CADTH Reimbursement Review
Etranacogene Dezaparvovec (Hemgenix)
Sponsor: CSL Behring Canada Inc.
Therapeutic area: Hemophilia B
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Clinical Review
Abbreviations
AAV
adeno-associated virus
AAV5
adeno-associated virus serotype 5
ABR
annualized bleeding rate
AE
adverse event
AHCDC
Association of Hemophilia Clinic Directors of Canada
AIR
annualized infusion rate
ALT
alanine transaminase
AST
aspartate aminotransferase
BMI
body mass index
CFC
coagulation factor concentrate
CHS
Canadian Hemophilia Society
CI
confidence interval
EHL
extended half-life
ESS
effective sample size
FIX
factor IX
gc
genome copy
GRADE
Grading of Recommendations Assessment, Development and Evaluation
Haem-A-QoL
Haemophilia Quality of Life Questionnaire for Adults
HJHS
Hemophilia Joint Health Score
HRQoL
health-related quality of life
IPTW
inverse probability of treatment weighting
ITC
indirect treatment comparison
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
nAb
neutralizing antibody
PROBE
Patient Reported Outcomes Burdens and Experiences
QoL
quality of life
rFIXFc
recombinant factor IX Fc fusion protein
rFIX-FP
recombinant factor IX albumin fusion protein
RR
relative risk
SD
standard deviation
SHL
standard half-life
SLR
systematic literature review
SMD
standardized mean difference
TEAE
treatment-emergent adverse event
TESAE
treatment-emergent serious adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information for the Application Submitted for Review
Item | Description |
|---|---|
Drug product | Etranacogene dezaparvovec (Hemgenix), 1 × 1013 gc/mL, suspension for IV infusion |
Sponsor | CSL Behring Canada Inc. |
Indication | For treatment of adults (aged 18 years or older) with hemophilia B (congenital factor IX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | October 23, 2023 |
Recommended dose | A single dose of 2 × 1013 gc/kg of body weight after dilution with 0.9% sodium chloride solution (normal saline) |
FIX = factor IX; gc = genome copy; NOC = Notice of Compliance.
Sources: Sponsor’s summary of clinical evidence1 and etranacogene dezaparvovec product monograph.2
Introduction
Hemophilia is a serious X chromosome–linked, lifelong genetic disorder that leaves patients vulnerable to blood loss and organ damage due to impaired functioning of the coagulation cascade.3,4 Hemophilia B is the second-most common type of hemophilia (after hemophilia A) and is characterized by an absence or shortage of coagulation factor IX (FIX) resulting from a mutation in the F9 gene.3,4 An FIX deficiency in hemophilia B prevents or reduces the ability of the coagulation cascade to produce fibrin.5 The severity of hemophilia B generally correlates with the degree of clotting factor deficiency.6 Patients with moderate and severe hemophilia B are defined by the World Federation of Hemophilia as having 1% to 5% and less than 1% of normal enzymatic FIX activity, respectively.6 However, according to the clinical experts consulted by CADTH, severity in clinical practice is defined by the patient’s phenotype (i.e., tendency to bleed) and not simply their factor activity levels; the decision to initiate prophylaxis with clotting factor concentrates takes into account their clinical phenotype and factor activity levels as well as lifestyle and professional activities. As of 2021, there were 704 patients with hemophilia B (with recorded severity) in Canada, 535 of whom were adult male patients. Of the adult male patients, 218 had moderate and 145 had severe hemophilia B.7 The estimated prevalence at birth per 100,000 males in Canada from 1991 to 2015 was 3.9 for all severities of hemophilia B and 1.3 for severe disease only.8
Current treatment strategies for hemophilia B are based on replacing the missing factor and can be done either as needed when bleeding episodes occur (on-demand therapy) or in a preventive manner (prophylaxis). FIX prophylaxis can be administered regularly, with the aim of keeping plasmatic FIX levels above a certain threshold (regular prophylaxis) or, occasionally, to increase plasmatic FIX levels in high-risk situations, such as during physical activity (situational prophylaxis). The goal of prophylaxis is to prevent bleeding in patients with hemophilia while allowing them to live an active life and achieve a quality of life comparable to that of people without hemophilia.9 According to the clinical experts consulted by CADTH, FIX prophylaxis therapy is the preferred management approach for patients with moderately severe or severe hemophilia.
Both recombinant FIX and plasma-derived FIX are used for FIX prophylactic therapy.6 Recombinant FIX products are manufactured using genetically engineered cells and recombinant technology, while plasma-derived FIX products are made from plasma donated by human blood donors in Canada. One plasma-derived product (Immunine) is reimbursed; however, recombinant FIX products are predominantly used to treat patients with hemophilia B.10
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of etranacogene dezaparvovec (Hemgenix) (1 × 1013 genome copies [gc]/mL, suspension for IV infusion) for the treatment of adults (aged 18 years or older) with hemophilia B (congenital FIX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes.
Stakeholder Perspectives
The information in this section is a summary of the input provided by the patient and clinician groups that responded to CADTH’s call for input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Patient input was gathered by the Canadian Hemophilia Society (CHS) from an online survey conducted from July 10 to 31, 2023. In total, 17 responses were gathered by the CHS. All respondents were affected by severe or moderately severe hemophilia B without inhibitors. In addition, in September 2022, the CHS conducted an online survey of people in Canada with severe hemophilia A and B. The CHS received 39 responses, including 31 with hemophilia A, 7 with hemophilia B, and 1 not specified.
Joint damage, primarily to knees, ankles, and elbows, caused by repeated internal hemarthroses, was reported to be the primary physical health impact of hemophilia B. Regarding currently available treatments, 5 patients who responded to the 2023 CHS survey reported being very satisfied, 17 said they were satisfied, 7 said they were neither satisfied nor dissatisfied, and 1 indicated they were very dissatisfied. The patients from this survey noted that treatments greatly complicate their everyday life, travel, and leisure activities. They also mentioned difficulty in receiving infusion treatments due to problems with vein visibility, issues with poor veins, and side effects. Patients also reported facing socioeconomic problems caused by, for example, having to miss work due to the need for regular visits to the hospital, travel and insurance issues, and access issues.
Patients hope gene therapy will lead to fewer FIX infusions, minimal needle injections, less stress, less bleeding, and fewer restrictions on activities, and make it easier to travel. In addition, about 63% of the respondents from the 2022 survey indicated they expect gene therapy to be effective in preventing bleeding for at least 10 years. The 2022 survey asked if people would choose to receive gene therapy knowing there would be frequent blood draws in the weeks and months following administration, and they would need to be followed up in a registry for 10 to 20 years. In response, 66% answered yes, 10% answered no, and 24% indicated they did not know.
The CHS mentioned that a small number of people in Canada (approximately 5) have undergone gene therapy for hemophilia B, but the organization knows nothing about the experience of these patients outside the preliminary data from the trials.
Clinician Input
Input From Clinical Experts Consulted by CADTH
According to the clinical experts consulted by CADTH, there are several unmet needs for hemophilia B. First, people with hemophilia B have a life disadvantage and quality of life disadvantage compared with the general population because there is no treatment available to reverse the course of disease. In addition, a therapy is needed that reduces the burden of treatment (e.g., need for recurrent IV injections, delayed or missed doses, and overall suboptimal treatment due to poor venous access and other difficulties related to the preparation for the FIX regimen) and improves adherence.
The clinical experts consulted by CADTH noted that the current standard of care in Canada for hemophilia B is IV replacement therapy with the missing clotting factor (i.e., FIX) and, unlike hemophilia A, there are currently no approved subcutaneous nonfactor therapies for patients with hemophilia B. The clinical experts noted that etranacogene dezaparvovec is a gene therapy for hemophilia B that would provide a potential curative option (i.e., a long-term phenotypic cure) by addressing the underlying disease process, which may represent a shift in the current treatment paradigm.
The clinical experts consulted by CADTH noted it is conceivable to give priority to those patients who have a severe bleeding phenotype, have difficult venous access and/or experience a high treatment burden with FIX prophylaxis, have recurrent bleeds despite prophylaxis or have difficulty being adherent to a prophylaxis regimen, or need to have sustained FIX levels because of comorbidities (e.g., joint disease or cardiovascular issues that require antiplatelets or anticoagulants). Eligible patients should meet criteria on levels of neutralizing antibodies (nAbs) against FIX and adeno-associated virus (AAV). The clinician would also complete an assessment of the patient’s eligibility based on their clinical judgment and lab tests (e.g., complete blood count and differential, liver and kidney functions, FIX activity, and FIX inhibitor status). The other tests that would be required would be for infectious diseases, including HIV, hepatitis B, and hepatitis C. According to the clinical experts consulted by CADTH, etranacogene dezaparvovec should not be given to pediatric patients with hemophilia B (aged < 18 years), while there is no concern using etranacogene dezaparvovec in patients aged 65 years and older with hemophilia B.15
The clinical experts consulted by CADTH noted that the most important assessment for treatment response is to monitor patients’ bleeding to observe whether etranacogene dezaparvovec prevents bleeding events and allows patients to live the lifestyle they want without concern for risk of bleeding. The clinical experts consulted by CADTH noted that FIX activity level may also be monitored for assessing response to treatment, which could allow clinicians to determine the degree to which the deficiency in FIX has been corrected by etranacogene dezaparvovec. The clinical experts consulted by CADTH noted that follow-up should focus on both efficacy and safety through clinical follow-ups (e.g., checking patients’ bleeding events and joint status via phone, virtual, or in-person check-ups) and lab tests (e.g., liver enzymes, FIX activity levels, liver ultrasound to detect hepatocarcinomas). The length of follow-up for hepatic function and FIX activity levels post infusion of etranacogene dezaparvovec should be lifelong.
To define treatment failure of etranacogene dezaparvovec, the clinical experts consulted by CADTH noted that the composite of FIX level (e.g., patient’s baseline FIX level before receiving etranacogene dezaparvovec) and return to prophylaxis with hemostatic therapy (e.g., return to regular administration of prohemostatic products to prevent any bleeding episode for at least 6 months per year) could be used to determine whether a treatment failure has occurred in patients treated with etranacogene dezaparvovec. According to the clinical experts consulted by CADTH, etranacogene dezaparvovec should be prescribed based on the judgment of a multidisciplinary team (e.g., consisting of specialists such as a hematologist with experience in treating patients with hemophilia, a physiotherapist to assess joint function, a hepatologist for liver-related issues, pharmacy support, and an HIV specialist if the patient is HIV positive), and can be administered in a specialty clinic in the outpatient setting, with longitudinal follow-up.
Clinician Group Input
A total of 8 clinicians from the Association of Hemophilia Clinic Directors of Canada (AHCDC) provided input for the CADTH review of etranacogene dezaparvovec. The AHCDC highlighted some unmet needs for patients with hemophilia with the severe bleeding phenotype and, specifically, hemophilia B. The AHCDC mentioned that with currently available treatments in Canada, patients with hemophilia B are dependent on regular IV infusions of FIX to prevent and treat bleeding for their whole life. In addition, the AHCDC noted there can be a major challenge with frequent venipuncture for routine prophylaxis for patients with poor venous access, as well as long-term complications with the placement of a central venous catheter, including risks of infection, bleeding, thromboembolism, and loss of function, requiring catheter removal.
The AHCDC noted that gene therapy provides the possibility of a long-term phenotypic cure for patients with hemophilia B. If effective, the new treatment option could provide a 1-time treatment leading to sustained FIX production. This may represent a paradigm shift in the treatment of hemophilia B. The AHCDC also mentioned that in contrast to patients with hemophilia A, who have the option of emicizumab, patients with hemophilia B have no current alternatives to coagulation factor concentrates (CFCs) outside of clinical trials, making the need for gene therapy greater for patients with hemophilia B.
The AHCDC indicated that eligible candidates for the gene therapy under review include those with a clinically severe bleeding phenotype requiring prophylaxis, no history of inhibitory antibodies, no significant comorbidities, and an anti-AAV nAb titre of less than 1:900. The group also added that patients with hemophilia who are not currently receiving prophylactic therapy (e.g., due to poor venous access or adherence issues with routine prophylaxis), but who experience repeated, serious spontaneous bleeding episodes, or have a history of life-threatening hemorrhage, are also candidates for gene therapy.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH Reimbursement Review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for etranacogene dezaparvovec:
relevant comparators
consideration for initiation of therapy
consideration of continuation or renewal of therapy
consideration for prescribing of therapy
generalizability
care provision issues
system and economic issues.
Clinical Evidence
Systematic Review
Description of Studies
One phase III, single-arm, open-label clinical trial, HOPE-B (N = 54), was included in the systematic literature review (SLR) conducted by the sponsor. HOPE-B consisted of a screening phase, lead-in phase, etranacogene dezaparvovec infusion phase, and posttreatment follow-up phase. The HOPE-B study included 67 adult male patients with moderately severe to severe hemophilia B (defined as a normal circulation FIX of ≤ 2%) who had been on a stable prophylaxis regimen for at least 2 months before screening. The patients were enrolled into the lead-in phase, during which they received continuous FIX prophylaxis and were followed up for at least 6 months (i.e., 26 weeks). Those with a history of FIX inhibitors or who tested positive for FIX inhibitors during the screening period and at the last visit of the lead-in period for the HOPE-B trial were excluded. Pre-existing nAbs against adeno-associated virus serotype 5 (AAV5) was not used as an exclusion criterion in the HOPE-B study. Thirteen patients discontinued or were excluded during the lead-in phase. A total of 54 patients from 33 study sites globally received etranacogene dezaparvovec and were followed for efficacy and safety.
The primary objective of the HOPE-B trial was to demonstrate the noninferiority of etranacogene dezaparvovec to reduce the annualized bleeding rate (ABR) for all bleeding events between month 7 and month 18 post infusion, compared with continuous routine FIX prophylaxis. Other efficacy end points included proportion of patients with no bleeds, ABR for spontaneous bleeds, ABR for joint bleeds, annualized infusion rate (AIR) of FIX replacement therapy, annualized consumption of FIX replacement therapy, Hemophilia Joint Health Score (HJHS), and Patient Reported Outcomes Burdens and Experiences (PROBE). Safety outcomes such as treatment-emergent adverse events (TEAEs), treatment-emergent serious adverse event (TESAEs), withdrawals due to adverse events (AEs), mortality, and notable harms (e.g., alanine transaminase [ALT] increased, aspartate aminotransferase [AST] increased) were also reported. Data collected during the lead-in phase served as comparison against etranacogene dezaparvovec for some safety and efficacy outcomes (e.g., ABR for all bleeding events, ABR for spontaneous bleeds, ABR for joint bleeds, AIR, and annualized FIX consumption).
In the 54 patients who received etranacogene dezaparvovec, the majority were white (74.1%) and had a mean age of 41.5 years (standard deviation [SD] = 15.8). Twenty-one patients (38.9%) had pre-existing nAbs against AAV5 before infusion of etranacogene dezaparvovec. The last testing before infusion of etranacogene dezaparvovec showed that 21 of the 54 patients (38.9%) had an anti-AAV5 nAb titre between 1:9 and 1:3,212 (median = 1:56.9). Excluding 1 patient with an anti-AAV5 titre greater than 1:3,000 (i.e., 1:3,212), the remaining 20 patients had a titre of between 1:9 and 1:678 (median = 1:49.1). There were 33 patients (62.3%) with an anti-AAV5 nAb titre below the lower limit of detection (i.e., 1:7).
The HOPE-B study is ongoing and expected to be completed in 2025. A postdose analysis at up to 36 months (data cut-off date of June 6, 2023) was used to support the sponsor’s present submission to CADTH.
Efficacy Results
Bleeding Outcomes
From month 7 to month 18 post etranacogene dezaparvovec infusion, 34 of the 54 patients (63.0%) treated with etranacogene dezaparvovec had no bleeds, compared with 14 of the same 54 patients (25.9%) who received FIX prophylaxis during the lead-in phase. The adjusted mean difference in ABR for all bleeding events between etranacogene dezaparvovec and routine FIX prophylaxis was −2.68 (95% confidence interval [CI], −3.81 to −1.55) from month 7 to month 18 post etranacogene dezaparvovec infusion, favouring etranacogene dezaparvovec. From month 7 to month 36 post infusion, 23 of the 54 patients (42.6%) treated with etranacogene dezaparvovec had no bleeds compared with 14 of the same 54 patients (25.9%) who received FIX prophylaxis during the lead-in phase. The adjusted mean difference in ABR for all bleeding events from month 7 to month 36 was −2.65 (95% CI, −3.83 to −1.47) in favour of etranacogene dezaparvovec.
The adjusted mean difference in ABR for spontaneous bleeds between etranacogene dezaparvovec and routine FIX prophylaxis was −1.08 (95% CI, −1.72 to −0.44) from month 7 to month 18 post etranacogene dezaparvovec infusion in favour of etranacogene dezaparvovec. The adjusted mean difference in ABR for spontaneous bleeds from month 7 to month 36 was −0.93 (95% CI, −1.62 to −0.25), in favour of etranacogene dezaparvovec.
The adjusted mean difference in ABR for joint bleeds between etranacogene dezaparvovec and routine FIX prophylaxis was −1.84 (95% CI, −2.54 to −1.13) from month 7 to month 18 post infusion in favour of etranacogene dezaparvovec. The adjusted mean difference in ABR for joint bleeds from month 7 to month 36 was −1.87 (95% CI,−2.54 to −1.20), favouring etranacogene dezaparvovec.
Use of FIX After Etranacogene Dezaparvovec Infusion
From month 7 to month 18 post etranacogene dezaparvovec infusion, the adjusted mean difference in AIR between etranacogene dezaparvovec and routine FIX prophylaxis was −69.96 (95% CI, −79.77 to −60.16), which favoured etranacogene dezaparvovec. Similarly, from month 7 to month 36 post infusion, the adjusted mean difference in AIR was −69.89 (95% CI, −79.70 to −60.08), which favoured etranacogene dezaparvovec. From month 7 to month 36 post etranacogene dezaparvovec infusion, the adjusted mean difference in annualized consumption of FIX replacement therapy between etranacogene dezaparvovec and routine FIX prophylaxis was −3,037.6 IU/kg (95% CI, −3,617.4 to −2,457.9) in favour of etranacogene dezaparvovec.
Hemophilia Joint Health Score
The mean change from baseline in HJHS score was −1.6 at month 12 (SD = 5.1), −2.6 at month 24 (SD = 5.0), and −3.0 at month 36 (SD = 7.4) post etranacogene dezaparvovec infusion. All patients treated with etranacogene dezaparvovec showed improvements in HJHS total score.
Patient Reported Outcomes Burdens and Experiences Score
Change from baseline at month 12 (mean = 0.040; SD = 0.097) and at month 24 (mean = 0.034; SD = 0.113) post infusion of etranacogene dezaparvovec both showed improvements in the PROBE summary score in patients treated with etranacogene dezaparvovec. Data from month 36 were not available.
Harms Results
The data cut-off date for harm results was June 6, 2023 (i.e., 36-month data cut-off); the harms results at the 24-month data cut-off were generally consistent.
At 36 months post infusion of etranacogene dezaparvovec, all patients had at least 1 TEAE. The system organ classes with the highest incidence of reported TEAEs were infections and infestations (87.0%), followed by musculoskeletal and connective tissue disorders (72.2%), and general disorders and administration-site conditions (59.3%). The TEAEs that were reported in more than 20% of the safety population in the HOPE-B trial were arthralgia (44.4%), headache (33.3%), nasopharyngitis (27.8%), fatigue (27.8%), ALT increased (24.1%), and back pain (22.2%). During the lead-in period (excluding discontinuers), 68.5% patients experienced at least 1 TEAE. The system organ classes with the highest incidence of reported TEAEs were infections and infestations (35.2%), followed by musculoskeletal and connective tissue disorders (22.2%) and gastrointestinal disorders (13.0%). The only AE reported in more than 10% of patients was nasopharyngitis (14.8%).
At 36 months post infusion of etranacogene dezaparvovec, 27.8% of the safety population had at least 1 TESAE. The system organ classes with the most frequently reported TESAEs were infections and infestations (7.4%, consisting of 5 events: infected biloma, COVID-19, cellulitis, device-related infection, and diverticulitis intestinal hemorrhagic) and musculoskeletal and connective tissue disorders (5.6%, consisting of 3 events: hemarthrosis, musculoskeletal chest pain, and osteoarthritis). During the lead-in period (excluding discontinuers), 7.4% of the patients experienced TESAEs, of which 5.6% were reported in the system organ classes of musculoskeletal and connective tissue disorders.
One patient discontinued infusion of the study drug due to an event of hypersensitivity after approximately 10% of the full dose of the study drug had been administered; this patient did not have FIX expression. One patient died due to a fatal event of cardiogenic shock 464 days (approximately 15 months) post infusion of etranacogene dezaparvovec. According to the product monograph,2 the patient, who had numerous cardiovascular and urologic risk factors and age 75 years at screening, died of urosepsis and cardiogenic shock at month 15 post dose (at age 77 years), an event that was determined to be not treatment related.
Post infusion of etranacogene dezaparvovec, an increase in ALT occurred in 24.1% (13 of 54) of the patients, followed by an increase in AST (16.7%, 9 of 54), anemia (9.3%, 5 of 54), and infusion-related reaction (5.6%, 3 of 54). Only 1 patient had anemia during the lead-in period when receiving FIX prophylaxis.
Critical Appraisal
Overall, the trial design of the pivotal HOPE-B trial (i.e., a nonrandomized, open-label, single-arm design) was considered appropriate and acceptable in the field of hemophilia B, although the interpretation of the study findings could be challenging. According to the clinical experts consulted by CADTH, the inclusion and exclusion criteria of the HOPE-B trial were appropriate and reflective of the patients they would have expected in clinical practice. It was noted that 67 patients were enrolled in the lead-in phase and only 54 patients were treated with etranacogene dezaparvovec and assessed for efficacy and safety, although it was determined by CADTH that the potential selection bias due to a considerable number of patients being excluded was low. Due to the single-arm, open-label design, reliable assessments of patient-reported outcomes, such as health-related quality of life (HRQoL) end points, could not be made. In the primary analyses, the documentation of bleeding events in the HOPE-B study relied on the use of an electronic diary (e-diary) by patients, which was also reviewed and assessed by the investigator. Based on details provided by the sponsor upon request, CADTH determined that the potential risk of bias that could lead to the exaggeration of treatment effects of etranacogene dezaparvovec (i.e., ABR outcomes) was likely low. According to the clinical experts consulted by CADTH, there were no serious concerns with the use of corticosteroids post infusion of etranacogene dezaparvovec. The conditions for the use of FIX post infusion of etranacogene dezaparvovec were generally considered appropriate and the definition of “return to routine FIX prophylaxis” in the context of the HOPE-B trial was acceptable. In the HOPE-B study, multiple statistical testing was conducted for several end points in a fixed sequence. However, multiplicity was controlled only for analyses that used data from the month 18 data cut-off and not for analyses that used data from the month 24 or month 36 data cut-offs, which might have resulted in potential inflation of the type I error rates. There were some concerns about the statistical models and assumptions adopted for bleeding outcomes in the HOPE-B study, which may pose challenges in interpreting the magnitude of the effect estimates of etranacogene dezaparvovec compared with FIX prophylaxis.
There are several considerations related to the generalizability of the HOPE-B trial. First, evidence from the currently available follow-up period (i.e., 36 months) in the HOPE-B study may not be adequate to inform long-term efficacy and safety, given the expectation of the long-lasting effects of etranacogene dezaparvovec. In addition, the HOPE-B trial included patients who had congenital hemophilia B with known severe or moderately severe FIX deficiency (≤ 2% of normal circulating FIX) and who had been on stable prophylaxis for at least 2 months before screening. However, the indication is not restricted to patients with severe or moderately severe hemophilia B (≤ 2% of normal circulating FIX), nor does it require eligible patients to have been on a stable FIX prophylaxis regimen for 2 months. According to the clinical experts consulted by CADTH, the eligibility criteria of patients in the HOPE-B study were generally aligned with the indication. However, the clinical experts consulted by CADTH noted that some patients, including those who have an FIX level greater than 2% and present severe clinical symptoms, and those who require but are not receiving stable FIX prophylaxis, may also benefit from etranacogene dezaparvovec.
According to the product monograph for etranacogene dezaparvovec, to be eligible to receive etranacogene dezaparvovec, the titre of pre-existing nAbs against AAV5 should be tested. However, patients enrolled in the pivotal HOPE-B trial were not selected based on their titre value for pre-existing nAbs against AAV5. In correspondence with CADTH, the sponsor claimed that a threshold for an acceptable AAV5 nAb, which is below 1:900, is expected to screen patients for eligibility to receive etranacogene dezaparvovec. According to the sponsor, there was no exclusion criterion in the HOPE-B study regarding the eligibility of patients with anti-AAV5 nAbs. In other words, all patients with detectable pre-existing AAV5 nAbs were enrolled. Regarding how the threshold (1:900) was determined, according to the sponsor, a cut-off at an AAV5 nAb titre of greater than 1:678 was selected based on the highest titre recorded in the subgroup of patients in the HOPE-B study with pre-existing AAV5 nAbs who showed clinically meaningful increases in FIX activity. The 1:678 titre was obtained from an in vitro cell-based assay custom-developed by the sponsor. The sponsor confirmed with Health Canada that the assay method was later validated to extend the linear measuring range with additional dilutions of the samples to be analyzed, with an improved test accuracy, especially at higher titres. The nAb titre value of 678 (rounding off to 1:700), is equivalent to a 9-point nAb titre value of 898 (rounding off to 1:900). The new 1:900 titre value is based on the updated nAb analytical validation assay with an extended linear measuring range (9-point dilution curve assay), versus the investigational clinical study assay at 7-point dilution. This does not represent a change in the concentration of the AAV5 nAb in the serum sample, but rather that the improved assay response curve of the validated method yields a comparatively higher titre.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: ABR for all bleeding events (including percentage of patients without any bleeds), ABR for spontaneous bleeds, ABR for joint bleeds, AIR, annualized FIX consumption, HJHS, PROBE, and harms. According to the GRADE guidance, nonrandomized comparative evidence starts at low certainty and noncomparative evidence starts at very low certainty. The GRADE summary of findings is presented in Table 2 and Table 3.
Table 2: Summary of Findings for Etranacogene Dezaparvovec for Patients With Hemophilia B (Outcomes With Comparative Data)
Outcome and follow-up | Patients (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|
Bleeding outcomes | ||||
ABR for all bleeding events Follow-up:
| N = 54 (1 single-arm study with intrapatient comparison) | Months 7 to 18 post infusion of etranacogene dezaparvovec Number (%) of patients with no bleeds:
Adjusted ABR (95% CI):
Adjusted mean difference in ABR (95% CI):
Months 7 to 36 post infusion of etranacogene dezaparvovec Number (%) of patients without any bleeds:
Adjusted ABR (95% CI):
Adjusted mean difference in ABR (95% CI):
| Lowa | Etranacogene dezaparvovec may result in a decrease in ABR for all bleeding events when compared with FIX prophylaxis. |
ABR for spontaneous bleeds Follow-up:
| N = 54 (1 single-arm study with intrapatient comparison) | Months 7 to 18 post infusion of etranacogene dezaparvovec Adjusted ABR (95% CI):
Adjusted mean difference in ABR (95% CI)
Months 7 to 36 post infusion of etranacogene dezaparvovec Adjusted ABR (95% CI):
Adjusted mean difference in ABR (95% CI):
| Lowa | Etranacogene dezaparvovec may result in a decrease in ABR for spontaneous bleeds when compared with FIX prophylaxis. |
ABR for joint bleeds Follow-up:
| N = 54 (1 single-arm study with intrapatient comparison) | Months 7 to 18 post infusion of etranacogene dezaparvovec Adjusted ABR (95% CI)
Adjusted mean difference in ABR (95% CI)
Months 7 to 36 post infusion of etranacogene dezaparvovec Adjusted ABR (95% CI)
Adjusted mean difference in ABR (95% CI)
| Lowa | Etranacogene dezaparvovec may result in a decrease in ABR for joint bleeds when compared with FIX prophylaxis. |
Use of FIX post infusion of etranacogene dezaparvovec | ||||
AIR Follow-up:
| N = 54 (1 single-arm study with intrapatient comparison) | Months 7 to 18 post infusion of etranacogene dezaparvovec Adjusted AIR (95% CI):
Adjusted mean difference in AIR (95% CI):
Months 7 to 36 post infusion of etranacogene dezaparvovec Adjusted AIR (95% CI):
Adjusted mean difference in AIR (95% CI):
| Lowa | Etranacogene dezaparvovec may result in a decrease in AIR when compared with FIX prophylaxis. |
Annualized FIX consumption (IU/kg) Follow-up:
| N = 54 (1 single-arm study with intrapatient comparison) | Months 7 to 36 post infusion of etranacogene dezaparvovec Adjusted mean difference in annualized FIX consumption (95% CI):
| Lowa | Etranacogene dezaparvovec may result in a decrease in total FIX consumption when compared with FIX prophylaxis. |
Harms | ||||
TESAEs Mortality ALT increased AST increased Anemia Infusion-related reaction Follow-up:
| N = 54 (1 single-arm study, with intrapatient comparison) | TESAEs:
Mortality, n (%):
ALT increased:
AST increased:
Anemia:
Infusion-related reaction increased:
| Very lowb | The evidence is uncertain about the effect of etranacogene dezaparvovec on harms outcomes.c |
ABR = annualized bleeding rate; AIR = annualized infusion rate; ALT = alanine transaminase; AST = aspartate aminotransferase; CI = confidence interval; FIX = factor IX; SD = standard deviation; TESAE = treatment-emergent serious adverse event.
aThe start point for the study design (single arm with comparative data) was low certainty. Risk of bias was not rated down. Although not optimal, the study design adopted by HOPE-B was considered to be of sufficiently low risk of confounding and sampling bias. The differences between patients in the proposed indication and patients in pivotal trial were not considered sufficient by the clinical experts consulted by CADTH to result in important differences in the observed effect. Imprecision was not rated down, as the improvement was considered clinically meaningful by the clinical experts consulted by CADTH.
bThe start point for the study design (single arm with comparative data) was low certainty. Rated down 1 level for imprecision due to the small sample size, although the safety profile was considered acceptable by the clinical experts consulted by CADTH.
cBased on a comparison between harms data from the lead-in period and harms data from post infusion of etranacogene dezaparvovec. The median duration of the lead-in phase was 7.129 months (range, 6.05 to 10.61). The data cut-off date for harm results post infusion of etranacogene dezaparvovec was June 6, 2023 (i.e., 36-month data cut-off).
Sources: HOPE-B Clinical Study Report29 and drug Reimbursement Review sponsor submission.55
Table 3: Summary of Findings for Etranacogene Dezaparvovec for Patients With Hemophilia B (Outcomes Without Comparative Data)
Outcome and follow-up | Patients, N | Effect | Certainty | What happens |
|---|---|---|---|---|
HJHS: 0 (best) to 124 (worst) Follow-up,:
|
(1 single-arm study) | Month 12 post infusion of etranacogene dezaparvovec Mean HJHS score
Month 24 post infusion of etranacogene dezaparvovec Mean HJHS score
Month 36 post infusion of etranacogene dezaparvovec Mean HJHS score
| Very lowa | The evidence is uncertain about the effect of etranacogene dezaparvovec on HJHS. |
PROBE summary score: 0 (worst) to 1 (best) Follow-up:
|
(1 single-arm study) | Month 12 post infusion of etranacogene dezaparvovec
Month 24 post infusion of etranacogene dezaparvovec Mean PROBE summary score
| Very lowb | The evidence is uncertain about the effect of etranacogene dezaparvovec on PROBE summary score. |
HJHS = Hemophilia Joint Health Score; MID = minimal important difference; PROBE = Patient Reported Outcomes Burdens and Experiences; SD = standard deviation.
aIn the absence of a comparator arm, certainty of evidence started at very low. The differences between patients in the proposed indication and patients in pivotal trial were not considered sufficient by the clinical experts consulted by CADTH to result in important differences in the observed effect. There was no MID identified. Imprecision was not rated down as the improvement was considered clinically meaningful by the clinical experts consulted by CADTH.
bIn the absence of a comparator arm, certainty of evidence started at very low. Rated down 1 level for risk of bias due to potential for bias arising from the open-label nature of the study and the subjective nature of the outcome. Indirectness was not rated down, as PROBE is commonly used in Canada. Imprecision was rated down 2 levels due to change from baseline was not considered clinically relevant by the clinical experts consulted by CADTH.
Sources: HOPE-B Clinical Study Report29 and drug Reimbursement Review sponsor submission.55
Indirect Comparisons
Description of Studies
The sponsor submitted an indirect treatment comparison (ITC) report containing a feasibility assessment and analysis of etranacogene dezaparvovec relative to 4 comparator therapies: recombinant FIX albumin fusion protein (rIX-FP, Idelvion), recombinant FIX Fc fusion protein (rFIXFc, Alprolix), pegylated nonacog beta pegol (Rebinyn), and nonacog alfa (BeneFIX), using a previously published SLR to identify studies. No information was provided with respect to the search strategy, data extraction process, or quality assessment of included studies. The sponsor concluded that no connected network of evidence could be established and assessed the feasibility of unanchored comparisons. For the comparison against rIX-FP, the sponsor had patient-level data and adopted an inverse probability of treatment weighting (IPTW) approach. For comparisons against rFIXFc and pegylated nonacog beta pegol, only aggregate-level data were available, and the sponsor opted for an unanchored matching-adjusted indirect comparison (MAIC) approach. Further, for rFIXFc and pegylated nonacog beta pegol, the primary analysis population of interest — patients receiving prophylaxis — limited information was available with respect to clinical outcomes of interest and clinically relevant covariates. Owing to challenges in the reporting of data for nonacog alfa, the sponsor noted that significant limitations may confound any conclusions drawn. Accordingly, the sponsor indicated these results as a sensitivity analysis, and comparisons with nonacog alfa are not summarized in this report.
Efficacy Results
For the comparison against rFIXFc, the ABR among the unadjusted etranacogene dezaparvovec population (ABR = 0.38; N = 51) was lower than patients receiving rFIXFc (ABR = 2.99; N = 32), corresponding to a relative risk (RR) of 0.13 (95% CI, 0.07 to 0.25). When adjusted for ABR, the sponsor reported a similar trend, with the ABR-adjusted MAIC population receiving etranacogene dezaparvovec (ABR = 0.43; effective sample size [ESS] = 28.2) being lower than that of patients receiving rFIXFc (ABR = 2.99; N = 32), corresponding to an RR of 0.14 (95% CI, 0.08 to 0.25). Other efficacy end points were not available in the primary analysis population.
For the comparison against pegylated nonacog beta pegol, the unadjusted ABR for etranacogene dezaparvovec (N = 51) was 0.36, which was lower than for pegylated nonacog beta pegol (N = 17), which had an ABR of 3.33 (RR = 0.11; 95% CI, 0.06 to 0.22). A similar trend was seen following univariable adjustment for prior ABR: the RR for etranacogene dezaparvovec (ESS = 8.5) relative to pegylated nonacog beta pegol (N = 17) was 0.24 (95% CI, 0.07 to 0.82); following univariable adjustment for prior FIX product class, the RR for etranacogene dezaparvovec (ESS = 21) relative to pegylated nonacog beta pegol (N = 17) was 0.10 (95% CI, 0.03 to 0.27). Other efficacy end points were not available in the primary analysis population.
Comparisons against rIX-FP demonstrated a consistent trend in favour of etranacogene dezaparvovec with respect to ABR, ABR for spontaneous bleeds, ABR for joint bleeds, proportion of patients with no bleeds, and FIX utilization.
Harms Results
Harms were not assessed in the ITC.
Critical Appraisal
With respect to indirect treatment efficacy, the sponsor-provided ITC reported favourable comparative efficacy for the available outcomes relative to rIX-FP, pegylated nonacog beta pegol, and rFIXFc. These comparisons should be considered uncertain owing to methodological limitations due to the lack of a common comparator, which necessitated unanchored comparisons. These comparisons rely on strong assumptions of complete reporting and statistical adjustment for all plausible characteristics, which may be effect modifiers or prognostic factors. This assumption cannot be tested, and for the comparison relative to pegylated nonacog beta pegol and rFIXFc, there was a substantial proportion of missing data on key covariates. Accordingly, the results of this ITC are subject to significant uncertainty.
Conclusions
One phase III, single-arm, open-label trial (HOPE-B) investigated the efficacy and safety of etranacogene dezaparvovec in 54 male patients who had moderately severe or severe hemophilia B (defined as a normal circulation FIX of ≤ 2%) and who had been on continuous routine FIX prophylaxis. Compared with the lead-in period when patients received FIX prophylaxis, etranacogene dezaparvovec may result in a decrease in ABR for all bleeding events, ABR for spontaneous bleeds, ABR for joint bleeds, AIR, and annualized FIX consumption post infusion of the gene therapy. The effects observed for all of these outcomes were considered clinically relevant by the clinical experts consulted by CADTH. However, there is uncertainty associated with interpretating the clinical significance of the magnitude of the treatment differences due to limitations such as the nonrandomized comparative design, potential risk of bias in self-recording bleeding events caused by the open-label design, multiplicity was not controlled for in the analyses using the 24 months and 36 months data cut-offs, and potential biases introduced by assumptions in the statistical models used to make the comparisons. The harms profile of etranacogene dezaparvovec during the follow-up period was considered acceptable by the clinical experts consulted by CADTH, despite that more harms events occurred post infusion of etranacogene dezaparvovec than those when patients were receiving FIX prophylaxis during the lead-in period. The harms evidence is limited, given the relatively short follow-up period and small sample size. A key gap in the pivotal trial evidence is that results remain unknown with respect to the long-term efficacy and safety of etranacogene dezaparvovec relative to FIX prophylaxis due to the current duration of follow-up (i.e., 36 months). One ITC provided efficacy data on the estimated effect of etranacogene dezaparvovec relative to rIX-FP (Idelvion), rFIXFc (Alprolix), and pegylated nonacog beta pegol (Rebinyn). No conclusions could be drawn on the relative efficacy from the ITC. The interpretation of the effect magnitude is uncertain and hindered by the lack of available connected evidence and potential confounding due to the lack of reporting of potentially influential prognostic and predictive factors. No safety data were reported in the sponsor-submitted ITC; therefore, no conclusions could be drawn on the comparative safety of etranacogene dezaparvovec versus other products based on this evidence.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of etranacogene dezaparvovec (Hemgenix) 1 × 1013 gc/mL suspension for IV infusion for the treatment of adults (aged 18 years or older) with hemophilia B (congenital FIX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
Hemophilia is a serious X chromosome–linked, lifelong genetic disorder that leaves patients vulnerable to blood loss and organ damage due to impaired functioning of the coagulation cascade.3,4 In general, hemophilia is inherited through an X chromosome with an F8 or F9 gene mutation, while approximately 30% of all cases result from spontaneous mutations.6 Hemophilia B is the second-most common type of hemophilia (after hemophilia A) and is characterized by an absence or shortage of FIX resulting from a mutation in the F9 gene.3,4 FIX is a vital component of the intrinsic coagulation cascade pathway, which is activated in response to vascular endothelium surface damage.11 Once initiated, the enzymes in the coagulation cascade activate in sequence until fibrin, a clot-forming protein, is produced.11,12 A FIX deficiency in hemophilia B prevents or reduces the ability of the coagulation cascade to produce fibrin.5
The severity of hemophilia B generally correlates with the degree of clotting factor deficiency.6 Moderate and severe hemophilia B cases are defined by the World Federation of Hemophilia as having 1% to 5% and less than 1% of normal enzymatic FIX activity, respectively.6 Moderately severe hemophilia has also been defined in previous clinical trials that have investigated treatment with prophylaxis as having a factor level of 1% to 2%.13 However, according to the clinical experts consulted by CADTH, in clinical practice, severity is determined based not only on FIX activity level but also on clinical phenotype. The development of FIX inhibitors is considered the most serious complication in patients with hemophilia B, since it renders current treatments ineffective and is associated with risks of anaphylaxis and nephrotic syndrome.6 The development of FIX inhibitors in hemophilia B is a rare event (1.5% to 3.0% of all patients) but is associated with significant morbidity.14
The clinical manifestations of hemophilia B are easy bruising and episodes of prolonged bleeding from surgery or trauma.4 In patients with moderate or severe hemophilia, spontaneous, serious, and life-threatening internal bleeding into joints, muscles, and vital organs may also occur.4 The frequency of spontaneous bleeding episodes is variable in patients with severe disease but may occur up to 20 or 30 times per year after minor trauma.4,15 The majority of spontaneous bleeds occur in the joints (70% to 80%) and muscles (10% to 20%).6 Less than 5% of bleeds occur in the central nervous system, e.g., intracranial hemorrhage, but these are particularly serious and debilitating, potentially leading to seizures, impaired motor function, or death in up to 20% of cases.6,16,17 Individuals with moderately severe to severe hemophilia frequently experience bleeding events and recurrent spontaneous bleeding into muscle, soft tissue, and joints (hemarthroses) starting in infancy and continuing throughout adulthood.4,18 However, bleeds can occur in any organ, and affected organs can include kidneys, stomach, intestines, and brain.4,6,19 Hemarthrosis is the most common manifestation of moderately severe to severe hemophilia B.4,6
As of 2021, there were 704 patients with hemophilia B (with recorded severity) in Canada, 535 of whom were adult males. Of the adult male patients, 218 had moderate and 145 had severe hemophilia B.7 The mean prevalence per 100,000 males in Canada from 1998 to 2006 was 3.23.20 The estimated prevalence at birth per 100,000 males in Canada from 1991 to 2015 was 3.9 for all severities of hemophilia B and 1.3 for severe disease only.8 According to a Statistics Canada report in 2021, around 30% of patients with hemophilia B had severe disease.21
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
Current treatment strategies for hemophilia B target symptom relief and prevention of permanent tissue damage and are based on replacing the missing coagulation factor, which can be done either as needed when bleeding episodes occur (on-demand therapy) or in a preventive manner (prophylaxis). The goal of prophylaxis is to prevent bleeding in patients with hemophilia while allowing them to live an active life and achieve a quality of life comparable to that of people without hemophilia.9 According to the clinical experts consulted by CADTH, FIX prophylaxis therapy, involving lifelong regular IV administration of FIX, is the preferred management approach for patients with moderately severe or severe hemophilia.
Both recombinant FIX and plasma-derived FIX are used for FIX prophylactic therapy.6 Recombinant FIX products are manufactured using genetically engineered cells and recombinant technology, while plasma-derived FIX products are made from plasma donated by human blood donors in Canada. One plasma-derived product (Immunine) is reimbursed; however, recombinant FIX products are predominantly used to treat patients with hemophilia B.10 The recombinant FIX products approved by Health Canada include rFIXFc (Alprolix), nonacog alfa (BeneFIX), rIX-FP (Idelvion), pegylated nonacog beta pegol (Rebinyn), and recombinant FIX nonacog gamma for injection (Rixubis).22 Among these products, rFIXFc, nonacog alfa, and pegylated nonacog beta pegol are funded by CADTH-participating drug programs; rIX-FP and recombinant FIX nonacog gamma for injection are only available in Quebec.22 Patients have access to FIX prophylaxis therapy through Canadian Blood Services.
Drug Under Review
Key characteristics of etranacogene dezaparvovec compared with other treatments available for adults (aged 18 years or older) with hemophilia B (congenital FIX deficiency) are summarized in Table 4.
Etranacogene dezaparvovec is a gene therapy designed to introduce a copy of the human FIX gene into hepatocytes to address the root cause of hemophilia B disease.2 Etranacogene dezaparvovec consists of a codon-optimized coding DNA sequence of the gain-of-function Padua variant of human FIX, under control of the liver-specific LP1 promoter, encapsulated in a nonreplicating recombinant AAV5.
Etranacogene dezaparvovec is indicated for the treatment of adults (aged 18 years or older) with hemophilia B (congenital FIX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes.23 Etranacogene dezaparvovec must be administered intravenously as a single dose of 2 × 1013 genome copies per kilogram (gc/kg) of body weight after dilution with 0.9% sodium chloride solution (normal saline).23 Following a single IV infusion, etranacogene dezaparvovec preferentially targets liver cells, where the vector DNA resides almost exclusively in episomal form. Subsequent to transduction, etranacogene dezaparvovec directs long-term liver-specific expression of the FIX Padua protein using a liver-specific promoter (LP1). As a result, etranacogene dezaparvovec partially or completely ameliorates the deficiency of circulating FIX procoagulant activity found in patients with hemophilia B, restoring the hemostatic potential and limiting bleeding episodes and the need for exogenous FIX treatment.23
Of note, according to the product monograph for etranacogene dezaparvovec, to be eligible to receive etranacogene dezaparvovec, the titre of pre-existing nAbs against AAV5 should be tested. However, patients enrolled in the pivotal HOPE-B trial were not selected based on a titre of pre-existing nAbs against AAV5. In correspondence with CADTH, the sponsor claimed that a threshold for an acceptable AAV5 nAb, which is below 1:900, is expected to be used to screen patients in clinical practice for eligibility to receive etranacogene dezaparvovec. According to the sponsor, there was no exclusion criterion in the HOPE-B study regarding the eligibility of patients with anti-AAV5 nAbs. In other words, all patients with detectable pre-existing AAV5 nAbs were enrolled. Regarding how the threshold (1:900) was determined, according to the sponsor, a cut-off of greater than 1:678 AAV5 nAbs was selected based on the highest titre recorded in the subgroup of patients in the HOPE-B study with pre-existing AAV5 nAbs who showed clinically meaningful increases in FIX activity. The 1:678 titre was obtained from an in vitro cell-based assay custom-developed by the sponsor. The sponsor confirmed with Health Canada that the assay method was later validated to extend the linear measuring range with additional dilutions of the samples to be analyzed, with an improved test accuracy, especially at higher titres. The nAb titre value of 678 (rounding off to 1:700), is equivalent to a 9-point nAb titre value of 898 (rounding off to 1:900). The new 1:900 titre value is based on the updated nAb analytical validation assay with an extended linear measuring range (9-point dilution curve assay), versus the investigational clinical study assay at 7-point dilution. This does not represent a change in the concentration of the AAV5 nAb in the serum sample, but rather that the improved assay response curve of the validated method yields a comparatively higher titre.
Table 4: Key Characteristics of Etranacogene Dezaparvovec, rFIXFc, Pegylated Nonacog Beta Pegol, Nonacog Alfa, and FIX Concentrate (Human)
Characteristic | Etranacogene dezaparvovec (Hemgenix) | rFIXFc (Alprolix) | Pegylated nonacog beta pegol (Rebinyn) | Nonacog alfa (BeneFIX) | FIX concentrate (human) (Immunine) |
|---|---|---|---|---|---|
Mechanism of action | After transduction, the drug directs the liver-specific expression of the FIX Padua protein using a liver-specific promoter (LP1) that helps to restore circulating FIX procoagulant activity. | Long-acting, fully recombinant, fusion protein comprising human coagulation FIX covalently linked to the Fc domain of human immunoglobulin G1 and produced by recombinant DNA technology. Recombinant FIX, extended half-life (rFIXFc). | The activation peptide, including the 40 kDa polyethylene-glycol moiety, is cleaved off, leaving the native FIX molecule. Recombinant FIX, extended half-life (glycopegylated). | Contains recombinant coagulation FIX, (nonacog alfa). FIX is activated by factor VII or tissue factor complex in the extrinsic pathway as well as FXIa in the intrinsic coagulation pathway. Recombinant FIX, standard half-life (nonacog alfa). | FIX concentrate (human) provides an increase in plasma levels of FIX and can temporarily correct the coagulation defect of patients with FIX deficiency. Plasma-derived FIX, standard half-life. |
Indicationa | Indicated for treatment of adults (aged 18 years or older) with hemophilia B (congenital FIX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes. | Indicated in adults and children with hemophilia B (congenital FIX deficiency or Christmas disease) for:
| Indicated in adults and children with hemophilia B (congenital FIX deficiency or Christmas disease) for:
| Indicated in patients with hemophilia B (congenital FIX deficiency or Christmas disease) for:
| Indicated in adults and children with hemophilia B (congenital FIX deficiency or Christmas disease) for:
|
Route of administrationb | Single IV infusion. | IV over several minutes after reconstitution. | IV bolus injection over several minutes after reconstitution. | IV infusion after reconstitution. | IV infusion. |
Recommended doseb | 2 × 1013 gc/kg of body weight after dilution with 0.9% sodium chloride solution (normal saline). | Treatment with all FIX products requires individualized dosage adjustment. | Treatment with all FIX products requires individualized dosage adjustment. | Treatment with all FIX products requires individualized dosage adjustment. | Treatment with all FIX products requires individualized dosage adjustment. |
Serious adverse effects or safety issues | Very common infusion-related reactions are fever, low blood pressure, chills, fast heartbeat, difficulty breathing, headache, dizziness, reddening of skin (flushing), abdominal pain, hives, chest discomfort. | Thromboembolic complications (e.g., pulmonary embolism, venous thrombosis, and arterial thrombosis). Inhibitors have been reported in previously untreated patients. Allergic-type hypersensitivity reactions are possible, including anaphylactic reactions. | Similar to rFIXFc. | Similar to rFIXFc. | Possibility of thromboembolic complications for patients at risk for thrombosis and/or receiving high-dose therapy. |
AAV = adeno-associated virus; Fc = fusion protein; FIX = factor IX; FXIa = activated factor XI; gc = genome copy; rFIXFc = recombinant factor IX Fc fusion protein.
aHealth Canada–approved indication.
bFor comparators, dose is for prophylaxis in adult patients.
Sources: CSL Behring Canada,23 Sanofi (2021),24 Novo Nordisk (2022),25 Pfizer (2017),26 Takeda Canada Inc.27
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient input received by CADTH has been included in the stakeholder section of this report.
Patient input was gathered by the CHS from an online survey conducted from July 10 to October 12, 2023. A total of 49 responses were gathered by the CHS. All respondents were affected by severe or moderately severe hemophilia B without inhibitors. Prior to this, in September 2022, the CHS conducted an online survey of people in Canada with severe hemophilia A or B. It received 39 responses: 31 patients reported having hemophilia A, 7 had hemophilia B, and 1 respondent did not specify their hemophilia type.
Joint damage, primarily to knees, ankles, and elbows, caused by repeated internal hemarthroses, was reported to be the primary physical health impact of hemophilia B. Regarding the currently available treatments, 5 patients who responded to the 2023 CHIS survey reported being very satisfied, 14 said they were satisfied, 7 said they were neither satisfied nor dissatisfied, and 1 indicated they were very dissatisfied. The patients from this survey noted that treatments greatly complicate their everyday life, travel, and leisure activities. They also mentioned the difficulty in receiving infusion treatments due to problems with vein visibility, issues with poor veins, and side effects. Patients also reported facing socioeconomic problems caused by, for example, having to miss work due to the need for regular visits, travel and insurance issues, and access issues.
When the patients with hemophilia B and caregivers who responded to the July 2023 CHS survey were asked how gene therapy could potentially change their lives, all respondents provided positive feedback. Patients hope gene therapy would lead to fewer FIX infusions, minimal needle injections, less stress, less bleeding, and fewer restrictions on activities, and enable them to travel more easily. In addition, more than 6 out of 10 respondents (63%) from the 2022 survey indicated they expected gene therapy to be effective in preventing bleeding for at least 10 years. The 2022 survey asked whether respondents would choose to receive gene therapy knowing there would be frequent blood draws in the weeks and months following administration, and they would need to be followed up in a registry for 10 to 20 years. In response, 66% answered yes, 10% answered no, and 24% indicated they did not know.
The CHS mentioned that a small number of people in Canada (approximately 5) have undergone gene therapy for hemophilia B, but the organization knows nothing about the experience of these patients outside the preliminary data from the trials.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the review of etranacogene dezaparvovec, a panel of 3 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with this condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
The goal of treatment of hemophilia B, according to the clinical experts consulted by CADTH, is to not only prevent major bleeds and long-term joint damage but also to give patients living with hemophilia B the freedom to participate in physical and social activities without restrictions, pursue the career or job to which they aspire, and provide patients with physical and mental well-being (essentially, to enable them to live a normal life). The clinical experts consulted by CADTH noted that the current standard of care in Canada for hemophilia B is IV replacement therapy with the missing clotting factor (i.e., FIX) and, unlike hemophilia A, there are currently no approved subcutaneous nonfactor therapies for patients with hemophilia B.
According to the clinical experts consulted by CADTH, there are several unmet needs for hemophilia B. First, people with hemophilia B have a life disadvantage and quality of life disadvantage compared with the general population, as there is no treatment available to reverse the course of the disease. In addition, a therapy that reduces the burden of treatment and improves adherence is needed. Although effective in restoring hemostasis, current FIX replacement therapy requires recurrent IV injections, which may result in a high burden of treatment and hinder adherence for people with hemophilia B (e.g., delayed or missed doses and overall suboptimal treatment due to poor venous access and other difficulties related to the preparation for the FIX regimen). Moreover, even with good adherence to treatment, bleeding events still happen in many patients because not all patients with hemophilia B respond to currently available treatments.
Place in Therapy
According to the clinical experts consulted by CADTH, etranacogene dezaparvovec is a gene therapy for hemophilia B that would provide a potential curative option (i.e., a long-term phenotypic cure) by addressing the underlying disease process, which may represent a shift in the current treatment paradigm.
The clinical experts consulted by CADTH noted that given the novel mechanism and the curative intent, etranacogene dezaparvovec should be offered to all patients who are eligible, and it is not necessary to reserve etranacogene dezaparvovec only for people with hemophilia B who are intolerant to other treatments or only if other treatments are contraindicated. According to the clinical experts consulted by CADTH, it is not necessary to recommend that patients try other treatments before initiating treatment with etranacogene dezaparvovec, given that the only current alternative for patients with hemophilia B with no FIX inhibitors is FIX replacement therapy, and given that etranacogene dezaparvovec is indicated for adult patients who would have been treated with FIX replacement therapy for many years. The clinical experts consulted by CADTH noted that patients’ values and preferences and considerations about how to manage health resources should play an important role in the use of etranacogene dezaparvovec, considering that the current efficacy and safety evidence on etranacogene dezaparvovec is limited (e.g., small number of participants in the pivotal HOPE-B trial, uncertainty about the long-term efficacy and safety).
Patient Population
Regarding the patients with hemophilia B who may be best suited for this therapy, the clinical experts consulted by CADTH noted it is conceivable to give priority to those patients who: have a severe bleeding phenotype, have difficult venous access or experience a high treatment burden with FIX prophylaxis, have recurrent bleeds despite prophylaxis or have difficulty being adherent to a prophylaxis regimen, or need to have sustained FIX levels because of comorbidities (e.g., joint disease, cardiovascular issues that require antiplatelets or anticoagulants). In addition to meeting criteria on nAbs against FIX and AAV, the clinical experts consulted by CADTH noted that the ideal patient should be willing and available to comply with requirements, such as the need to attend follow-up visits at the hemophilia treatment centre, allow laboratory monitoring, and abstain from alcohol post infusion.
According to the clinical experts consulted by CADTH, patients suitable for etranacogene dezaparvovec should be identified at a hemophilia treatment centre. The clinician would complete an assessment of eligibility based on their clinical judgment and lab tests (e.g., complete blood count and differential, liver and kidney functions, FIX activity, and FIX inhibitor status). The other tests that would be required would be for infectious diseases, including HIV, hepatitis B, and hepatitis C. The availability of results on pre-existing nAbs against AAV5 vectors is the first step in eligibility assessment, as the only patients who are eligible must have a pre-existing neutralizing AAV5 antibody titre below 1:900. In their assessment, the clinician may include the ABR; the HJHS, utilization of factor replacement therapy, and the annualized number of FIX infusions, the patient’s adherence to the prescribed treatment, the patient’s venous access, and HRQoL outcome measures. Imaging studies, such as abdominal ultrasound and liver ultrasound with elastography (i.e., FibroScan), will be needed to assess liver health before the infusion of this liver-directed gene therapy.
According to the clinical experts consulted by CADTH, etranacogene dezaparvovec should not be given to pediatric patients with hemophilia B (aged < 18 years), while there is no concern using etranacogene dezaparvovec in patients aged 65 years and older with hemophilia B. Additionally, patients with uncontrolled HIV infection or advanced liver fibrosis may not be eligible for etranacogene dezaparvovec.
The clinical experts consulted by CADTH noted there are currently no clinical or laboratory predictors of the treatment response. According to these experts, a recent study conducted by Shah et al. (2023)28 that was based on statistical modelling may provide some perspective: the models predicted that no more than 6 of 55 (10.91%) of observed participants would have FIX activity levels of less than 2% up to 25.5 years post etranacogene dezaparvovec infusion.
Assessing the Response Treatment
The clinical experts consulted by CADTH noted that the most important assessment for treatment response is to monitor patients’ bleeding to observe whether etranacogene dezaparvovec prevents bleeding events and allows patients to live the lifestyle they want without concern for the risk of bleeding. The clinical experts further noted that FIX activity level may also be monitored to assess response to treatment, which could allow clinicians to determine the degree to which the deficiency in FIX has been corrected by etranacogene dezaparvovec. Of note, the clinical experts consulted by CADTH noted that, in general, higher FIX activity level is associated with better bleeding outcomes (e.g., no bleeding). However, in some cases, there is a discrepancy between FIX activity level and bleeding outcomes.
The clinical experts consulted by CADTH noted that follow-up should focus on both efficacy and safety through clinical follow-ups (e.g., checking patients’ bleeding events and joint status via phone, virtual, or in-person check-ups) and lab tests (e.g., liver enzymes, FIX activity levels, liver ultrasound to detect hepatocarcinomas). The length of follow-up for hepatic function and FIX activity levels post infusion of etranacogene dezaparvovec should be lifelong. In terms of frequency, the clinical experts consulted by CADTH noted that postinfusion monitoring of etranacogene dezaparvovec will be more frequent in the short term (e.g., for the first 3 months post infusion, monitoring will include twice-a-week lab tests, mainly for liver enzymes and FIX level, starting around week 3 post infusion, or the lab tests will be conducted twice weekly initially and then once weekly) and less frequently over the long term (e.g., after the first 3 months, quarterly visits for the balance of the first year and then yearly visits for life, or monthly visits for the balance of the first year and then only as clinically indicated). The clinical experts consulted by CADTH noted that testing of FIX levels may not start immediately post infusion, given that the production of FIX by etranacogene dezaparvovec is unlikely to happen immediately after treatment, although it would be reasonable to monitor FIX activity level and liver function tests twice a week at the early postinfusion stage.
The clinical experts consulted by CADTH noted that post etranacogene dezaparvovec infusion, it will also be important to monitor changes in HJHS as well as in end points related to quality of life (QoL) (e.g., improvement in activities of daily living and physical activity and functioning, decrease in development of disability, improvement in psychosocial health and functioning). Of note, the clinical experts stated that, for measuring QoL among patients with hemophilia B in the Canadian setting, the PROBE tool is usually used instead of the Haemophilia Quality of Life Questionnaire for Adults (Haem-A-QoL) and Hemophilia Activities List, although these instruments are very much aligned in measuring QoL, and PROBE includes questions that cover activities of daily life.
Discontinuing Treatment
“Discontinuation of treatment” is not a concept that is applicable to gene therapy, which is a 1-time treatment. To define treatment failure of etranacogene dezaparvovec, the clinical experts consulted by CADTH further noted that the composite of FIX level (e.g., patient’s baseline FIX level before receiving etranacogene dezaparvovec) and return to prophylaxis with hemostatic therapy (e.g., per the definition provided in the HOPE-B trial) could be used to determine whether a treatment failure occurred in patients treated with etranacogene dezaparvovec. The clinical experts consulted by CADTH also noted that determining treatment failure should be done case by case and based on the judgment of the treating clinician.
Prescribing Considerations
The clinical experts consulted by CADTH noted that the prescribing of etranacogene dezaparvovec should be based on the judgment of a multidisciplinary team, organized by a comprehensive hemophilia treatment centre and consisting of specialists such as a hematologist with experience in treating patients with hemophilia, a physiotherapist to assess joint function, a hepatologist for liver-related issues, pharmacy support, and an HIV specialist if the patient is HIV positive.
According to the clinical experts consulted by CADTH, etranacogene dezaparvovec can be administered in a specialty clinic in the outpatient setting, with longitudinal follow-up. Currently, according to the clinical experts consulted by CADTH, all patients in Canada with clinically severe hemophilia are routinely followed in Canadian hemophilia treatment centres.
With respect to the roles of hemophilia treatment centres in administering etranacogene dezaparvovec and following up patients, the clinical experts consulted by CADTH did not reach consensus, noting 2 possible models. The first is a hub-and-spoke model in which selected hemophilia treatment centres in Canada would serve as infusion sites; patients would travel to the closest dosing site for infusion. The setting would likely be in the hospitals where the hemophilia clinics are located (outpatient clinics). Following infusion, the patients would then be monitored by their local hemophilia treatment centre.
Another model is that every hemophilia treatment centre in Canada would be able to infuse etranacogene dezaparvovec without the need for a hub-and-spoke model, given that these centres have always operated independently in incorporating innovative therapies into their clinical contexts and there is nothing special about the infusion of etranacogene dezaparvovec itself. Regarding the second model, the clinical experts consulted by CADTH acknowledged that it is the ideal scenario, as every hemophilia treatment centre would be able to administer etranacogene dezaparvovec. In reality, some hemophilia treatment centres will set up more quickly than others. The clinical experts consulted by CADTH noted that no matter which model is adopted, it should not create inequities between provinces and territories.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by the clinician groups. The full original clinician group input received by CADTH has been included in the stakeholder section of this report.
A total of 8 clinicians from the AHCDC provided input for this CADTH review of etranacogene dezaparvovec. The AHCDC highlighted some unmet needs for patients with hemophilia with the severe bleeding phenotype, specifically, hemophilia B. The AHCDC mentioned that the currently available treatments in Canada do not modify or alter the underlying disease process, thus making patients with hemophilia B dependent on regular IV infusions of FIX to prevent and treat bleeding for their whole life. In addition, the AHCDC highlighted the frequent venipuncture required for prophylactic CFC replacement. The group noted this can be a major challenge to routine prophylaxis for patients with poor venous access, and there can be long-term complications with the placement of a central venous catheter, including a risk of infection, bleeding, thromboembolism, and loss of function, requiring catheter removal. The group emphasized that all these factors lead to the need for patients with hemophilia B and a severe bleeding phenotype to restore the coagulation factor to clinically effective levels without the need for frequent venipunctures on a regular basis throughout their lifespan. The AHCDC also mentioned the variability of the efficacy of prophylaxis with CFCs across individuals, which makes some patients susceptible to breakthrough bleeding into joints and muscles. The group noted that these breakthrough bleeds result in pain, loss of function, absences from work or school, reduced QoL and, more importantly, disability from progressive joint damage. Lastly, the AHCDC highlighted that the FIX trough levels associated with prophylaxis regimens are often insufficient to allow for safe anticoagulation or dual antiplatelet therapy.
The AHCDC noted that gene therapy provides a possibility of long-term phenotypic cure for patients with hemophilia B. If effective, the new treatment option could provide a 1-time treatment leading to sustained FIX production, thus addressing the underlying disease process. This may represent a paradigm shift in the treatment of hemophilia B. The AHCDC also mentioned that in contrast to patients with hemophilia A, who have the option of emicizumab, patients with hemophilia B have no current alternatives to treatment with CFCs outside of clinical trials, making the need for gene therapy greater for patients with hemophilia B.
The AHCDC indicated that eligible candidates for the gene therapy under review include those with a clinically severe bleeding phenotype requiring prophylaxis, no history of inhibitory antibodies, no significant comorbidities, and an anti-AAV nAb titre of less than 1:900. The group also added that patients with hemophilia who are not currently receiving prophylactic therapy (e.g., due to poor venous access or adherence issues with routine prophylaxis) but who experience repeated, serious spontaneous bleeding episodes or have a history of life-threatening hemorrhage, are also candidates for gene therapy. The AHCDC highlighted that it is difficult to estimate the proportion of patients with hemophilia who would be eligible for gene therapy once it becomes commercially available due to the need for an anti-AAV antibody assay, a detailed liver assessment, and an assessment of the patient’s attitudes and perceptions.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Public drug plans do not fund the proposed comparators, which are FIX replacement products provided via Canadian Blood Services. Funding for these agents ultimately flows from separate provincial and territorial mechanisms and programs. | This is a comment from the drug plans to inform expert committee deliberations. |
Considerations for initiation of therapy | |
The indication includes patients “who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes.” In the pivotal trial (HOPE-B), patients had to have > 150 previous exposure days of treatment with FIX and to have been on stable prophylaxis for at least 2 months before screening. Questions for the clinical experts and/or CDEC:
| According to the clinical experts consulted by CADTH, the rationale for setting a minimum duration of treatment with FIX in the HOPE-B trial was mostly due to safety concerns regarding the development of inhibitors against FIX. The clinical experts noted that patients having > 150 previous exposure days of treatment with FIX over their lifetime is a reasonable duration, and the likelihood of excluding patients who do not meet this criterion but who would have benefited from etranacogene dezaparvovec treatment is very low. The clinical experts consulted by CADTH noted that while the indication is for patients who require prophylaxis, this does not necessarily mean patients have to be on stable prophylaxis treatment to be eligible for etranacogene dezaparvovec; thus, the concept of stable prophylaxis should not be introduced into the reimbursement criteria. The clinical experts noted that requiring patients to be on stable prophylaxis was reasonable for selecting participants in a clinical trial setting but not in a real-world setting because using stable prophylaxis as a reimbursement criterion in the real world would prevent some patients from benefiting from etranacogene dezaparvovec. For instance, although all patients with severe hemophilia B should be on prophylaxis, some of these patients may have difficulties complying with any prescribed stable or routine FIX prophylaxis treatment (e.g., due to difficulties with vein access or having trouble getting access to FIX therapies). These patients may benefit more from etranacogene dezaparvovec because they are not able to undergo stable or routine prophylaxis treatment. |
In the pivotal trial (HOPE-B), patients had to have severe or moderately severe FIX deficiency (defined as ≤ 2% of normal circulating FIX), and the indication notes there is no clinical experience in patients with FIX activity > 2%. Question for the clinical experts and/or CDEC:
| According to the clinical experts consulted by CADTH, the severity of hemophilia B in clinical practice is defined by the patient’s phenotype (i.e., tendency to bleed) and not only their factor activity levels; the decision to initiate prophylaxis with clotting factor concentrates takes into the account the patient’s clinical phenotype and factor activity levels as well as lifestyle and professional activities. In this context, there will be a small number of patients who may have a FIX level of > 2% but who would benefit from etranacogene dezaparvovec because of their severe bleeding phenotype and/or lifestyle. |
It is expected that a CADTH recommendation will be issued for another gene therapy for hemophilia B (fidanacogene elaparvovec) before etranacogene dezaparvovec is reviewed by CDEC. The drug plans request that CDEC consider alignment with the initiation criteria for fidanacogene elaparvovec, if applicable and appropriate. | This is a comment from the drug plans to inform expert committee deliberations. |
Considerations for continuation or renewal of therapy | |
The product is proposed as “a single-administration gene therapy that provides long-term prevention of hemophilia-related bleeds and eliminates the need for FIX prophylaxis therapy in most adult patients with hemophilia B.” Question for the clinical experts and/or CDEC:
| The clinical experts consulted by CADTH noted that nAbs against AAV5 will be developed from the first dose of etranacogene dezaparvovec, and that it is not possible under current technology to give a second dose to a patient. The clinical experts consulted by CADTH further noted that if, in the future, technology could offer solutions to the antibody response issue, a second dose might be useful for patients whose expression of FIX has been declining years after receiving the first dose of etranacogene dezaparvovec. |
Therapy will not be continued, per se, since it is a single-administration drug. However, there may be a need to confirm long-term response to therapy. Questions for the clinical experts and/or CDEC:
| The clinical experts consulted by CADTH noted that the objective parameters to assess treatment response in the trial included number of bleeds, FIX level (surrogate outcome), return to FIX prophylaxis, and FIX consumption. The clinical experts consulted by CADTH further noted that the composite of FIX level (e.g., patient’s baseline FIX level before receiving etranacogene dezaparvovec) and return to prophylaxis with hemostatic therapy (e.g., the definition provided by the HOPE-B trial) could be used to determine whether a treatment failure occurred in patients treated with etranacogene dezaparvovec. According to the clinical experts consulted by CADTH, ideally, the duration of follow-up should last a lifetime. The clinical experts consulted by CADTH further noted that 20 years may be a reasonable duration for confirming that a clinically meaningful response has been maintained. |
Considerations for prescribing of therapy | |
The sponsor noted the following:
| This is a comment from the drug plans to inform expert committee deliberations. |
Another gene therapy for hemophilia B (fidanacogene elaparvovec) is in the pipeline. Question for the clinical experts and/or CDEC:
| The clinical experts consulted by CADTH noted that, under current technology, it is impossible to give etranacogene dezaparvovec to a patient who has received fidanacogene elaparvovec or vice versa. Both etranacogene dezaparvovec and fidanacogene elaparvovec were developed using an AAV vector, which will cause patients to develop nAbs against the AAV vector post treatment. According to the clinical experts consulted by CADTH, although the AAV vectors used by etranacogene dezaparvovec and fidanacogene elaparvovec are not exactly the same, there still will be a very high proportion of cross reactivity between AAV vectors. |
Generalizability | |
The pivotal trial (HOPE-B) listed numerous exclusion criteria, but there are no related contraindications to therapy listed in the product monograph. The pivotal trial only included male patients, and the product monograph notes that “etranacogene dezaparvovec is not intended for administration in women.” Question for the clinical experts and/or CDEC:
| According to the clinical experts consulted by CADTH, many factors need to be considered before initiation of etranacogene dezaparvovec to identify patients who are likely to benefit from this drug. In general, the decision should be based on the judgment of the treating clinician through discussions with patients and their referring centres. The clinical experts consulted by CADTH highlighted several criteria that must be evaluated when determining a patient’s eligibility, such as anti-AAV5 nAb status, status of nAbs against FIX (FIX inhibitors), poor liver function, and allergy to corticosteroids. The clinical experts consulted by CADTH noted that some exclusion criteria used by the pivotal HOPE-B trial, which were reasonable in the clinical trial setting, may not be applicable in real-world clinical practice. For instance, the HOPE-B study excluded patients who had a history of an allergic reaction to FIX products. However, in the real world, these patients may be eligible for etranacogene dezaparvovec if they are allergic only to the components in the FIX products and not the FIX protein. Otherwise, according to the clinical experts consulted by CADTH, if patients are allergic to all available FIX products and in the meantime are ineligible for gene therapy, then there will be no treatment options to offer these patients. According to the clinical experts consulted by CADTH, another exclusion criterion of the HOPE-B study — having a history of nAbs against FIX — may not, by itself, serve in the real world as a basis for excluding patients to receive etranacogene dezaparvovec. In general, the clinical experts consulted by CADTH noted that treatment effects are not expected to be different between males and females due to the same underlying mechanism of disease, and female patients who would need etranacogene dezaparvovec are very rare. However, the clinical experts consulted by CADTH also noted that unless the safety risk of etranacogene dezaparvovec on female reproduction becomes clearer, it may not be appropriate for female patients of childbearing age to receive etranacogene dezaparvovec. |
The approved indication is specific to adults. The originally proposed indication, but not the approved indication, specified that patients should have a pre-existing neutralizing AAV5 antibody titre below 1:900. The product monograph notes, “Based on information obtained from the phase III CT-AMT-061-02 clinical study (HOPE-B), a threshold for an acceptable AAV5 neutralizing titre has been established to screen patients for eligibility to receive etranacogene dezaparvovec”; however, the product monograph does not appear to include a specific threshold. Questions for the clinical experts and/or CDEC:
| The clinical experts consulted by CADTH noted that etranacogene dezaparvovec should not be given to pediatric patients, given the lack of evidence. The clinical experts consulted by CADTH were aware of the sponsor’s clarification on the titre threshold of anti-AAV5 nAbs, which is < 1:900. The clinical experts consulted by CADTH agreed that selection of eligible patients, if etranacogene dezaparvovec were to be publicly reimbursed, should follow the 1:900 threshold. The clinical experts consulted by CADTH noted that the anti-AAV5 titre should be measured before, and as close as possible to, the infusion of etranacogene dezaparvovec. |
Care provision issues | |
The submission notes that continued hemostatic support with exogenous human FIX may be required during the first weeks after etranacogene dezaparvovec administration to provide sufficient FIX coverage for the initial days post treatment. Corticosteroid treatment is recommended for those who experience transaminitis after receiving etranacogene dezaparvovec. | This is a comment from the drug plans to inform expert committee deliberations. |
Neutralizing AAV5 antibody testing is required for eligibility. (The submission notes that a “validated assay for neutralizing AAV5 antibodies approved for etranacogene dezaparvovec should be used.”) It is unclear how widely such testing will be available or who will cover the associated costs. | This is a comment from the drug plans to inform expert committee deliberations. |
System and economic issues | |
The submission indicates reimbursement would result in an incremental pan-Canadian cost of $15.44 million in year 1, $24.70 million in year 2, and $22.62 million in year 3, for a 3-year total incremental cost of $62.72 million. The sensitivity analyses estimated the 3-year total incremental costs could range from $31.36 million to $94.08 million. | This is a comment from the drug plans to inform expert committee deliberations. |
Costs related to required laboratory testing should be considered. The submission notes that several tests are required for patient selection purposes, including neutralizing AAV5 antibody titre (as noted previously), assay for FIX inhibitor presence, liver enzymes, and hepatic ultrasound and elastography. In addition, regular monitoring is required after administration of etranacogene dezaparvovec, including monitoring of liver enzymes, FIX activity, and an assay for FIX inhibitor presence. Any related travel costs should also be considered. | This is a comment from the drug plans to inform expert committee deliberations. |
The sponsor notes:
| This is a comment from the drug plans to inform expert committee deliberations. |
AAV5 = adeno-associated virus serotype 5; CDEC = Canadian Drug Expert Committee; FIX = factor IX; nAb = neutralizing antibody.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of etranacogene dezaparvovec (Hemgenix) 1 × 1013 gc/mL suspension for IV infusion for the treatment of adults (aged 18 years or older) with hemophilia B (congenital FIX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes. The focus will be placed on comparing etranacogene dezaparvovec with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence provided by the sponsor for this review of etranacogene dezaparvovec is presented in 2 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and randomized controlled trials that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
1 pivotal phase III, open-label, single-arm study (including a lead-in phase before the infusion of etranacogene dezaparvovec to prospectively collect efficacy and safety data that served as a comparison) was identified in the systematic review
1 ITC containing analyses of etranacogene dezaparvovec relative to 4 comparator therapies: rIX-FP (Idelvion), rFIXFc (Alprolix), pegylated nonacog beta pegol (Rebinyn), and nonacog alfa (BeneFIX).
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
One study (HOPE-B)29 met the inclusion criteria of the sponsor-submitted SLR. The characteristics of the HOPE-B trial are summarized in Table 6.
HOPE-B is a phase III, open-label, single-dose, single-arm, multicentre trial investigating the use of etranacogene dezaparvovec for the treatment of adult male patients with moderately severe to severe hemophilia B (defined as normal circulation FIX ≤ 2%).1,30 A total of 54 patients from 33 study sites globally received etranacogene dezaparvovec and were followed for efficacy and safety. The primary objective of the HOPE-B trial was to demonstrate the noninferiority of etranacogene dezaparvovec to reduce ABR for all bleeding events during the 52 weeks following establishment of stable FIX expression (months 7 to 18) compared with standard of care (i.e., continuous routine FIX prophylaxis). The superiority of etranacogene dezaparvovec was also examined.
The HOPE-B study is ongoing and expected to be completed in 2025. An analysis performed 36 months post dose (data cut-off date of June 6, 2023) was used to support the sponsor’s present reimbursement request to CADTH.1 Of note, regardless of the date of initiation into the study, the data cut included up to 36 months of patient follow-up information.
Table 6: Details of Study Included in the Systematic Review
Detail | HOPE-B trial |
|---|---|
Designs and populations | |
Study design | Phase III, open-label, single-dose, single-arm, multicentre study to investigate etranacogene dezaparvovec administered to adult patients with severe or moderately severe hemophilia B |
Locations | 33 sites, including 17 sites in the US, 13 sites in the European Union, and 3 sites in the UK |
Key dates |
|
Randomized (N) | A total of 75 patients were screened; 67 patients were enrolled and were included in the lead-in period. A total of 54 patients received etranacogene dezaparvovec and were followed for efficacy and safety. |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Etranacogene dezaparvovec was administered at a dose of 2 × 1013 gc/kg as a 1-time IV infusion via a peripheral vein |
Comparator(s) | During the lead-in phase, which lasted for a minimum of 26 weeks (i.e., ≥ 6 months), patients recorded their use of FIX replacement therapy and bleeding episodes in their dedicated e-diary |
Study duration | |
Screening phase | Maximum 6 weeks |
Run-in phase | Minimum of 26 weeks (i.e., ≥ 6 months) |
Treatment phase | Patients received a single dose of etranacogene dezaparvovec at the dosing visit |
Follow-up phase |
|
Outcomes | |
Primary end point | ABR comparison between etranacogene dezaparvovec and prophylaxis between the lead-in phase and the 52 weeks following stable FIX expression (months 7 to 18 post treatment) |
Secondary and exploratory end points | Secondary
Exploratory
|
Publication status | |
Publications | Pipe et al. (2020)31 Thornburg (2021)32 Pipe et al. (2023)33 |
AAV5 = adeno-associated virus of serotype 5; ABR = annualized bleeding rate; AIR = annualized infusion rate; ALP = alkaline phosphatase; ALT = alanine transaminase; AST = aspartate aminotransferase; EQ-5D-5L = 5-Level EQ-5D; FIX = factor IX; gc = genome copy; Haem-A-QoL = Haemophilia Quality of Life Questionnaire for Adults; HAL = Hemophilia Activities List; HBV = hepatitis C virus; HCV = hepatitis C virus; HJHS = Hemophilia Joint Health Score; IMP = investigational medicinal product; nAb = neutralizing antibody; RNA = ribonucleic acid; ULN = upper limit of normal; VAS = visual analogue scale.
Note: Visit L-Final refers to the final visit of the lead-in phase.
Sources: HOPE-B Clinical Study Protocol Version 8.030 and the HOPE-B Clinical Study Report.29 Details included in the table are from the sponsor’s summary of clinical evidence.1
The study design schema of the HOPE-B trial is shown in Figure 1. In general, the HOPE-B study consists of the screening phase, lead-in phase, etranacogene dezaparvovec infusion phase, and posttreatment follow-up phase. During the lead-in phase, patients received continuous FIX prophylaxis and were followed up for at least 6 months (i.e., 26 weeks). A predefined washout period of 3 days for regular-acting FIX products and 10 days for extended half-life (EHL) FIX products occurred between screening and the lead-in phase. Outcome data collected from the lead-in phase served as the comparator for etranacogene dezaparvovec for some efficacy end points. For patients who completed the lead-in phase and received etranacogene dezaparvovec infusion (n = 54), the median duration of the lead-in phase was 7.129 months (range, 6.05 to 10.61). According to the sponsor’s clarification,34 monitoring of treatment compliance during the lead-in phase was captured in patient diaries, but no summary-level statistics were generated from these diaries.
Figure 1: Study Design Schema of the HOPE-B Study
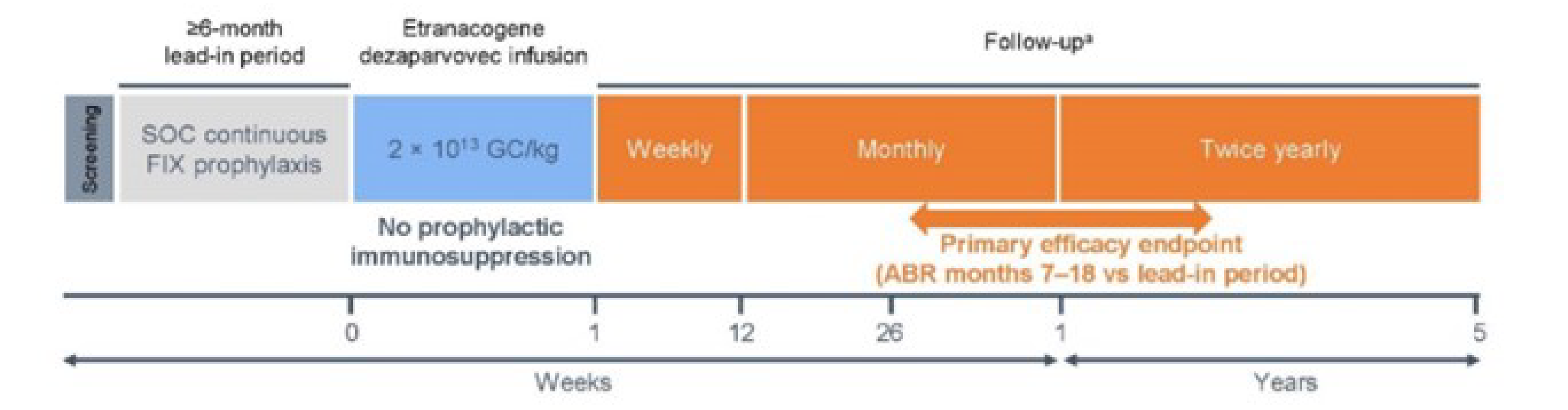
ABR = annualized bleeding rate; FIX = factor IX; GC = genome copy; SOC = standard of care; vs = versus.
a At least quarterly contact (± 2 weeks) between site staff and patients to monitor occurrence of adverse events.
Source: Sponsor’s summary of clinical evidence.1
Populations
Inclusion and Exclusion Criteria
Eligible patients in the HOPE-B study were male, aged 18 years or older, with a diagnosis of congenital hemophilia B that was classified as a moderately severe or severe FIX deficiency (defined as ≤ 2% of normal circulation FIX) for which patients had received continuous routine treatment with an FIX prophylaxis totalling more than 150 exposure days. Patients had to have been on stable prophylaxis treatment for at least 2 months before screening. Patients were excluded if they had a history of FIX inhibitors or tested positive for FIX inhibitors at the last visit of the lead-in period and during the screening period of the HOPE-B trial. Of note, according to the sponsor, pre-existing nAbs against AAV5 were not used as an exclusion criterion in the HOPE-B study.
Interventions
In the HOPE-B study, etranacogene dezaparvovec was administered as a single IV infusion at a dose of 2 × 1013 gc/kg of body weight over approximately 3 hours.
Outcomes
A list of the efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. The summarized end points are based on the outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review, according to the clinical experts consulted by CADTH and the input received from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team listed the end points considered to be most relevant to inform CADTH’s expert committee deliberations, in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Table 7: Outcomes Summarized From the HOPE-B Trial
Outcome measure | Time point post infusion of etranacogene dezaparvovec | HOPE-B |
|---|---|---|
ABR for all bleeding events (including proportion of patients with no bleeds) | Month 7 to Month 18 post infusion of etranacogene dezaparvovec | Primary |
Month 7 to month 36 | Exploratory | |
ABR for spontaneous bleeds | Month 7 to month 18 | Secondary |
Month 7 to month 36 | Exploratory | |
ABR for joint bleeds | Month 7 to month 18 | Secondary |
Month 7 to month 36 | Exploratory | |
AIR of FIX replacement therapy | Month 7 to month 18 | Secondary |
Month 7 to month 36 | Exploratory | |
Annualized consumption of FIX replacement therapy | Month 7 to month 36 | Exploratory |
HJHS | Month 12 | Exploratory |
Month 24 | Exploratory | |
Month 36 | Exploratory | |
PROBE summary scorea | Month 12 | Other |
Month 24 | Other |
ABR = annualized bleeding rate; AIR = annualized infusion rate; FIX = factor IX; HJHS = Hemophilia Joint Health Score; PROBE = Patient Reported Outcomes Burdens and Experiences; QoL = quality of life.
aPROBE data were obtained from patients who volunteered to participate in the optional PROBE questionnaire substudy. The objective of this substudy was to provide data complementary to the compendium of established patient-reported outcome tools regarding the impact of gene therapy on patient-relevant outcomes and QoL over time. According to the sponsor, the 36-month data for PROBE are not available.
Source: HOPE-B Clinical Study Protocol Version 8.0.30
Descriptions of efficacy and safety outcomes presented in the HOPE-B study and appraised in the CADTH Clinical Review are as follows.30,35
Efficacy Outcomes
Annualized Bleeding Rate
In its correspondence,36 the sponsor specified that ABR for all bleeding events refers to any untreated and treated bleeds regardless of bleeding types. Spontaneous bleeds are unprovoked bleeding events, which means there are no known reasons for a bleed.
The ABR is a ratio calculated as the number of bleeds divided by the length of observation (in years). Bleeding events were recorded in an e-diary by patients. According to clarification from the sponsor, the e-diary includes the infusion and bleeding diary and the daily evening diary. Patients were required to complete the infusion and bleeding diary to report any new information as it became available (e.g., reporting an FIX infusion, bleeding as soon as it occurred, or bleeding cessation). The patients were also required to complete the evening diary once a day. The purpose of the evening diary was to remind patients to enter new information, if they had forgotten to add necessary documentation to the infusion and bleeding diary during the day, or confirm that no new information needed to be entered. When a bleeding event was recorded, the study centre was contacted to review the record and assess the bleeding event. However, bleeds were counted irrespective of assessments by the investigator as to the trueness or newness of the bleed, except in the case of certain sensitivity analyses.
Annualized Infusion Rate
The yearly infusion rate of FIX replacement therapy, excluding replacement for invasive procedures, was determined for the lead-in and posttreatment periods. The number of infusions of exogenous FIX was counted for each patient during both the lead-in and postinfusion periods.
Annualized Consumption of FIX Replacement Therapy
The annualized consumption of FIX replacement therapy was calculated as the normalized amount of drug administered per baseline weight (before the lead-in period); these data were extrapolated, when necessary, for any time period that was less than or greater than 1 year. “Therapy administered” included the total amount of FIX given as replacement therapy, excluding any FIX administered for invasive procedures. Having no record of exogenous FIX use meant zero consumption.
Descriptions of the HJHS and PROBE measures are shown in Table 8.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
HJHS | The HJHS (version 2.1) comprises an assessment of specific features, or items, of the 6 index joints (i.e., elbows, knees, and ankles) as well as an assessment of global gait. For each of the 6 joints, the following items are scored: swelling (scored 0 to 3), duration of swelling (0 to 1), muscle atrophy (0 to 2), crepitus on motion (0 to 2), flexion loss (0 to 3), extension loss (0 to 3), joint pain (0 to 2), and strength (0 to 4). The maximum score for an individual index joint is 20. Gait is scored 0 to 4 based on walking, stairs, running, and hopping on 1 leg. The total score is the sum of all joint and gait scores (range 0 to 124), with a higher number equating to more severe joint damage.37,38 | Validity: In a multicentre international study containing patients with hemophilia as well as healthy adults, HJHS total scores were correlated with WFH Gilbert scores (Spearman correlation, rs = 0.95). WFH Gilbert scores are the original WFH Orthopedic Joint Score and, according to the authors of the study, this correlation demonstrated convergent construct validity.37 Discriminant (known groups) construct validity was evaluated using the Kruskal-Wallis nonparametric analysis of variance. The HJHS total score differentiated between age groups (Kruskal-Wallis, t = 35.02; P < 0.001) and disease severity in patients with hemophilia.37 Reliability: In a study consisting of male patients with hemophilia in the US, the Cronbach alpha value was 0.97 for the HJHS total score, above the established threshold of 0.70 in previous studies, indicating sufficient internal consistency according to the authors of the study.39,40 All items on the HJHS had been reported to capture sufficient correlation with their respective joint total scores (r = 0.34 to 0.83), where r is the Pearson product-moment correlation coefficient.39,41 In another study consisting of male patients with hemophilia in the US, the HJHS ankle domain reached a correlation of r > 0.5 for several domain and summary scores related to physical function, including scores specific to activity of the lower extremities (HAL domains of lying, sitting, kneeling, standing; functions of the legs, use of transportation; complex lower extremity activities; and overall activity). HJHS total score also demonstrated similar correlation for similar domains and summary scores, except use of transportation. However, the HJHS global gait score did not reach a correlation of r > 0.5 with any patient-reported outcomes for any instrument domain or summary scores.38 In a multicentre international study containing patients with hemophilia as well as healthy adults, the HJHS v2.1 items had demonstrated adequate internal reliability (Cronbach alpha = 0.88) based on previously established threshold.40 Correlations between item scores and the total score demonstrated that almost all HJHS items (muscle atrophy, crepitus, flexion and extension loss, joint pain, and strength) were highly correlated (alpha > 0.70) according to the authors of the study and previously established threshold, except for 2 items, swelling and duration of swelling, which were only moderately correlated.37 Responsiveness: The HJHS is more sensitive to early joint changes than the Gilbert score, according to the authors of the study.42 It has been reported to capture relevant features that distinguish between different prophylactic strategies in young adults with severe hemophilia43 and between severe and nonsevere hemophilia in children,42,44 and is responsive to changes following physiotherapy treatment.45 However, it is so sensitive that it showed positive scores in 40% of unaffected young adults (total score ≤ 3 points).46,47 | No MID was identified in the sponsor’s literature search for this population. |
PROBE | The PROBE questionnaire is a novel, patient-developed tool specific to hemophilia and is intended to capture clinical outcomes that are considered relevant by patients.48 This tool covers 4 domains pertaining to patient demographics, general health status, hemophilia-related health status, and HRQoL (using the EQ-5D-5L and EQ VAS).48,49 The questionnaire comprises 29 items.48,49 Scores range from 0 to 1, with higher scores indicative of better health status.49 | Validity: In a study consisting of people with hemophilia A or B or people without a bleeding disorder, according to the authors, the PROBE items demonstrated moderate to strong correlations with corresponding EQ-5D-5L domains.50 The PROBE score also demonstrated strong correlation with the EQ-5D-5L utility index score (r = 0.67), based on previously established thresholds,51 thus showcasing convergent validity, according to the authors of this study.50 The PROBE questionnaire and score demonstrated some differentiation properties between several subgroups, thus indicating known groups validity, according to the authors.50 Reliability: In a study consisting of people with hemophilia and people without a bleeding disorder, the test–retest properties of the PROBE questionnaire were investigated.49 Cohen kappa coefficients ranged from 0.69 to 1.00 for general health questions, and from 0.5 to 1.0 for specific hemophilia-related questions, indicating acceptable reliability based on previously established thresholds.49,52 The correlation coefficient of total PROBE score between 2 time points and between paper-based and web-based versions was 0.95, indicating acceptable reliability properties, as per previously established thresholds (≥ 0.75).49,53,54 In another study consisting of people with hemophilia A or B or people without a bleeding disorder, the Cronbach alpha coefficient (0.84) indicated acceptable internal consistency reliability based on previously established thresholds.40,50 Responsiveness: No evidence of responsiveness was identified in the sponsor’s literature search for this population. | No MID was identified in the sponsor’s literature search for this population. |
EQ VAS = EQ visual analogue scale; HAL = Hemophilia Activities List; HJHS = Hemophilia Joint Health Score; HRQoL = health-related quality of life; MID = minimal important difference; PROBE = Patient Reported Outcomes Burdens and Experiences; WFH = World Federation of Hemophilia.
Harms Outcomes
The harms outcomes assessed in the HOPE-B study included TEAEs, TESAEs, withdrawals due to AEs, mortality, and notable harms (e.g., ALT increased, AST increased). An AE was considered to be a TEAE if the event occurred after the administration of etranacogene dezaparvovec, or if the AE worsened during the study after the infusion of etranacogene dezaparvovec (e.g., intensity and/or severity changed to a worse grade). AEs were coded using the Medical Dictionary for Regulatory Activities.
Statistical Analysis
In the HOPE-B trial, the primary objective was to test for noninferiority of etranacogene dezaparvovec against FIX prophylaxis for the treatment of ABR. Post treatment, all bleeding events between month 7 and month 18 post infusion of etranacogene dezaparvovec were counted toward the reported ABR. A simulation study of ABR was conducted using a negative binomial distribution with a yearly event rate of 2.4 for the lead-in period and a rate of 1.9 for the period post infusion of etranacogene dezaparvovec. An individual-level Pearson correlation of 0.05 was used to induce a relationship between the number of events occurring within the 2 periods and a common dispersion parameter of 1.5. The results of the simulation study informed a necessary sample size of at least 50 analyzable patients to reject a null hypothesis of noninferiority using a noninferiority margin of 1.8 for the rate ratio of ABR and a power of 82.0% at the 0.025 significance level.
For analyses with data from the month 18 data cut-off, formal statistical testing of the efficacy end points was performed using the closed testing principle for type I error control for multiple testing. Unless otherwise specified, each end point tested for statistical significance was tested for superiority at a 1-sided alpha level of 0.025. Fixed sequential testing was performed using a hierarchical approach that continued until a nonsignificant result was obtained. The following is the order of the fixed sequential tests:
ABR comparison between etranacogene dezaparvovec and prophylaxis for noninferiority between the lead-in and the 52 weeks following 7 to 18 months of stable FIX expression post treatment (etranacogene dezaparvovec).
Endogenous FIX activity 6 months after etranacogene dezaparvovec dosing.
Endogenous FIX activity 12 months after etranacogene dezaparvovec dosing.
Endogenous FIX activity 18 months after etranacogene dezaparvovec dosing.
Annualized consumption of FIX replacement therapy, excluding FIX replacement for invasive procedures, during the 52 weeks following 7 to 18 months of stable FIX expression post treatment compared with the lead-in phase.
AIR of FIX replacement therapy, excluding FIX replacement for invasive procedures, during the 52 weeks following 7 to 18 months of stable FIX expression post treatment compared with the lead-in phase.
Comparison of the percentage of patients with a trough FIX activity of less than 12% of normal between the lead-in phase and after treatment with etranacogene dezaparvovec 52 weeks after 7 to 18 months of stable FIX expression.
ABR comparison between etranacogene dezaparvovec and prophylaxis for superiority between the lead-in and the 52 weeks following 7 to 18 months of stable FIX expression post treatment.
Rate of spontaneous bleeding events during the 52 weeks following 7 to 18 months of stable FIX expression post treatment compared with the lead-in phase.
Rate of joint bleeding events during the 52 weeks following 7 to 18 months of stable FIX expression post treatment compared with the lead-in phase.
Patient-reported outcome questionnaire scores from the International Physical Activity Questionnaire (total physical activity score) during the 12 months following etranacogene dezaparvovec dosing compared with the lead-in phase.
Patient-reported outcome questionnaire scores from the EQ visual analogue scale during the 12 months following etranacogene dezaparvovec dosing compared with the lead-in phase.
Of note, according to the sponsor, no tabulations pertaining to type I error control were required for analyses using data generated from follow-up after month 18 post infusion of etranacogene dezaparvovec (e.g., the month 36 analyses), and all P values and associated statements of significance were not controlled for multiplicity. Details on the statistical analysis of efficacy end points in the HOPE-B study are presented in Table 9.
Table 9: Statistical Analysis of Efficacy End Points in the HOPE-B Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity or subgroup analyses |
|---|---|---|---|---|
ABR for all bleeding events |
| Treatment (i.e., period) | Maintained as missing (i.e., analyzed as observed without any explicit imputation of missing data) | Sensitivity analyses
Subgroup analyses
|
ABR for spontaneous bleeds |
| Treatment (i.e., period) | Maintained as missing (i.e., analyzed as observed without any explicit imputation of missing data) | Sensitivity analyses
Subgroup analyses
|
ABR for joint bleeds |
| Treatment (i.e., period) | Maintained as missing (i.e., analyzed as observed without any explicit imputation of missing data) | Sensitivity analyses
Subgroup analyses
|
Annualized infusion rate of FIX replacement therapy |
| Treatment (i.e., period) | Maintained as missing (i.e., analyzed as observed without any explicit imputation of missing data) | Sensitivity analyses
Subgroup analyses
|
Annualized consumption of FIX replacement therapy |
| None | Maintained as missing (i.e., analyzed as observed without any explicit imputation of missing data) | Sensitivity analyses
Subgroup analyses
|
HJHS | Mean HJHS scores at each visit were estimated using a linear mixed model with repeated measures. | Visit | Imputation | Sensitivity analyses
|
PROBE summary score | PROBE summary scores and individual item responses were summarized descriptively by treatment and visit. | NA | NR | NR |
AAV5 = adeno-associated virus serotype 5; ABR = annualized bleeding rate; AIR = annualized infusion rate; CI = confidence interval; FAS = full analysis set; FIX = factor IX; HJHS = Hemophilia Joint Health Score; NA = not applicable; NR = not reported; PP = per protocol; PROBE = Patient Reported Outcomes Burdens and Experiences.
Sources: HOPE-B Clinical Study Protocol Version 8.0,30 HOPE-B Statistical Analysis Plan, version 5.0,35 sponsor’s summary of clinical evidence.1
Analysis Populations
The analysis populations of the HOPE-B trial are summarized in Table 10.
Table 10: Analysis Populations of the HOPE-B Trial
Study | Population | Definition | Application |
|---|---|---|---|
HOPE-B | Screen failure population | All patients who were screened but never entered the lead-in phase. | NA |
Lead-in discontinuers population | All patients who entered the lead-in phase but discontinued from the study before etranacogene dezaparvovec dosing. | NA | |
Safety population | All patients who were enrolled in either the lead-in safety population or posttreatment safety population. | Safety analyses | |
Lead-in safety population | All patients who were enrolled into the lead-in phase. | Period-specific safety tabulations | |
Posttreatment safety population | All patients who received etranacogene dezaparvovec, irrespective of any protocol deviations. | Period-specific safety tabulations | |
FAS | All patients who were enrolled, entered the lead-in phase, were dosed with etranacogene dezaparvovec, and were assessed for at least 1 of the study’s efficacy end points subsequent to etranacogene dezaparvovec dosing. | All efficacy analyses | |
PP population | All patients from the FAS population who adhered to a stable and adequate prophylaxis regimen during the lead-in phase, completed at least 18 months of efficacy assessments, and had no major protocol deviations that impacted the interpretation of efficacy. | All efficacy analyses |
FAS = full analysis set; NA = not applicable; PP = per protocol.
Sources: HOPE-B Clinical Study Protocol Version 8.030 and HOPE-B Clinical Study Report.29 Details included in the table are from the sponsor’s summary of clinical evidence.1
Protocol Amendments and Deviations
In total, there were 8 versions of the study protocol, including the original protocol (February 16, 2018) and 7 amendments (the seventh amendment was made on June 21, 2021). In the original protocol, endogenous FIX activity at 6 months post infusion of etranacogene dezaparvovec was the only primary end point. In protocol amendment 4, ABR following 52 weeks post infusion of etranacogene dezaparvovec was added as a primary end point. In protocol amendment 6, FIX activity was removed from primary outcomes, and ABR following 52 weeks post infusion of etranacogene dezaparvovec was revised to ABR from months 7 to 18 post infusion of etranacogene dezaparvovec (i.e., 52 weeks following establishment of stable FIX expression). The majority of protocol deviations were related to the timing of study visits, questionnaire completion, or absence or incorrect performance of laboratory tests.
Results
Patient Disposition
A summary of patient disposition in the HOPE-B study is presented in Table 11. In the HOPE-B trial, 67 patients met the eligibility criteria and entered the lead-in phase. Of the 67 patients, 53 received etranacogene dezaparvovec, 1 patient received a partial dose (i.e., 10%) of etranacogene dezaparvovec due to hypersensitivity but remained for posttreatment follow-up, and 13 patients discontinued or were excluded before infusion of etranacogene dezaparvovec. The reasons for the discontinuation or exclusion of the 13 patients included ineligible due to screen failure (8 patients), withdrawal of consent (3 patients), and other (i.e., 2 patients withdrew due to COVID-19 pandemic-related concerns).
Overall, 52 of 54 patients completed 24 months of follow-up post administration of etranacogene dezaparvovec. Among the 2 patients who received the full dose etranacogene dezaparvovec but did not complete 24 months of follow-up, 1 patient died 464 days (approximately 15 months) post infusion and 1 patient remained on routine prophylaxis and withdrew consent 24 months post infusion of etranacogene dezaparvovec (month 24 visit not completed). As of the data cut-off date for the 36-month follow-up, 52 patients had been followed for at least 3 years post treatment and were continuing in the study.
Table 11: Summary of Patient Disposition From the HOPE-B Trial Included in the Systematic Review
Patient disposition | Etranacogene dezaparvovec |
|---|---|
Screened, N (%) | 75 (100) |
Completed, n | 67 (89.3) |
Screen failures, n | 8 (10.7) |
Lead-in period, N (%) | 67 (89.3) |
Lead-in discontinuers, n | 13 (19.4) |
Treated with etranacogene dezaparvovec, N (%) | 54 (80.6) |
Received a partial dose due to adverse event (hypersensitivity), n | 1 (1.9) |
Received full dose, n | 53 (98.1) |
Early withdrawal from study (post treatment), n | 2 (3.7) |
Adverse event, n | 1 (50.0) |
Patient withdrew consent, n | 1 (50.0) |
Analysis population, n of N (%) | |
Lead-in discontinuers (i.e., not treated with etranacogene dezaparvovec) | 13 of 67 (19.4) |
Safety | 67 of 75 (89.3) |
Lead-in safety population | 67 of 75 (89.3) |
Posttreatment safety population (i.e., treated with etranacogene dezaparvovec) | 54 of 67 (80.6) |
FAS | 54 of 54 (100) |
PP population | 53 of 54 (98.1) |
PROBE substudy, n of N (%) | 50 of 54 (92.6) |
FAS = full analysis set; NA = not applicable; PP = per protocol; PROBE = Patient Reported Outcomes Burdens and Experiences.
Sources: HOPE-B Clinical Study Protocol Version 8.030 and Clinical Study Report.29 Details included in the table are from the sponsor’s summary of clinical evidence.1
Baseline Characteristics
The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. All patients in the HOPE-B trial were adult males. Among the 54 patients who received etranacogene dezaparvovec (posttreatment safety population or full analysis set), the majority of patients were white (74.1%) with a mean age of 41.5 (SD = 15.8) years. Among the 54 patients at baseline, 21 (38.9%) had pre-existing nAbs against AAV5 before infusion of etranacogene dezaparvovec, while 33 (61.1%) did not.
The last testing before infusion of etranacogene dezaparvovec showed that 21 of the 54 patients (38.9%) had a titre between 1:9 and 1:3,212 (median = 1:56.9). Excluding 1 patient with an anti-AAV5 titre greater than 1:3,000 (i.e., 1:3,212), the remaining 20 patients had a titre between 1:9 and 1:678 (median = 1:49.1). There were 33 patients (62.3%) with an anti-AAV5 nAb titre below the lower limit of detection (i.e., 1:7).
Exposure to Study Treatments
In the HOPE-B study, of the 54 patients who received etranacogene dezaparvovec, 53 received a single full dose of 2 × 1013 gc/kg etranacogene dezaparvovec. One patient received a partial dose (about 10% of the expected dose) before the patient withdrew due to hypersensitivity that occurred during infusion.
Prior and Concomitant Treatments
Prior to the infusion of etranacogene dezaparvovec, among the 54 patients who completed the lead-in period, 4 patients (7.4%) were treated with corticosteroids. The mean number of days with corticosteroid treatment was 1.3 days (SD = 0.6 days), with a range of between 1 and 2 days.
Chronic administration of corticosteroids was not allowed during the HOPE-B study; however, the use of corticosteroids for conditions such as an increase in AST or ALT was permitted.30 During the 24-month postinfusion period, 20 out of 54 patients (37.0%) received systemic corticosteroids. The mean number of days with corticosteroid use was 41.6 days (SD = 45.6 days) and ranged from 1 to 130 days. Reasons for corticosteroid use included transaminase elevation (9 of 54 patients; 16.7%), infusion reactions (3 of 54; 5.6%), and other (11 of 54; 20.4%), such as dental rehabilitation, surgery prophylaxis, immune response against the vector, allergies, pain, preoperative anti-inflammatory use, nephrolithiasis, tendovaginitis stenosans, liver transplant, left mandibular fracture, shoulder pain, facet joint injection to the L4 or L5 vertebrae, and prevention of postoperative nausea and vomiting. The mean number of days with corticosteroid use was 81.4 days (SD = 28.6) and ranged from 51 to 130 days during the 36-month postinfusion period. Between 24 and 36 months after the infusion period, no additional patients received systemic corticosteroids for transaminase elevation.
Table 12: Summary of Baseline and Clinical Characteristics From the HOPE-B Trial
Characteristic | Etranacogene dezaparvovec | |
|---|---|---|
Lead-in safety population (N = 67) | Posttreatment safety population, FAS (N = 54) | |
Age (years),a n | 67 | 54 |
Mean (SD) | 42.8 (16.2) | 41.5 (15.8) |
Median (range) | 38.0 (19 to 78) | 37.0 (19 to 75) |
Sex, n (%) | ||
Male | 67 (100.0) | 54 (100.0) |
Race, n (%) | ||
White | 50 (74.6) | 40 (74.1) |
Asian | 3 (4.5) | 2 (3.7) |
Black or African-American | 2 (3.0) | 1 (1.9) |
Other | 7 (10.4) | 6 (11.1) |
Missing | 5 (7.5) | 5 (9.3) |
Height (cm), n | 66 | 54 |
Mean (SD) | 176.9 (7.9) | 176.5 (8.2) |
Median (range) | 176.5 (153 to 197) | 176.5 (153 to 197) |
Weight (kg), n | 66 | 54 |
Mean (SD) | 87.2 (20.0) | 85.1 (19.3) |
Median (range) | 85.5 (58 to 169) | 84.0 (58 to 169) |
BMI (kg/m2), n | 66 | 54 |
Mean (SD) | 27.7 (5.4) | 27.2 (5.1) |
Median (range) | 26.7 (21 to 51) | 26.2 (21 to 51) |
Duration of hemophilia B (years),b n | 65 | 53 |
Mean (SD) | 40.8 (15.7) | 39.7 (15.0) |
Median (range) | 36.4 (18 to 78) | 34.3 (18 to 74) |
Severity of hemophilia B at time of diagnosis, n (%) | ||
Severe (FIX < 1%) | 56 (83.6) | 44 (81.5) |
Moderately severe FIX (FIX ≥ 1% and ≤ 2%) | 11 (16.4) | 10 (18.5) |
Bleeding episodes in year before screening, n (%) [number of episodes] | ||
Any bleeding episodes | 53 (79.1) [258] | 44 (81.5) [215] |
Joint bleeding episodes | 33 (49.3) [155] | 30 (55.6) [132] |
Spontaneous bleeding episodes | 36 (53.7) [141] | 32 (59.3) [118] |
Traumatic bleeding episodes | 26 (38.8) [72] | 20 (37.0) [64] |
Unknownc | 14 (20.9) [45] | 11 (20.4) [33] |
Bleeding episodes in year before screening, n (%) | ||
0 | 14 (20.9) | 10 (18.5) |
1 | 11 (16.4) | 9 (16.7) |
2 | 14 (20.9) | 10 (18.5) |
3 | 8 (11.9) | 8 (14.8) |
4 | 4 (6.0) | 4 (7.4) |
5 | 2 (3.0) | 2 (3.7) |
6 | 2 (3.0) | 2 (3.7) |
7 | 2 (3.0) | 2 (3.7) |
8 | 2 (3.0) | 2 (3.7) |
10 | 1 (1.5) | 0 |
11 to 15 | 4 (6.0) | 3 (5.6) |
> 20 | 2 (3.0) | 2 (3.7) |
FIX replacement therapy type,d n (%) | ||
Prophylactic | 67 (100.0) | 54 (100.0) |
On demand | 5 (7.5) | 4 (7.4) |
Most recent prescreening FIX therapy category, n (%) | ||
Extended half-life | 40 (59.7) | 31 (57.4) |
Standard half-life | 27 (40.3) | 23 (42.6) |
HIV status, n (%) | ||
Negative | 63 (94.0) | 51 (94.4) |
Positive | 4 (6.0) | 3 (5.6) |
Hepatitis B infection, n (%) | ||
Prior resolvede | 13 (19.4) | 9 (16.7) |
Hepatitis C infection, n (%) | ||
Prior or ongoinge | 38 (56.7) | 31 (57.4) |
Prior resolved | 34 (50.7) | 28 (51.9) |
Ongoing | 4 (6.0) | 3 (5.6) |
Positive at screeningf | 1 (1.5) | 1 (1.9) |
BMI = body mass index; FAS = full analysis set; FIX = factor IX; SD = standard deviation.
aAge was the age at the time of informed consent.
bDuration was calculated based on the date the patient was initially diagnosed with hemophilia B according to the case report form.
cUnknown as to whether spontaneous or traumatic.
dFIX replacement therapy in the year before screening; patients may have received on-demand and then prophylactic FIX replacement therapy in this time period.
ePrior or ongoing per reported medical history. All patients tested negative predose.
fPatients positive at screening were given a rating of “hepatitis C virus RNA = detected” for hepatitis. Patients were positive at screening and negative at the final visit during the lead-in phase.
Sources: HOPE-B Clinical Study Report.29 Note details included in the table are from the sponsor’s summary of clinical evidence.1
Use of FIX Treatment Post Etranacogene Dezaparvovec Infusion
Patients were permitted to continue administration of their continuous routine FIX treatment on the day of etranacogene dezaparvovec infusion and in the first weeks following infusion to provide sufficient FIX coverage for the initial days post treatment. During the follow-up post infusion, if the endogenous FIX activity result was 5% or greater, continuous routine FIX prophylaxis was discontinued, and further management was based on the investigator’s clinical judgment and patient preference. Continuation or reinitiation of continuous routine FIX prophylaxis could be considered if the endogenous FIX activity was between 2% and 5% in at least 2 consecutive laboratory tests, based on the investigator’s clinical judgment and patient preference. If endogenous FIX activity was less than 2%, continuous routine prophylaxis was continued or reinstated. Additional on-demand and/or intermittent prophylactic FIX treatment could be given after treatment with etranacogene dezaparvovec, if considered necessary.30
Return to routine FIX prophylaxis was defined as having at least 80% of the time being “contaminated” by an exogenous FIX treatment administered during a contiguous 3-month period on or subsequent to the start of month 7 post infusion. At 24 months post infusion of etranacogene dezaparvovec, 52 of the 54 patients had not resumed FIX prophylaxis treatment (i.e., remained free of the previous FIX prophylaxis treatment they had received); as of the 36-month cut-off, 51 patients had not resumed FIX prophylaxis treatment.
Efficacy
Key efficacy results in the full analysis set of the HOPE-B trial are presented in Table 13.
ABR for All Bleeding Events
During the lead-in phase while they were being treated with FIX prophylaxis, 14 of the 54 patients (25.9%) in the study experienced no bleeds; from month 7 to month 18 post etranacogene dezaparvovec infusion, 34 of the same 54 patients (63.0%) experienced no bleeds. The adjusted mean difference in ABR for all bleeding events between etranacogene dezaparvovec and routine FIX prophylaxis was −2.68 (95% CI, −3.81 to −1.55) from month 7 to month 18 post etranacogene dezaparvovec infusion, favouring etranacogene dezaparvovec.
From month 7 to month 36 post infusion of etranacogene dezaparvovec, 23 of the 54 patients (42.6%) treated with etranacogene dezaparvovec had no bleeds compared with 14 of the same 54 patients (25.9%) who did not experience bleeds during the lead-in period while receiving FIX prophylaxis. The adjusted mean difference in ABR for all bleeding events from month 7 to month 36 was −2.65 (95% CI, −3.83 to −1.47) in favour of etranacogene dezaparvovec.
Results from the sensitivity analysis that included only FIX-treated bleeds showed that from month 7 to month 18 post infusion of etranacogene dezaparvovec, the adjusted ABR for etranacogene dezaparvovec and FIX prophylaxis was 0.84 (95% CI, 0.41 to 1.72) and 3.62 (95% CI, 2.79 to 4.71), respectively (rate ratio = 0.23; 95% CI, 0.12 to 0.45). From month 7 to month 36 post infusion of etranacogene dezaparvovec, the adjusted ABR for etranacogene dezaparvovec and FIX prophylaxis was 1.15 (95% CI, 0.53 to 2.51) and 3.62 (95% CI, 2.79 to 4.71), respectively (rate ratio = 0.32; 95% CI, 0.15 to 0.67).
Results from the sensitivity analysis that included only bleeds assessed to be new and true by the investigator showed that from month 7 to month 18 post etranacogene dezaparvovec infusion, the adjusted ABR for etranacogene dezaparvovec and FIX prophylaxis was 1.04 (95% CI, 0.52 to 2.09) and 3.83 (95% CI, 2.93 to 5.01), respectively (rate ratio = 0.27; 95% CI, 0.14 to 0.52). From month 7 to month 36 post etranacogene dezaparvovec infusion, the adjusted ABR for etranacogene dezaparvovec and FIX prophylaxis was 1.04 (95% CI, 0.57 to 1.89) and 3.83 (95% CI, 2.93 to 5.01), respectively (rate ratio = 0.27; 95% CI, 0.16 to 0.47).
One of the 54 patients had an anti-AAV5 titre greater than 1:3,000 during the last testing before the infusion of etranacogene dezaparvovec. In the 53 patients who had an anti-AAV5 nAb titre of less than 3,000 (median = 49.1; range, 1:9 to 1:678), ABR results showed that from months 7 to 18 post etranacogene dezaparvovec infusion, the adjusted ABR for etranacogene dezaparvovec and FIX prophylaxis was 1.07 (95% CI, 0.63 to 1.81) and 3.88 (95% CI, 2.90 to 5.17), respectively (rate ratio = 0.27; 95% CI, 0.17 to 0.43). From months 7 to 36 post infusion, the adjusted ABR for etranacogene dezaparvovec and FIX prophylaxis was 1.07 (95% CI, 0.65 to 1.76) and 3.88 (95% CI, 2.90 to 5.17), respectively (rate ratio = 0.28; 95% CI, 0.17 to 0.46).
ABR for Spontaneous Bleeds
The adjusted mean difference in ABR for spontaneous bleeds between etranacogene dezaparvovec and routine FIX prophylaxis was −1.08 (95% CI, −1.72 to −0.44) from month 7 to month 18 post etranacogene dezaparvovec infusion in favour of etranacogene dezaparvovec. The adjusted mean difference in ABR for spontaneous bleeds from month 7 to month 36 was −0.93 (95% CI, −1.62 to −0.25), in favour of etranacogene dezaparvovec.
ABR for Joint Bleeds
The adjusted mean difference in ABR for joint bleeds between etranacogene dezaparvovec and routine FIX prophylaxis was −1.84 (95% CI, −2.54 to −1.13) from month 7 to month 18 post etranacogene dezaparvovec infusion in favour of etranacogene dezaparvovec. The adjusted mean difference in ABR for joint bleeds from month 7 to month 36 was −1.87 (95% CI, −2.54 to −1.20), favouring etranacogene dezaparvovec.
AIR of FIX Replacement Therapy
From month 7 to month 18 post etranacogene dezaparvovec infusion, the adjusted mean difference in AIR between etranacogene dezaparvovec and routine FIX prophylaxis was −69.96 (95% CI, −79.77 to −60.16) in favour of etranacogene dezaparvovec. Similarly, from month 7 to month 36 post etranacogene dezaparvovec infusion, the adjusted mean difference in AIR was −69.89 (95% CI, −79.70 to −60.08), which favoured etranacogene dezaparvovec.
Annualized Consumption of FIX Replacement Therapy
From month 7 to month 36 post etranacogene dezaparvovec infusion, the adjusted mean difference in annualized consumption of FIX replacement therapy between etranacogene dezaparvovec and routine FIX prophylaxis was −3,037.6 IU/kg (95% CI, −3,617.4 to −2,457.9) in favour of etranacogene dezaparvovec.
Hemophilia Joint Health Score
All patients treated with etranacogene dezaparvovec showed improvement in total HJHS score, with a mean change from baseline of −1.6 (SD = 5.1) at month 12, −2.6 (SD = 5.0) at month 24, and −3.0 (SD = 7.4) at month 36 post infusion.
Patient Reported Outcomes Burdens and Experiences
Change from baseline at month 12 (mean = 0.040; SD = 0.097) and month 24 (mean = 0.034; SD = 0.113) post infusion of etranacogene dezaparvovec both showed improvements in the PROBE summary score in patients treated with etranacogene dezaparvovec. Data from month 36 were not available.
Table 13: Summary of Key Efficacy Results From the HOPE-B Trial (Full Analysis Set)
Outcome | Postinfusion perioda | Lead-in period |
|---|---|---|
Etranacogene dezaparvovec (N = 54) | FIX prophylaxis (N = 54) | |
ABR for all bleeding events (month 7 to month 18 post infusion of etranacogene dezaparvovec) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Number of participants without any bleeds, n (%) | 34 (63.0) | 14 (25.9) |
Cumulative number of bleeding episodes, n | 54 | 136 |
Unadjusted ABRb | 1.08 | 4.11 |
Adjusted ABRc (95% CI) | 1.51 (0.81 to 2.82) | 4.17 (3.20 to 5.44) |
Adjusted mean difference (95% CI) | −2.68 (−3.81 to −1.55) | |
Rate ratio (2-sided 95% Wald CI; P value) | 0.36 (0.20 to 0.64; P = 0.0002) | |
ABR for all bleeding events (month 7 to month 36 post infusion of etranacogene dezaparvovec) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Number of participants without any bleeds, n (%) | 23 (42.6) | 14 (25.9) |
Cumulative number of bleeding episodes, n | 111 | 136 |
Unadjusted ABRb | 0.90 | 4.11 |
Adjusted ABRc (95% CI) | 1.52 (0.81 to 2.85) | 4.17 (3.20 to 5.44) |
Adjusted mean difference (95% CI) | −2.65 (−3.83 to −1.47) | |
Rate ratio (2-sided 95% Wald CI; P value) | 0.36 (0.20 to 0.66; P = 0.0004) | |
ABR for spontaneous bleeds (month 7 to month 18 post infusion of etranacogene dezaparvovec) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Cumulative number of bleeding episodes, n | 14 | 50 |
Unadjusted ABRb | 0.28 | 1.51 |
Adjusted ABRc (95% CI) | 0.44 (0.17 to 1.12) | 1.52 (1.01 to 2.30) |
Adjusted mean difference (95% CI) | −1.08 (−1.72 to −0.44) | |
Rate ratio (2-sided 95% Wald CI; P value) | 0.29 (0.12 to 0.71; P = 0.0034) | |
ABR for spontaneous bleeds (month 7 to month 36 post etranacogene dezaparvovec infusion) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Cumulative number of bleeding episodes, n | 36 | 50 |
Unadjusted ABRb | 0.29 | 1.51 |
Adjusted ABRc (95% CI) | 0.59 (0.25 to 1.40) | 1.52 (1.01 to 2.30) |
Adjusted mean difference (95% CI) | −0.93 (−1.62 to −0.25) | |
Rate ratio (2-sided 95% Wald CI; P value) | 0.39 (0.16 to 0.90; P = 0.0141) | |
ABR for joint bleeds (month 7 to month 18 post etranacogene dezaparvovec infusion) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Cumulative number of bleeding episodes, n | 19 | 77 |
Unadjusted ABRb | 0.38 | 2.33 |
Adjusted ABRc (95% CI) | 0.51 (0.23 to 1.11) | 2.34 (1.74 to 3.16) |
Adjusted mean difference (95% CI) | −1.84 (−2.54 to −1.13) | |
Rate ratio (2-sided 95% Wald CI; P value) | 0.22 (0.10 to 0.46; P < 0.0001) | |
ABR for joint bleeds (month 7 to month 36 post etranacogene dezaparvovec infusion) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Cumulative number of bleeding episodes, n | 41 | 77 |
Unadjusted ABRb | 0.33 | 2.33 |
Adjusted ABRc (95% CI) | 0.47 (0.24 to 0.95) | 2.34 (1.74 to 3.16) |
Adjusted mean difference (95% CI) | −1.87 (−2.54 to −1.20) | |
Rate ratio (2-sided 95% Wald CI; P value) | 0.20 (0.10 to 0.39; P < 0.0001) | |
AIR of FIX replacement therapy (month 7 to month 18 post etranacogene dezaparvovec infusion) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Cumulative number of infusions of FIX therapy, n | 134 | 2,380 |
Unadjusted AIRd | 2.51 | 71.87 |
Adjusted AIRe (95% CI) | 2.52 (0.91 to 6.95) | 72.48 (63.51 to 82.70) |
Adjusted mean difference (95% CI) | −69.96 (−79.77 to −60.16) | |
Rate ratio (2-sided 95% Wald CI; P value) | 0.03 (0.01 to 0.10; P < 0.0001) | |
AIR of FIX replacement therapy (month 7 to month 36 post etranacogene dezaparvovec infusion) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Cumulative number of infusions of FIX therapy, n | 261 | 2,380 |
Unadjusted AIRd | 1.99 | 71.87 |
Adjusted AIRe (95% CI) | 2.59 (1.04 to 6.43) | 72.48 (63.51 to 82.70) |
Adjusted mean difference (95% CI) | −69.89 (−79.70 to −60.08) | |
Rate ratio (2-sided 95% Wald CI; P value) | 0.04 (0.01 to 0.09; P < 0.0001) | |
Annualized consumption of FIX replacement therapy (month 7 to month 36 post etranacogene dezaparvovec infusion) | ||
Number of participants who contributed to the analysis | 54 | 54 |
IU/kg | ||
Unadjusted mean difference (SD) | −3,037.6 (2,124.1) | |
Adjusted mean difference (95% CI; P value) | −3,037.6 (−3,617.4 to −2,457.9; P < 0.0001) | |
HJHSf | ||
Baseline | ||
n | 53 | — |
Mean (SD) | 20.9 (16.6) | — |
Median (range) | 19.0 (0 to 59) | — |
Month 12 post etranacogene dezaparvovec infusion | ||
n | 51 | — |
Mean (SD) | 19.5 (16.8) | — |
Median (range) | 18.0 (0 to 68) | — |
Change from baseline | ||
n | 50 | — |
mean (SD) | −1.6 (5.1) | — |
Median (range) | −0.5 (−19 to 15) | — |
Month 24 post etranacogene dezaparvovec infusion | ||
n | 46 | — |
Mean (SD) | 18.8 (16.3) | — |
Median (range) | 17.5 (0 to 63) | — |
Change from baseline | ||
n | 45 | — |
Mean (SD) | −2.6 (5.0) | — |
Median (range) | −2.0 (−14 to 10) | — |
Month 36 post etranacogene dezaparvovec infusion | ||
n | 42 | — |
Mean (SD) | 16.7 (14.1) | — |
Median (range) | 16.0 (0 to 61) | — |
Change from baseline | ||
n | 42 | — |
Mean (SD) | −3.0 (7.4) | — |
Median (range) | −1.5 (−26 to 13) | — |
PROBE summary scoreg | ||
Baseline | ||
n | 48 | — |
Mean (SD) | 0.769 (0.162) | — |
Median (range) | 0.770 (0.37 to 0.99) | — |
Month 12 post etranacogene dezaparvovec infusion | ||
n | 44 | — |
Mean (SD) | 0.803 (0.158) | — |
Median (range) | 0.845 (0.41 to 0.99) | — |
Change from baseline | ||
n | 43 | — |
Mean (SD) | 0.040 (0.097) | — |
Median (range) | 0.040 (−0.15 to 0.30) | — |
Month 24 post etranacogene dezaparvovec infusion | ||
n | 43 | — |
Mean (SD) | 0.801 (0.140) | — |
Median (range) | 0.800 (0.46 to 1.00) | — |
Change from baseline | ||
n | 41 | — |
Mean (SD) | 0.034 (0.113) | — |
Median (range) | 0.030 (−0.19 to 0.29) | — |
ABR = annualized bleeding rate; AIR = annualized infusion rate; CI = confidence interval; FIX = factor IX; HJHS = Hemophilia Joint Health Score; PROBE = Patient Reported Outcomes Burdens and Experiences; SD = standard deviation.
aPostinfusion period refers to the number of days of observation within the time interval, excluding information before day 21.
bUnadjusted ABR was calculated as the ratio of the number of bleeds to the time of observation (in years).
cAdjusted ABR (parametric model estimate) and comparison of ABR between the lead-in and posttreatment period were estimated from a repeated measures generalized estimating equations negative binomial regression model accounting for the paired design of the trial with an offset parameter to account for the differential collection periods. Treatment period was included as a categorical covariate.
dUnadjusted AIR was calculated as the ratio of the number of infusions of FIX to the time of observation (in years). Usage related to invasive procedures is not included.
eAdjusted AIR (parametric model estimate) and comparison of infusion rate between the lead-in and posttreatment period were estimated from a repeated measures generalized estimating equations negative binomial regression model accounting for the paired design of the trial with an offset parameter to account for the differential collection periods. Treatment period is included as a categorical covariate.
fThe HJHS total score ranges from 0 to 124. A higher score is considered unfavourable.
gThe PROBE summary score ranges from 0 to 1.00. A higher score indicates better health.
Sources: HOPE-B Clinical Study Report29 and drug Reimbursement Review sponsor submission.55
Harms
A summary of harms in the HOPE-B study is shown in Table 14. The data cut-off date for harm results was June 6, 2023 (i.e., 36-month data cut-off). Harms results at the 24-month data cut-off were generally consistent.
Adverse Events
At 36 months post infusion of etranacogene dezaparvovec, all patients had at least 1 TEAE. The system organ classes with the highest incidence of reported TEAEs were infections and infestations (87.0%), followed by musculoskeletal and connective tissue disorders (72.2%) and general disorders and administration-site conditions (59.3%). The TEAEs reported in more than 20% of the safety population of the HOPE-B trial were arthralgia (44.4%), headache (33.3%), nasopharyngitis (27.8%), fatigue (27.8%), ALT increased (24.1%), and back pain (22.2%).
During the lead-in period (excluding discontinuers), 68.5% patients experienced at least 1 TEAE. The system organ classes with the highest incidence of reported TEAEs were infections and infestations (35.2%), followed by musculoskeletal and connective tissue disorders (22.2%) and gastrointestinal disorders (13.0%). The only AE reported in more than 10% of patients was nasopharyngitis (14.8%).
Serious Adverse Events
At 36 months post infusion of etranacogene dezaparvovec, 27.8% of the safety population had at least 1 serious TESAE. The system organ classes with the highest incidence of reported TESAEs were infections and infestations (7.4% of patients), consisting of 5 events (biloma infected, COVID-19, cellulitis, device-related infection, diverticulitis intestinal hemorrhagic), and musculoskeletal and connective tissue disorders (5.6% of patients), consisting of 3 events (hemarthrosis, musculoskeletal chest pain, osteoarthritis).
During the lead-in period (excluding discontinuers), 7.4% of patients experienced TESAEs, of which 5.6% were reported in the system organ classes of musculoskeletal and connective tissue disorders.
Withdrawals Due to Adverse Events
One patient discontinued infusion of the study drug due to an event of hypersensitivity after approximately 10% of the full dose of study drug was administered; this patient did not have FIX expression.
Mortality
One patient died due to a fatal event of cardiogenic shock 464 days (approximately 15 months) post infusion of etranacogene dezaparvovec. According to the product monograph,2 the patient, who was aged 75 years at screening and had numerous cardiovascular and urologic risk factors, died of urosepsis and cardiogenic shock at month 15 post dose (aged 77 years), an event that was determined to be not treatment related.
Notable Harms
Post infusion of etranacogene dezaparvovec, an increase in ALT occurred in 24.1% of patients (13 of 54), followed by an increase in AST (9 of 54; 16.7%), anemia (5 of 54; 9.3%), and infusion-related reaction (3 of 54; 5.6%). Only 1 patient had anemia during the lead-in period when receiving FIX prophylaxis.
Table 14: Summary of Harms Results in the HOPE-B Study (Safety Population)
Harms | Lead-in perioda FIX prophylaxis (N = 54) | Post infusion of etranacogene dezaparvovec at 36 months (N = 54) |
|---|---|---|
Most common adverse events, n (%) | ||
≥ 1 adverse event | 37 (68.5) | 54 (100.0) |
Infections and infestations | 19 (35.2) | 47 (87.0) |
COVID-19 | 0 | 17 (31.5) |
Nasopharyngitis | 8 (14.8) | 15 (27.8) |
Musculoskeletal and connective tissue disorders | 12 (22.2) | 39 (72.2) |
Arthralgia | 4 (7.4) | 24 (44.4) |
Back pain | 1 (1.9) | 12 (22.2) |
Pain in extremity | 1 (1.9) | 10 (18.5) |
General disorders and administration-site conditions | 2 (3.7) | 32 (59.3) |
Fatigue | 0 | 15 (27.8) |
Influenza-like illness | 1 (1.9) | 7 (13) |
Gastrointestinal disorders | 7 (13.0) | 28 (51.9) |
Toothache | 2 (3.7) | 7 (13.0) |
Diarrhea | 1 (1.9) | 7 (13.0) |
Nausea | 2 (3.7) | 6 (11.1) |
Injury, poisoning, and procedural complications | 4 (7.4) | 26 (48.1) |
Ligament sprain | 1 (1.9) | 6 (11.1) |
Investigations | 0 | 28 (51.9) |
ALT increased | 0 | 13 (24.1) |
AST increased | 0 | 9 (16.7) |
Blood creatine phosphokinase increased | 0 | 8 (14.8) |
Nervous system disorders | 1 (1.9) | 26 (48.1) |
Headache | 0 | 18 (33.3) |
Dizziness | 0 | 6 (11.1) |
Respiratory, thoracic, and mediastinal disorders | 6 (11.1) | 23 (42.6) |
Cough | 0 | 9 (16.7) |
Oropharyngeal pain | 2 (3.7) | 7 (13.0) |
Vascular disorders | 2 (3.7) | 15 (27.8) |
Hypertension | 1 (1.9) | 8 (14.8) |
Metabolism and nutrition disorders | 0 | 11 (20.4) |
Blood and lymphatic system disorders | 4 (7.4) | 11 (20.4) |
Hepatobiliary disorders | 0 | 9 (16.7) |
Hepatic steatosis | 0 | 7 (13.0) |
Serious adverse events, n (%) | ||
Patients with ≥ 1 serious adverse event | 4 (7.4) | 15 (27.8) |
Infections and infestations | 0 | 4 (7.4) |
Musculoskeletal and connective tissue disorders | 3 (5.6) | 3 (5.6) |
Patients who stopped treatment due to adverse events, n (%) | ||
Patients who stopped | 0 | 1 (1.9) |
Hypersensitivity | 0 | 1 (1.9) |
Death, n (%) | ||
Patients who died | 0 | 1 (1.9) |
Cardiogenic shock | 0 | 1 (1.9) |
Notable harms, n (%) | ||
ALT increased | 0 | 13 (24.1) |
AST increased | 0 | 9 (16.7) |
Anemia | 1 (1.9) | 5 (9.3) |
Infusion-related reaction | 0 | 3 (5.6) |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; FIX = factor IX.
aDiscontinuers were excluded from the lead-in period.
Sources: HOPE-B Clinical Study Report29 and drug Reimbursement Review sponsor submission.55
Internal Validity
The only eligible study identified in the sponsor-conducted SLR was HOPE-B (N = 54), a phase III, nonrandomized, single-arm, open-label clinical trial that included a lead-in phase (i.e., patients were on FIX prophylaxis for at least 6 months) before the infusion of etranacogene dezaparvovec. The data collected from patients during the lead-in phase were used as a self-control to measure some etranacogene dezaparvovec safety and efficacy outcomes (e.g., ABR for all bleeding events, ABR for spontaneous bleeds, ABR for joint bleeds, AIR, and annualized FIX consumption). Overall, the trial design (e.g., nonrandomized, open label, single arm) was considered appropriate and acceptable in the field of hemophilia B, although the interpretation of the study findings could be challenging and limited.
According to the clinical experts consulted by CADTH, the inclusion and exclusion criteria for the HOPE-B trial were appropriate and generally reflected the patients they would expect to treat in clinical practice. It was noted that 67 patients were enrolled in the lead-in phase and only 54 patients were treated with etranacogene dezaparvovec, which was assessed for efficacy and safety. However, it was determined by CADTH that the potential selection bias, due to a considerable number of patients being excluded, was low. First, the reasons for excluding 13 patients during the lead-in phase were considered reasonable. Second, according to the clinical experts consulted by CADTH, the baseline and clinical characteristics for the 54 patients who received etranacogene dezaparvovec versus the 67 patients who entered the lead-in phase were generally similar, suggesting that the population receiving treatment was not expected to be systematically different from the lead-in study population.
Patient compliance with FIX prophylaxis treatment during the lead-in phase remained unclear, as the sponsor stated in its correspondence with CADTH that summary-level statistics on compliance were not available. Due to the single-arm, open-label design, reliable assessments of patient-reported outcomes (e.g., HRQoL end points) could not be made. In the primary analyses, the documentation of bleeding events in the HOPE-B study relied on the use of an e-diary by patients, the contents of which were reviewed and assessed by the investigator. Based on details provided by the sponsor upon request, CADTH determined that the potential risk of bias that might lead to the exaggeration of the treatment effects of etranacogene dezaparvovec (i.e., ABR outcomes) was likely low. Results from the sensitivity analyses, which included only bleeds assessed to be new and true by the investigator, were consistent with the results from the primary analyses.
In the HOPE-B trial, participants were allowed to receive corticosteroids post infusion of etranacogene dezaparvovec for conditions such as an increase in AST or ALT. According to the clinical experts consulted by CADTH, there were no serious concerns with the use of corticosteroids post infusion. In addition, the rate ratios from the sensitivity analysis for etranacogene dezaparvovec and FIX prophylaxis for ABR for all bleeding events, which excluded periods with systemic corticosteroid use post infusion of etranacogene dezaparvovec, were similar to the rate ratios from the primary analyses (data not shown).
According to the clinical experts consulted by CADTH, the conditions for use of FIX prophylaxis post infusion of etranacogene dezaparvovec were generally considered appropriate, and so was the definition of “return to routine FIX prophylaxis” in the context of the HOPE-B trial. A sensitivity analysis that included time intervals for exogenous FIX use during the postinfusion period shows similar results for ABR for all bleeding events, suggesting that a postinfusion FIX prophylaxis regimen may not modify the treatment effects of etranacogene dezaparvovec.
In the HOPE-B study, multiple statistical tests were conducted for several end points in a fixed sequence. However, multiplicity was controlled only for analyses using data from the month 18 data cut-off and not for analyses with data from the month 24 or month 36 data cut-offs, which might have resulted in potential inflation of the type I error rates. There were some concerns about the assumptions for the statistical model that were adopted to inform the relative efficacy of etranacogene dezaparvovec against FIX prophylaxis. The first assumption was that the rate of bleeding during FIX prophylaxis in the lead-in phase would be comparable to the bleeding rate post infusion of etranacogene dezaparvovec during the follow-up phase, provided FIX prophylaxis had not been discontinued and etranacogene dezaparvovec had not been given as an intervention. This assumption was considered reasonable by the clinical experts consulted by CADTH. This assumption is required to interpret the differences observed in bleeding rates before and after treatment with etranacogene dezaparvovec in the HOPE-B study. The second assumption — that the negative binomial mixed model implies a constant bleed rate within each period of study — can make it challenging to interpret the magnitude of the effect estimates of etranacogene dezaparvovec compared with FIX prophylaxis. Specifically, the reported estimates of the relative ABRs describe a weighted average of the rate of bleeding over time that is dependent on the observed censoring mechanism. This weighted average can be overly optimistic and fail to accurately capture waning efficacy over time.
External Validity
There are several considerations related to the generalizability of the HOPE-B trial. First, the evidence from the currently available follow-up period (i.e., 36 months) in the HOPE-B study may not be adequate to inform long-term efficacy and safety, given the expectation of the long-lasting effects of etranacogene dezaparvovec.
In addition, the HOPE-B trial included patients who had congenital hemophilia B with known severe or moderately severe FIX deficiency (≤ 2% of normal circulating FIX) and who had been on stable prophylaxis for at least 2 months before screening. However, the indication does not restrict treatment to patients with severe or moderately severe hemophilia B (≤ 2% of normal circulating FIX) or require eligible patients to have been on a stable FIX prophylaxis regimen for 2 months. According to the clinical experts consulted by CADTH, the eligibility criteria for patients in the HOPE-B study were generally aligned with the indication. However, the clinical experts also noted that some patients, including those who have a FIX level greater than 2% and present severe clinical symptoms, and patients who require but are not receiving stable FIX prophylaxis treatment, may also benefit from etranacogene dezaparvovec.
The indication does not specify sex or gender (i.e., it includes both men and women), while the product monograph states that etranacogene dezaparvovec is not intended for administration in women. The pivotal HOPE-B trial enrolled only male patients, per the trial protocol.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and randomized controlled trials identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:56,57
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
According to GRADE guidance, nonrandomized comparative evidence starts at low certainty and noncomparative evidence starts at very low certainty. The CADTH review team carefully assessed the risk of selection bias and the potential for unmeasured confounding of the pivotal intrapatient single-arm trial, which compared bleeding pre- and post intervention. The GRADE report captures the study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of assessment of the certainty of the evidence was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the assessment of the certainty of the evidence for ABR for all bleeding events were set according to the presence or absence of an important effect based on thresholds informed by the sponsor and agreed upon by the clinical experts consulted by CADTH for this review. The target of the assessment of the certainty of the evidence was the presence or absence of any (non-null) effect for ABR for spontaneous bleeds, ABR for joint bleeds, AIR, and annualized FIX consumption due to the lack of a formal MID estimate. The certainty of the evidence was summarized narratively for HJHS, PROBE, and harms outcomes either due to lack of comparators or lack of formal statistical testing.
Results of GRADE Assessments
Table 2 and Table 3 present the GRADE summary of findings in the HOPE-B study for etranacogene dezaparvovec versus FIX prophylaxis in adult patients with hemophilia B.
Indirect and Other Comparative Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
Evidence for the efficacy of etranacogene dezaparvovec was assessed using the pivotal single-arm HOPE-B trial. Accordingly, head-to-head comparative efficacy has not been established against products that may be relevant to patients in Canada with hemophilia B. As such, the sponsor provided an ITC and other comparative evidence using patient-level data as evidence of the comparative efficacy of etranacogene dezaparvovec versus other therapies.
Description of the Indirect Treatment Comparison
The sponsor submitted 1 ITC report for review that used a previously conducted SLR published in a peer-reviewed journal in 2019 as its source of information on available trial evidence within the same analysis population.58 Owing to a lack of connected evidence, the sponsor performed a feasibility assessment on conducting an ITC between etranacogene dezaparvovec and the following comparator therapies and their associated clinical trials: rIX-FP (Idelvion) from the PROLONG-9FP study,59 rFIXFc (Alprolix) from the B-LONG study,60 pegylated nonacog beta pegol (Rebinyn) from the Paradigm 2 study,61 and nonacog alfa (BeneFIX) from the study NCT00093171.62 The sponsor noted that none of the identified trials had shared treatment nodes, and therefore indicated that approaches such as network meta-analysis were not feasible. Owing to limitations in reporting data, the sponsor did not conduct indirect comparisons relative to nonacog alfa, as the sponsor was unable to identify sufficient information on patient baseline characteristics or outcome definitions for comparison. For the setting in which patient-level data were available for both etranacogene dezaparvovec and rIX-FP, the sponsor used an inverse-weighted estimator of group differences; for rFIXFc and pegylated nonacog beta pegol, the sponsor used an unanchored MAIC approach to overcome the absence of patient-level data.
Indirect Treatment Comparison Design
Objectives
The objective of the sponsor-submitted ITC was to determine the comparative efficacy of etranacogene dezaparvovec against rIX-FP, rFIXFc, pegylated nonacog beta pegol, and nonacog alfa for the prophylactic treatment of severe or moderately severe hemophilia B.
Study Selection Methods
The sponsor utilized a published SLR58 for its evidence generation (Table 15). The sponsor reported that this published review accessed PubMed and Embase for data, but no information was provided on the review process with respect to duplicate reviews or adjudications. No further details were provided regarding the process used to extract data or to assess quality.
Table 15: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Patients with severe or moderately severe hemophilia B (FIX levels ≤ 2%) aged ≥ 12 years (adolescent or adult) receiving prophylactic rFIX treatment. |
Intervention |
|
Comparator | Any, not required |
Outcome | ABR, ABR for spontaneous bleeds, ABR for joint bleeds |
Study designs | Phase III trials |
Publication characteristics | Original full-text articles published between 1966 and October 17, 2018 |
Exclusion criteria |
|
Databases searched | PubMed, Embase |
Selection process | Publications underwent an initial screening based on the title and abstract using these inclusion and exclusion criteria. Potentially relevant publications then underwent a second screening based on the full text of the article. |
Data extraction process | The relevant data from all eligible publications were collected and aggregated to allow further analysis. Data were extracted using a singular data extraction form according to the following outcome measures: ABR, ABR for spontaneous bleeds, and ABR for joint bleeds |
Quality assessment | Quality assessment was not described. |
ABR = annualized bleeding rate; FIX = factor IX; gc = genome copy; ITC = indirect treatment comparison; pdFIX = plasma-derived factor IX; rFIX = recombinant factor IX; rFIXFc = recombinant factor IX Fc fusion protein; rIX-FP = recombinant factor IX albumin fusion protein.
Sources: Sponsor-submitted ITC;63 details included in the table are from the sponsor’s summary of clinical evidence.1
ITC and Other Comparative Analysis Methods
The ITC and other comparative analysis methods are summarized in Table 16. The sponsor noted that among the included studies, no common comparator was identified. As unanchored comparisons were necessary, the sponsor described a process to identify potentially clinically relevant covariates that could be considered as part of the feasibility assessment and adjusted for in subsequent analyses. To identify the relevant covariates to adjust for in treating bleeding rates in patients with hemophilia B, the sponsor conducted a ranked order questionnaire. The questionnaire was completed by 5 clinicians, 3 from the sponsor’s organization and 2 external physicians from unspecified UK organizations outside of the sponsor’s organization. The physicians were asked to consider which covariates were relevant prognostic factors or treatment-effect modifiers and to rank them in order of importance for each of the efficacy outcomes considered: ABR, ABR for spontaneous bleeds, and ABR for joint bleeds. The top 5 ranked factors for ABR were severity of hemophilia B, prior ABR, prior FIX regimen (prophylactic versus on demand), prior presence of target joints, and age. For FIX consumption, the top 5 ranked factors were severity of hemophilia B, prior FIX regimen (prophylactic versus on demand), prior ABR, prior presence of target joints, and prior FIX product class (EHL versus standard half-life [SHL]). For patient-reported outcomes, the top 5 ranked factors were severity of hemophilia B, prior ABR, prior FIX regimen (prophylactic versus on demand), prior presence of target joints, and age. No quantitative data or references were provided with respect to the impact of any of the listed covariates of potential interest.
For ABR, ABR for spontaneous bleeds, and ABR for joint bleeds, the remaining clinical factors that were assessed were, in order: prior FIX product class (EHL versus SHL), body mass index (BMI), weight, prior exposure days of treatment with an FIX protein, prior FIX product use, ALT threshold, AST threshold, history of FIX inhibitor antibodies, HIV status, total bilirubin threshold, family member with FIX inhibitor antibodies, and duration of diagnosed hemophilia B. For FIX consumption, the remaining clinical factors assessed were, in order: BMI, age, prior exposure days of treatment with an FIX protein, weight, ALT threshold, prior FIX product use, total bilirubin threshold, history of FIX inhibitor antibodies, family member with FIX inhibitor antibodies, HIV status, AST threshold, and duration of diagnosed hemophilia B.
The order of assessment was considered during model selection. The sponsor provided univariable adjustments performed sequentially, with MAIC or IPTW analyses run separately, adjusting for 1 additional factor at a time as per the ranked importance scale noted previously. The sponsor indicated it had attempted to balance ESS against improvements in the between-treatment standardized mean differences, although the criteria for what qualified as balance were not provided.
For ABR, ABR for spontaneous bleeds, and ABR for joint bleeds, the remaining clinical factors that were assessed were, in order: prior FIX product class (EHL versus SHL), BMI, weight, prior exposure days of treatment with an FIX protein, prior FIX product use, ALT threshold, AST threshold, history of FIX inhibitor antibodies, HIV status, total bilirubin threshold, family member with FIX inhibitor antibodies, and duration of diagnosed hemophilia B. For FIX consumption, the remaining clinical factors that were assessed were, in order: BMI, age, prior exposure days of treatment with an FIX protein, weight, ALT threshold, prior FIX product use, total bilirubin threshold, history of FIX inhibitor antibodies, family member with FIX inhibitor antibodies, HIV status, AST threshold, and duration of diagnosed hemophilia B. During the sponsor’s feasibility assessment, it was noted that the trial data for nonacog alfa were not considered appropriate for use in an unanchored MAIC analysis. Specifically, the sponsor noted that within the nonacog alfa trial, patient demographic details were available only for age, race, prior FIX product class status, and family members with a history of FIX inhibitor antibodies. As such, many of the identified prognostic and treatment effect–modifying factors were not available to be incorporated within the sponsor’s model. Further, the sponsor noted that these characteristics were reported only for the total trial population and not just the population utilizing nonacog alfa in the prophylactic setting. With respect to outcomes, the identified trial assessing nonacog alfa prophylaxis was not noted as having a definition for how bleeds were counted for ABR. As such, the sponsor did not conduct any formal analyses for the comparison of etranacogene dezaparvovec versus nonacog alfa. The sponsor did provide unadjusted comparisons within the methods section for nonacog alfa but, owing to the methodological issues identified by the sponsor, these comparisons are not reported within this CADTH review.
For the inverse-weighted comparison of etranacogene dezaparvovec versus rIX-FP, the sponsor excluded patients from the rIX-FP trial to ensure the population included in the analysis aligned with the enrolment criteria of the HOPE-B trial. Specifically, patients from the PROLONG-9FP study were removed if: they were aged 12 to 17 years (n = 7), had an ALT level greater than twice the upper limit of normal (n = 2), or had an AST level greater than twice the upper limit of normal (n = 2).
For the ITCs comparing etranacogene dezaparvovec with rFIXFc and pegylated nonacog beta pegol, each analysis consisted of different patient populations. For the rFIXFc trial, the primary analysis population was restricted to a subset of patients receiving weekly prophylaxis. For the pegylated nonacog beta pegol trial, the primary analysis population was restricted to patients who had received background prophylaxis with FIX products before the trial. In both cases, the primary analysis population lacked detailed patient demographics. Secondary analyses were conducted in a broader patient population without focusing solely on patients who received prophylaxis with FIX products before the trial. This was done because there was more comprehensive reporting on patient baseline characteristics for the broader patient population. Two patients out of 53 (4%) from the HOPE-B trial (assessing etranacogene dezaparvovec) were not included in any of the ITCs. One patient was excluded due to data protection requirements, and 1 patient was excluded because they had an anti-AAV5 nAb titre greater than 3,000 at baseline, whereas the planned etranacogene dezaparvovec label will exclude patients with anti-AAV5 nAb titres greater than 700. The sponsor noted that, before reviewing any data from the submitted analyses, both patients were excluded by study teams that were outside of those involved in the generation of the report.
Statistical Methods for rIX-FP Comparison
For the IPTW approach comparing etranacogene dezaparvovec with rIX-FP, the sponsor utilized entropy balancing64 using first and second (mean and variance) moments for patient weights, an equivalent methods-of-moment estimator. Weighted generalized linear regression with negative binomial distribution and a log link were used for rate outcomes, or a logit link for binary outcomes, and linear regression was used for continuous outcomes. A binomial distribution with a logit link function (i.e., logistic regression) was used for binary outcomes containing the proportion of patients with no events during the randomized period (i.e., percentage with 0 ABR events, percentage with 0 ABR events for spontaneous bleeds, and percentage with 0 ABR events for joint bleeds). Standard errors were estimated using the sandwich estimator. A Gaussian (normal) distribution with an identity link function (i.e., linear regression) was used for the continuous annualized FIX consumption outcome. Standard errors were estimated using the sandwich estimator.65 These sandwich standard errors were used to construct 2-sided 95% Wald CIs and the associated testwise P values for comparisons. Relative treatment effects were transformed into the natural scale (e.g., RR, odds ratio, mean difference) after estimation.
Statistical Methods for rFIXFc Comparison
For the comparison of etranacogene dezaparvovec versus rFIXFc, the propensity score was estimated using a method-of-moments estimator. The comparative estimate from the HOPE-B study was derived using a weighted, intercept-only generalized linear model. A negative binomial distribution with a log link function was used for the following rate outcomes: ABR (total), ABR for spontaneous bleeds, and ABR for joint bleeds. the model-reported intercept is an estimate of the ABR-related outcome on the log scale in a hypothetical scenario in which patients from the respective comparator trial received etranacogene dezaparvovec. Specifically, a binomial distribution with logit link function (i.e., logistic regression) was used for a binary outcome and the percentage with 0 ABR events (i.e., the proportion of patients who did not experience any ABR events in the trial period), where the intercept represents the log odds of the outcome of interest had patients from the respective comparator trial received etranacogene dezaparvovec. Standard errors were estimated using the sandwich estimator.65 These sandwich standard errors were used to construct 2-sided 95% Wald CIs and the associated testwise P values for comparisons.
Statistical Methods for Pegylated Nonacog Beta Pegol Comparison
For the comparison of etranacogene dezaparvovec versus pegylated nonacog beta pegol, the propensity score was estimated using a method-of-moments estimator. The comparative estimate from the HOPE-B study was derived using a weighted, intercept-only generalized linear model, and appropriate distributions and link functions were used to ensure that a suitable scale was used for estimation per outcome, applying the patient weights. The model-reported intercept is an estimate of the ABR-related outcome on the log scale in a hypothetical scenario in which patients from the respective comparator trial received etranacogene dezaparvovec. Specifically, a binomial distribution with a logit link function (i.e., logistic regression) was used for binary outcomes containing the percentage of patients with 0 ABR events (i.e., the proportion of patients who did not experience any ABR events in the trial period), where the intercept represents the log odds of the outcome of interest had patients from the respective comparator trial received etranacogene dezaparvovec. A Poisson distribution with log link function was used for ABR (total) and ABR for spontaneous bleeds outcomes. The choice of model was based on the assumed model of the reported estimates within the pegylated nonacog beta pegol trial. Standard errors were estimated using the sandwich estimator.65 These sandwich standard errors were used to construct 2-sided 95% Wald CIs and the associated testwise P values for comparisons.
Table 16: ITC Analysis Methods
Methods | Etranacogene dezaparvovec versus rIX-FP (Idelvion) | Etranacogene dezaparvovec versus rFIXFc (Alprolix) | Etranacogene dezaparvovec versus pegylated nonacog beta pegol (Rebinyn) | ||
|---|---|---|---|---|---|
Primary | Secondary | Primary | Secondary | ||
Propensity score estimation |
|
| |||
Data sources |
|
|
|
|
|
Outcomes | Rate:
Binary:
Continuous:
| Rate:
Continuous:
| Rate:
Binary:
Continuous:
| Rate:
| Rate:
Binary:
Continuous:
|
Outcome model | Weighted (ATT) generalized linear model with negative binomial distribution and log link (rate outcomes) or logit link (binary outcomes), or Gaussian distribution and identity link (continuous outcomes) | Weighted (ATC), intercept-only generalized linear model with negative binomial distribution and log link (rate outcomes) or logit link (binary outcomes), or Gaussian distribution with identity link (continuous outcomes) | Weighted (ATC), intercept-only generalized linear model with Poisson distribution and log link (rate outcomes), binomial distribution and logit link (binary outcomes), or Gaussian distribution with identity link (continuous outcomes) | ||
Covariates included | Matched only:
Univariable:a
Multivariable:
| Naive:
Univariable:
| Naive:
Univariable:a
Multivariable:
| Naive:
Univariable:a
| Naive:
Univariable:a
Multivariable:
|
Assessment of distribution or overlap in propensity score or patient weights |
| ||||
Balance assessment | Absolute SMDs before and after weighting were compared to assess covariate balance between populations | ||||
Sensitivity analysis | None | Base-case analyses with alternative imputation of standard errors for rate outcomes: 1. Equivalent variance of the log rates from the HOPE-B and B-LONG studies for all outcomes 2. Poisson model estimate of variance of the log rate from the total number of bleeds and total exposure time from the B-LONG study. | None | ||
Subgroup analysis | None | None | None | None | None |
ABR = annualized bleeding rate; ALT = alanine aminotransferase; AST = aspartate aminotransferase; ATC = average treatment effect in the comparator; ATT = average treatment effect in the treated; BMI = body mass index; ESS = effective sample size; FIX = factor IX; Haem-A-QoL = Haemophilia Quality of Life Questionnaire for Adults; IPD = individual patient data; IPTW = inverse probability of treatment weighting; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; rFIXFc = recombinant factor IX Fc fusion protein; rIX-FP = recombinant factor IX albumin fusion protein; SMD = standardized mean difference.
aFor univariable analysis with multiple covariates, the sponsor provided multiple univariable-adjusted comparisons individually.
Sources: Sponsor-submitted ITC;63 details included in the table are from the sponsor’s summary of clinical evidence.1
Results of Sponsor-Submitted ITC
Summary of Included Studies
The assessment of homogeneity for the sponsor-submitted ITC is shown in Table 17. None of the included trials were found to have shared treatment regimens. Trials were broadly similar with respect to hemophilia B severity, though there was noted variability in ABR before randomization and treatment across the trials, ranging from 3.35 (PROLONG-9FP study) through to 7.49 (Paradigm 2 study). Clinical definitions were broadly similar across the trials, although the PROLONG-9FP study’s primary analysis definitions for bleeding events were noted by the sponsor to be more restrictive compared with other studies. The duration of follow-up and design varied, predominantly because the PROLONG-9FP study had a treatment run-in period that was absent in other trials.
Table 17: Assessment of Homogeneity for Sponsor-Submitted ITC
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | In the HOPE-B, PROLONG-9FP, Paradigm 2, and B-LONG studies, the proportion of patients with severe hemophilia B ranged from 72.7% (PROLONG-9FP) to 82.8% (Paradigm 2), with remaining patients classified as moderate and proportions ranging from 27.5% (PROLONG-9FP) to 17.2% (Paradigm 2). Baseline ABR in the primary analysis populations was similar overall but was lowest in the PROLONG-9FP study at 3.35, followed by 4.1 in the HOPE-B study and 5.5 in the B-LONG study, and were highest in the Paradigm 2 study at 7.49. |
Treatment history |
|
Trial eligibility criteria |
|
Dosing of comparators | Etranacogene dezaparvovec in the HOPE-B study was administered as a single, 1-time infusion of 2 × 1013 gc/kg; rIX-FP in the PROLONG-9FP study was administered as a 35 IU/kg to 50 IU/kg weekly infusion. rFIXFc in the B-LONG study was administered as a 50 IU/kg weekly infusion. Pegylated nonacog beta pegol in the Paradigm 2 study was administered as a 40 IU/kg weekly infusion. |
Definitions of end points | Differences were noted with respect to definitions for bleeding-related end points. The HOPE-B trial reported several sensitivity analyses with varied definitions, and the sponsored proposed the use of a sensitivity analysis that repeats the main analysis for ABR using the FAS population while considering only bleeds treated with exogenous FIX that are assessed to be new and true by the investigator. HOPE-B study:
PROLONG-9FP study:
B-LONG study:
Paradigm 2 study:
|
Timing of end point evaluation | All bleeding-related end points and annualized FIX consumption were calculated as annualized rates based on the length of follow-up. The HOPE-B study had a longer follow-up than the other trials (24 months) but required 3 to 6 months following infusion to reach full efficacy. The B-LONG and Paradigm 2 trials had follow-ups of 52 weeks. The HOPE-B study had a median follow-up of 18 months and the PROLONG-9FP study had a follow-up of 19 months. Median follow-up for the B-LONG and Paradigm 2 studies was not reported. Assessment of change from baseline Haem-A-QoL was at 24 months for the HOPE-B study (etranacogene dezaparvovec) compared with 12 months for the B-LONG (rFIXFc) trial. Patients in the HOPE-B study with missing 24-month data (number not provided by sponsor) for Haem-A-QoL were treated under an LOCF framework, with values taken from the 6- or 12-month assessments post treatment. Assessment of change from baseline in Haem-A-QoL and EQ-5D results in the Paradigm 2 trial took place at an unspecified time point. Data from the HOPE-B trial were assessed at 24 months. Patients in the HOPE-B study with missing 24-month data (number not provided by sponsor) for the Haem-A-QoL or EQ-5D were treated under an LOCF framework, with values taken from the 6- or 12-month assessments post treatment. |
Study design | All trials were phase III, multicentre trials. All trials were international, and all were single-arm, open-label trials except for Paradigm 2, a single-blind trial in which patients were randomized to receive either 10 IU/kg or 40 IU/kg weekly prophylaxis. The HOPE-B study included a 6-month lead-in phase in which patients received prophylactic FIX replacement therapy before etranacogene dezaparvovec infusion. No other trials included such a lead-in phase. |
ABR = annualized bleeding rate; ALT = alanine aminotransferase; AST = aspartate aminotransferase; BMI = body mass index; FAS = full analysis set; FIX = factor IX; gc/kg = genome copy per kilogram; ITC = indirect treatment comparison; LOCF = last observation carried forward; rFIXFc = recombinant factor IX Fc fusion protein; rIX-FP = recombinant factor IX albumin fusion protein; ULN = upper limit of normal.
Sources: Sponsor-submitted ITC;63 details included in the table are from the sponsor’s summary of clinical evidence.1
Results
The methods, analysis populations, outcome definitions, and covariate balancing varied across the 3 comparative analyses considered in this report. This section is divided into results for each individual comparison.
Etranacogene Dezaparvovec Versus rIX-FP (Idelvion)
For the comparison of etranacogene dezaparvovec versus rIX-FP, the sponsor provided a summary of the baseline characteristics of the 2 trials of interest, presented in Table 18. Before adjustment, the following characteristics were imbalanced (defined as a standardized mean difference [SMD] > 0.2): severity of hemophilia B, prior ABR, age, prior FIX product class (EHL versus SHL), BMI, weight, HIV status, ALT and AST thresholds, prior FIX product use, total bilirubin threshold, family members with FIX inhibitor antibodies, and duration of diagnosed hemophilia B. Following IPTW, the following characteristics remained imbalanced (defined as an SMD > 0.2): prior FIX product class (EHL versus SHL), BMI, weight, HIV status, ALT and AST thresholds, prior FIX product use, total bilirubin threshold, family members with FIX inhibitor antibodies, and duration of diagnosed hemophilia B. For the PROLONG-9FP study, the ESS of the ITC analysis population following IPTW was 18.2.
Table 18: Baseline Characteristics of Patients in the HOPE-B Study and PROLONG-9FP Study (Base-Case Analysis)
Characteristics | Etranacogene dezaparvovec (HOPE-B study) ITC analysis set (N = 51) | rIX-FP (Idelvion, PROLONG-9FP study) efficacy population analysis set (N = 40) | ||
|---|---|---|---|---|
Before IPTW (unadjusted) (N = 38) | Before IPTW (matched only) (N = 28; 73.7%) | After IPTW (adjusted) (ESS = 18.2; 47.9%) | ||
Severity of hemophilia Ba | ||||
< 1 IU/dL | 41 (80.4) | 27.0 (71.1) | 20.0 (71.4) | 22.5 (80.4) |
1 IU/dL to 2 IU/dL | 10 (19.6) | 11.0 (28.9) | 8.0 (28.6) | 5.5 (19.6) |
SMD | — | 0.219 | 0.211 | 0 |
Prior ABRb | ||||
Mean (SD) | 4.10 (4.11) | 3.21 (3.70) | 2.89 (3.28) | 4.10 (4.15) |
SMD | — | 0.226 | 0.323 | 0 |
Prior FIX regimen,b n (%) | ||||
Prophylaxis | 51.0 (100.0) | 38.0 (100.0) | 28.0 (100.0) | 28.0 (100.0) |
On demand | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
SMD | — | 0 | 0 | 0 |
Agec (years) | ||||
Mean (SD) | 40.41 (14.97) | 31.18 (14.96) | 34.64 (13.91) | 40.41 (15.10) |
SMD | — | 0.617 | 0.399 | 0 |
Prior FIX product classb | ||||
EHL | 30.0 (58.8) | 12.0 (31.6) | 9.0 (32.1) | 9.0 (32.1) |
SHL | 21.0 (41.2) | 26.0 (68.4) | 19.0 (67.9) | 19.0 (67.9) |
SMD | — | 0.569 | 0.556 | 0.556 |
BMIa (kg/m2) | ||||
Mean (SD) | 27.29 (5.16) | 23.12 (4.14) | 23.43 (4.19) | 23.65 (4.21) |
SMD | — | 0.892 | 0.822 | 0.773 |
Weighta (kg) | ||||
Mean (SD) | 86.18 (19.27) | 69.78 (14.80) | 71.58 (14.22) | 72.99 (15.57) |
SMD | — | 0.954 | 0.862 | 0.753 |
Prior exposure days of treatment with FIX proteina | ||||
> 150 days | 51.0 (100.0) | 38.0 (100.0) | 28.0 (100.0) | 28.0 (100.0) |
≤ 150 days | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
SMD | — | 0 | 0 | 0 |
Prior FIX product,b n (%) | ||||
rFIX | 21.0 (41.2) | 17.0 (44.7) | 11.0 (39.3) | 9.9 (35.4) |
rIX-FP | 8.0 (15.7) | 12.0 (31.6) | 9.0 (32.1) | 9.0 (32.1) |
rFIXFc | 21.0 (41.2) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
Pegylated nonacog beta pegol | 1.0 (2.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
pdFIX | 0.0 (0.0) | 9.0 (23.7) | 8.0 (28.6) | 9.1 (32.4) |
SMD | — | 1.505 | 1.589 | 1.661 |
ALT threshold,a (IU/L) | ||||
Mean (SD) | 21.37 (13.81) | 31.07 (22.17) | 28.05 (14.18) | 25.57 (13.22) |
SMD | — | 0.525 | 0.477 | 0.311 |
AST threshold,a (IU/L) | ||||
Mean (SD) | 19.92 (7.81) | 29.93 (23.81) | 25.55 (10.60) | 23.98 (7.98) |
SMD | — | 0.565 | 0.605 | 0.514 |
History of FIX inhibitor antibodiesa | ||||
No | 51.0 (100.0) | 38.0 (100.0) | 28.0 (100.0) | 28.0 (100.0) |
Yes | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
SMD | — | 0 | 0 | 0 |
HIV statusa | ||||
Positive | 3.0 (5.9) | 6.0 (15.8) | 5.0 (17.9) | 6.5 (23.2) |
Negative | 48.0 (94.1) | 32.0 (84.2) | 23.0 (82.1) | 21.5 (76.8) |
SMD | — | 0.323 | 0.377 | 0.508 |
Total bilirubin thresholda (µmol/L) | ||||
Mean (SD) | 11.55 (5.95) | 17.17 (12.95) | 16.27 (9.22) | 16.26 (8.83) |
SMD | — | 0.557 | 0.608 | 0.625 |
Family with FIX inhibitor antibodies,a n (%) | ||||
No | 43.0 (84.3) | 38.0 (100.0) | 28.0 (100.0) | 28.0 (100.0) |
Yes | 7.0 (13.7) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
Missing | 1.0 (2.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
SMD | — | 0.610 | 0.610 | 0.610 |
Duration of diagnosed hemophilia Bc (years) | ||||
Mean (SD) | 39.59 (14.58) | 27.19 (13.80) | 29.73 (14.13) | 33.92 (15.26) |
SMD | — | 0.874 | 0.687 | 0.380 |
ABR = annualized bleeding rate; ALT = alanine aminotransferase; AST = aspartate aminotransferase; BMI = body mass index; EHL = extended half-life; ESS = effective sample size; FIX = factor IX; IPTW = inverse probability of treatment weighting; ITC = indirect treatment comparison; pdFIX = plasma-derived factor IX; rFIX = recombinant factor IX; rFIXFc = recombinant factor IX Fc fusion protein; rIX-FP = recombinant factor IX albumin fusion protein; SD = standard deviation; SHL = standard half-life; SMD = standardized mean difference.
aThe data for the covariate were taken at screening for the HOPE-B study and the comparator trial.
bThe data for the covariate were taken during the lead-in period for the HOPE-B study and were taken at screening for the comparator trial.
cThe data for the covariate were taken after the lead-in period for the HOPE-B study and were taken at screening for the comparator trial.
Source: Sponsor-submitted ITC.63
Results for the comparison of etranacogene dezaparvovec versus rIX-FP are summarized in Table 19; both an unadjusted and an IPTW-adjusted analysis are presented. In both cases, results are provided for a subset of patients who met the eligibility criteria of the HOPE-B trial. The multivariable-adjusted analysis was conducted among matched patients. For ABR, ABR for spontaneous bleeds, and proportion of patients with 0 ABR events was adjusted for prior ABR, severity of hemophilia B, and age. For ABR for joint bleeds, the multivariable propensity score analysis was adjusted for (in order): severity of hemophilia B, prior ABR, and age. For annualized FIX consumption (excluding surgical FIX consumption), a multivariable-adjusted analysis was conducted among matched patients and sequentially adjusted for (in order): severity of hemophilia B, prior ABR, and prior FIX product class (EHL versus SHL).
In the multivariable-adjusted model, when comparing ABR, the rate for etranacogene dezaparvovec (ABR rate = 0.38) versus rIX-FP (ABR rate = 1.97) had an associated RR of 0.19 (95% CI, 0.09 to 0.41). When comparing ABR for joint bleeds, the rate for etranacogene dezaparvovec (ABR for joint bleeds = 0.14) versus rIX-FP (ABR for joint bleeds = 1.61) had an associated RR of 0.09 (95% CI, 0.03 to 0.25). When comparing FIX consumption, mean total FIX consumption for etranacogene dezaparvovec was 44.34 IU/kg/year when compared against rIX-FP, where mean consumption was 2,619.49 IU/kg/year with an associated mean difference of −2,575.15 IU/kg/year (95% CI, −2,990.83 IU/kg/year to −2,159.46 IU/kg/year).
Table 19: Comparative Efficacy of Etranacogene Dezaparvovec Versus rIX-FP (Base-Case Analysis)
Detail | Etranacogene dezaparvovec | rIX-FP (Idelvion, unadjusted, unmatched) | rIX-FP (Idelvion, IPTW-weighted, multivariable-adjusted) |
|---|---|---|---|
Number of patients, N or ESS | N = 51 | N = 38 | ESS = 18.2 |
Etranacogene dezaparvovec versus comparator | |||
Annualized bleeding rate | 0.38 | 1.56 | 1.97 |
Comparative annualized bleeding rate, RR (95% CI) | NA | 0.24 (0.13 to 0.45) | 0.19 (0.09 to 0.41) |
Annualized spontaneous bleeding rate | 0.08 | 0.71 | 1.01 |
Comparative annualized spontaneous bleeding rate, RR (95% CI) | NA | 0.12 (0.04 to 0.32) | 0.08 (0.03 to 0.23) |
Annualized joint bleeding rate | 0.14 | 1.12 | 1.61 |
Comparative annualized joint bleeding rate, RR (95% CI) | NA | 0.12 (0.05 to 0.31) | 0.09 (0.03 to 0.25) |
Proportion of patients with 0 annualized bleeding rate | 0.725 | 0.263 | 0.131 |
Comparative proportion of patients with 0 annualized bleeding rate, OR (95% CI) | NA | 7.40 (2.87 to 19.1) | 17.60 (4.77 to 64.88) |
Analysis population for annualized total FIX consumption (IU/kg/year), N | 51 | 40 | 18.6 |
Annualized total FIX consumption (IU/kg/year) | 44.34 | 2,523.9 | 2,619.49 |
Mean difference in annualized total FIX consumption, IU/kg/year (95% CI) | NA | −2,479.56 (−2,660.76 to −2,298.36) | −2,575.15 (−2,990.83 to −2,159.46) |
CI = confidence interval; ESS = effective sample size; FIX = factor IX; IPTW = inverse probability of treatment weighting; NA = not applicable; OR = odds ratio; rIX-FP = recombinant factor IX albumin fusion protein; RR = relative risk.
Source: Sponsor-submitted indirect treatment comparison.63
Etranacogene Dezaparvovec Versus rFIXFc (Alprolix)
For the comparison of etranacogene dezaparvovec versus rFIXFc, the sponsor provided an overview of baseline characteristics among the group 1 population of the B-LONG trial, corresponding to those patients who received prior prophylaxis. The sponsor noted that only 3 covariates could be assessed among this population and, of these, prior ABR was imbalanced (SMD > 0.2) in the unadjusted population. Following MAIC, all 3 covariates were balanced (SMD < 0.2). A summary of differences is provided in Table 20.
Table 20: Baseline Characteristics of Patients in the HOPE-B Study and B-LONG Study (Etranacogene Dezaparvovec Versus rFIXFc, Primary Analysis)
Characteristics | B-LONG (group 1, prior prophylaxis) (N = 33) | HOPE-B ITC analysis set (N = 51) | |
|---|---|---|---|
Before MAIC (naive) (N = 51) | After MAIC (primary) (ESS = 28.2) | ||
Severity of hemophilia B,a n (%) | |||
< 1 IU/dL | NR | 41 (80.4) | 33.1 (80.1) |
1 IU/dL to 2 IU/dL | NR | 10 (19.6) | 8.2 (19.9) |
SMD | — | NA | NA |
Prior ABRb | |||
Mean (SD) | 5.5 (6.4) | 4.1 (4.1) | 5.5 (6.4) |
SMD | — | 0.262 | 0 |
Prior FIX regimen,b n (%) | |||
Prophylaxis | 33 (100) | 51 (100) | 41.4 (100) |
On demand | 0 (0) | 0 (0) | 0.0 (0) |
SMD | — | 0 | 0 |
Prior presence of target joints,b n (%) | |||
No | NR | 49 (96.1) | 40.3 (97.3) |
Yes | NR | 2 (3.9) | 1.1 (2.7) |
SMD | — | NA | NA |
Agec (years) | |||
Mean (SD) | NR | 40.4 (15.0) | 40.4 (14.8) |
SMD | — | NA | NA |
Prior FIX product class,b n (%) | |||
EHL | NR | 30 (58.8) | 26.9 (65.0) |
SHL | NR | 21 (41.2) | 14.5 (35.0) |
SMD | — | NA | NA |
BMIa (kg/m2) | |||
Mean (SD) | NR | 27.3 (5.2) | 27.4 (4.6) |
SMD | — | NA | NA |
Weighta (kg) | |||
Mean (SD) | NR | 86.2 (19.3) | 86.4 (18.1) |
SMD | — | NA | NA |
Prior exposure days of treatment with FIX protein,a n (%) | |||
> 150 days | NR | 51 (100) | 41.4 (100) |
≤ 150 days | NR | 0 (0) | 0.0 (0) |
SMD | — | NA | NA |
Prior FIX product,b n (%) | |||
rFIX | NR | 21 (41.2) | 14.5 (35.0) |
rIX-FP | NR | 8 (15.7) | 5.6 (13.5) |
rFIXFc | NR | 21 (41.2) | 20.3 (49.0) |
Pegylated nonacog beta pegol | NR | 1 (2.0) | 1.0 (2.4) |
SMD | — | NA | NA |
ALT thresholda (IU/L) | |||
Mean (SD) | NR | 21.4 (13.8) | 22.2 (13.6) |
SMD | — | NA | NA |
AST thresholda (IU/L) | |||
Mean (SD) | NR | 19.9 (7.8) | 19.9 (7.5) |
SMD | — | NA | NA |
History of FIX inhibitor antibodies,a n (%) | |||
No | 33 (100) | 51 (100) | 41.4 (100) |
Yes | 0 (0) | 0 (0) | 0.0 (0) |
SMD | — | 0 | 0 |
HIV status,a n (%) | |||
Positive | NR | 3 (5.9) | 1.8 (4.4) |
Negative | NR | 48 (94.1) | 39.6 (95.6) |
SMD | — | NA | NA |
Total bilirubin thresholda (µmol/L) | |||
Mean (SD) | NR | 11.6 (6.0) | 11.3 (5.7) |
SMD | — | NA | NA |
Family with FIX inhibitor antibodies,a n (%) | |||
No | NR | 43 (84.3) | 35.4 (85.6) |
Yes | NR | 7 (13.7) | 5.5 (13.3) |
Missing | NR | 1 (2.0) | 0.5 (1.1) |
SMD | — | NA | NA |
Duration of diagnosed hemophilia Bc (years) | |||
Mean (SD) | NR | 39.6 (14.6) | 39.8 (14.3) |
SMD | — | NA | NA |
ABR = annualized bleeding rate; ALT = alanine transaminase; AST = aspartate aminotransferase; BMI = body mass index; EHL = extended half-life; ESS = effective sample size; FIX = factor IX; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; NA = not available; NR = not reported; rFIX = recombinant factor IX; rFIXFc = recombinant factor IX Fc fusion protein; rIX-FP = recombinant factor IX albumin fusion protein; SD = standard deviation; SHL = standard half-life; SMD = standardized mean difference.
aThe data for the covariate were taken at screening for the HOPE-B study and the comparator trial.
bThe data for the covariate were taken during the lead-in period in the HOPE-B study and at screening in the comparator trial.
cThe data for the covariate were taken after the lead-in period in the HOPE-B study and at screening in the comparator trial.
Source: Sponsor-submitted ITC.63
In terms of comparative efficacy, results for ABR were available only in the primary analysis population. The sponsor noted that the ABR among the unadjusted etranacogene dezaparvovec population (ABR = 0.38; N = 51) was lower than among patients receiving rFIXFc (ABR = 2.99; N = 32), corresponding to an RR of 0.13 (95% CI, 0.07 to 0.25) for etranacogene dezaparvovec versus rFIXFc. When adjusted for ABR, the sponsor reported a similar trend, with the ABR-adjusted MAIC population of patients receiving etranacogene dezaparvovec having a lower ABR (ABR = 0.43; ESS = 28.2) than patients receiving rFIXFc (ABR = 2.99; N = 32), corresponding to an RR of 0.14 (95% CI, 0.08 to 0.25) for etranacogene dezaparvovec versus rFIXFc.
Etranacogene Dezaparvovec Versus Pegylated Nonacog Beta Pegol
For the comparison of etranacogene dezaparvovec versus pegylated nonacog beta pegol (Rebinyn), the sponsor provided an overview of baseline characteristics among the prior prophylaxis population of the Paradigm 2 trial. The sponsor noted that this population, while otherwise being a closer fit for the HOPE-B ITC analysis population, had limited information with respect to patient baseline characteristics. A summary of the pre- and post-MAIC population characteristics is provided in Table 21. Prior to MAIC, the following covariates were noted to be imbalanced (SMD ≥ 0.2) between trials: prior ABR, prior FIX product class, and prior FIX product use. Following MAIC, 2 characteristics were noted to still be imbalanced (SMD ≥ 0.2): prior FIX product class and prior FIX product use. The ESS following MAIC was 8.5 for the HOPE-B trial’s ITC analysis population.
Table 21: Baseline Characteristics of Patients in the HOPE-B Study and Paradigm 2 Study (Etranacogene Dezaparvovec Versus Pegylated Nonacog Beta Pegol, Base-Case Analysis)
Characteristics | Paradigm 2 (prior prophylaxis group) (N = 17) | HOPE-B ITC analysis set (N = 51) | |
|---|---|---|---|
Before MAIC (naive) (N = 51) | After MAIC (primary) (ESS = 8.5) | ||
Severity of hemophilia B,a n (%) | |||
< 1 IU/dL | NR | 41 (80.4) | 4.2 (38.8) |
1 IU/dL to 2 IU/dL | NR | 10 (19.6) | 6.7 (61.2) |
SMD | — | NA | NA |
Prior ABRb | |||
Mean (SD) | 7.5 (1.2)d | 4.1 (4.1) | 7.5 (1.3) |
SMD | — | 0.262 | 0 |
Prior FIX regimen,b n (%) | |||
Prophylaxis | 17 (100) | 51 (100) | 10.9 (100) |
On demand | 0 (0) | 0 (0) | 0.0 (0) |
SMD | — | 0 | 0 |
Prior presence of target joints,b n (%) | |||
No | NR | 49 (96.1) | 10.6 (97.3) |
Yes | NR | 2 (3.9) | 0.3 (2.7) |
SMD | — | NA | NA |
Agec (years) | |||
Mean (SD) | NR | 40.4 (15.0) | 37.2 (13.1) |
SMD | — | NA | NA |
Prior FIX product class,b n (%) | |||
EHL | 0 (0) | 30 (58.8) | 6.2 (57.0) |
SHL | 17 (100) | 21 (41.2) | 4.7 (43.0) |
SMD | — | 1.690 | 1.628 |
BMIa (kg/m2) | |||
Mean (SD) | NR | 27.3 (5.2) | 24.2 (3.4) |
SMD | — | NA | NA |
Weighta (kg) | |||
Mean (SD) | NR | 86.2 (19.3) | 77.6 (15.5) |
SMD | — | NA | NA |
Prior exposure days of treatment with FIX protein,a n (%) | |||
> 150 days | 17 (100) | 51 (100) | 10.9 (100) |
≤ 150 days | 0 (0) | 0 (0) | 0.0 (0) |
SMD | — | 0 | 0 |
Prior FIX product,b n (%) | |||
rFIX | 10 (58.8) | 21 (41.2) | 4.7 (43.0) |
rIX-FP | 0 (0) | 8 (15.7) | 1.7 (15.3) |
rFIXFc | 0 (0) | 21 (41.2) | 4.6 (41.6) |
Pegylated nonacog beta pegol | 0 (0) | 1 (2.0) | 0.0 (0.0) |
pdFIX | 7 (41.2) | 0 (0) | 0.0 (0) |
SMD | — | 2.064 | 2.013 |
ALT thresholda (IU/L) | |||
Mean (SD) | NR | 21.4 (13.8) | 19.0 (9.9) |
SMD | — | NA | NA |
AST thresholda (IU/L) | |||
Mean (SD) | NR | 19.9 (7.8) | 21.3 (7.0) |
SMD | — | NA | NA |
History of FIX inhibitor antibodies,a n (%) | |||
No | 17 (100) | 51 (100) | 10.9 (100) |
Yes | 0 (0) | 0 (0) | 0.0 (0) |
SMD | — | 0 | 0 |
HIV status,a n (%) | |||
Positive | NR | 3 (5.9) | 1.5 (13.3) |
Negative | NR | 48 (94.1) | 9.5 (86.7) |
SMD | — | NA | NA |
Total bilirubin thresholda (µmol/L) | |||
Mean (SD) | NR | 11.6 (6.0) | 12.1 (6.1) |
SMD | — | NA | NA |
Family with FIX inhibitor antibodies,a n (%) | |||
No | NR | 43 (84.3) | 9.9 (90.2) |
Yes | NR | 7 (13.7) | 0.2 (1.4) |
Missing | NR | 1 (2.0) | 0.9 (8.3) |
SMD | — | NA | NA |
Duration of diagnosed hemophilia Bc (years) | |||
Mean (SD) | NR | 39.6 (14.6) | 37.0 (12.6) |
SMD | — | NA | NA |
ABR = annualized bleeding rate; ALT = alanine transaminase; AST = aspartate aminotransferase; BMI = body mass index; EHL = extended half-life; ESS = effective sample size; FIX = factor IX; IPTW = inverse probability of treatment weighting; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; NA = not applicable; NR = not reported; pdFIX = plasma-derived factor IX; rFIX = recombinant factor IX; rFIXFc = recombinant factor IX Fc fusion protein; rIX-FP = recombinant factor IX albumin fusion protein; SD = standard deviation; SHL = standard half-life; SMD = standardized mean difference.
aThe data for the covariate were taken at screening in the HOPE-B study and the comparator trial.
bThe data for the for covariate were taken during the lead-in period in the HOPE-B study and at screening in the comparator trial.
cThe data for the for covariate were taken after the lead-in period in the HOPE-B study and at screening in the comparator trial.
dSD imputed from variance of ABR outcome to facilitate adjustment.
Source: Sponsor-submitted ITC.63
Among the population that had received prior prophylaxis treatment, data were available only for the comparison of ABR; data on ABR for spontaneous bleeds and ABR for joint bleeds were not available. The unadjusted ABR was lower for etranacogene dezaparvovec (0.36; N = 51) than for pegylated nonacog beta pegol (3.33; N = 17). The RR for etranacogene dezaparvovec relative to pegylated nonacog beta pegol was 0.11 (95% CI, 0.06 to 0.22). A similar trend was seen following univariable adjustment for prior ABR: the RR for etranacogene dezaparvovec (ESS = 8.5) relative to pegylated nonacog beta pegol (N = 17) was 0.24 (95% CI, 0.07 to 0.82); following univariable adjustment for prior FIX product class, the RR for etranacogene dezaparvovec (ESS = 21) relative to pegylated nonacog beta pegol (N = 17) was 0.10 (95% CI, 0.03 to 0.27).
Critical Appraisal of the ITC
A key limitation shared among the sponsor-submitted ITC is the absence of a common comparator, which requires methods that accommodate for unanchored comparisons. The use of unanchored MAIC as well as IPTW analyses generally requires an assumption that the propensity score model includes all treatment-effect modifiers and prognostic factors. This assumption cannot be tested and is often dependent on the completeness of the included covariates.66 In several of these analyses, this assumption is unlikely to hold due to the lack of available data from the covariates that were considered to be of the highest relevance. Failing to adjust for all relevant covariates introduces an unknown amount of bias in the reported effect estimates. In the analyses relative to rIX-FP, the sponsor noted that between-population differences (defined as an SMD threshold > 0.2) remained following inverse weighting of the HOPE-B patient population, which is an indication that the propensity score models did not include sufficient complexity to remove the potential bias.
The sponsor noted heterogeneity in the outcome definitions relating to bleeding events. The lack of a common comparator across the evidence base means that the impact of outcome definition differences cannot be assessed. Despite this, the clinical experts consulted by CADTH for this analysis did not feel that the definitions were substantially different and may be sufficient to explain the differences in observed effect sizes.
For comparisons of etranacogene dezaparvovec relative to rFIXFc and pegylated nonacog beta pegol, the interpretation of the primary analysis is hindered by the number of nonevaluable covariates among the study population receiving prophylaxis. This is further compounded by the lack of outcome reporting from these trials; the prophylaxis-receiving population had a limited number of outcomes reported; therefore, clinically relevant outcomes such as ABR for spontaneous bleeds and ABR for joint bleeds cannot be assessed. The sponsor did provide an exploratory sensitivity analysis of the mixed prophylaxis and on-demand populations, for which greater information was available on covariates and outcomes of interest. However, these comparisons are confounded by the use of a mixed-eligibility population, which cannot be accounted for in these analyses.
A significant limitation of the included ITC is the lack of comparative harms data. Accordingly, there is a gap in the indirect evidence with respect to the estimated harms of etranacogene dezaparvovec relative to other products.
As the sponsor did not provide full details of the SLR used to populate data for the report, uncertainty exists with respect to the potential for selection and reporting bias. Further, no information was provided on the extraction process for the included studies. Finally, the SLR that was used was published 4 years ago; therefore, uncertainty exists with respect to whether new relevant evidence, such as longer-term follow-up or subgroup analyses, may be available. As such, substantial uncertainty exists with respect to the totality of the evidence and the potential for missing data or subgroup data of relevance to this review.
In many analyses, the ESS of the comparison was particularly low. For example, the comparison of ABR for etranacogene dezaparvovec relative to pegylated nonacog beta pegol utilized an ESS of 8.5 for the RR for the etranacogene dezaparvovec population. A small ESS increases the uncertainty of the reported results, and the significance of the findings may be imprecise.
Discussion
Summary of Available Evidence
One ongoing phase III, single-arm, open-label clinical trial, HOPE-B (N = 54), was identified in the sponsor’s SLR. The primary objective of the HOPE-B trial was to demonstrate the noninferiority of etranacogene dezaparvovec in reducing ABR for all bleeding events between month 7 and month 18 post infusion compared with continuous routine FIX prophylaxis treatment. Other efficacy end points included proportion of patients with no bleeds, ABR for spontaneous bleeds, ABR for joint bleeds, AIR for FIX replacement therapy, annualized consumption of FIX replacement therapy, HJHS, and PROBE score. Safety outcomes such as TEAEs, TESAEs, withdrawals due to AEs, mortality, and notable harms (e.g., ALT increased, AST increased) were also reported. The HOPE-B study included a lead-in phase before the infusion of etranacogene dezaparvovec. Data collected during the lead-in phase served as a comparison against etranacogene dezaparvovec for some safety and efficacy outcomes (e.g., ABR for all bleeding events, ABR for spontaneous bleeds, ABR for joint bleeds, AIR, and annualized FIX consumption). The HOPE-B trial enrolled male patients who had moderately severe or severe hemophilia B (defined as a normal circulation FIX ≤ 2%) and were on continuous routine FIX prophylaxis treatment. Patients were excluded from the study if they had a history of FIX inhibitors or tested positive for FIX inhibitors at the last visit of the lead-in period and during the screening period of the HOPE-B trial. Pre-existing nAbs against AAV5 was not used as an exclusion criterion in the HOPE-B trial. Of the 54 patients who received etranacogene dezaparvovec, the majority were white (74.1%), with a mean age of 41.5 years (SD = 15.8 years); 21 patients (38.9%) had pre-existing nAbs against AAV5 before infusion of etranacogene dezaparvovec.
To evaluate the comparative efficacy of etranacogene dezaparvovec against other products, the sponsor submitted an ITC that included data from the rIX-FP (Idelvion), rFIXFc (Alprolix), pegylated nonacog beta pegol (Rebinyn), and nonacog alfa (BeneFIX) trials. Owing to a lack of connected evidence, the sponsor evaluated the feasibility of unanchored approaches using individual patient data to construct an IPTW estimator for comparisons against rIX-FP, and summary statistics for an unanchored MAIC estimator for rFIXFc, pegylated nonacog beta pegol, and BeneFIX. Owing to limitations in reporting, the sponsor determined that comparisons against BeneFIX may be inappropriate and considered its inclusion as a sensitivity analysis. Comparisons against rIX-FP were in favour of etranacogene dezaparvovec with respect to ABR, ABR for joint bleeds, ABR for spontaneous bleeds, proportion of patients with 0 bleeding events, and FIX utilization. Comparisons against rFIXFc demonstrated results in favour of etranacogene dezaparvovec for ABR; other efficacy end points were not available for the prophylaxis-receiving (primary analysis) population. Comparisons against pegylated nonacog beta pegol demonstrated results in favour of etranacogene dezaparvovec for ABR; other efficacy end points were not available for the prophylaxis-receiving (primary analysis) population. No safety data were presented. As no common comparator is available, the MAIC specifications required an assumption that all potentially relevant prognostic factors and treatment-effect modifiers had been included in the propensity score model. Particularly for comparisons against the rFIXFc and pegylated nonacog beta pegol prophylaxis-receiving populations, this was not possible owing to a lack of reporting baseline covariates for the prophylaxis-receiving (primary analysis) population. When using the individual patient data to compare the efficacy of etranacogene dezaparvovec against rIX-FP, all possible confounding variables should have been included.
Interpretation of Results
Efficacy
Overall, the efficacy evidence in the pivotal HOPE-B trial was in favour of etranacogene dezaparvovec over FIX prophylaxis in adult male patients who had moderately severe or severe hemophilia B (defined as normal circulation FIX ≤ 2%) and were on stable prophylaxis for at least 2 months before screening. This conclusion was based on the results from several efficacy end points, including bleeding-related end points (e.g., ABR for all bleeds, number of patients without any bleeds, ABR for spontaneous bleeds, ABR for joints bleeds), end points related to the use of FIX post infusion of etranacogene dezaparvovec (e.g., AIR), joint health–related end points (e.g., HJHS), as well as patient-reported outcomes (e.g., PROBE score). These efficacy end points were selected as areas of focus in the CADTH Clinical Review Report based on input from the clinical experts consulted by CADTH, patient and clinician groups, and expert committee members.
In the HOPE-B trial, the overall results for bleeding-related end points indicated that treatment with etranacogene dezaparvovec may result in a decrease in bleeding when compared with FIX prophylaxis treatment. For instance, between months 7 and 18 as well as months 7 and 36 post infusion, etranacogene dezaparvovec led to a decrease in ABR for all bleeding events compared with FIX prophylaxis received during the lead-in phase. The magnitude of the effect size, according to the clinical experts consulted by CADTH, was a clinically relevant improvement. Results from other bleeding outcomes (i.e., ABR for spontaneous bleeds, ABR for joint bleeds, ABR for traumatic bleeds) (Appendix 1) or the sensitivity analyses were generally consistent with ABR for all bleeding events, favouring etranacogene dezaparvovec over FIX prophylaxis. Moreover, along with the improvement in ABR for all bleeding events, there was a sustained increase in the FIX activity level percentage, as measured by 1 one-stage assay and 1 chromogenic assay (Appendix 1).
However, there is uncertainty associated with the interpretation of the efficacy findings due to the nonrandomized, open-label study design which, according to the GRADE guidance used by the CADTH review team, starts at low certainty. There was a potential risk of bias that may have resulted in the underestimation of ABR estimates or overestimation of the percentage of patients with no bleeds in patients treated with etranacogene dezaparvovec because of the open-label design and because bleeding events were self-reported. The risk of bias, although relatively low, could not be ruled out despite the sponsor implementing several measures to ensure patients’ correct use of the e-diary and compliance with its requirements in the HOPE-B trial. Another source of potential risk of bias in determining the magnitude of treatment effect might come from the assumptions of the models used to compare observations between the postinfusion phase and the lead-in phase in the HOPE-B study. Specifically, the probability of no bleeds did not account for the differences in follow-up time during the 2 phases and might bias results, although the direction is unknown.
In addition, there is uncertainty regarding the long-term efficacy of etranacogene dezaparvovec due to the relatively short duration of follow-up (i.e., 36 months), given that more than 60% of the respondents from the patient group input indicated they would expect a gene therapy to be effective in preventing bleeding for at least 10 years. To make any definite determinations on the overall long-term efficacy of etranacogene dezaparvovec, it was noted by the clinical experts consulted by CADTH that a longer follow-up of 20 to 25 years may be warranted.
The patient group input also reported that patients hope gene therapy would lead to fewer FIX infusions and minimal needle injections. Based on the results of the HOPE-B trial, etranacogene dezaparvovec may result in a decrease in AIR and FIX consumption when compared with FIX prophylaxis. The patient input also highlighted the importance of having a treatment for hemophilia B that maintains QoL (e.g., less stress, fewer restrictions on activities, easier to travel). According to the clinical experts consulted by CADTH, PROBE is a tool that is a commonly used in Canada to measure HRQoL outcomes in patients with hemophilia B. The changes from baseline at month 12 and month 24 post infusion of etranacogene dezaparvovec both showed improvements in the PROBE summary score in patients treated with etranacogene dezaparvovec. Improvements from baseline were also seen in other HRQoL outcomes in the HOPE-B study, such as in Haem-A-QoL scores, and the Haem-A-QoL instrument was considered by the clinical experts consulted by CADTH as generally aligned with PROBE. However, given the open-label design of the HOPE-B trial, the subjective nature of outcomes, and the lack of a comparator group, no valid inferences can be made on HRQoL outcomes. Similarly, no conclusion could be drawn with certainty on the HJHS results due to these limitations, despite improvements from baseline being observed.
In addition to the pivotal HOPE-B trial, the sponsor provided 1 ITC, which provided efficacy data on the estimated indirect effect of etranacogene dezaparvovec relative to rIX-FP, rFIXFc, and pegylated nonacog beta pegol. Owing to the lack of a common comparator, estimates required strong assumptions on the completeness of prognostic factors, treatment-effect modifiers, and potential confounders. In particular, comparisons relative to rFIXFc and pegylated nonacog beta pegol in the primary analysis are subject to further uncertainty owing to the lack of covariates to be adjusted and restricted to a limited subset of outcomes. The sponsor also noted differences that could not be adjusted for, such as in the definition of bleeding events between trials. As such, significant uncertainty remains with respect to the conclusions of ITC efficacy. Conversely, with these limitations in mind, the submitted evidence does show etranacogene dezaparvovec to have a consistent pattern of favourable efficacy, regardless of comparator.
Harms
According to the clinical experts consulted by CADTH, in the pivotal HOPE-B trial, the safety profiles of the patients treated with etranacogene dezaparvovec were considered acceptable overall, despite that more harms events occurred post infusion of etranacogene dezaparvovec than occurred when patients were receiving FIX prophylaxis treatment during the lead-in period. There was 1 death due to cardiogenic shock (according to the product monograph,2 this event was not treatment related) and 1 withdrawal due to hypersensitivity post infusion of etranacogene dezaparvovec. The evidence on harms in the HOPE-B study was limited due to the small number of patients involved and the relatively short duration of follow-up. No comparative assessment was made with respect to harms in the sponsor-submitted ITC and, as such, an evidence gap exists for the safety of etranacogene dezaparvovec relative to other prophylactic treatments.
Conclusion
One phase III, single-arm, open-label trial (HOPE-B) investigated the efficacy and safety of etranacogene dezaparvovec in 54 male patients who had moderately severe or severe hemophilia B (defined as normal circulation FIX ≤ 2%) and who had been on continuous routine FIX prophylaxis treatment. Compared with the lead-in period when patients received FIX prophylaxis, etranacogene dezaparvovec may result in a decrease in ABR for all bleeding events, ABR for spontaneous bleeds, ABR for joint bleeds, AIR, and annualized FIX consumption post infusion of the gene therapy. The effects observed for all of these outcomes were considered clinically relevant by the clinical experts consulted by CADTH. However, there is uncertainty associated with interpretating the clinical significance of the magnitude of the treatment differences due to limitations such as the nonrandomized comparative design, potential risk of bias in the self-recording of bleeding events caused by the open-label design, multiplicity was not controlled for in the analyses using the months 24 and 36 data cut-offs, and potential biases introduced by assumptions in the statistical models used to make the comparisons. The harms profile for etranacogene dezaparvovec during the follow-up period was considered acceptable by the clinical experts consulted by CADTH, despite that more harms events occurred post infusion of etranacogene dezaparvovec than when patients were receiving FIX prophylaxis during the lead-in period. The harms evidence is limited, given the relatively short follow-up period and small sample size. A key gap in the pivotal trial evidence is that results remain unknown with respect to the long-term efficacy and safety of etranacogene dezaparvovec relative to FIX prophylaxis due to the current duration of follow-up (i.e., 36 months). One ITC provided efficacy data on the estimated effect of etranacogene dezaparvovec relative to rIX-FP (Idelvion), rFIXFc (Alprolix), and pegylated nonacog beta pegol (Rebinyn). No conclusions could be drawn on relative efficacy from the ITC. Interpretation of the effect magnitude is uncertain and hindered by the lack of connected evidence available, and potential confounding due to the lack of reporting of potentially influential prognostic and predictive factors. No safety data were reported in the sponsor-submitted ITC; therefore, no conclusions could be drawn on the comparative safety of etranacogene dezaparvovec versus other products, based on this evidence.
References
1.Sponsor's clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: etranacogene dezaparvovec (Hemgenix), 1 × 1013 genome copies/mL, intravenous infusion [internal sponsor's package]. Ottawa (ON): CSL Behring Canada Inc.; 2023 Oct 4.
2.Etranacogene dezaparvovec (Hemgenix): 1 × 1013 genome copies/mL, intravenous infusion [product monograph]. Ottawa (ON): CSL Behring Canada Inc.; 2023 Oct 23.
3.Swystun LL, James P. Using genetic diagnostics in hemophilia and von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2015;2015:152-159. PubMed
4.National Organization for Rare Disorders. Hemophilia B. Rare Disease Database 2023; https://rarediseases.org/rare-diseases/hemophilia-b/. Accessed 2023 Jun 29.
5.Mann DM, Stafford KA, Poon MC, Matino D, Stafford DW. The Function of extravascular coagulation factor IX in haemostasis. Haemophilia. 2021;27(3):332-339. PubMed
6.Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1-158.
7.Association of Hemophilia Clinic Directors of Canada. CHR 2021 Data Summaries: Individuals with Factor IX Deficiency. 2023; https://ahcdc.ca/chr-2021. Accessed 2023 Jun 6.
8.Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann Intern Med. 2019;171(8):540-546. PubMed
9.Carcao M, VandenBerg HM, Gouider E, et al. Prophylaxis in hemophilia: WFH Guidelines for the Management of Hemophilia. 3 ed. Montreal (QC): World Federation of Hemophilia; 2020: https://www1.wfh.org/publications/files/pdf-1870.pdf. Accessed 2023 Oct 6.
10.Eversana. Canadian Clinical Experts Interview Report. In: Drug Reimbursement Review sponsor submission: etranacogene dezaparvovec (Hemgenix), 1 × 1013 genome copies/mL, intravenous infusion [internal sponsor's package]. Ottawa (ON): CSL Behring Canada Inc.; 2023 Oct 4. 2023.
11.Grover SP, Mackman N. Intrinsic pathway of coagulation and thrombosis. Arterioscler Thromb Vasc Biol. 2019;39(3):331-338. PubMed
12.Perez-Pujol S, Aras O, Escolar G. Factor V Leiden and inflammation. Thrombosis. 2012;2012:594986. PubMed
13.Windyga J, Lissitchkov T, Stasyshyn O, et al. Pharmacokinetics, efficacy and safety of BAX326, a novel recombinant factor IX: a prospective, controlled, multicentre phase I/III study in previously treated patients with severe (FIX level <1%) or moderately severe (FIX level ≤2%) haemophilia B. Haemophilia. 2014;20(1):15-24. PubMed
14.DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br J Haematol. 2007;138(3):305-315. PubMed
15.Mannucci PM, Tuddenham EG. The hemophilias--from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773-1779. PubMed
16.Witmer C, Presley R, Kulkarni R, Soucie JM, Manno CS, Raffini L. Associations between intracranial haemorrhage and prescribed prophylaxis in a large cohort of haemophilia patients in the United States. Br J Haematol. 2011;152(2):211-216. PubMed
17.Zanon E, Iorio A, Rocino A, et al. Intracranial haemorrhage in the Italian population of haemophilia patients with and without inhibitors. Haemophilia. 2012;18(1):39-45. PubMed
18.Konkle BA, Nakaya Fletcher S. GeneReviews® [Internet]: Hemophilia B. 2000; https://www.ncbi.nlm.nih.gov/books/NBK1495/. Accessed 2023 Jun 30.
19.Miesbach W, Reitter-Pfoertner SE, Klamroth R, et al. Co-morbidities and bleeding in elderly patients with haemophilia–a survey of the German, Austrian and Swiss Society of Thrombosis and Haemostasis Research (GTH). Haemophilia. 2017;23(5):721-727. PubMed
20.Stonebraker JS, Bolton-Maggs PH, Michael Soucie J, Walker I, Brooker M. A study of variations in the reported haemophilia B prevalence around the world. Haemophilia. 2012;18(3):e91-94. PubMed
21.Factor FIX Deficiency. Ottawa (ON): Statistics Canada: https://www.ahcdc.ca/storage/pdf/21/FIX-2021.pdf. Accessed 2023 Sep 23.
22.Canadian Hemophilia Society. Coagulation Products for Hemophilia B: Recombinant Factor IX Comparison Chart. 2023; https://www.hemophilia.ca/wp-content/uploads/2023/02/Coagulation-products-for-hemophilia-B.pdf. Accessed 2023 Jul 11.
23.Hemgenix® (etranacogene dezaparvovec): 1 × 1013 genome copies/mL, suspension for intravenous infusion [product monograph]. Ottawa (ON): CSL Behring Canada Inc.; 2023 Oct 23.
24.Sanofi Aventis Canada Inc. Alprolix® Coagulation Factor IX (Recombinant), Fc Fusion Protein [product monograph]. 2021.
25.Novo Nordisk Canada Inc. Rebinyn® Coagulation Factor IX (Recombinant), Pegylated nonacog beta pegol [product monograph]. 2022.
26.Pfizer Canada ULC. BeneFIX® Coagulation Factor IX (Recombinant) [product monograph]. 2017.
27.Takeda Canada Inc. Immunine® VH [product monograph]. 2021.
28.Shah J, Kim H, Sivamurthy K, Monahan PE, Fries M. Comprehensive analysis and prediction of long-term durability of factor IX activity following etranacogene dezaparvovec gene therapy in the treatment of hemophilia B. Curr Med Res Opin. 2023;39(2):227-237. PubMed
29.Clinical Study Report: Phase III, open-label, single-dose, multi-center multinational trial investigating a serotype 5 adeno-associated viral vector containing the Padua variant of a codonoptimized human Factor IX gene (AAV5-hFIXco-Padua, AMT-061) administered to adult subjects with severe or moderately severe hemophilia B [internal sponsor's report]. King of Prussia (PA): CSL Behring LLC; 2022 Jun 20.
30.Clinical Study Protocol Version 8.0 [internal sponsor's report]. Ottawa (ON): CSL Behring Canada Inc; 2022 Feb 7.
31.Pipe SW, Recht M, Key NS, et al. First Data from the Phase 3 HOPE-B Gene Therapy Trial: Efficacy and Safety of Etranacogene Dezaparvovec (AAV5-Padua hFIX variant; AMT-061) in Adults with Severe or Moderate-Severe Hemophilia B Treated Irrespective of Pre-Existing Anti-Capsid Neutralizing Antibodies. Blood. 2020;136(Supplement_2):LBA-6-LBA-6.
32.Thornburg CD. Etranacogene dezaparvovec for hemophilia B gene therapy. Ther Adv Rare Dis. 2021;2:26330040211058896. PubMed
33.Pipe SW, Leebeek FWG, Recht M, et al. Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. N Engl J Med. 2023;388(8):706-718. PubMed
34.CSL Behring Canada Inc. response to Oct 18, 2023 CADTH request for additional information regarding etranacogene dezaparvovec CADTH review [internal additional sponsor's information]. Ottawa (ON): CSL Behring Canada Inc.; 2023 Nov 1.
35.Statistical Analysis Plan Version 5.0 [internal sponsor's report]. Ottawa (ON): CSL Behring Canada Inc; 2022 Apr 15.
36.CSL Behring Canada Inc. response to Dec 1, 2023 CADTH request for additional information regarding etranacogene dezaparvovec CADTH review [internal additional sponsor's information]. Ottawa (ON): CSL Behring Canada Inc.; 2023 Dec 7.
37.St-Louis J, Abad A, Funk S, et al. The Hemophilia Joint Health Score version 2.1 Validation in Adult Patients Study: A multicenter international study. Res Pract Thromb Haemost. 2022;6(2):e12690. PubMed
38.Batt K, Recht M, Cooper DL, Iyer NN, Kempton CL. Construct validity of patient-reported outcome instruments in US adults with hemophilia: results from the Pain, Functional Impairment, and Quality of life (P-FiQ) study. Patient Prefer Adherence. 2017;11:1369-1380. PubMed
39.Wang M, Batt K, Kessler C, et al. Internal consistency and item-total correlation of patient-reported outcome instruments and hemophilia joint health score v2.1 in US adult people with hemophilia: results from the Pain, Functional Impairment, and Quality of life (P-FiQ) study. Patient Prefer Adherence. 2017;11:1831-1839. PubMed
40.Nunnally JC. Psychometric Theory (2nd ed.). New York (NY): McGraw-Hill; 1978.
41.P K. Computing test-reliability. A Handbook of Test Construction: Introduction to Psychometric Design. New York (NY): Methuen & Co; 1986:118–132.
42.Feldman BM, Funk SM, Bergstrom BM, et al. Validation of a new pediatric joint scoring system from the International Hemophilia Prophylaxis Study Group: validity of the hemophilia joint health score. Arthritis Care Res (Hoboken). 2011;63(2):223-230. PubMed
43.Fischer K, Steen Carlsson K, Petrini P, et al. Intermediate-dose versus high-dose prophylaxis for severe hemophilia: comparing outcome and costs since the 1970s. Blood. 2013;122(7):1129-1136. PubMed
44.Bladen M, Main E, Hubert N, Koutoumanou E, Liesner R, Khair K. Factors affecting the Haemophilia Joint Health Score in children with severe haemophilia. Haemophilia. 2013;19(4):626-631. PubMed
45.Groen W, van der Net J, Lacatusu AM, Serban M, Helders PJ, Fischer K. Functional limitations in Romanian children with haemophilia: further testing of psychometric properties of the Paediatric Haemophilia Activities List. Haemophilia. 2013;19(3):e116-125. PubMed
46.Fischer K, Poonnoose P, Dunn AL, et al. Choosing outcome assessment tools in haemophilia care and research: a multidisciplinary perspective. Haemophilia. 2017;23(1):11-24. PubMed
47.Sluiter D, Foppen W, de Kleijn P, Fischer K. Haemophilia Joint Health Score in healthy adults playing sports. Haemophilia. 2014;20(2):282-286. PubMed
48.Skinner MW, Chai-Adisaksopha C, Curtis R, et al. The Patient Reported Outcomes, Burdens and Experiences (PROBE) Project: development and evaluation of a questionnaire assessing patient reported outcomes in people with haemophilia. Pilot Feasibility Stud. 2018;4:58. PubMed
49.Chai-Adisaksopha C, Skinner MW, Curtis R, et al. Test-retest properties of the Patient Reported Outcomes, Burdens and Experiences (PROBE) questionnaire and its constituent domains. Haemophilia. 2019;25(1):75-83. PubMed
50.Chai-Adisaksopha C, Skinner MW, Curtis R, et al. Psychometric properties of the Patient Reported Outcomes, Burdens and Experiences (PROBE) questionnaire. BMJ Open. 2018;8(8):e021900. PubMed
51.Evans JD. Straightforward Statistics for the Behavioral Sciences. Vol Los Angeles (CA): Brooks/Cole Publishing Company; 1996.
52.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159-174. PubMed
53.Shrout PE, Fleiss JL. Intraclass correlations: uses in assessing rater reliability. Psychol Bull. 1979;86(2):420-428. PubMed
54.Cicchetti DV. Guidelines, criteria, and rules of thumb for evaluating normed and standardized assessment instruments in psychology. Psychol Assess. 1994;6(4):284-290.
55.Drug Reimbursement Review sponsor submission: etranacogene dezaparvovec (Hemgenix), 1 × 1013 genome copies/mL, intravenous infusion [internal sponsor's package]. Ottawa (ON): CSL Behring Canada Inc.; 2023 Oct 4.
56.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
57.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
58.Davis J, Yan S, Matsushita T, Alberio L, Bassett P, Santagostino E. Systematic review and analysis of efficacy of recombinant factor IX products for prophylactic treatment of hemophilia B in comparison with rIX-FP. J Med Econ. 2019;22(10):1014-1021. PubMed
59.Santagostino E, Martinowitz U, Lissitchkov T, et al. Long-acting recombinant coagulation factor IX albumin fusion protein (rIX-FP) in hemophilia B: results of a phase 3 trial. Blood. 2016;127(14):1761-1769. PubMed
60.Powell JS, Pasi KJ, Ragni MV, et al. Phase 3 Study of Recombinant Factor IX Fc Fusion Protein in Hemophilia B. N Engl J Med. 2013;369(24):2313-2323. PubMed
61.Collins PW, Young G, Knobe K, et al. Recombinant long-acting glycoPEGylated factor IX in hemophilia B: a multinational randomized phase 3 trial. Blood. 2014;124(26):3880-3886. PubMed
62.Lambert T, Recht M, Valentino LA, et al. Reformulated BeneFix: efficacy and safety in previously treated patients with moderately severe to severe haemophilia B. Haemophilia. 2007;13(3):233-243. PubMed
63.EtranaDez Indirect Treatment Comparisons versus Recombinant Factor IX Products for Hemophilia B [internal sponsor's report]. Drug Reimbursement Review sponsor submission: etranacogene dezaparvovec (Hemgenix), 1 × 1013 genome copies/mL, intravenous infusion [internal sponsor's package]. Ottawa (ON): CSL Behring Canada Inc. 2022 Jul 29.
64.Zhao Q, Percival D. Entropy Balancing is Doubly Robust. J Causal Inference. 2017;5(1).
65.Joffe MM, Have TRT, Feldman HI, Kimmel SE. Model Selection, Confounder Control, and Marginal Structural Models: Review and New Applications. Am Stat. 2004;58(4):272-279.
66.Phillippo D, Ades T, Dias S, Palmer S, Abrams KR, Welton N. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. (Technical Support Documents). Sheffield (UK): NICE Decision Support Unit; 2016: http://www.nicedsu.org.uk/Populationadjusted-ICs-TSD(3026862).htm. Accessed 2024 Jan 5.
Appendix 1: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 22: FIX Activity by Visit in the Posttreatment Period in the HOPE-B Trial (Full Analysis Set)
FIX activity (%) | Baseline | Month 7 post infusion | Month 18 post infusion | Month 24 post infusion | Month 36 post infusion |
|---|---|---|---|---|---|
One-stage assay based on aPTT | |||||
n | 54 | 47 | 50 | 50 | 48 |
Mean (SD) | 1.19 (0.39) | 41.20 (20.28) | 36.90 (21.40) | 36.66 (18.96) | 38.59 (17.82) |
Chromogenic assay | |||||
n | 54 | 47 | 50 | 50 | 49 |
Mean (SD) | 1.19 (0.39) | 17.27 (9.39) | 19.66 (11.72) | 20.50 (12.20) | 19.65 (10.02) |
aPTT = activated partial thromboplastin time; FIX:C = circulating factor IX; SD = standard deviation.
Source: Drug reimbursement review sponsor submission.55
Figure 2: FIX Activity by 1-Stage Assay Based on aPTT in the HOPE-B Trial (Full Analysis Set)
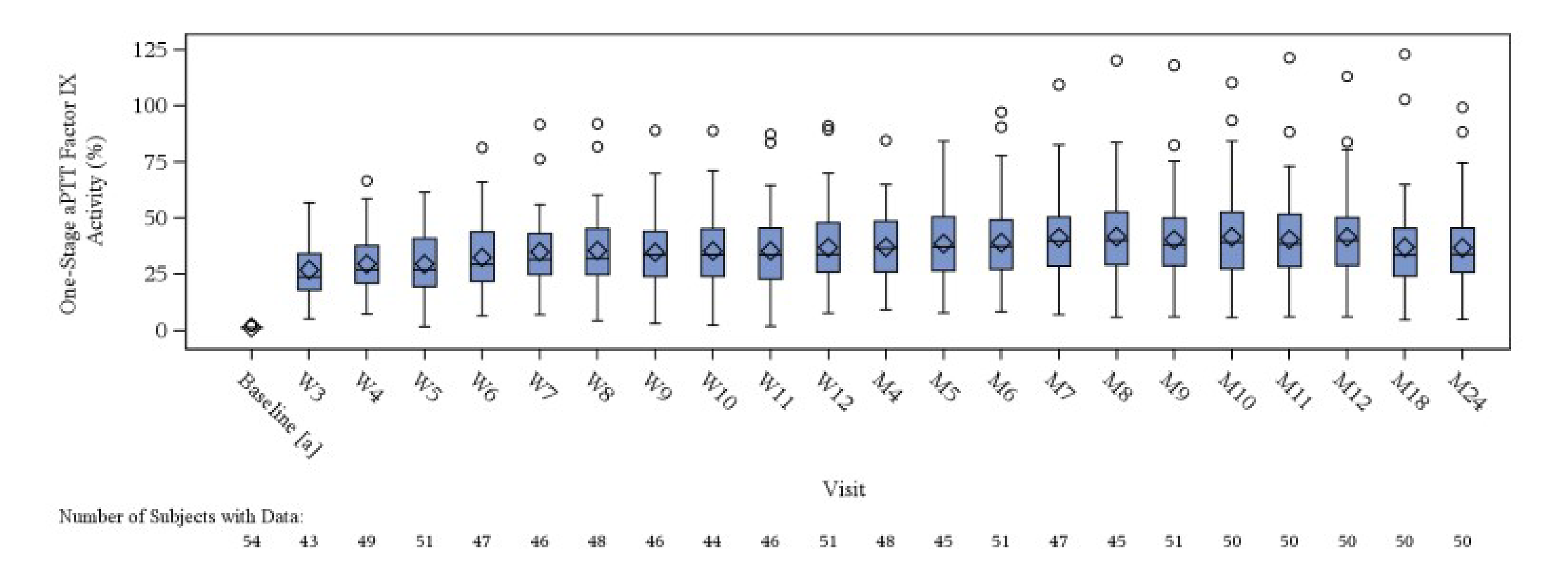
aPTT = activated partial thromboplastin time; FIX = factor IX; FIX:C = circulating factor IX; M = month; W = week.
Note: Based on a 24-month data cut-off (February 28, 2022). The lower and upper edges of the box correspond to the interquartile range, the 25th and 75th percentile. The line at the middle of the box corresponds to the median. The whiskers (horizontal lines connected to vertical lines) show the lowest and highest observation within 1.5 × interquartile range of the bottom and top of the box, respectively. The diamond is the arithmetic mean. Any points outside of the whiskers are plotted individually.
a Baseline FIX was imputed based on patient’s historical hemophilia B severity. If the patient had documented severe FIX deficiency (FIX plasma level < 1%), their baseline FIX activity level was imputed as 1%. If the patient had documented moderately severe FIX deficiency (FIX plasma level ≥ 1% and ≤ 2%), their baseline FIX activity level was imputed as 2%. The standard error was not provided at baseline.
Source: HOPE-B Clinical Study Report.29
Table 23: ABR for Traumatic Bleeds in the HOPE-B Study (Full Analysis Set)
Outcome | Postinfusion perioda Etranacogene dezaparvovec (N = 54) | Lead-in period FIX prophylaxis (N = 54) |
|---|---|---|
ABR for traumatic bleeds (month 7 to month 18 post infusion of etranacogene dezaparvovec) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Cumulative number of bleeding episodes, n | 30 | 70 |
Unadjusted ABRb | 0.60 | 2.11 |
Adjusted ABR (95% CI)c | 0.62 (0.31 to 1.23) | 1.96 (1.25 to 3.07) |
Rate ratio (2-sided 95% Wald CI; P value) | 0.30 (0.17 to 0.52; P < 0.0001) | |
ABR for traumatic bleeds (month 7 to month 36 post infusion of etranacogene dezaparvovec) | ||
Number of participants who contributed to the analysis | 54 | 54 |
Cumulative number of bleeding episodes, n | 57 | 70 |
Unadjusted ABRb | 0.46 | 2.11 |
Adjusted ABR (95% CI)c | 0.49 (0.28 to 0.86) | 1.96 (1.25 to 3.07) |
Rate ratio (2-sided 95% Wald CI; P value) | 0.25 (0.15 to 0.41; P < 0.0001) | |
ABR = annualized bleeding rate; CI = confidence interval; FIX = factor IX.
Note: Traumatic bleeds are those bleeding events provoked, which means there is a known reason for a bleed. However, traumatic bleeds do not include bleeds that were provoked for a medical/dental/other reason, so traumatic bleeds are a subset of provoked bleeds.
aPostinfusion period refers to the number of days of observation within the time interval, excluding information before day 21.
bUnadjusted ABR was calculated as the ratio of the number of bleeds to the time of observation (in years).
cAdjusted ABR and comparison of ABR between lead-in and posttreatment period were estimated from a repeated measures generalized estimating equations negative binomial regression model accounting for the paired design of the trial with an offset parameter to account for the differential collection periods. Treatment period was included as a categorical covariate.
Source: Drug reimbursement review sponsor submission.55
Pharmacoeconomic Review
Abbreviations
ABR
annualized bleed rate
AE
adverse event
AjBR
annualized joint bleed rate
BIA
budget impact analysis
CBS
Canadian Blood Services
CHS
Canadian Hemophilia Society
FIX
factor IX
ITC
indirect treatment comparison
MAIC
matching-adjusted indirect comparison
nAb
neutralizing antibody
QALY
quality-adjusted life-year
rFIX
recombinant factor IX
rFIXFc
recombinant factor IX Fc fusion protein
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Etranacogene dezaparvovec (Hemgenix), suspension for intravenous infusion |
Submitted price | Etranacogene dezaparvovec, 1 × 1013 vector genomes/mL: $4,690,000.00 per administration |
Indication | For treatment of adults (aged 18 years or older) with hemophilia B (congenital factor IX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes. |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | October 23, 2023 |
Reimbursement request | As per indication |
Sponsor | CSL Behring Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults (aged 18 years or older) with hemophilia B (congenital FIX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes. |
Treatment | Etranacogene dezaparvovec |
Comparators | rFIXFc (Alprolix) Nonacog alfa (BeneFIX) Pegylated nonacog beta pegol (Rebinyn) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (59 years) |
Key data sources | The effectiveness of etranacogene dezaparvovec was informed by the HOPE-B trial; the effectiveness of rFIX prophylaxis treatments was informed by sponsor-conducted ITCs. |
Submitted results | Etranacogene dezaparvovec was dominant (more effective and less costly) compared with the rFIX prophylaxis comparator treatments. |
Key limitations |
|
CADTH reanalysis results |
|
FIX = factor IX; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; nAb = neutralizing antibody; QALY = quality-adjusted life-year; rFIX = recombinant factor IX; rFIXFc = recombinant factor IX Fc fusion protein; WTP = willingness to pay.
Conclusions
The CADTH Clinical Review concluded that etranacogene dezaparvovec may reduce bleeds in adult patients with moderately severe to severe hemophilia B relative to treatment with factor IX (FIX) prophylaxis, based on observations from the pivotal HOPE-B trial. CADTH judged the certainty of the evidence to be low for most outcomes examined in the CADTH report and noted that there was uncertainty in the magnitude of differences in bleeding outcomes between etranacogene dezaparvovec and FIX prophylaxis owing to limitations, including the nonrandomized comparative design, potential risk of bias in self-recording bleeding events caused by the open-label design, lack of control for multiplicity in the analyses using the month 24 and month 36 data cut-offs, and potential biases introduced by assumptions in the statistical models used to make the comparisons. CADTH’s Clinical Review indicated there is considerable uncertainty in the indirect treatment comparison (ITC) with respect to the comparative efficacy of etranacogene dezaparvovec versus recombinant FIX (rFIX) prophylaxis treatments due to assumptions on the completeness of adjusting for prognostic and treatment-effect modifiers which cannot be tested, and differences in the definition of bleeding events between trials. The long-term efficacy and safety of etranacogene dezaparvovec are highly uncertain owing to limited long-term follow-up beyond the pivotal HOPE-B trial period.
CADTH undertook reanalyses to address 2 limitations in the sponsor’s base case and found that etranacogene dezaparvovec was less costly and more effective than rFIXFc and pegylated nonacog beta pegol. The incremental cost savings associated with etranacogene dezaparvovec are driven by the treatment acquisition costs associated with rFIX prophylaxis. List prices for rFIX prophylaxis are not available; therefore, the prices used in the CADTH base case are not reflective of a list price or any confidential pricing. If the true price of rFIX prophylaxis is approximately 48% and 61% less than the prices used in the sponsor’s model and in the CADTH base case for rFIXFc and pegylated nonacog beta pegol, respectively, etranacogene dezaparvovec will no longer be cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY) gained. CADTH further notes that the cost savings predicted by the model may be overestimated, as they rely on bleed rates for which the magnitude of benefit is highly uncertain, as is the assumption that these rates remain stable over the patient’s lifetime.
The incremental benefit for etranacogene dezaparvovec is driven by a lower lifetime number of bleeds compared with rFIX prophylaxis. CADTH’s base case suggests that etranacogene dezaparvovec will be more effective over a patient’s lifetime (i.e., 59 years) at reducing bleeding events, which leads to a QALY gain of 0.06 compared with rFIXFc and a QALY gain of 0.01 compared with pegylated nonacog beta pegol, but no predicted change in life expectancy. CADTH’s base case maintains the sponsor’s assumption that bleed rates for etranacogene dezaparvovec will be unchanged until patients return to rFIX prophylaxis. Should a waning in bleed rates before return to rFIX prophylaxis be observed in actual clinical use, the incremental benefit (and incremental savings) of etranacogene dezaparvovec would be overestimated. Additionally, if more people return to FIX prophylaxis than predicted by the durability models (which were more optimistic than the return-to-prophylaxis data observed in the HOPE-B trial) then total costs would be underestimated and total QALYs would be overestimated in the CADTH base case. As CADTH was unable to address uncertainty related to the comparative clinical data, including the magnitude and duration of benefit for etranacogene dezaparvovec compared with rFIX prophylaxis, the magnitude of the incremental benefit is highly uncertain.
In CADTH’s base case, etranacogene dezaparvovec becomes less costly than rFIXFc and pegylated nonacog beta pegol at 10.6 years and 10.8 years post infusion, respectively. As such, the bleed rates and treatment durability of etranacogene dezaparvovec relative to rFIX comparators would need to be maintained for at least 10 years and the price of rFIX treatments would need to be equal to those used in the model for cost savings to be realized.
CADTH was unable to resolve uncertainty related to the model structure. Due to the sponsor’s model structure, long-term quality-of-life changes and impacts of joint surgeries are not captured in either the sponsor’s or CADTH’s results. Finally, CADTH was unable to address the costs and consequences of neutralizing antibody (nAb) testing. Not including nAb testing in the CADTH base case underestimates the total costs of etranacogene dezaparvovec, but the magnitude of this is unknown due to a paucity of data on testing costs. As several key limitations remained unresolved, the reanalysis performed by CADTH is highly uncertain.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received from the Canadian Hemophilia Society (CHS) from an online survey. A total of 49 responses were gathered by the CHS and all respondents had moderately severe or severe hemophilia B without inhibitors. Additionally, the CHS conducted an online survey of people in Canada with severe hemophilia A and B and received 39 responses; 7 of those who responded had hemophilia B. Regarding the current availability of treatments for hemophilia B, the majority of respondents (52%) were satisfied with current treatments; 1 respondent (4%) noted that they were very dissatisfied. The socioeconomic aspects reported by patients included the time commitment to visit clinics, including taking time off work, access to clinics, and travel and insurance issues. When asked how gene therapy could potentially change the lives of patients and caregivers with hemophilia B, all patients noted that positive changes would include fewer infusions and injections, fewer bleeds, less joint damage (primarily knees, ankles, and elbows), fewer surgeries, fewer restrictions on activities, and the ability to travel more easily. Approximately 5 people in Canada have undergone gene therapy for hemophilia B; however, the CHS is unaware of their experience outside of the preliminary data obtained from the full trials.
Clinician input was received from the Association of Hemophilia Clinic Directors of Canada (AHCDC). The clinician group noted that the current standard of care in Canada for hemophilia B includes prophylactic replacement with clotting factor concentrates or nonfactor replacement therapy. Patients with hemophilia B have no other treatment alternatives to clotting factor concentrates. The clinician input noted that patients with poor venous access may require placement of a central venous catheter to receive rFIX prophylaxis, which is associated with a risk of infection, bleeding, thromboembolism, and loss of function, requiring the removal of the catheter. The AHCDC highlighted that gene therapy could provide a long-term phenotypic cure for patients with hemophilia B and could provide a 1-time treatment leading to sustained FIX production. The clinician group highlighted that eligible candidates for gene therapy include patients with a clinically severe bleeding phenotype requiring prophylaxis, with no history of inhibitory antibodies, no significant comorbidities, and an anti-AAV nAb titre of less than 1:900. Additionally, patients with hemophilia who are not currently receiving prophylactic therapy (e.g., due to poor venous access or adherence issues with routine prophylaxis), but who experience repeated, serious spontaneous bleeding episodes or have a history of life-threatening hemorrhage, are also candidates for gene therapy.
The drug plan input for this review noted that public drug plans do not fund the proposed comparators, as they are FIX replacement products provided by Canadian Blood Services (CBS). The public drug plans sought input on the minimum duration of time patients should be on FIX therapy before being eligible for treatment with gene therapies. Additionally, the plans sought input as to whether there are any instances where treating individuals with mild or moderate disease would be considered appropriate. As the product is proposed as a single-administration gene therapy, the drug plans sought input on whether there are any instances where a second dose would be considered appropriate. The plans noted that the costs related to laboratory testing should be considered, as several tests are required for patient selection purposes, including an anti–adeno-associated virus serotype 5 (AAV5) nAb titre, an assay for the presence of FIX inhibitor liver enzymes, and a hepatic ultrasound.
Several of these concerns were addressed in the sponsor’s model:
In the model, bleed rates and the need for FIX prophylaxis therapy were lower for patients who received etranacogene dezaparvovec compared with those receiving rFIX prophylaxis therapy.
CADTH addressed some of these concerns as follows:
CADTH removed the rFIX costs in the budget impact analysis (BIA) to report the incremental budget impact from the drug plan payer perspective.
Aligning with the Health Canada indication, a reanalysis was performed to include patients with mild or moderate disease who will receive FIX prophylaxis therapy and are eligible for treatment with etranacogene dezaparvovec.
CADTH was unable to address the following concerns raised from stakeholder input:
The explicit impact of fewer injections or joint health and joint-related surgeries could not be addressed owing to the structure of the submitted model.
The clinician input highlighted that patients with hemophilia who are not currently receiving prophylactic therapy (e.g., due to poor venous access, or adherence issues with routine prophylaxis), but who experience repeated, serious spontaneous bleeding episodes or have a history of life-threatening hemorrhage, are also candidates for gene therapy. This could not be addressed in the reanalysis owing to a lack of clinical data in this population.
The use of etranacogene dezaparvovec in patients with mild or moderate hemophilia B could not be addressed due to a lack of clinical efficacy data in those populations.
Costs associated with baseline testing for patient selection (e.g., nAb testing, FIX inhibitor presence, liver health tests) were not included in the submitted model or BIA.
Economic Review
The current review is for etranacogene dezaparvovec (Hemgenix) for the treatment of hemophilia B patients 18 years and older who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of etranacogene dezaparvovec compared with rFIX prophylaxis treatments for adult patients with moderately severe to severe hemophilia B.1 In the model, the sponsor compared etranacogene dezaparvovec with rFIXFc, nonacog alfa, and pegylated nonacog beta pegol.
Etranacogene dezaparvovec is available as a suspension for IV infusion (1 × 1013 vector genomes in 10 mL vials). The recommended dose of etranacogene dezaparvovec is a single IV infusion of 2 × 1013 genome copies per kg of body weight.2 The sponsor-submitted price for etranacogene dezaparvovec is $4,690,000.00 per administration per patient, regardless of the number of vials required. The sponsor estimated that the annual per-patient cost of rFIXFc, nonacog alfa, and nonacog beta pegol would be $492,557.19, $364,571.94, and $492,557.19, respectively.
The analysis was conducted from the perspective of the Canadian public health care payer. Cost and clinical outcomes (QALYs, life-years) were estimated over a lifetime horizon (59 years; 1-week cycle length). Discounting (1.5% per annum) was applied for both costs and outcomes.1
Model Structure
The sponsor submitted a Markov model consisting of 3 health states based on bleeding events (no bleed, joint bleed, nonjoint bleed) and death (Figure 1). All patients entered the model in the no bleed health state and received treatment with either etranacogene dezaparvovec or rFIX prophylaxis. Each week (i.e., each cycle) patients could remain in the no bleed health state or transition to the joint bleed or nonjoint bleed health state, where they accrue costs and a utility decrement associated with the type of bleed for that model cycle, after which they transition back to the no bleed health state. Patients could transition to the death health state from any alive health state aligned with the general population mortality rates for people in Canada (i.e., no additional risk of mortality was assumed for patients with hemophilia B or related to bleeds).3
Model Inputs
The baseline population characteristics used to inform the model were based on the HOPE-B trial, which enrolled adult men (mean age = 41.5 years; mean weight = 85.1 kg) with moderately severe to severe hemophilia B (defined by the sponsor as FIX activity levels of 2% or less). The HOPE-B study, an open-label, single-arm trial to demonstrate the efficacy and safety of etranacogene dezaparvovec, enrolled 54 patients who had been on stable FIX prophylaxis for at least 2 months before screening and had more than 150 previous exposure days of treatment with FIX protein.
In the absence of head-to-head evidence, the sponsor conducted ITCs to estimate the comparative efficacy of etranacogene dezaparvovec versus rFIXFc, nonacog alfa, and pegylated nonacog beta pegol using the relevant phase III comparator trials (B-LONG,4 NCT0009317,5 and Paradigm 2,6 respectively). The sponsor used rate ratios for annualized bleeding rates (ABRs) and annualized joint bleed rates (AjBRs) from the secondary analysis of the sponsor-conducted ITC to inform transition probabilities in the submitted model.
The long-term durability of etranacogene dezaparvovec (defined as the proportion of patients remaining FIX prophylaxis–free) was based on a Bayesian model–based prediction using the 36-month data from the HOPE-B trial, which was an update to the original extrapolation completed by Shah and colleagues using the 24-month data.7 This model aimed to predict the number of patients who would be free from FIX prophylaxis after etranacogene dezaparvovec infusion, assuming that patients would return to FIX prophylaxis when their FIX activity level dropped below 2%.
The model included adverse events (AEs) of grade 3 and above that occurred in 5% or more of patients in the HOPE-B trial. The probability of AEs was available only for nonacog alfa, and the sponsor assumed that the probability of AEs for the other rFIX prophylaxis therapies was equivalent. AE-related disutilities were estimated using disutility values sourced from published literature, including past National Institute for Health and Care Excellence appraisals,8-12 and assuming that each AE has an annual duration of 7 days.
The sponsor applied treatment-specific health state utility values in its base-case analysis. Patients who received etranacogene dezaparvovec were assigned a utility that was informed by EQ-5D-5L data collected from the HOPE-B trial at the 24-month cut-off point. They were then mapped to EQ-5D-3L using the Van Hout et al. mapping function.13 For the comparator treatments, the sponsor used a health state utility value from the HOPE-B clinical trial that was measured at the end of the lead-in prophylaxis phase. The sponsor included disutilities associated with bleed events that were sourced from an incremental cost-effectiveness ratio final evidence report.14 The disutility values associated with bleeds were measured in patients with hemophilia A with inhibitors.
The model included costs associated with the acquisition of etranacogene dezaparvovec and rFIX prophylaxis treatments, administration of etranacogene dezaparvovec, AEs, health care resource use, and disease management. The acquisition cost of etranacogene dezaparvovec was based on the sponsor’s submitted price. The sponsor assumed that patients who received etranacogene dezaparvovec would receive rFIX prophylaxis for 3 weeks following etranacogene dezaparvovec infusion and would receive routine follow-up for 5 years. Costs associated with baseline testing for patient selection (e.g., nAb testing, FIX inhibitor presence testing, liver health tests) were not included in the submitted model. Comparator acquisition costs were estimated based on the dosing regimen reported in the Canadian product monographs and the cost per pack derived from the annual costs reported in a prior CADTH review.15 The sponsor assumed that the annual cost of pegylated nonacog beta pegol was equivalent to rFIXFc. The sponsor assumed that rFIX prophylaxis was self-infused and thus did not include administration costs for comparator treatments. Patients who experienced a bleed event received 1 dose of the rFIX product they used for prophylaxis for all treatments included in the model. The sponsor included costs associated with subsequent treatment for patients whose FIX level falls below 2% following treatment with etranacogene dezaparvovec and assumed that all of these patients receive subsequent treatment with nonacog beta pegol. The frequency of disease management and health care resource use was informed by clinical expert opinion obtained by the sponsor, with unit costs obtained from the Ontario Schedule of Benefits: Physician Services and the Schedule of Benefits for Laboratory Services16,17 and the Canadian Institute for Health Information Patient Cost Estimator.18 Unit costs associated with AEs were captured during the first year of the model time horizon only and sourced from Ontario Schedule of Benefits: Physician Services,16,17 Public Health Agency of Canada,19 Canadian Institute for Health Information,20 British Columbia Medical Services Commission Payment Schedule,21 and published literature.22
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 500 iterations. The deterministic results were aligned with the submitted probabilistic results. The probabilistic findings are presented subsequently. The submitted analysis was based on the submitted price for etranacogene dezaparvovec and prices from a previous CADTH report for rFIX prophylaxis treatments.15
Base-Case Results
In the sponsor’s base-case analysis, etranacogene dezaparvovec was more effective and less costly (dominant) compared with rFIXFc, nonacog alfa, and pegylated nonacog beta pegol. Etranacogene dezaparvovec was estimated to be associated with a gain of 2.04 to 2.13 QALYs over the 59-year time horizon, with incremental cost savings of $4,331,305 to $8,517,139 (Table 3).
Results were driven by the acquisition cost of etranacogene dezaparvovec and the predicted cost savings from reduced bleeds and a reduced need for FIX prophylaxis (Table 9, Appendix 3). The acquisition costs of etranacogene dezaparvovec represent 68% of the total costs associated with etranacogene dezaparvovec. There was no life-year gain associated with etranacogene dezaparvovec; however, the sponsor’s model predicts that patients who received etranacogene dezaparvovec will spend more time in the no bleed health state compared with rFIX prophylaxis. As bleed events are associated with disutilities, the QALY gain associated with etranacogene dezaparvovec is driven by lower bleed rates along with treatment-specific utilities. Approximately 90% of the predicted QALYs to be gained with etranacogene dezaparvovec were accrued after the first 3 years of treatment (i.e., beyond the observed time in the HOPE-B trial).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Etranacogene dezaparvovec | 6,902,810 | 25.540 | Reference |
Nonacog alfa (rFIX) | 11,252,115 | 23.407 | Dominated by etranacogene dezaparvovec |
Pegylated nonacog beta pegol (rFIX) | 15,161,816 | 23.505 | Dominated by etranacogene dezaparvovec |
rFIXFc | 15,437,949 | 23.449 | Dominated by etranacogene dezaparvovec and nonacog beta pegol |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; rFIX = recombinant factor IX; rFIXFc = recombinant factor IX Fc fusion protein.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses, including adopting alternative modelling assumptions (i.e., time horizon, discount rate) as well as alternative assumptions related to the utility value for etranacogene dezaparvovec, alternative FIX activity thresholds to return to prophylaxis, and alternative efficacy assumptions for the rFIX therapies. In all scenarios, etranacogene dezaparvovec remained dominant over rFIX prophylaxis.
The sponsor conducted a scenario analysis from a societal perspective. This analysis included additional costs associated with patient productivity measured using the human capital approach. In this analysis, relative to rFIX therapies, etranacogene dezaparvovec was the dominant strategy. This result was the same as the sponsor’s base case using a health care payer perspective.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations of the sponsor’s analysis that have notable implications for the economic analysis:
The comparative efficacy and safety of etranacogene dezaparvovec with rFIX prophylaxis is uncertain. In the absence of head-to-head evidence for etranacogene dezaparvovec versus rFIX prophylaxis comparator treatments, the sponsor estimated the comparative effectiveness using an ITC. The sponsor used unanchored matching-adjusted indirect comparisons (MAICs) to inform the comparative efficacy of etranacogene dezaparvovec with rFIXFc and pegylated nonacog beta pegol. Data from the pivotal HOPE-B trial was used for the etranacogene dezaparvovec efficacy data; however, CADTH’s Clinical Review team noted limitations due to the open-label design and self-reported bleeds in the trial. CADTH’s Clinical Review team additionally noted several methodological limitations with the MAIC owing to the lack of comparators in the available evidence network. The MAICs were also limited by the completeness of adjusting for prognostic and treatment-effect modifiers, which cannot be tested, and differences in the definition of bleeding events between trials, which likely would favour etranacogene dezaparvovec. The sponsor-submitted ITC reported favourable comparative efficacy for etranacogene dezaparvovec for the available outcomes relative to rFIXFc and pegylated nonacog beta pegol; however, CADTH’s Clinical Review notes there is substantial uncertainty associated with these comparisons, and that the results should be treated with caution. Further, the ABRs estimated using the MAICs for etranacogene dezaparvovec are considerably different than those reported from the HOPE-B trial directly. Additionally, the sponsor-submitted ITC lacked a comparison of safety data; thus, CADTH’s Clinical Review team reported that no conclusions could be drawn regarding the comparative safety of etranacogene dezaparvovec and comparator rFIX prophylaxis.
With regard to the comparative efficacy of nonacog alfa and etranacogene dezaparvovec, the sponsor’s ITC feasibility assessment described several key limitations in reporting in the key nonacog alfa trial and concluded that an ITC would be severely limited and, thus, provided this comparison only as an addendum.
The use of separate ITCs to derive comparative efficacy between treatments precluded CADTH from conducting a sequential analysis. A sequential analysis assumes that the populations informing each comparator are homogenous. Thus, CADTH presented only pairwise results for rFIXFc and pegylated nonacog beta pegol.
Given the inability to draw conclusions from the ITC for etranacogene dezaparvovec versus nonacog alfa, CADTH presented the pairwise comparison of etranacogene dezaparvovec versus nonacog alfa as an exploratory analysis in Appendix 4.
The long-term effectiveness of etranacogene dezaparvovec is uncertain. The maximum duration of follow-up in the HOPE-B trial was 36 months; as such, evidence to support the duration and magnitude of benefit associated with etranacogene dezaparvovec compared with FIX prophylaxis is unavailable. Given that approximately 90% of the incremental QALYs predicted by the sponsor’s model to be gained with etranacogene dezaparvovec were derived on the basis of extrapolated findings rather than observed benefit, the lack of comparative long-term data introduces considerable uncertainty into the analysis. The sponsor assumed that the bleed rates for those who received etranacogene dezaparvovec and remained prophylaxis-free would be constant over their lifetime. The CADTH Clinical Review noted that the lack of long-term clinical efficacy data remains a critical gap in the pivotal evidence and indicated that no definite conclusion could be drawn with respect to the long-term efficacy and safety of etranacogene dezaparvovec. In its submitted model, the sponsor based the durability extrapolation on a previously published statistical model conducted by Shah and colleagues, using the 36-month data from the HOPE-B trial.7 In the statistical model, the sponsor assumed that patients would return to prophylaxis if their FIX activity levels fell below 2%. The sponsor’s durability model predicted that 100% and 99.9% of patients would be prophylaxis-free at 24 and 36 months, respectively. In the full analysis set of the HOPE-B trial at 24 and 36 months post etranacogene dezaparvovec infusion, 96% and 94% of patients were prophylaxis-free, respectively. According to CADTH’s Clinical Review Report, the full analysis set included 1 patient with an antibody titre greater than 1:3,000, and 1 patient who received an incomplete dose of etranacogene dezaparvovec. The clinical experts consulted by CADTH indicated they would not expect patients being treated with etranacogene dezaparvovec in a real-world setting to perform better than those in the trial. With regard to the reliability of the Shah study, CADTH notes the following sources of uncertainty: the statistical model was based on a small number of patients and nonresponders were excluded, and the duration of follow-up time informing the statistical model was short (i.e., 36 months). Additionally, the expert opinion solicited by CADTH indicated that FIX activity levels are not the primary driver of return to prophylaxis; instead, this will likely be determined by bleed rates and patients’ physical activities. The clinical experts also noted that patients may return to prophylaxis when their FIX activity levels are higher than 2% and indicated that the activity level thresholds and treatment goals may change over the modelled time horizon.
The CADTH reanalysis assumed that patients would reinitiate FIX prophylaxis when their FIX activity was below 5%. CADTH notes that a high degree of uncertainty remains regarding the long-term durability of etranacogene dezaparvovec. CADTH was unable to address treatment-waning assumptions in terms of long-term bleed rates for patients who had received etranacogene dezaparvovec.
CADTH conducted a scenario analysis using a 3-year time horizon, the time for which there are data from the HOPE-B trial.
The submitted model population does not align with the indicated population. The approved indication for etranacogene dezaparvovec is for adults with hemophilia B who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes. The pivotal HOPE-B trial was restricted to patients with moderately severe to severe hemophilia B (defined as normal circulation FIX ≤ 2%), receiving stable routine FIX prophylaxis for at least 2 months. As a result, there is no direct comparative evidence for the use of etranacogene dezaparvovec in patients with an FIX level greater than 2% but with a severe bleeding phenotype, nor in patients requiring but not receiving stable rFIX prophylaxis. The clinical experts consulted by CADTH and the clinician input received for this review indicated that some patients who were excluded from the HOPE-B trial are included in the indicated population may benefit from treatment with etranacogene dezaparvovec, specifically, patients with a severe bleeding phenotype and those who require but are unable to remain on stable prophylaxis (e.g., due to difficult venous access).
CADTH was unable to address this limitation owing to a lack of clinical data. As noted in the CADTH Clinical Review, the clinical efficacy of etranacogene dezaparvovec in patients who require but are not receiving prophylaxis, and patients with FIX activity above 2% but with severe bleed phenotype, is unknown, as is the cost-effectiveness of etranacogene dezaparvovec in the full Health Canada–indicated population.
The use of treatment-specific health state utility values is inappropriate. The sponsor applied treatment-specific utilities for etranacogene dezaparvovec and rFIX prophylaxis treatments. The sponsor applied a lower utility value to patients who received rFIX prophylaxis (0.78 for all comparators) and a higher utility value (0.85) for patients who were treated with etranacogene dezaparvovec. The use of treatment-specific utility values contradicts CADTH’s recommendation that utilities should reflect the health states in the economic model.23 Instead, all outcomes associated with treatment, along with their impact on patient utility, should be explicitly modelled rather than captured using a treatment-specific utility value. Therefore, including treatment-specific utilities to capture a difference in consequences between treatments that has not been modelled is inappropriate. Additionally, the application of treatment-specific utilities as well as differences in bleed rates (i.e., transition probabilities) may result in double counting of the impact of a treatment and thus overestimate the incremental QALYs associated with etranacogene dezaparvovec.
The CADTH reanalysis applied utilities by health state rather than treatment-specific utilities, applying the utility value for rFIX prophylaxis derived from the HOPE-B trial. CADTH notes that following this change, the driver of QALY differences is disutilities associated with bleed events, and that any quality-of-life benefit over and above that associated with bleed rates and AE disutilities is not captured in the submitted model.
The model structure does not appropriately capture the current treatment experience of patients with hemophilia B. The sponsor submitted a Markov model with health states associated with bleed activity per cycle (no bleed, nonjoint bleed, and joint bleed). Patients could move between each of these health states each cycle (i.e., each week); however, the utility value applied for these health states was the same (i.e., changes in quality of life were captured as disutilities associated with bleeds and not as differences in health state utilities). As such, following a nonjoint or joint bleed, the sponsor assumed that patients would not have long-term changes in their quality of life. The clinical experts consulted by CADTH indicated it is unlikely that patients will always return to their baseline quality of life following bleed events, and that an increasing number of bleeds (particularly joint bleeds) may further impact patients’ quality of life long-term. CADTH notes that the sponsor’s model structure likely results in underestimating the quality-of-life benefit for etranacogene dezaparvovec, the treatment with the lowest number of bleeds in the submitted model.
CADTH additionally notes that joint health and joint-related surgeries were not included in the submitted model. The patient input received by CADTH for this review indicated that joint damage to the knees, ankles, and elbows results in the greatest physical health and quality-of-life impact of hemophilia B. The clinical expert feedback received by CADTH indicated that surgical intervention in hemophilia B patients represents an important health event with regard to costs and quality of life.
CADTH was unable to address this limitation in its reanalysis.
Failure of parameter uncertainty to accurately reflect uncertainty around the incremental cost-effectiveness ratio. The incorporation of parameter uncertainty did not follow CADTH guidelines, as the sponsor did not source uncertainty estimates for most of the parameters. Instead, most model parameters used an arbitrary standard deviation of 20% of the mean. Further, the standard deviations applied to key efficacy parameters (i.e., ABR and AjBR) were inaccurate in the submitted model and did not align with the standard deviations reported in the sponsor’s pharmacoeconomic report. It is unclear whether the cost-effectiveness outcomes are underestimated or overestimated; however, improper incorporation of uncertainty biases the cost-effectiveness outcomes.
CADTH corrected the standard errors for ABRs and AjBRs in the sponsor’s model.
nAb testing was not included in the submitted model. The product monograph for etranacogene dezaparvovec indicates that nAb testing is required for patient selection. The sponsor did not include the costs or consequences of nAb testing in the submitted model. CADTH’s Guidelines for the Economic Evaluation of Health Technologies: Canada (4th edition), Appendix — Specific Guidance for Treatments with Companion Diagnostics indicates that “the consequence of a false-positive companion diagnostic result should be fully modelled” due to the potential reduction in treatment effectiveness, harm from treatment, and associated resource consumption.23 If a false-negative result is received (i.e., the test suggests that the patient does not have the predetermined nAb threshold when they do have it), the patient would incur the cost of etranacogene dezaparvovec but likely derive no benefit.
CADTH was unable to address this limitation in its reanalysis. CADTH notes that the exclusion of nAb testing costs results in underestimating the costs associated with treatment with etranacogene dezaparvovec; however, the cost of the test, along with the proportion of people who will be tested and determined to be ineligible for treatment, is unknown. Further, the impact on the clinical outcomes of treating ineligible patients is not captured and the magnitude of impact is unknown.
The price of rFIX prophylaxis is unknown. Both the sponsor’s and CADTH’s reanalyses are based on assumed costs of rFIX prophylaxis treatments; actual treatment costs are unknown and list prices are unavailable. Depending on the actual price paid for rFIX prophylaxis treatments, the cost-effectiveness of etranacogene dezaparvovec compared with rFIX prophylaxis treatments may be higher or lower.
CADTH was unable to address this limitation in its reanalysis. CADTH conducted a threshold analysis to determine the price of rFIX prophylaxis treatments at which etranacogene dezaparvovec is no longer a cost saving.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The sponsor assumed that patients with hemophilia B follow general population mortality (i.e., there was no additional risk of mortality associated with hemophilia B or bleeds). | Uncertain. Feedback received from the clinical experts consulted by CADTH noted that patients with hemophilia may have higher mortality than the general male population.24,25 Further, mortality was not an outcome of interest in the HOPE-B trial. |
Following etranacogene dezaparvovec infusion, the sponsor assumed patients would receive follow-up care for 5 years. | Inappropriate. The clinical experts consulted by CADTH indicated that lifetime follow-up will likely take place. However, the results of the model are not sensitive to assumptions around duration of follow-up. |
Patients on FIX prophylaxis treatments experience constant ABR and AjBR over the modelled time horizon. | Uncertain. The clinical experts consulted by CADTH indicated this may be a reasonable assumption for some patients; however, they noted there are other factors that may influence bleed rates, including changes in physical activity levels and treatment adherence. |
AjBR rate ratios of pegylated nonacog beta pegol relative to etranacogene dezaparvovec are not available. Therefore, the sponsor used the ABR rate ratio to serve as an approximation. | Uncertain. It is unlikely that the ABR and AjBR for pegylated nonacog beta pegol are the same. However, in the absence of data, it is uncertain what the bleed rates may be. |
The disutility associated with bleeds was measured in patients with hemophilia A with inhibitors. | Uncertain. The clinical experts consulted by CADTH indicated that bleeds in patients with hemophilia A with inhibitors may have larger quality-of-life impacts than patients with hemophilia B without inhibitors. Given the lower bleed rates associated with etranacogene dezaparvovec, overestimating the impact of a bleed will favour this therapy compared with rFIX prophylaxis. |
All patients who were treated with etranacogene dezaparvovec and reinitiated FIX prophylaxis were treated with pegylated nonacog beta pegol. | Inappropriate. The feedback received from the clinical experts indicated it is unlikely that all patients will be treated with pegylated nonacog beta pegol. However, the results of the model are not sensitive to assumptions around which FIX prophylaxis treatment is used. |
Patients treated with etranacogene dezaparvovec who experience a bleed used pegylated nonacog beta pegol to treat the bleed. | Inappropriate. The feedback received from the clinical experts indicated that patients will likely treat bleed events with the product they used before gene therapy. However, the results of the model are not sensitive to assumptions around which FIX prophylaxis treatment is used to treat bleeds. |
The sponsor assumed that patients would receive nonacog alfa every 3 days. | Inappropriate. The product monograph for nonacog alfa indicates that dosing can be every 3 or 4 days. The clinical experts consulted by CADTH indicated that most patients would receive treatment every 4 days. As a result of the sponsor’s dosing assumption, the treatment cost of nonacog alfa was overestimated. |
ABR = annual bleed rate; AjBR = annual joint bleed rate; FIX = factor IX; rFIX = recombinant factor IX.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with the clinical experts. These changes, summarized in Table 5, include alternative assumptions around the initiation of FIX prophylaxis following therapy with etranacogene dezaparvovec and applying health state utilities. The reanalysis is based on pairwise comparisons between etranacogene dezaparvovec and comparator treatments.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
1. BR and AjBR parameter uncertainty | Standard deviations for ABR and AjBR in the model did not align with those in the sponsor’s pharmacoeconomic report. | Corrected the standard deviations to those in the sponsor’s pharmacoeconomic report. |
Changes to derive the CADTH base case | ||
1. FIX activity level at which prophylaxis is reinitiated | 2% | 5% |
2. Health state utilities | Treatment-specific | Health state–specific |
CADTH base case | — | 1 + 2 |
ABR = annual bleed rate; AjBR = annual joint bleed rate; FIX = factor IX.
The CADTH base case focuses on pairwise comparisons of etranacogene dezaparvovec versus rFIXFc and pegylated nonacog beta pegol. The CADTH base case resulted in etranacogene dezaparvovec dominating both comparator treatments (incremental cost savings = $7,434,726 and $7,158,389 for rFIXFc and pegylated nonacog beta pegol, respectively; incremental QALYs = −0.06 and −0.01 for rFIXFc and pegylated nonacog beta pegol, respectively) (Table 6). The summary of CADTH’s stepped reanalysis results is presented in Table 10. Cost-effectiveness was driven by the 1-time cost of treatment with etranacogene dezaparvovec compared with the cost of frequent injections with comparator rFIX prophylaxis over the lifetime time horizon, and reduction in bleeds for patients treated with etranacogene dezaparvovec (Table 11). Etranacogene dezaparvovec becomes less costly than rFIXFc and pegylated nonacog beta pegol 10.6 years and 10.8 years post infusion, respectively. Approximately 90% of the predicted QALYs to be gained with etranacogene dezaparvovec were accrued after the first 3 years of treatment (i.e., beyond the observed time in the HOPE-B trial).
Table 6: Summary of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|---|
Etranacogene dezaparvovec vs. rFIXFc | ||||||
Sponsor’s base case | Etranacogene dezaparvovec | 6,925,878 | −8,498,989 | 25.538 | 2.092 | Reference |
rFIXFc | 15,424,867 | Reference | 23.446 | Reference | Dominated | |
CADTH base case (1 + 2; probabilistic) | Etranacogene dezaparvovec | 8,003,296 | −7,434,726 | 23.514 | 0.065 | Reference |
rFIXFc | 15,438,022 | Reference | 23.449 | Reference | Dominated | |
Etranacogene dezaparvovec vs. pegylated nonacog beta pegol | ||||||
Sponsor’s base case | Etranacogene dezaparvovec | 6,925,878 | −8,269,332 | 25.538 | 2.040 | Reference |
Pegylated nonacog beta pegol | 15,195,210 | Reference | 23.498 | Reference | Dominated | |
CADTH base (1 + 2; probabilistic) | Etranacogene dezaparvovec | 8,003,296 | −7,158,389 | 23.514 | 0.009 | Reference |
Pegylated nonacog beta pegol | 15,161,685 | Reference | 23.505 | Reference | Dominated | |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; rFIXFc = recombinant factor IX Fc fusion protein.
Scenario Analysis Results
As the confidentially negotiated prices of comparator rFIX prophylaxes are unknown, CADTH conducted threshold analyses to determine the price of comparators where etranacogene dezaparvovec would no longer be considered cost-effective. If the prices for rFIXFc and pegylated nonacog beta pegol are 48.2% and 60.5% less than those used in the CADTH base case, etranacogene dezaparvovec will no longer be cost-effective at a WTP threshold of $50,000 per QALY gained over the lifetime time horizon.
Given the inability to draw conclusions from the ITC for etranacogene dezaparvovec versus nonacog alfa, CADTH presented the pairwise comparison of etranacogene dezaparvovec versus nonacog alfa as an exploratory analysis (Table 12). As part of this analysis, CADTH explored the implications of an alternative dosing assumption for nonacog alfa. In both dosing scenarios, the results were the same (i.e., nonacog alfa was dominated by etranacogene dezaparvovec).
CADTH conducted a scenario analysis using a 3-year time horizon to reflect the period for which there are trial data for etranacogene dezaparvovec. The results of this analysis found that etranacogene dezaparvovec was dominated by rFIXFc and pegylated nonacog beta pegol (i.e., was less effective and more expensive), with an incremental cost of $2,947,013 and $3,237,681, respectively (Table 13). While etranacogene dezaparvovec was associated with the fewest number of nonjoint and joint bleeds over 3 years, the disutilities associated with treatment AEs over the first year were not offset by the benefit associated with a reduction in bleeds.
Issues for Consideration
The price of rFIX prophylaxis in the sponsor’s submitted model was based on a previous CADTH review on the treatment cost and budget impact of rFIXFc and does not reflect any confidential pricing that may have been negotiated by CBS. The true acquisition costs paid by CBS may differ from those included in the sponsor’s cost-effectiveness and BIAs.
At the time of writing this report, fidanacogene elaparvovec is undergoing review by CADTH for the treatment of adults with moderately severe to severe hemophilia B.26 The cost-effectiveness of etranacogene dezaparvovec compared with fidanacogene elaparvovec is unknown.
Overall Conclusions
The CADTH Clinical Review concluded that etranacogene dezaparvovec may reduce bleeds in adult patients with moderately severe to severe hemophilia B relative to treatment with FIX prophylaxis, based on observations from the pivotal HOPE-B trial. CADTH judged the certainty of the evidence to be low for most of the outcomes examined in the CADTH report and noted there was uncertainty in the magnitude of differences in bleeding outcomes between etranacogene dezaparvovec and FIX prophylaxis owing to limitations, including the nonrandomized comparative design, potential risk of bias in self-recording bleeding events caused by the open-label design, lack of control for multiplicity in the analyses using the month 24 and month 36 data cut-offs, and potential biases introduced by assumptions in the statistical models used to make the comparisons. CADTH’s Clinical Review indicated there is considerable uncertainty in the indirect ITC with respect to the comparative efficacy of etranacogene dezaparvovec versus rFIX prophylaxis treatments due to assumptions on the completeness of adjusting for prognostic and treatment-effect modifiers, which cannot be tested, and differences in the definition of bleeding events between trials. The long-term efficacy and safety of etranacogene dezaparvovec are highly uncertain owing to limited long-term follow-up beyond the pivotal HOPE-B trial period.
CADTH undertook reanalyses to address 2 limitations in the sponsor’s base case by using alternative assumptions around the initiation of FIX prophylaxis following therapy with etranacogene dezaparvovec and applying health state utilities. CADTH’s base-case analysis found that etranacogene dezaparvovec was less costly and more effective than rFIXFc and pegylated nonacog beta pegol. The incremental cost savings associated with etranacogene dezaparvovec are driven by the treatment acquisition costs associated with rFIX prophylaxis. List prices for rFIX prophylaxis are not available, and the prices used in the CADTH base case are therefore not reflective of list price or any confidential pricing. If the true price of rFIX prophylaxis is approximately 48% and 61% less than the prices used in the sponsor’s model and in the CADTH base case for rFIXFc and pegylated nonacog beta pegol, respectively, etranacogene dezaparvovec will no longer be cost-effective at a WTP threshold of $50,000 per QALY gained. CADTH further notes that the cost savings predicted by the model may be overestimated, as they rely on bleed rates for which the magnitude of benefit is highly uncertain, as is the assumption that these rates remain stable over the patient’s lifetime.
The incremental benefit for etranacogene dezaparvovec is driven by a lower lifetime number of bleeds compared with rFIX prophylaxis. CADTH’s base case suggests that etranacogene dezaparvovec will be more effective over a patient’s lifetime (i.e., 59 years) at reducing bleeding events, which leads to a QALY gain of 0.06 compared with rFIXFc and a QALY gain of 0.01 compared with pegylated nonacog beta pegol, but no predicted change in life expectancy. CADTH’s base case maintains the sponsor’s assumption that bleed rates for etranacogene dezaparvovec will be unchanged until patients return to rFIX prophylaxis. Should a waning in bleed rates before the return to rFIX prophylaxis be observed in actual clinical use, the incremental benefit (and incremental savings) of etranacogene dezaparvovec would be overestimated. Additionally, if more people return to FIX prophylaxis than predicted by the durability models (which were more optimistic than the return-to-prophylaxis data observed in the HOPE-B trial), then total costs would be underestimated and total QALYs would be overestimated in the CADTH base case. As CADTH was unable to address uncertainty related to the comparative clinical data, including the magnitude and duration of benefit for etranacogene dezaparvovec compared with rFIX prophylaxis, the magnitude of the incremental benefit is highly uncertain.
In CADTH’s base case, etranacogene dezaparvovec becomes less costly than rFIXFc and pegylated nonacog beta pegol 10.6 years and 10.8 years post infusion, respectively. As such, the bleed rates and treatment durability of etranacogene dezaparvovec relative to rFIX comparators would need to be maintained for at least 10 years, and the price of rFIX treatments would need to be equal to those used in the model in order for cost savings to be realized.
CADTH was unable to resolve uncertainty related to the model structure. Due to the sponsor’s model structure, long-term quality-of-life changes and the impacts of joint surgeries are not captured in either the sponsor’s or CADTH’s results. Finally, CADTH was unable to address the costs and consequences of nAb testing. Not including nAb testing in the CADTH base case underestimates the total costs of etranacogene dezaparvovec, but the magnitude of this is unknown due to a paucity of data on testing costs. As several key limitations remained unresolved, the reanalysis performed by CADTH is highly uncertain.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: etranacogene dezaparvovec. Burlington (ON): CSL Behring; 2023 Oct 3.
2.Hemgenix (etranacogene dezaparvovec): 1 x 1013 genome copes / mL [product monograph]. Ottawa (ON): CSL Behring Canada Inc.; 2023.
3.Statistics Canada. Table 13-10-0837-01 Life expectancy and other elements of the complete life table, single-year estimates, Canada, all provinces except Prince Edward Island. 2023; 10.25318/1310083701-eng. Accessed 2023 Sep 23.
4.Powell JS, Pasi KJ, Ragni MV, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. N Engl J Med. 2013;369(24):2313-2323. PubMed
5.Lambert T, Recht M, Valentino LA, et al. Reformulated BeneFix: efficacy and safety in previously treated patients with moderately severe to severe haemophilia B. Haemophilia. 2007;13(3):233-243. PubMed
6.Collins PW, Young G, Knobe K, et al. Recombinant long-acting glycoPEGylated factor IX in hemophilia B: a multinational randomized phase 3 trial. Blood. 2014;124(26):3880-3886. PubMed
7.Shah J, Kim H, Sivamurthy K, Monahan PE, Fries M. Comprehensive analysis and prediction of long-term durability of factor IX activity following etranacogene dezaparvovec gene therapy in the treatment of hemophilia B. Curr Med Res Opin. 2022:1-11. PubMed
8.National Institute for Health and Care Excellence. [TA561] Venetoclax in combination with rituximab for treating relapsed or refractory chronic lymphocytic leukaemia [ID1097] Committee Papers. (NICE Single Technology Appraisal) 2019; https://www.nice.org.uk/guidance/ta561/evidence/appraisal-consultation-committee-papers-pdf-6715163053. Accessed 2023 Sep 7.
9.Sullivan PW, Slejko JF, Sculpher MJ, Ghushchyan V. Catalogue of EQ-5D scores for the United Kingdom. Med Decis Making. 2011;31(6):800-804. PubMed
10.National Institute for Health and Care Excellence. [TA533] Ocrelizumab for treating relapsing multiple sclerosis [ID937] Committee Papers. (NICE Single Technology Appraisal) 2018; https://www.nice.org.uk/guidance/ta533/documents/committee-papers. Accessed 2023 Sep 7.
11.Hagiwara Y, Shiroiwa T, Shimozuma K, et al. Impact of Adverse Events on Health Utility and Health-Related Quality of Life in Patients Receiving First-Line Chemotherapy for Metastatic Breast Cancer: Results from the SELECT BC Study. Pharmacoeconomics. 2018;36(2):215-223. PubMed
12.Matza LS, Deger KA, Vo P, Maniyar F, Goadsby PJ. Health state utilities associated with attributes of migraine preventive treatments based on patient and general population preferences. Qual Life Res. 2019;28(9):2359-2372. PubMed
13.van Hout B, Janssen MF, Feng YS, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health. 2012;15(5):708-715. PubMed
14.Tice JA, Walton S, Herce-Hagiwara B, et al. Gene Therapy for Hemophilia B and An Update on Gene Therapy for Hemophilia A: Effectiveness and Value; Draft Evidence Report. Boston (MA): Institute for Clinical and Economic Review; 2022: https://icer.org/wp-content/uploads/2022/05/ICER_Hemophilia_Draft-Evidence-Report_091322.pdf. Accessed 2023 Sep 13.
15.Coagulation Factor IX FC Fusion Protein (Alprolix): Treatment Cost Comparison and Budget Impact Analysis. Ottawa (ON): CADTH; 2015: https://www.cadth.ca/coagulation-factor-ix-fc-fusion-protein-alprolix-treatment-cost-comparison-and-budget-impact. Accessed 2023 Aug 1.
16.Ontario Ministry of Health. Schedule of Benefits: Physician Services Under the Health Insurance Act. 2023; https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 Sep 7.
17.Ontario Ministry of Health. Schedule of Benefits for Laboratory Services. 2023; https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/sob_lab_2023.pdf. Accessed 2023 Sep 7.
18.Canadian Institute for Health Information. Patient cost estimator. 2023; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2023 Oct 6.
19.Public Health Agency of Canada. Weekly influenza reports. 2024; https://www.canada.ca/en/public-health/services/diseases/flu-influenza/influenza-surveillance/weekly-influenza-reports.html. Accessed 2023 Sep 7.
20.Canadian Institute for Health Information. Cost of a Standard Hospital Stay. 2024; https://yourhealthsystem.cihi.ca/hsp/inbrief?lang=en#!/indicators/015/cost-of-a-standard-hospital-stay-cshs/;mapC1;mapLevel2;provinceC5001;trend(C1);/. Accessed 2023 Sep 7.
21.Medical Services Commission Payment Schedule. Vancouver (BC): British Columbia Ministry of Health; 2022: https://www2.gov.bc.ca/assets/gov/health/practitioner-pro/medical-services-plan/msc_payment_schedule_-_may_2022.pdf. Accessed 2023 Sep 7.
22.Liyanarachi KV, Solligård E, Mohus RM, Åsvold BO, Rogne T, Damås JK. Incidence, recurring admissions and mortality of severe bacterial infections and sepsis over a 22-year period in the population-based HUNT study. PLOS ONE. 2022;17(7):e0271263. PubMed
23.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2023 Jan 8.
24.Hassan S, Monahan RC, Mauser-Bunschoten EP, et al. Mortality, life expectancy, and causes of death of persons with hemophilia in the Netherlands 2001-2018. J Thromb Haemost. 2021;19(3):645-653. PubMed
25.Plug I, Van Der Bom JG, Peters M, et al. Mortality and causes of death in patients with hemophilia, 1992-2001: a prospective cohort study. J Thromb Haemost. 2006;4(3):510-516. PubMed
26.CADTH. fidanacogene elaparvovec. 2023; https://www.cadth.ca/fidanacogene-elaparvovec. Accessed 2023 Dec 13.
27.HOPE-B Study Results Overview: 36-month data [data on file]. In: Drug Reimbursement Review sponsor submission: stranacogene dezaparvovec. Burlington (ON): CSL Behring; 2023 Oct 3.
28.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: etranacogene dezaparvovec. Burlington (ON): CSL Behring; 2023 Oct 3.
29.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1st, by age and sex. 2023; 10.25318/1710000501-eng. Accessed 2023 Sep 23.
30.Iorio A, Stonebraker JS, Chambost H, et al. Establishing the Prevalence and Prevalence at Birth of Hemophilia in Males. Ann Intern Med. 2019;171(8). PubMed
31.Factor FIX Deficiency. Ottawa (ON): Statistics Canada; 2021: https://www.ahcdc.ca/storage/pdf/21/FIX-2021.pdf. Accessed 2023 Sep 23.
32.Hem B Canada Market Assessment Fiscal Year 2023. In: Drug Reimbursement Review sponsor submission: etranacogene dezaparvovec. Burlington (ON): CSL Behring; 2023 Oct 3.
33.Procedures for CADTH Reimbursement Reviews. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/Drug_Review_Process/CADTH%20Drug%20Reimbursement%20Review%20Procedures.pdf. Accessed 2024 Jan 1.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost Comparison Table for the Treatment of Adults With Hemophilia B
Treatment | Strength | Form | Price ($)a | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Etranacogene dezaparvovec (Hemgenix) | 1 × 1013 genome copies/mL | Suspension for IV infusion | 4,690,000.0000 | Single dose of 2 × 1013 genome copies per kg | NA | NA |
Recombinant FIX therapies | ||||||
rFIXFc (Alprolix) | 250 IU 500 IU 1,000 IU 2,000 IU 3,000 IU 4,000 IU | Lyophilized powder for solution | 2.1332 per IU | 50 IU/kg once weekly or 100 IU/kg once every 10 to 14 days | 1,296.68 to 1,815.35 | 473,613 to 663,058 |
Coagulation factor IX [recombinant], Nonacog alfa (BeneFIX) | 250 IU 500 IU 1,000 IU 1,500 IU 2,000 IU 3,000 IU | Lyophilized powder for solution | 0.8796 per IU | 40 IU/kg every 3 or 4 days | 748.54 to 998.05 | 273,404 to 364,539 |
GCoagulation Factor IX [Recombinant] glycopegylated (Rebinyn) | 500 IU 1,000 IU 2,000 IU 3,000 IU | Lyophilized powder for solution | 2.6665 per IU | 40 IU/kg once weekly | 1,296.68 | 473,613 |
Plasma-derived FIX therapy | ||||||
FIX concentrate (human) (Immunine) | 480 to 720 IU/5 mL | Sterile powder for solution | 0.8796 per IU | 20 to 40 IU/kg every 3 or 4 days | 374.27 to 998.05 | 136,702 to 364,539 |
IV = intravenous; FIX = factor IX; Fc fusion protein
Note: All prices do not include dispensing fees. Annual costs were calculated assuming 365.25 days per year (51.18 weeks) and a patient weight of 85.1 kg based on the HOPE-B posttreatment safety population.27 Daily and annual costs are based on price per IU; wastage has not been accounted for.
aSponsor-submitted price for etranacogene dezaparvovec.28 Prices of FIX treatments are not available in CADTH-participating drug formularies; as such, the price for FIX comparators was adopted from the sponsor’s submission28 in which the price per IU for coagulation factor IX (recombinant) nonacog alfa and coagulation factor IX (recombinant) rFIXFc were derived from the CADTH report on a treatment cost comparison and budget impact analysis of coagulation factor IX (recombinant) rFIXFc.15 The sponsor assumed that the annual cost of coagulation factor IX (recombinant) Fc fusion protein is representative coagulation factor IX (recombinant) glycopegylated and that the cost per IU of coagulation factor IX (recombinant) nonacog alfa is representative of FIX concentrate (human).
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing. | No | Refer to limitation: The submitted model does not align with the indicated population. Additionally, while female patients with moderately severe to severe hemophilia B are captured in the Health Canada indication, they were excluded from the HOPE-B trial. The impact of this on the cost-effectiveness of etranacogene dezaparvovec is not expected to be significant but is unknown. |
Model has been adequately programmed and has sufficient face validity . | Yes | No comment. |
Model structure is adequate for decision problem. | No | Refer to limitation: Model structure does not appropriately capture the current treatment experience of patients with hemophilia B. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis). | No | Refer to limitation: Failure of parameter uncertainty to accurately reflect uncertainty around the ICER. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem. | No | Refer to limitation: Failure of parameter uncertainty to accurately reflect uncertainty around the ICER. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough detail). | No | The sponsor’s pharmacoeconomic report did not include all of the references used to inform model parameters (as included in the references sheet of the submitted model file), discrepancies between the pharmacoeconomic report and the submitted model (e.g., standard errors), difficult to locate exact analyses that informed some parameters from the ITC. |
ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 9: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Etranacogene dezaparvovec | rFIXFc | Pegylated nonacog beta pegol | Nonacog alfa |
|---|---|---|---|---|
Discounted life-years | ||||
Total | 30.091 | 30.091 | 30.091 | 30.091 |
By health state | ||||
No bleeds | 29.804 | 28.941 | 29.371 | 27.723 |
Non-joint bleed | 0.182 | 0.599 | 0.456 | 1.898 |
Joint bleed | 0.106 | 0.551 | 0.264 | 0.471 |
Discounted QALYs | ||||
Total | 25.540 | 23.449 | 23.505 | 23.407 |
By health state | ||||
No bleeds | 25.321 | 22.665 | 23.004 | 21.715 |
Non-joint bleed | 0.146 | 0.442 | 0.336 | 1.399 |
Joint bleed | 0.073 | 0.343 | 0.264 | 0.471 |
Discounted costs ($) | ||||
Total | 6,920,810 | 15,437,949 | 15,161,816 | 11,252,115 |
Treatment | 6,763,932 | 14,834,802 | 14,789,975 | 10,855,646 |
Follow-up | 1,134 | 0 | 0 | 0 |
Disease monitoring | 13,448 | 13,377 | 13,455 | 13,378 |
Disease management | 142,276 | 589,150 | 357,765 | 382,468 |
Adverse event | 20 | 620 | 621 | 622 |
FIX = factor IX; QALY = quality-adjusted life-year; rFIXFc = recombinant coagulation factor IX Fc fusion protein.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 10: Summary of the Stepped Analysis of the CADTH Reanalysis Results (Deterministic)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Etranacogene dezaparvovec vs. Coagulation FIX (recombinant) Fc fusion protein | ||||
Sponsor’s base case | Etranacogene dezaparvovec | 6,925,878 | 25.538 | Reference |
Coagulation FIX (recombinant) Fc fusion protein | 15,424,867 | 23.446 | Dominated | |
CADTH reanalysis 1 | Etranacogene dezaparvovec | 8,010,488 | 25.535 | Reference |
Coagulation FIX (recombinant) Fc fusion protein | 15,424,867 | 23.446 | Dominated | |
CADTH reanalysis 2 | Etranacogene dezaparvovec | 6,925,878 | 23.516 | Reference |
Coagulation FIX (recombinant) Fc fusion protein | 15,424,867 | 23.446 | Dominated | |
CADTH base case (1 + 2) | Etranacogene dezaparvovec | 8,010,488 | 23.513 | Reference |
Coagulation FIX (recombinant) Fc fusion protein | 15,424,867 | 23.446 | Dominated | |
CADTH base case (1 + 2; probabilistic) | Etranacogene dezaparvovec | 8,003,296 | 23.514 | Reference |
Coagulation FIX (recombinant) Fc fusion protein | 15,438,022 | 23.449 | Dominated | |
Etranacogene dezaparvovec vs. coagulation FIX (recombinant) pegylated nonacog beta pegol | ||||
Sponsor’s base case | Etranacogene dezaparvovec | 6,925,878 | 25.538 | Reference |
Coagulation FIX (recombinant) pegylated nonacog beta pegol | 15,195,210 | 23.498 | Dominated | |
CADTH reanalysis 1 | Etranacogene dezaparvovec | 8,010,488 | 25.535 | Reference |
Coagulation FIX (recombinant) pegylated nonacog beta pegol | 15,195,210 | 23.498 | Dominated | |
CADTH reanalysis 2 | Etranacogene dezaparvovec | 6,925,878 | 23.516 | Reference |
Coagulation FIX (recombinant) pegylated nonacog beta pegol | 15,195,210 | 23.498 | Dominated | |
CADTH base case (1 + 2) | Etranacogene dezaparvovec | 8,010,488 | 23.513 | Reference |
Coagulation FIX (recombinant) pegylated nonacog beta pegol | 15,195,210 | 23.498 | Dominated | |
CADTH base case (1 + 2; probabilistic) | Etranacogene dezaparvovec | 8,003,296 | 23.514 | Reference |
Coagulation FIX (recombinant) pegylated nonacog beta pegol | 15,161,685 | 23.505 | Dominated | |
FIX = factor IX; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Table 11: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Etranacogene dezaparvovec | Coagulation FIX (recombinant) Fc fusion protein | Coagulation FIX (recombinant) pegylated nonacog beta pegol |
|---|---|---|---|
Discounted life-years | |||
Total | 30.091 | 30.091 | 30.091 |
By health state | |||
No bleeds | 29.768 | 28.940 | 29.372 |
Non-joint bleed | 0.204 | 0.602 | 0.455 |
Joint bleed | 0.119 | 0.549 | 0.264 |
Discounted QALYs | |||
Total | 23.514 | 23.449 | 23.505 |
By health state | |||
No bleeds | 23.289 | 22.664 | 23.004 |
Non-joint bleed | 0.150 | 0.444 | 0.336 |
Joint bleed | 0.074 | 0.342 | 0.264 |
Discounted costs ($) | |||
Total | 8,003,296 | 15,438,022 | 15,161,685 |
Treatment | 7,828,541 | 14,834,802 | 14,789,975 |
Follow-up | 1,134 | 0 | 0 |
Disease monitoring | 13,448 | 13,377 | 13,455 |
Disease management | 160,150 | 589,224 | 357,634 |
Adverse event | 23 | 620 | 621 |
FIX = factor IX; QALY = quality-adjusted life-year.
Scenario Analyses
Table 12: Summary of CADTH’s Exploratory Analysis for Etranacogene Dezaparvovec Versus Nonacog Alfa
Intervention | Total costs ($) | Total QALYs | ICER vs. etranacogene dezaparvovec |
|---|---|---|---|
Etranacogene dezaparvovec | 8,003,296 | 23.514 | Reference |
Nonacog alfa, administered every 3 days | 11,097,082 | 23.408 | Dominated |
Nonacog alfa, administered every 4 days | 8,336,843 | 23.408 | Dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Table 13: Summary of Scenario Analyses Conducted on the CADTH Base Case
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Etranacogene dezaparvovec vs. rFIXFc | ||||
CADTH base case | Etranacogene dezaparvovec | 8,003,296 | 23.514 | Reference |
rFIXFc | 15,438,022 | 23.449 | Dominated | |
Scenario analysis: 3-year time horizon | rFIXFc | 1,796,908 | 2.298 | Reference |
Etranacogene dezaparvovec | 4,743,921 | 2.281 | Dominated | |
Etranacogene dezaparvovec vs. pegylated nonacog beta pegol | ||||
CADTH base case | Etranacogene dezaparvovec | 8,003,296 | 23.514 | Reference |
Pegylated nonacog beta pegol | 15,161,685 | 23.505 | Dominated | |
Scenario analysis: 3-year time horizon | Pegylated nonacog beta pegol | 1,506,240 | 2.287 | Reference |
Etranacogene dezaparvovec | 4,743,921 | 2.281 | Dominated | |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; rFIXFc = recombinant coagulation factor IX Fc fusion protein.
CADTH additionally conducted a threshold analysis to determine the price of comparators where etranacogene dezaparvovec would no longer be considered cost-effective at a $50,000 and $100,000 per QALY gained threshold. If the prices for rFIXFc and pegylated nonacog beta pegol are 48.2% and 60.5% less than those used in the CADTH base case, etranacogene dezaparvovec will no longer be cost-effective at either a $50,000 or $100,000 per QALY gained WTP threshold over the lifetime time horizon.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 14: Summary of Key Takeaways
Key takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted BIA assessed the budgetary impact resulting from reimbursing etranacogene dezaparvovec for the treatment of adults with hemophilia B who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes.28 The BIA was conducted from the perspective of the Canadian public drug plans over a 3-year time horizon (2024 to 2026) using an epidemiological approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec). Key inputs to the BIA are documented in Table 15.
The sponsor compared a reference scenario in which patients received rFIX prophylaxis to a new-drug scenario in which etranacogene dezaparvovec was reimbursed. Market share for rFIX prophylaxis in the reference scenario was based on feedback the sponsor obtained from CBS. In the new-drug scenario, the uptake of etranacogene dezaparvovec was assumed to be 3%, 8%, and 13% in year 1, year 2, and year 3, respectively, based on sponsor internal forecasts and interviews with Canadian clinicians. Wastage and administration costs were not included. rFIX costs were based on a CADTH report on coagulation FIX (recombinant) Fc fusions protein (Alprolix) from 2015. The annual drug acquisition costs for pegylated nonacog beta pegol (Rebinyn) and FIX concentrate (human) (Immunine) were assumed to be equivalent to rFIXFc (Alprolix) and nonacog alfa (BeneFIX), respectively.
The following key assumptions were made by the sponsor:
The sponsor assumed that while both males and females are eligible for treatment with etranacogene dezaparvovec, hemophilia B primarily impacts males and only males in Canada were included in the analysis.
The size of the target population will decline each year as etranacogene dezaparvovec is administered as a 1-time infusion.
Market shares were captured proportionally from all comparators except FIX concentrate (human) (Immunine), where no market share was captured.
All patients were assumed to have public coverage.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Proportion of Canadian males Prevalence of hemophilia B Proportion of adults in Canada Proportion of patients with moderately severe to severe disease Proportion of patients who will receive FIX prophylaxis | 49.7%29 3.9/100,000 males30 81%31 30%31 80%a |
Number of patients eligible for drug under review | 121 / 122 / 124 |
Market uptake (3 years)b | |
Uptake (reference scenario) rFIXFc (Alprolix) Nonacog alfa (BeneFIX) Pegylated nonacog beta pegol (Rebinyn) FIX concentrate (human) (Immunine) | 24% / 24% / 24% 39% / 39% / 39% 36% / 36% / 36% 1% / 1% / 1% |
Uptake (new-drug scenario) Etranacogene dezaparvovec (Hemgenix) rFIXFc (Alprolix) Nonacog alfa (BeneFIX) Pegylated nonacog beta pegol (Rebinyn) FIX concentrate (human) (Immunine) | 3% / 8% / 13% 22% / 22% / 21% 38% / 36% / 34% 35% / 33% / 31% 1% / 1% / 1% |
Cost of treatment (per patient) | |
Cost of treatment annually Etranacogene dezaparvovec (Hemgenix) rFIXFc (Alprolix) Nonacog alfa (BeneFIX) Pegylated nonacog beta pegol (Rebinyn) FIX concentrate (human) (Immunine) | $4,690,000 $473,289 $364,289 $473,289 $364,289 |
FIX = Factor IX; rFIXFc = recombinant coagulation factor IX Fc fusion protein.
aAssumption informed by clinical expert opinion.
bThe sponsor submitted an updated reference and new market shares for certain comparators to CADTH after submission.28 Coagulation FIX (recombinant) Fc fusions protein: 22%; nonacog alfa: 37%; pegylated nonacog beta pegol: 39%, years 1, 2, and 3. In the new-drug scenario, etranacogene dezaparvovec market uptake remained the same, however market shares for the other comparators differed. rFIXFc: 21%, 20%, 19%; nonacog alfa: 36%, 34%, 32%; pegylated nonacog beta pegol: 38%, 36%, 34%, and FIX concentrate (human): 2%, years 1, 2, and 3, respectively.28
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement of etranacogene dezaparvovec for the indicated population would lead to an incremental budget of $15,439,323 in year 1, $24,659,684 in year 2, and $22,621,322 in year 3 for a 3-year incremental budget impact of $62,720,329.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Eligible patient population does not align with the Health Canada indication: The sponsor’s submitted BIA models the reimbursement of etranacogene dezaparvovec in adult male hemophilia B patients with moderately severe to severe disease who receive FIX prophylaxis.28 This is narrower than the Health Canada indication, which does not specify that the population eligible for treatment with etranacogene dezaparvovec is sex-specific.2 According to clinical expert feedback received for this review, experts would consider using etranacogene dezaparvovec in female patients but noted that the number who would be eligible to receive the treatment in Canada was likely small.
Additionally, the sponsor assumed that only patients with moderately severe to severe disease would be eligible for treatment, an epidemiological assumption that does not align with the Health Canada indication. Instead, the Health Canada indication would include all patients who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes. Based on a market assessment report submitted by the sponsor, a proportion (39.7% and 21.3%) of treated moderate and mild patients were also on a prophylaxis regimen.32 This was confirmed by clinical expert feedback obtained by CADTH which indicated that some patients who are classified as having mild or moderate disease who had a severe bleeding phenotype would require routine prophylaxis to prevent or reduce the frequency of bleeding episodes. Therefore, some patients with mild or moderate disease may be considered for treatment with etranacogene dezaparvovec, which would align with the Health Canada indication.
To address this limitation, CADTH undertook a reanalysis using epidemiological information provided in the market assessment report provided by the sponsor. In this report, 27% of adult males presented with severe disease, 40% presented with moderate disease and 32% presented with mild disease. Additionally, 84% of severe patients, 40% of moderate patients and 19% of mild patients received FIX prophylaxis therapy.
CADTH was unable to address the exclusion of females from the BIA due to uncertainty of the prevalence of hemophilia B in this group. As such, the budget impact of reimbursing etranacogene dezaparvovec in females is unknown but is likely to be small.
The number of patients expected to receive etranacogene dezaparvovec in the next 3 years is uncertain: The sponsor assumed that, should etranacogene dezaparvovec be reimbursed, the uptake of the treatment will be 3%, 8%, and 13% in years 1, 2, and 3, respectively.28 As etranacogene dezaparvovec is a 1-time treatment, the sponsor assumed that the cumulative uptake over the first 3 years of reimbursement (2025 to 2027) would result in an estimated 14 patients receiving treatment with etranacogene dezaparvovec by the end of 2027. Clinical expert feedback indicated that the uptake is uncertain and may be higher than estimated by the sponsor.
Due to the uncertainty surrounding the uptake in etranacogene dezaparvovec, CADTH was unable to address this limitation. CADTH explored decreasing the uptake in a scenario analysis to align more with the sponsor’s submitted uptake estimates.
Market share estimates of currently available FIX prophylaxis are not reflective of Canadian clinical practice: In the reference scenario of the sponsor’s submitted BIA, it assumed that 24%, 39%, 36% and 1% of eligible patients would be receiving rFIXFc (Alprolix), nonacog alfa, pegylated nonacog beta pegol, and FIX concentrate (human) (Immunine), respectively, in years 1, 2, and 3, based on a market assessment report and the sponsor’s feedback from CBS.28,32 CADTH obtained clinical expert feedback that suggested the distribution of FIX market shares in the current and new treatment landscapes is not reflective of Canadian clinical practice. Instead, currently, approximately 20% of patients are receiving rFIXFc, 15% are receiving nonacog alfa, and 64% are receiving pegylated nonacog beta pegol.
Additionally, within the sponsor’s submitted market assessment report, CADTH noted that the market share of each relevant comparator did not align with the market shares applied in the sponsor’s base case. Notably, the market assessment report highlighted that approximately 64% of severe adult prophylaxis patients were on pegylated nonacog beta pegol, approximately 5% were on nonacog alfa and approximately 10% were on rFIXFc fusions protein.32 The market shares of the latter 2 comparators were reported to be lower in the market assessment report compared with CADTH clinical expert feedback; however, the report also included that approximately 20% of adult prophylaxis patients were on rFIX (Rixubis), a product that is not typically prescribed outside of Quebec.32
To address this limitation, CADTH undertook a reanalysis by revising the market shares of the comparators in both the reference and new-drug scenario to align with clinical expert feedback and the sponsor’s market assessment report.
Drug plan payer perspective: According to the Procedures for CADTH Reimbursement Reviews, the BIA base case should be undertaken from the perspective of a pan-Canadian drug plan program.33 As such, costs relating to the use of blood products (i.e., rFIX prophylaxis) are not paid by jurisdictional drug plan budgets and should thus be excluded from the drug plan perspective.
Costs associated with the use of blood products were excluded from the drug plan perspective. Blood product costs were included within a health care system perspective, which also included disease monitoring and administration costs.
Costing in the model is highly uncertain. The cost of FIX prophylaxis in the sponsor’s submitted model was based on assumed prices that were informed by a previous CADTH review for rFIXFc fusions protein as list prices for rFIX are not available.28 Additionally, these costs do not reflect any confidential pricing that may have been negotiated by CBS. As such, the estimated acquisition costs for FIX prophylaxis treatments are uncertain.
CADTH was unable to address this limitation.
Neutralizing antibody testing coverage status is uncertain. The sponsor excluded costs related to nAb testing from the BIA, indicating that the sponsor would cover the costs. However, if the cost of nAb testing is not covered by the sponsor, the costs incurred by the payer will be higher than estimated by the sponsor.
CADTH was unable to address this limitation due to a paucity of data regarding the cost of nAb testing.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s submitted analyses by aligning the eligible population with the Health Canada indication, removing the cost of rFIX prophylaxis from the drug plan perspective, and revising rFIX prophylaxis market shares from the health care system perspective. The changes applied to derive the CADTH base-case and key scenario analyses for both perspectives are described in Table 16.
Table 16: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption | ||
|---|---|---|---|---|
Corrections to the sponsor’s base case | ||||
None | — | — | ||
Changes to derive the CADTH base case | ||||
1. Eligible population | Proportion of adult males with hemophilia B with moderately severe to severe disease: 30% Proportion of moderately severe to severe patients that receive FIX prophylaxis therapy: 80% | Proportion of adult males with moderately severe to severe disease: 100%a Proportion of patients that are treated for hemophilia B: 44.5%b | ||
2. Perspective | Health care payer | Drug plan | ||
3. Reference market shares of some comparators are uncertain.c | Reference scenario (year 1 / 2 / 3) rFIXFc fusions protein: 24% / 24% / 24% Nonacog alfa: 39% / 39% / 39% Pegylated nonacog beta pegol: 36% / 36% / 36% | Reference scenario (year 1 / 2 / 3) rFIXFc: 20% / 20% / 20% Nonacog alfa: 15% / 15% / 15% Pegylated nonacog beta pegol: 64% / 64% / 64% | ||
CADTH base case | Reanalysis 1 + 2 | |||
CADTH health care system perspective scenario | Reanalysis 1 + 3 | |||
CBS = Canadian Blood Services; FIX = factor IX; rFIXFc = recombinant coagulation factor IX Fc fusion protein.
aTo apply the percentage of patients that are treated for hemophilia B by disease severity.
bCalculated based on the following: Distribution of severity of hemophilia B: 27% severe disease, 40% moderate disease and 32% mild disease (1% unknown, not applicable and normal) multiplied by the proportion that receives FIX prophylaxis therapy: 84% of severe patients, 40% of moderate patients and 19% of mild patients (assume 0% of unknown, not applicable and normal receive prophylaxis).32
cMarket shares for FIX concentrate (human) (Immunine) did not change.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18. Based on the CADTH base case, the budget impact associated with the reimbursement of etranacogene dezaparvovec for the treatment of adults with hemophilia B who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes would be associated with a budgetary increase of $31,520,232 in year 1, $53,523,195 in year 2, $54,760,039 in year 3, with a 3-year total incremental cost of $139,803,466.
Table 17: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base casea | 62,720,329 |
Reanalysis 1: eligible population | 116,315,601 |
Reanalysis 2: public drug plan perspective | 75,385,583 |
Reanalysis 3: market shares | 61,949,258 |
CADTH base caseb | 139,803,466 |
CADTH health care system scenario | 114,855,673 |
BIA = budget impact analysis.
aSponsor's submitted base case adopted a health care system perspective. This analysis includes acquisition costs for etranacogene dezaparvovec and FIX prophylaxis treatment.
bCADTH’s base case adopts the perspective of CADTH-participating drug plans. This analysis includes acquisition costs for etranacogene dezaparvovec. Acquisition costs for FIX are borne by the Canadian Blood Services and were excluded from this analysis.
CADTH conducted the following scenario analysis (Table 18) to highlight uncertainty associated with the potential budget impact.
Adopting the perspective of the public health care payer, in which the costs related to FIX prophylaxis, treatment administration, and disease monitoring costs (i.e., specialist visits, joint scans, viral screening) were included.
Conducting a scenario analysis where the market uptake for etranacogene dezaparvovec is halved.
Table 18: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 51,295,222 | 51,906,258 | 52,517,293 | 53,128,329 | 157,551,880 |
New drug | 51,295,222 | 67,345,580 | 77,176,977 | 75,749,651 | 220,272,209 | |
Budget impact | 0 | 15,439,323 | 24,659,684 | 22,621,322 | 62,720,329 | |
CADTH base case (drug plan perspective) | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 31,520,232 | 53,523,195 | 54,760,039 | 139,803,466 | |
Budget impact | 0 | 31,520,232 | 53,523,195 | 54,760,039 | 139,803,466 | |
CADTH scenario analysis 1: Health care system perspective | Reference | 100,919,081 | 102,121,243 | 103,323,404 | 104,525,566 | 309,970,213 |
New drug | 100,919,081 | 130,577,847 | 148,580,742 | 145,697,297 | 424,855,886 | |
Budget impact | 0 | 28,456,604 | 45,257,338 | 41,171,731 | 114,885,673 | |
CADTH scenario analysis 2: Halved market uptake of etranacogene dezaparvovec | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | 15,760,116 | 26,761,598 | 27,380,019 | 69,901,733 | |
Budget impact | 0 | 15,760,116 | 26,761,598 | 27,380,019 | 69,901,733 |
BIA = budget impact analysis.
Ethics Review
Abbreviations
AAV
adeno-associated virus
ABR
annualized bleeding rate
APM
alternative payment model
FIX
coagulation factor IX
HTC
hemophilia treatment centre
nAb
neutralizing antibody
Summary
Hemophilia B is a congenital, recessive bleeding disorder that is characterized by a deficiency of coagulation factor IX (FIX). It results in susceptibility to prolonged bleeding and subsequent organ or joint damage.
CADTH reviewed the patient group, clinician group, clinical expert, and drug program input gathered during this review, as well as relevant literature, to identify ethical considerations related to the use of etranacogene dezaparvovec for the treatment of adults (aged 18 years or older) with hemophilia B (congenital factor IX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes.
Ethical considerations identified in this review included those related to the following:
Treatment and experiences of hemophilia B: The standard of care is FIX prophylaxis, which is physically and psychosocially burdensome for individuals with moderate to severe hemophilia B. This therapy requires frequent IV infusions, which impact quality of life and lead to varying FIX activity levels despite adherence. Additionally, the therapeutic effect of FIX prophylaxis wanes between infusions, which leaves individuals vulnerable to bleeds and associated joint damage. This can impact peoples’ sense of freedom to fully engage in daily activities. There is an unmet need for a therapeutic option that can reduce the burden of treatment associated with FIX prophylaxis and provide a sustained therapeutic effect that limits the long-term risk of experiencing a bleed.
Clinical and economic evidence used in the evaluation of etranacogene dezaparvovec: The clinical trial evidence indicated that treatment with etranacogene dezaparvovec may result in a clinically relevant reduction in the annualized bleeding rate (ABR) for all bleeds. Similarly, as of the 36-month data cut provided by the sponsor, 51 of 54 HOPE-B trial participants remained free of FIX prophylaxis. However, there is uncertainty regarding interpretations of the magnitude of benefit as well as long-term safety and efficacy. This uncertainty challenges clinical and shared decision-making and will require rigorous informed consent. Uncertainty is further exacerbated for females and patients with FIX activity greater than 2% because they were excluded from the HOPE-B trial. Similarly, the clinical experts consulted by CADTH indicated that people reported as “Black/African American” were underrepresented in the trial. Although the clinical experts assumed the trial outcomes would be generalizable to all people with hemophilia B, there is uncertainty about who might benefit beyond the population reflected in the trial. Finally, uncertainty around long-term safety, efficacy, and comparative effectiveness limits the ability to accurately model cost-effectiveness and understand the opportunity costs associated with reimbursement.
Clinical use and implementation of etranacogene dezaparvovec as a gene therapy: As with other gene therapies, the use of etranacogene dezaparvovec poses potential risks, including transaminitis (9 of 54 HOPE-B trial participants experienced elevated transaminase levels) and theoretical concerns of long-term genotoxicity, resulting in cancer. As a 1-time infusion that cannot be reversed, clinicians will need to facilitate a thorough consent process that is supportive of shared decision-making and helps patients weigh the potential benefits and harms. These conversations will need to include: the consideration of the uncertainty regarding long-term safety and efficacy, the possibility of waning treatment effect resulting in a return to FIX prophylaxis, the ambiguity surrounding determinations of treatment failure and the development of cross-reactive anti–adeno-associated virus (AAV) vector neutralizing antibodies (nAbs) that may render individuals ineligible for future gene therapies. As it is presently unclear who is most likely to benefit from treatment, determining who should receive etranacogene dezaparvovec may be ethically challenging for providers. In particular, the absence of some populations from the HOPE-B trial (e.g., those with FIX activity greater than 2% and females) may incidentally lead to disparities in access if treatment is prioritized for those populations for which some safety and efficacy data are available. Ensuring equitable access to etranacogene dezaparvovec will also require addressing geographic barriers to accessing specialist care and monitoring.
Health systems: Ethical considerations related to the implementation of etranacogene dezaparvovec highlight challenges in fairly allocating limited resources for expensive therapies for rare diseases. Uncertainty around the long-term efficacy and safety of etranacogene dezaparvovec may prompt consideration of alternative payment models (APMs) to manage and redistribute the risks and benefits associated with reimbursing a highly expensive therapy of uncertain benefit for payers and manufacturers. The design of an APM has ethical implications, as it impacts the distribution of risks and benefits among parties. It is also necessary to consider the availability and costs of the data and clinical infrastructure required to effectively implement an APM. In particular, the clinical experts flagged that personnel shortages at hemophilia treatment centres (HTCs) across Canada may impact capacity to deliver therapy and to collect robust registry data. Uncertainty regarding who is most likely to benefit from therapy, potential shortages of the AAV vector used in etranacogene dezaparvovec, or limited delivery capacity at some HTCs, may necessitate clear prioritization criteria to facilitate fair and equitable access. Moreover, geographic challenges may require some patients to cross jurisdictions for access, leading to complexities in determining the jurisdictions responsible for reimbursement of the therapy and associated costs.
Objective
To identify and describe the ethical considerations associated with the use of etranacogene dezaparvovec for the treatment of adults (aged 18 years or older) with hemophilia B (congenital factor IX deficiency) who require routine prophylaxis to prevent or reduce the frequency of bleeding episodes, including considerations related to the context of hemophilia B, evidentiary basis, use of etranacogene dezaparvovec, and health systems.
Research Questions
This report addresses the following research questions:
What ethical considerations arise in the context of hemophilia B in adults?
What ethical considerations arise related to the evidence (e.g., clinical and economic data) used to evaluate etranacogene dezaparvovec?
What ethical considerations arise in the use of etranacogene dezaparvovec (as a gene therapy) for patients, their caregivers, and clinicians?
What ethical considerations for health systems are involved in the context of etranacogene dezaparvovec as a gene therapy?
Methods
To identify ethical considerations relevant to the use of etranacogene dezaparvovec in the treatment of hemophilia B, this ethics report was driven by relevant questions identified in the EUnetHTA Core Model 3.0,1 Ethics Analysis Domain and supplemented by relevant questions from the Equity Checklist for Health Technology Assessment (ECHTA).2 These guiding questions were organized to respond to the research questions posed and investigated ethical considerations related to the following:
the patients living with hemophilia B and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges related to diagnosis or clinical care; factors that might prevent patients from gaining access to therapies)
the evidence used to demonstrate the benefits, harms, and value of etranacogene dezaparvovec (i.e., ethical considerations in relevant clinical trials, including their representativeness, choice of outcome measures, appropriateness of analytical methods and models to all population groups, and ethical considerations related to the data or assumptions in the economic evaluation)
the use of etranacogene dezaparvovec (as a gene therapy), including considerations related to benefits and harms to patients, relatives, caregivers, clinicians or society, and considerations related to access to these therapies
the uptake of etranacogene dezaparvovec (as a gene therapy) in health systems, including considerations related to the distribution of health care resources.
Data Collection: Review of Project Inputs and Literature
Data to inform this ethics report were drawn from an identification of ethical considerations (e.g., values, norms, or implications related to the harms, benefits, and implications for equity, justice, resource allocation, and ethical considerations in the evidentiary basis) in the input from patient and clinician groups, clinical experts, and drug programs collected by CADTH to inform this review, as well as a complementary search of the published literature. Ongoing collaboration and communication with CADTH reviewers working on the clinical and economic reviews for this submission also assisted in identifying and clarifying the ethical considerations raised.
Review of Project Inputs
During this CADTH review, a single reviewer collected and considered input from 6 main sources for content related to ethical considerations relevant to addressing the research questions guiding this ethics report. In addition to published literature, this report considered the following sources:
the sponsor submission, including noting relevant information and external references or sources relevant to each of the research questions driving this report
the clinician group input received by CADTH from the Association of Hemophilia Clinic Directors of Canada
the patient input received by CADTH from the Canadian Hemophilia Society
the drug program input received by CADTH from the drug programs participating in the CADTH Reimbursement Review process
discussion with the clinical experts (n = 3) directly engaged by CADTH over the course of this Reimbursement Review, including through 2 clinical and economic consultation meetings involving 2 experts and 1 panel meeting involving 3 experts. During each of these meetings, the clinical experts were asked targeted questions related to ethical considerations corresponding to the research questions driving this report. All of the clinical experts were practising hematologists and had experience treating adult patients with hemophilia B, and 2 had experience with gene therapy for hemophilia B
engagement with CADTH clinical and economic reviewers to identify domains of ethical interest arising from their respective reviews as well as relevant questions and sources to further pursue in this report.
Literature Search Methods
The information specialist updated and supplemented a previous literature search (conducted August 28, 2023) on key resources including MEDLINE through Ovid, the Philosopher’s Index through Ovid, the Cumulative Index to Nursing and Allied Health Literature (CINAHL) through EBSCO, PsycInfo, and Scopus. A targeted Google Scholar search was also performed. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings) and keywords. The main search concepts were etranacogene dezaparvovec and hemophilia B.
CADTH-developed search filters were applied to the searches conducted in MEDLINE, CINAHL, and Scopus to limit retrieval to citations related to ethical concepts or considerations, equity concepts or considerations, or qualitative studies. Due to the limited number of results, no filters were applied to the searches conducted in PsycInfo and Philosopher’s Index to limit the retrieval by study type. Duplicates were removed by manual deduplication in EndNote. Retrieval was limited to the English language. The search was completed on October 26, 2023. The search strategy is available on request.
Literature Screening and Selection
Literature retrieved according to the search and selection methods detailed previously was screened in 2 stages. First, the titles and abstracts of the citations retrieved were screened for relevance by a single reviewer. Articles were identified and retrieved for full-text review by a single reviewer if their titles or abstracts identified ethical considerations or provided normative analysis (i.e., focusing on “what ought to be” through argumentation), or presented empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to the experiences, incidence, diagnosis, treatment, or outcomes of hemophilia B, or the evidence on, use of, or implications of etranacogene dezaparvovec for adults with hemophilia B. In the second stage, full-text publications categorized as “retrieve” were reviewed by the same reviewer. Texts that included substantive information meeting the aforementioned criteria were included in the review, and reports that did not meet these criteria were excluded. As a parallel process, other sources drawn from relevant bibliographies and relevant key concepts, in consultation with experts or other CADTH reviewers, were retrieved and reviewed using the selection criteria listed previously.
Data Analysis
The data analysis was driven by the 4 research questions guiding this report and included the collection, coding, and thematic analysis of data drawn from the literature and project inputs. The reviewer conducted 2 iterative cycles of coding and analysis to abstract, identify, and synthesize relevant ethical considerations in the literature and from relevant project inputs.
In the initial coding phase, publications and input sources were reviewed for ethical content (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis). Once identified, claims related to ethical content were coded using methods of qualitative description.3 In the second coding phase, major themes and subcodes were identified through repeated readings of the data3 and summarized in thematic categories within each guiding domain or research question. Where ethical content did not fit into the categories or domains outlined in the research questions, this was noted, as were discrepancies or conflicts between the ethical considerations or values identified between project sources or within thematic categories. The data analysis was iterative, and the themes identified — in the literature, in project inputs, and during consultations with clinical experts — were used to further refine and reinterpret the identified ethical considerations.
The data collected and analyzed from these sources were thematically organized and described according to the 4 research questions and domains driving this report. The results of this analysis and its limitations and conclusions are described subsequently.
Results
Description of Included Sources
Data to inform this ethics report drew from a review of patient group input, clinician group input, drug program input, and consultation with the clinical experts engaged by CADTH for this review. All clinical experts were hematologists with experience treating people living with hemophilia B. A description and summary of these sources is included in the Clinical Review Report.
The literature search identified 41 novel results. Following title and abstract screening, 28 citations were excluded and 13 potentially relevant publications from the electronic searches were retrieved for full-text review. Of the potentially relevant publications, 7 publications were excluded, as they did not discuss any ethical considerations of etranacogene dezaparvovec or hemophilia B. Six publications met the inclusion criteria and were included in this report. Nineteen additional publications about hemophilia B or gene therapy that had been identified in a recent CADTH review of another gene therapy for hemophilia B were also included in this report.
A total of 25 publications were used to inform this report. Of these publications, 5 discussed ethical considerations in the context of hemophilia B, including those related to diagnosis and treatment; 3 discussed patient and/or family and caregiver experiences in the context of hemophilia B; and 9 discussed the use of gene therapies (such as etranacogene dezaparvovec) for hemophilia. The remaining 8 publications were selected to provide a broader understanding of health systems considerations related to the costs of gene therapies or other expensive treatments for rare diseases.
Key Ethical Considerations
Treatment and Experiences of Hemophilia B
Hemophilia B is a rare, congenital bleeding disorder that is characterized by a deficiency of FIX due to variants or mutations in the F9 gene on the X chromosome.4,5 Whether due to a shortage or complete absence of FIX activity, people living with hemophilia B are susceptible to prolonged bleeding episodes.4 While bleeding events may occur as the result of trauma or injury, people with moderate to severe hemophilia B may also experience spontaneous internal bleeding into joints, muscles, and organs.4 Bleeding events may not only be life-threatening and painful in the short-term, but frequent bleeds into the joints may also lead to permanent joint damage and long-term mobility complications.4
The World Federation of Hemophilia (WFH) defines hemophilia B severity according to FIX activity levels, with moderate indicating 1% to 5% of normal and severe less than 1% of normal.5 Although not included in WFH guidelines, clinical trials in hemophilia B (including the HOPE-B study of etranacogene dezaparvovec) have also used the term “moderately severe” (FIX activity 1% to 2% of normal) to further specify their target population. The clinician input and input from the clinical experts engaged by CADTH indicated that clinical phenotype (i.e., tendency to bleed) is typically used in conjunction with FIX activity levels to determine severity. This input highlighted these 2 criteria as an important feature of clinical decision-making, given their impact on eligibility for and access to current and future treatment options (including gene therapies like etranacogene dezaparvovec).
Current Treatment Options for Hemophilia B
Prophylactic factor replacement therapy, known as FIX prophylaxis, is the current standard of care for people living with moderate to severe hemophilia B (and a bleeding phenotype).5 FIX prophylaxis is aimed at increasing FIX activity to levels that prevent or reduce bleeding events, particularly joint bleeds.5 Ideally, effective FIX prophylaxis treatment gives people with hemophilia B more control over their condition.5 Additionally, it allows them a degree of autonomy over daily activities that is similar to that of the unaffected population.5 FIX prophylaxis is done by IV infusion of FIX coagulation factor concentrates at home by the patient or their caregiver.
Clinician input, patient input, and clinical experts indicated that a limiting feature of FIX concentrates is the frequency at which they must be administered. Depending on the FIX concentrate used, people with hemophilia B undergo IV infusion as often as 2 to 3 times per week for standard half-life products, or once every 1 to 2 weeks for extended half-life products. While FIX prophylaxis can successfully increase FIX activity levels for most patients with hemophilia B, it is a lifelong, demanding therapy that can present long-term adherence challenges. For example, the clinician and patient input indicated that poor venous access and pain at infusion sites can impact adherence to the strict infusion regimen. This impacts patients’ ability to benefit from existing treatment options and may leave them with a significant unmet need. Further, as the effect of FIX prophylaxis wanes over time, even people achieving strict adherence to their dosing regimen will experience trough FIX activity levels. As a result, people remain susceptible to bleeding events (and associated joint damage) and may need to restrict their activity as FIX levels wane. Although the patient input suggested that many people with hemophilia in Canada are satisfied with their current treatment options, it also indicated that FIX prophylaxis can greatly complicate their daily lives.
Experiences of Hemophilia B
Patient input, input from the clinical experts, and the published literature all described the serious psychosocial and physical burden of living with or caring for someone with moderate to severe hemophilia (A or B).6 The patient input reported that a primary physical burden associated with hemophilia B is the joint damage caused by repeated episodes of internal bleeding. This is not only painful, but it was described as reducing people’s ability to participate in daily household and workplace activities.7 Furthermore, it contributed to an increased need for mobility support and joint replacement procedures later in life. This input indicated that susceptibility to bleeding limited people’s sense of autonomy regarding participation in daily activities such as sports and, as a consequence, severely affected quality of life.
As an X chromosome–linked condition, hemophilia (A or B) has historically been understood as a disease that primarily affects males and is carried largely asymptomatically by females.8 This understanding has fostered the development and implementation of treatments for males with hemophilia (A or B) to the general exclusion of females.8 Although females have been recognized as carrying the potential to be diagnosed with hemophilia (A or B) since 2012, some of the published literature has suggested that hemophilia in females remains underrecognized and underdiagnosed.8 As a result, females living with moderate to severe hemophilia B, though rare, may experience disparities in access to care for this condition. This is particularly salient for gene therapies for hemophilia B, which aim to address the underlying genetic cause of the bleeding disorder.
Ethics of Evidence and Evaluation of Etranacogene Dezaparvovec
The clinical evidence used to assess etranacogene dezaparvovec for the treatment of adults with hemophilia B is drawn from the phase III, single-arm, open-label HOPE-B trial (N = 54). The primary objective of the HOPE-B trial is to determine whether etranacogene dezaparvovec is noninferior to existing FIX prophylaxis, as measured through reductions in the ABR for all bleeding events between months 7 to 18 post infusion. The HOPE-B trial included a preliminary lead-in phase, during which participants received continuous FIX prophylaxis and were followed for at least 6 months. Data collected from the lead-in period were used to compare participants’ response to FIX prophylaxis, and response to etranacogene dezaparvovec for some outcomes. The details of the HOPE-B trial are discussed further in the Clinical Review and Pharmacoeconomic Review reports. This trial is ongoing (expected completion is in 2025), and the data discussed throughout this larger Reimbursement Review reflects the June 6, 2023, data cut submitted by the sponsor.
Ethical Considerations in Trial Data
The clinical expert and clinician group input suggested that the preliminary results of the HOPE-B trial are promising, with a clinically relevant decrease in ABR for all bleeding events. However, as described in the Clinical Review Report, there is uncertainty in the interpretation of the magnitude of effect due to limitations with the trial design. The sponsor provided 1 indirect treatment comparison evaluating the indirect effect of etranacogene dezaparvovec relative to 3 FIX replacement therapies: Idelvion, Alprolix, and Rebinyn. Although the indirect treatment comparison results suggested a benefit with etranacogene dezaparvovec compared with FIX replacement therapies, the CADTH Clinical Review notes that no definitive conclusions could be drawn from the results due to the methodological limitations of the unanchored comparisons. Similarly, the durability of effect and long-term safety of etranacogene dezaparvovec is presently uncertain, given that the current data cut for the HOPE-B trial extends to only 36 months post infusion. As an intervention that is meant to be effective over the course of one’s entire life, the uncertainty regarding the durability of effect and long-term safety of etranacogene dezaparvovec presents challenges for shared decision-making regarding its use and for accurately modelling and assessing its cost-effectiveness and opportunity costs.
Ethical Considerations of Trial Representativeness
The clinical experts considered the trial population to be broadly generalizable to the Canadian context, with some caveats. The HOPE-B trial enrolled males with moderately severe to severe hemophilia B according to FIX activity (i.e., ≤ 2%). This means that females and those with an FIX activity greater than 2%, regardless of bleeding phenotype, were excluded from the trial population. Although the clinical experts assumed etranacogene dezaparvovec would work similarly for included and excluded populations, given the consistent mechanism of action, it is possible that the lack of safety and efficacy data may impact future treatment decisions or reimbursement criteria, as discussed subsequently.
Similarly, the clinical experts indicated that “Black/African American” (n = 1) participants (as identified by the investigator) were underrepresented in the HOPE-B trial relative to clinical practice. These experts felt trial outcomes would be broadly generalizable to all people with hemophilia B. However, the published literature flagged underrepresentation as an issue of “genomic justice.”9 As with the HOPE-B trial (74.1% white, 3.7% Asian, 1.9% Black or African American, 11.1% other, and 9.3% missing), study populations for hemophilia gene therapies have largely failed to build a strong dataset for racialized people and those of non-European descent.9 As such, it is unclear whether the potential benefits or risks of gene therapies will apply equally to all racialized people and those of non-European descent.9
Ethical Considerations in the Use of Etranacogene Dezaparvovec
Weighing Potential Benefits and Harms
The proposed value of gene therapies (like etranacogene dezaparvovec) for hemophilia B lies in their potential to be a 1-time infusion that can trigger sustained endogenous FIX production. This would represent a substantial shift from the current standard of care (i.e., prophylactic FIX concentrate infusions) and may help address an existing unmet need for sustained FIX production and lessen the burden of care associated with FIX prophylaxis. According to the sponsor, successful treatment with etranacogene dezaparvovec may eliminate the need for FIX prophylaxis altogether. A total of 51 of 54 participants remained free of FIX prophylaxis at the 36-month cut-off; the durability of this effect beyond 36 months is uncertain. While some participants still experienced breakthrough bleeds requiring on-demand factor replacement therapy, the clinical experts indicated that early results are promising.
These early results are well aligned with patients’ expectations for improved control over their condition and freedom from the restrictions hemophilia places on daily activities. Given the challenges of frequent IV infusions described previously, the patient input reported that the possibility of eliminating IV infusions would be life-changing. Although patients indicated being generally satisfied with the current standard of care, they clearly valued the potential for gene therapies like etranacogene dezaparvovec to result in fewer spontaneous joint bleeds and to provide a greater sense of freedom to participate in daily activities (e.g., exercise, physical labour, and travel). Similarly, the published literature increasingly suggests that gene therapies represent a paradigm shift in hemophilia (A or B) care.10-12 Some have even suggested that gene therapies can provide a functional cure for people with hemophilia B, as they can enable continuous expression of FIX following infusion.13
However, while gene therapies (including etranacogene dezaparvovec) may offer a novel therapeutic opportunity for patients with hemophilia B, they are not without risks. Limited long-term efficacy and safety data regarding the use of gene therapies like etranacogene dezaparvovec means that some risks remain theoretical. In particular, the published literature has suggested there is a theoretical risk of genotoxicity associated with all gene therapies, which can lead to cancer.9,11,13-16 Ongoing monitoring through postmarket surveillance mechanisms like patient registries will be necessary to understand the long-term safety of gene therapies like etranacogene dezaparvovec.
People undergoing treatment with etranacogene dezaparvovec will also be exposed to the risk of developing transaminitis following infusion (16.7% of the HOPE-B trial population reported this), which can prevent successful facilitation of endogenous FIX production. This is treatable through immunosuppressive therapy; however, published accounts of patient experiences describe immunosuppressive therapy as the most challenging aspect of undergoing gene therapy.12 One study participant suggested their course of immunosuppressive therapy led to increased experiences of depression and anger, which would prevent them from pursuing a second course of gene therapy, were it offered.12 As 37% of participants in the HOPE-B trial underwent immunosuppressive therapy following infusion of etranacogene dezaparvovec, clinical providers will need to ensure that patients are aware of this risk and understand the challenge of immunosuppressive therapy as part of the informed consent process.
Overall, the clinical experts and the clinician group described being satisfied with the current safety profile of etranacogene dezaparvovec. They indicated that uncertainty regarding the durability of effect and long-term safety was not uncommon to gene therapies, and that it would not prevent them from offering it to eligible patients.
Determining Treatment Failure
The published literature10,13 and the clinical experts reported it would be challenging to determine treatment failure for etranacogene dezaparvovec. As a 1-time infusion with potential lifelong benefit, clarifying the parameters of treatment failure has ethical implications for longer-term determinations of clinical benefit, use, and health system value. Transgene expression of AAV vectors is expected to diminish over time, which would result in a reduced treatment response and may require some patients to return to FIX prophylaxis.11,13 As some patients may be under the impression that gene therapies (like etranacogene dezaparvovec) represent a lifelong cure,14,16,17 navigating potential treatment failure later in life could be challenging.
Additionally, the clinical experts suggested that treatment response should be determined through a process of shared decision-making between patients and clinical care teams. For example, the literature suggests patients and clinicians may need to adjust how they understand and respond to potential bleeds.10 Prior to gene therapy, patients are encouraged to assume that all joint pain or worsened chronic pain is likely a new bleed.10 However, following gene therapy, there is the assumption that patients will experience fewer bleeds, which expands the possibility that novel joint pain, or aggravated chronic pain, may be related to something other than a new bleed.10 This points to a potentially shifted treatment paradigm and the novel challenges that patients, their families, and their health care providers will need to navigate in the context of gene therapies such as etranacogene dezaparvovec.
Eligibility
The clinical experts highlighted 2 overarching ethically salient challenges associated with the establishment of eligibility criteria for reimbursement of etranacogene dezaparvovec: the need to provide equitable access based on ability to benefit rather than FIX activity levels alone, and considerations related to anti-AAV nAb testing.
FIX Activity and Phenotype
The HOPE-B trial used the designation of moderately severe to severe hemophilia B (defined as FIX activity ≤ 2% of normal) to determine participant eligibility. The clinical experts noted that using the same criterion to determine treatment eligibility could unfairly exclude patients with a similar potential to benefit from etranacogene dezaparvovec. Instead, they suggested that eligibility should be determined according to bleeding phenotype and whether patients require prophylactic FIX replacement therapy, in conjunction with FIX activity level. This approach would better support providing equitable access to individuals with a comparable potential benefit from etranacogene dezaparvovec.
Neutralizing Antibodies
Many people with hemophilia B may remain ineligible for treatment with etranacogene dezaparvovec due to the presence of nAbs,16 which raises ethical considerations related to false hope. The presence of anti-AAV nAbs may prevent successful delivery of the genetic material into hepatocytes and has typically served as an exclusion criterion in clinical trials for gene therapies.
Notably, however, the HOPE-B trial did not exclude prospective participants based on anti-AAV5 nAb positivity. In fact, 21 of 54 participants (38.9%) had a titre between 1:9 and 1:3,212. Nonetheless, the product monograph for etranacogene dezaparvovec has indicated that a titre threshold of 1:900 should be used to determine treatment eligibility. As only 1 HOPE-B study participant had a titre above this threshold, our ability to anticipate how many people are likely to be ineligible for etranacogene dezaparvovec based on nAb positivity is currently limited. The use of titre thresholds is presumably meant to protect people who are unlikely to benefit from treatment from unnecessary exposure to treatment-associated risks and to support resource stewardship by limiting access to those with a reasonable expectation of benefit. However, the clinical experts and published literature indicate that the impact of nAbs on the effectiveness of gene therapies is debated,18 which has ethical implications as to which patients will be deemed eligible to access therapy.
Regardless, being deemed ineligible for gene therapy could be a challenge for people with hemophilia in the context of the curative discourse surrounding gene therapies. As such, the published literature has indicated there will be a need to provide psychological support for those deemed ineligible to minimize the harm due to unmet expectations.17
Barriers to Access
The clinical experts discussed that geography may be a barrier to equitable access to etranacogene dezaparvovec (or other gene therapies) due to limited infusion centres across Canada. While the sponsor has indicated its plan to leverage existing HTCs, it is unclear how many HTCs have capacity and expertise to take on this novel treatment option. In fact, the clinical experts indicated there is still some uncertainty regarding whether Canada will follow a hub-and-spoke model (i.e., a few specialized infusion centres, with people’s local HTCs conducting postinfusion follow-up) or have all current HTCs become infusion centres. Regardless, although the number of infusion sites is expected to grow if etranacogene dezaparvovec is reimbursed, they still may not be located across Canada. This may result in disproportionate travel-associated burdens and costs for patients who reside farther from infusion centres and present geographic barriers to equitable access.
There may also be access challenges for those populations that were not represented in the HOPE-B trial. While the product monograph notes that etranacogene dezaparvovec is not “intended for administration in women,” they would ostensibly remain eligible for consideration given the use of “adults” in the indication.19 Nonetheless, their exclusion from the trial means there is currently no data available for them. The clinical experts indicated they would not expect there to be a difference in treatment outcomes between males and females. However, they expressed discomfort with prescribing etranacogene dezaparvovec to females who were pregnant or could become pregnant due to the absence of safety data regarding potential repercussions to reproduction. The clinical experts noted they would prescribe etranacogene dezaparvovec for females who have reached menopause.
Informed Consent
The challenge of weighing the potential benefits and harms of etranacogene dezaparvovec, coupled with the difficulty in determining treatment failure and eligibility, emphasizes the critical role of robust informed consent and shared decision-making.13,16,18,20,21 Informed consent should be understood as an ongoing process wherein providers place patients’ values at the forefront when examining potential treatment options. This is particularly important in the context of gene therapies like etranacogene dezaparvovec, as they cannot be discontinued once administered.13,16 Additionally, recognizing the anticipated decrease in the effectiveness of gene therapies (including etranacogene dezaparvovec) over time,11,13 clinical providers will need to clearly advise their patients that these therapies are not currently known to be curative. While the preliminary results from the HOPE-B study may be positive, having this conversation with patients will be necessary to help establish reasonable long-term expectations.13,14,16,17
Moreover, following infusion of gene therapy, patients develop nAbs against the AAV serotype used in that therapy.18 These nAbs not only preclude receiving the same gene therapy or another 1 using the same AAV vector, but they might also restrict eligibility for future gene therapies due to cross-reactivity with other AAV serotypes.21 Open and clear discussions about this possibility could mitigate patients’ potential regret if another gene therapy is found to be more effective in the future.21
Informed consent will also be an important component in the context of nAb testing, as there is currently no such testing capacity in Canada. As a result, nAb testing will need to be conducted by US-based laboratories, which will require conversations related to privacy and confidentiality requirements.
Health Systems Considerations
The use of etranacogene dezaparvovec as a gene therapy for hemophilia B raises ethical considerations related to sustainable funding, fair priority setting and resource allocation, and manufacturing and health system capacity in Canada.
Sustainability of Funding Gene Therapies
The introduction of gene therapies raises concerns regarding the ability of health care systems to sustainably manage the costs associated with these expensive, single-administration therapies.13,16,22 Etranacogene dezaparvovec’s long-term clinical effectiveness and cost-effectiveness are currently uncertain, which limits assessments of its long-term value and the opportunity costs of reimbursement.23 While this uncertainty will be familiar to decision-makers in the context of expensive drugs for rare diseases, it complicates reimbursement and resource allocation decisions. This is particularly true in the context of etranacogene dezaparvovec’s proposed value as a 1-time therapy with lifelong benefit.
Funding reforms for 1-time, high-cost therapies with uncertain long-term benefit, such as gene therapy and chimeric antigen receptor (CAR) T-cell therapy, have been suggested in the Canadian context, such as through some form of risk-sharing agreement.24-26 When choosing whether to implement an APM, a jurisdiction would need to consider how its design can present both novel infrastructural (e.g., clinical and data) requirements and have ethical implications for the distribution of potential risks and benefits of reimbursement. In an outcomes-based model, jurisdictions would need to agree upon clear parameters of success (i.e., clinical outcome cut-offs) with manufacturers, while also establishing postmarket surveillance mechanisms that could support data collection and management.22,24 There would also need to be agreement on the validity of data collection and a transparent process for value adjudication and contracting between parties.22,24 As such, the design of an outcome-based model (e.g., how parameters of success or “value” are defined) has ethical implications for the distribution of the potential benefits and burdens of such risk-sharing arrangements for manufacturers, payers, patients, and the public.25,26
In the context of hemophilia, some published literature has suggested that the benefits of APMs may be outstripped by challenges associated with developing the necessary infrastructure to support their implementation in such a small population.22 Nonetheless, hemophilia registries may be an ideal choice for the collection of much of the postmarket data required as part of any APM.13,18,20 Although some of the literature suggests that many existing registries may not have the infrastructure to begin collecting long-term data for gene therapies,27 clinical experts indicated the Canadian Hemophilia Registry is prepared.
Manufacturing and Health Systems Capacity
Beyond the challenges that gene therapies present for health system sustainability, potential manufacturing shortages and limited delivery capacity at some HTCs across Canada may require the development of fair and transparent prioritization criteria to allocate limited therapies. The clinical experts and published literature noted that manufacturing the AAV vectors used for AAV-mediated gene therapies is highly specialized and cumbersome.28,29 While some literature suggests that manufacturers have been able to keep up with demand for phase III and market authorization trials,28 more needs to be done to keep up with increased demand and to grow process efficiencies.29
Beyond potential manufacturing shortages, it is possible that existing hemophilia treatment infrastructure in Canada will be challenged by the addition of gene therapies like etranacogene dezaparvovec. For instance, the clinical experts indicated that some HTC-based pharmacies may have limited capacity to store and reconstitute etranacogene dezaparvovec. This may limit the geographic distribution of infusion sites and make accessing etranacogene dezaparvovec more challenging for people living outside the catchment areas of existing infusion sites. The clinical experts noted that determining jurisdictional responsibility for reimbursement of the various costs associated with delivering etranacogene dezaparvovec may be especially complicated when patients travel outside their province or territory to access therapy.
Published literature indicated that primary care providers, nurses, physiotherapists, and hepatologists are all likely to experience increased and shifting demands on their time in the wake of gene therapies like etranacogene dezaparvovec.13 For example, as nurses are the primary points of contact between HTCs and patients with hemophilia, their responsibilities with respect to patient care, monitoring, and administrative tasks are expected to increase with these therapies.13 The clinical experts expressed concern that these increased resource demands may be challenging for HTCs in Canada to meet, as many are dealing with nursing (and other human resourcing) shortages and limited budgets. The limited capacity to provide gene therapies like etranacogene dezaparvovec to all individuals seeking treatment highlights the importance of developing fair and transparent prioritization criteria.
However, the clinical experts maintained that if prioritization criteria are developed, this should be done in addition to ensuring that the HTCs involved in the delivery and/or follow-up of gene therapy are fully resourced. They warned that without adequate staffing, HTCs would have limited capacity to deliver gene therapy and to conduct the requisite long-term monitoring post infusion. This could similarly affect their ability to provide registries or other postmarket surveillance mechanisms that have the data required to understand the long-term safety and effectiveness of gene therapies such as etranacogene dezaparvovec.
Limitations
There is very little published literature that discusses the ethical considerations related to the use of etranacogene dezaparvovec for the treatment of hemophilia B, given both the rarity of the disease and the novelty of the therapy under review. Nonetheless, this does not imply that ethical considerations in the context of etranacogene dezaparvovec for hemophilia B are absent. This review of ethical considerations related to the use of etranacogene dezaparvovec for the treatment of hemophilia B was augmented by drawing from additional resources, including patient group, clinician group, and drug program input, and discussion with the clinical experts consulted during this review, as well as engagement with CADTH clinical and pharmacoeconomic review teams.
Although this Ethics Review Report drew on and considered patient group, clinician group, drug program, and clinical expert input, it is possible that more direct engagement with additional groups (e.g., direct interviews with patients, caregivers, family members, decision-makers) regarding their specific experiences with hemophilia B and/or etranacogene dezaparvovec could have offered additional, relevant ethical considerations or domains of analysis.
Conclusion
Input from patient and clinician groups and drug programs and relevant published literature were reviewed, and information was obtained through direct engagement with clinical experts, to identify and describe ethical considerations relevant to the use of etranacogene dezaparvovec for the treatment of adults living with hemophilia B. Ethical considerations in the context of hemophilia B underscore the significant treatment burden faced by individuals with moderate to severe hemophilia B. FIX prophylaxis, though effective, requires frequent IV infusions and leaves patients vulnerable to breakthrough bleeds during trough periods between infusions. Females with hemophilia B face potential inequities in access to care due to misdiagnosis or underdiagnosis. Despite promising clinical trial evidence regarding the effectiveness and safety of etranacogene dezaparvovec, the magnitude of benefit remains uncertain due to the risk of biases associated with the design of the HOPE-B trial. Similarly, the long-term safety and efficacy of etranacogene dezaparvovec are presently uncertain, as current data are limited to 36 months post infusion. This uncertainty may be further exacerbated for females and people with FIX activity greater than 2%, as they were excluded from the HOPE-B trial. Similarly, the clinical experts indicated that people identified as Black or African American were underrepresented in the trial population. Although the clinical experts assumed the trial outcomes would be generalizable to all patients with hemophilia B, limited clarity regarding who is most likely to benefit from etranacogene dezaparvovec may result in treatment decisions privileging people more reflective of the trial population.
The use of etranacogene dezaparvovec would open patients to some known risks, like developing transaminitis, and some theoretical risks, such as potential genotoxicity leading to cancer. Though the preliminary results from the HOPE-B trial are promising, the clinical experts indicated that many people with hemophilia B would still not pursue therapy due to limited certainty regarding long-term efficacy and safety. As etranacogene dezaparvovec is a 1-time infusion that cannot be reversed, providers will need to facilitate a thorough consent process that is supportive of shared decision-making that places their patients’ values in the forefront when determining treatment. This process will need to involve deliberate consideration of the uncertainty surrounding long-term safety and efficacy, the risk of treatment failure that would prompt a return to FIX prophylaxis, and the likelihood that patients would be rendered ineligible for future gene therapies due to cross-reactive anti-AAV nAbs. As it is presently unclear who is most likely to benefit from treatment, determining who should receive etranacogene dezaparvovec may present an ethical challenge for providers. In particular, the absence or underrepresentation of some populations in the HOPE-B trial (e.g., those with FIX activity greater than 2% and females) may lead to inequitable access if clinical decision-making prioritizes those patients for whom some safety and efficacy data are available. As treatment with etranacogene dezaparvovec will require access to multidisciplinary care and specialized treatment centres, ensuring equitable access will need to involve addressing any geographic barriers hindering access to specialist care and monitoring.
The potential implementation of etranacogene dezaparvovec carries ethical implications for health systems that highlight challenges in allocating limited resources for expensive therapies for rare diseases. Uncertainty regarding the long-term safety and efficacy of gene therapies like etranacogene dezaparvovec has prompted the consideration of APMs to mitigate the risks associated with reimbursing highly expensive therapies with unknown long-term benefits. While well-designed APMs may support a fair distribution of risks and benefits among parties (e.g., payers, manufacturers, patients, and the public), their need for ongoing surveillance may also require changes to clinical and data infrastructures that could be challenging or resource-intensive to implement. In Canada, it is possible that the Canadian Hemophilia Registry could support postmarket surveillance efforts. However, the clinical experts flagged that personnel shortages at HTCs across Canada may impact the ability to collect robust registry data. In addition to the need to navigate the pressures that this high-cost therapy may place on system sustainability, the potential for manufacturing shortages and limited capacity at some HTCs across Canada warrants the development of fair and transparent prioritization criteria. Moreover, geographic challenges, including limited availability of etranacogene dezaparvovec in treatment centre pharmacies, may compel some patients to cross jurisdictions for access. This may lead to increasing complexities in determining the jurisdictions responsible for reimbursement of the therapy and associated costs.
References
1.EUnetHTA. EUnetHTA Joint Action 2 Work Package 8, HTA Core Model version 3.0 2016; http://www.htacoremodel.info/BrowseModel.aspx. Accessed 2023 Sep 29.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37(1). PubMed
3.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
4.National Organization for Rare Disorders. Hemophilia B. 2023; https://rarediseases.org/rare-diseases/hemophilia-b/. Accessed 2023 Jun 29.
5.Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1-158.
6.Arya S, Siad FM, Wilton P, et al. Invisible bleeds: Lived experiences and barriers to care for men with hemophilia. J Thromb Haemost. 2022;20(2):296-306. PubMed
7.Cutter S, Molter D, Dunn S, et al. Impact of mild to severe hemophilia on education and work by US men, women, and caregivers of children with hemophilia B: The Bridging Hemophilia B Experiences, Results and Opportunities into Solutions (B-HERO-S) study. Eur J Haematol. 2017;98 Suppl 86:18-24. PubMed
8.Weyand AC, James PD. Sexism in the management of bleeding disorders. Res Pract Thromb Haemost. 2021;5(1):51-54. PubMed
9.Sanchez-Lara PA, Nathanson J, Valentino LA. Gene Therapy in Hemophilia: Latest Developments. Advances in Hemophilia Treatment: From Genetics to Joint Health: Springer; 2022:155-165.
10.Yamaguti-Hayakawa GG, Ozelo MC. Gene therapy for hemophilia: looking beyond factor expression. Exp Biol Med. 2022;247(24):2223-2232. PubMed
11.Ohmori T. Advances in gene therapy for hemophilia: basis, current status, and future perspectives. Int J Hematol. 2020;111(1):31-41. PubMed
12.Fletcher S, Jenner K, Pembroke L, Holland M, Khair K. The experiences of people with haemophilia and their families of gene therapy in a clinical trial setting: regaining control, the Exigency study. Orphanet J Rare Dis. 2022;17(1):155. PubMed
13.Hermans C, Gruel Y, Frenzel L, Krumb E. How to translate and implement the current science of gene therapy into haemophilia care? Ther Adv Hematol. 2023;14. PubMed
14.Fletcher S, Jenner K, Holland M, Chaplin S, Khair K. An exploration of why men with severe haemophilia might not want gene therapy: The exigency study. Haemophilia. 2021;27(5):760-768. PubMed
15.Vasquez-Loarte TC. Beliefs and Values about gene therapy and gene editing in patients with hemophilia and their family members [thesis]. Seattle (Washington): University of Washington; 2019: https://digital.lib.washington.edu/researchworks/handle/1773/45268. Accessed 2023 Sep 29.
16.Baas L, van der Graaf R, van Hoorn ES, Bredenoord AL, Meijer K. The ethics of gene therapy for hemophilia: A narrative review. J Thromb Haemost. 2023. PubMed
17.Fletcher S, Jenner K, Holland M, Khair K. Expectation and loss when gene therapy for haemophilia is not an option: An exigency sub‐study. Haemophilia. 2023. PubMed
18.Batty P, Lillicrap D. Gene therapy for hemophilia: Current status and laboratory consequences. Int J Lab Hematol. 2021;43(S1):117-123. PubMed
19.Etranacogene dezaparvovec (HEMGENIX): 1 × 1013 genome copies/mL, intravenous infusion [product monograph]. Ottawa (ON): CSL Behring Canada Inc.; 2023 Oct 23.
20.Valentino LA, Kaczmarek R, Pierce GF, et al. Hemophilia gene therapy: first, do no harm. J Thromb Haemost. 2023;21(9):2354-2361. PubMed
21.George LA. Hemophilia gene therapy: ushering in a new treatment paradigm? Hematology. 2021;2021(1):226-233. PubMed
22.Goodman C, Berntorp E, Wong O. Alternative payment models for durable and potentially curative therapies: The case of gene therapy for haemophilia A. Haemophilia. 2022;28(S2):27-34. PubMed
23.Kacetl J, Marešová P, Maskuriy R, Selamat A. Ethical Questions Linked to Rare Diseases and Orphan Drugs - A Systematic Review. Risk Manag Healthc Policy. 2020;13:2125-2148. PubMed
24.From Research to Reality: The Expert Panel on the Approval and Use of Somatic Gene Therapies in Canada. Ottawa (ON): Council of Canadian Academies; 2020: https://cca-reports.ca/wp-content/uploads/2019/08/Report-From-Research-to-Reality-EN.pdf. Accessed 2023 Sep 29.
25.CADTH Reimbursement Review: axicabtagene ciloleucel (Yescarta). Can J Health Technol. 2023;3(6). https://www.cadth.ca/sites/default/files/DRR/2023/PG0293-Yescarta.pdf. Accessed 2023 Sep 29.
26.CADTH Reimbursement Review: ciltacabtagene autoleucel (Carvykti). Can J Health Technol. 2023;3(8). https://www.cadth.ca/sites/default/files/DRR/2023/PG0302-Carvykti_combined.pdf. Accessed 2023 Sep 29.
27.Duda H, Hesse J, Haschberger B, Hilger A, Keipert C. The german hemophilia registry: Growing with its tasks. J Clin Med. 2020;9(11):1-11. PubMed
28.Nathwani AC. Gene therapy for hemophilia. Hematology (United States). 2022;2022(1):569-578. PubMed
29.Young P. Treatment to cure: Advancing AAV gene therapy manufacture. Drug Discov Today. 2023;28(7). PubMed
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third-party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.