CADTH Reimbursement Review
Bimekizumab (Bimzelx)
Sponsor: UCB Canada Inc.
Therapeutic area: Psoriatic arthritis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ACR
American College of Rheumatology
ACR20
American College of Rheumatology 20% improvement in rheumatoid arthritis
ACR50
American College of Rheumatology 50% improvement in rheumatoid arthritis
ACR70
American College of Rheumatology 70% improvement in rheumatoid arthritis
AE
adverse event
b/tsDMARD
biologic or targeted synthetic disease-modifying antirheumatic drug
BASDAI
Bath Ankylosing Spondylitis Disease Activity Index
bDMARD
biologic disease-modifying antirheumatic drug
BSA
body surface area
cDMARD
conventional nonbiologic disease-modifying antirheumatic drug
CI
confidence interval
COX-2
cyclooxygenase-2
CrI
credible interval
DAPSA
Disease Activity index in PSoriatic Arthritis
DB
double-blind
DMARD
disease-modifying antirheumatic drug
GRADE
Grading of Recommendations Assessment, Development and Evaluation
GRAPPA
Group for Research and Assessment of Psoriasis and Psoriatic Arthritis
HAQ-DI
Health Assessment Questionnaire–Disability Index
HRQoL
health-related quality of life
IBD
inflammatory bowel disease
IGA
Investigator’s Global Assessment
IL-12/23i
interleukin-12 and interleukin-23 inhibitor
IL-17A
interleukin-17A
IL-17AF
interleukin-17AF
IL-17F
interleukin-17F
IL-17i
interleukin-17 inhibitor
IL-17RA/IL-17RC
interleukin-17RA and interleukin-17RC
IL-23i
interleukin-23 inhibitor
ITC
indirect treatment comparison
JAKi
Janus kinase inhibitor
LDI
Leeds Dactylitis Index
LEI
Leeds Enthesitis Index
LSM
least squares mean
MAIC
matching-adjusted indirect comparison
MDA
minimal disease activity
MID
minimal important difference
NEC
not elsewhere classified
NMA
network meta-analysis
NSAID
nonsteroidal anti-inflammatory drug
NRI
nonresponder imputation
OLE
open-label extension
OR
odds ratio
PASI
Psoriasis Area Severity Index
PASI 75
75% reduction in Psoriasis Area Severity Index score
PASI 90
90% reduction in Psoriasis Area Severity Index score
PASI 100
100% reduction in Psoriasis Area Severity Index score
PCS
physical component summary
PDE4i
phosphodiesterase type 4 inhibitor
PsA
psoriatic arthritis
PtAAP
Patient’s Assessment of Arthritis Pain
RCT
randomized controlled trial
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
SE
standard error
SF-36
Short Form (36) Health Survey
SJC
swollen joint count
SLR
systematic literature review
SMD
standardized mean difference
TEAE
treatment-emergent adverse event
TJC
tender joint count
TNFi
tumour necrosis factor inhibitor
tsDMARD
targeted synthetic disease-modifying antirheumatic drug
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Bimekizumab (Bimzelx), 160 mg/mL, solution for SC injection |
Sponsor | UCB Canada Inc. |
Indication | The treatment of adult patients with active PsA. Bimzelx can be used alone or in combination with a cDMARD (e.g., methotrexate). |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | February 23, 2024 |
Recommended dosage | The recommended dosage for adult patients with active PsA is 160 mg (given as 1 SC injection of 160 mg) q.4.w. |
cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; NOC = Notice of Compliance; PsA = psoriatic arthritis; q.4.w. = every 4 weeks; SC = subcutaneous.
Sources: Sponsor’s submission package for review of bimekizumab and bimekizumab product monograph.1,2
Introduction
Psoriatic arthritis (PsA) is a chronic inflammatory, immune-mediated disease with heterogenous presentation and disease course. Patients present with a variety of disease manifestations,3-6 with peripheral arthritis and psoriasis as the most common disease manifestations of PsA.7 The joint inflammation associated with PsA is known to worsen over time and, if left untreated, can lead to permanent joint damage and long-term disability.4,8-10 The prevalence estimate for PsA is variable depending on the case definition and geography,11 and is estimated to be approximately 1 to 2 per 1,000 people in the general population, globally.12 A population-based Canadian study, conducted in Ontario, estimated the age- and sex-standardized cumulative prevalence of PsA in Ontario to range from 0.9 per 1,000 people in 2008 to 1.5 per 1,000 people in 2015.13
The goals of treatment for managing PsA include achieving the lowest level of disease activity (with a target of disease remission), maximizing functional status and health-related quality of life (HRQoL), preventing further disease progression, controlling symptoms, and avoiding complications (from untreated disease or adverse effects from treatment).14-16 First-line, pharmacological treatment of PsA typically includes disease-modifying antirheumatic drugs (DMARDs) such as conventional nonbiologic disease-modifying antirheumatic drugs (cDMARDs) (e.g., methotrexate, sulfasalazine, leflunomide, cyclosporine). Later-line, targeted treatments are usually reserved for patients who have an inadequate response to cDMARDs and include biologic disease-modifying antirheumatic drugs (bDMARDs) (e.g., tumour necrosis factor inhibitor [TNFi], interleukin-17 inhibitor [IL-17i], interleukin-12 and interleukin-23 inhibitor [IL-12/23i], interleukin-23 inhibitor [IL-23i]), and targeted synthetic disease-modifying antirheumatic drugs [tsDMARDs]) (e.g., Janus kinase inhibitor [JAKi] and phosphodiesterase type 4 inhibitor [PDE4i]).15,16 The clinical expert noted that the continuation of cDMARDs may be necessary until effectiveness of the targeted therapies is confirmed.
Bimekizumab is a humanized immunoglobulin G1 kappa monoclonal antibody with 2 identical, high-affinity antigen-binding regions that neutralize interleukin-17A (IL-17A), interleukin-17F (IL-17F), and interleukin-17AF (IL-17AF) cytokines, blocking their interaction with the interleukin-17RA and interleukin-17RC (IL-17RA/IL-17RC) receptor complex, which plays a role in PsA-related inflammation.2 The recommended dose of bimekizumab for adult patients with active PsA is 160 mg (given as 1 subcutaneous [SC] injection of 160 mg) every 4 weeks.2 The sponsor has requested reimbursement as per the approved Health Canada indication. Bimekizumab was previously reviewed by CADTH in April 2022 for the indication of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy, and a final recommendation for reimbursement with conditions was issued.17,18 In addition to the current CADTH review for the indication of adults with active PsA (with or without cDMARDs), bimekizumab is also under CADTH review for the indication of adults with active ankylosing spondylitis.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of bimekizumab 160 mg every 4 weeks for SC injection in the treatment of adult patients with active PsA, alone or in combination with a cDMARD.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Input was submitted for this CADTH review by 7 different patient groups. Input was received from Arthritis Consumer Experts and, in a joint submission, from the Canadian Psoriasis Network, Arthritis Society Canada, the Canadian Arthritis Patient Alliance, the Canadian Association of Psoriasis Patients, the Canadian Spondyloarthritis Association, and CreakyJoints. Arthritis Consumer Experts gathered information from patients via email on October 16, 2023 (n = 2), and through an online survey available from December 18, 2020, to January 26, 2021 (n = 5). The joint input obtained responses (n = 214) through an online survey that was available through the 6 collaborating organizations from September 27 to October 11, 2023. The latter survey was also sent to clinics in Canada that conducted bimekizumab trials for PsA to capture the experience of patients living with PsA or ankylosing spondylitis and the experiences of their caregivers.
The symptoms described as being the most challenging to manage included joint stiffness, fatigue, and pain, among others. According to the respondents, PsA interfered with their physical activity, sleep, work, social life, mental health, intimacy, and self-esteem. It was also noted that caregivers had to take on additional tasks or daily chores due to patients’ reduced mobility and the impact the disease has on patients’ mental and social health.
Survey results showed that 32% of respondents found nonsteroidal anti-inflammatory drugs (NSAIDs) effective, 39% found DMARDs effective, 40% found biologic therapies effective (with 24% rating them as very effective), and 44% found steroids effective. Respondents indicated that efficacious treatments are costly and accessing them was challenging, and adverse effects can be very difficult to manage. Among the 3 respondents who had experience with bimekizumab for PsA, 1 respondent noted that treatment was easy to use and effective in improving HRQoL without adverse effects.
Patients seek treatments that improve disease symptoms and HRQoL, have fewer adverse effects, are easier to administer, and are accessible and affordable.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
Despite there being various treatment options for managing PsA, the clinical expert stated that not all patients respond to available therapies and that treatments tend to improve disease in some domains but have variable or suboptimal efficacy in others. There is also concern over safety with all DMARDs, including the increased risk of infection and new onset or worsening of existing comorbidities. According to the expert, few patients attain a state of low disease activity, and it is important to have safe, well-tolerated treatments that are effective in all domains.
The clinical expert indicated that bimekizumab would be used after the failure of cDMARDs and, in accordance with its Health Canada indication, with or without a cDMARD. Additionally, it was noted that without good quality evidence to support combination therapy, bimekizumab would not likely be used concomitantly with other bDMARDs or tsDMARDs at this time.
The clinical expert was of the opinion that any patient with active PsA could receive bimekizumab, and that those with coexisting severe psoriasis may also benefit. They added that the drug would be avoided in those with inflammatory bowel disease (IBD), severe uveitis, or active infection. The expert also noted that patients with an inadequate response to targeted DMARDs are most in need of new treatments.
According to the clinical expert, treatment response is typically assessed at 3 months based on improvements in the number of tender and swollen joints, enthesitis, dactylitis, and skin psoriasis, and sometimes using composite indices (e.g., minimal disease activity [MDA], Disease Activity index in PSoriatic Arthritis [DAPSA]). Improvements in physical function, pain, fatigue, and lack of radiographic progression (the last of which is not typically used in clinical practice) may take longer than 3 months in patients with longstanding PsA. Moreover, due to the heterogeneity of the disease, response can differ across patients, though the clinical expert suggested that assessments are not likely to vary among rheumatologists.
The clinical expert stated that lack of response in musculoskeletal or skin domains, disease relapse, intolerance, and patient choice are the most important factors when considering the discontinuation of bimekizumab. Moreover, it was explained that some amount of disease activity can be considered acceptable, but that recurrent infections and IBD would require discontinuation.
The expert noted that diagnosis by primary care physicians and dermatologists can be challenging, and misdiagnosis is a possibility; therefore, a PsA diagnosis should be made by a rheumatologist trained in identifying inflammatory arthritis. Patients are typically treated in an outpatient setting in community clinics and clinics attached to community and academic hospitals, though severe skin and joint disease may require hospital admission. The expert noted that the treatment and management of PsA involves a rheumatologist and can include a dermatologist to manage the associated psoriasis.
Clinician Group Input
One clinician group, the Canadian Rheumatology Association (whose input was authored by the therapeutics committee), responded to CADTH’s call for clinician group input. The treatment goals, unmet needs, patient population, and reasons for discontinuation described by the clinician group largely aligned with that of the clinical expert consulted by CADTH. The group suggested that measures of treatment response also include improvement in axial disease, Psoriasis Area Severity Index (PASI), and body surface area (BSA) affected by psoriasis. They also noted that composite measures, such as the Composite Psoriatic Disease Activity Index, DAPSA, and Psoriatic Arthritis Disease Activity Score, can be used but are not practical for everyday use in clinics. For axial disease, the most frequently used measure for disease activity in Canada is the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI). The group indicated that a specialist would be required to prescribe bimekizumab and monitor for adverse events (AEs), but because the drug is administered as an SC injection, a patient could self-administer it.
Drug Program Input
The drug programs asked if the initiation, continuation, discontinuation, prescribing, and combination criteria from the most recent Canadian Drug Expert Committee recommendation for guselkumab for the treatment of PsA should be applied to bimekizumab. The programs also asked for clarification if ever there is a potential indication for bimekizumab to treat juvenile PsA, what an adequate trial of DMARDs would be, what type of specialist should treat adults with PsA, if bimekizumab should be used in combination with other DMARDs, and if a patient could be re-treated with bimekizumab.
The clinical expert consulted by CADTH agreed that the same initiation, continuation, discontinuation, and prescribing criteria from the Canadian Drug Expert Committee recommendation for guselkumab for the treatment of PsA can be applied to bimekizumab. They also noted that, with evidence from good quality trials in a younger population, bimekizumab could be used to treat juvenile PsA. According to the expert, an adequate trial of 3 months at the recommended therapeutic dose is necessary to determine if a patient is responding to a cDMARD, bDMARD, or tsDMARD before switching to or adding another therapy. The expert indicated that patients with PsA should be diagnosed, treated, and managed by a rheumatologist, and when 1 cannot be accessed, internists or dermatologists may be acceptable specialists. Regarding combination therapy, the Health Canada indication states that bimekizumab can be used alone or in combination with a cDMARD (e.g., methotrexate),2 and there is currently limited evidence for concomitant use with bDMARDs or tsDMARDs for the treatment of PsA. The expert was of the opinion that after switching from bimekizumab to another therapy, a patient could be re-treated with bimekizumab in the future.
Clinical Evidence
Systematic Review
Description of Studies
Two double-blind (DB), randomized controlled trials (RCTs) of adults with active PsA who had no prior exposure to biologic therapies (the BE OPTIMAL trial, N = 852) or who had a history of inadequate response or intolerance to 1 TNFi or 2 TNFis (the BE COMPLETE trial, N = 400) assessed whether bimekizumab 160 mg for SC injection every 4 weeks increased the proportion of patients attaining at least an American College of Rheumatology 50% improvement in rheumatoid arthritis (ACR50) response compared to placebo at 16 weeks.19,20 ACR50 response is defined as an improvement of at least 50% in both swollen joint counts (SJCs) and tender joint counts (TJCs) and at least 3 of 5 additional disease criteria. Other clinically relevant outcomes included the measurement of MDA, musculoskeletal response, skin response, and changes in function and symptom scores. Patients received either bimekizumab or placebo during the 16-week DB phase of each trial. After 16 weeks in the BE OPTIMAL trial, patients randomized to placebo were reallocated to bimekizumab for the 36-week active treatment–blind phase.
In the BE OPTIMAL trial, the mean age of patients ranged from 48.5 (standard deviation [SD] = 12.6) years to 48.7 (SD = 11.7) years and demographic characteristics were generally similar between the bimekizumab and placebo groups. The mean time since diagnosis of PsA was approximately 6 years and the mean time since diagnosis of psoriasis was approximately 15 years. Baseline clinical characteristics were generally balanced between the 2 treatment groups, except for the presence of enthesitis being higher in the bimekizumab group (33.2%) compared to the placebo group (24.9%). In the BE COMPLETE trial, the mean age of patients ranged from 50.1 (SD = 12.4) years to 51.3 (SD = 12.9) years and demographic characteristics were generally similar between the bimekizumab and placebo groups. The mean time since diagnosis of PsA was more than 9 years and the mean time since diagnosis of psoriasis was more than 17 years across the treatment groups. More than 76% of patients had an inadequate response to at least 1 TNFi and more than 11% of patients to at least 2 TNFis, and approximately 12% of patients had an intolerance to TNFis. Baseline clinical characteristics were imbalanced between the 2 treatment groups for the presence of enthesitis (higher in the bimekizumab group), NSAID therapy (lower in the bimekizumab group), and methotrexate use (higher in the bimekizumab group).
Efficacy Results
Signs and symptoms of disease activity were measured by American College of Rheumatology (ACR) response, MDA, Patient’s Assessment of Arthritis Pain (PtAAP), and SJC.
ACR Response
In the BE OPTIMAL trial, a greater proportion of patients in the bimekizumab treatment group reached the primary end point of ACR50 at week 16 than did those in the placebo group; the difference between treatment groups was 31.2% (95% confidence interval [CI], 25.2% to 37.3%; P < 0.001) for ACR50. Similarly, a greater proportion of patients in the bimekizumab treatment group reached an American College of Rheumatology 20% improvement in rheumatoid arthritis (ACR20) and an American College of Rheumatology 70% improvement in rheumatoid arthritis (ACR70) response (which are improvements of at least 20% and 70%, respectively, in both SJC and TJC and at least 3 of 5 additional disease criteria) at week 16 than did those in the placebo group. The difference between treatment groups was 37.2% (95% CI, 30.5% to 44.0%) for ACR20, and 19.3% (95% CI, 14.1% to 24.5%) for ACR70. Week 52 ACR results during the active treatment–blind period at all 3 ACR thresholds indicated that response rates increased in both groups and that there was a similar response between patients who crossed over from placebo to bimekizumab and those originally randomized to bimekizumab.
In the BE COMPLETE trial, a greater proportion of patients in the bimekizumab treatment group reached the primary end point of ACR50 at week 16 than did those in the placebo group; the difference between treatment groups was 29.0% (95% CI, 21.9% to 36.2%; P < 0.001) for ACR50. Similarly, a greater proportion of patients in the bimekizumab treatment group reached the ACR20 and ACR70 responses at week 16 than did those in the placebo group; the difference between treatment groups was 52.8% (95% CI, 43.6% to 61.9%) for ACR20 and 17.2% (95% CI, 12.2% to 22.2%) for ACR70. A larger proportion of patients in the bimekizumab group compared to those in the placebo group attained an ACR50 response for each of the subgroup categories of inadequate response to 1 TNFi (47.8% versus 6.8%, respectively), inadequate response to 2 TNFis (20.0% versus 0%, respectively), and intolerance to TNFis (38.2% versus 13.3%, respectively).
Minimal Disease Activity
Signs and symptoms of disease activity were also measured by MDA, where MDA is a composite end point and is considered to be attained if at least 5 of 7 criteria are reached: TJC of 0 or 1, SJC of 0 or 1, PASI of 1 or lower or affected BSA of 3% or less, a pain visual analogue scale score of 15 or lower, a Patient’s Global Assessment of PsA visual analogue scale score of 20 or lower, a Health Assessment Questionnaire–Disability Index (HAQ-DI) score of 0.5 or lower, and tender entheseal points of 0 or 1.
In the BE OPTIMAL trial, for clinical responses measured with the MDA criteria, patients treated with bimekizumab had higher response rates compared with placebo at week 16. The difference between treatment groups was 31.0% (95% CI, 24.5% to 37.5%; P < 0.001). MDA results at week 52 of the active treatment–blind period indicated that response rates increased in both groups and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab.
In the BE COMPLETE trial, patients treated with bimekizumab had higher response rates compared with placebo at week 16. The difference between treatment groups was 34.2% (95% CI, 26.1% to 42.2%; P < 0.001).
Patient’s Assessment of Arthritis Pain
PtAAP is a 100-point visual analogue scale for patients to record their arthritis pain from 0 (no pain) to 100 (most severe pain). The minimal important difference (MID) from the literature is estimated to be a 10-point decrease from baseline.21
In the BE OPTIMAL trial, the bimekizumab group had a greater mean decrease from baseline (i.e., improvement) in PtAAP compared with the placebo group at week 16. The mean difference between treatment groups was –19.1 (95% CI, –22.7 to –15.5). PtAAP results at week 52 indicated that the response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, the mean difference between treatment groups was –25.0 (95% CI, –30.0 to –20.0).
Swollen Joint Count
SJC evaluation includes 6 joints of the upper body, 34 joints of the upper extremities, and 26 joints of the lower extremities for a total of 66 joints. Each joint is assessed using a 2-point scale: 0 for no swelling and 1 for swollen joints.
In the BE OPTIMAL trial, the mean reduction at week 16 was greater in the bimekizumab group compared to the placebo group, with the mean difference between treatment groups being –4.0 joints (95% CI, –4.8 joints to –3.1 joints). SJC results at week 52 indicated that response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, the mean reduction at week 16 was greater in the bimekizumab group compared to the placebo group, with the mean difference between treatment groups being –5.3 joints (95% CI, –6.5 joints to –4.2 joints).
Measurement of Other Musculoskeletal Disease
The impact of treatment on musculoskeletal disease was assessed by measuring the resolution of enthesitis (with the Leeds Enthesitis Index [LEI]), and the resolution of dactylitis (with the Leeds Dactylitis Index [LDI]).
Enthesitis-Free State Based on LEI: For the pooled population of patients with enthesitis at baseline in the BE OPTIMAL and BE COMPLETE trials, a greater proportion of patients in the bimekizumab treatment group had resolution of enthesitis (LEI = 0) at week 16 than did those in the placebo group; the difference between treatment groups was 14.9% (95% CI, 4.0% to 25.9%; P = 0.008).
Dactylitis-Free State Based on LDI: For the pooled population of patients with dactylitis at baseline in the BE OPTIMAL and BE COMPLETE trials, a greater proportion of patients in the bimekizumab treatment group had resolution of dactylitis (LDI = 0) at week 16 than did those in the placebo group; the difference between treatment groups was 29.4% (95% CI, 11.7% to 47.1%; P = 0.002).
Measurement of Skin Disease
The extent and severity of skin disease was measured in both studies using PASI and Investigator’s Global Assessment (IGA).
PASI grades the extent and severity of psoriatic lesions and combines an assessment of the BSA affected with the severity of desquamation, erythema, and plaque induration or infiltration. It is scored from 0 to 72, with higher scores representing more severe disease. A 90% reduction in Psoriasis Area Severity Index score (PASI 90) is a dichotomous (yes or no) scale indicating whether a patient attained at least 90% improvement from baseline PASI score. In both trials, only patients who had psoriasis involving at least 3% BSA at baseline were assessed for PASI 90 at week 16.
The IGA is a 5-point composite physician assessment of the overall severity of the patient’s psoriatic lesions, where 0 is clear, 1 is almost clear, 2 is mild, 3 is moderate, and 4 is severe. In both trials, only patients who had an IGA score of at least 2 and psoriasis involving at least 3% BSA at baseline were evaluated for the outcome of at least a 2-grade reduction in IGA score at week 16.
90% Reduction in PASI Score: In the BE OPTIMAL trial, for PASI 90, patients treated with bimekizumab had higher response rates compared with placebo at week 16. The difference between treatment groups was 56.5% (95% CI, 48.6% to 64.3%; P < 0.001). PASI 90 results at week 52 indicated that response rates increased in both groups and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, for PASI 90, patients treated with bimekizumab had higher response rates compared with placebo at week 16. The difference between treatment groups was 57.6% (95% CI, 47.6% to 67.6%; P < 0.001). A larger proportion of patients in the bimekizumab group compared to the placebo group attained a PASI 90 response for each of the subgroup categories of inadequate response to 1 TNFi (difference versus placebo = 64.4% [95% CI, 53.6% to 75.3%]), inadequate response to 2 TNFis (difference versus placebo = 25.0% [95% CI, –16.2% to 66.1%]), and intolerance to TNFis (difference versus placebo = 49.7% [95% CI, 18.5% to 80.9%]).
IGA Score of 0 or 1 and At Least 2-Grade Reduction From Baseline: In the BE OPTIMAL trial, a larger proportion of patients in the bimekizumab group compared to the placebo group attained at least a 2-grade reduction from baseline in the IGA score at week 16. The difference between treatment groups was 46.0% (95% CI, 37.1% to 55.0%). IGA results at week 52 indicated that the response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, a larger proportion of patients in the bimekizumab group compared to the placebo group attained at least a 2-grade reduction from baseline in the IGA score at week 16. The difference between treatment groups was 58.2% (95% CI, 46.7% to 69.8%).
Physical Function
The improvement in physical function at week 16 was assessed using the HAQ-DI and Short Form (36) Health Survey (SF-36) physical component summary (PCS). HAQ-DI is a self-assessment questionnaire of 8 domains (dressing and grooming, arising, eating, walking, hygiene, reach, grip, and activities); patients’ difficulty in performing these activities is scored from 0 (without any difficulty) to 3 (unable to do). SF-36 is a 36-item, general health status instrument consisting of 8 health domains: physical functioning, pain, vitality, social functioning, psychological functioning, general health perceptions, role limitations due to physical challenges, and role limitations due to emotional challenges. The PCS ranges from 0 to 100, with higher scores indicating better health status.
Health Assessment Questionnaire–Disability Index: In the BE OPTIMAL trial, the bimekizumab group had a greater mean decrease from baseline (i.e., improvement) in HAQ-DI compared with the placebo group at week 16; the least squares mean (LSM) difference between treatment groups was –0.19 (95% CI, –0.25 to –0.13; P < 0.001). HAQ-DI results at week 52 indicated that the response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, the bimekizumab group had a greater mean decrease from baseline (i.e., improvement) in HAQ-DI compared with the placebo group at week 16; the LSM difference between treatment groups was –0.33 (95% CI, –0.42 to –0.23; P < 0.001). A larger proportion of patients in the bimekizumab group compared to the placebo group attained a HAQ-DI score decrease of at least 0.35 for each of the subgroup categories of inadequate response to 1 TNFi (difference versus placebo = 40.8% [95% CI, 28.6% to 53.1%]), inadequate response to 2 TNFis (difference versus placebo = 11.5% [95% CI, –19.9% to 42.9%]), and intolerance to TNFis (difference versus placebo = 24.7% [95% CI, –6.0% to 55.5%]).
SF-36 PCS: In the BE OPTIMAL trial, the bimekizumab group had a greater mean increase from baseline (i.e., improvement) in the SF-36 PCS compared with the placebo group at week 16; the LSM difference between treatment groups was 4.3 (95% CI, 3.2 to 5.4; P < 0.001). SF-36 PCS results at week 52 indicated that the response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, the bimekizumab group had a greater mean increase from baseline (i.e., improvement) in the SF-36 PCS compared with the placebo group at week 16; the LSM difference between treatment groups was 6.0 (95% CI, 4.4 to 7.7; P < 0.001).
Harms Results
Patients reporting at least 1 AE during the DB treatment periods of the BE OPTIMAL and BE COMPLETE trials ranged from 40.4% to 59.6% of patients in the bimekizumab groups and from 33.3% to 49.5% of patients in the placebo groups. Nasopharyngitis was the most common AE (3.7% to 9.3% in the bimekizumab groups versus 0.8% to 4.6% in the placebo groups), followed by upper respiratory tract infection (2.2% to 5.1% in the bimekizumab groups versus 1.5% to 6.4% in the placebo groups). During the BE OPTIMAL trial’s active treatment–blind period, 72.0% of patients in the bimekizumab group and 70.5% of patients in the placebo-bimekizumab crossover group reported at least 1 AE, with the most common being nasopharyngitis (7.0% and 8.5% in the bimekizumab and crossover groups, respectively).
The frequency of serious adverse events (SAEs) was 1.9% in the bimekizumab groups and ranged from 0 to 1.1% in the placebo groups during the DB period of both trials. During the BE OPTIMAL trial’s active treatment–blind period, 5.6% of patients in the bimekizumab group and 5.9% of patients in the placebo-bimekizumab crossover group reported at least 1 SAE.
Withdrawals from treatment due to AEs ranged from 0.7% to 1.9% of patients in the bimekizumab groups and from 0 to 1.1% of patients in the placebo groups during the DB period of both trials. During the BE OPTIMAL trial’s active treatment–blind period, 2.7% of patients in the bimekizumab group and 1.8% of patients in the placebo-bimekizumab crossover group reported an AE leading to drug discontinuation.
There were no deaths during the DB periods of the trials and 1 death in the placebo-bimekizumab crossover group (due to traumatic shock from a motorcycle accident) during the BE OPTIMAL trial’s active treatment–blind period.
During the DB period of both trials, few notable harms were reported among either the bimekizumab or placebo group (i.e., 0 or 1 patient per treatment group) for liver dysfunction based on Hy’s law, opportunistic infection, a major cardiovascular event, malignancy, anaphylaxis, or IBD. In the trials, approximately 4.5% of patients in the bimekizumab groups and 1% of patients in the placebo groups reported any fungal infection. Of these, 2.6% of patients in the bimekizumab groups and less than 1% of patients in the placebo groups reported a candida infection, approximately 2% of patients in the bimekizumab groups and less than 1% of patients in the placebo groups reported a fungal infection not elsewhere classified (NEC), and less than 1% of patients in the bimekizumab groups and no patients in the placebo groups reported a tinea infection.
During the BE OPTIMAL trial’s active treatment–blind period, there were few reports (< 5 patients in the treatment groups) of liver dysfunction based on Hy’s law, opportunistic infection, a major cardiovascular event, malignancy, anaphylaxis, or IBD. Of the 11.4% of patients who reported any fungal infection in the bimekizumab group, 6.8% reported a candida infection, 4.6% reported a fungal infection NEC, and 1.2% reported a tinea infection. Of the 9.2% of patients who reported any fungal infection in the placebo-bimekizumab crossover group, 7.0% reported a candida infection, 2.6% reported a fungal infection NEC, and 0.7% reported a tinea infection.
Liver toxicity, reactivation of tuberculosis infection, and serious injection-related AEs were not reported in either of the trials.
Critical Appraisal
There were some imbalances in baseline characteristics between the bimekizumab and placebo groups for the presence of enthesitis (both trials), the proportion of patients with 10% or greater BSA affected by psoriasis, PASI, and methotrexate use (the BE COMPLETE trial), which may have biased the results in favour of patients who start out with low disease activity at baseline, though the direction of bias for the overall treatment groups is less certain. Results during the active treatment–blind period of the BE OPTIMAL trial may have been confounded by the lack of a placebo comparison group (patients randomized to placebo were reallocated to bimekizumab) and any rescue therapies used (these were permitted after the 16-week DB period and use ranged from 4.8% to 7.0% of patients). Moreover, it is possible that permitted concomitant therapies (particularly cDMARDs) may not have reached full effect during the minimum 8 weeks that defined a stable dose, making it difficult to attribute treatment effects and harms to either bimekizumab or a concomitant drug. Four outcomes relevant to the CADTH review (PASI 90, IGA, LEI, and LDI) used subsets of the randomized set and it is uncertain if the known and unknown treatment effect modifiers were still balanced between the groups.
In general, both trials had limited racial diversity, which is not necessarily reflective of patients with PsA across Canada. Patients with other bDMARD or tsDMARD experience (other than TNFis) were not included and it is uncertain if the trial results are generalizable to these patients. However, based on the reported baseline characteristics, the clinical expert consulted by CADTH indicated that the patients in the trials were generally similar to those who are treated in clinical practice and could receive bimekizumab in Canada. Also, of the permitted concomitant medications, it was noted that hydroxychloroquine is rarely used in Canadian practice and apremilast is a targeted therapy accessed after cDMARDs. It was noted that ACR and some patient-reported outcomes are not typically used in clinical practice, though they may still provide important information to patients and clinicians.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development and Evaluation (GRADE) tool was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.22,23
Following the GRADE approach, evidence from RCTs started as high certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance was unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of an important effect based on thresholds identified in the literature for HAQ-DI, SF-36 PCS, and PtAAP. The target of the certainty of evidence assessment was the presence or absence of an important effect based on thresholds informed by the clinical expert consulted for this review for ACR, MDA, LEI, LDI, SJC, PASI 90, and IGA.
For the GRADE assessments, findings from the BE OPTIMAL and BE COMPLETE trials were assessed together per outcome because these studies were similar in population, intervention, design, and outcome measures.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence,24 consultation with the clinical expert, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: composite measures of disease activity (ACR50, ACR20, ACR70, and MDA), musculoskeletal-related outcomes (LEI, LDI, and SJC), skin-related outcomes (PASI 90 and IGA), and patient-reported outcomes for physical functioning and symptoms (HAQ-DI, SF-36 PCS, and PtAAP).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for bimekizumab versus placebo for patients with PsA.
Table 2: Summary of Findings for Bimekizumab Versus Placebo for Patients With PsA
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Bimekizumab | Difference | |||||
Composite measures of disease activity | |||||||
ACR50 response: patients with no prior exposure to biologics Follow-up: 16 weeks | 712 (1 RCT) | OR = 7.1 (4.6 to 10.9) | 85 per 1,000 | 397 (339 to 459) per 1,000 | 312 (252 to 373) more per 1,000 | Higha | Bimekizumab results in an increase in the proportion of patients who attain ACR50 when compared with placebo. |
ACR50 response: patients with a history of inadequate response or intolerance to 1 or 2 TNFis Follow-up: 16 weeks | 400 (1 RCT) | OR = 11.1 (5.4 to 23.0) | 43 per 1,000 | 333 (248 to 431) per 1,000 | 290 (219 to 362) more per 1,000 | Higha | Bimekizumab results in an increase in the proportion of patients who attain ACR50 when compared with placebo. |
ACR20 response: patients with no prior exposure to biologicsb Follow-up: 16 weeks | 712 (1 RCT) | OR = 5.4 (3.8 to 7.5) | 200 per 1,000 | 572 (512 to 631) per 1,000 | 372 (305 to 440) more per 1,000 | Highc | Bimekizumab results in an increase in the proportion of patients who attain ACR20 when compared with placebo. |
ACR20 response: patients with a history of inadequate response or intolerance to 1 or 2 TNFisb Follow-up: 16 weeks | 400 (1 RCT) | OR = 12.2 (7.0 to 21.1) | 144 per 1,000 | 672 (579 to 753) per 1,000 | 528 (436 to 619) more per 1,000 | Highc | Bimekizumab results in an increase in the proportion of patients who attain ACR20 when compared with placebo. |
ACR70 response: patients with no prior exposure to biologicsb Follow-up: 16 weeks | 712 (1 RCT) | OR = 7.2 (3.9 to 13.4) | 40 per 1,000 | 233 (185 to 289) per 1,000 | 193 (141 to 245) more per 1,000 | Highd | Bimekizumab results in an increase in the proportion of patients who attain ACR70 when compared with placebo. |
ACR70 response: patients with a history of inadequate response or intolerance to 1 or 2 TNFisb Follow-up: 16 weeks | 400 (1 RCT) | OR = 50.6 (6.9 to 370.0) | 4 per 1,000 | 176 (111 to 269) per 1,000 | 172 (122 to 222) more per 1,000 | Highd | Bimekizumab results in an increase in the proportion of patients who attain ACR70 when compared with placebo. |
MDA response: patients with no prior exposure to biologics Follow-up: 16 weeks | 712 (1 RCT) | OR = 5.4 (3.7 to 8.1) | 123 per 1,000 | 433 (374 to 494) per 1,000 | 310 (245 to 375) more per 1,000 | Highe | Bimekizumab results in an increase in the proportion of patients who attain MDA when compared with placebo. |
MDA response: patients with a history of inadequate response or intolerance to 1 or 2 TNFis Follow-up: 16 weeks | 400 (1 RCT) | OR = 13.1 (6.1 to 28.0) | 46 per 1,000 | 388 (296 to 488) per 1,000 | 342 (261 to 422) more per 1,000 | Highe | Bimekizumab results in an increase in the proportion of patients who attain MDA when compared with placebo. |
Musculoskeletal-related outcomes | |||||||
Enthesitis-free state based on the LEI in patients with enthesitis at baseline: pooled population of patients with no prior exposure to biologics and patients with a history of inadequate response or intolerance to 1 or 2 TNFis Follow-up: 16 weeks | 355 (1 RCT) | OR = 1.9 (1.2 to 3.1) | 300 per 1,000 | 450 (379 to 522) per 1,000 | 149 (40 to 259) more per 1,000 | Lowf,g | Bimekizumab may result in an increase in the proportion of patients who attain an enthesitis-free state when compared with placebo. |
Dactylitis-free state based on the LDI in patients with dactylitis at baseline: pooled population of patients with no prior exposure to biologics and patients with a history of inadequate response or intolerance to 1 or 2 TNFis Follow-up: 16 weeks | 137 (1 RCT) | OR = 3.4 (1.6 to 7.6) | 415 per 1,000 | 710 (590 to 806) per 1,000 | 294 (117 to 471) more per 1,000 | Lowf,h | Bimekizumab may result in an increase in the proportion of patients who attain a dactylitis-free state when compared with placebo. |
SJC (0 [best] to 66 [worst]) LSM change from baseline, joints: patients with no prior exposure to biologicsb Follow-up: 16 weeks | 712 (1 RCT) | NA | –2.3 | –6.3 (SE = 0.3) | –4.0 (–4.8 to –3.1) | Lowi | Bimekizumab may result in a decrease in the number of swollen joints when compared with placebo. |
SJC (0 [best] to 66 [worst]), LSM change from baseline, joints: patients with a history of inadequate response or intolerance to 1 or 2 TNFisb Follow-up: 16 weeks | 400 (1 RCT) | NA | –1.7 | –7.1 (SE = 0.5) | –5.3 (–6.5 to –4.2) | Moderatej | Bimekizumab likely results in a decrease in the number of swollen joints when compared with placebo. |
Skin-related outcomes | |||||||
PASI 90 response in patients with psoriasis involving ≥ 3% BSA at baseline: patients with no prior exposure to biologics Follow-up: 16 weeks | 357 (1 RCT) | OR = 63.0 (22.2 to 178.9) | 22 per 1,000 | 587 (487 to 679) per 1,000 | 565 (486 to 643) more per 1,000 | Moderatef,k | Bimekizumab likely results in an increase in the proportion of patients who attain PASI 90 when compared with placebo. |
PASI 90 response in patients with psoriasis involving ≥ 3% BSA at baseline: patients with a history of inadequate response or intolerance to 1 or 2 TNFis Follow-up: 16 weeks | 264 (1 RCT) | OR = 30.2 (12.4 to 73.9) | 53 per 1,000 | 629 (495 to 746) per 1,000 | 576 (476 to 676) more per 1,000 | Lowf,k,l | Bimekizumab may result in an increase in the proportion of patients who attain PASI 90 when compared with placebo. |
IGA score of 0 or 1 and ≥ 2-grade reduction from baseline in patients with psoriasis involving ≥ 3% BSA at baseline: patients with no prior exposure to biologicsb Follow-up: 16 weeks | 333 (1 RCT) | OR = 27.1 (10.6 to 69.5) | 35 per 1,000 | 495 (394 to 597) per 1,000 | 460 (371 to 550) more per 1,000 | Lowf,m | Bimekizumab may result in an increase in the proportion of patients who attain an IGA score of 0 or 1 when compared with placebo. |
IGA score of 0 or 1 and ≥ 2-grade reduction from baseline in patients with psoriasis involving ≥ 3% BSA at baseline: patients with a history of inadequate response or intolerance to 1 or 2 TNFisb Follow-up: 16 weeks | 245 (1 RCT) | OR = 40.9 (12.3 to 135.6) | 39 per 1,000 | 621 (483 to 742) per 1,000 | 582 (467 to 698) more per 1,000 | Lowf,m | Bimekizumab may result in an increase in the proportion of patients who attain an IGA score of 0 or 1 when compared with placebo. |
Patient-reported outcomes for physical functioning and symptoms | |||||||
HAQ-DI score (0 [best] to 3 [worst]) LSM change from baseline, points: patients with no prior exposure to biologics Follow-up: 16 weeks | 712 (1 RCT) | NA | –0.07 | –0.26 (SE = 0.03) | –0.19 (–0.25 to –0.13) | Highn | Bimekizumab results in a reduction in HAQ-DI score when compared with placebo. |
HAQ-DI score (0 [best] to 3 [worst]) LSM change from baseline, points: patients with a history of inadequate response or intolerance to 1 or 2 TNFis Follow-up: 16 weeks | 400 (1 RCT) | NA | 0.02 | –0.31 (SE = 0.04) | –0.33 (–0.42 to –0.23) | Highn | Bimekizumab results in a reduction in HAQ-DI score when compared with placebo. |
SF-36 PCS score LSM change from baseline, points: patients with no prior exposure to biologics Follow-up: 16 weeks | 712 (1 RCT) | NA | 1.9 | 6.3 (SE = 0.5) | 4.3 (3.2 to 5.4) | Higho | Bimekizumab results in an increase in SF-36 PCS score when compared with placebo. |
SF-36 PCS score LSM change from baseline, points: patients with a history of inadequate response or intolerance to 1 or 2 TNFis Follow-up: 16 weeks | 400 (1 RCT) | NA | 0.1 | 6.2 (SE = 0.7) | 6.0 (4.4 to 7.7) | Higho | Bimekizumab results in an increase in SF-36 PCS score when compared with placebo. |
PtAAP (0 [best] to 100 [worst]) LSM change from baseline, points: patients with no prior exposure to biologicsb Follow-up: 16 weeks | 712 (1 RCT) | NA | –4.6 | –23.8 (SE = 1.4) | –19.1 (–22.7 to –15.5) | Highp | Bimekizumab results in a reduction in PtAAP score when compared with placebo. |
PtAAP (0 [best] to 100 [worst]) LSM change from baseline, points: patients with a history of inadequate response or intolerance to 1 or 2 TNFisb Follow-up: 16 weeks | 400 (1 RCT) | NA | –1.6 | –26.6 (SE = 2.1) | –25.0 (–30.0 to –20.0) | Highp | Bimekizumab results in a reduction in PtAAP score when compared with placebo. |
ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; BSA = body surface area; CI = confidence interval; HAQ-DI = Health Assessment Questionnaire–Disability Index; IGA = Investigator’s Global Assessment; LDI = Leeds Dactylitis Index; LEI = Leeds Enthesitis Index; LSM = least squares mean; MDA = minimal disease activity; MID = minimal important difference; NA = not available; OR = odds ratio; PASI = Psoriasis Area Severity Index; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; PCS = physical component summary; PsA = psoriatic arthritis; PtAAP = Patient’s Assessment of Arthritis Pain; RCT = randomized controlled trial; SE = standard error; SF-36 = Short Form (36) Health Survey; SJC = swollen joint count; TNFi = tumour necrosis factor inhibitor.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aA difference of 20% between groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
bAnalysis of this outcome was not adjusted for multiplicity. The results are considered as supportive evidence.
cA difference of 30% to 40% between groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
dA difference of 10% to 15% between groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
eA difference of 15% to 20% between groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
fRated down 1 level for study limitations due to the loss of randomization in the population used for outcome analysis and the results being at a higher risk of bias.
gRated down 1 level for serious imprecision. The 95% CI for difference between groups includes the possibility of no benefit compared to the threshold of clinical importance that the clinical expert suggested for achieving an enthesitis-free state (150 more per 1,000 patients).
hRated down 1 level for serious imprecision. The 95% CI for difference between groups includes the possibility of no benefit compared to the lower threshold of clinical importance that the clinical expert suggested for achieving a dactylitis-free state (150 to 200 more per 1,000 patients).
iRated down 2 levels for very serious imprecision. Both boundaries of the 95% CI for difference between groups exclude the threshold of clinical importance that the clinical expert suggested for improvement in SJC (5 fewer swollen joints).
jRated down 1 level for serious imprecision. The 95% CI for difference between groups includes the possibility of no benefit compared to the threshold of clinical importance that the clinical expert suggested for improvement in SJC (5 fewer swollen joints).
kDid not rate down for serious imprecision. The 95% CI for difference between groups includes the possibility of no benefit compared to the threshold of clinical importance that the clinical expert suggested for PASI 90 (500 more per 1,000 patients); however, the lower bound of the 95% CI was close to the threshold and bimekizumab has previously been reviewed and approved for the treatment of patients with moderate-to-severe plaque psoriasis.
lRated down 1 level for study limitations. The increased risk of bias is due to an imbalance in baseline characteristics between treatment groups (a higher proportion of patients with a larger percentage of BSA affected by psoriasis and higher PASI scores in the bimekizumab group).
mRated down 1 level for serious imprecision. The 95% CI for difference between groups includes the possibility of no benefit compared to the threshold of clinical importance that the clinical expert suggested for IGA (500 more per 1,000 patients).
nA difference of −0.35 to −0.13 points between groups was identified from the literature as an MID for this outcome.
oA difference of 3.74 points between groups was identified from the literature as an MID for this outcome.
pA difference of –10 points between groups was identified from the literature as an MID for this outcome.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24
Long-Term Extension Studies
Description of Studies
Two long-term extension studies were submitted by the sponsor that evaluated bimekizumab for the treatment of adult patients with PsA. The completed BE ACTIVE 2 study25 was a 104-week, phase II, open-label extension (OLE) study that aimed to assess the long-term safety, tolerability, and efficacy of bimekizumab in 184 adult patients with PsA who completed the preceding phase IIb BE ACTIVE study.26 The BE ACTIVE study, not included in the systematic review, was a 48-week, randomized, DB, placebo-controlled, parallel-group, dose-ranging study that included adults with PsA who had previously been exposed to 1 TNFi. Patients were randomized to receive placebo or 1 of several bimekizumab dosing regimens for the initial 12-week placebo-controlled period, and then rerandomized to receive bimekizumab 160 mg every 4 weeks or 320 mg every 4 weeks up to week 48.
The BE VITAL study27,28 is an ongoing (estimated completion date: May 25, 2026), phase III, OLE study of the BE COMPLETE and BE OPTIMAL trials that is evaluating the long-term efficacy (up to week 140) and long-term safety (up to week 212) of bimekizumab in 1,131 patients with PsA who received bimekizumab 160 mg every 4 weeks.
The inclusion and exclusion criteria for the BE ACTIVE 225 and BE VITAL27,28 studies were consistent with those of the preceding trials (the BE ACTIVE,26 BE OPTIMAL,20 and BE COMPLETE19 trials). In both the BE ACTIVE 2 and BE VITAL studies, bimekizumab 160 mg every 4 weeks SC was administered. All data from the extension studies were analyzed descriptively using summary statistics.
Efficacy Results
In the BE ACTIVE 2 study, rates of ACR20, ACR50, and ACR70 response were similar at baseline and at week 104 with continued bimekizumab treatment. The proportion of patients who had attained MDA response was 58.6% at week 104 of the BE ACTIVE 2 study. For a 75% reduction in Psoriasis Area Severity Index score (PASI 75), PASI 90, and a 100% reduction in Psoriasis Area Severity Index score (PASI 100), data were limited at several visits due to an error in the original study protocol, which was later amended. For visits with what the sponsor considered to be a meaningful sample size of data collected, the proportion of patients who attained PASI 75, PASI 90, and PASI 100 at week 104 were 79.2%, 73.3%, and 65.8%, respectively. The SF-36 PCS score in the BE ACTIVE 2 study was sustained with continued bimekizumab treatment up to week 104 with a mean PCS change of 9.5 (standard error [SE] = 0.8).
In the BE VITAL study, sustained efficacy was observed with bimekizumab from week 16 to week 52 across clinical and patient-reported outcomes. At week 52, 51.7% of patients originally randomized to bimekizumab and 40.6% of patients randomized to placebo who crossed over to bimekizumab at week 16 had an ACR50 response. ACR20 and ACR70 responses similarly improved over time for these groups.
MDA was attained by 47.2% of bimekizumab patients and 33.1% of placebo-bimekizumab crossover patients at week 52. The proportions of patients attaining PASI 75, PASI 90, and PASI 100 increased out to week 52 in both those who were initially placebo-randomized patients and those who were bimekizumab-randomized patients with psoriasis affecting at least 3% BSA at baseline. Data for the SF-36 PCS were only reported up to week 40. The mean SF-36 PCS change from baseline was 7.3 (SE = 0.9) at week 40 for patients who switched from placebo to bimekizumab. For those who were originally randomized to bimekizumab, the mean SF-36 PCS change from baseline was 8.4 (SE = 0.6) at week 40.
Harms Results
The total time at risk was 392.3 patient-years during the BE ACTIVE 2 study. Most patients (80.9%) reported treatment-emergent adverse events (TEAEs), which were most commonly infections and infestations (55.2%). Overall, 7.7% of patients reported an SAE, 4.9% of patients discontinued bimekizumab due to TEAEs, and there were no deaths reported.
In the BE VITAL study, at least 1 TEAE was reported by 243 of 388 (62.6%) patients while receiving bimekizumab up to week 52. The most frequently reported TEAEs were hypersensitivity (4.9%), SARS-CoV-2 (COVID-19) infections (7.2%), fungal infections (9.5%), nasopharyngitis (5.9%), and urinary tract infection (5.9%). Serious infections occurred among 1.8% of patients, and 1.3% of patients had neutropenia. The proportion of patients who reported SAEs was 5.9%. The discontinuation of bimekizumab treatment due to TEAEs was among 4.1% of patients. One death was reported, which the sponsor deemed was unrelated to study treatment.
Critical Appraisal
The open-label design of the BE ACTIVE 2 and BE VITAL studies could bias the magnitude of the treatment effect due to unblinded exposure to the study medication during the treatment period, though the direction of bias is uncertain. In addition to that, the absence of control groups in both studies and the lack of data beyond week 52 in the BE VITAL study make interpretation of the findings challenging. Only those who completed the BE ACTIVE, BE OPTIMAL, and BE COMPLETE studies moved on to the BE ACTIVE 2 and BE VITAL studies, and there may have been selection bias involved.
As the BE ACTIVE 2 and BE VITAL studies consisted of patients who took part in the pivotal studies (the BE ACTIVE, BE OPTIMAL, and BE COMPLETE studies), it is reasonable to expect that the same strengths and limitations related to generalizability apply to the extension studies. The patient population of those studies may not be reflective of the wider, more heterogeneous clinical population in terms of demographic and clinical characteristics; therefore, the results presented may differ from those observed in a real-world clinical setting.
Indirect Comparisons
Description of Studies
The sponsor submitted a network meta-analysis (NMA) and a matching-adjusted indirect comparison (MAIC) for the indirect treatment comparison (ITC).29,30 The NMA assessed ACR20, ACR50, ACR70, MDA, PASI 90, and safety outcomes at week 12 to week 24, while the MAIC assessed ACR20, ACR50, ACR70, and MDA at week 52. Included trials were phase II to phase IV RCTs conducted in patients with adult-onset PsA treated with 1 drug from a set of specified interventions and dosing regimens that included IL-17is, IL-23is, and IL-12/23is (NMA and MAIC), as well as specific TNFis, CTLA immunoglobulin, JAKis, and PDE4is (NMA only).
Efficacy Results
The NMA for the biologic or targeted synthetic disease-modifying antirheumatic drug (b/tsDMARD)–naive population indicated that bimekizumab was more efficacious than most IL-12/23is and IL-23is, and abatacept for ACR outcomes, but may be similar to IL-17is, TNFis, or JAKis, with a few exceptions. The results for the TNFi-experienced population indicated that bimekizumab was favoured over interleukin-17 comparators for ACR20 response, but favourability varied relative to comparators for both ACR50 and ACR70 outcomes, and wide credible intervals (CrIs) suggest high imprecision in this subpopulation. The results for PASI 90 indicated that bimekizumab was favoured over most TNFi comparators and may be similar to other classes, but fewer comparisons were made in TNFi-experienced patients. Results for MDA indicated that bimekizumab was favoured for the comparisons of the combined interleukin-12 and interleukin-23, as well as interleukin-23, made in b/tsDMARD-naive patients, but favourability varied in TNFi-experienced patients and overall, there were fewer comparisons made for this outcome. Golimumab was favoured over bimekizumab for ACR20 and ACR50. The MAIC was subject to important limitations that preclude drawing firm conclusions about efficacy.
Harms Results
Comparison of specific harms was not possible due to a lack of specific information from the trials. Overall, bimekizumab was neither favoured, nor was it less favoured than most comparators for AEs, SAEs, or discontinuations due to AEs. One exception was that bimekizumab was favoured over ustekinumab for discontinuations due to AEs.
Critical Appraisal
While methods to mitigate sources of uncertainty were implemented, the results of the NMA are subject to some uncertainty due to the unmeasurable limitations of baseline risk adjustment and uncertainty over the extent to which it accounts for patient heterogeneity, as well as differences in study design and model selection that impact the comparability of the studies across the network. The results from the MAIC are highly uncertain, at risk of unmeasured bias, and also of limited applicability to the clinical context due to the inclusion of only some treatment options being available in the Canadian context.
Studies Addressing Gaps in the Evidence From the Systematic Review
No additional relevant studies addressing gaps in the evidence of the systematic review were submitted by the sponsor.
Conclusions
Based on the evidence from 2 DB RCTs, adults with PsA who received bimekizumab 160 mg every 4 weeks were more likely to demonstrate clinically meaningful improvements in composite measures of disease activity, dermatological manifestations (i.e., psoriasis), physical function, HRQoL, and pain at week 16. Results were generally consistent whether patients were biologic-naive or TNFi-experienced. Evidence that bimekizumab reduces the number of musculoskeletal manifestations (e.g., enthesitis, dactylitis, swollen joints) was less certain, but demonstrated a consistent direction of effect. The trial outcomes address some of the needs patients have for new PsA treatments as well as treatment goals that patients and clinicians have when managing PsA. Longer-term results indicated that treatment effects were maintained with the continued use of bimekizumab for up to 2 years for patients who had had prior TNFis; data for biologic-naive patients are forthcoming. There was no direct evidence comparing bimekizumab and other targeted DMARDs for PsA available for review. The indirect evidence suggests that bimekizumab is superior or similar to other bDMARDs for composite measures of disease activity (e.g., ACR and MDA) and plaque psoriasis outcomes; however, inconsistency in results across comparators, imprecision in results, study and patient characteristic heterogeneity, and methodological flaws reduce the certainty in these results. No new safety signals were identified with extended bimekizumab use in the trials and reports of patients experiencing notable harms were low. Given the available direct and indirect comparative evidence and its approval as a treatment for psoriasis, bimekizumab is an option for adults with PsA, including those with concurrent psoriasis.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of bimekizumab 160 mg every 4 weeks for SC injection in the treatment of adult patients with active PsA, alone or in combination with a cDMARD.
Disease Background
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
PsA is a heterogeneous, chronic inflammatory, immune-mediated disease characterized by musculoskeletal inflammation, in which patients present with a variety of different disease manifestations across multiple tissues and clinical domains, including psoriasis (and nail psoriasis), peripheral arthritis, axial disease, enthesitis, and dactylitis.3-6 The variable involvement of a number of disease domains among patients makes it hard to establish clear-cut stages and to assess disease severity.31 Therefore, composite outcome measures like MDA and ACR response are often used to measure low disease activity state and treatment response, respectively, in PsA.31
Peripheral arthritis (the involvement of peripheral joints, assessed by SJC and TJC) and psoriasis (skin disease) are the most common disease manifestations of PsA, occurring in 86% to 90.8% of patients and 82% of patients with PsA, respectively.7,32 Joint inflammation is known to worsen over time and, if left untreated, can lead to permanent joint damage, deformities, and long-term disability.4,8-10 Up to 80% of patients with PsA present with psoriasis for several years before developing joint problems4 or being diagnosed with PsA; however, for some, arthritis symptoms may occur simultaneously or before the onset of psoriasis. PsA is typically associated with psoriasis and has been reported in up to 30% of patients with psoriasis but can also occur in individuals who do not have skin disease.4,6 The most prevalent type of psoriasis in patients with PsA is plaque psoriasis; however, other types (e.g., pustular psoriasis, guttate psoriasis, nail psoriasis, erythrodermic psoriasis, inverse psoriasis) have also been reported.4
In addition to peripheral arthritis and psoriasis, PsA manifestations include axial involvement (inflammation in the spine and sacroiliac joints),4,5 occurring in 25% to 70% of patients with PsA,4 as well as dactylitis (swelling of the whole digit) and enthesitis (tenderness and swelling at the insertion of tendons and ligaments into bone), which occur in 40% to 50% of patients and 30% to 40% of patients with PsA, respectively.4,33 In addition to musculoskeletal manifestations, patients with PsA can often experience immune-mediated extra-articular manifestations such as uveitis and IBD (including ulcerative colitis and Crohn disease).4,34 Other comorbidities commonly observed in PsA include depression, cardiovascular disease, fibromyalgia, diabetes, metabolic syndrome, osteoporosis, and obesity, which further increase the burden of disease.4,35
Patients with PsA can experience debilitating pain and fatigue that result in decreased physical function and social participation and contribute to overall poor emotional and mental well-being.8,36-38 Moreover, longer disease duration, worse physical, emotional, and social aspects associated with PsA, work absenteeism due to sickness, and decreased household productivity are all associated with a profound negative impact on HRQoL.39-41
PsA symptoms usually start between the ages of 30 years and 50 years, and PsA affects both men and women equally.4,42 Prevalence and incidence estimates for PsA are variable depending on the case definition and geography,11 with an estimated incidence of 6 per 100,000 people and a prevalence of approximately 1 to 2 per 1,000 people in the general global population.12 A population-based Canadian study estimated the age- and sex-standardized cumulative prevalence of PsA in Ontario to range from 0.9 per 1,000 people in 2008 to 1.5 per 1,000 people in 2015.13 The same study estimated the age- and sex-standardized incidence in 2015 to be 14 per 100,000 people.13
With the lack of definitive diagnostic tests for PsA and the heterogeneous presentation and course of the disease, diagnosis can be complex.3,43,44 In Canada, PsA is typically diagnosed by a rheumatologist.45 The tools used in the diagnosis of PsA (i.e., blood testing for CRP levels and erythrocyte sedimentation rate, X-ray, ultrasound, and MRI scans) are widely available across Canada, as they are common diagnostic tools used in the Canadian health care setting.44,46,47
Standards of Therapy
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
According to clinical practice guidelines available for the management of PsA and input from the clinical expert consulted by CADTH, treatment goals include achieving the lowest level of disease activity (with a target of disease remission), maximizing functional status and HRQoL, preventing further disease progression, controlling symptoms, and avoiding complications (from untreated disease or adverse effects from treatment).14-16 The guidelines recommend that the choice of treatment should be a shared discussion and decision made between a patient and clinician and should consider the following factors: disease domain(s) affected, disease activity, existing comorbidities, prior treatments, and patient preferences.14,16 The 2021 Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) guidelines recommend that treatment be guided by the domain(s) that are most impacted by the disease as well as relevant comorbidities, which include peripheral arthritis, axial disease, enthesitis, dactylitis, skin psoriasis, psoriatic nail disease, uveitis, and IBD.16 Other health considerations include obesity, metabolic syndrome, cardiovascular disease, liver disease, mood disorders, chronic infections, malignancies, bone health, central sensitization, and reproductive health.16 The clinical expert also noted that the impact of disease on pain, function, HRQoL, and structural damage should be assessed and considered when managing PsA. In discussion with the patient, treatment decisions may be further impacted by personal goals, preferences, adverse effects, route of administration, convenience, and cost.16,48
The management of PsA can be nonpharmacological (e.g., physical therapy, exercise) or pharmacological, where first-line treatment typically includes cDMARDs, such as methotrexate, sulfasalazine, leflunomide, or cyclosporine.15,16 According to the guidelines and clinical expert consulted by CADTH, additional treatment can be included to address joint symptoms (e.g., NSAIDs, intra-articular glucocorticoid injections) or psoriasis and nail disease (e.g., topical therapies) as required.15,16 Later-line, targeted treatments are usually reserved for patients who have an inadequate response to cDMARDs and include bDMARDs (e.g., TNFi, IL-17i, IL-12/23i, IL-23i) and tsDMARDs (e.g., JAKi, PDE4i).16 The clinical expert noted that continuation of cDMARDs may be necessary until effectiveness of the targeted therapies is confirmed. The guidelines also indicate that the failure or intolerance of a treatment can lead to switching drugs within a class or to another class of medication.14-16 The clinician group that provided input also considered adverse effects and patient preference as reasons to discontinue a treatment. As per the clinical expert and the literature, surgical intervention (to correct joint deformities or severe joint destruction and to improve pain and function) may be an option later in the disease course.48 The 2019 European Alliance of Associations for Rheumatology guidelines suggest that cautious tapering of treatments can be considered when a patient attains complete and sustained remission for at least 6 months.14 Regular monitoring is necessary for treatment response, comorbidities, and AEs, and may take place at 3-month intervals during periods of active disease or changes in treatment.48 Patients with stable disease may be monitored at 6-month intervals, though some treatments may require more frequent lab monitoring.48
Drug Under Review
Key characteristics of bimekizumab and other treatments available for PsA are summarized in Table 3.
Bimekizumab is a humanized immunoglobulin G1 kappa monoclonal antibody with 2 identical, high-affinity, antigen-binding regions that bind and neutralize IL-17A, IL-17F, and IL-17AF cytokines, blocking their interaction with the IL-17RA/IL-17RC receptor complex, which plays a role in PsA-related inflammation.2 IL-17F is produced by innate immune cells and can be independent of IL-23. In vitro, dual neutralization of both IL-17A and IL-17F with bimekizumab suppresses the expression of inflammation-related genes and proteins and inhibits the migration of inflammatory cells and pathological osteogenesis to a greater extent than inhibition of IL-17A alone.2,49
Bimekizumab is currently being reviewed by Health Canada for the treatment of adult patients with active PsA. Bimekizumab can be used alone or in combination with a cDMARD (e.g., methotrexate). The recommended dose of bimekizumab for adult patients with active PsA is 160 mg (given as 1 SC injection of 160 mg) every 4 weeks.2 For patients with PsA with coexistent moderate-to-severe plaque psoriasis, the recommended dose is the same as for plaque psoriasis (i.e., 320 mg given as 2 SC injections of 160 mg each every 4 weeks for the first 16 weeks, and then every 8 weeks thereafter); regular assessment of efficacy is recommended after 16 weeks and if a sufficient clinical response in joints cannot be maintained, a switch to the recommended dose for PsA can be considered.2 After proper training in SC injection technique, patients may be able to self-administer bimekizumab at home.2 The sponsor has requested reimbursement as per the approved Health Canada indication.
Bimekizumab was previously reviewed by CADTH in April 2022 for the indication of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy and a final recommendation for reimbursement with conditions was issued.17,18 In addition to the current CADTH review for the indication of adults with active PsA (with or without cDMARDs), bimekizumab is also being reviewed for the indication of adults with active ankylosing spondylitis.
Table 3: Key Characteristics of Bimekizumab and Drug Comparators for the Treatment of PsA
Drug | Indicationa | Route of administration | Recommended dosage | Safety issues |
|---|---|---|---|---|
IL-17 inhibitor | ||||
Bimekizumab (IL-17A/F inhibitor) | The treatment of adult patients with active PsA. Bimekizumab can be used alone or in combination with a cDMARD (e.g., methotrexate). | SC | 160 mg (given as 1 SC injection) q.4.w. For patients with PsA with coexistent moderate-to-severe plaque psoriasis: 320 mg (given as 2 SC injections of 160 mg) q.4.w. for the first 16 weeks, and then q.8.w. thereafter | Infections (TB and serious infection), hypersensitivity reactions, and IBD (exacerbations or new onset) |
Ixekizumab (IL-17A inhibitor) | The treatment of adult patients with active PsA who have responded inadequately or are intolerant to 1 or more DMARD. Ixekizumab can be used alone or in combination with a cDMARD (e.g., methotrexate). | SC | 160 mg SC injection at week 0, followed by 80 mg SC injections q.4.w. thereafter | Infections (TB and serious infection), hypersensitivity reactions, and IBD (exacerbations or new onset) |
Secukinumab (IL-17A inhibitor) | The treatment of adult patients with active PsA when the response to previous DMARD therapy has been inadequate. Secukinumab can be used alone or in combination with methotrexate. | SC | 150 mg SC injection at week 0, week 1, week 2, week 3, and week 4, followed by 150 mg SC injections q.4.w. thereafter | Infections (TB and serious infection), hypersensitivity reactions, and IBD (exacerbations or new onset) |
IL-12/23 inhibitor | ||||
Ustekinumab | The treatment of adult patients with active PsA. Ustekinumab can be used alone or in combination with methotrexate. | SC | 45 mg SC injection at week 0 and week 4, followed by 45 mg SC injections q.12.w. thereafter | Infections and reactivation of latent infections (TB and serious infections), hypersensitivity reactions, malignancies, and RPLS |
IL-23 inhibitor | ||||
Guselkumab | The treatment of adult patients with active PsA. Guselkumab can be used alone or in combination with a cDMARD (e.g., methotrexate). | SC | 100 mg SC injection at week 0 and week 4, followed by 100 mg SC injections q.8.w. thereafter | Infections (TB and serious infection) and hypersensitivity reactions |
Risankizumab | The treatment of adult patients with active PsA. Risankizumab can be used alone or in combination with a cDMARD (e.g., methotrexate). | SC | 100 mg SC injection at week 0 and week 4, followed by 100 mg SC injections q.8.w. thereafter | Infections (TB and serious infection) and hypersensitivity reactions |
TNF inhibitor | ||||
Adalimumab | Reducing the signs and symptoms of active arthritis and inhibiting the progression of structural damage and improving the physical function in adult patients with PsA. Adalimumab can be used in combination with methotrexate in patients who do not respond adequately to methotrexate alone. | SC | 40 mg SC injection q.2.w. | Serious infections, malignancies, hypersensitivity reactions, neurologic events (e.g., demyelinating disease), and congestive heart failure |
Certolizumab pegol | Alone or in combination with methotrexate to reduce signs and symptoms and inhibit the progression of structural damage, as assessed by X-ray, in adult patients with moderately to severely active PsA who did not experience improvement with ≥ 1 DMARD | SC | Loading dose of 400 mg SC injection at week 0, week 2, and week 4. Maintenance dose of 200 mg SC injection q.2.w. or 400 mg SC injection q.4.w. | Serious infections, malignancies, hypersensitivity reactions, neurologic events (e.g., demyelinating disease), and congestive heart failure |
Etanercept | Reducing signs and symptoms, inhibiting the progression of structural damage of active arthritis, and improving physical function in adult patients with PsA. Etanercept can be used in combination with methotrexate in adult patients who do not respond adequately to methotrexate alone. | SC | 50 mg SC injection weekly | Serious infections, malignancies, hypersensitivity reactions, neurologic events (e.g., demyelinating disease), and congestive heart failure |
Golimumab | Reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active PsA. Golimumab can be used in combination with methotrexate in patients who do not respond adequately to methotrexate alone. | SC | 50 mg SC injection q.4.w. | Serious infections, malignancies, hypersensitivity reactions, neurologic events (e.g., demyelinating disease), and congestive heart failure |
Infliximab | The treatment of adult patients with active PsA. Infliximab can be used alone or in combination with methotrexate. | IV | IV infusion of 5 mg/kg at week 0, week 2, and week 6, followed by infusions q.8.w. thereafter | Serious infections, malignancies, hypersensitivity reactions, neurologic events (e.g., demyelinating disease), and congestive heart failure |
PDE4 inhibitor | ||||
Apremilast | Alone or in combination with methotrexate, for the treatment of active PsA in adult patients who have had an inadequate response, intolerance, or contraindication to a prior DMARD | Oral | 30 mg by mouth, twice daily (Initial titration schedule: 10 mg on day 1 in the a.m., day 2 in the a.m. and the p.m., and day 3 in the a.m.; 20 mg on day 3 in the p.m., day 4 in the a.m. and the p.m., and day 5 in the a.m.; 30 mg on day 5 in the p.m., and day 6 in the a.m. and the p.m.) | Infections and reactivation of latent infections (TB and serious infections), hypersensitivity reactions, malignancies, and RPLS |
JAK inhibitor | ||||
Tofacitinib | In combination with methotrexate or another cDMARD, for reducing the signs and symptoms of PsA in adult patients with active PsA when the response to previous DMARD therapy has been inadequate. The use of tofacitinib in combination with bDMARDs or potent immunosuppressants, such as azathioprine and cyclosporine, is not recommended. | Oral | 5 mg by mouth, twice daily | Serious infections (TB, invasive fungal infections, opportunistic infections), malignancies, thrombosis, and liver enzyme elevation |
Upadacitinib | The treatment of adults with active PsA who have had an inadequate response or intolerance to methotrexate or other DMARDs. Upadacitinib may be used as monotherapy or in combination with methotrexate. | Oral | 15 mg by mouth, once daily, to a maximum of 30 mg by mouth | Serious infections (TB, invasive fungal infections, opportunistic infections), malignancies, thrombosis, and liver enzyme elevation |
Other | ||||
Abatacept | The treatment of adult patients with active PsA when the response to previous DMARD therapy has been inadequate; can be used with or without non-bDMARDs | SC, IV | 125 mg SC, once weekly without the need for an IV loading dose Weight-based IV dosing (500 mg to 1 g) given at week 0, week 2, and week 4, followed by 125 mg SC injections q.4.w. thereafter | Infections, COPD exacerbations |
bDMARD = biologic disease-modifying antirheumatic drug; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; COPD = chronic obstructive pulmonary disease; DMARD = disease-modifying antirheumatic drug; IBD = inflammatory bowel disease; IL-12/23 = interleukin-12 and interleukin-23; IL-17 = interleukin-17; IL-17A = interleukin-17A; IL-17A/F = interleukin-17A and interleukin-17F; IL-23 = interleukin-23; JAK = Janus kinase; PDE4 = phosphodiesterase type 4; PsA = psoriatic arthritis; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; RPLS = reversible posterior leukoencephalopathy syndrome; SC = subcutaneous; TB = tuberculosis; TNF = tumour necrosis factor.
aHealth Canada–approved indication.
Sources: Product monographs for bimekizumab, ixekizumab, secukinumab, ustekinumab, guselkumab, risankizumab, adalimumab, certolizumab pegol, etanercept, golimumab, infliximab, apremilast, tofacitinib, upadacitinib, and abatacept.2,50-63
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the 2 submissions provided by 7 patient groups. The full original patient input received by CADTH has been included in the Stakeholder section of this report.
Patient group input was submitted for this CADTH review by 7 different patient groups: 1 submission from Arthritis Consumer Experts and 1 joint input from the Canadian Psoriasis Network, Arthritis Society Canada, Canadian Arthritis Patient Alliance, Canadian Association of Psoriasis Patients, Canadian Spondyloarthritis Association, and CreakyJoints.
Arthritis Consumer Experts gathered information from patients via email on October 16, 2023 (n = 2), and through an online survey available from December 18, 2020, to January 26, 2021 (n = 5). The joint input obtained responses (n = 214) through an online survey that was available through the 6 collaborating organizations’ communication channels from September 27 to October 11, 2023. The latter survey was also sent to clinics in Canada that conducted bimekizumab trials for PsA to capture the experience of patients living with PsA or ankylosing spondylitis and the experiences of their caregivers.
Of the 214 respondents to the joint survey, 100 (47%) participants identified as living with PsA and 3 participants had experience taking bimekizumab. A total of 92 respondents described the many disease symptoms that are challenging to manage, including joint stiffness (95%), fatigue (87%), hip pain (64%), changes in fingernails and toes (61%), difficulties concentrating (58%), sore heels (50%), stress (44%), and anxiety (38%). Regarding the most significant impacts on their everyday HRQoL, respondents reported that PsA interfered with their physical activity (80%), sleep (75%), work (59%), social life (51%), mental health (50%), and intimacy and self-esteem (48%). Because the disease reduces their ability to participate in activities and affects their mental and social health, respondents indicated that caregivers have to take on additional tasks or daily chores and help patients get to and from medical appointments. Patients from the Arthritis Consumer Experts input indicated that pain and lack of mobility affect their daily activities and HRQoL.
Among the 85 joint survey respondents who shared their experience with currently available treatments, 32% found NSAIDs effective, 39% found DMARDs effective, 40% found biologic therapies effective (with 24% rating them as very effective), and 44% found steroids effective. According to both patient inputs, respondents indicated that efficacious treatments are costly, accessing them can be challenging, and adverse effects are very difficult to manage.
Among the 3 survey respondents in the joint input who had experience with bimekizumab for PsA, 1 respondent answered questions about the drug, indicating the treatment was easy to use and effective in improving HRQoL without experiencing adverse effects.
Patients seek treatments that improve disease symptoms, improve HRQoL, have fewer adverse effects, are easier to administer, are accessible, and are affordable.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by a clinical specialist with expertise in the diagnosis and management of PsA.
Unmet Needs
Despite there being various treatment options for managing PsA, the clinical expert stated that not all patients respond to available therapies or that treatments may be effective initially but can fail over time. The expert also noted that currently available treatments tend to be effective for treating skin disease but have variable or suboptimal efficacy with other domains, including musculoskeletal symptoms. Because it is rare to reverse existing joint damage, the expert highlighted the importance of early diagnosis and treatment before damage occurs. There is also concern over safety with all DMARDs, including an increased risk of infection and new onset or worsening of existing comorbidities. According to the clinical expert, few patients attain a state of low disease activity; therefore, it is important to have safe, well-tolerated treatments that are effective with all domains.
Place in Therapy
The clinical expert indicated that bimekizumab would be used after the failure of cDMARDs and, in accordance with its Health Canada indication, with or without a cDMARD. It was also noted that, without good quality evidence to support combination therapy, bimekizumab would not likely be used concomitantly with other bDMARDs or tsDMARDs at this time.
Patient Population
Although there are currently no biomarkers to indicate which patients would respond better to a particular drug, the expert was of the opinion that any patient with active PsA could receive bimekizumab and that those with coexisting severe psoriasis may also benefit. They added that the drug would be avoided in those who have IBD or severe uveitis, as this class of medications has not been very effective in these disease areas, and patients with active infection (especially fungal infection) would be cautioned against using bimekizumab. As per the expert, patients with an inadequate response to targeted DMARDs are most in need of new treatments.
Assessing the Response Treatment
According to the clinical expert, treatment response is typically assessed at 3 months based on improvements in TJC and SJC, enthesitis, dactylitis, and skin psoriasis, and sometimes using composite indices (e.g., MDA or DAPSA, which are used in clinical trials). The clinical expert added that improvements in physical function, pain, fatigue, and lack of radiographic progression (not typically used in clinical practice) may take longer than 3 months in patients with longstanding PsA. Moreover, due to the heterogeneity of the disease, response can differ across patients, though the clinical expert suggested that assessments are not likely to vary among rheumatologists.
Discontinuing Treatment
The clinical expert stated that lack of response in musculoskeletal or skin domains, disease relapse, intolerance, and patient choice are the most important factors when considering the discontinuation of bimekizumab. Moreover, it was explained that some amount of disease activity can be considered acceptable, but that recurrent infections and IBD would require discontinuation.
Prescribing Considerations
The expert explained that due to the heterogeneous nature of the disease, the lack of biomarkers, and the similarity of symptoms with other musculoskeletal conditions, diagnosis by primary care physicians and dermatologists can be challenging, and misdiagnosis is a possibility. Therefore, diagnosis, management, and treatment should be led by a rheumatologist trained in identifying inflammatory arthritis and can include a dermatologist to manage the associated psoriasis. Patients are typically treated in an outpatient setting in community clinics and clinics attached to community and academic hospitals, though severe skin and joint disease may require hospital admission.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by 1 clinician group. The full original clinician group input received by CADTH has been included in the Stakeholder section of this report.
One clinician group, the Canadian Rheumatology Association, provided input for this CADTH review (3 clinicians from the association contributed to the input).
According to the clinician group, PsA is a complex disease with varied manifestations. Current treatment regimens include nonpharmacologic therapies (such as graded exercise programs, occupational therapy, diet, weight loss, and smoking cessation) and pharmaceutical treatments (such as topical emollients, corticosteroids, vitamin D analogues, coal tar, phototherapy, nonbiologic DMARDs, PDE4i, and biologic therapies).
As per the clinician group, the goals of treatment are to maximize HRQoL by controlling disease symptoms and inflammation, improving function, and preventing structural damage. The clinician group highlighted unmet patient needs similar to those described by the clinical expert consulted by CADTH: some patients do not respond to treatment and treatments have variable effect on the PsA disease domains. The clinician group also noted that current biologic treatments are associated with a secondary loss of effect that can lead to dose creep, adverse effects, and the persistence of active extra-articular manifestations despite improvement in musculoskeletal symptoms.
The clinician group expressed that bimekizumab may be used for patients who have not experienced improvement as a result of treatment with cDMARDs and continue to have high measures of disease activity. Measures of treatment response were consistent with what the clinical expert consulted by CADTH outlined and included improvement in axial disease, and MDA. The clinician group also noted that composite measures, such as Composite Psoriatic Disease Activity Index, DAPSA, and Psoriatic Arthritis Disease Activity Score, can be used as well, though they are not practical for everyday use in clinics. For skin disease, PASI and percentage of BSA affected by psoriasis are also used. For axial disease activity, the most frequently used measure in Canada is the BASDAI.
The clinician group highlighted the same reasons for the discontinuation of therapy that were noted by the clinical expert. The group indicated that a specialist would be required to prescribe bimekizumab and monitor for AEs, but because the drug is administered as an SC injection, a patient could self-administer it.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
The most recent recommendation for a bDMARD for the treatment of PsA (guselkumab) has the following initiation criterion: eligibility for this drug should be based on the criteria used by each of the public drug plans for reimbursement of bDMARDs for the treatment of adult patients with active PsA. Should the initiation of therapy criteria for PsA biologic drugs and JAKis be applied to bimekizumab? | The clinical expert consulted by CADTH agreed that the same initiation criteria can be applied to bimekizumab. |
Is there a potential indication for bimekizumab to treat juvenile PsA? | The expert expects that, with adequate evidence from good quality trials in a younger population, bimekizumab could be a treatment for juvenile PsA. |
What is an adequate trial for other DMARDs before accessing bimekizumab (or other advanced PsA treatments)? | The clinical expert stated that, in general, an adequate trial of 3 months at the recommended therapeutic dose is necessary to determine if a patient is responding to a cDMARD, bDMARD, or tsDMARD before switching to or adding another therapy. |
Considerations for continuation or renewal of therapy | |
The most recent recommendation for a bDMARD for the treatment of PsA (guselkumab) has the following renewal criterion: this drug should be renewed in a similar manner to other bDMARDs currently reimbursed for the treatment of adult patients with active PsA. Should the continuation of therapy criteria for PsA biologic drugs and JAKis be applied to bimekizumab? | The clinical expert consulted by CADTH agreed that the same renewal criteria can be applied to bimekizumab. |
Considerations for discontinuation of therapy | |
The most recent recommendation for a bDMARD for the treatment of PsA (guselkumab) has the following discontinuation criterion: this drug should be discontinued in a similar manner to other bDMARDs currently reimbursed for the treatment of adult patients with active PsA. Should the discontinuation of therapy criteria for PsA biologic drugs and JAKis be applied to bimekizumab? | The clinical expert consulted by CADTH agreed that the same discontinuation criteria can be applied to bimekizumab. |
Considerations for prescribing of therapy | |
The most recent recommendation for a bDMARD for the treatment of PsA (guselkumab) has the following prescribing criterion: patients should be under the care of a rheumatologist or a clinician who has experience treating adult patients with active PsA. Should the same prescribing criteria for PsA biologic drugs and JAKis be applied to bimekizumab? If a rheumatologist is not accessible (e.g., in a remote area), which health care providers should be able to prescribe bimekizumab and manage patients receiving the drug? | The clinical expert consulted by CADTH agreed that the same prescribing criteria can be applied to bimekizumab. The expert indicated that a patient should be diagnosed, treated, and managed by a rheumatologist. In situations where a rheumatologist cannot be accessed, the expert suggested that acceptable specialists can include internists or dermatologists. |
The most recent recommendation for a bDMARD for the treatment of PsA (guselkumab) has the following combination criterion: this drug should not be reimbursed when used in combination with bDMARDs or tsDMARDs for active PsA. Should the same criteria for PsA biologic drugs and JAKis be applied to bimekizumab? Can bimekizumab be used concomitantly with other DMARDs (e.g., cDMARDs, bDMARDs, tsDMARDs)? | The clinical expert consulted by CADTH agreed that the same criteria can be applied to bimekizumab. The Health Canada indication for adults with active PsA states that bimekizumab can be used alone or in combination with a cDMARD (e.g., methotrexate).2 However, there is currently limited evidence supporting concomitant use of bimekizumab with bDMARDs or tsDMARDs for the treatment of PsA. |
If treatment with bimekizumab fails and a patient in switched to another treatment, would a patient ever be re-treated with bimekizumab? | The expert was of the opinion that, from a clinical practice standpoint, it would be possible to return to a drug that was previously used. |
bDMARD = biologic disease-modifying antirheumatic drug; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; DMARD = disease-modifying antirheumatic drug; JAKi = Janus kinase inhibitor; PsA = psoriatic arthritis; tsDMARD = targeted synthetic disease-modifying antirheumatic drug.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of bimekizumab on adult patients with active PsA, alone or in combination with a cDMARD. The focus will be placed on comparing bimekizumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor for the review of bimekizumab is presented in 4 sections with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence; however, no relevant studies were submitted.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
Two pivotal RCTs identified in the systematic review
Two long-term OLE studies
Two ITCs (1 NMA and 1 MAIC).
Systematic Review
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5. Figure 1 and Figure 2 show the study designs for the BE OPTIMAL and BE COMPLETE trials, respectively.
Two phase III, DB, placebo-controlled RCTs were included in the systematic review: the BE OPTIMAL trial (N = 852) and the BE COMPLETE trial (N = 400).19,20 The 2 trials were conducted to evaluate the efficacy and safety of bimekizumab in adults with active PsA. The BE OPTIMAL trial consisted of a 2-week to 5-week screening phase, a 16-week DB treatment period, a 36-week active treatment–blind period, and a 20-week safety follow-up phase. During the 36-week period, all patients who received placebo during the DB phase were reallocated to receive bimekizumab and patients in the active treatment groups continued their respective treatments. The BE COMPLETE trial was similarly designed, but without the 36-week active treatment–blind period. Following treatment completion, eligible patients from both trials could enrol in the BE VITAL OLE study.
Table 5: Details of Studies Included in Systematic Review
Detail | BE OPTIMAL study | BE COMPLETE study |
|---|---|---|
Designs and populations | ||
Study design | Phase III, multicentre, randomized, DB, placebo-controlled, active reference trial | Phase III, multicentre, randomized, DB, placebo-controlled trial |
Locations | 135 sites in Australia, Belgium, Canada (4 sites), Czechia, France, Germany, Hungary, Italy, Japan, Poland, Russia, Spain, UK, and US | 92 sites in Australia, Canada (3 sites), the Czech Republic, Germany, Hungary, Italy, Japan, Poland, Russia, UK, and US |
Patient enrolment dates | First patient enrolled: May 2, 2019 Final patient’s final visit: July 11, 2022 | First patient enrolled: March 28, 2019 Final patient’s final visit: February 14, 2022 |
Randomized (N) | N = 852 patients. The patients were randomized to receive:
| N = 400 patients. The patients were randomized to receive:
|
Inclusion criteria |
| |
|
| |
Exclusion criteria |
| |
Drugs | ||
Intervention |
|
|
Comparator(s) |
|
|
Study duration | ||
Screening phase | 2 weeks to 5 weeks before treatment | 2 weeks to 5 weeks before treatment |
Treatment phase | 16-week DB treatment period followed by a 36-week active treatment–blind period | 16-week DB treatment period |
Follow-up phase | 20 weeks after final dose of IMP Entry into BE VITAL OLE study was permitted | 20 weeks after final dose of IMP Entry into BE VITAL OLE study was permitted |
Outcomes | ||
Primary end point | ACR50 response at week 16 | ACR50 response at week 16 |
Secondary and exploratory end points | Secondary:
Exploratory:
| Secondary:
Exploratory:
|
Publication status | ||
Publications | ||
ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; BSA = body surface area; CASPAR = ClASsification criteria for Psoriatic ARthritis; CCP = cyclic citrullinated peptide; COX-2 = cyclooxygenase-2; DAS28(CRP) = Disease Activity Score 28 based on CRP; DB = double-blind; HAQ-DI = Health Assessment Questionnaire–Disability Index; HCQ = hydroxychloroquine; hs-CRP = high-sensitivity CRP; IBD = inflammatory bowel disease; IGA = Investigator’s Global Assessment; IMP = investigational medicinal product; LDI = Leeds Dactylitis Index; LEF = leflunomide; LEI = Leeds Enthesitis Index; MCS = mental component summary; MDA = minimal disease activity; MTX = methotrexate; NSAID = nonsteroidal anti-inflammatory drug; OLE = open-label extension; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; PCS = physical component summary; PsA = psoriatic arthritis; PsAID-12 = Psoriatic Arthritis Impact of Disease-12; PsAQoL = Psoriatic Arthritis Quality of Life instrument; PSO = psoriasis; PtAAP = Patient’s Assessment of Arthritis Pain; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RA = rheumatoid arthritis; SC = subcutaneous; SF-36 = Short Form (36) Health Survey; SJC = swollen joint count; SSZ = sulfasalazine; TJC = tender joint count; TNFi = tumour necrosis factor inhibitor; vdHmTSS = van der Heijde modified total Sharp score.
aAdalimumab was included as an active reference treatment but was not included in any statistical testing in the trial.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Figure 1: Study Design for the BE OPTIMAL Trial
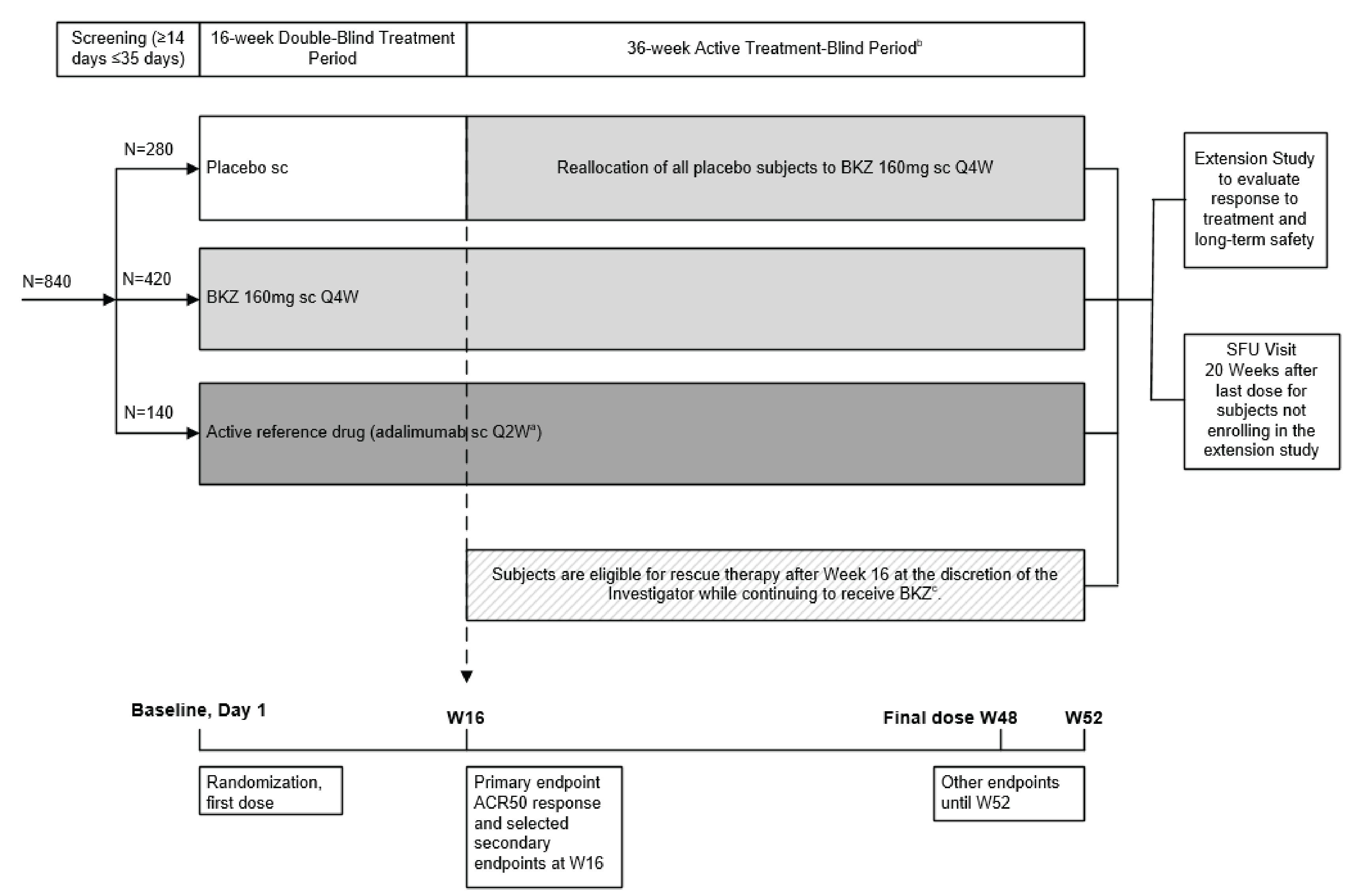
ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; BKZ = bimekizumab; DB = double-blind; OLE = open-label extension; Q2W = every 2 weeks; Q4W = every 4 weeks; sc = subcutaneous; SFU = safety follow-up; W = week.
a Adalimumab 40 mg sc Q2W.
b After 16 weeks of DB treatment, patients entered the active treatment–blind period. All patients randomized to placebo were reallocated to receive BKZ 160 mg Q4W. Patients randomized to BKZ or adalimumab continued to receive their respective treatments.
c Permitted rescue therapy as defined in the study.
Source: BE OPTIMAL Clinical Study Report.20
Populations
Inclusion and Exclusion Criteria
To be eligible for the trials, patients must have been 18 years or older with a documented diagnosis of adult-onset PsA and must have had at least 1 active plaque psoriatic lesion or a history of plaque psoriasis. Patients in the BE COMPLETE trial must have had a history of inadequate response (lack of efficacy after at least 3 months of treatment at an approved dose) or intolerance to 1 TNFi or 2 TNFis for PsA or psoriasis. Individuals were not eligible for either study if they had current or prior exposure to any biologic therapies (except TNFis in the BE COMPLETE trial) for the treatment of PsA or psoriasis, or for another inflammatory condition other than PsA or psoriasis, or had a form of psoriasis other than chronic plaque type.
Figure 2: Study Design for BE COMPLETE Trial
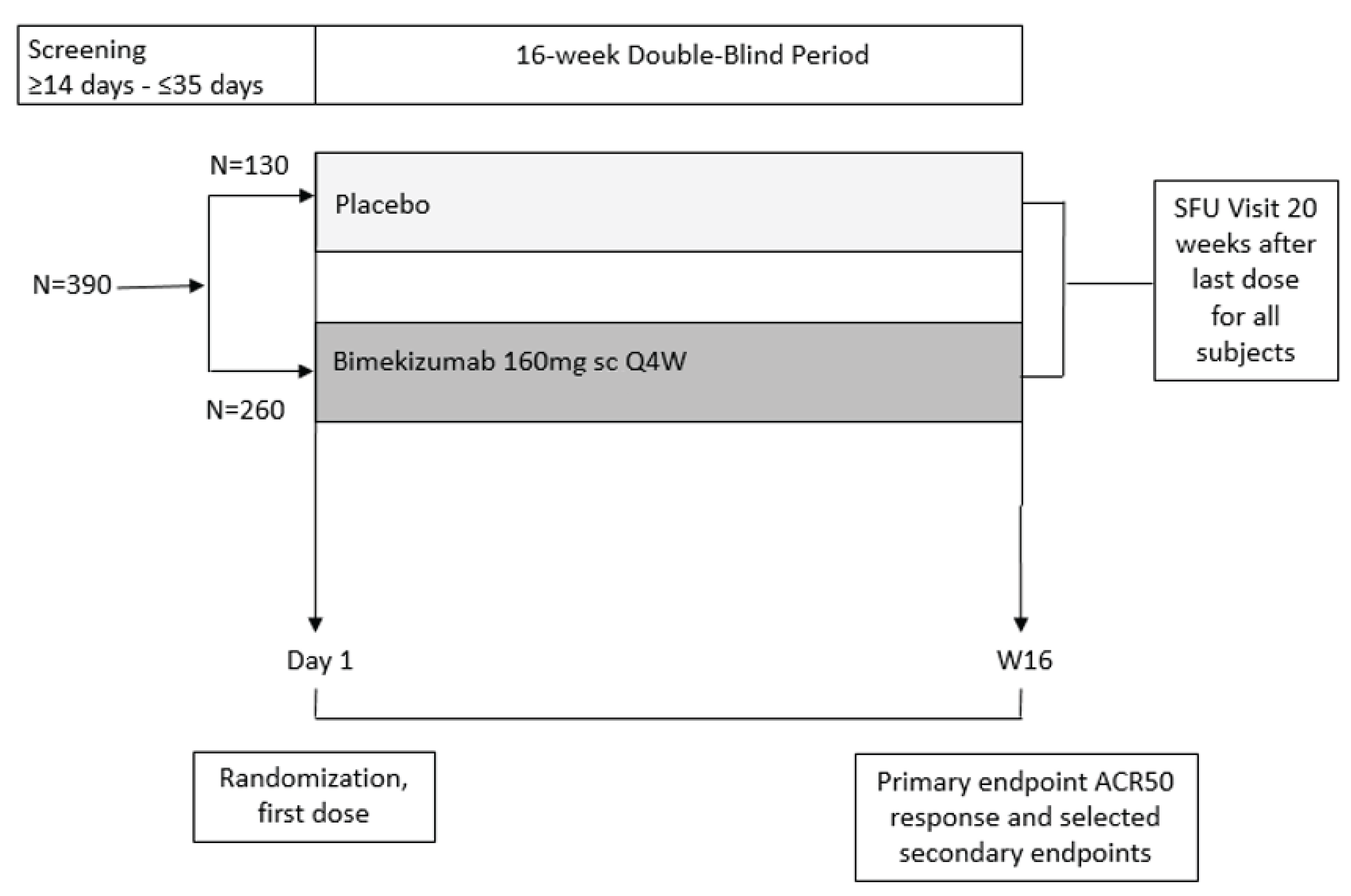
ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; DB = double-blind; OLE = open-label extension; Q4W = every 4 weeks; sc = subcutaneous; SFU = safety follow-up; W = week.
Source: BE COMPLETE Clinical Study Report.19
Interventions
Patients in the BE OPTIMAL trial were randomized 3:2:1 (stratified by region and bone erosion status) to receive bimekizumab 160 mg every 4 weeks SC via 1 mL injection (n = 431), matching 1 mL SC placebo (n = 281), or adalimumab 40 mg every 2 weeks SC via 0.8 mL or 0.4 mL injection (n = 140). Due to adalimumab being administered every 2 weeks, placebo every 4 weeks was also administered to the bimekizumab group on a staggered schedule to maintain the blinded treatment assignment. Adalimumab was included as an active reference treatment, but the trial was not powered for statistical comparisons between adalimumab and either bimekizumab or placebo and will not be further discussed in the direct evidence section of the CADTH review.
Patients in the BE COMPLETE trial were randomized 2:1 (stratified by region and TNFi exposure) to receive bimekizumab 160 mg every 4 weeks (n = 267) or matching placebo (n = 133). All administrations of the study drug were performed in the clinic by site staff.
Patients in both trials were permitted to continue NSAIDs, cyclooxygenase-2 (COX-2) inhibitors, oral corticosteroids, methotrexate, sulfasalazine, leflunomide, hydroxychloroquine, or apremilast for PsA if they were on a stable dose before baseline and during the study. Stable doses were required for at least 2 weeks for NSAIDs, COX-2 inhibitors, and oral corticosteroids (up to 10 mg per day prednisone or equivalent); for at least 8 weeks for methotrexate (up to 25 mg per week) and leflunomide (up to 20 mg per day); and for at least 4 weeks for sulfasalazine (up to 4 g per day), hydroxychloroquine (up to 400 mg per day), and apremilast (up to 60 mg per day). Changes in concomitant medications were not permitted — except for decreases in drug dose or frequency due to intolerance or AEs — during the first 16 weeks of the trials. Patients also could not use phototherapy, topical corticosteroids, vitamin D analogues, topical retinoids, keratolytics, coal tar, fumaric acid esters, or systemic retinoids for treatment of psoriasis during the first 16 weeks of the trials.
Rescue treatment was permitted after completion of the 16-week DB period in the BE OPTIMAL trial for patients who did not respond to treatment and patients continued to receive bimekizumab (either randomized to the treatment or crossed over from placebo after week 16) for the remainder of the trial. Rescue treatment included changes in a patient’s background therapy and consisted of NSAIDs, COX-2 inhibitors, methotrexate, sulfasalazine, leflunomide, hydroxychloroquine, apremilast, and up to 2 joint injections (for a total dose of 80 mg prednisone or equivalent, but not in the same joint). Doses were allowed up to the respective maximum permitted doses that were considered for stable treatment during the trial.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures in Table 7. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence24 as well as any outcomes identified as important to this review according to the clinical expert consulted by CADTH and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered to be most relevant to inform CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE.
The following were considerations that went into the selection of outcomes summarized in the report and assessed using GRADE:
the assessment of disease activity was an important clinical outcome according to the clinician group and clinical expert
response in the joints and evaluation of concurrent skin disease was important to patients and clinicians
improving HRQoL and symptoms were identified as being high priorities according to input provided by the patient group, clinician group, and clinical expert.
The primary end point for both trials was an ACR50 response at week 16. Relevant secondary end points for both trials included in the statistical testing hierarchy consisted of HAQ-DI, a PASI 90 response in patients with psoriasis involving at least 3% BSA at baseline, SF-36 PCS, and an MDA response. The BE OPTIMAL trial’s statistical testing hierarchy also included an enthesitis-free state based on LEI and a dactylitis-free state based on LDI in patients who had enthesitis or dactylitis at baseline, respectively, whose data were pooled from the BE OPTIMAL and BE COMPLETE trials. Other relevant secondary and exploratory outcomes in both trials included ACR20, ACR70, IGA, PtAAP, and SJC.
ACR Response
The ACR response rate is the percentage improvement relative to baseline (e.g., ACR50 is a 50% improvement in the ACR response) for a set of outcome measures.19,20 These include the TJC (based on 68 joints), the SJC (based on 66 joints), and at least 3 of the 5 remaining core measures: Patient’s Global Assessment of PsA, Physician’s Global Assessment of PsA, PtAAP, HAQ-DI, and high-sensitivity CRP.
Patient’s Global Assessment of Psoriatic Arthritis and Physician’s Global Assessment of Psoriatic Arthritis are evaluated using a 100-point visual analogue scale ranging from 0 (no symptoms) to 100 (severe symptoms).19,20 SJC, HAQ-DI, and PtAAP are described as follows.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | BE OPTIMAL study | BE COMPLETE study |
|---|---|---|---|
ACR50 response | At 16 weeks | Primarya | Primarya |
ACR20 response | At 16 weeks | Secondary | Secondary |
ACR70 response | At 16 weeks | Secondary | Secondary |
MDA response | At 16 weeks | Secondarya | Secondarya |
Enthesitis-free state based on LEI in patients with enthesitis at baseline in the pooled population of the BE OPTIMAL and BE COMPLETE trials | At 16 weeks | Secondarya | Secondary |
Dactylitis-free state based on LDI in patients with dactylitis at baseline in the pooled population of the BE OPTIMAL and BE COMPLETE trials | At 16 weeks | Secondarya | Secondary |
Change from baseline in SJC | At 16 weeks | Exploratory | Exploratory |
Reduction of PASI 90 in patients with psoriasis involving ≥ 3% BSA at baseline | At 16 weeks | Secondarya | Secondarya |
Proportion of patients with an IGA score of 0 or 1 and ≥ 2-grade reduction from baseline in patients with psoriasis involving ≥ 3% BSA at baseline | At 16 weeks | Secondary | Secondary |
Change from baseline HAQ-DI | At 16 weeks | Secondarya | Secondarya |
Change from baseline in SF-36 PCS | At 16 weeks | Secondarya | Secondarya |
Change from baseline in PtAAP | At 16 weeks | Secondary | Secondary |
ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; BSA = body surface area; HAQ-DI = Health Assessment Questionnaire–Disability Index; IGA = Investigator’s Global Assessment; LDI = Leeds Dactylitis Index; LEI = Leeds Enthesitis Index; MDA = minimal disease activity; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; PCS = physical component summary; PtAAP = Patient’s Assessment of Arthritis Pain; SF-36 = Short Form (36) Health Survey; SJC = swollen joint count.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchical testing).
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Minimal Disease Activity
MDA is a composite end point and is considered to be attained if at least 5 of the 7 criteria are reached: TJC of 0 or 1, SJC of 0 or 1, PASI of 1 or lower or affected BSA of 3% or less, a pain visual analogue scale score of 15 or lower, a Patient’s Global Assessment of Psoriatic Arthritis visual analogue scale score of 20 or lower, a HAQ-DI score of 0.5 or lower, and tender entheseal points of 0 or 1.19,20
Enthesitis-Free State Based on LEI
Enthesitis (inflammation where the tendon, ligament, or joint capsule meets bone) was measured using LEI.20 Assessments for enthesitis are performed by bilateral palpation on the lateral epicondyles of the humerus (elbows), medial femoral epicondyles (knees), and Achilles tendons (heels). Each location is scored as 0 (no pain) or 1 (pain) and ranges from 0 (no enthesitis) to 6 (severe enthesitis).
Dactylitis-Free State Based on LDI
Dactylitis (swelling of the entire digit related to articular and periarticular inflammation) was measured using LDI.20 The ratio of the circumference of the affected digit to the circumference of the digit on the contralateral hand or foot is measured and a minimum difference of 10% is used to define a dactylitic digit.68,69 If the contralateral digit is also affected, normative values based on population averages are used for comparison. The ratio is multiplied by the tenderness score (0 for absent and 1 for present), and the results from each digit with dactylitis are summed for a final score.
Swollen Joint Counts
SJC evaluation includes 6 joints of the upper body, 34 joints of the upper extremities, and 26 joints of the lower extremities for a total of 66 joints.19,20 Each joint is assessed using a 2-point scale: 0 for no swelling and 1 for swollen joints.
Psoriasis Area Severity Index
PASI assesses the severity and extent of psoriasis along with the percentage of BSA involved across 4 body areas (head [10%], upper limbs [20%], trunk [30%], and lower limbs [40%]).19,20 The redness, thickness, and scaliness of lesions for each area are evaluated using a 5-point scale from 0 (none) to 4 (very marked) for a total score ranging from 0 to 72 points, where higher scores indicate greater disease severity.
Investigator’s Global Assessment
IGA measures severity of psoriasis using a 5-point scale, where 0 is clear (no psoriasis, postinflammatory hyperpigmentation present), 1 is almost clear (no thickening, normal to pink colouration, no to minimal focal scaling), 2 is mild (detectable to mild thickening, pink to light red colouration, fine scaling), 3 is moderate (distinguishable to moderate thickening, dull to bright red colouration, moderate scaling), and 4 is severe (severe thickening with hard edges, bright to dark red colouration, severe and/or coarse scaling).70 In the pivotal trials, only patients with at least 3% BSA affected by psoriasis with an IGA score of at least 2 at baseline (i.e., at least mild psoriasis) were evaluated for this outcome.19,20 Treatment response was defined as a score of 0 or 1 (clear or almost clear) and at least a 2-category improvement from baseline.
Health Assessment Questionnaire–Disability Index
HAQ-DI consists of 20 items from 8 domains: dressing and grooming, arising, eating, walking, hygiene, reach, grip, and common daily activities.19,20 Each domain is rated on a 4-point scale from 0 (able to do without difficulty) to 3 (unable to do) and patients indicate if they typically use a mobility aid for the activity. The sum of the highest score in each domain (0 to 24) is divided by the number of domains in which at least 1 question is answered. The score is considered missing if fewer than 6 domains are completed. The overall score ranges from 0 to 3, with lower scores indicating better function. The MID from the literature is estimated to range from 0.13 to 0.35.71,72
SF-36 PCS
SF-36 is a generic HRQoL instrument consisting of 8 domains: physical functioning (10 items), role physical (4 items), bodily pain (2 items), general health (5 items), vitality (4 items), social functioning (2 items), role emotional (3 items), mental health (5 items), and 1 item for perceived stability or change in health (health transition) during the last year.19,20 Two summary scores (PCS and mental component summary) are calculated from the domains. Norm-based t scores for the summary and domain scores are standardized to a population (e.g., general US population) and higher scores indicate better health. The MID from the literature is estimated to be 3.74 for the PCS.73
Patient’s Assessment of Arthritis Pain
PtAAP is a 100-point visual analogue scale for patients to record their arthritis pain from 0 (no pain) to 100 (most severe pain).19,20 The MID from the literature is estimated to be a 10-point decrease from baseline.21
Safety
Safety was assessed by the proportion of patients who reported AEs (coded using Medical Dictionary for Regulatory Activities, version 19.0), SAEs, withdrawals from treatment due to AEs, and deaths during the DB treatment period and active treatment–blind period.19,20 Liver dysfunction based on Hy’s law, liver toxicity, opportunistic infection, fungal infection, candida infection, reactivation of tuberculosis infection, a major cardiovascular event, malignancy, a serious injection-related AE, anaphylaxis, and IBD were notable harms included in the CADTH review.
Statistical Analysis
The statistical analysis methods are summarized in Table 8.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
ACR20, ACR50, and ACR70 | The ACR20, ACR50, and ACR70 response rates represent an improvement of ≥ 20%, ≥ 50%, and ≥ 70%, respectively, in tender and swollen joint counts and in 3 of 5 additional criteria: patient global assessment of disease activity; physician global assessment of disease activity; patient assessment of pain; HAQ; CRP or ESR.19,20,74 | Validity: Content validity was acceptable and tender and swollen joint counts have been deemed appropriate for disease activity for patients with rheumatoid arthritis by the ACR expert committee.75 Construct validity was assessed using the known-group method in a study of patients with PsA and peripheral arthritis using data from 2 RCTs (n = 164) of TNFis. The number of patients who attained ACR20, ACR50, and ACR70 in both active drug groups was significantly higher than in the placebo groups.76 Reliability: Data are lacking for ACR response. Responsiveness: In patients with PsA and peripheral arthritis from 2 RCTs of TNFis (n = 164), the SRM ranged from –1.98 to –0.61 and the ES ranged from –2.84 to –0.57 for the active drug groups while the SRM ranged from –0.40 to 0.02 and the ES ranged from –0.37 to 0.15 in the placebo groups.76 | ACR20 is generally used to define improvement, indicating a response to treatment.76-78 An MID for ACR has not been estimated in patients with PsA. |
MDA | A composite outcome measure developed as a target of treatment for patients with PsA that encompasses the different aspects of disease domains | Validity: The kappa coefficient between MDA and patients’ rating of whether they were in a minimal disease state was 0.30.79 Data on reliability and responsiveness are lacking. | An MID for MDA has not been estimated in patients with PsA. |
LEI | An enthesitis index designed for use in PsA RCTs assessing lateral humeral epicondyles (elbows), medial femoral epicondyles (knees), and Achilles tendons (heels) bilaterally and scored as 0 (no pain) and 1 (painful).80 | Validity: The correlation results from Healy and Helliwell suggest that the associations are very robust for LEI.80 Reliability: Data are lacking for LEI. Responsiveness: The LEI index showed a large ES at 6 months and significant response to change.80 | An MID for LEI has not been estimated in patients with PsA. |
LDI | LDI evaluates for at least a 10% difference in the circumference of the digit compared to the opposite digit. | Measurement properties not assessed in patients with PsA | An MID for LDI has not been estimated in patients with PsA. |
PASI 75, PASI 90, and PASI 100 | The PASI is a disease-specific composite severity index based on an average score of erythema, scaling, and thickness of psoriatic lesions, weighted by the area of involvement on a 5-point scale of involvement (0 = none, 1 = slight, 2 = moderate, 3 = marked, and 4 = very marked). The total PASI ranges from 0 to 72 points, where a higher score indicates greater disease severity.19,20 | Validity: Construct validity was demonstrated through correlation of the PASI and DLQI scores (0.36 ≤ r ≤ 0.54) using data from an RCT (n = 445) of calcineurin inhibitors in patients with psoriasis.81 There was a correlation between the PASI and the LS-IGA and IGA (Spearman rank correlation coefficient of 0.92 and 0.73, respectively) in 1 study of 16 patients with plaque psoriasis.82 A study of 9 adult patients with plaque-type psoriasis assessed the correlations of the PASI with other commonly used instruments in psoriasis, including BSA and IGA, and reported a strong correlation with both measures (Pearson correlation coefficient > 0.78 and > 0.61, respectively).83 Content validity was assessed using the relative impact of the individual components of the measures on HRQoL. BSA was most consistently associated with DLQI scores, followed by plaque induration and erythema, while the scaling score was found to be minimally and inconsistently associated with DLQI scores.81 Reliability: Excellent intrarater and inter-rater reliability for the PASI have been observed in studies of patients with plaque psoriasis or psoriasis (ICC ranged from 0.72 to 0.97).82-85 Responsiveness: Based on data from a systematic review, the PASI score was found to have moderate sensitivity to change in psoriasis patients (ICC ranged from 0.5 to 0.8).86 The validity, reliability, and responsiveness of the PASI instrument have not been evaluated in patients with PsA. | Based on a systematic review of 13 RCTs evaluating biologics in psoriasis, a ≥ 75% reduction in the PASI score translates to clinically significant HRQoL improvement in patients assessed using the DLQI.87 An MID for the PASI has not been estimated in patients with PsA. |
HAQ-DI | The disability assessment component of the HAQ, a self-reported assessment of functional status.88 It contains 20 items divided into 8 domains: dressing and grooming, arising, eating, walking, hygiene, reach, grip, and common daily activities. Patients indicate the degree of difficulty they experienced in each domain on a 4-point scale that ranges from 0 (without difficulty) to 3 (unable to do), and specify whether they usually use an aid or device for each activity.19,20 | Validity: Convergent validity was assessed in a systematic review of 31 RCTs; strong to weak correlations were observed between HAQ-DI and PCS12, PsAID-FC, patient global assessments for arthritis, pain, tender and swollen joint counts, DAPSA, and patient global assessment for skin (rho ranged from 0.19 to 78).89,90 Reliability: Data from a longitudinal study in 14 countries of 414 patients with PsA assessed internal consistency; the Cronbach alpha for HAQ-DI was 0.92.89,90 HAQ-DI demonstrated almost perfect test-retest reliability in patients with PsA (ICC ranged from 0.90 to 0.94).91 Responsiveness: In a systematic review of 31 RCTs of patients with PsA, moderate ESs (Cohen’s d) were seen in HAQ-DI, PCS12, and PsAID-FC in the measurement of worsening (HAQ-DI = 0.46 [95% CI, 0.12 to 0.78]; PCS12 = −0.57 [95% CI, −0.92 to −0.24]; and PsAID-FC = 0.51 [95% CI, 0.16 to 0.87]). The SRM for worsening for HAQ-DI, PCS12, and PsAID-FC was 0.37 (95% CI, 0.10 to 0.61), −0.45 (95% CI, −0.74 to −0.19), and 0.38 (95% CI, 0.12 to 0.66), respectively.89 | In 1 systematic review and 3 unique studies, MID estimates for HAQ-DI ranged from −0.35 to −0.13 for improvement, and 0.13 to 0.30 for worsening in patients with PsA.71,72,89,90 The MIDs were estimated using the anchor-based approach. Reported MIDs for the HAQ-DI ranged between 0.13 and 0.35.71,72 |
SF-36 | A 36-item general health status instrument consisting of 8 domains — physical functioning (10 items), role physical (4 items), bodily pain (2 items), general health (5 items), vitality (4 items), social functioning (2 items), role emotional (3 items), and mental health (5 items) — and 1 item for perceived stability or change in health (health transition) during the last year. In addition to domain scores, the PCS and MCS scores are also calculated from the 8 domains.92 All domain and component scores are based on a scale of 0 to 100, with higher scores indicating higher health status.93,94 The norm-based t scores for the SF-36 PCS, MCS, and 8 domains are standardized with a mean of 50 and an SD of 10 in the general US population; scores outside the range of 45 to 55 are considered abnormal for the US general population.92 | Validity: In 1 study of 168 patients with PsA, construct validity was assessed using the known-group approach; patients with PsA were grouped under severe disease based on HAQ > 1.0, BASDAI > 50, and DAS28 (in 28 joints) > 5.1. All 8 scales and summary scores in the severe groups were significantly worse than the less severe groups.73,95 Reliability: With regard to internal consistency, substantial to moderate levels of agreement were observed for most of the SF-36 scales (the Cronbach alpha ranged from 0.33 to 0.70) in patients with PsA.73,95 SF-36 physical function domain demonstrated almost perfect test-retest reliability in patients with PsA (ICC = 0.96 [95% CI, 0.92 to 0.98]).91 Responsiveness: In a systematic review of 31 RCTs, the median ES was 0.77 (95% CI, 0.60 to 0.93) and 0.23 (95% CI, 0.09 to 0.36) for intervention and control groups in patients with PsA, respectively.89 | MIDs for either the PCS or MCS are typically between 2.5 points and 5 points.96-98 In another study of 20 patients with PsA, the MIDs were estimated as 3.74 points and 1.77 points for the PCS and MCS subsections, respectively.73 |
PtAAP | For the PtAAP using VAS or “pain VAS,” patients assess their arthritis pain using a VAS where 0 is “no pain” and 100 is “most severe pain.”19,20 The patient’s assessment of pain is part of the ACR core set of measures in arthritis.75 | PtAAP items have been relatively understudied in PsA. Data on reliability and validity are lacking.99-101 | The reported MID for the PtAAP is a 10-point decrease from baseline.21 |
ACR = American College of Rheumatology; ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BSA = body surface area; CI = confidence; DAPSA = Disease Activity index in PSoriatic Arthritis; DAS28 = Disease Activity Score 28; DLQI = Dermatology Life Quality Index; ES = effect size; ESR = erythrocyte sedimentation rate; HAQ = Health Assessment Questionnaire; HAQ-DI = Health Assessment Questionnaire–Disability Index; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; IGA = Investigator’s Global Assessment; LDI = Leeds Dactylitis Index; LEI = Leeds Enthesitis Index; LS-IGA = Lattice System Investigator’s Global Assessment; MCS = mental component summary; MDA = minimal disease activity; MID = minimal important difference; PASI = Psoriasis Area Severity Index; PASI 75 = 75% reduction in Psoriasis Area Severity Index score; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; PASI 100 = 100% reduction in Psoriasis Area Severity Index score; PCS = physical component summary; PCS12 = physical component summary score of the 12-Item Short Form Survey; PsA = psoriasis arthritis; PsAID-FC = Psoriatic Arthritis Impact of Disease instrument functional capacity score; PtAAP = Patient’s Assessment of Arthritis Pain; RCT = randomized controlled trial; SD = standard deviation; SF-36 = Short Form (36) Health Survey; SRM = standardized response mean; TNFi = tumour necrosis factor inhibitor; VAS = visual analogue scale.
Table 8: Statistical Analysis of Efficacy End Points for the BE OPTIMAL and BE COMPLETE Trials
End point | Statistical model | Adjustment factor | Handling of missing data | Sensitivity analysis |
|---|---|---|---|---|
BE OPTIMAL and BE COMPLETE trials | ||||
ACR50 response at week 16 | Logistic regression model Comparison of bimekizumab vs. placebo made using the 2-sided Wald test (alpha of 0.05) | Region (both trials) and bone erosion (the BE OPTIMAL trial) or prior TNFi exposure (the BE COMPLETE trial) | NRI: missing data at week 16 that were not preceded by an intercurrent event (i.e., treatment discontinuation), and any data after an intercurrent event, were imputed as nonresponders | Evaluation in the PP set and FAS (only if the FAS and RS had different numbers of patients) |
Change from baseline in HAQ-DI at week 16a | ANCOVA | Treatment, region, bone erosion at baseline, and baseline value | MI (MCMC, monotone regression): missing data at week 16 that were not preceded by an intercurrent event (i.e., treatment discontinuation) were imputed. | None |
PASI 90 at week 16 in patients with psoriasis involving ≥ 3% of BSA at baselinea | Logistic regression model Comparison of bimekizumab vs. placebo made using the 2-sided Wald test (alpha of 0.05) | The same as for the primary end point | NRI as for the primary end point | None |
Change from baseline in SF-36 PCS at week 16a | ANCOVA | The same as for continuous secondary end points | MI (MCMC, monotone regression) as for continuous secondary end points | None |
MDA response at week 16a | Logistic regression model Comparison of bimekizumab vs. placebo made using the 2-sided Wald test (alpha of 0.05) | The same as for the primary end point | NRI as for the primary end point | None |
Enthesitis-free state (LEI) at week 16 in patients with enthesitis at baseline in the pooled population of the BE OPTIMAL and BE COMPLETE trialsb | Logistic regression model Comparison of bimekizumab vs. placebo made using the 2-sided Wald test (alpha of 0.05) | The same as for the primary end point | NRI as for the primary end point In cases where ≥ 4 items were missing, LEI was set to missing. If ≥ 3 items were available, the available items were assessed and scored and then weighted by the number of the assessed items. | None |
Dactylitis-free state (LDI) at week 16 in patients with dactylitis at baseline in the pooled population of the BE OPTIMAL and BE COMPLETE trialsb | Logistic regression model Comparison of bimekizumab vs. placebo made using the 2-sided Wald test (alpha of 0.05) | The same as for the primary end point | NRI as for the primary end point | An analysis based on the population with dactylitis at baseline only from the BE OPTIMAL trial |
ACR20 and ACR70 responses at week 16c | Logistic regression model Comparison of bimekizumab vs. placebo made using the 2-sided Wald test (alpha of 0.05) | The same as for the primary end point | NRI as for the primary end point | None |
Proportion of patients with an IGA of 0 or 1 and a ≥ 2-grade reduction from baseline in patients with psoriatic skin lesions at baselinec | Logistic regression model Comparison of bimekizumab vs. placebo made using the 2-sided Wald test (alpha of 0.05) | The same as for the primary end point | NRI as for the primary end point | None |
Change from baseline in PtAAP | ANCOVA | The same as for continuous secondary end points | MI (MCMC, monotone regression) as for continuous secondary end points | None |
Change from baseline in SJC | ANCOVA | The same as for continuous secondary end points | MI (MCMC, monotone regression) as for continuous secondary end points | None |
ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; ANCOVA = analysis of covariance; BSA = body surface area; FAS = full analysis set; HAQ-DI = Health Assessment Questionnaire–Disability Index; IGA = Investigator’s Global Assessment; LDI = Leeds Dactylitis Index; LEI = Leeds Enthesitis Index; MCMC = Markov chain Monte Carlo; MDA = minimal disease activity; MI = multiple imputation; NRI = nonresponder imputation; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; PCS = physical component summary; PP = per-protocol; PtAAP = Patient’s Assessment of Arthritis Pain; RS = randomized set; SF-36 = Short Form (36) Health Survey; SJC = swollen joint count; TNFi = tumour necrosis factor inhibitor; vs. = versus.
aSequentially tested in the BE OPTIMAL and BE COMPLETE trials.
bSequentially tested in the BE OPTIMAL trial only.
cDescriptive analyses in the BE OPTIMAL and BE COMPLETE trials.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sample Size and Power Calculation
The sample size to demonstrate statistical superiority of bimekizumab over placebo used a 2-sided 2-sample chi-square test with continuity correction.102 In the BE OPTIMAL trial, a sample of 420 patients in the bimekizumab group and 280 patients in the placebo group provided more than 99% power to demonstrate superiority of bimekizumab in the primary end point (ACR50 response at week 16) based on a true treatment difference of 27.8% (odds ratio [OR] = 4.09). The placebo response rate in the primary end point was assumed to be 16%, based on data from the BE ACTIVE study (6.1% at week 12, biologic therapy–naive population),26 the FUTURE 2 study (15.9%, biologic therapy–naive population),103 the FUTURE 3 study (11.8%, biologic therapy–naive population),104 and the FUTURE 5 study (8.1% at week 16, mixed tumour necrosis factor exposure population).105 In the BE COMPLETE trial, a sample of 260 patients in the bimekizumab group and 130 patients in the placebo group provided 96% power to demonstrate the superiority of bimekizumab in the primary end point (ACR50 response at week 16) based on a true treatment difference of 16% (OR = 3.16). The placebo response rate in the primary end point was assumed to be 10%, based on data from the BE ACTIVE study (11.1% at week 12, population whose disease responded inadequately to TNFi)26 and the SPIRIT-P2 study (< 10% at week 16, population whose disease responded inadequately to TNFi).106
Statistical Testing
Statistical tests of efficacy results were calculated using 2-sided P values (alpha of 0.05). The primary end point in the BE OPTIMAL trial was adjusted for, using 2 stratification variables: region (North America, Western Europe, Eastern Europe, and Asia or North America, Eastern Europe, and Western Europe with Asia if less than 10% of patients was randomized in either the Asia or Western Europe regions) and bone erosion (0 or at least 1) at baseline. The primary end point in the BE COMPLETE trial was similarly adjusted for, using 2 stratification variables: region (the same as previously listed) and prior TNFi exposure (inadequate response to 1 or 2 prior TNFis, or intolerance to TNFis). Stratification variable suitability was assessed using the Pearson and Deviance and Hosmer-Lemeshow goodness-of-fit tests,107 where if the logistic regression model was unable to converse, the stratification variables were dropped from both the primary analyses and all supportive analysis and imputed models.
A sequential testing procedure was used to control the familywise type I error rate at an alpha of 0.05 (2-sided) for the primary end point and some secondary end points in both trials. End points were tested as outlined in Table 9. Statistical testing of an end point occurred only if the null hypothesis for the previous end point was rejected (i.e., bimekizumab was superior to placebo, P < 0.05).
Table 9: Sequential Testing Procedure for the BE OPTIMAL and BE COMPLETE Trials
End point | BE OPTIMAL study | BE COMPLETE study |
|---|---|---|
ACR50 response at week 16 | 1 | 1 |
Change from baseline HAQ-DI at week 16 | 2 | 2 |
Reduction of PASI 90 in patients with psoriasis involving ≥ 3% of BSA at baseline at week 16 | 3 | 3 |
Change from baseline in SF-36 PCS at week 16 | 4 | 4 |
MDA response at week 16 | 5 | 5 |
Change from baseline in vdHmTSS in patients with elevated hs-CRP and/or ≥ 1 bone erosion at baseline at week 16 | 6 | — |
Enthesitis-free state based on LEI in patients with enthesitis at baseline in the pooled population of the BE OPTIMAL and BE COMPLETE trials at week 16 | 7 | — |
Dactylitis-free state based on LDI in patients with dactylitis at baseline in the pooled population of the BE OPTIMAL and BE COMPLETE trials at week 16 | 8 | — |
Change from baseline in vdHmTSS at week 16 | 9 | — |
ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; BSA = body surface area; HAQ-DI = Health Assessment Questionnaire–Disability Index; hs-CRP = high-sensitivity CRP; LDI = Leeds Dactylitis Index; LEI = Leeds Enthesitis Index; MDA = minimal disease activity; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; PCS = physical component summary; SF-36 = Short Form (36) Health Survey; vdHmTSS = van der Heijde modified total Sharp score.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Data Imputation Methods
Both trials used nonresponder imputation (NRI) for missing primary end point data at week 16. Patients were considered nonresponders if they had missing data at week 16, had discontinued treatment before week 16 but remained in the trial, or were missing the relevant baseline value. For ACR50, nonresponse was imputed based on available ACR components when the value could not be derived (i.e., was missing).
Where multiple imputation methods were used, a missing value was replaced with a set of plausible values. Each value was a Bayesian draw from the conditional distribution of the missing data given the observed data. The Markov chain Monte Carlo method was used to impute intermittent missing data, then regression for monotone missing data. It was assumed that data were missing at random.
Reference-based multiple imputation methods were used for outcomes in the testing hierarchy and the ACR response components. Missing data were imputed based on data from only the placebo group.108 It was assumed that the statistical behaviour of patients who discontinued treatment (from either group) was the same as those in the placebo group. Data for all time points after treatment discontinuation were considered missing. Multiple imputation methods involved multiple draws from the posterior predictive distribution estimated from the placebo group.
Subgroup Analyses
Prespecified subgroup analyses were performed on the ACR50, HAQ-DI, and PASI 90 end points at week 16, and were not adjusted for multiplicity in the trials. Most subgroups were assessed at baseline, except concomitant cDMARDs, concomitant methotrexate, and antibimekizumab antibody status, which were assessed during the 16-week DB treatment period. A logistic regression was fitted using the same terms that were retained when running the primary analysis model, plus a term for the subgroup and the subgroup by treatment interaction for each analysis, except for analyses by region, by bone erosion at baseline, by high-sensitivity CRP value combined with bone erosion at baseline, and by antidrug antibody status. Of the subgroups assessed in the trials, prior treatment TNFi exposure (TNFi intolerance, inadequate response to at least 1 TNFi, or inadequate response to at least 2 TNFis), which was conducted in the BE COMPLETE trial only, was identified as relevant to the CADTH review.
Sensitivity Analyses
In the trials, sensitivity analyses included using the per-protocol set (to evaluate the impact of important protocol deviations), using the full analysis set (to evaluate the consistency between the randomized set and full analysis set if the populations differed), and assessing the individual ACR components.
Secondary Outcomes of the Studies
Efficacy results for the BE OPTIMAL trial were collected at baseline and week 2, week 4, week 8, week 12, week 16, week 20, week 24, week 28, week 32, week 36, week 44, and week 52, while safety was assessed at every study visit (i.e., every 2 weeks). Study visits for the BE COMPLETE trial took place every 4 weeks.
Power calculation assumptions for secondary end points in the statistical testing hierarchy were based on results from the BE ACTIVE,26 FUTURE 1,109 FUTURE 2,103 FUTURE 5,105 and SPIRIT-P1110 studies. Power calculations were based on the 2-sided 2-sample chi-square test with continuity correction (binary end points)102 or a 2-sided 2-group Satterthwaite t test (continuous end points).111
The BE OPTIMAL trial had more than 99% power to detect a difference of –0.24 for the mean change from baseline in HAQ-DI at week 16, more than 99% power to detect a difference for PASI 90 response at week 16 in patients with at least 3% BSA affected by psoriasis, more than 99% power to detect a difference in means of 5.83 for the change from baseline in SF-36 PCS at week 16, more than 99% power to detect an absolute difference of 32% (OR = 5.17) for MDA response at week 16, 69% power to detect a true treatment difference of 16% (OR = 1.91) for an enthesitis-free state at week 16, and 66% power to detect a true treatment difference of 24% (OR = 2.71) for a dactylitis-free state at week 16.
The BE COMPLETE trial had 85% power to detect a difference of 0.23 in the mean change from baseline in HAQ-DI at week 16, 99% power to detect a treatment difference of 28% (OR = 5.52) for PASI 90 response at week 16 in patients with at least 3% BSA affected by psoriasis, 89% power to detect a difference in means of 4.4 in the change from baseline in SF-36 PCS at week 16, and 92% power to detect an absolute difference of 12% (OR = 3.892) for MDA response at week 16.
As with the primary efficacy end point, secondary binary end points used the same estimand structure, with a nonresponder approach to handling missing data and the same analysis model. Secondary continuous end points used an analysis of covariance model with treatment, region, and bone erosion at baseline as fixed effects and the baseline value as covariate, and missing data were imputed using the Markov chain Monte Carlo (monotone regression) technique.
Analysis Populations
The analysis populations of the included studies are summarized in Table 10.
Results
Patient Disposition
The patient disposition of the included studies is summarized in Table 11.
For the BE OPTIMAL trial, 1,158 individuals were screened, of whom 306 were screened out mostly due to eligibility criteria. Subsequently, 431 patients were randomized to receive bimekizumab and 281 patients were randomized to receive placebo. More than 96% of patients in either group completed the 16-week DB treatment period; the most common reasons for discontinuing were due to AEs (1.9%) in the bimekizumab group and patient choice (1.4%) in the placebo group. Following the DB period, 414 patients continued to receive bimekizumab in the 36-week active treatment–blind period and 271 patients were reallocated from placebo to bimekizumab. More than 92% of patients in the treatment groups completed the active treatment–blind period, where the most common reason for discontinuing was due to AEs (2.2%). Finally, 379 patients and 254 patients originally randomized to bimekizumab and placebo, respectively, entered the BE VITAL OLE study.
For the BE COMPLETE trial, 556 individuals were screened, of whom 156 were screened out mostly due to ineligibility. Afterward, 267 patients were randomized to receive bimekizumab and 133 patients were randomized to receive placebo. Of these patients, 98.5% of those in the bimekizumab group and 94.0% of those in the placebo group completed the DB treatment period. The most common reason for discontinuing was due to AEs (0.7%) in the bimekizumab group and patient choice (3.0%) in the placebo group. Finally, 256 patients and 122 patients from the bimekizumab and placebo groups, respectively, entered the BE VITAL OLE study.
Table 10: Analysis Populations of the BE OPTIMAL and BE COMPLETE Trials
Study | Population | Definition | Application |
|---|---|---|---|
BE OPTIMAL and BE COMPLETE trials | RS | Enrolled patients who were randomized | Most efficacy analyses |
FAS | All randomized patients who received ≥ 1 dose of IMP and had valid measurements of all the components of the primary efficacy variable at baseline | Supportive analysis of the primary efficacy variable | |
PP | All patients in the RS who had no important protocol deviations or nonprotocol deviations related to prohibited medications affecting the primary efficacy variable (i.e., patients were excluded from the PP set if they had deviations related to prohibited medications observed before week 16) | Supportive analysis of the primary efficacy variable | |
SS | All patients who received ≥ 1 dose of IMP | Demographic tables, study treatment compliance, exposure, and safety variables | |
BE OPTIMAL trial only | Active treatment–blind set | All patients who received ≥ 1 dose of active IMP (bimekizumab or adalimumab) during the active treatment–blind period (week 16 and later) | Safety summaries and select efficacy end points in the active treatment–blind period |
FAS = full analysis set; IMP = investigational medicinal product; PP = per-protocol; RS = randomized set; SS = safety set.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 11: Summary of Patient Disposition From the BE OPTIMAL and BE COMPLETE Trials
Patient disposition | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo | Bimekizumab | Placebo | Bimekizumab | |
Screened, N | 1,158 | 556 | ||
Screening failure, n | 306 | 156 | ||
Ineligibility | 247 | 124 | ||
Withdrawal of consent | 30 | 6 | ||
Lost to follow-up | 4 | 2 | ||
Other | 25 | 24 | ||
Randomized, N (%) | 281 (100) | 431 (100) | 133 (100) | 267 (100) |
Completed DB treatment period, n (%) | 271 (96.4) | 414 (96.1) | 125 (94.0) | 263 (98.5) |
Discontinued DB treatment period, n (%) | 10 (3.6) | 16 (3.7) | 8 (6.0) | 4 (1.5) |
Adverse event | 2 (0.7) | 8 (1.9) | 0 (0.0) | 2 (0.7) |
Withdrawal by patient | 4 (1.4) | 6 (1.4) | 4 (3.0) | 1 (0.4) |
Lack of efficacy | 2 (0.7) | 0 (0.0) | 2 (1.5) | 1 (0.4) |
Lost to follow-up | 2 (0.7) | 0 (0.0) | 1 (0.8) | 0 (0.0) |
Other | 0 (0.0) | 2 (0.5) | 1 (0.8) | 0 (0.0) |
Started active treatment–blind period, N (%)a | 271 (100) | 414 (100) | — | — |
Completed active treatment–blind period, n (%) | 255 (94.1) | 383 (92.5) | — | — |
Discontinued active treatment–blind period, n (%) | 14 (5.2) | 27 (6.5) | — | — |
Adverse event | 6 (2.2) | 9 (2.2) | — | — |
Withdrawal by patient | 4 (1.5) | 9 (2.2) | — | — |
Lack of efficacy | 1 (0.4) | 6 (1.4) | — | — |
Lost to follow-up | 1 (0.4) | 3 (0.7) | — | — |
Other | 2 (0.7) | 0 (0.0) | — | — |
Entered OLE, n (%) | 254 (93.7) | 379 (91.5) | 122 (91.7) | 256 (95.9) |
FAS, n (%) | 280 (99.6) | 426 (98.8) | 132 (99.2) | 267 (100) |
PP set, n (%) | 265 (94.3) | 409 (94.9) | 125 (94.0) | 255 (95.5) |
SS, n (%) | 281 (100) | 431 (100) | 132 (99.2) | 267 (100) |
AMS, n (%) | 271 (96.4) | 431 (100) | — | — |
ATS, n (%) | 271 (96.4) | 414 (96.1) | — | — |
AMS = active medication set; ATS = active treatment–blind set; DB = double-blind; FAS = full analysis set; OLE = open-label extension; PP = per-protocol; q.4.w. = every 4 weeks; RS = randomized set; SS = safety set.
aPatients were summarized according to randomized treatment at baseline in the DB treatment period. Patients who were randomized to placebo switched to bimekizumab 160 mg q.4.w. after week 16.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics summarized in Table 12 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
In the BE OPTIMAL trial, the mean age of patients ranged from 48.5 (SD = 12.6) years to 48.7 (SD = 11.7) years and demographic characteristics were generally similar between the bimekizumab and placebo groups. There were 5 patients and 4 patients in the bimekizumab and placebo groups, respectively, from Canada. The mean time since diagnosis of PsA was approximately 6 years and the mean time since diagnosis of psoriasis was approximately 15 years. Half of patients had at least 3% BSA affected by psoriasis and nearly 80% of patients reported prior use of cDMARDs. Baseline clinical characteristics were generally balanced between the 2 treatment groups, except for enthesitis being higher in the bimekizumab group (33.2%) than in the placebo group (24.9%).
In the BE COMPLETE trial, the mean age of patients ranged from 50.1 (SD = 12.4) years to 51.3 (SD = 12.9) years and demographic characteristics were generally similar between the bimekizumab and placebo groups. There were 6 patients and 3 patients in the bimekizumab and placebo groups, respectively, from Canada. The mean time since diagnosis of PsA was more than 9 years and the mean time since diagnosis of psoriasis was more than 17 years across the treatment groups. More than 76% of patients had an inadequate response to at least 1 TNFi, more than 11% of patients had an inadequate response to at least 2 TNFis, and approximately 12% of patients had an intolerance to TNFis. More than 65% of patients had at least 3% of their BSA affected by psoriasis and more patients had a higher PASI in the bimekizumab group. Baseline clinical characteristics were imbalanced between the groups for enthesitis (higher in the bimekizumab group, 39.7%, versus the placebo group, 27.1%), NSAID therapy (lower in the bimekizumab group, 53.6%, versus the placebo group, 60.2%), and methotrexate use (higher in the bimekizumab group, 44.6%, versus the placebo group, 38.3%).
Table 12: Summary of Baseline Characteristics From the BE OPTIMAL and BE COMPLETE Trials (Randomized Set)
Characteristic | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 281) | Bimekizumab (N = 431) | Placebo (N = 133) | Bimekizumab (N = 267) | |
Demographic characteristics | ||||
Age (years) | ||||
Mean (SD) | 48.7 (11.7) | 48.5 (12.6) | 51.3 (12.9) | 50.1 (12.4) |
Median (range) | 50.0 (22.0 to 77.0) | 48.0 (20.0 to 81.0) | 51.0 (25.0 to 85.0) | 50.0 (20.0 to 78.0) |
Sex, n (%) | ||||
Female | 154 (54.8) | 230 (53.4) | 73 (54.9) | 137 (51.3) |
Male | 127 (45.2) | 201 (46.6) | 60 (45.1) | 130 (48.7) |
Race, n (%) | ||||
White | 270 (96.1) | 410 (95.1) | 128 (96.2) | 256 (95.9) |
Asian | 7 (2.5) | 17 (3.9) | 4 (3.0) | 9 (3.4) |
American Indian or Alaskan | 0 (0.0) | 1 (0.2) | 0 (0.0) | 0 (0.0) |
Black | 0 (0.0) | 1 (0.2) | 0 (0.0) | 0 (0.0) |
Other or mixed | 4 (1.4) | 1 (0.2) | 1 (0.8) | 2 (0.7) |
Missing | 0 (0.0) | 1 (0.2) | 0 (0.0) | 0 (0.0) |
Mass (kg) | ||||
Mean (SD) | 85.2 (18.8) | 84.8 (20.3) | 83.1 (17.9) | 87.4 (19.9) |
Median (range) | 83.5 (47.6 to 160.0) | 81.7 (40.0 to 166.5) | 82.2 (49.5 to 139.7) | 86.4 (40.6 to 169.7) |
Clinical characteristics | ||||
Time since first PsA diagnosis (years) | ||||
Mean (SD) | 5.6 (6.5) | 6.0 (7.3) | 9.2 (8.1) | 9.6 (9.9) |
PsA subtype, n (%) | ||||
Polyarticular symmetric arthritis | 181 (64.4) | 271 (62.9) | 86 (64.7) | 168 (62.9) |
Oligoarticular asymmetric arthritis | 76 (27.0) | 118 (27.4) | 32 (24.1) | 62 (23.2) |
DIP joint, predominant | 12 (4.3) | 17 (3.9) | 7 (5.3) | 13 (4.9) |
Spondylitis, predominant | 10 (3.6) | 15 (3.5) | 7 (5.3) | 15 (5.6) |
Arthritis mutilans | 1 (0.4) | 8 (1.9) | 0 (0.0) | 8 (3.0) |
Missing | 1 (0.4) | 2 (0.5) | 1 (0.8) | 1 (0.4) |
Time since first psoriasis diagnosis (years) | ||||
Mean (SD) | 15.4 (13.0) | 14.9 (13.4) | 17.9 (11.8) | 17.2 (13.4) |
Prior TNFi exposure | ||||
Inadequate response to 1 TNFi | NA | NA | 103 (77.4) | 203 (76.0) |
Inadequate response to 2 TNFis | NA | NA | 15 (11.3) | 30 (11.2) |
Intolerance to TNFis | NA | NA | 15 (11.3) | 34 (12.7) |
≥ 1 bone erosion at baseline, n (%) | ||||
Yes | 210 (74.7) | 341 (79.1) | NA | NA |
No | 59 (21.0) | 79 (18.3) | NA | NA |
Missing | 12 (4.3) | 11 (2.6) | NA | NA |
≥ 1 bone erosion and/or hs-CRP ≥ 6 mg/mL, n (%) | ||||
Yes | 236 (84.0) | 365 (84.7) | NA | NA |
No | 45 (16.0) | 66 (15.3) | NA | NA |
BSA affected by psoriasis, n (%) | ||||
< 3% | 141 (50.2) | 214 (49.7) | 45 (33.8) | 91 (34.1) |
≥ 3% to ≤ 10% | 92 (32.7) | 144 (33.4) | 63 (47.4) | 109 (40.8) |
> 10% | 48 (17.1) | 73 (16.9) | 25 (18.8) | 67 (25.1) |
PASIa | ||||
Mean (SD) | 7.9 (5.6) | 8.2 (6.8) | 8.5 (6.6) | 10.2 (9.1) |
PASI, n (%)a | ||||
< 10 | 102 (36.3) | 160 (37.1) | 67 (50.4) | 112 (41.9) |
10 to 20 | 34 (12.1) | 44 (10.2) | 16 (12.0) | 52 (19.5) |
> 20 | 4 (1.4) | 13 (3.0) | 5 (3.8) | 12 (4.5) |
TJC | ||||
Mean (SD) | 17.1 (12.5) | 16.8 (11.8) | 19.3 (14.2) | 18.4 (13.6) |
SJC | ||||
Mean (SD) | 9.5 (7.3) | 9.0 (6.2) | 10.3 (8.2) | 9.7 (7.5) |
HAQ-DI | ||||
Mean (SD) | 0.89 (0.61) | 0.82 (0.59) | 1.04 (0.69) | 0.97 (0.59) |
Enthesitis (LEI), n (%) | ||||
Yes | 70 (24.9) | 143 (33.2) | 36 (27.1) | 106 (39.7) |
No | 211 (75.1) | 282 (65.4) | 96 (72.2) | 161 (60.3) |
Missing | 0 (0.0) | 6 (1.4) | 1 (0.8) | 0 (0.0) |
Dactylitis, n (%) | ||||
Yes | 33 (11.7) | 56 (13.0) | 14 (10.5) | 34 (12.7) |
No | 248 (88.3) | 368 (85.4) | 118 (88.7) | 233 (87.3) |
Missing | 0 (0.0) | 7 (1.6) | 1 (0.8) | 0 (0.0) |
NSAID therapy at baseline, n (%) | ||||
Yes | 166 (59.1) | 255 (59.2) | 80 (60.2) | 143 (53.6) |
No | 115 (40.9) | 176 (40.8) | 53 (39.8) | 124 (46.4) |
Prior cDMARDs, n (%) | ||||
0 | 60 (21.4) | 92 (21.3) | 50 (37.6) | 98 (36.7) |
1 | 185 (65.8) | 270 (62.6) | 68 (51.1) | 150 (56.2) |
≥ 2 | 36 (12.8) | 69 (16.0) | 15 (11.3) | 19 (7.1) |
cDMARDs at baseline, n (%) | ||||
Yes | 194 (69.0) | 301 (69.8) | 63 (47.4) | 139 (52.1) |
No | 87 (31.0) | 130 (30.2) | 70 (52.6) | 128 (47.9) |
Methotrexate at baseline, n (%) | ||||
Yes | 163 (58.0) | 252 (58.5) | 51 (38.3) | 119 (44.6) |
No | 118 (42.0) | 179 (41.5) | 82 (61.7) | 148 (55.4) |
Oral corticosteroids at baseline, n (%) | ||||
Yes | 61 (21.7) | 80 (18.6) | 21 (15.8) | 38 (14.2) |
No | 220 (78.3) | 351 (81.4) | 112 (84.2) | 229 (85.8) |
BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; DIP = distal interphalangeal; HAQ-DI = Health Assessment Questionnaire–Disability Index; hs-CRP = high-sensitivity CRP; LEI = Leeds Enthesitis Index; NA = not available; NSAID = nonsteroidal anti-inflammatory drug; PASI = Psoriasis Area Severity Index; PsA = psoriatic arthritis; SD = standard deviation; SJC = swollen joint count; TJC = tender joint count; TNFi = tumour necrosis factor inhibitor.
aFor patients with psoriasis involving at least 3% of BSA at baseline.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The main differences between the trials were that in the BE COMPLETE trial, patients with an inadequate response or intolerance to TNFis were included and patients had a longer time since PsA and psoriasis diagnoses, a higher percentage of BSA affected by psoriasis and PASI, and a lower number of prior cDMARDs and methotrexate use at baseline.
Exposure to Study Treatments
Exposure to study treatments is summarized in Table 13.
During the DB treatment period of the BE OPTIMAL trial, patients had a mean exposure to study drug of 110.2 (SD = 10.3) days in the bimekizumab group and 108.9 (SD = 14.2) days in the placebo group. Mean adherence was 97.0% in the 2 groups. During the active treatment–blind period, patients had a mean exposure of 239.4 (SD = 44.0) days in the bimekizumab group and 243.5 (SD = 32.0) days in the placebo group. Mean adherence was 95.0% in the 2 groups.
In the BE COMPLETE trial, patients had a mean exposure of 110.7 (SD = 8.7) days in the bimekizumab group and 109.2 (SD = 14.6) days in the placebo group. Mean adherence was more than 98.1% in the 2 groups.
In the BE OPTIMAL trial, 4.8% of patients and 7.0% of patients in the bimekizumab and placebo groups, respectively, used rescue therapy during the active treatment–blind period. The most commonly used medications were systemic hormonal preparations (1.4% to 2.6%), musculoskeletal system drugs (1.2% to 1.8%), and antineoplastic and immunomodulating drugs (1.2% to 1.5%).
Table 13: Summary of Patient Exposure From the BE OPTIMAL and BE COMPLETE Trials
Exposure | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo | Bimekizumab | Placebo | Bimekizumab | |
DB treatment period: SS | N = 281 | N = 431 | N = 132 | N = 267 |
IMP duration, patient-yearsa | 83.8 | 130.1 | 39.5 | 81.0 |
Duration (days), mean (SD) | 108.9 (14.2) | 110.2 (10.3) | 109.2 (14.6) | 110.7 (8.7) |
Duration (days), median (range) | 112.0 (14 to 121) | 112.0 (26 to 125) | 112.0 (28 to 121) | 112.0 (55 to 133) |
Adherence (%), mean (SD) | 97.0 (9.3) | 97.0 (8.5) | 99.1 (6.5) | 98.1 (7.9) |
Active treatment–blind period: ATS | N = 271 | N = 414 | — | — |
IMP duration, patient-yearsa | 180.7 | 271.3 | — | — |
Duration (days), mean (SD) | 243.5 (32.0) | 239.4 (44.0) | — | — |
Duration (days), median (range) | 252.0 (25 to 274) | 252.0 (14 to 286) | — | — |
Adherence (%), mean (SD) | 95.0 (9.1) | 95.3 (9.7) | — | — |
ATS = active treatment–blind set; DB = double-blind; IMP = investigational medicinal product; SD = standard deviation; SS = safety set.
aTotal IMP duration was a subset of total time at risk excluding nontreated periods.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Protocol Deviations
In the BE OPTIMAL trial, approximately 23% of patients in both the bimekizumab and placebo groups had an important protocol deviation during the DB treatment period, where procedural noncompliance was the most common deviation for both groups (16.7% to 18.6%). Approximately 5% of patients in the treatment groups were excluded from the per-protocol set, mostly due to prohibited concomitant medication use.
In the BE COMPLETE trial, approximately 9% of patients in both treatment groups had an important protocol deviation, where inclusion criteria deviation (4.5% to 4.9%) and procedural noncompliance (3.4% to 3.8%) were the main reasons. Overall, 4.5% of patients in the treatment groups were excluded from the per-protocol set, mostly due to inclusion criteria deviation.
Efficacy
ACR Response
ACR50, ACR20, and ACR70 response results are summarized in Table 14.
In the BE OPTIMAL trial, a greater proportion of patients in the bimekizumab treatment group reached the primary end point of ACR50 at week 16 than did those in the placebo group; the difference between treatment groups for ACR50 was 31.2% (95% CI, 25.2% to 37.3%; P < 0.001). ACR50 results at week 52 indicated that response rates increased in both groups and that there was a similar response between patients who crossed over from placebo to bimekizumab and patients originally randomized to bimekizumab during the active treatment–blind period. Results for the sensitivity analyses using the per-protocol set supported those of the primary analysis.
In the BE OPTIMAL trial, a greater proportion of patients in the bimekizumab treatment group reached the secondary end points of ACR20 and ACR70 at week 16 than did those in the placebo group; the difference between treatment groups was 37.2% (95% CI, 30.5% to 44.0%) for ACR20 and 19.3% (95% CI, 14.1% to 24.5%) for ACR70. ACR20 and ACR70 results at week 52 indicated that response rates increased in both groups and that there was a similar response between placebo-bimekizumab crossover patients and bimekizumab patients during the active treatment–blind period.
In the BE COMPLETE trial, a greater proportion of patients in the bimekizumab treatment group reached the primary end point of ACR50 at week 16 than did those in the placebo group; the difference between treatment groups for ACR50 was 29.0% (95% CI, 21.9% to 36.2%; P < 0.001). Subgroup analyses for ACR50 at week 16 for prior TNFi exposure are summarized in Table 49 in Appendix 1. A larger proportion of patients in the bimekizumab group compared to those in the placebo group attained ACR50 response for each of the subgroup categories of inadequate response to 1 TNFi (47.8% versus 6.8%), inadequate response to 2 TNFis (20.0% versus 0%), and intolerance to TNFis (38.2% versus 13.3%) in the bimekizumab and placebo groups, respectively.
Table 14: ACR Response at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Randomized Set)
Outcome | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 281) | Bimekizumab (N = 431) | Placebo (N = 133) | Bimekizumab (N = 267) | |
ACR50 response: Primary outcome | ||||
n (%) | 28 (10.0) | 189 (43.9) | 9 (6.8) | 116 (43.4) |
Adjusted responder rate, % (95% CI)a | 8.5 (5.7 to 12.5) | 39.7 (33.9 to 45.9) | 4.3 (2.0 to 8.9) | 33.3 (24.8 to 43.1) |
Difference vs. placebo, % (95% CI)a | 31.2 (25.2 to 37.3) | 29.0 (21.9 to 36.2) | ||
OR vs. placebo (95% CI)a | 7.1 (4.6 to 10.9) | 11.1 (5.4 to 23.0) | ||
P valuea,b | < 0.001 | < 0.001 | ||
ACR20 response: Secondary outcome | ||||
n (%) | 67 (23.8) | 268 (62.2) | 21 (15.8) | 179 (67.0) |
Adjusted responder rate, % (95% CI)a | 20.0 (15.3 to 25.6) | 57.2 (51.2 to 63.1) | 14.4 (8.8 to 22.8) | 67.2 (57.9 to 75.3) |
Difference vs. placebo, % (95% CI)a | 37.2 (30.5 to 44.0) | 52.8 (43.6 to 61.9) | ||
OR vs. placebo (95% CI)a | 5.4 (3.8 to 7.5) | 12.2 (7.0 to 21.1) | ||
P valuea | < 0.001 | < 0.001 | ||
ACR70 response: Secondary outcome | ||||
n (%) | 12 (4.3) | 105 (24.4) | 1 (0.8) | 71 (26.6) |
Adjusted responder rate, % (95% CI)a | 4.0 (2.2 to 7.2) | 23.3 (18.5 to 28.9) | 0.4 (0.1 to 3.1) | 17.6 (11.1 to 26.9) |
Difference vs. placebo, % (95% CI)a | 19.3 (14.1 to 24.5) | 17.2 (12.2 to 22.2) | ||
OR vs. placebo (95% CI)a | 7.2 (3.9 to 13.4) | 50.6 (6.9 to 370.0) | ||
P valuea | < 0.001 | < 0.001 | ||
ACR = American College of Rheumatology; ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; CI = confidence interval; IMP = investigational medicinal product; OR = odds ratio; TNFi = tumour necrosis factor inhibitor; vs. = versus.
Note: Patients who had missing ACR50 data at week 16 or who discontinued the IMP before week 16, regardless of whether they had data or not, were considered nonresponders.
aCalculated using a logistic regression model with factors for treatment, bone erosion at baseline, and region in the BE OPTIMAL trial. Calculated using a logistic regression model with factors for treatment, prior TNFi exposure at baseline, and region in the BE COMPLETE trial.
bP value adjusted for multiple testing. Outcomes in both trials were sequentially tested to control the familywise type I error rate at alpha of 0.05 (2-sided).
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
In the BE COMPLETE trial, a greater proportion of patients in the bimekizumab treatment group reached the secondary end points of ACR20 and ACR70 at week 16 than did those in the placebo group, the difference between treatment groups was 52.8% (95% CI, 43.6% to 61.9%) for ACR20 and 17.2% (95% CI,12.2% to 22.2%) for ACR70.
Minimal Disease Activity
MDA results are summarized in Table 15.
In the BE OPTIMAL trial, for clinical responses measured with the MDA criteria, patients treated with bimekizumab had higher response rates compared with placebo at week 16. The difference between treatment groups was 31.0% (95% CI, 24.5% to 37.5%; P < 0.001). MDA results at week 52 indicated that response rates increased in both groups and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, patients treated with bimekizumab had higher response rates compared with placebo at week 16; the difference between treatment groups was 34.2% (95% CI, 26.1% to 42.2%; P < 0.001).
Enthesitis-Free State Based on LEI
LEI results for the pooled population of patients with enthesitis at baseline in the BE OPTIMAL and BE COMPLETE trials are summarized in Table 16.
A greater proportion of patients in the bimekizumab treatment group had resolution of enthesitis at week 16 (LEI = 0) than did those in the placebo group; the difference between treatment groups was 14.9% (95% CI, 4.0% to 25.9%; P = 0.008).
Table 15: MDA Response at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Randomized Set)
Outcome | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 281) | Bimekizumab (N = 431) | Placebo (N = 133) | Bimekizumab (N = 267) | |
Responders, n (%) | 37 (13.2) | 194 (45.0) | 8 (6.0) | 118 (44.2) |
Adjusted responder rate, % (95% CI)a | 12.3 (8.7 to 17.1) | 43.3 (37.4 to 49.4) | 4.6 (2.1 to 9.7) | 38.8 (29.6 to 48.8) |
Difference vs. placebo, % (95% CI)a | 31.0 (24.5 to 37.5) | 34.2 (26.1 to 42.2) | ||
OR vs. placebo (95% CI)a | 5.4 (3.7 to 8.1) | 13.1 (6.1 to 28.0) | ||
P valuea,b | < 0.001 | < 0.001 | ||
CI = confidence interval; MDA = minimal disease activity; OR = odds ratio; TNFi = tumour necrosis factor inhibitor; vs. = versus.
aCalculated using a logistic regression model with factors for treatment, bone erosion at baseline, and region in the BE OPTIMAL trial. Calculated using a logistic regression model with factors for treatment, prior TNFi exposure at baseline, and region in the BE COMPLETE trial.
bP value adjusted for multiple testing. Outcomes in both trials were sequentially tested to control the familywise type I error rate at alpha of 0.05 (2-sided).
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 16: Enthesitis-Free State at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Pooled Randomized Set With Enthesitis at Baseline)
Outcome | Pooled population for BE OPTIMAL and BE COMPLETE studies | |
|---|---|---|
Placebo (N = 106) | Bimekizumab (N = 249) | |
Responders, n (%) | 37 (34.9) | 124 (49.8) |
Adjusted responder rate, % (95% CI)a | 30.0 (21.6 to 40.0) | 45.0 (37.9 to 52.2) |
Difference vs. placebo, % (95% CI)a | 14.9 (4.0 to 25.9) | |
OR vs. placebo (95% CI)a | 1.9 (1.2 to 3.1) | |
P valuea,b | 0.008 | |
CI = confidence interval; ID = identification; OR = odds ratio; vs. = versus.
aCalculated using a logistic regression model with factors for treatment, study ID, and region.
bP value adjusted for multiple testing. Outcomes in both trials were sequentially tested to control the familywise type I error rate at alpha of 0.05 (2-sided).
Sources: BE OPTIMAL Clinical Study Report and sponsor’s summary of clinical evidence.20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Dactylitis-Free State Based on LDI
LDI results for the pooled population of patients with dactylitis at baseline from the BE OPTIMAL and BE COMPLETE trials are summarized in Table 17.
A greater proportion of patients in the bimekizumab treatment group had resolution of dactylitis at week 16 (LDI = 0) than did those in the placebo group. The difference between treatment groups was 29.4% (95% CI, 11.7% to 47.1%; P = 0.002).
Table 17: Dactylitis-Free State at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Pooled Randomized Set With Dactylitis at Baseline)
Outcome | Pooled population for BE OPTIMAL and BE COMPLETE studies | |
|---|---|---|
Placebo (N = 47) | Bimekizumab (N = 90) | |
Responders, n (%) | 24 (51.1) | 68 (75.6) |
Adjusted responder rate, % (95% CI)a | 41.5 (26.5 to 58.4) | 71.0 (59.0 to 80.6) |
Difference vs. placebo, % (95% CI)a | 29.4 (11.7 to 47.1) | |
OR vs. placebo (95% CI)a | 3.4 (1.6 to 7.6) | |
P valuea,b | 0.002 | |
CI = confidence interval; ID = identification; OR = odds ratio; vs. = versus.
aCalculated using a logistic regression model with factors for treatment, study ID, and region.
bP value adjusted for multiple testing. Outcomes in both trials were sequentially tested to control the familywise type I error rate at alpha of 0.05 (2-sided).
Sources: BE OPTIMAL Clinical Study Report and the sponsor’s summary of clinical evidence.20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Swollen Joint Count
SJC results are summarized in Table 18.
Table 18: Swollen Joint Count at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Randomized Set)
Outcome | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 281) | Bimekizumab (N = 431) | Placebo (N = 133) | Bimekizumab (N = 267) | |
Change from baseline, mean (SE) | –3.0 (0.5) | –6.6 (0.3) | –2.0 (0.5) | –7.0 (0.4) |
LSM (SE)a | –2.3 (0.4) | –6.3 (0.3) | –1.7 (0.6) | –7.1 (0.5) |
Mean difference vs. placebo (95% CI) | –4.0 (–4.8 to –3.1) | –5.3 (–6.5 to –4.2) | ||
P value | < 0.001 | < 0.001 | ||
ANCOVA = analysis of covariance; CI = confidence interval; LSM = least squares mean; SE = standard error; TNFi = tumour necrosis factor inhibitor; vs. = versus.
aCalculated using ANCOVA with treatment, bone erosion at baseline, and region as fixed effects, and baseline value as a covariate in the BE OPTIMAL trial. Calculated using ANCOVA with treatment, prior TNFi exposure at baseline, and region as fixed effects, and baseline value as a covariate in the BE COMPLETE trial.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
In the BE OPTIMAL trial, at week 16, the mean reduction at week 16 was greater in the bimekizumab group compared to the placebo group, with the mean difference between treatment groups being –4.0 joints (95% CI, –4.8 joints to –3.1 joints). SJC results at week 52 indicated that response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, the mean reduction at week 16 was greater in the bimekizumab group compared to the placebo group, with the mean difference between treatment groups being –5.3 joints (95% CI, –6.5 joints to –4.2 joints).
90% Reduction in PASI Score
PASI 90 results for patients who had psoriasis involving at least 3% BSA at baseline in the trials are summarized in Table 19.
In the BE OPTIMAL trial, for PASI 90, patients treated with bimekizumab had higher response rates compared with those treated with placebo at week 16. The difference between treatment groups was 56.5% (95% CI, 48.6% to 64.3%; P < 0.001). PASI 90 results at week 52 indicated that response rates increased in both groups and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, for PASI 90, patients treated with bimekizumab had higher response rates compared with those treated with placebo at week 16. The difference between treatment groups was 57.6% (95% CI, 47.6% to 67.6%; P < 0.001). Subgroup analyses for PASI 90 (patients with at least 3% BSA affected by psoriasis at baseline) at week 16 for prior TNFi exposure is summarized in Table 49 in Appendix 1. A larger proportion of patients in the bimekizumab group compared to those in the placebo group attained a PASI 90 response for each of the subgroup categories of inadequate response to 1 TNFi (difference versus placebo = 64.4% [95% CI, 53.6% to 75.3%]), inadequate response to 2 TNFis (difference versus placebo = 25.0% [95% CI, –16.2% to 66.1%]), and intolerance to TNFis (difference versus placebo = 49.7% [95% CI, 18.5% to 80.9%]).
Table 19: PASI 90 Response at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Randomized Set With Psoriasis Involving At Least 3% BSA at Baseline)
Outcome | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 140) | Bimekizumab (N = 217) | Placebo (N = 88) | Bimekizumab (N = 176) | |
PASI 90 response, n (%) | 4 (2.9) | 133 (61.3) | 6 (6.8) | 121 (68.8) |
Adjusted responder rate, % (95% CI)a | 2.2 (0.8 to 6.0) | 58.7 (48.7 to 67.9) | 5.3 (2.1 to 12.8) | 62.9 (49.5 to 74.6) |
Difference vs. placebo, % (95% CI)a | 56.5 (48.6 to 64.3) | 57.6 (47.6 to 67.6) | ||
OR vs. placebo (95% CI)a | 63.0 (22.2 to 178.9) | 30.2 (12.4 to 73.9) | ||
P valuea,b | < 0.001 | < 0.001 | ||
BSA = body surface area; CI = confidence interval; OR = odds ratio; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; TNFi = tumour necrosis factor inhibitor; vs. = versus.
aCalculated using a logistic regression model with factors for treatment, bone erosion at baseline, and region in the BE OPTIMAL trial. Calculated using a logistic regression model with factors for treatment, prior TNFi exposure at baseline, and region in the BE COMPLETE trial.
bP value adjusted for multiple testing. Outcomes in both trials were sequentially tested to control the familywise type I error rate at alpha of 0.05 (2-sided).
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
IGA Score of 0 or 1 and At Least 2-Grade Reduction From Baseline
IGA results for the patients who had an IGA score of at least 2 and psoriasis involving at least 3% BSA at baseline are summarized in Table 20.
In the BE OPTIMAL trial, a larger proportion of patients in the bimekizumab group compared to those in the placebo group attained at least a 2-grade reduction from baseline in the IGA score at week 16. The difference between treatment groups was 46.0% (95% CI, 37.1% to 55.0%). IGA results at week 52 indicated that the response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, a larger proportion of patients in the bimekizumab group compared to those in the placebo group attained at least a 2-grade reduction from baseline in the IGA score at week 16. The difference between treatment groups was 58.2% (95% CI, 46.7% to 69.8%).
Table 20: IGA Response at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Randomized Set With IGA Score of At Least 2 and Psoriasis Involving At Least 3% BSA at Baseline)
Outcome | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 129) | Bimekizumab (N = 204) | Placebo (N = 82) | Bimekizumab (N = 163) | |
Responders, n (%)a | 5 (3.9) | 103 (50.5) | 3 (3.7) | 99 (60.7) |
Adjusted responder rate, % (95% CI)b | 3.5 (1.4 to 8.6) | 49.5 (39.4 to 59.7) | 3.9 (1.2 to 12.0) | 62.1 (48.3 to 74.2) |
Difference vs. placebo, % (95% CI)b | 46.0 (37.1 to 55.0) | 58.2 (46.7 to 69.8) | ||
OR vs. placebo (95% CI)b | 27.1 (10.6 to 69.5) | 40.9 (12.3 to 135.6) | ||
P valueb | < 0.001 | < 0.001 | ||
BSA = body surface area; CI = confidence interval; IGA = Investigator’s Global Assessment; OR = odds ratio; TNFi = tumour necrosis factor inhibitor; vs. = versus.
aResponders were patients with a score of 0 or 1 and at least a 2-grade reduction from baseline.
bCalculated using a logistic regression model with factors for treatment, bone erosion at baseline, and region in the BE OPTIMAL trial. Calculated using logistic regression with factors for treatment, prior TNFi exposure at baseline, and region in the BE COMPLETE trial.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Health Assessment Questionnaire–Disability Index
HAQ-DI results are summarized in Table 21.
In the BE OPTIMAL trial, the bimekizumab group had a greater mean decrease from baseline (i.e., improvement) in HAQ-DI compared with the placebo group at week 16; the LSM difference between treatment groups was –0.19 (95% CI, –0.25 to –0.13; P < 0.001). HAQ-DI results at week 52 indicated that the response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, the bimekizumab group had a greater mean decrease from baseline (i.e., improvement) in HAQ-DI compared with the placebo group at week 16; the LSM difference between treatment groups was –0.33 (95% CI, –0.42 to –0.23; P < 0.001). Subgroup analyses for HAQ-DI (patients who had a score decrease from baseline of at least 0.35) at week 16 for prior TNFi exposure are summarized in Table 49 in Appendix 1. A larger proportion of patients in the bimekizumab group compared to those in the placebo group attained a HAQ-DI score decrease of at least 0.35 for each of the subgroup categories of inadequate response to 1 TNFi (difference versus placebo = 40.8% [95% CI, 28.6% to 53.1%]), inadequate response to 2 TNFis (difference versus placebo = 11.5% [95% CI, –19.9% to 42.9%]), and intolerance to TNFis (difference versus placebo = 24.7% [95% CI, –6.0% to 55.5%]).
Table 21: HAQ-DI at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Randomized Set)
Outcome | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 281) | Bimekizumab (N = 431) | Placebo (N = 133) | Bimekizumab (N = 267) | |
Baseline, mean (SD) | 0.89 (0.61) | 0.82 (0.59) | 1.04 (0.69) | 0.97 (0.59) |
Change from baseline, mean (SE) | –0.09 (0.03) | –0.26 (0.02) | –0.07 (0.04) | –0.38 (0.03) |
LSM (SE)a | –0.07 (0.03) | –0.26 (0.03) | 0.02 (0.05) | –0.31 (0.04) |
Mean difference vs. placebo (95% CI)a | –0.19 (–0.25 to –0.13) | –0.33 (–0.42 to –0.23) | ||
P valuea,b | < 0.001 | < 0.001 | ||
ANCOVA = analysis of covariance; CI = confidence interval; HAQ-DI = Health Assessment Questionnaire–Disability Index; LSM = least squares mean; SD = standard deviation; SE = standard error; TNFi = tumour necrosis factor inhibitor; vs. = versus.
aCalculated using ANCOVA with treatment, bone erosion at baseline, and region as fixed effects, and baseline value as a covariate in the BE OPTIMAL trial. Calculated using ANCOVA with treatment, prior TNFi exposure at baseline, and region as fixed effects, and baseline value as a covariate in the BE COMPLETE trial.
bP value adjusted for multiple testing. Outcomes in both trials were sequentially tested to control the familywise type I error rate at alpha of 0.05 (2-sided).
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
SF-36 PCS
SF-36 PCS results are summarized in Table 22.
In the BE OPTIMAL trial, the bimekizumab group had a greater mean increase from baseline (i.e., improvement) in the SF-36 PCS compared with the placebo group at week 16; the LSM difference between treatment groups was 4.3 (95% CI, 3.2 to 5.4; P < 0.001). SF-36 PCS results at week 52 indicated that the response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, the bimekizumab group had a greater mean increase from baseline (i.e., improvement) in the SF-36 PCS compared with the placebo group at week 16; the LSM difference between treatment groups was 6.0 (95% CI, 4.4 to 7.7; P < 0.001).
Patient’s Assessment of Arthritis Pain
PtAAP results are summarized in Table 23.
In the BE OPTIMAL trial, the bimekizumab group had a greater mean decrease from baseline (i.e., improvement) in PtAAP compared with the placebo group at week 16. The mean difference between treatment groups was –19.1 (95% CI, –22.7 to –15.5). PtAAP results at week 52 indicated that the response was maintained in the bimekizumab group and that there was a similar response between placebo-bimekizumab crossover patients and patients originally randomized to bimekizumab during the active treatment–blind period.
In the BE COMPLETE trial, the bimekizumab group had a greater mean decrease from baseline (i.e., improvement) in PtAAP compared with the placebo group at week 16. The mean difference between treatment groups was –25.0 (95% CI, –30.0 to –20.0).
Table 22: SF-36 PCS at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Randomized Set)
Outcome | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 281) | Bimekizumab (N = 431) | Placebo (N = 133) | Bimekizumab (N = 267) | |
Baseline, mean (SE) | 36.9 (0.6) | 38.1 (0.5) | 35.9 (0.9) | 36.4 (0.5) |
Change from baseline, mean (SE) | 2.3 (0.5) | 6.2 (0.4) | 1.4 (0.7) | 7.3 (0.5) |
LSM (SE)a | 1.9 (0.5) | 6.3 (0.5) | 0.1 (0.9) | 6.2 (0.7) |
Mean difference vs. placebo (95% CI)a | 4.3 (3.2 to 5.4) | 6.0 (4.4 to 7.7) | ||
P valuea,b | < 0.001 | < 0.001 | ||
ANCOVA = analysis of covariance; CI = confidence interval; LSM = least squares mean; PCS = physical component summary; SE = standard error; SF-36 = Short Form (36) Health Survey; TNFi = tumour necrosis factor inhibitor; vs. = versus.
aCalculated using ANCOVA with treatment, bone erosion at baseline, and region as fixed effects, and baseline value as a covariate in the BE OPTIMAL trial. Calculated using ANCOVA with treatment, prior TNFi exposure at baseline, and region as fixed effects, and baseline value as a covariate in the BE COMPLETE trial.
bP value adjusted for multiple testing. Outcomes in both trials were sequentially tested to control the familywise type I error rate at alpha of 0.05 (2-sided).
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Harms results during the 16-week DB treatment period are summarized in Table 24 while harms results during the 36-week active treatment–blind period are summarized in Table 50 in Appendix 1.
Adverse Events
During the DB treatment period of the BE OPTIMAL trial, 59.6% of patients in the bimekizumab group and 49.5% of patients in the placebo group reported at least 1 AE. The most common AEs were nasopharyngitis (9.3% and 4.6% in the bimekizumab and placebo groups, respectively) and upper respiratory tract infection (5.1% and 6.4% in the bimekizumab and placebo groups, respectively). During the active treatment–blind period, 72.0% of patients in the bimekizumab group and 70.5% of patients in the placebo-bimekizumab crossover group reported at least 1 AE. The most common AE was nasopharyngitis (7.0% and 8.5% in the bimekizumab and crossover groups, respectively).
During the DB treatment period of the BE COMPLETE trial, 40.4% of patients in the bimekizumab group and 33.3% of patients in the placebo group reported at least 1 AE. The most common AE was nasopharyngitis (3.7% and 0.8% in the bimekizumab and placebo groups, respectively).
Table 23: Patient’s Assessment of Arthritis Pain at Week 16 in the BE OPTIMAL and BE COMPLETE Trials (Randomized Set)
Outcome | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 281) | Bimekizumab (N = 431) | Placebo (N = 133) | Bimekizumab (N = 267) | |
Baseline, mean (SD) | 56.8 (23.2) | 53.6 (24.3) | 61.7 (24.6) | 58.3 (24.2) |
Change from baseline, mean (SE) | –6.3 (1.5) | –23.6 (1.3) | –4.5 (2.1) | –27.7 (1.7) |
LSM (SE)a | –4.6 (1.7) | –23.8 (1.4) | –1.6 (2.6) | –26.6 (2.1) |
Mean difference vs. placebo (95% CI)a | –19.1 (–22.7 to –15.5) | –25.0 (–30.0 to –20.0) | ||
P valuea | < 0.001 | < 0.001 | ||
ANCOVA = analysis of covariance; CI = confidence interval; LSM = least squares mean; SD = standard deviation; SE = standard error; TNFi = tumour necrosis factor inhibitor; vs. = versus.
aCalculated using ANCOVA with treatment, bone erosion at baseline, and region as fixed effects, and baseline value as a covariate in the BE OPTIMAL trial. Calculated using ANCOVA with treatment, prior TNFi exposure at baseline, and region as fixed effects, and baseline value as a covariate in the BE COMPLETE trial.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Serious Adverse Events
During the DB treatment period of the BE OPTIMAL trial, 1.9% of patients in the bimekizumab group and 1.1% of patients in the placebo group reported at least 1 SAE. No SAEs occurred in more than 2 patients per treatment group. During the active treatment–blind period, 5.6% of patients in the bimekizumab group and 5.9% of patients in the placebo-bimekizumab crossover group reported at least 1 SAE. The only SAE to occur in at least 2 patients per treatment group was osteoarthritis (0.3% in the bimekizumab group and no patients in the crossover group).
During the DB treatment period of the BE COMPLETE trial, 1.9% of patients in the bimekizumab group and no patients in the placebo group reported an SAE. No SAEs occurred in more than 2 patients per treatment group.
Withdrawals Due to Adverse Events
During the DB treatment period of the BE OPTIMAL trial, 1.9% of patients in the bimekizumab group and 1.1% of patients in the placebo group reported an AE leading to drug discontinuation. During the active treatment–blind period, 2.7% of patients in the bimekizumab group and 1.8% of patients in the placebo-bimekizumab crossover group reported an AE leading to drug discontinuation. No AEs leading to drug discontinuation occurred in more than 2 patients per treatment group throughout the trial.
During the DB treatment period of the BE COMPLETE trial, 0.7% of patients in the bimekizumab group and no patients in the placebo group reported an AE leading to drug discontinuation. No AEs leading to drug discontinuation occurred in more than 2 patients per treatment group.
Mortality
There were no deaths reported during the DB treatment period of the BE OPTIMAL trial. During the active treatment–blind period, 1 death in the placebo-bimekizumab crossover group occurred due to traumatic shock from a motorcycle accident.
There were no deaths reported during the DB treatment period of the BE COMPLETE trial.
Notable Harms
Liver toxicity, reactivation of tuberculosis infection, and serious injection-related AEs were not reported on in either of the trials.
During the 16-week treatment period of the BE OPTIMAL trial, there was 1 patient in the bimekizumab group and no patients in the placebo group who reported liver dysfunction based on Hy’s law. Of the 20 (4.6%) patients who reported any fungal infection in the bimekizumab group, 11 patients reported a candida infection and 9 patients reported a fungal infection NEC. Of the 4 (1.4%) patients who reported any fungal infection in the placebo group, 2 patients reported a candida infection and 2 patients reported a fungal infection NEC. There was 1 patient in each treatment group who reported a malignancy event. There were no reports of opportunistic infection, a major cardiovascular event, anaphylaxis, or IBD during the 16-week DB treatment period.
During the active treatment–blind period of the BE OPTIMAL trial, there was 1 patient in the bimekizumab group and no patients in the placebo-bimekizumab crossover group who reported liver dysfunction based on Hy’s law. There were 5 patients in the bimekizumab group and 4 patients in the crossover group who reported opportunistic infection. Of the 47 (11.4%) patients who reported any fungal infection in the bimekizumab group, 28 patients reported a candida infection, 19 patients reported a fungal infection NEC, and 5 patients reported a tinea infection. Of the 25 (9.2%) patients who reported any fungal infection in the crossover group, 19 patients reported a candida infection, 7 patients reported a fungal infection NEC, and 2 patients reported a tinea infection. There were 3 patients in the bimekizumab group and 1 patient in the crossover group who reported major cardiovascular events. There were 3 patients in the bimekizumab group and 4 patients in the crossover group who reported a malignancy event. There were 2 patients in the bimekizumab group and no patients in the crossover group who reported experiencing IBD. There were no reports of anaphylaxis during the active treatment–blind period.
Table 24: Summary of Harms Results During the Double-Blind Treatment Period From the BE OPTIMAL and BE COMPLETE Trials (Safety Set)
Harm | BE OPTIMAL study | BE COMPLETE study | ||
|---|---|---|---|---|
Placebo (N = 281) | Bimekizumab (N = 431) | Placebo (N = 132) | Bimekizumab (N = 267) | |
AEsa | ||||
Patients who experience ≥ 1 AE, n (%) | 139 (49.5) | 257 (59.6) | 44 (33.3) | 108 (40.4) |
Nasopharyngitis, n (%) | 13 (4.6) | 40 (9.3) | 1 (0.8) | 10 (3.7) |
Upper respiratory tract infection, n (%) | 18 (6.4) | 22 (5.1) | 2 (1.5) | 6 (2.2) |
SAEsb | ||||
Patients who experience ≥ 1 SAE, n (%) | 3 (1.1) | 8 (1.9) | 0 (0.0) | 5 (1.9) |
Patients who stopped treatment due to AEsb | ||||
Patients who stopped treatment, n (%) | 3 (1.1) | 8 (1.9) | 0 (0.0) | 2 (0.7) |
Deaths | ||||
Patients who died, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Notable harms | ||||
Liver dysfunction based on Hy’s law, n (%)c | 0 (0.0) | 1 (0.2) | 0 (0.0) | 0 (0.0) |
Liver toxicity, n (%) | NR | NR | NR | NR |
Opportunistic infection, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Any fungal infection, n (%) | 4 (1.4) | 20 (4.6) | 0 (0.0) | 12 (4.5) |
Candida infection, n (%) | 2 (0.7) | 11 (2.6) | 0 (0.0) | 7 (2.6) |
Fungal infection NEC, n (%) | 2 (0.7) | 9 (2.1) | 0 (0.0) | 4 (1.5) |
Tinea infection, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.4) |
Reactivation of tuberculosis infection, n (%) | NR | NR | NR | NR |
Major cardiovascular event, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Malignancy, n (%) | 1 (0.4) | 1 (0.2) | 1 (0.8) | 0 (0.0) |
Serious injection-related AE, n (%) | NR | NR | NR | NR |
Anaphylaxis, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Inflammatory bowel disease, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
AE = adverse event; NEC = not elsewhere classified; NR = not reported; SAE = serious adverse event.
aOccurring in at least 5% of patients.
bOccurring in at least 2 patients in any treatment group.
cHy’s law is defined as an alanine transaminase or aspartate transaminase level at least 3 times the upper limit of normal with coexisting total bilirubin at least 2 times the upper limit of normal in the absence of alkaline phosphatase at least 2 times the upper limit of normal.
Sources: BE OPTIMAL Clinical Study Report, BE COMPLETE Clinical Study Report, and the sponsor’s summary of clinical evidence.19,20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
In the BE COMPLETE trial, there were 12 (4.5%) patients in the bimekizumab group who reported any fungal infection, of whom 7 patients reported a candida infection, 4 patients reported a fungal infection NEC, and 1 patient reported a tinea infection. There were no reports of any fungal infection in the placebo group. There were no patients in the bimekizumab group and 1 patient in the placebo group who reported a malignancy. There were no reports of liver dysfunction based on Hy’s law, opportunistic infection, a major cardiovascular event, anaphylaxis, or IBD in the trial.
Critical Appraisal
Internal Validity
For both trials, there appeared to be a low risk of bias arising from the randomization process. Randomization was conducted using an interactive voice or web response system and the treatment groups of interest (bimekizumab and placebo) were generally balanced for most baseline characteristics. In the BE OPTIMAL trial, the proportion of patients with enthesitis at baseline was higher in the bimekizumab group than in the placebo group. In the BE COMPLETE trial, the proportion of patients with 10% or greater BSA affected by psoriasis, PASI, enthesitis, and methotrexate use at baseline was higher in the bimekizumab group than the placebo group. These imbalances could bias the results for musculoskeletal-related and skin-related outcomes as well as composite outcomes such as ACR and MDA, which include musculoskeletal and skin assessments. The bias is likely in favour of patients who start out with low disease activity at baseline, though the direction of bias for the overall treatment groups is less certain.
The risk of bias due to deviations from intended interventions was likely low. A bimekizumab-matching placebo was used in both trials for the first 16 weeks of treatment. The number of protocol deviations was similar between the treatment groups in each trial and sensitivity analyses using the per-protocol set were performed to investigate the impact of important protocol deviations, whose results supported the primary analysis. Adherence was greater than 97% during the 16-week DB treatment period for the trials and more than 95% during the BE OPTIMAL trial’s active treatment–blind period. After the 16-week DB period in the BE OPTIMAL trial, patients randomized to placebo began bimekizumab treatment for the remaining 36 weeks. During this time, all patients were permitted to use rescue therapy, which was used by 4.8% of patients and 7.0% of patients in the bimekizumab and placebo groups, respectively. Patients who accessed rescue therapy remained in the trial and there was no indication if their data were handled differently (e.g., imputed as nonresponders). As a result, the longer-term efficacy and harms results may be confounded by the lack of placebo group and by any rescue medication that was used. The use of stable concomitant therapies was permitted in the trials and the minimum stable duration ranged from 2 weeks to 8 weeks. These medications (particularly the cDMARDs) could take more time than the aforementioned minimum durations to reach full efficacy, making it difficult to attribute treatment effects and harms to either bimekizumab or a concomitant drug.
The risk of bias due to missing outcome data was low. Conservative data imputation methods were used in the trials. The number of discontinuations was low and balanced (1.4%) between the bimekizumab and placebo groups in the BE OPTIMAL trial. In the BE COMPLETE trial, 1 (0.4%) patient in the bimekizumab group withdrew by choice compared to 4 (3.0%) patients in the placebo group. Although the numbers are small and it is unlikely to be an important source of bias, it may suggest that data were not missing at random and/or treatment assignment may have been unblinded. There were 4 outcomes relevant to the CADTH review that did not use the full randomized populations. PASI 90 assessments included patients with at least 3% BSA affected by psoriasis at baseline for each trial and IGA response included patients who had at least 3% BSA affected by psoriasis and an IGA score of 2 or higher at baseline for each trial. The LEI and LDI outcomes included pooled data from the 2 trials for patients with enthesitis and dactylitis, respectively, at baseline. Because these 4 outcomes used subsets of the randomized population, it is uncertain if the known and unknown treatment effect modifiers were still balanced between the groups and there may have been unmeasurable bias that influenced the results.
There was likely a low risk of bias resulting from the measurement of outcomes. Outcomes in the trials were prespecified and physicians were blinded to treatment assignments. Assignment to adalimumab could have been unblinded due to the smaller injection volume (0.4 mL or 0.8 mL) compared to bimekizumab or placebo (1 mL), but the impact on the results was likely low because adalimumab was not included in the statistical comparisons.
The risk of bias resulting from selective reporting of results is low. The statistical analysis plan, protocol, and statistical testing hierarchy to control for type I error were prespecified in both trials. Subgroups were also prespecified but were not adjusted for multiplicity in the trials. Furthermore, 2 of the subgroups (patients with an inadequate response to 2 TNFis and patients with an intolerance to TNFis) had fewer patients, making it challenging to draw conclusions from these analyses with certainty.
External Validity
There were 9 patients in each of the trials who were from Canada and received either bimekizumab or placebo. There was limited racial diversity in the trials with most patients being white, which the clinical expert consulted by CADTH indicated does not reflect the diversity of patients with PsA across Canada. Patients were either biologic-naive (the BE OPTIMAL trial) or had a history of inadequate response or intolerance to 1 TNFi or 2 TNFis (the BE COMPLETE trial). Patients with experience with other bDMARDs (e.g., interleukin inhibitors) were not included and there were few patients with prior tsDMARD use; thus, it is uncertain if the results from the trials would be generalizable to patients who have experience with these targeted treatments. Patients were also excluded if they had active, symptomatic IBD (including Crohn disease and ulcerative colitis) or forms of psoriasis other than chronic plaque type (e.g., pustular, erythrodermic, and guttate psoriasis, drug-induced psoriasis). The clinical expert agreed that bimekizumab would be avoided in patients with a history of or current IBD; however, they noted that patients with other forms of psoriasis could be treated with bimekizumab in practice. Based on the reported baseline characteristics, the expert indicated that the patients in the trials were generally similar to those who are treated in clinical practice and could receive bimekizumab in Canada.
Patients could continue using other medications for PsA if they were on a stable dose before baseline. The clinical expert confirmed that the types of medications and doses are reasonable for treating PsA, and noted that methotrexate, sulfasalazine, and leflunomide are cDMARDs commonly used in practice while NSAIDs, COX-2 inhibitors, and corticosteroids are for immediate symptom relief (and are not disease-modifying). The expert also indicated that hydroxychloroquine is rarely used in Canadian practice and apremilast is a targeted therapy typically accessed after cDMARDs.
According to the clinical expert, treatment response is often assessed using TJC, SJC, enthesitis, dactylitis, and skin psoriasis improvement, and may include composite outcomes such as MDA or DAPSA. Although the other outcomes in the trials are not commonly used in practice, they may provide information important to clinicians, patients, and decision-makers. There was evidence of validity, reliability, and responsiveness for some outcome measures and only the patient-reported outcomes (HAQ-DI, SF-36 PCS, and PtAAP) had MIDs identified in the literature (though it was unclear if these were for between-group differences). The expert also noted that treatment response can be assessed at 3 months, suggesting that there should have been adequate time to observe a response for most outcomes relevant to the CADTH review during the first 16 weeks of the trials.
Although bimekizumab was administered in a clinic during the trials, it may be possible for patients to self-administer the drug at home after proper training and if the physician determines it is appropriate, as is described in the Health Canada product monograph for bimekizumab for the treatment of adults with moderate-to-severe plaque psoriasis.2
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:22,23
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of an important effect based on thresholds identified in the literature for HAQ-DI, SF-36 PCS, and PtAAP. The target of the certainty of evidence assessment was the presence or absence of an important effect based on thresholds informed by the clinical expert consulted for this review for ACR, MDA, LEI, LDI, SJC, PASI 90, and IGA.
For the GRADE assessments, findings from the BE OPTIMAL and BE COMPLETE trials were assessed together per outcome because these studies were similar in population, intervention, design, and outcome measures.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for bimekizumab versus placebo for patients with PsA.
Long-Term Extension Studies
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
The sponsor submitted 2 long-term extension studies that evaluated bimekizumab for the treatment of adult patients with PsA. The BE ACTIVE 2 study25 is a now-completed, 104-week, phase II OLE study that aimed to assess the long-term safety, tolerability, and efficacy of bimekizumab in 184 adults with PsA who completed the preceding phase IIb BE ACTIVE study. The BE ACTIVE study,26 not included in the systematic review, was a 48-week, randomized, DB, placebo-controlled, parallel-group, dose-ranging study that included adults with PsA who had previously been exposed to 1 TNFi. Patients were randomized to receive placebo or 1 of several bimekizumab dosing regimens for the initial 12-week placebo-controlled period, and then rerandomized to receive bimekizumab 160 mg every 4 weeks or 320 mg every 4 weeks up to week 48; during the BE ACTIVE 2 trial,25 all patients received the same bimekizumab 160 mg every 4 weeks dosing regimen used in the BE OPTIMAL20 and BE COMPLETE trials.19
The BE VITAL study27,28 is an ongoing, phase III OLE study of the BE COMPLETE and BE OPTIMAL trials that is evaluating the long-term efficacy (up to week 140) and long-term safety (up to week 212) of bimekizumab in 1,131 patients with PsA who received bimekizumab 160 mg every 4 weeks. This study is ongoing (the estimated completion date is May 25, 2026); therefore, the summary within this CADTH report will only cover data up to week 52 from 377 patients who completed 16 weeks of treatment in the BE COMPLETE trial.
Populations
The inclusion and exclusion criteria for the BE ACTIVE 225 and BE VITAL27,28 studies were consistent with those of the preceding trials (the BE ACTIVE,26 BE OPTIMAL,20 and BE COMPLETE19 trials). Namely, the studies included adults aged 18 years or older who had a clinical diagnosis of active PsA with symptom duration of at least 6 months at baseline, and baseline values of at least 3 of 68 for TJC and at least 3 of 66 for SJC. Patients who met the withdrawal criteria for the preceding trials were excluded from the extension studies. In cases with an ongoing SAE or history of serious infection, the medical monitor was consulted before patients were allowed to enrol in the extension studies.
Interventions
In both the BE ACTIVE 2 and BE VITAL studies, bimekizumab 160 mg every 4 weeks was administered subcutaneously. Patients were not allowed to use concomitant biologics while receiving bimekizumab but were permitted to use concomitant NSAIDs and/or cDMARDs at the discretion of the investigator; only 1 NSAID or COX-2 inhibitor was permitted at a given time. Steroid tapers were permitted for disease activity or disease-related symptoms (up to 20 mg prednisone or the equivalent for a maximum of 14 days). One injection per joint for up to 3 discrete joints was permitted per 12-month period.
Outcomes
The safety outcomes for the BE ACTIVE 225 and BE VITAL27,28 studies included incidences of TEAEs, SAEs, and withdrawal due to TEAEs. The efficacy outcomes for the BE ACTIVE 2 and BE VITAL studies were generally consistent with those of the preceding studies (i.e., the BE ACTIVE,26 BE OPTIMAL,20 and BE COMPLETE19 studies). The efficacy outcomes summarized in this section include:
measures of ACR50 and ACR70 response at various time points
measures of MDA response
measures of PASI 75, PASI 90, and PASI 100 response at various time points for patients with psoriasis affecting at least 3% BSA at the preceding study’s baseline
change from baseline in LDI and LEI (LEI was only evaluated in the BE VITAL study)
change from baseline in SF-36 PCS scores.
Measures of response and change from baseline values were calculated during the extension studies using the baseline values from the preceding studies. IGA response data were only collected during the BE VITAL study and have not yet been reported.
Statistical Analysis
All data from the extension studies were analyzed descriptively using summary statistics. No formal sample size calculations were performed. Analyses were based on a safety set (patients who received at least 1 dose of bimekizumab) and a full analysis set (patients who received at least 1 dose of bimekizumab and had at least 1 valid efficacy measurement).
Intermittent missing data were imputed using the Markov chain Monte Carlo method, followed by regression for monotone missing data. For the multiple imputation procedures, it was assumed that data were missing at random.
Results
Baseline Characteristics
Demographic and disease characteristics based on the baseline of the preceding studies for patients enrolled in the BE ACTIVE 2 and BE VITAL studies are presented in Table 25 and Table 26, respectively.
In the BE ACTIVE 2 study, the study population had a mean age of 49 (SD = 12.2) years, with 52.5% of patients being male and 47.5% being female. The mean time since PsA diagnosis was 8.0 years. Thirteen (7.1%) patients had psoriasis affecting at least 3% BSA. Additionally, the mean TJC of 68 joints was 4.9 (SD = 9.0) joints and the mean SJC of 66 joints was 1.7 (SD = 4.1) joints.
In the BE VITAL study, the study population had a mean age of 50.5 (SD = 12.5) years, with 47.5% males and 52.5% females. The mean time since PsA diagnosis was 9.5 years. Overall, 76.8% of patients had an inadequate response to 1 TNFi, 11.0% of patients had an inadequate response to 2 TNFis, and 12.3% of patients had an intolerance to TNFis. Additionally, 66.0% of patients had psoriasis affecting at least 3% BSA. The mean TJC was 18.7 (SD = 13.8) joints and the mean SJC was 9.9 (SD = 7.7) joints.
Table 25: Summary of Demographic Characteristics at Baseline of Preceding Studies (Safety Set)
Characteristic | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 183) | BE VITAL study, placebo in BE COMPLETE study (N = 133) | BE VITAL study, bimekizumab 160 mg q.4.w. in BE COMPLETE study (N = 267) |
|---|---|---|---|
Age (years) | |||
Mean (SD) | 49.0 (12.2) | 51.3 (12.9) | 50.1 (12.4) |
Median (range) | 49.0 (23 to 78) | NR | NR |
Sex, n (%) | |||
Male | 96 (52.5) | 60 (45.1) | 130 (48.7) |
Female | 87 (47.5) | 73 (54.9) | 137 (51.3) |
Race, n (%) | |||
Black | 4 (2.2) | NR | NR |
White | 179 (97.8) | 128 (96.2) | 256 (95.9) |
Other or mixed | 0 | NR | NR |
Body mass, kg | |||
Mean (SD) | 86.2 (18.8) | NR | NR |
Median (range) | 85.9 (51.0 to 150.8) | NR | NR |
BMI, kg/m2 | |||
Mean (SD) | 29.8 (6.1) | 29.0 (5.4) | 30.1 (6.5) |
Median (range) | 29.0 (19.0 to 54.1) | NR | NR |
Geographic region, n (%) | |||
Europe | 148 (80.9) | NR | NR |
North America | 35 (19.1) | NR | NR |
BMI = body mass index; NR = not reported; q.4.w. = every 4 weeks; SD = standard deviation.
Sources: BE ACTIVE 2 Clinical Study Report25 and BE COMPLETE Clinical Study Report.19
Table 26: Summary of Baseline Clinical Characteristics at Entry Into Extension Studies (Safety Set)
Characteristic | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 183) | BE VITAL study, placebo in BE COMPLETE study (N = 133) | BE VITAL study, bimekizumab 160 mg q.4.w. in BE COMPLETE study (N = 267) |
|---|---|---|---|
Time since PsA diagnosis, years | |||
Mean (SD) | 8.0 (7.9) | 9.2 (8.1) | 9.6 (9.9) |
Median (range) | 5.2 (1.0 to 51.3) | NR | NR |
< 2 years, n (%) | 31 (16.9) | NR | NR |
≥ 2 years, n (%) | 152 (83.1) | NR | NR |
TJC | |||
Mean (SD) | 4.9 (9.0) | 19.3 (14.2) | 18.4 (13.5) |
Median (range) | 1.0 (0.0 to 52.0) | NR | NR |
SJC | |||
Mean (SD) | 1.7 (4.1) | 10.3 (8.2) | 9.7 (7.5) |
Median (range) | 0.0 (0.0 to 29.0) | NR | NR |
CRP, mg/L | |||
Mean (SD) | 4.2 (5.3) | NR | NR |
Median (range) | 2.5 (0.1 to 38.0) | NR | NR |
≥ 6 mg/L, n (%) | NR | 59 (44.4) | 118 (44.2) |
BSA affected by psoriasis, n (%) | |||
< 3% | 169 (92.3) | NR | NR |
≥ 3% | 13 (7.1) | 88 (66.2) | 176 (65.9) |
Dactylitis at baseline, n (%) | |||
Yes | 50 (27.3) | 14 (10.5) | 34 (12.7) |
No | 133 (72.7) | 119 (89.5) | 233 (87.3) |
Enthesitis at baseline, n (%) | |||
Yes | 96 (52.5) | 36 (27.1) | 106 (39.7) |
No | 86 (47.0) | NR | NR |
Missing | 1 (0.5) | NR | NR |
Past TNFi therapy, n (%) | |||
Yes | 35 (19.1) | 133 (100) | 267 (100) |
Inadequate response to 1 TNFi | NR | 103 (77.4) | 204 (76.4) |
Inadequate response to 2 TNFis | NR | 15 (11.3) | 29 (10.9) |
Intolerance to TNFi | NR | 15 (11.3) | 34 (44.6) |
NSAID therapy, n (%) | |||
Past, n (%) | 52 (28.4) | NR | NR |
Current, n (%) | |||
Yes | 110 (60.1) | NR | NR |
1 NSAID | 107 (58.5) | NR | NR |
2 NSAIDs | 3 (1.6) | NR | NR |
≥ 3 NSAIDs | 0 | NR | NR |
No | 73 (39.9) | NR | NR |
Current synthetic DMARDs, n (%) | |||
Yes | 115 (62.8) | 63 (47.4) | 139 (52.1) |
Methotrexate | 107 (58.5) | 51 (38.3) | 119 (44.6) |
Sulfasalazine | 3 (1.6) | NR | NR |
Hydroxychloroquine | 0 | NR | NR |
Other | 7 (3.8) | NR | NR |
No | 68 (37.2) | 70 (52.6) | 128 (47.9) |
BSA = body surface area; DMARD = disease-modifying antirheumatic drug; NR = not reported; NSAID = nonsteroidal anti-inflammatory drug; PsA = psoriatic arthritis; q.4.w. = every 4 weeks; SD = standard deviation; SJC = swollen joint count; TJC = tender joint count; TNFi = tumour necrosis factor inhibitor.
Sources: BE ACTIVE 2 Clinical Study Report25 and BE COMPLETE Clinical Study Report.19
Patient Disposition
The patient disposition of the BE ACTIVE 2 and BE VITAL studies is summarized in Table 27. Among 184 patients who completed the BE ACTIVE trial and subsequently enrolled in the BE ACTIVE 2 study, 1 patient discontinued due to an AE (tongue fungal infection). Among the 183 patients who were dosed with bimekizumab in the BE ACTIVE 2 study (i.e., the safety set), most patients completed the study (88.0%). Discontinuation was reported for 12.0% of patients; the reason for discontinuation was most commonly related to TEAEs or consent withdrawal.
Among 388 patients who completed 16 weeks in the BE COMPLETE trial, 377 patients entered the BE VITAL long-term extension study. In total, 347 (86.8%) patients remained in the study at week 52. As per the protocol, patients withdrawn from study treatment could return for all remaining study visits. Of the patients who remained in the study at week 40 and week 52, 4 (1.0%) patients were not on randomized treatment. Ten patients discontinued due to TEAEs in the BE VITAL study. Discontinuation was reported in 18 patients; the reason for discontinuation was most commonly related to TEAEs or consent withdrawal.
Table 27: Patient Disposition for BE ACTIVE and BE VITAL Studies
Patient disposition | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 183) | BE VITAL study, placebo in BE COMPLETE study (N = 133) | BE VITAL study, bimekizumab 160 mg q.4.w. in BE COMPLETE study (N = 267) |
|---|---|---|---|
Started study, N (%) | 183 (100.0) | 121 (91.0) | 256 (95.9) |
Completed study, N (%) | 161 (88.0) | 111 (83.5) | 236 (88.4) |
Discontinued study, n (%) | 22 (12.0) | 10 (7.5) | 18 (6.7) |
Adverse event | 9 (4.9) | 4 (3.0) | 6 (2.2) |
Consent withdrawn | 9 (4.9) | 1 (0.8) | 7 (2.6) |
Lack of efficacy | 2 (1.1) | 3 (2.3) | 3 (1.1) |
Lost to follow-up | 1 (0.5) | 1 (0.8) | 2 (0.7) |
Others | 1 (0.5) | 1 (0.8) | 0 |
NR = not reported; q.4.w. = every 4 weeks.
Sources: BE ACTIVE 2 Clinical Study Report25 and Coates et al. (2024).27
Concomitant Medications and Cointerventions
The concomitant use of nonbiologic medication for PsA was permitted and common during the BE ACTIVE 2 study (90.2% of patients). The concomitant use of nonbiologic medication for PsA was not reported in the BE VITAL study. Biologic treatments for PsA were not permitted in either study. In the BE ACTIVE 2 study, the most common treatments were folic acid and methotrexate, and concomitant rescue therapy was reported for 24 (13.1%) patients (Table 28).
Table 28: Concomitant Medications Used During BE ACTIVE 2 Study (Safety Set)
Concomitant medication | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 183) |
|---|---|
Any PsA concomitant medications, n (%) | 165 (90.2) |
Folic acid | 95 (51.9) |
Methotrexate | 89 (48.6) |
Meloxicam | 29 (15.8) |
Methotrexate sodium | 23 (12.6) |
Diclofenac sodium | 23 (12.6) |
Methylprednisolone | 22 (12.0) |
Celecoxib | 11 (6.0) |
Diclofenac | 11 (6.0) |
PsA = psoriatic arthritis; q.4.w. = every 4 weeks.
Source: BE ACTIVE 2 Clinical Study Report.25
Exposure to Study Treatments
The total time at risk was 392.7 years in the BE ACTIVE 2 study (Table 29).
Because some patients switched from placebo in the BE COMPLETE trial to bimekizumab in the BE VITAL study, exposure is reported separately for patients randomized to bimekizumab or with any exposure to bimekizumab. The total time at risk was 259.5 years among 267 patients who were originally randomized to receive bimekizumab during the BE COMPLETE trial, and this time increased to 339.8 years among the 388 patients who received bimekizumab during the BE VITAL study (Table 29).
Table 29: Patient Exposure During the Extension Studies (Safety Set)
Exposure | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 183) | Week 0 to week 52 from the BE COMPLETE trial Patients randomized to bimekizumab 160 mg q.4.w. (N = 267) | Week 0 to week 52 from the BE COMPLETE trial Any exposure to bimekizumab 160 mg q.4.w., including switch from placebo (N = 388) |
|---|---|---|---|
Duration of exposure, days | |||
Mean (SD) | 678.7 (158.4) | NR | NR |
Median (range) | 728.0 (28.0 to 771.0) | NR | NR |
Time at risk, days | |||
Mean (SD) | 783.8 (168.8) | NR | NR |
Median (range) | 841.0 (1.0 to 856.0) | NR | NR |
Total time at risk, years | 392.7 | 259.5 | 339.8 |
NR = not reported; q.4.w. = every 4 weeks; SD = standard deviation.
Note: Data were not reported for the subgroups categorized according to previous treatment in the BE ACTIVE study.
Sources: BE ACTIVE 2 Clinical Study Report25 and Coates et al. (2024).27
Efficacy
ACR Response
ACR20, ACR50, and ACR70 response rates in the BE ACTIVE 2 study at various study time points relative to the baseline values from the BE ACTIVE study are summarized in Table 30. Rates of ACR20, ACR50, and ACR70 response were similar at baseline and at week 104 with continued bimekizumab treatment in the BE ACTIVE 2 study.
During the BE VITAL trial, patients originally randomized to bimekizumab showed an ACR50 response of 51.7% at week 52, by NRI analysis (Table 31). Patients who were initially randomized to placebo showed an ACR50 response of 40.6% at week 52 (Table 31). ACR20 and ACR70 similarly improved over time for these groups.
Table 30: ACR Response Rates in BE ACTIVE 2 Study Relative to BE ACTIVE Study Baseline (Full Analysis Set, NRI and OC)
Visit | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 181) | ||
|---|---|---|---|
ACR20 | ACR50 | ACR70 | |
Visit 1 (EV for the extension study) | |||
n (%) (NRI) | 145 (80.1) | 115 (63.5) | 82 (45.3) |
n of Nsub (%) (OC) | 145 of 181 (80.1) | 115 of 181 (63.5) | 82 of 181 (45.3) |
Visit 13 (week 104) | |||
n (%) (NRI) | 132 (72.9) | 109 (60.2) | 81 (44.8) |
n of Nsub (%) (OC) | 132 of 157 (84.1) | 109 of 157 (69.4) | 81 of 157 (51.6) |
ACR = American College of Rheumatology; ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; EV = entry visit; NRI = nonresponder imputation; Nsub = N subgroup; OC = observed case; q.4.w. = every 4 weeks.
Note: In the n (%) row (primary analysis), patients for whom ACR could not be derived due to missing data were counted as nonresponders, as this row corresponds to the NRI analysis. In the n of Nsub (%) row (OC sensitivity analysis), Nsub represented the number of patients with a nonmissing measurement at the given week, and percentages were calculated accordingly.
Source: BE ACTIVE 2 Clinical Study Report.25
Table 31: Efficacy Results in BE VITAL Study at Week 16 and Week 52 (Full Analysis Set, NRI and OC)
End Point | Placebo (week 0 to week 16) (N = 133) | Bimekizumab 160 mg q.4.w. (week 16 to week 52) (N = 133) | Bimekizumab 160 mg q.4.w. (N = 267) | |||||
|---|---|---|---|---|---|---|---|---|
Week 16 NRI % (n of N) | Week 16 OC % (n of N) | Week 52 NRI % (n of N) | Week 52 OC % (n of N) | Week 16 NRI % (n of N) | Week 16 OC % (n of N) | Week 52 NRI % (n of N) | Week 52 OC % (n of N) | |
ACR20 | 15.8 (NR) | 16.8 (21 of 125) | 60.2 (NR) | 71.4 (80 of 112) | 67.0 (NR) | 68.1 (179 of 263) | 68.2 (NR) | 78.4 (182 of 232) |
ACR50a | 6.8 (NR) | 7.2 (9 of 125) | 40.6 (NR) | 48.6 (54 of 111) | 43.4 (NR) | 44.1 (116 of 263) | 51.7 (NR) | 59.7 (138 of 231) |
ACR70 | 0.8 (NR) | 0.8 (1 of 125) | 25.6 (NR) | 30.6 (34 of 111) | 26.6 (NR) | 27.0 (71 of 263) | 35.6 (NR) | 40.8 (95 of 233) |
MDA | 6.0 (NR) | 6.4 (8 of 125) | 33.1 (NR) | 39.3 (44 of 112) | 44.2 (NR) | 44.9 (118 of 263) | 47.2 (NR) | 54.3 (126 of 232) |
Complete resolution of enthesitis (LEI)b | 22.2 (8 of 36) | 23.5 (8 of 34) | 58.3 (21 of 36) | 72.4 (21 of 29) | 49.1 (52 of 106) | 50.0 (52 of 104) | 56.6 (60 of 106) | 68.2 (60 of 88) |
Complete resolution of dactylitis (LDI)c | 42.9 (6 of 14) | 42.9 (6 of 14) | 85.7 (12 of 14) | 92.3 (12 of 13) | 70.6 (24 of 34) | 72.7 (24 of 33) | 85.3 (29 of 34) | 93.5 (29 of 31) |
PASI 75d | 10.2 (NR) | 11.4 (9 of 79) | 80.7 (NR) | 97.3 (71 of 73) | 82.4 (NR) | 83.3 (145 of 174) | 84.1 (NR) | 94.9 (148 of 156) |
PASI 90d | 6.8 (NR) | 7.6 (6 of 79) | 73.9 (NR) | 89.0 (65 of 73) | 68.8 (NR) | 69.5 (121 of 174) | 74.4 (NR) | 84.0 (131 of 156) |
PASI 100d | 4.5 (NR) | 5.1 (4 of 79) | 60.2 (NR) | 72.6 (53 of 73) | 58.5 (NR) | 59.2 (103 of 174) | 65.9 (NR) | 74.4 (116 of 156) |
ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; BSA = body surface area; LDI = Leeds Dactylitis Index; LEI = Leeds Enthesitis Index; MDA = minimal disease activity; NRI = nonresponder imputation; OC = observed case; PASI 75 = 75% reduction in Psoriasis Area Severity Index score; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; PASI 100 = 100% reduction in Psoriasis Area Severity Index score; q.4.w. = every 4 weeks.
Note: Table 31 has been adapted from “Table 2 Efficacy outcomes at week 16 and week 52” Coates et al. (2024)27 under the Creative Commons license agreement CC BY-NC 4.0 (https://creativecommons.org/licenses/by-nc/4.0/). Adaptations consist of the inclusion and reordering of only the rows present (e.g., ACR20, PASI 100) and removal of rows from the original table (e.g., VLDA response).
aACR50 at week 16 was the primary end point in the BE COMPLETE trial.
bPatients with enthesitis at baseline defined by LEI greater than 0.
cPatients with dactylitis at baseline defined by LDI greater than 0.
dIn patients with psoriasis affecting at least 3% BSA at baseline.
Source: Coates et al. (2024).27
MDA Response
The proportion of patients who had attained an MDA response was 58.6% at week 104 of the BE ACTIVE 2 study, and similar results were observed in secondary analyses using observed case data (Table 32).
In the BE VITAL study, the proportion of bimekizumab-randomized patients attaining the MDA composite measures of disease activity was 47.2% at week 52. Of initial placebo-randomized patients, after switching to bimekizumab, 33.1% of patients attained MDA (NRI) at week 52 (Table 31).
Table 32: MDA Response Rates by Visit in BE ACTIVE 2 Study Relative to BE ACTIVE Study Baseline (Full Analysis Set, NRI and OC)
Visit | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 181) |
|---|---|
Previous study baseline | |
n (%) (NRI) | 5 (2.8) |
n of Nsub (%) (OC) | 5 of 181 (2.8) |
Visit 1 (EV for the extension study) | |
n (%) (NRI) | 101 (55.8) |
n of Nsub (%) (OC) | 101 of 180 (56.1) |
Visit 13 (week 104) | |
n (%) (NRI) | 106 (58.6) |
n of Nsub (%) (OC) | 106 of 157 (67.5) |
EV = entry visit; MDA = minimal disease activity; NRI = nonresponder imputation; Nsub = N subgroup; OC = observed case; q.4.w. = every 4 weeks.
Note: In the n (%) row (primary analysis), patients for whom MDA could not be derived due to missing data were counted as nonresponders, as this row corresponds to the NRI analysis. In the n of Nsub (%) row (OC sensitivity analysis), Nsub represented the number of patients with a nonmissing measurement at the given week, and percentages were calculated accordingly.
Source: BE ACTIVE 2 Clinical Study Report.25
PASI Response
According to the sponsor summary, an error in the original protocol resulted in a limited number of patients being assessed for PASI response at select midstudy time points. However, this error was addressed in a protocol amendment and nearly all patients had PASI data available for the week 104 visit.
The BE ACTIVE 2 study evaluated PASI 75, PASI 90, and PASI 100 response rates at various time points, which were calculated relative to the baseline values from the BE ACTIVE study (Table 33). At week 104, the proportion of patients who attained PASI 75, PASI 90, and PASI 100 was 79.2%, 73.3%, and 65.8%, respectively.
For the BE VITAL trial, the proportions of patients attaining PASI 75, PASI 90, and PASI 100 increased out to week 52 in both those who were initially placebo-randomized patients and those who were bimekizumab-randomized patients with psoriasis affecting at least 3% BSA at baseline (Table 31).
Table 33: PASI Response Rates in BE ACTIVE 2 Study Relative to BE ACTIVE Study Baseline in Patients With Psoriasis Affecting At Least 3% BSA at Baseline (Full Analysis Set, NRI and OC)
Visit | BE ACTIVE 2 study, PASI 75 (N = 120) | BE ACTIVE 2 study, PASI 90 (N = 120) | BE ACTIVE 2 study, PASI 100 (N = 120) |
|---|---|---|---|
Visit 1 (EV for the extension study) | |||
n (%) (NRI) | 106 (88.3) | 96 (80.0) | 85 (70.8) |
n of Nsub (%) (OC) | 106 of 120 (88.3) | 96 of 120 (80.0) | 85 of 120 (70.8) |
Visit 13 (week 104) | |||
n (%) (NRI) | 95 (79.2) | 88 (73.3) | 79 (65.8) |
n of Nsub (%) (OC) | 95 of 107 (88.8) | 88 of 107 (82.2) | 79 of 107 (73.8) |
BSA = body surface area; EV = entry visit; NRI = nonresponder imputation; Nsub = S nubgroup; OC = observed case; PASI = Psoriasis Area Severity Index; PASI 75 = 75% reduction in Psoriasis Area Severity Index score; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; PASI 100 = 100% reduction in Psoriasis Area Severity Index score.
Note: In the n (%) row (primary analysis), patients for whom PASI could not be derived due to missing data were counted as nonresponders as this row corresponds to the NRI analysis. In the n of Nsub (%) row (OC sensitivity analysis), Nsub represented the number of patients with a nonmissing measurement at the given week, and percentages were calculated accordingly.
Source: BE ACTIVE 2 Clinical Study Report.25
LEI and LDI Response
Data regarding the LEI were only collected during the BE VITAL study among patients with enthesitis at baseline. For patients who switched from placebo to bimekizumab, the enthesitis resolution rate for LEI was 58.3% at week 52. Of the patients originally randomized to bimekizumab, 56.6% had enthesitis resolution for LEI at week 52 (Table 31).27
Due to the low number of patients with dactylitis in the BE ACTIVE 2 study, and the corresponding low number of data points for analysis, convergence issues were observed with the multiple imputation model. Therefore, analysis of LDI scores was only based on observed case data (Table 34). Compared to the baseline from the BE ACTIVE study, the mean change from baseline for LDI scores was –47.2 (SD = 78.2) at patients’ entry visit for the BE ACTIVE 2 study and was sustained to week 104 (–60.7 [SD = 71.5]) with continued bimekizumab treatment.
Among patients with dactylitis at baseline in the BE VITAL study, for patients who switched from placebo to bimekizumab, the rate of dactylitis resolution was 85.7% at week 52. For patients originally randomized to bimekizumab, the rate of dactylitis resolution was 85.3% at week 52 (Table 31).27
Table 34: Changes in LDI Scores by Visit in Extension Studies Relative to Preceding Study Baseline (Full Analysis Set, OC Data, No Imputation)
Visit | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 181) |
|---|---|
Previous study baseline | |
Mean (SD) | 56.8 (66.4) |
Visit 1 (EV for the extension study) | |
CFB, mean (SD) | –47.2 (78.2) |
Visit 13 (week 104) | |
CFB, mean (SD) | –60.7 (71.5) |
CFB = change from baseline; EV = entry visit; LDI = Leeds Dactylitis Index; OC = observed case; q.4.w. = every 4 weeks; SD = standard deviation.
Source: BE ACTIVE 2 Clinical Study Report.25
SF-36
The mean SF-36 PCS change score was sustained with continued bimekizumab treatment up to week 104, with mean PCS change of 9.5 (SE = 0.8) (Table 35).
In the BE VITAL study, data regarding the SF-36 PCS were only reported up to week 40. The mean SF-36 PCS change from baseline was 7.3 (SE = 0.9) at week 40 for patients who switched from placebo to bimekizumab. For those who were originally randomized to bimekizumab, the mean SF-36 PCS change from baseline was 8.4 (SE = 0.6) at week 40 (Table 36).27
Table 35: Changes in SF-36 Scores by Visit in Extension Studies Relative to Preceding Study Baseline (Full Analysis Set, MI)
Visit | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 181) SF-36 PCS |
|---|---|
Previous study baseline | |
Mean (SE) | 36.51 (0.66) |
Visit 1 (EV for the extension study) | |
CFB, mean (SE) | 9.45 (0.67) |
Visit 13 (week 104) | |
CFB, mean (SE) | 9.50 (0.76) |
CFB = change from baseline; EV = entry visit; MI = multiple imputation; PCS = physical component summary; q.4.w. = every 4 weeks; SE = standard error; SF-36 = Short Form (36) Health Survey.
Note: Missing values were replaced by 1 of a set of plausible values, where each value is a Bayesian draw from the conditional distribution of the missing data given the observed data. This was performed 100 times and summaries of the resulting datasets were combined for reporting.
Source: BE ACTIVE 2 Clinical Study Report.25
Table 36: Efficacy Results in BE VITAL Study at Week 16 and Week 40 (Full Analysis Set)
TEnd point | Placebo (week 0 to week 16) (N = 133) | Bimekizumab 160 mg q.4.w. (week 16 to week 52) (N = 133) | Bimekizumab 160 mg q.4.w. (N = 267) | |
|---|---|---|---|---|
SF-36 PCS CFB | Week 16 | Week 40 | Week 16 | Week 40 |
MI, mean (SE) | 1.4 (0.7) | 7.3 (0.9) | 7.3 (0.5) | 8.4 (0.6) |
CFB = change from baseline; MI = multiple imputation; PCS = physical component summary; q.4.w. = every 4 weeks; SE = standard error; SF-36 = Short Form (36) Health Survey.
Note: Table 36 has been adapted from “Table 2 Efficacy outcomes at week 16 and week 52” Coates et al. (2024)27 under the Creative Commons license agreement CC BY-NC 4.0 (https://creativecommons.org/licenses/by-nc/4.0/). Adaptations consist of the inclusion and reordering of only the rows present (e.g., SF-36 PCS) and removal of rows from the original table (e.g., ACR20 response).
Source: Coates et al. (2024).27
Harms
Harms results for the BE ACTIVE 2 and BE VITAL studies are displayed in Table 37 and Table 38, respectively. The total time at risk was 392.3 patient-years during the BE ACTIVE 2 study. Most patients reported TEAEs (80.9%), which were most commonly infections and infestations (55.2%). The proportion of patients who reported SAEs was 7.7%. The discontinuation of bimekizumab treatment due to TEAEs occurred among 4.9% of patients, mainly due to oral fungal infection (1.6%). No deaths were reported.
In the BE VITAL study, at least 1 TEAE was reported by 243 of 388 (62.6%) patients while receiving bimekizumab up to week 52. The most frequently reported TEAEs were hypersensitivity (4.9%), SARS-CoV-2 (COVID-19) infections (7.2%), fungal infections (9.5%), nasopharyngitis (5.9%), and urinary tract infection (5.9%). Serious infections occurred among 1.8% of patients, and 1.3% of patients had neutropenia. The proportion of patients who reported SAEs was 5.9%. The discontinuation of bimekizumab treatment due to TEAEs occurred among 4.1% of patients. One death was reported and deemed unrelated to study treatment.
Table 37: Summary of Harms in BE ACTIVE 2 Study (Safety Set)
Adverse event | BE ACTIVE 2 study, bimekizumab 160 mg q.4.w. (N = 183) |
|---|---|
Most common adverse events, n (%) | |
Patients with ≥ 1 TEAE | 148 (80.9) |
TEAEs reported for ≥ 3% of patients | |
Infections and infestations | 101 (55.2) |
Upper respiratory tract infection | 20 (10.9) |
Nasopharyngitis | 19 (10.4) |
Oral candidiasis | 13 (7.1) |
Bronchitis | 11 (6.0) |
Pharyngitis | 10 (5.5) |
Sinusitis | 10 (5.5) |
Oral fungal infection | 9 (4.9) |
Gastroenteritis | 6 (3.3) |
Tonsillitis | 6 (3.3) |
Urinary tract infection | 6 (3.3) |
Conjunctivitis | 6 (3.3) |
Skin and subcutaneous tissue disorders | 35 (19.1) |
Psoriasis | 14 (7.7) |
Musculoskeletal and connective tissue disorders | 35 (19.1) |
Psoriatic arthropathy | 12 (6.6) |
Investigations | 18 (9.8) |
Alanine aminotransferase, increased | 6 (3.3) |
Aspartate aminotransferase, increased | 6 (3.3) |
Gamma-glutamyltransferase, increased | 6 (3.3) |
SAEs, n (%) | |
Patients with ≥ 1 SAE | 14 (7.7) |
Gastrointestinal disorders | 2 (1.1) |
Anal fistula | 1 (0.5) |
Inguinal hernia | 1 (0.5) |
Umbilical hernia | 1 (0.5) |
Hepatobiliary disorders | 2 (1.1) |
Cholelithiasis | 2 (1.1) |
Injury, poisoning, and procedural complications | 2 (1.1) |
Meniscus injury | 1 (0.5) |
Ligament rupture | 1 (0.5) |
Musculoskeletal and connective tissue disorders | 3 (1.6) |
Osteochondrosis | 1 (0.5) |
Foot deformity | 2 (1.1) |
Reproductive system and breast disorders | 2 (1.1) |
Ovarian cyst | 1 (0.5) |
Uterine polyp | 1 (0.5) |
Patients who stopped bimekizumab treatment due to TEAEs, n (%) | |
Patients who stopped bimekizumab treatment | 9 (4.9) |
Eye disorders | 1 (0.5) |
Cataract | 1 (0.5) |
Gastrointestinal disorders | 2 (1.1) |
Paresthesia, oral | 1 (0.5) |
Aphthous ulcer | 1 (0.5) |
Infections and infestations | 3 (1.6) |
Oral fungal infection | 2 (1.1) |
Cellulitis | 1 (0.5) |
Folliculitis | 1 (0.5) |
Pregnancy, puerperium, and perinatal conditions | 1 (0.5) |
Pregnancy on contraceptive | 1 (0.5) |
Skin and subcutaneous tissue disorders | 2 (1.1) |
Dermatitis, allergic | 1 (0.5) |
Psoriasis | 1 (0.5) |
Deaths, n (%) | |
Patients who died | 0 |
Adverse events of special interest, n (%) | |
Potential case of liver dysfunction based on Hy’s lawa | 0 |
Q.4.w. = every 4 weeks; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aHy’s law is defined as an alanine transaminase or aspartate transaminase level at least 3 times the upper limit of normal with coexisting total bilirubin at least 2 times the upper limit of normal in the absence of alkaline phosphatase at least 2 times the upper limit of normal.
Source: BE ACTIVE 2 Clinical Study Report.25
Table 38: Summary of Harms in BE VITAL Study (Safety Set)
Adverse event | BE VITAL study, bimekizumab 160 mg q.4.w., week 0 to week 52 (N = 388) |
|---|---|
Most common adverse events, n (%) | |
Patients with ≥ 1 TEAE | 243 (62.6) |
TEAEs reported for ≥ 5% of patients | |
SARS-CoV-2 (COVID-19) infections | 28 (7.2) |
Oral candidiasis | 24 (6.2) |
Nasopharyngitis | 23 (5.9) |
Urinary tract infection | 23 (5.9) |
SAEs, n (%) | |
Patients with ≥ 1 SAE | 23 (5.9) |
Patients who stopped bimekizumab treatment due to TEAEs, n (%) | |
Patients who stopped bimekizumab treatment | 16 (4.1) |
Deaths, n (%) | |
Patients who died | 1 (0.3)a |
Q.4.w. = every 4 weeks; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aThere was a sudden death in a 54-year-old patient with a history of hypertension, aortic regurgitation, and electrocardiogram changes reflective of coronary artery disease; no further information was available.
Source: Coates et al. (2024).27
Critical Appraisal
Internal Validity
The open-label designs of the BE ACTIVE 2 and BE VITAL studies could bias the magnitude of the efficacy of outcomes due to unblinded exposure to the study medication during the treatment period. In addition to that, the absence of control groups in both studies and the lack of data beyond week 52 in the BE VITAL study make interpretation of the findings challenging. Only those who completed the BE ACTIVE, BE OPTIMAL, and BE COMPLETE studies moved on to the BE ACTIVE 2 and BE VITAL studies; there may have been selection bias involved. Given the ongoing nature of the BE VITAL study, the data presented in the CADTH summary are based on only a subset of the total eligible population for the OLE studies — namely, patients who have previous exposure to TNFi(s). Longer-term evidence for bDMARD-naive patients is forthcoming.
External Validity
As the BE ACTIVE 2 and BE VITAL studies consisted of patients who took part in the pivotal studies (the BE ACTIVE, BE OPTIMAL, and BE COMPLETE studies), it is reasonable to expect that the same strengths and limitations related to generalizability apply to the extension studies. The patient population of those studies may not be reflective of the wider, more heterogeneous clinical population in terms of demographic and clinical characteristics; therefore, the results presented may differ from those observed in a real-world clinical setting.
Indirect Evidence
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
A review of the indirect evidence was required as none of the pivotal trials or OLEs conducted comparisons between bimekizumab and the active control group, and the pivotal trials did not contain any active comparators other than adalimumab. An appraisal of the ITCs submitted by the sponsor was also necessary as data from the NMA were used to inform the clinical pharmacoeconomic model.
Description of Indirect Comparisons
The sponsor submitted an NMA and MAIC to assess the relative efficacy and safety of bimekizumab and other licensed biologic DMARDs for the treatment of adult-onset PsA. The NMA assessed efficacy and safety outcomes at week 12 to week 24 while the MAIC assessed efficacy outcomes at week 52.
ITC Design for NMA and MAIC
Objectives
The NMA was performed to generate evidence regarding the efficacy and safety of bimekizumab relative to relevant comparators at week 12 to week 24, while the objective of the MAIC was to generate comparative evidence regarding the 52-week clinical efficacy of bimekizumab relative to other key comparators.
Study Selection Methods
Both the NMA and the MAIC were informed by the same systematic literature review (SLR). RCTs of comparators in the PsA setting were identified via an SLR that was initially conducted on December 3, 2015, and then updated on January 7, 2020; May 2, 2022; and January 1, 2023. Table 39 outlines the study population, interventions, comparators, outcomes of interest, and study designs included in the sponsor’s SLR.
Briefly, the SLR for both the NMA and the MAIC included phase II to phase IV RCTs conducted in patients with adult-onset PsA treated with 1 of a set of specified interventions from drug classes. The MAIC specified IL-17is, IL-23is, and IL-12/23is only, while the NMA further included specific TNFis, CTLA immunoglobulin, JAKis, and a PDE4i. Biosimilars were not included and the submission did not specify why the drug classes of interest and the outcomes were different between the NMA and the MAIC. The comparators of interest for the NMA and the MAIC were standard of care or placebo.
Specific dosages for the interventions of interest (listed in Table 39) were excluded on the basis of not being licensed or relevant for PsA. The submission did not specify criteria for relevance to PsA or geographical reference for licensing. Study quality was assessed using the Cochrane Risk of Bias 2 checklist and summary results presented; no preplanned sensitivity analyses on the basis of study quality were specified.
Table 39: Study Selection Criteria and Methods for NMA and MAIC Submitted by Sponsor
Characteristic | NMA | MAIC |
|---|---|---|
Population | Adult-onset PsA, including subgroups of patients who were:
| |
Intervention |
|
|
Comparator | Standard of care (e.g., cDMARDs, NSAIDs, methotrexate), PBO | |
Outcome | At 12 weeks, 16 weeks, and 24 weeks (± 2 weeks):
At any time point:
| At week 52 (week 48 for 1 study):
|
Study designs | Randomized controlled trials, phase II, phase II or phase III, and phase III and phase IV | |
Publication characteristics | Original publications, including conference abstracts | |
Exclusion criteria |
|
|
Databases searched |
| |
Selection process | Screening was conducted independently and in duplicate by 2 researchers working in parallel. Disagreements on a publication’s eligibility were resolved by discussion or, if an agreement could not be reached, by a third reviewer providing arbitration. | |
Data extraction process | Data extraction was completed by a single reviewer with a second reviewer verifying the data against the original data source(s). In any cases of disagreement, a third reviewer was involved. A higher-level quality review by a senior reviewer followed. | |
Quality assessment | The Cochrane Risk of Bias 2 tool was used to evaluate study quality. | |
ACP = American College of Physicians; ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; b/tsDMARD = biologic or targeted synthetic disease-modifying antirheumatic drug; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; IL-12/23i = interleukin-12 and interleukin-23 inhibitor; IL-17i = interleukin-17 inhibitor; IL-23i = interleukin-23 inhibitor; JAKi = Janus kinase inhibitor; MAIC = matching-adjusted indirect comparison; MDA = minimal disease activity; NHS = National Health Service; NMA = network meta-analysis; NSAID = nonsteroidal anti-inflammatory drug; PASI 50 = 50% reduction in Psoriasis Area Severity Index score; PASI 75 = 75% reduction in Psoriasis Area Severity Index score; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; PASI 100 = 100% reduction in Psoriasis Area Severity Index score; PBO = placebo; PDE4i = phosphodiesterase type 4 inhibitor; PsA = psoriatic arthritis; PsARC = Psoriatic Arthritis Response Criteria; q.2.w. = every 2 weeks; SC = subcutaneous; TNFi = tumour necrosis factor inhibitor; vdHmTSS = van der Heijde modified total Sharp score; VLDA = very low disease activity.
Source: Sponsor’s summary of clinical evidence.24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
NMA Analysis Methods
Using studies from the SLR, networks were constructed only of treatments that were deemed relevant to clinical practice (i.e., recommended by key clinical guidelines, licensed by regulatory bodies, and/or routinely used). The submission did not specify specific guidelines, regulatory bodies, or clinical practice settings that may have been used as references for treatment selection. Trials were only included in the network if they fulfilled 1 of the following 3 criteria:
comprised 2 or more licensed or recommended treatment group
had a licensed or recommended group if that trial was needed to form a complete network
was a potential future comparator of bimekizumab at launch (time frame and launch date not specified in the submission).
Specific dosing regimens alone or in combination were listed in the submission and analyzed as separate nodes. Treatment groups of studies that were not relevant to the proposed comparisons were not included in the networks. Placebo was used as a common comparator in all networks. A total of 31 studies had a crossover design at the 24-week mark or earlier, and precrossover trial data were used when possible.
Detailed analysis methods are described in Table 40. Briefly, 2 sets of primary analyses were conducted for efficacy end points: 1 for a b/tsDMARD-naive PsA population and 1 for a TNFi-experienced PsA population. For safety end points, NMAs were run on the combined population following discussion with clinicians. The main analysis consisted of univariate fixed-effect and random-effects Bayesian NMAs on individual binary and continuous outcomes. The submission did not provide a justification for the use of NMA over other methodologies.
Baseline risk adjustment using meta-regression was undertaken to account for heterogeneity. Clinician input was not explicitly reported for all characteristics in the NMA but was referenced for certain characteristics as part of the discussion on whether instances of observed heterogeneity were important. Model fit was explored for fixed-effect, random-effects, baseline risk-adjusted, and baseline risk-unadjusted analyses. Model fit results from all 4 runs were reported, but for each outcome, the model selected for discussion and results reporting was chosen on the basis of the following criteria:
if the 95% CrI of baseline risk did not contain 0, the baseline-adjusted model was selected; otherwise, the unadjusted model was selected
for fixed-effect versus random-effects models, if the deviance information criterion for the random-effects model was lower than the deviance information criterion for the fixed-effect model by at least 3 points, then the random-effects model was selected; otherwise, the fixed-effect model was selected.
The possibility of rare events in the NMAs was handled by either using imputed data from a lower threshold for which there were fewer non-0 respondents, or via a continuity correction. If missing, SEs were calculated from SDs or by imputation if SD was also missing.
NMAs were run separately for each outcome and for the 2 populations of interest. For efficacy outcomes, 2 scenarios were run in each group:
base case: studies where data were available at the preferred time point of 16 weeks or, where not available (or earlier crossover occurred), were available at 12 weeks, 14 weeks, or 24 weeks
scenario 1: studies where data were available at the preferred time point of 16 weeks or, where not available (or earlier crossover occurred), were available at 12 weeks or 14 weeks without including week 24 data.
For safety outcomes (SAEs, discontinuations, and discontinuations due to AEs), data from the b/tsDMARD-naive and TNFi-experienced populations were pooled and a single set of analyses run as follows:
base case: studies where data were available at the preferred time point of 16 weeks or, where not available (or earlier crossover occurred), were available at 12 weeks, 14 weeks, or 24 weeks.
The base-case results for safety and efficacy end points are summarized in this report for the ACR20, ACR50, ACR70, PASI 90, and MDA outcomes.
Table 40: NMA Analysis Methods
Method | Description |
|---|---|
Analysis methods | Bayesian approach:
|
Priors | Random-effects NMAs used a vaguely informative normal prior with a uniform SD equivalent to 1.0 for all binary outcomes, and U (0, 2X) for continuous outcomes, where X is the median SD of the outcome within the study. Unconstrained parameters (such as treatment effects) used a prior of N (0, 10,000), though this was decreased to N (0, 1,000) in cases where convergence was imperfect. Similarly, where convergence was problematic, a distribution of U (0.001, 1) was used for the square root of the random-effects variance as opposed to U (0, 1). |
Assessment of model fit | For each of the 4 models, model fit statistics were reported, including:
|
Assessment of consistency | Consistency of the results was assessed using qualitative assessment between inputs and outputs via matrix of ORs or mean change (not provided in submission). |
Assessment of convergence |
|
Outcomes | Efficacy:
Safety:
|
Follow-up time points | Base case: Outcomes assessed at week 12 to week 24 Scenario 1: Outcomes assessed at week 12, week 14, and week 16
|
Construction of nodes | The analysis networks only include treatments that are relevant to clinical practice (i.e., recommended by key clinical guidelines, licensed by key regulatory bodies, and/or routinely used) or are in late-stage development and could be a potential future competitor. Each dose was a separate node in the network. Trials were only included in the network if 1 of the following applied:
|
Sensitivity analyses | All NMAs were rerun with data from scenario 1 described previously, excluding data from the 24-week mark |
Subgroup analysis | b/tsDMARD-naive population; TNFi-experienced population |
Methods for pairwise meta-analysis | Not reported |
ACR = American College of Rheumatology; AE = adverse event; b/tsDMARD = biologic or targeted synthetic disease-modifying antirheumatic drug; DIC = deviance information criterion; MDA = minimal disease activity; NMA = network meta-analysis; OR = odds ratio; PASI = Psoriasis Area Severity Index; PBO = placebo; PsARC = Psoriatic Arthritis Response Criteria; SAE = serious adverse event; SD = standard deviation; TNFi = tumour necrosis factor inhibitor; VLDA = very low disease activity.
Sources: Sponsor’s summary of clinical evidence and network meta-analysis technical document.24,30
MAIC Analysis Methods
The justification for an MAIC was to provide long-term comparative efficacy data; the submission did not provide any justification for an MAIC over other methodologies. As many of the studies employed a crossover design, patients in all trials who were initially randomized to placebo were excluded from the MAIC due to the fact that they would not have received placebo treatment for the entirety of the 52-week treatment period.
Analyses were undertaken in 2 populations of interest: bDMARD-naive patients and TNFi-experienced patients. The source of the RCTs included in the MAIC was the same SLR as for the NMA, and the submission contained a list of excluded studies with rationales for the exclusion of each one. Missing data were imputed for bimekizumab and comparators using NRI.
To generate matching factors, there were 2 previous MAICs referenced in patients with PsA112,113 and to shortlist potential matching characteristics, qualitative comparisons of baseline characteristics across the comparator trials, consultation with clinicians (n = 5), as well as an assessment of shortlisted variables quantitatively via univariate analyses measuring the impact on each outcome of interest and in the effective sample size when a characteristic was controlled were undertaken. This was carried out for every comparator treatment for both populations of interest. The final matching factors are shown in Table 41. Matching trial samples on race, weight, and DMARD use at baseline, the proportion of patients with dactylitis, and the proportion of patients with enthesitis at baseline was not undertaken.
Table 41: Included and Excluded Matching Factors in MAIC
Matching factor (all outcomes) | Excluded matching factor (with sponsor justification) |
|---|---|
Age | DMARD use at baseline (not reported in several studies; MTX adjusted instead) |
Sex | CRP (not usually adjusted for in MAICs and not consistently reported) |
MTX use | Weight or BMI (minor impact; similar across trials and not available from 1 trial) |
HAQ-DI score | Enthesitis at baseline (ESS too small after adjustment) |
BSA ≥ 3% with psoriasis | Dactylitis at baseline (ESS too small after adjustment) |
SJC based on 66 joints | Race (percentage of white participants very similar across trials; minor impact of adjustment) |
TJC based on 68 joints | — |
Disease duration | — |
BMI = body mass index; BSA = body surface area; DMARD = disease-modifying antirheumatic drug; ESS = effective sample size; HAQ-DI = Health Assessment Questionnaire–Disability Index; MAIC = matching-adjusted indirect comparison; MTX = methotrexate; SJC = swollen joint count; TJC = tender joint count.
Source: Sponsor’s matching-adjusted indirect comparison technical document.29
Following the selection of matching factors, patients from the relevant bimekizumab trial (the BE OPTIMAL trial for bDMARD-naive patients and the BE COMPLETE trial for TNFi-experienced patients) were reweighted to match the baseline characteristics of the relevant comparator trial patients, with weights determined via logistic regression. NRI was used for both comparators and bimekizumab.
For the comparison with secukinumab, 3 analyses were undertaken in both the bDMARD-naive and TNFi-experienced populations. In the first analysis, patients from the BE OPTIMAL (or BE COMPLETE) trial were compared with the relevant subgroup from the FUTURE 2 trial. In the second analysis, patients from the BE OPTIMAL (or BE COMPLETE) trial were compared to the relevant subgroup from the FUTURE 5 trial. In the third analysis, patients from the BE OPTIMAL (or BE COMPLETE) trial were compared to the relevant subgroup data from the FUTURE 2, FUTURE 3, FUTURE 4 (efficacy and safety of secukinumab 150 mg), and FUTURE 5 trials pooled together. For the remaining comparisons, published data from the entire comparator study population was used with the exception of secukinumab, guselkumab, and ustekinumab, which were trials with multiple doses of the same comparator; here, subgroups of the different dosing regimens were also compared in separate analyses.
The 52-week outcomes for ACR20, ACR50, ACR70, and MDA were calculated by applying weights from the matching logistic regression, and the recalculated outcomes were compared to comparator outcomes via unanchored, non–placebo-adjusted comparisons. Results were reported as ORs with 95% CIs (95% CI based on ESS).
Results of NMA
Summary of Included Studies
The initial and updated searches identified 1,160 records of potential interest; after deduplication and screening, 540 records were included, comprising 66 RCTs. Of these, 41 trials were deemed feasible to be included in the NMA based on the study selection criteria. Twenty-five unique trials were excluded for the following reasons: biosimilar product and study design (n = 2), study design issue (n = 3), irrelevant population (n = 13), cDMARD-naive population (n = 4), and wrong or unlicensed dose (n = 3). The 41 trials reported outcomes at 12 weeks, 16 weeks, or 24 weeks and met the criteria for inclusion in the NMA in either a b/tsDMARD-naive population, a TNFi-experienced PsA population, or a mixed population.
Domain-level summaries of the Cochrane Risk of Bias 2 assessments were presented for the included trials. Overall, 6 studies were given an “unclear” risk of bias rating due to unclear randomization methods. Several studies were given a bias rating of “some concerns,” all due to the outcome measurement domain; the submission notes that in some cases, outcome reporting was not always available at the desired time points and the blinding of outcome assessments was unclear in 21 studies. There were no sensitivity analyses to ascertain the impact of these biases on the NMA results.
A summary of the assessment of homogeneity for the NMA is described in Table 42. Age, sex, ethnicity, the mean time since diagnosis, concomitant use of methotrexate, concomitant use of NSAIDs, concomitant use of steroids, PASI, and a DAS28 score were shortlisted in the submission for heterogeneity assessment. Heterogeneity was assessed by plotting the mean values from individual studies against the pooled mean for each characteristic; a Grubbs’ Test to check for outliers across studies was followed by a brief qualitative description and conclusion of whether heterogeneity was high or low. There was no information on how the list of characteristics was generated, nor were there systematic tests or justifications for the submission’s conclusions of high or low heterogeneity.
Results
b/tsDMARD-Naive Population, Base Case
The evidence networks for the b/tsDMARD-naive patient populations are shown in Appendix 1, Figure 3 to Figure 7, and the efficacy results of the NMA analyses for ACR20, ACR50, ACR70, PASI 90, and MDA over week 12 to week 24 (base case) in b/tsDMARD-naive patients are presented in Table 43. For all outcomes presented in this patient population, the results from fixed-effect, baseline-adjusted models were reported. There were 19 comparator treatment regimens for each ACR outcome, 11 comparator treatment regimens for MDA, and 13 comparator treatment regimens for PASI 90.
Table 42: Assessment of Homogeneity for NMA
Characteristic | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Not evaluated |
Treatment history | Subgroups of b/tsDMARD-naive patients and TNFi-experienced patients considered in separate analyses; concomitant use of MTX, NSAIDs, and steroids also considered separately as potential sources of heterogeneity |
Trial eligibility criteria | No specific requirements for the SLR screening. Standard disease definition (CASPAR criteria) was observed for the inclusion criteria of most studies, with 3 to 5 affected joints (each swollen and tender) required for active disease. A total of 26 studies also mentioned psoriasis or a history of psoriasis in the inclusion criteria. Exclusion criteria were variable across studies and a potential source of heterogeneity. |
Dosing of comparators | Each dosing regimen was considered as a separate node in the network, even if the active drug was the same (with the exception of certolizumab pegol). The regimens were limited to recommended and/or licensed dosing regimens but there was no specification of geographic locations for licensure. |
Placebo response | Not evaluated; analysis methods included baseline adjustment |
Definitions of end points | ACR, PASI, and MDA are end points with standardized criteria |
Timing of end point evaluation | Variable; ranged ± 2 weeks from week 12, week 16, and week 24 |
Withdrawal frequency | Not evaluated |
Clinical trial setting | Not evaluated |
Study design | All were English language phase II to phase IV, DB, RCTs apart from 1 open-label, assessor-blinded RCT; 31 studies had a crossover design between week 12 and week 24, and 23 studies had an early escape design at week 12 or week 16. NMA data were taken from precrossover time points where possible. Study design was a possible source of heterogeneity for outcome assessment due to variation in crossover time points and early escape designs. |
Age | The number of studies that reported age as well as the range of median ages was reported. The mean age from each study was plotted relative to the pooled mean and Grubbs’ Test was used to check for outliers. Age is a potential source of heterogeneity as there was a 10-year age range across studies; no outliers were identified. |
Sex | The number of studies that reported sex as well as the range of median proportion of males was reported. The mean proportion of males from each study was plotted relative to the pooled mean and Grubbs’ Test was used to check for outliers. Heterogeneity present among the included studies was based on the reported ranges across studies (41.9% to 61.5% male), with no single outlier study identified. |
Ethnicity | The number of studies that reported on ethnicity as well as the range of median proportion of white participants was reported. The mean proportion of white participants from each study was plotted relative to the pooled mean and Grubbs’ Test was used to check for outliers. While no outlier was identified, there was a wide range in the proportion of white participants (76.5% to 98%), and trial participants were majority white in general, impacting generalizability. |
Mean time since diagnosis | The number of studies that reported time since diagnosis as well as the range of mean times since diagnosis was reported. The mean time since diagnosis from each study was plotted relative to the pooled mean and Grubbs’ Test was used to check for outliers. This is likely a source of heterogeneity based on the range of mean time since diagnosis (3.5 years to 11.4 years). |
Concomitant use of MTX | The number of studies that reported MTX use as well as the range of mean MTX use was reported. The mean proportion of MTX use from each study was plotted relative to the pooled mean to check for outliers. Ranging from 29.1% to 84.0%, the concomitant use of MTX is a likely source of heterogeneity. |
Concomitant use of NSAIDs | The number of studies that reported the use of NSAIDs as well as the range of NSAID use was reported. The mean proportion of NSAID use from each study was plotted relative to the pooled mean to check for outliers. The range of use was wide (33.3% to 100%), suggesting a notable source of heterogeneity. |
Concomitant use of steroids | The number of studies that reported concomitant steroid use as well as the range of steroid use was reported. The mean proportion of steroid use from each study was plotted relative to the pooled mean and Grubbs’ Test was used to check for outliers. One study was identified as an outlier with a much greater use of concomitant steroids. Concomitant steroid use is a likely a source of heterogeneity due to the range in steroid use (9.2% to 30.0%). |
PASI | The number of studies that reported a PASI as well as the range of PASI was reported. The mean score from each study was plotted relative to the pooled mean and Grubbs’ Test was used to check for outliers. The mean PASI baseline score is likely a source of heterogeneity due to the range (4.7 to 13.0). |
DAS28 | The number of studies that reported a DAS28 score (in 28 joints) as well as the range of scores was reported. The mean score from each study was plotted relative to the pooled mean and Grubbs’ Test was used to check for outliers. DAS28 did not range widely but was not widely reported (fewer than half of studies reported it); therefore, its heterogeneity cannot be qualified. |
ACR = American College of Rheumatology; b/tsDMARD = biologic or targeted synthetic disease-modifying antirheumatic drug; CASPAR = ClASsification criteria for Psoriatic ARthritis; DAS28 = Disease Activity Score 28; DB = double-blind; MDA = minimal disease activity; MTX = methotrexate; NMA = network meta-analysis; NSAID = nonsteroidal anti-inflammatory drug; PASI = Psoriasis Area Severity Index; PsA = psoriatic arthritis; RCT = randomized controlled trial; SLR = systematic literature review; TNFi = tumour necrosis factor inhibitor.
Source: Sponsor’s summary of clinical evidence, NMA Technical Report, and a summary of trial inclusion and exclusion criteria provided by the sponsor.24,30
Briefly, bimekizumab was favoured for ACR20, ACR50, and ACR70 over IL-12/23is or IL-23is (with the exception of guselkumab 100 mg every 8 weeks and ustekinumab 90 mg, which were favoured for ACR70). Bimekizumab was also favoured over abatacept for ACR20, ACR50, and ACR70, and apremilast for ACR20. Bimekizumab was not significantly favoured over IL-17is, TNFis, or JAKis across any other ACR outcomes, with the exception of ixekizumab 80 mg every 4 weeks and etanercept 25 mg, where it was favoured for ACR70. Golimumab 2 mg IV was favoured over bimekizumab for ACR20 and ACR50.
For the MDA outcome, bimekizumab was favoured over guselkumab and risankizumab (other IL-12/23i and IL-23i comparisons were not analyzed), as well as over adalimumab 40 mg. For the PASI 90 outcome, bimekizumab was favoured over TNFis with the exception of infliximab 5 mg IV, although CrIs were generally wide, suggesting less precision in the magnitude of the effect estimates. Bimekizumab was also favoured over an IL-17i (secukinumab 150 mg), an IL-12/23i (risankizumab 150 mg), and a JAKi (upadacitinib 15 mg).
The alternate analysis (scenario 1) included only data from week 12 to week 16 and followed the same model selection criteria. The scenario 1 analysis results used the same model in scenario 1 as was selected for the base case for the ACR20, ACR50, ACR70, and PASI 90 outcomes; for MDA, a random-effects, unadjusted model was selected instead. Notable differences between the base case and scenario 1 were that bimekizumab became favourable over secukinumab 150 mg for ACR20, became favourable over apremilast 30 mg for ACR50, was no longer favoured over guselkumab 100 mg every 4 weeks for ACR70, and was no longer favoured over adalimumab 40 mg for MDA.
TNFi-Experienced Population, Base Case
The evidence networks for the NMAs conducted in TNFi-experienced patients are presented in Appendix 1, Figure 8 to Figure 12, and the efficacy results for the NMAs in TNFi-experienced patients are presented in Table 44. The choice of model for each outcome varied due to the model selection criteria, but results from a random-effects, baseline-unadjusted model were reported for ACR20, ACR50, and MDA, while results from a fixed-effect, baseline-adjusted model were reported for ACR70 and results from a fixed-effect, baseline-unadjusted model were reported for PASI 90. There were 14 comparator treatment regimens for ACR20, ACR50, and ACR70, 9 comparator regimens for MDA, and 8 comparator regimens for PASI 90.
Briefly, bimekizumab was favoured over ixekizumab, secukinumab, guselkumab 100 mg every 8 weeks, risankizumab, ustekinumab, tofacitinib, upadacitinib, and abatacept for ACR20; secukinumab, tofacitinib, and abatacept for ACR50; and guselkumab 100 mg every 8 weeks and ustekinumab for ACR70. For MDA, bimekizumab was favoured over tofacitinib. For PASI 90, bimekizumab was favoured over upadacitinib. CrIs for most outcomes were wide, suggesting reduced precision in the magnitude of the effect estimate and possibly the direction of effect in certain comparisons.
The alternate analysis (scenario 1) included only data from week 12 to week 16 and followed the same model selection criteria. The same models were selected for scenario 1 as had been selected in the base case. Briefly, the main changes between the base case and scenario 1 were that bimekizumab was no longer favourable over secukinumab 150 mg for ACR50. Estimates also increased and CrIs widened dramatically for all comparators for ACR70 so that bimekizumab became favoured over secukinumab 150 mg, secukinumab 150 mg no loading dose, secukinumab 300 mg, and tofacitinib 5 mg with high imprecision.
Table 43: Key NMA Efficacy Results for b/tsDMARD-Naive Patients
Comparator | ACR20 | ACR50 | ACR70 | PASI 90 | MDA |
|---|---|---|---|---|---|
OR (95% CrI)a | OR (95% CrI)a | OR (95% CrI)a | OR (95% CrI)a | OR (95% CrI)a | |
Model | Fixed-effect, baseline-adjusted univariateb | Fixed-effect, baseline-adjusted univariateb | Fixed-effect, baseline-adjusted univariateb | Fixed-effect, baseline-adjusted univariateb | Fixed-effect, baseline-adjusted univariateb |
Number of studies | 31 | 23 | 20 | 14 | 16 |
Number of patients | 12,100 | 9,718 | 8,329 | 4,177 | 7,920 |
IL-17 inhibitor | |||||
IXE 80 mg q.4.w. | 1.23 (0.86 to 1.76) | 1.40 (0.97 to 2.01) | 1.60 (1.03 to 2.47) | 1.14 (0.73 to 1.76) | 1.32 (0.86 to 1.99) |
SEC 150 mg | 1.33 (0.98 to 1.78) | 1.05 (0.73 to 1.49) | 1.12 (0.69 to 1.76) | 2.78 (1.69 to 4.68) | 1.41 (0.83 to 2.33) |
SEC 150 mg noL | 1.14 (0.80 to 1.65) | 1.42 (0.92 to 2.12) | 1.50 (0.86 to 2.65) | NA | NA |
SEC 300 mg | 1.13 (0.85 to 1.49) | 1.17 (0.85 to 1.58) | 1.03 (0.64 to 1.59) | 1.41 (0.94 to 2.15) | 1.34 (0.90 to 1.95) |
IL-12/23 inhibitor or IL-23 inhibitor | |||||
GUS 100 mg q.4.w. | 1.45 (1.04 to 2.03) | 2.18 (1.48 to 3.20) | 1.86 (1.06 to 3.08) | 1.01 (0.66 to 1.57) | 2.06 (1.29 to 3.10) |
GUS 100 mg q.8.w. | 1.66 (1.18 to 2.32) | 1.76 (1.19 to 2.57) | 1.39 (0.82 to 2.23) | 0.89 (0.58 to 1.39) | 1.76 (1.09 to 2.69) |
RIS 150 mg | 1.56 (1.16 to 2.10) | 2.10 (1.52 to 2.89) | 1.93 (1.32 to 2.80) | 1.99 (1.37 to 2.96) | 1.99 (1.40 to 2.76) |
UST 45 mg | 2.51 (1.67 to 3.71) | 2.16 (1.36 to 3.33) | 2.07 (1.19 to 3.49) | NA | NA |
UST 90 mg | 1.92 (1.27 to 2.81) | 1.71 (1.10 to 2.58) | 1.69 (0.97 to 2.76) | NA | NA |
TNF inhibitor | |||||
ADA 40 mg | 1.09 (0.85 to 1.40) | 1.18 (0.90 to 1.54) | 1.11 (0.79 to 1.51) | 2.91 (2.09 to 4.11) | 1.41 (1.01 to 1.93) |
CZP pooled | 1.46 (0.96 to 2.19) | 1.15 (0.68 to 1.82) | 1.19 (0.66 to 1.93) | 4.62 (2.68 to 7.70) | 1.28 (0.54 to 2.38) |
ETA 25 mg | 0.72 (0.42 to 1.19) | 0.54 (0.15 to 1.29) | 2.98 (1.00 to 8.12) | 24.26 (8.86 to 88.77) | NA |
GOL 50 mg | 0.87 (0.45 to 1.48) | 0.76 (0.28 to 1.45) | 1.69 (0.64 to 3.34) | 4.78 (2.21 to 8.85) | NA |
GOL 2 mg IV | 0.42 (0.27 to 0.64) | 0.62 (0.40 to 0.92) | 0.65 (0.35 to 1.06) | 2.46 (1.63 to 3.79) | 1.11 (0.49 to 1.92) |
IFX 5 mg | 0.63 (0.35 to 1.04) | 0.56 (0.23 to 1.03) | 0.80 (0.34 to 1.49) | 1.87 (0.98 to 3.24) | NA |
JAK inhibitor | |||||
TOF 5 mg | 1.75 (1.08 to 2.83) | 1.72 (0.99 to 3.00) | 1.49 (0.80 to 2.78) | NA | 1.45 (0.76 to 2.64) |
UPA 15 mg | 0.97 (0.69 to 1.35) | 1.06 (0.77 to 1.47) | 1.01 (0.69 to 1.47) | 2.72 (1.76 to 4.25) | 1.35 (0.90 to 1.92) |
Other | |||||
APR 30 mg | 2.28 (1.66 to 3.11) | 2.24 (1.00 to 4.54) | 2.57 (0.61 to 7.16) | NA | NA |
ABA 125 mg | 1.94 (1.12 to 3.33) | 2.77 (1.50 to 5.23) | 2.80 (1.39 to 6.28) | NA | NA |
ABA = abatacept; ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; ADA = adalimumab; APR = apremilast; b/tsDMARD = biologic or targeted synthetic disease-modifying antirheumatic drug; CrI = credible interval; CZP = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; IFX = infliximab; IL-12/23 = interleukin-12 and interleukin-23; IL-17 = interleukin-17; IL-23 = interleukin-23; IXE = ixekizumab; JAK = Janus kinase; MDA = minimal disease activity; NA = not available; NMA = network meta-analysis; noL = no loading; OR = odds ratio; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TNF = tumour necrosis factor; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Note: Bolded values represent effect estimates favourable to bimekizumab (i.e., those that do not include the null).
aORs greater than 1 signify a response association favouring bimekizumab.
bAdjusted for baseline risk using meta-regression.
Source: Sponsor’s network meta-analysis technical document.30
Table 44: Key NMA Efficacy Results for TNFi-Experienced Patients
Comparator | ACR20 | ACR50 | ACR70 | PASI 90 | MDA |
|---|---|---|---|---|---|
OR (95% CrI)a | OR (95% CrI)a | OR (95% CrI)a | OR (95% CrI)a | OR (95% CrI)a | |
Model | Random-effects, baseline-unadjusted univariate | Random-effects, baseline-unadjusted univariate | Fixed-effect, baseline-adjusted univariateb | Fixed-effect, baseline-unadjusted univariate | Random-effects, baseline-unadjusted univariate |
Number of studies | 16 | 10 | 7 | 5 | 10 |
Number of patients | 3,076 | 2,215 | 1,719 | 734 | 2,133 |
IL-17 inhibitor | |||||
IXE 80 mg q.4.w. | 2.72 (1.13 to 7.37) | 1.28 (0.36 to 4.53) | 1.61 (0.22 to 4.92) | 2.69 (0.59 to 10.86) | 2.63 (0.71 to 9.37) |
SEC 150 mg | 3.78 (1.83 to 8.80) | 3.09 (1.10 to 9.37) | 2.10 (0.12 to 8.07) | 6.62 (0.20 to 69.73) | 4.60 (0.35 to 41.33) |
SEC 150 mg noL | 4.81 (1.86 to 11.78) | 4.19 (1.43 to 14.07) | 2.18 (0.14 to 10.14) | NA | NA |
SEC 300 mg | 2.57 (1.22 to 6.15) | 1.95 (0.68 to 5.94) | 1.24 (0.08 to 4.48) | 3.32 (0.09 to 40.65) | 2.28 (0.17 to 18.38) |
IL-12/23 inhibitor or IL-23 inhibitor | |||||
GUS 100 mg q.4.w. | 2.16 (0.73 to 6.84) | 1.52 (0.34 to 5.80) | 1.12 (0.15 to 4.19) | 1.48 (0.31 to 6.73) | 0.91 (0.15 to 5.08) |
GUS 100 mg q.8.w. | 2.67 (1.18 to 6.52) | 2.53 (0.71 to 8.19) | 5.00 (1.21 to 24.73) | 2.32 (0.64 to 8.10) | 1.77 (0.35 to 7.43) |
RIS 150 mg | 4.77 (1.94 to 12.96) | 2.56 (0.55 to 8.91) | 5.63 (0.58 to 22.47) | 2.22 (0.54 to 8.74) | 3.05 (0.67 to 12.29) |
UST 45 mg | 4.68 (1.24 to 16.48) | 3.91 (0.88 to 17.72) | 7.95 (1.25 to 118.50) | NA | NA |
UST 90 mg | 4.98 (1.40 to 17.81) | 3.66 (0.86 to 17.15) | 7.23 (1.27 to 90.04) | NA | NA |
TNF inhibitor | |||||
ADA 40 mg | NA | NA | NA | NA | NA |
CZP pooled | 1.07 (0.24 to 4.19) | 0.34 (0.02 to 3.00) | 2.02 (0.12 to 9.66) | 1.44 (0.04 to 12.25) | 1.46 (0.05 to 12.97) |
ETA 25 mg | NA | NA | NA | NA | NA |
GOL 50 mg | NA | NA | NA | NA | NA |
GOL 2 mg IV | NA | NA | NA | NA | NA |
IFX 5 mg | NA | NA | NA | NA | NA |
JAK inhibitor | |||||
TOF 5 mg | 3.57 (1.38 to 9.32) | 4.10 (1.45 to 12.96) | 1.41 (0.04 to 7.48) | NA | 6.81 (2.14 to 21.35) |
UPA 15 mg | 2.39 (1.09 to 6.01) | 1.20 (0.39 to 3.72) | 2.11 (0.73 to 10.88) | 4.78 (1.60 to 15.16) | 2.27 (0.71 to 7.22) |
Other | |||||
APR 30 mg | 2.46 (0.91 to 7.16) | NA | NA | NA | NA |
ABA 125 mg | 7.29 (3.11 to 19.63) | 6.68 (2.09 to 21.34) | 3.02 (0.17 to 12.75) | NA | NA |
ABA = abatacept; ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; ADA = adalimumab; APR = apremilast; CrI = credible interval; CZP = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; IFX = infliximab; IL-12/23 = interleukin-12 and interleukin-23; IL-17 = interleukin-17; IL-23 = interleukin-23; IXE = ixekizumab; JAK = Janus kinase; MDA = minimal disease activity; NA = not available; NMA = network meta-analysis; noL = no loading; OR = odds ratio; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TNF = tumour necrosis factor; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Note: Bolded values represent effect estimates favourable to bimekizumab (i.e., those that do not include the null).
aORs greater than 1 signify a response association favouring bimekizumab.
bAdjusted for baseline risk using meta-regression.
Source: Sponsor’s network meta-analysis technical document.30
Mixed Population Safety Analysis, Base Case
Full results of the NMAs looking at SAEs, trial discontinuations, and discontinuations due to AEs are contained in Table 45. Two additional comparators (brodalumab 140 mg and brodalumab 210 mg) were included in the safety NMA analyses relative to the efficacy analyses. Results from random-effects, baseline-unadjusted models were reported for discontinuation and discontinuation due to AEs, and results from a fixed-effect, baseline-unadjusted model were reported for SAEs.
The submission did not contain any information on specific SAEs or AEs; therefore, comparison across trials is limited. Briefly, bimekizumab was not associated with increased or reduced odds of SAEs, discontinuation, or discontinuation due to AEs with the exception of having higher odds of discontinuation due to an AE versus ustekinumab (45 mg and 90 mg), and higher odds of discontinuation versus etanercept 25 mg.
Results of MAIC
Summary of Included Studies
The SLR identified 13 trials that were included in the MAIC analyses; these comprised RCTs for secukinumab, ixekizumab, guselkumab, risankizumab, and ustekinumab. Patients who were originally randomized to placebo were excluded from the MAIC as they were not on placebo treatment for the entirety of the 52-week duration in some trials. Table 46 and Table 47 include details of the studies.
Three comparator datasets (the FUTURE 2 trial, the FUTURE 5 trial, and the pooled FUTURE 2 to FUTURE 5 trials) contained both TNFi-experienced and bDMARD-naive patients, and subgroups were reported for each — only the randomization in the FUTURE 5 trial was stratified on TNFi exposure. Among the TNFi-experienced population, subgroup data from the FUTURE 2, FUTURE 5, pooled FUTURE 2 to FUTURE 5, KEEPsAKE 2, and PSUMMIT 2 trials were used in the MAIC rather than the whole RCT sample. The COSMOS trial reported outcomes at week 48 whereas the remaining studies reported outcomes at week 52.
Table 45: Key NMA Safety Results for a Mixed (b/tsDMARD-Naive and TNFi-Experienced) Population
Comparator | SAE | Discontinuation | Discontinuation due to AE |
|---|---|---|---|
OR (95% CrI)a | OR (95% CrI)a | OR (95% CrI)a | |
Model | Fixed-effect, baseline-unadjusted univariate | Random-effects, baseline-unadjusted univariate | Random-effects, baseline-unadjusted univariate |
Number of studies | 39 | 29 | 38 |
Number of patients | 15,722 | 11,604 | 16,005 |
IL-17 inhibitor | |||
BRO 140 mg | 1.81 (0.52 to 8.07) | NA | 3.26 (0.80 to 14.72) |
BRO 210 mg | 0.88 (0.29 to 3.83) | NA | 2.44 (0.66 to 8.58) |
IXE 80 mg q.4.w. | 2.12 (0.69 to 7.33) | 1.08 (0.45 to 2.49) | 1.50 (0.49 to 5.27) |
SEC 150 mg | 2.45 (0.87 to 8.65) | 0.65 (0.25 to 1.67) | 1.14 (0.36 to 3.72) |
SEC 150 mg noL | 2.90 (0.80 to 12.67) | 1.46 (0.31 to 8.50) | 1.46 (0.36 to 5.99) |
SEC 300 mg | 2.05 (0.73 to 6.91) | 0.83 (0.30 to 2.37) | 1.58 (0.47 to 5.76) |
IL 12/23 or IL-23 inhibitor | |||
GUS 100 mg q.4.w. | 2.27 (0.69 to 10.00) | 1.03 (0.36 to 2.73) | 1.16 (0.34 to 4.53) |
GUS 100 mg q.8.w. | 2.29 (0.72 to 7.55) | 0.93 (0.36 to 2.28) | 1.71 (0.48 to 5.98) |
RIS 150 mg | 2.28 (0.77 to 7.64) | 1.31 (0.48 to 3.47) | 2.08 (0.46 to 10.91) |
UST 45 mg | 2.63 (0.69 to 11.14) | 2.27 (0.87 to 6.10) | 4.99 (1.32 to 24.01) |
UST 90 mg | 3.01 (0.75 to 14.36) | 1.66 (0.66 to 4.28) | 4.29 (1.19 to 16.30) |
TNF inhibitor | |||
ADA 40 mg | 1.25 (0.47 to 3.86) | 1.24 (0.61 to 2.58) | 0.80 (0.31 to 2.12) |
CZP pooled | 0.82 (0.18 to 3.80) | 0.84 (0.28 to 2.34) | 0.35 (0.03 to 1.86) |
ETA 25 mg | 2.10 (0.46 to 11.65) | 3.89 (1.30 to 11.68) | 0.98 (0.05 to 14.17) |
GOL 50 mg | 5.63 (0.97 to 41.59) | 1.41 (0.37 to 5.35) | 4.60 (0.62 to 38.47) |
GOL 2 mg IV | 1.79 (0.44 to 8.46) | NA | 0.73 (0.12 to 4.66) |
IFX 5 mg | 1.17 (0.19 to 5.15) | 0.97 (0.29 to 3.17) | 0.39 (0.05 to 1.92) |
JAK inhibitor | |||
TOF 5 mg | 1.25 (0.23 to 7.82) | NA | 1.16 (0.32 to 5.56) |
UPA 15 mg | 1.05 (0.38 to 3.71) | 1.41 (0.65 to 3.05) | 1.17 (0.44 to 3.44) |
Other | |||
APR 30 mg | 1.89 (0.62 to 6.13) | 0.62 (0.28 to 1.42) | 0.71 (0.26 to 1.99) |
ABA 125 mg | 2.55 (0.62 to 11.95) | NA | 1.35 (0.17 to 11.11) |
ABA = abatacept; ADA = adalimumab; AE = adverse event; APR = apremilast; b/tsDMARD = biologic or targeted synthetic disease-modifying antirheumatic drug; BRO = brodalumab; CrI = credible interval; CZP = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; IFX = infliximab; IL-12/23 = interleukin-12 and interleukin-23; IL-17 = interleukin-17; IL-23 = interleukin-23; IXE = ixekizumab; NA = not available; NMA = network meta-analysis; noL = no loading; OR = odds ratio; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; RIS = risankizumab; SAE = serious adverse event; SEC = secukinumab; TNF = tumour necrosis factor; TNFi = tumour necrosis factor inhibitor; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Notes: No model could converge for the serious infections outcome due to a low number of events.
Bolded values represent effect estimates not favourable to bimekizumab (i.e., those that do not include the null).
aORs greater than 1 signify a response association favouring bimekizumab.
Source: Sponsor’s network meta-analysis technical document.30
Table 46: Summary of Trial Groups Included in MAIC — bDMARD-Naive Patients
Study | Study design | N | Intervention | Control |
|---|---|---|---|---|
BE OPTIMAL trial | Double-blind; placebo-controlled to 16 weeks; active controlled-blind at week 52 | 431 | BKZ 160 mg q.4.w. | Placebo until week 16 |
FUTURE 2 studya | Double-blind; placebo-controlled to 24 weeks; dose blind at week 52 | 63 | SEC 150 mg | Placebo until week 24 |
67 | SEC 300 mg | |||
FUTURE 5 studyb | Double-blind; placebo-controlled to 24 weeks, dose blind at week 52 | 155 | SEC 150 mg LD | Placebo until week 24 |
154 | SEC 300 mg LD | |||
Pooled populations of FUTURE 2 to FUTURE 5 studiesa,b | Double-blind, placebo-controlled to 24 weeks; dose blind at week 52 | 643 | SEC 150 mg | Placebo until week 24 |
316 | SEC 300 mg | |||
SPIRIT-P1 study | Double-blind; placebo-controlled to 24 weeks; dose blind from week 24 to week 52 | 107 | IXE q.4.w. | Placebo until week 24 |
DISCOVER-2 study | Double-blind; placebo-controlled to 24 weeks | 248 | GUS q.8.w. | Placebo until week 24 |
245 | GUS q.4.w. | |||
KEEPsAKE 1 study | Double-blind; placebo-controlled to week 24; open-label at week 52 | 483 | RIS | Placebo until week 24 |
PSUMMIT 1 study | Double-blind; placebo-controlled to week 52 | 205 | UST 45 mg | Placebo until week 52 |
204 | UST 90 mg |
bDMARD = biologic disease-modifying antirheumatic drug; BKZ = bimekizumab; GUS = guselkumab; IXE = ixekizumab; LD = loading dose; MAIC = matching-adjusted indirect comparison; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TNF = tumour necrosis factor; TNFi = tumour necrosis factor inhibitor; UST = ustekinumab.
aThe study population was mixed bDMARD-naive and TNFi-experienced; results from the relevant subgroup were reported.
bThe randomization of the FUTURE 5 study was stratified on TNF exposure.
Source: Sponsor’s matching-adjusted indirect comparison technical report.29
Table 47: Summary of Trial Groups Included in MAIC — TNFi-Experienced Patients
Study | Study design | N | Intervention | Control |
|---|---|---|---|---|
BE COMPLETE study | Double-blind; placebo-controlled to 16 weeks; open-label at week 52 | 267 | BKZ 160 mg q.4.w. | Placebo until week 16 |
FUTURE 2 studya | Double-blind; placebo-controlled to 24 weeks; dose blind at week 52 | 37 | SEC 150 mg | Placebo until week 24 |
33 | SEC 300 mg | |||
FUTURE 5 studyb | Double-blind; placebo-controlled to 24 weeks; dose blind at week 52 | 65 | SEC 150 mg LD | Placebo until week 24 |
68 | SEC 300 mg LD | |||
Pooled FUTURE 2 to FUTURE 5 studiesa, b | Double-blind; placebo-controlled to 24 weeks; dose blind at week 52 | 264 | SEC 150 mg | Placebo until week 24 |
145 | SEC 300 mg | |||
SPIRIT-P2 study | Double-blind; placebo-controlled to 24 weeks; dose blind from week 24 to week 52 | 122 | IXE q.4.w. | Placebo until week 24 |
COSMOS study | Double-blind; placebo-controlled to 24 weeks; active treatment at week 48 | 189 | GUS q.8.w. | Placebo until week 24 |
KEEPsAKE 2 studya | Double-blind; placebo-controlled to week 24; open-label at week 52 | 105 | RIS | Placebo until week 24 |
PSUMMIT 2 studya | Double-blind; placebo-controlled to week 52 | 60 | UST 45 mg | Placebo until week 52 |
58 | UST 90 mg |
BKZ = bimekizumab; GUS = guselkumab; IXE = ixekizumab; LD = loading dose; MAIC = matching-adjusted indirect comparison; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TNF = tumour necrosis factor; TNFi = tumour necrosis factor inhibitor; UST = ustekinumab.
aThe study population was mixed bDMARD-naive and TNFi-experienced; results from the relevant subgroup were reported.
bThe randomization of the FUTURE 5 study was stratified on TNF exposure.
Source: Sponsor’s matching-adjusted indirect comparison technical report.29
Results
bDMARD-Naive Population
Results of the baseline characteristics prematching and postmatching for all trial comparisons are shown in Appendix 1, Table 52 to Table 62. Briefly, while no standardized mean differences (SMDs) or other statistical testing for differences were reported, the baseline characteristics that were included in the propensity score calculations appeared balanced postmatching. Of the baseline characteristics not included in the matching procedure, there were some that remained unbalanced after matching; most frequently, proportions of patients with dactylitis and enthesitis were notably different between the comparator trial and the bimekizumab trial.
For each ACR outcome, 8 unanchored MAIC comparisons were undertaken and for the MDA outcome, 5 unanchored MAIC comparisons were undertaken. Full results are in Table 48. Briefly, at week 52, for ACR20 response, bimekizumab was favoured over 2 comparators: ustekinumab 45 mg and ustekinumab 90 mg every 12 weeks. For ACR50 response, bimekizumab was favoured over 4 comparators: guselkumab 100 mg every 4 weeks, risankizumab 150 mg every 12 weeks, and ustekinumab 45 mg and ustekinumab 90 mg every 12 weeks. For ACR70 response, bimekizumab was favoured over 7 comparators: secukinumab 150 mg and secukinumab 300 mg, guselkumab 100 mg every 4 weeks and every 8 weeks, risankizumab 150 mg every 12 weeks, and ustekinumab 45 mg and ustekinumab 90 mg every 12 weeks. For MDA, bimekizumab was favoured over guselkumab 100 mg every 4 weeks and every 8 weeks.
Table 48: Key Results for MAIC — bDMARD-Naive Patients at Week 52
Comparator | Comparator study | Unadjusted results | MAIC results | ||
|---|---|---|---|---|---|
N (BKZ vs. comparator) | BKZ vs. comparator OR (95% CI) | ESS | BKZ vs. comparator OR (95% CI) | ||
ACR20 | |||||
SEC 150 mg q.4.w. | FUTURE 2 | 431 vs. 63 | 0.64 (0.34 to 1.23) | 236.15 | 0.64 (0.32 to 1.26) |
SEC 300 mg q.4.w. | FUTURE 2 | 431 vs. 67 | 1.13 (0.65 to 1.98) | 236.15 | 1.12 (0.62 to 2.03) |
IXE 160 mg q.4.w. | SPIRIT-P1 | 431 vs. 107 | 1.48 (0.95 to 2.31) | 229.55 | 1.49 (0.91 to 2.45) |
GUS 100 mg q.8.w. | DISCOVER-2 | 431 vs. 248 | 0.84 (0.59 to 1.20) | 142.04 | 0.86 (0.53 to 1.40) |
GUS 100 mg q.4.w. | DISCOVER-2 | 431 vs. 245 | 1.03 (0.73 to 1.46) | 155.08 | 1.09 (0.68 to 1.74) |
RIS 150 mg q.12.w. | KEEPsAKE 1 | 431 vs. 483 | 1.06 (0.80 to 1.41) | 231.00 | 1.02 (0.72 to 1.45) |
UST 45 mg q.12.w. | PSUMMIT 1 | 267 vs. 60 | 2.22 (1.57 to 3.14) | 146.65 | 2.14 (1.35 to 3.40) |
UST 90 mg q.12.w. | PSUMMIT 1 | 431 vs. 67 | 1.95 (1.38 to 2.77) | 152.63 | 1.98 (1.24 to 3.16) |
ACR50 | |||||
SEC 150 mg q.4.w. | FUTURE 2 | 431 vs. 63 | 1.24 (0.73 to 2.11) | 236.15 | 1.20 (0.69 to 2.10) |
SEC 300 mg q.4.w. | FUTURE 2 | 431 vs. 67 | 1.10 (0.65 to 1.84) | 236.15 | 1.06 (0.62 to 1.83) |
IXE 160 mg q.4.w. | SPIRIT-P1 | 431 vs. 107 | 1.18 (0.77 to 1.80) | 229.55 | 1.17 (0.74 to 1.87) |
GUS 100 mg q.8.w. | DISCOVER-2 | 431 vs. 248 | 1.28 (0.93 to 1.75) | 142.04 | 1.38 (0.90 to 2.09) |
GUS 100 mg q.4.w. | DISCOVER-2 | 431 vs. 245 | 1.42 (1.04 to 1.95) | 155.08 | 1.62 (1.07 to 2.44) |
RIS 150 mg q.12.w. | KEEPsAKE 1 | 431 vs. 483 | 1.57 (1.21 to 2.04) | 231.00 | 1.52 (1.11 to 2.09) |
UST 45 mg q.12.w. | PSUMMIT 1 | 267 vs. 60 | 2.83 (1.98 to 4.04) | 146.65 | 2.74 (1.75 to 4.29) |
UST 90 mg q.12.w. | PSUMMIT 1 | 431 vs. 67 | 2.30 (1.62 to 3.25) | 152.63 | 2.29 (1.48 to 3.55) |
ACR70 | |||||
SEC 150 mg | FUTURE 2 | 431 vs. 63 | 2.06 (1.12 to 3.81) | 236.15 | 2.39 (1.26 to 4.53) |
SEC 300 mg | FUTURE 2 | 431 vs. 67 | 1.76 (0.99 to 3.12) | 236.15 | 2.03 (1.11 to 3.72) |
IXE 160 mg q.4.w. | SPIRIT-P1 | 431 vs. 107 | 1.12 (0.72 to 1.75) | 229.55 | 1.11 (0.69 to 1.79) |
GUS 100 mg q.8.w. | DISCOVER-2 | 431 vs. 248 | 1.68 (1.19 to 2.35) | 142.04 | 2.08 (1.34 to 3.22) |
GUS 100 mg q.4.w. | DISCOVER-2 | 431 vs. 245 | 1.82 (1.29 to 2.58) | 155.08 | 2.20 (1.43 to 3.38) |
RIS 150 mg q.12.w. | KEEPsAKE 1 | 431 vs. 483 | 1.85 (1.39 to 2.45) | 231.00 | 1.80 (1.29 to 2.51) |
UST 45 mg q.12.w. | PSUMMIT 1 | 267 vs. 60 | 3.13 (2.07 to 4.74) | 146.55 | 3.33 (2.04 to 5.46) |
UST 90 mg q.12.w. | PSUMMIT 1 | 431 vs. 67 | 2.64 (1.78 to 3.94) | 236.15 | 3.05 (1.89 to 4.91) |
MDA | |||||
SEC 150 mg | FUTURE 2 | 431 vs. 63 | 2.12 (1.23 to 3.68) | 236.15 | 1.77 (1.00 to 3.15) |
SEC 300 mg | FUTURE 2 | 431 vs. 67 | 1.83 (1.07 to 3.12) | 236.15 | 1.53 (0.87 to 2.68) |
IXE 160 mg q.4.w. | SPIRIT-P1 | NA | NA | NA | NA |
GUS 100 mg q.8.w. | DISCOVER-2 | 431 vs. 248 | 2.72 (1.95 to 3.78) | 142.04 | 2.07 (1.35 to 3.17) |
GUS 100 mg q.4.w. | DISCOVER-2 | 431 vs. 245 | 2.34 (1.69 to 3.24) | 155.08 | 1.82 (1.20 to 2.76) |
RIS 150 mg q.12.w. | KEEPsAKE 1 | 431 vs. 483 | 2.00 (1.54 to 2.61) | 231.00 | 1.34 (0.98 to 1.84) |
UST 45 mg q.12.w. | PSUMMIT 1 | NA | NA | NA | NA |
UST 90 mg q.12.w. | PSUMMIT 1 | NA | NA | NA | NA |
ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; bDMARD = biologic disease-modifying antirheumatic drug; BKZ = bimekizumab; CI = confidence interval; ESS = effective sample size; GUS = guselkumab; IXE = ixekizumab; MAIC = matching-adjusted indirect comparison; MDA = minimal disease activity; NA = not available; OR = odds ratio; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; RIS = risankizumab; SEC = secukinumab; UST = ustekinumab; vs. = versus.
Note: Bolded values represent effect estimates favourable to BKZ (i.e., those that do not include the null).
Source: Sponsor’s matching-adjusted indirect comparison technical document.29
TNFi-Experienced Population
Results of the baseline characteristics prematching and postmatching for all trial comparisons are shown in Appendix 1, Table 63 to Table 72. Briefly, while no SMDs or other statistical testing for differences were reported, the baseline characteristics that were included in the propensity score calculations appeared balanced postmatching, with some exceptions. Of the characteristics not included in the matching procedure, dactylitis, enthesitis, pain visual analogue scale scores, and race were the 4 characteristics most commonly observed to have notable differences between the comparator and bimekizumab trials.
For each ACR outcome, 7 unanchored MAIC comparisons were undertaken and for the MDA outcome, 5 unanchored MAIC comparisons were undertaken. Full results are in Table 49. Briefly, for ACR20 response, bimekizumab was favoured over 5 comparators: secukinumab 150 mg every 4 weeks, guselkumab 100 mg every 8 weeks, risankizumab 150 mg every 12 weeks, and ustekinumab 45 mg and ustekinumab 90 mg every 12 weeks. For ACR50 response, bimekizumab was favoured over 6 comparators: secukinumab 150 mg and secukinumab 300 mg every 4 weeks, guselkumab 100 mg every 8 weeks, risankizumab 150 mg every 12 weeks, and ustekinumab 45 mg and ustekinumab 90 mg every 12 weeks. For ACR70 response, bimekizumab was favoured over 5 comparators: secukinumab 150 mg, guselkumab 100 mg every 8 weeks, risankizumab 150 mg every 12 weeks, and ustekinumab 45 mg and ustekinumab 90 mg every 12 weeks. For MDA, bimekizumab was favoured over 4 comparators: secukinumab 150 mg and secukinumab 300 mg every 4 weeks, guselkumab 100 mg every 8 weeks, and risankizumab 150 mg every 12 weeks. There were wider CIs noted for several of these comparisons, suggesting imprecision in the estimate of the effects.
Table 49: Indirect Comparison Results for ACR and MDA Outcomes — TNFi-Experienced Patients at Week 52
Comparator | Comparator study | Unadjusted results | MAIC results | ||
|---|---|---|---|---|---|
N (BKZ vs. comparator) | BKZ vs. comparator OR (95% CI) | ESS | BKZ vs. comparator OR (95% CI) | ||
ACR20 | |||||
SEC 150 mg q.4.w. | FUTURE 2 | 267 vs. 37 | 3.52 (1.72 to 7.21) | 145.50 | 3.50 (1.64 to 7.49) |
SEC 300 mg q.4.w. | FUTURE 2 | 267 vs. 33 | 1.78 (0.85 to 3.73) | 145.50 | 1.78 (0.82 to 3.87) |
IXE 160 mg q.4.w. | SPIRIT-P2 | 267 vs. 122 | 1.34 (0.86 to 2.10) | 179.89 | 1.15 (0.71 to 1.88) |
GUS 100 mg q.8.w. | COSMOS | 267 vs. 189 | 1.57 (1.07 to 2.32) | 180.84 | 1.77 (1.15 to 2.72) |
RIS 150 mg q.12.w. | KEEPsAKE 2 | 267 vs. 105 | 2.22 (1.40 to 3.53) | 161.91 | 1.78 (1.08 to 2.96) |
UST 45 mg q.12.w. | PSUMMIT2 | 267 vs. 60 | 4.28 (2.35 to 7.81) | 101.72 | 4.17 (2.13 to 8.16) |
UST 90 mg q.12.w. | PSUMMIT2 | 267 vs. 33 | 3.50 (1.93 to 6.36) | 70.52 | 4.19 (2.07 to 8.49) |
ACR50 | |||||
SEC 150 mg | FUTURE 2 | 267 vs. 37 | 3.88 (1.70 to 8.84) | 145.50 | 3.32 (1.41 to 7.80) |
SEC 300 mg | FUTURE 2 | 267 vs. 33 | 2.85 (1.27 to 6.40) | 145.50 | 2.44 (1.06 to 5.65) |
IXE 160 mg q.4.w. | SPIRIT-P2 | 267 vs. 122 | 1.49 (0.96 to 2.30) | 179.89 | 1.26 (0.79 to 2.01) |
GUS 100 mg q.8.w. | COSMOS | 267 vs. 189 | 1.66 (1.14 to 2.43) | 180.84 | 1.56 (1.03 to 2.36) |
RIS 150 mg q.12.w. | KEEPsAKE 2 | 267 vs. 105 | 3.86 (2.29 to 6.51) | 161.91 | 3.05 (1.74 to 5.32) |
UST 45 mg q.12.w. | PSUMMIT 2 | 267 vs. 60 | 5.35 (2.59 to 11.05) | 101.72 | 5.00 (2.26 to 11.05) |
UST 90 mg q.12.w. | PSUMMIT 2 | 267 vs. 33 | 4.57 (2.26 to 9.26) | 70.52 | 3.86 (1.70 to 8.79) |
ACR70 | |||||
SEC 150 mg | FUTURE 2 | 267 vs. 37 | 3.53 (1.32 to 9.44) | 145.50 | 2.95 (1.08 to 8.07) |
SEC 300 mg | FUTURE 2 | 267 vs. 33 | 2.49 (0.98 to 6.27) | 145.50 | 2.08 (0.80 to 5.37) |
IXE 160 mg q.4.w. | SPIRIT-P2 | 267 vs. 122 | 1.55 (0.96 to 2.50) | 179.89 | 1.32 (0.79 to 2.19) |
GUS 100 mg q.8.w. | COSMOS | 267 vs. 189 | 1.77 (1.16 to 2.69) | 180.84 | 1.66 (1.05 to 2.61) |
RIS 150 mg q.12.w. | KEEPsAKE 2 | 267 vs. 105 | 4.77 (2.43 to 9.38) | 161.91 | 3.69 (1.82 to 7.46) |
UST 45 mg q.12.w. | PSUMMIT 2 | 267 vs. 60 | 10.49 (3.17 to 34.74) | 145.50 | 9.85 (2.79 to 34.79) |
UST 90 mg q.12.w. | PSUMMIT 2 | 267 vs. 33 | 7.46 (2.59 to 21.44) | 70.52 | 6.29 (1.98 to 20.04) |
MDA | |||||
SEC 150 mg | FUTURE 2 | 267 vs. 37 | 4.62 (1.85 to 11.50) | 145.50 | 3.52 (1.38 to 8.99) |
SEC 300 mg | FUTURE 2 | 267 vs. 33 | 3.82 (1.61 to 9.06) | 145.50 | 2.92 (1.20 to 7.09) |
IXE 160 mg q.4.w. | SPIRIT-P2 | 267 vs. 122 | 1.70 (1.09 to 2.66) | 179.89 | 1.17 (0.73 to 1.87) |
GUS 100 mg q.8.w. | COSMOS | 267 vs. 189 | 2.42 (1.62 to 3.62) | 180.84 | 1.95 (1.27 to 3.02) |
RIS 150 mg q.12.w. | KEEPsAKE 2 | 267 vs. 105 | 3.84 (2.23 to 6.63) | 161.91 | 2.43 (1.37 to 4.32) |
UST 45 mg q.12.w. | PSUMMIT 2 | NA | NA | NA | NA |
UST 90 mg q.12.w. | PSUMMIT 2 | NA | NA | NA | NA |
ACR = American College of Rheumatology; ACR20 = American College of Rheumatology 20% improvement in rheumatoid arthritis; ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; ACR70 = American College of Rheumatology 70% improvement in rheumatoid arthritis; BKZ = bimekizumab; CI = confidence interval; ESS = effective sample size; GUS = guselkumab; IXE = ixekizumab; MAIC = matching-adjusted indirect comparison; MDA = minimal disease activity; NA = not available; OR = odds ratio; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; RIS = risankizumab; SEC = secukinumab; TNFi = tumour necrosis factor inhibitor; UST = ustekinumab; vs. = versus.
Note: Bolded values represent effect estimates favourable to BKZ (i.e., those that do not include the null).
Source: Sponsor’s matching-adjusted indirect comparison technical document.29
Critical Appraisal of NMA
The selection of studies used to inform the NMA was based on an SLR that was performed using standard methods. The study selection used multiple databases and conference proceedings to find relevant trials. A study flow chart was provided as well as a list of excluded studies with rationale for their exclusion. Efforts were made not only to include all valid comparators but also to align with recommended treatment guidelines and geographic relevance, although the exact guidance and geographic regions used for reference were not defined. The NMA included a comprehensive list of treatment options according to the clinical expert consulted by CADTH. It was also conducted in 2 populations based on their exposure to TNFis and bDMARDs or tsDMARDs; previous treatment is an important effect modifier noted by the clinical expert.
A little more than half of the studies were rated as having a low risk of bias, though the remainder were rated as having some concerns over outcome ascertainment due to the time points available and unclear blinding during outcome assessment. The impact of this variability is unclear as there were no sensitivities included that assessed the impact of the more biased studies on the reliability of the results. Furthermore, owing to the designs of the included studies, there is a lack of trial level comparative data that preserves randomization past 24 weeks, leaving a gap in knowledge of the longer-term outcomes for these comparators, which is noteworthy given that PsA is a lifelong disease.
The submission included a homogeneity analysis with a list of patient demographic and clinical characteristics assessed for possible differences across trials, including statistical tests for detecting outliers. Sources of heterogeneity have been identified, although the clinical expert noted that weight, or the presence of obesity, would also be an important effect modifier in that obesity impacts treatment response; this was not included in the NMA heterogeneity discussion. Furthermore, there are differences in study design between the included trials. Some studies included an early escape between week 12 and week 16, which is a possible source of heterogeneity and a possible loss of data for the analysis if trial data from after the early escape were used. Similarly, crossover designs differed at the time in which they crossed over — ranging from week 12 to week 24 — and also differed in the way crossover treatments were blinded. Furthermore, no information was provided on study year or clinical setting, and secular trends could contribute to practice changes and patient population changes over time. Lastly, the source trials for each NMA comparison were not defined explicitly and it was not clear if, in the process of defining the b/tsDMARD-naïve and TNFi-experienced subgroups, subgroup data or data from an entire trial population contributed to each comparator. If randomization of the RCTs was broken to use subgroup data, there is the additional possibility of unmeasured confounding for those comparisons. These factors could all affect the comparability of the studies and potentially impact the NMA estimates.
The submission applied several techniques to account for sources of heterogeneity. First, baseline risk adjustment was used to control for heterogeneity in the NMA. Adjusting for the variation in baseline risk across trials is an accepted method as the placebo rate and the relative effect versus placebo may be related.114 This method assumes that study and patient characteristics that are effect modifiers of the relative treatment effect are also differences in the placebo groups. However, this adjustment is a surrogate for specific characteristics or effect modifiers, and the method assumes that covariates have an equal weight across the distribution of studies; the extent to which these assumptions are true cannot be measured. As such, there remains uncertainty in this analysis. In addition, not all results of the NMA were adjusted for baseline risk in the TNFi-experienced population, and therefore some of the results reported are not otherwise controlled for heterogeneity.
The submission also used data from precrossover time points where available and applied methods such as imputation to count missing data as nonresponse. While this would allow for the control of some differences in the study crossover design and reduce possible bias due to missing data, this does not control for the range in the timing of outcome ascertainment and affects the ability of these results to be confidently applied to 24 weeks postinitiation, as many trials employed a crossover design at an earlier time point. As a sensitivity, the scenario 1 analyses were run, which only included week 12 to week 16 data. This would help mitigate the potential heterogeneity in the timing of outcome ascertainment by assessing the impact of using the wider window in the base-case analysis. However, the scenario 1 analysis was not available for all comparators and in the case of MDA for b/tsDMARD-naive patients, used a different model for scenario 1 than for the base case. This limits the ability to make firm conclusions about the impact of the data window in the NMAs for all viable treatment options in PsA.
The submission notes that random-effects models are appropriate when generalizations are to be made to a greater group of studies and patients and/or where there is heterogeneity across studies, methods, and researchers;30 however, per the selection criteria, fixed-effect models would be chosen unless the random-effects model deviance information criterion was smaller by a threshold that was not justified in the submission. While the model selection criteria follow guidance from the National Institute for Health and Care Excellence,114,115 the numbers of studies and numbers of patients included in the networks was sizable, which permitted the convergence of random-effects models for most outcomes. Reporting a fixed-effect NMA does not account for uncertainty in the estimates due to study variance, which is more likely captured in a random-effects model.
Finally, NMAs were run separately for b/tsDMARD-naive patients and TNFi-experienced patients, reflecting that treatment history is an important effect modifier for treatment response. However, the model selection criteria limits comparability of the results between different patient populations and between different outcomes for the same treatment. Since the model fit was not specified a priori, model results reported in the b/tsDMARD-naive patients were fixed-effect models, while in the TNFi-experienced populations, the model results reported were random-effects, baseline-unadjusted models (ACR20, ACR50, and MDA), fixed-effect, baseline-adjusted (PASI 90), and fixed-effect, baseline-unadjusted (ACR70). As outcomes are not defined with the same NMA model, the stability of the treatment effect across outcomes is more difficult to assess in TNFi-experienced patients, which affects the certainty of the estimates for the treatment benefit as a whole.
Overall, the results of the NMA are subject to some uncertainty due to the unmeasurable baseline risk adjustment and uncertainty over the extent to which it accounts for patient heterogeneity, as well as differences in study design and model selection, which impact the comparability of the studies across the network.
Critical Appraisal of MAIC
Since the same SLR informed the MAIC as the NMA, the same methods and limitations for the SLR are also applicable here. It was performed using standard methods and using multiple databases and conference proceedings to find relevant trials. Similar to the NMA, it was conducted separately in bDMARD-naive and TNFi-experienced patients, which accounts for this effect modifier. While the NMA included a comprehensive list of treatment comparators per the clinical expert consulted by CADTH, the MAIC was more restricted to IL-17is, IL-12/23is, and IL-12is. The submission noted that these are key comparators but did not provide further justification for the choice. This limits the applicability of the MAIC to the wider clinical context as certain drug comparators that would be options in Canadian clinical practice were not included in this comparison.
A key limitation to the current MAIC comparisons is the exclusion of placebo-treated patients and the fact that this MAIC is therefore unanchored, with disconnected treatment networks. While this meant the same treatment was consistently received over the 52-week time period, an unanchored MAIC requires the assumption that all prognostic factors and treatment effect modifiers are accounted for, which is a strong assumption largely considered impossible to meet — failure of this assumption leads to an unknown amount of bias in the effect estimate.116 The unanchored comparison should ideally provide sufficient evidence on the likely extent of error due to unaccounted-for covariates,116 which this analysis has not done. Cross-validation methods or other sensitivity analyses are suggested methods to explore the impact of the lack of anchoring.116 However, these were not reported in the current submission, which imparts a considerable amount of uncertainty in the results and remains an important impediment to inference.
The submission leveraged prior publications, expert input, and statistical methods to determine the characteristics to match on for all comparisons; of note, an unanchored MAIC assumes all prognostic factors as well as potential effect modifiers have been accounted for.116 Apart from unmeasured effect modifiers, the submission did not match on dactylitis, an important prognostic factor according to the clinical expert and 1 that was qualitatively imbalanced after matching in most comparisons. While it is noted in the submission that this is due to limitations in the dataset, it is nonetheless an important source of bias in the results as the MAIC is unanchored and requires matching on prognostic factors to reduce bias. In addition, the submission did not report any SMDs after matching to confirm balance of the characteristics used in matching as well as those not used in matching. This adds uncertainty to the estimates as the matching process was not quantitatively assessed to confirm the comparability of the studies after matching.
In addition, the effective sample size for all comparisons was small; in the bDMARD-naive population, it ranged from 33.0% to 70.9% for all comparisons and in the TNFi-experienced population, it ranged from 26.5% to 70.7%. This suggests variability in the estimates and contributes to the uncertainty in the results, although this is somewhat mitigated by the fact that extreme weights were not noted in the propensity score weight distributions provided with the submission. The source of the data may also contain unmeasured confounders as the comparisons with secukinumab in the bDMARD-naive populations, as well as secukinumab, risankizumab, and ustekinumab in the TNFi-experienced populations, used subgroup data from trials and the submission did not comment on whether this involved breaking randomization of the trials. This is an important consideration as the RCT design and control for measured and unmeasured confounders would no longer be applicable, which adds further uncertainty to the results. Lastly, no commentary was provided on study design differences in the post–placebo-controlled portion of the studies when it came to open-label or dose-blinded designs at week 52.
Overall, the results from this MAIC are considered to be highly uncertain, at risk of unmeasured bias, and also of limited applicability to the clinical context due to the inclusion of only some treatment options available in the Canadian context.
Studies Addressing Gaps in the Systematic Review Evidence
No additional relevant studies addressing gaps in the evidence of the systematic review were submitted by the sponsor.
Discussion
Summary of Available Evidence
Two DB, placebo-controlled RCTs of bimekizumab for the treatment of adults with active PsA who had no prior exposure to biologic therapies (the BE OPTIMAL trial, N = 852) or who had a history of inadequate response or intolerance to 1 TNFi or 2 TNFis (the BE COMPLETE trial, N = 400) were submitted by the sponsor for this systematic review.20,19,20 Patients in both trials were randomized to 16 weeks of treatment with bimekizumab 160 mg every 4 weeks or placebo. The BE OPTIMAL trial also included an adalimumab reference group, but because it was not powered for statistical testing and no statistical comparison was performed, the results are not discussed in the CADTH report. After 16 weeks of treatment in the BE OPTIMAL trial, patients who were randomized to placebo were reallocated to receive bimekizumab 160 mg every 4 weeks for a 36-week active treatment–blind period and patients randomized to bimekizumab continued their treatment. Rescue treatment was permitted during the active treatment–blind period. In both trials, patients could remain on a stable dose of concomitant treatments, such as NSAIDs, COX-2 inhibitors, oral corticosteroids, methotrexate, leflunomide, sulfasalazine, hydroxychloroquine, or apremilast. The primary end point of both trials was the proportion of patients who attained an ACR50 response at week 16. Secondary and exploratory end points relevant to the CADTH review included outcomes for MDA, musculoskeletal changes, skin disease, and patient-reported functioning and symptoms.
The mean age of patients ranged from 48.5 (SD = 12.6) years to 51.3 (SD = 12.9) years across the treatment groups in the 2 trials, and demographic characteristics were generally balanced between treatment groups and were similar across trials. In the BE OPTIMAL trial, half of patients had psoriasis that affected at least 3% of their BSA, 25% to 33% of patients had enthesitis, 80% of patients reported prior use of cDMARDs, and 70% of patients were using cDMARDs at baseline. In the BE COMPLETE trial, two-thirds of patients had psoriasis that affected at least 3% of their BSA, 27% to 40% of patients reported enthesitis, approximately 63% of patients reported prior use of cDMARDs, and 47% to 52% of patients were using them at baseline. Additionally, more than 76% of patients had an inadequate response to at least 1 TNFi, more than 11% of patients had an inadequate response to at least 2 TNFis, and approximately 12% had an intolerance to TNFis.
Longer-term safety and efficacy data for a total of 1 year to 2 years of treatment were available from the OLE studies (the BE ACTIVE 2 study and the BE VITAL study), which enrolled eligible patients who completed the BE ACTIVE, BE OPTIMAL, and BE COMPLETE studies, and all patients who received bimekizumab 160 mg every 4 weeks.25,117 At this time, only data for TNFi-experienced patients were available and have been reviewed.
Indirect evidence was provided from 2 sponsor-submitted ITCs that investigated the efficacy and safety of bimekizumab versus other licensed, targeted DMARDs available in Canada for the treatment of adult-onset PsA.29,30 The NMA presented data for a comprehensive list of comparators at week 12 to week 24 and the MAIC presented comparisons for IL-17is, IL-12/23is, and IL-12is at week 52.
Interpretation of Results
Efficacy
High certainty evidence showed that treatment with bimekizumab results in a clinically meaningful percentage of patients attaining ACR50 response at 16 weeks compared to placebo for both biologic-naive and TNFi-experienced populations based on the BE OPTIMAL and BE COMPLETE trials, respectively. Prespecified subgroup analysis results were consistent with the primary analysis in the BE COMPLETE trial among patients who had an inadequate response to 1 TNFi or 2 TNFis or an intolerance to TNFis, though the analysis was underpowered and there were wide CIs that suggest imprecision. ACR20 has often been used as the primary end point in trials for PsA, though the current pivotal trials applied a higher threshold (ACR50) for bimekizumab. The clinical expert consulted by CADTH suggested that the higher threshold is a more clinically important goal for improvement in PsA compared to ACR20. The results for ACR20 and ACR70 supported the primary outcome, though these end points were not controlled for multiplicity in the trials and 16 weeks of treatment is not a sufficient treatment period to observe a meaningful percentage of patients attaining an ACR70 response. In the pivotal trials, both biologic-naive and TNFi-experienced populations attained a clinically meaningful MDA response with bimekizumab over placebo. Based on the evidence in the pivotal trials at week 16, bimekizumab resulted in an increase in the proportion of patients who attained ACR50, ACR20, ACR70, and MDA when compared with placebo. The treatment effects appeared to be maintained during the 36-week active treatment–blind period in the BE OPTIMAL trial (biologic-naive patients). Results from the OLE studies indicated that ACR and MDA responses were maintained for patients receiving bimekizumab for up to 2 years; however, data are only available for TNFi-experienced patients and results for biologic-naive patients are forthcoming from the BE VITAL study.
In general, the results from the NMA for the b/tsDMARD-naive population indicated that bimekizumab was more efficacious than most IL-12/23is and IL-23is, and abatacept for ACR outcomes, but may be similar to IL-17is, TNFis, or JAKis, with a few exceptions. The results for the TNFi-experienced population indicated that bimekizumab was superior to some comparators for ACR20 response, but variably superior for both the ACR50 and ACR70 outcomes; however, the wide CrIs suggested imprecision in the results and low certainty in the comparative efficacy of bimekizumab in this subpopulation. There were fewer comparisons made for MDA for bimekizumab versus other PsA drugs, though the results suggest that b/tsDMARD-naive patients on bimekizumab may be more likely to attain MDA over those on IL-23is and adalimumab, and TNFi-experienced patients on bimekizumab may be more likely to attain MDA over those on tofacitinib. The lack of consistent effect across comparators or even drug classes, combined with some lingering uncertainties inherent in the analysis methods and their application to each NMA, make it challenging to draw conclusions for the relative efficacy of bimekizumab versus other classes of DMARDs. The results for the NMA were limited to 12-week to 24-week RCT follow-up durations. Longer-term evidence from the unanchored MAIC had a number of limitations that preclude conclusions from being made with certainty.
The clinical expert consulted by CADTH indicated that although ACR response is commonly used in clinical trials for PsA treatments, it is not typically measured in clinical practice. Instead, the clinical expert and the clinicians who provided input for the CADTH review noted that attainment of low disease activity (e.g., MDA) is a more common approach to measuring treatment response. Therefore, while the ACR results are considered by CADTH to be clinically meaningful and support the efficacy of bimekizumab, the MDA results may be more clinically relevant to practice.
In addition to ACR and MDA, the results for other musculoskeletal outcomes in the pivotal trials showed a consistent direction of effect in the patient populations (biologic-naive and TNFi-experienced), supporting the superior treatment effect of bimekizumab over placebo. The outcomes of LEI and LDI were assessed in subsets of the trial populations and due to the pooling of the 2 different trial populations, it was not possible to compare how prior TNFi treatment (or lack thereof) impacted these outcomes. Moreover, the certainty of the evidence is lower for the LEI and LDI outcomes due to the loss of randomized populations. For the 3 musculoskeletal-related outcomes (LEI, LDI, and SJC), the 95% CIs for the between-group differences included the possibility of no effect compared to the MIDs estimated by the clinical expert, indicating that there is imprecision in the treatment effect of bimekizumab on the proportion of patients who attained an enthesitis-free state or a dactylitis-free state, and a decrease in the number of swollen joints (for TNFi-experienced patients) when compared with placebo. SJC results for biologic-naive patients were low certainty due to the 95% CI for the between-group difference excluding the estimated MID (5 fewer swollen joints). Additionally, SJC was not controlled for multiplicity in either pivotal trial. The longer-term results from the OLE studies indicated that LEI and LDI response was maintained for TNFi-experienced patients receiving bimekizumab for up to 2 years. No longer-term data were presented for SJC and there was no evidence to support comparative efficacy for joint-related outcomes in the ITCs. Input from the patient groups, clinician group, and clinical expert stated that addressing musculoskeletal manifestations was important when treating PsA.
Upward of 82% of patients with PsA have concurrent psoriasis32 and having a treatment that addresses other aspects of clinical disease, such as skin disease, was noted as being important to the patient groups, clinician group, and clinical expert. Both skin-related outcomes were assessed in a subset of the study populations (patients must have had psoriasis affecting at least 3% BSA [51% to 66% of the randomized population for PASI 90] and at least a 2-grade reduction in IGA from baseline [47% to 61% of the randomized population for IGA]). Because these were no longer randomized populations and there was an increased risk of bias, the evidence was less certain. The results for the TNFi-experienced population in the BE COMPLETE trial were further at risk of bias due to an imbalance in baseline characteristics between treatment groups (a higher proportion of patients with a greater percentage of BSA affected by psoriasis and a higher PASI in the bimekizumab group), which reduced the certainty of the PASI 90 results. Therefore, bimekizumab likely results in an increase in the proportion of biologic-naive patients who attain PASI 90 and may result in an increase in the proportion of TNFi-experienced patients who attain PASI 90. For IGA, the 95% CIs for the between-group differences included the possibility of no effect compared to the MID estimated by the clinical expert, indicating that bimekizumab may result in an increase in the proportion of patients who attained an IGA score of 0 or 1, though this outcome was not controlled for multiplicity in either trial. Prespecified subgroup analysis results for PASI 90 were consistent with the primary analysis in the BE COMPLETE trial with the same limitations described previously. It may be important to consider that bimekizumab has already been reviewed and approved for the treatment of moderate-to-severe plaque psoriasis.2,17 The treatment effects for both PASI 90 and IGA appeared to be maintained during the 36-week active treatment–blind period in the BE OPTIMAL trial (biologic-naive patients). The longer-term data from the OLE studies indicate that PASI response was maintained for TNFi-experienced patients receiving bimekizumab for up to 2 years. Results from the NMA indicated that bimekizumab was better than some comparators for achieving PASI 90, but imprecision due to relatively wide CrIs make the results uncertain for both b/tsDMARD-naive and TNFi-experienced patients. There was no evidence for PASI response from the MAIC. Based on the clinical expert’s experience treating PsA, it was noted that interleukin inhibitors (including bimekizumab) tend to be effective for improving skin disease and that bimekizumab could be an option for patients with PsA and concurrent psoriasis, but not if they have IBD, severe uveitis, or active infections.
Patients and clinicians noted that it is important that new treatments address aspects of clinical disease that impact physical function, HRQoL, and symptoms. HAQ-DI and SF-36 PCS measure a person’s ability to perform daily tasks and their physical HRQoL. Results from the pivotal trial demonstrated that, compared to placebo, bimekizumab results in a reduction in HAQ-DI scores and an increase in SF-36 PCS scores, suggesting that physical ability and HRQoL improved with bimekizumab treatment. Both outcomes were controlled for multiplicity in the trials and when compared to MIDs identified from the literature, both were clinically significant. Prespecified subgroup analysis results for HAQ-DI were consistent with the primary analysis in the BE COMPLETE trial with the same limitations described previously. In the BE OPTIMAL and BE COMPLETE trials, the 95% CIs exceeded the MID, suggesting that bimekizumab reduces pain when compared to placebo for patients in both the biologic-naive and TNFi-experienced populations. The treatment effects for all 3 outcomes appeared to be maintained during the 36-week active treatment–blind period in the BE OPTIMAL trial (biologic-naive patients). Data from the OLE studies indicated that improvements in SF-36 PCS were maintained for TNFi-experienced patients receiving bimekizumab for up to 2 years. There was no longer-term evidence for the HAQ-DI or PtAAP in the OLE studies and no indirect evidence for any patient-reported outcomes from the ITCs. Although these were deemed as being important outcomes to patients, the clinical expert noted that symptoms from longstanding PsA may be harder to change over a short period of time and that the need for long-term evidence is necessary to better understand how bimekizumab impacts HRQoL and symptoms.
Evidence from the pivotal trials addressed a number of treatment goals for managing PsA (e.g., achieving MDA, controlling symptoms, and improving functional status and HRQoL) in patients who are biologic-naive or TNFi-experienced; however, there is a lack of evidence for patients who have used targeted DMARDs, such as other non-TNFi bDMARDs and tsDMARDs. Few patients in the trials reported prior use of apremilast (3% to 5% of patients across trials), tofacitinib (1% of patients across trials), and filgotinib (0% to 1% of patients across trials). This is an important consideration given that the clinical expert identified prior targeted DMARD use as a treatment effect modifier, there are various DMARDs for PsA available (besides TNFis), and it is likely that many current patients would have some experience with targeted DMARDs before receiving bimekizumab. Additionally, results after week 16 of the trials are difficult to interpret because of crossover from placebo to treatment in the BE OPTIMAL trial, the lack of a comparator, and the permitted use of rescue therapy (4.8% to 7.0% across treatment groups in the BE OPTIMAL trial). There is no direct evidence for how bimekizumab compares to other treatments for PsA and results from the ITCs were limited by heterogeneity in study and patient characteristics that may not have been completely adjusted for, as well as methodological flaws in the MAIC. Furthermore, the submitted evidence assessed efficacy and safety for a relatively short period of time (considering that PsA is a chronic disease) and more evidence is needed to make conclusions on long-term benefits and harms. Racial diversity was limited in the direct and indirect clinical evidence, which may prevent broad generalizability to patients with PsA in Canada who may receive bimekizumab treatment.
Harms
Overall, reports of AEs were higher in the bimekizumab groups compared to the placebo groups for both the BE OPTIMAL and BE COMPLETE trials and AEs were higher among biologic-naive patients compared to TNFi-experienced patients. Once patients crossed over from placebo to bimekizumab in the BE OPTIMAL trial at week 16, rates of AEs were not surprisingly similar across groups. There were low frequencies of SAEs, withdrawals from treatment due to AEs, and deaths during the active treatment–blind period of the BE OPTIMAL trial. Longer-term results from the OLE studies (for a total of 1 year to 2 years of treatment) were generally consistent with the pivotal trials, though these data are for TNFi-experienced patients only. Data for the biologic-naive population are anticipated as the BE VITAL study is ongoing. ITC results indicated that bimekizumab was not associated with either increased or reduced odds of SAEs, discontinuation, or discontinuation due to AEs, with the exception of having higher odds of discontinuation due to an AE versus ustekinumab (45 mg and 90 mg), and higher odds of discontinuation versus etanercept 25 mg. However, the findings should be interpreted with consideration of the notable heterogeneity in the NMA. Harms outcomes were not included in the MAIC.
Nearly all patients in the trials were receiving concomitant medications (with 44% to 66% being immunosuppressants, such as methotrexate) and it is possible AEs reported during the trials were attributed to these drugs. According to the clinical expert consulted by CADTH, the harms in the trials were considered manageable and the increased risk of fungal infections is expected for this class of drugs. IBD was a notable harm in the trials and although incidence of it was low, this may have been due to the exclusion of patients with active, symptomatic disease from the trials and that there were very few patients who reported a history of any Crohn disease, ulcerative colitis, or IBD in the sponsor-provided data. The expert indicated that bimekizumab would be avoided in patients with IBD, severe uveitis, or active infections.
Conclusion
Based on the evidence from 2 DB RCTs, adults with PsA who received bimekizumab 160 mg every 4 weeks were more likely to demonstrate clinically meaningful improvements in composite measures of disease activity, dermatological manifestations (i.e., psoriasis), physical function, HRQoL, and pain at week 16. Results were generally consistent whether patients were biologic-naive or TNFi-experienced. Evidence that bimekizumab reduces the number of musculoskeletal manifestations (e.g., enthesitis, dactylitis, swollen joints) was less certain, but demonstrated a consistent direction of effect. The trial outcomes address some of the needs patients have for new PsA treatments as well as treatment goals that patients and clinicians have when managing PsA. Longer-term results indicated that treatment effects were maintained with the continued use of bimekizumab for up to 2 years for patients who had had prior TNFis; data for biologic-naive patients are forthcoming. There was no direct evidence comparing bimekizumab and other targeted DMARDs for PsA available for review. The indirect evidence suggests that bimekizumab is superior or similar to other bDMARDs for composite measures of disease activity (e.g., ACR and MDA) and plaque psoriasis outcomes; however, inconsistency in results across comparators, imprecision in results, study and patient characteristic heterogeneity, and methodological flaws reduce the certainty in these results. No new safety signals were identified with extended bimekizumab use in the trials and reports of patients experiencing notable harms were low. Given the available direct and indirect comparative evidence and its approval as a treatment for psoriasis, bimekizumab is an option for adults with PsA, including those with concurrent psoriasis.
References
1.Drug Reimbursement Review sponsor submission: Bimzlex (bimekizumab), 160 mg/mL, solution for SC injection [internal sponsor's package]. UCB Canada Inc.; 2023 Oct 5.
2.Bimzelx (bimekizumab injection): 160 mg/mL solution for injection, subcutaneous use [product monograph]. UCB Canada Inc; 2024 Feb 23.
3.Hackett S, Ogdie A, Coates LC. Psoriatic arthritis: prospects for the future. Ther Adv Musculoskelet Dis. 2022;14:1759720X221086710. eCollection 2022.
4.Kishimoto M, Deshpande GA, Fukuoka K, et al. Clinical features of psoriatic arthritis. Best Pract Res Clin Rheumatol. Jun 2021;35(2):101670. Epub 2021 Mar 17. PubMed
5.Gladman DD. Clinical, radiological, and functional assessment in psoriatic arthritis: is it different from other inflammatory joint diseases? Ann Rheum Dis. Nov 2006;65 Suppl 3(Suppl 3):iii22-4.
6.Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. May 25 2017;376(21):2095-6.PubMed
7.Mease PJ, O'Brien J, Middaugh N, et al. Real-world evidence assessing psoriatic arthritis by disease domain: an evaluation of the CorEvitas psoriatic arthritis/spondyloarthritis registry. ACR Open Rheumatol. Aug 2023;5(8):388-398. PubMed
8.Picchianti-Diamanti A, Germano V, Ferlito C, Migliore A, D'Amelio R, Lagana B. Health-related quality of life and disability in patients with rheumatoid, early rheumatoid and early psoriatic arthritis treated with etanercept. Qual Life Res. Aug 2010;19(6):821-6. Epub 2010 Apr 6. PubMed
9.Wervers K, Vis M, Tchetveriko I, et al. Burden of psoriatic arthritis according to different definitions of disease activity: comparing minimal disease activity and the disease activity index for psoriatic arthritis. Arthritis Care Res (Hoboken). Dec 2018;70(12):1764-1770. PubMed
10.Leung Y-Y, Ho K-W, Li EK, et al. Predictors of functional deterioration in Chinese patients with psoriatic arthritis: a longitudinal study. BMC Musculoskelet Disord. 2014/08/26 2014;15(1):284.
11.Damiani G, Bragazzi NL, Karimkhani Aksut C, et al. The global, regional, and national burden of psoriasis: results and insights from the global burden of disease 2019 study. Front Med (Lausanne). 2021;8:743180. PubMed
12.Singh JA, Guyatt G, Ogdie A, et al. Special article: 2018 American College of Rheumatology/National Psoriasis Foundation guideline for the treatment of psoriatic arthritis. Arthritis Care Res (Hoboken). Jan 2019;71(1):2-29. PubMed
13.Eder L, Haddad A, Rosen CF, et al. The incidence and risk factors for psoriatic arthritis in patients with psoriasis: a prospective cohort study. Arthritis Rheumatol. Apr 2016;68(4):915-23. PubMed
14.Gossec L, Baraliakos X, Kerschbaumer A, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis. Jun 2020;79(6):700-712. PubMed
15.Singh JA, Guyatt G, Ogdie A, et al. Special article: 2018 American College of Rheumatology/National Psoriasis Foundation Guideline for the treatment of psoriatic arthritis. Arthritis Rheumatol. Jan 2019;71(1):5-32. PubMed
16.Coates LC, Soriano ER, Corp N, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA): updated treatment recommendations for psoriatic arthritis 2021. Nat Rev Rheumatol. Aug 2022;18(8):465-479. PubMed
17.CADTH reimbursement recommendation: bimekizumab (Bimzelx - UCB Canada Inc.). Can J Health Technol. 2022;2(4). Accessed 2023 Nov 6. https://www.cadth.ca/sites/default/files/DRR/2022/SR0698%20Bimzelx%20-%20CADTH%20Final%20Rec%20Final.pdf
18.CADTH reimbursement review clinical guidance report: bimekizumab (Bimzelx) for psoriasis, moderate to severe plaque. Can J Health Technol.; 2022;2(5). Accessed 2023 Nov 6. https://www.cadth.ca/sites/default/files/DRR/2022/SR0698-Bimzelx.pdf
19.Clinical Study Report: A multicenter, phase 3, randomized, double-blind, placebo controlled study evaluating the efficacy and safety of bimekizumab in the treatment of subjects with active psoriatic arthritis [internal sponsor's report]. UCB Biopharma SRL; 2022.
20.Clinical Study Report: A phase 3, multicenter, randomized, double-blind, placebo controlled, active-reference (adalimumab) study evaluating the efficacy and safety of bimekizumab in the treatment of study participants with active psoriatic arthritis [internal sponsor's report]. UCB Biopharma SRL; 2022.
21.Dworkin RH, Turk DC, Wyrwich KW, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain. 2008;9(2):105-121. PubMed
22.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. Mar 2020;119:126-135. Epub 2019 Nov 9. PubMed
23.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. Apr 2011;64(4):401-6. Epub 2011 Jan 5. PubMed
24.Sponsor's clinical summary report BIMZELX® (bimekizumab) for the treatment of adult patients with active psoriatic arthritis. BIMZELX® can be used alone or in combination with a conventional non-biologic disease-modifying antirheumatic drug (cDMARD) (e.g., methotrexate).[internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL, solution for SC injection. UCB Canada Inc.; 2023.
25.Clinical Study Report: A multicenter, open-label, follow-up study to evaluate the long-term safety and efficacy of bimekizumab in subjects with psoriatic arthritis [internal sponsor's report]. UCB Biopharma SRL; 2021.
26.Mease PJ, Asahina A, Gladman DD, et al. Effect of bimekizumab on symptoms and impact of disease in patients with psoriatic arthritis over 3 years: results from BE ACTIVE. Rheumatology (Oxford). Feb 1 2023;62(2):617-628. PubMed
27.Coates LC, Landewé R, McInnes IB, et al. Bimekizumab treatment in patients with active psoriatic arthritis and prior inadequate response to tumour necrosis factor inhibitors: 52-week safety and efficacy from the phase III BE COMPLETE study and its open-label extension BE VITAL. RMD Open. Feb 22 2024;10(1) PubMed
28.Coates LC, Landewe R, McInnes I, et al. POS0231 - sustained efficacy and safety of bimekizumab in patients with active psoriatic arthritis and prior inadequate response to tumour necrosis factor inhibitors: results from the phase 3 BE COMPLETE study and its open-label extension up to 1 year. Ann Rheum Dis. 2023;82:346-347.
29.Matching-adjusted indirect comparison (MAIC) in psoriatic arthritis (PsA) with Bimzelx week 52 Phase 3 data [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL solution for injection. UCB Canada Inc; 2023.
30.Network meta-analysis of efficacy and safety outcomes of Bimzelx in psoriatic arthritis (PsA) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL solution for injection. UCB Canada Inc; 2023.
31.Day J, Antony A, Tillett W, Laura CC. The state of the art—psoriatic arthritis outcome assessment in clinical trials and daily practice. Lancet Rheumatol. 2022;4(3):E220-E228. PubMed
32.López-Medina C, Molto A, Sieper J, et al. Prevalence and distribution of peripheral musculoskeletal manifestations in spondyloarthritis including psoriatic arthritis: results of the worldwide, cross-sectional ASAS-PerSpA study. RMD Open. Jan 2021;7(1) PubMed
33.Ohara Y, Kishimoto M, Takizawa N, et al. Prevalence and clinical characteristics of psoriatic arthritis in Japan. J Rheumatol. Aug 2015;42(8):1439-42. Epub 2015 Jun 15. PubMed
34.Pittam B, Gupta S, Harrison NL, Robertson S, Hughes DM, Zhao SS. Prevalence of extra-articular manifestations in psoriatic arthritis: a systematic review and meta-analysis. Rheumatology (Oxford). Sep 1 2020;59(9):2199-2206. PubMed
35.Kaine J, Song X, Kim G, Hur P, Palmer JB. Higher Incidence rates of comorbidities in patients with psoriatic arthritis compared with the general population using U.S. administrative claims data. J Manag Care Spec Pharm. Jan 2019;25(1):122-132. PubMed
36.Coates LC, Conaghan PG, D'Agostino MA, et al. Remission in psoriatic arthritis - where are we now? Rheumatology (Oxford). 2018;57(8):1321-1331. PubMed
37.de Vlam K, Steinfeld S, Toukap AN, et al. The burden of psoriatic arthritis in the biologics era: data from the Belgian epidemiological psoriatic arthritis study. Rheumatology (Oxford). Dec 1 2021;60(12):5677-5685. PubMed
38.Haugeberg G, Lund Nilsen TI, Kavanaugh A, Thomsen RS, Gulati AM, Hoff M. Physical and psychosocial burden of psoriatic arthritis: longitudinal data from a population-based study in Norway. Arthritis Care Res (Hoboken). Jan 2021;73(1):138-145. Epub 2020 Nov 30. PubMed
39.Kavanaugh A, Helliwell P, Ritchlin CT. Psoriatic arthritis and burden of disease: patient perspectives from the population-based Multinational Assessment of Psoriasis and Psoriatic Arthritis (MAPP) survey. Rheumatol Ther. Jun 2016;3(1):91-102. Epub 2016 Feb 29. PubMed
40.Tillett W, Coates LC, Kiri S, Taieb V, Willems D, Mease PJ. Achievement of more stringent disease control is associated with reduced burden on workplace and household productivity: results from long-term certolizumab pegol treatment in patients with psoriatic arthritis. Ther Adv Musculoskelet Dis. 2022;14:1759720x221140846.
41.Wallenius M, Skomsvoll JF, Koldingsnes W, et al. Work disability and health-related quality of life in males and females with psoriatic arthritis. Ann Rheum Dis. May 2009;68(5):685-9. PubMed
42.National Psoriasis Foundation. About psoriatic arthritis. Updated April 2022. Accessed 2023 Feb 27, https://www.psoriasis.org/about-psoriatic-arthritis/
43.Ogdie A, Weiss P. The epidemiology of psoriatic arthritis. Rheum Dis Clin North Am. Nov 2015;41(4):545-68. PubMed
44.Dures E, Bowen C, Brooke M, et al. Diagnosis and initial management in psoriatic arthritis: a qualitative study with patients. Rheumatol Adv Pract. 2019;3(2):rkz022. PubMed
45.Gladman DD, Starr M, Cividino A, et al. Canadian rheumatologists' perspectives on moderate psoriatic arthritis and oligoarticular psoriatic arthritis. J Rheumatol. Nov 2021;48(11):1692-1697. PubMed
46.Arthritis Society of Canada. Psoriatic arthritis. Accessed 2023 Feb 27, https://arthritis.ca/about-arthritis/arthritis-types-(a-z)/types/psoriatic-arthritis
47.Coates LC, Soriano ER, Corp N, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA): updated treatment recommendations for psoriatic arthritis 2021. Nat Rev Rheumatol. Aug 2022;18(8):465-479. Epub 2022 Jun 27. PubMed
48.Gladman DD, Orbai AM. Post TW, ed. Treatment of psoriatic arthritis. UpToDate; 2023. Accessed 2023 Oct 6. http://www.uptodate.com/
49.Maroof A, Okoye R, Smallie T, et al. Bimekizumab dual inhibition of IL-17A and IL-17F provides evidence of IL-17F contribution to chronic inflammation in diease-relevant cells [abstract]. Arthritis Rheumatol. November 6 2017;69(suppl 10)
50.Taltz (ixekizumab): 80 mg / 1.0 mL solution for injection, subcutaneous use [product monograph]. Eli Lilly Canada, Inc; 2023 Mar 7. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00069828.PDF
51.Cosentyx (secukinumab): 75 mg/0.5 mL solution for injection, 150 mg/1 mL solution for injection, 300 mg/2 mL solution for injection [product monograph]. Novartis Pharmaceutical Canada, Inc; 2023 Aug 17. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00072128.PDF
52.Wezlana (ustekinumab): solution for subcutaneous injection, 45 mg/0.5 mL and 90 mg/mL and solution for intravenous infusion, 130 mg/26 mL (5 mg/mL) [product monograph]. Amgen Canada Inc; 2023 Dec 27. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00073953.PDF
53.Tremfya (guselkumab): solution for injection, 100 mg/1 mL pre-filled syringe [product monograph]. Janssen Inc; 2022 Nov 8. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00068091.PDF
54.Skyrizi (risankizumab): 150 mg in 1 mL sterile solution (150 mg/mL) subcutaneous injection, 360 mg in 2.4 mL sterile solution (150 mg/mL) subcutaneous injection, 75 mg in 0.83 mL sterile solution (90 mg/mL) subcutaneous injection, 600 mg in 10 mL sterile solution (60 mg/mL) for intravenous infusion [product monograph]. AbbVie Corporation; 2023 May 15. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00070735.PDF
55.Hyrimoz (adalimumab): 20 mg in 0.4 mL sterile solution (50 mg/mL) subcutaneous injection in pre-filled syringe, 40 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection in a pre-filled syringe, 40 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection in a pre-filled autoinjector, 20 mg in 0.2 mL sterile solution (100 mg/mL) subcutaneous injection in a pre-filled syringe, 40 mg in 0.4 mL sterile solution (100 mg/mL) subcutaneous injection in pre-filled syringe, 80 mg in 0.8 mL sterile solution (100 mg/mL) subcutaneous injection in pre-filled syringe, 40 mg in 0.4 mL sterile solution (100 mg/mL) subcutaneous injection pre-filled autoinjector, 80 mg in 0.8 mL sterile solution (100 mg/mL) subcutaneous injection pre-filled autoinjector [product monograph]. Sandoz Canada Inc; 2023 Oct 31. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00073497.PDF
56.Cimzia (certolizumab pegol): solution for injection 200 mg/mL [product monograph]. UCB Canada Inc; 2019 Nov 13. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00053920.PDF
57.Erelzi (etanercept): solution for injection in a prefilled syringe 50 mg/mL, solution for injection in a prefilled autoinjector 50 mg/mL [product monograph]. Sandoz Canada Inc; 2023 Jan 18. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00069556.PDF
58.Simponi (golimumab): solution for subcutaneous injection, 50 mg/0.5 mL, 100 mg/1.0 mL and solution for intravenous infusion 50 mg/4.0 mL [product monograph]. Janssen Inc; 2023 May 4. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00070523.PDF
59.Renflexis (infliximab for injection): powder for solution, sterile, lyophilized, 100 mg /vial, intravenous infusion [product monograph]. Organon Canada Inc; 2023 Oct 4. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00072737.PDF
60.Apo-Apremilast tablets (apremilast): 10 mg, 20 mg, and 30 mg for oral use [product monograph]. Apotex Inc; 2023 Sep 26. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00072618.PDF
61.Xeljanz (tofacitinib): oral 5 mg tofacitinib, 10 mg tofacitinib, 11 mg tofacitinib [product monograph]. Pfizer Canada ULC; 2023 Aug 11. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00071982.PDF
62.Rinvoq (upadacitinib): extended-release tablets oral, 15 mg upadacitinib, 30 mg upadacitinib, 45 mg upadacitinib [product monograph]. AbbVie Corporation; 2023 Oct 31. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00073108.PDF
63.Orencia (abatacept): lyophilized powder for solution for infusion (250 mg/vial), solution for subcutaneous injection (125 mg/mL) [product monograph]. Bristol-Myers Squibb Canada; 2023 May 19. Accessed 2023 Nov 3. https://pdf.hres.ca/dpd_pm/00070861.PDF
64.McInnes IB, Asahina A, Coates LC, et al. Bimekizumab in patients with psoriatic arthritis, naive to biologic treatment: a randomised, double-blind, placebo-controlled, phase 3 trial (BE OPTIMAL). Lancet. Jan 7 2023;401(10370):25-37. PubMed
65.UCB Biopharma SRL. NCT03895203: A Study to test the efficacy and safety of bimekizumab in the treatment of subjects with active psoriatic arthritis (BE OPTIMAL) U.S. National Library of Medicine; 2023. Accessed 2023 Nov 7. https://clinicaltrials.gov/study/NCT03895203?term=NCT03895203&rank=1
66.Merola JF, Landewé R, McInnes IB, et al. Bimekizumab in patients with active psoriatic arthritis and previous inadequate response or intolerance to tumour necrosis factor-α inhibitors: a randomised, double-blind, placebo-controlled, phase 3 trial (BE COMPLETE). Lancet. Jan 7 2023;401(10370):38-48. PubMed
67.UCB Biopharma SRL. NCT03896581: A study to evaluate the efficacy and safety of bimekizumab in the treatment of subjects with active psoriatic arthritis (BE COMPLETE) U.S. National Library of Medicine; 2023. Accessed Nov 7, 2023. https://clinicaltrials.gov/study/NCT03896581?term=NCT03896581&rank=1
68.Healy PJ, Helliwell PS. Measuring dactylitis in clinical trials: which is the best instrument to use? J Rheumatol. Jun 2007;34(6):1302-6. PubMed
69.Helliwell PS, Firth J, Ibrahim GH, Melsom RD, Shah I, Turner DE. Development of an assessment tool for dactylitis in patients with psoriatic arthritis. J Rheumatol. Sep 2005;32(9):1745-50. PubMed
70.Langley RG, Feldman SR, Nyirady J, van de Kerkhof P, Papavassilis C. The 5-point Investigator's Global Assessment (IGA) scale: a modified tool for evaluating plaque psoriasis severity in clinical trials. J Dermatolog Treat. Feb 2015;26(1):23-31. Epub 2013 Dec 20. PubMed
71.Mease PJ, Woolley JM, Bitman B, Wang BC, Globe DR, Singh A. Minimally important difference of Health Assessment Questionnaire in psoriatic arthritis: relating thresholds of improvement in functional ability to patient-rated importance and satisfaction. J Rheumatol. Nov 2011;38(11):2461-5. PubMed
72.Kwok T, Pope JE. Minimally important difference for patient-reported outcomes in psoriatic arthritis: Health Assessment Questionnaire and pain, fatigue, and global visual analog scales. J Rheumatol. May 2010;37(5):1024-8. Epub 2010 Mar 15. PubMed
73.Leung YY, Zhu TY, Tam LS, Kun EW, Li EK. Minimal important difference and responsiveness to change of the SF-36 in patients with psoriatic arthritis receiving tumor necrosis factor-α blockers. J Rheumatol. Sep 2011;38(9):2077-9. PubMed
74.van Gestel AM, Haagsma CJ, van Riel PL. Validation of rheumatoid arthritis improvement criteria that include simplified joint counts. Arthritis Rheum. Oct 1998;41(10):1845-50. PubMed
75.Felson DT, Anderson JJ, Boers M, et al. The American College of Rheumatology preliminary core set of disease activity measures for rheumatoid arthritis clinical trials. The Committee on Outcome Measures in Rheumatoid Arthritis Clinical Trials. Arthritis Rheum. Jun 1993;36(6):729-40. PubMed
76.Fransen J, Antoni C, Mease PJ, et al. Performance of response criteria for assessing peripheral arthritis in patients with psoriatic arthritis: analysis of data from randomised controlled trials of two tumour necrosis factor inhibitors. Ann Rheum Dis. Oct 2006;65(10):1373-8. PubMed
77.Albert DA, Huang G, Dubrow G, Brensinger CM, Berlin JA, Williams HJ. Criteria for improvement in rheumatoid arthritis: alternatives to the American College of Rheumatology 20. J Rheumatol. May 2004;31(5):856-66. PubMed
78.Felson DT, Anderson JJ, Boers M, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. Jun 1995;38(6):727-35. PubMed
79.Coates LC, Strand V, Wilson H, et al. Measurement properties of the minimal disease activity criteria for psoriatic arthritis. RMD Open. 2019;5(2):e001002. PubMed
80.Healy PJ, Helliwell PS. Measuring clinical enthesitis in psoriatic arthritis: assessment of existing measures and development of an instrument specific to psoriatic arthritis. Arthritis Rheum. May 15 2008;59(5):686-91. PubMed
81.Simpson MJ, Chow C, Morgenstern H, Luger TA, Ellis CN. Comparison of three methods for measuring psoriasis severity in clinical studies (Part 2 of 2): use of quality of life to assess construct validity of the Lattice System Physician's Global Assessment, Psoriasis Area and Severity Index and Static Physician's Global Assessment. J Eur Acad Dermatol Venereol. Jul 2015;29(7):1415-20. PubMed
82.Berth-Jones J, Grotzinger K, Rainville C, et al. A study examining inter- and intrarater reliability of three scales for measuring severity of psoriasis: Psoriasis Area and Severity Index, Physician's Global Assessment and Lattice System Physician's Global Assessment. Br J Dermatol. Oct 2006;155(4):707-13. PubMed
83.Bożek A, Reich A. The reliability of three psoriasis assessment tools: psoriasis area and severity index, body surface area and physician global assessment. Adv Clin Exp Med. Aug 2017;26(5):851-856. PubMed
84.Langley RG, Ellis CN. Evaluating psoriasis with psoriasis area and severity index, psoriasis global assessment, and lattice system physician's global assessment. J Am Acad Dermatol. Oct 2004;51(4):563-9. PubMed
85.Fink C, Alt C, Uhlmann L, Klose C, Enk A, Haenssle HA. Intra- and interobserver variability of image-based PASI assessments in 120 patients suffering from plaque-type psoriasis. J Eur Acad Dermatol Venereol. Aug 2018;32(8):1314-1319. PubMed
86.Puzenat E, Bronsard V, Prey S, et al. What are the best outcome measures for assessing plaque psoriasis severity? A systematic review of the literature. J Eur Acad Dermatol Venereol. Apr 2010;24 Suppl 2:10-6. PubMed
87.Mattei PL, Corey KC, Kimball AB. Psoriasis Area Severity Index (PASI) and the Dermatology Life Quality Index (DLQI): the correlation between disease severity and psychological burden in patients treated with biological therapies. J Eur Acad Dermatol Venereol. Mar 2014;28(3):333-7. PubMed
88.Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. Feb 1980;23(2):137-45. PubMed
89.Leung YY, Holland R, Mathew AJ, et al. Clinical trial discrimination of physical function instruments for psoriatic arthritis: a systematic review. Semin Arthritis Rheum. Oct 2020;50(5):1158-1181. PubMed
90.Leung YY, Orbai AM, de Wit M, et al. Comparing the patient-reported physical function outcome measures in a real-life international cohort of patients with psoriatic arthritis. Arthritis Care Res (Hoboken). Apr 2021;73(4):593-602. PubMed
91.Leung YY, Tillett W, Hojgaard P, et al. Test-retest reliability for HAQ-DI and SF-36 PF for the measurement of physical function in psoriatic arthritis. J Rheumatol. Oct 2021;48(10):1547-1551. PubMed
92.Maruish M, Kosinski M, Bjorner J, Gandek B, Turner-Bowker DM, Ware JE. User's manual for the SF36v2 Health Survey. Quality Metric Incorporated; 2011.
93.Ware JE, Jr., Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. Jun 1992;30(6):473-83. PubMed
94.Frendl DM, Ware JE, Jr. Patient-reported functional health and well-being outcomes with drug therapy: a systematic review of randomized trials using the SF-36 health survey. Med Care. May 2014;52(5):439-45. PubMed
95.Leung YY, Ho KW, Zhu TY, Tam LS, Kun EW, Li EK. Testing scaling assumptions, reliability and validity of medical outcomes study short-form 36 health survey in psoriatic arthritis. Rheumatology (Oxford). Aug 2010;49(8):1495-501. PubMed
96.Hays RD, Morales LS. The RAND-36 measure of health-related quality of life. Ann Med. Jul 2001;33(5):350-7. PubMed
97.Strand V, Singh JA. Improved health-related quality of life with effective disease-modifying antirheumatic drugs: evidence from randomized controlled trials. Am J Manag Care. Dec 2007;13 Suppl 9:S237-51. PubMed
98.Samsa G, Edelman D, Rothman ML, Williams GR, Lipscomb J, Matchar D. Determining clinically important differences in health status measures: a general approach with illustration to the Health Utilities Index Mark II. Pharmacoeconomics. Feb 1999;15(2):141-55. PubMed
99.Mease PJ. Fibromyalgia, a missed comorbidity in spondyloarthritis: prevalence and impact on assessment and treatment. Curr Opin Rheumatol. Jul 2017;29(4):304-310. PubMed
100.Højgaard P, Klokker L, Orbai AM, et al. A systematic review of measurement properties of patient reported outcome measures in psoriatic arthritis: A GRAPPA-OMERACT initiative. Semin Arthritis Rheum. Apr 2018;47(5):654-665. PubMed
101.Ogdie A, Coates LC, Mease P. Measuring outcomes in psoriatic arthritis. Arthritis Care Res (Hoboken). Oct 2020;72 Suppl 10(Suppl 10):82-109.
102.Fleiss JL, Tytun A, Ury HK. A simple approximation for calculating sample sizes for comparing independent proportions. Biometrics. Jun 1980;36(2):343-6. PubMed
103.McInnes IB, Mease PJ, Kirkham B, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Sep 19 2015;386(9999):1137-46. Epub 2015 Jun 28. PubMed
104.Nash P, Mease PJ, McInnes IB, et al. Efficacy and safety of secukinumab administration by autoinjector in patients with psoriatic arthritis: results from a randomized, placebo-controlled trial (FUTURE 3). Arthritis Res Ther. Mar 15 2018;20(1):47. PubMed
105.Mease P, van der Heijde D, Landewe R, et al. Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: primary results from the randomised, double-blind, phase III FUTURE 5 study. Ann Rheum Dis. Jun 2018;77(6):890-897. Epub 2018 Mar 17. PubMed
106.Nash P, Kirkham B, Okada M, et al. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. Jun 10 2017;389(10086):2317-2327. Epub 2017 May 24. PubMed
107.Hosmer DW, Jr., Lemeshow S. Applied logistic regression. 2nd ed. John Wiley & Sons; 2000.
108.Mallinckrodt CH. Preventing and treating missing data in longitudinal clinical trials - a practical guide. Cambridge University Press; 2013.
109.Mease PJ, Kavanaugh A, Reimold A, et al. Secukinumab provides sustained improvements in the signs and symptoms of psoriatic arthritis: final 5-year results from the phase 3 FUTURE 1 study. ACR Open Rheumatol. Jan 2020;2(1):18-25. PubMed
110.Mease PJ, van der Heijde D, Ritchlin CT, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. Jan 2017;76(1):79-87. Epub 2016 Aug 23. PubMed
111.Moser BK, Stevens RG, Watts CL. The two-sample t test versus satterthwaite's approximate f test. research-article. Commun Stat Theory Methods. 27 Jun 2007 2007;18(11):3963-3975. doi:Communications in Statistics - Theory and Methods, Vol. 18, No. 11, 1989, pp. 3963–3975.
112.Nash P, McInnes IB, Mease PJ, et al. Secukinumab versus adalimumab for psoriatic arthritis: comparative effectiveness up to 48 weeks using a matching-adjusted indirect comparison. Rheumatol Ther. Jun 2018;5(1):99-122. PubMed
113.Strand V, McInnes I, Mease P, et al. Matching-adjusted indirect comparison: secukinumab versus infliximab in biologic-naive patients with psoriatic arthritis. J Comp Eff Res. May 2019;8(7):497-510. Epub 2019 Feb 26. PubMed
114.Dias S, Sutton AJ, Welton NJ, Ades AE. NICE DSU technical support document 3: heterogeneity: subgroups, meta-regression, bias and bias adjustment. University of Sheffield; 2012. Accessed 2023 Dec 5. https://www.ncbi.nlm.nih.gov/books/NBK395886/pdf/Bookshelf_NBK395886.pdf
115.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU technical support document 2: a generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. University of Sheffield; 2011. Accessed 2023 Jun 26. https://www.ncbi.nlm.nih.gov/books/NBK310366/pdf/Bookshelf_NBK310366.pdf
116.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams K, Welton NJ. NICE DSU technical support document 18: methods for population-adjusted indirect comparisons in submissions to NICE. University of Sheffield; 2016. Accessed 2023 Dec 5. https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf
117.UCB Canada Inc response to October 12, 2023 CADTH request for additional information regarding Bimzlex (bimekizumab) CADTH review [internal sponsor's report]. UCB Canada Inc; 2023 Oct 20.
Appendix 1: Detailed Outcome Data
Note this appendix has not been copy-edited.
Data Related to the Pivotal Trials
Table 50: Subgroup Analysis for Prior TNFi Exposure in BE COMPLETE Trial (Randomized Set)
Subgroup | BE COMPLETE study | |
|---|---|---|
Placebo | Bimekizumab | |
ACR50 | ||
Inadequate response to 1 TNFi, N | 103 | 203 |
n/Nsub (%) | 7/98 (7.1) | 97/200 (48.5) |
n (%) | 7 (6.8) | 97 (47.8) |
Adjusted responder rate, % (95% CI)a | NEb | NEb |
Difference vs. placebo, % (95% CI)a | NEb | |
OR vs. placebo (95% CI)a | NEb | |
Inadequate response to 2 TNFis, N | 15 | 30 |
n/Nsub (%) | 0/12 | 6/29 (20.7) |
n (%) | 0 | 6 (20.0) |
Adjusted responder rate, % (95% CI)a | NEb | NEb |
Difference vs. placebo, % (95% CI)a | NEb | |
OR vs. placebo (95% CI)a | NEb | |
Intolerance to TNFis, N | 15 | 34 |
n/Nsub (%) | 2/15 (13.3) | 13/34 (38.2) |
n (%) | 2 (13.3) | 13 (38.2) |
Adjusted responder rate, % (95% CI)a | NEb | NEb |
Difference vs. placebo, % (95% CI)a | NEb | |
OR vs. placebo (95% CI)a | NEb | |
HAQ-DI for patients who had a score decrease from baseline ≥ 0.35 | ||
Inadequate response to 1 TNFi, N | 82 | 177 |
n/Nsub (%) | 18/79 (22.8) | 109/174 (62.6) |
n (%) | 18 (22.0) | 109 (61.6) |
Adjusted responder rate, % (95% CI)a | 22.4 (14.1 to 33.7) | 63.2 (54.0 to 71.5) |
Difference vs. placebo, % (95% CI)a | 40.8 (28.6 to 53.1) | |
OR vs. placebo (95% CI)a | 6.0 (3.2 to 11.0) | |
Inadequate response to 2 TNFis, N | 14 | 28 |
n/Nsub (%) | 3/11 (27.3) | 9/27 (33.3) |
n (%) | 3 (21.4) | 9 (32.1) |
Adjusted responder rate, % (95% CI)a | 24.7 (8.2 to 54.5) | 36.2 (20.0 to 56.2) |
Difference vs. placebo, % (95% CI)a | 11.5 (–19.9 to 42.9) | |
OR vs. placebo (95% CI)a | 1.7 (0.4 to 7.9) | |
Intolerance to TNFis, N | 14 | 26 |
n/Nsub (%) | 3/14 (21.4) | 12/26 (46.2) |
n (%) | 3 (21.4) | 12 (46.2) |
Adjusted responder rate, % (95% CI)a | 21.6 (7.0 to 50.4) | 46.3 (27.8 to 65.9) |
Difference vs. placebo, % (95% CI)a | 24.7 (–6.0 to 55.5) | |
OR vs. placebo (95% CI)a | 3.1 (0.7 to 14.0) | |
PASI 90 for patients with ≥ 3% BSA affected by psoriasis at baseline | ||
Inadequate response to 1 TNFi, N | 67 | 142 |
n/Nsub (%) | 3/60 (5.0) | 101/141 (71.6) |
n (%) | 3 (4.5) | 101 (71.1) |
Adjusted responder rate, % (95% CI)a | 3.6 (1.1 to 11.3) | 68.1 (56.4 to 77.9) |
Difference vs. placebo, % (95% CI)a | 64.4 (53.6 to 75.3) | |
OR vs. placebo (95% CI)a | 56.5 (16.7 to 191.8) | |
Inadequate response to 2 TNFis, N | 11 | 12 |
n/Nsub (%) | 2/9 (22.2) | 6/11 (54.5) |
n (%) | 2 (18.2) | 6 (50.0) |
Adjusted responder rate, % (95% CI)a | 22.8 (5.7 to 59.0) | 47.8 (22.3 to 74.5) |
Difference vs. placebo, % (95% CI)a | 25.0 (–16.2 to 66.1) | |
OR vs. placebo (95% CI)a | 3.1 (0.4 to 21.9) | |
Intolerance to TNFis, N | 10 | 22 |
n/Nsub (%) | 1/10 (10.0) | 14/22 (63.6) |
n (%) | 1 (10.0) | 14 (63.6) |
Adjusted responder rate, % (95% CI)a | 8.4 (1.1 to 43.0) | 58.2 (35.4 to 77.9) |
Difference vs. placebo, % (95% CI)a | 49.7 (18.5 to 80.9) | |
OR vs. placebo (95% CI)a | 15.1 (1.6 to 143.1) | |
ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; BSA = body surface area; CI = confidence interval; HAQ-DI = Health Assessment Questionnaire–Disability Index; NE = Not Evaluable; OR = odds ratio; PASI 90 = 90% reduction in Psoriasis Area Severity Index score; TNFi = tumour necrosis factor inhibitor; vs. = versus.
Notes: In the n/Nsub (%) row, n represents the number of responders at week 16 whose response was observed and who did not prematurely discontinue study treatment before week 16. Nsub represents the number of patients with a nonmissing measurement for the outcome at week 16 and who did not prematurely discontinue study treatment before week 16, and percentages were calculated accordingly.
In the n (%) row, patients who have missing outcome data at week 16 or who discontinued study treatment before week 16 regardless of whether they have data or not are considered nonresponders. The n represents the number of responders at week 16, and percentages are calculated by treatment group on the number of patients in the referenced population.
aCalculated using a logistic regression with factors for treatment, prior TNFi exposure at baseline, region, and treatment by prior TNFi exposure at baseline interaction.
bResults are not evaluable where the model did not converge or where all the responses were the same in a subgroup.
Source: BE COMPLETE Clinical Study Report.19
Table 51: Summary of Harms Results During Active Treatment–Blind Period From the BE OPTIMAL Trial (Active Treatment–Blind Set)
Harm | BE OPTIMAL study | |
|---|---|---|
Placebo to bimekizumaba (N = 271) | Bimekizumab (N = 414) | |
AEsb | ||
≥ 1 AE, n (%) | 191 (70.5) | 298 (72.0) |
Nasopharyngitis, n (%) | 23 (8.5) | 29 (7.0) |
Upper respiratory tract infection, n (%) | 12 (4.4) | 21 (5.1) |
Urinary tract infection, n (%) | 15 (5.5) | 20 (4.8) |
Oral candidiasis, n (%) | 14 (5.2) | 19 (4.6) |
SAEsc | ||
≥ 1 SAE, n (%) | 16 (5.9) | 23 (5.6) |
Osteoarthritis, n (%) | 0 (0.0) | 2 (0.3) |
Patients who stopped treatment due to AEsc | ||
Patients who stopped treatment, n (%) | 5 (1.8) | 11 (2.7) |
Deaths | ||
Patients who died, n (%) | 1 (0.4) | 0 (0.0) |
Traumatic shock, n (%) | 1 (0.4) | 0 (0.0) |
AEs of special interest | ||
Liver dysfunction based on Hy’s law, n (%)d | 0 (0.0) | 1 (0.2) |
Liver toxicity, n (%) | NR | NR |
Opportunistic infection, n (%) | 4 (1.5) | 5 (1.2) |
Any fungal infection, n (%) | 25 (9.2) | 47 (11.4) |
Candida infection, n (%) | 19 (7.0) | 28 (6.8) |
Fungal infection NEC, n (%) | 7 (2.6) | 19 (4.6) |
Tinea infection, n (%) | 2 (0.7) | 5 (1.2) |
Reactivation of tuberculosis infection, n (%) | NR | NR |
Major cardiovascular event, n (%) | 1 (0.4) | 3 (0.7) |
Malignancy, n (%) | 4 (1.5) | 3 (0.7) |
Serious injection-related AE, n (%) | NR | NR |
Anaphylaxis, n (%) | 0 (0.0) | 0 (0.0) |
Inflammatory bowel disease, n (%) | 0 (0.0) | 2 (0.4) |
AE = adverse event; NEC = not elsewhere classified; NR = not reported; SAE = serious adverse event.
aPatients who were randomized to receive placebo during the 16-week DB period were reallocated to receive bimekizumab during the active treatment–blind period.
bOccurring in at least 5% of patients.
cOccurring in at least 2 patients in any treatment group.
dHy’s law is defined as an alanine transaminase or aspartate transaminase level at least 3 times the upper limit of normal with coexisting total bilirubin at least 2 times the upper limit of normal in the absence of alkaline phosphatase at least 2 times the upper limit of normal.
Sources: BE OPTIMAL Clinical Study Report and sponsor’s summary of clinical evidence.20,24 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Data Related to the ITC
NMA Networks, week 12 to week 24
Figure 3: NMA Network for ACR20 — b/tsDMARD-Naive Patients
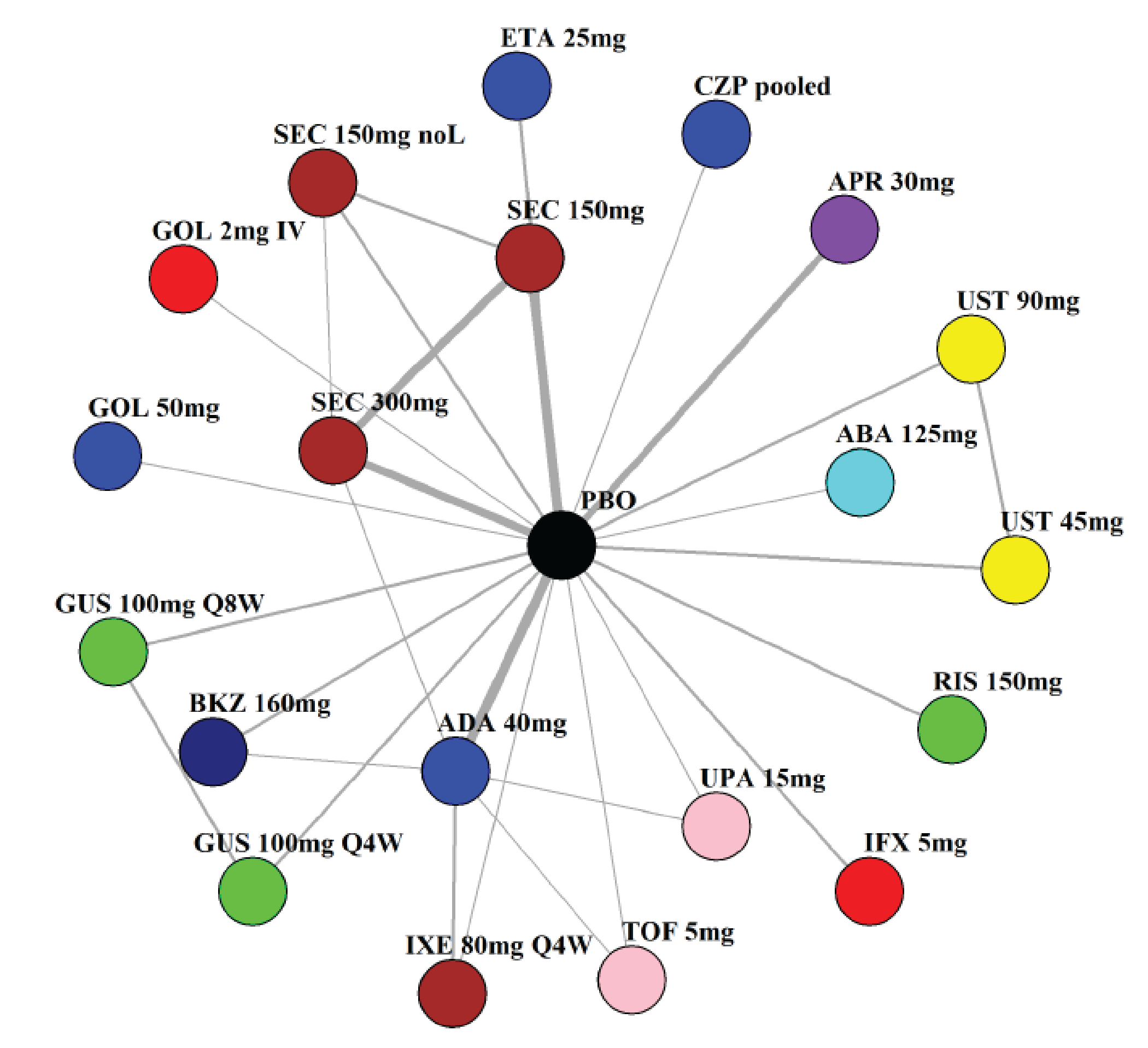
ABA = abatacept; ADA = adalimumab; APR = apremilast; BKZ = bimekizumab; CZP = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; IFX = infliximab; IXE = ixekizumab; noL = no loading; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Source: Sponsor’s network meta-analysis technical document.30
Figure 4: NMA Network for ACR50 — b/tsDMARD-Naive Patients
ABA = abatacept; ADA = adalimumab; APR = apremilast; BKZ = bimekizumab; CZP = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; IFX = infliximab; IXE = ixekizumab; noL = no loading; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Source: Sponsor’s network meta-analysis technical document.30
Figure 5: NMA Network for ACR70 — b/tsDMARD-Naive Patients
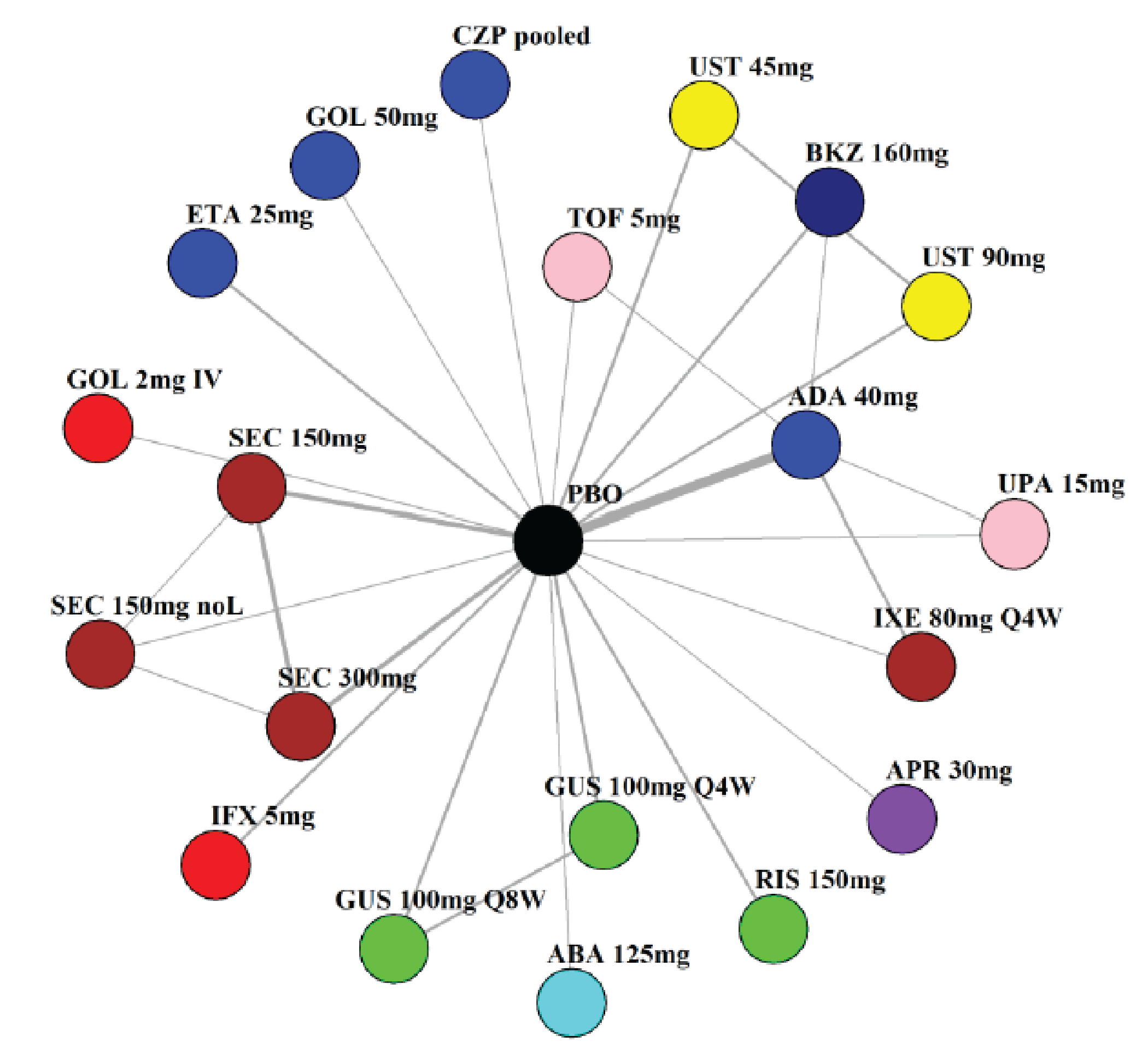
ABA = abatacept; ADA = adalimumab; APR = apremilast; BKZ = bimekizumab; CZP = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; IFX = infliximab; IXE = ixekizumab; noL = no loading; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Source: Sponsor’s network meta-analysis technical document.30
Figure 6: NMA Network for PASI 90 — b/tsDMARD-Naive Patients
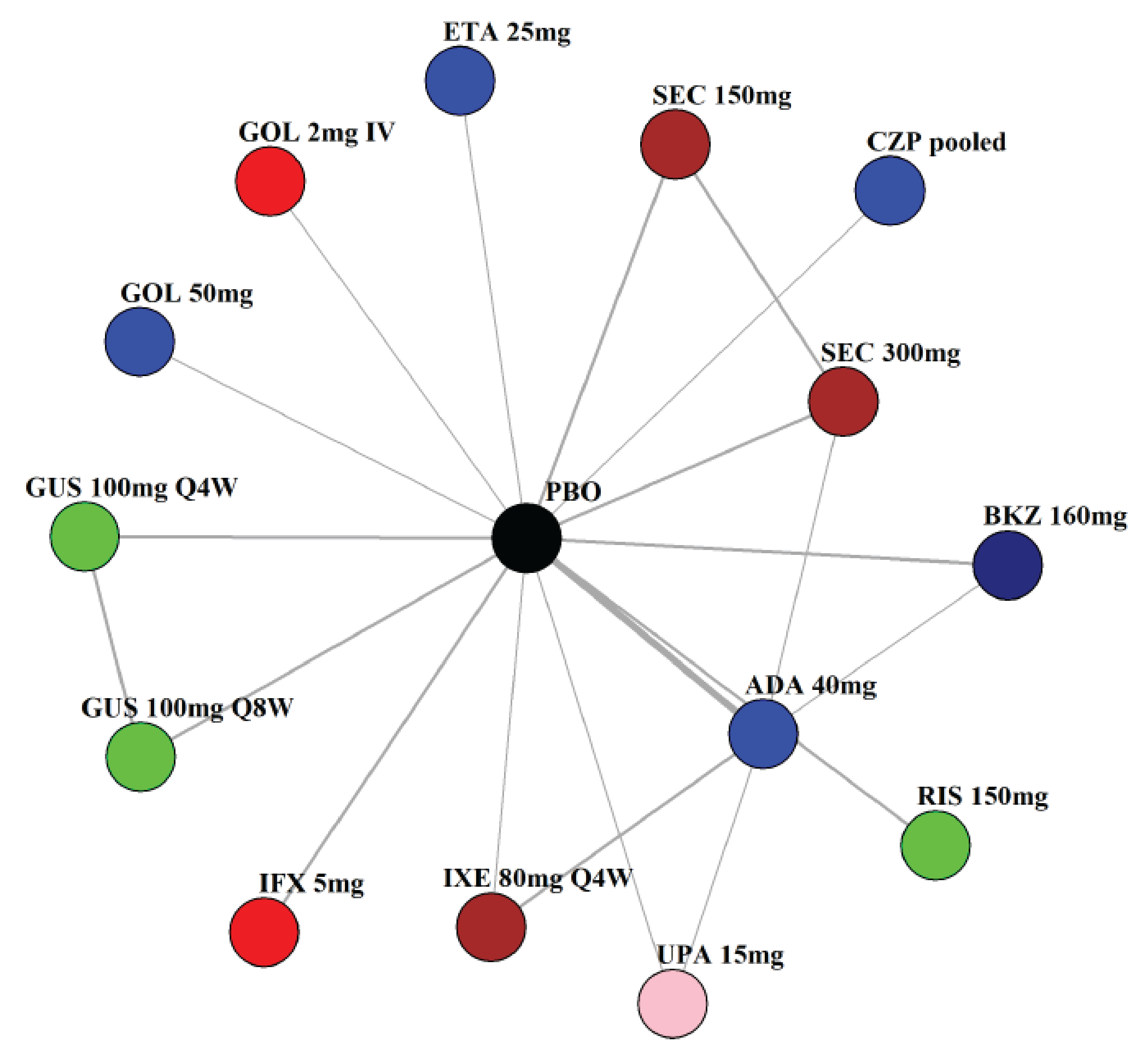
ADA = adalimumab; BKZ = bimekizumab; CZP = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; IFX = infliximab; IXE = ixekizumab; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; UPA = upadacitinib.
Source: Sponsor’s network meta-analysis technical document.30
Figure 7: NMA Network for MDA — b/tsDMARD-Naive Patients
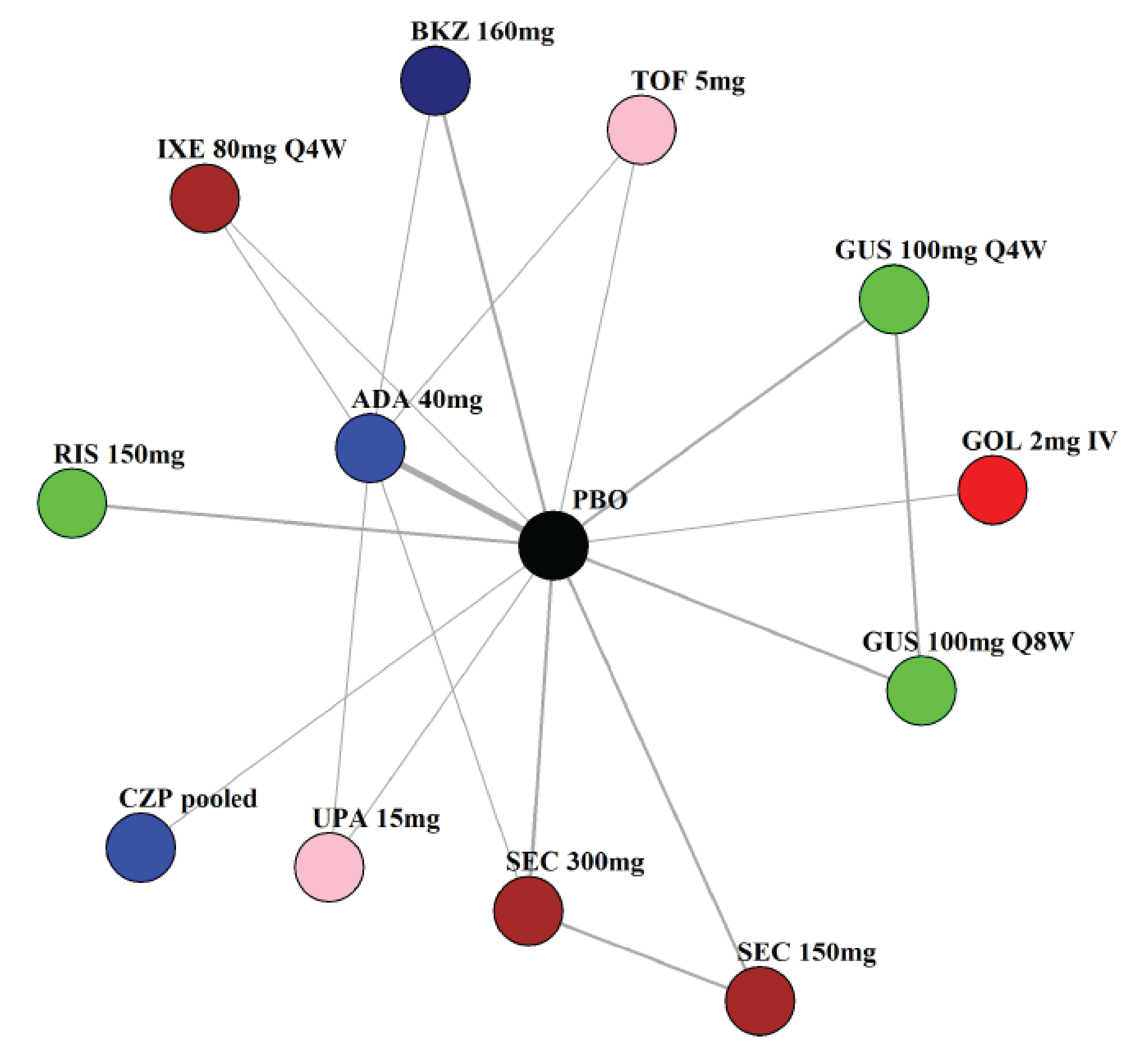
ADA = adalimumab; APR = apremilast; BKZ = bimekizumab; CZP = certolizumab pegol; GOL = golimumab; GUS = guselkumab; IXE = ixekizumab; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib.
Source: Sponsor’s network meta-analysis technical document.30
Figure 8: NMA Network for ACR20 — TNFi-Experienced Patients
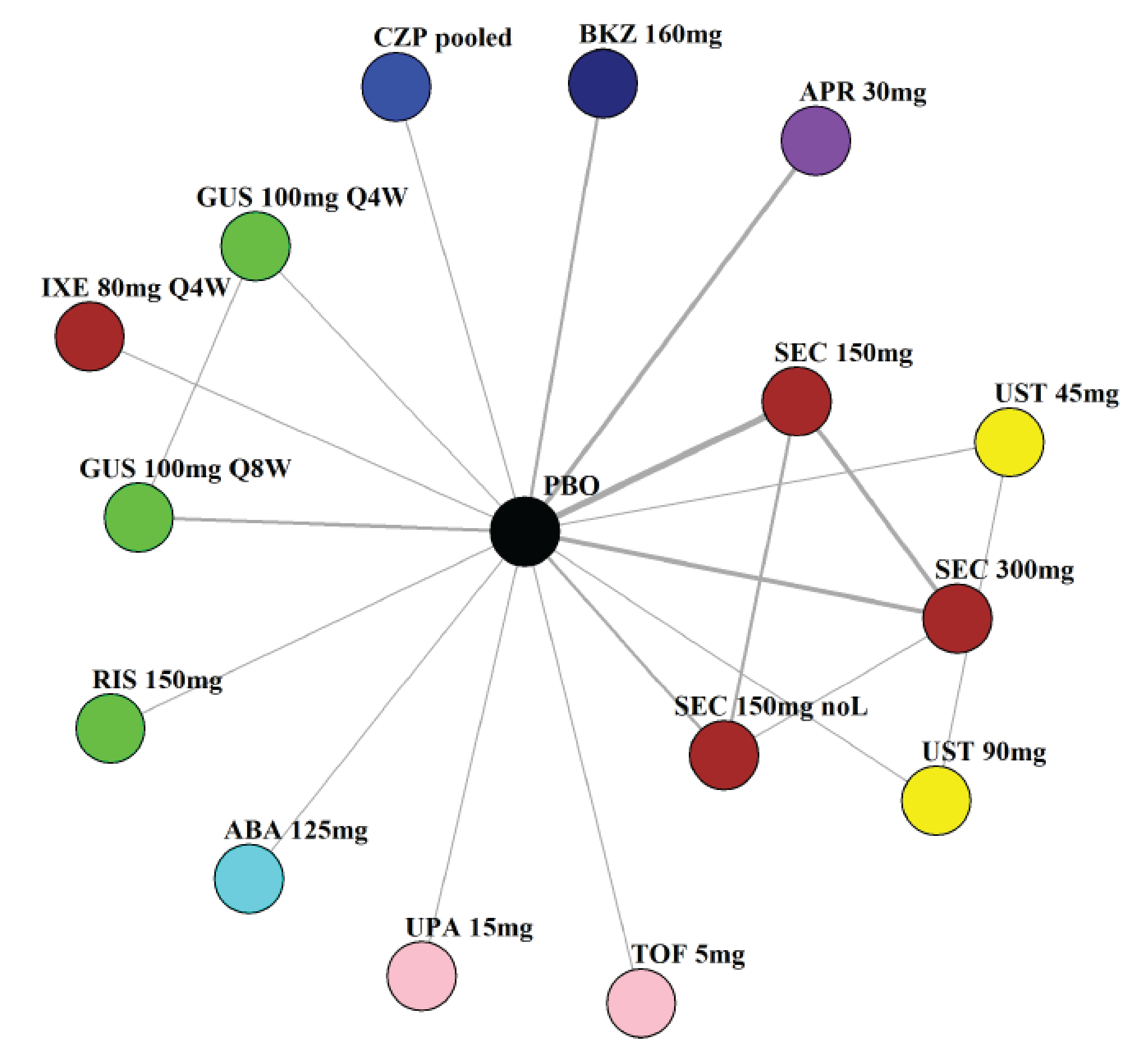
ABA = abatacept; APR = apremilast; BKZ = bimekizumab; CZP = certolizumab pegol; GUS = guselkumab; IXE = ixekizumab; noL = no loading; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Source: Sponsor’s network meta-analysis technical document.30
Figure 9: NMA Network for ACR50 — TNFi-Experienced Patients
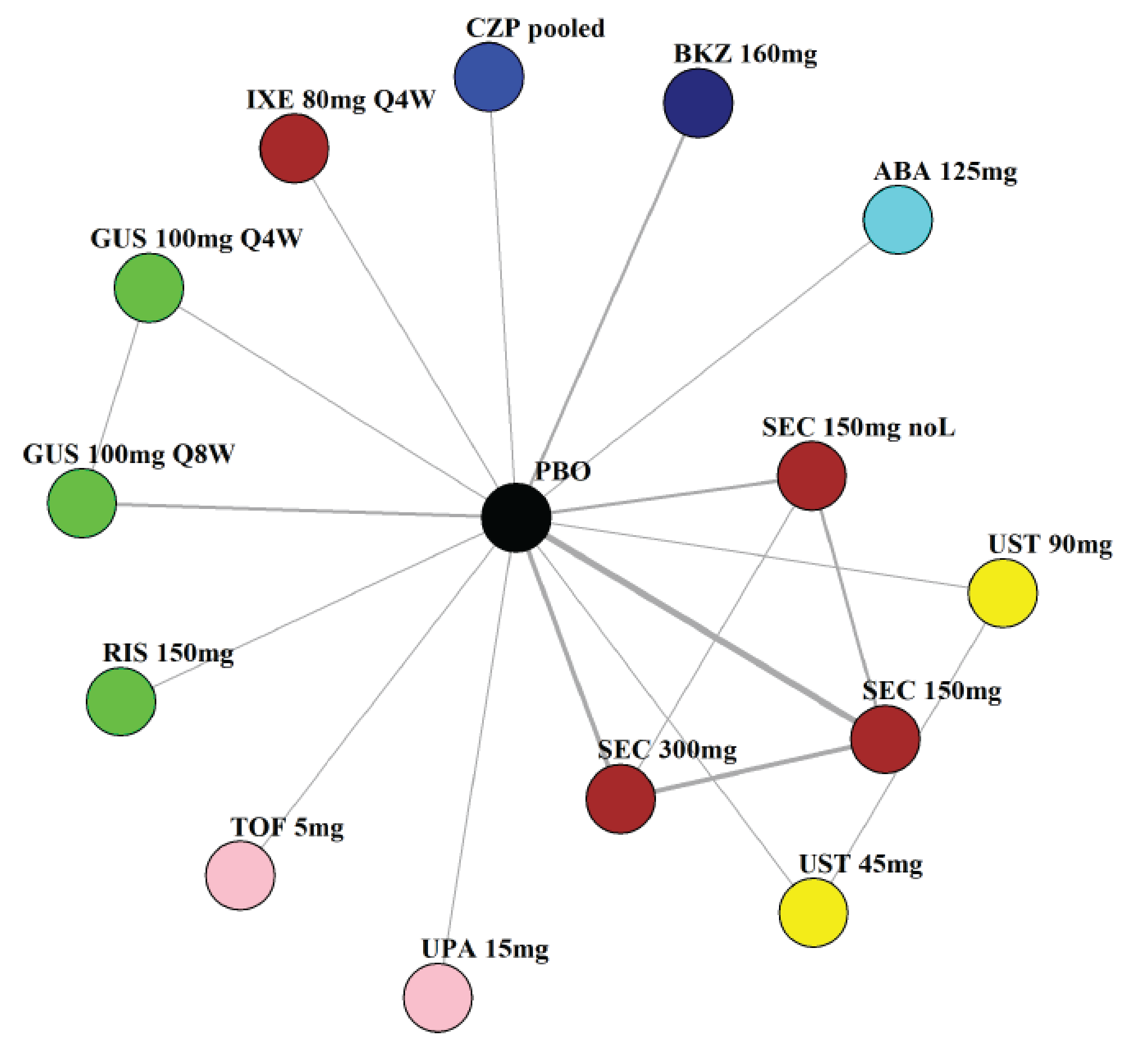
ABA = abatacept; BKZ = bimekizumab; CZP = certolizumab pegol; GUS = guselkumab; IXE = ixekizumab; noL = no loading; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Source: Sponsor’s network meta-analysis technical document.30
Figure 10: NMA Network for ACR70 — TNFi-Experienced Patients
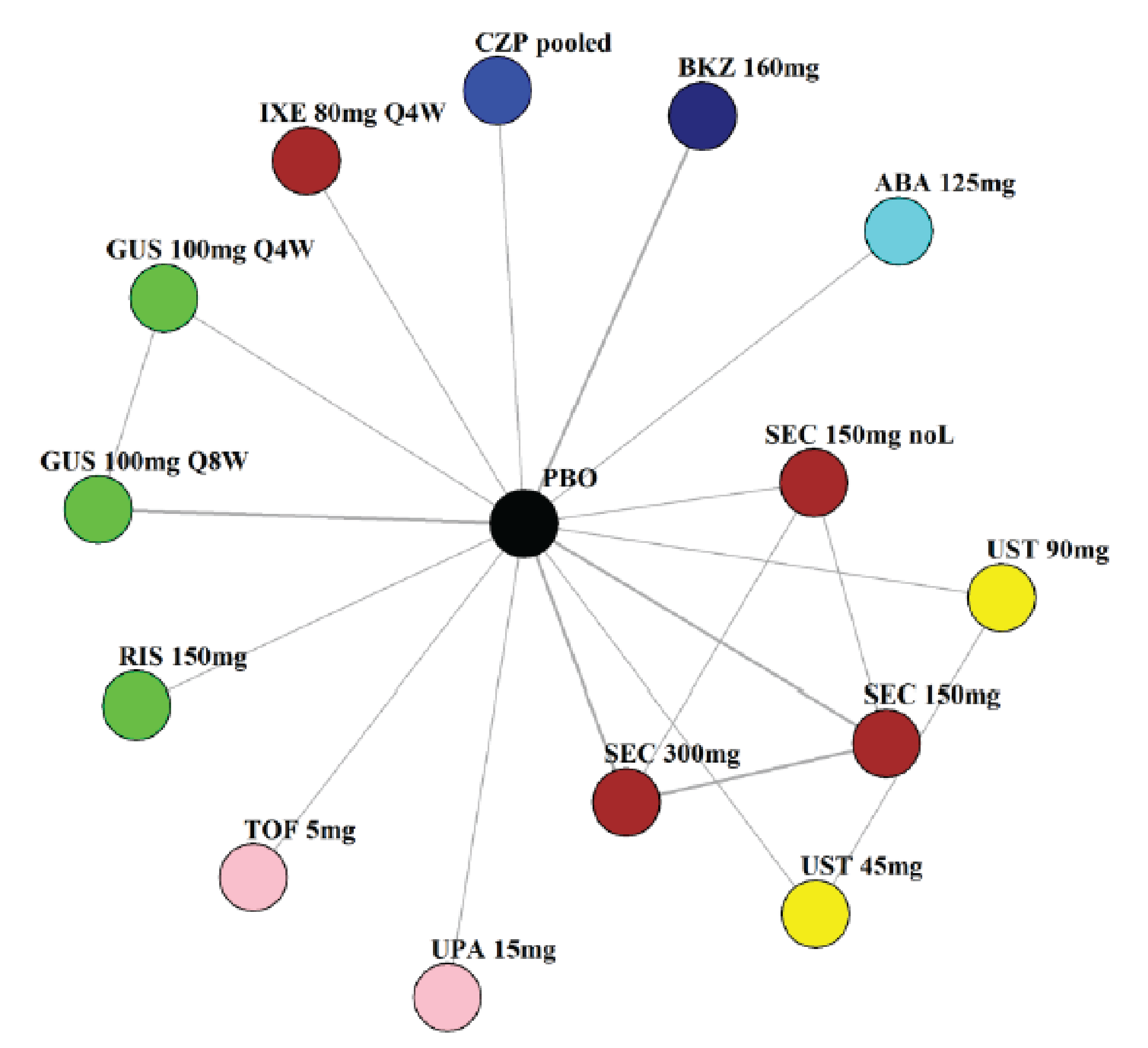
ABA = abatacept; BKZ = bimekizumab; CZP = certolizumab pegol; GUS = guselkumab; IXE = ixekizumab; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Source: Sponsor’s network meta-analysis technical document.30
Figure 11: NMA Network for PASI 90 — TNFi-Experienced Patients
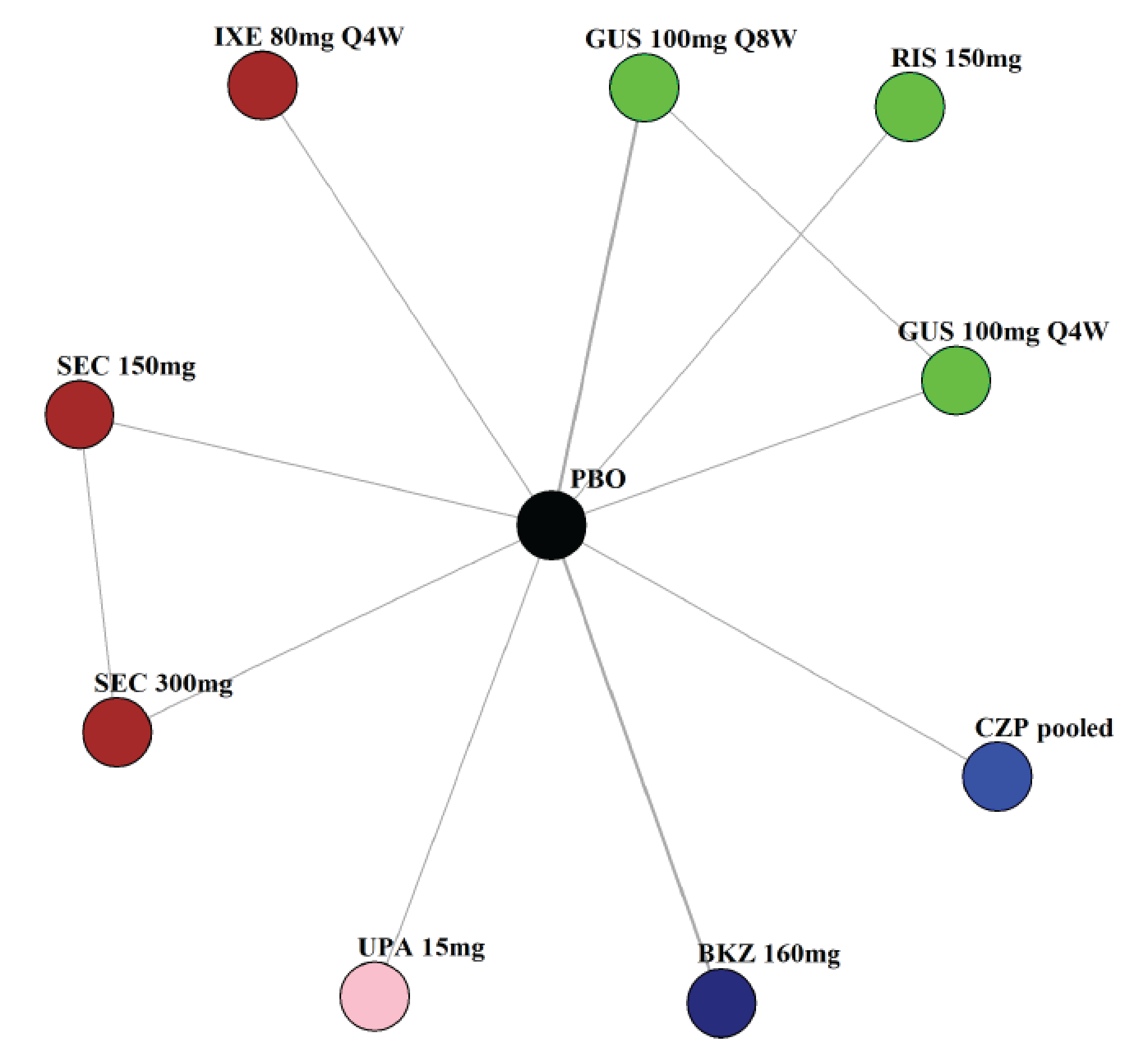
BKZ = bimekizumab; CZP = certolizumab pegol; GUS = guselkumab; IXE = ixekizumab; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; UPA = upadacitinib.
Source: Sponsor’s network meta-analysis technical document.30
Figure 12: NMA Network for MDA — TNFi-Experienced Patients
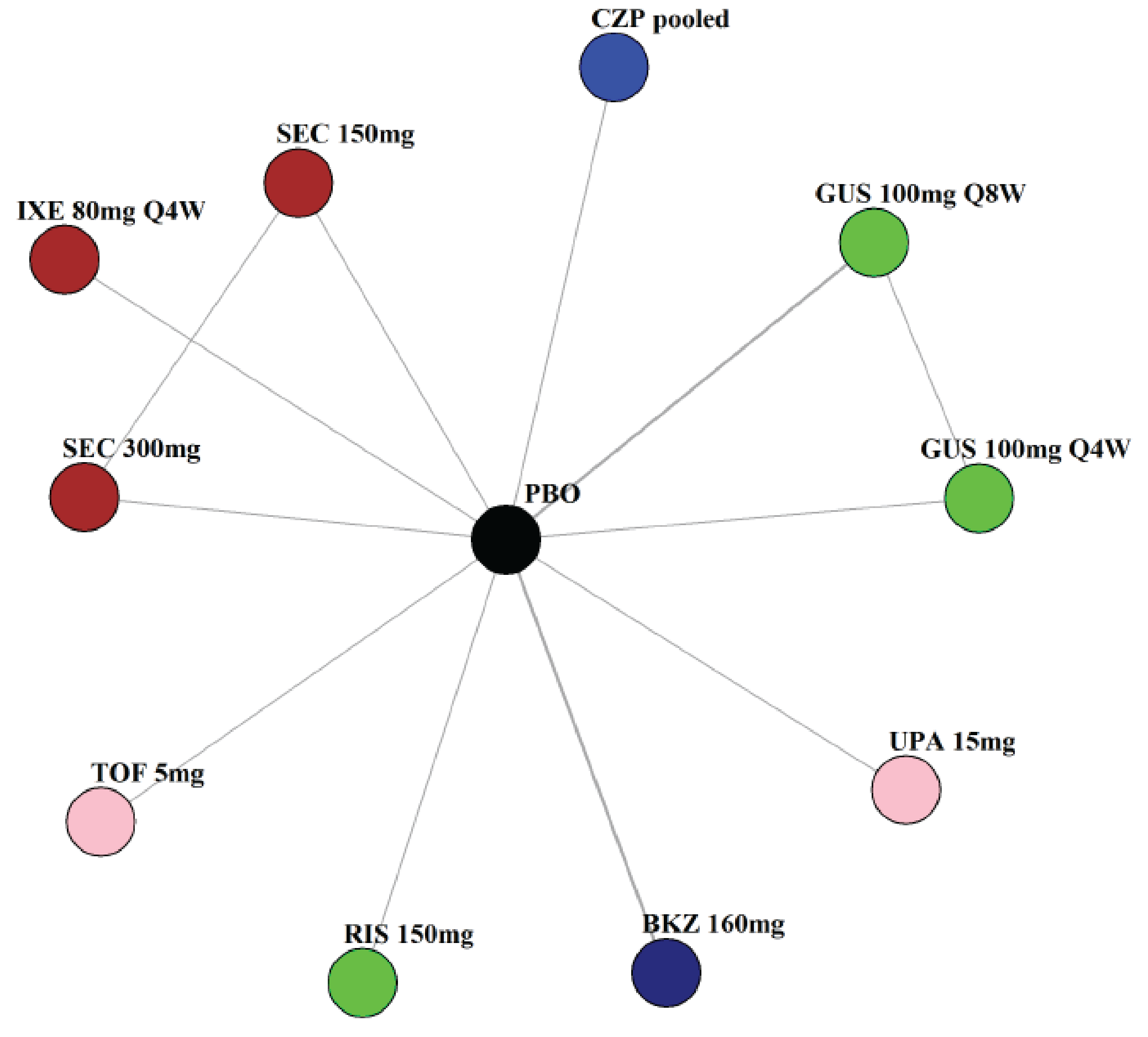
BKZ = bimekizumab; CZP = certolizumab pegol; GUS = guselkumab; IXE = ixekizumab; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib.
Source: Sponsor’s network meta-analysis technical document.30
MAIC Results of Population Matching: bDMARD-Naive Patient Comparisons
Table 52: Bimekizumab vs. Secukinumab Comparison (FUTURE 2 Study)
Variable | FUTURE 2 studya | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)b |
|---|---|---|---|
Age (SD), years | 47.1 (11.7) | 48.5 (12.6) | 47.1 (11.7) |
Males, % | 51 | 47 | 51 |
Race, % white | 94 | 95 | 96 |
Weight (SD), kg | 85.8 (19.1) | NA | 96.5 (21.4) |
BMI (SD), kg/m2 | 29.7 (5.9) | 29.2 (6.8) | 29.9 (7.9) |
Time since diagnosis (SD), years | NA | 6.0 (7.3) | 5.6 (6.8) |
MTX use, % | 49 | 59 | 49 |
cDMARD use, % | NA | 70 | 63 |
SJC (SD) | 10.8 (8.6) | 9.0 (6.2) | 9.4 (6.2) |
TJC (SD) | 20.3 (14.7) | 6.8 (11.8) | 17.7 (11.7) |
Pain VAS score (SD) | NA | 53.6 (24.3) | 61.8 (21.0) |
HAQ-DI score (SD) | 1.20 (0.6) | 0.82 (0.59) | 1.2 (0.6) |
Patients with BSA ≥ 3% affected by psoriasis, % | 51 | 50 | 51 |
Patients with dactylitis, % | 40 | 13 | 14 |
Patients with enthesitis, % | 61 | 26 | 37 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aFor the FUTURE 2 trial, patient baseline characteristics were assumed by the sponsor to be similar among bDMARD-naive patients and TNFi-experienced patients randomized to either secukinumab 150 mg or secukinumab 300 mg.
bSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 53: Bimekizumab vs. Secukinumab 150 mg Comparison (FUTURE 5 Study)
Variable | FUTURE 5 study | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 47.1 (11.7) | 48.5 (12.6) | 47.4 (13.2) |
Males, % | 53 | 47 | 50 |
Race, % white | 83 | 95 | 96 |
Weight (SD), kg | NA | NA | 87.0 (21.3) |
BMI (SD), kg/m2 | NA | 29.2 (6.8) | 30.1 (7.9) |
Time since diagnosis (SD), years | 5.8 | 6.0 (7.3) | 5.9 (7.4) |
MTX use, % | 54 | 59 | 56 |
cDMARD use, % | NA | 70 | 68 |
SJC (SD) | NA | 9.0 (6.2) | 9.4 (6.0) |
TJC (SD) | NA | 6.8 (11.8) | 19.2 (13.0) |
Pain VAS score (SD) | 53.4 | 53.6 (24.3) | 61.8 (21.1) |
HAQ-DI score (SD) | 1.20 | 0.82 (0.59) | 1.2 (0.6) |
Patients with BSA ≥ 3% affected by psoriasis, % | 59 | 50 | 61 |
Patients with dactylitis, % | NA | 13 | 12 |
Patients with enthesitis, % | NA | 26 | 36 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 54: Bimekizumab vs. Secukinumab 300 mg Comparison (FUTURE 5 Study)
Variable | FUTURE 5 study | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 48.3 (12.1) | 48.5 (12.6) | 48.3 (12.1) |
Males, % | 53 | 47 | 53 |
Race, % white | 87 | 95 | 96 |
Weight (SD), kg | NA | NA | 86.8 (21.2) |
BMI (SD), kg/m2 | NA | 29.2 (6.8) | 29.8 (7.5) |
Time since diagnosis (SD), years | 5.2 | 6.0 (7.3) | 5.7 (7.1) |
MTX use, % | 52 | 59 | 52 |
cDMARD use, % | NA | 70 | 65 |
SJC (SD) | NA | 9.0 (6.2) | 9.3 (6.2) |
TJC (SD) | NA | 6.8 (11.8) | 18.7 (12.8) |
Pain VAS score (SD) | 50.6 | 53.6 (24.3) | 60.0 (21.9) |
HAQ-DI score (SD) | 1.10 | 0.82 (0.59) | 1.1 (0.6) |
Patients with BSA ≥ 3% affected by psoriasis, % | 49 | 50 | 49 |
Patients with dactylitis, % | NA | 13 | 13 |
Patients with enthesitis, % | NA | 26 | 37 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 55: Bimekizumab vs. Secukinumab 150 mg Comparison (FUTURE 2 to FUTURE 5 Studies)
Variable | FUTURE 2 to FUTURE 5 studies | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 47.9 (12.2) | 48.5 (12.6) | 47.9 (12.2) |
Males, % | 49 | 47 | 49 |
Race, % white | 90 | 95 | 96 |
Weight (SD), kg | NA | NA | 86.5 (21.3) |
BMI (SD), kg/m2 | 29.2 (6.2) | 29.2 (6.8) | 29.9 (7.6) |
Time since diagnosis (SD), years | NA | 6.0 (7.3) | 5.9 (7.2) |
MTX use, % | 51 | 59 | 51 |
cDMARD use, % | NA | 70 | 64 |
SJC (SD) | 9.7 (7.7) | 9.0 (6.2) | 9.7 (6.6) |
TJC (SD) | 18.1 (13.4) | 6.8 (11.8) | 18.1 (12.1) |
Pain VAS score (SD) | 53.2 (23.0) | 53.6 (24.3) | 60.3 (21.8) |
HAQ-DI score (SD) | 1.12 (0.63) | 0.82 (0.59) | 1.1 (0.6) |
Patients with BSA ≥ 3% affected by psoriasis, % | 55 | 50 | 55 |
Patients with dactylitis, % | 37 | 13 | 14 |
Patients with enthesitis, % | 59 | 26 | 36 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 56: Bimekizumab vs. Secukinumab 300 mg Comparison (FUTURE 2 to FUTURE 5 Studies)
Variable | FUTURE 2 to FUTURE 5 studies | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 48.2 (12.4) | 48.5 (12.6) | 48.2 (12.4) |
Males, % | 53 | 47 | 53 |
Race, % white | 92 | 95 | 96 |
Weight (SD), kg | NA | NA | 86.3 (21.0) |
BMI (SD), kg/m2 | 28.7 (5.7) | 29.2 (6.8) | 29.8 (7.6) |
Time since diagnosis (SD), years | NA | 6.0 (7.3) | 5.6 (6.8) |
MTX use, % | 51 | 59 | 51 |
cDMARD use, % | NA | 70 | 64 |
SJC (SD) | 8.5 (5.7) | 9.0 (6.2) | 8.5 (5.3) |
TJC (SD) | 16.5 (11.6) | 6.8 (11.8) | 16.5 (11.1) |
Pain VAS score (SD) | 52.2 (23.2) | 53.6 (24.3) | 59.8 (21.9) |
HAQ-DI score (SD) | 1.12 (0.62) | 0.82 (0.59) | 1.1 (0.6) |
Patients with BSA ≥ 3% affected by psoriasis, % | 47 | 50 | 47 |
Patients with dactylitis, % | 37 | 13 | 13 |
Patients with enthesitis, % | 62 | 26 | 35 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 57: Bimekizumab vs. Ixekizumab Comparison (SPIRIT-P1 Study)
Variable | SPIRIT-P1 study | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 49.1 (10.1) | 48.5 (12.6) | 49.1 (12.1) |
Males, % | 45 | 47 | 42 |
Race, % white | 95 | 95 | 96 |
Weight (SD), kg | 85.5 (23.0) | NA | 87.0 (21.4) |
BMI (SD), kg/m2 | 30.2 (8.4) | 29.2 (6.8) | 30.2 (7.4) |
Time since diagnosis (SD), years | 6.2 (6.4) | 6.0 (7.3) | 6.2 (7.7) |
MTX use, % | 53 | 59 | 53 |
cDMARD use, % | 68 | 70 | 66 |
SJC (SD) | 11.4 (8.2) | 9.0 (6.2) | 11.4 (7.7) |
TJC (SD) | 20.5 (13.7) | 6.8 (11.8) | 20.5 (13.6) |
Pain VAS score (SD) | 60.1 (19.4) | 53.6 (24.3) | 62.8 (20.5) |
HAQ-DI score (SD) | 1.20 (0.54) | 0.82 (0.59) | 1.20 (0.6) |
Patients with BSA ≥ 3% affected by psoriasis, % | 73 | 50 | 73 |
Patients with dactylitis, % | 51 | 13 | 13 |
Patients with enthesitis, % | 65 | 26 | 37 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 58: Bimekizumab vs. Guselkumab q.4.w. Comparison (DISCOVER-2 Study)
Variable | DISCOVER-2 study | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 45.9 (11.5) | 48.5 (12.6) | 45.9 (11.5) |
Males, % | 58 | 47 | 58 |
Race, % white | 99 | 95 | 96 |
Weight (SD), kg | 85.8 (19.5) | NA | 89/5 (21.4) |
BMI (SD), kg/m2 | NA | 29.2 (6.8) | 30.3 (7.7) |
Time since diagnosis (SD), years | 5.5 (5.9) | 6.0 (7.3) | 5.5 (7.0) |
MTX use, % | 60 | 59 | 60 |
cDMARD use, % | 69 | 70 | 71 |
SJC (SD) | 12.9 (7.8) | 9.0 (6.2) | 12.9 (9.1) |
TJC (SD) | 22.4 (13.5) | 6.8 (11.8) | 22.4 (14.2) |
Pain VAS score (SD) | 62.0 (20.0) | 53.6 (24.3) | 64.1 (19.4) |
HAQ-DI score (SD) | 1.20 (0.60) | 0.82 (0.59) | 1.20 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 75 | 50 | 75 |
Patients with dactylitis, % | 49 | 13 | 14 |
Patients with enthesitis, % | 69 | 26 | 37 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; q.4.w. = every 4 weeks; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 59: Bimekizumab vs. Guselkumab q.8.w. Comparison (DISCOVER-2 Study)
Variable | DISCOVER-2 study | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 44.9 (11.9) | 48.5 (12.6) | 44.9 (11.9) |
Males, % | 52 | 47 | 52 |
Race, % white | 97 | 95 | 97 |
Weight (SD), kg | 83.0 (19.3) | NA | 88.0 (21.9) |
BMI (SD), kg/m2 | NA | 29.2 (6.8) | 30.3 (8.4) |
Time since diagnosis (SD), years | 5.1 (5.2) | 6.0 (7.3) | 5.1 (6.3) |
MTX use, % | 57 | 59 | 57 |
cDMARD use, % | 69 | 70 | 69 |
SJC (SD) | 11.7 (6.8) | 9.0 (6.2) | 11.7 (8.2) |
TJC (SD) | 19.8 (11.9) | 6.8 (11.8) | 19.8 (12.5) |
Pain VAS score (SD) | 63.0 (20.0) | 53.6 (24.3) | 65.0 (18.7) |
HAQ-DI score (SD) | 1.30 (0.60) | 0.82 (0.59) | 1.30 (0.50) |
Patients with BSA ≥ 3% affected by psoriasis, % | 71 | 50 | 71 |
Patients with dactylitis, % | 45 | 13 | 14 |
Patients with enthesitis, % | 64 | 26 | 34 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; q.8.w. = every 8 weeks; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 60: Bimekizumab vs. Risankizumab Comparison (KEEPsAKE 1 Study)
Variable | KEEPsAKE 1 study | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 52 | 48.5 (12.6) | 51.3 (12.2) |
Males, % | 52 | 47 | 52 |
Race, % white | 94 | 95 | 96 |
Weight (SD), kg | NA | NA | 87.5 (20.6) |
BMI (SD), kg/m2 | 30.7 (6.4) | 29.2 (6.8) | 30.1 (6.9) |
Time since diagnosis (SD), years | 7.1 (7.0) | 6.0 (7.3) | 7.1 (9.1) |
MTX use, % | 65 | 59 | 65 |
cDMARD use, % | 11 | 70 | 74 |
SJC (SD) | 12.1 (7.8) | 9.0 (6.2) | 12.1 (8.7) |
TJC (SD) | 20.8 (14.1) | 6.8 (11.8) | 20.8 (13.4) |
Pain VAS score (SD) | 57.1 (22.6) | 53.6 (24.3) | 61.6 (20.5) |
HAQ-DI score (SD) | 1.15 (0.66) | 0.82 (0.59) | 1.20 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 57 | 50 | 56 |
Patients with dactylitis, % | 31 | 13 | 15 |
Patients with enthesitis, % | 62 | 26 | 39 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 61: Bimekizumab vs. Ustekinumab 45 mg Comparison (PSUMMIT 1 Study)
Variable | PSUMMIT 1 study | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 48 | 48.5 (12.6) | 48.0 (12.5) |
Males, % | 52 | 47 | 52 |
Race, % white | NA | 95 | 96 |
Weight (SD), kg | NA | NA | 88.6 (23.2) |
BMI (SD), kg/m2 | 29.4 | 29.2 (6.8) | 30.3 (7.8) |
Time since diagnosis (SD), years | 3.4 | 6.0 (7.3) | 3.4 (4.1) |
MTX use, % | 49 | 59 | 48 |
cDMARD use, % | NA | 70 | 62 |
SJC (SD) | 10 | 9.0 (6.2) | 10.0 (6.6) |
TJC (SD) | 18 | 6.8 (11.8) | 18.0 (11.7) |
Pain VAS score (SD) | NA | 53.6 (24.3) | 64.1 (19.2) |
HAQ-DI score (SD) | 1.30 | 0.82 (0.59) | 1.30 (0.50) |
Patients with BSA ≥ 3% affected by psoriasis, % | 70.7 | 50 | 71 |
Patients with dactylitis, % | 49.3 | 13 | 13 |
Patients with enthesitis, % | 69.3 | 26 | 34 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 62: Bimekizumab vs. Ustekinumab 90 mg Comparison (PSUMMIT 1 Study)
Variable | PSUMMIT 1 study | BE OPTIMAL study (unmatched) | BE OPTIMAL study (matched)a |
|---|---|---|---|
Age (SD), years | 47 | 48.5 (12.6) | 47.0 (12.4) |
Males, % | 57 | 47 | 57 |
Race, % white | NA | 95 | 97 |
Weight (SD), kg | NA | NA | 89.7 (22.4) |
BMI (SD), kg/m2 | 30.0 | 29.2 (6.8) | 30.7 (8.4) |
Time since diagnosis (SD), years | 4.9 | 6.0 (7.3) | 4.9 (6.1) |
MTX use, % | 50 | 59 | 50 |
cDMARD use, % | NA | 70 | 64 |
SJC (SD) | 10 | 9.0 (6.2) | 10.0 (6.3) |
TJC (SD) | 20 | 6.8 (11.8) | 20.0 (13.1) |
Pain VAS score (SD) | NA | 53.6 (24.3) | 64.2 (19.5) |
HAQ-DI score (SD) | 1.30 | 0.82 (0.59) | 1.30 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 73 | 50 | 73 |
Patients with dactylitis, % | 48.5 | 13 | 13 |
Patients with enthesitis, % | 75.5 | 26 | 34 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
MAIC Results of Population Matching: TNFi-Experienced Patient Comparisons
Table 63: Bimekizumab vs. Secukinumab Comparison (FUTURE 2 Study)
Variable | FUTURE 2 studya | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)b |
|---|---|---|---|
Age (SD), years | 47.7 (12.3) | 50.1 (12.4) | 47.7 (12.3) |
Males, % | 52 | 49 | 52 |
Race, % white | 89 | 96 | 98 |
Weight (SD), kg | 90.4 (20.6) | NA | 90.2 (21.5) |
BMI (SD), kg/m2 | 31.3 (6.7) | 30.1 (6.5) | 3.9 (7.0) |
Time since diagnosis (SD), years | NA | 9.6 (9.9) | 9.1 (9.0) |
MTX use, % | 40 | 45 | 40 |
cDMARD use, % | NA | 52 | 46 |
SJC (SD) | 12.4 (9.7) | 9.7 (7.5) | 10.8 (8.7) |
TJC (SD) | 25.6 (19.1) | 18.4 (13.5) | 22.3 (15.7) |
Pain VAS score (SD) | NA | 58.3 (24.2) | 65.7 (21.3) |
HAQ-DI score (SD) | 1.30 (0.60) | 0.97 (0.59) | 1.30 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 48 | 66 | 48 |
Patients with dactylitis, % | 32 | 13 | 17 |
Patients with enthesitis, % | 66 | 46 | 44 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aFor the FUTURE 2 trial, patient baseline characteristics were assumed by the sponsor to be similar among bDMARD-naive patients and TNFi-experienced patients randomized to either secukinumab 150 mg or secukinumab 300 mg.
bSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 64: Bimekizumab vs. Secukinumab 150 mg Comparison (FUTURE 5 Study)
Variable | FUTURE 5 study | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)a |
|---|---|---|---|
Age (SD), years | 52.7 (11.8) | 50.1 (12.4) | 50.7 (11.9) |
Males, % | 55 | 49 | 51 |
Race, % white | 77 | 96 | 98 |
Weight (SD), kg | NA | NA | 90.7 (21.8) |
BMI (SD), kg/m2 | NA | 30.1 (6.5) | 31.5 (7.1) |
Time since diagnosis (SD), years | 7.1 | 9.6 (9.9) | 10.3 (10.0) |
MTX use, % | 58 | 45 | 34 |
cDMARD use, % | NA | 52 | 40 |
SJC (SD) | NA | 9.7 (7.5) | 10.6 (7.9) |
TJC (SD) | NA | 18.4 (13.5) | 25.2 (16.9) |
Pain VAS score (SD) | 57.3 | 58.3 (24.2) | 68.4 (20.2) |
HAQ-DI score (SD) | 1.40 | 0.97 (0.59) | 1.50 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 38 | 66 | 46 |
Patients with dactylitis, % | NA | 13 | 16 |
Patients with enthesitis, % | NA | 46 | 46 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 65: Bimekizumab vs. Secukinumab 300 mg Comparison (FUTURE 5 Study)
Variable | FUTURE 5 study | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)a |
|---|---|---|---|
Age (SD), years | 50.3 (14.3) | 50.1 (12.4) | 50.3 (14.3) |
Males, % | 40 | 49 | 40 |
Race, % white | 74 | 96 | 98 |
Weight (SD), kg | NA | NA | 88.7 (21.1) |
BMI (SD), kg/m2 | NA | 30.1 (6.5) | 31.3 (7.0) |
Time since diagnosis (SD), years | 10.1 | 9.6 (9.9) | 10.1 (9.9) |
MTX use, % | 49 | 45 | 49 |
cDMARD use, % | NA | 52 | 54 |
SJC (SD) | NA | 9.7 (7.5) | 11.1 (9.0) |
TJC (SD) | NA | 18.4 (13.5) | 24.0 (15.9) |
Pain VAS score (SD) | 57.9 | 58.3 (24.2) | 66.9 (21.7) |
HAQ-DI score (SD) | 1.50 | 0.97 (0.59) | 1.50 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 51 | 66 | 51 |
Patients with dactylitis, % | NA | 13 | 14 |
Patients with enthesitis, % | NA | 46 | 46 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 66: Bimekizumab vs. Secukinumab 150 mg Comparison (FUTURE 2 to FUTURE 5 Studies)
Variable | FUTURE 2 to FUTURE 5 studies | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)a |
|---|---|---|---|
Age (SD), years | 50.9 (11.7) | 50.1 (12.4) | 50.9 (11.7) |
Males, % | 49 | 49 | 49 |
Race, % white | 86 | 96 | 98 |
Weight (SD), kg | NA | NA | 90.8 (21.6) |
BMI (SD), kg/m2 | 30.9 (6.5) | 30.1 (6.5) | 31.3 (6.9) |
Time since diagnosis (SD), years | NA | 9.6 (9.9) | 10.2 (9.6) |
MTX use, % | 44 | 45 | 44 |
cDMARD use, % | NA | 52 | 49 |
SJC (SD) | 12.2 (10.2) | 9.7 (7.5) | 12.2 (9.7) |
TJC (SD) | 23.5 (6.5) | 18.4 (13.5) | 23.5 (15.6) |
Pain VAS score (SD) | 61.1 (20.7) | 58.3 (24.2) | 67.1 (20.9) |
HAQ-DI score (SD) | 1.43 (0.63) | 0.97 (0.59) | 1.40 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 47 | 66 | 47 |
Patients with dactylitis, % | 35 | 13 | 16 |
Patients with enthesitis, % | 71 | 46 | 47 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 67: Bimekizumab vs. Secukinumab 300 mg Comparison (FUTURE 2 to FUTURE 5 Studies)
Variable | FUTURE 2 to FUTURE 5 studies | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)a |
|---|---|---|---|
Age (SD), years | 49.4 (13.6) | 50.1 (12.4) | 49.4 (13.6) |
Males, % | 41 | 49 | 41 |
Race, % white | 83 | 96 | 98 |
Weight (SD), kg | NA | NA | 88.6 (20.9) |
BMI (SD), kg/m2 | 30.7 (6.3) | 30.1 (6.5) | 31.0 (6.9) |
Time since diagnosis (SD), years | NA | 9.6 (9.9) | 9.5 (9.3) |
MTX use, % | 46 | 45 | 46 |
cDMARD use, % | NA | 52 | 51 |
SJC (SD) | 10.5 (8.3) | 9.7 (7.5) | 10.5 (8.4) |
TJC (SD) | 22.2 (15.0) | 18.4 (13.5) | 22.2 (15.4) |
Pain VAS score (SD) | 59.4 (23.1) | 58.3 (24.2) | 65.0 (21.9) |
HAQ-DI score (SD) | 1.34 (0.65) | 0.97 (0.59) | 1.30 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 46 | 66 | 46 |
Patients with dactylitis, % | 39 | 13 | 15 |
Patients with enthesitis, % | 61 | 46 | 46 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 68: Bimekizumab vs. Ixekizumab Comparison (SPIRIT-P2 Study)
Variable | SPIRIT-P2 study | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)a |
|---|---|---|---|
Age (SD), years | 52.6 (13.6) | 50.1 (12.4) | 52.6 (12.3) |
Males, % | 52 | 49 | 52 |
Race, % white | 91 | 96 | 97 |
Weight (SD), kg | 89.9 (22.0) | NA | 89.9 (20.6) |
BMI (SD), kg/m2 | 30.9 (7.1) | 30.1 (6.5) | 30.9 (6.5) |
Time since diagnosis (SD), years | 11.0 (9.6) | 9.6 (9.9) | 11.0 (10.7) |
MTX use, % | 39 | 45 | 39 |
cDMARD use, % | 49 | 52 | 46 |
SJC (SD) | 13.1 (11.2) | 9.7 (7.5) | 13.1 (10.4) |
TJC (SD) | 22.0 (14.1) | 18.4 (13.5) | 22.0 (15.5) |
Pain VAS score (SD) | 63.9 (21.4) | 58.3 (24.2) | 63.3 (23.1) |
HAQ-DI score (SD) | 1.20 (0.60) | 0.97 (0.59) | 1.20 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 56 | 66 | 61 |
Patients with dactylitis, % | 23 | 13 | 16 |
Patients with enthesitis, % | 56 | 46 | 42 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 69: Bimekizumab vs. Guselkumab Comparison (COSMOS Study)
Variable | COSMOS study | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)a |
|---|---|---|---|
Age (SD), years | 49.0 (12.0) | 50.1 (12.4) | 49.0 (12.0) |
Males, % | 46 | 49 | 46 |
Race, % white | NA | 96 | 98 |
Weight (SD), kg | 84.0 (17.0) | NA | 89.1 (21.6) |
BMI (SD), kg/m2 | 29.0 (6.0) | 30.1 (6.5) | 30.8 (7.0) |
Time since diagnosis (SD), years | 8.3 (7.8) | 9.6 (9.9) | 8.3 (8.1) |
MTX use, % | 56 | 45 | 56 |
cDMARD use, % | NA | 52 | 62 |
SJC (SD) | 10.0 (7.0) | 9.7 (7.5) | 10.0 (7.5) |
TJC (SD) | 21.0 (13.0) | 18.4 (13.5) | 21.0 (14.3) |
Pain VAS score (SD) | 65.0 (19.0) | 58.3 (24.2) | 64.1 (21.9) |
HAQ-DI score (SD) | 1.30 (0.60) | 0.97 (0.59) | 1.30 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 70 | 66 | 70 |
Patients with dactylitis, % | 36 | 13 | 12 |
Patients with enthesitis, % | 67 | 46 | 45 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 70: Bimekizumab vs. Risankizumab Comparison (KEEPsAKE 2 Study)
Variable | KEEPsAKE 2 study | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)a |
|---|---|---|---|
Age (SD), years | 54.2 (12.7) | 50.1 (12.4) | 54.2 (12.7) |
Males, % | 44 | 49 | 44 |
Race, % white | NA | 96 | 96 |
Weight (SD), kg | NA | NA | 88.9 (19.9) |
BMI (SD), kg/m2 | 30.9 (6.7) | 30.1 (6.5) | 31.0 (6.3) |
Time since diagnosis (SD), years | 10.5 (9.2) | 9.6 (9.9) | 10.5 (10.2) |
MTX use, % | 45 | 45 | 43 |
cDMARD use, % | 14 | 52 | 49 |
SJC (SD) | 13.4 (8.9) | 9.7 (7.5) | 13.4 (10.6) |
TJC (SD) | 22.8 (14.9) | 18.4 (13.5) | 22.8 (15.8) |
Pain VAS score (SD) | 56.8 (24.9) | 58.3 (24.2) | 62.2 (23.4) |
HAQ-DI score (SD) | 1.20 (0.60) | 0.97 (0.59) | 1.20 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 55 | 66 | 55 |
Patients with dactylitis, % | 22 | 13 | 14 |
Patients with enthesitis, % | 71 | 46 | 43 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 71: Bimekizumab vs. Ustekinumab 45 mg Comparison (PSUMMIT 2 Study)
Variable | PSUMMIT 2 study | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)a |
|---|---|---|---|
Age (SD), years | 49 | 50.1 (12.4) | 49.0 (14.0) |
Males, % | 38 | 49 | 38 |
Race, % white | NA | 96 | 97 |
Weight (SD), kg | NA | NA | 90.1 (21.4) |
BMI (SD), kg/m2 | 30.2 | 30.1 (6.5) | 31.5 (7.0) |
Time since diagnosis (SD), years | 7.3 | 9.6 (9.9) | 7.3 (6.8) |
MTX use, % | 52.4 | 45 | 48 |
cDMARD use, % | NA | 52 | 54 |
SJC (SD) | 15 | 9.7 (7.5) | 14.5 (11.9) |
TJC (SD) | 24 | 18.4 (13.5) | 24.0 (15.8) |
Pain VAS score (SD) | NA | 58.3 (24.2) | 63.2 (22.9) |
HAQ-DI score (SD) | 1.40 | 0.97 (0.59) | 1.40 (0.60) |
Patients with BSA ≥ 3% affected by psoriasis, % | 77.7 | 66 | 60 |
Patients with dactylitis, % | 46.6 | 13 | 14 |
Patients with enthesitis, % | 69.9 | 46 | 46 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Table 72: Bimekizumab vs. Ustekinumab 90 mg Comparison (PSUMMIT 2 Study)
Variable | PSUMMIT 2 study | BE COMPLETE study (unmatched) | BE COMPLETE study (matched)a |
|---|---|---|---|
Age (SD), years | 48 | 50.1 (12.4) | 48.0 (13.3) |
Males, % | 38 | 49 | 38 |
Race, % white | NA | 96 | 99 |
Weight (SD), kg | NA | NA | 90.5 (22.5) |
BMI (SD), kg/m2 | 30.3 | 30.1 (6.5) | 31.6 (7.4) |
Time since diagnosis (SD), years | 5.7 | 9.6 (9.9) | 5.7 (5.4) |
MTX use, % | 49.5 | 45 | 46 |
cDMARD use, % | NA | 52 | 53 |
SJC (SD) | 13 | 9.7 (7.5) | 12.5 (10.4) |
TJC (SD) | 26 | 18.4 (13.5) | 25.5 (15.4) |
Pain VAS score (SD) | NA | 58.3 (24.2) | 65.7 (21.2) |
HAQ-DI score (SD) | 1.60 | 0.97 (0.59) | 1.60 (0.50) |
Patients with BSA ≥ 3% affected by psoriasis, % | 77.1 | 66 | 69 |
Patients with dactylitis, % | 39 | 13 | 11 |
Patients with enthesitis, % | 72.4 | 46 | 50 |
BMI = body mass index; BSA = body surface area; cDMARD = conventional nonbiologic disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire–Disability Index; MTX = methotrexate; NA = not available; SD = standard deviation; SJC = swollen joint count; SMD = standardized mean difference; TJC = tender joint count; VAS = visual analogue scale; vs. = versus.
Note: Rows in grey were variables included in matching.
aSMDs were not reported in the submission.
Source: Sponsor’s MAIC Technical Document and additional information provided by the sponsor.29
Pharmacoeconomic Review
Abbreviations
ACR
American College of Rheumatology
ACR50
American College of Rheumatology 50% improvement in rheumatoid arthritis
BIA
budget impact analysis
BSC
best supportive care
DMARD
disease-modifying antirheumatic drug
HAQ-DI
Health Assessment Questionnaire–Disability Index
ICER
incremental cost-effectiveness ratio
NIHB
Non-Insured Health Benefits
NMA
network meta-analysis
PASI
Psoriasis Area Severity Index
PASI 75
75% reduction in Psoriasis Area Severity Index score
PASI 90
90% reduction in Psoriasis Area Severity Index score
PsA
psoriatic arthritis
PsO
psoriasis
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Bimekizumab (Bimzelx), prefilled syringe or autoinjector (160 mg/mL) |
Indication | The treatment of adult patients with active psoriatic arthritis, used alone or in combination with a conventional nonbiologic cDMARD |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | February 23, 2024 |
Reimbursement request | As per indication |
Sponsor | UCB Canada Inc. |
Submission history | Yes Indication: For the treatment of moderate-to-severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy Recommendation date: March 30, 2022 Recommendation: Reimburse with clinical criteria and/or conditions |
DMARD = disease-modifying antirheumatic drug; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target populations | Adult patients with active PsA; biologic-naive and biologic-experienced subpopulations explored separately |
Treatment | Bimekizumab |
Dose regimen | 160 mg (given as 1 subcutaneous injection) every 4 weeks |
Submitted price | Bimekizumab: $1,625 per 1 mL of 160 mg bimekizumab syringe or autoinjector |
Submitted treatment cost | $22,042 for a biologic-naive patient or $22,563 for a biologic-experienced patient in year 1, respectively. The annual maintenance cost is $21,198 per patient. |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (50 years) |
Key data source | Comparative clinical efficacy was derived from a sponsor-submitted NMA based on data obtained from the BE OPTIMAL, BE COMPLETE, and respective comparator treatment trials to inform the probability of ACR50 and PASI response at 12 weeks to 16 weeks. |
Submitted results | Biologic-naive
Biologic-experienced
|
Key limitations |
|
CADTH reanalysis results |
|
ACR50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; BSC = best supportive care; DMARD = disease-modifying antirheumatic drug; ICER = incremental cost-effectiveness ratio; LY = life-year; NMA = network meta-analysis; PASI = Psoriasis Area Severity Index; PsA = psoriatic arthritis; QALY = quality-adjusted life-year.
Conclusions
Evidence from the BE OPTIMAL and BE COMPLETE trials in adult patients with psoriatic arthritis (PsA) showed that patients who received bimekizumab were more likely to demonstrate clinically meaningful improvement in composite measures of disease activity, physical function, health-related quality of life, and pain at week 16 versus those on placebo. In the absence of head-to-head data comparing bimekizumab to other targeted disease-modifying antirheumatic drugs (DMARDs) for PsA, the sponsor submitted a network meta-analysis (NMA) that suggested that bimekizumab may be better than some comparators for composite measures of disease activity (i.e., American College of Rheumatology [ACR] Criteria, minimal disease activity, and 90% reduction in Psoriasis Area Severity Index score [PASI 90]), but may not be different from other comparators for these outcomes. Results remain uncertain due to inconsistency in estimates across comparators and inherent uncertainty in the methods used to control for study and patient characteristic heterogeneity.
CADTH was unable to address uncertainty related to comparative clinical data and the long-term efficacy of bimekizumab compared to other advanced DMARDs for the treatment of adult patients with PsA. As such, CADTH was unable to derive a more reliable base-case estimate of the cost-effectiveness of bimekizumab. The sponsor's analyses indicated that bimekizumab is not cost-effective at the submitted price. Results of the sponsor’s base-case sequential analysis suggest that for biologic-naive patients, bimekizumab is not on the cost-effectiveness frontier and is extendedly dominated by adalimumab and infliximab, while it was associated with an incremental cost-effectiveness ratio (ICER) of $69,876 per quality-adjusted life-year (QALY) gained (incremental costs = $49,923; incremental QALYs = 0.71) versus tofacitinib for biologic-experienced patients. A price reduction of approximately 40% is required for bimekizumab to be considered an optimal treatment (i.e., to get on the cost-effectiveness frontier) for biologic-naive patients. To be considered cost-effective at a willingness-to-pay threshold of $50,000, a price reduction of 52% or 24% is required for the biologic-naive and biologic-experienced populations, respectively.
The sponsor’s results were based on efficacy inputs obtained from the sponsor’s submitted NMA; however, CADTH notes that the relative effect obtained from the sponsor’s NMA for effectiveness parameters represents a source of uncertainty. Therefore, while the sponsor’s base case suggests differences in treatment benefit compared to some therapies for the treatment of adult patients with PsA, these results will only be realized should the numerical differences observed in the NMA occur in clinical practice and lead to meaningful improvements for patients. Should these results not be realized, then there is insufficient evidence to suggest that bimekizumab should be priced higher than other biologic DMARD treatments for PsA.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received as a joint submission from the Canadian Psoriasis Network, Arthritis Society Canada, the Canadian Arthritis Patient Alliance, the Canadian Association of Psoriasis Patients, the Canadian Spondyloarthritis Association, and CreakyJoints. Information in the joint submission was primarily gathered through an online Canadian survey conducted from September 27 to October 11, 2023. Of the survey respondents, 100 identified as living with PsA where 52% and 19% of respondents rated the severity of their PsA as “moderate” and “severe,” respectively. Patients noted that joint stiffness and fatigue were the biggest symptoms and that PsA has a large impact on their quality of life (impacting their ability to exercise, sleep, work, and enjoy family life). Patient input on the effectiveness of current treatment options varied, with biologics having the highest proportion of “very effective” or “effective” ratings. Overall, nearly 30% of responses disagreed or strongly disagreed with the statement “my needs are met with the treatment I receive.” One survey participant previously used bimekizumab for PsA and commented that it was easier to use than other therapies and helped them return to daily activities. Additional patient input was received by Arthritis Consumer Experts’ JointHealth, where information from 7 patients with PsA was gathered via email or online survey. Feedback was aligned with input from the joint submission where patients felt that their PsA had many impacts on their day-to-day life and the experience with currently available treatments varied from patient to patient. None of the participants from the JointHealth input had experience with bimekizumab.
Registered clinician input was received from the Canadian Rheumatology Association. It noted that the predominant goal of PsA treatment is to maximize health-related quality of life through the control of symptoms, improved function, and the prevention of structural damage, which is often done through controlling inflammation. Clinician input noted that current therapies have limitations, including only treating certain elements of a patient’s symptoms (e.g., improving arthritis but not skin, or vice versa), the fact that not all patients respond to treatment, the secondary loss of effect with biologics, and side effects. The clinician input commented that due to the limited number of advanced therapies, bimekizumab represents an alternative treatment option for patients with PsA.
CADTH-participating drug plans noted that the comparators used in the trials (i.e., placebo and adalimumab) may not represent the best comparators to assess the efficacy of bimekizumab in PsA. They further raised concerns about adverse events as bimekizumab was associated with higher rates of infection and hypertension compared to placebo and adalimumab. Finally, the drug plans noted the presence of confidential negotiated prices for comparators and commented on the recent listing of similar biologics for the treatment of PsA.
Several of these concerns were addressed in the sponsor’s model:
The impact of PsA on patients’ quality of life was captured via health state utility values.
CADTH was unable to address the following concerns raised from stakeholder input:
Analyses were based on publicly available prices and therefore may not reflect the confidential prices for drugs.
Adverse events were not included due to the structure of the submitted model.
Economic Review
The current review is for bimekizumab (Bimzelx) for the treatment of adult patients with active PsA. Bimekizumab can be used alone or in combination with a nonbiologic cDMARD (e.g., methotrexate).
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of bimekizumab against biologic or targeted synthetic DMARDs and best supportive care (BSC) available in Canada for the treatment of patients with active PsA. The model population aligns with the Health Canada indication.1
Bimekizumab is available as prefilled syringes or autoinjectors, each containing 1 mL of 160 mg bimekizumab.2 The recommended dose is 160 mg (given as 1 subcutaneous injection) every 4 weeks.2 For patients with coexistent moderate-to-severe plaque psoriasis (PsO), the recommended dose is 320 mg every 4 weeks for the first 16 weeks, and every 8 weeks thereafter.2 The submitted price for bimekizumab is $1,625 per syringe or autoinjector.1 The annual cost of bimekizumab calculated by the sponsor was $22,042 or $22,563 for the biologic-naive and biologic-experienced populations in year 1, respectively. The annual maintenance cost was $21,198 in both groups. The comparators for this analysis included adalimumab, apremilast, certolizumab pegol, etanercept, golimumab, guselkumab, infliximab, ixekizumab, risankizumab, secukinumab, tofacitinib, upadacitinib, ustekinumab, and BSC. BSC was defined as a mix of nonbiologic cDMARDs and supportive and/or palliative care.
Outcomes of the model included QALYs and life-years over a lifetime horizon of 50 years. Discounting (1.5% per annum) was applied for both costs and outcomes. Different cycle lengths were used in the model for the initial response period versus the long-term follow-up period with a half-cycle correction applied.1
Model Structure
The sponsor submitted a Markov model to track a cohort of patients with PsA. The model consisted of 2 time periods (initial and maintenance) where patients entered at induction with the first treatment in sequence in either the no PsO health state (Psoriasis Area Severity Index [PASI] < 2.5), the mild PsO health state (PASI 2.5 to 10), or the moderate-to-severe PsO health state (PASI > 10).1 An American College of Rheumatology 50% improvement in rheumatoid arthritis (ACR50) response was measured at the end of the induction period, where responders remained on treatment and transitioned into the long-term follow-up period (i.e., maintenance phase) and nonresponders progressed to a subsequent line of therapy.1 Patients could transition to death at any point.1
Patients who experienced a treatment response in the initial period were assumed to maintain response in the maintenance period until death or treatment discontinuation (due to loss of response or tolerability).1 Patients who were nonresponders in the initial period transitioned to subsequent treatment. While the model allowed for up to 3 lines of active therapy before BSC, in the sponsor’s base-case analysis, patients were assumed to move directly to BSC after first-line therapy. It was assumed that treatment discontinuation does not occur during the initial response period.1 Refer to Figure 1 for an overview of the model structure.
Model Inputs
Data from the BE OPTIMAL and BE COMPLETE trials were used to inform the demographic characteristics of the biologic-naive and biologic-experienced populations, respectively (for the naive population, the mean age was 49 years and the proportion of females was 53.2%; for the experienced population, the mean age was 51 years and the proportion of females was 52.5%).
Efficacy in the model was informed by ACR50 and PASI responses. The comparative efficacy estimates in the model were obtained from the sponsor-conducted NMA (separate NMAs were done for biologic-naive and biologic-experienced patients).1 Analyses were conducted based on data obtained from the BE OPTIMAL, BE COMPLETE, and respective comparator treatment trials at 16 weeks, when available, and were modelled as having an estimated probability of attaining ACR50 and PASI responses. For PASI, it was assumed that 75% reduction in Psoriasis Area Severity Index score [PASI 75] response rates were dependent on ACR50 response; therefore, PASI 75 and ACR50 responses were correlated using a formula from Rodgers et al. (2011).3 Health Assessment Questionnaire–Disability Index (HAQ-DI) scores were also included in the analysis, modelled in relation to treatment response. Efficacy inputs were informed by data available up to week 52 in the BE OPTIMAL trial and to week 16 in the BE COMPLETE trial and were assumed to be equal for all active comparators. In the base-case analysis, HAQ-DI scores were assumed to remain stable over time beyond the initial treatment period.1
Adverse events were not considered in the model.
Mortality was based on the general population mortality obtained from life tables available from Statistics Canada and was adjusted to reflect an increased risk of mortality for patients with PsA using a standardized mortality rate of 1.34.4,5 The probability of death was assumed to be the same across health states and interventions.1
Health state utility estimates in the model were calculated as a function of HAQ-DI scores, PASI scores, and age.1 The algorithm was derived by mapping HAQ-DI and PASI scores to utility values based on EQ-5D-3L data from the BE OPTIMAL and BE COMPLETE trials with a UK value set.6
The economic model included drug acquisition costs, treatment administration costs, and disease management costs.1 Treatment costs were estimated using dosing schedules based on each treatment’s respective Health Canada product monograph. Drug unit costs were obtained from the Ontario Drug Benefit Formulary with the exception of guselkumab, whose cost was obtained from the guselkumab CADTH report.7,8 For adalimumab, apremilast, etanercept, and infliximab, it was assumed that all patients received biosimilars.1 Treatment administration costs were only incurred for IV therapies.1 Lastly, disease management costs were assumed to capture all monitoring and health care resource use and were based on absolute HAQ-DI scores, PsO severity, and the proportion of patients attaining a PASI 75 response.1
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (3,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented as follows.
Base-Case Results
In the sponsor’s base-case analysis for biologic-naive patients, bimekizumab was associated with an estimated cost of $174,766 and 10.60 QALYs over a lifetime horizon (Table 3). Based on the sponsor’s reported sequential analysis, bimekizumab was dominated by upadacitinib (more costly and less effective). This means that at the submitted price, bimekizumab would not be chosen as the optimal treatment strategy, regardless of a decision-maker’s willingness-to-pay threshold. In reference to treatments on the efficiency frontier, bimekizumab is extendedly dominated by adalimumab and infliximab. The ICER for bimekizumab compared to BSC was $59,130 per QALY gained (incremental costs = $45,199; incremental QALYs = 0.76).
Table 3: Summary of the Sponsor’s Economic Evaluation Results — Biologic-Naive Patients
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | $129,567 | 9.84 | Reference |
Tofacitinib | $136,807 | 10.31 | $15,440 |
Adalimumab | $153,291 | 10.56 | $65,793 |
Infliximab | $176,226 | 10.76 | $114,065 |
Dominated treatments | |||
Bimekizumab | $174,766 | 10.60 | Extended dominance by adalimumab and infliximab |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only treatments that are on the efficiency frontier as well as the drug under review are reported in the main body of this report. Full results are available in Appendix 3. The sponsor reported that bimekizumab was dominated by upadacitinib; however, as upadacitinib is not on the efficiency frontier, it is not reported in this table.
Source: Sponsor’s pharmacoeconomic submission.1
In the sponsor’s base-case analysis for biologic-experienced patients, bimekizumab was associated with an estimated cost of $189,106 and 9.35 QALYs over a lifetime horizon (Table 4). Approximately 93% of these QALYs were accrued after the trial assessment point (16 weeks) based on extrapolated long-term data. In the sequential analysis, 3 treatments were on the cost-effectiveness frontier, including BSC, tofacitinib, and bimekizumab. Bimekizumab had an ICER of $69,876 per QALY gained (incremental costs = $49,923; incremental QALYs = 0.71) versus tofacitinib. At a willingness-to-pay threshold of $50,000 per QALY, bimekizumab had a 4.3% chance of being cost-effective.
Table 4: Summary of the Sponsor’s Economic Evaluation Results — Biologic-Experienced Patients
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | $131,829 | 8.23 | Reference |
Tofacitinib | $139,183 | 8.64 | $17,841 vs. BSC |
Bimekizumab | $189,106 | 9.35 | $69,876 vs. tofacitinib |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: Only treatments that are on the efficiency frontier as well as the drug under review are reported in the main body of this report. Full results are available in Appendix 3.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses. They included alternative responder definitions, alternative algorithms for utility values and HAQ-DI, second-line treatment, apremilast as a comparator, HAQ-DI scores over time, and different discounting rates (i.e., 0% and 3%). For biologic-naive patients, results of the scenario analyses were similar to base-case analyses where bimekizumab was not on the cost-effectiveness plane. In biologic-experienced patients, the model was most sensitive to assumptions related to the responder definition where the ICER for bimekizumab versus tofacitinib was $91,848.
The sponsor conducted scenario analyses from a societal perspective; this analysis included additional costs associated with loss of productivity. In the analysis examining biologic-naive patients, bimekizumab was dominated by upadacitinib and infliximab. This was similar to the sponsor’s base-case analysis on biologic-naive patients using a health care payer perspective as bimekizumab was not on the cost-effectiveness plane. In the scenario examining biologic-experienced patients, the ICER was $53,625 per QALY gained relative to tofacitinib. This was similar to the sponsor’s base-case analysis using a health care payer perspective.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Relative treatment effects are uncertain. Relative treatment effects in the model are derived from the sponsor-conducted NMAs comparing bimekizumab to other biologic DMARDs for the treatment of adult patients with PsA. Outcomes included ACR response and PASI improvement. As noted in the CADTH clinical review, in the biologic-naive population, the output of the NMA suggests that bimekizumab was favoured over an interleukin 12 and interleukin 23 inhibitor or an interleukin 23 inhibitor for most ACR outcomes. For PASI 90, the NMA suggested that for PsO symptoms in the biologic-naive population, bimekizumab was more favourable compared to tumour necrosis factor–alpha inhibitors (excluding infliximab 5 mg IV), secukinumab 150 mg, risankizumab 150 mg, and upadacitinib 15 mg. Similar trends were observed in the biologic-experienced population where bimekizumab had favourable results over some comparators for ACR50 (secukinumab, tofacitinib, and abatacept) and PASI 90 (favoured over upadacitinib). However, most comparisons between bimekizumab and other active comparators across outcomes were not conclusively favourable or unfavourable for bimekizumab. The NMAs were associated with several limitations, including inherent uncertainty in the methods used to control for patient heterogeneity across studies, differences in model choice between the 2 patient populations, and uncertainty in the methods used to control for heterogeneity in study design (e.g., time of crossover and early escape). Lastly, while clinical expert feedback obtained by CADTH noted that treatment history is an important effect modifier for treatment response and the NMA analyses were done in separate populations on this basis, model fit was not specified a priori.
CADTH was unable to adequately address the uncertainty associated with estimates derived from the sponsor’s NMA.
The long-term treatment efficacy of bimekizumab is uncertain. The sponsor’s model is based on an NMA comparing treatment efficacy over the short-term (12 weeks to 24 weeks). Responders were assumed to continue treatment until discontinuation and receive the full efficacy benefit while on treatment. This implicitly assumed that treatment waning would not occur for modelled outcomes. Longer-term efficacy and the safety of bimekizumab were examined in the open-label extension trials (the BE ACTIVE 2 and BE VITAL studies); however, as noted in the CADTH clinical review, the open-label designs and the absence of control arms may bias the magnitude of efficacy outcomes. Thus, the long-term treatment efficacy of bimekizumab remains uncertain.
CADTH was unable to address this limitation. The impact of this limitation on the cost-effectiveness of bimekizumab is unknown.
Disease management costs are uncertain and may include double-counting. Disease management costs were calculated to capture the impact of both arthritis and PsO severity. Costs were derived based on absolute HAQ-DI scores and PsO severity plus the proportion of patients attaining a PASI 75 response. Therefore, the method assumed that the cost of treating a patient with PsA (excluding biologic therapies) is the same as the cost of treating a patient with arthritis plus the cost of treating a patient with PsO. Clinical expert feedback received by CADTH noted that while it may be reasonable to consider the cost of both rheumatoid arthritis and PsO, patients with severe PsO would be treated as PsO patients. Furthermore, by accounting for arthritis and PsO costs separately, the sponsor’s calculations may result in double-counting as some elements of patient care overlap such as pain control, hospitalization, and community care.
CADTH explored the uncertainty in the disease management costs by reducing the HAQ-DI-based coefficient and difference in costs between mild-to-moderate PsA and moderate-to-severe PsA by half in a scenario analysis.
Table 5: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients who discontinue their initial biologic DMARD transition to BSC and remain on BSC for the remainder of their lives. | Reasonable as a simplifying assumption. In clinical practice, patients who have inadequate response, lose response, or cannot tolerate their current biologic DMARD are likely to switch to alternative biologic DMARDs. However, in the absence of efficacy and safety data to inform treatment sequences, the sponsor used the simplifying assumption that patients transition to BSC after their initial biologic DMARD. |
BSC is defined as a mix of a nonbiologic cDMARD and supportive and/or palliative care (i.e., methotrexate, leflunomide, sulfasalazine, and hydroxychloroquine) as informed by the placebo arms of the BE OPTIMAL and BE COMPLETE trials. | Reasonable. However, clinical expert feedback received by CADTH noted that hydroxychloroquine is not commonly used in clinical practice across Canada. |
Patients who discontinue their biologic DMARD treatment (i.e., nonresponders on BSC) have their HAQ-DI scores revert to baseline and deteriorate at a rate of 0.018 every 3 months. | Uncertain. Clinical expert feedback received by CADTH indicated that a patient’s HAQ-DI score is likely to decrease gradually over 2 months to 3 months. |
PASI 75 response rates vary by treatment response. | Reasonable but uncertain. Clinical expert feedback received by CADTH noted that while there may be a correlation between PASI and ACR, it is considered moderate and dependent on the drug. |
AEs were similar across other biologic and targeted DMARD groups. | Likely reasonable. While drug plan input noted that bimekizumab had higher rates of infection and hypertension vs. placebo and adalimumab from the BE OPTIMAL and BE COMPLETE trials, clinical expert feedback received by CADTH noted that AEs across other biologic and targeted DMARD groups are often similar. |
All active treatments had an annual discontinuation rate of 16.5% in the maintenance phase. Patients on BSC do not discontinue BSC. | Reasonable. |
Responders maintain their improved HAQ-DI score until death or treatment discontinuation (due to loss of response or AEs). | Uncertain. While it is expected that responders would experience an improved HAQ-DI score and maintain their improvement for the short-term while on therapy, clinical expert feedback received by CADTH noted that there is uncertainty in the duration of this improvement over the long-term, particularly for biologic-experienced patients. The impact of this assumption on the results is unknown. |
AE = adverse event; ACR = American College of Rheumatology, BSC = best supportive care, DMARD = disease-modifying antirheumatic drug, HAQ-DI = Health Assessment Questionnaire–Disability Index, PASI = Psoriasis Area Severity Index; PASI 75 = 75% reduction in Psoriasis Area Severity Index score; vs. = versus.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Given the limitations CADTH identified with the sponsor’s economic submission, CADTH was unable derive a more reliable estimate of the cost-effectiveness of bimekizumab treatment of adult patients with active PsA. CADTH notes that the sponsor’s analysis remains uncertain. The estimates of relative effect obtained from the sponsor’s NMAs for effectiveness parameters informing the model represent the largest source of uncertainty in the decision model. The outputs of the sponsor’s base case suggest differences in treatment benefits between advanced therapies for the treatment of adult patients with PsA in a biologic-experienced population. These results will only be realized should the numerical differences observed in the NMA be deemed valid.
When reviewing the sponsor’s base-case results, the probability that bimekizumab is cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained was 0% and 4.3% for the biologic-naive and biologic-experienced populations, respectively.
Scenario Analysis Results
To explore the uncertainty surrounding the disease management costs included in the model, CADTH conducted scenario analyses reducing the HAQ-DI-based coefficient and difference in costs between mild-to-moderate PsA and moderate-to-severe PsA by half. In the biologic-naive population, bimekizumab was associated with an estimated cost of $148,296 and 10.58 QALYs over a lifetime horizon. In the sequential analysis, bimekizumab was extendedly dominated by etanercept and infliximab. In the biologic-experienced population, bimekizumab was associated with an estimated cost of $160,906 and 9.37 QALYs over a lifetime horizon. In the sequential analysis, 3 treatments were on the cost-effectiveness frontier, including BSC, tofacitinib, and bimekizumab. Bimekizumab had an ICER of $71,542 (incremental costs = $51,181; incremental QALYs = 0.72) versus tofacitinib.
Additionally, CADTH undertook price reduction analysis based on the sponsor’s base case to explore the needed price reduction to obtain an ICER for bimekizumab below the $50,000 per QALY threshold. These analyses demonstrated that for bimekizumab to be on the cost-effectiveness frontier in a biologic-naive population, a price reduction of approximately 40% would be required (ICER = approximately $75,000). To be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained, 52% and 24% price reductions are required for bimekizumab for the biologic-naive and biologic-experienced populations, respectively.
Table 6: CADTH Price Reduction Analyses
Analysis | Unit drug cost | Sponsor’s base-case ICERs (cost per QALY) | |
|---|---|---|---|
Price reduction | $ | Biologic-naive population | Biologic-experienced population |
No price reduction | 1,625 | Extendedly dominated by adalimumab and infliximab | $69,769 (vs. tofacitinib) |
10% | 1,463 | Extendedly dominated by adalimumab and infliximab | $61,645 (vs. tofacitinib) |
20% | 1,300 | Extendedly dominated by adalimumab and infliximab | $53,526 (vs. tofacitinib) |
30% | 1,138 | Extendedly dominated by adalimumab and infliximab | $45,408 (vs. tofacitinib) |
40% | 975 | $74,684 (vs. adalimumab) | $37,289 (vs. tofacitinib) |
50% | 813 | $51,813 (vs. tofacitinib) | $29,171 (vs. tofacitinib) |
60% | 650 | $36,481 (vs. tofacitinib) | $21,052 (vs. tofacitinib) |
70% | 488 | $21,149 (vs. tofacitinib) | $14,651 (vs. BSC) |
80% | 325 | $11,720 (vs. BSC) | $9,531 (vs. BSC) |
90% | 163 | $5,794 (vs. BSC) | $4,411 (vs. BSC) |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: Price reduction calculations for the biologic-experienced population were conducted on a new probabilistic run of the sponsor’s biologic-experienced base case. As such, results may differ slightly from the sponsor-submitted biologic-experienced results.
Issues for Consideration
Bimekizumab was previously reviewed by CADTH for the treatment of moderate-to-severe plaque PsO in 2022 and received a positive recommendation.9 Bimekizumab is currently under review by CADTH for the treatment of active ankylosing spondylitis.10
Publicly available list prices may not reflect actual acquisition costs incurred by public plans. The true acquisition costs paid by Canadian public drug plans may be lower than those listed on public formularies. CADTH’s cost-effectiveness and budget impact analysis (BIA) is sensitive to this issue, as drugs included in this review may have negotiated prices with various health care jurisdictions in Canada.
For patients with PsA with coexistent moderate-to-severe PsO, the recommended bimekizumab dose is 320 mg every 4 weeks for the first 16 weeks, and every 8 weeks thereafter (i.e., aligned with the dosing for PsO). Based on the BE OPTIMAL and BE COMPLETE trials, the sponsor estimated that approximately 17% of patients with PsA have coexistent moderate-to-severe PsO.11 Should the proportion in Canadian clinical practice be different, the increased drug cost associated with PsO bimekizumab dosing may impact the budget impact of reimbursing bimekizumab for PsA.
Overall Conclusions
Evidence from the BE OPTIMAL and BE COMPLETE trials in adult patients with PsA showed that patients who received bimekizumab were more likely to demonstrate clinically meaningful improvement in composite measures of disease activity, physical function, health-related quality of life, and pain at week 16 versus those on placebo. In the absence of head-to-head data comparing bimekizumab to other targeted DMARDs for PsA, the sponsor submitted an NMA that suggested that bimekizumab may be better than some comparators for composite measures of disease activity (i.e., ACR, minimal disease activity, and PASI 90), but may not be different from other comparators for these outcomes. Results remain uncertain due to inconsistency in estimates across comparators and inherent uncertainty in the methods used to control for study and patient characteristic heterogeneity.
CADTH was unable to address uncertainty related to comparative clinical data and the long-term efficacy of bimekizumab compared to other advanced DMARDs for the treatment of adult patients with PsA. As such, CADTH was unable to derive a more reliable base-case estimate of the cost-effectiveness of bimekizumab. The sponsor's analyses indicated that bimekizumab is not cost-effective at the submitted price. Results of the sponsor’s base-case sequential analysis suggest that for biologic-naive patients, bimekizumab is not on the cost-effectiveness frontier and is extendedly dominated by adalimumab and infliximab, while for biologic-experienced patients, bimekizumab is associated with an ICER of $69,876 per QALY gained (incremental costs = $49,923; incremental QALYs = 0.71) versus tofacitinib. A price reduction of approximately 40% is required for bimekizumab to be considered an optimal treatment (i.e., to get on the cost-effectiveness frontier) for biologic-naive patients. To be considered cost-effective at a willingness-to-pay threshold of $50,000, a price reduction of 52% or 24% is required for the biologic-naive and biologic-experienced populations, respectively.
The sponsor’s results were based on efficacy inputs obtained from the sponsor’s submitted NMA; however, CADTH notes that the relative effect obtained from the sponsor’s NMA for effectiveness parameters represents a source of uncertainty. Therefore, while the sponsor’s base case suggests differences in treatment benefit compared to some therapies for the treatment of adult patients with PsA, these results will only be realized should the numerical differences that were observed in the NMA occur in clinical practice and lead to meaningful improvements for patients. Should these results not be realized, there is insufficient evidence to suggest that bimekizumab should be priced higher than other biologic DMARD treatments for PsA.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL solution for SC injection. Oakville (ON): UCB Canada Inc; 2023 Oct 4.
2.Bimzelx (bimekizumab): 160 mg/mL solution for injection, subcutaneous use [product monograph]. Oakville (ON): UCB Canada Inc; 2024 Feb 23.
3.Rodgers M, Epstein D, Bojke L, et al. Etanercept, infliximab and adalimumab for the treatment of psoriatic arthritis: a systematic review and economic evaluation. Health Technol Assess. 2011;15(10):i-xxi, 1-329. PubMed
4.Colaco K, Widdifield J, Luo J, et al. Trends in mortality and cause-specific mortality among patients with psoriasis and psoriatic arthritis in Ontario, Canada. J Am Acad Dermatol. 2021;84(5):1302-1309. PubMed
5.Statistics Canada. Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2023 Dec 13.
6.Dolan P. Modeling valuations for EuroQol health states. Med Care. 1997;35(11):1095-1108. PubMed
7.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Nov 10.
8.CADTH drug reimbursement review pharmacoeconomic report: guselkumab (Tremfya) for psoriatic arthritis. Can J Health Technol 2022;3(2). https://www.cadth.ca/sites/default/files/DRR/2023/SR0733-Tremfya.pdf. Accessed 2023 Nov 14.
9.Updated CADTH reimbursement recommendations from a streamlined drug class or therapeutic review: biologics for plaque psoriasis. Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/TSDrugClas/TS0001/TS0001-PsO-Updated-Streamlined-Rec.pdf. Accessed 2023 Dec 13.
10.CADTH. Bimekizumab for ankylosing spondylitis. 2023; https://www.cadth.ca/bimekizumab-1. Accessed 2023 Dec 13.
11.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL solution for SC injection. Oakville (ON): UCB Canada Inc; 2023 Oct 5.
12.Humira (adalimumab injection): 40 mg in 0.8 mL, 10 mg in 0.1 mL, 20 mg in 0.2 mL, 40 mg in 0.4 mL, 80 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022 Sep 16: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/HUMIRA_PM_EN.pdf. Accessed 2023 Oct 24.
13.Cimzia (certolizumab pegol pegol): solution for injection 200 mg/mL [product monograph]. Oakville (ON) UCB Canada Inc; 2019 Feb 8: https://pdf.hres.ca/dpd_pm/00049574.PDF. Accessed 2023 Oct 24.
14.Enbrel (etanercept): solution in a prefilled syringe, 50 mg/mL and lyophilized powder in a vial for reconstitution, 25 mg/vial, subcutaneous injection [product monograph]. Mississauga (ON): Amgen Canada Inc; 2021 Mar 19: https://www.amgen.ca/-/media/Themes/CorporateAffairs/amgen-ca/amgen-ca/documents/products/en/enbrel_pm.pdf. Accessed 2023 Oct 24.
15.Plaquenil (hydroxychloroquine sulfate tablets): 200 mg [product monograph]. Laval (QC): sanofi-aventis Canada Inc; 2019 Aug 26: https://pdf.hres.ca/dpd_pm/00052851.PDF. Accessed 2023 Oct 24.
16.Salazopyrin (sulfasalazine): 500 mg & Enema 3 g/100 mL [product monograph]. Kirkland (QC): Pfizer Canada Inc; 2003 Sep 23: https://pdf.hres.ca/dpd_pm/00000465.PDF. Accessed 2023 Oct 24.
17.Methotrexate (methotrexate): tablet and injection [product monograph]. Kirkland (QC): Pfizer Canada Inc; 2011 Apr 21: https://pdf.hres.ca/dpd_pm/00013052.PDF. Accessed 2023 Oct 24.
18.Otezla (apremilast): tablets 10 mg, 20 mg, and 30 mg [product monograph]. Mississauga (ON): Amgen Canada Inc; 2020 Aug 5: https://pdf.hres.ca/dpd_pm/00057499.PDF. Accessed 2023 Oct 24.
19.Rinvoq (upadacitinib): oral extended-release tablets, 15 mg, 30 mg, and 45 mg [product monograph]. St-Laurent (QC): AbbVie Corporation; 2023 Oct 31: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/RINVOQ_PM_EN.pdf. Accessed 2023 Oct 24.
20.Stelara (ustekinumab): solution for subcutaneous injection 45 mg/0.5 mL and 90 mg/ 1.0 mL [product monograph]. Toronto (ON): Janssen Inc; 2018 Nov 9: https://pdf.hres.ca/dpd_pm/00048246.PDF. Accessed 2023 Oct 24.
21.Tremfya (guselkumab): 100 mg/1mL solution for injection [product monograph]. Toronto (ON): Janssen Inc; 2017 Nov 10: https://pdf.hres.ca/dpd_pm/00042101.PDF. Accessed 2023 Oct 24.
22.Taltz (ixekizumab): 80 mg/ 1mL solution for injection [product monograph]. Toronto (ON): Eli Lilly Canada Inc; 2018 Mar 29: https://pdf.hres.ca/dpd_pm/00044542.PDF. Accessed 2023 Oct 24.
23.Cosentyx (secukinumab): 75 mg/ 0.5 mL, 150 mg/ 1mL, 300 mg/ 2mL solution for injection [product monograph]. Montreal (QC): Novartis Pharmaceuticals Canada Inc.; 2023 Aug 17: https://www.ask.novartispharma.ca/download.htm?res=cosentyx_scrip_e.pdf&resTitleId=990. Accessed 2023 Oct 24.
24.Remicade (infliximab): 100 mg per vial powder for solution [product monograph]. Toronto (ON): Janssen Inc; 2017 Aug 4: https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/REMICADE-MONOGRAPH.PDF. Accessed 2023 Oct 24.
25.Simponi (golimumab): 50 mg/ 0.5 mL, 100 mg/ 1 mL solution for injection [product monograph]. Toronto (ON): Janssen Inc; 2018 Jan 15: https://pdf.hres.ca/dpd_pm/00043422.PDF. Accessed 2023 Oct 24.
26.Xeljanz (tofacitinib): 5 mg, 10 mg tablets [product monograph]. Kirkland (QC): Pfizer Canada; 2024 Apr 12: https://webfiles.pfizer.com/file/177102d8-752d-40dc-ab60-4597fe78565f?referrer=ccb731e5-4f2d-4f4a-b2dc-e5e912145fc6. Accessed 2023 Oct 24.
27.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2023: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 Oct 24.
28.Government of Saskatchewan. Saskatchewan drug plan: search formulary. 2023; http://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2023 Oct 24.
29.List of medications. Québec (QC): Régie de l’assurance maladie du Québec (RAMQ); 2023: https://www.ramq.gouv.qc.ca/sites/default/files/documents/non_indexes/liste_med_2023-11-08_en.pdf. Accessed 2023 Oct 24.
30.Statistics Canada. Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2023 Dec 13.
31.Eder L, Haddad A, Rosen CF, et al. The incidence and risk factors for psoriatic arthritis in patients with psoriasis: a prospective cohort study. Arthritis Rheumatol. 2016;68(4):915-923. PubMed
32.Canadian physician questionnaire and interview report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bimzelx (bimekizumab), 160 mg/mL, solution for SC injection. Oakville (ON): UCB Canada Inc; 2023.
Appendix 1: Cost Comparison Table
Table 7: CADTH Cost Comparison Table for Psoriatic Arthritis
Treatment | Strength/concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Bimekizumab (Bimzelx) | 160 mg/mL | Prefilled syringe or autoinjector | 1,625.0000a | 160 mg SC injection every 4 weeks 320 mg every 4 weeks for the first 16 weeks, and every 8 weeks afterb | First year: 58.04 to 75.83 Subsequent: 58.04 | First year: 21,198 to 27,698 Subsequent: 21,198 |
TNF inhibitors | ||||||
Adalimumab (Humira) | 40 mg/0.8 mL | Prefilled syringe or pen | 794.1000 | 40 mg every 2 weeks | 56.72 | 20,718 |
SEB adalimumab (biosimilars) | 20 mg/0.4 mL 40 mg/0.8 mL | Prefilled syringe or pen | 235.6350 471.2700 | 33.66 | 12,295 | |
Certolizumab pegol (Cimzia) | 200 mg/mL | Single-use prefilled syringe | 664.5100 | Loading dose: 400 mg SC injection at weeks 0, 2, and 4. Maintenance dose: 200 mg every 2 weeks or 400 mg every 4 weeks | First year: 52.92 Subsequent: 47.47 | First year: 19,330 Subsequent: 17,337 |
Etanercept (Enbrel) | 25 mg per vial | Vial | 202.9300 | 50 mg weekly | 57.98 | 21,177 |
50 mg/mL | Prefilled syringe or autoinjector | 405.9850 | 58.00 | 21,184 | ||
SEB etanercept (Erelzi, Brenzys) | 25 mg/0.5 mL | Vial | 120.5000 | 34.43 | 12,575 | |
50 mg/mL | Prefilled syringe or autoinjector | 241.0000 | ||||
Golimumab SC (Simponi) | 50 mg/0.5 mL 100 mg/mL | Prefilled syringe or autoinjector | 1,555.1700 | 50 mg monthly | 51.09 | 18,662 |
Infliximab (Remicade) | 100 mg/vial | Vial | 987.5600c | 5 mg/kg initial dose followed with additional similar doses at 2 and 6 weeks after the first infusion then every 8 weeks thereafter | First year: 105.07 Subsequent: 88.18 | First year: 38,378 Subsequent: 32,206 |
SEB infliximab (Inflectra) | 100 mg/vial | Vial | 525.0000 | First year: 55.86 Subsequent: 46.88 | First year: 20,402 Subsequent: 17,121 | |
SEB (infliximab Renflexis/ Avsola) | 100 mg/vial | Vial | 493.0000 | First year: 52.45 Subsequent: 44.02 | First year: 19,159 Subsequent: 16,078 | |
IL-17A inhibitors | ||||||
Secukinumab (Cosentyx) | 75 mg/0.5 mL 150 mg/mL 300 mg/mL | Prefilled syringe or vial | 882.5900 | 150 mg SC at weeks 0, 1, 2, 3, and 4 followed by monthly dosing | First year: 35.88 Subsequent: 29.00 | First year: 13,107 Subsequent: 10,591 |
Ixekizumab (Taltz) | 80 mg/mL | Prefilled syringe or pen | 1,723.8900 | 160 mg SC at week 0, followed by 80 mg every 4 weeks 160 mg SC at week 0, followed by 80 mg at weeks 2, 4, 6, 8, 10, and 12, then 80 mg every 4 weeksb | First year: 66.29 to 80.45 Subsequent: 61.57 | First year: 24,211 to 29,383 Subsequent:22,488 |
IL-23 inhibitors | ||||||
Guselkumab (Tremfya) | 100 mg/mL | Prefilled syringe or patient-controlled injector | 3,059.7400d | 100 mg at weeks 0 and 4 then every 8 weeks thereafter | First year: 56.73 Subsequent: 54.64 | First year: 20,722 Subsequent: 19,957 |
IL-12/23 inhibitors | ||||||
Ustekinumab (Stelara) | 45 mg/0.5 mL 90 mg/mL | Prefilled syringe or vial | 4,593.1400 | Patients < 100 kg: 45 mg at weeks 0 and 4, then every 12 weeks thereafter Patients > 100 kg: 90 mg at weeks 0 and 4, then every 12 weeks thereafter | First year: 63.06 Subsequent: 54.68 | First year: 23,034 Subsequent: 19,972 |
Janus kinase inhibitors | ||||||
Tofacitinib (Xeljanz) | 5 mg 10 mg | Tablet | 5.9897 21.1718 | 5 mg twice daily | 11.98 | 4,375 |
Upadacitinib (Rinvoq) | 15 mg 30 mg 45 mg | Tablet | 51.6810 | 15 mg daily | 51.68 | 18,876 |
Phosphodiesterase type 4 inhibitors | ||||||
Apremilast (Otezla) | 10 mg 20 mg 30 mg | Tablet | 18.9041e | 30 mg twice daily, after titration | 37.81 | 13,809 |
Conventional synthetic disease-modifying antirheumatic drugs | ||||||
Methotrexate (generics) | 2.5 mg 10 mg | Tablet | 0.2513 2.7983d | 10 to 25 mg per week until adequate response is attained Dose adjusted to optimal clinical response; 25 mg/ week not ordinarily exceeded | 0.09 to 0.40 | 34 to 146,465 |
20 mg/2 mL 50 mg/2 mL | Vial | 12.5000 8.9200 | ||||
Leflunomide (generics) | 10 mg 20 mg | Tablet | 2.0000 | Loading: 100 mg daily for 3 days Maintenance: 20 mg | First year: 2.07 Subsequent: 2.00 | First year: 755 Subsequent: 731 |
Sulfasalazine (generics) | 500 mg | Tablet | 0.2533 | Week 1: 500 mg/ daily Week 2: 1,000 mg daily Week 3: 1,500 mg daily Maintenance: 2,000 mg daily | First year: 0.98 Subsequent: 1.01 | First year: 359 Subsequent: 370 |
500 mg | EC tablet | 0.3863 | First year: 1.50 Subsequent: 1.55 | First year: 370 Subsequent year: 564 | ||
Hydroxychloroquine (generics) | 200 mg | Tablet | 0.1576 | 400 to 600 mg daily until good response is obtained (usually 4 to 12 weeks), then dosage reduced by 50% and continued at 200 to 400 mg daily | First year: 0.17 to 0.46 Subsequent: 0.16 to 0.32 | First year: 61 to 168 Subsequent: 58 to 115 |
EC = enteric coated; SC = subcutaneous; SEB = subsequent entry biologic; TNF = tumour necrosis factor.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Note: Recommended dosing informed by respective product monographs unless otherwise stated.12-26 All prices are from the Ontario Drug Benefit Formulary (accessed October 2023), unless otherwise indicated, and do not include dispensing fees. The annual cost is based on a 365.25-day year. All weight-based doses assume an average patient weight of 85 kg and wastage of excess medication in vials.7
aSponsor-submitted price.1
bFor patients with PsA with coexisting moderate-to-severe plaque psoriasis.
cOntario Drug Benefit Exceptional Access Program (accessed October 2023).27
dSaskatchewan formulary (accessed October 2023).28
eRégie de l’assurance maladie du Québec formulary (accessed October 2023).29
Note this table has not been copy-edited.
Appendix 2: Submission Quality
Note this table has not been copy-edited.
Description | Yes/no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
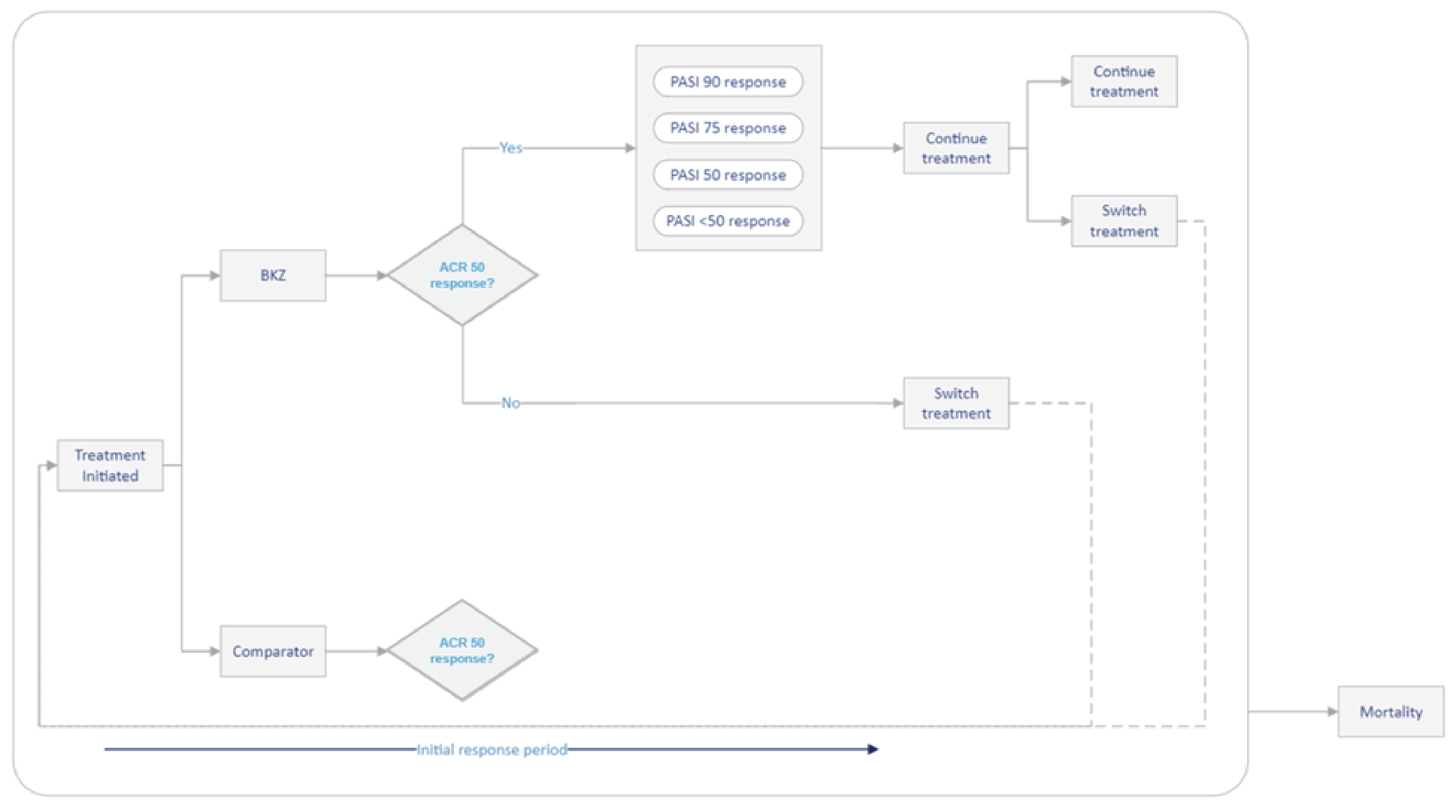
ACR 50 = American College of Rheumatology 50% improvement in rheumatoid arthritis; BKZ = bimekizumab; PASI = Psoriasis Area Severity Index.
Source: Sponsor’s pharmacoeconomic evaluation.1
Detailed Results of the Sponsor’s Base Case
Table 9: Sponsor’s Economic Evaluation Results — Biologic-Naive Patients
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | $129,567 | 9.84 | Reference |
Tofacitinib | $136,807 | 10.31 | $15,440 |
Adalimumab | $153,291 | 10.56 | $65,793 |
Infliximab | $176,226 | 10.76 | $114,065 |
Dominated treatments | |||
Etanercept | $156,893 | 10.59 | Extendedly dominated by INF |
Ustekinumab | $160,954 | 10.24 | Dominated by TOF, ADA, ETA |
Guselkumab | $162,011 | 10.32 | Dominated by ADA, ETA |
Certolizumab | $163,821 | 10.46 | Dominated by ADA, ETA |
Secukinumab | $164,924 | 10.46 | Dominated by ADA, ETA, CERT |
Risankizumab | $165,529 | 10.30 | Dominated by TOF, ADA, ETA, GUS, CERT, SEC |
Golimumab | $168,199 | 10.57 | Dominated by ETA |
Ixekizumab | $169,648 | 10.38 | Dominated by ADA, ETA, CERT, SEC, GOL |
Upadacitinib | $169,784 | 10.61 | Extendedly dominated by INF |
Bimekizumab | $174,766 | 10.60 | Extendedly dominated by INF |
ADA = adalimumab; BSC = best supportive care; CERT = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; ICER = incremental cost-effectiveness ratio; INF = infliximab; QALY = quality-adjusted life-year; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib.
Note: The sponsor reported that bimekizumab was dominated by upadacitinib; however, as upadacitinib is not on the efficiency, results are reported with respect to treatments that are on the efficiency frontier.
Table 10: Sponsor’s Economic Evaluation Results — Biologic-Experienced Patients
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | $131,829 | 8.23 | Reference |
Tofacitinib | $139,183 | 8.64 | $17,841 |
Bimekizumab | $189,106 | 9.35 | $69,876 |
Dominated treatments | |||
Certolizumab | $153,486 | 8.50 | Dominated by TOF |
Ustekinumab | $157,341 | 8.50 | Dominated by TOF, CERT |
Guselkumab | $164,394 | 8.74 | Extendedly dominated by SEC, UPA, BKZ |
Secukinumab | $164,398 | 8.78 | Extendedly dominated by UPA, BKZ |
Upadacitinib | $169,482 | 8.96 | Extendedly dominated by BKZ |
Risankizumab | $170,953 | 8.80 | Dominated by UPA |
Ixekizumab | $177,405 | 8.92 | Dominated by UPA |
BKZ = bimekizumab; BSC = best supportive care; CERT = certolizumab pegol; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib.
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Biologic-Naive Patients
Parameter | BKZ | BSC | ADA | CERT | ETA | GOL | GUS | INF | IXE | RIS | SEC | TOF | UST | UPA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Discounted LYs | ||||||||||||||
Total | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 | 25.88 |
Discounted QALYs | ||||||||||||||
Total | 10.60 | 9.84 | 10.56 | 10.46 | 10.59 | 10.57 | 10.32 | 10.76 | 10.38 | 10.30 | 10.46 | 10.31 | 10.24 | 10.61 |
First-line | 1.60 | 0.98 | 1.51 | 1.38 | 1.58 | 1.55 | 1.16 | 1.84 | 1.26 | 1.14 | 1.37 | 1.15 | 1.05 | 1.61 |
Final-line | 9.00 | 8.85 | 9.05 | 9.08 | 9.01 | 9.02 | 9.16 | 8.92 | 9.12 | 9.16 | 9.09 | 9.16 | 9.20 | 9.00 |
Discounted costs ($) | ||||||||||||||
Total | 174,766 | 129,567 | 153,291 | 163,821 | 156,893 | 168,199 | 162,011 | 176,226 | 169,648 | 165,529 | 164,924 | 136,807 | 160,954 | 169,784 |
b/tsDMARD drug | 45,285 | 0 | 23,548 | 33,615 | 26,096 | 38,022 | 32,380 | 39,525 | 40,072 | 35,769 | 35,155 | 6,700 | 31,060 | 39,910 |
cDMARD drug | 3,010 | 2,936 | 3,003 | 2,999 | 3,009 | 3,007 | 2,983 | 3,021 | 2,993 | 2,987 | 3,000 | 2,988 | 2,984 | 3,010 |
Administration | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7,139 | 0 | 0 | 0 | 0 | 0 | 0 |
Disease management | 126,471 | 126,631 | 126,740 | 127,207 | 127,788 | 127,170 | 126,648 | 126,540 | 126,582 | 126,773 | 126,769 | 127,119 | 126,910 | 126,864 |
ADA = adalimumab; BKZ = bimekizumab; BSC = best supportive care; b/tsDMARD = biologic or targeted synthetic disease-modifying antirheumatic drug; cDMARD = conventional disease-modifying antirheumatic drug; CERT = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; ICER = incremental cost-effectiveness ratio; INF = infliximab; IXE = ixekizumab; QALY = quality-adjusted life-year; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Source: Sponsor’s pharmacoeconomic submission.1
Table 12: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Biologic-Experienced Patients
Parameter | BKZ | BSC | CERT | GUS | IXE | RIS | SEC | TOF | UST | UPA |
|---|---|---|---|---|---|---|---|---|---|---|
Discounted LYs | ||||||||||
Total | 24.96 | 24.96 | 24.96 | 24.96 | 24.96 | 24.96 | 24.96 | 24.96 | 24.96 | 24.96 |
Discounted QALYs | ||||||||||
Total | 9.35 | 8.23 | 8.50 | 8.74 | 8.92 | 8.80 | 8.78 | 8.64 | 8.50 | 8.96 |
First-line | 2.04 | 0.98 | 0.81 | 1.15 | 1.41 | 1.24 | 1.22 | 1.01 | 0.80 | 1.47 |
Final-line | 7.32 | 7.24 | 7.69 | 7.59 | 7.51 | 7.56 | 7.57 | 7.63 | 7.69 | 7.49 |
Discounted costs ($) | ||||||||||
Total | $189,106 | $131,829 | $153,486 | $164,394 | $177,405 | $170,953 | $164,398 | $139,183 | $157,341 | $169,482 |
b/tsDMARD drug | $58,175 | $0 | $21,459 | $32,612 | $45,432 | $39,017 | $32,357 | $6,048 | $25,316 | $37,017 |
cDMARD drug | $1,976 | $1,910 | $1,935 | $1,935 | $1,955 | $1,949 | $1,949 | $1,941 | $1,935 | $1,955 |
Administration | $0 | $0 | $0 | $0 | $0 | $0 | $0 | $0 | $0 | $0 |
Disease management | $128,955 | $129,919 | $130,092 | $129,847 | $130,018 | $129,987 | $130,092 | $131,194 | $130,091 | $130,509 |
BKZ = bimekizumab; BSC = best supportive care; b/tsDMARD = biologic or targeted synthetic disease-modifying antirheumatic drug; cDMARD = conventional disease-modifying antirheumatic drug; CERT = certolizumab pegol; GUS = guselkumab; IXE = ixekizumab; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note this appendix has not been copy-edited.
Scenario Analyses
Table 13: CADTH Scenario Analysis Results — Disease Management Costs, Biologic-Naive Patients
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Best supportive care | $102,158 | 9.81 | Reference |
Tofacitinib | $109,466 | 10.26 | $16,249 |
Adalimumab (biosimilar) | $126,235 | 10.52 | $64,968 |
Etanercept (biosimilar) | $129,661 | 10.55 | $101,094 |
Infliximab (biosimilar) | $149,520 | 10.73 | $107,896 |
Dominated treatments | |||
Ustekinumab mixed dose | $133,751 | 10.21 | Dominated by tofacitinib, adalimumab (biosimilar), etanercept (biosimilar) |
Guselkumab | $134,937 | 10.29 | Dominated by adalimumab (biosimilar), etanercept (biosimilar) |
Certolizumab pegol | $136,466 | 10.42 | Dominated by adalimumab (biosimilar), etanercept (biosimilar) |
Risankizumab | $138,031 | 10.26 | Dominated by adalimumab (biosimilar), etanercept (biosimilar), guselkumab, certolizumab pegol pegol |
Secukinumab mixed dose | $138,198 | 10.43 | Dominated by adalimumab (biosimilar), etanercept (biosimilar) |
Golimumab | $140,444 | 10.52 | Dominated by etanercept (biosimilar) |
Ixekizumab | $142,042 | 10.34 | Dominated by adalimumab (biosimilar), etanercept (biosimilar), certolizumab pegol pegol, secukinumab mixed dose, golimumab |
Upadacitinib | $142,538 | 10.57 | Extendedly dominated by bimekizumab, infliximab (biosimilar) |
Bimekizumab | $148,296 | 10.58 | Extendedly dominated by infliximab (biosimilar) |
ADA = adalimumab; BSC = best supportive care; CERT = certolizumab pegol; ETA = etanercept; GOL = golimumab; GUS = guselkumab; ICER = incremental cost-effectiveness ratio; INF = infliximab; QALY = quality-adjusted life-year; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib.
Table 14: CADTH Scenario Analysis Results — Disease Management Costs, Biologic-Experienced Patients
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Best supportive care | $102,286 | 8.23 | Reference |
Tofacitinib | $109,725 | 8.66 | $17,450 |
Bimekizumab | $160,906 | 9.37 | $71,542 |
Dominated treatments | |||
Certolizumab pegol | $121,744 | 8.44 | Dominated by tofacitinib |
Ustekinumab mixed dose | $127,966 | 8.51 | Dominated by tofacitinib |
Guselkumab | $134,674 | 8.74 | Extendedly dominated by secukinumab mixed dose, upadacitinib, bimekizumab |
Secukinumab mixed dose | $135,312 | 8.80 | Extendedly dominated by upadacitinib, bimekizumab |
Upadacitinib | $140,993 | 8.99 | Extendedly dominated by bimekizumab |
Risankizumab | $142,320 | 8.82 | Dominated by upadacitinib |
Ixekizumab | $147,958 | 8.92 | Dominated by upadacitinib |
BKZ = bimekizumab; BSC = best supportive care; CERT = certolizumab pegol; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib.
Appendix 5: Submitted BIA and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 15: Summary of Key Takeaways
Key takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA to estimate the 3-year budget impact of reimbursing bimekizumab for the treatment of adult patients with active PsA. The analysis was taken from the perspective of the Canadian public drug plans. A 3-year time horizon was used from 2025 to 2027, with 2024 as the base year. The target population size was derived with an epidemiological approach. Key inputs to the BIA are documented in Table 16.
The BIA compared 2 scenarios to determine the incremental budget impact of reimbursing bimekizumab. The reference case scenario assumed that patients were on currently available PsA treatments. The new drug scenario included bimekizumab. In the sponsor’s base case, costs related to drug acquisition were considered. Vial sharing was not included.
State the key assumptions:
The proportion of patients with PsA requiring biologic/advanced therapy is 45%.
45% of patients would be eligible for public coverage.
17% of patients with PsA would have coexistent moderate-to-severe PsO that would use the higher dose of bimekizumab.
61% of patients with PsA would be tumour necrosis factor–alpha inhibitor inadequate responders and require a higher secukinumab dose; the 61% further includes patients receiving the PsO dose due to coexistent moderate-to-severe plaque PsO.
All CADTH-participating jurisdictions have implemented or will implement a biosimilar switching policy; therefore, patients in an induction year receive the biosimilar if available.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) | |
|---|---|---|
Target population | ||
Population of pan-Canada in base year PsA prevalence PsA incidence % of adult patients with PsA requiring biologic/advanced therapy % eligible for public coverage % existing cases in an induction year | 32,050,27930 0.149%31 0.014%31 45.0%32 45.0%11 16.5%8 | |
Number of patients eligible for drug under review | 9,670 / 9,784 / 9,897 | |
Market uptake (3 years) | ||
Induction | Maintenance | |
Uptake (reference scenario) Bimekizumab (Bimzelx) Certolizumab (Cimzia) Etanercept (Enbrel) Etanercept biosimilars Adalimumab (Humira) Adalimumab biosimilars Infliximab (Remicade) Infliximab biosimilars Golimumab (Simponi) Secukinumab (Cosentyx) Ixekizumab (Taltz) Ustekinumab (Stelara) Risankizumab (Skyrizi) Guselkumab (Tremfya) Upadacitinib (Rinvoq) Tofacitinib (Xeljanz) Apremilast (Otezla) | 0.00% / 0.00% / 0.00% 5.29% / 5.64% / 6.00% 0.00% / 0.00% / 0.00% 14.59% / 14.29% / 14.00% 0.00% / 0.00% / 0.00% 23.65% / 23.32% / 23.00% 0.00% / 0.00% / 0.00% 4.68% / 4.59% / 4.50% 8.02% / 8.08% / 8.15% 22.79% / 22.89% / 23.00% 8.19% / 8.60% / 9.00% 3.96% / 4.48% / 5.00% 0.47% / 0.64% / 0.80% 0.03% / 0.04% / 0.05% 4.94% / 4.97% / 5.00% 2.61% / 1.81% /1.00% 0.79% / 0.64% / 0.50% | 0.00% / 0.00% / 0.00% 5.29% / 5.64% / 6.00% 2.48% / 1.74% / 1.00% 12.11% / 12.56% / 13.00% 5.24% / 3.62% / 2.00% 18.41% / 19.71% / 21.00% 1.21% / 0.86% / 0.50% 3.47% / 3.73% / 4.00% 8.02% / 8.08% / 8.15% 22.79% / 22.89% / 23.00% 8.19% / 8.60% / 9.00% 3.96% / 4.48% / 5.00% 0.47% / 0.64% / 0.80% 0.03% / 0.04% / 0.05% 4.94% / 4.97% / 5.00% 2.61% / 1.81% / 1.00% 0.79% / 0.64% / 0.50% |
Uptake (new drug scenario) Bimekizumab (Bimzelx) Certolizumab (Cimzia) Etanercept (Enbrel) Etanercept biosimilars Adalimumab (Humira) Adalimumab biosimilars Infliximab (Remicade) Infliximab biosimilars Golimumab (Simponi) Secukinumab (Cosentyx) Ixekizumab (Taltz) Ustekinumab (Stelara) Risankizumab (Skyrizi) Guselkumab (Tremfya) Upadacitinib (Rinvoq) Tofacitinib (Xeljanz) Apremilast (Otezla) | 3.00% / 5.00% / 8.00% 5.13% / 5.36% / 5.52% 2.40% / 1.65% / 0.92% 11.75% / 11.93% / 11.96% 5.08% / 3.44% / 1.84% 17.86% / 18.72% / 19.32% 1.18% / 0.81% / 0.46% 3.37% / 3.55% / 3.68% 7.78% / 7.68% / 7.50% 22.11% / 21.75% / 21.16% 7.95% / 8.17% / 8.28% 3.84% / 4.25% / 4.60% 0.46% / 0.61% / 0.74% 0.02% / 0.04% / 0.05% 4.79% / 4.72% / 4.60% 2.54% / 1.72% / 0.92% 0.76% / 0.61% / 0.46% | 3.00% / 5.00% / 8.00% 5.13% / 5.36% / 5.52% 0.00% / 0.00% / 0.00% 14.15% / 13.58% / 12.88% 0.00% / 0.00% / 0.00% 22.94% / 22.16% / 21.16% 0.00% / 0.00% / 0.00% 4.54% / 4.36% / 4.14% 7.78% / 7.68% / 7.50% 22.11% / 21.75% / 21.16% 7.95% / 8.17% / 8.28% 3.84% / 4.25% / 4.60% 0.46% / 0.61% / 0.74% 0.02% / 0.04% / 0.05% 4.79% / 4.72% / 4.60% 2.54% / 1.72% / 0.92% 0.76% / 0.61% / 0.46% |
Cost of treatment per patients (induction year/maintenance year) | ||
Bimekizumab (Bimzelx) Certolizumab (Cimzia) Etanercept (Enbrel) Etanercept biosimilars Adalimumab (Humira) Adalimumab biosimilars Infliximab (Remicade) Infliximab biosimilars Golimumab (Simponi) Secukinumab (Cosentyx) Ixekizumab (Taltz) Ustekinumab (Stelara) Risankizumab (Skyrizi) Guselkumab (Tremfya) Upadacitinib (Rinvoq) Tofacitinib (Xeljanz) Apremilast (Otezla) | $22,303 / $21,198 $19,935 / $17,277 $21,184 $12,575 $20,718 $12,295 $32,920 / $24,476 $16,434 / $12,219 $20,287 $24,457 / $17,052 $39,425 / $24,203 $27,627 / $19,972 $29,683 / $21,458 $24,546 / $19,957 $18,876 $4,375 $13,678 | |
PsA = psoriatic arthritis.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analysis suggest that the reimbursement of bimekizumab for adults with PsA will be associated with an incremental cost of $1,300,605 in year 1, $2,199,792 in year 2, and $3,510,675 in year 3. Therefore, the 3-year total incremental budget impact is $7,011,073.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The total number of eligible patients was inaccurately estimated. The sponsor’s BIA used population numbers informed by Statistics Canada and Non-Insured Health Benefits annual reports to inform the pan-Canadian starting population. However, the values used did not appropriately limit the population to individuals aged 18 years and older, and therefore did not reflect the Health Canada indication for bimekizumab which is limited to adults with active PsA.
CADTH considered only the adult population in its reanalysis.
The Non-Insured Health Benefits (NIHB) population was inappropriately calculated. The sponsor calculated the total population of CADTH-participating drug plans by adding the population of the provinces, excluding Quebec, to the population of NIHB clients. NIHB clients living within the borders of a province are counted within provincial population data as reported by Statistics Canada; thus, the NIHB population was double counted in the sponsor’s analysis.
CADTH did not adjust for this limitation in reanalysis. The impact on pan-Canadian model results is expected to be minimal.
The proportion of adult patients with PsA requiring biologic/advanced therapies is uncertain. The sponsor estimated that 45% of adult patients with PsA require biologic or advanced therapy, informed by clinician feedback obtained by the sponsor. Clinical expert feedback received by CADTH noted that 45% may be on the lower end of the proportion of patients who required biologic or advanced therapies and estimated that it may be closer to 50%.
CADTH conducted a scenario analysis assuming 50% of adult patients with PsA require biologic/advanced therapy.
Market uptake and displacement for bimekizumab is uncertain. The sponsor’s submitted BIA indicated that bimekizumab would result in a market uptake of 3.00% in year 1, 5.00% in year 2, and 8.00% in year 3 based on internal forecasts conducted by the sponsor. It was assumed that bimekizumab would take market share from each comparator proportionately. Clinical expert feedback received by CADTH considered these estimates plausible, but noted bimekizumab market share may be more likely to come from other IL-17 inhibitors (i.e., secukinumab and ixekizumab) due to clinician preference and familiarity with drugs within the same class.
CADTH conducted a scenario analysis assuming that 70% of bimekizumab market share was obtained from secukinumab and ixekizumab using the sponsor’ provided option.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by correcting the population values used to inform the analysis to only consider adults 18 years and older.
Table 17: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Jurisdiction population corrections | Populations for all jurisdictions using the total population | Updated values to only consider the 18+ population as informed by Statistics Canada and the NIHB annual reports |
CADTH base case | Reanalysis 1 | |
NIHB = Non-Insured Health Benefits.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19. In the CADTH base case, the estimated 3-year incremental budget impact of reimbursing bimekizumab is expected to be $5,742,058 ($1,062,138 in year 1, $1,800,715 in year 2, and $2,879,205 in year 3).
Table 18: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | 3-year total |
|---|---|
Submitted base case | $7,011,073 |
CADTH reanalysis 1 | $5,742,058 |
CADTH base case | $5,742,058 |
BIA = budget impact analysis.
CADTH conducted the following scenario analyses. Results are provided in Table 19.
Assumed 50% of patients required biologic/advanced therapies.
Assumed 70% of bimekizumab market share was obtained from secukinumab and ixekizumab using the sponsor’s provided option to do so.
Table 19: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $164,562,781 | $166,845,673 | $169,136,070 | $171,433,971 | $507,415,714 |
New drug | $164,562,781 | $168,146,278 | $171,335,862 | $174,944,646 | $514,426,786 | |
Budget impact | $0 | $1,300,605 | $2,199,792 | $3,510,675 | $7,011,073 | |
CADTH base case | Reference | $133,877,899 | $136,021,121 | $138,171,593 | $140,329,314 | $414,522,029 |
New drug | $133,877,899 | $137,083,260 | $139,972,308 | $143,208,519 | $420,264,086 | |
Budget impact | $0 | $1,062,138 | $1,800,715 | $2,879,205 | $5,742,058 | |
CADTH scenario analysis 1: 50% advanced therapies | Reference | $148,753,221 | $151,134,579 | $153,523,992 | $155,921,460 | $460,580,032 |
New drug | $148,753,221 | $152,314,733 | $155,524,787 | $159,120,577 | $466,960,096 | |
Budget impact | $0 | $1,180,154 | $2,000,794 | $3,199,116 | $6,380,064 | |
CADTH scenario analysis 2: alternative bimekizumab market share sources | Reference | $133,877,899 | $136,021,121 | $138,171,593 | $140,329,314 | $414,522,029 |
New drug | $133,877,899 | $136,095,401 | $138,320,078 | $140,556,412 | $414,971,890 | |
Budget impact | $0 | $74,280 | $148,485 | $227,097 | $449,861 | |
CADTH scenario analysis 3: 24% price reduction | Reference | $133,877,899 | $136,021,121 | $138,171,593 | $140,329,314 | $414,522,029 |
New drug | $413,647,381 | $413,647,381 | $413,647,381 | $413,647,381 | $413,647,381 | |
Budget impact | $0 | –$156,389 | –$258,234 | –$460,025 | –$874,647 | |
CADTH scenario analysis 4: 52% price reduction | Reference | $133,877,899 | $136,021,121 | $138,171,593 | $140,329,314 | $414,522,029 |
New drug | $133,877,899 | $134,443,117 | $135,511,253 | $135,973,523 | $405,927,892 | |
Budget impact | $0 | –$1,578,004 | –$2,660,340 | –$4,355,792 | –$8,594,137 |
BIA = budget impact analysis.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.