Drugs, Health Technologies, Health Systems
Reimbursement Review
Bimekizumab (Bimzelx)
Sponsor: UCB Canada Inc.
Therapeutic area: Ankylosing spondylitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AS
ankylosing spondylitis
ASAS
Assessment of SpondyloArthritis International Society
ASAS-PR
partial remission in the Assessment of SpondyloArthritis International Society
ASAS20
improvement of 20% or more in the Assessment of SpondyloArthritis International Society
ASAS40
improvement of 40% or more in the Assessment of SpondyloArthritis International Society
ASDAS
Ankylosing Spondylitis Disease Activity Score
ASDAS-ID
Ankylosing Spondylitis Disease Activity Score inactive disease state
ASQoL
Ankylosing Spondylitis Quality of Life
axSpA
axial spondyloarthritis
BASDAI
Bath Ankylosing Spondylitis Disease Activity Index
BASDAI50
a reduction of at least 50% in the Bath Ankylosing Spondylitis Disease Activity Index score
BASFI
Bath Ankylosing Spondylitis Functional Index
bDMARD
biologic disease-modifying antirheumatic drug
BMI
body mass index
CI
confidence interval
CrI
credible interval
DMARD
disease-modifying antirheumatic drug
ESS
effective sample size
HLA-B27
human leukocyte antigen B27
IBD
inflammatory bowel disease
IL-17
interleukin-17
JAK
Janus kinase
LS
least squares
MAIC
matching-adjusted indirect comparison
MASES
Maastricht Ankylosing Spondylitis Enthesitis Score
MID
minimal important difference
mNY criteria
modified New York criteria
NMA
network meta-analysis
nr-axSpA
nonradiographic axial spondyloarthritis
NRI
nonresponder imputation
NRS
numeric rating scale
NSAID
nonsteroidal anti-inflammatory drug
NSP
nocturnal spinal pain
OR
odds ratio
PGADA
Patient’s Global Assessment of Disease Activity
QoL
quality of life
r-axSpA
radiographic axial spondyloarthritis
RCT
randomized controlled trial
SAE
serious adverse event
SE
standard error
SF-36 MCS
Short Form (36) Health Survey mental component summary
SF-36 PCS
Short Form (36) Health Survey physical component summary
SLR
systematic literature review
TEAE
treatment-emergent adverse event
TNF
tumour necrosis factor
WPAI-SHP
Work Productivity and Activity Impairment–Specific Health Problem
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Bimekizumab (BIMZELX), 160 mg/mL, solution for injection, subcutaneous injection |
Sponsor | UCB Canada Inc. |
Indication | The treatment of adult patients with active ankylosing spondylitis who have responded inadequately or are intolerant to conventional therapy. |
Reimbursement request | The treatment of adult patients with active ankylosing spondylitis who have responded inadequately or are intolerant to conventional therapy. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 11, 2024 |
Recommended dose | 160 mg (given as 1 subcutaneous injection) every 4 weeks |
NOC = Notice of Compliance.
Introduction
Ankylosing spondylitis (AS) is a chronic, inflammatory, and heterogeneous disease that places a significant burden on patients that is driven by pain, fatigue, and stiffness.1,2 Axial spondyloarthritis (axSpA) encompasses radiographic axial spondyloarthritis (r-axSpA, also known as AS) and nonradiographic axial spondyloarthritis (nr-axSpA). Although nr-axSpA shares several features with AS, advanced sacroiliac joint damage and spine ankylosis are absent.3 Patients with uncontrolled inflammation may progress to irreversible axial structural damage,4 spinal fractures, and severe spinal cord injury.5,6 Patients may also experience extramusculoskeletal manifestations, such as uveitis.6,7 A population-based study of the incidence and prevalence of AS in 2010 using Ontario provincial health administrative databases found age- and sex-standardized prevalence and incidence rates of 0.213% and 0.015%, respectively.6,7 AS was estimated to affect 300,000 patients in Canada in 2019.8
Nonsteroidal anti-inflammatory drugs (NSAIDs) are the first-line treatment for adult patients with active AS.4,9,10 After NSAIDs, advanced therapy consists of biologic disease-modifying antirheumatologic drugs (bDMARDs) or targeted DMARDs, respectively. There are currently 2 classes of bDMARDs available in Canada for AS, tumour necrosis factor (TNF) inhibitors and interleukin-17A (IL-17A) inhibitors. Janus kinase (JAK) inhibitors are the only class of targeted DMARD available for the treatment of AS in Canada, and are indicated after a patient’s condition has shown an inadequate response to a bDMARD. Many patients with AS receiving advanced therapy will experience treatment failure.11-13 When failure of advanced therapies occurs, it is recommended to switch to another advanced therapy, either within the same class or to another class.4,10 There is very little evidence to guide switching between advanced therapies; therefore, when treatment failure occurs, guidelines recommend switching to a different therapy that is either within or between treatment classes.4,10
Bimekizumab is a humanized immunoglobulin G1 kappa monoclonal antibody that neutralizes IL-17A, IL-17F, and IL-17AF cytokines, thereby potentially blocking proinflammation and pathological bone formation of axSpA.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of bimekizumab (Bimzelx) 160 mg/mL solution for subcutaneous injection in the treatment of adult patients with active AS.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
A total of 2 inputs were received for this review. One was submitted by Arthritis Consumer Experts (ACE), and the other was a joint submission by 4 patient groups, the Canadian Spondyloarthritis Association, Canadian Arthritis Patient Alliance, Arthritis Society Canada, and Creaky Joints. ACE conducted an online survey between 2019 and 2022 to gather information from patients (n = 4) with AS. The joint input from the 4 patient groups was prepared based on an online survey conducted from September to October 2023 among patients with AS (n = 109).
According to the joint input from the 4 patient groups, the majority of patients with AS experience back pain (90.48%), joint stiffness (79.05%), fatigue (77.14%), and hip pain (71.43%); have difficulties exercising or being active (80.77%); have challenges with sleep (73.08%); and have an impaired ability to work (57.69%) and make social connections (53.85%). In addition, patients living with AS require help with daily activities and emotional support from caregivers. The input from ACE echoes the patient experiences reported in the joint input, and added flare-ups, deconditioning, anxiety, and mood changes as other impacts of AS on patients’ daily lives. Outcomes of interest to patients mentioned in the joint input were improved symptoms (71%), such as less fatigue, pain, and stiffness; better quality of life (QoL) (67%), including an ability to socialize more and better mental well-being; affordability in managing AS (66%); reduced side effects of medications (48%); and convenience (36%) in terms of drug-dosing schedules, route of administration, or formulations. The ACE input agrees with these outcomes of interest and added that ease of movement, ability to exercise more, control of back spasms and inflammation, and less weight gain are other outcomes of interest.
The joint input emphasized that approximately half of patients become resistant to their treatments within 5 years; therefore, access to new treatment options is essential. Of note, the 4 patient groups pointed out that for patients in Canada, it takes an average of 7 to 10 years from the onset of symptoms to be diagnosed with AS. Delayed diagnosis and treatment almost always lead to irreversible damage and a negative impact on mental health. According to the input, patients with AS experience significantly impacted QoL and frustration during this time.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The clinical expert consulted by CADTH indicated that the goals of treatment are to control pain and inflammation and prevent radiographic damage and disability related to AS. The clinical expert stated that the treatment of AS is tailored according to the current manifestations of the disease (axial, peripheral, entheseal, extra-articular symptoms and signs), level of current symptoms, clinical findings and prognostic indicators, disease activity, pain, physical function, structural damage of joints (especially hip involvement and spinal deformities), comorbidities, concomitant drugs, and the wishes and expectations of the patient. The clinical expert consulted by CADTH indicated that unmet needs in the management of AS included the following:
a lack of response to available treatments once initiated (primary failure) in some patients
many patients developing active disease after initially experiencing a response to treatment (secondary failure)
limited access to early diagnosis and treatment
choosing the right drug for the right patient at the right time (precision medicine) due to the availability of relatively few targeted therapies (TNF, IL-17A, and JAK inhibitors)
safety concerns for most DMARDs as well as NSAIDs.
According to the clinical expert, these safety concerns include infections with most drugs, new onset or worsening of associated diseases (uveitis, inflammatory bowel disease [IBD], and psoriasis), and comorbidities; thus, treatments that are safe, effective for all manifestations, and well tolerated by most patients are needed. Though the efficacy of various drugs on the musculoskeletal manifestations are similar, no drug is equally effective for all manifestations. A drug’s effect on associated diseases may vary, according to the feedback from the clinical expert.
The clinical expert consulted by CADTH indicated that in clinical practice, bimekizumab would be used after failure of NSAIDs, either by itself or in combination with NSAIDs. The clinical expert stated they would not reserve bimekizumab for patients with refractory disease or patients who are intolerant to other therapies, as no other drugs target both IL-17A and IL-17F cytokines. The clinical expert stated that given bimekizumab’s efficacy in both musculoskeletal and skin disease, it may be the drug of choice following treatment with NSAIDs in patients with severe skin psoriasis who do not have IBD.
Patients with a personal or family history of IBD may not be candidates for treatment with bimekizumab. This is because, according to the clinical expert consulted by CADTH, the use of IL-17 inhibitors would increase the risk of IBD flares, based on the expert’s experience in using DMARDs that target IL-17A in patients with IBD. The clinical expert consulted by CADTH stated that patients who experience an inadequate response to currently available DMARDs most need an additional treatment option. The clinical expert indicated that the patients best suited for treatment with bimekizumab are generally identified by clinician examination and judgment.
According to the clinical expert consulted by CADTH, clinical response is determined by change in severity of back pain as assessed by patient-reported questionnaires, including total back pain score and the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI). More objective measures, such as the Ankylosing Spondylitis Disease Activity Score (ASDAS), are used in tertiary care centres. Other measures include improvements in enthesitis counts, in the number of tender and swollen joints, and in skin psoriasis. These measures align with the assessments used in the clinical trials. The clinical expert consulted by CADTH indicated that a BASDAI score at 3 to 6 months would be used to assess response. A reduction of at least 50% in the BASDAI score (BASDAI50), or an absolute reduction of at least 2 points in the BASDAI score, is usually required to suggest clinically significant improvement.
According to the clinical expert consulted by CADTH, a lack of response in back pain (given that other causes of back pain are excluded) and secondary treatment failure (relapse) are the most important factors to consider when deciding to discontinue treatment with bimekizumab. The clinical expert indicated that recurrent infections and the occurrence of IBD would require discontinuation of bimekizumab. The clinical expert indicated that discontinuing treatment with bimekizumab is determined by clinical evaluation by a rheumatologist, sometimes involving MRI.
The clinical expert stated that because rheumatologists are trained to identify inflammatory sacroiliitis and spondylitis, they should make the diagnosis. According to the clinical expert consulted by CADTH, patients with AS are usually treated in an outpatient setting, including community clinics and clinics attached to community and academic hospitals. In rare instances, severe disease, including skin, eye, and bowel disease, may warrant admission to a hospital. A rheumatologist is required to diagnose, treat, and monitor patients with AS. Since uveitis, IBD, and skin psoriasis are present with AS, ophthalmologists, gastroenterologists, and dermatologists are also relevant to disease management.
The clinical expert consulted by CADTH stressed that the treatment options for patients with active AS are limited; thus, bimekizumab provides an additional treatment option for such patients.
Clinician Group Input
No clinician group input was submitted for this review.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a CADTH recommendation for bimekizumab: relevant comparators, consideration for initiation of therapy, considerations for continuation or renewal of therapy, considerations for discontinuation of therapy, considerations for prescribing of therapy, generalizability, care provision issues, and system and economic issues.
The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
One pivotal trial (BE MOBILE 2) was included in the sponsor’s systematic review. The BE MOBILE 2 trial was a phase III, multicentre, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of bimekizumab in patients with active AS compared with placebo. This study enrolled adults with active AS (i.e., r-axSpA) and fulfilled the modified New York (mNY) criteria. Eligible study participants (N = 332) were randomized 2:1 to receive bimekizumab (n = 221) 160 mg/mL or placebo (n = 111) subcutaneously every 4 weeks. The mean age of all study participants was 40.4 years with a range of 19 to 80 years. Treatment groups were generally well balanced with respect to AS-related and other baseline disease characteristics. At baseline, the majority of study participants were using NSAID therapies (79.8%), and prior anti-TNF therapy was used by 16.3% of all study participants. The primary objective of the BE MOBILE 2 trial was to demonstrate the efficacy of bimekizumab administered subcutaneously every 4 weeks compared with placebo in the treatment of patients with active AS. The primary end point of the study was an improvement of 40% or more in the Assessment of SpondyloArthritis International Society (ASAS40). Secondary end points included scores for the BASDAI, Bath Ankylosing Spondylitis Functional Index (BASFI), nocturnal spinal pain (NSP) based on a numeric rating scale (NRS), the Maastricht Ankylosing Spondylitis Enthesitis Score (MASES), and health-related quality of life (HRQoL) scores using the Ankylosing Spondylitis Quality of Life (ASQoL) and Work Productivity and Activity Impairment–Specific Health Problem (WPAI-SHP) scales.
Efficacy Results
Improvement of 40% or More in the Assessment of SpondyloArthritis International Society
At week 16 of the double-blind treatment period, patients in the bimekizumab group reported a higher adjusted ASAS40 response rate (41.5%) compared with the placebo group (19.8%), with a between-group difference of 21.8% (95% confidence interval [CI], 11.4% to 32.1%). This corresponded to an odds ratio (OR) of 2.88 (95% CI, 1.71 to 4.87; P < 0.001) in favour of bimekizumab. No estimate of a between-group minimal important difference (MID) was identified by CADTH, but the clinical expert input suggested the absolute difference between groups was clinically important based on a 15% threshold. The ASAS40 response in the bimekizumab group was also observed at weeks 24, 36, and 52. Prespecified subgroup analyses of ASAS40 response rate at week 16 were generally consistent with the primary analysis. At week 16 of the double-blind treatment period, patients in the bimekizumab group who were either TNF alpha inhibitor–naive or –experienced reported a higher adjusted ASAS40 response rate compared with those in the placebo group (45.7% and 40.5%, respectively, for bimekizumab versus 23.4% and 17.6%, respectively, for placebo). The results of sensitivity and supportive analyses, including the tipping-point analyses, were in line with the primary efficacy results.
Bath Ankylosing Spondylitis Disease Activity Index
At week 16, patients in the bimekizumab group had a greater reduction from baseline in the least squares (LS) mean (reductions reflect improvement) in BASDAI score compared with patients in the placebo group (LS mean of −2.7 for bimekizumab versus −1.7 for placebo). An estimated median MID of 1.4 points (range, 0.9 to 1.8) was identified in the literature.14 The clinical expert consulted by CADTH indicated they would consider a 1-point difference between groups as clinically meaningful. The difference in LS mean between treatment groups was −1.04 (95% CI, −1.5 to −0.6; P < 0.001) in favour of bimekizumab. Generally, the treatment effects of bimekizumab on the BASDAI were observed at weeks 24, 36, and 52.
Bath Ankylosing Spondylitis Functional Index
At week 16, patients in the bimekizumab group had a greater reduction from baseline in the LS mean (reductions reflect improvement in physical function) in BASFI score compared with patients in the placebo group, which worsened (LS mean of −1.9 for bimekizumab versus −1.0 for placebo). An estimated median MID of 1.1 points (range, 1.0 to 1.4) was identified in the literature.14 The difference in LS mean between treatment groups was −1.1 (95% CI, −1.5 to −0.6; P < 0.001) in favour of bimekizumab. The clinical expert consulted by CADTH suggested an MID of 1 point for between-group difference. Generally, the treatment effects of bimekizumab on the BASFI were observed at weeks 24, 36, and 52.
Nocturnal Spinal Pain
At week 16, patients in the bimekizumab group had a greater reduction from baseline in LS mean (reductions reflect improvement) in NSP (based on an NRS) score compared with patients in the placebo group, which worsened (LS mean of −3.2 for bimekizumab versus −1.7 for placebo). An estimated median MID of 1.5 points (range, 1.1 to 2.3) was identified in the literature.14 The difference in LS mean between treatment groups was −1.5 (95% CI, −2.0 to −1.0; P < 0.001) in favour of bimekizumab. The clinical expert consulted by CADTH identified an MID of 1 point for between-group difference. Generally, the treatment effects of bimekizumab on the NSP were observed at weeks 24, 36, and 52.
Enthesitis-Free State Based on the MASES in Patients With Enthesitis at Baseline
At week 16 of the double-blind treatment period, patients with enthesitis at baseline in the bimekizumab group reported a higher adjusted enthesitis-free rate (43.8%) compared with those in the placebo group (23.9%), with a between-group difference of 19.8% (95% CI, 6.3% to 33.4%). This corresponded to an OR of 2.47 (95% CI, 1.30 to 4.68) in favour of bimekizumab. No estimate of a between-group MID was identified by CADTH, but the clinical expert suggested a 15% difference would be clinically important; therefore, the absolute difference between groups was clinically important. Generally, the treatment effects of bimekizumab on the enthesitis-free rate were observed at weeks 24 and 52. The outcome for enthesitis-free state was not controlled for type I error; thus, these data should be interpreted as supportive evidence only.
Ankylosing Spondylitis Quality of Life
At week 16, patients in the bimekizumab group had a greater reduction from baseline in LS mean (reductions reflect improvement) in ASQoL score compared with patients in the placebo group, which worsened (LS mean of −4.6 for bimekizumab versus −3.1 for placebo, respectively). An MID of 1 unit of worsening (i.e., + 1) or 2 units of improvement (i.e., −2) was identified in the literature.15 The difference in LS mean between treatment groups was −1.5 (95% CI, −2.4 to −0.7; P < 0.001) in favour of bimekizumab. The clinical expert consulted by CADTH identified an MID of 2 points for between-group difference. Generally, the treatment effects of bimekizumab on the ASQoL were observed at weeks 24, 36, and 52.
Work Productivity and Activity Impairment–Specific Health Problem
At week 16, the bimekizumab group compared with the placebo group had a greater mean reduction (improvement) from baseline in the WPAI-SHP score for the following:
Percent time missed due to disease-related problems: –5.5 for bimekizumab versus −1.2 for placebo, with a between-group difference of −2.9 (95% CI, –6.9 to 1.1)
Percent impairment while working due to disease-related problems: –20.8 for bimekizumab versus −6.1 for placebo, with a between-group difference of −12.5 (95% CI, –18.2 to −6.9)
Percent overall work impairment due to disease-related problems: –22.2 for bimekizumab versus –6.7 for placebo, with a between-group difference of −12.8 (95% CI, –18.7 to −6.9)
Percent activity impairment due to disease-related problems: –23.3 for bimekizumab versus −14.4 for placebo, with a between-group difference of −9.4 (95% CI, –13.9 to −4.9).
No MIDs for WPAI-SHP were identified in the literature. Generally, the treatment effects of bimekizumab on the WPAI-SHP domains were observed at weeks 24, 36, and 52, except for percent activity impairment due to disease-related problems where patients reported similar results between groups. The WPAI-SHP outcome was not controlled for type I error; thus, these data should be interpreted as supportive evidence only.
Harms Results
An adverse event (AE) was reported among 54.3% of patients in the bimekizumab group and 43.2% of patients in the placebo group at week 16. The most commonly reported AEs (i.e., reported by ≥ 5% of patients in either group) were infections and infestations (28.1% for bimekizumab versus 22.5% for placebo), gastrointestinal disorders (13.1% versus 9.9%), nervous system disorders (8.1% versus 4.5%), upper respiratory tract infection (2.7% versus 7.2%), and eye disorders (2.3% versus 6.3%).
Serious adverse events (SAEs) were reported among 2.3% of patients in the bimekizumab group and 0.9% of patients in the placebo group at week 16. The following SAEs were commonly reported in the bimekizumab group but not in any patients in the placebo group: goitre (0.5%), colitis ulcerative (0.5%), Crohn disease (0.5%), cholelithiasis (0.5%), and hepatitis A (0.5%).
Discontinuation due to AEs was reported among 2.7% of patients in the bimekizumab group but among no patients in the placebo group at week 16. The most commonly reported AEs that led to study discontinuation in the bimekizumab group were abnormal psychiatric evaluation (0.9%), lymphoid tissue hyperplasia (0.5%), Crohn disease (0.5%), oral candidiasis (0.5%), and rash (0.5%). No deaths due to AEs were reported during the double-blind treatment period in the BE MOBILE 2 trial.
Serious infections, fungal infections, opportunistic infections, malignancies, major adverse cardiac events, neutropenia, suicidal ideation and behaviours, IBD, hypersensitivity reactions, and liver injuries or disorders were considered notable harms by the sponsor and/or the clinical expert consulted by CADTH. The commonly reported notable harms were hypersensitivity reactions (7.7% for bimekizumab versus 1.8 for placebo), fungal infections (6.3% versus 0), liver injuries or disorders (4.5% versus 3.6%), IBD (1.8% versus 0), neutropenia (0.5% versus 0), and serious infections (0.5% versus 0.9%).
Critical Appraisal
Internal Validity
The CADTH review team noted there were no comparative data available beyond week 16, as patients in the placebo group were reallocated to receive bimekizumab during the 36-week maintenance period after finishing all assessments at the end of the 16-week double-blind treatment period; therefore, the direct comparative efficacy and safety of bimekizumab after week 16 are uncertain. For the analysis of the primary and key secondary end points, a fixed sequence testing procedure was employed to adjust for multiple comparisons across multiple end points, thereby controlling the type I error. The CADTH review team noted that the analyses of the enthesitis-free state based on the MASES index were not included in the fixed sequence testing hierarchy; thus, the results should be considered as supportive evidence. Although the subgroup analyses were prespecified, the BE MOBILE 2 trial was not powered to detect any change in the ASAS40 response rate between bimekizumab and placebo in subgroup analyses, except for the subgroup of patients who are TNF inhibitor–naive; additionally, no formal statistical tests for interaction between subgroups were conducted. There were 2 protocol amendments regarding eligibility criteria made after the enrolment of the first patient (April 25, 2019). The CADTH review team considered that these 2 protocol amendments may increase patient heterogeneity and introduce bias. The direction of the bias is uncertain, as there were no data reported on the numbers of patients with psoriatic arthritis or patients who had experienced treatment failure with NSAIDs other than the 2 NSAIDs included in the trial. HRQoL is considered a relevant outcome by patients with active AS and the clinical expert consulted by CADTH. However, the assessment of the ability to return to normal activities and/or functioning using the WPAI-SHP was not controlled for multiplicity; thus, it should be considered as exploratory and supportive.
External Validity
The BE MOBILE 2 trial used placebo as the comparator group. According to the clinical expert consulted by CADTH, an anti-TNF biosimilar monoclonal antibody would be an appropriate comparator for bimekizumab. The clinical expert indicated placebo is not an appropriate comparator; head-to-head studies with an active drug would be ideal. The BE MOBILE 2 trial excluded patients who had been treated with more than 1 TNF alpha inhibitor and/or more than 2 additional non–TNF alpha biological-response modifiers, or any IL-17 biological-response modifier at any time. The clinical expert indicated that these patients should be considered eligible for bimekizumab. Although the response rate might be lower, some patients’ conditions do respond to bDMARDs after previously failing to respond to TNF inhibitors and IL-17 inhibitors; therefore, the clinical expert stated they would switch treatments within the same class due to the relatively limited treatment options. According to the clinical expert, the study results would not be generalizable to the previously mentioned patients, as it is expected that response rates will be lower in that patient population, which tends to have lower response rates with subsequent treatments in clinical practice. In addition, there was no study site in Canada in the BE MOBILE 2 trial, which may compromise the generalizability of the study results to clinical practice in Canada.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal BE MOBILE 2 trial identified in the sponsor’s systematic review, the Grading of Recommendations, Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.16,17 Following the GRADE approach, evidence from randomized controlled trials (RCTs) started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, or publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with a clinical expert, and the input received from the patient groups and the public drug plans. The following outcomes were finalized in consultation with expert committee members: ASAS40, BASDAI, BASFI, NSP, MASES, HRQoL (ASQoL and WPAI-SHP), and SAEs.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty-of-evidence assessment was the presence or absence of a clinically important effect based on thresholds informed by the clinical expert consulted by CADTH for this review for ASAS40, BASDAI, BASFI, NSP, MASES, ASQoL, and SAEs. There is no established MID for the WPAI-SHP, and the clinical expert consulted by CADTH could not provide an MID threshold, so the target of the certainty-of-evidence assessment was the presence or absence of any (non-null) effect.
Table 2: Summary of Findings for Bimekizumab Versus Placebo for Patients With Active Ankylosing Spondylitis
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Bimekizumab 160 mg/mL | Difference | |||||
Disease activity and symptom | |||||||
Adjusted ASAS40 response rate at week 16 Follow-up: 16 weeks | 332 (1 RCT) | OR: 2.88 (1.71 to 4.87) | 198 per 1,000 | 415 per 1,000 (333 to 503 per 1,000) | 218 more per 1,000 (114 to 321 more per 1,000) | Moderatea | Bimekizumab likely results in a clinically important increase in the adjusted ASAS40 response rate at week 16 when compared with placebo. |
Change from baseline in BASDAI total score at week 16 Follow-up: 16 weeks | 332 (1 RCT) | NR | 1.7 fewer | 2.7 fewer (NR) | 1.0 fewer (1.5 fewer to 0.6 fewer) | Moderateb | Bimekizumab likely results in a clinically important difference in the change from baseline in BASDAI total score at week 16 when compared with placebo. |
Change from baseline in BASFI at week 16 Follow-up: 16 weeks | 332 (1 RCT) | NR | 1.0 fewer | 2.0 fewer (NR) | 1.1 fewer (1.5 fewer to 0.6 fewer) | Moderatec | Bimekizumab likely results in a clinically important reduction in BASFI at week 16 when compared with placebo. |
Change from baseline in NSP score (based on an NRS) at week 16 Follow-up: 16 weeks | 332 (1 RCT) | NR | 1.7 fewer | 3.2 fewer (NR) | 1.5 fewer (2.0 fewer to 1.0 fewer) | Highd | Bimekizumab results in a clinically important reduction in NSP score (based on an NRS) at week 16 when compared with placebo. |
Adjusted enthesitis-free rate based on the MASES index at week 16 in study participants with enthesitis at baseline Follow-up: 16 weeks | 199 (1 RCT) | OR: 2.47 (1.30 to 4.68) | 239 per 1,000 | 438 per 1,000 (331 to 550 per 1,000) | 198 more per 1,000 (63 to 334 more per 1,000) | Moderatea,e | Bimekizumab likely results in a clinically important increase in the adjusted enthesitis-free rate based on the MASES index at week 16 when compared with placebo. |
Health-related quality of life | |||||||
Change from baseline in ASQoL total score at week 16 Follow-up: 16 weeks | 332 (1 RCT) | NR | 3.1 fewer | 4.6 fewer (NR) | 1.5 fewer (2.4 fewer to 0.7 fewer) | Moderatef | Bimekizumab likely results in a clinically important reduction in ASQoL total score at week 16 when compared with placebo. |
Change from baseline in WPAI-SHP at week 16: Percent time missed due to disease-related problems Follow-up: 16 weeks | 239 (1 RCT) | NR | 1.2 fewer | 5.5 fewer (NR) | 2.9 fewer (6.9 fewer to 1.1 more) | Lowe,g | Bimekizumab may result in a reduction in the WPAI-SHP in percent time missed due to disease-related problems at week 16 when compared with placebo. The clinical importance of the reduction is unclear. |
Change from baseline in WPAI-SHP at week 16: Percent impairment while working due to disease-related problems Follow-up: 16 weeks | 225 (1 RCT) | NR | 6.1 fewer | 20.8 fewer (NR) | 12.5 fewer (18.1 fewer to 6.8 fewer) | Highe,g | Bimekizumab results in a reduction in the WPAI-SHP in percent impairment while working due to disease-related problems at week 16 when compared with placebo. The clinical importance of the reduction is unclear. |
Change from baseline in WPAI-SHP at week 16: Percent overall work impairment due to disease-related problems Follow-up: 16 weeks | 225 (1 RCT) | NR | 6.7 fewer | 22.2 fewer (NR) | 12.8 fewer (18.7 fewer to 6.9 fewer) | Highe,g | Bimekizumab results in a reduction in the WPAI-SHP in percent overall work impairment due to disease-related problems at week 16 when compared with placebo. The clinical importance of the reduction is unclear. |
Change from baseline in WPAI-SHP at week 16: Percent activity impairment due to disease-related problems Follow-up: 16 weeks | 318 (1 RCT) | NR | 14.4 fewer | 23.3 fewer (NR) | 9.4 fewer (13.9 fewer to 4.9 fewer) | Highe,g | Bimekizumab results in a reduction in the WPAI-SHP in percent activity impairment due to disease-related problems. The clinical importance of the reduction is unclear. |
Harms | |||||||
Proportion of patients who experienced any serious adverse event(s) at week 16 Follow-up: 16 weeks | 332 (1 RCT) | NR |
| Lowh | Bimekizumab may result in an increase in the proportion of patients who experienced a serious adverse event at week 16 when compared with placebo. | ||
ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASQoL = Ankylosing Spondylitis Quality of Life; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; CI = confidence interval; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; MID = minimal important difference; NR = not reported; NRS = numeric rating scale; NSP = nocturnal spinal pain; OR = odds ratio; RCT = randomized controlled trial; WPAI-SHP = Work Productivity and Activity Impairment Questionnaire–Specific Health Problem.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision. There is no established MID but the clinical expert consulted by CADTH considered that a 15% difference between groups in the adjusted ASAS40 response rate and the adjusted enthesitis-free rate at week 16 could be considered a threshold of clinical importance. For both outcomes, the point estimate and the upper bound of the 95% CI for the between-group difference suggested a clinically important difference for bimekizumab vs. placebo, while the lower bound of the 95% CI suggested no clinically important difference between the 2 groups.
bRated down 1 level for serious imprecision. There is no established between-group MID but the estimated median MID for the change from baseline is 1.4 points (range, 0.9 to 1.8). The clinical expert consulted by CADTH considered that a 1-point difference between groups in the change from baseline in BASDAI total score at week 16 could be considered a threshold of clinical importance. The point estimate and the upper bound of the 95% CI for the between-group difference suggested a clinically important difference for bimekizumab vs. placebo based on a 1-point threshold, while the lower bound of the 95% CI suggested no clinically important difference between the 2 groups.
cRated down 1 level for serious imprecision. There is no established MID for between-group difference but the estimated median MID for the change from baseline is 1.1 points (range, 1.0 to 1.4). The clinical expert consulted by CADTH considered that a 1-point difference between groups in the change from baseline in BASFI at week 16 could be considered a threshold of clinical importance. The point estimate and the upper bound of the 95% CI for the between-group difference suggested a clinically important difference for bimekizumab vs. placebo based on a 1-point threshold, while the lower bound of the 95% CI suggested no clinically important difference between the 2 groups.
dImprecision was not rated down. There is no established between-group MID, but the estimated median MID for the change from baseline is 1.5 points (range, 1.1 to 2.3). The clinical expert consulted by CADTH considered that a 1-point difference between groups in the change from baseline in NSP score (based on an NRS) at week 16 could be considered a threshold of clinical importance. The point estimate and the 95% CI for the between-group difference suggested a clinically important difference for bimekizumab vs. placebo based on a 1-point threshold.
eThe statistical testing was not adjusted for multiplicity in the trial and should be considered as supportive evidence.
fRated down 1 level for serious imprecision. There is no established between-group MID, but the estimated median MID for the change from baseline is −2 points for improvement. The clinical expert consulted by CADTH considered that a 2-point difference between groups in the change from baseline in ASQoL total score at week 16 could be considered a threshold of clinical importance. The point estimate and the upper bound of the 95% CI for the between-group difference suggested no clinically important difference for bimekizumab vs. placebo, while the lower bound of the 95% CI suggested a clinically important difference between the 2 groups based on a 2-point threshold.
gThere is no established MID and the clinical expert consulted by CADTH could not provide a threshold of important difference, so the target of the certainty appraisal was any effect for the change from baseline in the WPAI-SHP at week 16. For percent time missed due to problems (related to disease), impression was rated down for 2 levels, as the CADTH review team judged that the point estimate suggested a possibility of benefit, but the 95% CI for the between-group difference included the possibility of both benefit and harm (fewer benefits) for bimekizumab vs. placebo. For percent impairment while working due to (disease-related) problems, percent overall work impairment due to problems, and percent activity impairment due to problems, impression was not rated down, as the CADTH review team judged that the point estimate and the 95% CI for the between-group difference suggested no clinically important difference for bimekizumab vs. placebo.
hRated down 2 levels for very serious imprecision. The CADTH review team considered the 16-week double-blind follow-up period to not be long enough to assess comparative long-term harms. The lower bound of the 95% CI for the between-group difference was below zero while the upper bound was above zero, suggesting no clinically important difference between the 2 groups. Additionally, the rate of SAEs was relatively low in both treatment groups based on a small sample size.
Sources: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
Long-Term Extension Studies
Description of Studies
One single-arm, phase II, open-label extension study, BE AGILE 2, was submitted by the sponsor as supporting evidence. Patients who had been enrolled in and completed the BE AGILE trial were rolled over to the BE AGILE 2 study (N = 255), which was conducted in European countries and the US. All patients in the BE AGILE 2 trial received open-label bimekizumab 160 mg every 4 weeks for up to 204 weeks, which meant a total possible exposure of 252 weeks for those who received bimekizumab in the parent trial, BE AGILE.
Efficacy Results
ASAS40 response was sustained up to week 208 in the BE AGILE 2 trial, with response rates of 59% (147 out of 249) using nonresponder imputation (NRI) and 73.1% (147 out of 201) using observed case data. The mean BASDAI score (n = 249) decreased from baseline and was sustained at week 208 (decrease of −4.01; standard error [SE] of 0.13 versus an MID range of 0.9 to 1.8 points). The mean BASFI score (n = 249) decreased from baseline (−3.1; SE = 0.15) and was sustained at week 208. Relative to baseline, the NSP score (n = 249) decreased and was maintained at week 208 (−4.55; SE = 0.16) versus an MID range of 1.1 to 2.3 points. Also, the mean ASQoL score (n = 249) decreased from baseline (−5.9; SE = 0.3) and was maintained through week 208 versus an MID ranging from an increase of 1 (worsening) to a decrease of 2 (improvement) units. Among patients with enthesitis at baseline, the mean MASES (n = 164) decreased by −0.37 (SE = 0.23) and maintained improvement up to week 208 in the BE AGILE 2 trial. WPAI-SHP was not assessed in the BE AGILE 2 study.
Harms Results
A total of 237 (92.9%) study participants reported a treatment-emergent adverse event (TEAE) during the BE AGILE 2 trial. The most commonly reported TEAEs were nasopharyngitis (18%), upper respiratory tract infection and COVID-19 infection (12.9% each), and bronchitis (8.6%). There were 46 patients (18.0%) who experienced at least 1 SAE, with COVID-19 infection and pneumonia being the most common (1.2% each). Twenty-one patients (8.2%) discontinued study treatment due to a TEAE, mostly due to alanine aminotransferase (1.2%) and aspartate aminotransferase (0.8%) elevation. Two fatal TEAEs were reported during the study, 1 incident due to a road traffic accident and another incident due to cardiorespiratory arrest. Fungal infection (18.4%) and hypersensitivity (11.4%) were the most common AEs of special interest reported during the BE AGILE 2 study, where the vast majority of fungal infections did not lead to treatment discontinuation. (A single patient discontinued due to perirectal abscess.)
Critical Appraisal
The lack of a control group, open-label design, and selective patient population are the major limitations of the BE AGILE 2 extension study. An open-label design without a comparator arm can overestimate results for efficacy outcomes, especially patient-reported outcomes. Moreover, a risk of selection bias was noted for the BE AGILE 2 study, since patients who have experienced a response to bimekizumab and tolerated any side effects are more likely to continue the extension period.
Indirect Comparisons
Description of Studies
Network meta-analyses (NMAs) were performed to determine the clinical efficacy and safety of bimekizumab at weeks 12 and 16 compared with other relevant interventions for the treatment of patients with AS. The NMAs were conducted on 3 different networks: purely naive (100% bDMARD-naive; 24 studies, 4,145 patients), predominantly naive (approximately 90% bDMARD-naive; 26 studies, 5,271 patients), and purely experienced (100% bDMARD-experienced; 9 studies, 1,048 patients).
Unanchored matching-adjusted indirect comparisons (MAICs) were performed to establish the long-term relative clinical efficacy of bimekizumab compared with other IL-17A inhibitors in patients with AS at week 52.
Efficacy Results
Network Meta-Analyses
In the bDMARD purely naive network, for most comparisons between bimekizumab versus TNF, IL-17, or JAK inhibitors, there were no clear differences observed. The exceptions to this were 2 findings in which bimekizumab showed statistically significant improvement in the Short Form (36) Health Survey physical component summary (SF-36 PCS) results compared with adalimumab and compared with secukinumab, but the differences observed were not clinically significant and the credible intervals (CrIs) were wide, indicating uncertainty.
In the bDMARD predominantly naive network, for most comparisons between bimekizumab versus TNF, IL-17, or JAK inhibitors, there were no clear differences observed. There were some exceptions to this general observation that were statistically significant differences, but these were not clinically significant and the CrIs were wide, indicating uncertainty. Bimekizumab improved the SF-36 PCS and partial remission in the Assessment of SpondyloArthritis International Society (ASAS-PR) results compared with secukinumab. Results favoured etanercept compared with bimekizumab for the BASDAI50 and BASFI. Results favoured golimumab IV compared with bimekizumab for BASFI and ASQoL. Results favoured adalimumab and certolizumab over bimekizumab for Ankylosing Spondylitis Disease Activity Score inactive disease state (ASDAS-ID). Results favoured tofacitinib over bimekizumab for the Bath Ankylosing Spondylitis Metrology Index. Results favoured upadacitinib over bimekizumab for ASDAS-ID.
In the bDMARD purely experienced network, for most comparisons between bimekizumab versus TNF, IL-17, or JAK inhibitors, there were no clear differences observed. The exceptions to this were 2 findings in which results favoured certolizumab over bimekizumab for the ASQoL and SF-36 PCS. In these 2 instances, the difference may be clinically significant, but the CrIs were wide, indicating uncertainty, and these results were not confirmed in the other networks.
Matching-Adjusted Indirect Comparison
The MAIC analyses suggested that bimekizumab 160 mg every 4 weeks had statistically significantly better results at week 52 compared with ixekizumab 80 mg every 4 weeks for the following: improvement of 20% or more in the Assessment of SpondyloArthritis International Society (ASAS20), ASAS40, BASDAI change from baseline, and BASDAI50. Results also favoured bimekizumab over secukinumab 150 mg every 4 weeks for ASAS40 and BASDAI change from baseline. However, there were significant limitations to the MAIC that preclude making claims of superiority of bimekizumab over comparators.
Harms Results
Network Meta-Analyses
The sponsor conducted NMAs of bimekizumab compared with other medications in the context of axSpA for 2 harms outcomes, discontinuation due to any reason and SAEs. The comparators of interest with data available for this NMA were 2 IL-17A inhibitors (ixekizumab and secukinumab); TNF alpha inhibitors adalimumab, certolizumab pegol, etanercept, golimumab (subcutaneous or IV routes), and infliximab (IV); and JAK inhibitors tofacitinib and upadacitinib.
The network for analysis of discontinuation due to any reason contained 18 studies. Study discontinuation rates were low in all trials (range, 0 to 14 patients per treatment arm). Time points between 12 and 16 weeks were used for this analysis. The CrIs were very wide for most estimates. There were no clear differences observed between bimekizumab and any other treatment. There was 1 finding in which bimekizumab had a higher risk of study discontinuation compared with tofacitinib; however, the uncertainty around this estimate was high, as reflected by a wide CrI.
The network for analysis of SAEs contained 18 studies. SAE rates were low in all studies (range, 0 to 10 SAEs per treatment arm). Time points between 12 and 16 weeks were used for this analysis. The CrIs were very wide for most estimates. There were no clear differences between bimekizumab and any other treatments in the network.
Matching-Adjusted Indirect Comparison
There were no harms outcomes assessed in the MAIC.
Critical Appraisal
Network Meta-Analyses
The sponsor conducted an NMA using a Bayesian approach. This was a reasonable method to apply, given the common comparator of placebo. The sponsor’s decision to perform 3 separate NMA analyses based on the potential effect modifier of prior exposure to bDMARDs was appropriate. Some networks had a large number of trials and a large number of patients, which was considered a strength of the NMA analyses. The sponsor did not perform sensitivity analyses in the NMA and did not attempt to identify and adjust for effect modifiers, despite the availability of a large number of trials for some of the networks. The time point of 12 to 16 weeks that was selected for the outcome analyses was reasonable and clinically relevant for efficacy but was not as meaningful for harms since an assessment of long-term harms was lacking.
The CIs and CrIs were wide for many estimates in the NMA. Despite the large number of trials, the number of patients and events in some analyses were small, precluding the possibility of detecting a difference between treatments. For example, the incidence of harms outcomes was small, resulting in very wide CrIs around the estimates. For this reason, the results of the harms analyses were not informative and did not serve to illuminate the risk of harms for bimekizumab relative to other treatments.
Matching-Adjusted Indirect Comparison
The sponsor performed an unanchored MAIC because of the lack of a placebo arm beyond week 16 for bimekizumab and comparators. This was an adequate justification for performing an MAIC. The selection of comparators from the same pharmacologic group (IL-17A inhibitors) was a rational approach, but comparisons with other biologics would also have been of interest. The MAIC allowed a comparison of 52 weeks of clinical data. The MAIC analyses suggested there were some differences favouring bimekizumab compared with secukinumab and ixekizumab for ASAS20, ASAS40, BASDAI change from baseline, and BASDAI50, but several limitations of the MAIC prevent drawing strong conclusions regarding the comparative effectiveness of bimekizumab. For example, there were important differences between the studies included in the MAIC that did not account for several of the prognostic factors that were deemed important by the authors of the MAIC and were not used in the weighting adjustments of the MAIC. There were notable differences in the study populations before and after adjustment. In the MAIC analyses, the effective sample size (ESS) for the bimekizumab group was reduced to 80% for the comparison with secukinumab 150 mg, to 51% for the comparison with secukinumab 300 mg, and to 20% for the comparison with ixekizumab. Not all matching variables that were deemed important were used in the weighting adjustments of the MAIC analyses. Regarding the MAIC analyses, the sponsor noted that “the amount of bias in the indirect comparisons is likely to be substantial,”19 and the CADTH reviewers agree with this assessment.
Summary
The results of the sponsor’s NMA did not show consistent differences for efficacy or harms outcomes between bimekizumab and comparators in the networks. While differences were reported in a small number of comparisons in some populations, these were associated with wide 95% CrIs for many comparisons, indicating imprecision of the results.
Results of the sponsor’s MAIC favoured bimekizumab for some outcomes, but there were significant limitations. The limitations include differences in study design and providing models with partial adjustments of prognostic and effect modifiers rather than fully adjusted models. These limitations, in addition to the substantial reduction in the ESSs, undermine any claims of superior performance of bimekizumab over comparators in the MAIC.
Neither the NMA nor the MAIC provided clear evidence of a difference in efficacy or harm outcomes for bimekizumab compared to comparators.
Studies Addressing Gaps in the Evidence From the Systematic Review
The BE MOBILE 1 trial was a phase III, multicentre, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of bimekizumab in patients with active nr-axSpA. The sponsor identified BE MOBILE 1 as a study addressing the gap in the evidence on the efficacy and safety of bimekizumab. However, the CADTH review team considered the BE MOBILE 1 trial irrelevant to this review, as patients with active nr-axSpA are different from patients with active AS, the population for the indication being reviewed. Therefore, the CADTH review team found that no studies addressing gaps in the systematic review evidence had been identified for this review.
Conclusions
Patients and clinicians highlighted the need for new effective treatments for active AS that control disease and symptoms and improve QoL compared with current treatments.
One phase III, multicentre, randomized, double-blind, placebo-controlled trial (BE MOBILE 2) comparing bimekizumab with placebo in treating patients with moderate to severe AS demonstrated that bimekizumab increased the adjusted ASAS40 response rate at week 16. Likewise, the results of the analysis of BASDAI total score indicated that patients treated with bimekizumab had a greater improvement in disease control than patients who received placebo at week 16. A GRADE assessment of the sponsor-submitted systematic review, which included only the BE MOBILE 2 trial, suggested that bimekizumab likely results in a clinically important improvement in the ASAS40 response rate and in BASDAI total score compared with placebo.
Compared with placebo, bimekizumab results in a clinically important reduction in NSP score (based on an NRS) and likely results in a clinically important reduction in the BASFI, the adjusted enthesitis-free rate based on the MASES index, and the ASQoL total score. With regard to the WPAI-SHP, bimekizumab may result in a reduction in percent time missed due to problems (related to disease) and does result in a reduction in percent impairment while working due to problems, percent overall work impairment due to problems, and percent activity impairment due to problems. The CADTH review team noted that the clinical importance of the reduction in the WPAI-SHP is unclear because a clinically meaningful threshold could not be determined.
Compared with placebo, there is low-certainty evidence that bimekizumab may result in an increase in the percentage of patients who experienced SAEs at week 16. No new safety signals were identified in the long term BE AGILE and BE AGILE 2 trials in patients with AS.
The sponsor-submitted indirect treatment comparison analyses did not provide clear evidence of a difference in efficacy or harms outcomes for bimekizumab relative to other treatments.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of bimekizumab (Bimzelx) 160 mg/mL solution for subcutaneous injection in the treatment of adult patients with active AS.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
AS is a chronic, inflammatory, and heterogeneous disease that places a significant burden on patients that is driven by pain, fatigue, and stiffness.1,2 Axial spondyloarthritis encompasses r-axSpA (also known as AS) and nr-axSpA. AS is characterized by inflammatory back pain, sacroiliitis, a high prevalence of human leukocyte antigen B27 (HLA-B27), and excess bone formation that can lead to spinal ankylosis.4 Key symptoms of AS include fatigue and stiffness with loss of physical function.2,3,20
The symptoms and progression of AS can vary between patients.21 The initial symptoms of AS often begin in young adults (45 years and younger), with the peak onset between the ages of 20 and 30.2,22 Patients with AS typically present first with chronic back pain (≥ 3 months in duration) of insidious onset and an inflammatory nature that improves with exercise.1,2 This chronic back pain is caused by inflammation in the sacroiliac joints and/or spine. Over several years, as the disease progresses and inflammation continues, affected joints can become damaged and new bone formation can develop, leading to ankylosis of the sacroiliac joints and formation of syndesmophytes in the spine.21,23 (Syndesmophytes are bony growths in ligaments in the intervertebral joints that cause irreversible impairment of spinal mobility.23) Patients with uncontrolled inflammation may progress to irreversible axial structural damage,5,6 spinal fractures, and severe spinal cord injury,24 which is associated with uncontrolled inflammation.25 Therefore, inflammation is predictive of structural progression in patients with axSpA.25 Beyond the key axial symptoms, patients with AS can experience enthesitis, dactylitis, peripheral arthritis, and extramusculoskeletal manifestations, such as uveitis, IBD, and psoriasis and hidradenitis suppurativa.6,7
As a result of both the axial and nonaxial manifestations, patients with AS experience significant disease burden that impacts their daily lives, reduces their QoL, and leads to a reduction in work productivity.26 Because AS usually starts before age 45, the disease has a considerable effect on different aspects of life (career, family, and social life).27
A clinical diagnosis of AS is based on clinical presentation, in combination with laboratory and imaging tests of the spine and/or the sacroiliac joints.2,3,28 While there are no official diagnostic criteria for AS, the mNY classification criteria for AS or Assessment of SpondyloArthritis International Society (ASAS) classification criteria for axSpA are often referenced to aid in diagnosis.2,3 To determine whether patients meet the ASAS axSpA criteria, clinicians rely on physical examinations (to identify dactylitis, enthesitis, and psoriasis), blood testing (to measure C-reactive protein levels and detect HLA-B27), and imaging (such as X-rays or MRI scans to view structural damage or inflammation of the sacroiliac joint, a hallmark feature of AS).2,3,10 Elevated C-reactive protein is indicative of inflammation, which can be a sign of AS.2 HLA-B27 is considered to be a biomarker for AS, as 80% to 90% of patients with AS have tested positive for HLA-B27; therefore, testing for HLA-B27 presence is a key tool in AS diagnosis.3 A diagnosis of AS is confirmed once the patient exhibits radiographic abnormalities consistent with sacroiliitis.2,3,28 According to the recommendations of the American College of Rheumatology, Spondylitis Association of America, and Spondyloarthritis Research and Treatment Network, patients are considered to have active AS when symptoms are unacceptably bothersome to the patient and judged by the examining clinician to be due to inflammation.4 According to the clinical expert’s input, the patient is judged to have active AS based on a BASDAI score of 4 or greater and a rheumatologist assessment.
A population-based study of the incidence and prevalence of AS in 2010 using Ontario provincial health administrative databases found age- and sex-standardized prevalence and incidence rates of 0.213% and 0.015%, respectively.29 AS was estimated to affect 300,000 patients in Canada in 2019.8
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
International guidelines from the American College of Rheumatology, Spondylitis Association of America, and Spondyloarthritis Research and Treatment Network (published in 2019)4 and the Assessment of SpondyloArthritis International Society and European Alliance of Associations for Rheumatology (updated in 2022)10 are available to guide the treatment of AS.30 Updated Canadian guidelines from the Canadian Rheumatology Association and Spondyloarthritis Research Consortium of Canada were published in July 2024 were published in July 2024.32
According to the previously mentioned guidelines, treatment goals for patients with AS are to control symptoms and inflammation, prevent progressive structural damage, preserve or normalize physical function and social participation, decrease disease complications, and maintain ability to work.4,10 Similarly, the clinical expert consulted by CADTH stated that treatment goals include controlling pain and inflammation and preventing radiographic damage and disability.
The choice of treatment should be individualized to each patient to meet treatment goals.10 Clinicians in Canada select treatment based on the current signs and symptoms of disease (axial, peripheral, extra-articular manifestations) and patient preference (e.g., oral versus injection, frequency of injection) to ensure treatment goals can be met.30 The clinical expert consulted by CADTH agrees that treatment of AS is tailored according to current manifestations of the disease (axial, peripheral, entheseal, extra-articular symptoms and signs; level of current symptoms; clinical findings and prognostic indicators; disease activity, pain, and physical function; and structural damage at a joint, especially hip involvement and spinal deformities), comorbidities, concomitant drugs, and the wishes and expectations of the patient.
According to the clinical expert consulted by CADTH, physical therapy and NSAIDs are usually recommended to patients after the diagnosis is made. In Canada, several drug classes are used in the pharmacologic therapy of AS. NSAIDs are the first line of treatment for adult patients with active AS.4,9,10 If a patient experiences an inadequate response to NSAIDs, or if there are contraindications, advanced therapies are the next line of treatment.4,9,10 Advanced therapy consists of bDMARDs or targeted DMARDs. Currently, 2 classes of bDMARDs are available in Canada for AS, TNF inhibitors and IL-17A inhibitors. JAK inhibitors are the only class of targeted DMARDs available for the treatment of AS in Canada, and they are indicated after a patient has experienced an inadequate response to a bDMARD (Figure 1). In the Assessment of SpondyloArthritis International Society–European Alliance of Associations for Rheumatology 2022 update, TNF, IL-17, and JAK inhibitors are all recommended for patients when their condition has failed to respond to first-line NSAID therapy, with a preference to start advanced therapy with a TNF or IL-17 inhibitor.10 The clinical expert consulted by CADTH has a different opinion in that, unless contraindicated, a TNF inhibitor biosimilar should be the first biologic of choice for AS followed by another TNF inhibitor, then an IL-17 inhibitor, and then a JAK inhibitor. For patients with a history of recurrent uveitis or active IBD, a monoclonal antibody TNF inhibitor (adalimumab, certolizumab pegol, golimumab, or infliximab) is preferred, while for patients with significant psoriasis, an IL-17 inhibitor is preferred.10 The clinical expert CADTH consulted added that these drugs (especially the TNF inhibitors) improve the symptoms and signs of AS and, with prolonged treatment, lead to reduced spinal ankylosis (reduced progression as assessed by the modified Stoke Ankylosing Spondylitis Spinal Score), especially in those with early disease.
Many patients with AS receiving advanced therapy will experience treatment failure.11-13 When advanced therapies fail, it is recommended that patients switch to another advanced therapy, either within the same class or to another class.4,10 There is very little evidence to guide switching between advanced therapies; therefore, when treatment failure occurs, guidelines recommend switching to a different therapy that is either within or between treatment classes.4,10
Figure 1: Treatment Paradigm for Patients With Ankylosing Spondylitis in Canada
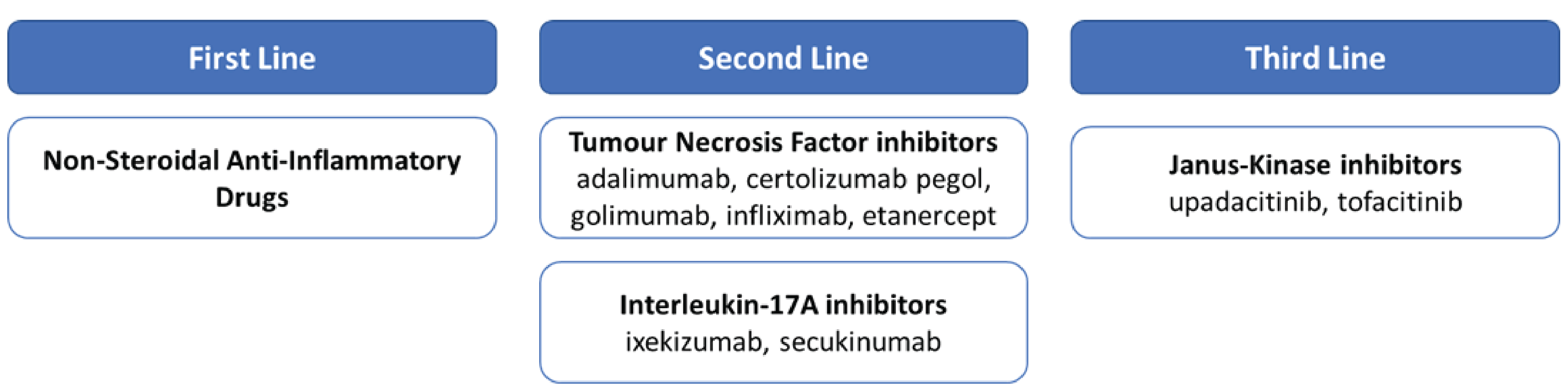
Source: The sponsor’s summary of clinical evidence.19
Drug Under Review
Key characteristics of bimekizumab and other treatments available for adult patients with active AS are summarized in Table 3.
Bimekizumab (Bimzelx) 160 mg/mL solution for subcutaneous injection is available in a prefilled syringe or autoinjector for single use only. The recommended dosage for adult patients with active axSpA (including AS and nr-axSpA) is 160 mg given as 1 subcutaneous injection every 4 weeks.33 After proper training in subcutaneous injection technique, patients may self-inject, if their physician determines it is appropriate, with medical follow-up as necessary.33
Bimekizumab was approved by Health Canada for the treatment of adult patients with active AS and the treatment of adult patients with active nr-axSpA on March 11, 2024. The reimbursement request for the review by CADTH is for the treatment of adult patients with active AS. Bimekizumab was previously approved by Health Canada on February 14, 2022, for moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy. On April 18, 2022, CADTH recommended bimekizumab for reimbursement with conditions for this indication.34
Bimekizumab is a humanized immunoglobulin G1 kappa monoclonal antibody. It has 2 identical antigen binding regions that bind and neutralize IL-17A, IL-17F, and IL-17AF cytokines, blocking their interaction with the IL-17RA and IL-17RC receptor complex.33 Elevations of IL-17A and IL-17F levels are each independent pivotal drivers of inflammation and pathological bone formation in in vitro models of axSpA. Therefore, inhibiting IL-17F in addition to IL-17A is expected to provide greater resolution of inflammation and more inhibition of pathological bone formation than blocking IL-17A alone.
Table 3: Key Characteristics of Bimekizumab, TNF Inhibitors, Other IL-17 Inhibitors, and JAK Inhibitors
Drug | Mechanism of action | Indication(s)a | Route and dosage | Serious adverse effects or safety issues | Other |
|---|---|---|---|---|---|
Bimekizumab (Bimzelx) | A humanized IgG1 kappa monoclonal antibody that inhibits IL-17A, IL-17F, and IL-17AF | Treatment of adult patients with active AS | 160 mg SC injection every 4 weeks | Infections, neutropenia, dermatitis | Potential for immunogenicity, i.e., antidrug antibody formation |
TNF inhibitors | |||||
Adalimumab36 (Humira and biosimilars) | A recombinant, fully human IgG1 kappa monoclonal antibody that selectively binds TNF alpha | Reducing signs and symptoms in adult patients with active AS who have had an inadequate response to conventional therapy | 40 mg SC injection every 2 weeks | Serious infections, neurologic events, malignancies | Formation of anti-adalimumab antibodies |
Certolizumab pegol37 (Cimzia) | Selectively binds and neutralizes TNF alpha. Does not contain the Fc region, which is normally present in a complete antibody | Reducing signs and symptoms in adult patients with active AS who have had an inadequate response to conventional therapy | Loading dosage: 400 mg SC injection at weeks 0, 2, and 4 Maintenance dosage: 200 mg every 2 weeks or 400 mg every 4 weeks | Serious infections, malignancies, heart failure | Formation of autoantibodies |
Etanercept38 (Enbrel and biosimilars) | A dimeric fusion protein linked to human IgG1 antibody that binds and blocks both TNF (alpha and beta) and lymphotoxin alpha | Reducing the signs and symptoms of active AS | 50 mg SC injection every week | Serious infections and malignancies, neurologic and hematologic events, heart failure | Formation of autoantibodies, which rarely can result in a lupus-like syndrome or autoimmune hepatitis (reversible) |
Golimumab39 (Simponi) | A human monoclonal antibody that binds TNF to prevent its interaction with its receptors | Reducing signs and symptoms in adult patients with active AS who have had an inadequate response to conventional therapies | 50 mg SC injection once a month, on the same date each month | Serious infections, malignancies, congestive heart failure, hematologic reactions | Formation of autoantibodies and may result in the development of a lupus-like syndrome |
Infliximab40 (Remicade and biosimilars) | A chimeric (human constant and murine variable regions) IgG1 kappa monoclonal antibody that specifically binds and neutralizes TNF alpha | Reducing the signs and symptoms of active AS | 5 mg/kg IV infusion at weeks 0, 2, and 6, then every 6 to 8 weeks thereafter | Serious infections, carcinogenesis and mutagenesis, and cardiovascular and hematologic events | Formation of autoantibodies and may result in the development of a lupus-like syndrome |
IL-17 inhibitors | |||||
Ixekizumab41 (Taltz) | A humanized IgG4 monoclonal antibody that binds and inhibits release of IL-17A | Treatment of adult patients with active AS who have had an inadequate response to or are intolerant to conventional therapy | 80 mg SC injection every 4 weeks. May start at an initial dose of 160 mg | Infections, IBD, skin reactions | A potential for immunogenicity. Detection of antibody formation |
Secukinumab42 (Cosentyx) | A human IgG1 kappa antibody that selectively binds to and neutralizes IL-17A | For the treatment of adult patients with active AS who have had an inadequate response to conventional therapy | 150 mg SC injection at weeks 0, 1, 2, 3, and 4 followed by 150 mg SC injection every 4 weeks thereafter; if a patient continues to have active AS, a monthly maintenance dose of 300 mg should be considered | Infections, IBD, hypersensitivity reactions | Treatment-emergent antidrug antibodies |
JAK inhibitors | |||||
Tofacitinib43 (Xeljanz) | Inhibits JAK1, JAK2, JAK3 and, to a lesser extent, TyK2 | Treatment of adults with active AS who have had an inadequate response to a bDMARDs or when the use of those therapies is inadvisable | 5 mg by mouth twice daily | Serious infections, malignancies, thrombosis, MACE | Drug–drug interactions with potent CYP3A4 inhibitors and inducers, potent CYP2C19 inhibitors |
Upadacitinib44 (Rinvoq) | Inhibits JAK1 at a greater inhibitory potency relative to JAK2, JAK3, and TyK2 | Treatment of adults with active AS who have had an inadequate response to a bDMARD or when the use of those therapies is inadvisable | 15 mg by mouth once daily | Serious infections, malignancies, thrombosis, MACE | Drug–drug interactions with strong CYP3A4 inhibitors and inducers |
AS = ankylosing spondylitis; bDMARD = biologic disease-modifying antirheumatologic drug; CYP = cytochrome; Fc region = fragment crystallizable region; IBD = inflammatory bowel disease; IgG1 = immunoglobulin G1; IL = interleukin; JAK = Janus kinase; MACE = major adverse cardiovascular event; SC = subcutaneous; TNF = tumour necrosis factor; TyK2 = tyrosine kinase 2.
aHealth Canada–approved indication.
Sources: Sponsor’s summary of clinical evidence19 and product monographs for Bimzelx, Humira, Cimzia, Enbrel, Simponi, Remicade, Taltz, Cosentyx, Xeljanz, and Rinvoq.45-54
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient inputs received by CADTH have been included in the stakeholder section of this report.
A total of 2 inputs were submitted for this review. One was from ACE, Canada’s largest and longest-running national arthritis patient organization. The other was a joint submission by 4 patient groups:
the Canadian Spondyloarthritis Association, the only patient-led organization in Canada dedicated solely to people living with spondyloarthritis
the Canadian Arthritis Patient Alliance, a grassroots, patient-driven, independent, and national education and advocacy organization
Arthritis Society Canada, the largest charitable funder of cutting-edge arthritis research
Creaky Joints, a digital community for arthritis patients and caregivers worldwide.
ACE conducted an online survey between 2019 and 2022 to gather information from patients with AS (n = 4). The joint input by the 4 other patient groups was prepared based on an online survey conducted from September to October 2023 among patients with AS (n = 109).
According to the joint input from the 4 patient groups, the majority of patients with AS experience back pain (90.48%), joint stiffness (79.05%), fatigue (77.14%), and hip pain (71.43%); have difficulties exercising or being active (80.77%); challenges with sleep (73.08%); and have an impaired ability to work (57.69%) and make social connections (53.85%). In addition, patients living with AS require help with daily activities and emotional support from caregivers. Even though 30% of respondents stated they do not typically miss school or work due to AS symptoms or side effects of medications, 18% of patients stated they do not work or attend school due to AS and 22% of patients missed an average of 1 to 5 days of work per month due to AS. Even though 73.4% of participants said they did not require support from caregivers, others needed help with climbing, carrying, long-distance walking, shopping and housework, meal preparation, and needed emotional support from family, friends, or a home-care worker. The input from ACE echoes the patient experiences reported in the joint input, and added flare-ups, deconditioning, anxiety, and mood changes as other impacts of AS on patients’ daily lives. Outcomes of interest mentioned in the joint input were improved symptoms (71%), such as less fatigue, pain, and stiffness; better QoL (67%), including an ability to socialize more and better mental well-being; affordability in managing AS (66%); reduced side effects of medications (48%); and convenience (36%) in terms of drug-dosing schedules, route of administration, or formulations. The ACE input agrees with these outcomes of interest and added that ease of movement and ability to exercise more, control of back spasms and inflammation, and less weight gain are other outcomes of interest.
The joint input emphasized that approximately half of patients become resistant to their treatments within 5 years; therefore, access to new treatment option is essential. Of note, the 4 patient groups pointed out that for patients in Canada, it takes an average of 7 to 10 years from the onset of symptoms to be diagnosed with AS. Delayed diagnosis and treatment almost always lead to irreversible damage and a negative impact on mental health. According to the input, patients with AS experience significantly impacted QoL and frustration during this time.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management adult patients with active AS.
Unmet Needs
The clinical expert consulted by CADTH indicated that the goals of treatment are to control pain and inflammation and prevent radiographic damage and disability related to AS. The clinical expert stated that the treatment of AS is tailored according to current manifestations of the disease (axial, peripheral, entheseal, extra-articular symptoms and signs); level of current symptoms; clinical findings and prognostic indicators; disease activity, pain, and physical function; and structural damage at a joint, especially hip involvement and spinal deformities), comorbidities, concomitant drugs, and the wishes and expectations of the patient. In addition, the clinical expert commented that the optimal management of AS requires a combination of nonpharmacological and pharmacological treatments; nonpharmacological treatment of AS should include patient education and regular exercise. Individual and group physical therapy should be considered.
The clinical expert consulted by CADTH indicated that unmet needs in the management of AS included the following:
a lack of response to available treatments once initiated (primary failure) in some patients
many patients developing active disease after initially experiencing a response to treatment (secondary failure)
limited access to early diagnosis and treatment
choosing the right drug for the right patient at the right time (precision medicine) due to the availability of relatively few targeted therapies (TNF, IL-17A, and JAK inhibitors)
safety concerns for most DMARDs as well as NSAIDs.
According to the clinical expert, these safety concerns include infections with most drugs, new onset or worsening of associated diseases (uveitis, IBD, and psoriasis), and comorbidities; thus, treatments that are safe, effective for all manifestations, and well tolerated by most patients are needed. Current therapies include those administered orally, subcutaneously, and intravenously; patients thus have options when deciding which therapy to choose. Though the efficacy of various drugs on musculoskeletal manifestations are similar, no drug is equally effective for all manifestations and a drug’s effect on associated diseases may vary, according to the feedback from the clinical expert.
Place in Therapy
According to the clinical expert consulted by CADTH, current drugs used for the treatment of AS include NSAIDs and DMARDs including TNF inhibitors (i.e., etanercept, infliximab, adalimumab, golimumab and certolizumab), IL-17A inhibitors (i.e., secukinumab, ixekizumab) and JAK inhibitors (i.e., upadacitinib). These drugs (especially the TNF inhibitors) improve the symptoms and signs of AS and, with prolonged treatment, lead to reduced spinal ankylosis (reduced progression as assessed by the modified Stoke Ankylosing Spondylitis Spinal Score), especially in those with early disease. After the diagnosis is made, physical therapy and NSAIDs are recommended to patients. If the patient continues to have active disease, as judged by a BASDAI score greater than 4 and by a rheumatologist, then treatment with DMARDs is indicated. DMARDs have been shown to improve signs and symptoms, reduce spinal damage progression, and improve HRQoL and function. These drugs may also reduce the need for joint replacement, especially of hip joints.
The clinical expert consulted by CADTH indicated that in clinical practice, bimekizumab would be used after the failure of NSAIDs, either by itself or in combination with NSAIDs. The clinical expert stated they would not reserve bimekizumab for patients with refractory disease or patients who are intolerant to other therapies, as there are no other drugs targeting both IL-17A and IL-17F cytokines. The clinical expert stated that given bimekizumab’s efficacy in both musculoskeletal and skin disease, it may be the drug of choice following treatment with NSAIDs in patients with severe skin psoriasis who do not have IBD.
Patient Population
Patients with a personal or family history of IBD may not be candidates for treatment with bimekizumab. This is because, according to the clinical expert consulted by CADTH, the use of IL-17 inhibitors increase the risk of IBD flares, based on the expert’s experience in using DMARDs that target IL-17A in patients with IBD. However, one should note that the incidence of flares or new-onset IBD was very low, as patients with active IBD were excluded from the clinical trials with bimekizumab. The presence of IBD or severe uveitis and active infections, especially fungal infections, would be contraindications for treatment with bimekizumab.
The clinical expert consulted by CADTH stated that patients experiencing an inadequate response to currently available DMARDs are most in need of an additional treatment option. The clinical expert indicated that the patients best suited for treatment with bimekizumab are generally identified by clinician examination and judgment. There are no biomarkers for treatment response or AEs. The clinical expert indicated that patients with objective evidence of inflammation, such as a positive MRI or abnormal C-reactive protein level, are most likely to respond to DMARD therapy.
Assessing the Response to Treatment
According to the clinical expert consulted by CADTH, clinical response is determined by a change in the severity of back pain as assessed by patient-reported questionnaires, including total back pain score and the BASDAI. More objective measures, such as the ASDAS, are used in tertiary care centres. Other measures include improvements in enthesitis counts, in the number of tender and swollen joints, and in skin psoriasis. These measures are aligned with the assessments used in clinical trials.
The clinical expert consulted by CADTH indicated that a BASDAI score at 3 to 6 months would be used to assess response. A reduction in the BASDAI50 score, or an absolute reduction of at least 2 points in the BASDAI score, is usually required to suggest clinically significant improvement. The clinical expert stated that an improvement in physical function may be desired but may be delayed. The clinical expert mentioned that patients also report improvement in pain and fatigue, but such improvement may be difficult to achieve in longstanding disease. According to the clinical expert, the response magnitude is unlikely to vary between rheumatologists. The clinical expert commented that, with longer follow-up assessments, the lack of spinal radiographic progression may be demonstrated, but that is not usual clinical practice.
Discontinuing Treatment
According to the clinical expert consulted by CADTH, the lack of response in back pain (given that other causes of back pain are excluded) and secondary failure (relapse) are the most important factors to consider when deciding to discontinue treatment with bimekizumab. The clinical expert indicated that recurrent infections and the occurrence of IBD would require discontinuation of bimekizumab. The clinical expert indicated that discontinuing treatment with bimekizumab is determined through a clinical evaluation by a rheumatologist and sometimes involves MRI scans.
Prescribing Considerations
The clinical expert consulted by CADTH indicated that, currently, there are no markers that will help predict which patients will do well or not when using any drug, including bimekizumab. Diagnosing AS by primary care practitioners is difficult due to the lack of biomarkers, difficulty in interpreting MRI scans, and the overlap of symptoms with common musculoskeletal symptoms (such as back pain). The clinical expert stated that rheumatologists are trained to identify inflammatory sacroiliitis and spondylitis; therefore, they should make the diagnosis. Misdiagnosis is more likely in primary care than in rheumatology clinics.
According to the clinical expert consulted by CADTH, patients with AS are usually treated in an outpatient setting, including community clinics and clinics attached to community and academic hospitals. In rare instances, severe disease, including skin, eye, and bowel disease, may warrant admission to a hospital. A rheumatologist is required to diagnose, treat, and monitor patients with AS. Since uveitis, IBD, and skin psoriasis are present with AS, ophthalmologists, gastroenterologists, and dermatologists are also relevant to disease management.
Additional Considerations
The clinical expert consulted by CADTH stressed that the treatment options for patients with active AS are limited; thus, bimekizumab provides an additional treatment option for such patients.
Clinician Group Input
No clinician group input was submitted for this review.
Drug Program Input
Drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The comparator drug is placebo (BE MOBILE 1 trial was for nonradiographic axSpA and the BE MOBILE 2 trial was for radiographic axSpA). The biologics that CDEC has reviewed for use in AS are golimumab (March 17, 2010), ixekizumab (March 24, 2020), etanercept (October 25, 2016), certolizumab pegol (April 17, 2015), secukinumab (August 23, 2016), adalimumab (June 27, 2007), infliximab (December 19, 2014), and upadacitinib (May 11, 2023). Noted: Bimekizumab dually inhibits IL-17A and IL-17F. According to the 2022 update of the ASAS-EULAR recommendations for the management of SpA, the medications used for axSpA are secukinumab, ixekizumab, tofacitinib, and TNFi biosimilars. These recommendations suggest that NSAIDs and TNFi drugs remain primary classes of medications for radiographic and nonradiographic axSpA. What is an appropriate comparator for patients with axSpA, given the biosimilar or biologic space (e.g., secukinumab [Cosentyx])? | According to the clinical expert consulted by CADTH, an anti-TNF biosimilar monoclonal antibody would be an appropriate comparator for bimekizumab. The clinical expert indicated placebo is not an appropriate comparator; head-to-head studies with an active drug would be ideal, but such trials are few and far between. |
Would bimekizumab be an option for patients with peripheral symptoms of AS (no axial involvement)? | The clinical expert CADTH consulted indicated there are no studies for pure peripheral SpA. Extrapolation from psoriatic arthritis studies would indicate that bimekizumab is likely effective. |
Both trials enrolled patients who had experienced prior failure of ≥ 2 NSAIDs or a history of intolerance or contraindications to NSAIDs. Patients were excluded if they had received > 1 TNFi, > 2 additional biologic response modifiers, or any IL-17 response modifier. Should bimekizumab be first line in treating patients with AS, as NSAIDs are for symptomatic control of pain and do not modify the disease? Should the CDEC reimbursement criteria align with the enrolment criteria where patients need to experience the failure of 2 or more NSAIDs? If yes, could you define the duration of an adequate trial of NSAIDs? | The clinical expert consulted by CADTH indicated that unless there are strong contraindications against NSAIDs use, the use of DMARDs (e.g., bimekizumab) would be second line after NSAIDs. The clinical expert confirmed that failure of 2 or more NSAIDs is a fair criterion for reimbursement. Many patients are adequately controlled with NSAIDs and physical therapy and do not need tDMARDs. According to the clinical expert, the duration of an adequate trial of NSAIDs would be about 1 month. This is because NSAIDs are quick-acting, and the clinical expert would not wait as, AS is a systemic inflammatory disease. |
Would patients access this medication at the same level as TNFi and IL-17 modifiers, despite no head-to-head comparison data? How about in relation to JAK inhibitors, such as upadacitinib? | The clinical expert consulted by CADTH would suggest that unless contraindicated, a TNFi biosimilar should be the first biologic of choice for AS followed by another TNFi, then an IL-17i (including bimekizumab), and a JAKi, such as upadacitinib. The clinical expert stated that for patients with active AS who have contraindications to NSAIDs, such as advanced cardiovascular disease, renal disease, IBD, or Crohn disease, tDMARD therapy (i.e., JAKi drugs) would be appropriate. |
Can the medication be used as monotherapy, i.e., without methotrexate? | The clinical expert consulted by CADTH confirmed that bimekizumab can be used as monotherapy. |
Medications such as secukinumab and ixekizumab are not preferred for patients with extramusculoskeletal manifestations (e.g., IBD and uveitis) according to the 2022 ASAS-EULAR recommendations for the management of axSpA. What is the place in therapy of bimekizumab regarding its use in patients exhibiting these manifestations? | According to the clinical expert consulted by CADTH, until it is adequately proven that bimekizumab improves uveitis and does not lead to de novo IBD or IBD flares, bimekizumab should not be preferred in patients with these manifestations. |
Would treatment goals include reducing structural damage progression? Does bimekizumab help in reducing structure damage progression in axial AS? | The clinical expert consulted by CADTH indicated that treatment goals should not include reducing structural damage progression, as this would require early and prolonged treatment, which is difficult to implement in routine clinical practice. According to the clinical expert consulted by CADTH, there is no evidence to prove that bimekizumab helps reduce the progression of structure damage in axial AS. |
What advantages and disadvantages does bimekizumab hold over other medications in this space? | According to the clinical expert consulted by CADTH, the advantage of bimekizumab over other medications in treating patients with active AS is that it is an option in patients with severe psoriasis or those experiencing failure with a TNFi or other IL-17i (although such patients were excluded from clinical trials) or a JAKi. The disadvantages of bimekizumab were the risk of IBD and fungal infections, especially mucocutaneous candidiasis, but that is easily treated with oral and topical antifungals. |
It almost feels like a class review is required to form consistent criteria with all these drugs. | Comment from the drug programs to inform CDEC deliberations. |
CADTH has reviewed bimekizumab before for more moderate to severe psoriasis. The medication received a positive reimbursement recommendation (March 30, 2022). | Comment from the drug programs to inform CDEC deliberations. |
There are other medications in this space that align with the criteria for the biologics in place in the various drug plans for AS, instead of being specific to IL-17i drugs. | Comment from the drug programs to inform CDEC deliberations. |
Considerations for initiation of therapy | |
Is there a standardized definition for active AS? The trials use BASDAI ≥ 4 and spinal pain (BASDAI item 2) ≥ 4. Is this definition used in practice? Or is this definition for active axial AS? | The clinical expert CADTH consulted confirmed that the definition for active AS used in the BE MOBILE 2 trial (i.e., BASDAI ≥ 4 and spinal pain [BASDAI item 2] ≥ 4) is used in practice but includes judgment by a rheumatologist and might include CRP and MRI evaluation. |
Patients enrolled in the BE MOBILE 1 and 2 trials were aged 18 years and older. Is this a medication that can be used in the pediatric population? Most of the patients enrolled in the MOBILE 1 and MOBILE 2 trials had high or very high disease activity measured by ASDAS-CRP. Would you be able to comment on the efficacy of bimekizumab in patients with mild or moderate disease activity? Should this medication not be offered to these patients? | The clinical expert CADTH consulted confirmed that bimekizumab might be used in the pediatric population, even though the drug is not approved for this population. Other IL-17i drugs have been used in related diseases. The clinical expert consulted by CADTH indicated that patients with mild to moderate disease are also likely to respond, especially if they have objective measures of inflammation such as elevated CRP and MRI changes. |
In what situations would a clinician start bimekizumab right away without the requirement of an NSAID trial? Would it be possible to include discussion on comorbidities? Would you be able to comment on onset of action and response relative to other comparators? When presented with a patient whose condition has failed to respond (to specific) TNFi or IL-17i drugs, what is the efficacy of the switch to bimekizumab? | According to the clinical expert consulted by CADTH, bimekizumab may be used directly if NSAIDs are contraindicated, especially in the presence of bleeding disorders, peptic ulcer disease, renal disease, hypertension, and atherosclerotic vascular disease. The clinical expert consulted by CADTH commented that bimekizumab has a relatively quick onset of action and is comparable to TNFi drugs. According to the clinical expert consulted by CADTH, the response rate is likely to be lower in patients whose condition has failed to respond to TNFi drugs, and may be even lower when treatment with other IL-17i drugs has failed. After NSAIDs, these drugs could be used as first-line tDMARDs. The clinical expert consulted by CADTH indicated that the choice is based on the presence of comorbidities and risk of side effects. After NSAIDs, a TNFi biosimilar may be preferred, followed by any of the other DMARDs for AS. |
There are other medications in this space. There should be alignment with the criteria for the biologics in place in the various drug plans for AS, instead of being specific to IL-17i drugs. | Comment from the drug programs to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
What is an appropriate tool to monitor disease activity? For example, ASDAS high disease activity defined as ≥ 2.1 vs. ASAS40 vs. BASDAI (which is a tool used as criteria in jurisdictions to show beneficial effects of treatment for renewal). Could you comment on subjective vs. objective tools? | According to the clinical expert consulted by CADTH, ASDAS or BASDAI may be used to monitor disease activity. The BASDAI is simple to use and is preferred, but ASDAS is more objective since it includes CRP. ASAS40 is a response criterion and not a measure of disease state; hence, it is not preferred for long-term monitoring. The clinical expert consulted by CADTH indicated that physician judgment should also be considered. The clinical expert consulted by CADTH commented that the tools are inherently subjective since the main manifestation is back pain. The ASDAS is more objective than the BASDAI. |
There are other medications in this space. There should be alignment with the criteria for the biologics in place in the various drug plans for AS, instead of being specific to IL-17i drugs. | Comment from the drug programs to inform CDEC deliberations. |
Considerations for discontinuation of therapy | |
What definition would you use for loss of response, absence of clinical benefit, and disease progression in clinical practice? Based on what parameters? | The clinical expert consulted by CADTH indicated that the lack of response should be based on BASDAI response as stated earlier or using the ASDAS. Worsening back pain, as judged by the rheumatologist to be due to ongoing inflammation, would define the absence of clinical benefit and disease progression. The clinical expert stated that CRP testing and MRI may help the rheumatologist inform their judgment. |
For renewal and subsequent renewal for this medication, it would be good to understand the tools used and the targets for these tools (pretreatment vs. during treatment vs. after stabilization) to help jurisdictions with adjudication. | Comment from the drug programs to inform CDEC deliberations. |
Considerations for prescribing of therapy | |
The current dosage is 160 mg/mL SC every 4 weeks (no loading dose, based on dose-response studies). Is there any evidence for increasing or decreasing the frequency of medication administration for this indication? | According to the clinical expert consulted by CADTH, there is no evidence for increasing or reducing the frequency of medication administration for this indication; however, this may be required in patients with severe disease or frequent flares between doses, which the clinical expert expected to be infrequent. |
Generalizability | |
Patients with inflammatory conditions other than nonradiographic axSpA or radiographic axSpA were excluded. What is the incidence of inflammatory conditions in patients with nonradiographic axSpA or radiographic axSpA? Is this generalizable to the axSpA population if this population has concomitant inflammatory conditions? | According to the clinical expert consulted by CADTH, peripheral arthritis, psoriasis, uveitis, and IBD are often present in patients with AS. The clinical expert commented that these were not exclusion criteria, except for active IBD and recent flare of uveitis. The clinical expert consulted by CADTH confirmed that the data presented in the BE MOBILE 2 trial would be generalizable to patients with active AS and concomitant inflammatory conditions. The clinical expert stated that psoriasis, IBD, and peripheral arthritis are inflammatory conditions related to the disease and patients in their clinical practice with AS rarely have comorbid inflammatory conditions such as rheumatoid arthritis. |
Care provision issues | |
The screening period included LTBI treatment (additional health intervention). What is the incidence or prevalence of fungal infections while using bimekizumab? | The clinical expert stated that inhibition of IL-17 is associated with mucocutaneous candidiasis, including oropharyngeal, vaginal, and esophageal candidiasis. The clinical expert indicated that the incidence rate ranges from 2% to 21% and would be higher with higher doses, as observed in psoriasis and psoriatic arthritis trials. |
Depending on the incidence or prevalence, this may add out-of-pocket costs, costs to patients, and/or costs to the health care system to acquire antifungal therapy. | Comment from the drug programs to inform CDEC deliberations. |
System and economic issues | |
Presence of confidential negotiated prices for comparators. | Comment from the drug programs to inform CDEC deliberations. |
AS = ankylosing spondylitis; ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASAS-EULAR = Assessment of SpondyloArthritis International Society–European Alliance of Associations for Rheumatology; ASDAS = Ankylosing Spondylitis Disease Activity Score; axSpA = axial spondyloarthritis; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CDEC = Canadian Drug Expert Committee; CRP = C-reactive protein; DMARD = disease-modifying antirheumatic drug; IBD = inflammatory bowel disease; IL-17 = interleukin-17; IL-17i = interleukin-17 inhibitor; JAKi = Janus kinase inhibitor; LTBI = latent tuberculosis infection; NSAID = nonsteroidal anti-inflammatory drug; SpA = spondyloarthritis; tDMARD = targeted disease-modifying antirheumatic drug; TNFi = tumour necrosis factor inhibitor.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects bimekizumab 160 mg/mL solution for subcutaneous injection in the treatment of active AS in adult patients. The focus of this report is on comparing bimekizumab with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of bimekizumab is presented in 3 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section uses the GRADE approach and follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
One pivotal study: One randomized, double-blind, placebo-controlled, multicentre phase III trial (BE MOBILE 2) identified in the systematic review
One long-term extension study: 1 single-arm phase II trial (BE AGILE) and its open-label extension study (BE AGILE 2)
Four indirect treatment comparisons: 3 NMAs and 1 MAIC.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
One pivotal trial (BE MOBILE 2) was included in the sponsor’s systematic review. The BE MOBILE 2 trial was a phase III, multicentre, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of bimekizumab in patients with active AS compared with placebo. The primary objective of the BE MOBILE 2 trial was to demonstrate the efficacy of bimekizumab administered subcutaneously every 4 weeks compared with placebo in the treatment of patients with active AS. This study enrolled adults who had active AS (i.e., r-axSpA) and fulfilled the mNY criteria.
Table 5: Details of Studies Included in the Systematic Review
Details | BE MOBILE 2 study |
|---|---|
Designs and populations | |
Study design | Randomized, double-blind, placebo-controlled, multicentre phase III trial of adult patients with active AS |
Locations | 93 clinical sites in Belgium, Bulgaria, China, the Czech Republic, France, Germany, Hungary, Japan, Netherlands, Poland, Spain, Turkey, the UK, and the US |
Patient enrolment dates |
|
Randomized (N) | A total of 332 patients were enrolled and randomized (221 patients to the bimekizumab group and 111 patients to the placebo group) |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Bimekizumab (160 mg) via subcutaneous injection at week 0 (baseline) and then q.4.w. |
Comparator | Placebo via subcutaneous injection at week 0 (baseline) and then q.4.w. at weeks 4, 8, and 12. After week 16, participants in the placebo group were reallocated to receive bimekizumab (160 mg) subcutaneously q.4.w. |
Study duration | |
Screening phase | 14 days to 35 days |
Treatment phase | The treatment phase (52 weeks) consisted of:
|
Follow-up phase | The BE MOBILE 2 study included a 20-week safety follow-up after the final dose of bimekizumab. Participants who completed week 52 and were not withdrawn may have been eligible for enrolment in an ongoing open-label extension study (AS0014; NCT04436640). |
Outcomes | |
Primary end point | ASAS40 response at week 16 |
Secondary and Exploratory end points | Secondary (fixed sequence testing)
Secondary (outside fixed sequence testing)
Exploratory analyses of the primary and secondary end points were conducted at various time points up to week 52. |
Publication status | |
Publications | NCT03928743 Van der Heijde et al. (2023)55 |
AS = ankylosing spondylitis; ASAS5/6 = an improvement of at least 20% in 5 of 6 Assessment of SpondyloArthritis International Society domains; ASAS20 = Improvement of 20% or more in the Assessment of SpondyloArthritis International Society; ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASAS-PR = partial remission in the Assessment of SpondyloArthritis International Society; ASDAS-MI = major improvement in Ankylosing Spondylitis Disease Activity Score; ASQoL = Ankylosing Spondylitis Quality of Life; AxSpA = axial spondyloarthritis; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index, BASFI = Bath Ankylosing Spondylitis Functional Index, BASMI = Bath Ankylosing Spondylitis Metrology Index; IL-17 = interleukin-17; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; mNY = modified New York criteria; NRS = numeric rating scale; NSAID = nonsteroidal anti-inflammatory drug; NSP = nocturnal spinal pain; q.4.w. = every 4 weeks; SF-36 PCS = Short Form (36) Health Survey physical component summary; TNF = tumour necrosis factor.
Sources: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
Patients were enrolled between April 25, 2019, and August 8, 2022. The final data cut-off date was September 9, 2022. Eligible study participants (N = 332) were randomized 2:1 to receive bimekizumab 160 mg/mL (n = 221) or placebo (n = 111) subcutaneously every 4 weeks. Randomization was performed via an interactive voice or web response system and stratified according to region and prior exposure to TNF alpha inhibitors (yes versus no).
The BE MOBILE trial included a screening period, a 16-week double-blind treatment period, a 36-week maintenance period, and a 20-week safety follow-up period (Figure 2). Assessments of eligibility were initiated during the screening period with a minimum duration of approximately 14 days and a maximum duration of up to 35 days. The screening period also enabled the washout of medications not permitted for use during the study, allowed initiation of treatment for latent tuberculosis where necessary, and allowed completion of the imaging assessments required to determine eligibility, including time for the reading of test results by a central laboratory.
At the end of the 16-week double-blind treatment period, after all assessments had been completed, patients in the placebo group were reallocated to receive bimekizumab 160 mg subcutaneous every 4 weeks during the 36-week maintenance period. Starting at week 20, nonbiologic rescue therapy for axSpA could have been adjusted or added. The maximum study duration per study participant was up to 73 weeks. Patients who completed the BE MOBILE 2 trial (52 weeks) were potentially eligible to be enrolled in a separate ongoing open-label extension study (AS0014; NCT04436640).
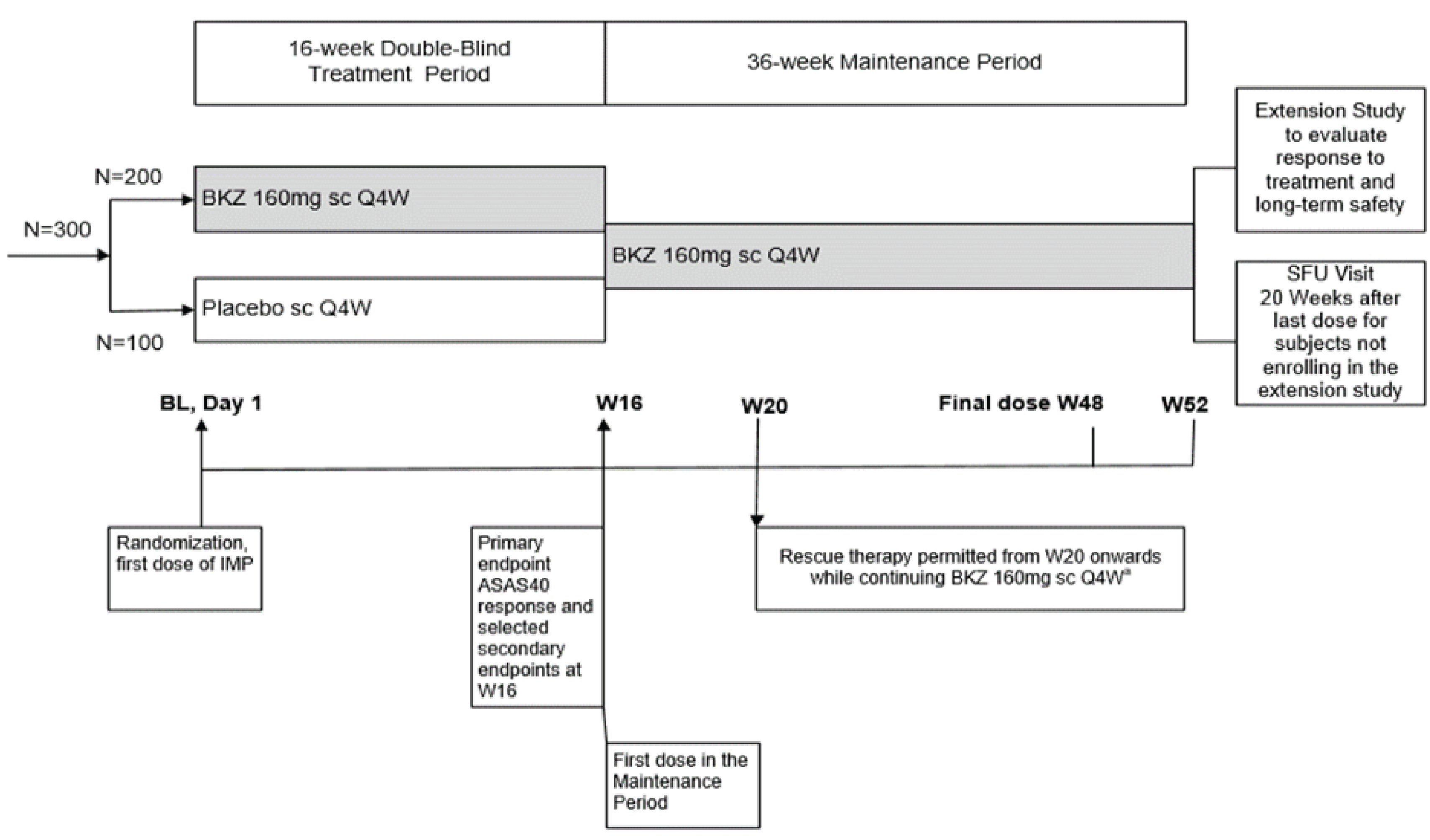
ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; BKZ = bimekizumab; BL = baseline; IMP = investigational medicinal product; q.4.w. = every 4 weeks; sc = subcutaneous; SFU = safety follow-up; W = week.
Note: The planned enrolment was approximately 300 participants.
a Study participants were eligible for nonbiologic rescue therapy starting at week 20 with treatment at the discretion of the investigator while continuing to receive BKZ. Treatment with non-BKZ biologics or prohibited treatment led to BKZ discontinuation, if applicable, per withdrawal criteria.
Source: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18
Populations
Inclusion and Exclusion Criteria
Study participants were required to be adults with a diagnosis of active AS based on radiologic evidence fulfilling the mNY criteria for AS, including experiencing 3 or more months of symptoms and younger than age 45 at symptom onset, with moderate to severe active disease at baseline (defined as BASDAI ≥ 4 plus spinal pain ≥ 4 on a 0 to 10 NRS from BASDAI item 2) and intolerant to NSAIDs or with disease that has failed to respond to NSAIDs. Patients were allowed to have previously received treatment with a TNF alpha inhibitor or to be biologic treatment–naive; the enrolment of patients experienced with TNF alpha inhibitors was limited to 30% of the total study population. Patients were excluded if they had received more than 1 TNF alpha inhibitor and/or more than 2 additional non–TNF alpha inhibitor biological-response modifiers, or any IL-17 biological-response modifiers at any time. Patients were also excluded if they were diagnosed with an inflammatory condition other than axSpA (e.g., rheumatoid arthritis). Patients with a diagnosis of Crohn disease, or ulcerative colitis or another IBD were allowed to participate if they did not have active symptomatic disease at baseline.
Interventions
Eligible patients were randomized 2:1 to receive bimekizumab (160 mg subcutaneously every 4 weeks) or placebo (subcutaneously every 4 weeks) during the double-blind treatment period. Patients in the placebo group were then reassigned to receive bimekizumab treatment (160 mg subcutaneously every 4 weeks) during the 36-week maintenance period. Bimekizumab was supplied in a 1 mL prefilled syringe at a nominal formulation of 160 mg/mL for subcutaneous injection. Placebo (preservative-free physiological saline) was also supplied in a 1 mL prefilled syringe.
Special precaution was taken during the double-blind treatment period up to week 16; given the differences in presentation between the delivery systems, the bimekizumab and placebo treatments were administered at the investigational sites by unblinded study personnel. The unblinded personnel were not involved in the study in any other way; to preserve patient and physician blinding, these staff members did not assess the patients or communicate the treatments being administered.
Patients were allowed to remain on their background medication if the dose and regimen was stable before baseline and was to be maintained until week 16. No medication increases or additions for treating AS were permitted until after the week 20 visit assessments. However, a decrease in the dose or dosing frequency of any medication was permitted at any time for reasons of intolerance, AEs, and/or side effects. All concomitant medications, including over-the-counter products, herbal and traditional remedies, vitamin and mineral supplements, other dietary supplements, “nutraceuticals,” and hormones were recorded in the study participant’s source documentation (e.g., clinical chart) and on the electronic case report form. Nonbiologic rescue therapies (e.g., acetaminophen, paracetamol, NSAIDs, cyclooxygenase-2 inhibitors, corticosteroids, methotrexate, leflunomide, hydroxychloroquine, sulfasalazine, and apremilast) could be used as add-on therapy to bimekizumab at any time from week 20 or later at the investigator’s discretion.
Protocol Amendment
A total of 4 protocol amendments were reported in the BE MOBILE 2 trial. Of these, 2 amendments were particularly noteworthy and impactful. Protocol amendment 1, made September 11, 2019, removed eligibility for patients with psoriatic arthritis from study enrolment. Protocol amendment 2, made October 17, 2019, restricted eligibility to patients whose disease had failed to respond to 2 different NSAIDs that had been taken at the maximum tolerated dose for a total of 4 weeks.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. The summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical expert consulted by CADTH and the input from the patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered the most relevant to inform CADTH’s expert committee deliberations, and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Table 6: Outcomes Summarized From the Studies Included in the Sponsor’s Systematic Review
Outcome measure | Time point | BE MOBILE 2 trial |
|---|---|---|
Efficacy outcomes | ||
Adjusted ASAS40 response rate | At 16 weeks | Primarya |
Change from baseline in BASDAI total score | Secondarya | |
Change from baseline in BASFI | Secondarya | |
Change from baseline in NSP score (based on an NRS) | Secondarya | |
Adjusted enthesitis-free rate based on the MASES index in patients with enthesitis at baseline | Secondary | |
Health-related quality of life | ||
Change from baseline in ASQoL total score | At 16 weeks | Secondarya |
Change from baseline in WPAI-SHP | Exploratory | |
Safety outcomes | ||
Serious adverse events | At 16 weeks | Exploratory |
ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASQoL = Ankylosing Spondylitis Quality of Life; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; NRS = numeric rating scale, NSP = nocturnal spinal pain; WPAI-SHP = Work Productivity and Activity Impairment Questionnaire–Specific Health Problem.
aStatistical testing for these end points was adjusted for multiple comparisons (i.e., sequential testing procedure).
Sources: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
A summary of instruments used in the BE MOBILE 2 trial are provided in Table 7.
Improvement of 40% or More in the ASAS
The primary end point in the BE MOBILE 2 trial was the ASAS40 response rate at week 16. An ASAS40 response is defined as a relative improvement of at least 40% and an absolute improvement from baseline of at least 2 units (range of 0 to 10 units on an NRS) in at least 3 of the 4 main domains (Patient’s Global Assessment of Disease Activity [PGADA], pain assessment, BASFI, and BASDAI) without any worsening in the remaining domains.18,19 ASAS40 response is considered important to patients, according to patient group input.
Bath Ankylosing Spondylitis Disease Activity Index
BASDAI was assessed as a secondary outcome in the BE MOBILE 2 trial. The BASDAI consists of 6 horizontal NRSs and was used to measure the disease activity of AS from the study participant’s perspective. Each NRS contains 10 units to measure the severity and duration of the 5 major symptoms: fatigue, spinal pain, peripheral joint pain and swelling, enthesitis, and morning stiffness over the last week. Scores range from 0 to 10, with lower scores indicating lower disease activity; a reduction in the BASDAI score is considered improvement. The ASAS inflammation component is calculated as the average of the 2 scores relating to morning stiffness measurements (i.e., question 5, “How would you describe the overall level of morning stiffness you have had from the time you wake up?” and question 6, “How long does your morning stiffness last from the time you wake up?”). The ASAS inflammation score ranges from 0 to 10. If 1 of the 2 morning stiffness measurements was missing, the other was used as the ASAS inflammation score. If both measurements were missing, the ASAS inflammation score was set to missing. An estimated median MID of 1.4 points (range, 0.9 to 1.8) has been reported for patients with active AS using anchor-based and distribution-based methods.14 The clinical expert consulted by CADTH indicated they would consider a 2-point improvement from baseline as clinically meaningful for within-group difference. The BASDAI was commonly used in clinical practice to assess patients with AS and considered an important outcome by patients. In addition, BASDAI data were the source of key inputs to the pharmacoeconomic model that was submitted to CADTH.
Bath Ankylosing Spondylitis Functional Index
BASFI was assessed as a secondary outcome in the BE MOBILE 2 trial.
The BASFI contains 10 questions and was measured by the study participant to assess their physical functions during the past week. The first 8 questions evaluate activities related to functional anatomic limitations due to the course of AS. The final 2 questions evaluate the study participants’ ability to cope with everyday life. An NRS ranging from 0 (easy) to 10 (impossible) was used to answer the questions on the test. The arithmetic mean of the 10 scores gives the BASFI score, which is a value between 0 and 10, with lower scores indicating better physical function. In case of missing answers to 1 or 2 of the single items within the questionnaire, the BASFI score was calculated by imputing the missing items using the mean of the completed items. Then, the BASFI score was calculated as described previously. If more than 2 of the items were missing, the BASFI score was shown as missing. An estimated median MID of 1.1 points (range, 1.0 to 1.4) has been reported for patients with AS using anchor-based and distribution-based methods.14 According to patient input, the BASFI has been commonly used in clinical practice to assess patients with AS and is considered an important outcome by patients.
Nocturnal Spinal Pain
NSP (based on an NRS) was assessed as a secondary outcome in the BE MOBILE 2 trial. The NSP score was analyzed as a secondary end point. The spinal pain experienced by participants was measured using 2 separate questions: total pain in the spine due to AS, and pain in the spine at night (i.e., the NSP) due to AS. Participants considered the average amount of pain in the preceding week and reported it on a scale of 0 (no pain) to 10 (most severe pain).18,19 Using anchor-based and distribution-based methods, an estimated median MID of 1.5 points (range, 1.1 to 2.3) has been reported for patients with active AS.14 According to patient input, the NSP has been commonly used in clinical practice to assess patients with AS and is considered an important outcome by patients.
Maastricht Ankylosing Spondylitis Enthesitis Score
MASES was assessed as a secondary outcome in the BE MOBILE 2 trial. The MASES is an index that measures the severity (i.e., intensity and extent) of enthesitis through the assessment of 13 entheses (bilateral costochondral [1], costochondral [7], anterior superior iliac spine, posterior iliac spine, iliac crest and proximal insertion of the Achilles tendon sites, and the fifth lumbar vertebral body spinous process). Each result is scored as 0 or 1 and then summed for a possible score of 0 to 13, with higher scores indicating worse enthesitis. The enthesitis-free state is based on the MASES index and is defined as study participants having achieved a MASES index of 0 at week 16.18,19 According to patient input, the MASES has been commonly used in clinical practice to assess patients with AS and is considered an important outcome by patients.
Ankylosing Spondylitis Quality of Life
ASQoL was assessed as a secondary outcome in the BE MOBILE 2 trial. The ASQoL is an 18-item questionnaire that was developed to measure HRQoL in patients with active AS. Each item on the questionnaire is given a score of 1 (yes, QoL impaired) or 0 (no, QoL not impaired). Total scores range from 0 to 18, with higher scores indicating worse HRQoL.18,19 An estimated MID of 1 unit of worsening (i.e., change of + 1) or 2 units of improvement (i.e., change of −2) in patients with AS was identified in the literature.15 The ASQoL is considered an important outcome by patients with AS, according to the patient input..
Work Productivity and Activity Impairment Questionnaire–Specific Health Problem
The WPAI-SHP (version 2.0) is a patient-reported questionnaire that assesses patient’s employment status, work absenteeism, work impairment while working (presenteeism), overall work, and daily activity impairment attributable to a specific health problem.56,57 Five out of the 6 items of the WPAI-SHP are regrouped into 4 dimensions, with scores expressed as percentages, where higher numbers indicate greater impairment and less productivity, i.e., worse outcomes, as described in the WPAI-SHP scoring rules. The WPAI-SHP is considered an important outcome by patients with AS, according to the patient input.
Safety Outcomes
In the BE MOBILE 2 trial, patients who experienced serious and nonserious AEs during the double-blind treatment period had their experiences recorded and reported using the Medical Dictionary for Regulatory Activities (version 19.0). Every AE was followed until it resolved or stabilized, the investigator determined it was no longer clinically significant, or the patient was lost to follow-up. If a patient’s AE was ongoing at the end of the study, follow-up was to be provided until resolution or a stable level of sequelae was achieved, the investigator no longer deemed the AE clinically significant, or the patient was lost to follow-up. If no follow-up was provided, the investigator had to justify. The follow-up was usually continued for 20 weeks after the patient discontinued treatment with bimekizumab. Notable harms included serious infection, fungal infection, opportunistic infection, malignancy, major adverse cardiac event, neutropenia, suicidal ideation and behaviour, IBD, hypersensitivity reaction, and liver injury or disorder.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
ASAS | A composite set of response criteria that is commonly used in AS trials; contains 6 domains. ASAS40: ≥ 40% relative and absolute improvement from baseline of ≥ 2 units (range, 0 to 10 units on an NRS) in ≥ 3 of 4 domains (PGADA, pain assessment, BASFI, BASDAI), without any worsening in the remaining domain.19 | The ASAS40 has a good discriminatory capacity between treatment (with infliximab) and placebo (χ2 = 26.5; 95% CI, 13.3 to 41.1).58 The criteria for the ASAS20 were identified as the best-performing criteria out of 20 different ASAS-based criteria, with strong discriminatory performance (χ2 = 54.2 based on 923 observations).59 | Unknown |
ASQoL | An 18-item questionnaire was developed for measuring HRQoL in patients with active AS. Each item on the questionnaire is given a score of 1 (yes, QoL impaired) or 0 (no, QoL not impaired). Total scores range from 0 to 18, with higher scores indicating worse HRQoL.19 | Content validity has been ensured during the development stage by incorporating interviews with patients with AS.60 Evidence of excellent internal consistency (alpha = 0.89 to 0.92), test-retest reliability (Spearman r and ICC = 0.91 to 0.92), convergent validity (Spearman r = 0.72 to 0.75 with BASFI), and construct validity (differentiated disease activity, perceived general health, perceived severity, P < 0.001) in patients with AS.60 High test–retest reliability (r > 0.9), a good correlation with BASDAI (r = 0.79). Responsiveness to self-perceived change in health was noted in another study.61 | One unit of worsening (i.e., + 1) or 2 units of improvement (i.e., −2) in patients with AS.15 |
BASDAI | Self-reported, disease-specific Questionnaire that consists of six 10-unit horizontal NRSs to measure severity of fatigue, spinal and peripheral joint pain and swelling, enthesitis, and morning stiffness (both severity and duration) over the last week. Scores range from 0 to 10, with lower scores indicating lower disease activity.19 | Test–retest (median, 7 days; range, 4 to 10 days) reliability (r > 0.9) has been demonstrated. Internal consistency (alpha = 0.83 to 0.94) and construct validity were acceptable.62 BASDAI appeared to be sensitive to change, reflecting a 16% (mean) improvement in patient scores after 3 weeks of intensive physiotherapy treatment.63 Demonstrated responsiveness during treatment with TNF alpha inhibitors in patients with SpA (effect size = 1.86).64 | 10 mm in absolute value or 22.5% in relative value.65 0.7 units (1.1 units for active AS) for improvement.66 A median estimated MID of 1.4 points (range, 0.9 to 1.8) in patients with AS using anchor-based and distribution-based methods.14 |
BASFI | Self-administered instrument addressing physical function and patient’s ability to cope with everyday life. The mean of the scores (ranging from 0 to 10 points) for 10 items is calculated, and lower scores indicate better physical function.19 | Test–retest (median 7 days; range 4 to 10 days) reliability (r > 0.9) has been demonstrated. Internal consistency (alpha = 0.93 to 0.97) and construct validity were acceptable.62,67 BASFI is 1 of 3 AS assessment instruments with the most extensive evidence for validity through comparison with instruments that measure similar or related constructs, and/or with measures of mobility.68 | 7 mm on VAS or 17.5% of the baseline score.65 0.4 units (0.6 for active AS) for improvement.66 A median estimated MID of 1.1 points (range, 1.0 to 1.4) in patients with AS using anchor-based and distribution-based methods.14 |
MASESa | An index that measures the severity (i.e., intensity and extent) of enthesitis through the assessment of 13 entheses (bilateral costochondral 1, costochondral 7, anterior superior iliac spine, posterior iliac spine, iliac crest and proximal insertion of the Achilles tendon sites, and the fifth lumbar vertebral body spinous process).19 Each result is scored as 0 or 1 and then summed for a possible score of 0 to 13, with higher scores indicating worse enthesitis.69 | Weak positive correlations with BASDAI, BASFI, and fatigue.70 MASES was not significantly correlated with BASMI or ASQoL.70 | Unknown. |
NSP score (based on an NRS) | Patient-reported questionnaire consisting of 2 questions:19
Score is calculated based on the average amount of pain experienced by the patient in the preceding week on a scale of 0 (no pain) to 10 (most severe pain).19 | In patients with AS (n = 91), test–retest-reliability (ICC = 0.9), internal consistency (alpha = 0.93 to 0.97), known-group validity based on clinical outcomes (PGADA and ASDAS), and responsiveness to change based on PGADA criteria have been demonstrated. Moderate to high correlations with PGADA (Pearson r = 0.38 to 0.64) and ASDAS (r = 0.62 to 0.77) have been noted.14 | A median estimated MID of 1.5 points (range, 1.1 to 2.3) in patients with AS using anchor-based and distribution-based methods.14 |
WPAI-SHP | A patient-reported questionnaire that assesses employment status, work absenteeism, work impairment while working (presenteeism), overall work, and daily activity impairment attributed to a specific health problem.56,57 In the BE MOBILE studies, 5 out of 6 items were regrouped into the 4 dimensions, with scores expressed as percentages, where higher numbers indicated greater impairment and less productivity, i.e., worse outcomes. | No evidence for measurement properties was found in patients with AS. | Unknown. |
AS = ankylosing spondylitis; ASAS = Assessment of SpondyloArthritis International Society; ASAS20 = improvement of 20% or more in the Assessment of SpondyloArthritis International Society; ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASQoL = Ankylosing Spondylitis Quality of Life; axSpA = axial spondylarthritis; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; BASMI = Bath Ankylosing Spondylitis Metrology Index; CI = confidence interval; ES = effect size; HRQoL = health-related quality of life; ICC = intraclass coefficient; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; MEI = Mander Enthesitis Index; MID = minimal important difference; NRS = numeric rating scale; NSP = nocturnal spinal pain; PGADA = Patient’s Global Assessment of Disease Activity; QoL = quality of life; VAS = visual analogue scale; WPAI-SHP = Work Productivity and Activity Impairment Questionnaire–Specific Health Problem.
aAnalyses of the MASES index were restricted to the subset of study participants with enthesitis at baseline (i.e., a MASES index score > 0). If ≥ 7 items were available, MASES was derived by dividing the sum score by the number of available assessments and multiplying the result by 13. If < 7 items were available, MASES was treated as missing. An enthesitis-free state based on the MASES index was a categorical status defined as achieving a MASES index score of 0 (i.e., complete resolution of enthesitis) at week 16 in the BE MOBILE 2 trial.
Source: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18
Statistical Analysis
Details of the statistical analysis methods used in the BE MOBILE 2 trial are summarized in Table 8.
Sample Size and Power Calculation
The sample size and power calculations were done at a significance level of 0.05 in a 2-sided 2-sample Chi square test with continuity correction, in addition to using software (nQuery Advisor 7.0). Approximately 300 study participants were planned to be randomly assigned (2:1 ratio) to either the treatment group (200 patients) or the placebo group (100 patients). The following assumptions were made regarding sample size and power calculations:
Using the ASAS40 response data from the phase IIb bimekizumab study (AS0008), it was assumed there would be a 46.7% rate of response to bimekizumab 160 mg every 4 weeks at week 12.71
It was assumed the treatment responses at week 12 and week 16 would be the same.
The ASAS40 response rate for placebo at week 16 was assumed to be 15%.
In addition, the observed ASAS40 response rates were adjusted to account for a higher number of study participants with prior exposure to TNF alpha inhibitors and a higher number of study participants with early withdrawal.
Of note, the BE MOBILE 2 trial was not powered to detect any change in the ASAS40 response rate between bimekizumab and placebo in subgroup analyses, except for the subgroup of patients who are TNF inhibitor–naive.
Statistical Test or Model
The primary end point of ASAS40 response at week 16 was analyzed for all study participants in the randomized set. The statistical null hypothesis for the ASAS40 response at week 16 was that there is no difference between the bimekizumab treatment and placebo groups in the proportion of study participants with an ASAS40 response (i.e., the conditional OR for ASAS40 response in the bimekizumab treatment compared with placebo was equal to 1). The alternative hypothesis was that there is a difference between bimekizumab treatment and placebo. A logistic regression model was used to assess the treatment effect on ASAS40 response at week 16. The model included fixed effects for treatment, and prior exposure to TNF alpha inhibitors (yes versus no) and region as stratification factors. The suitability of including these variables in the model was assessed using the Hosmer-Lemeshow goodness-of-fit test.
Multiple Testing Procedure
A fixed sequence testing procedure was applied for the primary end point of ASAS40 and the key secondary end points. The testing procedure accounted for multiplicity and control of the familywise type I error rate at a 2-sided alpha of 0.05. According to this strategy, the statistical testing of an end point can be investigated only if the null hypothesis for the previous end point has been rejected (i.e., if P < 0.05). Figure 3 shows the testing order for the primary and key secondary end points in the BE MOBILE 2 trial.
Figure 3: Sequential Testing Procedure of Primary and Key Secondary Efficacy End Points From the BE MOBILE 2 Trial

Source: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18
Data Imputation Methods
The primary efficacy analysis evaluated the composite estimand using NRI that combines the clinically meaningful improvement from baseline in ASAS40 response at week 16 and the intercurrent event of not discontinuing early from study treatment for any reason before week 16.
The following 4 attributes describe the composite estimand that was used to define the treatment effect of interest for the primary efficacy analysis:
Population: Study participants were enrolled according to the protocol-specified inclusion and exclusion criteria and randomized to bimekizumab.
Study participant-level outcome: ASAS40 at week 16.
Intercurrent event handling: An intercurrent event was defined as discontinuation of study treatment before week 16. A composite strategy was implemented in which a positive clinical outcome was defined as achieving ASAS40 at week 16 and not discontinuing study treatment through week 16.
Population-level summary measure: Conditional OR comparing bimekizumab with placebo.
Intercurrent events were acknowledged as an unfavourable outcome for the composite because study participants with intercurrent events were categorized as not having experienced a response to the study treatment (nonresponder). Consequently, if the date of an intercurrent event occurred before or at week 16, study participants were considered nonresponders at week 16. Additionally, missing data at week 16 that were not preceded by an intercurrent event were imputed as nonresponders.
Other methods were also used to assess the effects of missing data as defined in the sensitivity analyses.
Subgroup Analyses
Subgroup analyses were performed on the primary efficacy end point of ASAS40 response at week 16 on the randomized set. Subgroups analyses (except for the subgroup of patients who were TNF inhibitor–naive) did not take multiplicity into account. The following subgroups are considered relevant based on input from the clinical expert consulted by CADTH:
gender (male, female)
disease duration (< 2 years, ≥ 2 years)
body mass index (BMI) (< 18.5, ≥ 18.5 to < 25, ≥ 25 to < 30, ≥ 30)
high-sensitivity C-reactive protein level (≤ upper limit of normal of 5 mg/L, > upper limit of normal of 5 mg/L)
prior exposure to TNF alpha inhibitors (yes, no)
HLA-B27 positivity (yes, no)
For each subgroup analysis, a logistic regression was fitted that involved the same terms from the primary analysis model and additional terms for the subgroup and the subgroup by treatment interaction. However, for the analysis by stratification variables (i.e., prior exposure to TNF alpha inhibitors and region), the analysis model contained the same terms from the primary analysis model plus the subgroup by treatment interaction only. The same imputation method as the 1 used for the primary analysis (i.e., NRI) was used to handle missing data. The covariates were provided in the same order as for the primary analysis model, with the terms for subgroup and for subgroup by treatment interaction added at the end of the model statement. For each subgroup category and each treatment group, the number of responders, the adjusted responder rate with the associated 95% CI, and the adjusted OR (for the comparison of bimekizumab and placebo) and associated 95% CI were provided. The ORs and associated 95% CIs for each subgroup category are displayed on a single forest plot in Appendix 1 (Figure 7).
Sensitivity Analyses
Several planned sensitivity analyses were conducted for the primary outcome and other outcomes using additional analysis sets (per-protocol set, full analysis set, and COVID-19–free set) and alternative methods for handling missing data (multiple imputation, treatment policy strategy, observed case, or tipping-point analysis), as presented in Table 8.
Secondary Outcomes
The change from baseline in BASFI at week 16, change from baseline in NSP score at week 16, and change from baseline in ASQoL total score at week 16 were analyzed using the analysis of covariance with treatment, region, and prior exposure to TNF alpha inhibitors (yes versus no) as fixed effects and the respective baseline value as covariate. Missing data were imputed using reference-based multiple imputation.
The adjusted enthesitis-free rate (based on the MASES index) at week 16 among the study participants with enthesitis at baseline was analyzed using logistic regression for treatment effect and adjusted for prior TNF alpha inhibitor exposure (yes versus no) and region; the comparison was based on the 2-sided Wald test (alpha = 0.05). Missing data were imputed using multiple imputation.
The change from baseline in WPAI-SHP at week 16 was analyzed using the analysis of covariance with treatment, region, and prior TNF alpha inhibitor exposure (yes versus no) as fixed effects and the baseline value as covariate. Work productivity and activity impairment scores were based on 1 item related to activity impairment while working (presenteeism), 2 items related to absenteeism, and multiple items related to overall work productivity. A score could not be calculated if a response to a corresponding item was missing. Because questions 2 to 6 were only relevant when the answer to question 1 (Are you currently employed?) was yes, and because the imputation of results was questionable without knowing the answer to question 1, only observed case data were analyzed for this end point.
Table 8: Statistical Analysis of Efficacy End Points From the BE MOBILE 2 Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Adjusted ASAS40 response rate at week 16 | Logistic regression for treatment effect, comparison based on the 2-sided Wald test (alpha = 0.05) | Stratification factors: prior TNF alpha inhibitor exposure (yes vs. no) and region | Nonresponder imputation |
|
Change from baseline at week 16 in: | — | — | NR | |
BASDAI total score | ANCOVA | Reference-based MI | ||
BASFI score | ||||
NSP score | ||||
ASQoL total score | ||||
WPAI-SHP | OC | |||
Adjusted enthesitis-free rate based on the MASES index at week 16 among study participants with enthesitis at baseline | Logistic regression, 2-sided Wald test (alpha = 0.05) | MI |
ANCOVA = analysis of covariance; ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASQoL = Ankylosing Spondylitis Quality of Life; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; FAS = full analysis set; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; MI = multiple imputation; NR = not reported; NSP = nocturnal spinal pain; OC = observed case; PPS = per-protocol set; TNF = tumour necrosis factor; WPAI-SHP = Work Productivity and Activity Impairment Questionnaire–Specific Health Problem.
Sources: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
Analysis Populations
A summary of analysis populations used in the BE MOBILE 2 trial is provided in Table 9.
Table 9: Analysis Populations of the BE MOBILE 2 Trial
BE Mobile 2 trial populations | Definition | Application |
|---|---|---|
Randomized set | Consists of all randomized study participants | Primary and secondary efficacy analyses |
Safety set | Consists of all randomized study participants who received at least 1 dose of bimekizumab | Safety analyses, demographic characteristics |
Maintenance set | Consists of all study participants who received at least 1 dose of bimekizumab treatment in the maintenance treatment period | Supportive efficacy analyses, safety analyses, demographic characteristics |
Full analysis set | Consists of all randomized study participants who received at least 1 dose of bimekizumab and had a valid measurement for all components of the primary efficacy variable at baseline | Primary and supportive efficacy analyses |
Per-protocol set | Consists of all study participants in the randomized set who had no important protocol deviations affecting the primary efficacy variable | Supportive efficacy analyses |
Observed case set | Consists of observed data for study participants who were still on the randomized treatment at week 16 | Sensitivity analyses |
Sources: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
Results
Patient Disposition
A total of 612 patients were screened; of these, 280 patients (45.8%) did not pass screening. Ineligibility due to not meeting inclusion criterion 4 (diagnosis of AS per mNY criteria, including documented radiologic evidence based on central laboratory reading) was the most common reason (42.2%) for screen failure. A total of 332 patients were randomized and started the double-blind treatment period, 221 patients in the bimekizumab group and 111 patients in the placebo group. The percentages of study participants who completed the double-blind treatment period were similar between the treatment groups: 213 patients (96.4%) in the bimekizumab group and 109 patients (98.2%) in the placebo group. During the double-blind treatment period, a higher percentage of patients in the bimekizumab group (3.6%) discontinued from the study compared with the placebo group (1.8%). The most common primary reasons for discontinuation during the double-blind treatment period were due to an AE (1.4% for bimekizumab versus 0 for placebo) and withdrawal by study participant (1.4% versus 0.9%).
Important Protocol Deviations
The incidence of important protocol deviations was similar between the treatment groups (5.0% for bimekizumab versus 4.5% for placebo) (Table 11). Overall, the most common important protocol deviation was prohibited concomitant medication use (1.4% versus 2.7%), which was reported more frequently in the placebo group compared with the bimekizumab group.
Table 10: Summary of Patient Disposition From the BE MOBILE 2 Trial (Enrolled Set)
Patient disposition | BE MOBILE 2 trial | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
Screened, N | 612 | |
Screen failure, n | 280 | |
Reason for screening failure, n | — | |
Ineligibilitya | 258 | |
Otherb | 11 | |
Withdrawal by study participant | 9 | |
Adverse event | 1 | |
Lost to follow-up | 1 | |
Started double-blind treatment period, N (%) | 221 (100) | 111(100) |
Completed double-blind treatment period, n (%) | 213 (96.4) | 109 (98.2) |
Discontinued from study during double-blind treatment period, n (%) | 8 (3.6) | 2 (1.8) |
Reason for discontinuation, n (%) | ||
Adverse events | 3 (1.4) | 0 |
Withdrawal by study participant | 3 (1.4) | 1 (0.9) |
Lack of efficacy | 1 (0.5) | 0 |
Other | 1 (0.5) | 1 (0.9) |
Randomized set, N | 221 | 111 |
FAS, N | 220 | 111 |
PP, N | 204 | 106 |
Safety set, N | 221 | 111 |
AS = ankylosing spondylitis; FAS = full analysis set; mNY criterial = modified New York criteria; PP = per protocol.
aMost study participants were ineligible because they did not meet inclusion criterion 4 (diagnosis of AS per mNY criteria, including documented radiologic evidence [X-ray] read by a central laboratory).
bThe cases with “other” as the reason for screening failure relate mainly to the COVID-19 situation, such as hospital closures and UCB Inc.’s decision to stop enrolment during the first pandemic wave.
Sources: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
Table 11: Summary of Important Protocol Deviations From the BE MOBILE 2 Trial (Randomized Set)
Important protocol deviation | BE MOBILE 2 trial | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
Patients with at least 1 important protocol deviation, n (%) | 11 (5.0) | 5 (4.5) |
Prohibited concomitant medication use | 3 (1.4) | 3 (2.7) |
COVID-19 treatment deviation | 3 (1.4) | 1 (0.9) |
Treatment nonadherence | 3 (1.4) | 0 |
Procedural noncompliance | 1 (0.5) | 1 (0.9) |
Incorrect treatment or dose | 1 (0.5) | 0 |
COVID-19 visit deviation | 1 (0.5) | 1 (0.9) |
Source: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18
Baseline Characteristics
Overall, demographic characteristics were well balanced between treatment groups. The mean age of all study participants was 40.4 years with a range of 19 to 80 years. The majority of study participants were male (72.3%) and white (80.4%), 27.7% of participants were female, 0.3% of participants were Black, 0.9% participants were from another or a mixed racial group, and the racial group information was missing for 1.2% of participants. Study participants were most commonly enrolled in the following regions: Eastern Europe (49.1%), Western Europe (29.8%), Asia (18.4%), and North America (2.7%).
The treatment groups were generally well balanced at baseline with respect to AS-related and other disease characteristics. At baseline, the majority of all study participants were using NSAID therapies (79.8%) and anti-TNF therapy had previously been used by 16.3% of study participants. Generally, the baseline disease burden factors were well balanced across treatment groups.
The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or were considered to affect the outcomes or interpretation of the study results.
Table 12: Summary of Baseline Characteristics From the BE MOBILE 2 Trial (Safety Set)
Characteristic | BE MOBILE 2 trial | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
Demographic characteristics | ||
Age, years | ||
Mean (SD) | 41.0 (12.1) | 39.2 (12.6) |
Median (range) | 40.0 (19.0 to 80.0) | 38.0 (19.0 to 75.0) |
Age, n (%) | ||
18 to < 65 years | 212 (95.9) | 109 (98.2) |
65 to < 85 years | 9 (4.1) | 2 (1.8) |
≥ 85 years | 0 | 0 |
Gender, n (%) | ||
Male | 160 (72.4) | 80 (72.1) |
Female | 61 (27.6) | 31 (27.9) |
Racial group, n (%) | ||
White | 177 (80.1) | 90 (81.1) |
Asian | 37 (16.7) | 20 (18.0) |
Other or mixed | 3 (1.4) | 0 |
Black | 0 | 1 (0.9) |
Missing | 4 (1.8) | 0 |
Region, n (%) | ||
Asia | 40 (18.1) | 21 (18.9) |
Eastern Europe | 108 (48.9) | 55 (49.5) |
North America | 6 (2.7) | 3 (2.7) |
Western Europe | 67 (30.3) | 32 (28.8) |
Weight (kg) | ||
Mean (SD) | 80.0 (19.1) | 81.3 (18.5) |
Median (range) | 77.5 (37.0 to 159.0) | 78.7 (42.6 to 130.3) |
BMI (kg/m2) | ||
Mean (SD) | 26.8 (5.7) | 27.08 (5.8) |
Median (range) | 26.0 (15.2 to 56.0) | 26.1 (17.5 to 45.7) |
Disease characteristics | ||
Prior TNF alpha inhibitor exposure, n (%) | ||
Yes | 37 (16.7) | 17 (15.3) |
No | 184 (83.3) | 94 (84.7) |
Time since first diagnosis of AS (years) | ||
Mean (SD) | 6.7 (8.3) | 5.7 (6.9) |
Median (range) | 4.0 (0.1 to 37.7) | 3.4 (0.1 to 41.0) |
Time since first symptoms of AS (years) | ||
Mean (SD) | 14.2 (11.01) | 11.9 (8.6) |
Median (range) | 10.9 (0.6 to 59.1) | 10.6 (0.4 to 41.0) |
Modified NY clinical and radiological criteria, n (%) | ||
Clinical criterion Aa | 221 (100.0) | 111 (100.0) |
Clinical criterion Bb | 195 (88.2) | 93 (83.8) |
Clinical criterion Cc | 143 (64.7) | 59 (53.2) |
Radiologic criteriond | 221 (100) | 111 (100) |
HLA-B27, n (%) | ||
Positive | 191 (86.4) | 93 (83.8) |
Negative | 30 (13.6) | 18 (16.2) |
Past anti-TNF therapy, n (%) | ||
Yes | 37 (16.7) | 17 (15.3) |
No | 184 (83.3) | 94 (84.7) |
Current NSAID therapies,e n (%) | ||
Yes | 180 (81.4) | 85 (76.6) |
No | 41 (18.6) | 26 (23.4) |
Current conventional synthetic DMARDs,e n (%) | ||
Yes | 47 (21.3) | 20 (18.0) |
No | 174 (78.7) | 91 (82.0) |
Current conventional synthetic DMARD type,e n (%) | ||
Methotrexate | 7 (3.2) | 5 (4.5) |
Sulfasalazine | 38 (17.2) | 15 (13.5) |
Otherf | 2 (0.9) | 0 |
Current oral corticosteroid use,e n (%) | ||
Yes | 15 (6.8) | 8 (7.2) |
No | 206 (93.2) | 103 (92.8) |
Current analgesic/opioid therapies,e n (%) | ||
Yes | 31 (14.0) | 14 (12.6) |
No | 190 (86.0) | 97 (87.4) |
Baseline disease activity and burden | ||
PGADAg | ||
Mean (SD) | 6.6 (2.0) | 6.7 (1.8) |
Median (range) | 7.0 (0 to 10.0) | 7.0 (0 to 10.0) |
Total spinal pain NRS scoreg | ||
Mean (SD) | 7.1 (1.6) | 7.2 (1.2) |
Median (range) | 7.0 (2.0 to 10.0) | 7.0 (4 to 10) |
BASFI scoreg | ||
Mean (SD) | 5.3 (2.2) | 5.2 (2.0) |
Median (range) | 5.5 (0.0 to 9.6) | 5.4 (0.4 to 9.2) |
Mean of BASDAI questions 5 and 6g | ||
Mean (SD) | 6.7 (1.9) | 6.8 (1.6) |
Median (range) | 6.5 (2.0 to 10.0) | 6.5 (2.5 to 10.0) |
BASDAI total score | ||
Mean (SD) | 6.5 (1.3) | 6.5 (1.3) |
Median (range) | 6.4 (3.7 to 9.4) | 6.5 (4.0 to 9.3) |
BASDAI spinal pain question 2 score | ||
Mean (SD) | 7.4 (1.4) | 7.3 (1.3) |
Median (range) | 8.0 (4.0 to 10.0) | 7.0 (4.0 to 10.0) |
hs-CRP (mg/L) | ||
Geometric mean (geoCV %) | 6.5 (275.0) | 6.7 (197.4) |
Median (range) | 8.2 (0.1 to 105.4) | 6.3 (0.3 to 104.3) |
hs-CRP, n (%)h | ||
≤ ULN | 84 (38.0) | 44 (39.6) |
> ULN | 137 (62.0) | 67 (60.4) |
ASDAS-CRP, n (%) | ||
Number of patients contributing to the analysis | 220 | 111 |
Mean (SD) | 3.7 (0.8) | 3.7 (0.8) |
Median (range) | 3.7 (1.8 to 6.0) | 3.7 (2.1 to 5.4) |
ASDAS-CRP status,i n (%) | ||
Inactive disease | 0 | 0 |
Low disease activity | 3 (1.4) | 0 |
High disease activity | 84 (38.0) | 47 (42.3) |
Very high disease activity | 133 (60.2) | 64 (57.7) |
Missing | 1 (0.5) | 0 |
AS = ankylosing spondylitis; ASAS = Assessment of SpondyloArthritis International Society; ASDAS = Ankylosing Spondylitis Disease Activity Score; ASDAS-CRP = Ankylosing Spondylitis Disease Activity Score using C-reactive protein; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; BMI = body mass index; DMARD = disease-modifying antirheumatic drug; geoCV = geometric coefficient of variation; HLA-B27 = human leukocyte antigen B27; hs-CRP = high-sensitivity C-reactive protein; NRS = numeric rating scale; NSAID = nonsteroidal anti-inflammatory drug; NY = New York; PGADA = Patient's Global Assessment of Disease Activity; SD = standard deviation; TNF = tumour necrosis factor; ULN = upper limit of normal.
aCriterion A referred to low back pain and stiffness for more than 3 months that improved with exercise but was not relieved by rest.
bCriterion B referred to limitation of motion of the lumbar spine in both the sagittal and frontal planes.
cCriterion C referred to limitation of chest expansion relative to normal values corrected for age and sex.
dRadiologic criterion referred to sacroiliitis grade ≥ 2 bilaterally or grade 3 to 4 unilaterally.
eCurrent medications were medications concomitant at baseline.
fThe other category includes participants receiving a concomitant combination of the above conventional synthetic DMARDs.
gThis was a component of the primary outcome measure of ASAS.
hThe ULN value for hs-CRP was 5 mg/L.
iASDAS-CRP status definition: Inactive disease is when ASDAS-CRP < 1.3; low disease activity is when ASDAS-CRP ≥ 1.3 to < 2.1; high disease activity is when ASDAS-CRP ≥ 2.1 to ≤ 3.5; and very high disease activity is when ASDAS-CRP > 3.5.
Sources: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
Prior and Concomitant Disease
In the final analysis safety set (data cut-off date: September 9, 2022), a slightly higher proportion of patients in the bimekizumab group reported a previous and ongoing medical history condition compared with the placebo group (93.2% for bimekizumab versus 90.1% for placebo) (Table 13). Generally, the most frequently reported conditions or diseases (≥ 15% of patients in either treatment group) at baseline were similar between treatment groups, except for tendinitis (27.1% versus 19.8%), which was reported more frequently in the bimekizumab group, and uveitis (10.4% versus 18.9%), which was reported more frequently in the placebo group.
History of Peripheral and Extra-Articular Manifestations
In the final analysis safety set (data cut-off date: September 9, 2022), generally, the most frequently reported disease history of peripheral and extra-articular manifestations at screening or baseline were similar between treatment groups, except for enthesitis, which was reported more frequently in the bimekizumab group (29.0% for bimekizumab versus 21.6% for placebo), and uveitis, which was reported more frequently in the placebo group (14.9% versus 21.6%) (Table 14).
Table 13: Summary of Prior and Concomitant Disease From the BE MOBILE 2 Trial (Safety Set)
Disease | BE MOBILE 2 trial | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
Patients with any previous and ongoing medical history conditions, n (%) | 206 (93.2) | 100 (90.1) |
Commonly reported prior and concomitant disease (≥ 15% of patients), n (%) | ||
Musculoskeletal and connective tissue disorders | 134 (60.6) | 67 (60.4) |
Tendinitis | 60 (27.1) | 22 (19.8) |
Arthritis | 48 (21.7) | 22 (19.8) |
Peripheral arthritis | 29 (13.1) | 17 (15.3) |
Metabolism and nutrition disorders | 74 (33.5) | 23 (20.7) |
Gastrointestinal disorders | 64 (29.0) | 26 (23.4) |
Infections and infestations | 64 (29.0) | 26 (23.4) |
Vascular disorders | 58 (26.2) | 26 (23.4) |
Hypertension | 52 (23.5) | 23 (20.7) |
Eye disorders | 46 (20.8) | 25 (22.5) |
Uveitis | 23 (10.4) | 21 (18.9) |
Skin and subcutaneous tissue disorders | 35 (15.8) | 20 (18.0) |
Immune system disorders | 24 (10.9) | 19 (17.1) |
Source: BE MOBILE 2 trial final Clinical Study Report (data cut-off date: September 9, 2022).18
Exposure to Study Treatments
The median time on bimekizumab during the double-blind treatment period was 112 days for both the bimekizumab and placebo groups (Table 15). Nonbiologic rescue therapy was at the investigator’s discretion as an add-on therapy to bimekizumab at any time from week 20; therefore, during the 16-week double-blind treatment period, no patients received rescue therapy. Treatment adherence was similar between treatment groups (97.2% for bimekizumab versus 97.3% for placebo) during the double-blind treatment period.
Table 14: Summary of History of Peripheral and Extra-Articular Manifestations From the BE MOBILE 2 Trial (Safety Set)
Manifestations | BE MOBILE 2 trial, n (%) | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
Peripheral arthritis | 85 (38.5) | 40 (36.0) |
Enthesitis | 64 (29.0) | 24 (21.6) |
Heel enthesitis | 43 (19.5) | 19 (17.1) |
Nonheel enthesitis | 24 (10.9) | 7 (6.3) |
Uveitis | 33 (14.9) | 24 (21.6) |
Psoriasis | 16 (7.2) | 10 (9.0) |
Dactylitis | 12 (5.4) | 6 (5.4) |
Inflammatory bowel disease | 3 (1.4) | 1 (0.9) |
Source: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18
Table 15: Summary of Treatment Exposure During Double-Blind Treatment Period From the BE MOBILE 2 Trial
Exposure | BE MOBILE 2 trial | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
Duration, days | ||
Mean (standard deviation) | 108.1 (15.0) | 110.4 (11.7) |
Median (range) | 112.0 (28.0 to 117.0) | 112.0 (16.0 to 133.0) |
Sources: BE MOBILE 2 trial final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
Concomitant Medications
The use of concomitant medications was similar between the bimekizumab and placebo groups in the double-blind treatment period (95.0% for bimekizumab versus 93.7% for placebo) (Table 16). The most common concomitant medications (≥ 50% of patients) in the bimekizumab group versus the placebo group were musculoskeletal system medications (86.0% versus 79.3%), nonsteroidal anti-inflammatory and antirheumatic products (85.1% versus 79.3%), and drugs used to treat the alimentary tract and metabolism, which were reported more frequently in the bimekizumab group compared with the placebo group (56.6% versus 50.5%).
The most common concomitant axSpA-related medications used during the double-blind treatment period were NSAIDs (81.9% for bimekizumab versus 78.4% for placebo) and conventional synthetic DMARDs, which were reported more frequently in the bimekizumab group compared with the placebo group (21.3% versus 18.0%). Use of analgesics and opioids was reported by a slightly higher proportion of patients in the placebo group compared with the bimekizumab group (14.9% versus 15.3%) (Table 17). The most common concomitant NSAIDs used during the double-blind treatment period were etoricoxib (17.2% for bimekizumab versus 15.3% for placebo) and meloxicam (15.4% versus 11.7%), which were both reported more frequently in the bimekizumab group compared with the placebo group, and diclofenac sodium, which was reported more frequently in the placebo group compared with the bimekizumab group (7.2% versus 11.7%). No study participants used concomitant bDMARDs (TNF inhibitors or non–TNF inhibitors).
Table 16: Summary of Concomitant Medications During Double-Blind Treatment Period the BE MOBILE 2 Trial (Safety Set)
Concomitant medications | BE MOBILE 2 trial | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
Patients who received any concomitant medications, n (%) | 210 (95.0) | 104 (93.7) |
Commonly reported concomitant medications (≥ 15% of patients), n (%) | ||
Drugs used in musculoskeletal system | 190 (86.0) | 88 (79.3) |
Anti-inflammatory and antirheumatic products (nonsteroids) | 188 (85.1) | 88 (79.3) |
Etoricoxib | 38 (17.2) | 17 (15.3) |
Sulfasalazine | 38 (17.2) | 15 (13.5) |
Meloxicam | 34 (15.4) | 13 (11.7) |
Drugs used in alimentary tract and metabolism | 125 (56.6) | 56 (50.5) |
Drugs for peptic ulcer and gastroesophageal reflux disease | 85 (38.5) | 45 (40.5) |
Omeprazole | 34 (15.4) | 19 (17.1) |
Anti-infective for systemic use | 57 (25.8) | 28 (25.2) |
Drugs used in cardiovascular system | 56 (25.3) | 29 (26.1) |
Drugs used in nervous system | 44 (19.9) | 20 (18.0) |
Source: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18
Table 17: Summary of Concomitant axSpA-Related Medications During the Double-Blind Treatment Period of the BE MOBILE 2 Trial (Safety Set)
Concomitant AxSpA-related medications | BE MOBILE 2 trial | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
Concomitant axSpA-related medications (≥ 5% of patients), n (%) | ||
NSAIDs | 181 (81.9) | 87 (78.4) |
Etoricoxib | 38 (17.2) | 17 (15.3) |
Meloxicam | 34 (15.4) | 13 (11.7) |
Diclofenac sodium | 16 (7.2) | 13 (11.7) |
Celecoxib | 21 (9.5) | 7 (6.3) |
Diclofenac | 18 (8.1) | 5 (4.5) |
Naproxen | 14 (6.3) | 6 (5.4) |
Ibuprofen | 10 (4.5) | 7 (6.3) |
Aceclofenac | 7 (3.2) | 9 (8.1) |
Analgesics or opioids | 33 (14.9) | 17 (15.3) |
Mild opioids | 15 (6.8) | 6 (5.4) |
Nonmild opioids | 1 (0.5) | 0 |
Unclassifieda | 15 (6.8) | 9 (8.1) |
csDMARDs | 47 (21.3) | 20 (18.0) |
Corticosteroids | 15 (6.8) | 8 (7.2) |
Oral | 15 (6.8) | 8 (7.2) |
axSpA = axial spondyloarthritis; csDMARD = conventional synthetic disease-modifying antirheumatic drug; NSAID = nonsteroidal anti-inflammatory drug.
aThe unclassified category included the following medications: tizanidine hydrochloride, tolperisone hydrochloride, baclofen, tizanidine, gabapentin, pregabalin, and metamizole.
Source: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18
Efficacy
Details of key efficacy data in the randomized set during the double-blind treatment period are summarized in Table 18.
Improvement of 40% or More in the ASAS
At week 16 of the double-blind treatment period, patients in the bimekizumab group reported a higher adjusted ASAS40 response rate (41.5%) compared with the placebo group (19.8%), with a between-group difference of 21.8% (95% CI, 11.4% to 32.1%). This corresponded to an OR of 2.88 (95% CI, 1.71 to 4.87; P < 0.001) in favour of bimekizumab.
In the exploratory analyses, the ASAS40 response rate for patients in the bimekizumab group increased from week 16 (44.8%) to week 52 (58.4%). In patients who were switched from placebo to bimekizumab at week 16, the adjusted ASAS40 response rate increased from week 16 (22.5%) to week 24 (56.8%), and further increased by week 52 (68.5%). Refer to Appendix 1 for the detailed exploratory analyses data.
Table 18: Summary of Key Efficacy Results From the BE MOBILE 2 Trial (Randomized Set)
End points | BE MOBILE 2 trial | |
|---|---|---|
Bimekizumab 160 mg/mL N = 221 | Placebo N = 111 | |
ASAS40 response at week 16a | ||
Number of responders, n (%) | 99 (44.8) | 25 (22.5) |
Adjusted response rate,b % (95% CI) | 41.5 (33.3 to 50.3) | 19.8 (12.9 to 29.2) |
Absolute difference between study groups in adjusted response rate,b % (95% CI) | 21.8 (11.4 to 32.1) | |
Odds ratiob (95% CI) | 2.88 (1.71 to 4.87) | |
P valueb | P < 0.001 | |
Change from baseline in BASDAI total score at week 16c | ||
Mean (SE) | –2.9 (0.1) | –1.9 (0.2) |
Median (range) | –2.9 (−9.1 to 2.6) | –1.9 (−8.4 to 2.5) |
LS mean (SE) | –2.7 (0.2) | –1.7 (0.2) |
Difference between study groups in LS mean (95% CI) | –1.0 (−1.5 to −0.6) | |
P valueb | P < 0.001 | |
Change from baseline in BASFI at week 16c | ||
Mean (SE) | –2.2 (0.1) | –1.1 (0.2) |
Median (range) | –1.9 (−7.5 to 3.2) | –1.0 (−5.5 to 2.8) |
LS mean (SE) | –2.0 (0.2) | –1.0 (0.2) |
Difference between study groups in LS mean (95% CI) | –1.1 (−1.5 to −0.6) | |
P valueb | P < 0.001 | |
Change from baseline in NSP score (based on an NRS) at week 16c | ||
Mean (SE) | –3.3 (0.2) | –1.9 (0.2) |
LS mean (SE) | –3.2 (0.2) | –1.7 (0.2) |
Difference between study groups in LS mean (95% CI) | –1.5 (−2.0 to −1.0) | |
P valueb | P < 0.001 | |
Enthesitis-free state based on the MASES index at week 16 in study participants with enthesitis at baselined | ||
Number of patients contributing to the analysis, n | 132 | 67 |
Number of patients who were enthesitis-free, n (%) | 68 (51.5) | 22 (32.8) |
Adjusted enthesitis-free rateb (95% CI) | 43.8 (33.1 to 55.0) | 23.9 (14.5 to 36.9) |
Absolute difference between study groups in adjusted enthesitis-free rate, % (95% CI) | 19.8 (6.3 to 33.4) | |
Odds ratio (95% CI) | 2.47 (1.30 to 4.68) | |
Nominal P valueb | 0.006 | |
Change from baseline in ASQoL total score at week 16c | ||
Mean (SE) | –4.9 (0.3) | –3.2 (0.3) |
LS mean (SE) | –4.6 (0.3) | –3.1 (0.4) |
Difference between study groups in LS mean (95% CI) | –1.5 (−2.4 to −0.7) | |
P valueb | P < 0.001 | |
Change from baseline in WPAI-SHP at week 16e | ||
Number of patients contributing to the analysis, n | 156 | 83 |
Percent time missed due to disease-related problems, mean (SD) | –5.5 (17.7) | –1.2 (18.3) |
Difference in means between study groups (95% CI) | –2.9 (−6.9 to 1.1) | |
Number of patients contributing to the analysis, n | 148 | 77 |
Percent impairment while working due to disease-related problems, mean (SD) | –20.8 (23.5) | –6.1 (22.0) |
Difference in means between study groups, (95% CI) | –12.5 (−18.1 to −6.8) | |
Number of patients contributing to the analysis, n | 148 | 77 |
Percent overall work impairment due to disease-related problems, mean (SD) | –22.2 (23.9) | –6.7 (23.3) |
Difference in means between study groups (95% CI) | –12.8 (−18.7 to −6.9) | |
Number of patients contributing to the analysis, n | 210 | 108 |
Percent activity impairment due to disease-related problems, mean (SD) | –23.3 (22.9) | –14.4 (21.6) |
Difference in means between study groups (95% CI) | –9.4 (−13.9 to −4.9) | |
ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASQoL = Ankylosing Spondylitis Quality of Life; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; CI = confidence interval; LS = least squares; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; NRS = numeric rating scale; NSP = nocturnal spinal pain; SD = standard deviation; SE = standard error; WPAI-SHP = Work Productivity and Activity Impairment Questionnaire–Specific Health Problem.
aAt week 16, missing data for ASAS40 were imputed as nonresponse.
bAdjusted response rate, odds ratio, and P values for the comparison of bimekizumab vs. placebo were calculated using logistic regression with factors for treatment, prior TNF alpha inhibitor exposure, and region.
cMissing data of change from baseline at week 16 for the BASDAI total score, BASFI, NSP score, and ASQoL total score were imputed using reference-based multiple imputation.
dMissing data for MASES index change from baseline at week 16 and enthesitis-free state based on the MASES index at week 16 in the subgroup of study participants with enthesitis at baseline were imputed using multiple imputation.
eThe analyses of change from baseline in WPAI-SHP at week 16 used observed cases.
Sources: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
Prespecified subgroup analyses of the ASAS40 response rate at week 16 were generally consistent with the primary analysis across all prespecified subgroups, except for subgroups of patients with a BMI of at least 30 kg/m2 and patients with a BMI of less than 18.5 kg/m2. It is noted that the sample sizes in these subgroups were smaller. At week 16 of the double-blind treatment period, patients in the bimekizumab group who were TNF alpha inhibitor–naive or –experienced reported a higher adjusted ASAS40 response rate compared with those in the placebo group (45.7% and 40.5% for bimekizumab versus 23.4% and 17.6% placebo). This corresponded to ORs of 2.79 (95% CI, 1.59 to 4.91) and 3.48 (95% CI, 0.84 to 14.40) in favour of bimekizumab in the TNF alpha inhibitor–naive and –experienced subgroups, respectively. In the exploratory analyses, the ASAS40 response rate for patients who were TNF alpha inhibitor–naive in the bimekizumab group increased from week 16 (45.7%) to week 52 (58.7%). In patients who were TNF alpha inhibitor–naive and were switched from placebo to bimekizumab at week 16, the response rate in TNF alpha inhibitor–naive participants increased from week 16 (23.4%) to week 24 (59.6%) and further increased at week 52 (71.3%).
The results of sensitivity and supportive analyses, including the tipping-point analyses, were in line with the primary efficacy results. Patients in the bimekizumab group had higher ASAS40 response rates compared with patients in the placebo group. In the mean change from baseline in the individual components of the ASAS40, at week 16, patients in the bimekizumab group reported a greater reduction from baseline in LS mean (reductions reflect improvement) compared with patients in the placebo group in each of the ASAS40 components: PGADA (−2.7 for bimekizumab versus −1.4 for placebo), total spinal pain (−3.1 versus −1.7), BASFI physical function score (−2.0 versus −1.0), and BASDAI mean score for questions 5 and 6 (inflammation and morning stiffness) (−3.0 versus −1.9).
Bath Ankylosing Spondylitis Disease Activity Index
In the BE MOBILE 2 final analysis, at baseline, the mean BASDAI score was 6.45 in the bimekizumab group and 6.51 in the placebo group. At week 16, patients in the bimekizumab group had a greater reduction from baseline in the LS mean (reductions reflect improvement) in BASDAI score compared with patients in the placebo group (LS mean of −2.7 for bimekizumab versus −1.7 for placebo). An estimated median MID of 1.4 points (range, 0.9 to 1.8) was identified in the literature.14 The clinical expert consulted by CADTH indicated they would consider a 1-point difference between groups to be clinically meaningful. The difference in LS mean between treatment groups was −1.04 (95% CI, −1.5 to −0.6; P < 0.001) in favour of bimekizumab.
In the exploratory analyses, the mean change from baseline in BASDAI total score for patients in the bimekizumab group decreased up to week 16 (−2.9) and decreased further to week 52 (−3.6) for patients in the bimekizumab group. In patients who were switched from placebo to bimekizumab at week 16, the mean change in BASDAI total score decreased from baseline to week 16 (−1.9) to week 24 (−3.26), and further decreased to week 52 (−4.0). Refer to Appendix 1 for the detailed exploratory analyses data.
Bath Ankylosing Spondylitis Functional Index
In the BE MOBILE 2 final analysis, at baseline, the mean BASFI score was 5.3 in the bimekizumab group and 5.2 in the placebo group. At week 16, patients in the bimekizumab group had a greater reduction from baseline in LS mean (reductions reflect improvement) in BASFI score compared with patients in the placebo group, which worsened (LS mean of −1.9 for bimekizumab versus −1.0 for placebo). An estimated median MID of 1.1 points (range, 1.0 to 1.4) was identified in the literature.14 The difference in LS mean between treatment groups was −1.1 (95% CI, −1.5 to −0.6; P < 0.001) in favour of bimekizumab. The clinical expert consulted by CADTH identified an MID of 1 point for between-group difference.
In the exploratory analyses, the mean reduction in BASFI score further decreased from baseline to week 16 (−2.2) to week 52 (−2.8) for patients in the bimekizumab group. In patients who were switched from placebo to bimekizumab at week 16, the mean change in BASFI score decreased from baseline to week 16 (−1.1) to week 24 (−2.2), and further decreased at week 52 (−2.8). Refer to Appendix 1 for the detailed exploratory analyses data.
Nocturnal Spinal Pain (Based on an NRS)
In the BE MOBILE 2 final analysis, at baseline, the mean NSP score (based on an NRS) was 6.6 in the bimekizumab group and 6.8 in the placebo group. At week 16, patients in the bimekizumab group had a greater reduction from baseline in LS mean (reductions reflect improvement) in NSP score (based on an NRS) compared with patients in the placebo group, which worsened (LS mean of −3.2 for bimekizumab versus −1.7 for placebo). An estimated median MID of 1.5 points (range, 1.1 to 2.3) was identified in the literature.14 The difference in LS mean between treatment groups was −1.5 (95% CI, −2.0 to −1.0; P < 0.001) in favour of bimekizumab. The clinical expert consulted by CADTH identified an MID of 1 point between groups.
In the exploratory analyses, the mean reduction in NSP (based on an NRS) further decreased −3.3 at week 16 and −4.1 at week 52 for patients in the bimekizumab group. In patients who were switched from placebo to bimekizumab at week 16, the mean change in NSP (based on an NRS) decreased from −1.9 at week 16 to −3.7 at week 24, and further decreased −4.6 at week 52. Refer to Appendix 1 for the detailed exploratory analyses data.
Enthesitis-Free State Based on MASES in Patients with Enthesitis at Baseline
At week 16, the enthesitis-free state based on a MASES index assessment was performed only in patients with enthesitis at baseline, including 132 patients in the bimekizumab group and 67 patients in the placebo group. At week 16 of the double-blind treatment period, patients with enthesitis at baseline in the bimekizumab group reported a higher adjusted enthesitis-free rate compared with those in the placebo group (43.8% for bimekizumab versus 23.9% for placebo), with a between-group difference of 19.8% (95% CI, 6.3% to 33.4%). This corresponded to an OR of 2.47 (95% CI, 1.30 to 4.68; nominal P = 0.006) in favour of bimekizumab. No estimate of a between-group MID was identified by CADTH, but clinical expert input suggested a 15% difference would be clinically important; therefore, the absolute difference between groups was clinically important. The enthesitis-free state outcome was not controlled for type I error rate; thus, these data should be interpreted as supportive evidence only.
In the exploratory analyses, the enthesitis-free rate for patients with enthesitis at baseline in the bimekizumab group at week 16 and week 52 was 51.5% and 50.8%, respectively. In patients with enthesitis at baseline who were switched from placebo to bimekizumab at week 16, the enthesitis-free rate at week 16 was 32.8% and 49.3% at week 24, and decreased to 46.3% at week 52. Refer to Appendix 1 for the detailed exploratory analyses data.
Ankylosing Spondylitis Quality of Life
In the BE MOBILE 2 final analysis, at baseline, the mean ASQoL score was 9.0 in the bimekizumab group and 8.5 in the placebo group. At week 16, patients in the bimekizumab group had a greater reduction from baseline in LS mean (reductions reflect improvement) in ASQoL score compared with patients in the placebo group, which worsened (LS mean of −4.6 for bimekizumab versus −3.1 for placebo). An MID of 1 unit of worsening (i.e., change of + 1) or 2 units of improvement (i.e., change of −2) was identified in the literature.15 The difference in LS mean between treatment groups was −1.5 (95% CI, −2.4 to −0.7; P < 0.001) in favour of bimekizumab. The clinical expert consulted by CADTH identified an MID of 2 points between groups.
In the exploratory analyses, the mean reduction in ASQoL further decreased −5.0 from baseline to week 16 and −5.7 from baseline to week 52 for patients in the bimekizumab group. In patients who were switched from placebo to bimekizumab at week 16, the mean change in ASQoL decreased −3.2 from baseline to week 16 and −4.8 from baseline to week 24, and further decreased −5.6 from baseline to week 52. Refer to Appendix 1 for the detailed exploratory analyses data.
Work Productivity and Activity Impairment Questionnaire–Specific Health Problem
At baseline, the mean WPAI-SHP scores were similar between the bimekizumab group and the placebo group for percent time missed due to disease-related problems (11.5 for bimekizumab versus 10.9 for placebo), percent impairment while working due to disease-related problems (46.1 versus 42.3), percent overall work impairment due to disease-related problems (49.2 versus 43.9), and percent activity impairment due to disease-related problems (53.3 versus 54.1). At week 16, the bimekizumab group compared with the placebo group had a greater mean reduction from baseline in the following:
WPAI-SHP score for percent time missed due to disease-related problems (−5.5 versus −1.2), with a between-group difference of −2.9 (95% CI, –6.9 to 1.1)
percent impairment while working due to disease-related problems (−20.8 versus −6.1), with a between-group difference of −12.5 (95% CI, –18.2 to −6.9)
percent overall work impairment due to disease-related problems (−22.2 versus −6.7), with a between-group difference of −12.8 (95% CI, –18.7 to −6.9)
percent activity impairment due to disease-related problems (−23.3 versus −14.4), with a between-group difference of −9.4 (95% CI, –13.9 to −4.9).
No MIDs for WPAI-SHP were identified in the literature. The WPAI-SHP outcome was not controlled for type I error; thus, these data should be interpreted as supportive evidence only.
In the exploratory analyses, generally, compared with the placebo group, the bimekizumab group had a greater mean reduction from baseline in WPAI-SHP score at weeks 24, 26, and 52 for percent time missed due to problems (related to disease), percent impairment while working due to problems, and percent overall work impairment due to problems. Patients in the bimekizumab and placebo groups reported similar reductions in percent activity impairment due to problems at weeks 24, 26, and 52. Refer to Appendix 1 for the detailed exploratory analyses data.
Harms
Details of AEs, SAEs, withdrawals due to AEs, mortality, and notable harms data in the safety set during the double-blind treatment period are summarized in Table 19.
Adverse Events
During the double-blind treatment period, an AE was reported for 54.3% of patients in the bimekizumab group and 43.2% in the placebo group. The most common AEs (i.e., reported by ≥ 5% of patients in either group) were infections and infestations (28.1% for bimekizumab versus 22.5% for placebo), including nasopharyngitis (7.7% versus 3.6%), gastrointestinal disorders (13.1% versus 9.9%), nervous system disorders (8.1% versus 4.5%), upper respiratory tract infections (2.7% versus 7.2%), and eye disorders (2.3% versus 6.3%).
Serious Adverse Events
During the double-blind treatment period, SAEs were reported for 2.3% of patients in the bimekizumab group and 0.9% of patients in the placebo group. The following SAEs were commonly reported in the bimekizumab group but not reported by any patients in the placebo group: goitre (0.5%), colitis ulcerative (0.5%), Crohn disease (0.5%), cholelithiasis (0.5%), and hepatitis A (0.5%).
Withdrawals Due to Adverse Events
During the double-blind treatment period, 2.7% of patients in the bimekizumab group and no patients in the placebo group discontinued study due to AEs. The commonly reported AEs that led to study discontinuation were abnormal psychiatric evaluation (0.9%), lymphoid tissue hyperplasia (0.5%), Crohn disease (0.5%), oral candidiasis (0.5%), and rash (0.5%).
Mortality
No deaths due to AEs were reported during the double-blind treatment period in the BE MOBILE 2 trial.
Notable Harms
Serious infections, fungal infections, opportunistic infections, malignancies, major adverse cardiac events, neutropenia, suicidal ideation and behaviours, IBD, hypersensitivity reactions, and liver injuries or disorders were considered notable harms by the sponsor and/or the clinical expert consulted by CADTH. The most commonly reported notable harms were hypersensitivity reactions (7.7% for bimekizumab versus 1.8% for placebo), fungal infections (6.3% versus 0%), liver injuries or disorders (4.5% versus 3.6%), IBD (0.9% versus 0%), neutropenia (0.5% versus 0%), and serious infections (0.5% versus 0.9%). No patients in either group reported any opportunistic infection, malignancy, major adverse cardiac event, or suicidal ideation and behaviour.
Table 19: Summary of Harms Results During From the BE MOBILE 2 Trial (Safety Set)
Adverse events | BE MOBILE 2 trial (data cut-off date: September 9, 2022) | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
Most common adverse events (≥ 5% of patients), n (%) | ||
Patients with ≥ 1 adverse events | 120 (54.3) | 48 (43.2) |
Infections and infestations | 62 (28.1) | 25 (22.5) |
Nasopharyngitis | 17 (7.7) | 4 (3.6) |
Upper respiratory tract infection | 6 (2.7) | 8 (7.2) |
Gastrointestinal disorders | 29 (13.1) | 11 (9.9) |
Nervous system disorders | 18 (8.1) | 5 (4.5) |
Eye disorders | 5 (2.3) | 7 (6.3) |
Serious adverse events (≥ 0.5% of patients), n (%) | ||
Patients with ≥ 1 serious adverse events | 5 (2.3) | 1 (0.9) |
Goitre | 1 (0.5) | 0 |
Colitis ulcerative | 1 (0.5) | 0 |
Crohn disease | 1 (0.5) | 0 |
Cholelithiasis | 1 (0.5) | 0 |
Hepatitis A | 1 (0.5) | 0 |
Viral infection | 0 | 1 (0.9) |
Depression | 0 | 1 (0.9) |
Patients who stopped treatment due to adverse events, n (%) | ||
Patients who stopped | 6 (2.7) | 0 |
Abnormal psychiatric evaluation | 2 (0.9) | 0 |
Lymphoid tissue hyperplasia | 1 (0.5) | 0 |
Crohn disease | 1 (0.5) | 0 |
Oral candidiasis | 1 (0.5) | 0 |
Rash | 1 (0.5) | 0 |
Deaths, n (%) | ||
Patients who died | 0 | 0 |
Notable harms, n (%) | ||
Hypersensitivity reactionsa | 17 (7.7) | 2 (1.8) |
Fungal infections | 14 (6.3) | 0 |
Liver injury or disordersb | 10 (4.5) | 4 (3.6) |
Inflammatory bowel diseasec | 2 (0.9) | 0 |
Serious infections | 1 (0.5) | 1 (0.9) |
Neutropenia | 1 (0.5) | 0 |
aIncluding anaphylaxis.
bIncluding drug related hepatic disorders, excluding liver neoplasms, benign (including cysts and polyps) and liver neoplasms, malignant and unspecified.
cAdjudicated definite and probable inflammatory bowel disease.
Sources: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18 The details included in the table are from the sponsor’s summary of clinical evidence.19
Critical Appraisal
Internal Validity
The BE MOBILE 2 trial was a phase III, multicentre, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of bimekizumab 160 mg/mL administered subcutaneously every 4 weeks compared with placebo in treating patients with active AS. Eligible patients were assigned on a 2:1 ratio to a treatment group using an interactive voice or web response system with stratification by region and prior exposure to TNF alpha inhibitors (yes versus no). Generally, baseline characteristics were balanced between the treatment groups, which indicated that the randomization was successful. According to the clinical expert consulted by CADTH, the baseline characteristics of patients in the BE MOBILE 2 trial were reflective of the general population of patients with active AS. More patients discontinued due to AEs in the bimekizumab group compared with the placebo group (1.4% versus 0%). The clinical expert confirmed this is reflective of clinical practice, as they would expect more side effects with an immunomodulatory therapy. The CADTH review team agreed with the clinical expert that the number of patients who discontinued due to AEs was small in the bimekizumab group, and the difference between treatment groups was not significant, but noted there may be a potential risk of unblinding due to the elevated risk of AEs in the bimekizumab group.
The CADTH review team noted there were no comparative data available beyond week 16, as patients in the placebo group were reallocated to receive bimekizumab during the 36-week maintenance period after finishing all assessments at the end of the 16-week double-blind treatment period. Therefore, it is uncertain what the direct comparative efficacy and safety of bimekizumab is after week 16.
The sponsor assumed that treatment response at week 12 and week 16 would be the same in the sample size and power calculations. The clinical expert consulted by CADTH confirmed that the assumption is reflective of clinical practice, as they do not observe much of a difference in treatment effect between week 12 and week 16.
Multiple data imputation methods, such as NRI, reference-based multiple imputation, and observed case, were used in the efficacy analysis based on variable type (i.e., binary and continuous). In the analysis of the primary end point, ASAS40, a composite estimand using NRI was applied, considering the ASAS40 is based on 4 different component scores. Therefore, the CADTH review team considered that missing data for the primary analysis in the BE MOBILE 2 trial were adequately imputed. For the analysis of the primary and key secondary end points, a fixed sequence testing procedure was employed to adjust for multiple comparisons across multiple end points, thereby controlling the type I error. The CADTH review team noted that the analyses of enthesitis-free state based on MASES index was not included in the fixed sequence testing hierarchy; thus, the results should be considered exploratory and supportive. Although the subgroup analyses were prespecified, the BE MOBILE 2 trial was not powered to detect any change in the ASAS40 response rate between bimekizumab and placebo in subgroup analyses, except for the subgroup of patients who are TNF inhibitor–naive; additionally, no formal statistical tests for interaction between subgroups were conducted. More patients in the bimekizumab group used concomitant NSAIDs, conventional synthetic DMARDs, and drugs to treat the alimentary tract and metabolism compared with the placebo group. The CADTH review team agreed with the clinical expert that the differences in the use of concomitant medications between treatment groups were small and less likely to impact treatment effect. In addition, the clinical expert confirmed that the proportions of patients using the previously mentioned concomitant medications were aligned with clinical practice.
In the BE MOBILE 2 final protocol, dated November 26, 2018, psoriatic arthritis was not listed in the inflammatory conditions that excluded patients from the trial, and patients who had intolerance to administration of at least 1 NSAID were eligible for the trial. Protocol amendment 1, made September 11, 2019, removed eligibility for patients with psoriatic arthritis from study enrolment. Protocol amendment 2, made October 17, 2019, restricted eligibility to patients who had failed to respond to 2 different NSAIDs taken at the maximum tolerated dose for a total of 4 weeks. Of note, protocol amendments 1 and 2 were made after the enrolment of the first patient (April 25, 2019). Therefore, the CADTH review team considered that these 2 protocol amendments could increase patient heterogeneity and introduce bias. The direction of the bias is uncertain, as there were no data reported on the numbers of patients in the trial with psoriatic arthritis or whose disease had failed to respond to more than 2 NSAIDs.
HRQoL is considered a relevant outcome by both patients with active AS and the clinical expert consulted by CADTH. However, the assessment of work productivity using the WPAI-SHP was not controlled for multiplicity and thus should be considered only as exploratory and supportive.
External Validity
The BE MOBILE 2 trial used placebo as the comparator group. According to the clinical expert consulted by CADTH, an anti-TNF biosimilar monoclonal antibody would be an appropriate comparator for bimekizumab. The clinical expert indicated placebo is not an appropriate comparator; head-to-head studies with an active drug would be ideal. The BE MOBILE 2 trial excluded patients who had been treated with more than 1 TNF alpha inhibitor and/or more than 2 additional non–TNF alpha biological-response modifiers, or any IL-17 biological-response modifier at any time. The clinical expert indicated that these patients should be considered eligible for bimekizumab. According to the clinical expert, although the response rate might be lower, some patients do response to bDMARDs after the failure of treatment with TNF inhibitors and IL-17 inhibitors; therefore, the clinical expert would switch treatments within the same class due to limited treatment options. According to the clinical expert, the study results would not be generalizable to these previously mentioned patients, as it is expected that the response rates will be lower in this patient population, which tends to have lower response rates with subsequent treatments in clinical practice.
The clinical expert consulted by CADTH stated that IBD is a common inflammatory condition that happens in patients with AS. According to the clinical expert, about 10% to 15% of patients with AS would have active IBD and 60% of patients would have subclinical IBD based on biopsies. Patients with active IBD were not eligible for the BE MOBILE 2 trial; thus, the number of patients with IBD flares in the trial may be lower compared with the general population of patient with AS. The clinical expert indicated that patients with a personal or family history of IBD may not be candidates for treatment with bimekizumab based on their experience with bDMARDs targeting IL-17A in patients with IBD. The presence of IBD or severe uveitis and active infections, especially fungal infections, would be contraindications for treatment with bimekizumab, as per the feedback from the clinical expert. The CADTH review team noted that IBD is a warning in the proposed product monograph. The clinical expert stated that the exclusion of patients with active IBD would not significantly compromise the generalizability of the study results, as most patients with AS would be eligible for treatment with bimekizumab in clinical practice.
In addition, there was no study site in Canada in the BE MOBILE 2 trial, which may compromise the generalizability of the study results to the clinical practice in Canada.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.16,17
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, the evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty-of-evidence assessment was the presence or absence of a clinically important effect based on thresholds informed by the clinical expert consulted by CADTH for this review for ASAS40, BASDAI, BASFI, NSP, MASES, ASQoL, and SAEs. For WPAI-SHP, there is no established MID and the clinical expert consulted by CADTH could not provide a threshold of important difference, so the target of the certainty-of-evidence assessment was the presence or absence of any (non-null) effect.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for bimekizumab versus placebo.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following has summarized and validated by the CADTH review team.
Description of Studies
BE AGILE 2 (AS0009, NCT03355573) is a single-arm phase II long-term extension study, which followed the preceding study, BE AGILE (AS0008, NCT02963506), which was a dose-ranging, randomized, placebo-controlled phase IIb trial.
Of the 303 adult patients living with active AS who had been enrolled in the BE AGILE trial, 255 (84.5%) entered the BE AGILE 2 study, which took place at 50 sites in 10 European countries and the US. The data presented in this report were collected for up to approximately 5 years for the final analyses (data cut-off: October 19, 2022), which combined 48 weeks of treatment in the BE AGILE study and an additional 204 weeks of treatment in the BE AGILE 2 trial.72
Most of the key outcomes measured in the BE MOBILE 2 trial were also evaluated in the BE AGILE 2 trial, such as ASAS40, BASDAI, BASFI, NSP, ASQoL, Bath Ankylosing Spondylitis Metrology Index, MASES, and safety outcomes.
Populations
Patients were eligible for the BE AGILE 2 trial if they had completed the BE AGILE study and, in the opinion of the investigator, were expected to benefit from continued treatment with bimekizumab in the BE AGILE 2 trial. Consistent with the inclusion criteria for the BE MOBILE 2 trial, patients were eligible for the BE AGILE 2 study if they were adults with a diagnosis of active AS based on radiographic evidence fulfilling the mNY criteria for AS, including 3 or more months of symptoms, younger than age 45 at symptom onset, with moderate to severe active disease at baseline (defined as BASDAI ≥ 4 plus spinal pain ≥ 4 on a 0 to 10 NRS from BASDAI item 2), and intolerant to or with AS that has failed to respond to NSAIDs. Patients were allowed to have previously received treatment with a TNF alpha inhibitor.
Patients were excluded from the BE AGILE 2 study if they met the withdrawal criteria in the BE AGILE trial or if they had signs or symptoms that could indicate a medically significant active infection or had an infection requiring systemic antibiotics within 2 weeks of study entry.
Interventions
All patients in the BE AGILE 2 trial received open-label treatment with bimekizumab 160 mg every 4 weeks, regardless of their prior dosing regimen in BE AGILE, the parent trial. During the long-term extension period, NSAIDs, DMARDs (methotrexate, sulfasalazine, leflunomide), and/or joint injections (e.g., intra-articular hyaluronic acid and intrabursal corticosteroid) were permitted. However, a concomitant biologic drug was not allowed under any circumstances during the long-term extension study. Medication changes, additions, and a decrease in dose or dosing frequency of any drug (except for bimekizumab) were permitted at any time after enrolment into the open-label extension study.
Outcomes
Most of the key outcomes measured in the BE MOBILE 2 trial were also evaluated in the BE AGILE 2 trial. These outcomes were defined and evaluated in the same manner as in the BE MOBILE 2 trial. For a full description of these outcomes, refer to Table 6.
Statistical Analysis
All outcomes in the BE AGILE 2 trial were descriptively evaluated compared with baseline from the BE AGILE trial (e.g., BE AGILE 2 trial week 208 [total exposure = 252 weeks] versus BE AGILE study week 0 [total exposure = 0 weeks]). As in the BE MOBILE 2 trial, missing data for binary outcomes in the BE AGILE 2 trial were imputed using NRI, while missing data for continuous outcomes were imputed using multiple imputation based on the assumption that data were missing at random. No formal hypothesis testing was conducted in the BE AGILE 2 trial and no adjustment was made for multiple comparisons.
Results
Patient Disposition
Among the 303 patients who were randomized in the BE AGILE trial, 297 patients (98.0%) completed the double-blind period, and 265 patients (87.5%) completed the dose-blind period at week 48. Nine patients did not enter the BE AGILE 2 trial and 1 patient entered the BE AGILE 2 trial but did not receive bimekizumab. A total of 256 study participants enrolled in the BE AGILE 2 trial and 255 (99.6%) participants received at least 1 dose of bimekizumab in the BE AGILE 2 trial. A majority of patients (78.9%) completed the study. The discontinuation rate for BE AGILE 2 was 20.7% and was most frequently related to TEAEs (7.4%) or withdrawal of consent (9.0%) (Table 20).
Patient disposition | BE AGILE 2 trial |
|---|---|
Enrolled in the BE AGILE study, N | 303 |
Enrolled in the BE AGILE 2 study, N | 256 |
Completed BE AGILE 2 study, N (%) | 202 (78.9) |
Discontinued BE AGILE 2 study, n (%) | 53 (20.7) |
Reason for discontinuing | — |
TEAE | 19 (7.4) |
Lack of efficacy | 2 (0.8) |
Lost to follow-up | 4 (1.6) |
Consent withdrawal (not due to AE) | 23 (9.0) |
Other | 5 (2.0) |
Full analysis set, N | 249 |
Safety analysis set, N | 255 |
AE = adverse event; TEAE = treatment-emergent adverse event.
aAttended week 208 visit.
Sources: BE AGILE 2 Clinical Study Report (Table 7 to 1).72 The details included in the table are from the sponsor’s summary of clinical evidence.19
Exposure to Study Treatments
In the BE AGILE 2 trial safety set, 96.1% of all study participants reported having greater than 75% treatment compliance. The extent of exposure to bimekizumab during the open-label extension period is summarized in Table 21.
Table 21: Summary of Extent of Exposure
Detail | BE AGILE 2 safety set (N = 255) |
|---|---|
Duration of exposure, days | — |
Mean (SD) | 1,283.7 (404.6) |
Median (range) | 1,456.0 (28 to 1,492) |
Time at risk, days | — |
Mean (SD) | 1,389.6 (412.3) |
Median (range) | 1,569.0 (59 to 1,605) |
Total time at risk during BE AGILE 2, patient-years | 970.2 |
SD = standard deviation.
Sources: BE AGILE 2 Clinical Study Report.72 The details included in the table are from the sponsor’s summary of clinical evidence.19
A total of 25 (9.8%) study participants received rescue medication during the BE AGILE 2 trial. The rescue medications used were NSAIDs (6.7%, with the majority having a change in NSAID type or dose), DMARDs (2.4%), analgesics (2.0%), oral corticosteroids (0.8%), and intra-articular corticosteroids and ”unclassified” (0.4%).
Most patients (83.5%) were receiving concomitant during the BE AGILE 2 trial, as described in Table 22.
Table 22: Concomitant Medications
Detail | BE AGILE 2 safety set (N = 255) |
|---|---|
Current NSAID therapy, n (%) | |
Yes | 213 (83.5) |
1 | 209 (82.0) |
2 | 4 (1.6) |
≥ 3 | 0 |
No | 42 (16.5) |
Current synthetic DMARDs, n (%) | |
Yes | 61 (23.9) |
Methotrexate | 21 (8.2) |
Sulfasalazine | 42 (16.5) |
Hydroxychloroquine | 0 |
No | 194 (76.1) |
Current oral corticosteroids, n (%) | |
Yes | 24 (9.4) |
DMARD = disease-modifying antirheumatic drug; NSAID = nonsteroidal anti-inflammatory drug.
Sources: The details included in the table are from the sponsor’s summary of clinical evidence19 and the BE AGILE 2 Clinical Study Report.72
Baseline Characteristics
Refer to Table 23 for baseline characteristics.
In the BE AGILE 2 trial, most patients were male (85.1%) and were positive for HLA-B27 (91.0%). Mean symptom duration was approximately 14 years (SD = 9.41) and mean disease duration (time since AS diagnosis) was approximately 7.8 years (SD = 8.5). Mean BASDAI score was approximately 7.4 points (SD = 1.4), reflecting moderately severe patient-reported disease activity. Most patients (79.2%) had received NSAIDs for their active AS in the past. Approximately 11% of patients had previously been exposed to treatment with TNF alpha inhibitors.
Table 23: Baseline Characteristics
Characteristic | BE AGILE 2 safety set (N = 255) |
|---|---|
Age | |
Mean (SD), years | 41.8 (11.4) |
18 to < 65 years, n (%) | 245 (96.1) |
Sex, n (%) | |
Male | 217 (85.1) |
Female | 38 (14.9) |
Racial group, n (%) | |
White | 253 (99.2) |
Other or mixed | 2 (0.8) |
Weight, kg | |
Mean (SD) | 80.99 (16.75) |
Median (range) | 79.50 (47.8 to 153.0) |
BMI, n (%) | |
< 25 kg/m2 | 95 (37.3) |
25 to < 30 kg/m2 | 101 (39.6) |
≥ 30 kg/m2 | 59 (23.1) |
Geographic region, n (%) | |
Eastern Europe | 229 (89.8) |
North America | 8 (3.1) |
Western Europe | 18 (7.1) |
Age at first diagnosis, mean (SD), years | 34.5 (10.2) |
Disease characteristics | |
Time since first symptoms of AS, years | |
Mean (SD) | 14.02 (9.41) |
Median (range) | 12.1 (0.2 to 47.2) |
Time since diagnosis of AS, years | |
Mean (SD) | 7.79 (8.46) |
Median (range) | 4.6 (0.0 to 37.3) |
Age at first diagnosis of AS, years | |
Mean (SD) | 34.5 (10.2) |
Median (range) | 33.5 (16 to 74) |
Positive for HLA-B27a | 232 (91.0) |
BASDAI spinal pain, mean (SD) | 7.4 (1.4) |
PGADA, mean (SD) | 6.9 (1.7) |
Total spinal pain score, mean (SD) | 7.0 (1.8) |
BASFI, mean (SD) | 5.7 (1.9) |
ASDAS-CRP, mean (SD) | 3.9 (0.8) |
hs-CRP, mg/Lb (n = 254) | |
Mean (SD) | 19.5 (21.5) |
Median (range) | 12.1 (0.3 to 130.1) |
Prior medications | |
Past TNFi therapy, n (%) | 29 (11.4) |
Past NSAID therapy, n (%) | 202 (79.2) |
AS = ankylosing spondylitis; ASDAS-CRP = Ankylosing Spondylitis Disease Activity Score using C-reactive protein; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Function Index; BMI = body mass index; HLA-B27 = human leukocyte antigen B27; hs-CRP = high-sensitivity C-reactive protein; NSAID = nonsteroidal anti-inflammatory drug; PGADA = Patient’s Global Assessment of Disease Activity; SD = standard deviation; TNFi = tumour necrosis factor inhibitor.
Sources: The details included in the table are from the sponsor’s summary of clinical evidence,19 the BE AGILE 2 Clinical Study Report,72 and Baraliakos et al. (2022).71
Efficacy
ASAS40 response was sustained up to week 208 in the BE AGILE 2 trial, with response rates of 59% (147 out of 249) using NRI, and 73.1% (147 out of 201) using observed case data. During the BE AGILE 2 trial, the mean BASDAI score (n = 249) decreased from baseline and was sustained at week 208 (decrease of −4.01; SE = 0.13) versus an MID of −2.0 points. The mean BASFI score (n = 249) decreased from baseline by −3.1 (SE = 0.15) and was sustained at week 208. Relative to baseline, the NSP score (n = 249) decreased −4.55 (SE = 0.16) and was maintained at week 208 versus an MID of 1.5 points. Also, the mean ASQoL score (n = 249) decreased from baseline by −5.9 (SE = 0.3) and was maintained at all times by week 208 versus an MID of −5.1 points. Among patients with enthesitis at baseline, the mean MASES (n = 164) decreased −0.36 (SE = 0.23) and maintained improvement up to week 208 in the BE AGILE 2 trial. WPAI-SHP was not assessed in the BE AGILE 2 study.
Harms
Refer to Table 24 for harms data.
A total of 237 (92.9%) study participants reported a TEAE during the BE AGILE 2 study. The most commonly reported TEAEs were nasopharyngitis (18%), upper respiratory tract infection and COVID-19 infection (12.9% each), and bronchitis (8.6%).
There were 46 patients (18.0%) who experienced at least 1 SAE, with COVID-19 infection and pneumonia being the most common (1.2% each). Twenty-one patients (8.2%) discontinued study treatment due to a TEAE, mostly due to alanine aminotransferase (1.2%) and aspartate aminotransferase (0.8%) elevation. Two fatal TEAEs were reported during the study. One study participant experienced a fatal TEAE of road traffic accident, which was considered by study investigators to not be related to bimekizumab. One study participant who had pre-existing cardiovascular risk factors, including hypertension, type 2 diabetes, and left bundle branch block, experienced a fatal TEAE of cardiorespiratory arrest, which was not considered related to bimekizumab, per the investigator’s assessment.
Of note, fungal infection (18.4%) and hypersensitivity (11.4%) were the most common AEs of special interest reported in the safety set during the BE AGILE 2 trial. The fungal infection cases were all judged by the investigators to be mild to moderate in intensity and the vast majority did not lead to treatment discontinuation (a single patient discontinued due to perirectal abscess).
Table 24: Summary of Harms Results From Long-Term Extension Studies
Adverse events | BE AGILE 2 safety set (N = 255) |
|---|---|
Most common adverse events, n (%)a | |
Patients with ≥ 1 TEAE | 237 (92.9) |
Nasopharyngitis | 46 (18.0) |
Upper respiratory tract infection | 33 (12.9) |
COVID-19 infection | 33 (12.9) |
Bronchitis | 22 (8.6) |
ALT increased | 19 (7.5) |
Pharyngitis | 18 (7.1) |
Oral candidiasis | 17 (6.7) |
Hypercholesterolemia | 16 (6.3) |
Tonsillitis | 15 (5.9) |
Sinusitis | 15 (5.9) |
Arthralgia | 15 (5.9) |
Hypertension | 14 (5.5) |
Psoriasis | 13 (5.1) |
Headache | 13 (5.1) |
AST increased | 13 (5.1) |
SAEs, n (%)b | |
Patients with ≥ 1 SAE | 46 (18.0) |
COVID-19 infection | 3 (1.2) |
Pneumonia | 3 (1.2) |
Colitis ulcerative | 2 (0.8) |
Perirectal abscess | 2 (0.8) |
Osteoarthritis | 2 (0.8) |
Benign prostatic hyperplasia | 2 (0.8) |
Patients who stopped treatment due to adverse events, n (%)b | |
Patients who stopped | 21 (8.2) |
ALT increased | 3 (1.2) |
AST increased | 2 (0.8) |
Deaths, n (%) | |
Patients who died | 2 (0.8) |
Road traffic accident | 1 (0.4) |
Cardiorespiratory arrest | 1 (0.4) |
Adverse events of special interest, n (%) | |
Fungal infections | 47 (18.4) |
Hypersensitivity | 29 (11.4) |
Serious infections | 14 (5.5) |
IBD | 9 (3.5) |
Malignant or unspecified tumour | 3 (1.2) |
Neutropenia | 3 (1.2) |
Opportunistic infections | 2 (0.8) |
Adjudicated MACE | 2 (0.8) |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; IBD = inflammatory bowel disease; MACE = major adverse cardiovascular event; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aFrequency > 5%.
bCategory reported in ≥ 2 people.
Sources: The details included in the table are from the sponsor’s summary of clinical evidence19 and the BE AGILE 2 Clinical Study Report.72
Critical Appraisal
Internal Validity
In general, the results of the efficacy end points assessed in the BE AGILE 2 trial appear to support the long-term effectiveness of bimekizumab. However, the findings from the BE AGILE 2 extension study (exposure of up to 252 weeks with the BE AGILE and BE AGILE 2 trials combined) were limited due to the lack of a control group and the nature of an open-label study. In clinical trials, the efficacy magnitude (particularly in patients’ self-reported outcomes) is often overestimated due to the nature of an open-label trial and the absence of a control group. The long-term outcome efficacy should therefore be interpreted while considering this limitation. Moreover, there is a risk of selection bias, as patients who have responded to bimekizumab and who tolerated side effects, if any, during the 2 parent studies, are more likely to continue during the extension period. This bias would also increase the chance of overestimation of the efficacy end point measures. Even though detailed information on the use of concomitant medications (NSAIDs and conventional synthetic DMARDs) and rescue medications have been provided in the open-label period, the impact of concomitant and rescue medications on the long-term efficacy assessment still remains unclear. Of note, no new safety signals or increased risks were identified with up to 5 years of bimekizumab treatment between the BE AGILE and BE AGILE 2 studies.
External Validity
No Canadian study site was included in the BE AGILE and BE AGILE 2 studies. However, the clinical expert consulted by CADTH did not raise any issues regarding the generalizability of study results to real-world clinical practice in Canada.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
The objective of this section is to summarize and critically appraise the methods and findings of the indirect evidence submitted by the sponsor. The sponsor submitted indirect evidence in the form of an NMA and an MAIC to address gaps in the evidence.
Description of Indirect Comparisons
The sponsor conducted a systematic literature review (SLR) to identify evidence for the NMAs and the MAIC in October 2013 and included update searches to January 2023. The searches identified 3,104 records that were potentially of interest and, of these, 286 publications were included that described 65 RCTs. Of the 65 unique trials identified, 28 and 8 trials were deemed feasible to be included in the NMA and MAIC, respectively, based on the study selection criteria described in Table 25.
Objectives
Network Meta-Analyses
The NMAs were performed to determine the clinical efficacy and safety of bimekizumab compared with other relevant interventions at weeks 12 to 16 for the treatment of patients with AS. The NMAs were conducted on 3 different networks: purely naive (100% bDMARD-naive), predominantly naive (approximately 90% bDMARD-naive), and purely experienced (100% bDMARD-experienced).
Matching-Adjusted Indirect Comparisons
The MAICs were performed to establish the long-term relative clinical efficacy of bimekizumab, where data availability allowed, compared with other IL-17A inhibitors in patients with AS at week 52.
Indirect Comparison Methods
Study Selection Methods
Network Meta-Analyses
A feasibility assessment was performed to determine which of the 65 unique RCTs identified by the SLR were suitable for inclusion in the NMA. The eligibility criteria for including studies identified by the SLR in the NMA were in addition to the eligibility criteria of the clinical SLR. The NMA eligibility criteria were applied to ensure that the trial data could be synthesized within a meta-analysis framework. Treatments were restricted to doses and schedules with marketing authorization. Of the 65 unique RCTs included in the SLR, 37 met the additional NMA eligibility criteria.
Matching-Adjusted Indirect Comparison
The NMA and MAIC analyses were informed by the same SLR. Studies reporting efficacy outcomes for RCTs of IL-17A inhibitors in AS were identified through the SLR. Of the 65 unique RCTs included in the SLR, 8 were included in the MAIC analyses.
Table 25: Study Selection Criteria and Methods for the NMAs and MAIC Submitted by the Sponsor
Characteristics | NMA criteria | MAIC criteria |
|---|---|---|
Population | Adult patients (aged ≥ 18 years) with either AS (r-axSpA) or nr-axSpA, who have had any of the following:
Note: While the systematic review included all patients with AS (r-axSpA) or nr-axSpA, the indirect comparisons only considered the network of evidence among patients with AS (r-axSpA) (i.e., the population of interest for the present submission). | |
Interventions |
|
|
Comparators |
|
|
Outcomes | Note: While a broad spectrum of outcomes was evaluated in the NMAs, a smaller list of key outcomes of interest (based on the sponsor’s systematic review) is presented in the sponsor’s submission. At week 12 to 16 Composite and disease activity outcomes:
Enthesitis:
Inflammation (including before and after treatment and change from baseline):
HRQoL (including before and after treatment and change from baseline):
Pain (including before and after treatment and change from baseline):
Discontinuation due to any reason at week 12 to 16 SAEs at week 12 to 16 | At week 52:
|
Study designs |
| |
Publication characteristics | Original English-language publications (entire publication must be available in English, not just the abstract) | |
Exclusion criteria | Population:
Study design
Languages:
| |
Databases searched |
| |
Selection process | Titles and/or abstracts were screened by 2 independent reviewers against predefined selection criteria. Any conflicts regarding eligibility were resolved through discussion between the 2 reviewers. Where necessary, arbitration was provided by a third, more senior reviewer. Full-text papers were screened by 2 independent reviewers against the selection criteria, with any disputes regarding eligibility resolved through dialogue between the 2 reviewers. Again, arbitration was provided by a third, more senior reviewer if required. A record was kept of all publications excluded at this stage, along with a clear justification for their exclusion (based on the predefined eligibility criteria). | |
Data extraction process | All relevant data from the studies identified were populated into a data extraction table. Data extraction was completed by 1 reviewer and quality-checked by a second independent reviewer to ensure that the final data extraction table was of the highest quality. Where data gaps, errors, or inconsistencies were identified in data extracted during previous iterations of the SLR, these were corrected. These corrections were performed by 1 reviewer and quality-checked by a second independent reviewer. | |
Quality assessment | The Cochrane Risk of Bias 2 tool was used to evaluate study quality. | |
AS = ankylosing spondylitis; ASAS = Assessment of SpondyloArthritis International Society; ASAS20 = improvement of 20% or more in the Assessment of SpondyloArthritis International Society; ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASAS5/6 = an improvement of at least 20% in 5 of 6 Assessment of SpondyloArthritis International Society domains; ASDAS = Ankylosing Spondylitis Disease Activity Score; ASDAS-CII = clinically important improvement in Ankylosing Spondylitis Disease Activity Score; ASDAS-CRP = Ankylosing Spondylitis Disease Activity Score using C-reactive protein; ASDAS-ID = Ankylosing Spondylitis Disease Activity Score; ASDAS-MI = major improvement in Ankylosing Spondylitis Disease Activity Score; ASQoL = Ankylosing Spondylitis Quality of Life; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASDAI50 = a reduction of at least 50% in the Bath Ankylosing Spondylitis Disease Activity Index score; BASFI = Bath Ankylosing Spondylitis Functional Index; BASMI = Bath Ankylosing Spondylitis Metrology Index; CRP = C-reactive protein; DMARD = disease-modifying antirheumatic drug; G-BA = Gemeinsamer Bundesausschuss; HAS = Haute Autorité de Santé; HRQoL = health-related quality of life; IL-17 = interleukin-17; IQWiG = Institute for Quality and Efficiency in Health Care; JAK = Janus kinase; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; MCS = mental component summary; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; nr-axSpA = nonradiographic axial spondyloarthritis; NRS = numerical rating scale; NSAID = nonsteroidal anti-inflammatory drug; PBAC = Pharmaceutical Benefits Advisory Committee; PCS = physical component summary; PICOS = population, intervention, comparator(s), outcomes, and study design; PGADA = Patient’s Global Assessment of Disease Activity; r-axSpA = radiographic axial spondyloarthritis; RCT = randomized controlled trial; SF-36 = Short Form (36) Health Survey; SLR = systematic literature review; SMC = Scottish Medicines Consortium; SPARCC = Spondyloarthritis Research Consortium of Canada; TNF alpha = tumour necrosis factor alpha.
aUnless the publication explicitly states that patients are NSAID-naive or still receiving their first NSAID, it was assumed that patients participating in RCTs for IL-17, TNF alpha, or JAK inhibitors experienced treatment failure with at least 1 NSAID.
bThese outcomes were listed as relevant outcomes in the eligibility criteria table of the previous SLR, but there is no evidence they were ever extracted; where present, they were extracted from publications included in the April 2022 and January 2023 clinical SLR updates.
cThese outcomes are predefined primary or secondary outcomes from the BE MOBILE 1 and BE MOBILE 2 trials, but there is no evidence they were extracted during the previous SLR; where present, they were extracted from publications included in the 2022 clinical SLR update.
dPublications pooling data across multiple trials from the same clinical trial program were eligible for inclusion.
eRelevant SLRs and NMAs were included at the title- or abstract-screening stage so their bibliographic reference lists could be hand-searched for relevant studies; they were then excluded at the full-text screening stage unless they presented novel data.
Source: Sponsor’s summary of clinical evidence.19
Analysis Methods
Network Meta-Analyses
A Bayesian NMA was conducted and the summary for the methodology can be found in Table 26. The following types of NMA model were employed:
binomial model with logit link
binomial model with logit link with log odds of response on placebo arm as interaction term
normal model with identity link
normal model with identity link with change from baseline in placebo are as interaction term.
The trial arm data were fitted to a generalized linear model via Bayesian Markov Chain Monte Carlo methods. The WinBUGs model was run for a minimum burn-in of 10,000 iterations to maximize convergence. Subsequently, 3 chains of 1,000 samples were drawn from the posterior distributions. The mean residual deviance (total residual deviance divided by number of data points) and the deviance information criterion output from WinBUGs provided an overall estimate of how well the predicted values fit the observed dataset.
The primary output from WinBUGs is the relative treatment effect compared with the reference treatment, which is placebo for this analysis. From these outputs, other estimates can be calculated, such as the relative treatment effects comparing all treatments in the network. The relative treatment estimates from the logit model are the (log) OR. The relative treatment effect estimates from the continuous models are mean differences in the change from baseline.
Both random-effects models and fixed-effects models were tested. A random-effects NMA allows the true treatment effect (e.g., OR between 2 treatments) to vary between studies due to heterogeneity. In these random-effects models, a uniform (uninformative) prior was used for the between-studies standard deviation, assuming that the heterogeneity is the same across all comparisons.
For the bDMARD-naive networks and for most outcomes, the fixed-effect placebo-adjusted model was favoured, unless the random-effects placebo-adjusted model was clearly better. For the bDMARD-experienced networks and for most outcomes, the fixed-effect model was favoured, unless the fixed effect with placebo adjustment was clearly better. The decision to recommend the random-effects placebo considered a balance of factors, including whether the 95% CrI was within a realistic range of values.
Rankings are provided, as well as the surface under the cumulative ranking curves, to express the percentage of efficacy for each treatment compared with an ideal treatment ranked first without uncertainty. In addition, the ranking was used to calculate the direct probability of bimekizumab ranking better than a comparator, that is, the proportion of simulations where the rank for bimekizumab was better than the comparator. To account for placebo response and the potential impact on the relative effects, a placebo-adjusted analysis was conducted, where feasible. The main analysis was conducted with a separate node for each treatment.
Table 26: Indirect Comparison Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Bayesian approach:
|
Priors | Not reported. |
Assessment of model fit | The trial arm data were fitted to a generalized linear model via Bayesian Markov Chain Monte Carlo methods. The mean residual deviance (total residual deviance divided by number of data points) and the DIC output from WinBUGs provide an overall estimate of how well the predicted values fit the observed dataset. The DIC is used to compare different models for the same likelihood and data. An alternative form of the DIC was also estimated (alt DIC = total residual deviance + posterior variance). The model with the lowest DIC or alt DIC is deemed to best predict a replicate dataset of the same structure to that observed. Differences in DIC of more than 10 should rule out the model with the highest DIC, whereas if the difference is less than 5, then other criteria, such as average residual deviance and CrI range, are used to judge model fit. While a random-effects model should be preferred in most cases, there are some exceptions where the fixed-effect model may provide more robust estimates, e.g., if the dataset does not contain more than 1 study for each contrast or if there are zero events in the common control arm. |
Assessment of consistency | Not reported |
Assessment of convergence | The WinBUGs model was run for a minimum burn-in of 10,000 iterations to maximize convergence. |
Outcomes | Relevant outcomes for the present submission: ASAS20, ASAS40, ASAS5/6, ASAS-PR, ASDAS-MI, BASDAI, ASQoL, BASFI, BASMI, MASES, NSP, SF-36 PCS |
Follow-up time points | ≥ 12 weeks (differs between studies) |
Construction of nodes | Nodes within the network were constructed by combining trial groups for each molecule. Similar but distinct dosing schedules of the same molecule were combined in the node for that molecule (e.g., 25 mg twice per week and 50 mg once per week were combined for etanercept; 200 mg every 2 weeks and 400 mg every 4 weeks were combined for certolizumab pegol). |
Sensitivity analyses | Not reported |
Subgroup analysis | bDMARD-naive population, predominantly bDMARD-naive population, bDMARD-experienced population |
ASAS-PR = partial remission in the Assessment of SpondyloArthritis International Society; ASAS20 = improvement of 20% or more in the Assessment of SpondyloArthritis International Society; ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASAS5/6 = an improvement of at least 20% in 5 of 6 Assessment of SpondyloArthritis International Society domains; ASDAS = Ankylosing Spondylitis Disease Activity Score; ASDAS-MI = Ankylosing Spondylitis Disease Activity Score–Major Improvement; ASQoL = Ankylosing Spondylitis Quality of Life; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; BASMI = Bath Ankylosing Spondylitis Metrology Index; bDMARD, biologic disease-modifying antirheumatic drug; CrI = credible interval; DIC = deviation information criterion; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; SF-36 = Short Form (36) Health Survey physical component summary.
Source: Clinical evidence summary submitted by the sponsor.19
Matching-Adjusted Indirect Comparison
The MAIC analyses were conducted in accordance with the National Institute for Health and Care Excellence Decision Support Unit Technical Support Document 18.73
To adjust for cross-trial differences, patients from the bimekizumab trials were reweighted to match the baseline characteristics of patients in the comparator trials; weights were determined using a logistic regression based on matching variables selected through reviewing patient characteristics, reviewing existing relevant literature, validation from 2 key opinion leaders, and sensitivity analyses. The following key matching variables were tested for use in this MAIC analysis: age, BASFI at baseline, ASDAS at baseline, proportion male, proportion with prior exposure to TNF inhibitors, BMI or weight, time from diagnosis, time from symptom onset, BASDAI at baseline, PGADA at baseline, proportion white, and proportion who had used sulfasalazine.
Bimekizumab 52-week outcomes were calculated by applying weights from the matching logistic regression with the bimekizumab trial data. These recalculated outcomes were then compared with comparator outcomes via unanchored (non–placebo adjusted) comparisons and were reported as ORs with 95% CIs (based on ESS) for binary outcomes and as differences in means with 95% CIs for continuous outcomes based on ESS. No time points other than week 52 were considered.
All available IL-17 inhibitors used to treat AS were included as comparators due to their similar mechanism of action. Outcomes from the bimekizumab trials at week 52 were compared with outcomes from comparator trials at week 52. The following outcomes were included in the MAIC: ASAS20, ASAS40, ASAS-PR, BASDAI change from baseline, BASDAI50, and an ASDAS of less than 2.1. For binary end points, each MAIC was conducted on NRI data, derived where necessary from the number of observed responses and the number of randomized patients. For continuous end points, published comparator data were reported using a mixed model for repeated measures or observed case data; missing data from the bimekizumab trials were imputed using last observation carried forward. The recalculated bimekizumab week 52 outcomes were compared with those for secukinumab and ixekizumab using unanchored (non–placebo adjusted) comparisons and were reported as ORs or mean differences with 95% CIs.
Results
Summary of Included Studies
Network Meta-Analysis
The SLR identified 28 trials that reported data for the AS population that were deemed feasible to be included in the AS NMA. The NMA included a mixed set of studies in relation to prior bDMARD experience:
Fifteen studies included 100% bDMARD-naive patients: ATLAS,74 Bao (2014),75 Calin (2004),76 Canadian AS trial (Lambert et al. [2007]),77 COAST-V,78 Davis (2003),79 ETN Study 314,80 GO-RAISE,81 Gorman (2002),82 Hu (2012),83 Huang (2014),84 Leeds ETN Study,85 SELECT-AXIS 1,86 SPINE,87 van der Heijde (2017).88
Two studies included 100% bDMARD-experienced patients: COAST-W,89 SELECT-AXIS 2 (Study 1).90
Eleven studies included patients who had some experience with bDMARDs (between 12% and 39% of the patients in the trial): ASSERT (actual percentage is unknown; assumed to be less than 50% due to the age of the study and to align with previous meta-analyses),91 ASTRUM,92 BE AGILE,93 BE MOBILE 294, Deodhar (2021),95 GO-ALIVE,96 MEASURE 297, MEASURE 4,98 MEASURE 5,99 RAPID-axSpA,100 Xue (2022).101
Evidence networks were constructed in 3 scenarios:
A purely bDMARD-naive network with 24 studies and 4,145 patients (Figure 4). This network includes either studies where 100% of patients are bDMARD-naive or studies that reported separate data for the bDMARD-naive subgroup:
Fifteen studies where 100% of the included patients were bDMARD-naive: ATLAS, Bao (2014), Calin (2004), Canadian AS trial, COAST-V, Davis (2003), ETN Study 314, GO-RAISE, Gorman (2002), Hu (2012), Huang (2014), Leeds ETN Study, SELECT-AXIS 1, SPINE, van der Heijde (2017).
Nine studies reporting data for the bDMARD-naive subgroup: ASTRUM, BE AGILE, BE MOBILE 2, Deodhar (2021), MEASURE 2, MEASURE 4, MEASURE 5, RAPID-axSpA, Xue (2022).
A predominantly bDMARD-naive network with 26 studies and 5,271 patients (Figure 5). This network includes studies where it was either confirmed or can be assumed that more than 50% of the included patients were bDMARD-naive:
Fifteen studies where 100% of patients were bDMARD-naive: ATLAS, Bao (2014), Calin (2004), Canadian AS trial, COAST-V, Davis (2003), ETN Study 314, GO-RAISE, Gorman (2002), Hu (2012), Huang (2014), Leeds ETN Study, SELECT-AXIS 1, SPINE, van der Heijde (2017).
Ten studies where more than 50% of patients were bDMARD-naive: ASTRUM (71% bDMARD-naive), BE AGILE (89%), BE MOBILE 2 (82%), Deodhar (2021) (76%), GO-ALIVE (85%), MEASURE 2 (61%), MEASURE 4 (72%), MEASURE 5 (79%), RAPID-axSpA (85%), Xue (2022) (88%).
One study with an unknown percentage of bDMARD-naive patients, but for which it is reasonable to assume that more than 50% of patients were bDMARD-naive: ASSERT.
A purely bDMARD-experienced network with 9 studies and 1,048 patients (Figure 6). This network includes either studies where 100% of the included patients were bDMARD-experienced or studies that reported separate data for the bDMARD-experienced subgroup:
Two studies where 100% of the included patients were bDMARD-experienced: COAST-W and SELECT-AXIS 2 (Study 1).
Seven studies reporting data for the bDMARD-experienced: ASTRUM, BE MOBILE 2, Deodhar (2021), MEASURE 2, MEASURE 4, MEASURE 5, and RAPID-axSpA.
All studies were RCTs with a duration of at least 12 weeks. Study dates were not provided but it was evident that some of the older included studies were performed in the late 1990s or early 2000s. The size of the individual treatment arms ranged from 20 to 305 patients. The majority of patients in the studies were male, representing approximately 63% to 100% of the patients enrolled across treatment arms. The interventions were administered subcutaneously, intravenously, or by mouth. The frequency of the administration of the treatments varied and included daily, weekly, every 2 weeks, and every 4 weeks regimens.
The outcome time frame used for this NMA was 12 to 16 weeks. Preference was given to 16-week data if studies reported measurements at more than 1 time point within this time frame, as this aligned with the follow-up time frame for the primary and secondary end points in BE MOBILE 1 and 2, the phase III trials in which patients with AS (nr-axSpA) received bimekizumab, the key treatment of interest in the networks. Randomization was conducted in appropriately 16 of 28 trials but the randomization methods were unclear in 12 trials. Allocation concealment techniques were adequate for most trials but were unclear for 8 trials. Most studies employed blinding techniques (26 of 28 trials). Five of 28 trials had imbalances in prognostic factors at baseline. All studies had a placebo group. No studies were deemed unsuitable by the sponsor for inclusion in the NMA based on concerns regarding risk of bias. In general, the evaluated trials had a low risk of bias, though the level of risk was unclear for some items.
The majority of the studies included patients with axSpA who have active disease that aligns with the Assessment in Spondyloarthritis International Society–European Alliance of Associations for Rheumatology (ASAS-EULAR) or British Society of Rheumatology criteria for starting TNF inhibitor treatment after NSAID failure. Of the 28 AS studies included in the NMA, enrolment of patients in the majority of trials was based on the 1984 mNY criteria. However, according to the sponsor, the entry criteria for 3 studies aligned with the ASAS classification criteria, which used the mNY criteria for r-axSpA plus additional criteria. Therefore, some patients included in the other trials might not have been eligible for enrolment in the COAST-V, COAST-W, or Xue (2022) studies. It was assumed by the sponsor that these populations were sufficiently similar to enable these studies to be directly compared in the NMA.
The range of the proportion of patients in the trials with HLA-B27–positive status was approximately 75% to 95%. C-reactive protein scores were not consistently reported, with some data missing and other studies reporting measures of mean, geometric mean, or median values, making it difficult to compare between studies. Where C-reactive protein means were reported, the range was approximately 12 mg/L to 32 mg/L. The range in mean baseline values was 5.5 to 7.5 for the BASDAI and 3.4 to 4.2 for the ASDAS using C-reactive protein.
Matching-Adjusted Indirect Comparison
Bimekizumab was compared with secukinumab 150 mg every 4 weeks, secukinumab 300 mg every 4 weeks, and ixekizumab 80 mg every 4 weeks for this analysis. In the analysis for bimekizumab versus secukinumab 150 mg, individual patient data from the BE MOBILE 2 trial were matched to the pooled secukinumab 150 mg loading dose and no-loading summary data from the MEASURE 1, MEASURE 2, MEASURE 3 and MEASURE 4 trials for pairwise comparisons. In the analysis for bimekizumab versus secukinumab 300 mg, individual patient data from the BE MOBILE 2 trial were matched to the secukinumab 300 mg summary data from the MEASURE 3 study for pairwise comparisons. It should be noted that in the MEASURE 3 trial, patients received 3 IV-loading doses of secukinumab 300 mg every 2 weeks, which is off label, before being switched at week 4 to subcutaneous secukinumab 300 mg every 4 weeks; the impact of this on the comparisons could not be determined. For pairwise comparisons in the bimekizumab versus ixekizumab analysis, individual patient data from patients randomized to bimekizumab in the BE MOBILE 2 trial were matched to the summary data from the patients in the COAST-V and COAST-W trials who were randomized to bimekizumab 80 mg every 4 weeks.
Summary of Results
Efficacy
Network Meta-Analysis
In the bDMARD purely naive network, the results favoured bimekizumab over placebo for most outcomes except for MASES and the SF-36 mental component summary (SF-36 MCS). For most comparisons between bimekizumab versus TNF, IL-17, or JAK inhibitors, there were no clear differences observed. The exceptions to this were 2 findings in which bimekizumab showed improvement in SF-36 PCS results compared with adalimumab and secukinumab.
In the bDMARD predominantly naive network, results favoured bimekizumab over placebo for most outcomes except for MASES and SF-36 MCS. For most comparisons between bimekizumab versus TNF, IL-17, or JAK inhibitors, there were no clear differences observed. There were some exceptions to this general observation. Bimekizumab showed improvement in SF-36 PCS and ASAS-PR results compared with secukinumab. Results favoured etanercept compared with bimekizumab for BASDAI50 and BASFI. Results favoured golimumab IV compared with bimekizumab for BASFI and ASQoL. Results favoured adalimumab and certolizumab over bimekizumab for ASDAS-ID. Results favoured tofacitinib over bimekizumab for Bath Ankylosing Spondylitis Metrology Index. Results favoured upadacitinib over bimekizumab for ASDAS-ID.
In the bDMARD purely experienced network, results favoured bimekizumab over placebo for ASAS-40, ASDAS using C-reactive protein, and BASFI. There were no clear differences between bimekizumab and placebo for ASAS20, major improvement in the ASDAS, BASDAI50, BASDAI, and NSP. For most comparisons between bimekizumab versus TNF, IL-17, or JAK inhibitors, there were no clear differences observed. The exceptions to this were 2 findings in which the results favoured certolizumab over bimekizumab for ASQoL and SF-36 PCS.
In the 3 networks, there were no clear differences observed between bimekizumab and other active comparators for the outcomes identified for the GRADE analysis in this report (ASAS40, BASDAI, BASFI, NSP, enthesitis state, ASQoL, WPAI-SHP, and SAEs).
Harms
Network Meta-Analysis
The sponsor conducted NMAs of bimekizumab versus relevant comparators in the context of axSpA for 2 harms outcomes, discontinuation due to any reason and SAEs. The comparators of interest with data available for this NMA were the IL-17A inhibitors ixekizumab and secukinumab; the TNF alpha inhibitors adalimumab, certolizumab pegol, etanercept, golimumab (subcutaneous or IV routes) and infliximab (IV); and the JAK inhibitors tofacitinib and upadacitinib.
The network for the analysis of discontinuation due to any reason contained 18 studies. Study discontinuation rates were low in all trials (range of 0 to 14 patients per treatment arm). Time points between 12 and 16 weeks were used for this analysis. The CrIs were very wide for most estimates. There were no clear differences observed between bimekizumab and any other treatment. There was 1 finding in which bimekizumab had a higher risk of study discontinuation compared with tofacitinib; however, the uncertainty around this estimate was high, as reflected by a wide CrI.
The network for the analysis of SAEs contained 18 studies. SAE rates were low in all studies (range of 0 to 10 SAEs per treatment arm). Time points between 12 and 16 weeks were used for this analysis. The CrIs were very wide for most estimates. There were no clear differences between bimekizumab and any other treatments in the network.
Evidence Networks
Figure 4: Network 1 — Purely bDMARD-Naivea
ADA = adalimumab; bDMARD = biologic disease-modifying antirheumatic drug; BID = twice a day; BIW = twice a week; BKZ = bimekizumab; CZP = certolizumab pegol; ETN = etanercept; GOL = golimumab; IFX = infliximab; IR = inadequate response; IXE = ixekizumab; NSAID = nonsteroidal anti-inflammatory drug; PBO = placebo; Q2W = twice a week; Q4W = 4 times a week; Q6W = 6 times a week; Q8W = 8 times a week; QD = once daily; QW = once weekly; SC = subcutaneous; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib.
a Study population is IR to ≥ 1 NSAIDs and either 100% bDMARD-naive patients or data available for the subset of patients that are naive to bDMARDs (> 9 patients); studies in grey are studies excluded from this network but included in network 2 (predominantly naive). Light blue indicates interleukin-17A inhibitors, dark blue indicates interleukin-17A and interleukin-17F inhibitors (i.e., bimekizumab), red indicates Janus kinase inhibitors, and green indicates tumour necrosis factor inhibitors.
Source: Clinical evidence summary submitted by the sponsor.19
Figure 5: Network 2 — Predominantly bDMARD-Naive
ADA = adalimumab; axSpA = axial spondyloarthritis; bDMARD = biologic disease-modifying antirheumatic drug; BID = twice a day; BIW = twice a week; BKZ = bimekizumab; CZP = certolizumab pegol; ETN = etanercept; GOL = golimumab; IFX = infliximab; IR = inadequate response; IXE = ixekizumab; NSAID = nonsteroidal anti-inflammatory drug; PBO = placebo; Q2W = twice a week; Q4W = 4 times a week; Q6W = 6 times a week; Q8W = 8 times a week; QD = once daily; QW = once weekly; SC = subcutaneous; SEC = secukinumab; TOF = tofacitinib; TNF = tumour necrosis factor; UPA = upadacitinib.
Note: Study population is IR to ≥ 1 NSAID studies and 61% to 100% bDMARD-naive patients. Approximately 90% of patients included in this network were bDMARD-naive. Light blue indicates interleukin-17A inhibitors, dark blue indicates interleukin-17A or −17F inhibitors (i.e., bimekizumab), red indicates Janus kinase inhibitors, and green indicates tumour necrosis factor inhibitors.
Source: Clinical evidence summary submitted by the sponsor.19
Figure 6: Network 3 — Purely bDMARD-Experienceda
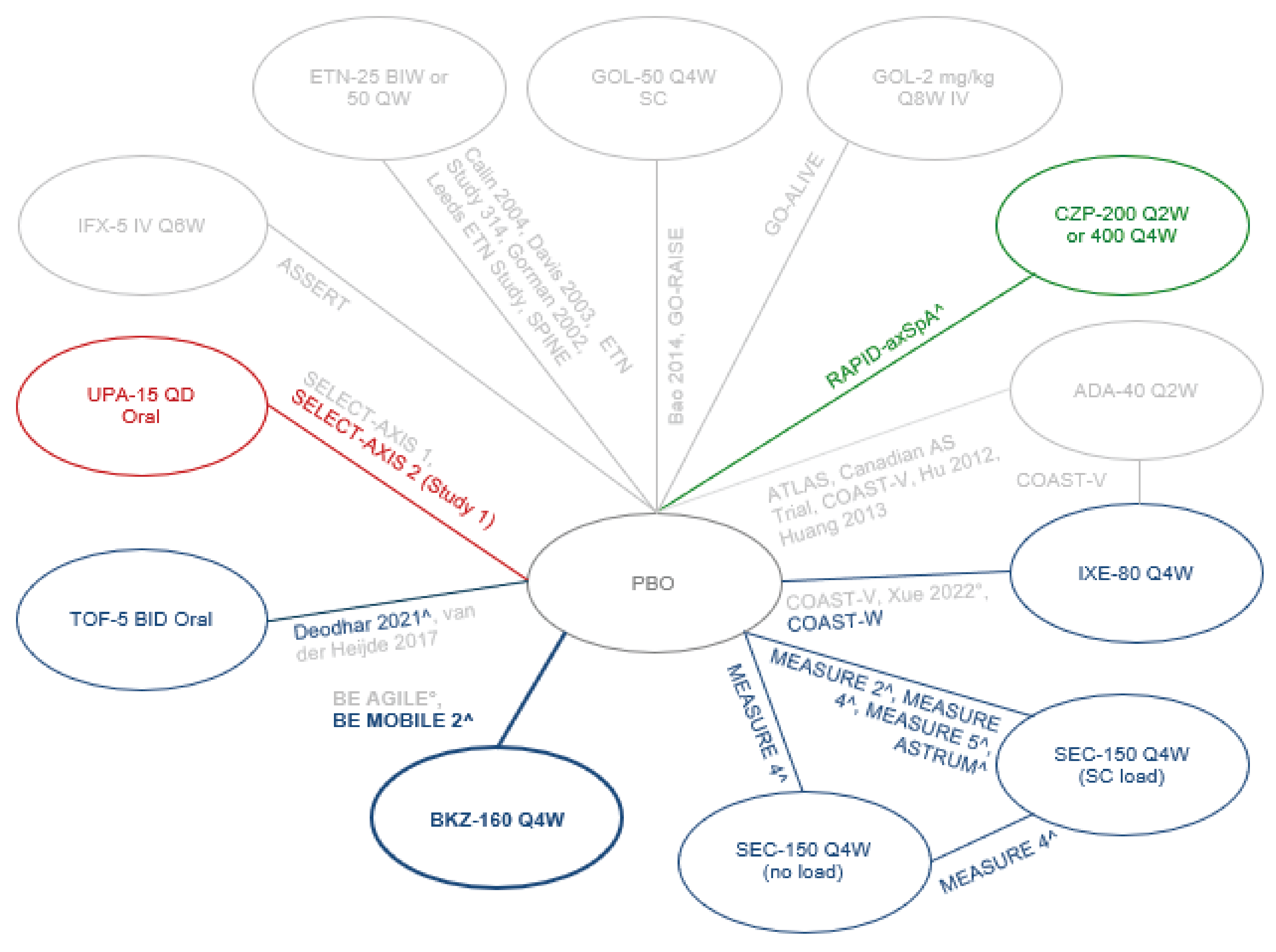
ADA = adalimumab; ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASDAS-CRP = Ankylosing Spondylitis Disease Activity Score using C-reactive protein; BASDAI50 = a reduction of at least 50% in the Bath Ankylosing Spondylitis Disease Activity Index score; BASFI = Bath Ankylosing Spondylitis Functional Index; bDMARD = biologic disease-modifying antirheumatic drug; BID = twice a day; BIW = twice a week; BKZ = bimekizumab; CZP = certolizumab pegol; ETN = etanercept; GOL = golimumab; IFX = infliximab; IR = inadequate response; IXE = ixekizumab; NSAID = nonsteroidal anti-inflammatory drug; PBO = placebo; Q2W = twice a week; Q4W = 4 times a week; Q6W = 6 times a week; Q8W = 8 times a week; QD = once daily; QW = once weekly; SC = subcutaneous; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib.
Note: Studies in grey are those excluded from this network but included in network 2 (predominantly naive). Light blue indicates interleukin-17A inhibitors, dark blue indicates interleukin-17A and −17F inhibitors (i.e., bimekizumab), red indicates Janus kinase inhibitors, and green indicates tumour necrosis factor inhibitors.
a Study population of patients who experienced an IR to ≥ 1 NSAIDs who were either 100% bDMARD-experienced or who had had 1 or more prior bDMARDs (> 9 patients).
° Data not included in network, as there were < 10 patients in study arm.
^ Data available for the subset of patients who had received 1 or more prior bDMARD (> 9 patients).
Source: Clinical evidence summary submitted by the sponsor.19
Table 27: Summary of Patient Characteristics for Studies Included in the NMA (N = 28)
Study | Treatment arm | N, ITT | Age, mean (SD) | Male (%) | HLA-B27 positive (%) | CRP mg/L mean (SD) | BASDAI, mean (SD) | ASDAS-CRP, mean (SD) | Total spine pain, mean (SD) | Prior bDMARDs (%) |
|---|---|---|---|---|---|---|---|---|---|---|
ASSERT91 | IFX 5 mg IV q.6.w. | 201 | 40 | 78.1 | 86.5 | 15 | 6.6 | NR | NR | NR |
ASSERT91 | PBO | 78 | 41 | 87.2 | 88.5 | 17 | 6.5 | NR | NR | NR |
ASTRUM92 | SEC-150 mg q.4.w. (SC load) | 71 | 46.2 (13.4) | 57.7 | NR | NR | 6.0 (1.4) | 3.4 (0.7) | NR | 28.2 |
ASTRUM92 | PBO | 70 | 45.4 (12.6) | 55.7 | NR | NR | 6.2 (1.3) | 3.4 (0.7) | NR | 28.6 |
ATLAS74 | ADA 40 mg q.2.w. | 208 | 41.7 (11.69) | 75.5 | 78.4 | 18 (22) | 6.3 (1.7) | NR | 64.4 (20.9) | 0 |
ATLAS74 | PBO | 107 | 43.4 (11.32) | 73.8 | 79.4 | 22 (29) | 6.3 (1.7) | NR | 67.2 (21.5) | 0 |
Bao (2014)75 | GOL 50 mg q.4.w. SC | 108 | 30.5 (10.27) | 83.3 | NR | 20.6 (21.23) | 6.6 (1.31) | NR | NR | 0 |
Bao (2014)75 | PBO | 105 | 30.6 (8.6) | 82.9 | NR | 18.6 (19.89) | 6.5 (1.54) | NR | NR | 0 |
BE AGILE93 | BKZ 160 mg q.4.w. | 60 | 42.4 (13.1) | 86.7 | 86.7 | 20.5 (19.3) | 6.3 (1.3) | 3.9 (0.8) | NR | 11.7 |
BE AGILE93 | PBO | 60 | 39.7 (10.3) | 81.7 | 95 | 17.6 (24.6) | 6.5 (1.4) | 3.8 (0.9) | NR | 11.7 |
BE MOBILE 294 | BKZ 160 mg q.4.w. | 221 | 41 (12.1) | 72.4 | 86.4 | 8.2 | 6.5 (1.3) | 3.7 (0.8) | 7.1 (1.6) | 17.6 |
BE MOBILE 294 | PBO | 111 | 39.2 (12.6) | 72.1 | 83.8 | 6.3 | 6.5 (1.3) | 3.7 (0.8) | 7.2 (1.2) | 18.0 |
Calin (2004)76 | ETN 25 mg b.i.w. | 45 | 45.3 (9.5) | 80 | NR | 154 | 6.1 | NR | NR | 0 |
Calin (2004)76 | PBO | 39 | 40.7 (11.4) | 77 | NR | 97 | 5.86 | NR | NR | 0 |
Canadian AS trial77 | ADA 40 mg q.2.w. | 38 | 41.9 (11.14) | 76.3 | 86.8 | 18 (17) | 6.2 (1.7) | NR | 67.2 (16.7) | 0 |
Canadian AS trial77 | PBO | 44 | 40 (10.87) | 81.8 | 81.8 | 23 (26) | 6.5 (1.6) | NR | 71.7 (14.8) | 0 |
COAST-V78 | ADA 40 mg q.2.w. | 90 | 41.8 (11.4) | 81 | 91 | 12.5 (17.6) | 6.7 (1.5) | 3.7 (0.8) | NR | 0 |
COAST-V78 | IXE 80 mg q.4.w. | 81 | 41 (12.1) | 84 | 93 | 12.2 (13.3) | 6.8 (1.3) | 3.7 (0.7) | NR | 0 |
COAST-V78 | PBO | 87 | 42.7 (12) | 83 | 89 | 16 (21) | 6.8 (1.2) | 3.9 (0.7) | NR | 0 |
COAST-W89 | IXE 80 mg q.4.w. | 114 | 47.4 (13.4) | 79.8 | NR | NR | 7.5 (1.3) | 4.2 (0.9) | NR | 100 |
COAST-W89 | PBO | 104 | 46.6 (12.7) | 83.7 | NR | NR | 7.3 (1.3) | 4.1 (0.8) | NR | 100 |
Davis (2003)79 | ETN 25 mg b.i.w. | 138 | 42.1 | 76 | 84 | 19 | 5.81 | NR | 61.1 | 0 |
Davis (2003)79 | PBO | 139 | 41.9 | 76 | 84 | 20 | 5.96 | NR | 63.5 | 0 |
Deodhar (2021)95 | TOF 5 mg b.i.d. oral | 133 | 42.2 (11.9) | 87.2 | 83.5 | 16.4 (17.3) | 6.4 (1.5) | 3.8 (0.9) | 6.9 (1.5) | 23.3 |
Deodhar (2021)95 | PBO | 136 | 40 (11.1) | 79.4 | 85.3 | 18 (19.7) | 6.5 (1.4) | 3.9 (0.8) | 6.9 (1.6) | 22.8 |
ETN study 31480 | ETN 50 q.w. | 155 | 41.5 (11) | 69.7 | NR | 21.7 (24.6) | 6.24 (1.7) | NR | 63.9 (19.2) | 0 |
ETN study 31480 | ETN 25 b.i.w. | 150 | 39.8 (10.7) | 76 | NR | 19.8 (20.8) | 5.94 (1.67) | NR | 63.5 (21.1) | 0 |
ETN study 31480 | PBO | 51 | 40.1 (10.9) | 78.4 | NR | 22 (22.9) | 6.11 (1.37) | NR | 63.1 (18.4) | 0 |
GO-ALIVE96 | GOL 2 mg/kg q.8.w. IV | 105 | 38.4 (10.1) | 81.9 | 89.5 | 20 (18.2) | 7 (1.2) | 4.2 (0.7) | 7.2 (1.3) | 15.2 |
GO-ALIVE96 | PBO | 103 | 39.2 (10.8) | 74.8 | 90.3 | 19.3 (16.7) | 7.1 (1.2) | 4.1 (0.8) | 7.3 (1.5) | 13.6 |
GO-RAISE81 | GOL 50 mg q.4.w. SC | 138 | 39.2 (12.46) | 73.9 | 81.8 | 18 (18) | 6.5 (1.6) | NR | 7.1 (1.5) | 0 |
GO-RAISE81 | PBO | 78 | 40.6 (12.71) | 70.5 | 84.6 | 19 (23) | 6.6 (1.5) | NR | 7.5 (1.6) | 0 |
Gorman (2002)82 | ETN 25 mg b.i.w. | 20 | 38 (10) | 65 | 95 | 20 (18) | NR | NR | NR | 0 |
Gorman (2002)82 | PBO | 20 | 39 (10) | 90 | 90 | 15 (12) | NR | NR | NR | 0 |
Hu (2012)83 | ADA 40 mg q.2.w. | 26 | 28.2 (6.9) | 92.3 | 96.2 | 24.6 (23.2) | 5.9 (1.4) | 3.7 (0.8) | NR | 0 |
Hu (2012)83 | PBO | 20 | 27.4 (7.2) | 100 | 95 | 32.1 (29.1) | 6.2 (1.1) | 4 (0.9) | NR | 0 |
Huang (2014)84 | ADA 40 mg q.2.w. | 229 | 30.1 (8.7) | 80.8 | 95.6 | 22.4 (24) | 6 (1.4) | 3.7 (0.9) | 6.8 (1.5) | 0 |
Huang (2014)84 | PBO | 115 | 29.6 (7.5) | 82.6 | 94.8 | 23 (30) | 6.2 (1.4) | 3.7 (1) | 6.7 (1.6) | 0 |
Leeds ETN study85 | ETN 25 mg b.i.w. | 20 | 40.8 (9.7) | 75 | NR | NR | 6.05 (1.71) | NR | NR | 0 |
Leeds ETN study85 | PBO | 20 | 39.4 (10.1) | 85 | NR | NR | 5.46 (1.74) | NR | NR | 0 |
MEASURE 297 | SEC 150 mg q.4.w. (SC load) | 72 | 41.9 (12.5) | 64 | 79 | 25.8 (50.1) | 6.59 (1.5) | 3.73 (0.89) | 66.2 (16.7) | 38.9 |
MEASURE 297 | PBO | 74 | 43.6 (13.2) | 76 | 78 | 15.71 (18.5) | 6.78 (1.3) | 3.82 (0.76) | 69.2 (18.8) | 39.2 |
MEASURE 498 | SEC 150 mg q.4.w. (SC load) | 116 | 44.5 (11.62) | 69.8 | 86.2 | 6.25 | 7 (1.23) | NR | 74.9 (13.07) | 26.7 |
MEASURE 498 | SEC 150 mg q.4.w. (no load) | 117 | 41.2 (11.07) | 70.9 | 84.6 | 6.2 | 7.0 (1.31) | NR | 74.2 (14.18) | 27.3 |
MEASURE 498 | PBO | 117 | 43.4 (12.46) | 65 | 79.5 | 5.4 | 7.1 (1.27) | NR | 75 (13.8) | 29 |
MEASURE 599 | SEC 150 mg q.4.w. (SC load) | 305 | 35.1 (10.38) | 82.6 | 90.5 | 7.5 | 6.91 (1.38) | NR | 71.6 (14.51) | 21.3 |
MEASURE 599 | PBO | 153 | 33 (10.02) | 86.3 | 92.8 | 7.8 | 6.87 (1.25) | NR | 70.5 (13.44) | 20.3 |
RAPID-axSpA100 | CZP 200 mg q.2.w. | 65 | 41 (10.8) | 72.3 | 81.5 | 20.53 (27.19) | 6.52 (1.67) | NR | NR | 16.9 |
RAPID-axSpA100 | CZP 400 mg q.4.w. | 56 | 41.9 (11.5) | 73.2 | 78.6 | 18.26 (22.98) | 6.18 (1.29) | NR | NR | 16.1 |
RAPID-axSpA100 | PBO | 57 | 41.6 (12.8) | 71.9 | 84.2 | 25.22 (26.7) | 6.44 (1.85) | NR | NR | 28.1 |
SELECT-AXIS 186 | UPA 15 mg q.d. oral | 93 | 47 (12.8) | 68 | 75 | 9.6 (12.6) | 6.3 (1.8) | 3.5 (0.8) | 6.8 (1.8) | 0 |
SELECT-AXIS186 | PBO | 94 | 43.7 (12.1) | 73 | 78 | 11.7 (11.1) | 6.5 (1.6) | 3.7 (0.7) | 6.7 (1.8) | 0 |
SELECT-AXIS 2 (Study 1)90 | UPA 15 mg q.d. oral | 211 | 42.6 (12.4) | 73 | 85 | 15.8 (17.7) | 6.8 (1.3) | 3.9 (0.8) | 7.5 (1.5) | 100 |
SELECT-AXIS 2 (Study 1)90 | PBO | 209 | 42.2 (11.8) | 76 | 81 | 14.5 (17.8) | 6.8 (1.3) | 3.9 (0.8) | 7.4 (1.4) | 99.5 |
SPINE87 | ETN 50 mg q.w. | 39 | 46 (11) | 95 | 79 | 25 (31) | 6.4 (1.2) | 3.9 (0.7) | 7.0 (1.6) | 0 |
SPINE87 | PBO | 43 | 48 (10) | 91 | 86 | 17 (19) | 5.8 (1.5) | 3.6 (0.8) | 6.1 (2.0) | 0 |
van der Heijde (2017)88 | TOF 5 mg b.i.d. oral | 52 | 41.2 (10.3) | 75 | 84.6 | NR | 6.5 (1.9) | 3.7 (0.9) | NR | 0 |
van der Heijde (2017)88 | PBO | 51 | 41.9 (12.9) | 62.7 | 86.3 | NR | 6.3 (1.9) | 3.7 (0.8) | NR | 0 |
Xue (2022)101 | IXE 80 mg q.4.w. | 74 | 33.5 (8.89) | 86.5 | NR | NR | NR | NR | NR | 89 |
Xue (2022)101 | PBO | 73 | 34.4 (8.98) | 89.0 | NR | NR | NR | NR | NR | 88 |
ASDAS-CRP = Ankylosing Spondylitis Disease Activity Score using C-reactive protein; b.i.d. = twice a day; b.i.w. = twice a week; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BKZ = bimekizumab 160 mg q.4.w. SC; Crl = credible interval; CRP = C-reactive protein; CZP = certolizumab pegol 200 mg q.2.w. SC or 400 mg q.4.w. SC; ETN = etanercept 50 mg q.w. SC or 25 mg b.i.w. SC; GOL = golimumab 50 mg q.4.w SC or golimumab 2 mg/kg q.8.w. IV; HLA-B27 = human leukocyte antigen B27; IFX = infliximab 5 mg IV q.6.w.; IL = interleukin; IXE = ixekizumab 80 mg q.4.w. SC; JAK = Janus kinase; MD = mean difference; NA = not applicable (not included in network); NR = not reported; OR = odds ratio; PBO = placebo; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; q.8.w. = every 8 weeks; q.d. = once daily; q.w. = every week; SC = subcutaneous; SEC (no load) = secukinumab 150 mg q.4.w. SC (no loading); SEC (SC load) = secukinumab 150 mg q.4.w. SC (with SC loading); TNF = tumour necrosis factor; TOF = tofacitinib 5 mg b.i.d. oral; UPA = upadacitinib 15 mg q.d. oral.
Source: Clinical evidence summary submitted by the sponsor.19
Table 28: Summary of Outcomes at 12 to 16 Weeks —bDMARD Purely Naive Network (1 of 2)a
Outcomes | BKZ vs.b | ASAS20 | ASAS40 | ASDAS-MI | BASDAI50 | ASDAS-CRP | BASDAI | BASFI | NSP |
|---|---|---|---|---|---|---|---|---|---|
Binomial: OR (95% CrI)c | Continuous: MD (95% Crl)c | ||||||||
NA | PBO | 3.24 (2.17 to 4.52) | 3.96 (3.02 to 5.11) | 7.53 (1.03 to 23.58) | 4.87 (2.42 to 9.79) | −0.89 (−1.65 to −0.22) | −1.36 (−1.68 to −0.99) | −1.24 (−1.56 to −0.91) | −1.69 (−2.12 to −0.90) |
TNFi | ADA | 0.91 (0.55 to 1.37) | 1.16 (0.81 to 1.60) | 0.64 (0.10 to 2.28) | 1.05 (0.47 to 2.40) | 0.28 (−0.77 to 1.21) | 0.06 (−0.32 to 0.52) | −0.05 (−0.42 to 0.35) | −0.11 (−0.69 to 1.08) |
CZP | 1.17 (0.60 to 2.23) | 1.11 (0.68 to 1.81) | NE | 0.94 (0.19 to 3.29) | 0.19 (−1.03 to 1.33) | −0.12 (−0.71 to 0.54) | −0.19 (−0.77 to 0.41) | −0.37 (−1.20 to 0.60) | |
ETN | 0.79 (0.48 to 1.21) | 0.83 (0.60 to 1.14) | 0.58 (0.07 to 176.60) | 0.76 (0.36 to 2.16) | 0.10 (−1.15 to 1.43) | 0.20 (−0.30 to 0.75) | 0.43 (−0.03 to 0.89) | 0.68 (−0.77 to 3.58) | |
GOL SC | 1.04 (0.51 to 1.75) | 1.18 (0.78 to 1.74) | 0.70 (0.06 to 2.31) | 1.00 (0.31 to 3.30) | NA | 0.61 (−0.11 to 1.43) | −0.14 (−0.70 to 0.50) | NA | |
GOL IV | NA | NA | NA | NA | NA | NA | NA | NA | |
IFX | NA | NA | NA | NA | NA | NA | NA | NA | |
IL-17i | IXE | 0.96 (0.48 to 1.75) | 0.99 (0.61 to 1.52) | 0.60 (0.08 to 2.32) | 0.94 (0.33 to 2.64) | 0.24 (−0.91 to 1.36) | 0.36 (−0.29 to 1.04) | 0.25 (−0.38 to 0.85) | 0.49 (−0.19 to 1.78) |
SEC (SC load) | 1.26 (0.83 to 1.90) | 1.21 (0.88 to 1.65) | NA | NA | 0.05 (−1.48 to 1.26) | −0.19 (−0.63 to 0.24) | 0.75 (−0.07 to 1.49) | 0.51 (−0.46 to 1.62) | |
SEC (no load) | 1.33 (0.73 to 2.34) | 1.47 (0.88 to 2.53) | NA | NA | NA | −0.32 (−0.93 to 0.28) | NA | NA | |
JAK inhibitor | TOF | 0.79 (0.45 to 1.30) | 1.01 (0.66 to 1.52) | 2.07 (0.04 to 12.48) | 1.27 (0.41 to 4.07) | −0.04 (−1.17 to 0.95) | −0.09 (−0.88 to 0.69) | 0.22 (−0.27 to 0.68) | NA |
UPA | 1.04 (0.56 to 1.90) | 0.85 (0.53 to 1.37) | 0.99 (0.19 to 3.59) | 1.18 (0.39 to 3.53) | 0.05 (−1.14 to 1.44) | NA | 0.11 (−0.49 to 0.68) | 0.11 (−0.99 to 1.12) | |
ADA = adalimumab 40 mg q.2.w. SC; AS = ankylosing spondylitis; ASAS20 = improvement of ≥ 20% in the Assessment of SpondyloArthritis International Society; ASAS40 = improvement of ≥ 40% in the Assessment of SpondyloArthritis International Society; ASDAS-MI = major improvement in Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; bDMARD = biological disease-modifying antirheumatic drug; b.i.d. = twice a day; b.i.w. = twice a week; BKZ = bimekizumab 160 mg q.4.w. SC; Crl = credible interval; CRP = C-reactive protein; CZP = certolizumab pegol 200 mg q.2.w. SC or 400 mg q.4.w. SC; ETN = etanercept 50 mg q.w. SC or 25 mg b.i.w. SC; GOL = golimumab 50 mg q.4.w. SC or golimumab 2 mg/kg q.8.w. IV;IFX = infliximab 5 mg IV q.6.w.; IL-17i = interleukin-17 inhibitor; IXE = ixekizumab 80 mg q.4.w. SC; JAK = Janus kinase; MD = mean difference; NA = not applicable (not included in network); NE = not estimated; NSP = nocturnal spine pain; OR = odds ratio; PBO = placebo; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; q.8.w. = every 8 weeks; q.d. = once daily; SC = subcutaneous; SEC (no load) = secukinumab 150 mg q.4.w. SC (no loading); SEC (SC load) = secukinumab 150 mg q.4.w. SC (with SC loading); TNFi = tumour necrosis factor inhibitor; TOF = tofacitinib; UPA = upadacitinib 15 mg q.d. oral.
Note: Bold font indicates significant difference, meaning that the CrI does not cross the null.
aHorizontal separation lines indicate placebo and different drug classes.
bPresented models are suggested base case results, on balance based on model fit parameters and 95% CrI.
cSignificant difference based on 95% CrI: for binomial outcomes, OR = 1 is the line of no effect; for continuous outcomes, difference = 0 is the line of no effect.
Source: Full network meta-analysis report submitted by the sponsor.102
Table 29: Summary of Outcomes at 12 to 16 Weeks —bDMARD Purely Naive Network (2 of 2)a
Outcomes | BKZ vs.b | ASAS-5/6 | ASAS-PR | ASDAS < 2.1 | ASDAS-ID | ASDAS-CII | ASQoL | BASMI | Fatigue NRS | MASES | PGADA | SF-36 MCS | SF-36 PCS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Binomial: OR (95% CrI)c | Continuous: MD (95% Crl)c | ||||||||||||
NA | PBO | 5.48 (1.57 to 10.72) | 4.95 (3.25 to 7.18) | 4.80 (1.96 to 56.23) | 5.81 (3.05 to 17.78) | 5.59 (1.10 to 28.87) | −2.26 (−2.93 to −1.32) | −0.29 (−0.41 to −0.17) | −1.21 (−1.55 to −0.81) | −0.36 (−1.33 to 3.55) | −1.44 (−1.81 to −1.04) | 0.97 (−0.72 to 3.85) | 5.52 (3.94 to 6.86) |
TNFi | ADA | 0.79 (0.31 to 3.58) | 1.33 (0.80 to 2.48) | 1.35 (0.29 to 583.80) | 0.43 (0.19 to 1.37) | 0.74 (0.09 to 5.90) | −0.66 (−2.01 to 1.50) | 0.05 (−0.10 to 0.20) | −0.16 (−0.64 to 0.40) | 0.36 (−1.02 to 5.10) | 0.22 (−0.23 to 0.72) | −0.25 (−3.06 to 5.54) | 2.31 (0.50 to 3.87) |
CZP | 1.06 (0.31 to15.58) | 1.38 (0.48 to 5.99) | 0.81 (0.13 to 13.42) | NE | 0.64 (0.04 to 8.70) | −0.51 (−1.80 to 1.37) | 0.04 (−0.28 to 0.35) | −0.38 (−1.05 to 0.35) | −0.43 (−2.30 to 2.90) | 0.41 (−0.32 to 1.17) | 2.32 (−2.61 to 13.79) | 1.20 (−1.16 to 3.30) | |
ETN | 0.97 (0.48 to 8.71) | 0.96 (0.55 to 2.11) | 1.17 (0.16 to 18.85) | 1.83 (0.38 to 11.13) | 0.83 (0.04 to 16.28) | −0.16 (−2.79 to 3.64) | 0.13 (−0.12 to 0.38) | 0.40 (−0.13 to 1.02) | NA | −0.02 (−1.37 to 1.33) | −6.82 (−22.55 to 0.61) | −2.03 (−6.82 to 3.16) | |
GOL SC | 0.95 (0.35 to 5.14) | 1.09 (0.56 to 2.30) | NA | 0.66 (0.09 to 2.30) | NA | NA | −0.07 (−0.23 to 0.09) | NA | 0.56 (−1.61 to 8.64) | NA | 2.13 (−6.93 to 0.37) | 1.44 (−0.89 to 3.47) | |
GOL IV | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
IFX | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
IL-17i | IXE | NA | 1.74 (0.79 to 3.93) | 1.09 (0.23 to 411.0) | 0.53 (0.15 to 1.88) | 0.69 (0.05 to 8.77) | NA | 0.07 (−0.15 to 0.31) | 0.10 (−0.50 to 0.73) | NA | −0.08 (−0.70 to 0.55) | NA | 2.01 (−0.25 to 4.16) |
SEC (SC load) | 1.79 (0.38 to 9.53) | 1.72 (0.95 to 2.99) | NA | NA | NA | −0.14 (−1.18 to 0.77) | NA | 0.01 (−0.78 to 0.84) | NA | NA | NA | 2.01 (0.57 to 3.30) | |
SEC (no load) | 1.77 (0.21 to 16.56) | NA | NA | NA | NA | 0.28 (−1.13 to 1.53) | NA | NA | NA | NA | NA | 1.64 (−0.47 to 3.77) | |
JAKi | TOF | 1.16 (0.16 to 10.22) | 1.28 (0.46 to 3.27) | 0.72 (0.05 to 4.67) | 2.50 (0.77 to 13.76) | 0.88 (0.04 to 15.22) | 0.22 (−0.83 to 1.35) | 0.12 (−0.05 to 0.31) | NA | NA | 0.29 (−0.27 to 0.90) | −0.78 (−5.09 to 7.38) | 1.70 (−0.21 to 3.39) |
UPA | NA | 1.68 (0.42 to 16.61) | 0.85 (0.11 to 872.70) | NE | 1.31 (0.09 to 19.54) | −0.52 (−1.76 to 0.81) | −0.04 (−0.25 to 0.19) | NA | 0.38 (−1.13 to 3.30) | 0.38 (−0.59 to 1.38) | NA | NA | |
ADA = adalimumab 40 mg q.2.w. SC; AS = ankylosing spondylitis; ASAS-PR = partial remission in the Assessment of SpondyloArthritis International Society; ASAS-5/6 = improvement of 20% or more in at least 5 of the 6 domains of the Assessment of SpondyloArthritis International Society; ASDAS-CII = clinically important improvement in Ankylosing Spondylitis Disease Activity Score; ASDAS-ID = Ankylosing Spondylitis Disease Activity Score inactive disease state; ASQoL = Ankylosing Spondylitis Quality of Life; BASMI = Bath Ankylosing Spondylitis Metrology Index; bDMARD = biological disease-modifying antirheumatic drug; b.i.d. = twice a day; b.i.w. = twice a week; BKZ = bimekizumab 160 mg q.4.w. SC; CrI = credible interval; CZP = certolizumab pegol 200 mg q.2.w. SC or 400 mg q.4.w. SC; ETN = etanercept 50 mg q.w. SC or 25 mg b.i.w. SC; GOL = golimumab 50 mg q.4.w. SC or golimumab 2 mg/kg q.8.w. IV; IFX = infliximab 5 mg IV q.6.w.; i = inhibitor; IL = interleukin; IXE = ixekizumab 80 mg q.4.w. SC; JAK = Janus kinase; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; MD = mean difference; NA = not applicable (not included in network); NE = not estimated; NRS = numeric rating scale; OR = odds ratio; PBO = placebo; PGADA = Patient’s Global Assessment of Disease Activity; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; q.8.w. = every 8 weeks; q.d. = once daily; SC = subcutaneous; SEC (no load) = secukinumab 150 mg q.4.w. SC (no loading); SEC (SC load) = secukinumab 150 mg q.4.w. SC (with SC loading); SF-36 MCS = Short Form (36) Health Survey mental component summary; SF-36 PCS = Short Form (36) Health Survey physical component summary; TNF = tumour necrosis factor; TOF = tofacitinib 5 mg b.i.d. oral; UPA = upadacitinib 15 mg q.d. oral.
Note: Bold font indicates significant difference, meaning that the CrI does not cross the null.
aHorizontal separation lines indicate placebo and different drug classes.
bPresented models are suggested base case results, on balance based on model fit parameters and 95% CrI.
cSignificant difference based on 95% CrI; for binomial outcomes, OR = 1 is the line of no effect; for continuous outcomes, difference = 0 is the line of no effect.
Source: Full network meta-analysis report submitted by the sponsor.
Table 30: Summary of Outcomes at 12 to 16 Weeks, bDMARD Predominantly Naive Network (1 of 3)a
Outcomes | BKZ vs.b | ASAS20 | ASAS40 | ASDAS-MI | BASDAI50 | ASDAS-CRP | BASDAI | BASFI | NSP |
|---|---|---|---|---|---|---|---|---|---|
Binomial: OR (95% CrI)c | Continuous: MD (95% Crl)c | ||||||||
NA | PBO | 3.33 (2.10 to 5.34) | 4.09 (3.15 to 5.26) | 6.23 (1.25 to 38.31) | 3.50 (2.52 to 4.70) | −0.86 (−1.34 to −0.43) | −1.32 (−1.61 to −1.00) | −1.23 (−1.53 to −0.91) | −1.64 (−2.19 to −0.91) |
TNFi | ADA | 0.97 (0.54 to 1.83) | 1.09 (0.76 to 1.49) | 0.59 (0.09 to 4.47) | 0.86 (0.58 to 1.24) | 0.34 (−0.30 to 0.90) | 0.12 (−0.25 to 0.50) | −0.01 (−0.38 to 0.37) | −0.01 (−0.67 to 0.82) |
CZP | 1.14 (0.53 to 2.42) | 1.01 (0.65 to 1.59) | 0.55 (0.06 to 6.11) | 0.81 (0.38 to 1.45) | 0.26 (−0.50 to 0.99) | 0.03 (−0.60 to 0.68) | −0.11 (−0.74 to 0.57) | −0.11 (−0.87 to 0.82) | |
ETN | 0.79 (0.43 to 1.36) | 0.78 (0.57 to 1.07) | 0.60 (0.06 to 7.47) | 0.59 (0.38 to 0.89) | 0.15 (−0.74 to 0.95) | 0.26 (−0.23 to 0.74) | 0.47 (0.05 to 0.96) | 0.82 (−0.64 to 2.86) | |
GOL SC | 1.06 (0.54 to 2.18) | 1.11 (0.73 to 1.61) | 0.65 (0.06 to 7.88) | 0.84 (0.46 to 1.36) | NA | 0.67 (−0.03 to 1.44) | −0.02 (−0.56 to 0.60) | NA | |
GOL IV | 0.50 (0.20 to 1.18) | 0.79 (0.45 to 1.30) | NA | 0.95 (0.52 to 1.64) | 0.72 (−0.12 to 1.56) | NA | 0.63 (0.09 to 1.20) | 0.78 (−0.34 to 2.24) | |
IFX | 0.70 (0.30 to 1.68) | 0.80 (0.50 to 1.19) | NA | NA | NA | NA | NA | NA | |
IL-17i | IXE | 1.02 (0.53 to 2.01) | 0.99 (0.62 to 1.50) | 0.67 (0.07 to 8.00) | 0.88 (0.46 to 1.59) | 0.25 (−0.49 to 0.95) | 0.19 (−0.29 to 0.76) | −0.09 (−0.57 to 0.39) | 0.59 (−0.15 to 1.49) |
SEC (SC load) | 1.26 (0.71 to 2.11) | 1.13 (0.85 to 1.53) | 1.07 (0.12 to 16.34) | 1.53 (0.89 to 2.49) | −0.20 (−0.86 to 0.44) | −0.20 (−0.58 to 0.16) | 0.46 (−0.16 to 1.12) | 0.52 (−0.48 to 1.83) | |
SEC (no load) | 1.25 (0.58 to 2.60) | 1.56 (1.00 to 2.51) | NA | NA | NA | −0.26 (−0.81 to 0.28) | NA | NA | |
JAKi | TOF | 0.86 (0.41 to 1.57) | 1.07 (0.70 to 1.57) | 1.07 (0.14 to 11.52) | 0.99 (0.63 to 1.54) | 0.00 (−0.69 to 0.62) | 0.04 (−0.39 to 0.50) | 0.11 (−0.33 to 0.54) | 0.00 (−0.91 to 1.15) |
UPA | 1.02 (0.45 to 2.27) | 0.81 (0.50 to 1.33) | 0.78 (0.06 to 10.57) | 1.00 (0.58 to 1.72) | 0.08 (−0.76 to 0.82) | NA | 0.09 (−0.50 to 0.69) | 0.19 (−0.79 to 1.14) | |
ADA = adalimumab 40 mg q.2.w. SC; AS = ankylosing spondylitis; ASAS20 = Assessment of SpondyloArthritis International Society-improvement of ≥ 20%; ASAS40 = Assessment of SpondyloArthritis International Society-improvement of ≥ 40%; ASDAS-MI = Ankylosing Spondylitis Disease Activity Score – major improvement; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; bDMARD = biological disease-modifying antirheumatic drug; b.i.d. = twice a day; b.i.w. = twice a week; BKZ = bimekizumab 160 mg q.4.w. SC; Crl = credible interval; CRP = C-reactive protein; CZP = certolizumab pegol 200 mg q.2.w. SC or 400 mg q.4.w. SC; ETN = etanercept 50 mg q.w. SC or 25 mg b.i.w. SC; GOL = golimumab 50 mg q.4.w. SC or golimumab 2 mg/kg q.8.w. IV; I = inhibitor; IFX = infliximab 5 mg IV q.6.w.; IL = interleukin; IXE = ixekizumab 80 mg q.4.w. SC = JAK = Janus kinase; MD = mean difference; NA = not applicable (not included in network); NE = not estimated; NSP = nocturnal spine pain; OR = odds ratio; PBO = placebo; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; q.8.w. = every 8 weeks; q.d. = once daily; SC = subcutaneous; SEC (no load) = secukinumab 150 mg q.4.w. SC (no loading); SEC (SC load) = secukinumab 150 mg q.4.w. SC (with SC loading); TNF = tumour necrosis factor; TOF = tofacitinib 5 mg b.i.d. oral; UPA = upadacitinib 15 mg q.d. oral.
Note: Bold font indicates significant difference, meaning that the Crl does not cross the null.
aHorizontal separation lines indicate placebo and different drug classes.
bPresented models are suggested base case results, on balance based on model fit parameters and 95% CrI.
cSignificant difference based on 95% CrI: for binomial outcomes, OR = 1 is the line of no effect; for continuous outcomes, difference = 0 is the line of no effect.
Source: Clinical evidence summary submitted by the sponsor.19
Table 31: Summary of Outcomes at 12 to 16 Weeks — bDMARD Predominantly Naive Network (2 of 3)
Outcomes | BKZ vs.a | ASAS-5/6 | ASAS-PR | ASDAS < 2.1 | ASDAS-ID | ASDAS-CII | ASQoL | BASMI | Fatigue NRS |
|---|---|---|---|---|---|---|---|---|---|
Binomial: OR (95% CrI)b | Continuous: MD (95% Crl)b | ||||||||
NA | PBO | 6.24 (3.76 to 12.52) | 5.01 (3.60 to 6.97) | 4.32 (1.99 to 6.58) | 4.68 (3.04 to 7.77) | 5.88 (1.30 to 42.53) | −2.08 (−2.78 to −1.09) | −0.29 (−0.40 to −0.15) | −1.27 (−1.61 to −0.83) |
TNFi | ADA | 0.88 (0.50 to 2.75) | 1.13 (0.72 to 1.74) | 1.05 (0.19 to 2.24) | 0.35 (0.21 to 0.62) | 0.79 (0.12 to 8.69) | −0.19 (−1.43 to 1.73) | 0.07 (−0.06 to 0.24) | −0.16 (−0.65 to 0.41) |
CZP | 1.17 (0.44 to 9.60) | 1.03 (0.24 to 2.98) | 0.70 (0.19 to 2.10) | 0.00 (0.00 to 0.68)c | 0.60 (0.02 to 9.76) | 0.45 (−0.94 to 2.61) | 0.02 (−0.26 to 0.29) | −0.32 (−0.95 to 0.53) | |
ETN | 0.99 (0.57 to 3.78) | 0.84 (0.53 to 1.38) | 0.84 (0.09 to 2.38) | 1.42 (0.46 to 5.25) | 0.83 (0.04 to 222.80) | 0.50 (−2.11 to 4.42) | 0.13 (−0.12 to 0.39) | 0.37 (−0.13 to 1.01) | |
GOL SC | 1.03 (0.51 to 4.32) | 0.93 (0.51 to 1.75) | NA | 0.66 (0.26 to 1.40) | NA | NA | −0.05 (−0.19 to 0.11) | NA | |
GOL IV | 0.50 (0.22 to 2.31) | 1.45 (0.72 to 3.06) | NA | 0.71 (0.32 to 1.52) | 0.33 (0.03 to 56.32) | 1.48 (0.12 to 3.18) | −0.01 (−0.18 to 0.20) | NA | |
IFX | NA | NA | NA | NA | NA | NA | NA | NA | |
IL-17i | IXE | NA | 1.66 (0.77 to 3.53) | 0.84 (0.15 to 1.79) | 0.54 (0.19 to 1.41) | 0.74 (0.08 to 11.73) | NA | 0.02 (−0.18 to 0.28) | 0.08 (−0.45 to 0.70) |
SEC (SC load) | 1.43 (0.33 to 2.56) | 1.64 (1.06 to 2.60) | NA | NA | 1.58 (0.01 to 18.70) | −0.09 (−0.98 to 0.84) | NA | −0.27 (−0.91 to 0.44) | |
SEC (no load) | 1.56 (0.22 to 3.22) | NA | NA | NA | NA | 0.08 (−1.14 to 1.43) | NA | NA | |
JAKi | TOF | 1.04 (0.51 to 6.81) | 1.46 (0.84 to 2.53) | 0.76 (0.19 to 1.58) | 1.28 (0.56 to 2.96) | 1.02 (0.12 to 28.20) | 0.08 (−0.95 to 1.38) | 0.15 (0.00 to 0.33) | −0.02 (−0.57 to 0.59) |
UPA | NA | 1.01 (0.23 to 3.65) | 0.55 (0.08 to 1.22) | 0.01 (0.00 to 0.63)c | 1.28 (0.05 to 15.50) | −0.33 (−1.69 to 1.05) | −0.01 (−0.21 to 0.23) | NA | |
ADA = adalimumab 40 mg q.2.w. SC; AS = ankylosing spondylitis; ASAS-PR = partial remission in the Assessment of SpondyloArthritis International Society; ASAS-5/6 = improvement of 20% or more in at least 5 of the 6 domains of the Assessment of SpondyloArthritis International Society; ASDAS-CII = clinically important improvement in Ankylosing Spondylitis Disease Activity Score; ASDAS-ID = Ankylosing Spondylitis Disease Activity Score inactive disease state; ASQoL = Ankylosing Spondylitis Quality of Life; BASMI = Bath Ankylosing Spondylitis Metrology Index; bDMARD = biological disease-modifying antirheumatic drug; b.i.d. = twice a day; b.i.w. = twice a week; BKZ = bimekizumab 160 mg q.4.w. SC; CrI = credible interval; CZP = certolizumab pegol 200 mg q.2.w. SC or 400 mg q.4.w. SC; ETN = etanercept 50 mg q.w. SC or 25 mg b.i.w. SC; GOL = golimumab 50 mg q.4.w. SC or golimumab 2 mg/kg q.8.w. IV; IFX = infliximab 5 mg IV q.6.w.; I = inhibitor; IL = interleukin; IXE = ixekizumab 80 mg q.4.w. SC; JAK = Janus kinase; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; MD = mean difference; NA = not applicable (not included in network); NE = not estimated; NRS = numeric rating scale; NSP = nocturnal spine pain; OR = odds ratio; PBO = placebo; PGADA = Patient’s Global Assessment of Disease Activity; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; q.8.w. = every 8 weeks; q.d. = once daily; SC = subcutaneous; SEC (no load) = secukinumab 150 mg q.4.w. SC (no loading); SEC (SC load) = secukinumab 150 mg q.4.w. SC (with SC loading); SF-36 MCS = Short Form (36) Health Survey mental component summary; SF-36 PCS = Short Form (36) Health Survey physical component summary; TNF = tumour necrosis factor; TOF = tofacitinib 5 mg b.i.d. oral; UPA = upadacitinib 15 mg q.d. oral.
Note: Bold font indicates significant difference, meaning that the credible interval does not cross the null.
aPresented models are suggested base case results, on balance based on model fit parameters and 95% CrI.
bSignificant difference based on 95% CrI; for binomial outcomes, OR = 1 is the line of no effect; for continuous outcomes, difference = 0 is the line of no effect.
cHorizontal separation lines indicate placebo and different drug classes.
Source: Clinical Evidence Summary submitted by the sponsor.19
Table 32: Summary of Outcomes at 12 to 16 Weeks — bDMARD Predominantly Naive Network (3 of 3)a
Outcomes | BKZ vs.b | MASES | PGADA | SF-36 MCS | SF-36 PCS |
|---|---|---|---|---|---|
Continuous: MD (95% CrI)c | |||||
NA | PBO | −0.04 (−1.51 to 2.99) | −1.42 (−1.80 to −1.01) | 0.82 (−0.74 to 2.42) | 4.86 (2.93 to 6.24) |
TNFi | ADA | 0.22 (−1.01 to 2.89) | 0.28 (−0.17 to 0.77) | −0.85 (−3.52 to 2.41) | 1.39 (−0.66 to 2.84) |
CZP | −0.38 (−2.06 to 2.80) | 0.51 (−0.14 to 1.23) | −0.33 (−4.53 to 3.51) | −0.57 (−3.28 to 1.69) | |
ETN | NA | 0.05 (−1.29 to 1.44) | −5.56 (−15.00 to 3.34) | −2.92 (−8.28 to 2.21) | |
GOL SC | 0.30 (−1.47 to 6.06) | NA | −1.96 (−4.91 to 0.65) | 0.08 (−2.51 to 2.07) | |
GOL IV | NA | NA | −4.82 (−8.73 to −1.63) | −0.61 (−3.24 to 1.51) | |
IFX | NA | NA | NA | NA | |
IL-17i | IXE | 0.33 (−1.21 to 3.25) | −0.03 (−0.64 to 0.61) | 0.57 (−3.17 to 4.35) | 2.26 (−0.51 to 4.28) |
SEC (SC load) | 0.20 (−1.69 to 4.35) | NA | −0.24 (−4.62 to 5.21) | 1.67 (0.02 to 3.06) | |
SEC (no load) | NA | NA | NA | 1.42 (−0.74 to 3.42) | |
JAKi | TOF | −0.10 (−1.75 to 1.93) | 0.05 (−0.51 to 0.70) | −0.91 (−3.48 to 2.32) | 1.27 (−1.01 to 3.11) |
UPA | 0.26 (−1.45 to 2.79) | 0.43 (−0.54 to 1.42) | NA | NA | |
ADA = adalimumab 40 mg q.2.w. SC; AS = ankylosing spondylitis; ASAS-PR = partial remission in the Assessment of SpondyloArthritis International Society; ASAS-5/6 = improvement of 20% or more in at least 5 of the 6 domains of the Assessment of SpondyloArthritis International Society; ASDAS-CII = clinically important improvement in Ankylosing Spondylitis Disease Activity Score; ASDAS-ID = Ankylosing Spondylitis Disease Activity Score inactive disease state; ASQoL = Ankylosing Spondylitis Quality of Life; BASMI = Bath Ankylosing Spondylitis Metrology Index; bDMARD = biological disease-modifying antirheumatic drug; b.i.d. = twice a day; b.i.w. = twice a week; BKZ = bimekizumab 160 mg q.4.w. SC; CrI = credible interval; CZP = certolizumab pegol 200 mg q.2.w. SC or 400 mg q.4.w. SC; ETN = etanercept 50 mg q.w. SC or 25 mg b.i.w. SC; GOL = golimumab 50 mg q.4.w. SC or golimumab 2 mg/kg q.8.w. IV; IFX = infliximab 5 mg IV q.6.w.; I = inhibitor; IL = interleukin; IXE = ixekizumab 80 mg q.4.w. SC; JAK = Janus kinase; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; MD = mean difference; NA = not applicable (not included in network); NE = not estimated; NRS = numeric rating scale; NSP = nocturnal spine pain; OR = odds ratio; PBO = placebo; PGADA = Patient’s Global Assessment of Disease Activity; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; q.8.w. = every 8 weeks; q.d. = once daily; SC = subcutaneous; SEC (no load) = secukinumab 150 mg q.4.w. SC (no loading); SEC (SC load) = secukinumab 150 mg q.4.w. SC (with SC loading); SF-36 MCS = Short Form (36) Health Survey mental component summary; SF-36 PCS = Short Form (36) Health Survey physical component summary; TNF = tumour necrosis factor; TOF = tofacitinib 5 mg b.i.d. oral; UPA = upadacitinib 15 mg q.d. oral.
Note: Bold font indicates significant difference, meaning that the credible interval does not cross the null.
aHorizontal separation lines indicate placebo and different drug classes.
bPresented models are suggested base case results, on balance based on model fit parameters and 95% CrI.
cSignificant difference based on 95% CrI; for binomial outcomes, OR = 1 is the line of no effect; for continuous outcomes, difference = 0 is the line of no effect.
Source: Clinical evidence summary submitted by the sponsor.19
Table 33: Summary of Outcomes at 12 to 16 Weeks — bDMARD-Experienced Network (1 of 2)a
Outcomes | BKZ vs.b | ASAS20 | ASAS40 | ASDAS-MI | BASDAI50 | ASDAS-CRP | BASDAI | BASFI | NSP |
|---|---|---|---|---|---|---|---|---|---|
Binomial: OR (95% CrI)c | Continuous: MD (95% Crl)c | ||||||||
NA | PBO | 2.91 (0.93 to 8.51) | 3.46 (1.67 to 6.88) | 2.17 (0.45 to 15.69) | 1.00 (0.32 to 3.46) | −0.77 (−1.26 to −0.25) | −0.48 (−3.59 to 2.55) | −1.25 (−2.25 to −0.24) | −0.68 (−2.39 to 1.08) |
TNFi | ADA | NA | NA | NA | NA | NA | NA | NA | NA |
CZP | 1.30 (0.11 to 6.50) | 0.70 (0.17 to 2.35) | 0.28 (0.01 to 4.01) | 0.13 (0.01 to 1.13) | 0.41 (−0.36 to 1.16) | 1.60 (−4.49 to 6.98) | 0.06 (−1.76 to 1.90) | 1.40 (−0.94 to 3.66) | |
ETN | NA | NA | NA | NA | NA | NA | NA | NA | |
GOL SC | NA | NA | NA | NA | NA | NA | NA | NA | |
GOL IV | NA | NA | NA | NA | NA | NA | NA | NA | |
IFX | NA | NA | NA | NA | NA | NA | NA | NA | |
IL-17i | IXE | 1.50 (0.35 to 4.75) | 1.94 (0.83 to 4.43) | 0.43 (0.06 to 3.84) | 0.38 (0.09 to 1.68) | 0.23 (−0.32 to 0.81) | 0.84 (−3.85 to 4.47) | −0.26 (−1.36 to 0.91) | 0.92 (−0.90 to 2.79) |
SEC (SC load) | 1.13 (0.36 to 3.93) | 0.86 (0.39 to 1.87) | NA | NA | NA | 0.53 (−2.77 to 3.22) | NA | 1.21 (−1.09 to 3.60) | |
SEC (no load) | 1.05 (0.28 to 4.51) | 1.87 (0.65 to 5.30) | NA | NA | NA | 0.67 (−2.66 to 3.58) | NA | NA | |
JAKi | TOF | 1.85 (0.07 to 11.39) | 1.71 (0.50 to 5.21) | NA | NA | 0.06 (−0.70 to 0.78) | NA | −0.53 (−1.76 to 0.69) | NA |
UPA | 0.82 (0.28 to 2.95) | 0.85 (0.40 to 1.78) | NA | 0.27 (0.08 to 1.01) | 0.25 (−0.29 to 0.83) | 0.66 (−2.57 to 3.59) | −0.12 (−1.24 to 1.10) | 0.96 (−0.83 to 2.93) | |
ADA = adalimumab 40 mg q.2.w. SC; AS = ankylosing spondylitis; ASAS-PR = partial remission in the Assessment of SpondyloArthritis International Society; ASAS-5/6 = improvement of 20% or more in at least 5 of the 6 domains of the Assessment of SpondyloArthritis International Society; ASDAS-CII = clinically important improvement in Ankylosing Spondylitis Disease Activity Score; ASDAS-ID = Ankylosing Spondylitis Disease Activity Score inactive disease state; ASQoL = Ankylosing Spondylitis Quality of Life; BASMI = Bath Ankylosing Spondylitis Metrology Index; bDMARD = biological disease-modifying antirheumatic drug; b.i.d. = twice a day; b.i.w. = twice a week; BKZ = bimekizumab 160 mg q.4.w. SC; CrI = credible interval; CZP = certolizumab pegol 200 mg q.2.w. SC or 400 mg q.4.w. SC; ETN = etanercept 50 mg q.w. SC or 25 mg b.i.w. SC; GOL = golimumab 50 mg q.4.w. SC or golimumab 2 mg/kg q.8.w. IV; IFX = infliximab 5 mg IV q.6.w.; I = inhibitor; IL = interleukin; IXE = ixekizumab 80 mg q.4.w. SC; JAK = Janus kinase; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; MD = mean difference; NA = not applicable (not included in network); NE = not estimated; NRS = numeric rating scale; NSP = nocturnal spine pain; OR = odds ratio; PBO = placebo; PGADA = Patient’s Global Assessment of Disease Activity; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; q.8.w. = every 8 weeks; q.d. = once daily; SC = subcutaneous; SEC (no load) = secukinumab 150 mg q.4.w. SC (no loading); SEC (SC load) = secukinumab 150 mg q.4.w. SC (with SC loading); SF-36 MCS = Short Form (36) Health Survey mental component summary; SF-36 PCS = Short Form (36) Health Survey physical component summary; TNF = tumour necrosis factor; TOF = tofacitinib 5 mg b.i.d. oral; UPA = upadacitinib 15 mg q.d. oral.
Note: Bold font indicates significant difference, meaning that the credible interval does not cross the null.
aHorizontal separation lines indicate placebo and different drug classes.
bPresented models are suggested base case results, on balance based on model fit parameters and 95% CrI.
cSignificant difference based on 95% CrI; for binomial outcomes, OR = 1 is the line of no effect; for continuous outcomes, difference = 0 is the line of no effect.
Source: Clinical evidence summary submitted by the sponsor.19
Table 34: Summary of Outcomes at 12 to 16 Weeks — bDMARD-Experienced Network (2 of 2)a
Outcomes | BKZ vs.b | ASQoL | BASMI | Fatigue NRS | MASES | PGADA | SF−36 MCS | SF−36 PCS |
|---|---|---|---|---|---|---|---|---|
Continuous: MD (95% CrI)c | ||||||||
NA | PBO | −1.23 (−3.34 to 0.86) | −0.22 (−0.74 to 0.31) | −0.67 (−2.07 to 0.79) | −0.38 (−1.99 to 1.13) | −1.20 (−2.44 to 0.07) | NA | 2.79 (−2.78 to 8.38) |
TNFi | ADA | NA | NA | NA | NA | NA | NA | NA |
CZP | 4.24 (0.38 to 8.15) | 0.50 (−0.39 to 1.37) | 1.43 (−0.54 to 3.42) | −0.28 (−3.10 to 2.44) | 0.83 (−1.15 to 2.70) | NA | −6.89 (−12.89 to −0.36) | |
ETN | NA | NA | NA | NA | NA | NA | NA | |
GOL SC | NA | NA | NA | NA | NA | NA | NA | |
GOL IV | NA | NA | NA | NA | NA | NA | NA | |
IFX | NA | NA | NA | NA | NA | NA | NA | |
IL−17i | IXE | NA | 0.08 (−0.48 to 0.68) | 0.63 (−0.89 to 2.22) | NA | 0.49 (−0.84 to 1.94) | NA | −2.03 (−8.21 to 3.93) |
SEC (SC load) | 0.26 (−2.21 to 2.75) | NA | −0.05 (−2.08, 2.01) | NA | NA | NA | −0.56 (−6.53 to 5.57) | |
SEC (no load) | 0.29 (−2.54 to 3.26) | NA | NA | NA | NA | NA | −0.85 (−7.27 to 5.59) | |
JAKi | TOF | −0.33 (−3.11 to 2.36) | 0.32 (−0.24 to 0.90) | NA | NA | 0.42 (−1.13 to 1.94) | NA | 0.93 (−5.35 to 7.09) |
UPA | 1.81 (−0.52 to 4.24) | 0.08 (−0.50 to 0.71) | 0.41 (−1.07 to 2.04) | 1.10 (−1.04 to 3.08) | 0.35 (−1.11 to 1.91) | NA | NA | |
ADA = adalimumab 40 mg q.2.w. SC; AS = ankylosing spondylitis; ASAS-PR = partial remission in the Assessment of SpondyloArthritis International Society; ASAS-5/6 = improvement of 20% or more in at least 5 of the 6 domains of the Assessment of SpondyloArthritis International Society; ASDAS-CII = clinically important improvement in Ankylosing Spondylitis Disease Activity Score; ASDAS-ID = Ankylosing Spondylitis Disease Activity Score inactive disease state; ASQoL = Ankylosing Spondylitis Quality of Life; BASMI = Bath Ankylosing Spondylitis Metrology Index; bDMARD = biological disease-modifying antirheumatic drug; b.i.d. = twice a day; b.i.w. = twice a week; BKZ = bimekizumab 160 mg q.4.w. SC; CrI = credible interval; CZP = certolizumab pegol 200 mg q.2.w. SC or 400 mg q.4.w. SC; ETN = etanercept 50 mg q.w. SC or 25 mg b.i.w. SC; GOL = golimumab 50 mg q.4.w. SC or golimumab 2 mg/kg q.8.w. IV; IFX = infliximab 5 mg IV q.6.w.; I = inhibitor; IL = interleukin; IXE = ixekizumab 80 mg q.4.w. SC; JAK = Janus kinase; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; MD = mean difference; NA = not applicable (not included in network); NE = not estimated; NRS = numeric rating scale; NSP = nocturnal spine pain; OR = odds ratio; PBO = placebo; PGADA = Patient’s Global Assessment of Disease Activity; q.4.w. = every 4 weeks; q.6.w. = every 6 weeks; q.8.w. = every 8 weeks; q.d. = once daily; SC = subcutaneous; SEC (no load) = secukinumab 150 mg q.4.w. SC (no loading); SEC (SC load) = secukinumab 150 mg q.4.w. SC (with SC loading); SF-36 MCS = Short Form (36) Health Survey mental component summary; SF-36 PCS = Short Form (36) Health Survey physical component summary; TNF = tumour necrosis factor; TOF = tofacitinib 5 mg b.i.d. oral; UPA = upadacitinib 15 mg q.d. oral.
Note: Bold font indicates significant difference, meaning that the Crl does not cross the null.
aHorizontal separation lines indicate placebo and different drug classes.
bPresented models are suggested base case results, on balance based on model fit parameters and 95% CrI.
cSignificant difference based on 95% CrI; for binomial outcomes, OR = 1 is the line of no effect; for continuous outcomes, difference = 0 is the line of no effect.
Source: Clinical evidence summary submitted by the sponsor.19
Efficacy
Matching-Adjusted Indirect Comparison
MAIC analyses suggested that bimekizumab 160 mg every 4 weeks had statistically significantly better results at week 52 compared with ixekizumab 80 mg every 4 weeks for ASAS20, ASAS40, and BASDAI change from baseline, and BASDAI50. Results also favoured bimekizumab over secukinumab 150 mg every 4 weeks for ASAS40 and BASDAI change from baseline. A summary of the MAIC results is provided in Table 35.
Table 35: Summary of MAIC Results of Bimekizumab in Patients With AS at Week 52
BKZ BE MOBILE 2 trial vs. comparator | ASAS20a OR (95% CI) | ASAS40a OR (95% CI) | ASAS-PR OR (95% CI) | BASDAI-CfB MD (95% CI) | BASDAI50 OR (95% CI) | ASDAS OR < 2.1 (95% CI) |
|---|---|---|---|---|---|---|
SEC 150 mg q.4.w. | 1.44 (0.99 to 2.09) | 1.49 (1.05 to 2.11) | 1.40 (0.69 to 2.85) | −0.43 (−0.83 to −0.03) | NA | NA |
SEC 300 mg q.4.w. | 1.03 (0.52 to 2.01) | 1.08 (0.59 to1.97) | 1.07 (0.55 to 2.09) | NA | NA | NA |
IXE 80 mg q.4.w. | 2.14 (1.11 to 4.12) | 2.03 (1.05 to 3.91) | 0.74 (0.36 to 1.53) | −0.74 (−1.46 to −0.03) | 2.13 (1.10 to 4.12) | 1.49 (0.73 to 3.01) |
AS = ankylosing spondylitis; ASAS = Assessment of SpondyloArthritis International Society; ASAS20 = improvement of 20% or more in the Assessment of SpondyloArthritis International Society; ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASAS-PR = partial remission in the Assessment of SpondyloArthritis International Society; BASDAI50 = a reduction (improvement) of at least 50% in the Bath Ankylosing Spondylitis Disease Activity Index score; BASDAI-CfB = change from baseline in the Bath Ankylosing Spondylitis Disease Activity Index; BKZ = bimekizumab; CI = confidence interval; IXE = ixekizumab; NA = not applicable; OR = odds ratio; q.4.w. = every 4 weeks; SEC = secukinumab.
Note: Bold font indicates statistically significant difference, favouring bimekizumab.
aPlus other ASAS improvement criteria.
Source: Clinical evidence summary submitted by the sponsor.19
Critical Appraisal
Network Meta-Analysis
The sponsor conducted an NMA using a Bayesian approach. This was a reasonable method to apply, given the common comparator of placebo. The NMA was informed by an SLR of relevant databases. Study selection was based on appropriate criteria and 2 reviewers completed the study selection and data extraction. A quality assessment was performed by 2 reviewers for each included study. The sponsor’s assessment of study quality was that the studies were of reasonable quality and no study was excluded based on this quality assessment, despite some trials having used unclear methods of randomization and allocation concealment. It was not clear how the results of the quality assessment were incorporated into the analyses.
The sponsor’s decision to perform 3 separate NMA analyses based on the potential effect modifier of prior exposure to bDMARDs was appropriate. Some networks had a large number of trials and a large number of patients, which was considered a strength of the NMA analyses. The sponsor did not perform sensitivity analyses in the NMA and did not attempt to identify and adjust for effect modifiers, despite the availability of a large number of trials for some of the networks. The outcomes selected by the sponsor were relevant, and a smaller number of these outcomes were selected for the GRADE assessment in this report (ASAS40, BASDAI, BASFI, NSP, enthesitis state, ASQoL, WPAI-SHP, and SAEs). The time point of 12 to 16 weeks that was selected for the outcome analyses was reasonable and clinically relevant for efficacy but not as meaningful for harms since an assessment of long-term harms was lacking. There were closed loops in the networks but there was no assessment for consistency reported.
CIs and CrIs were wide for many estimates in the NMA. For most outcomes, the fixed-effects placebo-adjusted models were used and this may have underestimated uncertainty in the treatment effects. For some of the binomial outcomes, there were zero or a low number of events in the placebo arm and too few studies reporting the outcome of interest. Despite the large number of trials, the number of patients and events in some analyses were small, precluding the possibility of detecting a difference between treatments. For example, the incidence of harms outcomes was small, resulting in very wide CrIs around the estimates. For this reason, the results of the harms analyses were not informative and did not serve to illuminate the risk of harms for bimekizumab relative to other treatments.
The predominantly bDMARD-naive network provided the most complete set of results across outcomes and comparators, whereas, for the purely bDMARD-naive network, subgroup data were not reported for some outcomes and comparators. In the predominantly bDMARD-naive network, the proportion of bDMARD-naive patients included in each study ranged from 61% to 100%. Across the 26 studies included in the predominantly bDMARD-naive network, the proportion of bDMARD-naive patients was more than 90%. The number of bDMARD-experienced patients in the BE MOBILE 2 trial may have been too low to detect a difference between bimekizumab and placebo in this network (37 patients in the bimekizumab group and 17 patients in the placebo group were bDMARD-experienced). The sponsor offered this as an explanation for the lack of any difference observed between bimekizumab and placebo in this network.102 Nine studies reported data for this subgroup, leading to a network of 8 treatments (including placebo), whereas 26 studies reported data for the bDMARD predominantly naive network comprising 13 treatments.
Matching-Adjusted Indirect Comparison
The sponsor performed an unanchored MAIC because of the lack of a placebo arm beyond week 16 for bimekizumab and comparators. This was an adequate justification for performing an MAIC. The selection of comparators from the same pharmacologic group (IL-17A inhibitors) was a rational approach, but comparisons with other biologics would also have been of interest. The MAIC allowed a comparison of 52-week clinical data. The MAIC analyses suggested there were some differences favouring bimekizumab compared with secukinumab and ixekizumab for ASAS20, ASAS40, BASDAI change from baseline, and BASDAI50, but several limitations of the MAIC prevent drawing strong conclusions regarding the comparative effectiveness of bimekizumab.
There may have been important differences between the studies included in the MAIC that were not accounted for. The sponsor did not report how these differences were handled. Study design differences cannot be adjusted for in the MAIC weighting procedures. The sponsor did not incorporate the results of the quality assessment of the studies into the analysis and this could be a factor that would limit the comparability of the clinical data. In the sponsor’s NMA, the networks were separated into 3 categories based on previous exposure to bDMARDs. In the MAIC, there was an attempt to match for prior TNFi exposure, but no explanation of how differences in prior exposure to other bDMARDs was controlled for in the MAIC.
In the MAIC, there were notable differences in study populations before and after adjustment. In the MAIC analyses, the ESS for the bimekizumab group was reduced to 80% for the comparison with secukinumab 150 mg, to 51% for the comparison with secukinumab 300 mg, and to 20% for the comparison with ixekizumab.
A list of prognostic factors and effect modifiers were selected through a review of published data, reviewing another previously published MAIC in AS, and soliciting expert input. The 12 matching variables were age, BASFI at baseline, ASDAS at baseline, percent male, percent with prior exposure to TNF inhibitors, BMI or weight, time from diagnosis, time from symptom onset, BASDAI at baseline, PGADA at baseline, percent white, and percent with sulfasalazine use; however, not all matching variables were used for each comparison. The number of variables used for a given comparison was from 6 to 8 variables. This means that up to 6 of the prognostic factors that were deemed important were not used in the weighting adjustments of the MAIC analyses. Variables were excluded for several reasons. Some were excluded because there were no aggregate data available from the published studies. ASDAS was excluded as a matching factor because of its significant reduction impact on the ESS. If ASDAS caused a significant reduction in the ESS, this would suggest there would be a strong possibility of residual bias in the MAIC comparisons in which ASDAS was excluded. This would also be expected in the cases where other prognostic factors and effect modifiers were excluded in the MAIC analyses, limiting the comparability of the populations. Regarding the MAIC analyses, the sponsor noted that “the amount of bias in the indirect comparisons is likely to be substantial”19 and CADTH reviewers agree with this assessment.
Summary
The results of the sponsor’s NMA did not show consistent differences between bimekizumab and comparators in the networks for efficacy or harms outcomes. While differences were reported in a small number of comparisons in some populations, these were associated with wide 95% CrIs for many of the comparisons, indicating imprecision of the results.
The results of the sponsor’s MAIC favoured bimekizumab for some outcomes but there were significant limitations. These limitations include differences in the study design and patient populations, and heterogeneity in baseline characteristics across studies. These factors, in addition to the substantial reduction in ESSs, undermine any claims of bimekizumab having superior performance over the comparators in the MAIC.
Neither the NMA nor the MAIC provided clear evidence of a difference in efficacy or harms outcomes for bimekizumab versus comparators.
Studies Addressing Gaps in the Systematic Review Evidence
The BE MOBILE 1 trial was a phase III, multicentre, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of bimekizumab in patients with active nr-axSpA. The sponsor identified BE MOBILE 1 as a study that addresses the gap in the evidence on the efficacy and safety of bimekizumab. However, the CADTH review team considered the BE MOBILE 1 trial to be not relevant to this review, as patients with active nr-axSpA are different from patients with active AS, which is the population for the indication being reviewed. Therefore, the CADTH review team found that no studies addressing gaps in the systematic review evidence had been identified for this review.
Discussion
Summary of Available Evidence
One pivotal trial (BE MOBILE 2) was included in the sponsor’s systematic review. The BE MOBILE 2 trial was a phase III, multicentre, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of bimekizumab in patients with active AS compared with placebo. This study enrolled adults who had active AS (i.e., r-axSpA) and fulfilled the mNY criteria. Eligible study participants (N = 332) were randomized 2:1 to receive bimekizumab (n = 221) 160 mg/mL or placebo (n = 111) subcutaneously every 4 weeks. The mean age of all study participants was 40.4 years with a range of 19 to 80 years. The majority of study participants were male (72.3%) and white (80.4%), 27.7% of participants were female, 0.3% of participants were Black, 0.9% of participants were from another or a mixed racial group, and the racial group information was missing for 1.2% of participants. Study participants were most commonly enrolled in the following regions: Eastern Europe (49.1%), Western Europe (29.8%), Asia (18.4%), and North America (2.7%). The treatment groups were generally well balanced with respect to AS-related and other baseline disease characteristics. At baseline, the majority of all study participants were using NSAID therapies (79.8%) and anti-TNF therapy had previously been used by 16.3% of study participants. The primary objective of the BE MOBILE 2 trial was to demonstrate the efficacy of bimekizumab administered subcutaneously every 4 weeks compared with placebo in the treatment of patients with active AS. The primary end point of the study was ASAS40, and secondary end points included BASDAI, BASFI, NSP, MASES, and HRQoL using ASQoL and WPAI-SHP.
BE AGILE 2 is a single-arm phase II open-label extension study that followed the preceding study, BE AGILE, a dose-ranging, randomized, placebo-controlled phase IIb trial. Of the 303 adult patients living with active AS who had been enrolled in the BE AGILE trial, 255 (84.5%) entered the BE AGILE 2 study, which took place at 50 sites in 10 European countries and the US. The data presented in this report combined 48 weeks of treatment in the BE AGILE trial and an additional 204 weeks of treatment in the BE AGILE 2 trial. Most of the key outcomes measured in the BE MOBILE 2 study were also evaluated in the BE AGILE 2 trial, such as ASAS40, BASDAI, BASFI, NSP, ASQoL, Bath Ankylosing Spondylitis Metrology Index, MASES, and safety outcomes.
The sponsor conducted NMAs to determine the clinical efficacy and safety of bimekizumab compared with IL-17 inhibitors (i.e., ixekizumab, secukinumab, and brodalumab), TNF alpha inhibitors (i.e., adalimumab, certolizumab pegol, etanercept, golimumab, infliximab, and biosimilars), JAK inhibitors (i.e., tofacitinib, upadacitinib, and filgotinib), conventional DMARDs (i.e., cyclosporine, sulfasalazine, methotrexate, leflunomide, and hydroxychloroquine), and NSAIDs (i.e., celecoxib, etoricoxib, ibuprofen, naproxen, ketoprofen, flurbiprofen, indomethacin [indometacin], etodolac, diclofenac, aceclofenac, sulindac, piroxicam, meloxicam, and tenoxicam) at weeks 12 to16, for the treatment of patients with AS. The NMAs were conducted on 3 different networks: purely naive (100% bDMARD-naive), predominantly naive (more than 90% bDMARD-naive), and purely experienced (100% bDMARD-experienced). The MAIC were performed to establish long-term relative clinical efficacy of bimekizumab, where data availability allowed, compared with other IL-17A inhibitors (i.e., ixekizumab, secukinumab, and brodalumab) in patients with AS at week 52.
Interpretation of Results
Efficacy
Bimekizumab 160 mg/mL administered subcutaneously every 4 weeks is indicated for the treatment of patients with active AS. The pivotal BE MOBILE 2 trial assessed the comparative efficacy and safety of the bimekizumab versus placebo. Region and prior TNF were used as stratification factors in the randomization of treatment.
As the aim of treatment for patients with AS is to control pain and inflammation and prevent radiographic damage and disability, symptom and disease control (i.e., reducing inflammation, stiffness, pain, and fatigue), and HRQoL were highlighted by patients and the clinical expert consulted by CADTH to be important outcomes. As such, the symptom and disease outcomes of the ASAS40, BASDAI, BASFI, NSP, and MASES, as well as the HRQoL outcomes of the ASQoL and WPAI-SHP, which were captured in the BE MOBILE 2 trial, are relevant to assessing the clinical benefit of bimekizumab treatment.
The BE MOBILE 2 trial included patients with moderate to severe active AS (defined as a BASDAI score of ≥ 4 and spinal pain of ≥ 4 on a 0 to 10 NRS [from BASDAI item 2]). The clinical expert confirmed that the threshold used in the trial is aligned with clinical practice for patient with moderate to severe AS, who are eligible for escalating therapy. Patients with a BASDAI score of less than 4 would be considered to have relatively manageable disease that could be treated with physiotherapy and anti-inflammatory therapies.
Patients with nonactive IBD were allowed to enrol in the BE MOBILE 2 trial. The clinical expert indicated that the use of IL-17 inhibitors such as secukinumab and ixekizumab would increase the risk of IBD flares; as a result, many clinicians would consider patients with a personal or family history of IBD not ideal for treatment with IL-17 inhibitors due to the increased risk of IBD flares. The clinical expert speculated that patients receiving bimekizumab may not have an elevated risk of IBD, as bimekizumab blocks both IL-17A and IL-17F cytokines and thus may result in more significant IL-17 blockage compared with other IL-17 inhibitors; however, long-term extension studies may be required to determine whether the risk of IBD would be different between bimekizumab and other IL-17 inhibitors. Patients with previous experience with more than 1 TNF alpha inhibitor and/or more than 2 additional non–TNF alpha biological-response modifiers, or any IL-17 biological-response modifier at any time were excluded from the BE MOBILE 2 trial. The clinical expert indicated that those patients should be considered eligible for bimekizumab, albeit with lower response rates.
The generalizability of the study results is uncertain considering that the treatments included in the comparator group do not align with clinical practice in Canada. Of note, commonly prescribed treatments and medications for AS, such as an anti-TNF biosimilar monoclonal antibody, identified by the clinical expert for this review, were not included in the comparator group. Moreover, patients in the BE MOBILE 2 trial were mainly recruited from Europe; there were no study sites in Canada, which may compromise the generalizability of the study results in clinical practice in Canada.
Based on results from the BE MOBILE 2 trial, there is moderate certainty that bimekizumab likely results in a clinically important increase, based on a 15% threshold, in the adjusted ASAS40 response rate at week 16 when compared with placebo, with a between-group difference of 21.8% (95% CI, 11.4% to 32.1%). There was a notable response observed in the placebo group, with nearly 20% achieving ASAS40 at week 16. Therefore, even though the risk difference and associated upper 95% CI exceeded the threshold of 15% suggested by the clinical expert, there is the possibility that the observed 41.5% of bimekizumab-treated patients who achieved ASAS40 at week 16 may overestimate what might be observed in practice settings.
Generally, subgroup and sensitivity analyses were consistent with the primary analysis of ASAS40, except for subgroups of patients with a BMI of at least 30 kg/m2 and patients with a BMI of less than 18.5 kg/m2. The small number of patients in the subgroups may have resulted in the inconsistent results observed in some subgroups. Patients in the bimekizumab group who were TNF alpha inhibitor–naive reported a higher adjusted ASAS40 response rate compared with those in the placebo group (44.4% for bimekizumab versus 22.2% for placebo). Similar results were reported in the TNF alpha inhibitor–experienced group (40.5% versus 17.6%); however, the sample size in this subgroup was much smaller. The clinical expert consulted by CADTH commented that the subgroup with prior exposure (yes versus no) to TNF alpha inhibitors was clinically relevant, as patients who have experienced treatment failure with TNF alpha inhibitors may have disease that is more treatment resistant compared with the patients who were TNF alpha inhibitor–naive. Also, the treatment effect and response rate would be lower in patients who were TNF alpha inhibitor–experienced. The CADTH review team noted that the BE MOBILE 2 trial was not specifically powered to test statistical inferences between bimekizumab and placebo in these subgroups, except for the subgroup of patients who were TNF inhibitor–naive. In addition, although the subgroup analyses were prespecified, patient randomization was not stratified according to all subgroup factors, except for region and prior exposure (yes versus no) to TNF alpha inhibitors; it was unclear whether the balance of known and unknown factors between treatment groups that was achieved by randomization was preserved in the subgroups. As a result, chance cannot be ruled out in subgroup effects between treatment groups, given that no formal statistical tests for interaction between the TNF subgroups were done. Moreover, the subgroup analyses were not controlled for multiple comparison except for the TNF alpha inhibitor–naive subgroup. Generally, the treatment effects of bimekizumab on the ASAS40 were observed up to 52 weeks in the BE MOBILE 2 trial maintenance period and up to 208 weeks in the BE AGILE 2 trial.
The results of the analysis of BASDAI total score indicated that patients treated with bimekizumab reported a greater improvement in disease control (change of −2.9) at week 16 than patients who received placebo (change of −1.9), with a risk difference of −1.0 (95% CI, −1.5 to −0.6). An estimated median MID for the change from baseline of 1.4 points (range, 0.9 to 1.8) was identified in the literature,14 but a between-group MID was not identified. As is typical of assessments for reimbursement, the within-group MID may be a proxy in the absence of an established between-group estimate to determine the magnitude of the treatment effect of the intervention. The clinical expert consulted by CADTH indicated they would consider a 1-point difference between groups as clinically meaningful. Given the range of potential thresholds, the CADTH reviewers used the lower end of the range in line with the clinical expert’s opinion (i.e., a 1-point difference between groups). Therefore, bimekizumab likely results in a clinically important difference in the change from baseline in BASDAI total score at week 16 when compared with placebo. However, as noted in the GRADE assessment, there is uncertainty in the benefit because the upper 95% CI exceeded the 1-point threshold, indicating that some patients will experience a change in the BASDAI that may not be clinically important. Moreover, given there is a range of potential thresholds for clinical significance in the BASDAI, using the median or the higher end of the MID range estimate would mean that the observed difference from the BE MOBILE 2 trial may not be clinically important. Nonetheless, as a secondary outcome, the results for the BASDAI may be considered as supportive of the primary ASAS40 results. Additionally, the clinical expert commented that they prefer a state (status) measure (e.g., BASDAI) rather than a response measure (e.g., ASAS40) in clinical practice. This is because the aim of treatment is to get patients into a state of low disease activity and the most practical way of measuring it would be achieving a BASDAI score of less than 4. The clinical expert stated that ASAS40 is a commonly used measure in clinical trials but not in clinical practice, as it does not directly reflect change from baseline in terms of disease activity in its 4 components.
The sponsor performed an NMA to understand the efficacy and harms of bimekizumab relative to TNF, IL-17, and JAK inhibitors in patients with AS after 12 to 16 weeks of treatment. The sponsor also performed an unanchored MAIC analysis to examine the efficacy of bimekizumab compared with IL-17A inhibitors after 52 weeks of treatment. The results of the sponsor’s NMA did not show consistent differences between bimekizumab and comparators in the networks for efficacy outcomes. While differences were reported in a small number of comparisons in some populations, these were associated with wide 95% CrIs, indicating imprecision of the results. The results of the sponsor’s MAIC favoured bimekizumab for some efficacy outcomes but there were significant limitations. The limitations included differences in study design, patient populations, and heterogeneity in baseline characteristics across studies. These factors, in addition to the substantial reduction in ESSs, undermine any claims of superior performance of bimekizumab over comparators in the MAIC. The results of the sponsor’s NMA did not show consistent differences between bimekizumab and comparators in the networks for harms outcomes. The time point of 12 to 16 weeks that was used for the harms outcome analyses was not long enough to assess long-term harms.
With regard to other end points, generally, the presented totality of efficacy results suggested that, compared with placebo, bimekizumab results in a clinically important reduction in NSP score (based on an NRS), and likely results in a clinically important reduction in the BASFI and in the adjusted enthesitis-free rate based on the MASES index. Patients and clinicians consider HRQoL and return to usual daily activities such as working to be relevant outcomes and expect the treatment of interest would be effective on improving these. Generally, compared with placebo, bimekizumab likely results in a clinically important improvement in HRQoL as measured with the reduction in ASQoL total score from baseline. With regard to working as measured with the WPAI-SHP, bimekizumab may result in a reduction in percent time missed due to (disease-related) problems, and does result in a reduction in percent impairment while working due to problems, percent overall work impairment due to problems, and percent activity impairment due to problems. There were several limitations regarding the assessment of HRQoL in the BE MOBILE 2 trial. For example, a higher proportion of patients received concomitant medications for the management of AS and pain in the bimekizumab group than in the placebo group. No multiplicity adjustments were performed in the analysis of the WPAI-SHP. Moreover, there are no published MID estimates for the WPAI-SHP in patients with AS, and the clinical expert consulted was unsure of what a clinically important difference between groups would be. Thus, there is uncertainty as to whether the results observed for the WPAI-SHP in the BE MOBILE 2 trial were meaningful.
Harms
Slightly more patients in the bimekizumab group experienced at least 1 AE or SAE compared with the placebo group at week 16. Given that bimekizumab is an immunomodulatory therapy, the clinical expert would expect an increased risk of AEs. According to the clinical expert, AEs that appear to occur more often in the bimekizumab group, i.e., infections and nasopharyngitis, are expected in patients with IL-17 inhibitors. More patients in the bimekizumab group reported gastrointestinal disorders and nervous system disorders compared with patients in the placebo group. The clinical expert commented that the elevated risk of gastrointestinal disorders highlighted that IL-17 inhibitors would increase the risk of AEs in the gastrointestinal system and that the elevated risk of nervous system disorders was of concern and needed to be monitored along with well-known potential side effects, including mucocutaneous candidiasis and IBD. With respect to notable harms, hypersensitivity reactions, fungal infections, liver injuries or disorders, IBD, and neutropenia were reported more frequently in the bimekizumab group. The clinical expert commented that neutropenia was expected with IL-17 inhibitors and anti-TNF therapies, and IBD and fungal infections were clearly related to the drug. The clinical expert highlighted that liver issues are not commonly observed in patients with IL-17 inhibitors. They also noted that concomitant NSAIDs can cause liver injury, especially when there are underlying liver problems. Given that slightly more patients used concomitant NSAIDs in the bimekizumab group, overall, the imbalance in liver injuries or disorders was not significant. No new safety signals were identified in the long term BE AGILE and BE AGILE 2 trials in patients with AS.
Conclusion
Both patients and clinicians highlighted the need for new effective treatments for active AS that control disease and symptoms and improve QoL compared with current treatments.
One phase III, multicentre, randomized, double-blind, placebo-controlled trial (BE MOBILE 2) comparing bimekizumab with placebo in treating patients with moderate to severe AS demonstrated that bimekizumab increased the adjusted ASAS40 response rate at week 16. Likewise, the results of the analysis of BASDAI total score indicated that patients treated with bimekizumab had a greater improvement in disease control than patients who received placebo at week 16. A GRADE assessment of the sponsor-submitted systematic review, which included only the BE MOBILE 2 trial, suggested that bimekizumab likely results in a clinically important improvement in ASAS40 response rate and in the BASDAI total score compared with placebo.
Compared with placebo, bimekizumab results in a clinically important reduction in NSP score (based on an NRS), likely results in a clinically important reduction in the BASFI, the adjusted enthesitis-free rate based on the MASES index, and the ASQoL total score. With regard to WPAI-SHP, bimekizumab may result in a reduction in percent time missed due to problems (related to disease) and does result in a reduction in percent impairment while working due to problems, percent overall work impairment due to problems, and percent activity impairment due to problems. The CADTH review team noted that the clinical importance of the reduction in WPAI-SHP score is unclear because a clinically meaningful threshold could not be determined.
Compared with placebo, there is low-certainty evidence that bimekizumab may have resulted in an increase in the percentage of patients who experienced SAEs at week 16. No new safety signals were identified in the long term BE AGILE and BE AGILE 2 trials in patients with AS.
The sponsor-submitted indirect treatment comparison analyses did not provide clear evidence of a difference in efficacy or harms outcomes for bimekizumab relative to other treatments.
References
1.de Koning A, Schoones JW, van der Heijde D, van Gaalen FA. Pathophysiology of axial spondyloarthritis: consensus and controversies. Eur J Clin Invest. 2018;48(5). PubMed
2.Navarro-Compan V, Sepriano A, El-Zorkany B, van der Heijde D. Axial spondyloarthritis. Ann Rheum Dis. 2021;80(12):1511-1521. PubMed
3.Sieper J, Braun J, Dougados M, Baeten D. Axial spondyloarthritis. Nat Rev Dis Primers. 2015;1:15013. PubMed
4.Ward MM, Deodhar A, Gensler LS, et al. 2019 update of the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol. 2019;71(10):1599-1613. PubMed
5.Protopopov M, Poddubnyy D. Radiographic progression in non-radiographic axial spondyloarthritis. Expert Rev Clin Immunol. 2018;14(6):525-533. PubMed
6.Michelena X, Lopez-Medina C, Marzo-Ortega H. Non-radiographic versus radiographic axSpA: what's in a name? Rheumatology (Oxford). 2020;59(Suppl4):iv18-iv24. PubMed
7.de Winter JJ, van Mens LJ, van der Heijde D, Landewe R, Baeten DL. Prevalence of peripheral and extra-articular disease in ankylosing spondylitis versus non-radiographic axial spondyloarthritis: a meta-analysis. Arthritis Res Ther. 2016;18(1):196. PubMed
8.Arthritis Society of Canada. Ankylosing spondylitis. 2019; https://arthritis.ca/about-arthritis/arthritis-types-(a-z)/types/ankylosing-spondylitis. Accessed 2023 Jun 07.
9.Inc EC. Clinician Interview Report for Ankylosing Spondylitis. 2023.
10.Ramiro S, Nikiphorou E, Sepriano A, et al. ASAS-EULAR recommendations for the management of axial spondyloarthritis: 2022 update. Ann Rheum Dis. 2023;82(1):19-34. PubMed
11.Deodhar A, Sandoval D, Holdsworth E, Booth N, Hunter T. Use and switching of biologic therapy in patients with non-radiographic axial spondyloarthritis: a patient and provider survey in the United States. Rheumatol Ther. 2020;7(2):415-423. PubMed
12.Michelsen B, Lindstrom U, Codreanu C, et al. Drug retention, inactive disease and response rates in 1860 patients with axial spondyloarthritis initiating secukinumab treatment: routine care data from 13 registries in the EuroSpA collaboration. RMD Open. 2020;6(3). PubMed
13.Yi E, Dai D, Piao OW, Zheng JZ, Park Y. Health care utilization and cost associated with switching biologics within the first year of biologic treatment initiation among patients with ankylosing spondylitis. J Manag Care Spec Pharm. 2021;27(1):27-36. PubMed
14.van Tubergen A, Black PM, Coteur G. Are patient-reported outcome instruments for ankylosing spondylitis fit for purpose for the axial spondyloarthritis patient? A qualitative and psychometric analysis. Rheumatology (Oxford). 2015;54(10):1842-1851. PubMed
15.Richard N, Haroon N, Tomlinson G, Sari I, Touma Z, Inman RD. Establishing the Minimal Clinically Important Difference (MCID) for the Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL) [abstract]. Arthritis Rheumatol. 2018;70(suppl 9).
16.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
17.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
18.Clinical Study Report: AS0011, BE MOBILE 2. A phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of bimekizumab in subjects with active ankylosing spondylitis [internal sponsor's report]. Brussels (BE): UCB Biopharma SRL; 2023 Jan 25.
19.Bimzelx (bimekizumab) for the treatment of adult patients with active ankylosing spondylitis: clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL solution in a single use pre-filled syringe or autoinjector, for subcutaneous injection. Oakville (ON): UCB Canada Inc.; 2023 Oct.
20.Reveille JD, Witter JP, Wesiman MH. Prevalence of axial spondylarthritis in the united states: estimates from a cross-sectional survey. Arthritis Care Res. 2012;64(6):905-910. PubMed
21.Feintuch S. Ankylosing spondylitis prognosis: what patients need to know. Upper Nyack (NY): CreakyJoints; 2020: https://creakyjoints.org/about-arthritis/axial-spondyloarthritis/axspa-overview/ankylosing-spondylitis-prognosis/. Accessed 2023 Jun 07.
22.Arthritis Society of C. Ankylosing Spondylitis. https://arthritis.ca/about-arthritis/arthritis-types-(a-z)/types/ankylosing-spondylitis. Accessed June 7, 2023.
23.de Koning A, Schoones JW, van der Heijde D, van Gaalen FA. Pathophysiology of axial spondyloarthritis: consensus and controversies. Eur J Clin Invest. 2018;48(5):e12913. PubMed
24.Taurog JD, Chhabra A, Colbert RA. Ankylosing spondylitis and axial spondyloarthritis. N Engl J Med. 2016;374(26):2563-2574. PubMed
25.Dougados M, Sepriano A, Molto A, et al. Sacroiliac radiographic progression in recent onset axial spondyloarthritis: the 5-year data of the DESIR cohort. Ann Rheum Dis. 2017;76(11):1823-1828. PubMed
26.Boonen A, van der Linden SM. The burden of ankylosing spondylitis. J Rheumatol Suppl. 2006;78:4-11. PubMed
27.Packham J. Optimizing outcomes for ankylosing spondylitis and axial spondyloarthritis patients: a holistic approach to care. Rheumatology (Oxford). 2018;57(suppl_6):vi29-vi34. PubMed
28.Robinson PC, van der Linden S, Khan MA, Taylor WJ. Axial spondyloarthritis: concept, construct, classification and implications for therapy. Nat Rev Rheumatol. 2021;17(2):109-118. PubMed
29.Haroon NN, Paterson JM, Li P, Haroon N. Increasing proportion of female patients with ankylosing spondylitis: a population-based study of trends in the incidence and prevalence of AS. BMJ Open. 2014;4(12):e006634. PubMed
30.Eversana Canada Inc. Clinician Interview Report for Ankylosing Spondylitis. 2023 [sponsor provided reference].
31.Rohekar S, Chan J, Tse SM, et al. 2014 update of the Canadian Rheumatology Association/spondyloarthritis research consortium of Canada treatment recommendations for the management of spondyloarthritis. Part I: principles of the management of spondyloarthritis in Canada. J Rheumatol. 2015;42(4):654-664. PubMed
32.Rohekar S, Pardo Pardo J, Mirza R, et al. Canadian Rheumatology Association/Spondyloarthritis Research Consortium of Canada Living Treatment Recommendations for the Management of Axial Spondyloarthritis. The Journal of Rheumatology. 2024:jrheum.2023-1237.
33.Bimzelx (bimekizumab injection): 160 mg solution for injections in pre-filled syringe; Bimzelx (bimekizumab injection): 160 mg solution for injections in autoinjector [product monograph]. UCB Canada Inc.: Oakville (ON); 2023 March 24.
34.Drug Reimbursement Expert Review Committee final recommendation: bimekizumab (Bimzelx) for plaque psoriasis. Can J Health Technol. 2022;2(4). https://www.canjhealthtechnol.ca/index.php/cjht/article/view/sr0698/sr0698. Accessed 2023 Nov 20.
35.UCB's Global Corporate Website. Positive top-line results for Bimzelx (bimekizumab) in phase 3 ankylosing spondylitis trial. 2023: https://www.ucb.com/stories-media/Press-Releases/article/Positive-Top-Line-Results-for-BIMZELXRVbimekizumab-in-Phase-3-Ankylosing-Spondylitis-Trial. Accessed 2023 Nov 20.
36.Humira (adalimumab injection): 50 mg/mL or 100 mg/mL sterile solution for subcutaneous injection, in a pen or pre-filled syringe [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022.
37.Cimzia (certolizumab pegol): 200 mg/mL solution for injection in a single-use prefilled syringe; Cimzia (certolizumab pegol): 200 mg/mL solution for injection in a single-use prefilled Autoinjector [product monograph]. UCB Canada Inc.: Oakville (ON); 2019.
38.Enbrel (etanercept): 50 mg/mL solution in a prefilled syringe; Enbrel (etanercept): 25 mg/vial lyophilized powder in a vial for reconstitution subcutaneous injection [product monograph]. Mississauga (ON): Amgen Canada Inc.; 2021.
39.Simponi (golimumab injection): 50 mg/0.5 mL or 100 mg/1.0 mL single-use SmartJect autoinjector for subcutaneous injection; Simponi (golimumab injection): 50 mg/0.5 mL or 100 mg/1.0 mL single-use pre-filled syringe for subcutaneous injection; Simponi I.V. (golimumab for injection): 50 mg/4.0 mL vial single-use vial sterile solution for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc.; 2022.
40.Remicade (infliximab) 100 mg/vial sterile lyophilized powder for solution for intravenous injection [product monograph]. Toronto (ON): Janssen Inc.; 2017.
41.Taltz (ixekizumab): 80 mg/1.0 mL solution for subcutaneous injection in prefilled auto injector or prefilled syringe [product monograph]. Toronto (ON): Eli Lilly Canada Inc.; 2022.
42.Cosentyx (secukinumab): 75 mg/0.5 mL or 150 mg/1 mL solution in prefilled glass syringes or prefilled SensoReady pen for subcutaneous injection; Cosentyx (secukinumab): 150 mg yophilized powder in single use vial solution for injection [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2022.
43.Xeljanz (tofacitinib as tofacitinib citrate): 5 mg or 10 mg oral tablets; Xeljanz XR (tofacitinib as tofacitinib citrate): 11 mg extended-release oral tabelts [product monographs]. Kirkland (QC): Pfizer Canada ULC; 2023.
44.Rinvoq (upadacitinib): 15 mg, 30 mg, and 45 mg, extended-release oral tablets [product monographs]. St-Laurent (QC): AbbVie Corporation; 2022.
45.Inc UCBC. BIMZELX (bimekizumab) DRAFT Product Monograph. 2023.
46.AbbVie C. Product Monograph: HUMIRA (adalimumab). 2022.
47.Inc UCBC. Product Monograph: CIMZIA (certolizumab pegol). 2019.
48.Immunex C. Product Monograph: ENBREL (etanercept). 2021.
49.Janssen I. Product Monograph: SIMPONI (golimumab). 2022.
50.Janssen I. Product Monograph: REMICADE (infliximab). 2017.
51.Eli Lilly Canada I. Product Monograph: TALTZ (ixekizumab). 2022.
52.Novartis Pharmaceutical Canada I. Product Monograph: COSENTYX (secukinumab). 2022.
53.Pfizer Canada ULC. Product Monograph: XELJANZ (tofacitinib). 2023.
54.AbbVie C. Product Monograph: RINVOQ (upadacitinib). 2022.
55.van der Heijde D, Deodhar A, Baraliakos X, et al. Efficacy and safety of bimekizumab in axial spondyloarthritis: results of two parallel phase 3 randomised controlled trials. Ann Rheum Dis. 2023;82(4):515-526. PubMed
56.Reilly MC, Gooch KL, Wong RL, Kupper H, van der Heijde D. Validity, reliability and responsiveness of the Work Productivity and Activity Impairment Questionnaire in ankylosing spondylitis. Rheumatology (Oxford). 2010;49(4):812-819. PubMed
57.Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5):353-365. PubMed
58.Brandt J, Listing J, Sieper J, Rudwaleit M, van der Heijde D, Braun J. Development and preselection of criteria for short term improvement after anti-TNF alpha treatment in ankylosing spondylitis. Ann Rheum Dis. 2004;63(11):1438-1444. PubMed
59.Anderson JJ, Baron G, van der Heijde D, Felson DT, Dougados M. Ankylosing spondylitis assessment group preliminary definition of short-term improvement in ankylosing spondylitis. Arthritis Rheum. 2001;44(8):1876-1886. PubMed
60.Doward LC, Spoorenberg A, Cook SA, et al. Development of the ASQoL: a quality of life instrument specific to ankylosing spondylitis. Ann Rheum Dis. 2003;62(1):20-26. PubMed
61.Haywood KL, A MG, Jordan K, Dziedzic K, Dawes PT. Disease-specific, patient-assessed measures of health outcome in ankylosing spondylitis: reliability, validity and responsiveness. Rheumatology (Oxford). 2002;41(11):1295-1302. PubMed
62.Madsen OR, Rytter A, Hansen LB, Suetta C, Egsmose C. Reproducibility of the Bath Ankylosing Spondylitis Indices of disease activity (BASDAI), functional status (BASFI) and overall well-being (BAS-G) in anti-tumour necrosis factor-treated spondyloarthropathy patients. Clin Rheumatol. 2010;29(8):849-854. PubMed
63.Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. J Rheumatol. 1994;21(12):2286-2291. PubMed
64.Pedersen SJ, Sørensen IJ, Hermann KG, et al. Responsiveness of the Ankylosing Spondylitis Disease Activity Score (ASDAS) and clinical and MRI measures of disease activity in a 1-year follow-up study of patients with axial spondyloarthritis treated with tumour necrosis factor alpha inhibitors. Ann Rheum Dis. 2010;69(6):1065-1071. PubMed
65.Pavy S, Brophy S, Calin A. Establishment of the minimum clinically important difference for the bath ankylosing spondylitis indices: a prospective study. J Rheumatol. 2005;32(1):80-85. PubMed
66.Kviatkovsky MJ, Ramiro S, Landewé R, et al. The Minimum Clinically Important Improvement and Patient-acceptable Symptom State in the BASDAI and BASFI for patients with ankylosing spondylitis. J Rheumatol. 2016;43(9):1680-1686. PubMed
67.van Tubergen A, Black PM, Coteur G. Are patient-reported outcome instruments for ankylosing spondylitis fit for purpose for the axial spondyloarthritis patient? A qualitative and psychometric analysis. Rheumatology. 2015;54(10):1842-1851. PubMed
68.Haywood KL, Garratt AM, Dawes PT. Patient-assessed health in ankylosing spondylitis: a structured review. Rheumatology (Oxford). 2005;44(5):577-586. PubMed
69.Heuft-Dorenbosch L, Spoorenberg A, van Tubergen A, et al. Assessment of enthesitis in ankylosing spondylitis. Ann Rheum Dis. 2003;62(2):127-132. PubMed
70.Rezvani A, Bodur H, Ataman S, et al. Correlations among enthesitis, clinical, radiographic and quality of life parameters in patients with ankylosing spondylitis. Mod Rheumatol. 2014;24(4):651-656. PubMed
71.Baraliakos X, Deodhar A, Dougados M, et al. Safety and efficacy of bimekizumab in patients with active ankylosing spondylitis: three-year results from a phase IIb randomized controlled trial and its open-label extension study. Arthritis Rheumatol. 2022;74(12):1943-1958. PubMed
72.Clinical Study Report: AS0009, BE AGILE 2. A multicenter, open-label extension study to evaluate the long term safety and efficacy of bimekizumab in subjects with ankylosing spondylitis. Belgium (BE): UCB Biopharma SRL; 2023 May 15.
73.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Making. 2018;38(2):200-211. PubMed
74.van der Heijde D, Kivitz A, Schiff MH, et al. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2006;54(7):2136-2146. PubMed
75.Bao C, Huang F, Khan MA, et al. Safety and efficacy of golimumab in Chinese patients with active ankylosing spondylitis: 1-year results of a multicentre, randomized, double-blind, placebo-controlled phase III trial. Rheumatology (Oxford). 2014;53(9):1654-1663. PubMed
76.Calin A, Dijkmans BA, Emery P, et al. Outcomes of a multicentre randomised clinical trial of etanercept to treat ankylosing spondylitis. Ann Rheum Dis. 2004;63(12):1594-1600. PubMed
77.Lambert RG, Salonen D, Rahman P, et al. Adalimumab significantly reduces both spinal and sacroiliac joint inflammation in patients with ankylosing spondylitis: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2007;56(12):4005-4014. PubMed
78.van der Heijde D, Cheng-Chung Wei J, Dougados M, et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet. 2018;392(10163):2441-2451. PubMed
79.Davis JC, Jr., Van Der Heijde D, Braun J, et al. Recombinant human tumor necrosis factor receptor (etanercept) for treating ankylosing spondylitis: a randomized, controlled trial. Arthritis Rheum. 2003;48(11):3230-3236. PubMed
80.van der Heijde D, Da Silva JC, Dougados M, et al. Etanercept 50 mg once weekly is as effective as 25 mg twice weekly in patients with ankylosing spondylitis. Ann Rheum Dis. 2006;65(12):1572-1577. PubMed
81.Inman RD, Davis JC, Jr., Heijde D, et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis: results of a randomized, double-blind, placebo-controlled, phase III trial. Arthritis Rheum. 2008;58(11):3402-3412. PubMed
82.Gorman JD, Sack KE, Davis JC, Jr. Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor alpha. N Engl J Med. 2002;346(18):1349-1356. PubMed
83.Hu Z, Xu M, Li Q, et al. Adalimumab significantly reduces inflammation and serum DKK-1 level but increases fatty deposition in lumbar spine in active ankylosing spondylitis. Int J Rheum Dis. 2012;15(4):358-365. PubMed
84.Huang F, Gu J, Zhu P, et al. Efficacy and safety of adalimumab in Chinese adults with active ankylosing spondylitis: results of a randomised, controlled trial. Ann Rheum Dis. 2014;73(3):587-594. PubMed
85.Barkham N, Coates LC, Keen H, et al. Double-blind placebo-controlled trial of etanercept in the prevention of work disability in ankylosing spondylitis. Ann Rheum Dis. 2010;69(11):1926-1928. PubMed
86.van der Heijde D, Song IH, Pangan AL, et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): a multicentre, randomised, double-blind, placebo-controlled, phase 2/3 trial. Lancet. 2019;394(10214):2108-2117. PubMed
87.Dougados M, Braun J, Szanto S, et al. Efficacy of etanercept on rheumatic signs and pulmonary function tests in advanced ankylosing spondylitis: results of a randomised double-blind placebo-controlled study (SPINE). Ann Rheum Dis. 2011;70(5):799-804. PubMed
88.van der Heijde D, Deodhar A, Wei JC, et al. Tofacitinib in patients with ankylosing spondylitis: a phase II, 16-week, randomised, placebo-controlled, dose-ranging study. Ann Rheum Dis. 2017;76(8):1340-1347. PubMed
89.Deodhar A, Poddubnyy D, Pacheco-Tena C, et al. Efficacy and Safety of Ixekizumab in the Treatment of Radiographic Axial Spondyloarthritis: Sixteen-Week Results From a Phase III Randomized, Double-Blind, Placebo-Controlled Trial in Patients With Prior Inadequate Response to or Intolerance of Tumor Necrosis Factor Inhibitors. Arthritis Rheumatol. 2019;71(4):599-611. PubMed
90.Van der Heijde D, Baraliakos X, Sieper J, et al. POS0306 Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis refractory to biologic therapy: a double-blind, randomized, placebo-controlled phase 3 trial. Ann Rheum Dis. 2022;81(Suppl 1):402-403.
91.van der Heijde D, Dijkmans B, Geusens P, et al. Efficacy and safety of infliximab in patients with ankylosing spondylitis: results of a randomized, placebo-controlled trial (ASSERT). Arthritis Rheum. 2005;52(2):582-591. PubMed
92.Kiltz U, Baraliakos X, Brandt-Jrgens J, et al. Evaluation of the nonsteroidal anti-inflammatory drug-sparing effect of secukinumab in patients with ankylosing spondylitis: multicenter, randomised, double-blind, phase IV study. Arthritis Rheumatol. 2021;73(SUPPL 9):1886-1888.
93.van der Heijde D, Gensler LS, Deodhar A, et al. Dual neutralisation of interleukin-17A and interleukin-17F with bimekizumab in patients with active ankylosing spondylitis: results from a 48-week phase IIb, randomised, double-blind, placebo-controlled, dose-ranging study. Ann Rheum Dis. 2020;79(5):595-604. PubMed
94.Clinical Study Report: AS0011, BE MOBILE 2, week 24 interim analysis. A phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of bimekizumab in subjects with active ankylosing spondylitis [internal sponsor's report]. UCB Biopharma SRL. Brussels (BE)2022 Jul 15.
95.Deodhar A, Sliwinska-Stanczyk P, Xu H, et al. Tofacitinib for the treatment of ankylosing spondylitis: a phase III, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2021;80(8):1004-1013. PubMed
96.Reveille JD, Deodhar A, Caldron PH, et al. Safety and efficacy of intravenous golimumab in adults with ankylosing spondylitis: results through 1 year of the GO-ALIVE study. J Rheumatol. 2019;46(10):1277-1283. PubMed
97.Baeten D, Sieper J, Braun J, et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N Engl J Med. 2015;373(26):2534-2548. PubMed
98.Kivitz AJ, Wagner U, Dokoupilova E, et al. Efficacy and safety of secukinumab 150 mg with and without loading regimen in ankylosing spondylitis: 104-week results from MEASURE 4 study. Rheumatol Ther. 2018;5(2):447-462. PubMed
99.Huang F, Sun F, Wan W, et al. FRI0414 Secukinumab provides rapid and significant improvement in the signs and symptoms of ankylosing spondylitis: Primary (16-week) results from a phase 3 china-centric study, measure 5. Ann Rheum Dis. 2019;78:894-895.
100.Landewe R, Braun J, Deodhar A, et al. Efficacy of certolizumab pegol on signs and symptoms of axial spondyloarthritis including ankylosing spondylitis: 24-week results of a double-blind randomised placebo-controlled Phase 3 study. Ann Rheum Dis. 2014;73(1):39-47. PubMed
101.Xue Y, Hu J, Liu D, et al. Efficacy and Safety of Ixekizumab in Chinese Patients with Radiographic Axial Spondyloarthritis: 16-Week Results from a Phase 3 Study [abstract]. Arthritis Rheumatol. 2022;74(S9).
102.Bimekizumab for the treatment of ankylosing spondylitis: 2023 SLR and NMA update [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL solution in a single use pre-filled syringe or autoinjector, for subcutaneous injection. Oakville (ON): UCB Canada Inc.; 2023 Apr 28.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 36: Summary of Exploratory Efficacy Results From the BE MOBILE 2 Trial (Randomized Set)
End points | BE MOBILE 2 (Data cut-off date: September 9, 2022) | |
|---|---|---|
Bimekizumab 160 mg/mL (N = 221) | Placebo (N = 111) | |
ASAS40 (NRI) | ||
Response rate at week 24 | 119.0 (53.8) | 63.0 (56.8) |
Response rate at week 36 | 123.0 (55.7) | 64.0 (57.7) |
Response rate at week 52 | 129.0 (58.4) | 76.0 (68.5) |
Change from baseline in BASDAI total score (MI) | ||
Change from baseline at week 24, mean (SE) | –3.3 (0.1) | –3.3 (0.2) |
Change from baseline at week 36, mean (SE) | –3.4 (0.1) | –3.5 (0.2) |
Change from baseline at week 52, mean (SE) | 3.6 (0.1) | –4.0 (0.2) |
Change from baseline in BASFI (MI) | ||
Change from baseline at week 24, mean (SE) | –2.5 (0.2) | –2.2 (0.2) |
Change from baseline at week 36, mean (SE) | –2.6 (0.2) | –2.5 (0.2) |
Change from baseline at week 52, mean (SE) | –2.8 (0.1) | –2.8 (0.2) |
Change from baseline in NSP score (based on an NRS, MI) | ||
Change from baseline at week 24, mean (SE) | –3.8 (0.2) | –3.7 (0.3) |
Change from baseline at week 36, mean (SE) | –3.9 (0.2) | –4.1 (0.2) |
Change from baseline at week 52, mean (SE) | –4.1 (0.2) | –4.6 (0.3) |
Change from baseline in ASQoL total score (MI) | ||
Change from baseline at week 24, mean (SE) | –5.4 (0.3) | –4.8 (0.4) |
Change from baseline at week 36, mean (SE) | –5.5 (0.3) | –5.2 (0.4) |
Change from baseline at week 52, mean (SE) | –5.7 (0.3) | –5.6 (0.4) |
Enthesitis-free state based on the MASES index in study participants with enthesitis at baseline (NRI) | ||
Participants achieving an enthesitis-free state at week 24, n (%) | 70 (53.0) | 33 (49.3) |
Participants achieving an enthesitis-free state at week 52, n (%) | 67 (50.8) | 31 (46.3) |
Change from baseline in WPAI-SHP (observed case) | ||
Percent time missed due to disease-related problems at week 24 | ||
Number of participants contributing to the analysis, n | 152 | 79 |
Change from baseline, mean (SD) | –4.9 (23.4) | –4.7 (19.5) |
Percent time missed due to disease-related problems, at week 36 | ||
Number of participants contributing to the analysis, n | 154 | 80 |
Change from baseline, mean (SD) | –4.7 (22.8) | –2.8 (21.8) |
Percent time missed due to disease-related problems at week 52 | ||
Number of participants contributing to the analysis, n | 153 | 81 |
Change from baseline, mean (SD) | –4.7 (24.6) | –2.8 (19.6) |
Percent impairment while working due to disease-related problems at week 24 | ||
Number of participants contributing to the analysis, n | 136 | 75 |
Change from baseline, mean (SD) | –21.2 (26.0) | –16.6 (23.2) |
Percent impairment while working due to disease-related problems at week 36 | ||
Number of participants contributing to the analysis, n | 143 | 70 |
Change from baseline, mean (SD) | –22.5 (25.9) | –20.2 (22.0) |
Percent impairment while working due to disease-related problems at week 52 | ||
Number of participants contributing to the analysis, n | 143 | 75 |
Change from baseline, mean (SD) | –26.5 (21.5) | –24.6 (22.9) |
Percent overall work impairment due to disease-related problems at week 24 | ||
Number of participants contributing to the analysis, n | 136 | 75 |
Change from baseline, mean (SD) | –21.9 (28.2) | –17.3 (23.4) |
Percent overall work impairment due to disease-related problems at week 36 | ||
Number of participants contributing to the analysis, n | 143 | 70 |
Change from baseline, mean (SD) | –23.5 (27.5) | –20.7 (24.4) |
Percent overall work impairment due to disease-related problems at week 52 | ||
Number of participants contributing to the analysis, n | 143 | 75 |
Change from baseline, mean (SD) | –27.0 (23.1) | –23.6 (24.8) |
Percent activity impairment due to disease-related problems at week 24 | ||
Number of participants contributing to the analysis, n | 201 | 107 |
Change from baseline, mean (SD) | –24.3 (23.8) | –26.4 (26.0) |
Percent activity impairment due to disease-related problems at week 36 | ||
Number of participants contributing to the analysis, n | 197 | 105 |
Change from baseline, mean (SD) | –26.2 (23.7) | –26.4 (24.9) |
Percent activity impairment due to disease-related problems at week 52 | ||
Number of participants contributing to the analysis, n | 196 | 102 |
Change from baseline, mean (SD) | –28.8 (22.8) | –31.9 (24.8) |
ASAS40 = improvement of 40% or more in the Assessment of SpondyloArthritis International Society; ASQoL = Ankylosing Spondylitis Quality of Life; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; CI = confidence interval; LS = least squares; MASES = Maastricht Ankylosing Spondylitis Enthesitis MI = multiple imputation; NRI = nonresponder imputation; NRS = numeric rating scale; NSP = nocturnal spinal pain; SD = standard deviation; SE = standard error; WPAI-SHP = Work Productivity and Activity Impairment Questionnaire–Specific Health Problem.
Source: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18
Figure 7: Forest Plot of ASAS40 Odds Ratio at Week 16 by Subgroups (Randomized Set; Data Cut-Off Date: September 9, 2022)

ASAS = Assessment of SpondyloArthritis International Society; ASDAS = Ankylosing Spondylitis Disease Activity Score; BKZ = bimekizumab; CI = confidence interval; csDMARD = conventional synthetic disease-modifying; HLA-B27 = human leukocyte antigen B27; hs-CRP = high-sensitivity C-reactive protein; NE = not evaluable; PBO = placebo; TNF = tumour necrosis factor; ULN = upper limit of normal.
Note: The ULN value for hs-CRP was 5 mg/L.
Model-adjusted response rates are presented.
Missing data were imputed using nonresponder imputation.
Source: BE MOBILE 2 final Clinical Study Report (data cut-off date: September 9, 2022).18
Pharmacoeconomic Review
Abbreviations
AE
adverse event
AS
ankylosing spondylitis
ASAS20
improvement of 20% or more in the Assessment of SpondyloArthritis International Society
ASAS40
improvement of 40% or more in the Assessment of SpondyloArthritis International Society
BASDAI
Bath Ankylosing Spondylitis Disease Activity Index
BASDAI50
a reduction of at least 50% in the Bath Ankylosing Spondylitis Disease Activity Index score
BASFI
Bath Ankylosing Spondylitis Functional Index
BSC
best supportive care
DMARD
disease-modifying antirheumatic drug
NMA
network-meta-analysis
NSAID
nonsteroidal anti-inflammatory drug
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Bimekizumab (Bimzelx), prefilled syringe or autoinjector |
Indication | The treatment of adult patients with active ankylosing spondylitis |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 11, 2024 |
Reimbursement request | As per indication |
Sponsor | UCB Canada Inc. |
Submission history |
|
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree plus Markov model |
Target population | Adult patients with active AS |
Treatment | Bimekizumab |
Dose regimen | 160 mg (given as 1 subcutaneous injection) every 4 weeks |
Submitted price | Bimekizumab 160 mg/mL subcutaneous injection: $1,625.00 |
Submitted treatment cost | $21,198 annually |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (75 years) |
Key data sources | Comparative clinical efficacy was derived from a sponsor-submitted NMA based on data from the BE MOBILE 2 and comparator treatment trials to inform the probability of BASDAI50 and difference in mean change from baseline in clinical scores for BASDAI and BASFI response at 12 to 16 weeks. |
Submitted results |
|
Key limitations | The efficacy and safety of bimekizumab relative to other biologic DMARDs for the treatment of active AS is uncertain owing to a lack of head-to-head trials and limitations with the sponsor’s NMAs. The indirect evidence submitted by the sponsor did not show clear differences in the efficacy or safety of bimekizumab compared with other currently available treatments for active AS. Findings were inconsistent in the NMA and confidence intervals were wide. |
CADTH reanalysis results | There is insufficient clinical evidence to justify a price premium for bimekizumab relative to currently available treatments for active AS. |
AS = ankylosing spondylitis; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index score; BASDAI50 = a reduction of at least 50% in the Bath Ankylosing Spondylitis Disease Activity Index score; BASFI = Bath Ankylosing Spondylitis Functional Index; DMARD = disease-modifying antirheumatic drug; LY = life-year; NMA = network meta-analysis; QALY = quality-adjusted life-year.
Conclusions
Based on the CADTH Clinical Review of the BE MOBILE 2 trial, the available evidence suggests that bimekizumab is associated with an increase in the adjusted improvement of 40% or more in the Assessment of SpondyloArthritis International Society (ASAS40) response rate at week 16 and a greater improvement in the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) total score when compared with placebo. While results of the sponsor’s network meta-analysis (NMA) reported differences in a small number of outcomes in some populations, these were associated with wide 95% credible intervals. Similar findings were found in the sponsor's matching-adjusted indirect comparison; however, limitations such as differences in study design, patient population, and heterogeneity in baseline characteristics across studies undermine any claims of superior performance of bimekizumab over comparators in the analysis. Therefore, the CADTH Clinical Review team concluded that the sponsor’s indirect treatment comparisons did not provide evidence of a difference in efficacy or harms outcomes for bimekizumab versus comparators.
In the sponsor’s economic submission, tofacitinib, etanercept, adalimumab, infliximab, upadacitinib, golimumab, and certolizumab pegol are associated with improved clinical effects in terms of quality-adjusted life-years (QALYs) and lower costs when compared with bimekizumab. However, given the uncertainty in the clinical evidence, there is insufficient evidence to suggest that bimekizumab should be priced higher than other biologic disease-modifying antirheumatic drug (DMARD) treatments for ankylosing spondylitis (AS).
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received from 2 groups, Arthritis Consumer Experts and a joint submission by the Canadian Spondylarthritis Association, Canadian Arthritis Patient Alliance, Arthritis Society Canada, and Creaky Joints. Input for both was gathered from online surveys from a total of 118 participants. Based on the joint input, the most common symptoms identified by surveyed participants included back pain, joint stiffness, fatigue, and hip pain, which patients reported as having the greatest impact on their ability to exercise and sleep. When asked about experiences with currently available treatments, the joint submission noted that biologics were rated as highly effective, while the effectiveness of NSAIDs varied among patients. The most commonly reported side effects included nausea, gastrointestinal issues, and fatigue. Two patients from the joint input indicated they used bimekizumab, 1 of whom noted they had no issues. Respondents noted that treatment resistance commonly occurs; therefore, additional treatment options that improve symptoms and result in an earlier diagnosis are wanted. Similar feedback was expressed in the Arthritis Consumer Experts input.
No registered clinician input was received for this review.
The CADTH-participating drug plans noted concerns with the use of placebo as the comparator in the clinical trials, given the availability of other biologic therapies for the treatment of AS in Canada. The drug plan input noted the current dose of bimekizumab for AS is 160 mg subcutaneously every 4 weeks and inquired about increased or reduced administration frequency. Lastly, the plans noted the presence of confidential negotiated prices for comparators.
Economic Review
The current review is for bimekizumab (Bimzelx) for adult patients with active AS.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of bimekizumab against DMARDs and conventional care for the treatment of adult patients with active AS. The model population represents the reimbursement request and aligns with the Health Canada indication.1
Bimekizumab is available as prefilled syringes or autoinjectors, each containing 1 mL of 160 mg bimekizumab.2 The recommended dose is 160 mg (given as 1 subcutaneous injection) every 4 weeks.2 The submitted price for bimekizumab is $1,625 per syringe or autoinjector.1 The comparators for this analysis included adalimumab, etanercept, golimumab, infliximab, secukinumab, certolizumab pegol, ixekizumab, upadacitinib, tofacitinib, and conventional care. Conventional care was defined as first-line treatment of AS, including non-pharmacological management and nonsteroidal anti-inflammatory drugs (NSAIDs).
The outcomes of the model included QALYs and life-years over a lifetime horizon of 75 years. Discounting (1.5% per annum) was applied for both costs and outcomes. For the Markov potion of the model, a 3-month cycle length was used with a half-cycle correction applied.1
Model Structure
The sponsor’s submitted model incorporated 2 parts, a decision tree for the first year of the model and a Markov model for the remainder of the time horizon (Figure 1 and Figure 2, respectively).1
At model entry, patients initiated bimekizumab or a comparator and entered the decision tree. Patients who achieved a reduction of at least 50% in BASDAI score (BASDAI50) at week 12 to 16 continued to maintenance treatment until the end of year 1.1 Patients who did not respond, and a proportion of responders who discontinued treatment due to lack of efficacy or other reasons, switched to the next line of treatment (i.e., conventional care).1 Response evaluation for conventional care is assessed the same way as initial treatment, and patients who did not respond or discontinued treatment transitioned to best supportive care (BSC).1 At the end of the first year, patients entered 1 of 7 health states within the Markov model: on initial DMARD maintenance, just discontinued initial DMARD, on next DMARD induction, on next DMARD maintenance, just discontinued next DMARD, on BSC, and death.1
Patients in the “on initial DMARD maintenance” health state could remain, discontinue (i.e., transition to “just discontinued initial DMARD”) or begin treatment with the next DMARD (i.e., transition to “on next DMARD induction”).1 Patients in the “on next DMARD induction” health state could experience no response or respond and transition to the “just discontinued next DMARD” or the “on next DMARD maintenance” health states, respectively.1 Patients in the “on next DMARD maintenance” health state could remain or transition to “just discontinued next DMARD.”1 “Just discontinued initial DMARD” and “just discontinued next DMARD” are tunnel states where patients could remain for 1 cycle only before transitioning to “on BSC.” Patients could transition to death at any point.1
Model Inputs
Data from the BE MOBILE 2 trial were used to inform baseline characteristics (mean age = 40.4; percent male = 72.3%).3
Comparative efficacy data for bimekizumab and comparators were informed by the sponsor-conducted NMA. Analyses were conducted based on data obtained from the BE MOBILE 2 trial and respective comparator treatment trials for BASDAI50 at 12 to 16 weeks. As no BASDAI50 data were available for infliximab, the NMA of tumour necrosis factor (TNF) inhibitors was used to derive a relative risk.
In the initial assessment period (i.e., decision tree), the sponsor assumed that all patients who were nonresponders after the initial DMARD and the next DMARD would experience treatment discontinuation. During maintenance treatment, an 11% annual risk of biologic DMARD discontinuation was applied, aligned with the value used in the York model.4 The same rate of discontinuation was applied to all comparators due to limitations in the availability of comparative data.1 It was assumed that 50% of discontinuation was due to lack of efficacy rather than other causes.1
Two types of adverse events (AEs) were applied in the model: tuberculosis reactivation and other serious infections. Bimekizumab rates were informed by the BE MOBILE trial, while conventional care was assumed to have no AEs. The AE rates of the remaining comparators were informed by a previous AS technology appraisal for the ixekizumab or upadacitinib summary of product characteristics.5
Background mortality was included in the model and was based on Canadian general life tables.6 To account for the increased risk of mortality for patients with AS, a standardized mortality ratio of 1.63 and 1.38 was applied for men and women, respectively.7
The model estimated EQ-5D utilities as a function of BASDAI and Bath AS Functional Index (BASFI) scores. BASDAI and BASFI scores of patients in the model were converted to EQ-5D utility values using a mapping algorithm derived from the bimekizumab phase III trial.1
The economic model included drug acquisition costs, treatment administration costs, biologic initiation and monitoring costs, and disease-management costs.1 Treatment costs were estimated using dosing schedules based on each treatment’s respective Health Canada product monograph.1 Drug unit costs were obtained from the Ontario Drug Benefit Formulary.8 The lowest-cost biosimilar available was used for adalimumab, etanercept, and infliximab.1 The proportion of patients treated with each therapy included in the conventional-care arm was based on feedback obtained by the clinical experts consulted by the sponsor. Drug administration costs were assumed to be based on an hour’s wage for a nurse for training on subcutaneous injection.1 The cost of IV injection was informed by the published literature.9 The frequency and cost of resource use associated with biologic initiation and monitoring were informed by the published literature and validated by the clinical experts consulted by the sponsor.1,4,10,11 Lastly, disease-management costs were calculated as a function of BASFI score for each health state where the intercept value ($2,785.79) was informed by Boonen et al., and considered drug costs (excluding treatment with TNF inhibitors), administration, monitoring, hospitalization, and health care visits, aids, and appliances.12
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
In the sponsor’s probabilistic base-case analysis, bimekizumab was associated with a cost of $653,172 and 17.26 QALYs over a lifetime horizon. In the sequential analysis, bimekizumab was not on the frontier and was dominated (more costly and less effective) by adalimumab, etanercept, golimumab, infliximab, upadacitinib, certolizumab pegol, and tofacitinib. At the submitted price, bimekizumab has a 0% probability of being cost-effective at a $50,000 willingness-to-pay threshold.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Conventional care | 405,774 | 15.083 | Reference |
Tofacitinib | 593,682 | 17.283 | 85,392 vs. conventional care |
Etanercept | 621,470 | 17.559 | 100,638 vs. tofacitinib |
Dominated treatments | |||
Adalimumab | 622,301 | 17.328 | Dominated |
Infliximab | 631,032 | 17.391 | Dominated |
Secukinumab 150 mg | 635,490 | 17.132 | Dominated |
Secukinumab 300 mg | 644,390 | 17.130 | Dominated |
Upadacitinib | 646,052 | 17.272 | Dominated |
Golimumab | 646,263 | 17.417 | Dominated |
Certolizumab pegol | 647,747 | 17.313 | Dominated |
Bimekizumab | 653,172 | 17.261 | Dominated |
Ixekizumab | 664,331 | 17.315 | Dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses, including alternate response assessments (i.e., improvement of 20% or more in the ASAS [ASAS20], ASAS40, and the Ankylosing Spondylitis Disease Activity Score–Major Improvement), a lower discounting rate, using general population mortality, assuming brand name pricing for adalimumab, etanercept, and infliximab, and alternative discounting rates (i.e., 0% and 3%). Generally, the results of the scenario analyses were similar to the base case, where bimekizumab was not on the cost-effectiveness frontier.
Additionally, the sponsor conducted a scenario analysis from a societal perspective. This analysis included additional costs associated with indirect costs associated with work productivity loss. This was similar to the sponsor’s base-case analysis using a health care payer perspective.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative and clinical efficacy and safety of bimekizumab versus other biologic and targeted DMARDs is uncertain. There is a lack of head-to-head evidence comparing bimekizumab with biologic and targeted DMARDs. In the absence of head-to-head evidence for most comparators, the sponsor conducted NMAs to inform various parameters in the economic model for all treatments, including BASDAI and BASFI. As noted in the CADTH Clinical Review, the sponsor’s NMA did not show clear differences between bimekizumab and comparators for BASDAI and change in BASFI. The CADTH Clinical Review team concluded that the comparative findings of the sponsor’s NMAs were insufficient to support claims of differences in efficacy or harms of bimekizumab relative to other biologic DMARDs because of the presence of substantial imprecision and unresolved heterogeneity.
Given the lack of direct evidence for bimekizumab relative to biologic and targeted DMARDs and limitations with the sponsor’s NMA, it is uncertain whether bimekizumab provides a net benefit exceeding that of any of the currently available treatments for active AS.
Issues for Consideration
Bimekizumab was previously reviewed by CADTH for the treatment of moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy and received a “reimburse with conditions” recommendation.13 Bimekizumab is also undergoing a review by CADTH for the treatment of adult patients with active psoriatic arthritis.14
The sponsor’s analyses rely on publicly accessible list prices and do not reflect the existing confidential prices negotiated by public plans. Given that infliximab, certolizumab pegol, etanercept, secukinumab, and adalimumab have successfully undergone price negotiations for the treatment of AS, it is likely that the current unit cost paid by public drug plans for these treatments is lower than the values used in the sponsor’s analyses.15-19
Overall Conclusions
Based on the CADTH Clinical Review of the BE MOBILE 2 trial, the available evidence suggests that bimekizumab is associated with an increase in the adjusted ASAS40 response rate at week 16 and a greater improvement in BASDAI total score compared to placebo. While results of the sponsor’s NMA reported differences in a small number of comparisons in some populations, these were associated with wide 95% credible intervals. Similar findings were found in the sponsor’s matching-adjusted indirect comparison; however, limitations such as differences in study design, patient population, and heterogeneity in baseline characteristics across studies undermine any claims of superior performance of bimekizumab over comparators in the analysis. Therefore, the CADTH Clinical Review team concluded that the sponsor’s indirect treatment comparisons did not provide evidence of a difference in efficacy or harms outcomes for bimekizumab versus comparators.
In the sponsor’s economic submission, tofacitinib, etanercept, adalimumab, infliximab, upadacitinib, golimumab, and certolizumab pegol are associated with improved clinical effects (QALYs) and lower costs when compared with bimekizumab. Given the uncertainty in the sponsor-submitted indirect evidence, there is insufficient evidence to suggest that bimekizumab should be priced higher than other biologic DMARD treatments for AS. Thus, to ensure cost-effectiveness, bimekizumab should be priced no more than the lowest-cost biologic DMARD that is funded for the treatment of AS.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL solution in a single use pre-filled syringe or autoinjector, for subcutaneous injection. Oakville (ON): UCB Canada Inc.; 2023 Oct 19.
2.Bimzelx (bimekizumab): 160 mg/mL solution in a single use pre-filled syringe or autoinjector, for subcutaneous injection [product monograph]. Oakville (ON): UCB Canada Inc.; 2024 Mar 11.
3.van der Heijde D, Deodhar A, Baraliakos X, et al. Efficacy and safety of bimekizumab in axial spondyloarthritis: results of two parallel phase 3 randomised controlled trials. Ann Rheum Dis. 2023;82(4):515-526. PubMed
4.Corbett M, Soares M, Jhuti G, et al. Tumour necrosis factor-α inhibitors for ankylosing spondylitis and non-radiographic axial spondyloarthritis: a systematic review and economic evaluation. Health Technol Assess. 2016;20(9):1-334, v-vi. PubMed
5.Ixekizumab for treating axial spondyloarthritis after NSAIDs [ID1532]. Supporting evidence: committee papers. London (UK): National Institute for Health and Care Excellence (NICE); 2021: https://www.nice.org.uk/guidance/ta718/evidence/appraisal-consultation-committee-papers-pdf-9194641741. Accessed 2023 Nov 23.
6.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2023 Nov 16.
7.Bakland G, Gran JT, Nossent JC. Increased mortality in ankylosing spondylitis is related to disease activity. Ann Rheum Dis. 2011;70(11):1921-1925. PubMed
8.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Nov 16.
9.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
10.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2023 Nov 16.
11.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 23, 2023)). Toronto (ON): Ontario Ministry of Health; 2023: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 Nov 16.
12.Boonen A, van der Heijde D, Landewé R, et al. Direct costs of ankylosing spondylitis and its determinants: an analysis among three European countries. Ann Rheum Dis. 2003;62(8):732-740. PubMed
13.Drug Reimbursement Expert Review Committee updated recommendation: biologics for plaque psoriasis. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/TSDrugClas/TS0001/TS0001-PsO-Updated-Streamlined-Rec.pdf. Accessed 2023 Dec 18.
14.Drug Reimbursement Review: bimekizumab (Bimzelx) for psoriatic arthritis. Ottawa (ON): CADTH; 2024: https://www.cadth.ca/bimekizumab-0. Accessed 2023 Dec 18.
15.pan-Canadian Pharmaceutical Alliance. Inflectra (infliximab). 2015; https://www.pcpacanada.ca/negotiation/20858. Accessed 2023 Dec 11.
16.pan-Canadian Pharmaceutical Alliance. Cimzia (certolizumab pegol). 2016; https://www.pcpacanada.ca/negotiation/20863. Accessed 2023 Dec 11.
17.pan-Canadian Pharmaceutical Alliance. Rymti (etanercept). 2023; https://www.pcpacanada.ca/negotiation/22082. Accessed 2023 Dec 11.
18.pan-Canadian Pharmaceutical Alliance. Cosentyx (secukinumab). 2017; https://www.pcpacanada.ca/negotiation/20952.
19.pan-Canadian Pharmaceutical Alliance. Abrilada (adalimumab). 2022; https://www.pcpacanada.ca/negotiation/21637.
20.Humira (adalimumab injection): 50 mg/mL or 100 mg/mL sterile solution for subcutaneous injection, in a pen or pre-filled syringe [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022 Sep 16: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/HUMIRA_PM_EN.pdf. Accessed 2023 Nov 22.
21.Cimzia (certolizumab pegol): 200 mg/mL solution for injection in a single-use prefilled syringe; Cimzia (certolizumab pegol): 200 mg/mL solution for injection in a single-use prefilled Autoinjector [product monograph]. Oakville (ON): UCB Canada Inc.; 2019 Feb 08: https://pdf.hres.ca/dpd_pm/00049574.PDF. Accessed 2023 Nov 22.
22.Enbrel (etanercept): 50 mg/mL solution in a prefilled syringe; Enbrel (etanercept): 25 mg/vial lyophilized powder in a vial for reconstitution subcutaneous injection [product monograph]. Mississauga (ON): Amgen Canada Inc.; 2021 Mar 19: https://www.amgen.ca/-/media/Themes/CorporateAffairs/amgen-ca/amgen-ca/documents/products/en/enbrel_pm.pdf. Accessed 2023 Nov 22.
23.Plaquenil (hydroxychloroquine sulfate tablets): 200 mg tablets [product monograph]. Laval (QC): sanofi-aventis Canada Inc.; 2019 Aug 26: https://pdf.hres.ca/dpd_pm/00052851.PDF. Accessed 2023 Nov 22.
24.Salazopyrin (sulfasalazine): 500 mg tablets and 3 g/100 mL enema; Salazopyrin EN-tabs (sulfasalazine delayed-release tablets): 500 mg delayed-release tablets [product monograph]. Kirkland (QC): Pfizer Canada Inc.; 2003 Sep 23: https://pdf.hres.ca/dpd_pm/00000465.PDF. Accessed 2023 Nov 22.
25.Methotrexate (methotrexate): 2.5 mg oral tablets; Methotrexate (methotrexate): 25 mg/mL intramuscular, intravenous, or intraarterial injection [product monograph]. Kirkland (QC): Pfizer Canada Inc.; 2011 Apr 21: https://pdf.hres.ca/dpd_pm/00013052.PDF. Accessed 2023 Nov 22.
26.Rinvoq (upadacitinib): 15 mg, 30 mg, and 45 mg, extended-release oral tablets [product monographs]. St-Laurent (QC): AbbVie Corporation; 2023 Oct 31: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/RINVOQ_PM_EN.pdf. Accessed 2023 Nov 22.
27.Taltz (ixekizumab): 80 mg/1.0 mL solution for subcutaneous injection in prefilled auto injector or prefilled syringe [product monograph]. Toronto (ON): Eli Lilly Canada Inc.; 2018 Mar 29: https://pdf.hres.ca/dpd_pm/00044542.PDF. Accessed 2023 Nov 22.
28.Cosentyx (secukinumab): 75 mg/0.5 mL or 150 mg/1 mL solution in prefilled glass syringes or prefilled SensoReady pen for subcutaneous injection; Cosentyx (secukinumab): 150 mg yophilized powder in single use vial solution for injection [product monograph]. Montreal (QC): Novartis Pharmaceuticals Canada Inc.; 2023 Aug 17: https://www.ask.novartispharma.ca/download.htm?res=cosentyx_scrip_e.pdf&resTitleId=990. Accessed 2023 Nov 22.
29.Remicade (infliximab) 100 mg/vial sterile lyophilized powder for solution for intravenous injection [product monograph]. Toronto (ON): Janssen Inc.; 2017 Aug 04: https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/REMICADE-MONOGRAPH.PDF. Accessed 2023 Nov 22.
30.Simponi (golimumab injection): 50 mg/0.5 mL or 100 mg/1.0 mL single-use SmartJect autoinjector for subcutaneous injection; Simponi (golimumab injection): 50 mg/0.5 mL or 100 mg/1.0 mL single-use pre-filled syringe for subcutaneous injection; Simponi I.V. (golimumab for injection): 50 mg/4.0 mL vial single-use vial sterile solution for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc.; 2018 Jan 15: https://pdf.hres.ca/dpd_pm/00043422.PDF. Accessed 2023 Nov 22.
31.Xeljanz (tofacitinib as tofacitinib citrate): 5 mg or 10 mg oral tablets; Xeljanz XR (tofacitinib as tofacitinib citrate): 11 mg extended-release oral tabelts [product monographs]. Kirkland (QC): Pfizer Canada Inc.; 2019 Feb 04: https://pdf.hres.ca/dpd_pm/00049539.PDF. Accessed 2023 Nov 22.
32.Accel-Leflunomide (leflunomide): 10 mg and 20 mg film-coated oral tablets [product monograph]. Pointe-Claire (QC): Accel Pharma Inc.; 2018 Jul 12: https://pdf.hres.ca/dpd_pm/00046765.PDF. Accessed 2023 Nov 22.
33.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2023: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 Nov 22.
34.Saskatchewan Drug Plan: search formulary. 2023; http://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2023 Nov 02.
35.Haroon NN, Paterson JM, Li P, Haroon N. Increasing proportion of female patients with ankylosing spondylitis: a population-based study of trends in the incidence and prevalence of AS. BMJ Open. 2014;4(12):e006634. PubMed
36.Bohn R, Cooney M, Deodhar A, Curtis JR, Golembesky A. Incidence and prevalence of axial spondyloarthritis: methodologic challenges and gaps in the literature. Clin Exp Rheumatol. 2018;36(2):263-274. PubMed
37.Magrey M, Walsh JA, Flierl S, et al. The International Map of axial spondyloarthritis survey: a US patient perspective on diagnosis and burden of disease. ACR Open Rheumatol. 2023;5(5):264-276. PubMed
38.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL solution in a single use pre-filled syringe or autoinjector, for subcutaneous injection. Oakville (ON): UCB Canada Inc.; 2023 Oct 19.
39.Drug Reimbursement Review pharmacoeconomic report: ixekizumab (Taltz) for the treatment of adult patients with active ankylosing spondylitis who have responded inadequately to, or are intolerant to conventional therapy. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0630-taltz-pharmacoeconomic-review-report.pdf. Accessed 2023 Dec 19.
Appendix 1: Cost Comparison Table
Note this appendix has not been copy-edited.
The comparators presented in Table 4 have been deemed appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table, and as such the table may not represent the actual costs to public drug plans.
Table 4: CADTH Cost Comparison Table for AS
Treatment | Strength or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Bimekizumab (Bimzelx) | 160 mg/mL | Prefilled syringe or autoinjector | 1,625.0000a | 160 mg every 4 weeks | 58.04 | 21,198 |
TNF alpha inhibitors | ||||||
Adalimumab (Humira) | 40 mg/0.8 mL | Prefilled syringe or pen | 794.1000 | 40 mg every 2 weeks | 56.72 | 20,718 |
SEB: adalimumab (Biosimilars) | 20 mg/0.4 mL 40 mg/0.8 mL | Prefilled syringe or pen | 235.6350 471.2700 | 33.66 | 12,295 | |
Certolizumab pegol (Cimzia) | 200 mg/mL | Single-use prefilled syringe | 664.5100 | Loading dose: 400 mg SC injection at weeks 0, 2, and 4. Maintenance dose: 200 mg every 2 weeks or 400 mg every 4 weeks | First year: 52.92 Subsequent: 47.47 | First year: 19,330 Subsequent: 17,337 |
Etanercept (Enbrel) | 25 mg per vial | Vial | 202.9300 | 50 mg per week | 57.98 | 21,177 |
50 mg/mL | Prefilled syringe or autoinjector | 405.9850 | 58.00 | 21,184 | ||
SEB: etanercept (Erelzi, Brenzys) | 25 mg/0.5 mL | Vial | 120.5000 | 34.43 | 12,575 | |
50 mg/mL | Prefilled syringe or autoinjector | 241.0000 | ||||
Golimumab SC (Simponi) | 50 mg/0.5 mL 100 mg/mL | Prefilled syringe or autoinjector | 1,555.1700 | 50 mg monthly | 51.09 | 18,662 |
Infliximab (Remicade)b | 100 mg per vial | Vial | 987.5600c | 5 mg/kg initial dose followed with additional similar doses at 2 and 6 weeks after the first infusion then every 6 to 8 weeks thereafter | First year: 105.07 to 126.58 Subsequent: 88.18 to 117.57 | First year: 38,378 to 46,233 Subsequent: 32,206 to 42,941 |
SEB Infliximab (Inflectra)b | 100 mg per vial | Vial | 525.0000 | First year: 55.86 to 67.29 Subsequent: 46.88 to 62.50 | First year: 20,402 to 25,578 Subsequent: 17,121 to 22,828 | |
SEB (Infliximab Renflexis/ Avsola) b | 100 mg per vial | Vial | 493.0000 | First year: 52.45 to 63.19 Subsequent: 44.02 58.69 | First year: 19,159 to 23,080 Subsequent: 16,078 to 21,437 | |
IL-17A inhibitors | ||||||
Secukinumab (Cosentyx) | 75 mg/0.5 mL 150 mg/ mL 300 mg/mL | Prefilled syringe or vial | 882.5900 | 150 mg at weeks 0, 1, 2, 3, and 4, then 150 mg to 300 mg monthly | First year: 35.88 to 59.69 Subsequent: 29.00 to 57.99 | First year: 13,107 to 21,800 Subsequent: 10,591 to 21,182 |
Ixekizumab (Taltz) | 80 mg/ mL | Prefilled syringe or pen | 1,723.8900 | 80 mg every 4 weeks | 61.57 | 22,448 |
Janus kinase inhibitors | ||||||
Tofacitinib (Xeljanz) | 5 mg 10 mg | Tablet | 5.9897 21.1718 | 5 mg twice daily | 11.98 | 4,375 |
Upadacitinib (Rinvoq) | 15 mg | Tablet | 51.6810 | 15 mg once daily | 51.68 | 18,876 |
Conventional synthetic DMARDs | ||||||
Methotrexate (generics) | 2.5 mg 10 mg | Tablet | 0.5013 2.7983d | 10 to 25 mg per week until adequate response is achieved | 0.4 to 1.27 | 146 to 465 |
20 mg/2 mL 50 mg/2 mL | Vial | 12.5000 8.9200 | ||||
Leflunomide (generics) | 10 mg 20 mg | Tablet | 2.0000 | Loading: 100 mg daily for 3 days Maintenance: 20 mg | First year: 2.07 Subsequent: 2.00 | First year: 755 Subsequent: 731 |
Sulfasalazine (generics) | 500 mg | Tablet | 0.2533 | Week 1: 500 mg daily Week 2: 1,000 mg daily Week 3: 1,500 mg daily Maintenance: 2,000 mg daily | First year: 0.98 Subsequent: 1.01 | First year: 359 Subsequent: 370 |
500 mg | EC tablet | 0.3863 | First year: 1.50 Subsequent: 1.55 | First year: 370 Subsequent year: 564 | ||
Hydroxy-chloroquine (generics) | 200 mg | Tablet | 0.1576 | 400 to 600 mg daily until a good response is obtained (usually 4 to 12 weeks), then dosage reduced by 50% and continued at 200 to 400 mg daily | First year: 0.17 to 0.46 Subsequent: 0.16 to 0.32 | First year: 61 to 168 Subsequent: 58 to 115 |
DMARD = disease-modifying antirheumatic drug; EC = enteric coated; IL-17 = interleukin 17; SEB = subsequent entry biologic; TNF = tumour necrosis factor.
Note: Recommending dosing informed by respective product monographs unless otherwise stated.20-32 All prices are from the Ontario Drug Benefit Formulary (accessed November 2023), unless otherwise indicated, and do not include dispensing fees. Annual cost is based on 365.25 days per year. All weight-based doses assume an average patient weight of 85 kg and wastage of excess medication in vials.
aSponsor-submitted price.1
bInfliximab costs calculated assuming an 8-week period after the initial 3 doses.
cOntario Drug Benefit Exceptional Access Program (accessed November 2023).33
dSaskatchewan Formulary (accessed November 2023).34
Appendix 2: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
Figure 1: Model Structure (Decision Tree)
axSpA = axial spondyloarthritis; BSC = best supportive care; BKZ = bimekizumab; BL1 = first b/tsDMARD received in the model; BL2 = next b/tsDMARD received in the model; b/ts = biologic/targeted synthetic; Comp = comparator; DMARD = disease-modifying antirheumatic drug; maint = maintenance; QALY = quality-adjusted life-years = SC: standard care; wk = week.
Source: Sponsor’s pharmacoeconomic report.1
Figure 2: Model Structure (Markov Model)
axSpA = Axial spondyloarthritis; BL1 = First b/tsDMARD received in the model; BL2 = Second b/tsDMARD received in the model; BSC = Best supportive care
Source: Sponsor’s pharmacoeconomic report.1
Appendix 3: Submitted Budget Impact Analysis (BIA) and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 5: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA to estimate the three-year budget impact of reimbursing bimekizumab for the treatment of adult patients with AS. The analysis was taken from the perspective of the Canadian public drug plans. A three-year time horizon was used from 2025 to 2027, with 2024 as the base year. The target population size was derived with an epidemiological approach. Key inputs to the BIA are documented in Table 6.
State the key assumptions:
Proportion of adult patients with active AS is 78.9%
Proportion of patients with AS requiring biologic/advanced therapy is 45%
Proportion of existing cases in an induction year is 11%
53% of patients with AS would be inadequate responders and require a higher secukinumab dose
All CADTH-participating jurisdictions have implemented or will implement a biosimilar switching policy; therefore, patients in an induction year receive the biosimilar if available.
Table 6: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) | |
|---|---|---|
Target population | ||
AS prevalence AS incidence % of adult patients with active AS % of adult patients with AS requiring biologic or advanced therapy % eligible for public coverage % existing cases in an induction year | 0.213%35 0.015%36 78.9%37 45.0%38 45.0%38 11.0%39 | |
Number of patients eligible for the drug under review | 10,907 / 11,035 / 11,163 | |
Market uptake (3 years) | First year (induction) | Subsequent years (maintenance) |
Uptake (reference scenario) Bimekizumab (Bimzelx) Certolizumab (Cimzia) Etanercept (Enbrel) Etanercept biosimilars Adalimumab (Humira) Adalimumab biosimilars Infliximab (Remicade) Infliximab biosimilars Golimumab (Simponi) Secukinumab (Cosentyx) Ixekizumab (Taltz) Upadacitinib (Rinvoq) Tofacitinib (Xeljanz) | 0.00% / 0.00% / 0.00% 6.52% / 6.41% / 6.30% 0.00% / 0.00% / 0.00% 18.78% / 18.64% / 18.50% 0.00% / 0.00% / 0.00% 39.89% / 39.69% / 39.50% 0.00% / 0.00% / 0.00% 5.38% / 5.19% / 5.00% 18.18% / 18.09% / 18.00% 10.09% / 10.14% / 10.20% 0.22% / 0.26% / 0.30% 0.82% / 1.41% / 2.00% 0.13% / 0.16% / 0.20% | 0.00% / 0.00% / 0.00% 6.52% / 6.41% / 6.30% 4.20% / 3.85% / 3.50% 14.57% / 14.79% / 15.00% 10.18% / 9.59% / 9.00% 29.71% / 30.10% / 30.50% 1.50% / 1.25% / 1.00% 3.88% / 3.94% / 4.00% 18.18% / 18.09% / 18.00% 10.09% / 10.14% / 10.20% 0.22% / 0.26% / 0.30% 0.82% / 1.41% / 2.00% 0.13% / 0.16% / 0.20% |
Uptake (new-drug scenario) Bimekizumab (Bimzelx) Certolizumab (Cimzia) Etanercept (Enbrel) Etanercept biosimilars Adalimumab (Humira) Adalimumab biosimilars Infliximab (Remicade) Infliximab biosimilars Golimumab (Simponi) Secukinumab (Cosentyx) Ixekizumab (Taltz) Upadacitinib (Rinvoq) Tofacitinib (Xeljanz) | 2.00% / 4.00% / 6.00% 6.39% / 6.15% / 5.92% 0.00% / 0.00% / 0.00% 18.40% / 17.89% / 17.39% 0.00% / 0.00% / 0.00% 39.09% / 38.11% / 37.13% 0.00% / 0.00% / 0.00% 5.27% / 4.98% / 4.70% 17.82% / 17.37% / 16.92% 9.89% / 9.74% / 9.59% 0.22% / 0.25% / 0.28% 0.80% / 1.35% / 1.88% 0.13% / 0.16% / 0.19% | 2.00% / 4.00% / 6.00% 6.39% / 6.15% / 5.92% 4.12% / 3.70% / 3.29% 14.28% / 14.19% / 14.10% 9.98% / 9.21% / 8.46% 29.12% / 28.90% 28.67% 1.47% / 1.20% / 0.94% 3.80% / 3.78% / 3.76% 17.82% / 17.37% / 16.92% 9.89% / 9.74% / 9.59% 0.22% / 0.25% / 0.28% 0.80% / 1.35% / 1.88% 0.13% / 0.16% / 0.19% |
Annual cost of treatment per patient (induction year / maintenance year) | ||
Bimekizumab (Bimzelx) Certolizumab (Cimzia) Etanercept (Enbrel) Etanercept biosimilars Adalimumab (Humira) Adalimumab biosimilars Infliximab (Remicade) Infliximab biosimilars Golimumab (Simponi) Secukinumab (Cosentyx) Ixekizumab (Taltz) Upadacitinib (Rinvoq) Tofacitinib (Xeljanz) | $21,198 $19,935 / $17,337 $21,184 $12,575 $20,718 $12,295 $33,775 / $24,476 $16,861 / $12,219 $20,287 $20,624 / $16,151 $22,488 $18,876 $4,375 | |
AS = ankylosing spondylitis.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analysis suggest that the reimbursement of bimekizumab for adults with AS will be associated with an incremental cost of −$796,372 in year 1, $442,308 in year 2, and $1,731,697 in year 3. Therefore, the 3-year total incremental budget impact is $1,377,633.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Total number of eligible patients was inaccurately estimated. The sponsor’s BIA used population numbers informed by Statistics Canada and Non-Insured Health Benefit (NIHB) annual reports to inform the pan-Canadian starting population. However, the values used did not appropriately limit the population to individuals aged 18 years and older and therefore did not reflect the Health Canada indication for bimekizumab, which is limited to adults with active AS.
CADTH considered only the adult population in its reanalysis.
The NIHB population was inappropriately calculated. The sponsor calculated the total population of CADTH-participating drug plans by adding the population of the provinces, excluding Quebec, to the population of NIHB clients. NIHB clients living within the borders of a province are counted within provincial population data as reported by Statistics Canada, thus the NIHB population was double counted in the sponsor’s analysis.
CADTH did not adjust for this limitation in reanalysis. The impact on pan-Canadian model results is expected to be minimal.
Population size is uncertain. The sponsor’s base-case analysis was conducted based on adult patients with AS, aligned with the Health Canada indication. However, as the BE MOBILE 2 trial was restricted to patients with moderate to severe AS only, clinical expert feedback received by CADTH noted that bimekizumab may only be used in patients with moderate to severe AS which is approximately 50% of adult patients in clinical practice.
CADTH conducted a scenario analysis where the patient population was aligned with the trial population (i.e., adult patients with active moderate to severe AS).
The proportion of adult patients with AS requiring biologic/advanced therapies is uncertain. The sponsor estimated that 45% of adult patients with AS require biologic or advanced therapy, informed by clinician feedback obtained by the sponsor. Clinical expert feedback received by CADTH noted that 45% may be on the lower end of the proportion of patients who required biologic or advanced therapies and estimated that it may be closer to 50%.
CADTH conducted a scenario analysis assuming that 50% of adult patients with psoriatic arthritis require biologic or advanced therapy.
CADTH Reanalyses of the BIA
Table 7: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Jurisdiction population corrections | Populations for all jurisdictions using the total population | Updated values to only consider the 18+ population as informed by Statistics Canada and the NIHB annual reports |
CADTH base case | Reanalysis 1 | |
NIHB = Non-Insured Health Benefit.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 8 and a more detailed breakdown is presented in Table 9. In the CADTH base case, the estimated 3-year incremental budget impact of reimbursing bimekizumab is expected to be $1,464,006 (−$533,456 in year 1, $473,163 in year 2, and $1,524,299 in year 3).
Table 8: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 1,377,633 |
CADTH reanalysis 1 | 1,464,006 |
CADTH base case | 1,464,006 |
BIA = budget impact analysis.
CADTH conducted the following scenario analyses. Results are provided in Table 9.
Assumed bimekizumab reimbursement is restricted to adult patients with moderate to severe AS only where 50% of cases are moderate to severe AS.
Assumed 50% of patients required biologic/advanced therapies.
Table 9: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 171,987,816 | 173,332,546 | 174,660,930 | 175,972,971 | 523,966,447 |
New drug | 171,987,816 | 172,536,174 | 175,103,238 | 177,704,668 | 525,344,079 | |
Budget impact | 0 | −796,372 | 442,308 | 1,731,697 | 1,377,633 | |
CADTH base case | Reference | 139,918,439 | 141,311,426 | 142,688,712 | 144,050,298 | 428,050,437 |
New drug | 139,918,439 | 140,777,970 | 143,161,875 | 145,574,597 | 429,514,442 | |
Budget impact | 0 | −533,456 | 473,163 | 1,524,299 | 1,464,006 | |
CADTH scenario analysis 1: moderate to severe AS | Reference | 85,993,908 | 86,666,273 | 87,330,465 | 87,986,485 | 261,983,223 |
New drug | 85,993,908 | 86,581,492 | 87,857,052 | 89,146,544 | 263,585,087 | |
Budget impact | 0 | −84,781 | 526,586 | 1,160,058 | 1,601,864 | |
CADTH scenario analysis 2: 50% biologic/advanced therapy | Reference | 191,097,573 | 192,591,717 | 194,067,701 | 195,525,523 | 582,184,941 |
New drug | 191,097,573 | 191,637,214 | 194,491,280 | 197,384,251 | 583,512,744 | |
Budget impact | 0 | −954,503 | 423,579 | 1,858,728 | 1,327,804 |
BIA = budget impact analysis.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.