CADTH Reimbursement Review
Inebilizumab (Uplizna)
Sponsor: Horizon Therapeutics Canada
Therapeutic area: Neuromyelitis optica spectrum disorders
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AAR
annualized attack rate
AC
adjudication committee
AE
adverse event
AQP4
aquaporin-4
AQP4-IgG
anti-aquaporin-4 immunoglobulin G
CI
confidence interval
CNMSC
Canadian Network of Multiple Sclerosis Clinics
CNS
central nervous system
EDSS
Expanded Disability Status Scale
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IPD
individual patient data
ITT
intention to treat
IVIG
IV immunoglobulin
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
MS
multiple sclerosis
NMA
network meta-analysis
NMO
neuromyelitis optica
NMOSD
neuromyelitis optica spectrum disorder
NRS
numerical rating scale
OLP
open-label period
RCP
randomized controlled period
RCT
randomized controlled trial
RR
relative risk
SAE
serious adverse event
SF-36
Short Form (36) Health Survey
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Inebilizumab (Uplizna), 10 mg/mL, single-dose vials containing 100 mg inebilizumab in 10 mL solution, IV infusion. |
Sponsor | Horizon Therapeutics Canada |
Indication | As monotherapy for the treatment of adult patients with NMOSD who are seropositive for AQP4-IgG. Treatment should be administered under the supervision of a qualified healthcare professional. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | December 15, 2023 |
Recommended dose | The recommended dosage is:
|
AQP4-IgG = anti-aquaporin-4 immunoglobulin G; NMOSD = neuromyelitis optica spectrum disorder; NOC = Notice of Compliance.
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a rare, chronic disorder of the central nervous system (CNS) that is characterized by acute attacks or relapses that cause inflammation in the optic nerve (optic neuritis) and spinal cord (myelitis).1 A defining feature of NMOSD is the presence of pathogenic serum autoantibodies against aquaporin-4 (AQP4), which led to its differentiation from multiple sclerosis (MS).2-4 This disease has a relapsing nature in which patients experience acute attacks and relapses characterized by new or worsening signs and symptoms. These attacks are unpredictable and can lead to accruing disabilities and, often, to permanent impairment.5-9 The clinical presentation of an NMSOD attack typically involves optic neuritis that causes ocular pain and vision loss. Myelitis causes sensory loss, weakness, or paralysis in the legs or arms, painful spasms,10-13 and bladder and bowel dysfunction.3,5 At its worst, severe high-cervical myelitis and brainstem lesions can lead to fatal respiratory failure.3,11,13 The consequences of NMOSD extend beyond clinical settings and include physical, functional, and psychological effects that alter every aspect of patients’ and caregivers’ lives and impact their health-related quality of life (HRQoL).9,14-16
NMOSD disproportionately affects females.5,17 Systematic reviews based on data from several countries have estimated there are 0.053 to 0.4 incident cases per 100,000 people and 0.51 to 4.4 prevalent cases per 100,000 people.18,19 No Canadian-specific estimates were identified in these studies.
Inebilizumab is a humanized afucosylated monoclonal antibody that binds to CD19 for the treatment of NMOSD.20-22 Inebilizumab has a Health Canada indication as monotherapy for the treatment of adult patients with NMOSD who are seropositive for anti-AQP4 immunoglobulin G (AQP4-IgG).23 The reimbursement request is as per the approved indication. The recommended dosage for inebilizumab is an initial 300 mg dose via IV infusion followed 2 weeks later by a second 300 mg dose via IV infusion; subsequent doses (starting 6 months from the first infusion) are administered as single 300 mg doses via IV infusion every 6 months.23
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, MS Canada and the Sumaira Foundation (TSF) responded to CADTH’s call for patient input on this topic.
MS Canada gathered information for this submission through an online survey in 2023 that included 13 respondents. TSF gathered information in 2023 through 2 online surveys and videoconferencing interviews with patients and caregivers, and through TSF’s experience working in the NMOSD communities. The TSF survey included data from 51 patients and 9 caregivers.
The 2 patient groups indicated that NMOSD follows a relapsing-remitting disease course and is initiated with a severe attack and continues with subsequent devastating attacks that affect the patient’s vision and lead to mobility issues and chronic pain. The disease has a tremendous impact on all aspects of patients’ and caregivers’ lives, including a negative effect on their quality of life, their independence and employment, and on their social, family, and school life.
The patient inputs stated that treatment for NMOSD involved IV steroids, IV immunoglobulin (IVIG), or plasmapheresis or plasma exchange, as well as the use of off-label drugs with varying levels of therapeutic benefit due to worsening symptoms and/or challenging side effects experienced by the patient while cycling through different therapies.
According to the 2 patient inputs, access is very limited for therapies such as eculizumab, satralizumab, and rituximab, and the administration schedule for eculizumab can be too arduous for some patients. According to these inputs, patients need to have access to therapy options that can reduce the risk of future attacks, maintain the current level of physical ability, and slow disease progression.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of NMOSD.
Unmet Needs
The clinical experts indicated that the treatment of NMOSD includes a series of treatment goals relating to 3 broad areas: prevention of relapses (disease modification), treatment of relapses, and treatment of residual symptoms. Although ideal, it is unlikely that any single treatment would cover all 3 areas. Of these 3 areas, preventive treatment was described to be of special interest because relapses are the major source of disability accumulation for people with NMOSD. As a result, preventive treatment is expected to result in downstream desirable effects, including better HRQoL, an increased ability to maintain independence and employment, and a reduced reliance on caregivers. The clinical experts highlighted that it is important to control progression as early as possible because damage leading to neurologic disability, including paralysis and blindness, may be irreversible after an attack.
The clinical experts agreed that most patients with NMOSD still have relapses despite their current treatment regimens, and current first-line therapies (e.g., azathioprine and mycophenolate mofetil) are not considered particularly effective in preventing NMOSD attacks. Additionally, these therapies are associated with significant adverse effects, especially if used in conjunction with corticosteroids. The greatest unmet need in the treatment of NMOSD is for therapies that prevent relapses more effectively without intolerable side effects.
Place in Therapy
The clinical experts agreed that the current clinical practice for treatment and attack prevention in NMOSD is suboptimal and inconsistent due to the low efficacy of the off-label treatments used in the first-line setting and barriers to treatment access, which vary by province. The newer therapies with indications for NMOSD, such as satralizumab and eculizumab, present especially significant barriers to access for most patients due to inconsistent coverage or the lack of public reimbursement, and the onerous dosing schedule of eculizumab. Due to the impact of NMOSD on a person’s ability to maintain employment, patients with NMOSD are more likely to lack private insurance.
As the vast majority of patients with NMOSD will relapse and relapses may lead to permanent disability or death, all people with NMOSD should be on relapse-preventive treatment.
The experts agreed that because the impact of relapses on people with NMOSD is devastating and preventing as many relapses as possible is critical to the prevention of significant disability, inebilizumab should be available as first- and later-line treatment for patients diagnosed with AQP4-IgG–seropositive NMOSD. The experts indicated that inebilizumab would be used as a monotherapy.
Patient Population
NMOSD is a rare disease. The clinical experts indicated that patients diagnosed with NMOSD who are seropositive for AQP4-IgG should be candidates for treatment with inebilizumab. It is the standard of care in Canada to assess patients with NMOSD for AQP4-IgG; the experts noted there are no major challenges in this regard in Canada, other than that the test results may be delayed in some locations. The specificity of AQP4-IgG is very high, so the risk of misdiagnosis is very low.
The patient eligibility criteria in the pivotal study, N-MOmentum, were considered by the experts to be broadly representative of patients with NMOSD in Canada, with the exception that the study excluded patients with recent steroidal treatment. Although appropriate from the perspective of clinical trial design, in real-world practice there are many comorbidities that may require steroid treatment, and these patients should not necessarily be excluded from treatment with inebilizumab; such a decision would need to be considered by the expert clinician managing a patient’s particular case. Patients who have received IVIG or have concomitant diseases should not necessarily be excluded from receiving inebilizumab in the real world.
Within the population of patients who have NMOSD and are AQP4-IgG seropositive, it is unknown which patients are more likely to benefit from inebilizumab.
It is possible that patients who are seronegative for AQP4-IgG may also benefit from inebilizumab. Fulfillment of seronegative NMOSD criteria would be necessary to establish the diagnosis to allow appropriate access.
Assessing the Response to Treatment
The experts indicated that a clinically meaningful response to treatment relates to the reduction of the relapse rate and prolongation of times to relapse. Although the absence of relapse is indicative of a clinically meaningful response, this may not be realistic, as the number and severity of relapses that patients experience differ on an individual level (e.g., some patients may have several relapses per year); thus, a reduction in the number of relapses is still a reasonable goal. The determination of relapses is fairly objective; however, it is not the only factor, and assessment of treatment response is based on a combination of patient-reported symptoms, a clinical exam, clinical tools, and patient history. Other important outcome measures include evaluation of attack severity and degree of recovery from attack, as well as accumulation of disability.
The experts noted there is a lack of formal guidance on how to assess treatment response, but it would be reasonable to assess initial treatment response 3 months after the initial infusion then every 6 months until stability is achieved, and then every year for patients with stable NMOSD. However, it was noted that within the first 6 months of treatment, the attack rate may still be higher than when stability is achieved.
MRIs are not routinely conducted for patients with NMOSD outside of initial diagnosis and so would not be used in assessing the response to treatment.
Discontinuing Treatment
Discontinuation of treatment should occur if the patient is completely dependent and unable to leave their bed (Expanded Disability Status Scale [EDSS] score of 9.0 and above).
Discontinuation of treatment should be considered on a case-by-case basis in the event of a severe relapse (e.g., requiring intubation and support on a ventilator) or if the patient has experienced 2 or more relapses within 2 years, has severe or unacceptable adverse events (AEs), or has contraindications for therapy.
Prescribing Considerations
The clinical experts agreed that treatment should be supervised by a neurologist with expertise in this area. Although NMOSD and MS are not the same disease, the populations and medications are similar and patients with NMOSD are often cared for in MS clinics. The clinical experts stated that neurologists with experience or expertise in related subspecialties should prescribe inebilizumab, including clinicians with experience in MS neurology, and/or neurologists working in an MS clinic or in neuroimmunology, autoimmune neurology, and/or neuro-ophthalmology. However, patients in remote areas may have issues with access to subspecialists. For patients living in remote areas, local neurologists without subspecialty expertise could work by distance in conjunction with neurologists who are experts in a relevant subspecialty.
Inebilizumab is expected to be used as a monotherapy and not combined with other monoclonal antibodies indicated for the treatment of NMOSD. However, there may be situations in which it is combined with classical immunosuppressants. There is a lack of data regarding combination therapies.
Clinician Group Input
One clinician group, the Canadian Network of Multiple Sclerosis Clinics (CNMSC), responded to CADTH’s call for clinician group input (input authored by 1 clinician).
According to the CNMSC, a variety of off-label therapies are used for the treatment of NMOSD in Canada, including corticosteroids, azathioprine, mycophenolate mofetil, and rituximab. However, breakthrough NMOSD attacks are reported on all of these drugs, and government drug program funding varies by province and territory. Recently, 2 monoclonal antibodies, eculizumab and satralizumab, were approved by Health Canada. However, access to these therapies is extremely limited due to their high cost and stringent funding criteria. All of the therapies in use for NMOSD work by suppressing the immune system to prevent attacks, with variable efficacy. Failure of treatment, resulting in even just 1 relapse, can lead to a profound, permanent disability, including blindness and paralysis.
The clinician group input noted there is a large unmet need in Canada for high-efficacy, well-tolerated therapies for NMOSD that have a significant impact on preventing and/or reducing attacks. Use of some off-label therapies is limited by many side effects, and many patients continue to have attacks despite treatment with drugs such as azathioprine and mycophenolate mofetil and, to a lesser extent, rituximab. Also, eculizumab is given by IV infusion every 2 weeks, which is too onerous for some patients to tolerate.
According to the clinician group, the main treatment goals include the use of an efficacious, safe, and tolerable therapy administered immediately after the first attack to ideally avoid all future relapses, reduce the severity of attacks and the cumulative disability associated with them, and minimize AEs related to therapies. In particular, there is a major unmet need among patients who have a breakthrough attack on their first therapy, as it can be challenging to identify a subsequent therapy that will be effective at preventing attacks and will be tolerated by the patient. The best approach for patients is to use a product after an attack that is as highly efficacious as possible so as to avoid potentially catastrophic subsequent attacks and, thus, optimize patient outcomes. The clinician group input noted that inebilizumab could be used as first-line treatment and as subsequent treatments for patients who have had breakthrough attacks on other therapies or who were intolerant of other therapies. Inebilizumab would be expected to be used as a monotherapy based on the available clinical evidence and to avoid cumulative immunosuppressive effects. The clinician group also noted that, although rituximab and inebilizumab both suppress B cells, there is some evidence that patients with polymorphisms in the FCGR3A gene may have an incomplete response to rituximab but not to inebilizumab. There is a lack of head-to-head data to compare inebilizumab with rituximab. The CNMSC also noted there is no clear preferred drug among the novel monoclonal antibodies for the treatment of NMOSD (e.g., eculizumab, inebilizumab, ravulizumab, and satralizumab) and that the best mechanism of action may vary by patient; however, there is generally limited access to these therapies in Canada at this time.
According to the CNMSC, the key outcome measure is a new attack, which is marked by new neurologic symptoms such as vision loss, weakness, sensory impairment, or dysfunction of the bladder or bowel. Although usually marked by a new enhancing lesion on MRI, this is not necessary to diagnose an attack. The clinician group indicated that the drug renewal process should consider the occurrence of any relapse in the previous year and the number of relapses, EDSS score or results of other disability measures, and any change from baseline (note that the EDSS is not validated in NMOSD). The CNMSC recommended discontinuing the drug if the patient has a new attack, a serious AE related to the therapy, or an EDSS score of 8 or higher.
The CNMSC stated that the treatment of patients with NMOSD should be assessed and managed by neurologists specialized in demyelinating diseases through an MS or demyelinating disease centre, and inebilizumab can be administered in a hospital or private clinic. Patients eligible for treatment with inebilizumab should have a confirmed diagnosis of NMOSD and a positive serum test for AQP4-IgG.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH Reimbursement Review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for inebilizumab:
relevant comparators
considerations for:
the initiation of therapy
continuation or renewal of therapy
discontinuation of therapy
prescribing of therapy
generalizability.
The clinical experts consulted by CADTH provided advice on the potential implementation issues raised by the drug program. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
One double-blind, randomized, placebo-controlled phase II and III study (N-MOmentum) was included in this review. The N-MOmentum trial consisted of a 197-day randomized controlled period (RCP) and a single-arm open-label period (OLP) that had a minimum duration of 2 years. N-MOmentum randomized 231 patients who were adults with NMOSD, had a documented history of either 1 or more acute NMOSD attacks in the prior year or 2 or more attacks in the prior 2 years that required rescue therapy, and had an EDSS score of 7.5 or less or 8.0 in special circumstances (i.e., if the investigator and medical monitor assessed that the patient was reasonably able to complete the study). The EDSS is a measure of disability validated for use in patients with MS that has been applied in NMOSD due to the similarities in disability caused by these distinct conditions and due to the lack of an NMOSD-specific tool for assessing disease-related disability. The EDSS ranges from 0 to 10, where 0 represents no disability, 9 represents a complete lack of independent mobility, and 10 represents death. The majority of patients were seropositive for AQP4-IgG, and subgroup data were available for the seropositive population. The primary end point was the time in days from day 1 to the onset of an NMOSD attack (determined by an adjudication committee [AC]) on or before day 197. Key secondary end points included the proportion of patients with a worsening in EDSS score from baseline to last visit during the RCP, change in binocular low-contrast visual acuity score from baseline to last visit during the RCP, and the number of NMOSD-related inpatient hospitalizations during the RCP. Other secondary or exploratory outcomes included the NMOSD attack rate in inebilizumab-treated patients, safety outcomes, and HRQoL, which was measured using the Short Form (36) Health Survey (SF-36). The binocular low-contrast visual acuity score was measured using a low-contrast Landolt C broken ring chart; the scoring of this assessment is based on the number of characters on the chart that the patient is able to identify, from 0 to 70 inclusive, where 70 indicates the patient was able to correctly identify all characters on the chart (i.e., best visual acuity score), and 0 indicates they were not able to identify any characters correctly (i.e., poorest visual acuity score). The SF-36 is a generic HRQoL questionnaire that yields a physical component score and a mental component score, in which higher scores represent better HRQoL.
At baseline, the patients included in the N-MOmentum trial were mostly female (> 90%), had received prior acute or maintenance therapies for NMOSD (> 98%), had disease that was seropositive for AQP4-IgG (> 93%), and had a mean age of approximately 43 years. The median EDSS score at baseline was 4 in the placebo group and 3.5 in the inebilizumab group (range, 0.0 to 8.0); 29% and 24% had an EDSS score of greater than 5 points at baseline, respectively.
Efficacy Results
NMOSD Attacks
Among the patients with AQP4-IgG–seropositive NMOSD in the randomized period, treatment with inebilizumab (versus placebo) was associated with a 77.3% reduction in the risk of an AC-determined NMOSD attack (Kaplan-Meier hazard ratio [HR] = 0.227; 95% confidence interval [CI], 0.1214 to 0.4232; P < 0.0001). During the RCP, a larger proportion of patients were attack-free in the inebilizumab group (87.6%) than in the placebo group (56.6%). Treatment with inebilizumab likely results in a clinically important increase in the probability of having no attack at day 197 compared with placebo.
The results were similar in the overall intention-to-treat (ITT) population and were also consistent across prespecified subgroups. Additionally, the results were similar based on investigator-determined NMOSD attacks. During the RCP among patients with seropositive NMOSD, major attacks occurred in 6 of 18 attacks (33.3%) in patients treated with inebilizumab and in 10 of 22 attacks (45.5%) among patients treated with placebo. Recovery from attacks was graded by the AC based on improvements in the attack criteria. As a proportion of patients with attacks, “no attack recovery” was reported for 27.8% of patients in the inebilizumab group and 40.9% of patients in the placebo group. Most attacks were myelitis (11 of 18 in the inebilizumab arm and 14 of 22 in the placebo arm), followed by optic neuritis (8 and 10, respectively), and few occurred in the brainstem (0 and 1, respectively). When calculated across the RCP and OLP, the annualized rate of AC-determined NMOSD attacks in any patient treated with inebilizumab was 0.086 attacks per year in the total population and 0.09 attacks per year in the population with AQP4-IgG–seropositive NMOSD.
Proportion of Patients with a Worsening in EDSS Score
During the RCP among patients with AQP4-IgG–seropositive NMOSD, treatment with inebilizumab likely resulted in a clinically important reduction in the proportion of patients who experienced a worsening from baseline in EDSS score compared with placebo at 197 days (odds ratio = 0.355; 95% CI, 0.1704 to 0.7252; P = 0.0047) ||| ||||||||||||| ||||||||||| |||||| ||||||| ||| ||||||| ||||||||. Results were similar in the overall ITT population.
Change From Baseline in Low-Contrast Visual Acuity Score
The change in binocular low-contrast visual acuity score from baseline to the last RCP visit did not appear to differ by treatment group within the population with AQP4-IgG–seropositive NMOSD ||| |||| ||||||||||| ||||||| ||| ||| ||||||| || |||||||||||||||. Results were similar in the overall ITT population. Based on these results, inebilizumab likely does not result in a clinically important difference in low-contrast visual acuity compared with placebo at 197 days.
Number of NMOSD-Related Inpatient Hospitalizations
|||||| ||| ||| ||||| ||| |||||||| |||||||||||| ||||||||||| ||| |||| ||||| || ||||||||||||| ||||||||| |||||||||||||||| ||| ||||| |||| ||| |||||| || ||||||||||||| ||| ||| ||||||||||||| |||||||| |||||||||| ||| ||||| |||||| ||| |||||| |||||| ||||||| |||| ||||||| || ||| ||||||| ||| ||||||||||| || ||| ||||||||| || ||| |||||||| |||||||| |||||||||| ||| |||||||||| |||||||||| ||||||| ||| ||||| ||| ||||||| ||||| ||| ||| |||||||| || ||| ||||| ||| || |||||||||||| || |||||| ||||||||||| || ||||||||||||| ||||||||| ||||||||||||||||| ||| ||| || || ||| |||||||| |||||||||| |||||| |||| ||||||| || ||||||||| ||| ||| ||||||| |||||. The clinical experts consulted by CADTH indicated that any benefit would be clinically meaningful. In summary, inebilizumab may be associated with a benefit in this outcome but it is uncertain at the time frame assessed.
Short Form (36) Health Survey
|||||| ||| ||| ||||| |||||||| |||||||||||| |||||||| ||||||| |||| |||||||||||| ||| |||||||| ||| |||| |||||| |||| |||||||| || ||| |||||| ||||||||| ||||| ||| ||||| ||||||||| ||||||||| ||||| |||||| ||| ||||| |||| |||||| || |||| ||| ||||||||||||| ||| |||| |||||| |||| |||||||| || ||| |||||||| ||||||||| ||||| ||| ||||| |||| |||||| ||| ||||| |||| |||||| || |||| ||| ||||||||||||. No statistical test results were reported, but it was reported that there were no numerical differences between treatment arms.
Pain Numerical Rating Scale
|||||| ||| |||| ||| |||| ||||||| |||| |||||||| || |||| || || ||| ||||||| |||| ||||| ||| ||| |||| ||||||||| |||| ||||||| |||||| ||||||||| |||||| ||| ||||||| ||| |||||||| |||||||||||| ||| ||||||| ||| |||||||||||| |||||||||| ||| ||||||| |||| |||||| ||| ||| |||| ||||||||| |||||||| |||||||||| |||||||| |||||| ||| ||| || ||| |||||||| |||||||||||| ||| ||||||| ||| |||||||||||| |||||||||| || ||||||||| |||||||||| |||||| ||| |||| ||||||||| |||| |||||||||||| |||||| ||||||| || || |||||||||| |||||||||| |||||| || |||| ||| |||| |||||||| || || ||||| |||||||| || ||||||||
Harms Results
Nearly all patients with AQP4-IgG–seropositive NMOSD experienced at least 1 AE during the study (87.2% of patients treated with placebo and 86.4% of patients treated with inebilizumab, respectively) and the results were similar in the ITT subpopulation. Among patients with AQP4-IgG–seropositive NMOSD, the rate of serious AEs (SAEs) was 4.3% in the inebilizumab group and 11.5% in the placebo group during the RCP. Over the entire duration of the study, 20.4% of patients who received any dose of inebilizumab experienced an SAE. Withdrawals due to AEs were uncommon: including both the RCP and OLP, withdrawals due to AE occurred in 1 patient (who had AQP4-IgG–seropositive disease) treated with placebo only, and in 4 patients who received inebilizumab (of which 3 had NMOSD that was AQP4-IgG seropositive, and 1 that was seronegative). There were no deaths during the RCP. During the entire study, among patients treated with any dose of inebilizumab, 3 patients died due to NMOSD, pneumonia, and COVID-19 pneumonia (1 case each).
In the RCP among patients with AQP4-IgG–seropositive NMOSD, 50.0% in the placebo group and 49.1% in the inebilizumab group experienced at least 1 treatment-emergent AE (TEAE) of special interest, most commonly infections (44.2% and 40.4%, respectively) followed by infusion-related reactions (9.6% and 9.3%, respectively), hepatic function abnormality (3.8% and 5.0%), and cytopenia (0% and 5.0%). Results were similar in the overall ITT population.
During the OLP, among patients with AQP4-IgG–seropositive NMOSD, most patients experienced at least 1 TEAE of special interest (85.1% of patients in the placebo-to-inebilizumab group and 71.4% in the inebilizumab-to-inebilizumab group). Similar to the RCP, the most common TEAE of special interest was infection, followed by infusion-related reaction, hepatic function abnormality, and then cytopenia. In addition, a few patients experienced hypersensitivity (0% and 1.3%, respectively) |||||||||| || ||||||||| ||||||||||| |||||||||| |||||||||||||||||||| || ||| |||||||||||||||||||||||||||| |||||| ||||||||||||||||| ||||||||| ||||| ||| || ||||||||||||. Again, results were similar in the overall ITT population. The infections that occurred were generally mild and did not lead to treatment discontinuation in the OLP or RCP. However, higher rates of infection were observed during the OLP (versus during the RCP), which may be related to the prolonged duration of treatment and follow-up. Cytopenias were more common in inebilizumab-treated patients, which is consistent with inebilizumab’s mechanism of action and the class effects of B-cell depletion.
Critical Appraisal
The N-MOmentum phase II and III trial was the only study included in this review. The N-MOmentum study included an initial period that was randomized, placebo-controlled, and double-blind, with a duration of up to 197 days (the RCP) in which patients received inebilizumab 300 mg IV or placebo IV on day 1 and day 15. Patients could proceed to an open-label, single-arm period (the OLP) with a minimum duration of 2 years, during which patients received inebilizumab 300 mg IV every 6 months starting 6 months after the first infusion. In the RCP, there were no major concerns with regard to internal validity related to the study design in terms of, for example, the method of randomization, concealment of allocation, maintenance of blinding, or balance of patient characteristics between treatment arms. As the trial was stopped early based on the recommendation of an independent data-monitoring committee, which found that the efficacy of inebilizumab had been established and, as there was no justification to keep exposing patients to placebo, there may be a risk of overestimating the true effect due to a slightly low information fraction (40 of 57 planned NMOSD attack events in the seropositive subpopulation). The end points in the trial were appropriately defined and were considered important to patients and clinicians, according to group inputs and clinical expert consultation. There was a high number of censored patients in the primary outcome of time to first NMOSD attack, especially in the inebilizumab treatment arm. However, this was considered unlikely to introduce bias because of the low number of early withdrawals and was considered likely to be due to the early cessation of the trial and the high proportion of attack-free patients at day 197. The key secondary outcome of change in EDSS score was an appropriate and important outcome, but the EDSS measure has some weaknesses, including over-reliance on ambulation as a metric of disability and a lower sensitivity to change in other types of disability at some ranges in the scale. EDSS is validated in MS but has not been validated in NMOSD; however, there are no superior scales for measuring disability in this population.
The eligibility criteria and baseline patient characteristics of the N-MOmentum trial were considered by the consulted clinical experts to be a reasonable approximation of patients with NMOSD in Canadian clinical practice, with the minor exception that some patients excluded for concomitant immunosuppressive or steroid therapy or prior IVIG would potentially be candidates for inebilizumab in real-world practice. All aspects of treatment management, including the steroid taper, rescue therapy, and preinfusion medications adequately reflected clinical practice, according to the clinical experts. The N-MOmentum trial had a high proportion of screen failures (236 screen failures of 467 patients screened) and only 5 of these were the result of the study’s early cessation. The clinical experts consulted by CADTH indicated that approximately one-third of the screen failures were due to tuberculosis testing because of the global nature of the study, which would be expected to be lower in Canadian clinical practice. As such, the CADTH team considered this not to be a major concern for generalizability.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) instrument was used to assess the certainty of the evidence for outcomes considered most relevant to inform the deliberations of the CADTH expert committee, and a final certainty rating was determined as outlined by the GRADE Working Group.24,25
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The selection of outcomes for the GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and the input received from the patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with members of the expert committee:
time to first NMOSD attack (assessed in GRADE as probability of no attack at day 168 and 197)
disability (proportion with a worsening in EDSS score)
low-contrast visual acuity (change from baseline to last visit)
number of NMOSD-related inpatient hospitalizations
HRQoL (SF-36 mean change from baseline)
pain numerical rating scale (NRS) (mean change from baseline)
proportion of patients with SAEs.
Long-Term Extension Studies
No long-term extension studies were submitted to CADTH.
Indirect Comparisons
Description of Studies
The sponsor-submitted indirect treatment comparisons (ITCs) included comparisons against satralizumab and eculizumab using published study data and matching-adjusted indirect comparison (MAIC) methodology to adjust for between-trial differences, and a comparison against rituximab using individual patient data (IPD). Other therapies (azathioprine and mycophenolate mofetil) were also of interest and were included in the study selection criteria, but ITCs against these therapies were ultimately not considered feasible. Additionally, there was a published network meta-analysis (NMA) comparing eculizumab with satralizumab and inebilizumab, which was summarized briefly for comparison but not formally assessed.28
Efficacy Results
In the sponsor-submitted anchored MAICs, the result for time to NMOSD attack was assessed by comparing inebilizumab with each of satralizumab monotherapy and eculizumab. The results of the indirect comparison between inebilizumab and satralizumab were inconclusive due to wide 95% CIs that cross the null value and suggest imprecision (HR = 0.666; 95% CI, 0.182 to 2.435). The risk of an NMOSD attack trended toward being higher with inebilizumab than with eculizumab (HR = 3.947; 95% CI, 0.917 to 17.0), which agreed with the published NMA in terms of the direction of effect.
The sponsor also submitted unanchored IPD analyses comparing inebilizumab with rituximab for the outcomes of annualized attack rate (AAR) and EDSS. For AAR, the results lacked 95% CIs and could not be interpreted due to missing critical information in the reporting of methodology and results. For EDSS, no relative effect estimates were reported, so the results could not be interpreted.
Table 2: Summary of Findings for Inebilizumab Versus Placebo for Patients With AQ4P-IgG+ NMOSD in the N-MOmentum Study
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | New drug | Difference | |||||
Time to AC-determined NMOSD attack | |||||||
Proportion of patients with no attack (by AC) during randomized controlled period Follow-up: Day 169 | 213 (1 RCT) | NR | 592 per 1,000 patients | 901 per 1,000 patients (95% CI, 840 to 939) | 309 more per 1,000 patients (95% CI, 162 to 455 more) | Moderatea | Inebilizumab likely results in a clinically important increase in the probability of having no attack at day 169 compared with placebo |
Proportion of patients with no attack (by AC) during randomized controlled period Follow-up: Day 197 | 213 (1 RCT) | KM HR for time to first attack = 0.227 (95% CI, 0.1214 to 0.4232) | 566 per 1,000 patients | 876 per 1,000 patients (95% CI, 810 to 920) | 310 more per 1,000 patients (95% CI, 158 to 461 more) | Moderatea | Inebilizumab likely results in a clinically important increase in the probability of having no attack at day 197 compared with placebo |
Worsening in EDSS score | |||||||
Proportion with worsening from baseline in EDSS Follow-up: Last visit (up to day 197) | 213 (1 RCT) | OR = 0.355 (0.1704 to 7,252) | 346 per 1,000 patients | 149 per 1,000 patients (95% CI, 94 to 204) | 197 fewer per 1,000 patients (|||||| ||| ||| ||||| || || |||||) | Moderateb | Inebilizumab likely results in a clinically important reduction in the proportion of patients who have worsening from baseline in EDSS compared with placebo at 197 days |
Visual acuity | |||||||
Change from baseline in low-contrast visual acuity score Follow-up: Last visit (up to day 197) | 213 (1 RCT) | NR | Observed mean = 0.846 (SE = 1.405) LS mean = 0.600 (SE = 0.999) | Observed mean, 0.481 (SE = 0.486) LS mean, 0.562 (SE = 0.572) | LS mean difference, −0.038 (95% CI, −2.3122 to 2.2357) | Lowc | Inebilizumab may not result in a clinically important difference in low-contrast visual acuity compared with placebo at 197 days |
Number of NMOSD-related inpatient hospitalizations | |||||||
Number of patients with NMOSD-related inpatient hospitalizations Follow-up: 197 days | 213 (1 RCT) | RR = 0.291 (0.1054 to 0.8017) | 7 of 52 patients with a mean of 1.4 events (SD = 0.8) and median of 1 event (range, 1 to 3) | 9 of 161 patients with a mean of 1.0 events (SD = 0.0) and median of 1 (range, 1 to 1) event | 0.37 fewer hospitalizations (||||| ||| |||| ||||| || |||| ||||) | Lowd | Inebilizumab may result in a reduction in NMOSD-related inpatient hospitalizations compared with placebo over 197 days |
HRQoL | |||||||
SF-36 mean change from baseline Follow-up: Week 28 | 133 (1 RCT) | NR | Mental CS = 3.303 (SD = 9.372) Physical CS = 0.364 (SD = 6.632) | Mental CS = 1.719 (SD = 8.057) Physical CS = 0.710 (SD = 7.421) | NR | NAe | The effect of inebilizumab on HRQoL cannot be determined |
Pain NRS | |||||||
Pain NRS mean change from baseline Follow-up: Week 28 | 213 (1 RCT) | NR | Observed mean = 0.514 (SE = 0.304) LS mean, 0.567 (SE = 0.229) | Observed mean = 0.296 (SE = 0.119) LS mean, 0.279 (SE = 0.130) | LS mean difference = −0.288 (95% CI, −0.8080 to 0.2318) | Moderatef | Inebilizumab likely results in no clinically meaningful difference in the change in pain NRS from baseline compared with placebo at 28 weeks |
Harms | |||||||
Proportion of patients with SAEs during the randomized period | 213 (1 RCT) | NR | 115 per 1,000 patients (NR) | 43 per 1,000 patients (NR) | 72 fewer per 1,000 patients (NR) | Moderateg | Inebilizumab likely results in a lower proportion of patients with SAEs at 197 days compared with placebo; there is some uncertainty about the clinical importance of the estimates |
AC = adjudication committee; AQP4-IgG+ = anti-aquaporin-4 immunoglobulin G seropositive; CI = confidence interval; CS = component score; EDSS = Expanded Disability Status Scale; HR = hazard ratio; HRQoL = health-related quality of life; KM = Kaplan-Meier; LS = least squares; MID = minimal important difference; NA = not applicable; NMOSD = neuromyelitis optica spectrum disorder; NR = not reported; NRS = numerical rating scale; OR = odds ratio; RCT = randomized controlled trial; RR = rate ratio; SAE = serious adverse event; SD = standard deviation; SE = standard error; SF-36 = Short Form (36) Health Survey.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level due to early cessation of the trial. Relatedly, there was a high degree of censoring that was imbalanced between treatment arms. However, the reasons for censoring did not appear concerning, so the CADTH review team judged that it was not likely to present an additional serious concern for bias; ergo, it was not rated down a second time. No threshold of importance could be determined, and the experts consulted by CADTH indicated that any benefit is clinically important, so potential benefits and harms were determined relative to the null value.
bNo minimally important between-arm difference could be established, so the optimal information size approach was used to rate down 1 level in imprecision.
cRated down 2 levels for very serious imprecision because the CI included both potential benefit and potential harm. No MID was identified so potential benefits and harms were determined relative to the null value.
dRated down 1 level for serious imprecision because the CI included both potential benefit and potential harm. No threshold for clinically important differences was identified and the experts consulted by CADTH indicated that any benefit is clinically important, so potential benefits and harms were determined relative to the null value. The time frame of the study may be inadequate to determine a clinically meaningful difference in hospitalizations over a patient’s life, so the certainty was rated down 1 additional level for indirectness.
eNo 95% CIs or between-group differences were reported for SF-36.
fRated down 1 level for serious imprecision because the CI included both potential benefit and potential harm. No minimally important difference was established so potential benefits and harms were determined relative to the null value. Note that this outcome was not controlled for multiple comparisons.
gRated down 1 level for serious concerns regarding imprecision; no 95% CI of the difference was available, so the optimal information size approach was used to judge imprecision. No minimally important threshold of difference was established, but the CADTH review team judged that the effect estimate might include an important between-group difference.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence and from the sponsor’s responses to additional information requested by CADTH.26,27
Harms Results
No harms outcomes were assessed in the ITCs.
Critical Appraisal
In the MAICs comparing inebilizumab with satralizumab monotherapy and eculizumab, there was unresolved between-trial heterogeneity with respect to patient populations and outcome definitions that were not mitigated by the MAIC methodology. Additionally, the factors selected for adjustment were not informed by clinical expert opinion or the literature regarding important treatment-effect modifiers in NMOSD. The MAICs may have been over-adjusted for clinically unimportant factors that were selected in an inappropriate manner, without consulting the literature or a clinical expert, and were based only on a statistical analysis of the N-MOmentum trial. The results of both comparisons had wide 95% CIs, suggesting substantial imprecision in the effect estimates, as well as small effective sample sizes, which results in the effects being overly influenced by small subgroups of patients and highlights poor overlap.
The submitted methodology for the MAIC analyses of inebilizumab versus rituximab is insufficient for critical appraisal. The 4 studies informing the rituximab data were small observational studies (N ≤ 32) in geographic regions likely to differ from Canadian demographics and clinical practice. The sponsor noted it was not feasible to conduct ITCs comparing inebilizumab with other off-label treatments such as azathioprine and mycophenolate mofetil due to small sample sizes and the use of observational study designs. However, the same limitations exist for the available rituximab data, and the use of the MAIC methodology cannot correct for these limitations. Despite having access to the IPD for the rituximab studies, the sponsor-submitted MAICs only adjusted the N-MOmentum trial data to reflect the rituximab-treated populations, which are less similar to the population of patients in Canada. Additionally, no relative effect estimates were reported for EDSS, and no 95% CIs were reported for AAR. There was no justification provided for why the sponsor selected single-arm observational studies to inform the ITCs instead of using an available published, placebo-controlled, double-blind, randomized trial comparing rituximab with placebo in seropositive NMOSD.29 No conclusions can be drawn from the indirect comparisons with rituximab.
In all of the submitted MAICs, the submitted technical reports were missing critical details of the methods and results, which limited our ability to appraise the evidence and raised concerns about the validity of the analyses.
No safety-related outcomes were assessed.
There are no direct or indirect data available for the efficacy and safety of inebilizumab compared with azathioprine, mycophenolate mofetil, or ravulizumab.
Studies Addressing Gaps in the Evidence From the Systematic Review
No additional studies were submitted to CADTH.
Conclusions
NMOSD is a rare, disabling, and life-threatening inflammatory disorder of the CNS characterized by acute attacks with a relapsing pattern that cause potentially irreversible damage to the optic nerve and spinal cord. There is a large unmet need for high-efficacy, well-tolerated therapies for NMOSD that have a significant impact on preventing and/or reducing attacks. Patients and clinicians highlighted that the main treatment goals include the use of an efficacious, safe, and tolerable therapy administered as soon as possible after the first attack to avoid all relapses, reduce the severity of attacks and the cumulative disability associated with them, and minimize AEs related to therapies. One phase II and III, double-blind, randomized, placebo-controlled study (N-MOmentum) was included in this review, which had a duration of 197 days followed by an open-label, single-arm period of at least 2 years.
The data that were submitted to CADTH and the end points assessed in the study were considered clinically relevant for the treatment of patients with NMOSD. The results from the randomized period of the N-MOmentum trial demonstrated that treatment with inebilizumab likely results in a clinically meaningful benefit in time to first relapse and the proportion of patients with a worsening in EDSS score compared with placebo at 197 days. Other secondary or exploratory outcomes were less conclusive and did not show a clearly meaningful benefit of inebilizumab over placebo, including change in low-contrast visual acuity score, NMOSD-related inpatient hospitalizations, HRQoL, and pain. The reasons for this incongruence are uncertain, but it may be that the duration of the randomized study was insufficient to detect clinically meaningful differences in these outcomes.
The ITCs included MAICs comparing inebilizumab with satralizumab and eculizumab, and IPD analyses comparing inebilizumab with rituximab. Results were inconclusive for the comparison with satralizumab. The ITCs suggest the risk of an NMOSD attack is higher with inebilizumab treatment than with eculizumab treatment, but the magnitude of benefit is uncertain due to wide 95% CIs, imprecision, and unresolved between-trial heterogeneity. The comparison with rituximab could not be interpreted due to limitations in the analysis inherent to the data available, as well as sparsely reported methodology and results and inappropriate methodological decisions. There are no direct or indirect data available for the efficacy and safety of inebilizumab compared with azathioprine, mycophenolate mofetil, or ravulizumab.
Relative to previous studies in the clinical development program for inebilizumab, no new safety signals or concerns with the drug were identified in either study period, and long-term inebilizumab treatment was well tolerated after a median exposure of more than 3.2 years during the study. Almost all patients experienced at least 1 AE during the study regardless of treatment assignment, but the results suggested that patients treated with inebilizumab may have fewer SAEs than patients treated with placebo at 197 days. Cytopenias were more common in patients treated with inebilizumab compared with placebo, which is consistent with inebilizumab’s mechanism of action and the class effects of B-cell depletion.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of inebilizumab 300 mg IV in the treatment of adults with NMOSD in who are seropositive for autoantibodies against AQP4-IgG.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
NMOSD is a rare, life-threatening, and chronic inflammatory disorder of the CNS that is characterized by acute attacks or relapses that cause potentially irreversible damage to the optic nerve and spinal cord. A defining feature of NMOSD is the presence of pathogenic serum autoantibodies against AQP4, which is detected in about 80% to 90% of NMOSD patients and which led to its differentiation from MS.2-4 The clinical presentation typically involves inflammation of the optic nerve (optic neuritis) and/or spinal cord (transverse myelitis), but some cases may involve diencephalic, brainstem, and cerebral hemisphere attacks.1 The consequences of NMOSD extend beyond clinical settings and include physical, functional, and psychological effects, causing loss of independence, reduced ability to work or perform activities of daily living, reduced sexual function, and reduced emotional well-being due to impaired HRQoL.9,14-16
NMOSD attacks are unpredictable, lasting from days to weeks, and recovery from attacks is often incomplete, leading to an accumulation of morbidity, including permanent visual and motor disabilities such as blindness and/or paralysis.5-9 Even a single attack can be so severe that, despite rescue treatment, patients may lose their ability to walk without assistance or become functionally blind in at least 1 eye. For example, almost 80% of patients will experience legal blindness (visual acuity ≤ 0.1) during their first NMOSD attack.6,7 Unilateral or bilateral optic neuritis, including central vision loss with ocular pain, is often the initial event of relapsing NMOSD. Clinical manifestations of myelitis may include limb weakness, quadriplegia or paraplegia, sensory loss, bladder dysfunction, spasms, and pain.13 Brainstem involvement may manifest with nausea, vomiting, hiccups, vertigo, hearing loss, facial weakness, trigeminal neuralgia, diplopia, ptosis, or nystagmus; myelitis that extends into the brainstem may cause respiratory failure and death.3,11,13 Neurologic deficits in patients with NMOSD accumulate after each inflammatory attack;30,31 76% of patients have incomplete recovery after their first relapse, and 100% of patients have incomplete recovery after their sixth relapse.7,32 At least 90% of NMOSD patients suffer from recurrent attacks,33 and 94% of patients experienced 3 to 4 attacks over a median period of 5 years, according to 1 study.34,35 As a result, prevention of relapse is the key goal of therapy and paramount in the overall management of patients with NMOSD to avoid increasing neurodegeneration and functional disability.3-5
Estimated mortality rates due to NMOSD have varied considerably, from 7% to 32%, depending on age, relapse rate, and recovery from attacks.31,36,37 A retrospective US-based study estimated an overall mortality rate of 7% and an annual mortality rate of 0.68 deaths per 100 patient-years for patients with NMOSD, with a mean disease duration of 6.9 years at the time of death.36 In a study of 63 patients with late-onset NMOSD (≥ 50 years of age) from France, Germany, Turkey, and the UK, with a mean follow-up period of 4.6 years, the annual mortality rate was 2.8% and the 5-year survival rate was 86.7%; older age at onset and the AAR have independently predicted death.37
NMOSD disproportionately affects females, with a reported female-to-male ratio of 9:1 to 12:1 in patients with AQP4-IgG–seropositive NMOSD.5,17 No Canadian epidemiological studies specific to NMOSD are currently available, although systematic reviews based on data from several countries have estimated 0.053 to 0.4 incident cases per 100,000 people and 0.51 to 4.4 prevalent cases per 100,000 people.18,19
In Canada, NMOSD is diagnosed using the criteria developed by the International Panel for Neuromyelitis Optica Diagnosis5 in patients with AQP4-IgG–seropositive disease and 1 or more of 6 core clinical characteristics (i.e., optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical MRI lesions, or symptomatic cerebral syndrome with NMOSD-typical brain lesions).5
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
Currently, there is no cure for NMOSD. The goals of treatment for NMOSD relate to 3 broad areas: the prevention of relapses, the treatment of relapses, and the treatment of residual symptoms following an episode. The focus of this review is on the prevention of relapses based on the indication and place in therapy for inebilizumab. According to the clinician group input submitted to CADTH, relapse prevention is an important goal due to the permanent, life-altering disability that often arises from an attack despite acute management of attacks. Secondary goals of preventive therapy include reducing the severity of attacks, thereby reducing cumulative disability associated with attacks, and minimizing the AEs associated with therapy.
Current preventive treatment of NMOSD in Canada involves off-label therapies, including corticosteroids, azathioprine, mycophenolate mofetil, and rituximab, although public funding of these therapies varies by province and the clinical data for these treatments in NMOSD are absent or limited. Rituximab may exert its therapeutic effect on patients with NMOSD through B-cell–mediated humoral immunity,38,39 and was shown to be superior to azathioprine for NMOSD in 1 open-label RCT38 and superior to placebo in 1 Japanese double-blind RCT.29 Azathioprine is a purine analogue that interferes with the DNA synthesis of rapidly proliferating cells and has been widely used as a first-line immunosuppressant medication for autoimmune diseases.40 Azathioprine was first studied in 1998 in patients with NMOSD, where it was found to have a benefit in reducing disability,40 but gastrointestinal and hematological side effects are associated with its use. Mycophenolate mofetil was developed to be a specific immunosuppressant drug with limited side effects by targeting guanosine more than adenosine and is used widely for the treatment of autoimmune diseases and NMOSD, with more adverse effects than azathioprine.40,41
Two biologics, eculizumab and satralizumab, were recently approved in Canada for the treatment of patients with NMOSD who are AQP4-IgG seropositive. Eculizumab was the first drug approved in Canada for the prevention of NMOSD relapses based on data from a phase III RCT in adults with AQP4-IgG–positive NMOSD.42 In August 2020, the CADTH Canadian Drug Expert Committee (CDEC) recommended that eculizumab be reimbursed for the treatment of adult patients with AQP4-IgG–positive NMOSD if specific clinical and pricing criteria were met.43 However, negotiations with the pan-Canadian Pharmaceutical Alliance concluded without agreement, so it is assumed that eculizumab is not a benefit on any public plans across Canada. In April 2021, CDEC recommended that satralizumab be reimbursed for the treatment of AQP4-IgG–seropositive NMOSD in patients aged 12 years and older who have had an inadequate response to at least 1 other preventive treatment, provided specific clinical and pricing criteria have been met, but public coverage of satralizumab varies by province.44 The input received from patient groups indicated that financial barriers to access were a major concern among patients with NMOSD. Additionally, the clinical experts consulted by CADTH highlighted that reliance on private insurance is difficult in a population with high levels of unpredictable and accumulating disability that may prevent them from working as a result of their NMOSD. In addition to financial barriers to access, the patient and clinician group input and the clinical experts consulted by CADTH noted that the administration schedule of eculizumab requires infusions every 2 weeks, which is a practical barrier that may disproportionately affect patients living in rural areas, patients with disabilities as a result of NMOSD-related motor or visual difficulties who therefore cannot drive or use transit independently, and patients who otherwise have limited access to specialist treatment facilities.
Another monoclonal antibody, ravulizumab (Ultomiris), is also under review by Health Canada and CADTH for preventive treatment in adult patients with AQP4-IgG–seropositive NMOSD.
Drug Under Review
Key characteristics of inebilizumab and other treatments available for NMOSD are summarized in Table 3.
Inebilizumab is a humanized afucosylated monoclonal antibody treatment for NMOSD.20-22 The precise mechanism by which inebilizumab exerts its therapeutic effects in NMOSD is unknown but is presumed to involve binding to CD19 on pre-B and mature B lymphocytes, which leads to antibody-dependent cellular cytolysis.23 By depleting circulating CD19-positive B cells as well as AQP4-IgG–producing plasmablasts and plasma cells, inebilizumab acts upstream in the disease process by downregulating the production of AQP4-IgG, which represents a new mechanism to target the pathways that contribute to the pathogenesis of NMOSD.2
Inebilizumab has a Health Canada indication as monotherapy for the treatment of adult patients with NMOSD who are AQP4-IgG seropositive.23 The reimbursement request is as per the approved indication. Treatment should be administered under the supervision of a qualified health care professional. The recommended dosage for inebilizumab is an initial 300 mg dose via IV infusion followed 2 weeks later by a second 300 mg dose via IV infusion; subsequent doses (starting 6 months from the first infusion) are administered as single 300 mg doses via IV infusion every 6 months.23 The prepared infusion solution should be at room temperature before starting the infusion. Inebilizumab should be administered under the close supervision of an experienced health care professional with access to appropriate medical support to manage potential severe reactions, such as serious infusion reactions.23
Although there are several comparators listed in Table 3, there are important caveats for each. Ravulizumab is not yet approved by Health Canada. The clinical trials of satralizumab and eculizumab were concurrent with the clinical trial for inebilizumab, which precluded using either therapy as an active control. Rituximab, azathioprine, and mycophenolate mofetil are the only listed comparators that were marketed in Canada at the time of the initiation of the inebilizumab phase III clinical trials, but each is used off-label, without a Health Canada–approved indication in NMOSD and with little to no robust clinical data supporting their use in NMOSD; additionally, based on the clinical expert input, azathioprine and mycophenolate mofetil are not considered highly effective therapies in NMOSD.
Table 3: Key Characteristics of Inebilizumab, Ravulizumab, Satralizumab, Eculizumab, Rituximab, Azathioprine, and MMF
Characteristic | Inebilizumab (Uplizna) | Ravulizumab (Ultomiris) | Satralizumab (Enspryng) | Eculizumab (Soliris) | Rituximab (Rituxan) | Azathioprine (Imuran) | MMF (Myfortic, CellCept) |
|---|---|---|---|---|---|---|---|
Mechanism of action | Monoclonal antibody that specifically binds to CD19 on B lymphocytes. | Monoclonal antibody that specifically binds to the human terminal complement protein C5. | Monoclonal antibody that blocks interleukin 6 receptor. | Monoclonal antibody that specifically binds to the complement protein C5. | Monoclonal antibody that specifically binds to the transmembrane antigen CD20. | Immunosuppressant | Immuno-suppressant |
Indicationa | As monotherapy for the treatment of adults with NMOSD who are AQP4-IgG seropositive. | For the treatment of adults with AQP4-positive NMOSD. | As monotherapy or in combination with IST for the treatment of NMOSD in adults and adolescents who are anti-AQP4 seropositive. | For the treatment of NMOSD in adults who are anti-AQP4 antibody positive. | No Health Canada indication for the treatment of NMOSD. | No Health Canada indication for the treatment of NMOSD. | No Health Canada indication for the treatment of NMOSD. |
Route of administration | IV | IV | SC | IV | IV | Oral | Oral, IV |
Recommended dose | An initial 300 mg dose via IV infusion, followed 2 weeks later by a second 300 mg dose via IV infusion; subsequent doses (starting 6 months from the first infusion) are administered as single 300 mg doses via IV infusion every 6 months. | The recommended ravulizumab IV maintenance dose in adult patients (≥ 18 years) with NMOSD with a body weight ≥ 40 kg is based on the patient’s body weight. Maintenance doses are administered every 8 weeks, starting 2 weeks after the loading dose:
| 120 mg at weeks 0, 2, and 4 for the first 3 administrations, followed by a maintenance dose of 120 mg every 4 weeks. | Eculizumab should be administered by IV infusion. The therapy consists of 900 mg weekly for the first 4 weeks, followed by 1,200 mg for the fifth dose 1 week later, then 1,200 mg every 2 weeks thereafter. | RA protocol Premedication: An analgesic or antipyretic drug, antihistaminic drug, and glucocorticoids should always be administered before each infusion of rituximab. The recommended dosage of rituximab is a 1,000 mg IV infusion followed 2 weeks later by a second 1,000 mg IV infusion. Re-treatment in Patients with RA: The need for further courses should be evaluated 24 weeks following the previous course with re-treatment. Patients may receive further courses no sooner than 16 weeks following the previous course. | RA protocol: The initial dose should be approximately 1.0 mg/kg (50 mg to 100 mg) given as a single dose or on a twice-daily schedule. The dose may be increased, beginning at 6 to 8 weeks and thereafter by steps at 4-week intervals. Dose increments should be 0.5 mg/kg daily up to a maximum dose of 2.5 mg/kg/day. Maintenance therapy should be at the lowest effective dose, and the dose given can be lowered, with decremental changes of 0.5 mg/kg or approximately 25 mg daily every 4 weeks, while other therapy is kept constant. The optimum duration of maintenance azathioprine has not been determined. | Myfortic: 720 mg administered orally twice daily, for a daily dose of 1.440 g. CellCept: 1 g to 3 g daily, administered orally or via IV twice a day. |
Serious adverse effects or safety issues | Infusion reactions, infections, reduction in immunoglobulins, fetal risk (based on animal data). | Serious meningococcal infections. | Infections; monitor liver enzymes and neutrophils. | Serious or fatal meningococcal infections. | Infusion reactions, progressive multifocal leuko-encephalopathy, hepatitis B virus, mucocutaneous reactions, infections, cardiovascular events. | Leukopenia, thrombocytopenia, macrophage activation syndrome, infection, carcinogenicity, hepatoxicity, fetal harm. | Infection, lymphoma, fetal harm. |
AQP4 = aquaporin-4; AQP4-IgG = anti-aquaporin-4 immunoglobulin G; IST = immunosuppressive therapy; MMF = mycophenolate mofetil; NMOSD = neuromyelitis optica spectrum disorder; RA = rheumatoid arthritis; SC = subcutaneous.
aHealth Canada–approved indication.
Sources: Product monographs for inebilizumab (Uplizna),23 ravulizumab (Ultomiris),45 satralizumab (Enspryng),46 Eculizumab (Soliris),47 rituximab (Rituxan),48 azathioprine (Imuran),49 and MMF (Myfortic50 and CellCept51).
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient inputs received by CADTH have been included in the stakeholder section of this report.
Two patient groups, MS Canada and TSF, responded to CADTH’s call for patient input for the current review of inebilizumab as a monotherapy for the treatment of adult patients with NMOSD who are AQP4-IgG seropositive.
MS Canada gathered information for this submission through a survey that was conducted from August 4 to 14, 2023, that targeted Canadians living with NMOSD and their caregivers. The MS survey included data from 13 respondents; most of them were female (83%) and ranged in age from 25 years to older than 65 years.
TSF gathered information through 2 online surveys, 1 for patients and 1 for caregivers, between July 13 and 31, 2023. Additionally, TSF gathered information through videoconference interviews with 4 patients and 1 caregiver on August 8, 2023; 1 interviewee had direct experience with inebilizumab. The TSF survey included data from 51 patients and 9 caregivers from around the world, including Canada. Around 54% of the respondents were between 35 and 54 years of age, approximately 32% were between 55 and 74 years of age, and 25% of the patients were between the ages of 25 and 34 years.
The 2 patient groups indicated that NMOSD is a rare autoimmune syndrome of the CNS characterized by a relapsing-remitting disease course. NMOSD attacks are also called relapses, flare-ups, or exacerbations, and may include optic neuritis (affecting eye function), transverse myelitis (affecting limb function), and/or area postrema syndrome (episodes of otherwise unexplained hiccups or nausea and vomiting). Attacks may result in permanent neurologic damage and disability. Symptoms may include loss of vision, numbness, mobility issues, chronic pain, muscle cramps or painful spasms, fatigue, bowel and bladder dysfunction, and difficulty falling and staying asleep. The disease has a tremendous impact on all aspects of patients’ and caregivers’ lives, including a negative effect on their quality of life, their independence and employment, and on their social, family, and school life. Patients with NMOSD have a substantially elevated risk of mortality compared with the general population, and NMOSD-related deaths are typically attributed to respiratory failure following an attack. The patient inputs estimated that within the first 5 years of the disease, approximately 30% of NMOSD patients die and approximately 50% become either blind and/or require the use of a wheelchair.
The patient inputs stated that the standard treatment for NMOSD may involve IV steroids, IVIG, or plasmapheresis or plasma exchange. Off-label immunosuppressants may be used to help prevent further attacks, with varying levels of therapeutic benefit. Recently, 2 medications were approved by Health Canada with indications for the treatment of adults with AQP4-IgG–seropositive NMOSD (eculizumab and satralizumab), but access to these medications is limited. Symptoms such as neuropathy, pain, stiffness, muscle spasms, and bladder and bowel control problems can be managed with various medications and therapies. Many patients suffer significant additional attacks and additional disability while cycling through different therapies, and may experience challenging side effects of treatment.
Some respondents reported treatment experience with eculizumab, satralizumab, or rituximab, and felt these medications were effective or somewhat effective in managing their NMOSD. There were numerous barriers to access associated with these therapies, including out-of-pocket costs, lack of insurance coverage and, for eculizumab, scheduling time-consuming infusions too often and planning life around these appointments. According to the patient input groups, there is an unmet need for accessible therapy options that are able to reduce the risk of future attacks, maintain the current level of physical ability, and slow disease progression.
One respondent from the MS survey and the 5 patients and 2 caregivers who shared their experience using inebilizumab in the TSF input indicated that using inebilizumab has effectively reduced attacks, improved their quality of life and, because of the drug’s easy-to-manage schedule and manageable side effects, they could be involved with their family, friends, and workplace.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of NMOSD.
Unmet Needs
The clinical experts indicated that treatment of NMOSD includes a series of treatment goals relating to 3 broad areas: prevention of relapses (disease modification), treatment of relapses, and treatment of residual symptoms. Although ideal, it is unlikely that any single treatment would cover all 3 areas. Of these 3 areas, preventive treatment was described as being of special interest because relapses are the major source of disability accumulation for people with NMOSD. As a result, preventive treatment is expected to result in downstream desirable effects, including better HRQoL, increased ability to maintain independence and employment, and a reduced reliance on caregivers. The clinical experts highlighted that it is important to control progression as early as possible because damage leading to neurologic disability, including paralysis and blindness, may be irreversible after an attack.
The clinical experts agreed that most patients with NMOSD still have relapses despite their current treatment regimens, and current first-line therapies (e.g., azathioprine and mycophenolate mofetil) are not considered particularly effective in preventing NMOSD attacks. Additionally, these therapies are also associated with significant adverse effects, especially if used in conjunction with corticosteroids. The greatest unmet need in the treatment of NMOSD is for therapies that prevent relapses more effectively without intolerable side effects.
Place in Therapy
The clinical experts agreed that current clinical practice for treatment and attack prevention in NMOSD is suboptimal and inconsistent due to the low efficacy of the off-label treatments used in the first-line setting and barriers to treatment access, which vary by province. The newer therapies with indications for NMOSD, such as satralizumab and eculizumab, present especially significant barriers to access for most patients due to inconsistent coverage or the lack of public reimbursement, and the onerous dosing schedule of eculizumab. Due to the impact of NMOSD on a person’s ability to maintain employment, patients with NMOSD are more likely to lack private insurance.
As the vast majority of patients with NMOSD will relapse and relapses may lead to permanent disability or death, all people with NMOSD should be on relapse-preventive treatment.
The experts agreed that because the impact of relapses on people with NMOSD is devastating and preventing as many relapses as possible is critical to the prevention of significant disability, inebilizumab should be available as first- and later-line treatment for patients diagnosed with NMOSD who are AQP4-IgG seropositive. The experts indicated that inebilizumab would be used as a monotherapy.
Patient Population
NMOSD is a rare disease. The clinical experts indicated that patients diagnosed with NMOSD who are seropositive for AQP4-IgG should be candidates for treatment with inebilizumab. It is the standard of care in Canada to assess patients with NMOSD for AQP4-IgG; the experts noted there are no major challenges in this regard in Canada, other than that the test results may be delayed in some locations. The specificity of AQP4-IgG is very high, so the risk of misdiagnosis is very low.
The patient eligibility criteria in the pivotal study, N-MOmentum, were considered by the experts to be broadly representative of patients with NMOSD in Canada, with the exception that the study excluded patients with recent steroidal treatment. Although appropriate from the perspective of a clinical trial design, in real-world practice there are many comorbidities that may require steroid treatment, and these patients should not necessarily be excluded from treatment with inebilizumab; such a decision would need to be considered by the expert clinician managing a patient’s particular case. Patients who have received IVIG or have concomitant diseases should also not necessarily be excluded from receiving inebilizumab in the real world.
Within the population of patients who have NMOSD and are AQP4-IgG seropositive, it is unknown which patients are more likely to benefit from inebilizumab.
It is possible that patients who are seronegative for AQP4-IgG may also benefit from inebilizumab. Fulfillment of seronegative NMOSD criteria would be necessary to establish the diagnosis to allow appropriate access.
Assessing the Response to Treatment
The experts indicated that a clinically meaningful response to treatment relates to the reduction of the relapse rate and prolongation of times to relapse. Although the absence of relapse is indicative of a clinically meaningful response, this may not be realistic, as the number and severity of relapses that patients experience differ on an individual level (e.g., some patients may have several relapses per year); thus, a reduction in the number of relapses is still a reasonable goal. The determination of relapses is fairly objective; however, it is not the only factor, and assessment of treatment response is based on a combination of patient-reported symptoms, a clinical exam, clinical tools, and patient history. Other important outcome measures include evaluation of attack severity and degree of recovery from attack, as well as accumulation of disability.
The experts noted there is a lack of formal guidance on how to assess treatment response, but it would be reasonable to assess initial treatment response 3 months after the initial infusion then every 6 months until stability is achieved, and then every year for patients with stable NMOSD. However, it was noted that within the first 6 months of treatment, the attack rate may still be higher than when stability is achieved.
MRIs are not routinely conducted for patients with NMOSD outside of initial diagnosis and so would not be used in assessing the response to treatment.
Discontinuing Treatment
Discontinuation of treatment should occur if the patient is completely dependent and unable to leave their bed (EDSS score of 9.0 or above).
Discontinuation of treatment should be considered on a case-by-case basis in the event of a severe relapse (e.g., requiring intubation and support on a ventilator) or if the patient has experienced 2 or more relapses within 2 years, has severe or unacceptable AEs, or has contraindications for therapy.
Prescribing Considerations
The clinical experts agreed that treatment should be supervised by a neurologist with expertise in this area. Although NMOSD and MS are not the same disease, the populations and medications are similar and patients with NMOSD are often cared for in MS clinics. The clinical experts stated that neurologists with experience or expertise in related subspecialties should prescribe inebilizumab, including clinicians with experience in MS neurology, and/or neurologists working in an MS clinic or in neuroimmunology, autoimmune neurology, and/or neuro-ophthalmology. However, patients in remote areas may have issues with access to subspecialists. For patients living in remote areas, local neurologists without subspecialty expertise could work by distance in conjunction with neurologists who are experts in a relevant subspecialty.
Inebilizumab is expected to be used as a monotherapy and not combined with other monoclonal antibodies indicated for the treatment of NMOSD. However, there may be situations in which it is combined with classical immunosuppressants. There is a lack of data regarding combination therapies.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the stakeholder section of this report.
One clinician group, the CNMSC (input authored by 1 clinician), responded to CADTH’s call for clinician group input.
Clinician perspectives from the CNMSC were obtained through clinical experience, knowledge of the medical literature, and from clinicians across the country who specialize in this therapeutic area.
According to the clinician group, a variety of off-label therapies are used for the treatment of NMOSD in Canada, including corticosteroids, azathioprine, mycophenolate mofetil, and rituximab. However, breakthrough NMOSD attacks are reported for all of these drugs, and government drug program funding varies by province and territory. Recently, 2 monoclonal antibodies, eculizumab and satralizumab, were approved by Health Canada. However, access to these therapies is extremely limited due to their high cost and stringent funding criteria. All of the therapies in use for NMOSD work by suppressing the immune system to prevent attacks, with variable efficacy. Failure of treatment, resulting in even just 1 relapse, can lead to a profound, permanent disability, including blindness and paralysis.
As per the CNMSC, there is a large unmet need in Canada for high-efficacy, well-tolerated therapies for NMOSD that have a significant impact on preventing and/or reducing attacks. Use of some off-label therapies is limited by many side effects, and many patients continue to have attacks despite treatment with drugs such as azathioprine and mycophenolate mofetil and, to a lesser extent, rituximab. Also, eculizumab is given by IV infusion every 2 weeks, which is too onerous for some patients to tolerate.
According to the clinician group, the main treatment goals include the use of an efficacious, safe, and tolerable therapy administered immediately after the first attack to ideally avoid all future relapses, reduce the severity of attacks and the cumulative disability associated with them, and minimize AEs related to therapies. In particular, there is a major unmet need among patients who have a breakthrough attack on their first therapy, as it can be challenging to identify a subsequent therapy that will be effective at preventing attacks and will be tolerated by the patient. The best approach for patients is to use a product after an attack that is as highly efficacious as possible so as to avoid potentially catastrophic subsequent attacks and, thus, optimize patient outcomes. The clinician group input noted that inebilizumab could be used as first-line treatment and as subsequent treatments for patients who have had breakthrough attacks on other therapies or who were intolerant of other therapies. Inebilizumab would be expected to be used as a monotherapy based on the available clinical evidence and to avoid cumulative immunosuppressive effects. The clinician group also noted that, although rituximab and inebilizumab both suppress B cells, there is some evidence that patients with polymorphisms in the FCGR3A gene may have an incomplete response to rituximab but not to inebilizumab.52 There is a lack of head-to-head data comparing inebilizumab and rituximab. The CNMSC also noted there is no clear preferred drug among the novel monoclonal antibodies for the treatment of NMOSD (e.g., eculizumab, inebilizumab, ravulizumab, and satralizumab) and that the best mechanism of action may vary by patient; however, there is generally limited access to these therapies in Canada at this time.
According to the CNMSC, the key outcome measure is a new attack, which is marked by new neurologic symptoms such as vision loss, weakness, sensory impairment, or dysfunction of the bladder or bowel. Although usually marked by a new enhancing lesion on MRI, this is not necessary to diagnose an attack. The clinician group indicated that the drug renewal process should consider the occurrence of any relapse in the previous year and the number of relapses, EDSS score or results of other disability measures, and any change from baseline (note that the EDSS is not validated in NMOSD). The CNMSC recommended discontinuing the drug if the patient has a new attack, a serious AE related to the therapy, or an EDSS score of 8 or higher.
The CNMSC stated that the treatment of patients with NMOSD should be assessed and managed by neurologists specialized in demyelinating diseases through an MS or demyelinating disease centre, and inebilizumab can be administered in a hospital or private clinic. Patients eligible for treatment with inebilizumab should have a confirmed diagnosis of NMOSD and a positive serum test for AQP4-IgG.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The N-MOmentum pivotal trial, a phase II and III multicentre, multinational, double-blind randomized, placebo-controlled study. Is placebo an appropriate comparator, or should inebilizumab be compared with other drugs (such as eculizumab, satralizumab, ravulizumab, tocilizumab, rituximab) or other immunosuppressive treatments (such as azathioprine, mycophenolate mofetil, methotrexate) for maintenance treatment of NMOSD? | The clinical experts indicated that placebo was a reasonable comparator because at the time of the study initiation, the other monoclonal antibodies indicated for the treatment of NMOSD (e.g., eculizumab, satralizumab) were not available, and there were no other targeted disease-modifying therapies with an indication in NMOSD. The remaining therapies such as rituximab and immunosuppressive treatments are used off-label. Additionally, the immunosuppressive drugs are not considered particularly effective in this population based on clinical practice, are associated with significant side effects, and have very little data available in patients with NMOSD. |
Given the N-MOmentum trial compares inebilizumab with placebo and not with current standards of therapy, where would inebilizumab be placed in treatment? Should inebilizumab be considered as a last line of approved therapies? Do patients need to trial satralizumab first? The sponsor indicated that inebilizumab is expected to have a place in therapy as an important new treatment option for patients who are intolerant to or whose disease has failed to respond to off-label immunosuppressive therapy (rituximab, azathioprine, or mycophenolate mofetil) in the same manner as satralizumab. Do you agree with the proposed place in therapy? | The clinical experts indicated that patients with NMOSD should be able to access inebilizumab as either a first-line or later-line therapy. The clinical experts also noted that patients should not be required to trial immunosuppressive therapies before having access to inebilizumab. The listed immunosuppressive treatments (rituximab, azathioprine, mycophenolate mofetil) are used off-label, are not considered particularly effective in this population based on clinical practice, are associated with significant side effects, and have very little data available in patients with NMOSD. Additionally, broad immunosuppressant therapies are considered to be symptomatic treatments only, not disease-modifying therapies, in contrast with the targeted monoclonal antibodies such as inebilizumab that target underlying pathophysiology. As any attack may have permanently disabling consequences, it is clinically inappropriate to require patients with this disease to trial ineffective therapies that have little supportive evidence. The clinical experts also indicated that patients should not be required to trial satralizumab or eculizumab before having access to inebilizumab due to heterogenous issues with inequitable access to these therapies across Canada, and because different patients may have different responses to and/or preferences toward the different targeted therapies now available for NMOSD. For example, the differences in infusion scheduling may make some therapies inaccessible for practical reasons for some patients, especially those with disabilities, those who live in a remote area, or those who otherwise cannot manage the financial or travel burden to access infusion therapies frequently, such as every 2 weeks. Patients with NMOSD are more likely to have a disability and experience unemployment because of their condition. |
Rituximab is used off-label for NMOSD, and its mechanism of action relates to inebilizumab. Given the similarity in the mechanism of action and lack of head-to-head trials between rituximab and inebilizumab, would using rituximab instead of inebilizumab be more cost-effective and achieve a similar response? | The clinical experts indicated that due to a lack of head-to-head data and very uncertain indirect data, it is unknown how the efficacy and safety of inebilizumab compares with rituximab. However, based on the differences in mechanism of action, inebilizumab (anti-CD19) is associated with a broader immunosuppression than rituximab (anti-CD20) because it targets B-cell lineage earlier in the cells’ evolution; thus, it is expected to theoretically be more effective than rituximab. Additionally, rituximab has very limited clinical evidence in the treatment of NMOSD, while there are phase III data supporting the efficacy of inebilizumab for this condition. Given the severity of the disease, even 1 attack may be permanently disabling; ergo, it is considered inappropriate to use rituximab in place of inebilizumab. The clinical experts also noted that although rituximab and inebilizumab both suppress B cells, there is some evidence that patients with F allele polymorphism at amino acid 158 of the FCGR3A gene (F158) may have an incomplete response to rituximab (anti-CD20) but not to inebilizumab (anti-CD19).52 |
Considerations for initiation of therapy | |
The SAkuraSky and SAkuraStar trials, which assessed the efficacy and safety of satralizumab, enrolled patients with an EDSS score of ≤ 6.5 points, while the N-MOmentum trial assessed the efficacy and safety of inebilizumab-enrolled patients with an EDSS score of ≤ 7.5, with potential to include patients with a score of 8. This difference in EDSS score will result in a larger population being eligible for treatment, which needs to be considered. | This was a comment from the drug programs to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
One of the methods to diagnose NMOSD is by MRI. Will this be needed for reassessment for renewal of therapy? This may pose a limitation for access to patients. | The clinical experts indicated that an MRI would not be needed for initiation or reassessment of therapy and is not routinely conducted in the management of NMOSD. |
Considerations for discontinuation of therapy | |
What parameters should be assessed to monitor for loss of response, absence of clinical benefit, or disease progression? | The clinical experts indicated that frequency, severity, and recovery from NMOSD attacks are the primary metrics of efficacy. It should be noted that a single attack does not necessarily indicate drug failure, as targeted therapies such as inebilizumab may also result in fewer attacks, milder attacks, or better recovery from attacks, even if they are not completely prevented. Additionally, efficacy should not be evaluated before 6 months of treatment with inebilizumab; ergo, patients should not necessarily discontinue therapy as a result of a single NMOSD attack or due to attacks occurring in the first 6 months of treatment. Similarly, hospitalization may not necessarily be informative with regard to attack severity, as hospitalization may be required for standard treatment of attacks (e.g., plasmapheresis). |
The recommendation for satralizumab is to discontinue with an EDSS score of 8 or greater. Should inebilizumab follow the same criteria? | The clinical experts indicated that inebilizumab should be discontinued if a patient reaches an EDSS score of 9, and this should be a medical decision rather than a coverage decision due to the complexities of measuring disability in patients with NMOSD. The clinical experts further noted that the EDSS is not validated in NMOSD and has general weaknesses even in the measurement of MS-related disability, as it focuses primarily on ambulation as a measure of disability; for patients with NMOSD, it is particularly insensitive to other types of paralysis and to losing visual acuity. A patient with an EDSS score of 8 requires the use of a wheelchair, but may still otherwise be independent in day-to-day life, and future NMOSD attacks could cause a loss of independence and/or cause losses in visual acuity or permanent blindness, which are clinically important outcomes to prevent. |
Considerations for prescribing of therapy | |
Dosing of inebilizumab provides the least frequent dosing compared with others in this space. Would there be a more considerable uptake of inebilizumab than other drugs, such as satralizumab and ravulizumab? | The clinical experts indicated that more uptake of inebilizumab is likely due to the less frequent dosing schedule. |
Who should prescribe inebilizumab? Is it a neurologist, ophthalmologist, or others? How do patients living in remote areas access such specialties? | The clinical experts indicated that neurologists with experience or expertise in a related subspecialty should prescribe inebilizumab. Relevant subspecialties include MS neurology, neuroimmunology, autoimmune neurology, and neuro-ophthalmology; however, patients in remote areas may have issues with access to subspecialists. For patients living in remote areas, local neurologists without subspecialty expertise could work by distance in conjunction with neurologists who are experts in a relevant subspecialty. The clinical experts also noted that ophthalmologists should not prescribe inebilizumab, although they may be involved in the diagnosis of optic neuritis in patients with NMOSD. |
Would you prescribe a combination of therapies if they have different mechanisms of action (i.e., eculizumab, ravulizumab, satralizumab)? | The clinical experts indicated that Inebilizumab would not be combined with eculizumab, satralizumab, or other monoclonal antibodies for the treatment of NMOSD due to higher risks and lack of data. The clinical experts further noted there could potentially be circumstances in which inebilizumab is combined with classical immunosuppressants, such as azathioprine. |
Given that rituximab has a similar mechanism of action, would patients be eligible for inebilizumab (CD19 inhibitor) if they have already trialled rituximab (CD20 inhibitor)? | The clinical experts indicated there are similarities in the mechanism of action between rituximab and inebilizumab, as both target B cells. However, based on the differences in mechanism of action, inebilizumab (anti-CD19) is associated with broader immunosuppression than rituximab (anti-CD20) due to targeting B-cell lineage earlier in the cells’ evolution; thus, it is expected to theoretically be more effective than rituximab. Patients who experience treatment failure with rituximab may still experience clinical benefit with inebilizumab due to these differences in mechanism. The clinical experts also noted that although rituximab and inebilizumab both suppress B cells, there is some evidence that patients with F allele polymorphism at amino acid 158 of the FCGR3A gene (F158) may have an incomplete response to rituximab (anti-CD20) but not to inebilizumab (anti-CD19).52 |
Generalizability | |
Would patients with optic neuritis benefit from inebilizumab? | The clinical experts indicated that inebilizumab is not expected to be used off-label in this population. |
CDEC = Canadian Drug Expert Committee; EDSS = Expanded Disability Status Scale; MS = multiple sclerosis; NMOSD = neuromyelitis optica spectrum disorder.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of inebilizumab IV infusion in the treatment of adult patients with NMOSD who are AQP4-IgG seropositive. The focus will be placed on comparing inebilizumab with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of inebilizumab is presented in 4 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
1 RCT with an open-label single-arm period identified in a systematic review
1 ITC.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of the Included Study (N-MOmentum)
Detail | The N-MOmentum Trial |
|---|---|
Designs and populations | |
Study design | Double-blind, randomized, placebo-controlled phase II and III study |
Locations | Patients were randomized at 99 investigational sites in 25 countries ||||||||||||||||| |||||||| |||||||||| ||||| |
Key dates |
|
Randomized (N) | At the time enrolment was terminated, 231 of a planned 252 patients had been randomized to receive inebilizumab (n = 175) or placebo (n = 56) |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Inebilizumab 300 mg IV on day 1 and day 15 |
Comparator(s) | Placebo IV on day 1 and day 15 |
Study duration | |
Screening phase | 28 days |
Randomized controlled period | 197 days |
Open-label period | Minimum 2 years |
Outcomes | |
Primary end point | Time (days) from day 1 to the onset of an AC-determined NMOSD attack on or before day 197 |
Secondary and exploratory end points | Key secondary (alpha controlled):
Secondary (not alpha controlled):
Exploratory:
|
Publication status | |
Publications | Aktas et al. Ann Neurol. 2021;89(5):895-910. Bennett et al. eBioMedicine. 2022; doi: 10.1016/j.ebiom.2022.104321. Cree et al. Mult Scler J. 2021;27(13):2052-2061. Cree et al. Lancet. 2019;394(10206):1352-63. Flanagan et al. Mult Scler Relat Disord. 2022; doi: 10.1016/j.msard.2021.103352. Marignier et al. Neurology. 2021; doi: 10.1212/NXI.0000000000000978. Marignier et al. Mult Scler Relat Disord. 2022; doi: 10.1016/j.msard.2021.103356. Rensel et al. Mult Scler J. 2022;28(6):925-932. |
Clinical trial record number | NCT02200770 |
AC = adjudication committee; AQP4-IgG = anti-aquaporin-4 immunoglobulin G; EDSS = Expanded Disability Status Scale; Gd = gadolinium; IDMC = independent data-monitoring committee; IVIG = IV immunoglobulin; LLN = lower level of normal; MCS = mental component score; MS = multiple sclerosis; NMO = neuromyelitis optica; NMSOD = neuromyelitis optica spectrum disorder; OLP = open-label period; PCS = physical component score; RCP = randomized controlled period; SF-36 = Short Form (36) Health Survey.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.
N-MOmentum was a multicentre, international, double-blind, placebo-controlled study with a 197-day RCP to compare the efficacy and safety of inebilizumab versus placebo, followed by a minimum 2-year single-arm OLP. Patients who experienced an NMOSD attack, as identified by a blinded AC, or who completed the day 197 visit without an attack, exited the RCP and had the option to enrol into an OLP that lasted a minimum of 2 years for each patient. During the OLP, patients initiated or continued treatment with inebilizumab in a manner that did not unblind their RCP treatment.
The primary objective of the study was to compare the efficacy of inebilizumab with placebo in reducing the risk of an NMOSD attack in adult patients diagnosed with NMOSD. A total of 467 patients entered the 28-day screening period, and 231 eligible patients were randomized 3:1 thereafter to receive either inebilizumab or placebo. Of the randomized patients, 213 had AQP4-IgG– seropositive NMOSD and 18 had seronegative NMOSD. Patients were stratified according to AQP4-IgG status (seropositive versus seronegative) and region (Japan versus non-Japan). The study included 99 investigational sites in 25 countries, including 1 site in Canada that enrolled 2 patients.
The purpose of the screening period was to establish patients’ eligibility to participate in the study based on the inclusion and exclusion criteria. AQP4-IgG serostatus was verified by a central laboratory. For patients with disease that was found in the screening period to be AQP4-IgG negative, relevant data documenting their neuromyelitis optica (NMO) disease were assessed by an independent eligibility committee to ensure the data were consistent with the diagnosis of NMO. Of the 467 patients who entered the study, 236 experienced screening failure. ||| ||||||| ||||| ||||||||| |||||||| ||||| ||||| |||||| |||||||||. The most common reason for screening failure (n = 204) was not meeting the inclusion or exclusion criteria. |||| |||||| ||||||| ||| ||| ||||||| ||| ||||||||||||||||||| |||||||| |||||||| |||| || ||||||| |||||||||||| ||||||| || ||||| ||| |||||||| ||| |||||||| ||||||| || |||||||||| |||||||||||| || ||| |||||| |||| |||||| ||||||||| |||||| ||||||||||||| |||||||| || ||||||||||||| |||||||||||| |||| |||||||| || |||||||||||| |||||| || |||||| ||||||||.
Patients were not allowed to receive other immunosuppressive therapies concomitantly during the N-MOmentum trial, which differed from the phase III study of eculizumab (PREVENT) and 1 of the 2 satralizumab phase III studies (the SAkuraSky trial evaluated combination therapy with immunosuppressants, while the SAkuraStar trial evaluated satralizumab monotherapy). A direct comparison of inebilizumab versus eculizumab or satralizumab was not possible due to their concurrent clinical development programs, and direct comparison with off-label treatments (rituximab, azathioprine, or mycophenolate mofetil) was considered by the sponsor to be infeasible due to uncertainty regarding their efficacy and safety.
The trial was designed with the intention of reducing risks to placebo-treated patients, including by establishing a relatively short duration for the placebo-controlled period (6.5 months), randomizing patients in a 3:1 ratio (allows for an enriched safety database while minimizing the number of required events and at-risk patients in the placebo arm), allowing patients to enter the OLP and transition to receive treatment after a single attack, providing close monitoring by an independent unblinded data-monitoring committee, and allowing liberal use of rescue therapies for patients who experienced an attack.
The N-MOmentum trial was stopped early on the recommendation of an independent data-monitoring committee, which found that the efficacy of inebilizumab had been established and that there was no justification to continue exposing placebo-treated patients to an increased risk of NMOSD attack. While still blinded to the study results, the sponsor offered all patients in the RCP the option to enter the OLP or exit to a safety follow-up period, per the recommendation of the independent data-monitoring committee. ||| ||||| ||||||| ||||||||| ||| ||||| ||| ||||| || |||||||| || |||||.
Figure 1: N-MOmentum Study Flow Diagram
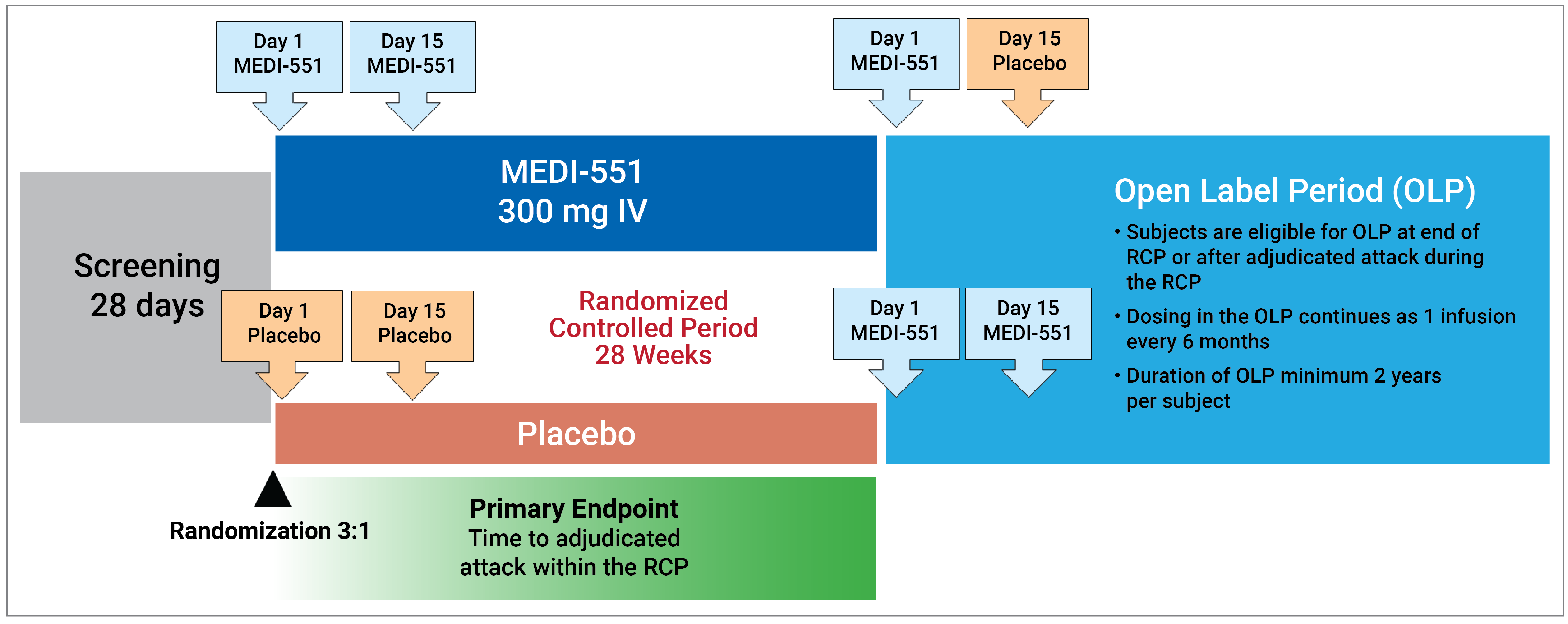
MEDI-551 = inebilizumab; OLP = open-label period; RCP = randomized controlled period.
Source: Clinical Study Report for N-MOmentum.53
Populations
Inclusion and Exclusion Criteria
Eligible patients were adults with NMOSD (AQP4-IgG seropositive or seronegative) who had a documented history of acute NMOSD attacks (1 or more attacks in the prior year or 2 or more attacks in the prior 2 years), required rescue therapy, and had an EDSS score at randomization of 7.5 or less, or 8.0 if the investigator and medical monitor agreed the patient was reasonably able to participate. Patients were excluded from the study if they had AQP4-IgG–seronegative NMOSD and a brain MRI abnormality that met the diagnostic criteria for MS, had an active infection, or had a known history of primary immunodeficiency, malignancy, or any concomitant disease other than NMOSD that required steroids at doses of greater than 20 mg/day for more than 21 days and that had been administered 6 months or less before screening. Patients were also excluded if they had received certain monoclonal antibodies or immunosuppressive drugs within 3 months before randomization; rituximab within 6 months before screening; IVIG within 1 month before randomization; or any of alemtuzumab, total lymphoid irradiation, bone marrow transplant, or T-cell vaccination therapy at any time.
Interventions
On days 1 and 15 at the start of the RCP, patients received either a 300 mg dose of inebilizumab or matched-administration placebo via a 90-minute IV infusion at the study site. Vials of inebilizumab and placebo were identically labelled and indistinguishable in appearance. After the first dose of inebilizumab or placebo during the RCP, all patients received a 2-week course of corticosteroids (prednisone 20 mg/day or equivalent) plus a 1-week taper to provide a measure of protection against attacks until the full pharmacodynamic effect of inebilizumab was reached, as all patients had discontinued prior immunosuppressive therapy before day 1.
During the OLP, all patients received inebilizumab. However, to maintain the blinding of the study, patients who were randomized to inebilizumab in the RCP received 300 mg IV inebilizumab on day 1 of the OLP, blinded IV placebo on day 15 of the OLP, then 300 mg IV inebilizumab every 26 weeks thereafter. Patients randomized to placebo in the RCP received 300 mg IV inebilizumab on day 1 of the OLP, blinded 300 mg IV inebilizumab on day 15 of the OLP, then 300 mg IV inebilizumab every 26 weeks thereafter.
Rescue therapy (e.g., IV corticosteroids, IVIG, and/or plasma exchange) was initiated as needed for NMOSD attacks at the discretion of the site investigator. The treatment of an attack was preferably initiated after completion of the attack assessments and the determination as to whether the protocol attack criteria were met. However, the investigator could initiate rescue therapy at any time before the full assessment was completed. Rescue therapy was given as directed by the investigator and could include IV corticosteroids, IVIG, and/or plasma exchange.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. The summarized end points are based on the outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review, according to the clinical experts consulted by CADTH and the stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered to be most relevant to inform the deliberations of the CADTH expert committee and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing the deliberations of CADTH’s expert committee were also assessed using GRADE.
Table 6: Outcomes Summarized From the N-MOmentum Trial
Outcome measure | Time point | N-MOmentum end point |
|---|---|---|
Time to AC-determined NMOSD attack | The primary analysis was conducted at the end of the randomized controlled period (i.e., day 197) Descriptive results for all outcomes are also presented through the end of the open-label period (> 2 years) | Primarya |
Proportion of patients with worsening in EDSS score | Key secondarya | |
Change from baseline in low-contrast visual acuity score | Key secondarya | |
Number of NMOSD-related inpatient hospitalizations | Key secondarya | |
SF-36 | Exploratory | |
Pain NRS | Exploratory | |
Safety outcomes | Exploratory |
AC = adjudication committee; EDSS = Expanded Disability Status Scale; NMSOD = neuromyelitis optica spectrum disorder; NRS = numerical rating scale; SF-36 = Short Form (36) Health Survey.
aStatistical testing of these end points was designed to control the overall 2-sided type I error at an alpha of 0.05 based on the Bonferroni chain procedure. The primary end point was first tested in the seropositive cohort and, if significant, further tested in the intention-to-treat population (both alpha = 0.05). If the difference was statistically significant in the intention-to-treat population, each secondary end point was tested via the same sequential strategy (first seropositive group then intention-to-treat population) at an alpha of 0.05 divided by 4 (0.0125).
Sources: Clinical Study Report for N-MOmentum.53 Details included in the table are from the sponsor’s summary of clinical evidence.
Time to AC-Determined NMOSD Attack
The primary efficacy outcome in the N-MOmentum trial was time (days) from day 1 to the onset of an AC-determined NMOSD attack on or before day 197. This end point was chosen to reflect the most significant and expected clinical impact of treatment, which is reducing the risk of NMOSD attacks. The primary analysis included only AC-determined attacks with an assessment visit scheduled within 120 hours (5 days) of reporting symptoms ||| ||| ||||||||||| ||||||||| |||||| || |||| || || |||||||||| |||||. The AC consisted of 3 NMOSD experts (2 neurologists and 1 neuro-ophthalmologist) who performed blinded assessments and then determined by majority vote whether an event qualified as an NMOSD attack. Supportive analyses were also conducted using other AC-determined attacks that were not included in the primary analysis.
At the time of the study design, there was no widely accepted set of criteria for the diagnosis of an NMOSD attack. Therefore, an objective set of attack criteria was needed to ensure the uniform and consistent diagnosis of attacks across heterogeneous participating sites and diverse investigator diagnosis practices. The trial sponsor, working closely with a panel of NMOSD disease experts and with input from the FDA, developed a set of NMOSD attack criteria that recognizes attacks in all domains affected by NMOSD (optic neuritis, myelitis, brain, and brainstem). These criteria include substantial clinical manifestations alone, as well as MRI-augmented findings in cases with more modest clinical manifestations. The definition of an NMO or NMOSD attack was the presence of a new symptom(s) or worsening of an existing symptom(s) related to NMO or NMOSD that met at least 1 of the protocol-defined criteria for an NMO or NMOSD attack (refer to Table 7 for criteria).
As supportive analyses, secondary and exploratory outcomes related to attacks and attack rates are discussed in this report, but not formally assessed using GRADE. Attack severity and recovery were also considered as exploratory analyses in the N-MOmentum trial. Attack severity was graded using 2 scales: the Opticospinal Impairment Scale (OSIS) and a scale based on the 18 protocol-defined attack criteria. Higher scores on the OSIS indicate poorer functioning across domains of visual acuity (0 to 8 points), motor function (0 to 5 points), sensory function (0 to 5 points), and sphincter function (0 to 5 points). The second scale assigned severity categories (mild, moderate, or severe) based on the degree of change for the applicable criterion or criteria. Attack recovery was also graded based on the domain-specific change in OSIS scores and graded as major or minor.
Additionally, a descriptive summary of the rate of AC-determined NMOSD attacks over various lengths of exposure to inebilizumab was chosen as an end point in the N-MOmentum trial to provide an approximate rate of attacks on an annualized basis. The AAR was defined as the total number of AC-determined attacks divided by total person-years of inebilizumab exposure. |||||||| || |||||||||| |||||| |||| ||| ||| ||||||| ||||||||| |||||| ||||| ||| || |||||||||| ||||||| |||||||| |||| ||||||| |||| ||| |||||||||||||||||| ||||||| ||||| || |||||||||||||| ||||||| ||||| ||||| ||||||||| |||| ||| ||||||||||| |||||||||||| ||| |||||| ||||.
Table 7: Protocol-Defined Criteria for an NMOSD Attack in the N-MOmentum Trial
Example symptoms of an NMOSD attacka | Attack typeb | Protocol-defined attack criteria |
|---|---|---|
| Optic neuritis | 1. > 15-character drop in high-contrast Landolt C broken ring chart from last visit as measured in a previously affected eye and no other ophthalmological explanation 2. ≥ 2-step dropc in CF to NLP from last visit as measured in a previously affected eye and no other ophthalmological explanation 3. ≥ 7-character drop in low-contrast Landolt C broken ring chart from last visit as measured in either eye alone (monocular) AND a new RAPD in affected eye 4. ≥ 7-character drop in low-contrast Landolt C broken ring chart from last visit as measured in either eye alone (monocular) AND loss of a previously documented RAPD in fellow eye 5. ≥ 5-character drop in high-contrast Landolt C broken ring chart from last visit as measured in either eye alone (monocular) AND a new RAPD in affected eye 6. ≥ 5-character drop in high-contrast Landolt C broken ring chart from last visit as measured in either eye alone (monocular) AND loss of a previously documented RAPD in fellow eye 7. ≥ 1-step dropd in CF to NLP from last visit as measured in a previously affected eye AND a new RAPD in affected eye 8. ≥ 1-step dropd in CF to NLP from last visit as measured in a previously affected eye AND loss of a previously documented RAPD in fellow eye 9. ≥ 7-character drop in low-contrast Landolt C broken ring chart from last visit as measured in either eye alone (monocular) AND a new Gd-enhancing or new or enlarging T2 MRI lesion in the corresponding optic nervee 10. ≥ 5- or more character drop in high-contrast Landolt C broken ring chart from last visit as measured in either eye alone (monocular) AND a new Gd-enhancing or new or enlarging T2 MRI lesion in the corresponding optic nervee 11. ≥ 1-step dropd in CF to NLP from last visit as measured in a previously affected eye AND a new Gd-enhancing or new or enlarging T2 MRI lesion in the corresponding optic nerve |
| Myelitisf | 12. ≥ 2-point worsening in 1 or more of the relevant (pyramidal, bladder or bowel, sensory) FSS compared with last visit 13. ≥ 1-point worsening in EDSS score compared with last visit if the previous EDSS score was 5.5 or more 14. ≥ 1-point worsening in 2 or more of the relevant (pyramidal, bladder or bowel, sensory) FSS compared with last visit when the last visit score was 1 or greater AND a new Gd-enhancing or new or enlarging T2 MRI lesion in the spinal cord 15. ≥ 0.5-point worsening in EDSS compared with last visit if previous EDSS was 5.5 or more AND a new Gd-enhancing or new or enlarging T2 MRI lesion in the spinal cord |
| Brainstem | 16. Isolated (not present at last visit) intractable nausea, vomiting, and/or hiccups lasting for greater than 48 hours AND a new Gd-enhancing or new or enlarging T2 MRI lesion in the brainstem 17. ≥ 2-point worsening in 1 or more of the relevant (brainstem, cerebellar) FSS compared with last visit AND a new Gd-enhancing or new or enlarging T2 MRI lesion in the brainstem |
| Brain | 18. ≥ 2-point worsening in 1 or more of the relevant (cerebral, sensory, pyramidal) FSS (with a score of 3 or more at the current visit) compared with last visit AND a new Gd-enhancing or new or enlarging T2 MRI lesion in the brain consistent with the clinical presentation |
CF = counting fingers; EDSS = Expanded Disability Status Scale; FSS = functional system score; Gd = gadolinium; HM = hand motion; LP = light perception; NLP = no light perception; NMOSD = neuromyelitis optica spectrum disorder; ON = optic neuritis; RAPD = relative afferent pupillary defect.
aThe symptoms listed are examples and are not inclusive of all NMOSD symptoms.
bFour major areas of the body may be affected by an attack: the optic nerve, resulting in ON; the spinal cord, resulting in myelitis; the brainstem, resulting in a number of outcomes; and the brain.
cAt least a 2-step drop (worsening) on the Landolt C broken ring chart (e.g., a drop from CF to LP or NLP, or from HM to NLP).
dAt least a 1-step drop (worsening) on the Landolt C broken ring chart (e.g., a drop from CF to HM, LP, or NLP; from HM to LP or NLP; or from LP to NLP).
eLesions seen in the optic chiasm also count toward these criteria.
fA 1-point change in a single FSS without a change in the EDSS, with or without a new Gd-enhancing or new or enlarging T2 MRI lesion in the spinal cord, is not considered a clinically significant change and did not count as an attack per this protocol.
gOther neurologic signs could include double vision, dysarthria, dysphagia, vertigo, oculomotor palsy, weakness, nystagmus, or other cranial nerve abnormality.
Sources: Clinical Study Report for N-MOmentum.53 Details included in the table are from the sponsor’s summary of clinical evidence.
Proportion of Patients With Worsening in EDSS Score
One of the key secondary end points in the N-MOmentum trial was worsening in baseline EDSS score at last visit during the RCP. The EDSS is a validated tool for MS-related disability that ranges from 0 (normal neurologic exam) to 10 (death from MS), with half steps from 1.0 to 9.5; higher values reflect greater impairment and disability. Although MS and NMOSD are not the same condition, they share features in clinical presentation. No minimal important difference (MID) was identified that was specific to NMOSD, but estimates from patients with MS suggest that a single 1.5-point change in EDSS score could be considered a meaningful deterioration from the patient’s perspective,54 and that important changes were considered, i.e., a change of 1.5 points or more from a baseline of 0 points, a change of 1.0 point or more from a baseline of 1 to 5.5 points, or a change of 0.5 points or more from a baseline of 6 points or greater.55 The clinical experts consulted by CADTH agreed that patients with a higher baseline EDSS score would have a lower threshold for EDSS changes that would be considered clinically meaningful compared with patients who had a lower baseline EDSS score. In the N-MOmentum trial, trained and certified neurologists (EDSS raters), who were independent of study investigators and patients, conducted the assessments. A worsening in the overall EDSS score during the N-MOmentum study was defined based on the following criteria:
a worsening in EDSS score of 2 or more points in patients with a baseline EDSS score of 0 points
a worsening in EDSS score of 1 or more points in patients with a baseline EDSS score of 1 to 5 points
a worsening in EDSS score of 0.5 points or more in patients with a baseline EDSS score of 5.5 points or greater.
Change From Baseline in Low-Contrast Visual Acuity Score
Another key secondary end point during the RCP was the change from baseline to the last visit in the binocular low-contrast visual acuity score measured using the Landolt C broken ring chart. This chart uses a standardized format to be equally detectable by observers with no sight impairment and comprises a series of incomplete rings resembling the letter C. The break in the ring can be oriented in any direction and the patient must indicate where the break is located for each ring when the chart is held at a distance of 3 m, with the result based on the smallest rings that can be correctly read on the chart. Trained, experienced, and blinded independent ophthalmologists or ophthalmology technicians conducted all examinations using standardized retro-illuminated light cabinets, which eliminated any inter-site variability in height and room lighting. An MID has not been identified for the change in the low-contrast visual acuity score in NMOSD. The low-contrast visual acuity score is commonly used in MS and the clinical experts consulted by CADTH indicated this is an appropriate outcome for patients with NMOSD, especially as a complement to the EDSS, which may be less sensitive to changes in visual acuity. The scoring of this assessment is based on the number of characters on the chart that the patient is able to identify, from 0 to 70 inclusive, where 70 indicates the patient was able to correctly identify all characters on the chart (i.e., best visual acuity score), and 0 indicates they were not able to identify any characters correctly (i.e., poorest visual acuity score).
Number of NMOSD-Related Inpatient Hospitalizations
The number of NMOSD-related inpatient hospitalizations was a key secondary end point. An NMOSD-related inpatient hospitalization was defined as a hospital stay |||| |||| |||||| |||||||| || ||| ||||| ||| || ||||||||| || lasted for more than 1 day ||||||||||| || ||| |||||||||| ||||||| ||||||||| |||| ||| ||||||||| |||||| |||| |||||||| |||| ||||| || || ||||||||| |||||||||| |||| |||||||||| ||||||||| |||||||||| ||||| ||| |||||||| |||||||||||||||| |||| |||| |||| ||| ||| |||||||||||||| || ||||||||||| ||||||||||| || ||||||||||.
Short Form (36) Health Survey
An exploratory end point was the change from baseline to the last RCP visit in the 4-week recall in the SF-36 physical component score and mental component score. The SF-36 measures 8 dimensions of a patient’s functional health and well-being and also provides 2 summary scores that characterize a patient’s mental and physical health status (the mental component score and physical component score, respectively), with higher global scores reflecting better HRQoL. The sponsor is not aware of a reported MID for the SF-36 specific to NMOSD. The SF-36 has been validated in patients with MS and neurologic disabilities.56,57 Indirect evidence from patients with MS suggests that the MID ranges for the SF-36 domains are 4 to 9 points for physical functioning, 6 to 8 points for role physical, 6 to 7 points for social functioning, and 9 points for the physical component score.57
Pain NRS
An exploratory end point was the change from baseline to the last RCP visit in the pain NRS in 5 anatomic locations (eyes, upper back, lower back, arms, and legs). Patients were asked to rate pain at each location |||| ||| ||||||||, with scores ranging from 0 (no pain) to 10 (worst pain imaginable). No MID for the NRS specific to NMOSD was identified.
Safety Outcomes
Safety data were collected and recorded from the time of enrolment, through the treatment periods, and through the end of the safety follow-up period. TEAEs were reported separately for each of the treatment periods and according to patient disposition. However, because they were already reported as the primary outcome, NMOSD attacks were not recorded as TEAEs during the RCP and OLP, |||||||| ||||| ||||||| |||| |||||||| || ||||| |||||| ||| ||||||||| ||||||| |||||| ||||||||| ||||||| || || ||| ||||||||| |||||||| ||||||| ||| ||||| ||| |||||||||| ||| ||| |||||||| ||||||||||). All TEAEs were coded according to system organ class and preferred term using the Medical Dictionary for Regulatory Activities (version 21.0). The severity of TEAEs was assessed by the investigator and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events. AEs of special interest were defined as:
infusion-related reactions, anaphylactic reactions, and hypersensitivities
infections
hepatic function abnormalities
cytopenias
progressive multifocal leukoencephalopathy.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Change in pain NRS | Pain NRS in 5 anatomic locations (eyes, upper back, lower back, arms, and legs) is used to capture patient’s worst pain levels experienced. Patients are asked to rate pain at each location |||| ||| ||||||||, with scores ranging from 0 (no pain) to 10 (worst pain imaginable).58 | No studies on NMOSD or MS. The reliability and validity of the NRS have been documented for chronic pain intensity measures.59-61 | None identified for patients with NMOSD or MS. |
SF-36 | A generic self-reported questionnaire consisting of 8 domains: physical functioning, role physical, bodily pain, general health, vitality, social functioning, role emotional, and mental health. The SF-36 also yields 2 summary measures of physical health (the PCS measure) and mental health (the MCS measure) derived from scale aggregates. Higher global scores are associated with better quality of life. | No studies on patients with NMOSD were found. The instrument has been validated in patients with MS and neurologic disabilities. One HTA (systematic review)56 with 7 studies and 3,142 patients showed proper reliability (Cronbach alpha 0.70 for all subscales) and validity (with correlations ranging from 0.5 to 0.81) for all domains. Two studies showed good-to-excellent internal consistency for the total instrument and for all subscales (within PCS and MCS) with the exception of social function. Correlations between SF-36 subscales and impairment measures were weak. Inter-rater reliability between patients with disabilities and their caretakers was moderate. | No MID studies were found for patients with NMOSD. Indirect evidence from patients with MS was obtained. Only the physical functioning, role physical, social functioning, and PCS from the SF-36 were included in 1 study.57 MID ranges for the SF-36 domains were as follows: 4 to 9 points for physical functioning, 6 to 8 for role physical, and 6 to 7 for social functioning; for the PCS score, the MID was consistently 6. |
HTA = health technology assessment; HRQoL = health-related quality of life; MCS = mental component score; MID = minimal important difference; mRS = modified Rankin Scale; MS = multiple sclerosis; NMOSD = neuromyelitis optica spectrum disorder; NRS = numerical rating scale; PCS = physical component score; SF-36 = Short Form (36) Health Survey.
Statistical Analysis
End Points
The primary end point (time to the onset of an NMOSD attack) was assessed using a Cox proportional hazards model in both the population with AQP4-IgG–seropositive NMOSD and the ITT population. The sponsor did not report any assessment of whether the proportional hazards assumption was appropriate. || |||||||||| ||| ||||||||| ||| ||||||| |||| ||| ||| ||||||| ||||||||| |||||||| |||| |||||||| ||||||||| || ||||| ||||||| ||||||||||||| |||||||||| || |||||| ||||||||| ||||||||.
Worsening in EDSS score was assessed using a logistic regression model, with treatment and baseline EDSS as explanatory variables. The percentage of patients meeting the end point and the 95% CIs and P values for the odds ratios are presented. Missing values due to dropout or missing data were conservatively considered as “worsening” according to the nonresponder imputation rule.
Change from baseline in low-contrast visual acuity was assessed using an analysis of covariance model that used treatment and baseline Landolt C broken ring chart binocular score as explanatory variables. The last nonmissing value was used for the analysis in instances of missing values due to dropout or missing data.
Sample Size and Power Calculation
The N-MOmentum trial was planned to detect a reduction in relative risk (RR) of 60% in the primary outcome (time from day 1 to the onset of an AC-determined NMOSD attack on or before day 197) with 90% power and a 2-sided alpha of 0.05. A total of 67 AC-determined NMOSD attacks were assumed for the ITT population. Patients were randomized in a 3:1 ratio to either inebilizumab or placebo within AQP4-IgG seropositive and seronegative strata, assuming approximately 80% of patients had seropositive NMOSD and 20% had seronegative NMOSD. ||||| || |||| ||||||||||||| || ||| |||||||||||| |||||| ||| ||| || ||| |||||||| ||| ||||| ||||| |||| ||||||||||||| ||| ||||| || |||||||| |||||||| |||| |||||||||.
The plan was to randomize and treat 252 patients, which was based on a blinded review of the attack rate for the first 78 patients to complete the RCP and a simulation based on these patients that indicated a 90% probability of achieving the required 67 AC-determined attacks with 252 patients. However, the data-monitoring committee recommended halting enrolment based on a clear demonstration of efficacy and condition power of more than 99%,20 despite the targets of 252 patients and 67 adjudicated attacks not being met.
Statistical Testing
Unless otherwise stated, all efficacy analyses were performed with a 2-sided test at a 5% significance level. This summary contains the final analyses from the RCP as well as the descriptive data throughout the OLP |||||| ||||||||| ||||| |||||| |||||||| || ||||||.
The study was designed to strongly control the overall 2-sided type I error rate at an alpha of 0.05 based on the Bonferroni-based chain procedure. The primary null hypothesis was to be hierarchically tested first at an alpha of 0.05 in the AQP4-IgG–seropositive cohort and, if significant, would be further tested in the ITT population at an alpha of 0.05. If and only if the treatment-group comparison was statistically significant within the ITT population, then the secondary hypotheses would be tested.
The null hypotheses for the 4 key secondary end points followed the same sequential testing strategy as the primary analysis (testing first within patients with seropositive disease followed by the ITT population if the comparison within the first group of patients was statistically significant). Each secondary hypothesis was initially tested based on the Bonferroni method at an alpha of 0.05 divided by 4 (0.0125). If the null hypothesis for a particular secondary end point was rejected across both the seropositive and ITT populations, the type I error saved was to be propagated equally to other non-rejected sets of secondary null hypotheses. The testing procedure was to be repeated until all null hypotheses were rejected or no further null hypothesis could be rejected.
Subgroup Analyses
Prespecified subgroup analyses were performed for the efficacy end points in the ITT population:
sex (male versus female)
baseline EDSS score (< 5 points versus ≥ 5 points)
number of prior NMOSD relapses (< 2 versus ≥ 2)
disease duration category (< 5 years versus ≥ 5 years)
AQP4-IgG serostatus at screening (positive versus negative).
Additional analyses were conducted post hoc according to previous rituximab treatment (yes versus no) and previous conventional immunosuppressant use as maintenance therapy (azathioprine or mycophenolate mofetil, yes versus no, distinct from rescue therapy).
Safety results were also reported separately according to AQP4-IgG serostatus (positive versus negative) and according to the sex (male versus female) of the patients participating in the RCP. |||||||||| |||| ||| || |||| |||||||| |||||||| |||| ||||||||| || ||||| |||| ||||||| ||| |||||||| ||||||||| ||| ||| |||||||||| || ||| ||||||| || ||| ||||||| |||||||| ||| ||||||| ||||||| || ||||| |||||| ||| |||.
Table 9: Statistical Analysis of Efficacy End Points in the N-MOmentum Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
|||| || ||||| |||||| | ||| |||||||||||| ||||||| ||||| | |||||||||| |||| |||||||||| ||||| | || |||||||||| ||| ||||||||| ||| ||||||| |||| ||| ||| ||||||| |||||||| | |||| ||||||||||| |||||||| |||| ||||||||| ||| ||| ||||||| ||||||||| ||||| ||||||||| ||| ||||||||| ||||||||||||||||| || |||||||||| ||||| ||||| |||||||| ||| |||||||| |||| |||||||||| || ||||||||||||| |||||| ||||| |||| ||||||||| ||||||||||||||||||| |||||||| ||| |||||||||||| ||| ||| |||||| |||||||||||| || ||||| |||||||||||| || |||||||| |||||||| ||||||||| || ||||||||||||| |||||| || |||||| |||||||||||| || ||||||||||||||||||||||| ||||| |||||||||||||||| ||||||| |||| ||| ||| ||| || ||| |||| ||| |||||||| ||| ||||||||||| |||||||||||| ||| ||||||||||||| || ||| |||| || |||||| ||| |||||||| ||| ||||||||||| ||||||||||||| ||||||| |||||| || || ||| ||||||| || |||||||||| || |||||||||||||||| |||||||| |
||||||||| || |||| ||||| | |||||||| |||||||||| | |||| | ||||||| |||||||||| || ||||||||||| ||||||||| || ||||||||||||| |||||||||| |||| | || |
|||||||||||| |||||| |||||| ||||| | |||||| | |||||||| ||||||||||||| ||||| ||||| ||||||||| ||||| | ||| |||| ||||||||||| ||||| |||| || |||||||||| ||| |||||||| | || |
||||||||||||| ||||||||| |||||||||||||||| | |||||||| |||||||| |||||||||| | |||| | || | || |
|||||||||| |||||| |||| | |||| |||||||||||| ||||||| ||||| | ||| | ||| | ||| |
||||| | |||||| | |||||||| ||||| ||| || ||| ||||| | || | || |
|||| ||| | |||||| | |||||||| |||| ||| ||||| | ||| |||| ||||||||||| ||||| |||| || |||||||||| ||| |||||||| | || |
AC = adjudication committee; ANCOVA = analysis of covariance; EDSS = Expanded Disability Status Scale; IDMC = independent data-monitoring committee; ITT = intention to treat; MCS = mental component score; mRS = modified Rankin Scale; NMOSD = neuromyelitis optica spectrum disorder; NR = not reported; NRS = numerical rating scale; PCS = physical component score; PD = pharmacodynamic; RCP = randomized controlled period; SF-36 = Short Form (36) Health Survey; SFP = safety follow-up period.
Sources: Clinical Study Report for N-MOmentum.53 Details included in the table are from the sponsor’s summary of clinical evidence.
Analysis Populations
The analysis sets for each study are summarized in Table 10.
Table 10: Analysis Populations of the N-MOmentum Trial
Study | Population | Definition | Application |
|---|---|---|---|
N-MOmentum | ITT | Patients randomized and who received any IP were included in the ITT population. These patients were analyzed according to their randomized treatment group regardless of whether they received an intervention other than the one that was planned. | Efficacy analyses |
As-treated | Patients who received any IP were included in the as-treated population. Specifically, patients randomized to inebilizumab who received all placebo doses were included in the placebo group; conversely, patients randomized to placebo who received at least 1 dose of inebilizumab were included in the active treatment group. | Safety analyses | |
Open-label | Patients who received at least 1 dose of inebilizumab during the OLP. These patients had baseline data from the RCP. | To evaluate the long-term safety and efficacy of inebilizumab |
IP = investigational product; ITT = intention to treat; OLP = open-label period; RCP = randomized controlled period.
Sources: Clinical Study Report for N-MOmentum.53 Details included in the table are from the sponsor’s summary of clinical evidence.
Results
Patient Disposition
Patient disposition is summarized in Table 11. Although only the patients with AQP4-IgG–seropositive NMOSD are relevant for this review, this table reflects the total population, including patients with AQP4-IgG–seronegative NMOSD.
Of 467 screened patients, 175 were randomized to treatment with inebilizumab and 56 to placebo. Screening failures occurred primarily as a result of not meeting the inclusion or exclusion criteria (n = 204), while a minority was lost to follow-up, withdrew consent, or were excluded due to other unspecified reasons. ||||| ||||||| ||| ||||| ||| ||||| ||||| || |||||||||| |||||||||| |||||||| || ||| ||||||| || ||||||||| || |||| |||| |||| |||||||| || |||||| ||||||||| || || ||||||| |||| |||||| || |||||||| |||| ||||||| ||.
Of the patients randomized to treatment with inebilizumab, 1 did not receive treatment. The majority of patients in both arms completed the randomized period of the study, while fewer than 4% in each arm did not complete it due to AEs, withdrawal of consent, or other unspecified reasons. |||||| ||| |||||||||| ||||||| ||||||||||||| ||| || |||||||| ||| |||||||| ||| ||| || |||||||||||| ||| ||| || ||||||| ||| ||| |||||||| ||| |||| || ||| ||||| ||||||||| |||||||||| || |||||||||||| ||| ||| ||| |||||||| ||| |||| ||| |||| |||||| ||||||| |||| |||||||||| || |||||||| || ||||||||||| ||||| |||||||| ||||||||||| |||||||| ||||||||||| ||| || |||| || ||| ||||| ||||||||| |||||||||| || ||||||| ||| ||| ||| |||||||| ||| |||| ||| |||| |||||| |||||| ||| |||||||| |||||||||||| |||||||| ||| || || ||||||||.
Table 11: Summary of Patient Disposition in the N-MOmentum Trial
Patient disposition | N-MOmentum | |
|---|---|---|
Inebilizumab | Placebo | |
Screened, n | 467 | |
Reason for screening failure, n (% of screened) | 236 (50.5) | |
Did not meet inclusion or exclusion criteria (% of screening failure) | 204 (86.4) | |
Lost to follow-up (% of screening failure) | 2 (0.8) | |
Withdrawal of consent (% of screening failure) | 7 (3.0) | |
Other (% of screening failure) | 23 (9.7) | |
Randomized, n | 175 | 56 |
Not treated (% of randomized) | 1 (0.6) | 0 (0) |
ITT population, N (% of randomized) | 174 (99.4) | 56 (100) |
Completed RCP, n (% of ITT) | 169 (97.1) | 54 (96.4) |
Did not complete RCP, n (% of ITT) | 6 (3.4) | 2 (3.6) |
Adverse events (% of did not complete) | 2 (33.3) | 0 (0) |
Withdrawal of consent (% of did not complete) | 1 (0.6) | 1 (50.0) |
Death (% of did not complete) | 0 (0) | 0 (0) |
Other (% of did not complete) | 3 (50.0) | 1 (50.0) |
Completed RCP but did not roll over to OLP, n (% of ITT) | 4 (2.4) | 3 (5.6) |
Rolled over to OLP, n (% of ITT) | 165 (94.8) | 51 (91.1) |
||||||||| |||||| || |||||| |||| || |||| | ||| |||||| | || |||||| |
||| ||| |||||||| |||||| || |||||| |||| || |||| | || |||||| | |||||| |
||||||| |||||| || || ||| ||| ||||||||| | ||||| | |||||| |
|||| || ||||||||| || || ||| ||| ||||||||| | ||||| | ||| |
|||||||||| || ||||||| || || ||| ||| ||||||||| | || |||||| | ||| |
Death (% of did not complete) | 2 (5.9) | 1 (12.5) |
||||| || || ||| ||| ||||||||| | || |||||| | |||||| |
Other study populations | ||
As-treated, n | 174 | 56 |
Open-label, n | 165 | 51 |
Non–open-label, n | 9 | 5 |
Any inebilizumab, n | 174 | 51 |
ITT = intention to treat; OLP = open-label period; RCP = randomized controlled period.
Sources: Clinical Study Report for N-MOmentum.53 Details included in the table are from the sponsor’s summary of clinical evidence.
Baseline Characteristics
The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Although only the patients with AQP4-IgG–seropositive NMOSD are relevant for this review, baseline data for both patients with seropositive NMOSD and the total population (including patients with seropositive and seronegative NMOSD) are summarized in the table for comprehensiveness. This is also the case for the safety section of this review.
Among those with seropositive NMOSD, the patients were approximately 43 years old on average and the vast majority were female (approximately 94%). Approximately half of patients were white in both treatment arms, but the distribution of other races differed between the arms. In the placebo arm, the next most common race was “other” (approximately 19%), followed by Asian (15%), American Indian or Alaskan Native (10%), and Black or African American (10%). In the inebilizumab arm, the next most common race after white was Asian (approximately 23%) followed by Black or African American (9%), “other” (7.5%), American Indian or Alaskan Native (7%), and multiple categories (< 1%). || |||||||| |||| |||||| |||||||| || ||||| ||||||| |||||||| || |||||| ||||||||| |||| ||||||||||||| ||| || |||||||| |||| ||||||| || ||| || ||||| ||||||||||||| ||| || |||||||| |||| || ||||| ||||||||||.
||||| |||||||||||| ||||||||| ||| |||| |||||||||||| ||| ||||||||||||| |||||| |||||| ||||| || |||||||| |||| ||||||||||| |||||||| ||||| || ||| ||||||| ||| |||| ||| |||||||||||| |||| |||||||||| the mean number of gadolinium-enhancing lesions was slightly lower among patients randomized to placebo.
Nearly all patients (> 98%) had received prior acute or maintenance treatments for NMOSD. A higher proportion of patients assigned to placebo (50%) than inebilizumab (36%) had previously been treated with plasmapheresis as an acute therapy for NMOSD; a minority of patients had previously been treated with IVIG (6% and 5%, respectively). Approximately 60% of patients in both arms had a treatment history with maintenance therapies for NMOSD, including azathioprine and mycophenolate mofetil, and the proportions were similar between treatment arms. Approximately 14% of patients in both treatment arms had prior exposure to a biologic drug for NMOSD.
As very few patients included in the study had AQP4-IgG–seronegative NMOSD (18 out of 230 patients randomized), the baseline characteristics of the total population (including those with either seropositive or seronegative NMOSD) were very similar to the baseline characteristics of the seropositive population.
Table 12: Summary of Baseline Characteristics in the N-MOmentum Trial
Characteristic | AQP4-IgG–sero+ population (N = 213) | Total population (N = 230) | ||
|---|---|---|---|---|
Placebo (N = 52) | Inebilizumab (N = 161) | Placebo (N = 56) | Inebilizumab (N = 174) | |
Demographics | ||||
Mean age, years (SD) | 42.4 (14.3) | 43.2 (11.6) | 42.6 (13.9) | 43.0 (11.6) |
Median age, years (range) | 43 (18 to 74) | 43 (18 to 73) | 42.5 (18 to 74) | 43 (18 to 73) |
< 65 years, n (%) | 48 (92.3%) | 155 (96.3%) | 52 (92.9%) | 168 (96.6%) |
Female sex, n (%) | 49 (94.2%) | 151 (93.8%) | 50 (89.3%) | 159 (91.4%) |
Mean BMI, kg/m2 (SD) | 27.3 (6.9) | 25.3 (5.6) | 27.0 (6.7) | 25.2 (5.5) |
Race, n (%) | ||||
American Indian or Alaskan Nativea | 5 (9.6%) | 11 (6.8%) | 5 (8.9%) | 14 (8.0%) |
Asian | 8 (15.4%) | 37 (23.0%) | 8 (14.3%) | 39 (22.4%) |
Black or African American | 5 (9.6%) | 14 (8.7%) | 5 (8.9%) | 15 (8.6%) |
|||||| |||||||| || ||||| ||||||| |||||||| | |||||| | |||||| | |||||| | |||||| |
White | 24 (46.2%) | 86 (53.4%) | 28 (50.0%) | 92 (52.9%) |
Other | 10 (19.2%) | 12 (7.5%) | 10 (17.9%) | 13 (7.5%) |
Multiple categories checked | 0 | 1 (0.6%) | 0 | 1 (0.6%) |
Region, n (%) | ||||
|| | || ||||||| | || ||||||| | || ||||||| | || ||||||| |
|||||| | || ||||||| | ||| ||||||| | || ||||||| | ||| ||||||| |
EDSS | ||||
Mean (SD) | 4.35 (1.63) | 3.81 (1.77) | 4.19 (1.68) | 3.81 (1.81) |
Median (range) | 4.00 (1.0 to 8.0) | 3.50 (0.0 to 8.0) | 4.00 (1.0 to 8.0) | 3.5 (0.0 to 8.0) |
1 to 5 points, n (%) | 36 (69.2%) | 122 (75.8%) | 40 (71.4%) | 129 (74.1%) |
> 5 points, n (%) | 16 (30.8%) | 37 (23.0%) | 16 (28.6%) | 41 (23.6%) |
|||||||||||| |||||| |||||| ||||| |||||||||| | ||||
|||| |||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| |
||||||||||||| |||||| |||||| ||||| ||||||||||| | ||||
|||| |||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| |
Total number of Gd-enhancing lesions | ||||
Mean (SD) | 0.8 (0.9) | 1.2 (1.2) | 0.9 (0.9) | 1.2 (1.2) |
Median (range) | 1 (0 to 4) | 1 (0 to 5) | 1 (0 to 4) | 1 (0 to 5) |
Prior treatments for NMOSD | ||||
Any acute or maintenance treatment, n (%) | 51 (98.1%) | 159 (98.8%) | 55 (98.2%) | 172 (98.9%) |
Acute treatments, n (%) | ||||
Plasmapheresis, n (%) | 26 (50.0%) | 58 (36.0%) | 27 (48.2%) | 67 (38.5%) |
IVIG, n (%) | 3 (5.8%) | 8 (5.0%) | 3 (5.4%) | 8 (4.6%) |
||| ||||||||||| ||||||||||||||||| | || ||||||| | || ||||||| | || ||||||| | || ||||||| |
Azathioprine, n/N (%) | 21/26 (80.8%) | 63/81 (77.8%) | 22/27 (81.5%) | 64/83 (77.1%) |
Mycophenolate mofetil, n/N (%) | 8/26 (30.8%) | 26/81 (32.1%) | 8/27 (29.6%) | 27/83 (32.5%) |
Any biologic drug, n (%) | 7 (13.5%) | 23 (14.3%) | 7 (12.5%) | 25 (14.4%) |
AQP4-IgG = anti-aquaporin-4 immunoglobulin G; BMI = body mass index; EDSS = Expanded Disability Status Scale; Gd = gadolinium; IVIG = IV immunoglobulin; NMOSD = neuromyelitis optica spectrum disorder; SD = standard deviation; sero− = seronegative; sero+ = seropositive.
aIncludes patients from South and Central America.
|||| ||| ||||||||||||| |||||| |||||| |||||| ||| ||| |||||| || |||||||||| |||| ||| || |||| |||||||||||||| |||||||||||| ||||| ||||| |||| |||||||| |||||||| ||||| ||| ||| ||||||||||||| |||||||||| ||||||||||||.
Sources: Clinical Study Report for N-MOmentum.53 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
Exposure to study treatments is summarized in Table 13. No major differences were observed between the patients with AQP4-IgG–seropositive NMOSD and the patients in the overall population.
||||| |||||||||| ||||| |||||| ||| |||| ||||| |||| |||||||| || ||| |||| |||||||| || |||||||| || ||| |||||||||||| ||| ||||||| ||| ||| ||||||| ||| |||||||| ||| |||| |||| |||||||||||| |||| ||| ||||| |||||| ||| |||| |||||| ||||||||| || ||||||||| |||||||| |||| |||| |||| |||| |||| || ||| |||||||||||| ||| ||||||| ||||| ||||| ||| ||||| |||||||||||| || |||||||||||| |||||||| |||||| ||| ||||||||||| ||| |||| |||||| |||||||| || ||||||||| |||||||| ||| ||||||| |||| || ||| |||||||||||||||||||||||||||| ||||| ||| ||||||| |||| || ||| ||||||||||||||||||||||| |||||| ||| |||| |||| |||||| || |||||||||||| ||||| |||||| ||| ||| ||| ||||| || ||| |||||||||||||||||||||||||||| ||||| |||| ||||| |||||| |||| || ||| ||||||||||||||||||||||| ||||| |||| ||||| ||||||| || |||||||||||||||||||||||||||| ||||| |||||||||||||| |||| || ||| ||| || || |||||||| ||||||||.
Total inebilizumab exposure was 730.4 person-years among all patients who received inebilizumab across the RCP and OLP. The median duration of inebilizumab treatment was 3.2 years and the maximum duration of exposure was 5.4 years at the data cut-off (November 6, 2020).
During the RCP, all patients received a protocol-mandated 2-week course of oral corticosteroids plus a 1-week taper following the first administration of the study drug (active or placebo). Mean total corticosteroid use from day 1 to day 14 was 19.71 mg (range, 1.4 mg to 25.0 mg) in the inebilizumab arm and 19.78 mg (range, 11.4 mg to 25.0 mg) in the placebo arm, with appropriate tapering from day 15 to 21 for most patients in the placebo arm (78.6%) and in the inebilizumab arm (82.2%).
Rescue therapies are summarized in Table 14. More patients treated with placebo required rescue therapies during the trial compared with patients who received inebilizumab.
Table 13: Summary of ||||||| || ||| ||||| || |||||||||| [Redacted]
|||||||| | |||||||| |||||||||||| | ||||| |||||||||| | ||
|---|---|---|---|---|
||||||| ||||| | |||||||||||||||||| | ||||||||||||| | ||||||||||||||||||| | |
|||||||| |||||| ||| ||| ||||||||||| ||||||||||| | ||||
||||||||||| || ||||||| || |||||||||||||| | || ||||||| | ||| ||||||| | || ||||||| | ||| ||||||| |
|||||||||||| |||| ||||||||||||| |||| || |||| | ||| | ||||| |||||| | ||| | ||||| |||||| |
|||||||| || ||||||| || ||||||||||||| |||| |||| |||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| |
||||| |||||||||||| | ||||| | ||||| | ||||| | ||||| |
|||||||| |||||| ||| ||| |||| ||||||||||| | ||||||| || ||||||| | ||||||| || ||||||| | ||||||| || ||||||| | ||||||| || ||||||| |
||||| ||||||||| |||| |||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| |
|||||||| || ||||||||||||| |||| |||| |||| | |||||| ||||||| | |||||| ||||||| | |||||| ||||||| | |||||| ||||||| |
||||| |||||||||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||||||||||||||| ||||||| |||||||||||| |||||||||||| ||||||| ||||||||||||||||||||||| ||||||| ||||||||| |||||||||||||||||| |||||||| ||||| |||||| ||| |||||||||||||||||| |||||||| || ||| ||||| ||| |||| ||| ||||||||| ||||||| || |||||||| |||||||||
Table 14: Summary of ||||||| || ||| ||||| || |||||||||| [Redacted]
|||||||||| | |||||||||||| | ||||||||||||||||||| |
|---|---|---|
||||||||||||||||||||||||||||||||||| | ||
|||||||| |||||| |||||||||||| | || ||||||| | || ||||||| |
|||||||||||||||||| | || ||||||| | || ||||||| |
|||||||||||||||||| | ||||| | |||||| |
|||||||| | || | || |
||| | ||||||| || ||||||||||||||||| | |||||||||||| || |||||||||||| ||||||| |
|||||||| |||||| |||||||||||| | || ||||||| | || ||||||| |
|||||||||||||||||| | || ||||||| | || ||||||| |
|||||||||||||||||| | |||||| | |||||| |
||||||||| | ||||| | || |
|||||||||||| ||||||| |||||||||||||||||||||||| ||||||||||||||| |||||||| ||||| |||||| ||| ||||||||||||||||| |||||||| || ||| ||||| ||| |||| ||| ||||||||| ||||||| || |||||||| |||||||||
Efficacy
NMOSD Attacks
Time to NMOSD Attack During the RCP
Among patients with AQP4-IgG–seropositive NMOSD in the randomized period, treatment with inebilizumab (versus placebo) was associated with a 77.3% reduction in the risk of an AC-determined NMOSD attack (HR = 0.227; 95% CI, 0.1214 to 0.4232; P < 0.0001). During the RCP, a larger proportion of patients were attack-free in the inebilizumab group (87.6%) than in the placebo group (56.6%). Results were similar in the overall ITT population and were also consistent across prespecified subgroups. Additionally, the results were similar based on investigator-determined NMOSD attacks. Assessment of the proportional hazards assumption was not reported. The results were consistent across subgroups (Figure 3).
Time to NMOSD Attack During Open-Label Period
Descriptive results were reported for NMOSD attacks during the single-arm OLP. ||||| |||| || ||||||||| ||||||| || || |||||||| |||||||||| || ||| ||||||||||||| ||| |||||||||| ||||||||| |||| |||||||||||||||| |||||||| |||||||| |||| ||| || || ||||| ||||| ||| |||| ||| ||| || || ||||| |||||| ||||| ||| || ||||||||||||||||||||||| |||||||| ||| || |||||||||| ||||||||||||||||||| |||||| ||| |||||||| || || |||||| ||| || |||| |||||||||||||| |||| |||||||||||||||| |||||| |||| ||| ||||||||||||| ||| |||||| ||| ||| |||| ||| |||||| |||||||||
Figure 2: KM Plot of Time to AC-Determined NMOSD Attack in the N-MOmentum Trial, RCP (Patients With AQP4-IgG–Seropositive NMOSD)
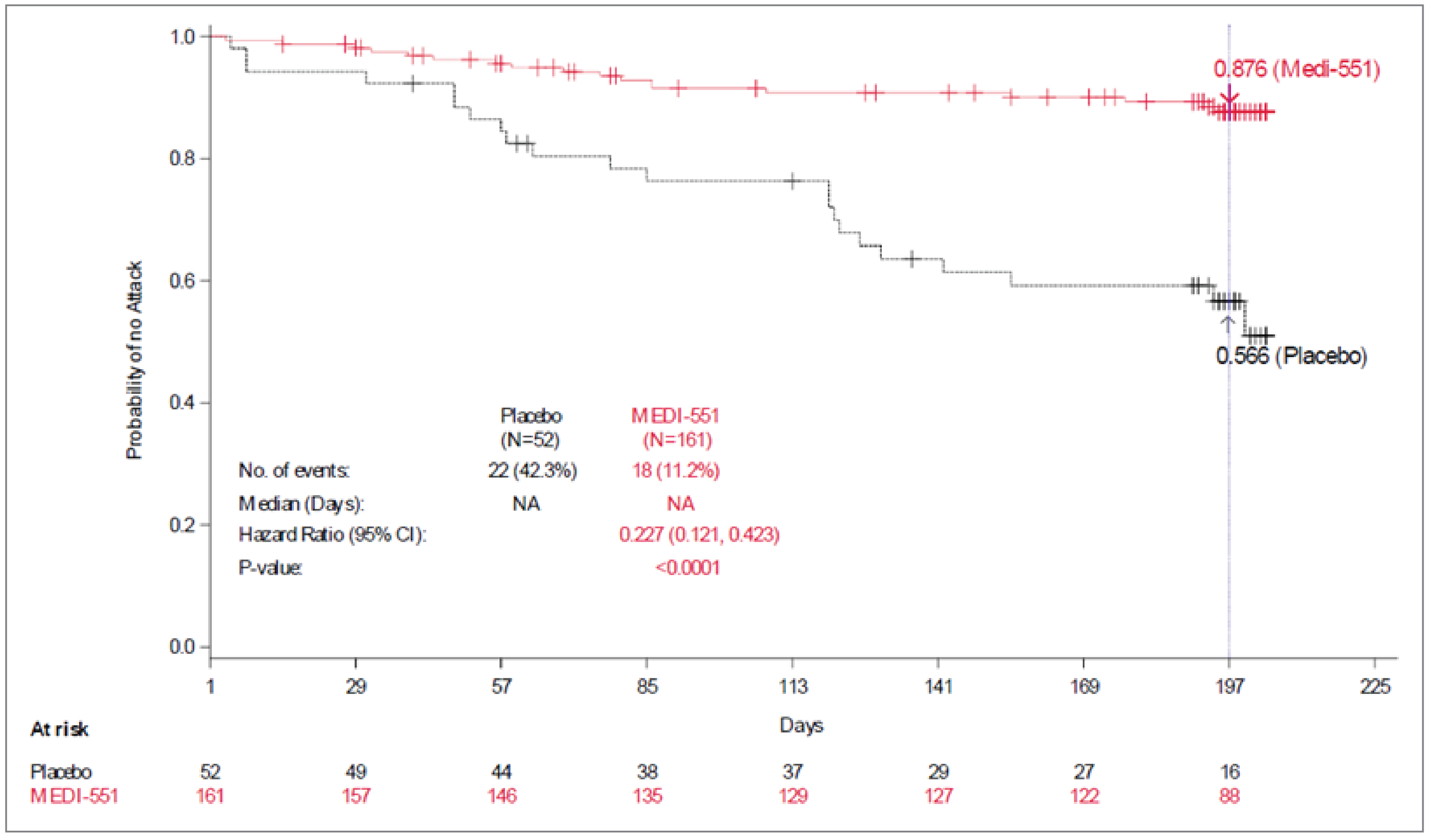
AC = adjudication committee; anti-aquaporin-4 immunoglobulin G; CI = confidence interval; KM = Kaplan-Meier; MEDI-551 = inebilizumab; NA = not applicable; NMOSD = neuromyelitis optica spectrum disorder; RCP = randomized controlled period.
Sources: Clinical Study Report for N-MOmentum53 and publication by Cree et al. (2019).20


Severity of Attacks During Randomized Period
As an exploratory analysis, attack severity was graded by investigators using a predefined exploratory scale based on the degree of neurologic worsening since the prior assessment. During the RCP among patients with seropositive NMOSD, major attacks occurred in 6 of 18 attacks (33.3%) in patients treated with inebilizumab and in 10 of 22 attacks (45.5%) among patients treated with placebo.
Recovery From Attacks During Randomized Period
Recovery from attacks was graded by the AC based on improvements in the attack criteria. As a proportion of patients with attacks, “no attack recovery” was reported for 27.8% of patients in the inebilizumab group and 40.9% of patients in the placebo group.
Domain of Attacks During Randomized Period
In the cohort of patients with AQP4-IgG–seropositive NMOSD who were treated with inebilizumab, there were 18 AC-determined attacks in 161 patients (11.2%). Of these 18 attacks, none were located in the brainstem, 11 were myelitis, and 8 were optic neuritis. One of the attacks was both myelitis and optic neuritis.
In the cohort with AQP4-IgG–seropositive NMOSD who were treated with placebo, there were 22 AC-determined attacks in 54 patients (42.3%). Of these 22 attacks, 1 was located in the brainstem, 14 were myelitis, and 10 were optic neuritis. These counts are not mutually exclusive because some attacks occurred in 2 domains and were counted under each: 1 was both brainstem and optic neuritis, and 1 was both myelitis and optic neuritis.
Annualized Attack Rate
An analysis of the “any inebilizumab” population (i.e., patients who received inebilizumab at any point during the RCP and/or OLP) identified probabilities of being attack-free that ranged from 84.5% at 1 year to 77.1% at 4 years, and the AAR ranged from 0.185 events per year in year 1 to 0.019 events per year in year 4.
When calculated across the RCP and OLP, the annualized rate of AC-determined NMOSD attacks in any patient treated with inebilizumab was 0.086 attacks per year, and a similar rate was observed in the population with AQP4-IgG–seropositive NMOSD (0.09 attacks per year).
A post hoc analysis of 75 patients with AQP4-IgG–seropositive NMOSD who received inebilizumab for 4 years or more in the RCP and OLP demonstrated that most attacks during inebilizumab treatment occurred within the first 6 months, with few attacks occurring after 6 months of inebilizumab treatment.
Note that data were not available for placebo-treated patients because the study design allowed such patients to move into the OLP after the first NMOSD attack.
Proportion of Patients With Worsening in EDSS Score
During the RCP, worsening in the EDSS score was less common in the inebilizumab group than in the placebo group among patients with AQP4-IgG–seropositive NMOSD (odds ratio = 0.355; 95% CI, 0.1704 to 0.7252; P = 0.0047) || ||||||||||||| ||||||||||| |||||| ||||||| ||| |||||| || ||||||| ||| ||||||| |||| ||||||| || ||| ||||||| ||| |||||||||||.
During the OLP, the overall inebilizumab-to-inebilizumab group had low proportions of patients who experienced worsening from baseline in EDSS score, varying from 8.1% at week 13 of the OLP to 10.8% at week 104 of the OLP. In the overall placebo-to-inebilizumab group, a higher proportion of patients experienced worsening from baseline in EDSS score at week 13 of the OLP (22.4%), although this proportion decreased to 7.0% at week 104 of the OLP. Consistent trends were observed in the population of patients with AQP4-IgG–seropositive NMOSD.
Post hoc analyses of EDSS worsening during the RCP in patients with AQP4-IgG–seropositive NMOSD with or without prior azathioprine or mycophenolate mofetil use revealed numerical benefits with inebilizumab regardless of patients’ prior use of conventional immunosuppressants.
Change From Baseline in Low-Contrast Visual Acuity Score
The change in binocular low-contrast visual acuity score from baseline to the last RCP visit did not appear to differ by treatment group within the AQP4-IgG–seropositive population (least squares |||| |||| ||||||||||| ||||||| ||| ||| ||||||| || |||||||||||||||. The results were similar in the overall ITT population.
No major changes in the binocular low-contrast visual acuity score were observed during the OLP in the placebo-to-inebilizumab group or in the inebilizumab-to-inebilizumab group ||||| |||||| |||| ||||||||| |||| || |||| ||||||||.
Number of NMOSD-Related Inpatient Hospitalizations
During the RCP among the population of patients with AQP4-IgG–seropositive NMOSD, the rate ratio of NMOSD-related inpatient hospitalizations was 0.291 (95% CI, 0.1054 to 0.8017; P = 0.0170) ||| ||| ||||||||||||| |||||||| |||||||||| ||| ||||| |||||| ||| |||||| ||||||. The results were similar in the overall ITT population ||| ||| ||||||||| || ||| |||||||| |||||||||| ||| |||||||||| |||||||||| ||||||| ||| ||||| || ||||||| ||||| ||| ||| |||||||| || ||| ||||| ||| || |||||||||||| || |||||| ||||||||||| || ||||||||||||| ||||||||| ||||||||||||||||.
|||||| ||| |||| ||| |||| || ||||||||||||| ||||||||| |||||||||||||||| || ||| |||||||||||||||||||||||||||| ||||| ||| |||||| ||| || ||| ||||||||||||||||||||||| ||||| ||| |||||.
Short Form (36) Health Survey
|||||| ||| ||| ||||| |||||||| |||||||||||| |||||||| ||||||| |||| |||||||||||| ||| |||||||| ||| |||| |||||| |||| |||||||| || ||| |||||| ||||||||| ||||| ||| ||||| |||| |||||| ||| ||||| |||| |||||| || |||| ||| ||||||||||||| ||| |||| |||||| |||| |||||||| || ||| |||||||| ||||||||| ||||| ||| ||||| |||| |||||| ||| ||||| |||| |||||| || |||| ||| ||||||||||||| || ||||||||||| |||| ||||||| |||| ||||||||| ||| || ||| |||||||| |||| ||||| |||| || ||||||||| ||||||||||| ||||||| ||||||||| |||||
Pain NRS
During the RCP, the mean changes from baseline to week 28 in the average pain score for all body locations were similar across treatment groups and between the population of patients with AQP4-IgG–seropositive NMOSD and the overall ITT populations. Similarly, the average pain scores for all body locations remained relatively constant during the OLP in the AQP4-IgG–seropositive and overall ITT populations, regardless of treatment assignment during the RCP.
Table 15: Summary of Key Efficacy Results From the N-Momentum Trial
Outcome | AQP4 − IgG–sero+ population (N = 213) | |
|---|---|---|
Placebo (N = 52) | Inebilizumab (N = 161) | |
Time to NMOSD attacka | ||
Number of patients with an attack, n (%) | 22 (42.3%) | 18 (11.2%) |
|||||| || |||||||| ||||||||||| | || ||||||| | ||| ||||||| |
Hazard ratio (95% CI) | 0.227 (0.1214 to 0.4232) | |
P valueb | < 0.0001 | |
Proportion of patients with worsening in EDSS scorea | ||
Proportion with worsening in EDSS score from baseline to last visit, n (%) | 18 (34.6%) | 24 (14.9%) |
Odds ratio (95% CI) | 0.352 (0.1704 to 0.7252) | |
||||| | |||||| | |
||||||||||||| |||||||||| ||||||| ||| | |||||| |||||||| |||||| | |
Change from baseline in low-contrast visual acuity scorea | ||
Patients evaluated | 52 | 158 |
|||||||| |||| |||| | ||||| ||||||| | ||||| ||||||| |
LS mean (SE) | 0.600 (0.999) | 0.562 (0.572) |
LS mean difference (SE) | −0.038 (1.153) | |
95% CI | (−2.3122 to 2.2357) | |
P valued | 0.9736 | |
NMOSD-related inpatient hospitalizationsa | ||
Affected patients | 7 | 9 |
Mean (SD) | 1.4 (0.8) | 1.0 (0.0) |
|||||| ||||||| | |||| | |||| |
Rate ratio (95% CI) | 0.291 (0.1054 to 0.8017) | |
P valuee | 0.0170 | |
||||||||||||| |||||||||| |||||| ||| | ||||| ||||||| ||||| | |
||||| |||||| ||||||||| ||||| | ||
|||||||| | || | ||| |
|||||||| |||| |||| | |||||| |||||||| | |||||| |||||||| |
|||| || | || | ||| |
|||| || |||| |||||| |||| |||||||| |||| | ||||| ||||||| | ||||| ||||||| |
||||| |||||||| ||||||||| ||||| | ||
||||||| | || | ||| |
|||||||| |||| |||| | |||||| ||||||| | |||||| |||||||| |
|||| || | || | ||| |
|||| || |||| |||||| |||| |||||||| |||| | ||||| ||||||| | ||||| ||||||| |
|||||| |||| |||||||| || |||| ||| |||||||| |||| ||||| |||| ||| |||| |||||||||| | ||
| |||| |||| | || | ||| |
|||||||| |||| |||| | ||||| ||||||| | ||||| ||||||| |
|| |||| |||| | ||||| ||||||| | ||||| ||||||| |
|| |||| |||||||||| |||| ||| ||||||| | |||||| ||||||| | |
||| || || || |||| |||||||||| ||| ||||||| | ||||||||| ||||||| | |
|||| | |||||| | |
AC = adjudication committee; anti-aquaporin-4 immunoglobulin G; CI = confidence interval; EDSS = Expanded Disability Status Scale; LS = least squares; ITT = intention to treat; N = total number; n = sample size; NA = not applicable; LS = least squares; NA = not applicable; NMOSD = neuromyelitis optica spectrum disorder; NRS = numerical rating scale; SD = standard deviation; SE = standard error; sero− = seronegative; sero+ = seropositive; SF-36 = Short Form (36) Health Survey.
aBased on Cox regression, logistic regression, or negative binomial models, with placebo as the reference group.
bStatistically significant at an alpha threshold of 0.05.
cStatistically significant at an alpha threshold of 0.0125.
dNot statistically significant at an alpha threshold of 0.05.
eStatistically significant at an alpha threshold of 0.025 (nor an alpha of 0.05; data not shown).
||||||| |||| ||||| |||| ||| |||| ||||||||| ||| |||||||||| |||| |||| ||||||||| |||| ||||||||||| |||||||.
||||||| || |||| ||||||||||| ||| ||| ||| |||||||| ||| ||||||||| || ||||| || |||||||| || |||||||||| |||||| ||||| |||| ||||||||||| |||||| ||| |||| ||||||||||| ||||||||||||| ||||||||||| ||||||||| |||||| ||||||| ||| ||| |||||||||| ||| |||||||| ||||||||||||.
Sources: Clinical Study Report for N-MOmentum.53 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
Refer to Table 16 and Table 17 for harms data during the RCP and OLP, respectively.
Adverse Events
Among the population of patients with AQP4-IgG–seropositive NMOSD, most patients experienced at least 1 AE during the RCP (71.2% of patients treated with placebo and 73.9% of patients treated with inebilizumab) and during the entire study duration (||||| ||| |||||| ||||||||||||). Results were similar for the overall ITT population. In the “any inebilizumab” population, 92.3% of patients with AQP4-IgG–seropositive NMOSD ||| ||||| || ||| |||||||| ||||||||||| || |||||||.
Relative to previous studies in the clinical development program, no new safety signals or concerns with the drug were identified in either study period, and long-term inebilizumab treatment was well tolerated after a median exposure of approximately 3.2 years during the study. Across both treatment periods, the highest severity TEAE experienced by most patients was a mild (grade 1) or moderate (grade 2) event.
During the RCP, most patients in the total as-treated population experienced 1 or more TEAEs (inebilizumab: ||||||; placebo: 73.2%) and similar TEAE incidence rates were observed in both treatment groups |||||||||||||| |||||| |||||||||| |||||||||||||| |||||||| |||||| |||||||||| |||||||||||||||.
No major differences were observed in the safety profile of inebilizumab in subgroups of patients with or without prior azathioprine or mycophenolate mofetil use || || |||||||| ||||||||||||||| |||||||.
Serious Adverse Events
Among patients with AQP4-IgG–seropositive NMOSD, the rate of SAEs was 4.3% in the inebilizumab group and 11.5% in the placebo group during the RCP.
|||||| ||| |||||| ||||| ||||||||| ||| |||| || |||| ||| ||||| || ||| |||||||||||||||||||||||||||| |||||| ||| ||||| || ||| ||||||||||||||||||||||| |||||| Among the “any inebilizumab” total ITT population, 20.4% experienced an SAE. Results were similar in the overall ITT population.
Withdrawals Due to Adverse Events
During the RCP, withdrawals due to AEs occurred in 0 patients treated with placebo (in either the AQP4-IgG–seropositive or overall ITT population), and in 2 patients treated with inebilizumab ||||| ||||||||||| ||| |||||||| ||| || |||||||||||||| |||||||| ||||||||| ||||||||||||| |||||||||| |||||||
|||||| ||| |||||| ||||| ||||||||| ||||||||||| ||| || || |||||||| ||||||||| ||||||| |||| ||||||| |||| ||| |||||||| |||||||||||||| ||| ||||||||| ||||||| |||| |||||||||||| ||| |||||||| |||||||| ||||||||||||| ||||| |||||||||||||| || ||| |||| ||||||||||||| |||||||||||||||| || ||| |||||||||||| |||||||||| |||||||| || ||| ||||||| ||| |||||||||| |||||||| ||| || ||||
Mortality
There were no deaths during the RCP. During the entire study, among the “any inebilizumab” total ITT population, 3 patients died due to NMOSD, pneumonia, and COVID-19 pneumonia (1 case each).
Notable Harms
In the RCP among patients with AQP4-IgG–seropositive NMOSD, 50.0% in the placebo group and 49.1% in the inebilizumab group experienced at least 1 TEAE of special interest, most commonly infections (44.2% and 40.4%, respectively) followed by infusion-related reactions (9.6% and 9.3%, respectively), hepatic function abnormality (3.8% and 5.0%), and cytopenia (0% and 5.0%). Results were similar in the overall ITT population.
During the entire study duration, among patients with AQP4-IgG–seropositive NMOSD, most patients experienced at least 1 TEAE of special interest (85.1% in the placebo-to-inebilizumab group and 71.4% in the inebilizumab-to-inebilizumab group). Similar to the RCP, the most common TEAE of special interest was infection, followed by infusion-related reaction, hepatic function abnormality, and then cytopenia. In addition, a few patients experienced hypersensitivity (0% and 1.3%, respectively) and ||||||| || ||||||||| ||||||||||| |||||||||| |||||||||||||||||||| || ||| |||||||||||||||||||||||||||| |||||| |||||||||||||||| ||||||||| ||||| ||| || |||||||||||. Again, results were similar in the overall ITT population. Cytopenias were more common in patients treated with inebilizumab compared with placebo, which is consistent with inebilizumab’s mechanism of action and the class effects of B-cell depletion.
Table 16: Key Harms During the RCP of the N-MOmentum Trial (As-Treated Population)
Adverse events | AQP4-IgG–sero+ population (N = 213) | Total (N = 230) | ||
|---|---|---|---|---|
Placebo (N = 52) | Inebilizumab (N = 161) | Placebo (N = 56) | Inebilizumab (N = 174) | |
Most common adverse events (≥ 5% in total inebilizumab group), n (%) | ||||
≥ 1 adverse event | 37 (71.2) | 119 (73.9) | 41 (73.2) | 127 (73.0) |
Nasopharyngitis | 6 (11.5) | 12 (7.5) | 6 (10.7) | 13 (7.5) |
Urinary tract infection | 5 (9.6) | 18 (11.2) | 5 (8.9) | 20 (11.5) |
Infusion-related reaction | 5 (9.6) | 15 (9.3) | 6 (10.7) | 16 (9.2) |
Arthralgia | 3 (5.8) | 17 (10.6) | 3 (5.4) | 18 (10.3) |
Back pain | 2 (3.8) | 11 (6.8) | 2 (3.6) | 13 (7.5) |
Headache | 4 (7.7) | 14 (8.7) | 4 (7.1) | 14 (8.0) |
Serious adverse events, n (%) | ||||
Patients with ≥ 1 SAE | 6 (11.5) | 7 (4.3) | 6 (10.7) | 9 (5.2) |
Patients who stopped treatment due to adverse events, n (%) | ||||
Patients who stopped | 0 | 2 (1.2) | 0 | 2 (1.1) |
|||||||| ||||||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||| |||||| | |||||| | |||||| | |||||| | |||||| |
Deaths, n (%) | ||||
Patients who died | 0 | 0 | 0 | 0 |
Adverse events of special interest, n (%) | ||||
Patients with at least 1 TEAE of special interest | 26 (50.0) | 79 (49.1) | 27 (48.2) | 83 (47.7) |
Infusion-related reaction | 5 (9.6) | 15 (9.3) | 6 (10.7) | 16 (9.2) |
Anaphylactic reaction | 0 | 0 | 0 | 0 |
Hypersensitivity | 0 | 0 | 0 | 0 |
Infections | 23 (44.2) | 65 (40.4) | 23 (41.1) | 68 (39.1) |
Hepatic function abnormality | 2 (3.8) | 8 (5.0) | 2 (3.6) | 8 (4.6) |
Cytopenia | 0 | 8 (5.0) | 0 | 8 (4.6) |
Opportunistic infection | 0 | 0 | 0 | 0 |
Progressive multifocal leukoencephalopathy | 0 | 0 | 0 | 0 |
AQP4-IgG = anti-aquaporin-4 immunoglobulin G; RCP = randomized controlled period; SAE = serious adverse event; sero− = seronegative; sero+ = seropositive; TEAE = treatment-emergent adverse event.
Sources: Clinical Study Report for N-MOmentum.53 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 17: Overall Summary of ||||||| || ||| ||||| || |||||||||| ||||||||||| |||||| ||| |||||||||||||||| ||||||||||| [Redacted]
||||||| |||||| | |||||||| |||||||||||| | |||||||||||| | ||| |||||||||||| | |||
|---|---|---|---|---|---|---|
||||||| || ||||||| | ||||||| || ||||||| | ||||||| || ||||||| | ||||||| || ||||||| | ||||||||||| | |||||||| | |
|||| |||||| ||||||| |||||| || ||| || ||||| ||| |||||||||||| ||||||||| | ||||||
||||||| ||||| | || ||||||| | ||| ||||||| | || ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
||||||||||||||| | ||||||| | || ||||||| | ||||||| | || ||||||| | || ||||||| | || ||||||| |
||||||| ||||| ||||||||| | || ||||||| | || ||||||| | || ||||||| | || ||||||| | || ||||||| | || ||||||| |
||||| ||||||||||| ||||| ||||||||| | ||||||| | || ||||||| | ||||||| | || ||||||| | || ||||||| | || ||||||| |
|||||||| | ||||||| | || ||||||| | ||||||| | || ||||||| | || ||||||| | || ||||||| |
|||||||||| | ||||||| | || ||||||| | || ||||||| | || ||||||| | || ||||||| | || ||||||| |
|||| |||| | |||||| | || |||||| | ||||||| | || |||||| | || ||||||| | || ||||||| |
||||||| ||||||| |||||| |||| ||||||||| || ||| || |||||||| || ||||| ||| |||||||||||| |||||||||| | ||||||
|||||||| |||||| | || ||||||| | || ||||||| | || ||||||| | || ||||||| | || ||||||| | || ||||||| |
|||||||||||||||| ||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||| ||| |||||||||||| | || ||||||| | || |||||| | || ||||||| | || |||||| | || ||||||| | || ||||||| |
||||||| |||||| ||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||||||||||||| ||| ||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||||| ||||||||| ||| ||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| ||| ||||||| ||||||||| ||| || ||||||| |||||||||| | ||||||
|||||||| ||| ||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||||| |||||||||| |||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||||| ||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||| |||||||| |||| ||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||| |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||| |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||| | ||||||
|||||||| ||| |||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||||| |||||| || ||||||| |||||||||||| | ||||||
|||||||| |||| || ||||| ||| || | || ||||||| | ||| ||||||| | || ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
|||||||||||||||| |||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||||| |||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||| | || ||||||| | ||| ||||||| | || ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
||||||| |||||||| ||||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
||||||||||||| |||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| |||| ||||||| |||| ||| |||| |||||||| |||||||||| || ||| |||||| || ||||||||| |||| || ||||||||| |||| ||||||||| |||||| |||||||||||| |||||||||| |||||||||||||||||||| |||||||||| ||||| ||| ||||||||| ||||||||||||||||||||||| ||||||| |||||||||||| ||||||||||||| |||||||||| |||||||||||||||||||| ||||||||| ||||||| |||||| ||||||||||||||||| |||||||| ||||| |||||| ||||||||||||||||||||| ||||||| |||||||||||||| |||||||| ||||| |||||| ||| ||||||||||||||||| |||||||| || ||| ||||| ||| |||| |||||||||||| ||||||| || |||||||| |||||||||
Critical Appraisal
Internal Validity
The N-MOmentum phase II and III trial was the only study included in this review. The N-MOmentum trial included an initial period that was randomized, placebo-controlled, and double-blind, with a duration of up to 197 days (the RCP) in which patients received an IV infusion of inebilizumab 300 mg or placebo on day 1 and day 15. Patients could proceed to an open-label, single-arm period (the OLP) with a minimum duration of 2 years, during which patients received inebilizumab 300 mg IV every 6 months starting 6 months after the first infusion. Blinding of initial treatment allocation was maintained during the OLP by mimicking the initial administration schedule (i.e., day 1 and day 15 IV) using matched-administration placebo. All key outcomes were assessed during the RCP, so this critical appraisal does not focus on the OLP; however, in general, an open-label and single-arm study design in the OLP has implications for the overall strength and interpretability of the results, and there is an increased risk of bias in the estimation of treatment effects due to potential confounding related to natural history and any other known or unknown prognostic factors; therefore, the data from the OLP are considered supportive. One strength of including an OLP in the study design is a longer duration of exposure for the assessment of safety outcomes.
In the RCP, there were no major concerns with regard to internal validity related to the study design in terms of, for example, the method of randomization, concealment of allocation, maintenance of blinding, or balance of patient characteristics between treatment arms. Although there were some minor between-arm differences in the distribution of races, median EDSS score, baseline visual acuity scores, and the rates of prior plasmapheresis, these differences were not expected to cause bias or confound interpretation of the results of the study. Based on consultations with the clinical experts, these factors were not expected to influence the relapse rate or time to first NMOSD attack, which are the most important drivers of all other outcomes (e.g., disability worsening, decline in visual acuity) in NMOSD. Regarding the maintenance of blinding, there were no particular AEs that were more frequent in the treatment arm that would be expected to unblind patients. The patient baseline characteristics and the results of the study end points were similar between the AQP4-IgG–seropositive subpopulation (n = 213) and the total population (N = 230), which was expected, given that few patients had seronegative NMOSD (n = 17). Randomization was stratified by serotype, followed by Japan versus non-Japan regions. The subgroup results of the seropositive population therefore retained the study design benefits of randomization.
The trial was designed to reduce potential risks to patients treated with placebo by limiting the RCP duration, implementing a 3:1 randomization ratio, and allowing patients to enter the OLP after a single attack. The N-MOmentum trial was stopped early on the recommendation of an independent data-monitoring committee, which found that the efficacy of inebilizumab had been established (conditional power > 99%) and that there was no justification to continue exposing placebo-treated patients to an increased risk of NMOSD attack. While still blinded to the study results, the sponsor offered all patients in the RCP the option to enter the OLP or exit to a safety follow-up period, per independent data-monitoring committee recommendation. No information fraction for the analysis relative to the preplanned analysis was reported. Because the study was originally powered for 67 NMOSD attack events in the seropositive subgroup and there were only 42 attacks during the RCP as of the end of the study, the information fraction would be somewhat low, which implies there is some risk of overestimating the true effect. However, statistical testing of the primary and key secondary outcomes was designed to control 2-sided type I error at an alpha of 0.05 based on the Bonferroni chain procedure.
The primary end point of time to AC-determined NMOSD attack was considered appropriate by the clinical experts consulted by CADTH, and this was supported by the patient and clinician group inputs, which stated that prevention of attacks was the most critical outcome due to all other symptoms and the potential for disability stemming from the damage that occurs and accumulates during attacks. The secondary end points of the N-MOmentum trial, including EDSS score, low-contrast visual acuity score, and NMOSD-related inpatient hospitalizations, also addressed outcomes considered valuable to patients and clinicians.
A small proportion of patients did not complete the RCP: 6 out of 174 patients assigned to inebilizumab, and 2 out of 56 assigned to placebo. There were no concerns regarding imbalanced withdrawals or the reasons for withdrawals. || ||| |||| || |||||||| |||||||| || |||||||||||| |||||||||| || ||||||| ||| ||| |||||||| ||| |||||| ||||||||| ||| || ||||||||||| || |||||||| ||| |||||||| ||| |||||| || ||||| |||||||| ||| ||| ||||||| ||||||||||.
Despite low withdrawals, a high number of patients were censored in the primary analysis of time to first NMOSD attack, especially in the inebilizumab arm. However, this was not considered concerning because the primary reason for censoring patients appeared to be the early termination of the study, given that withdrawals were generally low. As a result, the CADTH team concluded that censoring was unrelated to prognosis or other confounding factors. The sponsor did not report any evaluation of the proportional hazards assumption in the context of the Cox model used for time to first NMOSD attack, but it was considered unlikely to represent a major concern, based on visual appraisal of the Kaplan-Meier curves.
The proportion of patients who had a worsening in EDSS score was evaluated as a key secondary outcome. The usefulness and validity of the EDSS in NMOSD was discussed with the clinical experts consulted by CADTH. EDSS is a validated scale for disability in MS but has limitations, including an over-reliance on ambulation as a metric of disability, when other motor challenges in MS can contribute to loss of independence and impair the patient’s ability to do daily tasks of living and working. As a result, EDSS is known to become less sensitive to a change in disability at some ranges. This weakness is exacerbated when applied to NMOSD, for which the EDSS was neither designed nor validated. A strength of the EDSS is its ubiquity and familiarity among clinicians and its resulting ease of interpretation and, moreover, there is no more robust or valid scale to replace it with. Ultimately, despite its weaknesses in general and in its application to NMOSD instead of MS, the EDSS was an appropriate choice for the purpose of the clinical trial, and some of its insensitivities to change are overcome by the trial’s inclusion of low-contrast visual acuity as a key secondary outcome. The thresholds for “worsening” are the same as those commonly used in MS trials although, notably, there is some minor inter-trial variability and subjectivity in how worsening is defined across trials of MS. Nonetheless, the thresholds chosen were considered appropriate by the clinical experts consulted by CADTH.
External Validity
The eligibility criteria and baseline patient characteristics of the N-MOmentum trial were considered by the consulted clinical experts to be a reasonable approximation of patients with NMOSD in Canadian clinical practice. The exclusion criteria related to concurrent immunosuppressive or steroid therapy, prior IVIG therapy, and other prior therapies were appropriate for the context of the clinical trial design, but it was noted by the clinical experts that these patients would not necessarily be excluded from accessing inebilizumab for the treatment of NMOSD in clinical practice. For instance, it would not be appropriate in clinical practice to exclude patients who require concomitant steroid treatment for comorbid conditions such as asthma or inflammatory bowel disease, or to exclude patients who have received prior IVIG. Patients in the trial were also required to receive a 2-week course of corticosteroids followed by a mandatory taper, with the intention of protecting them from attack until the development of effect from the study drug; the clinical experts consulted by CADTH indicated that this adequately reflects real-world clinical practice. Premedication for infusions and practices related to rescue medication were also reflective of real-world practice in Canada.
The N-MOmentum trial had a high proportion of screen failures that was considered potentially concerning for external validity. Of 467 patients who entered the N-MOmentum study, 236 were screen failures, and only 5 of these were the result of the study’s early cessation. The most common reason was not meeting the inclusion or exclusion criteria (n = 204). Some common reasons, in no order, included unacceptable biomarker metrics for organ function, low B cells, recent receipt of prohibited therapeutics before screening (e.g., rituximab in the prior 6 months), positive or indeterminate tuberculosis test results, or insufficient number of recent NMOSD attacks.27 A clinical expert consulted by CADTH indicated that this is a high rate of screen failures for clinical practice but less surprising for clinical trials, which are always more restrictive. |||||||||||||||| || ||| |||||| |||||||| |||| ||| || |||||||||||| |||||||| ||||| ||| |||||||| |||||| ||||| ||| ||||||||| |||| || ||| ||||||| || |||||||| |||||||| ||| ||||| || ||| || |||||| ||||| || |||||||||||| || ||||||| ||||| ||||||||| |||||||| || ||| |||||.
The assessment of NMOSD attacks may differ in clinical practice, as there are no standardized guidelines. The criteria for NMOSD attacks in the N-MOmentum trial were more selective and specific than assessment in clinical practice but were not considered generally inappropriate by the experts consulted by CADTH.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the deliberations of CADTH’s expert committee, and a final certainty rating was determined as outlined by the GRADE Working Group:24,25
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for inebilizumab versus placebo.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
There is a lack of head-to-head data comparing inebilizumab with other active therapies for adults with NMOSD. The objective of this section is to summarize and critically appraise the ITC submitted by the sponsor, which assessed the relative efficacy of inebilizumab versus each of satralizumab, eculizumab, and rituximab in patients with NMOSD. This summary also informs the pharmacoeconomic evaluation.
Description of Indirect Comparisons
The sponsor-submitted ITCs included comparisons of inebilizumab against satralizumab and eculizumab using published study data and MAIC methodology to adjust for between-trial differences, and a comparison against rituximab using IPD. Other therapies (azathioprine and mycophenolate mofetil) were also of interest and were included in the study selection criteria, but ITCs against these therapies were ultimately not considered feasible. The study selection criteria are presented in Table 18.
Table 18: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adult patients with NMOSD |
Intervention | Inebilizumab |
Comparator | Relevant comparators per CADTH’s criteria:
Additional comparators were considered in the global review, but are not considered “relevant comparators” per CADTH’s criteria |
Outcome |
|
||||| ||||||| | ||||||||||||| |||||||| |||||||| ||||||| ||| |||||||||| ||||||| |||| |||||||| ||| ||||||||||||||||||| |||||||||| |||||||||| ||||||| |||| ||| |||||||| || ||||| |
||||||||||| ||||||||||||||| | ||||||||| ||||||| ||| ||||| ||||||||| |||| |||||||| ||||||||| ||||||| |
||||||||| |||||||| | ||||||| ||| ||||| || |||||||||||| ||||||| ||||||||||||||||||||||||| ||||||||||||||||||||| ||||||| ||| |||||||| || |||||||| ||||| |||||||| |||| ||||||| ||||||||| ||||||||||| |||||||| ||||||||||||| ||||||||||||| |||||||||| |||||||||||| |||||||||||||| ||||||| || ||||| |||||||| |||||||||||||||| ||||||| |
||||||||| |||||||| | |||||| |||||||||||||| ||||||||||||||||||||| ||||| || ||||||| ||| |||||| |||||| |||||||||| |||||||||||||||||||||||| |||||||| ||| |||||| ||| ||||||| ||| ||||||||||||| ||||||||| || ||||||| |||||||| |||||||| ||||||||||||||| |||||| ||||||| |||||||| |||||||||| |||||||||||||| |||||| |||||||| |||||||||| ||| |||||| |||||||||| ||||||| |
||||||||| ||||||| | |||||| ||| ||||||||| |||| ||||||||||||| |||||||| || ||| |||||||| ||| ||||||||| |||||||| || ||||||||||| |||||||| |||||||| |||| ||||||||||||| ||||||||| ||| ||||||||||||| |||| |||||||| ||||||| |||||||||| ||||| ||||||||| ||| |||||||| |
|||| |||||||||| ||||||| | |||| |||| ||||||||| ||| |||||||| ||||||| ||||| || ||||||||||| |||||||| || |||||| |||||||||||| ||||||||||| ||| |||| ||||||||||| |||| |||||||||| ||| ||||||||| || ||| ||||||||||| |||| || ||||||| |||||| || ||||||||||| ||||| ||||||||| |
||||||| |||||||||| | ||||| |||||||| ||||||||| |||||||| ||| ||||||| || |||| |||||||| ||||| ||||| ||| |||||||| |||||||||||| ||||| ||| |||||||||| |||||| ||| |||||||||||||| |||||||| || |||| || ||| |||||||| ||||||||||||||| |||||||||| ||||||| ||||||||| ||| |||||| |||||||| ||| ||||||| |||| ||||||| |||||| ||||||||||| ||||||||| |
AAR = annualized attack rate; AC = adjudication committee; ITC = indirect treatment comparison; NMOSD = neuromyelitis optica spectrum disorder; SLR = systematic literature review; vs. = versus.
Sources: Technical reports for SLR62 and for the indirect comparisons of inebilizumab with satralizumab and eculizumab63 and rituximab.64 Details included in the table are from the sponsor’s summary of clinical evidence.
ITC Design
Objectives
At the time of this submission, there were no direct comparisons of inebilizumab with relevant comparator treatments for adult patients with NMOSD.
The sponsor was aware of a published NMA that attempted to compare the efficacy of eculizumab, satralizumab, and inebilizumab in adult patients with AQP4-IgG–seropositive NMOSD,28 but noted that the analysis had several weaknesses: small sample sizes in the monotherapy-based comparisons; significant differences in patient baseline data such as prior attack history, baseline disability, and disease duration; substantial variation in the definitions of attack criteria across the included trials; and no attack events were detected for patients who received eculizumab monotherapy, so an imputed HR was selected by the authors for use in the NMA. Additionally, the trials differed in the age range of eligible patients and whether patients could receive concurrent immunotherapy during the study.
The sponsor therefore conducted indirect comparisons with the stated objective of generating more robust evidence. Comparisons were conducted against satralizumab and eculizumab using published summary-level data from placebo-controlled trials, and against (off-label) rituximab using IPD from a collection of small observational studies.
Although azathioprine and mycophenolate mofetil were also off-label comparators of interest, the sponsor noted that the studies available for these therapies in NMOSD were predominantly retrospective or prospective observational studies; only 1 small unblinded randomized trial was identified, which compared azathioprine with rituximab in patients in Iran.38 Considerable heterogeneity was noted across all studies that evaluated azathioprine or mycophenolate mofetil (e.g., different study designs, variable dosing, different definitions of NMOSD attack) and therefore it was unfeasible to conduct a robust indirect comparison of inebilizumab versus azathioprine or mycophenolate mofetil. Notably, this was also the case for rituximab (i.e., data selected by the sponsor were from small observational studies in narrow populations); however, the sponsor had access to IPD for these studies.
Study Selection Methods
To inform the comparisons, relevant publications were identified through a global systematic literature review that was conducted to identify clinical and economic studies published between 2010 and June 2021, with an updated targeted literature search for azathioprine or mycophenolate mofetil studies conducted in July 2023.
||| |||||||| ||| ||||||||| ||||| || ||| ||||||||| ||||||||| ||| |||||||||| ||||||| ||| ||||||||||||| |||||||| ||||||||| ||| |||||||||| |||||| || ||| |||||||| ||| |||||||| ||||||||||||| ||| |||||||||| ||||||||| ||| ||||||| |||||||||| |||||||| || |||||||||| |||||||| ||||||| ||| ||||||| ||| ||||||||||||| ||||||| |||||||| ||| ||||||||||| ||||||| || |||||| |||||| |||||||| |||||||| ||| |||||||||| ||||||| || |||||||||||| ||||||||||||||||||||| |||||||||| |||||||| ||| ||||| ||||||||||||| ||||||| ||||||||| |||||||| ||||||||||||| |||||| |||||||| |||||||||| |||| |||||||||||| ||||| ||| ||||| |||||||| || ||| ||||||||||||| ||| ||||||||| |||| || ||||||||| || ||| ||||||||||| ||||||||| |||| |||||||||| || ||||||| ||| || || |||||||||| ||| |||||||||| |||||||| |||||||| |||| ||| || || ||||||| || |||||||||||| |||||| ||||||| |||| |||||||| |||| |||||||| ||||||||||| |||||||||||| ||||. Table 18 || ||| ||||| |||||||| |||||||||||| || || |||||||| ||| ||||| |||||||| |||||||| || ||||||||| ||| ||||||| ||||||||||||| |||||||| || |||||||| |||| |||| || ||||| || || ||| || ||||| ||||||| ||| |||| ||||||| |||| |||||||| ||| ||||||||| || ||| ||||||||||| |||||||| ||||| || ||| ||||| ||| ||||||||| ||| |||||||||| ||||| || ||| |||| ||||| |||| ||||||||| ||||||| |||||| ||| ||||||||||||| || ||||||||| ||||||| ||| ||| |||||||| |||| |||||||| ||||||| |||||||||| |||||||||||| ||| |||||||| |||| |||||||||| ||| ||||||||| || ||| ||||||||||| ||||||||| || ||||||||||| |||||| ||||||||| ||| ||| ||| ||||||| || ||||||||| ||| ||||.
The studies from the systematic literature review were used to inform the indirect comparisons of inebilizumab versus rituximab, satralizumab, and eculizumab. To supplement missing data from the trial publications (especially regarding the AQP4-IgG–seropositive subpopulation), data to guide the satralizumab comparisons were also obtained from the FDA Center for Drug Evaluation and Research’s review of satralizumab in NMOSD69 and CADTH’s Clinical Review Report for satralizumab in NMOSD.70 Data to guide the eculizumab comparisons were also obtained from the CADTH Clinical Review Report for eculizumab in NMOSD.71|||| ||||||| || |||| |||||||| ||||| ||| |||||||| || ||| |||||||| ||| |||| ||||||| ||||||| ||||||||| ||||||| ||| |||||||| |||||||||||| |||| ||| |||||||||| ||||| |||| |||||||| ||| ||||||| || ||||| ||| ||||||| |||||||||| ||||||||| |||| ||||||| |||| ||| ||||||||||||| |||||||| |||| ||| || |||||||||| |||| |||||||| |||||||||||||| |||| ||| || ||||||| ||||||| ||||| |||| || ||||||||||| || |||||||||| ||| |||| || ||||||||| || ||| |||||||| ||||||| ||| |||| || ||||||| |||| ||| ||||| || |||| |||| ||||||||| || ||||| ||| |||||||| |||| || |||| || |||||||||||||| ||||||| || ||||||||||||| |||||||||| |||| |||||| |||||| ||| ||||||| || |||||||||||||| ||||||| |||| |||||||| |||||| ||||||| || ||| || |||| |||||||||||||| ||| |||||| ||||||| |||| |||||||| ||||| ||| |||||||| ||||||||||||||| |||||||||| ||||||| |||||| ||||||||| ||||||||| |||||||.
ITC Analysis Methods
Comparison Against Satralizumab and Eculizumab
Because only summary-level data were available for the comparators satralizumab and eculizumab, anchored indirect comparisons were conducted using a Bucher indirect comparison75 (i.e., unadjusted) and anchored MAIC methodology76 (i.e., adjusted for between-trial heterogeneity) using placebo as the common comparator. The MAIC reweighted the IPD of the N-MOmentum study to match the average baseline characteristics of the comparator trial based on summary-level data. With the weights from the MAIC, the results of the inebilizumab trial were re-estimated as if the patients had the same select baseline characteristics as in the comparator trial.
The MAIC approach approximates a propensity score–weighting approach, where the patients in the trial with IPD are weighted by their inverse odds of being in that trial compared with the comparator trial. The weights can be estimated using the generalized method of moments.77 HRs of weighted Cox regressions were generated using the weights from the MAIC analyses. ||||||||| |||||| ||||| |||| |||||||| || ||| |||||| || ||| |||||| ||||||| ||||||| || ||| ||| || ||| ||||||| |||||||.
The baseline characteristics selected for adjustment were those identified by the sponsor as having statistically significant differences between the trials in the ITT populations. No literature or clinical expert opinion was sought to inform potentially important treatment-effect modifiers.
The only outcome assessed was the time to NMOSD attack.
Comparison Against Rituximab
||| ||||||| ||| |||||| || ||| |||| ||||||||||||||| |||||||||| ||||||||| |||||||. An unanchored MAIC was conducted using entropy balancing78 to reweight the IPD data from the N-MOmentum trial on patients receiving inebilizumab to achieve covariate balance with IPD data for patients treated with rituximab. |||| |||||||||||||| |||||||| ||| |||| ||||| || ||||||| ||||||||| ||||||| |||||||| || ||||||| |||||||| |||| |||||||| ||||| ||| ||||||| ||||||||||||||| ||||||||||||||| || ||||| || ||||||| ||| ||||||||||||| ||||| |||||||| ||||||||||||| || ||||||||||| || ||| ||||| |||| |||||||| |||| ||| ||||||||||. The analyses were unanchored. The technical report states that AAR and EDSS were both assessed; however, only results for AAR were provided by the sponsor.
Table 19: ITC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | An unanchored MAIC approach was used for the comparison of inebilizumab vs. rituximab, while an anchored MAIC approach was used for the comparisons of inebilizumab vs. satralizumab and inebilizumab vs. eculizumab. Efficacy was compared across the different studies by reweighting the inebilizumab-treated patients to match the populations from studies of comparators based on patient and trial characteristics. Relative efficacy was estimated using the reweighted population. Various models were used for each comparison that involved matching based on different sets of baseline characteristics. |
Priors | NR |
Assessment of model fit | NR |
Assessment of consistency | NR |
Assessment of convergence | NR |
Outcomes |
|
Follow-up time points | NR |
Sensitivity analyses | Analyses were conducted where different sets of baseline characteristics were used for matching. Justifications for these particular sensitivity analyses were not provided.
|
Subgroup analysis | Only the AQP4-IgG–seropositive subpopulation was relevant for the purposes of this assessment. ||| ||||||| ||||||||| |||| || ||| ||| ||||||||||| |||||| ||| ||||||||||| ||||||| || ||| |||||||||||| ||||||||||| || |||||. |
Methods for pairwise meta-analysis | NR |
AAR = annualized attack rate; AC = adjudication committee; AQP4-IgG = anti-aquaporin-4 immunoglobulin G; EDSS = Expanded Disability Status Scale; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; NMOSD = neuromyelitis optica spectrum disorder; NR = not reported; vs. = versus..
Sources: Technical reports for the systematic literature review62 and for the indirect comparisons of inebilizumab with satralizumab and eculizumab63 and rituximab.64 Details included in the table are from the sponsor’s summary of clinical evidence.
Results of ITC
Summary of Included Studies
Data for inebilizumab were based on IPD from the N-MOmentum trial, as previously described.
Data for the comparators were identified from the following sources:
Inebilizumab versus eculizumab: Summary-level data from 1 double-blind, randomized, placebo-controlled, time-to-event trial (PREVENT) restricted to patients with AQP4-IgG–seropositive NMOSD with a baseline EDSS score of 7 points or less.
Inebilizumab versus satralizumab: Summary-level data from 2 double-blind, randomized, placebo-controlled, time-to-event trials (SAkuraSky and SAkuraStar) that enrolled adolescent and adult patients with an EDSS score of 6.5 points or less. One trial, SAkuraStar, evaluated satralizumab monotherapy. The other trial, SAkuraSky, evaluated satralizumab added onto stable immunosuppressive therapy. An additional dataset was available to facilitate a comparison of inebilizumab versus satralizumab monotherapy in the subgroup of patients with AQP4-IgG–seropositive NMOSD. Results for the comparison with SAkuraSky are not discussed in this report due to the difference in study design (i.e., combination therapy).
Inebilizumab versus rituximab: IPD from 4 small observational studies evaluating different induction and maintenance dosing regimens with variable follow-up times.
For the eculizumab and satralizumab comparisons, the included studies and their key design elements are summarized in Table 20, and the baseline characteristics of the study populations are summarized in Table 21. There were considerable differences in the study populations (e.g., the SAkuraSky trial uniquely included adolescent patients) and the outcome definition of NMO or NMOSD “attacks” or “relapses.” Additionally, the trials differed in the age range of eligible patients and whether patients could receive concurrent immunotherapy during the study. The N-MOmentum and SAkuraStar trials disallowed concurrent immunotherapy, while the SAkuraSky trial required it and the PREVENT trial allowed it. In the PREVENT trial, the majority of patients were taking concomitant immunotherapy during the trial. Patient baseline characteristics differed between the trials in terms of race, region, and prior maintenance therapy (N-MOmentum versus PREVENT trial only; prior maintenance therapy status was not reported by the satralizumab trials), and whether recent attacks were first attacks or relapses. The sponsor also identified that there were differences in disease duration between the trials, but these data were not reported.
The comparator studies informing the IPD analyses of inebilizumab compared with rituximab are summarized in Table 22 and the baseline characteristics are summarized in Table 23. All of the included rituximab studies were small, uncontrolled, regional observational studies with 18 to 32 patients. The published, placebo-controlled, double-blind RCT of rituximab conducted by Tahara et al. (2020)29 in adult patients with AQO4-IgG–seropositive NMOSD was not discussed by the sponsor as a potential source of data for ITCs. It is unclear why this study was not selected, as justification was not reported.
Table 20: Summary of Studies Included in the MAICs Comparing Inebilizumab With Satralizumab and Eculizumab
Characteristic | N-MOmentum | SAkuraStar | SAkuraSky | PREVENT |
|---|---|---|---|---|
NCT number | NCT02200770 | NCT02073279 | NCT02028884 | NCT01892345 |
Study design | Phase II and III, randomized, double-blind, placebo-controlled study | Phase III, randomized, double-blind, placebo-controlled study | Phase III, randomized, double-blind, placebo-controlled study | Phase III, randomized, double-blind, placebo-controlled, time-to-event study |
Setting | International (Canada, US, Europe, Asia, South America, New Zealand) | International (US, Europe, Asia) | International (US, Europe, Asia) | International (US, South America, Australia, Europe, Asia) |
Population | Adults with NMOSD, either AQP4-IgG seropositive or seronegative | Adults with NMOSD, either AQP4-IgG seropositive or seronegative | Adolescents and adults with NMOSD, either AQP4-IgG seropositive or seronegative | Adults with AQP4-IgG–seropositive NMOSD |
Key eligibility criteria |
|
|
|
|
Sample size | 231 (213 of whom were seropositive for AQP4-IgG) | 95 (64 of whom were seropositive for AQP4-IgG) | 83 (55 of whom were seropositive for AQP4-IgG) | 143 |
Intervention (dose) | Inebilizumab (300 mg IV on day 1 and day 15 during the randomized period) | Satralizumab monotherapy (120 mg SC at weeks 0, 2, and 4 and every 4 weeks thereafter) | Satralizumab in combination with stable immunosuppressant treatment (120 mg SC at weeks 0, 2, and 4 and every 4 weeks thereafter) | Eculizumab (900 mg IV weekly for the first 4 doses starting on day 1, followed by 1,200 mg every 2 weeks starting at week 4) |
Comparator | Placebo | Placebo | Placebo in combination with stable immunosuppressant treatment | Placebo |
Concomitant immunotherapy: Allowed or disallowed | Disallowed | Disallowed | Required | Allowed but not required; most patients did receive |
Duration | Randomized controlled period was 197 days in duration | Up to week 216 | The median treatment duration with satralizumab in the double-blind period was 107.4 weeks | The median treatment duration with eculizumab was 89.43 weeks |
Primary end point | Time to first AC-determined NMOSD attack | First protocol-defined relapse in a time-to-event analysis | First protocol-defined relapse in a time-to-event analysis | First adjudicated relapse |
Key outcomes assessed in AQP4– seropositive population |
|
|
|
|
Definition of “attack” or “relapse” | The definition of an NMO or NMOSD attack was the presence of a new symptom(s) or worsening of an existing symptom(s) related to NMO or NMOSD that met at least 1 of the protocol-defined criteria for an NMO or NMOSD attack, such as:
| An increase of at least 1 of the following:
| An increase of at least 1 of the following:
|
|
AC = adjudication committee; AQP4 = aquaporin-4; AQP4-IgG = anti-aquaporin-4 immunoglobulin G; AZA = azathioprine; EDSS = Expanded Disability Status Scale; FSS = functional system score; HAI = Hauser Ambulation Index; KFS = Kurtzke Functional System; MAIC = matching-adjusted indirect comparison; MMF = mycophenolate mofetil; mRS = modified Rankin Scale; NCT = National Clinical Trial; NMO = neuromyelitis optica; NMOSD = neuromyelitis optica spectrum disorder; SC = subcutaneous.
Sources: Clinical Study Report for N-MOmentum53 and publications of the SAkuraStar,79 SAkuraSky,80 and PREVENT42 trials. Technical report for indirect treatment comparisons comparing inebilizumab with satralizumab and eculizumab.63 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 21: Baseline Characteristics in Studies Included in the MAICs Comparing Inebilizumab With Satralizumab and Eculizumab (Anti-AQP4–Seropositive Population)
Category | N-MOmentum | SAkuraStar | SAkuraSky | PREVENT | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Inebilizumab | PBO | Total | Satralizumab | PBO | Total | P value | Satralizumab | PBO | Total | P value | Eculizumab | PBO | Total | P value | |
N | 161 | 52 | 213 | 41 | 23 | 64 | NA | 26 | 26 | 52 | NA | 96 | 47 | 143 | NA |
Mean age, years | 43.17 | 42.42 | 42.99 | 46.05 | 40.13 | 43.92 | 0.169 | 45.62 | 45.62 | 45.6 | 0.277 | 43.9 | 45 | 44.3 | 0.658 |
Age SD, years | 11.59 | 14.33 | 12.32 | 12.00 | 11.52 | 11.83 | NA | 10.45 | 14.71 | 14.16 | NA | 13.32 | 13.29 | 13.27 | NA |
Female (%) | 94 | 94 | 94 | 76 | 96 | 83 | 0.001 | 100 | 100 | 100 | 0.191 | 92 | 89 | 91 | 0.519 |
Race | |||||||||||||||
White (%) | 53 | 46 | 52 | 46 | 57 | 50 | 0.041 | 46 | 46 | 46 | 0.007 | 48 | 51 | 49 | 0.010 |
Non-white (%) | 47 | 54 | 48 | 54 | 43 | 50 | NR | 54 | 54 | 54 | NR | 52 | 49 | 51 | NR |
Asian (%) | 23 | 15 | 21 | 17 | 26 | 20 | NR | 54 | 50 | 52 | NR | 39 | 32 | 36 | NR |
Black or African American (%) | 9 | 10 | 9 | 27 | 13 | 22 | NR | 0 | 0 | 0 | NR | 9 | 17 | 12 | NR |
American Indian or Native American (%) | 7 | 10 | 8 | 5 | 0 | 3 | NR | 0 | 0 | 0 | NR | 4 | 0 | 3 | NR |
Other (%) | 7 | 19 | 10 | 5 | 4 | 5 | NR | 0 | 4 | 2 | NR | 48 | 51 | 49 | NR |
Region | |||||||||||||||
European Union (%) | 41 | 31 | 38 | 20 | 22 | 20 | 0.000 | 50 | 50 | 50 | 0.003 | 33 | 40 | 36 | 0.000 |
North America (%) | 18 | 21 | 19 | 68 | 52 | 63 | 0 | 0 | 0 | 30 | 32 | 31 | |||
Asia-Pacific (%) | 25 | 17 | 23 | 12 | 26 | 17 | 50 | 50 | 50 | 36 | 28 | 34 | |||
Rest of world (%) | 16 | 31 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||
AAR | |||||||||||||||
Mean | 1.68 | 1.46 | 1.63 | NR | NR | NR | NR | 1.62 | 1.35 | 1.5 | 0.696 | 1.94 | 2.07 | 2.0 | 0.085 |
SD | 1.49 | 1.36 | 1.46 | NR | NR | NR | 0.55 | 0.42 | 0.49 | 0.90 | 1.04 | 0.94 | |||
EDSS | |||||||||||||||
Mean | 3.81 | 4.35 | 3.94 | 4.00 | 3.40 | 3.78 | 0.509 | 4.21 | 3.72 | 3.97 | 0.231 | 4.15 | 4.26 | 4.19 | 0.121 |
SD | 1.77 | 1.63 | 1.74 | 1.60 | 1.50 | 1.56 | 1.54 | 1.49 | 1.52 | 1.65 | 1.51 | 1.60 | |||
Prior maintenance therapy | |||||||||||||||
Yes (%) | 68 | 69 | 68 | NR | NR | NR | NA | NR | NR | NR | NA | 92 | 96 | 93 | 0.000 |
Recent relapse | |||||||||||||||
First attack (%) | 15 | 25 | 17 | 12 | 17 | 14 | 0.533 | 42 | 58 | 50 | 0.001 | NR | NR | NR | NA |
Relapse (%) | 85 | 75 | 83 | 88 | 83 | 86 | 58 | 42 | 50 | NR | NR | NR | |||
AAR = annualized attack rate; AQP4 = aquaporin-4; EDSS = Expanded Disability Status Scale; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; PBO = placebo; NA = not applicable; NR = not reported; SD = standard deviation.
Sources: Clinical Study Report for N-MOmentum53 and publications of the SAkuraStar,79 SAkuraSky,80 and PREVENT42 trials. Technical report for ITCs comparing inebilizumab with satralizumab and eculizumab.63 Details included in the table are from the sponsor’s summary of clinical evidence.
Table 22: Summary of Comparator Studies Included in the IPD Analyses Comparing Inebilizumab With Rituximab
Category | Cabre et al. (2018) | Kim et al. (2011) | Li et al. (2018) | Seyed Ahadi et al. (2020) |
|---|---|---|---|---|
Design | Two-year open prospective multicentre study | Two-year prospective open-label study | Prospective cohort study over a 1- to 5-year follow-up period | Prospective cohort study with a mean follow-up of 13 years |
Population | 32 | 30 | 19 | 18 |
Region | French Caribbean region | Korea | China | Iran |
Dose |
|
|
|
|
Concomitant immunosuppressives | None reported | None reported | None reported | None reported |
IPD = individual patient data.
Sources: Clinical Study Report for N-MOmentum53 and technical report for indirect comparison with rituximab.63
Table 23: Baseline Characteristics in Studies Included in the IPD Analyses Comparing Inebilizumab With Rituximab (Total Population)
Characteristics | Inebilizumab arm of N-MOmentum trial | Cabre et al. (2018) | Kim et al. (2011) | Li et al. (2018) | Seyed Ahadi et al. (2020) | Rituximab studies combined |
|---|---|---|---|---|---|---|
Mean age (SD) | 42.99 (11.56) | 39.59 (12.70) | 38.52 (10.75) | 38.06 (14.21) | 32.78 (9.09) | 37.73 (11.95) |
Proportion female (SD) | 0.91 (0.28) | 0.94 (0.25) | 0.90 (0.31) | 0.84 (0.37) | 0.94 (0.24) | 0.91 (0.29) |
Proportion seropositive (SD) | 0.93 (0.26) | 0.62 (0.49) | 0.69 (0.47) | 0.89 (0.32) | 0.50 (0.51) | 0.67 (0.47) |
Baseline AAR (SD) | 1.73 (1.52) | 1.34 (0.93) | 2.05 (0.74) | 2.91 (2.20) | 1.80 (1.34) | 1.94 (1.40) |
Baseline EDSS (SD) | 3.81 (1.81) | 5.92 (1.83) | 4.40 (2.08) | 4.05 (1.89) | 4.17 (1.70) | 4.79 (2.03) |
AAR = annualized attack rate; EDSS = Expanded Disability Status Scale; IPD = individual patient data; SD = standard deviation.
Sources: Clinical Study Report for N-MOmentum53 and technical report for indirect comparison with rituximab.63
Table 24: Assessment of Homogeneity for ITC
Characteristics | Description and handling of potential effect modifiers | |
|---|---|---|
MAICs vs. satralizumab and eculizumab | IPD analyses vs. rituximab | |
Disease severity | There is no single metric of disease severity; however, EDSS can be used as a metric of disability. Eligibility criteria for maximum EDSS differed by trial, and there was minor variability in mean and median EDSS scores at baseline. In the comparisons with satralizumab, patients from N-MOmentum with an EDSS score > 7 were excluded from the IPD. Mean or median EDSS score was not adjusted in the MAICs. Ultimately, this was not expected to introduce bias in the outcome of time to first NMOSD attack. | There is no single metric of disease severity; however, EDSS can be used as a metric of disability. Baseline EDSS score differed across trials but was adjusted in the analyses. Ultimately, this was not expected to introduce bias in the outcome of AAR. The studies were not limited to patients with seropositive NMOSD and the analyses were conducted using the total population, although adjusted for. Based on clinical expert input, this was not expected to introduce bias in the analyses. |
Treatment history | This was not reported in the satralizumab trials. Between the N-MOmentum and eculizumab trials, baseline characteristics differed significantly with regard to the percent of patients who had prior maintenance therapy. This was adjusted for in the MAIC analysis. | NR |
Trial eligibility criteria | Refer to disease severity regarding the ranges of EDSS scores allowed in each trial. One satralizumab trial (SAkuraSky) included adolescent patients in addition to adults, while all other trials included only adults. This could not be adjusted in the MAICs. Additionally, 1 satralizumab trial (SAkuraSky) required stable immunotherapy; the eculizumab trial allowed it and most patients were receiving it (PREVENT), and N-MOmentum and the other satralizumab trial (SAkuraStar) disallowed immunotherapy. This could not be adjusted in the MAICs. | NR |
Dosing of comparators | No concerns raised for homogeneity. | Not applicable; single-arm trials, unanchored comparison. |
Placebo response | No concerns raised for homogeneity. | Not applicable; single-arm trials, unanchored comparison. |
Definitions of end points | The definition of an NMO or NMOSD attack (also called relapse) significantly differed between trials. This could not be adjusted for in the MAICs. | NR |
Timing of end point evaluation | This varied substantially, but the end point “time to NMOSD attack or relapse” is a time-to-event analysis. | This varied substantially, but the end point of AAR is a rate. Nonetheless, this may introduce heterogeneity if there are differences in attack rate over time. |
Withdrawal frequency | No concerns raised for homogeneity. | NA |
Clinical trial setting | No concerns raised for homogeneity. | Small, regional studies in single locations. None of the studies were conducted in Canada or other parts of North America. |
Study design | No concerns raised for homogeneity. | Rituximab data are available only from very small, nonrandomized, single-arm, regional studies located outside of North America. |
AAR = annualized attack rate; AQP4 = aquaporin-4; EDSS = Expanded Disability Status Scale; IPD = individual patient data; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; NA = not applicable; NMO = neuromyelitis optica; NMOSD = neuromyelitis optica spectrum disorder; NR = not reported; vs. = versus.
Sources: Clinical Study Report for N-MOmentum53 and publications of the SAkuraStar,79 SAkuraSky,80 and PREVENT42 trials. Technical report for ITCs comparing inebilizumab with satralizumab and eculizumab63 and technical report for indirect comparison with rituximab.63 Details included in the table are from the sponsor’s summary of clinical evidence.
Results
Inebilizumab Versus Satralizumab Monotherapy
For the comparison of inebilizumab versus satralizumab, the AQP4-IgG–seropositive subpopulations were used based on data from an FDA Center for Drug Evaluation and Research clinical review69 and the CADTH Clinical Review Report.70 |||||||| |||| |||||||||| |||| ||||||| |||||||| |||| ||| ||| || ||||| ||| ||||||||||| |||||||| || ||| |||||||||||| ||||||| The sponsor identified that the comparison against SAkuraStar was more relevant due to its evaluation of monotherapy being more similar to the trial design of N-MOmentum, which also disallowed concomitant immunotherapy; in contrast, SAkuraSky evaluated satralizumab plus stable immunotherapy. The results of the comparison with SAkuraSky are not assessed in this review for the same reason.
In the comparison with satralizumab, the following characteristics were adjusted in the MAIC because the sponsor identified that they differed significantly at baseline:
sex
geographical region
race.
Additional analyses were conducted in which different combinations of the previously mentioned characteristics were adjusted (e.g., sex and geography but not race, or sex only).
When all of the selected characteristics were adjusted (model 4), the result for time to first NMOSD attack (inebilizumab versus satralizumab) was an HR of 0.666 (95% CI, 0.182 to 2.435), suggesting a numerically lower risk of attack with inebilizumab. However, the result was not statistically significant because the 95% CI crossed 1. The effective sample sizes were very low: in the placebo arm (N = 52), the effective sample size was 26.8 (approximately 52%) and in the inebilizumab arm (N = 161), it was 38.7 (approximately 24%).
The results differed numerically by model but were not statistically significant in any case.
|||||||||| |||||||| ||| ||||||||| || ||||| ||| |||||||||| |||| ||||||||| |||||||||||||||||||||||| ||||||| ||| ||| |||||| ||| |||||||| |||| ||||| |||||| ||| ||| ||||| || |||||| ||||| |||| ||||||| ||||||||||| |||| || |||||| |||||||||| |||| |||||||||||| ||| |||| |||| ||||||||||| || ||| ||||||||| ||||||||||.
Table 25: Time to First NMOSD Attack, Inebilizumab Versus Satralizumab Monotherapy (N-MOmentum and SAkuraStar Trials, Anchored MAIC)
Result | Model 4 |
|---|---|
Adjusted HR (95% CI), inebilizumab vs. placebo | 0.171 (0.067 to 0.451) |
Effective sample size | |
Inebilizumab, N | 38.7 |
Placebo, N | 26.8 |
Matched baseline characteristics | |
AQP4 seropositive statusa | Yes |
Gender | Yes |
Geography | Yes |
Race | Yes |
HR (SE), satralizumab vs. placebo | 0.261 (0.449) |
Indirect comparison | |
HR (95% CI), inebilizumab vs. satralizumab | 0.666 (0.182 to 2.435) |
AQP4 = aquaporin-4; CI = confidence interval; HR = hazard ratio; ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; NMOSD = neuromyelitis optica spectrum disorder; SE = standard error; vs. = versus.
aFor studies that included patients with seronegative NMOSD, only the seropositive subgroup was included in the analyses.
Sources: Clinical Study Report for N-MOmentum.53 Technical report for ITCs comparing inebilizumab with satralizumab and eculizumab.63 Details included in the table are from the sponsor’s summary of clinical evidence.
Inebilizumab Versus Eculizumab
The PREVENT trial only enrolled patients with AQP4-IgG–seropositive NMOSD and patients with a baseline EDSS of 7 points or less, so patients with seronegative NMOSD and those with a baseline EDSS of greater than 7 were excluded from the inebilizumab IPD to facilitate the MAIC. The eculizumab trial, PREVENT, allowed immunotherapy. The majority of patients did have concomitant immunotherapy during the PREVENT trial, and more attacks were required in the previous 1 to 2 years before enrolment than were required for the N-MOmentum trial; these differences could not be adjusted for in the MAICs.
In the comparison with eculizumab, the following characteristics were adjusted in the MAIC because the sponsor identified that they differed significantly at baseline:
race
geographical region
prior maintenance therapy experience.
There are differences in the definition of NMOSD attack between the N-MOmentum and PREVENT trials that could not be accounted for by the MAIC methodology.
In model 4, which matched for all 4 characteristics (race, geographical region, experience with prior maintenance therapy, and AQP4 seropositive status), the risk of a committee-adjudicated NMOSD attack was higher with inebilizumab than with eculizumab (HR = 3.947; 95% CI, 0.917 to 17.0). The effective sample sizes were low; in the placebo arm (N = 52), the effective sample size was 24.5 (approximately 47%) and in the inebilizumab arm (N = 161), it was 97.0 (approximately 60%).
The process for the adjudication of Committee-adjudicated attacks in the PREVENT trial were less stringent than in the N-MOmentum trial and committee adjudication was conducted retrospectively.42 Additionally, the results of the PREVENT trial differed based on whether attacks were committee-adjudicated (HR = 0.06; 95% CI, 0.02 to 0.20; P < 0.001) or identified by physicians (HR = 0.18; 95% CI, 0.10 to 0.34; P < 0.001).42 Comparing the investigator-assessed NMOSD attacks from both trials results in an RR of 1.507 (95% CI, 0.700 to 3.245). Regardless of approach, there was a higher risk of attack associated with inebilizumab than eculizumab, although the results were only statistically significant using the committee-adjudicated values in some but not all models. The 95% CIs were very wide in all analyses, with the majority of the interval falling within a range that would imply a benefit of eculizumab.
Table 26: Risk of Attack With Inebilizumab Versus Eculizumab (N-MOmentum and PREVENT Trials, Anchored MAIC)
Result | Model 4 |
|---|---|
HR (95% CI), inebilizumab vs. placebo | 0.237 (0.104 to 0.541) |
Effective sample size | |
Inebilizumab, N | 97.0 |
Placebo, N | 24.9 |
Matched baseline characteristics | |
AQP4 seropositive status | Yes |
Race | Yes |
Geography | Yes |
Treatment experience | Yes |
HR (SE), eculizumab vs. placebo | 0.208 (0.653) |
Indirect comparison | |
HR (95% CI), inebilizumab vs. eculizumab | 3.947 (0.917 to 17.0) |
AQP4 = aquaporin-4; CI = confidence interval; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; SE = standard error; vs. = versus.
Sources: Clinical Study Report for N-MOmentum.53 Technical report for indirect treatment comparisons comparing inebilizumab with satralizumab and eculizumab.63 Details included in the table are from the sponsor’s summary of clinical evidence.
Inebilizumab Versus Rituximab
The MAICs were based on the RR of lowering the AAR in patients with NMOSD with inebilizumab compared with rituximab. In all comparisons, including the combined rituximab IPD (RR = 1.32; standard error = 0.12), the RR was greater than 1 for all comparisons, which implies a higher risk of attack for patients with NMOSD who received rituximab compared with inebilizumab. However, no 95% CI was provided, which limits the ability to assess the uncertainty around these estimates. Although an effective sample size was reported for each comparison, it is unclear what these numbers represent, as they far exceed the populations of each of the rituximab trials alone or in combination, and the sponsor did not appear to adjust the rituximab trial data. Most of the reported effective sample sizes also exceeded the number of patients treated with inebilizumab in N-MOmentum. There was therefore no effective sample size reported for the N-MOmentum IPD after MAIC adjustment.
Additionally, no relative effect estimates were provided for the outcome of EDSS, so the results cannot be assessed.
Table 27: Inebilizumab Versus Rituximab for Lowering the AAR (N-MOmentum and Rituximab Studies With IPD, MAIC)
Rituximab study | ESSa | Rate of AAR change (SE) | RR (SE) | |
|---|---|---|---|---|
Inebilizumab | Rituximab | |||
Cabre et al. (2018) | 118 | −0.90 (0.05) | −0.51 (0.05) | 1.75 (0.21) |
Kim et al. (2011) | 150 | −0.90 (0.08) | −0.75 (0.08) | 1.20 (0.16) |
Li et al. (2018) | 137 | −0.91 (0.04) | −0.81 (0.04) | 1.12 (0.07) |
Seyed Ahadi et al. (2020) | 92 | −0.93 (0.04) | −0.67 (0.04) | 1.38 (0.10) |
Combined rituximab | 225 | −0.91 (0.05) | −0.67 (0.05) | 1.35 (0.12) |
AAR = annualized attack rate; ESS = effective sample size; IPD = individual patient data; MAIC = matching-adjusted indirect comparison; RR = relative risk; SE = standard error.
Note: RR was estimated via ordinary least squares regression using the weights from the entropy balancing. A value > 1 favours inebilizumab.
aIt is unclear from the submitted report what population this ESS refers to, as it greatly exceeds the population of the rituximab trials, and most values exceed the population of the inebilizumab arm in the N-MOmentum trial.
Sources: Technical report for indirect comparison with rituximab.63 Details included in the table are from the sponsor’s summary of clinical evidence.
Published NMA
The methods and results of the published NMA28 were not assessed in detail for this review. However, for the purpose of critically appraising the sponsor-submitted ITCs, the published NMA will be summarized briefly. The published NMA compared eculizumab with inebilizumab, satralizumab monotherapy, and satralizumab plus stable immunotherapy using a fixed-effects Bayesian NMA approach using the same evidence base as the sponsor (i.e., the N-MOmentum, SAkuraStar, SAkuraSky, and PREVENT trials) but with access to the PREVENT trial’s IPD, and only summary-level data from the N-MOmentum study. It was the opinion of the study authors that the studies included in the NMA were sufficiently similar in study design and patient population, although no detailed justification was provided. The authors highlighted that background immunotherapy was the most important difference between the included studies, and conducted separate NMAs for monotherapies and combination therapies, respectively, using IPD from the PREVENT trial to separate patients into those with or without background immunotherapy. The published NMA therefore reported on 3 comparisons that were conducted:
Analysis 1: Combined monotherapy and combination therapy in the PREVENT, SAkuraStar, SAkuraSky, and N-MOmentum trials.
Analysis 2: Monotherapy in the PREVENT trial monotherapy subgroup (n = 21 out of 96) and the SAkuraStar and N-MOmentum trials.
Analysis 3: Combination therapy in the PREVENT trial combination therapy subgroup (n = 75 out of 96) and the SAkuraSky and N-MOmentum trials.
The results of the published NMA, briefly, were as follows:
Analysis 1: Eculizumab (monotherapy or combination therapy) was superior to satralizumab (monotherapy or combination therapy) for time to first NMOSD attack; inebilizumab was not reported, as no data were present for combination therapy.
Analysis 2: Eculizumab monotherapy was superior to both satralizumab monotherapy and inebilizumab monotherapy for time to first NMOSD attack.
Analysis 3: Due to a wide credible interval, the results were too uncertain to draw conclusions in the comparison between eculizumab combination therapy and satralizumab combination therapy. The point estimate was considered by the authors to be numerically in favour of eculizumab combination therapy.
Critical Appraisal of the Sponsor-Submitted ITCs
Indirect Comparison With Satralizumab and Eculizumab
Both the sponsor-submitted ITCs and the published NMAs were anchored analyses. The sponsor-submitted ITCs and the published NMA disagreed on whether there was important between-trial heterogeneity in the evidence base. Neither provided robust justification for their conclusions, although both agreed that background immunotherapy was an important complicating factor, which affects any comparisons between the N-MOmentum (no background immunotherapy) and PREVENT (allowed immunotherapy, and most patients received it) trials.
The sponsor identified the following weaknesses of the published NMA: small sample sizes in the monotherapy-based comparisons; significant differences in patient baseline information, such as data on prior attack history, baseline disability, and disease duration; substantial variation in the definitions of attack criteria across the included trials; and no attack events were detected for patients who received eculizumab monotherapy, so an imputed HR was selected by the authors for use in the NMA. In addition to these factors, the trials differed in the age range of eligible patients and whether patients could receive concurrent immunotherapy during the study, although the published NMA was able to control for the latter by selecting subgroups from the PREVENT trial’s IPD. Although these are valid limitations to discuss, the majority of these concerns were not addressed by the methodology used in the sponsor-submitted ITCs, with the exception of the adjustment for seropositive status, baseline disability and, in 1 case (the comparison with eculizumab), treatment experience, which may be a rough proxy for other disease-related factors such as attack history.
Using MAIC methodology and the IPD from the N-MOmentum trial, the sponsor adjusted for differences in some eligibility and/or patient baseline characteristics such as race, geography, and treatment experience (eculizumab) or sex (satralizumab). Importantly, the sponsor selected these characteristics based only on whether they differed between the trials, without any external input from the published literature or clinical expert opinion as to whether they were important treatment-effect modifiers. The clinical experts consulted by CADTH considered that demographic factors such as sex, race, or geographic location were not likely to be important treatment-effect modifiers in NMOSD, which suggests that all of the submitted MAICs may be over-adjusting for these factors; accordingly, the effective sample sizes for the MAIC models became quite low, indicating poor overlap between the populations and reducing precision. Furthermore, identifying a factor based on statistical significance alone is not an appropriate approach since effect modifiers with small imbalances or even no imbalances could introduce bias if not included in the weighted adjustment of the MAIC.81 Thus, it is likely that other important effect-modifying factors were not accounted for using this approach.
The sponsor identified that the comparison with the SAkuraSky trial was not relevant because the patients enrolled in that trial were required to receive background immunotherapy, in contrast with the N-MOmentum trial, which disallowed concurrent immunotherapy due to the introduction of important between-trial heterogeneity. The results for this comparison are not included in this report for the same reason, as the clinical expert consulted by CADTH agreed this was an important confounding factor. However, the majority of patients in the PREVENT trial also received background immunotherapy, which could not be adjusted for in the sponsor-submitted ITCs. This therefore represents an important source of remaining between-trial heterogeneity despite the MAIC methodology. The result of the published NMA comparing eculizumab with inebilizumab may be marginally more accurate because the publication authors were able to exclude the patients in the PREVENT trial who were on concurrent immunotherapy, although there is still important uncertainty introduced because the monotherapy group in the PREVENT trial is a nonrandomized post hoc subgroup with a low sample size.
The ITCs only evaluated time to first NMOSD attack, which the clinical experts consulted by CADTH agreed is the most critical outcome in NMOSD. No safety outcomes were evaluated. The definitions of NMOSD attacks differed substantially between the trials: the N-MOmentum trial had the most strict and specific criteria, whereas the comparator trials had more general and relaxed criteria. As there are no well-defined, widely accepted criteria for defining an attack, no single approach is more or less appropriate than the other. However, it does introduce between-trial heterogeneity that was not resolved using the MAIC methodology reported by the sponsor.
The results of the comparison between inebilizumab and satralizumab were too imprecise to draw conclusions from due to wide 95% CIs that included the null value and the limitations of the methodology, as previously described. The sponsor-submitted MAIC comparing inebilizumab with eculizumab, although also imprecise, agreed with the published, unadjusted NMA that there was a higher risk of an NMOSD attack associated with inebilizumab compared with eculizumab. However, the magnitude of effect is uncertain due to imprecision and the limitations of both the submitted MAIC and published NMA, as previously described.
The sponsor-provided technical report lacked significant detail on the methodology and the results of the analyses and did not appear to adhere to best practices for conducting MAICs. The poor reporting substantially limited CADTH’s ability to appraise the evidence and raises concerns about the validity of the analyses.
Indirect Comparison With Rituximab
The sponsor-submitted MAICs comparing inebilizumab with rituximab were based on 4 small (N = 18 to 30) single-arm observational studies conducted in specific regions with populations that likely differ from Canadian clinical practice. Unanchored analyses were conducted due to the lack of a comparator arm in the rituximab studies, which raises the threshold of the assumptions required to interpret the ITC results as unbiased, as it must be assumed that all prognostic factors and treatment-effect modifiers have been accounted for. Importantly, the sponsor did not report any effort to identify important prognostic factors and treatment-effect modifiers from a clinical perspective. The sponsor noted it was not feasible to conduct ITCs comparing inebilizumab with other off-label treatments such as azathioprine and mycophenolate mofetil due to small sample sizes and observational study designs. However, the same limitations exist for the available rituximab data, and the use of IPD methodology cannot correct for these limitations. Additionally, adjusting the N-MOmentum data to reflect these studies may result in the target population becoming more dissimilar to Canadian clinical practice. Furthermore, such an adjustment was unnecessary, given the availability of IPD from the rituximab studies. Given that IPD were available for all studies in the comparison, a comparison that utilized all available data to target the treatment effect of interest, such as an appropriately specified propensity score–based comparison, would have been more appropriate.
The published, placebo-controlled, double-blind RCT of rituximab in adult patients with NMOSD conducted by Tahara et al. (2020)29 was not discussed by the sponsor as a potential source of data for ITCs. It is unclear why this study was not selected, as justification was not reported. Due to the inherent weaknesses of unanchored analyses, it would have been important to assess the feasibility of using higher-quality evidence in the MAICs.
Only the outcome of AAR was assessed. There is an evidence gap for time to first NMOSD relapse. Some of the methodologies in the sponsor-submitted ITCs suggested that an analysis was also conducted for EDSS. However, no relative effect estimates were reported. No safety outcomes were evaluated.
The submitted methodology for the IPD analyses of inebilizumab versus rituximab is insufficient for critical appraisal because of poor reporting and omission of key details in both the methodology and results, in addition to the limitations noted previously. No conclusions can be drawn from the indirect comparisons with rituximab.
Discussion
Summary of Available Evidence
One completed, double-blind, randomized, placebo-controlled phase II and III study (N-MOmentum) was included in this review. The N-MOmentum trial consisted of a 197-day RCT phase (RCP) and a single-arm OLP that had a minimum duration of 2 years. The N-MOmentum trial randomized 231 patients who were adults with NMOSD, had a documented history of either 1 or more acute NMOSD attacks in the prior year or 2 or more attacks in the prior 2 years that required rescue therapy, and had an EDSS score of 7.5 or less, or 8.0 in special circumstances. The majority of patients had NMOSD that was seropositive for AQP4-IgG, and subgroup data were available for the seropositive population. The primary end point was the time in days from day 1 to the onset of an AC-determined NMOSD attack on or before day 197. The key secondary end points included the proportion of patients with a worsening in EDSS score from baseline to last visit during the RCP, change in binocular low-contrast visual acuity score from baseline to last visit during the RCP, and the number of NMOSD-related inpatient hospitalizations during the RCP. Other secondary or exploratory outcomes included the NMOSD attack rate in inebilizumab-treated patients, safety outcomes, and HRQoL (SF-36).
At baseline, the patients included in the N-MOmentum trial were mostly female (> 90%), had received prior acute or maintenance therapies for NMOSD (> 98%), were seropositive for AQP4-IgG (> 92%), and had a mean age of approximately 43 years. The median EDSS score at baseline was 4 in the placebo group (range, 1.0 to 8.0) and 3.5 in the inebilizumab group (range, 0.0 to 8.0); 29% and 24% had an EDSS score of greater than 5 points at baseline, respectively.
A sponsor-submitted ITC was summarized and critically appraised that consisted of anchored MAICs comparing the N-MOmentum trial results (inebilizumab versus placebo) indirectly with phase III RCTs of satralizumab and eculizumab, and unanchored IPD analyses comparing inebilizumab indirectly with rituximab in several small observational studies. A published NMA was briefly summarized to compare with the sponsor-submitted ITC but was not formally appraised.
Interpretation of Results
Efficacy
Evidence from the N-MOmentum trial demonstrated that treatment with inebilizumab likely resulted in a clinically important reduction in NMOSD attacks and worsening in EDSS score among patients with AQP4-IgG–seropositive NMOSD compared with placebo during the RCP of the study. Little to no differences were observed between treatment arms for the secondary outcomes of change from baseline in low-contrast visual acuity score, number of NMOSD-related inpatient hospitalizations, and change from baseline in pain NRS, nor were differences observed in the exploratory outcome of change from baseline in SF-36. There may also be a clinical benefit of inebilizumab in the exploratory outcomes of attack severity and attack recovery, but these outcomes were not formally assessed and are supportive only.
NMOSD is a rare, severe, debilitating, and life-threatening disease with no cure, and patients have limited access to targeted treatment options across Canada due to financial barriers and, for some therapies, arduous IV administration schedules. According to the patient and clinician group input and the clinical experts consulted by CADTH, the main goal of preventive treatment in NMOSD is the delay, reduction, or elimination of NMOSD-related attacks, and secondary goals include reducing the severity and improving recovery from attacks, and reducing disability accumulated from incomplete recovery after attacks. The outcomes included in the trial were therefore considered to be of clinical importance to patients and clinicians and inebilizumab demonstrated a clinically significant benefit in some of the most important outcomes.
There was incongruence between NMOSD attacks, for which there was an observed clinically meaningful benefit of inebilizumab, and the secondary outcomes related to visual acuity, pain, and NMOSD-related inpatient hospitalizations, for which there was no clear clinically important benefit observed and which are also important to patients and clinicians. The development of worsening visual acuity and pain occurs as a result of attacks, particularly those that are incompletely resolved, and so it was expected that a reduction in attacks, the occurrence of milder attacks, and better recovery from attacks as observed with inebilizumab compared with placebo may lead to improvements in these outcomes. The domain of attacks (i.e., optical neuritis, myelitis, or brainstem) may also affect these outcomes, but there was no difference between treatment arms. The clinical experts consulted by CADTH noted that in the setting of a clinical trial, there may be earlier rigorous intervention with rescue therapy in the acute treatment of attacks that may mask differences in residual symptoms that could occur in real-world treatment. Patients treated with placebo also more commonly received rescue therapy. It is also possible that the duration of the RCP was insufficient to detect a clinically meaningful difference in pain and visual acuity, as these symptoms are accumulated over time because of repeat attacks, as the clinical experts consulted by CADTH noted that the result may be due to the overall low incidence of optical neuritis over the trial duration in either treatment arm. The estimated AAR for any inebilizumab-treated patient with AQP4-IgG–positive NMOSD in the N-MOmentum trial (RCP and OLP) was 0.09 attacks per year. A large proportion of patients were attack-free at day 197 (87.6% in the inebilizumab group and 56.6% in the placebo group), which may similarly suggest that a longer study duration would be needed to observe the impact of repeated attacks on visual acuity and pain. Although the OLP had a minimum duration of 2 years, the lack of a control arm precludes making judgments on the benefit of inebilizumab over placebo. The clinical experts consulted by CADTH noted that other factors that could explain this may include a floor effect in the placebo group (15 of 56 participants [27%] read 0 of 70 characters on a binocular test using a low-contrast visual acuity chart at baseline), or preserved fellow-eye visual acuity, obscuring the effect of monocular optic neuritis on binocular visual activity.
The duration of the trial was also considered insufficient to observe meaningful differences in NMOSD-related inpatient hospitalizations. There was also no important difference identified for HRQoL-related outcomes, which was not statistically analyzed in the submission to CADTH.
In the absence of direct comparative evidence between inebilizumab and other preventive therapies for the treatment of NMOSD, the sponsor submitted indirect comparisons with eculizumab, satralizumab, and rituximab. The indirect evidence submitted to CADTH suggested that treatment with eculizumab may be associated with a clinically meaningful benefit in time to first NMOSD attack when compared indirectly with inebilizumab. No other outcomes were assessed and there were substantial unresolved sources of between-trial heterogeneity in the analysis, such as differences in the outcome definition and patient populations, small effective sample sizes, and imprecision. The results were consistent between the submitted MAIC and a published NMA in terms of the direction of effect (i.e., suggesting a benefit of eculizumab compared with inebilizumab), although the precise magnitude and clinical importance of the benefit of eculizumab over inebilizumab are uncertain due to significant limitations and imprecision in both analyses. Despite evidence of a benefit of eculizumab over inebilizumab, the clinical experts consulted by CADTH noted that inebilizumab would still have an important place in therapy due to patient preferences, differences in mechanism of action, and barriers to access.
Results were more uncertain for the indirect comparison of inebilizumab with satralizumab monotherapy for the outcome of time to first NMOSD attack because the CIs were wide, including the null value and including potentially important harms and benefits. Additionally, there was unresolved between-trial heterogeneity in the outcome definitions, and the selected treatment-effect modifiers were not selected or validated by clinical experts and may have been over-matched, causing small effective sample sizes and imprecision.
The indirect comparison of inebilizumab with rituximab was an unanchored analysis of AAR and EDSS scores, and the rituximab data were informed by 4 single-arm observational studies with small numbers of patients (≤ 30 per trial) that were conducted in specific regions that are likely to differ from the population in Canada. The indirect comparison appeared to use MAIC methodology in which the N-MOmentum trial data were adjusted to be more similar to the less relevant, international observational study data, despite access to IPD for these studies, which would have allowed a much more appropriate propensity-scoring approach. The sponsor noted that it was not feasible to conduct ITCs comparing with other off-label treatments such as azathioprine and mycophenolate mofetil due to small sample sizes and observational study designs. However, the same limitations exist for the observational studies selected by the sponsor to inform the rituximab ITCs, and the use of IPD and MAIC methodologies cannot correct for these limitations. There was no justification provided for why the sponsor did not assess the feasibility of using an available published, double-blind, placebo-controlled RCT of rituximab in the treatment of adult patients with NMOSD as a source for rituximab trial data in the ITCs. Additionally, as the comparisons were unanchored due to the selection of single-arm observational studies, it would have been imperative to adjust for all prognostic factors and treatment-effect modifiers, but the sponsor did not report an effort to identify these. No methodology or justification was provided to support the selected adjustment factors in the sponsor-submitted MAIC. In the results of the MAICs, the CIs for the relative effect estimates of AAR were not reported, and relative effect estimates for the EDSS were not reported in the submission to CADTH. Overall, the reporting of both the methodology and results was insufficient to allow for critical appraisal. As a result, no conclusions can be drawn about the comparative efficacy of inebilizumab versus rituximab.
No direct or indirect evidence exists to compare inebilizumab with azathioprine, mycophenolate mofetil, or ravulizumab. Based on clinical experience and the current trial data for inebilizumab, inebilizumab is expected to be more effective than azathioprine and mycophenolate mofetil, which are considered generally ineffective therapies in this population and are not considered disease-modifying therapies. The results of the ravulizumab trial are concurrently under review in a separate CADTH assessment at the time of this report. As no patients experienced an NMOSD attack in the single-arm ravulizumab study,82 it may be speculated that an indirect comparison between ravulizumab and inebilizumab could resemble the results of the eculizumab comparison previously described. Ravulizumab has a different mechanism of action than inebilizumab and has an administration schedule of IV infusions every 8 weeks. The clinical experts consulted by CADTH, in considering whether ravulizumab and inebilizumab were both hypothetically accessible, noted that patients may respond to, prefer, or tolerate 1 therapy better than the other, even if there is a difference in clinical efficacy for time to first NMOSD attack. Additionally, patients in clinical practice may be treated with ravulizumab and inebilizumab in sequence after the failure of 1 drug to prevent attacks, if both are accessible.
Harms
Over the duration of the study, nearly all patients experienced at least 1 AE, but the majority were mild to moderate. No new safety signals were identified. SAEs were more common in the placebo group than in the inebilizumab group during the study. Withdrawals due to AEs were uncommon. Most patients experienced an AE of special interest, most commonly infection, followed by infusion-related reaction, hepatic function abnormality, and cytopenia. Cytopenias were more common in patients treated with inebilizumab, which is consistent with inebilizumab’s mechanism of action and the class effects of B-cell depletion. In addition, a few patients experienced hypersensitivity ||| ||||| |||||||||| || ||||||||| ||||||||||| |||||||||| |||||||||||||||||||| || ||||||| ||||||| |||| ||||||||||||| ||||||||||||||| ||||||||| ||||| ||| || |||||||||||. The infections that occurred were generally mild and did not lead to treatment discontinuation in the OLP or RCP.
Conclusion
NMOSD is a rare, disabling, and life-threatening inflammatory disorder of the CNS characterized by acute attacks with a relapsing pattern that cause potentially irreversible damage to the optic nerve and spinal cord. There is a large unmet need for high-efficacy, well-tolerated therapies for NMOSD that have a significant impact on preventing and/or reducing attacks. Patients and clinicians highlighted that the main treatment goals include the use of an efficacious, safe, and tolerable therapy administered as soon as possible after the first attack to avoid all relapses, reduce the severity of attacks and the cumulative disability associated with them, and minimize AEs related to therapies. One phase II and III, double-blind, randomized, placebo-controlled study (N-MOmentum) was included in this review, which had a duration of 197 days followed by an open-label, single-arm period of at least 2 years.
The data submitted to CADTH and the end points assessed in the study were considered clinically relevant for the treatment of patients with NMOSD. Results from the N-MOmentum trial randomized period demonstrated that treatment with inebilizumab likely results in a clinically meaningful benefit in time to first relapse, and in the proportion of patients with a worsening in EDSS score compared with placebo at 197 days. Other secondary or exploratory outcomes were less conclusive and did not show a clearly meaningful benefit of inebilizumab over placebo, including change in low-contrast visual acuity score, NMOSD-related inpatient hospitalizations, HRQoL, and pain. The reasons for this incongruence are uncertain, but it may be that the duration of the randomized study was insufficient to detect clinically meaningful differences in these outcomes.
The ITCs included MAICs comparing inebilizumab with satralizumab and eculizumab, and IPD analyses comparing inebilizumab with rituximab. Results were inconclusive for the comparison with satralizumab. The ITCs suggest that the risk of NMOSD attack is higher with inebilizumab treatment than with eculizumab, but the magnitude of benefit is uncertain due to wide 95% CIs, imprecision, and unresolved between-trial heterogeneity. The comparison with rituximab could not be interpreted due to limitations in the analysis inherent to the data available, as well as sparsely reported methodology and results and inappropriate methodological decisions. There are no direct or indirect data available for the efficacy and safety of inebilizumab compared with azathioprine, mycophenolate mofetil, or ravulizumab.
Relative to previous studies in the clinical development program, no new safety signals or concerns with inebilizumab were identified in either study period and long-term inebilizumab treatment was well tolerated, with a median exposure of greater than 3.2 years during the study. Almost all patients experienced at least 1 AE during the study regardless of treatment assignment, but the results suggested that patients treated with inebilizumab may have fewer SAEs at 197 days than patients treated with placebo. Cytopenias were more common in patients treated with inebilizumab compared with placebo, which is consistent with inebilizumab’s mechanism of action and the class effects of B-cell depletion.
Reduction in Cumulative Total Active MRI Lesions
The reduction in cumulative total active MRI lesions (new gadolinium-enhancing or new or enlarging T2) during the RCP was a key secondary end point. To establish eligibility and a baseline value, all patients underwent a CNS (neuroaxis) MRI scan that included the optic nerve, spinal cord, and brain. ||||||||| ||| |||| ||| |||||||| || || ||||| ||| |||||||| ||| ||||||||| ||| ||| ||||||| || ||||| ||||||. The images were read by 2 independent neuroradiologists at the central reading site, with any discrepancies resolved via a consensus re-read. To reduce the risk of bias, the MRI findings were not to be reviewed by the site staff. Although an MID has not been established for changes in MRI findings, health care providers may use these findings to diagnose and manage NMOSD in practice.
The cumulative number of active MRI lesions in the AQP4-IgG–seropositive population was lower in patients treated with inebilizumab compared with placebo (1.7 versus 2.3 lesions; rate ratio = 0.568; 95% CI, 0.3851 to 0.8363).
|| ||| ||| || ||| |||| ||| ||||||||||||||||||||||| ||||| ||||||| || ||| ||||||| ||| ||| |||||||||||||||||||||||||||| ||||| |||||| || ||| |||||||.
Table 29: Cumulative Total Active MRI Lesions in the N-MOmentum Trial AQP4-IgG–Seropositive Population
Outcome | AQP4-IgG–seropositive population (N = 213) | |
|---|---|---|
Placebo (N = 52) | Inebilizumab (N = 161) | |
Cumulative total active MRI lesionsa | ||
Patients evaluated | 31 | 74 |
Mean (SD) | 2.3 (1.3) | 1.7 (1.0) |
Median (range) | ||||| | ||||| |
Rate ratio (95% CI) | 0.568 (0.3851 to 0.8363) | |
Nominal P valuea | 0.0042 | |
Adjusted P valuea | |||||| | |
AQP4-IgG = anti-aquaporin-4 immunoglobulin G; CI = confidence interval; SD = standard deviation.
aBased on Cox regression, logistic regression, or negative binomial models, with placebo as the reference group.
References
1.Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53(5):1107-1114. PubMed
2.Weinshenker BG, Wingerchuk DM. Neuromyelitis spectrum disorders. Mayo Clin Proc. 2017;92(4):663-679. PubMed
3.Wingerchuk DM, Lucchinetti CF. Neuromyelitis optica spectrum disorder. N Engl J Med. 2022;387(7):631-639. PubMed
4.Carnero Contentti E, Correale J. Neuromyelitis optica spectrum disorders: from pathophysiology to therapeutic strategies. J Neuroinflammation. 2021;18(1):208. PubMed
5.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. PubMed
6.Seok JM, Cho EB, Lee HL, et al. Clinical characteristics of disabling attacks at onset in patients with neuromyelitis optica spectrum disorder. J Neurol Sci. 2016;368:209-213. PubMed
7.Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14. PubMed
8.Borisow N, Mori M, Kuwabara S, Scheel M, Paul F. Diagnosis and treatment of NMO spectrum disorder and MOG-encephalomyelitis. Front Neurol. 2018;9:888. PubMed
9.Eaneff S, Wang V, Hanger M, et al. Patient perspectives on neuromyelitis optica spectrum disorders: data from the PatientsLikeMe online community. Mult Scler Relat Disord. 2017;17:116-122. PubMed
10.Zarei S, Eggert J, Franqui-Dominguez L, et al. Comprehensive review of neuromyelitis optica and clinical characteristics of neuromyelitis optica patients in Puerto Rico. Surg Neurol Int. 2018;9:242. PubMed
11.Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17(8):1019-1032. PubMed
12.Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. 2014;176(2):149-164. PubMed
13.Ajmera MR, Boscoe A, Mauskopf J, Candrilli SD, Levy M. Evaluation of comorbidities and health care resource use among patients with highly active neuromyelitis optica. J Neurol Sci. 2018;384:96-103. PubMed
14.Mealy MA, Boscoe A, Caro J, Levy M. Assessment of patients with neuromyelitis optica spectrum disorder using the EQ-5D. Int J MS Care. 2019;21(3):129-134. PubMed
15.Beekman J, Keisler A, Pedraza O, et al. Neuromyelitis optica spectrum disorder: patient experience and quality of life. Neurol Neuroimmunol Neuroinflamm. 2019;6(4):e580. PubMed
16.Mutch K, Methley A, Hamid S, Moore P, Jacob A. If they are OK, we are OK: the experience of partners living with neuromyelitis optica. Disabil Rehabil. 2017;39(13):1279-1286. PubMed
17.Papp V, Illes Z, Magyari M, et al. Nationwide prevalence and incidence study of neuromyelitis optica spectrum disorder in Denmark. Neurology. 2018;91(24):e2265-e2275. PubMed
18.Marrie RA, Gryba C. The incidence and prevalence of neuromyelitis optica: a systematic review. Int J MS Care. 2013;15(3):113-118. PubMed
19.Etemadifar M, Nasr Z, Khalili B, Taherioun M, Vosoughi R. Epidemiology of neuromyelitis optica in the world: a systematic review and meta-analysis. Mult Scler Int. 2015;2015:174720. PubMed
20.Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394(10206):1352-1363. PubMed
21.Schiopu E, Chatterjee S, Hsu V, et al. Safety and tolerability of an anti-CD19 monoclonal antibody, MEDI-551, in subjects with systemic sclerosis: a phase I, randomized, placebo-controlled, escalating single-dose study. Arthritis Res Ther. 2016;18(1):131. PubMed
22.Chen D, Gallagher S, Monson NL, Herbst R, Wang Y. Inebilizumab, a B cell-depleting Anti-CD19 antibody for the treatment of autoimmune neurological diseases: insights from preclinical studies. J Clin Med. 2016;5(12). PubMed
23.Uplizna (inebilizumab for injection): 100 mg / 10 mL (10 mg / mL) solution for intravenous infusion [product monograph]. Vaughan (ON): Horizon Therapeutics Canada; 2023 Dec 15.
24.Rugge B, Balshem H, Sehgal R, Relevo R, Gorman P, Helfand M. Appendix A, lipid conversion factors. Screening and treatment of subclinical hypothyroidism or hyperthyroidism Rockville (MD): Agency for Healthcare Research and Quality (US); 2011.
25.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
26.Horizon Therapeutics Canada response to September 5, 2023 CADTH request for additional information regarding Uplizna review [internal sponsor’s report]. Vaughan (ON): Horizon Therapeutics Canada; 2023 Sep 15.
27.Horizon Therapeutics Canada response to September 29, 2023 CADTH request for additional information regarding Uplizna review [internal sponsor’s report]. Vaughan (ON): Horizon Therapeutics Canada; 2023 Oct 5.
28.Wingerchuk DM, Zhang I, Kielhorn A, et al. Network meta-analysis of Food and Drug Administration-approved treatment options for adults with aquaporin-4 immunoglobulin G-positive neuromyelitis optica spectrum disorder. Neurol Ther. 2021;11(1):123-135. PubMed
29.Tahara M, Oeda T, Okada K, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2020;19(4):298-306. PubMed
30.Kitley J, Leite MI, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain. 2012;135(Pt 6):1834-1849. PubMed
31.Jasiak-Zatonska M, Kalinowska-Lyszczarz A, Michalak S, Kozubski W. The immunology of neuromyelitis optica-current knowledge, clinical implications, controversies and future perspectives. Int J Mol Sci. 2016;17(3):273. PubMed
32.Cabre P, Gonzalez-Quevedo A, Bonnan M, et al. Relapsing neuromyelitis optica: long term history and clinical predictors of death. J Neurol Neurosurg Psychiatry. 2009;80(10):1162-1164. PubMed
33.Sherman E, Han MH. Acute and chronic management of neuromyelitis optica spectrum disorder. Curr Treat Options Neurol. 2015;17(11):48. PubMed
34.Oh J, Levy M. Neuromyelitis optica: an antibody-mediated disorder of the central nervous system. Neurol Res Int. 2012;2012:460825. PubMed
35.Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol. 2012;69(9):1176-1180. PubMed
36.Mealy MA, Kessler RA, Rimler Z, et al. Mortality in neuromyelitis optica is strongly associated with African ancestry. Neurol Neuroimmunol Neuroinflamm. 2018;5(4):e468. PubMed
37.Collongues N, Marignier R, Jacob A, et al. Characterization of neuromyelitis optica and neuromyelitis optica spectrum disorder patients with a late onset. Mult Scler. 2014;20(8):1086-1094. PubMed
38.Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. 2017;264(9):2003-2009. PubMed
39.Etemadifar M, Salari M, Mirmosayyeb O, et al. Efficacy and safety of rituximab in neuromyelitis optica: review of evidence. J Res Med Sci. 2017;22(18). PubMed
40.Kessler RA, Mealy MA, Levy M. Treatment of neuromyelitis optica spectrum disorder: acute, preventative, and symptomatic. Curr Treat Options Neurol. 2016;85(1). PubMed
41.Kümpfel T, Giglhuber K, Aktas O, et al. Update on the diagnosis and treatment of neuromyelitis optica spectrum disorders (NMOSD) - revised recommendations of the Neuromyelitis Optica Study Group (NEMOS). Part II: attack therapy and long-term management. J Neurol. 2024;271(1):141-176. PubMed
42.Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in aquaporin-4–positive neuromyelitis optica spectrum disorder. New Engl J Med. 2019;381(7):614-625. PubMed
43.CADTH Canadian Drug Expert Committee recommendation: eculizumab (Soliris-Alexion Pharma Canada Corp). Ottawa (ON): CADTH; 2020 Aug 24: https://www.cadth.ca/sites/default/files/cdr/complete/SR0640%20Soliris%20-%20CDEC%20Final%20Recommendation%20August%2024%2C%202020%20for%20posting.pdf. Accessed 2023 Apr 18.
44.CADTH reimbursement recommendation: satralizumab (Enspryng) for treatment of neuromyelitis optica spectrum disorder. Ottawa (ON): CADTH; 2021 Apr: https://www.cadth.ca/sites/default/files/cdr/complete/SR0663%20Enspryng%20-%20CDEC%20Final%20Recommendation%20April%2023%2C%202021_for%20posting.pdf. Accessed 2023 Apr 18.
45.Soliris (eculizumab): 30 mL parenteral solution (10 mg/mL) for injection [product monograph]. Zurich (CH): Alexion Pharma GmbH; 2021 Mar 25.
46.Enspryng (satralizumab injection): solution, 120 mg/mL, subcutaneous [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2021 Apr 21.
47.Ultomiris (ravulizumab for injection): 10 mg/mL & 100 mg/mL concentrate for solution for intravenous infusion [product monograph]. Mississauga (ON): Alexion Pharma Canada Corp; 2023 Oct 30.
48.Rituxan (rituximab): 10 mg/mL intravenous infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2023 Jun 2.
49.Imuran (azathioprine): azathioprine tablets (50 mg); azathioprine sodium for injection (50 mg azathioprine per vial) [product monograph]. Toronto (ON): Aspen Pharmacare Canada, Inc; 2018 Nov 8.
50.Myfortic (myciphenolic acid enteric-coated tablets): 180 mg, 360 mg (as mycophenolate sodium) [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc; 2018 May 10.
51.CellCept (myciphenolic mofetil): capsules – 250 mg; film-coated tablets – 500 mg; powder for oral suspension – 200 mg/mL (when reconstituted) [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2018 Nov 14.
52.Kim HJ, Aktas O, Patterson KR, et al. Inebilizumab reduces neuromyelitis optica spectrum disorder risk independent of FCGR3A polymorphism. Ann Clin Transl Neurol. 2023. PubMed
53.Clinical Study Report: CD-IA-MEDI-551-1155. Clinical Study Report for CD-IA-MEDI-551-1155, a double-masked, placebo-controlled study with open-label period to evlauate the efficacy and safety of MEDI-551 in adult subjects with neuromyelitis optia and neuromyelitis optica spectrum disorders [internal sponsor’s report]. Gaithersburg (MD): MedImmune, LLC; 2021 Jun 9.
54.de Groot V, Beckerman H, Uitdehaag BM, et al. The usefulness of evaluative outcome measures in patients with multiple sclerosis. Brain. 2006;129(Pt 10):2648-2659. PubMed
55.Goldman MD, LaRocca NG, Rudick RA, et al. Evaluation of multiple sclerosis disability outcome measures using pooled clinical trial data. Neurology. 2019;93(21):e1921-e1931. PubMed
56.Riemsma RP, Forbes CA, Glanville JM, Eastwood AJ, Kleijnen J. General health status measures for people with cognitive impairment: learning disability and acquired brain injury. Health Technol Assess. 2001;5(6):1-100. PubMed
57.Robinson D, Jr., Zhao N, Gathany T, Kim LL, Cella D, Revicki D. Health perceptions and clinical characteristics of relapsing-remitting multiple sclerosis patients: baseline data from an international clinical trial. Curr Med Res Opin. 2009;25(5):1121-1130. PubMed
58.Jensen MP, Karoly P, Braver S. The measurement of clinical pain intensity: a comparison of six methods. Pain. 1986;27(1):117-126. PubMed
59.Jensen MP, Turner JA, Romano JM, Fisher LD. Comparative reliability and validity of chronic pain intensity measures. Pain. 1999;83(2):157-162. PubMed
60.Sharrack B, Hughes RA, Soudain S, Dunn G. The psychometric properties of clinical rating scales used in multiple sclerosis. Brain. 1999;122 (Pt 1):141-159. PubMed
61.Shahrbanian S, Duquette P, N EM. Impairment, disability and fatigue in multiple sclerosis. Caspian J Intern Med. 2018;9(3):244-251. PubMed
62.Systematic literature review of clinical and economic evidence of current treatments for neuromyelitis optica spectrum disorder [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Uplizna (inebilizumab), 10 mg/mL solution for intravenous infusion. Vaughan (ON): Horizon Therapeutics Canada; 2021 Oct.
63.Technical note for comparative analyses of the efficacy of inebilizumab compared to satralizumab and eculizumab [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Uplizna (inebilizumab), 10 mg/mL solution for intravenous infusion. Vaughan (ON): Horizon Therapeutics Canada; 2022 Aug 19.
64.Efficacy of inebilizumab compared to rituximab for the indication neuromyelitis optica spectrum disorder - IPD analyses [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Uplizna (inebilizumab), 10 mg/mL solution for intravenous infusion. Vaughan (ON): Horizon Therapeutics Canada; 2022 Dec 30.
65.Shamseer L, Moher D, Clarke M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ. 2015;350:g7647. PubMed
66.CRD’s guidance for undertaking reviews in health care York (GB): Centre for Reviews and Dissemination; 2009: https://www.york.ac.uk/media/crd/Systematic_Reviews.pdf. Accessed 2020 May 11.
67.Higgins J, Thomas J. Cochrane handbook for systematic reviews of interventions 2019: https://training.cochrane.org/handbook/current Accessed 2020 May 11.
68.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. PubMed
69.Center for Drug Evaluation Research. Clinical review(s). Enspryng (satralizumab) injection: 120mg/mL in a single-dose prefilled syringe. Company: Genentech, Inc. Application No.:761149. Approval date: 8/14/2020 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2020: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761149Orig1s000MedR.pdf. Accessed 2023 Aug 22.
70.CADTH common drug review. Clinical review report: satralizumab (Enspryng) for neuromyelitis optica spectrum disorder. Ottawa (ON): CADTH; 2021 Jun: https://www.cadth.ca/sites/default/files/cdr/clinical/sr0663-enspryng-clinical-review-report.pdf. Accessed 2023 Mar 16.
71.CADTH common drug review. Clinical review report: eculizumab (Soliris) for neuromyelitis optica spectrum disorder. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/clinical/sr0640-soliris-nmosd-clinical-review-report.pdf. Accessed 2023 Mar 16.
72.Sterne JAC, Savović J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898. PubMed
73.Sterne JA, Hernán MA, Reeves BC, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ. 2016;355:i4919. PubMed
74.Scottish Intercollegiate Guidelines Network (SIGN) checklist for cohort studies. Edinburgh (GB): SIGN; 2012: https://www.sign.ac.uk/what-we-do/methodology/checklists/. Accessed 2020 May 11.
75.Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol. 1997;50(6):683-691. PubMed
76.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials. PharmacoEcon. 2010;28(10):935-945. PubMed
77.Signorovitch JE, Sikirica V, Erder MH, et al. New tool for timely comparative effectiveness research. Value Health. 2012;15(6):940-947. PubMed
78.Phillippo DM, Dias S, Ades AE, Welton NJ. Equivalence of entropy balancing and the method of moments for matching‐adjusted indirect comparison. Res Synth Methods. 2020;11(4):568-572. PubMed
79.Traboulsee A, Greenberg BM, Bennett JL, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19(5):402-412. PubMed
80.Yamamura T, Kleiter I, Fujihara K, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. New Engl J Med. 2019;381(22):2114-2124. PubMed
81.Phillippo D, Ades T, Dias S, Palmer S, Abrams KR, Welton N. NICE DSU technical support document 18: Methods for population-adjusted indirect comparisons in submissions to NICE Sheffield (GB): NICE Decision Support Unit; 2016: https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf. Accessed 2023 Nov 7.
82.Pittock SJ, Barnett M, Bennett JL, et al. Ravulizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. Ann Neurol. 2023;93(6):1053-1068. PubMed
Appendix 1: Detailed Outcome Data
Note that this appendix has not be copy-edited.
Table 28: Absolute ||||||| ||| |||||||| || |||||||||| [Redacted]
||||||| | ||||||||| | |||||||||||| |||||||| ||||||| |||| ||| | ||||||||||||| |||||||| ||||||||||| |||| ||| | ||||
|---|---|---|---|---|---|---|---|
||| |||||||||| | |||||||| |||||||||||| |||||||||| | ||| |||||||||| | |||||||| ||||||||| | ||||
||||||| | |||||||||||| | ||||||| | |||||||||||| | ||||
||||||||||| || || |||||| ||| |||||||||||| |||||||||| |||||| |||||||||||||||| | ||| || | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||||| |||||| | ||||| |||||||| |||||| |
||| || | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||||| |||||| | ||||| ||||||| |||||| | |
||| || | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | |
||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||||| |||||| | ||||| ||||||| |||||| | |
||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | |
||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | |
||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | |
||||||||||| || || |||||| ||| ||||||||||||||||| | ||| || | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||||| |||||| | ||||| |||||||| |||||| |
||| || | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||||| |||||| | ||||| |||||||| |||||| | |
||| || | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | |
||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||||| |||||| | ||||| ||||||| |||||| | |
||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | |
||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | |
||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | |
|||||||||| |||| ||||||||| |||| |||||||| || |||| || | ||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | |||||| |||||||| |||||| | |||||| |||||||| |||||| |
|||||||||| ||||| ||||| | ||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| |
||| |||||||||||| ||| ||||||| | ||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| |
||||||||||||| || ||| ||||||| | ||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| ||||| | ||||| ||||||| |||||| |
||||||||||||||||| |||||||||||||||| | ||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
||||||| ||||||| ||||||||| || |||||||||| ||||||||||| ||||||||| || |||||
Pharmacoeconomic Review
Abbreviations
AE
adverse event
AQP4-IgG
anti-aquaporin-4 immunoglobulin G
BIA
budget impact analysis
BSC
best supportive care
EDSS
Expanded Disability Status Scale
ITC
indirect treatment comparison
MAIC
matching-adjusted indirect comparison
NMOSD
neuromyelitis optica spectrum disorder
QALY
quality-adjusted life-year
TTAA
time to first adjudicated attack
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Inebilizumab (Uplizna), 10 mg/mL solution for injection |
Submitted price | Inebilizumab, 10 mg/mL: $25,623 per 10 mL vial |
Indication | For the treatment of adult patients with neuromyelitis optica spectrum disorders who are anti-aquaporin-4 immunoglobulin G seropositive |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | December 15, 2023 |
Reimbursement request | As per indication |
Sponsor | Horizon Therapeutics Canada |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with NMOSD who are AQP4-IgG seropositive |
Treatment | Inebilizumab |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (60 years) |
Key data sources |
|
Submitted results | The ICER for inebilizumab compared with satralizumab was calculated to be $1,033,466 per QALY gained (incremental cost = $503,207; incremental QALYs = 0.49). |
Key limitations |
|
CADTH reanalysis results | CADTH was unable to address the identified limitations of the submitted economic evaluation through reanalysis. A CADTH base case could therefore not be specified. The cost-effectiveness of inebilizumab in adults with NMOSD who are AQP4-IgG seropositive is unknown relative to all included comparator treatments. |
AQP4-IgG = anti-aquaporin-4 immunoglobulin G; BSC = best supportive care; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; NMOSD = neuromyelitis optica spectrum disorder; QALY = quality-adjusted life-year; TTAA = time to first adjudicated attack.
Conclusions
The CADTH Clinical Review found that treatment with inebilizumab produces a meaningful benefit over best supportive care (BSC) but not over eculizumab in terms of time to first adjudicated attack (TTAA). Conclusions could not be drawn for the comparison of inebilizumab versus satralizumab or off-label rituximab. The indirect comparisons were subject to considerable between-trial heterogeneity with respect to the definition of the TTAA outcome. Because the populations represented in each indirect treatment comparison (ITC) were different, the efficacy of different treatments could not be compared, adding additional uncertainty to estimates of the relative clinical effectiveness of inebilizumab.
CADTH identified several additional limitations of the sponsor’s submitted economic evaluation that could not be addressed through reanalysis. Most prominently, the submitted model did not adhere to best practices for Markov models; in this case, it failed to incorporate the general population mortality risk as a competing risk to other transitions in the model, and transition probabilities did not always consider the correct treatment-specific risk of a neuromyelitis optica spectrum disorder (NMOSD) attack. CADTH’s review of the pharmacoeconomic model led to the identification of several errors that CADTH could not correct within the period of this review. This limitation prevented CADTH from assessing the cost-effectiveness of inebilizumab relative to all specified comparators, including BSC.
At the sponsor's submitted price, inebilizumab is expected to cost $421 per patient per day or $153,738 annually. At publicly available list prices for the comparators, inebilizumab is more costly than off-label rituximab ($16 daily, $5,940 annually) and satralizumab ($336 daily, $122,850 annually), but less costly than eculizumab ($1,900 daily, $694,231 annually). According to the sponsor’s submitted base case, inebilizumab was associated with increased quality-adjusted life-years (QALYs) at an increased cost compared with satralizumab. In the sponsor’s base case, 98% of the incremental cost was attributable to drug acquisition costs. The sponsor’s estimate suggests that inebilizumab would require a substantial price reduction to be considered cost-effective compared with satralizumab at a willingness-to-pay threshold of $50,000 per QALY gained.
Conclusions regarding the trade-off between these treatments are difficult to draw, as they depend on the administration costs of treatment, differences in relative efficacy, the amount of time on treatment, the risk of adverse events (AEs), and the impact of treatment on health-related quality of life. In the absence of a valid model to synthesize this evidence, the relative cost-effectiveness of inebilizumab in adults with NMOSD who are seropositive for anti-aquaporin-4 immunoglobulin G (AQP4-IgG) is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input for this review was obtained from the Sumaira Foundation and MS Canada. Information for both submissions was collected through an online survey. Intended respondents included patients living with NMOSD or caregivers of patients living with NMOSD. While the MS Canada survey was targeted at respondents in Canada, the submission from the Sumaira Foundation included responses from Canada and the US as well as a range of African and European countries. Respondents in the MS Canada submission reported experience with eculizumab (n = 2), satralizumab (n = 2), and rituximab (n = 4). Similarly, respondents in the Sumaira Foundation submission reported experience with the same medications as well as with tocilizumab, prednisone, azathioprine, and mycophenolate mofetil. Both submissions identified treatment gaps relating to the number of side effects, maintenance of physical ability, and pain management. In the MS Canada submission, 1 patient had experience with inebilizumab through enrolment in a clinical trial. Following the conclusion of the trial and 18 months of treatment, the patient was forced to switch to another therapy. In the Sumaira Foundation submission, 5 patients had experience with inebilizumab that had been made available through private insurance or enrolment in a clinical trial. Private access to inebilizumab was facilitated after their condition failed to respond to other treatments, and respondents reported an effective response in terms of attack reduction.
Registered clinician input was received from the Canadian Network of Multiple Sclerosis Clinics. The submission defined NMOSD as an inflammatory disease of the central nervous system marked by distinct attacks that most often include transverse myelitis and optic neuritis. Active NMOSD can cause severe progressive damage in the brain, optic nerves, and spinal cord. Relapses are unpredictable and can result in rapid progression as well as permanent neurologic damage and disability. The primary objective of treatment is to prevent NMOSD attacks. Secondary objectives include a reduction in attack severity and cumulative disability from attacks, and the minimization of AEs. The authors suggested that an ideal therapy would have a good safety and tolerability profile and completely prevent attacks following diagnosis. Many of the current therapies in use for NMOSD attacks are prescribed off-label. Of the drugs specifically indicated for NMOSD, patients rarely qualify for satralizumab, and public coverage for eculizumab is not available. The clinician input submission suggested that inebilizumab could be used as a first-line treatment or as a treatment for patients who have a breakthrough attack after starting another therapy.
In their input, the drug plans sought clarification on inebilizumab’s place in therapy and the need for a direct comparison with other therapies used to treat NMOSD. Concerns were raised about the cost-effectiveness compared with other treatments (including off-label rituximab) and with respect to the feasibility of adopting inebilizumab due to its budget impact.
Several of these concerns were addressed in the sponsor’s model:
Inebilizumab was compared against current treatment options, including off-label rituximab.
The effect of AEs on patient health-related quality of life was considered.
CADTH was unable to address the following concerns raised from stakeholder input:
The submitted model failed to satisfy the requirements for the desired model structure and could not produce valid results. Therefore, CADTH was unable to assess the relative cost-effectiveness of inebilizumab against other available treatment options.
Economic Review
The current review is for inebilizumab (Uplizna) for the treatment of adults with NMOSD who are seropositive for AQP4-IgG.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted an economic evaluation comparing inebilizumab with treatments currently reimbursed by at least 1 participating drug plan for the indication under review, treatments that are used off-label in Canadian practice, or those that have received a recommendation in favour of reimbursement from CADTH for the indication under review.1 This target population was aligned with the Health Canada indication and reimbursement request.
Inebilizumab is available as a 10 mL vial for IV administration with a concentration of 10 mg/mL. The submitted price was $25,623 per vial. For the indicated population, the recommended dosage is an initial dose of 300 mg at weeks 1 and 3 followed by a maintenance dose of 300 mg every 6 months (beginning with the first dose). At the submitted price, the maintenance dose will cost $421 per day and $153,738 per year.1
A total of 4 alternative treatments to inebilizumab were considered in the economic evaluation: eculizumab (Soliris), satralizumab (Enspryng), off-label rituximab, and BSC. Eculizumab and satralizumab are available as a 30 mL vial for IV administration at a concentration of 10 mg/mL. The recommended dosage for eculizumab is 900 mg per week for 4 weeks, followed by 1,200 mg in week 5 and 1,200 mg every 2 weeks thereafter. At a unit cost of $6,675 per vial, the maintenance dose for eculizumab will cost $1,900 per day and $694,231 per year. The recommended dosage for satralizumab is 120 mg at weeks 0, 2, and 4 followed by a maintenance dose of 120 mg every 4 weeks. At a unit cost of $9,450 per vial, the maintenance dose for satralizumab will cost $336 per day and $122,850 per year. Rituximab is available as a 10 mL vial at a concentration of 10 mg/mL. The recommended dosage is 1,000 mg at week 0 and 2, followed by a maintenance dose of 1,000 mg every 6 months. At a unit cost of $30, the maintenance dose of rituximab will cost $16 per day and $5,940 per year. Lastly, BSC was defined as the absence of any active therapy and relied on data from the placebo arm of the relevant trials.1
Modelled outcomes included life-years and QALYs. Costs were estimated from the perspective of the Canadian public health care payer. Model outputs were generated over a lifetime horizon of 60 years with a cycle length of 1 month. Costs and outcomes were discounted at 1.5%.1
Model Structure
The sponsor submitted a Markov model that tracked a hypothetical cohort of patients across health states defined as discrete values on the Expanded Disability Status Scale (EDSS). This outcome measure records values between 0 to 10, with 0 representing full function and 10 being equivalent to death. A total of 21 mutually exclusive health states were included in the model, representing 0.5-point increments in the full range of possible EDSS scores. Specific estimates of the number of patients in each health state were influenced by the predicted change in EDSS score, as well as time-dependent transition probabilities reflecting the risk of an NMOSD attack, the risk of death, and the risk of treatment discontinuation.1
A summary of the model structure is presented in Figure 1. At model entry, patients initiated 1 of the eligible first-line treatments for NMOSD. At the beginning of each model cycle, patients faced a risk of discontinuing active treatment and switching to BSC. Afterward, patients faced a treatment-specific risk of an NMOSD attack. This outcome meant that patients could remain in their current health state (no change in EDSS score) or transition to a worse health state (higher EDSS score). Meanwhile, patients who did not experience an NMOSD attack (had stable disease) would have no change in the EDSS score and therefore would remain in their current health state. Throughout the model time horizon, patients were subject to an all-cause mortality risk that increased with age.1
Model Inputs
Costs and effects were estimated using a homogeneous baseline population. All data summarizing baseline characteristics of the cohort were obtained from the N-MOmentum trial.1-3 This was a randomized phase II and III trial that involved the direct comparison of inebilizumab with placebo in adult patients with AQP4-IgG–seropositive or –seronegative NMOSD. Data of interest included baseline age (mean = 42 years), sex (93.9% female), baseline EDSS score, and changes in EDSS score following an NMOSD attack.1-3
Estimates of relative efficacy for TTAA were obtained from 3 pairwise ITCs. This approach was justified due to the absence of a direct comparison of inebilizumab with every comparator specified in the economic evaluation.1 The first ITC estimated the hazard ratio of inebilizumab relative to satralizumab in an anchored matching-adjusted indirect comparison (MAIC) using data from the N-MOmentum and SAkuraStar (satralizumab versus placebo) trials.2,4 The second ITC estimated the hazard ratio of inebilizumab relative to eculizumab in an anchored MAIC using data from the N-MOmentum and PREVENT (eculizumab versus placebo) trials.2,5 The final ITC estimated the hazard ratio of inebilizumab relative to rituximab in an unanchored MAIC. Unlike the preceding MAICs, efficacy data for rituximab were compiled from 2 prospective open-label studies and 2 prospective cohort studies.6-9 The following estimated hazard ratios were used in the economic evaluation:
inebilizumab versus satralizumab: 0.666 (95% CI, 0.182 to 2.435)
inebilizumab versus eculizumab: 3.947 (95% CI, 0.917 to 17.0)
inebilizumab versus rituximab: 0.741 (95% CI, 0.585 to 0.937).1
While the sponsor acknowledged the availability of an existing network meta-analysis, this method of generating estimates of relative efficacy was not included as a source of relative efficacy in the economic evaluation.1
The model relied on treatment-specific survival functions to predict the risk of a first NMOSD attack.1 For inebilizumab and placebo, parametric survival curves were fitted to data representing the TTAA outcome in the N-MOmentum trial. Independent parametric models for each treatment were fitted using the exponential, log-logistic, log-normal, Weibull, Gompertz, gamma, and generalized gamma distributions. Based on the sponsor’s assessment of model fit statistics, the submitted base case assumed a generalized gamma and exponential distribution for inebilizumab and placebo, respectively. To generate the survival curves for the remaining treatments, the estimates of relative efficacy obtained from the MAICs were applied to the inebilizumab reference curve.1
To reflect the risk of experiencing more than 1 attack, the number of attacks in a group of 1,000 patients within the time horizon of the model was simulated. For each of the 1,000 patients, the sponsor completed 80 random draws from the uniform distribution to represent the risk of 1 to 80 subsequent attacks. The fundamental assumption for this procedure was that patients draw from the same distribution of time to attack, independent of the number of attacks. Quantile functions from the generated survival curves were subsequently used to determine the number of days until a patient would experience 1 to 80 subsequent attacks. This step allowed the function to look up the time point on the survival curve that corresponded to the sampled attack risk. From this, the proportion of patients expected to experience a subsequent attack was calculated.1
In each model cycle, patients faced a risk of discontinuing active treatment and initiating BSC. This treatment-specific parameter was estimated by applying the annual risk of discontinuation for each treatment to the corresponding cycles where the treatment was administered. Annual discontinuation risks were estimated from the randomized period of the N-MOmentum (inebilizumab), PREVENT (eculizumab), and SAkuraStar and SAkuraSky (satralizumab) trials.2,4,5,10 Values for rituximab were calculated as a weighted average from 3 observational studies that reported discontinuation data.11-13
All-cause mortality was incorporated as a time-dependent risk parameter in the economic evaluation. This input was estimated as the sex-weighted general population mortality risk.1 Values were calculated from Statistics Canada life tables published in January 2022.14
In addition to tracking the proportion of the cohort in each health state, the model also tracked the occurrence of AEs. The specific AEs considered in the model included: anemia, diarrhea, headache, hepatotoxicity, infection, influenza, leukopenia, nausea, pain in an extremity, thrombosis, upper respiratory tract infection, urinary tract infection, and urticaria. The treatment-specific risk of each AE was obtained from the relevant arm of the N-MOmentum trial or the relevant treatment arm from 1 of the studies in the submitted systematic review.2,4-7,9-13,15-30 Data for the treatments included from multiple studies were calculated as weighted averages.1
Health-related quality of life was captured in the model by combining health state utilities with disutilities associated with NMOSD attacks and AEs. The health state utility values were obtained from the indirect measurement of patient preferences in the N-MOmentum trial using the Short Form (36) Health Survey version 2 questionnaire.1,2 In the base case, values for specific EDSS ranges were converted to EQ-5D utilities using the algorithm by Rowen et al.1,31 Additional scenarios considered alternate procedures by Brazier and Hummert.1,32,33 The Rowen and Brazier procedures resulted in the calculation of the NMOSD attack disutility of −0.1994 and −0.0241, respectively. Values for AE-specific disutilities were sourced from a range of published studies identified by the sponsor.34-40 The methods used to identify AE disutility values were not reported.1
The submission considered costs associated with the acquisition, administration, and monitoring of treatment as well as those associated with the management of AEs and end-of-life care. Treatment acquisition costs were estimated by applying treatment prices to the dosing schedule for each alternative treatment considered in the model. While the price of inebilizumab reflected the sponsor’s submitted price, the price for each alternative therapy was sourced from the Ontario Drug Benefit Formulary.1,41 Dosing was established from the recommended dosage listed in the relevant product monographs. Treatment administration costs were assumed to include physician administration, chair time, premedications (for inebilizumab only), and vaccinations (for eculizumab only). Both BSC and satralizumab (which is self-administered by the patient) were assumed to have no administration costs. Monitoring costs reflected the resource use associated with stable NMOSD and NMOSD attacks. This included inpatient hospitalization (general and intensive care unit), visits to the emergency department, ambulance use, primary care, home visits (nurse or physician), and other health care services.1 Annual health care resource use for stable and unstable NMOSD was obtained from the N-MOmentum trial. The proportion of stable patients consuming each service was categorized into 3 ranges of EDSS scores: 0 to 3.5, 3.5 to 6, and 6 to 10.1 Unit costs were obtained from the Canadian Institute for Health Information, the Ontario Schedule of Benefits: Physician Services, Ontario Ambulance Services Billing, and the median hourly wage for a registered nurse in Canada.42-45 Costs to treat each AE were based on the resource use needed for treatment and were informed by clinical experts consulted by the sponsor.1 End-of-life costs were assumed to be $12,813 and were applied upon entry to the death state (EDSS score of 10).1,46
Summary of Sponsor’s Economic Evaluation Results
The costs and QALYs of each alternative were generated using a Monte Carlo simulation with 500 iterations. Results from the submitted base case were aligned with those generated deterministically. Results from the probabilistic base case are presented subsequently.
Base-Case Results
The submitted analysis was based on the publicly available prices of the comparator treatments. Results from the base case of the submitted economic evaluation are presented in Table 3.
The expected costs and QALYs for inebilizumab were $2,386,939 and 11.05. Due to the sponsor’s strategy for the ITC, a complete incremental analysis of all treatments could not be performed. Instead, a stratified approach was necessary, as each relative effect estimate represented a distinct subpopulation. Relative to eculizumab, inebilizumab was more effective and less costly. In every other pairwise comparison, inebilizumab was the more costly and more effective alternative.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Comparison 1: Inebilizumab vs. BSC | |||
Best supportive care | 97,961 | 2.54 | Reference |
Inebilizumab | 2,386,939 | 11.05 | 268,757 |
Comparison 2: Inebilizumab vs. rituximab | |||
Rituximab | 250,200 | 7.34 | Reference |
Inebilizumab | 2,386,939 | 11.05 | 575,049 |
Comparison 3: Inebilizumab vs. satralizumab | |||
Satralizumab | 1,883,732 | 10.57 | Reference |
Inebilizumab | 2,386,939 | 11.05 | 1,033,466 |
Comparison 4: Inebilizumab vs. eculizumab | |||
Inebilizumab | 2,386,939 | 11.05 | Reference |
Eculizumab | 6,472,153 | 8.01 | Dominated by inebilizumab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
In addition to the base case, 12 distinct scenario analyses were considered. While each of the considered scenarios had a slight impact on the estimated costs and effects, none led to a different conclusion regarding the cost-effectiveness of inebilizumab. In 1 of the scenarios, the sponsor conducted the economic evaluation from a societal perspective. This involved additional costs associated with patient time use, transportation, and direct and indirect nonmedical costs. In this scenario, the incremental cost-effectiveness ratio of satralizumab compared with inebilizumab was estimated to be $975,810. While this was lower than the submitted base case, which used a Canadian health care payer perspective, it had no impact on the conclusion that inebilizumab would not be cost-effective at a threshold of $50,000 per QALY.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Estimates of relative treatment efficacy are highly uncertain. The submitted economic evaluation compared inebilizumab with relevant alternatives that could be used as first-line treatment in the indicated population. These included eculizumab, satralizumab, off-label rituximab, and BSC (placebo). In the absence of a direct comparison between every treatment, the evidence of relative treatment efficacy considered in the economic evaluation was derived from the N-MOmentum trial and 3 independent ITCs. The CADTH Clinical Review of the direct evidence from the N-MOmentum trial concluded that inebilizumab did offer a benefit in terms of TTAA relative to BSC. The indirect evidence included only eculizumab and satralizumab. Conclusions regarding the comparison against rituximab could not be drawn, as the analysis used in the economic evaluation was not included in the clinical submission. Of the evidence that was available, the CADTH Clinical Review concluded that inebilizumab did not offer a benefit relative to eculizumab, and that imprecision in the estimated hazard ratio prevented a definitive conclusion from being reached in the comparison with satralizumab. Furthermore, considerable heterogeneity was identified with respect to the definition of the TTAA outcome and the populations of the included trials. These limitations have meaningful consequences for how the estimates of relative efficacy could be incorporated into the economic evaluation. The use of separate ITCs means that comparative efficacy conclusions cannot be made between all comparators, given the heterogeneity of the trials from which the ITCs are derived.
CADTH was unable to address the limitations in the comparative evidence against satralizumab or rituximab. The cost-effectiveness of inebilizumab against these comparators is unknown.
Methods to calculate health-state membership did not reflect the declared model structure. The submitted economic evaluation sought to track a hypothetical cohort of patients across mutually exclusive health states, characterized as EDSS scores ranging from 0 (perfect health) to 10 (death).
Further inspection of the formulas used to determine health-state membership revealed calculations that did not reflect the intended model structure. In its submitted report, the sponsor claimed that a Markov model was used to generate all estimates of health-state membership.1 This method involves the iterative application of transition probabilities to the estimates of health-state membership from the preceding cycle.47,48 Membership in a specific health state must be calculated by multiplying the proportion of a cohort in each state by the probability of moving to a new state (transition probability) and adding the results together.47,48 A review of the sponsor’s approach to calculating health-state membership revealed that the mortality risk in the general population was not included as a competing risk in the transition between health states (EDSS scores). Instead, the sponsor’s model considered the risk of death from an NMOSD attack to be independent of the risk of death among the general population. This assumption produces a survival bias in favour of treatments that prevent NMOSD attacks that is not a reflection of the efficacy of treatment. Modelling a competing risk would require the transition probabilities associated with changes in EDSS score to factor in general population mortality as it changes with age; however, the sponsor’s model did not do this. This was inconsistent with the sponsor’s claim that transitions to the death state (EDSS = 10) were affected by the risk of death following an NMOSD attack or the general population mortality risk.1
The submitted model also did not apply the treatment-specific risk of an NMOSD attack in an appropriate manner. In the first cycle on BSC, following the discontinuation of active therapy, the submitted model assumed that the risk of an NMOSD attack was specific to the discontinued treatment rather than to BSC. The correct implementation of this treatment and time-dependent parameter required the use of an NMOSD attack risk that was specific to the treatment the patient was receiving in a specific cycle of the model. In response to CADTH’s initial identification of this limitation, the sponsor disclosed its assumption that the efficacy of the initial dose of inebilizumab (or eculizumab or satralizumab) would likely carry through the duration of a model cycle (1 month) after discontinuation within that cycle. Further, the sponsor stated its belief that the differences in frequency of administration between inebilizumab (every 6 months) and eculizumab or satralizumab (every 2 and 4 weeks, respectively), meant that the persistence of the assumption of effect for 1 month was a conservative assumption that favoured the comparators. No evidence was provided to substantiate this assumption.
Given the nature of the identified methodological concerns, the sponsor’s model likely overestimates the incremental QALYs for inebilizumab compared with BSC by an unknown amount.
CADTH was unable to address this limitation. Correcting the calculation of health-state membership would have involved a complete redevelopment of the economic model. Such activity is beyond the scope of CADTH reviews.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
As described earlier, the inability to address several key limitations prevented CADTH from specifying a base case or conducting a reanalysis of the economic evaluation. First, the estimates of relative efficacy were not suited to the determination of relative cost-effectiveness with multiple comparators. This was attributed to an ITC strategy that generated estimates of relative efficacy from independent pairwise comparisons, using trials where considerable heterogeneity concerns were identified. Based on the submitted relative efficacy data, the only appropriate comparison would have been inebilizumab and BSC. Second, the specified model did not use a valid approach to calculate health-state membership. Inspection of the formulas used to calculate health-state membership revealed an implementation that did not reflect the declared model structure. The model failed to treat the general population mortality risk as a competing outcome in the calculation of the transition probabilities, treating it instead as an independent outcome. Lastly, in the first cycle following discontinuation of active therapy, it was assumed patients would continue to benefit from a treatment they were no longer receiving. Given these limitations, CADTH was unable to generate estimates of costs, life-years, or QALYs for any of the comparators specified for this submission. As a consequence, the cost-effectiveness of inebilizumab in adults with NMOSD who are AQP4-IgG seropositive is unknown.
Issues for Consideration
The sponsor’s model defined health states according to categorized EDSS scores. The effect of inebilizumab on patient outcomes was the direct product of anticipated changes in EDSS as patients move between categories. The clinical experts raised concerns about the appropriateness of this method. First, EDSS is not a continuous scale but rather a set of discrete scores representing different levels and types of disability. Second, higher levels of EDSS are primarily associated with impairments to mobility, whereas blindness (a key NMOSD symptom) is not directly considered within the EDSS scoring system. The appropriateness of EDSS as a proxy for the severity of NMOSD adds an unknown level of uncertainty to the estimates of cost-effectiveness.
Overall Conclusions
The CADTH Clinical Review found that treatment with inebilizumab produces a meaningful benefit over BSC in terms of TTAA. In the absence of direct comparisons with every other alternative, conclusions regarding relative efficacy were drawn from a series of independent ITCs. From these estimates of relative efficacy, the CADTH Clinical Review concluded that inebilizumab did not offer a TTAA benefit relative to eculizumab. Meanwhile, no conclusions could be drawn for the comparison of inebilizumab relative to satralizumab or off-label rituximab. The indirect comparisons were subject to considerable between-trial heterogeneity with respect to the definition of the TTAA outcome and the compared populations. As a result, the use of independent MAICs yielded estimates of relative efficacy that did not represent the same population of patients and could not be compared. Given that the TTAA parameters affected estimates of health-state membership through predicted changes in EDSS score, estimates of relative efficacy were required to represent a homogeneous patient population. Therefore, the submitted evidence was not suited for the objective of determining relative cost-effectiveness among multiple comparators.
CADTH identified several additional limitations with the sponsor’s submitted economic evaluation that could not be addressed through reanalysis. Most prominently, the submitted model did not adhere to accepted standards for the development of a Markov model with time-dependent and treatment-specific parameters.47,48 This was reflected by formulas that failed to adjust the changes in EDSS scores for the risk of mortality in the general population and may not accurately reflect the treatment-specific risk of an NMOSD attack. This produced a bias in favour of inebilizumab compared with BSC of a magnitude that could not be estimated. Consequently, CADTH was unable to assess the cost-effectiveness of inebilizumab at the submitted price.
At the sponsor’s submitted price, inebilizumab is expected to cost $421 per day and $153,738 per year. At the public list price for all comparators, inebilizumab is more costly than off-label rituximab ($16 per day, $5,940 per year) and satralizumab ($338 per day, $123,188 per year), but less costly than eculizumab ($1,907 per day, $696,138 per year). Conclusions regarding the trade-off between these treatments are difficult to draw, as they depend on the administration costs of treatment, differences in relative efficacy, the amount of time on treatment, the risk of AEs, and the impact of treatment on health-related quality of life. In the absence of a valid model to synthesize this evidence, the relative cost-effectiveness of inebilizumab in adults with NMOSD who are AQP4-IgG seropositive is unknown.
According to the sponsor’s submitted base case, inebilizumab was associated with increased QALYs at an increased cost compared with satralizumab. Drug acquisition costs accounted for 98% of the incremental cost in the sponsor’s base case. The sponsor’s estimate suggests that inebilizumab would require a substantial price reduction to be considered cost-effective compared with satralizumab at a willingness-to-pay threshold of $50,000 per QALY gained. CADTH’s analysis of the submitted economic evidence found that the sponsor’s assumptions and other methodological issues within the pharmacoeconomic model likely overestimated the incremental effectiveness of inebilizumab. CADTH is unable to comment on the specific price reduction necessary to reach a desired threshold, given the inability of the model to accurately reflect incremental costs or effectiveness against any comparator.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Uplizna (inebilizumab), 10 mg/ML single-use vial for injection. Vaughan (ONTARIO): Horizon Therapeutics Canada; 2023 Aug 16. In.
2.Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394(10206):1352-1363. PubMed
3.Clinical Study Report for CD-IA-MEDI-551-1155, A Double-Masked, Placebo-Controlled Study with Open-Label Period to Evaluate the Efficacy and Safety of MEDI-551 in Adult Subjects with Neuromyelitis Optica and Neuromyeltitis Optica Spectrum Disorders, (2021).
4.Traboulsee A, Greenberg BM, Bennett JL, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19(5):402-412. PubMed
5.Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N Engl J Med. 2019;381(7):614-625. PubMed
6.Cabre P, Mejdoubi M, Jeannin S, et al. Treatment of neuromyelitis optica with rituximab: a 2-year prospective multicenter study. J Neurol. 2018;265(4):917-925. PubMed
7.Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68(11):1412-1420. PubMed
8.Li T, Zhang LJ, Zhang QX, et al. Anti-Rituximab antibody in patients with NMOSDs treated with low dose Rituximab. J Neuroimmunol. 2018;316:107-111. PubMed
9.Seyed Ahadi M, Naser Moghadasi A, Asgari N, Sahraian MA. Efficacy and safety of rituximab in patients with refractory neuromyelitis optica spectrum disorders: A prospective observation in Iranian cases. Caspian J Intern Med. 2020;11(2):155-162. PubMed
10.Yamamura T, Kleiter I, Fujihara K, et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N Engl J Med. 2019;381(22):2114-2124. PubMed
11.Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. 2017;264(9):2003-2009. PubMed
12.Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler. 2011;17(10):1225-1230. PubMed
13.Uzunkopru C, Tutuncu M, Gunduz T, et al. The efficacy of rituximab in patients with neuromyelitis optica spectrum disorder: A real-world study from Turkey. Int J Clin Pract. 2021;75(7):e14158. PubMed
14.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. In: Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 1800 Jan 1.
15.Annovazzi P, Capobianco M, Moiola L, et al. Rituximab in the treatment of Neuromyelitis optica: a multicentre Italian observational study. J Neurol. 2016;263(9):1727-1735. PubMed
16.Correa-Diaz EP, Torres-Herran GE, Mino Zambrano JE, Paredes-Gonzalez V, Caiza-Zambrano FJ. Impact of Rituximab on relapse rate and disability in an Ecuadorian cohort of patients with neuromyelitis optica spectrum disorders. Mult Scler Relat Disord. 2021;48:102683. PubMed
17.Gomez-Figueroa E, Noriega-Morales G, Casallas-Vanegas A, et al. Effect of rituximab on disease activity in latin American patients with anti-aquaporin-4 (+) neuromyelitis optica spectrum disorder. Clin Neurol Neurosurg. 2020;196:106007. PubMed
18.Jeong IH, Park B, Kim SH, Hyun JW, Joo J, Kim HJ. Comparative analysis of treatment outcomes in patients with neuromyelitis optica spectrum disorder using multifaceted endpoints. Mult Scler. 2016;22(3):329-339. PubMed
19.Kim SH, Jeong IH, Hyun JW, et al. Treatment Outcomes With Rituximab in 100 Patients With Neuromyelitis Optica: Influence of FCGR3A Polymorphisms on the Therapeutic Response to Rituximab. JAMA Neurol. 2015;72(9):989-995. PubMed
20.Lin J, Li X, Xue B, et al. Low-dosage of rituximab in Chinese patients with neuromyelitis optica spectrum disorder. J Neuroimmunol. 2018;317:1-4. PubMed
21.Lu Q, Luo J, Hao H, et al. A long-term follow-up of rituximab treatment in 20 Chinese patients with neuromyelitis optica spectrum disorders. Mult Scler Relat Disord. 2020;40:101933. PubMed
22.Novi G, Bovis F, Capobianco M, et al. Efficacy of different rituximab therapeutic strategies in patients with neuromyelitis optica spectrum disorders. Mult Scler Relat Disord. 2019;36:101430. PubMed
23.Radaelli M, Moiola L, Sangalli F, et al. Neuromyelitis optica spectrum disorders: long-term safety and efficacy of rituximab in Caucasian patients. Mult Scler. 2016;22(4):511-519. PubMed
24.Shaygannejad V, Fayyazi E, Badihian S, et al. Long-term tolerability, safety and efficacy of rituximab in neuromyelitis optica spectrum disorder: a prospective study. J Neurol. 2019;266(3):642-650. PubMed
25.Torres J, Pruitt A, Balcer L, Galetta S, Markowitz C, Dahodwala N. Analysis of the treatment of neuromyelitis optica. J Neurol Sci. 2015;351(1-2):31-35. PubMed
26.Trewin BP, Adelstein S, Spies JM, et al. Precision therapy for neuromyelitis optica spectrum disorder: A retrospective analysis of the use of class-switched memory B-cells for individualised rituximab dosing schedules. Mult Scler Relat Disord. 2020;43:102175. PubMed
27.Xiao H, Zeng W, Li L, et al. Retrospective Observation of Low-Dose Rituximab Treatment in Chinese Patients With Neuromyelitis Optica Spectrum Disorders in a Real-World Setting. Front Neurol. 2020;11:642. PubMed
28.Yang Y, Wang CJ, Wang BJ, Zeng ZL, Guo SG. Comparison of efficacy and tolerability of azathioprine, mycophenolate mofetil, and lower dosages of rituximab among patients with neuromyelitis optica spectrum disorder. J Neurol Sci. 2018;385:192-197. PubMed
29.Zhang M, Zhang C, Bai P, Xue H, Wang G. Effectiveness of low dose of rituximab compared with azathioprine in Chinese patients with neuromyelitis optica: an over 2-year follow-up study. Acta Neurol Belg. 2017;117(3):695-702. PubMed
30.Zhao S, Zhou H, Xu Q, Dai H, Wei S. Efficacy of Low-Dose Rituximab on Neuromyelitis Optica-Associated Optic Neuritis. Front Neurol. 2021;12:637932. PubMed
31.Rowen D, Brazier J, Roberts J. Mapping SF-36 onto the EQ-5D index: how reliable is the relationship? Health Qual Life Outcomes. 2009;7:27. PubMed
32.Brazier J, Usherwood T, Harper R, Thomas K. Deriving a preference-based single index from the UK SF-36 Health Survey. J Clin Epidemiol. 1998;51(11):1115-1128. PubMed
33.Hummert MW, Schoppe LM, Bellmann-Strobl J, et al. Costs and Health-Related Quality of Life in Patients With NMO Spectrum Disorders and MOG-Antibody-Associated Disease: CHANCE(NMO) Study. Neurology. 2022;98(11):e1184-e1196. PubMed
34.Matza LS, Sapra SJ, Dillon JF, et al. Health state utilities associated with attributes of treatments for hepatitis C. Eur J Health Econ. 2015;16(9):1005-1018. PubMed
35.Stein EM, Yang M, Guerin A, et al. Assessing utility values for treatment-related health states of acute myeloid leukemia in the United States. Health Qual Life Outcomes. 2018;16(1):193. PubMed
36.Shiroiwa T, Noto S, Fukuda T. Japanese Population Norms of EQ-5D-5L and Health Utilities Index Mark 3: Disutility Catalog by Disease and Symptom in Community Settings. Value Health. 2021;24(8):1193-1202. PubMed
37.Wehler E, Storm M, Kowal S, Campbell C, Boscoe A. A Health State Utility Model Estimating the Impact of Ivosidenib on Quality of Life in Patients with Relapsed/Refractory Acute Myeloid Leukemia. In:2018.
38.Nafees B, Stafford M, Gavriel S, Bhalla S, Watkins J. Health state utilities for non small cell lung cancer. Health Qual Life Outcomes. 2008;6:84. PubMed
39.Shingler S, Fordham B, Evans M, et al. Utilities for treatment-related adverse events in type 2 diabetes. J Med Econ. 2015;18(1):45-55. PubMed
40.Hawe E, McBride D, Balp MM, Tian H, Halliday A, Stull DE. EQ-5D Utilities in Chronic Spontaneous/Idiopathic Urticaria. Pharmacoeconomics. 2016;34(5):521-527. PubMed
41.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. https://www.formulary.health.gov.on.ca/formulary/. Published 2023. Accessed 2023 Sep 6.
42.Canadian Institute for Health I. Care in Canadian ICUs. Ottawa, ON2016.
43.Ontario Ministry of H, Long Term C. Schedule of Benefits, Physician Services Under the Health Insurance Act. In:2023.
44.Ontario Ministry of H, Long Term C. Ambulance Billing Services. https://www.health.gov.on.ca/en/public/publications/ohip/amb.aspx. Accessed March 2023.
45.Government of C. Labour Market Information: Wages for Registered nurses and registered psychiatric nurses. https://www.jobbank.gc.ca/wagereport/occupation/993. Accessed.
46.Isenberg SR, Meaney C, May P, et al. The association between varying levels of palliative care involvement on costs during terminal hospitalizations in Canada from 2012 to 2015. BMC Health Serv Res. 2021;21(1):331. PubMed
47.Sonnenberg FA, Beck JR. Markov models in medical decision making: a practical guide. Medical decision making. 1993;13(4):322-338. PubMed
48.Briggs A, Sculpher M, Claxton K. Decision modelling for health economic evaluation. Oup Oxford; 2006.
49.Government of O. Ontario Drug Benefit Formulary. https://www.formulary.health.gov.on.ca/formulary/. Published 2023. Updated April 2023. Accessed May 2018.
50.Exceptional Access Program (EAP). In: Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2023: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 1800 Jan 1.
51.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Uplizna (inebilizumab), 10 mg/ML single-use vial for injection. Vaughan (ONTARIO): Horizon Therapeutics Canada; 2023 Aug 16. In. Accessed September 22, 2023.
52.Marrie RA, Gryba C. The incidence and prevalence of neuromyelitis optica: a systematic review. Int J MS Care. 2013;15(3):113-118. PubMed
53.Etemadifar M, Nasr Z, Khalili B, Taherioun M, Vosoughi R. Epidemiology of neuromyelitis optica in the world: a systematic review and meta-analysis. Mult Scler Int. 2015;2015:174720. PubMed
54.Canada S. Table 17-10-0005-01 Population estimates on July 1st, by age and sex. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Updated December 21, 2022. Accessed June 14, 2023.
55.Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017;264(10):2088-2094. PubMed
56.Cadth. Satralizumab (Enspryng) for the treatment of neuromyelitis optica spectrum disorder. 2021.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: CADTH Cost Comparison Table for NMOSD
Treatment | Strength or concentration | Form | Price | Recommended dosage | Daily cost | Annual cost |
|---|---|---|---|---|---|---|
Inebilizumab (Uplizna) | 10 mg/mL | 10 mL vial | $25,623.0000 |
| Year 1: $631.80 Year 2+: $421.20 | Year 1: $230,607 Year 2+: $153,738 |
Indicated for NMOSD | ||||||
Eculizumab (Soliris) | 10 mg/mL | 30 mL vial | $6,675.3000 |
| Year 1: $1,973.81 Year 2+: $1,900.70 | Year 1: $720,932 Year 2+: $694,231 |
Satralizumab (Enspryng) | 10 mg/mL | 30 mL vial | $9,450.0000b |
| Year 1: $362.22 Year 2+: $336.35 | Year 1: $132,300 Year 2+: $122,850 |
Off-label treatmentsc | ||||||
ISTs | ||||||
Azathioprine (generic) | 50 mg | Tablet | $0.2405 | 3 to 5 mg/kg daily | $0.96 | $351 |
Mycophenolate mofetil (generic) | 250 mg 500 mg | Tablet | $0.3712 $0.7423 | 2,000 mg daily | $2.97 | $1,084 |
Rituximab (generic) | 10 mg/mL | 10 mL vial | $29.7000 |
| Year 1: $24.41 Year 2+: $16.27 | Year 1: $8,910 Year 2+: $5,940 |
Rituximab (Truxima) | 100 mg/mL 500 mg/mL | 10 mL vial pack 50 mL vial pack | $297.0000 $1,485.0000 |
| Year 1: $24.41 Year 2+: $16.27 | Year 1: $8,910 Year 2+: $5,940 |
IST = immunosuppressive therapy; NMOSD = neuromyelitis optica spectrum disorder.
Note: Prices do not include dispensing fees. Costs assume a body weight of 67 kg and include wastage of unused medication in vials. All prices are from the Ontario Drug Benefit Formulary (accessed September 2023), unless otherwise indicated.49
aSponsor’s submitted price: $76,869.0000 per package of 3 vials.1
bUnit price obtained from the Ontario Exceptional Access Program (accessed September 2023).50
cOff-label treatment dosing derived from the sponsor’s submission and validated by clinical expert feedback obtained by CADTH.1
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing. | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity. | No | The calculations used to estimate state membership did not reflect the conceptualized Markov model. Refer to limitation: Methods to calculate state membership did not reflect the declared model structure. |
Model structure is adequate for decision problem. | No | The calculations used to estimate state membership did not reflect the conceptualized Markov model. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis). | No | Due to the errors in calculating state membership, the model overestimated the costs and benefits (LYs and QALYs) in absolute and relative terms. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem. | No | Characterization of parameter uncertainty requires the specification of a valid mechanism to estimate state membership. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details). | No | No comment. |
LY = life-year; QALY = quality-adjusted life-year.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
EDSS = Expanded Disability Status Scale; NMOSD = neuromyelitis optica spectrum disorder.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
The sponsor’s base case results do not represent valid estimates of costs, life-years, or QALYs. As detailed in the key limitations section of the report, the submitted model relied on calculations to estimate state membership which did not reflect the conceptualized Markov model. The identified errors resulted in a cohort simulation which could not keep track of every patient and, as a result, decreased in size over time. This resulted from formulas that removed dead patients from the cohort trace, failed to incorporate the general population mortality risk as a competing risk to changes in EDSS scores, and did not correctly incorporate the time- and treatment-dependent risk of an NMOSD attack. Therefore, detailed results from the sponsor’s base case where not reported.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Given the limitations within the submitted pharmacoeconomic model, CADTH was unable to conduct any additional analyses to assess the relative cost-effectiveness of inebilizumab for the treatment of adults with NMOSD who are AQP4-IgG seropositive.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 6: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
AQP4-IgG = anti-aquaporin-4 immunoglobulin G; BIA = budget impact analysis; NMOSD = neuromyelitis optica spectrum disorder.
Summary of Sponsor’s BIA
The sponsor-submitted budget impact analysis (BIA) assessed the budgetary impact resulting from reimbursing inebilizumab for the treatment of adults with NMOSD that is AQP4-IgG seropositive.51 The BIA was conducted from the perspective of the Canadian public drug plans over a 3-year horizon (2025 to 2027) with 2024 as the base year using epidemiological approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits Program. The analysis was performed using jurisdiction-specific values by summing up individual provincial results to obtain consolidated results. Key inputs to the BIA are documented in Table 8.
The following key assumptions were made by the sponsor:
As no epidemiological studies specific to NMOSD in Canada exist, the sponsor used the midpoint of the reported prevalence range from systemic reviews.
The sponsor used a blended method to determine drug acquisition costs for inebilizumab and relevant comparators. In each year, it was assumed that one-third of the patients are in the induction year of treatment (year 1), and two-thirds of patients are in the maintenance years of treatment (year 2 or later).
The sponsor assumed no market shares would be captured from ISTs if inebilizumab were reimbursed.
Table 7: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Prevalence of NMOSD Proportion of patients who are adults Proportion of patients with AQP4-IgG–seropositive NMOSD Patients who are actively managed and receiving maintenance therapy due to risk of relapse Proportion of patients covered under public drug plans | 81%54 73%55 100%a 32%56 |
Number of patients eligible for drug under review | 149 / 151 / 152 |
Market uptake (3 years) | |
Uptake (reference scenario): ISTs Rituximab Satralizumab Eculizumab | 23% / 18% / 16% 66% / 63% / 60% 11% / 19% / 24% 0% / 0% / 0% |
Uptake (new drug scenario): Inebilizumab ISTs Rituximab Satralizumab Eculizumab | 6% / 11% / 13% 23% / 18% / 16% 61% / 55% / 51% 10% / 16% / 20% 0% / 0% / 0% |
Cost of treatment (per patient)b | |
Cost of treatment over 1 year: Inebilizumab ISTs Rituximab Satralizumab Eculizumab | $230,607 $1,435 $8,910 $132,300 $721,251 |
AQP4-IgG = anti-aquaporin-4 immunoglobulin G; IST = immunosuppressive therapy; NMOSD = neuromyelitis optica spectrum disorders.
aSponsor obtained clinician input.
bCost of treatment over 1 year refers to year 1 costs. Cost may be higher than in subsequent years due to differences between induction and maintenance doses.
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement of inebilizumab for the treatment of adults with NMOSD who are AQP4-IgG seropositive would result to an incremental budget impact of $1,846,631 in year 1, $2,400,637 in year 2, and $2,742,699 in year 3. The total 3-year incremental cost of reimbursing inebilizumab is $6,989,967. Sensitivity analyses were completed to (i) include an alternative proportion of patients with NMOSD, (ii) adjust for a population in Canada, (ii) include an alternative proportion of patients with AQP4-IgG seropositivity, (iv) include an alternative proportion of patients covered under public drug plans, and (v) adjust for optimistic and pessimistic market shares. These sensitivity analyses impacted the 3-year incremental budget impact, which varied from $1,452,091 to $12,527,843.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Prevalence of NMOSD in the pan-Canadian population is underestimated: The sponsor’s submitted budget impact estimated that the proportion of NMOSD patients in pan-Canadian jurisdictions was approximately 2.46 per 100,000 people. As no epidemiological data for this indication exists for the pan-Canadian population, this estimate was derived from the midpoint range (between 0.51 and 4.4 per 100,000) from a systematic review that noted the epidemiology of NMOSD in the world.52 Clinical expert feedback obtained by CADTH highlighted that a prevalence of 2.46 per 100,000 people is lower than what is observed in clinical practice. The clinical expert feedback indicated that the prevalence of NMOSD may be closer to 4.4 per 100,000 people.
To address this limitation, CADTH undertook a reanalysis with a prevalence of 4.4 per 100,000 people as part of the base case.
Total treatment costs are uncertain: In the sponsor’s submitted BIA, the sponsor used a blended cost method to combine induction and maintenance costs for all treatments to simplify calculations in the model. In the model, it was assumed that one-third of patients are in the induction year of treatment (year 1) and two-thirds of patients are in the maintenance phase (years 2+). The use of blended cost method adds uncertainty to the total treatment costs, as the assumption may not represent clinical practice.
CADTH could not undertake reanalysis to address this limitation as the sponsor’s BIA model lacked flexibility to incorporate treatment costs separately during induction and maintenance phases.
CADTH Reanalyses of the BIA
Table 8: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption | |
|---|---|---|---|
Corrections to sponsor’s base case | |||
None | — | — | |
Changes to derive the CADTH base case | |||
1. Prevalence of patients with NMOSD in Canada | 2.46 per 100,000 people (midpoint between range of 0.51 to 4.4 per 100,000 people) | 4.4 per 100,000 people | |
CADTH base case | Reanalysis 1 | ||
NMOSD = neuromyelitis optica spectrum disorder.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 9 and a more detailed breakdown is presented in Table 10. Based on the CADTH base case, the budget impact associated with the reimbursement of inebilizumab in the indicated target population is expected to be $3,309,644 in year 1, $4,302,566 in year 2, $4,915,632 in year 3, with a 3-year total of $12,527,843.
Table 9: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 6,989,967 |
CADTH base case | 12,527,843 |
BIA = budget impact analysis.
Table 10: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 1,869,102 | 2,794,339 | 4,303,900 | 5,278,206 | 12,376,445 |
New drug | 1,869,102 | 4,640,970 | 6,704,536 | 8,020,905 | 19,366,412 | |
Budget impact | 0 | 1,846,631 | 2,400,637 | 2,742,699 | 6,989,967 | |
CADTH base case | Reference | 3,349,919 | 5,008,184 | 7,713,710 | 9,459,922 | 22,181,816 |
New drug | 3,349,919 | 8,317,829 | 12,016,277 | 14,375,554 | 34,709,659 | |
Budget impact | 0 | 3,309,644 | 4,302,566 | 4,915,632 | 12,527,843 |
BIA = budget impact analysis.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.