CADTH Reimbursement Review
Tralokinumab (Adtralza)
Sponsor: LEO Pharma Inc.
Therapeutic area: Atopic dermatitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AD
atopic dermatitis
AE
adverse event
ANCOVA
analysis of covariance
BSA
body surface area
CDLQI
Children’s Dermatology Life Quality Index
CI
confidence interval
Crl
credible limit
DLQI
Dermatology Life Quality Index
EASI
Eczema Area Severity Index
EASI-50
reduction of at least 50% in Eczema Area Severity Index score from baseline
EASI-75
reduction of at least 75% in Eczema Area Severity Index score from baseline
EASI-90
reduction of at least 90% in Eczema Area Severity Index score from baseline
ESS
effective sample size
HADS
Hospital Anxiety and Depression Scale
HRQoL
health-related quality of life
ICER
Institute for Clinical and Economic Review
IGA
Investigator’s Global Assessment
IL
interleukin
IQR
interquartile range
ITC
indirect treatment comparison
ITT
intention to treat
JAKi
Janus kinase inhibitor
LTE
long-term extension
LOCF
last observation carried forward
MAIC
matching adjusted indirect comparison
MID
minimal important difference
mNRI
modified nonresponder imputation
NMA
network meta-analysis
NRS
numeric rating scale
OR
odds ratio
PDE-4
phosphodiesterase type 4
PGA
Physician’s Global Assessment
PICO
patient, intervention, comparison, and outcome
POEM
Patient-Oriented Eczema Measure
PP-NRS
peak pruritus numeric rating scale
PYE
patient-years of exposure
QoL
quality of life
RCT
randomized controlled trial
RR
relative risk
RD
risk difference
SAE
serious adverse event
SCORAD
Scoring Atopic Dermatitis
SD
standard deviation
TCS
topical corticosteroids
TCI
topical calcineurin inhibitor
TEAE
treatment-emergent adverse event
URTI
upper respiratory tract infection
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Tralokinumab (Adtralza), 150 mg per 1 mL prefilled syringe and 300 mg per 2 mL prefilled pen,a solution for subcutaneous injection |
Sponsor | LEO Pharma Inc. |
Indication | Tralokinumab is indicated for the treatment of moderate-to-severe AD in adult and adolescent patients 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable; tralokinumab can be used with or without topical corticosteroids |
Reimbursement request | For the treatment of patients aged 12 years and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an adequate trial or are ineligible for each of the following therapies: phototherapy (where available) and off-label immunosuppressants |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | February 3, 2023 |
Recommended dose | An initial dose of 600 mg followed by 300 mg administered every other week as subcutaneous injection. At prescriber’s discretion, dosing every fourth week may be considered for some patients who achieve clear or almost clear skin after 16 weeks of treatment |
AD = atopic dermatitis; NOC = Notice of Compliance.
aA 300 mg per 2 mL prefilled pen is currently not marketed in Canada.
Introduction
Atopic dermatitis (AD), also referred to as eczema, is a chronic, heterogeneous inflammatory relapsing-remitting skin condition that occurs most frequently in early childhood.1 It is estimated that the prevalence of eczema in Canada is 8.9% in adolescents (aged 13 to 14 years), and 3.5% in adults.2,3 Acute worsening of AD, commonly referred to as flares, presents as dry, red, itchy skin that can lead to lesions that blister, ooze, and crust. An intense and debilitating itch and chronically relapsing eczematous lesions are the key clinical hallmarks of moderate-to-severe disease4 and could lead to sleep disturbances, psychosocial distress, and reduced quality of life (QoL) in patients and caregivers.
Conventional treatment options for moderate-to-severe AD include topical therapies, phototherapy, and off-label systemic immunosuppressants. Newer systemic treatments, including dupilumab (a biologic), abrocitinib and upadacitinib (oral small molecules, each of which is a Janus kinase inhibitor [JAKi]), are effective options that are currently available for patients who did not progress on conventional treatments, although some patients do not achieve an adequate response to dupilumab and JAKi treatments. Dupilumab is associated with conjunctivitis, which may necessitate treatment discontinuation for some patients.5 Upadacitinib and abrocitinib treatments require baseline and routine laboratory monitoring and have black-box warnings in the product monograph related to infections, malignancies, thrombosis, and major adverse cardiovascular events.6,7
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of tralokinumab, 150 mg per 1 mL prefilled syringe and 300 mg per 2 mL prefilled pen, administered by subcutaneous injection, in the treatment of moderate-to-severe AD in patients aged 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Tralokinumab can be used with or without topical corticosteroids (TCS). Tralokinumab was previously reviewed by CADTH for the treatment of adults with moderate-to-severe AD and received a recommendation not to reimburse.8
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Two separate patient group inputs were received — 1 from the Eczema Society of Canada and another from Eczema Québec and the Canadian Skin Patient Alliance. The Eczema Society of Canada’s input was based on a survey of 3,000 patients, caregivers and family members; questionnaires; and 1-on-1 interviews (number not reported) with patients and caregivers. Eczema Québec’s input was based on patient testimonials (n = 6), interviews (n = 10), and 2 group discussions (n = 13 in total), as well as insights gleaned from the McGill University Health Centre’s Centre of Excellence for Atopic Dermatitis and a report (The Skin I’m in: 2022 Update) from 2021 to 2023. The groups noted that symptoms of moderate-to-severe AD include inflamed, red, and dry skin that cracks, oozes, bleeds, and in some cases involves thickening and/or infections of the skin. Often, patients experience “flare-ups’ of worsening symptoms. Some patients experience remission, but others never experience relief. The input noted that itch is frequently reported as the most burdensome symptom and has been described as “incapacitating,” “debilitating,” or “bugs crawling all over,” leading to disrupted sleep, fatigue, decreased functionality, and significant impacts on daily life, work, and school. Skin rashes were reported to be not only painful but a source of embarrassment and stigmatization, affecting self-esteem and social relationships. Family members and/or caregivers shared that they experience negative impacts on intimacy, family dynamics, and relationships, as well as feelings of anxiety, depression, and sleep loss. Patients with moderate-to-severe AD also reported that their choices of work, clothing, foods, environments, hobbies, regular activities, travel, and hygiene routines are limited due to AD. Some patients reported contemplating suicide due to uncontrollable AD. The joint input by Eczema Québec and the Canadian Skin Patient Alliance quoted data from the Canadian Institute for Health Information showing that patients sometimes end up in the emergency department or become hospitalized when AD is not well controlled. Patients expressed a need for treatments that can result in improvement in symptoms (dryness, flaking, inflammation, blistering, and cracking), reduction in itch frequency and/or intensity, long-term improvement in QoL (sleep, prevention of flares, discomfort, psychological burden), and ability to carry out daily activities (work, school, leisure, personal hygiene), and that are safe (reducing infections with minimal short-term and long-term adverse effects), affordable, flexible, and easy to administer.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts noted that there is an unmet need for more treatment options for moderate-to-severe AD that are effective and safe, given that some patients do not respond or are refractory to the newer systemic treatments (dupilumab, upadacitinib, abrocitinib) and that JAKi options are associated with safety concerns. One clinical expert also noted that there is a need for treatment options that could improve adherence and convenience of drug administration for patients who are averse to needles (dupilumab is administered as a subcutaneous injection) or have difficulty adhering to daily administration of oral upadacitinib and abrocitinib.
The clinical experts expected tralokinumab to have the same place in therapy as dupilumab, serving as an additional biologic option for the treatment of moderate-to-severe AD after failure of off-label immunosuppressants. In the clinical experts’ opinion, any patient with moderate-to-severe AD could be a candidate for tralokinumab treatment. The clinical experts noted that tralokinumab would most likely be used in patients with AD in the absence of comorbid conditions such as asthma, chronic rhinosinusitis with nasal polyposis, and eosinophilic esophagitis, as these patients could benefit from dupilumab treatment instead, given that dupilumab is also indicated for the treatment of these conditions.
The clinical experts noted that disease improvement is assessed in clinical practice using instruments, such as the Physician’s Global Assessment (PGA; also referred to as the Investigator’s Global Assessment [IGA] in clinical trials), Eczema Area and Severity Index (EASI), Dermatology Life Quality Index (DLQI), Children’s Dermatology Life Quality Index (CDLQI), and worst daily pruritus numeric rating scale (NRS). In the clinical experts’ clinical experience, it takes approximately 6 months to observe optimal benefits from tralokinumab treatment. They noted that significant improvements in QoL and ability to perform daily activities are indicators of a meaningful response to treatment even if the skin is not completely clear of all erythema or lichenification. The clinical experts noted that it would be appropriate to consider switching therapy in patients who experience no improvement in clinical or patient-reported outcomes, or in those who have intolerable side effects. Tralokinumab could be prescribed by a dermatologist, allergist, immunologist, and pediatrician with expertise in the diagnosis, treatment, and monitoring of patients with AD, in the clinical experts’ opinion.
Clinician Group Input
Three clinician groups, the Atlantic Specialist Group Managing AD (7 clinicians), Dermatology Association of Ontario (16 clinicians), and Canadian Dermatology Association (unknown number of clinicians) provided separate inputs. The 3 clinician groups and the clinical experts consulted by CADTH agreed that the goals of therapy are to improve symptoms (long-term and durable relief of chronic itch; minimization of dry and inflamed skin; clear or almost clear skin’ less oozing, scaling, cracking, or fissures; and improved QoL (better sleep) and function (focus on work and school). The clinical experts added that a reduction in anxiety or depressive symptoms and caregiver burnout are goals of therapy. As for unmet needs, the clinician groups and the clinical experts consulted by CADTH all agreed that not all patients respond to or tolerate the existing systemic treatments. Each JAKi has safety and contraindication issues (black-box warnings for patients with risk factors for cardiovascular events, cancers, and infections), and dupilumab is associated with conjunctivitis. New treatments are therefore needed to provide more options for patients whose AD is not well controlled with existing systemic therapies. The clinician groups stated that tralokinumab would have the same place in therapy as dupilumab, after phototherapy and/or off-label systemic therapies (if required by insurance or public plans) and may be trialled if patients fail to respond to dupilumab and oral JAKi treatment. The clinician groups reported that the suitable patient population aligns with the reimbursement request. They also noted that those who did not respond to biologics and/or JAKi therapy; have a history of conjunctivitis and/or risk factors associated with cardiovascular events, thrombosis, malignancy, serious infections and/or significant drug-drug interactions; or find it challenging to adhere to stricter dosing schedules, and those over the age of 65 years would be best suited for tralokinumab treatment. The clinical experts added that tralokinumab would most likely be used in patients with “pure” AD without comorbid asthma or eosinophilic esophagitis and those with special site involvement. The 3 clinician groups and the clinical experts consulted by CADTH indicated that they would assess response to treatment based on body surface area (BSA) affected, pruritus NRS, PGA (in clinical practice) and/or EASI, if required by an insurance company or payers, at 6 months after initiation of tralokinumab. According to the clinician groups, a lack of response or efficacy, worsening disease, deterioration of QoL, increased affected BSA, presence of adverse events (AEs), unacceptable intolerance, and allergies would prompt clinicians to consider discontinuation of tralokinumab treatment. Last, the clinician groups and the clinical experts agreed that a dermatologist, allergist, pediatrician, or immunologist well versed in managing moderate-to-severe AD should be allowed to prescribe tralokinumab. The 3 clinician groups raised concerns regarding differential access to tralokinumab, which is currently only funded by private insurance, and the need to try off-label immunosuppressants with lower efficacy and increased risk before accessing newer systemic agents.
Drug Program Input
The drug programs identified recent safety warnings for abrocitinib and upadacitinib that may preclude these drugs from being true comparators for adolescent patients. The clinical experts noted that these treatments are appropriate comparators for tralokinumab given that JAKi therapies are used to treat AD in adolescent patients in clinical practice, although most clinicians generally prefer to prescribe biologics first due to a better safety profile in this patient population.
The drug programs expressed interest in understanding whether tralokinumab should be reimbursed when used in patients who lost response to, or never achieved clinical benefit from, a trial of dupilumab. The clinical experts described these patients as reasonable candidates for tralokinumab treatment. The CADTH review team noted that the benefits of tralokinumab in patients who had prior systemic dupilumab and/or JAKi treatments were inconclusive in the 2 sponsor-submitted observational studies due to important limitations of the studies, including the open-label, retrospective, and noncomparative study designs, and small sample sizes.
The drug programs noted that consideration might be given to aligning the initiation, renewal, and prescribing criteria for tralokinumab with the existing criteria for dupilumab.
Clinical Evidence
Pivotal Studies and Randomized Controlled Trial Evidence
Description of Studies
Five phase III, double-blind, randomized controlled trials (RCTs) assessed whether tralokinumab increased the proportion of patients with an IGA score of 0 (clear) or 1 (almost clear) and the proportion of patients with a reduction of at least 75% in an Eczema Area and Severity Index score from baseline (EASI-75) at week 16 compared to placebo in patients with moderate-to-severe AD, were included in the submission; 1 of which included adolescent patients (ECZTRA 6, N = 301)9 and 4 of which included adult patients (ECZTRA 1, N = 802; ECZTRA 2, N = 794, ECZTRA 3, N = 380; ECZTRA 7, N = 277).10-12 The 4 studies in adults were previously reviewed by CADTH, and no new data from these studies were submitted for the current review. All enrolled patients had previously not progressed with topical therapy for AD. Patients in the ECZTRA 7 study had previously experienced uncontrolled disease on, or were not candidates for, systemic cyclosporine A treatment. Tralokinumab was compared with placebo, as monotherapy in the ECZTRA 1, 2, and 6 studies, and tralokinumab with TCS was compared with placebo plus TCS in the ECZTRA 3 and 7 studies. The proportion of patients with a reduction of at least 4 points in worst daily pruritus NRS, change from baseline in Scoring Atopic Dermatitis (SCORAD) score, and change from baseline in DLQI or CDLQI score were assessed at week 16 as key secondary end points in the ECZTRA 1, 2, 3, and 6 studies. In the ECZTRA 7 study, these were assessed as secondary end points at weeks 16 and 26. The mean age of the study population was 14.6 years (standard deviation [SD] = 1.7) in the ECZTRA 6 study and ranged between 36.5 years (SD = 14.1) and 39.1 years (SD = 15.2) in the ECZTRA 1, 2, 3, and 7 studies. The majority of patients were white and male in all studies. In the ECZTRA 6 study, prior systemic immunosuppressant, monoclonal antibody, and phototherapy treatments for AD were reported in 21.1%, 2.4%, and 25.6% of patients, respectively. In the ECZTRA 1, 2, 3, and 7 studies, prior phototherapy was noted in 43.7% to 58.8% of patients. Prior systemic immunosuppressant treatment was more common in the ECZTRA 7 study than in other studies in adults, with cyclosporine A being the most frequently used across studies (74.7% in the ECZTRA 7 study and 31.1% to 36.4% in the ECZTRA 1, 2, and 3 studies). Small proportions of patients in the ECZTRA 3 and 7 studies received prior monoclonal antibody treatment for AD (6.3% and 7.6%, respectively).
Efficacy Results — Initial Treatment Period
The key efficacy results in the initial treatment period of the ECZTRA 6 study (adolescents) and the ECZTRA 1, 2, 3, and 7 studies (adults) are summarized in Table 2 and Table 3, respectively. Results presented in this section pertaining to the primary estimand (i.e., the COVID-19–modified composite in the ECZTRA 7 study, the composite estimand in other studies for binary end points, and the hypothetical estimand for continuous end points in all studies), unless otherwise specified.
Investigator’s Global Assessment of 0 or 1
Adolescents (Aged 12 to < 18 Years): In the ECZTRA 6 study, the difference between the tralokinumab 300 mg every 2 weeks group and the placebo group in the coprimary end point of an IGA of 0 or 1 (i.e., the proportion of patients achieving an IGA score of 0 [clear] or 1 [almost clear]) at week 16 was 13.8% (95% confidence interval [CI], 5.3% to 22.3%; P = 0.002), in favour of tralokinumab.
Adults: The between-group differences in the coprimary end point of an IGA of 0 or 1 at week 16 were 8.6% (95% CI, 4.1% to 13.1%; P = 0.002) in the ECZTRA 1 study and 11.1% (95% CI, 5.8% to 16.4%; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 12.4% (95% CI, 2.9% to 21.9%; P = 0.015) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS; all favouring tralokinumab (or tralokinumab plus TCS).
In the ECZTRA 7 study, the between-group difference in the secondary end point of an IGA score of 0 or 1 was ||||| |||| ||| |||| || |||||| at week 16, and ||||| |||| ||| |||| || |||||| at week 26 when comparing tralokinumab every 2 weeks plus TCS with placebo plus TCS. Neither end point was tested for superiority due to prior failure in the testing hierarchy (i.e., reduction of worst daily pruritus NRS of at least 4 points from baseline).
Eczema Area and Severity Index
Adolescents: In the ECZTRA 6 study, the between-group difference in the coprimary end point of EASI-75 at week 16 was 22.0% (95% CI, 12.0% to 32.0%; P < 0.001), in favour of tralokinumab 300 mg every 2 weeks over placebo. Analyses of a reduction of at least 90% in the Eczema Area and Severity Index score from baseline (EASI-90), a reduction of at least 50% in the Eczema Area and Severity Index score from baseline (EASI-50), and change from baseline in EASI also showed results in favour of tralokinumab; however, these end points were not adjusted for multiplicity and were therefore at an increased risk of type I error (false-positive results).
Adults: The between-group differences in the coprimary end point of EASI-75 at week 16 were 12.1% (95% CI, 6.5% to 17.7%; P < 0.001) in the ECZTRA 1 study and 21.6% (95% CI, 15.8% to 27.3%; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 20.2% (95% CI, 9.8% to 30.6%; P < 0.001) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS with placebo plus TCS; all favouring tralokinumab (or tralokinumab plus TCS).
In the ECZTRA 7 study, the between-group difference in the primary end point of EASI-75 at week 16 was 14.1% (95% CI, 2.5% to 25.7%; P = 0.018), in favour of tralokinumab every 2 weeks plus TCS over placebo plus TCS. The between-group difference in the secondary end point of EASI-75 at week 26 was 14.1% (95% CI, 2.9% to 25.35%), for which superiority testing was not conducted due to prior failure in the testing hierarchy.
In the ECZTRA 1, 2, and 3 studies, EASI-90, EASI-50, and change from baseline in EASI at week 16 were secondary end points. In the ECZTRA 7 study, EASI-90 scores at weeks 16 and 26 were exploratory end points, and change from baseline in EASI at week 16 and week 26 were secondary end points. Results of these outcomes were in favour of tralokinumab (or tralokinumab plus TCS); however, they were not adjusted for multiplicity and were therefore at an increased risk of type I error (false-positive results).
Scoring Atopic Dermatitis
Adolescents: In the ECZTRA 6 study, the between-group difference in the key secondary end point of adjusted mean change from baseline in SCORAD at week 16 was −19.7 (95% CI, −27.1 to −12.2; P < 0.001), in favour of tralokinumab 300 mg every 2 weeks over placebo. Results of the secondary (treatment policy) and tertiary (composite) estimands were consistent with those of the primary (hypothetical) estimand.
Adults: The between-group differences in the key secondary end point of adjusted mean change from baseline in SCORAD at week 16 were −10.4% (95% CI, −14.4% to −6.5%; P < 0.001) in the ECZTRA 1 study and −14.0% (95% CI, −18.0% to −10.1%; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and −10.8% (95% CI, −15.2% to −6.5%; P < 0.001) in the ECZTRA 3 study when comparing between tralokinumab every 2 weeks plus TCS with placebo plus TCS; all favouring tralokinumab (or tralokinumab plus TCS). Results of the secondary (treatment policy) and tertiary (composite) estimands were consistent with those of the primary (hypothetical) estimand.
In the ECZTRA 7 study, the between-group differences in the secondary end point of adjusted mean change from baseline in SCORAD were −8.6 (95% CI, −13.0 to −4.2) at week 16 and −8.9 (95% CI, −13.2 to −4.6) at week 26 when comparing tralokinumab every 2 weeks plus TCS with placebo plus TCS. Results of the secondary (treatment policy) and tertiary (COVID-19–modified composite) estimands were consistent with those of the primary estimand at weeks 16 and 26. Neither end point was tested for superiority due to prior failure in the testing hierarchy.
Worst Daily Pruritis Numeric Rating Scale and Adolescent Worst Pruritis Numeric Rating Scale
Adolescents: In the ECZTRA 6 study, the between-group difference in the key secondary end point of the proportion of patients with a reduction of at least 4 points in adolescent worst pruritus NRS at week 16 was 21.7% (95% CI, 12.3% to 31.1%; P < 0.001), favouring tralokinumab 300 mg every 2 weeks over placebo.
Results of the responder analysis based on a 3-point reduction threshold (secondary end point) also favoured tralokinumab. The between-group difference with respect to the secondary end point of adjusted mean change from baseline in adolescent worst pruritus NRS at week 16 was −1.5 (95% CI, −2.4 to −0.6). Neither end point was adjusted for multiplicity and was therefore at increased risk of type I error (false-positive results).
Adults: The between-group differences in the key secondary end point of proportion of patients with a reduction of at least 4 points in worst pruritus NRS at week 16 were 9.7% (95% CI, 4.4% to 15.0%; P = 0.002) in the ECZTRA 1 study and 15.6% (95% CI, 10.3% to 20.9%; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 11.3% (95% CI, 0.9% to 21.6%; P = 0.037) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS; all were in favour of tralokinumab (or tralokinumab plus TCS). Results of the responder analysis based on a 3-point reduction threshold (a secondary end point) were also in favour of tralokinumab (or tralokinumab plus TCS); however, this end point was not adjusted for multiplicity and was at increased risk of producing false-positive results.
In the ECZTRA 7 study, the proportions of patients with a reduction of at least 4 points in worst pruritus NRS at week 16 and at week 26 were secondary end points. The between-group difference at week 16 was 9.7% (95% CI, −2.0% to 21.4%; P = 0.106) at week 16, which did not indicate a difference between tralokinumab every 2 weeks plus TCS and placebo plus TCS. Results of the secondary (composite) estimand were consistent with those of the primary estimand. The between-group difference at week 26 was 7.3% (95% CI, −4.6% to 19.2%) and was not tested for superiority due to prior failure in the testing hierarchy.
The between-group differences in the secondary end point of adjusted mean change from baseline in worst pruritus NRS at week 16 were −0.9 (95% CI, −1.4 to −0.4) in the ECZTRA 1 study and −1.3 (95% CI, −1.7 to −0.8) in the ECZTRA 2 study when comparing tralokinumab with placebo, and −1.2 (95% CI, −1.7 to −0.7) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. In the ECZTRA 7 study, the between-group differences (exploratory end points) were −0.9 (95% CI, −1.4 to −0.4) at week 16 and −0.9 (95% CI, −1.4 to −0.3) at week 26 when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. These end points were not adjusted for multiplicity and were at increased risk of type I error (false-positive results).
Dermatology Life Quality Index and Children’s Dermatology Life Quality Index
Adolescents: In the ECZTRA 6 study, the between-group difference in the key secondary end point of adjusted mean change from baseline in CDLQI at week 16 was −2.6 (95% CI, −4.5 to −0.7; P = 0.007), in favour of tralokinumab 300 mg every 2 weeks over placebo. Results of the secondary (treatment policy) and tertiary estimands (composite) were consistent with those of the primary (hypothetical) estimand.
Results of the responder analysis of proportion of patients with a reduction of at least 6 points in CDLQI from baseline at week 16 (secondary end point) were in favour of tralokinumab; however, this end point was not adjusted for multiplicity and was therefore at increased risk of type I error (false-positive results).
Adults: The between-group differences in the key secondary end point of change from baseline in DLQI at week 16 were −2.1 (95% CI, −3.4 to −0.8; P = 0.002) in the ECZTRA 1 study and −3.9 (95% CI, −5.2 to −2.6; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo and −2.9 (95% CI, −4.3 to −1.6; P < 0.001) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS; all were in favour of tralokinumab (or tralokinumab plus TCS). Results of the composite estimand were consistent with those of the primary (hypothetical) estimand.
In the ECZTRA 7 study, changes from baseline in DLQI at weeks 16 and 26 were secondary end points. The between-group difference at week 16 was −1.5 (95% CI, −2.6 to −0.4). Results of the secondary (treatment policy) and tertiary (COVID-19–modified composite) estimands were not consistent with the primary (hypothetical) estimand and did not suggest a difference between the treatment groups. At week 26, the between-group difference was −1.6 (95% CI, −2.7 to −0.5). Results of the composite estimand were consistent with those of the primary estimand. Neither end point was tested for superiority due to prior failure of the testing hierarchy.
The proportion of patients with a reduction of at least 4 points in DLQI from baseline was a secondary end point (at week 16) in the ECZTRA 1, 2, and 3 studies, and an exploratory end point in the ECZTRA 7 study. Results favoured tralokinumab (or tralokinumab plus TCS) in the ECZTRA 1, 2, and 3 studies ||| ||| ||| ||||||||||||||| ||||||| |||||||||||| ||||||| ||| ||||||||||| || ||||| || ||| || || |||||| ||. These end points were not adjusted for multiplicity.
Other Efficacy End Points
Adolescents: In the ECZTRA 6 study, results of change from baseline in eczema-related sleep NRS, an exploratory end point; Patient-Oriented Eczema Measure (POEM), a secondary end point; and Hospital Anxiety and Depression Scale (HADS) anxiety scores, an exploratory end point at week 16, were in favour of tralokinumab 300 mg every 2 weeks over placebo. However, these end points were not adjusted for multiplicity and were at increased risk of producing false-positive results.
The results do not suggest a difference between treatment groups in change from baseline in HADS depression scores (an exploratory end point) at week 16. The 95% CI in the between-group difference in proportion of patients with a HADS anxiety or depression score of less than 8 (an exploratory end point) was wide, crossing the null.
Use of TCS and number of days without topical treatment were not assessed in the ECZTRA 6 study.
Adults: Results of change from baseline in eczema-related sleep NRS and POEM (exploratory end points) were in favour of tralokinumab (or tralokinumab plus TCS) across the ECZTRA 1, 2, 3, and 7 trials; however, these end points were not adjusted for multiplicity and were at increased risk of type I error (false-positive results).
Results did not consistently suggest a difference between tralokinumab (or tralokinumab plus TCS) and placebo (or placebo plus TCS) across studies with respect to change from baseline in HADS anxiety and depression scores, proportion of patients with HADS anxiety or depression scores of less than 8 (exploratory end points in the ECZTRA 1, 2, 3, and 7), amount of TCS used, and number of days without topical treatment (secondary end points in the ECZTRA 3 study and exploratory end points in the ECZTRA 7 study). These end points were not adjusted for multiplicity.
Efficacy Results — Maintenance (or Continuous) Treatment Period
IGA Score of 0 or 1 at Week 52 (ECZTRA 1, 2, and 6) or Week 32 (ECZTRA 3) Among Patients With an IGA of 0 or 1 at Week 16
Adolescents: In the ECZTRA 6 study, the proportions of patients receiving tralokinumab 300 mg every 2 weeks with an IGA 0 or 1 at week 16 who maintained their IGA of 0 or 1 response at week 52 were 37.5% (3 out of 8 patients; 95% CI, 13.7% to 69.4%) in the tralokinumab 300 mg every 2 weeks then every 2 weeks group and 87.5% (7 out of 8 patients; 95% CI, 52.9% to 97.8%) in the tralokinumab 300 mg every 2 weeks then every 4 weeks group. No statistical analysis was conducted to assess the between-group difference.
Adults: In the ECZTRA 1 and 2 studies, the proportion of patients with an IGA of 0 or 1 at week 16 (without use of rescue medication) who maintained their IGA of 0 or 1 (without use of rescue medication) at week 52 was included in the statistical hierarchy. In the ECZTRA 1 study, the difference between the tralokinumab every 2 weeks group and the placebo group was 6.0% (95% CI, −21.8% to 33.7%; P = 0.68). Due to failure of this end point, no superiority testing was conducted for the difference between the tralokinumab every 4 weeks group and the placebo group (lower in the testing hierarchy), which was −9.5% (95% CI, −37.1% to 18.0%). In the ECZTRA 2 study, the difference between the tralokinumab every 2 weeks group and the placebo group was 34.1% (95% CI, 13.4% to 54.9%; P = 0.004). The difference between the tralokinumab every 4 weeks group and the placebo group was 19.9% (95% CI, −1.2 to 40.9; P = 0.084). Due to failure of this end point, no superiority testing was conducted for the end point lower in the testing hierarchy (i.e., EASI-75 at week 52 between tralokinumab 300 mg every 4 weeks and placebo).
In the ECZTRA 3 study, the proportions of patients with an IGA of 0 or 1 at week 16 who maintained their IGA of 0 or 1 response at week 32 were 89.6% (95% CI not reported) in the tralokinumab every 2 weeks plus TCS group and 77.6% (95% CI not reported) in the tralokinumab every 4 weeks plus TCS group. No statistical analysis was conducted to assess the between-group difference. This end point was not assessed in the ECZTRA 7 study.
EASI-75 at Week 52 (ECZTRA 1, 2, and 6) or Week 32 (ECZTRA 3) Among Patients With EASI-75 at Week 16
Adolescents: In the ECZTRA 6 study, the proportions of patients with EASI-75 at week 16 (without use of rescue medication) who maintained their EASI-75 response at week 52 (without use of rescue medication) were 44.4% (4 out of 9 patients; 95% CI, 18.9% to 73.3%) in the tralokinumab 300 mg every 2 weeks (week 0 to 16) then every 2 weeks group (week 17 to 52) and 53.8% (7 out of 13 patients; 95% CI, 29.1% to 76.8%) in the tralokinumab 300 mg every 2 weeks (week 0 to 16) then every 4 weeks (week 17 to 52) group. No statistical analysis was conducted to assess the between-group difference on these end points.
Adults: In the ECZTRA 1 study, the proportion of patients with EASI-75 at week 16 (without use of rescue medication) who maintained their EASI-75 response (without use of rescue medication) at week 52 was not tested for superiority due to prior failure in the testing hierarchy (the proportion of patients with an IGA of 0 or 1 at week 16 who maintained their IGA of 0 or 1 at week 52). The difference between the tralokinumab every 2 weeks group and the placebo group was 21.2% (95% CI, −0.2% to 42.6%). The difference between the tralokinumab every 4 weeks group and the placebo group was 11.7% (95% CI, −8.7% to 32.0%).
In the ECZTRA 2 study, the difference in the proportion of patients with EASI-75 at week 16 who maintained their EASI-75 response at week 52 between the tralokinumab 300 mg every 2 weeks and placebo groups was included in the statistical testing hierarchy and was 33.7% (95% CI, 17.3% to 50.0%; P < 0.001). The difference in the proportion of patients with EASI-75 at week 16 who maintained their EASI-75 response at week 52 between the tralokinumab 300 mg every 4 weeks and placebo groups was not tested for superiority due to failure of a prior end point in the statistical testing hierarchy (i.e., IGA of 0 or 1 at week 52 between tralokinumab 300 mg every 4 weeks and placebo).
In the ECZTRA 3 study, the proportions of patients with an IGA of 0 or 1 at week 16 who maintained their IGA of 0 or 1 at week 32 were 92.5% (95% CI not reported) in the tralokinumab every 2 weeks plus TCS group and 90.8% (95% CI not reported) in the tralokinumab every 4 weeks plus TCS group. No statistical analysis was conducted to assess the between-group difference. This end point was not assessed in the ECZTRA 7 study.
Harms Results — Initial Treatment Period
The key harms results in the initial treatment period of ECZTRA 6 (adolescents) and ECZTRA 1, 2, 3, and 7 (adults) are summarized in Table 2 and Table 3, respectively.
Treatment-Emergent Adverse Events
In the initial treatment period of the ECZTRA 1, 2, 3, 6, and 7 studies, the proportion of patients with at least 1 treatment-emergent adverse event (TEAE) ranged between 61.5% and 77.5% in the tralokinumab (or tralokinumab plus TCS) group and between 61.7% and 78.8% in the placebo (or placebo plus TCS) group. No notable between-group difference in the proportion of patients who reported at least 1 TEAE in the initial treatment period was observed across studies. The most common TEAEs reported in the tralokinumab group (in at least 10% of patients) were upper respiratory tract infection (URTI), viral URTI, AD, conjunctivitis, and headache.
Serious Treatment-Emergent Adverse Events
The frequency of serious TEAEs in the initial treatment period ranged between 0.7% and in the tralokinumab (or tralokinumab plus TCS) group, and between 2.5% and 5.3% in the placebo (or placebo plus TCS) group in all pivotal studies.
Withdrawal due to Adverse Events
No treatment withdrawal due to AEs or death was reported in adolescent patients. In the ECZTRA 1, 2, 3, and 7 studies, the proportions of adult patients who withdrew from treatment due to AEs ranged from 0.7% to 3.3% in the tralokinumab (or tralokinumab 300 mg every 2 weeks plus TCS) group and from 0.8% to 4.1% in the placebo (or placebo plus TCS) group.
Mortality
Two deaths (related to an unknown cause and myocardial infarction) were reported in the tralokinumab group in the ECZTRA 1 study, and 1 death (related to metastatic squamous cell carcinoma) was reported in the tralokinumab group in the ECZTRA 2 study. No deaths were reported in all other studies.
Notable Harms
There was no notable difference between the tralokinumab group and the placebo group in the frequency of eczema herpeticum, malignancies, skin infection requiring systemic treatment, and eye disorders reported in adolescents and adults, except that conjunctivitis was consistently reported more frequently in the tralokinumab group (3.0% to 11.1%) than in the placebo group (1.5% to 4.4%) across the studies in adults.
Harms Results — Maintenance (or Continuous) Treatment Period
The overall results in the maintenance (or continuous) treatment period of the ECZTRA 1, 2, and 3 studies were consistent with those of the initial treatment period.
Critical Appraisal
The randomization and allocation concealment methods were adequate; although there were some baseline imbalances in the ECZTRA 3 and 6 studies, these may have been due to chance and did not appear to consistently favour either treatment group. The trials were adequately blinded; however, there is a small potential for bias in measurement of patient-reported outcomes (i.e., adolescent worst daily pruritus NRS, eczema-related sleep NRS, POEM, DLQI or CDLQI, and HADS) leading to inflated efficacy of tralokinumab due to possible unblinding in patients becoming aware of their assignments based on treatment response; however, the presence and extent of such potential bias is unknown. In the initial treatment period, an IGA of 0 or 1, EASI-75, reduction of at least 4 points in adolescent worst daily pruritus NRS from baseline, change from baseline in SCORAD and DLQI outcomes were controlled for multiplicity, while the other end points (secondary and exploratory) were not controlled and were at an increased risk of type I error (false-positive results). Continuous secondary and exploratory end points (change from baseline in EASI, POEM, worst daily pruritus NRS, eczema-related sleep NRS, and HADS scores) were at a high risk of bias due to a large amount of missing data that were not appropriately accounted for in the statistical analysis. No conclusion can be drawn on subgroup analyses due to the lack of sample-size consideration and control for multiplicity. In the maintenance (or continuous) treatment period, the IGA of 0 or 1 and EASI-75 outcomes were adjusted for multiplicity in the ECZTRA 1 and 2 trials; however, results were uncertain due to a sizable reduction in sample sizes, wide CIs for IGAs of 0 or 1 and EASI-75 outcomes, and inconsistent results between the ECZTRA 1 and 2 studies.
Table 2: Key Results From ECZTRA 6 — Initial Treatment Period (Adolescents)
Outcomes at week 16 in the initial treatment period | Tralokinumab 300 mg q.2.w. (N = 97) | Placebo q.2.w. (N = 94) |
|---|---|---|
IGA score of 0 or 1 (full analysis set) | ||
n/N (%) | 17/97 (17.5) | 4/94 (4.3) |
Difference vs. placebo, % (95% CI)a | 13.8 (5.3 to 22.3; P = 0.002) | |
EASI-75 (full analysis set) | ||
n/N (%) | 27/97 (27.8) | 6/94 (6.4) |
Difference vs. placebo, % (95% CI)a | 22.0 (12.0 to 32.0; P < 0.001) | |
SCORAD (full analysis set) | ||
Number of patients contributing to the analysis | 66 | 35 |
Baseline SCORAD, mean (SD) | 68.3 (13.7) | 67.4 (14.9) |
Change from baseline, adjusted mean change (SE) | −29.1 (2.4) | −9.5 (3.0) |
Difference vs. placebo, (95% CI)b | −19.7 (−27.1 to −12.2; P < 0.001) | |
Adolescent worst pruritus NRS (weekly average) (full analysis set) | ||
Number of patients who contributed to the analysis | 62 | 31 |
Baseline adolescent worst pruritus NRS | 7.8 (1.5) | 7.5 (1.7) |
Change from baseline, adjusted mean change (SE) | −3.0 (0.3) | −1.5 (0.3) |
Difference vs. placebo, (95% CI)b,c | −1.5 (−2.4 to −0.6; P < 0.001) | |
Reduction of ≥ 4 from baseline, n/N (%) | 24/96 (25.0) | 3/90 (3.3) |
Difference vs. placebo, % (95% CI)a | 21.7 (12.3 to 31.1; P < 0.001) | |
CDLQI score (full analysis set) | ||
Number of patients contributing to the analysis | 84 | 89 |
Baseline CDLQI, mean (SD) | 13.4 (7.3) | 13.3 (6.0) |
Change from baseline, adjusted mean change (SE) | −6.7 (0.6) | −4.1 (0.7) |
Difference vs. placebo, (95% CI)b | −2.6 (−4.5 to −0.7; P = 0.007) | |
Harms, n (%) (safety analysis set) | ||
TEAEs | 63 (64.9) | 58 (61.7) |
Serious TEAEs | 1 (1.0) | 5 (5.3) |
Treatment withdrawal due to AE | 0 (0) | 0 (0) |
Deaths | 0 (0) | 0 (0) |
Notable harms, n (%) (safety analysis set) | ||
Eczema herpeticum | 0 (0) | 1 (1.1) |
Malignancies diagnosed after randomization | 0 (0) | 0 (0) |
Skin infection requiring systemic treatment | 2 (2.1) | 2 (2.1) |
Eye disorders | ||
Conjunctivitis | 0 (0) | 0 (0) |
Bacterial conjunctivitis | 1 (1.0) | 0 (0) |
Allergic conjunctivitis | 2 (2.1) | 2 (2.1) |
Viral keratitis | 1 (1.0) | 0 (0) |
AE = adverse event; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; q.2.w. = every 2 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; SE = standard error; TEAE = treatment-emergent adverse event.
Notes: The key outcomes summarized in this table include the coprimary efficacy end points, the key secondary efficacy end points, and other end points that were noted by the clinical experts consulted by CADTH to be of high importance for clinical decision-making. Unless otherwise specified, AEs are reported based on the Medical Dictionary for Regulatory Activities preferred term.
aThe analysis was conducted using a Cochran-Mantel-Haenszel test stratified by baseline IGA and region based on the composite estimand.
bThe analysis was conducted using the repeated measurements model, with baseline IGA, region, and treatment-by-week interaction as factors and interaction between week and baseline value as a covariate, based on the hypothetical estimand.
cThe end point was not adjusted for multiplicity and was at an increased risk of type I error (false-positive result).
Sources: Clinical Study Report for ECZTRA 613 and the sponsor’s Summary of Clinical Evidence.14
Table 3: Key Results From ECZTRA 1, 2, 3, and 7 — Initial Treatment Period (Adults; Original Review)
Outcomes | ECZTRA 1 (follow-up at 16 weeks) | ECZTRA 2 (follow-up at 16 weeks) | ECZTRA 3 (follow-up at 16 weeks) | ECZTRA 7 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
Follow-up at 16 weeks | Follow-up at 26 weeks | |||||||||
Tralokinumab q.2.w. N = 601 | Placebo N = 197 | Tralokinumab q.2.w. N = 591 | Placebo N = 201 | Tralokinumab q.2.w. + TCS N = 252 | Placebo + TCS N = 126 | Tralokinumab q.2.w. + TCS N = 138 | Placebo + TCS N = 137 | Tralokinumab q.2.w. + TCS N = 138 | Placebo + TCS N = 137 | |
IGA score of 0 or 1 (full analysis set) | ||||||||||
n/N (%) | 95/601 (15.8) | 14/197 (7.1) | 131/591 (22.2) | 22/201 (10.9) | 98/252 (38.9) | 33/126 (26.2) | |||||| |||||| | |||||| |||||| | |||| |||||| | |||| |||||| |
Difference, % (95% CI) | 8.6 (4.1 to 13.1; P = 0.002)a | 11.1 (5.8 to 16.4; P < 0.001)a | 12.4 (2.9 to 21.9; P = 0.015)a | |||| |||| || |||||||||| | |||| |||| || ||||||||| | |||||
EASI-75 (full analysis set) | ||||||||||
EASI-75, n/N (%) | 150/601 (25.0) | 25/197 (12.7) | 196/591 (33.2) | 23/201 (11.4) | 141/252 (56.0) | 45/126 (35.7) | 88.6/138 (64.2) | 69.2/137 (50.5) | 95.0 (68.8) | 75.7 (55.3) |
Difference, % (95% CI) | 12.1 (6.5 to 17.7; P < 0.001)a | 21.6 (15.8 to 27.3; P < 0.001)a | 20.2 (9.8 to 30.6; P < 0.001)a | 14.1 (2.5 to 25.7; P = 0.018)b | 14.1 (2.9 to 25.3; P = 0.014)b,c | |||||
SCORAD (full analysis set) | ||||||||||
n | 353 | 96 | 430 | 98 | 229 | 107 | 117 | 110 | 116 | 104 |
Baseline score, mean (SD) | 70.3 (13.0) | 71.7 (12.5) | 70.0 (13.4) | 70.5 (12.2) | 67.0 (13.3) | 68.9 (13.2) | 70.2 (12.0) | 70.8 (12.8) | 70.2 (12.0) | 70.8 (12.8) |
Change from baseline, adjusted mean (SE) | −25.2 (0.94) | −14.7 (1.80) | −28.1 (0.92) | −14.0 (1.79) | −37.7 (1.25) | −26.7 (1.83) | −42.7 (1.6) | −34.1 (1.6) | −46.3 (1.5) | −37.3 (1.6) |
Difference, (95% CI) | −10.4 (−14.4 to −6.5; P < 0.001)d | −14.0 (−18.0 to −10.1; P < 0.001)d | −10.9 (−15.2 to −6.5; P < 0.001)d | −8.6 (−13.0 to −4.2; P < 0.001)c,e | −8.9 (−13.2 to −4.6; P < 0.001)c,e | |||||
Worst daily pruritus NRS (weekly average) (full analysis set) | ||||||||||
n | 325 | 88 | 401 | 94 | 221 | 100 | 115 | 112 | 111 | 101 |
Baseline score, mean (SD) | 7.7 (1.4) | 7.7 (1.4) | 7.9 (1.5) | 8.0 (1.4) | 7.7 (1.5) | 7.9 (1.5) | 7.3 (1.5) | 7.5 (1.4) | 7.3 (1.5) | 7.5 (1.4) |
Change from baseline, adjusted mean (SE) | −2.6 (0.11) | −1.7 (0.21) | −2.9 (0.11) | −1.6 (0.21) | −4.1 (0.15) | −2.9 (0.21) | −4.0 (0.2) | −3.1 (0.2) | −4.3 (0.2) | −3.4 (0.2) |
Difference, (95% CI) | −0.9 (−1.4 to −0.4; P < 0.001)d,f | −1.3 (−1.7 to −0.8; P < 0.001)d,f | −1.2 (−1.7 to −0.7; P < 0.001)d,f | −0.9 (−1.4 to −0.4; P < 0.001)e,f | −0.9 (−1.4 to −0.3; P = 0.002)e,f | |||||
Reduction from baseline ≥ 4, n/N (%)b | 119/594 (20.0) | 20/194 (10.3) | 144/575 (25.0) | 19/200 (9.5) | 113/249 (45.4) | 43/126 (34.1) | 61/134 (45.5) | 48/135 (35.6) | 63/134 (47.2) | 54/135 (39.7) |
Difference, % (95% CI) | 9.7 (4.4 to 15.0; P = 0.002)a | 15.6 (10.3 to 20.9; P < 0.001)a | 11.3 (0.9 to 21.6; P = 0.037)a | 9.7 (−2.0 to 21.4; P = 0.106)b | 7.3 (−4.6 to 19.2; P = 0.228)b,c | |||||
DLQI (full analysis set) | ||||||||||
n | 335 | 95 | 419 | 97 | 226 | 104 | 112 | 106 | 107 | 97 |
Baseline score, mean (SD) | 16.8 (7.1) | 17.0 (6.6) | 17.7 (7.1) | 17.8 (7.3) | 17.6 (7.1) | 17.2 (7.2) | 15.9 (6.5) | 16.4 (6.3) | 15.9 (6.5) | 16.4 (6.3) |
Change from baseline, adjusted mean (SE) | −7.1 (0.31) | −5.0 (0.59) | −8.8 (0.30) | −4.9 (0.60) | −11.7 (0.39) | −8.8 (0.56) | −11.2 (0.40) | −9.6 (0.40) | −11.5 (0.40) | −9.9 (0.40) |
Difference, (95% CI) | −2.1 (−3.4 to −0.8; P = 0.002)d | −3.9 (−5.2 to −2.6; P < 0.001)d | −2.9 (−4.3 to −1.6; P < 0.001)d | −1.5 (−2.6 to −0.4; P = 0.009)c,e | −1.6 (−2.7 to −0.5; P = 0.005)c,e | |||||
Harms, n (%) (safety analysis set) | ||||||||||
TEAEs | 460 (76.4) | 151 (77.0) | 364 (61.5) | 132 (66.0) | 180 (71.4) | 84 (66.7) | NR | NR | 107 (77.5) | 108 (78.8) |
Serious TEAEs | 23 (3.8) | 8 (4.1) | 10 (1.7) | 5 (2.5) | 2 (0.8) | 4 (3.2) | NR | NR | 1 (0.7) | 5 (3.6) |
Treatment withdrawal due to AE | 20 (3.3) | 8 (4.1) | 9 (1.5) | 3 (1.5) | 6 (2.4) | 1 (0.8) | NR | NR | 1 (0.7) | 3 (2.2) |
Deaths | 2 (0.3) | 0 (0) | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | NR | NR | 0 (0) | 0 (0) |
Notable harms, n (%) (safety analysis set) | ||||||||||
Eczema herpeticum | 3 (0.5) | 2 (1.0) | 2 (0.3) | 5 (2.5) | 1 (0.4) | 1 (0.8) | NR | NR | 1 (0.7) | 0 (0) |
Malignancies diagnosed after randomization | 0 (0) | 0 (0) | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | NR | NR | 0 (0) | 0 (0) |
Skin infections requiring systemic treatment | 13 (2.2) | 4 (2.0) | 21 (3.5) | 22 (11.0) | 4 (1.6) | 7 (5.6) | NR | NR | 1 (0.7) | 8 (5.8) |
Eye disorders | NR | NR | ||||||||
Conjunctivitis | 43 (7.1) | 4 (2.0) | 18 (3.0) | 3 (1.5) | 28 (11.1) | 4 (3.2) | NR | NR | 13 (9.4) | 6 (4.4) |
Bacterial conjunctivitis | 2 (0.3) | 0 (0) | 2 (0.3) | 1 (0.5) | 0(0) | 0(0) | NR | NR | 0(0) | 0(0) |
Viral conjunctivitis | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (0.8) | NR | NR | 0 (0) | 0 (0) |
Allergic conjunctivitis | 16 (2.7) | 3 (1.5) | 12 (2.0) | 2 (1.0) | 5 (2.0) | 2 (1.6) | NR | NR | 0 (0) | 0 (0) |
Keratoconjunctivitis | 1 (0.2) | 0 (0) | 2 (0.3) | 0 (0) | 1 (0.4) | 0 (0) | NR | NR | 1 (0.7) | 0 (0) |
Keratitis | 3 (0.5) | 0 (0) | 1 (0.2) | 1 (0.5) | 0 (0) | 0 (0) | NR | NR | 1 (0.7) | 1 (0.7) |
AE = adverse event; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI-75 = reduction of at least 75% in Eczema Area Severity Index score from baseline; IGA = Investigator’s Global Assessment; NR = not reported; NRS = numeric rating scale; q.2.w. = every 2 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; SE = standard error; TCS = topical corticosteroids; TEAE = treatment-emergent adverse event.
Note: Unless otherwise specified, AEs are reported based on the Medical Dictionary for Regulatory Activities preferred term.
aThe analysis was conducted using a Cochran-Mantel-Haenszel test stratified by baseline IGA and region based on the composite estimand.
bThe analysis was conducted using a Cochran-Mantel-Haenszel test stratified by prior cyclosporine A use, country, and baseline disease severity based on the COVID-19–modified composite estimand.
cThis end point was included in the statistical testing hierarchy; however, no superiority testing was conducted for this end point due to prior failure in the statistical testing hierarchy.
dThe analysis was conducted using the repeated measurements model, with baseline IGA, region, and treatment-by-week interaction as factors and interaction between week and baseline value as a covariate, based on the hypothetical estimand (primary estimand).
eThe analysis was conducted using the repeated measurements model, with baseline IGA, country, prior cyclosporine A use and treatment-by-week interaction as factors, and interaction between week and baseline value as a covariate, based on the hypothetical estimand (primary estimand).
fThe end point was not adjusted for multiplicity and was at an increased risk of type I error (false-positive result).
Sources: Clinical Study Reports for ECZTRA 1, 2, 3, and 715-18 and the sponsor’s Summary of Clinical Evidence.14
The study population of the ECZTRA 7 trial (i.e., adults who had uncontrolled disease or were not deemed to be candidates for topical therapy and cyclosporine A) was more reflective of the anticipated place in therapy of tralokinumab compared with other included RCTs in patients who had uncontrolled disease with topical therapy alone. The study interventions of the ECZTRA 3 and 7 studies (i.e., tralokinumab in combination with TCS) were also more reflective of the real-world use of tralokinumab compared with the ECZTRA 1, 2, and 6 studies (i.e., tralokinumab monotherapy) based on clinical expert input that patients typically use biologics in combination with TCS for active lesions. The clinical relevance of SCORAD, POEM, HADS outcomes is unclear given that these instruments are not routinely used in clinical practice. Based on their experience, the clinical experts consulted by CADTH considered the duration of follow-up in the initial treatment period (16 weeks) to be insufficient to adequately assess efficacy, as most patients would require at least 6 months of tralokinumab treatment to achieve an optimal response. Results of the maintenance treatment period (up to 52 weeks) are likely more generalizable but inconclusive due to issues with internal validity. The absence of direct comparative evidence between tralokinumab and relevant comparators (dupilumab, upadacitinib, and abrocitinib) represents a gap in pivotal trial evidence in the treatment of patients with moderate-to-severe AD.
Long-Term Extension Studies
Description of Study
One ongoing, open-label, single-arm, multicentre, long-term extension (LTE) trial, ECZTEND, was submitted by the sponsor. This study involved patients with moderate-to-severe AD who previously participated in clinical trials of tralokinumab (i.e., ECZTRA 1 to 8 and TraSki).14 Patients were eligible to participate in the ECZTEND trial if they had completed the treatment period(s) in 1 of the parent trials, regardless of the type of previous treatment (i.e., tralokinumab or placebo) or treatment response. All patients received tralokinumab with dosing administered by self-injection as prescribed by the product monograph. Patients were permitted to use concomitant TCS or topical calcineurin inhibitors (TCIs) and were required to apply an emollient at least twice daily for at least 14 days before the ECZTEND trial baseline and continue throughout the trial. The primary outcome was long-term safety, specifically the number of AEs experienced during the study. The secondary outcomes were based on efficacy and included achieving an IGA of 0 or 1 and EASI-75, each measured at weeks 16, 56, 88, 104, 136, 152, 184, 216, and 248. All analyses were descriptive and based on observed cases, with sensitivity analyses using last observation carried forward (LOCF) or modified nonresponder imputation (mNRI) to account for missing data. The 2 major cohorts used for the outcomes analyses were adults and adolescents. The data cut-off dates for the adult cohort for the reported interim analyses were April 30, 2021 (all participants from the ECZTRA 1, 2, 3, 4, 5, and 7 studies enrolled in the ECZTEND study, n = 1,442, with up to 3.5 years of follow-up; 3-year subgroup containing participants from the ECZTRA 1 and 2 studies, n = 347) and April 30, 2022 (4-year subgroup containing participants from the ECZTRA 1 and 2 studies, n = 347). The data cut-off date for the adolescent cohort was April 30, 2022 (participants from the ECZTRA 6 study, up to 3 years of follow-up, n = 127).
Efficacy Results
EASI-75
EASI-75 was assessed relative to the baseline in the parent trials. EASI-75 was achieved in 85.1% of patients (411 of 483, observed data) at week 104 in the ECZTEND study (i.e., up to 3 years of cumulative exposure to tralokinumab in the parent trials and the ECZTEND study) in the all-participants adult cohort; in 84.5% of patients (147 of 174, observed data) at week 152 in the ECZTEND study in the 4-year adult subgroup; and in 84.4% of patients (92 of 109, observed data) at week 56 in the ECZTEND study (i.e., 2 years of cumulative exposure to tralokinumab in the parent trials and the ECZTEND study) in the adolescent cohort. The results of the sensitivity analyses were consistent with those of the primary analysis using observed data.
IGA of 0 or 1
IGA of 0 or 1 was achieved in 50.5% of patients (244 of 483, observed data) at week 104 in the ECZTEND study (i.e., up to 3 years of cumulative exposure to tralokinumab in the parent trials and the ECZTEND study) in the all-participants adult cohort; in 52.6% of patients (92 of 175, observed data) at week 152 in the ECZTEND study in the 4-year adult subgroup; and in 61.5% of patients (67 of 109, observed data) at week 56 in the ECZTEND study (i.e., 2 years of cumulative exposure to tralokinumab in the parent trials and the ECZTEND study) in the adolescent cohort. The results of the sensitivity analyses were consistent with those of the primary analysis using observed data.
Harms Results
In the adult cohort (all participants, n = 1,442), 1,127 patients (78.2%) experienced at least 1 TEAE. In the 3-year adult subgroup (n = 347), 295 patients (85.0%) experienced at least 1 TEAE. In the adolescent cohort (n = 127), 83 patients (65.4%) experienced at least 1 TEAE. In all cohorts, the 3 most common AEs were a viral URTI (13.4% to 28.8%), AD (10.2% to 19.6%), and URTI (7.0% to 10.1%). Between 2.4% and 8.9% of patients reported a serious adverse event (SAE) in these cohorts. Conjunctivitis was reported in 77 patients (5.3%) and 7 patients (3.6%) from the all-participants adult cohort and the adolescent cohort, respectively. Frequency of treatment discontinuation was reported to be between 0.8% to 2.6%. No deaths were reported in the adult cohorts. However, 1 death (0.8%) due to an accident occurred in the adolescent cohort.
Critical Appraisal
Similar to other LTE studies, in the ECZTEND study, it is uncertain if the observed long-term effects can be attributed to tralokinumab treatment due to the lack of a comparison group and no adjustment for potential confounding. A risk of selection bias that favours tralokinumab is also possible, given that patients who perceived the treatment to be benefiting them during the parent trials were more likely to transfer to the extension study. Similarly, long-term safety concerns may be underestimated, as those who had experienced intolerable AEs in the parent trials were excluded from the ECZTEND trial. Given the open-label design of the study, there is also a risk of bias in the measurement of patient-reported outcomes (worst weekly pruritus NRS and DLQI), potentially favouring tralokinumab. The results related to benefits are at risk of being overestimated as they are interim findings.
The ECZTEND trial included patients who completed 1 of the parent trials regardless of treatment response. This is different from clinical practice, in which patients are expected to continue tralokinumab treatment only if they demonstrate objective improvement of disease after an adequate trial of treatment. It is unknown how many patients enrolled in the ECZTEND trial were nonresponders in the parent trial, potentially affecting the generalizability of the study population, because it is unclear what proportion of patients had experienced prior failure of immunosuppressant therapy, which is the likely place in therapy of tralokinumab. Further, the use of concomitant TCS and rescue medications could influence treatment response; however, utilization of such medications was not reported in the study and the impact on generalizability of study findings is therefore unclear.
Indirect Comparisons
In the absence of head-to-head evidence comparing tralokinumab to other relevant therapies used to manage AD, the sponsor submitted 4 indirect treatment comparisons (ITCs) of the effects of tralokinumab and other treatments in patients with moderate-to-severe AD. Of the ITCs submitted, 2 were network meta-analyses (NMAs), including 1 in adults and 1 in adolescents, and 2 were matching adjusted indirect comparisons (MAICs), both in adults.19-22
Network Meta-Analyses
Description of Studies
The sponsor submitted an NMA conducted by the Institute for Clinical and Economic Review (ICER) that aimed to evaluate the relative efficacy and safety of treatment with tralokinumab versus other therapies in adult patients with moderate-to-severe AD.19 It is not clear if this NMA was identified by a systematic literature search, and, if so, how it was selected from the available literature. The ICER NMA was used to inform the sponsor-submitted economic model for the treatment effect of tralokinumab up to week 16. A sponsor-commissioned NMA, the LEO Pharma NMA, ||||| || |||||||| ||| |||||||| |||||||| ||| |||||| || ||||||||| |||| |||||||||||| |||||| ||||| ||||||||| || |||||||||| |||||||| ||||||| ||| |||| || || ||| || ||||| |||| |||||||||||||||||| || ||||||| ||||| || ||| |||
Efficacy Results
Efficacy results of the NMA are presented for monotherapy and combination therapy by population (i.e., adults and adolescents). A pairwise comparison against baricitinib is not presented as the treatment is not currently approved for use in Canada.
EASI-50
Adult Population (ICER Network Meta-Analysis): The EASI-50 treatment responses to all included monotherapy interventions in adult patients were greater than those to placebo. Treatments with upadacitinib 30 mg (relative risk [RR] = 1.75; 95% credible interval [CrI], 1.50 to 2.10), abrocitinib 200 mg (RR = 1.59; 95% CrI, 1.31 to 1.95), upadacitinib 15 mg (RR = 1.53; 95% CrI, 1.20 to 1.84), and dupilumab 300 mg (RR = 1.40; 95% CrI, 1.18 to 1.69) were favoured for achievement of EASI-50 compared to tralokinumab 300 mg. The point estimate for EASI-50 favoured abrocitinib 100 mg over tralokinumab 300 mg, but the CrI also included the potential of little-to-no difference between the treatments (RR = 1.21; 95% CrI, 0.95 to 1.53).
The treatment responses to all included combination-therapy interventions on EASI-50 in adult patients exceeded those to placebo. Treatments with upadacitinib 30 mg (RR = 1.45; 95% CrI, 1.27 to 1.71), abrocitinib 200 mg (RR = 1.32; 95% CrI, 1.14 to 1.57), upadacitinib 15 mg (RR = 1.32; 95% CrI, 1.15 to 1.57), dupilumab 300 mg (RR = 1.26; 95% CrI, 1.09 to 1.49), and abrocitinib 100 mg (RR = 1.20; 95% CrI, 1.02 to 1.43) were favoured for achievement of EASI-50 compared to tralokinumab 300 mg.
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || ||||||| || || ||||| || |||||||||| |||||||| |||| |||||||| |||| |||||||| || |||||||||| ||||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| ||||||| ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
EASI-75
Adult Population (ICER Network Meta-Analysis): The EASI-75 treatment responses to all included monotherapy interventions in adult patients were greater than those to placebo. Treatments with upadacitinib 30 mg (RR = 2.77; 95% CrI, 1.77 to 2.77), abrocitinib 200 mg (RR = 1.89; 95% CrI, 1.45 to 2.49), upadacitinib 15 mg (RR = 1.79; 95% CrI, 1.42 to 2.29), and dupilumab 300 mg (RR = 1.58; 95% CrI, 1.25 to 2.03) were favoured for achievement of EASI-75 compared to tralokinumab 300 mg. The point estimate for EASI-75 favoured abrocitinib 100 mg over tralokinumab 300 mg, but the CrI also included the potential of little-to-no difference between the treatments (RR = 1.29; 95% CrI, 0.93 to 1.76).
The treatment response to all included combination-therapy interventions on EASI-75 in adult patients exceeded those to placebo. Treatments with upadacitinib 30 mg (RR = 1.90; 95% CrI, 1.53 to 2.45), abrocitinib 200 mg (RR = 1.58; 95% CrI, 1.25 to 2.07), upadacitinib 15 mg (RR = 1.48 95% CrI, 1.26 to 2.07), dupilumab 300 mg (RR = 1.46; 95% CrI, 1.15 to 1.90), and abrocitinib 100 mg (RR = 1.34; 9% Crl 1.03 to 1.76) were favoured for achievement of EASI-75 compared to tralokinumab 300 mg.
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || ||||||| || |||||||||| |||||||| |||| |||||||| |||| |||||||| ||||||||| |||| |||||||||||| || || || ||| |||||||| ||| ||||||||||| || ||||||| |||||||| || |||||||||||| ||| || ||||| ||||| ||||| ||||| ||| |||| |||| || |||||| ||| || |||||||||||| ||| || |||| ||||| ||| ||| |||| || ||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| ||||||| ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
EASI-90
Adult Population (ICER Network Meta-Analysis): The EASI-90 treatment responses to all included monotherapy interventions in adult patients were greater than those to placebo. Treatments with upadacitinib 30 mg (RR = 2; 95.89% CrI, 2.19 to 3.95), abrocitinib 200 mg (RR = 2.36; 95% CrI, 1.65 to 3.39), upadacitinib 15 mg (RR = 2.17; 95% CrI, 1.60 to 3.00), and dupilumab 300 mg every 2 weeks (RR = 1.83; 95% CrI, 1.34 to 2.54) were favoured for achievement of EASI-90 compared to tralokinumab 300 mg. The point estimate for EASI-90 favoured abrocitinib 100 mg over tralokinumab 300 mg, but the CrI also included the potential of little-to-no difference between the treatments (RR = 1.39; 95% CrI, 0.91 to 2.09).
The EASI-90 treatment responses to all included combination-therapy interventions in adult patients were superior to those to placebo. Treatments with upadacitinib 30 mg (RR = 2.74; 95% CrI, 1.98 to 3.97), abrocitinib 200 mg (RR = 2.01; 95% CrI, 1.41 to 2.98), upadacitinib 15 mg (RR = 2.01; 95% CrI, 1.43 to 2.96), dupilumab 300 mg (RR = 1.76; 95% CrI, 1.24 to 2.57), and abrocitinib 100 mg (RR = 1.54; 95% CI, 1.05 to 2.31) were favoured for achievement of EASI-90 compared to tralokinumab 300 mg.
Adolescent Population (LEO Pharma Network Meta-Analysis): The EASI-90 treatment responses to all included monotherapy interventions in adolescent patients were superior to those to placebo. Treatment with upadacitinib 15 mg was favoured for achievement of EASI-90 compared to tralokinumab 150 mg (odds ratio [OR] = 15.82; 95% Crl, 1.60 to 734.24) and tralokinumab 300 mg (OR = 17.95; 95% Crl, 1.74, 843.75). Treatment with upadacitinib 30 mg once daily was favoured for achievement of EASI-90 compared to tralokinumab 150 mg (OR = 47.05; 95% CrI, 4.82 to 2,297.05) and to tralokinumab 300 mg (OR = 53.95; 95% Crl, 5.45 to 2,620.94). The Crls for comparisons were too wide to draw any conclusions of certainty in in achieving EASI-90 between tralokinumab and the other active comparators.
Investigation’s Global Assessment
Adult Population (ICER Network Meta-Analysis): The IGA treatment responses to all included monotherapy interventions in adult patients were greater than those to placebo. Treatments with upadacitinib 30 mg (RR = 3.97; 95% CrI, 2.54 to 6.31), upadacitinib 15 mg (RR = 3.07; 95% CrI, 1.88 to 4.99), abrocitinib 200 mg (RR = 2.75; 95% CI, 1.54 to 4.95), and dupilumab 300 mg (RR = 2.15; 95% CrI, 1.31 to 3.60) were favoured for achievement of an IGA of 0 or 1 compared to tralokinumab 300 mg. The Crls for the comparison between tralokinumab and abrocitinib 100 mg were too wide to draw any conclusions of certainty in IGA responses in adult patients receiving monotherapy for AD.
The IGA treatment responses to all included combination interventions in adult patients were superior to those to placebo. Treatments with upadacitinib 30 mg (RR = 2.83; 95% CrI, 1.90 to 4.27), abrocitinib 200 mg (RR = 2.24; 95% CI, 1.44 to 3.49), upadacitinib 15 mg (RR = 2.08; 95% CrI, 1.35 to 3.25), dupilumab 300 mg (RR = 1.85; 95% CrI, 1.20 to 2.88), and abrocitinib 100 mg (RR = 1.66; 95% CI, 102 to 2.68) were favoured for achievement of an IGA of 0 or 1 compared to tralokinumab 300 mg.
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || ||| ||| || |||||||||| |||||||| |||| |||||||| |||| |||||||| ||||||||| |||| |||||||||||| || || || ||| |||||||| ||| ||||||||||| || ||| ||| |||||||| || |||||||||||| ||| || |||| ||||| ||| |||| |||| || |||||| ||| || |||||||||||| ||| || |||| ||||| ||| ||| |||| || ||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || || ||||||||| ||| ||| ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Peak Puritis NRS Improvement of 4 Points or Greater
Adult Population (ICER Network Meta-Analysis): The treatment responses to all included monotherapy interventions as measured by an improvement of 4 points or greater on the peak pruritis numeric rating scale (PP-NRS) in adult patients were greater that those to placebo. Treatments with upadacitinib 30 mg (RR = 2.16; 95% CrI, 1.14 to 4.58), dupilumab 300 mg (RR = 2.12; 95% CrI, 1.06 to 4.43), and upadacitinib 15 mg (RR = 1.97; 95% CrI, 1.01 to 4.28) were favoured for achievement of an improvement of at least 4 points in PP-NRS compared to tralokinumab 300 mg. The Crls for the remaining comparisons were too wide to draw any conclusions of certainty in improvement of at least 4 points in PP-NRS between tralokinumab and other active comparators among adult patients.
The treatment responses to all included combination-therapy interventions as measured by an improvement of at least 4 points in PP-NRS in adult patients were greater that those to placebo. Treatments with upadacitinib 30 mg (RR = 2.37; 95% CrI, 1.75 to 3.29), abrocitinib 200 mg (RR = 2.04; 95% Crl, 1.47 to 2.89), upadacitinib 15 mg (RR = 1.91; 95% CrI, 1.34 to 2.74), and dupilumab 300 mg (RR = 1.79; 95% CrI, 1.28 to 2.55) were favoured for achievement of improvement of at least 4 points in PP-NRS compared to tralokinumab 300 mg. The point estimate for improvement of at least 4 points in PP-NRS favoured abrocitinib 100 mg over tralokinumab 300 mg, but the CrI also included the potential of little-to-no difference between the treatments (RR = 1.40; 95% Crl, 0.93 to 2.10).
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || |||||||| ||| ||||||||||||| |||| || || |||||||||| |||||||| |||| |||||||| |||| |||||||| || |||||||||| ||||||||| ||| |||| ||| |||||||||||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || |||||||| ||| |||||||||||| |||| || ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Children’s Dermatology Life Quality Index
A network meta-analysis of the CDLQI was not reported in the ICER NMA.
||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || ||||| ||||||||||||| |||||||| || |||| || || |||||||||| |||||||| |||| |||||||| |||| |||||||| ||||||||| |||| ||||||||||| ||| || ||| |||||||| ||| ||||||||||| || ||||| ||||||||||||| |||||||| || |||| || |||||||| || |||||||||||| ||| || |||| ||||| ||| ||| |||| || ||||||| ||| ||||| |||||||| ||| ||||| |||||||||||||| |||||||| || |||| || |||||||| ||||||||| ||||||| || ||| |||| |||||||||||| ||| ||| ||| ||| ||| |||| |||||||| ||| ||||||||| || |||||||||||| |||||||||| ||||||| ||| |||||||||| |||| ||||| ||| |||| |||| || |||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||| ||||||||||||||| |||||||| || |||| || ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Patient-Oriented Eczema Measure
A network meta-analysis of POEM was not reported in the ICER NMA.
||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || |||| |||||||||||||| |||||||| || |||| || || |||||||||| |||||||| |||| |||||||| |||| |||||||| |||||| ||| ||||||||||| ||| || ||| ||||||||| |||| |||||||||||| ||| || ||| |||||||| ||| ||||||||||| || |||| |||||||||||||| |||||||| || |||| || |||||||| || ||||||||||| ||| || |||| ||||| ||| ||| |||| || |||||| ||| ||||| |||||||| ||| |||| ||||||||||||| |||||||| || |||| || |||||||| ||||||||| ||||||| || ||| |||| |||||||||||| ||| ||| ||| ||| ||| |||| |||||||| ||| ||||||||| || |||||||||||| |||||||||| ||||||| ||| |||||||||| |||| ||||| ||| |||| |||| || ||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || |||| ||||||||||||||| |||||||| || |||| || ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Harms Results
Adverse Events
A network meta-analysis of harms data was not reported in the ICER NMA.
|| |||||||||| ||||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||| || |||| || ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Critical Appraisal
ICER Network Meta-Analysis
The ICER NMA was based on studies identified from a systematic literature review of relevant randomized evidence of treatments for adults and adolescent with AD. The systematic literature search was based on a patient, intervention, comparison, and outcome (PICO) model defined a priori, with efficacy and safety outcomes predefined. The systematic literature search was comprehensive. The selection process was not clearly defined, and data extraction was conducted by a single reviewer, increasing the risk of bias and error. While the risk of bias of the comparator trials was assessed, the method used was not reported, and risk of bias was not assessed by outcome. Several sources of clinical and heterogeneity were identified, which challenged the plausibility of the underlying transitivity assumption. These included variations in patient age, duration of disease, disease severity, length of the washout period, time point of follow-up (12 to 16 weeks), and methods of imputation for missing data. To account for differences in corticosteroid use across trials, separate NMAs were conducted for monotherapy and combination therapies. However, the treatment of patients in the control group (placebo plus TCS) were not consistent across the combination-therapy trials. Statistical heterogeneity and consistency were not tested, despite the availability of several closed loops.
The networks were sparse (several comparisons with relatively few studies), and all comparisons to tralokinumab were indirect, which increased the uncertainty in the findings. No sensitivity analysis exploring possible assumptions made by the reviewers were reported. Moreover, there was no indication of model adjustment to account for the correlation in the 3 arm trials. Harms outcomes were not evaluated.
LEO Pharma Network Meta-Analysis
The LEO Pharma NMA was based on studies identified from a systematic review of relevant randomized evidence of treatment for moderate-to-severe AD in adolescent patients. The systematic literature search was based on a PICO model defined a priori, with efficacy and safety outcomes predefined. The systematic literature search was comprehensive. The reasons for study exclusions were reported; and the selection and data-extraction processes were adequate to minimize the risk of bias and error. While the risk of bias of the comparator trials was assessed, the methods used were not reported and the risk of bias was not assessed by outcome. Several sources of heterogeneity were identified across the included studies. These included variation in the time point of follow-up, the predetermined duration of AD for study inclusion, exclusion criterion related to prior use of biologics, and protocol for investigational drug discontinuation for rescue treatment.
No information was given on model fit, and assessment of statistical consistency despite the presence of closed loops. No sensitivity analysis exploring possible assumptions made by the reviewers was reported. All comparisons to tralokinumab were indirect, which introduces increased uncertainty in the findings. Due to the small sample sizes, the CrIs were wide for several comparisons, which precluded drawing conclusions about comparative efficacy and safety for those outcomes.
Matching Adjusted Indirect Comparisons
Description of Studies
The sponsor submitted 2 MAICs conducted on its behalf by a third party comparing the relative efficacy of tralokinumab versus dupilumab in adults with moderate-to-severe AD.21,22 In both MAICs, evidence for tralokinumab was based on individual patient data, while evidence for dupilumab was based on published aggregate data. ||| |||||||| |||| ||||| || ||| |||||||| ||||||| || |||| |||||| ||||| || ||||||| |||||||||||| ||||||| || |||||||||||| |||| ||||||||| ||||| || ||||||||||| |||| ||||||| ||||||||||||||| |||||| ||||||||| |||||||| || ||||| |||||||||||| ||| || ||||||||||| |||| ||||||| ||||||||||||||| || || ||||| || ||||||||| || |||||| |||||||| |||| || ||| ||| ||||||||| ||||||||| ||||||||| |||||||||||| |||||||||||| The unanchored MAIC based on the ECZTRA 3 and LIBERTY AD CHRONOS trials aimed to assess the long-term efficacy outcomes for tralokinumab 300 mg (ECZTRA 3) administered every 2 weeks and 300 mg every 4 weeks against dupilumab (LIBERTY AD CHRONOS) every 2 weeks at 32 to 52 weeks of follow-up in adult patients with moderate-to-severe AD.21
Efficacy Results
ECZTRA 7 Versus LIBERTY AD CAFÉ
||||| ||||||||||| || |||||||| ||| |||||||| |||||||| ||||||||||||||| || ||| |||||||| ||||||| |||||||||| || ||||||||| ||||||| |||| ||||||| || |||||||||| || ||| |||||||| |||| |||||||| || ||| ||||||||| |||| ||||||| ||||||||||||||| ||||||||| |||||| ||| ||||||||| |||||| |||| ||||| ||||||||| |||||||||||||||| ||| |||| ||| |||||||||||| ||| |||| ||||||| ||||||||||||||| ||||||||| ||| |||| || ||| |||||||| ||||||||||| ||| |||| ||| ||| ||||||| |||| ||||||| ||||||||||||||| ||||||||| ||| |||||| || ||| |||||||| |||||||||||| |||| ||||||| || ||| ||||||||| ||||||| || |||| |||||||| |||||||| |||| |||||||| ||||||| |||||||||||| |||| ||||||| ||||||||||||||| ||| ||||||||| |||| ||||||| |||||||||||||| ||| || |||||| || ||||||||| |||| ||||||| ||||||||||||||| ||| ||||||| |||| |||||| ||| ||| ||||| || |||||| ||| ||||| |||||||| ||| |||| ||||||||| || || |||||||| |||||||| ||||||||| |||| ||||||| ||||||||||||||| |||| |||||||||||| |||| ||||||| |||||||||||||||| ||| ||| || |||||||| ||| ||||||||| || |||||||||||| |||||||||| ||||||| ||| |||||||||| |||| |||||| ||| ||| ||||| || ||||| ||| ||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||| ||||||||| |||||||| |||||| |||||||| ||| |||| |||||||| |||||||| |||| |||||||||| ||||| ||||| ||||||| |||||||||||| |||| ||||||| ||||||||||||||| ||| ||||||||| |||| ||||||| |||||||||||||||| ||||||||||| |||||||| |||| ||||||||| |||||||||| |||| ||| |||| |||||||| |||| |||||||||
ECZTRA 3 Versus LIBERTY AD CHRONOS
After matching, the reported baseline characteristics of the weighted patient population of the ECZTRA 3 study were matched with those of the LIBERTY AD CHRONOS study. A total of 106 patients were included in the dupilumab treatment group. The effective sample size (ESS) following match adjustment was 123.4 for the tralokinumab treatment arm (49.36% of the original population).
The results of the unanchored efficacy MAIC analysis of the ECZTRA 3 versus LIBERTY AD CHRONOS studies between tralokinumab and dupilumab was in favour of tralokinumab for an IGA of 0 or 1 (risk difference [RD] = 13.9; 95% CI, 0.6 to 27.3) and change in DLQI (mean difference = −1.7; 95% CI, −3.0 to −0.3) at week 52. The CI were too wide to draw any conclusions of certainty about the remaining outcomes between tralokinumab and dupilumab (at week 32: EASI-75, EASI-50, EASI-90, and IGA of 0 or 1; at week 52:% change in EASI, change in worse daily pruritis NRS, percent change in SCORAD, change in POEM; at week 53: EASI-75, EASI-50, EASI-90, worst daily pruritis NRS improvement of at least 4 points, POEM improvement of at least 4 points, DLQI improvement of at least 4 points).
Harms Results
||| ||||||| || ||| ||||||||| ||||||| || |||| |||||||| |||||| |||| |||||||| ||||||| |||||||||||| |||| ||||||| ||||||||||||||| ||| ||||||||| |||| ||||||| |||||||||||||| |||||||||||| |||| ||| ||||| |||||||| ||| |||||||||||||| ||| || ||||||| ||||||||| |||||||| |||||||||||| |||| ||||||| ||||||||||||||| |||| ||||||||| |||| ||||||| |||||||||||||||| ||| ||| || |||||||| ||| ||||||||| || |||||||||||| |||||||||| ||||||| ||| |||||||||| |||| |||||| ||| ||| ||||| || ||||| ||| ||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||| ||||||||| |||||| |||||||| |||||| ||| |||| |||||||||||||| |||||||||| |||||| ||||||||| |||| |||||||| ||| |||| ||||||| ||||||| |||||||||||| |||| ||||||| ||||||||||||||| ||| ||||||||| |||| ||||||| ||||||||||||||||
No harms end points were evaluated in the ECZTRA 3 versus LIBERTY AD CHRONOS MAIC.
Critical Appraisal
ECZTRA 7 Versus LIBERTY AD CAFÉ
A comparison of the ECZTRA 7 study versus the LIBERTY AD CAFÉ study was chosen after a review of | trials evaluating the treatment of tralokinumab or dupilumab in patients with moderate-to-severe AD. There was no description of a literature search or selection criteria, or any indication of how the 8 trials were located. The sponsor noted that the decision to conduct a MAIC was based on substantial heterogeneity that precluded standard indirect comparisons (e.g., an NMA or Bucher comparison). How the matching variables were selected for the MAIC was not described. Baseline characteristics postmatching were well balanced, with almost perfect matching of the covariates included in the MAIC. However, complete baseline demographic and disease characteristics for patients in both trials were not reported. The application of weights resulted in a reduced ESS of ||||, in which ||||| of enrolled patients in the ECZTRA 7 study were lost. The reduction in sample size in the primary analysis resulted in imprecision, leading to uncertainty of the results. Sensitivity analysis using a larger population by way of an unadjusted indirect comparison was generally consistent with the primary MAIC, but with a narrower CI favouring dupilumab. There was no assessment of residual confounding in the analysis.
ECZTRA 3 Versus CHRONOS
The ECZTRA 3 versus CHRONOS MAIC lacked a description of a literature search or selection criteria, or any indication of how the trials were selected for the MAIC. There was also a lack of transparency in the data-extraction process and quality assessment. Although both the ECZTRA 3 and LIBERTY AD CHRONOS trials included a placebo, an unanchored MAIC was conducted. The decision to conduct an unanchored MAIC was appropriately justified due to differences in trial design (re-randomized versus treat-through) that may have resulted in differences in the treatment of placebo across the ECZTRA 3 and LIBERTY AD CHRONOS trials. Nonetheless, the ECZTRA 3 versus LIBERTY AD CHRONOS MAIC was limited by heterogeneity between the dupilumab target population and the analysis set. First, the dupilumab target population in the LIBERTY AD CHRONOS study was not the same analysis set used in the results reported at week 32 and week 52. Consequently, the matched tralokinumab population may not be completely representative of the dupilumab population results in reports at week 52. Next, the time point at which tralokinumab (week 32) and dupilumab (week 52) were compared were different. The magnitude and direction of bias related to differences in the analysis time point is uncertain. However, because the clinical experts suggested that superior results are expected for tralokinumab at week 52 versus week 32, the analysis may be at risk of a bias in favour of treatment with dupilumab. Unadjusted and match-adjusted baseline covariates were reported. Baseline characteristics postmatching were well balanced, with almost perfect matching of the covariates included in the MAIC. However, complete baseline demographic and disease characteristics for patients in both trials were not reported. The application of weights resulted in a reduced ESS of 123.4, in which 50.64% of enrolled patients in the ECZTRA 3 study were lost. The reduction of sample size in the primary analysis resulted in imprecision, leading to uncertainty of the results. There was no assessment of residual confounding in the analysis.
Studies Addressing Gaps in the Pivotal and RCT Evidence
Description of Studies
Two observational studies were submitted by the sponsor to address gaps in evidence. There was no description of the search or selection methods used to identify these studies. Pezzolo and Naldi was an open-label, retrospective, 12-week case series conducted in Italy (N = 12) and published as a letter to the editor. This study included 12 adult patients whose disease was uncontrolled with dupilumab treatment.23 Pereyra-Rodriguez et al. (N = 85) was a retrospective, 16-week study conducted in Spain.24 This study assessed 85 adult patients, including those who had previously been treated with either dupilumab (29.4%) or upadacitinib (8.2%). These 2 studies also assessed clinical experience with tralokinumab in real-world settings.
Efficacy Results
In the study by Pezzolo and Naldi, the mean EASI score at baseline before any systemic therapy was 36.58 (range = 21 to 47). All 12 adult patients with AD reached EASI-75 within 8 weeks, with continuing improvement at 12 weeks. The mean EASI scores were 27.58 (range = 20 to 35) at study baseline and 4.67 (range = 0 to 13) at week 12. The mean itch NRS scores were 8.42 (range = 7 to 10) at baseline and 2.92 (range = 0 to 7) at week 12. The mean sleep NRS scores were 7.0 (range = 3 to 10) at baseline and 1.92 (range = 0 to 5) at week 12. In the study by Pereyra-Rodriguez et al., the mean EASI scores at baseline were 25.4 (SD = 8.1) and 7.5 (SD = 6.9) at week 16. The mean SCORAD scores were 55.8 (SD = 13.3) at baseline and 20.0 (SD = 14.78) at week 16. The mean PP-NRS scores were 8.1 (SD = 1.8) at baseline and 3.5 (SD = 2.4) at week 16. At baseline, 47 patients (55.3%) had an IGA of 4. At the end of the follow-up period, 18.8% of patients (the absolute number was not reported) had an IGA of 0 or 1.
Harms Results
In the study by Pezzolo and Naldi, no serious AEs were reported. Also, the conjunctivitis that had been observed in 4 patients during the previous treatment with dupilumab did not recur. In the study by Pereyra-Rodriguez et al., the most frequent AEs were conjunctivitis and red face (5 patients, 5.9% each) with 1 patient having both events at the same time. Of those 5 patients, 2 had experienced conjunctivitis with prior dupilumab treatment, and 3 were naive to advanced therapy with no prior eye-related AEs. Moreover, 3 patients (3.5%) experienced worsening and generalized AD lesions, 2 patients (2.4%) developed reactions at the injection site, and 2 patients (2.4%) reported anxiety-depressive syndrome. One patient discontinued treatment due to severe conjunctivitis.
Critical Appraisal
It is not clear how the studies addressing gaps were selected, creating a potential for study selection bias (i.e., relevant studies may have been left out). There is a high level of uncertainty in the results due to the following study limitations common to both studies: small sample sizes (N = 12 in Pezzolo and Naldi; N = 85 in Pereyra-Rodriguez et al.); potential selection bias in the absence of a clear description of patient selection methods; noncomparative study design with a lack of adjustment for confounding; a lack of clarity on whether the studies were designed a priori; and if retrospective data were collected in a systematic way. As well, no formal hypothesis testing was performed in the study by Pezzolo and Naldi. There was no control for multiple comparisons in Pereyra-Rodriguez et al., which resulted in an increased risk of false-positive results. The durations of follow-up (12 weeks in Pezzolo and Naldi and 16 weeks in Pereyra-Rodriguez et al.) were also inadequate for assessing response to tralokinumab treatment, according to the input of clinical experts consulted by CADTH. As neither of the studies included adolescent patients, the treatment effects in adolescents who had prior dupilumab and/or JAKi treatments were not addressed by these studies.
Conclusions
Evidence from 3 pivotal double-blind RCTs demonstrated that 16 weeks of treatment with tralokinumab resulted in improvements in the severity and extent of AD as measured by EASI, IGA or SCORAD; severity of itching (worst daily pruritus NRS); and health-related quality of life (HRQoL) as measured by DLQI in adults with moderate-to-severe AD who had an inadequate response to topical AD therapy, compared to placebo, when used as monotherapy (in the ECZTRA 1 and 2 studies) and in combination with TCS (the ECZTRA 3 study); however, either the magnitude of improvement in SCORAD and DLQI scores did not meet the literature-reported minimal important difference (MID) estimates or the 95% CI included the potential of a difference falling below conservative MID estimates. Analyses of other clinically important outcomes, including sleep disturbance and symptoms of anxiety and depression, also favoured tralokinumab, although, due to a large amount of missing data and a lack of adjustment for multiplicity, these results are likely to be biased. Similar results were observed in 1 pivotal double-blind RCT (ECZTRA 6) conducted with tralokinumab monotherapy in adolescents who had an inadequate response to topical AD therapy; MID estimates were reached for the improvement in SCORAD score but not for the improvement in CDLQI scores at week 16. The anticipated place of therapy of tralokinumab is in patients with moderate-to-severe AD with inadequate response to topical AD therapy and phototherapy, as well as systemic immunosuppressants. One double-blind RCT (ECZTRA 7) of adults with severe AD who had an inadequate response to topical AD therapy and cyclosporine A provided supporting evidence for the use of tralokinumab in combination with TCS in such patients. The ECZTRA 7 trial found that, compared to placebo, 16 weeks of tralokinumab was associated with a higher proportion of patients with an EASI-75 response but it did not find a difference in reducing the severity of itching (worst daily pruritus NRS). The results of the analysis of other outcomes were inconclusive due to a prior failure in the statistical testing hierarchy. Overall, interpretation of the clinical meaningfulness of findings at week 16 from the RCTs was hindered by the insufficient duration of follow-up given that an optimal response to tralokinumab is usually observed at 6 months in clinical practice, according to clinical expert input. No conclusion can be drawn on the efficacy of tralokinumab beyond week 16 based on the submitted evidence due to important limitations of the included studies, including inconsistent results between trials and evidence of imprecision in the longer-term results in RCTs, and risks of selection bias and confounding due to the noncomparative trial design of the LTE study (ECZTEND), similar to other long-term extension studies.
Evidence from 4 ITCs comparing tralokinumab to other advanced therapies for the treatment of moderate-to-severe AD suggests that tralokinumab performs worse than or similar to its main comparator, dupilumab, in adults. The lone exception to this trend was noted in the ECZTRA 3 versus LIBERTY AD CHRONOS unanchored MAIC, in which the results favoured tralokinumab versus dupilumab for an IGA of 0 or 1 and change in DLQI at week 52. Comparisons of the efficacy and safety of tralokinumab versus abrocitinib and upadacitinib are considered uncertain. The combined ITC evidence for adults is associated with important uncertainty due to the potential for intransitivity in the NMA, and potential residual confounding and lack of precision in the MAICs. Results of the lone NMA evaluating the efficacy of tralokinumab in adolescents were imprecise and potentially affected by intransitivity that precludes any conclusion of certainty about the comparative efficacies of tralokinumab versus dupilumab, abrocitinib, or upadacitinib. Evidence from 2 observational studies on the use of tralokinumab in patients who had uncontrolled disease with prior dupilumab and/or JAKi treatment was inconclusive given the small sample sizes, and the open-label, noncomparative study designs.
Tralokinumab appeared to be well tolerated in adults and adolescents in the RCTs and remained so beyond 52 weeks based on evidence from the ECZTEND trial. However, based on the indirect evidence, no conclusion about the safety of tralokinumab compared with other advanced therapies for moderate-to-advance AD can be drawn with any certainty, due to imprecision in all relevant analyses (2 MAICs in adults and 1 NMA in adolescents).
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of tralokinumab, 150 mg per 1 mL prefilled syringe and 300 mg per 2 mL prefilled pen, administered by subcutaneous injection for the treatment of moderate-to-severe AD in patients aged 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Tralokinumab can be used with or without TCS.
Disease Background
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
Atopic dermatitis, also known as eczema, is a chronic, heterogeneous, inflammatory, relapsing-remitting skin condition.1 The pathogenesis of AD involves a complex interplay between genetic predisposition, the environment, skin barrier dysfunction, and immune dysregulation.4,25-27 The immune dysregulations in AD are predominantly driven by T-helper 2 lymphocytes and their derived cytokines.4,26 Interleukin (IL)-13 is the primary cytokine responsible for the signs and symptoms of moderate-to-severe AD, and its levels in the serum have shown to be correlated to disease severity.4,26,28-33
Cases of AD most frequently occur in early childhood, with studies estimating that the prevalence of eczema symptoms across age groups in Canada is around 12% in children aged 6 to 7 years, 8.9% in adolescents aged 13 to 14 years, and 3.5% in adults (aged 18 years and older).2,3 The majority outgrow the disease later in life, while 25% to 40% of cases will persist into adulthood.34 Although AD is more common in adolescents, adulthood disease tends to be more severe, with 8% to 17% of affected adults experiencing severe disease.3,35,36 Nonetheless, adolescents who suffer from moderate-to-severe AD are challenged to endure significant burdens that also affect their caregivers during a formative and transitional phase of their life.
Patients with AD often experience acute worsening of their condition, commonly referred to as flares. During flares, patients with AD can experience extremely dry, red, itchy skin that can lead to lesions that can blister, ooze, and crust. Excessive scratching can lead to skin infections, as well as long-term changes to the skin, including skin thickening.37 An intense and debilitating itch (pruritus) and chronically relapsing eczematous lesions are the key clinical hallmarks of moderate-to-severe disease.4 In a 2013 Global Burden of Disease study, 41.5% of patients with moderate-to-severe AD had itch for at least 18 hours a day.38 The presence of itch is estimated to be significantly higher in adolescents aged 12 to 15 years than in older patients with AD (98.0% versus 87.3%, respectively; P < 0.05).39 As a result of this itching, patients experience sleep disturbances (e.g., difficulty falling asleep and frequent awakenings), as reported by the Eczema Society of Canada Quality of Life Report for 2016 to 2017, in which 79% of survey respondents reported interrupted or loss of sleep.37,40
Chronic itching and sleep disturbance are associated with mental health instability.38,41-43 Individuals with AD reportedly exhibit higher rates of depression, anxiety, suicidal ideation, attention deficit or hyperactivity disorder, and autism spectrum disorder.4,44-46 Psychosocial impacts such as avoidance of social activities, avoidance of exercise, missing of work and important life events, and the need to change careers or give up certain activities were reported in 48%, 47%, 32% and 30% of adult patients, respectively.37 Similarly, anxiety, difficulty in participating in physical activities, avoidance of social activities, and bullying were reported in 30%, 30%, 29%, and 14% of Canadian children and caregivers, respectively.37 Individuals who suffer from AD and their families or caregivers experience a negative impact on the quality of life due to the psychological distress associated with the disease.
The pan-Canadian (excluding Québec) prevalence of moderate-to-severe AD in adult and adolescent populations who have been treated with off-label immunosuppressants was estimated to be 82,506.47,14
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
The foundation of AD therapy for all patients relies on basic skin care, which involves liberally applying moisturizers and emollients to the skin and avoiding aggravating factors that may be physical, environmental, or chemical.48 For patients whose disease is moderate to severe in nature, conventional treatment options may include phototherapy, off-label immunosuppressants, and systemic corticosteroids.48 However, these therapies are limited by accessibility and/or safety challenges. Phototherapy is not widely available across Canada and may be impractical for patients and caregivers to access given the frequency of treatments required at specialty clinics (2 to 3 times per week).48-50 Off-label systemic immunosuppressants such as cyclosporine A and methotrexate are typically used for the shortest duration and at the lowest doses possible due to the lack of long-term efficacy, risk of significant side effects, and burdensome laboratory-monitoring requirements.49,51-55 The clinical experts consulted by CADTH also mentioned that methotrexate is not recommended in women of childbearing potential given that it is an abortifacient and can be teratogenic. Systemic corticosteroids provide a quick relief of AD, but are associated with short- and long-term safety issues, such that the American Academy of Dermatology has recommended that these therapies be avoided or used in short-term durations only as a bridge to other drugs.37,52
More recent novel therapies that have been reviewed by CADTH for adults and adolescents with moderate-to-severe AD include biologics (dupilumab) and oral small molecules, including upadacitinib and abrocitinib, both of which are a JAKi.6,7,56 Despite these important advancements in therapy for AD patients, there are still patients who do not achieve an adequate response to dupilumab and JAKi treatments. While dupilumab is generally well tolerated, drug-induced conjunctivitis has been observed in clinical trials and in clinical practice, necessitating discontinuation for some patients.57 Upadacitinib and abrocitinib treatments require baseline and routine laboratory monitoring and have black-box warnings in the product monograph related to infections, malignancies, thrombosis, and major adverse cardiovascular events.6,7
According to the clinical experts consulted by CADTH for this review, the order of escalation of therapy is as follows: TCS and/or topical nonsteroids (a TCI or phosphodiesterase type 4 [PDE-4] inhibitor), followed by newer systemic treatments, including biologics and a JAKi, and off-label systemic immunosuppressant treatment (typically methotrexate). Most jurisdictions in Canada currently reimburse the cost of dupilumab for patients aged 12 years and older with moderate-to-severe AD. Upadacitinib and abrocitinib are currently undergoing negotiation by the pan-Canadian Pharmaceutical Alliance for patients aged 12 years and older with moderate-to-severe AD. The clinical experts consulted by CADTH noted that physicians prefer to err on the side of a superior safety profile; for this reason, among patients with moderate-to-severe AD who were considered for newer systemic treatments (biologics or JAKi treatment), biologics tend to be trialled first, reserving JAKi options for patients who have an inadequate response to biologics (although there are rare cases in which a JAKi may be trialled before biologics). The clinical experts added that adolescent patients are much more likely to trial a biologic before a JAKi, given the possible longer-term safety implications that come with a JAKi.
The clinical experts consulted by CADTH stated that reducing the severity of symptoms, minimizing adverse effects, improving HRQoL (including improvement in sleep quality anxiety or depressive symptoms), increasing the ability to maintain employment and independence, and reducing caregiver burden are important goals of treatment of patients with moderate-to-severe AD.
Drug Under Review
The key characteristics of tralokinumab are summarized in Table 4, along with other treatments available for patients with moderate-to-severe AD.
Tralokinumab is available in a 150 mg per 1 mL prefilled syringe and 300 mg per 2 mL prefilled pen, both of which can be self-administered as subcutaneous injections.58 The 300 mg per 2 mL prefilled pen is not currently marketed in Canada. The recommended dose for adult and adolescent patients 12 years and older is an initial 600 mg (four 150 mg injections with the 150 mg per 1 mL prefilled pen or 2 300 mg injections with the 300 mg per 2 mL prefilled pen) followed by 300 mg (two 150 mg injections with the 150 mg per 1 mL prefilled pen or 1 300 mg injections with the 300 mg per 2 mL prefilled pen) administered every 2 weeks) as a subcutaneous injection. At the prescriber’s discretion, every-fourth-week dosing may be considered for some patients who achieve clear or almost clear skin after 16 weeks of treatment. Some patients with an initial partial response may subsequently improve with continued treatment every 2 weeks beyond 16 weeks.58
Tralokinumab is indicated for the treatment of moderate-to-severe AD in adult and adolescent patients 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Tralokinumab can be used with or without TCS or TCIs.58 Tralokinumab was previously reviewed by CADTH for treatment of moderate-to-severe AD in adult patients and received a recommendation not to reimburse.8
Tralokinumab is a fully human immunoglobulin G4 monoclonal antibody that binds specifically to type 2 cytokine IL-13 and inhibits its interaction with IL-13 receptors alpha 1 and alpha 2. Tralokinumab neutralizes the activity of IL-13 by blocking its interaction with the IL-13 receptor alpha 1/IL-4 receptor alpha complex. IL-13 is a major driver of type 2 inflammation in AD, with skin showing overexpression of IL-13. IL-13 signals via the IL-13 receptor alpha 1/IL-4 receptor alpha complex and stimulates inflammatory responses, contributes to itch induction, and impairs the expression of proteins necessary for a normal skin barrier.58
The sponsor-requested reimbursement criteria differ from the Health Canada–approved indication but align with the February 2023 CADTH Canadian Drug Expert Committee recommendation in response to the Request for Advice for dupilumab,59 i.e., for the treatment of patients aged 12 years and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable, and those who had an adequate trial or are ineligible for each of the following therapies: phototherapy (where available) and off-label immunosuppressants.
Table 4: Key Characteristics of Tralokinumab, Dupilumab, Upadacitinib, and Abrocitinib
Characteristic | Tralokinumab (Adtralza) | Dupilumab (Dupixent) | Upadacitinib (Rinvoq) | Abrocitinib (Cibinqo) |
|---|---|---|---|---|
Mechanism of action | A fully human IgG4 monoclonal antibody that neutralizes the activity of IL-13 by blocking its interaction with IL-13R-alpha-1/IL-4R-alpha receptor complex. IL-13 is a major driver of type 2 inflammation of AD. | A recombinant human IgG4 monoclonal antibody that inhibits IL-4 signalling via blocking type I receptor (IL-4R-alpha/gamma chain) and both IL-4 and IL-13 signalling through blocking type II receptor (IL-4R-alpha/IL-13R-alpha). IL-4 and IL-13 are key type 2 (including T-helper 2) cytokines involved in AD. | A JAK inhibitor that modulates the signalling pathway at the point of JAKs, preventing the phosphorylation and activation of STATs. Pro-inflammatory cytokines (including IL-4, IL-13, IL-22, TSLP, IL-31 and interferon-gamma) transduce signals via the JAK1 pathway and are involved in AD pathogenesis. | A selective JAK1 inhibitor that modulates cytokine signalling pathway at the point of JAK1, preventing phosphorylation and activation of STATs that modulate intracellular activity including gene expression. Both the parent compound and the active metabolite (M1 and M2) inhibit cytokine signalling with similar selectivity. |
Indicationa | For the treatment of moderate-to-severe AD in adult and adolescent patients 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Tralokinumab can be used with or without topical corticosteroids. | For the treatment of patients aged 6 months and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Dupilumab can be used with or without topical corticosteroids. | For the treatment of adults and adolescents 12 years of age and older with refractory moderate-to-severe AD who are not adequately controlled with a systemic treatment (e.g., a steroid or biologic) or when use of those therapies is inadvisable. Upadacitinib can be used with or without topical corticosteroids. | For the treatment of patients 12 years and older with refractory moderate-to-severe AD, including the relief of pruritus, who have had an inadequate response to other systemic drugs (e.g., steroid or biologic), or for whom these treatments are not advisable. Abrocitinib can be used with or without medicated topical therapies for AD. |
Route of administration | Subcutaneous | Subcutaneous | Oral | Oral |
Recommended dose | For adult and adolescent patients 12 years and older, an initial dose of 600 mg is followed by 300 mg administered q.2.w. as a subcutaneous injection. At the prescriber’s discretion, q.4.w. dosing may be considered for some patients who achieve clear or almost clear skin after 16 weeks of treatment. | Adults: Initial dose of 600 mg (two 300 mg injections), followed by 300 mg q.2.w. Children and adolescent (aged 6 to 17 years):
Pediatrics (aged 6 months to 5):
| Adults: Starting dose of 15 mg orally once daily. If an adequate response (e.g., EASI-75) is not achieved, consider increasing dosage to 30 mg once daily. For some patients, such as those with severe disease, a starting dose of 30 mg once daily may be appropriate. Discontinue if an adequate response is not achieved with the 30 mg dose after 16 weeks of treatment. Use the lowest effective dose needed to maintain response. For patients aged > 65 years, a 30 mg dose once daily is not recommended. Adolescents (aged 12 to 17 years):
| The recommended dose is 100 mg or 200 mg orally once daily for adolescents and adults aged under 65 years, based on the individual goal of therapy and potential risk for adverse reactions. For patients using the 200 mg once-daily dosage, after symptom control is achieved by week 12, consider dose reduction to 100 mg once daily. Relative to patients who maintained the 200 mg dose, the risk of occurrence of serious adverse reactions decreased in patients who reduced their dose to 100 mg beyond 12 weeks in clinical studies. If symptom control is lost after dose reduction, the dose can be increased to 200 mg. Exceeding a daily dose of 200 mg is not recommended. |
Serious adverse effects or safety issues | Conjunctivitis, eosinophilia, injection-site reactions | Conjunctivitis, eosinophilia, injection-site reaction, blepharitis, oral herps, eye pruritus, dry eye, herpes simplex, keratitis | Serious warnings and precautions box (“black box warning”): serious infections, malignancy, thrombosis, MACE | Serious warnings and precautions box (“black box warning”): serious infections, malignancy, thrombosis, MACE |
Other | Full treatment effect, i.e., maximal improvement in symptom, may not be achieved until closer to 6 months according to clinician group input | Higher rates of conjunctivitis and rash are reported compared to tralokinumab in the RCT and real-world clinical setting according to clinician group input | Drug-drug interaction with strong CYP3A4 inhibitors and inducers | Drug-drug interaction with moderate to strong CYP2C9 inhibitors and strong inducers of CYP enzymes |
AD = atopic dermatitis; EASI-75 = reduction of at least 75% in Eczema Area Severity Index score from baseline; IgG4 = immunoglobin G4; IL = interleukin; JAK1 = Janus kinase 1; MACE = major adverse cardiovascular event; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; STAT = signal transducers and activators of transcriptions; TSLP = thymic stromal lymphopoietin (cytokine family protein).
aHealth Canada–approved indication.
Sources: Sponsor’s clinical evidence summary14 and Adtralza,58 Dupixent,56 Rinvoq,6 and Cibinqo7 Health Canada product monographs.
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient inputs received by CADTH are included in the stakeholder section at the end of this report.
The Eczema Society of Canada and Eczema Québec in collaboration with the Canadian Skin Patient Alliance submitted 2 separate patient group inputs. The Eczema Society of Canada’s input was based on a survey of 3,000 patients or caregivers and family members; questionnaires; and 1-on-1 interviews (number not reported) with patients and caregivers. Eczema Québec’s input was based on patient testimonials (n = 6), interviews (n = 10), and 2 group discussions (n = 13 in total), as well as insights gleaned from the McGill University Health Centre’s Centre of Excellence for Atopic Dermatitis and a report (The Skin I’m In: 2022 Update) from 2021 to 2023. The groups noted that symptoms of moderate-to-severe AD include inflamed, red, and dry skin that cracks, oozes, bleeds and in some cases involves thickening and/or infections of the skin. Often, patients experience “flare-ups” that are periods of worsening symptoms. Some patients experience remission, but others never experience relief. The input noted that itch is frequently reported as the most burdensome symptom and has been described as “incapacitating,” “debilitating,” and “bugs crawling all over,” leading to disrupted sleep, fatigue, decreased functionality, and significant impacts on daily life, work, and school. Skin rashes were reported to be not only painful but a source of embarrassment and stigmatization affecting self-esteem and social relationships. Family members and/or caregivers reported impacts on intimacy, family dynamics, and relationships, and experience feelings of anxiety, depression, and sleep loss. Patients with moderate-to-severe AD also reported that AD limits their choices of work, clothing, foods, environments, hobbies, regular activities, travel, and hygiene routines. Some patients reported contemplating suicide due to uncontrollable AD. The joint input by Eczema Québec and the Canadian Skin Patient Alliance quoted data from the Canadian Institute for Health Information indicating that patients sometimes end up in the emergency department or become hospitalized when AD is not well controlled. Patients expressed a need for treatments that can result in improvement in symptoms (dryness, flaking, inflammation, blistering, cracking), reduction in itch frequency and/or intensity, long-term improvement in quality of life (sleep, prevention of flares, discomfort, psychological burden), and improved ability to carry out daily activities (work, school, leisure, personal hygiene), and that are safe (reducing infection and minimizing short- and long-term adverse effects), affordable, flexible, and easy to administer.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of AD.
Unmet Needs
The clinical experts noted that none of the available treatment options can reverse the course of the disease. They added that not all patients respond to the newer systemic treatments (dupilumab, upadacitinib, and abrocitinib) for moderate-to-severe AD and that some patients can become refractory to current treatment options. There is therefore an unmet need for more effective and safe treatment options for moderate-to-severe AD. Currently, many patients with AD have access to only 1 biologic drug (dupilumab). In the event of inadequate treatment response or side effects to dupilumab, patients are limited to receiving JAKi treatments, which are associated with more safety concerns compared with biologics. One clinical expert also identified a need for treatment options that can improve adherence to and the convenience of drug administration for patients who are averse to needles (dupilumab is administered as subcutaneous injection) or have trouble adhering to daily administration of oral upadacitinib and abrocitinib.
Place in Therapy
According to the clinical experts, the addition of tralokinumab would likely not result in a paradigm shift. The clinical experts expected tralokinumab to occupy the same place in therapy as dupilumab, serving as an additional biologic option for the treatment of moderate-to-severe AD after failure of off-label immunosuppressants. In the clinical experts’ opinion, the potential toxicities of off-label immunosuppressants and the need for close monitoring make biologics ideal first-line treatments in patients who require a systemic treatment, although they noted that most jurisdictions currently require patients to try an immunosuppressant treatment before receiving dupilumab, and this requirement would likely be applicable to tralokinumab.
Patient Population
In the clinical experts’ opinion, any patient with moderate-to-severe AD could be a candidate for tralokinumab treatment. The clinical experts noted that tralokinumab would most likely be used in patients with AD in the absence of comorbid conditions such as asthma, chronic rhinosinusitis with nasal polyposis, and eosinophilic esophagitis, because these patients could benefit from dupilumab treatment, given that dupilumab is also indicated for the treatment of these conditions. The clinical experts noted that no specific diagnostic test is available to help identify responders to treatment, and clinicians generally use clinical assessments and reviews of medical history to determine if tralokinumab treatment is appropriate.
Assessing the Response Treatment
The clinical experts noted that the effectiveness of therapy is determined by clinical outcomes (e.g., percent BSA involved and number of active areas) and patient-reported outcomes (e.g., itch, sleep, and QoL) that are typically assessed every 3 to 6 months. The clinical experts noted that, in their experience, it takes approximately 6 months to observe the optimal benefits of tralokinumab treatment, and they noted that a lack of response before 6 months should not be considered evidence of treatment failure. The clinical experts described significant improvement in QoL and ability to perform daily activities (e.g., work, school, and household activities) as indicators of a meaningful response to treatment even if the skin is not completely clear of all erythema or lichenification. Measurements of disease improvement such as a PGA (or IGA in clinical trials) and EASI, and patient-reported outcomes, including DLQI (or CDLQI) and worst daily pruritus NRS, are commonly used in clinical practice to assess response to treatment.
Discontinuing Treatment
The clinical experts noted that it would be appropriate to consider a switch of therapy in patients who have no improvement in clinical outcomes or patient-reported outcomes, or who have intolerable side effects (e.g., injection-site reactions, ocular side effects, and recurrent herpetic infections).
Prescribing Considerations
The clinical experts noted that it would be appropriate to limit the authority to prescribe tralokinumab to dermatologists, allergists, immunologists, and pediatricians with expertise in the diagnosis, treatment, and monitoring of patients with AD.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group inputs received by CADTH are included in the stakeholder section at the end of this report.
Three clinician groups, the Atlantic Specialist Group Managing AD (7 clinicians), Dermatology Association of Ontario (16 clinicians), Canadian Dermatology Association (an unknown number of clinicians) provided 3 separate inputs. The 3 groups and the clinical experts consulted by CADTH agreed that the goals of therapy are to improve symptoms (long-term and durable relief of chronic itch, minimization of dry and inflamed skin, clear or almost clear skin, less oozing, scaling, cracking, or fissures), and improved QoL (better sleep) and function (ability to focus on work and school). The clinical experts added reduction in anxiety or depressive symptoms and caregiver burnout to this list. As for unmet needs, the clinician groups and the clinical experts consulted by CADTH all agreed that not all patients respond to or tolerate existing systemic treatments. JAKi treatments have safety and contraindication issues (black-box warnings for patients with risk factors for cardiovascular events, cancers, and infections), and dupilumab is associated with conjunctivitis. New treatments are therefore needed to provide more options for patients whose AD is not well controlled with existing systemic therapies. The clinician groups stated that tralokinumab would have the same place in therapy as dupilumab after phototherapy and/or off-label systemic therapies (if required by insurance or public plans) and may be trialled if patients fail to respond to dupilumab and an oral JAKi. The clinician groups said the suitable patient population aligns with the reimbursement request. They also noted that those who did not respond to biologics and/or JAKi treatments or have a history of conjunctivitis, risk factors associated with cardiovascular events, thrombosis, malignancy, serious infections, and/or significant drug-drug interactions or difficulty adhering to stricter dosing schedules, and those over the age of 65 years would be best suited for tralokinumab treatment. The clinical experts added that tralokinumab would most likely be used in patients with “pure” AD without comorbid asthma or eosinophilic esophagitis and those with special site involvement. The 3 clinician groups and the clinical experts consulted by CADTH indicated that they would assess response to treatment based on BSA affected, pruritus NRS, PGA (in clinical practice) and/or EASI, if required by an insurance company or payers, at 6 months after initiation of tralokinumab. According to the clinician groups, a lack of response or efficacy, worsening disease, deterioration of QoL, increased BSA affected, presence of AEs or unacceptable intolerance, and allergies would prompt clinicians to consider discontinuing tralokinumab treatment. Last, the clinician groups and the clinical experts agreed that a dermatologist, allergist, pediatrician, or immunologist well versed in managing moderate-to-severe AD should be allowed to prescribe tralokinumab. The 3 clinician groups raised concerns regarding differential access to tralokinumab, which is currently only funded by private insurance, and the requirement of trying off-label immunosuppressants with lower efficacy and increased risk before accessing newer systemic agents.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
All pivotal trials were placebo-controlled, so there is no evidence comparing tralokinumab with other biologic drugs funded for AD. | For CDEC consideration. |
Dupilumab is another biologic drug funded for AD. The most recent CADTH recommendation was that dupilumab be reimbursed for the treatment of patients aged 12 years and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable, only if certain conditions are met. Another CADTH reimbursement review of dupilumab for AD is under way, focusing on the 6-months through 12-years age group. Two JAKi drugs — Cibinqo (abrocitinib) and Rinvoq (upadacitinib) — received positive CADTH recommendations for patients aged 12 years and older with AD and are under active negotiation at the pCPA. However, recent safety warnings may preclude these drugs from being true comparators for adolescent patients as many clinicians would be reluctant to prescribe them. | For CDEC consideration. JAKi treatments are relevant comparators for tralokinumab in both the adult and adolescent populations. They are prescribed in clinical practice for the treatment of AD in adolescent patients, although most clinicians generally prefer to prescribe biologics first due to a superior safety profile in this patient population. |
Considerations for initiation of therapy | |
The sponsor noted in the submission that Adtralza has also demonstrated efficacy in patients who had uncontrolled diseasewith prior systemic treatment with dupilumab and/or JAKi in real-world studies. Should reimbursement be provided for patients who lost response to, or never achieved clinical benefit from, a trial of dupilumab? | For CDEC consideration. Some patients with psoriasis respond well to 1 biologic treatment but not another, and this is expected to be similar in patients with AD. Patients who lost response to or never achieved a clinical benefit from a trial of dupilumab or a JAKi would be reasonable candidates for tralokinumab. Two observational studies of patients who had prior experience with systemic dupilumab and/or JAKi treatments were included in the submission. While the results of these studies suggest that tralokinumab was associated with reductions in the severity and extent of AD, itch symptoms, and sleep disruptions from baseline, the results are uncertain due to the open-label, retrospective, and noncomparative study design, along with small sample sizes. |
Consider alignment with initiation criteria for dupilumab in AD, including definitions regarding moderate-to-severe AD, refractory disease, and adequate trials for different prerequisite therapies. | For CDEC consideration. |
Considerations for continuation or renewal of therapy | |
Consider alignment with renewal criteria for dupilumab in AD. | For CDEC consideration. |
Considerations for prescribing of therapy | |
Standard maintenance dosing is 300 mg q.2.w., but the product monograph notes, “At prescriber’s discretion, every fourth week dosing may be considered for some patients who achieve clear or almost clear skin after 16 weeks of treatment; however, the probability of maintaining clear or almost clear skin may be decreased with dosing every fourth week.” Should q.4.w. dosing be mandated in any situations based on clinical trial results? | For CDEC consideration. |
Consider alignment with prescribing criteria for dupilumab in AD. | For CDEC consideration. |
System and economic issues | |
Dupilumab successfully completed pCPA negotiations for patients ≥ 12 years of age with moderate-to-severe AD | For CDEC consideration. |
AD = atopic dermatitis; CDEC = CADTH Canadian Drug Expert Committee; JAKi = Janus kinase inhibitor; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; pCPA = pan-Canadian Pharmaceutical Alliance.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of tralokinumab 150 mg/mL solution for subcutaneous injection for the treatment of moderate-to-severe AD in adult and adolescent patients aged 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. The focus is on comparing tralokinumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of tralokinumab is presented in 4 sections, and CADTH’s critical appraisal of the evidence is included after each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The second section includes a sponsor-submitted LTE study. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the pivotal and RCT evidence.
Included Studies
Clinical evidence addressed in the CADTH review and appraised in this document includes:
Five pivotal studies or RCTs identified in systematic review (ECZTRA 1, 2, 3, 6, and 7)9-12
Four indirect treatment comparisons (2 NMAs19,20 and 2 MAICs21,22)
Two additional studies addressing gaps in evidence (Pezzolo and Naldi [2023]23 and Pereyra-Rodriguez et al. [2023]24).
Pivotal Studies and RCT Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6 (adolescents) and Table 7 (adults).
Adolescents
One randomized, double-blind, placebo-controlled, pivotal phase III, 52-week trial (ECZTRA 6,9 N = 301) in the adolescent population met the inclusion criteria of the sponsor-conducted systematic review. The ECZTRA 6 trial evaluated the efficacy of tralokinumab compared with placebo in adolescent patients (aged 12 to less than 18 years) with moderate-to-severe AD. The trial was conducted at 72 study sites across North America, Europe, Asia, and Australia, with 11 sites located in Canada. Patients were enrolled between June 19, 2018, and November 4, 2019. The study is now complete. The study design is shown in Figure 1. It consisted of the following study periods:
Screening phase (week −6 to week 0): Patients were assessed for study eligibility, and systemic and topical treatments for AD were washed out (TCS, TCIs, and topical PDE-4 inhibitors had a 2-week washout while off-label immunosuppressants and systemic corticosteroids had a 4-week washout). The patient or the patient’s legally acceptable representative signed and dated an informed consent form to participate in the trial.
Initial treatment period (day 0 to week 16): Enrolled patients were randomized in a 1:1:1 ratio to receive tralokinumab 600 mg for 1 dose then 300 mg every 2 weeks, tralokinumab 300 mg for 1 dose then 150 mg every 2 weeks, or placebo every 2 weeks. Randomization was conducted using a central interactive response system and stratified by region (North America, Europe, Japan, and Australia) and baseline disease severity (IGA of 3 or 4).
Maintenance treatment period (week 16 to week 52): Patients in the tralokinumab group who achieved either primary end point (an IGA of 0 or 1 or EASI-75) at week 16 without the use of rescue medication were considered responders and re-randomized in a 1:1 ratio to receive tralokinumab every 2 weeks or every 4 weeks at their original dose (150 mg or 300 mg). Patients receiving placebo who met at least 1 of the 2 primary end points at week 16 without the use of rescue medication continued to receive blinded placebo every 2 weeks until week 52. All other patients at week 16, and those who lost response or received rescue medication during maintenance, were transferred to open-label treatment with tralokinumab 300 mg every 2 weeks with optional use of mild-to-moderate-potency TCS or TCIs. Patients were transferred from maintenance to open-label treatment at any visit from week 16 if they met any of the following criteria and transfer was considered appropriate by the investigator:
IGA of 0 at week 16: IGA of at least 2 and not achieving EASI-75 over at least a 4-week period
IGA of 1 at week 16: IGA of at least 3 and not achieving EASI-75 over at least a 4-week period
IGA above 1 at week 16: not achieving EASI-75 over at least a 4-week period
Received rescue treatment (from week 16 or later).
Safety follow-up period (week 52 to week 66): Safety assessments were conducted for those who did not enter the ECZTEND extension trial.
Adults
Three pivotal phase III studies (ECZTRA 1, N = 802; ECZTRA 2,10 N = 794; ECZTRA 3,11 N = 380) and 1 phase IIIb study (ECZTRA 7;12 N = 277) in the adult population met the inclusion criteria of the sponsor-conducted systematic review. These double-blind RCTs were included in the original CADTH reimbursement review of tralokinumab in adults. The ECZTRA 1 and 2 studies were designed to compare the efficacy of tralokinumab against placebo in patients with moderate-to-severe AD over 52 weeks of treatment. The ECZTRA 3 and 7 studies were designed to demonstrate that tralokinumab in combination with TCS is superior to placebo in combination with TCS; the ECZTRA 3 study assessed patients with moderate-to-severe AD over 32 weeks of treatment and the ECZTRA 7 study assessed patients with severe AD who were not adequately controlled with or had contraindications to oral cyclosporine A over 26 weeks of treatment. They were multicentre studies that included study sites in North America, Europe, Asia, and Australia; specifically, the ECZTRA 2 and 3 studies included 19 and 10 study sites, respectively, in Canada. All studies are now complete. The study designs are shown in Figure 2 (ECZTRA 1 and 2), Figure 3 (ECZTRA 3), and Figure 4 (ECZTRA 3). The studies consisted of the following study periods:
Figure 1: ECZTRA 6 Trial Design (Adolescents)
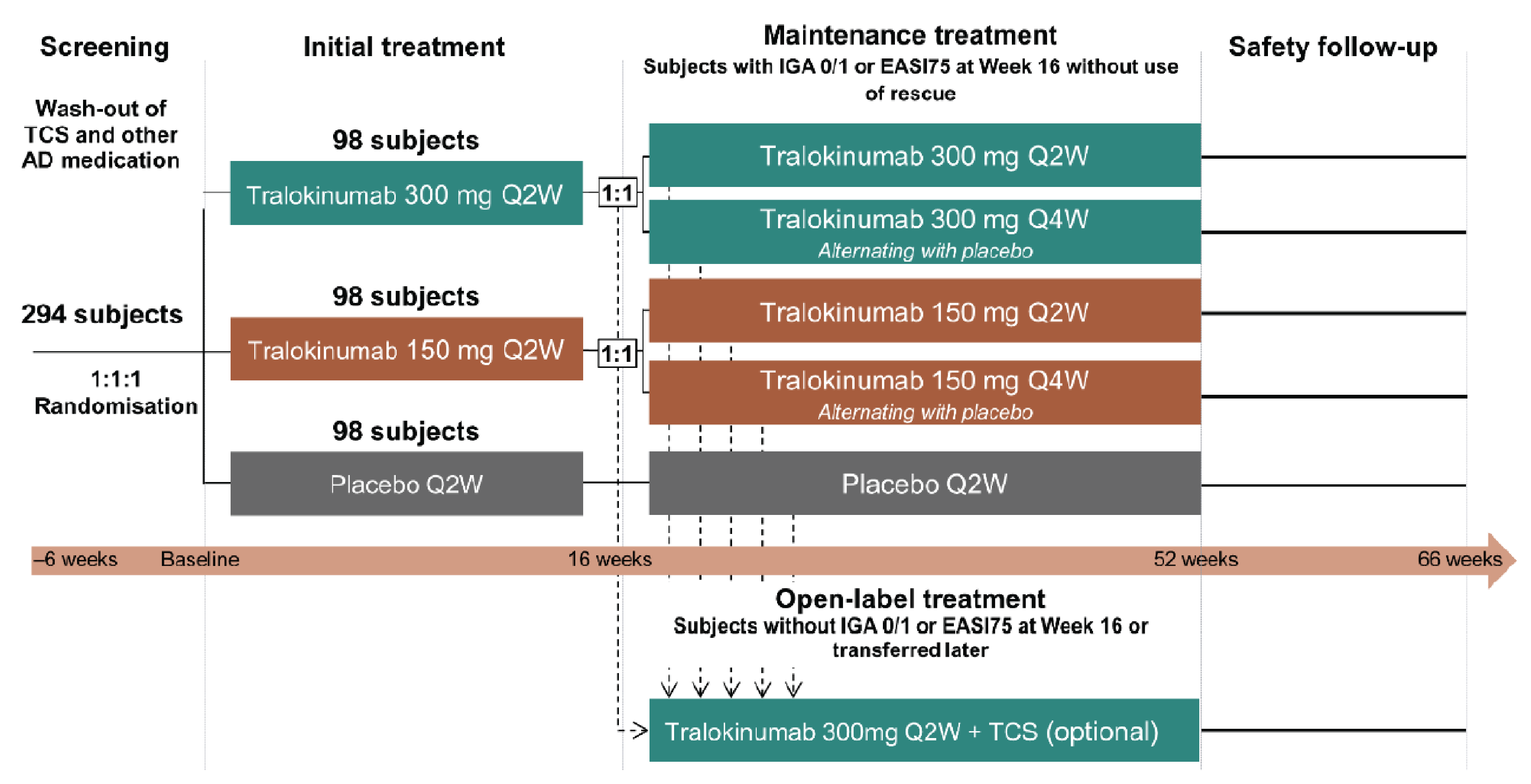
AD = atopic dermatitis; EASI75 = reduction of at least 75% in Eczema Area Severity Index score from baseline; IGA = Investigator’s Global Assessment; Q2W = every 2 weeks; Q4W = every 4 weeks; TCS = topical corticosteroids.
Note: Transfer criteria were patients with an IGA of 0 at week 16 who over 3 consecutive visits had an IGA of 2 or greater and did not achieve EASI-75; patients with an IGA of 1 at week 16 who over 3 consecutive visits had an IGA of 3 or higher and did not achieve EASI-75; patients with an IGA above 1 at week 16 who over 3 consecutive visits did not achieve EASI-75; and patients who received rescue treatment during the maintenance phase.
Source: Clinical Study Report for ECZTRA 6.13
ECZTRA 1 and 2
Screening phase (2 to 6 weeks): Patients were assessed for study eligibility and systemic and topical treatments for AD were washed out (TCS, TCIs, and topical PDE-4 inhibitors had a 2-week washout while off-label immunosuppressants and systemic corticosteroids had a 4-week washout).
Initial treatment period (day 0 to week 16): Enrolled patients were randomized in a 3:1 ratio to either tralokinumab (600 mg for 1 dose then 300 mg every 2 weeks) or placebo every 2 weeks. Randomization was conducted using a computer-generated randomization schedule and stratified by region (North America, Japan, and Europe) and baseline disease severity (an IGA of 3 or 4).
Maintenance treatment period (week 16 to week 52): Patients in the tralokinumab group who achieved a clinical response at week 16 were re-randomized at a ratio of 2:2:1 to receive either tralokinumab every 2 weeks, tralokinumab every 4 weeks (alternating dose administrations of placebo and tralokinumab every 2 weeks), or placebo every 2 weeks. Patients who did not achieve a clinical response at week 16, as well as patients who did not maintain an adequate clinical response during the maintenance treatment period were transferred to open-label tralokinumab every 2 weeks with optional use of TCS. Patients randomized to the placebo group in the initial treatment period who achieved a clinical response at week 16 continued to receive placebo every 2 weeks in the maintenance treatment period while maintaining blinding.
Safety follow-up period (week 52 to week 66): Safety assessments were conducted.
Figure 2: ECZTRA 1 and 2 Trial Design (Adults, Original Review)
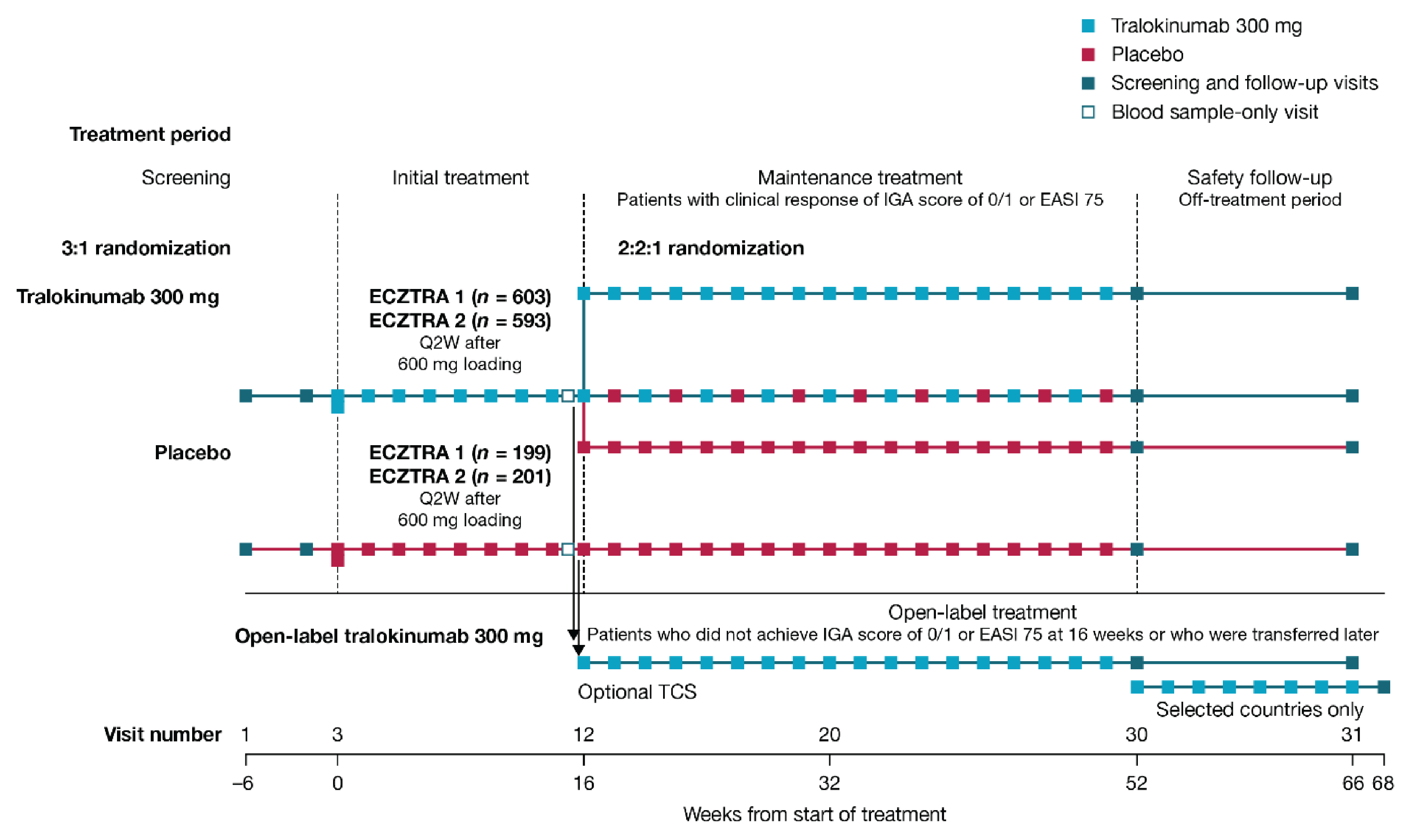
EASI 75 = reduction of at least 75% in Eczema Area Severity Index score from baseline; IGA = Investigator’s Global Assessment; Q2W = every 2 weeks; TCS = topical corticosteroids.
ECZTRA 3
Screening phase (2 to 6 weeks): Patients were assessed for study eligibility and systemic and topical treatments for AD were washed out (TCS, TCIs, and topical PDE-4 inhibitors had a 2-week washout while off-label immunosuppressants and systemic corticosteroids had a 4-week washout).
Initial treatment period (day 0 to week 16): Enrolled patients were randomized 2:1 to receive either tralokinumab (600 mg for 1 dose then 300 mg every 2 weeks) or placebo every 2 weeks. Randomization was conducted using a central interactive voice-response system and stratified by region (Europe and North America) and baseline disease severity (an IGA of 3 or 4). All patients were instructed to use supplied TCS once daily as needed.
Continuation treatment period (week 16 to week 32): Patients who were assigned to the tralokinumab group in the initial treatment period and had a clinical response at week 16 were re-randomized in 1:1 ratio to either tralokinumab every 2 weeks, or tralokinumab every 4 weeks (alternating dose administrations of placebo and tralokinumab every 2 weeks). Randomization was stratified by region (Europe and North America) and IGA response at week 16 (an IGA of 0 or 1, or above 1). Patients who received placebo in the initial treatment period and had a clinical response at week 16 continued to receive placebo every 2 weeks. Patients randomized to tralokinumab or placebo in the initial treatment period who did not have a clinical response at week 16 were given tralokinumab every 2 weeks in the continuation treatment period. All patients continued to stay on the TCS regimen as needed.
Follow-up period (week 32 to week 46): All patients who did not enter the ECZTEND trial after completion of the continuation treatment period were followed for safety assessments until week 46.
ECZTRA 7
Screening period (2 to 6 weeks): Patients were assessed for study eligibility and systemic and topical (except for TCS and TCIs) treatments for AD were washed out (topical PDE-4 inhibitors had a 2-week washout while off-label immunosuppressants and systemic corticosteroids had a 4-week washout).
Treatment period (day 0 to week 26): Enrolled patients were randomized 1:1 to receive either tralokinumab (600 mg for 1 dose then 300 mg every 2 weeks) or placebo. Randomization was conducted using interactive response technology and stratified by prior cyclosporine A use (yes or no), country (Germany: yes or no), and baseline disease severity (an IGA of 3 or 4). All patients were instructed to use a supplied TCS once daily as needed.
Safety follow-up period (week 27 to week 40): All patients who did not enter the ECZTEND trial after completion of the treatment period were followed for safety assessments until week 40.
Figure 3: ECZTRA 3 Trial Design (Adults, Original Review)
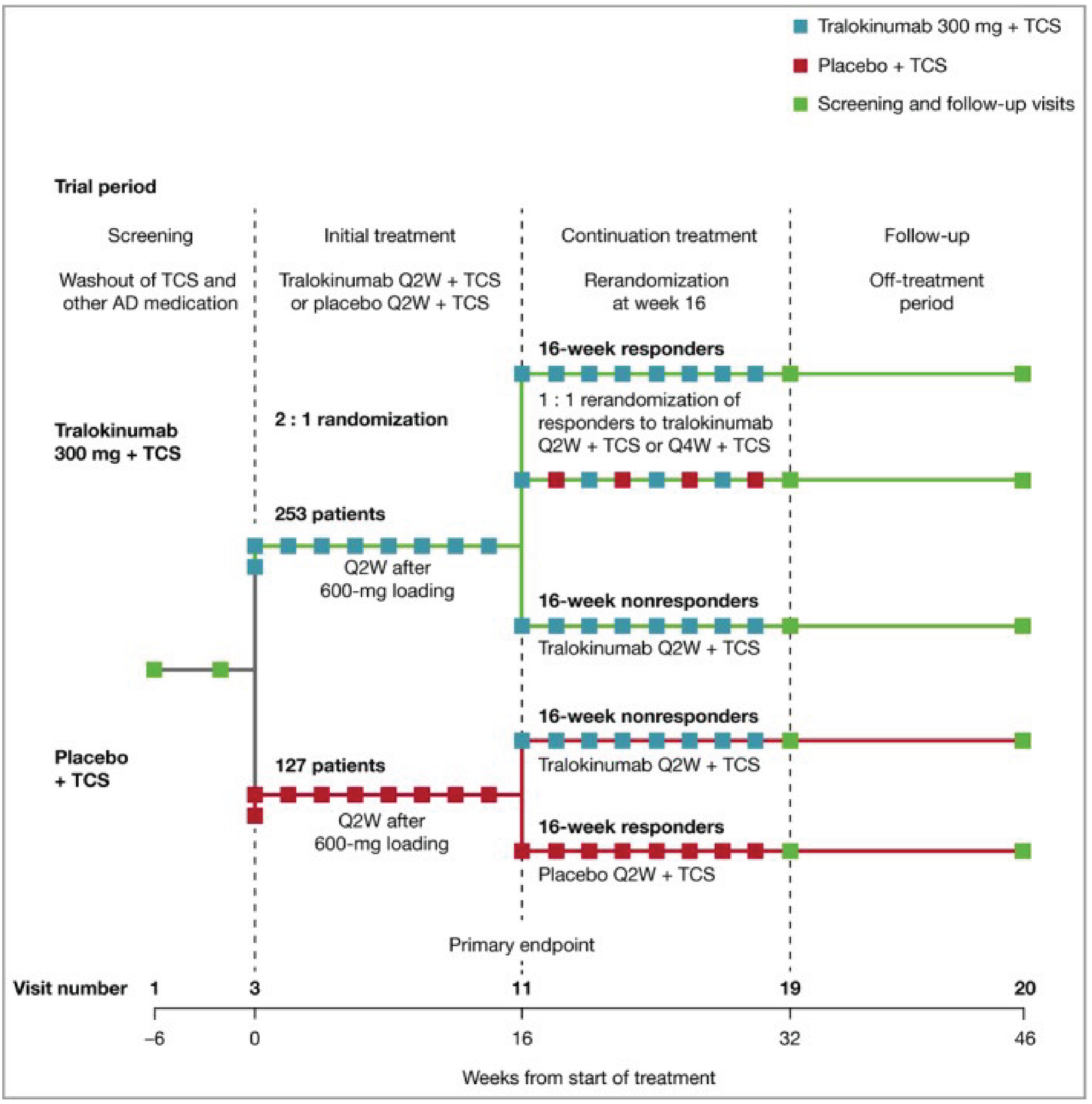
AD = atopic dermatitis; Q2W = every 2 weeks; Q4W = every 4 weeks; TCS = topical corticosteroids.
Note: Clinical response is defined as patients achieving an Investigator’s Global Assessment of 0 or 1 or at least 75% reduction in the Eczema Area and Severity Index.
Source: Clinical Study Report for ECZTRA 3.17
Figure 4: ECZTRA 7 Trial Design (Adults, Original Review)
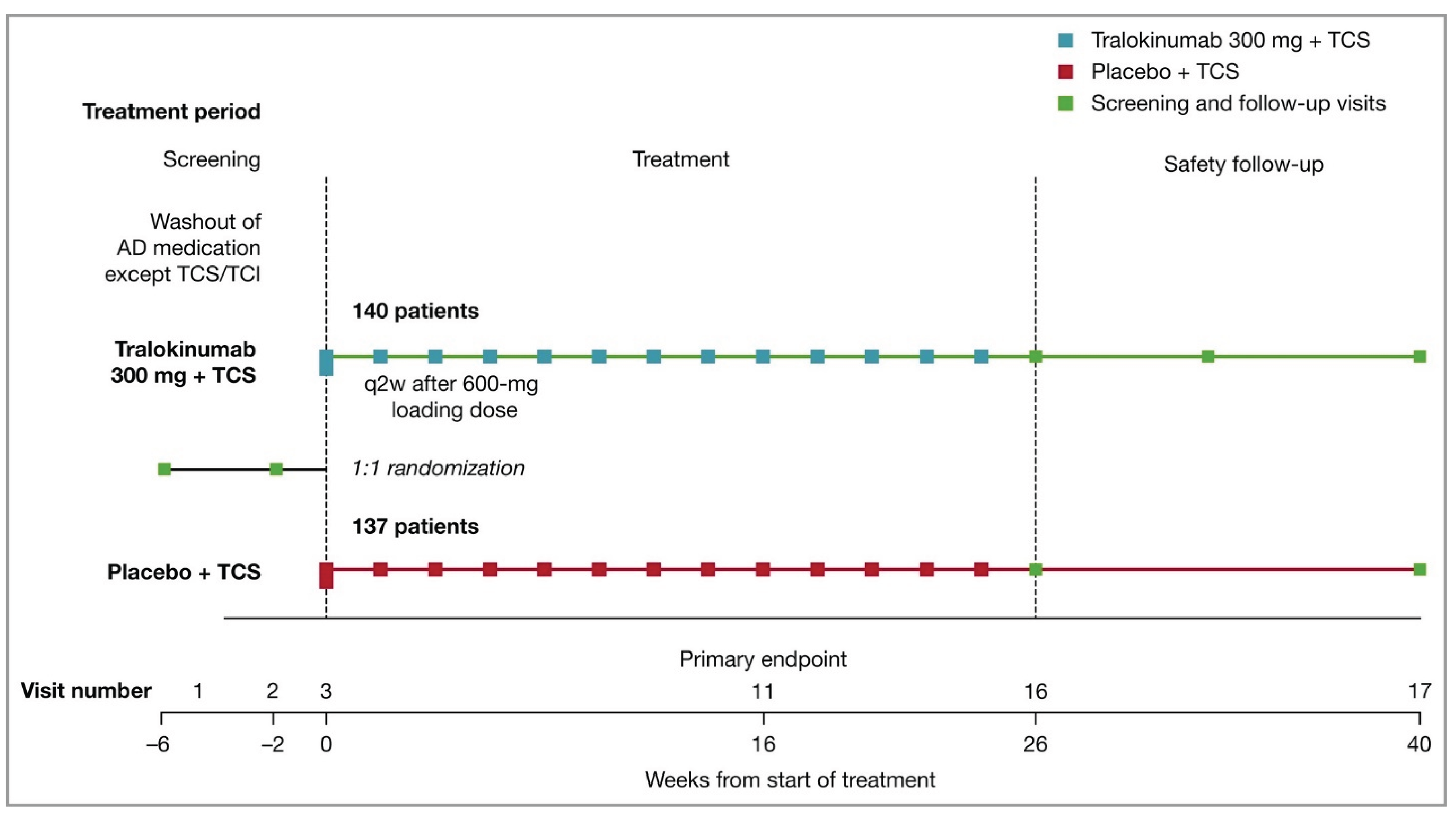
AD = atopic dermatitis; q2w = every 2 weeks; TCI = topical calcineurin inhibitor; TCS = topical corticosteroids.
Source: Clinical Study Report for ECZTRA 7.18
Table 6: Study Design of ECZTRA 6 (Adolescents)
Study design | ECZTRA 6 |
|---|---|
Designs and populations | |
Study design | Phase III, double-blind, placebo-controlled RCT |
Locations | 87 sites in Europe, North America (including 10 in Canada), Australia, and Asia |
Patient enrolment dates | June 19, 2018, to November 4, 2019 |
Randomized (N) | 301 |
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Initial treatment period (week 0 to 16):
Maintenance treatment period (week 17 to 52):
|
Comparator(s) | Placebo q.2.w. |
Study duration | |
Screening | 2 to 6 weeks |
Initial treatment period | 16 weeks |
Maintenance treatment period | 36 weeks |
Follow-up phase | 14 weeks |
Outcomes | |
Primary end points |
|
Secondary and exploratory end points | Key secondary end points:
Other secondary end points:
Exploratory end points:
Maintenance end points:
|
Publication status | |
Publication | Paller et al. (2023)9 |
AD = atopic dermatitis; AE = adverse event; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; EASI-90 = reduction of at least 90% in Eczema Area and Severity Index score from baseline; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; PDE-4 = phosphodiesterase type 4; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = 50% decrease in Scoring Atopic Dermatitis; SCORAD-75 = 75% decrease in Scoring Atopic Dermatitis; TCI = topical calcineurin inhibitor; TCS = topical corticosteroids.
aTralokinumab 150 mg q.2.w. and q.4.w. regimens are not of interest to this review as these regimens are not approved by Health Canada.
Sources: Clinical Study Report for ECTZRA 613 and sponsor’s Summary of Clinical Evidence.14
Table 7: Study Designs of ECZTRA 1, 2, 3, and 7 (Adults, Original Review)
Study design | ECZTRA 1 and 2 | ECZTRA 3 | ECZTRA 7 |
|---|---|---|---|
Designs and populations | |||
Study design | Phase III, double-blind, RCT | Phase III, double-blind, RCT | Phase IIIb, double-blind, RCT |
Locations | ECZTRA 1: approximately 130 sites in the US, Europe, and Japan ECZTRA 2: approximately 130 sites in North America (including 19 in Canada), Europe, Australia, and Korea | 63 sites in North America (including 10 in Canada) and Europe | 68 sites in Europe |
Patient enrolment dates | ECZTRA 1: May 30, 2017, to March 5, 2018 ECZTRA 2: June 29, 2017, to April 26, 2018 | March 19, 2018, to November 14, 2018 | December 13, 2018, to September 28, 2020 |
Randomized (N) | ECZTRA 1: 802; ECZTRA 2: 794 | 380 | 277 |
Key inclusion criteria |
| Same as for ECZTRA 1 and 2 |
|
Key exclusion criteria |
| Same as for ECZTRA 1 and 2 | Same as for ECZTRA 1 and 2, except that enrolment of patents who received TCS or TCI within 2 weeks prior were allowed |
Drugs | |||
Intervention | Initial treatment period (week 0 to 16):
Maintenance treatment period (week 17 to 52) Patients who received tralokinumab and achieved a clinical response at week 16:
| Initial treatment period (week 0 to 16):
Continuation treatment period (week 17 to 32): Patients who received tralokinumab treatment and achieved a clinical response at week 16:
| Tralokinumab 600 mg at day 0, then 300 mg SC q.2.w. + TCSd |
Comparator(s) | Placebo q.2.w. | Placebo q.2.w. + TCSd | Placebo q.2.w. + TCSd |
Study duration | |||
Screening | Up to 6 weeks | Up to 6 weeks | Up to 6 weeks |
Double-blind (initial treatment) | 16 weeks | 16 weeks | 26 weeks |
Double-blind (maintenance or continuation treatment period) | Maintenance treatment period: 36 weeks | Continuous treatment period: 16 weeks | NA |
Follow-up | 14 weeks | 14 weeks | 14 weeks |
Outcomes | |||
Primary end points |
|
|
|
Secondary and exploratory end points | Key secondary end points:
Other secondary end points:
Exploratory end points:
| Key secondary end points:
Other secondary end points:
Exploratory end points:
| Key secondary end points:
Other secondary end points:
Exploratory end points:
|
Notes | |||
Publications | Wollenberg et al. (2021)10 | Silverberg et al. (2021)11 | Gutermuth et al. (2022)12 |
AD = atopic dermatitis; AE = adverse event; BSA = body surface area; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; EASI-90 = reduction of at least 90% in Eczema Area and Severity Index score from baseline; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NA = not applicable; NRS = numeric rating scale; PDE-4 = phosphodiesterase type 4; POEM = Patient-Oriented Eczema Measure; PGI-B = Patient Global Impression of Bother; PGI-S = Patient Global Impression of Severity; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SC = subcutaneous; SAE = serious adverse event; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = 50% decrease in Scoring Atopic Dermatitis; SCORAD-75 = 75% decrease in Scoring Atopic Dermatitis; SF-36 = Short Form (36) Health Survey; TCI = topical calcineurin inhibitor; TCS = topical corticosteroids; TSQM = Treatment Satisfaction Questionnaire for Medication; VAS = visual analogue scale; WPAI-GH = Work Productivity and Activity Impairment – General Health.
aAt screening and baseline.
bDuring the week before baseline.
cPatients were permitted to continue using stable doses of such emollients if initiated before the screening visit.
dPatients were instructed to apply a thin layer of supplied TCS once daily to areas with active lesions as needed in the treatment period.
Sources: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 and 7,18 CADTH Reimbursement Review for Adtralza (2022),65 and sponsor’s Summary of Clinical Evidence.14
Populations
Key inclusion and exclusion criteria of included studies are summarized in Table 6 (adolescents) and Table 7 (adults).
Inclusion and Exclusion Criteria
Adolescents
The ECZTRA 6 study included adolescents aged 12 to 17 years who were diagnosed with AD for at least 1 year and had an inadequate response or intolerance to TCS and/or TCIs, or for whom topical medications were medically inadvisable. Patients were required to have an EASI of at least 12 at screening and at least 16 at baseline, an IGA score of at least 3 at screening and at baseline, AD involvement of at least 10% of BSA, and an adolescent pruritus NRS average score of at least 4 during the week before baseline. Patients who had been treated with a topical PDE-4 inhibitor, TCS, or TCI within 2 weeks prior, or used systemic immunosuppressive or immunomodulating drugs, systemic corticosteroids, or 3 or more bleach baths within the past 4 weeks were excluded, as were those who had used tanning beds or phototherapy within the preceding 6 weeks.
Adults
The inclusion and exclusion criteria of the ECZTRA 1, 2, 3, and 7 studies were the same as for the ECZTRA 6 study, except that they were conducted in adult patients. Inadequate response was explicitly defined as failure to achieve and maintain remission or a low disease-activity state (comparable to an IGA of 0 [clear] to 2 [mild]) despite treatment with a daily regimen of TCS of medium to high potency. As well, the pruritus NRS average score was used to assess the severity of pruritus (as opposed to adolescent pruritis NRS in the ECZTRA 6 study). Patients enrolled in the ECZTRA 7 study had a history of inadequate response, intolerance, or unacceptable toxicity to cyclosporine A, or were not deemed to be candidates for cyclosporine A treatment and an EASI of at least 20 at screening and baseline (as opposed to an EASI of at least 12 at screening and at least 16 at baseline in other studies), and were allowed to use TCS and TCIs within 2 weeks before enrolment (which was not permitted in other studies).
Interventions
Adolescents
Initial Treatment Period
In the initial treatment period of the ECZTRA 6 study, patients were randomly assigned to receive 1 of the following until week 16:
Tralokinumab 600 mg (i.e., 4 injections of 150 mg, 1 mL each) subcutaneously on day 0, followed by tralokinumab 300 mg (i.e., 2 injections of 150 mg, 1 mL each) subcutaneously every 2 weeks
Tralokinumab 300 mg (i.e., 2 mL) and placebo 2 mL subcutaneously on day 0, followed by tralokinumab 150 mg (1 mL) and placebo (1 mL) subcutaneously every 2 weeks
Placebo 4 mL on day 0 then 2 mL subcutaneously every 2 weeks.
Maintenance Treatment Period
In the maintenance treatment period of the ECZTRA 6 study, patients who achieved a clinical response at week 16 (defined as an IGA of 0 or 1 or EASI-75 without the use of rescue treatment) while receiving tralokinumab in the initial treatment period were re-randomized to receive tralokinumab every 2 weeks or every 4 weeks at the same dose as in the initial treatment period until week 52.
Patients initially randomized to tralokinumab 300 mg every 2 weeks were re-randomized to either tralokinumab 300 mg every 2 weeks in a 2 mL injection or tralokinumab 300 mg every 4 weeks in alternating 2 mL injections of 300 mg of tralokinumab and placebo.
Among patients initially randomized to tralokinumab 150 mg, patients re-randomized to tralokinumab 150 mg every 2 weeks received tralokinumab 150 mg (1 mL) + placebo (1 mL). Patients re-randomized to tralokinumab 150 mg every 4 weeks received alternating 1 mL doses of tralokinumab 150 mg and placebo or a 2 mL dose of placebo.
Patients receiving placebo every 2 weeks who met the primary end points at week 16 without the use of rescue medication continued to receive placebo every 2 weeks until week 52.
Patients who transferred to open-label treatment received tralokinumab 300 mg (2 mL) every 2 weeks with optional use of TCS or TCIs of mild-to-moderate potency.
Blinding
Tralokinumab and placebo were visually distinct. All patients, study investigators, and study personnel who were involved in the treatment, clinical evaluation, and monitoring of patients were blinded from treatment assignment in the treatment period of all studies. All treatment groups received the same number of injections at each visit. The packaging and labelling of tralokinumab and placebo contained no evidence of their identity. The interventions were administered by a health care provider, the patient, or the patient’s caregiver (open-label treatment only).
Rescue Medications and Concomitant Medications
All patients were to use an emollient twice daily (or more often, as needed) for at least 14 days before randomization and continue this treatment throughout the trial. Rescue treatment for AD could be provided to patients at the discretion of the investigator. Investigators were encouraged to try rescue topical treatments first for at least 14 days before escalating to systemic medications. TCS of any WHO class could be used as topical rescue treatment (it was unclear if any restrictions were imposed on frequency or dose of topical rescue treatment). Systemic rescue treatment with corticosteroids or nonsteroidal immunosuppressive drugs (e.g., cyclosporine A or methotrexate) required immediate discontinuation of tralokinumab. After the treatment with these drugs was completed, tralokinumab could be resumed if deemed appropriate by the investigator and sponsor’s medical expert. Use of biological rescue treatment was not allowed for the entire trial duration. The following concomitant medications related to AD treatment were permitted from screening through safety follow-up: oral antibiotics, antiviral, or antifungal therapy for skin infections, and oral antihistamines.
Adults
Initial Treatment Period
In the initial treatment period of the ECZTRA 1 and 2 trials, patients were randomized to receive either of the following until week 16:
tralokinumab 600 mg (i.e., 4 injections of 150 mg, 1 mL each) subcutaneously on day 0, followed by tralokinumab 300 mg (i.e., 2 injections of 150 mg, 1 mL each) subcutaneously every 2 weeks until week 16
placebo 4 mL on day 0, and then 2 mL every 2 weeks.
The same interventions were also used in the initial treatment period of the ECZTRA 3 study and the treatment period of the ECZTRA 7 study. In addition, all patients in the ECZTRA 3 and 7 studies were instructed to initiate treatment once daily with a supplied TCS (mometasone furoate 0.1% cream) on active lesions as needed throughout the treatment period. In the ECZTRA 7 study, patients received these interventions until week 26.
Maintenance (or Continuous) Treatment Period (ECZTRA 1, 2, and 3)
In the maintenance treatment period of ECZTRA 1 and ECZTRA 2, patients randomized to tralokinumab in the initial treatment period who had a clinical response (i.e., those who were in tralokinumab group during the initiation treatment period and had a clinical response, defined as an IGA of 0 or 1 or EASI-75 with rescue medication use) at week 16 were re-randomized to receive 1 of the following until week 52:
tralokinumab 300 mg (2 mL) every 2 weeks
tralokinumab 300 mg every 4 weeks (alternating dose administrations of 300 mg tralokinumab and 2 mL placebo every 2 weeks to maintain blinding)
placebo (2 mL) every 2 weeks.
In the continuous treatment period of the ECZTRA 3 study, patients who had clinical response to tralokinumab at week 16 were re-randomized to receive either of the following until week 32:
tralokinumab 300 mg (2 mL) every 2 weeks in combination with an TCS on an as-needed basis
tralokinumab 300 mg (2 mL) every 4 weeks, in combination with an TCS on an as-needed basis.
In the ECZTRA 1, 2, and 3 studies, patients randomized to the placebo group in the initial treatment period who achieved a clinical response at week 16 continued to receive placebo every 2 weeks until the end of the maintenance treatment period. Patients who did not achieve a clinical response to tralokinumab at week 16 (as well as patients who did not maintain an adequate clinical response during the maintenance treatment period of ECZTRA 1 and 2) were transferred to tralokinumab every 2 weeks with optional use of TCS.
The blinding procedures in these trials were the same as those in the ECZTRA 6 study. The protocol for rescue and concomitant medication use was the same as in the ECZTRA 6 study, except that in the ECZTRA 3 and 7 studies, only higher-potency TCS (Europe Class above 3 or below 4) could be used as topical rescue treatment.
Outcomes
A list of efficacy end points assessed in this clinical review report is provided in Table 8 and summarized in the text that follows. Summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence as well as any identified as important to this review according to stakeholders (e.g., clinical experts, clinician groups, or patient groups).
Unless otherwise specified, the baseline value was defined as the latest predose assessment.
Investigator’s Global Assessment
The IGA score is an investigator-reported assessment used to rate the severity of AD.15 It is based on a 5-point scale ranging from 0 to 4, in which “0” indicates clear, and “4” indicates severe AD. Evidence for validity and reliability of this instrument is summarized in Table 9. No MID was identified in adult or adolescent patients with AD.15
Table 8: Outcomes Summarized from ECZTRA 1, 2, 3, and 7
Outcome measure | Time point | ECZTRA 1 and 2 | ECZTRA 3 | ECZTRA 6 | ECZTRA 7 |
|---|---|---|---|---|---|
Initial treatment period | |||||
Severity of AD and AD lesions | |||||
IGA of 0 or 1 |
| Primarya | Primarya | Primarya | Secondarya |
EASI | |||||
EASI-75 |
| Primarya | Primarya | Primarya | Primarya (week 16) Secondarya (week 26) |
EASI-90 |
| Secondary | Secondary | Secondary | Exploratory |
EASI-50 |
| Secondary | Secondary | Secondary | NA |
Change from baseline |
| Secondary | Secondary | Secondary | Secondary |
Change from baseline in SCORAD |
| Key secondarya | Key secondarya | Key secondarya | Secondarya |
Symptom reduction | |||||
Worst daily pruritus NRS | |||||
Reduction of ≥ 4 points from baseline |
| Key secondarya | Key secondarya | Key secondarya,b | Secondarya |
Change from baseline |
| Secondary | Secondary | Secondaryb | Exploratory |
Reduction of ≥ 3 points from baseline |
| Secondary | Secondary | Secondaryb | NA |
Change in eczema-related sleep NRS from baseline |
| Exploratory | Exploratory | Exploratory | Exploratory |
Change from baseline in POEM |
| Exploratory | Exploratory | Secondary | Exploratory |
HRQoL or anxiety/depression symptoms | |||||
DLQI (or CDLQI) | |||||
Change from baseline |
| Key secondarya | Key secondarya | Key secondarya,c | Secondarya |
Reduction of ≥ 4 points (or CDLQI ≥ 6 points) from baseline |
| Secondary | Secondary | Secondary | ||||||||||| |
HADS | |||||
Change from baseline |
| Exploratory | Exploratory | Exploratory | ||||||||||| |
HADS anxiety and depression scores < 8 |
| Exploratory | Exploratory | Exploratory | ||||||||||| |
Use of topical therapy | |||||
Amount of TCS used |
| NA | Secondary | NA | Exploratory |
Number of days without topical treatment |
| NA | Secondary | NA | Exploratory |
Maintenance treatment period | |||||
IGA of 0 or 1, among patients with IGA of 0 or 1 at week 16 achieved without rescue medication after initial randomization to tralokinumab |
| Maintenancea | Maintenance | Maintenance | NA |
EASI-75, among patients with EASI-75 at week 16 achieved without rescue medication after initial randomization to tralokinumab |
| Maintenancea | Maintenance | Maintenance | NA |
IGA of 0 or 1, among patients with EASI-75 and IGA ≥ 2 at week 16 |
| Maintenance | NA | Maintenance | NA |
IGA 0 or 1 or EASI-75, among patients with IGA of 0 or 1 or EASI-75 at week 16 achieved without rescue medication |
| Maintenance | NA | Maintenance | NA |
Harms outcomes | |||||
Adverse events (AE, SAE, WDAE, death, and notable harms: eye disorder (e.g., conjunctivitis), eczema herpeticum, malignancies, skin infections requiring systemic treatment, injection-site reactions, oral herpes, upper respiratory infection, and acne | Baseline to end of safety follow-up | Secondary | Secondary | Secondary | Secondary |
AD = atopic dermatitis; AE = adverse event; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; EASI-90 = reduction of at least 90% in Eczema Area and Severity Index score from baseline; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NA = not applicable; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; SAE = serious adverse event; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroids; WDAE = withdrawal due to adverse event.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
bAdolescent worst pruritis NRS, an age-appropriate version of the worst daily pruritus NRS, was used to assess itch in adolescents in the ECZTRA 6 study.
cCDLQI was used in the adolescent population in the ECZTRA 6 study.
Sources: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 6,13 and 718 and sponsor’s Summary of Clinical Evidence.14
Eczema Area and Severity Index
The EASI is an investigator-reported scale that is used to assess the severity and extent of AD.15 The severity of 4 disease characteristics of AD (erythema, infiltration and/or papulation, excoriations, and lichenification) are assessed on a 4-point scale ranging from “0” (none or absent) to “3” (severe). The total EASI score ranges from 0 to 72 points, with higher values indicating more severe and/or more extensive condition. EASI-50, EASI-75, and EASI-90 indicate improvements of at least 50%, 75%, and 90% from baseline in the EASI score, respectively.15 Evidence for validity, reliability and responsiveness of this instrument is summarized in Table 9. The MID was estimated to be 6.6 in a study that included adults with AD.66 No MID for adolescents was identified.
Scoring Atopic Dermatitis
Scoring Atopic Dermatitis (SCORAD) is an investigator-administered tool to measure the extent and severity of AD lesions, along with subjective symptoms. The instrument assesses 3 components of AD: the extent of affected BSA, severity, and symptoms. The total SCORAD is calculated based on the 3 components, with a maximum possible total score of 103, in which a higher score indicates more severe disease. Evidence for validity and reliability, and responsiveness of this instrument are summarized in Table 9. The MID was estimated to be 8.7 points in a study that included adults and children with AD.66
Worst Pruritus Numeric Rating Score and Adolescent Worst Pruritus Numeric Rating Score
The pruritus NRS is a tool used for patients to self-report the worst itch over the past 24 hours using an 11-point scale, with 0 indicating “no itch” and 10 indicating “worst itch possible.” Evidence for reliability, validity, and responsiveness in patients with AD is summarized in Table 9. A study by Simpson et al. (2017), estimated that improvement on the pruritus NRS of at least 3 to 4 points from baseline is a clinically meaningful change, which was calculated using anchor- and distribution-based methods based on data from a study in adults with moderate-to-severe AD.67 Another study, by Yosipovitch et al. (2019), estimated the MID to be between 2 and 4 points based on anchors (EASI, IGA, and Pruritus Categorical Scale), and 1.0 based on distribution methods.68
The adolescent worst pruritis NRS tool is an age-appropriate version of the worst daily pruritus NRS tool developed for adults. Evidence for reliability, validity, responsiveness, and MID of adolescent worst pruritus NRS in patients with AD is not available.
Eczema-Related Sleep Numeric Rating Scale
The eczema-related sleep NRS is used by the patient to rate how much their eczema had interfered with their sleep the last night using an 11-point NRS, with 0 indicating that it “did not interfere” and 10 indicating that it “completely interfered.”69 Evidence for validity is summarized in Table 9. No MID estimate was identified in adult or adolescent patients with AD.
Patient-Oriented Eczema Measure
The POEM is a 7-item, AD-specific, symptom questionnaire.70 Based on the self-reported frequency of occurrence during the past week, the 7 items (itching, sleep, bleeding, weeping, cracking, flaking, and dryness) are assessed using a 5-point scale (0 = “no days,” 1 = “1 to 2 days,” 2 = “3 to 4 days,” 3 = “5 to 6 days,” and 4 = “every day”). The maximum total score is 28; with a higher score indicative of worse disease severity.70 Evidence for validity, reliability, and responsiveness of the POEM is summarized in Table 9. The MID has been established in AD as 3.4 points in adults66 and from 3.0 to 3.9 points in children.71,72 Another study established 5 points as the MID for adults using global severity of AD as anchor.70
Dermatology Life Quality Index and Children Dermatology Life Quality Index
The DLQI is a widely used dermatology-specific HRQoL instrument for use in adults. It is a 10-item questionnaire in which patients self-report 6 different aspects that may affect QoL (symptoms and feelings, daily activities, leisure, work and school performance, personal relationships, and treatment).15 The DLQI score ranges between 0 and 30. The higher the score, the greater QoL is impaired.15 Evidence for reliability, validity, and responsiveness of the DLQI instrument in patients with moderate-to-severe AD was not identified. Estimates of the MID for a variety of skin conditions range from 2.2 to 6.9 points, but no information about MID had been found for adults with AD.73-75
The CDLQI questionnaire is based on the adult version (DLQI) and is designed and validated in patients with dermatological conditions aged 3 to 16.13 The CDLQI is available in text and cartoon versions; the text version was used in the ECZTRA 6 study. The questionnaire consists of 10 items addressing the patient’s perception of the impact of their skin disease on various aspects of their QoL over the last week, including dermatology-related symptoms and feelings, leisure, school, friendships, sleep, and the impact of treatment. The total score ranges from 0 to 30, with a higher score indicative of a poor QoL.13 Evidence for validity, reliability, and responsiveness of this instrument is summarized in Table 9. In adolescents, a reduction in the CDLQI of 6 to 8 points has been suggested as the clinically relevant threshold for within-person change in CDLQI in moderate-to-severe AD.76
Hospital Anxiety and Depression Scale
The HADS is a widely used patient-reported questionnaire designed to identify anxiety disorders and depression.88 The HADS questionnaire contains 14 items that are used to assess symptoms experienced in the previous week, among which 7 items are related to anxiety and 7 items are related to depression. The score ranges between 0 and 21 for each subscale (anxiety and depression); a high score is indicative of a poor state.88 Evidence for reliability, validity, responsiveness, and MID of HADS in adult or adolescent patients with AD is not available.
Harms Outcomes
Harms outcomes were assessed as secondary end points. This includes AEs, SAEs, withdrawal due to AEs, and death, as well as notable harms of interest to this review, including eye disorder (e.g., conjunctivitis), eczema herpeticum, malignancies, skin infections requiring systemic treatment, injection-site reactions, oral herpes, upper respiratory infection, and acne.
Table 9: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | Minimal important difference |
|---|---|---|---|
IGA of AD severity | Investigator-reported assessment instrument used in clinical trials to rate AD severity. 5-point scale, ranging from 0 (clear) to 4 (severe) with distinct, morphological descriptors for each category.a,77 | Validity: Moderate to strong correlation with EASI (r = 0.66 to 0.72) in adult patients with AD.77 Reliability: Moderate intrarater (ICC = 0.54, SD = 0.28) and interrater reliability (CV = 33.0, SD = 12.3) in adult patients with AD.77 Responsiveness: No evidence identified. | No MID has been identified in adult or adolescent patients with AD. |
EASI | A physician-administered, composite index that assesses the severity and extent of AD.b,78 The severity of 4 AD disease characteristics (erythema, induration/papulation, excoriation, and lichenification) on the 4 body regions (head and neck, trunk, upper extremities, and lower extremities) is assessed by the investigator on a 4-point scale ranging from 0 (none or absent) to 3 (severe). The EASI score equals the sum of the weighted scores obtained for each body region. Scores range from 0 to 72, with higher values indicating more severe and/or more extensive condition.78 EASI-50, EASI-75, and EASI-90 represent a ≥ 50%, ≥ 75%, and ≥ 90% reduction from baseline in EASI score, respectively.78 | Validity: In adult patients with AD, moderate to strong correlation with SCORAD (r = 0.84 to 0.93) was found.66,79,80 In pediatric patients with AD including those over 12 years old, EASI was correlated strongly with IGA (r > 0.8 at day 43 and 6 months).78 Reliability: The internal consistency of the EASI is adequate in adult patients with AD, with Spearman and Cronbach alpha values of 0.86 and 0.94, respectively.80 Test-retest reliability was also adequate (intra- and interrater reliability kappa = 0.76),80 whereas the reliability of each component of the EASI ranged from 0.38 (ICC, lichenification) to 0.75 (ICC, area), indicating poor to good intrarater reliability.77 No evidence of reliability in adolescent patients with AD was identified. Responsiveness: In a study of adult patients with AD (MAcAD trial), responsiveness to improvement and decline in global severity based on IGA over 24 weeks was demonstrated (AUC = 0.67, 95% CI = 0.60 to 0.76).66 In pediatric patients with AD, sensitivity to change was judged as adequate (P < 0.001, n = 1,068) to detect improvement in disease status from baseline after 8 days of treatment.78 | In adults with AD, the overall MID has been reported to be 6.6.66 No MID has been identified for adolescents with AD. |
SCORAD | A physician-administered tool to measure the extent and severity of AD lesions, along with subjective patient-reported symptoms.66 The total maximum score ranges from 0 to 103, with higher values indicating more severe disease. The assessment consists of 3 componentsc: A = extent, B = intensity, C = subjective symptoms.66 | Validity: Two systematic reviews found agreement between objective SOCARD and global assessments of disease severity (r = 0.62 with OSAAD, r = 0.18 to 0.69 with VAS itch) in adult patients with AD.80,81 Reliability: In adult patients with AD, adequate correlation of items within objective SCORAD score has been demonstrated (ICC = 0.64 to 0.86); however, intra-observer reliability (test-retest) was unclear. Inter-observer reliability between 10 trained observers has been deemed adequate, except for edema/papulation (poorest agreement by 2-way analysis of variance).80 Responsiveness: In the MAcAD trial, responsiveness to improvement and decline in global severity as measured by IGA over 24 weeks was noted in mainly adult and unknown number of adolescent patients with AD (AUC = 0.73, 95% CI = 0.70 to 0.77).66 | The MID has been estimated using mean change scores of SCORAD of patients that showed a relevant improvement based on IGA, defined as an “improvement” or “decline” of ≥ 1 point in NRS and IGA;, a difference of 8.7 points in SCORAD was estimated as the MID for adults and children (17% of analyzed population) with AD.66 |
DLQI | A patient-reported, dermatology-specific health-related QoL instrument for use in adults. Consists of 10 items addressing the patient’s perception of the impact of their skin disease on 5 different aspects of QoL, each scored on a 4-point Likert scale (0 = “not at all,” 1 = “only a little,” 2 = “quite a lot,” 3 = “very much”).
The total score is the sum of the 10 items (0 to 30), with a higher score indicative of a poorer QoL (0 to 1 = no effect; 2 to 5 = small effect; 6 to 10 = moderate effect; 11 to 20 = very large effect; 21 to 30 = extremely large effect). Recall period is the past 1 week.73-75 | Validity: During the development phase, input from adult patients with AD (n = 9; other eczema n = 10) ensured content validity.82 Construct validity was demonstrated by a strong correlation with POEM (r = 0.78) and a moderate correlation with SCORAD (r = 0.42). Reliability: In patients with stable AD, test-retest reliability was adequate (ICC > 0.7). Among adult patients with mixed skin diseases including AD, internal consistency was acceptable (Cronbach alpha = 0.75 to 0.92).83-85 Responsiveness: In patients aged over 16 years with a variety of skin conditions including AD (n = 192, patients with eczema = 12.5%); improved DLQI score was observed in those whose disease severity decreased over a 1 to 3 month period (P < 0.0001).74 | Estimates of the MID have ranged from 2.2 to 6.9, but no information about MID was found specifically for adult patients with AD.73-75 |
CDLQI | A patient-reported, dermatology-specific questionnaire. Based on the adult version of the DLQI, designed for patients with dermatological conditions from 3 to 16 years of age. Available in text and cartoon versions.d Consists of 10 items addressing the patient’s perception of the impact of their skin disease on various aspects of their health-related QoL over the last week, including dermatology-related symptoms and feelings, leisure, school, friendships, sleep, and the impact of treatment.76,86,87 Each question is scored on a 4-point Likert scale (0 = “not at all,” 1 = “only a little,” 2 = “quite a lot,” 3 = “very much”). The total score is the sum of the 10 items (0 to 30), with a higher score indicative of a worse health-related QoL.76,86,87 | Validity: Three studies demonstrated concurrent validity, 2 between the CDLQI and Cardiff Acne Disability Index and 1 between the CDLQI and Childhood Atopic Dermatitis Impact Scale.87 Convergent construct validity and divergent construct validity of the CDLQI were demonstrated in 45 and 6 studies, respectively.87 Reliability: The CDLQI (examined in 6 studies) has good internal consistency, with Cronbach alpha values ranging from 0.82 to 0.92.86,87 Test-retest reliability is adequate, with Spearman rank order correlation coefficient calculated in 4 studies (range 0.74 to 0.97).86,87 One study examined the ICC with finding of 0.80.86,87 Responsiveness: Examined in 26 studies, which demonstrated responsiveness to change in the CDLQI.87 | In adolescent patients with moderate-to-severe AD, a reduction of 6.0 to 8.0 points has been suggested as the clinically relevant threshold for within-person change corresponding to improvement in anchors.76 |
Worst pruritus NRS, adult and adolescent | Patient-reported worst itch over the past 24 hours using an 11-point NRS, with 0 indicating “no itch” and 10 indicating “worst itch possible.”68 The adolescent worst pruritus NRS tool is an age-appropriate version of the worst daily pruritus NRS tool developed for the assessment of itch in adults. The adolescent version uses the same rating scale as the adult version with wording that is more appropriate for the younger population.13 | Psychometric assessment was performed in adult patients with moderate-to-severe AD from clinical trial populations (SOLO 1 and SOLO 2).68 Validity: Content validity was ensured through concept elicitation during development and in-depth, 1-to-1 patient interviews (n = 14). Construct validity with similar constructs (PCS, DLQI itch item, SCORAD itch VAS) was strong (Pearson r = 0.61 to 0.77), whereas with those with dissimilar constructs (EASI, IGA) was weak to moderate (r = 0.09 to 0.24). Known-group validity was established: patients with “absent” or “mild” itch based on PCS, “no impact” on DLQI or “excellent” on PGADS had a significantly lower score on NRS (P < 0.0001).68 Reliability: Test-retest reliability over 1 week was adequate. (ICC = 0.95 to 0.96).68 Responsiveness: Change from baseline at week 16 in NRS was correlated well with those with PCS (Pearson r = 0.71), DLQI itch item (r = 0.66), and SCORAD itch VAS (r = 0.77), less well with EASI (r = 0.50) and IGA (r = 0.50).68 Psychometric assessment in adolescent population with AD has not been identified. | In adults, a change from baseline of ≥ 2 to 4 points may be considered to be an important within-person change.67,68 Evidence for an MID in adolescents was not identified. |
Eczema-related sleep NRS | Patient-reported sleep interference caused by eczema last night using an 11-point NRS, with 0 indicating that it “did not interfere” and 10 indicating that it “completely interfered.”69 | Validity: Content validity has been ensured by concept elicitation, cognitive debriefing interviews, and additional interviews in adolescent and adult patients with moderate-to-severe AD.69 Evidence of reliability, other validity, and responsiveness in patients with AD is not available. | No MID has been identified in adult or adolescent patients with AD. |
POEM | A patient-reported, AD-specific, symptom questionnaire, with the assessment period being the past week.70 Consists of 7 items (itching, sleep, bleeding, weeping, cracking, flaking, and dryness), each assessed on a 5-point categorical response scale (0 = “no days,” 1 = “1 to 2 days,” 2 = “3 to 4 days,” 3 = “5 to 6 days,” and 4 = “every day”). The total score is the sum of the 7 items (ranging from 0 to 28) and reflects disease-related morbidity, with a higher score indicative of worse symptoms.70 | Validity: In adult patients, concurrent validity was reported in those with moderate-severe self-reported AD severity (Spearman r = 0.53); however, weak correlation (r = 0.39) with clear-to-mild AD was found. Convergent validity with DLQI (r = 0.59), correlation with EASI (r = 0.52), weaker correlation with worst itch NRS (r = 0.45) were found in adult patients with AD.70 Reliability: Internal consistency was acceptable (Cronbach alpha = 0.88), and test-retest reliability was acceptable, with 95% of scores falling within 2.6 points on repeat testing (mean score difference = 0.04, SD = 1.32) in adult patients with AD.70 Responsiveness: In the Prove trial conducted in adult patients with AD, responsiveness to improvement and decline in global severity as measured by IGA over 18 weeks was noted.66 | The MID was established in AD as 3.4 points in adults and from 3.0 to 3.9 points in children.66,71,72 Another study established 5 points as the MID for adults using global severity of AD as anchor.70 |
HADS | Patient-reported, hospital-setting questionnaire used to detect states of anxiety and depression. Consists of 14 items that assess the patient’s anxiety (7 items) and depression (7 items) during the last week.88 Each question is scored on a 4-point Likert scale ranging from 0 (best) to 3 (worst); a person can score between 0 and 21 for each subscale (anxiety and depression), with higher scores indicating a poorer state.88 | Evidence of reliability, validity, and responsiveness in patients with AD is not available. | No MID has been identified in adult or adolescent patients with AD. |
AD = atopic dermatitis; AUC = area under the curve; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; EASI-90 = reduction of at least 90% in Eczema Area and Severity Index score from baseline; HADS = Hospital Anxiety and Depression Scale; ICC = intraclass correlation coefficient; IGA = Investigator’s Global Assessment; MID = minimal important difference; NRS = numeric rating scale; OSAAD = Objective Severity Assessment of Atopic Dermatitis; PCS = Pruritus Categorical Scale; PGADS = Patient Global Assessment of Disease Status; POEM = Patient-Oriented Eczema Measure; QoL = quality of life; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; VAS = visual analogue scale.
aIGA categories:
0 (clear): No inflammatory signs of AD; No erythema and no elevation (papulation and/or infiltration).
1 (almost clear): Just perceptible erythema, and just perceptible papulation and/or infiltration; barely perceptible erythema and/or minimal lesion elevation (papulation and/or infiltration) that is not widespread.
2 (mild disease): Mild erythema and mild papulation and/or infiltration; visibly detectable, light pink erythema and very slight elevation (papulation and/or infiltration).
3 (moderate disease): Moderate erythema and moderate papulation and/or infiltration; dull red, clearly distinguishable erythema and clearly perceptible but not extensive elevation (papulation and/or infiltration).
4 (severe disease): Severe erythema and severe papulation and/or infiltration; deep or dark-red erythema, marked and extensive elevation (papulation and/or infiltration).
bThe EASI is a composite index, including an assessment of disease extent and percent of BSA involved, converted to a proportional factor (scale of 0 to 6), in 4 body regions (head and neck, lower limbs, upper limbs, and trunk). The proportion allocated to each body region depends on the patient’s age. In patients aged 8 years or older, the proportions are 10% for head and neck, 20% for upper extremities, 30% for trunk, and 40% for lower extremities; in patients aged 7 years or younger, the proportions are 20% for head and neck, 20% for upper extremities, 30% for trunk, and 30% for lower extremities. The EASI also includes an assessment of erythema (E), infiltration and/or papulation (I), excoriation (Ex) and lichenification (L), each on a scale of 0 to 3. The algorithm for calculating the EASI uses, for each body region, the sum of the clinical sign scores (E + I + Ex + L) multiplied by the area, multiplied by the proportional factor. The total EASI score is the sum of the 4 body-region scores.
cThe SCORAD is calculated as: (A)/5 + 7 × (B)/2 + (C):
Extent (A) is assessed as a percentage of each defined body area and reported as the sum of all areas, with a maximum score of 100%.
Intensity (B) of 6 specific symptoms of AD (erythema, edema and/or papulation, oozing and/or crusting, excoriation, lichenification, and dryness) is assessed on an average representative area using a 4-point scale ranging from 0 (none or absent) to 3 (severe). The sum of the intensity score of the 6 symptoms is reported, with a maximum score of 18.
Subjective symptoms (C) involve a subjective assessment of the average itch and sleeplessness over the last 3 days or nights, recorded for each symptom by the patient on a VAS ranging from 0 (no itch or no sleeplessness) to 10 (worst imaginable itch or sleeplessness), with a maximum combined possible score of 20.
dThe text version was used in the ECZTRA 6 study.
Sources: Sponsor’s clinical evidence summary, ECZTRA 6 Clinical Study Report,13 Bozek and Reich (2017),78 Barbier et al. (2004),78 Hanifin et al. (2001),79 Schram et al. (2012),66 Schmitt et al. (2013),80 Rehal and Armstrong (2011),81 Lewis-Jones and Finlay (1995),86 Salek et al. (2013),87 Simpson et al. (2019),76 Yosipovitch et al. (2019),68 Simpson et al. (2017),51 Dias-Barbosa et al. (2020),69 Silverberg et al. (2020),70 Howells et al. (2018),72 Zigmond and Snaith (1983),88 Basra et al. (2008),83 Basra et al. (2015),74 Shikiar et al. (2005),73 Heinl et al. (2016),75 Lewis and Finlay (2004),84 and Badia et al. (1999).85
Statistical Analysis
Statistical analysis methods of the primary estimand of efficacy end points are summarized in Table 10.
Sample-Size Considerations
ECZTRA 1 and 2: A sample-size calculation determined that approximately 780 randomized patients were required (3:1 ratio, i.e., 585 patients to tralokinumab and 195 patients to placebo) to demonstrate a statistically significant difference between tralokinumab and placebo with respect to both primary end points (an IGA of 0 or 1 at week 16 and EASI-75 at week 16) at a 2-sided significance level of 0.05 with over 99% power, assuming the IGA of 0 or 1 response rates in tralokinumab and placebo groups were 30% and 10%, respectively, and EASI-75 response rates in tralokinumab and placebo groups were an IGA 0 or 1, and 40% and 15%, respectively.
With an IGA response rate of 30% at week 16, 175 IGA responders initially treated with tralokinumab were expected to be re-randomized into the maintenance treatment period (2:2:1 ratio, i.e., 70 patients on tralokinumab every 2 weeks, 70 patients on tralokinumab every 4 weeks, and 35 patients on placebo). Assuming IGA response rates at week 52 of 80%, 50%, and 5%, respectively, for tralokinumab every 2 weeks, tralokinumab every 4 weeks, and placebo, the nominal power to show a difference at the 4% significance level would be greater than 99% between tralokinumab every 2 weeks and placebo, and greater than 99% between tralokinumab every 4 weeks and placebo.
With an EASI-75 response rate of 40% at week 16, 235 EASI-75 responders initially treated with tralokinumab were expected to enter the maintenance treatment period (94 patients on tralokinumab every 2 weeks, 94 patients on tralokinumab every 4 weeks, and 47 patients on placebo). Assuming EASI-75 response rates at week 52 of 90%, 55%, and 5%, respectively, for tralokinumab every 2 weeks, tralokinumab every 4 weeks, and placebo, the nominal power to show a difference at the 4% significance level would be greater than 99% between tralokinumab every 2 weeks and placebo and greater than 99% between tralokinumab every 4 weeks and placebo.
ECZTRA 3: Approximately 369 randomized patients were required in the initial treatment period (2:1 ratio, 246 patients to tralokinumab every 2 weeks plus TCS and 123 patients to placebo plus TCS) to demonstrate a statistically significant difference between tralokinumab plus TCS and placebo plus TCS with respect to an IGA of 0 or 1 at week 16 (primary end point) at a 2-sided significance level of 0.05 with 90% power, assuming the IGA of 0 or 1 response rates in tralokinumab plus TCS and placebo plus TCS groups were 30% and 15%, respectively. For the primary end point of EASI-75 at week 16, a sample size of 369 patients provided a power exceeding 99.9% to detect a difference between the 2 groups, assuming response rates of 40% and 15%, respectively.
ECZTRA 6: Approximately 294 randomized patients were required in the initial treatment period (1:1:1 ratio, i.e., 98 patients to tralokinumab 300 mg, 98 patients to tralokinumab 150 mg, and 98 patients to placebo) to demonstrate a statistically significant difference between tralokinumab 300 mg and placebo with respect to an IGA of 0 or 1 at week 16 (primary end point) at a 2-sided significance level of 0.05 with approximately 94% power, assuming response rates for tralokinumab 300 mg and placebo groups of 30% and 10%, respectively. For the primary end point of EASI-75 at week 16, a sample size of 294 patients would provide a power of approximately 98% to detect a difference between tralokinumab 300 mg and placebo, assuming response rates of 40% and 15%, respectively.
ECZTRA 7: A sample size of 250 patients would provide 99% power to detect a treatment difference for the primary end point at a significance level of 0.05, assuming EASI-75 response rates at week 16 of 40% and 15% for tralokinumab plus TCS and placebo plus TCS, respectively. Assuming a response rate of 30% versus 15% in reduction of worst daily pruritus NRS (weekly average) score of at least 4 from baseline to week 16 for tralokinumab plus TCS and placebo plus TCS, respectively, the sample size would provide at least 80% power to reject the hypotheses related to the primary and secondary end point evaluating pruritus at week 16.
Multiplicity Adjustment
ECZTRA 1 and 2: To control the overall type I error rate, the primary analyses for the primary and secondary end points for the initial and maintenance treatment followed the testing procedures shown in Figure 5. IGAs of 0 or 1 at week 16, followed by EASI-75 at week 16, were evaluated at a 5% significance level. If both tests were statistically significant, the significance level (alpha) was split between the analyses of the 3 secondary end points at week 16 (alpha = 1%) and the analyses of the 2 maintenance end points at week 52 (alpha = 4%). A different testing procedure was used for the US regulatory submission but will not be summarized in this report.
Figure 5: Testing Procedures for Primary, Secondary, and Maintenance End Points in ECZTRA 1 and 2 (Global, Non-US Submission)
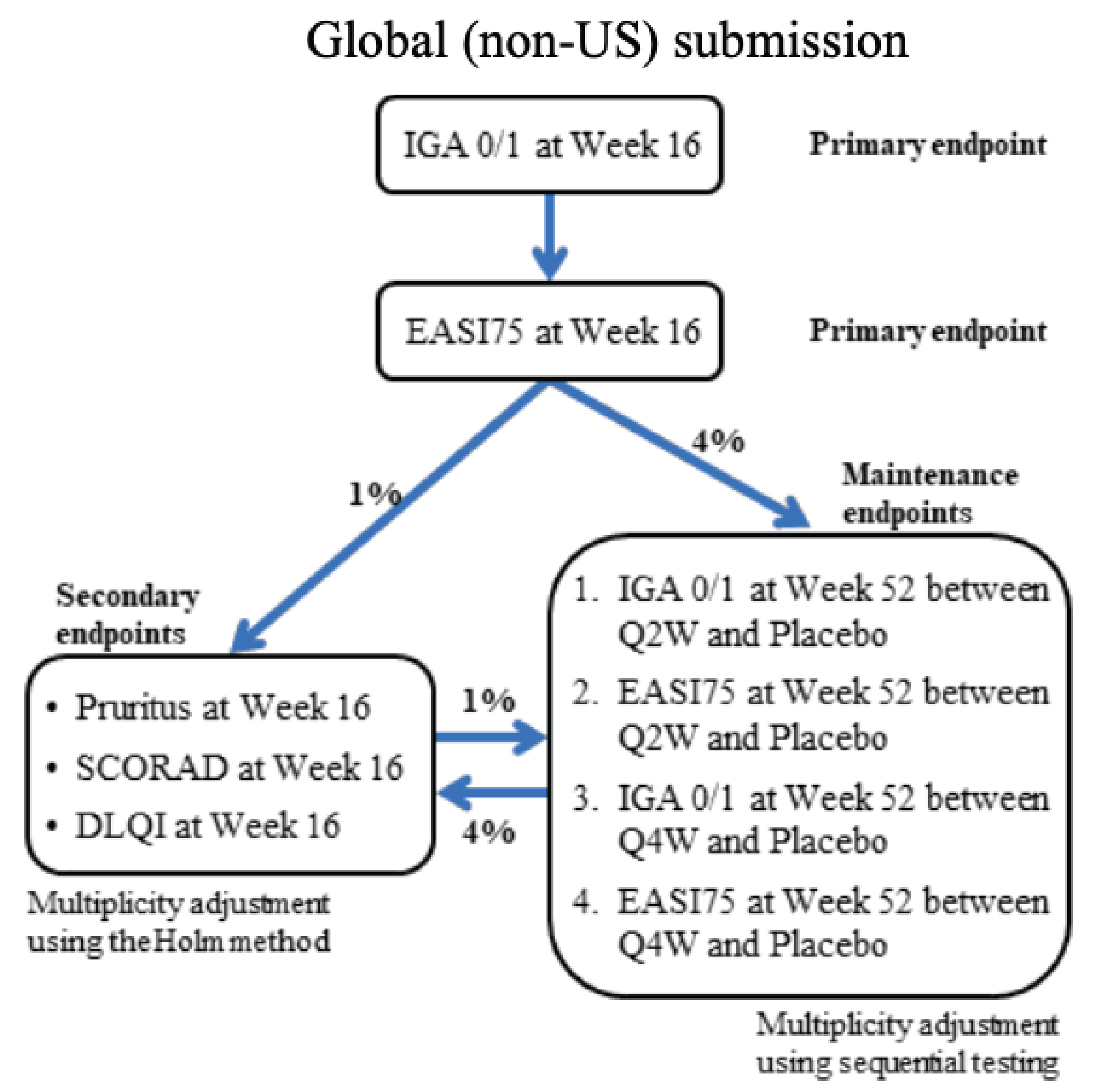
DLQI = Dermatology Life Quality Index; EASI75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; IGA = Investigator’s Global Assessment; SCORAD = Scoring Atopic Dermatitis; Q2W = every 2 weeks; Q4W = every 4 weeks.
ECZTRA 3: The overall type I error rate for the primary analysis of the primary estimands for the primary and confirmatory secondary end points was controlled by a combination of hierarchical testing and Holm-Bonferroni multiplicity adjustment as outlined in Figure 6. The testing procedure was similar to that in the ECZTRA 1 and 2 studies, except that the maintenance treatment end points were not included in the testing hierarchy in the ECZTRA 3 study. A different testing procedure was used for the US regulatory submission but will not be summarized in this report.
Figure 6: Testing Procedures for Primary and Confirmatory Secondary End Points in ECZTRA 3 (Global, Non-US Submission)
DLQI = Dermatology Life Quality Index; EASI75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; IGA = Investigator’s Global Assessment; SCORAD = Scoring Atopic Dermatitis.
Note: Arrows indicate order of testing when the hypothesis is rejected for an end point within a box.
Source: Clinical Study Report for ECZTRA 3.17
ECZTRA 6: To control the overall type I error rate, the primary analyses of the primary estimands for the primary and confirmatory secondary end points followed the testing procedures outlined in Figure 7. An IGA of 0 or 1 at week 16, followed by EASI-75 at week 16, between tralokinumab 300 mg every 2 weeks and placebo were evaluated at a 5% significance level. If both tests were significant, the significance level (alpha) was split between the analyses of the 3 secondary end points between tralokinumab 300 mg every 2 weeks and placebo at week 16 (alpha = 2.5%), and the analyses of the 2 primary end points and 3 secondary end points between tralokinumab 150 mg every 2 weeks and placebo at week 16 (alpha = 2.5%). A different testing procedure was used for the US regulatory submission but it will not be summarized in this report.
Figure 7: Testing Procedure for Primary and Secondary End Points in ECZTRA 6 (Global, Non-US Submission)
CDLQI = Children’s Dermatology Life Quality Index; EASI75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; IGA = Investigator’s Global Assessment; SCORAD = Scoring Atopic Dermatitis.
Notes: The numbers in parentheses indicate significance levels that have been passed on from rejected hypotheses for the other tralokinumab dose levels.
Source: Clinical Study Report for ECZTRA 6.13
ECZTRA 7: The primary and secondary end points were evaluated hierarchically as shown in Figure 8. The hypothesis relating to a specific end point was rejected only if all hypotheses relating to end points earlier in the hierarchy were also rejected at the 5% significance level. Hypothesis testing was based on the primary analysis of the primary estimand for each associated end point.
Statistical Analysis for Primary Efficacy End Points
ECZTRA 1, 2, 3, and 6
Statistical model: The difference in response rates between treatment groups with respect to the 2 primary outcomes was analyzed at a 2-sided significance level of 5% using the Cochran-Mantel-Haenszel test (single imputation analyses) or using combined inference from multiple Mantel-Haenszel risk differences and associated standard errors using Rubin’s rule (multiple imputation analyses). The stratification factors included region and baseline disease severity. The primary estimand (composite strategy) assessed treatment differences in response rates of IGA 0 or 1 and EASI-75 after 16 weeks achieved without rescue medication, regardless of treatment discontinuation. Patients who had received rescue medication before the week 16 visit were considered nonresponders. Patients with missing data at week 16 and where rescue medication had not been used before week 16 were imputed as nonresponders.
Figure 8: Testing Hierarchy for Primary and Secondary End Points in ECZTRA 7

Alpha = statistical significance level; DLQI = dermatology life quality index; EASI75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; IGA = investigator’s global assessment; NRS = numeric rating scale; SCORAD = scoring atopic dermatitis.
Source: Clinical Study Report for ECZTRA 7.18
Sensitivity analyses: Three sensitivity analyses were conducted:
All patients who permanently discontinued the investigational medicinal product before week 16 were considered nonresponders, even if no rescue medication had been used.
Missing data at week 16 was imputed using the LOCF rather than nonresponder imputation for patients who did not receive rescue medication and did not withdraw due to an AE or lack of efficacy.
Tipping-point analysis using multiple imputations with patients who, before the week 16 visit, had received rescue medication and were considered nonresponders. Missing week 16 responses were imputed from a Bernoulli distribution with a varying parameter, P, for patients in the placebo group who did not use rescue medication. Patients in the tralokinumab group with missing week 16 data were imputed as nonresponders. Different percentages of placebo patients were considered responders for the different P values. The tipping point is the P value that changed the conclusion from significant to nonsignificant.
Subgroup analyses: According to the clinical experts, this review is interested in 1 subgroup analysis: baseline IGA.
Two additional estimands were defined, including:
Secondary hypothetical estimand: All values were censored after permanent discontinuation of the investigational product or initiation of rescue medication, and multiple imputations of missing values were applied within each treatment group, assuming data were missing at random.
Tertiary treatment policy estimand: All data were used as observed, and multiple imputation of missing values was applied, assuming data were missing at random within the 4 groups defined according to the randomized treatment group and whether patients permanently discontinued the investigational product before week 16.
ECZTRA 7
Statistical model: The difference in EASI-75 between treatment groups was analyzed at a 2-sided significance level of 5% using the Cochran-Mantel-Haenszel test (single imputation analyses) or multiple Mantel-Haenszel risk differences and associated standard errors to allow for a combined inference using Rubin’s rule (multiple imputation analyses). The stratification factors included prior cyclosporine A use (yes or no) and baseline disease severity (IGA of 3 or 4). The primary estimand (COVID-19–modified composite) assessed treatment difference in response rates of EASI-75 after 16 weeks achieved without rescue medication or treatment discontinuation, as if the COVID-19 pandemic had not happened. Patients who received rescue treatment or permanently discontinued the investigational product before the week 16 visit, without prior patient-onset of COVID-19, were considered nonresponders at all visits after the relevant event occurred. Any missing or collected data from patients who had patient-onset of COVID-19 as their first prior intercurrent event were handled differently and instead were imputed as missing at random following the start of patient-onset of COVID-19. Data missing before any intercurrent event were handled as nonresponses, unless data were missing due to the COVID-19 pandemic, in which case they were imputed as missing at random.
Sensitivity analysis: To examine the deviations from the missing-at-random assumption in the primary analysis, a tipping-point analysis, which assumed data were not missing at random, was performed.
Subgroup analyses: The following subgroup analyses were of interest to this review: prior cyclosporine A use (yes or no) and disease severity at baseline (IGA of 3 or 4). Interaction between subgroups and treatment effect were tested using a conditional logistic regression model.
Three additional estimands were defined for the primary end point, including:
secondary estimand (“composite”), which assessed the treatment difference in response rates of EASI-75 after 16 weeks achieved without either rescue treatment or treatment discontinuation, regardless of the occurrence of the COVID-19 pandemic
tertiary estimand (“treatment policy”), which assessed the treatment difference in response rate of EASI-75 after 16 weeks between tralokinumab plus TCS and placebo plus TCS regardless of rescue treatment and IMP discontinuation, as if the COVID-19 pandemic did not happen
quaternary estimand (“hypothetical”), which assessed the treatment difference in response rates of EASI-75 after 16 weeks if all patients adhered to the treatment regimen in the sense that they did not discontinue the investigational product permanently, no rescue treatment was prescribed, and as if the COVID-19 pandemic did not happen before week 16.
Statistical Analysis for Secondary Efficacy End Points
ECZTRA 1, 2, 3, and 6
Statistical model: Binary secondary outcomes were conducted as described for the primary end points. In the primary estimand (hypothetical strategy) of continuous secondary outcomes, data collected after permanent discontinuation of the investigational product or after initiation of rescue medication were excluded. Analyses were conducted using a repeated measurements model as follows: change in measurement value = (treatment × week) + (baseline value × week) + region + baseline IGA. For patients for whom no postbaseline data were collected before initiation of rescue medication, the week 2 change was imputed as 0. Unless otherwise stated, all significance tests were 2-sided using a 5% significance level.
Sensitivity analysis: Multiple imputations of missing values were applied based on regression models fitted on observed data from the placebo group. The following analysis of covariance (ANCOVA) model at week 16 was used: change in measurement value = treatment + baseline value + region + baseline IGA. Estimates and standard errors from analyses of multiple imputed datasets were combined using Rubin’s rule.
Two additional estimands were defined for the key continuous secondary end points, including the treatment policy estimand and the composite policy estimand. The treatment policy estimand was as described in the preceding section for the primary end points. In the composite policy estimand, patients who had received rescue medication before the week 16 visit were considered nonresponders by using the worst observation carried forward and multiple imputation of missing values was applied using the ANCOVA model.
ECZTRA 7
Statistical model: Binary secondary outcomes were analyzed as described for the primary end points. In the primary estimand (hypothetical strategy) of continuous secondary outcomes, data collected after permanent discontinuation of the investigational product, after initiation of rescue treatment, or after patient-onset of COVID-19 were excluded. Analyses were conducted using a repeated measurements model as follows: change in measurement value = (treatment × week) + (baseline value × week) + prior cyclosporine A use + country + baseline IGA, assuming the data were missing at random.
Sensitivity analysis: Imputation of missing data at week 16 and week 26 was performed using a pattern-mixture model in which missing data in the tralokinumab plus TCS group as well as the placebo plus TCS group were imputed using data from the placebo plus TCS group and the so-called copy-reference approach. In the ANCOVA model, change from baseline in measurement value = treatment + baseline value + prior cyclosporine A use + country + baseline IGA. The estimates and standard errors from analyses of multiple imputed datasets were combined using Rubin’s rule.
Subgroup analysis: Subgroup analyses for the key secondary end points were the same as described for the primary end point.
Two additional estimands were defined for the key continuous secondary end points using the treatment policy strategy and the COVID-19–modified composite strategy.
Statistical Analysis for Maintenance Efficacy End Points
ECZTRA 1, 2, and 6
Statistical model: Two binary maintenance end points were assessed: an IGA of 0 or 1 at week 52 among patients with an IGA of 0 or 1 at week 16 achieved without rescue medication after initial randomization to tralokinumab, and EASI-75 at week 52 among patients with EASI-75 at week 16 achieved without rescue medication after initial randomization to tralokinumab. The difference in response rates between treatment groups was analyzed using the Cochran-Mantel-Haenszel test stratified by region in the ECZTRA 1 and 2 studies, a binomial model (providing response rates), and corresponding 95% CIs based on the Wilson score method in the ECZTRA 6 study. All patients who had received rescue medication before the week 52 visit, including TCS, and/or those who had been transferred to open-label treatment with tralokinumab, were considered nonresponders. Patients with missing data at week 52 were imputed as nonresponders.
Sensitivity analysis: Data missing at week 52 for patients who did not receive rescue medication did not transfer to open-label treatment, and did not withdraw from the trial due to AE or lack of efficacy were imputed using LOCF instead of nonresponder imputation.
ECZTRA 3
Descriptive statistics were used for end points in the continuation treatment period (an IGA of 0 or 1 and EASI-75 at week 32).
Analysis Populations
Analysis Populations of the ECZTRA 1, 2, 3, 6, and 7 studies are summarized in Table 11.
Results
This section summarizes the results of the pivotal and RCT evidence identified by the sponsor. The results of the tralokinumab 150 mg groups (every 2 weeks and every 4 weeks) in the ECZTRA 6 study are not of interest to this review and are not presented in this report as the dosing does not align with the recommended dosing stated in the product monograph.
Patient Disposition
Patient disposition in the ECZTRA 6 study (adolescents) is presented in Table 12 (initial treatment period) and Table 13 (maintenance treatment period). Patient disposition in the ECZTRA 1, 2, 3, and 7 (adults) studies is presented in Table 14 (initial treatment period), Table 15 (maintenance treatment period in the ECZTRA 1 and 2 studies), and Table 16 (continuous treatment period in the ECZTRA 3 study).
Table 10: Statistical Analysis of Efficacy End Points (Primary Estimand)
End point | Statistical model | Adjustment factors | Censoring and handling of missing data | Sensitivity analyses | ECZTRA 1 and 2 | ECZTRA 3 | ECZTRA 6 | ECZTRA 7 |
|---|---|---|---|---|---|---|---|---|
End points included in the statistical testing hierarchy | ||||||||
Initial treatment period | ||||||||
| CMH test or using multiple Mantel-Haenszel risk differences and associated standard errors to produce combined inferences using Rubin’s rule. | Region and baseline disease severity | Patients who had received rescue medication before the week 16 visit were considered nonresponders Patents with missing data at week 16 and where rescue medication had not been used before week 16 were imputed as nonresponders |
| Yes | Yes | Yes | — |
Prior CsA use, country, and baseline disease severity | Patients who before the week 16 visit received rescue treatment or permanently discontinued IMP, without prior patient-onset of COVID-19, were considered nonresponders Any missing or collected data from patients who had patient-onset of COVID-19 as their first prior intercurrent event were instead imputed as MAR following the start of patient-onset of COVID-19 Data missing before any intercurrent event were handled as nonresponse, unless if data were missing due to the COVID-19 pandemic, in which case it was imputed assuming MAR | Tipping-point analysis | — | — | — | — | ||
Change from baseline at week 16 in:
| Repeated measurements modelb | Baseline IGA, region and treatment-by-week interaction as factors, and interaction between week and baseline value as a covariate | Data collected after permanent discontinuation of tralokinumab or placebo or after initiation of rescue medication were not included in the analysis For patients who did not have any postbaseline data collected before initiation of rescue medication, the week 2 change was imputed as 0 | Multiple imputation using ANCOVA modelc | Yes | Yes | Yes | — |
Change from baseline at weeks 16 and 26 in:
| Repeated measurements modeld | Baseline IGA, country, prior CSA use and treatment-by-week interaction as factors, and interaction between week and baseline value as a covariate | Data collected after permanent discontinuation of IMP, after initiation of rescue treatment, or after patient-onset of COVID-19 were excluded | Imputation of missing data at week 16/week 26 used a pattern-mixture model where missing data in the tralokinumab + TCS group as well as the placebo + TCS group were imputed using data from the placebo + TCS groupe | — | — | — | Yes |
Maintenance (or continuous) treatment period | ||||||||
| CMH test | Region | All patients who before the week 52 visit had received rescue medication, including TCS, and/or been transferred to open-label treatment with tralokinumab were considered nonresponders Patients with missing data at week 52 were imputed as nonresponders | Data missing at week 52 for patients who did not receive rescue medication, did not transfer to open-label, and did not withdraw from the trial due to AE and/or lack of efficacy were imputed using LOCF | Yes | — | — | — |
Descriptive statistics | NA | NA | NA | — | Yes | — | — | |
Binomial model | NA | All patients who used rescue treatment between week 16 and week 52, permanently discontinued treatment, or transferred to open-label treatment were considered nonresponders Missing data for patients who did not attend the week 52 visit and who did not use rescue treatment between week 16 and week 52 were imputed as nonresponders | NA | — | — | Yes | — | |
End points not included in the statistical testing hierarchy | ||||||||
Categorical outcomes:
| Same as IGA of 0 or 1 | NA | Yes | Yes | Yes | Yesf | ||
Continuous outcomes: Change from baseline at week 16 (all trials) and week 26 (ECZTRA 7) in:
| Same as change from baseline at weeks 16 (and 26) in SCORAD and DLQI | NA | Yes | Yes | Yes | Yes | ||
| Repeated measurements model with an unstructured covariance matrixg | NA | Results obtained after initiation of rescue treatment were excluded | NA | — | Yes | — | Yes |
AE = adverse event; ANCOVA = analysis of covariance; CDLQI = Children's Dermatology Life Quality Index; CMH = Cochran-Mantel-Haenszel; CsA = cyclosporine A; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator's Global Assessment; IMP = investigational medicinal product; LOCF = last observation carried forward; MAR = missing at random; NA = not applicable; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring Atopic Dermatitis; SCORAD-50 = 50% reduction in Scoring Atopic Dermatitis; SCORAD-75 = 75% reduction in Scoring Atopic Dermatitis; TCS = topical corticosteroids.
aDLQI in the ECZTRA 1, 2, and 3 studies; CDLQI in the ECZTRA 6 study.
bModel used: change in (SCORAD or DLQI, or CDLQI) = treatment × week + baseline (SCORAD or DLQI, or CDLQI) × week + region + baseline IGA.
cModel used: change from baseline in (SCORAD or DLQI, or CDLQI) = treatment + baseline value + region + baseline IGA.
dModel used: change from baseline in SCORAD or DLQI = (treatment × visit) + (baseline SCORAD or DLQI × visit) + prior CSA use + country (Germany: yes or no) + baseline IGA.
eANCOVA model: change from baseline in SCORAD or DLQI = treatment + baseline SCORAD or DLQI + prior CSA use + country (Germany: yes or no) + baseline IGA.
fExcluding EASI-50, SCORAD-75, SCORAD-50, and reduction in worst daily pruritus NRS of 3 or more points from baseline (not assessed in the study).
gModel used: the amount of TCS used (or a number of days without topic treatment use) = (treatment × week) + region + baseline IGA.
Sources: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 6,13 and 718 and the sponsor’s Summary of Clinical Evidence.14
Table 11: Analysis Populations of ECZTRA 1, 2, 3, 6, and 7
Population | Definition | Application | ECZTRA 1 and 2 | ECZTRA 3 | ECZTRA 6 | ECZTRA 7 |
|---|---|---|---|---|---|---|
Full analysis set | All patients randomized to initial treatment who were exposed to the IMP (tralokinumab or placebo) | Analyzed for efficacy up to week 16 (ECZTRA 1, 2, 3, 6) or week 26 (ECZTRA 7) | Yes | Yes | Yesa | Yes |
Maintenance analysis set | All patients who received tralokinumab in the initial treatment period and who were re-randomized to maintenance treatment; patients who were not exposed to maintenance treatment were not re-randomized and were excluded | Analyzed for efficacy from week 16 to week 52 | Yes | — | Yesa | — |
Continuation treatment analysis set | Patients in the full analysis set who did not withdraw from the trial before or at the week 16 visit and who were exposed to at least 1 dose of the IMP in the continuation treatment period | Analyzed for efficacy from week 16 to 32 | — | Yes | — | — |
Safety analysis set | All patients randomized to initial treatment who were exposed to the IMP (identical to the full analysis set) | Analyzed for safety up to week 16 (ECZTRA 1, 2, and 6) or week 26 (ECZTRA 7) | Yes | — | Yesa | Yes |
All patients randomized to initial treatment who were exposed to the IMP and for whom postbaseline safety data were available | Analyzed for safety up to week 16 | — | Yes | — | — | |
Maintenance safety analysis set | All patients assigned to the maintenance treatment period and received at least 1 dose of maintenance treatment | Analyzed for safety from week 16 to week 52 | Yes | — | Yesa | — |
Continuation treatment safety analysis set | Patients in the full analysis set who did not withdraw from the trial before or at the week 16 visit and who were exposed to at least 1 dose of the IMP in the continuation treatment period | Analyzed for safety from week 16 to 32 | — | Yes | — | — |
IMP = investigational medicinal product.
Note: In addition to the analysis populations, a safety follow-up analysis set (included in all studies), a per-protocol analysis set (included in the ECZTRA 1, 2, 3, 6), and an open-label safety analysis set (included in the ECZTRA 1, 2, and 6 studies) were defined in the studies but they were not summarized as they are not of interest to this review.
aPatients from site 340 (n = 2) and site 341 (n = 7) were excluded due to several issues that involved noncompliance with good clinical practice.
Source: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 6,13 and 718 and the sponsor’s Summary of Clinical Evidence.14
Adolescents
Initial Treatment Period
Of 347 screened patients in the ECZTRA 6 study, 46 (13.3%) were screening failures, mostly due to failure to meet eligibility criteria (10.1%). A total of 301 patients were randomized to the tralokinumab 300 mg every 2 weeks group (n = 101), tralokinumab 150 mg every 2 weeks group (n = 100) (results not presented in this report), and the placebo group (n = 100) in the initial treatment period. Study treatment discontinuation was reported in 3% and 8% of patients receiving tralokinumab and placebo, respectively, with the most common reason being withdrawal by parents or guardians in both groups. The full analysis set and the safety analysis set included 97 patients (96.0%) in the tralokinumab group and 94 patients (94.0%) in the placebo group.
Maintenance Treatment Period
In the ECZTRA 6 study, 27 patients were week 16 tralokinumab responders assigned to maintenance treatment with tralokinumab 300 mg every 2 weeks (n = 13) or tralokinumab 300 mg every 4 weeks (n = 14); 6 patients were week 16 placebo responders who continued to receive placebo as maintenance treatment. One patient discontinued study treatment in each of the tralokinumab groups, and no patient discontinued study treatment in the placebo group.
Adults
Initial Treatment Period
Screening failure rates ranged between 12.9% to 25.0% across the ECZTRA 1, 2, 3, and 7 studies, most due to failure to meet eligibility criteria. The respective numbers of patients randomized to tralokinumab (or tralokinumab plus TCS) and placebo (or placebo plus TCS) were 603 and 199 in the ECZTRA 1 study, 593 and 201 in the ECZTRA 2 study, 253 and 127 in the ECZTRA 3 study, and 140 and 137 in the ECZTRA 7 study. No notable between-group imbalance in study treatment discontinuation was noted across studies (ranging between 4.4% and 10.9%), except in the ECZTRA 2 study, in which more patients in the placebo group (10.9%) discontinued from study treatment than from the tralokinumab group (5.6%). The full analysis set and safety analysis set included all or close to all randomized patients (at least 98.6%) in these studies.
Maintenance (or Continuous) Treatment Period
ECZTRA 1: 185 patients were week 16 tralokinumab responders assigned to maintenance treatment (71 in the tralokinumab every 2 weeks group, 78 in the tralokinumab every 4 weeks group, and 36 in the placebo group) and 29 patients were week 16 placebo responders who continued to receive placebo as maintenance treatment.
ECZTRA 2: 227 patients were week 16 tralokinumab responders assigned to maintenance treatment (91 in the tralokinumab every 2 weeks group, 90 in the tralokinumab every 4 weeks group, and 46 in the placebo group) and 31 patients were week 16 placebo responders who continued to receive placebo as maintenance treatment.
ECZTRA 3: Of the 353 patients assigned to continuation treatment, 138 were week 16 tralokinumab responders (evenly split between tralokinumab every 2 weeks plus TCS and tralokinumab every 4 weeks plus TCS), 95 were week 16 tralokinumab nonresponders who received tralokinumab every 2 weeks plus TCS maintenance treatment, 79 were week 16 placebo nonresponders who received tralokinumab every 2 weeks plus TCS maintenance treatment, and 41 were week 16 placebo responders who continued to receive placebo as maintenance treatment.
Table 12: Patient Disposition for the Initial Treatment Period in ECZTRA 6 (Adolescents)
Patient disposition | Tralokinumab 300 mg q.2.w. | Placebo q.2.w. |
|---|---|---|
Screened, N | 347a | |
Reason for screening failure,a n (%) | ||
Failure to meet eligibility criteria | 35 (10.1) | |
Withdrawal of consent (patient) | 3 (0.9) | |
Withdrawal of consent (parent or guardian) | 5 (1.4) | |
Lost to follow-up | 1 (0.3) | |
Other | 2 (0.6) | |
Randomized, N (%) | 301 (86.7)b | |
101 | 100 | |
Discontinued from study treatment, n (%) | 3 (3.0) | 8 (8.0) |
Reason for study treatment discontinuation, n (%) | ||
Adverse events | 0 (0) | 0 (0.0) |
Lost to follow-up | 0 (0) | 2 (2.0) |
Withdrawal by patient | 0 (0) | 0 (0.0) |
Withdrawal by parent or guardian | 2 (2.0) | 3 (3.0) |
Lack of efficacy | 0 (0) | 1 (1.0) |
Other | 1 (1.0) | 2 (2.0) |
Full analysis set, n (%) | 97 (96.0)c | 94 (94.0)d |
Safety analysis set, n (%) | 97 (96.0) | 94 (94.0) |
Completed week 16 on treatment, n (%) | 94 (93.1) | 86 (86.0) |
q.2.w. = every 2 weeks.
aRefers to all screened patients, including those who were eventually randomized to tralokinumab 150 mg q.2.w. (not summarized in this table).
bRefers to all randomized patients, including those who were randomized to tralokinumab 150 mg q.2.w. (not summarized in this table).
cFour patients were excluded from the full analysis set due to good clinical practice noncompliance issues at the study site (n = 3) and not receiving study intervention (n = 1).
dSix patients were excluded from the full analysis set due to good clinical practice noncompliance issues at the study site (n = 5) and not receiving study intervention (n = 1).
Sources: Clinical Study Reports for ECZTRA 613 and the sponsor’s Summary of Clinical Evidence.14
Table 13: Patient Disposition for Maintenance Treatment Period in ECZTRA 6 (Adolescents)
Patient disposition | Week 16 tralokinumab 300 mg q.2.w. responders | Week 16 placebo responders | |
|---|---|---|---|
Tralokinumab 300 mg q.2.w. (N = 13) | Tralokinumab 300 mg q.4.w. (N = 14) | Placebo q.2.w. (N = 6) | |
Assigned maintenance treatment, N | 13 | 14 | 6 |
Discontinued from study treatment, N (%) | 1 (7.7) | 1 (7.1) | 0 (0.0) |
Reason for discontinuation of study treatment, N (%) | |||
Lost to follow-up | 0 (0.0) | 1 (7.1) | 0 (0.0) |
Patient withdrawal | 1 (7.7) | 0 (0.0) | 0 (0.0) |
Transferred to open-label, N (%) | 5 (38.5) | 4 (28.6) | 2 (33.3) |
With no use of rescue medication after re-randomization | 3 (23.1) | 3 (21.4) | 1 (16.7) |
With use of rescue medication after re-randomization | 2 (15.4) | 1 (7.1) | 1 (16.7) |
Maintenance analysis set, N (%) | 11 (84.6) | 13 (92.9) | 0 (0.0) |
Maintenance safety analysis set, N (%) | 11 (84.6) | 13 (92.9) | 6 (100.0) |
Completed maintenance period, N (%) | 5 (38.5) | 8 (57.1) | 4 (66.7) |
q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
Source: Clinical Study Reports for ECZTRA 613 and the sponsor’s Summary of Clinical Evidence.14
Table 14: Patient Disposition for Initial Treatment Period in ECZTRA 1, 2, 3, and 7 (Adults, Original Review)
Patient disposition | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7 | ||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab q.2.w. | Placebo | Tralokinumab q.2.w. | Placebo | Tralokinumab q.2.w. + TCS | Placebo + TCS | Tralokinumab + TCS | Placebo + TCS | |
Screened, n | 991 | 1,028 | 507 | 318 | ||||
Randomized, n | 603 | 199 | 593 | 201 | 253 | 127 | 140 | 137 |
Discontinued from study treatment, n (%) | 51 (8.5) | 18 (9.0) | 33 (5.6) | 22 (10.9) | 17 (6.7) | 6 (4.7) | ||||| | ||||| |
Reason for study treatment discontinuation before week 16, n (%) | ||||||||
Adverse events | 12 (2.0) | 6 (3.0) | 7 (1.2) | 4 (2.0) | 5 (2.0) | 1 (0.8) | ||||| | ||||| |
Lost to follow-up | 11 (1.8) | 2 (1.0) | 3 (0.5) | 2 (1.0) | 4 (1.6) | 0 (0.0) | ||||| | ||||| |
Withdrawal by patient | 9 (1.5) | 6 (3.0) | 9 (1.5) | 4 (2.0) | 6 (2.4) | 1 (0.8) | ||||| | ||||| |
Lack of efficacy | 6 (1.0) | 2 (1.0) | 5 (0.8) | 4 (2.0) | 1 (0.4) | 1 (0.8) | ||||| | ||||| |
Other | 13 (2.2) | 2 (1.0) | 9 (1.5) | 8 (4.0) | 1 (0.4) | 3 (2.4) | ||||| | ||||| |
COVID-19 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | ||||| | ||||| |
Reason for study treatment discontinuation before week 26, n (%) | ||||||||
Adverse events | NA | NA | NA | NA | NA | NA | 1 (0.7) | 3 (2.2) |
Withdrawal by patient | NA | NA | NA | NA | NA | NA | ||||| | ||||| |
Lack of efficacy | NA | NA | NA | NA | NA | NA | ||||| | ||||| |
Other | NA | NA | NA | NA | NA | NA | ||||| | ||||| |
COVID-19 | NA | NA | NA | NA | NA | NA | ||||| | ||||| |
Full analysis set, n (%) | 601 (99.7) | 197 (99.0) | 591 (99.7) | 201 (100.0) | 252 (99.6) | 126 (99.2) | 138 (98.6) | 137 (100) |
Safety analysis set, n (%) | 602 (99.8) | 196 (98.5) | 592 (99.8) | 200 (99.5) | 252 (99.6) | 126 (99.2) | 138 (98.6) | 137 (100) |
NA = not applicable; q.2.w. = every 2 weeks; TCS = topical corticosteroids.
Sources: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 and 718 and the sponsor’s Summary of Clinical Evidence.14
Table 15: Patient Disposition for Maintenance Treatment Period in ECZTRA 1 and 2 (Adults, Original Review)
Patient disposition | ECZTRA 1 | ECZTRA 2 | ||||||
|---|---|---|---|---|---|---|---|---|
Week 16 tralokinumab responders | Week 16 placebo responders | Week 16 tralokinumab responders | Week 16 placebo responders | |||||
Tralokinumab q.2.w. | Tralokinumab q.4.w. | Placebo | Placebo | Tralokinumab q.2.w. | Tralokinumab q.4.w. | Placebo | Placebo | |
Assigned maintenance treatment, n | 71 | 78 | 36 | 29 | 91 | 90 | 46 | 31 |
Not dosed, n (%) | 3 (4.2) | 2 (2.6) | 1 (2.8) | 0 (0.0) | 0 (0.0) | 1 (1.1) | 0 (0.0) | 0 (0.0) |
Permanently discontinued study treatment, n (%) | 4 (5.6) | 6 (7.7) | 4 (11.1) | 4 (13.8) | 9 (9.9) | 13 (14.4) | 5 (10.9) | 7 (22.6) |
Reason for discontinuation of study treatment, n (%) | ||||||||
Adverse events | 1 (1.4) | 1 (1.3) | 0 (0.0) | 0 (0.0) | 2 (2.2) | 1 (1.1) | 0 (0.0) | 0 (0.0) |
Lost to follow-up | 1 (1.4) | 0 (0.0) | 2 (5.6) | 1 (3.4) | 2 (2.2) | 1 (1.1) | 1 (2.2) | 1 (3.2) |
Withdrawal by patient | 1 (1.4) | 2 (2.6) | 0 (0.0) | 2 (6.9) | 2 (2.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Lack of efficacy | 0 (0.0) | 1 (1.3) | 0 (0.0) | 0 (0.0) | 1 (1.1) | 2 (2.2) | 0 (0.0) | 0 (0.0) |
Other | 1 (1.4) | 2 (2.6) | 2 (5.6) | 1 (3.4) | 2 (2.2) | 9 (10.0) | 4 (8.7) | 6 (19.4) |
Transferred to open-label, n (%) | 23 (32.4) | 19 (24.4) | 10 (27.8) | 10 (34.5) | 29 (31.9) | 27 (30.0) | 26 (56.5) | 8 (25.8) |
With no use of rescue medication after re-randomization | 17 (23.9) | 15 (19.2) | 6 (16.7) | 7 (24.1) | 20 (22.0) | 22 (24.4) | 23 (50.0) | 7 (22.6) |
With use of rescue medication after re-randomization | 6 (8.5) | 4 (5.1) | 4 (11.1) | 3 (10.3) | 9 (9.9) | 5 (5.6) | 3 (6.5) | 1 (3.2) |
Maintenance analysis set, n (%) | 68 (95.8) | 76 (97.4) | 35 (97.2) | NR | 91 (100.0) | 89 (98.9) | 46 (100.0) | NR |
Maintenance safety analysis set, n (%) | 68 (95.8) | 76 (97.4) | 35 (97.2) | 29 (100.0) | 91 (100.0) | 89 (98.9) | 46 (100.0) | 31 (100.0) |
Completed maintenance period, n (%) | 44 (62.0) | 53 (67.9) | 21 (58.3) | 15 (51.7) | 52 (57.1) | 50 (55.6) | 15 (32.6) | 16 (51.6) |
NR = not reported; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
Source: Clinical Study Reports for ECZTRA 115 and ECZTRA 216 and the sponsor’s Summary of Clinical Evidence.14
Table 16: Patient Disposition for Continuation Treatment Period in ECZTRA 3 (Adults, Original Review)
Patient disposition | Week 16 tralokinumab responders | Week 16 tralokinumab nonresponders | Week 16 placebo nonresponders | Week 16 placebo responders | |
|---|---|---|---|---|---|
Tralokinumab q.2.w. + TCS | Tralokinumab q.4.w. + TCS | Tralokinumab q.2.w. + TCS | Tralokinumab q.2.w. + TCS | Placebo + TCS | |
Assigned continuation treatment, n | 69 | 69 | 95 | 79 | 41 |
Permanently discontinued study treatment, n (%) | 1 (1.4) | 3 (4.3) | 7 (7.4) | 7 (8.9) | 3 (7.3) |
Reason for study treatment discontinuation, n (%) | |||||
Adverse events | 0 (0) | 1 (1.4) | 1 (1.1) | 2 (2.5) | 1 (2.4) |
Withdrawal by patient | 0 (0) | 1 (1.4) | 1 (1.1) | 2 (2.5) | 1 (2.4) |
Lack of efficacy | 0 (0) | 0 (0) | 3 (3.2) | 1 (1.3) | 0 (0) |
Other | 1 (1.4) | 1 (1.4) | 2 (2.1) | 2 (2.5) | 1 (2.4) |
Continuation treatment analysis set, n (%) | 69 (100) | 69 (100) | 95 (100) | 79 (100) | 41 (100) |
Continuation treatment safety analysis set, n (%) | 69 (100) | 69 (100) | 95 (100) | 79 (100) | 41 (100) |
Completed continuation period (week 32), n (%) | 68 (98.6) | 65 (94.2) | 87 (91.6) | 72 (91.1) | 38 (92.7) |
q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; TCS = topical corticosteroids.
Source: Clinical Study Report for ECZTRA 317 and the sponsor’s Summary of Clinical Evidence.14
Baseline Characteristics
Baseline patient characteristics of the included studies are summarized in Table 17 (adolescents) and Table 18 (adults). The baseline characteristics outlined in the Table 17 and Table 18 are limited to those most relevant to this review or considered to affect the outcomes or interpretation of the study results.
Adolescents
The study population of ECZTRA 6 (n = 191) had a mean age of 14.6 (SD = 1.7) years. There was about an equal proportion of males (51.5%) and females (48.5%). The majority of patients were white (57.5%). Patients had a mean duration of AD diagnosis of 12.3 years (SD = 3.6), a mean BSA involvement of 50.9% (SD = 23.0%), and a mean EASI score of 31.68 (SD = 13.60). Moderate disease and severe disease (as measured by an IGA) were present in 54.0% and 46.0% of patients, respectively. The proportions of patients who had prior TCS, systemic immunosuppressant, monoclonal antibody, and phototherapy treatment for AD were 100%, 21.1%, 2.4%, and 25.6%, respectively.
The baseline patient characteristics were generally balanced between the tralokinumab 300 mg every 2 weeks group and the placebo group, except that a notably higher proportion of patients in the placebo group had received prior systemic corticosteroid treatment (52.1%), wet wraps (30.9%), and phototherapy (30.9%) compared with the tralokinumab 300 mg every 2 weeks group (34.0%, 21.6%, and 16.5%, respectively).
Adults
In the ECZTRA 1 study (n = 802), 2 (n = 794), 3 (n = 380), and 7 (n = 277), the mean age of the overall study population ranged between 36.5 years (SD = 14.1) and 39.1 years (SD = 15.2). More than half of patients were male (55.0% to 59.6%). At baseline, the mean duration of AD diagnosis ranged from 26.2 years (SD = 13.9) to 28.3 years (SD = 14.7). The mean BSA involvement ranged from 48.1% (SD = 24.2%) to 54.7% (SD = 22.2%). The mean EASI score ranged between 29.35 (SD = 12.25) to 32.95 (SD = 12.54). The proportion of patients with severe disease (as assessed by IGA) was between 46.3% and 50.7%. The proportion of patients who received prior TCS was between 98.0% to 99.6%. In the ECZTRA 3 and 7 studies, in which prior use of monoclonal antibodies was reported, the proportions were 6.3% and 7.6%, respectively. There was generally no notable difference in the baseline patient characteristics across the studies, except that the ECZTRA 7 study enrolled a higher proportion of patients who are white (98.2%), received prior cyclosporine A treatment (74.7%), and received prior phototherapy (58.8%), compared with the ECZTRA 1, 2, and 3 studies (with respective ranges of 55.0% to 59.6%, 31.1% to 36.4%, and 43.7% to 48.1%).
No notable between-group difference in baseline patient characteristics was present in the ECZTRA 1 study. In the ECZTRA 2 study, a higher proportion of patients received prior systemic corticosteroids in the tralokinumab group (69.1%) compared with the placebo group (62.2%). In the ECZTRA 3 study, several imbalances between treatment groups were noted, including a lower proportion of patients who were male (49.4%) and who had received prior systemic corticosteroids (58.5%) and prior methotrexate treatment (11.5%) in the tralokinumab every 2 weeks plus TCS group compared with the placebo plus TCS group (66.1%, 67.7%, and 23.6% respectively). As well, a higher proportion of patients were white (80.2%) and had received prior phototherapy (48.2%) in the tralokinumab every 2 weeks plus TCS group compared with the placebo plus TCS group (66.9% and 41.7%, respectively). In the ECZTRA 7 study, a lower proportion of patients received prior phototherapy in the tralokinumab every 2 weeks plus TCS group (56.4%) compared with the placebo plus TCS group (61.3%)
Table 17: Summary of Baseline Characteristics of ECZTRA 6 — Full Analysis Set (Adolescents)
Characteristic | Tralokinumab 300 mg q.2.w. N = 97 | Placebo N = 94 |
|---|---|---|
Age (years), mean (SD) | 14.6 (1.7) | 14.3 (1.6) |
Male, n (%) | 47 (48.5) | 51 (54.3) |
Race, n (%) | ||
White | 56 (57.7) | 53 (56.4) |
African American or African | 14 (14.4) | 11 (11.7) |
Asian | 20 (20.6) | 23 (24.5) |
American Indian or Alaska Native | 0 (0) | 1 (1.1) |
Native Hawaiian or other Pacific Islander | 2 (2.1) | 2 (2.1) |
Other or missing data | 5 (5.2) | 4 (4.3) |
Duration of AD (years), mean (SD) | 12.1 (3.7) | 12.1 (3.5) |
EASI score, mean (SD) | 31.76 (13.91) | 31.21 (14.47) |
Baseline BSA (%), mean (SD) | 49.6 (23.3) | 51.4 (23.9) |
IGA score, n (%) | ||
Moderate disease | 49 (50.5) | 51 (54.3) |
Severe disease | 48 (49.5) | 43 (45.7) |
SCORAD mean (SD) | 68.31 (13.71) | 67.36 (14.91) |
Adolescent worst pruritus NRS (electronic diary), mean (SD) | 7.83 (1.53) | 7.49 (1.65) |
Mean CDLQI score (SD) | 13.40 (7.26) | 13.34 (6.04) |
Prior AD treatment, n (%) | ||
Topical corticosteroids | 97 (100) | 94 (100) |
Topical calcineurin inhibitors | 60 (61.9) | 56 (59.6) |
Systemic corticosteroids | 33 (34.0) | 49 (52.1) |
Systemic immunosuppressants | 19 (19.6) | 20 (21.3) |
Mycophenolate | 1 (1.0) | 3 (3.2) |
Cyclosporine | 15 (15.5) | 12 (12.8) |
Methotrexate | 6 (6.2) | 10 (10.6) |
Other immunosuppressants | 0 (0) | 1 (1.1) |
Monoclonal antibodies (type not specified) | 2 (2.1) | 3 (3.2) |
Wet wraps | 21 (21.6) | 29 (30.9) |
Phototherapy | 16 (16.5) | 29 (30.9) |
AD = atopic dermatitis; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; q.2.w. = every 2 weeks; NRS = numeric rating scale; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation.
Source: Clinical Study Report for ECTZRA 613 and the sponsor’s Summary of Clinical Evidence.14
Table 18: Baseline Characteristics in ECZTRA 1, 2, 3, and 7 — Full Analysis Set (Adult Population, Original Review)
Characteristic | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7 | ||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab q.2.w. N = 603 | Placebo N = 199 | Tralokinumab q.2.w. N = 593 | Placebo N = 201 | Tralokinumab q.2.w. + TCS N = 253 | Placebo + TCS N = 127 | Tralokinumab q.2.w. + TCS N = 140 | Placebo + TCS N = 137 | |
Mean (SD) age, years | 38.6 (13.7) | 39.4 (15.2) | 37.2 (14.7) | 35.1 (14.0) | 39.8 (15.3) | 37.7 (14.8) | |||| |||||| | |||| |||||| |
Male, n (%) | 351 (58.2) | 123 (61.8) | 359 (60.5) | 114 (56.7) | 125 (49.4) | 84 (66.1) | 82 (58.6) | 83 (60.6) |
Race, n (%) | ||||||||
White | 426 (70.6) | 138 (69.3) | 374 (63.1) | 123 (61.2) | 203 (80.2) | 85 (66.9) | 137 (97.9) | 135 (98.5) |
African American or African | 41 (6.8) | 18 (9) | 43 (7.3) | 17 (8.5) | 23 (9.1) | 12 (9.4) | 0 (0) | 1 (0.7) |
Asian | 120 (19.9) | 40 (20.1) | 154 (26.0) | 52 (25.9) | 17 (6.7) | 24 (18.9) | 0 (0) | 1 (0.7) |
American Indian or Alaska Native | 1 (0.2) | 0 (0.0) | 2 (0.3) | 0 (0.0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Native Hawaiian or other Pacific Islander | 5 (0.8) | 0 (0.0) | 1 (0.2) | 0 (0) | 1 (0.4) | 1 (0.8) | 1 (0.7) | 0 (0) |
Other or missing data | 10 (1.6) | 3 (1.5) | 20 (3.4) | 9 (4.5) | 9 (3.6) | 5 (3.9) | 2 (1.4) | 0 (0) |
Mean duration of atopic dermatitis, years (SD) | 27.9 (14.5) | 29.6 (15.1) | 28.3 (15.9) | 27.5 (14.7) | 28.0 (16.5) | 28.7 (15.0) | |||| |||||| | |||| |||||| |
EASI score mean (SD) | 32.2 (13.7) | 32.9 (13.9) | 32.1 (14.3) | 32.6 (13.9) | 28.8 (12.0) | 30.4 (12.8) | |||| |||||| | |||| |||||| |
Baseline BSA mean (SD) | 52.7 (24.1) | 54.2 (25.6) | 52.6 (25.6) | 53.0 (25.0) | 47.6 (23.3) | 49.0 (25.9) | |||| |||||| | |||| |||||| |
IGA score, n (%) | ||||||||
Moderate disease | 296 (49.1) | 95 (47.7) | 305 (51.4) | 100 (49.8) | 136 (53.8) | 66 (52.0) | 68 (49.3) | 70 (51.1) |
Severe disease | 305 (50.6) | 102 (51.3) | 286 (48.2) | 101 (50.2) | 116 (45.8) | 60 (47.2) | 70 (50.7) | 67 (48.9) |
SCORAD mean (SD) | 70.3 (13.0) | 71.7 (12.5) | 70.0 (13.4) | 70.5 (12.2) | 67.0 (13.3) | 68.9 (13.2) | |||| |||||| | |||| |||||| |
Weekly average worst daily pruritus NRS mean (SD) | 7.7 (1.4) | 7.7 (1.4) | 7.9 (1.5) | 8.0 (1.4) | 7.7 (1.5) | 7.9 (1.5) | ||| ||||| | ||| ||||| |
Mean DLQI score (SD) | 16.8 (7.1) | 17.0 (6.6) | 17.7 (7.1) | 17.8 (7.3) | 17.6 (7.1) | 17.2 (7.2) | |||| ||||| | |||| ||||| |
Patients receiving beforepical corticosteroids, n (%) | 591 (98.0) | 195 (98.0) | 584 (98.5) | 200 (99.5) | 251 (99.2) | 122 (96.1) | 140 (100.0) | 136 (99.3) |
Patients receiving prior systemic corticosteroids, | 357 (59.2) | 119 (59.8) | 410 (69.1) | 125 (62.2) | 148 (58.5) | 86 (67.7) | 98 (70.0) | 91 (66.4) |
Patients receiving prior systemic nonsteroidal immunosuppressants, n (%) | ||||||||
Azathioprine | 39 (6.5) | 7 (3.5) | 72 (12.1) | 25 (12.4) | 13 (5.1) | 12 (9.4) | 18 (12.9) | 18 (13.1) |
Cyclosporine | 227 (37.6) | 65 (32.7) | 204 (34.4) | 65 (32.3) | 75 (29.6) | 43 (33.9) | 105 (75.0) | 102 (74.5) |
Methotrexate | 77 (12.8) | 26 (13.1) | 127 (21.4) | 38 (18.9) | 29 (11.5) | 30 (23.6) | 23 (16.4) | 26 (19.0) |
Mycophenolate | 27 (4.5) | 9 (4.5) | 37 (6.2) | 14 (7.0) | 7 (2.8) | 5 (3.9) | 3 (2.1) | 5 (3.6) |
Other immunosuppressant | 29 (4.8) | 11 (5.5) | 31 (5.2) | 10 (5.0) | 6 (2.4) | 0 (0) | 16 (11.4) | 12 (8.8) |
Prior monoclonal antibody treatment or dupilumab, n (%) | NR | NR | NR | NR | 14 (5.5) | 10 (7.9) | 9 (6.4) | 12 (8.8) |
Prior phototherapy, n (%) | 291 (48.3) | 95 (47.7) | 258 (43.5) | 89 (44.3) | 122 (48.2) | 53 (41.7) | 79 (56.4) | 84 (61.3) |
BSA = body surface area; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = investigator’s global assessment; NR = not reported; NRS = numeric rating scale; SCORAD = Scoring Atopic Dermatitis; q.2.w. = every 2 weeks; SD = standard deviation; TCS = topical corticosteroids.
Sources: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 and 7,18 CADTH Reimbursement Review for Adtralza (2022),65 and the sponsor’s Summary of Clinical Evidence.14
Exposure to Study Treatments
Study treatment exposure in the ECZTRA 6 study (adolescents) is presented in Table 19 (initial treatment period) and Table 20 (maintenance treatment period). Patient disposition in the ECZTRA 1, 2, 3, and 7 (adults) is presented in Table 21 (initial treatment period) and Table 22 (maintenance [or continuous] treatment period).
Adolescents
In the initial treatment period of ECZTRA 6, the majority of patients in both treatment groups received all doses of the study treatment (93.8% in the tralokinumab 300 mg every 2 weeks group and 90.4% in the placebo group). The mean durations of exposure were 0.304 patient-years of exposure (PYE) (SD = 0.030) in the tralokinumab group, and 0.297 PYE (SD = 0.054) in the placebo group.
In the maintenance treatment period of ECZTRA 6, the proportions of patients who received all doses of study treatment were 63.6% in the tralokinumab 300 mg every 2 weeks group, 84.6% in the tralokinumab 300 mg every 4 weeks group, and 50.0% in the placebo group. The mean durations of exposure were 0.510 PYE (SD = 0.232) in the tralokinumab 300 mg every 2 weeks group, 0.522 PYE (SD = 0.244) in the tralokinumab 300 mg every 4 weeks group, and 0.497 PYE (SD = 0.316) in the placebo group.
Adults
More than 70% of patients received all doses of the study treatment in the initial treatment period across the ECZTRA 1, 2, 3, and 7 studies. No notable between-group difference in the mean duration of exposure was noted in the initial treatment period across these studies. In the ECZTRA 1, 2, and 3 studies, the mean duration of exposure in both treatment groups was approximately 0.30 PYE. || |||||| || ||| |||| |||||||| || |||||||| ||| |||| ||||||| ||| || ||| |||||||||||||||| ||| |||| |||||| ||| || ||| ||||||||||| ||||||
In the maintenance (or continuous) treatment period of the ECZTRA 1, 2, and 3 studies, between 76.9% and 94.2% of patients who were week 16 tralokinumab responders received all doses of study treatment. No notable between-group difference in the mean duration of exposure was noted in the ECZTRA 3 study. In the ECZTRA 1 study, the mean duration of exposure was lower in patients who transitioned from tralokinumab to placebo (0.52 PYE; SD = 0.24) compared with the tralokinumab every 2 weeks group (0.56 PYE; SD = 0.22) and tralokinumab every 4 weeks group (0.57 PYE; SD = 0.21). In the ECZTRA 2 study, the mean duration of exposure was also lower in in the patients who transitioned from tralokinumab to placebo (0.44 PYE; SD = 0.22) compared with other treatment groups (tralokinumab every 2 weeks group [0.52 PYE; SD = 0.24]; tralokinumab every 4 weeks group [0.50 PYE; SD = 0.24]).
Table 19: Patient Exposure for ECZTRA 6 in Initial Treatment Period — Safety Analysis Set (Adolescents)
Exposure | Tralokinumab 300 mg q.2.w. (N = 97) | Placebo q.2.w. (N = 94) |
|---|---|---|
Total, PYE | 29.48 | 27.93 |
Duration of exposure, mean PYE (SD) | 0.304 (0.030) | 0.297 (0.054) |
Duration of exposure, median PYE (IQR) | 0.307 (0.304 to 0.309) | 0.307 (0.304 to 0.309) |
Missed dose, n (%) | ||
0 | 91 (93.8) | 85 (90.4) |
1 | 3 (3.1) | 7 (7.4) |
2 | 2 (2.1) | 1 (1.1) |
> 3 | 1 (1.0) | 1 (1.1) |
IQR = interquartile range; PYE = patient-years of exposure; q.2.w. = every 2 weeks; SD = standard deviation.
Sources: Clinical Study Report for ECZTRA 613 and the sponsor’s Summary of Clinical Evidence.14
Table 20: Patient Exposure for ECZTRA 6 in Maintenance Treatment Period — Safety Analysis Set (Adolescents)
Exposure | Week 16 tralokinumab 300 mg q.2.w. responders | Week 16 placebo responders | |
|---|---|---|---|
Tralokinumab 300 mg q.2.w. (N = 11) | Tralokinumab 300 mg q.4.w. (N = 13) | Placebo q.2.w. (N = 6) | |
Total, PYE | 5.61 | 6.78 | 2.98 |
Duration of exposure, mean PYE (SD) | 0.510 (0.232) | 0.522 (0.244) | 0.497 (0.316) |
Duration of exposure, median PYE (IQR) | 0.654 (0.364 to 0.692) | 0.686 (0.376 to 0.691) | 0.693 (0.150 to 0.705) |
Missed dose, n (%) | |||
0 | 7 (63.6) | 11 (84.6) | 3 (50.0) |
1 | 3 (27.3) | 0 (0) | 2 (33.3) |
2 | 0 (0) | 0 (0) | 1 (16.7) |
3 | 0 (0) | 2 (15.4) | 0 (0) |
> 3 | 1 (9.1) | 0 (0) | 0 (0) |
IQR = interquartile range; PYE = patient-years of exposure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SD = standard deviation.
Source: Clinical Study Report for ECZTRA 613 and the sponsor’s Summary of Clinical Evidence.14
Table 21: Patient Exposure from ECZTRA 1, 2, 3, and 7 in Initial Treatment Period — Safety Analysis Set (Adults, Original Review)
Exposure | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7a | ||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab 300 mg q.2.w. (N = 602) | Placebo (N = 196) | Tralokinumab 300 mg q.2.w. (N = 592) | Placebo (N = 200) | Tralokinumab 300 mg q.2.w. + TCS (N = 252) | Placebo + TCS (N = 126) | Tralokinumab 300 mg q.2.w. + TCS (N = 138) | Placebo + TCS (N = 137) | |
Total, PYE | 177.56 | 57.13 | 176.90 | 57.35 | 75.0 | 37.9 | 65.35 | 65.41 |
Duration of exposure, mean PYE (SD) | 0.29 (0.05) | 0.29 (0.06) | 0.30 (0.04) | 0.29 (0.07) | 0.30 (0.05) | 0.30 (0.04) | |||| |||||| | |||| |||||| |
Duration of exposure, median PYE (IQR) | 0.31 (0.30 to 0.31) | 0.31 (0.30 to 0.31) | 0.31 (0.30 to 0.31) | 0.31 (0.30 to 0.31) | 0.307 (0.304 to 0.309) | 0.307 (0.306 to 0.309) | ||||| |||||| || |||||| | ||||| |||||| || |||||| |
Missed dose, n (%) | ||||||||
0 | 540 (89.7) | 171 (87.2) | 549 (92.7) | 167 (83.5) | 236 (93.7) | 113 (89.7) | ||| |||||| | || |||||| |
1 | 45 (7.5) | 18 (9.2) | 31 (5.2) | 17 (8.5) | 12 (4.8) | 9 (7.1) | || |||||| | || |||||| |
2 | 6 (1.0) | 2 (1.0) | 7 (1.2) | 3 (1.5) | 3 (1.2) | 1 (0.8) | || ||||| | || ||||| |
3 | 2 (0.3) | 3 (1.5) | 2 (0.3) | 6 (3.0) | 0 (0) | 3 (2.4) | ||||| | ||||| |
> 3 | 9 (1.5) | 2 (1.0) | 3 (0.5) | 7 (3.5) | 1 (0.4) | 0 (0) | ||||| | ||||| |
IQR = interquartile range; PYE = patient-years of exposure; q.2.w. = every 2 weeks; SD = standard deviation; TCS = topical corticosteroids.
aRefers to the patient exposure in the 26-week treatment period.
Sources: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 and 718 and the sponsor’s Summary of Clinical Evidence.14
Table 22: Patient Exposure from ECZTRA 1, 2, and 3 in Maintenance (Continuous Treatment) Period — Maintenance (or Continuous) Safety Analysis Set (Adults, Original Review)
Exposure | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | |||||
|---|---|---|---|---|---|---|---|---|
Week 16 tralokinumab responders | Week 16 tralokinumab responders | Week 16 tralokinumab responders | ||||||
Tralokinumab 300 mg q.2.w. (N = 68) | Tralokinumab 300 mg q.4.w. (N = 76) | Placebo (N = 35) | Tralokinumab 300 mg q.2.w. (N = 91) | Tralokinumab 300 mg q.4.w. (N = 89) | Placebo (N = 46) | Tralokinumab 300 mg q.2.w. + TCS (N = 69) | Tralokinumab 300 mg q.4.w. + TCS (N = 69) | |
Total, PYE | 37.80 | 42.99 | 18.16 | 46.93 | 44.65 | 20.07 | 21.46 | 20.70 |
Duration of exposure, mean PYE (SD) | 0.56 (0.22) | 0.57 (0.21) | 0.52 (0.24) | 0.52 (0.24) | 0.50 (0.24) | 0.44 (0.22) | 0.311 (0.011) | 0.300 (0.043) |
Duration of exposure, median PYE (IQR) | 0.69 (0.47 to 0.69) | 0.69 (0.48 to 0.69) | 0.69 (0.23 to 0.69) | 0.68 (0.27 to 0.69) | 0.69 (0.27 to 0.69) | 0.44 (0.23 to 0.69) | 0.308 (0.308 to 0.316) | 0.308 (0.306 to 0.313) |
Missed dose, n (%) | ||||||||
0 | 54 (79.4) | 62 (81.6) | 29 (82.9) | 70 (76.9) | 73 (82.0) | 37 (80.4) | 64 (92.8) | 65 (94.2) |
1 | 11 (16.2) | 10 (13.2) | 4 (11.4) | 14 (15.4) | 9 (10.1) | 5 (10.9) | 5 (7.2) | 3 (4.3) |
2 | 2 (2.9) | 2 (2.6) | 1 (2.9) | 2 (2.2) | 3 (3.4) | 2 (4.3) | 0 (0) | 1 (1.4) |
3 | 0 (0) | 1 (1.3) | 0 (0) | 4 (4.4) | 1 (1.1) | 1 (2.2) | 0 (0) | 0 (0) |
> 3 | 1 (1.5) | 1 (1.3) | 1 (2.9) | 1 (1.1) | 3 (3.4) | 1 (2.2) | 0 (0) | 0 (0) |
IQR = interquartile range; PYE = patient-years of exposure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SD = standard deviation; TCS = topical corticosteroids.
Sources: Clinical Study Reports for ECZTRA 1,15 2,16 and 317 and the sponsor’s Summary of Clinical Evidence.14
Rescue Treatment Exposure
Rescue treatment exposure in the ECZTRA 6 study (adolescents) is presented in Table 23. Patient disposition in the ECZTRA 1, 2, 3, and 7 studies (adults) is presented in Table 24 (initial treatment period) and Table 25 (maintenance [or continuous] treatment period).
Adolescents
In the initial treatment period of the ECZTRA 6 study, 29.9% of patients in the tralokinumab 300 mg every 2 weeks group and 56.4% of patients in the placebo group received rescue medication. The most frequently used rescue medication was TCS in both groups (29.9% in the tralokinumab 300 mg every 2 weeks group and 54.3% in the placebo group).
In the maintenance treatment period of the ECZTRA 6 study, 2 patients (18.2%) in the tralokinumab 300 mg every 2 weeks group and 1 patient (7.7%) in the tralokinumab 300 mg every 4 weeks group received rescue medication.
Adults
In the initial treatment period of the ECZTRA 1, 2, 3, and 7 studies, the frequency of rescue medication use was consistently lower in the tralokinumab (or tralokinumab plus TCS) group compared with the placebo (or placebo plus TCS) group (ECZTRA 1: 35.8% versus 46.2%; ECZTRA 2: 22.8% versus 44.3%; ECZTRA 3: 2.8% versus 10.2%; ECZTRA 7: 5.7% versus 13.9%).
In the ECZTRA 1 and 2 studies, among week 16 tralokinumab responders who were re-randomized to receive maintenance treatment, the proportion of patients who received rescue medication was the highest in the placebo group (ECZTRA 1: 41.7%; ECZTRA 2: 19.6%), followed by the tralokinumab 300 mg every 4 weeks group (ECZTRA 1: 33.3%; ECZTRA 2: 17.8%), and the tralokinumab 300 mg every 2 weeks group (ECZTRA 1: 28.2%; ECZTRA 2: 16.5%). In the ECZTRA 3 study, among week 16 tralokinumab responders who were re-randomized to receive continuous treatment, no patients in the tralokinumab 300 mg every 2 weeks plus TCS group and 1 patient (1.4%) in the tralokinumab 300 mg every 4 weeks plus TCS group received rescue medication.
In both the initial and maintenance (or continuous) treatment periods, the most frequently used rescue medication was TCS in all treatment groups across ECZTRA 1, 2, and 3.
Table 23: Rescue Medication Exposure from ECZTRA 6 (Adolescents)
Rescue medication, n (%) | Initial treatment period (full analysis set) | Maintenance treatment period (maintenance full analysis set) | ||
|---|---|---|---|---|
Tralokinumab 300 mg q.2.w. (N = 97) | Placebo (N = 94) | Week 16 tralokinumab 300 mg q.2.w. responders | ||
Tralokinumab 300 mg q.2.w. (N = 11) | Tralokinumab 300 mg q.4.w. (N = 13) | |||
Any rescue medication | 29 (29.9) | 53 (56.4) | 2 (18.2) | 1 (7.7) |
Topical | ||||
Corticosteroids | 29 (29.9) | 51 (54.3) | 2 (18.2) | 1 (7.7) |
Other | 5 (5.2) | 8 (8.5) | 0 (0) | 1 (7.7) |
Systemic | ||||
Corticosteroids | 1 (1.0) | 5 (5.3) | 0 (0) | 0 (0) |
Immunosuppressants | 1 (1.0) | 1 (1.1) | 0 (0) | 0 (0) |
q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
Source: Clinical Study Report for ECZTRA 6.13
Table 24: Rescue Medication Exposure from ECZTRA 1, 2, 3, and 7 in Initiation Treatment Period (Adults, Original Review)
Rescue medication, n (%) | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7 | ||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab 300 mg q.2.w. (N = 603) | Placebo q.2.w. (N = 199) | Tralokinumab q.2.w. (N = 593) | Placebo (N = 201) | Tralokinumab q.2.w. + TCS (N = 253) | Placebo + TCS (N = 127) | Tralokinumab q.2.w. + TCS (N = 140) | Placebo + TCS (N = 137) | |
Any rescue medication | 216 (35.8) | 92 (46.2) | 135 (22.8) | 89 (44.3) | 7 (2.8) | 13 (10.2) | 8 (5.7) | 19 (13.9) |
Topical | ||||||||
Corticosteroids | 203 (33.7) | 90 (45.2) | 115 (19.4) | 74 (36.8) | 5 (2.0) | 10 (7.9) | 6 (4.3) | 16 (11.7) |
Other | 29 (4.8) | 13 (6.5) | 24 (4.0) | 11 (5.5) | 1 (0.4) | 0 (0) | 0 (0) | 0 (0) |
Systemic | ||||||||
Corticosteroids | 18 (3.0) | 13 (6.5) | 9 (1.5) | 18 (9.0) | 3 (1.2) | 3 (2.4) | 3 (2.1) | 8 (5.8) |
Immunosuppressants | 6 (1.0) | 3 (1.5) | 6 (1.0) | 15 (7.5) | 0 (0) | 3 (2.4) | 0 (0) | 2 (1.5) |
Other | 2 (0.3) | 1 (0.5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (0.7) |
q.2.w. = every 2 weeks; TCS = topical corticosteroids.
Note: These analyses included all randomized patients.
Source: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 and 7.18
Table 25: Rescue Medication Exposure from ECZTRA 1, 2, and 3 in Maintenance Treatment Period (Adults, Original Review)
Rescue medication, n (%) | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | |||||
|---|---|---|---|---|---|---|---|---|
Week 16 tralokinumab responders | Week 16 tralokinumab responders | Week 16 tralokinumab responders | ||||||
Tralokinumab 300 mg q.2.w. (N = 71) | Tralokinumab 300 mg q.4.w. (N = 78) | Placebo (N = 36) | Tralokinumab 300 mg q.2.w. (N = 91) | Tralokinumab 300 mg q.4.w. (N = 90) | Placebo (N = 46) | Tralokinumab 300 mg q.2.w. + TCS (n = 69) | Tralokinumab 300 mg q.4.w. + TCS (n = 69) | |
Any rescue medication | 20 (28.2) | 26 (33.3) | 15 (41.7) | 15 (16.5) | 16 (17.8) | 9 (19.6) | 0 (0) | 1 (1.4) |
Topical | ||||||||
Corticosteroids | 20 (28.2) | 26 (33.3) | 15 (41.7) | 13 (14.3) | 13 (14.4) | 9 (19.6) | 0 (0) | 0 (0) |
Other | 3 (4.2) | 3 (3.8) | 4 (11.1) | 3 (3.3) | 3 (3.3) | 2 (4.3) | 0 (0) | 0 (0) |
Systemic | ||||||||
Corticosteroids | 1 (1.4) | 1 (1.3) | 1 (2.8) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (1.4) |
Immunosuppressants | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Other | 0 (0) | 0 (0) | 1 (2.8) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; TCS = topical corticosteroids.
Note: These analyses included all patients who were assigned maintenance (or continuous) treatment.
Source: Clinical Study Reports for ECZTRA 1,15 2,16 and 3.17
Efficacy — Initial Treatment Period
Results in the initial treatment period are summarized in Table 26 (adolescents) and Table 27 (adults).
Investigator’s Global Assessment of 0 or 1
Adolescents
In the ECZTRA 6 study, the difference between the tralokinumab 300 mg every 2 weeks group and the placebo group with respect to the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) at week 16 (coprimary end point) in the primary estimand (composite) was 13.8% (95% CI, 5.3% to 22.3%; P = 0.002), in favour of tralokinumab. Results of the sensitivity analyses were consistent the primary analysis. Subgroup analyses showed that there appeared to be a higher proportion of IGA 0 or 1 responders in the tralokinumab group compared with the placebo group across baseline IGA subgroups (moderate versus severe); however, no statistical testing on the between-group difference was conducted.
Adults
The proportion of patients achieving an IGA score of 0 or 1 at week 16 was a coprimary end point in the ECZTRA 1, 2, and 3 studies. In the primary estimand (composite), the between-group differences were 8.6% (95% CI, 4.1% to 13.1%; P = 0.002) in the ECZTRA 1 study and 11.1% (95% CI, 5.8% to 16.4%; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 12.4% (95% CI, 2.9% to 21.9%; P = 0.015) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS; all were in favour of tralokinumab (or tralokinumab plus TCS). Results of the sensitivity analyses were consistent with those of the primary analysis in all studies. Subgroup analyses showed that there appeared to be a higher proportion of IGA of 0 or 1 responders in the tralokinumab group compared with the placebo group across baseline IGA subgroups; however, no statistical testing on the between-group difference was conducted.
In the ECZTRA 7 study, the proportion of patients achieving an IGA score of 0 or 1 at week 16 and week 26 were secondary end points. || ||| ||||||| |||||||| ||||||||| |||||||| ||||||||||| ||| ||||||||||||| |||||||||| ||| ||||| |||| ||| |||| || |||||| || |||| ||| ||| ||||| |||| ||| |||| || |||||| || |||| ||| |||| ||||||||| |||||||||||| ||||||| |||| |||||||||||| ||||||| || ||| ||||||||| |||||||| ||||||||||||||| |||||||||| |||| ||| ||||||| |||||||| || ||||| || ||| ||| Neither end point was tested for superiority due to prior failure in the testing hierarchy (i.e., reduction of worst daily pruritus NRS of at least 4 points from baseline).
Eczema Area and Severity Index
Adolescents
EASI-75: In the ECZTRA 6 study, the difference between the tralokinumab 300 mg every 2 weeks group and the placebo group with respect to EASI-75 (i.e., the proportion of patients with EASI-75, a coprimary end point) was 22.0% (95% CI, 12.0% to 32.0%; P < 0.001) in the primary estimand (composite), in favour of tralokinumab. Results of the sensitivity analyses were consistent with those of the primary analysis. Subgroup analyses showed results in favour of tralokinumab across baseline IGA subgroups; however, no statistical testing on the between-group difference was conducted.
EASI-90, EASI-50, and Change From Baseline in EASI: In the ECZTRA 6 study, the difference between tralokinumab 300 mg every 2 weeks and placebo with respect to adjusted mean change in EASI from baseline at week 16 (secondary end point) was −9.4 (95% CI, −13.5 to −5.3) in the hypothetical estimand and the between-group differences in EASI-90 and EASI-50 (secondary end points) were 13.7% (95% CI, 5.2% to 22.2%) and 38.5% (95% CI, 26.8% to 50.2%), respectively, in the composite estimand. These secondary end points were not adjusted for multiplicity.
Adults
EASI-75: An EASI-75 response at week 16 was a coprimary end point in the ECZTRA 1, 2, and 3 studies. In the primary estimand (composite), the between-group differences were 12.1% (95% CI, 6.5% to 17.7%; P < 0.001) in the ECZTRA 1 study and 21.6% (95% CI, 15.8% to 27.3%; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 20.2% (95% CI, 9.8% to 30.6%; P < 0.001) in the ECZTRA 3 study when comparing between tralokinumab every 2 weeks plus TCS with placebo plus TCS; all were in favour of tralokinumab (or tralokinumab plus TCS). Results of the sensitivity analyses were consistent the primary analysis in all studies. Subgroup analyses showed results in favour of tralokinumab across baseline IGA subgroups (moderate versus severe); however, no statistical testing on the between-group difference was conducted.
In the ECZTRA 7 study, EASI-75 at week 16 was the primary end point and EASI-75 at week 26 was a secondary end point. In the primary estimand (COVID-19–modified composite), the between-group difference at week 16 was 14.1% (95% CI, 2.5% to 25.7%; P = 0.018) in favour of tralokinumab every 2 weeks plus TCS. The between-group difference at week 26 was 14.1% (95% CI, 2.9% to 25.35%), for which no superiority testing was conducted due to prior failure in the testing hierarchy. Results of the secondary estimand (composite) were consistent with those of the primary estimand at weeks 16 and 26. |||||||| |||||||| |||||||||||| |||||||||| || |||||| |||||||||| || |||| || ||| |||| || || ||| |||||||||||| ||||||| ||||| |||||||| |||| ||| ||||||||||| ||||| |||||| ||||||||| || |||||||| ||| |||||||| ||| ||||| ||| |||||||||||| || ||||||| |||||||| ||||| || ||||||||||| |||||| ||| |||||| || |||| || ||| |||||||| ||||||| |||||||||||||| |||||||||| ||||||||| |||||||||| || |||||||| |||| |||||||||| || ||||| |||||||||
EASI-90 and EASI-50: The secondary end points in the ECZTRA 1, 2, and 3 studies were EASI-90 and EASI-50 at week 16. The between-group differences (COVID-19–modified composite) in EASI-90 at week 16 were 10.3% (95% CI, 6.4% to 14.1%) in the ECZTRA 1 study and 12.7% (95% CI, 8.3% to 17.0%) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 11.4% (95% CI, 2.1% to 20.7%) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. The between-group differences in EASI-50 at week 16 were 20.1% (95% CI, 13.3% to 26.8%) in the ECZTRA 1 study and 29.3% (95% CI, 22.5% to 36.1%) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 21.3% (95% CI, 11.3% to 31.3%) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. Neither secondary end point was adjusted for multiplicity.
In the ECZTRA 7 study, EASI-90 at weeks 16 and 26 were exploratory end points. The between-group differences (COVID-19–modified composite) in EASI 90 were 12.3% (95% CI, 1.1% to 23.6%) at week 16 and 12.9% (95% CI, 1.4% to 24.4%) at week 26 when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. Neither end point was adjusted for multiplicity. EASI-50 was not assessed in the study.
Change From Baseline in EASI: Change from baseline in EASI at week 16 was a secondary end point in the ECTRA 1, 2, and 3 studies and was not adjusted for multiplicity. The between-group differences (hypothetical estimand) were −6.4 (95% CI, −8.8 to −4.1) in the ECZTRA 1 study and −9.9 (95% CI, −12.2 to −7.5) in the ECZTRA 2 study when comparing tralokinumab with placebo, and −5.4 (95% CI, −7.7 to −3.1) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS.
In the ECZTRA 7 study, changes from baseline in EASI at week 16 and week 26 were secondary end points and were not adjusted for multiplicity. The between-group differences (hypothetical estimand) were −3.9 (95% CI, −6.3 to −1.6) at week 16 and −3.5 (95% CI, −5.7 to −1.3) at week 26 when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS.
Scoring Atopic Dermatitis
Adolescents
In the ECZTRA 6 study, the between-group difference between the tralokinumab 300 mg every 2 weeks group and the placebo group with respect to adjusted mean change from baseline in SCORAD at week 16 (key secondary end point) was −19.7 (95% CI, −27.1 to −12.2; P < 0.001) in the primary estimand (hypothetical) in favour of tralokinumab. Results of the sensitivity analysis were consistent with those of the primary analysis. Results of the secondary (treatment policy) and tertiary (composite) estimands were consistent with those of the primary estimand.
Adults
Change from baseline in SCORAD at week 16 was a key secondary end point in the ECZTRA 1, 2, and 3 studies. In the primary estimand (hypothetical), the between-group differences were −10.4% (95% CI, −14.4% to −6.5%; P < 0.001) ECZTRA 1 and −14.0% (95% CI, −18.0% to −10.1%; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and −10.8% (95% CI, −15.2% to −6.5%; P < 0.001) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS; all were in favour of tralokinumab (or tralokinumab plus TCS). The results of the sensitivity analysis were consistent with those of the primary analysis. Results of the secondary (treatment policy) and tertiary (composite) estimands were consistent with those of the primary estimand.
Changes from baseline in SCORAD at weeks 16 and 26 were secondary end points in the ECZTRA 7 study. In the primary estimand (hypothetical), the adjusted mean between-group difference was −8.6 (95% CI, −13.0 to −4.2) at week 16 and −8.9 (95% CI, −13.2 to −4.6) at week 26 when comparing tralokinumab every 2 weeks plus TCS with placebo plus TCS. The results of the sensitivity analysis were consistent with those of the primary analysis at weeks 16 and 26. Results of the secondary (treatment policy) and tertiary (COVID-19–modified composite) estimands were consistent with those of the primary estimand at weeks 16 and 26. Neither end point was tested for superiority due to prior failure of the testing hierarchy.
Worst Daily Pruritis NRS and Adolescent Worst Pruritis NRS
Adolescents
Reduction of at Least 4 Points in Adolescent Worst Pruritus NRS: In the ECZTRA 6 study, the between-group difference between the tralokinumab 300 mg every 2 weeks group and the placebo group with respect to the proportion of patients with at least 4 points of reduction in adolescent worst pruritus NRS at week 16 (a key secondary end point) was 21.7% (95% CI, 12.3% to 31.1%; P < 0.001) in the primary estimand (composite) in favour of tralokinumab. Results of the sensitivity analyses were consistent with those of the primary analysis.
Change From Baseline in Adolescent Worst Pruritus NRS and Proportion of Patients With a Reduction of at Least 3 Points From Baseline in Adolescent Worst Pruritus NRS: In the ECZTRA 6 study, the between-group difference (hypothetical estimand) between the tralokinumab 300 mg every 2 weeks group and the placebo group with respect to adjusted mean change from baseline in adolescent worst pruritus NRS at week 16 (a secondary end point) was −1.5 (95% CI, −2.4 to −0.6). The between-group difference with respect to the proportion of patients with a reduction of at least 3 points from baseline in adolescent worst pruritus NRS at week 16 (secondary end point) was 20.3% (95% CI, 9.7% to 31.0%). Neither secondary end point was adjusted for multiplicity.
Adults
Reduction of at Least 4 Points in Worst Pruritus NRS: The proportion of patients with a reduction of at least 4 points in worst pruritus NRS at week 16 was a key secondary end point of the ECZTRA 1, 2, and 3 studies. In the primary estimand (composite), the between-group differences were 9.7% (95% CI, 4.4% to 15.0%; P = 0.002) in the ECZTRA 1 study and 15.6% (95% CI, 10.3% to 20.9%; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 11.3% (95% CI, 0.9% to 21.6%; P = 0.037) in the ECZTRA 3 study when comparing between tralokinumab every 2 weeks plus TCS against placebo plus TCS; all were in favour of tralokinumab (or tralokinumab plus TCS). Results of the sensitivity analyses were consistent with those of the primary analysis.
In the ECZTRA 7 study, the proportion of patients with reduction of at least 4 points in worst pruritus NRS at week 16 and at week 26 were secondary end points. In the primary estimand (COVID-19–modified composite), the between-group difference at week 16 was 9.7% (95% CI, −2.0% to 21.4%; P = 0.106) at week 16, favouring neither tralokinumab every 2 weeks plus TCS nor placebo plus TCS. Results of the secondary (composite) estimand were consistent with those of the primary estimand. The between-group difference at week 26 was 7.3% (95% CI, −4.6% to 19.2%) in the primary estimand but was not tested for superiority due to prior failure in the testing hierarchy.
Reduction of at Least 3 Points in Worst Pruritus NRS: The proportion of patients with a reduction of at least 3 points in worst pruritus NRS at week 16 was a secondary end point in the ECZTRA 1, 2, and 3 studies and was not adjusted for multiplicity. The between-group differences (composite estimand) were 15.2% (95% CI, 9.2% to 21.3%) in the ECZTRA 1 study and 20.1% (95% CI, 13.9% to 26.2%) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 19.3% (95% CI, 8.8% to 29.9%) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. This end point was not assessed in the ECZTRA 7 study.
Change From Baseline in Worst Pruritus NRS: Change from baseline in worst daily pruritis NRS at week 16 was a secondary end point in the ECZTRA 1, 2, and 3 studies and was not adjusted for multiplicity. The between-group differences (hypothetical estimand) were −0.9 (95% CI, −1.4 to −0.4) in the ECZTRA 1 study and −1.3 (95% CI, −1.7 to −0.8) in the ECZTRA 2 study when comparing tralokinumab with placebo, and −1.2 (95% CI, −1.7 to −0.7) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS and placebo plus TCS.
In the ECZTRA 7 study, changes from baseline in worst daily pruritis NRS at week 16 and week 26 were exploratory end points and were not adjusted for multiplicity. The between-group differences (hypothetical estimand) were −0.9 (95% CI, −1.4 to −0.4) at week 16 and −0.9 (95% CI, −1.4 to −0.3) at week 26 when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS.
Eczema-Sleep–Related NRS
Adolescents
In the ECZTRA 6 study, the change from baseline in eczema-related sleep NRS was an exploratory end point and was not adjusted for multiplicity; the between-group difference between the tralokinumab 300 mg every 2 weeks group and the placebo group was −1.3 (95% CI, −2.2 to −0.4) in the hypothetical estimand.
Adults
Change from baseline in eczema-related sleep NRS at week 16 was an exploratory end point in the ECZTRA 1, 2, and 3 studies and was not adjusted for multiplicity. The between-group differences (hypothetical estimand) were −0.7 (95% CI, −1.2 to −0.2) in the ECZTRA 1 study and −1.4 (95% CI, −1.9 to −0.9) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and −1.3 (95% CI, −1.8 to −0.8) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS.
In the ECZTRA 7 study, changes from baseline in eczema-related sleep NRS at weeks 16 and 26 were exploratory end points and were not adjusted for multiplicity. The between-group differences (hypothetical estimand) were −0.8 (95% CI, −1.3 to −0.2) at week 16 and −0.6 (95% CI, −1.1 to −0.00) at week 26 when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS.
Patient-Oriented Eczema Measure
Adolescents
In the ECZTRA 6 study, the between-group difference between tralokinumab 300 mg every 2 weeks group and the placebo group in adjusted mean change from baseline in POEM at week 16 (secondary end point) was −6.0 (95% CI, −8.4 to −3.6) in the hypothetical estimand. This end point was not adjusted for multiplicity.
Adults
Change from baseline in POEM score was an exploratory end point in the ECZTRA 1, 2, 3, and 7 studies and was not adjusted for multiplicity. The between-group differences (hypothetical estimand) at week 16 were −4.6 (95% CI, −6.0 to −3.1) in the ECZTRA 1 study and −5.1 (95% CI, −6.5 to −3.6) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, −4.0 (95% CI, −5.6 to −2.4) in the ECZTRA 3 study, and −3.4 (95% CI, −5.0 to −1.8) in the ECZTRA 7 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. In the ECZTRA 7 study, the between-group difference (hypothetical estimand) at week 26 was −3.6 (95% CI, −5.3 to −1.9) when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS.
Dermatology Life Quality Index and Children’s Dermatology Life Quality Index
Adolescents
Change From Baseline in CDLQI: In the ECZTRA 6 study, the between-group difference between the tralokinumab 300 mg every 2 weeks group and the placebo group with respect to the adjusted mean change from baseline in CDLQI at week 16 (key secondary end point) was −2.6 (95% CI, −4.5 to −0.7; P = 0.007) in the primary estimand (hypothetical), in favour of tralokinumab. The results of the sensitivity analysis were consistent with those of the primary analysis. Results of the secondary (treatment policy) and tertiary estimands (composite) were consistent with those of the primary estimands.
Reduction of at Least 6 Points in CDLQI: In the ECZTRA 6 study, the proportion of patients with a reduction of at least 6 points in CDLQI from baseline at week 16 was a secondary end point and was not adjusted for multiplicity; the between-group difference between the tralokinumab 300 mg every 2 weeks group and the placebo group was 23.9% (95% CI, 11.0% to 36.7%) in the composite estimand.
Adults
Change From Baseline in DLQI: Change from baseline in DLQI at week 16 was a key secondary end point in the ECZTRA 1, 2, and 3 studies. In the primary estimand (hypothetical), the between-group differences were −2.1 (95% CI, −3.4 to −0.8; P = 0.002) in the ECZTRA 1 study and −3.9 (−5.2 to −2.6; P < 0.001) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and −2.9 (95% CI, −4.3 to −1.6; P < 0.001) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS; all favouring tralokinumab (or tralokinumab plus TCS). In all studies, the results of the sensitivity analysis were consistent with those of the primary analysis. Results of the composite estimand were consistent with those of the primary estimand.
In the ECZTRA 7 study, changes from baseline in DLQI at weeks 16 and 26 were key secondary end points. In the primary estimand (hypothetical), the between-group difference at week 16 was −1.5 (95% CI, −2.6 to −0.4). The results of the sensitivity analysis were consistent with those of the primary analysis. Results of the secondary (treatment policy) and tertiary (COVID-19–modified composite) estimands were not consistent with the primary estimand and the evidence was insufficient to show a difference between the treatment groups. At week 26, the between-group difference was −1.6 (95% CI, −2.7 to −0.5). The results of the sensitivity analysis were consistent with those of the primary analysis. Results of composite estimand were consistent with those of the primary estimand. Neither end point was tested for superiority due to prior failure of the testing hierarchy.
Reduction of at Least 4 Points in DLQI: The proportion of patients with a reduction of at least 4 points in DLQI from baseline at week 16 was a secondary end point in the ECZTRA 1, 2, and 3 studies and was not adjusted for multiplicity. The between-group differences (composite estimand) were 13.0% (95% CI, 5.4% to 20.5%) in the ECZTRA 1 study and 28.9% (95% CI, 21.4% to 36.3%) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and 17.6% (95% CI, 8.0% to 27.1%) in the ECZTRA 3 study when comparing tralokinumab every 2 weeks against TCS with placebo plus TCS.
In the ECZTRA 7 study, the proportion of patients with a reduction of at least 4 points in DLQI from baseline at week 16 and week 26 were exploratory end points and were not adjusted for multiplicity. ||| ||||||||||||| |||||||||| ||||||||| |||||||| ||||||||| ||||||||| ||| |||| |||| ||| ||||| || |||||| || |||| ||| ||| |||| |||| ||| ||||| || |||||| || |||| ||| |||| ||||||||| ||||||| |||||||||||| ||||||| |||| ||||||||||||
Hospital Anxiety and Depression Scale
Adolescents
In the ECZTRA 6 study, anxiety and depression scores were assessed as exploratory end points and were not adjusted for multiplicity. The between-group differences (hypothetical estimand) between the tralokinumab 300 mg every 2 weeks group and the placebo group with respect to change from baseline in HADS anxiety and depression scores were −1.2 (95% CI, −2.4 to −0.1) and −1.0 (95% CI, −2.2 to 0.2), respectively. The between-group difference (composite estimand) in the proportion of patients with either a HADS anxiety or depression score of less than 8 (n = 191) at week 16 was 7.1% (95% CI, −12.4% to 26.6%).
Adults
The HADS scores were assessed as exploratory end points and were not adjusted for multiplicity in the ECZTRA 1, 2, 3, and 7 studies.
Change From Baseline in HADS Anxiety Score: The between-group differences (hypothetical estimand) in change from baseline in HADS anxiety score at week 16 were −0.2 (95% CI, −0.8 to 0.4) in the ECZTRA 1 study and −0.7 (95% CI, −1.3 to 0.1) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and −1.3 (95% CI, −2.1 to −0.6) in the ECZTRA 3 study and 0.16 (95% CI, −0.56 to 0.87) in the ECZTRA 7 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. || |||||| || ||| ||||||||||||| |||||||||| || |||||| |||| |||||||| || |||| ||||||| ||||| || |||| || ||| |||| |||| ||| ||||| || ||||| |||| ||||||||| ||||||| |||||||||||| ||||||| |||| ||||||||||||
Change From Baseline in HADS Depression Score: The between-group differences (hypothetical estimand) in change from baseline in HADS depression score at week 16 were −0.3 (95% CI, −0.9 to 0.4) in the ECZTRA 1 study and −1.1 (95% CI, −1.8 to −0.5) in the ECZTRA 2 study when comparing tralokinumab every 2 weeks with placebo, and −0.8 (95% CI, −1.5 to −0.2) in the ECZTRA 3 study and 0.02 (95% CI, −0.67 to 0.71) in the ECZTRA 7 study when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. || |||||| || ||| ||||||||||||| |||||||||| || |||| || ||| ||||| |||| ||| ||||| || ||||| |||| ||||||||| ||||||| |||||||||||| ||||||| |||| ||||||||||||
HADS Anxiety or Depression Score Less Than 8: The between-group differences (composite estimand in the ECZTRA 1, 2, and 3 studies; COVID-19–modified composite estimand in the ECZTRA 7 study) in the proportion of patients with either a HADS anxiety or depression score of less than 8 at week 16 were 3.0% (95% CI, −5.8% to 11.8%) in the ECZTRA 1 study (n = 798) and 21.2% (95% CI, 11.8% to 30.5%) in the ECZTRA 2 study (n = 792) when comparing tralokinumab every 2 weeks with placebo, and 24.8% (95% CI, 9.3% to 40.4%) in the ECZTRA 3 study (n = 378) and 6.6% (95% CI, −12.4% to 25.5%) in the ECZTRA 7 study (n = 109), when comparing tralokinumab every 2 weeks plus TCS against placebo plus TCS. || |||||| || ||| ||||||||||||| |||||||||| || |||| || ||| |||| |||| ||| ||||| || |||||| |||| ||||||||| ||||||| |||||||||||| ||||||| |||| ||||||||||| ||||||
Use of Topical Corticosteroids
Adolescents
This outcome was not assessed in the ECZTRA 6 study.
Adults
The amount of TCS used was a secondary end point in the ECZTRA 3 study and an exploratory end point in the ECZTRA 7 study and was not adjusted for multiplicity. The outcome was not measured in the ECZTRA 1 and 2 studies. In the ECZTRA 3 study, the between-group difference in the amount of TCS used at week 15 to 16 was −8.6 g (95% CI, −14.1 to −3.2). In the ECZTRA 7 study, the between-group differences in the amount of TCS used were −11.5 g (95% CI, −22.9 to −0.2) at week 15 to 16, and −11.7 g (95% CI, −23.9 to 0.4) at week 25 to 26.
Number of Days Without Topical Treatment
Adolescents
This outcome was not assessed in the ECZTRA 6 study.
Adults
The number of days without topical treatment was a secondary end point in the ECZTRA 3 study and an exploratory end point in the ECZTRA 7 study and was not adjusted for multiplicity. The between-group differences at week 16 were 0.5 days (95% CI, −0.2 to 1.1) in the ECZTRA 3 study and 1.9 days (95% CI, 1.2 to 2.5) in the ECZTRA 7 study.
Table 26: Key Efficacy Results from ECZTRA 6 at Week 16 — Full Analysis Set (Adolescents)
Outcomes at week 16 | Tralokinumab 300 mg q.2.w. (N = 97) | Placebo q.2.w. (N = 94) |
|---|---|---|
IGA score of 0 or 1 | ||
n/N (%) | 17/97 (17.5) | 4/94 (4.3) |
Difference vs. placebo, % (95% CI)a | 13.8 (5.3 to 22.3, P = 0.002) | |
EASI | ||
Number of patients contributing to the analysis of change from baseline in EASI score | 66 | 35 |
Baseline EASI, mean (SD) | 31.8 (13.9) | 31.2 (14.5) |
Change from baseline, adjusted mean change (SE) | −18.1 (1.3) | −8.7 (1.6) |
Difference vs. placebo, (95% CI)b,c | −9.4 (−13.5 to −5.3; P < 0.001) | |
EASI-75, n/N (%) | 27/97 (27.8) | 6/94 (6.4) |
Difference vs. placebo, % (95% CI)a | 22.0 (12.0 to 32.0; P < 0.001) | |
EASI-90, n/N (%; 95% CI) | 17/97 (17.5; 11.2 to 26.3) | 4/94 (4.3; 1.7 to 10.4) |
Difference vs. placebo, % (95% CI)a,c | 13.7 (5.2 to 22.2; P = 0.002) | |
EASI-50, n/N (%; 95% CI) | 50/97 (51.5; 41.7 to 61.2) | 13/94 (13.8; 8.3 to 22.2) |
Difference vs. placebo, % (95% CI)a,c | 38.5 (26.8 to 50.2; P < 0.001) | |
SCORAD | ||
Number of patients contributing to the analysis of change from baseline in SCORAD | 66 | 35 |
Baseline SCORAD, mean (SD) | 68.3 (13.7) | 67.4 (14.9) |
Change from baseline, adjusted mean change (SE) | −29.1 (2.4) | −9.5 (3.0) |
Difference vs. placebo, (95% CI)b | −19.7 (−27.1 to −12.2, P < 0.001) | |
Adolescent worst pruritus NRS (weekly average) | ||
Number of patients that contributed to the analysis of change from baseline in adolescent worst pruritis NRS (weekly average) | 62 | 31 |
Baseline adolescent worst pruritus NRS, mean (SD) | 7.8 (1.5) | 7.5 (1.7) |
Change from baseline, adjusted mean change (SE) | −3.0 (0.3) | −1.5 (0.3) |
Difference vs. placebo, (95% CI)b,c | −1.5 (−2.4 to −0.6; P < 0.001) | |
Reduction of ≥ 4 from baseline, n/N (%) | 24/96 (25.0) | 3/90 (3.3) |
Difference vs. placebo, % (95% CI)a | 21.7 (12.3 to 31.1; P < 0.001) | |
Reduction of ≥ 3 from baseline, n/N, (%; 95% CI) | 28/96 (29.2; 21.0 to 38.9) | 8/91 (8.8; 4.5 to 16.4) |
Difference vs. placebo, % (95% CI)a,c | 20.3 (9.7 to 31.0; P < 0.001) | |
Eczema-related sleep NRS | ||
Number of patients contributing to the analysis | 62 | 31 |
Baseline eczema-related sleep NRS, mean (SD) | 6.8 (2.1) | 6.8 (2.1) |
Change from baseline, adjusted mean change (SE) | −3.1 (0.3) | −1.8 (0.4) |
Difference vs. placebo, % (95% CI)b,c | −1.3 (−2.2 to −0.4; P = 0.005) | |
POEM | ||
Number of patients contributing to the analysis of change from baseline in POEM | 65 | 33 |
Baseline POEM, mean (SD) | 20.1 (5.8) | 20.8 (5.6) |
Change from baseline, adjusted mean change (SE) | −8.4 (0.8) | −2.4 (1.0) |
Difference vs. placebo, adjusted mean change (95% CI)b,c | −6.0 (−8.4 to −3.6; P < 0.001) | |
CDLQI | ||
Number of patients contributing to the analysis of change from baseline in CDLQI score | 84 | 89 |
Baseline CDLQI, mean (SD) | 13.4 (7.3) | 13.3 (6.0) |
Change from baseline, adjusted mean change (SE) | −6.7 (0.6) | −4.1 (0.7) |
Difference vs. placebo, (95% CI)b | −2.6 (−4.5 to −0.7; P = 0.007) | |
Reduction ≥ 6 from baseline, n/N (%; 95% CI) | 32/81 (39.5; 29.6 to 50.4) | 13/82 (15.9; 9.5 to 25.3) |
Difference vs. placebo, % (95% CI)a,c | 23.9 (11.0 to 36.7; P < 0.001) | |
HADS | ||
Number of patients contributing to the analysis | 65 | 33 |
Baseline HADS anxiety score | 6.7 (4.5) | 6.9 (4.5) |
Change from baseline in HADS anxiety score, adjusted mean change (SE) | −2.7 (0.3) | −1.5 (0.4) |
Difference vs. placebo, (95% CI)b,c | −1.2 (−2.4 to −0.1; P = 0.028) | |
Baseline HADS depression score | 4.8 (3.9) | 4.4 (3.6) |
Change from baseline in HADS depression score, adjusted mean change (SE) | −1.7 (0.4) | −0.6 (0.5) |
Difference vs. placebo, (95% CI)b,c | −1.0 (−2.2 to 0.2; P = 0.087) | |
HADS anxiety and HADS depression scores < 8 at week 16, n/N (%; 95% CI)a | 13/43 (30.2; 18.6 to 45.1) | 9/39 (23.1; 12.6 to 38.3) |
Difference vs. placebo, % (95% CI)a,c | 7.1 (−12.4 to 26.6; P = 0.48) | |
CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; EASI = Eczema Area and Severity Index; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; EASI-90 = reduction of at least 90% in Eczema Area and Severity Index score from baseline; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; SE = standard error.
aThe analysis was conducted using a Cochran-Mantel-Haenszel test stratified by baseline IGA and region based on the composite estimand.
bThe analysis was conducted using the repeated measurements model, with baseline IGA, region, and treatment-by-week interaction as factors and interaction between week and baseline value as a covariate, based on the hypothetical estimand.
cThe end point was not adjusted for multiplicity and was at an increased risk of type I error (false-positive result).
Sources: Clinical Study Report for ECZTRA 613 and the sponsor’s Summary of Clinical Evidence.14
Table 27: Key Efficacy Results From ECZTRA 1, 2, 3, and 7 at Week 16 or 26 — Full Analysis Set (Adults, Original Review)
Outcomes | ECZTRA 1 (follow-up at 16 weeks) | ECZTRA 2 (follow-up at 16 weeks) | ECZTRA 3 (follow-up at 16 weeks) | ECZTRA 7 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Follow-up at 16 weeks | Follow-up at 26 weeks | ||||||||||||
Tralokinumab q.2.w. N = 601 | Placebo N = 197 | Tralokinumab q.2.w. N = 591 | Placebo N = 201 | Tralokinumab q.2.w. + TCS N = 252 | Placebo + TCS N = 126 | Tralokinumab q.2.w. + TCS N = 138 | Placebo + TCS N = 137 | Tralokinumab q.2.w. + TCS N = 138 | Placebo + TCS N = 137 | ||||
IGA score of 0 or 1 | |||||||||||||
n/N (%) | 95/601 (15.8) | 14/197 (7.1) | 131/591 (22.2) | 22/201 (10.9) | 98/252 (38.9) | 33/126 (26.2) | |||||| |||||| | |||||| |||||| | |||| |||||| | |||| |||||| | |||
Difference, % (95% CI) | 8.6 (4.1 to 13.1; P = 0.002)a | 11.1 (5.8 to 16.4; P < 0.001)a | 12.4 (2.9 to 21.9; P = 0.015)a | |||| |||| || ||||||||||| | |||| |||| || ||||||||||| | ||||||||
EASI | |||||||||||||
n | 353 | 96 | 430 | 98 | 229 | 108 | 117 | 110 | 116 | 104 | |||
Baseline EASI, mean (SD) | 32.2 (13.7) | 32.9 (13.9) | 32.1 (14.3) | 32.6 (13.9) | 28.8 (12.0) | 30.4 (12.8) | 32.1 (11.5) | 33.8 (13.5) | 32.1 (11.5) | 33.8 (13.5) | |||
Change from baseline, adjusted mean (SE) | −15.5 (0.55) | −9.0 (1.05) | −16.9 (0.55) | −7.0 (1.06) | −21.0 (0.67) | −15.6 (0.96) | −26.4 (0.8) | −22.4 (0.8) | −27.2 (0.8) | −23.7 (0.8) | |||
Difference, (95% CI) | −6.4 (−8.8 to −4.1; P < 0.001)d,e | −9.9 (−12.2 to −7.5; P < 0.001)d,e | −5.4 (−7.7 to −3.1; P < 0.001)d,e | −3.9 (−6.3 to −1.6; P < 0.001)e,f | −3.5 (−5.7 to −1.3; P = 0.002)e,f | ||||||||
EASI-75, n/N (%) | 150/601 (25.0) | 25/197 (12.7) | 196/591 (33.2) | 23/201 (11.4) | 141/252 (56.0) | 45/126 (35.7) | 88.6/138 (64.2) | 69.2/137 (50.5) | 95.0 (68.8) | 75.7 (55.3) | |||
Difference, % (95% CI) | 12.1 (6.5 to 17.7; P < 0.001)a | 21.6 (15.8 to 27.3; P < 0.001)a | 20.2 (9.8 to 30.6; P < 0.001)a | 14.1 (2.5 to 25.7; P = 0.018)b | 14.1 (2.9 to 25.3; P = 0.014)b,c | ||||||||
EASI-90, n/N (%) | 87/601 (14.5) | 8/197 (4.1) | 108/591 (18.3) | 11/201 (5.5) | 83/252 (32.9) | 27/126 (21.4) | 56.7/138 (41.1) | 40.2/137 (29.3) | 67.1/138 (48.6) | 49.8/137 (36.4) | |||
Difference, % (95% CI) | 10.3 (6.4 to 14.1; P < 0.001)a,e | 12.7 (8.3 to 17.0; P < 0.001)a,e | 11.4 (2.1 to 20.7; P = 0.022)a,e | 12.3 (1.1 to 23.6; P = 0.032)b,e | 12.9 (1.4 to 24.4; P = 0.027)b,e | ||||||||
EASI-50, n/N (%) | 250/601 (41.6) | 42/197 (21.3) | 295/591 (49.9) | 41/201 (20.4) | 200/252 (79.4) | 73/126 (57.9) | 110.4/138 (80.0) | 110.4/137 (80.0) | 111.2/138 (80.5) | 91.9/137 (67.1) | |||
Difference, % (95% CI) | 20.1 (13.3 to 26.8; P < 0.001)a,e | 29.3 (22.5 to 36.1; P < 0.001)a,e | 21.3 (11.3 to 31.3; P < 0.001)a,e | 10.6 (0.3 to 20.8; P = 0.043)b,e | 13.7 (3.5 to 23.9; P = 0.008)b,e | ||||||||
SCORAD | |||||||||||||
n | 353 | 96 | 430 | 98 | 229 | 107 | 117 | 110 | 116 | 104 | |||
Baseline SCORAD, mean (SD) | 70.3 (13.0) | 71.7 (12.5) | 70.0 (13.4) | 70.5 (12.2) | 67.0 (13.3) | 68.9 (13.2) | 70.2 (12.0) | 70.8 (12.8) | 70.2 (12.0) | 70.8 (12.8) | |||
Change from baseline, adjusted mean (SE) | −25.2 (0.94) | −14.7 (1.80) | −28.1 (0.92) | −14.0 (1.79) | −37.7 (1.25) | −26.7 (1.83) | −42.7 (1.6) | −34.1 (1.6) | −46.3 (1.5) | −37.3 (1.6) | |||
Difference, (95% CI) | −10.4 (−14.4 to −6.5; P < 0.001)d | −14.0 (−18.0 to −10.1; P < 0.001)d | −10.9 (−15.2 to −6.5; P < 0.001)d | −8.6 (−13.0 to −4.2; P < 0.001)c,f | −8.9 (−13.2 to −4.6; P < 0.001)c,f | ||||||||
Worst daily pruritus NRS (weekly average) | |||||||||||||
n | 325 | 88 | 401 | 94 | 221 | 100 | 115 | 112 | 111 | 101 | |||
Baseline worst daily pruritus NRS, mean (SD) | 7.7 (1.4) | 7.7 (1.4) | 7.9 (1.5) | 8.0 (1.4) | 7.7 (1.5) | 7.9 (1.5) | 7.3 (1.5) | 7.5 (1.4) | 7.3 (1.5) | 7.5 (1.4) | |||
Change from baseline, adjusted mean (SE) | −2.6 (0.11) | −1.7 (0.21) | −2.9 (0.11) | −1.6 (0.21) | −4.1 (0.15) | −2.9 (0.21) | −4.0 (0.2) | −3.1 (0.2) | −4.3 (0.2) | −3.4 (0.2) | |||
Difference, (95% CI) | −0.9 (−1.4 to −0.4; P < 0.001)d,e | −1.3 (−1.7 to −0.8; P < 0.001)d,e | −1.2 (−1.7 to −0.7; P < 0.001)d,e | −0.9 (−1.4 to −0.4; P < 0.001)e,f | −0.9 (−1.4 to −0.3; P = 0.002)e,f | ||||||||
Reduction from baseline ≥ 4, n/N (%)b | 119/594 (20.0) | 20/194 (10.3) | 144/575 (25.0) | 19/200 (9.5) | 113/249 (45.4) | 43/126 (34.1) | 61/134 (45.5) | 48/135 (35.6) | 63/134 (47.2) | 54/135 (39.7) | |||
Difference, % (95% CI) | 9.7 (4.4 to 15.0; P = 0.002)a | 15.6 (10.3 to 20.9; P < 0.001)a | 11.3 (0.9 to 21.6; P = 0.037)a | 9.7 (−2.0 to 21.4; P = 0.106)b | 7.3 (−4.6 to 19.2; P = 0.228)b,c | ||||||||
Reduction from baseline ≥ 3, n/N (%)b | 177/597 (29.6) | 28/195 (14.4) | 199/583 (34.1) | 28/200 (14.0) | 150/251 (59.8) | 51/126 (40.5) | NR | NR | NR | NR | |||
Difference,% (95% CI) | 15.2 (9.2 to 21.3; P < 0.001)a,e | 20.1 (13.9 to 26.2; P < 0.001)a,e | 19.3 (8.8 to 29.9; P < 0.001)a,e | NR | NR | ||||||||
Eczema-related sleep NRS (weekly average) | |||||||||||||
n | 325 | 88 | 401 | 94 | 221 | 100 | 115 | 112 | 111 | 101 | |||
Baseline eczema-related Sleep NRS | 6.9 (2.0) | 6.8 (1.9) | 7.2 (2.0) | 7.3 (2.1) | 6.9 (2.1) | 7.1 (2.2) | 6.3 (2.1) | 6.9 (1.6) | 6.3 (2.1) | 6.9 (1.6) | |||
Change from baseline, adjusted mean (95% CI or SE) | −2.6 (95% CI, −2.9 to −2.4) | −1.9 (95% CI, −2.4 to −1.5) | −2.9 (95% CI, −3.1 to −2.7) | −1.5 (95% CI, −1.9 to −1.1) | −4.3 (SE = 0.15) | −3.1 (SE = 0.22) | −4.1 (SE = 0.2) | −3.4 (SE = 0.2) | −4.3 (SE = 0.2) | −3.7 (SE = 0.2) | |||
Difference, (95% CI) | −0.7 (−1.2 to −0.2; P = 0.007)d,e | −1.4 (−1.9 to −0.9; P < 0.001)d,e | −1.3 (−1.8 to −0.8; P < 0.001)d,e | −0.8 (−1.3 to −0.2; P = 0.005)e,f | −0.6 (−1.1 to −0.0; P = 0.037)e,f | ||||||||
POEM | |||||||||||||
n | 334 | 93 | 418 | 97 | 226 | 103 | 110 | 106 | 105 | 97 | |||
Baseline score | 22.8 (5.1) | 23.0 (4.6) | 22.8 (4.9) | 22.9 (5.1) | 22.3 (5.1) | 22.4 (5.6) | 21.3 (5.1) | 20.9 (5.7) | 21.3 (5.1) | 20.9 (5.7) | |||
Change from baseline, adjusted mean (95% CI or SE) | −7.6 (95% CI, −8.3 to −7.0) | −3.0 (95% CI, −4.3 to −1.8) | −8.8 (95% CI, −9.4 to −8.1) | −3.7 (95% CI, −5.0 to −2.4) | −11.8 (95% CI, −12.7 to −10.9) | −7.8 (95% CI, −9.1 to −6.5) | −11.7 (SE = 0.6) | −8.3 (SE = 0.6) | −12.6 (SE = 0.6) | −9.1 (SE = 0.6) | |||
Difference, (95% CI) | −4.6 (−6.0 to −3.1; P < 0.001)d,e | −5.1 (−6.5 to −3.6; P < 0.001)d,e | −4.0 (−5.6 to −2.4; P < 0.001)d,e | −3.4 (−5.0 to −1.8; P < 0.001)e,f | −3.6 (−5.3 to −1.9; P < 0.001)e,f | ||||||||
DLQI | |||||||||||||
n | 335 | 95 | 419 | 97 | 226 | 104 | 112 | 106 | 107 | 97 | |||
Baseline DLQI, mean (SD) | 16.8 (7.1) | 17.0 (6.6) | 17.7 (7.1) | 17.8 (7.3) | 17.6 (7.1) | 17.2 (7.2) | 15.9 (6.5) | 16.4 (6.3) | 15.9 (6.5) | 16.4 (6.3) | |||
Change from baseline, adjusted mean (SE) | −7.1 (0.31) | −5.0 (0.59) | −8.8 (0.30) | −4.9 (0.60) | −11.7 (0.39) | −8.8 (0.56) | −11.2 (0.40) | −9.6 (0.40) | −11.5 (0.40) | −9.9 (0.40) | |||
Difference, (95% CI) | −2.1 (−3.4 to −0.8; P = 0.002)d | −3.9 (−5.2 to −2.6; P < 0.001)d | −2.9 (−4.3 to −1.6; P < 0.001)d | −1.5 (−2.6 to −0.4; P = 0.009)c,f | −1.6 (−2.7 to −0.5; P = 0.005)c,f | ||||||||
Reduction from baseline ≥ 4, n/N (%) | 258/578 (44.6) | 60/190 (31.6) | 325/577 (56.3) | 54/198 (27.3) | 207/278 (83.5) | 81/123 (65.9) | ||||||| |||| | ||||||| |||| | ||| |||| | || |||| | |||
Difference, (95% CI) | 13.0 (5.4 to 20.5; P = 0.001)a,e | 28.9 (21.4 to 36.3; P < 0.001)a,e | 17.6 (8.0 to 27.1; P < 0.001)a,e | ||| ||||| || ||||||||||| | ||| ||||| || |||||||||| | ||||||||
HADS | |||||||||||||
HADS anxiety score | |||||||||||||
n | 333 | 95 | 417 | 97 | 226 | 104 | 109 | 106 | ||| | || | |||
Baseline score | 6.9 (4.1) | 7.1 (4.0) | 7.1 (4.2) | 7.1 (4.4) | 6.7 (4.2) | 6.7 (4.3) | 6.1 (3.8) | 6.7 (4.2) | ||| ||||| | ||| ||||| | |||
Change from baseline, adjusted mean (95% CI or SE) | −1.4 (95% CI, −1.7 to −1.1) | −1.2 (95% CI,−1.7 to −0.7) | −1.8 (95% CI, −2.0 to −1.5) | −1.0 (95% CI, −1.6 to −0.5) | −2.3 (SE = 0.21) | −1.0 (SE = 0.30) | −1.94 (SE = 0.25) | −2.10 (SE = 0.26) | ||||| |||||||| | ||||| |||||||| | |||
Difference, (95% CI) | −0.2 (−0.8 to 0.4; P = 0.45)d,e | −0.7 (−1.3 to −0.1; P = 0.014)d,e | −1.3 (−2.1 to −0.6; P < 0.001)d,e | 0.16 (−0.56 to 0.87; P = 0.663)e,f | |||| |||||| || |||||||||| | ||||||||
HADS depression score | |||||||||||||
n | 333 | 95 | 417 | 97 | 226 | 104 | 109 | 106 | ||| | || | |||
Baseline score | 5.6 (4.2) | 6.0 (4.3) | 5.8 (4.3) | 6.0 (4.1) | 5.0 (3.9) | 5.2 (4.1) | 4.6 (3.7) | 5.0 (3.9) | ||| ||||| | ||| ||||| | |||
Change from baseline, adjusted mean (95% CI or SE) | −0.9 (95% CI, −1.2 to −0.6) | −0.6 (95% CI, −1.2 to −0.1) | −1.6 (95% CI, −1.9 to −1.3) | −0.4 (95% CI, −1.0 to 0.2) | −2.1 (SE = 0.19) | −1.2 (SE = 0.28) | −1.53 (SE = 0.25) | −1.55 (SE = 0.25) | ||||| ||||||| | ||||| ||||||| | |||
Difference, (95% CI) | −0.3 (−0.9 to 0.4; P = 0.42)d,e | −1.1 (−1.8 to −0.5; P < 0.001)d,e | −0.8 (−1.5 to −0.2; P = 0.015)d,e | 0.02 (−0.67 to 0.71; P = 0.959)e,f | ||||| |||||| || |||||||||| | ||||||||
HADS anxiety and depression scores < 8 | |||||||||||||
n/N (%) | 63/289 (21.8) | 19/103 (18.4) | 112/292 (38.4) | 15/93 (16.1) | 55/102 (53.9) | 16/54 (29.6) | 25/51 (49.1) | 25/58 (43.2) | ||||| |||||| | ||||| |||||| | |||
Difference, (95% CI) | 3.0 (−5.8 to 11.8; P = 0.52)a,e | 21.2 (11.8 to 30.5; P < 0.001)a,e | 24.8 (9.3 to 40.4; P = 0.003)a,e | 6.6 (−12.4 to 25.5; P = 0.498)b,e | ||| ||||| || ||||||||| | ||||||||
TCS use | |||||||||||||
Amount of TCS used (g) at week 15 to 16 or week 25 to 26, adjusted mean (SE) | NR | NR | NR | NR | 11.6 (1.57) | 20.2 (2.27) | 27.3 (4.1) | 38.8 (4.1) | 29.1 (4.3) | 40.9 (4.4) | |||
Difference vs. placebo, % (95% CI) | NR | NR | −8.6 (−14.1 to −3.2, P = 0.002)e,g | −11.5 (−22.9 to −0.2; P = 0.047) e,g | −11.7 (−23.9 to 0.4; P = 0.059) e,g | ||||||||
Number of days without topical treatment at week 16 or week 26, adjusted mean (SE) | NR | NR | NR | NR | 3.4 (0.19) | 3.0 (0.27) | 4.2 (0.2) | 2.4 (0.2) | 4.0 (0.2) | 2.7 (0.2) | |||
Difference vs. placebo, (95% CI) | NR | NR | 0.5 (−0.2 to 1.1; P = 0.17)e,g | 1.9 (1.2 to 2.5; P < 0.001)e,g | 1.3 (0.6 to 2.0; P < 0.001)e,g | ||||||||
CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; EASI-90 = reduction of at least 90% in Eczema Area and Severity Index score from baseline; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator’s Global Assessment; NR = not reported; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; SE = standard error; TCS = topical corticosteroids.
aThe analysis was conducted using a Cochran-Mantel-Haenszel test stratified by baseline IGA and region based on the composite estimand.
bThe analysis was conducted using a Cochran-Mantel-Haenszel test stratified by prior cyclosporine A use, country, and baseline disease severity based on the COVID-19–modified composite estimand.
cThis end point was included in the statistical testing hierarchy; however, no superiority testing was conducted for this end point due to prior failure in the statistical testing hierarchy.
dThe analysis was conducted using the repeated measurements model, with baseline IGA, region, and treatment-by-week interaction as factors and interaction between week and baseline value as a covariate, based on the hypothetical estimand (primary estimand).
eThe end point was not adjusted for multiplicity and was at an increased risk of type I error (false-positive result).
fThe analysis was conducted using the repeated measurements model, with baseline IGA, country, prior cyclosporine A use and treatment-by-week interaction as factors, and interaction between week and baseline value as a covariate based on the hypothetical estimand (primary estimand).
gThis analysis was conducted using repeated measurements model with an unstructured covariance matrix.
Sources: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 and 718 and the sponsor’s Summary of Clinical Evidence.14
Efficacy — Maintenance Treatment Period
Key results in the maintenance treatment period are summarized in Table 28 for adolescents and Table 29 for adults.
Table 28: Key Efficacy Results in Maintenance Treatment Period in ECZTRA 6 — Efficacy Analysis Set (Adolescents)
Outcomes | Week 16 tralokinumab 300 mg q.2.w. responders | |
|---|---|---|
Tralokinumab 300 mg q.2.w. | Tralokinumab 300 mg q.4.w. | |
IGA of 0 or 1 at week 52 among patients with an IGA score of 0 or 1 at week 16 | ||
Responder, n/N (%; 95% CI)a,b | 3/8 (37.5; 13.7 to 69.4) | 7/8 (87.5; 52.9 to 97.8) |
EASI-75 at week 52 among patients with EASI-75 at week 16 | ||
Responder, n/N (%; 95% CI)a,b | 4/9 (44.4; 18.9 to 73.3) | 7/13 (53.8; 29.1 to 76.8) |
IGA of 0 or 1 at week 52 among patients with EASI-75 and IGA score ≥ 2 at week 16 | ||
Responder, n/N (%; 95% CI)a,b | 1/3 (33.3; NR) | 0/5 (0; not reported) |
IGA of 0 or 1 at week 52 among patients with IGA score of 0 or 1 or EASI-75 at week 16 | ||
Responder, n/N (%; 95% CI)a,b | 4/11 (36.4; 15.2 to 64.6) | 7/13 (53.8, 29.1 to 76.8) |
EASI-75 at week 52 among patients with IGA score of 0 or 1 or EASI-75 at week 16 | ||
Responder, n/N (%; 95% CI)a,b | 5/11 (45.5; 21.3 to 72.0) | 7/13 (53.8; 29.1 to 76.8) |
CI = confidence interval; EASI-75 = reduction of at least 50% in Eczema Area and Severity Index from baseline score; IGA = Investigator’s Global Assessment; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
aThe CI was calculated based on Wilson score method.
bThe end point was not adjusted for multiplicity and was at an increased risk of type I error (false-positive result).
Source: Clinical Study Report for ECZTRA 613 and the sponsor’s Summary of Clinical Evidence.14
IGA Score of 0 or 1 at Week 52 (ECZTRA 1, 2, and 6) or Week 32 (ECZTRA 3) Among Patients With an IGA Score of 0 or 1 at week 16
Adolescents
In the ECZTRA 6 study, the proportions of patients receiving tralokinumab 300 mg every 2 weeks with an IGA of 0 or 1 at week 16 (without use of rescue medication) who maintained their IGA of 0 or 1 response at week 52 (without use of rescue medication) were 37.5% (3 out of 8 patients, 95% CI, 13.7% to 69.4%) in the tralokinumab 300 mg every 2 weeks (week 0 to 16) then every 2 weeks (week 17 to 52) group and 87.5% (7 out of 8 patients, 95% CI, 52.9% to 97.8%) in the tralokinumab 300 mg every 2 weeks then every 4 weeks group. No statistical analysis was conducted to assess the between-group difference.
Adults
In the ECZTRA 1 and 2 studies, the proportion of patients with an IGA of 0 or 1 at week 16 (without use of rescue medication) who maintained their IGA of 0 or 1 response (without use of rescue medication) at week 52 was included in the statistical hierarchy. In the ECZTRA 1 study, the difference between the tralokinumab
Table 29: Key Efficacy Results in Maintenance Treatment Period in ECZTRA 1 and 2 and Continuous Treatment Period in ECZTRA 3 — Maintenance Analysis Set in ECZTRA 1 and 2 and Continuous Treatment Analysis Set in ECZTRA 3 (Adults, Original Review)
Outcomes | ECZTRA 1a (follow-up at 52 weeks) | ECZTRA 2a (follow-up at 52 weeks) | ECZTRA 3a (follow-up at 32 weeks) | |||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab q.2.w. | Tralokinumab q.4.w. | Placebo | Tralokinumab q.2.w. | Tralokinumab q.4.w. | Placebo | Tralokinumab q.2.w. + TCS | Tralokinumab q.4.w. + TCS | |
IGA of 0 or 1 at week 52 (ECZTRA 1 and 2) or week 32 (ECZTRA 3) | ||||||||
Responder, n/N (%) | 20/39 (51.3) | 14/36 (38.9) | 9/19 (47.4) | 32/54 (59.3) | 22/49 (44.9) | 7/28 (25.0) | 43/48 (89.6) | 38/49 (77.6) |
Difference vs. placebo, % (95% CI) | 6.0 (−21.8 to 33.7; P = 0.68)b | −9.5 (−37.1 to 18.0; P = 0.50)b,c | Reference | 34.1 (13.4 to 54.9; P = 0.004)b | 19.9 (−1.2 to 40.9; P = 0.084)b | Reference | NA | |
EASI-75 at week 52 (ECZTRA 1 and 2) or week 32 (ECZTRA 3) | ||||||||
Responder, n/N (%) | 28/47 (59.6) | 28/57 (49.1) | 10/30 (33.3) | 43/77 (55.8) | 38/74 (51.4) | 9/42 (21.4) | 62/67 (92.5) | 59/65 (90.8) |
Difference vs. placebo, % (95% CI)b | 21.2 (−0.2 to 42.6; P = 0.056)b,c | 11.7 (−8.7 to 32.0; P = 0.27)b,c | Reference | 33.7 (17.3 to 50.0; P < 0.001)b | 30.0 (13.7 to 46.4; P = 0.001)b,c | Reference | NA | |
IGA of 0 or 1 at week 52 (ECZTRA 1 and 2) or week 32 (ECZTRA 3) | ||||||||
Responder, n/N (%) | 1/22 (4.5) | 5/29 (17.2) | 0/15 (0.0) | 4/33 (12.1) | 6/32 (18.8) | 0/17 (0) | 4/20 (20.0) | 8/17 (47.1) |
Difference vs. placebo, % (95% CI)b | 4.3 (−4.2 to 12.8; P = 0.43)b,d | 17.3 (3.6 to 31.1; P = 0.082)b,d | Reference | 12.5 (1.2 to 23.8; P = 0.14)b,d | 18.7 (5.2 to 32.3; P = 0.063)b,d | Reference | NA | |
IGA of 0 or 1 or EASI-75 at week 52 (ECZTRA 1 and 2) or week 32 (ECZTRA 3) | ||||||||
Responder, n/N (%) | 28/50 (56.0) | 29/58 (50.0) | 1/30 (36.7) | 45/80 (56.3) | 38/76 (50.0) | 9/43 (20.9) | NA | NA |
Difference vs. placebo, % (95% CI)b | 14.1 (−7.1 to 35.3; P = 0.020)b,d | 9.4 (−11.2 to 30.0; P = 0.38)b,d | Reference | 34.3 (18.3 to 50.3; P < 0.001)b,d | 28.8 (12.7 to 44.9; P = 0.002)b,d | Reference | NA | |
CI = confidence interval; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; NA = not applicable; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; TCS = topical corticosteroids.
aPatients who achieved a clinical response with tralokinumab (ECZTRA 1 and 2) or tralokinumab plus TCS (ECZTRA 3) at week 16 were eligible to continue maintenance treatment (ECZTRA 1 and 2) or continuation treatment (ECZTRA 3) and were included in this dataset, and the outcomes reported were achieved without rescue medication.
bMantel-Haenszel risk difference compared to placebo, stratified by region.
cThis end point was included in the statistical testing hierarchy; however, no superiority testing was conducted due to prior failure in the statistical testing hierarchy.
dThe end point was not adjusted for multiplicity and was at an increased risk of type I error (false-positive result).
Sources: Clinical Study Reports for ECZTRA 1, 2, and 315-17 and the sponsor’s Summary of Clinical Evidence.14
every 2 weeks group and the placebo group was 6.0% (95% CI, −21.8% to 33.7%; P = 0.68). Due to failure of this end point, no superiority testing was conducted for the difference between the tralokinumab every 4 weeks group and the placebo group (lower in the testing hierarchy), which was −9.5% (95% CI, −37.1% to 18.0%). In the ECZTRA 2 study, the difference between the tralokinumab every 2 weeks group and the placebo group was 34.1% (95% CI, 13.4% to 54.9%; P = 0.004). The difference between the tralokinumab every 4 weeks group and the placebo group was 19.9% (95% CI, −1.2 to 40.9; P = 0.084); due to failure of this end point, no superiority testing was conducted for the end point lower in the testing hierarchy (i.e., EASI-75 at week 52 between tralokinumab 300 mg every 4 weeks and placebo).
In the ECZTRA 3 study, the proportions of patients with an IGA of 0 or 1 at week 16 who maintained their IGA of 0 or 1 response at week 32 were 89.6% (95% CI not reported) in the tralokinumab every 2 weeks plus TCS group and 77.6% (95% CI not reported) in the tralokinumab every 4 weeks plus TCS group. No statistical analysis was conducted to assess the between-group difference.
This end point was not assessed in the ECZTRA 7 study.
EASI-75 at Week 52 (ECZTRA 1, 2, and 6) or Week 32 (ECZTRA 3) Among Patients With EASI-75 at Week 16
Adolescents
In the ECZTRA 6 study, the proportion of patients with EASI-75 at week 16 (without the use of rescue medication) who maintained their EASI-75 response at week 52 (without use of rescue medication) was 44.4% (4 out of 9 patients, 95% CI, 18.9% to 73.3%) in the tralokinumab 300 mg every 2 weeks (at week 16) then every 2 weeks (at week 52) group and 53.8% (7 out of 13 patients, 95% CI, 29.1% to 76.8%) in the tralokinumab 300 mg every 2 weeks then every 4 weeks group. No statistical analysis was conducted to assess the between-group difference on these end points.
Adults
In the ECZTRA 1 study, the proportion of patients with EASI-75 at week 16 (without use of rescue medication) who maintained their EASI-75 response (without use of rescue medication) at week 52 was not tested for superiority due to prior failure of the statistical hierarchy (the proportion of patients with IGA 0 or 1 at week 16 who maintained their IGA of 0 or 1 response at week 52). The difference between the tralokinumab every 2 weeks group and the placebo group was 21.2% (95% CI, −0.2% to 42.6%). The difference between the tralokinumab every 4 weeks group and the placebo group was 11.7% (95% CI, −8.7% to 32.0%).
In the ECZTRA 2 study, the 33.7% difference (95% CI, 17.3% to 50.0%; P < 0.001) in the proportion of patients with EASI-75 at week 16 who maintained their EASI-75 response at week 52 between tralokinumab 300 mg every 2 weeks and placebo was included in the statistical testing hierarchy. The difference in proportion of patients with EASI-75 at week 16 who maintained their EASI-75 response at week 52 between tralokinumab 300 mg every 4 weeks and placebo was not tested for superiority due to failure of a prior end point in the statistical testing hierarchy (i.e., an IGA 0 or 1 at week 52 between tralokinumab 300 mg every 4 weeks and placebo).
In the ECZTRA 3 study, the proportions of patients with an IGA of 0 or 1 at week 16 who maintained their IGA of 0 or 1 response at week 32 were 92.5% (95% CI not reported) in the tralokinumab every 2 weeks plus TCS group and 90.8% (95% CI not reported) in the tralokinumab every 4 weeks plus TCS group. No statistical analysis was conducted to assess the between-group difference.
This end point was not assessed in the ECZTRA 7 study.
IGA Score 0 or 1 at Week 52 (ECZTRA 1, 2, and 6) or Week 32 (ECZTRA 3) Among Patients With EASI-75 and an IGA Score of 2 or Higher at Week 16
Adolescents
In the ECZTRA 6 study, the proportions of patients with EASI-75 and an IGA of at least 2 at week 16 who achieved an IGA of 0 or 1 at week 52 was 33.3% (1 out of 3 patients, 95% CI not reported) in the tralokinumab 300 mg every 2 weeks (at week 16) then every 2 weeks (at week 52) group and 0% (0 out of 5 patients, 95% CI not reported) in the tralokinumab 300 mg every 2 weeks then every 4 weeks group. No statistical analysis was conducted to assess the between-group difference.
Adults
In the ECZTRA 1 and 2 studies, the proportion of patients with EASI-75 and IGA of at least 2 at week 16 who achieved an IGA 0 or 1 response at week 52 was an exploratory outcome and not adjusted for multiplicity. The differences between the tralokinumab every 2 weeks group and the placebo group were 4.3% (95% CI, −4.2% to 12.8%) in the ECZTRA 1 study and 12.5% (95% CI, 1.2% to 23.8%) in the ECZTRA 2 study. The differences between the tralokinumab every 4 weeks group and the placebo group were 17.3% (95% CI, 3.6% to 31.1%) in the ECZTRA 1 study and 18.7% (95% CI, 5.2% to 32.3%) in the ECZTRA 2 study.
In the ECZTRA 3 study, the proportions of patients with an IGA of 0 or 1 at week 16 who maintained their IGA of 0 or 1 response at week 32 were 89.6% (95% CI not reported) in the tralokinumab every 2 weeks plus TCS group and 77.6% (95% CI not reported) in the tralokinumab every 4 weeks plus TCS group. No statistical analysis was conducted to assess the between-group difference.
This end point was not assessed in the ECZTRA 7 study.
IGA Score of 0 or 1 at Week 52 (ECZTRA 1, 2, and 6) or Week 32 (ECZTRA 3) Among Patients With an IGA Score of 0 or 1 or EASI-75 at Week 16
Adolescents
In the ECZTRA 6 study, the proportions of patients with EASI-75 or an IGA of 0 or 1 at week 16 who achieved an IGA of 0 or 1 at week 52 was 36.4% (4 out of 11 patients, 95% CI, 15.2% to 64.6%) in the tralokinumab 300 mg every 2 weeks (at week 16) then every 2 weeks (at week 52) group, and 53.8% (7 out of 13 patients, 95% CI, 29.1% to 76.8%) in the tralokinumab 300 mg every 2 weeks then every 4 weeks group. No statistical analysis was conducted to assess the between-group difference.
Adults
This end point was not assessed in the ECZTRA 1, 2, 3, and 7.
EASI-75 at Week 52 (ECZTRA 1, 2, and 6) or week 32 (ECZTRA 3) Among Patients With an IGA Score of 0 or 1 or EASI-75 at Week 16
Adolescents
In the ECZTRA 6 study, the proportion of patients with EASI-75 or an IGA score of 0 or 1 at week 16 who achieved an EASI-75 response at week 52 was 45.5% (5 out of 11 patients, 95% CI, 21.3% to 72.0%) in the tralokinumab 300 mg every 2 weeks (at week 16) then every 2 weeks (at week 52) group, and 53.8% (7 out of 13 patients, 95% CI, 29.1% to 76.8%) in the tralokinumab 300 mg every 2 weeks then every 4 weeks group. No statistical analysis was conducted to assess the between-group difference.
Adults
This end point was not assessed in the ECZTRA 1, 2, 3, and 7 studies.
IGA Score of 0 or 1 or EASI-75 at Week 52 (ECZTRA 1 and 2) or Week 32 (ECZTRA 3) Among Patients With an IGA Score of 0 or 1 or EASI-75 at Week 16
Adolescents
This end point was not assessed in the ECZTRA 6 study.
Adults
In the ECZTRA 1 and 2 studies, the proportion of patients with EASI-75 or an IGA of 0 or 1 at week 16 who achieved EASI-75 or an IGA of 0 or 1 at week 52 was an exploratory outcome and not adjusted for multiplicity. The differences between the tralokinumab every 2 weeks group and the placebo group were 14.1% (95% CI, −7.1% to 35.3%) in the ECZTRA 1 study and 34.3% (95% CI, 18.3% to 50.3%) in the ECZTRA 2 study. The differences between the tralokinumab every 4 weeks group and the placebo group were 9.4% (95% CI, −11.2% to 30.0%) in the ECZTRA 1 and 2 studies and 8.8% (95% CI, 12.7% to 44.9%) in the ECZTRA 2 study. This end point was not assessed in the ECZTRA 3 and 7 studies.
Other Efficacy End Points
Adolescents
Other study end points, including SCORAD, adolescent worst pruritus NRS, eczema-related sleep NRS, CDLQI, HADS, and POEM scores, were assessed up to week 52 in the ECZTRA 6 study. Results at weeks 16 and 52 are presented in Table 74 in Appendix 1. In general, a within-group reduction in mean score was evident for most end points (i.e., suggesting improvement) from week 16 to week 52; however, the analysis was based on a small sample size. No statistical analysis was conducted to assess between-group differences on these end points.
Adults
Other study end points, including SCORAD, worst daily pruritus NRS, eczema-related sleep NRS, DLQI, HADS, POEM, Short Form (36) Health Survey, EQ-5D-5L, and Work Productivity and Activity Impairment – General Health scores, were assessed up to week 52 in the ECZTRA 1 and 2 studies and up to week 32 in the ECZTRA 3 study. No statistical testing was conducted on these end points to assess the between-group difference. Results at weeks 16 and 52 are presented in Table 75 in Appendix 1. In general, there appeared to be a within-group reduction in mean score for most end points (i.e., suggesting improvement) from week 16 to week 52; however, no statistical analysis was conducted to assess between-group differences in these end points.
Harms
Harms outcomes in the initial treatment period are summarized in Table 30 for adolescents and Table 31 for adults. Harms outcomes in the maintenance (or continuous) treatment period are summarized in Table 32 for adolescents and Table 33 for adults.
Adverse Events
Adolescents
In the ECZTRA 6 study, of the 97 patients in the tralokinumab group and 94 patients in the placebo group, 64.9% and 61.7% of patients, respectively, experienced at least 1 TEAE in the initial treatment period. The most common TEAEs (reported in at least 10% of patients) in the tralokinumab group were viral URTI (12.4%) and URTI (11.3%), both of which were more frequently reported compared with the placebo group (8.5% and 4.3%, respectively). In the maintenance treatment period, TEAE findings in the tralokinumab groups were generally consistent with those of the initial treatment period.
Adults
No notable between-group difference in the proportion of patients who reported at least 1 TEAE in the initial treatment period was observed across the ECZTRA 1, 2, 3, and 7 studies. The proportion of patients with at least 1 TEAE ranged from 61.5% to 77.5% in the tralokinumab group (i.e., tralokinumab 300mg every 2 weeks or tralokinumab 300 mg every 2 weeks plus TCS) and between 66.0% to 78.8% in the placebo group (i.e., placebo-only or placebo plus TCS). The most common TEAEs reported in the tralokinumab group (at least 10% in any study) were viral URTI, URTI, conjunctivitis, and headache; conjunctivitis was consistently more frequently reported in the tralokinumab group (3.0% to 11.1%) than the placebo group (1.5% to 4.4%) across studies. Results in the maintenance (or continuous) treatment period of the ECZTRA 1, 2, and 3 studies were generally consistent with the initial treatment period. The frequency of TEAEs was consistently lower in tralokinumab every 4 weeks group (ranging between 59.4% and 69.7%) compared with tralokinumab every 2 weeks group (ranging between 68.1% and 79.4%) across the studies.
Serious Adverse Events
Adolescents
In the initial treatment period of ECZTRA 6, an SAE related to radius fracture was reported in 1 patient (1.0%) in the tralokinumab 300 mg every 2 weeks group. Five patients (5.3%) in the placebo group reported SAEs. In the maintenance treatment period, no SAE was reported in any of the treatment groups.
Adults
In the initial treatment period of the ECZTRA 1, 2, 3, and 7 studies, the proportion of patients who reported SAEs ranged from 0.7% to 3.8% in the tralokinumab group (i.e., tralokinumab 300 mg every 2 weeks or tralokinumab 300 mg every 2 weeks plus TCS), and 2.5% to 4.1% in the placebo group (i.e., placebo-only or placebo plus TCS). In the maintenance treatment period, a similarly low frequency of SAEs was reported in all treatment groups in the ECZTRA 1, 2, and 3 studies.
Withdrawal Due to Adverse Events
Adolescents
In the ECZTRA 6 study, no patient withdrew from study treatment due to AEs in any treatment group in the initial treatment period or the maintenance treatment period.
Adults
In the ECZTRA 1, 2, 3, and 7 studies, the proportions of patients who withdrew from treatment due to AEs ranged from 0.7% to 3.3% in the tralokinumab group (i.e., tralokinumab 300 mg every 2 weeks or tralokinumab 300 mg every 2 weeks plus TCS) and 0.8% to 4.1% in the placebo group (i.e., placebo-only or placebo plus TCS). In the maintenance (or continuous) treatment period of the ECZTRA 1, 2, and 3 studies, a similarly low frequency of treatment withdrawal due to AEs was observed in the tralokinumab groups and no treatment withdrawal due to an AE was observed in the placebo groups.
Mortality
Adolescents
In the ECZTRA 6 study, no death was reported in any treatment group in the initial treatment period and the maintenance treatment period.
Adults
In the initial treatment period, 2 patients (0.3%) in the tralokinumab 300 mg every 2 weeks group died in the ECZTRA 1 study (due to 1 unknown cause and 1 myocardial infarction) and 1 patient (0.2%) in the tralokinumab 300 mg every 2 weeks group died in the ECZTRA 2 study (due to metastatic squamous cell carcinoma). No deaths were reported in the tralokinumab group in the ECZTRA 3 and 7 studies, or in the placebo groups for all studies. In the maintenance (or continuous) treatment period of the ECZTRA 1, 2, and 3 studies, no deaths were observed in any treatment group.
Notable Harms
Adolescents
In the initial treatment period of ECZTRA 6, there was no notable difference between the tralokinumab group and the placebo group in the proportion of patients who reported eczema herpeticum, malignancies, skin infection requiring systemic treatment, eye disorders, injection-site reactions, oral herpes, and acne. The frequency of URTI was higher in the tralokinumab group (11.3%) than in the placebo group (4.3%), although the absolute difference in number of events was small. Findings in the maintenance treatment period were similar to those in the initial treatment period.
Adults
In the initial treatment period of the ECZTRA 1, 2, 3, and 7 studies, there was generally no notable difference between the tralokinumab group and the placebo group with respect to the frequency of eczema herpeticum, malignancies, eye disorders (except conjunctivitis), oral herpes, upper respiratory infection, and acne. Conjunctivitis was consistently more frequently reported in the tralokinumab group (3.0% to 11.1%) compared with the placebo group (1.5% to 4.4%) across studies. In the ECZTRA 2 study, skin infections requiring systemic treatment were notably less common in the tralokinumab group (3.5%) compared with the placebo group (11%). In the ECZTRA 3 study, injection-site reactions were more common in the tralokinumab group (6.7%) compared with the placebo group (0%). However, such between-group differences in the frequency of skin infections and injection-site reactions were not consistent across studies.
In the maintenance (or continuous) treatment period of the ECZTRA 1, 2, and 3 studies, no notable difference was reported for most notable harms, except that the frequency of injection-site reactions was consistently higher in the tralokinumab groups (4.4% to 9.2%) compared with the placebo groups (0% to 2.9%) in both the ECZTRA 1 and 2 studies. In the ECZTRA 2 study, the frequency of URTIs was higher in the tralokinumab groups (15.4% in the every 2 weeks group and 10.1% in the every 4 weeks group) compared with the placebo groups (6.5%), but such differences was not observed in the ECZTRA 1 study.
Table 30: Harms Outcomes for ECZTRA 6 in the Initial Treatment Period — Safety Analysis Set (Adolescents)
Adverse events | Tralokinumab 300 mg q.2.w. (N = 97) | Placebo (N = 94) |
|---|---|---|
Patients with ≥ 1 TEAE, n (%) | ||
Patients with any TEAE | 63 (64.9) | 58 (61.7) |
Most common TEAEa | ||
Viral upper respiratory tract infection | 12 (12.4) | 8 (8.5) |
Upper respiratory tract infection | 11 (11.3) | 4 (4.3) |
Atopic dermatitis | 7 (7.2) | 12 (12.8) |
SAE, n (%) | ||
Patients with an SAE | 1 (1.0) | 5 (5.3) |
Radius fracture | 1 (1.0) | 0 (0) |
Infectious mononucleosis | 0 (0) | 1 (1.1) |
Atopic dermatitis | 0 (0) | 1 (1.1) |
Acute respiratory failure | 0 (0) | 1 (1.1) |
Asthma | 0 (0) | 1 (1.1) |
Anaphylactic reaction | 0 (0) | 1 (1.1) |
Treatment withdrawal due to AE, n (%) | ||
Patients who stopped | 0 (0) | 0 (0) |
Deaths, n (%) | ||
Patients who died | 0 (0) | 0 (0) |
Notable harms, n (%) | ||
Eczema herpeticum | 0 (0) | 1 (1.1) |
Malignancies diagnosed after randomization | 0 (0) | 0 (0) |
Skin infection requiring systemic treatment | 2 (2.1) | 2 (2.1) |
Eye disorders | ||
Conjunctivitis | 0 (0) | 0 (0) |
Bacterial conjunctivitis | 1 (1.0) | 0 (0) |
Allergic conjunctivitis | 2 (2.1) | 2 (2.1) |
Viral keratitis | 1 (1.0) | 0 (0) |
Injection-site reactions | 2 (2.1) | 0 (0) |
Oral herpes | 0 (0) | 1 (1.1) |
Upper respiratory tract infection | 11 (11.3) | 4 (4.3) |
Acne | 3 (3.1) | 4 (4.3) |
AE = adverse event; q.2.w. = every 2 weeks; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Note: Unless otherwise specified, AEs are reported based on the Medical Dictionary for Regulatory Activities preferred term.
a10% or greater of patients in any group.
Sources: Clinical Study Report for ECZTRA 613 and the sponsor’s Summary of Clinical Evidence.14
Critical Appraisal
Internal Validity
ECZTRA 6, as well as all the included trials in adults, were randomized, double-blinded, and placebo-controlled. The method used for the initial randomization and re-randomization across trials consisted of a central interactive web-response system, which enabled the concealment of the allocation sequence. The baseline demographics and disease characteristics were generally balanced in the ECZTRA 1 and 2 studies, suggesting that the randomization was successful in these studies. In the ECZTRA 6 study, a notably higher proportion of patients in the placebo group had received prior systemic corticosteroid treatment, wet wraps, and phototherapy, compared with the tralokinumab 300 mg every 2 weeks group. In the ECZTRA 3 study, the tralokinumab every 2 weeks plus TCS group had a lower proportion of patients who were male and received prior systemic corticosteroid and prior methotrexate treatment, as well as a higher proportion of patients who were white and received prior phototherapy compared with the placebo plus TCS group. The baseline imbalances did not appear to systematically favour either treatment group, and although the clinical experts noted that the higher use of prior systemic corticosteroids could be suggestive of patients with more severe disease, in their opinion, none of these imbalances raised concerns about potential bias in the study results.
Table 31: Harms Outcomes for ECZTRA 1, 2, 3, and 7 in Initial Treatment Period — Safety Analysis Set (Adults, Original Review)
Adverse events | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7 | ||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab 300 mg q.2.w. (n = 602) | Placebo (n = 196) | Tralokinumab 300 mg q.2.w. (n = 592) | Placebo (n = 200) | Tralokinumab 300 mg q.2.w. + TCS (n = 252) | Placebo + TCS (n = 126) | Tralokinumab 300 mg q.2.w. + TCS (n = 138) | Placebo + TCS (n = 137) | |
Patients with ≥ 1 TEAE, n (%) | ||||||||
Patients with any TEAE | 460 (76.4) | 151 (77.0) | 364 (61.5) | 132 (66.0) | 180 (71.4) | 84 (66.7) | 107 (77.5) | 108 (78.8) |
Most common TEAEa | ||||||||
Atopic dermatitis | 156 (25.9) | 75 (38.3) | 98 (16.6) | 67 (33.5) | 6 (2.4) | 10 (7.9) | 7 (5.1) | 16 (11.7) |
Viral upper respiratory tract infection | 139 (23.1) | 41 (20.9) | 49 (8.3) | 17 (8.5) | 49 (19.4) | 14 (11.1) | 37 (26.8) | 35 (25.5) |
Upper respiratory tract infection | 9 (1.5) | 2 (1.0) | 59 (10.0) | 17 (8.5) | 19 (7.5) | 6 (4.8) | 10 (7.2) | 10 (7.3) |
Conjunctivitis | 43 (7.1) | 4 (2.0) | 18 (3.0) | 3 (1.5) | 28 (11.1) | 4 (3.2) | 6 (4.3) | 2 (1.5) |
Headache | 28 (4.7) | 10 (5.1) | 16 (2.7) | 6 (3.0) | 22 (8.7) | 6 (4.8) | 21 (15.2) | 13 (9.5) |
SAE, n (%) | ||||||||
Patients with an SAE | 23 (3.8) | 8 (4.1) | 10 (1.7) | 5 (2.5) | 2 (0.8) | 4 (3.2) | 1 (0.7) | 5 (3.6) |
Most common serious adverse eventsb | ||||||||
Atopic dermatitis | 4 (0.7) | 1 (0.5) | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (0.7) |
Exfoliative generalized dermatitis | 2 (0.3) | 1 (0.5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Treatment withdrawal due to AE, n (%) | ||||||||
Patients who stopped | 20 (3.3) | 8 (4.1) | 9 (1.5) | 3 (1.5) | 6 (2.4) | 1 (0.8) | 1 (0.7) | 3 (2.2) |
Most common reason for treatment withdrawalb | ||||||||
Atopic dermatitis | 5 (0.8) | 5 (2.6) | 2 (0.3) | 2 (1.0) | 0 (0) | 1 (0.8) | 0 (0) | 1 (0.7) |
Injection-site reaction | 4 (0.7) | 0 (0) | 0 (0) | 0 (0) | 1 (0.4) | 0 (0) | 0 (0) | 0 (0) |
Deaths, n (%) | ||||||||
Patients who died | 2 (0.3) | 0 (0) | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Unknown | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Myocardial infarction | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Metastatic squamous cell carcinoma | 0 (0) | 0 (0) | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Notable harms, n (%) | ||||||||
Eczema herpeticum | 3 (0.5) | 2 (1.0) | 2 (0.3) | 5 (2.5) | 1 (0.4) | 1 (0.8) | 1 (0.7) | 0 (0) |
Malignancies diagnosed after randomization | 0 (0) | 0 (0) | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Skin infections requiring systemic treatment | 13 (2.2) | 4 (2.0) | 21 (3.5) | 22 (11.0) | 4 (1.6) | 7 (5.6) | 1 (0.7) | 8 (5.8) |
Eye disorders | ||||||||
Conjunctivitis | 43 (7.1) | 4 (2.0) | 18 (3.0) | 3 (1.5) | 28 (11.1) | 4 (3.2) | 13 (9.4) | 6 (4.4) |
Bacterial conjunctivitis | 2 (0.3) | 0 (0) | 2 (0.3) | 1 (0.5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Viral conjunctivitis | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (0.8) | 0 (0) | 0 (0) |
Allergic conjunctivitis | 16 (2.7) | 3 (1.5) | 12 (2.0) | 2 (1.0) | 5 (2.0) | 2 (1.6) | 0 (0) | 0 (0) |
Keratoconjunctivitis | 1 (0.2) | 0 (0) | 2 (0.3) | 0 (0) | 1 (0.4) | 0 (0) | 1 (0.7) | 0 (0) |
Keratitis | 3 (0.5) | 0 (0) | 1 (0.2) | 1 (0.5) | 0 (0) | 0 (0) | 1 (0.7) | 1 (0.7) |
Injection-site reaction | 24 (4.0) | 0 (0) | 15 (2.5) | 2 (1.0) | 17 (6.7) | 0 (0) | 2 (1.4) | 0 (0) |
Oral herpes | 6 (1.0) | 4 (2.0) | 3 (0.5) | 4 (2.0) | 4 (1.6) | 1 (0.8) | 5 (3.6) | 6 (4.4) |
Upper respiratory infection | 9 (1.5) | 2 (1.0) | 59 (10.0) | 17 (8.5) | 19 (7.5) | 6 (4.8) | 10 (7.2) | 10 (7.3) |
Acne | 3 (0.5) | 1 (0.5) | 2 (0.3) | 0 (0) | 1 (0.4) | 1 (0.8) | 1 (0.7) | 2 (1.5) |
AE = adverse event; q.2.w. = every 2 weeks; SAE = serious adverse event; TEAE = treatment-emergent adverse event; TCS = topical corticosteroids.
Note: The table presents harms outcomes between week 0 and week 16 in the ECZTRA 1, 2, and 3 studies, and between week 0 and week 26 in the ECZTRA 7 study. Unless otherwise specified, AEs are reported based on the Medical Dictionary for Regulatory Activities preferred term.
a10% or greater of patients in any group.
bTwo more patients in any group.
Sources: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 and 718 and the sponsor’s Summary of Clinical Evidence.14
Table 32: Harms Outcomes for ECZTRA 6 in the Maintenance Treatment Period —Maintenance Safety Analysis Set (Adolescents)
Adverse events | Tralokinumab 300 mg q.2.w. responders | Week 16 placebo responders | |
|---|---|---|---|
Tralokinumab 300 mg q.2.w. (n = 11) | Tralokinumab 300 mg q.4.w. (n = 13) | Placebo (n = 6) | |
Patients with ≥ 1 TEAE, n (%) | |||
Patients with any treatment-emergent AE | 7 (63.6) | 6 (46.2) | 4 (66.7) |
Most common TEAE,a n (%) | |||
Viral upper respiratory tract infection | 2 (18.2) | 1 (7.7) | 0 (0) |
Upper respiratory tract infection | 2 (18.2) | 0 (0) | 0 (0) |
Atopic dermatitis | 0 (0) | 0 (0) | 1 (16.7) |
SAE, n (%) | |||
Patients with a SAE | 0 (0) | 0 (0) | 0 (0) |
Treatment withdrawal due to AE, n (%) | |||
Patients who stopped | 0 (0) | 0 (0) | 0 (0) |
Deaths, n (%) | |||
Patients who died | 0 (0) | 0 (0) | 0 (0) |
Notable harms, n (%) | |||
Eczema herpeticum | 0 (0) | 0 (0) | 0 (0) |
Malignancies diagnosed after randomization | 0 (0) | 0 (0) | 0 (0) |
Skin infection requiring systemic treatment | 0 (0) | 0 (0) | 1 (16.7) |
Conjunctivitis | 0 (0) | 1 (7.7) | 0 (0) |
Injection-site reactions | 0 (0) | 1 (7.7) | 0 (0) |
Oral herpes | 1 (9.1) | 0 (0) | 0 (0) |
Upper respiratory infection | 2 (18.2) | 0 (0) | 0 (0) |
Acne | 0 (0) | 0 (0) | 0 (0) |
AE = adverse event; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
a10% or greater of patients in any group.
Sources: Clinical Study Report for ECZTRA 613 and the sponsor’s Summary of Clinical Evidence.14
Table 33: Harms Outcomes for ECZTRA 1, 2, 3 in Maintenance (or Continuous) Treatment Period — Maintenance (Continuous) Safety Analysis Set (Adults, Original Review)
Adverse events | ECZTRA 1 (follow-up: 52 weeks) | ECZTRA 2 (follow-up: 52 weeks) | ECZTRA 3 (follow-up: 32 weeks) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Week 16 tralokinumab responders | Week 16 placebo responders | Week 16 tralokinumab responders | Week 16 placebo responders | Week 16 tralokinumab responders | Week 16 tralokinumab nonresponders | ||||||
Tralokinumab q.2.w. N = 68 | Tralokinumab q.4.w. N = 76 | Placebo N = 35 | Placebo N = 29 | Tralokinumab q.2.w. N = 91 | Tralokinumab q.4.w. N = 89 | Placebo N = 46 | Placebo N = 31 | Tralokinumab q.2.w. + TCS N = 69 | Tralokinumab q.4.w.+ TCS N = 69 | Tralokinumab q.2.w. + TCS N = 95 | |
Patients with ≥ 1 TEAE, n (%) | |||||||||||
Patients with any TEAE | 54 (79.4) | 53 (69.7) | 25 (71.4) | 19 (65.5) | 62 (68.1) | 56 (62.9) | 32 (69.6) | 15 (48.4) | 48 (69.6) | 41 (59.4) | 62 (65.3) |
Most common eventsa | |||||||||||
Atopic dermatitis | 11 (16.2) | 14 (18.4) | 13 (37.1) | 6 (20.7) | 13 (14.3) | 14 (15.7) | 9 (19.6) | 2 (6.5) | 1 (1.4) | 1 (1.4) | 8 (8.4) |
Viral upper respiratory tract infection | 14 (20.6) | 18 (23.7) | 4 (11.4) | 2 (6.9) | 9 (9.9) | 6 (6.7) | 7 (15.2) | 4 (12.9) | 12 (17.4) | 9 (13.0) | 20 (21.1) |
Upper respiratory tract infection | 1 (1.5) | 2 (2.6) | 1 (2.9) | 1 (3.4) | 14 (15.4) | 9 (10.1) | 3 (6.5) | 2 (6.5) | 7 (10.1) | 3 (4.3) | 6 (6.3) |
SAE, n (%) | |||||||||||
Patients with an SAE | 1 (1.5) | 3 (3.9) | 0 (0) | 1 (3.4) | 0 (0) | 3 (3.4) | 0 (0) | 0 (0) | 3 (4.3) | 0 (0) | 2 (2.1) |
Treatment withdrawal due to AE, n (%) | |||||||||||
Patients who stopped | 1 (1.5) | 1 (1.3) | 0 (0) | 0 (0) | 2 (2.2) | 1 (1.1) | 0 (0) | 0 (0) | 0 (0) | 1 (1.4) | 1 (1.1) |
Deaths, n (%) | |||||||||||
Patients who died | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Harms of special interest, n (%) | |||||||||||
Eczema herpeticum | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (1.1) | 0 (0) | 0 (0) | 1 (3.2) | 0 (0) | 0 (0) | 1 (1.1) |
Malignancies diagnosed after randomization | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (1.1) | 0 (0) | 0 (0) | 0 (0) | 1 (1.4) | 0 (0) |
Skin infections requiring systemic treatment | 2 (2.9) | 2 (2.6) | 0 (0) | 0 (0) | 2 (2.2) | 1 (1.1) | 1 (2.2) | 1 (3.2) | 0 (0) | 0 (0) | 1 (1.1) |
Eye disorders | |||||||||||
Conjunctivitis | 3 (4.4) | 4 (5.3) | 0 (0) | 0 (0) | 5 (5.5) | 1 (1.1) | 2 (4.3) | 1 (3.2) | 3 (4.3) | 0 (0) | 2 (2.1) |
Allergic conjunctivitis | 3 (4.4) | 1 (1.3) | 2 (5.7) | 0 (0) | 2 (2.2) | 3 (3.4) | 1 (2.2) | 0 (0) | 0 (0) | 1 (1.4) | 2 (2.1) |
Keratoconjunctivitis | 2 (2.9) | 0 (0) | 0 (0) | 0 (0) | 1 (1.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Keratitis | 1 (1.5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Viral conjunctivitis | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (1.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Bacterial conjunctivitis | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (1.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Injection-site reactions | 5 (7.4) | 7 (9.2) | 1 (2.9) | 0 (0) | 4 (4.4) | 4 (4.5) | 0 (0) | 0 (0) | 5 (7.2) | 4 (5.8) | 5 (5.3) |
Oral herpes | 1 (1.5) | 3 (3.9) | 0 (0) | 1 (3.4) | 1 (1.1) | 0 (0) | 1 (2.2) | 2 (6.5) | 3 (4.3) | 4 (5.8) | 4 (4.2) |
Upper respiratory infection | 1 (1.5) | 2 (2.6) | 1 (2.9) | 1 (3.4) | 14 (15.4) | 9 (10.1) | 3 (6.5) | 2 (6.5) | 7 (10.1) | 3 (4.3) | 6 (6.3) |
Acne | 1 (1.5) | 0 (0) | 1 (2.9) | 1 (3.4) | 0 (0) | 2 (2.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
AE = adverse event; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SAE = serious adverse event; TEAE = treatment-emergent adverse event; TCS = topical corticosteroids.
a10% or greater of patients in any group.
Source: Clinical Study Reports for ECZTRA 1,15 2,16 and 317 and the sponsor’s Summary of Clinical Evidence.14
The blinding of patients and study personnel was appropriately maintained. However, given that a placebo was used in these studies, it is possible that patients may have potentially become unblinded or aware of their assignments through improvement or lack of improvement (placebo) in their AD symptoms over the study period, which could have biased the results in favour of tralokinumab for patient-reported outcomes (i.e., [adolescent] worst daily pruritus NRS, eczema-related sleep NRS, POEM, DLQI, CDLQI, and HADS); however, the presence and extent of such potential bias is unknown. The investigator-assessed coprimary end points (EASI-75 and an IGA of 0 or 1) were semi-objective measures, resulting in some potential for bias, but the risk is probably low.
Efficacy analyses were conducted in the full analysis set, which included all patients randomized to treatment who were exposed to tralokinumab or placebo, which is different from the ideal approach to assess the effect of assignment to the intervention; that is, the intention-to-treat (ITT) approach in which all randomized patients were included. The exclusion of randomized patients from the analysis set could potentially affect randomization and study results. In the ECZTRA 6 study, 4.9% of randomized patients were excluded from the full analysis set in the initial treatment period. However, given that the proportion of excluded patients was small and balanced between the treatment groups and the baseline patient characteristics in the full analysis set were also balanced between treatment groups, the impact on the study results is likely small. Among the adult studies, the full analysis set included all or almost all randomized patients.
The primary analysis of binary outcomes was assessed based on the composite (or COVID-modified composite) estimand, in which patients who had received rescue medication before the week 16 visit were considered nonresponders. This estimand was considered appropriate because it is able to maintain ITT principles while accounting for confounding effects of rescue medication on treatment response, as noted in the Biologics Safety and Efficacy Assessment Report by Health Canada.89 In the ECZTRA 6 study, an IGA of 0 or 1, EASI-75, and HADS depression and anxiety scores of less than 8 at week 16 were assessed based on a small number of events in both treatment groups, which could lead to instability of the treatment-effect estimates (i.e., the results could change substantially with small changes in event rates in either group). For continuous outcomes, the primary estimand was conducted using the hypothetical approach, in which intercurrent events (e.g., discontinuation of study intervention or initiation of rescue medication) were censored and imputed using multiple imputations and assuming data were missing at random. However, this assumption is unlikely to hold as the use of rescue medication was driven by the lack of efficacy of study intervention. Nonetheless, for key secondary continuous end points, the treatment policy estimand (i.e., including all observed data regardless of intercurrent events) and the composite estimand (i.e., patients who received rescue medication were censored and imputed by using worst observation carried forward), which likely yielded more conservative results compared with the hypothetical estimand, were in general consistent with the hypothetical estimand in all studies (except for change from baseline in DLQI at week 16 in the ECZTRA 7 study), increasing the certainty of the results. Continuous secondary and exploratory end points (change from baseline in EASI, POEM, worst daily pruritus NRS, eczema-related sleep NRS, and HADS scores) were at a high risk of bias due to a large amount of missing data that were not appropriately accounted for in the statistical analysis (i.e., absence of additional estimands). Sensitivity analyses were conducted for all primary and key secondary outcomes to assess the impact of different imputation methods, which showed results consistent with the primary analyses.
A hierarchal testing procedure was appropriately used to account for multiplicity in the coprimary and key secondary end points (as well as an IGA of 0 or 1 and EASI-75 maintenance end points in the ECZTRA 1 and 2 studies). Analyses of other secondary and exploratory end points were not part of the statistical hierarchy and are at an increased risk of type I error (false-positive results). Although subgroup analyses were specified a priori, the lack of sample-size consideration and control for multiplicity render the findings exploratory.
Longer-term efficacy results in those who responded to tralokinumab initially at week 16 were available in the ECZTRA 1, 2, and 6 studies (52 weeks), and ECZTRA 3 study (32 weeks); however, the sample sizes were reduced considerably at these later time points for all studies (91 or fewer patients in each treatment group) and the wide CIs for the IGA of 0 or 1 and EASI-75 outcomes impeded drawing definitive conclusions. The results for an IGA of 0 or 1 and EASI-75 were inconsistent in the identically designed ECZTRA 1 and 2 trials at week 52, which increases the uncertainty of the longer-term efficacy of tralokinumab.
Instruments used for measuring the coprimary and key secondary end points (IGA, EASI, SCORAD, worst pruritus NRS, and POEM) were shown to be both reliable and valid in adults with AD, except for DLQI, for which evidence regarding reliability and validity in patients with AD was not identified. Evidence for the validity and reliability of these instruments in the adolescent population was limited.
External Validity
The clinical experts noted that the inclusion criteria were generally reflective of the clinical practice in Canada, although the experts anticipated that patients who previously had uncontrolled diseaseor were deemed not to be candidates for systemic immunosuppressants would be eligible for tralokinumab treatment. This is more closely reflected in the ECZTRA 7 trial, in which patients were required to have had uncontrolled disease or deemed not to be candidates for cyclosporine A treatment to allow for study enrolment. The populations of other studies (ECZTRA 1, 2, 3, and 6) are less generalizable to clinical practice, given that these studies involved a relatively small proportion of patients with prior immunosuppressant treatment. In addition, patients who had recently used phototherapy and systemic immunosuppressants were excluded, which the clinical experts noted would be unlikely in clinical practice when considering eligibility for tralokinumab treatment. The clinical experts also noted that proportionally more trial patients were white and had received prior systemic corticosteroids at baseline compared with what is seen in clinical practice. However, the clinical experts did not expect that any of these differences in treatment history and demographics would have a significant effect on the generalizability of the study findings.
The ECZTRA 3 and 7 trials assessed the use of tralokinumab in combination with TCS, and this is more reflective of real-world practice compared with other included studies (tralokinumab monotherapy), based on clinical expert input that patients typically use biologics in combination with TCS for active lesions. While some patients use biologics as monotherapy (without TCS), as in the ECZTRA 1, 2, and 6 studies, the clinical expert noted that this group represents a small proportion of patients receiving systemic treatments. Furthermore, maintenance regimens of tralokinumab 300 mg every 2 weeks and every 4 weeks were assessed in the studies, although the clinical experts commented that an every 4 weeks regimen is not commonly prescribed in current practice.
The efficacy outcomes assessed in the study, including severity and extent of AD, symptoms, and HRQoL, were of clinical importance to patients and clinicians. The clinical experts considered the IGA, EASI, DLQI, and worst daily pruritus NRS to be clinically relevant as they are commonly used in clinical practice to assess response to treatment. SCORAD and POEM, which are AD-specific symptom scales commonly used in clinical trials, and HADS are not used in clinical practice, according to the clinical experts. Patients expressed a need for treatments that are easy to administer. Although ease of use of treatment is not a standalone outcome in the included studies, this outcome was captured in the DLQI questionnaire.
The clinical experts noted that most patients would require at least 6 months of treatment to achieve an optimal response after initiation of tralokinumab treatment. It is possible that the duration of the follow-up in the initial treatment period (16 weeks) of included studies might not have been long enough to fully capture the treatment effects of tralokinumab. The results of the maintenance treatment period (up to 52 weeks of tralokinumab treatment) are likely more generalizable; however, these longer-term results are inconclusive due to issues with internal validity.
The included placebo-controlled trials were the only phase III RCTs of tralokinumab available to date. The clinical experts consulted by CADTH noted that biologic (dupilumab) and JAKi (abrocitinib and upadacitinib) treatments are also available for the treatment of moderate-to-severe AD in adolescent and adult patients and are considered relevant comparators of tralokinumab. The absence of head-to-head evidence comparing tralokinumab and these comparator drugs is an evidence gap in the treatment of moderate-to-severe AD. No pivotal studies or RCTs that assess the use of tralokinumab after the failure of dupilumab or JAKi options were identified by the sponsor’s systematic review.
Long-Term Extension Studies
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Description of Study
One ongoing, open-label, single-arm, multicentre, LTE trial (ECZTEND) was summarized to provide additional evidence on the efficacy and safety of tralokinumab in patients with moderate-to-severe AD who previously participated in the clinical trials for tralokinumab (i.e., ECZTRA 1 to 8 and TraSki).14 Patients were eligible to participate in the ECZTEND study if they completed the treatment period(s) in 1 of the parent trials, regardless of the type of previous treatment (tralokinumab or placebo) or treatment response. The study is being conducted at approximately 330 sites in Canada, Europe, Japan, and the US. The trial includes a screening period of 2 weeks (week −2 to week 0), which is expected to overlap with the last period in the parent trial for the majority of the patients; a long-term treatment period of approximately 0.5 to 5 years; and a safety follow-up period of 14 weeks, starting 2 weeks after the last dose of tralokinumab (i.e., the final safety follow-up visit will be 4 to 16 weeks after the last dose). The primary outcome was long-term safety, specifically the number of AEs experienced during the study. The secondary outcomes are for efficacy and included achieving an IGA of 0 or 1 and achieving EASI-75 in the parent trial, measured at weeks 16, 56, 88, 104, 136, 152, 184, 216, and 248. Blinding of previous treatment allocation was maintained for patients who continued from a blinded arm of a parent trial (i.e., ECZTRA 1 to 8) and entered the LTE study.
The most recent interim analyses summarized are from 4 distinct datasets:
Adult cohort:
All participants from the ECZTRA 1, 2, 3, 4, 5, and 7 studies enrolled in the ECZTEND trial, with patients having received up to 42 months of total tralokinumab treatment at data cut-off on April 30, 2021 (≤ 2.5 years in the open-label extension ECZTEND trial and ≤ 1 year in the parent trial; n = 1,442).64 These are referred to as “all participants.”
All participants from the ECZTRA 1 and 2 studies enrolled in the ECZTEND trial who completed 4 years of tralokinumab treatment at data cut-off on April 30, 2022 (52 weeks in the ECZTRA 1 or 2 study plus 152 weeks in the ECZTEND study; n = 347; efficacy data only).90 These are referred to as the “4-year subgroup.”
All participants from the ECZTRA 1 and 2 studies enrolled in the ECZTEND study who completed 3 years of tralokinumab treatment at data cut-off on April 30, 2021 (52 weeks in the ECZTRA 1 study or 2 plus 104 weeks in the ECZTEND study; n = 347; safety data only).62 These are referred to as the “3-year subgroup.”
Adolescent cohort:
All participants from ECZTRA 6 enrolled in the ECZTEND study who had up to 3 years of tralokinumab treatment at data cut-off on April 30, 2022 (52 weeks in the ECZTRA 6 study plus up to 104 weeks in the ECZTEND study; n = 127).60
Populations
Eligibility Criteria
Individuals were eligible to participate in the ECZTEND study if they met the following inclusion criteria:91
completed the treatment period(s) of 1 of the parent trials: ECZTRA 1 to 8, or TraSki
able and willing to self-administer tralokinumab treatment (or have it administered by a caregiver) at home after the initial 3 injection visits at the trial site
use of a stable dose of emollients twice daily (or more often, as needed) for at least 14 days before the baseline and continuing throughout the study and follow-up periods.
Individuals were excluded from participating in the ECZTEND study if they met the following exclusion criteria:91
any condition that required permanent discontinuation of trial treatment in the parent trial
more than 26 weeks have elapsed since the patient received the last injection of the investigational medicinal product in the parent trial
patients who, during their participation in the parent trial, developed an AE leading to temporary discontinuation or a serious SAE deemed related to tralokinumab by the investigator, which in the opinion of the investigator, could indicate that continued treatment with tralokinumab may present an unreasonable safety risk for the patient
treatment with systemic immunosuppressive and/or immunomodulating drugs and/or systemic corticosteroids within 5 half-lives before the baseline
treatment with a topical PDE-4 inhibitor or a topical JAKi within 2 weeks before the baseline
receipt of any marketed biological therapy (i.e., immunoglobulin or anti-immunoglobulin E), including dupilumab or investigational biologic agents:
any cell-depleting agents, including, but not limited to, rituximab, within 6 months before the baseline, or until the lymphocyte count returns to normal, whichever is longer
other biologics: within 3 months or 5 half-lives, whichever is longer, before the baseline.
Baseline Characteristics
The baseline demographic and clinical characteristics for each ECZTEND analysis cohort is summarized in Table 34 and Table 35.
The median age of the adult population (all participants, N = 1,442) was 38 years (interquartile range [IQR] = 27 to 50 years) with a median age of AD onset of 3 years (IQR = 1 to 15 years). The adult population had a median duration of AD of 27 years (IQR = 18 to 39). The median age of the adolescent population (N = 127) was 16 years (IQR = 14 to 17) with a median age of AD onset of 1 year (IQR = 0 to 2). At the ECZTEND baseline, the median duration of AD in the adolescent population was 15 years (IQR = 13 to 17). In the adult population, more males than females were enrolled (57.6% versus 42.4%). The majority of patients in the adult and adolescent populations were white (75.9% in adults; 73.2% in adolescents). For the adult population (all-participants set), most patients were enrolled following participation in the ECZTRA 1, ECZTRA 2, or ECZTRA 3 study (71.1%). All adolescents completed ECZTRA 6 (100%) as the parent trial.
At baseline of the parent trials, the adult population all had an IGA of 3 (53.1%) or 4 (46.9%), a median EASI score of 26.8 (IQR = 20.5 to 37.6), a median SCORAD score of 67.7 (IQR = 60.0 to 77.9), a median DLQI score of 16 (IQR = 11 to 22). The median score on the worst weekly pruritus NRS for the adult population at parent trial baseline was 7.9 (IQR = 6.8 to 8.8).
The adolescent population showed an IGA of 3 (65.4%) or 4 (34.6%) at parent trial baseline. The median EASI score was 25.6 (IQR = 19.2 to 36.9), the median SCORAD score was 66 (IQR = 57.4 to 75.5), the median CDLQI score was 14 (IQR = 8.5 to 18.5), and the median POEM score was 21 (IQR = 16 to 24) at parent trial baseline in the adolescent population. Less than half (44.1%) of adolescent patents were enrolled in North America. The median number of days since the last dose in parent trial was 50 (IQR = 21 to 110 days).
Table 34: Patient Demographics at ECZTEND Baseline
Characteristic | ECZTEND | |||
|---|---|---|---|---|
Adult safety analysis set (all participants) N = 1,442 | Adult efficacy analysis set (all participants) N = 616 | Adult efficacy analysis (4-year subgroup) or safety analysis set (3-year subgroup) N = 347 | Adolescent efficacy or safety analysis set N = 127 | |
Age, median years (IQR) | 38.0 (27.0 to 50.0) | 40.0 (27.0 to 51.0) | 42.0 (30.0 to 53.0) | 16.0 (14.0 to 17.0) |
Age at onset of AD, median years (IQR) | 3.0 (1.0 to 15.0) | 3.0 (1.0 to 14.5) | 3.0 (1.0 to 15.0) | 1.0 (0.0 to 2.0) |
Duration of AD, median years (IQR) | 27.0 (18.0 to 39.0) | 29.0 (19.0 to 42.0) | 29.0 (19.0 to 43.0) | 15.0 (13.0 to 17.0) |
Sex, n (%) | ||||
Male | 831 (57.6) | 369 (59.9) | 205 (59.1) | 65 (51.2) |
Female | 611 (42.4) | 247 (40.1) | 142 (40.9) | 62 (48.8) |
Race, n (%) | ||||
White | 1,093 (75.9) | 445 (72.5) | 259 (74.6) | 93 (73.2) |
Black | 108 (7.5) | 41 (6.7) | 20 (5.8) | 12 (9.4) |
Asian | 203 (14.1) | 106 (17.3) | 56 (16.1) | 14 (11.0) |
Parent trial, n (%) | ||||
ECZTRA 1 | 450 (31.2) | 326 (52.9) | 224 (64.6) | NA |
ECZTRA 2 | 293 (20.3) | 168 (27.3) | 123 (35.4) | NA |
ECZTRA 3 | 282 (19.6) | 85 (13.8) | NA | NA |
ECZTRA 4 | 31 (2.1) | NA | NA | NA |
ECZTRA 5 | 149 (10.3) | 37 (6.0) | NA | NA |
ECZTRA 6 | NA | NA | NA | 127 (100) |
ECZTRA 7 | 237 (16.4) | NA | NA | NA |
AD = atopic dermatitis; IQR = interquartile range; NA = not applicable.
Note: In the adolescent population, CDLQI was administered. In parent trials, worst pruritus NRS was assessed daily; in the ECZTEND study, worst pruritus NRS was assessed based on recall of the previous week before the visit.
Sources: Blauvelt et al. (2022),64 Blauvelt et al. (2023),63 Langley et al. (2022),62 Simpson et al. (2023),60 and the sponsor’s Sumary of Clinical Evidence.14
Table 35: Baseline Disease Characteristics — ECZTEND
Disease characteristics | Adult safety analysis set (all participants) N = 1,442 | Adult efficacy analysis set (all participants) N = 616 | Adult efficacy analysis (4-year subgroup) or safety analysis set (3-year subgroup) N = 347 | Adolescent efficacy or safety analysis set N = 127 | ||||
|---|---|---|---|---|---|---|---|---|
Parent trial baseline | ECZTEND baseline | Parent trial baseline | ECZTEND baseline | Parent trial baseline | ECZTEND baseline | Parent trial baseline | ECZTEND baseline | |
IGA severity, n (%) | ||||||||
0 or 1 | NA | 442 (30.6) | NA | 179 (29.1) | NA | 98 (28.2) | NA | 55 (43.3) |
2 | NA | 524 (36.3) | NA | 211 (34.3) | NA | 123 (35.4) | NA | 43 (33.9) |
3 | 765 (53.1) | 391 (27.1) | 315 (51.1) | 185 (30.0) | 172 (49.6) | 106 (30.5) | 83 (65.4) | 26 (20.5) |
4 | 677 (46.9) | 85 (5.9) | 301 (48.9) | 41 (6.7) | 175 (50.4) | 20 (5.8) | 44 (34.6) | 3 (2.4) |
EASI, median (IQR) | 26.8 (20.5 to 37.6) | 4.8 (1.7 to 12.0) | 26.9 (19.7 to 37.5) | 4.8 (2.0 to 12.5) | 26.7 (19.7 to 38.4) | 4.7 (2.2 to 12.4) | 25.6 (19.2 to 36.9) | 2.6 (0.6 to 7.8) |
SCORAD, median (IQR) | 67.7 (60.0 to 77.9) | 30.2 (18.7 to 45.0) | 67.4 (60.2 to 77.0) | 32.0 (19.8 to 46.1) | 68.1 (60.8 to 78.1) | 32.8 (20.6 to 46.9) | 66.0 (57.4 to 75.5) | 25.8 (13.5 to 37.5) |
DLQI,a n | 1,391 | 1,400 | 608 | 595 | 343 | 332 | 120 | 106 |
Median (IQR) | 16.0 (11.0 to 22.0) | 5.0 (2.0 to 10.0) | 17.0 (11.0 to 22.0) | 5.0 (2.0 to 9.0) | 17.0 (11.0 to 230) | 5.0 (2.0 to 10.0) | 14.0 (8.5 to 18.5) | 4.0 (NR) |
Worst weekly pruritus NRS,b n | 1,257 | 1,440 | 576 | 615 | 346 | 347 | NR | NR |
Median (IQR) | 7.9 (6.8 to 8.8) | 5.0 (3.0 to 7.0) | 7.9 (6.9 to 8.9) | 5.0 (3.0 to 7.0) | 7.9 (6.9 to 8.9) | 5.0 (3.0 to 8.0) | NR | NR |
POEM, n | NR | NR | NR | NR | NR | NR | 122 | 122 |
Median (IQR) | NR | NR | NR | NR | NR | NR | 21.0 (16.0 to 24.0) | 10.0 (5.0 to 15.0) |
BSA, median (IQR) | NR | NR | NR | NR | NR | NR | 46.0 (29.0 to 58.0) | NR |
Region, n (%) | ||||||||
North America | NR | NR | NR | 56 (44.1) | ||||
Europe | NR | NR | NR | 71 (55.9) | ||||
Time from last dose in parent trial | ||||||||
Median days (IQR) | NR | NR | NR | 50.0 (21.0 to 110.0) | ||||
BSA = body surface area; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; IQR = interquartile range; NA = not applicable; NR = not reported; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring Atopic Dermatitis.
aIn the adolescent population, the CDLQI was administered.
bIn parent trials, worst pruritus NRS was assessed daily; in the ECZTEND study, worst pruritus NRS was assessed based on recall of the previous week before the visit.
Sources: Blauvelt et al. (2022),64 Blauvelt et al. (2023),63 Langley et al. (2022),62 Simpson et al. (2023),60 and the sponsor’s Summary of Clinical Evidence.14
Interventions
Following the screening period, and at least 2 weeks after the last dose in the parent trial, patients from the ECZTRA 6 (adolescents) parent trial received a dose of tralokinumab 300 mg (2 mL) administered by subcutaneous injection at baseline of ECZTEND. Patients from all other parent trials (adults) received an initial loading dose of tralokinumab 600 mg (4 mL) at baseline. For the rest of the treatment period, all patients received doses of tralokinumab 300 mg every 2 weeks (2 mL).91
Patients self-injected tralokinumab or had tralokinumab injected by a caregiver in their home after adequate training by site staff during the first 3 treatment visits (weeks 0, 2, and 4). Patients who already had experience with home use of tralokinumab from the open-label arms of parent trials (ECZTRA 1 or 2) were able to begin self-injecting at baseline without training. At the trial visits, tralokinumab was to be injected at the trial site, preferably by the patient or their caregiver, or alternatively by site staff. The first 3 treatment visits were also used for postdose monitoring for immediate drug reactions, as some patients will have received a placebo in the parent trial and would therefore be naive to tralokinumab. Patients who transferred from the open-label arms or trials (ECZTRA 1, 2, 4, or 6 or TraSki) and received at least 3 doses of tralokinumab in the parent trial were exempt from this monitoring.91
Patients were permitted to use concomitant TCS (US Class ≥ 4 or Europe Class ≤ 3) or TCIs at the investigator’s discretion. If TCS were used, the patient was to be monitored for signs of local or systemic TCS toxicity, and the safety and appropriateness of continued or repeated courses of TCS therapy was to be evaluated by site staff.91 Systemic rescue treatments could include systemic corticosteroids or nonsteroidal systemic immunosuppressive drugs (e.g., cyclosporine A, methotrexate, mycophenolate mofetil, or azathioprine), and treatment with the investigational medicinal product was to be immediately discontinued. After systemic treatment was completed, tralokinumab could be resumed if deemed appropriate by the investigator, but not sooner than 5 half-lives after the last dose of systemic rescue treatment.
All patients were required to use an emollient twice daily (or more often, as needed) for at least 14 days before baseline, and continue their background emollient treatment throughout the trial (including the safety follow-up). Other permitted concomitant medications included oral antibiotics, antiviral, antifungal therapies for skin infections, and oral antihistamines.91
Prohibited concomitant therapies, in addition to those listed in the exclusion criteria and as rescue medications, were phototherapy and 3 or more bleach baths per week.91
Outcomes
Data used for the interim analyses for the adult cohort include safety and efficacy data collected during the treatment period from the ECZTEND baseline up to week 152, reflecting up to 4 years of total tralokinumab exposure (and week 104, up to 3 years of tralokinumab exposure for a subgroup) that includes the parent trial duration.61,62 Data used for the interim analyses for the adolescent cohort include safety data collected during the treatment period from the ECZTEND baseline up to week 104 and efficacy data up to week 56.61,62 Efficacy analyses for the adolescent cohort were not performed at week 104, as few patients had reached this time point at the time of data cut-off.
The primary objective of the study was to evaluate the long-term safety of tralokinumab, and has been summarized as AEs, SAEs, AEs of special interest, withdrawals due to AEs, and discontinuations.60,62
The secondary objective of the study was to evaluate the effectiveness of tralokinumab given as continuous treatment, re-treatment, or introduced for the first time in tralokinumab-naive patients. Effectiveness has been summarized as the percentage of patients achieving an IGA score of 0 or 1 and EASI-75 at weeks 16, 56, 88, 104, and 152 for the adult cohort (and weeks 16 and 56 for the adolescent cohort).60-62
Other exploratory end points summarized include the worst weekly pruritus NRS score, worst weekly pruritus NRS score of 3 or lower, eczema-related weekly sleep NRS scores, DLQI score of 5 or lower, and median improvement in EASI from baseline.60,62
Statistical Analysis
Adult Cohort
At the time of data cut-off on April 30, 2021, a total of 1,442 adult patients who had completed the ECZTRA 1, 2, 3, 4, 5, or 7 pivotal trials were enrolled in the ECZTEND study. All 1,442 patients who received tralokinumab were included in the safety analysis set, with patients having received up to 42 months of total tralokinumab exposure. The efficacy analysis set included 616 patients, all of whom had reached the 2-year (week 104) time point in the ECZTEND study or would have reached that time point had they not discontinued earlier, before the data cut-off. Demographic and baseline characteristics were presented as descriptive statistics. For the primary safety end point, the number and proportion of patients experiencing AEs during the treatment period were presented. Results for the secondary efficacy end points (an IGA of 0 or 1 and EASI-75) and exploratory efficacy end points (worst weekly pruritus NRS of 3 or lower and DLQI of 5 or lower) were presented descriptively as the proportion of patients achieving the end point at week 104 relative to baseline (as-observed, LOCF, and mNRI). The LOCF method imputes the value recorded at the participant’s last visit for subsequent missed time points. The mNRI method considers participants who discontinue from the trial due to AEs or lack of efficacy as nonresponders, and other missing data are imputed with LOCF. No formal sample size or power calculations were performed, and missing data were not imputed for safety results.61
Furthermore, 347 of the 1,442 patients (24.1%) who were enrolled in the ECZTEND study were from the ECZTRA 1 and 2 pivotal trials (similarly designed, 52-week, monotherapy RCTs). This subgroup of patients was analyzed to evaluate the 3-year efficacy and 4-year safety of tralokinumab treatment (hereafter the 3-year and 4-year subgroups, respectively), with all patients having been enrolled in the ECZTEND study for 2 years (104 weeks) and 3 years (152 weeks) at data cut-off in addition to the 52 weeks in the parent trial. All 347 patients who received tralokinumab were included in the efficacy analysis set, which was identical to the safety analysis set, except for the data cut-off date (duration of study: 3 years for the efficacy analysis and 4 years for the safety analysis).62
Adolescent Cohort
At the time of data cut-off on April 30, 2022, 127 patients who had completed week 52 in the ECZTRA 6 study were enrolled in the ECZTEND study. All 127 patients who received tralokinumab were included in the safety analysis set, which was identical to the efficacy analysis set. Demographic and baseline characteristics were presented as descriptive statistics. For the safety end points, the number of patients with any TEAEs, deaths, SAEs, and withdrawals from the trial due to AEs were presented. Efficacy end points presented were the proportion of patients achieving an IGA of 0 or 1, EASI-75 (as-observed, LOCF, and mNRI), and percent EASI improvement from pivotal trial baseline at week 56 (as observed). In mNRI analysis, discontinuation due to AEs or lack of efficacy were considered nonresponsive. Other missing data were imputed with the LOCF. No formal sample size or power calculations were performed, and missing data were not imputed for safety results.60
Results
Patient Disposition
The patient disposition for each ECZTEND analysis cohort is summarized in Table 36.
In the adult cohort, 1,442 patients were rolled over from the parent trials and 77.1% of them are still enrolled in the ongoing ECZTEND trial. For the 22.9% of the adult patients who discontinued the study, a lack of efficacy (5.5%), other (5.2%), and patient withdrawal (3.4%) were the most common reasons. In the adolescent cohort, 168 patients were screened, 127 were enrolled, and 81.1% of those enrolled remain in the ECZTEND trial. For the 18.9% of adolescent patients who discontinued the study, other (7.1%), lost to follow-up (3.9%), lack of efficacy (2.4%), and parent or guardian withdrawal (2.4%) were the most common reasons. One death (0.8%) due to a car accident was reported as the reason for discontinuation of study in adolescent cohort.
Table 36: Patient Disposition — ECZTEND
Patient disposition | ECZTEND | |||
|---|---|---|---|---|
Adult cohort (all participants) | Adult cohort (4-year subgroup) | Adult cohort (3-year subgroup) | Adolescent cohort | |
Screened, N | NR | ||| | ||| | 168 |
Enrolled, N | 1,442 | 347 | 347 | 127 |
Ongoing, n (%) | ||||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Discontinued study, n (%) | 330 (22.9) | || |||||| | 75 (21.6) | 24 (18.9) |
Adverse event | || ||||| | || ||||| | || ||||| | 1 (0.8) |
Lost to follow-up | || ||||| | || ||||| | || ||||| | 5 (3.9) |
Patient withdrawal | 49 (3.4) | || ||||| | 11 (3.2) | 2 (1.6) |
Parent or guardian withdrawal | 0 | || ||||| | 0 | 3 (2.4) |
Lack of efficacy | 80 (5.5) | || ||||| | 20 (5.8) | 3 (2.4) |
Other | 75 (5.2) | || ||||| | 18 (5.2) | 9 (7.1) |
Unknown | || ||||| | || ||||| | || ||||| | 0 |
Death | 0 | 0 | 0 | 1 (0.8) |
Full analysis set, N | 616 (42.7) | 347 (100) | 347 (100) | 127 (100) |
Safety analysis set, N | 1,442 (100) | 347 (100) | 347 (100) | 127 (100) |
NR = not reported.
Source: Blauvelt et al. (2022),64 Blauvelt et al. (2023),63 Langley et al. (2022),62 Simpson et al. (2023),60 and the sponsor’s Summary of Clinical Evidence.14
Exposure to Study Treatments
In the adult cohort, the median duration of exposure was 131.5 weeks (IQR = 83.4 to 161.8). In the adolescent cohort, the median duration of exposure was |||| ||||| ||||||| || |||| ||||||| The adherence rate (%) was not reported in either population.
Table 37: Patient Exposure — ECZTEND
Exposure | ECZTEND | |||
|---|---|---|---|---|
Adult cohort (all participants) | Adult cohort (4-year subgroup) | Adult cohort (3-year subgroup) | Adolescent cohort | |
Tralokinumab 300 mg q.2.w. ± optional TCS (N = 1,442) | Tralokinumab 300 mg q.2.w. ± optional TCS (N = 347) | Tralokinumab 300 mg q.2.w. ± optional TCS (N = 347) | Tralokinumab 300 mg q.2.w. ± optional TCS (N = 127) | |
Total, patient-years | 2,446.2 | NR | 707.7 | 201.5 |
Duration, mean number of weeks (SD) | ||||| |||||| | NR | ||||| |||||| | |||| |||||| |
Duration, median number of weeks (IQR) | ||||| ||||| || |||||| | NR | ||||| |||||| || |||||| | |||| ||||| || ||||| |
Adherence, % | NR | NR | NR | NR |
IQR = interquartile range; NR = not reported; q.2.w. = every 2 weeks; SD = standard deviation; TCS = topical corticosteroids.
Source: Sponsor’s clinical evidence summary.14
Efficacy
Secondary and exploratory outcomes for efficacy are summarized separately for each analysis cohort. Results are presented relative to baseline in the parent trials.
Efficacy Outcomes in the ECZTEND Study — Adult Cohort (All Participants, n = 616)
EASI-75
EASI-75 was achieved by 411 out of 483 (85.1%) patients at week 104 (observed data, relative to parent trial baseline), as shown in Figure 9.
IGA of 0 or 1
An IGA of 0 or 1 was achieved by 244 out of 483 patients (50.5%) at week 104 (observed data), as shown in Figure 9.
Patient-Reported Outcomes
A worst weekly pruritus NRS score of 3 or lower was achieved by 292 out of 482 patients (60.6%) and a DLQI score of 5 or lower was achieved by 360 out of 471 patients (76.4%) at week 104 (observed data) as shown in Figure 9.
Figure 9: ECZTEND Adult Cohort (All Participants, n = 616) — Proportion of Patients Achieving EASI-75, IGA of 0 or 1, Worst Weekly Pruritus NRS Score of 3 or Lower and DLQI Score of 5 or Lower
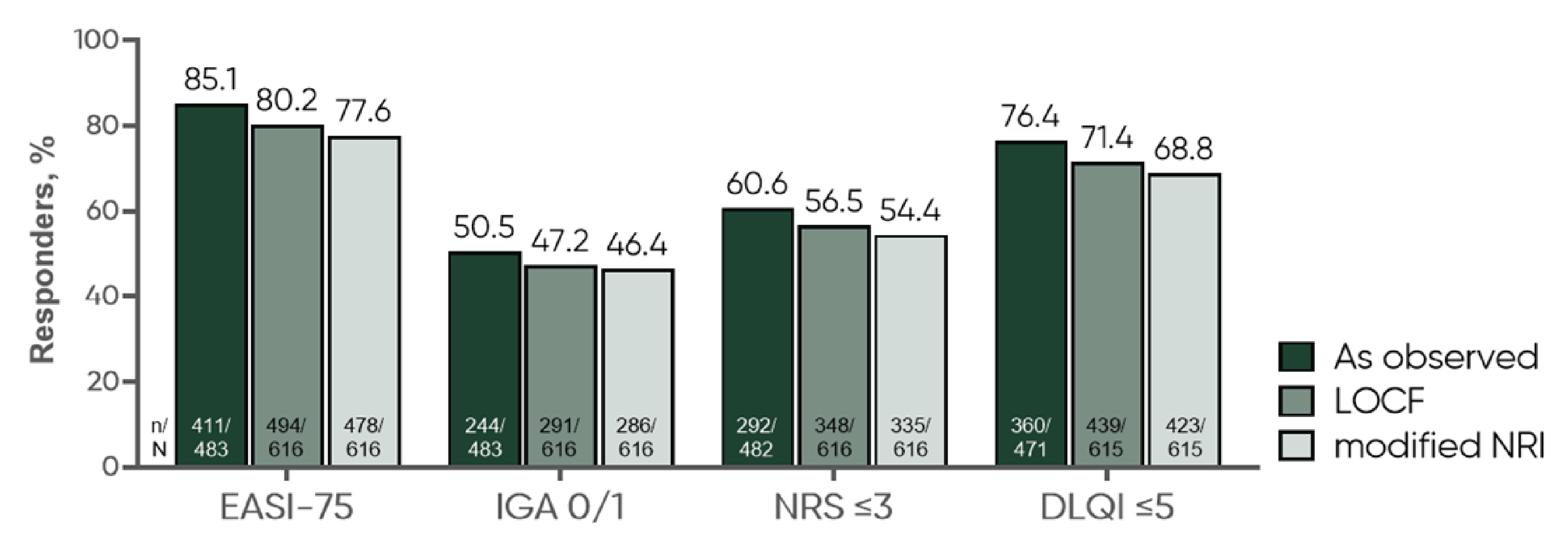
DLQI = Dermatology Life Quality Index; EASI-75 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; IGA = Investigator’s Global Assessment; LOCF = last observation carried forward; NRI = nonresponder imputation; NRS = numeric rating scale.
Note: The observed analysis includes data for all participants with a valid measurement at the indicated time point.
Source: Blauvelt et al. (2022).61
Efficacy Outcomes — Adult Cohort (4-Year Subgroup, n = 347)
The number of patients observed and/or imputed after week 104 was reduced due to some patients declining to re-consent to stay in the study (The original consent patients signed indicated that the ECZTEND trial would run for 3 years. However, this was later amended to 5 years and required re-consent).
IGA of 0 or 1
An IGA score of 0 or 1 was observed in 92 of 175 patients (52.6%) at week 152 in the ECZTEND trial. Results were consistent with sensitivity analyses (LOCF and mNRI) presented in Table 38.
EASI-75
An EASI-75 result was observed in 147 out of 174 patients (84.5%) at week 152 in the ECZTEND trial, relative to parent trial baseline. Results were consistent with sensitivity analyses (LOCF and mNRI) presented in Table 38.
Median EASI
At 4 years of tralokinumab treatment, the median EASI improvement was greater than 90% (exact percentage not reported), relative to parent trial baseline. Median EASI improvement was regained within 12 weeks for patients with 5 weeks of interruption between the parent trial and ECZTEND; the interruption is not expected to affect the efficacy over the long-term. The 4-year cohort subgroup included patients who were receiving tralokinumab monotherapy (every 2 weeks then every 4 weeks or every 2 weeks plus optional TCS in open-label arm) for 52 weeks in the ECZTRA 1 and 2 studies, followed by 152 weeks of treatment in the LTE in the ECZTEND trial. Patients had a variable time between last treatment in the parent trial and first treatment (maximum 26 weeks) (Figure 10).
Figure 10: EASI Response in Patients Treated for 1 Year With Monotherapy and 3 Years in ECZTEND
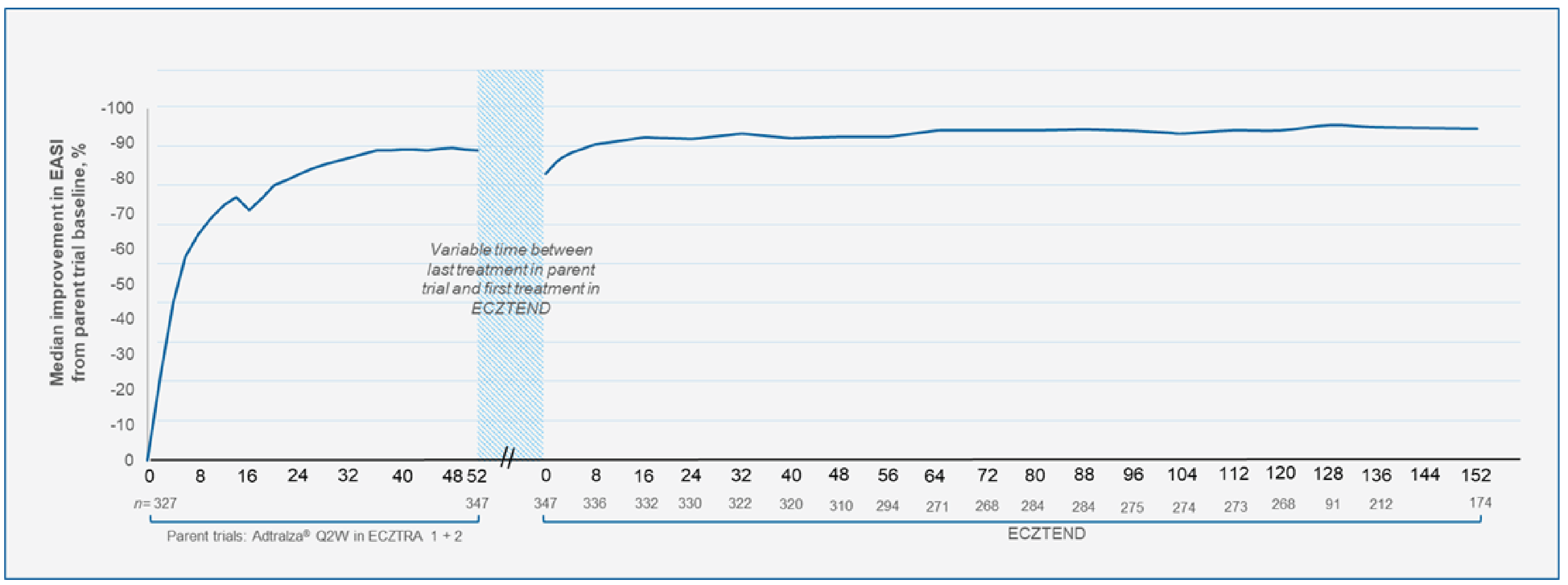
Source: Sponsor’s Summary of Clinical Evidence, ECZTEND.63,91
Worst Weekly Pruritus NRS Score of 4 or Lower (No to Mild Itch)
A worst weekly pruritus NRS score of 4 or lower was observed in 68.0% of patients (absolute number of patients is not reported) at week 152 in the ECZTEND trial.
DLQI Score of 5 or Lower (No to Small Effect of AD on Quality of Life)
A DLQI score of 5 or lower was observed in 79.0% of patients (absolute number of patients was not reported) at week 152 in the ECZTEND study.
Efficacy Outcomes in the ECZTEND Study — Adolescent Cohort (n = 127)
Efficacy outcomes for the adolescent cohort are presented for week 56 (reflecting a total of 2 years of tralokinumab exposure). Week 104 efficacy data were not included in the interim analysis, as few patients had reached 104 weeks in the ECZTEND study at the data cut-off.
EASI-75
At week 56 in the ECZTEND study, 92 out of 109 participants (84.4%) achieved EASI-75 (observed data; relative to ECZTRA 6 baseline) (Figure 11).
IGA of 0 or 1
At week 56 in the ECZTEND study, 67 out of 109 (61.5%) participants achieved an IGA of 0 or 1 (observed data), as shown in Figure 11.
Table 38: Proportion of Patients Achieving an IGA of 0 or 1 or EASI-75 After 152 Weeks in ECZTEND (4-Year Adult Subgroup)
Week | Observed cases | LOCFa | mNRIa | ||||
|---|---|---|---|---|---|---|---|
Patients observed, N | Responders, n (%) | Patients observed/ imputed, N/m | Responders, n (%) | Patients observed/ imputed, N/n | Responders, n (%) | ||
Proportion of patients achieving an IGA of 0 or 1 after 152 weeks | |||||||
Parent trial | 16 | 347 | 93 (26.8) | 347/0 | 93 (26.8) | 347/0 | 93 (26.9) |
52 | 347 | 153 (44.1) | 347/0 | 153 (44.1) | 347/0 | 153 (44.1) | |
ECZTENDb | 0 | 347 | 98 (28.2) | 347/0 | 98 (28.2) | 347/0 | 98 (28.2) |
56 | 294 | 139 (47.3) | 294/53 | 155 (44.7) | 294/53 | 154 (44.4) | |
104 | 274 | 131 (47.8) | 274/73 | 154 (44.4) | 274/73 | 151 (43.4) | |
152 | 175 | 92 (52.6) | 175/89 | 150 (53.0) | 175/89 | 137 (51.9) | |
Proportion of patients achieving EASI-75 after 152 weeks | |||||||
Parent trial | 16 | 347 | 159 (45.8) | 347/0 | 159 (45.8) | 347/0 | 159 (45.8) |
52 | 347 | 281 (81.0) | 347/0 | 281 (81.0) | 347/0 | 281 (81.0) | |
ECZTENDb | 0 | 347 | 210 (60.5) | 347/0 | 210 (60.5) | 347/0 | 210 (60.5) |
56 | 294 | 240 (81.6) | 294/53 | 273 (76.9) | 294/53 | 267 (76.9) | |
104 | 274 | 227 (82.8) | 274/73 | 271 (75.8) | 274/73 | 263 (75.8) | |
152 | 174 | 147 (84.5) | 174/90 | 226 (85.6) | 174/90 | 221 (83.7) | |
EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; LOCF = last observation carried forward; mNRI = modified nonresponder imputation with discontinuations due to adverse event(s) or lack of efficacy set as nonresponse and other missing data imputed with LOCF.
aImputations are only performed on parts of the trial patients who have consented to. Therefore, only patients who consented to continue in the ECZTEND study following a protocol amendment in May 2021 prolonging the trial from 3 to 5 years have been imputed beyond May 2021.
bVariable time between last treatment in parent trial and first treatment in the ECZTEND trial.
Source: Sponsor’s Summary of Clinical Evidence.14
Figure 11: ECZTEND Adolescent Cohort — EASI Score per Visit (A), and IGA 0 or 1 and EASI-75 at Week 56 (B)
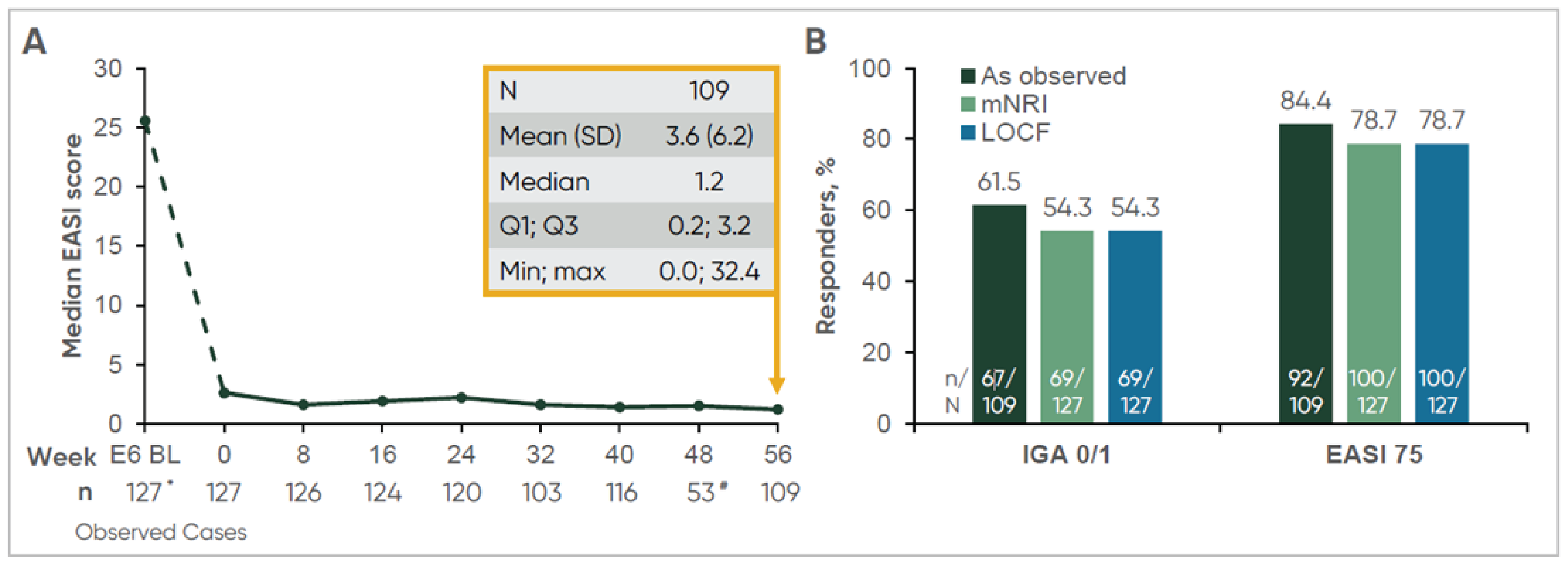
BL = baseline; E6 = ECZTRA 6; EASI 75 = EASI 75 = reduction of at least 75% in Eczema Area Severity Index score from baseline; IGA = Investigator’s Global Assessment; LOCF = last observation carried forward; max = maximum; min = minimum; NRI = nonresponder imputation; SD = standard deviation.
* The 127 participants who entered ECZTEND were not the full ECZTRA 6 population.
# Low n due to changes in the visit schedule after May 2021.
Source: Simpson et al. (2023).60
Harms
Harms outcomes are the primary end point for the ECZTEND study and are summarized separately for each analysis cohort. Table 39 provides harms data.
Harms Outcomes in the ECZTEND Study — Adult Cohort (All Participants, n = 1,442)
Overall, 1,127 patients (78.2%) experienced at least 1 TEAE, with the 3 most common AEs being viral URTI (a common cold, 20.5%), AD (17.8%), and URTI (7.0%). A total of 101 patients (7.0%) reported an SAE, with AD occurring in 6 patients (0.4%), and asthma, coronavirus infection, and eczema herpeticum each occurring in 3 patients (0.2%). Harms of special interest included eye disorders (9.2%), eczema herpeticum (1.2%), malignancy (0.6%), and skin infections requiring systemic treatment (2.5%). Thirty-four patients (2.4%) discontinued treatment due to AE, the most common reason being AD in 8 patients (0.6%). No deaths were reported.
Harms Outcomes in the ECZTEND Study — Adult Cohort (3-Year Subgroup, n = 347)
Overall, 295 patients (85%) experienced at least 1 TEAE, with the most common AEs being viral URTI (a common cold, 28.8%), AD (19.6), and URTI (10.1%). Thirty-one patients (8.9%) reported an SAE, with most reported as single events without any clustering of SAE types. Harms of special interest included eye disorders (10.1%), eczema herpeticum (0.9%), malignancy (0.9%), skin infections requiring systemic treatment (4.0%). Nine patients (2.6%) discontinued treatment due to AE; the most common being AD in 5 (1.4%) patients. No deaths were reported.
Harms Outcomes in the ECZTEND Study — Adolescent Cohort (n = 127)
Overall, 83 patients (65.4%) experienced at least 1 TEAE, with the 3 most common AEs being viral URTI (a common cold, 13.4%), AD (10.2%), and URTI (5.5%). Three patients (2.4%) reported an SAE. Conjunctivitis was reported in 7 patients (3.6%). No other harms of special interest (keratitis or keratoconjunctivitis, eczema herpeticum skin infections requiring systemic treatment, or malignancies) were reported. One patient (0.8%) discontinued treatment due to an AE of AD. One death (0.8%) due to a car accident was reported.
Table 39: Summary of Harms — ECZTEND
Adverse events | ECZTEND | ||
|---|---|---|---|
Adult cohorts | Adolescent cohort | ||
All participants tralokinumab q.2.w. + optional TCS N = 1,442 | 3-year subgroup treatment q.2.w. + optional TCS N = 347 | Tralokinumab q.2.w. + optional TCS N = 127 | |
Most common adverse events, n (%) | |||
≥ 1 adverse event | 1,127 (78.2) | 295 (85.0) | 83 (65.4) |
Viral URTIa | 295 (20.5)a | 100 (28.8) | 17 (13.4) |
Atopic dermatitis | 257 (17.8) | 68 (19.6) | 13 (10.2) |
URTI | 101 (7.0) | 35 (10.1) | 7 (5.5) |
Headache | 79 (5.5) | 20 (5.8) | 5 (3.9) |
Conjunctivitis | 77 (5.3) | NR | 7 (3.6) |
Pruritus | NR | 18 (5.2) | NR |
Asthma | NR | NR | 1 (0.8) |
Serious adverse events, n (%) | |||
Patients with ≥ 1 SAE | 101 (7.0) | 31 (8.9) | 3 (2.4) |
Atopic dermatitis | 6 (0.4) | NR | 0 |
Asthma | 3 (0.2) | NR | 0 |
Corona virus infection | 3 (0.2) | NR | 0 |
Eczema herpeticum | 3 (0.2) | NR | 0 |
Retinal attachment | NR | NR | 1 (0.8) |
Psychotic disorder | NR | NR | 1 (0.8) |
Hypertension | NR | NR | 1 (0.8) |
Patients who stopped treatment due to adverse events, n (%) | |||
Patients who stopped | 34 (2.4) | 9 (2.6) | 1 (0.8) |
Atopic dermatitis | 8 (0.6) | || ||||| | 1 (0.8) |
Breast cancer | 2 (0.1) | NR | NR |
Invasive ductal breast carcinoma | 2 (0.1) | NR | NR |
Prostate cancer | 2 (0.1) | || ||||| | NR |
Conjunctivitis | 2 (0.1) | NR | NR |
Allergic conjunctivitis | 2 (0.1) | NR | NR |
Deaths, n (%) | |||
Patients who died | 0 | 0 | 1 (0.8)b |
Adverse events of special interest, n (%) | |||
Conjunctivitis | ||| ||||| | || ||||| | 7 (3.6) |
Keratoconjunctivitis | || ||||| | || ||||| | 0 |
Keratitis | || ||||| | || ||||| | 0 |
Eczema herpeticum | || ||||| | || ||||| | 0 |
Skin infections requiring systemic treatment | || ||||| | || ||||| | 0 |
Malignancies | || ||||| | || ||||| | 0 |
q.2.w. = every 2 weeks; NR = not reported; SAE = serious adverse event; TCS = topical corticosteroid; URTI = upper respiratory tract infection.
aMost commonly reported as common cold.
bDue to car accident.
Sources: Blauvelt et al. (2022),61 Blauvelt et al. (2023),63 Langley et al. (2022),62 Simpson et al. (2023),60 and the sponsor’s Summary of Clinical Evidence.14
Critical Appraisal
Internal Validity
The sponsor submitted an LTE study that included adult and adolescent patients with AD who received tralokinumab treatment for 4 and 3 years, respectively. Even though the data provided additional information regarding long-term safety and supportive evidence for efficacy of tralokinumab, there are many study limitations, similar to other LTE studies. First, because patients could only enrol after completing the parent trial, there is a risk of selection bias that favours tralokinumab given that patients who perceived the treatment to be benefiting them were more likely to enrol in the extension study. Similarly, long-term safety concerns may be underestimated as those who had experienced intolerable AEs in the parent trials were excluded from the ECZTEND trial. Further, there is a risk of bias in the measurement of patient-reported outcomes (worst weekly pruritus NRS and DLQI), potentially favouring tralokinumab, given the open-label study design. Also, the single-arm study design does not allow for definitive conclusions about the long-term effects attributable to tralokinumab, due to the lack of a comparison group and no adjustment for potential confounding. There is also a sizable proportion of patients who discontinued from the ECZTEND study (18.9% to 28.5%), resulting in missing data, although the results of sensitivity analyses using the LOCF and the mNRI methods were consistent with those of the primary analysis based on observed data. Last, the study findings are at risk of being overestimated given that they are interim findings; only 42.7% (n = 616) of the 1,442 initially enrolled patients were included in the all-participant analysis at the data cut-off.
External Validity
The ECZTEND trial included patients who completed 1 of the parent trials regardless of treatment response. This is different from clinical practice, in which patients are expected to continue tralokinumab treatment only if they demonstrate objective improvement of disease after an adequate trial of treatment. It is unclear what proportion of patients enrolled in the ECZTEND trial represented nonresponders in the parent trial and the extent to which this could affect the generalizability of the study population. Further, the patient populations of the parent trials were heterogeneous in terms of treatment history. It is unclear what proportion of patients received tralokinumab treatment after failure of prior systemic immunosuppressant therapy, which is the likely place of therapy of tralokinumab. The generalizability of the ECZTEND study population is therefore uncertain. Further, while the use of concomitant TCS and rescue medications could influence treatment response, utilization of such medications was not reported in the study and the impact on generalizability of study findings is therefore unclear. The assessment scales included in the study are used in clinical practice and were considered clinically relevant by the clinical experts consulted by CADTH. The experts commented that the duration of follow-up ECZTEND was sufficient to assess the long-term efficacy and safety of tralokinumab.
Indirect Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
In the absence of head-to-head evidence comparing tralokinumab against other relevant advanced therapies used to manage AD, the sponsor submitted 4 ITCs of the treatment effects of tralokinumab and other treatments in patients with moderate-to-severe AD. There was no indication of how the submitted ITCs were selected (e.g., how the NMAs were chosen among those available in the literature). Of the ITCs submitted, 2 were NMAs and 2 were MAICs.19-22
Description of the Network Meta-Analyses
Objectives
The sponsor submitted an NMA conducted by the ICER that aimed to evaluate the relative efficacy and safety of treatment with tralokinumab versus other therapies in adults and children with moderate-to-severe AD.19 However, only data relevant to adults were suitable for the NMA. The ICER NMA was used to inform the sponsor-submitted economic model for the treatment effect of tralokinumab up to week 16.
A sponsor-commissioned NMA, the LEO Pharma NMA, ||||| || |||||||| ||| |||||||| |||||||| ||| |||||| || ||||||||| |||| |||||||||||| |||||| ||||| ||||||||| || |||||||||| |||||||| ||||||| ||| |||| || || ||| || ||||| |||| |||||||||||||||||| |
Study Selection Methods
The comparator studies eligible for inclusion in the submitted NMAs were selected according to systematic reviews specific to each. The systematic reviews for both NMAs were defined by the PICO model described in Table 40.
Clinical evidence for the systematic reviews informing both NMAs were identified using multiple electronic databases, trial registries, and other sources as listed in Table 41. The literature searches for the ICER NMA and the LEO Pharma NMA were current to February 27, 2023, and |||||||| ||| ||||, respectively. In the ICER NMA, full-text articles were screened by single reviewer. The data-extraction process was not specified. In the ICER NMA, an assessment of risk of bias of included articles was conducted using criteria published by the US Preventive Services Task Forces,92 the level of certainty in the available evidence of a net health benefits among each of the intervention of focus was evaluated using the ICER evidence rating matrix, and publication bias was evaluated using the clinicaltrials.gov database of trials. || ||| ||| |||||| |||| ||||||||| ||| |||||| |||| ||||||||||||| |||||||| |||||||||||| |||||||||| ||||||||| |||||||||||||| ||||||| |||||||| |||| |||||||| || |||| ||| ||||||||| |||||||| |||| ||||||| ||||||||| ||| |||| || |||| || ||||||| |||||||| || ||| |||||||||| |||||| ||||||||| ||| ||| |||||| ||| |||| |||||||| ||||| ||| |||||||| |||| || |||| |||| ||||||| ||| || |||| |||||||||| ||| ||||||| ||| |||||||| ||| |||||||| |||||| |||||||||
For the purposes of the ICER NMA, the population of interest was based on the adults aged 18 years and older with moderate-to-severe AD. Analysis of results did not include the adolescent population for tralokinumab, and therefore was not summarized in the sponsor’s submission. Different dosing arms of the same medication were treated as individual comparators (nodes). In the ICER NMA, evidence of intervention efficacy was derived from studies at least 4 weeks in duration. Outcomes evaluated in the ICER NMA included IGA, EASI-50, EASI-75, EASI-90, and PP-NRS scores demonstrating an improvement of more than 4 points at weeks 12 and 16. ||| ||||||| || ||| ||| |||||| |||| ||| |||||||||| || |||||||| |||| ||||||||||| ||||||| ||| |||| || || || || ||||| |||| |||||||||||||||||| ||| ||||||||| |||||| |||| || ||| |||| ||||||||||| |||| ||||||| || |||||||||| ||||||||||||| ||||||||| || ||| ||| |||||| ||| |||||||| ||| |||| |||| ||| |||| ||| |||| ||| |||||||| |||| ||||| ||| ||| ||||| || || ||||||||||||| ||| || ||||| |||||||||||||| ||||||||| ||| ||||||||||||||
Among the comparators eligible for inclusion in the NMA, baricitinib is currently not approved for use in Canada.
Table 40: PICO for the Systematic Reviews Contributing to the Sponsor-Submitted NMAs
PICO component | ICER NMA | LEO Pharma NMA |
|---|---|---|
Population | Adults and children with moderate-to-severe AD whose disease has either not responded adequately to topical therapies or for whom topical therapies have not been tolerated or are medically inadvisable | |||||||| |||| ||||||| || || || ||||| |||| |||||||||||||||||| | |
Interventions |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |
Comparators |
| |||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||||||||||||||| |
Outcome | Efficacy outcomes:
Safety outcomes:
| |||||||| ||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| || |||||||| ||| |||||||| |||||| |||||||||||| ||| ||||||||||||||||||||||| ||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||||||| ||||||||||| ||| || |||||||||||||||||| |||||| |||||||| || |||||||| |||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||| ||||||||||| |
Study designs | RCTs | |||| |
Last updated | February 27, 2023 | ||||| ||| |||| |
AD = atopic dermatitis; AE = adverse event; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; GI = gastrointestinal perforation; HADS = Hospital Anxiety and Depression Scale; ICER = Institute for Clinical and Economic Review; IGA = Investigator’s Global Assessment; IPD = individual patient data; JAK = Janus kinase; MACE = major adverse cardiovascular event; NMA = network meta-analysis; PICO = population, intervention, comparator, and outcome; POEM = Patient-Oriented Eczema Measure; PP-NRS = peak pruritus numeric rating scale; PRO = patient-reported outcome; QoL = quality of life; RCT = randomized controlled trials; SAE = severe adverse event; SCORAD = Scoring Atopic Dermatitis; SF-36 = Short Form (36) Health Survey; TEAE = treatment-emergent adverse event; TCS = topical corticosteroids; VTE = venous thromboembolism event; WPAI = Work Productivity and Activity Impairment.
aIncludes studies of adolescents with moderate AD or severe AD alone.
Sources: ICER NMA technical document19 and LEO Pharma NMA technical document.20
Table 41: Study Selection and Methods for NMAs Submitted by the Sponsor
Characteristics | ICER NMAa | LEO Pharma NMA |
|---|---|---|
Population | Adults > 18 years old with moderate-to-severe AD | ||||||||||| ||||||| ||| |||| || || || || ||||| |||| |||||||||||||||| |
Intervention |
| ||||||||||| |||| || ||||||| || ||| |
Comparator |
| |||||||||| |||| || |||||| || |||||||||||| |||| || |||| ||| || ||| ||| ||| || |||||||||||||||| ||| || ||||| || ||||||||||||||||||| |
Outcomes | Efficacy:
HRQoL and PRO:
Safety:
| ||||||||||||| ||| ||| || ||||||||||| ||| ||||||||||||||||||||||| ||||||||||||||||||| |
Study design | Randomized controlled trials | |||||||||||||||||||||| |
Exclusion criteria | Studies not reporting at least 1 outcome of interest | |||||| ||| ||||||||| || ||||||||||| || ||||||||| |||||||| |||| ||| ||| |||| || |||||| ||||||||| ||||| || || || || ||||| |
Sources searched | Databases:
Grey literature:
| |||||||||||||||||||||||||||||||||||||||||| ||||||||||||| |||||||||||||||||||||| ||||||||||||||||||| ||||| ||||||||||||| |||||||||||| |
Search limits |
| ||||||| |||| |
Selection process | Full-text articles screened by single reviewer, providing justification for exclusions | |||||| ||| ||||||||| |||| ||||||||||||| |||||||| || ||| ||||||||||| |||||||||| ||||||||| ||||||| |||||||||||| ||||||| |||||||| |||| |||||||| || |||| ||| ||||||||| |||||||| |||| |||||||| |||||||| |
Data-extraction process | Not specified | |||| |||||||||| ||| ||||||||| || ||| |||||||| ||| ||||||| |||||||| |||||||| |
Risk-of-bias assessment | USPSTF criteria using the categories “good,” “fair,” and “poor”; procedure used to assess risk of bias was not reported | |||||||| |||| || |||| |||| ||||||| || |||| || |||| |||||||||| ||| ||||||| ||| || ||| |||||||| ||| ||||||| |||||| ||||||||| |
Evidence certainty assessment | ICER evidence rating matrix; procedure used to assess evidence certainty assessment was not reported | ||| |||||||| |
AD = atopic dermatitis; AE = adverse event; CENTRAL = Cochrane Central Register of Controlled Trials; CDLQI = Children’s Dermatology Life Quality Index; CDSR = Cochrane Database of Systematic Reviews; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; EASI-90 = reduction of at least 90% in Eczema Area and Severity Index score from baseline; HRQoL = health-related quality of life; HTA = health technology assessment; ICER = Institute for Clinical and Economic Review; IGA = Investigator’s Global Assessment; NMA = network meta-analysis; PP-NRS = peak pruritus numeric rating scale; POEM = Patient-Oriented Eczema Measure; PRO = patient-reported outcome; q.2.w. = every 2 weeks; q.d. = once per day; SC = subcutaneous; TCS = topical corticosteroids; TEAE = treatment-emergent adverse event; USPSTF = US Preventive Services Task Force.
aThe frequency of administration for the intervention and comparators was not recorded in the ICER NMA technical report nor the sponsor's summary of the clinical evidence.
Sources: ICER NMA technical document,19 LEO Pharma NMA technical document,20 and the sponsor’s Summary of Clinical Evidence.14
Indirect Treatment Comparison Analysis Methods for the ICER and LEO Pharma NMA
ICER Network Meta-Analysis
Indirect comparisons of abrocitinib, baricitinib, dupilumab, tralokinumab, and upadacitinib in the ICER NMA were made using a Bayesian NMA with a noninformative prior distribution for all model parameters. IGAs and PP-NRS scores demonstrating an improvement of at least 4 points were analyzed as dichotomous outcomes (yes or no) using a binomial likelihood and log link. EASI outcomes were analyzed as ordered categorical data with up to 4 distinct groups (i.e., EASI < 50, EASI-50, EASI-75, and EASI-90) representing a reduction in the EASI of less than 50%, at least 50%, at least 75% and at least 90%, respectively. Using the EASI outcomes reported in the included studies, mutually exclusive groups were created by reclassifying the data as less than 50, 50 to 74, 75 to 89, and 90 or greater. Accordingly, a multinomial likelihood model with a probit link with methods from the National Institute for Health and Clinical Excellence Decision Support Unit was used.94
Separate networks for monotherapy trials and combination trials were developed. Methods of analysis are summarized in Table 42. Both random- and fixed-effects models for each network were explored, and the model with the lowest deviance information criterion was considered to have the best fit to the data. Convergence was assessed via visual examination of the Brook-Gelman-Rubin diagnostic and historical plots.95,96 Placebo-adjusted models were presented when these provided a better fit to the data (i.e., regression coefficient was statistically significant and there was a reduction in between-trial heterogeneity).
Only the analysis for the primary model was reported. No information was given on details of the assessment of statistical heterogeneity, statistical consistency, or clinical and methodological similarity across studies.
LEO Pharma Network Meta-Analysis
|||||||| |||||||||| || |||||||||||| |||||||||| |||||||||||| ||| |||||||||||| || ||| ||| |||||| ||| ||| |||| |||||||||||| ||| |||| ||||| ||||||| |||||| ||||||| ||| ||| ||||||||||||||| || |||||||| ||| |||||||||| || ||||| ||| ||| |||||||| ||||||| ||||| |||||| ||||||||||| |||||| ||| ||| ||||||||| |||| || ||||||| ||||||||| ||||||| |||| |||||||| ||| |||||| ||||||||| ||| ||| |||||||| |||| ||| |||||||||| |||||||||||| ||| ||||| ||| ||||||||| ||||||| ||| ||||| ||||||||||| |||||| ||||||| ||||| ||| |||||||| ||| ||| ||||||| ||||| ||| ||||||||| || ||||||||||| ||| ||||| || ||| |||||||||| || ||||| ||| ||| |||||||||| || ||||||||||| |||||||||||| || |||||||| || |||||||||||||| |||||||||| |||||| |||||||| ||||||||||| ||| ||||||||||| |||||||| |||| ||| ||||||||
Table 42: Network Meta-Analyses Methods
Methods | ICER NMA | LEO Pharma NMA |
|---|---|---|
Analysis methods | NMA powered by a Bayesian model; fixed or random effects chosen based on DIC | ||| ||||||| ||||||||| |||||| ||||| ||||||| |
Priors | Unspecified, noninformative prior distributions for all model parameters | |||| ||||||| ||||| ||| ||||||| ||| ||| |||||||||| |
Assessment of model fit | Deviance information criterion | NR |
Assessment of consistency | NR | NR |
Assessment of convergence | Brook-Gelman-Rubin diagnostic and historical plots | |||||||||| ||||| || |||||| |||||||||| || ||||||||| |||||||| |||||| ||||| ||| ||||||| |||||||| |||||| |
Outcomes |
| |||||||||||||||| ||||||||||||||| |||||||||||||||||||| |||||||| |||||||||||||||| ||||||||||| ||||||||||||||| |
Follow-up time points | 12 to 16 weeksa | || || || ||||| |
Construction of nodes | Each treatment and dose was a node in the NMA | |
Sensitivity analyses | NR | |||| |
Subgroup analysis | Not presentedb | NR |
Methods for pairwise meta-analysis | NR | NR; ||||| ||||||| ||||| |
AE = adverse event; CDLQI = Children’s Dermatology Life Quality Index; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; EASI-90 = reduction of at least 90% in Eczema Area and Severity Index score from baseline; ICER = Institute for Clinical and Economic Review; IGA = Investigator’s Global Assessment; NMA = network meta-analysis; NR = not reported; POEM = Patient-Oriented Eczema Measure; PP-NRS = peak pruritus numeric rating scale; RCT = randomized controlled trial.
aThe primary end points of the abrocitinib trials (JADE MONO-1, JADE MONO-2, and JADE COMPARE) were measured at 12 weeks, while the primary end points in the remaining studies were measured at 16 weeks.
bDescriptive presentation of subgroups results by age (children, adolescents, and adults) and disease severity (moderate or severe) only; not analyzed in the NMA.
Sources: ICER NMA technical document,19 LEO Pharma NMA technical document,20 and the sponsor’s Summary of Clinical Evidence.14
Results of the ICER and LEO Pharma Network Meta-Analyses
Summary of Included Studies
ICER Network Meta-Analysis
Overall, 62 reports of 16 studies met the inclusion criteria for the moderate-to-severe patient population. Of these, 21 were included in the NMA. Reasons for exclusion were not specified. An overview of the included studies is summarized in Table 43.
Of the included trials, 14 were placebo-controlled monotherapy trials and 6 were placebo-controlled combinations with topical therapy trials; only 2 trials (HEADS Up and JADE COMPARE) included active comparator groups. All trials were conducted over 12 to 16 weeks and used stable doses. Although dupilumab was tested at different doses, only the FDA-approved dose of 300 mg once every 2 weeks was included in the NMA. All studies were of parallel design and assessed to be of “good” quality according to the US Preventive Services Task Force rating scale. All studies used some form of imputation, but the methods varied across studies. Multiple imputation, LOCF, and nonresponse imputation were used in various combinations to account for missing data. ITT analysis was only used in studies evaluating upadacitinib.
The trial populations varied with respect to age (mean age range = 31 to 41 years), duration of disease (mean duration = 21 to 28 years) and disease severity (IGA score of 4 range = 32% to 55%).
Potential sources of heterogeneity across the included studies are summarized in Table 45. Different washout periods were applied across trials. The washout period was 2 weeks in the tralokinumab trials (ECZTRA 1, 2, and 3) compared to 1 week in the corresponding dupilumab trials (SOLO 1 and 2 and LIBERTY AD CHRONOS), and as brief as 72 hours in the abrocitinib trials (JADE MONO-1, JADE MONO-2, and JADE COMPARE). Another source of potential heterogeneity was the time point at which primary efficacy outcomes were measured across studies. The primary end points were measured at 12 weeks in the abrocitinib trials (JADE MONO-1, JADE MONO-2, and JADE COMPARE), while the primary end points were measured at 16 weeks in the remaining studies. For combination-therapy trials, treatments of patients in the control group (placebo plus TCS) were not consistent. Of the combination-therapy trials, only the ECZTRA 3 study standardized combination TCS with mometasone furoate 0.1% cream. Other trials allowed a variety of TCS options and TCS potency levels. Whereas ECZTRA 3 supplied TCS free of charge by trial sites, TCS combination therapy was not provided by the trial sponsor of the JADE and COMPARE studies. Access to TCS combination therapy was not detailed in the remaining trials.
LEO Pharma Network Meta-Analysis
|| |||||| ||| ||||||| || || |||||| ||| ||| |||||||||| |||||| ||||||||| ||||||||| |||||||| ||||||| |||| |||||||||||| |||||||| || ||| |||||||||||| ||||| |||||| ||||||| || |||||||||| ||||| ||| ||| |||||| |||||||| |||| ||| ||||||||||| ||||| || ||| ||| ||||||| || |||||||| ||||||||| ||||| |||| || ||| |||| ||||||| |||| |||||||| |||||||| |||||||| ||| |||||| |||| ||| |||||||||||||||||| || || ||||||||||| ||||||||||||| |||||| |||| |||||||| || ||| |||||||||| |||||| ||||||||| ||| |||| || |||||| ||||||||||||| |||| ||||| ||||| ||| ||||||||||||| ||||||| |||||||| ||| ||| |||| |||||||| ||| |||||||||||||||||| |||||| || || ||||||| || |||||||| |||||||||| ||||||| || ||| || |||||| ||| ||||||| || ||| ||||||| || |||||||| ||||||| ||||||| |||||| ||||| |||||| || ||||||| |||||||||| |||| || ||||||||| ||||||||||||||||| |||| |||||||| || ||| |||||| ||||||||| ||||||||||| |||||||||| || ||| |||||||||| |||| |||||||| || ||| ||| |||||| |||| || |||||||| || ||| ||||||| || || ||||| ||| ||| ||| ||||||| |||||||| || ||| ||| |||||| ||||||||||||| ||| ||||||| || |||| ||||| |||| ||||||||| |||||||||||| ||||||| || |||| || || |||| |||| || |||||| |||||||| ||| ||||||||| |||||| ||||||||| |||| ||||||||||| ||| ||||||| |||| ||| |||||||||| ||||||||| |||| |||||||| || ||| |||| ||| |||||| |||| ||||||||| ||||||||| |||||| ||| ||||||| || |||| ||||| ||||| ||||||||| |||||||||||| || ||||| ||||||| ||||| ||||||||| |||||||| |||||| |||| || ||||| ||||||||||||| || || ||||| |||||||||||||| ||||||||| ||| |||||||||||||||||| ||||||| ||| ||||| ||||||| || |||| |||||| || |||||| ||| |||| ||| ||||||||| || ||||||| || |||| ||||| ||||| |||| ||| |||||||| ||||||| || ||||||||||| ||||||||| |||||||||||||||||||||| |||| |||||||||||||||||| || |||| |||||||| ||| |||||||||| ||||||| || || ||||| ||| || |||| ||||||| |||||||| || ||| |||| |||| ||| ||| ||| |||||| ||||||| |||||| || || ||||||||| ||| |||||| ||||||||||||||| || |||||||| || || ||||||||| |||||||||| ||||| |||||| ||||||||| |||||||||||||| |||||||||| |||||||||||| |||||| ||||||||||||||||||||| ||| ||| ||||||||| ||||| |||||||||||| |||| |||| ||||||| |||| ||| |||| ||||| |||| |||| ||| ||||| ||||||||||||| ||| || || ||||| |||| ||||| ||||||| ||| |||||||||| || |||| |||||||||||| ||| ||||||||| ||||||| |||||| |||||| |||||| ||| || ||||| |||||| || ||||||||| ||||||||||||||| |||||||||| || ||||| |||||| ||| || |||| ||| ||||||||| |||||||||||| ||||||| |||||| || ||| |||||||||| ||||||||| |||| ||||| |||| || |||| |||| ||||||||| ||||||||||||| ||| |||||||| || || |||||| |||| |||| || |||| |||||| ||| ||| |||||||||| || ||||||||||| |||| |||||||| ||||||| ||||| || |||||| ||| |||||| |||| || |||||| || ||| |||||| |||||| ||||||||||||| ||||||||| ||||||| || ||||||||||||| |||||| ||| |||||||| ||||||| ||| |||||||||| || ||||| ||| ||| |||| ||||| || ||||| ||||||| |||||||| |||||||| |||| |||||||| |||||| ||||||| ||||||| ||| ||||||| ||||||||| |||| |||||||| || || ||||| || ||| ||||||||||| |||||| ||||| |||| || |||| |||| || ||||| ||| ||||||| ||||||||| |||| |||||||| || || ||||| || ||| ||||||||| ||||||| ||||||| || ||||||| || ||||| ||||||| || ||| ||||||| || ||| ||| |||||||||||||| |||||||| || || ||| ||||| ||||||||| ||| || ||||||||| ||| |||||| |||||||||| |||||||||||| |||||||| || || ||||||| || || ||| || ||||||| ||| ||| ||||||||| |||||| ||||||| || ||||||| || ||||| |||| |||| || |||| |||| ||| ||||||| |||||| || ||||||||||||| |||||| |||||| ||| ||| ||||||||| ||||||||| ||||||| || ||||| ||| || |||||||||| ||||| |||||||| ||| ||||||||| ||||||||| |||||||| ||||||||||||| |||||| |||||||| || |||| ||||| || ||||||||||||| || ||||| |||||||||| ||||| ||||||| || |||||| ||| ||||| ||||||||| |||||| || |||| |||||||||| || ||||||||||||| ||||||| || || ||| |||||||||||||| |||||| |||||||| || |||| ||||| || ||||||||||||| || ||||| |||||||||| ||||| ||| || ||| ||||||||||| ||||| ||||||| || |||||| ||| ||||| ||||||||| |||||| || |||| ||||||||||| ||||||| |||||||| || ||||| || ||| || ||||||||| || ||||| || |||| || ||||| |||| || ||||||||||||||| ||||||| ||||||||||| ||| || ||||| |||||||| || ||||||||| |||||||| |||| ||||||| || || || || ||||| || |||||||| || ||||||||||||| || |||| || ||||| |||| || ||||||||||||||| ||||||| ||||| |||| || |||| |||| ||| |||||||| |||||||| ||| |||| ||| ||||||||||||||| |||| ||||||||||||||| ||| |||||| ||||||||| |||||| |||||| ||||||| |||||||| ||| || |||||| ||||||| |||||||||| ||| |||||||| |||||||||| |||||| ||||||| |||||||| || ||||| ||||||| || || ||||||| || || || || |||| ||||||| ||| ||||||||||||||| || ||| ||||||||||||||| ||||||| |||||||| || |||||||| ||||||||||||||| || ||||||||||||| |||||||| ||||||||||||||||| ||||| |||| |||| || ||| ||||||| ||||||| || ||||||| || |||||| |||||||||| |||||| ||||||||| ||||||||||||||| ||||| ||||| || |||||||| ||||||||||||||| || ||||||||||||| ||||||||||||||||| |||| |||| |||| |||||| |||| |||| ||||| |||||||| || || ||||||| || ||| |||||||| ||| ||| ||| ||||||||||||||| |||| ||||||||||||||| ||| |||||| ||||||||| ||| ||| |||||||| || |||| ||||| |||| |||| ||
Table 43: Overview of Trials Included in the ICER Network Meta-Analysis
Trial | Treatment arms | N | Measured at baseline | |||
|---|---|---|---|---|---|---|
Mean EASI | Mean age, years | Mean disease duration, years | IGA score of 4, % | |||
Monotherapy | ||||||
JADE MONO-1100 |
| 387 | 30.2 | 32.4 | 23.4 | 40.7 |
JADE MONO-2101 |
| 391 | 28.5 | 35.1 | 21.0 | 32.2 |
Gooderham (2019)102 |
| 167 | 25.6 | 40.8 | 23.0 | 40.8 |
BREEZE-AD 15 |
| 624 | 31.0 | 35.7 | 25.7 | 41.8 |
BREEZE-AD 25 |
| 615 | 33.5 | 34.5 | 24.0 | 50.5 |
BREEZE-AD 5103 |
| 440 | 27.1 | 39.7 | 23.7 | 41.7 |
ECZTRA 110 |
| 802 | 29.3 | 37.0 | 27.5 | 50.9 |
ECZTRA 210 |
| 794 | 28.9 | 32.0 | 25.3 | 49.2 |
MEASURE UP 1104,a |
| 847 | 29.5 | 34.0 | NR | 45.2 |
MEASURE UP 2104,a |
| 836 | 29.1 | 33.6 | NR | 54.9 |
Heads Up90 |
| 692 | NR | NR | NR | NR |
Guttman-Yassky (2020)105 |
| 167 | 25.6 | 40.8 | 23.0 | 40.8 |
LIBERTY AD SOLO 1106 |
| 671 | 30.7 | 38.7 | 26.7 | 48.3 |
LIBERTY AD SOLO 2106 |
| 708 | 29.4 | 34.7 | 24.8 | 48.3 |
Thaci (2016)107 |
| 379 | 31.9 | 37.0 | 28.0 | 47.3 |
Combination therapy | ||||||
JADE COMPARE108 |
| 837 | 30.9 | 37.7 | 22.7 | 35.4 |
BREEZE-AD 7109 |
| 329 | 29.57 | 33.8 | 24.03 | 45.0 |
Guttman-Yassky (2018)110 |
| 104 | 21.23 | 36.5 | 22.03 | NR |
ECZTRA 311 |
| 380 | 25.5 | 36.0 | 26.0 | 46.3 |
AD-UP111 |
| 907 | 29.6 | 34.1 | NR | 52.9 |
LIBERTY AD CHRONOS112 |
| 740 | 29.8 | 31.2 | 26.7 | 47.7 |
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI = Eczema Area Severity Index; ICER = Institute for Clinical and Economic Review; IGA = Investigator’s Global Assessment; NMA = network meta-analysis; NR = not reported; q.w. = once weekly; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; TCS = topical corticosteroids; TRA = tralokinumab; UPA = upadacitinib.
Note: All time points at 16 weeks except JADE MONO-1 and JADE MONO-2 (12 weeks) and COMPARE (12 and 16 weeks).
aIncluded in pooled baseline values presented in the table but not included in comparative clinical efficacy.
Note: Pooled estimates from JADE MONO-1, JADE MONO-2, MEASURE UP 1, and MEASURE UP 2 were in patients aged 12 years and older.
Sources: ICER NMA technical document19 and the sponsor’s Summary of Clinical Evidence.14
Table 44: Overview of Trials Included in the LEO Pharma Network Meta-Analysis
Trial | Treatment arms | N | Mean age, years (SD) | AD severitya | Mean EASI (SD) | Mean disease duration, years (SD) | |
|---|---|---|---|---|---|---|---|
Moderate | Severe | ||||||
|||| ||||| |||| |||| | |||| ||| || || | ||| | |||||| || |||| | || | || | |||| |||||| | |||| ||||| |
|||| ||| || || | ||| | |||||| || |||| | || | || | |||| |||||| | |||| ||||| | |
||| | ||| | |||||| || |||| | || | || | |||| |||||| | |||| ||| | |
||||| | ||| ||| || ||| | ||| | |||| ||||| | |||| | |||| | |||| ||||||| | |||| ||||| |
||| ||| || ||| | ||| | |||| ||||| | |||| | |||| | |||| ||||||| | |||| ||||| | |
||| | ||| | |||| ||||| | |||| | |||| | |||| ||||||| | |||| ||||| | |
||||||| | | ||| || || || | ||| | || | |||| | |||| | |||| |||||| | |||| ||||| |
||| || || || | |||||||
||| | |||||||
||||||| || | ||| || || || | ||| | || | |||| | |||| | |||| |||||| | |||| ||||| |
||| || || || | |||||||
||| | |||||||
||||||| || ||| | ||||||| || || ||| | ||| | |||| ||||| | |||| | |||| | |||| |||||| | |||| ||| |
||| ||| || || ||| | ||| | |||| ||||| | |||| | |||| | |||| |||||| | |||| ||||| | |
||||||| | ||| | |||| ||||| | |||| | |||| | |||| |||| | |||| ||||| | |
|||||||||||||||| ||||||||||||| |||||||||| |||| |||||||| |||||| |||||| |||||||| |||| ||||||||||||||| |||||| |||||||||| |||||||||||||||||||||| ||||||| ||||||||||| ||||||| |||||||| |||||||||| ||||||||||| ||||||||||||||| |||||| ||||| ||||||||||| |||| ||||||||||||||||||| ||||||||||| ||||||||| ||||||||| |||||||| ||||||||| |||||| |||||| ||||||||| |||||||| ||| |||||| |||||||| |||| |||||| |||||||| ||||||||||||| ||||||||| ||||||||||||||| ||||||||||||||| ||||||||||||||||||||||| |||| ||| || ||||||||||||||||||||||||||||| |||| |||| |||||||| || |||||| |||| ||| |||| ||||||||| ||||||||||||| ||||||||||||||| |||| |||| |||||||| ||| ||| |||||| |||||| || |||||||||||| ||| ||| ||| ||||||||| ||||||| ||||||||| |||| ||||| || ||||||||||| ||||||||| || |||| || ||| |||| |||||||||||| ||||||| |||||||||| ||||||||||||| |||| ||||||||||| ||| |||||| ||| ||||||||| ||||||||||||||| |||||||| || ||| ||||| ||| |||| ||| ||||||||| ||||||| || |||||||| |||||||
Table 45: Assessment of Clinical and Methodological Homogeneity for the ICER and LEO Pharma Network Meta-Analyses
Characteristics | Description and handling of potential effect modifiers | |
|---|---|---|
ICER NMA | LEO Pharma NMA | |
Definition of disease severity | Definition of disease severity used across the included trials was not reported | ||| ||||||| ||| ||||| ||||||||||| |||| || |||||| || || ||| |||||| |||||| || ||||||| || ||||| ||||| |||| ||| |||||||| ||||||||||| ||||||||| ||||||||||| |
Disease severity | Proportion of patients with IGA score 4 ranged from a low of 32.2% (JADE MONO-2) to a high of 52.9% | |||||||||| || |||||||| |||| |||||||| || |||||| ||||| || ||||| ||||||| || || |||| || ||||| |||||||| || ||| ||| ||| |||||| || |||||| |||||| || ||| ||||| ||||||| |||| ||||||| ||| || ||||| |||||||| || ||| |
Washout period | 72 hours in the abrocitinib trials (JADE MONO-1, JADE MONO-2, and JADE COMPARE); 1 week in the dupilumab trials (SOLO 1, 2, and LIBERTY AD CHRONOS); and 2 weeks in the tralokinumab trials (ECZTRA 1, 2, and 3) | |||| ||| |||||| || ||||| ||| ||||||||| |
Concomitant medications | Both monotherapy and combination therapies were eligible for inclusion; separate networks were for monotherapy and combination therapies were developed | ||| |||||||| ||||||| |||| ||||||||||| |
Inclusion criterion: duration of AD | Per-protocol inclusion criteria of duration of AD across trials were not reported | || |||||||| || ||||||| |||| ||||||| || || ||| || |||||||| ||| ||| ||||||||| |||||| ||||||| || ||||||| || ||||| |||| |||| || |||| |||| || |
Exclusion criterion: prior use of biologics | Per-protocol exclusion criteria based on prior use of biologics across trials were not reported | ||||||||| ||||||||| ||||||| ||||| ||| |||||||||||||| |||||| ||||||||| || |||| ||||| || ||||||||||||| || ||||| |||||||||| ||||| ||||||| || |||||| ||| ||||| ||||||||| |||||| || |||||||||||| || ||||||||||||| ||||||| ||| ||| |||||||||||||| |||||| ||||||||| || |||| ||||| || ||||||||||||| || ||||| |||||||||| ||||| ||| || ||| ||||||||||| ||||| ||||||| || |||||| ||| ||||| ||||||||| |||||| || |||| ||||||||||| ||||||| |||||||| || |||||| ||| || ||||||||| || ||||| || |||| || ||||| |||| || ||||||||||||||| ||||||| ||||||||||| ||| || ||||| |||||||| || ||||||||| |||||||| |||| ||||||| || || || || ||||| || |||||||| || ||||||||||||| || |||| || ||||| |||| || ||||||||||||||| ||||||| ||||| |||| || |||| |||| ||| |
Per-protocol use of rescue therapy | Per-protocol use of rescue therapy across trials was not reported | |||||||| ||| || |||||| ||||||| |||||||||| ||| |||||||| |||||||||| |||||| |||||||| |||||||| || ||||| ||||||| |||| ||||||| || || || || |||| ||||||| ||| |
Discontinuation of investigational product | Per-protocol criteria for discontinuation of investigational products across trials were not reported | ||| ||| || |||||||| ||||||||||||||| || ||||||||||||| |||||||| ||||||||||||||||| ||||| |||||||| ||||||| || ||||||| || |||||| ||||||||| |||||| ||||||||||||||| |||||||| ||||| ||| || |||||||| ||||||||||||||| || ||||||||||||| ||||||||||||||||| |||| ||| |||| ||||| |||||||| || || ||||||| || ||| |
Access and dosing of TCS combination therapy | Dosing: ECZTRA 3 standardized combination TCS with mometasone furoate 0.1% cream, whereas other trials allowed a subset of TCS options and potency levels Access: ECZTRA 3 supplied TCS free of charge by trial sites, TCS combination therapy was not provided by the trial sponsor of JADE and COMPARE, and access to TCS combination therapy was not detailed in the remaining trials | || |
Timing of end point evaluation | Primary end points were measured at 12 weeks in the abrocitinib trials (JADE MONO-1, JADE MONO-2, and JADE COMPARE) and at 16 weeks in the remaining studies | ||||||| ||||||||| |||| |||||||| || || ||||| || ||| ||||||||||| |||||| ||| || ||||| || ||| ||||||||| ||||||| |
Approach to missing data | Methods used to account for missing data varied across studies from use of MI (JADE MONO-1, MONO-2, Gooderham [2019], and LIBERTAY AD CHRONOS), NRI (JADE COMPARE), NRI and MI (ECZTRA 1, 2, 3, MEASURE UP-1, −2, and AD-UP), LOCF and NRI (Guttman-Yassky [2020]), and MI, LOCF and NRI (LIBERTY AD SOLO-1 and SOLO-2), LOCF and NRI (Tachi [2016]) | ||||||| |||| || ||||||| ||| ||||||| |||| |||||| ||||||| |||| ||| ||||||||| |
Application of ITT principal | ITT analysis was used only in studies evaluating upadacitinib | ||||||| ||| |||||||| ||| |||||||| |||||| ||| |||||||| ||||||| ||| ||| ||||||||| |
AD = atopic dermatitis; NA = not applicable; ICER = Institute for Clinical and Economic Review; ITT = indirect treatment comparison; LOCF = last observation carried forward; MI = multiple imputation; NMA = network meta-analysis; NR = not reported; NRI = nonresponder imputation; TCS = topical corticosteroids.
Sources: ICER NMA technical document,19 LEO Pharma NMA technical document,20 and the sponsor’s Summary of Clinical Evidence.14
Evidence Networks
ICER Network Meta-Analysis
The evidence networks for both monotherapy and combination trials in adult patients with AD included in the ICER NMA are illustrated in Figure 12 and Figure 13, respectively.
The overall NMA evidence for monotherapy trials consisted of 15 trials evaluating 5 interventions, including abrocitinib, baricitinib, dupilumab, tralokinumab, and upadacitinib across 9 dosing regiments, connected by comparisons to placebo. Five closed loops were formed between the connections of abrocitinib 100 mg, abrocitinib 200 mg and placebo; baricitinib 1 mg, baricitinib 2 mg, and placebo; dupilumab 300 mg every 2 weeks, upadacitinib 30 mg, and placebo; upadacitinib 30 mg, upadacitinib 15 mg and placebo; and upadacitinib 30 mg, upadacitinib 15 mg, dupilumab 200 mg every 2 weeks, and placebo. The model of best fit for the NMA of EASI was determined to be multinomial with a probit link and consisted of 15 trials; for NMA of IGA and PP-NRS scores of at least 4 points; the model of best fit for each was binomial with a log link and consisted of 14 trials.
The overall NMA evidence for combination trials consisted of 6 trials evaluating 5 interventions, including abrocitinib, baricitinib, dupilumab, tralokinumab, and upadacitinib across 6 dosing regiments, connected by comparisons to placebo. Six closed loops were formed between the connections of: abrocitinib 100 mg, abrocitinib 200 mg and placebo; abrocitinib 100 mg, dupilumab 300 mg q.2.w. and placebo; abrocitinib 200, dupilumab 300 mg q.2.w., and placebo; abrocitinib 100 mg, abrocitinib 200 mg, and dupilumab 300 mg q.2.w.; abrocitinib 100 mg, abrocitinib 200 mg, dupilumab 300 mg q.2.w. and placebo; and upadacitinib 15 mg, upadacitinib 30 mg, and placebo. The model of best for the NMA of EASI was multinominal with a probit link and consisted of 6 trials. For the NMA of the IGA the model of best fit was binomial with a log link and consisted of 6 trials, and for the NMA of PP-NRS of at least 4 points the model of best fit was binomial with a log link and consisted of 5 trials.
LEO PHARMA Network Meta-Analysis
||| |||||||| ||||||| ||| ||||||||||| || |||||||||| |||||||| |||| || |||||||| || ||| ||| |||||| ||| || ||||||||||| || |||||| ||| ||| ||||||| ||| |||||||| ||| ||||||||||| |||||| ||||||||| ||||||| |||||||||||||||||||||| ||||||||| |||||||||||| |||||||||| |||||||||||| ||| |||||||||||| ||||||||||| |||||||||| ||||||||| || ||| |||||||||| || |||||||| |||| |||||| ||||| |||| |||||| ||||||| ||| ||||||||||| ||| ||| ||||||||||| ||| ||||||||||| ||| || ||| |||||||| ||| ||||||||| ||| || |||| ||||||||| ||||||| ||| ||| |||||||| ||| |||||||||||| ||| || |||| |||||||||||| ||| ||| ||| |||||||| ||| ||| |||||||||||| || || ||| |||||||||||| || || || ||| |||||||| ||| |||| |||||||| |||||||| |||||| ||||||||| ||||||||| ||| |||||| |||||||| || ||||||||| ||| ||||||||| ||| ||||||||||||| ||| ||||| |||| |||||||| || ||| ||||| |||||||| ||| ||||||||| || ||||||||||||||| |||| ||||||||||| ||| ||| |||||||| |||||| |||||||| ||| ||||||||||||| |||||||| ||| ||||||||| || |||||||||||||||| ||| ||||||||||| ||| |||| ||| |||||||| ||| |||||
Figure 12: Evidence Network Diagram for Included Studies for Monotherapy in Adults in the ICER Network Meta-Analysis
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; PBO = placebo; q.2.w. = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib.
Note: Numbers in nodes are doses in milligrams.
Sources: ICER NMA technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 13: Evidence Network Diagram for Included Studies for Combination Therapy in Adults in the ICER Network Meta-Analysis
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; PBO = placebo; q.2.w. = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib.
Note: Numbers in nodes are doses in milligrams.
Sources: ICER NMA technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 14: Evidence Network Diagram for Included Studies for Monotherapy in Adolescents in the LEO Pharma Network Meta-Analysis

Figure was redacted at the sponsor’s request.
Results
Efficacy results of the NMA are presented for monotherapy and combination therapy by population (e.g., adults and adolescents). A pairwise comparison against baricitinib is not presented as the treatment is currently not approved for use in Canada.
EASI-50
Adult Population (ICER Network Meta-Analysis): Relative risks and 95% CrIs across various comparisons for EASI-50 among adult patients who received monotherapy are summarized in Figure 15. The treatment response of all included monotherapy interventions on EASI-50 in adult patients were favoured over placebo. Treatments with upadacitinib 30 mg (RR = 1.75; 95% CrI, 1.50 to 2.10), abrocitinib 200 mg (RR = 1.59; 95% CrI, 1.31 to 1.95), upadacitinib 15 mg (RR = 1.53; 95% CrI, 1.20 to 1.84), and dupilumab 300 mg (RR = 1.40; 95% CrI, 1.18 to 1.69) were favoured for achievement of EASI-50 compared to tralokinumab 300 mg. The point estimate for EASI-50 favoured abrocitinib 100 mg over tralokinumab 300 mg, but the CrI also included the potential of little-to-no difference between the treatments (RR = 1.21; 95% CrI, 0.95 to 1.53).
The RRs and 95% CrIs across various comparisons for achievement of EASI-50 among adult patients who received combination therapy are summarized in Figure 16. The treatment response of all included combination-therapy interventions on EASI-50 in adult patients were favoured over placebo. Treatments with upadacitinib 30 mg (RR = 1.45; 95% CrI, 1.27 to 1.71), abrocitinib 200 mg (RR = 1.32; 95% CrI, 1.14 to 1.57), upadacitinib 15 mg (RR = 1.32; 95% CrI, 1.15 to 1.57), dupilumab 300 mg (RR = 1.26; 95% CrI, 1.09 to 1.49), and abrocitinib 100 mg (RR = 1.20; 95% CrI, 1.02 to 1.43) were favoured for achievement of EASI-50 compared to tralokinumab 300 mg.
Adolescent Population (LEO Pharma Network Meta-Analysis A): ||| |||| ||||| |||| ||| ||| ||| ||| ||||||| |||||| ||||||| ||||||||||| ||| ||||||||||| || ||||||| || || ||||| ||||| |||||||||| |||||||| ||| |||||||| ||||||||||| ||| |||||||||| || ||||| ||| ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || ||||||| || || ||||| || |||||||||| |||||||| |||| |||||||| |||| |||||||| || |||||||||| ||||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| ||||||| ||||||| |||||||||||| ||| ||| ||||| |||||| |||||||||||||
Figure 15: Relative Risk (95% Credible Interval) Across Comparisons for EASI-50 in Monotherapy RCTs in Adults (ICER Network Meta-Analysis)
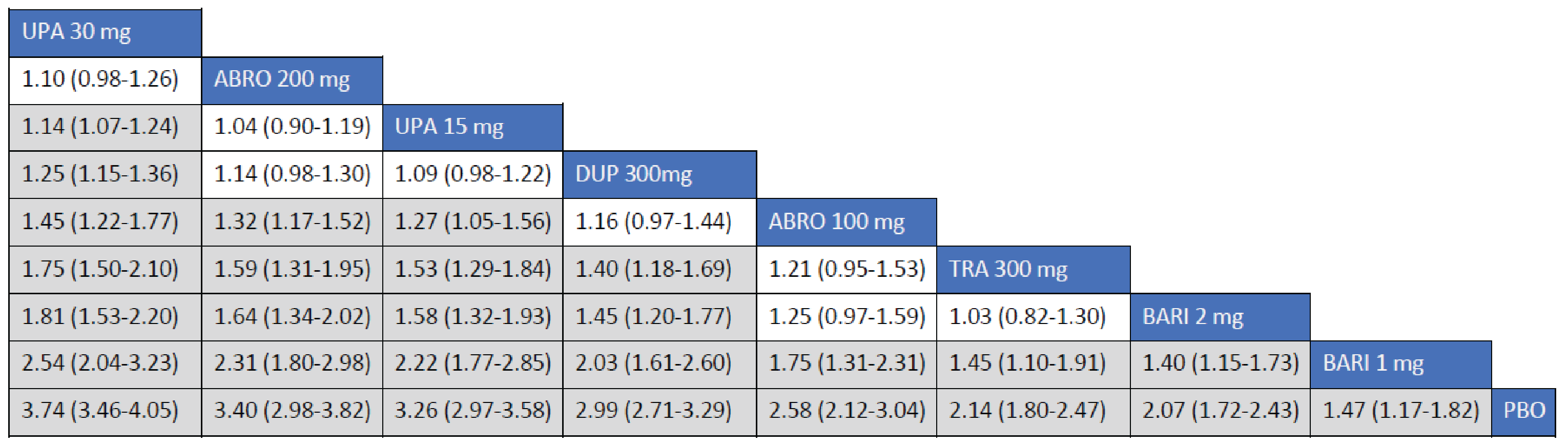
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-50 = reduction of at least 50% in Eczema Area Severity Index score from baseline; ICER = Institute for Clinical and Economic Review; NMA = network meta-analysis; PBO = placebo; RCT = randomized controlled trial; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Sources: ICER NMA technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 16: Relative Risk (95% Credible Interval) Across Comparisons for EASI-50 in Combination RCTs in Adults (ICER Network Meta-Analysis)
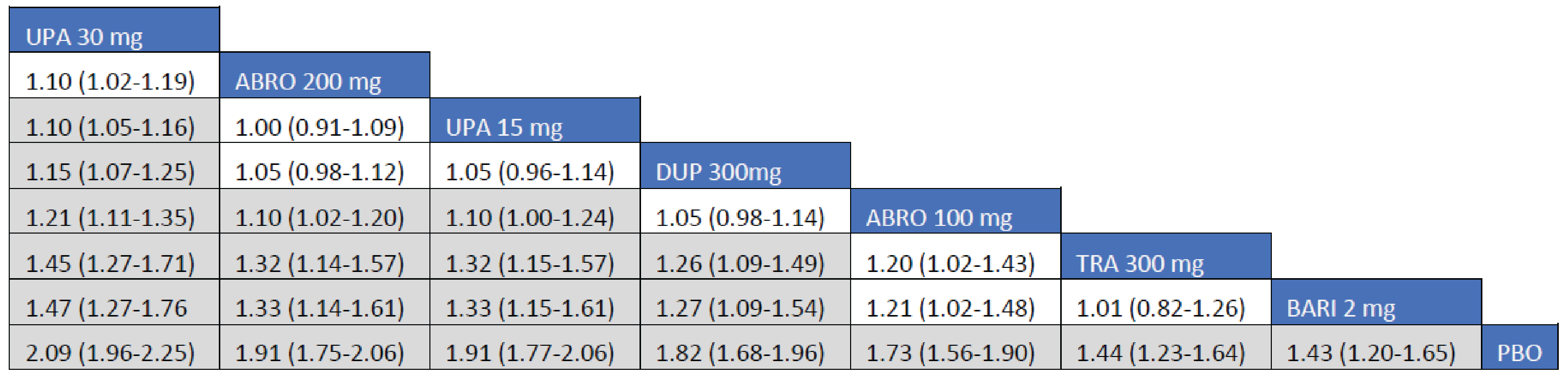
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-50 = reduction of at least 50% in Eczema Area Severity Index score from baseline; ICER = Institute for Clinical and Economic Review; NMA = network meta-analysis; PBO = placebo; RCT = randomized controlled trial; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Sources: ICER NMA technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 17: Odds Ratio (95% Credible Interval) for EASI-50 at 16 Weeks Across Pairwise Comparisons of Monotherapy RCTs in Adolescents (LEO Pharma Network Meta-Analysis)

Figure was redacted at the sponsor’s request.
EASI-75
Adult Population (ICER Network Meta-Analysis): The RRs and 95% CrIs across various comparisons for achievement of EASI-75 among adult patients who received monotherapy are summarized in Figure 18. The treatment response of all included monotherapy interventions on EASI-75 in adult patients were favoured over placebo. Treatments with upadacitinib 30 mg (RR = 2.77; 95% CrI, 1.77 to 2.77), abrocitinib 200 mg (RR = 1.89; 95% CrI, 1.45 to 2.49), upadacitinib 15 mg (RR = 1.79; 95% CrI, 1.42 to 2.29), and dupilumab 300 mg (RR = 1.58; 95% CrI, 1.25 to 2.03) were favoured for achievement of EASI-75 compared to tralokinumab 300 mg. The point estimate for EASI-75 favoured abrocitinib 100 mg over tralokinumab 300 mg, but the CrI also included the potential of little-to-no difference between the treatments (RR = 1.29; 95% CrI, 0.93 to 1.76).
The RRs and 95% CrIs across various comparisons for achievement of EASI-75 among adult patients who received combination therapy are summarized in Figure 19. The treatment response of all included combination-therapy interventions on EASI-75 in adult patients were favoured over placebo. Treatments with upadacitinib 30 mg (RR = 1.90; 95% CrI, 1.53 to 2.45), abrocitinib 200 mg (RR = 1.58; 95% CrI, 1.25 to 2.07), upadacitinib 15 mg (RR = 1.48 95% CrI, 1.26 to 2.07), dupilumab 300 mg (RR = 1.46; 95% CrI, 1.15 to 1.90), and abrocitinib 100 mg (RR = 1.34; 9% Crl 1.03 to 1.76) were favoured for achievement of EASI-75 compared to tralokinumab 300 mg.
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| || ||| ||| ||| |||||| ||||||| |||||||||| ||| ||||||||||| || ||||||| ||||| |||||||||| |||||||| ||| |||||||| ||||||||||| ||| |||||||||| || ||||| ||| ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || ||||||| || |||||||||| |||||||| |||| |||||||| |||| |||||||| ||||||||| |||| |||||||||||| || || || ||| |||||||| ||| ||||||||||| || ||||||| |||||||| || |||||||||||| ||| || |||| ||||| ||| |||| |||| || |||||| ||| || |||||||||||| ||| || |||| ||||| ||| ||| |||| || ||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || || ||||||||| ||||||| ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Figure 18: Relative Risk (95% Credible Interval) Across Comparisons for EASI-75 in Monotherapy RCTS in Adults (ICER Network Meta-Analysis)
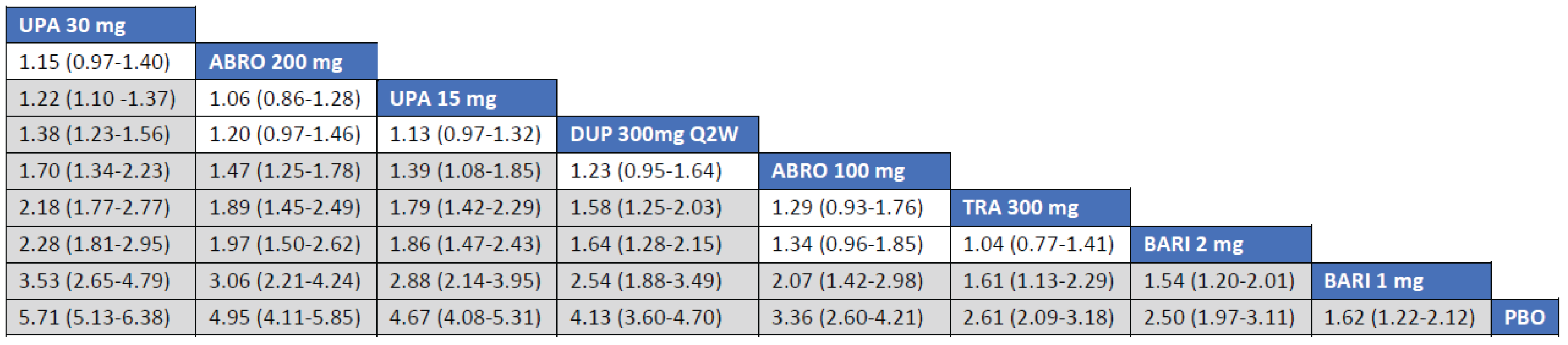
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-75 = reduction of at least 75% in Eczema Area Severity Index score from baseline; ICER = Institute for Clinical and Economic Review; PBO = placebo; Q2W = every 2 weeks; RCT = randomized controlled trial; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Sources: ICER network meta-analysis technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 19: Relative Risk (95% Credible Interval) Across Comparisons for EASI-75 in Combination-Therapy RCTs in Adults (ICER Network Meta-Analysis)
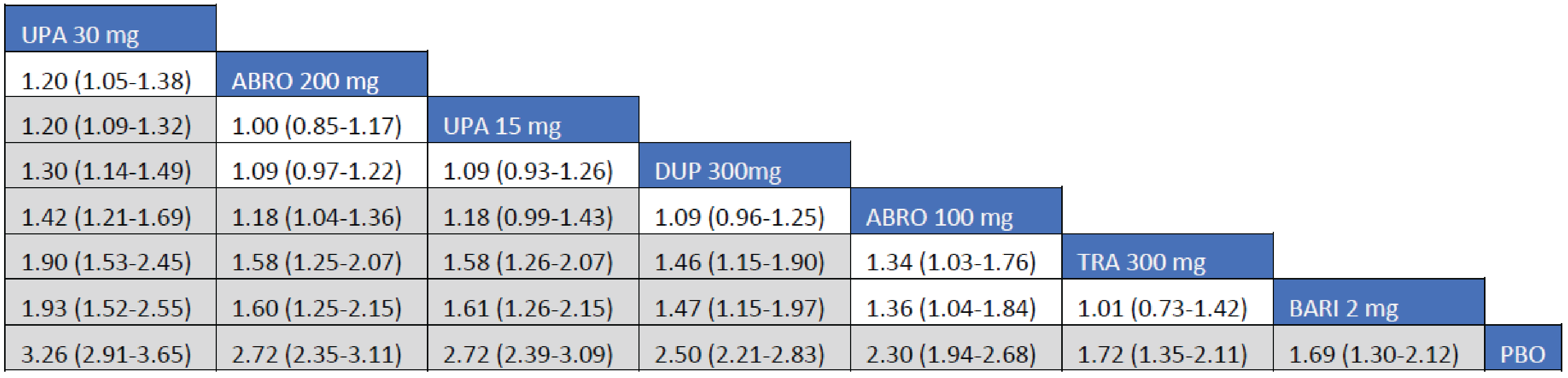
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-75 = reduction of at least 75% in Eczema Area Severity Index score from baseline; ICER = Institute for Clinical and Economic Review; PBO = placebo; RCT = randomized controlled trial; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Sources: ICER network meta-analysis technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 20: Odds Ratio (95% Credible Interval) for EASI-75 Across Pairwise Comparisons of Monotherapy RCTs in Adolescents (LEO Pharma Network Meta-Analysis)

Figure was redacted at the sponsor’s request.
EASI-90
Adult Population (ICER Network Meta-Analysis): The RRs and 95% CrIs across comparisons for achievement of EASI-90 among adult patients who received monotherapy are summarized in Figure 21. The treatment response of all included monotherapy interventions on the EASI-90 in adult patients were favoured over placebo. Treatments with upadacitinib 30 mg (RR = 2; 95.89% CrI, 2.19 to 3.95), abrocitinib 200 mg (RR = 2.36; 95% CrI, 1.65 to 3.39), upadacitinib 15 mg (RR = 2.17; 95% CrI, 1.60 to 3.00), and dupilumab 300 mg every 2 weeks (RR = 1.83; 95% CrI, 1.34 to 2.54) were favoured for achievement of EASI-90 compared to tralokinumab 300 mg. The point estimate for EASI-90 favoured abrocitinib 100 mg over tralokinumab 300 mg, but the CrI also included the potential of little-to-no difference between the treatments (RR = 1.39; 95% CrI, 0.91 to 2.09).
The RRs and 95% CrIs across comparisons for achievement of EASI-90 among adult patients who received combination therapy are summarized in Figure 22. The treatment response of all included combination-therapy interventions on the EASI-90 in adult patients were favoured over placebo. Treatments with upadacitinib 30 mg (RR = 2.74; 95% CrI, 1.98 to 3.97), abrocitinib 200 mg (RR = 2.01; 95% CrI, 1.41 to 2.98), upadacitinib 15 mg (RR = 2.01; 95% CrI, 1.43 to 2.96), dupilumab 300 mg (RR = 1.76; 95% CrI, 1.24 to 2.57), and abrocitinib 100 mg (RR = 1.54; 95% CI, 1.05 to 2.31) favoured EASI-90 compared to tralokinumab 300 mg.
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| || ||| ||| ||| |||||| ||||||||||| ||| ||||||||||| || ||||||| ||||| |||||||||| |||||||| ||| |||||||| ||||||||||| ||| |||||||||| || ||||| ||| ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || ||||||| || |||||||||| |||||||| |||| |||||||| |||| |||||||| ||||||||| |||| |||||||||||| || || ||| |||||||| ||| ||||||||||| || ||||||| |||||||| || |||||||||||| ||| || |||| |||||| ||| |||| |||| || ||||||| ||| |||||||||||| ||| || |||| |||||| ||| |||| ||||| |||||||| ||||||||| |||| |||||||||||| || || || ||| |||||||||| |||| |||||||| ||||||| |||||||| || |||||||||||| ||| || |||| |||||| ||| |||| |||| || |||||||| ||| || |||||||||||| ||| || |||| |||||| ||| ||| |||| || ||||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || || ||||||||| ||||||| ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Figure 21: Relative Risk (95% Credible Interval) Across Comparisons for EASI-90 in Monotherapy RCTs in Adults (ICER Network Meta-Analysis)
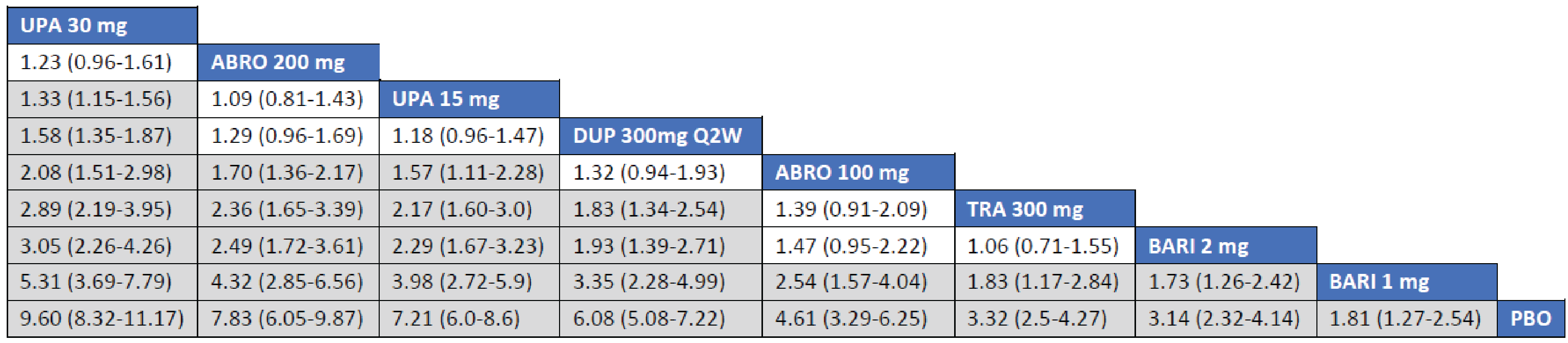
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-90 = reduction of at least 90% in Eczema Area Severity Index score from baseline; ICER = Institute for Clinical and Economic Review; PBO = placebo; Q2W = every 2 weeks; RCT = randomized controlled trial; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Sources: ICER network meta-analysis technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 22: Relative Risk (95% Credible Interval) Across Comparisons for EASI-90 in Combination-Therapy RCTs in Adults (ICER Network Meta-Analysis)
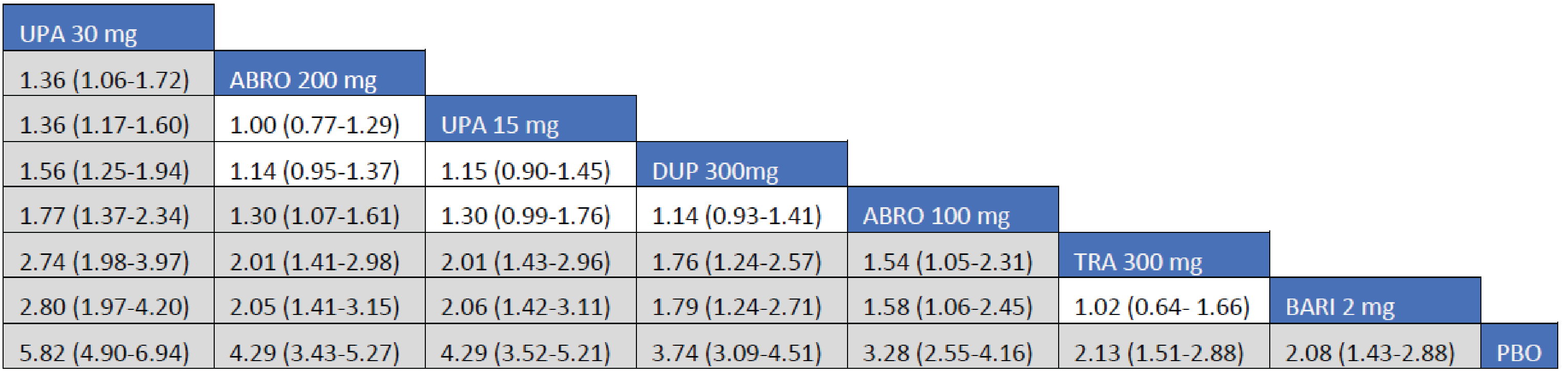
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI-90 = reduction of at least 90% in Eczema Area Severity Index score from baseline; ICER = Institute for Clinical and Economic Review; PBO = placebo; RCT = randomized controlled trial; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Sources: ICER network meta-analysis technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 23: Odds Ratio (95% Credible Interval) for EASI-90 Across Pairwise Comparisons of Monotherapy RCTs in Adolescents (LEO Pharma Network Meta-Analysis)

Figure was redacted at the sponsor’s request.
Investigation’s Global Assessment
Adult Population (ICER Network Meta-Analysis): The RRs and 95% CrIs across various comparisons for IGAs among adult patients who received monotherapy are summarized in Figure 24. The treatment response of all included monotherapy interventions on the IGA in adult patients were favoured over placebo. Treatments with upadacitinib 30 mg (RR = 3.97; 95% CrI, 2.54 to 6.31), upadacitinib 15 mg (RR = 3.07; 95% CrI, 1.88 to 4.99), abrocitinib 200 mg (RR = 2.75; 95% CI, 1.54 to 4.95), and dupilumab 300 mg (RR = 2.15; 95% CrI, 1.31 to 3.60) were associated with favoured IGAs compared to tralokinumab 300 mg. The Crls for the comparison between tralokinumab and abrocitinib 100 mg were too wide to draw any conclusions of certainty in IGAs in adult patients receiving monotherapy for AD.
The RR and 95% CrI across various comparisons for IGAs among adult patients who received combination therapy are summarized in Figure 25. The treatment response of all included combination interventions on the IGA in adult patients were favoured over placebo. Upadacitinib 30 mg (RR = 2.83; 95% CrI, 1.90 to 4.27), abrocitinib 200 mg (RR = 2.24; 95% CI, 1.44 to 3.49), upadacitinib 15 mg (RR = 2.08; 95% CrI, 1.35 to 3.25), dupilumab 300 mg (RR = 1.85; 95% CrI, 1.20 to 2.88), and abrocitinib 100 mg (RR = 1.66; 95% CI, 102 to 2.68) were associated with favourable IGAs compared to tralokinumab 300 mg.
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| || ||| ||| ||| |||||| ||||||| ||||||||||| ||| ||| ||| ||||| |||||||||| |||||||| ||| |||||||| ||||||||||| ||| |||||||||| || ||||| ||| ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || ||| ||| || |||||||||| |||||||| |||| |||||||| |||| |||||||| ||||||||| |||| |||||||||||| || || || ||| |||||||| ||| ||||||||||| || ||| ||| |||||||| || |||||||||||| ||| || |||| ||||| ||| |||| |||| || |||||| ||| || |||||||||||| ||| || |||| ||||| ||| ||| |||| || ||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || || ||||||||| ||| ||| ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Figure 24: Relative Risk (95% Credible Interval) Across Comparisons for IGA in Monotherapy RCTs in Adults (ICER Network Meta-Analysis)
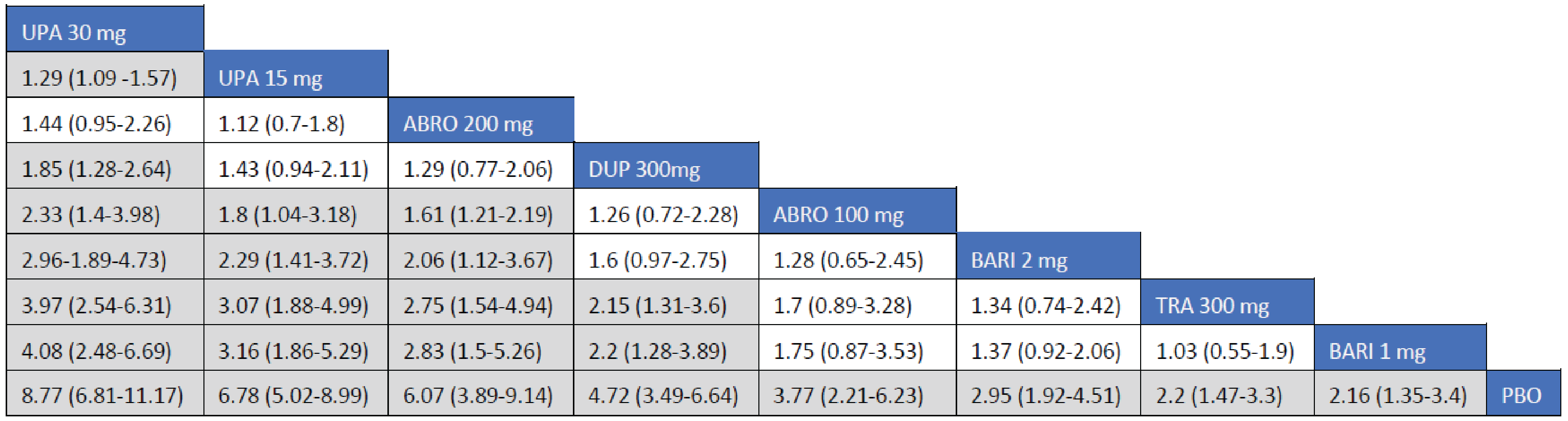
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; IGA = Investigator’s Global Assessment; PBO = placebo; RCT = randomized controlled trials; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Sources: ICER network meta-analysis technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 25: Relative Risk (95% Credible Interval) Across Comparisons for IGA in Combination RCTs in Adults (ICER Network Meta-Analysis)
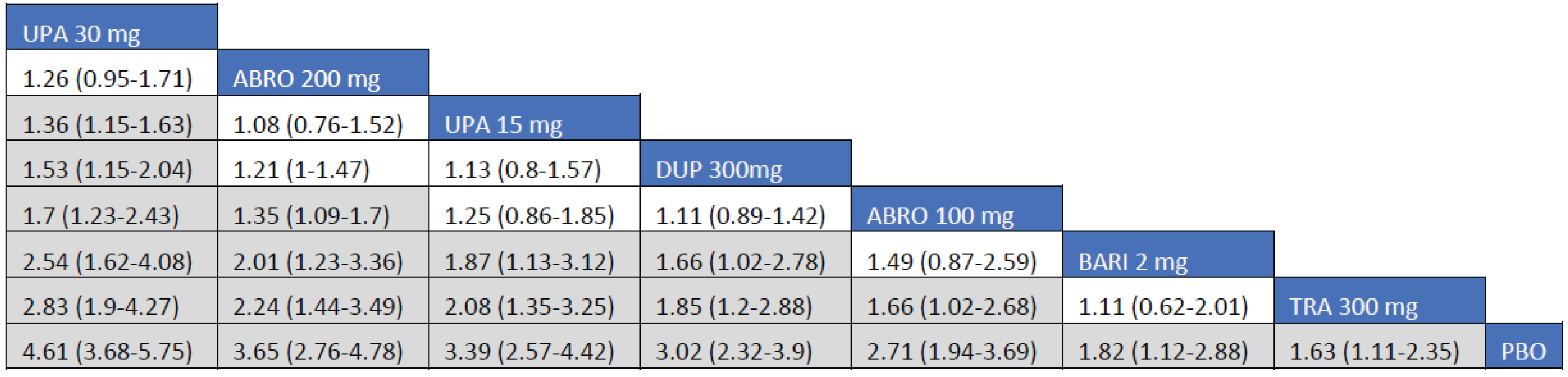
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; IGA = Investigator’s Global Assessment; PBO = placebo; RCT = randomized controlled trials; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Sources: ICER network meta-analysis technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 26: Odds Ratio (95% Credible Interval) for IGA Scores of 0 or 1 Across Pairwise Comparisons of Monotherapy RCTs in Adolescents (LEO Pharma Network Meta-Analysis)

Figure was redacted at the sponsor’s request.
PP-NRS With an Improvement of at Least 4 Points
Adult Population (ICER Network Meta-Analysis): The RRs and 95% CrIs across comparisons for PP-NRS scores demonstrating an improvement of at least 4 points among adult patients who received monotherapy are summarized in Figure 27. The treatment response of all included monotherapy interventions on an improved PP-NRS score of at least 4 points in adult patients was favoured over placebo. Treatments with upadacitinib 30 mg (RR = 2.16; 95% CrI, 1.14 to 4.58), dupilumab 300 mg (RR = 2.12; 95% CrI, 1.06 to 4.43), and upadacitinib 15 mg (RR = 1.97; 95% CrI, 1.01 to 4.28) were favoured for an improved PP-NRS score of at least 4 points compared to tralokinumab 300 mg. The Crls for the remaining comparisons were too wide to draw any conclusions of certainty in an improved PP-NRS score of at least 4 points between tralokinumab and other active comparators among adult patients.
The RRs and 95% CrIs across comparisons for an improved PP-NRS score of at least 4 points among adult patients who received combination therapy are summarized in Figure 28. The treatment response of all included combination-therapy interventions on an improved PP-NRS score of at least 4 points in adult patients was favoured over placebo. Treatments with upadacitinib 30 mg (RR = 2.37; 95% CrI, 1.75 to 3.29), abrocitinib 200 mg (RR = 2.04; 95% Crl, 1.47 to 2.89), upadacitinib 15 mg (RR = 1.91; 95% CrI, 1.34 to 2.74), and dupilumab 300 mg (RR = 1.79; 95% CrI, 1.28 to 2.55) were favoured for an improved PP-NRS score of at least 4 points compared to tralokinumab 300 mg. The point estimate for an improved PP-NRS score of at least 4 points favoured abrocitinib 100 mg over tralokinumab 300 mg, but the CrI also included the potential of little-to-no difference between the treatments (RR = 1.40; 95% Crl, 0.93 to 2.10).
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| || ||| ||| ||| |||||| ||||||||||| ||| |||||||| ||| |||||||||||| |||| || ||||| |||||||||| |||||||| ||| |||||||| ||||||||||| ||| |||||||||| || ||||| ||| ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || |||||||| ||| |||||||||||| |||| || || |||||||||| |||||||| |||| |||||||| |||| |||||||| || |||||||||| ||||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || |||||||| ||| ||||||||||||| |||| || ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Figure 27: Relative Risk (95% Credible Interval) Across Comparisons for an Improved PP-NRS Score of at Least 4 Points in Monotherapy RCTs in Adults (ICER Network Meta-Analysis)
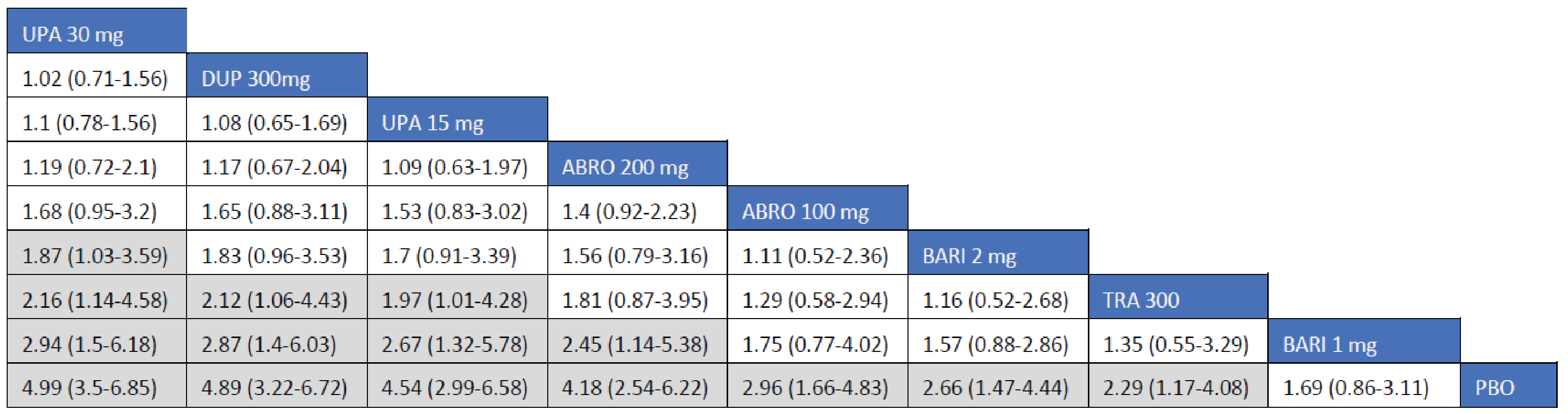
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; PBO = placebo; PP-NRS = peak pruritus numeric rating scale; RCT = randomized controlled trial; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Source: ICER network meta-analysis technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 28: Relative Risk (95% Credible Interval) Across Comparisons for an Improved PP-NRS Score of at Least 4 Points in Combination RCTs in Adults (ICER Network Meta-Analysis)
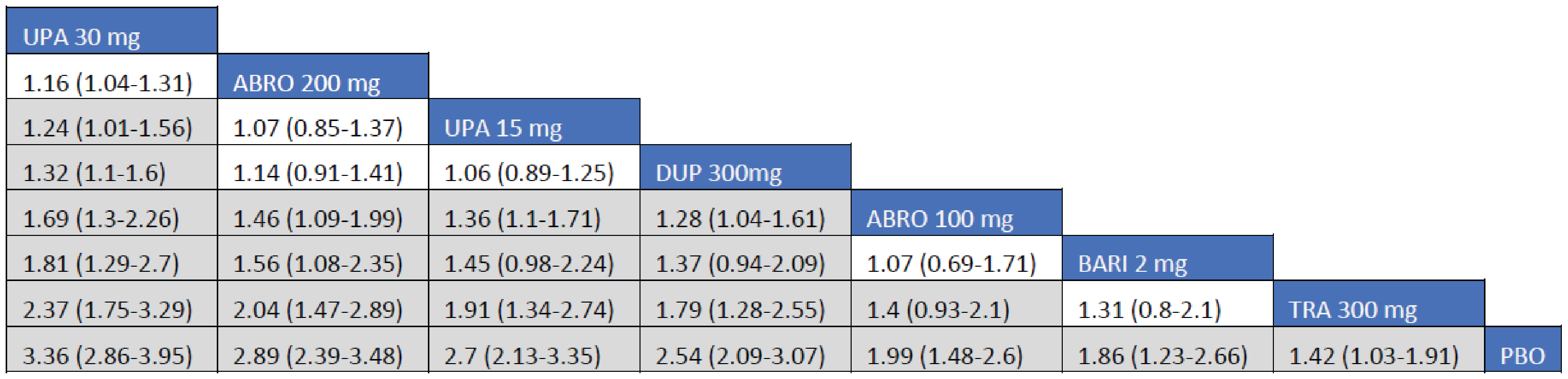
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; PBO = placebo; PP-NRS = peak pruritus numeric rating scale; RCT = randomized controlled trial; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not cross over 1.
Source: ICER network meta-analysis technical document19 and the sponsor’s Summary of Clinical Evidence.14
Figure 29: Odds Ratio (95% Credible Interval) for an Improved PP-NRS Score of at Least 4 Points Across Pairwise Comparisons of Monotherapy RCTs in Adolescents (LEO Pharma Network Meta-Analysis)

Figure was redacted at the sponsor’s request.
Children’s Dermatology Life Quality Index
Adult Population (ICER Network Meta-Analysis): A network meta-analysis of the CDLQI was not reported in the ICER NMA.
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| || ||| ||| ||| |||||| ||||||||||| ||| ||||| |||||||||||||| |||||||| || |||| || |||||||||||||| |||||||| ||| |||||||| ||||||||||| ||| |||||||||| || ||||| ||| ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || ||||| ||||||||||||| |||||||| || |||| || || |||||||||| |||||||| |||| |||||||| |||| |||||||| ||||||||| |||| ||||||||||| ||| || ||| |||||||||| |||| |||||||| ||||| ||||||||| |||||||| || |||| || |||||||| || |||||||||||| ||| || |||| ||||| ||| ||| |||| || ||||||| ||| ||||| |||||||| ||| ||||| |||||||||||||| |||||||| || |||| || |||||||| ||||||||| ||||||| || ||| |||| |||||||||||| ||| ||| ||| ||| ||| |||| |||||||| ||| ||||||||| || |||||||||||| |||||||||| ||||||| ||| |||||||||| |||| ||||| ||| |||| |||| || |||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||| |||||||||||||| |||||||| || |||| || ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Figure 30: Odds Ratio (95% Credible Interval) for CDLQI Across Pairwise Comparisons of Monotherapy RCTs in Adolescents (LEO Pharma Network Meta-Analysis)

Figure was redacted at the sponsor’s request.
POEM
Adult Population (ICER Network Meta-Analysis): A network meta-analysis of POEM scores was not reported in the ICER NMA.
Adolescent Population (LEO Pharma Network Meta-Analysis): ||| || ||| ||| ||| |||||| ||||||||||| ||| |||| ||||||||||||||| |||||||| || |||| || ||||| |||||||||| |||||||| ||| |||||||| ||||||||||| ||| |||||||||| || ||||| ||| ||| ||||||||| |||||||| || ||| |||||||| ||||||||||| ||||||||||||| || |||| |||||||||||||| |||||||| || |||| || || |||||||||| |||||||| |||| |||||||| |||| |||||||| |||||| ||| ||||||||||| ||| || ||| ||||||||| |||| |||||||||||| ||| || ||| |||||||| ||| |||| |||||||||||||| |||||||| || |||| || |||||||| || ||||||||||| ||| || |||| ||||| ||| ||| |||| || |||||| ||| ||||| |||||||| ||| |||| ||||||||||||||| |||||||| || |||| || |||||||| ||||||||| ||||||| || ||| |||| |||||||||||| ||| ||| ||| ||| ||| |||| |||||||| ||| ||||||||| || |||||||||||| |||||||||| ||||||| ||| |||||||||| |||| ||||| ||| |||| |||| || ||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || |||| |||||||||||||| |||||||| || |||| || ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Figure 31: Odds Ratio (95% Credible Interval) for POEM Across Pairwise Comparisons of Monotherapy RCTs in Adolescents (LEO Pharma NMA)

Figure was redacted at the sponsor’s request.
Harms
Adverse Events
Adults (ICER Network Meta-Analysis): A network meta-analysis of harms data was not reported in the ICER NMA.
Adolescents (LEO Pharma Network Meta-Analysis): ||| || ||| ||| ||| |||||| ||||||||||| ||| ||| || |||| || ||||| |||||||||| |||||||| ||| |||||||| ||||||||||| ||| |||||||||| || ||||| ||| || |||||||||| ||||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||| || |||| || ||||||| |||||||||||| ||| ||| ||||| |||||| ||||||||||||
Figure 32: Odds Ratio (95% Credible Interval) for Adverse Events Across Pairwise Comparisons of Monotherapy RCTs in Adolescents (LEO Pharma Network Meta-Analysis)

Figure was redacted at the sponsor’s request.
Critical Appraisal of ICER and LEO Pharma Network Meta-Analyses
ICER Network Meta-Analysis
The ICER NMA was based on studies identified from a systematic literature review of relevant randomized evidence of treatments for adults and adolescent with AD. The systematic literature search was based on a PICO model defined a priori, with efficacy and safety outcomes predefined. The systematic literature search was comprehensive, involving multiple electronic databases, clinical registries, and supplementary sources. However, the reasons for study exclusions were not reported and the selection process was not clearly defined, and relevant studies could have been missed. Data extraction was conducted by a single reviewer, resulting in a potential for errors or omissions. While the risk of bias of the comparator trials was assessed, the method used was not reported, and risk of bias was not assessed by outcome.
Several sources of clinical and methodological heterogeneity were identified. The patient population varied in age (mean age range = 31 to 41 years), duration of disease (mean duration = 21 to 28 years) and disease severity (IGA score of 4 range = 32% to 55%). According to feedback from the clinical experts consulted by CADTH for the purpose of this review, the range of IGA scores of 4 may suggest that the patient populations were different across the included studies. The washout period ranged from 1 to 2 weeks, and was as short as 72 hours in the abrocitinib trials (JADE MONO-1, JADE MONO-2, and JADE COMPARE). A longer washout period may lead to exacerbation before trials; however, the clinical experts were uncertain if it would lead to more use of rescue medication during the trial. Measurement of the primary end points ranged from 12 weeks to 16 weeks in the remaining studies. To account for differences in corticosteroid use across trials, separate NMAs were conducted for monotherapy and combination therapies. However, the treatment of patients in the control group (placebo plus TCS) was not consistent across the combination-therapy trials. Of the combination-therapy trials, only 1 (ECZTRA 3) standardized the combination of TCS with mometasone furoate 0.1% cream. Other trials allowed a subset of TCS options and potency levels. Whereas 1 trial supplied TCS free of charge by trial sites, TCS combination therapy was not provided by the trial sponsor of 2 studies; access to TCS combination therapy was not detailed in the remaining trials. Based on input from the clinical experts, more-potent TCS can lead to better responses in the placebo group. Finally, all trials included in the review used imputation to adjust for missing data (combinations of multiple imputation, nonresponder imputation, or LOCF), although there was no systematic difference in imputation methods across end points. It is unknown what effect the different methods of imputation may have had on the results. The sources of clinical and methodological heterogeneity could have introduced intransitivity, which would result in biased effect estimates.
No information was given on details of the assessment of statistical heterogeneity or statistical consistency. The networks were sparse (several comparisons with relatively few studies). While the lack of head-to-head comparisons among active treatments would make tests for consistency difficult, the network consisted of several closed loops that could have been tested. No sensitivity analysis exploring possible assumptions made by the reviewers were reported. Moreover, there was no indication of model adjustment to account for the correlation in the 3 arm trials. All comparisons to tralokinumab were indirect, which introduced increased uncertainty into the findings.
NMA results were presented only for EASI, IGA, and PP-NRS outcomes; harms outcomes and other outcomes of relevance to patients (e.g., HRQoL) were not reported.
LEO Pharma Network Meta-Analysis
The LEO Pharma NMA was based on studies identified from a systematic review of relevant randomized evidence of treatment for moderate-to-severe AD in adolescent patients. The systematic literature search was based on a PICO model defined a priori, with efficacy and safety outcomes predefined. The systematic literature search was comprehensive, involving multiple electronic databases, clinical registries, and supplementary sources. The reasons for study exclusions were reported; and the selection and data-extraction processes were defined. Overall, || trials were excluded because studies were either ongoing or recruiting patients. It is unknown if those trials have since been published, and evidence may be missing from the NMA. Data extraction was conducted by 2 reviewers in a double-blinded fashion. While the risk of bias of the comparator trials was assessed, the methods used were not reported and the risk of bias was not assessed by outcome.
Several sources of heterogeneity were identified across the included studies. First, the time point at which primary efficacy outcomes were measured across studies |||||| ||||||| || || || ||||| || ||| ||||||||||| ||||||. The predetermined duration of AD for study inclusion was || ||||||| ||| |||||| |||||||||| |||||||||||| ||| || ||||||| ||| ||| ||||||||| ||||||. Based on input from the clinical experts, a predetermined duration of AD may not significantly influence treatment response. However, the clinical experts did add that patients with longer disease duration are more likely to have lichenification and other chronic changes, suggesting a more stubborn disease that has been treated with more options. Another source of heterogeneity across trials was the exclusion criterion related to prior use of biologics. Based on feedback from the clinical experts, prior use of biologics for AD is not likely to affect treatment response. Finally, protocol use and investigational drug discontinuation for rescue treatment varied across trials.
No information was given on model fit and assessment of statistical consistency. The networks were sparse (several comparisons with relatively few studies). While the lack of head-to-head comparisons among active treatments would make tests for consistency difficult, the network consisted of several closed loops that could have been tested. No sensitivity analysis exploring possible assumptions made by the reviewers were reported. All comparisons to tralokinumab were indirect, which introduces increased uncertainty to the findings. Due to small sample sizes, the CrIs for several comparisons were wide, which precluded drawing any conclusions about comparative efficacy and safety.
Description of Sponsor-Submitted Matching Adjusted Indirect Comparisons
Objectives
The sponsor submitted 2 MAICs, conducted on its behalf by a third party, comparing the relative efficacy of tralokinumab versus dupilumab in adults with moderate-to-severe AD.21,22
||| |||||||| |||| ||||| || ||| |||||||| ||||||| || |||| |||||| ||||| || ||||||| |||||||||||| ||||||| || |||||||||||| |||| |||||||| ||||| || ||||||||||| |||| ||||||| ||||||||||||||| ||||||| ||||||||| |||||||| || ||||| |||||||||||| ||| || ||||||||||| |||| ||||||| ||||||||||||||| || || ||||| || ||||||||| || ||||| |||||||| |||| || ||| ||| ||||||||| ||||||||| ||||||||| |||||||||||| ||||||||
The unanchored MAIC based on the ECZTRA 3 and LIBERTY AD CHRONOS trials was designed to assess the long-term efficacy outcomes for tralokinumab 300 mg (ECZTRA 3) administered every 2 weeks and 300 mg every 4 weeks against dupilumab (LIBERTY AD CHRONOS) every 2 weeks at 32 to 52 weeks of follow-up in adult patients with moderate-to-severe AD.21
Study Selection Methods
The study selection criteria and methods for the sponsor-submitted MAICs are summarized in Table 64.
ECZTRA 7 Versus LIBERTY AD CAFÉ
|||| ||||||||||||||||||| |||||| || |||||||||||| ||||||| || || || ||| || ||| ||||||||| |||||| || ||||||| || ||||| ||||||| || |||||||| |||| |||||||||| ||| |||||||||| ||||| ||| ||||||||||| || |||||||||||||||||| ||| ||| ||||||||||| || ||| |||||||| ||| || |||||||| ||||||| || |||||||| ||||||||||||| ||||||| || ||||||||||| || ||||||| |||||||| ||||||| ||| ||| || ||||||||||| ||| ||| ||| || |||||| |||||||||| |||||| ||||||| |||||||| ||||||| || |||| |||| |||||||| ||| ||| |||| ||| || ||||| |||||||||| || ||||||| |||||||||| ||||||||||||||| ||| ||||||||| |||| ||| ||||||||||||| ||||||| ||||||||| || ||||| ||||||||| ||||||||||||||||| ||||
ECZTRA 3 Versus LIBERTY AD CHRONOS
The trial selection process for the ECZTRA 3 versus LIBERY AD CHRONOS MAIC was not reported.
Table 46: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | ECZTRA 7 vs. LIBERTY AD CAFÉ | ECZTRA 3 vs. CHRONOS |
|---|---|---|
Population | ||||| |||||||||| |||| |||||| || || ||| |||||||||| |||||||||| |||||||| | Adult patients with moderate-to-severe AD |
Intervention | |||||||||||| |||| ||| | Tralokinumab plus TCS as needed |
Comparator | ||||||||| |||| ||| | Dupilumab plus TCS as needed |
Outcome | || |||| |||||||||||| |||| ||||||||||||||||||||||||||||||||||||| |||||| |||||||| | Binary analyses Dupilumab week 32 and week 52 vs. tralokinumab week 32:
Dupilumab week 52–only vs. tralokinumab week 32:
Continuous analyses Dupilumab week 52–only vs. tralokinumab week 32:
|
Study designs | |||| | RCTs |
Publication characteristics | NR | NR |
Exclusion criteria | NR | NR |
Databases searched | NR | NR |
Selection process | NR | NR |
Data-extraction process | NR | NR |
Quality assessment | NR | NR |
AD = atopic dermatitis; CsA = cyclosporine A; DLQI = Dermatology Life Quality Index; EASI-50 = reduction of at least 50% in Eczema Area Severity Index score from baseline; EASI-75 = reduction of at least 75% in Eczema Area Severity Index score from baseline; EASI-90 = reduction of at least 90% in Eczema Area Severity Index score from baseline; IGA = Investigator's Global Assessment; ITC = indirect treatment comparison; NA = not applicable; NR = not reported; POEM = Patient-Oriented Eczema Measure; PP-NRS = Peak Pruritus Numeric Rating Scale; RCT = randomized control trial; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroids.
Sources: ECZTRA 3 and LIBERTY AD CHRONOS MAIC technical document;21 ECZTRA 7 vs. LIBERY AD CAFÉ MAIC technical document,22 and the sponsor’s Summary of Clinical Evidence.14
Indirect Treatment Comparison Design for Sponsor-Submitted MAICs
ITC Analysis Methods
A summary of the analysis methods for the MAIC is presented in Table 65.
ECZTRA 7 Versus LIBERTY AD CAFÉ
|| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| |||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| ||||| |||||||| |||| |||||||| ||| |||||||| ||| ||| |||||||| |||||||||| ||||||| |||||||||||| ||| |||||||||| ||| ||||||| ||||||||| |||| |||| |||||||| ||| ||||||| || |||||||||||| || |||| |||| ||||||||||||| |||||||| |||
ECZTRA 3 Versus LIBERTY AD CHRONOS
An unanchored MAIC approach was selected for the indirect comparison between tralokinumab and dupilumab, which relied on individual patient data from ECZTRA-3 and aggregate data from LIBERTY AD CHRONOS.
The selection process to choose variables for matching was not reported. The following covariates were used for matched adjustment: age, gender, race (percent white), AD duration, and body mass index, EASI, SCORAD, DLQI, and IGA, at baseline. Matching was performed by weighing the baseline characteristics from the individual patient data from ECZTRA 3 such that their weighted mean baseline characteristics matched those of the population of LIBERTY AD CHRONOS. Propensity-score weighting in which patients with individual patient data (i.e., those in ECZTRA 3) were weighted by their inverse odds of being in that trial as opposed to being in LIBERTY AD CHRONOS.117 The aforementioned covariates were entered into logistic regression to estimate propensity scores for the unanchored MAIC.115,117 Variables that were not considered important effect modifiers were excluded from the analysis to avoid unnecessary erosion of accuracy. Weights were calculated by each treatment arm separately according to methods detailed by Signorovitch et al.114 The weighting was such that individual patient data of the ECZTRA-3 population resembled the LIBERTY AD CHRONOS population according to the mean values of baseline variables used in the matching.
The following binary efficacy end points were analyzed at both week 32 and week 52 for dupilumab and week 32 for tralokinumab: EASI-50, EASI-75, EASI-90, and an IGA of score 0 or 1. The following binary efficacy end points were analyzed at week 52 for dupilumab and week 32 for tralokinumab: DLQI improvement of at least 4, POEM improvement of at least 4, and worst daily pruritus NRS improvement of at least 4. The following continuous outcomes were analyzed at week 52 for dupilumab and week 32 for tralokinumab: percent change in EASI, percent change in SCORAD, the weekly average change in worse daily pruritus score, change in DLQI and change in POEM.
To estimate the comparative efficacy of tralokinumab and dupilumab, individual patient-level data from ECZTRA 3 (tralokinumab 300 mg every 2 weeks and every 4 weeks combined)11,118 and published aggregate data from LIBERTY AD CHRONOS (dupilumab every 2 weeks) were used.112 The risk difference of tralokinumab combined with TCS versus placebo combined with TCS was estimated using a Cochran-Mantel-Haenszel analysis. Results were presented as RDs or mean differences with 95% CIs on forest plots. For both the ECZTRA 3 and LIBERTY AD CHRONOS trials, patients who used rescue medication or had missing values were imputed as nonresponders for binary outcomes. Continuous efficacy end points were analyzed with an ANCOVA model that included the baseline measurement of the end point and randomization strata (region and IGA baseline) as covariates, using an LOCF approach after rescue treatment initiation or dropout.
Table 47: Matching Adjusted Indirect Treatment Comparison Analysis Methods
Methods | ECZTRA 7 vs. LIBERTY AD CAFÉ | ECZTRA 3 vs. CHRONOS |
|---|---|---|
Analysis methods | |||||||| |||| | Unanchored MAIC |
Outcomes | |||||||| |||||||| |||||||| || |||| ||||||| ||||||||||| |||||||||||||||||||||||||| |||| |||||||| || ||||| ||||| |||||||| ||| || || ||||| |||||||| || ||| ||||| ||| ||||||||||| || |||||| ||||| |||| |||||||||||||| ||||||||| || || ||||| |||||| ||||||||| || || ||||| ||||| |||| |||||||||| ||||||| |||||||| |||| ||||||| |||| || ||||| ||| |||| |||||| |||||||||| |||| |||||||||| ||||||||||||||||||||||||||| |||| |||| ||||||| |||| |||||||||||||||||||||||||| |||||||||||||||||||||||||||||| ||||| ||||| |||||| ||||| |||||||||| |||| |||||||| |||||||||| ||| ||||||||| |||||| ||| ||||| || ||||| |||| |||||||||| ||| |||||| |||| |||| || |||||| |||| ||| ||||||||| || ||| ||||||| || |||| |||||||| | Binary analyses Dupilumab week 32; week 52 vs. tralokinumab week 32:
Dupilumab week 52 vs. tralokinumab week 32:
Continuous analyses Dupilumab week 52 vs. tralokinumab week 32:
|
Follow-up time points | || ||||| | Week 32 and week 52 |
Sensitivity analyses | || ||||||||| |||||||| || ||| ||||||||||| |||||||| |||| ||||||||||||||| ||||||||| || ||| |||| ||||||||| ||| ||||||||| |||||||| |||||||||| || ||||| |||||||| ||| ||||||| |||||||||||||| ||||| ||||||| ||| ||||||| ||| |||||||| ||||||||| ||||||||||||||||| ||||||||| ||||||| |||| || ||||||| || || ||| |||| ||||||||||||||||||| ||||||||| || ||| |||| ||||||||| ||| ||||| |||||||| ||||||||| ||||||| || ||||||||||||| ||||||||| || ||| |||| ||||||||| ||| ||||| ||||||||| ||||||||| ||||||| || ||||||||||| ||| |||||||| ||||||||||| | NR |
AD = atopic dermatitis; AE = adverse event; AESI = adverse event of special interest; CMH = Cochran-Mantel-Haenszel; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = investigator global assessment, MAIC = matching adjusted indirect comparison; NR = not reported, NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; PT = preferred term; SCORAD = Scoring Atopic Dermatitis.
Sources: ECZTRA 3 and LIBERTY AD CHRONOS MAIC technical document;21 ECZTRA 7 vs. LIBERY AD CAFÉ MAIC technical document,22 and the sponsor’s Summary of Clinical Evidence.14
Results of Sponsor-Submitted MAICs
Summary of Included Studies
The trials included in the ECTRA 3 and LIBERTY AD CHRONOS studies and the ECTRA 7 and LIBERTY AD CAFÉ MAICs are summarized in Table 66, and baseline characteristics of the included studies are presented in Table 67.
ECZTRA 7 Versus LIBERTY AD CAFÉ
||| |||||||| ||||||| || |||| |||| |||||||| |||||||||| ||||||||||||| |||| ||| |||||||| ||| |||||||| |||||||||||| || ||||||||||| |||| ||||||| ||||||||||||||| ||||| |||||| ||||||| || ||||||||||| |||| ||||||| ||||||||||||||| |||| || ||||||||||||| ||||||||| ||||||||| |||| |||| |||||||| ||| |||||||| ||||||||| || ||||||||||| |||| ||||||| ||||||||||||||| ||||| |||||| ||||||| || ||||||||||| |||| ||||||| ||||||||||||||| |||| || ||||||| || |||| ||||||||||| |||||| |||| |||||||||||||| ||||||||||| |||| |||||| |||| ||||||||| || ||||| |||||||| |||| |||||| || ||| |||| ||||||||||| || ||| ||| || ||||||||||| || |||||||||||||||| || |||||||||||||||||||| ||||| |||| ||||||| ||||||||||| || ||| ||||||||| |||||||| ||||||| ||||| ||| |||| |||||| ||| |||||||||| |||||||| || ||||||| ||||||||| |||||| || ||| ||||||||||| || ||| ||||||| || |||| ||||||| ||||||||| ||| |||||||||||| |||||||| || |||||||||| |||| || ||||||| ||||||||| || ||||| ||||| ||||| ||| || |||| || |||| ||||| || ||||||||||||| ||| |||||||||| ||| |||||| ||||||| ||||||| || |||| |||||||| |||||||| || |||||||||| |||| || ||||||| ||||||||| ||||| ||||| ||| || |||||||| || |||| ||||| || |||||||| |||||| ||| ||||||||||| ||| || ||||||| ||||||||||||||| ||| |||||| |||||||||| |||||| ||| ||||| |||||| |||| |||||||| ||||||| ||| ||||||| ||||||||||| ||||||| |||||||| |||| || |||||| || |||||||||||| || ||| |||||| |||||| ||| || || |||| || |||||| ||||| || ||| ||||||| || |||| |||||| ||||||||||| ||||||| ||||||||||||||| ||| || |||||| ||||||| ||| ||| || || ||||||| |||| ||||| |||| |||||| ||| ||||| ||||||||| |||||| || |||||| |||||| |||||| || |||||| || |||||| ||||||||||| |||| |||||||| || ||| |||| ||||||||| |||||||||||||| ||||||| |||||| |||||||||| ||| |||||||| ||||| |||| ||||||| || ||||| |||| ||||||||||||| |||||| || |||||||| || ||||| |||||||| || || ||||| || |||||| || ||||| || |||||||| |||| ||||||| || ||| |||||||||| || ||||| ||||||| |||| ||||||| |||| |||||||| || |||| || || ||| ||||||||| |||| |||||||| || ||| |||| ||||||||| || |||| ||||||| ||| ||||||| |||||||| |||||||| ||| ||||||| || |||| ||| |||||||| || ||||||| |||||||| |||||| ||||||||| || ||||||||||| |||||||||||| ||| ||||||||||||||| ||||||| ||| ||||||| ||| ||||||| || ||||||| ||||| || ||||||||| |||| |||||||||| ||||||||||||||| |||||||| ||| |||||||| |||||| ||||||||| |||| |||||||||| |||||||||||||| || ||||||| || ||||| || ||||| ||||||| |||| || |||| |||||| |||| ||||||| || ||||||||||||||||||||||| |||||| |||||| |||||||| ||||||| || |||| |||| |||||||||| ||||||| || ||||| || ||| |||||||||| || |||||||| |||| || ||| ||||| || || ||| |||||| |||| ||| ||||||| ||| ||||| ||||| |||||||| ||| || ||||||||| |||||| ||||| ||||| ||| ||| ||||| |||||||| ||||||| |||||| |||||| ||| ||||||||| ||||||| |||||| |||| || |||| ||||||||| |||||| || ||| |||||||||| ||| |||| |||||||||||||| ||||| |||||| |||| || ||||| ||| |||||| |||| |||| ||||| ||| || ||||| || ||| ||||||||| |||| ||||||| ||||||||||||||| ||||||||| ||||| || |||| ||||| ||| || ||||| || ||| ||||||| |||| ||||||| |||||||||||||| ||||||||| ||||| || ||| ||||||| || |||| |||||| ||||| ||| ||| |||||| ||||||||||||| ||| || ||| |||||||||| ||| |||||| ||||||| ||||| || ||||| || ||| ||||||| || |||| |||||| ||| ||| ||| |||||||||| ||| ||||||||||| || ||||||||||||||||||| || |||||||| || ||| |||||| ||||| ||||| ||| || |||| ||||| || ||| ||||||| || |||| |||||| ||||| |||||||| |||||||| ||| || |||||||| ||| |||||||| || ||||| ||| ||||| || |||||||| || ||| |||||||||||| |||| ||||||| ||||||||||||||| ||| ||||||| |||| ||||||| |||||||||||||| ||||||||| ||||| |||||||||||| || ||| |||||||||| ||| ||| ||||| ||| ||||| || ||| ||||||||| |||| ||||||| |||||||||||||| ||| ||||||| |||| ||||||| |||||||||||||| ||||||||| |||||| |||||||||||| || ||| ||||||| || |||| ||||||
ECZTRA 3 Versus LIBERTY AD CHRONOS
The ECZTRA 3 versus LIBERY AD CHRONOS MAIC included individual patient-level data for patients who received tralokinumab 300 mg every 2 weeks and every 4 weeks combined (n = 250)11,118 versus published aggregate data from patients who received dupilumab every 2 weeks (n = 106) in the LIBERTY AD CHRONOS study.112 Both trials were double-blinded RCTs.
In the ECZTRA 3 study, patients who achieved a clinical response (an IGA of 0 or 1 or EASI-75 at week 16) were re-randomized 1:1 to tralokinumab every 2 weeks or every 4 weeks while those who did not achieve a clinical response from either tralokinumab or placebo received tralokinumab every 2 weeks from week 16; the remaining patients who achieved a clinical response to placebo continued to receive placebo. In the LIBERTY AD CHRONOS study, patients received their randomly assigned treatment (dupilumab 300 mg once weekly or every 2 weeks or placebo) for the entire 52-week duration of the study.
Both trials were conducted in adult patients with moderate-to-severe AD who had an inadequate response to or who had intolerance or contraindication to topical treatments. The trials differed on the minimum time period for inadequate response to topical treatment (1 year in the ECZTRA 3 study and 6 studies months in the LIBERTY AD CHRONOS study). The concomitant use of topical corticosteroids and rescue medication during the study period also differed between the trials. Concomitant topical steroids were of potent to medium strength in the ECZTRA 3 study, and to be used as needed while in the LIBERTY AD CHONOS study, and concomitant TCS were of medium potency in the LIBERTY AD CHONOS study. In the ECZTRA 3 study rescue medications were provided at any time following randomization, whereas rescue medication was provided after week 2 in the LIBERTY AD CHONOS study. Rescue medications were taken as needed in both trials.
The observational period of the ECZTRA 3 study was 32 weeks compared to 52 weeks in the LIBERTY AD CHRONOS study. Missing data in both trials were managed as nonresponses. Mean EASI scores between the ECZTRA 3 and LIBERTY AD CHRONOS studies were generally similar at 28.7 (SD = 11.8) and 33.6 (SD = 13.3), respectively. Mean baseline DLQI scores were higher in the ECZTRA 3 study (67.0 = SD, 13.2) relative to LIBERTY AD CHRONOS 14.5 (SD = 7.3). A greater proportion of patients reported prior use of systemic steroids in the ECZTRA 3 study (58.5%) compared to the LIBERTY AD CHRONOS study (38.2%).
Table 48: Comparison of Trials Included in the Sponsor-Submitted MAICs
Details | ECZTRA 7 and LIBERTY AD CAFÉ | ECZTRA 3 and LIBERTY AD CHRONOS | ||
|---|---|---|---|---|
Tralokinumab (ECZTRA 7) N = 325 | Dupilumab (LIBERTY AD CAFÉ) N = 325 | Tralokinumab (ECZTRA 3) | Dupilumab (LIBERTY AD CHRONOS) | |
Overall design | Randomized, double-blind, placebo-controlled, parallel-group clinical trial | Randomized, double-blind, placebo-controlled, phase III trial | ||
Study duration | 26 weeks | 16 weeks initial | 32 weeks | 52 weeks |
Duration of placebo-controlled period | 26 weeksa | 16 weeks | 16 weeks followed by re-randomized responder maintenance phase for another 16 weeks | 52 weeks (16 and 52 weeks) |
Primary end points | EASI-75 at week 16 | EASI-75 at week 16 | NR | NR |
Primary analysis for primary end points | Patients were considered nonresponders after initiation of rescue treatment or permanent discontinuation of the study drug before week 16; missing data imputed as nonresponses | Patients were considered nonresponders after initiation of rescue treatment; missing data imputed as nonresponses | NR | NR |
Key inclusion criteria |
|
| Adults aged 18 years and older with inadequate response to topical treatment within 1 year or for whom topical treatments were inadvisable | Adults aged 18 years and older with inadequate response to TCS with or without TCI or systemic treatment in previous 6 months |
Washout of TCS or TCI |
|
| 2 weeks | 1 week |
Concomitant TCS |
|
|
|
|
Rescue medication use |
|
| Provided at any time following randomization | Provided after week 2 |
AD = atopic dermatitis; CsA = cyclosporine A; IP = investigational product; MAIC = matched adjusted indirect comparison; NIMP = noninvestigational medicinal product; NR = not reported; TCI = topical calcineurin inhibitor; TCS = topical corticosteroids.
aOutcomes were also reported after week 16.
Sources: ECZTRA 3 and LIBERTY AD CHRONOS MAIC technical document,21 ECZTRA 7 vs. LIBERY AD CAFÉ MAIC technical document,22 and the sponsor’s Summary of Clinical Evidence.14
Table 49: Comparison of Baseline Characteristics of Studies in the Sponsor-Submitted MAICs Before Matching
Baseline characteristics | ECZTRA 7 vs. LIBERTY AD CAFÉ | ECZTRA 3 vs. LIBERTY AD CHRONOS | ||||
|---|---|---|---|---|---|---|
Tralokinumab (ECZTRA 7) N = 325 | Dupilumab (LIBERTY AD CAFÉ) N = 325 | Tralokinumab (ECZTRA 3) N = 250 | Dupilumab (CHRONOS) N = 106 | |||
Treatment arm | TRA + TCS | PLC + TCS | DUPI + TCS | PLC + TCS | TRA + TCS | DUPI + TCS |
IGA score of 4 (%) | 50.7 | 48.9 | 46.7 | 48.1 | NR | NR |
Median EASI (IQR)a | 28.60 (22.40 to 38.00) | 29.10 (22.80 to 40.15) | 31.6 (25.2 to 39.2) | 31.7 (24.2 to 40.7) | 28.7 (11.8) | 33.6 (13.3) |
Median SCORAD (IQR)a | 69.20 (61.50 to 76.50) | 68.90 (61.20 to 81.00) | 66.7 (61.1 to 76.2) | 67.5 (58.5 to 76.6) | NR | NR |
Median worst daily pruritus NRS | 7.43 (6.43 to 8.29) | 7.50 (6.59 to 8.37) | 7.0 (5.4 to 8.0) | 6.9 (4.9 to 8.1) | NR | NR |
Median DLQIa | 16.00 (11.0 to 21.00) | 16.00 (11.00 to 21.00) | 14.0 (8.0 to 22.0) | 13.0 (7.0 to 19.5) | 17.6 (7.1) | 14.5 (7.3) |
Prior CsA use (%) | 75.0 | 74.5 | 64.5 | 66.7 | NR | NR |
CsA considered not appropriate (%) | 25.0 | 25.5 | 35.5 | 33.3 | NR | NR |
Prior systemic steroids (%) | 70.0 | 66.4 | 31.8 | 37.0 | 58.5 | 38.2 |
CsA = cyclosporine A; DLQI = Dermatology Life Quality Index; DUPI = dupilumab; EASI = Eczema Area Severity Index; IGA = Investigator’s Global Assessment; IQR = interquartile range; MAIC = matching adjusted indirect comparison; NR = not reported; NRS = numeric rating scale; PLC = placebo; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroids; TRA = tralokinumab.
aValues presented for ECZTRA 3 vs. LIBERTY AD CHRONOS represent mean (standard deviation).
Sources: ECZTRA 3 and LIBERTY AD CHRONOS MAIC technical document,21 ECZTRA 7 vs. LIBERY AD CAFÉ MAIC technical document,22 and the sponsor’s Summary of Clinical Evidence.14
Results
|||||||||| ||| ||||||||||||||||| ||||||| ||||||||||||||| |||| |||||||| ||||||| || |||| ||| |||||||||| || ||||| ||| ||||| ||||||||||| || |||||||| ||| |||||||| ||||||||||||||| || ||| |||||||| ||||||| |||||||||| || ||||||||| ||||||| |||| ||||||| || |||||||| || ||| |||||||| |||| |||||||| || ||| ||||||||| |||| ||||||| ||||||||||||||| ||||||||| |||||| ||| ||||||||| |||||| |||| ||||| ||||||||| |||||||||||||||| ||| |||| ||| |||||||||||| ||| |||| ||||||| ||||||||||||||| ||||||||| ||| |||| || ||| |||||||| ||||||||||| ||| |||| ||| ||| ||||||| |||| ||||||| ||||||||||||||| ||||||||| ||| |||||| || ||| |||||||| |||||||||||| |||||||||| || ||||||| ||| || |||||||| ||||||||||| || ||| ||||||| ||||||||| || ||| ||| || ||||| |||||||| ||||||||| || ||||||| ||| ||||||| |||||||| ||||||| || |||||
Unadjusted and matched adjusted patient characteristics from the ECZTRA 3 and LIBERTY AD CHRONOS trials are summarized in Table 69. After matching, the baseline characteristics of the weighted patient population of the ECZTRA 3 trial were matched with those of the LIBERTY AD CHRONOS trial. A total of 106 patients were included in the dupilumab treatment group. The ESS following match-adjustment was 123.4 for the tralokinumab treatment arm (49.36% of the original population).
Table 50: Baseline Characteristics in ECZTRA 7 Versus LIBERTY AD CAFÉ MAIC Before and After Matching, Main Analysis [Redacted]
|||||||| ||||||||| | ||||||| | |||||||||||| | ||||||||| | |||
|---|---|---|---|---|---|---|
||||||| || |||||||||||| | |||||| ||||||||| | |||||| ||||||||| | ||||||| || |||||||||||| | |||
|||||||| | |||||||| | |||||||||| | |||||||| | |||||||||| | |||||||| | |||||||| |
||||| | ||| | ||| | |||| | ||| | |||| | ||| |
|||| |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|| ||||||||| |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||| |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||| |||| | |||| | |||| | |||| | |||| | |||| | |||| |
|||| |||| | |||| | |||| | |||| | |||| | ||| | ||| |
||||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||||| |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||||| |||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||| ||||| | |||| | |||| | |||| | |||| | |||| | |||| |
||||||| |||| |||||| ||||||||||||| |||| ||||||| |||||| ||||||||| |||| ||| |||||||| |||||| |||||||||||||| |||||| ||||||||||| |||||||||||| |||||| |||||||||| |||||||| |||||||| |||||| || |||| |||| ||||||||| |||||||
Table 51: Baseline Characteristics in ECZTRA 3 Versus LIBERY AD CHRONOS MAIC Before and After Matching
Baseline characteristics | Dupilumab | Tralokinumab unweighted | Tralokinumab weighted |
|---|---|---|---|
N (ESS) | 106 | 250 | 123.5 |
Age (years), mean (SD) | 39.6 (14.0) | 39.8 (15.3) | 39.6 (16.0) |
Male (%) | 58.5 | 49.2 | 58.5 |
Body mass index (kg/m2), mean (SD) | 25.5 (5.8) | 27.6 (6.) | 25.5 (5.6) |
Disease duration (years), mean (SD) | 30.1 (15.5) | 27.9 (16.4) | 30.1 (17.6) |
White (%) | 69.8 | 80.4 | 69.8 |
EASI score, mean (SD) | 33.6 (13.3) | 28.7 (11.8) | 33.6 (13.9) |
IGA score, mean (SD) | 3.5 (0.5) | 3.5 (0.5) | 3.5 (0.5) |
DLQI score, mean (SD) | 14.5 (7.3) | 17.6 (7.1) | 14.5 (6.6) |
SCORAD score, mean (SD) | 69.3 (15.2) | 67.0 (13.2) | 69.3 (14.3) |
EASI = Eczema Area and Severity Index; ESS = effective sample size; DLQI = Dermatology Life Quality Index; IGA = Investigators Global Assessment SCORAD = Scoring Atopic Dermatitis; SD = standard deviation
Sources: ECZTRA 3 and LIBERTY AD CHRONOS MAIC technical document21 and the sponsor’s Summary of Clinical Evidence.14
Efficacy Outcomes
ECZTRA 7 Versus LIBERTY AD CAFÉ
||| |||| |||||||| |||||||| ||||||| || |||| |||| |||||||| ||| ||||||||| || |||||| ||| |||| ||||||| || ||| ||||||| ||||||| || |||| |||||||| |||||||| |||| |||||||| ||||||| |||||||||||| |||||||||||||||||||||| ||| ||||||||| |||| ||||||| |||||||||||||| ||| || |||||| || ||||||||| |||| ||||||| ||||||||||||||| ||| ||||||| |||| |||||| ||| ||| ||||| || |||||| ||| ||||| |||||||| ||| |||| ||||||||| || || |||||||||| |||||||| ||||||||| |||| ||||||| ||||||||||||||| |||| |||||||||||| |||| ||||||| |||||||||||||||| ||| ||| || |||||||| ||| ||||||||| || |||||||||||| |||||||||| ||||||| ||| |||||||||| |||| |||||| ||| ||| ||||| || ||||| ||| || ||| |||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||| ||||||||| |||||||| |||||| |||||||| ||| |||| |||||||| |||||||| |||| |||||||||| ||||| ||||| ||||||| |||||||||||| |||| ||||||| ||||||||||||||| ||| ||||||||| |||| ||||||| |||||||||||||||| ||||||||||| |||||||| |||| ||||||||| |||||||||| |||| ||| |||| |||||||| |||| |||||||||
ECZTRA 3 Versus LIBERTY AD CHRONOS
The main efficacy ECZTRA 3 versus LIBERTY AD CHRONOS MAIC analyses for binary and continuous outcomes are presented in Figure 16 and Figure 17, respectively.
The results of the ECZTRA 3 versus LIBERTY AD CHRONOS unanchored efficacy MAIC analysis between tralokinumab and dupilumab were in favour of tralokinumab for IGAs of 0 or 1 (RD = 13.9; 95% CI, 0.6 to 27.3) and change in DLQI (mean difference = −1.7; 95% CI, −3.0 to −0.3) at week 52. The CIs for comparisons were too wide to draw any conclusions of certainty on the remaining outcomes between tralokinumab and dupilumab.
Figure 33: Main Efficacy MAIC Analysis of ECZTRA 7 Versus LIBERTY AD CAFÉ

Figure was redacted at the sponsor’s request.
Figure 34: Efficacy MAIC Analysis of ECZTRA 3 Versus LIBERTY AD CHRONOS; Risk Difference of Achieving Binary Efficacy End Points (Week 32 and Week 52)
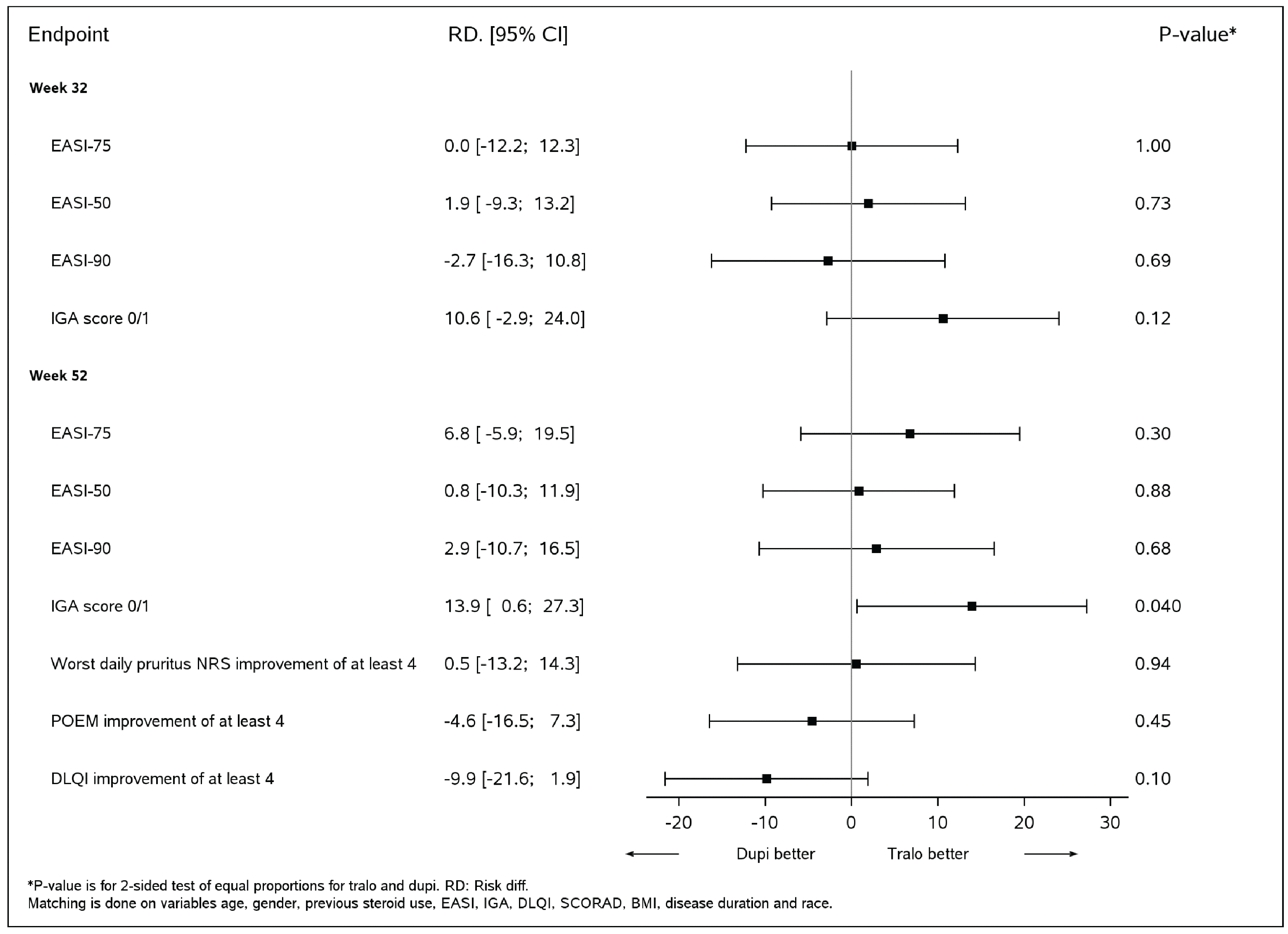
BMI = body mass index; CI = confidence interval; DLQI = Dermatology Life Quality Index; Dupi = dupilumab; EASI-50 = reduction of at least 50% in Eczema Area and Severity Index score from baseline; EASI-75 = reduction of at least 75% in EASI score from baseline; EASI-90 = reduction of at least 90% in Eczema Area and Severity Index score from baseline; IGA = Investigator’s Global Assessment; MAIC = matching adjusted indirect comparison; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; RD = risk difference; SCORAD = Scoring Atopic Dermatitis; Tralo = tralokinumab.
Source: ECZTRA 3 and LIBERTY AD CHRONOS MAIC technical document.21
Figure 35: Efficacy MAIC Analysis of ECZTRA 3 Versus LIBERTY AD CHRONOS Mean Difference for Continuous Outcomes for Tralokinumab (Week 32) and Dupilumab (Week 52)
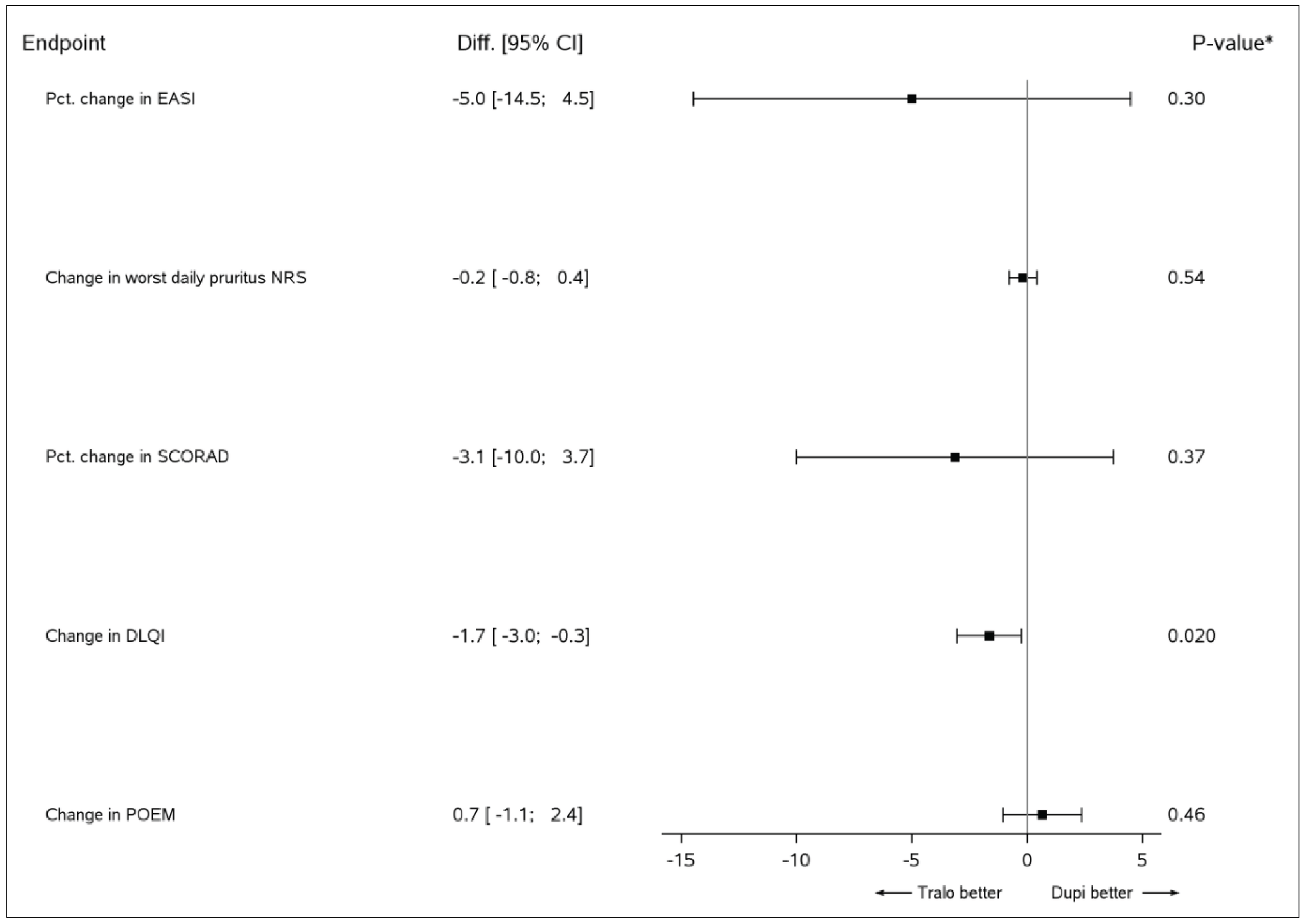
CI = confidence interval; DLQI = Dermatology Life Quality Index; Diff. = mean difference; Dupi = dupilumab; EASI = Eczema Area and Severity Index; NRS = numeric rating scale; MAIC = matching adjusted indirect comparison; POEM = patient-oriented eczema measure; SCORAD = Scoring Atopic Dermatitis; Tralo = tralokinumab.
Source: ECZTRA 3 and LIBERTY AD CHRONOS MAIC technical document.21
Harms Outcomes
ECZTRA 7 Versus LIBERTY AD CAFÉ
||| |||| |||||| |||||||| ||||||| || |||| |||| |||||||| ||| ||||||||| |||||| ||||||| ||||||| || ||| ||||||| ||||||| || |||| |||||||| |||||| |||| |||||||| ||||||| |||||||||||| |||| ||||||| ||||||||||||||| ||| ||||||||| |||| ||||||| |||||||||||||| |||||||||||| |||| ||| ||||| |||||||| ||| |||||||||||||| ||| || ||||||| ||||||||| |||||||| |||||||||||| |||| ||||||| ||||||||||||||| |||| ||||||||| |||| ||||||| |||||||||||||||| ||| |||||| ||| ||||||||| || |||||||||||| |||||||||| ||||||| ||| |||||||||| |||| |||||| ||| ||| ||||| || ||||| ||| ||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||| ||||||||| |||||| |||||||| |||||| ||| ||||||||||||| |||||||||| |||||| ||||||||| |||| |||||||| ||| |||| ||||||| ||||||| |||||||||||| |||| ||||||| ||||||||||||||| ||| ||||||||| |||| ||||||| ||||||||||||||||
ECZTRA 3 Versus LIBERTY AD CHRONOS
No harms end points were evaluated in the ECZTRA 3 versus LIBERTY AD CHRONOS MAIC.
Figure 36: Main Safety MAIC Analysis of ECZTRA 7 Versus LIBERTY AD CAFÉ

Figure was redacted at the sponsor’s request.
Critical Appraisal of Sponsor-Submitted Matching Adjusted Indirect Comparisons
ECZTRA 7 Versus LIBERTY AD CAFÉ
The comparison of ECZTRA 7 versus LIBERTY AD CAFÉ was chosen after a review of | trials evaluating the treatment of tralokinumab or dupilumab in patients with moderate-to-severe AD. There were no description of a literature search or selection criteria, or any indication of how the | trials were located. Based on input from the clinical experts consulted by CADTH for the purpose of this review, and the search results from the ICER NMA, the likelihood of that relevant trials were excluded from consideration is low. There was also a lack of transparency in the data-extraction process, and no quality assessment of the trials was presented.
The sponsor noted that the choice to conduct a MAIC was based on substantial clinical and methodological heterogeneity that precluded the conduct of a standard indirect comparisons (e.g., NMA or Bucher comparison). The choice of selecting ECZTRA 7 and LIBERTY AD CAFÉ for inclusion in the MAIC was based on the anticipation that tralokinumab and dupilumab would likely be used in combination with TCS (as in these trials). The choice of an anchored MAIC was appropriate because the trials included a common comparator — placebo plus TCS. Anchoring the MAIC by the placebo arm of each trial may mitigate potential biases associated with a MAIC, providing the transitivity assumption holds and the placebo populations are relatively exchangeable.
Unbiased estimates from an anchored MAIC require that all potential effect modifiers be balanced across the groups being compared. How the matching variables were selected for the MAIC was not described. It is unknown whether the selection of the matching variables was based on a statistical model, clinical expertise, or some combination of the 2. Characteristics postmatching were well balanced, with almost perfect matching of the covariates included in the MAIC. The clinical experts consulted by CADTH for this review noted that prior use of other agents, such as steroidal immunosuppressive, UV B exposure, and phototherapy, as a matching variable should have been explored. However, the complete baseline demographic and disease characteristics for patients in both trials were not reported after matching; only the balance of patient characteristics relevant to the covariates used in the matching were reported. It is therefore unclear what effect the matching had on the balance of other relevant patient characteristics. There was no assessment of residual confounding in the analysis. The application of weights resulted in a reduced ESS of ||||, in which ||||| of enrolled patients in the ECZTRA 7 study were lost. The reduction of sample size in the primary analysis contributes to imprecision (the CIs for most comparisons included the null) and indicates that the effect estimates are driven by a subset of less than half the original population. However, sensitivity analyses using a larger population by way of an unadjusted indirect comparisons were generally consistent with the primary MAIC, but with narrower CIs favouring dupilumab.
ECZTRA 3 Versus CHRONOS
The ECZTRA 3 versus CHRONOS MAIC lacked description of a literature search or selection criteria. The sponsor indicated that these trials were selected for the MAIC as both were reflective of real-world practice (i.e., used in combination with TCS) and had data at time points that were aligned with the initiation criteria for dupilumab in most jurisdictions. There was also a lack of transparency in the data-extraction process, and no quality assessment of the trials was presented.
Although both the ECZTRA 3 and LIBERTY AD CHRONOS studies included a placebo, an unanchored MAIC was conducted. The choice of an unanchored MAIC was appropriate due to differences in trial design (re-randomized versus treat-through) that may have resulted in differences in treatment of placebo across the 2 studies. In this scenario, an anchored MAIC would have introduced bias.
Unbiased estimates from an unanchored MAIC require that all potential effect modifiers and prognostic variables be balanced across the groups being compared. How the matching variables were selected for the MAIC was not described. It is unknown whether the selection of the matching variables was based on a statistical model, clinical expertise, or some combination of the 2. The clinical experts consulted by CADTH for this review noted that prior use of other agents, such as steroidal immunosuppressive, UV B exposure, and phototherapy, as a matching variable should have been explored. Nonetheless, the ECZTRA 3 versus LIBERTY AD CHRONOS MAIC was limited by heterogeneity in populations between the 2 studies. First, the results reported at week 32 and week 52 did not use the same dupilumab target population in the LIBERTY AD CHRONOS study. Of the 106 dupilumab patients at baseline, 80% to 84% were included in the subset of patients analyzed at week 52. Consequently, the matched tralokinumab population may not be completely representative of the dupilumab population results reported at week 52. Next, the time points at which tralokinumab (week 32) and dupilumab (week 52) were compared were different. Based on input from the clinical expert, better results are expected for tralokinumab at week 52 versus week 32, and the analysis may therefore be at risk of a bias that favours treatment with dupilumab. In the ECZTRA 3 and LIBERTY AD CHRONOS studies, the minimum times period for an adequate response to topical treatment were 1 year and 6 months, respectively. Based on clinical expert input, this difference may result in different reporting of the number of patients with inadequate response. There was no assessment of residual confounding in the analysis. Also, the timing and potency of TCS differed between the trials. Based on input from the clinical expert, the timing and use of different potency of TCS would not influence the patient population; however, they could influence the clinical severity of AD symptoms. Finally, the crossover to a lower dose of tralokinumab every 4 weeks after 16 weeks in the ECZTRA 3 may have underestimated the efficacy of tralokinumab compared to dupilumab in the MAIC based on input from the clinical expert.
Unadjusted and match-adjusted baseline covariates were reported for age, sex, body mass index, disease duration, and race, as well as EASI, IGA, DLQI, and SCOARD scores at baseline. Baseline characteristics postmatching were well balanced, with almost perfect matching of the covariates included in the MAIC. However, the complete baseline demographic and disease characteristics for patients in both trials were not reported after matching; only the balance of patient characteristics relevant to the covariates used in the matching were reported. The effect the matching on the balance of other relevant patient characteristics is therefore unclear. The application of weights resulted in a reduced ESS of 123.4, in which 50.64% of enrolled patients in the ECZTRA 3 study were lost. The reduction in the sample size in the primary analysis contributes to imprecision (the CIs for most comparisons included the null) and indicates that the effect estimates are driven by a subset of half the original population. Sensitivity analyses to verify any of the assumptions of the analysis were not provided.
Studies Addressing Gaps in the Pivotal and RCT Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Description of Studies
Patients with resistant or severe AD who have had uncontrolled disease with dupilumab or JAKi treatment were not included in the pivotal studies. Also, the efficacy and safety of tralokinumab in real-world settings cannot be studied in clinical trials. To address these gaps in evidence, 2 observational studies (Pezzolo and Naldi23 and Pereyra-Rodriguez et al.24) submitted by the sponsor are summarized in this report. There was no description of the search or selection methods used to identify these studies.
Real-World Evidence of Tralokinumab in the Treatment of Resistant AD: An Open-Label, Retrospective Case Series Study
Study Design and Objectives
A retrospective case series study by Pezzolo and Naldi reported as a letter to the editor was conducted in San Bortolo Hospital, Vicenza, Italy.23 Given that there are limited data available for patients who had an inadequate response to previous systemic AD therapies, this case series examined the effect of tralokinumab treatment in patients for whom first-line biologic dupilumab was not successful (i.e., no response after 16 weeks). Tralokinumab was administered to all patients (N = 12) over 12 weeks. Response data were collected at weeks 4, 8, and 12.
Populations
A total of 12 adult patients (aged ≥ 18 years) eligible for treatment with tralokinumab according to the approved indications were included in the analysis. Patients accessed tralokinumab through a compassionate-use program in an open-label, retrospective study after the failure of the first-line approved biologic treatment (dupilumab).
Interventions
Tralokinumab was administered to all 12 patients for 12 weeks according to the labelled loading dose of 600 mg followed by 300 mg every 2 weeks. No comparator was included in this study. Concomitant treatments or potential confounders were not reported.
Outcomes
Outcomes were neither clearly prespecified nor defined. It is not clear how they were retrospectively collected. Outcomes reported in the study included:
EASI-75
change from baseline in EASI score
change from baseline in itch NRS
change from baseline in sleep NRS
adverse effects.
Statistical Analysis
No statistical analysis was performed. All data were presented with descriptive statistics.
Results
Baseline Characteristics
The baseline characteristics of the participants in the study are summarized in Table 70.
Table 52: Baseline Characteristics (All-Patients Population)
Characteristics | Case series N = 12 |
|---|---|
Male, n (%) | 6 (50%) |
Mean age, years (range) | 42.58 (19 to 82) |
Mean BMI, kg/m2 (range) | 24.49 (18.57 to 35.59) |
EASI at baseline before any systemic therapy, mean (range) | 36.58 (21 to 47) |
Co-presentation, n (%) | |
Asthma and/or rhino-conjunctivitis | 6 (50%) |
Ulcerative colitis | 1 (8.3%) |
BMI = body mass index; EASI = Eczema Area and Severity Index.
Sources: Pezzolo and Naldi23 and the sponsor’s Summary of Clinical Evidence.14
Efficacy
EASI Score
All patients on tralokinumab reached EASI-75 within 8 weeks, with continued decreases in EASI scores until week 12. The mean EASI scores were 27.58 (range = 20 to 35) at baseline and 4.67 (range = 0 to 13) at week 12.23
Itch Numeric Rating Scale
The itch NRS scores were 8.42 (range = 7 to 10) at baseline, 3.67 (range = 0 to 7) at week 4, and 2.92 (range = 0 to 5) at week 12.23
Sleep Numeric Rating Scale
The sleep NRS scores were 7.00 (range = 3 to 10) at baseline and 1.92 (range = 0 to 5) at week 12.23
Harms
No serious adverse effects were reported. The conjunctivitis that had been observed in 4 patients during the previous treatment with dupilumab did not recur.23
Real-World Evidence of Tralokinumab in the Treatment of Severe AD: Short-Term, Retrospective Effectiveness, and Safety Results
Study Design and Objectives
A retrospective cohort study by Pereyra-Rodriguez et al. was conducted in adult patients with moderate-to-severe AD who initiated tralokinumab treatment between April 1 and June 30, 2022, in 16 hospitals in Spain.24 Data collected included age, duration of the disease, medical history (comorbidities), and previous systemic and biologic and/or JAKi treatments. Patients who had never been exposed to a biologic or JAKi were defined as naive to advanced therapy.
Populations
All patients who had received a confirmed diagnosis of AD from an experienced dermatologist, with an EASI score of at least 21 and an inadequate response or intolerance to cyclosporine A were included (n = 85). Patients who received concomitant systemic treatment for AD were excluded.
Interventions
Tralokinumab was prescribed according to the Spanish Medicines Agency access protocol. Information about patients who had received approved doses (a 600 mg loading dose at week 0 followed by 300 mg every 2 weeks) for 16 weeks in the past was collected. No washout period was required for previous medications as per data collection criteria. The use of TCS was allowed during the 16-week treatment period.
Outcomes
Disease severity was measured by SCORAD, EASI, BSA, IGA, and PP-NRS at the baseline visit and at weeks 4 and 16 of follow-up. HRQoL was assessed using the DLQI and AEs.
Statistical Analysis
Descriptive statistics were calculated for each demographic and clinical variable using frequencies and percentages for categoric variables and mean ± SD for continuous variables. A D’Agostino-Pearson normality test of the quantitative variables was performed. When the distribution was non-normal, the differences observed on the different scales were compared with a Wilcoxon test. A P value of less than 0.05 was considered statistically significant. To compare the differences in the scales between naive and non-naive patients in the different weeks, Dunn's test was used. There was no control for multiplicity.
Results
Disposition
Of 85 patients included, 1 discontinued treatment due to severe conjunctivitis, despite the treatment prescribed by an ophthalmologist. In addition, a female patient suspended treatment at week 15 due to a desire for pregnancy, which occurred 11 weeks after the last administration.
Baseline Characteristics
The baseline characteristics of the participants at the baseline visit of the study are summarized in Table 71.
A total of 85 patients (43 males [50.6%]) were included in the study. The mean age was 39.0 years (SD = 16.1). The mean duration of the disease was 16.4 years (SD = 12.2), based on almost 72% of patients experiencing onset before the age of 18 (early-onset AD), compared to 28% experiencing onset in adulthood. All included patients had severe disease, with the following mean baseline values: SCORAD, 55.8 ± 13.3; EASI, 25.4 ± 8.1; DLQI, 15.8 ± 5.4; and PP-NRS, 8.1 ± 1.8. Two-thirds (65.3%) of the patients had an IGA of 4.
Patients had a history of multiple previous treatment failures: 77.2% had received cyclosporine A. Also, 58 patients (68.2%) were naive to advanced therapy (biologic or JAKi drugs), while 15 (17.6%) had used 1 drug, 11 (12.9%) used 2 drugs, and 1 had used 3. About one-third (29.4%) of patients had used dupilumab; 18 (72.0%) had discontinued it due to ineffectiveness, and 7 (28.0%) due to adverse effects (6 conjunctivitis and 1 acute toxic hepatitis). Furthermore, 7 patients (8.2%) had previously used upadacitinib (6 patients discontinued due to ineffectiveness and 1 due to recurrent ocular herpes), and 6 (7.1%) previously had uncontrolled disase with baricitinib.
Table 53: Demographic and Clinical Characteristics at Baseline (All-Patients Population)
Characteristics | Retrospective study, N = 85 |
|---|---|
Age (years), mean (SD) | 39.0 (16.1) |
Sex (male), n (%) | 43 (50.6) |
Disease duration (years), mean (SD) | 16.4 (12.2) |
Body mass index (kg/m2), mean (SD) | 24.6 (4.6) |
AD pattern, n (%) | |
Early onset (< 18 years) | 61 (71.8) |
Adult onset (≥ 18 years) | 24 (28.2) |
Comorbidities, n (%) | |
Obesity | 13 (15.3) |
Ischemic heart disease | 4 (4.7) |
Cancer in the past 5 years | 2 (2.4) |
Arterial hypertension | 8 (9.4) |
Dyslipidemia | 11 (12.9) |
Nasal polyps | 0 |
Conjunctivitis | 26 (30.6) |
Extrinsic asthma | 31 (36.5) |
Allergic rhinitis | 37 (43.5) |
Alopecia areata | 10 (11.8) |
Eosinophilic esophagitis | 1 (1.2) |
Food allergy | 8 (9.4) |
Previous treatments, n (%) | |
Systemic corticosteroids | 75 (88.2) |
Oral cyclosporine A | 66 (77.6) |
Phototherapy | 33 (38.8) |
Dupilumab | 25 (29.4) |
Baricitinib | 6 (7.1) |
Upadacitinib | 7 (8.2) |
Abrocitinib | 1 (1.2) |
Baseline severity scores | |
SCORAD, mean (SD) | 55.8 (13.3) |
EASI, mean (SD) | 25.4 (8.1) |
Pruritus NRS, mean (SD) | 8.1 (1.8) |
DLQI, mean (SD) | 15.8 (5.4) |
PGA score of 4, n (%) | 47 (55.3) |
AD = atopic dermatitis; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; NRS = Numeric Rating Scale; PGA = Patient Global Assessment; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation.
Source: Pereyra-Rodriguez, et al.24 and the sponsor’s Summary of Clinical Evidence.14
Efficacy
EASI Score
The mean EASI score was 25.4 (SD = 8.1) at baseline and 7.5 (SD = 6.9) at week 16 (P < 0.0001). In addition, 82.4% and 57.6% of the patients achieved EASI-50 and EASI-75, respectively, at week 16. Patients naive to advanced therapy appeared to have lower baseline scores and EASI response levels at weeks 4 and 16 compared to non-naive patients (24.6 versus 27.2 at baseline; 14.2 versus 18.9 at week 4; 6.3 versus 10.2 at week 16, but this was not tested statistically. The proportions of EASI-75 responders were 67.2% in the naive group and 40.7% in the non-naive group.
SCORAD
The mean SCORAD was 55.8 (SD = 13.3) and 20.0 (SD = 14.78) at week 16 (P < 0.0001).
PP-NRS
The mean PP-NRS was 8.1 (SD = 1.8) at baseline and 3.5 (SD = 2.4) at week 16 (P < 0.0001).
IGA 0 or 1
Forty-seven of the patients (55.3%) had an IGA of 4 at baseline; 18.8% (absolute number of patients not reported) showed an IGA of 0 or 1 at the end of the follow-up period.
Harms
The most frequent AEs were conjunctivitis and red face (5 cases, 5.9% each); 1 patient presented with both AEs at the same time. Two of the 5 patients with conjunctivitis had previously suffered conjunctivitis while receiving dupilumab. The other 3 patients, naive to advanced therapy, had never previously presented with eye problems. Three cases of worsening and generalized AD lesions (3.5%), 2 cases of reaction at the injection point (indurated red plaque of more than 24 hours) (2.4%), and 2 of anxiety-depressive syndrome (2.4%) were reported. Other reported adverse effects included arthralgia, corneal herpes, menstrual-related pain, cough, and syncopal episodes (1.2% each). One patient discontinued treatment due to severe conjunctivitis.
Critical Appraisal
Internal Validity
It is not clear how the studies addressing gaps were selected, creating a potential for study selection bias (i.e., relevant studies may have been left out). Pivotal trials and the LTE trial (ECZTEND) did not directly evaluate the safety and effectiveness of tralokinumab in patients previously treated with newer systemic therapies (including dupilumab, upadacitinib, and abrocitinib). These 2 observational studies provided additional information regarding this patient population and context regarding the use of tralokinumab in real-world clinical practice. However, there is a high level of uncertainty in the results given that the sample sizes were small (N = 12 for Pezzolo and Naldi and N = 85 for Pereyra-Rodriguez et al.). There is also a risk of selection bias as it is unclear how patients were selected for enrolment in both studies. Furthermore, these studies are noncomparative and included no adjustment for confounding, making it impossible to draw conclusions about benefits or harms attributable to tralokinumab versus any comparator. The study by Pezzolo and Naldi appears to lack a study protocol and the methods were not clearly described. As well, no formal hypothesis was tested and only descriptive findings were available. Pereyra-Rodriguez et al. appear to have established a study protocol, but it was not available for review by the CADTH review team; it is therefore unclear what part of the study was planned a priori. The protocol also did not control for multiple comparisons, resulting in an increased risk of false-positive results. In both studies, data were collected retrospectively, and it is unclear if they were collected systematically, which increases the likelihood of bias and error. Last, the duration of follow-up in both studies (12 weeks in Pezzolo and Naldi and 16 weeks in Pereyra-Rodriguez et al.) were inadequate for assessing response to tralokinumab treatment, according to the input of clinical experts consulted by CADTH. Taken together, a firm conclusion cannot be drawn from these studies.
External Validity
The Pezzolo and Naldi study was conducted in Italy, while the Pereyra-Rodriguez et al. study was conducted in Spain. The clinical experts consulted by CADTH did not expect any significant differences in the treatment approach in Italy and Spain compared with Canada. The difference in study location is, therefore, unlikely to affect the generalizability of study findings. Finally, because neither study included adolescent patients, the treatment effects in adolescents who had received prior treatment with dupilumab and/or a JAK inhibitor were not addressed.
Discussion
Summary of Available Evidence
This section summarizes the evidence regarding tralokinumab in the treatment of moderate-to-severe AD based on 5 phase III RCTs, 1 LTE study, 4 ITCs, and 2 observational studies.
Five pivotal phase III, double-blind, randomized, placebo-controlled trials met the inclusion criteria for the systematic review conducted by the sponsor. The ECZTRA 6 study (N = 301) was conducted with tralokinumab as monotherapy for 52 weeks in adolescents with moderate-to-severe AD who had uncontrolled disease with topical therapy.9 The ECZTRA 1 (N = 802), ECZTRA 2 (N = 794), and ECZTRA 3 (N = 380) studies were conducted using tralokinumab as monotherapy for 52 weeks (ECZTRA 1 and 2) or in combination with TCS for 32 weeks (ECZTRA 3), in adults with moderate-to-severe AD who had uncontrolled disease with topical therapy.10,11 The ECZTRA 7 study was conducted with tralokinumab in combination with TCS for 26 weeks in adults with severe AD who had uncontrolled disease with topical therapy and systemic cyclosporine A treatment.12 In the ECZTRA 6 study, prior systemic immunosuppressant treatment for AD was reported in 21.1% of patients. Prior systemic immunosuppressant treatment was more common in the ECZTRA 7 study than in the ECZTRA 1, 2, and 3 studies, with cyclosporine A being the most frequently used across studies (74.7% in the ECZTRA 7 study and 31.1% to 36.4% in the ECZTRA 1, 2, and 3 studies). Prior monoclonal antibody exposure was low in the pivotal trials that reported such information (between 2.4% and 7.6% in the ECZTRA 3, 6, and 7 studies). Evidence from these studies was supplemented with the results from 1 ongoing LTE study (ECZTEND) with 4 and 3 years of data in adults and adolescents, respectively.60-64
In the absence of head-to-head evidence comparing tralokinumab against other relevant advanced therapies used to manage AD, the sponsor submitted 4 ITCs of the treatment effect of tralokinumab and other treatments in patients with moderate-to-severe AD. Of the submitted ITCs, 2 were NMAs and 2 were MAICs.19-22 The ICER NMA evaluated the relative efficacy and safety of treatment with tralokinumab versus other therapies in adult patients with moderate-to-severe AD19 and was used to inform the sponsor-submitted economic model for the treatment effect of tralokinumab up to week 16. ||| ||| |||||| ||| ||||||||| ||| |||||||| |||||||| ||| |||||| || ||||||||| |||| |||||||||||| |||||| ||||| ||||||||| || |||||||||| |||||||| ||||||| ||| |||| || || ||| || ||||| |||| |||||||||||||||||| || || || |||| || || |. Both MAICs evaluated tralokinumab against dupilumab in adult patients with moderate-to-severe AD || ||| ||||| |||||||||| ||| ||||||||||| ||| || || |||||||table 55
Two retrospective observational studies designed to address the evidence gap in the use of tralokinumab in patients with prior exposure to a biologic and/or JAKi in real-world clinical practice were submitted by the sponsor. The studies included adults with moderate-to-severe AD. All 12 patients in the study by Pezzolo and Naldi previously had uncontrolled disease with dupilumab treatment.23 Pereyra-Rodriguez et al. included 85 patients who were naive to both biologic and JAKi treatments (68.2%) and those who previously received a biologic and/or JAKi (dupilumab, 29.4%; upadacitinib, 8.2%; abrocitinib, 1.2%).24
The included studies evaluated a range of outcomes that are important in the management of AD, including the severity and extent of AD (e.g., an IGA of 0 or 1, EASI-75), symptoms (e.g., worst daily pruritus NRS, eczema-related sleep NRS, and POEM), HRQoL (e.g., DLQI and CDLQI), patient-reported anxiety and depression (e.g., HADS), and use of topical therapy.
Interpretation of Results
Efficacy
Evidence from ECZTRA 6, a trial involving adolescents with moderate-to-severe AD who had uncontrolled disease with topical therapy, supported the superiority of tralokinumab over placebo with respect to an IGA of 0 or 1, EASI-75, and change from baseline in SCORAD at week 16, which addresses a key treatment outcome of severity and extent of AD noted by patients and clinicians. Similar results were observed in adults with moderate-to-severe AD and prior failure of topical therapy who received tralokinumab with TCS (in the ECZTRA 3 study) or without TCS (in the ECZTRA 1 and 2 studies). The between-group difference in change from baseline in SCORAD at week 16 met the literature-reported MID estimate of 8.7 in adolescent patients66 (the point estimate and the entire 95% CI for the between-group difference were greater than 8.7 in the ECZTRA 6 study) but did not consistently do so in adult patients. (The point estimate and the entire 95% CI were greater than 8.7 in the ECZTRA 2 study; the point estimate was greater than 8.7 but the 95% CI indicates that a difference falling below 8.7 in the ECZTRA 1 and 3 studies is also compatible with the data. This means that the data in the ECZTRA 1 study and 3 are most compatible with an effect exceeding the MID, but also includes the possibility that the true effect falls below the MID.) The clinical experts commented that, while the magnitude of benefit of the point estimates for tralokinumab versus placebo at week 16, in general, appeared to be modest, the duration of the follow-up was insufficient to adequately assess efficacy given that, in their clinical experience, tralokinumab typically achieves optimal treatment effects in approximately 6 months after initiation. EASI-50 was considered a more clinically relevant end point compared with EASI-75 given the early follow-up at week 16, according to the clinical experts and clinician groups. The results for EASI-50 at week 16 were generally in favour of tralokinumab; however, this outcome was not adjusted for multiplicity and presents an increased risk of type I error (false-positive results). No conclusion can be drawn on subgroup effects due to the lack of sample-size consideration and control for multiplicity.
Reduction of AD symptoms, an important goal in the treatment of AD, was assessed in these studies using patient-reported outcomes, including (adolescent) worst daily pruritus NRS, eczema-related sleep NRS, and POEM scales. According to the clinical experts, worst daily pruritus NRS is an instrument routinely used in clinical practice, while eczema-related sleep NRS and POEM are not, although they are frequently used in clinical trials. The studies used a responder analysis based on the proportion of patients who achieved an improvement from baseline in worst daily pruritus NRS of at least 4 points, which was considered clinically important in previous studies.67,68 Results showed a greater proportion of patients achieved a reduction of at least 4 points in (adolescent) worst daily pruritus NRS at week 16 with tralokinumab treatment compared to placebo in both adolescents (in the ECZTRA 6 study) and adults (in the ECZTRA 1, 2, and 3 studies). Results of change from baseline in worst daily pruritus NRS, eczema-related sleep NRS, and POEM in these trials also favoured tralokinumab compared with placebo; however, they are at risk of bias due to a large amount of missing data that were not appropriately accounted for (i.e., absence of additional estimands), and are at an increased risk of type I error without adjustment for multiplicity. There is also a potential risk of bias in measurements of these patient-reported outcomes, potentially resulting in inflated efficacy of tralokinumab.
The DLQI (or CDLQI), a dermatology-specific HRQoL instrument commonly applied in clinical practice, was used to assess the HRQoL of patients in the studies. The questionnaire captures the impact of AD on several important outcomes noted in the patient group input, including symptoms, psychological burden, ability to carry out daily activities, and ease of treatment. Results of the key secondary end point of change from baseline in DLQI (or CDLQI) at week 16 favoured tralokinumab over placebo in the adolescents included in the ECZTRA 6 study and the adults in the ECZTRA 1, 2, and 3 studies. An MID estimate for DLQI in adults with AD is not available. The MID was estimated to range between 2.2 and 6.9 in studies of various skin conditions,73,74 which could be reasonably applied to adults with AD in consultation with the clinical experts. The between-group difference in change from baseline in DLQI at week 16 did not meet the conservative MID estimate of 6.9 in the adult population in the ECZTRA 1, 2, and 3 studies (the point estimate and the entire 95% CI for the between-group difference were smaller than 6.9). Results in the adolescent population of the ECZTRA 6 study also did not meet the MID estimates of CDLQI (6 to 8) identified from the literature.76 Responder analysis based on a reduction of at least 4 points in DLQI (or 6 points in CDLQI) from baseline at week 16 produced results that favour tralokinumab; however, these findings are associated with an increased risk of type I error due to the lack of adjustment of multiplicity, which limits the usefulness of interpretation of results. Similarly, the effects of tralokinumab treatment on other important outcomes noted by the stakeholders, including anxiety and depression (as assessed by the HADS), amount of TCS used, and number of days without TCS use, were also inconclusive due to a large amount of missing data and the lack of adjustment for multiplicity. As with other patient-reported outcomes, it is possible that the efficacy of tralokinumab as assessed by DLQI (or CDLQI) and HADS was inflated due potential bias in measurement of the outcomes.
These results pertain to the ECZTRA 1, 2, 3, and 6 studies, which included a smaller proportion of patients with prior systemic immunosuppressants relative to the ECZTRA 7 study. Given that tralokinumab will likely be used following failure of topical therapy, phototherapy, and systemic immunosuppressants in clinical practice, the patient population of the ECZTRA 7 study (adults who have uncontrolled disease with or were deemed not to be candidates for topical therapy and cyclosporine A) was considered to be more generalizable to clinical practice compared with the other pivotal studies. The ECZTRA 7 trial met the primary end point of EASI-75 at week 16 but failed the first key secondary end point of the proportion of patients with a reduction of at least 4 points in worst daily pruritus NRS at week 16. Results of other key secondary end points at weeks 16 and 26 (based on an IGA of 0 or 1, EASI-75, SCORAD, and DLQI) suggested therapeutic benefits with tralokinumab over placebo, in combination with TCS; however, no conclusions on superiority can be drawn because no such statistical test was conducted due to prior failure in the testing hierarchy.
The longer-term efficacy of the tralokinumab every 2 weeks and every 4 weeks regimens beyond 16 weeks was assessed in the pivotal RCTs; however, the results are uncertain given the sizable reduction in sample sizes in the maintenance (or continuous) treatment period which resulted in wide CIs for the IGA of 0 or 1 and EASI-75 outcomes. As well, results for an IGA of 0 or 1 and EASI-75 were inconsistent between the identically designed ECZTRA 1 and 2 trials at week 52, which further increases the uncertainty of the results. Results of the LTE study of the pivotal trials (ECZTEND), suggest that the efficacy of tralokinumab was maintained in adults and adolescents for 4 and 3 years, respectively; however, these results were analyzed descriptively and were subject to uncertainty due to risks of bias in measurement of the outcome and confounding due to the open-label, noncomparative trial design, similar to other LTE studies.
All included pivotal studies were placebo-controlled and no direct comparative evidence between tralokinumab and relevant comparators, including dupilumab, upadacitinib, and abrocitinib, was identified. To address this evidence gap, the sponsor submitted 4 ITCs of tralokinumab and other advanced treatments for moderate-to-severe AD. It is not clear whether any systematic selection procedure was used to identify these 4 ITCs. Overall, the submitted indirect comparative evidence from an NMA and 2 MAICs suggested that tralokinumab performs worse than or similar to dupilumab in adults with moderate-to-severe AD. The lone exception to this trend was noted in the ECZTRA 3 versus LIBERTY AD CHRONOS unanchored MAIC, in which results favoured tralokinumab versus dupilumab for an IGA of 0.1 and change in DLQI at week 52. Moreover, the indirect comparative efficacy of tralokinumab versus abrocitinib and upadacitinib was considered uncertain based on NMA evidence. Based on clinical expert input, the 12- and 16-week assessment time points used in the NMAs may have been too early to measure the comparative efficacy of tralokinumab. The CADTH review team cannot make conclusions with any certainty about the efficacy of tralokinumab compared with other treatments for AD in the adolescent patient population because results of the submitted NMA were imprecise, although the effect estimates for the comparisons to dupilumab appear to be directionally aligned with findings in the adult population. The combined ITC evidence is also associated with important uncertainty due to the potential for intransitivity in the NMAs, potential residual confounding in the MAICs, and, for all but the ICER NMA in adults, small sample size (or effective ESS), rendering most estimates too imprecise for definitive conclusions.
Although the results of the sponsor-submitted retrospective observational studies were suggestive of a benefit from tralokinumab in adults who were either naive to or had previous exposure to and failed JAKi and/or dupilumab treatment, the results of these studies were associated with a high level of uncertainty due to several limitations, including the potential for selection bias; a lack of a comparator group without adjustment for confounding; the open-label design, which may introduce performance and detection biases; retrospective collection of data, which may be subject to bias and error; and small sample sizes (n = 12 and n = 85).
Harms
The safety profile of tralokinumab in adolescent patients was generally consistent with that in adult patients in the pivotal RCTs. There was no notable difference in the frequency of TEAEs between tralokinumab and placebo. The most frequently reported TEAEs associated with tralokinumab were URTIs (including viral causes), which the clinical experts noted to be uncommon AEs of tralokinumab in their clinician experience. Conjunctivitis was consistently more frequently reported in the tralokinumab group compared with the placebo group in the initial treatment period across studies in adults, although it was not serious in most patients who experienced it and did not lead to treatment discontinuation. The clinical experts noted no concerns with the safety profile of tralokinumab overall. No new safety signal was reported in the LTE trial (ECZTEND), as well as the 2 observational studies, including patients with prior JAKi or dupilumab treatment. Of the submitted indirect evidence, 2 MAICs provided harms data versus dupilumab in adults, but the findings were inconclusive due to imprecision. There was no evidence regarding safety versus other comparators in adults. Results of the NMA comparing the harms of tralokinumab with those of relevant comparators in adolescents were uncertain due to imprecision.
Conclusion
Evidence from 3 pivotal double-blind RCTs demonstrated that 16 weeks of treatment with tralokinumab resulted in improvements in the severity and extent of AD (as measured by EASI-75, an IGA of 0 or 1, and SCORAD), the severity of itching (worst daily pruritus NRS), and HRQoL (DLQI) in adults with moderate-to-severe AD who had an inadequate response to topical AD therapy, compared to placebo, when used as monotherapy (in the ECZTRA 1 and 2 studies) and in combination with TCS (in the ECZTRA 3 study); however, either the magnitude of improvement in SCORAD and DLQI scores did not meet the literature-reported MID estimates or the 95% CI included the potential of a difference falling below conservative MID estimates. Analyses of other clinically important outcomes, including sleep disturbance and symptoms of anxiety and depression, also favoured tralokinumab; although this was due to a large amount of missing data and a lack of adjustment for multiplicity, and these results are likely to be biased. Similar results were observed in 1 pivotal double-blind RCT (ECZTRA 6) conducted with tralokinumab monotherapy in adolescents who had an inadequate response to topical AD therapy; MID estimates were reached for the improvement in SCORAD score but not for the improvement in CDLQI scores at week 16. The anticipated place of therapy of tralokinumab is in patients with moderate-to-severe AD and an inadequate response to topical AD therapy, phototherapy, and systemic immunosuppressants. One double-blind RCT (ECZTRA 7) in adults with severe AD who had an inadequate response to topical AD therapy and cyclosporine A provided supportive evidence for the use of tralokinumab in combination with TCS in such patients. The ECZTRA 7 trial showed that, compared to placebo, 16 weeks of tralokinumab was associated with a higher proportion of patients with an EASI-75 but did not suggest a difference in reducing the severity of itching (worst daily pruritus NRS). Results of other outcomes were inconclusive due to a prior failure in the statistical testing hierarchy. Overall, interpretation of the clinical meaningfulness of findings at week 16 from the RCTs was hindered by the insufficient duration of follow-up, given that an optimal response to tralokinumab treatment is usually observed at 6 months in clinical practice, according to clinical expert input. No conclusion can be drawn about the efficacy of tralokinumab beyond week 16 based on the submitted evidence due to important limitations of the included studies, including inconsistent results between trials and evidence of imprecision in the longer-term results in RCTs; and risks of selection bias and confounding due to the noncomparative trial design of the LTE study (ECZTEND), similar to other LTE studies.
Evidence from 4 ITCs comparing tralokinumab to other advanced therapies for the treatment of moderate-to-severe AD suggest that tralokinumab performs worse than or similar to its main comparator, dupilumab, in adults. The lone exception to this trend was noted in the ECZTRA 3 versus LIBERTY AD CHRONOS unanchored MAIC, in which results favoured tralokinumab versus dupilumab for an IGA of 0 or 1 and change in DLQI at week 52. Comparisons about the efficacy and safety of tralokinumab versus abrocitinib and upadacitinib are considered uncertain. The combined ITC evidence for adults is associated with important uncertainty due to the potential for intransitivity in the NMA, and potential residual confounding and lack of precision in the MAICs. Results of the lone NMA evaluating the efficacy of tralokinumab in adolescents were imprecise and potentially affected by intransitivity, which precludes forming any conclusions of certainty about the comparative efficacy of tralokinumab versus dupilumab, abrocitinib, and upadacitinib. Evidence for the use of tralokinumab in patients who had uncontrolled disease with prior dupilumab and/or a JAKi based on 2 observational studies was inconclusive given the small sample sizes and the open-label, noncomparative study designs.
Tralokinumab appeared in the RCTs to be well tolerated in adults and adolescents and remained so beyond 52 weeks based on evidence from the ECZTEND trial. Based on the indirect evidence, no conclusion about the comparative safety of tralokinumab to other advanced therapies for moderate-to-advance AD can be drawn with any certainty, due to imprecision in all relevant analyses (2 MAICs in adults and 1 NMA in adolescents).
References
1.Avena-Woods C. Overview of atopic dermatitis. Am J Manag Care. 2017;23(8 Suppl):S115-S123. PubMed
2.Odhiambo JA, Williams HC, Clayton TO, Robertson CF, Asher MI, Group IPTS. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124(6):1251-1258 e1223. PubMed
3.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: Results from an international survey. Allergy. 2018;73(6):1284-1293. PubMed
4.Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387(10023):1109-1122. PubMed
5.Reich K, DeLozier AM, Nunes FP, et al. Baricitinib improves symptoms in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: patient-reported outcomes from two randomized monotherapy phase III trials. J Dermatolog Treat. 2022;33(3):1521-1530. PubMed
6.Rinvoq (upadacitinib extended-release tablets): extended-release tablets, 15 mg upadacitinib, oral; extended-release tablets, 30 mg upadacitinib, oral [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022 Aug 2: https://pdf.hres.ca/dpd_pm/00067922.PDF. Accessed 2023 Nov 3.
7.Cibinqo (abrocitinib): Tablets, 50 mg, 100 mg, 200 mg, oral [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2022.
8.CADTH Drug Reimbursement Expert Review Committee final recommendation: tralokinumab (Adtralza - LEO Pharma). Can J Health Technol. 2022;2(3). https://www.cadth.ca/sites/default/files/DRR/2022/SR0689%20Adtralza%20-%20Confidential%20Final%20CADTH%20Rec-Final.pdf. Accessed 2023 Jun 22.
9.Paller AS, Flohr C, Cork M, et al. Efficacy and Safety of Tralokinumab in Adolescents With Moderate to Severe Atopic Dermatitis: The Phase 3 ECZTRA 6 Randomized Clinical Trial. JAMA Dermatol. 2023;159(6):596-605. PubMed
10.Wollenberg A, Blauvelt A, Guttman-Yassky E, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. 2021;184(3):437-449. PubMed
11.Silverberg JI, Toth D, Bieber T, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol. 2021;184(3):450-463. PubMed
12.Gutermuth J, Pink AE, Worm M, Soldbro L, Bjerregard Oland C, Weidinger S. Tralokinumab plus topical corticosteroids in adults with severe atopic dermatitis and inadequate response to or intolerance of ciclosporin A: a placebo-controlled, randomized, phase III clinical trial (ECZTRA 7). Br J Dermatol. 2022;186(3):440-452. PubMed
13.Clinical Study Report: LP0162-1334. Tralokinumab monotherapy for adolescent subjects with moderate-to-severe atopic dermatitis ECZTRA 6 Version 1.0 Final [internal sponsor's report]. Ballerup (DK): LEO Pharma Inc; 2021 Aug 9.
14.CADTH Sponsor's Summary of Clinical Evidence. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5. Toronto (ON): LEO Pharma Inc.; 2023.
15.Clinical Study Report: LP0162-1325. Tralokinumab monotherapy for moderate-to-severe atopic dermatitis ECZTRA 1 Version 1.0 Final [internal sponsor's report]. Ballerup (DK): LEO Pharma Inc; 2020 Feb 11.
16.Clinical Study Report: LP0162-1326. Tralokinumab monotherapy for moderate-to-severe atopic dermatitis ECZTRA 2 Version 1.0 Final [internal sponsor's report]. Ballerup (DK): LEO Pharma Inc; 2020 Feb 20.
17.Clinical Study Report: LP0162-1339. Tralokinumab in combination with topical corticosteroids for moderate-to-severe atopic dermatitis ECZTRA 3 Version 1.0 Final [internal sponsor's report]. Ballerup (DK): LEO Pharma Inc; 2020 Feb 18.
18.Clinical Study Report: LP0162-1346. Tralokinumab in combination with topical corticosteroids in subjects with severe atopic dermatitis who are not adequately controlled with or have contraindications to oral cyclosporine A ECZTRA 7 Version 1.0 Final [internal sponsor's report]. Ballerup (DK): LEO Pharma Inc; 2021 Mar 4.
19.JAK Inhibitors and monoclonal antibodies for teh treatment of atopic dermatitis: Effectiveness and Value. Institute for Clinical and Economic Review. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5. 2021.
20.Network meta-analyis of tralokinumab in atopic dermatitis for adolescent patients [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5.
21.Matching-adjusted indirect comparison to assess long-term efficacy outcomes for tralokinumab (ECZTRA 3) versus dupilumab (LIBERTY AD CHRONOS) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5.
22.Matching-adjusted indirect comparison of tralokinumab versus dupilumab based on the ECZTRA 7 study and LIBERTY AD CAFE data for atopic dermatitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5.
23.Pezzolo E, Naldi L. Tralokinumab in the treatment of resistant atopic dermatitis: An open-label, retrospective case series study. J Eur Acad Dermatol Venereol. 2023. PubMed
24.Pereyra-Rodriguez J-J, Herranz P, Ruiz-Villaverde R, et al. Treatment of severe atopic dermatitis with Tralokinumab in real clinical practice. Short-term effectiveness and safety results. Clin Exp Dermatol. 2023. PubMed
25.Bos JD, Brenninkmeijer EE, Schram ME, Middelkamp-Hup MA, Spuls PI, Smitt JH. Atopic eczema or atopiform dermatitis. Exp Dermatol. 2010;19(4):325-331. PubMed
26.Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. 2019;143(1):1-11. PubMed
27.Guttman-Yassky E, Waldman A, Ahluwalia J, Ong PY, Eichenfield LF. Atopic dermatitis: pathogenesis. Semin Cutan Med Surg. 2017;36(3):100-103. PubMed
28.Bieber T, D'Erme AM, Akdis CA, et al. Clinical phenotypes and endophenotypes of atopic dermatitis: Where are we, and where should we go? J Allergy Clin Immunol. 2017;139(4S):S58-S64. PubMed
29.Furue K, Ito T, Tsuji G, et al. The IL-13-OVOL1-FLG axis in atopic dermatitis. Immunology. 2019;158(4):281-286. PubMed
30.Koppes SA, Brans R, Ljubojevic Hadzavdic S, Frings-Dresen MH, Rustemeyer T, Kezic S. Stratum Corneum Tape Stripping: Monitoring of Inflammatory Mediators in Atopic Dermatitis Patients Using Topical Therapy. Int Arch Allergy Immunol. 2016;170(3):187-193. PubMed
31.Szegedi K, Lutter R, Res PC, et al. Cytokine profiles in interstitial fluid from chronic atopic dermatitis skin. J Eur Acad Dermatol Venereol. 2015;29(11):2136-2144. PubMed
32.Tazawa T, Sugiura H, Sugiura Y, Uehara M. Relative importance of IL-4 and IL-13 in lesional skin of atopic dermatitis. Arch Dermatol Res. 2004;295(11):459-464. PubMed
33.Tsoi LC, Rodriguez E, Degenhardt F, et al. Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J Invest Dermatol. 2019;139(7):1480-1489. PubMed
34.Silvestre Salvador JF, Romero-Perez D, Encabo-Duran B. Atopic Dermatitis in Adults: A Diagnostic Challenge. J Investig Allergol Clin Immunol. 2017;27(2):78-88. PubMed
35.Silverberg JI, Barbarot S, Gadkari A, et al. Atopic dermatitis in the pediatric population: A cross-sectional, international epidemiologic study. Ann Allergy Asthma Immunol. 2021;126(4):417-428 e412. PubMed
36.Silverberg JI, Simpson EL. Association between severe eczema in children and multiple comorbid conditions and increased healthcare utilization. Pediatr Allergy Immunol. 2013;24(5):476-486. PubMed
37.Eczema Society of Canada. Atopic Dermatitis Quality of Life Report - Moderate-to-Severe Disease - 2016/2017 Survey Results. 2017; https://eczemahelp.ca/wp-content/uploads/2019/02/ESC_Quality-of-Life-Report_Nov-2017−1.pdf. Accessed 2022 Oct 21.
38.Simpson EL, Bieber T, Eckert L, et al. Patient burden of moderate to severe atopic dermatitis (AD): Insights from a phase 2b clinical trial of dupilumab in adults. J Am Acad Dermatol. 2016;74(3):491-498. PubMed
39.Stingeni L, Belloni Fortina A, Baiardini I, Hansel K, Moretti D, Cipriani F. Atopic Dermatitis and Patient Perspectives: Insights of Bullying at School and Career Discrimination at Work. J Asthma Allergy. 2021;14:919-928. PubMed
40.Jeon C, Yan D, Nakamura M, et al. Frequency and Management of Sleep Disturbance in Adults with Atopic Dermatitis: A Systematic Review. Dermatol Ther (Heidelb). 2017;7(3):349-364. PubMed
41.Medic G, Wille M, Hemels ME. Short- and long-term health consequences of sleep disruption. Nat Sci Sleep. 2017;9:151-161. PubMed
42.Sibbald C, Drucker AM. Patient Burden of Atopic Dermatitis. Dermatol Clin. 2017;35(3):303-316. PubMed
43.Silverberg JI. Associations between atopic dermatitis and other disorders. F1000Res. 2018;7:303. PubMed
44.Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338-351. PubMed
45.Brunner PM, Silverberg JI, Guttman-Yassky E, et al. Increasing Comorbidities Suggest that Atopic Dermatitis Is a Systemic Disorder. J Invest Dermatol. 2017;137(1):18-25. PubMed
46.Sandhu JK, Salame N, Ehsani-Chimeh N, Armstrong AW. Economic burden of cutaneous infections in children and adults with atopic dermatitis. Pediatr Dermatol. 2019;36(3):303-310. PubMed
47.British Columbia Adult AD Population Funnel [CONFIDENTIAL data on file]. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5.
48.Wong ITY, Tsuyuki RT, Cresswell-Melville A, Doiron P, Drucker AM. Guidelines for the management of atopic dermatitis (eczema) for pharmacists. Can Pharm J (Ott). 2017;150(5):285-297. PubMed
49.Common Drug Review Clinical Review Report: dupilumab (Dupixent - Sanofi Genzyme). Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/clinical/sr0636-dupixent-clinical-review-report.pdf. Accessed 2020 Dec 15.
50.Canadian Skin Patient Alliance. COVID-19 Frequently Asked Questions. 2020; https://canadianskin.ca/education/covid-19#i-am-receiving-phototherapy-for-my-skin-disease-are-dermatologists-continuing-to-offer-these-treatments-in-their-clinics. Accessed 2021 Apr 1.
51.Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? Recommendations from an expert panel of the International Eczema Council. J Am Acad Dermatol. 2017;77(4):623-633. PubMed
52.Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327-349. PubMed
53.Wollenberg A, Barbarot S, Bieber T, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part II. J Eur Acad Dermatol Venereol. 2018;32(6):850-878. PubMed
54.Boguniewicz M, Fonacier L, Guttman-Yassky E, Ong PY, Silverberg J, Farrar JR. Atopic dermatitis yardstick: Practical recommendations for an evolving therapeutic landscape. Ann Allergy Asthma Immunol. 2018;120(1):10-22 e12. PubMed
55.Gooderham M, Lynde CW, Papp K, et al. Review of Systemic Treatment Options for Adult Atopic Dermatitis. J Cutan Med Surg. 2017;21(1):31-39. PubMed
56.Duxipen (dupilumab injection): 300 mg single-use syringe (300 mg/2 mL); 300 mg single-use pen (300 mg/2 mL); 200 mg single-use syringe (200 mg/1.14 mL); 200 mg single-use pen (200 mg/1.14 mL); 100 mg single-use syringe (100 mg/0.67 mL) [product monograph]. Vol 2023. Mississauga (ON): Sanofi-aventis Canada Inc.; 2023 May 1: https://pdf.hres.ca/dpd_pm/00070465.PDF. Accessed 2023 Jun 21.
57.Wollenberg A, Beck LA, de Bruin Weller M, et al. Conjunctivitis in adult patients with moderate-to-severe atopic dermatitis: results from five tralokinumab clinical trials. Br J Dermatol. 2022;186(3):453-465. PubMed
58.Adtralza (tralokinumab): 150 mg/mL single-use pre-filled syringe and 300 mg/2mL single-use pre-filled pen, solution for subcutaneous injection [product monograph]. Toronto (ON): LEO Pharma Inc.; 2023 Feb 3.
59.CADTH Reimbursement Recommendation: dupilumab (Dupixent - Sanofi-Aventis Canada Inc). Can J Health Technol. 2023;3(2). https://www.cadth.ca/sites/default/files/DRR/2023/SF0754REC-Dupixent-RfA.pdf. Accessed 2023 Apr 6.
60.Simpson E, Paller A, Wollenberg A, et al. Long-term safety and efficacy of tralokinumab in adolescents with moderate-to-severe atopic dermatitis: an interim analysis of ECZTEND. American Academy of Dermatology 2023; March 17-21, 2023; New Orleans (LA).
61.Blauvelt A, Langley RG, Lacour JP, et al. Long-term 2-year safety and efficacy of tralokinumab in adults with moderate-to-severe atopic dermatitis: Interim analysis of the ECZTEND open-label extension trial. J Am Acad Dermatol. 2022;87(4):815-824. PubMed
62.Langley R, Reich K, Simpson E, et al. Long term improvements in disease severity, itch, and quality of life after 3 years of tralokinumab treatment in adults with moderate to severe atopic dermatitis. 4th Annual Revolutionizing Atopic Dermatitis Conference, April 9-11, 2022; 2022.
63.Blauvelt A, Langley R, Peris K, et al. Continuous tralokinumab treatment over 4 years in adults with moderate-to-severe atopic dermatitis provides long-term disease control [submitted abstract]. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5.
64.Blauvelt A, Langley R, Simpson E, et al. Long-term Safety and Efficacy of Tralokinumab in More Than 1400 Moderate-to-Severe Atopic Dermatitis Patients Treated for up to 42 Months: an Interim Analysis of ECZTEND. American Academy of Dermatology Annual Meeting; 2022 Mar 25-29; Seattle (WA).
65.Drug Reimbursement Review: tralokinumab (Adtralza). Can J Health Technol. 2022;2(6). https://www.cadth.ca/sites/default/files/DRR/2022/SR0689-Adtralza_combined.pdf. Accessed 2023 Jul 25.
66.Schram ME, Spuls PI, Leeflang MMG, Lindeboom R, Bos JD, Schmitt J. EASI, (objective) SCORAD and POEM for atopic eczema: responsiveness and minimal clinically important difference. Allergy. 2012;67(1):99-106. PubMed
67.Simpson E, Beck LA, Gadkari A, et al. Defining a responder on the Peak Pruritus Numerical Rating Scale (NRS) in patients with moderate-to-severe atopic dermatitis: Detailed analysis from randomized trials of dupilumab. J Am Acad Dermatol. 2017;76(6, Supplement 1):AB93.
68.Yosipovitch G, Reaney M, Mastey V, et al. Peak Pruritus Numerical Rating Scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol. 2019;181(4):761-769. PubMed
69.Dias-Barbosa C, Matos R, Vernon M, Carney CE, Krystal A, Puelles J. Content validity of a sleep numerical rating scale and a sleep diary in adults and adolescents with moderate-to-severe atopic dermatitis. J Patient Rep Outcomes. 2020;4(1):100. PubMed
70.Silverberg JI, Lei D, Yousaf M, et al. Comparison of Patient-Oriented Eczema Measure and Patient-Oriented Scoring Atopic Dermatitis vs Eczema Area and Severity Index and other measures of atopic dermatitis: A validation study. Ann Allergy Asthma Immunol. 2020;125(1):78-83. PubMed
71.CADTH Reimbursement Recommendation: upadacitinib (Rinvoq). Can J Health Technol. 2022;2(6). https://www.cadth.ca/sites/default/files/DRR/2022/SR0685REC-Rinvoq%20AD-KH_BF-KH-meta.pdf. Accessed 2023 Nov 3.
72.Howells L, Ratib S, Chalmers JR, Bradshaw L, Thomas KS. How should minimally important change scores for the Patient-Oriented Eczema Measure be interpreted? A validation using varied methods. Br J Dermatol. 2018;178(5):1135-1142. PubMed
73.Shikiar R, Harding G, Leahy M, Lennox RD. Minimal important difference (MID) of the Dermatology Life Quality Index (DLQI): results from patients with chronic idiopathic urticaria. Health Qual Life Outcomes. 2005;3:36. PubMed
74.Basra MKA, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the Minimal Clinically Important Difference and Responsiveness of the Dermatology Life Quality Index (DLQI): Further Data. Dermatology. 2015;230(1):27-33. PubMed
75.Heinl D, Prinsen CA, Deckert S, et al. Measurement properties of adult quality-of-life measurement instruments for eczema: a systematic review. Allergy. 2016;71(3):358-370. PubMed
76.Simpson EL, de Bruin-Weller M, Eckert L, et al. Responder Threshold for Patient-Oriented Eczema Measure (POEM) and Children’s Dermatology Life Quality Index (CDLQI) in Adolescents with Atopic Dermatitis. Dermatology and Therapy. 2019;9(4):799-805. PubMed
77.Bożek A, Reich A. Assessment of Intra- and Inter-Rater Reliability of Three Methods for Measuring Atopic Dermatitis Severity: EASI, Objective SCORAD, and IGA. Dermatology. 2017;233(1):16-22. PubMed
78.Barbier N, Paul C, Luger T, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150(1):96-102. PubMed
79.Hanifin JM, Thurston M, Omoto M, et al. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. Exp Dermatol. 2001;10(1):11-18. PubMed
80.Schmitt J, Langan S, Deckert S, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation. J Allergy Clin Immunol. 2013;132(6):1337-1347. PubMed
81.Rehal B, Armstrong AW. Health outcome measures in atopic dermatitis: a systematic review of trends in disease severity and quality-of-life instruments 1985-2010. PLoS One. 2011;6(4):e17520. PubMed
82.Barrett A, Hahn-Pedersen J, Kragh N, Evans E, Gnanasakthy A. Patient-Reported Outcome Measures in Atopic Dermatitis and Chronic Hand Eczema in Adults. Patient. 2019;12(5):445-459. PubMed
83.Basra MK, Fenech R, Gatt RM, Salek MS, Finlay AY. The Dermatology Life Quality Index 1994-2007: a comprehensive review of validation data and clinical results. Br J Dermatol. 2008;159(5):997-1035. PubMed
84.Lewis V, Finlay AY. 10 years experience of the Dermatology Life Quality Index (DLQI). J Investig Dermatol Symp Proc. 2004;9(2):169-180. PubMed
85.Badia X, Mascaró JM, Lozano R. Measuring health-related quality of life in patients with mild to moderate eczema and psoriasis: clinical validity, reliability and sensitivity to change of the DLQI. The Cavide Research Group. Br J Dermatol. 1999;141(4):698-702. PubMed
86.Lewis-Jones MS, Finlay AY. The Children's Dermatology Life Quality Index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132(6):942-949. PubMed
87.Salek MS, Jung S, Brincat-Ruffini LA, et al. Clinical experience and psychometric properties of the Children's Dermatology Life Quality Index (CDLQI), 1995-2012. Br J Dermatol. 2013;169(4):734-759. PubMed
88.Zigmond AS, Snaith RP. The Hospital Anxiety and Depression Scale. Acta Psychiatr Scand. 1983;67(6):361-370. PubMed
89.Health Canada reviewer's report: Adtralza (tralokinumab) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5. Ottawa (ON): LEO Pharma Inc.; 2023.
90.Blauvelt A, Teixeira HD, Simpson EL, et al. Efficacy and Safety of Upadacitinib vs Dupilumab in Adults With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatology. 2021;157(9):1047-1055. PubMed
91.Clinical Trial Report: LP0162-1337 ECZTEND - all subjects enrolled in the ECZTEND Study at least 60 weeks prior to data cutoff 30-Apr-2021 [CONFIDENTIAL data on file]. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5.
92.Ollendorf DA, Pearson SD. ICER Evidence Rating Matrix: A User's Guide. Boston (MA): Institute for Clinical and Economic Review; 2020: https://icer-review.org/methodology/icers-methods/icer-evidence-ratingmatrix/. Accessed 2023 Jul 4.
93.Higgins JP, Altman DG, Gøtzsche PC, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928. PubMed
94.NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5. 2011.
95.Brooks SP, Gelman A. General Methods for Monitoring Convergence of Iterative Simulations. J Comput Graph Stat. 1998;7(4):434-455.
96.Gelman A, and D. B. Rubin. Inference from Iterative Simulation Using Multiple Sequences. Stat Sci. 1992;7:457.
97.Quantics Biostatistics. The key assumptions of network meta-analysis. 2016; https://www.quantics.co.uk/blog/the-key-assumptions-of-network-meta-analysis/#:~:text=In%20network%20meta%2Danalysis%2C%20we,transitivity%20(similarity)%20and%20consistency.
98.Gelman A. Prior distributions for variance parameters in hierarchical models. Bayesian Analysis. 2006;1(3):515-533.
99.Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol. 1980:44-47.
100.Simpson EL, Sinclair R, Forman S, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet. 2020;396(10246):255-266. PubMed
101.Silverberg JI, Simpson EL, Thyssen JP, et al. Efficacy and Safety of Abrocitinib in Patients With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol. 2020;156(8):863-873. PubMed
102.Gooderham MJ, Forman SB, Bissonnette R, et al. Efficacy and Safety of Oral Janus Kinase 1 Inhibitor Abrocitinib for Patients With Atopic Dermatitis: A Phase 2 Randomized Clinical Trial. JAMA Dermatol. 2019;155(12):1371-1379. PubMed
103.Simpson EL, Forman S, Silverberg JI, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis: Results from a randomized monotherapy phase 3 trial in the United States and Canada (BREEZE-AD5). J Am Acad Dermatol. 2021;85(1):62-70. PubMed
104.Guttman-Yassky E, Teixeira HD, Simpson EL, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. 2021;397(10290):2151-2168. PubMed
105.Guttman-Yassky E, Thaçi D, Pangan AL, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;145(3):877-884. PubMed
106.Simpson EL, Bieber T, Guttman-Yassky E, et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med. 2016;375(24):2335-2348. PubMed
107.Thaçi D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. The Lancet. 2016;387(10013):40-52. PubMed
108.Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus Placebo or Dupilumab for Atopic Dermatitis. N Engl J Med. 2021;384(12):1101-1112. PubMed
109.Reich K, Kabashima K, Peris K, et al. Efficacy and Safety of Baricitinib Combined With Topical Corticosteroids for Treatment of Moderate to Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol. 2020;156(12):1333-1343. PubMed
110.Guttman-Yassky E, Silverberg JI, Nemoto O, et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: A phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J Am Acad Dermatol. 2019;80(4):913-921.e919. PubMed
111.Reich K, Teixeira HD, de Bruin-Weller M, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. 2021;397(10290):2169-2181. PubMed
112.Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287-2303. PubMed
113.Simpson EL, Paller AS, Siegfried EC, et al. Efficacy and Safety of Dupilumab in Adolescents With Uncontrolled Moderate to Severe Atopic Dermatitis: A Phase 3 Randomized Clinical Trial. JAMA Dermatol. 2020;156(1):44-56. PubMed
114.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative Effectiveness Without Head-to-Head Trials. Pharmacoeconomics. 2010;28(10):935-945. PubMed
115.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. 2016: http://www.nicedsu.org.uk.
116.de Bruin-Weller M, Thaçi D, Smith CH, et al. Dupilumab with concomitant topical corticosteroid treatment in adults with atopic dermatitis with an inadequate response or intolerance to ciclosporin A or when this treatment is medically inadvisable: a placebo-controlled, randomized phase III clinical trial (LIBERTY AD CAFÉ). Br J Dermatol. 2018;178(5):1083-1101. PubMed
117.Signorovitch JE, Sikirica V, Erder MH, et al. Matching-Adjusted Indirect Comparisons: A New Tool for Timely Comparative Effectiveness Research. Value Health. 2012;15(6):940-947. PubMed
118.Silverberg JI, Adam DN, Zirwas M, et al. Tralokinumab Plus Topical Corticosteroids as Needed Provides Progressive and Sustained Efficacy in Adults with Moderate-to-Severe Atopic Dermatitis Over a 32-Week Period: An the ECZTRA 3 studyPost Hoc Analysis. Am J Clin Dermatol. 2022;23(4):547-559. PubMed
Appendix 1: Detailed Outcome Data
Table 54: Sensitivity Analysis of the Primary Estimand in the Initial Treatment Period of ECZTRA 6 — Full Analysis Set (Adolescents)
Outcomes assessed at week 16 | Tralokinumab 300 mg q.2.w. (N = 97) | Placebo (N = 94) |
|---|---|---|
IGA score of 0 or 1 | ||
Sensitivity analysis — patients permanently discontinued study | ||
IGA score of 0 or 1, n/N (%) | 17/97 (17.5) | 4/94 (4.3) |
Difference,a % (95% CI) | 13.8 (5.3 to 22.3; P = 0.002) | |
Sensitivity analysis — LOCF | ||
IGA score of 0 or 1, n/N (%) | 17/97 (17.5) | 5/94 (5.3) |
Difference,a,b % (95% CI) | 12.8 (4.1 to 21.4; P = 0.005) | |
EASI-75 | ||
Sensitivity analysis — patients permanently discontinued study | ||
EASI-75, n/N (%) | 27/97 (27.8) | 5/94 (5.3) |
Difference,a % (95% CI) | 23.0 (13.1 to 32.9; P < 0.001) | |
Sensitivity analysis – LOCF | ||
EASI-75, n/N (%) | 27/97 (27.8) | 7/94 (7.4) |
Difference,a,b % (95% CI) | 21.0 (10.8 to 31.1; P < 0.001) | |
Worst daily pruritus NRS (weekly average) | ||
Sensitivity analysis — patients permanently discontinued study treatment | ||
Worst daily pruritus NRS (weekly average) reduction ≥ 4, n/N (%) | 24/96 (25.0) | 3/90 (3.3) |
Difference,a % (95% CI) | 21.7 (12.3 to 31.1; P < 0.001) | |
Sensitivity analysis – LOCF | ||
Worst daily pruritus NRS (weekly average) reduction ≥ 4, n/N (%) | 28/96 (29.2) | 3/90 (3.3) |
Difference,a,b % (95% CI) | 25.9 (16.1 to 35.7; P < 0.001) | |
SCORAD | ||
Change from baseline in SCORAD, adjusted mean (SE) | −26.0 (2.5) | −9.7 (3.3) |
Difference,c % (95% CI) | −16.3 (−23.9 to −8.7; P < 0.001) | |
CDLQI | ||
Change from baseline in CDLQI, adjusted mean (SE) | −6.2 (0.7) | −3.8 (0.9) |
Difference,c % (95% CI) | −2.4 (−4.4 to −0.4; P = 0.017) | |
CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; LOCF = last observation carried forward; NRS = numeric rating scale; q.2.w. = every 2 weeks; SCORAD = Scoring Atopic Dermatitis; SE = standard error.
aThis end point was analyzed with Cochran-Mantel-Haenszel risk difference, stratified by region and baseline IGA.
bMissing data at week 16 imputed using LOCF for patients who did not receive rescue medication and did not withdraw due to an AE or lack of efficacy.
cThis end point was analyzed using the analysis of covariance (ANCOVA) model at week 16: Change in SCORAD = Treatment + Baseline SCORAD + Region + Baseline IGA. Multiple imputation of missing values at week 16 was performed based on data from placebo group.
Note: This table has not been copy-edited.
Source: Clinical Study Report for ECZTRA 6.13
Table 55: Sensitivity Analysis of the Primary Estimand in the Initial Treatment Period of ECZTRA 1, 2, 3, and 7 – Full Analysis Set (Adults; Original Review)
Outcomes assessed at week 16 | ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ECZTRA 7 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Tralokinumab q.2.w. N = 601 | Placebo N = 197 | Tralokinumab q.2.w. N = 591 | Placebo N = 201 | Tralokinumab q.2.w. + TCS N = 252 | Placebo + TCS N = 126 | Tralokinumab q.2.w. + TCS N = 138 | Placebo + TCS N = 137 | ||||
IGA score of 0 or 1 | |||||||||||
Sensitivity analysis — patients permanently discontinued study treatment before week 16 were considered nonresponders | |||||||||||
IGA score of 0 or 1, n/N (%) | 95/601 (15.8) | 14/197 (7.1) | 131/591 (22.2) | 22/201 (10.9) | 98/252 (38.9) | 33/126 (26.2) | NR | NR | |||
Difference, % (95% CI) | 8.6 (4.1 to 13.1; P = 0.002) | 11.1 (5.8 to 16.4; P < 0.001) | 12.4 (2.9 to 21.9; P = 0.015) | NR | |||||||
Sensitivity analysis — LOCFb | |||||||||||
IGA score of 0 or 1, n/N (%) | 96/601 (16.0) | 15/197 (7.6) | 132/591 (22.3) | 23/201 (11.4) | 99/252 (39.3) | 33/126 (26.2) | NR | NR | |||
Difference,a % (95% CI) | 8.2 (3.7 to 12.8; P = 0.003) | 10.7 (5.4 to 16.1; P < 0.001) | 12.8 (3.3 to 22.3; P = 0.012) | NR | |||||||
EASI-75 | |||||||||||
Sensitivity analysis — patients permanently discontinued study treatment before week 16 were considered nonresponders | |||||||||||
EASI-75, n/N (%) | 148/601 (24.6) | 24/197 (12.2) | 196/591 (33.2) | 23/201 (11.4) | 140/252 (55.6) | 44/126 (34.9) | NR | NR | |||
Difference,a % (95% CI) | 12.3 (6.7 to 17.8; P < 0.001) | 21.6 (15.8 to 27.3; P < 0.001) | 20.6 (10.2 to 30.9; P < 0.001) | NR | |||||||
Sensitivity analysis — LOCFb | |||||||||||
EASI-75, n/N (%) | 154/601 (25.6) | 26/197 (13.2) | 197/591 (33.3) | 24/201 (11.9) | 143/252 (56.7) | 46/126 (36.5) | NR | NR | |||
Difference,a % (95% CI) | 12.2 (6.6 to 17.9; P < 0.001) | 21.2 (15.5 to 27.0; P < 0.001) | 20.2 (9.8 to 30.6; P < 0.001) | NR | |||||||
Worst daily pruritus NRS (weekly average) | |||||||||||
Sensitivity analysis – patients permanently discontinued study treatment before week 16 were considered nonresponders | |||||||||||
Worst daily pruritus NRS (weekly average) reduction ≥ 4, n/N (%) | 119/594 (20.0) | 20/194 (10.3) | 144/575 (25.0) | 19/200 (9.5) | 113/249 (45.4) | 43/126 (34.1) | NR | NR | |||
Difference,a % (95% CI) | 9.7 (4.4 to 15.0; P = 0.002) | 15.6 (10.3 to 20.9; P < 0.001) | 11.3 (0.9 to 21.6; P = 0.037) | NR | |||||||
Sensitivity analysis — LOCFb | |||||||||||
Worst daily pruritus NRS (weekly average) reduction ≥ 4, n/N (%) | 128/594 (21.5) | 21/194 (10.8) | 156/575 (27.1) | 20/200 (10.0) | 122/249 (49.0) | 46/126 (36.5) | NR | NR | |||
Difference,a % (95% CI) | 10.6 (5.2 to 16.0; P < 0.001) | 17.2 (11.7 to 22.6; P < 0.001) | 12.6 (2.1 to 23.0; P = 0.021) | NR | |||||||
SCORADc | |||||||||||
Change from baseline in SCORAD, adjusted mean (SE) | −24.9 (1.23) | −17.2 (1.98) | −26.9 (1.06) | −13.8 (2.00) | −37.5 (1.27) | −26.8 (1.80) | −42.2 (1.6) | −34.0 (1.6) | |||
Difference,a % (95% CI) | −7.7 (−11.4 to −3.9; P < 0.001) | −13.0 (−17.1 to −9.0; P < 0.001) | −10.9 (−15.2 to −6.6; P < 0.001) | −8.1 (−12.6 to −3.7; P < 0.001) | |||||||
DLQId | |||||||||||
Change from baseline in DLQI, adjusted mean (SE) | −7.5 (0.41) | −5.7 (0.63) | −8.6 (0.36) | −5.2 (0.68) | −11.6 (0.40) | −8.8 (0.57) | −11.1 (0.4) | −9.7 (0.4) | |||
Difference,a % (95% CI) | −1.8 (−3.0 to −0.6; P = 0.005) | −3.4 (−4.8 to −2.0; P < 0.001) | −2.8 (−4.2 to −1.5; P < 0.001) | −1.4 (−2.6 to −0.3; P = 0.017) | |||||||
DLQI = Dermatology Life Quality Index; CI = confidence interval; EASI = Eczema Area and Severity Index; IGA = Investigator’s Global Assessment; LOCF = last observation carried forward; NR = not reported; NRS = numeric rating scale; q.2.w. = every 2 weeks; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroid.
aMantel-Haenszel risk difference, stratified by region and baseline IGA.
bMissing data at week 16 imputed using LOCF for patients who did not receive rescue medication and did not withdraw due to an adverse event or lack of efficacy.
cThis analysis was conducted using the analysis of covariance (ANCOVA) model, where change in SCORAD = treatment + baseline SCORAD + region + baseline IGA. Multiple imputation of missing values at week 16 was performed based on data from placebo group.
dThis analysis was conducted using the ANCOVA model: Change in DLQI = treatment + baseline DLQI + region + baseline IGA. Multiple imputation of missing values at week 16 was performed based on data from placebo group.
Note: This table has not been copy-edited.
Source: Clinical Study Reports for ECZTRA 1,15 2,16 3,17 and 7.18
Table 56: Other Efficacy Results in Maintenance Treatment Period in ECZTRA 6 — Maintenance Analysis Set (Adolescents)
Outcomes | WEEK 16 tralokinumab 300 mg q.2.w. responders | |
|---|---|---|
Tralokinumab 300 mg q.2.w. (N = 13) | Tralokinumab 300 mg q.4.w. (N = 14) | |
Adolescent worst pruritus NRS | ||
Reduction of adolescent worst pruritus NRS (weekly average) ≥ 4 from baseline at week 16, n/N (%) | 5/11 (45.5) | 10/13 (76.9) |
Reduction of adolescent worst pruritus NRS (weekly average) ≥ 4 from baseline at week 52, n/N (%) | 2/11 (18.2) | 2/13 (15.4) |
Adolescent worst pruritus NRS (weekly average) at week 16, mean (SD) | 4.315 (2.198) | 2.470 (2.065) |
Adolescent worst pruritus NRS (weekly average) at week 52, mean (SD) | 1.950 (1.786) | 3.517 (2.510) |
SCORAD | ||
SCORAD at week 16, mean (SD) | 17.61 (10.68) | 18.69 (10.55) |
SCORAD at week 52, mean (SD) | 10.36 (11.05) | 14.09 (14.36) |
CDLQI | ||
Reduction of CDLQI score ≥ 6 from baseline at week 16, n/N (%) | 6/7 (85.7) | 5/10 (50.0) |
Reduction of CDLQI score ≥ 6 from baseline at week 52, n/N (%) | 3/7 (42.9) | 3/10 (30.0) |
CDLQI score at week 16, mean (SD) | 4.7 (3.0) | 3.4 (3.7) |
CDLQI score at week 52, mean (SD) | 2.3 (3.3) | 2.4 (2.8) |
POEM | ||
Reduction of POEM ≥ 6 from baseline at week 16, n/N (%) | 8/10 (80.0) | 10/13 (76.9) |
Reduction of POEM ≥ 6 from baseline at week 52, n/N (%) | 3/10 (30.0) | 6/13 (46.2) |
POEM at week 16, mean (SD) | 8.1 (5.0) | 6.5 (5.2) |
POEM at week 52, mean (SD) | 3.8 (6.2) | 5.8 (3.9) |
HADS | ||
HADS total score at week 16, mean (SD) | 7.6 (6.2) | 4.5 (5.7) |
HADS total score at week 52, mean (SD) | 3.0 (3.6) | 3.9 (7.5) |
Eczema-related sleep NRS | ||
Eczema-related sleep NRS (weekly average) at week 16, mean (SD) | 3.408 (2.185) | 1.668 (1.671) |
Eczema-related sleep NRS (weekly average) at week 52, mean (SD) | 1.477 (1.192) | 1.947 (2.793) |
CDLQI = Children’s Dermatology Life Quality Index; HADS = hospital anxiety and depression scale; NRS = Numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation.
Note: This table has not been copy-edited.
Source: Clinical Study Report for ECZTRA 6.13
Table 57: Other Efficacy End Points in Maintenance (or Continuous) Treatment Period in ECZTRA 1, 2, and 3 — Maintenance Analysis Set in ECZTRA 1 and 2; Continuous Treatment Analysis Set in ECZTRA 3 (Adults, Original Review)
Outcomes | ECZTRA 1a (Follow-up at 52 weeks) | ECZTRA 2a (Follow-up at 52 weeks) | ECZTRA 3a (Follow-up at 32 weeks) | |||||
|---|---|---|---|---|---|---|---|---|
Tralokinumab q.2.w. | Tralokinumab q.4.w. | Placebo | Tralokinumab q.2.w. | Tralokinumab q.4.w. | Placebo | Tralokinumab q.2.w. + TCS | Tralokinumab q.4.w. + TCS | |
SCORAD | ||||||||
Week 16, mean (SD) | 22.4 (12.0) | 21.4 (11.8) | 21.1 (13.8) | 21.2 (12.5) | 22.4 (10.8) | 23.2 (11.4) | 17.6 (10.1) | 18.1 (9.9) |
Week 52 (ECZTRA 1/2) or 32 (ECZTRA 3), mean (SD) | 16.0 (13.5) | 18.2 (11.4) | 19.0 (14.4) | 14.1 (12.7) | 16.3 (10.7) | 25.5 (17.9) | 13.5 (10.0) | 17.6 (10.7) |
Worst daily pruritus NRS | ||||||||
Week 16, mean (SD) | 3.6 (2.3) | 3.2 (2.0) | 3.2 (2.3) | 3.2 (2.1) | 3.5 (2.2) | 3.8 (2.4) | 2.6 (2.1) | 3.0 (1.9) |
Week 52 (ECZTRA 1/2) or 32 (ECZTRA 3), mean (SD) | 2.7 (2.4) | 2.6 (1.8) | 3.1 (2.5) | 1.9 (2.0) | 2.7 (2.3) | 3.0 (2.1) | 2.2 (2.0) | 2.7 (1.9) |
Eczema-related sleep NRS | ||||||||
Week 16, mean (SD) | 2.5 (2.3) | 2.4 (2.2) | 2.5 (2.5) | 2.5 (2.2) | 2.4 (2.1) | 3.1 (2.6) | 1.7 (1.9) | 2.0 (1.9) |
Week 52 (ECZTRA 1/2) or 32 (ECZTRA 3), mean (SD) | 1.5 (2.1) | 1.7 (1.8) | 2.2 (2.4) | 1.4 (2.2) | 2.1 (2.0) | 2.7 (2.6) | 1.3 (1.7) | 1.7 (1.9) |
DLQI | ||||||||
Week 16, mean (SD) | 4.5 (4.7) | 4.0 (4.5) | 4.6 (4.4) | 4.3 (4.2) | 4.8 (5.2) | 5.0 (4.2) | 3.4 (3.8) | 3.8 (4.7) |
Week 52 (ECZTRA 1/2) or 32 (ECZTRA 3), mean (SD) | 3.2 (4.6) | 2.7 (2.6) | 2.7 (2.3) | 2.4 (4.2) | 4.1 (4.9) | 3.9 (3.2) | 3.0 (3.6) | 4.0 (4.4) |
HADS | ||||||||
HADS total score, mean (SD) | ||||||||
Week 16 | 7.8 (7.1) | 7.1 (5.8) | 8.4 (6.3) | 6.4 (6.2) | 8.0 (6.6) | 6.9 (4.9) | 5.4 (5.9) | 7.3 (6.2) |
Week 52 (ECZTRA 1/2) or 32 (ECZTRA 3), mean (SD) | 7.6 (8.5) | 7.0 (6.3) | 6.5 (5.6) | 5.5 (6.2) | 7.0 (7.3) | 6.7 (6.0) | 4.2 (4.7) | 7.0 (5.9) |
HADS anxiety score, mean (SD) | ||||||||
Week 16 | 4.7 (3.9) | 4.3 (3.3) | 4.6 (3.4) | 3.8 (3.6) | 4.7 (3.9) | 4.0 (2.6) | 3.4 (3.9) | 4.7 (3.7) |
Week 52 (ECZTRA 1/2) or 32 (ECZTRA 3), mean (SD) | 4.6 (4.6) | 4.3 (3.7) | 3.4 (3.0) | 3.3 (3.6) | 4.1 (4.2) | 3.5 (2.9) | 2.8 (3.1) | 4.6 (3.9) |
HADS depression score, mean (SD) | ||||||||
Week 16 | 3.2 (3.7) | 2.9 (3.1) | 3.8 (3.4) | 2.6 (3.2) | 3.4 (3.5) | 2.9 (3.3) | 2.0 (2.7) | 2.6 (3.1) |
Week 52 (ECZTRA 1/2) or 32 (ECZTRA 3), mean (SD) | 3.1 (4.3) | 2.7 (3.2) | 3.1 (3.9) | 2.3 (3.0) | 2.9 (3.4) | 3.1 (4.0) | 1.5 (2.2) | 2.4 (2.6) |
POEM | ||||||||
Week 16, mean (SD) | 9.5 (6.0) | 8.7 (5.4) | 10.1 (6.7) | 8.5 (5.5) | 9.7 (6.9) | 9.8 (6.1) | 6.6 (6.3) | 8.0 (5.4) |
Week 52 (ECZTRA 1/2) or 32 (ECZTRA 3), mean (SD) | 7.1 (5.9) | 7.0 (4.7) | 10.3 (7.7) | 5.4 (5.8) | 8.1 (6.6) | 12.4 (7.4) | 6.5 (6.0) | 8.3 (5.2) |
DLQI = Dermatology Life Quality Index; HADS = Hospital Anxiety and Depression Scale; NRS = numeric rating scale; POEM = Patient-Oriented Eczema Measure; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation.
aPatients who achieved a clinical response with tralokinumab (ECZTRA 1/2) or tralokinumab +TCS (ECZTRA 3) at week 16 were eligible to continue maintenance treatment (ECZTRA 1/2) or continuation treatment (ECZTRA 3) and were included in this dataset, and the outcomes reported were achieved without rescue medication.
Note: This table has not been copy-edited.
Source: Clinical Study Reports for ECZTRA 1,15 2,16 and 3.17
Pharmacoeconomic Review
Abbreviations
AD
atopic dermatitis
AE
adverse event
BIA
budget impact analysis
BSC
best supportive care
DLQI
Dermatology Life Quality Index
EASI
Eczema Area and Severity Index
EASI-75
reduction of at least 75% in Eczema Area and Severity Index score from baseline
HRQoL
health-related quality of life
IGA
Investigator’s Global Assessment
ITC
indirect treatment comparison
JAK
Janus kinase
MAIC
matching adjusted indirect comparison
NMA
network meta-analysis
QALY
quality-adjusted life-year
TCS
topical corticosteroid
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Tralokinumab (Adtralza), 150 mg per 1 mL prefilled syringe, and 300 mg per 2 mL prefilled pena solution for subcutaneous injection |
Submitted price | Tralokinumab, 150 mg/1mL: $422.26 per syringe |
Indication | For the treatment of moderate-to-severe atopic dermatitis in adult and adolescent patients 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Tralokinumab can be used with or without topical corticosteroids. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | February 3, 2023 |
Reimbursement request | For the treatment of patients aged 12 years and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an adequate trial or be ineligible for each of the following therapies: phototherapy (where available) and off-label immunosuppressants |
Sponsor | LEO Pharma Inc. |
Submission history | Previously reviewed: Yes Indication: For the treatment of moderate-to-severe atopic dermatitis in adult patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable Recommendation date: March 7, 2022 Recommendation: Do not reimburse |
NOC = Notice of Compliance.
aA 300 mg per 2 mL prefilled pen is currently not marketed in Canada.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Hybrid decision tree and Markov model |
Target population | Patients aged 12 years and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an adequate trial or be ineligible for each of the following therapies: phototherapy (where available) and off-label immunosuppressants |
Treatment | Tralokinumab plus BSC (low-to-midpotency topical corticosteroids) |
Comparators | Dupilumab plus BSC Abrocitinib plus BSC Upadacitinib plus BSC |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (maximum age 110) |
Key data sources | ECZTRA 1, 2, and 3 for tralokinumab inputs; Institute for Clinical and Economic Review Evidence Report NMA for 16-week efficacy for all comparator treatments |
Submitted results | Sequential ICER for tralokinumab vs. abrocitinib = $55,701 per QALY gained (incremental costs = $5,785; incremental QALYs = 0.10); upadacitinib and dupilumab are more costly and more effective than tralokinumab |
Key limitations |
|
CADTH reanalysis results |
|
AD = atopic dermatitis; BSC = best supportive care; EASI-75 = reduction of at least 75% in Eczema Area and Severity Index score from baseline; ICER = incremental cost-effectiveness ratio; IGA = Investigator’s Global Assessment; JAK = Janus kinase; NMA = network meta-analysis; QALY = quality-adjusted life-year; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
Conclusions
The CADTH Clinical Review found that tralokinumab resulted in improvements in the severity and extent of atopic dermatitis (AD) as measured by a reduction of at least 50% in Eczema Area Severity Index score from baseline (EASI-75) and an Investigator’s Global Assessment (IGA) of 0 or 1in adolescents and adults with moderate-to-severe AD who had an inadequate response to topical AD therapy compared to placebo after 16 weeks of treatment. Overall, interpretation of the clinical meaningfulness of the findings from the submitted pivotal trials was hindered by the insufficient duration of the follow-up at week 16, given that an optimal response to tralokinumab treatment is usually observed at 6 months in clinical practice. No conclusions can be drawn about the efficacy of tralokinumab beyond week 16 based on the submitted evidence due to limitations of the included studies. Evidence from submitted indirect treatment comparisons (ITCs) of tralokinumab with other advanced therapies for the treatment of moderate-to-severe AD, on which the sponsor’s economic model was based, suggests that tralokinumab performs worse than or similar to its main comparator, dupilumab, in adults. The lone exception to this trend was noted in the ECZTRA 3 versus LIBERTY AD CHRONOS unanchored matching adjusted indirect comparison (MAIC), in which results favoured tralokinumab over dupilumab for an IGA of 0 or 1 and a change in the Dermatology Life Quality Index (DLQI) at week 52. However, these analyses are associated with important uncertainties due to several limitations with the submitted ITCs. The indirect comparative efficacy of tralokinumab versus abrocitinib and upadacitinib was also considered uncertain based on network meta-analysis (NMA) evidence. Results of the lone NMA evaluating the efficacy of tralokinumab in adolescents were imprecise and potentially affected by intransitivity, which precludes any conclusion of certainty about the comparative efficacy of tralokinumab versus relevant comparators. Based on the indirect evidence, no conclusion about the safety of tralokinumab compared with other advanced therapies for moderate-to-severe AD could be drawn with any certainty, and there was no comparative evidence for long-term efficacy outcomes or treatment discontinuation available for adults or adolescents.
CADTH undertook a reanalysis to address several limitations in the sponsor’s analysis, which included adopting alternate estimates for the 52-week conditional response rate of abrocitinib, revising the proportion of responders on tralokinumab switching to a dosing schedule of every 4 weeks after week 16, updating the health-state utility values, and altering the proportion of patients on the high dose of Janus kinase (JAK) inhibitor treatments. CADTH was unable to address limitations related to the uncertainty regarding initial treatment response at week 16 or the lack of comparative data for the reimbursement population beyond week 16.
In the CADTH base case, tralokinumab was less costly and less effective (fewer quality-adjusted life-years [QALYs]) than all comparator treatments. Uncertainty remains due to limitations with the evidence on the long-term effectiveness and safety of tralokinumab compared with relevant comparators. Furthermore, the generalizability of the results of the CADTH base case to adolescent patients is uncertain due to limitations with the available evidence in this population. When considering the Health Canda product monograph–recommended dosing for tralokinumab (every 2 weeks) and publicly available list prices, tralokinumab is less costly than dupilumab and more costly than abrocitinib and upadacitinib on an annual per-patient basis, based on drug costs alone and not accounting for potential differences in treatment efficacy and discontinuation.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was provided by 3 groups: the Eczema Society of Canada, the Canadian Skin Patient Alliance, and Eczema Québec. Survey respondents indicated that AD symptoms negatively affect daily life by causing dryness, red skin, intense itching, thickening of the skin, or skin infections. Itch was reported to be the most burdensome symptom, leading to disrupted sleep, fatigue, decreased functionality, and significant impacts on daily life, work, and school. Uncontrolled AD may lead to hospitalizations, primarily among adults. Patients also experience flare-ups of disease that involve periods of worsening condition and its symptoms. Patients reported using topical corticosteroids (TCS), topical calcineurin inhibitors, phosphodiesterase type 4 inhibitors, off-label oral systemic medications (e.g., cyclosporine, methotrexate, and azathioprine), phototherapy, oral corticosteroids (e.g., prednisone) for flare-ups, dupilumab, or JAK inhibitors. Common side effects associated with topical treatments include redness, skin thinness, and rashes, along with more rare side effects such as hormonal issues, cataracts, and skin cancers. Immunosuppressants and JAK inhibitors are associated with more severe long-term side effects, and dupilumab was noted to have the most favourable safety profile of all available treatments. Patient input indicated that the most important outcomes for new treatment options include improvement in symptoms, long-term improvement in quality of life (e.g., sleep, prevention of flares, and reduced psychological burden), safety (e.g., reduced infection and minimal short- and long-term adverse effects), ease of use, and affordability. Patient experience with tralokinumab indicated that treatment improved symptoms, allowed them to return to daily activities, and reduced the frequency and intensity of flares.
CADTH received registered clinician input from the Canadian Dermatology Association, Dermatology Association of Ontario, and Atlantic Specialist Group Managing Atopic Dermatitis. The clinician groups stated that initial treatment would consist of emollients and topical prescription therapies (TCS and topical calcineurin inhibitors). Patients whose disease remains uncontrolled may then receive phototherapy or off-label systemic immunosuppressants in addition to topical treatments. Newer systemic agents in the form of biologics and oral JAK inhibitors are approved for AD and are standard therapy for those who do not have disease control after topical therapies or phototherapy. Clinician groups stated that tralokinumab fits into the same treatment paradigm and line of therapy as dupilumab, but that ideally patients should only have to fail topical treatments and phototherapy before receiving biologics or JAK inhibitors.
The drug plans expressed concerns surrounding the lack of active comparators in all pivotal trials, as well as implementation issues related to relevant comparators such as dupilumab, abrocitinib, and upadacitinib. Specifically, the drug plans noted that recent safety warnings may preclude describing JAK inhibitors as true comparators due to clinician reluctance to prescribe them. It was noted that consistency with renewal criteria for dupilumab in AD should also be considered. The drug plans emphasized potential eligibility concerns regarding whether reimbursement should be provided for those who did not achieve a response or lost response to dupilumab. Drug plans also expressed uncertainty around patients potentially switching from a dosing schedule of every 2 weeks to every 4 weeks after achieving a response after 16 weeks of treatment, and whether dosing every 4 weeks should be mandated in any situations based on clinical trial results. Last, the drug plans noted that dupilumab successfully completed pan-Canadian Pharmaceutical Alliance negotiations for patients 12 years and older with moderate-to-severe AD.
Two of these concerns were addressed in the sponsor’s model:
Clinical effectiveness was based on treatment response, with the inclusion of costs related to flare-ups, adverse events (AEs), and monitoring.
Every 4 weeks dosing with tralokinumab was included as an option.
In addition, CADTH addressed the concerns regarding the impact of changing the dosing schedule of every 4 weeks for tralokinumab.
CADTH was unable to address the concern raised in stakeholder input regarding the lack of long-term comparative efficacy data after 16 weeks for tralokinumab versus comparator treatments.
Economic Review
The current review is for tralokinumab (Adtralza) for patients aged 12 years and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an adequate trial or would be ineligible for phototherapy (where available) and off-label immunosuppressants.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of tralokinumab plus best supportive care (BSC) compared with dupilumab plus BSC, abrocitinib plus BSC, and upadacitinib plus BSC. The target model population comprised patients aged 12 years and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an adequate trial or would be ineligible for phototherapy (where available) and off-label immunosuppressants.1 The target population for this review aligns with the sponsor’s reimbursement request. However, the target population and reimbursement request are narrower than the Health Canada indication, which states that tralokinumab is indicated for the treatment of moderate-to-severe AD in adult and adolescent patients 12 years and older whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. The sponsor’s request for a deviation to focus the review on the reimbursement request rather than the Health Canada–indicated population was granted by CADTH.
Tralokinumab is available in 150 mg (150 mg per 1 mL) single-use prefilled syringes for self-administered subcutaneous injection.1,2 The recommended dosage is an initial dose of 600 mg followed by 300 mg administered every other week, and the annual cost of treatment is $21,958 based on a unit cost of $422.26 per syringe (annual cost of $22,802 in the first year of treatment only).2 The annual costs of comparator treatments were $25,446 for dupilumab based on a unit cost of $978.70 per syringe (annual cost of $26,424 in the first year of treatment only); $17,763 to $19,880 for abrocitinib based on a unit cost of $48.67 per 50 mg tab or $54.47 per 100 mg or 200 mg tab; and $18,864 to $28,090 for upadacitinib based on a unit cost of $51.68 per 15 mg tablet or $76.96 per 30 mg tablet.1,3 The annual cost of BSC per patient comprised costs related to low-to-midpotency TCS, assumed to be mometasone furoate 0.1% ointment.1,3 Annual costs of TCS were estimated to be $88.37 for biologic and JAK inhibitor responders and $126.37 for BSC and nonresponders.1
The analysis was conducted from the perspective of the Canadian public health care payer. Costs and clinical outcomes (life-years and QALYs) were estimated over a lifetime time horizon (maximum age of 110; 1-year cycle length) and discounted at an annual rate of 1.5%.
Model Structure
The model structure included an induction period encompassing the first year of treatment (with 16-week and 52-week response assessments) and a maintenance period for the remainder of the lifetime horizon. The short-term phase was based on a decision tree (Figure 1),1 in which all patients start at baseline receiving tralokinumab, dupilumab, abrocitinib, or upadacitinib in combination with BSC. Response to treatment was first assessed at 16 weeks based on a reduction of at least 75% in EASI score from baseline (EASI-75).1 Patients who responded to treatment at 16 weeks continued to receive treatment until week 52, at which time patients were assessed for sustained response. Patients who did not respond to tralokinumab, dupilumab, abrocitinib, or upadacitinib at 16 weeks discontinued their active treatment and received BSC alone for the remainder of the model horizon. The long-term maintenance phase (Figure 2) consisted of a Markov model with 3 health states: maintenance treatment, BSC treatment, and death.1 Patients who began each active treatment and who had a sustained response at 52 weeks entered the Markov model in the maintenance treatment health state, while those receiving BSC alone at 52 weeks entered the BSC treatment health state. In each cycle, patients in the maintenance treatment health state could discontinue active treatment for any reason, such as lack of long-term efficacy, AEs, and patient or physician preference, and transition to the BSC treatment state or die. Patients in the BSC treatment health state remained on BSC alone until death.
Model Inputs
The baseline patient characteristics in the sponsor’s model were aligned with the ECZTRA 3 trial of patients 18 years and older with an inadequate response to topical medications or documented systemic treatment for AD in the past year (mean age = 39.1 years; 55% male).4
Clinical efficacy (i.e., treatment response) was based on the NMA results presented in the latest Evidence Report of the Institute for Clinical and Economic Review, which informed the proportion of patients achieving EASI-75 at week 16 for all active comparators using the following trials: ECZTRA 3 (tralokinumab), LIBERTY-AD CHRONOS (dupilumab), JADE COMPARE (abrocitinib), and AD-UP (upadacitinib).5 The sponsor assumed that patients who received rescue treatment were nonresponders who would discontinue treatment. The sustained response was assessed at 52 weeks based on the probability of a sustained response conditional on achieving a prior response at 16 weeks as defined by EASI-75 response criteria.1 These estimates for each treatment under consideration were based on unadjusted values identified from each respective trial (i.e., a naive comparison).4,6-8 The post-trial annual loss of effect was assumed to be 0 and clinical efficacy after week 52 was assumed to remain constant for the remainder of the time horizon.1 Patients could discontinue treatment after 52 weeks in the maintenance period of treatment, based on annual discontinuation rates from their respective clinical trial(s) (ECZTRA 1 and ECZTRA 2 for tralokinumab,9,10 SOLO continue for dupilumab,11 Measure Up for upadacitinib,7 and JADE Regimen for abrocitinib8).
Utilities in the sponsor’s model were assumed to vary by treatment received (biologics and JAK inhibitor therapy or BSC alone) and by treatment response (responder or nonresponder). Utility values were derived by the sponsor using a mixed-model regression approach and EQ-5D-3L data collected from the ECZTRA 3 trial, which enrolled patients aged 18 years and older.4 All patients entered the model with a baseline utility based on the ECZTRA 3 trial population. Patients then accrued health-utility gains relative to baseline based on response and treatment received. Responders on biologic or JAK inhibitor therapy experienced gains in health-related quality of life (HRQoL), whereas it was assumed that receiving BSC alone would not result in improved HRQoL and all nonresponders would revert to baseline utility levels. During the 52-week induction phase, patients receiving biologic or JAK inhibitor therapy were assumed to experience gains in HRQoL even if they were defined as nonresponders in comparison with the baseline BSC health-utility value. Disutilities associated with AEs, such as injection-site reactions, oral herpes, conjunctivitis, upper respiratory tract infections, or acne, were not included in the sponsor’s model.1
Costs considered in the model included drug acquisition costs for biologic or JAK inhibitor treatment, BSC, health care resource utilization associated with monitoring, flare-ups, and AEs. All biologic and JAK inhibitor drug acquisition costs were sourced from the IQVIA DeltaPA dataset.3 Costs related to BSC were calculated based on rates of TCS use from the ECZTRA 3 study and the unit costs of mometasone furoate 0.1% ointment from the Ontario Drug Benefit Formulary.4,12 The sponsor assumed that 40% of responders on tralokinumab would switch from a dosing schedule of every 2 weeks to every 4 weeks, subsequently halving the drug acquisition costs of tralokinumab for these responders.1 No training or administration costs were included in the model, as it was assumed that these would be covered by patient support programs for biologic therapies and are not applicable to JAK inhibitor therapies. Costs related to health care resource utilization were based on the frequency of resource use, which varied by treatment received (biologic, JAK inhibitor, or BSC and/or nonresponders) derived from a Régie de l’assurance maladie du Québec database analysis.13 Unit costs for health care resource use were derived from the Régie de l’assurance maladie du Québec database and Ontario Ministry of Health Schedule of Benefits.13,14 Costs related to AEs were associated with a unit cost per occurrence reflecting a follow-up virtual assessment with a dermatologist based on the Ontario Ministry of Health Schedule of Benefits.14 Costs related to flare-ups were calculated based on rescue therapy use, which varies based on the time of flare-up, and treatment received based on the ECZTRA 3 trial, with costs sourced from the Ontario Drug Benefit Formulary.4,12
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following section.
Base-Case Results
Tralokinumab was associated with an estimated cost of $158,697 and 19.80 QALYs over the lifetime time horizon. Based on a sequential analysis considering all comparators, tralokinumab was associated with an incremental cost-effectiveness ratio of $55,701 per QALY gained (incremental costs: $5,785; incremental QALYs: 0.10) compared to abrocitinib (Table 3). Dupilumab and upadacitinib remained on the cost-effectiveness frontier and were more costly and more effective. In the sponsor’s base case, tralokinumab had approximately a 41% probability of being cost-effective at a willingness-to-pay threshold of $50,000.
Results were driven by the reduced drug acquisition costs of tralokinumab following transition from a dosing schedule of every 2 weeks to every 4 weeks for responders and the small difference in incremental QALYs compared to abrocitinib and other active comparators derived from differences in week 16 response rates, sustained responses, and treatment discontinuation. The sponsor’s model estimated that approximately 1% of the total QALYs for tralokinumab accrued during the 16-week period for which comparative data were estimated from the NMA, and the remaining total QALYs accrued over the period for which there is no direct or indirect evidence.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Abrocitinib | 152,912 | 19.69 | Reference |
Tralokinumab | 158,697 | 19.80 | 55,701 |
Upadacitinib | 165,270 | 19.84 | 157,101 |
Dupilumab | 219,012 | 20.13 | 181,586 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Full results are reported in Appendix 3. The submitted analysis is based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses that examined the impact of alternative response definitions (an IGA of 0 or 1 and reduction of at least 50% in EASI score from baseline) and alternative discontinuation rates based on data from the ECZTEND trial for all comparators, reducing the proportion of patients on tralokinumab who switch from a dosing schedule of every 2 weeks to every 4 weeks after 16 weeks, restricting the target population to the adolescent population, and shorter time horizons of 5 or 10 years. The results were most sensitive to reducing the proportion of patients switching from a tralokinumab dosing schedule of every 2 weeks to every 4 weeks, alternative discontinuation rates using the ECZTEND trial for all comparators, and an alternative response definition (an IGA of 0 or 1).
The sponsor included a scenario analysis for the adolescent population (aged 12 to < 18 years), which used clinical data from the ECZTRA 6 study, 16-week response rates from the LEO Pharma adolescent NMA, and 52-week conditional response rates from various adult studies for each active comparator. In this analysis, tralokinumab was less costly and less effective (fewer QALYs) than all comparator treatments.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative effectiveness of tralokinumab to relevant comparators for initial response to treatment is uncertain: In the sponsor’s model, tralokinumab was compared to dupilumab, abrocitinib, and upadacitinib. In the absence of direct head-to-head evidence, several ITCs were included in the sponsor’s submission. For response at week 16 in the sponsor’s pharmacoeconomic model, an NMA conducted by the Institute for Clinical and Economic Review that assessed responses based on EASI-75 in adult patients only was used to estimate these values for tralokinumab and the other comparators.5 The CADTH Clinical Review identified several sources of heterogeneity, including variation in duration of disease, treatment of patients in the control group across the combination therapy trials, and disease severity across trials. Additional limitations with the NMA included differing time points of response measurement across included studies, inconsistent imputation for missing data across end points, and harms that were not evaluated. Overall, the CADTH Clinical Review found that treatment with dupilumab, abrocitinib, and upadacitinib was associated with favourable EASI-75 responses at week 16 compared to tralokinumab. In its summary, the CADTH Clinical Review reported that all sponsor-submitted ITCs comparing tralokinumab to comparators found that tralokinumab performs worse than or similar to its main comparator, dupilumab, in adults. The lone exception was the ECZTRA 3 versus LIBERTY AD CHRONOS unanchored MAIC, in which results favoured tralokinumab over dupilumab for an IGA of 0 or 1 and change in DLQI at week 52. Given the limitations with the submitted ITCs, the magnitude of the effect estimated from the NMA is associated with uncertainty. As such, the clinical effectiveness, and therefore the cost-effectiveness, of tralokinumab relative to other active comparators is associated with uncertainty; however, the ITC results generally suggest tralokinumab is inferior with regard to responses at week 16.
CADTH was unable to address this limitation.
The comparative durability of treatment response, discontinuation and safety is highly uncertain: After initial treatment response at week 16, conditional sustained response based on EASI-75 was assessed at week 52 in the sponsor’s model, after which patients could discontinue treatment due to loss of efficacy. This conditional sustained response was based on a naive and noncomparative estimate using the proportion of those who achieved a response at week 16 and sustained that response at week 52 from each individual trial for tralokinumab and comparators. However, according to the CADTH Clinical Review, no conclusion can be drawn on the efficacy of tralokinumab beyond week 16 based on the submitted evidence due to important limitations of the included studies, including inconsistent results between trials and evidence of imprecision in the longer-term results in randomized controlled trials. Furthermore, given that the inputs for the other comparators were derived from a naive comparison, the effectiveness of tralokinumab compared with relevant comparators beyond week 52 is highly uncertain. Upon assessment of the sustained response at 52 weeks, the clinical expert input obtained by CADTH indicated that some of the estimated 52-week conditional response rates did not meet face validity. Notably, the durability of response of abrocitinib was estimated to be markedly worse than that of comparators and did not align with clinical expectations. For example, the conditional 52-week responses were 46.5% and 65.8% for 100 mg and 200 mg abrocitinib, respectively, versus 87.3% and 89.6% for 15 mg and 30 mg upadacitinib, respectively. Based on clinical practice, it was expected that abrocitinib would have similar durability of response to upadacitinib. The long-term efficacy of tralokinumab and durability of treatment effect beyond the trial period, as well as in comparison with its relevant comparators, therefore, remains uncertain.
CADTH also noted that, following the assessment of response following 52 weeks, the treatment benefit was assumed to be sustained for the remainder of the model time horizon. Loss of efficacy was included as part of treatment-discontinuation rates, which were key drivers of cost-effectiveness after 52 weeks. Patients were subject to an annual risk of discontinuation based on naive data sourced from each comparator’s respective trial (i.e., ECZTRA 1, ECZTRA 2, SOLO CONTINUE, Measure Up 1, Measure Up 2, and JADE Regimen) and did not account for potential differences between trials. JAK inhibitor treatments had nearly twice the annual risk of discontinuation compared with biologics. The clinical experts consulted by CADTH for this review noted that the greater risk of discontinuation with JAK inhibitor treatment was reasonable; however, the magnitude of the difference was uncertain. Furthermore, according to the CADTH Clinical Review of the submitted indirect evidence, no conclusion about the safety of tralokinumab compared with other advanced therapies for moderate-to-advance AD could be made with any certainty. As such, the limitations of the discontinuation and harms data for all active treatments introduces substantial uncertainty into the estimates of cost-effectiveness of tralokinumab versus comparator treatments.
CADTH adjusted the 52-week conditional response rate of abrocitinib to equal that of upadacitinib to reflect clinical expert input and expectations of clinical practice. CADTH could not address issues related to the lack of comparative long-term efficacy data after week 16 and the uncertainty regarding comparative discontinuation and safety.
A scenario analysis was conducted that explored the impact of setting the annual risk of discontinuation of all comparator treatments to equal that of dupilumab.
Uncertainty surrounds the response assessment method: The sponsor’s model used the EASI-75 response definition in the base-case analysis. Alternative response measures such as an IGA of 0 or 1 and a reduction of at least 50% in EASI score from baseline were included in the model and were tested in scenario analyses. Clinical expert input obtained by CADTH noted that patient-reported outcomes (i.e., an IGA of 0 or 1) and gestalt assessments, rather than EASI-75, may be more reflective of how the response to treatment would be assessed in some instances and the likelihood of continuing treatment. Additionally, it was noted that symptoms of AD may vary from week to week, and EASI time points may not adequately reflect the benefit that patients are achieving (i.e., improved pruritis, sleep, and mental health) because any benefit is based on specific parameters such as the amount of erythema in the skin. This creates uncertainty surrounding the response definitions used and what may be most reflective of clinical practice.
CADTH assessed the impact of adopting the IGA of 0 or 1 response definition in a scenario analysis.
Maintenance dosing after 16 weeks for tralokinumab is highly uncertain: The sponsor assumed that, after 16 weeks of treatment on the initial dosing schedule of every 2 weeks for tralokinumab, 40% of patients achieving a response based on EASI-75 would switch to a schedule of every 4 weeks. The sponsor’s assumption is based on internal market research and could not be validated by CADTH reviewers. According to the product monograph text, “at prescriber’s discretion, every fourth week dosing may be considered for some patients who achieve clear or almost clear skin after 16 weeks of treatment; however, the probability of maintaining clear or almost clear skin may be decreased with dosing every fourth week.”2 Whether the sponsor’s assumption would be realized in clinical practice is uncertain, given that the product monograph states that the probability of a maintained response may decrease with altered dosing. Based on clinical expert input, the decision to switch to dosing every 4 weeks may differ on an individual-patient basis, and there is a reasonable likelihood of a response being lost on a dosing schedule of every 4 weeks. The clinical experts stated that patients who cease responding after switching to dosing every 4 weeks would resume dosing every 2 weeks, which was not an option in the sponsor’s submitted model. The sponsor assumed that 40% of all responders switch to every 4 weeks and remain on this schedule for the remainder of their lifetime or until complete treatment discontinuation. The sponsor’s assumption has notable impacts on the incremental costs between tralokinumab and comparators over the lifetime time horizon; a dosing schedule of every 4 weeks results in a 50% reduction in drug acquisition costs for tralokinumab after 16 weeks for responders who switch to the less-frequent dosing. However, this change in dosing schedule was assumed to result in no loss of efficacy over time for patients receiving tralokinumab every 4 weeks, meaning that the sponsor assumed the response would be sustained indefinitely despite switching to less-frequent dosing.
CADTH adjusted the proportion of patients receiving treatment every 4 weeks after achieving a treatment response at 16 weeks from 40% to 20% to account for the potential loss of response over time that was not modelled by the sponsor and to better reflect clinical expert feedback on the proportion of patients likely to start administering treatment every 4 weeks.
Additional scenario analyses assessing the impact of changing the dosing schedule after 52 weeks instead of 16 weeks, and alternative proportions of responders switching to every 4 weeks ranging from 5% to 40% were included by CADTH.
Health-state utility values are not aligned with expectations: The sponsor included health-state utility values for patients on BSC, denoted as the baseline utility value, responders on treatment, and nonresponders to JAK inhibitor or biologic therapies. The health-state utility values for biologic and JAK inhibitor nonresponders in the 52-week induction period were close to the value used for treatment responders despite these patients not achieving a treatment response and having discontinued treatment. According to clinical expert input, a proportion of patients may experience improvement despite not reaching the EASI-75 threshold for a response (i.e., partial responders); however, the proportion of patients expected to achieve this is unknown. It is unlikely the benefit would be large enough that these patients would achieve the majority of the same benefit, on average, as full responders when compared with the baseline utility value. Although clinical expert input obtained by CADTH indicated that partial responders may continue treatment after response assessment, this was not an option in the model, and nonresponders were assumed to benefit while not incurring any treatment costs. The sponsor did not include the possibility of benefiting from BSC alone.
CADTH set the nonresponse on biologic and JAK inhibitor treatment utility value during the 52-week induction period to be equal to the baseline utility value.
The model structure may not accurately reflect the clinical treatment pathway experienced by patients with AD: In the sponsor’s model, patients who receive treatment and do not achieve EASI-75 at week 16 are assumed to not receive any further treatment besides BSC alone for the remainder of their lifetime. Clinical expert input obtained by CADTH indicated that patients would continue to trial different treatments (i.e., cycle between currently available comparator treatments) and not remain on BSC alone for the remainder of their lifetime. The sponsor did not model the possibility of subsequent lines of treatment upon failing initial treatment, which introduces some uncertainty to the submitted economic evaluation results.
CADTH could not address this limitation in reanalysis.
The expected proportion of patients on the higher dose of JAK inhibitors is underestimated: The sponsor assumed that 2% of adult patients and 0% of adolescent patients on upadacitinib will receive the higher 30 mg dose. Similarly, the sponsor assumed that 2% of all patients on abrocitinib will receive the higher 200 mg dose. These estimates were both based on the sponsor’s internal estimates. The clinical experts consulted by CADTH expected that 20% of all patients on upadacitinib and abrocitinib would be receiving the higher dose of each respective treatment, based on current clinical practice in Canada.
CADTH adjusted the proportion of patients receiving the higher dose of upadacitinib and abrocitinib to 20% to better reflect clinical practice in Canada.
The majority of inputs in the pharmacoeconomic model are derived from adults: Tralokinumab is indicated for use in adult and adolescent patients aged 12 years and older. The sponsor used the ECZTRA 3 study to inform baseline patient characteristics in the economic model and clinical efficacy for tralokinumab at 16 weeks and 52 weeks. The ECZTRA 3 study included patients aged 18 years and older, and therefore no patients aged between 12 and 17 years informed these estimates in the base case. The sponsor conducted a subgroup analysis for adolescents (aged 12 to < 18 years only) based on ECZTRA 6 adolescent trial data and inputs from the LEO Pharma adolescent population NMA for treatment efficacy at 16 weeks. The CADTH Clinical Review concluded that the results of the lone NMA evaluating the efficacy of tralokinumab in adolescents were imprecise and potentially affected by intransitivity, which precludes making any conclusions of certainty about the comparative efficacy of tralokinumab versus relevant comparators. Additionally, the conditional response at 52 weeks was still informed by naive comparisons derived from data in adults for all comparators in this subgroup analysis. Results from the sponsor’s adolescent subgroup analysis appeared to be generally aligned with those of the CADTH reanalyses, which found tralokinumab was less costly and less effective than all comparators for adults and adolescents. However, this analysis was associated with uncertainty given the limitations of the submitted evidence and the lack of specificity of several key inputs to adolescents.
CADTH was unable to address limitations with the subgroup analysis specific to adolescent patients.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH undertook reanalyses that addressed key limitations within the submitted economic model, as summarized in Table 5. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. All CADTH probabilistic reanalyses were based on 1,000 iterations. CADTH was unable to address the other limitations of the model, including the lack of long-term comparative clinical data beyond 16 weeks.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients enrolled in the ECZTRA trials were assumed to be representative of patients in Canada who would be eligible for treatment with tralokinumab. | Uncertain: Clinical expert input obtained by CADTH noted that the characteristics of the participants in the pivotal trial were generally representative of the patients seen in clinical practice; however, the trials enrolled patients with an EASI score higher than that of patients who may be considered for treatment in clinical practice. The score cut-offs for EASI-75 in the pivotal trials are therefore not reflective of all adolescent and adult patients with moderate and severe AD. |
All-cause mortality was included in the model, with no additional risk of mortality associated with AD. | Appropriate: AD is not expected to influence survival. |
No utility impacts were associated with adverse events. | Uncertain: The sponsor’s model included a unit cost per occurrence of injection-site reactions, oral herpes, overall conjunctivitis, or upper respiratory tract infections; however, no health-related quality-of-life impacts were included. |
Efficacy can be reasonably assessed at 16 weeks. | Uncertain: Clinical expert input obtained by CADTH noted that 16 weeks is a relatively short period of time to assess a response to biologics, and more improvement may be expected at week 26. The trajectory of treatment benefit is expected to improve up to 52 weeks with biologics. |
AD = atopic dermatitis; EASI = Eczema Area and Severity Index.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Durability of treatment response at 52 weeks | Conditional response for abrocitinib at 52 weeks is 46.5% for 100 mg and 65.8% for 200 mg dosing, lower than upadacitinib. | Conditional response for abrocitinib at 52 weeks was set to be equal to that of upadacitinib.
|
2. Dosing schedule of tralokinumab after 16 weeks | After achieving response on every 2 weeks dosing at 16 weeks, 40% of responders will switch to q.4.w. dosing for the remainder of the time horizon. | After achieving response on q.2.w. dosing at 16 weeks, 20% of responders will switch to q.4.w. for the remainder of the time horizon. |
3. Health-state utility value for treatment nonresponders | Nonresponders to biologic and JAK inhibitor treatments received utility values higher than baseline during the initial 52-week induction period. | Nonresponders to biologic and JAK inhibitor treatments received the same utility value as baseline during the initial 52-week induction period. |
4. Proportion of patients on high-dose JAK inhibitors |
| 20% of all patients on JAK inhibitor treatments will receive the higher dose of their respective treatment. |
CADTH base case | — | Reanalysis 1 + 2 + 3 + 4 |
JAK = Janus kinase; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
CADTH undertook a stepped analysis, incorporating each change proposed in Table 5 to the sponsor’s base case to highlight the impact of each change (Table 9).
In the CADTH base case, tralokinumab was associated with an estimated total cost of $168,090 and 19.79 QALYs over the lifetime time horizon (Table 6). As a result, tralokinumab was less costly and less effective (fewer QALYs) than all comparator treatments. These findings align with the available indirect clinical evidence suggesting that tralokinumab is less effective than dupilumab at 16 weeks.
The key drivers of the cost-effectiveness estimates are the assumptions surrounding long-term comparative efficacy and drug acquisition costs of tralokinumab related to a dosing schedule of every 4 weeks.
Table 6: Summary of CADTH Reanalysis Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Tralokinumab | 168,090 | 19.79 | Reference |
Abrocitinib | 179,598 | 20.03 | 47,431 |
Dupilumab | 218,414 | 20.09 | 656,706 |
Upadacitinib | 175,488 | 19.85 | Extendedly dominated by tralokinumab and abrocitinib |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The submitted analysis is based on the public available prices of the comparator treatments.
Scenario Analysis Results
Given the results of the CADTH base case, which suggest tralokinumab is the least costly and least effective treatment option, price-reduction scenarios were not conducted. CADTH notes that, based on publicly available list prices, tralokinumab is less costly than dupilumab and more costly than abrocitinib and upadacitinib on an annual basis when considering the Health Canda product monograph–recommended dosing schedule for tralokinumab (i.e., every 2 weeks).
CADTH undertook a series of scenario analyses exploring the impacts of alternative assumptions on the cost-effectiveness of tralokinumab, which included:
evaluating the impact of using an IGA of 0 or 1 for the treatment-response definition
assessing the impact of implementing every 4 weeks dosing after 52 weeks instead of 16 weeks
assessing the impact of 5% of responders switching from every 2 weeks to every 4 weeks dosing after 16 weeks
assessing the impact of 40% of responders switching from every 2 weeks to every 4 weeks dosing after 16 weeks, aligning with the sponsor’s base case assumption
setting the annual risk of discontinuation of all active treatments to be equal to that of dupilumab
applying a modest utility benefit for nonresponders on biologic or JAK inhibitor treatments during the 52-week induction period using the midpoint value between responders and nonresponders.
The results of these analyses are presented in Table 12. Tralokinumab remained the least costly and least effective treatment option across almost all scenario analyses. However, in the scenario analysis assessing the impact of 5% of responders switching from a dosing schedule of every 2 weeks to every 4 weeks after 16 weeks, tralokinumab was found to be dominated by upadacitinib (i.e., tralokinumab was more costly and less effective). Across all scenarios, the parameters that had the largest impact on cost-effectiveness estimates were the proportion of responders on tralokinumab assumed to switch from dosing every 2 weeks to every 4 weeks and the annual risk of discontinuation.
Issues for Consideration
Tralokinumab was previously reviewed by CADTH for the treatment of adult patients with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and who had an adequate trial or be ineligible for each of the following therapies: phototherapy (where available), methotrexate, and cyclosporine. This prior submission included a cost-minimization analysis comparing tralokinumab to dupilumab. CADTH concluded that the validity of the cost-minimization analysis approach was uncertain and the final recommendation of the CADTH Canadian Drug Expert Committee indicated that tralokinumab should not be reimbursed. The current submission for tralokinumab included several key differences, including expansion of the target population to include adolescents aged 12 years and older, the use of a cost-utility analysis, and the inclusions of abrocitinib and upadacitinib as comparators.
The submitted price of dupilumab is uncertain, as it has been previously submitted for multiple indications including moderate-to-severe AD and severe asthma.15,16 The CADTH Canadian Drug Expert Committee reimbursement recommendations for dupilumab indicate that a price reduction is required and dupilumab has undergone negotiations at the pan-Canadian Pharmaceutical Alliance multiple times. However, the confidential negotiated price of dupilumab is unknown. Clinical expert input indicated that JAK inhibitor treatments may not be true comparators and that dupilumab is the most similar and relevant comparator to tralokinumab. Should the price paid for dupilumab be lower than public list prices, the cost-effectiveness of dupilumab versus tralokinumab is likely underestimated.
No training or administration costs were included in the model, as it was assumed that these would be covered by patient support programs for biologic therapies and are not applicable to JAK inhibitor therapies. The impact of this assumption on the cost-effectiveness estimates of tralokinumab versus comparators is unknown.
Overall Conclusions
The CADTH Clinical Review found that tralokinumab resulted in improvements in the severity and extent of AD (EASI-75 and an IGA of 0 or 1) in adolescents and adults with moderate-to-severe AD who had inadequate response to topical AD therapy compared to placebo after 16 weeks of treatment. Overall, the interpretation of clinical meaningfulness of findings from the submitted pivotal trials was hindered by insufficient duration of follow-up at week 16 given that an optimal response to tralokinumab treatment is usually observed at 6 months in clinical practice. No conclusion can be drawn on the efficacy of tralokinumab beyond week 16 based on the submitted evidence due to limitations of the included studies. Evidence from submitted ITCs comparing tralokinumab to other advanced therapies for the treatment of moderate-to-severe AD, on which the sponsor’s economic model was based, suggests that tralokinumab performs worse than or similar to its main comparator, dupilumab, in adults. The lone exception to this trend was noted in the ECZTRA 3 versus LIBERTY AD CHRONOS unanchored MAIC, in which results favoured tralokinumab over dupilumab for an IGA of 0 or 1 and change in DLQI at week 52. However, these analyses are associated with important uncertainty due to several limitations with the submitted ITCs. The indirect comparative efficacy of tralokinumab versus abrocitinib and upadacitinib was also considered uncertain based on NMA evidence. Results of the lone NMA evaluating the efficacy of tralokinumab in adolescents were imprecise and potentially affected by intransitivity, which precludes any conclusion of certainty about the comparative efficacy of tralokinumab versus relevant comparators. Based on the indirect evidence, no conclusion about the comparative safety of tralokinumab to other advanced therapies for moderate-to-advance AD could be drawn with any certainty, and no comparative evidence regarding long-term efficacy outcomes or treatment discontinuation was available for adults or adolescents.
CADTH identified several other limitations with the sponsor’s economic evaluation. This included uncertainty related to the most appropriate approach to defining treatment response; uncertainty in the assumed proportion of patients expected to maintenance dosing receive every 4 weeks rather than every 2 weeks; concerns with the validity of health-state utility values; underestimation of the proportion of patients expected to receive the higher dose of JAK inhibitor therapy; lack of modelling of subsequent treatment use following initial treatment discontinuation; and concerns regarding the inputs used to assess the cost-effectiveness of tralokinumab in the adolescent population.
CADTH undertook a reanalysis to address several limitations in the sponsor’s analysis. This included adopting alternative estimates for the 52-week conditional response rate of abrocitinib, revising the proportion of responders on tralokinumab switching to a dosing schedule of every 4 weeks after week 16, updating the health-state utility values, and altering the proportion of patients on a high dose of JAK inhibitor treatments. CADTH was unable to address limitations related to uncertainty with the initial treatment response at week 16 or the lack of comparative data for the reimbursement population beyond week 16.
In the CADTH base case, tralokinumab was less costly and less effective (fewer QALYs) than all comparator treatments. Uncertainty remains due to limitations of the evidence regarding the long-term effectiveness and safety of tralokinumab compared with relevant comparators. Furthermore, the generalizability of the results of the CADTH base case to adolescent patients is uncertain due to limitations with the available evidence in this population. When considering the Health Canda product monograph–recommended dosing for tralokinumab (i.e., every 2 weeks) and publicly available list prices, tralokinumab is less costly than dupilumab and more costly than abrocitinib and upadacitinib on an annual per-patient basis based on drug costs alone and not accounting for potential differences in treatment efficacy and discontinuation.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5.
2.Adtralza (tralokinumab): 150 mg/1 mL single use pre-filled syringe [product monograph]. Toronto (ON): LEO Pharma Inc; 2023 Feb 3.
3.DeltaPA Prices. June 2023. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5. 2023.
4.Clinical Study Report: LP0162-1339. Tralokinumab in combination with topical corticosteroids for moderate-to-severe atopic dermatitis ECZTRA 3 (ECZema TRAlokinumab trial no. 3) [internal sponsor's report]. Ballerup (DK): LEO Pharma A/S; 2020 Feb 18.
5.JAK Inhibitors and Monoclonal Antibodies for the Treatment of Atopic Dermatitis: Effectiveness and Value; Final Evidence Report. Boston (MA): Institute for Clinical and Economic Review; 2021: https://icer.org/wp-content/uploads/2020 or 12/Atopic-Dermatitis_Final-Evidence-Report_081721.pdf. Accessed 2021 Aug 10.
6.Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287-2303. PubMed
7.Guttman-Yassky E, Teixeira HD, Simpson EL, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. 2021;397(10290):2151-2168. PubMed
8.Blauvelt A, Silverberg JI, Lynde CW, et al. Abrocitinib induction, randomized withdrawal, and retreatment in patients with moderate-to-severe atopic dermatitis: Results from the JAK1 Atopic Dermatitis Efficacy and Safety (JADE) REGIMEN phase 3 trial. J Am Acad Dermatol. 2022;86(1):104-112. PubMed
9.Clinical Study Report: LP0162-1325. Tralokinumab monotherapy for moderate-to-severe atopic dermatitis ECZTRA 1 (ECZema TRAlokinumab trial no. 1 [internal sponsor's report]. Ballerup (DK): LEO Pharma A/S; 2020 Feb 11.
10.Clinical Study Report: LP0162-1326. Tralokinumab monotherapy for moderate-to-severe atopic dermatitis ECZTRA 2 (ECZema TRAlokinumab trial no. 2) [internal sponsor's report]. Ballerup (DK): LEO Pharma A/S; 2020 Feb 20.
11.Worm M, Simpson EL, Thaçi D, et al. Efficacy and Safety of Multiple Dupilumab Dose Regimens After Initial Successful Treatment in Patients With Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatology. 2020;156(2):131-143. PubMed
12.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario Drug Benefit Formulary/Comparative Drug Index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Jul 26.
13.PeriPharm Inc. Retrospective Database Analysis to Assess the Prevalence, Demographics and Healthcare Resource Utilization in Adults with Moderate-to-Severe Atopic Dermatitis in Quebec. Analysis of RAMQ Data for LEO Pharma Inc. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5. 2021.
14.Ontario Ministry of Health. Ontario Schedule of Benefits - Physician Services Under the Health Insurance Act (January 2022). 2022; https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20210314.pdf. Accessed 2022 Jul 26.
15.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: dupilumab (Dupixent - Sanofi Genzyme). Can J Health Technol. 2021;1(6). https://www.cadth.ca/sites/default/files/attachments/2021-06/CADTH_reimbursement_recommendation_dupilumab_%28dupixent%29_1.pdf. Accessed 2023 Aug 22.
16.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: dupilumab (Dupixent - Sanofi-Aventis Canada Inc). Ottawa (ON): CADTH; 2020 Apr 24: https://www.cadth.ca/sites/default/files/cdr/complete/SR0636%20Dupixent%20-%20CDEC%20Final%20%20Recommendation%20April%2024%2C%202020%20for%20posting.pdf. Accessed 2023 Aug 22.
17.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario Drug Benefit Formulary/Comparative Drug Index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Jun 29.
18.DeltaPA. Ottawa (ON): IQVIA; 2022: https://www.iqvia.com/. Accessed 2023 Jun 29.
19.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5.
20.Iqvia. Pharmastat. 2022; https://www.customerportal.iqvia.com/sites/portal/. Accessed 2023 Jan 23.
21.Data on file. DeltaPA Prices. March 2023. In: Drug Reimbursement Review sponsor submission: Adtralza (tralokinumab), 150 mg pre-filled (150 mg/1 mL) syringes for subcutaneous injection. Toronto (ON): LEO Pharma Inc; 2023 Jun 5. 2023.
Appendix 1: Cost-Comparison Table
Note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing product listing agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost-Comparison Table for Moderate-to-Severe Atopic Dermatitis
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosage | Average daily cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Tralokinumab | 150 mg/1 mL | Prefilled syringe | 422.2600a | 600 mg initially, followed by 300 mg every 2 weeksc | Year 1: 62.47 Year 2: 60.16 | Year 1: 22,802 Year 2: 21,958 |
Biologic treatments | ||||||
Dupilumab (Dupixent) | 300 mg/2 mL 200 mg/1.14 mL | Prefilled syringe | 978.7000 | For adults: 600 mg initially, followed by 300 mg every 2 weeks For adolescents 12 to 17 years of age less than 60 kg: 400 mg initially, followed by 200 mg every 2 weeks | Year 1: 72.40 Year 2+: 69.72 | Year 1: 26,424 Year 2+: 25,446 |
JAK-1 inhibitors | ||||||
Abrocitinib | 50 mg 100 mg 200 mg | Oral tablet | 48.6667b 54.4667b | 100 mg or 200 mg once daily Dose reduction to 100 mg once daily after symptom control is achieved by week 12 | 48.67 to 54.47 | 17,763 to 19,880 |
Upadacitinib | 15 mg 30 mg | ER oral tablet | 51.6810 76.9600b | 15 mg once daily Updose to 30 mg once daily for severe disease or if inadequate response is not achieved (e.g., EASI-75) | 51.68 to 76.96 | 18,864 to 28,090 |
ER = extended release; JAK-1 = Janus kinase 1.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed June 2023),17 unless otherwise indicated, and do not include dispensing fees. Recommended dosages are from the respective product monographs, unless otherwise indicated. Treatment course for all systemic therapies is based on an annual period of 52 weeks (or 365 days) unless otherwise indicated.
aSponsor’s submitted price.1
bIQVIA Delta PA list price (June 2023).18
cThe product monograph notes: At prescriber’s discretion, every fourth week dosing may be considered for some patients who achieve clear or almost clear skin after 16 weeks of treatment; however, the probability of maintaining clear or almost clear skin may be decreased with dosing every fourth week.
Appendix 2: Submission Quality
Note this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | Yes | No comment |
Model structure is adequate for decision problem | No | Refer to CADTH appraisal regarding model structure issue excluding evaluation of subsequent therapies |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | The sponsor’s probabilistic sensitivity analyses did not properly incorporate random draws for 52-week conditional response rates) in each iteration. Refer to CADTH appraisal regarding the omission of data informing efficacy and baseline characteristics of adolescent patients aged 12 to 17 |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 9: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | Abrocitinib | 151,626 | 19.68 | Reference |
Tralokinumab | 157,589 | 19.78 | 56,194 | |
Upadacitinib | 164,330 | 19.83 | 150,009 | |
Dupilumab | 214,677 | 20.09 | 190,465 | |
CADTH reanalysis 1: durability of treatment response at 52 weeks | Tralokinumab | 157,589 | 19.78 | Reference |
Abrocitinib | 178,117 | 20.07 | 71,326 | |
Dupilumab | 214,677 | 20.09 | 1,702,819 | |
Dominated treatments | ||||
Upadacitinib | 164,330 | 19.83 | Extendedly dominated by tralokinumab and abrocitinib | |
CADTH reanalysis 2: treatment dosing after 16 weeks | Abrocitinib | 151,626 | 19.68 | Reference |
Upadacitinib | 164,331 | 19.83 | 84,106 | |
Dupilumab | 214,677 | 20.09 | 190,465 | |
Dominated treatments | ||||
Tralokinumab | 166,283 | 19.82 | Dominated by upadacitinib | |
CADTH reanalysis 3: health-state utility value for treatment nonresponders | Abrocitinib | 151,626 | 19.64 | Reference |
Tralokinumab | 157,589 | 19.73 | 63,857 | |
Upadacitinib | 164,330 | 19.80 | 99,739 | |
Dupilumab | 214,677 | 20.06 | 194,598 | |
CADTH reanalysis 4: proportion of patients on high dose JAK inhibitors | Abrocitinib | 153,584 | 19.69 | Reference |
Tralokinumab | 157,589 | 19.78 | 43,263 | |
Dupilumab | 214,677 | 20.09 | 184,586 | |
Dominated treatments | ||||
Upadacitinib | 174,182 | 19.87 | Extendedly dominated by tralokinumab and abrocitinib | |
CADTH base case: reanalysis 1 + 2 + 3 + 4) — deterministic | Tralokinumab | 166,283 | 19.77 | Reference |
Abrocitinib | 177,789 | 20.01 | 47,332 | |
Dupilumab | 214,677 | 20.06 | 779,495 | |
Dominated treatments | ||||
Upadacitinib | 174,182 | 19.84 | Extendedly dominated by tralokinumab and abrocitinib | |
CADTH base case (reanalysis 1 + 2 + 3 + 4) — probabilistic | Tralokinumab | 168,090 | 19.79 | Reference |
Abrocitinib | 179,598 | 20.03 | 47,431 | |
Dupilumab | 218,414 | 20.09 | 656,706 | |
Dominated treatments | ||||
Upadacitinib | 175,488 | 19.85 | Extendedly dominated by tralokinumab and abrocitinib | |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The submitted analysis is based on the public available prices of the comparator treatments.
Table 10: Disaggregated Summary of CADTH’s Economic Evaluation Results — QALYs
Treatment | Component | Value |
|---|---|---|
Tralokinumab | Responder | 2.52 |
Nonresponder | 17.27 | |
Total | 19.79 | |
Abrocitinib | Responder | 3.51 |
Nonresponder | 16.52 | |
Total | 20.03 | |
Upadacitinib | Responder | 2.80 |
Nonresponder | 17.05 | |
Total | 19.85 | |
Dupilumab | Responder | 3.74 |
Nonresponder | 16.34 | |
Total | 20.09 |
QALY = quality-adjusted life-year.
Table 11: Disaggregated Summary of CADTH’s Economic Evaluation Results — Costs
Treatment | Component | Value |
|---|---|---|
Tralokinumab | Drug acquisition costs | $63,804 |
Monitoring costs | $100,366 | |
Background costs | $3,867 | |
Adverse event costs | $53 | |
Total | $168,090 | |
Abrocitinib | Drug acquisition costs | $76,972 |
Monitoring costs | $98,732 | |
Background costs | $3,825 | |
Adverse event costs | $70 | |
Total | $179,598 | |
Upadacitinib | Drug acquisition costs | $71,384 |
Monitoring costs | $100,190 | |
Background costs | $3,857 | |
Adverse event costs | $56 | |
Total | $175,488 | |
Dupilumab | Drug acquisition costs | $116,736 |
Monitoring costs | $97,791 | |
Background costs | $3,813 | |
Adverse event costs | $75 | |
Total | $218,414 |
Table 12: Scenario Analyses Conducted on the CADTH Reanalysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|---|
CADTH base case | Tralokinumab | 168,090 | 19.79 | Reference |
Abrocitinib | 179,598 | 20.03 | 47,431 | |
Dupilumab | 218,414 | 20.09 | 656,706 | |
Dominated treatments | ||||
Upadacitinib | 175,488 | 19.85 | Extendedly dominated by tralokinumab and abrocitinib | |
CADTH scenario 1: IGA of 0 or 1 response assessment | Tralokinumab | 145,177 | 19.51 | Reference |
Abrocitinib | 160,277 | 19.77 | 57,145 | |
Dupilumab | 188,127 | 19.81 | 704,771 | |
Dominated treatments | ||||
Upadacitinib | 160,409 | 19.68 | Extendedly dominated by tralokinumab and abrocitinib | |
CADTH scenario 2: q.4.w. maintenance dosing after 52 weeks | Tralokinumab | 169,298 | 19.79 | Reference |
Abrocitinib | 179,736 | 20.03 | 43,901 | |
Dupilumab | 219,200 | 20.09 | 611,419 | |
Dominated treatments | ||||
Upadacitinib | 175,294 | 19.85 | Extendedly dominated by tralokinumab and abrocitinib | |
CADTH scenario 3 to 5% of responders switch to q.4.w. maintenance dosing | Upadacitinib | 174,751 | 19.84 | Reference |
Abrocitinib | 179,677 | 20.03 | 20,974 | |
Dupilumab | 218,343 | 20.09 | 704,271 | |
Dominated treatments | ||||
Tralokinumab | 175,671 | 19.82 | Dominated by upadacitinib | |
CADTH scenario 4 to 40% of responders switch to q.4.w. maintenance dosing | Tralokinumab | 159,029 | 19.75 | Reference |
Abrocitinib | 179,679 | 20.03 | 72,404 | |
Dupilumab | 218,950 | 20.09 | 638,331 | |
Dominated treatments | ||||
Upadacitinib | 175,085 | 19.85 | Extendedly dominated by tralokinumab and abrocitinib | |
CADTH scenario 5: discontinuation of all treatments set equal to dupilumab | Tralokinumab | 168,421 | 19.77 | Reference |
Abrocitinib | 183,669 | 20.06 | 53,363 | |
Upadacitinib | 211,898 | 20.24 | 154,982 | |
Dominated treatments | ||||
Dupilumab | 215,206 | 20.04 | Dominated by upadacitinib | |
CADTH scenario 6: modest benefit for nonresponders on biologic or JAK inhibitor treatments for the 52-week induction period | Tralokinumab | 168,312 | 19.81 | Reference |
Abrocitinib | 179,332 | 20.05 | 47,028 | |
Dupilumab | 220,001 | 20.12 | 542,769 | |
Dominated treatments | ||||
Upadacitinib | 175,163 | 19.87 | Extendedly dominated by tralokinumab and abrocitinib | |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The submitted analysis is based on the publicly available prices of the comparator treatments.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 13: Summary of Key Takeaways
Key takeaways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) estimating the incremental budget impact of reimbursing tralokinumab for patients living with AD aged 12 and older.19 The BIA was undertaken from the perspective of a Canadian public payer over a 3-year time horizon (2025 to 2027) using a claims-based approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits Program. Key inputs to the BIA are documented in Table 14.
The sponsor compared a reference scenario in which patients received dupilumab, upadacitinib, or abrocitinib to a new drug scenario in which tralokinumab was reimbursed. The claims-based approach was based on public drug plan data derived from IQVIA Pharmastat claims data for dupilumab,20 as all other comparators are not yet publicly reimbursed. For provinces where public claims data were not available or erroneous (e.g., British Columbia, Alberta, Manitoba, PEI, Saskatchewan, and the Non-Insured Health Benefits Program), Ontario was used as a substitute and adjusted to account for province size differences.19 A linear forecasting approach was implemented to extrapolate the number of claims across the 2024 to 2027 time horizon using private claims data for all relevant treatment options. The sponsor assumed that market size for public claims would also grow due to the availability of JAK inhibitor treatments currently under pan-Canadian Pharmaceutical Alliance negotiations; it was assumed that oral JAK inhibitors would attain public reimbursement in Year 1. The sponsor’s analysis used drug acquisition costs for tralokinumab based on the sponsor’s submitted price.19 To derive drug acquisition costs, the sponsor estimated the number of units per dupilumab claim (including loading dose assumption) and assumed that each claim equated to a 28-day supply of treatment for dupilumab and all comparators. Drug acquisition costs for comparator treatments were estimated using IQVIA DeltaPA data.21
The sponsor assumed that the majority of tralokinumab market shares will be derived from the displacement of dupilumab. The sponsor also assumed that 40% of patients receiving tralokinumab will switch to maintenance dosing every 4 weeks after 16 weeks of treatment.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (Year 1 / Year 2 / Year 3) |
|---|---|
Target population | |
Number of total claims (pan-Canadian) | 25,800 / 36,441 / 52,28220 |
Market uptake (3 years) | |
Uptake (reference scenario) Tralokinumab Dupilumab Abrocitinib Upadacitinib | 0.00% / 0.00% / 0.00% 75.20% / 71.10% / 68.90% 1.80% / 2.20% / 2.40% 23.00% / 26.70% / 28.70% |
Uptake (new drug scenario) Tralokinumab Dupilumab Abrocitinib Upadacitinib | 25.00% / 30.00% / 35.00% 55.02% / 48.09% / 43.00% 2.50% / 2.80% / 2.85% 17.48% / 19.11% / 19.15% |
Cost of treatment (per claim) | |
Cost of treatment per claim (28-day course) Tralokinumab (300 mg q.2.w.) Tralokinumab (300 mg q.4.w.) Dupilumab Abrocitinib (100 mg) Abrocitinib (200 mg) Upadacitinib (15 mg) Upadacitinib (30 mg) | $2,044.82 $1,022.41 $2,369.71 $1,362.67 $2,725.34 $1,447.07 $2,154.88 |
q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
Note: Market shares were based on the sponsor’s internal estimates.
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget impact of funding tralokinumab for AD was cost savings of $2,969,431 in Year 1, $4,602,338 in Year 2, and $7,216,890 in Year 3, for a cumulative savings of $14,788,659 across the 3-year time horizon.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Maintenance dosing after 16 weeks for tralokinumab is highly uncertain: The sponsor assumed that after 16 weeks of treatment on a every 2 weeks dosing schedule, 40% of all patients would switch to a every 4 weeks dosing schedule. As per the product monograph for tralokinumab, “At prescriber’s discretion, every fourth week dosing may be considered for some patients who achieve clear or almost clear skin after 16 weeks of treatment; however, the probability of maintaining clear or almost clear skin may be decreased with dosing every fourth week.” As discussed in the critical appraisal of the cost-utility analysis, there is uncertainty surrounding whether such a high proportion of patients would switch to and remain on every 4 weeks dosing in clinical practice, as the monograph states that the probability of maintaining response may decrease with altered dosing. Notably, the sponsor does not include the option for patients to resume every 2 weeks dosing upon loss of response in the model.
The sponsor’s assumption has notable impacts on estimated cost savings associated with tralokinumab; every 4 weeks dosing schedule results in a 50% reduction in drug acquisition costs for tralokinumab after 16 weeks. In the BIA, this assumption is directly applied to 40% of all claims, which effectively assumes that all inducers respond to tralokinumab at 16 weeks and continue to receive every 4 weeks maintenance therapy. This assumption results in considerable impacts to the estimated cost savings associated with tralokinumab, since the model does not account for nonresponders or loss of response and patients are assumed to remain on every 4 weeks dosing for the remainder of the 3-year time horizon of the analysis. The number of patients assumed to receive every 4 weeks dosing is therefore likely overestimated by the sponsor and cost savings may also therefore be overestimated.
CADTH adjusted the proportion of patients switching to every 4 weeks dosing to reflect the changes in the cost-utility analysis, being 20% of all responders. CADTH could not address issues regarding lack of consideration for nonresponders due to the model structure, uncertainty in the claims-based approach, and lack of available claims data for tralokinumab.
CADTH conducted scenario analyses to reflect 5% and 40% of responders switching to every 4 weeks maintenance dosing, aligned with scenarios conducted with the cost-utility analysis.
Market uptake of tralokinumab is associated with uncertainty: The market uptake of tralokinumab was assumed to be 25% in Year 1, 30% in Year 2, and 35% in Year 3 based on the sponsor’s internal forecasting for the treatment of moderate-to-severe AD in the reimbursement population. The accuracy of the sponsor’s internal market shares could not be validated by CADTH. Clinical expert input obtained by CADTH indicated that the uptake of tralokinumab did not meet face validity and appeared to be overestimated, given that dupilumab is established in the market and is expected to retain the majority of market shares for biologic users and remain relatively stable. Clinical expert input estimated that it was more likely that tralokinumab would capture 25% of the market by Year 3, starting with 15% in Year 1 and 20% in Year 2. CADTH notes that uncertainty remains in these estimates.
CADTH adjusted the market shares of tralokinumab to capture 15% of the market in Year 1, 20% in Year 2, and 25% in Year 3 to reflect clinical expert input.
Market shares for JAK inhibitors are associated with uncertainty: The sponsor has estimated that JAK inhibitors will occupy approximately 25% (1.8% abrocitinib; 23% upadacitinib) of the total market shares for the treatment of moderate-to-severe AD in the reimbursement population in the absence of data on public JAK inhibitor claims. Clinical expert input obtained by CADTH noted that the total estimated market shares for JAK inhibitors appeared reasonable but that the market shares for abrocitinib appeared to be considerably underestimated. In clinical practice, it is estimated that approximately 45% of patients on JAK inhibitors are on abrocitinib with the remaining 55% receiving upadacitinib. Furthermore, clinical expert input expressed concerns with the high total proportion (25% of market shares) of adolescents receiving JAK inhibitors, as the majority of adolescents are expected to be receiving biologics in clinical practice due to the unfavourable safety profile of JAK inhibitors when compared to biologics.
CADTH adjusted the market shares of abrocitinib and upadacitinib to reflect clinical expert input indicating that 45% of patients on JAK inhibitors would receive abrocitinib, with the remainder on upadacitinib.
Uncertainty regarding claims-based approach to derive target population: The sponsor used a claims-based approach to estimate the budget impact of reimbursing tralokinumab for moderate-to-severe AD and there were concerns with this approach. Primarily, claims data were only available for dupilumab and the sponsor used various assumptions to estimate numbers for tralokinumab, abrocitinib, and upadacitinib. As tralokinumab, abrocitinib, and upadacitinib are not publicly reimbursed in Canada, a market growth function was used to simulate the effects of reimbursement of JAK inhibitors using private claims data, which could not be validated by CADTH. The sponsor also assumed that tralokinumab would not contribute to overall meaningful claims growth given its similarities to dupilumab, which could not be validated by clinical experts input obtained by CADTH. Furthermore, claims data from the IQVIA Pharmastat database were only available in select jurisdictions (Ontario, New Brunswick, Newfoundland and Labrador, and Nova Scotia) and for remaining jurisdictions, was estimated by applying a population adjustment factor to the total population for each province with missing data. The population adjustment factor was calculated by dividing the total population for each province with missing claims data by the total population of Ontario, but this method effectively assumes that the claims data for all missing provinces would be similar for Ontario, which is associated with uncertainty. There remains considerable uncertainty whether dupilumab claims data constitute an appropriate proxy estimate for tralokinumab and JAK inhibitor treatments.
CADTH could not address this limitation in reanalysis.
The expected proportion of patients on the higher dose of JAK inhibitors is underestimated: The sponsor assumed that 2% of adult patients and 0% of adolescent patients on upadacitinib will receive the higher 30 mg dose. Similarly, the sponsor assumed that 2% of all patients on abrocitinib will receive the higher 200 mg dose. These estimates were both based on the sponsor’s internal estimates. Based on clinical expert input, it was expected that 20% of all patients on upadacitinib and abrocitinib would be receiving the higher dose of each respective treatment based on current clinical practice in Canada.
CADTH adjusted the proportion of adult patients receiving the higher dose of upadacitinib and abrocitinib to 20% to better reflect clinical practice in Canada, aligned with the cost-utility analysis. Due to BIA model limitations, CADTH could not increase the proportion of adolescent patients on the higher dose of upadacitinib to 20%. The proportion of adolescent patients on the higher dose of abrocitinib was increased to 20%
CADTH Reanalyses of the BIA
Table 15: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Proportion of patients on tralokinumab receiving q.4.w. maintenance dosing | 40% of all claims are assumed to be for q.4.w. maintenance dosing after 16 weeks | 20% of claims are assumed to be for q.4.w. maintenance dosing, aligned with the cost-utility analysis |
2. Market shares – tralokinumab | 25% / 30% / 35% | 15% / 20% / 25% |
3. Market shares – JAK inhibitors | Abrocitinib: 2.50% / 2.80% / 2.85% Upadacitinib: 17.48% / 19.11% / 19.15% (Baseline market share of abrocitinib was 1.80% for upadacitinib it was 23.00%) | Abrocitinib: 8.99% / 9.86% / 9.90% Upadacitinib: 10.99% / 12.05% / 12.10% (Note: baseline market share of abrocitinib was 11.16% and upadacitinib was 13.64%) |
4. Proportion of patients on higher dose of JAK inhibitors |
|
|
CADTH base case | Reanalysis 1 + 2 + 3 + 4 | |
JAK = Janus kinase; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17.
CADTH’s reanalysis found that funding tralokinumab for patients 12 and older with AD resulted in cost savings of $1,418,549 in Year 1, $2,256,300 in year 2, and $3,625,310 in year 3, for a cumulative savings of $7,300,159 across the 3-year time horizon. CADTH’s reanalysis found that the reimbursement of tralokinumab is likely to result in substantially less cost savings than predicted by the sponsor’s model.
CADTH conducted additional scenario analyses to address remaining uncertainty using the CADTH base case. Results are provided in Table 17.
Assuming 5% of responders will switch to every 4 weeks maintenance dosing after 16 weeks of treatment.
Assuming 40% of responders will switch to every 4 weeks maintenance dosing after 16 weeks of treatment.
Table 16: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | −$14,788,659 |
CADTH reanalysis 1: proportion of patients on q.4.w. maintenance dosing | −$9,434,452 |
CADTH reanalysis 2: market shares tralokinumab | −$10,165,076 |
CADTH reanalysis 3: market shares JAK inhibitors | −$14,461,388 |
CADTH reanalysis 4: proportion of patients on high dose of JAK inhibitors | −$15,653,019 |
CADTH base case | −$7,300,159 |
JAK = Janus kinase; q.4.w. = every 4 weeks.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $39,302,475 | $58,897,537 | $81,737,448 | $116,150,626 | $256,785,612 |
New drug | $39,302,475 | $55,928,106 | $77,135,111 | $108,933,736 | $241,996,953 | |
Budget impact | $0 | −$2,969,431 | −$4,602,338 | −$7,216,890 | −$14,788,659 | |
CADTH base case | Reference | $39,947,550 | $59,864,228 | $83,331,325 | $118,612,682 | $261,808,235 |
New drug | $39,947,550 | $58,445,679 | $81,075,025 | $114,987,371 | $254,508,075 | |
Budget impact | $0 | −$1,418,549 | -$2,256,300 | −$3,625,310 | −$7,300,159 | |
CADTH scenario analysis: 5% q.4.w. maintenance dosing | Reference | $39,947,550 | $59,864,228 | $83,331,325 | $118,612,682 | $261,808,235 |
New drug | $39,947,550 | $58,881,216 | $81,895,265 | $116,458,371 | $257,234,852 | |
Budget impact | $0 | −$983,012 | −$1,436,060 | −$2,154,310 | −$4,573,382 | |
CADTH scenario analysis: 40% q.4.w. maintenance dosing | Reference | $39,947,550 | $59,864,228 | $83,331,325 | $118,612,682 | $261,808,235 |
New drug | $39,947,550 | $57,864,964 | $79,981,371 | $113,026,038 | $250,872,372 | |
Budget impact | $0 | −$1,999,264 | −$3,349,954 | −$5,586,644 | −$10,935,862 |
q.4.w. = every 4 weeks.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.