CADTH Reimbursement Review
Odevixibat (Bylvay)
Sponsor: Medison Pharma Canada Inc.
Therapeutic area: Progressive familial intrahepatic cholestasis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ALT
alanine aminotransferase
ANCOVA
analysis of covariance
AST
aspartate aminotransferase
BMI
body mass index
BRIC
benign recurrent intrahepatic cholestasis
BSEP
bile salt export pump
CI
confidence interval
CLF
Canadian Liver Foundation
CMH
Cochran-Mantel-Haenszel
CPHRG
Canadian Pediatric Hepatology Research Group
DFS
surgical biliary diversion–free survival
EFS
event-free survival
GGT
gamma-glutamyl transferase
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IPTW
inverse probability of treatment weighting
IWRS
interactive web response system
LS
least squares
MID
minimal important difference
NLS
native liver survival
ObsRO
observer-reported outcome
OS
overall survival
OvEC
Odevixibat Versus External Control
PedsQL
Pediatric Quality of Life Inventory
PFIC
progressive familial intrahepatic cholestasis
PFIC1
progressive familial intrahepatic cholestasis type 1
PFIC2
progressive familial intrahepatic cholestasis type 2
PFIC3
progressive familial intrahepatic cholestasis type 3
PRO
patient-reported outcome
PS
propensity score
RCT
randomized controlled trial
SAE
serious adverse event
sBA
serum bile acid
SBD
surgical biliary diversion
SD
standard deviation
SE
standard error
TEAE
treatment-emergent adverse event
UDCA
ursodeoxycholic acid
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on the Application Submitted for Review
Item | Description |
|---|---|
Drug product | Odevixibat (Bylvay) 200 mcg, 400 mcg, 600 mcg, 1,200 mcg capsules |
Sponsor | Medison Pharma Canada Inc. |
Indication | For the treatment of pruritus in patients aged 6 months or older with progressive familial intrahepatic cholestasis |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | October 30, 2023 |
Recommended dose | 40 mcg/kg administered orally once daily in the morning. If an adequate clinical response has not been achieved after 3 months of continuous therapy, the dose may be increased to 120 mcg/kg/day, with a maximum daily dose of 7,200 mcg. |
NOC = Notice of Compliance.
Introduction
Progressive familial intrahepatic cholestasis (PFIC) is a rare and life-shortening heterogeneous group of liver disorders of autosomal recessive inheritance that affects the production and/or composition of bile from the liver.1 PFIC is categorized based on genetic defect, clinical presentation, laboratory findings, and liver histology.1 At least 6 subtypes of PFIC have been described in the literature, although the nomenclature beyond types 1 to 3 is somewhat indeterminate. PFIC type 1 (PFIC1) and type 2 (PFIC2) represent approximately two-thirds of cases, and type 3 (PFIC3) represents a large portion of the remainder.1-5 Patients with PFIC1 and PFIC2 generally present with jaundice and severe pruritus in the first few months of life, with 78% developing jaundice before the age of 12 months.6 PFIC3 can occur during infancy, childhood, and even into young adulthood. Although the genetic mutations underlying the PFIC subtypes differ, the common feature of all subtypes is elevated serum bile acid (sBA) concentrations and severe pruritus. PFIC is a rare disease that is estimated to affect from 1 in every 50,000 to 100,000 children born worldwide.4,5 While global or country-specific prevalence estimates are not available for PFIC, it is believed to be responsible for approximately 10% to 15% of cholestatic liver diseases among children and 10% to 15% of liver transplant indications in children.4,5
PFIC is characterized by the early onset of cholestasis (usually during infancy) with severe pruritus and fat malabsorption that progresses rapidly and leads to liver failure.1 Elevated bile acid concentrations result in ongoing liver inflammation, fibrosis, cirrhosis and, eventually, liver failure. Intractable pruritus is the most troubling symptom of PFIC.
PFIC is a fatal disease. The survival rate in patients with PFIC who have not undergone surgical biliary diversion (SBD) or a liver transplant is 50% at age 10 and almost zero at age 20.6 PFIC may manifest with many symptoms, including jaundice, hepatomegaly, severe pruritus, splenomegaly, diarrhea, discoloured stools, failure to thrive, vitamin E deficiency, vitamin D deficiency, and pancreatitis (PFIC1).7
The clinical experts consulted by CADTH for this review stated that although there are numerous anti-itch medications, including antihistamines and other drugs like rifampicin that indirectly address itch, they may be effective for mild to moderate pruritus but are not effective therapies for severe pruritus. One clinical expert noted that accumulation of bile acids damages the liver; however, it is not clear whether a medication like ursodeoxycholic acid (UDCA) is able to address this key aspect of the pathophysiology of PFIC. Surgery is also a key nonpharmacological approach, although it is not always successful, carries a high risk of morbidity, and is not suitable for the subset of patients who have cirrhosis.
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of odevixibat at the approved doses compared with relevant comparators for the treatment of pruritus in patients aged 6 months or older with PFIC.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Input was received from the Canadian Liver Foundation (CLF). It surveyed patients and caregivers living with PFIC and received 14 responses (4 of these were from Canada).
Families expressed feelings of helplessness, anguish, and frustration, noting that a diagnosis of PFIC has severely impacted the lives of their loved ones and also their own daily activities. Respondents highlighted the significant impact that constant itch has on their daily lives, and how disrupted sleep leaves them and their loved ones chronically fatigued.
Respondents highlighted the importance of improving their quality of life as well as improving itch and sleep, achieving normal growth, maintaining energy, and slowing the progression of their disease. The CLF emphasized the need to ensure equitable access to therapies for PFIC across the country.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
According to the clinical experts consulted by CADTH for this review, there is a major unmet need in PFIC for a drug that can address the underlying pathophysiology of the disease, that can effectively control pruritus (particularly severe pruritus), and potentially slow progression of the disease.
The clinical experts did not identify a specific subtype of PFIC that is more likely to benefit from odevixibat; however, they did highlight the fact that randomized controlled trial (RCT) evidence is available only for the PFIC1 and PFIC2 subtypes. The clinical experts indicated that the severity of pruritus should be the main determinant of when to initiate therapy, with signs such as excoriations and significant lack of sleep as key indicators of severe itch. The clinical experts noted that the key indicator of treatment response is a reduction in itch, and this should be accompanied by improvement in sleep, feeding and, in older children, school performance, sports activities, and mood and/or energy levels. The clinical experts stressed that although sBA level can also be used to assess response, it does not always correlate well with itch and the assay is not widely available. According to the clinical experts, the main reason to discontinue odevixibat would be because the patient is undergoing a liver transplant. An additional consideration would be tolerability or safety issues.
Clinician Group Input
The Canadian Pediatric Hepatology Research Group (CPHRG), which functions under the aegis of the Canadian Association for the Study of the Liver, provided input for this review.
The CPHRG agreed with the clinical experts consulted by CADTH that current pharmacologic treatments have limited efficacy and do not address the underlying disease process, whereas surgical options carry a high risk of morbidity and mortality. They also agreed that a response to odevixibat would be indicated by improvement in pruritus and sleep, and indications for discontinuation would include continued progression of disease (e.g., liver transplant) and drug intolerance.
The CPHRG input did not state whether it had experience with odevixibat.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for odevixibat:
consideration for initiation of therapy
consideration of discontinuation of therapy
consideration for prescribing of therapy
system and economic issues.
The clinical experts consulted by CADTH provided advice on the potential implementation issues raised by the drug programs (Table 4).
Clinical Evidence
Systematic Review
Description of Studies
PEDFIC 1 (N = 62) was a phase III, multicentre (1 site in Canada), double-blind, randomized, placebo-controlled study to demonstrate the efficacy and safety of odevixibat 40 mcg/kg/day and 120 mcg/kg/day in children with PFIC1 or PFIC2.8,9 The study included up to an 8-week screening period, a 24-week treatment period, and a 4-week follow-up period. The primary outcome of PEDFIC 1 was the proportion of patients who experienced at least a 70% reduction in sBA concentration from baseline to the end of treatment or a lowering of sBA to 70 µmol/L or less after 24 weeks of treatment.
This was the primary outcome used for submission to regulatory bodies outside of the US, including Canada, and was therefore considered the primary outcome of interest for the purposes of this report. Secondary outcomes included the following:
proportion of positive pruritus assessments at the patient level over the 24-week treatment period based on the PRUCISION observer-reported outcome (ObsRO) instrument (this was the primary outcome for submission to the FDA)
change in growth from baseline to week 24
change from baseline in sleep parameters (awakenings) measured with the PRUCISION Patient-Reported Outcome (PRO) and ObsRO instruments at each 4-week interval over the 24-week treatment period
proportion of individual assessments meeting the definition of a positive pruritus assessment at the patient level from weeks 0 to 4 and weeks 0 to 12
number of patients undergoing biliary diversion surgery or liver transplant.
The median age of the patients in the PEDFIC 1 study was 3.2 years and ranged from 6 months to 15.9 years. Most patients (47 of 62; 76%) were between 6 months and 5 years of age; 12 (19%) were between 6 and 12 years of age, and 3 (5%) were between 13 and 18 years of age; a limited number of patients (10; 16%) were 8 years of age or older. Median height-for-age and weight-for-age z scores were −1.70 and −0.95, respectively, indicating the patients were below their age-matched peers for growth. Most (45 patients; 73%) had PFIC2 and 17 (27%) had PFIC1. According to the investigator, almost all patients (60; 97%) had a history of significant pruritus present and most (42; 68%) had sBA levels greater than 100 µmol/L (40.85 mcg/mL) within the 6 months before enrolment in the study. At study entry, 50 patients (81%) were on UDCA and 41 (66%) were on rifampicin. Overall, 8 patients (13%) reported prior biliary tract surgeries (all reports of biliary diversion). Median sBA levels were elevated at baseline at 228.0 µmol/L, 188.5 µmol/L, and 254.5 µmol/L in the odevixibat 40 mcg/kg/day, odevixibat 120 mcg/kg/day, and placebo groups, respectively. Median levels of hepatic biochemical parameters were elevated at baseline, including alanine aminotransferase (ALT) (approximately 2 × upper limit of normal [ULN]), aspartate aminotransferase (AST) (less than 2 × ULN), and total bilirubin (1.8 × ULN). Based on the Child-Pugh classification, 41 patients (66%) had mild hepatic impairment and 21 (34%) had moderate hepatic impairment; no patients had severe impairment.
Efficacy Results
Mortality
Mortality was reported as a safety outcome in the PEDFIC 1 study, and there were no deaths in that study.
Need for Surgery
The need for surgery was a secondary outcome in the PEDFIC 1 study, and there were no instances of surgeries for liver transplant or biliary diversion in that study.
Health-Related Quality of Life
Health-related quality of life (HRQoL) was assessed as an exploratory outcome using the Pediatric Quality of Life Inventory (PedsQL) instrument. It is scored on a 0 to 100 scale, with higher scores indicating improved quality of life. After 24 weeks, the least squares (LS) mean difference versus placebo for the odevixibat 40 mcg/kg/day group was ||||| |||| |||||||||| |||||||| ||||| ||||||| |||||| ||| ||| ||| ||| |||||||||| ||||| ||| |||| |||| ||| |||||| ||||||.
Pruritus
The sponsor designed its own instrument for assessing pruritus. The assessment of the proportion of positive pruritus responses at 24 weeks was a secondary outcome of the study. After 24 weeks of treatment with odevixibat, the between-group differences in the LS means for the comparisons of the 40 mcg/kg/day odevixibat group with placebo was 28.23% (95% confidence interval [CI], 9.83 to 46.64), and the 120 mcg/kg/day odevixibat group with placebo was 21.71% (95% CI, 1.87 to 41.54). ||||||||||| |||||| ||||||| |||| |||| |||||||| ||| ||||| |||| |||||| |||| |||||||||| |||||| ||||||| ||| ||| |||||||||| || |||||||||| ||||| || |||||| |||| ||| ||||| ||||||| ||| ||| ||| ||| ||||||||||||||| || |||||| |||| ||| ||||| |||||||| || ||||||| || ||| ||||||||||| |||||| ||||||| |||| |||||||| |||||||||||| || ||| |||||| ||||| |||||| |||| |||||||||| |||||| ||||||| ||| ||| |||||||||| || |||||||||| ||||| || ||||||||| ||| |||||| ||||||| ||| ||| ||| ||| |||||||||| ||||| || |||||| |||| ||| |||||| |||||||.
Serum Bile Acids
The primary outcome of PEDFIC 1 was the proportion of patients experiencing at least a 70% reduction in fasting sBA from baseline to end of treatment or a lowering of sBA to 70 µmol/L or less after 24 weeks. The adjusted difference in proportions between odevixibat 40 mcg/kg/day and placebo was 44.1% (95% CI, 23.6 to 64.6; P = 0.0015) and 21.6% (95% CI, −0.5% to 43.8%; P = 0.0174) between odevixibat 120 mcg/kg/day and placebo. ||||||||||||| ||||||||||| |||| ||||||| ||| ||| |||| ||||||| |||| |||||||| || || |||||| |||| || |||||||| |||||||||| || ||||||||||| ||||||| |||||||||| || |||||||||| ||| ||||||| ||| ||||| |||| ||| |||||| ||||||| ||| ||||||| |||||||||| ||| |||||||||| ||| ||||||| ||| ||||| |||| ||| ||||||| |||||||.
Growth
Improvement in growth (height, weight, body mass index [BMI]) was assessed as a secondary outcome by comparing changes from baseline in z scores relative to a typical pediatric growth chart. For height, the LS mean between-group difference for odevixibat versus placebo after 24 weeks was |||| |||| ||| ||||| ||||| ||| ||| || |||||||||| ||||| ||| |||| |||| ||| |||||| ||||| for the 120 mcg/kg/day group. For weight, the LS mean between-group difference was |||| |||| ||| |||||| ||||| ||| ||| || |||||||||| ||||| ||| |||| |||| ||| |||||| ||||| ||| ||| ||| |||||||||| |||||. For BMI, the LS mean between-group difference was |||| |||| ||| |||||| ||||| ||| ||| || |||||||||| ||||| ||| ||||| |||| ||| |||||| ||||| ||| ||| ||| |||||||||| |||||.
Number of Awakenings
The changes over time in sleep parameters, specifically awakenings, were assessed as a secondary outcome using data derived from the PRUCISION pruritus instruments developed by the sponsor. The LS mean between-group difference in number of awakenings from baseline to weeks 21 to 24 was |||| |||| ||| |||||| |||||| || ||| || |||||||||| ||||| ||| ||||| |||| ||| ||||||| ||||| || ||| ||| |||||||||| |||||.
Total Bilirubin
The change from baseline to week 24 in total bilirubin was an exploratory outcome. The LS mean between-group difference versus placebo in total bilirubin was |||||| |||||| |||||||| |||||| || ||| || |||||||||| ||||| ||| ||||| |||||| |||||||| |||||| || ||| ||| |||||||||| |||||.
Harms Results
Adverse Events
Overall, 35 of the 42 patients (83%) who received odevixibat experienced at least 1 treatment-emergent adverse event (TEAE), as did 17 of the 20 patients (85%) who received placebo; the overall incidence of TEAEs was similar in the odevixibat 40 mcg/kg/day and 120 mcg/kg/day treatment groups (83% and 84%, respectively). The most commonly reported types of events during the study were gastrointestinal disorders and infections. Overall, the most commonly reported TEAEs (≥ 10% overall) among patients who received odevixibat, with corresponding incidence for patients who received placebo, were diarrhea |||| || |||, pyrexia (29% versus 25%), upper respiratory tract infection (19% versus 15%), vomiting (17% versus 0%), ALT increased (14% versus 5%), and blood bilirubin increased (12% versus 10%).
Serious Adverse Events
Treatment-emergent serious adverse events (SAEs) were reported in 3 of the 42 patients (7%) who received odevixibat and in 5 of the 20 patients (25%) who received placebo. No treatment-emergent SAEs were reported in the 40 mcg/kg/day treatment group. The most commonly reported types of treatment-emergent SAEs were infections, reported in ||||||| of the 20 patients in the placebo group and in 1 of the 19 patients (5%) in the 120 mcg/kg/day group. The only event reported in more than 1 patient overall was urinary tract infection, which was reported in 1 patient each in the placebo and 120 mcg/kg/day groups. None of the treatment-emergent SAEs led to the discontinuation of treatment.
Withdrawals Due to Adverse Events
Dose interruptions due to TEAEs were reported at a higher incidence in patients who received odevixibat (9 of 42; 21%) compared with patients who received placebo (1 of 20; 5%). The highest incidence was reported among patients who received the 120 mcg/kg/day dose (6 of 19; 32%); while 3 of the 23 patients (13%) in the 40 mcg/kg/day group had treatment interruptions due to TEAEs. |||| ||| || |||||||| |||| ||||||||| ||||||||||||| ||| || |||||| ||| |||||| |||| ||||||| || |||||||||| || ||||||| ||||||||||| |||| ||||||| ||| ||||||||| ||| |||||||||||| |||||||| || ||| ||||||||| ||||| ||| ||||| |||| ||| ||||| |||| ||||||||||| ||| || ||||||| ||||||||||| |||| ||||||| ||||||||| |||||||||||| || ||| |||| |||||| |||||||||| ||||| ||| |||||||| ||| |||||| || ||||||| || ||| ||||||||| |||||||||| |||||||. All these patients completed the PEDFIC 1 study and rolled over to the extension (PEDFIC 2) to receive odevixibat, except 1 patient who discontinued the study due to the inability to attend clinic visits.
One patient receiving odevixibat 120 mcg/kg/day discontinued the study drug due to a TEAE of diarrhea.
Critical Appraisal
The PEDFIC 1 study was double-blinded, with steps taken to maintain blinding and allocation concealment during the randomization process. Despite randomization, there were imbalances in several baseline characteristics, suggesting that prognostic balance was not achieved; this is likely the result of the small sample size. Given the small size of the trial, a relatively large number of patients discontinued treatment and were rolled into the extension, where all patients were given the higher dose (120 mcg/kg/day) of odevixibat. Although steps were taken to account for these missing data points for outcomes such as pruritus and sBA, a number of key outcomes such as PedsQL had data missing for more than 20% of the population.
With respect to external validity, major issues included the fact that the enrolled population was limited to patients with PFIC1 or PFIC2, while the proposed indication is not restricted to any subtypes. Additionally, the PEDFIC 1 trial assessed 2 different doses of odevixibat, 40 mcg/kg/day and 120 mcg/kg/day, and this differs from the proposed labelling, which recommends that all patients begin at 40 mcg/kg/day and then titrate up to 120 mcg/kg/day if there is a lack of response at 12 weeks. The trial was not of sufficient size or duration to adequately assess key clinical outcomes such as mortality or the need for surgical intervention.
GRADE Summary of Findings and Certainty of the Evidence
CADTH’s selection of outcomes for a Grading of Recommendations Assessment, Development, and Evaluation (GRADE) assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and the public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
Clinical outcomes:
mortality
need for surgery (biliary diversion or liver transplant)
growth (change from baseline to week 24 in z scores for height, weight, and BMI)
Patient-reported outcomes:
PedsQL (change from baseline to week 24 in the PedsQL Parent Report and Family Impact Module)
Pruritus (proportion of positive pruritus assessments at the patient level at week 24, weeks 0 to 4, and weeks 0 to 12)
Sleep parameters (change from baseline to weeks 21 to 24 in number of awakenings)
Lab parameters:
sBA (proportion of patients with at least a 70% reduction in fasting sBA or an sBA of 70 µmol/L or less at week 24 and at week 12)
Liver function (change from baseline to week 24 in total bilirubin)
Harms:
Clinically significant diarrhea
Adjudicated hepatic events
Table 2: Summary of Findings for ODE Versus PLA for Patients With PFIC1 or PFIC2
Outcome measure | Patients (studies), N | Relative effect (95% CI) | Absolute effect (95% CI) | Difference | Certainty | What happens | |
|---|---|---|---|---|---|---|---|
PLA | ODE 40 mcg ODE 120 mcg | ||||||
Mortality | |||||||
Deaths (safety end point) Follow-up: 24 weeks | ODE 40 mcg (N = 23) PLA (N = 20) | NR | 0 | 0 | 0 | Very lowa | Both doses: The evidence is very uncertain about the effects of ODE on survival (mortality) when compared with placebo after 24 weeks of follow-up. |
ODE 120 mcg (N = 19) | — | — | 0 | 0 | Very lowa | ||
Need for surgery | |||||||
Liver transplants or biliary diversion surgery Follow-up: 24 weeks | ODE 40 mcg (N = 23) PLA (N = 20) | NR | 0 | 0 | 0 | Very lowb | Both doses: The evidence is very uncertain about the effects of ODE on the need for surgery (liver transplant or biliary diversion) when compared with placebo after 24 weeks of follow-up. |
ODE 120 mcg (N = 19) | — | — | 0 | 0 | Very lowb | ||
HRQoL | |||||||
PedsQL Family Impact Module, mean (SE) change from baseline (scores are linearly transformed to a 0 to 100 scale, where higher scores = improved HRQoL) Follow-up: 24 weeks | 40 mcg (N = 19) PLA (N = 17) | || | |||| || | |||||| |||||| ||||||| | ||||||||||| |||| ||||||| |||| || ||||| |||||| | Very lowc | Both doses: The evidence is very uncertain about the effects of ODE on parent or family HRQoL (PedsQL Family Impact Module) after 24 weeks of follow-up. |
120 mcg (N = 13) | — | — | ||||||| |||||| |||||| | ||||||| ||||| |||| ||||||| |||| || ||||| ||||| | Very lowc | ||
Pruritus assessments | |||||||
Proportion of positive pruritus assessments at the patient level (scratching score of ≤ 1 or at least a 1-point drop from baseline on the PRUCISION ObsRO instrument), mean (SE)d Follow-up: 24 weeks | ODE 40 mcg (N = 23) PLA (N = 20) | NR | 28.7 per 100 | 40 mcg: 58.3 per 100 (6.2 per 100) | 40 mcg: 28.2 more per 100 (9.8 to 46.6 more per 100) | Moderatee | Both doses: ODE likely results in a reduction in pruritus after 24 weeks of follow-up; the clinical importance of the reduction is unclear. |
ODE 120 mcg (N = 19) | NR | — | 120 mcg: 47.7 per 100 (8.1 per 100) | 120 mcg: 21.7 more per 100 (1.9 to 41.5 more per 100) | — | ||
Proportion of individual assessments meeting the definition of a positive pruritus assessment at the patient level from weeks 0 to 4, as reported on the PRUCISION ObsRO instrument, mean (SE)d Follow-up: 4 weeks | ODE 40 mcg (N = 23) PLA (N = 20) | NR | |||| ||| ||||| | ||||||||| ||| ||||| | |||||| ||||| |||| ||| ||| |||| ||||| || |||| |||| ||| | | 40 mcg: moderatef | 40 mcg: ODE likely results in a reduction in pruritus after 4 weeks of follow-up; the clinical importance of the reduction is unclear. |
ODE 120 mcg (N = 19) | NR | — | ||||||| ||||| ||| ||| | ||||||| ||||| |||| ||| ||| |||| ||||| || |||| |||| ||| | 120 mcg: lowg | 120 mcg: ODE may result in a reduction in pruritus after 4 weeks of follow-up; the clinical importance of the reduction is unclear. | |
Proportion of individual assessments meeting the definition of a positive pruritus assessment at the patient level from weeks 0 to 12, as reported on the PRUCISION ObsRO instrument, mean (SE)d Follow-up: 12 weeks | ODE 40 mcg (N = 23) PLA (N = 20) | NR | || | |||||| ||||| ||| ||| |||| | |||||| ||||| |||| ||| ||| |||| || |||| | 40 mcg: moderatee | Both doses: ODE likely results in a reduction in pruritus after 12 weeks of follow-up; the clinical importance of the reduction is unclear. |
ODE 120 mcg (N = 19) | — | |||| ||| ||| | ||||||| ||||| ||| |||| | ||||||| ||||| |||| ||| ||| |||| |||| | 120 mcg: moderatef | ||
Serum bile acid | |||||||
Proportion of patients experiencing at least a 70% reduction in fasting serum bile acid concentration from baseline to the end of treatment or reaching a level ≤ 70 µmol/L Follow-up: 24 weeks | ODE 40 mcg (N = 23) PLA (N = 20) | NR | 0 | 40 mcg: 43.5 per 100 | 40 mcg: 44.1 more per 100 (23.6 to 64.6 more per 100) | 40 mcg: lowh | Both doses: ODE may result in a reduction in sBA after 24 weeks of follow-up; the clinical importance of the reduction is unclear. |
ODE 120 mcg (N = 19) | NR | — | 120 mcg: 21.1 per 100 | 120 mcg: 21.6 more 100 (0.5 fewer to 43.8 more per 100) | 120 mcg: lowi | ||
Proportion of patients experiencing at least a 70% reduction in fasting serum bile acid concentration from baseline to the end of treatment or reaching a level ≤ 70 µmol/Ld Follow-up: 12 weeks | ODE 40 mcg (N = 23) PLA (N = 20) | NR | ||| ||| ||||||| | |||||| ||||| ||| |||| | |||||| ||||| |||| ||| ||| |||| || ||| | Lowi | Both doses: ODE may result in a reduction in sBA after 12 weeks of follow-up; the clinical importance of the reduction is unclear. |
ODE 120 mcg (N = 19) | NR | — | — | — | — | ||
Sleep parameters | |||||||
Mean (SE) change from baseline in sleep parameters (number of awakenings) measured with the PRUCISION PRO and ObsRO instruments at each 4-week interval over the 24-week treatment period Follow-up: 24 weeks | ODE 40 mcg (N = 19) PLA (N = 14) | NA | |||||| | |||||| ||||| ||||||| || | |||||| ||||| |||| ||||| |||| || ||||| | Very lowc | Both doses: The evidence is very uncertain about the effects of odevixibat on awakenings after 24 weeks of follow-up. |
ODE 120 mcg (N = 16) | — | — | ||||||| |||||| ||||||| | ||||||| ||||| |||| |||||| |||| || |||| | Very lowc | ||
Growth parameters | |||||||
Mean (SE) change from baseline in growth, height z score Follow-up: 24 weeks | ODE 40 mcg (N = 17) PLA (N = 12) | NA | ||||| | ||||||||||| |||||||| | |||||| ||||| |||| ||| |||||||||| || | 40 mcg: lowj | 40 mcg: ODE may result in an improvement in height z score compared to placebo after 24 weeks of follow-up; the clinical importance is uncertain. |
ODE 120 mcg (N = 15) | — | — | |||||||||||| |||||| | ||||||| ||||| |||| ||||| |||| || ||| | 120 mcg: very lowc | 120 mcg: The evidence is very uncertain about the effects of ODE on height after 24 weeks of follow-up. | |
Mean (SE) change from baseline in growth, weight z score Follow-up: 24 weeks | ODE 40 mcg (N = 18) PLA (N = 12) | NA | |||| | |||||| ||||| ||||| | |||||| ||||| |||| ||||| |||| || |||| ||||| || | 40 mcg: lowj | 40 mcg: ODE may result in an improvement in weight z score compared to placebo after 24 weeks of follow-up; the clinical importance is uncertain. |
ODE 120 mcg (N = 15) | — | — | ||||||| |||||| |||||| | ||||||| ||||| |||| ||||| |||| || |||| | 120 mcg: very lowc | 120 mcg: The evidence is very uncertain about the effects of ODE on weight after 24 weeks of follow-up. | |
Laboratory parameters | |||||||
Mean (SE) change from baseline in total bilirubin, µmol/L Follow-up: 24 weeks | ODE 40 mcg (N = 17) PLA (N = 11) | NA | −9.6 | 40 mcg: −23.7 (9.2) | |||||| |||||| |||| |||||| |||| || |||| | Very lowc | Both doses: The evidence is very uncertain about the effects of ODE on total bilirubin after 24 weeks of follow-up. |
ODE 120 mcg (N = 15) | — | — | 120 mcg: −19.3 (13.6) | ||||||| ||||| |||| |||||| |||| || ||||| | Very lowc | ||
Harms | |||||||
Clinically significant diarrhea | ODE 40 mcg (N = 23) PLA (N = 20) | NR | ||||| | ||||| | ||| |||| ||| ||| ||||| |||| || |||| | Lowk | Both doses: ODE may result in little to no difference in the risk of clinically significant diarrhea after 24 weeks of follow-up. |
ODE 120 mcg (N = 19) | — | — | ||||| | ||| |||| ||||| |||| |||| ||| | Lowk | ||
Adjudicated hepatic events | ODE 40 mcg (N = 23) PLA (N = 20) | NR | |||||| | |||||| | ||| |||| ||| ||| ||||| || |||| |||| | Lowl | Both doses: ODE may result in an increased risk of adj hepatic events after 24 weeks follow-up. |
ODE 120 mcg (N = 19) | — | — | |||||| | |||| ||| ||| ||||| |||| || |||| |||| | Lowl | ||
BMI = body mass index; CI = confidence interval; GRADE = Grading of Recommendations Assessment, Development, and Evaluation; HRQoL = health-related quality of life; MID = minimal important difference; NA = not applicable; NR = not reported; ObsRO = observer-reported outcome; ODE = odevixibat; PLA = placebo; PRO = patient-reported outcome; RCT = randomized controlled trial; SE = standard error.
Note: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 2 levels for very serious imprecision, as there were no events and a small sample size; rated down 1 level for serious indirectness, as the follow-up for this outcome was determined to be insufficient in consultation with clinical experts.
bRated down 2 levels for very serious imprecision, as there were no events and a small sample size; rated down 1 level for serious indirectness, as the follow-up for this outcome was determined to be insufficient in consultation with clinical experts.
cRated down 1 level for serious concerns about risk of bias due to missing outcome data. Rated down 2 levels for very serious concerns regarding imprecision; there was no published between-group MID identified, and the clinical experts consulted by CADTH were unable to estimate a threshold for clinically important effects; therefore, the null was used. The 95% CI for both doses overlapped with both benefit and harm.
dThese analyses were not adjusted for multiplicity, are at increased risk of false-positive findings, and therefore should be considered as supportive evidence.
eRated down 1 level for serious concerns about imprecision. No published between-group MID was identified, and the clinical experts consulted by CADTH were unable to estimate a threshold for clinically important effects; therefore, the null was used. Although the point estimate and entire CI excluded the null, the small sample size raises concern for potential overestimation of the true effect and there is evidence of prognostic imbalance. Because the effect appeared plausible, the CADTH review team rated it down only once.
fRated down 1 level for serious concerns about imprecision. No published between-group MID was identified, and the clinical experts consulted by CADTH were unable to estimate a threshold for clinically important effects; therefore, the null was used. Though the point estimate suggested a benefit, the 95% CI also included the potential for little to no difference.
gRated down 2 levels for very serious concerns about imprecision. No published between-group MID was identified, and the clinical experts consulted by CADTH were unable to estimate a threshold for clinically important effects; therefore, the null was used. Though the point estimate suggested a benefit, the 95% CI also included the potential for little to no difference and harm.
hRated down 1 level for serious concerns about imprecision. No published between-group MID was identified, and the clinical experts consulted by CADTH were unable to estimate a threshold for clinically important effects; therefore, the null was used. Though the point estimate and entire CI excluded the null, the small sample size raises concern for potential overestimation of the true effect and there is evidence of prognostic imbalance. Because the effect appeared plausible, the CADTH review team rated it down only once. Rated down 1 level for serious concerns about indirectness; this is a surrogate outcome with an unclear relationship to the clinical outcomes of interest.
iRated down 1 level for serious concerns about imprecision. No published between-group MID was identified, and the clinical experts consulted by CADTH were unable to estimate a threshold for clinically important effects; therefore, the null was used. Though the point estimate suggested a benefit, the 95% CI also included the potential for little to no difference (based on the judgment of the CADTH team). Rated down 1 level for serious concerns about indirectness; this is a surrogate outcome with an unclear relationship to the clinical outcomes of interest.
jRated down 1 level for serious concerns about risk of bias due to missing outcome data. Rated down 1 level for serious concerns regarding imprecision; there was no published between-group MID identified, and the clinical experts consulted by CADTH were unable to estimate a threshold for clinically important effects; therefore, the null was used. The point estimate suggests a benefit but the lower bound of the 95% CI includes the potential for little to no difference.
kRated down 2 levels for very serious imprecision, as there was only 1 event in each group and a very wide CI, which included the potential for both benefit and harm.
Rated down 2 levels for very serious imprecision, as the very wide CI included the potential for both benefit and harm.
lThese analyses were not part of the statistical analysis plan and were requested by CADTH to facilitate the GRADE assessment.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence and the Clinical Study Report for PEDFIC 1.
Long-Term Extension Studies
Description of Study
PEDFIC 2 is an ongoing phase III, multicentre, nonrandomized, open-label extension study to investigate the long-term efficacy and safety of a 120 mcg/kg/day dose of odevixibat in patients with PFIC (Figure 14).10,11 Cohort 1 (n = 56) consists of children with PFIC1 or PFIC2 who participated in the PEDFIC 1 study. Cohort 2 (n = 58) consists of patients with PFIC1 or PFIC2 who have elevated sBA and cholestatic pruritus and either did not meet eligibility criteria for the PEDFIC 1 study or were eligible for enrolment in the PEDFIC 2 study after recruitment for PEDFIC 1 was completed. The primary outcome of the PEDFIC 2 study was change from baseline in sBA after 24 (or 72) weeks of treatment. Secondary outcomes included proportion of positive pruritus assessments at the patient level over the 24-week (or 72-week) treatment period using the PRUCISION ObsRO instrument, change from baseline in sBA at various time points, proportion of individual assessments meeting the definition of a positive pruritus assessment at the patient level using the PRUCISION ObsRO instrument at various time points, proportion of individual morning and evening assessments meeting the definition of a positive pruritus assessment at the patient level using the PRUCISION ObsRO instrument at various time points, and the number of patients undergoing biliary diversion surgery or liver transplant.
Efficacy Results
Serum Bile Acids
Median changes in sBAs levels from baseline in the PEDFIC 2 study to weeks 22 to 24 were 5.8 µmol/L (range, −151.5 to 125.0) in patients who had received 40 mcg/kg/day in the PEDFIC 1 study, and |||| ||||||| ||||| ||||||| || |||||||| ||| ||| |||||||| ||| ||||||||||| ||| |||||||| ||| ||| |||||||| ||||||| || |||||| || |||||| ||||||| ||||||| || |||| ||||| ||||||||| ||| ||||| || ||||||||| |||| |||||||||| ||| |||||||||| ||| |||| |||||||| |||||| ||||||| ||| ||| |||||||| || |||||||| |||| |||||||| |||||| ||||||.
Median changes (range) in sBAs levels from the PEDFIC 2 study baseline to weeks 70 to 72 were ||| ||||||| |||||| ||||||| || |||||||| ||| ||| |||||||| || |||||||||| || |||||| || ||| ||||| |||||||| |||||| ||||||| || |||||||| ||| ||| |||||||| ||| ||||||||||| ||| |||||||| ||| ||| |||||||| ||||||| || |||||| || |||||| ||||||| ||||||| || |||| ||||| ||||||||| ||| ||||| || ||||||||| |||| |||||||||| ||| |||||||||| ||| |||| |||||||| |||||| ||||||| ||| ||| |||||||| || |||||||| ||||| |||||||| |||||| ||||||.
Surgical Intervention
|||||||| |||||||| || |||| ||||| ||||||||| |||||||| |||||||||||| |||||||||||||||| ||| ||||||||| ||||||| ||||||||| ||||||| ||| || |||||||| ||| ||||||||| ||||| ||||||||||||||| ||||||||. There was 1 patient who had their surgery before completing 24 weeks of treatment, |||||||| ||| ||||| ||||||| ||||||| ||||| || ||| || ||||||||||| ||| ||||| ||||||| ||||||| ||||| || ||| ||| |||| || ||| || |||||||| ||| ||||| ||||||| ||||| |||||||||| ||| || ||||| ||||||||| ||||||.
Pruritus
Among patients who had received active treatment in the PEDFIC 1 study and those who were treatment-naive at study entry, the median (range) proportion of positive pruritus assessments was ||||| ||| |||||| ||||| || ||||| || ||||||||| ||| ||||| ||| |||||| ||||| || ||||| || ||| |||||||||| || |||||| || ||| |||||| ||||||| |||||||||| || |||||||| |||||||| ||||||||||| ||| |||||||| ||| ||| |||||||| || |||||||||| || ||||||||| |||||||||||| || ||| |||||||||| || ||||| ||||||||| ||||| ||| |||||| ||||| || |||||| ||||| ||| |||||| ||||| || |||||| ||| |||||| ||||||| |||||||||| || |||||||| |||||||| ||||||||||| ||| |||||||| ||| ||| |||||||| ||| |||||||||| ||| ||||| ||| ||||| ||||| || |||||| ||||| ||| |||||| ||||| || |||||| |||||||||| |||| |||||||| ||||||||| |||||||||| |||| ||||||| || |||||| ||| |||||||||| || |||||| || ||| |||||| ||||||| |||||||||| || |||||||| |||||||| ||||||||||| || ||| ||||||| ||||| ||| ||||| |||||| |||||| |||| ||| ||||||| ||||||||| |||||| ||| ||||| |||||| |||||| |||| ||| ||||||| ||||||||| ||||||| || |||||| || ||| |||||| ||||||| |||||||||| || |||||||| |||||||| ||||||||||| || ||| ||||||| ||||| ||| ||||| |||| ||| ||||||| ||||||||| |||||| ||| ||||| |||||| |||||| |||| ||| ||||||| ||||||||| ||||||.
|||| |||| |||||||||| |||| ||| |||||||| ||| ||||||||| ||||| || || ||| || |||||| ||||||||||.
Harms Results
Overall, ||| ||||| || ||| ||| |||||||| |||||||||| |||||||| |||| ||||| ||||||||||| || ||||||||| |||||| ||| |||||| ||| |||| |||||||| |||||||| ||||| ||||| |||||||| |||| ||||| ||||||||||| ||||| ||||||||| |||||| ||||| |||||| ||| ||||||| ||| ||||| ||||||||| ||||||||| ||||| ||||| |||||||| ||| |||||||| |||| |||| |||||||| || || || ||| || ||||||||| || |||||||| ||| ||||||||| || ||||| |||||||| |||||||| |||||| ||| ||||||| |||||| ||| ||||||||| |||||| || |||||| ||||||||||| || ||||| || ||| ||| |||||||| |||||||||| |||||||| |||| ||||| ||||||||||||||| || || ||||||||| |||||||| ||||| |||||||||||||| |||||||| ||| ||| |||||||||| |||||||| |||||||||| || |||||| |||||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || |||||||| || ||||| |||||||| || |||||| || ||||| ||||||| |||| |||| |||||||| |||||||||| |||| ||||||||| ||||||| |||||||| || ||||||| ||| |||||||| |||||||||| || ||||||||| || ||||||||||||||| || |||||| || ||| ||||||||||| ||||| |||||||| |||| || |||||| ||| ||| ||||| |||| || ||||||||| |||| |||| |||||||| || |||||| |||||||| ||||||||||||||||||| |||||||| |||||||||||| ||||||| |||| ||| || ||||||||| |||||||||||||||| ||||||||||||||| ||||||| ||| |||||||| || ||||||||| || |||||| |||||||| |||||||| ||| |||||||| ||||||| || |||||| || ||||||||| |||||||| || |||||| ||||||||||| || |||||||||| ||||||||||| |||||||| |||||||| |||||| ||| |||||| ||||| ||||||| ||||| |||| |||||||| || || ||||||| |||||||| ||| ||||||||||| ||||||| ||||||| ||||||||| || ||||||| |||||||| ||| ||| |||||||||| |||||||| |||||||||| || |||||| || || ||||||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || |||||||| || ||||||| |||||||| || |||||| || || |||||||| |||||||| ||| ||||||||||||| |||||||||| ||||||||| || ||||||| |||||||| ||| ||| |||||||||| |||||||| |||||||||| || |||||| ||||||||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || |||||||||||||||| |||||||| || |||||| || || |||||||| |||||||| ||| ||||||||||||| |||||| ||||||||| || ||||||| |||||||| ||| ||| |||||||||| |||||||| |||||||||| || |||||| ||||||||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || |||||||||||||| |||||||| || ||||||.
No deaths occurred during the study.
Critical Appraisal
The PEDFIC 2 study was limited by its open-label and noncomparative design; since there is no comparator, it did not show the comparative benefit of odevixibat versus relevant comparators. Furthermore, the small sample size of PEDFIC 2 led to difficulties in drawing any firm conclusion on the efficacy and safety of odevixibat. Due to its open-label and nonblinding nature, the absence of blinding can lead to assessor bias, and the patient or caregiver would most likely be in favour of the intervention (i.e., odevixibat) for efficacy outcomes. Moreover, the subjective outcomes (e.g., pruritus assessments at the patient level and individual assessments meeting the definition of a positive pruritus assessment at the patient level) are at risk of bias, regardless of blinding.
Although there was an amendment to include a starting dose of 40 mcg/kg/day with the possibility to escalate the dose after 12 weeks to 120 mcg/kg/day if there is no improvement in pruritus, the rationale for selecting the optimal starting dose and titration strategy still remained unclear. As of July 31, 2022, the PEDFIC 2 study had not assessed the long-term efficacy and safety of the lower starting dose regimen of 40 mcg/kg/day.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Study
The Odevixibat Versus External Control (OvEC) study was conducted to evaluate the effect of odevixibat on clinical outcomes in children with SBD-naive PFIC1 or PFIC2 participating in the PEDFIC 1 and PEDFIC 2 studies (N = 69) compared with an external control cohort of children from the NAPPED study (the NAPPED cohort) with SBD-naive PFIC1 or PFIC2 (N = 80). The primary objective was to evaluate the effect of odevixibat on death, liver transplant, or SBD in children with PFIC1 or PFIC2. The primary end point was event-free survival (EFS), and the secondary end points included native liver survival (NLS), SBD-free survival (DFS), and overall survival (OS). The NAPPED study involved collecting retrospective data into a large database to investigate the natural history of PFIC.12,13 The OvEC study used inverse probability of treatment weighting (IPTW) methods to reduce the impact of confounding in comparing the clinical outcomes. A cohort of 69 odevixibat-treated patients was compared with 80 patients (controls) from the NAPPED study. The median study duration in the odevixibat cohort was 22.6 months (range, 1.9 to 39.2 months). The follow-up duration in the NAPPED cohort was truncated accordingly.
Efficacy Results
Results comparing efficacy outcomes between the odevixibat cohort and NAPPED cohort are summarized in Table 26.
EFS: In total, 6 patients (9%) in the odevixibat cohort had an EFS event versus 44 patients (55%) in the NAPPED cohort. The weighted hazard ratio (HR) was 0.20 (95% CI, 0.09 to 0.45; P = 0.0016).
NLS: In total, 4 patients (6%) in the odevixibat cohort had an NLS event versus 21 patients (26%) in the NAPPED cohort. The weighted HR was 0.33 (95% CI, 0.11 to 1.03; P = 0.0900).
DFS: In total, 2 patients (3%) in the odevixibat cohort had a DFS event versus 31 patients (39%) in the NAPPED cohort. The weighted HR was 0.13 (95% CI, 0.04 to 0.39; P = 0.0023).
OS: No patients died in the odevixibat cohort whereas 4 patients (5%) died in the NAPPED cohort. The weighted HR was 0 (95% CI, 0 to not estimable; P = 0.0845).
Critical Appraisal
Patients in the PEDFIC 1 and PEDFIC 2 studies were compared with the NAPPED cohort using IPTW methods in an attempt to minimize the impact of confounding on the results. It should be noted that this method cannot control for substantial differences resulting from the different study designs between the 2 cohorts (RCT versus retrospective registry review). Details of the NAPPED cohort were limited; it is not clear how patients were selected into the cohort (i.e., potential for selection bias is unknown), what their characteristics were before weighting, or what treatments they received. Similarly, the data-collection methods for the NAPPED cohort, how missing data were accounted for, the number of losses to follow-up, and outcome definitions have not been reported. The authors appropriately used eligibility criteria for the NAPPED cohort that were considered similar to those used for the PEDFIC studies; however, the characteristics of patients at baseline and the overlap in covariates before weighting were not described. Thereafter, the primary method to compare the 2 cohorts was based on using stabilized weights computed from the propensity score (PS) model. The dosing used in the PEDFIC 1 and PEDFIC 2 studies did not align with the proposed product monograph for all patients, as some started on 120 mcg/kg/day and others escalated to this dose despite responding to the lower dose. The treatments used among patients in the registry were not described; therefore, it is not clear whether these treatments would correspond to those currently used for PFIC in Canada (the date that patients were added to the registry is also unclear). For some outcomes, the follow-up time was likely to be too short and/or the sample size too small to capture relevant events. Numerous methodological limitations within the study limit the generalizability of the findings.
Conclusions
One pivotal, sponsor-funded multinational double-blind RCT was included in this review. The PEDFIC 1 study randomized 62 patients with either PFIC1 or PFIC2, in a 1:1:1 manner, to odevixibat 40 mcg/kg/day, 120 mcg/kg/day, or placebo over a treatment course of 24 weeks. The odevixibat 40 mcg/kg/day dose is the proposed starting dose for odevixibat, with a proposed dose escalation to 120 mcg/kg/day after 12 weeks if the patient’s condition does not respond to treatment; therefore, it is the 40 mcg/kg/day dose that is the focus of this review. It should be noted that there is limited clinical evidence to support dose escalation in the manner described in the proposed product monograph. Compared with placebo, treatment with odevixibat at a dose of 40 mcg/kg/day likely improves pruritus within 4 weeks, and this improvement is likely to be maintained to at least 24 weeks. Odevixibat 40 mcg/kg/day may elicit reductions in sBA at 12 weeks of therapy; however, the clinical significance and the impact of these reductions on mortality risk and risk of surgery are uncertain due to the sample size and limited duration of follow-up. Additionally, odevixibat may improve growth (height and weight z scores), but it is not clear whether the magnitude of these benefits is clinically important. The impact of odevixibat on HRQoL, sleep (number of awakenings), and total bilirubin is very uncertain, largely due to a wide variation in responses and the risk of bias due to missing data. There were no clear indications of any safety or tolerability issues with odevixibat in either the 24-week double-blind phase or the extension phase. It is important to note that only the extension phase included other PFIC subtypes aside from PFIC1 and PFIC2. This is consistent with the proposed indication, which is not restricted to any subtype. Data from the open-label extension phase suggest there are patients who may respond to a dose escalation from 40 mcg/kg/day to 120 mcg/kg/day; however, there are also patients who may not, and it is unclear whether patients’ conditions are responding to the increased dose or longer duration of therapy. Additionally, unlike the proposed dosing in the product monograph, which requires that the condition fail to respond after 12 weeks before undergoing dose escalation, all patients in the extension were escalated, regardless of the response after 24 weeks. There was no indirect comparison available that would compare odevixibat with other drugs used for PFIC, although the drugs used for PFIC are generally used off label. The sponsor did submit an IPTW that compared results from the odevixibat groups in the PEDFIC 1 and PEDFIC 2 studies with registry data in an effort to demonstrate the potential benefits of odevixibat for clinical outcomes such as EFS, NLS, DFS, and OS; however, due to multiple limitations with the sponsor’s analysis, no conclusions can be drawn from it.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of odevixibat administered orally as 40 mcg/kg or 120 mcg/kg once daily in the morning for the treatment of pruritus in patients aged 6 months or older with PFIC.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
PFIC comprises a rare and life-shortening heterogeneous group of liver disorders of autosomal recessive inheritance that affects the production and/or composition of bile from the liver. PFIC is categorized based on genetic defect, clinical presentation, laboratory findings, and liver histology.1 At least 6 subtypes of PFIC have been described in the literature, although the nomenclature beyond subtypes 1 to 3 is somewhat indeterminate. PFIC1 and PFIC2 represent approximately two-thirds of cases, and PFIC3 represents most of the remainder.1-5 Patients with PFIC1 and PFIC2 generally present with jaundice and severe pruritus in the first few months of life, with 78% developing jaundice before the age of 12 months.6 PFIC3 can occur during infancy, childhood, and even into young adulthood. Although the genetic mutations underlying the PFIC subtypes differ, the common feature among all subtypes is elevated sBA concentrations and severe pruritus. PFIC is a rare disease that is estimated to affect between 1 in every 50,000 to 100,000 children born worldwide.4,5 While global or country-specific prevalence estimates are not available for PFIC, it is believed to be responsible for about 10% to 15% of cholestatic liver diseases among children and 10% to 15% of liver transplant indications in children.4,5
PFIC is characterized by the early onset of cholestasis (usually during infancy) with severe pruritus and fat malabsorption that rapidly progresses and leads to liver failure.1 Elevated bile acid concentrations result in ongoing liver inflammation, fibrosis, cirrhosis and, eventually, liver failure. Intractable pruritus is the most troubling symptom of PFIC.
PFIC is a fatal disease. The survival rate in patients with PFIC who have not undergone SBD or a liver transplant is 50% at age 10 and almost zero at age 20.6 PFIC may manifest with many symptoms, including jaundice, hepatomegaly, severe pruritus, splenomegaly, diarrhea, discoloured stools, failure to thrive, vitamin E deficiency, vitamin D deficiency, and pancreatitis (PFIC1).7 In a qualitative study with patients and caregivers on the daily impacts associated with PFIC and other pediatric cholestatic liver diseases, severe pruritus was the most common and debilitating symptom, occurring most frequently at night, with pruritus-related sleep disturbance reported by 67% of patients with PFIC.14 Significant pruritus can lead to severe cutaneous mutilation (often drawing blood), loss of sleep, irritability, poor attention, and impaired school performance.3
Diagnostic Testing Requirements
Most patients with PFIC treated by pediatric hepatologists or gastroenterologists are referred by a pediatrician, family physician, or emergency department physician due to symptoms of a suspected liver problem. PFIC is generally suspected in children with a clinical history of cholestasis of unknown origin.1,5 Liver function tests, tests for sBA levels, and imaging studies help to rule out the other causes of liver disease.1 A high sBA concentration excludes primary bile acid synthesis disorders. Patients with PFIC1 or PFIC2 have normal serum gamma-glutamyl transferase (GGT) activity, while patients with PFIC3 have high serum GGT activity. Patients with PFIC3 can also be distinguished from PFIC1 and PFIC2 in that they rarely present with cholestatic jaundice at the neonatal period, but rather later in infancy, childhood, or young adulthood.
According to 1 of the clinical experts consulted by CADTH on this review, a biopsy in PFIC1 shows bland cholestasis, whereas in PFIC2 it shows severe hepatitis; in PFIC3, features consistent with bile duct obstruction or damage are present. Special staining methods (immunohistochemistry) can show the decreased expression or lack of a bile salt export pump (BSEP), which is diagnostic of PFIC2, or multidrug resistance protein 3 in the bile canalicular membrane, which is diagnostic of PFIC3.
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
According to the clinical experts consulted by CADTH for this review, the main treatment goals are to provide relief from itch, improve sleeping and quality of life for both the patient and caregiver(s), support normal growth and development, prolong adequate liver function, and avoid PFIC complications such as hepatocellular carcinoma.
The clinical experts emphasized the importance of managing pruritus, as severe itch impacts sleep, which then impacts nutrition, growth and development, and the school performance of children and has significant effects on caregivers. The clinical experts noted that current pharmacologic treatments for PFIC are intended to address symptoms and are not curative. The clinical experts stated that although there are numerous anti-itch medications, including antihistamines and other drugs such as rifampicin that indirectly address itch, they may be effective for mild to moderate pruritus but are not effective therapies for severe pruritus. One clinical expert noted that accumulation of bile acids damages the liver; however, it is not clear whether a medication like UDCA is able to address this key aspect of the pathophysiology of PFIC.
Nonpharmacologic measures, including maintaining an adequate diet, are an important part of meeting treatment goals, according to the clinical experts. Surgery is also a key nonpharmacological approach. The clinical experts noted that surgery is not always successful, carries a high morbidity risk, and is not suitable for the subset of patients who have cirrhosis. One expert noted that liver transplant outcomes are better for patients who have normal nutrition and development.
The clinical experts highlighted the limited efficacy of currently available treatments when addressing unmet needs and added that a medication that could modulate disease progression to, for example, fibrosis, cirrhosis, or liver failure, and is able to address cholestatic pruritus would be a welcome addition to the treatment armamentarium.
Drug Under Review
The recommended dosage for odevixibat is 40 mcg/kg administered orally once daily in the morning. If an adequate clinical response has not been achieved after 3 months of continuous therapy, the dose may be increased to 120 mcg/kg/day, with a maximum daily dose of 7,200 mcg. Odevixibat is a reversible and selective inhibitor of the ileal bile acid transporter. This transporter, expressed mainly in the distal ileum, is a key element in the enterohepatic circulation of bile acids and is responsible for the reabsorption back to the liver of 95% of the intestinal bile acids; therefore, blockade of this transporter is thought to reduce the reuptake of bile acids, facilitating their excretion.
Odevixibat was approved by Health Canada for the treatment of pruritus in patients with PFIC aged 6 months or older. The sponsor is requesting reimbursement for odevixibat per the Health Canada indication. It has been approved by the FDA, with an indication for the treatment of pruritus in patients with PFIC who are 3 months of age or older, and by the European Medicines Agency, with an approved indication identical to the proposed indication in Canada.
Key characteristics of odevixibat and other treatments available for PFIC are summarized in Table 3.
Table 3: Key Characteristics of Odevixibat, UDCA, Rifampicin, and Cholestyramine
Characteristic | Odevixibat | UDCA | Rifampicin | Cholestyramine |
|---|---|---|---|---|
Mechanism of action | Inhibits the ileal bile acid transporter | UDCA is a naturally occurring hydrophilic bile acid that displaces hydrophobic bile acids whose accumulation may contribute to the pathophysiology of cholestatic liver diseases | Rifamycin antibiotic; mechanism of action in managing PFIC is not established | Bile acid–binding resin; facilitates excretion of bile acids |
Indicationa | For the treatment of PFIC in patients aged 6 months or older | For the management of cholestatic liver diseases, such as primary biliary cirrhosis | Not officially indicated for hepatobiliary disorders |
|
Route of administration | Oral | Oral | Oral | Oral |
Recommended dose | 40 mcg/kg administered orally once daily in the morning; if an adequate clinical response has not been achieved after 3 months of continuous therapy, the dose may be increased to 120 mcg/kg/day, with a maximum daily dose of 7,200 mcg | 10 mg/kg/day to 30 mg/kg/day | 5 mg/day to 10 mg/day | 1 g/day to 4 g/day |
Serious adverse effects or safety issues |
| No major safety issues |
| Hyperchloremic acidosis |
Other | — | — | High risk of drug interactions (CYP450 inducer) | Risk of drug interactions (binds negatively charged drugs in gut) |
CYP450 = cytochrome P450; DRESS = drug reaction with eosinophilia and systemic symptoms; PFIC = progressive familial intrahepatic cholestasis; UDCA = ursodeoxycholic acid.
aHealth Canada–approved indication.
Sources: Gunaydin et al. (2018)1 and PEDFIC 1 Clinical Study Report (2020).8
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient input received by CADTH has been included in the stakeholder section of this report.
CADTH received 1 patient group submission from the CLF. CLF is an organization dedicated to promoting liver health, increasing public awareness and understanding of liver disease, and providing support to individuals affected by liver disease. CLF advocates for all people in Canada affected by liver disease, from newborns to seniors, including patients and their caregivers.
CLF conducted an online survey of patients and caregivers between June 27 and July 11, 2023. A total of 14 people responded, including 4 patients and caregivers in Canada. The patients and caregivers provided first-hand qualitative input regarding their experience with respect to the patient’s PFIC diagnosis, experience as the caregiver and/or loved one of someone with PFIC, experience with the disease, experience with previous therapies, and experience with the therapy under review.
Most families affected by PFIC expressed feelings of helplessness, anguish, and frustration. Respondents indicated how a PFIC diagnosis has severely impacted the lives of their loved ones and their day-to-day activities while adding physical and emotional stressors and worries.
Some of the respondents explained the impact of PFIC using the following phrases: constant itch, lack of concentration, lack of sleep, tiredness, weakness, bathroom issues, frequent hospitalization, emotional disorder due to fear and stress, and emotionally draining for both the caregiver and child with the disease.
In terms of current therapy options, some of the respondents identified the following unmet needs: available medications did not decrease itching or improve overall feelings of health, comfort, and vitality; while the medications do help, they do not halt the disease’s progress, nor do they particularly ease symptoms.
Regarding the important outcomes, the patient group identified improving HRQoL as a key priority. Additionally, the patients and caregivers indicated that continuous and peaceful sleep, reduction in itching, normal growth and weight gain, good energy levels, and slowing down the progression of the disease are valued outcomes. They also mentioned they hoped to have a treatment that is effective in the short-term and has no side effects in the long term.
Five respondents indicated having experience with odevixibat through a clinical trial. Respondents reported that this medication was the only thing that worked for them and that it was life-changing for both the patient and the whole family. One patient reported not experiencing side effects for 1 year then experienced diarrhea, which was managed with dose reductions.
CLF believes it is important to ensure greater and more equitable access to important treatments for patients with PFIC while expanding therapeutic options for patients and health care professionals. CLF indicated it is crucial that patients across the country have equitable access to all treatments for liver disease and that provincial borders should not be a barrier.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the review of odevixibat, a panel of 4 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with PFIC, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows. This discussion applies specifically to PFIC1 (FIC1 deficiency) and PFIC2 (BSEP deficiency), conditions for which clinical research data are under consideration.
Unmet Needs
According to the clinical experts, the current anti-itch therapies are used off label and address only mild to moderate itch; therefore, effective pharmacological therapies for pruritus are oral medications. One clinical expert noted that the pruritus associated with PFIC is often debilitating and is highly distracting, akin to chronic pain. In children, pruritus has a negative impact on feeding, learning, and sleeping, as well as a negative impact on family functioning.
Additionally, there are no therapies that modify disease progression (i.e., fibrosis, cirrhosis, liver failure) and this is an unmet need for patients with PFIC and related biliary diseases. The clinical experts noted that surgical interventions carry a high morbidity and are not always successful. They are not suitable for the subset of patients with cirrhosis. One clinical expert went on to note that although partial external biliary diversion is an option, it necessitates the creation of a stoma in the front of the abdomen, which means the patient must wear a bag over it, and dehydration can occur. This clinical expert also noted that they consider liver transplant to be a last resort to be reserved for patients whose disease has failed to respond to other treatments. Additionally, the clinical expert noted that clearing bile acids, which tend to damage the liver when retained, may reduce liver damage, and it is unclear whether treatment with UDCA is able to accomplish this. This clinical expert noted that UDCA has other beneficial effects, most notably that it replaces the more toxic hydrophobic bile acids, potentially reducing intrahepatic damage, but it does not reduce the bile acid pool, nor does it relieve itch. Another clinical expert added that UDCA may qualify, according to some experts, as the standard of care irrespective of pruritus, and they also noted that it does not always improve things for patients with PFIC1. One clinical expert summarized the issues as follows:
Not all patients respond to available treatments.
If they do respond, their condition may become refractory to those treatments.
Treatments may have untoward side effects (e.g., sedation due to phenobarbital) and they may be unpalatable, (e.g., cholestyramine, which is notoriously bad tasting).
Surgical interventions can be problematic.
The clinical experts noted that none of the available medical treatments clearly reverse the course of the liver disease, and this would be a key outcome of interest. It is desirable to have medical interventions to address key outcomes (in this case, normal nutrition, neurodevelopment, and sleeping).
Place in Therapy
The clinical experts agreed that since none of the other off-label treatments are effective for severe pruritus, odevixibat would likely become the first treatment for front-line therapy for PFIC in patients with severe pruritus. One clinical expert believed there may be some flexibility on when to use odevixibat, given the spectrum of pruritus severity. The clinical experts noted, for example, that if a patient were doing well on their existing therapy, they would likely not switch them to odevixibat until their pruritus became more severe. This clinical expert stated that for pruritus that is mild or not incapacitating, other treatments would likely be tried first, and also noted there is some limited evidence of possible synergy between therapies, such as rifampicin and odevixibat. This clinical expert noted that if odevixibat proves to be highly effective, then it would likely become the treatment of choice. They also noted that, because the clinical situations in PFIC1 and PFIC2 are so varied, there should not be any artificial restrictions imposed (such as intolerance or contraindications to available treatments, or the character of the underlying disease, or requiring that the patient’s condition fail to respond to current conventional therapies). The clinical expert also noted that it is not clear whether odevixibat will reverse established liver disease, although this is theoretically possible. The clinical experts appeared to agree that odevixibat will shift the treatment paradigm in PFIC.
Patient Population
One clinical expert noted that the patients most in need of odevixibat are those with severe itch related to elevated sBA, and the other clinical expert mentioned that these were the patients who were the focus of research (although the patients in the studies specifically had PFIC1 or PFIC2).
One clinical expert mentioned that identifying patients with severe itch would be straightforward (some of these patients have chronic excoriations, for example); however, there are various “itching scales” that could be used to help. The other clinical expert noted that patients with cholestasis and itch do not necessarily require sBA testing, as long as no other cause of itch is clearly identified, although the clinical experts agreed that a high sBA level does support the diagnosis of PFIC. This clinical expert went on to mention that underdiagnosis of PFIC rarely occurs but has been found in older patients with cryptogenic liver disease but with no pruritus. One clinical expert went on to note specific lab findings that can also aid in diagnosis, such as low or normal serum GGT along with obvious cholestatic liver disease. GGT can be assessed with a simple lab test. PFIC3 can pose some problems for diagnosis because serum GGT is elevated, but these issues can be sorted out through standard diagnostic algorithms. This clinical expert went on to state it is unclear to them whether PFIC subtypes like PFIC1 or PFIC2 can be used to predict a favourable response.
The clinical experts agreed with the sponsor’s estimate of a global prevalence of 1 in 50,000 for PFIC, although they added that with improvements in diagnostics, particularly in the availability of genetic testing and a better understanding of the various genotypes involved, this number may rise. The clinical experts also noted that given the severity of the disease and the high mortality rate, a successful therapy could and should result in an increase in the number of patients with PFIC who live into adulthood.
One clinical expert noted that adult patients with PFIC are relatively rare, more challenging to diagnose, and tend to have less severe disease. These adults with PFIC may or may not have pruritus, and this clinical expert noted that if they had an adult patient with cholestatic pruritus of any cause (mainly comprising patients with primary biliary cholangitis or primary sclerosing cholangitis, which has a prevalence of approximately 1 in 3,000) they would consider starting them on odevixibat if they thought the patient would benefit. This clinical expert also noted that in decades of practice, they had encountered only 1 adult with PFIC, and this patient did not have pruritus.
Assessing the Response to Treatment
The clinical experts noted that improvement in pruritus would be the outcome of most importance for assessing response, with 1 clinical expert adding that improvement in the condition of the skin and general well-being would also be important. The clinical experts agreed that routine monitoring of sBA is of less importance for assessing response, and that pruritus should be readily assessed because it is a prominent symptom and may be measured more objectively. One clinical expert added that in older children, additional variables of potential interest would include school performance, ability to participate in sports activities, and mood and energy levels. One clinical expert added that slowing disease progression and reducing the need for partial external biliary diversion or liver transplant are other relevant outcomes that would also be of importance, while the other clinical expert was unsure whether avoiding surgical interventions (mainly liver transplant) should be a treatment outcome. One clinical expert mentioned that features of a clinically meaningful response would include decreased pruritus to nonproblematic levels (able to sleep, focus on school and play, able to socialize) improved skin condition, and good nutrition, whereas stabilization of symptoms would be clinically apparent but important.
The clinical experts agreed that response could be assessed early, likely earlier than the 12 weeks suggested in the proposed product monograph. The clinical experts reiterated that itch is such a bothersome symptom for many patients that a positive response would be easily detected. The clinical experts were also clear that pruritus is the obvious outcome of interest, and that if a patient had a clear improvement in itch but not a commensurate improvement in sBA, they should still be kept on the drug because it is treating the itch that is of paramount importance.
Discontinuing Treatment
The clinical experts agreed that liver transplant would be an indication for discontinuing odevixibat, and 1 clinical expert also added that adverse events (AEs) may lead to modulation of treatment, depending on their severity. One clinical expert added that it is unclear to them whether odevixibat would serve as a bridge to partial external biliary diversion, as that has not been established by current research, and they would consider odevixibat to be a means for avoiding this intervention. This clinical expert also noted that surgery represents some degree of permanent alteration, and its efficacy drops off over time.
Although elevations in liver enzymes was a discontinuation criterion in the PEDFIC 1 study, the clinical experts did not consider this to necessarily be a criterion for discontinuation in real-world use. The clinical experts believe that these elevations in liver enzymes due to odevixibat are likely transient, and it is also difficult to determine whether any elevations in hepatic enzymes are due to the underlying disease process or to the drug itself.
Otherwise, the clinical experts noted diarrhea and associated abdominal cramping as a potential tolerability issue. An additional concern with the cramping is that it may negatively impact feeding in younger children.
Prescribing Considerations
The clinical experts agreed that specialist intervention would be required for diagnosis, treatment, and monitoring by, specifically, a pediatric hepatologist or gastroenterologist for pediatric patients. One clinical expert added that odevixibat treatment could likely be managed by a general pediatrician in rural areas, with supervision at a distance by a pediatric hepatologist or gastroenterologist. Adult patients would be managed by an adult hepatologist or gastroenterologist.
The clinical experts did not rule out the possibility of using odevixibat as a combination therapy with 1 of the existing therapies that are currently being used off label for PFIC, and they noted that many of the patients in the pivotal trial were on 1 of these therapies; however, the clinical utility of adding 1 or more of these off-label therapies to odevixibat may be difficult to establish without data from large registries, according to the clinical experts. The clinical experts did note that monotherapy would be desirable for patient adherence. One clinical expert also noted that the capsule formulation would not work with infants and, in such cases, an elixir might be needed. Additionally, this clinical expert pointed out that rounding rules will need to be established, for example, whether to round up or down to a convenient dose. This clinical expert also pointed out that response will need to be assessed frequently in infants, as significant changes over relatively short periods of time are common in this age cohort.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the stakeholder section of this report.
CADTH received 1 clinician group submission from the CPHRG, which operates under the aegis of the Canadian Association for the Study of the Liver, a nonprofit organization that seeks to eliminate liver disease through research, education, and advocacy. CPHRG gathered data and information about the drug under review from a review of the published literature about PFIC, from attendance at conferences, and from abstract presentations.
CPHRG stated there are currently no curative medical therapies for PFIC liver disease, and the management strategies described are all standard of care in Canada. CPHRG reported there are no practice guidelines due to the rarity of the disease, and limited published data that meet the standards for a guideline; however, multiple review articles encompass this information.
According to CPHRG, some of the unmet needs of standard-of-care treatments are lack of efficacy or being only transiently effective for patients, so that surgical options such as external biliary diversion must be considered. CPHRG noted that this surgical option leaves the child with a stoma, which is considered unacceptable to most families. CPHRG reported that another unmet need in this field is reducing significant mortality and morbidity from major liver transplant surgery and lifelong immune suppression, which is the only option for patients with PFIC whose SBD fails or who will not accept this treatment approach. CPHRG believed that a clinically meaningful response would be patients and their families reporting an improvement in pruritus and improvement in sleep duration, which can be measured by asking how often the child wakes at night or by documenting improvements in skin excoriations. CPHRG noted that sBA levels can also be used; however, in clinical practice, this is not done routinely due to cost and logistics (i.e., this test is often sent to specialized laboratories and is not readily available in all gastroenterology practice settings).
CPHRG indicated that patients with PFIC and cholestatic pruritus that is persistent on standard-of-care treatments would be eligible for the drug under review. CPHRG reported that patients with adequately controlled pruritus would also be eligible for treatment to improve liver disease outcomes and prevent or delay the need for a liver transplant. The clinician group noted that since the mechanism of action of the drug under review is inhibiting the ileal bile acid transporter to lower sBAs, they thought it reasonable to anticipate that patients with elevated sBAs are most likely to respond to treatment. CPHRG mentioned that the most likely reason to discontinue treatment would be if the liver disease of a patient with PFIC progresses and they undergo a liver transplant. Other factors that CPHRG indicated should be considered when deciding to discontinue treatment would be treatment-associated AEs. CPHRG noted that the drug under review should be prescribed and monitored by a pediatric gastroenterologist or hepatologist in a specialty clinic setting.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
PEDFIC 1 had a baseline requirement for elevated serum bile acid levels. Would this be considered a clinical criterion when considering treatment initiation? | The clinical panel indicated that elevated serum bile acids would not be a critical criterion for initiating treatment with odevixibat. The clinical panel emphasized that cholestatic pruritus is more important to consider. Furthermore, the panel noted there can be challenges in accessing tests for serum bile acids, and serum bile acid levels do not always correlate with pruritus. |
PEDFIC 1 included patients from age 6 months to 18 years. Would there be an upper limit to the age at which to initiate therapy? What other criteria would be considered for initiation of therapy? | The clinical panel agreed there should not be an upper age limit at which to initiate therapy. However, the panel reported that few patients with PFIC1 or PFIC2 with cholestatic pruritus requiring treatment with odevixibat would be older than the age of 18 years at the time of treatment initiation. The clinical panel highlighted the need for patients to have continued access to treatment with odevixibat as they become older (i.e., from adolescent to adult) if they continue to benefit from the treatment. |
In the PEDFIC 1 study, most patients were on other therapies such as UDCA, rifampicin, and cholestyramine (off label for symptomatic relief). Would the clinical experts suggest that the criteria include a trial of other therapies prior to starting odevixibat? | The clinical panel indicated that patients should not be required to try other therapies before starting odevixibat. The clinical experts noted that the therapies currently used off label to treat cholestatic pruritus associated with PFIC have limitations to their efficacy (e.g., they provide only symptomatic relief to patients with mild to moderate itch). In contrast with these therapies, the panel noted that odevixibat directly and mechanistically addresses the pruritus in PFIC. The clinical panel also noted that since odevixibat is the only treatment for severe itch and it can reverse some of the disease pathology within the liver, it would not be appropriate to require patients to try other treatments before initiating odevixibat. |
Considerations for discontinuation of therapy | |
What level of response would be considered clinically meaningful with respect to serum bile acid and pruritus? What other assessments would be relevant to drug coverage? | The clinical panel noted that improvement in pruritus would be the outcome of most importance for assessing response. The panel noted that improvement in the condition of the skin and general well-being would also be important. In older children, additional variables of interest would include school performance, ability to participate in sports activities, and improvements in mood and energy levels. The clinical experts indicated that features of a clinically meaningful response to treatment would include decreasing pruritus to nonproblematic levels (i.e., able to sleep, able to focus on play and school, greater socialization), improved condition of skin, and good nutrition. The clinical experts noted that stabilization of symptoms would also be an acceptable outcome. The clinical experts agreed that routine monitoring of serum bile acids is of less importance for assessing response. |
Considerations for prescribing of therapy | |
What is the optimal starting dose and how would the dose be titrated? | The clinical panel noted that, as per the draft product monograph, the recommended dose of odevixibat is 40 mcg/kg/day. The draft product monograph also states that if an adequate clinical response (improvement in pruritus and reduction of serum bile acid levels) has not been achieved after 3 months of continuous therapy, the dose may be increased to 120 mcg/kg/day, with a maximum daily dose of 7,200 mcg. The clinical experts indicated they would start the patients at the 40 mcg/kg but they would not triple the dose to 120 mcg/kg at 12 weeks. Instead, they would assess response sooner and increase the dose in smaller increments if the patient was not deriving benefit at the 40 mcg/kg/day dose. The clinical experts reported they would first try doubling the dose (e.g., to 80 mcg/kg/day) or use an incremental approach to titrate the dose, noting that large dose increases can be associated with adverse effects (e.g., diarrhea, abdominal pain). |
System and economic issues | |
Price per 30-capsule pack is as follows:
Assuming 95% of eligible patients with PFIC will be treated with odevixibat by year 3, the model estimated that 69 children and 14 adults would receive odevixibat by year 3 in Canada. The estimated net budget impact of odevixibat over the model time horizon of 3 years is $137.9 million. | This is a comment from the drug plans to inform CDEC deliberations. |
Should the potential to require surgical intervention be considered? How comfortable are you with the end point of lowering serum bile acid and its potential correlation to increased native liver survival? | The clinical panel agreed with considering the potential to require surgical intervention as part of the cost analysis. The clinical panel noted that preventing bile duct diversion surgery or liver transplant should be considered. The clinical panel noted that lower serum bile acids do not necessarily correlate with itching, but they may correlate with increased native liver survival. |
Do you agree with the decision to not include partial external biliary diversion in the cost comparison? | This question is addressed in the Pharmacoeconomic Report. |
CDEC = Canadian Drug Expert Committee; PFIC = progressive familial intrahepatic cholestasis; UDCA = ursodeoxycholic acid.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of odevixibat at approved doses in the treatment of PFIC in patients aged 6 months and older. The focus will be placed on comparing odevixibat with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of odevixibat is presented in 4 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes a sponsor-submitted long-term extension study. The third section would normally include indirect evidence from the sponsor; however, none was submitted. The fourth section includes an additional study that was considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
1 pivotal RCT identified in the systematic review
1 long-term extension study
1 additional study addressing gaps in evidence.
Systematic Review
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Study
Characteristics of the included study are summarized in Table 5.
Table 5: Details of the Study Included in the Systematic Review (PEDFIC 1)
Detail | PEDFIC 1 study |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, randomized, double-blind, placebo-controlled study. |
Locations | 45 study centres with patients enrolled at 33 centres: Europe (17 centres), US (8), Turkey (4), Australia (1), Canada (1), Israel (1), Saudi Arabia (1). |
Patient enrolment dates | First patient enrolled: May 16, 2018 Last patient completed: July 28, 2020. |
Randomized (N) | 62 patients: 23 to odevixibat 40 mcg/kg/day, 19 to odevixibat 120 mcg/kg/day, 20 to placebo. |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention |
|
Comparator(s) | Placebo orally once daily for 24 weeks |
Study duration | |
Screening phase | 8 weeks |
Treatment phase | 24 weeks |
Follow-up phase | 4 weeks |
Outcomes | |
Primary end point | Proportion of patients who experienced at least a 70% reduction in concentration of serum bile acids from baseline to the end of treatment or a lowering of serum bile acids to ≤ 70 µmol/L after 24 weeks of treatment. |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publication status | |
Publications | Thompson RJ, Arnell H, Artan R, et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2022;7(9):830-842. |
ALT = alanine aminotransferase; APRI = AST to platelet ratio index; AST = aspartate aminotransferase; BSEP = bile salt export pump; FIB-4 = Fibrosis-4 Index for Liver Fibrosis; GGT = gamma-glutamyl transferase; GI = gastrointestinal; GIC = Global Impression of Change; GIS = Global Impression of Symptoms; iBAT = ileal bile acid transporter; INR = international normalized ratio; MELD = model for end-stage liver disease; p-C4 = plasma 7 alpha-hydroxy-4-cholesten-3-one; PedsQL = Pediatric Quality of Life Inventory; PELD = pediatric end-stage liver disease; PFIC = progressive familial intrahepatic cholestasis; ObsRO = observer-reported outcome; PRO = patient-reported outcome; ULN = upper limit of normal.
Note: Two additional reports were included (Clinical Study Report for PEDFIC 1, sponsor’s submission).
Source: PEDFIC 1 Clinical Study Report (2020).8
PEDFIC 1 (N = 62) was a phase III, multicentre (1 site in Canada), double-blind, randomized, placebo-controlled study that aimed to demonstrate the efficacy and safety of odevixibat 40 mcg/kg/day and 120 mcg/kg/day in children with PFIC1 and PFIC2 (Figure 1).8,9 The study included up to an 8-week screening period, a 24-week treatment period, and a 4-week follow-up period.
Two screening visits were conducted: visit 1 occurred within 35 to 56 days before the first dose of the study drug, and visit 2 was within 7 to 28 days before the first dose. The screening procedures included a review of medical and surgical history and concomitant medications, genetic confirmation for PFIC1 or PFIC2, a physical examination, a skin examination, measurement of vital signs, clinical chemistry tests, and an sBA assessment. At visit 1, all patients and/or their caregivers were given an electronic diary (eDiary) and instructed on its use. Data were to be entered twice daily, once in the morning and again in the evening. The eDiary included patient-reported and observer-reported outcome items from the PRUCISION PRO and ObsRO instruments for the evaluation of itching, scratching, and sleep disturbance throughout the study. The eDiary was used by patients 8 years of age and older (PRO) and by caregivers for patients of all ages (ObsRO). The diaries could be completed by the patient’s caregiver or the caregiver’s designee. Ideally, the same caregiver completed the eDiary for a given patient throughout the study. If a new caregiver began entering ObsRO on the PRUCISION instrument, they were trained in how to use the diary before they began making entries. Caregivers also used the eDiary to report the time at which each dose of the study drug was administered. At the second screening visit (visit 2), additional laboratory samples, including sBA and liver function tests, were collected for eligibility assessments.
After completion of the screening period, eligible patients were randomized on day 0 (visit 3) in a 1:1:1 fashion to receive odevixibat 40 mcg/kg/day, odevixibat 120 mcg/kg/day, or matching placebo. Note that under the proposed dosing in the draft product monograph, all patients would begin on odevixibat 40 mcg/kg/day and would escalate to 120 mcg/kg/day if they were not responding after 12 weeks; therefore, the odevixibat 120 mcg/kg/day group in the PEDFIC 1 study is not relevant to the proposed dosing; however, it was agreed that data for the odevixibat 120 mcg/kg/day dose should be reported so that decision-makers could gauge how well the dose performs with respect to efficacy and harms. Eligibility for randomization was determined using the eDiary pruritus data obtained in the 14 consecutive days before visit 3, a clinical genetic confirmation of the PFIC diagnosis, and the liver biochemistry evaluations, including sBA levels, from the previous screening visits. Randomization was stratified according to PFIC type (type 1 and type 2) and age (6 months to 5 years, 6 to 12 years, and 13 to ≤ 18 years).
The design of the study was discussed with the FDA and European Medicines Agency; based on these discussions, different primary end points were chosen for the US (change in pruritus) and for Europe and rest of the world (rate of sBA response).
Patients who completed the PEDFIC 1 treatment period (week 24) could continue into an optional 72-week open-label extension study (PEDFIC 2) in which all patients received odevixibat (described in more detail in the long term extension section).
Inclusion and Exclusion Criteria
Eligible patients were between the ages of at least 6 months and no more than 18 years and had a genetically confirmed diagnosis of PFIC1 or PFIC2. Based on the level of reduction from baseline for the primary sBA analysis (reduced by at least 70% or to 70 µmol/L or less), the eligibility criteria required patients in the trial to have elevated sBA levels of at least 100 µmol/L at baseline. Patients were to demonstrate significant pruritus based on a caregiver-observed scratching average, as reported in the eDiary, of at least 2 (on a 0 to 4 scale) in the 2 weeks before randomization. Patients with PFIC2 with variations of the ABCB11 gene that predicted a complete absence of the BSEP protein were excluded, as odevixibat may not be effective in these patients. Patients were also excluded if they had a past medical history or presence of other liver disease, clinically significant infection, suspected or confirmed cancers, inflammatory bowel disease, chronic kidney disease, or other primary pruritic skin diseases. Eligible patients could not have undergone SBD within the 6 months before the start of the screening period or a previous liver transplant or planned liver transplant within 6 months of randomization. The original protocol was amended on March 1, 2019, to allow for the inclusion of patients post SBD.
Figure 1: PEDFIC 1 Study Design
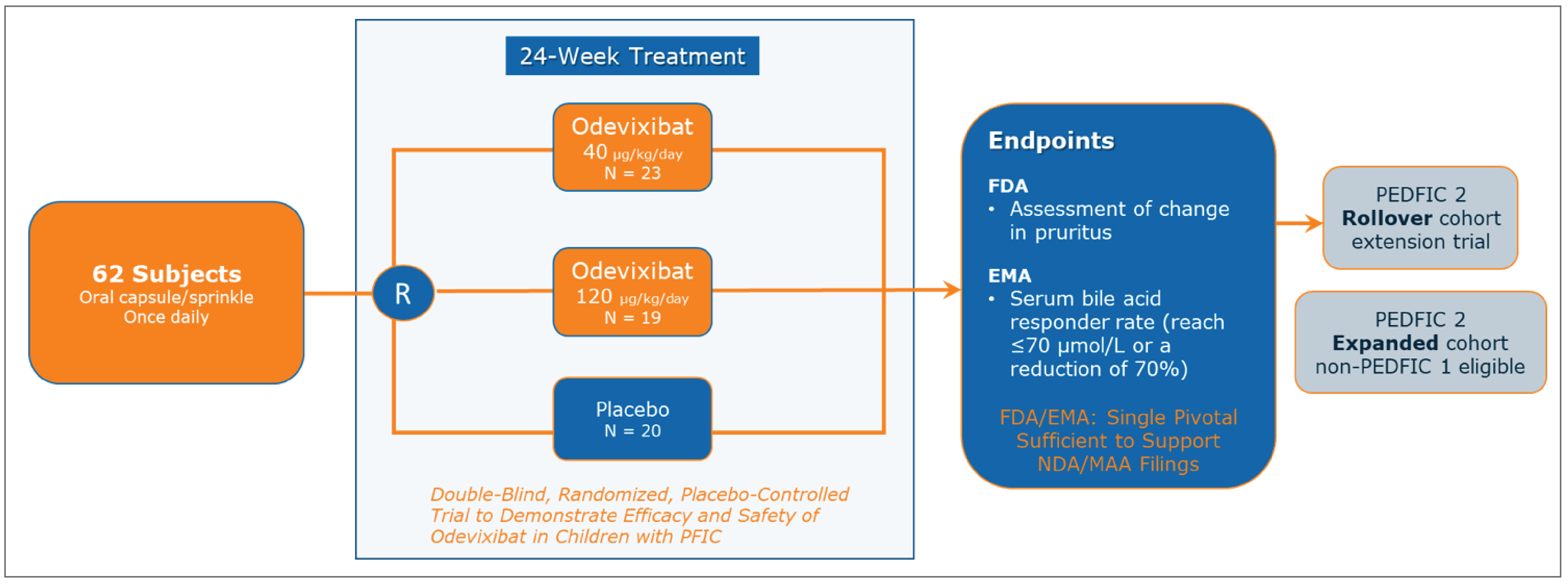
EMA = European Medicines Agency; MAA = Marketing Authorization Application; NDA = New Drug Application; PFIC = progressive familial intrahepatic cholestasis.
Sources: Thompson et al. (2022),9 PEDFIC 1 Clinical Study Report (2020).8
Interventions
Odevixibat (40 mcg/kg/day or 120 mcg/kg/day) or placebo was administered orally once daily by patients or caregivers. The number and type of capsules that were administered varied by the body weight of the patient and the randomized dose. Patients were to take their daily dose in the morning with food.
To facilitate blinding of the treatment assignment, the study drug and matching placebo had the same shape and size. Labels on the study drug containers did not identify the randomized treatment assignment. The traceability of the treatment was ensured by the study drug number, which corresponded to the randomization arm and was assigned by an interactive web response system (IWRS).
Treatment was to be interrupted if a patient developed diarrhea with at least 1 of the following concomitant signs or symptoms: grossly bloody stools, vomiting, electrolyte imbalances and/or dehydration requiring treatment with oral or IV rehydration, fever (38°C or greater), and/or the diarrhea persisted for 7 or more days. If the symptoms were resolved, the patient was allowed to restart the treatment. If the diarrhea reoccurred within 1 week with no alternate etiology, dosing was to be permanently discontinued.
Treatment was also interrupted if any of the following criteria were met:
ALT or AST level 3 times greater than baseline or more, or 800 IU/L or greater, whichever came first, and total bilirubin greater than 2 times the ULN
ALT or AST level greater than 10 times the ULN or 5 times greater than baseline, or an absolute threshold of 800 IU/L or greater, whichever came first, in the presence of normal lactate dehydrogenase and creatine phosphokinase
total bilirubin increased, unrelated to hemolysis (elevated reticulocyte count) or established genetic diseases such as Gilbert syndrome:
doubling of total bilirubin if it was less than 3 mg/dL (equivalent to 51.3 µmol/L) at baseline, or
increase by more than 3 mg/dL (equivalent to 51.3 µmol/L) if total bilirubin was 3 mg/dL or greater (equivalent to 51.3 µmol/L) at baseline
INR increase refractory to vitamin K administration:
INR greater than 1.5 if INR was normal at baseline, or
increase by greater than 0.4 if INR was abnormal at baseline
any increase in total bilirubin and transaminases if accompanied by either a symptom of clinical hepatitis (e.g., vomiting, nausea, pain in right upper quadrant) or immunological reaction (rash or greater than 5% eosinophilia).
If any of the criteria were met, a drug-induced liver injury work-up for alternative etiologies was initiated, including a liver profile (AST, ALT, total bilirubin, direct bilirubin), and prothrombin time or INR was repeated within 48 to 72 hours and the patient was monitored using close observation. If underlying cholestatic liver disease variability or another alternative etiology was identified and liver tests returned to baseline, a rechallenge could be considered after consultation with the medical monitor. If ALT or total bilirubin elevations were observed after a rechallenge, then a repeat rechallenge was discouraged. In the case of a possible or probable drug-induced liver injury and/or if a decompensation event had occurred (e.g., variceal hemorrhage, ascites, hepatic encephalopathy), dosing was to be permanently discontinued.
From the first day of screening to the last day of the treatment period, medications with effects on the concentration of bile acids in the gastrointestinal tract (e.g., cholestyramine, colesevelam, colestipol), drugs with known effects on gastrointestinal motility (e.g., sucralfate, loperamide, codeine, erythromycin), and other investigational products used to treat PFIC (e.g., 4-phenylbutyrate) were not allowed.
Other drugs or natural products with possible effects on gastrointestinal motility (e.g., selective serotonin reuptake–inhibiting drugs, tetracyclic antidepressants, fibre supplementation, yogourt variants) were allowed provided there was stable usage of the product at least 4 weeks before enrolment until treatment discontinuation.
Treatment with UDCA, rifampicin, and/or antihistamines was also allowed provided the patient was on a stable dosage at least 4 weeks before enrolment and no dosage changes were planned during the entire study period. Topical treatment was allowed without restriction.
Outcomes
Mortality
Deaths were reported under harms.
Need for Surgery
The number of patients undergoing biliary diversion surgery or liver transplant was a secondary outcome of the study.
Pediatric Quality of Life Inventory
The PedsQL is designed to examine problems within 4 functional domains: physical, emotional, social, and school.15 Different versions of the PedsQL were used, depending on the age of the patient: child and parent report core modules for 5-to-7-year-olds, 8-to-12-year-olds, and 13-to-18-year-olds, and a parent report core module for toddlers (2 to 4 years old). The caregiver was also asked to complete the PedsQL Family Impact Module (domains: physical, emotional, social, cognitive, communication, worry, daily activities, family relationships) designed to measure the impact of pediatric chronic health conditions on parents and the family.
The PedsQL questionnaire is scored based on the questionnaire taken (i.e., the report for toddlers is scored differently than the report for young children). The scoring scale for the PedsQL is based on the publication by Mapi Research Trust16 and is summarized in Table 4. The raw scores are then reverse scored on a scale from 0 to 100, where higher scores indicate improved HRQoL.
The minimal important difference (MID) for the PedsQL total score for the Child Self-Report and Parent-Proxy Report has been reported to be 4.4 and 4.5 for within-group differences, respectively.17
Pruritus
Albireo Pharma conducted a literature review with the objective of identifying any instruments that are currently used to measure pruritus in adolescents and adults but did not identify any publicly available instruments that adequately assess the symptoms and impact of the disease on the pediatric patient with PFIC and/or provide the caregiver perspective. Therefore, Albireo developed the novel (PRUCISION) PRO and ObsRO instruments for pediatric patients with cholestatic liver disease to assess itching, scratching, and sleep disturbance.18,19 The quantitative measurement characteristics of these instruments, including the assessment of the performance and psychometric properties (reliability, validity, and sensitivity to change) of individual items, were established through an analysis of the final data from the PEDFIC 1 study, which was conducted by a group independent of the sponsor.19 The small sample size (n = 10) precluded the psychometric validation of the PRO instrument.19 The ObsRO (n = 62) was found to have acceptable reliability (i.e., moderate to strong interitem correlations and moderate to good test–retest reliability, depending on the item).19 Construct validity was demonstrated by moderate to strong correlations with some of the scales within other tools (i.e., GIS and PedsQL); additionally, known-groups analyses versus the GIS severity categories showed ObsRO scores changing in the expected direction.19 Similarly, analyses of sensitivity to change showed PRO and ObsRO scores changing in the expected direction for patients (“improved” versus “not improved”) and for most scales on related tools (GIS and PedsQL).19 Using distribution and anchor-based methods, the minimally clinically important within-person change was determined to be a reduction of 1 point.19 Further analyses in other populations are needed to confirm the reliability and validity of the tool.
The final ObsRO and PRO instruments focused on the key symptoms of pruritus, sleep disturbance and associated tiredness, and used pictorial response scales from 0 to 4, where each response was distinguished by a unique facial expression, verbal anchor, number, and colour code.
The PRO consisted of 7 questions that included 2 response formats:
rating scales for morning diary questions 1, 2, 3, and 5, and evening diary questions 1 and 2 (e.g., 0 = no itching, 1 = a little itching, 2 = medium itching, 3 = a lot of itching, and 4 = the worst itching)
binary responses for morning diary question 4 (i.e., no, yes).
For the rating scales, higher scores indicate a greater amount of itching, sleep disturbance, or tiredness.
The ObsRO consisted of 9 questions that included 3 response formats:
rating scales for morning diary question 1 and evening diary questions 1 and 2 (e.g., 0 = no scratching, 1 = a little scratching, 2 = medium scratching, 3 = a lot of scratching, and 4 = worst possible scratching)
binary responses for morning diary questions 2, 3, 4, 5 and 7 (i.e., no, yes)
numeric responses of 0 to 99 for morning diary question 6.
For the rating scales, higher scores indicate a greater amount of scratching, sleep disturbance, or tiredness.
Completion of the PRO items was required only for patients 8 to 18 years of age. For patients 8 to 12 years of age, the caregivers were to read the PRO items along with the child and record the child’s response. A guide was to be provided to the caregivers that had standardized explanations of the PRO items in case the patient was confused or required clarification. The ObsRO items were completed by the caregivers of all patients. Itching (PRO), observed scratching (ObsRO), and sleep disturbance (PRO and ObsRO) were recorded twice daily in the eDiary. Patients and/or caregivers were to complete the eDiary every day in the morning and in the evening. The morning diary was to be completed shortly after the patient woke up and was used to record nighttime itching and scratching severity, aspects of sleep disturbance, and tiredness upon waking (morning scores). The evening (bedtime) diary was to be completed just before the patient went to bed to record the severity of the patient’s itching, scratching, and tiredness during the day (evening scores). Both morning and bedtime diaries included PRUCISION ObsRO and PRO items.
For the US, the primary efficacy end point was the proportion of positive pruritus assessments at the patient level over the 24-week treatment period. This was a secondary end point for Europe and the rest of the world. A positive pruritus assessment was defined as a scratching score of 1 or less, or at least a 1-point drop from baseline on the PRUCISION ObsRO instrument. Both morning and evening pruritus assessments were included in the analysis of this end point. All morning scores from the 14 days before or on the first dose day of the study medication were averaged to provide the morning baseline score. All evening scores from the 14 days before the first dose of the study medication were averaged to provide an evening baseline. The baseline score was rounded to an integer to evaluate the positive pruritus assessments for the primary analysis. For the primary end point analysis, if a patient’s baseline average score was 1 or less, then only the criterion of a 1-point drop from baseline on the PRUCISION ObsRO instrument was used to determine whether or not a pruritus assessment was positive.
Serum Bile Acid
The primary efficacy end point was the proportion of patients experiencing at least a 70% reduction in the concentration of sBAs from baseline to the end of treatment, or a lowering of the sBA level to 70 µmol/L or less after 24 weeks of treatment. Blood samples for the analysis of sBAs were drawn at all visits. Patients were to fast (only water intake was permissible) for at least 4 hours before the collection of samples. Exceptions could be made for infants younger than 12 months of age if they were unable to fast for the full 4 hours. For any visit at which the result of a bile acids sample was unreportable, an additional unscheduled visit for a repeat sample was requested. All sBA results during the treatment period and at follow-up were blinded. A central laboratory performed the quantitative assessment of the sBA levels, utilizing a validated commercial enzyme cycling assay.
Sleep Parameters
This outcome was assessed using the data gathered from the PRUCISION ObsRO and PRO instruments.
Growth
Growth was measured as height and weight using a certified weight scale. BMI was calculated as weight (kg) divided by height (m2). Change in growth parameters was assessed using linear growth deficit (weight, height, and BMI for age) compared with a standard growth curve (z score, standard deviation [SD] from the 50th percentile). For children aged 2 years and older, the software and/or methods used were from the Centers for Disease Control and Prevention; for children younger than 2 years of age, the WHO website was used. It is not clear why different sources were used for these 2 age groups.
Total Bilirubin
Total bilirubin was assessed at weeks 4, 12, and 24. No further details were provided by the sponsor.
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6 followed by descriptions of the outcome measures. The summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review by the clinical experts consulted by CADTH and in the stakeholder input from the patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered to be most relevant to inform the deliberations of CADTH’s expert committee and finalized this list of end points in consultation with members of that committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | PEDFIC 1 trial |
|---|---|---|
Mortality | Week 24 | Included under harms |
Surgical intervention | ||
Number of patients undergoing biliary diversion surgery or liver transplant | Week 24 | Secondary |
Health-related quality of life | ||
Change from baseline in PedsQL Family Impact Module | Week 24 | Exploratory |
Change from baseline in PedsQL Parent Report | Week 24 | Exploratory |
Pruritus assessments | ||
Proportion of positive pruritus assessments at the patient level (scratching score of ≤ 1 or at least a 1-point drop from baseline on the PRUCISION ObsRO instrument) | Week 24 | Secondary |
Proportion of individual assessments meeting the definition of a positive pruritus assessment at the patient level from weeks 0 to 4, 0 to 8, 0 to 12, 0 to 18, and 0 to 24, respectively, as reported on the PRUCISION ObsRO instrument | Weeks 0 to 4 weeks 0 to 12 | Secondary |
Serum bile acid | ||
Proportion of patients experiencing at least a 70% reduction in fasting serum bile acid concentration from baseline to the end of treatment or reaching a level ≤ 70 µmol/L | Week 12 week 24 | Primary |
Sleep parameters | ||
Change in sleep parameters from baseline measured with the PRUCISION PRO and ObsRO instruments at each 4-week interval over the 24-week treatment period | Week 24 | Secondary |
Growth parameters | ||
Change from baseline in growth (BMI, height, weight) | Week 24 | Secondary |
Laboratory parameters | ||
Change from baseline in total bilirubin | Week 24 | Exploratory |
BMI = body mass index; ObsRO = observer-reported outcome; PedsQL = Pediatric Quality of Life Inventory; PRO = patient-reported outcome; sBA = serum bile acid.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.
Of the various outcomes used to assess sBA, in consultation with the clinical experts consulted by CADTH, it was decided that assessments at weeks 12 and 24 would be informative; the week 12 data were included to help determine the extent of response at a time point that is consistent with the proposed dosing instructions (i.e., to consider a dose increase if there is no response after 12 weeks). The proportions of positive pruritus assessments at 4, 12, and 24 weeks were also included. The 12-week time point is consistent with the proposed label, and the clinical experts believed that patients would be more appropriately assessed for pruritus response at this time point rather than simply using sBA level. It was also agreed that data for week 4 would be included, as the clinical experts believed it was important for decision-makers to know how early a response occurs.
The HRQoL outcomes reported by the sponsor in its summary of evidence were assessed using GRADE (for both the PedsQL Parent Report and Family Impact Module), as it was clear in the patient and clinician input that PFIC has a significant impact on HRQoL, both on patients and their families. The global assessment of symptoms was not assessed using GRADE because it was believed to be more important to focus on HRQoL and pruritus, which is by far the most important symptom of PFIC. Of the numerous sleep parameters reported by the sponsor in its summary of clinical evidence and in consultation with the clinical experts, it was decided that awakenings would be the most relevant and objective measure; thus, it was included in the review. The clinical experts also agreed that all growth parameters were relevant (BMI, height, and weight); therefore, all 3 were assessed using GRADE. After discussion with the clinical experts, it was decided that total bilirubin was the most relevant biochemical assessment of liver function for this disorder, as it relates directly to the pathophysiology of PFIC; therefore, outcomes related to liver enzymes were not included. The clinical experts were also skeptical of the value and/or generalizability of other biochemical assays to assess liver function and/or damage, such as the AST to platelet ratio index, Fibrosis-4 Index for Liver Fibrosis, and FibroScan; therefore, these were not included.
After consultation with the review team, the notable harms, hepatic and gastrointestinal AEs, were assessed using GRADE. The clinical experts believed that gastrointestinal AEs were likely to be the primary tolerability issue with odevixibat.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
HRQoL | |||
PedsQL Parent Report | Parent-reported outcome with 4 domains:
Each domain is scored on a 4-point scale:
Scores obtained are then reverse scored such that a score of 0 = 100, 1 = 75, 2 = 50, 3 = 25, and 4 = 0, with higher scores indicating improvement Different versions are used, depending on the age of the child | Not reported | The MID is 4.5 points for a within-group change;17 no MID has been identified for a between-group difference |
PedsQL Family Impact Module | Completed by the caregiver, with 8 domains:
4-point scale:
The scores obtained are then reverse scored such that a score of 0 = 100, 1 = 75, 2 = 50, 3 = 25, and 4 = 0, with higher scores indicating improvement | Not reported | The MID is 4.4 points for a within-group change;17 no MID for a between-group difference has been identified |
Pruritus assessments | |||
PRUCISION ObsRO and PRO instruments Sleep parameters were assessed using this instrument | The final ObsRO and PRO instruments focused on the symptoms of pruritus and sleep disturbance and associated tiredness, and used 0 to 4 pictorial response scales, where each response was distinguished by a unique facial expression, verbal anchor, number, and colour code. The PRO consisted of 7 questions that included 2 response formats: rating scales for 4 morning diary questions (questions 1, 2, 3, and 5) and 2 evening diary questions (questions 1 and 2), e.g., 0 = no itching, 1 = a little itching, 2 = medium itching, 3 = a lot of itching, 4 = the worst itching, and binary responses (i.e., no, yes) for morning diary question 4. The ObsRO consisted of 9 questions that included 3 response formats: rating scales for morning diary question 1 and evening diary questions 1 and 2 (e.g., 0 = no scratching, 1 = a little scratching, 2 = medium scratching, 3 = a lot of scratching, 4 = worst possible scratching), binary responses (i.e., no, yes) for 5 of the morning diary questions (questions 2, 3, 4, 5, and 7), and numeric response for morning diary question 6 (i.e., 0 to 99). For the rating scales, higher scores indicate a greater amount of scratching, sleep disturbance, and tiredness. | Anchor-based, distribution-based, and receiver operating characteristic approaches were used. The primary estimate for the meaningful within-patient improvement was based on an anchor-based approach using the GIC and GIS as anchors for the ObsRO and PRO instruments, with CaGIS and CaGIC as the primary anchors for the ObsRO pruritus measures. Thresholds were established for change from baseline to weeks 12 and 24 using monthly and biweekly scratching scores, with the week 24 monthly scores considered primary. Analyses were conducted for daily scores (morning and evening diaries), as well as nighttime (evening diary) and daytime (morning diary) scores. Strong correlations (> 0.50) were observed between the ObsRO pruritus measure and the GIS and GIC anchors. There was limited variability observed (< 1.0) in the baseline pruritus values, with the distribution-based analysis and the lower bound of the 95% CI for the ObsRO pruritus measure in stable anchor groups (i.e., GIS change from baseline of zero, and GIC answer of “no change”) was less than 1.0. | None identified |
BMI = body mass index; CaGIC = Caregiver Global Impression of Change; CaGIS = Caregiver Global Impression of Symptoms; CI = confidence interval; GIC = Global Impression of Change; GIS = Global Impression of Symptoms; HRQoL = health-related quality of life; MID = minimal important difference; ObsRO = observer-reported outcome; PedsQL = Pediatric Quality of Life Inventory; PRO = patient-reported outcome; sBA = serum bile acid; SD = standard deviation.
Statistical Analysis
For the primary end point used in Canada and the rest of the world outside of the US (proportion of patients experiencing at least a 70% reduction in fasting sBA concentration from baseline to the end of treatment or a lowering of sBA to 70 µmol/L or less), statistical analyses were performed using the Cochran-Mantel-Haenszel (CMH) test stratified by PFIC type and age category to compare the proportion of the responders at the end of treatment in the active and placebo groups. The stratification factors used in the analysis were based on actual strata and not as recorded in the IWRS. The proportion, together with the corresponding Clopper-Pearson exact 95% CI, odds ratio and corresponding 95% CI, and P value from the CMH test, were presented. The proportion difference with corresponding exact unconditional 95% CI without adjusting for stratification factors was also presented. The Miettinen-Nurminen (score) CI adjusting for the stratification factors was reported for common risk difference (i.e., the proportion difference) and the exact CI was reported for the common odds ratio by using an algorithm based on Vollset, Hirji, and Elashoff.20 The concentration of sBAs at baseline was calculated as the average of the last 2 values before the first dose. If only 1 nonmissing value was available, it was used as the baseline. If a patient’s baseline value was 70 µmol/L or less, then only the criterion of at least a 70% reduction in fasting sBA concentration was used to determine whether or not the patient was a responder for the analysis. The end value was calculated as the average of the values at weeks 22 and 24 after the start of treatment. If 1 value was missing, then the nonmissing value was used as the end value. If both values were missing, then the end value was considered missing. Patients who dropped out or who completed treatment but had a missing average at the end of treatment, and those in need of surgical rescue (i.e., biliary diversion and/or liver transplant) were classified as having a nonresponsive condition. Sensitivity analyses were performed that excluded patients with a baseline sBA value of 70 µmol/L or less; used a logistic regression model that included treatment arm, baseline value, and stratification factors; and included a tipping point analysis.
For the primary end point for the US (proportion of positive pruritus assessments at the patient level at week 24, which was the secondary end point in Europe and the rest of the world), the data were analyzed using an analysis of covariance (ANCOVA) model that included treatment arm, rounded morning and evening baseline pruritus scores, and stratification factors (i.e., PFIC type and age category). At each assessment, the morning score was compared with the rounded baseline morning average, and the evening score was compared with the rounded baseline evening average. LS mean and standard error (SE), LS mean difference and SE, 95% CIs, and P values (where applicable) between treatments versus placebo were summarized. If there were concerns on model assumptions, normality was checked based on a Shapiro-Wilk test, and the homogeneity of variances was checked based on a Levene test. For normality testing, a Shapiro-Wilk test was performed for each arm. The P value from each arm was combined to get an overall P value based on a Fisher combined probability test. All intermittently missing assessments were classified as negative pruritus assessments.
Change in sBAs, ALT, and growth were analyzed using a mixed-model for repeated measures (MMRM), including terms for baseline, PFIC type, age category, treatment, visit, treatment-by-baseline interaction, and treatment-by-visit interaction. For sBAs, the MMRM analysis may be performed based on log-transformed values, if deemed appropriate.
The proportion of responders at weeks 12 and 24 based on the ObsRO instrument was analyzed using the same model specified for the primary analysis of sBAs. The proportion of individual assessments meeting the definition of a positive pruritus assessment at the patient level from weeks 0 to 4, 0 to 8, 0 to 12, 0 to 18, and 0 to 24, and the proportion of positive pruritus assessments at each 4-week interval over the 24-week treatment period for morning or evening, were analyzed using the same model specified for the primary analysis for the proportion of positive pruritus assessments at the patient level at week 24. This analysis was performed separately for the PRO and ObsRO instruments. The proportion of patients achieving a positive pruritus assessment more than 50% of the time was analyzed using the same model specified for the primary analysis for the proportion of positive pruritus assessments at the patient level at week 24. This analysis was also performed separately for morning-only and evening-only pruritus assessments.
The number and percent of patients undergoing SBD and/or liver transplant were summarized using descriptive statistics. Kaplan-Meier curves were used when appropriate for the time-to-event data. Median event-free times and associated 95% CIs were calculated for each treatment group using Brookmeyer and Crowley methodology and a log–log transformation for constructing CIs.
Exploratory parameters, including total bilirubin, additional PRUCISION PRO and ObsRO sleep parameters, and PedsQL scores were analyzed descriptively. For continuous data, the change from baseline was analyzed in addition to the presentation of actual visit values. For categorical data, shift tables or frequency and percentages of patients are presented, as appropriate. A line graph of PRUCISION PRO and ObsRO itching and scratching daily severity scores over time for each patient was produced. Mortality was reported under Harms, and there were no analyses planned for this data.
Comparisons of the change from baseline at week 24 in the PedsQL total score (calculated as the average score of all answered items) between the treatment groups were conducted using an ANCOVA. The model included terms for baseline, PFIC type, age category, and treatment. The analysis was conducted based on the total scores reported by child (≥ 5 years of age) and by parent (including only parents of patients ≥ 5 years of age) separately. If the data reported by children aged 5 years or older was based on a sample size that was less than 10, then the ANCOVA was not conducted. The PedsQL total score for the Family Impact Module was analyzed similarly. The total score and the domain scores were summarized descriptively.
Sample Size and Power Calculation
The study planned to enrol 60 to 70 patients to obtain at least 20 evaluable patients in each arm. For each primary end point, simulations with 5,000 iterations using 20 patients per arm were conducted to estimate the power after multiplicity adjustment, resulting in an SE of less than 0.7% for each estimated power.
Based on the data from the phase II study (A4250-003), both low- and high-dose groups were assumed to have the same positive treatment effects in both the sBA and pruritus end points in the simulation. For sBAs, binomial distributions were used to simulate the proportion of responders to estimate the power. The simulated proportions were analyzed using the CMH test to generate 1-sided P values for the following comparisons: both odevixibat arms pooled versus placebo, low dose versus placebo, and high dose versus placebo. Assuming a response rate of 60% in the odevixibat arms and 10% in the placebo arm, the power to claim significance for a particular odevixibat arm after multiplicity adjustment was approximately 94%. The probability to claim significance for at least 1 arm and for both arms was approximately 99% and 91%, respectively. If the response rates were 50% in the odevixibat arms and 10% in the placebo arm, with 20 patients per arm, the probability to claim significance for a particular odevixibat arm after multiplicity adjustment was approximately 82%. The probability to claim significance for at least 1 arm and for both arms was approximately 91% and 73%, respectively.
For the proportion of positive pruritus assessments at the patient level in pruritus scores, beta-binomial distributions were used for power simulations. The effect size was 1.0526 from the original sample size calculation using change from baseline as the end point. The same effect size was assumed for the current end point for the low and high dose versus the control. Differences of 15%, 20%, 25%, and 30% in the proportion of positive pruritus assessments were considered in the power simulation. Within each difference, proportions of positive assessments in the placebo arm ranging from 15% to 35% were considered. Subsequently, the proportion of positive assessments in an active arm, the SD, and the corresponding beta-binomial parameters were calculated to satisfy the assumed effect size. These parameters were used to simulate correlated binary results for each patient. The simulated proportions were analyzed using an ANCOVA to generate 1-sided P values for the following comparisons: both odevixibat arms pooled versus placebo, low dose versus placebo, and high dose versus placebo. The simulation in each scenario was repeated for 5,000 iterations using the current sample size of 20 patients per arm. The simulated power to claim significance for a particular arm after multiplicity adjustment was quite consistent under different scenarios and was approximately 89%. The probability to claim significance for at least 1 arm and for both arms was approximately 95% and 83%, respectively.
Multiplicity Control
For the primary end point, a pooled analysis for the closed testing procedure was applied to control the 1-sided overall type I error rate for the 2 treatment comparisons versus placebo at the 0.025 level. In the closed testing procedure, the first comparison was the pooled result from the low- and high-dose odevixibat groups compared with placebo. If the 1-sided P value was 0.025 or less, the 1-sided P values for the low-dose group versus placebo and the high-dose group versus placebo were calculated. If both individual P values were 0.025 or less, a significant treatment effect was declared for both dose groups. If only 1 was 0.025 or less, a significant treatment effect was declared on the corresponding dose group.
Analyses of the secondary and exploratory end points were intended to provide supportive efficacy and safety information regarding the differences between the treatment groups. No adjustments were performed for multiple comparisons when testing these secondary and exploratory end points.
Subgroup Analyses
Subgroup efficacy analyses on the primary end point and selected secondary end points (changes from baseline to each visit in sBA, ALT, and growth) were performed by age group (6 months to 5 years, 6 to 12 years, and 13 to 18 years), by PFIC type (1 and 2), baseline sBAs level (250 µmol/L or greater and less than 250 µmol/L), Child-Pugh classification (A, B, C), patients with the BSEP type of PFIC2, and the use of UDCA and rifampicin (alone or either). A statistical analysis was performed only when the sample size was 10 or greater in each treatment group. If the sample size was less than 10 in any treatment group, only summary statistics are provided; the P value is not reported. Forest plots were also produced. Due to the anticipated small sample size in these subgroups, analyses by subgroups did not include the stratification factors.
Table 8: Statistical Analysis of Efficacy End Points From the PEDFIC 1 Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Mortality | NA | NA | NA | NA |
Need for surgery | NA | NA | NA | NA |
Change from baseline in PedsQL Parent Report and Family Impact Module | ANCOVA | Baseline total scores of the PedsQL as covariate, and treatment group and stratification factors (PFIC type and age category) as fixed effects. | None | None |
sBA response | CMH test; the Miettinen-Nurminen (score) CI is reported, adjusting for stratification factors. | Stratified by PFIC type and age category. | Patients who dropped out of treatment, or who completed treatment but had a missing average at the end of the treatment, were categorized as nonresponders. Patients in need of surgical rescue (i.e., biliary diversion and/or liver transplant) were also classified as nonresponders. |
|
Proportion of positive pruritus assessments at the patient level at week 24 | ANCOVA | Rounded baseline scores as covariates, and treatment group and stratification factors (PFIC type and age category) as fixed effects. | All intermittently missing assessments were classified as negative pruritus assessments. All planned assessments after the intercurrent events (premature treatment discontinuation, death, or initiation of rescue treatments such as biliary diversion surgery or liver transplant) were counted as negative pruritus assessments. | If there were concerns about model assumptions (i.e., normality and homogeneity of variances), a nonparametric ANCOVA based on the rank scores was used as a sensitivity analysis. To address concerns about low baseline morning or evening scores (< 2), an additional analysis was performed that excluded baseline scores that rounded to 0 or 1. |
Change in growth | MMRM model | Including terms for baseline, PFIC type, age category, treatment, visit, treatment-by-baseline interaction, and treatment-by-visit interaction. | NA | None |
Sleep parameters (awakenings), total bilirubin | Descriptive statistics | NA | NA | NA |
ANCOVA = analysis of covariance; CI = confidence interval; CMH = Cochrane-Mantel-Haenszel; MMRM = mixed-model for repeated measures; NA = not applicable; PedsQL = Pediatric Quality of Life Inventory; PFIC = progressive familial intrahepatic cholestasis; sBA = serum bile acid.
Sources: Details included in the table are from the sponsor’s summary of clinical evidence and from the Clinical Study Report for PEDFIC 1.
Analysis Populations
Table 9: Analysis Populations of PEDFIC 1
Population | Definition | Application |
|---|---|---|
FAS | All randomized patients who received at least 1 dose of the study treatment. Patients were analyzed as randomized. | The FAS was the primary analysis set for efficacy analyses. |
Safety analysis set | All randomized patients who received at least 1 dose of the study drug. In the event an incorrect study drug was dispensed to a patient in error, the treatment arm of the most commonly administered study drug for that patient was assigned for analysis of the patient’s data. | The safety analysis set was used for safety analyses. |
PP analysis set | All patients in the FAS who did not have any important protocol deviations. | The PP analysis set provided supportive data for the primary and selected secondary efficacy end points. |
FAS = full analysis set; PP = per protocol.
Source: PEDFIC 1 Clinical Study Report (2020).8
Results
Patient Disposition
A total of 107 pediatric patients were screened and 62 were enrolled into the study (Table 10). The specific reasons for the 45 screening failures were not reported; however, the sponsor noted that the majority were due to patients not having significant pruritus. Overall, 49 patients (79%) completed the planned 24-week treatment period, 11 patients rolled over to the long-term extension trial before completion of 24 weeks of treatment per protocol due to intolerable symptoms after completing between 12 and 18 weeks, 1 patient discontinued treatment due to an AE of diarrhea, and 1 patient discontinued for other reasons (noncompliance or inability to travel to the site).
There were numerically more patients who withdrew from treatment in the placebo group (25%) compared with the odevixibat 120 mcg/kg group (16%), and all patients in the placebo group discontinued due to lack of efficacy or intolerable symptoms.
Baseline Characteristics
The baseline characteristics of the PEDFIC 1 study are summarized in Table 11. The median age of the patients was 3.2 years and ranged from 6 months to 15.9 years. Patients treated with odevixibat 120 mcg/kg/day were older (median age 4.9 years) compared with patients in the placebo group (2.8 years) and patients in the 40 mcg/kg/day group (3.2 years). Most patients (47 of 62; 76%) were between 6 months and 5 years of age, 12 (19%) were between 6 and 12 years of age, and 3 (5%) were between 13 and 18 years of age; a limited number of patients (n = 10; 16%) were 8 years of age or older and were eligible to complete the PRUCISION PRO instrument.
Table 10: Summary of Patient Disposition for the PEDFIC 1 Study
Patient disposition | PEDFIC 1 study | ||
|---|---|---|---|
Odevixibat 40 mcg/kg | Odevixibat 120 mcg/kg | Placebo | |
Screened, N | 107 | ||
Randomized, N (%) | 23 (100) | 19 (100) | 20 (100) |
Discontinued from treatment, n (%) | 5 (21.7) | 3 (15.8) | 5 (25.0) |
Reason for discontinuation, n (%) | |||
Adverse events | 0 | 1 (5.3) | 0 |
Lack of efficacy or intolerable symptoms | 4 (17.4) | 2 (10.5) | 5 (25.0) |
Other | 1 (4.3) | 0 | 0 |
Continued to PEDFIC 2 extension study, N (%) | 21 (91.3) | 16 (84.2) | 19 (95.0) |
FAS, N | 23 (100) | 19 (100) | 20 (100) |
PP, N | 21 (91.3) | 17 (89.5) | 18 (90.0) |
Safety, N | 23 (100) | 19 (100) | 20 (100) |
FAS = full analysis set; PP = per protocol.
Source: PEDFIC 1 Clinical Study Report (2020).8
Consistent with patients with PFIC having impaired growth, the median height-for-age and weight-for-age z scores were −1.70 and −0.95, respectively, indicating the patients were below their age-matched peers for growth. A review of the z scores across the treatment groups indicates that patients in the placebo and 120 mcg/kg/day groups had more impaired growth, including in both height and weight, compared with patients in the 40 mcg/kg/day group.
All 62 patients had genetic confirmation of PFIC based on central reader review. Patients with PFIC1 or PFIC2 were included in the study, most (45 patients; 73%) had PFIC2 and 17 (27%) had PFIC1. Almost all patients (60; 97%) had a history of significant pruritus present per the investigator, and most (42 patients; 68%) had levels of sBAs greater than 100 µmol/L within 6 months before enrolment in the study.
The majority of patients (55; 89%) were receiving UDCA and/or rifampicin at study entry, with 50 patients (81%) on UDCA and 41 (66%) on rifampicin. At baseline, a smaller proportion of patients randomized to the 120 mcg/kg/day group were receiving UDCA (68%) compared with patients randomized to the 40 mcg/kg/day group (83%) and to the placebo group (90%). For rifampicin, a smaller proportion of patients in both active treatment groups were receiving this medication at baseline (57% and 58% in the 40 mcg/kg/day and 120 mcg/kg/day groups, respectively) compared with the placebo group (85%). Overall, 8 patients (13%) reported prior biliary tract surgeries (all reports of biliary diversion).
Median levels of sBAs were elevated at baseline at 228.0 µmol/L (93.1 mcg/mL), 188.5 µmol/L (77.0 mcg/mL), and 254.5 µmol/L (104.0 mcg/mL) in the odevixibat 40 mcg/kg/day, odevixibat 120 mcg/kg/day, and placebo groups, respectively. Median levels of hepatic biochemical parameters were elevated at baseline, including ||| ||| |||| ||||||||||||||||| ||| ||||| |||| |||| ||||||||| ||| ||||| ||||||||| ||||| ||||||| ||| |||||| |||||||. Based on the Child-Pugh classification, 41 patients (66%) had mild hepatic impairment and 21 (34%) had moderate hepatic impairment; no patients had severe impairment.
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Table 11: Summary of Baseline Characteristics From the PEDFIC 1 Study (FAS)
Characteristic | PEDFIC 1 study | ||
|---|---|---|---|
Odevixibat 40 mcg/kg (N = 23) | Odevixibat 120 mcg/kg (N = 19) | Placebo (N = 20) | |
Demographic | |||
Age (years) | |||
Mean (SD) | 3.9 (3.7) | 5.2 (4.2) | 3.8 (3.9) |
Median (range) | 3.2 (0.6 to 15.9) | 4.9 (1.0 to 13.2) | 2.8 (0.5 to 15.0) |
6 months to 5 years, n (%) | 17 (73.9) | 14 (73.7) | 16 (80.0) |
6 to 12 years, n (%) | 5 (21.7) | 4 (21.1) | 3 (15.0) |
13 to 18 years, n (%) | 1 (4.3) | 1 (5.3) | 1 (5.0) |
Sex, n (%) | |||
Female | 12 (52.2) | 11 (57.9) | 8 (40.0) |
Male | 11 (47.8) | 8 (42.1) | 12 (60.0) |
Race, n (%) | |||
Asian | 0 | 1 (5.3) | 1 (5.0) |
Black or African American | 2 (8.7) | 0 | 0 |
White | 18 (78.3) | 17 (89.5) | 17 (85.0) |
Other | 3 (13.0) | 1 (5.3) | 2 (10.0) |
Anthropometric | |||
Height z scores | |||
Mean (SD) | −1.5 (1.3) | −2.1 (1.6) | −2.3 (1.5) |
Median (range) | |||| |||||| |||| | |||| |||||| |||| | |||| |||||| |||| |
Weight z scores | |||
Mean (SD) | −0.7 (1.3) | −1.2 (1.5) | −1.5 (1.4) |
Median (range) | |||| |||||| |||| | |||| |||||| |||| | |||| |||||| |||| |
BMI z scores | |||
Mean (SD) | 0.4 (0.9) | 0.3 (1.2) | 0.1 (1.4) |
Median (range) | ||| |||||| |||| | ||| |||||| |||| | ||| |||||| |||| |
Disease | |||
PFIC type, n (%) | |||
PFIC1 | 7 (30.4) | 5 (26.3) | 5 (25.0) |
PFIC2 | 16 (69.6) | 14 (73.7) | 15 (75.0) |
Years since diagnosis | |||
Mean (SD) | 2.3 (2.6) | 3.7 (3.8) | 2.8 (3.6) |
Median (range) | 1.5 (0 to 9.0) | 1.6 (−0.1 to 11.9) | 1.1 (−0.1 to 13.3) |
History of significant pruritus per investigator report, n (%) | 22 (95.7) | 19 (100) | 19 (95.0) |
Baseline medication use, n (%) | |||
UDCA | 19 (82.6) | 13 (68.4) | 18 (90.0) |
Rifampicin | 13 (56.5) | 11 (57.9) | 17 (85.0) |
UDCA and/or rifampicin | 21 (91.3) | 15 (78.9) | 19 (95.0) |
Patients with prior biliary diversion, n (%) | ||||| | |||||| | |||||| |
Serum bile acid (µmol/L) | |||
Mean (SE) | ||||| |||||| | ||||| |||||| | ||||| |||||| |
Median (range) | 228.0 (76.0 to 605.0) | 188.5 (36.0 to 599.5) | 254.5 (56.5 to 435.0) |
ALT (IU/L) | |||
Mean (SD) | 127.7 (165.8) | 89.1 (86.9) | 76.9 (56.2) |
Median (range) | 83.0 (21.0 to 798.0) | 59.0 (16.0 to 314.0) | 55.5 (19.0 to 236.0) |
AST (IU/L) | |||
Mean (SD) | 114.2 (82.7) | 96.0 (70.3) | 90.2 (51.9) |
Median (range) | 90.0 (37.0 to 405.0) | 83.0 (38.0 to 320.0) | 75.5 (32.0 to 219.0) |
Total bilirubin (µmol/L) | |||
Mean (SD) | 52.2 |||||| | 57.0 |||||| | 53.3 |||||| |
Median (range) | |||| ||||| |||||| | |||| ||||| |||||| | |||| ||||| |||||| |
Child-Pugh classification, n (%) | |||
A (mild) | 15 (65.2) | 14 (73.7) | 12 (60.0) |
B (moderate) | 8 (34.8) | 5 (26.3) | 8 (40.0) |
C (severe) | 0 | 0 | 0 |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; BMI = body mass index; FAS = full analysis set; PFIC = progressive familial intrahepatic cholestasis; SD = standard deviation; SE = standard error; UDCA = ursodeoxycholic acid.
Source: PEDFIC 1 Clinical Study Report (2020).8
Exposure to Study Treatments
The median duration of exposure was approximately 24 weeks in all treatment groups and ranged from 4 to 27.6 weeks (Table 12). The majority of patients, including 34 of 42 (81%) in the overall odevixibat group and 15 of 20 (75%) in the placebo group, received 20 or more weeks of study treatment. Study drug compliance was checked through eDiary responses. Additionally, the study-site staff were to count all of the unused study drug that the patients returned at visits 4 through 9 (week 4 to week 24) and record details in the electronic case report form. Overall, adherence with the daily dosing of the study drug was high, with a median overall adherence rate, calculated from the eDiary, of 93% and 99% for the overall odevixibat and placebo groups, respectively. Compliance as calculated from the case report form was also high, with a median overall compliance of ||| ||| ||||, respectively.
All 62 patients took at least 1 concomitant medication during the treatment period. Most patients received concomitant medications for the treatment of pruritus and vitamin supplementation (Table 10). Note that for patients receiving medication for pruritus, the dose and/or regimen was not to change. Generally, the types and use of these medications were similar across the treatment groups, with the exception that a higher percentage of patients in the placebo group reported using other bile acids and derivatives (UDCA) and other antibacterials (rifampicin). Two patients (1 patient each in the placebo and odevixibat 120 mcg/kg/day groups) initiated treatment with rifampicin during the treatment period, which was prohibited by the protocol.
Efficacy
Mortality
There were no deaths in the study.
Surgical Intervention
None of the 62 patients underwent biliary diversion surgery or liver transplant during the study.
Table 12: Summary of Patient Exposure and Concomitant Use From the PEDFIC 1 Study (Safety Analysis Set)
Exposure | PEDFIC 1 study | ||
|---|---|---|---|
Odevixibat 40 mcg/kg (N = 23) | Odevixibat 120 mcg/kg (N = 19) | Placebo (N = 20) | |
Duration (weeks), mean (SD) | 21.7 (5.0) | 21.7 (5.8) | 21.6 (4.6) |
Duration (weeks), median (range) | 23.9 (10.7 to 25.9) | 23.9 (4.0 to 27.6) | 23.7 (11.7 to 29.1) |
Adherence by eDiary (%), mean (SD) | 91.3 (8.8) | 90.5 (8.2) | 95.2 (8.5) |
Adherence by eDiary (%), median (range) | 94.0 (72.6 to 100) | 91.5 (64.3 to 100) | 98.6 (64.9 to 100) |
Adherence by case report form (%), mean (SD) | |||| ||||| | |||| |||||| | |||| |||||| |
Adherence by case report form (%), median (range) | |||| |||||| |||||| | |||| |||||| |||||| | ||||| |||||| |||||| |
Concomitant medications used, n (%) | |||
|||| ||||| ||| ||||||||||| | || |||||| | || |||||| | || |||||| |
|||||||||| ||||||||| | || |||||| | || |||||| | || |||||| |
||||||| | || |||||| | || |||||| | || |||||| |
||||| |||||||||||||| | || |||||| | || |||||| | || |||||| |
||||| ||||| ||||||| |||||||||||| | || |||||| | || |||||| | || |||||| |
|||||||| | || |||||| | || |||||| | || |||||| |
||||||| || ||||| | || |||||| | || |||||| | || |||||| |
|||||||||||||| ||||| | || |||||| | || |||||| | || |||||| |
|||||||||| ||||||||||| | || |||||| | || |||||| | || |||||| |
|||||||||||| || |||||||| | || |||||| | || |||||| | || |||||| |
|||| ||||||||| |||| |||||||||||| | || |||||| | || |||||| | || |||||| |
|||||||||||| || |||||||||||| ||||| |||||||||||||| | || |||||| | || |||||| | || |||||| |
||||||||||||| ||||||||||| | || |||||| | || |||||| | || |||||| |
||||||||| |||| ||||||||||| | || |||||| | || |||||| | || |||||| |
||||||| | || |||||| | || |||||| | || |||||| |
|||||||||| |||||| | || |||||| | || |||||| | || |||||| |
||||||||| ||||| |||||||||||| | || |||||| | || |||||| | || |||||| |
||||||||| ||||||||||||||||||||| |||||||| | || |||||| | || |||||| | || |||||| |
|||||||||||| ||| ||||||||||| | || |||||| | || |||||| | || |||||| |
||||||||||| |||| |||||||| |||||||| | || |||||| | || |||||| | || |||||| |
||||||||| |||||| ||||||||||| | || |||||| | || |||||| | || |||||| |
||||||||||| ||||||||||| | || |||||| | || |||||| | || |||||| |
||||||||||||||| | || |||||| | || |||||| | || |||||| |
||||||||||| |||||| ||||||||| | || |||||| | || |||||| | || |||||| |
SD = standard deviation.
Source: PEDFIC 1 Clinical Study Report (2020).8
Health-Related Quality of Life
Pediatric Quality of Life Inventory
HRQoL was assessed as an exploratory outcome using the PedsQL instrument, with higher scores indicating improved quality of life (Table 13). After 24 weeks, the LS mean difference versus placebo for the odevixibat 40 mcg/kg/day group was ||||| |||| |||||||||| |||||||| ||||| ||||||| |||||| ||| ||| ||| ||| |||||||||| ||||| ||| |||| |||| ||| |||||| ||||||.
Table 13: Summary of Change From Baseline to Week 24 in the PedsQL (Full Analysis Set)
Detail | Odevixibat 40 mcg/kg (N = 23) | Odevixibat 120 mcg/kg (N = 19) | Placebo (N = 20) |
|---|---|---|---|
PedsQL Parent Report, mean (SE)a | |||
N (baseline) | || | || | || |
Baseline | ||||| |||||| | ||||| |||||| | ||||| |||||| |
N (Week 24) | || | || | || |
Mean change (SE) to week 24 | |||| |||||| | ||||| |||||| | |||| |||||| |
LS mean difference (95% CI) vs. placebob | ||||| |||||||| |||||| | |||| ||||||| |||||| | — |
PedsQL Family Impact Module, mean (SE) | |||
N (baseline) | || | || | || |
Baseline | ||||| |||||| | ||||| |||||| | ||||| |||||| |
N (week 24) | || | || | || |
Mean change (SE) to week 24 | ||||| |||||| | ||||| |||||| | |||| |||||| |
LS mean change to week 24 | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
LS mean difference (95% CI) vs. placebo | |||| |||||||| |||||| | |||| |||||||| |||||| | — |
CI = confidence interval; GRADE = Grading of Recommendations Assessment, Development, and Evaluation; LS = least squares; PedsQL = Pediatric Quality of Life Inventory; SE = standard error; vs. versus.
aA Parent Report was completed only for patients who were 2 years of age or older.
bThese analyses were not conducted a priori as part of PEDFIC 1 but were instead requested by CADTH to facilitate GRADE assessment.
Source: PEDFIC 1 Clinical Study Report (2020).8
Serum Bile Acid
Proportion of Patients With at Least a 70% Reduction From Baseline in the Concentration of sBAs or Reaching a Level of 70 µmol/L or Less After 24 Weeks of Treatment
After 24 weeks of treatment, for the proportion of patients with at least a 70% reduction in sBA concentration from baseline or reaching a level of 70 µmol/L or less, the between-group differences compared with placebo were 0.441% for odevixibat 40 mcg/kg/day (95% CI, 0.236 to 0.646; adjusted P = 0.0015), and 0.216% for odevixibat 120 mcg/kg/day (95% CI, −0.005 to 0.438; adjusted P = 0.0174) (Table 14). Results in the per-protocol analysis set were consistent with the full analysis set.
||||| ||||||||||| |||||||| |||| ||||||||| ||| ||| ||||||| |||||||| || ||||| |||| ||||| ||||||||||||||||| |||||||||| ||||||||| || |||||||| ||||||||| |||||||| ||||| |||||||| ||||| ||| ||| ||||||| |||||||||| ||||| ||||||||| || ||||| |||| || ||||| |||| ||||| |||||||||| || ||| ||||||| |||||| ||| |||||||| |||||||||| ||||| ||| ||| ||||||||| ||| ||| |||||| ||||||||||| |||||||||||||||||||||| || ||| ||||||| ||| |||||||||||||| ||||||| ||| |||||||| ||||| |||| ||||| |||||| ||| ||||||| ||||||| || ||| |||||||| || ||| |||||||||| || ||||| |||| ||||| |||||||||| ||||||||| ||||||||||||| |||| |||||||||| |||| ||| ||||||| ||||||||| ||| ||| ||||||| ||||| ||||||||| |||| ||||||||||||| |||||||| |||| ||||||| |||| || |||| ||||| |||| |||||||| || || ||||||| || ||||||||| |||||||| |||| ||||||||||||| |||||||| || ||||| ||| ||| |||||||| |||||| || || ||||||||||||||| ||| ||| ||||||| |||||||||| ||||| || |||| |||||||||| ||||||.
Subgroup
In patients aged 6 months to 5 years (n = 47) and patients between 6 and 12 years of age (n = 12), the proportion of patients who received odevixibat and met the sBAs end point was ||||||| |||||| ||| |||||| ||| |||||||||||||| ||||||| ||| |||||||||| || |||||| ||||||||||||| ||| ||| |||||||||| || ||| ||||||| |||||||||| |||||||| ||| |||||||||| ||| |||||||| |||||||||| ||| |||| ||| ||||| || |||| ||||||| ||||||||| || ||| ||| |||||||||| ||||| ||| ||| |||| ||| |||||||||| |||||||| |||| |||| |||||||||| ||| ||||| |||||||| ||||||| ||||||||| || ||| || |||||||||| |||||| |||||||||||| || ||||| |||| ||||| |||| |||| ||||| ||| ||||||||| ||| |||||.
For patients receiving odevixibat, the proportion of sBA responders was higher for patients with PFIC2 (12 of 30 patients; 40.0%) compared with patients with PFIC1 (2 of 12 patients; 16.7%), although the comparison of each group with placebo had widely overlapping CIs.
The proportion of sBA responders in the odevixibat groups was ||| |||| ||| |||||||| |||| |||||||| ||||| |||| |||| ||||||||||||| |||| |||||| || || || ||||||||| |||||| ||| |||| |||||| || || || ||||||||| ||||||| |||||||| |||| || |||||||| || ||| ||||||| ||||||.
Subgroup analyses based on concomitant use of UDCA and/or rifampicin were not informative due to the small number of patients who did not use concomitant medication during the study.
Table 14: Summary of Key Efficacy Results From the PEDFIC 1 Study (FAS)
Detail | Odevixibat 40 mcg/kg (N = 23) | Odevixibat 120 mcg/kg (N = 19) | Placebo (N = 20) |
|---|---|---|---|
Serum bile acid | |||
Proportion of patients experiencing at least a 70% reduction in fasting serum bile acid concentration | |||
Responders, n (%) | 10 (43.5) | 4 (21.1) | 0 |
95% CI | 23.2 to 65.5 | 6.1 to 45.6 | 0 to 16.8 |
Proportion difference vs. placebo without adjusting for stratification factors (95% CI)a | 0.435 (0.220 to 0.655) | 0.211 (0.021 to 0.456) | — |
Proportion difference vs. placebo adjusting for stratification factors (95% CI)b | 0.441 (0.236 to 0.646) | 0.216 (−0.005 to 0.438) | — |
1-sided unadjusted P valuec | 0.0003 | 0.0174 | — |
1-sided adjusted P valued | 0.0015 | 0.0174 | — |
Week 12 | |||
|||||||||||||| | |||||| | |||||| | ||||| |
|||||||||| |||||||||| || ||||||| ||||||| ||||||||| ||| |||||||||||||| ||||||| |||| || | ||||| |||||||| ||||||| | ||||| |||||||| ||||||| | — |
|||||||||| |||||||||| || ||||||| ||||||||| ||| |||||||||||||| ||||||| |||| || | ||||| |||||||| ||||||| | ||||| ||||||||| ||||||| | — |
Pruritus assessments at week 24 based on the PRUCISION ObsRO instrument | |||
Proportion of positive pruritus assessments at the patient level | |||
Mean (SE) | 58.3 (6.2) | 47.7 (8.1) | 28.7 (5.2) |
Median (range) | 60.1 (1.8 to 97.0) | 45.5 (0 to 91.3) | 23.4 (0.9 to 79.2) |
LS mean difference vs. placebo (95% CI)e | 28.23 (9.83 to 46.64) | 21.71 (1.87 to 41.54) | — |
1-sided unadjusted P valuee | 0.0016f | 0.0163f | — |
|| | || | || | || |
|||| ||||| ||||| ||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
|| |||| |||||||||| || ||||||| |||| ||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | — |
|| | || | || | || |
|||| ||||| ||||| |||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
|| |||| |||||||||| || ||||||| |||| ||| | ||||| |||||| |||||| | ||||| |||||| |||||| | — |
ANCOVA = analysis of covariance; CI = confidence interval; FAS = full analysis set; LS = least squares; ObsRO = observer-reported outcome; PFIC = progressive familial intrahepatic cholestasis; SD = standard deviation; SE = standard error; vs. = versus.
aClopper-Pearson exact CI is reported for the percentage of responders, and the exact unconditional CI is reported for the proportion difference without adjusting for stratification factors.
bThe Miettinen-Nurminen (score) CI is reported, adjusting for stratification factors.
cBased on the Cochran-Mantel-Haenszel test adjusting for stratification factor (PFIC type).
dFor an individual dose, the adjusted P value was calculated as the maximum value of the unadjusted P value for all doses of odevixibat and the unadjusted P value for the individual dose.
eThe analysis was based on an ANCOVA model with rounded baseline scores as covariates, and treatment group and stratification factors (PFIC type and age category) as fixed effects.
fThese P values were not adjusted for multiplicity.
Source: PEDFIC 1 Clinical Study Report (2020).8
Pruritus Assessments
Proportion of Positive Pruritus Assessments at the Patient Level (ObsRO Instrument)
Based on the results of the ANCOVA model, the between-group difference in the LS means for the comparisons of the 40 mcg/kg/day odevixibat group with placebo was 28.23 (95% CI, 9.83 to 46.64; 1-sided adjusted P = 0.0016), and the 120 mcg/kg/day odevixibat group with placebo was 21.71 (95% CI, 1.87 to 41.54; 1-sided adjusted P = 0.0163) (Table 14). Data were consistent in the per-protocol analysis set for the 40 mcg/kg/day groups, and although the results were numerically higher for the 120 mcg/kg/day group compared with placebo, the difference did not reach statistical significance.
Proportion of Positive Pruritus Assessments at the Patient Level Over Time (ObsRO Instrument)
Figure 2 presents the proportion of positive pruritus assessments at the patient level over time on treatment by grouped weeks for the morning and evening pruritus scores combined.

Sleep Parameters
Sleep Parameters Based on the ObsRO
The changes over time in sleep parameters, specifically awakenings, was assessed as a secondary outcome using data derived from the PRUCISION pruritus instrument developed by the sponsor (Table 15). The LS mean between-group difference in number of awakenings from baseline to weeks 21 to 24 was |||| |||||||||| |||| ||| |||||| |||||| || ||| || ||||| ||||| ||| ||||| |||||||||| |||| ||| ||||||| ||||| || ||| ||| |||||||||| |||||.
Table 15: Summary of Sleep Parameters From the PEDFIC 1 Study (FAS)
Detail | Odevixibat 40 mcg/kg (N = 23) | Odevixibat 120 mcg/kg (N = 19) | Placebo (N = 20) |
|---|---|---|---|
Mean (SE) number of awakenings (ObsRO) | |||
Baseline | ||||| ||||||| | |||| ||||||| | 8.98 ||||||| |
N | || | || | || |
Mean (SE) change from baseline to weeks 21 to 24 | |||| ||||||| | ||||| ||||||| | ||||| ||||||| |
LS mean difference (95% CI) vs. placeboa | |||| ||||||| |||||| | ||||| |||||||| ||||| | — |
CI = confidence interval; FAS = full analysis set; GRADE = Grading of Recommendations Assessment Development and Evaluation; LS = least squares; ObsRO = observer-reported outcome; PFIC = progressive familial intrahepatic cholestasis; SE = standard error; vs. = versus.
aCalculated from an analysis of covariance model with baseline total scores for the number of awakenings (ObsRO) as covariate, and treatment group and stratification factors (PFIC type and age category) as fixed effects. These analyses were not conducted a priori as part of PEDFIC 1 but were instead requested by CADTH to facilitate GRADE assessment.
Growth Parameters
Improvement in growth (height, weight, BMI) was assessed as a secondary outcome by comparing changes from baseline in z scores relative to a typical pediatric growth chart. For height, the LS mean between-group difference between odevixibat and placebo after 24 weeks was |||| |||| ||| ||||| ||||| ||| ||| || |||||||||| ||||| ||| |||| |||| ||| |||||| ||||| ||| ||| ||| |||||||||| ||||| (Table 16). For weight, the LS mean between-group difference was |||| |||| ||| |||||| ||||| ||| ||| || |||||||||| ||||| ||| |||| |||| ||| |||||| ||||| ||| ||| ||| |||||||||| |||||. For BMI, the LS mean between-group difference was |||| |||| ||| |||||| ||||| ||| ||| || |||||||||| ||||| ||| ||||| |||| ||| |||||| ||||| ||| ||| ||| |||||||||| |||||.
Laboratory Parameters
The change from baseline to week 24 in total bilirubin was an exploratory outcome. The LS mean between-group difference versus placebo in total bilirubin was |||||| |||||| |||| ||| ||||||| |||||| || ||| || |||||||||| ||||| ||| ||||| |||||| |||| ||| ||||||| |||||| || ||| ||| |||||||||| ||||| (Table 17).
Harms
Refer to Table 18 for harms data.
Table 16: Summary of Change From Baseline to Week 24 in Growth Parameters (Full Analysis Set)
Parameters | ODE 40 mcg/kg (N = 23) | ODE 120 mcg/kg (N = 19) | PLA (N = 20) |
|---|---|---|---|
Height (z score), mean (SE) | |||
Baseline | −1.45 (0.27) | −2.09 (0.37) | −2.26 (0.34) |
N (week 24) | 17 | 15 | 12 |
Change to week 24 | 0.05 (0.11) | 0.00 (0.16) | −0.16 (0.10) |
LS mean difference between the ODE and PLA groups (95% CI) | |||| |||||| ||||| | |||| ||||||| ||||| | — |
Weight (z score), mean (SE) | |||
Baseline | −0.74 (0.27) | −1.19 (0.35) | −1.52 (0.32) |
N (week 24) | 18 | 15 | 12 |
Change to week 24 | 0.29 (0.11) | 0.15 (0.12) | 0.10 (0.10) |
LS mean difference between the ODE and PLA groups (95% CI) | |||| ||||||| ||||| | |||| ||||||| ||||| | — |
BMI (z score), mean (SE) | |||
Baseline | 0.41 (0.19) | 0.28 (0.27) | 0.10 (0.31) |
N (week 24) | 17 | 15 | 12 |
Change to week 24 | 0.36 (0.11) | 0.20 (0.20) | 0.26 (0.16) |
LS mean difference between ODE and PLA groups (95% CI) | |||| ||||||| ||||| | ||||| ||||||| ||||| | — |
BMI = body mass index; CI = confidence interval; LS = least squares; ODE = odevixibat; PLA = placebo; SE = standard error.
Source: PEDFIC 1 Clinical Study Report (2020).8
Table 17: Summary of Change From Baseline to Week 24 in Biochemical Parameters (Full Analysis Set)
Detail | Odevixibat 40 mcg/kg (N = 23) | Odevixibat 120 mcg/kg (N = 19) | Placebo (N = 20) |
|---|---|---|---|
Total bilirubin (µmol/L), mean (SE) | |||
Baseline | 52.2 (10.1) | 57.0 (18.1) | 53.3 (13.0) |
N (week 24) | 17 | 15 | 11 |
Change to week 24 | −23.7 (9.2) | −19.3 (13.6) | −9.6 (15.2) |
LS mean difference (95% CI) vs. placeboa | |||||| |||||||| |||||| | ||||| |||||||| |||||| | — |
GRADE = Grading of Recommendations Assessment Development and Evaluation; LS = least squares; PFIC = progressive familial intrahepatic cholestasis; SE = standard error; vs. = versus.
aCalculated from an analysis of covariance model with baseline total bilirubin (µmol/L) as covariate, and treatment group and stratification factors (PFIC type and age category) as fixed effects. These analyses were not conducted a priori as part of PEDFIC 1 but were instead requested by CADTH to facilitate GRADE assessment.
Source: PEDFIC 1 Clinical Study Report (2020).8
Table 18: Summary of Harms Results From the PEDFIC 1 Study (SAS)
Harms | Odevixibat 40 mcg/kg (N = 23) | Odevixibat 120 mcg/kg (N = 19) | Placebo (N = 20) |
|---|---|---|---|
Most common AEs (10% or more patients in any group), n (%) | |||
≥ 1 AE | 19 (82.6) | 16 (84.2) | 17 (85.0) |
Diarrhea | 9 (39.1) | 4 (21.1) | 1 (5.0) |
Pyrexia | 7 (30.4) | 5 (26.3) | 5 (25.0) |
Upper respiratory tract infection | 3 (13.0) | 5 (26.3) | 3 (15.0) |
Vomiting | 4 (17.4) | 3 (15.8) | 0 |
Alanine aminotransferase increased | 3 (13.0) | 3 (15.8) | 1 (5.0) |
Blood bilirubin increased | 3 (13.0) | 2 (10.5) | 2 (10.0) |
SAEs, n (%) | |||
Patients with ≥ 1 SAE | 0 | 3 (15.8) | 5 (25.0) |
Urinary tract infection | 0 | 1 (5.3) | 1 (5.0) |
Dehydration | 0 | 1 (5.3) | 0 |
Supraventricular tachycardia | 0 | 1 (5.3) | 0 |
Liver function test increased | 0 | 1 (5.3) | 0 |
Patients who stopped treatment due to AEs, n (%) | |||
Patients who stopped | 3 (13.0) | 6 (31.6) | 1 (5.0) |
Deaths, n (%) | |||
Patients who died | 0 | 0 | 0 |
AEs of special interest, n (%) | |||
Clinically significant diarrhea | ||||| | ||||| | ||||| |
RR (95% CI) vs. placeboa | ||||| ||||||| ||||||| | ||||| ||||||| ||||||| | — |
RD (95% CI) vs. placeboa | |||||| ||||||||| ||||||| | |||||| ||||||||| ||||||| | — |
Adjudicated hepatic events | ||||| | ||||| | ||||| |
RR (95% CI) vs. placeboa | ||||| ||||||| |||||| | ||||| ||||||| |||||| | — |
RD (95% CI) vs. placeboa | ||||| ||||||||| ||||||| | |||||| ||||||||| ||||||| | — |
Hepatobiliary disorders | ||||| | ||||| | ||||| |
RR (95% CI) vs. placeboa | || | || | — |
RD (95% CI) vs. placeboa | ||||| ||||||||| ||||||| | ||||| ||||||||| ||||||| | — |
Liver-related TEAEs | ||||| | ||||| | ||||| |
RR (95% CI) vs. placeboa | ||||| ||||||| |||||| | ||||| ||||||| |||||| | — |
RD (95% CI) vs. placeboa | ||||| ||||||||| ||||||| | |||||| ||||||||| ||||||| | — |
AE = adverse event; CI = confidence interval; GRADE = Grading of Recommendations Assessment, Development, and Evaluation; NC = not calculable; RD = risk difference; RR = relative risk; SAE = serious adverse event; SAS = safety analysis set; TEAE = treatment-emergent adverse event; vs. = versus.
aThese analyses were not conducted a priori as part of PEDFIC 1 but were instead requested by CADTH to facilitate GRADE assessment.
Source: PEDFIC 1 Clinical Study Report (2020).8
Adverse Events
Overall, 35 of the 42 patients (83%) who received odevixibat experienced at least 1 TEAE, as did 17 of 20 patients (85%) who received placebo; the overall incidence of TEAEs was similar in the odevixibat 40 mcg/kg/day and 120 mcg/kg/day treatment groups (83% and 84%, respectively). The most commonly reported types of events during the study were gastrointestinal disorders and infections. Overall, the most commonly reported TEAEs (≥ 10% overall) among patients who received odevixibat versus those who received placebo were diarrhea (31% versus 5%), pyrexia (29% versus 25%), upper respiratory tract infection (19% versus 15%), vomiting (17% versus 0%), ALT increased (14% versus 5%), and blood bilirubin increased (12% versus 10%).
Treatment-Emergent Serious Adverse Events
Treatment-emergent SAEs were reported in 3 of 42 patients (7%) who received odevixibat and in 5 of 20 patients (25%) who received placebo. No TESAEs were reported in the 40 mcg/kg/day treatment group. The most commonly reported types of TESAEs were infections, reported in 4 of 20 patients (20%) in the placebo group and in 1 of 19 patients (5%) in the 120 mcg/kg/day group. The only event reported in more than 1 patient was urinary tract infection, reported in 1 patient each in the placebo and 120 mcg/kg/day groups. None of the TESAEs led to discontinuation of treatment.
Withdrawals Due to Adverse Events
Dose interruptions due to TEAEs were reported at a higher incidence in patients who received odevixibat (9 of 42; 21%) compared with patients who received placebo (1 of 20; 5%). The highest incidence was reported among patients who received the 120 mcg/kg/day dose (6 of 19; 32%); in the 40 mcg/kg/day group, 3 of the 23 patients (13%) had treatment interruptions due to TEAEs. In || ||| || |||||||| with treatment interruptions due to TEAEs, the events were related to elevations in hepatic biochemical test results and treatment was interrupted, as required by the protocol. All | of the cases where the study drug was interrupted due to hepatic biochemical test results underwent adjudication by the study’s data safety monitoring board, and all such events were assessed as being related to the patient’s underlying disease. ||| ||||| ||||| |||||||| ||||||||| ||||||||| |||||| |||| || |||||||| ||||||| ||||||||||. Patient ||||||||| discontinued from the study due to the inability to attend clinic visits.
One patient receiving odevixibat 120 mcg/kg/day discontinued the study drug due to a TEAE of diarrhea.
Mortality Due to Adverse Events
There were no deaths in the study.
Notable Harms
Clinically Significant Diarrhea
A medical review of all cases of diarrhea was conducted to determine if any met the criteria for clinically significant events as follows:
diarrhea with duration of at least 21 days without other etiology
diarrhea of severe intensity or reported as an SAE
diarrhea with concurrent dehydration requiring treatment with rehydration and/or other treatment intervention.
Based on medical review, |||||||| ||| ||| |||||||| ||| |||||||||| ||||||||||| ||||||||| |||||||||||||||| || |||| ||||||||| |||||.
Hepatic Adverse Events
|||||||| |||| |||| || |||||||| ||||||||| |||||| ||| |||||||||||| || ||| |||| |||||| |||||||||| ||||| ||| ||||||| |||||| |||||| ||| |||||| ||||||||| || ||||| || || |||||||| ||| |||||||| |||||||||| |||||||| || || |||||||| ||| |||||||| |||||||| ||| ||||||||| ||| ||| || || || ||||||||| || ||| || |||||||||| ||||| ||| ||| || || || ||||||||| || ||| ||| |||||||||| |||||| |||||| ||| || ||||||||| || |||||| ||| || ||||||||||||| ||||||||| || |||||| |||||||||| || ||| || |||||||||| |||||| || |||||| || |||||||||| || ||| ||| |||||||||| |||||| ||| || |||||| |||||||||| || ||| ||||||| |||||| ||||| || ||||||||||| ||||||| ||| ||||| |||| ||||||||||| || ||| || ||| ||||||||| |||||||||| ||||||| || ||| || ||||| ||||| |||||| ||||||||||||||| |||||||| |||||||||.
Two patients, 1 in each of the odevixibat dose groups, had TEAEs in the hepatobiliary disorders System Organ Class, ||||||||| |||| |||||||||||| ||| |||||||||||||| |||||||| |||||||| || ||| |||||||||||||| ||||| ||| |||| |||||||| ||||||||| || ||| || |||||||||| ||||||.
A total of 11 of the 42 patients (26%) in the overall odevixibat group experienced liver-related TEAEs, as did 4 of the 20 patients (20%) in the placebo group. The incidence rate was 22% (5 of 23 patients) in the 40 mcg/kg/day group and 32% (6 of 19 patients) in the 120 mcg/kg/day group. ||| |||| |||||| ||||||||||||| ||||| || |||||||| || ||| ||||||||||||| |||| ||| ||||||||| || ||||||||| || ||| |||||||||| || || || ||| ||||||| |||||| ||| ||||| ||||||||| ||||||||| |||||||||||| || ||| |||||||||| |||||||||| || || ||| ||||||| ||||||| ||| ||||| ||||||||||||||||||||| ||||||||||||| ||||| |||| |||||||| ||||||||||||||||||| |||||||.
None of the 62 patients had a liver decompensation event.
Critical Appraisal
Internal Validity
In the PEDFIC 1 study, adequate methods were used for randomization and to maintain allocation concealment (use of an IWRS). Despite this, there were imbalances in several baseline characteristics that suggest prognostic balance across the groups was not achieved. These imbalances may have resulted from chance, given that the sample size was small and, based on the available characteristics, did not appear to be systematically favouring any treatment group, according to the experts consulted by CADTH. The investigators took adequate measures to facilitate adequate blinding of the participants and personnel involved in the trial (use of a matched placebo). Diarrhea was a relatively frequent event (39% of patients in the odevixibat 40 mcg/kg/day group versus 5% in the placebo group) and is a known and anticipated adverse effect of treatment, given the mechanism of the drug; therefore, it is possible that this may have resulted in unblinding for some patients, their caregivers, and trial investigators. There is a risk that this could have introduced bias (likely favouring odevixibat) in the care provided to patients and the measurement of the subjective outcomes.
There was no control for multiple comparisons outside of the primary outcome, which was controlled for multiple comparisons (2 doses of odevixibat versus placebo). The lack of multiplicity control for all subsequent outcomes increases the risk of finding a statistically significant difference between odevixibat and placebo where, in reality, none exists. However, aside from the pruritus assessment, other outcomes included in this report failed to reach statistical significance based on a conventional alpha of 0.05.
There was a relatively large proportion of patients who discontinued treatment during the PEDFIC 1 trial, 21% overall, although 11 of these patients (18% overall) continued into the extension phase (PEDFIC 2) where they all received the higher dose of odevixibat (120 mcg/kg/day). The discontinuations were highest in the placebo group (25%) and lowest in the odevixibat group (16%). For the primary outcome of sBA responders and the secondary outcome of pruritus responses, patients with missing values at week 24 were treated as nonresponders. Given that most patients, including all patients in the placebo group, discontinued due to lack of efficacy or intolerable symptoms, the impact of this missing data for assessing week 24 responses might have been mitigated. For most other outcomes, however, there was no attempt to account for missing data. Therefore, for outcomes such as the PedsQL, growth, awakenings, and total bilirubin, there is a high risk of bias due to missing outcome data.
The PedsQL scale was used to assess HRQoL and appears to be a well-established and validated scale in pediatrics, according to the clinical experts consulted by CADTH for this review. Due to the young age of many patients in the PEDFIC 1 study, the sponsor relied heavily on proxy reports, notably the PedsQL Parent Report, to gather data. Although there is evidence supporting the validity of using this proxy approach with the PedsQL in pediatric populations,21 it is still important to note that there may be a difference in what the proxy thinks and the child thinks with respect to their HRQoL and symptoms. ||| ||||||| ||||||||| || ||||||||||||||||||||| ||| ||| ||| |||||| |||||; therefore, the clinical significance of the between-group differences for the Parent Report and the Family Impact Module cannot be determined. The PRUCISION pruritus scale used to assess pruritus and its impact on other outcomes, such as sleep, was developed by the sponsor, and that includes the MID they used to indicate a “positive” response. The sponsor’s rationale for creating its own instrument was that there was no publicly available pruritus scale at the time of study initiation; however, it remains unclear whether a new instrument was necessary when it was known that another was already developed, and it is not ideal that the instrument used by the sponsor has only been validated within its own trial, rather than being externally validated in another separate trial that they were not sponsoring. MIDs were also not available for other assessment measures, such as growth, number of awakenings, and total bilirubin, making it difficult to assess the clinical significance of the reported differences between odevixibat and placebo.
There were prespecified subgroup analyses planned by the sponsor; however, the findings from these analyses are limited by the small sample size in the PEDFIC 1 study. For example, for some of the smaller subgroups (e.g., those based on age), the sponsor had to resort to reporting individual patient data, and samples that small are not useful for drawing conclusions about efficacy or harms.
External Validity
The dosing used in the PEDFIC 1 trial is not consistent with that proposed in the draft product monograph. The current proposed dosing recommendations are for patients to begin on 40 mcg/kg/day and then escalate to 120 mcg/kg/day after 12 weeks if there is a lack of response. However, in the pivotal trial, PEDFIC 1, patients began on either 40 mcg/kg/day or 120 mcg/kg/day, and there was no group that underwent this up-titration in the proposed dosing. As a result, information is lacking on the efficacy of odevixibat if it were used as intended in the draft product monograph in terms of dose escalation (it is unclear if it would differ from the findings herein). Most patients in the PEDFIC 1 study were on either UDCA or rifampicin or both, and these are the key standards of care for patients with PFIC1 and PFIC2, although both have limited efficacy, according to the clinical experts. The clinical experts consulted by CADTH for this review believe it is possible that odevixibat might be used in combination with some of these therapies.
PEDFIC 1 was not of sufficient size or duration to adequately assess key clinical outcomes, such as mortality or the need for surgical intervention (liver transplant or biliary diversion). Both of these limitations are understandable, given the rarity of the disease and the fact that this was a placebo-controlled trial in a pediatric population in a disease with a very severe clinical course. Nevertheless, both outcomes are of interest to patients and the lack of data for each is a limitation of the PEDFIC 1 study.
PEDFIC 1 enrolled patients with PFIC1 and PFIC2; however, the current proposed indication does not restrict the use of odevixibat based on PFIC subtype. According to the clinical experts consulted by CADTH for this review, there is no way to determine for certain whether odevixibat will work in other subtypes; however, there is no reason to think it would not work. That said, the clinical experts were clear that if clinicians were to follow evidence-based practice, they would limit prescribing odevixibat to PFIC subtypes 1 and 2.
The primary outcome of PEDFIC 1 for jurisdictions outside of the US, and the only outcome controlled for multiplicity, was the proportion of patients experiencing an sBA response. The precise relationship between sBA levels and disease progression and pruritus has not been established, and it is clear from patient input that pruritus and disease progression (need for surgery, mortality) are the outcomes that are most important to patients. Additionally, the clinical experts consulted by CADTH for this review believed the use of a cut-off of a 70% reduction in sBA for response was too conservative, and questioned how that threshold was chosen. In its comments on the Clinical Review Report, the sponsor revealed that this threshold was determined from a systematic review and meta-analysis that evaluated the relationship between liver biochemistry and improvement in pruritus. The sponsor reported that it found that sBA levels of less than 70 µmol/L or a decrease of more than 70% from baseline predicted a pruritus response with good sensitivity and specificity.22 The clinical experts also noted it is unlikely that sBA would be routinely used to monitor treatment in patients with PFIC, as the assay is not widely available and pruritus is a much more readily monitored and clinically relevant outcome to patients. Similarly, HRQoL is a key outcome for patients, but was only an exploratory outcome in the PEDFIC 1 study and, due to missing data, the findings were difficult to interpret.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE working group:23,24
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
For RCTs: Following the GRADE approach, evidence from RCTs starts as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of the evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for odevixibat versus placebo.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the CADTH review team.
Description of Study
PEDFIC 2 is an ongoing phase III, multicentre, nonrandomized, open-label extension study to investigate the long-term efficacy and safety of a 120 mcg/kg/day dose of odevixibat in patients with PFIC (Figure 3).10,11 The study includes an 8-week screening period (cohort 2 only), a 72-week treatment period, and a 4-week follow-up period. An optional extension period for continued treatment follows the 72-week treatment period. Patients who complete treatment through week 72 and elect to continue receiving treatment with odevixibat in the optional extension period return to the clinic for visits every 16 weeks.
PEDFIC 2 is currently ongoing, and interim data were presented by the sponsor based on a cut-off date of July 31, 2022. This planned interim analysis was completed for regulatory submission purposes to supplement the efficacy and safety data from the PEDFIC 1 study. The planned final analysis for the PEDFIC 2 study will occur once all patients complete the week 72 assessment or are off study, whichever occurs first.

Populations
Cohort 1 consists of children with PFIC types 1 and 2 who participated in the PEDFIC 1 study. Cohort 2 consists of patients with PFIC who have elevated sBAs and cholestatic pruritus and who either did not meet the eligibility criteria for the PEDFIC 1 study or were eligible for enrolment in the PEDFIC 2 study after recruitment to PEDFIC 1 was completed.
For cohort 1, patients were eligible if they completed 24 weeks of treatment in the PEDFIC 1 study (note that before amendment 6, [June 24, 2019], patients who withdrew early from the PEDFIC 1 study after a minimum of 12 weeks of treatment due to intolerable symptoms were also eligible to enter this study). Patients who withdrew from the PEDFIC 1 study due to a study drug–related AE or who were noncompliant with treatment in the PEDFIC 1 study were not eligible.
For cohort 2, patients of any age with a clinical diagnosis of PFIC and with a body weight of 5 kg or greater at screening and clinical genetic confirmation of PFIC were eligible. The study was amended on December 21, 2021, to provide access to odevixibat for patients with an episodic form of PFIC (i.e., benign recurrent intrahepatic cholestasis). Up to 40 patients post biliary diversion surgery could participate in cohort 2. Other eligibility criteria for cohort 2 were identical to the criteria for the PEDFIC 1 study (Table 3), except the following exclusions were removed:
patient with a past medical history of chronic kidney disease with impaired renal function and a glomerular filtration rate of less than 70 mL/min/1.73 m2
patient with a surgical history of disruption of the enterohepatic circulation (biliary diversion surgery) within 6 months before the start of the screening period
patient who has been previously treated with an ileal bile acid transporter inhibitor whose pruritus has not responded to treatment.
Interventions
For all patients who entered the study, odevixibat therapy was initiated at a dosage of 120 mcg/kg taken orally once daily. However, this dosing regimen did not align with the proposed product monograph, which recommends dose escalation for lack of response. In the event a patient was unable to tolerate the odevixibat dose of 120 mcg/kg/day after a minimum of 1 week, for reasons other than new liver findings or severe diarrhea, the dose could be down-titrated to 40 mcg/kg/day. Patients who were down-titrated could return to the higher dose as soon as the investigator considered it appropriate. More than 1 upward-dose titration (from 40 mcg/kg/day directly to 120 mcg/kg/day) for the same event was not recommended.
The study protocol was amended on December 21, 2021, to include a starting dose of 40 mcg/kg/day with the possibility to escalate to 120 mcg/kg/day after 12 weeks if there is no improvement in pruritus, based on investigator judgment.
Permitted concomitant therapies were the same as what was described for the PEDFIC 1 study.
Outcomes
The outcomes assessed in the PEDFIC 2 study were similar to PEDFIC 1 (Table 19). The time point for analysis for this interim study is after 24 weeks of treatment; the final analyses will be based on the week 72 time point. This report will focus only on the relevant PEDFIC 2 outcomes that align with those from the PEDFIC 1 study.
Table 19: Outcomes Assessed in the PEDFIC 2 Study
Type | End points |
|---|---|
Primary | Change from baseline in serum bile acid after 24 (or 72) weeks of treatment. |
Secondary |
|
Exploratory |
|
ALT = alanine aminotransferase; APRI = AST to platelet ratio index; AST = aspartate aminotransferase; FIB-4 = Fibrosis-4 Index for Liver Fibrosis; GGT = gamma-glutamyl transferase; GIC = Global Impression of Change; GIS = Global Impression of Symptoms; INR = international normalized ratio; MELD = model for end-stage liver disease; p-C4 = plasma 7 alpha-hydroxy-4-cholesten-3-one concentration; PedsQL = Pediatric Quality of Life Inventory; PELD = pediatric end-stage liver disease; ObsRO = observer-reported outcome; PRO = patient-reported outcome.
Source: PEDFIC 2 interim Clinical Study Report (2022).25
Statistical Analysis
All analyses for this report were conducted on the full analysis set, defined as all patients who received at least 1 dose of the study drug in the PEDFIC 2 study.
Descriptive statistics were used unless otherwise specified. Data were summarized by cohort or treatment subgroup and overall in tabular format. Continuous variables were summarized using descriptive statistics, including the number of patients with nonmissing values (n), mean, median, SD or SE, minimum, and maximum. For categorical variables, summaries include counts of patients (frequencies) and percentages. Descriptive summaries of change from baseline in categorical variables were provided using shift tables, as applicable.
No imputations were conducted for missing data. Any assessments after intercurrent events (death or initiation of rescue treatments such as biliary diversion surgery or liver transplant) or follow-up assessments (≥ the last dose day plus 15 days) are excluded from analysis. For the analysis of the eDiary data, the data after premature treatment discontinuation (i.e., the last dose of study treatment) are excluded from analysis.
Results
Baseline Characteristics
The baseline characteristics in the PEDFIC 2 study are summarized in Table 20, excluding || patients enrolled with benign recurrent intrahepatic cholestasis (BRIC) (due to the small number of patients with BRIC enrolled at the time of data cut-off, no results are presented for these patients). The median age at study entry was ||| ||||| ||| |||||| |||| ||| || |||| |||||| |||| |||||||| |||| ||||| ||||||| |||| ||||||| |||||||| |||||||||||| || |||| ||||||| ||| ||||| |||||| ||||| ||||| ||| |||| |||||| ||| |||| |||||| |||||||| || ||||||||| |||||||| ||||| ||||| ||| ||| |||||| || |||||||| |||| |||||||| |||||||||||| ||| || ||||||| || ||||| || |||||||| ||||| ||||| |||||||| |||| ||||||| || || |||||||| || |||| ||||||| ||| |||||||| || |||||||| ||||||| |||| ||||||||| |||| |||||| |||||||||| || ||||| ||||| |||| ||||| || |||| ||| ||||| || ||||||||||| |||||| |||||||| ||||||| ||| ||||| ||||||| ||||||||| |||||||.
Patient Disposition
Patient disposition at the time of the data cut-off for the PEDFIC 2 study is summarized in Table 21. A total of ||| patients were enrolled in the PEDFIC 2 study as of the data cut-off of July 31, 2022; all patients had received treatment as of the data cut-off (|||||||||| || |||||| |||||| |||| |||| |||||| || || |||||||| || |||||| || ||| |||||||| |||||||||| ||||||||| || ||||| |||| ||| ||||| ||| ||||||||| ||||| || ||||| |||||||| ||| |||||||||| |||||||| ||| |||||| ||||||| |||||||| |||||||||||||| || ||||| |||| |||| |||||||||| |||||||| |||| |||||| || ||| || |||||||| || ||||||||| ||| |||||| |||| |||| |||||| || || ||| |||||||||| |||| ||||||| |||| |||||||||| ||| || ||| |||||||| |||||||| ||||| || || ||| ||| |||||||| ||| |||||||||| |||| |||||||.
As of the data cut-off date, of the ||| |||||||| || ||| |||||| || |||||||| |||||||||| || |||||||| || ||||||||| || |||||||| || |||||| || ||| ||||||||| ||| || |||| ||||||||| ||||||| || |||||||| ||||||||||||||||| || ||||||||| || |||||||| || |||||| || |||| |||||||||| ||| ||| || |||||||| ||| || ||||| ||||||||| |||||| ||| || |||||||| |||||||||| || |||||||| || ||||||||| || |||||||| || |||||| || ||| |||||||||||| ||||||||| ||||| || |||| ||| ||||| ||| || |||||||| ||| ||||||||| || ||||| || |||||||||| || |||||||| ||||||| ||| |||||||| ||||||||| |||||| |||||| |||| || || ||| |||| |||||| |||||||||| || || |||||||| |||||||||| || |||||||| ||| ||||| |||||||||| ||||||||| |||||| ||| || |||||||| ||| |||| |.
Exposure to Study Treatments
Study Treatments
Median overall duration of exposure to odevixibat 120 mcg/kg/day in the PEDFIC 2 study was 80.9 weeks and ranged from 4.3 weeks to 189.3 weeks at the time of the data cut-off (Table 22).
Median duration of exposure was approximately |||| ||| ||| ||| weeks in patients who had received 40 mcg/kg/day, 120 mcg/kg/day, or placebo in the PEDFIC 1 study, respectively. In cohort 2, which started enrolment approximately 1 year after the first patient in cohort 1 was rolled over to PEDFIC 2, the median exposure was |||| weeks.
Overall, adherence with daily dosing of odevixibat was high, with a median overall adherence of ||||| || |||||||||| |||| ||| |||||| ||| ||||| || |||||||||| |||| ||| |||| |||||| ||||. The rate of adherence was similar across study groups in cohort 1 and cohort 2.
Table 20: Baseline Characteristics in the PEDFIC 2 Study (Full Analysis Set)
Characteristic | Odevixibat 120 mcg/kg, once-daily dosing | ||||||
|---|---|---|---|---|---|---|---|
Cohort 1a | Cohort 2 (||||)b | Cohort 2 + placeboc (||||) | Overall (||||) | ||||
Odevixibat 40 mcg/kg (||||) | Odevixibat 120 mcg/kg (||||) | Odevixibat All doses (||||) | Placebo (||||) | ||||
Demographic | |||||||
Age, years | |||||||
Mean (SD) | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||||||||| | ||| ||||| |
Median (range) | ||| ||||| ||||| | ||| ||||| ||||| | ||| ||||| ||||| | ||| ||||| ||||| | ||| ||||| ||||| | ||| ||||| ||||| | ||| ||||| ||||| |
< 6 months | || | || | || | || | || | || | || |
6 months to 5 years, n (%) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
6 to 12 years, n (%) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
13 to 18 years, n (%) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
> 18 years | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Sex, n (%) | |||||||
Female | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Male | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Race, n (%) | |||||||
Asian | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Black or African American | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
White | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Other | || | || | || | || | || | || | || |
Anthropometric | |||||||
Height z scores | |||||||
Mean (SD) | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| |
Median (range) | |||| |||||| |||| | |||| |||||| |||| | |||| |||||| |||| | |||| |||||| | | |||| |||||| | | |||| |||||| |||| | |||| ||||| |||| |
Weight z scores | |||||||
Mean (SD) | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| |
Median (range) | |||| |||||| |||| | |||| |||||| |||| | |||| |||||| |||| | |||| |||||| | | |||| |||||| | | |||| |||||| |||| | |||||||||| |||| |
BMI z scores | |||||||
Mean (SD) | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | |||| ||||| | |||| ||||| | ||| ||||| |
Median (range) | ||| |||||| |||| | ||| |||||| |||| | ||| |||||| |||| | ||| |||||| | |||| |||||| | | ||| |||||| |||| | ||| |||||| |||| |
Disease | |||||||
PFIC type, n (%) | |||||||
Type 1 | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Type 2 | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Type 3 | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Other | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Years since diagnosis | |||||||
Mean (SD) | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| |
Median (range) | ||| ||||| |||| | ||| ||||| ||||| | ||| ||||| ||||| | ||| ||||| ||||| | ||| ||| ||||| | ||| ||| ||||| | ||| ||| ||||| |
Baseline medication use, n (%) | |||||||
UDCA | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Rifampicin | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
UDCA and/or rifampicin | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Patients with prior biliary diversion, n (%) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Hepatic and renal function | |||||||
Serum bile acid, µmol/L | |||||||
Mean (SE) | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | || |
Median (range) | |||| ||||| |||||| | ||||| ||||| |||||| | ||||| ||||| |||||| | ||||| |||||| || | ||||| |||||| | | ||||| |||||| |||||| | || |
ALT, IU/L | |||||||
Mean (SD) | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| |
Median (range) | |||| ||||| |||||| | |||| |||||| |||||| | |||| ||||| |||||| | |||| |||||| | | |||| |||||| | | |||| |||||| |||||| | |||| ||||| |||||| |
AST, IU/L | |||||||
Mean (SD) | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| |
Median (range) | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| | | |||| |||||| | | |||| |||||| |||||| | |||| |||||| || |
Total bilirubin, µmol/L | |||||||
Mean (SD) | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
Median (range) | ||| ||||| |||||| | |||| ||||| |||||| | |||| ||||| |||||| | |||| ||||| || | |||| ||||| ||| | |||| ||||| ||||||| | |||| ||||| ||| |
Child-Pugh classification, n (%) | |||||||
A (mild) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
B (moderate) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
C (severe) | || | || | || | || | || | || | || |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; BMI = body mass index; BRIC = benign recurrent intrahepatic cholestasis; GGT = gamma-glutamyl transferase; PFIC = progressive familial intrahepatic cholestasis; SD = standard deviation; SE = standard error; UDCA = ursodeoxycholic acid.
aFor patients in cohort 1, the dose indicated is the dose administered during their participation in the PEDFIC 1 study.
bExcluding || patients with BRIC who were enrolled in cohort 2.
cCohort 2 (excluding patients with BRIC) + placebo refers to patients enrolled in cohort 2 plus the patients who were assigned to placebo during their participation in the PEDFIC 1 study.
Source: PEDFIC 2 interim Clinical Study Report (2022).25
Concomitant Medications and Co-Interventions
All patients took at least 1 concomitant medication during the treatment period. Most patients received concomitant medications for the treatment of pruritus and for vitamin supplementation. Generally, the types and use of these medications were similar across the study groups, with the exception that a |||||| |||||||||| || |||| || ||||||||| ||||| |||||||| |||||||| ||| ||| || |||||||||| ||||||||| |||| || ||||| ||||||||||| || ||||| ||| ||||| |||||||||||||| |||||||||||| |||| || |||| compared with patients who received odevixibat in the PEDFIC 1 study.
|||| |||||||| ||||||||| ||||||||| |||| |||||||||| |||||| ||| ||||||||| ||||||.
Efficacy
Serum Bile Acid
Median changes (range) in sBAs levels from the PEDFIC 2 study at baseline to week 22 and 24 were ||| |||||||| |||||| ||||||| || |||||||| ||| ||||||||||| || |||||||||| || |||||| || ||| |||| ||||||| ||||| ||||||| || |||||||| ||| ||| |||||||| ||| ||||||||||| ||| |||||||| ||| ||| |||||||| ||||||| || |||||| || |||||| ||||||| ||||||| || |||| ||||| ||||||||| ||| ||||| || ||||||||| |||| |||||||||| ||| |||||||||| ||| |||| |||||||| |||||| ||||||| ||| ||| |||||||| || |||||||| |||| |||||||| |||||| ||||||.
Median changes (range) in sBAs levels from the PEDFIC 2 study at baseline to week 70 and 72 were ||| ||||||| |||||| ||||||| || |||||||| ||| ||||||||||| || |||||||||| || |||||| || ||| ||||| |||||||| |||||| ||||||| || |||||||| ||| ||| |||||||| ||| ||||||||||| ||| |||||||| ||| ||| |||||||| ||||||| || |||||| || |||||| ||||||| ||||||| || |||| ||||| ||||||||| ||| ||||| || ||||||||| |||| |||||||||| ||| |||||||||| ||| |||| |||||||| |||||| ||||||| ||| |||.
Table 21: Patient Disposition in the PEDFIC 2 Study
Disposition category n (%) | Odevixibat 120 mcg/kg, once-daily dosing | ||||||
|---|---|---|---|---|---|---|---|
Cohort 1a | Cohort 2b | Cohort 2 + placeboc | Overall | ||||
Odevixibat 40 mcg/kg | Odevixibat 120 mcg/kg | Odevixibat All doses | Placebo | ||||
Screened | || | || | || | || | ||| | || | || |
Enrolled | || | || | || | || | || | || | ||| |
Dosed | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | ||| ||||| |
72-week treatment period | |||||||
Completed treatment | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Ongoing on treatmentd | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | ||| ||||| |
Discontinued treatment early | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | ||| ||||| |
Primary reason for treatment discontinuation | |||||||
Adverse event | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | ||| ||||| |
Withdrawal of consent or assent | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | ||| ||||| |
Physician decision | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | ||| ||||| |
Other | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | ||| ||||| |
Optional extension period | |||||||
Did not enter | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | ||| ||||| |
Entered | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
Ongoingd | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
BRIC = benign recurrent intrahepatic cholestasis.
Note: Cohort 1 patients entered from the PEDFIC 1 study and therefore did not undergo screening. ||||||| ||||| ||| ||| ||||| ||| |||||||||||| ||||||||| || ||| || |||| ||||||||| |||||| ||| |||| ||||| ||||||| || ||| ||||| || || |||.
aFor patients in cohort 1, the dose indicated is the dose administered during their participation in the PEDFIC 1 study.
bCohort 2 enrolled 2 patients with BRIC; they are currently ongoing in the 72-week treatment period.
cCohort 2 (excluding patients with BRIC) plus placebo refers to the patients enrolled in cohort 2 plus the patients who were assigned to placebo during their participation in the PEDFIC 1 study.
dContinuing on treatment and in the study as of the data cut-off date of July 31, 2022.
e||||||| ||||| ||| ||||||||| ||| ||||||| ||||||||| ||||||| ||||||| ||| ||||||| || |||||||| || ||||||| ||||||| ||||| ||| |||| ||| ||| ||||||||||||||| ||||| ||| ||||||||| ||| ||||||| ||||||||| ||||||| ||||||| ||| ||||||| ||||||||| ||||| ||| ||||||||| ||| ||||||| ||||||||| ||||||| ||||||| ||| ||||||| ||||||||| ||||| ||| ||||||||| ||| ||||||| ||||||||| ||||||| ||||||| ||| ||||||| ||||||||| ||||| ||| ||||||||| ||| ||||||| ||||||||| ||||||| ||||||| ||| ||||||| ||.
Source: PEDFIC 2 interim Clinical Study Report (2022).25

Table 22: Exposure to Study Drug in the PEDFIC 2 Study (Full Analysis Set)
Exposure | Odevixibat 120 mcg/kg, once-daily dosing | ||||||
|---|---|---|---|---|---|---|---|
Cohort 1a | Cohort 2 (||||)b | Cohort 2 + placeboc (||||) | Overall (||||) | ||||
Odevixibat 40 mcg/kg (||||) | Odevixibat 120 mcg/kg (||||) | Odevixibat All doses (||||) | Placebo (||||) | ||||
Duration (weeks), mean (SD) | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| |
Duration (weeks), median (range) | ||||| |||||| |||||| | ||||| |||||| |||||| | ||||| |||||| |||||| | ||||| |||||| |||||| | |||| ||||| |||||| | |||| ||||| |||||| | |||| ||||| |||||| |
Adherence by eDiary (%), mean (SD) | |||| |||||| | |||| |||||| | |||| |||||| | |||||||||| | |||| |||||| | ||||||||||| | ||||||||||| |
Adherence by eDiary (%), median (range) | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| | |||| ||||| |||||| | |||| ||||| |||||| | |||| ||||| |||||| |
Adherence by case report form (%), mean (SD) | ||||| |||||| | |||| |||||| | |||| |||||| | ||||| |||||| | |||| |||||| | ||||||||||| | ||||||||||| |
Adherence by case report form (%), median (range) | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| | |||| |||||| |||||| |
BRIC = benign recurrent intrahepatic cholestasis; SD = standard deviation.
aFor patients in cohort 1, the dose indicated is the dose administered during their participation in the PEDFIC 1 study.
bExcluding | patients enrolled with BRIC.
cCohort 2 (excluding patients with BRIC) + placebo refers to patients enrolled in cohort 2 plus the patients who were assigned to placebo during their participation in the PEDFIC 1 study.
Source: PEDFIC 2 interim Clinical Study Report (2022).25
Table 23: Summary of Change in Serum Bile Acids After 24 Weeks and 72 Weeks of Treatment (Full Analysis Set)
Detail | Odevixibat 120 mcg/kg, once-daily dosing | |||||
|---|---|---|---|---|---|---|
Cohort 1a | Cohort 2 (||||) | Cohort 2 + placebob (||||) | ||||
Odevixibat 40 mcg/kg (||||) | Odevixibat 120 mcg/kg (||||) | Odevixibat all doses (||||) | Placebo (||||) | |||
Baseline,c n | || | || | || | || | || | || |
Mean (SE) (µmol/L) | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| |
Median (range) (µmol/L) | |||| ||||| |||||| | ||||| ||||| |||||| | ||||| ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| |
Week 22 and 24, n | || | || | || | || | || | || |
Mean (SE) (µmol/L) | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| |
Median (range) (µmol/L) | |||| ||||| |||||| | ||||| ||||| |||||| | |||| ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| |
Change from baseline, n | || | || | || | || | || | || |
Mean (SE) (µmol/L) | ||||| |||||| | ||||| |||||| | ||||| ||||| | |||||| |||||| | |||||||||||| | ||||| |||||| |
Median (range) (µmol/L) | |||| |||||||| |||||| | ||||| ||||||| ||||| | ||||| |||||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||| |
% change from baseline, n | || | || | || | || | || | || |
Mean (SE) (µmol/L) | |||| |||||| | ||||| |||||| | |||| |||||| | |||| |||||| | |||||||||||| | ||||| |||||| |
Median (range) (µmol/L) | ||||| |||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| |
Week 70 and 72, n | || | || | || | || | || | || |
Mean (SE) (µmol/L) | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| |
Median (range) (µmol/L) | |||| ||||| ||||| | |||| ||||| |||||| | |||| ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||| | |
Change from baseline, n | || | || | || | || | || | || |
Mean (SE) (µmol/L) | |||| |||||| | |||| |||||| | ||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| |
Median (range) (µmol/L) | ||| ||||||| |||||| | ||||| |||||||| |||||| | |||| |||||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| ||||| | |
% change from baseline, n | || | || | || | || | || | || |
Mean (SE) (µmol/L) | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| |
Median (range) (µmol/L) | ||||| |||||| | ||||| ||||||| ||||||| | |||| ||||||| ||||||| | ||||| |||||| | ||||| |||||| | ||||| ||||||| | |
BRIC = benign recurrent intrahepatic cholestasis; SE = standard error.
aFor patients in cohort 1, the dose indicated is the dose administered during their participation in the PEDFIC 1 study.
bCohort 2 (excluding patients with BRIC) + placebo refers to patients enrolled in cohort 2 plus patients who were assigned to placebo during their participation in the PEDFIC 1 study.
cBaseline for the PEDFIC 2 study; end of treatment for the PEDFIC 1 study.
Source: PEDFIC 2 interim Clinical Study Report (2022).25
Pruritus Assessments
The effect of treatment with odevixibat 120 mcg/kg/day on pruritus severity over 72 weeks in all study groups in cohort 1 and in cohort 2 is summarized in Table 24 and Figure 5.
Among patients who had received active treatment in the PEDFIC 1 study and those who were treatment-naive at study entry, the median (range) proportion of positive pruritus assessments was ||||| ||| |||||| ||||| || ||||| || ||||||||| ||| ||||| ||| |||||| ||||| || ||||| || ||| |||||||||| || |||||| || ||| |||||| ||||||| |||||||||| || |||||||| |||||||| ||||||||||| ||| |||||||| ||| ||| |||||||| || |||||||||| || ||||||||| |||||||||||| || ||| |||||||||| || ||||| ||||||||| ||||| ||| |||||| ||||| || |||||| ||||| ||| |||||||| ||||||||| |||||||||| |||| ||||||| || |||||| ||| |||||||||| || |||||| || ||| |||||| ||||||| |||||||||| || |||||||| |||||||| ||||||||||| || ||| ||||||| ||||| ||| ||||| |||||| |||||| |||| ||| ||||||| ||||||||| |||||| ||| ||||| |||||| |||||| |||| ||| ||||||| ||||||||| ||||||| || |||||| || ||| |||||| ||||||| |||||||||| || |||||||| |||||||| ||||||||||| || ||| ||||||| ||||| ||| ||||| |||| ||| ||||||| ||||||||| |||||| ||| ||||| |||||| |||||| |||| ||| ||||||.
Data were consistent when the analysis was performed based on morning and evening scores separately.
Table 24: Summary of Proportion of Positive Pruritus Assessments at the Patient Level (Combined Morning and Evening PRUCISION ObsRO Scores) Over the 24-Week Treatment Period (Full Analysis Set)
Detail | Odevixibat 120 mcg/kg, once-daily dosing | |||||
|---|---|---|---|---|---|---|
Cohort 1a | Cohort 2 ||||| | Cohort 2 + placebob ||||| | ||||
Odevixibat 40 mcg/kg ||||| | Odevixibat 120 mcg/kg ||||| | Odevixibat All doses ||||| | Placebo ||||| | |||
Week 0 to 24 | || | || | || | || | || | || |
n | || | || | || | || | || | || |
Mean (SE) | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| |
Median (range) | |||| ||| ||||| | |||| ||| ||||| | |||| ||| ||||| | |||| ||||| ||||| | |||| ||||| ||||| | |||| ||||| ||||| |
Week 0 to 72 | ||||||
n | || | || | || | || | || | || |
Mean (SE) | |||| ||||| | |||| ||||| | |||| ||||| | |||| |||||| | |||| ||||| | |||| ||||| |
Median (range) | |||| ||| ||||| | |||| ||| ||||| | |||| ||| ||||| | |||| ||||| ||||| | |||| ||||| ||||| | |||| ||||| ||||| |
BRIC = benign recurrent intrahepatic cholestasis; ObsRO = observer-reported outcome; SE = standard error.
aFor patients in cohort 1, the dose indicated is the dose administered during their participation in the PEDFIC 1 study.
bCohort 2 (excluding patients with BRIC) + placebo refers to patients enrolled in cohort 2 plus patients who were assigned to placebo during their participation in the PEDFIC 1 study.
Source: PEDFIC 2 interim Clinical Study Report (2022).25

Sleep Parameters
Treatment with odevixibat to improve patient’s sleep based on observer-reported information was consistent with what was observed in pruritus. For patients who had previously received odevixibat during the PEDFIC 1 study, mean changes from baseline to weeks 21 to 24 in patients who had received 40 mcg/kg/day versus those who had received 120 mcg/kg/day in the PEDFIC 1 study was || ||| ||||| ||| |||||| || ||||||||||| ||| ||||||||| ||||| ||||||||| ||| ||| |||||||| ||||||| || ||||| ||||||||| ||| ||||||||||||||| |||| ||||||| |||| |||||||| || ||||| ||||| |||| || ||| || ||| |||||| || ||||||||||.
Growth Parameters
Changes in height and weight scores were noted during treatment with odevixibat 120 mcg/kg/day (Figure 6 and Figure 7).
For patients in cohort 1 who had previously received odevixibat in the PEDFIC 1 study, the mean (SE) change from baseline to week 24 in height z score was |||| ||||||||| |||| ||||||| ||| ||| |||||||| ||| |||||||||| |||||| ||||||||| ||| ||||||| ||| ||| |||||||| || |||||||||| |||||| |||||||||| |||| |||| ||||||| |||| |||||||| || |||| || || |||||||||||| |||| ||||| |||||||| ||| ||||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| || |||||||||| ||| ||| ||||||||||| ||||||||||||| |||| |||| ||||||| |||| |||||||| || |||| || || ||||||||| |||| ||||| |||||||| ||| ||||| ||||||||| ||| ||||| ||||||| ||||||||||||.
For patients in cohort 1 who had received placebo in the PEDFIC 1 study and in patients in cohort 2, the mean (SE) changes in height z score were ||||| ||||||||| ||| ||||| ||||||||| ||||||||||||| |||| |||| ||||||| |||| |||||||| || |||| || || |||||||||||| |||| ||||| |||||||| ||| ||||| |||||||| ||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || |||||||| ||| |||||||| || |||||| || ||||||||||||| ||| |||| |||| ||||||| |||| |||||||| || ||||||||| |||| ||||| |||||||| ||| ||||| ||||||||| ||||||||||||.
For patients in cohort 1 who had previously received odevixibat in the PEDFIC 1 study, the mean (SE) change from baseline to week 72 in height z score was ||||| ||||||||| |||| ||||||| ||| ||| |||||||| ||| |||||||||| |||||| ||||||||| ||| ||||||| ||| ||| |||||||| || |||||||||| |||||| |||||||||| |||| |||| ||||||| |||| |||||||| || |||| || || ||||||||||| |||| ||||| |||||||| ||| ||||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| || |||||||||| ||| ||| ||||||||||| ||||||||||||| |||| |||| ||||||| |||| |||||||| || |||| || || ||||||||| |||| ||||| |||||||| ||| ||||| ||||||||| ||| ||||| ||||||| ||||||||||||.
For patients in cohort 1 who had received placebo in the PEDFIC 1 study and in patients in cohort 2, mean (SE) changes in height z score were ||||| |||||||| ||| ||||| ||||||||| ||||||||||||| |||| |||| ||||||| |||| |||||||| || |||| || || |||||||||||| |||| ||||| |||||||| ||| ||||| |||||||| ||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || ||||||||| ||| |||||||| || |||||| || ||||||||||||| ||| |||| |||| ||||||| |||| |||||||| || |||||||| |||| ||||| |||||||| ||| ||||| ||||||||| ||||||||||||.


Surgical Intervention
|||||||| |||||||| || |||| ||||| ||||||||| |||||||| |||||||||||| |||||||||||||||| ||| ||||||||| ||||||| ||||||||| ||||||| ||| || |||||||| ||| ||||||||| ||||| ||||||||||||||| ||||||||| ||||| |||||||||| ||| ||| ||||| ||||||| |||||| |||||||||| || ||||| || |||||||||||||||||| ||| ||||| ||||||| ||||||| ||||| || ||| || ||||||||||| ||| ||||| ||||||| ||||||| ||||| || ||| ||| |||| || ||| || |||||||| ||| ||||| ||||||| ||||| |||||||||| ||| || |||| ||||||||| ||||||.
Laboratory Parameters
For patients in cohort 1 who had received placebo, mean change from baseline to week 24 was ||||| |||||| || ||||| |||||||||| ||| ||||||||||||||| |||| |||||| ||| ||||| |||||| || ||||| |||||||||.
Health-Related Quality of Life
A summary of the total score for the PedsQL Parent Report and Family Impact Module is presented in Table 25.
For patients who had previously received odevixibat in the PEDFIC 1 study, mean (SE) change of total scores on the PedsQL Parent Report from baseline to week 24 in the PEDFIC 2 study for patients who had received 40 mcg/kg/day and 120 mcg/kg/day were |||| ||||||| ||| |||| |||||||| ||||||||||||.
For patients in cohort 1 who received odevixibat in the PEDFIC 1 study, the mean (SE) changes from baseline to week 24 were |||| ||||||| ||| |||| ||||||| ||| |||||||| ||| ||| |||||||||| |||||||| ||| || |||||||||| ||| ||| |||||||||| ||||| ||||||||||||| ||| |||||||| || ||||||||| |||||||| |||||||| ||| |||| |||| |||||| ||| ||||| ||||||| ||| ||| |||||||||||||| ||| ||||| ||| |||| |||| |||||| ||| ||||| |||||||.
Table 25: Summary of Change From Baseline to Week 24 and Week 72 in PedsQL Total Score (Full Analysis Set)
Detail | Odevixibat 120 mcg/kg, once-daily dosing (|||||)a,b | |||||
|---|---|---|---|---|---|---|
Cohort 1a | Cohort 2 (||||||) | Cohort 2 + placebob (||||||) | ||||
Odevixibat 40 mcg/kg (||||||) | Odevixibat 120 mcg/kg (||||||) | Odevixibat All doses (||||||) | Placebo (||||||) | |||
PedsQL Parent Report, mean (SE) | ||||||
N (baseline) | || | || | || | || | || | || |
Baseline | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
N (week 24) | || | || | || | || | || | || |
Mean change to week 24 | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
N (week 72) | || | || | || | || | || | || |
Mean change to week 72 | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
PedsQL Family Impact Module, mean (SE) | ||||||
N (baseline) | || | || | || | || | || | || |
Baseline | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
N (week 24) | || | || | || | || | || | || |
Mean change to week 24 | |||| ||||||| | |||| ||||||| | |||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
N (week 72) | || | || | || | || | || | || |
Mean change to week 72 | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | |||| ||||||| | ||||| ||||||| | |||| ||||||| |
BRIC = benign recurrent intrahepatic cholestasis; PedsQL = Pediatric Quality of Life Inventory; SE = standard error.
Note: Baseline was the last available assessment before the first dose of the study drug.
aFor patients in cohort 1, the dose indicated is the dose administered during their participation in the PEDFIC 1 study.
bCohort 2 (excluding patients with BRIC) + placebo refers to patients enrolled in cohort 2 plus patients who were assigned to placebo during their participation in the PEDFIC 1 study.
Source: PEDFIC 2 interim Clinical Study Report (2022).25
Results in Additional PFIC Subtypes
||||| |||||| |||||||| |||| ||||| |||| |||||||| || |||||| || ||||| |||| |||||| |||||| || |||||| ||| |||| ||| ||| ||||| |||| |||||||||| ||||||| |||||||||||| ||| ||||||||| |||||||||||||||||||| ||||||||||| || |||||||| ||| ||||||||| || ||||| |||| |||| ||||||| |||| || ||||||||||| ||| ||| ||||| |||| ||||| ||||||||| |||||||||| || |||||||| ||||||||||||| ||| |||||| ||||| ||||||||| |||| |||||||||.
NLS in Odevixibat sBA Responders
NLS in patients whose disease responded to odevixibat in the PEDFIC studies was determined in a pooled analysis from the patients’ first dose of odevixibat to a cut-off date of January 31, 2022.26 Given that a clinically meaningful reduction in sBA levels likely includes reductions smaller than the strict 70% threshold, additional analyses were performed to evaluate NLS in patients classified as partial sBA responders (defined as patients with a reduction in sBA of ≥ 30% to < 70%). || || |||||||| |||| || ||||| || ||||| || |||||||||| ||||||||| ||||| ||||||||| |||||||| || |||||| || ||||||||| |||||| || || ||||| |||| ||| |||||||||| ||| ||| |||||||| ||||||||||| || ||||| |||| ||||||| ||| |||||||||| ||||| ||| |||| |||||||||| ||| || ||||| |||| ||||||||||||||| |||| || ||| || ||| |||||||| ||| |||||||||| ||||||| ||||||||||||| ||| || ||||||| ||| |||||||||| ||||||| |||||||||| |||||| ||| || ||| |||||||||| ||| ||||| |||||||||||. Figure 8 || ||| |||||||| ||| ||||||||| ||||| ||||||||||||||| ||||||||||| ||| |||||||||| || ||| |||| |||| ||| |||| ||||||||| ||| |||| |||||||||| || ||| ||||||| |||| ||| ||| ||| || ||| |||||||||| ||| ||||||| |||||||||| |||||||||||| |||||||| |||| ||| |||| |||||||| || ||| || ||| |||||||||||||| |||||||||||.

Harms
A summary of harms results from the PEDFIC 2 study is presented in Table 26.
Overall, ||| ||||| || ||| ||| |||||||| |||||||||| |||||||| |||| ||||| ||||||||||| || ||||||||| |||||| ||| |||||| ||| |||| |||||||| |||||||| ||||| ||||| |||||||| |||| ||||| ||||||||||| ||||| ||||||||| |||||| ||||| |||||| ||| ||||||| ||| ||||| ||||||||| ||||||||| ||||| ||||| || |||||||| ||| ||||||||| || ||||| |||||||| |||||||| |||||| ||| ||||||| |||||| ||| ||||||||| |||||| || |||||| ||||||||||| || ||||| || ||| ||| |||||||| |||||||||| |||||||| |||| ||||| |||||||||||||||| || || ||||||||| |||||||| ||||| |||||||||||||| |||||||| ||| ||| |||||||||| |||||||| |||||||||| || |||||| ||||||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || ||||||||| || ||||| |||||||| || |||||| || ||||| ||||||| |||| |||| |||||||| |||||||| |||| ||||||||| ||||||| |||||||| || ||||||| ||| |||||||| |||||||||| || |||||||||| || |||||||||||||| || |||||| || ||| ||||||||||| ||||| |||||||| |||| || |||||| ||| ||| ||||| |||| || ||||||||| |||| |||| |||||||| || |||||| |||||||| ||||||||||||||||||| |||||||| |||||||||||| |||||| |||| ||| || ||||||||| |||||||||||||||| ||||||||||||||| ||||||| ||| |||||||| || |||||||||| || |||||| |||||||| |||||||| ||| |||||||| ||||||| || |||||| || ||||||| |||||||| || |||||| ||||||||||| || |||||||||| ||||||||||| |||||||| |||||||| |||||| ||| |||||| ||||| ||||||| ||||| |||| |||||||| || || ||||||| |||||||| ||| ||||||||||| ||||||| ||||||| ||||||||| || ||||||| |||||||| ||| ||| |||||||||| |||||||| |||||||||| || |||||| || || ||||||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || |||||||| || ||||||| |||||||| || |||||| || || |||||||| |||||||| ||| ||||||||||||| |||||||||| ||||||||| || ||||||| |||||||| ||| ||| |||||||||| |||||||| |||||||||| || |||||| ||||||||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || ||||||||||||||| |||||||| ||||||| || || |||||||| |||||||| ||| ||||||||||||| |||||| ||||||||| || ||||||| |||||||| ||| ||| |||||||||| |||||||| |||||||||| || |||||| |||||||| |||||||| ||| ||| |||||||||| |||||||| ||||||| || ||||||||||||||| |||||||| || |||||.
No deaths occurred during the study.
Critical Appraisal
Internal Validity
The PEDFIC 2 study was limited by its open-label and noncomparative design. Since there is no comparator, it did not show the comparative benefit of odevixibat versus relevant comparators. Furthermore, the small sample size and large losses to follow-up in the PEDFIC 2 study led to difficulties in drawing any firm conclusions on the efficacy and safety of odevixibat, as the effect estimates were associated with wide variation. Due to its open-label and nonblinded nature, the absence of blinding can lead to assessor bias, and the patient and/or caregiver would be most likely in favour of the intervention, i.e., odevixibat, for efficacy outcomes. Moreover, the subjective outcomes (e.g., pruritus assessments at the patient level and individual assessments meeting the definition of a positive pruritus assessment at the patient level) are at risk of bias, regardless of blinding.
Table 26: Summary of Harms Results From the PEDFIC 2 Study (Full Analysis Set)
Harms | Odevixibat 120 mcg/kg, once-daily dosing (|||||)a,b | |||||
|---|---|---|---|---|---|---|
Cohort 1a | Cohort 2 (||||||) | Cohort 2 + placebob (||||||) | ||||
Odevixibat 40 mcg/kg (||||||) | Odevixibat 120 mcg/kg (||||||) | Odevixibat All doses (||||||) | Placebo (||||||) | |||
Most common AEs, n (%) | ||||||
≥ 1 AE | || ||||| | || ||||| | || ||||| | || |||||| | || |||||| | || |||||| |
Occurring in > 10% overall population | ||||||
Increased blood bilirubin | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Cough | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Pyrexia | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Upper respiratory tract infection | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Nasopharyngitis | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Diarrhea | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
SAEs, n (%) | ||||||
Patients with ≥ 1 SAE | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Patients who stopped treatment due to AEs, n (%) | ||||||
Patients who stopped | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Deaths, n (%) | ||||||
Patients who died | 0 | 0 | 0 | 0 | 0 | 0 |
AEs of special interest, n (%) | ||||||
Clinically significant diarrhea | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Adjudicated hepatic events | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Hepatobiliary disorders | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Liver-related TEAEs | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
AE = adverse event; BRIC = benign recurrent intrahepatic cholestasis; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aFor patients in cohort 1, the dose indicated is the dose administered during their participation in the PEDFIC 1 study.
bCohort 2 (excluding patients with BRIC) + placebo refers to patients enrolled in cohort 2 plus patients who were assigned to placebo during their participation in the PEDFIC 1 study.
Source: PEDFIC 2 interim Clinical Study Report (2022).25
PEDFIC 2 did not assess the lower-dose regimen, 40 mcg/kg/day, as a starting dose for its efficacy and safety in the long-term. There was an amendment to include a starting dose of 40 mcg/kg/day with the possibility of escalating the dose to 120 mcg/kg/day after 12 weeks if there is no improvement in pruritus; however, there was no data provided to assess the efficacy and safety for the lower starting dose in the long term study. Therefore, the rationale for selecting the optimal starting dose and titration strategy remained unclear.
External Validity
Because the patients from cohort 1 of the PEDFIC 2 study were originally from the pivotal PEDFIC 1 trial, and the population enrolled in cohort 2 of the PEDFIC 2 study had demographic and clinical characteristics that were consistent with their characteristics at entry into the PEDFIC 1 study, with some exceptions, it is reasonable to expect that the same limitations to generalizability are relevant to the open-label long-term safety extension phase. Moreover, there were other patients with different PFIC types included in the PEDFIC 2 trial, but the sample size for this subgroup was too small to draw any conclusion. Given the nature of the noncomparative study design, it is not possible to compare the effectiveness and tolerability of odevixibat with standard of care. In terms of the optimal starting dose and titration strategy of PEFIC 2, there is a high degree of uncertainty regarding generalizability due to unclear or unaddressed rationale, which was also not aligned with current clinical practice and the proposed product monograph. Due to the lack of evidence on the optimal starting dose and titration strategy, it is hard to draw a conclusion on the efficacy and safety of the lower starting dose in the long term.
Studies Addressing Gaps in the Systematic Review Evidence
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Table 27: Summary of Gaps in the Evidence
Gap in pivotal and RCT evidence | Studies that address the gaps | |
|---|---|---|
Study description | Summary of key results | |
There is a lack of longer-term data in the PEDFIC 1 study, as this study was limited to 24 weeks in duration. | The OvEC study was a matched cohort study comparing the clinical outcomes in odevixibat-treated patients from the PEDFIC 1 and PEDFIC 2 studies vs. data from a retrospective natural history study (NAPPED). | In the OvEC study, results comparing efficacy outcomes between the odevixibat cohort and the NAPPED cohort are as follows:
|
CI = confidence interval; DFS = surgical biliary diversion–free survival; EFS = event-free survival; HR = hazard ratio; NE = not estimable; NLS = native liver survival; OS = overall survival; OvEC = odevixibat versus external control; RCT = randomized controlled trial; vs. = versus.
Description of Studies
The OvEC study was conducted to compare clinical outcomes (SBD, liver transplant, death) in patients with PFIC without prior SBD who were treated with odevixibat in the PEDFIC 1 and PEDFIC 2 studies (N = 69) with external controls from matched NAPPED data (N = 80), a retrospective database investigating the natural history of PFIC.12,13
Study Design and Objectives
The PEDFIC 1 and PEDFIC 2 studies have been previously described in this report. The NAPPED study includes real-world data from 50 treatment centres in Europe, North America, Asia, and Australia.
The objectives for OvEC were to evaluate the effect of odevixibat on clinical outcomes in children with SBD-naive PFIC1 or PFIC2 participating in the PEDFIC 1 and PEDFIC 2 clinical studies compared with an external control cohort of SBD-naive children from the NAPPED study. The primary objective was to evaluate the effect of odevixibat on the first clinical event (death, liver transplant, or SBD) in children with PFIC1 or PFIC2; the primary end point was EFS. The OvEC study used IPTW, a PS matching method, in an attempt to reduce the potential for confounding when comparing the cohorts. A cohort of 69 odevixibat-treated patients was compared with 80 patients (controls) in the NAPPED study. The median study duration in the odevixibat cohort was 22.6 months (range, 1.9 to 39.2 months). The follow-up duration in the NAPPED cohort was truncated accordingly.
Figure 9: Schematic of the OvEC Cohort Populations
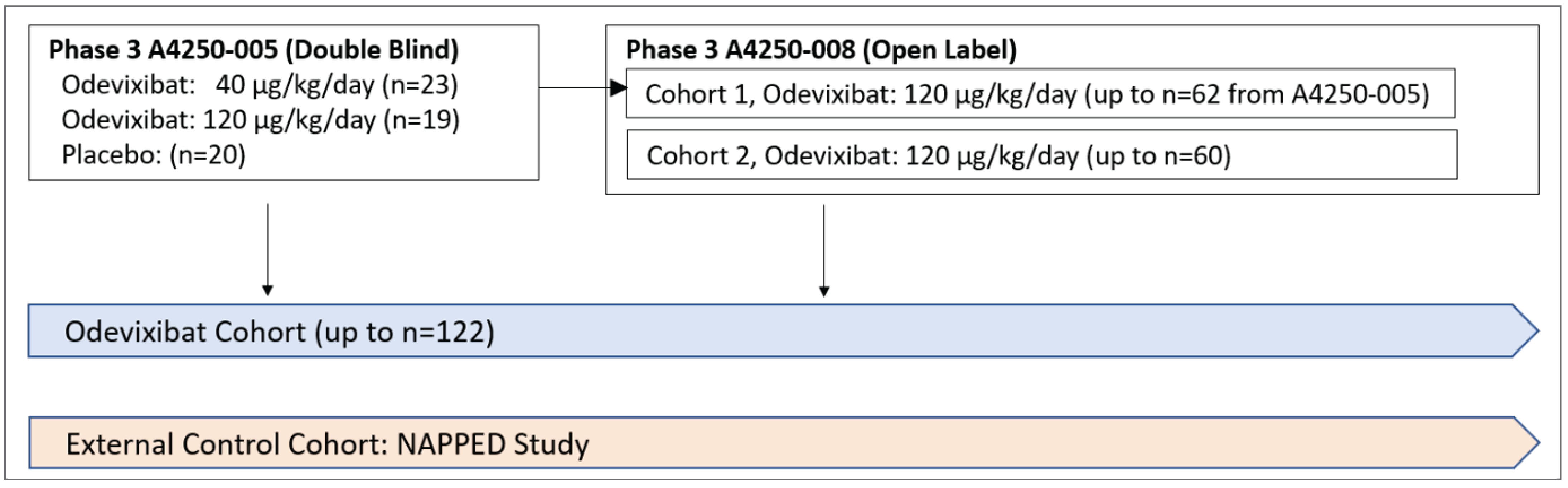
Note: A4250-005 = the PEDFIC 1 study;8 A4250=008 = the PEDFIC 2 study.25
Populations
Patients in the odevixibat cohort were selected from the PEDFIC 1 and PEDFIC 2 trials according to the following inclusion criteria:
Patients treated with odevixibat in the PEDFIC 1 study with at least 1 post odevixibat assessment and who did not have a prior SBD
OR
Patients treated with placebo in the PEDFIC 1 study who first received odevixibat in cohort 1 of the PEDFIC 2 study and had at least 1 post odevixibat assessment and who meet the following additional eligibility criteria:
had elevated sBA concentration (≥ 100 μmol/L, taken as the average of the last 2 samples at least 7 days but less than 57 days apart and before the first dose of odevixibat)
did not have a liver transplant or SBD before treatment with odevixibat
did not develop suspected or proven hepatocellular carcinoma in the PEDFIC 1 study
had a serum ALT level that is 10 times the ULN or less, or an ALT level of 50 IU/L or less (if ULN is unavailable) at the last visit within 6 months on or before the first dose of odevixibat
had a total bilirubin level of 10 times the ULN or less, or a total bilirubin of 20 µmol/L or less (if ULN is unavailable) at the last visit or within 6 months or before the first dose of odevixibat
OR
Patients treated with odevixibat in cohort 2 of the PEDFIC 2 study who had at least 1 post odevixibat assessment and who meet the following additional eligibility criteria:
aged 6 months to 18 years at the first dose of odevixibat
had a clinical diagnosis of PFIC1 or PFIC2, excluding pathologic variations of the ABCB11 gene that predict complete absence of the BSEP protein
did not have SBD before treatment with odevixibat.
Patients for the external cohort of SBD-naive children (NAPPED cohort) were selected from the NAPPED database according to the following inclusion criteria:
A male or female patient in the NAPPED study with a clinical diagnosis of PFIC1 or PFIC2 and not enrolled in the PEDFIC 1 or PEDFIC 2 studies.
Patient must have clinical genetic confirmation of PFIC1 or PFIC2 through identification of biallelic pathogenic variants in either the ATP8B1 or ABCB11 gene, excluding pathologic variations of the ABCB11 gene that predict complete absence of the BSEP protein.
The patient had at least 1 visit in the NAPPED study (the first of which becomes the day 1 visit for this cohort) and meets the following eligibility criteria:
aged 6 months to 18 years with a body weight above 5 kg on day 1
elevated sBA concentration, specifically measured to be 100 μmol/L or greater, taken as the average of the last 2 samples at least 7 days but less than 57 days apart after day 1; if only 1 sample is available, the sBA must be at least 100 μmol/L
does not have BRIC
did not undergo a liver transplant or SBD on or before day 1
did not have hepatocellular carcinoma on or before day 1
serum ALT level of 10 times the ULN or less, or an ALT of 50 IU/L or less (if ULN is unavailable) at last ALT assessment within 6 months on or before day 1
total bilirubin of 10 times the ULN or less, or a total bilirubin of 20 µmol/L or less (if ULN is unavailable) at last bilirubin assessment within 6 months on or before day 1
has at least 1 assessment after day 1.
Outcomes
The end points assessed in the OvEC study are presented in Table 28. The primary end point was EFS. Secondary end points included NLS, DFS, and OS.
Statistical Analysis
Sample Size
The sample size for the OvEC study was based on the number of available patients who met cohort eligibility.28 Sample size calculations were based on the log-rank statistic to test for a treatment difference in EFS between the 2 cohorts.
It was expected that there would be at least 68 SBD-naive patients in the odevixibat cohort and at least 75 SBD-naive patients in the external control cohort for the OvEC primary analysis. Additionally, power calculations for the primary analysis were based on the following assumptions:
Patients in the odevixibat cohort were enrolled at a uniform rate for a period of approximately 113 weeks and followed for approximately 72 weeks for the primary analysis. No loss to follow-up other than administrative censoring was assumed for the odevixibat cohort. EFS after day 1 was assumed to be exponentially distributed.
Patients in the external control cohort will be administratively censored at approximately 185 weeks to align their follow-up for clinical outcomes with the odevixibat cohort for the primary analysis. The 2-year EFS rate is expected to be approximately 70% but not higher than 80%. A 1-year loss-to-follow-up rate of 10% was assumed for the external control cohort. Loss to follow-up and EFS after day 1 were assumed to be exponentially distributed.
Under these assumptions, the analysis would have at least 93% power to detect an HR of 0.30 for EFS with a type I error rate of 0.05 when the 2-year EFS rate is 70% for the external control cohort. Power would be at least 79% when the 2-year EFS rate is 80% for the external control cohort.
Table 28: End Points in the OvEC Study
End points | Description |
|---|---|
Primary | EFS, defined as time from day 1 (primary analysis) and from birth (supportive analysis) to the first occurrence of any of the following events: death, liver transplant, SBD |
Secondary |
|
Exploratory |
|
ALT = alanine aminotransferase; APRI = aspartate aminotransferase to platelet ratio index; AST = aspartate aminotransferase; DFS = surgical biliary diversion–free survival; EFS = event-free survival; GGT = gamma-glutamyl transferase; NLS = native liver survival; OS = overall survival; SBD = surgical biliary diversion.
Source: Albireo Pharma data on file.11,27
Propensity Score and Matching Methods
The impact of selection bias and confounding (i.e., heterogeneity in effect modifiers and prognostic factors between the 2 cohorts) on estimates of treatment differences were minimized by the use of:
cohort eligibility criteria to induce a core set of common eligibility criteria on the 2 OvEC cohorts
IPTW PS matching methods to balance the 2 treatment cohorts with respect to important baseline covariates.28
The primary method to compare the 2 cohorts was based on using stabilized weights computed from the PS. To assess the robustness of results obtained using IPTW methods, matching techniques were used.
The PS is the probability of receiving the experimental treatment (odevixibat), conditional on a set of observed baseline covariates. Conditional on the PS, the distribution of measured baseline covariates is expected to be the same in both cohorts.
The following important baseline covariates were used to compute a PS model:
age (years) at day 1
age group at diagnosis (< 2 years of age; 2 to 4 years of age; 5 to 12 years of age; > 12 years of age; and unknown)
sex at birth
genetic severity–based PFIC type and BSEP genotype (PFIC1, PFIC2 with a BSEP1 genotype, and PFIC2 with a BSEP2 genotype)
sBAs (μmol/L) at baseline
height-for-age z score
weight-for-age z score
ALT, AST, total bilirubin, GGT
geographical region (North America, Europe, and rest of world).
Only baseline covariates with less than 15% missing data were considered eligible for the PS model. Logistic regression was used to compute a PS model for eligible patients in both cohorts with the terms for the eligible baseline covariates.
The PS model was used to compute the following for each patient: PS, IPTW, and stabilized weight.
Weighted analyses, based on stabilized weights, were used to compute an average treatment effect.
The following matching methods were used:
full matching based on minimizing the total absolute difference in the logit of the PS
full matching based on minimizing the total absolute difference in the logit of the PS with exact matching on genetic severity
full matching based on minimizing the total absolute difference in the logit of the PS with exact matching on age quantiles (defined from the odevixibat cohort)
1:1 matching based on minimizing the total absolute difference in the logit of the PS.
With full matching, patients in both cohorts are allocated to matched sets that consist of 1 treated patient and at least 1 control patient or 1 control patient and at least 1 treated patient. With 1:1 matching, all patients are assigned a weight of 1 (i.e., no weighting). However, the sponsor did not specify which matching method corresponds to the results generated.
Balance diagnostics will be produced before and after applying weights for each matching method. If the matched sets for a matching method are deemed inadequate, the PS model may be modified using predefined strategies.
Weighted analyses, based on weights obtained for each full matching method, were used to compute an average treatment effect. Unweighted analyses, for the 1:1 matching method, were used to compute an average treatment effect for the treated. Patients with matched identifiers from each matching method were used to compute the matching-method summaries.
Subgroups
Analyses of EFS, NLS, and DFS were conducted in the genetic severity subgroups and in the subgroup of patients born on or after the year 2000.
Subgroup analyses were to be conducted only for analyses based on IPTW methods.
Multiple Comparisons
Type I error is 5% for the primary efficacy analysis. No corrections were made for multiplicity in the secondary and exploratory analyses.
Analysis Sets
The full analysis set includes all patients in both cohorts. The includes all patients in both cohorts with nonmissing PSs.
The primary efficacy analysis was based on stabilized weights and performed in the evaluable analysis set for the cohorts. Sensitivity analyses were performed based on: different matching methods; time to event from birth; and the exclusion of patients in the NAPPED cohort with an event within 4 weeks, 8 weeks, or 12 weeks after day 1. The secondary end points (survival, NLS, and DFS) were analyzed using the same methods as the primary efficacy end point, EFS.
Results
Results comparing efficacy outcomes between the odevixibat cohort and NAPPED cohort are summarized in Table 29.
EFS: In total, 6 patients (9%) in the odevixibat cohort had an EFS event versus 44 patients (55%) in the NAPPED cohort. The weighted HR was 0.20 (95% CI, 0.09 to 0.45; P = 0.0016).
NLS: In total, 4 patients (6%) in the odevixibat cohort had an NLS event versus 21 patients (26%) in the NAPPED cohort. The weighted HR was 0.33 (95% CI, 0.11 to 1.03; P = 0.0900).
DFS: In total, 2 patients (3%) in the odevixibat cohort had a DFS event versus 31 patients (39%) in the NAPPED cohort. The weighted HR was 0.13 (95% CI, 0.04 to 0.39; P = 0.0023).
OS: In total, no patients died in the odevixibat cohort while 4 patients (5%) died in the NAPPED cohort. The weighted HR was 0 (95% CI, 0 to not estimable; P = 0.0845).
Results were consistent when different sensitivity analyses were performed.
Table 29: Survival Outcomes in Odevixibat-Treated Patients and External Controls in the OvEC Study
Detail | Odevixibat-treated cohort (N = 69) | NAPPED control cohort (N = 80) |
|---|---|---|
Event-free survival | ||
Events, n (%) | 6 (9%) | 44 (55%) |
P value | 0.0016 | — |
HR (95% CI) | 0.20 (0.09 to 0.45) | — |
Native liver survival | ||
Events, n (%) | 4 (6%) | 21 (26%) |
P value | 0.0900a | — |
HR (95% CI) | 0.33 (0.11 to 1.03) | — |
DFS | ||
Events, n (%) | 2 (3%) | 31 (39%) |
P value | 0.0023a | — |
HR (95% CI) | 0.13 (0.04 to 0.39) | — |
OS | ||
Events, n (%) | 0 (0%) | 4 (5%) |
P value | 0.0845a | — |
HR (95% CI) | 0 (0 to NE) | — |
CI = confidence interval; DFS = surgical biliary diversion–free survival; HR = hazard ratio; NE = not estimable; OS = overall survival; OvEC = odevixibat versus external control.
Note: The sample sizes and events are unweighted; the HRs and P values for odevixibat versus control cohorts are weighted.
aP values are not adjusted for multiplicity.
Source: Albireo data on file.11,27
Critical Appraisal
Internal Validity
Patients in the PEDFIC 1 and PEDFIC 2 studies were compared with the cohort from the NAPPED study using IPTW methods in an attempt to minimize the impact of confounding on the results. It should be noted that this method cannot control for the substantial differences resulting from the different study designs between the 2 cohorts (RCT versus retrospective registry review). Details of the cohort from the NAPPED study were limited; it is not clear how patients were selected into the cohort (i.e., potential for selection bias is unknown), their characteristics before weighting, or what treatments they received. Similarly, data-collection methods for the NAPPED cohort, how missing data were accounted for, the number of losses to follow-up, and outcome definitions have not been reported. The authors appropriately used eligibility criteria for the NAPPED study cohort that were considered similar to those used for the PEDFIC studies; however, the characteristics of patients at baseline and the overlap in covariates before weighting were not described. Thereafter, the primary method to compare the 2 cohorts was based on using stabilized weights computed from the PS model. To do so, a list of important baseline covariates was used; however, it is not clear how this list was derived. Consultation with the clinical experts indicated that some effect modifiers or prognostic factors were left out of the matching, such as measure of liver disease, degree of decompensation, and evidence of portal hypertension and cirrhosis. Additionally, factors with more than 15% missing data were excluded from the model, but it is not clear which these were. There is no evidence that covariate balance was assessed post matching. As a result, the likelihood of residual confounding is high. The treatments used among patients in the registry were not described, and the degree to which concomitant therapies contributed to the treatment effect is unknown. Aside from the primary outcome, there was no control for multiple comparisons; therefore, there is an increased risk of false-positive findings for DFS and OS. The sample size of both cohorts was small, and the analyses are based on few events, which renders the results unstable and further reduces confidence in the findings.
External Validity
Though IPTW methods were used to match the cohorts for analysis, the initial characteristics of patients in the NAPPED study were not described. The dosing used in the PEDFIC 1 and PEDFIC 2 studies did not align with the proposed product monograph for all patients, as some started on 120 mcg/kg/day, and others escalated to this dose despite responding to the lower dose. The treatments used among patients in the registry were not described; therefore, it is not clear whether these would correspond to the treatments currently used for PFIC in Canada (the date of inclusion of patients in the registry is also unclear). For some outcomes, the follow-up time was likely to be too short and/or the sample size too small to capture relevant events. Numerous methodological limitations within the study limit the generalizability of the findings.
Discussion
Summary of Available Evidence
There was 1 double-blind RCT included in the sponsor’s systematic review. The PEDFIC 1 study was a pivotal, multinational (28 sites, including 1 site in Canada), sponsor-funded, double-blind RCT that randomized 62 patients with PFIC1 or PFIC2 1:1:1 to odevixibat 40 mcg/kg/day, odevixibat 120 mcg/kg/day, or matching placebo over a treatment course of 24 weeks. The primary outcome was the percentage of patients who experienced a reduction in sBA at 24 weeks, and key secondary outcomes included the need for surgery (biliary diversion or liver transplant), and the percentage of positive pruritus assessments at 24 weeks. HRQoL was also reported as an exploratory outcome, and mortality was reported under harms.
Additional evidence was available from the PEDFIC 2 study, an open-label extension to the PEDFIC 1 study with a follow-up to 72 weeks, which also enrolled || additional patients with any PFIC subtype, and || of these patients had PFIC3. All patients in the PEDFIC 2 study received odevixibat 120 mcg/kg/day. There were 11 patients in the PEDFIC 1 study who stopped dosing early (4 from the odevixibat 40 mcg/kg/day group, 2 from the odevixibat 120 mcg/kg/day group, and 5 from the placebo group) and were rolled into the extension study, PEDFIC 2, at that higher dose. Additional evidence was available from the OvEC study, where the sponsor compared clinical outcomes from 69 patients enrolled in the PEDFIC 1 and PEDFIC 2 studies who had not had biliary diversion surgery with a cohort of patients from the NAPPED study, comprising a registry of patients with PFIC, using IPTW methods.
The median age of the patients in the PEDFIC 1 study was 3.2 years and ranged from 6 months to 15.9 years. Most patients (47 of 62; 76%) were between 6 months and 5 years of age; 12 (19%) were between 6 and 12 years of age, and 3 (5%) were between 13 and 18 years of age; a limited number of patients (10; 16%) were 8 years of age or older. The median height-for-age and weight-for-age z scores were −1.70 and −0.95, respectively, indicating the patients were below their age-matched peers for growth. Most (45 patients; 73%) had PFIC2 and 17 (27%) had PFIC1. Almost all patients (60; 97%) had a history of significant pruritus present per the investigator and most (42; 68%) had levels of sBA greater than 100 µmol/L within 6 months before enrolment in the study. The majority of patients (55; 89%) were receiving UDCA and/or rifampicin at study entry with 50 patients (81%) on UDCA and 41 (66%) on rifampicin. Overall, 8 patients (13%) reported prior biliary tract surgeries (all reports of biliary diversion).
Interpretation of Results
Efficacy
PFIC is a rare disease (occurs in 1 of 50,000 to 1 of 100,000 births worldwide6) associated with morbidity and mortality. It is clear from the patient, caregiver, and clinician input provided to CADTH that PFIC has a significant impact on a patient’s HRQoL. Families noted that the most bothersome symptom of PFIC is pruritus, which interferes with sleep (patients’ and caregivers’), feeding, social activities, and school performance, in addition to the physical damage incurred by constant scratching. The clinical experts consulted by CADTH on this review also noted the subsequent impact of lack of sleep and interference with feeding on growth and development. Families are also fearful of the long-term prognosis for their children, as the survival rate in patients with PFIC who do not undergo surgical treatments (SBD or liver transplant) is 50% at age 10 and almost zero at age 20.6 Families and clinical experts were in agreement that the current treatment options for PFIC are very limited and are ineffective once pruritus becomes more severe, with limited impact on disease progression. The PEDFIC 1 study focused on pruritus and levels of sBA as outcomes, with sBA used as a surrogate for mortality and need for surgery, outcomes which were reported but needed a larger study with longer follow-up to adequately assess. Other outcomes reported included HRQoL (PedsQL), sleep (awakenings), growth, and total bilirubin. Overall, there is evidence that the 40 mcg/kg/day dose of odevixibat likely improves pruritus within 4 weeks and may reduce sBA after 12 weeks of therapy; however, the impact of odevixibat on mortality and the need for surgery is uncertain, as is its impact on HRQoL, and these limitations and others are addressed in more detail subsequently.
Given there were no deaths and no events of surgery (either liver transplant or biliary diversion) during the PEDFIC 1 study, there is no comparative evidence suggesting that odevixibat improves these outcomes versus placebo. The lack of events is likely due to the trial’s small sample size and relatively short follow-up (24 weeks). With its 72-week follow-up and larger sample size, the PEDFIC 2 study had a number of patients who underwent surgery, ||| |||||||| || ||||| |||||||| ||| || ||| ||||||||| ||||| ||||||||||. With the lack of a control group in the PEDFIC 2 study, there is no way to know whether this represents an improvement over placebo or usual care. The sponsor also submitted its OvEC study, an analysis it performed using IPTW methods to compare 69 patients with PFIC1 or PFIC2 from the PEDFIC 1 and 2 studies with a cohort of patients from the NAPPED registry. Although the results of the sponsor’s analysis suggest there may be a reduced risk of negative clinical outcomes in patients taking odevixibat, the analysis has numerous methodological issues that reduce certainty in these results. The potential impact of odevixibat on these important clinical outcomes therefore relies largely on the surrogate outcome of sBA and, more specifically, the lowering of sBA (considered a response), which was the primary outcome used for regulatory approval in countries outside the US. Based on the GRADE assessment, there was evidence of low certainty from the PEDFIC 1 study that treatment with odevixibat may reduce sBA when compared with placebo. The sponsor presented results from an analysis from the PEDFIC 1 and PEDFIC 2 studies that correlated sBA with NLS and found that none of the patients who experienced a positive sBA response underwent a liver transplant, while 8 patients who did not experience a response underwent a liver transplant. These findings suggest there may be a relationship between elevated sBA and liver transplant; however, the precise nature of this relationship has not been established. As noted by a clinical expert consulted by CADTH on this review, it is the parenchymal sBA levels that are important, and sBA is simply a surrogate for that more valuable surrogate measure.
Although PEDFIC 1 compared both the 40 mcg/kg/day and 120 mcg/kg/day doses of odevixibat with placebo, it is only the 40 mcg/kg/day dose that is being proposed as a starting dose, with up-titration to 120 mcg/kg/day as a potential option for patients not experiencing a response to the lower dose. Only the PEDFIC 2 study reported outcomes for this up-titration to the 120 mcg/kg/day dose. The sponsor reported that the percentage of positive pruritus responders increased in patients who transitioned from 40 mcg/kg/day to 120 mcg/kg/day from the PEDFIC 1 to PEDFIC 2 studies (||||| ||||| || ||||| || ||||| ||||| || |||||); however, there was wide variation in responses. An increase in positive response in the patients from the PEDFIC 1 to PEDFIC 2 studies (||||| ||||| || ||||| || ||||| ||||| || |||||) who continued on 120 mcg/kg/day suggests that improvement in symptoms due to odevixibat may be delayed in some patients; it is also possible that some of the improvement shown is due to natural variation in their symptoms with time, and this was also echoed in the FDA integrated review.29 It is also important to note that, unlike the PEDFIC 1 study, PEDFIC 2 is an open-label trial, and it is not known whether knowledge of the intervention they were receiving may have biased patient responses on the pruritus scales. Ultimately, the FDA decided to approve the 120 mcg/kg/day dose for a few reasons, including the fact that it did perform statistically better than placebo in the PEDFIC 1 study, and that it is possible that patients with severe disease may respond to a higher dose. The FDA also noted, however, that an initial dose escalation to 80 mcg/kg/day may be more prudent, even though that dose was not studied in the PEDFIC 1 study.29 The clinical experts consulted by CADTH for this review also believed they would favour a more gradual dose titration than is currently proposed in the draft product monograph. Therefore, in summary, there is limited clinical evidence to support dose escalation in the manner described in the draft product monograph.
An additional complication of the dosing in the PEDFIC 1 study is that a dose-response effect was largely lacking, despite a 3-fold difference between the low and high dose. In fact, there were instances where there appeared to be a reverse dose-response effect, where the lower odevixibat 40 mcg/kg/day dose appeared to perform better than the odevixibat 120 mcg/kg/day dose. One of the more obvious examples of this was the rate of patients experiencing a response in sBA at 24 weeks, which was the primary outcome of PEDFIC 1. Looking at the unadjusted data, sBA responses occurred in 44% of patients at the lower dose and 21% of patients at the higher dose (no patients taking placebo experienced a response in sBA). This is particularly puzzling because this outcome is directly related to the mechanism of the drug (further supported by the placebo response of zero) and is measured objectively, versus a PRO assessment, which can be more easily biased. A similar, but not quite as obvious, trend was evident in the proportion of positive pruritus responses at week 24, which occurred 58% of the time in the odevixibat 40 mcg/kg/day group and 48% of the time in the odevixibat 120 mcg/kg/day group. Although more subjectively measured, this is clearly a critical outcome for patients and the 10% difference in response in favour of the lower over the higher dose is difficult to explain. Possible explanations include that this is a rare disease with an expected relatively small sample size, which contributes to wide within-group variations in patient characteristics, including the course of the underlying disease, and therefore results in considerable within-group variability in responses to the drug. Other outcomes where the lower odevixibat 40 mcg/kg/day dose appeared to perform better than the higher dose included all growth parameters (height, weight, BMI) and change in total bilirubin from baseline to week 24.
The combination of a small study population and the relatively large number of patients who discontinued treatment meant that no conclusions could be drawn about several important outcomes such as PedsQL, awakenings, growth, and total bilirubin. For example, the low sample size likely contributed to the ||||||||| |||| |||||||||| ||||||||| ||| |||||||| ||||||| |||||||||| || ||||||||| |||||| || ||| |||| ||||||||||||||| |||| ||||||| || ||| |||||||||||| ||| ||| || |||| |||||||||||| ||||||||||||| || ||| ||||||||||||| |||| ||| |||||||||| |||||||| ||| ||| ||||||||||||| ||||||||||| ||||||| |||||||||| ||| ||||||| |||||| |||||||| |||| |||||||||| ||||||||||| ||||||| ||| |||||||||| ||||||||||| ||||. This high degree of uncertainty is not uncommon when assessing drugs for rare diseases, and it is important to remember that a lack of evidence of a benefit is not equivalent to evidence of no benefit. Although there is uncertainty about the impact of odevixibat on HRQoL, there is evidence that odevixibat likely improves pruritus, and pruritus is clearly the major symptom experienced by patients with PFIC.
The clinical experts consulted by CADTH agreed that it is a major generalizability issue that the only comparative data for odevixibat is in patients with PFIC1 or PFIC2 subtypes. The clinical experts also noted the challenges in trying to speculate on how efficacious odevixibat would be on other subtypes, but they also believed that the mechanism of action would suggest that it could be efficacious in any form of cholestatic pruritus. According to the clinical experts, this issue with generalizability is further complicated by the continual evolution of PFIC, largely due to advances in genotyping, which currently has the number of PFIC subtypes at 10 and counting. PFIC1, PFIC2, and PFIC3 seem to be by far the most common subtypes in a very rare disease; therefore, it might not be practical to expect there to be convincing evidence of efficacy for the more uncommon subtypes at this time. Additionally, the clinical experts would not be surprised if the current system of nomenclature for PFIC subtypes is completely revised in the future, as more is understood about the genetics of the disease.
Harms
The most common harm in the PEDFIC 1 study was diarrhea, and this is an expected side effect of odevixibat, given its mechanism of action, according to the clinical experts consulted by CADTH for this review. There is no indication from the PEDFIC 1 study that the risk of diarrhea increases with dose (39% of patients experienced diarrhea with odevixibat 40 mcg/kg/day, 21% with odevixibat 120 mcg/kg/day). There was no indication that the diarrhea progresses to a point where it becomes an SAE, or “clinically significant” diarrhea, and there was little to no difference in risk between patients in the odevixibat 40 mcg/kg/day dose, odevixibat 120 mcg/kg/day dose and placebo (|| |||||| || |||||| ||, respectively). In their input to CADTH, a patient’s family who had experience with odevixibat noted diarrhea as a side effect, which they were simply dealing with by adjusting dosing. Additionally, liver-related AEs were also common; however, this was across all groups. There may be an increased risk of adjudicated hepatic events, as they occurred in ||| || |||||||| || ||| |||||||||| || |||||||||| |||||| ||| || |||||||| || ||| |||||||||| ||| |||||||||| |||||| ||| ||| || |||||||| || ||| ||||||| |||||. Although, according to the product monograph, odevixibat can increase liver enzymes, elevation in liver enzymes is also considered by the clinical experts to be part of the underlying condition in PFIC in that it may be occurring due to damage to the liver rather than because of a transient drug-induced elevation in hepatic enzymes. There was no evidence of an increased risk of SAEs with either odevixibat dose compared with placebo and no patients died during the study. One patient in the odevixibat 120 mcg/kg/day group discontinued treatment due to recurrent diarrhea; otherwise, no other patients discontinued due to an AE.
Conclusion
One pivotal, sponsor-funded multinational double-blind RCT was included in this review. The PEDFIC 1 study randomized 62 patients with either PFIC1 or PFIC2 in a 1:1:1 manner to odevixibat 40 mcg/kg/day, 120 mcg/kg/day, or placebo over a treatment course of 24 weeks. The odevixibat 40 mcg/kg/day dose is the proposed starting dose for odevixibat, with a proposed dose escalation to 120 mcg/kg/day after 12 weeks for patients who do not experience a response; therefore, it is the 40 mcg/kg/day dose that is the focus of this review. It should be noted there is limited clinical evidence to support dose escalation in the manner described in the proposed product monograph. Compared with placebo, treatment with odevixibat at a dose of 40 mcg/kg/day likely improves pruritus within 4 weeks, and this improvement is likely to be maintained to at least the end of the 24-week treatment period. Odevixibat 40 mcg/kg/day may elicit reductions in sBA at 12 weeks of therapy; however, the clinical significance and the impact of these reductions on mortality risk and risk of surgery are uncertain due to the small sample size and limited duration of follow-up. Additionally, odevixibat may improve growth (height and weight z scores); it is not clear whether the magnitude of these benefits is clinically important. The impact of odevixibat on HRQoL, sleep (number of awakenings), and total bilirubin is very uncertain, largely due to wide variation in responses and the risk of bias due to missing data. There were no clear indications of any safety or tolerability issues with odevixibat in either the 24-week double-blind phase or the extension phase. It is important to note that only the extension phase included other PFIC subtypes aside from PFIC1 and PFIC2; this is consistent with the proposed indication, which is not restricted by subtype. Data from the open-label extension phase suggest there are patients who may respond to a dose escalation from 40 mcg/kg/day to 120 mcg/kg/day; however, there are also patients who may not, and it is unclear whether patients are responding to the increased dose or the longer duration of therapy. Additionally, unlike the dosing proposed in the product monograph, which requires a lack of clinical response after 12 weeks before undergoing dose escalation, all patients in the extension were escalated, regardless of the response they experienced after 24 weeks. There was no indirect comparison available that would compare odevixibat with other drugs used for PFIC, although the drugs used for PFIC are generally used off label. In an effort to demonstrate the potential benefits of odevixibat for clinical outcomes such as EFS, NLS, DFS, and OS, the sponsor did submit an IPTW that compared the results from the odevixibat groups in the PEDFIC 1 and PEDFIC 2 studies with registry data; however, due to multiple limitations with their analysis, no conclusions could be drawn from it.
References
1.Gunaydin M, Cil ATB. Progressive familial intrahepatic cholestasis: diagnosis, management, and treatment. Hepat Med. 2018;10:95. PubMed
2.Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012;36:S26-S35. PubMed
3.Mehl A, Bohorquez H, Serrano M-S, Galliano G, Reichman TW. Liver transplantation and the management of progressive familial intrahepatic cholestasis in children. World J Transplant. 2016;6(2):278. PubMed
4.Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4(1):25-36. PubMed
5.Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4(1):1. PubMed
6.Pawlikowska L, Strautnieks S, Jankowska I, et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol. 2010;53(1):170-178. PubMed
7.Baker A, Kerkar N, Todorova L, Kamath BM, Houwen RHJ. Systematic review of progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2019;43(1):20-36. PubMed
8.Albireo. Clinical Study Report. PEDFIC 1. A double-blind, randomized, placebo-controlled, phase 3 study to demonstrate efficacy and safety of A4250 in children with progressive familial intrahepatic cholestasis types 1 and 2. 2020 Oct 30. Gӧteborg, Sweden. [Internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg, and 1200 mcg, oral. Toronto (ON): Medison Pharma Canada Inc.; 2023.
9.Thompson RJ, Arnell H, Artan R, et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2022;7(9):830-842. PubMed
10.Thompson RJ, Artan R, Baumann U, et al. Interim results from an ongoing, open-label, single-arm trial of odevixibat in progressive familial intrahepatic cholestasis. JHEP Reports. 2023:100782.
11.Albireo. Data on File: Odevixibat vs External Control Study. Primary and secondary endpoint analysis (Part A and Part B). October [Internal sponsor's report]. 2022. In: Drug Reimbursement Review sponsor submission: Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg, and 1200 mcg, oral. Toronto (ON): Medison Pharma Canada Inc.; 2023.
12.van Wessel DBE, Thompson RJ, Gonzales E, et al. Impact of genotype, serum bile acids, and surgical biliary diversion on native liver survival in FIC1 deficiency. Hepatology. 2021. PubMed
13.van Wessel DBE, Thompson RJ, Gonzales E, et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J Hepatol. 2020. PubMed
14.Torfgard K, Gwaltney C, Paty J, Mattsson JP, Soni PN. Symptoms and daily impacts associated with progressive familial intrahepatic cholestasis and other pediatric cholestatic liver diseases: a qualitative study with patients and caregivers. Abstract H-P-088. General Hepatology. 2018;67(suppl 1):S208-S209.
15.Varni JW, Seid M, Rode CA. The PedsQL: measurement model for the pediatric quality of life inventory. Med Care. 1999;37(2):126-139. PubMed
16.Mapi Research Trust. The Peds QL™ scoring algorithm: scoring the Pediatric Quality Of Life Inventory™. 2017; https://www.pedsql.org/score.html. Accessed 2023 Feb 14.
17.Varni JW, Burwinkle TM, Seid M, Skarr D. The PedsQL 4.0 as a pediatric population health measure: feasibility, reliability, and validity. Ambul Pediatr. 2003;3(6):329-341. PubMed
18.Albireo. Patient- and observer-reported outcome pruritus measures: Summary of measurement characteristics. Evidence dossier. Odevixibat Progressive Familial Intrahepatic Cholestasis Development Programme. Draft 1.0 [Internal sponsor's report]. 2020. In: Drug Reimbursement Review sponsor submission: Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg, and 1200 mcg, oral. Toronto (ON): Medison Pharma Canada Inc.; 2023.
19.Gwaltney C, Ivanescu C, Karlsson L, Warholic N, Kjems L, Horn P. Validation of the PRUCISION instruments in pediatric patients with progressive familial intrahepatic cholestasis. Adv Ther. 2022;39(11):5105-5125. PubMed
20.Vollset SE, Hirji KF, Elashoff RM. Fast computation of exact confidence limits for the common odds ratio in a series of 2 × 2 tables. J Am Stat Assoc. 1991;86(414):404-409.
21.Varni JW, Limbers CA, Burwinkle TM. Parent proxy-report of their children's health-related quality of life: an analysis of 13,878 parents' reliability and validity across age subgroups using the PedsQL™ 4.0 Generic Core Scales. Health Qual Life Outcomes. 2007;5(1):2. PubMed
22.Verkade HJ, Thompson RJ, Arnell H, et al. Systematic review and meta-analysis: Partial external biliary diversion in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2020. PubMed
23.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
24.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
25.Albireo. Clinical Study Report. PEDFIC 2. An Open label Extension Study to Evaluate Long term Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2. 2022 Dec 16. Gӧteborg, Sweden. [Internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg, and 1200 mcg, oral. Toronto (ON): Medison Pharma Canada Inc.; 2023.
26.Clemson C, Valcheva V, Sturm E, et al. Native liver survival in odevixibat SBA responders: data from the PEDFIC studies in patients with progressive familial intrahepatic cholestasis. Hepatology. 2022;76:S206.
27.Hansen B, Valcheva V, Yu Q, et al. Analysis of long-term treatment effects of odevixibat on clinical outcomes in children with progressive familial intrahepatic cholestasis in odevixibat clinical studies vs external controls from the NAPPED database. Abstract submitted: EASL Congress; 2023 Jun 21-24; Vienna, Austria. J Hepatol. 2023;75(suppl 1):S969-S970. https://pure.rug.nl/ws/portalfiles/portal/762940328/1-s2.0-S0168827823030222-main.pdf. Accessed 2024 Jan 23.
28.Albireo. Operational statistical analysis plan: analysis of the effect of odevixibat (A4250) versus external controls on clinical outcomes in children with PFIC [Internal sponsor's report]. 2022 Apr 29. In: Drug Reimbursement Review sponsor submission: Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg, and 1200 mcg, oral. Toronto (ON): Medison Pharma Canada Inc.; 2023.
29.Center for Drug Evaluation and Research. Integrated review. BYLVAY (odevixibat) oral. Company: Albireo AB. Application No.: 215498. Approval date: 7/19/2021 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2021: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/215498Orig1s000IntegratedR.pdf. Accessed 2023 Sep 18.
Appendix 1: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Albireo gathered evidence from multiple sources to establish the content validity of these instruments. The evidence included information on PFIC specifically, as well as from closely related cholestatic liver diseases (Alagille syndrome, biliary atresia, and sclerosing cholangitis). First, a literature review was conducted to identify the most relevant signs and symptoms of cholestatic liver disease and to identify clinical outcome assessment instruments. Subsequently, discussions with expert clinicians and concept elicitation interviews with patients with chronic liver disease and/or their caregivers were conducted to confirm and add to the initial findings. The findings from the literature review and interviews were used to develop the initial versions of the PRO and ObsRO instruments. Subsequently, cognitive interviews were conducted with patients and/or their caregivers to evaluate their ability to comprehend the instructions, items, and response scales of PRO and ObsRO pruritus measures.
Over the course of the interviews, the minimum age at which the PRO could be administered was assessed. This was based upon multiple inputs, including the patient’s ability to pay attention over the course of the interview, ability to read the items, ability to answer the items, ability to explain what the items meant to them, ability to complete the card-sorting task (ranking cards based on severity of symptoms), and ability to answer the responder definition questions. As a result, it was decided that the PRO would be administered only in patients ages 8 years and older; the caregiver-completed ObsRO instrument could be used in all patients.
PRO morning diary:
How bad was your worst itching since you went to bed last night?
How hard was it to fall asleep last night because of your itching?
How hard was it to stay asleep last night because of your itching?
Did you wake up last night because of itching?
How tired do you feel this morning?
PRO evening diary:
How bad was your worst itching since you woke up this morning?
How tired were you since you woke up this morning?
ObsRO morning diary:
How bad was your child’s worst scratching since he/she went to bed last night?
Since your child went to bed last night, did you see blood due to scratching?
Did your child need a caregiver to help him/her fall asleep last night due to his/her itching?
Did your child need a caregiver to soothe him/her at some time during the night last night due to his/her itching?
Did your child need a caregiver to sleep with him/her at some time during the night last night due to his/her itching?
How many times did you notice that your child woke up last night?
Did your child take any prescribed or over-the-counter medicines before going to bed last night that may have made him/her sleepy?
ObsRO evening diary:
How bad was your child’s worst scratching since he/she woke up this morning?
How tired did your child seem to be today?
Pharmacoeconomic Review
Abbreviations
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
HRQoL
health-related quality of life
iBAT
ileal bile acid transporter
ICER
incremental cost-effectiveness ratio
LT
liver transplant
PEBD
partial external biliary diversion
PedsQL
Pediatric Quality of Life Inventory
PFIC
progressive familial intrahepatic cholestasis
PFIC1
progressive familial intrahepatic cholestasis type 1
PFIC2
progressive familial intrahepatic cholestasis type 2
PFIC3
progressive familial intrahepatic cholestasis type 3
QALY
quality-adjusted life-year
sBA
serum bile acid
SOC
standard of care
UDCA
ursodeoxycholic acid
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Odevixibat (Bylvay), 200 mcg, 400 mcg, 600 mcg, and 1,200 mcg oral capsules |
Submitted price |
|
Indication | For the treatment of progressive familial intrahepatic cholestasis (PFIC) in patients aged 6 months or older |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | October 30, 2023 |
Reimbursement request | As per indication |
Sponsor | Medison Pharma Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance; PFIC = progressive familial intrahepatic cholestasis.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Patients aged 6 months or older with PFIC |
Treatment | Odevixibat plus SOC |
Comparator | SOC alone (defined as off-label use of UDCA, rifampicin, antihistamines, and naltrexone) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (99 years) |
Key data sources | PEDFIC 1, PEDFIC 2, NAPPED natural history study |
Submitted results | ICER = $2,000,828 per QALY gained (incremental costs = $9,601,944; incremental QALYs = 4.80) |
Key limitations |
|
CADTH reanalysis results |
|
ICER = incremental cost-effectiveness ratio; LT = liver transplant; PEBD = partial external biliary diversion; PFIC = progressive familial intrahepatic cholestasis; QALY = quality-adjusted life-year; sBA = serum bile acid; SOC = standard of care; UDCA = ursodeoxycholic acid.
Conclusions
Evidence from the PEDFIC 1 trial indicates that odevixibat 40 mcg/kg likely improves pruritus within 4 weeks, and this improvement is likely to be maintained for at least 24 weeks compared with standard of care (SOC). Using Grading of Recommendations Assessment, Development, and Evaluation (GRADE), CADTH categorized this evidence as having moderate certainty, but noted it was unclear what the clinical importance of the reduction was. Similarly, odevixibat 40 mcg/kg may elicit reductions in serum bile acid (sBA) levels after 24 weeks of follow-up; however, the clinical significance and the impact of these reductions on mortality risk and risk of surgery is uncertain due to the sample size and limited duration of follow-up. CADTH categorized the evidence for a reduction in sBA levels as being low certainty. Importantly, sBA levels were determined to be a surrogate outcome for the more important outcome for patients, namely pruritus, which raises concerns about the appropriateness of the sponsor’s model structure, which was based on sBA levels. Data from the open-label extension trial (PEDFIC 2) suggest there are patients who may respond to a dose escalation from 40 mcg/kg/day to 120 mcg/kg/day; however, it is unclear whether patients are responding to the increased dose or the longer duration of therapy.
Results from the CADTH base case were aligned with the sponsor’s: the incremental cost-effectiveness ratio (ICER) for odevixibat plus SOC exceeds conventional thresholds for cost-effectiveness. In the CADTH base case, odevixibat plus SOC is associated with higher costs (incremental costs = $9,688,198) and higher quality-adjusted life-years (QALYs) (incremental QALYs = 2.80) compared with SOC alone over a lifetime time horizon, resulting in an ICER of $3,462,139 per QALY gained. The main difference between the CADTH base-case results and the sponsor-submitted results is a reduction in life expectancy associated with odevixibat (1.27 additional life-years versus the sponsor estimate of 3.39). The main difference is that life expectancy for those receiving a transplant is expected to be longer in the CADTH base case, based on large registry databases.
For the CADTH base case, a price reduction of 98.6% for odevixibat would be required for odevixibat plus SOC to be cost-effective compared with SOC alone at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained. This would reduce annual drug costs for odevixibat from $771,078 to $10,795 for adult patients receiving the low dose and from $2,313,233 to $32,385 for adult patients receiving the high dose.
CADTH notes the results were driven by the high drug acquisition cost for odevixibat and the dose escalation of patients from 40 mcg/kg to 120 mcg/kg after nonresponse. For example, dose escalation to 120 mcg/kg increases the annual drug acquisition costs of odevixibat from $771,078 to $2,313,234 per patient in the adult population compared with the 40 mcg/kg dose. Based on the CADTH Clinical Review and the clinical experts consulted by CADTH, there is limited clinical justification to support the proposed 3-fold dose escalation. A scenario analysis was conducted to exclude dose escalation, which decreased the drug costs associated with odevixibat by more than $4 million, resulting in a decreased ICER of $2,237,178 per QALY.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
CADTH received patient group input from the Canadian Liver Foundation based on an online survey of 14 respondents, 4 of whom were patients and caregivers in Canada. Respondents indicated that the symptoms of progressive familial intrahepatic cholestasis (PFIC) severely impact daily life by decreasing quality of life, affecting sleep and emotional health, impacting the ability to perform daily activities, and causing debilitating itch symptoms. Respondents noted there are no effective therapies for PFIC that target the underlying bile flow deficiencies or slow disease progression, and respondents described using pharmacological treatments for symptom relief such as ursodeoxycholic acid (UDCA), rifampicin, hydroxyzine, cholestyramine, antihistamines, and naloxone. Patients may also undergo medical procedures such as biliary diversion (e.g., partial external biliary diversion [PEBD]), liver transplant (LT), ileal exclusion, or cholecystectomy. Patients and caregivers indicated that the most important outcomes for new treatment options include symptom reduction and prevention of disease progression, specifically as it relates to quality of life, reduction in itch, growth, and sleep. Five respondents had experience with odevixibat and described symptom alleviation, improved quality of life, and minimal side effects. Reported adverse events were described as minor and included diarrhea.
Clinician input was received from the Canadian Association for the Study of the Liver. This input indicated that there are no currently curative medical therapies for PFIC, and that SOC is restricted to treatments aimed at managing cholestasis and its associated complications, such as nutritional support, fat-soluble vitamin deficiency, and pruritus. Pharmacological treatments to manage symptoms include UDCA, antihistamines, cholestyramine, rifampin, sertraline, and naltrexone. Surgical procedures such as surgical biliary diversion and LT are also considered. The clinicians noted that odevixibat would be used in combination with currently available medications, as it is an effective symptomatic therapy for treating cholestatic pruritus by lowering sBA levels and, further, may delay or prevent LT.
The drug plan input expressed concerns surrounding baseline requirements for elevated sBA levels, whether a trial of other therapies should be required before initiation of therapy, and whether there is an upper limit to the age at which therapy should be initiated. The plans noted uncertainty surrounding loss of response, the level of sBA, and the pruritus response that would be considered clinically meaningful, particularly in the context of sBA being a proxy measure with a potential correlation to increased native liver survival. Additionally, the drug plans expressed concern surrounding the optimal starting dose and method of dose titration in clinical practice.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s model incorporated health states defined by sBA response and included key surgical events in the treatment of PFIC, such as PEBD and LT.
Health-related quality of life (HRQoL) was included in the model for patients with PFIC via health state utility values.
In addition, CADTH addressed some of these concerns as follows:
CADTH assessed the impact of excluding dose escalation of odevixibat from 40 mcg/kg to 120 mcg/kg on drug acquisition costs in a scenario analysis.
CADTH was unable to address the following concerns raised from stakeholder input:
CADTH was unable to address the uncertainty surrounding the appropriateness of sBA levels and pruritus response used to assess the treatment efficacy of odevixibat in the sponsor’s submitted model.
Economic Review
The current review is for odevixibat (Bylvay) for patients 6 months or older with PFIC.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of odevixibat plus SOC compared with SOC alone. The model population comprised patients with PFIC aged 6 months or older.1 The target population for this review is aligned with the sponsor’s reimbursement request.
Odevixibat is available in 200 mcg, 400 mcg, 600 mcg, and 1,200 mcg oral capsules to be swallowed whole or sprinkled on food based on the ease of administration for the individual patient.1,2 The recommended dosage is 40 mcg/kg administered once daily in the morning. If an adequate clinical response has not been achieved after 3 months of continuous therapy, the dose may be increased to 120 mcg/kg per day, with a maximum daily dose of 7,200 mcg per day.2 The annual cost of odevixibat ranges from $64,256 to $771,078 based on the 40 mcg/kg dosing for a patient weight range of 4 kg to 77 kg. The annual cost of odevixibat ranges from $192,769 to $2,313,233 based on the 120 mcg/kg dosing for a patient weight range of 4 kg to greater than 55 kg, after which the maximum daily dose of 7,200 mcg per day is met. The costs for SOC included costs of UDCA, cholestyramine, rifampicin, and naltrexone, which all patients were assumed to receive.1
The analysis was conducted from the perspective of the Canadian public health care payer. Costs and clinical outcomes (life-years and QALYs) were estimated over a lifetime time horizon (99 years; 1-year cycle length), discounted at an annual rate of 1.5% per annum.
Model Structure
The model structure consisted of a Markov model with 7 health states based on the potential elimination of PEBD and delayed time to LT, driven by response to treatment (Figure 1).1 The model health states included: no PEBD, no response; no PEBD, response; PEBD, no response; PEBD, response; LT; post-LT; and death.1 All patients enter the model in the no PEBD, no response state and may transition to the no PEBD, response state, remain in the same state, or progress further to the PEBD and LT health states. Patients may progress to LT before and after PEBD, and a proportion of patients require a second LT, which is assumed to occur within the same year as the first LT; however, patients receiving odevixibat do not progress to PEBD health states, as it is assumed that odevixibat is a pharmacological alternative to PEBD. Patients who do not achieve response on odevixibat therefore directly undergo LT without prior PEBD. In each cycle, patients may discontinue odevixibat due to a lack of response after 6 months of continuous treatment. Patients are at risk of death in each model cycle.
Model Inputs
Baseline patient characteristics in the model were aligned with the PEDFIC 1 trial, which enrolled patients 6 months or older with PFIC1 or PFIC2 (mean age = 4.25 years; 50% female; 27% with PFIC1).3
The pharmacoeconomic model was informed by inputs from the PEDFIC 1 and PEDFIC 2 clinical trials3,4 and the NAPPED natural history study.5,6 Clinical efficacy (i.e., treatment response) for patients receiving odevixibat 40 mcg/kg after 3 months was based on the PEDFIC 1 trial, and the 6-month response for patients up-titrating from the 40 mcg/kg to 120 mcg/kg dosing after nonresponse at 3 months was derived from the PEDFIC 2 study.3,4 Response was defined as achieving a reduction of 70% or greater in sBA, which was assumed to be an adequate proxy measure for a decrease in pruritus.1 The sponsor estimated an unadjusted overall response rate by combining the response rate for patients on the 40 mcg/kg initial dosing with those who escalated to the 120 mcg/kg dosing after 3 months and achieved response on the higher dose after a cumulative 6 months of treatment.1 Response to off-label SOC was 0% based on the PEDFIC 1 trial.3 The annual loss of response on odevixibat was estimated to be 3.53%, based on discontinuation data from the PEDFIC 2 trial.4
All inputs related to PEBD and LT were sourced from the NAPPED study5,6 or based on sponsor assumption. It was assumed that 0% of patients receiving odevixibat would undergo a PEBD and would instead directly proceed to LT. The probability of LT without prior PEBD was derived from the NAPPED study using native liver survival curves for those who did not undergo PEBD.5,6 For patients receiving SOC alone, the probability of transitioning to PEBD was derived from the NAPPED study and stratified by patients under age 3 and patients aged 3 years and older.5,6 Response to PEBD for patients receiving SOC alone was based on the postprocedural sBA response rates observed in the NAPPED study, defined as a 75% or greater reduction in sBA for PFIC1 and achieving an sBA level of less than 65 μmol/L for PFIC2.5,6 The annual probability of LT after PEBD was estimated using native liver survival curves for patients with PEBD in the NAPPED study5,6 whose condition did not respond to treatment. For a proportion of patients, a second transplant may be required and is assumed to occur in the same year as the initial transplant, based on a weighted average from the published literature.7
To model the survival of patients with PFIC, the sponsor assumed there is an increased risk of mortality among patients with PFIC based on treatment response status and LT status. Patients in health states with no treatment response (i.e., “no PEBD, no response” and “PEBD, no response”) experienced an annual probability of death before the surgical procedure of 0.27%.5,6 For acute post-LT mortality occurring in the first year, the sponsor conducted a meta-analysis and estimated a joint PFIC1 and PFIC2 mortality rate of 13%.8-10 For long-term post-LT mortality, a pooled estimate for annual probability of death was calculated to be 1.91%.8,11 Patients achieving treatment response were assumed to experience the same risk of mortality as the general population in Canada.
The sponsor’s model included health state utility values for patients estimated from a published study reporting HRQoL in children with liver disease (including half of chronic intrahepatic cholestasis children with a confirmed PFIC diagnosis) using the Pediatric Quality of Life Inventory (PedsQL) mapped to the EQ-5D.12 The published study did not differentiate between patients with and without a response to treatment, and the sponsor assumed that utility values for responders are equal to healthy patients and the utility values for nonresponders are equal to patients with chronic intrahepatic cholestasis. The previously described values are applied to the “before PEBD” health states. To obtain values for the “after PEBD” health states, a disutility associated with a stoma bag was applied.13 For patients experiencing a loss of response in the “before PEBD” or “after PEBD” health states, a disutility associated with short stature is applied based on an HRQoL study in children with chronic kidney disease.14 Utilities for the LT and post-LT health states were also derived from the literature. In the year of their transplant (acute post-LT period), patients are assumed to have the utility associated with severe pruritus.15 The long-term post-LT health state data from a systematic review of children who underwent LT were derived by mapping PedsQL scores to the EQ-5D.16
Costs included in the model were drug acquisition costs, PEBD costs, LT costs, health care resource utilization costs, and adverse event costs. Drug acquisition costs for odevixibat were based on the sponsor’s submitted price,1 while acquisition costs for drugs included as SOC were obtained from the Ontario Drug Benefit Formulary.17 Drug acquisition costs for odevixibat were based on the product monograph, where patients received 40 mcg/kg per day for 3 months after which the dose was escalated to 120 mcg/kg per day for a maximum of 3 additional months if there was no initial response.2 The sponsor only included dose escalation costs of odevixibat for those who experienced a response after 3 additional months of treatment. The cost of SOC for all patients was based on the proportion of use for each treatment observed from the PEDFIC 1 trial3 and the National Institute for Health and Care Excellence (NICE) submission of odevixibat.18 PEBD costs, LT costs, and post-LT complication costs were sourced from the Ontario Case Costing Initiative.7,19,20 Costs related to annual post-LT maintenance and monitoring21 and for immunosuppressants were also included.22 Health care resource use was based on the frequency of resource use and tests from the sponsor’s burden-of-illness study1 and included visits to a general practitioner, pediatrician, hepatologist, gastroenterologist, emergency medicine specialist, orthopedist, endocrinologist, and nurse. Resource utilization also included the costs for stoma care.23 Unit costs for health care resource use were derived from the Ontario Schedule of Benefits,24 Ontario Nurses’ Association Collective Agreement,25 and the Ontario government’s Assistance for Children with Severe Disabilities program for ostomy supplies.23
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
In the sponsor’s base-case analysis, odevixibat plus SOC was associated with an estimated cost of $9,853,023 and 27.60 QALYs over the 99-year horizon, resulting in an ICER of $2,000,828 per QALY gained (incremental costs = $9,601,944; incremental QALYs = 4.80) compared with SOC alone (Table 3). In the sponsor’s analysis, odevixibat plus SOC had a 0% probability of being cost-effective at a WTP threshold of $50,000.
Results were driven by the drug acquisition costs for odevixibat plus SOC (incremental costs = $9,601,944) and the predicted gain in QALYs (incremental QALYs = 4.80). The sponsor’s model estimated that less than 3% of the total QALYs with odevixibat plus SOC were accrued during the 24-week PEDFIC 1 trial period, indicating that more than 97% of the total QALYs for odevixibat were accrued in the posttrial period.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|
SOC | 251,078 | Reference | 22.80 | Reference | Reference |
Odevixibat + SOC | 9,853,023 | 9,601,944 | 27.60 | 4.80 | 2,000,828 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The submitted analysis is based on the publicly available prices of all treatments, including comparator treatments. SOC was assumed by the sponsor to comprise symptomatic treatment and to include ursodeoxycholic acid, cholestyramine, rifampicin, and naltrexone.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
The sponsor provided several scenario analyses, including allowing patients on odevixibat to undergo a PEBD, alternate dosing assumptions regarding dose escalation, caregiver utilities, and adopting an alternate time horizon of 25 years. Across all scenarios, odevixibat plus SOC was not cost-effective at a WTP threshold of $50,000, with estimated ICERs for odevixibat plus SOC versus SOC alone ranging from $1,322,267 to $2,781,339.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Model structure does not adequately capture key aspects of PFIC in clinical practice: In the sponsor’s model, health states are defined by sBA levels, as sBA levels are assumed to be associated with a decrease in pruritus; however, sBA levels are a proxy measure of response since pruritus control was described to be the primary goal of treatment by patients and clinicians. Furthermore, reductions in sBA levels were assumed to lead to delays in progression to LT, but the clinical experts noted there is limited evidence to support the link between a reduction in sBA levels and delayed LT. The clinical experts also noted there is uncertainty in the assertion that surgical interventions such as PEBD may be avoided if a reduction in sBA levels is maintained.
Treatment response in the model was defined as reaching a 70% or greater reduction in sBA levels; however, the clinical experts expressed concerns with the sBA level cut-off for determining a clinically meaningful response. The clinical experts stated that physician preference in clinical practice would be to assess treatment response and potentially up-titrate the dose of odevixibat according to pruritus response rather than according to a set reduction in sBA. Importantly, sBA levels may also not adequately capture all of the disease-related aspects most relevant to patients, including key outcomes that impact HRQoL such as neurodevelopmental progress, growth maintenance, and avoidance of hepatocellular carcinoma.
The clinical experts also noted that a patient’s severity of disease before starting treatment would greatly influence the efficacy of iBAT inhibitors such as odevixibat. For example, patients who require LT may be too far along in the disease process for odevixibat to prevent severe pruritus. The clinical experts commented that there is also limited evidence to support that patients who do not experience a response to odevixibat would similarly not respond to PEBD and, therefore, would not undergo this surgical procedure.
CADTH was unable to address this limitation owing to the structure of the sponsor’s economic model.
The impact of odevixibat on survival is highly uncertain: The sponsor’s model predicts a survival advantage with odevixibat plus SOC relative to SOC alone (incremental life-years = 3.39), which is not supported by the clinical trial data. Based on the CADTH Clinical Review, the effects of odevixibat on survival are highly uncertain compared with placebo after 24 weeks of follow-up during the PEDFIC 1 trial. No deaths occurred in the PEDFIC 1 trial and the CADTH Clinical Review determined that the trial was not of sufficient size or duration to adequately assess key clinical outcomes such as mortality or the need for surgical intervention (PEBD or LT). Using GRADE, CADTH categorized the evidence pertaining to mortality as very low certainty.
In the sponsor’s model, the predicted survival benefit was driven by varying risks of mortality assigned to different health states based on presurgical and postsurgical status. Patients were expected to experience increased mortality associated with acute LT occurring in the first year of transplant (11.3%) and over the long-term the first year after transplant (1.9%); however, these mortality estimates are uncertain, according to the clinical experts consulted by CADTH, and appeared to be overestimated due to data quality issues in the sponsor-identified sources used to parameterize the model. For example, for mortality associated with acute LT, the sponsor conducted a meta-analysis of 3 studies8-10 that had limited sample sizes and high heterogeneity, with mortality estimates ranging from 0% to 37% across the studies. For long-term mortality risk post LT, the sponsor naively pooled data from 2 studies8,11 that also suffered from limited sample sizes and high heterogeneity, with mortality estimates ranging from 1.60% to 3.57%. Further limitations with these identified sources include the variation in the country where the study was conducted, the use of older studies (generally preceding 2010), and the lack of justification to restrict the data to LT for PFIC disorders. The clinical experts expressed concerns with the quality of the data used to inform mortality inputs in the model. They stated there is no clinical justification to restrict the mortality data to LT for PFIC disorders, and that large registry data exist for pediatric and infant mortality post LT, which are preferred over the dated and limited sources used by the sponsor.
Furthermore, patients in pre-LT health states (i.e., patients with and without PEBD) experiencing a loss of response were assumed to have an increased risk of mortality compared with the general population. This was deemed to be uncertain, according to the clinical experts, who stated there is a lack of clinical data to support a higher risk of mortality for patients in pre-LT health states who lose response.
Overall, the overestimation of mortality risk for the pre-LT and post-LT health states appeared to overestimate the incremental life-years estimated for odevixibat plus SOC relative to SOC alone, which likely biased the estimates of cost-effectiveness in favour of odevixibat plus SOC. The impact of odevixibat on survival remains highly uncertain due to the lack of clinical evidence in support of a survival benefit.
CADTH adjusted the risk of mortality associated with the acute LT and long-term post-LT health states to reflect more recent data from a large registry of pediatric LT recipients from 2013 to 2018, as follows:
CADTH removed the increased risk of mortality associated with a loss of response in pre-LT health states.
The anticipated dosing and drug acquisition costs of odevixibat are highly uncertain: According to the product monograph, the recommended dose of odevixibat is 40 mcg/kg administered orally once daily in the morning.2 If adequate clinical response has not been achieved after 3 months, dosing may be increased to 120 mcg/kg/day, with a maximum daily dose of 7,200 mcg per day.2 Although this dosing recommendation reflects what occurred in the PEDFIC 1 and PEDFIC 2 trials, the CADTH Clinical Review concluded there is limited clinical evidence to support dose escalation in the manner it is described in the product monograph. The CADTH Clinical Review found there was no apparent dose-response effect observed with odevixibat in the PEDFIC 1 trial, meaning that increasing the dose of odevixibat did not appear to increase the efficacy of treatment. This was also echoed by the clinical experts consulted for this review. As such, there is considerable uncertainty surrounding the dosing proposed in the product monograph and whether this would be reflective of clinical practice.
According to the clinical experts consulted by CADTH, there is uncertainty in both the definition of treatment response and the proposed dose escalation to 120 mcg/kg. Primarily, the clinical experts indicated that treatment response based on pruritus response is preferred in clinical practice, as it better reflects outcomes meaningful to patients as opposed to sBA levels, which may not be correlated consistently with pruritus. Second, the clinical experts indicated that response assessment would likely occur before 12 weeks and, if dose escalation were to occur, it would likely occur incrementally (i.e., from 40 mcg/kg to 80 mcg/kg) instead of tripling the dose upon a lack of response. Furthermore, although dose escalation may be considered in clinical practice, the reasons why a patient did not experience the desired response on the initial 40 mcg/kg dose would be examined on a case-by-case basis before making the decision to dose escalate.
CADTH also notes that in the sponsor’s model, the sponsor only accounts for additional drug acquisition costs for patients who respond on the 120 mcg/kg dosing schedule, which is estimated to be 25% according to the data from the PEDFIC 2 trial; however, the product monograph suggests that every patient on the 40 mcg/kg dose who does not achieve adequate response after 3 months of initial treatment would escalate to the 120 mcg/kg dose for a minimum of 3 additional months. Therefore, the drug acquisition costs are underestimated in the model because the costs associated with patients who escalate to the 120 mcg/kg dose and who do not achieve response after 3 additional months were not accounted for in the calculations for drug acquisition costs.
Ultimately, there is considerable uncertainty surrounding how dose escalation may occur in clinical practice, which has impacts on the estimated drug acquisition costs for odevixibat and, therefore, on the cost-effectiveness estimates. Based on the estimated annual costs for odevixibat, the cost for an adult weighing 77 kg (i.e., the average weight of an adult in Canada) is $771,078 if they receive 40 mcg/kg per day and is capped at $2,313,233 if they receive 120 mcg/kg per day. There is an additional cost of $1,542,156 per patient per year associated with tripling the dose of odevixibat, which would occur for the remainder of the lifetime of the patients for whom odevixibat is effective. The estimated incremental differences in costs due to the dose escalation of odevixibat from 40 mcg/kg to 120 mcg/kg is a key driver of cost-effectiveness estimates.
CADTH assessed the impact of excluding the dose escalation of odevixibat from 40 mcg/kg to 120 mcg/kg in a scenario analysis.
Comparative clinical efficacy of odevixibat is highly uncertain: In the economic model, the definition of response was based on achieving a reduction in sBA levels of 70% or greater. As per the proposed dosing in the product monograph, patients who do not achieve response in line with that definition after 12 weeks of initial treatment were expected to dose escalate to 120 mcg/kg for an additional 12 weeks,2 which was assessed in the PEDFIC 2 open-label extension study. The sponsor estimated an unadjusted overall response rate that combined the response rates for patients on the initial 40 mcg/kg dose with those who dose-escalated and achieved response on the 120 mcg/kg dose. According to the CADTH Clinical Review, the interim results from the PEDFIC 2 study suggest there are patients who may respond to dose escalation but also patients who may not, and it was unclear whether patients were responding to dose escalation or the longer duration of therapy. The CADTH Clinical Review also noted that all patients in the PEDFIC 2 trial were dose-escalated, regardless of their response status at the end of the PEDFIC 1 study’s 24-week follow-up period, which is not aligned with the dosing proposed in the product monograph. Furthermore, the sponsor’s base case used response at 24 weeks for the initial 40 mcg/kg response rate from the PEDFIC 1 trial instead of 12 weeks as per the product monograph in their calculation of overall response. Therefore, the naive, unadjusted overall response rate is associated with uncertainty due to limitations with the trial data (e.g., misalignment between the proposed dosing in the product monograph and what occurred in the PEDFIC 2 study) and the sponsor’s methods for estimating overall response.
Furthermore, the sponsor assumes that the key clinical impact of odevixibat for treating patients with PFIC would be avoidance of PEBD and delaying LT. Notably, the CADTH Clinical Review determined that the pivotal trials could not adequately assess key clinical outcomes such as mortality or the need for surgical intervention (PEBD or LT) and, thus, no conclusions could be drawn about the proposed correlation between sBA reduction and avoidance of PEBD or LT. CADTH notes that all data pertaining to PEBD and LT were obtained from the NAPPED natural history study and were noncomparative, observational data.5,6 The use of external observational data in addition to naive, unadjusted response data to parameterize the economic model therefore results in high uncertainty surrounding the estimates of cost-effectiveness.
CADTH could not address limitations related to the lack of comparative clinical evidence.
In a scenario analysis, CADTH adjusted the response rate for the 40 mcg/kg dose to reflect the 12-week data from the PEDFIC 1 study, which aligns with the product monograph dosing schedule.
Uncertainty in long-term treatment effectiveness of odevixibat: Comparative clinical efficacy data for odevixibat versus placebo are available up to the total 24 weeks of follow-up from the PEDFIC 1 trial. There are no further clinical data beyond an additional 24 weeks of interim results from the open-label single-arm extension study (PEDFIC 2). In the model, the sponsor assumed that the naively estimated overall response rate pooled from the PEDFIC 1 and PEDFIC 2 trials, as described previously, would be maintained indefinitely beyond the study period unless treatment discontinuation or mortality occurred. The potential waning of treatment effect over time was not explored in the sponsor’s model. Although the clinical experts consulted by CADTH noted it may be feasible for treatment benefit to be sustained, there is limited clinical evidence to support the assumption that it would persist for the remainder of the model time horizon (up to 99 years). CADTH notes that more than 97% of the incremental QALYs gained with odevixibat plus SOC relative to SOC alone were accrued on the basis of extrapolation (i.e., in the posttrial period), which highlights the impact of assumptions regarding long-term relative treatment efficacy on cost-effectiveness estimates.
CADTH was unable to address this limitation owing to the lack of long-term effectiveness data for odevixibat.
The impact of odevixibat on quality of life is uncertain: The sponsor predicted an incremental gain of 4.80 QALYs with odevixibat plus SOC compared with SOC alone; however, the CADTH Clinical Review concluded that the impact of odevixibat on HRQoL is uncertain due to wide variation in responses and risk of bias due to missing data. The sponsor stated similar conclusions in its submission and therefore opted to use values adopted from various sources published in the literature that were not specific to PFIC patients.12 For example, the base utility values were derived from a cohort of children with liver disease, where half had chronic intrahepatic cholestasis with confirmed PFIC.12 For health states associated with disease progression (e.g., PEBD health states), the sponsor included various disutilities from the published literature (e.g., stoma bag, short stature).13,14 It is therefore uncertain whether these values reflect the preferences of patients in Canada with PFIC.
In particular, certain utility values did not appear to meet face validity according to the clinical experts consulted by CADTH. For example, the sponsor assumed that a patient’s quality of life would decrease substantially upon receiving PEBD regardless of whether they respond or not. The predicted utility value for a patient who does not experience a response without PEBD is therefore estimated by the sponsor to be higher than that of a patient who experiences a response after PEBD. The clinical experts consulted by CADTH indicated that patients who experience a response after PEBD would likely experience an increase in utility and that the sponsor’s estimates did not meet face validity.
In its reanalysis, CADTH set the utility value for a patient experiencing a response after receiving PEBD equal to that of a patient without PEBD who does not experience a response.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Efficacy can be reasonably assessed at 12 weeks. | Uncertain. The clinical expert feedback obtained by CADTH noted that a pruritus response is likely to occur within 4 weeks and that reductions in sBA levels may occur as early as 12 weeks; however, the clinical significance and impact of sBA reduction on mortality risk and surgical outcomes is uncertain. Clinical experts indicated that response would likely be assessed in practice earlier than 12 weeks. |
Patients enrolled in the PEDFIC 1 and PEDFIC 2 trials were assumed to be representative of patients in Canada who would be eligible for odevixibat. | Uncertain. The CADTH Clinical Review noted that only patients with PFIC1 and PFIC2 were included in the PEDFIC 1 study, whereas no restrictions were placed based on PFIC subtype for the PEDFIC 2 trial. The clinical expert input obtained by CADTH noted that the pivotal trial participant characteristics were generally representative of patients seen in clinical practice since the majority of patients present with PFIC1 or PFIC2; however, PFIC3 was noted to have a different disease mechanism and to present differently in practice. |
PFIC1 = progressive familial intrahepatic cholestasis type 1; PFIC2 = progressive familial intrahepatic cholestasis type 2; PFIC3 = progressive familial intrahepatic cholestasis type 3; sBA = serum bile acid.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH undertook reanalyses that addressed key limitations within the submitted economic model, as summarized in Table 5. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with the clinical experts. CADTH was unable to address the other limitations of the model, including the model structure and lack of available long-term clinical data.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Overestimation of acute LT mortality and long-term post-LT mortality. | Overestimated mortality associated with acute LT and long-term post LT due to data quality issues in sponsor-identified sources.
| Adjusted acute LT and long-term post-LT mortality to reflect large registry data for pediatric LT recipients from a 2013 to 2018 cohort. |
2. Additional risk of mortality associated with loss of response in pre-LT health states. | Patients experiencing a loss of response in pre-LT health states (i.e., PEBD and no PEBD) have an increased risk of mortality. | Patients experiencing a loss of response in pre-LT health states do not experience an increased risk of mortality. |
3. Face validity of PEBD responder utility value. | The utility value for a patient without PEBD without response is higher than that of a patient responding after receiving PEBD. | The utility value for a patient without PEBD without response is equal to that of a patient responding after receiving PEBD. |
CADTH base case. | — | Reanalysis 1 + 2 + 3. |
LT = liver transplant; NHS = National Health Service; PEBD = partial external biliary diversion.
CADTH undertook a stepped analysis, incorporating each change to the sponsor’s base case proposed in Table 5 to highlight the impact of each change (Table 6; disaggregated results are presented in Appendix 4, Table 11). All of CADTH’s probabilistic reanalyses were based on 1,000 iterations.
Results from the CADTH base case suggest that odevixibat plus SOC was associated with higher costs (incremental costs = $9,688,198) and higher QALYs (incremental QALYs = 2.80) compared with SOC alone over a lifetime time horizon, resulting in an ICER of $3,462,139 per QALY gained. In the CADTH base case, odevixibat plus SOC had a 0% probability of being cost-effective at a WTP threshold of $50,000 per QALY.
In the CADTH base case, the results were driven by the drug acquisition costs of odevixibat and the predicted incremental gain of 2.80 QALYs with odevixibat plus SOC. The most impactful driver of cost-effectiveness results is allowing for dose escalation from 40 mcg/kg to 120 mcg/kg dosing if there is no response after 3 months of initial treatment, which increases the annual drug acquisition costs for odevixibat (to treat those for whom the drug is effective) from $64,256 to $192,769 for pediatric patients and from $771,078 to $2,313,233 for adult patients. Consistent with the sponsor’s submission, the CADTH base case predicts that more than 97% of the total QALYs gained with odevixibat plus SOC are accrued after the trial period of 24 weeks.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | SOC | 258,665 | 22.66 | Reference |
Odevixibat + SOC | 10,050,230 | 27.59 | 1,986,617 | |
CADTH reanalysis 1: Acute LT and post-LT mortality | SOC | 271,316 | 28.44 | Reference |
Odevixibat + SOC | 10,060,041 | 31.90 | 2,825,908 | |
CADTH reanalysis 2: pre-LT mortality | SOC | 264,872 | 23.17 | Reference |
Odevixibat + SOC | 10,055,621 | 28.01 | 2,024,767 | |
CADTH reanalysis 3: PEBD responder utility value | SOC | 258,665 | 23.20 | Reference |
Odevixibat + SOC | 10,050,230 | 27.59 | 2,230,369 | |
CADTH base case (1 + 2 + 3) | SOC | 277,803 | 29.62 | Reference |
Odevixibat + SOC | 10,065,648 | 32.41 | 3,504,291 | |
CADTH base case (1 + 2 + 3): Probabilistic | SOC | 266,872 | 29.59 | Reference |
Odevixibat + SOC | 9,955,070 | 32.39 | 3,462,139 |
ICER = incremental cost-effectiveness ratio; LT = liver transplant; PEBD = partial external biliary diversion; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The CADTH reanalyses are based on the publicly available prices of comparator treatments and do not reflect confidential negotiated prices. All results are presented deterministically, unless otherwise stated.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s results and CADTH’s base case. The CADTH base case suggested that a 98.6% price reduction for odevixibat would be required to achieve cost-effectiveness for odevixibat plus SOC relative to SOC alone at a $50,000 per QALY threshold (Table 7).
Table 7: CADTH Price Reduction Analyses
Price reduction | ICERs for odevixibat + SOC vs. SOC alone ($/QALY) | |
|---|---|---|
Sponsor base case | CADTH reanalysis | |
No price reduction | 1,986,617 | 3,504,291 |
10% | 1,788,035 | 3,153,862 |
20% | 1,589,453 | 2,803,433 |
30% | 1,390,871 | 2,453,005 |
40% | 1,192,289 | 2,102,576 |
50% | 993,707 | 1,752,148 |
60% | 795,125 | 1,401,719 |
70% | 596,543 | 1,051,290 |
80% | 397,961 | 700,862 |
90% | 199,379 | 350,433 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
CADTH undertook scenario analyses to explore the impact of alternative assumptions on the cost-effectiveness of odevixibat, which are outlined as follows:
Patients are initiated on a dose of 40 mcg/kg and if they do not experience a response after 3 months, they do not escalate to 120 mcg/kg dosing.
Treatment response on the initial 40 mcg/kg dose was adjusted to reflect assessment at 12 weeks instead of 24 weeks, as per the product monograph. After 12 weeks, patients who did not experience a response could escalate to 120 mcg/kg dosing.
Treatment response was based on pruritus score (using PRUCISION patient-reported outcome and observer-reported outcome instruments). Using these instruments, the response to odevixibat 40 mcg/kg and 120 mcg/kg was 58.31% and 47.69% respectively. An assumption was made that responding to 120 mcg/kg after failing to respond to 40 mcg/kg would be |||||| based on data from the PEDFIC 2 study. Finally, 28.74% of those who received SOC alone (placebo in the trial) also responded, based on this instrument. An assumption had to be made regarding the rate of loss of response for those receiving SOC alone. Given this, 2 analyses were conducted. In 1 analysis, CADTH assumed the same loss of response as was assumed for odevixibat (approximately a 4% loss of response each year); in a separate analysis, a 50% loss of response each year was assumed for those receiving SOC alone.
The results of these analyses are presented in Appendix 4 (Table 12). In the scenario analysis where patient dose escalation to 120 mcg/kg did not occur, the ICER comparing odevixibat plus SOC with SOC alone decreased to $2,237,178 per QALY. When treatment response was adjusted to reflect the product monograph suggestion to assess treatment at 12 weeks instead of 24 weeks, the ICER comparing odevixibat plus SOC with SOC alone increased to $3,778,582 per QALY. The scenario analyses conducted by CADTH highlight the considerable impact of dose escalation on odevixibat drug acquisition costs and, therefore, the cost-effectiveness of odevixibat.
When response is assessed using pruritus results, the QALYs increase for both SOC and the odevixibat arm. It is now assumed that improvement in pruritus (rather than sBA) will delay the need for surgical intervention (PEBD) and LT. In the trial, 28.74% of patients receiving SOC (placebo) achieved a response. It is highly uncertain whether this response will be maintained over time. If patients who achieved a response in the placebo arm maintain that response, then this reduces the incremental benefit of odevixibat relative to the base case (which assessed response based on sBA results). If the patients who experience a response in the placebo arm lose response quickly, then the incremental benefit of odevixibat is slightly higher than the base case, although costs are also higher, as patients remain on therapy longer. In both analyses, the ICER remains above $3 million; therefore, the conclusions regarding cost-effectiveness are broadly similar. This is because the incremental drug costs remain above $10 million over the patient lifetime, yet the incremental QALYs for odevixibat versus SOC are approximately 2 to 3.
Issues for Consideration
CADTH notes that the current Health Canada indication does not restrict the use of odevixibat based on PFIC subtype; however, the pivotal trial (PEDFIC 1) enrolled patients with PFIC1 or PFIC 2 only. The cost-effectiveness of odevixibat in other PFIC subtypes is therefore highly uncertain due to the lack of clinical evidence.
The clinical experts indicated that odevixibat may also be used in patients who have received an LT for conditions such as recurrent primary biliary cholangitis, primary sclerosing cholangitis, and chronic rejection. The cost-effectiveness of odevixibat for the treatment of conditions other than PFIC is unknown at this time due to the lack of available efficacy data for off-label use.
Overall Conclusions
Evidence from the PEDFIC 1 trial indicates that odevixibat 40 mcg/kg likely improves pruritus within 4 weeks, and this improvement is likely to be maintained for at least 24 weeks compared with SOC. Using GRADE, CADTH categorized this evidence as having moderate certainty, but noted that the clinical importance of the reduction was unclear. Similarly, odevixibat 40 mcg/kg may elicit reductions in sBA levels after 24 weeks of follow-up; however, the clinical significance and the impact of these reductions on mortality risk and risk of surgery are uncertain due to the sample size and limited duration of follow-up. CADTH categorized the evidence for a reduction in sBA levels as being low certainty. Importantly, sBA levels were determined to be a surrogate outcome for the more important outcome for patients, namely pruritus, which raises concerns about the appropriateness of the sponsor’s model structure, which was based on sBA levels. Data from the open-label extension trial (PEDFIC 2) suggest there are patients who may respond to a dose escalation from 40 mcg/kg/day to 120 mcg/kg/day; however, it is unclear whether patients are responding to the increased dose or the longer duration of therapy.
CADTH undertook reanalyses to address limitations in the sponsor’s economic evaluation, including adjusting the risk of mortality associated with acute LT and long-term post LT, removing the additional risk of mortality associated with loss of response in pre-LT health states, and adjusting the utility value for patients achieving a response after PEBD. CADTH was unable to address the lack of long-term comparative clinical data and concerns with the model structure.
Results from the CADTH base case were aligned with the sponsor’s: the ICER for odevixibat plus SOC exceeds conventional thresholds for cost-effectiveness. In the CADTH base case, odevixibat plus SOC is associated with higher costs (incremental costs = $9,688,198) and higher QALYs (incremental QALYs = 2.80) compared with SOC alone over a lifetime time horizon, resulting in an ICER of $3,462,139 per QALY gained. The gain in QALYs is driven by large improvements in utility from avoiding PEBD and life expectancy increases (a gain of 1.27 life-years) from avoiding LT, for those on odevixibat plus SOC. The main difference between the CADTH base-case results and the sponsor-submitted results is a reduction in life expectancy associated with odevixibat (1.27 additional life-years versus the sponsor estimate of 3.39). The main difference is that life expectancy for those receiving a transplant is expected to be longer in the CADTH base case, based on the data in large registry databases.
For the CADTH base case, a price reduction of 98.6% for odevixibat would be required for odevixibat plus SOC to be cost-effective compared with SOC alone at a WTP threshold of $50,000 per QALY gained. This would reduce the annual drug costs for odevixibat from $771,078 to $10,795 for adult patients receiving the low dose and from $2,313,233 to $32,385 for adult patients receiving the high dose.
CADTH notes that results were driven by the high drug acquisition cost of odevixibat and the dose escalation of patients from 40 mcg/kg to 120 mcg/kg after nonresponse. For example, dose escalation to 120 mcg/kg increases the annual drug acquisition costs for odevixibat from $771,078 to $2,313,233 per patient in the adult population compared with the 40 mcg/kg dose. Based on the CADTH Clinical Review and clinical experts consulted by CADTH, there is limited clinical justification to support the proposed 3-fold dose escalation. A scenario analysis was conducted to exclude dose escalation, which decreased the drug costs associated with odevixibat by more than $4 million, resulting in a decreased ICER of $2,237,178 per QALY.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg, and 1200 mcg, oral. Toronto (ON): Medison Pharma Canada Inc.; 2023 June 23.
2.Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg and 1200 mcg, oral [product monograph]. Toronto (ON): Medison Pharma Canada Inc; 2023 Oct 30.
3.Albireo. Clinical Study Report. PEDFIC 1. A double-blind, randomized, placebo-controlled, phase 3 study to demonstrate efficacy and safety of A4250 in children with progressive familial intrahepatic cholestasis types 1 and 2 [internal sponsor's report]. 2020. In: Drug Reimbursement Review sponsor submission: Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg, and 1200 mcg, oral. Toronto (ON): Medison Pharma Canada Inc.; 2023.
4.Albireo. Clinical Study Report. PEDFIC 2. An open label extension study to evaluate long term efficacy and safety of A4250 in children with progressive familial intrahepatic cholestasis types 1 and 2 [internal sponsor's report]. 2022 Dec 16. In: Drug Reimbursement Review sponsor submission: Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg, and 1200 mcg, oral. Toronto (ON): Medison Pharma Canada Inc.; 2023. Accessed 2022 Dec 16.
5.van Wessel DBE, Thompson RJ, Gonzales E, et al. Impact of genotype, serum bile acids, and surgical biliary diversion on native liver survival in FIC1 deficiency. Hepatology. 2021;74(2):892-906. PubMed
6.van Wessel DBE, Thompson RJ, Gonzales E, et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J Hepatol. 2020;73(1):84-93. PubMed
7.Bull LN, Pawlikowska L, Strautnieks S, et al. Outcomes of surgical management of familial intrahepatic cholestasis 1 and bile salt export protein deficiencies. Hepatol Commun. 2018;2(5):515-528. PubMed
8.Wanty C, Joomye R, Van Hoorebeek N, et al. Fifteen years single center experience in the management of progressive familial intrahepatic cholestasis of infancy. Acta gastro-enterologica Belgica. 2004;67(4):313-319. PubMed
9.Valamparampil JJ, Rinaldhy K, Reddy MS, Shanmugam N, Rela M. 5. Outcomes of liver transplantation for pediatric recipients with progressive familial intrahepatic cholestasis. Paper presented at: 1st National Meeting of the Liver Transplantation Society of India (LTSICON); 2018; India. Published in J Clin Exp Hepatol.
10.Aydogdu S, Cakir M, Arikan C, et al. Liver transplantation for progressive familial intrahepatic cholestasis: clinical and histopathological findings, outcome and impact on growth. Pediatr Transplant. 2007;11(6):634-640. PubMed
11.Hori T, Egawa H, Takada Y, et al. Progressive familial intrahepatic cholestasis: a single‐center experience of living‐donor liver transplantation during two decades in Japan. Clin Transplant. 2011;25(5):776-785. PubMed
12.Kamath BM, Chen Z, Romero R, et al. Quality of life and its determinants in a multicenter cohort of children with Alagille syndrome. J Pediatr. 2015;167(2):390-396 e393. PubMed
13.Hornbrook MC, Wendel CS, Coons SJ, et al. Complications among colorectal cancer survivors: SF-6D preference-weighted quality of life scores. Med Care. 2011;49(3):321-326. PubMed
14.Al-Uzri A, Matheson M, Gipson DS, et al. The impact of short stature on health-related quality of life in children with chronic kidney disease. J Pediatr. 2013;163(3):736-741 e731. PubMed
15.Kini SP, DeLong LK, Veledar E, McKenzie-Brown AM, Schaufele M, Chen SC. The impact of pruritus on quality of life: the skin equivalent of pain. Arch Dermatol. 2011;147(10):1153-1156. PubMed
16.Parmar A, Vandriel SM, Ng VL. Health-related quality of life after pediatric liver transplantation: a systematic review. Liver Transpl. 2017;23(3):361-374. PubMed
17.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Mar 2.
18.National Institute for Health and Care Excellence (NICE). Odevixibat for treating progressive familial intrahepatic cholestasis. (Highly specialised technologies guidance HST17) 2022; https://www.nice.org.uk/guidance/hst17. Accessed 2024 Jan 23.
19.Ontario Ministry of Health and Long Term Care. Ontario Case Costing Initiative (OCCI). https://hsim.health.gov.on.ca/hdbportal/. Accessed 2023 Mar 2.
20.Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4(1):1-12. PubMed
21.Siddiq S, Jimenez-Rivera C, Kuenzig ME, et al. Direct health care costs, health services utilization, and outcomes of biliary atresia: a population-based cohort study. J Pediatr Gastroenterol Nutr. 2020;70(4):436-443. PubMed
22.National Institute for Health and Care Excellence (NICE). Everolimus for preventing organ rejection in liver transplantation. (Technology appraisal guidance TA348) 2015; https://www.nice.org.uk/guidance/ta348. Accessed 2024 Jan 23.
23.Canada wide reimbursement programs. Mississauga (ON): Ostomy Canada Society; 2020: https://www.ostomycanada.ca/wp-content/uploads/2020/05/Canada-Wide-Reimbursement-ProgramsR.pdf. Accessed 2024 Jan 23.
24.Ontario Ministry of Health. Schedule of benefits for laboratory services. 2020 Jul 1; https://www.health.gov.on.ca/en/pro/programs/ohip/sob/. Accessed by sponsor, no date provided.
25.Collective agreement [Hospital central agreement]. Toronto (ON): Ontario Nurses Association; 2023: https://www.ona.org/wp-content/uploads/6-20230331_hospcentralagreement-draft.pdf. Accessed by sponsor, no date provided.
26.NHS Blood and Transplant. Annual report on liver transplantation. Report for 2017/2018 (1 April 2008 – 31 March 2018). 2018; https://nhsbtdbe.blob.core.windows.net/umbraco-assets-corp/13974/nhsbt-liver-transplantation-annual-report-2017-2018.pdf. Accessed 2024 Jan 23.
27.Bowring MG, Massie AB, Chu NM, et al. Projected 20- and 30-year outcomes for pediatric liver transplant recipients in the United States. J Pediatr Gastroenterol Nutr. 2020;70(3):356-363. PubMed
28.Shields M, Connor Gorber S, Janssen I, Tremblay MS. Bias in self-reported estimates of obesity in Canadian health surveys: an update on correction equations for adults. Health Rep. 2011;22(3):35-45. PubMed
29.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bylvay (odevixibat): capsules, 200 mcg, 400 mcg, 600 mcg, and 1200 mcg, oral. Toronto (ON): Medison Pharma Canada Inc.; 2023 June 23.
30.World Health Organisation. WHO growth charts for Canada. 2014; https://www.cpeg-gcep.net/content/who-growth-charts-canada. Accessed 2023 Apr 15.
31.Canadian Pediatric Endocrine Group (CPEG). WHO weight chart for Canada. 2014 revision; https://www.cpeg-gcep.net/content/who-growth-charts-canada. Accessed 2024 Jan 23.
32.Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4(1):25-36. PubMed
33.Pawlikowska L, Strautnieks S, Jankowska I, et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol. 2010;53(1):170-178. PubMed
34.Statistics Canada. Canadian Community Health Survey (CCHS) - 2019. 2019; https://www23.statcan.gc.ca/imdb/p3Instr.pl?Function=assembleInstr&lang=en&Item_Id=1207185. Accessed 2023 Jan 13.
35.Canadian Institute for Health Information (CIHI). Report on transplant, waiting list, and donor statistics. Table 1A (2017-2022). 2023 Jun; https://www.cihi.ca/en/summary-statistics-on-organ-transplants-wait-lists-and-donors. Accessed 2023 Aug 30.
36.Nikeghbalian S, Malekhosseini SA, Kazemi K, et al. The largest single center report on pediatric liver transplantation: experiences and lessons learned. Ann Surg. 2021;273(2):e70-e72. PubMed
37.Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012;36:S26-S35. PubMed
38.Degiorgio D, Colombo C, Seia M, et al. Molecular characterization and structural implications of 25 new ABCB4 mutations in progressive familial intrahepatic cholestasis type 3 (PFIC3). Eur J Hum Genet. 2007;15(12):1230-1238. PubMed
39.Gonzales E, Gardin A, Almes M, et al. Outcomes of 38 patients with PFIC3: Impact of genotype and of response to ursodeoxycholic acid therapy. JHEP Rep. 2023;5(10):100844. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Progressive Familial Intrahepatic Cholestasis
Treatment | Strength/ concentration | Form | Price | Recommended dosage | Daily cost ($)a | Course or annual cost ($)a |
|---|---|---|---|---|---|---|
Odevixibat (Bylvay) | 200 mcg 400 mcg 600 mcg 1,200 mcg | Oral capsule | 175.9247b 351.8493 527.7740 1,055.5480 | 40 mcg/kg administered orally once daily in the morning. If adequate clinical response has not been achieved after 3 months of continuous therapy, the dose may be increased to 120 mcg/kg/day, with a maximum daily dose of 7,200 mcg per day. | Low dose for patients 4 kg to 77 kg: 176 to 2,111 High dose for patients 4 kg to > 55 kg: 528 to 6,333 | Low dose for patients 4 kg to 77 kg: 64,256 to 771,078 High dose for patients 4 kg to > 55 kg: 192,769 to 2,313,233 |
Note: Recommended dosages are from the respective product monographs2 unless otherwise indicated. CADTH assumed a patient weight ranging from 4 kg to 77 kg, representing a pediatric patient of 6 months of age and the average adult patient in Canada, respectively.28
aLow dose refers to the initiation dose of odevixibat, 40 mcg/kg. High dose refers to the indicated dose escalation dosing of 120 mcg/kg as per the product monograph.2
bSponsor-submitted price.1
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing. | No | Refer to the issue for consideration regarding the patient population of PFIC and lack of data for subtypes outside of PFIC1 and PFIC2. |
Model has been adequately programmed and has sufficient face validity. | Yes | No comment. |
Model structure is adequate for decision problem. | No | Refer to the critical appraisal point pertaining to model structure regarding the definition of response assessment and lack of clinical evidence to support several assumptions used to define the movement of patients between the model health states. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis). | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem. | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough detail). | No | The submission lacked clarity and detail in the technical report (e.g., calculation of drug acquisition costs, calculations for proportion of patients expected to dose escalate). When adapting the original model to the Canadian submission, there were inconsistencies in the labelling of response and the key used to define health states in the “Transitions” sheet was unclear. |
PFIC = progressive familial intrahepatic cholestasis.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
LT = Liver transplant; PEBD = Partial external biliary diversion
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Odevixibat + SOC | SOC alone | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 35.26 | 31.87 | 3.39 |
Response | 11.02 | 0.00 | 11.02 |
Loss of response | 9.05 | 7.08 | 1.96 |
PEBD response | 0.00 | 3.27 | –3.27 |
PEBD nonresponse | 0.00 | 2.85 | –2.85 |
LT | 0.71 | 0.84 | –0.13 |
Post-LT | 14.48 | 17.82 | –3.34 |
Discounted QALYs | |||
Total | 27.60 | 22.80 | 4.80 |
Response | 9.74 | 0.00 | 9.74 |
Loss of response | 7.22 | 5.81 | 1.41 |
PEBD response | 0.00 | 2.08 | –2.08 |
PEBD nonresponse | 0.00 | 1.63 | –1.63 |
LT | 0.49 | 0.59 | –0.10 |
Post-LT | 10.16 | 12.70 | –2.54 |
Discounted costs ($) | |||
Total | 9,853,023 | 251,078 | 9,601,944 |
Response | 9,538,519 | 0 | 9,538,519 |
Loss of response | 137,935 | 21,951 | 115,984 |
PEBD | 0 | 20,010 | –20,010 |
LT | 129,724 | 153,370 | –23,646 |
Post-LT | 35,306 | 42,077 | –6,771 |
Immunosuppression | 8,742 | 10,355 | –1,612 |
Adverse events | 2,796 | 3,315 | –520 |
ICER ($/QALY) | 2,000,828 | ||
ICER = incremental cost-effectiveness ratio; LT = liver transplant; LY = life-year; PEBD = partial external biliary diversion; QALY = quality-adjusted life-year.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 11: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Odevixibat + SOC | SOC alone | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 42.23 | 40.96 | 1.27 |
Response | 11.01 | 0.00 | 11.01 |
Loss of response | 9.29 | 7.21 | 2.08 |
PEBD response | 0.00 | 3.36 | −3.36 |
PEBD nonresponse | 0.00 | 2.92 | −2.92 |
LT | 0.73 | 0.87 | −0.13 |
Post-LT | 21.20 | 26.61 | −5.41 |
Discounted QALYs | |||
Total | 32.39 | 29.59 | 2.80 |
Response | 9.74 | 0.00 | 9.74 |
Loss of response | 7.43 | 5.93 | 1.50 |
PEBD response | 0.00 | 2.68 | −2.68 |
PEBD nonresponse | 0.00 | 1.66 | −1.66 |
LT | 0.50 | 0.60 | −0.10 |
Post-LT | 14.71 | 18.71 | −3.99 |
Discounted costs ($) | |||
Total | 9,955,070 | 266,872 | 9,688,198 |
Response | 9,626,819 | 22,456 | 9,626,819 |
Loss of response | 140,085 | 20,404 | 117,629 |
PEBD | 0 | 155,231 | −20,404 |
LT | 131,017 | 49,919 | −24,214 |
Post-LT | 41,610 | 15,576 | −8,308 |
Immunosuppression | 12,764 | 3,287 | −2,812 |
Adverse events | 2,774 | 22,456 | −513 |
ICER ($/QALY) | 3,462,139 | ||
ICER = incremental cost-effectiveness ratio; LT = liver transplant; LY = life-year; PEBD = partial external biliary diversion; QALY = quality-adjusted life-year.
Scenario Analyses
Table 12: Scenario Analyses Conducted on the CADTH Reanalysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CADTH base case | SOC alone | 266,872 | 29.59 | Reference |
Odevixibat + SOC | 9,955,070 | 32.39 | 3,462,139 | |
CADTH scenario 1: No dose escalation to 120 mcg/kg | SOC alone | 268,875 | 29.54 | Reference |
Odevixibat + SOC | 5,220,705 | 31.76 | 2,237,178 | |
CADTH scenario 2: Initial treatment response at 12 weeks | SOC alone | 273,270 | 29.58 | Reference |
Odevixibat + SOC | 9,945,724 | 32.14 | 3,778,582 | |
CADTH scenario 3: Response assessment based on pruritis, assume patients who respond to SOC maintain response | SOC alone | 276,263 | 30.90 | Reference |
Odevixibat + SOC | 12,222,051 | 33.11 | 5,401,698 | |
CADTH scenario 4: Response assessment based on pruritis, assume patients who respond to SOC lose response quickly | SOC alone | 277,604 | 29.75 | Reference |
Odevixibat + SOC | 12,222,051 | 33.11 | 3,557,248 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; ref = reference; SOC = standard of care.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA) estimating the incremental budget impact of reimbursing odevixibat for the treatment of progressive familial intrahepatic cholestasis (PFIC) in patients aged 6 months or older.29 The BIA was undertaken from the perspective of a Canadian public payer over a 3-year time horizon (2025 to 2027) using an epidemiologic approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits Program. Data to inform the model were obtained from various sources, including the published literature, the sponsor’s internal data, and input from clinical experts consulted by the sponsor. Key inputs to the BIA are documented in Table 15.
The sponsor compared a reference scenario in which patients received SOC alone to a new drug scenario in which odevixibat was reimbursed as an add-on therapy to SOC. The sponsor’s analysis included drug acquisition costs for odevixibat based on the sponsor’s submitted price. SOC was assumed by the sponsor to comprise nonspecific therapy of symptoms such as off-label use of UDCA, rifampicin, antihistamines, and naltrexone. No costs for SOC were included in the model as it was received by all patients regardless of odevixibat use. The annual costs of odevixibat estimated by the sponsor were based on the product monograph, and all patients started on 40 mcg/kg dosing.2 Patient weight was estimated to be 16.2 kg for pediatric patients and 64.6 kg for adult patients, based on published World Health Organization (WHO) weight and height tables.30,31 Nonresponders to the initial 40 mcg/kg dosing switched to 120 mcg/kg dosing as per the proposed dosing in the product monograph after 3 months. In the base case of the analysis, all patients on either dosing regimen were assumed to continue treatment through the time horizon of the BIA.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Incidence of PFIC Infant (< 1 year) | 1 in 50,00020 |
Prevalence of PFIC Pediatric (1 to 19 years) Adult (> 20 years) | 1 in 75,000a 1 in 100,000a |
Proportion without LT Pediatric (1 to 19 years) Adult (> 20 years) | 85.0%32 4.3%33 |
Proportion experiencing post-LT recurrence Pediatric (1 to 19 years) Adult (> 20 years) | 7.2%a |
Proportion of patients with public coverage | Jurisdiction and age-specific: 25% to 100%34 |
Number of patients eligible for the drug under review | 70 / 78 / 83 |
Market uptake (3 years) | |
Uptake (reference scenario) Odevixibat plus SOC SOC | 0% / 0% / 0% 100% / 100% / 100% |
Uptake (new drug scenario) Odevixibat plus SOC SOC | 90% / 95% / 95% 10% / 5% / 5% |
Cost of treatment (per patient) | |
Cost of treatment over 1 year Odevixibat pediatric (low dose) Odevixibat pediatric (high dose) Odevixibat adult (low dose) Odevixibat adult (high dose) SOC | $192,769b $578,308b $771,078b $2,313,233b $0c |
LT = liver transplant; PFIC = progressive familial intrahepatic cholestasis; SOC = standard of care.
aBased on the sponsor’s assumption, clinical expert opinion, or internal estimates.
bBased on an average weight of 16.2 kg for pediatric patients and 64.6 kg for adult patients, estimated using published WHO weight and height tables.30,31
cSOC assumed to be UDCA, rifampicin, antihistamines, and naltrexone.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the 3-year budget impact of reimbursing odevixibat for the treatment of PFIC in patients aged 6 months or older would be $137,944,244 (year 1: $38,747,085; year 2: $48,475,248; year 3: $50,721,911).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the results of the BIA:
Uncertainty regarding the sponsor’s epidemiological approach to calculate target population: The sponsor used the incidence of PFIC and various assumptions surrounding prevalence to estimate the target population. The reported incidence of PFIC is estimated to be between 1 in 50,000 and 1 in 100,000.20 The sponsor stated that estimates of PFIC prevalence do not exist due to the rarity of the disease, and it was therefore assumed that incidence was 1 in 50,000 for patients younger than 1 year old, 1 in 75,000 for pediatric patients aged 1 to 19 years, and 1 in 100,000 for adult patients aged 20 years or older). The assumption of decreasing prevalence was attributed to disease mortality; however, the clinical experts consulted by CADTH suggested that the estimate of incidence in patients younger than 1 year did not meet face validity. CADTH took an alternate approach to provide an updated estimate using the average number of pediatric LTs occurring each year in Canada (46),35 the proportion of pediatric LTs that are for PFIC (13%),36 and the proportion of PFIC patients under 18 who receive an LT annually based on NAPPED data (6%).5,6 If 6 LTs a year are for pediatric PFIC patients (aged 0 to 17) and 6% of pediatric PFIC patients are expected to receive an LT each year then from this a prevalence of 100 patients can be estimated. This translates into approximately 1 in 73,000. Accounting for mortality an incidence estimate of approximately 1 in 70,000 was assumed, which was validated by clinical experts. CADTH notes this value is also roughly the midpoint of the published incidence estimates making it the most robust number to consider.
Additionally, the sponsor used native liver survival to estimate the proportion of patients without LT. It was assumed that patients receiving successful LT would no longer require treatment with odevixibat. Native liver survival was estimated to be 85% in pediatric patients32 and 4.3% in adult patients.33 Clinical experts indicated that the proportion of pediatric patients with native liver survival appeared to be overestimated and CADTH estimated this would likely be approximately 60% based on available data from NAPPED,5,6 which is supported by the sponsor’s pharmacoeconomic model predictions across the pediatric cohort. For adults, clinical experts indicated that nearly all patients will have received an LT, and native liver survival was estimated to be closer to 1%.
CADTH adjusted the estimate of PFIC incidence to 1 in 70,000 to reflect updated estimates based on available Canadian data.
Estimates of native liver survival in pediatric and adult patients were adjusted to reflect clinical expert input and data from NAPPED.
Treatment with odevixibat due to post-LT recurrence is likely overestimated in adults: In the derivation of the target population, the sponsor assumed patients who underwent successful LT would not experience disease recurrence and therefore not require further treatment with odevixibat, as described above. However, a small proportion of patients with PFIC2 are expected to experience disease recurrence after LT. The sponsor estimated that 10% to 20% of patients with PFIC2 would recur, and 50% of recurring patients would be eligible for odevixibat based on the approximate breakdown of PFIC1 and PFIC2 subtypes in the PEDFIC 1 trial, resulting in an overall estimate of 7.2% of patients who will experience disease recurrence and be eligible for odevixibat. This estimate of recurrence was applied to both pediatric and adult patients. According to the clinical experts consulted by CADTH, however, this estimate was likely overestimated for adult patients. Clinical experts stated that adult PFIC patients experiencing disease recurrence after LT are extremely rare and have not been observed in clinical practice across 4 transplant centres in Canada. Instead, it was noted that treatment burden in adults would likely be driven by chronic rejection and other diseases related to cholestatic LTs. There is considerable uncertainty surrounding the true estimate of recurrence in adult and pediatric patients; however, this was likely to be relatively infrequent according to clinical experts consulted by CADTH.
CADTH adjusted post-LT recurrence requiring treatment with odevixibat to be 0% for adult patients.
The anticipated dose escalation of odevixibat and drug acquisition costs are highly uncertain: The drug acquisition costs of odevixibat are highly influential and are based on patient weight and the proposed dose escalation from 40 mcg/kg to 120 mcg/kg upon failure to achieve treatment response after an initial 3 months of treatment.2 As stated in the critical appraisal section of the cost-utility analysis, the CADTH Clinical Review concluded that there is limited clinical evidence to support dose escalation in the manner it is described in the product monograph. Based on clinical experts consulted by CADTH, it is uncertain how dose escalation may occur in clinical practice given the limited clinical rationale to establish the initial and up-titrated dose for odevixibat. If dose escalation were to occur, clinical experts indicated that it may be on a case-by-case basis and would also occur incrementally (i.e., from 40 mcg/kg to 80 mcg/kg).
Therefore, there is considerable uncertainty surrounding how dose escalation may occur in clinical practice, which has sizable impacts on the estimated drug acquisition costs of odevixibat, and therefore the cost-effectiveness estimates. Based on the estimated annual costs of odevixibat, the cost for an adult weighing 77 kg (i.e., average weight of an adult in Canada) is $771,078 if they receive 40 mcg/kg per day and is capped at $2,313,233 if they receive 120 mcg/kg per day. There is an additional cost of $1,542,156 per patient per year associated with tripling the dose of odevixibat, which would occur for the remainder of the patient’s lifetime in those for whom odevixibat is effective. The estimated incremental differences in costs due to dose escalation of odevixibat from 40 mcg/kg to 120 mcg/kg is a key driver of budget impact estimates.
CADTH assessed the impact of excluding dose escalation of odevixibat from 40 mcg/kg to 120 mcg/kg in a scenario analysis.
Uncertainty in the definition of PFIC and variation across disease subtypes: The pivotal PEDFIC 1 trial only included patients with PFIC1 or PFIC2. However, the clinical experts indicated there are considerable differences in disease mechanism between PFIC1 and PFIC2 compared with PFIC3. Based on available literature, the disease mechanism of PFIC3 is related to toxic bile production, whereas PFIC1 and PFIC2 are related to bile production itself.37,38 According to published literature, PFIC1 and PFIC2 make up approximately two-thirds of all PFIC cases, with PFIC3 representing the remaining third of cases.37 The clinical experts indicated that PFIC1 and PFIC2 are associated with cholestatic pruritus, however, the pruritus associated with PFIC3 appears to be more variable, which results in uncertainty surrounding whether iBAT inhibitors would be as relevant for treatment as they are for PFIC1 and PFIC2.39 For example, literature supports that UDCA is a first-line treatment for PFIC3 and can result in improved pruritus.39 However, despite differences in disease mechanism and clinical presentation, the clinical experts did suggest that iBAT inhibitors may be still considered as treatment for PFIC3 patients if pruritus is severe.
CADTH conducted a scenario analysis that removes PFIC3 patients from treatment eligibility based on clinical expert input, such that the budget impact of treating PFIC1 and PFIC2, aligned with the available PEDFIC trial data, may be assessed separately.
The proportion of patients eligible for public drug plan coverage is uncertain: The sponsor estimated jurisdiction-specific public coverage for treatment eligibility using the Statistics Canada Canadian Community Health Survey 201934 and assuming that Maritime provinces with 2 or fewer patients with PFIC would have 100% coverage. However, public coverage estimates for select provinces appeared uncertain according to clinical experts consulted by CADTH. Clinical experts indicated that the expected coverage rate for adults generally appeared to meet face validity, but it was unlikely that pediatric coverage rates would be lower than 100% given the rarity of the disease and high drug acquisition costs of odevixibat.
CADTH adjusted the public coverage rates for Alberta, Manitoba, and Ontario to reach 100% in a scenario analysis.
CADTH Reanalyses of the BIA
Table 15: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Adjusted incidence of PFIC | 1 in 50,000 | 1 in 70,000 |
2. Native liver survival for pediatrics and adults | Pediatric: 85% Adult: 4.3% | Pediatric: 60% Adult 1% |
3. Post-LT disease recurrence requiring odevixibat in adults | 7.2% | 0% |
CADTH base case | Reanalysis 1 + 2 + 3 | |
LT = liver transplant; PFIC = progressive familial intrahepatic cholestasis.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17.
CADTH’s reanalysis found that funding odevixibat for the treatment of PFIC patients aged 6 months or older resulted in a budget impact of $16,531,305 in year 1, $21,046,984 in year 2, and $22,429,894 in year 3, for a cumulative budget impact of $60,008,183 across the 3-year time horizon.
CADTH conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 17.
Assuming dose escalation from 40 mcg/kg to 120 mcg/kg does not occur if response is not achieved after 3 months.
Assuming public coverage of 100% for all patients across all jurisdictions.
Assessing a target population that only includes patients with PFIC1 or PFIC2, aligned with the available PEDFIC trial data.
Assuming an incidence of 1 in 200,000.
Assume assessment of response is based on pruritus (assume 58.31% respond and remain on 40 mcg/kg and nonresponders move onto 120 mcg/kg).
The budget impact of funding odevixibat ranged from $29,573,995 to $79,601,429 across CADTH scenario analyses. The scenario analysis that excluded dose escalation from 40 mcg/kg to 120 mcg/kg resulted in a decreased 3-year budget impact of $29,573,995. CADTH notes that this decrease in the estimated budget impact is due to patients remaining on the initial 40 mcg/kg dose of odevixibat for the full 3-year time horizon instead of dose-escalating to 120 mcg/kg upon nonresponse. Alternatively, a scenario analysis in which more patients respond to the 40 mcg dose (58.31% versus 43.50%) results in a lower budget impact as fewer patients are escalated to a higher 120 mcg/kg dose.
Table 16: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 137,944,244 |
CADTH reanalysis 1: Incidence of PFIC | 133,915,054 |
CADTH reanalysis 2: Native liver survival in pediatrics and adults | 104,489,141 |
CADTH reanalysis 3: Post-LT recurrence | 98,827,511 |
CADTH base case | 60,008,183 |
BIA = budget impact analysis; LT = liver transplant; PFIC = progressive familial intrahepatic cholestasis.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|
Submitted base case | Reference | 0 | 0 | 0 | 0 |
New drug | 38,747,085 | 48,475,248 | 50,721,911 | 137,944,244 | |
Budget impact | 38,747,085 | 48,475,248 | 50,721,911 | 137,944,244 | |
CADTH base case | Reference | 0 | 0 | 0 | 0 |
New drug | 16,531,305 | 21,046,984 | 22,429,894 | 60,008,183 | |
Budget impact | 16,531,305 | 21,046,984 | 22,429,894 | 60,008,183 | |
CADTH scenario analysis: 98.6% price reduction | Reference | 0 | 0 | 0 | 0 |
New drug | 231,438 | 294,658 | 314,019 | 840,115 | |
Budget impact | 231,438 | 294,658 | 314,019 | 840,115 | |
CADTH scenario analysis 1: No dose escalation to 120 mcg/kg | Reference | 0 | 0 | 0 | 0 |
New drug | 8,946,353 | 10,021,867 | 10,605,775 | 29,573,995 | |
Budget impact | 8,946,353 | 10,021,867 | 10,605,775 | 29,573,995 | |
CADTH scenario analysis 2: 100% public coverage | Reference | 0 | 0 | 0 | 0 |
New drug | 21,974,202 | 27,924,671 | 29,702,556 | 79,601,429 | |
Budget impact | 21, 974,202 | 27, 924,671 | 29,702,556 | 79,601,429 | |
CADTH scenario analysis 3: Restrict to patients with PFIC1 or PFIC2 patients only | Reference | 0 | 0 | 0 | 0 |
New drug | 11,020,870 | 14,031,323 | 14,953,263 | 40,005,455 | |
Budget impact | 11,020,870 | 14,031,323 | 14,953,263 | 40,005,455 | |
CADTH scenario analysis 4: Incidence of 1 in 200,000 | Reference | 0 | 0 | 0 | 0 |
New drug | 15,251,975 | 18,816,253 | 19,392,520 | 53,460,749 | |
Budget impact | 15,251,975 | 18,816,253 | 19,392,520 | 53,460,749 | |
CADTH scenario analysis 5: Assessment of response based on pruritus | Reference | 0 | 0 | 0 | 0 |
New drug | 14,540,955 | 18,153,908 | 19,327,154 | 52,022,018 | |
Budget impact | 14,540,955 | 18,153,908 | 19,327,154 | 52,022,018 |
BIA = budget impact analysis; PFIC = progressive familial intrahepatic cholestasis.
Ethics Review
Abbreviations
PFIC
progressive familial intrahepatic cholestasis
PFIC1
progressive familial intrahepatic cholestasis type 1
PFIC2
progressive familial intrahepatic cholestasis type 2
sBA
serum bile acid
Summary
Progressive familial intrahepatic cholestasis (PFIC) comprises a rare group of inherited heterogeneous genetic disorders characterized by anomalies in bile acid secretion or transport and a spectrum of life-threatening critical liver complications. The resulting buildup of bile in the liver can have damaging effects, such as pruritis (itching), which can be severe enough to disrupt daily life and sleep.
Patient group, clinician group, clinical expert, and drug program input as well as relevant literature was gathered in the course of this CADTH review and were reviewed to identify ethical considerations relevant to the use of odevixibat for PFIC.
Ethical considerations identified in this review include those related to the following:
Diagnosis, treatment, and experiences of PFIC: Ethical considerations in the context of PFIC highlighted patients and caregivers experience a tremendous physical, psychosocial, and financial burden from the unremitting pruritis (itching) that is associated with this disease. Addressing referral bias, where referrals may be influenced by illness severity, and ensuring early diagnosis of PFIC where possible, is important in preventing needless suffering and reducing the burden on the health care system. There is an unmet need for an effective disease-modifying treatment for pruritis in PFIC, given its devastating impacts on patients and their families. Surgical treatment alternatives, such as a liver transplant, are invasive and life-altering.
Clinical and economic evidence used in the evaluation of odevixibat: Some clinically meaningful outcomes were identified in the trials used to evaluate odevixibat, including a significant reduction in serum bile acids (sBAs) and symptom relief. These studies also exhibit considerable evidentiary uncertainty. Specifically, uncertainty arises when: attempting to extend efficacy results beyond the study population of patients with PFIC subtypes 1 (PFIC1) and 2 (PFIC2), correlating sBA to pruritis, and considering durations exceeding the study’s 24-week time frame. Ensuring that patients are adequately informed of these evidentiary uncertainties in a shared decision-making process and that health care resources are distributed fairly and equitably, are important steps in addressing these ethical concerns.
Clinical use and implementation of odevixibat: The clinical experts indicated odevixibat has some promise, given its potential to address some unmet needs for the treatment of PFIC-associated pruritis with a favourable safety profile. However, it is essential to emphasize the importance of equitable access with regard to continuity of care and access as pediatric patients become adults.
Health systems: The reimbursement of odevixibat brings to the forefront a complex array of ethical considerations, including those related to opportunity costs and resource allocation in the context of uncertain evidence, as well as those related to equitable access and ensuring sufficient infrastructures to support continuity of care and access.
Objective
To identify and describe ethical considerations associated with the use of odevixibat for the treatment of pruritis in patients aged 6 months or older with PFIC, including considerations related to the context of PFIC, evidentiary basis, use of odevixibat, and health systems considerations.
Research Questions
This report addresses the following research questions:
What ethical considerations arise in the context of PFIC?
What ethical considerations arise related to the evidence (e.g., clinical and economic data) used to evaluate odevixibat?
What ethical considerations arise in the use of odevixibat for patients, their caregivers, and clinicians?
What are the ethical considerations for health systems involved in the context of odevixibat?
Methods
To identify ethical considerations relevant to the use of odevixibat in the treatment of PFIC, this Ethics Review was driven by relevant questions identified in the EUnetHTA Core Model 3.0, Ethics Analysis Domain1 and supplemented by relevant questions from the Equity Checklist for Health Technology Assessments (ECHTA).2 These guiding questions were organized to respond to the research questions posed, and the investigated ethical considerations related to the following:
The patients living with PFIC and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges related to diagnosis or clinical care; factors that might prevent patients from gaining access to therapies).
The evidence used to evaluate the benefits, harms, and value of odevixibat (i.e., ethical considerations in relevant clinical trials, including their representativeness, choice of outcome measures, and appropriateness of analytical methods and models to all population groups, and ethical considerations related to the data or assumptions in the economic evaluation).
The use of odevixibat, including considerations related to the benefits and harms to patients, relatives, caregivers, clinicians, or society, and considerations related to access to these therapies.
The uptake of odevixibat in health systems, including considerations related to the distribution of health care resources.
Data Collection: Review of Project Inputs and Literature
The data informing this Ethics Review Report drew from the ethical considerations (e.g., values, norms, or implications related to the harms, benefits, and implications for equity, justice, resource allocation, and ethical considerations in the evidentiary basis) identified in the patient and clinician group, clinical expert, and drug program input collected by CADTH to inform this review, as well as a complementary search of the published literature. Ongoing collaboration and communication with CADTH reviewers working on the clinical and economic reviews for this submission also assisted in the clarification and identification of the ethical considerations raised.
Review of Project Inputs
During this CADTH review, a single reviewer collected and considered input from 6 main sources of content related to ethical considerations relevant to addressing the research questions guiding this Ethics Review. In addition to published literature, this report considered the following sources:
The sponsor submission, including noting relevant information and external references or sources relevant to each of the research questions driving this report.
One clinician group input received by CADTH from the Canadian Association for the Study of the Liver.
One patient input received by CADTH from the Canadian Liver Foundation.
Drug program input received by CADTH from drug programs participating in the CADTH Reimbursement Review process.
Discussion with 4 clinical experts directly engaged by CADTH over the course of this Reimbursement Review, including through 1 clinical and 1 economic consultation meeting involving 2 experts and 1 panel meeting involving 4 experts. During each of these meetings, the clinical experts were asked targeted questions related to ethical considerations corresponding to the research questions driving this report. All the clinical experts were practising pediatric hepatology, liver transplants, and gastroenterology.
Engagement with the CADTH clinical and economic reviewers to identify domains of ethical interest arising from their respective reviews and to identify relevant questions and sources to further pursue in this report.
Literature Search Methods
An information specialist conducted a literature search on key resources including MEDLINE through Ovid, Philosopher’s Index through Ovid, APA PsycINFO through Ovid, the Cumulative Index to Nursing and Allied Health Literature (CINAHL) through EBSCO, Scopus, and Google Scholar. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings) and keywords. The main search concepts were Bylvay (odevixibat) and progressive familial intrahepatic cholestasis.
CADTH-developed search filters were applied to limit retrieval to citations related to ethical concepts or considerations. Duplicates were removed by manual deduplication in EndNote. The search was completed on July 21, 2023. The search strategy is available on request.
Literature Screening and Selection
The literature retrieved according to the search and selection methods detailed previously was screened in 2 stages. First, titles and abstracts of the retrieved citations were screened by a single reviewer for relevance. Articles were identified and retrieved for full-text review by a single reviewer if their titles or abstracts identified ethical considerations or provided normative analyses (i.e., focusing on “what ought to be” through argumentation), or presented empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to: the experiences, incidence, diagnosis, treatment, or outcomes of PFIC; or the evidence on, use of, or implications of odevixibat for patients with PFIC. In the second stage, full-text publications categorized as “retrieve” were reviewed by the same reviewer. Texts that included substantive information meeting the aforementioned criteria were included in the review, and reports that did not meet these criteria were excluded. As a parallel process, other sources drawn from relevant bibliographies were retrieved and reviewed using the selection criteria listed previously.
Data Analysis
Data analysis was driven by the 4 research questions guiding this report and included the collection, coding, and thematic analysis of data drawn from the literature and project inputs. The reviewer conducted 2 iterative cycles of coding and analysis to abstract, identify, and synthesize relevant ethical considerations from the literature and from relevant project inputs.
In the initial coding phase, publications and input sources were reviewed for ethical content (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis). Once identified, claims related to ethical content were coded using methods of qualitative description.3 In the second coding phase, major themes and subcodes were identified through repeated readings of the data3 and summarized into thematic categories within each guiding domain or research question. Where ethical content did not fit into these categories or the domains outlined in the research questions, this was noted, as were discrepancies or conflicts between ethical considerations or values identified between project sources or within thematic categories. The data analysis was iterative, and the themes identified in the literature, in project inputs, and during consultations with the clinical experts were used to further refine and reinterpret the ethical considerations identified.
The data collected and analyzed from these sources were thematically organized and described according to the 4 research questions and domains driving this report. The results of this analysis and its limitations and conclusions are described subsequently.
Results
Description of Included Sources
The data to inform this Ethics Review were drawn from a review of patient group input, clinician group input, drug program input, and consultation with clinical experts engaged by CADTH for this review. All the clinical experts were active in relevant clinical roles in Canada and have experience treating patients (pediatric and adult) with PFIC and/or using odevixibat. A description and summary of these sources are included in the Clinical Review Report.
The literature search identified 70 results. Following title and abstract screening, 18 citations were excluded and 52 potentially relevant publications from the electronic searches were retrieved for full-text review. Of the potentially relevant publications, 32 publications were excluded, as they did not discuss the ethical considerations of odevixibat or PFIC (n = 30) or were not published in English (n = 2). Twenty publications met the inclusion criteria and were included in this report. One additional publication was retrieved from backward searching of the included publications’ reference lists.
A total of 21 publications were used to inform this report. Of these, 19 publications discussed ethical considerations in the context of PFIC, including those related to diagnosis and treatment, and 2 publications discussed patient and/or family and caregiver experiences in the context of PFIC. Details regarding the characteristics of the included publications are reported in Table 1.
Key Ethical Considerations
Diagnosis, Treatment, and Experiences of PFIC
Diagnosis
As noted in the Clinical Report, PFIC comprises a rare, progressive, and fatal group of inherited heterogeneous genetic disorders. PFIC is estimated to affect between 1 in every 50,000 to 100,000 children born worldwide.4,5 While global or country-specific prevalence estimates are not available for PFIC, it is believed to be responsible for approximately 10% to 15% of children with cholestatic liver diseases and 10% to 15% of liver transplant indications in children.4,5 This disorder is characterized by anomalies in bile acid secretion or transport.6 Bile, a liquid made in the liver, is released into the gut to aid digestion. In cholestatic liver diseases such as PFIC, there is an interruption in bile flow that can lead to buildup. A buildup of bile can have damaging effects, such as pruritis (itching), which can be severe enough to disrupt daily life and sleep.7 Other common clinical manifestations include cholestasis and jaundice, which typically emerge during infancy or early childhood, although symptoms can emerge in adulthood as well.6 As reported in the Clinical Report, without surgical intervention, 50% of patients with PFIC live beyond the age of 10 years, and almost none live to the age of 20 years.8 PFIC is also associated with a spectrum of life-threatening critical liver complications, including portal hypertension, liver failure, cirrhosis, and hepatocellular carcinoma, along with additional manifestations beyond the liver.6
Symptom onset commonly occurs during infancy and early childhood. Pediatricians, general practitioners, and emergency department physicians will typically refer the child to a pediatric hepatologist or gastroenterologist if they suspect liver complications. However, the availability of pediatric gastroenterologists is limited in Canada, with several provinces lacking capacity and access to this specialized medical field, posing concerns regarding delays in accessing a timely diagnosis and routine treatment and care.9 A combination of clinical, laboratory, and biochemical approaches and exclusion of other causes of congenital cholestasis have historically been used to diagnose PFIC; however, molecular genetic testing has recently become the gold standard and, if appropriate referrals are made, is available across Canada.6,10
Early diagnosis is essential for initiating appropriate interventions and treatments for PFIC. Timely identification not only improves the quality of life for affected individuals but also helps to manage health care costs by avoiding more complex and costly interventions that may become necessary as the disease advances. One challenge to early diagnosis is referral bias, which occurs in clinical practice when the choice to refer patients for a medical intervention is influenced by the severity of their illness. The clinical experts noted that referral bias may be present in the diagnosis of PFIC. Additionally, expert clinicians noted they believe the incidence of PFIC to be higher than reported, as there are patients with milder or less severe forms of PFIC that do not get referred to specialists. If health care providers primarily refer patients with more severe symptoms, those with early or less severe symptoms might not receive timely evaluation and diagnosis. This delay can lead to symptoms worsening and disease progression. Addressing referral bias and ensuring early diagnosis of PFIC is therefore important in preventing unnecessary suffering and reducing the burden on the health care system.
The clinical experts also noted that a limitation in molecular genetic testing for PFIC is that it may not always detect all variants or subtypes. The type of PFIC is identified based on genetic defect, clinical presentation, laboratory findings, and liver histology.8 To date, there are 6 known subtypes of PFIC, with the most common being PFIC 1 to 3, although novel genetic mutations are continually being discovered.10 As noted in the Clinical Report, patients with PFIC1 and PFIC2 generally present with jaundice and severe pruritus in the first few months of life, with 78% developing jaundice before the age of 12 months. PFIC type 3 can occur during infancy, childhood, and even into young adulthood. While the genetic mutations causing the various PFIC subtypes vary, a shared characteristic among all subtypes is the presence of elevated sBAs levels and severe pruritus. As noted in the CADTH Clinical Review Report, adult patients with PFIC are relatively rare, more challenging to diagnose, and experience less severe disease. These adults may or may not have pruritus.
As PFIC is inherited from both parents, siblings of an affected individual have a 25% likelihood of being affected as well. Due to the inherited nature of the disease, it has been recommended that when a new case is identified, both parents and siblings undergo screening for the disease.6,11 Researchers have also recommended antenatal diagnosis for families affected by PFIC.11 Additionally, there are regions where there are more frequent cases of PFIC, such as areas in Greenland.12 According to the literature and the clinical experts consulted by CADTH, genetic counselling and carrier screening are strongly recommended in populations that have a higher prevalence of PFIC.12
Current Treatment Landscape
Historically, treatments for PFIC were predominantly supportive in approach. These included nutritional support, addressing symptoms (e.g., antipruritis medications, medications to reduce bile salts), and employing complementary and alternative methods.7,10 Various surgical techniques have been introduced, including interventions that redirect bile flow. Early biliary diversion surgery became the standard approach for managing drug-resistant pruritus and postponing liver transplant in cases of PFIC.10 Other surgical interventions that avoid the creation of a permanent stoma involve establishing an internal diversion for bile. Examples of such techniques include ileal bypass, ileal exclusion, and cholecystocolostomy.13,14 These surgical procedures may improve the elimination of bile acids, ameliorate pruritus, and/or diminish the need for a liver transplant, although there are important risks and side effects associated with these approaches (e.g., nutritional deficiencies, diarrhea, bowel obstruction). Nevertheless, there is an important need for pharmaceutical and disease-modifying treatments that optimize symptom alleviation and limit the advancement of liver disease.14
Input from the patient group revealed that the currently available off-label medications (i.e., without specific, official indication for PFIC) and therapies have limited effectiveness in reducing pruritis and enhancing overall well-being, comfort, and vitality. While these off-label medications offer some symptomatic relief, they do not stop disease progression or significantly alleviate the symptoms. This is in line with insights provided by the clinical experts consulted during CADTH’s review, who state there is an unmet need within PFIC treatments for targeted, disease-modifying drugs. Specifically, there is a demand for a drug capable of tackling the fundamental pathophysiology of the disease, effectively managing pruritus — especially severe cases — while also slowing the disease’s progression. Both the clinical experts and the Canadian Association for the Study of the Liver have conveyed that currently available medications for PFIC exhibit limited effectiveness and that surgical alternatives carry a high risk of morbidity and mortality.
Patient and Caregiver Experiences of Pruritis and PFIC
The patient and clinician group input, clinical expert input, and the published literature reported that living with PFIC is physically, psychosocially, and financially burdensome for patients and their caregivers.6,7,15,16 According to the patient group input, families conveyed sentiments of powerlessness, distress, and frustration, underscoring how a PFIC diagnosis has profoundly altered the well-being of both their family members and their daily routines. The clinical experts stated that observing their patients with severe pruritus, scratching themselves to the point of bleeding, is deeply distressing. They likened severe pruritus to a form of torture and emphasized the substantial psychosocial impact it can have on families, including marital breakdown from the strain of caring for a medically complex child. Although there is limited literature on patient and caregiver perspectives regarding experiences of pruritus and PFIC,6 researchers have noted that significant pruritus can result in substantial skin damage, resulting in sleep disturbances, heightened irritability, reduced focus, and impaired school performance.8 The clinical experts also noted observing instances where caregivers may believe their child with PFIC is functioning well but are dismayed to learn they performed quite poorly on educational tests because they are so chronically fatigued. The experts also highlighted the financial strain on caregivers caused by missing work to attend medical appointments or provide care for their child. The feedback from the patient group highlighted the importance of improving overall quality of life for patients and caregivers. The group stressed the importance of addressing concerns such as pruritis and sleep disturbances, promoting healthy growth, sustaining energy levels, and slowing disease progression. Simultaneously, the group advocated for the enhancement of therapeutic alternatives available to both patients and health care practitioners.
Ethics of Evidence and Evaluation of Odevixibat
Odevixibat belongs to the drug class of intestinal bile acid transport inhibitors, which operate by hindering the reabsorption of bile acids, thereby diminishing the overall bile acid volume in the bloodstream. This is accomplished by impeding the apical sodium-dependent bile acid transporter in the terminal ileum.10 Odevixibat functions by inhibiting the reabsorption of bile acids within the intestines and can therefore potentially aid in reducing the buildup of harmful bile acids in the liver, leading to the possible alleviation of symptoms, such as pruritus as well as potential improvement in liver function.14 As described in the Clinical Report, the pivotal trial to evaluate odevixibat is PEDFIC 1, a multicentre (1 site in Canada), 24-week, phase III, double-blind, randomized, placebo-controlled study examining the efficacy and safety of this drug in doses of 40 mcg/kg/day and 120 mcg/kg/day in children (n = 62) aged 6 months or older with PFIC1 or PFIC2.15 The median age of the patients was 3.2 years (range, 6 months to 15.9 years). There are also interim results from PEDFIC 2, an ongoing, open-label, 72-week study that evaluates odevixibat in children from PEDFIC 1 (cohort 1) and new patients with PFIC (any age), all of whom received 120 mcg/kg per day.17 However, the evidence presented in these trials used to evaluate odevixibat faces several uncertainties and limitations, which have implications for the clinical and health system adoption of this therapy.
As detailed in the Clinical Report, the primary outcome of the PEDFIC 1 trial was the proportion of patients who experienced at least a 70% reduction in sBA concentration from baseline to the end of treatment or reached a level of 70 μmol/L or lower after 24 weeks of treatment. Secondary outcomes included the proportion of positive pruritus assessments at the patient level over the 24-week treatment period based on the Albireo observer-reported outcome (ObsRO) instrument, growth changes from baseline to week 24, changes in awakenings from baseline in sleep measured with the Albireo patient-reported outcome (PRO) and ObsRO instruments at each 4-week interval over the 24-week treatment period, the proportion of individual assessments meeting the definition of a positive pruritus assessment at the patient level from weeks 0 to 4 and weeks 0 to 12, and the number of patients undergoing biliary diversion surgery or liver transplant.
Findings indicate this drug may effectively reduce both pruritus and sBAs in children with both PFIC subtypes compared with placebo.15 However, the clinical experts emphasized that employing sBAs as a trial end point poses a challenge, as sBA does not always correlate well with pruritis. Specifically, using sBA becomes problematic when considering dose adjustments, particularly when a patient continues to suffer from severe pruritus despite a decrease in their sBA, which would meet the primary metric of clinical effectiveness. Another issue with this end point that experts raised is that certain regions face challenges in obtaining timely sBA testing and results, often necessitating the need to send samples for external analysis. For these reasons, there are limitations in using sBA as an end point to establish the efficacy of odevixibat in this patient population.
As noted in the Clinical Report, the clinical experts state the main treatment goals are to provide relief from pruritis and improve sleeping and quality of life for patients and caregivers to support normal growth and development, prolong adequate liver function (thereby avoiding or delaying time to liver transplant), and avoid PFIC complications such as hepatocellular carcinoma. A key objective in patients with PFIC is to reduce the experience of unremitting pruritis, which can ultimately lead to the need for a liver transplant. The Clinical Report notes that the effect of odevixibat on mortality and the necessity for surgical intervention, however, remains uncertain, as neither outcome was observed during the 24-week study period. While odevixibat may demonstrate an improvement in pruritus, its effect on health-related quality of life remains uncertain due to significant variability in responses and a relatively high amount of missing data. Although sBA decreased as early as 12 weeks of therapy, the clinical significance and the impact of these reductions on mortality risk and risk of surgery is uncertain. Finally, the impact of odevixibat on growth, the frequency of awakenings, and total bilirubin levels is also uncertain, primarily due to the wide variation in individual responses.
Highlighting the limitations of the findings in the PEDFIC 1 study, it is important to acknowledge that the trial’s eligibility criteria, which excluded patients with significant hepatic parameter disruptions (e.g., those with more advanced stages of hepatic impairment), can limit the generalizability of the outcomes to all PFIC patients displaying such traits. PEDFIC 1 enrolled participants exclusively with PFIC subtypes 1 or 2, and although PEDFIC 2 included various subtypes, those outside PFIC1 and PFIC2 were underrepresented. Consequently, evidentiary uncertainty is apparent when attempting to extend efficacy results beyond the confines of the subtype 1 and 2 study population.
Limitations in PEDFIC 2 include a lack of a comparator arm. Further investigation involving the less common subtypes could shed light on the potential advantages of odevixibat for all individuals affected by PFIC, especially as recent findings from a compilation of cases and a literature review suggest odevixibat may not be suitable for all patients across the various subtypes of PFIC.10
Additionally, research into the sustainability of odevixibat’s benefits and risks in long-term use is required.6 There is evidentiary uncertainty when considering the extrapolation of findings beyond the trial’s 24-week duration. While this uncertainty concerning efficacy and safety, especially in the long-term, is not uncommon in the context of drugs for rare diseases, it can hinder the assessment of the balance of harms and benefits of using or forgoing odevixibat, which can impact clinical decision-making. Furthermore, the lack of long-term efficacy data and limitations in assessments of effectiveness6,18 have implications for the pharmacoeconomic assessment of odevixibat because it limits the ability to accurately model and assess its cost-effectiveness. This limitation, which may also impact cost-effectiveness analyses for drugs for rare diseases more broadly, presents challenges for assessing the opportunity costs — or foregone benefits — associated with reimbursing and resourcing a particular intervention over others.19
Ethical Considerations in the Use of Odevixibat
Informed Consent
The clinical experts indicated that despite long-term safety and effectiveness uncertainty, they considered the available evidence sufficient to prescribe odevixibat, especially in the absence of effective alternatives to alleviate suffering associated with unremitting pruritis. However, as previously mentioned, there are children experiencing pruritis who have rarer PFIC subtypes who may not benefit from odevixibat. Despite the clinical promise ascribed to odevixibat, it is important for prescribing clinicians to acknowledge the uncertainties in the evidence as part of informed consent processes. The informed consent process should include both children and their families in a process of shared decision-making that transparently acknowledges possible benefits and adverse effects for all subtypes, as well as the current state of evidence (including uncertainty in safety and efficacy data, especially in the long-term). These conversations should continue in a process of shared decision-making as the evidence base grows. Reobtaining informed consent is also imperative once the child transitions to autonomously making their own health decisions.
Equity of Access
The clinical experts indicated potential challenges to implementing odevixibat equitably for all eligible patients if it were reimbursed in Canada, as well as challenges in ensuring continuity of care and access as pediatric patients become adults. Specifically, according to sponsor-submitted estimates for budget impact analysis, there are some provinces where coverage is anticipated to be minimal for adult populations (e.g., ≤ 30% in Alberta, Ontario, and Manitoba). Many adult patients in these situations would be required to get additional private insurance for coverage, which may cause challenges for child-age patients who receive odevixibat but later lose coverage as they become adults. Although the number of adult patients with PFIC has been noted as minimal, the clinical experts noted that these numbers may increase as more children and adults with PFIC are identified and experience longer survival outcomes. The clinical experts advised that in cases where pediatric patients continue to experience sustained benefits from odevixibat into adulthood, coverage levels should remain consistent. Ensuring fair and equitable access to effective treatments for both pediatric and adult patients is essential, as denying or reducing coverage for adults benefiting from childhood treatments can harm their health and autonomy. Continuity of care is particularly important during the transition from specialized pediatric to adult care settings, especially when services may vary in different geographic locations due to factors such as adults relocating for study, work, or settling in new areas.
The clinical experts indicated that many of their patients, including those who do not have a PFIC diagnosis but experience severe pruritis, will want to gain access to odevixibat. This raises questions about the applicability of odevixibat beyond those with PFIC subtypes 1 and 2, as well as obligations to develop and deliver effective therapies for these groups.
Health Systems Considerations
The reimbursement of odevixibat for pruritis in PFIC raises several ethical considerations relating to health systems and resource allocation. The clinical experts noted that this medication could potentially reduce health care resource utilization often linked to more invasive approaches, such as liver transplant, which is considered a last-resort measure reserved for patients who have exhausted other treatment options. Additionally, they suggested that in pediatric populations, odevixibat may offer the potential for improvements in nutrition, weight gain, concentration, and academic performance.
However, the noted uncertainties in both the incidence rates for PFIC and the evidence used to evaluate odevixibat yield several challenges for health system uptake. An increase in the identification of PFIC patients, and an increasing number of patients surviving to adulthood may yield a higher budget impact of this drug. Further, the noted evidentiary uncertainty concerning the efficacy and safety of odevixibat, especially in the long-term and across patient groups, can hinder the assessment of the balance of harms and benefits of using or forgoing odevixibat, which can impact health system decision-making and resource allocation. These limitations, often encountered in the analysis of drugs for rare diseases, pose challenges in determining the opportunity costs, or the potential foregone benefits, when allocating resources and reimbursement for a specific intervention compared with others.
Limitations
There is very little published literature that discusses the ethical considerations related to the use of odevixibat for the treatment of PFIC, particularly from patient and caregiver perspectives, given both the rarity of the disease and the novelty of the drug under review.
Nonetheless, this does not imply that ethical considerations in the context of odevixibat for PFIC are absent, and this review of ethical considerations was augmented by drawing from additional resources collected in the course of this Reimbursement Review, including patient group, clinician group, and drug program input, and discussion with clinical experts, as well as engagement with CADTH clinical and pharmacoeconomic review teams, to provide a more comprehensive understanding of the ethical considerations related to the use of odevixibat for the treatment of PFIC.
Although this Ethics Review Report drew on and considered patient group, clinician group, drug program, and clinical expert input, it is possible that more direct engagement with key stakeholders (e.g., direct interviews with patients, caregivers, family members, and decision-makers) on their specific experiences with PFIC and/or odevixibat could have offered additional relevant ethical considerations or domains of analysis.
Conclusion
The input from patient groups, clinician groups, and provincial drug programs; direct engagement with clinical experts; and the published literature were reviewed for ethical considerations relevant to the use of odevixibat for the treatment of pruritis in patients with PFIC aged 6 months and older. Severe pruritis associated with PFIC results in a tremendous physical, psychosocial, and financial burden experienced by patients and their families and there is an unmet need for the availability of effective disease-modifying therapies. The input from both the clinical experts and the patient groups conveyed that current off-label medications exhibit limited effectiveness and that surgical alternatives carry a high risk of morbidity and mortality.
Odevixibat is a potential prospective first-line drug that may address at least a portion of these unmet requirements for patients, and clinical trial evidence indicates that this therapy may result in a clinically meaningful decrease in pruritis and sBA levels. There is, however, evidentiary uncertainty concerning its safety and long-term treatment outcomes and quality of life, which limits the assessment of clinical benefits and harms associated with its use as well as the pharmacoeconomic assessment of cost-effectiveness.
Additionally, the implementation and health system uptake of odevixibat raises ethical and equity concerns for families with children afflicted by rarer subtypes of PFIC who may face disparities in accessing treatments like odevixibat, as well as equity-of-access challenges for continuity of access among adult patients who have benefited from odevixibat as children but who may no longer meet public funding eligibility criteria.
References
1.EUnetHTA joint action 2 work package 8, HTA core model version 3.0. Diemen (NL): EUnetHTA 2016: http://www.htacoremodel.info/BrowseModel.aspx. Accessed 2023 Nov 3.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37(1). PubMed
3.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
4.Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4:1-12. PubMed
5.Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4(1):25-36. PubMed
6.Baker A, Kerkar N, Todorova L, Kamath BM, Houwen RH. Systematic review of progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2019;43(1):20-36. PubMed
7.Gwaltney C, Bean S, Venerus M, et al. Development of the Patient- and Observer-Reported PRUCISION instruments to assess pruritus and sleep disturbance in pediatric patients with cholestatic liver diseases. Adv Ther. 2022;39(11):5126-5143. PubMed
8.Jones-Hughes T, Campbell J, Crathorne L. Epidemiology and burden of progressive familial intrahepatic cholestasis: a systematic review. Orphanet J Rare Dis. 2021;16(1):255. PubMed
9.Leddin D, Carroll M, Gillis C, Cehovin A. Gastroenterology practitioner and trainee numbers in Canada 2018: annual report from the Canadian Association of Gastroenterology. J Can Assoc Gastroenterol. 2021;4(2):52-56. PubMed
10.Hüpper MN, Pichler J, Huber W-D, et al. Surgical versus medical management of progressive familial intrahepatic cholestasis—case compilation and review of the literature. Children. 2023;10(6):949. PubMed
11.Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012;36:S26-S35. PubMed
12.Nielsen I, Eiberg H. Cholestasis Familiaris Groelandica: an epidemiological, clinical and genetic study. Int J Circumpolar Health. 2004;63(sup2):192-194.
13.Wassman S, Pfister E-D, Kuebler JF, et al. Quality of life in patients with progressive familial intrahepatic cholestasis: no difference between post-liver transplantation and post-partial external biliary diversion. J Pediatr Gastroenterol Nutr. 2018;67(5):643-648. PubMed
14.Baumann U, Sturm E, Lacaille F, et al. Effects of odevixibat on pruritus and bile acids in children with cholestatic liver disease: phase 2 study. Clin Res Hepatol Gastroenterol. 2021;45(5):101751. PubMed
15.Thompson RJ, Arnell H, Artan R, et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2022;7(9):830-842. PubMed
16.Kamath BM, Abetz-Webb L, Kennedy C, et al. Development of a novel tool to assess the impact of itching in pediatric cholestasis. Patient. 2018;11(1):69-82. PubMed
17.Thompson RJ, Artan R, Baumann U, et al. Interim results from an ongoing, open-label, single-arm trial of odevixibat in progressive familial intrahepatic cholestasis. JHEP Reports. 2023:100782.
18.Slavetinsky C, Sturm E. Odevixibat and partial external biliary diversion showed equal improvement of cholestasis in a patient with progressive familial intrahepatic cholestasis. BMJ Case Reports CP. 2020;13(6):e234185. PubMed
19.Kacetl J, Marešová P, Maskuriy R, Selamat A. Ethical questions linked to rare diseases and orphan drugs - a systematic review. Risk Manag Healthc Policy. 2020;13:2125-2148. PubMed
20.Abdelrhim H, Khan S, Heaton P, Peeka R. Balancing medical and non-accidental causes of multiple fractures in a child with progressive familial intrahepatic cholestasis. Am J Case Rep. 2017;18:1190-1193. PubMed
21.Alam S, Lal BB. Recent updates on progressive familial intrahepatic cholestasis types 1, 2 and 3: outcome and therapeutic strategies. World J Hepatol. 2022;14(1):98. PubMed
22.Al-Hussaini A, Lone K, Bashir MS, et al. ATP8B1, ABCB11, and ABCB4 genes defects: novel mutations associated with cholestasis with different phenotypes and outcomes. J Pediatr. 2021;236:113-123.e112. PubMed
23.Andersen S, Okkels H, Krarup H, Laurberg P. Geographical clustering and maintained health in individuals harbouring the mutation for Greenland familial cholestasis: a population-based study. Scand J Gastroenterol. 2006;41(4):445-450. PubMed
24.Odevixibat. In: LiverTox: Clinical and research information on drug-induced liver injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012. Last updated 2022: https://www.ncbi.nlm.nih.gov/books/NBK579037/. Accessed 2024 Jan 23.
25.Heo Y-A. Odevixibat in progressive familial intrahepatic cholestasis: a profile of its use. Drugs Ther Perspect. 2022;38(7):301-307.
26.Pfister ED, Dröge C, Liebe R, et al. Extrahepatic manifestations of progressive familial intrahepatic cholestasis syndromes: presentation of a case series and literature review. Liver Int. 2022;42(5):1084-1096. PubMed
27.Wehrman A, Lee CK. The cholestatic infant: updates on diagnosis and genetics. Curr Opin Pediatr. 2022;34(5):491-495. PubMed
Appendix 1: Details of Included Publications
Note that this appendix has not been copy-edited.
Table 1: Details of Included Publications
First author, year | Publication type | Objective | Key ethical considerations | Funding source |
|---|---|---|---|---|
Abdelrhim, 201720 | Case report | To illustrate a dilemma in a child with PFIC type 2 who presented with “red flag” fractures indicative of child abuse. |
| None reported. |
Alam, 202221 | Review | To provide an overview of PFIC types 1, 2, and 3. |
| None reported. |
Al-Hussaini, 202122 | Database study | To characterize the clinical, laboratory, histologic, and molecular features and outcome of gene-confirmed PFIC 1 to 3 among Arabs and to evaluate for “genotype-phenotype” correlations. | Genotype-phenotype correlations have been discerned among multiple mutations in the ABCB11 and ABCB4 genes. This emphasizes the significance of promptly confirming the diagnosis through genetic testing. Such confirmation can offer valuable guidance to both physicians and patients regarding the anticipated progression of the disease. | None reported. |
Anderson, 200623 | Database study | To assess the carrier frequency and the possible impact on health in populations in East Greenland. | Heterozygosity for Greenland familial cholestasis is prevalent among Inuit individuals in East Greenland. However, being a carrier does not constitute a risk factor for developing the disease. Screening may be considered in these communities. | Greenland Homerule; Aalborg City Christmas Lottery; the Obel Family Foundation; the Northern Jutland Research Foundation; the Danish Hospital Foundation for Medical Research. |
Anonymous, 201224 | Review | To provide an overview of Odevixibat. |
| None reported. |
Baker, 20196 | Systematic review | To consolidate the data obtained from small studies and analyses of patient records published over the past 35 years, so that the results can be used to help inform the management of patients with PFIC and the design of future clinical studies in patients with PFIC. |
| Shire International GmBH. |
Baumann, 202114 | RCT | To evaluate the safety, tolerability, and efficacy of single- and multiple-dose treatment with oral odevixibat in pediatric patients with cholestatic liver disease and pruritus. |
| Sponsored by Albireo AB. Editorial, and Albireo AB. |
Feldman, 2020 | Review | To outline the etiologies, diagnostic pathways and current and emerging management strategies for neonatal cholestasis. | Defining the genetic cause of cholestatic conditions in each infant has the potential to facilitate the implementation of novel therapies tailored to specific gene mutations, thereby achieving optimal outcomes for patients. | None reported |
Gwaltney, 20227 | Qualitative interview study | To develop PRO and ObsRO instruments for pediatric patients with CLDs and their caregivers that reflected the patient experience of CLD. |
| Albireo Pharma, Inc. Rapid Service |
Heo, 202225 | Review | To provide an evaluation of odevixibat in the treatment of progressive familial intrahepatic cholestasis. |
| None reported |
Hupper, 202310 | Review and case reports | To perform a literature survey on medical and surgical treatments for PFIC and review the charts of patients with PFIC at a tertiary hospital. | The literature indicates that medical treatment involving IBAT, such as odevixibat, is not effective or suitable for every patient across all subtypes of PFIC. | None reported |
Jacquemin, 201211 | Review | To provide an overview of PFIC. |
| None reported |
Jones-Hughes, 20218 | Systematic review | To explore current evidence for the epidemiology and natural history of PIFC and BRIC; to explore current evidence for the human and economic burden of PFIC and BRIC. | PFIC stands as a highly distressing condition for both children and their parents, with pruritus emerging as a central issue. In fact, pronounced pruritus can result in severe skin damage (sometimes leading to bleeding) and can impact various daily activities due to sleep disturbances, irritability, reduced focus, and compromised school performance. | Albireo Pharma, Inc. |
Kamath, 201816 | Database study and case study | To develop a clinical outcome assessment for itching in children with cholestatic pruritus. | Pruritis impacted growth, stamina, mood, fatigue, self-esteem, and job maintenance in the patient with PFIC. There was also a financial and emotional impact on the family. | Lumena Pharmaceuticals; National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases-sponsored ChiLDReN Network; Shire International GmbH |
Nielsen, 200412 | Database study | To provide a detailed genealogy, including a clinical description of Cholestasis Familiaris Groenlandica or PFIC type 1 in Indigenous Inuit families in Greenland. | Genetic counselling and carrier screening are strongly recommended for parents in Greenlandic society. | Greenland Homerule (Department of Health care) |
Pfister, 202226 | Review and case series | To outline the expression of the PFIC-associated gene products in various organs and provide an overview of extrahepatic manifestations. |
| BMBF through HiChol |
Slavetinsky, 202018 | Case report | To compare the effects of iBAT inhibition with PEBD surgery in a single patient with PFIC who received odevixibat. | Phase II trial results suggest odevixibat is a safe compound for long-term use in treating cholestatic liver disease; however, additional long-term studies are needed to evaluate long-term effects and safety in a pediatric setting. Such trials are currently ongoing. | Albireo AB |
Thompson, 202215 | RCT | To evaluate the effects of odevixibat, an ileal bile acid transporter inhibitor, vs. placebo in children with PFIC. | The alleviation of pruritus and lowering of serum bile acids may potentially lead to a decreased requirement for diversion surgery in patients undergoing odevixibat treatment. Avoiding surgical intervention and possible associated consequences can improve quality of life. | Albireo Pharma |
Thompson, 202317 | Interim results of an RCT | To present interim results from an ongoing, open-label extension study evaluating long-term efficacy and safety of odevixibat in patients with PFIC. | Patients with PFIC generally exhibited good tolerability to odevixibat, which was linked to sustained reductions in serum bile acids and pruritus. | Albireo Pharma |
Wehrman, 202227 | Review | To highlight recently published studies regarding diagnosis and treatment of cholestasis in infants. | Diarrhea, liver test abnormalities, vomiting, abdominal pain, and fat-soluble vitamin deficiencies were common adverse events in patients taking odevixibat. | None reported |
BRIC = benign recurrent intrahepatic cholestasis; CLD = cholestatic liver disease; iBAT = intestinal bile acid transport; PFIC = progressive familial intrahepatic cholestasis; PRO = patient-reported outcome; ObsRO = observer-reported outcome; RCT = randomized controlled trial.
Stakeholder Input
Patient Input
Canadian Liver Foundation
About Canadian Liver Foundation
The Canadian Liver Foundation (CLF) is a leading organization dedicated to promoting liver health, increasing public awareness and understanding of liver disease, and providing support to individuals affected by liver disease.
The CLF was founded in 1969 out of the passion and concern of a group of business leaders and doctors who believed that liver disease needed a champion. Since then, the CLF has relentlessly driven advancements in research, treatment, and support. We remain the only non-government organization in Canada focused on liver health and the main source of non-profit funding for all forms of liver disease, investing nearly 40 million dollars to date.
The CLF reaches millions of Canadians through our public and professional education programs, patient support programs, and outreach efforts. We advocate for all Canadians affected by liver from newborns to seniors, including patients and their caregivers.
Information Gathering
The CLF reached out to patients and caregivers who recently accessed our support services, including our National Help Line, regarding their Progressive Familial Intrahepatic Cholestasis (hereby referred to as PFIC) diagnosis. We anticipated that this submission could be challenging as it relates to the procurement of patient and caregiver input due to the rare nature of this disease, coupled with limited number of patients accessing the therapy under review who may not be available to respond to our request for patient input. On May 31, 2023, the CLF contacted the US- based patient advocacy group, The PFIC Advocacy and Resource Network Inc. (hereby referred to as The PFIC Network), to request assistance with patient recruitment. The PFIC Network were keen to conduct outreach through their networks and channels (June 27 to July 12). This outreach was conducted in both English and French.
Fortunately, our collaborative efforts to gather the experiences of patients and caregivers of those living with PFIC via an online survey resulted in fourteen respondents who provided their story. This outreach effort resulted in four Canadian patient and caregiver responses, four American respondents, and five respondents outside of North America (international), with five respondents having experience with the drug under review.
The data was gathered via online survey created by the Canadian Liver Foundation and circulated by both the CLF and The PFIC Network via e-blast, newsletter, and social media communications. The survey was made available to the CLF community and members of The PFIC Network between June 27 and July 11, 2023. The patients and caregivers provided firsthand compelling, and relevant qualitative input regarding their:
Experience with respect to their PFIC diagnosis
Experience as caregivers/loved ones for someone with PFIC
Disease experience
Experience with respect to previous therapies
Experience with respect to the therapy under review
The qualitative data from the survey will be referenced throughout this submission. Quotes from respondents from the survey are included in various sections of this submission.
Disease Experience
Progressive Familial Intrahepatic Cholestasis (PFIC) is a genetic disease primarily affecting the liver. For a child to be diagnosed, they would need to receive two mutated genes, one from each parent. PFIC is estimated to affect between one in 50,000 to one in 100,000 individuals at birth. It is commonly associated with mutations of the ATP8B1, ABCB11, ABCB4, TJP2 and NR1H4 genes seen in patients with PFIC type 1, type 2, type 3, 4 and 5 respectively.
PFIC occurs when the liver cells struggle to produce and drain bile. Therefore, in patients affected with PFIC, the lack of bile secretion also leads to unnecessary waste being kept in the blood stream, and the body not being able to properly absorb fats and fat-soluble vitamins (these are certain vitamins that are absorbed along with fats in the diet that can be stored in the body’s tissues). In addition, there is cholestasis which results in liver cell damage and can progress to more severe liver disease. The treatment considerations for PFIC are both medical and surgical. This is based on attempting to increase the flow of bile from the liver, maintain normal growth and development, and prevent or correct any of the specific nutritional deficiencies that often develop. Due to bile not flowing out of the liver in patients with PFIC, medications designed to increase the flow of bile are frequently prescribed. These medications can decrease the damage in the liver and may improve the digestion of fat and fat-soluble vitamins. The overall life expectancy for children with PFIC is variable based on if patients receive liver transplants, but also depends on the severity of the liver scarring as a result of the disease. There is presently no procedure that can correct the bile flow deficiencies in the liver, and liver transplantation which occurs in childhood is required once cirrhosis advances to a stage where the liver fails to perform its functions.
Most families affected by PFIC expressed feelings of helplessness, anguish, and frustration. When asking parents and/or caregivers of loved one’s living with PFIC, respondents indicated how a PFIC diagnosis has severely impacted the lives of their loved ones and affected their day-to-day activities, while adding physical and emotional stressors and worries:
“Constant itching, she cannot walk some days due to the pain of the cuts on her feet from scratching, my daughter is 14 and gets about 3-5 hours of sleep a night, she is moody from lack of sleep, can't concentrate at school.” — Parent
“Itching, lower immune system.” — Parent
“Pruritus, which leads to skin conditions, extreme sleep deprivation, reduced diet, low mood, self-harm (causing pain) to take away the itch.” — Parent
“Due to his inability to absorb vitamin K, he gets nose bleeds a lot, and it takes a long time to stop these (15-20 minutes) so he gets regular vitamin K injections. He is often jaundiced, and sometimes kids at school would ask about it, so he is conscious about his appearance.” — Parent
“My husband and I had to take time off work due to multiple hospital stays when his PFIC causes other medical complications. His siblings also experience mental stress and anxiety every time he is rushed to the ER and had stay in the hospital for days for treatment.” — Parent
“Itch, lack of concentration, lack of sleep, tiredness, weakness, emotional disorder due to fear and stress.” — Parent
“My daughter has ongoing PTSD and affects every part of her life every day. We are reminded all the time of her first and devastating episode.” — Parent
“The feeling of itchiness and the need to scratch. Emotionally draining for both the caregiver and child with disease. People not understanding. More itchiness when hot and sweaty. Bathroom issues.” — Parent
“Itch is the main symptoms and if affects the quality of life of my son as he can't sleep, play and enjoy his childhood properly and as such it does affect us parents...it affects our mental health and physical health also due to sleep deprivation.” — Parent
“My son suffers with fatigue, low vitamin levels, lack of appetite and constant severe itching. The latter is most detrimental to day-to-day life as it hurts his ability to engage in play and sleep. This means we as his parents suffer from a lack of sleep as we try and settle him. We shield from other people and most public places to avoid infections which will further hurt his liver function. He is also on the transplant list which means he needs to be infection-free, and we can't be more than an hour from home.” — Parent
“Itching is extremely debilitating, painful and takes a severe toll on my mental health which translates to my physical health. chronic fatigue makes functioning extremely hard. I am hospitalized for at least a week but usually longer at least twice a year for flare ups of my condition followed by weeks to months of further recovery and increased symptoms which severely affect my ability to maintain work/school/relationships”. — Patient
“The main symptoms my son has experienced are weight loss, sleep deprivation, mental health issues and extreme pruritis. The itch was so bad that he couldn’t concentrate enough to eat or even to play with his siblings. It caused him so much sleep deprivation that he would sometimes fall asleep going up the stairs or on the kitchen floor. His skin was torn to shreds all over his body no matter what we did to prevent him from scratching. His quality of life was horrific. All of these things affected us as parents greatly and affected our other kids. He often woke us up at night and he constantly needed comfort during the day.” — Parent
Experiences With Currently Available Treatments
The treatment for patients with PFIC is tailored to each individual, but all patients will need some form of treatment for their disease. This can include medication, surgical treatments, and diet changes, which are often prescribed or recommended to help address symptoms associated with PFIC, including itching, bile buildup, and nutrition issues like vitamin deficiencies and poor growth. Medications such as ursodeoxycholic acid can help to improve bile flow and reduce itching, while surgeries such as bile duct surgeries or even liver transplants can help to alleviate symptoms or get rid of them altogether. Ursodeoxycholic acid can also help to reduce blood cholesterol levels and jaundice, which are common symptoms for those with PFIC. In addition to these treatments, vitamin and caloric supplementation can help with growth and overall nutrition.
“Urso, hydroxyzine, cholestyramine, rifampin and others in the past I cannot remember They don't work - waiting for odevixibat but it is taking months.” — Parent
“Odevixibat is currently the only medication. I tried rifampicin, antihistamines, sleeping medication, cannabis oil, Cholestyramine, Naloxone.” — Parent
“We are currently on ursodiol and odevixibat as well as high doses of vitamin A, D, E and K. We are on Imodium for his diarrhea. And he is drinking electrolytes to keep hydrated. We just got back from a week stay at the hospital due to low potassium and other electrolyte imbalance, his body is unable to regulate it despite constant IV. It is frustrating; a treatment can work for a while, and then something like a viral infection (or unknown circumstances) can wreak havoc and send him to the hospital.” — Parent
“Current medications available did not work for my daughter. In fact, some of them made the itch worse.” — Parent
“Currently, it is ursodeoxycholic acid, rifampicin, and odevixibat.” — Parent
“Maralixibat, ursodeoxycholic acid, and rifampicin. [He] was on maralixibat but it didn't work; what he has is incurable and progressive. So, the medications he takes help but don't halt the disease's progress or particularly ease the symptoms.” — Parent
“I have tried (I believe) all potential drug therapies including, but not limited to ursodiol, rifampin, cholestyramine, Welchol, naltrexone, and more, as well as surgeries including cholecystectomy, ileal exclusion and Puestow procedure (not liver surgeries but all because of problems assumed to be related to my liver condition). My itching has not been really managed with therapies, nor have any of my symptoms really. I need something to decrease itching and increase overall feelings of health and comfort and vitality.” — Patient
“Currently he is part of a trial for an IBAT inhibitor. Previously he had taken sodium phenylbutyrate, hydroxyzine, Benadryl, cholestyramine, rifampin, and ursodiol. None of those helped him.” — Parent
Respondents also indicated the challenges relating to treatment access, follow-up care, and limited knowledge about PFIC in the health care community:
“We fear the return of the itch and currently have no access to new medications, episodic PFIC is unlikely to be transplanted or diversion surgery, the constant stress of no treatment is a huge and unnecessary burden.” — Parent
“I can't believe in our 21st century and with advancement in technologies and medicine, still there is no treatment that can reduce the level of bilirubin in blood. The treatment available only helps partially and not completely...” — Parent
“The most challenging is the mental anguish that occurs from lack of awareness and knowledge from a majority of healthcare providers. Even the most knowledgeable ones rarely are able to provide me with information or treatment or support. Lack of awareness and funding for research is scary and frustrating. It is extremely hard to navigate a world with a rare disease that no one can predict or cure, for both patients and caregivers.” — Patient
“He is currently on a drug trial and had a lot of issues getting into the trial. Our hospital wasn’t set up with the trial until I told them about it, so it took a long time to get approved and go through the ethics board.” — Parent
Improved Outcomes
Given the rare nature of PFIC and the liver damage that it causes, there is a strong need to prioritize patient centered outcomes such as quality of life. There is also a strong need for education, research, and awareness on PFIC among health care providers and investigators. According to the various patient and caregiver input received, the therapy under review addresses and provides these desired improvements.
“Stop the itch, even enough so that she can sleep.” — Parent
“Something that takes away the itch and allows my daughter to sleep and live a happy and healthy life.” — Parent
“Our expectation of successful treatment is that he can live a fairly normal life without having to constantly worry about itching, or having to go to the bathroom every two hours when he has hangouts with his friends. As well as slowing down the progression of the disease.” — Parent
“Useful in the present and no side effects in the long term.” — Parent
“Just stop the itch. The itch is so unbearable. People have described it as making them suicidal. A successful medication would reduce the itch and secondary to that slow or stop the progression of liver failure.” — Parent
“A process that makes sure the bile gets out of the body which makes the body less itchy.” — Parent
“TO STOP THE ITCH.” — Parent
“Improved liver values, continuous and peaceful sleep, significant reduction in itching.” — Parent
“No itch. Normal growth and weight gain. Good energy levels. A 'zest' for life and ability to play and enjoy life.” — Parent
“Helps symptomatically or helps with disease progression.” — Parent
“To decrease negative symptoms, mainly pruritis, by a noticeable degree.” — Patient
“I would expect a treatment to calm the itch enough that my son could focus enough to eat, sleep and have fun. Improve his quality of life. And/or show signs of slowing or stopping liver disease progression.” — Parent
Experience With Drug Under Review
Odevixibat (brand name: Bylvay) is a treatment that is specifically designed to treat cholestatic pruritus for those with PFIC. It is FDA approved for patients with PFIC and Alagille Syndrome who are 6 months of age and older. Odevixibat is an oral medication taken once daily in the morning with a meal in the form of a capsule or tablet. This medication works by targeting the ileal bile acid transporter protein in the intestine. By blocking this protein, odevixibat is able to reduce the quantity of bile acid that is retransported back to the liver from the intestinal tract, and therefore reduces damage to the liver.
Five respondents indicated having experience with the drug under review. Respondents indicated that the primary method of access to the drug under review was through clinical trial.
When asked about side-effects or symptoms with the drug under review, the following input was received from patients and caregivers:
“We were on Bylvay for one year without any side effects. Our doctor had thought his recent diarrhea issue were partly due to Bylvay, so we are trying to reduce the dosage right now to try to find the balance of finding relief for the itching and reduce the diarrhea.” — Parent
The survey respondents expressed the improvements in their overall quality of life once they began treatment and limited challenges with obtaining the treatment:
“It was the only thing that worked. Her bile salts returned to normal levels within days.” — Parent
“It has been completely life changing for my five-year-old and the whole family We all couldn’t have gone on much longer without it.” — Parent
“We don’t require anything better at this stage, odevixibat has improved itch completely.” — Parent
“He had mild itching with the external diversion, but when he converted to the internal diversion, the itching retuned to very severe, and when we got approved for the Bylvay, within in days, his itching went from a five (five being his itchiest) to a zero (no itching).” — Parent
“The improvements in liver values with Bylvay alone were not significant (however, the prescribed dosage was always very low, in some periods lower than what was indicated on the leaflet). In association with rifampicin, the quality of life improves markedly.” — Parent
“Can be very helpful for itching for some patients who do not have other options.” — Parent
“Yes, if it works to alleviate the symptoms of PFIC anyone suffering from it should have access to the medicine.” — Patient
Companion Diagnostic Test
Not applicable. This drug does not require a companion diagnostic test.
Anything Else?
The Canadian Liver Foundation believes that liver disease patients, their caregivers and health care providers should have access to the most effective treatment options regardless of geographical location, financial status, treatment status, or disease severity, in order to ensure the best possible outcomes.
The aim of treatment is to maximize effectiveness and minimize the adverse side effects with the hope for improved patient outcomes. It is important to ensure greater and more equitable access to important treatments for Progressive Familial Intrahepatic Cholestasis (PFIC) patients while expanding therapeutic options for patients and healthcare professionals. We think it is crucial that patients across the country have equitable access to all treatments for liver disease and that provincial borders should not be a barrier.
The hope is that access to odevixibat provides patients and caregivers with improved and additional treatment options. Furthermore, the hope is that the cost of treatment does not increase as this would place a significant and unexpected financial burden on families. However, if accessing odevixibat is not seamlessly and readily available as part of various provincial reimbursement programs, then patients will have less access to these treatments. We therefore strongly support and urge that a positive funding recommendation be issued for odevixibat for the treatment of cholestatic pruritis in patients with PFIC. We believe a positive funding recommendation aligns well with the identified patient need for a new, effective, and easily administered treatment option that is capable of maintaining a high quality of life and durable response.
Conflict of Interest Declaration — Canadian Liver Foundation
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No additional assistance was provided to complete this submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
Outside assistance from The PFIC Network was used to help collect data, as they were the organization we partnered with to circulate the survey. This submission itself was completed by CLF staff and volunteers.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
The CLF is committed to bringing liver research to life for all Canadians through liver research, education, patient support and advocacy. The CLF receives funding from a variety of sources with the majority coming from donations from individuals and corporations across the country. We use these funds to support the CLF’s liver health awareness and education initiatives, patient support services, and research grant programs.
The CLF receives some program funding in the form of unrestricted educational grants from pharmaceutical companies. Grant agreements are established in support of activities initiated by the CLF and prohibit the funder from having any input or influence in program objectives or deliverables.
Table 1: Financial Closures for Canadian Liver Foundation
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Clinician Group Input
Canadian Association for the Study of the Liver
About Canadian Association for the Study of the Liver
CASL is a non-profit organization that seeks to eliminate liver disease through research, education and advocacy. Our members are experts on liver disease in Canada: hepatologists, gastroenterologists, pediatricians, surgeons, radiologists, researchers, nurses, trainees, community advocates, and patients and family partners. The Canadian Paediatric Hepatology Research Group (CPHRG) is a committee within CASL which encompasses all the specialist paediatric hepatologists in Canada. Dr. Carolina Jiminez is the Chair of the CPHRG. https://hepatology.ca
Information Gathering
The data and information presented here are gathered from a review of the published literature about Progressive Familial Intrahepatic Cholestasis (PFIC) and Odevixibat and attendance at conferences and abstract presentations about Odevixibat. Further the information is based on collective expert opinion within the CPHRG drawn from decades of experience managing patients with PFIC.
Current Treatments and Treatment Goals
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of rare autosomal recessive liver disorders of childhood characterized by mutations in genes encoding proteins involved in the hepatocellular transport system. The incidence is approximately 1:100,000. The main clinical features of PFIC include cholestasis, jaundice and pruritus, with symptoms typically appearing in infancy or early childhood. PFIC is associated with a range of potentially fatal complications of the liver, including portal hypertension, liver failure, cirrhosis and hepatocellular carcinoma (HCC; BSEP deficiency (PFIC2)), as well as extrahepatic manifestations (FIC deficiency; PFIC1). The biochemical features of PFIC1 and PFIC2 are low levels of gamma-glutamyl transferase (GGT) with elevated serum bile acid and decreased primary bile acid concentrations, while PFIC3 (MDR3 deficiency) is associated with high levels of GGT. There are multiple other forms of PFIC which are better described by their genetic/protein defect, rather than a numerical categorization. Despite the multiple diseases encompassed by the term PFIC, they share common features of cholestasis, pruritus and fat malabsorption (fat soluble vitamin deficiency – FSVD).
Treatment strategies in PFIC aim at managing cholestasis and its associated complications, such as nutritional support, FSVD, and pruritus. There are currently no curative medical therapies for PFIC-liver disease, and the treatment paradigm described below is supportive and aims to ameliorate symptoms, however no therapies target the underlying disease mechanism of defective canalicular transport protein. Surgical biliary diversion to lower serum bile acid concentrations attempts to ameliorate the sequelae of abnormal hepatocellular transport and is associated with improved native liver survival in PFIC. However, this surgery is invasive and typically leaves children with a stoma which is cosmetically disfiguring and deeply impacts quality of life.
The management strategies described are all standard of care in Canada. There are no practice guidelines that outline this treatment paradigm due to the rarity of the disease and limited published data that meet the standards for a guideline, however multiple review articles encompass this information.
Nutritional Management
Children with PFIC require approximately 125% of the recommended daily allowance of calories and may need more for catch up growth. This is typically secondary to decreased oral intake and fat malabsorption. Medium chain triglyceride-rich foods are encouraged for ease of absorption, as well as other calorie dense foods. In children not being able to meet their caloric demands, tube feeding (nasogastric or via gastrostomy) is often required, especially in the context of progressive liver disease.
Supplementation with fat-soluble vitamins is crucial. To aid with adherence and cost, cholestasis-specific formulations are available in Canada (e.g., DEKAs) via the special access pharmacy and are the preferred strategy for supplementing vitamins. However, individual vitamin supplementation is acceptable if generic multivitamin preparations are the only available option.
Management of Pruritus
Pharmacological treatments
Treatment of cholestatic pruritus requires a stepwise approach. All the following medications are used off-label. Antihistamines are initiated first and are typically not effective but can be considered in mild cases and to augment sleep. Ursodeoxycholic acid promotes bile excretion rendering it more hydrophilic. Due to its attractive safety profile, it is typically used as early in the management of cholestasis. Cholestyramine, a bile salt-binding agent may also be considered. Cholestyramine decreases bile acid pool size by binding bile salts in the small intestine and hence preventing their reabsorption. However, poor palatability and interference with absorption of other drugs (specifically fat-soluble vitamins) limits its use and it is almost never used in clinical practice. Rifampin is much preferred to treat pruritus instead of cholestyramine. Through its enzymatic induction in the liver, it is thought to increase the metabolism of pruritogens. Opioid antagonists such as naltrexone, are sometimes added to the regimen if pruritus persists and may provide modest additional benefit. Opioid withdrawal symptoms which may occur in one-third of patients limits its use in clinical practice. Lastly, sertraline, a selective serotonin reuptake inhibitor (SSRI), has been used in refractory cases. Its mechanism of action is poorly understood. Limited pediatric studies support its use as adjunctive therapy intractable cholestatic pruritus and it is infrequently used in clinical practice.
Surgical interventions
It has been observed that patients with PFIC may benefit from surgical biliary diversion (SBD) procedures, such as partial external biliary diversion (PEBD) or ileal exclusion. Partial external biliary diversion where a jejunal conduit is used to drain the gallbladder externally to a stoma on the abdominal wall, is the most commonly performed procedure. SBD aims to decrease the size of the bile acid pool by interrupting the enterohepatic circulation. Many patients with PFIC experience relief or amelioration of pruritus with SBD.
Unfortunately, not all patients benefit from SBD and, at some point, the majority require a liver transplantation for refractory or recurrent pruritus or progressive liver disease. Severe BSEP deficiency has also been associated with the development of hepatocellular carcinoma at an early age, which by itself may necessitate liver transplant.
Beyond the management of pruritus, it has recently been discovered by the global NAPPED consortium that lowering serum bile acids with SBD actually improves overall liver health in PFIC, presumably by depleting intrahepatic bile acids. SBD is associated with increased native liver survival in patients with BSEP deficiency (excluding those patients with 2 severe mutations). Further, lowering serum bile acids below a certain threshold is associated with native liver survival. In these BSEP patients the interruption of the enterohepatic circulation seems to postpone or even remove the need for LT. Similar, though slightly less convincing data, exist for FIC1 deficiency.
Taken together, these data support a treatment goal of interrupting the enterohepatic circulation in PFIC to alleviate pruritus, and more importantly to improve long-term liver disease outcomes and postpone and event prevent the need for liver transplantation. It should be noted that some PFIC patients will have progressive liver disease and recurrence of pruritus following a SBD that had initially been successful. Fewer biliary diversion procedures are being performed in Canada in the last 5 years due to access to IBAT (intestinal bile acid transporter) inhibitors and families’ preference not to have a stoma with SBD.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
The treatment paradigm for cholestatic pruritus described above falls short for many patients with PFIC-associated cholestatic liver disease. Patients with even mild to moderate cholestasis typically suffer from severe, debilitating pruritus. Ursodeoxycholic acid is used as a choloretic and a treatment for cholestasis,but has no impact on pruritus. Antihistamines are rarely effective as anti-pruritucs, cholestyramine is unpalatable, and although rifampin does provide some symptomatic relief for pruritic patients, it is usually ineffective in substantially ameliorating or eradicating pruritus. Sertraline and naltrexone provide marginal additional benefit, if at all. Therefore, current medical treatment paradigms for pruritus are insufficient for many cholestatic patients with PFIC. Thus, surgical options have to be considered. An external biliary diversion can be offered to PFIC patients with pruritus that is refractory to medical therapies, and even to improve liver disease outcomes, however it leaves the child with a stoma which is unacceptable to most families. Internal biliary diversions for PFIC are reported but are considered generally less effective than external biliary diversions. For patients with PFIC whose SBD fails or who will not accept this treatment approach the only option is liver transplantation. Between 50-75% of PFIC patients end up requiring liver transplantation. Liver transplantation is, of course, associated with significant mortality and morbidity from major surgery and lifelong immune suppression.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
As described above, many patients with PFIC and cholestatic pruritus have inadequately treated pruritus with standard of care medical/surgical therapy. Odevixibat would be added to the current toolkit of available medical therapies. Odevixibat would be used in combination with the other available medications. None of the currently available therapies interrupt enterohepatic circulation of bile acids and lower serum bile acids by blocking bile acid uptake in the ileum. It is true that cholestyramine is a bile acid-binding resin and can also reduce bile acid return to the liver, however it is not as efficacious as blocking the intestinal bile acid transporter and more importantly it is unpalatable, limiting its utility. As a result cholestyramine is rarely used in clinical practice.
Odevixibat treats cholestatic pruritus which is very debilitating for patients. Pruritus disrupts sleep for children and the whole family with wide-ranging impacts on health-related quality of life. Odevixibat is an effective symptomatic treatment. The data demonstrating that SBD and lowering of serum bile acid levels is associated with improved native liver survival, suggests that odevixibat has an important role as a treatment for cholestasis associated with PFIC to delay or prevent liver transplantation. Odevixibat would be used in patients with PFIC who have persistent pruritus on ursodexycholic acid, antihistamines and rifampin and would also be considered in patients with PFIC and cholestasis, even if their pruritus is adequately controlled with existing medications. Odevixibat would be added into the treatment plan (rather than as a replacement for these other medications). It is possible that some patients may be able to wean off some of the standard medications once they are established on Odevixibat. Naltrexone and sertraline are rarely offered in clinical practice due to very limited efficacy and tenuous safety profiles and therefore we would NOT recommend that these be attempted prior to offering Odevixibat.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients with PFIC and cholestatic pruritus, which is persistent on standard of care medical treatment would be eligible for treatment. Patients with PFIC and cholestatic liver disease and adequately controlled pruritus would also be eligible for treatment to improve liver disease outcomes and prevent or delay the need for liver transplantation. Since the mechanism of action of Odexivibat is to lower serum bile acids, it is reasonable to anticipate that patients with elevated serum bile acids are most likely to respond to treatment. Patients with moderate to severe pruritus, as determined by clinician evaluation and parent/caregiver/patient report, would have the greatest need.
The diagnosis of PFIC requires low GGT cholestasis (except for MDR3 deficiency), some histologic features and in some cases, extrahepatic features. It is an autosomal recessive condition and making a genetic diagnosis is not always straightforward due to the likelihood of patients with novel variants in PFIC genes and compound heterozygotes (patients with 2 different variants on a disease gene). Further the ethnic diversity of Canadian patients with PFIC means that genetic testing often identifies variants which have not previously been reported in the literature and are hence difficult to evaluate. Thus, the diagnosis of PFIC generally requires a phenotypic diagnosis (consistent biochemistry, clinical picture and liver histology) and in addition, but not necessarily, a confirmatory genetic diagnosis.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The primary outcomes in the clinical trials of PFIC patients were patient-reported assessments of pruritus severity and serum bile acids. The exact tool to assess pruritus in the trials is not feasible in clinical practice as it requires twice daily scores over 2 weeks. In clinical practice pruritus severity is assessed by asking the patient/family about severity of pruritus, sleep disturbance and then examining the skin for excoriations. The physical examination can be scored according to the Clinician Scratch Scale and this was also included in the clinical trials. Serum bile acid levels can also be used, however in clinical practice this is not done routinely due to cost and logistics as this test is often sent to specialized laboratories and is not readily available in all gastroenterology practice settings.
A clinically meaningful response would be patients/families reporting an improvement in pruritus, improvement in sleep duration which can be objectively measured by asking how often the child wakes at night or by documenting improvements in skin excoriations.
What factors should be considered when deciding to discontinue treatment with the drug under review?
The most likely reason to discontinue treatment with Odevixibat would be if a PFIC patient’s liver disease progresses and they undergo liver transplantation. Patients with PFIC can be continued on Odevixibat while waiting for liver transplantation as it can improve pruritus and quality of life.
Other factors that should be considering when deciding to discontinue treatment with Odevixibat would be treatment associated adverse events. The safety profile of the drug is generally good and data from the clinical trials are summarized below.
Table 2: Clinical Trials Data From Patients Treated With Odevixibat
Patients, n (%) | Patients Treated With Odevixibat for ≥96 Weeks, n=36 |
|---|---|
Any TEAEs | |||||| ||||||||| ||||||| |
Drug-related TEAEs | |||||| ||||||||| ||||||| |
Severe TEAEs | |||||| ||||||||| ||||||| |
Serious TEAEs | |||||| ||||||||| ||||||| |
TEAEs leading to study treatment interruption | |||||| ||||||||| ||||||| |
TEAEs leading to study treatment discontinuation | |||||| ||||||||| ||||||| |
The most important reported adverse effects in the clinical trials were gastrointestinal upset (diarrhea, abdominal pain) and increased ALT. Some of these led to interruption of Odevixibat, but not discontinuation. It is certainly possible that, in clinical practice, that gastrointestinal upset or increased ALT may lead to discontinuation, though based on the available clinical trial data, we do not expect this to affect large numbers of patients with PFIC.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Odevixibat should be prescribed and monitored by a paediatric gastroenterologist or hepatologist in a specialty clinic setting.
Additional Information
Not applicable.
Conflict of Interest Declarations — Canadian Association for the Study of the Liver
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Binita M. Kamath
Position: Division Head (interim)
Division of Gastroenterology, Hepatology and Nutrition, The Hospital for Sick Children
Senior Associate Scientist, Research Institute; Professor, University of Toronto
Date: 09-07-2023
Table 3: COI Declaration for Canadian Association for the Study of the Liver — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Mirum: Consultant Unrestricted Educational Grant | — | — | Xa | Xb |
Albireo: Consultant Unrestricted Educational Grant | — | — | Xa | Xb |
Audentes (Astellas): Consultant | — | X | — | — |
aConsultant
bGrant
Declaration for Clinician 2
Name: Carolina Jiminez
Position: Associate Professor, Department of Pediatrics
Faculty of Medicine, University of Ottawa
Director of Liver Services Chief, Division of Gastroenterology, Hepatology and Nutrition
Department of Pediatrics, Children's Hospital of Eastern Ontario
Date: 12-07-2023
Table 4: COI Declaration for Canadian Association for the Study of the Liver — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Simon Lam
Position: Clinical Assistant Professor
Gastroenterology, Hepatology & Nutrition
Alberta Children's Hospital
Date: 12-07-2023
Table 5: COI Declaration for Canadian Association for the Study of the Liver — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Susan M. Gilmour
Position: Professor, Pediatric Gastroenterology/Nutrition
Department Pediatrics
Faculty of Medicine and Dentistry
University of Alberta
Stollery Children’s Hospital
Date: 12-07-2023
Table 6: COI Declaration for Canadian Association for the Study of the Liver — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Mirum | Xa | — | Xb | — |
aConsultant
bClinical Trial
Declaration for Clinician 5
Name: Quais Mujawar
Position: Assistant Professor, Department of Pediatrics, Health Sciences Centre Winnipeg, University of Manitoba
Date: 12-07-2023
Table 7: COI Declaration for Canadian Association for the Study of the Liver — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Mirum | Xa | — | — | — |
Medison/Albireo | Xa | — | — | — |
aConsultant
Declaration for Clinician 6
Name: Vicky Ng
Position: Professor of Pediatrics, University of Toronto
Medical Director, Pediatric Liver Transplantation
Staff Hepatologist, Division of Pediatric Gastroenterology, Hepatology and Nutrition, The Hospital for Sick Children
Date: 12-07-2023
Table 8: COI Declaration for Canadian Association for the Study of the Liver — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 7
Name: Fernando Alvarez
Position: Full Professor
Gastroenterology, Hepatology and Nutrition
Department of Pediatrics
Faculty of Medicine
Gastroenterology, Hepatology and Nutrition Department of the CHU Sainte-Justine
Date: 12-07-2023
Table 9: COI Declaration for Canadian Association for the Study of the Liver — Clinician 7
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 8
Name: Dhandapani Ashok
Position: Pediatric Gastroenterologist & Hepatologist
The Children’s Hospital at London Health Sciences Centre
Associate Professor, Department of Pediatrics
Schulich School of Medicine and Dentistry, Western University
Date: 12-07-2023
Table 10: COI Declaration for Canadian Association for the Study of the Liver — Clinician 8
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Mirum | — | Xa,b | — | — |
aConsultant
bUnrestricted Educational Grant
Declaration for Clinician 9
Name: Simon Ling
Position: Professor of Paediatrics, University of Toronto
Division of Gastroenterology, Hepatology & Nutrition, Hospital for Sick Children
Project Investigator, SickKids Research Institute
Date: 12-07-2023
Table 11: COI Declaration for Canadian Association for the Study of the Liver — Clinician 9
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | Xa | — | Xb | — |
Mirum | Xa | — | — | — |
Medison/Albireo | Xa | — | — | — |
aConsultant
bResearch Support
Declaration for Clinician 10
Name: Andrea Zizzo
Position: Associate Professor, Western University
Head, Division of Paediatric Gastroenterology & Hepatology
Chair, Resident Research Subcommittee
Director, PROGrS volunteer program
Children’s Hospital, London Health Sciences Centre
Date: 11-07-2023
Table 12: COI Declaration for Canadian Association for the Study of the Liver — Clinician 10
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 11
Name: Simone Kortbeek
Position: Clinical Assistant Professor
Section of Gastroenterology, Hepatology & Nutrition
Cumming School of Medicine, University of Calgary
Alberta Children’s Hospital
Date: 11-07-2023
Table 13: COI Declaration for Canadian Association for the Study of the Liver — Clinician 11
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 12
Name: Mohsin Rashid
Position: Professor of Pediatrics, Medicine & Medical Education
Department of Pediatrics
Gastroenterology & Nutrition
Dalhousie University
IWK Health Center
Date: 11-07-2023
Table 14: COI Declaration for Canadian Association for the Study of the Liver — Clinician 12
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 13
Name: Herbert Brill
Position: Associate Professor, Department of Pediatrics, University of Toronto
Associate Clinical Professor, Department of Pediatrics, McMaster University
Date: 11-07-2023
Table 15: COI Declaration for Canadian Association for the Study of the Liver — Clinician 13
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Medison/Albireo | Xa | — | — | — |
aAdvisory Board
Declaration for Clinician 14
Name: Pushpa Sathya
Position: Associate Professor
Pediatric GI & Hepatology
Discipline of Pediatrics, Faculty of Medicine
Memorial University of NL
Janeway Children’s Health Centre
Date: 11-07-2023
Table 16: COI Declaration for Canadian Association for the Study of the Liver — Clinician 14
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 15
Name: Rick Schreiber
Position: Clinical Professor, Division of Gastroenterology, Hepatology and Nutrition, Department of Pediatrics
Faculty of Medicine, University of British Columbia
BC Children’s Hospital
Date: 12-07-2023
Table 17: COI Declaration for Canadian Association for the Study of the Liver — Clinician 15
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 16
Name: Mohit Kehar
Position: Pediatric Gastroenterologist and Hepatologist
Assistant Professor
Division of Pediatric Gastroenterology, Hepatology and Nutrition
Children Hospital of Eastern Ontario, Ottawa
Date: 11-07-2023
Table 18: COI Declaration for Canadian Association for the Study of the Liver — Clinician 16
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 17
Name: Marie-Eve Chartier
Position: Pediatric Gastroenterologist-Hepatologist
Assistant Clinical Professor
Department of Pediatrics, Montreal University
Division of Gastroenterology, Hepatology and Nutrition
CHU Sainte-Justine
Date: 10-07-2023
Table 19: COI Declaration for Canadian Association for the Study of the Liver — Clinician 17
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 18
Name: Sylvie Lebel
Position: Medical Director, Liver transplant and Hepatology
BC Children’s Hospital
Associate Professor of Pediatrics
University of British Columbia
Date: 10-07-2023
Table 20: COI Declaration for Canadian Association for the Study of the Liver — Clinician 18
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 19
Name: Jeff Critch
Position: Associate Professor of Pediatrics, Memorial University of NL
Division of Gastroenterology
Janeway Children’s Health and Rehabilitation Centre
Date: 10-07-2023
Table 21: COI Declaration for Canadian Association for the Study of the Liver — Clinician 19
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 20
Name: Orlee Guttman
Position: Clinical Assistant Professor
GI Fellowship Program Director
Division of Gastroenterology, Hepatology and Nutrition
BC Children's Hospital
Date: 13-07-2023
Table 22: COI Declaration for Canadian Association for the Study of the Liver — Clinician 20
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.