.) Subsequent: 5,000 to 8,750 (4 to 7 inj.) |
Anti-VEGF inhibitors | ||||||
Aflibercept 2 mg (Eylea) | 40 mg/mL | 0.05 mL Solution for intravitreal injection | 1,418.0000 | 2 mg every 4 weeks for first 3 doses followed by 2 mg every 8 to 16 weeks | Year 1: 23.29 to 31.06 Subsequent: 15.53 to 27.18 | Year 1: 8,508 to 11,344 (6 to 8 inj.) Subsequent: 5,672 to 9,926 (4 to 7 inj.) |
Bevacizumab (Avastin) | 25 mg/mL | 4 mL 16 mL Solution for intravitreal injection | 519.1800c 2,076.7104c | 1.25 mg every 4 weeks for first 3 doses followed by 1.25 mg every 6 to 8 weeksd | Year 1: 0.38 to 0.47 Subsequent: 0.33 to 0.43 | Year 1: 138 to 173 (8 to 10 inj.) Subsequent: 121 to 156 (7 to 9 inj.) |
Bevacizumab (Mvasi) | 25 mg/mL | 4 mL 16 mL Solution for intravitreal injection | 347.0000c 1,388.0000c | 1.25 mg every 4 weeks for first 3 doses followed by 1.25 mg every 6 to 8 weeksd | Year 1: 0.25 to 0.32 Subsequent: 0.22 to 0.29 | Year 1: 93 to 116 (8 to 10 inj.) Subsequent: 81 to 104 (7 to 9 inj.) |
Brolucizumab (Beovu) | 120 mg/mL | 0.05 mL Solution for intravitreal injection | 1,390.0000 | 6 mg every 4 weeks for first 3 doses followed by 6 mg every 8 to 12 weeks | Year 1: 22.83 to 30.44 Subsequent: 19.03 to 26.64 | Year 1: 8,340 to 11,120 (6 to 8 inj.) Subsequent: 6,950 to 9,730 (5 to 7 inj.) |
Faricimab (Vabysmo) | 120 mg/mL | 0.05 mL Solution for intravitreal injection | 1,350.0000b | 6 mg every 4 weeks for first 4 doses followed by 6 mg at a dosing interval of 8 to 16 weeks | Year 1: 22.18 to 33.26 Subsequent: 14.78 to 25.87 | Year 1: 8,100 to 12,150 (6 to 9 inj.) Subsequent: 5,400 to 9,450 (4 to 7 inj.) |
Ranibizumab (Lucentis) | 10 mg/mL | 0.23 mL Solution for intravitreal injection | 1,616.5500 | 0.5 mg every month for first 3 doses followed by 0.5 mg every 1 or 3 months | Year 1: 26.56 to 53.11 Subsequent: 22.13 to 53.11 | Year 1: 9,699 to 19,399 (6 to 12 inj.) Subsequent: 8,083 to 19,399 (5 to 12 inj.) |
Ranibizumab (biosimilar) | 10 mg/mL | 0.23 mL | 995.0000 | 0.5 mg every month for first 3 doses followed by 0.5 mg every 1 or 3 months | Year 1: 16.34 to 32.69 Subsequent: 13.62 to 32.69 | Year 1: 5,970 to 11,940 (6 to 12 inj.) Subsequent: 4,975 to 11,940 (5 to 12 inj.) |
CDA-AMC = Canada’s Drug Agency; inj. = injections; nAMD = neovascular (wet) age-related macular degeneration; VEGF = vascular endothelial growth factor.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed November 2023),29 unless otherwise indicated, and do not include dispensing fees. Annual costs are based on 52 weeks per year. Annual and daily costs for aflibercept 8 mg includes a frequency of once every 12, 16, or 20 weeks, according to the PULSAR trial.
aRecommended doses are from the respective product monographs, unless otherwise indicated.
bSponsor submitted price.1
cPrice obtained from the IQVIA Delta PA database (accessed November 2023).29
dBevacizumab is used off-label in this population and, as such, does not have a recommended dosage for nAMD in the product monograph. Dosage and number of administrations per vial (30 per 4 mL vial) were obtained from a previous CADTH review;30 number of annual doses was based on clinical input received by CDA-AMC for the current review.
Appendix 2: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 5: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
CDA-AMC = Canada’s Drug Agency.
Summary of Sponsor’s BIA
In the submitted budget impact analysis (BIA), the sponsor estimated the incremental budget impact of reimbursing aflibercept 8 mg for the treatment of nAMD.31 The BIA was undertaken using an epidemiologic approach from the perspective of a Canadian public payer over a three-year time horizon (January 2024 to December 2026). The number of patients with nAMD eligible for aflibercept 8 mg was estimated based of the prevalence of AMD, the proportion of AMD that is neovascular, the proportion of patients diagnosed and undergoing treatment for nAMD, the proportion eligible for public coverage, and the rate of bilateral nAMD.31 The sponsor’s analysis included drug acquisition costs and excluded dispensing fees and markups.
The reference scenario included aflibercept 2 mg, faricimab, ranibizumab, ranibizumab (biosimilar), brolucizumab, and bevacizumab. The market share estimates for these products were informed by jurisdiction-specific market research and clinical expert consultation conducted by the sponsor. In the new drug scenario, aflibercept 8 mg was assumed to primarily displace aflibercept 2 mg and faricimab, with market share of aflibercept 8 mg based on clinician input solicited by the sponsor. Market share and injection frequency were considered separately for prevalent and incident patients, with the number of injections per year informed by a sponsor-conducted ITC. Key inputs to the BIA are documented in Table 6.
Table 6: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (Year 1 / Year 2 / Year 3) | |
|---|---|---|
Target population | ||
Pan-Canadian population aged 45+ (excluding Quebec) | 13,590,991 | |
Prevalence of AMD | 8.69%32 | |
Incidence of AMD | 1.4 per 1,00033 | |
Proportion of AMD that is nAMD | 10%34 | |
Proportion of patients whose nAMD is diagnosed | 90%b | |
Proportion of patients who receive treatment | 90%b | |
Proportion eligible for public coverage | 95%b | |
Rate of bilateral disease | 23%35 | |
Number of patients eligible for drug under review | 91,791 / 92,709 / 93,636 | |
Number of eyes eligible for drug under review | 112,903 / 114,032 / 115,172 | |
Market uptake (3 years) | Incident patients | Prevalent patients |
Uptake (reference scenario) | ||
Aflibercept 2 mg | 38% / 35% / 32% | 39% / 37% / 34% |
Ranibizumab | 0% / 0% / 0% | 0% / 0% / 0% |
Ranibizumab biosimilar | 6% / 6% / 6% | 13% / 11% / 11% |
Brolucizumab | 1% / 1% / 1% | 1% / 1% / 1% |
Faricimab | 22% / 25% / 29% | 14% / 17% / 20% |
Bevacizumab | 32% / 32% / 32% | 32% / 32% / 32% |
Uptake (new drug scenario) | ||
Aflibercept 8 mg | 15% / 19% / 22% | 9% / 16% / 20% |
Aflibercept 2 mg | 29% / 25% / 21% | 35% / 29% / 24% |
Ranibizumab | 0% / 0% / 0% | 0% / 0% / 0% |
Ranibizumab biosimilar | 6% / 5% / 5% | 12% / 10% / 10% |
Brolucizumab | 1% / 1% / 1% | 1% / 1% / 1% |
Faricimab | 17% / 18% / 19% | 10% / 11% / 12% |
Bevacizumab | 32% / 32% / 32% | 32% / 32% / 32% |
Annual cost of treatment per patient (induction year / subsequent years)c | ||
Aflibercept 8 mg | $6,500 / $3,750 | |
Aflibercept 2 mg | $11,131 / $7,969 | |
Ranibizumab | $14,856 / $13,531 | |
Ranibizumab biosimilar | $9,144 / $8,328 | |
Brolucizumab | $8,771 / $6,811 | |
Faricimab | $9,383 / $6,575 | |
Bevacizumabd | $4,246 / $3,011 | |
AMD = age-related macular degeneration; CDA-AMC = Canada’s Drug Agency; nAMD = neovascular (wet) age-related macular degeneration.
aThe sponsor assumed that the relevant population included adults over 45 years of age as it affects an older population and to align with epidemiology estimates used in the BIA.32
bBased on values used in the CDA-AMC reimbursement review of faricimab (Vabysmo).30
cAnnual cost was calculated by multiplying the cost per dose by the annual number of administrations predicted by the sponsor’s NMA.31
dSponsor assumed that the cost of bevacizumab was $519.18 per vial based on the cost of branded bevacizumab (Avastin) and that one dose would be obtained per vial. The total cost of bevacizumab would be lower if generic bevacizumab is used and/or vial sharing occurs in clinical practice.
Summary of the Sponsor’s BIA Results
Results of the sponsor analysis suggest that the reimbursement of aflibercept 8 mg for the treatment of nAMD will be associated with an incremental savings of $21,217,778 in year 1, $56,283,529 in year 2, and $80,657,606 in year 3, for a 3-year incremental budgetary savings of $158,158,913.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The frequency of anti-VEGF administration is uncertain. In the BIA, the annual cost of aflibercept 2 mg and comparators was estimated by use of the number of annual injections predicted by the sponsor’s noncomparative pairwise assessment. As noted in the CDA-AMC Clinical Review, the sponsor submitted naive (visual) pairwise comparisons and no comparative analyses were performed. As such, the uncertainty in the relative injection frequency described in the CDA-AMC Appraisal of the Sponsor’s Economic Evaluation also applies to the submitted BIA. The sponsor’s noncomparative assessment suggests that the number of annual injections with aflibercept 8 mg may be similar to that of other anti-VEGFs administered using a treat-and-extend or PRN schedule.
CDA-AMC conducted a scenario analysis that assumed equal frequency of injections for all comparators.
Multiple administrations from a single vial may be possible for some comparators. The sponsor assumed that 1 dose would be obtained from each vial of aflibercept 2 mg, ranibizumab, and bevacizumab. Clinical expert opinion obtained from CDA-AMC for this review and previous reviews have indicated that multiple administrations from a single vial may be possible for these anti-VEGFs. Given that the volume within a vial is greater than that required for a single dose, with the proper syringes multiple administrations can be obtained; however, this practice may be jurisdiction-specific. CDA-AMC additionally notes that, because the sponsor assumed that aflibercept would not displace bevacizumab, the assumption of 1 administration per vial did not impact the incremental results.
As part of a scenario analysis, CDA-AMC assumed that 3 administrations per vial of aflibercept 2 mg and ranibizumab and 30 administrations per vial of bevacizumab were possible.
The displacement of other anti-VEGFs by aflibercept 2 mg is uncertain. The sponsor estimated market shares of aflibercept 8 mg and comparators based on market research conducted by the sponsor and expert opinion solicited by the sponsor, with aflibercept 8 mg assumed to primarily displace aflibercept 2 mg and faricimab. Input received by CDA-AMC for this review indicated that aflibercept 8 mg is likely to predominantly displace aflibercept 2 mg, with a lesser impact on faricimab.
CDA-AMC explored uncertainty in the displacement of anti-VEGF comparators by aflibercept 8 mg in scenario analyses.
The price of drugs paid by public drug plans is uncertain: Both the sponsor and the CDA-AMC analyses are based on publicly available list prices for all comparators. Drug plan feedback received for this review indicated there are confidential negotiated prices for the comparators. Thus, the actual costs paid by the public drug plans for anti-VEGFs are unknown. Depending on the negotiated prices, reimbursing aflibercept 8 mg for the treatment of nAMD may lead to lower or no cost savings compared to other available anti-VEGFs.
CDA-AMC was unable to incorporate the presence of confidential negotiated prices in the reanalysis.
CDA-AMC Reanalyses of the BIA
In the absence of more reliable estimates to inform the key parameters of the BIA, the sponsor’s submitted base case was maintained. CDA-AMC expects that the budget impact of reimbursing aflibercept 8 mg for the treatment nAMD will be sensitive to more reliable inputs which may affect the comparator costs (e.g., administration frequency, vial sharing, availability of biosimilars). CDA-AMC conducted scenario analyses to explore the impact of uncertainty in the number of annual administrations of anti-VEGFs, the number of administrations per vial of aflibercept 2 mg and ranibizumab, displacement of anti-VEGFs by aflibercept 8 mg, and the introduction of a biosimilar for aflibercept 2 mg (Table 7).
The results of these scenario analyses suggest that the budgetary impact of reimbursing aflibercept 8 mg for the treatment of nAMD is sensitive to the administration frequency of each drug and the potential for vial sharing; results of these analyses suggested that the introduction of aflibercept 8 mg would not be cost saving, rather, it would result in $18.8 million and $21.5 million additional costs over 3 years, respectively, indicating that the cost savings associated with the reimbursement of aflibercept 8 mg may have been overestimated in the sponsor’s base case. In addition, the predicted cost savings associated with the introduction of aflibercept 8 mg was highly influenced by assuming a hypothetical price of an aflibercept 2 mg biosimilar with an estimated decline in cost savings of 67% compared to the sponsor’s base-case analysis. Thus, the budget impact of reimbursing aflibercept 8 mg for nAMD is sensitive to assumptions about comparator prices, and its reimbursement may lead to additional costs to the health care system rather than cost savings.
Table 7: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $774,215,620 | $754,565,561 | $757,343,567 | $760,528,867 | $2,272,437,995 |
New drug | $774,215,620 | $733,347,783 | $701,060,037 | $679,871,262 | $2,114,279,083 | |
Budget impact | $0 | −$21,217,778 | −$56,283,529 | −$80,657,606 | −$158,158,913 | |
CDA-AMC scenario 1: Equal administration frequencya | Reference | $415,805,691 | $417,988,489 | $424,170,817 | $428,865,796 | $1,271,025,103 |
New drug | $415,805,691 | $435,136,307 | $430,262,542 | $424,458,626 | $1,289,857,475 | |
Budget impact | $0 | $17,147,817 | $6,091,725 | −$4,407,170 | $18,832,372 | |
CDA-AMC scenario 2: Multiple administrations of aflibercept 2 mg, ranibizumab, and bevacizumabb | Reference | $285,039,017 | $308,506,110 | $328,702,283 | $349,635,638 | $986,844,031 |
New drug | $285,039,017 | $327,763,942 | $336,459,803 | $344,164,105 | $1,008,387,850 | |
Budget impact | $0 | $19,257,832 | $7,757,520 | -$5,471,534 | $21,543,818 | |
CDA-AMC scenario 3: Aflibercept 8 mg primarily displaced aflibercept 2 mgc | Reference | $774,215,620 | $754,565,561 | $757,343,567 | $760,528,867 | $2,272,437,995 |
New drug | $774,215,620 | $730,515,430 | $696,688,704 | $674,092,695 | $2,101,296,830 | |
Budget impact | $0 | −$24,050,131 | −$60,654,863 | −$86,436,172 | −$171,141,166 | |
CDA-AMC scenario 4: Biosimilar of aflibercept 2 mg is availabled | Reference | $598,581,740 | $587,536,170 | $598,307,849 | $614,048,793 | $1,799,892,812 |
New drug | $598,581,740 | $590,509,133 | $580,015,213 | $577,579,546 | $1,748,103,892 | |
Budget impact | $0 | $2,972,963 | −$18,292,636 | -$36,469,248 | −$51,788,920 | |
CDA-AMC scenario 5: Aflibercept 8 mg q.12.w. dosing frequencye | Reference | $774,215,620 | $754,565,561 | $757,343,567 | $760,528,867 | $2,272,437,995 |
New drug | $774,215,620 | $746,468,610 | $719,061,583 | $700,373,469 | $2,165,903,662 | |
Budget impact | $0 | −$8,096,951 | −$38,281,984 | -$60,155,398 | −$106,534,333 |
CDA-AMC = Canada’s Drug Agency.
aThe frequency of administration was set to be equal to aflibercept 8 mg frequency for all comparators.
bCDA-AMC assumed that 3 administrations per vial of aflibercept 2 mg and ranibizumab and 30 administrations per vial of bevacizumab were possible.
cCDA-AMC assumed that the displacement of faricimab was half that estimated by the sponsor, and the displacement was shifted to aflibercept 2 mg.
dThe cost per injection of aflibercept 2 mg biosimilar ($796 per injection) was obtained from the sponsor’s BIA report. The sponsor assumed hypothetical potential list prices for aflibercept 2 mg biosimilar, however, such a price may not be predictive of any actual price offered by a third party.
eCDA-AMC assumed that aflibercept 8 mg would follow a Q12w dosing regimen, and applied the number of injections in the first and subsequent years based on the sponsor-conducted NMA.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.