Drugs, Health Technologies, Health Systems
Reimbursement Review
Inclisiran (Leqvio)
Sponsor: Novartis Pharmaceuticals Canada Inc.
Therapeutic area: Primary hypercholesterolemia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ACS
acute coronary syndrome
ADA
antidrug antibodies
AE
adverse event
ANCOVA
analysis of covariance
ApoB
apolipoprotein B
ASCVD
atherosclerotic cardiovascular disease
ASCVD-RE
atherosclerotic cardiovascular disease risk equivalent
CABG
coronary artery bypass graft surgery
CAD
coronary artery disease
CCS
Canadian Cardiovascular Society
CDEC
CADTH Canadian Drug Expert Committee
CHD
coronary heart disease
CHPA
Canadian Heart Patient Alliance
CI
confidence interval
CrI
credible interval
CV
cardiovascular
CVD
cardiovascular disease
EAIR
exposure-adjusted incidence rate
ECG
electrocardiogram
EOS
end of study
FH
familial hypercholesterolemia
FRS
Framingham Risk Score
HDL-C
high-density-lipoprotein cholesterol
HeFH
heterozygous familial hypercholesterolemia
HoFH
homozygous familial hypercholesterolemia
HR
hazard ratio
HRQoL
health-related quality of life
IRT
Interactive Response Technology
ITC
indirect treatment comparison
ITT
intention to treat
LDL-C
low-density-lipoprotein cholesterol
LLT
lipid-lowering therapy
Lp(a)
lipoprotein (a)
LSM
least squares mean
MACE
major adverse cardiac events
MAR
missing at random
MedDRA
Medical Dictionary for Regulatory Activities
MI
myocardial infarction
mITT
modified intention to treat
MMRM
mixed model for repeated measures
MTD
maximally tolerated dose
nFH
nonfamilial hypercholesterolemia
NMA
network meta-analysis
PAD
peripheral arterial disease
PMM
pattern-mixture model
PY
patient-year
RCT
randomized controlled trial
RR
relative risk
SAE
serious adverse event
SC
subcutaneous
siRNA
small interfering ribonucleic acid
SLR
systematic literature review
TEAE
treatment-emergent adverse event
TESAE
treatment-emergent serious adverse event
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Inclisiran (Leqvio), 284 mg in 1.5 mL, solution for SC injection in prefilled syringe |
Sponsor | Novartis Pharmaceuticals Canada Inc. |
Indication | As an adjunct to lifestyle changes, including diet, to further reduce LDL‐C level in adults with the following conditions who are on a maximally tolerated dose of a statin, with or without other LDL‐C‐lowering therapies:
|
Reimbursement request | Per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 26, 2021 |
Recommended dose | Inclisiran 284 mg (inclisiran as inclisiran sodium) administered as a single SC injection: initially, again at 3 months, followed by every 6 months |
ASCVD = atherosclerotic cardiovascular disease; HeFH = heterozygous familial hypercholesterolemia; LDL-C = low-density-lipoprotein cholesterol; nFH = nonfamilial hypercholesterolemia; NOC = Notice of Compliance; SC = subcutaneous.
Introduction
In Canada, cardiovascular disease (CVD) is the second leading cause of death and accounted for almost 20% of all deaths in 2020.1 Despite its pathophysiological complexity, the 1 prerequisite for atherosclerotic plaque development is the presence of low-density-lipoprotein cholesterol (LDL-C).2 Hypercholesterolemia can be grouped into 2 forms: nonfamilial hypercholesterolemia (nFH), and familial hypercholesterolemia (FH, also referred to as acquired or genetic hypercholesterolemia). nFH is characterized by elevated LDL-C levels. Its etiology is likely a complex interplay between several genetic, environmental risk factors that increase the risk of nFH and include diet, smoking, physical inactivity, and other factors known to be associated with an increased risk of CVD (e.g., diabetes, chronic kidney disease, and hypertension).3-6 In Canada, the 1 year incidence rate for atherosclerotic cardiovascular disease (ASCVD) ranges between 7.2 and 8.8 per 1,000 person years, and the 5-year prevalence of ASCVD ranges between 6.91% and 8.55% in adults.7-9
Elevated LDL-C is directly associated with the development of atherosclerosis and ASCVD.10 The 3 main subcategories of ASCVD are coronary artery disease (CAD), cerebrovascular disease, and peripheral arterial disease (PAD). Individuals with hypercholesterolemia and a history of an atherosclerotic event are categorized as having established clinical ASCVD (i.e., they are secondary-prevention patients), whereas individuals with hypercholesterolemia at risk of developing ASCVD are considered to be primary-prevention patients. A subset of primary-prevention patients at greater risk of ASCVD is referred to an ASCVD risk-equivalent (ASCVD-RE) subset. Patient with ASCVD-RE are defined as those with type 2 diabetes mellitus, FH, or a 10-year risk of a cardiovascular (CV) event of at least 20% as assessed by the Framingham Risk Score (FRS) for CVD or equivalent.11 The proportion of the overall ASCVD population considered to be at high risk is estimated to be approximately 25%.9 In accordance with Canadian guidelines, published literature, and validation from clinicians in Canada, patients with high-risk nFH ASCVD are defined as having any of the following criteria in the past 12 months: diabetes, recurrent vascular events, PAD, or acute coronary syndrome (ACS); and LDL-C levels greater than 1.8 mmol/L despite maximally tolerated dose (MTD) statins with or without other lipid-lowering therapies (LLTs).9,12-17 Throughout this resubmission, patients with any of these criteria will be considered part of the high-risk ASCVD subgroup.
FH is 1 of the most common genetic disorders and is caused by mutations in the genes encoding the LDL receptor (LDLR), apolipoprotein B (ApoB), or proprotein convertase subtilisin/kexin type 9 (PCSK9), leading to high plasma levels of LDL-C.10 Depending on the number of mutant alleles, patients can be categorized as having homozygous FH (HoFH) or heterozygous FH (HeFH).18 HeFH has an estimated prevalence of approximately 1 in 25019 to 1 in 311 individuals.20,21 The clinical presentation of FH is variable, and is affected by the number and type of mutations together with other genetic factors. Individuals with FH have elevated LDL-C levels from a young age, and the ongoing exposure to elevated LDL-C results in a higher cumulative risk of developing ASCVD.22 Patients with FH may present with physical findings such as tendon xanthomata or xanthelasma.23 FH is associated with a higher risk of CV events than in the general population.24-26
Inclisiran (Leqvio) was previously reviewed by CADTH in February 2022 for the same indication, and the recommendation was to not reimburse.27 Key reasons for this recommendation included the fact that there was insufficient evidence that inclisiran reduces CV morbidity and mortality, or all-cause mortality, as the pivotal trials — ORION-9, ORION-10, and ORION-11 — were not designed to assess these outcomes.27 Additionally, the CDA-AMC Canadian Drug Expert Committee (CDEC) noted that the long-term efficacy and safety of inclisiran has not been determined, and that there were 2 ongoing studies — ORION-4 and ORION-8 — that are expected to provide evidence to better characterize the pertinent clinical outcomes, as well as provide long-term efficacy and safety data. CDEC also noted that there was no direct comparison of inclisiran with evolocumab or alirocumab, or other add-on drugs, and that there were limitations with the submitted indirect treatment comparison (ITC), including the relatively short follow-up (24 weeks) of patients with a chronic condition.
The sponsor outlined the basis for its resubmission. To address the lack of evidence for reductions in CV morbidity and/or mortality and all-cause mortality, the sponsor included a posthoc pooled analysis of major adverse cardiovascular events (MACE) in the pivotal ORION studies and, to address concerns about long-term efficacy and harms, the findings of the long-term extension studies (ORION-3 and ORION-8). To address the lack of long-term safety data, in addition to the ORION-3 and ORION-8 studies, the sponsor submitted a pooled analysis of 7 ORION trials. Finally, the sponsor submitted a revised budget impact model to address CDA-AMC’s concerns from the first recommendation.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of inclisiran in adults with primary hypercholesterolemia (HeFH or nFH).
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CDA-AMC’s call for input and from clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
Two patient groups — the Canadian Heart Patient Alliance (CHPA) and the HeartLife Foundation — provided input based on survey and interview responses (CHPA) and from executives of the HeartLife Foundation.
Patients describe a condition that is very difficult to manage, impacts their physical and mental well-being, has a significant financial burden on families, and impacts their quality of life.
Adherence and access to newer treatments, such as the PCSK9 inhibitors, were identified by patients as key challenges in managing their condition. Patients emphasized the importance of having a safe, tolerable, and effective treatment to maintain their LDL-C below recommended thresholds. Patients also noted the importance of having a less frequent dosing regimen to manage their condition.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
Nonadherence, intolerance to high-intensity statins, inability to reach recommended lipid targets despite MTD statins and ezetimibe, and lack of access to PCSK9 inhibitors are the major unmet needs identified by the clinical experts in treatment of patients with HeFH and with nFH with ASCVD. Accordingly, the clinical experts believed that in addition to being another PCSK9-targeting drug, inclisiran may help with nonadherence due to the less frequent dosing schedule.
The clinical experts believed that for patients with HeFH, in addition to those unable to reach their LDL-C target despite MTD statins, with or without ezetimibe, patients especially well suited to inclisiran would include those with other risk factors, such as smoking, diabetes, hypertension, and elevated lipoprotein (a) (Lp[a]). For patients with nFH with ASCVD, the clinical experts believed that well-suited patients would include those unable to tolerate high-intensity statins, those with early disease onset or recurrent disease, those whose LDL-C is far from the threshold, and those with the risk factors identified for patients with HeFH. The clinical experts also referenced the 2021 Canadian Cardiovascular Society (CCS) guidelines, which identified which secondary-prevention patients are likely derive the most benefit from the intensification of statin therapy with the additional use of a PCSK9 inhibitor. These included patients with recent ACS (in the past 52 weeks), diabetes mellitus or metabolic syndrome, polyvascular disease, symptomatic PAD, recurrent myocardial infarction (MI), MI in the past 2 years, previous coronary artery bypass graft (CABG) surgery, LDL-C of 2.6 mmol/L or greater or HeFH, or Lp(a) of 120 nmol/L or greater.
The clinical experts noted that genetic testing should not be required to confirm a diagnosis of HeFH due to the lack of availability of testing, and they also noted that HeFH is underdiagnosed in Canada. Various lipid parameters would be used to assess response to treatment in addition to LDL-C, including non–high-density-lipoprotein cholesterol (HDL-C) and ApoB. Although there is no recent guidance on how frequently to assess response, after the initial titration, response is typically assessed every 6 to 12 months.
Clinician Group Input
Fourteen clinician groups provided input: Alberta Cardiovascular Disease Prevention Collaborative (8 clinicians contributed to the input); BC Lipid Specialists (11 clinicians contributed to the input); le Centre hospitalier universitaire Dr.-Georges-L.-Dumont (CHUDGLD) (6 clinicians contributed to the input); Cambridge Cardiac Rehab Program (6 clinicians contributed to the input); CCS Dyslipidemia Guideline Committee (14 clinicians contributed to the input); Cardiology Association of Niagara (3 clinicians contributed to the input); Egyptian Cardiologists of Niagara (3 clinicians contributed to the input); Kawartha Cardiology Clinic (7 clinicians contributed to the input); Lipid Clinic of McMaster University and Hamilton Health Sciences (1 clinician contributed to the input); Mazankowski Alberta Heart Institute (3 clinicians contributed to the input); Oakville Cardiologists (9 clinicians contributed to the input); service of cardiology, Internal Medicine Department and Heart Failure Group at St. Thomas Elgin General Hospital (5 clinicians contributed to the input); Western University, Division of Cardiology, Cardiac Rehabilitation and Secondary Prevention Program (3 clinicians contributed to the input); and University of Toronto faculty and clinicians at St. Michael’s Hospital who are actively involved in the treatment of patients with ASCVD and/or lipid disorders (10 clinicians contributed to the input).
The clinician groups agreed that the major issues with managing hypercholesterolemia, whether it be HeFH or nFH with ASCVD, are adherence (as well as intolerance) and lack of accessibility to drug therapies, and that the main outcome of interest is a reduction in lipid parameters (LDL-C, non-HDL-C, and ApoB) at 6 months, initially, and assessed annually thereafter.
The clinician groups believed that inclisiran would be best suited for patients at risk of ASCVD or patients with FH who require additional LLT, who become refractory to statins and ezetimibe, along with those who struggle with adherence or tolerability.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for inclisiran:
relevant comparators
considerations for the initiation of therapy
considerations for continuation or renewal of therapy
considerations for discontinuation of therapy.
The clinical expert consulted by CDA-AMC provided advice on the potential implementation issues raised by the drug program. Refer Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
The major focus of this resubmission was a posthoc pooled analysis of MACE from the ORION-9, ORION-10, and ORION-11 trials. These trials, all included in the original submission, were phase III, double-blind, randomized controlled trials (RCTs) comparing inclisiran to placebo in adult patients with HeFH (ORION-9) or ASCVD (ORION-10 and ORION-11) and ASCVD-RE (ORION-11) (i.e., those with diabetes, FH, or a 10-year risk of a CV event of at least 20% as assessed by the FRS for CVD or equivalent) who were receiving MTD statins or who were statin intolerant. Patients in the ORION-9 trial had a history of HeFH, with a diagnosis of HeFH confirmed by genetic testing or phenotypic Simon Broome criteria, and/or a documented history of untreated LDL-C of greater than 190 mg/dL and a family history of FH; elevated cholesterol or early heart disease may indicate FH. In all 3 ORION studies, patients were randomized (1:1) to either inclisiran sodium 300 mg or placebo in addition to MTD statins. The ORION-9, ORION-10, and ORION-11 trials enrolled 482, 1,561, and 1,617 patients, respectively. The studies were all 18 months in duration, with patients receiving 4 300 mg doses of inclisiran sodium (on day 1, day 90, day 270, and day 450). The primary outcome of the ORION-9, ORION-10, and ORION-11 trials was the percent change in LDL-C from baseline to day 510. In all trials, the coprimary end point was the average percentage change in LDL-C from baseline to the period from after day 90 and up to day 540, reflecting the start of the biannual dosing regimen. Incidences of CV death, resuscitated cardiac arrest, nonfatal MI, and nonfatal stroke (ischemic and hemorrhagic) were exploratory outcomes in the ORION trials within the composite outcome of MACE, and total deaths was a secondary outcome reported as adverse events (AEs) in the ORION studies.
Baseline characteristics of the ORION trials were balanced between groups, and generally applicable to the population in Canada. The ORION-9 trial enrolled patients with a median age of 56 years — the ratio of men to women was relatively even (47.1% men, 52.9% women) — with either ASCVD (27.4%) or ASCVD-RE (72.6%), of whom 93.2% had HeFH. A total of 73.9% of patients were on high-intensity statins at baseline, with 25.3% either partially or completely intolerant to statins, and 52.3% were treated with ezetimibe. The ORION-10 trial enrolled mostly men (69.4%), with a median age of 67 years, all with ASCVD (91.1% with coronary heart disease [CHD]). Approximately two-thirds (69.4%) of patients were on a high-intensity statin at baseline, with 22.0% partially or completely intolerant. A total of 9.9% of patients were treated with ezetimibe. The ORION-11 trial enrolled patients with ASCVD (87.4%) and ASCVD-RE (12.6%). Patients were mostly men (71.7%), with a median age of 65 years. A total of 78.0% of patients were receiving high-intensity statins, 11.4% were considered partially or completely intolerant to statins, and 7.1% of patients were treated with ezetimibe.
Efficacy Results
Major Adverse Cardiac Events
In the ORION-9, ORION-10, and ORION-11 trials, the exploratory end point of MACE was defined as the composite of CV death, resuscitated cardiac arrest, nonfatal MI, and nonfatal stroke (hemorrhagic or nonhemorrhagic), using predefined Medical Dictionary for Regulatory Activities (MedDRA) search.
As part of their resubmission, the sponsor conducted a pooled analysis of clinical outcomes from the ORION-9, ORION-10, and ORION-11 trials and provided what it referred to as a sensitivity analysis that pooled data from the ORION-10 and ORION-11 studies. The pooled analysis of all 3 trials is not relevant for this review, as it combines the HeFH and the nFH with ASCVD populations; these 2 populations are viewed separately for this review, consistent with the indication. The sensitivity analysis that was conducted to assess the effects of inclisiran (n = 1,494) compared to placebo (n = 1,477) on MACE in the ASCVD and ASCVD-RE populations is relevant.
HeFH Population: The incidence of MACE in the inclisiran and placebo arms of the ORION-9 trial were 10 (4.1%) and 10 (4.2%), respectively; the absolute number of MACE in the inclisiran and placebo arms were 10 and 11 events, respectively; the corresponding relative risk (RR) was ███ ████ ███ ████ ████.
The exploratory end point of nonfatal MI occurred in 9 (3.7%) patients experiencing | events in the inclisiran arm compared to 10 (4.2%) patients experiencing ██ events in the placebo arm, and for the exploratory end point of nonfatal stroke, no patients in either the inclisiran or placebo treatment arms experienced an event.
nFH with ASCVD: Results from the posthoc pooled analysis of the ORION-10 and ORION-11 trials showed ███ ██████ patients in the inclisiran arm experienced ███ ██████, while ███ ███████ patients in the placebo arm experienced ███ ██████. The reported hazard ratio (HR) of ████ ████ ███ █████ █████ for MACE favoured inclisiran over placebo in the pooled ORION-10 and ORION-11 patient population.
The incidence of MACE in the inclisiran and placebo arms of the ORION-10 trial was 58 (7.4%) and 79 (10.2%), respectively; the absolute number of MACE in the inclisiran and placebo arms was 66 and 90 events, respectively; the corresponding RR was ███ ████ ███ ████ ████. The incidence of MACE in the inclisiran and placebo arms of the ORION-11 trial was 63 (7.8%) and 83 (10.3%), respectively; the absolute number of MACE in the inclisiran and placebo arms was 65 and 100 events, respectively; the corresponding RR was ███ ████ ███ ████ ████.
From the same posthoc pooled analysis of ORION-10 and ORION-11, the effects of inclisiran (n = 1,494) compared to placebo (n = 1,477) on fatal or nonfatal MI outcome in the ASCVD and ASCVD-RE population showed that ██ ██████ patients in the inclisiran arm experienced ██ ██████, while ██ ██████ patients in the placebo arm experienced ██ ██████. The reported HR was ████ ████ ███ █████ █████ for fatal or nonfatal MI. The fatal or nonfatal stroke outcome in the ASCVD and ASCVD-RE population showed that ██ ██████ patients in the inclisiran arm experienced ██ ██████, while ██ ██████ patients in the placebo arm experienced ██ ██████. The HR was ████ ████ ███ █████ █████ for fatal or nonfatal stroke in the pooled ORION-10 and ORION-11 patient population.
Low-Density-Lipoprotein Cholesterol
The coprimary end points of percent change in LDL-C from baseline to day 510 and time-average percent change in LDL-C from baseline to the period from after day 90 and up to day 540 was the same for the ORION-9, ORION-10, and ORION-11 trials.
Heterozygous Familial Hypercholesterolemia: The between-group difference for inclisiran and placebo in the percent reduction in LDL-C in the ORION-9 trial was –47.9% (95% confidence interval [CI], –53.5 to –42.3; P < 0.0001). For the time-average percent change in LDL-C from baseline to the period from after day 90 and up to day 540, the least squares mean (LSM) difference from placebo favoured inclisiran in the ORION-9 trial, at −44.30% (95% CI, −48.48 to −40.12; P < 0.0001). The results of the sensitivity analyses for both outcomes were consistent with the overall population.
nFH with ASCVD: The between-group difference for inclisiran and placebo in percent reduction in LDL-C in the ORION-10 trial was – 52.2% (95% CI, –55.7 to –48.8; P < 0.0001), and in the ORION-11 trial was –49.9% (95% CI, –53.1 to –46.6; P < 0.0001). For the time-average percent change in LDL-C from baseline to the period from after day 90 and up to day 540, the LSM difference from placebo favoured inclisiran in the ORION-10 trial, at −53.78% (95% CI, −56.23 to −51.33; P < 0.0001), and in the ORION-11 trial, at −49.17% (95% CI, −51.57 to −46.77; P < 0.0001). The results of the sensitivity analyses for both outcomes were consistent with the overall population.
Harms Results
Heterozygous Familial Hypercholesterolemia
In the ORION-9 trial, the most common AEs in the inclisiran and placebo groups were nasopharyngitis (11.6% versus 8.3%), influenza (5.4% versus 8.8%), upper respiratory tract infection (6.6% versus 6.7%), and back pain (7.1% versus 4.2%). There were 18 (7.5%) patients in the inclisiran arm and 33 (13.8%) patients in the placebo arm who experienced at least 1 serious adverse event (SAE). The most common SAEs were unstable angina, myocardial ischemia, acute MI, aortic valve stenosis, and back pain. Three (1.2%) patients in the inclisiran group withdrew due to an AE, whereas no patients in the placebo group did. ███████ ███████ ██ ██████████ ████ █████ ████ ████ ████████ ███████ █████████ ████ █████████ ███ ███ ████████ ███ ██████ ████ ██ ███ ████████ █████ ███ ███ █████████ ████ ████████ ██████ ████ ██████████ ███████████ ███████ ██ ██████████
nFH with ASCVD
The frequency of AEs was consistent in the inclisiran and placebo groups, as well as across trials, with 73.5% versus 74.8% of patients, respectively, experiencing at least 1 AE in the ORION-10 trial, and 82.7% versus 81.5% experiencing at least 1 AE in the ORION-11 trial. In the ORION-10 and ORION-11 trials, SAEs occurred in 22.4% and 22.3% of patients, respectively, treated with inclisiran compared to 26.3% and 22.5% of patients, respectively, treated with placebo. Withdrawal due to adverse events (WDAEs) was similar in the ORION-10 and ORION-11 trials, at 2.4% and 2.8% of patients, respectively, treated with inclisiran and 2.2% and 2.2% of patients, respectively, treated with placebo.
██ ██████████ ██ ██████████ ██████ ███ ██████████████ █████████ ███ ████████ ████ ██████████ ███ ███████ ██ ███ █████ ███████ ████████ ███ █████████ ███ ██████ ██ ███ ███████ ███████ In all trials, fewer patients treated with placebo than with inclisiran reported AEs at the injection site. Injection-site reactions were mild to moderate, and no severe reactions were seen across trials. █████ ████ ██ █████ ███ ██████████ ███████████ ███████ ██████████ ███ ███████ ███ █████ ███████ █████ ██ ████████████████ ██████████ █████ ███████ ██ ███████ ███████
Critical Appraisal
There are a number of issues associated with the posthoc pooled analysis provided by the sponsor for this resubmission. First of all, it is a posthoc analysis, which increases the potential for bias. The primary analysis includes all 3 pivotal trials (ORION-9, ORION-10, and ORION-11); however, this combines 2 separate populations of patients — those with HeFH and those with nFH with ASCVD — and these patients are being considered separately for this review. Importantly, the ORION-9, ORION-10, and ORION-11 trials were not powered to assess MACE, so the events were captured in the safety population, the definitions used may not be inclusive or specific enough, and there was no blinded, centralized assessment of events. Otherwise, the ORION-9, ORION-10, and ORION-11 trials appear to have been reasonably well conducted, with adequate measures to maintain blinding, a multiple testing procedure to reduce the risk of type I error, and low dropout rates.
With respect to external validity, key issues are that clinical outcomes such as CV mortality and morbidity were not assessed in the pivotal ORION trials, and there was no active comparator, such as a PCSK9 inhibitor. Additionally, health-related quality of life (HRQoL) was not assessed in any of the included trials.
GRADE Summary of Findings and Certainty of the Evidence
Grading of Recommendations Assessment, Development and Evaluation (GRADE) was not performed for this review because it is a resubmission.
Table 2: Summary of Efficacy and Harms Data for the HeFH and nFH With ASCVD Populations
Outcome | ORION-9 (HeFH) | |||
|---|---|---|---|---|
Inclisiran (N = 242) | Placebo (N = 240) | |||
MACEa | ||||
n (%) | 10 (4.1) | 10 (4.2) | ||
Events, n | 10 | 11 | ||
Cardiovascular deaths | ||||
n (%) | 1 (0.4) | 0 | ||
Nonfatal MI | ||||
n (%) | 9 (3.7) | 10 (4.2) | ||
Events, n | | | ██ | ||
Nonfatal stroke | ||||
n (%) | 0 | 0 | ||
Events, n | 0 | 0 | ||
Percent change in LDL-C from baseline to day 510 (coprimary outcome) | ||||
Number of patients contributing to the analysis | 242 | 240 | ||
Baseline, mg/dL, mean (SD) | 151.4 (50.4) | 154.7 (58.0) | ||
LSM change from baseline, % (95% CI) | –39.7 (–43.7 to –35.6) | 8.2 (4.3 to 12.2) | ||
LSM difference vs. control, % (95% CI)b | –47.9 (–53.5 to –42.3) | |||
P value | < 0.0001 | |||
Time-adjusted percent change in LDL-C from day 90 to day 540 (coprimary outcome) | ||||
Number of patients contributing to the analysis | 242 | 240 | ||
Baseline, mg/dL, mean (SD) | 151.4 (50.4) | 154.7 (58.0) | ||
LSM change from baseline, % (95% CI) | –38.1 (–41.0 to –35.1) | 6.2 (3.3 to 9.2) | ||
LSM difference vs. control, % (95% CI)b | –44.3 (–48.5 to –40.1) | |||
P value | < 0.0001 | |||
Harms | ||||
Patients with ≥ 1 AE | 185 (76.8) | 172 (71.7) | ||
Patients with ≥ 1 SAE | 18 (7.5) | 33 (13.8) | ||
Incidence of WDAEs | 3 (1.2) | 0 (0) | ||
████████ ██████████████ █████ | █████ | █████ | ||
Patients with ≥ 1 AE at the injection site | 41 (17.0) | 4 (1.7) | ||
████████████████ █████████ | ██ █████ | ██ █████ | ||
█████ ██████ | █████ | █████ | ||
███████ ██████ | █████ | █████ | ||
Pooled results | ORION-10 and ORION-11 (nFH with ASCVD) | |||
Inclisiran (N = 1,494) | Placebo (N = 1,477) | |||
Posthoc pooled analysis, MACEa | ||||
███ | ███ █████ | ███ ██████ | ||
███████ | ███ | ███ | ||
██ ████ ██ | ████ ██████ █████ | |||
Fatal and nonfatal MI | ||||
███ | ██ █████ | ██ █████ | ||
███████ | ██ | ██ | ||
██ ████ ███ | ████ ██████ █████ | |||
Fatal and nonfatal stroke | ||||
███ | ██ █████ | ██ █████ | ||
███████ | ██ | ██ | ||
██ ████ ██ | ████ ██████ █████ | |||
Outcome | ORION-10 | ORION-11 | ||
Inclisiran (N = 781) | Placebo (N = 780) | Inclisiran (N = 810) | Placebo (N = 807) | |
Percent change in LDL-C from baseline to day 510 (coprimary outcome) | ||||
Number of patients contributing to the analysis | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 104.5 (39.6) | 104.8 (37.0) | 107.2 (41.8) | 103.7 (36.4) |
LSM change from baseline, % (95% CI) | –51.3 (–53.8 to –48.8) | 1.0 (–1.5 to 3.4) | –45.8 (–48.2 to –43.5) | 4.0 (1.76 to 6.3) |
LSM difference vs. control, % (95% CI)b | –52.2 (–55.7 to –48.8) | –49.9 (–53.1 to –46.6) | ||
P value | < 0.0001 | < 0.0001 | ||
Time-adjusted percent change in LDL-C from day 90 to day 540 (coprimary outcome) | ||||
Number of patients contributing to the analysis | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 104.5 (39.6) | 104.8 (37.0) | 107.2 (41.8) | 103.7 (36.4) |
LSM change from baseline, % (95% CI) | –51.3 (–53.0 to –49.5) | 2.5 (0.77 to 4.25) | –45.8 (–47.5 to –44.1) | 3.4 (1.7 to 5.1) |
LSM difference vs. control, % (95% CI)b | –53.8 (–56.2 to –51.3) | –49.2 (–51.6 to –46.8) | ||
P value | < 0.0001 | < 0.0001 | ||
Harms, patients, n (%) | ||||
Patients with ≥ 1 AE | 574 (73.5) | 582 (74.8) | 671 (82.7) | 655 (81.5) |
Patients with ≥ 1 SAE | 175 (22.4) | 205 (26.3) | 181 (22.3) | 181 (22.5) |
Patients who WDAE | 19 (2.4) | 17 (2.2) | 23 (2.8) | 18 (2.2) |
████████ ████ ██ ██████████████ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
Patients with ≥ 1 AE at the injection site | 47 (6.0) | 15 (1.9) | 62 (7.6) | 14 (1.7) |
████████████████ █████████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
█████ ██████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
███████ ██████ | ██ █████ | ██ █ ████ | ██ █████ | ██ █████ |
AE = adverse events; ASCVD = atherosclerotic cardiovascular disease; CI = confidence interval; HeFH = heterozygous familial hypercholesterolemia; HR = hazard ratio; LDL-C = low-density-lipoprotein cholesterol; L = litre; LSM = least squares mean; MACE = major adverse cardiovascular events; MI = myocardial infarction; nFH = nonfamilial hypercholesterolemia; NR = not reported; SAE = serious adverse event; SD = standard deviation; WDAE withdrawal due to adverse event.
aObserved MACE counts include treatment-emergent and non–treatment-emergent adverse events.
bA control-based pattern-mixture model (PMM) was used for missing data imputation, with 100 total imputed datasets. A mixed model for repeated measures (MMRM) on each of the 100 datasets was performed by including fixed effects for treatment, visit, interaction between treatment and visit, and baseline LDL-C as a covariates.
c██████ ██████ █████████ ████ ███ ████████████ ██████ ██████ ████ █████████ ███ █████ ██ ████████ ███████ ██████ ████ ███████ █████ ████ ████ ████████ ████ ███ █████████████ █████████ ████████ ████ ████████ ████ ██ █████ █████ ███ ██ ██████████████████████ ███████ ██████ ████ ███ ██ ███████ ███ ████████
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report.30
Long-Term Extension Studies
ORION-331,32 and ORION-833 Trials
Description of Studies
The ORION-3 trial31,32 was a 4-year, open-label extension study of the phase II ORION-1 trial. The primary objective of this study was to assess the effect of long-term treatment with twice-yearly small interfering (si)RNA therapeutic inclisiran dosing on LDL-C reductions at day 210 compared to baseline in the ORION-1 trial. The secondary and exploratory objectives were to assess the effects of inclisiran on cholesterol and other lipids levels and PCSK9 levels for up to 4 years in each arm, as well as the long-term safety and tolerability of inclisiran. Another exploratory objective was to evaluate the effects of transitioning from evolocumab to inclisiran. A total of 382 participants were enrolled from 52 centres across 5 countries; 56 of those patients were enrolled from Canadian centres.
The ORION-8 trial is a global, open-label, long-term extension study of patients with ASCVD, ASCVD-RE, or HeFH and elevated LDL-C despite a MTD of LDL-C-lowering therapies who completed the phase II ORION-3 study, or any of the phase III ORION-9, ORION-10, or ORION-11 studies. The primary objectives of the study are to evaluate the effect of inclisiran treatment on the proportion of patients achieving prespecified LDL-C targets, and the safety and tolerability of long-term use of inclisiran. The secondary objectives are to evaluate the effect of inclisiran on LDL-C levels and other lipids and lipoproteins. The study has enrolled 3,275 participants from 268 centres in 13 countries, including Canada (3 centres).
Efficacy Results
Of the original ORION-1 cohort of 497 patients, 290 of 370 patients allocated to the drug continued into the inclisiran-only arm and 92 of 127 patients allocated to placebo entered the switching arm in the ORION-3 extension study conducted between March 24, 2017, and December 17, 2021. Overall, efficacy results were consistent and sustained up to the end of the study. In the inclisiran-only arm, LDL-C was reduced by 47.5% (95% CI, 50.7% to 44.3%) at day 210 and was sustained at that level over 1,440 days. During the 4 years of the open-label extension, the mean percentage change and mean absolute change in LDL-C concentrations in the inclisiran-only arm ranged between –34.3% and –53.8% and between –1.13 mmol/L and –1.76 mmol/L, respectively, with the upper limit of the 95% CI at all time points being lower than –30% and excluding zero. The mean percentage change and mean absolute change in LDL-C in the switching arm ranged between –38.2% and –65.7% and between –1.20 mmol/L and –2.00 mmol/L, respectively.
In the inclisiran-only arm, the mean percentage change in total cholesterol ranged from –21.1% to –30.2%, remaining relatively consistent throughout the follow-up period. Non-HDL-C, ApoB, and triglycerides also remained consistently decreased throughout the follow-up period. Lp(a) concentration decreased by 16.3% at day 30 with no meaningful changes thereafter.
In the ORION-8 trial, the proportion of patients who attained global lipid targets at day 1,080 was similar in the inclisiran-only group (78%), the switching group (79%), and the group of patients who rolled over from the ORION-3 trial (77%). Similarly, the percent of patients with ASCVD who attained global lipid targets (< 70 mg/dL) at day 1,080 was similar in the inclisiran-only group (79%), the switching group (80%), and the group of patients who were rolled over from the ORION-3 trial (77%). The percent of patients with ASCVD-RE who attained global lipid targets (< 100 mg/dL) was 73% in the inclisiran-only group, 75% in the switching group, and 77% in the group of patients who were rolled over from the ORION-3 trial.
The mean percentage change from baseline to day 1,080 in LDL-C was –49.0% (95% CI, –50.5% to –47.4%) in the inclisiran-only group, –49.7% (95% CI, –51.3% to –48.0%) in the switching group, and –50.0% (95% CI, –52.6% to –47.3%) in the group rolled over from the ORION-3 trial.
Harms Results
The most common AEs in the ORION-3 trial were infection, hypertension, arthralgia, and fatigue. In the inclisiran-only arm, 275 (96.8%) patients experienced at least 1 AE. A total of 104 (36.6%) patients experienced at least 1 SAE. Nineteen (6.7%) patients and 12 (4.2%) patients discontinued the study treatment due to AEs and SAEs, respectively.
Overall, of the 87 patients in the switching arm, 80 (92%) patients experienced at least 1 AE. Thirty (34.5%) patients experienced at least 1 SAE. Five (5.7%) patients and 3 (3.4%) patients discontinued the study treatment due to AEs and SAEs, respectively.
Over the 4-year study duration, 7 deaths (2.5%) were reported in the inclisiran group and 1 death was reported in the switching arm, and none of the deaths was assessed as drug-related.
In the ORION-8 trial, 79% of patients in each of the inclisiran-only and switching groups reported an AE, as did 64% of patients who rolled over from the ORION-3 trial. The number of patients who discontinued treatment due to an AE (██ ██ ████ █████) was similar in the inclisiran-only group and the switching group, versus ████ of patients who rolled over from the ORION-3 trial.
With respect to SAEs, 31% of patients in the inclisiran-only group, 33% of patients in the switching group, and 15% of patients who rolled over from the ORION-3 trial experienced an SAE.
With respect to AEs of special interest, the following occurred in the inclisiran-only group, the switching group, and the group of patients who rolled over from the ORION-3 trial: █████████ ████ ██████ ███ ██████ ██ ██████ ████ ███████ ██████ ███ ██████ ██ ██████ ████ ███ ███████████████ ████████ ████ ██████ ███ ██████ ████ █████ ██████ ███ ██████ ██ ██████ ███ ███ ████████████████ ███ ██ ████ ███████
████ ████████ ██ ██ ██ ████████ ██ ███ █████ ████ █████████ ██ ███████████ ███ ██ ████████ ███ ████████ ████ ███████ ██ ███████████ ███ ██ ██ ████████ ███ ██████ ████ ████ ████████
Critical Appraisal
The open-label design of the ORION-3 and ORION-8 trials is considered a limitation that could bias the results parameters. Furthermore, only those who completed the parent trials were eligible for participation in these extensions, which might have potentially led to a selection bias. The lack of a control and/or comparator arm is considered a key constraint that limits the interpretation of study outcomes.
Because the ORION-3 and ORION-8 studies consisted of patients who took part in the pivotal studies, it is reasonable to expect that the same strengths and limitations related to generalizability apply to the extension studies, with the additional caveat of potential selection bias due to the enrolment criteria.
Indirect Comparisons
Description of Studies
The sponsor submitted an ITC that compared the efficacy of inclisiran to relevant drug comparators in patients with HeFH or ASCVD (or ASCVD-RE). The objective of the sponsor-submitted report was to conduct a feasibility assessment via systematic review of the literature and, if possible, to conduct an indirect comparison evaluating the relative efficacy and safety of inclisiran versus relevant drug comparators, including ezetimibe, and other PCSK9 inhibitors in patients with HeFH or ASCVD (or ASCVD-RE).34
The sponsor-submitted ITC was informed by a systematic review of RCTs conducted in April 2020. Thirty-nine studies met the inclusion criteria for the review and feasibility assessment, and 24 studies were subselected for inclusion in the ITC based on network connectivity and the homogeneity of study characteristics, patient characteristics, or outcomes that were likely modifiers of the relative treatment effects.34
The analyses were conducted using a network meta-analysis (NMA). Selection of both fixed and random effects were conducted for outcomes of interest. Random-effects analyses were selected as the base case, given the number of studies per node and the observed heterogeneity in patient and trial characteristics. Three network scenarios were conducted: patients with HeFH on MTD statins, patients with ASCVD and ASCVD-RE on MTD statins, and patients with ASCVD and ASCVD-RE who are intolerant to statins. Efficacy outcomes included percent, absolute, and time-adjusted change from baseline in LDL-C, percent change from baseline in HDL-C, and safety outcomes (including total discontinuations and discontinuations due to AEs).34
Efficacy Results
A total of 7 trials were included in the network for the HeFH population on MTD statins, 13 studies were included in the base-case network for the ASCVD and ASCVD-RE populations on MTD statins, where 1 closed loop was formed, and 7 trials were included in the network for ASCVD and ASCVD-RE populations intolerant to statins. In the HeFH population on MTD statins network, there was no difference between inclisiran and alirocumab or evolocumab for any efficacy or safety outcomes. In the ASCVD and ASCVD-RE population on MTD statins network, inclisiran was favoured over ezetimibe for efficacy outcomes related to LDL-C, but there was no difference between inclisiran and alirocumab or evolocumab for any efficacy or safety outcomes. In the ASCVD and ASCVD-RE population intolerant to statins network, inclisiran was favoured over ezetimibe for efficacy outcomes related to LDL-C but not safety outcomes. There was no difference between inclisiran and alirocumab or evolocumab in any efficacy or safety outcomes.
Critical Appraisal
There were several limitations to the key assumptions made in the NMA approach about the background statin use and the time of assessment of outcomes, impacting clinical and methodological heterogeneity, which resulted in limited interpretability and generalizability of the results. Although not reported or accounted for, these assumptions likely impacted treatment effects and the results of each NMA and were a significant source of heterogeneity in the studies. It was assumed in the NMA that individual statins had similar efficacy as background therapy, regardless of dose, and would not bias the results of the NMA; however, based on discussions with the clinical expert consulted by CDA-AMC, this is not considered a reasonable assumption. It was also assumed that differences in CV risk and severity would not impact the relative effects on LDL-C, and therefore no attempt was made to adjust for differences in baseline characteristics due to the number of studies and inconsistent reporting of characteristics. The NMA used 24 weeks as the time of assessment, which is considered acceptable for lipid and lipoprotein outcomes. End-of-study (EOS) values for safety were used and considered comparable if the duration of follow-up was 24 weeks or longer. Variations in trial length are bound to influence the number of patients withdrawing for various reasons and given the 24-week time of assessment, may undermine the true treatment effects. Additionally, given the biannual dosing regimen of inclisiran, a 24-week time of assessment may be insufficient to assess safety outcomes compared to the 2-week dosing regimen of alirocumab and evolocumab.
Overall, the studies included in the NMA were believed to be statistically heterogeneous based on the considerable I2; however, it is unclear what the source of heterogeneity was. The observed heterogeneity was likely due to observed and unobserved differences in patient populations across the included studies, data imputation analysis methods, and the specific background treatments allowed and/or delivered. Unidentified or unknown clinical heterogeneity (particularly treatment-effect modifiers) and methodological heterogeneity need to be explored, as it is unclear if the transitivity assumption was appropriately met.
In general, all treatments were favoured over placebo for all outcomes in each network scenario; however, the results typically displayed exceedingly wide credible intervals (CrIs), challenging the precision of the results.
Studies Addressing Gaps in the Evidence From the Systematic Review
Pooled Safety Analysis of 7 ORION Trials35
Description of Studies
A posthoc analysis35 comprised patients treated with 300 mg inclisiran sodium or placebo in the completed (i.e., ORION-1, ORION-3, ORION-5, ORION-9, ORION-10 and ORION-11) and ongoing (ORION-8) trials was conducted. The objective was to obtain data regarding the long-term safety and tolerability of inclisiran for up to 6 years in a large, pooled dataset from 7 completed and ongoing trials and a diverse sample of patients at risk for CV events. Exposure-adjusted incidence rates (EAIRs) and Kaplan-Meier estimates of the cumulative incidence of reported treatment-emergent adverse events (TEAEs), abnormal laboratory measurements, and the incidence of antidrug antibodies (ADAs) were analyzed.
This analysis included 3,576 patients treated with inclisiran for up to 6 years and 1,968 patients treated with placebo for up to 1.5 years, with 9,982.1 and 2,647.7 patient-years (PYs) of exposure, respectively.
Harms Results
At least 1 SAE was reported in 32.2% and 22.1% of patients in the inclisiran and placebo groups, respectively. The most common SAEs were cardiac, reported in 11.6% and 9.0% of patients, respectively. At least 1 AE led to study drug discontinuation in 3.2% and 1.7% of patients in the inclisiran and placebo groups, respectively.
AEs at the injection site were more frequent with inclisiran (9.3%) than with placebo (1.8%). AEs at the injection site leading to study drug discontinuation were higher with inclisiran (0.1 per 100 PYs) than with placebo (0.0 per 100 PYs).
Kaplan-Meier analyses showed that AEs that were serious or that led to discontinuation; hepatic, muscle, and kidney events; incident diabetes; and elevations of creatine kinase or creatinine accrued at a comparable rate between groups for up to 1.5 years, with similar trends continuing for inclisiran beyond this period. Fewer major CV events reported as AEs occurred with inclisiran during this period. Treatment-induced ADAs were uncommon with inclisiran (4.6%), and few of these were persistent (1.4%).
Critical Appraisal
Internal Validity: The findings are derived from pooled data from 7 clinical trials with specific inclusion criteria; thus, patient populations enrolled at different times may have had different clinical characteristics not reflected in the tables of baseline characteristics and may not be fully reflective of a general population. Although EAIRs were calculated, no direct comparison of events between inclisiran and placebo is possible beyond the first 1.5 years, and only a few patients were exposed to inclisiran for more than 4 years, which limits the ability to draw meaningful conclusions.
External Validity: The pooled-data analysis consisted of patients who took part in the pivotal studies, so it is reasonable to expect that the same strengths and limitations related to generalizability apply to this study.
Conclusions
The major areas of focus for this resubmission were to address the lack of formal assessment of clinical outcomes data from the ORION-9, ORION-10, and ORION-11 trials, and the lack of longer-term safety and efficacy data for inclisiran. Given the indication, the 2 populations — patients with HeFH (ORION-9) and patients with nFH with ASCVD (ORION-10 and ORION-11) — should be viewed separately. There was no evidence that inclisiran reduced the risk of MACE in the HeFH population in ORION-9; however, according to the clinical experts, this type of data has not been available from clinical trials of other drugs, given that these events would be less frequent in this population over the typical follow-up period of a clinical trial, and that HeFH is less common than nFH. There was evidence of a reduced risk of MACE with inclisiran treatment in the nFH with ASCVD population when the results of ORION-10 and ORION-11 were pooled; however, this was a posthoc analysis and it is important to note that these trials were not designed to compare these outcomes between treatment groups. The conclusions about inclisiran regarding lipid parameters remain the same: inclisiran elicits a statistically and clinically significant reduction in LDL-C and a similar improvement in other lipid parameters, and this improvement in LDL-C appeared to be maintained throughout the 3 years of additional treatment with inclisiran during the open-label ORION-8 trial. There was no indication of any new safety or tolerability concerns with inclisiran during the long-term extensions, or when the results of various ORION trials were pooled. The ITC submitted by the sponsor did not provide conclusive evidence on the relative efficacy and safety of inclisiran compared to other PCSK9 inhibitors or ezetimibe in the context of HeFH or ASCVD. The ITC is of minimal value when comparing the efficacy of inclisiran with other PCSK9 inhibitors or ezetimibe, because it did not include an evaluation of clinical outcomes.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of inclisiran in the treatment of primary hypercholesterolemia in patients with HeFH or nFH with ASCVD.
Disease Background
The contents of this section have been informed by materials submitted by the sponsor and clinical expert input. The following information has been summarized and validated by the CDA-AMC review team.
In Canada, CVD is the second leading cause of death and accounted for almost 20% of all deaths in 2020.1 CVD encompasses multiple diseases and can be subdivided in different ways, including its atherosclerotic origin. Atherosclerosis is the build-up of cholesterol and fatty deposits (plaque) inside arteries, which can eventually narrow the vessel lumen.36,37 Despite its pathophysiological complexity, the 1 prerequisite for atherosclerotic plaque development is the presence of LDL-C.2 A clinical condition in which an individual has elevated levels of cholesterol, including LDL-C, is referred to as hypercholesterolemia. Hypercholesterolemia can be grouped into 2 forms: nFH and FH (also referred to as acquired or genetic hypercholesterolemia). nFH is characterized by elevated LDL-C levels. Its etiology is likely a complex interplay between several genetic and environmental risk factors, rather than a simple monoallelic disruption of the LDLR gene. Environmental risk factors that increase the risk of nFH include diet, smoking, physical inactivity, and other factors known to be associated with an increased risk of CVD (e.g., diabetes, chronic kidney disease, and hypertension).3-6 In Canada, the 1-year incidence rate for ASCVD ranges between 7.2 and 8.8 per 1,000 person years, and the 5 year prevalence of ASCVD ranges between 6.91% and 8.55% in adults.7-9
Elevated LDL-C is directly associated with the development of atherosclerosis and ASCVD.10 The 3 main subcategories of ASCVD are CAD, cerebrovascular disease, and PAD. To best manage their patients, clinicians stratify individuals based on their risk of a first (primary prevention) or a recurrent (secondary prevention) clinical event using their demographic and clinical pedigrees. Individuals with hypercholesterolemia and a history of an atherosclerotic event are categorized as having established clinical ASCVD (i.e., they are secondary-prevention patients), whereas individuals with hypercholesterolemia at risk of developing ASCVD are primary-prevention patients. A subset of primary-prevention patients at greater risk of ASCVD are referred to as having an ASCVD-RE. Patient with ASCVD-RE are defined as those with type 2 diabetes mellitus, FH, or a 10-year risk of a CV event of at least 20% as assessed by the FRS for CVD or equivalent.11 The proportion of the overall ASCVD population considered to be at high risk is estimated to be approximately 25%.9 In accordance with Canadian guidelines and published literature and validation from Canadian clinicians, patients at high-risk for nFH with ASCVD are defined as patients who meet any of the following criteria: diabetes, recurrent vascular events, PAD, or ACS in the past 12 months; and LDL-C levels greater than 1.8 mmol/L despite MTD statins with or without other LLTs.9,12-17 Throughout this resubmission, the high-risk ASCVD subgroup will refer to patients with any of these criteria.
FH is 1 of the most common genetic disorders and is caused by mutations in the genes encoding LDLR, ApoB, or PCSK9, leading to high plasma levels of LDL-C.10 Depending on the number of mutant alleles, patients can be categorized as having HoFH or HeFH.18 HeFH has an estimated prevalence of approximately 1 in 250 individuals;19 but a more recent worldwide meta-analysis suggests a prevalence of approximately 1 in 311.20,21 The clinical presentation of FH is variable, affected by the number and type of mutations together with other genetic factors. Individuals with 2 mutated LDLR alleles (HoFH or compound heterozygotes) have higher LDL-C levels than those with 1 mutant allele (HeFH).18 Individuals with FH have elevated LDL-C levels from a young age, and the ongoing exposure to elevated LDL-C results in a higher cumulative risk of developing ASCVD.22 Patients with FH may present with physical findings such as tendon xanthomata or xanthelasma.23 Multiple diagnostic criteria for FH exist, but the CCS recommends the proposed criteria developed by FH Canada. A diagnosis of FH should be considered in patients with a baseline LDL-C of at least 5 mmol/L for patients who are at least 40 years of age (or LDL-C ≥ 4.0 mmol/L for patients aged < 18 years; LDL-C ≥ 4.5 mmol/L for patients aged ≥ 18 years and < 40 years). The presence of 1 or more major criteria (DNA mutation, tendon xanthomas, LDL-C ≥ 8.5 mmol/L) establishes a diagnosis of definite FH.38 FH is associated with an increased risk of CV events compared with the general population.24-26 For example, a Canadian prospective observational study of 339 patients with definite or probable HeFH with a mean follow-up of 10.9 years documented a baseline ASCVD history of 12.1% and mean LDL-C level of 5.9 mmol/L.39 The incidence of adverse vascular outcomes were as follows: any CV event was 33.5 per 1,000 PYs; and MI, CABG, percutaneous coronary intervention, angina, and transient ischemic attack and/or ischemic stroke (IS) was 6.5, 6.5, 11.9, 7.6, and 1.1 per 1,000 PYs, respectively.39 Importantly, the probability of having a second CV event 1 year after the first CV event was 52%, increasing to 59% in the 5 years after the first event, indicating that most recurrent events occur within the first year in this population.39 Compared with the general population, all-cause mortality is reported to be 1.4-fold to 1.9-fold higher in patients with FH.40 Last, the impact on HRQoL is generally prolonged and follows the course of recovery. Some patients do not fully recover, so the event may have a prolonged impact on HRQoL, especially if subsequent complications develop; recurrent events have a particularly substantial humanistic burden because they can have a cumulative impact on a patient’s HRQoL, with studies demonstrating worse HRQoL in patients with recurrent events.41-43 Figure 1 is a proposed Canadian definition for HeFH that is being adopted by many cardiologists across Canada, according to the clinical experts consulted by CDA-AMC on this review.
Figure 1: Proposed Canadian Definition of FH
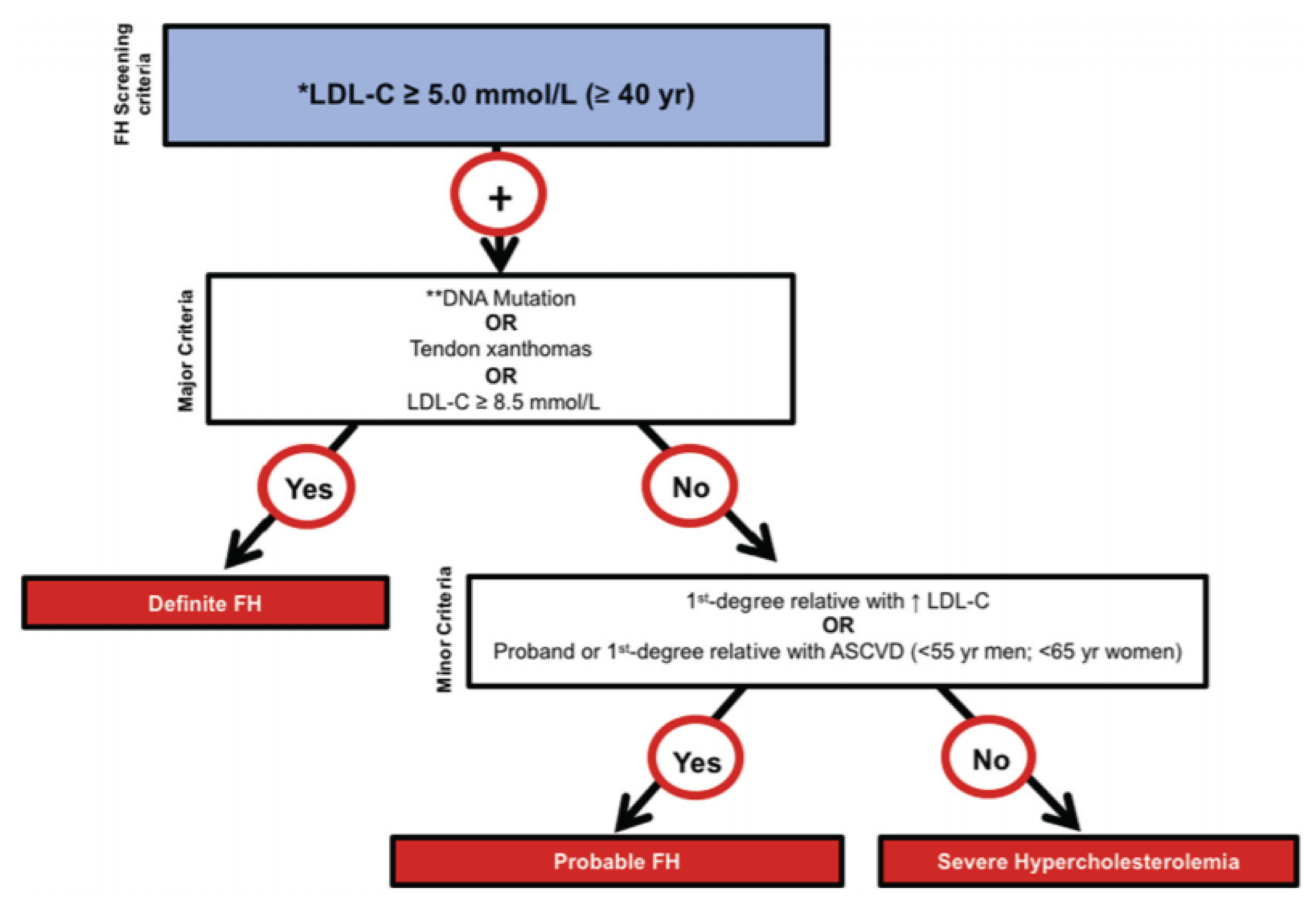
ASCVD = atherosclerotic cardiovascular disease; FH = familial hypercholesterolemia; LDL-C = low-density-lipoprotein cholesterol; yr = year.
*Secondary causes of high LDL-C should be ruled out (severe or untreated hypothyroidism, nephrotic syndrome, hepatic disease [biliary cirrhosis], medication especially antiretroviral drugs).
**Causal DNA mutation refers to the presence of a known FH-causing variant in the LDLR, APOB, or PCSK9 gene, based on the presence of the variant in the ClinVar, HGMD, or WDLV databases, in the proband or a first-degree relative.
Source: Ruel et al. Simplified Canadian definition for familial hypercholesterolemia, Can J Cardiol. 2018 Sep;34(9):1210 to 1214. Copyright 2018 by the authors. Available from: https://onlinecjc.ca/article/S0828-282X(18)30383-0/fulltext. Reprinted in accordance with Creative Commons Attribution 4.0 International Licence (CC BY 4.0): https://creativecommons.org/licenses/by/4.0/50.44
The impact of ASCVD on mortality must also be considered. For example, all-cause mortality was 5.5% in the year after a CV event in an Italian study,45 whereas 11.5% and 13.9% of patients died in the 6 months after a first and second CV event, respectively, in a UK-based study.46 CV-specific in-patient mortality was also high, and in-patient mortality was shown to be 11.4 per 1,000 PYs in a US-based study.47 CV mortality was shown to increase after a subsequent CV event (1 prior CV event: 4.7 per 100 PYs; ≥ 2 prior CV events: 6.7 per 100 PYs) in a Swedish study.48 Furthermore, rates of subsequent CV events are higher in patients with ASCVD. In the US, patients with ASCVD had a 7-fold higher rate of CV events over the 12 months after diagnosis than patients without ASCVD,49 and approximately a third of patients experienced a CV event over 5 years of follow-up after a first event.50 A Swedish population-based study assessed the risk of MACE, including MI, IS, and CV death in individuals with prior ASCVD (defined as having MI, IS, or PAD), identified in national population-based registers.48 Over a mean follow-up of 7.3 years, 44% of patients experienced a CV event. The MACE composite rate (defined as MI, IS, or CV death) was 6.3 per 100 PYs, while the ASCVD composite end point (defined as including MI, IS, unstable angina, coronary revascularization, or CV death) was 7.0 per 100 PYs.48
Standards of Therapy
The contents of this section have been informed by materials submitted by the sponsor and clinical expert input. The following information has been summarized and validated by the CDA-AMC review team.
The LDL-C goal for patients with FH without ASCVD (primary prevention) is a 50% reduction from baseline (i.e., untreated LDL-C) and attainment of an LDL-C of less than 2.5 mmol/L.12,23 For patients with FH and established ASCVD (secondary prevention), the approach recommended is the same as for patients with ASCVD; namely, the attainment of LDL-C levels consistently below 1.8 mmol/L.12
The 2021 CCS Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults recommend that CV risk assessment be conducted every 5 years for men and women aged 40 to 75 years, using the modified FRS or the Cardiovascular Life Expectancy Model to guide therapy and reduce major CV events.12 The CCS guidelines also provide specific guidance to mitigate CVD risk.12 The previous CCS guidelines, published in 2016, recommended maintenance of an LDL-C level of less than 2.0 mmol/L in patients with ASCVD (i.e., secondary prevention),51 but this was revised to a lower threshold of less than 1.8 mmol/L in the updated 2021 guidelines.12 Patients with ASCVD-RE (i.e., those with diabetes mellitus > 40 years) should be treated to maintain an LDL-C of less than 2.0 mmol/L.12
Nonpharmacological interventions for hypercholesterolemia promote a healthy diet and lifestyle, which can have beneficial lipid-lowering effects and CV outcomes.12,52 Guidelines highlight modifiable risk factors of weight control, dietary patterns, physical inactivity, smoking, alcohol consumption, and psychosocial factors (i.e., sleep patterns, mental health) that can be targeted for the primary prevention of CVD, with observed beneficial responses in LDL-C, HDL-C, and triglyceride levels, and reductions in CV events and mortality.12,39,51 Furthermore, for the secondary prevention of CV events, it is recommended that sedentary behaviour be limited, physical activity be encouraged, and previously identified modifiable lifestyle factors be addressed so that they have an additive effect on a reduction of ASCVD events.12
Per CCS guidelines, statins are first-line therapy for individuals with hypercholesterolemia.12 Statins are classified as low-to-moderate intensity (i.e., lowering LDL-C by 30% to < 50%) or high intensity (i.e., lowering LDL-C by > 50%; examples, according to the clinical experts, would include atorvastatin 40 mg to 80 mg daily, or rosuvastatin 20 mg to 40 mg daily).6 They are generally well tolerated, but may be uncommonly associated with myalgia and myopathy that may rarely progress to rhabdomyolysis, abnormalities in liver enzymes, and an increased risk of new-onset diabetes.53 Statin intolerance has been reported to occur in 10% to 20% of patients. Adherence to statin therapy ranges from 25% to 61% over 3 to 5 years in patients with ASCVD.54-59
Ezetimibe is a second-line therapy and is usually combined with statins in patients who do not achieve LDL-C goals with statins alone or administered as monotherapy in patients who are intolerant to statins.12 Ezetimibe is generally well tolerated but may be associated with gastrointestinal disorders, headache, fatigue, and myalgia.60 Importantly, LDL-C levels are only reduced by 10% to 20% from baseline with concomitant statin therapy, which may prove to be insufficient to optimally lower LDL-C in patients with HeFH or ASCVD, especially in patients with ASCVD whose LDL-C remains above 1.8 mmol/L despite MTD statin therapy.12,60,61
Bile acid sequestrants are also a second-line therapy (alternative to ezetimibe) and can be used alone or combined with statins and/or ezetimibe.12,51 Their use is currently limited by a high burden of AEs (particularly gastrointestinal side effects), limited efficacy, and drug-drug interactions.51,62 As such, they have only a very minor role in contemporary lipid management, according to the clinical experts consulted by CDA-AMC on this review.
PCSK9 inhibitors are recommended as an add-on therapy to lower LDL-C in patients whose LDL-C remains above target despite MTD statin therapy.12 Evolocumab and alirocumab have recently been introduced into clinical practice and can lower LDL-C levels by approximately 45% to 60% in patients taking MTD statins with or without ezetimibe.63 In large phase III clinical trials, PCSK9 inhibitors were shown to significantly reduce the risk of CV events when added to recommended LLT (MTD statins with or without ezetimibe).64,65 Safety data from clinical trials of evolocumab and alirocumab suggest that both therapies are well tolerated.64-66 Evolocumab and alirocumab are administered by subcutaneous (SC) self-injection every 2 weeks or monthly by the patient or their caregiver, following appropriate guidance provided by a health care professional on the proper SC injection technique.67,68 Evolocumab and alirocumab (referred to as PCSK9 inhibitor monoclonal antibodies in this resubmission) were recommended for public reimbursement by CDA-AMC as adjunct therapies to diet and MTD statins for the treatment of patients with HeFH or patients with clinical ASCVD who need additional LDL-C reduction.69,70 However, evolocumab and alirocumab are currently only reimbursed for patients with HeFH in most provinces, including Ontario,71,72 and are not reimbursed under typical circumstances for the treatment of ASCVD in any province, due to unsuccessful negotiations with the pan-Canadian Pharmaceutical Alliance.73,74
Despite the availability of statins and other LLTs, most patients with FH, as well as most patients with nFH with ASCVD, fail to achieve guideline-recommended LDL-C levels, thus remaining at high risk for preventable morbidity and mortality attributable to future CV events. Key reasons for failure to sufficiently lower LDL-C include tolerability issues, challenges with medication adherence, and, for ASCVD patients, lack of access to additional LLTs with sufficient LDL-C-lowering efficacy.
The 2021 CCS dyslipidemia guidelines highlight that within the ASCVD population with nFH, there exist subgroups of patients who are deemed to be at high risk for future events (more so than a general group of patients with ASCVD; e.g., patients with stable angina) and who derive the largest absolute benefit from intensification of LLTs with PSCK9 inhibitors.12 These subgroups include recent (defined as patients hospitalized for an index ACS event to 52 weeks postindex ACS), and patients with clinically evident ASCVD and any of the following: diabetes or metabolic syndrome, polyvascular disease (vascular disease in ≥ 2 arterial beds), symptomatic PAD, recurrent MI, MI in past 2 years, previous CABG surgery, LDL-C of at least 2.6 mmol/L or HeFH, and Lp(a) of at least 60 mg/dL (120 nmol/L) in patients whose LDL-C remains above 1.8 mmol/L, despite MTD statins (with or without another LLT).
Drug Under Review
Key characteristics of inclisiran are summarized in Table 3, along with other treatments available for patients with HeFH or patients with nFH with ASCVD.
Inclisiran is administered by SC injection at a dose of 284 mg per 1.5 mL, at baseline, 3 months, and every 6 months thereafter. It is indicated as an adjunct to lifestyle changes, including diet, to further reduce LDL‐C level in adults with the following conditions who are on MTD statins, with or without other LDL‐C‐lowering therapies:
HeFH
nFH with ASCVD.
The sponsor’s reimbursement request is consistent with the indication.
Inclisiran is an siRNA that reduces the expression of PCSK9. PCSK9 is a protein that binds to and inhibits the recycling of LDLR, therefore reducing the expression of PCSK9 and increasing the number of LDLRs, and this facilitates the clearance of LDL-C from the circulation.
Inclisiran was previously reviewed by CADTH in February 2022 for the same indication, and the recommendation was to not reimburse.27 Key reasons for this recommendation included the fact that there was insufficient evidence that inclisiran reduces CV morbidity and mortality, or all-cause mortality, as the pivotal trials — ORION-9, ORION-10, and ORION-11 — were not designed to assess these outcomes.27 Additionally, CDEC noted that the long-term efficacy and safety of inclisiran has not been determined, and that there are 2 ongoing studies — ORION-4 and ORION-8 — that are expected to provide evidence to better characterize the pertinent clinical outcomes, as well as provide long-term efficacy and safety data. CDEC also noted that there was no direct comparison of inclisiran to evolocumab or alirocumab, or to other add-on drugs, and that there were limitations to the submitted ITC, including the relatively short follow-up period (24 weeks) in patients with a chronic condition.
The sponsor outlined the basis for its resubmission. To address the lack of evidence for the reduction of CV morbidity and/or mortality and all-cause mortality, the sponsor included a pooled analysis of the ORION-9, ORION-10, and ORION-11 studies, focusing on the risk of MACE with inclisiran versus placebo across the 3 trials. In the posthoc analysis of this exploratory outcome from these 3 trials, inclisiran reduced the risk of MACE versus placebo. To address the issue of the lack of long-term safety data, the sponsor submitted the ORION-3 trial, as well as a pooled analysis of 7 ORION trials. From the ORION-3 study, the sponsor noted that most of the AEs were mild to moderate, and that 1% of patients experienced an SAE that was possibly related to inclisiran. With respect to the pooled analysis, the trajectory of SAEs or AEs did not change with additional years of inclisiran treatment. Finally, the sponsor submitted a revised budget impact model to address CDA-AMC’s concerns from the first recommendation. It focused on the population of patients with nFH with ASCVD who are at high risk and reported a reduction in budget impact of approximately one-third.
Table 3: Key Characteristics of Treatments for Patients With HeFH and With nFH and ASCVD
Drug | Indications | Route and dose |
|---|---|---|
Inclisiran (Leqvio) | Inclisiran is indicated as an adjunct to lifestyle changes, including diet, to further reduce LDL-C level in adults with HeFH or nFH with ASCVD who are receiving MTD statins ± other LDL-C-lowering therapies. | The recommended dose of inclisiran is 284 mg administered as a single subcutaneous injection initially, again at 3 months, followed by every 6 months thereafter. |
Statins (Using atorvastatin as an example) | Atorvastatin is indicated in adults as an adjunct to lifestyle changes, including diet, for:
| The optimal dose of atorvastatin ranges from 10 mg to 80 mg once daily orally. |
Ezetrol (ezetimibe) | Ezetimibe is indicated as an adjunct to lifestyle changes, including diet, when the response to diet and other nonpharmacological measures has been inadequate for patients with:
| The recommended dose of ezetimibe is 10 mg once daily orally, as monotherapy or combination therapy with a statin or fenofibrate. |
Evolocumab (Repatha) | Evolocumab is indicated as an adjunct to diet and standard-of-care therapy to reduce the risk of MI, stroke, and coronary revascularization in adult patients with ASCVD by further lowering LDL-C levels. Evolocumab is also indicated in patients with primary hyperlipidemia (including HeFH and ASCVD) as an adjunct to diet ± statin therapy ± other LLTs to provide additional lowering of LDL-C. | The recommended dose for evolocumab is either 140 mg every 2 weeks or 420 mg once monthly; both doses are clinically equivalent. |
Alirocumab (Praluent) | Alirocumab is indicated in combination with MTD statins ± other LLTs to reduce the risk of MI, ischemic stroke, and UA requiring hospitalization in adults with established CVD. Alirocumab is indicated for the reduction of LDL-C in adults with primary hyperlipidemia (heterozygous familial and nonfamilial) as an adjunct to diet ± statin therapy ± other LLTs. | The recommended starting dose of alirocumab is 75 mg once every 2 weeks administered subcutaneously. Alternatively, 300 mg once every 4 weeks may be administered for patients who prefer less frequent dosing. If the LDL-C response is inadequate, the dosage may be increased to 150 mg every 2 weeks. |
ApoB = apolipoprotein B; ASCVD = atherosclerotic cardiovascular disease; CVD = cardiovascular disease; ECG = electrocardiogram; HDL-C = high-density-lipoprotein cholesterol; HeFH = heterozygous familial hypercholesterolemia; LDL-C low-density-lipoprotein cholesterol; LLT = lipid-lowering therapy; MI = myocardial infarction; MTD = maximally tolerated dose; nHF = nonfamilial hypercholesterolemia; TG = triglycerides; total-C = total cholesterol; UA = unstable angina.
Sources: Product monograph for inclisiran,75 evolocumab,67 alirocumab,68 atorvastatin calcium,76 atorvastatin calcium.77
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups. The full original patient input(s) received by CDA-AMC have been included in the Stakeholder section of this report.
Two patient groups — the CHPA and the HeartLife Foundation — responded to CDA-AMC’s call for patient input for the current review of inclisiran as an adjunct to lifestyle changes, including diet and MTD statins, with or without other LDL-C-lowering therapies, in adult patients with HeFH or nFH with ASCVD who require additional lowering of LDL-C.
The information provided by CHPA was gathered through a survey questionnaire that was in effect from June 8 until July 10, 2023, targeting patients with or at high risk for ASCVD, followed by individual interviews with 10 individuals who had a diagnosis of ASCVD or had experienced a heart attack or stroke and had received inclisiran. The HeartLife Foundation submission was completed by its patient partner executives; it includes data from literature review, 1-on-1 interviews with patients living with the disease, and study review.
Among the 85 CHPA survey respondents, 58% reported having a diagnosis of ASCVD or having experienced a heart attack or stroke, 27% of respondents had symptoms of or were at high risk for ASCVD, 67% reported having a diagnosis of FH, and 25% reported having other lipid disorders.
The patient groups emphasized that living with ASCVD and high levels of LDL-C is very hard to manage, affects the physical and mental well-being, has a significant financial burden on families, and impacts their quality of life. Symptoms like shortness of breath, chest pain, and fatigue were stated by the respondents, who indicated the negative impact of a heart attack, bypass surgery, or stroke on themselves and their families. Many with a family history of heart disease and/or high cholesterol commented on their fear of following a family pattern of early death.
According to the 2 patient inputs, managing hypercholesterolemia requires a low-fat diet and medications including but not limited to statins, ezetimibe, and PCSK9 inhibitors. Although 100% of respondents from the CHPA input indicated that they ate a low-fat diet, more than half of them indicated this was not entirely effective. Among the 92% of respondents who used statins, 81% found them to be effective or somewhat effective to achieve target LDL-C levels; however, 64% experienced moderate-to-severe side effects. Of the 87% who used ezetimibe, only 40% indicated that it was effective, but it has fewer side effects than statins. More than half (57%) of respondents had been prescribed a PCSK9 inhibitor, with about 92% of those on the drug reporting that it worked well or very well to manage their cholesterol levels, and 14% said there were moderate side effects only. The HeartLife Foundation submission added that patient adherence to currently available and publicly reimbursed therapeutic options is acknowledged to be poor, and access to efficacious treatments like PCSK9 inhibitors is very limited.
The patient groups stated that patients seek a safe, tolerable, and effective treatment that can minimize the long-term health consequences of ASCVD in high-risk patients by effectively managing LDL-C levels below the recommended threshold. This, in turn, can reduce the occurrence of MACE, including myocardial (re)infarction, IS, the need for coronary revascularization, and CV death, and ultimately enhance the quality of life for individuals living with ASCVD. Patients also want an accessible therapy with a more affordable and manageable treatment regimen, less frequent dosing, fewer side effects, easier administration, and less disruption to work or daily life.
All the patient partners interviewed for the HeartLife input and patients interviewed for the CHPA input had received inclisiran, and almost all of them stated that the drug was highly effective and improved their overall well-being and quality of life.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 3 clinical specialists with expertise in the diagnosis and management of hypercholesterolemia.
Unmet Needs
The clinical experts agreed that the major issues that affect the treatment of patients with HeFH or with nFH with ASCVD are nonadherence, intolerance to high-intensity statins, inability to reach recommended lipid targets despite MTD statins and ezetimibe, and lack of access to PCSK9 inhibitors. Related to that, a large percentage of patients (much greater than 50%) with HeFH are not able to reach their target LDL-C of less than 2.5 mmol/L, and with nFH with ASCVD are not able to reach their target of less than 1.8 mmol/L. The clinical experts noted that in patients with HeFH, the addition of a PCSK9 inhibitor to statin therapy will help most patients achieve their target; however, only a small number of these patients will receive these drugs.
The clinical experts noted that the adherence issues are compounded by the side effects of the drugs used, and the fact that patients may be reluctant to take a medication on a chronic basis to prevent future events or symptoms. The clinical experts appeared to agree that statins are the major cause of side effects, and they noted that there is evidence suggesting that combining better-tolerated drugs like ezetimibe and PCSK9 inhibitors with statins may be 1 strategy for achieving targets. The clinical experts also noted that ezetimibe is not potent enough to achieve target LDL-C as monotherapy, and that access to the PCSK9 monoclonal antibodies is limited.
Place in Therapy
The clinical experts consider inclisiran as another option among the PCSK9-targeting drugs, equivalent to the monoclonal antibodies that target PCSK9, evolocumab, and alirocumab, which are currently used as adjunctive therapies combined with statins (with or without ezetimibe) in patients unable to reach their target LDL-C. The clinical experts all emphasized the importance of having another option for add-on therapy to statins, and that the infrequent dosing of inclisiran may be an advantage with respect to adherence. The clinical experts were also clear that inclisiran is unlikely to become a first-line therapy, ahead of statins, because of the large body of evidence supporting the use of statins. The clinical experts also noted that, per the indication, patients would be required to have reached the MTD of a statin (which may include 0 mg) before moving to other options like inclisiran. Statin intolerance should be documented, as recommended by CCS guidelines. None of the clinical experts believed that inclisiran will shift the treatment paradigm.
Patient Population
With respect to patients with HeFH, the clinical experts believed that in addition to those unable to reach their LDL-C target despite MTD statins, with or without ezetimibe, patients who would be especially well suited to inclisiran would include those with other risk factors, such as smoking, diabetes, hypertension, or elevated Lp(a).
The clinical experts believed that patients with nFH with ASCVD who meet the indication (LDL-C above threshold despite MTD statins, plus or minus ezetimibe) are particularly well suited to inclisiran and could include those unable to tolerate high-intensity statins, those with early disease onset or recurrent disease, those whose LDL-C is far from threshold, and those with the risk factors.
The clinical experts believed that the patients enrolled in the pivotal ORION trials are consistent with the target population for inclisiran.
Suitable patients would be identified based on laboratory tests (lipid panel and ApoB), according to the clinical experts. The clinical experts noted that although genetic testing could be used to confirm FH, this is not required, nor should it be, to gain access to inclisiran, given the limited availability of such tests in various regions of the country.
The clinical experts also noted that HeFH is underdiagnosed in Canada, with 1 clinical expert estimating that only 10% to 15% of patients with HeFH have been diagnosed.
Assessing the Response to Treatment
The clinical experts agreed that a reduction in lipid metrics, most notably LDL-C, but also non-HDL-C and ApoB, is used to assess response to treatment in the clinic. All these outcomes were assessed in the pivotal ORION trials. The clinical experts noted that clinical outcomes such as CV morbidity and mortality were of paramount importance overall; however, these are clearly not used to monitor treatment response in the clinic. One clinical expert drew attention to the fact that despite guidelines emphasizing that lipids need to be monitored regularly, there is no specific guidance as to how frequently lipid response should be monitored. This clinical expert noted that using 2003 guidance, which is still likely relevant today, the levels of lipids and lipoprotein are expected to reach steady state within 6 weeks of initiating drug therapy, and long-term follow-up after the initial titration phase can be performed every 6 to 12 months. The clinical experts emphasized that reducing LDL-C to below target and keeping it there should be the focus, rather than aiming for an arbitrary percent reduction (such as a 40% reduction, for example).
Discontinuing Treatment
The clinical experts agreed that medication intolerance, loss of coverage, and treatment that no longer aligns with a patient’s goals of care are reasons to discontinue inclisiran. Of course, according to the clinical experts, failure to meet an arbitrary percent reduction in LDL-C while on MTD statins, plus or minus ezetimibe, would also be a reason for stopping or switching treatment.
Prescribing Considerations
Although HeFH is more likely to be diagnosed by a specialist, none of the clinical experts believed that diagnosis requires a specialist, and for patients with nFH with ASCVD, a diagnosis can be made by a specialist or primary care physician. One of the clinical experts was very clear that requiring specialist intervention in diagnosis, management, or monitoring would be unnecessary and counterproductive, and could result in inequities in access to inclisiran, given the inequities in access to specialist care across the country.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by clinician groups. The full original clinician group input(s) received by CDA-AMC have been included in the Stakeholder section of this report.
Fourteen clinician groups — Alberta Cardiovascular Disease Prevention Collaborative (8 clinicians contributed to the input); BC Lipid specialists (11 clinicians contributed to the input); le Centre hospitalier universitaire Dr.-Georges-L.-Dumont (6 clinicians contributed to the input); Cambridge Cardiac Rehab Program (6 clinicians contributed to the input); CCS Dyslipidemia Guideline Committee (14 clinicians contributed to the input); Cardiology Association of Niagara (3 clinicians contributed to the input); Egyptian Cardiologists of Niagara (3 clinicians contributed to the input); Kawartha Cardiology Clinic (7 clinicians contributed to the input); Lipid Clinic of McMaster University and Hamilton Health Sciences (1 clinician contributed to the input); Mazankowski Alberta Heart Institute (3 clinicians contributed to the input); Oakville Cardiologists (9 clinicians contributed to the input); Service of cardiology, Internal Medicine Department and Heart Failure Group at St. Thomas Elgin General Hospital (STEGH; 5 clinicians contributed to the input); Western University, Division of Cardiology, Cardiac Rehabilitation and Secondary Prevention Program (3 clinicians contributed to the input); and University of Toronto faculty and clinicians at St. Michael’s Hospital who are actively involved in the treatment of patients with ASCVD and/or lipid disorders (10 clinicians contributed to the input) — responded to CDA-AMC’s call for clinician group input. Information for this input was gathered through a review of the relevant literature and publications, as well as experience and knowledge in this field.
According to the clinician groups, in the Canadian context, the current treatment paradigm for dyslipidemia or high cholesterol levels in patients with ASCVD and FH involves drug therapies, such as statins (first-line therapy), ezetimibe, and PCSK9 monoclonal antibodies, in addition to dietary modifications, consisting of reducing saturated fat intake, increasing fibre intake, and adopting a generally healthy and balanced diet. Other medications, such as fibrates, are not indicated for LDL-C lowering, and bile acid resins are seldom used.
The clinician group input stated that the CCS dyslipidemia guidelines recommend lifestyle modifications to improve lipid parameters and an LDL-C threshold of 1.8 mmol/L or less. However, a high proportion of patients, including those in the high-risk population with dyslipidemia, cannot achieve the recommended lipid threshold because of intolerance to currently available treatments (particularly statins), lack of compliance to treatment, variable response to currently available treatments, and lack of accessibility (i.e., cost) to highly effective PCSK9 inhibitors. Suboptimal therapy increases the risk of MACE. As such, the overarching treatment goals are to reduce the risk of MACE and CV mortality by optimizing the reduction in lipid profiles with efficacious, accessible therapies with fewer side effects and therapies with longer dosing intervals.
The clinician groups agreed that inclisiran can improve lipid profiles by inhibiting PCSK9 production, resulting in the increased expression of hepatic LDL receptors. The difference from other drugs that target the PCSK9 pathway is in the molecular mechanism (siRNA) that leads to the degradation of the messenger RNA encoding PCSK9, specifically in the cytoplasm of hepatocytes. It has the advantage of being administered by SC injection every 6 months rather than every 2 weeks. The drug under review would be used as an add-on therapy to MTD statins with or without additional LLTs in patients who require additional lipid-lowering, per the CCS dyslipidemia guidelines. Patients with or at risk of ASCVD or FH who require additional LLT and who become refractory to statins and ezetimibe, along with those who struggle with compliance, would be best suited for the drug under review. High-risk patients who cannot tolerate high-dose statin therapy due to side effects would also benefit from inclisiran. Furthermore, patients with HeFH who are at risk for ASCVD would benefit from the drug under review, according to BC Lipid specialists; le Centre hospitalier universitaire Dr.-Georges-L.-Dumont; Cambridge Cardiac Rehab Program; CCS; Oakville Cardiologists; and Western University, Division of Cardiology, Cardiac Rehabilitation and Secondary Prevention Program.
According to the clinician groups, the percentage of reduction in lipid parameters (LDL-C, non-HDL-C, and ApoB measurements) 6 months after treatment, and yearly thereafter, are the main outcomes of interest that can be assessed to determine whether a patient is responding to treatment. A clinically meaningful response to treatment would be indicated by at least a 30% reduction in LDL-C or non-HDL-C levels. The clinician groups stated that discontinuation of therapy should be considered in patients with significant intolerance, severe side effects, or a lack of response.
The clinician groups stated that inclisiran should be able to be appropriately used by both primary care and specialist physicians in specialty clinics, community settings, community outpatient clinics, and hospitals.
Drug Program Input
The drug programs provide input on each drug being reviewed through CDA-AMC’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
ORION-9, ORION-10, and ORION-11 were all placebo-controlled trials. There are no head-to-head trials comparing inclisiran to other therapies. | This was a comment from the drug programs to inform CDEC deliberations. |
The PCSK9 monoclonal antibody therapies are only reimbursed for HeFH by the public drug plans. Reimbursement for indications beyond HeFH (i.e., ASCVD) will be associated with a large budget impact due to the expected population size that would be eligible for treatment. | This was a comment from the drug programs to inform CDEC deliberations. |
Considerations for initiation of therapy | |
Is genetic testing required to make the diagnosis of HeFH, or is this determined only clinically? | The clinical experts agreed that genetic testing is not required to make the diagnosis of HeFH. It is also not available across Canada and making it a requirement would result in inequities in access. |
How is a diagnosis of HeFH confirmed? In some jurisdictions, a definite or probable diagnosis of HeFH, using Simon Broome or Dutch Lipid Clinic Network criteria or genetic testing, is used. Should a diagnosis of HeFH be confirmed in a similar manner before initiating inclisiran? | The clinical experts identified new criteria that have emerged in the past 3 years, based on work performed by Canadian researchers, which indicate that a diagnosis of FH should be considered in patients with a baseline LDL-C of 5 mmol/L or greater and who are at least 40 years of age (or LDL-C ≥ 4.0 mmol/L for age < 18 years, or LDL-C ≥ 4.5 mmol/L for age ≥ 18 years and < 40 years). The presence of 1 or more major criteria (DNA mutation, tendon xanthomas, LDL-C ≥ 8.5 mmol/L) establishes a diagnosis of definite FH. Genetic testing is not necessary for diagnosis, and approximately 30% of patients with a definitive diagnosis of HeFH do not display a monogenic variant.44 This is now accepted by Canadian guidelines committees.23 |
How is diagnosis of nFH with ASCVD confirmed? | The clinical experts noted that the threshold for LDL-C (currently 1.8 mmol/L on MTD statins) is likely going to continue going lower, as evidence continues to emerge that the lower the LDL-C the better in this population. |
How do you define patients with high-risk ASCVD? Are such patients expected to benefit the most from treatment with inclisiran? | The clinical experts referred to the Canadian guidelines12,51 to clearly identify patients with high-risk ASCVD as those who have been hospitalized for an index ACS in the past 52 weeks, and those with clinically evident ASCVD and any of the following:
The clinical experts emphasized the difficulties in requiring criteria that are time-sensitive, so, for example, in a fractured health care system, it may be very difficult for physicians to determine when an MI actually occurred. Similarly, according to the clinical experts, other requirements (such as determining PAD or polyvascular disease), create extra burden for the clinician, as these are time-consuming to assess. They also emphasized that it is often these high-risk patients who have the most limited access to the health care system and are thus in the worst position to obtain the assessments that allow them to access the drugs they need. Ultimately, according to the clinical experts, this creates more inequity in the health care system. |
Inclisiran is to be used as an adjunct to MTD statins
| The clinical experts agreed that inclisiran could be used as monotherapy if a patient is intolerant to statins. The clinical experts noted that the length of the trial of a statin before moving to inclisiran is somewhat arbitrary; however, 3 months would be a reasonable estimate. With respect to the question about whether statin intensity matters, the clinical experts noted that new data suggest that the better approach may be to use a moderate-intensity statin along with ezetimibe, rather than pushing for MTD statins. The clinical experts believed that the trial of a high-intensity statin may be worthwhile, but they noted that if a patient is far from their target, doubling the statin dose and adding ezetimibe is unlikely to be sufficient to achieve their target LDL-C. Yes, the clinical experts believed that patients trialling 2 different statins would be reasonable, and this is widely accepted. For MTD, the clinical experts believed that 1 needs to rely on patient testimony when it comes to intolerance, and adherence is essentially defined as whether the patient is taking the drug or not. |
Should ezetimibe or other nonstatin LLTs be used before starting inclisiran? | Reflecting the 2021 Canadian Cardiovascular Society guidelines, if LDL-C is little above threshold (1.8 mmol/L to 2.2 mmol/L), then, yes, adding ezetimibe makes sense, according to the clinical experts; however, if LDL-C is more than 2.2 mmol/L, then the ezetimibe step is a waste of time. The clinical experts believed that other LLTs, like bile acid binding resins, are not a viable option due to their tolerability issues. |
Many jurisdictions require a trial of ezetimibe before reimbursing PCSK9 inhibitors for the treatment of HeFH, including:
Should such criteria be applied before initiating treatment with inclisiran? | The clinical experts believed that these criteria should not be required before a patient is eligible for inclisiran. |
Is there a discrepancy between the Canadian guidelines for FH with established ASCVD (LDL-C target of < 2.6 mmol/L in the ORION-9 and ORION−11 trials and < 1.8 mmol/L in the ORION-10 trial)? | The clinical experts believed that the ORION-9, ORION-10, and ORION-11 trials are in line with the guidelines. |
Should the initiation criteria for inclisiran be aligned with the criteria for alirocumab and evolocumab in patients with HeFH? | The clinical experts agreed that the initiation criteria for inclisiran should be aligned with those for alirocumab and evolocumab in patients with HeFH. |
Considerations for continuation or renewal of therapy | |
Should the renewal criteria for inclisiran be aligned with the criteria for alirocumab and evolocumab in patients with HeFH? Note: Although CDA-AMC recommendations for alirocumab and evolocumab do not include renewal criteria, some of the jurisdictions do have renewal criteria. | The clinical experts noted that the current requirement is a 40% reduction in LDL-C after a 4-month trial; however, inclisiran is given every 6 months, so these do not align. The clinical experts were of the opinion that although a timeline of 12 months for renewal would make more sense than 4 months, the requirement for renewal creates an unnecessary administrative burden at multiple levels. The clinical experts also noted that the 40% threshold is not based on evidence; the more important target is for the patient to be reaching their targets for LDL-C. |
Considerations for discontinuation of therapy | |
If a patient using inclisiran for primary prevention experiences a heart attack or stroke, should that patient continue using inclisiran for secondary prevention? | This question is referring to the HeFH population. The clinical experts were clear that these patients should continue the drug, and likely need more aggressive intervention. |
Should inclisiran be initiated in patients with hypercholesterolemia who have had a prior heart attack or stroke (i.e., for secondary prevention)? | The clinical experts agreed that inclisiran should be initiated in these patients if they are not at their LDL-C target despite MTD statins plus or minus ezetimibe. |
Should the discontinuation criteria for inclisiran be aligned with the criteria for alirocumab and evolocumab in patients with HeFH? Note: Although CDA-AMC recommendations for alirocumab and evolocumab do not include discontinuation criteria, some of the jurisdictions do have discontinuation criteria. | The clinical experts stated that patients who do not respond to inclisiran should have the drug discontinued; however, a nonresponse to PCSK9 inhibitors is rare. In most cases, a nonresponse is usually due to an administration error. Otherwise, the clinical experts agreed that discontinuation should be considered for patients who are intolerant to inclisiran, or patients who have a competing illness that makes the use of inclisiran no longer necessary. |
ACS = acute coronary syndrome; ASCVD = atherosclerotic cardiovascular disease; CABG = coronary artery bypass graft; CDEC = Canadian Drug Expert Committee; FH = familial hypercholesterolemia; HeFH = heterozygous familial hypercholesterolemia; LDL-C = low-density-lipoprotein cholesterol; LLT = lipid-lowering therapy; Lp(a) = lipoprotein (a); MI = myocardial infarction; MTD = maximally tolerated dose; PAD = peripheral artery disease.
Clinical Evidence
The objective of CDA-AMC’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of inclisiran 284 mg in 1.5 mL for SC injection in the treatment of HeFH and nFH with ASCVD in adults. The focus will be placed on comparing inclisiran to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of inclisiran is presented in 4 sections, with CDA-AMC’s critical appraisal of the evidence included at the end of each section. The first section, the Systematic Review, includes pivotal studies and RCTs that were selected in accordance with the sponsor’s systematic review protocol. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the CDA-AMC review and appraised in this document:
Three pivotal studies or RCTs identified in the systematic review, as well as a posthoc pooled analysis
Two long-term extension studies
One ITC
One additional study addressing gaps in evidence.
Systematic Review
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | ORION-9 | ORION-10 | ORION-11 |
|---|---|---|---|
Designs and populations | |||
Study design | Phase III, double-blind, placebo-controlled RCT | Phase III, double-blind, placebo-controlled RCT | Phase III, double-blind, placebo-controlled RCT |
Locations | 47 sites in 8 countries: Canada (3), US (12), Czech Republic (3), Denmark (6), Netherlands (5), South Africa (9), Spain (6), and Sweden (3) | 146 sites in the US | 72 sites in 8 countries: Czech Republic (2), Germany (5), Hungary (4), Netherlands (1), Poland (19), South Africa (8), Ukraine (9), UK (24) |
Patient enrolment dates | Start date: December 12, 2017 End date: August 27, 2019 Study end date: September 17, 2019 | Start date: December 21, 2017 End date: September 9, 2019 Study end date: September 17, 2019 | Start date: November 1, 2017 End date: July 31, 2019 Study end date: August 27, 2019 |
Randomized (N) | Total N = 482 N (inclisiran) = 242 N (placebo) = 240 | Total N = 1,561 N (inclisiran) = 781 N (placebo) = 780 | Total N = 1,617 N (inclisiran) = 810 N (placebo) = 807 |
Inclusion criteria |
|
|
|
Exclusion criteria |
|
|
|
Drugs | |||
Intervention | Inclisiran sodium 300 mg (equivalent to 284 mg of inclisiran) administered as a SC injection on day 1, day 90, day 270, and day 450 | Inclisiran sodium 300 mg (equivalent to 284 mg of inclisiran) administered as a SC injection on day 1, day 90, day 270, and day 450 | Inclisiran sodium 300 mg (equivalent to 284 mg of inclisiran) administered as a SC injection on day 1, day 90, day 270, and day 450 |
Comparator(s) | Placebo saline injections on day 1, day 90, day 270, and day 450 | Placebo saline injections on day 1, day 90, day 270, and day 450 | Placebo saline injections on day 1, day 90, day 270, and day 450 |
Study duration | |||
Screening phase | 14 days (day –14 to day –1) | 14 days (day –14 to day –1) | 14 days (day –14 to day –1) |
Run-in phase | NA | NA | NA |
Treatment phase | 450 days (day 1 to day 450) | 450 days (day 1 to day 450) | 450 days (day 1 to day 450) |
Follow-up phase | 90 days (day 450 to day 540) | 90 days (day 450 to day 540) | 90 days (day 450 to day 540) |
Outcomes | |||
Primary end point |
|
|
|
Secondary and exploratory end points | Key secondary efficacy end points:
Other secondary end points:
Exploratory end points:
| Key secondary efficacy end points:
Other secondary end points:
Exploratory end points:
| Key secondary efficacy end points:
Other secondary end points:
Exploratory end points:
|
Publication status | |||
Publications | Publication: Raal et al. (2020).79 ClinicalTrials.gov entry: ClinicalTrials.gov [internet]. Bethesda (Maryland): National Library of Medicine (US). October 28, 2020. Identifier: NCT03397121, Trial to Evaluate the Effect of Inclisiran Treatment on Low-Density Lipoprotein Cholesterol (LDL-C) in Subjects With Heterozygous Familial Hypercholesterolemia (HeFH) (ORION-9); October 28, 2020 [cited January 5, 2023]. Available from: https://clinicaltrials.gov/ct2/show/study/NCT03397121 | Publication: Ray et al. (2020).11 ClinicalTrials.gov entry: ClinicalTrials.gov [internet]. Bethesda (Maryland): National Library of Medicine (US). October 5, 2020. Identifier: NCT03399370. Inclisiran for Participants With Atherosclerotic Cardiovascular Disease and Elevated Low-density Lipoprotein Cholesterol (ORION-10); October 5, 2020 [cited January 5, 2023]. Available from: https://clinicaltrials.gov/ct2/show/NCT03399370 | Publication: Ray et al. (2020).11 ClinicalTrials.gov entry: ClinicalTrials.gov [internet]. Bethesda (Maryland): National Library of Medicine (US). August 21, 2020. Identifier: NCT03400800, inclisiran for Subjects With ASCVD or ASCVD-Risk Equivalents and Elevated Low-density Lipoprotein Cholesterol (ORION-11); August 21, 2020 [cited January 5, 2023]. Available from: https://clinicaltrials.gov/ct2/show/record/NCT03400800 |
ApoB = apolipoprotein B; ASCVD = atherosclerotic cardiovascular disease; ASCVD-RE = atherosclerotic cardiovascular disease risk equivalent; CeVD = cerebrovascular disease; CHD = coronary heart disease; CV = cardiovascular; FH = familial hypercholesterolemia; FRS = Framingham Risk Score; HDL-C = high-density-lipoprotein cholesterol; HeFH = heterozygous familial hypercholesterolemia; LDL-C = low-density-lipoprotein cholesterol; LVEF = left ventricular ejection fraction; MACE = major adverse cardiovascular events; MI = myocardial infarction; NA = not applicable; NYHA = New York Heart Association; PAD = peripheral arterial disease; RCT = randomized controlled trial; SC = subcutaneous; TC = total cholesterol.
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report.30
A total of 3 studies were identified from the literature for inclusion in this systematic review: ORION-9, ORION-10, and ORION-11. A number of other studies were identified that are ongoing, with no data available yet, and therefore will not be reported in this review (VICTORION-INITIATE, ORION-18, ORION-4, VICTORION-2P, VICTORION-INCEPTION). As ORION-8 is a long-term extension study, it will be described in detail in Long-Term Extension Studies section below.
The ORION-9 Trial28
The primary objectives of the ORION-9 study were to evaluate the effect of inclisiran treatment on LDL-C levels at day 510 and time-adjusted percentage change in LDL-C levels from baseline to the period from after day 90 and up to day 540. Secondary objectives of the ORION-9 study were to evaluate the effect of inclisiran treatment on PCSK9, total cholesterol, ApoB, and non-HDL-C at day 510; LDL-C and PCSK9 levels over time to day 540; the maximum reduction in LDL-C levels; LDL-C and PCSK9 levels over time in individual patients; other lipids, lipoproteins, and apolipoproteins; and the proportion of patients achieving prespecified LDL-C targets; and to evaluate the safety and tolerability profile of inclisiran. Exploratory objectives were to evaluate the effect of inclisiran on CV effects such as CV death, resuscitated cardiac arrest, nonfatal MI, and nonfatal stroke; the proportion of patients in each group with any LDL-C reduction from baseline at any visit (i.e., responders); and the response of LDL-C reduction related to underlying causal mutations of HeFH. ORION-9 was an international, multicenter, phase III, placebo-controlled, double-blind, randomized study. Eligible patients were adults older than 18 years with a diagnosed history of HeFH and elevated LDL-C levels who were either on MTD statins or who had documented evidence of intolerance to at least 2 different statins. The study enrolled 482 patients, was conducted at 47 centres, and screened patients in 8 countries: Canada (3 centres), Czech Republic (3), Denmark (6), Netherlands (5), South Africa (9), Spain (6), Sweden (3), and the US (12). A total of 23 patients were enrolled from Canadian sites: 12 patients randomized to the inclisiran arm and 11 patients randomized to the placebo arm. Patients were randomized by an automated Interactive Response Technology (IRT) only after patient eligibility was confirmed. Treatment allocation was stratified by country and by current use of statins or other LLTs in block sizes of 4. All groups were studied concurrently. Patients were randomized (1:1) to receive either inclisiran sodium 300 mg (equivalent to inclisiran 284 mg) or placebo on top of MTD statins. The study was 18 months in duration, with patients receiving 4 300 mg doses of inclisiran sodium (on day 1, day 90, day 270, and day 450). The screening period occurred between day –14 and day –1 before randomization, and involved confirming eligibility and collecting baseline assessments, such as a physical examination, vital signs, 12-lead electrocardiogram (ECG), fasting lipid profile, limited serum chemistry, hematology, and coagulation, and collecting a pharmacogenetic sample. There was no run-in period in the study. The clinical cut-off date for receiving treatment for the final analysis was August 27, 2019.
The ORION-10 Trial29
The primary objectives of the ORION-10 study were to evaluate the effect of inclisiran treatment on LDL-C levels at day 510 and time-adjusted percentage change in LDL-C levels from baseline to the period from after day 90 and up to day 540. Secondary objectives of the ORION-10 study were to evaluate the effect of inclisiran on PCSK9, total cholesterol, ApoB, and non-HDL-C at day 510; LDL-C and PCSK9 levels over time to day 540; the maximum reduction in LDL-C levels; LDL-C and PCSK9 levels over time in individual patients; other lipids, lipoproteins, and apolipoproteins; and the proportion of patients achieving prespecified LDL-C targets; and to evaluate the safety and tolerability profile of inclisiran. The exploratory objective was to evaluate the effect of inclisiran on CV events such as CV death, resuscitated cardiac arrest, nonfatal MI, and nonfatal stroke (ischemic and hemorrhagic). The ORION-10 trial was a US-based, multicenter, phase III, placebo-controlled, double-blind, randomized study with 1,561 patients. Eligible patients were adults older than 18 years with a diagnosed history of ASCVD (CHD, cerebrovascular disease, or PAD) and elevated LDL-C levels who were either on MTD statins or who had documented evidence of intolerance to at least 2 different statins. The study was conducted in 146 centres in the US. Patients were randomized by an automated IRT only after patient eligibility was confirmed. Treatment allocation was stratified by the current use of statins or other LLTs in block sizes of 4. Patients were randomized (1:1) to receive either inclisiran sodium 300 mg or placebo on top of MTD statins. The study was 18 months in duration, with patients receiving 4 300 mg doses of inclisiran sodium or placebo (on day 1, day 90, day 270, and day 450). The screening period occurred between day –14 and day –1 before randomization and involved confirming eligibility and collecting baseline assessments, such as a physical examination, vital signs, 12-lead ECG, fasting lipid profile, limited serum chemistry, hematology, and coagulation. There was no run-in period in the study. The clinical cut-off date for receiving treatment for the final analysis was September 10, 2019.
The ORION-11 Trial78
The primary objectives of the ORION-11 study were to evaluate the effect of inclisiran treatment on LDL-C levels at day 510 and time-adjusted percentage change in LDL-C levels from baseline to the period from after day 90 and up to day 540. Secondary objectives of the ORION-11 study were to evaluate the effect of inclisiran on PCSK9, total cholesterol, ApoB, and non-HDL-C at day 510; LDL-C and PCSK9 levels over time to day 540; the maximum reduction in LDL-C levels; LDL-C and PCSK9 levels over time in individual patients; other lipids, lipoproteins, and apolipoproteins; and the proportion of patients achieving prespecified LDL-C targets; and to evaluate the safety and tolerability profile of inclisiran. The exploratory objective was to evaluate the effect of inclisiran on CV events such as CV death, resuscitated cardiac arrest, nonfatal MI, and nonfatal stroke (ischemic and hemorrhagic). ORION-11 was an international (non-US), multicenter, phase III, placebo-controlled, double-blind, randomized study with 1,617 patients. Eligible patients were adults older than 18 years with a diagnosed history of ASCVD (CHD, CVD, or PAD) or ASCVD-RE (type 2 diabetes, FH, or a 10-year risk of 20% or greater of having a CV event assessed by FRS or equivalent [target LDL-C of < 100 mg/dL]) and elevated LDL-C levels (≥ 1.8 mmol/L [≥ 70 mg/dL] for patients with ASCVD and ≥ 2.6 mmol/L [≥ 100 mg/dL] for patients with ASCVD-RE) who were either on MTD statins with or without other stable (≥ 30 days with no planned change) LLT or who had documented evidence of intolerance to at least 2 different statins. The study was conducted in 72 centres in 8 countries in Europe and South Africa: Czech Republic (2 centres), Germany (5), Hungary (4), Netherlands (1), Poland (19), South Africa (8), Ukraine (9), and the UK (24). Patients were randomized by an automated IRT only after patient eligibility was confirmed. Treatment allocation was stratified by country and by current use of statins or other LLTs in block sizes of 4. Patients were randomized (1:1) to receive either inclisiran sodium 300 mg or placebo on top of MTD statins. The study was 18 months in duration, with patients receiving 4 300 mg doses of inclisiran sodium or placebo (on day 1, day 90, day 270, and day 450). The screening period occurred between day –14 and day –1 before randomization and involved confirming eligibility and collecting baseline assessments, such as a physical examination, vital signs, 12-lead ECG, fasting lipid profile, limited serum chemistry, hematology, and coagulation. There was no run-in period in the study. The clinical cut-off date for receiving treatment for the final analysis was July 31, 2019.
The ORION-9, ORION-10, and ORION-11 trials shared an identical study design; the diagram is presented in Figure 2.
Figure 2: Diagram of Study Design for the ORION-9, ORION-10, and ORION-11 Trials
FUP = follow-up; V = visit.
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report.30
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria for the ORION-9, ORION-10, and ORION-11 trials were largely identical, apart from the studied population of interest. The ORION-9 trial was conducted in patients with HeFH, the ORION-10 trial was conducted in patients with ASCVD (CHD, cerebrovascular disease, or PAD), and the ORION-11 trial was conducted in patients with ASCVD and ASCVD-RE.28-30 Notably, the LDL-C cut-off values were different among the studies. For patients with clinical ASCVD, the LDL-C cut-off was 1.8 mmol/L or greater; for those with ASCVD-RE, the cut-off LDL-C was 2.6 mmol/L or greater.28-30
Interventions
The ORION-9, ORION-10, and ORION-11 trials employed the same interventions for the active and placebo arms.28-30 In each trial, patients were randomized (1:1) to receive either inclisiran sodium 300 mg or placebo via SC injection on top of MTD statins. Patients received 4 300 mg doses of inclisiran sodium or placebo (on day 1, day 90, day 270, and day 450). All study drugs were administered under aseptic conditions by qualified clinical study site staff and under the supervision of the investigator or a designee. The site of injection was the abdomen, with alternating sites for each injection. It is not clear what the plan was for patients who missed doses.
In each trial, the study drugs were blinded before distribution to each site. Each study drug vial (or prefilled syringe in the ORION-10 trial) contained either inclisiran or placebo and had a yellow shroud to blind the vial. The pharmacist or qualified designee prepared the blinded study drug under aseptic conditions to be administered to the patient on that day. Randomization via an automated IRT was used to assign patients to a blinded study drug. The clinical study site pharmacist maintained the double-blind, according to site-specific procedures and the pharmacy manual. Because inclisiran may be visually distinguishable from placebo, blinded syringes were provided to all study sites and used to maintain the blind.
All patients had the right to withdraw at any point during treatment without prejudice. The investigator could discontinue any patient at any time if medically necessary. Reasons for study discontinuation were considered to constitute 1 of the following:
AE
death
withdrawal of patient consent
physician decision
loss to follow-up
initiation of protocol-prohibited LLT (i.e., an approved PCSK9 inhibitor).
Prior and Concomitant Therapy
For eligible prior and concomitant medications for all 3 ORION trials — including statins and other LLTs (i.e., ezetimibe) — patients were required to have been on stable dose for a minimum of 30 days before screening. Patients receiving statins were to be receiving the MTD. Hormone replacement therapy, prescription medications to treat preexisting medical conditions (such as diabetes or hypertension), and prescription and nonprescription medications to treat AEs at the discretion of the investigator were also permitted.
Medications that were prohibited from being added during all trials included those that are prescribed to lower LDL-C, such as statins, ezetimibe, lomitapide, mipomersen, niacin, colesevelam, bile acid absorption inhibitors, monoclonal antibodies directed toward PCSK9, and any medications taken for the purpose of lipid-lowering, including over-the-counter or herbal therapies.
██ ███ ███████ ██████ ███ ████████ ███ ██ ███████ ██ █████ ███████████████ ███████ ██████████ ███ █████ ██████ █ █████ ██ ███████
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence, as well as any outcomes identified as important to this review by to the clinical expert(s) consulted by CDA-AMC and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to CDA-AMC’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. These outcomes were not assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | ORION-9 | ORION-10 | ORION-11 |
|---|---|---|---|---|
Incidence of CV death, resuscitated cardiac arrest, nonfatal MI, and nonfatal stroke | Not specified | Exploratory | Exploratory | Exploratory |
Percentage change in LDL-C | ORION-9, ORION-10, ORION-11: From baseline to day 510 | Primarya | Primarya | Primarya |
Time-adjusted percent change in LDL-C | ORION-9, ORION-10, ORION-11: From baseline to the period from after day 90 up to day 540 | Primarya | Primarya | Primarya |
Absolute change in LDL-C | ORION-9, ORION-10, ORION-11: From baseline to day 510 | Secondarya | Secondarya | Secondarya |
Time-adjusted absolute change in LDL-C | ORION-9, ORION-10, ORION-11: From baseline to the period from after day 90 and up to day 540 | Secondarya | Secondarya | Secondarya |
Percentage change in Lp(a) | ORION-9, ORION-10, ORION-11: From baseline to day 510 | Secondarya | Secondarya | Secondarya |
Percentage change in ApoB | ORION-9, ORION-10, ORION-11: From baseline to day 510 | Secondarya | Secondarya | Secondarya |
Percentage change in non-HDL-C | ORION-9, ORION-10, ORION-11: From baseline to day 510 | Secondarya | Secondarya | Secondarya |
Absolute change in non-HDL-C | ORION-9, ORION-10, ORION-11: From baseline to day 510 | Secondarya | Secondarya | Secondarya |
Absolute change in LDL-C | ORION-9, ORION-10, ORION-11: From baseline to each assessment time up to day 540 | Secondarya | Secondarya | Secondarya |
Percentage change in LDL-C | ORION-9, ORION-10, ORION-11: From baseline to each assessment time up to day 540 | Secondarya | Secondarya | Secondarya |
Achieving LDL-C < 100 mg/dL, < 70 mg/dL, < 50 mg/dL, and < 25 mg/dL | ORION-9, ORION-10, ORION-11: From baseline to day 510 | Secondarya | Secondarya | Secondarya |
Achieving ≥ 50% reduction from baseline in LDL-C | ORION-9, ORION-10, ORION-11: Not specified | Secondarya | Secondarya | Secondarya |
Achieving global lipid targets for the level of ASCVD risk | ORION-9, ORION-10, ORION-11: Not specified | Secondarya | Secondarya | Secondarya |
Safety and tolerability profile of inclisiran, measured by AEs, SAEs | Not specified | Secondary | Secondary | Secondary |
Notable harms: injection-site reactions, andhepatic, renal, and diabetic safety parameters | Not specified | Safety | Safety | Safety |
AE = adverse event; ApoB = apolipoprotein B; ASCVD = atherosclerotic cardiovascular disease; CV = cardiovascular; HDL-C = high-density-lipoprotein cholesterol; LDL-C = low-density-lipoprotein cholesterol; Lp(a) = lipoprotein(a); MI = myocardial infarction; SAE = serious adverse event.
aAll analyses of the primary, key secondary, and other secondary end points were imputed using various multiple imputation methods.
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report.30
Patients were in a fasted state for all efficacy laboratory assessments. Parameters assessed were total cholesterol, triglycerides, LDL-C, HDL-C, non-HDL-C, very-low-density-lipoprotein cholesterol, apolipoprotein A1, ApoB, Lp(a), high-sensitivity C-reactive protein, and PCSK9. All laboratory assessments and assays were performed by a central laboratory, except for urinalysis.
The primary outcome of the ORION-9, ORION-10, and ORION-11 trials was the percent change in LDL-C from baseline to day 510. All trials had a coprimary end point of time-adjusted percentage change in LDL-C from baseline to the period from after day 90 and up to day 540, defined as the average percentage change in LDL-C from baseline to the period from after day 90 and up to day 540, reflecting the start of the biannual dosing regimen.
Key secondary efficacy end points of the ORION trials included:
absolute change in LDL-C from baseline to day 510
time-adjusted absolute change in LDL-C from baseline to the period from after day 90 and up to day 540
percentage change from baseline to day 510 in PCSK9, total cholesterol, ApoB, and non-HDL-C.
Key secondary end points were not tested if either of the coprimary efficacy end points’ null hypotheses failed to be rejected.
Other secondary outcomes were consistent across the ORION trials, and included maximum percent change in LDL-C; absolute and percent change in LDL-C from baseline to each assessment time up to day 540; individual responsiveness (defined as the number of patients reaching on-treatment LDL-C levels of < 25 mg/dL, < 50 mg/dL, < 70 mg/dL, and < 100 mg/dL) at day 510; proportion of patients in each group with a greater than or equal to 50% LDL-C reduction from baseline; absolute change and percentage change in other lipids, lipoproteins, apolipoproteins, and PCSK9 from baseline at each subsequent visit to day 540; and the proportion of patients in each group who attain global lipid targets for their level of ASCVD risk.
Exploratory end points included the incidence of CV death, resuscitated cardiac arrest, nonfatal MI, and nonfatal stroke (ischemic and hemorrhagic), and the proportion of patients in each group with any LDL-C reduction from baseline at any visit (responders). In the ORION-9 trial, an additional exploratory outcome consisted of response of LDL-C reduction related to underlying causal mutations of HeFH.
Blood samples were taken at scheduled time points to determine LDL-C concentrations. Aliquots of plasma and serum were collected at each time point and stored for additional analyses, including future analysis of biomarkers of CV risk. Plasma samples were analyzed using a validated enzyme-linked immunosorbent assay to determine PCSK9 protein concentration.
The safety and tolerability of inclisiran was a secondary end point of the study and was measured by AEs, SAEs, vital signs, clinical laboratory values, ECG measurements, and the formation and characterization of ADAs. AEs were defined as any untoward medical occurrence in a patient administered a medicinal product that does not necessarily have a causal relationship with the treatment and were coded using MedDRA Version 20.1. The severity of AEs was assessed by the investigator on a 3-point scale (mild, moderate, or severe).
No patient-reported HRQoL outcomes were assessed in the ORION trials.
Statistical Analysis
Sample Size and Power Calculation
For the ORION-9, ORION-10, and ORION-12 trials, the sample-size calculation was performed with the assumption (based on the observed results from a phase II study) that the difference in change from baseline between the active-dose group and the placebo group for LDL-C will be no less than 30 mg/dL, with a standard deviation of 20 mg/dL. Assuming about a 5% drop out rate, the sample size was approximately 380 patients in the ORION-9 trial and 1,425 patients in the ORION-10 and ORION-11 trials, which is evaluable for efficacy across the placebo and inclisiran groups. The sample size of at least 380 patients provided more than 90% power to detect a 30% reduction of LDL-C in the inclisiran group compared to the placebo group, at 1-sided significance level of 0.025. In the ORION-9 trial, due to faster than expected enrolment, the actual enrolment was 482 patients. This increased sample size contributed additional safety data and did not appreciably affect power calculations.
Statistical Test or Model
The incidence of MACE was a prespecified exploratory end point in the ORION-9, ORION-10, and ORION-11 studies. Relevant AEs (i.e., MACE, MI, and stroke) were identified using standard nomenclature from MedDRA. A basket of MedDRA-defined CV terms was used to define the prespecified, nonadjudicated, exploratory end point of MACE (defined per protocol) as a composite of cardiac death, cardiac arrest, nonfatal MI, and fatal and nonfatal stroke. The analysis of MACE was based on the safety population (N = 3,655 patients representing 2,653 PYs of exposure to inclisiran); data were analyzed using Cox regression methods and Kaplan-Meier survival analysis; and the meta-analysis was conducted using the Mantel-Haenszel approach with the Peto method for study pooling, with the study as a fixed effect in the model. Odds ratios and corresponding 95% CIs were provided for the meta-analysis as overall effect estimates.
In the ORION trials, analysis of the first coprimary outcome (percentage change in LDL-C) was conducted on the intention-to-treat (ITT) population and was based on an analysis of covariance (ANCOVA) model on the percentage change in LDL-C from baseline to day 510 on each multiply imputed dataset (100 total). The model included the fixed effect of treatment group (and for the ORION-11 trial, the current use of statins or other LLTs) and baseline LDL-C as a covariate. Treatment effects from these 100 ANCOVA analyses were combined using Rubin’s method via the SAS PROC MIANALYZE procedure. The difference in LSM between treatment groups and the corresponding 2-sided 95% CI was provided for hypothesis testing.
Analysis of the second coprimary outcome (time-adjusted percentage change in LDL-C) was also conducted on the ITT population and was based on a mixed model for repeated measures (MMRM) on the percentage change in the LDL-C from baseline over all visits on each multiply imputed dataset (100 total). The model included fixed effects for treatment, visit, baseline value, and the interaction between treatment and visit. The restricted maximum likelihood estimation approach was used with the covariance structure set as unstructured. The time-adjusted percentage change in LDL-C from baseline to the period from after day 90 and up to day 540 was calculated from the MMRM. Linear combinations of the estimated means after day 90 and up to day 540 were used to compare treatment effects. Treatment effects from these 100 MMRM analyses were combined using Rubin’s method via the SAS PROC MIANALYZE procedure. The difference in LSM between treatment groups and the corresponding 2-sided 95% CI was provided for hypothesis testing.
Multiple Testing Procedure
The key secondary efficacy end points were not tested if either of the coprimary efficacy end points’ null hypotheses failed to be rejected. The Hochberg procedure was applied to control the familywise type I error rate at a 2-sided alpha significance level of 0.05 for the key secondary end points.
Data Imputation Methods
A control-based pattern-mixture model (PMM) was used to explore the possibility of data missing not at random for patients who discontinued the study. For patients who discontinued the study without further follow-up data, missing values after study discontinuation were imputed under the assumption that their outcome would be similar to those in the placebo group who had similar background characteristics. For patients who did not discontinue the study, intermittent missing values were imputed based on the missing-at-random (MAR) assumption. Multiple imputations were used to account for uncertainty in the imputation process and results from the imputed datasets were combined using Rubin’s method.
A key difference in the multiple imputation washout methodology that is unique to the ORION-11 trial was the addition of a modified multiple imputation washout model, which is believed to be the most appropriate method for the imputation of missing data, as it provides the best estimate of what the values of missing data points would have been if had they been measured. The modified method accounted for patients in the inclisiran group who received all 4 doses of the study drug, had the day 510 value missing, had evaluable data at day 540, and had their intermittent missing day 510 values imputed assuming the missing data were MAR.
Subgroup Analyses
Key prespecified subgroup analyses for the efficacy and safety end points included sex, age, body mass index, race, baseline statin use and intensity, other LLT, baseline triglyceride level, metabolic disease, ASCVD status (ORION-9 and ORION-11 trials only), renal impairment (based on estimated glomerular filtration rate), history of allergy, baseline LDL-C, study centre region, and postbaseline-LDL-C; phenotype and baseline ezetimibe use were only subgroups in the ORION-9 trial.
Sensitivity Analyses
The following sensitivity analyses were performed:
A control-based PMM, using the same imputed datasets and an MMRM that were used for the second coprimary end point primary analysis, was used to compare treatments at day 510. Multiple imputations were used to account for uncertainty in the imputation process, and results from the imputed datasets were combined using Rubin’s method.
For both coprimary outcomes, an MMRM analysis without multiple imputation that assumes that missing data are MAR was performed. The model included fixed effects for treatment, visit, baseline value, and the interaction between treatment and visit. The restricted maximum likelihood estimation approach was used with the covariance structure set as unstructured. A linear contrast at day 510 was used to compare treatment groups for the first coprimary outcome, percent change in LDL-C, and a linear combination of estimated means after day 90 and up to day 540 was used to compare treatment groups for the second coprimary outcome, time-adjusted percent change in LDL-C.
The impact of country and current use of statins or other LLTs was assessed by including country, the treatment-by-country interaction, and the current use of statins or other LLTs (yes or no) as fixed effects in the primary analysis ANCOVA (using multiple imputation washout model data).
An ANCOVA model without multiple imputation was performed using the modified intention-to-treat (mITT) population. The model included the fixed effect of treatment group and baseline LDL-C as a covariate.
A tipping-point analysis was performed to search for the tipping point that would reverse the study conclusion. In the tipping-point analysis, the deltas in the treatment group were varied independently (the inclisiran group is progressively worse while the placebo group is not impacted, and the inclisiran group is progressively worse while the placebo group progressively improves) until the hypothesis tests on the coprimary efficacy end points became statistically insignificant.
Secondary Outcomes of the Studies
The absolute change in LDL-C from baseline to day 510 and percentage change from baseline to day 510 in PCSK9, total cholesterol, ApoB, and non-HDL-C were analyzed using an MMRM with covariates. The time-adjusted absolute change in LDL-C from baseline to the period from after day 90 and up to day 540 was analyzed similarly to that of the time-adjusted percentage change in LDL-C from baseline to the period from after day 90 and up to day 540. Missing values were imputed using the control-based PMM on LDL-C, PCSK9, total cholesterol, ApoB, and non-HDL-C, and absolute change or percentage change from baseline was calculated based on imputed data before any analysis was performed. The MMRM without multiple imputation was used in sensitivity analyses for the key secondary end points.
Table 7: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
ORION-9 trial | ||||
Percentage change in LDL-C from baseline to day 510 (coprimary). | ANCOVA on the percentage change in LDL-C from baseline to day 510 on each multiply imputed dataset (100 total). Treatment effects from these 100 ANCOVA analyses were combined using Rubin’s method and the difference in the LSM between treatment groups and corresponding 2-sided 95% CI was provided for hypothesis testing. | The model included the fixed effect of treatment group and baseline LDL-C as a covariate. | Missing values imputed for LDL-C after a reflexive approach using a multiple imputation (100 total imputed datasets) washout model. The percentage change in LDL-C at each visit was calculated after missing data were imputed. |
|
Time-adjusted percent change in LDL-C from baseline to the period from after day 90 and up to day 510 (coprimary). | MMRM on the percentage change in LDL-C from baseline over all visits on each multiply imputed dataset (100 total). The model included fixed effects for treatment, visit, baseline value, and interaction between treatment and visit. Treatment effects from the 100 MMRM analyses were combined using Rubin’s method. The difference in the LSM between treatment groups and corresponding 2-sided 95% CI was provided for hypothesis testing. | Not specified. | Control-based PMM was used to explore the possibility of data MNAR for patients who discontinued the study. For patients who discontinued the study without any further follow-up data, missing values were imputed under the assumption that their outcome would be similar to those in the placebo group with similar background characteristics. For patients who did not discontinue the study, intermittent missing values were imputed based on the MAR assumption. |
|
Absolute change in LDL-C from baseline to day 510 (key secondary). | MMRM with covariates | Covariates and baseline characteristics included the baseline value of efficacy measurement, the observed value of efficacy measurement at day 90, day 150, day 270, day 330, day 450, day 510, and day 540, and the current use of statins or other LLTs. | Missing values were imputed using the control-based PMM. | MMRM without multiple imputation was used in the sensitivity analyses. |
Time-adjusted absolute change in LDL-C from baseline to the period from after day 90 and up to day 540 (key secondary). | MMRM similar to what was used for coprimary outcome #2 time-adjusted percent change in LDL-C from baseline to the period from after day 90 and up to day 510 (coprimary). | Not specified. | ||
Percentage change from baseline to day 510 in PCSK9, total cholesterol, ApoB, and non-HDL-C (key secondary). | Same as key secondary outcome #1. Absolute change in LDL-C from baseline to day 510 (key secondary). | Same as key secondary outcome #1. Absolute change in LDL-C from baseline to day 510 (key secondary). | ||
ORION-10 trial | ||||
Percentage change in LDL-C from baseline to day 510 (coprimary). | ANCOVA model on the percentage change in LDL-C from baseline to day 510 on each multiply imputed dataset (100 total). Treatment effects from these 100 ANCOVA analyses were combined using Rubin’s method, and the difference in the LSM between treatment groups and the corresponding 2-sided 95% CI was provided for hypothesis testing. | The model included the fixed effect of treatment group and baseline LDL-C as a covariate. | Missing values imputed for LDL-C after a reflexive approach using a multiple imputation (100 total imputed datasets) washout model. The percentage change in LDL-C at each visit was calculated after missing data were imputed. |
|
Time-adjusted percentage change in LDL-C from baseline to the period from after day 90 and up to day 540 (coprimary). | MMRM on the percentage change in LDL-C from baseline over all visits on each multiply imputed dataset (100 total). The model included fixed effects for treatment, visit, baseline value, and interaction between treatment and visit. Treatment effects from the 100 MMRM analyses were combined using Rubin’s method. The difference in the LSM between treatment groups and the corresponding 2-sided 95% CI was provided for hypothesis testing. | Not specified. | Control-based PMM was used to explore the possibility of data MNAR for patients who discontinued the study. For patients who discontinued the study without any further follow-up data, missing values were imputed under the assumption that their outcome would be similar to those in the placebo group with similar background characteristics. For patients who did not discontinue the study, intermittent missing values were imputed based on the MAR assumption. |
|
Absolute change in LDL-C from baseline to day 510 (key secondary). | MMRM with covariates. | Covariates and baseline characteristics included the baseline value of efficacy measurement, the observed value of efficacy measurement at day 90, day 150, day 270, day 330, day 450, day 510, and day 540, and the current use of statins or other LLTs. | Missing values were imputed using the control-based PMM. | MMRM without multiple imputation was used in the sensitivity analyses. |
Time-adjusted absolute change in LDL-C from baseline to the period from after day 90 and up to day 540 (key secondary). | MMRM, similar to what was used for coprimary outcome #2. time-adjusted percent change in LDL-C from baseline to the period from after day 90 and up to day 510 (coprimary). | Not specified. | ||
Percentage change from baseline to day 510 in PCSK9, total cholesterol, ApoB, and non-HDL-C (key secondary). | Same as key secondary outcome #1. Absolute change in LDL-C from baseline to day 510 (key secondary). | Same as key secondary outcome #1. Absolute change in LDL-C from baseline to day 510 (key secondary). | ||
ORION-11 trial | ||||
Percentage change in LDL-C from baseline to day 510 (coprimary). | ANCOVA model on the percentage change in LDL-C from baseline to day 510 on each multiply imputed dataset (100 total). Treatment effects from these 100 ANCOVA analyses were combined using Rubin’s method and the difference in the LSM between treatment groups and corresponding 2-sided 95% CI was provided for hypothesis testing. | The model included the fixed effects of treatment group and the current use of statins or other LLTs at baseline (yes or no) and baseline LDL-C as a covariate. | Missing values imputed for LDL-C after a reflexive approach using a multiple imputation (100 total imputed datasets) washout model. The percentage change in LDL-C at each visit was calculated after missing data were imputed. |
|
Time-adjusted percentage change in LDL-C from baseline to the period from after day 90 and up to day 540 (coprimary). | MMRM on the percentage change in LDL-C from baseline over all visits on each multiply imputed dataset (100 total). The model included fixed effects for treatment, visit, baseline value, interaction between treatment and visit, and the current use of statins or other LLTs. Treatment effects from the 100 MMRM analyses were combined using Rubin’s method. The difference in the LSM between treatment groups and corresponding 2-sided 95% CI was provided for hypothesis testing. | Not specified. | Control-based PMM was used to explore the possibility of data MNAR for patients who discontinued the study. For patients who discontinued the study without any further follow-up data, missing values were imputed under the assumption that their outcome would be similar to those in the placebo group with similar background characteristics. For patients who did not discontinue the study, intermittent missing values were imputed based on the MAR assumption. |
|
Absolute change in LDL-C from baseline to day 510 (key secondary). | MMRM with covariates. | Covariates and baseline characteristics included the baseline value of efficacy measurement, the observed value of efficacy measurement at day 90, day 150, day 270, day 330, day 450, day 510, and day 540, and the current use of statins or other LLTs. | Missing values were imputed using the control-based PMM | MMRM without multiple imputation was used in the sensitivity analyses. |
Time-adjusted absolute change in LDL-C from baseline to the period from after day 90 and up to day 540 (key secondary). | MMRM similar to what was used for coprimary outcome #2. time-adjusted percent change in LDL-C from baseline to the period from after day 90 and up to day 510 (coprimary). | Not specified. | ||
Percentage change from baseline to day 510 in PCSK9, total cholesterol, ApoB, and non-HDL-C (key secondary). | Same as key secondary outcome #1. Absolute change in LDL-C from baseline to day 510 (key secondary). | Same as key secondary outcome #1. Absolute change in LDL-C from baseline to day 510 (key secondary). | ||
ANCOVA = analysis of covariance; ApoB = apolipoprotein B; CI = confidence interval; HDL-C = high-density-lipoprotein cholesterol; LDL-C = low-density-lipoprotein cholesterol; LLT = lipid-lowering therapy; LSM = least squares mean; MAR = missing at random; mITT = modified intention to treat; MMRM = mixed model for repeated measures; MNAR = missing not at random; PMM = pattern-mixture model.
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report.30
Analysis Populations
Table 8: Analysis Populations of the ORION-9, ORION-10, and ORION-11 Trials
Study | Population | Definition | Application |
|---|---|---|---|
ORION-9 | ITT | All patients randomized, with treatment classification based on the randomized treatment. | Used for analyses of primary and secondary end points. |
Full analysis set | Patients who were randomized, took any study medication, and had at least 1 posttreatment lipid data measurement. | Population used in an additional efficacy analysis and sensitivity analysis for the coprimary end point. | |
mITT | All patients who received at least 1 dose of the investigational product and had baseline and day 510 follow-up LDL-C assessments. | Population used in an additional efficacy analysis and sensitivity analysis for the coprimary end point. | |
Safety | All patients who received at least 1 dose of the investigational product. A patient who received any amount of inclisiran during the study was analyzed within the inclisiran treatment group. | Primary population for safety analyses. | |
ORION-10 | ITT | All patients randomized, with treatment classification based on the randomized treatment. | Used for analyses of primary and secondary end points. |
Full analysis set | Patients who were randomized, took any study medication, and had at least 1 posttreatment lipid data measurement. | Population used in an additional efficacy analysis and sensitivity analysis for the coprimary end point. | |
mITT | All patients who received at least 1 dose of the investigational product and had baseline and day 510 follow-up LDL-C assessments. | Population used in an additional efficacy analysis and sensitivity analysis for the coprimary end point. | |
Safety | All patients who received at least 1 dose of the investigational product. A patient who received any amount of inclisiran during the study was analyzed within the inclisiran treatment group. | Primary population for safety analyses. | |
ORION-11 | ITT | All patients randomized, with treatment classification based on the randomized treatment. | Used for analyses of primary and secondary end points. |
Full Analysis Set | Patients who were randomized, took any study medication, and had at least 1 posttreatment lipid data measurement. | Population used in an additional efficacy analysis and sensitivity analysis for the coprimary end point. | |
mITT | All patients who received at least 1 dose of investigational product and had baseline and day 510 follow-up LDL-C assessments. | Population used in an additional efficacy analysis and sensitivity analysis for the coprimary end point. | |
Safety | All patients who received at least 1 dose of investigational product. A patient who received any amount of inclisiran during the study was analyzed within the inclisiran treatment group. | Primary population for safety analyses. |
ITT = intention to treat; LDL-C = low-density-lipoprotein cholesterol; mITT = modified intention to treat.
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report.30
Results
Patient Disposition
The ORION-9, ORION-10, and ORION-11 trials shared a similar study design and patient enrolment model.
Table 9 summarizes the patient flow of the ORION trials. In the ORION-9 trial, a total of 617 patients were screened and 482 were randomized. The most common reason for screening failure was that the inclusion or exclusion criteria were not met █████████ ███ ██ ███████████ ███████ █████████████ ████████ ██ █ ███ ███████ A total Of the 2,329 patients screened in the ORION-10 trial, 1,561 were randomized. In the ORION-11 trial, a total of 2,381 patients were screened and 1,617 were randomized. The primary reason for screening failure in the ORION-10 and ORION-11 trials was the patient not meeting the inclusion and/or exclusion criteria, ████████████ ███ ███████████ █████ █████ ████████ ██ █ ████ ██████ ██████ ██████. The proportion of patients that failed screening varied across trials but was more similar in the ORION-10 and ORION-11 trials (32.98% and 32.09%) compared to the ORION-9 trial (21.88%).
Across all studies, 94% of patients or more completed the study (i.e., had a day 540 EOS visit). The discontinuation rate was similar across studies, with the ORION-10 trial having the greatest proportion of patients discontinuing treatment. In all studies, the primary reason for discontinuation was withdrawal of consent. The proportion of patients discontinuing due to death was similar in the ORION-10 and ORION-11 trials but was lower in the ORION-9 trial.
Table 9: Summary of Patient Disposition From the Studies Included in the Systematic Review
Patient disposition | ORION-9 | ORION-10 | ORION-11 | |||
|---|---|---|---|---|---|---|
Inclisiran (N = 242) | Placebo (N = 240) | Inclisiran (N = 781) | Placebo (N = 780) | Inclisiran (N = 810) | Placebo (N = 807) | |
Screened, N | 617 | 2,329 | 2,381 | |||
Reason for screening failure, N | ||||||
Screen failures | 135 | 768 | 764 | |||
█████ ████████ ███ ███ | ███ | ███ | ███ | |||
████████ ███████ | ██ | ██ | ██ | |||
█████ | ██ | ██ | ██ | |||
Randomized, N | 482 | 1,561 | 1,617 | |||
Discontinued from study, n (%) | 7 (2.9) | 9 (3.8) | 60 (7.7) | 86 (11.0) | 38 (4.7) | 37 (4.6)a |
Reason for discontinuation, n | ||||||
Loss to follow-up | 1 | 2 | 10 | 24 | 6 | 3 |
Adverse event | 0 | 0 | 8 | 5 | 4 | 0 |
Withdrew consent | 0 | 4 | 24 | 34 | 13 | 17 |
Death | 1 | 1 | 12 | 11 | 14 | 15 |
Initiation of protocol-prohibited approved PCSK9 inhibitor | 0 | 1 | 0 | 9 | 0 | 0 |
Physician decision | 0 | 0 | 1 | 0 | 1 | 1 |
Other | 5 | 1 | 5 | 3 | 0 | 1 |
ITT, n | 242 | 240 | 781 | 780 | 810 | 807 |
FAS, n | 241 | 239 | 767 | 768 | 803 | 800 |
████ | ███ | ███ | ███ | ███ | ███ | ███ |
Safety, n | 241 | 240 | 781 | 778 | 811 | 804 |
FAS = full analysis set; ITT = intention to treat; mITT = modified intention to treat, NR = not reported.
aOne patient randomized to placebo was given inclisiran.
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report.30
Baseline Characteristics
The baseline characteristics outlined in Table 10 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Table 10: Summary of Baseline Characteristics From the ORION-9, ORION-10, and ORION-11 Trials (ITT Population)
Details | ORION-9 | ORION-10 | ORION-11 | |||
|---|---|---|---|---|---|---|
Inclisiran (N = 242) | Placebo (N = 240) | Inclisiran (N = 781) | Placebo (N = 780) | Inclisiran (N = 810) | Placebo (N = 807) | |
Age, mean (SD) | 54.4 (12.5) | 55.0 (11.8) | 66.4 (8.9) | 65.7 (8.9) | 64.8 (8.3) | 64.8 (8.7) |
Male sex, n (%) | 112 (46.3) | 115 (47.9) | 535 (68.5) | 548 (70.3) | 579 (71.5) | 581 (72.0) |
White, n (%) | 226 (93.4) | 227 (94.6) | 653 (83.6) | 685 (87.8) | 791 (97.7) | 796 (98.6) |
CV risk factors, n (%) | ||||||
ASCVD | 59 (24.4) | 73 (30.4) | 781 (100.0) | 780 (100.0) | 712 (87.9) | 702 (87.0) |
ASCVD risk equivalent | 183 (75.6) | 167 (69.6) | 0 | 0 | 98 (12.1)a | 105 (13)a |
Current smoker | 28 (11.6) | 28 (11.7) | 123 (15.7) | 111 (14.2) | 160 (19.8) | 132 (16.4) |
Hypertension | 102 (42.1) | 101 (42.1) | 714 (91.4) | 701 (89.9) | 640 (79.0) | 661 (81.9) |
Diabetes | 20 (8.3) | 28 (11.7) | 371 (47.5) | 331 (42.4) | 296 (36.5) | 272 (33.7) |
HeFH | ██ | ██ | 8 (1.0) | 12 (1.5) | 14 (1.7) | 14 (1.7) |
Concomitant lipid-modifying therapy, n (%) | ||||||
Statin | 219 (90.5) | 217 (90.4) | 701 (89.8) | 692 (88.7) | 766 (94.6) | 766 (94.9) |
High-intensity statin | 185 (76.4) | 171 (71.3) | 538 (68.9) | 546 (70.0) | 633 (78.1) | 628 (77.8) |
Ezetimibe | 135 (55.8) | 120 (50.0) | 80 (10.2) | 74 (9.5) | 52 (6.4) | 62 (7.7) |
Lipid measures, mg/dL, mean (SD) | ||||||
Total | 230.0 (54.6) | 232.4 (62.8) | 180.6 (46.1) | 180.6 (43.6) | 187.3 (48.2) | 183.3 (42.8) |
LDL-C | 151.4 (50.4) | 154.7 (58.0) | 104.5 (39.6) | 104.8 (37.0) | 107.2 (41.8) | 103.7 (36.4) |
Non-HDL-C | 178.5 (55.4) | 181.5 (62.5) | 134.0 (44.5) | 134.7 (43.5) | 137.6 (46.9) | 133.9 (41.0) |
HDL-C | 51.5 (15.1) | 50.8 (13.1) | 46.6 (14.3) | 45.9 (14.4) | 49.7 (15.5) | 49.3 (13.8) |
ApoB | 123.8 (33.2) | 124.5 (34.8) | 94.1 (25.6) | 94.6 (25.1) | 97.1 (28.0) | 95.1 (5.2) |
Lipoprotein (a), nmol/L | ||||||
Median | 57 | 54 | 57 | 56 | 42 | 35 |
IQR | 22 to 180 | 20 to 185 | 18 to 181 | 20 to 189 | 18 to 178 | 18 to 181 |
Triglycerides, mg/dL | ||||||
Median | 120 | 119 | 127 | 129 | 135 | 135 |
IQR | 82 to 167 | 85 to 166 | 92 to 181 | 96 to 182 | 99 to 181 | 102 to 185 |
High-sensitivity C-reactive protein, mg/L | ||||||
Median | 1.2 | 1.3 | ██ | ██ | ██ | ██ |
IQR | 0.5 to 2.9 | 0.6 to 3.2 | ██ | ██ | ██ | ██ |
PCSK9, mcg/L, mean (SD) | 452.2 (131.2) | 429.1 (135.3) | 422.1 (176.9) | 414.9 (145.7) | 355 (98.9) | 353 (97.4) |
Baseline eGFR (mL/min per 1.73m2) | ||||||
Mean (SD) | 86.3 (20.41) | 83.8 (19.33) | 75.6 (22.27) | 76.1 (22.05) | 80.0 (19.23) | 79.1 (19.54) |
████ | ████ | ████ | ████ | ████ | ████ | ████ |
████ | ||||||
███ | ████ | ████ | ████ | ████ | ████ | ████ |
████ | ████ | ████ | ████ | ████ | ████ | ████ |
████ | ████ | ████ | ████ | ████ | ████ | ████ |
████ | ████ | ████ | ████ | ████ | ████ | ████ |
████ | ||||||
████ | ████ | ████ | ████ | ████ | ████ | ████ |
████ | ████ | ████ | ████ | ████ | ████ | ████ |
████ | ████ | ████ | ████ | ████ | ████ | ████ |
████ | ||||||
███ | ████ | ████ | ████ | ████ | ████ | ████ |
██ | ████ | ████ | ████ | ████ | ████ | ████ |
ApoB = apolipoprotein B; ASCVD = atherosclerotic cardiovascular disease; CV = cardiovascular; eGRF = estimated glomerular filtration rate; HDL-C = high-density-lipoprotein cholesterol; HeFH = heterozygous familial hypercholesterolemia; IQR = interquartile range; LDL-C = low-density-lipoprotein cholesterol; LDLR = low-density-lipoprotein receptor; NA = not applicable; NR = not reported; SD = standard deviation.
aIn the ORION-11 trial, patients in this category had type 2 diabetes, FH, or a 10-year risk of a CV event of 20% or greater, as assessed by the FRS for CVD or equivalent.
| ██████ ███ ████████ ███ ███ █████ ████████ ████ ██ █ █████
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report,30 Raal FJ et al.,79 Ray KK et al. (2020).11
Exposure to Study Treatments
██ ████████ ████ ████████ ██ ████████ ███ ███ ██████████ ███ ███████ ████ ████ █████ ███ █████ █████ █████████████ ██ ████████ █ █████████ ███ ████ ████████ ██ ████████ ███ ██████████ ███ █████ ███ █████ █████ █████████████ ███ █████ ███ █████ ████ ███ ████████ █████████████ █████ █████████████ ██ ████████ ██ ███████ ████ █████ ███ █████ ██ ██████████ ███ ███████ █████ █████████████ ██ █████████ █████ █████████████ ██ ████████ ███ ██████ ██ ███ ██████████ ███ ███ ██████ ██ ███ ███████ ████ ███ ██████████ ███ ██ ████████ ███ ██████ █████████████ ██ ████████ █████ ███ ███████ ███ ███ ██████ █████████████ ██ █████████ █ ████████ ███████ ██ ███████ ████████ ██ █████████████ ██ ████████ █████████ ███ ████████ ███ █████████ ██ █████ ████████ ███ █████████ ██ Table 11.
Concomitant Medications
There were no medications or cointerventions required during the study. To be eligible for enrolment, participants must have been treated with any of the following statins: atorvastatin 40 mg or 80 mg once daily; rosuvastatin 20 mg or 40 mg once daily; or simvastatin 40 mg once daily, or, if patient was on 40 mg daily for more than 1 year, 80 mg once daily. Lower doses of statins were allowed in cases of partial statin intolerance, defined as intolerance to any of the previously described statins at any of the aforementioned doses. ██████ ███ ████████ ██ ███ ███ ██████████ ███████ ███ ██ █████ ███ ███ █ ███████████ ███████████ ██ █████████████ ████ ███ ███ █████ ███ █████████ ████████ ███████ ███ █████████ ██████.
In the ORION-9 trial, overall, █████ █████████ ██ ████████ ████████ ███, 90.5% (436/482) of patients received statin therapy, 55.4% (267/482) received other LLTs (58.3% [140/240] of patients in the placebo group and 52.5% [127/242] of patients in the inclisiran group), and 3.9% (19/482) received other LLTs and no statins. Overall, 73.9% (356/482) were on a high-intensity statin at baseline. A total of 14.5% (70/482) of patients received a moderate-intensity statin and 1.9% (9/482) patients received a low-intensity statin at baseline. Approximately half (52.3%; 252/482) of patients the were treated with ezetimibe. In the ORION-10 trial, overall, █████ ███████████ ██ ████████ ████████ ███, 89.2% (1,393/1,561) of patients received statin therapy and 31.3% (489/1,561) received other LLTs. Two-thirds (69.4%; 1,084/1,561) of patients were on a high-intensity statin at baseline. A total of 18.7% (292/1,561) of patients received a moderate-intensity statin and 0.8% (12/1,561) received a low-intensity statin at baseline. A total of 9.9% (156/1,561) patients were treated with ezetimibe. In the ORION-11 trial, overall, █████ ███████████ ██ ████████ ████████ ███, 94.7% (1,532/1,617) of patients received statin therapy and 11.8% (191/1,617) received other LLTs. A total of 78.0% (1,261/1,617) of patients were on a high-intensity statin at baseline, 15.5% (251/1,617) of patients were on a moderate-intensity statin at baseline, and 0.4% (6/1,617) were on a low-intensity statin at baseline. A total of 7.1% (114/1,617) patients were treated with ezetimibe.
██ ████████ ████████ █████ ██ ████████ █████████ ███ ██ ███ ██ ███████ ██ ███ ██████ ██████ ███ ██████ █ ███████ ██████ ██ ███ ███████ █████ █████ ███████ ████ █ █████ ██████████ ████████ █████████████ ██ ███████ ██ ███ ██████████ █████ █████ ███████ ████ █ █████ ██████████ ████████ ██████ ███ ██████ ██ █████████ █████ ███████████ ██ ████████ ███ ██ ███ ███ █████ ██ ███████ ██ ███ █████ ██████ ███ ██████ ██████ ██ ██████ █████████ ███ ███████ ██ ████████ ██████ ███████ ███████ ███████ ███ ████████ ██████ ██████████ ██████ ████████ ██████ ███ ██████ █ ████████ ██ ███ ███████ █████ ████ ████ █ █████ ██████████ ████████ ███ ████ █████████████ ██ █████████ ████████ █████ ███████████ ██ ████████ ███ ██ ███ ███ █████ ██ ███████ ██ ███ ███ ██████ ███ ██████ ████████ ████ ███████████████ ████████ ██████ █████████ ███████ ████ ██████████████████ ████████ █████ ████████ ███████████████ ████████ ██████ ████ ████████ ██████████████████ ██████████ ██ ████████ ████ ██████████ █████ ████ ████████
Table 11: Summary of Patient Exposure From the ORION-9, ORION-10, and ORION-11 Trials (Safety Population)
Exposure | ORION-9 | ORION-10 | ORION-11 | |||
|---|---|---|---|---|---|---|
Inclisiran (N = 241) | Placebo (N = 240) | Inclisiran (N = 781) | Placebo (N = 778) | Inclisiran (N = 811) | Placebo (N = 804) | |
██████████████ | █████ | █████ | ██████ | ██████ | ██████ | ██████ |
██████████████ | █████ | █████ | ██████ | ██████ | ██████ | ██████ |
██████████████ | █████ | █████ | ██████ | ██████ | ██████ | ██████ |
██████████████ | █████ | █████ | ██████ | ██████ | ██████ | ██████ |
IQR = interquartile range; NR = not reported; SD = standard deviation.
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report.30
Efficacy
Major Adverse Cardiovascular Events
In the ORION-9, ORION-10, and ORION-11 trials, the exploratory end point of MACE was defined as the composite of CV death, resuscitated cardiac arrest, nonfatal MI, and nonfatal stroke (hemorrhagic or nonhemorrhagic), using predefined MedDRA search.
As part of its resubmission, the sponsor conducted a pooled analysis of clinical outcomes from the ORION-9, ORION-10, and ORION-11 trials and also provided what was referred to as a sensitivity analysis that pooled data from the ORION-10 and ORION-11 studies. The pooled analysis of all 3 trials is not relevant for this review, as it combines the HeFH and the nFH with ASCVD populations, and these 2 populations are being viewed separately for this review, consistent with the indication. The sensitivity analysis that was conducted to assess the effects of inclisiran (n = 1,494) compared to placebo (n = 1,477) on MACE in the ASCVD and ASCVD-RE populations is relevant.80
HeFH Population
The incidence of MACE in the inclisiran and placebo arms of the ORION-9 trial was 10 (4.1%) and 10 (4.2%), respectively; the absolute number of MACE for the inclisiran and placebo arms was 10 and 11, respectively; the corresponding RR was ███ ████ ███ ████ █████
nFH With ASCVD
Results from the posthoc pooled analysis of the ORION-10 and ORION-11 trials showed ███ ██████ patients in the inclisiran arm experienced ███ ██████, while ███ ███████ patients in the placebo arm experienced ███ ██████. The reported HR of ████ ████ ███ █████ █████ for MACE favoured inclisiran over placebo in the ORION-10 and ORION-11 patient population.
The incidence of MACE in the inclisiran and placebo arms of the ORION-10 trial was 58 (7.4%) and 79 (10.2%), respectively; the absolute number of MACE in the inclisiran and placebo arms was 66 and 90, respectively; the corresponding RR was ███ ████ ███ ████ ████. The incidence of MACE in the inclisiran and placebo arms of the ORION-11 trial was 63 (7.8%) and 83 (10.3%), respectively; the absolute number of MACE for the inclisiran and placebo arms was 65 and 100, respectively; the corresponding RR was ███ ████ ███ ████ ████.
Myocardial Infarction:
Heterozygous Familial Hypercholesterolemia: In the ORION-9 trial, the exploratory end point of nonfatal MI occurred in 9 (3.7%) patients experiencing | events in the inclisiran arm compared to 10 (4.2%) patients experiencing ██ events in the placebo arm.
nFH With ASCVD: Using a posthoc pooled analysis of the ORION-10 and ORION-11 trials, the effects of inclisiran (n = 1,494) compared to placebo (n = 1,477) on fatal or nonfatal MI outcomes in the ASCVD and ASCVD-RE population showed that ██ ██████ patients in the inclisiran arm experienced ██ ██████, while ██ ██████ patients in the placebo arm experienced ██ ██████. The reported HR was ████ ████ ███ █████ █████ for fatal or nonfatal MI in the ORION-10 and ORION-11 patient population.
Stroke:
Heterozygous Familial Hypercholesterolemia: In the ORION-9 trial, for the exploratory end point of nonfatal stroke, no patients in either the inclisiran or placebo treatment arms experienced an event.
nFH With ASCVD: Using a posthoc pooled analysis of the ORION-10 and ORION-11 trials, the effect of inclisiran (n = 1,494) compared to placebo (n = 1,477) on fatal or nonfatal stroke outcomes in the ASCVD and ASCVD-RE population showed that ██ ██████ patients in the inclisiran arm experienced ██ ██████, while ██ ██████ patients in the placebo arm experienced ██ ██████. The HR was ████ ████ ███ █████ █████ for fatal or nonfatal stroke in the ORION-10 and ORION-11 patient populations.
Table 12: Posthoc Pooled Analysis of MACE From the ORION-9, ORION-10, and ORION-11 Trials
Outcome | Pooled analysis of CV event outcomes | |
|---|---|---|
Inclisiran (N = 1,833) | Placebo (N = 1,822) | |
All 3 ORION trials | ||
MACE | ||
n (%) | 131 (7.1) | 172 (9.4) |
Events, n | 141 | 201 |
Time at risk, per 100 PYs | 5.35 | 7.71 |
OR (95% CI) | 0.74 (0.58 to 0.94) | |
HR (95% CI)a | 0.75 (0.60 to 0.94) | |
Fatal or nonfatal MI | ||
n (%) | 33 (1.8) | 41 (2.3) |
Events, n | 34 | 45 |
OR (95% CI) | 0.80 (0.50 to 1.27) | |
HR (95% CI)a | 0.81 (0.51 to 1.29) | |
Fatal or nonfatal stroke | ||
n (%) | 13 (0.7) | 15 (0.8) |
Events, n | 14 | 16 |
OR (95% CI) | 0.86 (0.41 to 1.81) | |
HR (95% CI)a | 0.80 (0.39 to 1.67) | |
Outcome | ORION-10 and ORION-11 trialsb | |
Inclisiran (N = 1,494) | Placebo (N = 1,477) | |
MACEc | ||
███ | ███ █████ | ███ ██████ |
█████ | ███ | ███ |
██ ████ ██ | ████ ██████ █████ | |
Fatal and nonfatal MI | ||
███ | ██ █████ | ██ █████ |
███████ | ██ | ██ |
██ ████ ██ | ████ ██████ █████ | |
Fatal and nonfatal stroke | ||
███ | ██ █████ | ██ █████ |
███████ | ██ | ██ |
██ ████ ██ | ████ ██████ █████ | |
ASCVD = atherosclerotic cardiovascular disease; CI = confidence interval; CV = cardiovascular; HR = hazard ratio; MACE = major adverse cardiac events; MI = myocardial infarction; OR = odds ratio; PY = patient-year.
aThe HR and 95% CI are from a Cox model with treatment and study ID as factors.
bThe pooled analyses for CV events conducted by Ray et al. (2022)81 included all patients from the 3 pivotal ORION trials, where a proportion of patients had HeFH; thus, this analysis removed the ORION-9 population and included patients only from the ORION-10 and ORION-11 trials with ASCVD and ASCVD-RE, of whom > 98% had nFH.
c████████ ████ ██████ ███████ ██████████████████ ███ ██████████████████████ ██████████████████ ██████ █████████ ████ ███ ████████████ ██████ ██████ ████ █████████ ███ █████ ██ ████████ ███████ ██████ ████ ███████ █████ ████ ████ ████████ ████ ███ █████████████ ███████████████ ████ ████████ ████ ██ █████ █████ ███ ██ ██████████████████████ ███████ ██████ ████ ███ ██ ███████ ███ █████████
Sources: Novartis. (2023) Data on File. CKJX839A1 Inc-Pub084;80 Ray KK et al. (2022).81
Low-Density-Lipoprotein Cholesterol
Percent Change in LDL-C From Baseline to Day 510
The first coprimary key end point of percent change in LDL-C from baseline to day 510 was the same in the ORION-9, ORION-10, and ORION-11 trials.
Heterozygous Familial Hypercholesterolemia:
The between-group difference between inclisiran and placebo in percent reduction in LDL-C in the ORION-9 trial was –47.9% (95% CI, –53.5% to –42.3%; P < 0.0001).
nFH With ASCVD:
The between-group difference between inclisiran and placebo in percent reduction in LDL-C in the ORION-10 trial was – 52.2% (95% CI, –55.7% to –48.8%; P < 0.0001) and in the ORION-11 trial was –49.9% (95% CI, –53.1% to –46.6%; P < 0.0001).
Sensitivity analyses conducted using control-based PMM, MMRM, and ANCOVA multiple imputation washout model showed similar results. Of note, the modified multiple imputed washout model analysis was only performed for the ORION-11 trial. Similar statistically significant (P < 0.0001) placebo-adjusted differences were observed regardless of analysis population (ITT, full analysis set, or mITT) used.82
Subgroups:
Across the studies, placebo-adjusted LDL-C reductions favouring inclisiran were similar in most subgroups. A statistically significant interaction was observed for above and below the median and quartiles of baseline LDL-C and PCSK9 levels. In all subgroups, inclisiran lowered LDL-C more than placebo.
Time-Adjusted Percent Change in LDL-C From Baseline After Day 90 up to Day 540
The second coprimary key end point of time-average percent change in LDL-C from baseline to the period from after day 90 and up to day 540 was the same for the ORION-9, ORION-10, and ORION-11 trials.
Heterozygous Familial Hypercholesterolemia:
The LSM difference from placebo favoured inclisiran in the ORION-9 trial, at −44.30% (95% CI, −48.48% to −40.12%; P < 0.0001). The results of the sensitivity analyses for time-adjusted percent change in LDL-C from baseline to the period from after day 90 and up to day 540 were consistent with the overall population.
nFH With ASCVD:
The LSM difference from placebo favoured inclisiran in the ORION-10 trial, at −53.78% (95% CI, −56.23% to −51.33%; P < 0.0001), and in the ORION-11 trial, at −49.17% (95% CI, −51.57% to −46.77%; P < 0.0001). The results of the sensitivity analyses for time-adjusted percent change in LDL-C from baseline to the period from after day 90 and up to day 540 were consistent with the overall population.
Subgroups:
Subgroup analyses were provided for the coprimary outcome of time-adjusted percent change in LDL-C after day 90 up to day 540 for the ORION-9, ORION-10, and ORION-11 trials. Results of the subgroup analyses of interest to this review, including baseline LDL-C, baseline statin treatment, statin intensity, and LLT use, were consistent with the overall patient population.
Mean Absolute Change in LDL-C From Baseline to Day 510
Heterozygous Familial Hypercholesterolemia:
In the ORION-9 trial, the key secondary end point of LSM absolute change in LDL-C from baseline to day 510 was −68.89 mg/dL (95% CI, −77.11 to −60.67 mg/dL; P < 0.0001). Sensitivity analysis using an MMRM demonstrated similar results.
nFH with ASCVD:
The key secondary end point of mean absolute change in LDL-C from baseline to day 510 was −54.12 mg/dL (95% CI, −57.37 to −50.88 mg/dL; P < 0.0001) in the ORION-10 trial and −51.87 mg/dL (95% CI, −55.01 to −48.72 mg/dL; P < 0.0001) in the ORION-11 trial. Sensitivity analysis using an MMRM demonstrated similar results.
Time-Adjusted Absolute Change in LDL-C From Baseline After Day 90 up to Day 540
Heterozygous Familial Hypercholesterolemia:
Compared to placebo, the time-adjusted absolute change from baseline to the period from after day 90 and up to day 540 in the ORION-9 trial was −62.74 mg/dL (95% CI, −69.01 to −56.48 mg/dL; P < 0.0001). Sensitivity analysis using an MMRM demonstrated similar results.
nFH With ASCVD:
Compared to placebo, the time-adjusted absolute change from baseline to the period from after day 90 and up to day 540 in the ORION-10 trial was –53.28 mg/dL (–55.75 to –50.80 mg/dL; P < 0.0001) and in the ORION-11 trial was −48.94 mg/dL (95% CI, −51.39 to −46.48 mg/dL; P < 0.0001). Sensitivity analysis using an MMRM demonstrated similar results.
Other Lipid Parameters
Percent change in total cholesterol, non-HDL-C, and ApoB from baseline to day 510 is reported in Table 41 in Appendix 1.
Table 13: Summary of Key Lipid Parameters From the ORION-9, ORION-10, and ORION-11 Trials
Outcome | ORION-9 | ORION-10 | ORION-11 | |||
|---|---|---|---|---|---|---|
Inclisiran (N = 242) | Placebo (N = 240) | Inclisiran (N = 781) | Placebo (N = 780) | Inclisiran (N = 810) | Placebo (N = 807) | |
Primary outcomes | ||||||
Percent change in LDL-C from baseline to day 510a | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 151.4 (50.4) | 154.7 (58.0) | 104.5 (39.6) | 104.8 (37.0) | 107.2 (41.8) | 103.7 (36.4) |
LSM change from baseline, % (95% CI) | –39.7 (–43.7 to –35.6) | 8.2 (4.3 to 12.2) | –51.3 (–53.8 to –48.8) | 1 (–1.5 to 3.4) | –45.8 (–48.2 to –43.5) | 4 (1.76 to 6.3) |
LSM difference vs. control, % (95% CI) | –47.9 (–53.5 to –42.3) | –52.2 (–55.7 to –48.8) | –49.9 (–53.1 to –46.6) | |||
P value | < 0.0001 | < 0.0001 | < 0.0001 | |||
Time-adjusted percent change in LDL-C from day 90 to day 540b | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 151.4 (50.4) | 154.7 (58.0) | 104.5 (39.6) | 104.8 (37.0) | 107.2 (41.8) | 103.7 (36.4) |
LSM change from baseline, % (95% CI) | –38.1 (–41.0 to –35.1) | 6.2 (3.3 to 9.2) | –51.3 (–53.0 to –49.5) | 2.5 (0.77 to 4.25) | –45.8 (–47.5 to –44.1) | 3.4 (1.7 to 5.1) |
LSM difference vs. control, % (95% CI) | –44.3 (–48.5 to –40.1) | –53.8 (–56.2 to –51.3) | –49.2 (–51.6 to –46.8) | |||
P value | < 0.0001 | < 0.0001 | < 0.0001 | |||
Key secondary outcomes | ||||||
Mean absolute change in LDL-C from baseline to day 510 | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 151.4 (50.4) | 154.7 (58.0) | 104.5 (39.6) | 104.8 (37.0) | 107.2 (41.8) | 103.7 (36.4) |
LSM change from baseline, mg/dL (95% CI) | –59.0 (–64.8 to –53.2) | 9.9 (4.1 to 15.8) | –56.2 (–58.5 to –53.9) | –2.1 (–4.4 to 0.2) | 50.9 (–53.1 to –48.7) | 1.0 (–1.3 to 3.2) |
LSM difference vs. control, mg/dL (95% CI) | –68.9 (–77.1 to –60.7) | –54.1 (−57.4 to −50.9) | –51.9 (−55.0 to −48.7) | |||
P value | < 0.0001 | < 0.0001 | < 0.0001 | |||
Time-adjusted absolute change in LDL-C from day 90 to day 540 | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 151.4 (50.4) | 154.7 (58.0) | 104.5 (39.6) | 104.8 (37.0) | 107.2 (41.8) | 103.7 (36.4) |
LSM change from day 90, mg/dL (95% CI) | –56.6 (–61.0 to –51.2) | 6.2 (1.7 to 10.6) | –53.7 (–55.4 to –51.9) | –0.4 (–2.1 to 1.4) | –48.6 (–50.4 to –46.9) | 0.3 (–1.4 to 2.0) |
LSM difference vs. control, mg/dL (95% CI) | –62.7 (–69.0 to –56.5) | –53.3 (−55.8 to −50.8) | –48.9 (−51.4 to −46.5) | |||
P value | < 0.0001 | < 0.0001 | < 0.0001 | |||
CI = confidence interval; LDL-C = low-density-lipoprotein cholesterol; LSM = least squares mean; SD = standard deviation.
aData reported is from the prespecified washout model to account for missing data.
bData reported is from the prespecified control-based PMM to account for missing data.
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report,30 Raal FJ et al.,79 Ray KK et al. (2020).11
Harms
Refer to Table 14 for harms data.
Adverse Events
Heterozygous Familial Hypercholesterolemia
The incidence of AEs in patients treated with inclisiran and with placebo was 76.8% and 71.7% in the ORION-9 trial.
In the ORION-9 trial, the most common AEs in the inclisiran and placebo groups were nasopharyngitis (11.6% versus 8.3%), influenza (5.4% versus 8.8%), upper respiratory tract infection (6.6% versus 6.7%), and back pain (7.1% versus 4.2%).
nFH With ASCVD
The incidence of AEs was consistent in patients treated with inclisiran and those treated with placebo, as well as across trials, with 73.5% versus 74.8% patients experiencing at least 1 AE in the ORION-10 trial and 82.7% versus 81.5% experiencing at least 1 AE in the ORION-11 trial.
The most common AEs in the ORION-10 trial were diabetes mellitus (15.4% versus 13.9%), hypertension (5.4% versus 5.4%), back pain (5.0% versus 5.0%), bronchitis (5.9% versus 3.9%), and dyspnea (5.0% versus 4.2%). In the ORION-11 trial, the most common AEs were diabetes mellitus (10.9% versus 11.7%), nasopharyngitis (11.2% versus 11.2%), hypertension (6.5% versus 6.7%), and upper respiratory tract infection (6.4% versus 6.1%).
Serious Adverse Events
Heterozygous Familial Hypercholesterolemia
In the ORION-9 trial, 18 (7.5%) patients in the inclisiran arm and 33 (13.8%) patients in the placebo arm experienced at least 1 SAE. The most common SAEs were unstable angina, myocardial ischemia, acute MI, aortic valve stenosis, and back pain.
nFH With ASCVD
In the ORION-10 trial, 175 (22.4%) patients in the inclisiran arm and 205 (26.3%) patients in the placebo arm experienced at least 1 SAE. The most common SAEs were CAD, cardiac failure (congestive), acute MI, pneumonia, and noncardiac chest pain. In the ORION-11 trial, 181 (22.3%) patients in the inclisiran arm and 181 (22.5%) patients in the placebo arm experienced at least 1 SAE. The most common SAEs were angina pectoris, acute MI, unstable angina, CAD, and atrial fibrillation.
Withdrawal Due to Adverse Events
Heterozygous Familial Hypercholesterolemia
In the ORION-9 trial, 3 (1.2%) patients in the inclisiran group withdrew due to an AE. No patients in the placebo group withdrew due to an AE. ███████ ███████ ██ ██████████ ████ █████ ████ ████ ████████ ███████ █████████ ████ █████████ ███ ███ ████████ ███ ██████ ████ ██ ███ ████████ █████ ███ ███ █████████ ████ ████████ ██████ ████ ██████████ ███████████ ███████ ██ █████████.
nFH With ASCVD
The incidence of WDAEs in the ORION-10 trial was similar across groups, at 2.4% (19 patients) in the inclisiran group and 2.2% (17 patients) in the placebo group. ███ ████ ██████ ███ ██ ████ ███████████ ███ ███████████████ ████████ ███████ ██ ██████████ ████ ██████ █████████ ███ ██ ██████████ █████████████ ███ ████████ ███████████████ ███ ██████████ ██████ █████████ █████ ███ ██████ █████████ █████ ███ ██████ ███████ ██████ █████████ █████ ███ ██████ ███ ███████ █████████ ███ ███ █████████ █████ ██ ███ ██████ ████ █████ ████ ███ ███████ ██ █████████ ██ ██████ ██ ███ █████████████
The incidence of WDAEs in the ORION-11 trial was similar to that in the ORION-10 trial, with 23 (2.8%) and 18 (2.2%) patients in the inclisiran group and the placebo group, respectively, experiencing AEs that led to withdrawal. ███████ ██████ ██ ███ ██████████ ███ ███████ ██████ ███████ ██ ██████████ ██ █████████ ████████ ███████ █████████ █████ ███ ██████ ████████████████ █████████ ███ ███████ █████████ █████ ██████ ███████████████ ███ ██████████ ██████ █████████ █████ ███ ██████ ███ █████████ █████ ███ ██████
Mortality
Heterozygous Familial Hypercholesterolemia
In the ORION-9 trial, only 2 deaths occurred (0.4%), 1 in each trial arm.
nFH With ASCVD
A total of 23 patients died during the ORION-10 study, 12 (1.5%) in the inclisiran group and 11 (1.4%) in the placebo group. In total, 29 (1.8%) patients died during the ORION-11 study, 14 (1.7%) in the inclisiran group and 15 (1.9%) in the placebo group.
Notable Harms
Heterozygous Familial Hypercholesterolemia
██████████ ██████ ███ ██████████████ █████████ ████████ ██ █ ██████ ███ █ ██████ ████████ ██ ███ ██████████ ███ ███████ ██████ ██ ████████ The incidence of AEs at the injection site in the inclisiran and placebo groups was 17.0% (41 patients) versus 1.7% (4 patients) in the ORION-9 trial. Injection-site reactions were mild to moderate, and no severe reactions were seen. One patient withdrew from the study due to an injection-site reaction in the inclisiran group of the ORION-9 trial. ██ ████████ ████████ █████████ █████████ ███ ███ ████████████████ █████████ ████ ██████████ ████ ██ ████████ ██ █████ ██████████████ ███ █████████ ██ ████████████████ █████████ ██ ███████████ ███ ███████████████ ████████ ███ ██ ██████ ███ ██ ██████ ██ ████████ ███ █████████ ██ █████ ██████ ██ ███ ██████████ ███ █████████████ ██ ███████ ███ █ ██████ ███ █ ███████ ███ █████████ ██ ███████ ██████ ███ ██ █ ██████ ███ █ ██████ ██ ███████████ ███ ███████████████ ████████ ██ ████████ ███ ██████ ████ ████ ██ ████████.
nFH With ASCVD
██████████ ██████ ███ ██████████████ █████████ ████████ ██ ██ ██████ ███ ██ ██████ ████████ ██ ███ ██████████ ███ ███████ ██████ ██ █████████ ███ ██ ██████ ██ ██ ██████ ████████ ██ █████████ The incidence of AEs at the injection site in the inclisiran and placebo groups was 6.0% (47 patients) and 1.9% (15 patients), respectively, in the ORION-10 trial, and was 7.6% (62 patients) and 1.7% (14 patients), respectively, in the ORION-11 trial. Injection-site reactions were mild to moderate, and no severe reactions were seen across trials. One patient withdrew from the study due to an injection-site reaction in the inclisiran group in the ORION-10 trial, while 2 patients in the inclisiran group of the ORION-11 trial withdrew from the study due to injection-site reactions. ██ ████████ ████████ █████████ █████████ ███ ███ ████████████████ █████████ ████ ██████████ ████ ██ ████████ ██ █████ ██████████████ ███ █████████ ██ ████████████████ █████████ ██ ███████████ ███ ███████████████ ████████ ███ ██ ██████ ███ ██ ██████ ██ █████████ ███ ██ ██████ ███ ██ ██████ ██ █████████ ███ █████████ ██ █████ ██████ ██ ███ ██████████ ███ ███████ ██████ ██ ████████ ███ ███ ████ ██ ██████ ███ ██ ███████ ███ ██ ██████ ███ ██ ███████ █████████████ ████████ █████ ███ ███████ █████ ████ ██ ███ ███████ ██████ ███ ████ ██ ███ ██████████ ██████ ███ █████████ ██ ███████ ██████ ███ ██ ██████ ███ ██ ███████ ███ ██ ██████ ███ ██ ██████ ██ ███████████ ███ ███████████████ ████████ ██ █████████ ███ ████ █████████████ ███ ██████ ████ ████ ██ █████████
Table 14: Summary of Harms Results From the Studies Included in the Systematic Review
Harms | ORION-9 | ORION-10 | ORION-11 | |||
|---|---|---|---|---|---|---|
Inclisiran (N = 241) | Placebo (N = 240) | Inclisiran (N = 781) | Placebo (N = 778) | Inclisiran (N = 811) | Placebo (N = 804) | |
n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
Adverse events | ||||||
Patients with ≥ 1 AE | 185 (76.8) | 172 (71.7) | 574 (73.5) | 582 (74.8) | 671 (82.7) | 655 (81.5) |
Most common AEa | ||||||
Nasopharyngitis | 28 (11.6) | 20 (8.3) | 21 (2.7) | 24 (3.1) | 91 (11.2) | 90 (11.2) |
Injection-site reaction | 22 (9.1) | 0 (0) | ██ █████ | █████ | ██ █████ | ███ |
Back pain | 17 (7.1) | 10 (4.2) | 39 (5.0) | 39 (5.0) | 27 (3.3) | 28 (3.5) |
URTI | 16 (6.6) | 16 (6.7) | 37 (4.7) | 38 (4.9) | 52 (6.4) | 49 (6.1) |
Influenza | 13 (5.4) | 21 (8.8) | ██ | ██ | 19 (2.3) | 20 (2.5) |
Bronchitis | 9 (3.7) | 4 (1.7) | 46 (5.9) | 30 (3.9) | 23 (2.8) | 16 (2.0) |
Hypertension | 9 (3.7) | 8 (3.3) | 42 (5.4) | 42 (5.4) | 53 (6.5) | 54 (6.7) |
Arthralgia | 9 (3.7) | 7 (2.9) | 35 (4.5) | 33 (4.2) | 47 (5.8) | 32 (4.0) |
Diabetes mellitus | █████ | █████ | 120 (15.4) | 108 (13.9) | 88 (10.9) | 94 (11.7) |
Osteoarthritis | █████ | ████ | ██ ██ | ██ █ | 32 (3.9) | 40 (5.0) |
Dyspnea | █████ | █████ | 39 (5.0) | 33 (4.2) | ██ █████ | █████ |
Serious adverse events | ||||||
Patients with ≥ 1 SAE | 18 (7.5) | 33 (13.8) | 175 (22.4) | 205 (26.3) | 181 (22.3) | 181 (22.5) |
Most common SAEb | ||||||
CAD | ██ | ██ | 15 (1.9) | 22 (2.8) | 8 (1.0) | 11 (1.4) |
Cardiac failure, congestive | ██ | ██ | 7 (0.9) | 20 (2.6) | █████ | █████ |
Acute MI | 2 (0.8) | 1 (0.4) | 14 (1.8) | 12 (1.5) | 5 (0.6) | 18 (2.2) |
Unstable angina | 1 (0.4) | 4 (1.7) | 4 (0.5) | 10 (1.3) | 11 (1.4) | 11 (1.4) |
Myocardial ischemia | 1 (0.4) | 3 (1.3) | ██ | ██ | █████ | █████ |
Pneumonia | ██ | ██ | 11 (1.4) | 9 (1.2) | 9 (1.1) | 7 (0.9) |
Noncardiac chest pain | ██ | ██ | 10 (1.3) | 9 (1.2) | 4 (0.5) | 8 (1.0) |
Atrial fibrillation | ██ | ██ | 10 (1.3) | 8 (1.0) | 10 (1.2) | 6 (0.7) |
COPD | ██ | ██ | 8 (1.0) | 8 (1.0) | ██ | ██ |
Angina pectoris | ██ | ██ | █████ | ████ | 14 (1.7) | 13 (1.6) |
Occlusive PAD | ██ | ██ | ██ | ██ | 7 (0.9) | 8 (1.0) |
WDAE | ||||||
Incidence of WDAEs (SOC) | 3 (1.2) | 0 (0) | 19 (2.4) | 17 (2.2) | 23 (2.8) | 18 (2.2) |
███████ █████ | ██ | ██ | ███ | ███ | ███ | ███ |
███████ █████ | ██ | ██ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
Notable harms | ||||||
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
Patients with at least 1 AE at the injection siteb | 41 (17.0) | 4 (1.7) | 47 (6.0) | 15 (1.9) | 62 (7.6) | 14 (1.7) |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
███████ █████ | ███ | ███ | ███ | ███ | ███ | ███ |
AE = adverse event; ALP = alkaline phosphatase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CAD = coronary artery disease; COPD = chronic obstructive pulmonary disease; E = event count; GFR = glomerular filtration rate; GGT = gamma glutamyl transpeptidase; INR = international normalized ratio; MI = myocardial infarction; NR = not reported; PAD = peripheral artery disease; SAE = serious adverse event; SOC = system organ class; TEAE = treatment-emergent adverse event; URTI = upper respiratory tract infection; WDAE = withdrawal due to adverse event.
aOccurring in ≥ 5% of patients in one group.
bOccurring in ≥ 1% of patients in one group.
cOccurring in ≥ 0.5% of patients in one group.
Critical Appraisal
Internal Validity
The major piece of new evidence for the systematic review portion of this resubmission was a posthoc pooled analysis of MACE from the ORION-9, ORION-10, and ORION-11 trials. The main issues with this type of analysis are that MACE and its components were only an exploratory outcome from these ORION trials, it is a posthoc analysis, and the pooled analysis that includes all 3 ORION trials mixes the HeFH population from the ORION-9 trial with the nFH with ASCVD populations from ORION-10 and ORION-11 trials. The fact that MACE and its components was an exploratory outcome in these ORION trials also introduces the potential for bias. Sample sizes were not determined based on these outcomes; events were captured via the safety population and definitions may not be inclusive or specific enough; there was no blinded, centralized assessment of events; and the timing was likely insufficient to assess CV events. The use of a posthoc analysis introduces significant potential for bias, as an investigator may be biased by their ability to review the data when deciding what analyses to conduct and how to construct the composite outcome. For example, it is clear from the pooled analysis that an improvement for inclisiran versus placebo is driven entirely by the ORION-10 and ORION-11 trials, whereas there does not appear to be any difference in risk of MACE between the inclisiran and placebo groups in the ORION-9 trial. Therefore, pooling all 3 trials leads 1 to the erroneous conclusion that the risk of MACE was reduced with inclisiran treatment across the ORION-9, ORION-10, and ORION-11 trials, when that was not the case. Finally, combining results from all 3 ORION trials is not appropriate, as this ignores the fact that these trials feature 2 distinct populations, each separately identified within the indication. Additionally, there may even be issues with pooling the ORION-10 and ORION-11 trials, as there are some differences in baseline characteristics between these 2 study populations, most notably that all patients in the ORION-10 trial had ASCVD, whereas approximately 88% of patients in the ORION-11 trial had ASCVD, with the remaining categorized as ASCVD-RE. There was also a higher percentage of patients who discontinued treatment in the ORION-10 trial than in the ORION-11 trial, further reinforcing that these are, indeed, 2 distinct study populations. It is not clear whether the sponsor took steps to adjust for these sources of heterogeneity.
ORION-9, ORION-10, and ORION-11 were all phase III, double-blind, RCTs. Appropriate methods for randomization (using IRT), treatment allocation (stratified by country and the current use of statins or other LLTs in block sizes of 4), and maintenance of blinding to treatment assignment were used in all 3 trials, reducing selection, performance, and detection biases. ████████ ██ ███ ████ ███████ █████ ██ █████ █████████ ██ ███ ████████ █████ ███ █████ ████ ███ ███████ ███ ███ ███████ ███████ ███ █████ ███ ███████ ██ ██ ███████ ██ ██ ███ █████ ████████ ████████ ███ ███████ █████ ██ ███ ████████ ██████ There were no notable differences in baseline characteristics in the studies, and there was no imbalance in discontinuations, suggesting that attrition bias was limited. Moreover, unblinding was only permitted in the event of an emergency or AE for which it was necessary to know the study drug to determine an appropriate course of therapy. Injection-site reactions are known complications of PCSK9 inhibitors and were more frequent in the inclisiran groups across trials, which could have revealed treatment assignment, but overall, reactions were not common, and the effect they would have on the results and unblinding is unclear.
The primary analyses of the ORION trials were conducted in the ITT population. Low dropout rates were seen in all trials; however, the total number of missing data was not reported for any outcomes in the trials, and therefore the extent of missing data in each group at various time points and for each key outcome is unknown. Efforts were made to reduce the amount of missing data, including diligent follow-up. Missing data were imputed in the coprimary end point of percent change in LDL-C from baseline to day 510 using a multiple imputation washout model on actual values with baseline and observed efficacy measures, and the current use of statins or other LLTs as covariates. Missing values in the inclisiran group were imputed under the assumption that their outcome was similar to those in the placebo group with similar background characteristics. This method may be subject to bias, resulting in greater treatment effects in favour of inclisiran. For patients in the inclisiran group only missing day 510 data, values were imputed. For patients in the placebo group, missing values over all visits after early termination were imputed based on the MAR assumption. Again, this may impact the direction of the treatment effect in favour of inclisiran over time. Results of the coprimary and secondary outcomes were consistently large, with minimal differences between observed and imputed values. Numerous sensitivity analyses, with and without multiple imputations, were also employed. Therefore, it is unlikely that missing data would have impacted the LSM percent change in LDL-C.
The prespecified power and sample-size calculation for all ORION trials was identical and was based on the assumption that the difference in change from baseline in LDL-C between the inclisiran and placebo groups would be no less than 30 mg/dL, with a standard deviation of 20 mg/dL; however, the enrolled populations were much higher than the power and sample-size calculation defined. ███████████ ██████ █████ ████ █████████ ███ ████████ ███ ████████ ██ ████ ███ █████ ██████ ████████ █████████████ █████ ████ ████ ████████ ██ ██████ ████ ███ ██ ███████ ██████████ █ ███████ ██ ████ ███████████ ████ ██████████ ████████ ███████ ████████ ███ ████ ███ █████ ██████ ████ ██ ████ █████ ███ ██████ ██ ███ ██████████ ██ ██████████ ██████ ████ ██████ ████ ██████ █████████ The additional enrolled patients in the ORION trials could have led to an overpowering of study results, in which the higher number of patients enrolled could have increased the probability of seeing minuscule differences between groups. However, overpowering is unlikely to have affected the results of the ORION studies, given the large differences in efficacy observed between inclisiran and placebo and given the request by the FDA, so any ethical and resource allocation issues are of no concern for the ORION trials. The higher sample sizes contributed additional safety information for all trials; however, secondary outcomes and CV-related events considered to be of interest to this review were not accounted for.
Acceptable methods to account for multiplicity were used in all trials. The coprimary efficacy outcome was controlled for multiplicity using the familywise error rate and a nested testing procedure, first on the percent change in LDL-C from baseline to day 510, and then on the time-adjusted percentage change in LDL-C from baseline to the period from after day 90 and up to day 540. The Hochberg procedure was applied for key secondary end points.83-85 Other secondary end points and exploratory end points were not controlled for multiplicity, nor for missing data, including the composite outcome of MACE, which was considered most clinically important to this review. The proportion of patients reaching global lipid targets (LDL-C < 100 mg/dL or < 70 mg/dL) was a secondary end point that was also not controlled for multiplicity or missing data. The sponsor’s evaluation of these outcomes was conducted on the ITT population; however, there was a discrepancy between the number of patients in the ITT population and the reported number of patients at day 510, with the proportion of patients missing from the analysis ranging from 5% to 15% across trials. The resulting missing patients inflated the proportion of patients achieving global lipid targets in both the inclisiran and placebo arms.13,24,37
Subgroup analyses for efficacy and safety were prespecified in the statistical analysis plan, and missing data were accounted for using the MMRM method but were not adjusted for multiplicity. CIs for most subgroup analyses suggested precision; however, subgroups with a lower number of patients had wider, more imprecise CIs. Subgroups based on risk status (ASCVD or ASCVD-RE) were conducted for the ORION-9 and ORION-11 trials; however, the ASCVD-RE subgroup in the ORION-11 trial was not considered to be of interest, given that it was mostly made up of patients who were not part of the population for which reimbursement was requested by the sponsor (only 11 patients had HeFH and the rest had ASCVD-RE).
External Validity
The ORION-9, ORION-10, and ORION-11 trials aimed to enrol patients with ASCVD and/or ASCVD-RE and specific serum LDL-C and triglyceride cutoffs. ██ ████████ █████ ██ ████████ ████ ██████ █████████ ███ ███ ██ ███████ █████████████ ███ ████ █████ ██ ████████ ███ ████ █████ ███ █████ ██ ██████ ████████ ███ ███ ████ ███ ███████████ █████ █████ █████████ Screening failures and inclusion criteria were considered appropriate for the ORION-10 and ORION-11 trials, given the specified LDL-C cut-points of 1.8 mmol/L and 2.6 mmol/L, which are aligned with current CCS guidelines, and therefore the threat of sampling bias is estimated to be low, given the eligibility criteria. The included patient populations in the ORION studies were mostly reflective of the funding request; however, with the exception of the ORION-10 trial, the ORION trials were multinational trials, and the ORION-9 study was the only study that enrolled patients in Canada (n = 23). The proportion of patients receiving high-intensity statins at baseline was as expected in the HeFH population in the ORION-9 trial (73.9%), as well as in the ASCVD populations in the ORION-10 trial (69.4%) and in the ORION-11 trial (78.0%); however, the clinical expert believed that more patients with HeFH would be receiving ezetimibe than what was seen in the ORION-9 trial (52.3%). Partial or complete intolerance to statins at baseline ranged from 11.4% to 25.3%, which is in line with the proportion of 15% to 20% estimated by the clinical expert. ███ ██████████ ██ ████████ ██ ████ █████ ████ █████ ██ ██ ██████ ████ ████ ████████ ██████ ███████ ███ ███ ████████ ██ ██████ ████████ ███████ ████ ███ ████ █████ ██ ███ ██████████ ██ ███ ██████ The ORION trials excluded patients who experienced MACE in the 3 months before randomization, and the clinical experts believed this made sense, given that the primary outcome was LDL-C, not MACE; given the high risk of recurrence in the short-term (6 months), excluding these patients would eliminate this potential confounder. Additionally, 1 clinical expert believed it to be debatable whether LLT this soon after an event would have any impact on the subsequent risk of clinical events in the short-term. The inclusion criteria for the ORION-9, ORION-10, and ORION-11 studies excluded patients who were currently receiving treatment with PCSK9 monoclonal antibodies or who had received them in the 90 days before screening, and the baseline characteristics of patients in the ORION trials did not include prior treatment with PCSK9 inhibitors. The product monograph for inclisiran states that when transitioning from a PCSK9 inhibitor to inclisiran, the last dose of the PCSK9 inhibitor should be delivered and then, at the next scheduled date, inclisiran can be administered. The effect of switching from other PCSK9 inhibitors to inclisiran on the reduction in LDL-C, CV-related morbidity, and mortality remains uncertain. Despite this, and according to the clinical expert consulted by CDA-AMC, the baseline demographic and medical characteristics of the ORION trials reflect the HeFH and ASCVD populations expected to use inclisiran in Canada.
All ORION trials were placebo-controlled trials and did not include an active comparator, which allows for adequate evaluation of the treatment effect of inclisiran but may overestimate the treatment effects. A demonstration of comparative effectiveness to another PCSK9 inhibitor would have allowed for better interpretation of the efficacy results. Despite this, the clinical expert consulted by CDA-AMC noted that the incremental improvements in LDL-C and the differences between the inclisiran and placebo groups are still clinically meaningful, given that in both the clinical trial and real-world settings, patients with HeFH and/or ASCVD are heavily treated with background medications (e.g., statins, ezetimibe, blood pressure medication, diabetes medications), and therefore the differences seen were still notable. Given that there were no direct comparisons between inclisiran and alirocumab or evolocumab, the 2 available PCSK9 inhibitors available in Canada, the sponsor provided an ITC to address this gap.
The outcomes used to assess the efficacy of inclisiran were chosen based on validated laboratory assessments of lipids, are considered widely accepted surrogates for clinically relevant outcomes, and are reflective and important in guiding treatment decisions in Canadian clinical practice. The duration of the trial (18 months) was also considered appropriate for assessing these outcomes over time, given that trials for PCSK9 inhibitors of alirocumab and evolocumab were 12 weeks to 24 weeks in duration, and effects on lipids are rapidly seen.13 However, the included studies were not designed to assess important CV-related outcomes, including reductions in MACE and all-cause and CV-related mortality, as these outcomes were exploratory and not powered for statistical analysis. Across trials, there were no differences between groups and, therefore, there was no cause for concern with these outcomes. However, the impact of inclisiran on these outcomes remains uncertain, and the duration of the trial was considered too short for assessing reductions in these outcomes. Moreover, the product monograph for inclisiran states that the effect of inclisiran on CV morbidity and mortality has not been determined.17
No HRQoL or patient-reported outcomes were assessed in the ORION trials, and therefore the effect of inclisiran with respect to these outcomes remains unknown.
Long-Term Extension Studies
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
The ORION-3 Trial31,32
Description of Studies
ORION-331,32 is a 4-year, open-label, multicenter extension study of the phase II, 1-year ORION-1 study that was conducted across 52 study sites in 5 countries. The primary objective of this study was to assess the effect of long-term treatment with twice-yearly siRNA therapeutic inclisiran dosing on LDL-C reductions at day 210 compared to baseline. The secondary and exploratory objectives were to assess the effects of inclisiran on cholesterol levels, other lipids levels, and PCSK9 levels up to 4 years in each arm, as well as the long-term safety and tolerability of inclisiran. An additional exploratory objective was to evaluate the effects of transitioning from evolocumab to inclisiran. A total of 382 participants were enrolled from 52 centres in 5 countries: Canada (11), Germany (5), Netherlands (14), the UK (14), and the US (8). In total, 56 patients were enrolled in the study from Canadian centres.
Populations
Patients 18 years and older with prevalent ASCVD or high-risk primary prevention and elevated LDL-C despite MTD statins or other LDL-lowering treatments, or with documented statin intolerance, who had completed 1 year of observation in the ORION-1 trial were eligible for this study. Patients were excluded if they had any uncontrolled or serious medical or surgical condition that reduced life expectancy, had received or were receiving current treatment with a PCSK9 inhibitor, or had recent or planned use of any investigational medicinal products other than inclisiran.
Interventions
Patients who received inclisiran in the ORION-1 trial received twice-yearly 300 mg SC inclisiran sodium throughout the ORION-3 trial (inclisiran-only arm), whereas those who received placebo in the ORION-1 trial received SC evolocumab 140 mg every 2 weeks until day 360 and then transitioned to twice-yearly inclisiran for the remainder of the ORION-3 study (switching arm). Inclisiran was administered as a single SC injection of 300 mg of inclisiran sodium in a 1.5 mL solution by a health care professional at the study site. Evolocumab was self-administered by the patient as a single 1.0 mL SC injection, using a single-use autoinjector containing 140 mg of evolocumab.
Outcomes
The primary efficacy end point was the percentage change in LDL-C with inclisiran from baseline of the ORION-1 trial to day 210 of the ORION-3 trial in the inclisiran-only arm. Secondary and exploratory end points included changes in LDL-C and other lipid parameters up to 4 years in each arm, in addition to individual responses to inclisiran and safety and tolerability outcomes (AEs, SAEs, WDAEs, notable harms).
Statistical Analysis
There was no formal sample-size calculation for this trial. Descriptive statistics, reported as counts and proportions, were used to summarize data. Missing data were not imputed. Patients missing any data required for computing the end point were excluded from the analysis.
As a posthoc analysis, the mean percentage change from the ORION-1 trial baseline in LDL-C averaged over time for the mITT population in the inclisiran-only arm was analyzed for the entire 4-year duration of the ORION-3 trial and for each of the 4 years. The average over time was calculated as the arithmetic mean of the LSM at each study visit, estimated using a MMRM, which included study visits as the fixed effect. For the switching-arm ITT population, the same was analyzed from the ORION-3 trial baseline for year 1, the 3-year period from year 2 to year 4, and each of the previous 3 years. Analyses for the inclisiran-only arm were performed in the ITT and the mITT populations; mITT was the primary population used for presentation whenever available. All patients who received at least 1 dose of inclisiran were included in the safety analysis.
Results
Baseline Characteristics
Baseline characteristics of the inclisiran-only and switching-arm groups in the ORION-3 trial are comparable and are summarized in Table 15.
Table 15: Summary of Baseline Characteristics in the ORION-3 Study
Patient disposition | Inclisiran only (N = 290) | Switching arm (N = 92) |
|---|---|---|
Age, years, mean (SD) | 63.3 (11.1) | 61.9 (10.6) |
Age ≥ 65 years, n/N (%) | 145/290 (50) | 39/92 (42) |
Female, n/N (%) | 102/290 (35) | 37/92 (40) |
Male, n/N (%) | 188/290 (65) | 55/92 (60) |
Race, n/N (%) | ||
White | 271/289 (94) | 85/91 (93) |
Black or African American | 10/289 (3) | 5/91 (5) |
American Indian or Alaska Native | 3/289 (1) | 1/91 (1) |
Asian | 4/289 (1) | 0/91 (0) |
Country, n/N (%) | ||
Canada | ██████ ███████ | █████ ███████ |
Germany | ██████ ███████ | █████ ███████ |
Netherlands | ███████ ███████ | █████ ███████ |
UK | ██████ ███████ | █████ ███████ |
US | ██████ ███████ | █████ ███████ |
BMI | ||
n | 286 | 91 |
Mean, kg/m2 (SD) | 29.1 (5.6) | 29.7 (4.9) |
Baseline eGFR, mL/min per 1.73 m2 | ||
n | 284 | 90 |
mean (SD) | 79.4 (19.0) | 81 (18.5) |
Baseline efficacy evaluationsa | ||
n | 277 | 77 |
PCSK9, mcg/L, mean (SD) | 432.7 (141.8) | 413.1 (130.7) |
n | 277 | 90 |
LDL-C, mmol/L, mean (SD) | 3.33 (1.30) | 3.17 (1.35) |
Medical history and comorbiditiesb n/N (%) | ||
Hypertension | 187/283 (66) | 61/90 (68) |
Family history of coronary artery disease | 147/257 (57) | 52/82 (63) |
Family history of dyslipidemia | 122/231 (53) | 46/76 (61) |
Diabetes mellitus | 69/283 (24) | 18/89 (20) |
ASCVD | 185/284 (65) | 62/87 (71) |
ASCVD risk equivalent | 99/284 (35) | 25/87 (29) |
ASCVD = atherosclerotic cardiovascular disease; BMI = body mass index; eGFR = estimated glomerular filtration rate; ITT = intention to treat; LDL = low-density-lipoprotein; mITT = modified intention to treat; SD = standard deviation.
aFor the inclisiran-only arm, the efficacy baseline is for the mITT population; for the switching arm, the efficacy baseline is for the ITT population. The inclisiran-only arm uses the ORION-1 trial baseline and the switching arm uses the ORION-3 trial baseline.
bIn the safety population.
Sources: ORION-3 Clinical Study Report,31 Ray KK et al. (2023).32
Patient Disposition
Patient disposition in the treatment arms of the ORION-3 trial is summarized in Table 16. Based on data published for the ORION-1 trial,86 of the 483 patients who completed the ORION-1 trial, 382 (79.1%) were enrolled in the ORION-3 trial. Of the 290 patients enrolled in the inclisiran-only arm, 233 (80.3%) completed the study. The primary reason for discontinuation was withdrawal of consent, by 25 (8.6%) patients. Of the 92 patients enrolled in the switching arm, 80 (87.0%) completed the study. The primary reason for discontinuation was also the withdrawal of consent, by 5 (5.4%) patients.
Patient disposition | Inclisiran only (N = 290) | Switching arm (N = 92) |
|---|---|---|
Completed study, N (%) | 233 (80.3) | 80 (87.0) |
Discontinued study, n (%) | 57 (19.7) | 12 (13.0) |
Withdrew consent | 25 (8.6) | 5 (5.4) |
█████████ ████ | █████ | █████ |
████ ██ ███████ | █████ | █████ |
█████ | █████ | █████ |
███████ █████ | █████ | █████ |
██████████ | █████ | █████ |
██████ | █████ | █████ |
ITT, N | 290 | 92 |
mITT, N | 277 | 88 |
Safety population, N | 284 | 90 |
Safety population + patients switched to inclisiran, N | 284 | 87 |
ITT = intention to treat; mITT = modified intention to treat.
Sources: ORION-3 Clinical Study Report,31 Ray KK et al. (2023).32
Concomitant Medications and Cointerventions
The following medications were permitted during the study: hormone replacement therapy, lipid-lowering medications, prescription medications for the treatment of preexisting medical conditions, and medications to treat an AE at the discretion of the investigator.
There were no required concomitant medications and/or treatments in the ORION-3 trial; however, to be eligible for enrolment in the preceding ORION-1 study, participants must have either been receiving a stable dose of a statin or other LLT. Accordingly, 76% of patients in the inclisiran-only group and 78% of patients in the switching arm were taking a LLT at baseline (Table 17)
Table 17: Summary of LLT Usage During the ORION-3 Trial
Patient disposition | Inclisiran only (N = 284) | Switching arm (N = 90) |
|---|---|---|
Baseline concomitant LLT in the safety population | ||
Any lipid-lowering therapy, n/N (%) | 216/284 (76) | 70/90 (78) |
Any statin therapy, n/N (%) | 186/284 (66) | 63/90 (70) |
████ █████████ | ███ ██████ | ██ ██████ |
████████ █████████ | ██ ██████ | ██ ██████ |
███ █████████ | █████ | █████ |
█████████████ | █████ | █████ |
███████ ████ ███████ | ███ ██████ | ██ ██████ |
█████ ███ | ███ ██████ | ██ ██████ |
█████ ███ ████ ███████ | █████ | █████ |
New or changed concomitant LLT in the safety populationa | ||
██ ███ ███████████ ██ | ██ ██████ | ██ ██████ |
██ ███ ███████████ ██ | █████ | █████ |
██ ███ ███████████ ██ | █████ | █████ |
██ ███ ███████████ ██ | █████ | █████ |
██ ███ ███████████ ██ | █████ | █████ |
██ ███ ███████████ ██ | █████ | █████ |
██ ███ ███████████ ██ | █████ | █████ |
██ ███ ███████████ ██ | █████ | █████ |
██ ███ ███████████ ██ | █████ | █████ |
LLT = lipid-lowering therapy.
aNew or changed concomitant LLT is defined as any LLT with a start date after day 1 of the ORION-3 trial.
Source: ORION-3 Clinical Study Report.31
Exposure to Study Treatments
There were 1,045.5 PYs of exposure to inclisiran in the inclisiran-only arm during the open-label extension phase, resulting in total cumulative exposure to inclisiran from the beginning of the ORION-1 trial through to the end of the ORION-3 trial of 1,209.6 PYs. The exposure to inclisiran in the switching arm was 250.2 PYs (Table 18). ██ ███████ ██ ███████ █████████ ██ ██████ ███ ████ █████████
Table 18: Patient Exposure (Safety Population Plus Patients Switched to Inclisiran)
Patient disposition | Inclisiran only (N = 284) | Switching arm (N = 87) |
|---|---|---|
Total, patient-years | 1,045.5 | 250.2 |
█████████ ████ ████ | ██████ ████████ | ██████ ████████ |
█████████ ██████ █ | ██████ ██████ █████ | ██████ ██████ █████ |
█████████ | ██ | ██ |
IQR = interquartile range; NR = not reported; SD = standard deviation.
Sources: ORION-3 Clinical Study Report,31 Ray KK et al. (2023).32
Efficacy
Percentage Change in LDL-C From Baseline of the ORION-1 Trial to Day 210 of the ORION-3 Trial for the Inclisiran-Only Arm
In the inclisiran-only arm, LDL-C concentrations at day 210 were decreased by 1.56 mmol/L (95% CI, –1.68 to –1.44 mmol/L), reflecting a 47.5% reduction (95% CI, –50.7% to –44.3%) (Table 19). This result was observed approximately 570 days after the first inclisiran exposure in the ORION-1 trial.
Table 19: Change in LDL-C From Baseline in the ORION-1 Trial to Day 210 in the ORION-3 Trial for the Inclisiran-Only Arm
Patient disposition | Inclisiran only (N = 290) |
|---|---|
Number of patients contributing to the analysis | 277 |
Change from baseline, mmol/L (95% CI) | –1.56 (–1.68 to –1.44) |
Change from baseline, % (95% CI) | –47.5% (–50.7 to –44.3) |
P value | < 0.0001 |
CI = confidence interval; LDL-C = low-density-lipoprotein cholesterol.
Sources: ORION-3 Clinical Study Report,31 Ray KK et al. (2023).32
Secondary Efficacy End Points
Percentage and Absolute Change in LDL-C From Baseline
During the 4 years of the open-label extension, the mean percentage change and mean absolute change in LDL-C concentrations in the inclisiran-only arm ranged between –34.3% and –53.8%, and between –1.13 mmol/L and –1.76 mmol/L, respectively, with the upper limit of the 95% CI at all time points being lower than –30% and excluding 0 (Table 20). The highest reduction was observed for the day 870 to day 900 mean percentage change, at –53.81%.
The mean percentage change and mean absolute change in LDL-C in the switching arm ranged between –38.2% and –65.7%, and between –1.20 mmol/L and –2.00 mmol/L, respectively.
Table 20: Secondary and Exploratory Efficacy End Points of the ORION-3 Trial (Percentage and Absolute Change in LDL-C From Baseline by Treatment Arm and Visit Daya)
Day | N | Inclisiran onlyb (N = 277) percentage change, % (95% CI) | Inclisiran onlyb (N = 277) absolute change, mmol/L (95% CI) | N | Switching armb (N = 92) percentage change, % (95% CI) | Switching armb (N = 92) absolute change, mmol/L (95% CI) |
|---|---|---|---|---|---|---|
30 | 277 | −49.4 (−52.3 to −46.6) | −1.62 (−1.73 to −1.51) | 90 | −65.1 (−68.7 to −61.4) | −1.98 (−2.13 to −1.82) |
90 | 276 | −44.2 (−47.1 to −41.2) | −1.44 (−1.55 to −1.34) | 90 | −65.7 (−70.3 to −61.2) | −2.00 (−2.19 to −1.81) |
180 | 277 | −34.3 (−37.6 to −31.0) | −1.13 (−1.24 to −1.02) | 90 | −64.4 (−68.4 to −60.3) | −2.00 (−2.19 to −1.81) |
210 | 277 | −47.5 (−50.7 to −44.3) | −1.56 (−1.68 to −1.44) | 88 | −63.6 (−67.7 to −59.6) | −1.95 (−2.13 to −1.77) |
270 | 274 | −42.6 (−46.4 to −38.9) | −1.42 (−1.55 to −1.30) | 86 | −60.2 (−65.4 to −55.0) | −1.88 (−2.09 to −1.67) |
360 | 269 | −37.1 (−41.0 to −33.3) | −1.24 (−1.36 to −1.12) | 87 | −47.8 (−54.1 to −41.4) | −1.47 (−1.69 to −1.25) |
390 | 266 | −49.8 (−53.2 to −46.4) | −1.63 (−1.75 to −1.51) | 84 | −58.6 (−63.2 to −53.9) | −1.75 (−1.94 to −1.56) |
450 | 262 | −47.2 (−50.7 to −43.7) | −1.55 (−1.67 to −1.43) | 85 | −43.4 (−48.9 to −37.8) | −1.32 (−1.50 to −1.14) |
510 | NA | NA | NA | 81 | −49.1 (−53.4 to −43.8) | −1.45 (−1.62 to −1.28) |
540 | 264 | −38.2 (−41.9 to −34.5) | −1.26 (−1.38 to −1.15) | 82 | −48.6 (−53.4 to −44.3) | −1.45 (−1.61 to −1.28) |
570 | 264 | −48.0 (−51.1 to −44.9) | −1.59 (−1.71 to −1.48) | NA | NA | NA |
630 | 261 | −47.4 (−50.8 to −43.9) | −1.55 (−1.67 to −1.43) | 83 | −41.6 (−46.6 to −36.6) | −1.29 (−1.47 to −1.11) |
720 | 253 | −38.4 (−41.8 to −35.1) | −1.28 (−1.40 to −1.15) | 82 | −46.4 (−50.6 to −42.1) | −1.42 (−1.59 to −1.24) |
810 | 242 | −48.4 (−51.9 to −44.8) | −1.57 (−1.70 to −1.44) | 80 | −42.7 (−47.3 to −38.2) | −1.31 (−1.49 to −1.12) |
870 or 900c | 213 | −53.8 (−57.3 to −50.3) | −1.76 (−1.91 to −1.62) | 58 | −50.7 (−58.1 to −43.4) | −1.53 (−1.82 to −1.25) |
990 | 225 | −45.3 (−48.5 to −42.2) | −1.46 (−1.58 to −1.35) | 81 | −40.9 (−47.1 to −34.8) | −1.22 (−1.42 to −1.02) |
1,080 | 225 | −51.1 (−54.8 to −47.4) | −1.64 (−1.78 to −1.50) | 65 | −50.9 (−56.5 to −45.3) | −1.49 (−1.68 to −1.29) |
1,170 | 231 | −42.1 (−45.9 to −38.4) | −1.38 (−1.52 to −1.24) | 75 | −38.2 (−44.7 to −31.6) | −1.20 (−1.43 to −0.98) |
1,260 | 219 | −50.6 (−54.8 to −46.5) | −1.63 (−1.77 to −1.48) | 74 | −47.3 (−54.8 to −39.8) | −1.44 (−1.68 to −1.19) |
1,350 | 229 | −42.6 (−46.4 to −38.9) | −1.39 (−1.53 to −1.25) | 76 | −44.2 (−49.9 to −38.5) | −1.35 (−1.54 to −1.15) |
1,440 | 232 | −46.7 (−50.7 to −42.8) | −1.55 (−1.70 to −1.40) | 77 | −46.5 (−53.0 to −40.0) | −1.46 (−1.71 to −1.22) |
CI = confidence interval; LDL-C = low-density-lipoprotein cholesterol; NA = not applicable.
aData are n or mean (95% CI).
bFor the inclisiran-only arm, data were analyzed in the mITT population; for the switching arm, data were analyzed in the ITT population. The inclisiran-only arm uses the ORION-1 trial baseline and the switching arm uses the ORION-3 trial baseline.
cDay 870 for inclisiran-only arm and day 900 for the switching arm. Per the study protocol, patients in the inclisiran-only arm did not have a day 510 visit, and patients in the switching arm did not have the day 570 visit.
Sources: ORION-3 Clinical Study Report,31 Ray KK et al. (2023).32
Percentage and Absolute Change in Other Efficacy Parameters From Baseline for the Inclisiran-Only Arm
In the inclisiran-only arm, the mean percentage change in total cholesterol ranged from –21.1% to –30.2%, remaining relatively consistent throughout the follow-up period (Table 21). Non-HDL-C, ApoB, and triglycerides also remained consistently decreased throughout the follow-up period. Lp(a) concentration decreased by 16.3% at day 30, with no meaningful changes thereafter.
Harms
AE results are displayed in Table 22. Overall, of the 284 patients in the inclisiran-only arm, 275 (96.8%) patients experienced at least 1 TEAE and 79 (27.8%) patients experienced at least 1 TEAE possibly related to the study drug. A total of 104 (36.6%) patients experienced at least 1 treatment-emergent serious adverse event (TESAE). There were 7 deaths (2.5%) in the inclisiran group, all reported as having no reasonable possibility of being related to the treatment administration by the investigator. Nineteen (6.7%) patients and 12 (4.2%) patients discontinued the study treatment due to TEAEs and TESAEs, respectively.
Table 21: Secondary Efficacy End Points of the ORION-3 Trial (Percentage and Absolute Change in Other Efficacy Parameters From Baseline for the Inclisiran-Only Arm)
Day | N | Percentage change | Absolute change |
|---|---|---|---|
Total cholesterol, mmol/La | |||
30 | 277 | −30.2 (−32.1 to −28.3) | −1.64 (−1.76 to −1.52) |
90 | 276 | −27.1 (−29.1 to −25.1) | −1.47 (−1.59 to −1.35) |
180 | 277 | −21.1 (−23.2 to −18.9) | −1.14 (−1.26 to −1.02) |
210 | 277 | −29.8 (−32.0 to −27.6) | −1.61 (−1.73 to −1.48) |
270 | 277 | −26.3 (−28.8 to −23.8) | −1.44 (−1.58 to −1.30) |
540 | 265 | −22.9 (−25.2 to −20.6) | −1.26 (−1.39 to −1.13) |
1,440 | 232 | −28.6 (−31.6 to −25.7) | −1.59 (−1.75 to −1.42) |
Non-HDL-C, mmol/La | |||
30 | 277 | −41.7 (−44.3 to −39.1) | −1.72 (−1.84 to −1.60) |
90 | 276 | −37.8 (−40.4 to −35.1) | −1.55 (−1.67 to −1.43) |
180 | 277 | −30.0 (−32.9 to −27.1) | −1.23 (−1.35 to −1.11) |
210 | 277 | −41.7 (−44.5 to −38.9) | −1.70 (−1.83 to −1.57) |
270 | 277 | −37.4 (−40.7 to −34.1) | −1.55 (−1.69 to −1.41) |
540 | 265 | −32.9 (−35.9 to −29.9) | −1.36 (−1.49 to −1.23) |
1,440 | 232 | −40.4 (−44.2 to −36.6) | −1.69 (−1.85 to −1.52) |
HDL-C, mmol/La | |||
30 | 277 | 7.6 (5.5 to 9.6) | 0.08 (0.05 to 0.10) |
90 | 276 | 7.7 (5.7 to 9.7) | 0.08 (0.05 to 0.11) |
180 | 277 | 8.5 (6.4 to 10.5) | 0.09 (0.07 to 0.12) |
210 | 277 | 8.8 (6.8 to 10.8) | 0.09 (0.07 to 0.12) |
270 | 277 | 9.8 (7.7 to 11.9) | 0.10 (0.08 to 0.13) |
540 | 265 | 9.4 (7.1 to 11.6) | 0.10 (0.07 to 0.13) |
1,440 | 232 | 9.1 (6.3 to 11.9) | 0.10 (0.06 to 0.13) |
ApoB, mg/dLa | |||
30 | 277 | −40.4 (−42.6 to −38.3) | −42.6 (−45.2 to −40.0) |
90 | 275 | −35.6 (−38.1 to −33.2) | −37.6 (−40.5 to −34.7) |
180 | 267 | −26.5 (−29.1 to −24.0) | −27.9 (−30.7 to −25.2) |
210 | 277 | −36.7 (−39.1 to −34.2) | −38.4 (−41.2 to −35.5) |
270 | 170 | −32.5 (−36.3 to −28.6) | −34.5 (−38.5 to −30.4) |
1,440 | 226 | −33.7 (−37.2 to −30.2) | −36.3 (−40.0 to −32.5) |
Lp(a), nmol/Lb | |||
30 | 277 | −16.3 (−20.2 to −12.9) | −4.0 (−6.0 to −3.0) |
90 | 276 | −1.4 (−5.4 to 0.0) | −1.0 (−2.0 to 0.0) |
180 | 273 | 5.7 (1.3 to 9.4) | 2.0 (1.0 to 3.0) |
210 | 277 | 0.0 (−1.2 to 6.2) | 0.0 (−1.0 to 1.0) |
270 | 170 | 4.5 (0.0 to 15.5) | 2.0 (0.0 to 4.0) |
1,440 | 225 | −6.3 (−12.1 to 0.0) | −1.0 (−3.0 to 0.0) |
Triglycerides, mmol/Lb | |||
30 | 277 | −11.2 (−16.3 to −6.2) | −0.14 (−0.19 to −0.07) |
90 | 276 | −9.9 (−13.7 to −3.5) | −0.11 (−0.18 to −0.03) |
180 | 277 | −9.1 (−11.8 to −4.9) | −0.12 (−0.16 to −0.06) |
210 | 277 | −8.3 (−12.6 to −4.9) | −0.12 (−0.19 to −0.07) |
270 | 277 | −10.7 (−17.0 to −5.4) | −0.12 (−0.20 to −0.07) |
540 | 265 | −10.3 (−14.5 to −6.1) | −0.12 (−0.19 to −0.08) |
1,440 | 232 | −12.0 (−17.8 to −5.6) | −0.17 (−0.26 to −0.07) |
VLDL-C calculated, mmol/Lb | |||
30 | 272 | −11.5 (−15.8 to −5.0) | −0.05 (−0.08 to −0.03) |
90 | 270 | −9.3 (−14.3 to −4.4) | −0.05 (−0.08 to −0.03) |
180 | 272 | −9.1 (−11.8 to −5.9) | −0.05 (−0.08 to −0.03) |
210 | 271 | −8.3 (−12.5 to −5.0) | −0.05 (−0.08 to −0.03) |
270 | 272 | −11.8 (−18.2 to −5.3) | −0.05 (−0.10 to −0.03) |
540 | 262 | −10.1 (−14.3 to −5.3) | −0.05 (−0.08 to −0.03) |
1,440 | 226 | −12.3 (−18.4 to −6.3) | −0.08 (−0.13 to −0.03) |
VLDL-C measured, mmol/Lb | |||
30 | 276 | −8.4 (−12.7 to 0.0) | −0.05 (−0.10 to 0.0) |
90 | 274 | −9.9 (−17.2 to −4.0) | −0.05 (−0.10 to −0.03) |
180 | 238 | −14.6 (−21.1 to −7.1) | −0.10 (−0.13 to − 0.05) |
210 | 271 | −21.4 (−25.0 to −16.0) | −0.13 (−0.16 to −0.10) |
270 | 170 | −19.2 (−27.6 to −12.5) | −0.10 (−0.16 to −0.08) |
1,440 | 226 | −12.9 (−20.0 to −5.6) | −0.08 (−0.13 to −0.03) |
hsCRP, mg/Lb | |||
30 | 277 | 0.0 (−7.1 to 13.3) | 0.0 (−0.1 to 0.1) |
90 | 275 | 0.0 (−5.3 to 16.7) | 0.0 (0.0 to 0.1) |
180 | 239 | 6.7 (0.0 to 18.9) | 0.1 (0.0 to 0.2) |
210 | 165 | 0.0 (−6.5 to 12.5) | 0.0 (−0.1 to 0.1) |
270 | 170 | 0.0 (−16.7 to 14.3) | 0.0 (−0.1 to 0.2) |
1,440 | 226 | 0.0 (−9.8 to 20.0) | 0.0 (−0.1 to 0.2) |
ApoB = apolipoprotein B; HDL-C = high-density-lipoprotein cholesterol; hsCRP = high-sensitivity C-reactive protein; Lp(a) = lipoprotein (a); VLDL-C = very-low-density-lipoprotein cholesterol.
aData presented as mean (95% CI).
bData presented as median (95% CI).
Note: Lp(a), ApoB, hsCRP, and VLDL-C were not assessed on day 540, per protocol. Changes are relative to the ORION-1 trial baseline. Data were analyzed in the mITT population.
Sources: ORION-3 Clinical Study Report,31 Ray KK et al. (2023).32
Overall, of the 87 patients in the switching arm, 80 (92%) patients experienced at least 1 TEAE and 22 (25.3%) patients experienced at least 1 TEAE possibly related to the study drug after switching. Thirty (34.5%) patients experienced at least 1 TESAE. There was 1 death in this group, reported as having no reasonable possibility of being related to the treatment administration by the investigator. Five (5.7%) patients and 3 (3.4%) patients discontinued the study treatment due to TEAEs and TESAEs, respectively.
Table 22: Summary of Harms in the ORION-3 Trial
Adverse events | Inclisiran-only arm (N = 284) | Switching arma (N = 87) |
|---|---|---|
Most common AEs, n (%) | ||
≥ 1 TEAE | 275 (96.3) | 80 (92.0) |
≥ 1 TEAE possibly related to the study drug | 79 (27.8) | 22 (25.3) |
Nasopharyngitis | 55 (19.4) | 13 (14.9) |
Hypertension | 42 (14.8) | 17 (19.5) |
Arthralgia | 40 (14.1) | 10 (11.5) |
Urinary tract infection | 37 (13.0) | 7 (8.0) |
Influenza | 36 (12.7) | 10 (11.5) |
Diabetes mellitus | 32 (11.3) | 6 (6.9) |
Back pain | 28 (9.9) | 13 (14.9) |
Fatigue | 23 (8.1) | 10 (11.5) |
Influenza-like illness and/or influenza | 15 (5.3) | 10 (11.5) |
Serious adverse events, n (%) | ||
≥ 1 TESAE | 104 (36.6) | 30 (34.5) |
≥ 1 TESAE possibly related to the study drug | 3 (1.1) | 1 (1.1) |
Patients who stopped treatment due to AEs, n (%) | ||
Discontinued due to TEAE | 19 (6.7) | 5 (5.7) |
Deaths, n (%) | ||
Total number of deaths | 7 (2.5) | 1 (1.1) |
Angina pectoris | 1 (0.4) | 0 |
Coronary artery occlusion | 1 (0.4) | 0 |
COVID-19 | 1 (0.4) | 1 (1.1) |
Ischemic stroke | 1 (0.4) | 0 |
Dyspnea | 1 (0.4) | 0 |
Respiratory failure | 1 (0.4) | 0 |
Aortic aneurysm rupture | 1 (0.4) | 0 |
AE = adverse event; TEAE = treatment-emergent adverse event; TEAE = treatment-emergent serious adverse event.
aTEAEs occurring after receiving inclisiran at day 360.
Sources: ORION-3 Clinical Study Report,31 Ray KK et al. (2023).32
Critical Appraisal
Internal Validity
ORION-3 was designed as an open-label study, which could potentially lead to an underreporting of AEs. Furthermore, only those who completed the ORION-1 trial were eligible for participation in the ORION-3 trial and, of these, approximately 79% entered the extension study, which might have potentially led to a selection bias. In addition, the absence of a control arm makes the interpretation of safety challenging in such a high-risk population with multiple comorbidities taking concomitant medications. This study also did not provide a randomized comparison between evolocumab and inclisiran, but instead used switching arms to evaluate the efficacy and safety of treatment transition in a single arm.
External Validity
Because the ORION-3 study consisted of patients who took part in the pivotal study, it is reasonable to expect that the same strengths and limitations related to generalizability apply to the extension studies, with the additional caveat of potential selection bias due to the enrolment criteria.
The ORION-8 Trial87
ORION-8 is a global, open-label, long-term extension study of patients with ASCVD, ASCVD-RE, or HeFH and elevated LDL-C, despite an MTD of LDL-C-lowering therapies, who have completed the phase II ORION-3 study or any of the phase III ORION-9, ORION-10, or ORION-11 studies. A diagram of the study design for the ORION-8 trial is provided in Figure 3. The primary objectives of the study are to evaluate the effect of inclisiran treatment on the proportion of patients achieving prespecified LDL-C targets and the safety and tolerability of long-term use of inclisiran. The secondary objectives are to evaluate the effect of inclisiran on LDL-C levels and other lipids and lipoproteins. The study enrolled 3,275 participants from 268 centres in 13 countries, including Canada (3 centres).
Figure 3: Diagram of the Study Design for the ORION-8 Trial
EOS = end of study; V = visit.
*Patients from the open-label ORION-3 study will receive no drug administration on day 1.
Source: ORION-8 Clinical Study Report.87
Populations
The inclusion criteria in the ORION-8 trial are completion of the phase II ORION-3 study or 1 of the phase III ORION-9, ORION-10, or ORION-11 studies (patient received the last dose of study drug and completed the final study visit, per applicable protocol); being on current LLTs from the previous study with no planned medication or dose change during study participation; and being willing and able to give informed consent before initiation of any study-related procedures and willing to comply with all required study procedures.
Patients were excluded from the ORION-8 trial if they had any uncontrolled or serious disease or any medical or surgical condition that may interfere with participation (or with interpretation of the study results) or put the participant at significant risk; had severe non-CVD that reduces life expectancy to less than 3 years; or had planned use of any investigational medicinal products other than inclisiran.
Interventions
Inclisiran sodium 300 mg/1.5 mL (equivalent to 284 mg inclisiran) was administered as a single SC injection on day 1, day 90, and every 180 days thereafter until day 990. Treatment phases are double-blind from day 1 (blinded inclisiran or blinded placebo) to day 90 and open-label from day 90 (open-label inclisiran for all patients) and subsequent dosing visits with inclisiran every 180 days until the end of the study. Note that patients who received blind placebo in the preceding feeder study received blinded inclisiran, and patients who received blinded inclisiran in the feeder study received blinded placebo on day 1 in the ORION-8 trial, after which they received the first dose of study medication on day 90.
There were no required concomitant medications and/or treatments; however, to be eligible for enrolment in the ORION-8 trial, participants had to have received a stable dose of a statin or other LLT in 1 of the preceding studies, with no planned medication or dose alteration during the study.
Outcomes
The 2 primary outcome measures in the ORION-8 trial are the percentage of patients achieving prespecified LDL-C targets at day 1,080 and the incidence of AEs and SAEs from baseline to day 1,080. Secondary outcome measures are the absolute change and percentage change in LDL-C from baseline to day 1,080 and the absolute change and percentage change in other lipids and lipoproteins from baseline to day 1,080. Safety outcomes assessed include AEs, SAEs, and other AE-related events, such as injection-site reactions.
Statistical Analysis
The efficacy outcome results in the ORION-8 trial were summarized using descriptive statistics for the safety population (Table 26). ██████ ██████████ ███████ ████ ███ ███ ███████ ███ ███ ████████ ████ ███ ██████████ █████████ ███ ██████ ██████████ ███ ████ ███ ███ ████████ ███ ██████ █████████ The observational period for the study included day 1 (EOS visit in the previous study) and the treatment phase, which consists of the first dose (day 1), the day 90 dose, and subsequent dosing visits every 180 days until day 990 and the EOS (day 1,080) visit. ███ █████ █████████ ██████ ███ ███████ █████████████ ███████ ████ ██ █████████ ██ ███ ██████████ ████ ██████ ████ ███████ ███ ███ ████████ ██ ███ ███████████ ████████.
██ ███████ ████████ ███ █████████ ██ ████ ██████
Results
Table 23: Baseline Demographic and Clinical Characteristics
Characteristics | Phase III inclisiran-inclisiran (N = 1,512) | Phase III placebo-inclisiran (N = 1,478) | ORION-3 rollover (N = 284) |
|---|---|---|---|
Age, years, mean (SD) | 64.7 (9.8) | 64.6 (9.8) | 67.7 (10.0) |
███ █ ██ ██████ █ ███ | ███ ██████ | ███ ██████ | ███ ██████ |
Male, n (%) | 1,030 (68.1) | 1,002 (67.8) | 184 (64.8) |
White race, n (%) | 1,383 (91.5) | 1,387 (93.8) | 271 (95.4) |
Black or African American, n (%) | 103 (6.8) | 78 (5.3) | 7 (2.5) |
Asian, n (%) | 17 (1.1) | 7 (0.5) | 3 (1.1) |
Native Hawaiian or Other Pacific, n (%) | 6 (0.40) | 4 (0.3) | 0 |
American Indian or Alaska Native, n (%) | 3 (0.2) | 2 (0.1) | 2 (0.7) |
BMI, kg/m2 | |||
n | |||
████ █ ████ | ████ █████ | ████ █████ | ████ █████ |
Baseline eGFR, mL/min per 1.73 m2 | |||
n | |||
████ █ ████ | ████ ██████ | ████ ██████ | ████ ██████ |
ASCVD = atherosclerotic cardiovascular disease; BMI = body mass index; CV = cardiovascular; eGFR = estimated glomerular filtration rate; LLT = lipid-lowering therapy; SD = standard deviation.
Patient Disposition
In the cohort of patients who continued inclisiran and in the cohort of patients who switched from placebo to inclisiran, 17.6% and 18.8%, respectively, discontinued the study. The most common reasons for discontinuation were death and withdrawal of consent, reported by approximately 5% each across treatment groups.
Patient disposition | Phase III inclisiran-inclisiran (N = 1,512) | Phase III placebo-inclisiran (N = 1,478) | ORION-3 rollover (N = 284) |
|---|---|---|---|
Patients who completed parent trial, n | ORION-9: 235 ORION-10: 721 ORION-11: 772 Total: 1,728 | ORION-9: 231 ORION-10: 694 ORION-11: 770 Total: 1,695 | Inclisiran only: 233 Switching: 80 Total: 313 |
Patients enrolled, n | 1513 | 1,478 | 284 |
Patients enrolled and treated, n | 1,512 | 1,478 | 284 |
Completed study, N (%) | 1,246 (82.4) | 1,200 (81.1) | 0 |
Discontinued study, n (%) | 266 (17.6) | 278 (18.8) | 284 (100.0) |
Reasons for discontinuation, n (%) | |||
Sponsor’s administrative decision | 0 | 0 | 272 (95.8) |
Death | 80 (5.3) | 81 (5.5) | 4 (1.4) |
Withdrew consent | 77 (5.1) | 78 (5.3) | 2 (0.7) |
Lost to follow-up | 47 (3.1) | 52 (3.5) | 1 (0.4) |
Other | 34 (2.3) | 40 (2.7) | 2 (0.7) |
Adverse event | 22 (1.5) | 23 (1.6) | 0 |
Physician decision | 6 (0.4) | 4 (0.3) | 2 (0.7) |
Initiation of protocol-prohibited approved PCSK9 inhibitor | 0 | 0 | 1 (0.4) |
Consented to ORION-8, N (%) | 1,513 (100.0) | 1,478 (100.0) | 284 (100.0) |
Safety population, n (%) | 1,512 (99.9) | 1,478 (100.0) | 284 (100.0) |
Source: Clinical Study Report for ORION-8.87
Exposure to Study Treatments
███ ████ ████████ ██ ████████ ███ ███████ ███████ ███ █████ ██ ████████ ███ ████████ ██ ██████████ ██████████ █████ █████ ███ ███ ██████ ███ ████████ ████ ███████ ██ ██████████ ██ ███ █████████ ████ ██████ ███████ █████████ ███ ███ ████████ ███ ██ █ ███ █████ █████ ███ ██████ ████ ██████ ███ ████████ █████
Table 25: Patient Exposure (Safety Population Plus Patients Switched to Inclisiran)
Patient disposition | Phase III inclisiran-inclisiran (N = 1,512) | Phase III placebo-inclisiran (N = 1,478) | ORION-3 rollover (N = 284) |
|---|---|---|---|
██████ ██████ | ██████ | ██████ | █████ |
█████████ ████ ███ | ████ ███████ | █████ ███████ | █████ ██████ |
█████████ ██████ █ | ████ ███████ | █████ ███████ | █████ ██████ |
██████████ | ██ | ██ | ██ |
IQR = interquartile range; NR = not reported; SD = standard deviation.
Source: Clinical Study Report for ORION-8.87
Efficacy
Low-Density-Lipoprotein Cholesterol
The proportion of patients who attained global lipid targets at day 1,080 was similar in the patients who continued inclisiran in the extension (78%), patients who switched to inclisiran in the extension (79%), and patients who rolled over from the ORION-3 trial (77%). Similarly, the percent of patients with ASCVD who attained global lipid targets (< 70 mg/dL) at day 1,080 was similar in those who continued inclisiran (79%), those who switched to inclisiran (80%), and those who were rolled over from the ORION-3 trial (77%). The percent of patients with ASCVD-RE who attained global lipid targets (< 100 mg/dL) was 73% in the patients who continued inclisiran, 75% in patients who switched from placebo to inclisiran, and 77% in patients who were rolled over from the ORION-3 trial.
The mean percentage change from baseline to day 1,080 in LDL-C was –49.0% (95% CI, –50.5% to –47.4%) in the group that continued on inclisiran, –49.7% (95% CI, –51.3% to –48.0%) in the group that switched to inclisiran, and –50.0 (95% CI, –52.6 to –47.3) in the group rolled over from the ORION-3 trial.
Table 26: Efficacy Results From the ORION-8 Trial
Outcome | Phase III inclisiran-Inclisiran (N = 1,512) | Phase III placebo-Inclisiran (N = 1,478) | ORION-3 rollover (N = 284) |
|---|---|---|---|
Patients who attain global lipid targets at day 1,080 or EOS, n/N (%) | 978/1,251 (78.2) | 952/1,204 (79.1) | 212/276 (76.8) |
ASCVD patients who attain global lipid targets < 70 mg/dL at day 1,080 or EOS, n/N (%) | 822/1,036 (79.3) | 794/993 (80.0) | 135/176 (76.7) |
ASCVD-RE patients who attain global lipid targets of < 100 mg/dL at day 1,080 or EOS, n/N (%) | 156/215 (72.6) | 158/211 (74.9) | 77/100 (77.0) |
Percent change from baseline in LDL-C | |||
Mean (95% CI) baseline | –50.0 (–51.3 to –48.8) | 3.1 (1.4 to 4.9) | –48.2 (–51.2 to –45.2) |
Mean (95% CI) change at day 1,080 or EOS | –49.0 (–50.5 to –47.4) N = 1,251 | –49.7 (–51.3 to –48.0) N = 1,204 | –50.0 (–52.6 to –47.3) N = 276 |
ASCVD = atherosclerotic cardiovascular disease; ASCVD-RE = atherosclerotic cardiovascular disease risk equivalent; CI = confidence interval; EOS = end of study; LDL-C = low-density-lipoprotein cholesterol
Source: Clinical Study Report for ORION-8.87
Harms
AEs were reported by 79% of patients who continued inclisiran, 79% of those who switched from placebo to inclisiran, and 64% of patients who rolled over from the ORION-3 trial. There were also similar numbers of patients who discontinued treatment due to an AE (██ ██ ████ █████) in the group who continued inclisiran and in the group who switched from placebo to inclisiran, versus ████ of patients who rolled over from the ORION-3 trial.
SAEs were reported by 31% of patients who continued inclisiran, 33% of patients who switched from placebo to inclisiran, and 15% of patients who rolled over from ORION-3.
With respect to AEs of special interest, the following occurred in the group of patients who continued on inclisiran, the group who switched from placebo to inclisiran, and the group who rolled over from the ORION-3 trial: █████████ ████ ██████ ███ ██████ ██ ██████ ████ ███████ ██████ ███ ██████ ██ ██████ ████ ███ ███████████████ ████████ ████ ██████ ███ ██████ ████ █████ ██████ ███ ██████ ██ ██████ ███ ███ ████████████████ ███ ██ ████ ██████.
████ ████████ ██ ██ ██ ████████ ██ ███ █████ ████ █████████ ██ ███████████ ███ ██ ████████ ███ ████████ ████ ███████ ██ ███████████ ███ ██ ██ ████████ ███ ██████ ████ ████ ████████
Refer to Table 27 for harms data.
Table 27: Summary of Harms Results From the Long-Term Extension Studies
Adverse events | Phase III inclisiran-inclisiran (N = 1,512) | Phase III placebo-inclisiran (N = 1,478) | ORION-3 rollover (N = 284) |
|---|---|---|---|
Most common AEs, n (%) | |||
≥ 1 adverse event | 1,197 (79.2) | 1,170 (79.2) | 181 (63.7) |
████████████████████ | ███ ██████ | ███ ██████ | ██ ██████ |
██████████████ ██████ | ███ ██████ | ███ ██████ | ██ █████ |
████████████████████ | ███ ██████ | ███ ██████ | ██ ██████ |
SAEs, n (%) | |||
Patients with ≥ 1 SAE | 464 (30.7) | 482 (32.6) | 43 (15.1) |
Patients who stopped treatment due to AEs, n (%) | |||
████████ ███ ███████ | ██ █████ | ██ █████ | | █████ |
Deaths, n (%) | |||
Fatal SAE | 80 (5.3) | 81 (5.5) | 4 (1.4) |
Common causes (≥ 1% in any group) | |||
████████████████████ | ██ █████ | ██ █████ | █████ |
██████████████ ██████ | ██ █████ | ██ █████ | ██ █████ |
████████████████████ | ██ █████ | █████ | ██ █████ |
AEs of special interest, n (%) | |||
████████████████████ | ██ █████ | ███ █████ | ██ █████ |
██████████████ ██████ | ██ █████ | ██ █████ | ██ █████ |
████████████████████ | ███ ██████ | ███ ██████ | ██ █████ |
█████ ██████ | ██ █████ | ██ █████ | ██ █████ |
████████████████ | ███ █████ | ███ █████ | ██ █████ |
████ | ███ █████ | ███ ██████ | ██ █████ |
AE = adverse event; MACE = major adverse cardiovascular events; SAE = serious adverse event.
Source: Clinical Study Report for ORION-8.87
Critical Appraisal
Internal Validity
The open-label design of the ORION-8 trial increases the potential for bias in the reporting of AEs, and results for efficacy and harms are difficult to interpret due to the lack of a comparator group. A key limitation of the ORION-8 trial is the potential selection bias since only patients who completed the ORION-9, ORION-10, or ORION-11 trials or enrolled in the ORION-3 trial were eligible to enrol in this extension. Furthermore, it appears that not all patients who completed the ORION-9, ORION-10, or ORION-11 trials, approximately 87% of completers, enrolled in the extension, again suggesting that this may be a selected population.
External Validity
Because the ORION-8 study consisted of patients who took part in the pivotal studies, it is reasonable to expect that the same strengths and limitations related to generalizability apply to the extension studies, with the additional caveat of potential selection bias described previously.
Indirect Evidence
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
Due to the lack of direct evidence comparing inclisiran with other existing therapies as monotherapy, or as add-on therapy in the treatment of adult patients with HeFH or ASCVD, the sponsor submitted an ITC that was used to inform the pharmacoeconomic model.34 The objective of this section is to summarize and critically appraise the methods and findings of the sponsor-submitted NMA and ITC comparing inclisiran with relevant drug comparators for the treatment of adult patients with HeFH or ASCVD whose LDL-C is uncontrolled with MTD statins, with or without ezetimibe, or for the treatment of patients with uncontrolled LDL-C who have an intolerance or contraindication to statins, with or without ezetimibe.
A focused literature search for NMAs dealing with hypercholesterolemia was run-in MEDLINE All (1946–) and Embase (1974‒) on April 14, 2021. No limits were applied to the search. Retrieval was not limited by publication date or by language. Articles were screened by 1 researcher for ITCs that met the patient, intervention, comparator, and outcome criteria listed in Table 28. In addition, the sponsor-submitted ITC was reviewed.
The literature search for NMAs identified 103 articles; however, no studies evaluated the efficacy or safety of inclisiran in patients with HeFH or ASCVD against relevant comparators.
Description of Indirect Comparison(s)
The sponsor submitted an ITC that compared the efficacy of inclisiran to relevant drug comparators in patients with HeFH or ASCVD (or ASCVD-RE). The sponsor first conducted a systematic literature review (SLR) to evaluate various efficacy, safety, and HRQoL outcomes to assess the feasibility of conducting an NMA. Thirty-nine studies met the inclusion criteria of the systematic review and feasibility assessment, and 24 studies were subselected for inclusion in the NMA, based on network connectivity and whether there were any differences in study, patient, or outcome characteristics that were likely modifiers of the relative treatment effects.34 Table 28 summarizes the available selection criteria specific to the ITC and/or NMA, as well as the methods for study selection for the SLR.
Table 28: Study Selection Criteria and Methods for the NMA
Detail | Sponsor-Submitted NMA |
|---|---|
Population | Adults (≥ 18 years) with HeFH or ASCVD
|
Intervention | Inclisiran 284 mg |
Comparatora |
|
Outcome |
|
Study design | RCTs |
Publication characteristics | Full-text, peer-reviewed publications; conference abstracts and presentations; SLRs; and manufacturer data on file. |
Exclusion criteria | Patients with HoFH, low-intensity statin background or no prior statin treatment (unless intolerant and/or contraindicated), non-RCTs, < 12 weeks of follow-up, < 10 patients per group, editorials, press releases, expert opinion, and letters, and case studies. |
Databases searched | The following databases were searched: Ovid MEDLINE, Embase (Ovid), Cochrane Central Register of Controlled Trials, PubMed, CPCI-S. Hand searches of clinical trial registries and conferences were also conducted. |
Selection process | Titles and abstracts were screened using the Covidence online screening tool, followed by independent review of records by 2 researchers. Full-text citations were reviewed independently by 2 reviewers, according to the predefined inclusion criteria. |
Data extraction process | Two independent researchers extracted data to the predefined extraction forms. A single researcher collated the data from both researchers to identify discrepancies, and a third researcher was involved to resolve discrepancies. |
Quality assessment | The Cochrane risk of bias assessment tool was used to assess the quality of the included RCTs. |
AE = adverse event; ASCVD = atherosclerotic cardiovascular disease; CFB = change from baseline; HDL-C = high-density-lipoprotein cholesterol; HeFH = heterozygous familial hypercholesterolemia; HoFH = homozygous familial hypercholesterolemia; LDL-C = low-density-lipoprotein cholesterol; MTD = maximally tolerated dose; NMA = network meta-analysis; RCT = randomized controlled trial; SLR = systematic literature review.
aNote that only statins are used in the MTD statin and uncontrolled LDL-C ± ezetimibe population.
Source: Sponsor-submitted NMA report.34
Methods of ITC
Objectives
The objective of the sponsor-submitted report was to conduct a feasibility assessment via an SLR and, if possible, to conduct an indirect comparison evaluating the relative efficacy and safety of inclisiran versus relevant drug comparators, including ezetimibe and other PCSK9 inhibitors in patients with HeFH or ASCVD (or ASCVD-RE).34
Study Selection Methods
The sponsor-submitted NMA was informed by a systematic review of RCTs conducted in April 2020. The sponsor provided the protocol and plan for analyses in a separate report. Briefly, eligible publications were full-text, peer-reviewed RCTs. Planned methods for citations identification was through searches of the MEDLINE, Embase, and Cochrane CENTRAL databases, among others. Study selection and data extraction were planned to be conducted by 2 independent reviewers, with discrepancies resolved by discussion using the Covidence online screening tool.34 Assessment of the quality of included studies was planned using the Cochrane risk of bias assessment tool; however, no quality assessment was included in the NMA report.
Table 29 summarizes the predefined study selection criteria for the systematic review in the sponsor-submitted ITCNMA. The list of comparators and outcomes of interest included in the literature review was broader than that of the NMA; otherwise, the patient population was similar. No limitations on publication language were applied. The eligible patient population for the review included adults with HeFH or ASCVD with inadequate LDL-C control on MTD statins and those with an intolerance or contraindication to statins, which was identical to the NMA population. Multiple networks were constructed based on the HeFH, ASCVD, and ASCVD-RE population. The outcomes of interest to the NMA were percent change and absolute change in LDL-C at 24 weeks, total discontinuations and discontinuations due to AEs at 24 weeks, and percent change in HDL-C at 24 weeks.34
Table 29: PICOS Criteria of the SLR to Identify Trials for the Sponsor-Submitted NMA
PICOS component | Inclusion criteria | Exclusion criteria |
|---|---|---|
Population | ██████ ███ ██████████ ██ ██████████████ ████████████████████████ █████ ███████ ██ ███ ██████ ██ ███ ███ ████████████████ ████ ██████████ ███████ ██ █████████████ ██ ██ ██████ | ████████ █████████████ █████████████ ████████ ████████████████████████████████████████████ █████ █████████████████ ████████ ██████████████ █████ ██████ ██████████ ███ █████ ███████████ |
Intervention | ███████████ ██ ███████████ █████████ ██ ███ █████████████████████████████████ ██████████████ █████ | █████ █████████████████ ████ ███ ███ ███████████ ██ ████████ ██ ███ █████ |
Comparator | ██ █████████████ ██████ ███████████ ████████████ | ███ |
Outcomes | ████████ █████████ ███ ████ ████████ ███ ██ ████████████████ ██ ████████ ███████ █████ ████████████████████████████████ ███ ████ ███████████ ███████ ██████████ ██████ █████████ █████████████████████ ███ ██████████████ ███ ██████████████ ███████████████ | ██████ █████████ ████ █████ ████████ ███ ██████ |
Study type | ████ | ██████████ ██ █████ ██ ██████ ████ ██ ████████ ███ █████ |
Publications | ████ ████ ████ █████████████████████████ █████████ ███ ████████████████████████ ██████████ ███████████ ██████ | ███████████████████████████████████████████████████████████████ ████████ |
AE = adverse event; ASCVD = atherosclerotic cardiovascular disease; CFB = change from baseline; CV = cardiovascular; FH = familial hypercholesterolemia; HDL-C = high-density-lipoprotein cholesterol; HeFH = heterozygous familial hypercholesterolemia; HoFH = homozygous familial hypercholesterolemia; HRQoL = health-related quality of life; hsCRP = high-sensitivity C-reactive protein; HTA = health technology assessment; LDL-C = low-density-lipoprotein cholesterol; LLT = lipid-lowering therapy; MTD = maximally tolerated dose; NR = not reported; NYHA = New York Heart Association; PICOS = population, intervention, comparison, results, and study design; RCT = randomized controlled trial; SAE = serious adverse event; SLR = systematic literature review; TC = total cholesterol; TRAE = treatment-related adverse event; VLDL-C = very-low-density-lipoprotein cholesterol.
Source: Sponsor-submitted SLR protocol.34
Feasibility Assessment
A feasibility assessment was conducted to review the studies identified in the SLR, which included the following criteria: determination of a connected network comparing the treatments of interest regarding the outcomes of interest; and differences in study, patient, or outcome characteristics across comparisons that are likely modifiers of the relative treatment effects. The primary outcomes of interest for the NMA were percent, absolute, and time-adjusted change from baseline in LDL-C; percent change from baseline in HDL-C; total discontinuations; and discontinuations due to AEs.34 Several study design, patient, and intervention characteristics were identified a priori as potential treatment-effect modifiers. Key assumptions and recommendations from the feasibility assessment for the approach used in the sponsor-submitted NMA are summarized in Table 30 and include the following.
Background Ezetimibe and Statins
████████ ████ █████████ ██████ ███ ███████████ ██████████ ███ ███ ███████ ████ ██████████ █████████ ██████ ██ ████████ ██ █ █████████ ██████ ████████ ██ ████████ ███████ ████ ████████ ███ ████████ ███████ ██████ ████ ███ ████████████ █████ ████ ███ █████ ███████ ████ ███████ ██████████ ██████████ ████████ ██ ███ ███████ ████ █████████ ███████ █ ██████████ ███ ███ ███ ███ █████████ ██████████ ███ ██████████ ██ ████████ █████████ ██████████████ ███ ██████████████████ ███████ ████ ████████ ████ ███ █████ ██████ ██ █████████ ███████████████ ███ ████ █ █████ ██ ████ ██████ ████ ████████ ████ ███ ███ █████ ███████████ ██ ██████████ ██████ ████████ In the NMA, it was assumed that individual statins (atorvastatin, rosuvastatin, and simvastatin) had similar efficacy as background therapy, regardless of dose, and would not bias the results of the NMA. ██ ███ ███████ ████ ███ ███ ██████████ ██ ██████ ██████████ ████████ ██ ███ █████ ██████ █████ ███ ████ ███ ████████ ███ ███████████ ████████ ███ ██████ ██████████ ████████ ████ ██████████ ████ ██████ ████ ████████ ████ ███ ████████ ███ █████████ █████ ██████████ ████ ██████████ ██████ ███████ ██ █ ███████ ████ ██ ███ ██████████ ███████ ██ ████ █████ ████ ███ ███ ███ ██████ ███ ████████ █████████ ███████
CV Risk and Severity
Studies included in the feasibility assessment were inconsistent in their reporting of CV risk and severity, and definitions of risk equivalent varied across trials. Populations in the ORION trials included patients considered to be ASCVD-RE, defined by the presence of type 2 diabetes, FH, or a 20% or greater 10-year risk of a CV event, assessed by the FRS for CVD or equivalent. █████ ██████ ████████ ████ ███████████ ████████ ████████ ████ ████████ █████ ███████ █████ █████████ ███ ██ ████ ██ ███ ██████████ ████████ ███████ ██████████ ██████ ████████ ███████ ██████ ████████ ██ ████████ █████████ ███ ███ ██ ████ ██████████ ████ ███████
Treatment Dosing
████ ████ ███ ████████ ███ ████████ ██████ ███ ██████ ██ ███ █████ █████████ █████ ██ ██ ████████ █████ ██████████ ██ ███████████ ███ █████████ ████ ██ ██████████ ██ ███ ██ ████ ████████ ███ ██ ██ ████ ███ ████ █████████ ███ ████████████ ██ ███ ████ ██ ████ ███ ██████████ ███ ████ ██████████ ██ ██████ ████████ ██ ███ ████████ ███ ████████ █████████ ████ ███ ███ ███ ██████ ██████████ █████ ███ ██ ████ ██ ████ ███████ ████ █ ██████ ████ ██████████ ███ █████████ ██ █ ████ ██ ███ ██ ███ ██ ████ ███████ ████ ███ █████████ ██ ███ ███████ █████ █████ ███ ███ ████████████ ██████████ ██ █ ████ ██ ███ ██ ███ ██████ ████████ ████████ ████████ ████ █████████ ██ ███ ██████ ████████ ███ ████ ███ ██████████ ███ ████████ ██ ███████ ████ █████ ████████ ██ ███████ ███████ █████ ███ █████ ██████████ ██ ████████ █████████ ███ ███████ ████ ███████
Time of Assessment and Follow-Up:
Follow-up in the ORION trials was 540 days; however, other PCSK9 inhibitors had a much shorter duration of follow-up. Twenty-four weeks was selected as the preferred base-case time point for multiple reasons. ██████ █████ ██ ██ ████████ ███████ ██ █████ ████████ ██ █████ ███ ██████████ ██████ ████ █████ ██ ████ ██ ███ ████████████ ██ ██████████ █████████ ████████ ██ ████ ███ █████████████ ███ ████ ████████ ████████ ██████████ ███ ████████ ████████ ██ ████████ ████ ██ ███ ██ ██████ For safety outcomes, results were presented at the end of the study and, given the variation in follow-up, EOS outcomes were considered comparable if the duration of follow-up was 24 weeks or longer. ███████████ ████████ ████ █████████ ██ ████ ███ ██████ ██ █████████ █████████ ████ ███ █████ ███████ █████████ █ ████████ ████ ████████ ███ ███████ ██ ██ █████ ███ ███████ ████ ████████ █████████████ ████████ █████ ████████ ███ ██████ ███████ ████ ██████ ████████████
Table 30: Summary of Main Assumptions and Recommendations in the Sponsor-Submitted NMA
Potential effect modifier | Assumptions | Recommendation |
|---|---|---|
Population characteristics | ||
Background ezetimibe | ███████████ ██ ███ ████████████ ██ ████████ █████████ ██████████ █████████ █████ ███ ████ ███ ███████ ██ ███ ████ | ███████ ████████ ███████ █████████████ ██ ██████████ █████████ ██ ██ ██████ ████████ |
Background statins | ███ ██████ ███████ ████████ ████ █████████ ███ ██████ ██ ███████ █████████████ ██ █████████████ █████████ ███████, it was assumed that individual statins (e.g., atorvastatin, rosuvastatin, simvastatin) would have similar efficacy as background therapy, regardless of the specific statin and dosage. ███████████ ██ ██████████ ██████ ███████ █████ ████████ ██████ █████████ █████ █████ ███ ████ ███ ███████ ██ ███ █████Small proportion of statin-intolerant patients in the ORION trials (ORION-10, 22%;ORION-11, 12%; ORION-9, 25%) would not bias the NMA. | ████████ ██ ██ █████████ ████████ ███████ ██ █ ██████ ██████████ ██████████ ██ ██████████ ██████ ████████ ██████████████████ ████████ ██ ██ █████████ ███ ██████ ███████ ████████ ███ █████████ ███ ███████ ███ █████ ██ ████████ ███ ███ ██████ ███████████████████ ██████ ████ ███ ███ ███████ ████████ ██████████ ██ ███ ███████ ███ ████████ ███████ ██ ███████ ███████ ███ ███████ ████████ █████ ████████ ███████████ ████████████ ████ ███ ██████████ ████ ███ █████ ██████ ██████ ██ ████ ███ ███ █████████ █████████ ████ █ ██ █████ ██ █████████ ██ ████████ ████ ███ █████ ██████ ████ ███ ██████ █████████████████ ████ ██ ████████ ████ ███ █████████ ████████ ███ ████ █████████ ██ █ ████████████ █████ ██ ██████ ██████████ ███████████ ██████ ███████ ████████ ████ ████ █████ ████ ████████ ████ ████████ ███ █████████ ██████████ ███████ ███████ █████ ████ █████████ ██████████ ████████ ██ ███ █████ ███████ ████ █████████ ████████ ████████ ███ ███ ██████████ ██ █████ ████████ ███ ██ ████ ████ █████ ██████ ███████ ████ ████████ ████████ ████████ ████████ |
CV risk and severity | Differences in CV risk and severity of patients within each population strata of interest (i.e., HeFH and ASCVD or ASCVD-RE) would not impact the relative effects observed for efficacy outcomes focused on changes in LDL-C ███ ████████████████. █████████ ████████ ██████ ████ ██████ ███ ████████ ████ ████████ █████ ██████████ ██████ ██ ███████ ████ ███ ████ ██████ ████ ██ ██████████ ██████ ███ ████ ███████ ██ ██████ ██ █ █████ █████████ ██ █████ ████████ ██ ████████████ ██████ ████ █████ █ ██████ █████ ████████ █████ ███ ███ ██████ ██ ███████ █████████ ██████ █████ ██ ████████ ██████ █████████ ███ ██████ ██ ██████ ████████ █████ ██████ ██ ████████ ███ ██ ██████ ████████ ███ ████████████ ██████████ ████████ | ███████████████ ██ ███████ ██ ██████ ███ ███████████ ██ ████████ ███████████████ ███ ███ ████████████ █████ ███ ██████ ██ ███████ ████████ ██ ███ ████████ ███ ███████████████ ██ █████████ ██ ███████████████ ██████ ███ ████████ ████████████ ████ ███ ██████████ ████ ███ █████ ██████ ██████ ██ ████ ██ ███ █████ ███████████ ████████ ████████ █████ ██ ████████ █████ ███ ███ ████████████ █████ ███ ███████ ██████ ██ ██████ █████████ ████ ████ ███ ███ ██████ ████ ██ ███ ██████████████████ █ ██ ████ ███████ ███████ ███████ ███████ █████████ ███████ ████ ██ ███ ███ ████ ███████ ███ ███████ ████████ ███ ███ █████ ███ ██████ ███████ |
Treatment characteristics | ||
Inclisiran | ██ ███████████ ████ ████████ ███████ █████ ██████ ████ ███████ ██ ██████████ █████ ██ █████████ | ██ ██████ ████ ████████ ████ ███ ████████ █████ ██ ██████████ ███████ |
Alirocumab | ██ ███ ███████ ████ ███ ████ ███ ██ ████████ ██ ███ ██ ██ ████████ ███ ███ ██ ███ ████████ ████ ███████████ ██ ██ ██████████ ██ ███ ████ █████████ ██ ███ █████████ | █████ ███ ██████████ ████████████ ██ ███ ██ ██ █████ ████ ███████ ███ █████████ ████ ████ ██████ ████████ ███ ███ ██ ████████ ██████ ████████ ██ ███ ████████ ███ ████████ █████████ ████ █████████ ███ █████ ███ ██████ █████████ ██████████ █████████ ██ █ ████████ ██████████ ███ ██ ███ ████████ ██ ████ █████ ████ ██████ █ ██████ ███ ███ ███████ |
Evolocumab | ███ ███████ █████ ████████████ ███ █████████ ████████ ██ ███████████ ███ ██ ███ ██ ███ ██ ███ ████ █████████ ██████████ █████ ██ ███████ ██████████ ██████ ████ █████████ ███ ██ ██████ | ████ ██████ ████ ████████ ████ ███ ████████ ███ ████ ██████████ ███ ███ ██ ██ ████████████ ██ ████ ████████████ ███ ██████████ ██ ███████████ █ ████ ████████████ ████████ ███ █████ ██ ███████ ███ ████ ███ ██████████ ████ ███████ ███████ ████ ██████ █████ ████ ████████ ██ ███████ ████████ |
Ezetimibe | ██ ███████████ | ██ ██████ ████████ ████ ███ ████████ |
Placebo | ██████████ ██████ ███ █████████ █████████ ████ ██████████ ██████████ ██ ███████ █████████████ ████ ████████ ██████ ██████ ████ ██████ ██ ██████ █████████████████████ ██ █████ ██ ██████████ ███████ ██████ █████████ ████ ████ ██ ███████████ ███████ ████ ███████ ██ ████ ███ ███ ███ ██████ ███ ████████ █████████ ████████ | ████ ██████ ████ ████████ ████ ███ ████████ ███ █████████ █████ ██████████ ████ ██████████ ██████ ███████ ██ █ ███████ ████ ██ ███ ██████████ ███████ |
Outcome characteristics | ||
Time points | █████████ ██ ██ █████ ██ ███ ██████ █████████ ██ ████████ ███████ ████ ███████ ████████ ████ ████ ████ ███████ ███ ███ ███████████ ████████████ ██████████ █████ ███ ████ ████ ███████████ ████ ██ ██ ██ ███ ██ ██ ████ ███ | 24 weeks ████ ████ ███ ███████████ was selected as the preferred time point of interest for the base case, ████ ███████ ████ ████ ████████ ██ █████ ███████ ███████ ████ ██ ███ █████████████████ ███ ████ █████████ ██ ████ ███ ██████ ██ █████████ █████████ ████ ███ █████ ███████ █████████ █ ████████ █████ ████████ ███ ███████ ██ ██ █████ ███ ███████ ████ ████████ █████████████ ████████ █████ ████████ ███ ██████ ███████ ████ ██████ █████████████ |
For safety outcomes of interest, results are presented at the end of the study and, therefore, given the variation in follow-up, end-of-study outcomes were considered comparable if the duration of follow-up was 24 weeks or longer. | ██████ ████ █████ █████ ████████ ███████ ████ ██ █████ ████ ██ ████████ ████ ███ ████████ ███ █████████ █████████████████ | |
ASCVD = atherosclerotic cardiovascular disease; ASCVD-RE = atherosclerotic cardiovascular disease risk equivalent; CV = cardiovascular; FH = familial hypercholesterolemia; HeFH = heterozygous familial hypercholesterolemia; ITT = intention to treat; LDL-C = low-density-lipoprotein cholesterol; MTD = maximally tolerated dose; NMA = network meta-analysis; Q2W = every 2 weeks; QM = every month; SA = sensitivity analysis.
Source: Sponsor-submitted NMA report.34
ITC Analysis Methods
The NMA methods are briefly summarized in Table 31. The analyses were conducted within a Bayesian framework. Selection of both fixed and random effects was conducted for outcomes of interest. Random-effects analyses were selected as the base case, given the number of studies per node and the observed heterogeneity in patient and/or trial characteristics, ████████ ████ ███ █████████ ███ ██████ ████████ ██ ██████ ██████████ █████ █████████ █████ ███████ ████ ███ ████ ██ ███ ████ ████ ██ ███ ███████████ ██ ███████████ ████ █████ ██ ██ ███████████
███ ████████ ████ ███████ ███ ██ ██████ █████ ███████████ ██████ ████████████ █████ ███ █████ ███████ ███████████ ███████ ██ ███████ ████████ ████ ██████████ ███ ███ ██████████ ███████████████████ ██████████ ████████ ████ ████ ███ █████████ ███████████ ███████████ ███ █████████ ██ ██████████ ███ █████████████████████████ █████ ███ ██████████ ██ ███ ██████ ██ █████ █████ █████ ██ ███ ████████ ██████████ ██ ███ ███████████ ███ ██████ ███ ████ ██████████████ ██ ███ █████████ ███████ ███ ████ ██████ ████ ████ ██ ██ ████████ ██ ███ ██████ ███ ███ ███ ████████ ████████ ██████ ███████████████ ██████ ████ ████ ███ ████████ ███ █████████ ████████ ██████ ███████ ████ ████ █ ███████ ███████████ ██████ █████ ████ ███████ ████████ ███████████ ███ █████ ██ █████ ███ ██████████ ██ ███ ███ ███ ██████ █████████ ███ █ ███ ██ ███ ██████████ █████████ █████ █ ███ ███ ██████ ████████████ ████████ █████████ ██ ███ ████████ ██████ ████████ ███████████ ██ ██████ ███ █████████████ ██ ██████████ █████████ ██ ██████████ ███ ████████ ██ ████ █████████ ███████ ████ ████ ████ ██ ████ ███ ███████████ ██ █ █████████ ██ ██ ███ ████ ██████████ ███ ███████████ ██ ██████████ █████ ██████ ████ ████ ███████████ ███ ████████ ████ █████████ ██ ████████ ████████ ███████
█████ █████████ ███ ███ ███████████ ████ ████ ██ ██████ ███ ███ ████████ █████████ ███ ████ ██ █████████ ███ ███ ████████ ████████ ████ █████ ███ ███████ ████ ███████ ████ ██ ███ █████ ██ ████████████ █████████ ██████ ██ ████████ ██████████ ████████ ██████ ███ ██ █████ ███████ ██████████ ███ ██████ ████████ █████████ ██████ ███ █████ ██████ ████ █████████ ████ ██ ████ ████ ██████ ███ ████ ████████ ████ ████████ ██████████ ███ ██████████ ██ ██████ █████████ ██████ ████████ ███ ██████ █████████ ███ ██████ ███████████ ██████████ ███ ██████████ ████████ ███ ████████ ██ █████ ███ ████ ██ █████ █████ ████ █████ ███████ ███ ███ ████ ████ ███████ █████████ ██ ███ ████████ █████████ ███████ █████████ ██ █ ████ ███████████ ██ ████ █████ █████ █████ ████ ███ ███
███████████ ███ ███████ ███████ ██████████ ██ ███ ██████ ██ █████ █████ █████ ██ ███ ███ ██ ███ ███████████ ██ ██████ ████ ███████████ ████ █████████ █████ ███ ███ ████████ ██ █████████ ███ ████████ ███████████ █████████ █████ ███ ███ █████████ ████ ██ ███ ████████ ████████ ███ ███ █████ ███ ██████ ███████ █████████████ ███████████ █████████ ████ ███ ███████ ██ ████ ████ ████ ██████ ███ █████████ ██████████ █ ████ ██ █ █████████████ ██████ ███████ ██████
███████████ ██ █████ ███████ ███ ███████ ███████████████ ████ ████ ██ ██████ █████████████ ████████ ██ ███████████ ██ ████ ███████ ███ ████████ ██████ ████ ███ ██████████ Table 30 ███ ███ ███████████ ███████████ █████████████ ███ ████ ██████████ ███ ████████ ██████████ █████████ ████████ █████████████ ███ ███████████ ███████████ █████████████ ███ ██████ █████ ███ ███████ ███████████████ ████ ████████ █████ ████ █ ████████ ███████████ ██ █████████ ███████ ████ ███ ██████ ██ ██████ ███ ████████ ████████ ████████ ███ ██████████ ██ ███████████ ███ ████████ ███████ █████████████ ██ ██████ █████ ██ ███ █████████ ███ ████ ██████ █████ ██████ ████████ ███████ ███ ████████ ██ ██ ██████████ █████ ██ ████████ ████ ████████ ████ ███ ████████ ████████ ███████ █████ ██ ██████ ███████ ██████ ████ ██ ████ ██ █████████ █████ ██ ██████ ███████ ████ ███ ███ ████ █████████ ███████████ █████ ██████ █████ █████████████ ███ ████████ ██ ██████ ████ ██████ ██ ██████████ ██████ ████ ██████████ ██ ██ ████ ███ █████████ ███ ██████████ ███ ██████████ ███████ ██████ ███████████
███████ ███████████ ████████ ████ ██████████ ██████ ██ ████ ███████████ ███████ ███ █████ ██████ ███ ██████████ █████ █████████ ███████ ██ ████ ██ ███ ██████ ██ █████████ ███████ ██████████ ███████ ███████████ ████████ █ ████████ ███████ ██████ ██████████ ████████ ████ ███ ████ ███ ██████████ ███ ████████ ████████ ███ ████████ ██████ ██████████ ████████ ████ ███ █████ ███████████ █████ ████ ██████████ ██████ ███ ███ ███████ ██████ ██████████ █████████ ███████████ ████████ █ ███ █ ████ ████ ██ ██████ ███ ██████ ██ █████████ █████████ ████ ███ █████ ███████ ████████████ ██ ████ ███ ██████ ██ █████████ ██████ ██ █████████████ ███████ ███ █████ ████████ ██ █████████ ███████████ ████████ █ ███ ████ ██ ████ ███ ██████ ██ █████████ ████ ████ ███████ ██ ████████ ██ █████ ████ ███ ███████████ ████████ ██ ██ ████ ███ ██████ ██ █████████ ███████ ██████████ ██████ ████████ ████ ██ ███ █████ ███ ███████ ████████ ███ █████ █████████ ███ ████████ ██ ███████ ████ ██ ███ █████████ █████████ ████████ ██████ █ ███ ██████ ███ ██████ ████████ ██████ ███ █ ██████ ████ █████ █ ██████ █████ ████████ █████ ██ ███ ██████ ██ ███████ █████████ █████ ████████ ███████ ████████ ██ █████ ████████ █████ ██████ ██ ████████
Table 31: ITC Analysis Methods
Method | Sponsor-submitted ITC and NMA |
|---|---|
ITC methods | ██████ ███ ██████████████ ████████ ███ |
Priors | ██████ ███████████████ █████ █████████████ ████ ████ ███ ███ ████████ ███ █████████ ███████ |
Assessment of model fit | █████ ███ ████ ██ ████████ █████████ ███ ███ ███ █████████ ████ ██ ███ ████████ ████████ ███ ███ ██ ███ ██ ██████ |
Assessment of consistency | █████████████ ███ ████ ██████████ ████ ██ ████████ ██████████ █████████ ████████ █████████████ ███ ███████████ ████████ ██ ███ ██████████ ██ ███████████ ███ ██ █████████████ |
Assessment of convergence | ███████████ ████ █████ ██ ███████ ███████ ██████████ ██ ███ ██████ ██ █████ █████ █████ ██ ███ ███ ██ ███ ███████████ █████ ██████████ ███████████ ████ ██ ████ |
Outcomes | Outcomes included percent and absolute change in LDL-C from baseline at 24 weeks, total discontinuations and discontinuations due to AEs at 24 weeks, and percent change in HDL-C at 24 weeks. |
Follow-up time points | 24-week follow-up was chosen as the base case. |
Construction of nodes | ███████ ████ ███ ████████ ███ ███ ███████ ████ ██████ ██ ███ █████ █████████ ███ ████ ███ ██████████ ██ ███ ███████ █████ ███ ██████ ███ ████ █████ ██ ██████████ ████ |
Sensitivity analyses | ████ ███████████ ████████ ████ ██████████ ████ █████████ ██ ███████ ██████ ██████████ ████████ ████ ███ ████ ███████████ ███ ████████ ████████ ███ ████████ ██████ ██████████ ████████ ████ ███ █████ ███████████ █████ ████ ██████████ ██████ ███ ███ ███████ ██████ ██████████ █████████ ███ ███ ████ █████████ ██████ ██ █████████████ ███████ ███ █████ ████████ █████████ ████ █████████ ████ ████ ███████ ██ ████████ ██ ████████ |
Subgroup analysis | █████ |
Methods for pairwise meta-analysis | ██ |
ASCVD = atherosclerotic cardiovascular disease; DIC = deviance information criterion; FE = fixed effects; HeFH = heterozygous familial hypercholesterolemia; HDL-C = high-density-lipoprotein cholesterol; ITC = indirect treatment comparison; ITT = intention to treat; LDL-C = low-density-lipoprotein cholesterol; MTD = maximally tolerated dose; NMA = network meta-analysis; NR = not reported; Q2W = every 2 weeks; QM = every month; RE = random effects; SA = sensitivity analysis; SD = standard deviation.
Source: Sponsor-submitted NMA report.34
Results of ITC
Summary of Included Studies
The results of the systematic review were included in a separate report. █████ ██ ██ ████████ ████ ████ ████████ ███ ███ ███████████ ██ ██████████ ██ ████ ████████ ███████ ████ ████ █████████ ██ ███ ████████ ███ ██████ ██ ██████████ ██ █████ ██████████ █████ ██ ███ ████████ ████ ██ ███████ ██████████ ██████████████ █████████ ██████ ███████████ ██ ██████ ████ ███████████ █████ ██████ █████ █ ████ ██ ███ ██████ ████████ ███ ███ ████ ███ ██████ ███ █████████ ███ █████████ ████████ █ ████████ ███████████ ██ ███ ██████ ████████ ██ ███ ██████████ ██████ ███ ███ ███ ███ █████████ ████ ███████ ████ ██████████ ███ ███ ███ ███ ██ ███ ██████ ██ █████████████████████ ████████ ██████ █████████ █ ███████ ██ ████████ ███████████ ██ ████████ ███████ ███████████████ ████ ███ ████████ ██████ ████ ███ █████████ ███ █████████ ██████████ ██ █████████████ ███████ ██ █████████ █████████ ██ █████ ██████ ███ ███ ████████
Results
HeFH Populations
████████ ████ █████ ███ ██ ██ ████████ ███ ███ ███████████ ███ ███████████ ██ ████████ ██████ █████████████ ██ ██████████ ███████ ████████ ████████ ██ ████████ ███ ███ ████ ██████ ██████████ ███████████ ██ █ ███████ ████ ████ ████ ████████ ███ ████████ ████ ████ ███████ ████ ███ ███████
The network for the HeFH population on MTD statins is shown in Figure 4. A total of 7 trials were included in the network. ███ █████ █████████ ██████████ ██ ███████ ████████ ████ █████ ███ ████████ ██ ███ █████ ██ ████████ █████ ███ ███████ ████ ██ █████ ███ ████████ ██ ███ ███████████ ████████ ████████████ ████████ ██ ███ ████ ███████
Figure 4: Network Diagram for the HeFH Populations on MTD Statins
HeFH = heterozygous familial hypercholesterolemia; MTD = maximally tolerated dose; NMA = network meta-analysis.
*Subgroup data for patients with HeFH were used in the analysis.
Notes: Interventions and placebo arms are in addition to background statin with or without other LLTs; no network is feasible for statin-intolerant patients.
Red text indicates that the study was excluded from a sensitivity analysis.
Source: Sponsor-submitted NMA Report.34
Base-Case Results
Base-case results for the percent change in LDL-C at 24 weeks, absolute change in LDL-C at 24 weeks, total discontinuations at 24 or more weeks, discontinuations due to AEs at 24 or more weeks, and percent change in HDL-C at 24 weeks in the HeFH populations on MTD statins are summarized in Table 32.
████ ███████ ██ ███████ ██████ ██ ██████ ██████████ ███ ████████ ████ ███████ █████ ██████████ █████ ███████ ████ ████ ███████ █████████ ███ ███ ███ ████████ ████ ██████████ ████ █████ ████ ████ ███████ ███████ ███ ██████████ ████ █████ ████ ████ ███████ ████████ ███ ██████████ ████ ████████ ████ ██████
███ ████████ ██████ ██ █████ ██ ██ ██████ ███ ██████████ ██ ███ ███████ ████ ████████ ████ ████████ ██████████ ███ ███ ████████ ████ ███ ████████ ███████████ ██ ██████████ ████ ████ █████ ████ ████ ███████ ███████ ██ ██████████ ████ █████ █████ ████ ████ ███████ ████████
███████ ███ █████ ████████████████ ██ █ ██ █████ ███████████ ████ ███████ ██████████ ██ ██████████ ████ ████████ ████ ███████ ████ ████ ██████ █████ ███ ███ ████ ██████ ██████ ██████████████ ███ ██████████ ███ ███ ████████ ████ ██████████ ████ ████ ████ ████ █████ ███████ ███████████ ████ ██████████ ████ ███ ████████ ███ ████ ████████ █████ ████ ████ ████ ████ █████████ ███ █████████████ █████ ███ █ ███████ ██████
███ ████████████████ ███ ██ ███ ██ █ ██ ██████ ███████ ██████████ ██ ██████████ ████ ████████ ████ ███████ ████ █████ ████ ████ █████ █████████ ███ ███ ████ ████ ████ █████ ██████ ██████████████ ███ █████ ███ ██ ██████████ ███████ ██████████ ███ ██████████ ████ █████ ████ ████ █████ ██████████ ██ ██ ██████ ████ ███ █████ █████ █████ ████ ██████ ██ ████████████████ ███ ███ ██ ███ ██████████ ██████ ███████████ ██ ████ ███ ████████ █████████ ███████ █████ ███ ███████████ ████████ ██ ███ ███████ ██ ███ █████████ ██ ██████████ ███ ███████ ████████ ██ ███████████ ███████ ██████ ██ ███████████ ████ ████████ ███████████ ████ ██████████ ████ ███ ████████ ███ ████ ████████ █████ ████ ████ ████ ████ █████████ ███ █████████████ █████ ███ █ ███████ █████
███ ███████ ██████ ██ █████ ██ ██ ██████ █████████ ██ █████ ██████ █████████ ██ ████████████ ██████████ ███ ███ ████ █████████ ████ ██ ████████ ████ ███████ ████ ███████ ██ ███████ ██████ ██ █████ ██ ██ █████ ████ █████ ████ ████ █████ ████████ ██████████ ███ ██████████ ████ ██████ ██ ██████████ ████████ ██ ████████ ███ ██ ██████████ ███ ████████ ███████ ███ ██ ███ █████ ██████████
██ ███ █████████ ███████████ █████████████ ███ ██████████ ████████ ██ ████████ ██ ███ ██████ █ █████ ██ ███████ █████ █████████████ ███ ███ ███████ █████████ ██████████ ██ ███████ ███ ███ █████ ██████ ███ ███████ ██████ ██ █████ ██ ██ █████ █████ ████████████ ████████
Table 32: HeFH MTD Random-Effects NMA Results
Comparator | Random-effect difference (95% CrI) |
|---|---|
Percent change in LDL-C at 24 weeks (mean difference) | |
██████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ███████ | ███████ ████████ ███████ |
██████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ██████████ | ████ ████████ ██████ |
██████████ ███ ██████████ | ████ ████████ ██████ |
Absolute change in LDL-C at 24 weeks (mean difference) | |
██████████ ███ ███████ | ██████ █████████ ███████ |
██████████ ███ ███████ | ██████ █████████ ███████ |
██████████ ███ ███████ | ██████ █████████ ███████ |
██████████ ███ ██████████ | ████ ████████ ██████ |
██████████ ███ ██████████ | █████ ████████ ██████ |
Total discontinuations at ≥ 24 weeks (odds ratio) | |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ██████████ | ████ ██████ █████ |
Discontinuation due to AEs at ≥ 24 weeks (odds ratio) | |
██████████ ███ ███████ | █████ ██████ ████████ |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ██████████ | █████ ██████ ████████ |
Percent change in HDL-C at 24 weeks (mean difference) | |
██████████ ███ ███████ | ████ ███████ ██████ |
██████████ ███ ███████ | ████ ██████ ██████ |
██████████ ███ ███████ | ████ ███████ ██████ |
██████████ ███ ██████████ | █████ ████████ █████ |
██████████ ███ ██████████ | █████ ████████ █████ |
AE = adverse event; CrI = credible interval; HDL-C = high-density-lipoprotein cholesterol; HeFH = heterozygous familial hypercholesterolemia; LDL-C = low-density-lipoprotein cholesterol; MTD = maximally tolerated dose; NMA = network meta-analysis.
Source: Sponsor-submitted NMA report.34
Sensitivity Analysis
███████ ██ ███ ███████████ ████████ ███ ████████ ███ ████████ ██████ ██ █████ ██ ██ █████ ███ ██████████ ██ Table 33 ██ ███████████ ████████ ████ █████████ ███ █████ ████████████████ ██ █ ██ ██████ ████████████████ ███ ██ ███ ██ █ ██ ██████ ██ ███████ ██████ ██ █████ ██ ██ ██████ ████ █████ ███ ███████████ ████████ ██ ██ ██ ███ █ ████ █████████ ███ ███████ ██████ ██ █████ ██ ██ ██████ ████████ ████ ██ ████████ ████ ████ ████ ███████ ███ ███ ██████████ ███ █████████ ███████████ ████████ █ █████ █████ ███ ████ ████████ ██████████ ███ ███████ ████ ███████ ██ ███ ████ █████ ████ ██████████ █████████ ███████ ██ ███ ███████████ █████████ ███ ███ ██████████ ██ ██████████
Table 33: HeFH MTD — Sensitivity Analysis Results for the Difference in Percent and Absolute Change in LDL-C
Comparator | Difference in CFB (95% CrI) | |
|---|---|---|
Percent change in LDL-C | Absolute change in LDL-C | |
████ ████ | ||
██████████ | ████ ████████ ██████ | ████ ████████ ██████ |
██████████ | ████ ████████ ██████ | █████ ████████ ██████ |
███████ | ██████ ████████ ███████ | ██████ ████████ ███ |
████ ███████ ███████ ██████ ██████████ ████████ | ||
██████████ | ████ ████████ ██████ | ████ ████████ ██████ |
██████████ | ████ ████████ ██████ | █████ ████████ ██████ |
███████ | ██████ ████████ ███████ | ██████ ████████ ██ |
████ ███████ ██████ ███████ ████ | ||
██████████ | █████ ████████ █████ | █████ ████████ ██████ |
██████████ | █████ ████████ ██████ | █████ ████████ ██████ |
███████ | ██████ ████████ ███████ | ██████ ████████ ██ |
████ ███████ █████████████ ███████ ████ | ||
██████████ | █████ ███████ ██████ | ██ |
██████████ | █████ ████████ ██████ | ██ |
███████ | ██████ ████████ ███████ | ██ |
████ ███████ ███████ ████ ██ | ||
██████████ | ████ ████████ ██████ | ██ |
██████████ | ████ ████████ ██████ | ██ |
███████ | ██████ ████████ ██████ | ██ |
CFB = change from baseline; CrI = credible interval; HeFH = heterozygous familial hypercholesterolemia; LDL-C = low-density-lipoprotein cholesterol; MTD = maximally tolerated dose; NR = not reported; SA = sensitivity analysis.
Source: Sponsor-submitted NMA report.34
ASCVD and ASCVD-RE Populations on MTD Statins
The network for the ASCVD and ASCVD-RE populations on MTD statins is displayed in Figure 5. A total of 13 studies were included in the base-case network. ███ ███████ ██████ █ ██████ ████ ███████ ████████ ███████████ ███████████ ███ ██████████ ██ ██████████ ██████████ ███ ████ ██████ █████ ██████ ████████ ███████ ████ ████████ ████ ███ ████████ ████████ ███████ █████ ██ ██████ ███████ ██████ ████ ██ ████ ██ █████████ █████ ██ ██████ ███████ ████ ███ ███ ████ █████████ ██████████
Figure 5: Network Diagram for the ASCVD and ASCVD-RE Populations on MTD Statins
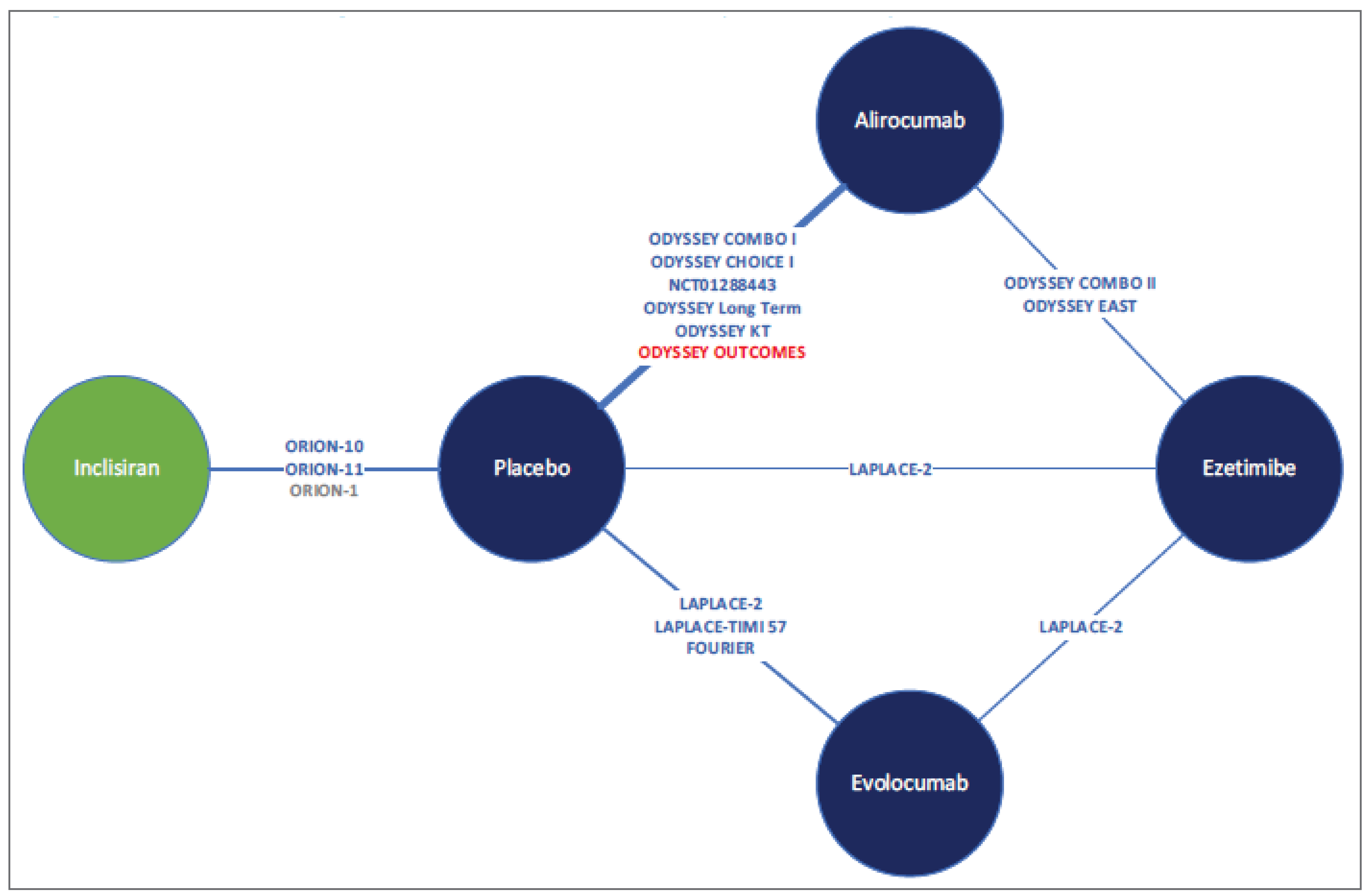
ASCVD = atherosclerotic cardiovascular disease; ASCVD-RE = atherosclerotic cardiovascular disease risk equivalent; MTD = maximally tolerated dose; NMA = network meta-analysis.
Notes: Interventions and placebo arms are in addition to background statins, with or without other LLTs.
Red text indicates that the study was excluded from a sensitivity analysis.
Grey text indicates that the study was only included in a sensitivity analysis.
Source: Sponsor-submitted NMA report.34
Base-Case Results
Base-case results for the percent change in LDL-C at 24 weeks, absolute change in LDL-C at 24 weeks, total discontinuations at 24 or more weeks, discontinuations due to AEs at 24 or more weeks, and percent change in HDL-C at 24 weeks in the ASCVD and ASCVD-RE population on MTD statins are summarized in Table 34.
████ ████ ███ ███ ███████████ ████████ ████ █████████ ███ ███ █████ ███ ████ ██████████ ███████████ ██ ███ ███████ ███ ███████ ██████ ██ █████ ██ ██ ██████ ████ ███████ ██ ███████ ██████ ██ █████ ██ ██ ██████ ██████████ ███ ████████ ████ ████ ███████ ████ ███████ ████ ████ ███████ ████████ ███ █████████ ████ ███████ ████ ███ ███████ █████████ ███ ███ ███ ████████ ████ ██████████ ███ ███████████ ███ ██████████ ████ ████████ ████ ███████
███ ███ ████████ ██████ ██ █████ ██ ██ ██████ ██████████ ███ ████████ ████ ███████ ████ ██████ █████ ████ ████ ███████ ████████ ███ █████████ ████ ██████ █████ ████ ████ ███████ ████████ ███ ██████████ ████ ████████ ████ ███████ ████ ███████ ██ ████████ ██████ ██ █████
████ ███ ████ ████ ████████ ████ █████████ ███ █████ █████████████████ ██ ██████████ ████████ ██ ███ ████████ ████ ████████ ████ ████████ ███ ███ ████████ ██████████ ██
██ ████ █████ █████████████████ ████████████████ ███ ██ ███ ████ ███ █████████ █████ ████████ ███████████ ███ ██ ██████████ ████ ████████ ████ ███████
███████ ██ ███ ████ ████ ████████ ███████ ████ ██████████ ██ ████████ ████ ███████ ████ █████ ████ ████ █████ ███████ ███ █████████ ████ █████ ████ ████ █████ ███████ ████████ ██ ███████ ██████ ██ █████ ██ ██ ██████ █████████ ████ ██████████ █████████████ ████ ████████ ████ ███████ ████ █████ ████ ████ █████ █████ ███ ███ █████ ████ ████ ████████ ████████ █████ ███ ██ ██████████ ███████ ███████████
Table 34: Random-Effects NMA Results for the ASCVD and ASCVD-RE Populations on MTD Statins
Comparator | Random-effect difference (95% CrI) |
|---|---|
Percent change in LDL-C at 24 weeks (mean difference) | |
██████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ███████ | ██████ ████████ ███████ |
█████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ██████████ | ████ ████████ ██████ |
██████████ ███ ██████████ | ████ ███████ ██████ |
██████████ ███ █████████ | ██████ ████████ ███████ |
Absolute change in LDL-C at 24 weeks (mean difference) | |
██████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ███████ | ██████ ████████ ███████ |
█████████ ███ ███████ | ██████ ████████ ██████ |
██████████ ███ ██████████ | ████ ████████ ██████ |
██████████ ███ ██████████ | █████ ███████ ██████ |
██████████ ███ █████████ | ██████ ████████ ██████ |
Total discontinuations at ≥ 24 weeks (odds ratio) | |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ███████ | ████ ██████ █████ |
█████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ██████████ | ████ ██████ █████ |
██████████ ███ ██████████ | ████ ██████ █████ |
██████████ ███ █████████ | ████ ██████ █████ |
Discontinuation due to AEs at ≥ 24 weeks (odds ratio) | |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ███████ | ████ ██████ █████ |
█████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ██████████ | ████ ██████ █████ |
██████████ ███ ██████████ | ████ ██████ █████ |
██████████ ███ █████████ | ████ ██████ █████ |
Percent change in HDL-C at 24 weeks (mean difference) | |
██████████ ███ ███████ | ████ ██████ ██████ |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ███████ | ████ ██████ ██████ |
█████████ ███ ███████ | █████ ███████ █████ |
██████████ ███ ██████████ | ████ ███████ █████ |
██████████ ███ ██████████ | █████ ███████ █████ |
██████████ ███ █████████ | ████ ██████ ██████ |
AE = adverse event; ASCVD = atherosclerotic cardiovascular disease; ASCVD = atherosclerotic cardiovascular disease risk equivalent; CrI = credible interval; HDL-C = high-density-lipoprotein cholesterol; LDL-C = low-density-lipoprotein cholesterol; MTD = maximally tolerated dose; NMA = network meta-analysis.
Source: Sponsor-submitted NMA report.34
Sensitivity Analysis
Results of the sensitivity analyses for the percent change and absolute change in LDL-C at 24 weeks are summarized in Table 35.
███████████ ████████ ████ ██████████ ████ ████ ████ ███████ ███ ███████ ██████ ██ ██████ ███████ ██ ███████████ ████████ █ ███ █ ██████████ ██ ████ ███ █████████████ ████████ ███ ████████ ██████ ██████████ ███ ████████ ████ ██████████ ████ █████ ████ ███ █████ ███████ ███ ███ █████ ████ ███ █████ ███████ ██████████████ █████████ ████████ ██████ ██████ ██ ███ █████████████ █████ ████████ ███ ████████ ███████ ████ ██████████ ████ █ ███████████ ███████ ██████████████ ███ ██████████ ████ ███████████ ███ ███ ███ ██████████ █████████
█████ ███████████ ████████ ████ █████████ ███ ████████ ██████ ██ ██████ ███████████ ████████ ███ ███ ████ ██ ██████ ██ ███ ██████████████ ██ ████████ ███ ████ ██████████ ████ ████ ████ ████████ ████ ██████████ ████████ ████ ███████ ███ █████████ ██ ███ ███████████ █████████ ███ ██ ██████████ ███████ ██████████ ███ ██████████ ███ ███████
Table 35: Sensitivity Analysis Random-Effects NMA Results for the ASCVD and ASCVD-RE Populations on MTD Statins
Comparator | Difference in CFB (95% CrI) | |
|---|---|---|
Percent change in LDL-C | Absolute change in LDL-C | |
████ ████ | ||
██████████ | ████ ████████ ███ | ████ ████████ ██████ |
██████████ | ████ ███████ █████ | █████ ███████ ██████ |
█████████ | ██████ ████████ ████ | ██████ ████████ ███ |
███████ | ██████ ████████ ████ | ██████ ████████ ████ |
████ ███████ ████████ ███ ████████ ██████ ██████████ ████████ | ||
██████████ | ████ ████████ ██████ | ████ ████████ ██████ |
██████████ | ████ ███████ ██████ | █████ ███████ ██████ |
█████████ | ██████ ████████ ███████ | ██████ ████████ ███ |
███████ | ██████ ████████ ███████ | ██████ ████████ ████ |
████ ███████ ██████ ████████ ███ ████████ ████ | ||
██████████ | ████ ███████ ██████ | ████ ████████ ██████ |
██████████ | █████ ██████ ███ | ████ ███████ ██████ |
█████████ | ██████ ████████ ████ | █████ ████████ ████ |
███████ | ██████ ████████ ████ | ██████ ████████ ████ |
████ ███████ █████████████ ██████ ██ ███ ████████ ████ | ||
██████████ | ████ ███████ ██ | ██ |
██████████ | █████ ██████ ██ | ██ |
█████████ | ██████ ████████ ███ | ██ |
███████ | ██████ ████████ ████ | ██ |
████ ███████ ███████ ████ | ||
██████████ | ████ ███████ ██████ | ██ |
██████████ | █████ ███████ ████ | ██ |
█████████ | ██████ ████████ ███ | ██ |
███████ | ██████ ████████ ███ | ██ |
████ ███████ ███████ ████████ | ||
██████████ | ████ ████████ ██████ | ██ |
██████████ | ████ ███████ ██████ | ██ |
█████████ | ██████ ████████ ███ | ██ |
███████ | ██████ ████████ ██ | ██ |
ASCVD = atherosclerotic cardiovascular disease; ASCVD = atherosclerotic cardiovascular disease risk equivalent; CFB = change from baseline; CrI = credible interval; LDL-C = low-density-lipoprotein cholesterol; MTD = maximally tolerated dose; NMA = network meta-analysis; NR = not reported; SA = sensitivity analysis.
Source: Sponsor-submitted NMA report.34
ASCVD and ASCVD-RE Populations Intolerant to Statins
A total of 7 trials were included in the network for the ASCVD and ASCVD-RE populations intolerant to statins (Figure 6). There were no closed loops.
Base-Case Results
Base-case results for the percent change in LDL-C at 24 weeks, absolute change in LDL-C at 24 weeks, total discontinuations at 24 or more weeks, discontinuations due to AEs at 24 or more weeks, and percent change in HDL-C at 24 weeks in the ASCVD and ASCVD-RE population intolerant to statins are summarized in Table 36.
Figure 6: Network Diagram for the ASCVD and ASCVD-RE Populations Intolerant to Statins
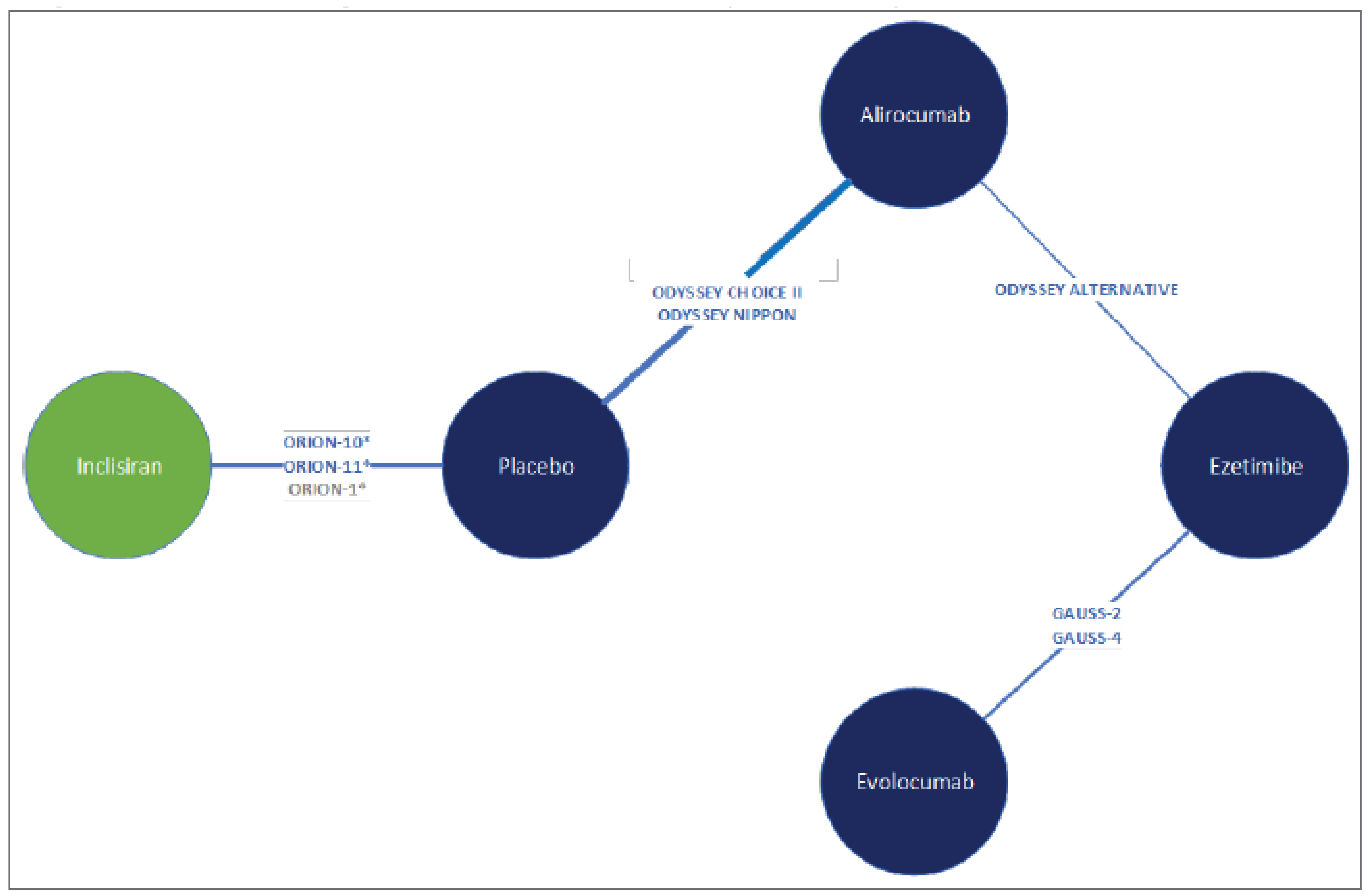
ASCVD = atherosclerotic cardiovascular disease; ASCVD = atherosclerotic cardiovascular disease risk equivalent; NMA = network meta-analysis.
*Subgroup data for statin-intolerant patients to be used in the analysis.
Notes: Interventions and placebo arms are in addition to background statins, with or without other LLTs.
Grey text indicates that the study was only included in a sensitivity analysis.
Source: Sponsor-submitted NMA report.34
████ █████ ███████████ ████████ █ ███ █ ████ █████████ ███ ███████ ██████ ██ ██████ ██ ███ ████ █████ ██████████ ███ ████████ ████ ███████ ████ ███████ ████ ████ ███████ █████████ ███ ███ ███ █████████ ████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██ █████████ ████ ███████ ████ ████ ███████ ███████ ███ ██████████ ████ ████████ ████ ███████ ████ ███████ ██ ██████ ██ █████ ██ ██ ██████
████ █████ ███████████ ████████ █ ███ █ ████ █████████ ███ ███████ ██████ ██ ██████ ██ ███ ████ █████ ██████████ ███ ████████ ████ ███████ ████ ███████ ████ ████ ███████ █████████ ███ ███ ███ █████████ ████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██ █████████ ████ ███████ ████ ████ ███████ ███████ ███ ██████████ ████ ████████ ████ ███████ ████ ███████ ██ ██████ ██ █████ ██ ██ ██████████ █████ ███████████ ████████ █ ███ █ ████ █████████ ███ ███████ ██████ ██ ██████ ██ ███ ████ █████ ██████████ ███ ████████ ████ ███████ ████ ███████ ████ ████ ███████ █████████ ███ ███ ███ █████████ ████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██ █████████ ████ ███████ ████ ████ ███████ ███████ ███ ██████████ ████ ████████ ████ ███████ ████ ███████ ██ ██████ ██ █████ ██ ██ ██████████ █████ ███████████ ████████ █ ███ █ ████ █████████ ███ ███████ ██████ ██ ██████ ██ ███ ████ █████ ██████████ ███ ████████ ████ ███████ ████ ███████ ████ ████ ███████ █████████ ███ ███ ███ █████████ ████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██ █████████ ████ ███████ ████ ████ ███████ ███████ ███ ██████████ ████ ████████ ████ ███████ ████ ███████ ██ ██████ ██ █████ ██ ██ ███████████ █████ ███████████ ████████ █ ███ █ ████ █████████ ███ ███████ ██████ ██ ██████ ██ ███ ████ █████ ██████████ ███ ████████ ████ ███████ ████ ███████ ████ ████ ███████ █████████ ███ ███ ███ █████████ ████ ██████████ ████ ██████ ████ ████ ███████ ████████
Table 36: Random-Effects and/or Fixed-Effects NMA Results for the ASCVD and ASCVD-RE Populations Intolerant to Statins
Comparator | Random-effects and/or fixed-effects differencea (95% CrI) |
|---|---|
Percent change in LDL-C at 24 weeks (mean difference) | |
██████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ███████ | ██████ ████████ ███████ |
█████████ ███ ███████ | ██████ ████████ ██████ |
██████████ ███ ██████████ | █████ ████████ ██████ |
██████████ ███ ██████████ | █████ ████████ ██████ |
██████████ ███ █████████ | ██████ ████████ █████ |
Absolute change in LDL-C at 24 weeks (mean difference) | |
██████████ ███ ███████ | ██████ ████████ ███████ |
██████████ ███ ███████ | ██████ █████████ ███████ |
██████████ ███ ███████ | ██████ █████████ ███████ |
█████████ ███ ███████ | ██████ ████████ ██████ |
██████████ ███ ██████████ | █████ ███████ ██████ |
██████████ ███ ██████████ | █████ ████████ ██████ |
██████████ ███ █████████ | ██████ ████████ ██████ |
Total discontinuations at ≥ 24 weeks (odds ratio)a | |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ███████ | ██ |
█████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ██████████ | ████ ██████ █████ |
██████████ ███ ██████████ | ██ |
██████████ ███ █████████ | ████ ██████ █████ |
Discontinuation due to AEs at ≥ 24 weeks (odds ratio)a | |
██████████ ███ ███████ | ████ ██████ ██████ |
██████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ███████ | ██ |
█████████ ███ ███████ | ████ ██████ █████ |
██████████ ███ ██████████ | ████ ██████ ███████ |
██████████ ███ ██████████ | ██ |
██████████ ███ █████████ | ████ ██████ ██████ |
Percent change in HDL-C at 24 weeks (mean difference) | |
██████████ ███ ███████ | █████ ██████ ██████ |
██████████ ███ ███████ | ████ ██████ ██████ |
██████████ ███ ███████ | █████ ███████ ██████ |
█████████ ███ ███████ | ████ ███████ ██████ |
██████████ ███ ██████████ | ████ ███████ ██████ |
██████████ ███ ██████████ | █████ ████████ ██████ |
██████████ ███ █████████ | ████ ████████ ██████ |
AE = adverse event; ASCVD = atherosclerotic cardiovascular disease; ASCVD-RE = atherosclerotic cardiovascular disease risk equivalent; CrI = credible interval; HDL-C = high-density-lipoprotein cholesterol; LDL-C = low-density-lipoprotein cholesterol; NMA = network meta-analysis.
aFixed effects were conducted for the total discontinuations and discontinuations due to AEs at ≥ 24 weeks.
Source: Sponsor-submitted NMA report.34
Sensitivity Analysis
████ █████ ███████████ ████████ █ ███ █ ████ █████████ ███ ███████ ██████ ██ ██████ ██ ███ ████ █████ ██████████ ███ ████████ ████ ███████ ████ ███████ ████ ████ ███████ █████████ ███ ███ ███ █████████ ████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██ █████████ ████ ███████ ████ ████ ███████ ███████ ███ ██████████ ████ ████████ ████ ███████ ████ ███████ ██ ██████ ██ █████ ██ ██ ██████████ █████ ███████████ ████████ █ ███ █ ████ █████████ ███ ███████ ██████ ██ ██████ ██ ███ ████ █████ ██████████ ███ ████████ ████ ███████ ████ ███████ ████ ████ ███████ █████████ ███ ███ ███ █████████ ████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██ █████████ ████ ███████ ████ ████ ███████ ███████ ███ ██████████ ████ ████████ ████ ███████ ████ ███████ ██ ██████ ██ █████ ██ ██ ██████████ █████ ███████████ ████████ █ ███ █ ████ █████████ ███ ███████ ██████ ██ ██████ ██ ███ ████ █████ ██████████ ███ ████████ ████ ███████ ████ ███████ ████ ████ ███████ █████████ ███ ███ ███ █████████ ████ ██████████ ████ ██████ ████ ████ ███████ ████████ ██████████ ████ ██████ ████ ████ ███████
Table 37: Sensitivity Analysis Random-Effects NMA Results for the ASCVD and ASCVD-RE Populations Intolerant to Statins
Comparator | Difference in CFB (95% CrI) | |
|---|---|---|
Percent change in LDL-C | Absolute change in LDL-C | |
████ ████ | ||
██████████ | █████ ████████ ██████ | █████ ███████ ██████ |
██████████ | █████ ████████ ██████ | ████ ███████ ██████ |
█████████ | ██████ ████████ █████ | ██████ ████████ ██ |
███████ | ██████ ████████ ███████ | ██████ ████████ |
████ ███████ ██████ ████████ ███ ████████ ████ | ||
██████████ | █████ ███████ ██████ | █████ ██████ ██████ |
██████████ | █████ ███████ ██████ | █████ ██████ ██████ |
█████████ | ██████ ████████ █████ | ██████ ███████ ███ |
███████ | ██████ ████████ ███████ | ██████ █████ |
████ ███████ █████████████ ██████ ██ ███ ████████ ████ | ||
██████████ | █████ ███████ ██████ | ██ |
██████████ | █████ ███████ ██████ | ██ |
█████████ | ██████ ████████ █████ | ██ |
███████ | ██████ ████████ ███████ | ██ |
ASCVD = atherosclerotic cardiovascular disease; ASCVD-RE = atherosclerotic cardiovascular disease risk equivalent; CFB = change from baseline; CrI = credible interval; LDL-C = low-density-lipoprotein cholesterol; NMA = network meta-analysis; NR = not reported; SA = sensitivity analysis.
Source: Sponsor-submitted NMA report.34
Critical Appraisal of ITC
The NMA was based on an adequately conducted systematic literature search that included planned searches of multiple databases, conference proceedings, clinical trial registries, as well as a regulatory and health technology assessment agency websites. Screening was conducted based on standard methods, with studies selected independently in duplicate, according to prespecified criteria. Although planned, no quality assessment of the included studies was reported in the sponsor’s NMA; however, a full quality assessment was included in the SLR report.
The population, interventions, and outcomes of the sponsor-submitted systematic review were relevant to Canadian clinical practice. The eligible studies included adults with HeFH or nFH (ASCVD or ASCVD-RE) whose LDL-C is inadequately controlled by MTD statins or who are statin intolerant. The interventions included in the review were broader than the NMA and included icosapent ethyl, which is not publicly funded, and bempedoic acid, which is not currently available in Canada, as of this report; however, no studies were included in the NMA that evaluated these treatments. The dosing regimens of included interventions reflected clinical practice. The outcomes included in the systematic review were also broader than those selected for the NMA. Outcomes were relevant and appropriate for the treatment of HeFH and ASCVD; however, outcomes important to patients and of critical importance to this review, including the reduction in MACE or other CV events, were not considered or included as part of the population, intervention, comparators, outcomes, and study designs (PICOS) for the NMA.
A feasibility assessment was conducted to determine whether to conduct an NMA based on network connectivity and differences in study, patient, and outcome characteristics, which were provided in the accompanying SLR report, and reasons for the exclusion of studies was provided in the NMA report. It was noted that visual inspection of study heterogeneity was conducted for treatment-effect modifiers, including background statin use, definition of CV risk and severity, and time points for assessment. As noted by the clinical expert consulted by CDA-AMC, the treatment-effect modifiers of age, sex, and baseline LDL-C levels — the main drivers of differences in this population — were not included as treatment-effect modifiers in the NMA. There were several limitations of the key assumptions made in the feasibility assessment about background statin use and the time of assessment of outcomes, impacting clinical and methodological heterogeneity.
The NMA aimed to include only studies with patients who were receiving MTD statins as background therapy (with or without ezetimibe); however, a definition of MTD from each included study was not provided, and only the proportion of patients taking low-density, moderate-density, and high-intensity statins was provided in the SLR report, which may result in unknown heterogeneity across populations. Moreover, the ORION trials (ORION-9, ORION-10, and ORION-11) included a small proportion of patients that were intolerant to statins (25%, 22%, and 11%, respectively), but the proportion of statin-intolerant patients in other trials was not noted, so heterogeneity may exist. Of note, the results of sensitivity analyses, which excluded patients from the ORION-10 and ORION-11 trials and were consistent with the base-case results, and therefore it is unlikely that these patients had a significant impact on the results. It was also assumed that individual moderate-intensity and high-intensity statins (atorvastatin, rosuvastatin, and simvastatin) had a similar efficacy as background therapy, regardless of dose, and would not bias the results of the NMA. It was considered reasonable to assume that the background statin therapy used in all clinical trials followed treatment guidelines and would be well balanced, and differences in treatment effect would likely be minimal; however, it is unclear what effect different dosages of moderate-intensity or high-intensity statins may have had, based on discussions with the clinical expert consulted by CDA-AMC. Moreover, it is worth noting that inclisiran would only be given after treatment with MTD statins, and it was unclear if this was the case in the included studies. It was also assumed that differences in CV risk and severity would not impact the relative effects on LDL-C, and therefore no attempt to adjust for differences in baseline characteristics was conducted due to the number of studies and inconsistent reporting of characteristics.
The ORION trials had the longest follow-up of all included trials, and the duration of follow-up varied significantly across trials, from 12 weeks to 18 months (approximately 77 weeks), resulting in heterogeneity across included studies. The NMA used 24 weeks as the time of assessment, which may underestimate and bias the results of trials with longer durations. Although 24 weeks is likely appropriate to assess lipid-related outcomes, including LDL-C and HDL-C changes, it may not be sufficient to assess safety outcomes. Given the variation in trial follow-ups and durations, the authors considered EOS values for safety outcomes to be comparable if the duration of follow-up was 24 weeks or longer. As a result of the longer duration of the ORION trials, it is likely that more total discontinuations and discontinuations due to AEs were recorded purely based on trial length. This assumption is also likely to result in variations of events, favouring trials with shorter durations and differences in dosing regimens. Given the biannual dosing regimen of inclisiran, a 24-week time of assessment may be insufficient to assess safety outcomes compared to the 2-week dosing regimen of alirocumab and evolocumab; hence, the EOS duration was used for comparisons involving inclisiran.
Both fixed-effects and random-effects models were conducted. Random effects were considered most appropriate, given the number of studies per node and the heterogeneity observed. Fixed-effects NMAs were only conducted for safety outcomes in the ASCVD and ASCVD-RE population of patients intolerant to statins, given the small number of included studies. Model fit using deviance information criterion was assessed; however, no results for model convergence or fit were reported. Data from the ORION-10 and ORION-11 studies were pooled for the ASCVD analyses, which was acceptable given the observable similarities in the included populations; however, the method of pooling was not specified. It was noted that statistical heterogeneity for each pairwise comparison was high, with varying significant and nonsignificant P values for Q, and I2 values ranging from 0% to more than 80%. The authors also assessed global statistical heterogeneity via tau, which considered heterogeneity to be moderate. The Cochrane handbook for systematic review of interventions indicates that an I2 value of 75% or higher represents considerable heterogeneity, with the caveat that it is dependent on the magnitude and direction of effects and the strength of evidence for heterogeneity.89 Overall, the studies included in the NMA were believed to be statistically heterogeneous, based on the considerable range of I2 values; however, the results were considered to be uncertain due to the small number of studies included in the analysis. Moreover, the source of heterogeneity is unclear, as it was not explored. The authors relied on visual inspection of heterogeneity, based on the statistical tests, and concluded that the observed heterogeneity is likely due to observed and unobserved differences in patient populations across the included studies, data imputation analysis methods, and the specific background treatments allowed and/or delivered. Unidentified or unknown clinical (particularly treatment-effect modifiers) or methodological heterogeneity needs to be explored via additional subgroup analyses or meta-regression, as it is unclear if the transitivity assumption was appropriately met.
In the HeFH network, 1 trial was noted to be based on a subgroup of patients with HeFH (ODYSSEY Long-term), resulting in broken randomization for the comparison of this study with others, which may bias the results of the alirocumab and placebo comparison. For all outcomes except total discontinuations at24 or more weeks, all treatments were generally favoured over placebo, yet there was no difference between PCKS9 inhibitors. Additionally, results for all outcomes except total discontinuations at 24 or more weeks displayed exceedingly wide CrIs, leading to imprecise estimates of the treatment effect. In the ASCVD and ASCVD-RE population on MTD statins, randomization was preserved across studies and a closed loop was formed, allowing for both direct and indirect comparisons, which demonstrated no inconsistency. Except for safety outcomes, CrIs were wide, resulting in uncertainty across results. In the ASCVD and ASCVD-RE population of patients’ intolerant to statins, subgroup data were used for the ORION trials, which resulted in broken randomization from the ORION trials. For all outcomes, mostly all treatments were favoured over placebo; however, the CrIs for each outcome except total discontinuations at 24 or more weeks were exceedingly wide, resulting in imprecision in the treatment effects. It is unclear what caused the wide CrIs for outcomes in the NMAs, but it is believed to be due to study heterogeneity, and low sample sizes for certain outcomes. Sensitivity analyses were appropriately conducted to evaluate differences between the ORION trials and comparator PSCK9 inhibitor trials, as well as the impact of excluding outlier comparator trials. Results of the sensitivity analyses were consistent with base-case analyses.
In general, the 3 network scenarios made several clinical and methodological assumptions concerning the included populations, which limit the ability to interpret the generalizability of the results. Key assumptions included the equivalence of background statins regardless of individual statin or dose, the assumption that background ezetimibe is not an effect modifier, no adjustment of baseline characteristics for CV risk and severity, and the limited time of assessment for efficacy and safety outcomes given the variation in study durations. Although not reported or accounted for, these assumptions likely impacted treatment effects and the results of each NMA. Despite the limitations, there was no difference reported between inclisiran and other PCSK9 inhibitors in the efficacy and safety outcomes evaluated, and the results of most outcomes in all network scenarios displayed exceedingly wide CrIs, further challenging the precision of the results.
Studies Addressing Gaps in the Systematic Review Evidence
The contents of this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CDA-AMC review team.
Pooled Safety Analysis of 7 ORION Trials35
Description of Studies
This posthoc analysis comprised patients treated with 300 mg inclisiran sodium or placebo in the completed (ORION-1, ORION-3, ORION-5, ORION-9, ORION-10, and ORION-11) and ongoing (ORION-8) trials. The objective was to obtain data regarding the long-term safety and tolerability of inclisiran for up to 6 years.
In the safety analysis from these 7 clinical trials, patient-level data were pooled with the posthoc analysis containing placebo-controlled and open-label extension data.35 Specifically, the placebo patient pool was composed of a combined 1,968 patients representing 2,647.7 PYs of placebo exposure from the ORION-1, ORION-5, and the ORION-9, ORION-10, ORION-11 studies; the inclisiran patient pool was composed of 3,576 patients from all 7 trials and represents 9,982.1 PYs of inclisiran exposure. Databases were locked before the conduct of this analysis for all trials except ORION-8.35,90
The end points for this pooled safety analysis include the cumulative incidence over time for up to 6 years and the EAIRs of SAEs, TEAEs leading to drug discontinuation, scientifically relevant AEs (including hepatic, muscle, and renal events, incident diabetes, and AEs at the injection site), and MACE (including CV death, cardiac arrest, nonfatal MI, and nonfatal stroke), and changes in related laboratory parameters, as well as the incidence of treatment-induced ADAs.
The cumulative incidence of SAEs, AEs leading to study drug discontinuation, and AEs were examined over time with Kaplan-Meier curves, using data from the placebo-controlled studies for up to 1.5 year through to open-label extension studies with up to 6 years of cumulative exposure. The cumulative incidence of ADAs was also monitored for up to 4 years.90 Samples for detecting ADAs were collected at every study visit during the ORION-8, ORION-9, ORION-10, and ORION-11 trials; on day 1 and every 90 days thereafter during the ORION-3 trial; and on day 1, day 90, day 330, and day 720 during the ORION-5 trial. Patients from the ORION-1 trial were excluded from ADA analysis because the laboratory assay used in the ORION-1 trial differed from the other studies.
Results
Baseline Characteristics
As of March 9, 2022, a total of 3,576 patients (with a total of 9,982.1 PYs of drug exposure) have been treated with inclisiran across 7 ongoing and completed clinical trials, and 1,968 patients have been treated with placebo for up to 1.5 years (with a total of 2,647.7 PYs of exposure). Baseline demographic and clinical characteristics were generally comparable between treatment arms (Table 38). Most patients in both groups had ASCVD (83.6% and 84.1%, respectively) and were treated with statins (91.1% and 90.7%, respectively).
Table 38: Baseline Demographic and Clinical Characteristics
Characteristic | Inclisiran pool (N = 3,576) | Placebo pool (N = 1,968) |
|---|---|---|
Age, years (mean ± SD) | 63.5 ± 10.32 | 63.6 ± 10.22 |
Age ≥ 75 years, n (%) | 451 (12.6) | 270 (13.7) |
Female, n (%) | 1,184 (33.1) | 645 (32.8) |
Male, n (%) | 2,392 (66.9) | 1,323 (67.2) |
White race, n (%) | 3,298 (92.2) | 1,838 (93.4) |
BMI, kg/m2 | ||
n | 3,570 | 1,966 |
mean (SD) | 30.3 (5.71) | 30.6 (5.78) |
Baseline eGFR, mL/min per 1.73 m2 | ||
n | 3,575 | 1,967 |
mean (SD) | 79.2 (20.84) | 78.9 (20.93) |
CV risk factors, n (%) | ||
ASCVD | 2,990 (83.6) | 1,655 (84.1) |
ASCVD-RE | 586 (16.4) | 313 (15.9) |
Coronary heart disease | 2,690 (75.2) | 1,504 (76.4) |
Cerebrovascular disease | 478 (13.4) | 255 (13.0) |
Peripheral artery disease | 319 (8.9) | 168 (8.5) |
Diabetes mellitus | 1,226 (34.3) | 660 (33.5) |
Hypertension | 2,793 (78.1) | 1,552 (78.9) |
Current smoker | 555 (15.5) | 288 (14.6) |
LLT, n (%) | ||
Statin only | 2,416 (67.6) | 1,316 (66.9) |
Nonstatin LLT only | 141 (3.9) | 69 (3.5) |
Statin and nonstatin LLT | 842 (23.5) | 468 (23.8) |
No LLT | 177 (4.9) | 115 (5.8) |
ASCVD = atherosclerotic cardiovascular disease; ASCVD = atherosclerotic cardiovascular disease risk equivalent; BMI = body mass index; CV = cardiovascular; eGFR = estimated glomerular filtration rate; LLT = lipid-lowering therapy; SD = standard deviation.
Source: Wright et al. Safety and Tolerability of Inclisiran for Treatment of Hypercholesterolemia in 7 Clinical Trials, J Am Coll Cardiol. 2023 December 12;82(24):2251 to 2261. Copyright 2023 by the authors. Available from: https://www.sciencedirect.com/science/article/pii/S0735109723077392?via%3Dihub. Reprinted in accordance with Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International Licence (CC BY 4.0-NC-ND): https://creativecommons.org/licenses/by-nc-nd/4.0/.35
Harms
At least 1 SAE was reported in 32.2% and 22.1% patients in the inclisiran and placebo groups, respectively, with corresponding EAIRs of 13.80 (95% CI, 13.01 to 14.62) and 18.14 (95% CI, 16.48 to 19.93) per 100 PYs, respectively. The most common SAEs were cardiac, reported in 11.6% and 9.0% of patients, respectively. When adjusted for exposure, cardiac EAIRs were 4.39 (95% CI,3.98 to 4.84) per 100 PYs for inclisiran and 6.90 (95% CI, 5.92 to 7.99) per 100 PYs for placebo.35
At least 1 AE led to study drug discontinuation in 3.2% and 1.7% of patients in the inclisiran and placebo groups, respectively. The most common AE leading to study drug discontinuation was neoplasm (1.0% for inclisiran and 0.5% for placebo).35 The difference in the EAIRs was −0.14 (95% CI, −0.68 to 0.29) (Table 39).
AEs at the injection site were more frequent with inclisiran (9.3%) than with placebo (1.8%) (Table 40). In the inclisiran group, AEs at the injection site were more common in women (14.4%) than in men (6.7%). AEs at the injection site leading to study drug discontinuation were higher with inclisiran (0.1 per 100 PYs) than with placebo (0.0 per 100 PYs).
Kaplan-Meier analyses of hepatic-related, muscle-related, and kidney-related events showed no differences between the inclisiran and placebo treatment arms for up to 1.5 years. In the inclisiran group, compared to the placebo group, Kaplan-Meier analyses showed no evidence of an excess incidence of incident diabetes, and numerically fewer MACE-related safety events were reported.35
Table 39: Difference in EAIRs Between the Inclisiran and Placebo Pools
Event | Difference in EAIR (95% CI) |
|---|---|
AE leading to study drug discontinuation | −0.14 (−0.68 to 0.29) |
Serious AE | −4.34 (−6.30 to −2.51) |
Injection-site AE | 2.23 (1.61 to 2.79) |
Injection-site AE leading to study drug discontinuation | 0.10 (−0.05 to 0.19) |
Hepatic events | −0.67 (−1.47 to 0.02) |
Muscle events | −0.03 (−0.24 to 0.05) |
Kidney events | −0.48 (−1.33 to 0.25) |
Incident diabetes | −3.04 (−4.53 to −1.74) |
MACE-related safety events | −2.96 (−4.10 to −1.94) |
Alanine aminotransferase > 3 × ULN | 0.01 (−0.29 to 0.21) |
Creatine kinase > 5 × ULN | −1.17 (−1.88 to −0.59) |
Creatinine ≥ 50% from baseline | −0.58 (−1.33 to 0.06) |
AE = adverse event; CI = confidence interval; EAIR = exposure-adjusted incidence rate; MACE = major adverse cardiovascular events; ULN = upper limit of normal.
Note: EAIR differences are per 100 PYs. Differences < 0 indicate lower EAIRs in the inclisiran pool.
Source: Wright et al. Safety and Tolerability of Inclisiran for Treatment of Hypercholesterolemia in 7 Clinical Trials, J Am Coll Cardiol. 2023 December 12;82(24):2251 to 2261. Copyright 2023 by the authors. Available from: https://www.sciencedirect.com/science/article/pii/S0735109723077392?via%3Dihub. Reprinted in accordance with Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International Licence (CC BY 4.0-NC-ND): https://creativecommons.org/licenses/by-nc-nd/4.0/.35
Overall, incident diabetes and MACE-related safety events were reported in 10.7% and 10.2% of patients, respectively, in the inclisiran group (5,819.2 and 9,595.9 PYs of exposure, respectively), compared with 9.2% and 8.9% of patients, respectively, in the placebo group (1,652.6 and 2,591.3 PYs of exposure, respectively) (Table 40). However, the EAIR for incident diabetes was 4.04 (95% CI, 3.54 to 4.59) per 100 PYs in the inclisiran group versus 7.08 (95% CI, 5.86 to 8.48) per 100 PYs in the placebo group, with a difference in EAIRs (95% CI) of −3.04 (95% CI, −4.53 to −1.74). Similarly, the EAIR for MACE was 3.79 (95% CI, 3.41 to 4.20) per 100 PYs in the inclisiran group versus 6.75 (95% CI, 5.79 to 7.83) per 100 PYs in the placebo group (Table 40).
Critical Appraisal
Internal Validity
The findings are derived from pooled data from 7 clinical trials with specific inclusion criteria, and, thus, patient populations enrolled at different times may have had different clinical characteristics not reflected in the tables of baseline characteristics and may not be fully reflective of a general population. Although EAIRs were calculated, no direct comparison of events with inclisiran versus placebo is possible beyond the first 1.5 years, and only a few patients were exposed to inclisiran for more than 4 years, which limits us to drawing a meaningful conclusion.
External Validity
The pooled data analysis consisted of patients who took part in the pivotal studies, so it is reasonable to expect that the same strengths and limitations related to generalizability apply to this study.
Table 40: TEAEs of Clinical Relevance
Adverse events | Inclisiran pool (N = 3,576) | Placebo pool (N = 1,968) | ||||
|---|---|---|---|---|---|---|
Patients, m/n (%) | Total exposure, years | EAIR (95% CI) | Patients, m/n (%) | Total exposure, years | EAIR (95% CI) | |
Liver | ||||||
Hepatic events (broad SMQ search) | 232/3,576 (6.5) | 9,725.8 | 2.39 (2.09 to 2.71) | 81/1,968 (4.1) | 2,649.1 | 3.06 (2.43 to 3.80) |
≥ 1 liver function test | 92/3,576 (2.6) | 9,981.0 | 0.92 (0.74 to 1.13) | 31/1,967 (1.6) | 2,689.8 | 1.15 (0.78 to 1.64) |
Alanine aminotransferase > 3 × ULN | 31/3,574 (0.9) | 10,070.7 | 0.31 (0.21 to 0.44) | 8/1,966 (0.4) | 2,699.8 | 0.30 (0.13 to 0.58) |
Alkaline phosphatase > 2 × ULN | 24/3,574 (0.7) | 10,072.0 | 0.24 (0.15 to 0.35) | 5/1,963 (0.3) | 2,697.5 | 0.19 (0.06 to 0.43) |
Aspartate aminotransferase > 3 × ULN | 30/3,575 (0.8) | 10,074.8 | 0.30 (0.20 to 0.43) | 11/1,965 (0.6) | 2,695.4 | 0.41 (0.20 to 0.73) |
Bilirubin > 2 × ULN | 32/3,567 (0.9) | 10,042.5 | 0.32 (0.22 to 0.45) | 12/1,961 (0.6) | 2,692.4 | 0.45 (0.23 to 0.78) |
Muscle | ||||||
Muscle events (narrow SMQ search) | 4/3,576 (0.1) | 10,105.1 | 0.04 (0.01 to 0.10) | 2/1,968 (0.1) | 2,705.5 | 0.07 (0.01 to 0.27) |
Creatine kinase > 5 × ULN | 122/3,568 (3.4) | 9,853.2 | 1.24 (1.03 to 1.48) | 64/1,961 (3.3) | 2,652.2 | 2.41 (1.86 to 3.08) |
Kidney | ||||||
Kidney events (broad SMQ search) | 283/3,576 (7.9) | 9,678.6 | 2.92 (2.59 to 3.29) | 90/1,968 (4.6) | 2,642.3 | 3.41 (2.74 to 4.19) |
Creatinine ≥ 50% increase from baseline | 209/3,576 (5.8) | 9,866.8 | 2.12 (1.84 to 2.43) | 72/1,967 (3.7) | 2,670.0 | 2.70 (2.11 to 3.40) |
Diabetes mellitus and MACE | ||||||
Incident diabetes | 235/2,195 a (10.7) | 5,819.2 | 4.04 (3.54 to 4.59) | 117/1,277 (9.2) | 1,652.6 | 7.08 (5.86 to 8.48) |
MACE-related safety events | 364/3,576 (10.2) | 9,595.9 | 3.79 (3.41 to 4.20) | 175/1,968 (8.9) | 2,591.3 | 6.75 (5.79 to 7.83) |
Cardiovascular death | 81/3,576 (2.3) | 10,108.9 | 0.80 (0.64 to 1.00) | 15/1,968 (0.8) | 2,706.1 | 0.55 (0.31 to 0.91) |
Cardiac arrest | 13/3,576 (0.4) | 10,105.9 | 0.13 (0.07 to 0.22) | 1/1,968 (0.1) | 2,705.5 | 0.04 (0.00 to 0.21) |
Nonfatal myocardial infarction | 239/3,576 (6.7) | 9,674.4 | 2.47 (2.17 to 2.80) | 145/1,968 (7.4) | 2,600.9 | 5.57 (4.70. 6.56) |
Nonfatal stroke | 61/3,576 (1.7) | 10,030.7 | 0.61 (0.47 to 0.78) | 18/,1968 (0.9) | 2,696.2 | 0.67 (0.40 to 1.06) |
CI = confidence interval; EAIR = exposure-adjusted incidence rate; MACE = major adverse cardiovascular events; SMQ = Standardized MedDRA Queries; TEAE = treatment-emergent adverse event; ULN = upper limit of normal.
Notes: The proportion of patients was calculated by dividing the number of patients with events (m) by the number of baseline at-risk patients (n). These included patients whose baseline values did not meet the threshold. For incident diabetes, baseline at-risk patients are defined as having no diabetes at baseline. For creatinine assessment, baseline at-risk patients are defined as having nonmissing baseline creatinine values. For reported TEAE analyses, the baseline at-risk population included all patients in the pool (N). Baseline was defined as the last available record before the first administration of the study drug in the pool.
Broad SMQs include all possible causes for the condition of interest; narrow SMQs are likely to represent the condition of interest.
aPatients who switched to inclisiran on day 360 of the ORION-3 trial were excluded from this analysis because hemoglobin s and fasting glucose levels were not assessed on that visit.
Source: Wright et al. Safety and Tolerability of Inclisiran for Treatment of Hypercholesterolemia in 7 Clinical Trials, J Am Coll Cardiol. 2023 December 12;82(24):2251 to 2261. Copyright 2023 by the authors. Available from: https://www.sciencedirect.com/science/article/pii/S0735109723077392?via%3Dihub. Reprinted in accordance with Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International Licence (CC BY 4.0-NC-ND): https://creativecommons.org/licenses/by-nc-nd/4.0/.35
Discussion
Summary of Available Evidence
The sponsor’s resubmission was targeted at key gaps in evidence identified by CDEC in the original submission of inclisiran. Note that for the purposes of this review, the 2 subpopulations listed in the indication, patients with HeFH and patients with ASCVD, are being treated separately. To address concerns over the lack of information on CV morbidity and mortality, the sponsor submitted a pooled analysis of MACE from the ORION-10 and ORION-11 trials, which both featured populations with nFH and ASCVD. The ORION-9, ORION-10, and ORION-11 studies were 540 days (18 months) in duration. The ORION-9 trial included adult patients with established HeFH or ASCVD, the ORION-10 trial included adult patients with ASCVD, and the ORION-11 trial included patients with ASCVD and ASCVD-RE. A total of 482, 1,561, and 1,617 patients were enrolled in the ORION-9, ORION-10, and ORION-11 trials, respectively. In all 3 ORION trials, patients were randomized to 284 mg of inclisiran or matching placebo. The coprimary end points of the ORION trials were the percent change in LDL-C from baseline to day 510 and the time-adjusted percent change in LDL-C from baseline to the period from after day 90 and up to day 540. Key secondary outcomes included the absolute change in LDL-C from baseline to day 510, the time-adjusted percent change from baseline to the period from after day 90 and up to day 540, and the percent change from baseline to day 510 in PCSK9, total cholesterol, ApoB, and non-HDL-C. To address other limitations related more to longer-term efficacy and safety, the sponsor submitted the ORION-3 trial, which was an open-label extension that enrolled patients from the ORION-1 trial, the ORION-8 trial (which was an extension that enrolled patients from the ORION-9, ORION-10, and ORION-11 trials), as well as patients from the ORION-3 trial. The sponsor also submitted a pooled safety analysis from 7 ORION trials (ORION-1, ORION-3, ORION-5, ORION-8, ORION-9, ORION-10, and ORION-11).
Baseline characteristics of the ORION trials were balanced between groups, and generally applicable to the population in Canada. The ORION-9 trial enrolled mostly white patients (94.0%), with a median age of 56 years and a relatively even ratio of men and women (47.1% versus 52.9%) with either ASCVD (27.4%) or ASCVD-RE (72.6%), 93.2% of whom had HeFH. A total of 73.9% of patients were on high-intensity statins at baseline, with 25.3% either partially or completely intolerant to statins, and 52.3% were treated with ezetimibe. The ORION-10 trial enrolled mostly white (85.7%) men (69.4%), with a median age of 67 years, and all had ASCVD (91.1% had CHD). Approximately two-thirds (69.4%) of patients were on a high-intensity statin at baseline, with 22.0% partially or completely intolerant. A total of 9.9% of patients were treated with ezetimibe. The ORION-11 trial enrolled patients with ASCVD (87.4%) and ASCVD-RE (12.6%). Patients were mostly white (98.1%) and men (71.7%), with a median age of 65 years. A total of 78.0% of patients were receiving high-intensity statins, 11.4% were considered partially or completely intolerant to statins, and 7.1% of patients were treated with ezetimibe.
Interpretation of Results
Efficacy
With respect to the HeFH population, there were very few MACE in the ORION-9 trial, and the incidence of MACE was almost identical in the inclisiran and placebo groups. This is not an unexpected finding, according to the clinical experts consulted by CDA-AMC on this review, as patients with HeFH are at lower risk of experiencing a MACE in the short to intermediate term than patients with ASCVD. Given the relatively lower risk of MACE in this population, and the fact that HeFH is far less common than nFH and ASCVD, the clinical experts were of the opinion that we are unlikely to find an RCT that will be able to demonstrate an improvement in MACE in this population. Therefore, for the HeFH population, LDL-C must be relied upon for determining the efficacy of inclisiran. According to the guidelines, a reduction of 1 mmol/L (approximately 38.67 mg/dL) in LDL-C is estimated to reduce the RR of ASCVD by 20% to 22%.3 Between-group differences in the ORION-9, ORION-10, and ORION-11 trials of −68.89 mg/dL, −54.12 mg/dL, and −51.87 mg/dL, respectively, indicate that long-term treatment with inclisiran may result in a reduction of these events.
Patients with ASCVD featured in the ORION-10 and ORION-11 trials are more likely to experience MACE than patients with HeFH; these trials were larger than the ORION-9 trial, which was apparent when comparing MACE results to the results from the ORION-9 trial. When the sponsor conducted a posthoc pooled analysis of the ORION-10 and ORION-11 trials, it showed that patients treated with inclisiran had a lower risk of MACE than those treated with placebo, with █ ██ ████ ████ ███ █████ █████. However, there are important limitations to this analysis, described earlier in this report, and the hypothesis that treatment with inclisiran reduces the risk of MACE in patients with ASCVD needs to be tested in a prospectively designed trial of adequate size and duration to assess this outcome. The currently ongoing ORION-4 trial will aim to address this important gap; however, results are not available yet.
The longer-term efficacy of inclisiran was assessed in the ORION-3 trial and, more importantly, the ORION-8 trial. Both trials were extensions; the ORION-3 trial was a 4-year, open-label extension of the phase II ORION-1 trial, whereas the ORION-8 trial was an open-label extension of the ORION-9, ORION-10, and ORION-11 trials, as well as the ORION-3 trial. Neither of the trials featured a comparator. The conclusions that can be drawn regarding efficacy are limited, given the lack of a comparator; however, data from the ORION-8 trial suggest that although there is no additional efficacy gained from longer treatment with inclisiran, there is also no reduction in efficacy over time. For example, the patients in the group that received inclisiran throughout the ORION-9, ORION-10, and ORION-11 trials had a 50% reduction in LDL-C after 510 days in their parent trial and a 49% reduction in LDL-C after another 1,080 days of inclisiran in the extension. Similarly, the patients who received placebo in the parent trials experienced a 50% reduction in LDL-C after 1,080 days of inclisiran in the extension trial (ORION-8), as did the patients who were rolled over from the ORION-3 trial, also demonstrating consistency of effect over time. Similar results were seen for patients achieving global lipid targets, with a similar proportion of patients achieving global lipid targets by day 1,080 (EOS) in the inclisiran-only group, in patients rolled over from the ORION-3 trial (77%), and in the patients switched to inclisiran (79%).
Although not an outcome of the study but an outcome important to patients, the biannual dosing regimen provides a more manageable dosing and administration schedule than the current PCSK9 inhibitors available; alirocumab and evolocumab require injection 26 times per year. The clinical experts consulted by CDA-AMC also noted that this dosing regimen aligns with routine patient follow-up in these populations, which would also improve adherence to treatment, as evidenced by the high proportion of patients completing the 18-month studies (89.0% to 97.1%), and ██████ ██ ████████ █████████ ███ ████ █████ ██ ███ █████ ████ ██████ ██ ███████ ███ █████████ █████████ ████ ███ ███████ ██████████ █████ ███████ ███ ███ █████ ████ ████████████ ████ ██ ████████ ███ ████████ █████ █████████ ████.
The sponsor-submitted ITC compared the efficacy and safety of inclisiran and relevant drug comparators (evolocumab and alirocumab) for the treatment of adult patients with HeFH or ASCVD with uncontrolled LDL-C by MTD statins with or without ezetimibe, or for those with uncontrolled LDL-C and an intolerance or contraindication to with or without ezetimibe. The results of the ITC suggest that there is no difference in efficacy between inclisiran and alirocumab or evolocumab. The considerable clinical, methodological, and statistical heterogeneity, coupled with the wide CrIs in each network scenario for comparisons between inclisiran and placebo, PCSK9 inhibitors, and ezetimibe, resulted in significant uncertainty about the comparative efficacy of inclisiran. In addition, important outcomes of all-cause mortality, CV-related mortality, and CV-related morbidity (MACE) were not included in the analysis.
Harms
To address the comment by CDEC that the longer-term safety profile of inclisiran requires further evaluation, the sponsor provided a pooled analysis of harms from 7 ORION trials (ORION-1, ORION-3, ORION-5, ORION-8, ORION-9, ORION-10, and ORION-11), as well as data from the ORION-8 long-term, open-label extension. There was no indication from the ORION-8 trial that further exposure to inclisiran led to a higher risk of AE over time, as an identical number of patients (79%) experienced an AE in the group that received inclisiran throughout the parent trial and the 1,080 days of the extension as received placebo in the parent trial and inclisiran in the extension. Similarly, there was no evidence of an increase in SAEs with longer duration of therapy; 31% of patients in the inclisiran-only group experienced an SAE by the end of the extension, as did 33% of patients in the switching group.
In the pooled analysis of 7 ORION trials, the percentage of patients with SAEs was numerically higher with inclisiran than with placebo (32% versus 22%), although exposure was higher in the inclisiran group; the EAIR of SAEs was 13.80 per 100 PYs (95% CI, 13.01 to 14.62) with inclisiran and 18.14 (95% CI, 16.48 to 19.93) with placebo.
The incidence of harms reported in the ORION-9, ORION-10, and ORION-11 trials was similar in the inclisiran and placebo groups, with the occurrence of AEs ranging from 71.7% to 82.7%. No important or consistent differences in SAEs, WDAEs, or most notable harms were evident between placebo and inclisiran groups across trials, except for harms related to administration (such as injection-site reactions), which were higher in the inclisiran arm in the ORION-9, ORION-10, and ORION-11 trials (17.0%, 6.0%, and 7.6%, respectively) than in the placebo arm (1.7%, 1.9%, and 1.7%, respectively).
Conclusion
The major areas of focus for this resubmission were to address the lack of formal assessment of clinical outcomes data from the ORION-9, ORION-10, and ORION-11 trials, and the lack of longer-term safety and efficacy data for inclisiran. Given the indication, the 2 populations, patients with HeFH (ORION-9) and nFH with ASCVD (ORION-10 and ORION-11), should be viewed separately. There was no evidence that inclisiran reduced the risk of MACE in the HeFH population in the ORION-9 trial; however, according to the clinical experts, this type of data has not been available from clinical trials of other drugs, given that these events would be less frequent in this population over the typical follow-up of a clinical trial and given that HeFH is less common than nFH. There was evidence of a reduced risk of MACE with inclisiran treatment in the nFH with ASCVD population when the results of the ORION-10 and ORION-11 trials were pooled; however, this was a posthoc analysis and it is important to note that these trials were not designed to compare treatment groups for these outcomes. The conclusions about inclisiran regarding lipid parameters remain the same: inclisiran elicits a statistically and clinically significant reduction in LDL-C and a similar improvement in other lipid parameters, and this improvement in LDL-C appeared to be maintained through the 3 years of additional treatment with inclisiran during the open-label ORION-8 trial. There was no indication of any new safety or tolerability concerns with inclisiran during long-term extensions, or when results of the various ORION trials were pooled. The ITC submitted by the sponsor did not provide conclusive evidence on the relative efficacy and safety of inclisiran compared to other PCSK9 inhibitors or ezetimibe in the context of HeFH or ASCVD. The ITC provides minimal value when comparing the efficacy of inclisiran with other PCSK9 inhibitors or ezetimibe, because it did not include an evaluation of clinical outcomes.
References
1.Statistics Canada. Leading causes of death, total population, by age group. 2022 Jan 24; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310039401. Accessed 2023 Mar 9.
2.Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161(1):161-172. PubMed
3.Yusuf S, Joseph P, Rangarajan S, et al. Modifiable risk factors, cardiovascular disease, and mortality in 155 722 individuals from 21 high-income, middle-income, and low-income countries (PURE): a prospective cohort study. Lancet. 2020;395(10226):795-808. PubMed
4.O'Donnell MJ, Chin SL, Rangarajan S, et al. Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): a case-control study. Lancet. 2016;388(10046):761-775. PubMed
5.Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188. PubMed
6.Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24):e285-e350. PubMed
7.Chen G, Farris MS, Cowling T, et al. Treatment and Low-Density Lipoprotein Cholesterol Management in Patients Diagnosed With Clinical Atherosclerotic Cardiovascular Disease in Alberta. Can J Cardiol. 2019;35(7):884-891. PubMed
8.Mackinnon ES, Har B, Champsi S, Wani RJ, Geyer L, et al. Guideline LDL-C Threshold Achievement in Acute Myocardial Infarction Patients: A Real-World Evidence Study Demonstrating the Impact of Treatment Intensification with PCSK9i. Cardiol Ther. 2023;12(2):327-338. PubMed
9.Novartis. Data on file. Novartis Cardiology Advisory Board. May 2023 [sponsor-provided reference]. 2023.
10.Roy S. Atherosclerotic Cardiovascular Disease Risk and Evidence-based Management of Cholesterol. N Am J Med Sci. 2014;6(5):191-198. PubMed
11.Ray KK, Wright RS, Kallend D, et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med. 2020;382(16):1507-1519. PubMed
12.Pearson GJ, Thanassoulis G, Anderson TJ, et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can J Cardiol. 2021;37(8):1129-1150. PubMed
13.Razek O, Cermakova L, Armani H, et al. Attainment of Recommended Lipid Targets in Patients With Familial Hypercholesterolemia: Real-World Experience With PCSK9 Inhibitors. Can J Cardiol. 2018;34(8):1004-1009. PubMed
14.Akioyamen LE, Chu A, Genest J, et al. Prevalence and Treatment of Familial Hypercholesterolemia and Severe Hypercholesterolemia in Older Adults in Ontario, Canada. CJC Open. 2022;4(9):739-747. PubMed
15.E. A. S. Familial Hypercholesterolaemia Studies Collaboration. Global perspective of familial hypercholesterolaemia: a cross-sectional study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Lancet. 2021;398(10312):1713-1725. PubMed
16.Gupta M, Mancini GBJ, Wani RJ, et al. Real-World Insights Into Evolocumab Use in Patients With Hyperlipidemia: Canadian Analysis From the ZERBINI Study. CJC Open. 2022;4(6):558-567. PubMed
17.Ryzhaya N, Cermakova L, Trinder M, et al. Sex Differences in the Presentation, Treatment, and Outcome of Patients With Familial Hypercholesterolemia. J Am Heart Assoc. 2021;10(11):e019286. PubMed
18.Dainis AM, Ashley EA. Cardiovascular Precision Medicine in the Genomics Era. JACC Basic Transl Sci. 2018;3(2):313-326. PubMed
19.Akioyamen LE, Genest J, Shan SD, et al. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open. 2017;7(9):e016461. PubMed
20.Hu P, Dharmayat KI, Stevens CAT, et al. Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: a systematic review and meta-analysis. Circulation. 2020;141(22):1742-1759. PubMed
21.Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J Am Coll Cardiol. 2020;75(20):2553-2566. PubMed
22.Ference BA, Graham I, Tokgozoglu L, Catapano AL. Impact of lipids on cardiovascular health: JACC health promotion series. J Am Coll Cardiol. 2018;72(10):1141-1156. PubMed
23.Brunham LR, Ruel I, Aljenedil S, et al. Canadian Cardiovascular Society Position Statement on Familial Hypercholesterolemia: Update 2018. Can J Cardiol. 2018;34(12):1553-1563. PubMed
24.Iyen B, Qureshi N, Kai J, Akyea RK, Leonardi-Bee J, et al. Risk of cardiovascular disease outcomes in primary care subjects with familial hypercholesterolaemia: A cohort study. Atherosclerosis. 2019;287:8-15. PubMed
25.Mundal LJ, Igland J, Veierod MB, et al. Impact of age on excess risk of coronary heart disease in patients with familial hypercholesterolaemia. Heart. 2018;104(19):1600-1607. PubMed
26.Emanuelsson F, Nordestgaard BG, Benn M. Familial Hypercholesterolemia and Risk of Peripheral Arterial Disease and Chronic Kidney Disease. J Clin Endocrinol Metab. 2018;103(12):4491-4500. PubMed
27.CADTH Reimbursement Review Recommendation: inclisiran (Leqvio - Novartis Pharmaceuticals Canada Inc.) [sponsor-provided reference]. Can J Health Technol. 2022;2(2). https://www.cadth.ca/sites/default/files/DRR/2022/SR0681%20Leqvio%20-%20Confidential%20Final%20CADTH%20Rec%20Final.pdf.
28.Clinical Study Report: ORION-9 (MMDC-PCS-17-03). A placebo-controlled, double-blind, randomized trial to evaluate the effect of 300 mg of inclisiran sodium given as subcutaneous injections in subjects with heterozygous familial hypercholesterolemia (HeFH) and elevated low density lipoprotein cholesterol (LDL-C) [sponsor-provided reference]. Parsippany-Troy Hills (NJ): The Medicines Company; 2019 Dec 2.
29.Clinical Study Report: ORION-10 (MDCO-PCS-17-04). A placebo-controlled, double-blind, randomized trial to evaluate the effect of 300 mg of inclisiran sodium given as subcutaneous injections in subjects with atherosclerotic cardiovascular disease (ASCVD) and elevated low density lipoprotein cholesterol (LDL-C) [sponsor-provided reference]. Parsippany-Troy Hills (NJ): The Medicines Company; 2019 Dec 5.
30.Clinical Study Report: ORION-11 (MDCO-PCS-17-08). A placebo-controlled, double-blind, randomized trial to evaluate the effect of 300 mg of inclisiran sodium given as subcutaneous injections in subjects with atherosclerotic cardiovascular disease (ASCVD) or ASCVD-risk equivalents and elevated low-density lipoprotein cholesterol (LDL-C) [sponsor-provided reference]. Parsippany-Troy Hills (NJ): The Medicines Company; 2019 Nov 20.
31.Clinical Study Report: ORION-3 (MDCO-PCS-16-01). An open label, active comparator extension trial to assess the effect of long term dosing of inclisiran and evolocumab given as subcutaneous injections in subjects with high cardiovascular risk and elevated LDL-C (ORION-3) [sponsor-provided reference]. Parsippany-Troy Hills (NJ): Novartis; 2022.
32.Ray KK, Troquay RPT, Visseren FLJ, et al. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol. 2023;11(2):109-119. PubMed
33.Clinical Study Report: ORION-8 [sponsor-provided reference]. 2023.
34.Network meta-analysis to support global submission for Inclisiran [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Leqvio (inclisiran), 284 mg in 1.5 mL (189 mg/mL) solution for subcutaneous injection. Dorval (QC): Novartis Pharmaceuticals Canada Inc; 2021.
35.Wright RS, Koenig W, Landmesser U, et al. Pooled Safety Analysis of Inclisiran in 3,576 Patients with Approximately 10,000 Person Years of Exposure from 7 Trials. J Am Coll Cardiol. 2023;81(8):1626-1626.
36.Mayo Clinic. Arteriosclerosis/atherosclerosis [sponsor-provided reference]. 2020; https://www.mayoclinic.org/diseases-conditions/arteriosclerosis-atherosclerosis/symptoms-causes/syc-20350569.
37.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317-325. PubMed
38.Brunham LR, Ruel I, Khoury E, et al. Familial hypercholesterolemia in Canada: Initial results from the FH Canada national registry. Atherosclerosis. 2018;277:419-424. PubMed
39.Brunham LR, Cermakova L, Lee T, et al. Contemporary Trends in the Management and Outcomes of Patients With Familial Hypercholesterolemia in Canada: A Prospective Observational Study. Can J Cardiol. 2017;33(3):385-392. PubMed
40.Jung KJ, Koh H, Choi Y, Lee SJ, Ji E, et al. Familial hypercholesterolemia and atherosclerotic cardiovascular mortality among Korean adults with low levels of serum cholesterol. Atherosclerosis. 2018;278:103-109. PubMed
41.De Smedt D, Clays E, Annemans L, et al. Health related quality of life in coronary patients and its association with their cardiovascular risk profile: results from the EUROASPIRE III survey. Int J Cardiol. 2013;168(2):898-903. PubMed
42.Lewis EF, Li Y, Pfeffer MA, et al. Impact of cardiovascular events on change in quality of life and utilities in patients after myocardial infarction: a VALIANT study (valsartan in acute myocardial infarction). JACC Heart Fail. 2014;2(2):159-165. PubMed
43.Munyombwe T, Hall M, Dondo TB, et al. Quality of life trajectories in survivors of acute myocardial infarction: a national longitudinal study. Heart. 2020;106(1):33-39. PubMed
44.Ruel I, Brisson D, Aljenedil S, et al. Simplified Canadian Definition for Familial Hypercholesterolemia. Can J Cardiol. 2018;34(9):1210-1214. PubMed
45.Roggeri A, Gnavi R, Dalmasso M, et al. Resource consumption and healthcare costs of acute coronary syndrome: a retrospective observational administrative database analysis. Crit Pathw Cardiol. 2013;12(4):204-209. PubMed
46.Danese MD, Gleeson M, Kutikova L, et al. Estimating the economic burden of cardiovascular events in patients receiving lipid-modifying therapy in the UK. BMJ Open. 2016;6(8):e011805. PubMed
47.Boklage SH, Malangone-Monaco E, Lopez-Gonzalez L, Ding Y, Henriques C, Elassal J. Statin utilization patterns and outcomes for patients with acute coronary syndrome during and following inpatient admissions. Cardiovasc Drugs Ther. 2018;32(3):273–280. PubMed
48.Lindh M, Banefelt J, Fox KM, et al. Cardiovascular event rates in a high atherosclerotic cardiovascular disease risk population: estimates from Swedish population-based register data. Eur Heart J Qual Care Clin Outcomes. 2019;5(3):225–232. PubMed
49.Ohsfeldt RL, Gandhi SK, Fox KM, Bullano MF, Davidson M. Medical and cost burden of atherosclerosis among patients treated in routine clinical practice. J Med Econ. 2010;13(3):500-507. PubMed
50.Toth PP, Danese M, Villa G, et al. Estimated burden of cardiovascular disease and value-based price range for evolocumab in a high-risk, secondary-prevention population in the US payer context. J Med Econ. 2017;20(6):555-564. PubMed
51.Anderson TJ, Gregoire J, Pearson GJ, et al. 2016 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in the Adult. Can J Cardiol. 2016;32(11):1263-1282. PubMed
52.Sebregts EH, Falger PR, Bar FW. Risk factor modification through nonpharmacological interventions in patients with coronary heart disease. J Psychosom Res. 2000;48(4-5):425-441. PubMed
53.Ward NC, Watts GF, Eckel RH. Response by Ward et al to Letter Regarding Article, “Statin Toxicity: Mechanistic Insights and Clinical Implications”. Circ Res. 2019;124(12):e121-e122. PubMed
54.Benner JS, Tierce JC, Ballantyne CM, et al. Follow-up lipid tests and physician visits are associated with improved adherence to statin therapy. Pharmacoeconomics. 2004;22 Suppl 3(3):13-23.
55.Caspard H, Chan AK, Walker AM. Compliance with a statin treatment in a usual-care setting: retrospective database analysis over 3 years after treatment initiation in health maintenance organization enrollees with dyslipidemia. Clin Ther. 2005;27(10):1639-1646. PubMed
56.Donnelly LA, Doney AS, Morris AD, Palmer CN, Donnan PT. Long-term adherence to statin treatment in diabetes. Diabet Med. 2008;25(7):850-855. PubMed
57.Banach M, Rizzo M, Toth PP, et al. Statin intolerance - an attempt at a unified definition. Position paper from an International Lipid Expert Panel. Arch Med Sci. 2015;11(1):1-23. PubMed
58.Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158(7):526-534. PubMed
59.Mancini GB, Baker S, Bergeron J, et al. Diagnosis, Prevention, and Management of Statin Adverse Effects and Intolerance: Canadian Consensus Working Group Update (2016). Can J Cardiol. 2016;32(7 Suppl):S35-65. PubMed
60.Menzin J, Aggarwal J, Boatman B, et al. Ezetimibe Use and LDL-C Goal Achievement: A Retrospective Database Analysis of Patients with Clinical Atherosclerotic Cardiovascular Disease or Probable Heterozygous Familial Hypercholesterolemia. J Manag Care Spec Pharm. 2017;23(12):1270-1276. PubMed
61.Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372(25):2387-2397. PubMed
62.Feingold KR. Cholesterol Lowering Drugs [sponsor-provided reference]. In: Feingold KR, Anawalt B, Boyce A, et al., eds. Endotext. South Dartmouth (MA): MDText; 2000.
63.Dicembrini I, Giannini S, Ragghianti B, Mannucci E, Monami M. Effects of PCSK9 inhibitors on LDL cholesterol, cardiovascular morbidity and all-cause mortality: a systematic review and meta-analysis of randomized controlled trials. J Endocrinol Invest. 2019;42(9):1029-1039. PubMed
64.Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379(22):2097-2107. PubMed
65.Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. PubMed
66.Zhang XL, Zhu QQ, Zhu L, et al. Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med. 2015;13(1):123. PubMed
67.Repatha® (evolocumab injection) [sponsor-provided reference]. Mississauga (ON): Amgen Canada, Inc; 2021. Accessed 2021 Dec 9.
68.PRALUENT® (alirocumab injection) [sponsor-provided reference]. Laval (QC): Sanofi-aventis Canada, Inc; 2023. Accessed 2023 Feb 17.
69.CADTH Canadian Drug Expert Committee final recommendation: Evolocumab (Repatha - Amgen Canada Inc) [sponsor-provided reference]. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/sites/default/files/cdr/complete/SR0515_Repatha_Resubmission_complete_Nov_24_17.pdf.
70.CADTH Canadian Drug Expert Committee final recommendation: Alirocumab (Praluent - Sanofi-aventis Canada Inc.) [sponsor-provided reference]. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/cdr/complete/SR0469_complete_Praluent_Jul-20-16.pdf.
71.Ontario Ministry of Health and Long-Term Care. Alirocumab Clinical Criteria [sponsor-provided reference]. Ontario drug benefit formulary/comparative drug index 2019; https://www.formulary.health.gov.on.ca/formulary/limitedUseNotes.xhtml?pcg9Id=240624004.
72.Ontario Ministry of Health and Long-Term Care. Evolocumab Clinical Criteria [sponsor-provided reference]. Ontario drug benefit formulary/comparative drug index 2019; https://www.formulary.health.gov.on.ca/formulary/limitedUseNotes.xhtml?pcg9Id=240600076.
73.Pan-Canadian Pharmaceutical Alliance. Praluent (alirocumab) [sponsor-provided reference]. 2019: https://www.pcpacanada.ca/negotiation/21075.
74.Pan-Canadian Pharmaceutical Alliance. Repatha (evolocumab) [sponsor-provided reference]. 2019: https://www.pcpacanada.ca/negotiation/21004.
75.LEQVIO™ (inclisiran) [sponsor-provided reference]. Dorval (QC): Novartis Pharmaceuticals Canada, Inc; 2021. Accessed 2021 Aug 12.
76.LIPITOR (atorvastatin calcium): 10 mg, 20 mg, 40 mg and 80 mg oral tablets (as atorvastatin calcium) [product monograph]. Kirkland (QC): Upjohn Canada ULC; 2023: https://pdf.hres.ca/dpd_pm/00069540.PDF. Accessed 2023 Nov 14.
77.ACH-Ezetimibe (ezetimibe): 10 mg tablets [product monograph]. Kirkland (QC): Accord Healthcare, Inc.; 2019: https://pdf.hres.ca/dpd_pm/00052024.PDF. Accessed 2023 Nov 14.
78.Clinical Study Protocol: ORION-11 (MDCO-PCS-17-08). A placebo-controlled, double-blind, randomized trial to evaluate the effect of 300 mg of inclisiran sodium given as subcutaneous injections in subjects with atherosclerotic cardiovascular disease (ASCVD) or ASCVD-risk equivalents and elevated low-density lipoprotein cholesterol (LDL-C) [sponsor-provided reference]. 2019 Jan 31.
79.Raal FJ, Kallend D, Ray KK, et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N Engl J Med. 2020;382(16):1520-1530. PubMed
80.Novartis. Data on file. CKJX839A1 Inc-Pub084 [sponsor-provided reference]. 2023.
81.Ray KK, Raal FJ, Kallend DG, et al. Inclisiran and cardiovascular events: a patient-level analysis of phase III trials. Eur Heart J. 2022;44(2):129-138. PubMed
82.Clinical Study Protocol: ORION-9 (MMDC-PCS-17-03). A placebo-controlled, double-blind, randomized trial to evaluate the effect of 300 mg of inclisiran sodium given as subcutaneous injections in subjects with heterozygous familial hypercholesterolemia (HeFH) and elevated low density lipoprotein cholesterol (LDL-C) [sponsor-provided reference]. 2019 Jan 31.
83.U. S. Food and Drug Administration. Product Details for NDA 214012: LEQVIO (inclisiran sodium). Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations 2021; https://www.accessdata.fda.gov/scripts/cder/ob/results_product.cfm?Appl_Type=N&Appl_No=214012#40467. Accessed 2023 Jul 10.
84.Novartis. Prescribing information: LEQVIO (inclisiran) injection, for subcutaneous use [sponsor-provided reference]. Silver Spring (MD): U.S. Food and Drug Administration; 2023: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/214012s009lbl.pdf.
85.Linton MF, Yancey PG, Davies SS. The Role of Lipids and Lipoproteins in Atherosclerosis [sponsor-provided reference]. 2019: https://www.ncbi.nlm.nih.gov/books/NBK343489/. Accessed 2020 Mar.
86.Ray KK, Landmesser U, Leiter LA, et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N Engl J Med. 2017;376(15):1430-1440. PubMed
87.Clinical Study Report: ORION-8 Global Amendment l (MDCO-PCS-17-05) [sponsor-provided reference]. 2018 Nov 1.
88.Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE DSU technical support document 4: inconsistency in networks of evidence based on randomised controlled trials. NICE Decision Support Unit technical support documents. London (GB): National Institute for Health and Care Excellence (NICE); 2014.
89.Higgins JPT, Green S, eds. Cochrane handbook for systematic reviews of interventions [sponsor-provided reference]. The Cochrane Collaboration; 2011.
90.Novartis. Data on file. Unpublished [sponsor-provided reference].
91.Wright RS, Ray KK, Raal FJ, Kallend DG, Jaros M, et al. Pooled Patient-Level Analysis of Inclisiran Trials in Patients With Familial Hypercholesterolemia or Atherosclerosis. J Am Coll Cardiol. 2021;77(9):1182-1193. PubMed
92.Ference BA, Cannon CP, Landmesser U, Luscher TF, Catapano AL, Ray KK. Reduction of low density lipoprotein-cholesterol and cardiovascular events with proprotein convertase subtilisin-kexin type 9 (PCSK9) inhibitors and statins: an analysis of FOURIER, SPIRE, and the Cholesterol Treatment Trialists Collaboration. Eur Heart J. 2018;39(27):2540-2545. PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 41: Other LDL-C Outcomes and MACE
Outcome | ORION-9 | ORION-10 | ORION-11 | |||
|---|---|---|---|---|---|---|
Inclisiran (N = 242) | Placebo (N = 240) | Inclisiran (N = 781) | Placebo (N = 780) | Inclisiran (N = 810) | Placebo (N = 807) | |
Key Secondary Outcomes | ||||||
Mean absolute change in LDL-C from baseline to Day 510 | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 151.4 (50.4) | 154.7 (58.0) | 104.5 (39.6) | 104.8 (37.0) | 107.2 (41.8) | 103.7 (36.4) |
LSM change from baseline, mg/dL (95% CI) | −59.0 (−64.8, −53.2) | 9.9 (4.1, 15.8) | −56.2 (−58.5, −53.9) | −2.1 (−4.4, 0.2) | −50.9 (−53.1, −48.7) | 1.0 (−1.3, 3.2) |
LSM difference vs. control, mg/dL (95% CI) | −68.9 (−77.1, −60.7) | −54.1 (−57.4, −50.9) | −51.9 (−55.0, −48.7) | |||
P value | < 0.0001 | < 0.0001 | < 0.0001 | |||
Time-adjusted absolute change in LDL-C from Day 90 to Day 540 | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 151.4 (50.4) | 154.7 (58.0) | 104.5 (39.6) | 104.8 (37.0) | 107.2 (41.8) | 103.7 (36.4) |
LSM change from day 90, mg/dL (95% CI) | −56.6 (−61.0, −51.2) | 6.2 (1.7, 10.6) | −53.7 (−55.4, −51.9) | −0.4 (−2.1, 1.4) | −48.6 (−50.4, −46.9) | 0.3 (−1.4, 2.0) |
LSM difference vs. control, mg/dL (95% CI) | −62.7 (−69.0, −56.5) | −53.3 (−55.8, −50.8) | −48.9 (−51.4, −46.5) | |||
P value | < 0.0001 | < 0.0001 | < 0.0001 | |||
Percent change in non-HDL-C from baseline to Day 510 | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 178.5 (55.4) | 181.5 (62.5) | 134.0 (44.5) | 134.7 (43.5) | 137.6 (46.9) | 133.9 (41.0) |
LSM change from baseline, % (95% CI) | −34.9 (−38.5, −31.4) | 7.4 (3.9, 10.9) | −47.4 (−49.4, −45.4) | −0.1 (−2.1, 2.0) | −41.2 (−43.1, −39.2) | 2.2 (0.2, 4.1) |
LSM difference vs. control, % (95% CI) | −42.4 (−47.3, −37.4) | −47.4 (−50.3, −44.5) | −43.3 (−46.0, −40.6) | |||
P value | < 0.0001 | < 0.0001 | < 0.0001 | |||
Percent change in ApoB from baseline to Day 510 | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 123.8 (33.2) | 124.5 (34.8) | 94.1 (25.6) | 94.6 (25.1) | 97.1 (28.0) | 95.1 (5.2) |
LSM change from baseline, % (95% CI) | −33.1 (−35.9, −30.4) | 2.9 (0.1, 5.7) | −44.8 (−46.5, −43.1) | −1.7 (−3.5, 0.0) | −38.2 (−39.8, −36.6) | 0.8 (−0.8, 2.4) |
LSM difference vs. control, % (95% CI) | −36.1 (−40.0, −32.1) | −43.1 (−45.5, −40.7) | −38.9 (−41.2, −36.7) | |||
P value | < 0.0001 | < 0.0001 | < 0.0001 | |||
Incidence of CV death, resuscitated cardiac arrest, nonfatal MI, and nonfatal strokei | ||||||
Prespecified exploratory cardiovascular end point/MACE | ||||||
n (%) | 10 (4.1) | 10 (4.2) | 58 (7.4) | 79 (10.2) | 63 (7.8) | 83 (10.3) |
Events, n | 10 | 11 | 66 | 90 | 65 | 100 |
Cardiovascular death | ||||||
n (%) | 1 (0.4) | 0 | █████ | 5 (0.6) | 9 (1.1) | ██ █████ |
Events, n | █████ | █████ | █████ | █████ | █████ | █████ |
Resuscitated Cardiac Arrest | ||||||
n (%) | 0 | 0 | 1 (0.1) | 1 (0.1) | 3 (0.4) | 0 |
Events, n | █████ | █████ | █████ | █████ | █████ | █████ |
Nonfatal MI | ||||||
n (%) | 9 (3.7) | 10 (4.2) | 40 (5.1) | 64 (8.2) | 47 (5.8) | 68 (8.5) |
Events, n | █████ | █████ | █████ | █████ | █████ | █████ |
Nonfatal stroke (ischemic or hemorrhagic) | ||||||
n (%) | 0 | 0 | ██ █████ | 10 (1.3) | 4 (0.5) | █████ |
Events, n | █████ | █████ | █████ | █████ | █████ | █████ |
ApoB = apolipoprotein B; CI = confidence interval; CV = cardiovascular; dL = decalitre; HDL-C = high-density-lipoprotein cholesterol; L = litre; LDL-C = low-density-lipoprotein cholesterol; LSM = least squares mean; MACE = major adverse cardiovascular event; mg = milligram; MI = myocardial infarction; NOC = Notice of Compliance; NR = not reported; SD = standard deviation.
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report;30 Raal FJ, Kallend D et al.;79 Ray KK et al.11
Table 42: Other Secondary and Exploratory Efficacy Outcomes
Outcome | ORION-9 | ORION-10 | ORION-11 | |||
|---|---|---|---|---|---|---|
Inclisiran (N = 242) | Placebo (N = 240) | Inclisiran (N = 781) | Placebo (N = 780) | Inclisiran (N = 810) | Placebo (N = 807) | |
Other secondary outcomes | ||||||
Percent change in triglyceride from baseline to Day 510 (median) | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, median (IQR) | 120 (82, 167) | 119 (85, 166) | 127 (92, 181) | 129 (96, 182) | 135 (99, 181) | 135 (102, 185) |
Change from baseline, % | −11.1 | −0.7 | −14.9 | −2.3 | −12.0 | −5.0 |
Treatment group difference vs. control, % | −11.8 | −12.6 | −7.0 | |||
P value | ██ | ██ | ██ | |||
Percent change in HDL-C from baseline to Day 510 | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, mg/dL, mean (SD) | 51.5 (15.1) | 50.8 (13.1) | 46.6 (14.3) | 45.9 (14.4) | 49.7 (15.5) | 49.3 (13.8) |
Change from baseline, % | 8.6 | 6.0 | 7.5 | 2.4 | 10.2 | 4.1 |
Treatment group difference vs. control, % | 2.6 | 5.1 | 6.1 | |||
P value | ██ | ██ | ██ | |||
Percent change in Lp(a) from baseline to Day 540 (median) | ||||||
Number of patients contributing to the analysis | 242 | 240 | 781 | 780 | 810 | 807 |
Baseline, nmol/L, median (IQR) | 57 (22, 180) | 54 (20, 185) | 57 (18, 181) | 56 (20, 189) | 42 (18, 178) | 35 (18, 181) |
Change from baseline, % | −13.5 | 3.7 | −21.9 | 3.7 | −18.6 | 0 |
Treatment group difference vs. control, % | −17.2 | −25.6 | −18.6 | |||
P value | ██ | ██ | ██ | |||
Proportion of patients in each group with any LDL-C reduction at Day 510 | ||||||
LDL-C target level | ||||||
█████ | ||||||
██ | ██ | ██ | ██ | ██ | ██ | ██ |
██ | ████ ██████ █████ | ███████████ █████ | ███████████ █████ | |||
██ | ██ | ██ | ██ | |||
< 25 mg/dLg | ||||||
n (%) | 2 (0.8) | 0 | 160 (20.5) | 4 (0.5) | 95 (11.7) | 1 (0.1) |
OR (95% CI) | ██ | 39.1 (15.0, 101.6) | 78.3 (12.1, 507.0) | |||
P value | ██ | ██ | ██ | |||
< 50 mg/dLg | ||||||
n (%) | 46 (19.0) | 2 (0.8) | 483 (61.8) | 19 (2.4) | 420 (51.9) | 19 (2.4) |
OR (95% CI) | ██ | 53.6 (34.0, 84.7) | 42.7 (26.7, 68.3) | |||
P value | ██ | ██ | ██ | |||
< 70 mg/dLg | ||||||
n (%) | 99 (40.9) | 3 (1.3) | 581 (74.4) | 119 (15.3) | 564 (69.6) | 104 (12.9) |
OR (95% CI) | ██ | 19.2 (14.7, 25.2) | 18.5 (14.2, 24.1) | |||
P value | ██ | ██ | ██ | |||
< 100mg/dLg | ||||||
n (%) | 158 (65.3) | 21 (8.8) | 651 (83.4) | 387 (49.6) | 661 (81.6) | 425 (52.7) |
OR (95% CI) | ██ | 9.6 (7.0, 13.4) | 6.8 (5.1, 9.0) | |||
P value | ██ | ██ | ██ | |||
≥ 100 mg/dLg | ||||||
n (%) | 73 (30.2) | 208 (86.7) | 40 (5.1) | 279 (35.8) | 63 (7.8) | 314 (38.9) |
OR (95% CI) | ██ | 0.1 (0.1, 0.1) | 0.1 (0.1, 0.2) | |||
P value | ██ | ██ | ██ | |||
██████ | ||||||
███ | ██████ | ██████ | ██████ | ██████ | ██████ | ██████ |
██ ████ | ████████ | ████████ | ██████ | |||
█████ | ██ | ██ | ██ | |||
Missing | ||||||
n (%) | 11 (4.5) | 11 (4.6) | 90 (11.5)h | 114 (14.6)h | 86 (10.6)h | 68 (8.4)h |
ApoB = apolipoprotein; CI = confidence interval; CV = cardiovascular; dL = decalitre; HDL-C = high-density-lipoprotein cholesterol; IQR = interquartile range; L = litre; LDL-C = low-density-lipoprotein cholesterol; LDLR = low-density-lipoprotein receptor; Lp(a) = lipoprotein(a); MACE = major adverse cardiac events; MedDRA = Medical Dictionary for Regulatory Activities; mg = milligram; MI = myocardial infarction; NA = not applicable; nmol = nanomole; NR = not reported; OR = odds ratio; RR = risk ratio; SD = standard deviation; mcg = microgram.a
a████████ ███ ███ ██████████████ ███ ███ █████ ███ █████ ████████ ██ █████ ███ ███ ████ ████ ███████
b███ ██████████ ██ ███████████ █████████ ████ ████████ ███ ███████ ███ ██████████ ████ █████████
c███ █████████ ██ ███ ███ ███ ███ ████ █ ████████ ██████████ ████████ ████ █████████ ██ ███████
d███ ███████ ███ █████████ ██ ███ ███ ███ ███ ████ █ ████████ ██████████ █
e██████ ███ ███ █████ ███ █████ ████████ ███ ████ █████ ███ ████████ █████████
f████ █████ ████████ ██████████ ██████ ████ ████████ ███████ ████ ███ ███████ ████████ ████ ████ ██
gPatients can be counted in multiple categories.
hReasons for missing data included patient discontinued study, a sample issue, or a missed visit; however, for all analyses of goal attainment, multiple imputation methods were used.
i████████ ██ ██ ██████ ████ █████████ ██ ███ ██████ ███████████ ██ ████████ ████ ███ ███ ████ ██████
j███████████ ██████████████ ██████ ████ ███████████████ ██████ █████████ █████ ██████████
Sources: ORION-9 Clinical Study Report;28 ORION-10 Clinical Study Report;29 ORION-11 Clinical Study Report;30 Raal FJ et al.,79 Ray KK et al.11
Table 43: Posthoc Pooled Analysis of the 3 ORION Trials
Outcome | Pooled analysis of CV event outcomes in all 3 trials | |
|---|---|---|
Inclisiran (N = 1,833) | Placebo (N = 1,822) | |
MACEa | ||
n (%) | 131 (7.1) | 172 (9.4) |
Events, n | 141 | 201 |
Time-at-risk, per 100 PYs | 5.35 | 7.71 |
OR (95% CI) | 0.74 (0.58, 0.94) | |
HR (95% CI)b | 0.75 (0.60, 0.94) | |
Fatal or nonfatal MI | ||
n (%) | 33 (1.8) | 41 (2.3) |
Events, n | 34 | 45 |
OR (95% CI) | 0.80 (0.50, 1.27) | |
HR (95% CI)b | 0.81 (0.51, 1.29) | |
Fatal or nonfatal stroke | ||
n (%) | 13 (0.7) | 15 (0.8) |
Events, n | 14 | 16 |
OR (95% CI) | 0.86 (0.41, 1.81) | |
HR (95% CI)b | 0.80 (0.39, 1.67) | |
ASCVD = atherosclerotic cardiovascular disease; CI = confidence interval; CV = cardiovascular; HR = hazard ratio; MACE = major adverse cardiac events; MI = myocardial infarction; OR = odds ratio; PYs = patient-years.
aObserved MACE counts include treatment-emergent and nontreatment-emergent adverse events.
bThe hazard ratio and 95% confidence interval are from a Cox model with treatment and study ID as factors.
Sources: Novartis. (2023) Data on File. CKJX839A1 Inc-Pub084,80 Ray KK et al.81
Table 44: Pooled Harms Results of the 3 ORION Trials
Adverse eventsa | Inclisiran (N = 1,833) | Placebo (N = 1,822) |
|---|---|---|
Most common TEAEs, n (%)b,c | ||
≥ 1 TEAE | 1,430 (78.0) | 1,409 (77.3) |
Diabetes mellitusd | 212 (11.6) | 207 (11.4) |
Nasopharyngitis | 140 (7.6) | 134 (7.4) |
Upper respiratory tract infection | 105 (5.7) | 103 (5.7) |
Hypertension | 104 (5.7) | 104 (5.7) |
Arthralgia | 91 (5.0) | 72 (4.0) |
Serious adverse events, n (%) | ||
Patients with ≥ 1 SAE | 374 (20.4) | 419 (23.0) |
New, worsening, or recurrent cancer | 44 (2.4) | 49 (2.7) |
Patients who stopped treatment due to adverse events, n (%) | ||
Patients who stopped | ||
≥ 1 TEAE leading to discontinuation | 45 (2.5) | 35 (1.9) |
Deaths, n (%) | ||
Total deaths | 27 (1.5) | 27 (1.5) |
Any cause | ORION-9: 1 (0.4) ORION-10: 12 (1.5) ORION-11: 14 (1.7) | ORION-9: 1 (0.4) ORION-10: 11 (1.4) ORION-11: 15 (1.9) |
Cardiovascular cause | ORION-9: 1 (0.4) █████████ █ ████████ | ORION-9: 0 (0.0) █████████ █ ████████ |
Cancer-related | ORION-10: 1 (0.1) ORION-11: 3 (0.4) | ORION-10: 3 (0.4) |
Adverse events of special interest, n (%) | ||
Clinically relevant TEAE at injection sitee | ||
Any reaction | 91 (5.0) | 12 (0.7) |
Mild | 67 (3.7) | 11 (0.6) |
Moderate | 24 (1.3) | 1 (0.1) |
Severe | 0 | 0 |
Persistent | 0 | 0 |
Liver function | ||
Alanine aminotransferase > 3x ULN | 9 (0.5) | 7 (0.4) |
Aspartate aminotransferase > 3x ULN | 8 (0.4) | 10 (0.5) |
Alkaline phosphatase > 2x ULN | 8 (0.4) | 5 (0.3) |
Bilirubin > 2x ULN | 14 (0.8) | 14 (0.8) |
Kidney function | ||
Creatinine > 2 mg/dL | 36 (2.0) | 42 (2.3) |
Muscle | ||
Creatine kinase > 5x ULN | 24 (1.3) | 22 (1.2) |
Hematology | ||
Platelet count < 75x109/L | 1 (0.1) | 2 (0.1) |
dL = decalitre; mg = milligram; MI = myocardial infarction; L = litre; SAE = serious adverse event; TEAE = treatment-emergent adverse event; ULN = upper level normal.
aReported adverse events for inclisiran were prospectively defined using the Medical Dictionary for Regulatory Activities standardized terms by system organ classification.
bMost common adverse events were defined with a threshold of ≥ 2% occurrence in the safety population.
cThe safety population included all participants who received at least 1 dose of inclisiran or placebo.
dDiabetes TEAE represents worsening of glycemic control as defined in the clinical protocol.
eClinically relevant TEAE at the injection site included the preferred terms injection-site erythema, injection-site hypersensitivity, injection-site pruritus, injection-site rash, and injection-site reaction. Among TEAEs, only those at the injection site were considered adverse drug reactions.
Sources: ORION-9 Clinical Study Report,28 ORION-10 Clinical Study Report,29 ORION-11 Clinical Study Report,30 Wright RS et al.91
Pharmacoeconomic Review
Abbreviations
ACS
acute coronary syndrome
ASCVD
atherosclerotic cardiovascular disease
CI
confidence interval
CMA
cost-minimization analysis
CPRD
Clinical Practice Research Database
CUA
cost-utility analysis
CV
cardiovascular
HeFH
heterozygous familial hypercholesterolemia
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LDL-C
low-density-lipoprotein cholesterol
MACE
major adverse cardiovascular event
MI
myocardial infarction
NMA
network meta-analysis
PAD
peripheral artery disease
QALY
quality-adjusted life-year
RR
relative risk
SOC
standard of care
UA
unstable angina
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Inclisiran (Leqvio), 189 mg/mL solution for injection |
Submitted price | Inclisiran, 284 mg/1.5 mL, prefilled syringe: $2,839.28 |
Indication | As an adjunct to lifestyle changes, including diet, to further reduce LDL-C in adults with the following conditions who are on the maximally tolerated dose of a statin, with or without other LDL-C-lowering therapies:
|
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 26, 2021 |
Reimbursement request | Per indication |
Sponsor | Novartis Pharmaceuticals Canada Inc. |
Submission history | Previously reviewed: Yes Indication: As an adjunct to lifestyle changes, including diet, to further reduce LDL-C levels in adults with the following conditions who are on the maximally tolerated dose of a statin, with or without other LDL-C-lowering therapies:
Recommendation date: February 7, 2022 Recommendation: Do not reimburse |
ASCVD = atherosclerotic cardiovascular disease; HeFH = heterozygous familial hypercholesterolemia; LDL-C = low-density-lipoprotein cholesterol; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Economic evaluation 1 (nFH with ASCVD population):
Economic evaluation 2 (HeFH population):
|
Target population | Adult patients with HeFH or nFH with ASCVD who require additional lowering of LDL-C despite maximally tolerated statin therapy. |
Treatment | Inclisiran + SOC (defined as the maximally tolerated dose of statin therapy ± ezetimibe) |
Comparators | nFH with ASCVD population:
HeFH population:
|
Perspective | Canadian publicly funded health care payer |
Outcomes | CUA: QALYs, LYs |
Time horizon | nFH with ASCVD population (CUA): Lifetime (40 years) HeFH population (CMA): 2, 3, 4, 10, 15, and 25 years |
Key data sources | ORION-10 and ORION-11, both randomized controlled trials vs. placebo Sponsor-submitted NMA |
Submitted results | nFH with ASCVD population:
HeFH population:
|
Key limitations |
|
CDA-AMC reanalysis results | nFH with ASCVD population:
HeFH population (CMA):
|
ASCVD = atherosclerotic cardiovascular disease; CMA = cost-minimization analysis; CUA = cost-utility analysis; HeFH = heterozygous familial hypercholesterolemia; ICER = incremental cost-effectiveness ratio; LDL-C = low-density-lipoprotein cholesterol; LY = life-year; MACE = major adverse cardiovascular events; nFH = nonfamilial hypercholesterolemia; NMA = network meta-analysis; NR = not reported; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Conclusions
The CDA-AMC clinical review concluded that inclisiran offers a meaningful reduction in LDL-C compared to standard of care (SOC) but is no more effective than existing proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors. Given the imprecision in the estimates of relative effect, the magnitude of the reduction in low-density-lipoprotein cholesterol (LDL-C) that can be expected in the heterozygous familial hypercholesterolemia (HeFH) and atherosclerotic cardiovascular disease (ASCVD) populations remains unclear. Conclusions could not be drawn for the assessment of relative efficacy using the major adverse cardiac events (MACE) outcome for either population. Although inclisiran appeared to lower the risk of MACE relative to SOC, there was a high risk of bias in the submitted analysis.
CDA-AMC identified 4 key limitations of the submitted economic evaluation for the ASCVD subpopulation. However, only 1 limitation was addressed through reanalysis: the characterization of all relevant sources of parameter uncertainty. In the CDA-AMC base case, inclisiran plus SOC was more costly (incremental costs = $59,990) and more effective (incremental quality-adjusted life-years [QALYs] = 0.77) than SOC. This resulted in an incremental cost-effectiveness ratio (ICER) of $77,705 per QALY gained. There was a 0% probability that inclisiran plus SOC was cost-effective at a threshold of $50,000 per QALY gained. A 32% price reduction would be required for inclisiran plus SOC to be considered cost-effective at that threshold. Although changes in the CDA-AMC base case did not affect the conclusion regarding cost-effectiveness, it did result in greater uncertainty in the total costs and QALYs associated with each treatment considered in the economic model.
In the HeFH population, the sponsor justified the use of a cost-minimization analysis (CMA) by assuming there was no difference in efficacy between inclisiran and the other PCSK9 inhibitors. Although this is consistent with the conclusions regarding reductions in LDL-C, the strength of the assumption was weakened by the imprecision of the estimates of relative effect. For the HeFH subpopulation, CDA-AMC did not undertake a reanalysis of the sponsor’s base-case CMA. A 1% or 2% price reduction may be required if patients are expected to be on inclisiran for 2 years or less. At time horizons beyond 2 years, inclisiran is expected to have the lowest total costs, which suggests that no price reduction is required. This is due to the higher upfront cost of inclisiran compared to alirocumab or evolocumab, which is offset by a lower cost in subsequent years. The longer a patient is assumed to remain on inclisiran versus other comparators, the higher the potential cost savings.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CDA-AMC review process.
Patient input for this review was obtained from the Canadian Heart Patient Alliance and the HeartLife Foundation. Information was collected from patients in Canada via online survey in the submission from the Canadian Heart Patient Alliance and via 1-on-1 interviews in the submission from the HeartLife Foundation. Patients reported experience with current drug therapies such as statins or PCSK9 inhibitors. Input suggested a desire for an accessible treatment with less frequent administration. Six respondents to the survey from the Canadian Heart Patient Alliance reported experience with the drug under review. Five indicated it was effective or very effective, whereas 1 said it was not effective in lowering cholesterol to target levels.
Registered clinician input was received from groups across Canada. These included the Mazankowski Alberta Heart Institute; the Alberta Cardiovascular Disease Prevention Collaborative; the Atlantic Cardiovascular Society; the Cambridge Cardiac Rehabilitation Program; Egyptian Cardiologists of Niagara; Cardiology Associates of Niagara; Lipid Specialists in British Columbia; the Internal Medicine Department and Heart Failure Group at St. Thomas Elgin General Hospital; the Service of Cardiology at Centre Hospitalier Universitaire in Moncton, New Brunswick; the Canadian Cardiovascular Society Dyslipidemia Guideline Committee; Cardiologists in Oakville, Ontario; and the Cardiac Rehabilitation and Secondary Prevention Program at Western University. The consensus among the submissions was that the current pathway of care should begin with behavioural modification interventions, such as smoking cessation, physical exercise, and dietary modifications. Statins are recommended as the initial pharmacological treatment to reduce ASCVD events. Concomitant ezetimibe therapy was suggested for patients who do not achieve adequate LDL-C control on a maximally tolerated statin dose. If adequate LDL-C is still not achieved, PCSK9 inhibitors such as evolocumab and alirocumab can be considered as third-line options. Clinical input submissions suggested that inclisiran would be another candidate for third-line treatment, along with existing PCSK9 inhibitors.
Drug-plan input raised concerns about the coverage status of comparators to inclisiran. PCSK9 inhibitors, such as evolocumab and alirocumab, and other comparators, such as ezetimibe, are covered in the HeFH population but not in the ASCVD population. Concerns were raised about the budget impact from the approval of inclisiran in the ASCVD population, which is substantially larger than the HeFH population.
Several of these concerns were addressed in the sponsor’s model:
Inclisiran was assumed to be used after statin therapy (with or without ezetimibe) or for those who are intolerant to statins.
The economic evaluation for the ASCVD population restricted comparators to SOC. Meanwhile, the economic evaluation for the HeFH population included comparators that are currently funded, such as evolocumab and alirocumab.
Economic Review
The current review is for inclisiran for patients with HeFH or nonfamilial hypercholesterolemia with ASCVD who require additional lowering of LDL-C despite maximally tolerated statin therapy. This is a resubmission; in February 2022, the Canadian Drug Expert Committee (CDEC) did not recommend the reimbursement of inclisiran in this patient population. As part of the resubmission, the sponsor submitted the findings of the ORION-3 study; the ORION-8 study; a pooled analysis of the ORION-9, ORION-10, and ORION-11 studies focusing on the risk of MACE in inclisiran versus placebo across the 3 trials;, and a pooled analysis of 7 ORION trials (ORION-1, ORION-3, ORION-5, ORION-8, ORION-9, ORION-10, ORION-11).
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted 2 independent economic evaluations specific to each subpopulation of the Health Canada–indicated population. For patients with nonfamilial hypercholesterolemia with ASCVD, the sponsor submitted a cost-utility analysis (CUA) that compared the cost-effectiveness of inclisiran plus SOC to SOC alone.1 For the purposes of this submission, SOC was defined as the maximally tolerated dose of statin therapy with or without ezetimibe. Additionally, the ASCVD population was assumed to be comprised of 5 distinct subgroups: patients with acute coronary syndrome (ACS) for 0 to 1 year, patients with ACS for 1 to 2 years, and patients with other coronary heart disease, ischemic stroke, or peripheral artery disease (PAD).1 For the population of patients with HeFH, the sponsor submitted a CMA that compared inclisiran with alirocumab and evolocumab.2
Inclisiran is available as a prefilled 284 mg/1.5 mL (189 mg/mL) syringe for administration as a subcutaneous injection by a health care professional. The recommended dosage for inclisiran is 284 mg administered initially, at month 3, and subsequently every 6 months. The annual cost of inclisiran is $5,679 ($8,518 in the initial year) based on a unit cost of $2,839 per syringe. The annual cost of SOC in the CUA was estimated by the sponsor to be $56 per patient. Alirocumab is available as a single-use prefilled pen or syringe at concentrations of 75 mg/mL and 150 mg/mL. At a unit cost of $268 per pen or syringe and a recommended dosage of 75 mg once every 2 weeks or 300 mg once every 4 weeks, the annual cost of alirocumab is $6,987. Evolocumab is available as a single-use prefilled cartridge (420 mg/3.5 mL; unit cost = $588) or as a single-use prefilled syringe or autoinjector (140 mg/1 mL; unit cost = $271). Depending on the formulation used, evolocumab will cost between $7,053 and $7,077 per year. Alirocumab and evolocumab were only included in the CMA for the HeFH subpopulation.
The clinical outcomes considered in the CUA were QALYs and life-years. The sponsor adopted a lifetime horizon (40 years) using yearly cycles and undertook the analysis from the perspective of the publicly funded health care payer.1 For the CMA, total costs associated with a patient continuously treated with inclisiran, evolocumab, or alirocumab were estimated for 2 years, 3 years, 4 years, 10 years, 15 years, and 25 years.2 Both the CUA and CMA applied a discount rate of 1.5% per year to costs and outcomes, where relevant.1,2
Model Structure
For the CUA (ASCVD population), the sponsor submitted a Markov model that tracked subsequent cardiovascular (CV) events after an initial CV event for a hypothetical cohort of patients (Figure 1 in Appendix 3).1 Health states in the model included an unspecified initial CV event, subsequent CV events (unstable angina [UA], myocardial infarction [MI], stroke, revascularization, and CV death), and death due to non-CV causes (non-CV death). The model used tunnel states to reflect the risk of a subsequent CV event as a parameter that is dependent on the time since a preceding CV event. For the initial CV event, UA, MI, and stroke, 3 tunnel states were used to capture the time since the preceding nonfatal subsequent CV event: 0 to 1 years, 1 to 2 years, and stable (2+ years).1
At model entry, patients were assumed to occupy 1 of the 3 initial health states: initial (0 to 1 years), initial (1 to 2 years), or initial stable. The specific allocation of patients to these states was dependent on the assumed subgroup of patients with ASCVD. At the beginning of each model cycle, patients in the inclisiran model arm faced a risk of treatment discontinuation and switching to SOC. As a result, transitions between health states occurred after the assessment of treatment status.1 After a subsequent nonfatal CV event, patients transitioned to the first tunnel state of the corresponding CV event. It was assumed that patients could experience a maximum of 4 subsequent CV events, and recurrent events of the same type were not permitted. Meanwhile, patients transitioned to the CV death or non-CV death after a fatal CV or non-CV event, respectively. Patients who did not experience a subsequent CV event or non-CV death transitioned to the next tunnel state, increasing the amount of time since the last CV event.1
For the CMA (HeFH population), it was assumed there was no difference in relative efficacy between treatments. As a result, it was unnecessary to specify a model to simulate clinical events for a hypothetical patient cohort.2
Model Inputs
The costs and effects used in the CUA were estimated assuming a homogeneous baseline population of ASCVD patients. All data summarizing the baseline population characteristics of the cohort were obtained from the ORION-10 and ORION-11 trials. Both studies were randomized, phase III, placebo-controlled, double-blind trials of inclisiran.1,3,4 The ORION-10 trial sought to evaluate the effect of inclisiran in patients with ASCVD and elevated LDL-C levels.1,3 Meanwhile, the objective of the ORION-11 trial was to evaluate the effect of inclisiran in patients with ASCVD or ASCVD risk-equivalent and elevated LDL-C.1,4 The ORION-9 pivotal trial was excluded from the CUA because it included patients from the HeFH population.1,5 Population characteristics of interest included baseline age (mean = ████ █████), sex (███ female), diabetic status (███ yes), and LDL-C (mean = ████ ████).1
Due to the heterogeneity of the ASCVD population, costs and effects were generated for each ASCVD subgroup and then combined using a weighted average. The subgroup weights applied in the base case were obtained from the National Institute for Health and Care Excellence (NICE) appraisal of alirocumab: 8.7% for patients with ACS for 0 to 1 year, 0.9% for patients with ACS for 1 to 2 years, 62.3% for patients with other coronary heart disease, 19.2% for patients with ischemic stroke, and 8.9% for patients with PAD.1,6 In addition, a separate scenario analysis was conducted that assumed subgroup weights specific to the Canadian population: 9.1% for patients with ACS for 0 to 1 year, 1.0% for patients with ACS for 1 to 2 years, 62.8% for patients with other coronary heart disease, 21.0% for patients with ischemic stroke, and 6.1% for patients with PAD.1,7 In both scenarios, it was assumed that there were no subgroup differences in terms of baseline population characteristics.1
Transition probabilities for the calculation of state membership were influenced by 3 distinct parameters: CV event risk, non-CV risk of death, and the risk of treatment discontinuation. The CV event risk represented a collection of treatment-specific values that were dependent on the amount of time since revascularization, UA, MI, stroke, or CV death. Estimation of these values involved a 2-step process. First, the baseline CV event risk was estimated for a specific subgroup of patients within the ASCVD population. These data were estimated using the Aurum cohort from the Clinical Practice Research Database (CPRD), a longitudinal research database compiled from primary care practices across the UK.1 Annual risks were estimated for patients with and without diabetes and then weighted using the prevalence of diabetes in the ASCVD population. It was assumed that the baseline CV event risk was 50% higher for all ASCVD subgroups except PAD (base case). Second, the baseline CV event risk was adjusted to reflect the risk from treatment.1 To obtain the CV event risk specific to SOC, a log-linear relationship was assumed between the baseline event risk and the relative risk (RR) of a CV event based on the change in LDL-C.1,8 For this procedure, the change in LDL-C was calculated as the difference between the baseline LDL-C value and the mean LDL-C expected from treatment with SOC. Data regarding the RR per 1 mmol/L reduction in LDL-C were obtained from a network meta-analysis (NMA) comparing the effect of statin therapy with placebo on CV risk outcomes.1,9,10 Values assumed for each CV event included 0.75 (95% confidence interval [CI], 0.92 to 0.78) for revascularization, 0.73 (95% CI, 0.70 to 0.76) for UA and MI, 0.79 (95% CI, 0.74 to 0.85) for stroke, and 0.84 (95% CI, 0.80 to 0.88) for CV death.1,9,10 An additional transformation was required to estimate the CV event risks specific to treatment with inclisiran. In the base case, the sponsor assumed this would involve the same log-linear relationship, based on the change in expected LDL-C levels between inclisiran and SOC. For the RR of a CV event for each 1 mmol/L, the sponsor assumed no difference in efficacy between inclisiran and SOC.1 Two additional scenario analyses were considered in which the inclisiran-specific CV event risk was calculated by multiplying some of the SOC event risks by a hazard ratio (HR) for MACE. In both cases, the inclisiran-specific risk of MI, stroke, and CV death were adjusted by an HR of 0.75 (95% CI, 0.60 to 0.94) calculated from an analysis of patient-level data in phase III trials.1,11 Values for the HR included 0.75 (95% CI, 0.60 to 0.94) for the ASCVD and HeFH populations and ████ ████ ███ ██████████ for the ASCVD population.1,11,12
The time-dependent non-CV risk of death parameter was calculated as the difference between the all-cause mortality risk and the CV-specific mortality risk in Canada. Both inputs represented the age-adjusted and sex-weighted mortality risks using data published by Statistics Canada.13,14 Finally, a 2% annual discontinuation risk was assumed in the inclisiran arm of the model.1
The sponsor estimated age-adjusted and sex-adjusted EQ-5D utility values for individuals with no history of CV disease.1,6,15 Baseline utilities for each health state were estimated by combining these values with utility multipliers for each CV event. Post–CV event disutilities (UA, nonfatal stroke, nonfatal MI) were applied multiplicatively to the health state utilities. Patients were assumed to experience acute disutility in the first year after an event, after which they would experience a chronic postevent utility. No adverse events were considered in the sponsor’s model.
The model included drug costs (acquisition, administration), as well as costs related to the management of CV events. Drug-acquisition costs for inclisiran were based on the sponsor’s submitted price, whereas prices for alirocumab, evolocumab, statins, and ezetimibe were obtained from the Ontario Drug Benefit Formulary.16 The treatment mix of individual statins and statin doses as part of SOC was obtained from IQVIA.1,17 Administration costs were included only for inclisiran, which the sponsor assumed would be administered by a nurse over a 10-minute period.1 Costs associated with CV events (MI, UA, stroke, revascularization) were obtained from the literature.1,18-22 The cost of CV death was based on a generic 1-month end-of-life cost in Canada.1
Summary of Sponsor’s Economic Evaluation Results
For the CUA (ASCVD population), the sponsor-submitted probabilistic analysis aligned with the reimbursement required for the ASCVD population. Results were generated from a Monte Carlo simulation of 500 iterations and were aligned with the deterministic analysis. The submitted analyses were based on the publicly available prices of the comparator treatments.
Base-Case Results
Results from the submitted CUA for the ASCVD population are reported in Table 3. The incremental costs and QALYs for inclisiran plus SOC relative to SOC alone were $60,541 and 0.81, respectively. This led to an ICER of $75,156 for inclisiran plus SOC relative to SOC. At a willingness-to-pay threshold of $50,000 per QALY, inclisiran plus SOC had a 0% probability of cost-effectiveness.
In the CMA for the HeFH population, the annual acquisition and administration cost of inclisiran was $8,538 in the first year of treatment and $5,692 thereafter. If a 2-year time horizon is assumed, a comparison of the predicted costs suggested that inclisiran would be more expensive than alirocumab or evolocumab. However, if a time horizon of at least 3 years was assumed, inclisiran was reported to be cost saving.
Table 3: Summary of the Sponsor’s CUA Results (ASCVD Population)
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
SOC | 41,278.89 | Reference | 8.60 | Ref. | Reference |
Inclisiran + SOC | 101,819.40 | 60,540.51 | 9.40 | 0.81 | 75,156 |
ASCVD = atherosclerotic cardiovascular disease; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Source: Sponsor’s pharmacoeconomic submission, CUA.1
Table 4: Summary of the Sponsor’s Cost-Minimization Analysis Results (HeFH Population)
Drug | Total drug costs ($) | Incremental drug costs ($) | Total costs ($) (drug + admin) | Incremental costs for inclisiran ($) |
|---|---|---|---|---|
Time horizon of 2 years | ||||
Inclisiran | 14,112.48 | Reference | 14,145.63 | Reference |
Alirocumab | 13,871.72 | 240.76 | 13,871.72 | 273.91 |
Evolocumab | 14,049.89 | 62.59 | 14,049.89 | 95.74 |
Evolocumab | 14,001.77 | 110.71 | 14,001.77 | 143.86 |
Time horizon of 3 years | ||||
Inclisiran | 19,541.76 | Reference | 19,587.67 | Reference |
Alirocumab | 20,552.48 | –1,010.72 | 20,552.48 | –964.81 |
Evolocumab | 20,816.45 | –1,274.69 | 20,816.45 | –1,228.78 |
Evolocumab | 20,745.15 | –1,203.39 | 20,745.15 | –1,157.49 |
Time horizon of 25 years | ||||
Inclisiran | 103,855.49 | Reference | 104,099.47 | Reference |
Alirocumab | 124,300.90 | –20,445.41 | 124,300.90 | –20,201.43 |
Evolocumab | 125,897.41 | –22,041.92 | 125,897.41 | –21,797.95 |
Evolocumab | 125,466.20 | –21,610.71 | 125,466.20 | –21,366.73 |
admin = administration; HeFH = heterozygous familial hypercholesterolemia.
Note: Results generated from time horizons of 4 years, 10 years, and 15 years are not reported.
Source: Sponsor’s pharmacoeconomic submission, cost-minimization analysis.2
Sensitivity and Scenario Analysis Results
For the ASCVD population, several additional scenarios were considered in the assessment of relative cost-effectiveness. Scenarios that considered alternate discount rates (3%, 0%), a shorter time horizon (30 years), and inclisiran discontinuation rates (10%) did not affect the conclusion regarding the relative cost-effectiveness of inclisiran. Likewise, the use of Canadian-specific ASCVD subgroup weights did not affect this conclusion, with an ICER of inclisiran plus SOC relative to SOC estimated to be $74,669 per QALY gained.1 Two additional scenarios were considered in which the inclisiran-specific CV event risk was calculated by multiplying the SOC event risks by a HR for MACE. Detailed results are reported in Appendix 3 (Table 12), which illustrate that this alternate assumption had no impact on the conclusion that inclisiran would not be cost-effective at a $50,000 per QALY threshold.
The sponsor conducted a scenario analysis from a societal perspective that included additional costs associated with productivity gains estimated using the human capital approach.1 In this scenario, the ICER of inclisiran plus SOC relative to SOC was estimated to be $63,783 per QALY gained.1 Although this was lower than the submitted base case, which used a Canadian health care payer perspective, it had no impact on the conclusion that inclisiran would not be cost-effective at a $50,000 per QALY threshold.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The relative clinical effectiveness of inclisiran is highly uncertain: The sponsor’s submission considered 2 distinct outcomes that could be used to draw conclusions for relative clinical effectiveness: the incidence of MACE and the change in LDL-C. For the MACE outcome, the sponsor conducted a post hoc analysis of pooled data from the ORION-9, ORION-10, and ORION-11 trials. In addition, the sponsor conducted a separate analysis using the ASCVD population only (ORION-10 and ORION-11 trials). Results from both analyses suggested that inclisiran had a lower risk of MACE than placebo (HR = 0.75; ██ █ ████).1,11,12 However, the potential for bias in the analysis prevented the CDA-AMC clinical review from drawing a definitive conclusion for this outcome. Concerns were raised that the ORION trials were not designed to make comparisons between treatment groups for this outcome. In addition, the decision to pool the data from all 3 ORION trials failed to distinguish the HeFH and ASCVD populations, which were separately identified in the indication for this review.
For the change in LDL-C, assessments of relative effectiveness were made using the sponsor-submitted indirect treatment comparison (ITC). The sponsor-submitted ITC did not include an evaluation of clinical efficacy outcomes. This ITC compared inclisiran with SOC (placebo), evolocumab, and alirocumab. The CDA-AMC clinical review concluded that there was no difference in relative efficacy between inclisiran and alirocumab or evolocumab in the HeFH and ASCVD populations, but that inclisiran offered a greater reduction in LDL-C than placebo. However, the findings were subject to considerable uncertainty, as reflected by wide CIs. The CDA-AMC clinical review suggested that the findings may be subject to even greater uncertainty because of considerable clinical and methodological heterogeneity in the evidence used in the submitted ITC.
In the context of the economic evaluation, the impact of the uncertainty with respect to both outcomes differed by subpopulation. For the HeFH population, the sponsor justified the use of a CMA by assuming there was no difference in efficacy between inclisiran, alirocumab, and evolocumab. This assumption could not be tested for the MACE outcome due to a lack of evidence. In terms of a reduction in LDL-C, the conclusion of the CDA-AMC clinical review using the mean values from the NMA were supportive of this assumption. However, the strength of this assumption is weakened by the wide CIs reflected in the ITC. For the ASCVD population, both outcomes were used as input parameters to the economic model to predict the total cost and QALYs for inclisiran and SOC. Details regarding the impact of the MACE data on the decision uncertainty are outlined in the next limitation.
CDA-AMC was unable to address this limitation.
The sponsor’s MACE scenarios were associated with uncertainty: The sponsor considered 2 additional scenario analyses to predict the CV event risk for inclisiran using an HR for MACE. Estimates for the HRs represented the combined HeFH and ASCVD populations (HR = 0.75) from the ORION-9, ORION-10, and ORION-11 trials, as well as the ASCVD subpopulation (██ █ ████) using data from the ORION-10 and ORION-11 trials.1,11,12 The purpose of these scenario analyses was to explore the impact of using new safety evidence on CV event risk instead of changes in LDL-C.
CDA-AMC identified 3 concerns with this approach. First, the sponsor considered an estimate of the MACE HR that was not specific to the specified population being modelled. Given the focus of the CUA on the ASCVD subpopulation, it was inappropriate to use an estimate of an HR (0.75) that reflected the combined HeFH and ASCVD subpopulations. The relevant scenario is the 1 that used the HR from the ASCVD subgroup (████). Second, the introduction of the MACE scenario did not remove the underlying relationship between changes in LDL-C and CV event risk. Instead of considering changes in LDL-C specific to inclisiran, the MACE HR was applied to SOC-specific CV event risks (e.g., MI, stroke, and CV death). In these scenarios, it was assumed there would be no difference in the risk of revascularization or UA between inclisiran and SOC. Therefore, the estimation of CV event risk was still dependent on changes in LDL-C for SOC, which was used as the input to the calculation of the CV event risks using the MACE HR. As such, the scenarios remain dependent on the LDL-C prediction of CV events. Third, the introduction of the MACE scenario did not have a meaningful impact on the decision uncertainty in the model. As detailed in Table 12, the MACE scenario using the ASCVD-specific HR (████) did not influence the conclusion regarding the cost-effectiveness of inclisiran at a threshold of $50,000 per QALY in the sponsor’s submission. Furthermore, the scenario suggests that the model is no longer dependent on uncertainty in 2 parameters: the inclisiran-specific percent change in LDL-C from the submitted NMA, and the RR of a CV event (refer to Appendix 3). As a result, reliance on fewer input parameters that affect the risk of some CV events is unlikely to lead to an increase in decision uncertainty.
CDA-AMC did not consider the MACE scenario to be relevant for the decision problem.
Generalizability of baseline risk data to a Canadian context: In the absence of Canadian-specific data, the sponsor used data from the Aurum cohort of the CPRD database in the UK to inform the baseline CV risk for patients with ASCVD. As noted in the original inclisiran submission and previous CDA-AMC reviews, transition probabilities obtained from CPRD might not be applicable to patients with ASCVD in Canada. In the absence of Canadian-specific data, the impact of this limitation on the results of the economic evaluation is unclear.
CDA-AMC was unable to address this limitation.
Failure to characterize all relevant sources of parameter uncertainty: To address the fact that the true value of a parameter may not be known, CDA-AMC guidelines require the probabilistic evaluation of economic models.23 This involves the repeated estimation of costs and QALYs for each alternative using values selected at random from an assumed distribution for each parameter.23-25 To effectively support decision-making in a context of uncertainty, it is critical to ensure that the costs and effects are generated in a manner that considers imprecision in all model input parameters.23-25 Failure to do so may result in different mean costs and QALYs and, in some circumstances, a different conclusion regarding the cost-effectiveness of the drug under review. A significant limitation of the sponsor’s submission was the assumption that there was no uncertainty with respect to 4 baseline characteristics: age, sex, diabetic status, and LDL-C.1 This assumption is problematic for 2 reasons. First, it is inconsistent with the uncertainty reflected in the submitted evidence from the ORION trials for the ASCVD subpopulation. Second, each of the baseline characteristics affected each CV event risk calculation and, by extension, the movement between health states in the model. Therefore, the failure to characterize all relevant sources of uncertainty may result in a different estimate for the ICER and, in some cases, a different conclusion regarding relative cost-effectiveness.23-25
CDA-AMC addressed this limitation by characterizing the uncertainty in all relevant input parameters. It was assumed the age and LDL-C values would follow a normal distribution. Standard deviation statistics were calculated from patient-level data from the ORION trials included in the submitted spreadsheet. Meanwhile, the sex and diabetic status parameters were assumed to follow a beta distribution, using the method-of-moments approach.24
Additionally, the key assumption detailed in Table 5 was made by the sponsor and has been appraised by CDA-AMC.
Table 5: Key Assumption of the Submitted Economic Evaluation
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
No between-treatment difference in the relative risk of a cardiovascular event per 1 mmol/L reduction in LDL-C. | The clinical experts consulted by CDA-AMC agreed that this is consistent with current evidence. |
LDL-C = low-density-lipoprotein cholesterol.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC conducted a reanalysis of the economic evaluation for the ASCVD subpopulation that addressed some of the key limitations identified in the sponsor’s submission. The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. A summary of each independent modification to the submitted economic evaluation is presented in Table 6. The costs and effects for the CDA-AMC base case were generated using a Monte Carlo simulation of 2,500 iterations.
Table 6: CDA-AMC Revisions to the Submitted CUA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Failure to characterize parameter uncertainty | Baseline characteristics (age, percent female, LDL-C, percent with diabetes) were not varied probabilistically. | Uncertainty in baseline characteristics needs to be characterized. Age and LDL-C involved a normal distribution; percent female and percent with diabetes involved a beta distribution. |
CDA-AMC base case | — | Reanalysis 1 |
LDL-C = low-density-lipoprotein cholesterol.
Results from the CDA-AMC base case are presented in Table 7. Consistent with the sponsor’s base case, the CDA-AMC reanalysis was based on publicly available prices of the comparator treatments. With expected costs and QALYs of $100,461 and 9.47, respectively, inclisiran plus SOC was more costly and more effective than SOC alone, which is reflected in the estimated ICER of $77,705. At a threshold of $50,000 per QALY gained, inclisiran had a 0% probability of being cost-effective.
Additional details summarizing the CDA-AMC base are presented in Appendix 4. Results from the CDA-AMC base case suggest that the changes had a small impact on the expected costs and QALYs, and by extension the ICER. The disaggregated results reported in Table 13 suggest that the acquisition cost of inclisiran was a meaningful contributor to the conclusion that inclisiran plus SOC is not cost-effective at a threshold of $50,000 per QALY gained. This finding is consistent with the sponsor’s base case. In both circumstances, the incremental benefits from treatment were not large enough to offset the relative increase in costs. The effect of the changes in the CDA-AMC base case was most pronounced in the distributions of costs and QALYs used to make conclusions about relative cost-effectiveness. As detailed in Figure 2 in Appendix 4, the CDA-AMC base case reflected a much different characterization of uncertainty for the costs and QALYs associated with each treatment.
Table 7: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results for the Submitted CUA
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | SOC | 41,278.89 | 8.60 | Reference |
Inclisiran + SOC | 101,819.40 | 9.40 | 75,156 | |
CDA-AMC reanalysis 1 and base case | SOC | 40,470.67 | 8.70 | Reference |
Inclisiran + SOC | 100,460.53 | 9.47 | 77,705 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
CDA-AMC did not identify any limitations with the sponsor-submitted CMA for HeFH and, therefore, did not conduct any reanalyses.
Scenario Analysis Results
For the economic evaluation of the ASCVD subpopulation, a series of scenario analyses were conducted to explore the price reductions required to obtain an ICER for inclisiran plus SOC below a $50,000 per QALY threshold. As summarized in Table 8, a 32% price reduction for inclisiran is required for the inclisiran plus SOC arm of the economic model to be considered cost-effective in the ASCVD subpopulation.
For the economic evaluation of the HeFH subpopulation, a scenario analysis was conducted to determine the price reduction needed to obtain total costs equivalent to the least and most expensive comparators in the CMA. As summarized in Table 9, a price reduction of 2% and 1%, respectively, was required for inclisiran to be less expensive than the least (alirocumab) or most (evolocumab 140 mg/mL) expensive comparator, assuming a 2-year time horizon. No price reduction was required when a time horizon of 3 years or greater was assumed.
Table 8: CDA-AMC Price Reduction Analyses for the ASCVD Population
Analysis | ICERs for Inclisiran + SOC vs. SOC ($/QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CDA-AMC reanalysis |
No price reduction | 75,156 | 77,705 |
10% | 66,993 | 69,027 |
20% | 58,532 | 60,350 |
30% | 50,070 | 51,673 |
40% | 41,608 | 42,996 |
50% | 33,146 | 34,319 |
60% | 24,685 | 25,641 |
70% | 16,223 | 16,965 |
80% | 7,762 | 8,287 |
90% | Dominated | Dominated |
ASCVD = atherosclerotic cardiovascular disease; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year SOC = standard of care; vs. = versus.
Table 9: CDA-AMC Price Reduction Analyses for the HeFH Population
Analysis | Submitted price ($) | Reduction needed (%) | Reduced price ($) | Savings relative to submitted pricea ($) |
|---|---|---|---|---|
Time horizon of 2 years | ||||
Price reduction required to equal the least expensive comparator (alirocumab)b | 2,839.28 | 2 | 2,782.49 | 56.79 |
Price reduction required to equal the most expensive comparator (evolocumab 140 mg/mL)b | 2,839.28 | 1 | 2,810.89 | 28.39 |
HeFH = heterozygous familial hypercholesterolemia.
Note: At a time horizon of 3 years and greater, inclisiran + SOC is less expensive than all other comparators and does not require a price reduction.
aSavings from the sponsor’s list price per patient per year.
bRelative to publicly available list prices of comparators.
Issues for Consideration
At the time of this writing, PCSK9 inhibitors are not reimbursed by CDA-AMC-participating drug plans for the treatment of ASCVD. Although evolocumab and alirocumab received conditional recommendations from the Canadian Drug Expert Committee, negotiations with the pan-Canadian Pharmaceutical Alliance concluded without agreement on either drug.26-29 Both evolocumab and alirocumab are reimbursed for the treatment of HeFH.
Overall Conclusions
The CDA-AMC clinical review concluded that inclisiran offers a meaningful reduction in LDL-C compared to SOC. However, there was no difference in relative efficacy between inclisiran and other PCSK9 inhibitors (i.e., alirocumab and evolocumab) in the HeFH or ASCVD subpopulations based on the sponsor’s NMA. The magnitude in reduction of LDL-C that can be expected from inclisiran is unclear due to the imprecision of the relative effect estimates obtained from the NMAs. Given the indirect estimation of relative treatment effect, such findings may be subject to additional unmeasured uncertainty attributable to the clinical and methodological heterogeneity of the trials used in the analysis. Meanwhile, the CDA-AMC clinical review could not reach definitive conclusions regarding the relative effectiveness of inclisiran for the MACE outcome. This was attributed to a high risk of bias from the conduct of a post hoc analysis of an outcome that was not considered in the original ORION trial designs.
The impact of both outcomes on the economic evaluation differed by population. In the HeFH population, the sponsor justified the use of a CMA by assuming there was no difference in efficacy between inclisiran and the other PCSK9 inhibitors. Although this could not be evaluated using the MACE outcome, it was supported by the evidence for changes in LDL-C. However, the strength of this assumption was weakened by uncertainty in the estimates obtained from the NMA. In the ASCVD population, both outcomes were included as input parameters to distinct scenarios of the cohort simulation used to estimate costs and QALYs. However, the MACE scenario did not represent a meaningful source of decision uncertainty.
CDA-AMC identified 4 key limitations of the submitted economic evaluation for the ASCVD subpopulation: uncertainty in the relative effectiveness of inclisiran, the specification of an inappropriate scenario, the absence of Canadian-specific baseline risk data, and a failure to characterize all relevant sources of parameter uncertainty. CDA-AMC attempted to address some of these limitations through reanalysis. These changes involved assigning distributions to 4 parameters: baseline age, baseline LDL-C, sex, and diabetic status.
For the ASCVD subpopulation, the CDA-AMC base case resulted in similar conclusions to the sponsor’s submission. Relative to SOC alone, inclisiran plus SOC was more costly (incremental costs = $59,990) and more effective (incremental QALYs = 0.77). This resulted in an ICER of $77,705 per QALY gained. There was a 0% probability that inclisiran plus SOC was cost-effective at a threshold of $50,000 per QALY gained. A 32% price reduction would be required for inclisiran plus SOC to be considered cost-effective at that threshold. Although the changes in the CDA-AMC base case did not affect the conclusion regarding cost-effectiveness, it did result in greater uncertainty in the total costs and QALYs associated with each treatment considered in the economic model.
For the HeFH subpopulation, CDA-AMC did not undertake a reanalysis of the sponsor’s base-case CMA. A 1% or 2% price reduction may be required if patients are expected to be on inclisiran for 2 years or less. For time horizons beyond 2 years, inclisiran was expected to have the lowest total costs, which suggests that no price reduction would be required. This is due to the higher upfront cost of inclisiran compared to alirocumab and evolocumab, which is offset by a lower cost in subsequent years. The longer a patient is assumed to remain on inclisiran versus other comparators, the higher the potential cost savings.
References
1.Cost utility analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Leqvio® (inclisiran), solution for subcutaneous injection, 284 mg in 1.5 mL (189 mg/mL). Montreal (QC): Novartis Pharmaceuticals Canada Inc.; 2023 Sep 19.
2.Cost minimization analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Leqvio® (inclisiran), solution for subcutaneous injection, 284 mg in 1.5 mL (189 mg/mL). Montreal (QC): Novartis Pharmaceuticals Canada Inc.; 2023 Sep 19.
3.Clinical Study Report: ORION-10 (MDCO-PCS-17-04). A placebo-controlled, double-blind, randomized trial to evaluate the effect of 300 mg of inclisiran sodium given as subcutaneous injections in subjects with atherosclerotic cardiovascular disease (ASCVD) and elevated low density lipoprotein cholesterol (LDL-C) [sponsor-provided reference]. Parsippany-Troy Hills (NJ): The Medicines Company; 2019 Dec 5.
4.Clinical Study Report: ORION-11 (MDCO-PCS-17-08). A placebo-controlled, double-blind, randomized trial to evaluate the effect of 300 mg of inclisiran sodium given as subcutaneous injections in subjects with atherosclerotic cardiovascular disease (ASCVD) or ASCVD-risk equivalents and elevated low-density lipoprotein cholesterol (LDL-C) [sponsor-provided reference]. Parsippany-Troy Hills (NJ): The Medicines Company; 2019 Nov 20.
5.Clinical Study Report: ORION-9 (MMDC-PCS-17-03). A placebo-controlled, double-blind, randomized trial to evaluate the effect of 300 mg of inclisiran sodium given as subcutaneous injections in subjects with heterozygous familial hypercholesterolemia (HeFH) and elevated low density lipoprotein cholesterol (LDL-C) [sponsor-provided reference]. Parsippany-Troy Hills (NJ): The Medicines Company; 2019 Dec 2.
6.Alirocumab for treating primary hypercholesterolaemia and mixed dyslipidaemia [sponsor-provided reference]. (Technology appraisal guidance TA393). London (GB): National Institute for Health and Care Excellence (NICE); 2016: https://www.nice.org.uk/guidance/ta393/resources/alirocumab-for-treating-primary-hypercholesterolaemia-and-mixed-dyslipidaemia-82602908493253.
7.Blais C, Rochette L, Ouellet S, Huynh T. Complex Evolution of Epidemiology of Vascular Diseases, Including Increased Disease Burden: From 2000 to 2015. Can J Cardiol. 2020;36(5):740-746. PubMed
8.Cholesterol Treatment Trialists Collaborators, Mihaylova B, Emberson J, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380(9841):581-590. PubMed
9.Cholesterol Treatment Trialists Collaborators, Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670-1681. PubMed
10.Cholesterol Treatment Trialists Collaboration. Efficacy and safety of statin therapy in older people: a meta-analysis of individual participant data from 28 randomised controlled trials. Lancet. 2019;393(10170):407-415. PubMed
11.Ray KK, Raal FJ, Kallend DG, Jaros MJ, Koenig W, et al. Inclisiran and cardiovascular events: a patient-level analysis of phase III trials. Eur Heart J. 2022;44(2)(2):129-138.
12.Novartis. (2023) Data on File. CKJX839A1 Inc-Pub084 [sponsor-provided reference].
13.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2023 Nov 1.
14.Statistics Canada. Table 13-10-0392-01. Deaths and age-specific mortality rates, by selected grouped causes. Release date: 2022-01-24 [sponsor-provided reference]. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310039201. Accessed 2023 May 25.
15.Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health. 2010;13(5):509-518. PubMed
16.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Oct 23.
17.IQVIA. IQVIA Compuscript National Trx Ext Units from Jan 2019-Nov 2020 [sponsor-provided reference]. 2020.
18.Tran DT, Welsh RC, Ohinmaa A, Thanh NX, Kaul P. Resource Use and Burden of Hospitalization, Outpatient, Physician, and Drug Costs in Short- and Long-term Care After Acute Myocardial Infarction. Can J Cardiol. 2018;34(10):1298-1306. PubMed
19.Padwal RS, So H, Wood PW, et al. Cost-effectiveness of home blood pressure telemonitoring and case management in the secondary prevention of cerebrovascular disease in Canada. J Clin Hypertens (Greenwich). 2019;21(2):159-168. PubMed
20.Yu AYX, Krahn M, Austin PC, Rashid M, Fang J, et al. Sex differences in direct healthcare costs following stroke: a population-based cohort study. BMC Health Serv Res. 2021;21:619. PubMed
21.Third Line Pharmacotherapy for Type 2 Diabetes - Update [sponsor-provided reference]. (CADTH optimal use report vol.3, no.1b). Ottawa (ON): CADTH; 2013: https://www.cadth.ca/sites/default/files/pdf/OP0512_Diabetes%20Update_Third-line_e.pdf. Accessed 2023 Nov 1.
22.Boczar KE, Beanlands R, Wells G, Coyle D. Cost-effectiveness of colchicine for recurrent cardiovascular events. CJC Open. 2023;5(5):348-356. PubMed
23.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2023 Nov 17.
24.Briggs A, Claxton K, Sculpher M. Decision Modelling for Health Economic Evaluation. New York: Oxford University Press; 2006.
25.Claxton K. Exploring uncertainty in cost-effectiveness analysis. Pharmacoeconomics. 2008;26(9):781-798. PubMed
26.CADTH Canadian Drug Expert Committee final recommendation: Evolocumab (Repatha - Amgen Canada Inc) [sponsor-provided reference]. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/sites/default/files/cdr/complete/SR0515_Repatha_Resubmission_complete_Nov_24_17.pdf.
27.CADTH Canadian Drug Expert Committee Final Recommendation: Alirocumab (Praluent - Sanofi-aventis Canada Inc.) [sponsor-provided reference]. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/cdr/complete/SR0469_complete_Praluent_Jul-20-16.pdf.
28.Pan-Canadian Pharmaceutical Alliance. Praluent (alirocumab) [sponsor-provided reference]. 2019: https://www.pcpacanada.ca/negotiation/21075.
29.Pan-Canadian Pharmaceutical Alliance. Repatha (evolocumab) [sponsor-provided reference]. 2019: https://www.pcpacanada.ca/negotiation/21004.
30.Government of Ontario. Exceptional Access Program product prices. 2023; https://www.ontario.ca/page/exceptional-access-program-product-prices. Accessed 2023 Oct 23.
31.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Leqvio® (inclisiran), solution for subcutaneous injection, 284 mg in 1.5 mL (189 mg/mL). Montreal (QC): Novartis Pharmaceuticals Canada Inc.; 2023 Sep 19.
32.Chiang CE, Schwartz GG, Elbez Y, et al. Alirocumab and Cardiovascular Outcomes in Patients With Previous Myocardial Infarction: Prespecified Subanalysis From ODYSSEY OUTCOMES. Can J Cardiol. 2022;38(10):1542-1549. PubMed
33.Canadian Chronic Disease Surveillance System (CCDSS). Ischemic heart disease prevalence. (2019-2020). https://health-infobase.canada.ca/ccdss/data-tool/. Accessed 2023 Oct 30.
34.Canadian Chronic Disease Surveillance System (CCDSS). Stroke prevalence. (2019-2020). https://health-infobase.canada.ca/ccdss/data-tool/. Accessed 2023 Oct 30.
35.Akioyamen LE, Genest J, Shan SD, et al. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open. 2017;7(9):e016461. PubMed
Appendix 1: Cost Comparison Table
Table 10: CDA-AMC Cost Comparison for the Treatment of Primary Hypercholesterolemia for Patients With HeFH or ASCVD
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Inclisiran (Leqvio) | 189 mg / mL | Single-use, prefilled syringe (284 mg / 1.5 mL) | 2,839.2800 | 284 mg administered initially at month 3, and subsequently every 6 months | Initial year: 23.34 Subsequent years: 15.56 | Initial year: 8,518 Subsequent years: 5,679 |
Anti-PCSK9 monoclonal antibody | ||||||
Alirocumab (Praluent) | 75 mg / mL 150 mg / mL | Single-use prefilled pen or syringe | 267.8300 per pen or syringe | 75 mg once every 2 weeks or 300 mg once every 4 weeks | 19.07 | 6,964 |
Evolocumab (Repatha) | 120 mg / mL 140 mg / mL | Single-use prefilled cartridge (420 mg / 3.5 mL) Single-use prefilled syringe or autoinjector (140 mg / 1 mL) | 587.75000 (per cartridge) 271.2700 (per autoinjector) | 420 mg monthly (cartridge) 140 mg every 2 weeks (syringe/autoinjector) | 19.31 19.38 | 7,053 7,077 |
Cholesterol absorption inhibitor | ||||||
Ezetimibe (Ezetrol) | 10 mg | Tablet | 1.9443 | 10 mg daily | 1.94 | 711 |
Ezetimibe (generic) | 10 mg | Tablet | 0.1811 | 10 mg daily | 0.18 | 67 |
Lipid-regulating drug | ||||||
Icosapent ethyl (Vascepa) | 1 g | Capsule | 2.4500a | 2 g twice daily | 9.80 | 3,580 |
Fibrates | ||||||
Bezafibrate | 400 mg | Tablet | 2.6688 | 400 mg daily | 2.67 | 975 |
Bezafibrate (generic) | 400 mg | Tablet | 1.7460 | 400 mg daily | 1.75 | 638 |
Fenofibrate (generic) | 67 mg 100 mg 200 mg | Capsule | 0.6025 0.6105 0.9257 | 67 to 200 mg daily | 0.60 0.61 0.93 | 221 223 339 |
Fenofibrate (Lipidil EZ) | 48 mg 145 mg | Tablet | 0.4799 1.2289 | 48 to 145 mg daily | 0.48 1.23 | 176 449 |
Gemfibrozil (generic) | 300 mg | Capsule | 0.1340 | 600 mg daily | 0.27 | 98 |
Micro-coated fenofibrate (Lipidil Supra) | 160 mg | Tablet | 1.3968 | 160 mg daily | 1.40 | 511 |
The comparators presented in the above table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed October 2023), unless otherwise indicated, and do not include dispensing fees.16
aUnit price obtained from the Ontario Exceptional Access Program (accessed October 2023).30
Note: This table has not been copy-edited.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | Yes | No comment |
Model structure is adequate for decision problem | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Failure to characterize uncertainty for parameters representing baseline characteristics. Refer to limitation in the CDA-AMC appraisal section of this report. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Failure to characterize uncertainty for parameters representing baseline characteristics. Refer to limitation in the CDA-AMC appraisal section of this report. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Calculation of Cardiovascular Event Risk
In the submitted base case, CV event risk for SOC and inclisiran was calculated as a function of changes in LDL-C. This involved a 2-step process. First, the SOC-specific risk of each cardiovascular event was calculated using the equation below. For each event, k, a log-linear relationship was assumed between the baseline event risk, and the relative risk of a CV event per 1 mmol / L reduction in LDL-C. Values for the RR parameter were obtained from a NMA comparing the effect of statin therapy and placebo on cardiovascular risk outcomes.
The baseline LDL-C value represented the baseline value from the ORION-10 and −11 trials, while the SOC-specific LDL-C value was calculated from the application of the parameter representing the percent change in LDL-C specific to SOC from the sponsor-submitted NMA.
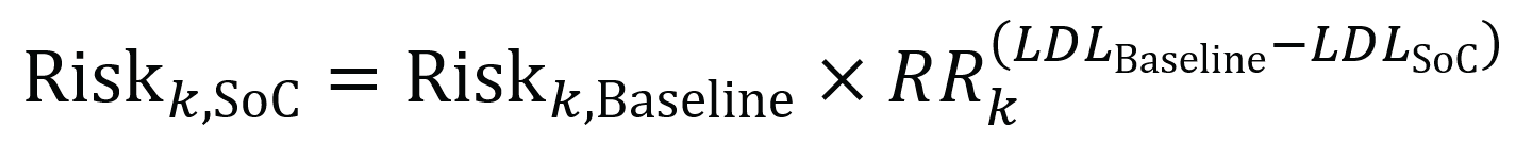
Additional calculations were required to estimate the inclisiran-specific risk of each cardiovascular event from the corresponding SOC-specific risk. In the submitted base case, a similar log-linear relationship was used to capture the change in expected LDL-C levels between inclisiran and SOC. The LDL-C values used in the equation represent the predicted change from baseline for SOC and inclisiran using the corresponding estimates of the percent change in LDL-C from the sponsor-submitted NMA.
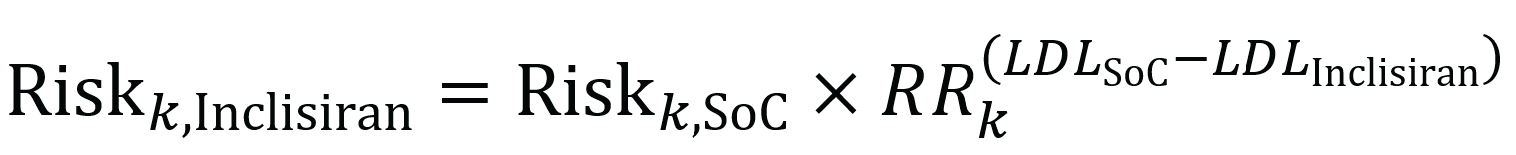
An additional scenario analysis was also included in the submission, where the risk of some cardiovascular events were calculated using a HR for major adverse cardiovascular events. As detailed in the equation below, this risk was only applied to MI, stroke, and cardiovascular death. The sponsor assumed no difference between inclisiran and SOC for the risk of revascularization or unstable angina. As a result, the inclisiran-specific event risks were dependent on changes in LDL-C between baseline and SOC and did not consider evidence relating to the percent change in LDL-C specific to inclisiran.
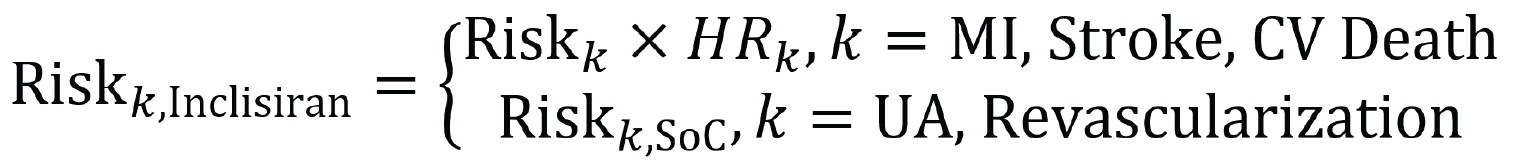
Detailed Results of the Sponsor’s Submission
Results from the sponsor’s base case along with the MACE scenarios are presented in Table 12. This is included to reflect the impact of the new safety evidence which was not present in the original inclisiran submission to CDA-AMC. Results from the MACE scenarios illustrate 2 key issues. First, as described above the MACE scenario only affected the risk of inclisiran-specific CV events. This is reflected by the fact that the SOC results were unchanged from the submitted base case. Second, the MACE scenario does not appear to be an important source of uncertainty for the decision problem. The introduction of this structural assumption did not affect the conclusion that inclisiran plus SOC would not be cost-effective at a $50,000 per QALY threshold. While it impacted the estimated ICER, the extent to which the MACE HR should be viewed as an influential parameter is unclear. For example, results from the first scenario may not be relevant to the decision problem as the data used to estimate the HR of 0.75 represented the combined HeFH and ASCVD subpopulation. Meanwhile, the effect of the ASCVD-specific HR on the ICER was much smaller – which suggests the impact of this parameter on the overall decision uncertainty is minimal.
Table 12: CUA Results — Base-Case and MACE Scenarios
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
Submitted Base Case | |||||
SOC | 41,278.89 | Ref. | 8.60 | Ref. | Ref. |
Inclisiran + SOC | 101,819.40 | 60,540.51 | 9.40 | 0.81 | 75,156 |
MACE Scenario 1: HR = 0.75 (HeFH + ASCVD) | |||||
SOC | 41,278.89 | Ref. | 8.60 | Ref. | Ref. |
Inclisiran + SOC | 106,087.97 | 64,809.08 | 9.27 | 0.67 | 96,709 |
MACE Scenario 2: HR = ████ (ASCVD Only) | |||||
SOC | 41,278.89 | Ref. | 8.60 | Ref. | Ref. |
Inclisiran + SOC | 106,520.81 | 65,241.92 | 9.41 | 0.82 | 79,825 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus; SOC = Standard of Care; ASCVD = atherosclerotic cardiovascular disease; HeFH = heterozygous familial hypercholesterolemia; MACE = major adverse cardiovascular event; HR = hazard ratio.
Source: Sponsor’s pharmacoeconomic submission - CUA.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
To address some of the key limitations from the sponsor’s submission, a series of changes were implemented to derive the CDA-AMC base case. Each revision listed in Table 6 was implemented independently and the corresponding results are presented in Table 7. A disaggregated summary of the CDA-AMC base-case simulation is presented in Table 13.
In addition, Figure 2 represents the base case probabilistic results plotted on the cost-effectiveness plane. Panel A represents the base case submitted by the sponsor, while Panel B represents the distributions generated in the CDA-AMC base case. This figure highlights how the characterization of uncertainty for the baseline characteristics had an important effect on the distribution of costs and QALYs generated by the economic model.
Detailed Results of CDA-AMC Base Case
Table 13: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Parameter | Inclisiran + SOC | SOC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 13.57 | 12.52 | 1.05 |
Discounted QALYs | |||
Total | 9.47 | 8.70 | 0.77 |
By health state or data source | |||
Initial | 6.63 | 5.42 | 1.20 |
Post UA | 0.50 | 0.58 | −0.09 |
Post MI | 0.58 | 0.73 | −0.15 |
Post Stroke | 0.57 | 0.67 | −0.10 |
Post Revascularization | 1.20 | 1.30 | −0.10 |
Discounted costs ($) | |||
Total | 100,460.53 | 40,470.67 | 59,989.86 |
Acquisition: Inclisiran | 67,268.48 | 0.00 | 67,268.48 |
Acquisition: SOC | 758.29 | 699.51 | 58.78 |
Administration | 158.03 | 0.00 | 158.03 |
CV Event Costs | 32,275.73 | 39,771.16 | -$7,495.43 |
ICER ($/QALY) | 77,705 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care; UA = unstable angina; MI = myocardial infarction; NA = not available.
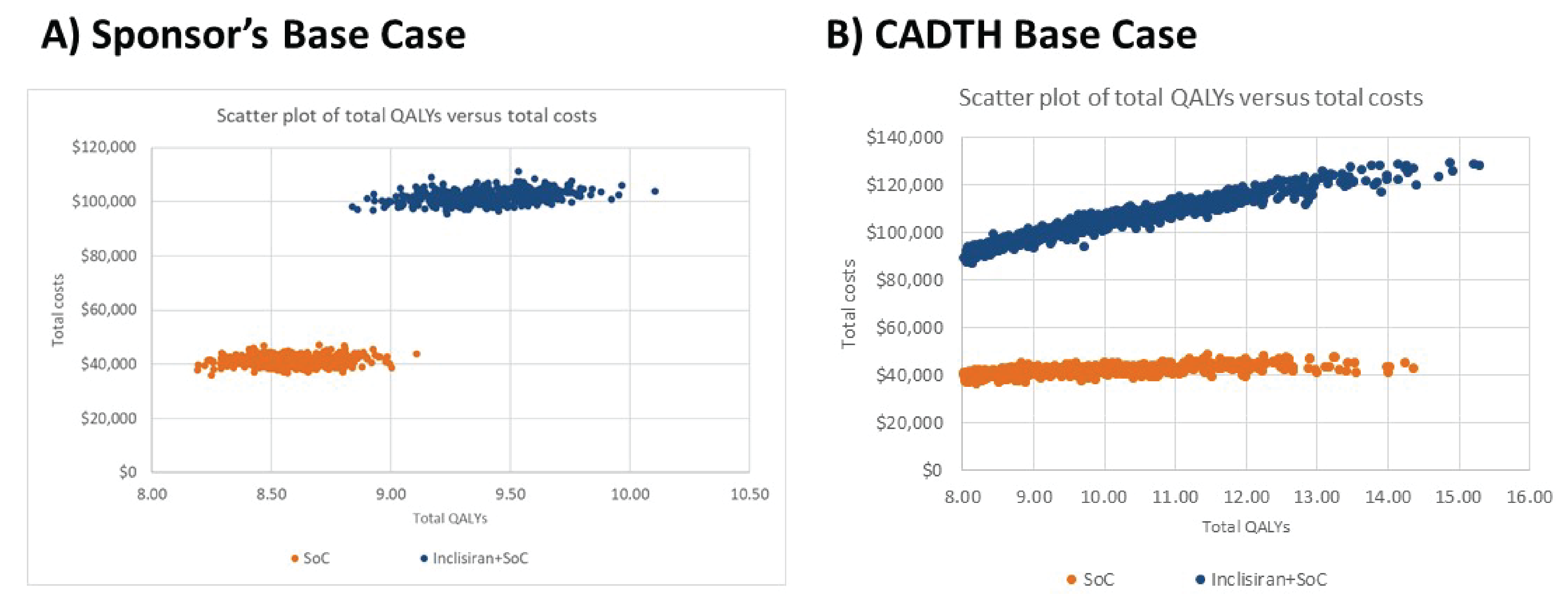
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 14: Summary of Key Take Aways
Key Take Aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted budget impact analysis (BIA) assessed the expected budget impact of reimbursing inclisiran as an adjunct to diet and maximally tolerated statin therapy, with or without other lipid-lowering therapies, in adult patients with HeFH or ASCVD who require additional lowering of LDL-C.31 The BIA was undertaken from the perspective of Canadian public drug plans (excluding Quebec) over a 3-year time horizon, stratified by population (HeFH or ASCVD). For each population, an epidemiological approach was used to estimate the eligible number of patients in each year of the BIA (Figure 3).31 Key inputs to the BIA are documented in Table 15.
In the reference scenario, it was assumed that patients eligible for treatment would receive 1 of the currently available alternatives to inclisiran. For the HeFH population, this was restricted to evolocumab and alirocumab. For the ASCVD population, alternatives to inclisiran included statins with or without ezetimibe. In the new drug scenario, it was assumed that inclisiran (as an add-on to statins used with or without ezetimibe) would displace market share from the treatments included in the reference scenario.31
Key assumptions:
The sponsor assumed a prevalence of 0.46% for HeFH and 8.55% for ASCVD.31
The percentage of patients diagnosed with HeFH was assumed to be constant, at 50%.31
The proportion of patients in the ASCVD subpopulation treated with a lipid-lowering therapy (66%) was assumed to follow data reported from an administrative study in Alberta.31,32
Eligibility for public coverage was assumed to be █████ of patients in the HeFH subpopulation and █████ in the ASCVD subpopulation. These estimates were obtained by multiplying the proportion of individuals with public coverage by the percentage of ASCVD or HeFH cases in predefined age groups. The age-stratified prevalence for HeFH and ASCVD were obtained from a published study and the Canadian Chronic Disease Surveillance System.31,33-35
In the HeFH subpopulation, it was assumed that inclisiran would take market share from evolocumab and alirocumab. Given that these treatments are not currently reimbursed in the ASCVD subpopulation, it was assumed inclisiran would only take market share from statins used with or without ezetimibe.31
Figure 3: Sponsor’s Estimation of the Size of the Eligible Population

Note: This figure has been redacted.
ASCVD = atherosclerotic cardiovascular disease; HeFH = heterozygous familial hypercholesterolemia; LDL = low-density lipoprotein; LLT = lipid-lowering therapy.
Source: sponsor’s pharmacoeconomic submission.31
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate | ||
|---|---|---|---|
Target population | |||
Pan-Canadian Population (excluding Quebec) | 26,445,908 / 26,867,659 / 27,275,276 | ||
Subpopulation | HeFH | ASCVD | |
Prevalence | 0.46% | 8.55% | |
Diagnosed (year 1 / year 2 / year 3) | 50% | NA | |
Treated with a lipid-lowering therapy | NA | 66.0% | |
Uncontrolled LDL | ██ | █████ | |
Public coverage | █████ | █████ | |
Number of patients eligible for drug under review | ██████ █ ██████ | ███████ █ █████ | |
Market Uptake (3 years) | |||
Uptake (reference scenario) | |||
statin ± ezetimibe | ██ | ████ | |
evolocumab | █████ █ █████ █ █ | ██ █ ██ █ ██ | |
alirocumab | █████ █ █████ █ █ | ██ █ ██ █ ██ | |
Uptake (new drug scenario) | |||
inclisiran | ███ █ ███ █ ███ | ██ █ ███ █ ███ | |
statin ± ezetimibe | ███ █ ███ █ ███ | ███ █ ███ █ ███ | |
evolocumab | ███ █ ███ █ ███ | ██ █ ██ █ ██ | |
alirocumab | ██ █ ██ █ ██ | ██ █ ██ █ ██ | |
Cost of treatment (per patient) | |||
Cost of treatment over: 1 Year | |||
inclisiran | $5,678.56 | ||
statin ± ezetimibe | With ezetimibe: $117.31; Without ezetimibe: $51.17 | ||
evolocumab | $7,077.24 | ||
alirocumab | $6,987.49 | ||
ASCVD = atherosclerotic cardiovascular disease; NA = Not available; HeFH = heterozygous familial hypercholesterolemia; LDL = low-density lipoprotein.
Source: Sponsor’s pharmacoeconomic submission.31
Summary of the Sponsor’s BIA Results
In the HeFH subpopulation, the net budget impact of inclisiran was $2,126,379 in Year 1, $474,051 in Year 2, and -$2,160,026 in Year 3. The three-year net budget impact of inclisiran was estimated to be $440,404.
In the ASCVD subpopulation, the net budget impact of inclisiran was $344,838,487 in Year 1, $676,139,138 in Year 2, and $826,213,367 in Year 3. The three-year net budget impact of inclisiran was estimated to be $1,847,190,991.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The comparator prices are uncertain: The results from the BIA are based on publicly available list prices for all comparator treatments (evolocumab and alirocumab). Drug-plan feedback received for this review indicated that there are confidential negotiated prices when prescribed in the HeFH subpopulation. As a result, the actual costs paid by the CDA-AMC-participating drug plans for PCSK9 inhibitors are unknown. If the PCSK9 inhibitor prices are lower than the public list prices, the budget impact of inclisiran in the HeFH subpopulation will increase. Conversely, if the PCSK9 inhibitor prices are higher than the public list prices, then the budget impact of inclisiran in the HeFH subpopulation will decrease. Given that the existing PCSK9 inhibitors are not funded in the ASCVD subpopulation, the high budget impact can be attributed to 2 factors: i) the difference in cost between inclisiran and statins ± ezetimibe; and ii) the predicted number of patients eligible for treatment.
This limitation could not be addressed. CDA-AMC does not have access to the confidential list prices negotiated by public drug plans.
CDA-AMC Reanalyses of the BIA
In the absence of more reliable estimates to inform the key parameters of the BIA, the sponsor’s submitted base case was maintained. A separate scenario analysis was conducted to determine the budget impact in both populations if a 32% price reduction, identified in the CUA, was applied to inclisiran. As with the submitted base case, the price reduction scenario was based on publicly available prices for the comparator treatments. A detailed breakdown of the budget impact results is presented in Table 16.
Table 16: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
HeFH Population | ||||||
Submitted base case | Reference | $59,890,269 | $72,027,370 | $84,164,238 | $96,300,510 | $252,492,118 |
New drug | $59,890,269 | $74,153,749 | $84,638,289 | $94,140,484 | $252,932,522 | |
Budget impact | $0 | $2,126,379 | $474,051 | -$2,160,026 | $440,404 | |
ASCVD Population | ||||||
Submitted base case | Reference | $29,313,942 | $29,795,268 | $30,270,427 | $30,729,797 | $90,795,492 |
New drug | $29,313,942 | $374,633,755 | $706,409,564 | $856,943,163 | $1,937,986,483 | |
Budget impact | $0 | $344,838,487 | $676,139,138 | $826,213,367 | $1,847,190,991 | |
CDA-AMC scenario analysis: 32% price reduction | Reference | $29,313,942 | $29,795,268 | $30,270,427 | $30,729,797 | $90,795,492 |
New drug | $29,313,942 | $264,285,440 | $490,045,040 | $592,554,886 | $1,346,885,366 | |
Budget impact | $0 | $234,490,171 | $459,774,614 | $561,825,089 | $1,256,089,874 | |
BIA = budget impact analysis; ASCVD = atherosclerotic cardiovascular disease; HeFH = heterozygous familial hypercholesterolemia.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.