CADTH Reimbursement Review
Efgartigimod Alfa (Vyvgart)
Sponsor: argenx Canada Inc.
Therapeutic area: Generalized myasthenia gravis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
Ab
antibody
Ab+
antibody positive
Ab–
antibody negative
AChEI
acetylcholinesterase inhibitor
AChR
acetylcholine receptor
AE
adverse event
AESI
adverse event of special interest
CDEC
Canadian Drug Expert Committee
CI
confidence interval
CMI
clinically meaningful improvement
CRD
Centre for Reviews and Dissemination
CrI
credible interval
EQ-5D-5L
5-Level EQ-5D
Fc
fragment crystallizable
FcRn
neonatal fragment crystallizable receptor
gMG
generalized myasthenia gravis
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
IgG
immunoglobulin G
HRQoL
health-related quality of life
ICU
intensive care unit
IST
immunosuppressive therapy
ITC
indirect treatment comparison
ITT
intention to treat
IVIg
intravenous immunoglobulin
LRP4
lipoprotein receptor-related protein 4
LSM
least squares mean
MAR
missing at random
MCSE
Monte Carlo standard error
MD
maintenance dose
MDC
Muscular Dystrophy Canada
MedDRA
Medical Dictionary for Regulatory Activities
MG
myasthenia gravis
MG-ADL
Myasthenia Gravis Activities of Daily Living
MGFA
Myasthenia Gravis Foundation of America
MGII
Myasthenia Gravis Impairment Index
MG-QoL15
Myasthenia Gravis Quality of Life 15-item
MG-QoL15r
Revised Myasthenia Gravis Quality of Life 15-item
MID
minimally important difference
mITT
modified intention to treat
MNAR
missing not at random
MuSK
muscle-specific kinase
NA
not applicable
NMA
network meta-analysis
NMD4C
Neuromuscular Disease Network for Canada
NR
not reported
NSIST
nonsteroidal immunosuppressive therapy
OR
odds ratio
PE
plasma exchange
PP
plasmapheresis
QMG
Quantitative Myasthenia Gravis
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SE
standard error
SEB
study baseline
SLR
systematic literature review
SSQ
single simple question
TEAE
treatment-emergent adverse event
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Efgartigimod alfa (Vyvgart), 20 mg/mL solution, 10 mg/kg administered as an IV infusion over 1 hour once weekly for 4 doses (i.e., weeks 0 to 3). For patients weighing 120 kg or more, the recommended dose of efgartigimod alfa is 1,200 mg (3 vials) per infusion. |
Sponsor | argenx Canada Inc. |
Indication | For the treatment of adult patients with generalized myasthenia gravis (gMG) who are anti-acetylcholine receptor (AChR) antibody positive. |
Reimbursement request | argenx Canada Inc. is requesting that efgartigimod alfa be reimbursed as an add-on therapy for adult patients with AChR antibody positive (gMG whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or nonsteroidal immunosuppressant therapy) |
Health Canada approval status | NOC issued |
Health Canada review pathway | Standard review |
NOC date | September 19, 2023 |
Recommended dose | 10 mg/kg administered as an IV infusion over 1 hour once weekly for 4 doses (i.e., weeks 0 to 3). For patients weighing 120 kg or more, the recommended dose of efgartigimod alfa is 1,200 mg (3 vials) per infusion. |
AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; gMG = generalized myasthenia gravis; NOC = Notice of Compliance.
Source: The sponsor’s submission.1
Introduction
Myasthenia gravis (MG) is a rare, chronic, neuromuscular autoimmune disease mediated by pathogenic autoantibodies that target structural components of the neuromuscular junction, impairing neuromuscular transmission and leading to muscle weakness and fatigue.2-4 Many patients initially present with symptoms affecting only the eye muscles (i.e., ocular MG). Approximately 85% of patients go on to develop generalized myasthenia gravis (gMG), with weakness affecting the neck, trunk, limbs, and bulbar and respiratory muscles. Patients with gMG experience symptoms that negatively impact health-related quality of life (HRQoL).5 The disease has a fluctuating natural history; MG exacerbation (an increase in symptoms in patients who were previously asymptomatic or minimally symptomatic, defined as a ≥ 3-point worsening in Quantitative Myasthenia Gravis [QMG] score versus baseline) and myasthenic crisis (muscle weakness causing life-threatening difficulties with breathing and swallowing and requiring ventilator support) can occur gradually or without warning.6
Approximately 85% of patients with gMG are acetylcholine receptor (AChR) antibody (Ab) seropositive (Ab+); as many as 15% of patients with gMG are seronegative for AChR-Ab (Ab–).7,8 An estimated 1% to 10% of patients do not have AChR antibodies but do have autoantibodies against muscle-specific kinase (MuSK) antibody seropositive (MuSK-Ab+) or autoantibodies against lipoprotein receptor-related protein-4 (LRP4) Ab seropositive (LRP4-Ab+), which also lead to a decrease in AChRs.3 The Myasthenia Gravis Foundation of America (MGFA) classification system is a tool used to categorize gMG based on clinical features and/or disease severity.9 The classification ranges from Class I (i.e., ocular weakness only), through Class II, Class III and Class IV (representing patients with gMG with mild, moderate, and severe muscle weakness, respectively9), to Class V (defined by intubation, with or without mechanical ventilation, except when employed during routine postoperative management, or myasthenic crisis). The incidence of MG in Canada is estimated at 23 cases per 1 million person-years, with a prevalence of 32 cases per 100,000 adults (0.032%) in Canada.10-12 Thus, there are approximately 8,121 patients with MG across the CADTH-participating drug programs (0.032% × 25,376,703 adult patients in CADTH-participating drug programs in 2023). Approximately 85% of adults with MG are anticipated to progress to gMG, which corresponds to approximately 6,903 adult patients with gMG in Canada.
The clinical experts that CADTH consulted for this review indicated that the goal of treatment in patients with gMG is to reduce disease symptoms and adverse effects of MG therapy and to allow the patient to function as they would normally with good HRQoL. Other goals of treatment include avoiding MG exacerbations and myasthenic crisis, minimizing hospitalizations and intensive care unit (ICU) admissions, and reducing the numbers and doses of therapies (e.g., especially corticosteroid use) required for symptom control. The available main therapies for gMG include acetylcholinesterase inhibitors (AChEIs), corticosteroids, nonsteroidal immunosuppressive therapies (NSISTs), rituximab, intravenous immunoglobulin (IVIg), plasma exchange (PE) or plasmapheresis (PP), and terminal complement inhibitors (i.e., ravulizumab and eculizumab). According to the clinical experts consulted by CADTH for this review, the first-line standard of care (i.e., the conventional therapy) for MG are AChEIs, corticosteroids, and NSISTs (e.g., azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine, tacrolimus, methotrexate). Mild to moderate gMG (MGFA Class II or IIIa) is initially treated symptomatically with AChEIs (usually pyridostigmine);13 the onset of benefit occurs in hours to days. If this provides insufficient symptom relief, immunosuppressive therapy (IST) with corticosteroids (usually prednisone) is administered;14 maximal responses typically occur 2 to 6 months later, after which slow tapering of corticosteroids is begun. In patients who do not respond to corticosteroids, who have significant comorbidities such that long-term corticosteroid treatment is contraindicated, or whose doses of corticosteroids cannot be tapered, treatment with NSISTs15 and/or immunomodulatory drugs, including rituximab, may be initiated.16 The clinical experts stated that the onset of benefit from NSISTs occurs in months to years (approximately 9 to 18 months for azathioprine and mycophenolate mofetil). While rituximab has not been approved as a gMG treatment by Health Canada, it is considered a treatment option in Canada for patients with refractory gMG who are AChR-Ab+, according to surveys conducted with clinical experts.16 According to the clinical experts, in patients with moderate to severe gMG, especially those who have respiratory or bulbar weakness, IVIg, PE, or PP may be administered17,18 in addition to rituximab, either at the time of IST initiation or to treat MG exacerbation or myasthenic crisis.
As MG symptoms improve, doses of AChEIs, corticosteroids, and NSISTs are reduced and the frequency of IVIg, PE, or PP are reduced until the minimal maintenance therapy required for remission is identified. Patients with refractory gMG who are AChR-Ab+ may be candidates for the complement inhibitor eculizumab. While eculizumab received a recommendation for reimbursement with conditions in 2020,19 price negotiations concluded without an agreement in December 2022.20 A survey of 7 expert clinicians from across 6 provinces indicated that ravulizumab would be another option, if it were to be approved and funded. Ravulizumab would be a treatment option for patients who have an inadequate response to conventional therapy.18 In April 2023, CADTH issued a draft “do not reimburse” recommendation for ravulizumab in this indication.21 The clinical experts consulted by CADTH for this review emphasized that most patients with gMG (more than 80%) respond well to currently available treatments; although these cannot cure the disease, excellent symptom control is achieved in most patients and prognosis is generally good in terms of muscle strength and function as well as HRQoL. Despite treatment with conventional therapy (AChEIs, corticosteroids, and/or NSISTs), many patients continue to experience disease burden and symptoms that impact their HRQoL22-27 and experience treatment-related side effects that may be severe.
Efgartigimod alfa is a first-in-class human immunoglobulin G1 (IgG1) antibody fragment crystallizable (Fc)-fragment that blocks the neonatal Fc receptor (FcRn).28,29 Efgartigimod alfa is supplied as a 20 mg/mL solution, with 10 mg/kg administered as an IV infusion over 1 hour once weekly for 4 doses (i.e., weeks 0 to 3). In patients weighing 120 kg or more, the recommended dose of efgartigimod alfa is 1,200 mg (3 vials) per infusion. Efgartigimod alfa reduces the levels of pathogenic IgG autoantibodies.29 Efgartigimod alfa received a Health Canada Notice of Compliance for the treatment of adult patients with gMG who are AChR-Ab+ on September 19, 2023. The sponsor’s reimbursement request is that efgartigimod alfa be reimbursed as an add-on therapy for adult patients with AChR-Ab+ gMG whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs, which is a subgroup of the approved Health Canada indication.1
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Input
CADTH received 1 patient group submission from the Muscular Dystrophy Canada (MDC). MDC identified and contacted adults living with MG and invited them to participate in a survey and semistructured interviews. Respondents indicated that MG has a significant impact on their productivity, levels of fatigue, energy levels, quality of sleep, respiratory health, mobility, strength, independence, relationships and social participation, eyes and vision, speech, and swallowing. They also explained that the impact of MG extends beyond physical symptoms and affects their mental health, quality of life, and the well-being of their families.
Some of the respondents indicated they feel that their lungs are weaker, they had to go on a ventilator in ICU, choking on food or saliva interferes with breathing, they cannot even walk inside their own house, they always keep a walker or cane nearby because they never knew when the MG would flare up, they cannot sleep at night because of aches, and they are unable to drive. Some indicated that they had slurred speech, frequently go cross-eyed, their double vision interferes with reading, and they have experienced multiple acute hospitalizations.
When asked about how their MG is being managed with available treatments, 3 main themes emerged from the analysis: negative experiences with steroids (e.g., adverse effects; costs), the slow onset of medication effects, and a feeling of trial and error with medications. Regarding improved outcomes, the patient group identified 3 aspects of MG that they wanted better controlled: decreased intensity of exacerbations and side effects, maintenance of independence, and fewer serious hospital admissions. The method, duration, and frequency, and convenience of treatment, as well as the cost are very important to the patients and caregivers. They preferred less travel, fewer hospital visits, and less invasive methods of treatment. HRQoL was noted as a key priority over convenience of a drug.
Respondents stated that they would be willing to deal with side effects of medications if these aspects of MG were better controlled. They also stated that although their current medications decreased the number of exacerbations, they do not have an impact on overall quality of life. They expected new treatments to help them become independent, stop the myasthenic crises, address the respiratory and general weakness, be easier to swallow (for pills), reduce pain, not lead to diabetes, be target treatment for MG instead of general immunosuppression, be less expensive, work quickly, be a single daily dose in the morning.
One respondent had received Vyvgart as a participant in a clinical trial and explained that this medication replaced their need for IVIg, the effects appeared quicker compared with other therapies, the infusion time was less than expected, treatment was received less frequently compared with other therapies, and they experienced fewer side effects compared with other therapies. This patient respondents highlighted that while diarrhea was a problem and not unique to Vyvgart, it was manageable after the first cycle of treatment.
All the respondents had experienced diagnostic blood testing, and many had undergone single-fibre electromyography to confirm diagnosis. A total of 80% of the respondents reported difficulty receiving a diagnosis. Based on early findings of the MG journey mapping project, MDC reported 7 years from time of first bothersome symptom to diagnosis, with a range up to 23 years. According to MDC, the majority of respondents found the process of testing and diagnosis cost-effective but lengthy, with many missed opportunities, delayed diagnosis, misdiagnosis (such as stroke and Bell palsy), and costs incurred. Those who were diagnosed during a crisis or hospitalization reported a smooth diagnosis (25%).
Clinician Input
Input From Clinical Experts Consulted by CADTH
The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of gMG.
The clinical experts indicated that approximately 90% of patients respond to current treatments; however, response is often partial, meaning that there are still symptoms that affect QoL and function. According to the clinical expert, besides prednisone and rescue treatments, ISTs take very long to act (e.g., azathioprine takes at least a year). The clinical experts explained that this means that patients may be exposed to higher doses of steroids for longer periods, and experience persistent symptoms for longer, before even knowing whether this medication will be effective or not. Current treatments are nontargeted, causing overall more diffuse immunosuppression, and there is an increased risk of cancer with long-term use. Also, the risk of all steroid-related adverse events (AEs) increases with prolonged doses. The clinical experts also indicated that the fraction of patients with refractory gMG varies according to definition, although a reasonable estimate would be 10% to 20% of the total population. According to the clinical experts consulted by CADTH for this review, patients with gMG usually start with pyridostigmine, but most patients will need disease-modifying treatment with immunosuppressants, most commonly prednisone. Depending on severity, age, and comorbidities, an NSIST (e.g., azathioprine, mycophenolate mofetil, and tacrolimus) may be started early after diagnosis, or later (for example, unable to reduce dose of steroids). Some patients with severe disease at onset (e.g., crisis or severe symptoms) may receive a rescue treatment such as IVIg or PE early to have fast improvement while immunosuppression begins. Few patients receive chronic IVIg or PE, and some of these patients are dependent on these treatments. According to the clinical experts, the treatment goals is to achieve minimal symptoms or remission, with the fewest AEs from treatments. Patients express a need to improve the ability to perform their daily life activities, reduce fatigue, and improve their ability to care for their family and work or home obligations.
The clinical experts stated that efgartigimod alfa has a specific mechanism of action related to the pathophysiology of MG (reduction of IgG levels, including AChR antibodies). Efgartigimod alfa reduces levels of IgG, but it does not affect the process of producing AChR antibodies. The clinical experts indicated that they do not foresee efgartigimod alfa being used as first-line treatment; rather, efgartigimod alfa would be suitable for individuals without adequate response to available treatments, those who are dependent on IVIg or PE, or those with very severe disease to bridge the gap of delayed action of standard immunosuppressive therapies. The clinical experts noted that patients should have received standard conventional treatments first because standard conventional treatments will be satisfactory in a large number of patients. The clinical experts also stated that most treatments for patients with gMG are given off label because of the lack of randomized controlled trial (RCT) evidence (e.g., prednisone, azathioprine, mycophenolate mofetil, tacrolimus, and rituximab). The clinical experts noted that IVIg or PE used as a rescue treatment is different from IVIg or PE used chronically (i.e., maintenance therapies, once a month); very few patients use IVIg or PE chronically. According to the clinical experts, eculizumab or ravulizumab are used too rarely at present to include them as comparators. One clinical expert indicated that, if cost was not an issue, one could argue that efgartigimod alfa could be tried as an initial therapy in patients for whom pyridostigmine alone was ineffective; however, they noted that the cost is likely to be a major barrier.
The clinical experts indicated that efgartigimod alfa will provide a new treatment for patients with gMG who are AChR-Ab+ and probably for patients who are MuSK-Ab+. However, whether patients with gMG who are AChR-Ab– would respond to efgartigimod alfa is unknown because few patients who are AChR-Ab– were included in the ADAPT trial.
The clinical experts indicated that, in most clinics (not academic), patients are not given standardized assessments. In academic settings, clinicians use validated measures. The clinical experts noted that the Myasthenia Gravis Activities of Daily Living (MG-ADL) scale used in trials is easy to use and can be easily incorporated into routine clinical practice and all settings (community, hospital, and academic). The clinical experts recommended using the MG-ADL for patients with active treatment at all visits, to be able to follow the clinical course. The clinical experts stated that an improvement (reduction) of 2 points is considered significant. Apart from symptom scores, overall function, and ability to return to work are also assessed. The clinical experts expressed that the ability to reduce or stop chronic use of corticosteroids, IVIg, or PE is an important outcome when considering the use of efgartigimod alfa. They explained that some patients are dependent on chronic IVIg or PE, weaning off these treatments is important. The clinical experts also highlighted that reduction or avoidance of hospitalization due to MG is an important outcome.
Frequency of assessments depends on symptoms and patient stability. Patients whose symptoms are well-controlled are typically seen by clinicians every 6 months, but can be seen more frequently (e.g., every 2 to 3 months) in case of worsening health, new medications, and so on. For efgartigimod alfa, most responses were fast, but a small proportion of responders lagged, and response could be seen at the second cycle. Therefore, the clinical experts suggested 2 to 3 months may be needed to assess response to cycle 1 and/or to assess for need of another cycle. An assessment at 6 months may be needed to determine which patients do not respond to treatment. For those who do respond, subsequent assessments could then be done every 3 to 6 months to determine if new cycles are needed.
The clinical experts indicated that efgartigimod alfa should be discontinued if a patient has no response to treatment (i.e., no improvement in symptoms and function), experiences a severe AE (e.g., severe infusion reaction), needs rescue treatment (IVIg or PE, or increased dose of steroids), or is unable to reduce chronic use of corticosteroids, IVIg, or PE. The clinical experts indicated that patients should be under the care of a neurologist with experience in diagnosing and treating MG, usually a neuromuscular specialist. The infusion itself can be arranged at infusion clinics.
Clinician Group Input
CADTH received 1 clinician group submission from the Neuromuscular Disease Network for Canada (NMD4C).
NMD4C stated that conventional treatment options for gMG have been based on symptomatic therapy, short-term rescue immunotherapy and long-term IST. Moreover, nonspecific immunosuppressants have been only partially effective and many patients do not attain stable remission, with 10% to 20% of patients not responding or intolerant to these drugs.
According to NMD4C, some of the unmet needs of the standard treatments are side effects, lack of effectiveness for all patients, long periods of treatment, and transient effectiveness. Another unmet need in this field is the lack of therapeutic options for seronegative patients.
NMD4C noted that patients who are AChR-Ab+ will most likely respond to the drug under review. Patients with MuSK antibodies and those who are double seronegative (i.e., AChR-Ab– and MuSK-Ab–) might respond. Patients who worsen quickly, particularly those who experience a myasthenic MG crisis are most in need of an intervention that works quickly, but patients who have symptoms restricted to only the ocular muscles are unlikely to require such rapid intervention with the drug under review. To identify the patients best suited for treatment with the drug under review, clinician examination and judgment supplemented by assessment of MG activities of daily living using scales that reflect severity of disease, such as the QMG score, Myasthenia Gravis Impairment Index (MGII), and the single simple question (SSQ). If scales are not available, then antibody testing needs to be done; but according to the clinician group, this can be delayed. It is not clear at this point how to predict which patients are more likely to respond to treatment, except by determining the presence of AChR antibodies.
NMD4C indicates that diagnosis of double seronegative patients is an issue since cluster antibodies to both acetylcholine and MuSK may be present but need to be tested specifically, which can take weeks.
The clinician group indicated that to determine patients’ response to therapy, scales such as the MG-ADL, QMG, MGII, and SSQ at 2 and 4 weeks are required and after that the assessment should be based on the patient’s status. The clinician group noted that a clinically meaningful response to treatment used in the clinical trials is 2 or more points on the ADL and 3 or more points on the QMG. For the SSQ, the clinician group suggested that levels above 72% indicate general satisfaction. In case of lack of response, discontinuation of treatment should be considered.
The clinician group mentioned that the usual Ig treatment for MG can be effective, but it places a significant burden on the Canadian health care system and supplies can be at risk in situations such as during a pandemic. They think the drug under review is likely to replace Ig therapies.
In summary, the clinician group’s input is aligned with the input provided by the clinical experts consulted by CADTH.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Clinical Evidence
Systematic Review
Description of Studies
One phase III, double-blind, placebo-controlled RCT (ADAPT, N = 167)30 is included in the systematic review.31 The objective of the ADAPT trial was to evaluate the efficacy and safety of efgartigimod alfa added on to conventional therapy versus placebo added to conventional therapy in adult patients with gMG whose symptoms persisted despite a stable dose of standard-of-care treatment (concomitant gMG treatment) with AChEIs, corticosteroids, and/or NSISTs. All patients had MGFA Class II to IV gMG and MG-ADL total score of 5 or more. The mean age was 44.7 years in efgartigimod alfa group and 49.2 years in placebo group, and most patients were white (83.1% in efgartigimod alfa group and 87.5% in placebo group). In the patient population who were AChR-Ab+, 129 (100%) patients had received 1 prior therapy, 124 (96.1%) had received 2 prior therapies, and 102 (79.1%) had received 3 or more prior therapies. The majority had previously received 2 or more (93.4%) or 3 or more (77.2%) different classes of conventional therapy medication (any combination of AChEIs, corticosteroids, and/or NSISTs at the physician’s discretion). In the patient population who were AChR-Ab+, patients who received 3 classes of prior therapy (steroid plus NSIST plus AChEI), 41 (63%) were in the efgartigimod alfa group and 37 (58%) were in the placebo group. Eleven (16.9%) patients in the efgartigimod alfa group and 23 (36%) in the placebo group had received any 2 of the 3 prior therapies (i.e., steroid, NSIST, and/or AChEI). In addition, of the patients who were AChR-Ab+, 63% had prior gMG treatment failures (also known as refractory gMG)32 and 37% had did not have treatment failure but responded inadequately to the existing standard-of-care gMG therapy.
Patients were randomized 1:1 to receive efgartigimod alfa or a matching placebo in cycle 1 (i.e., for first 8 weeks) followed by an individualized treat-as-needed regimen based on the patients’ MG-ADL response. All patients received a stable concomitant treatment during the trial. The primary outcome of the study was the percentage of patients with AChR-Ab+ who were “MG-ADL responders” (defined in the study as a patient with a ≥ 2-point improvement [reduction] in MG-ADL score) in the first treatment cycle. Key secondary outcomes included percentage of MG-ADL responders in cycle 1 in the overall population (i.e., AChR-Ab+ and AChR-Ab–), percentage of time patients with AChR-Ab+ showed a clinically meaningful improvement (CMI) in MG-ADL score (≥ 2-point reduction) up to day 126, time from week 4 to qualify for re-treatment in the AChR-Ab+ population; percentage of early MG-ADL responders in cycle 1 in the AChR-Ab+ population (i.e., MG-ADL ≥ 2 points occurred by week 2), and change from cycle baseline in MG-ADL total score in cycle 1 and cycle 2. Changes from cycle baseline in HRQoL (MG-QoL15r and EQ-5D visual analogue scale [VAS]) in cycle 1 and cycle 2 were assessed as tertiary or exploratory outcomes. Post hoc analysis was performed for gMG hospitalization, gMG exacerbation, and gMG crisis. It should also be noted that, although the ADAPT trial duration was designed for 26 weeks, the primary, key secondary, and the HRQoL outcomes at the end of the study (i.e., week 26) were not assessed. Instead, the outcomes were assessed at the end of cycle 1 and cycle 2.
Efficacy Results
Patients Who Are AChR-Ab+
Myasthenia Gravis Activities of Daily Living
MG-ADL responders during cycle 1 and cycle 2: MG-ADL responders during cycle 1 in patients who were AChR-Ab+ was the primary outcomes in the ADAPT trial. There were 38% (95% confidence interval [CI], 22% to 56%) more patients in the efgartigimod alfa group (those with AChR-Ab+ randomized to receive efgartigimod alfa) than in the placebo group achieved an improvement in MG-ADL of 2 points or more during cycle 1. The between-group difference was considered clinically meaningful by the clinical experts consulted by CADTH.
Various post hoc subgroup analyses were conducted for MG-ADL responders during cycle 1. Consistent with the primary analysis, these results demonstrated that efgartigimod alfa produces improvements in MG-ADL response compared to placebo, regardless of prior therapies, concomitant therapies, disease duration, thymectomy, and prior treatment failure;1,30,32,33 however, the trial was not powered to detect subgroup differences. In terms of the MG-ADL responders, similar benefit was observed in cycle 2.
Early MG-ADL responders: Early MG-ADL responders (i.e., those who responded at week 2 of cycle 1) in patients who were AChR-Ab+ was assessed as a fifth key secondary outcome. Because the statistical testing hierarchy was broken at the fourth secondary end point (i.e., time to qualify for re-treatment), the percentage of patients in the AChR-Ab+ population who were early MG-ADL responders was not statistically tested based on statistical plan in the protocol. Nevertheless, a higher proportion of patients in the efgartigimod alfa group than in the placebo group achieved an MG-ADL improvement of 2 or more points at week 2 (between-group difference = 31.9%; 95% CI, not reported [NR]) The between-group difference was considered clinically meaningful by the clinical experts consulted by CADTH. The percentage of MG-ADL early responders during cycle 2 was not assessed and not reported in the sponsor’s evidence summary.
Percentage time of the MG-ADL CMI up to day 126: Among patients who were AChR-Ab+, the percentage of time with a CMI in the MG-ADL total score up to day 126 was assessed as a third key secondary outcome and was included in the hierarchy test to control for type I error. According to the clinical experts, the percentage of time with a CMI in the MG-ADL total score in the efgartigimod alfa group was clinically meaningfully longer (between-group difference = 22.07%; 95% CI, 10.94% to 33.18%; P = 0.0001) than that in the placebo group.
MG-ADL change from cycle baseline: During cycle 1 and cycle 2, the changes from cycle baseline in MG-ADL score were assessed as exploratory outcomes for patients who were AChR-Ab+. At week 4 of cycle 1, the reduction (improvement) of MG-ADL total score in the efgartigimod alfa group was larger than that in the placebo group (between-group difference = –2.84; 95% CI, –3.8 to –1.9; P < 0.0001). This was assessed as an exploratory outcome with no multiplicity adjustment (i.e., it was not included in the hierarchy test); therefore, there is an increased risk of type I error. However, the findings were aligned with the early responder analysis and considered clinically meaningful by the clinical experts consulted by CADTH. It should be noted that the maximum MG-ADL change from cycle baseline with efgartigimod alfa appeared to occur at approximately week 4 of the cycle. The magnitude of the improvement and the comparative benefit of efgartigimod alfa compared with placebo tended to smaller at the end of the cycle. Similar results were observed in cycle 2.
Time to re-treatment: Time to qualify for re-treatment for patients who were AChR-Ab+ was assessed as the fourth key secondary outcome. The median time to qualify for re-treatment in the efgartigimod alfa group (median = 35 days; 95% CI, 29 to 43 days) was numerically but not significantly greater than the time in the placebo group (median = 8 days; 95% CI, 1 day to 30 days; log-rank P = 0.2604). The statistical hierarchy test was broken at this point. Since the week 4 visit, the proportion of patients who were qualified for re-treatment appeared to be similar in both groups (between-group difference: 1.4%; 95% CI, NR). The clinical experts that CADTH consulted for this review indicated that the results likely show that approximately half of patients would need re-treatment around week 6 of the treatment cycle.
Disease Severity (Assessed With QMG)
QMG responder during cycle 1: The percentage of QMG responders among patients who were AChR-Ab+ was assessed as the first key secondary outcome. It was reported that 49.0% (95% CI, 34.5% to 63.5%) more patients in the efgartigimod alfa group compared with the placebo group achieved a QMG response in cycle 1 (odds ratio [OR] = 7.1, 95% CI, 3.24 to 16.49; P < 0.0001). According to the clinical experts, this benefit of treatment with efgartigimod alfa compared with placebo was considered clinically meaningful.
Health-Related Quality of Life
The HRQoL (i.e., MG-QoL15r and EQ-5D VAS) was assessed as an exploratory outcome among patients who were AChR-Ab+. The change from cycle baseline in MG-QoL15r and EQ-5D VAS scores were assessed for cycle 1 and cycle 2. At week 4 of cycle 1, the reduction (improvement) in MG-QoL15r score in the efgartigimod alfa group was greater than that in the placebo group (between-group difference = –5.45; 95% CI, –7.221 to –3.685; P < 0.0001). The increase (improvement) in EQ-5D VAS score in the efgartigimod alfa group was greater than that in the placebo group (between-group difference = 13.28; 95% CI, 8.32 to 18.24; P < 0.0001). Because HRQoL was assessed as an exploratory outcome with no multiplicity adjustment (i.e., it was not included in the statistical hierarchy test), there is an increased risk of type I error; however, the results provide supportive evidence. Although there is no known minimally important difference (MID) for either the MG-QoL15r or EQ-5D VAS among patients with gMG, the clinical experts considered the results to be clinically meaningful. It should be noted that both maximum MG-QoL15r and EQ-5D VAS improvement with efgartigimod alfa occurred at approximately week 4 of the cycle. The magnitude of the improvement and the comparative benefit of efgartigimod alfa compared with placebo tended to be smaller at the end of the cycle. Similar results were observed in cycle 2.
Other Clinical Outcomes (MG Hospitalizations, MG Exacerbations, and MG Crisis)
In patients who were AChR-Ab+, during the 26-week double-blind period, the event rates for MG hospitalization and MG crisis were low in both groups. MG exacerbations were identified in 17 patients (26.2%) in the efgartigimod alfa group and 27 patients (44.3%) in the placebo group. The between-group absolute risk difference was –18.2 (95% CI, NR). The results of MG hospitalization, MG exacerbations, and MG crisis were based on post hoc analyses. Therefore, the results for these outcomes were inconclusive.
Harms Results
Reduction of side effects was identified in the patient input for this review as of interest for patients with gMG. The ADAPT trial, including its randomized controlled period and open-label extension, provided relevant information regarding the safety profile of efgartigimod alfa in the treatment of gMG. However, it did not provide direct comparative evidence regarding the adverse effects of efgartigimod alfa versus other active gMG therapies. In the AChR-Ab+ population, during the randomized controlled period, the proportion of patients with treatment-emergent adverse events (TEAEs) in the efgartigimod alfa group appeared to be similar to that in the placebo group (75.4% versus 84.4%, respectively). The proportion of patients with serious adverse events (SAEs) was low in both groups and appeared lower in the efgartigimod alfa group than in the placebo group (4.6% versus 9.4%) in the ADAPT trial. Withdrawals due to AEs occurred in similar proportions in both the efgartigimod alfa and placebo groups (3.1% and 4.7%, respectively) in the ADAPT trial. No deaths were reported during the double-blind period; however, the length of follow-up in the trial may not have been long enough to assess this outcome with certainty. The main notable harms (i.e., AEs of special interest [AESIs] for this review) were in the Medical Dictionary for Regulatory Activities (MedDRA, version 23.0) system organ class of “infections and infestations,” which were reported in a higher proportion of patients in the efgartigimod alfa group than in the placebo group (44.6% versus 34.4%). No meningococcal infections were reported. According to the clinical experts CADTH consulted for this review, the TEAEs reported in the ADAPT trial were expected and commonly seen in existing immunosuppressive treatments, such as complement C5-inhibitor treatment of gMG.
Critical Appraisal
Appropriate methods of randomization, blinding, and allocation concealment were reported. Outcomes were assessed using validated scales incorporating physician and patient assessments, and end points requiring a combination of CMI and sustained effect. However, minimally important between-group differences, that is, the thresholds used for the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) for all outcomes, were not available. Therefore, clinical experts’ opinion informed the thresholds to determine whether the between-group differences observed for each outcome were clinically meaningful. Appropriate statistical methods were used in the ADAPT trial. Multiplicity adjustments were made for the primary and 5 key secondary outcomes to control for family-wise type I error (the probability of making more than 1 type I error). Overall, the ADAPT trial was relatively well designed; however, a potentially key limitation of the ADAPT trial includes notable imbalance of baseline disease characteristics between groups. For example, the proportions of patients who had a total MG-ADL score of 10 or higher, prior combination use of steroid plus AChEI, and prior thymectomy were imbalanced between groups. Furthermore, the proportions of patients who used concomitant AChEI or concomitant steroid plus AChEI were also imbalanced between the 2 treatment groups. Whether these baseline imbalances introduced bias is uncertain. However, the clinical experts consulted by CADTH for this review indicated that these observed imbalances were unlikely to have significantly affected the study results. The efficacy outcome assessments for patients who were AChR-Ab– were assessed as sensitivity or subgroup analysis. In addition, the ADAPT trial was a placebo-controlled trial. The comparative efficacy information comparing efgartigimod alfa with existing gMG therapies (e.g., AChEIs, steroids, NSIST, IVIg, PE, complement C5 inhibitors) are unknown. Furthermore, MG-ADL total score change from cycle baseline, HRQoL, and all outcomes examined in cycle 2 were assessed as either tertiary or exploratory outcomes, which were not included in the statistical hierarchy test and were not controlled for type I error. Therefore, the results of all those tertiary and exploratory analysis should interpreted with the consideration of the lack of control for the type I error. Finally, reduction of steroid use and reduction of high-dose steroid use are treatment goals for efgartigimod alfa; however, these outcomes could not be assessed because of the study design — the concomitant treatments were not allowed to change unless it was used for rescue. The impact of efgartigimod alfa on changes in MG medications could not be evaluated because this was not allowed as per the study protocol.
According to clinical experts CADTH consulted for this review, the population included in the ADAPT trial well reflects the patients who experience unmet needs in the treatment of gMG in Canadian clinical settings. However, patients with gMG MGFA Class I (ocular MG) and Class V were excluded in the ADAPT trial. Whether the ADAPT trial findings can be generalized to patients with MGFA Class I (ocular MG) or Class V is uncertain. The clinical experts CADTH consulted for this review indicated that efgartigimod alfa will provide a new treatment for patients with gMG who are AChR-Ab+ and probably for those who are MuSK-Ab+. The clinician group input for this review also indicated that patients with MuSK antibodies might respond to efgartigimod alfa. It is uncertain whether the findings from the ADAPT trial can be generalized to patients who are AChR-Ab–, MuSK-Ab–, or double negative (AChR-Ab– and MuSK-Ab–). The number of patients who were AChR-Ab– was relatively small, and the ADAPT trial was not powered for testing the statistically significant between-group difference. Therefore, the comparative efficacy of efgartigimod alfa versus placebo for patients who are AChR-Ab– is inconclusive. In addition, although 6 (3.6%) patients in the ADAPT trial were MuSK-Ab+, there was no sensitivity or subgroup analysis for patients who were MuSK-Ab+. The comparative efficacy of efgartigimod alfa versus placebo in patients who are MuSK-Ab+ is also unknown. Therefore, findings for the overall population were mainly driven by the patients who were AChR-Ab+.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to informing CADTH’s expert committee deliberations. A final certainty rating was determined as outlined by the GRADE Working Group.34,35 Following the GRADE approach, evidence from RCTs was initially rated as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: activities of daily living (proportion of MG-ADL responders in cycle 1 and cycle 2; proportion of early MG-ADL responder in cycle 1; mean proportion of time with a clinically meaningful (≥ 2-point) improvement in MG-ADL (follow-up: 126 days); time to qualify for re-treatment (up to 168 days); MG-ADL total score change from cycle baseline at week 4 of cycle 1 and cycle 2; disease severity (measured using the QMG); HRQoL (MG-QoL15r and EQ-5D-5L VAS) change from cycle baseline at week 4 in cycle 1 and cycle 2); and other clinical outcomes (MG hospitalization, MG exacerbation, and MG crisis by week 26) and notable harms (i.e., infections and infestations).
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For this review, the target of the certainty of evidence assessment was based on the presence of absence of a clinically important effect, as informed by MIDs suggested by the sponsor and agreed upon by the clinical experts consulted by CADTH for this review (for change from baseline in MG-ADL score and QMG score), or by thresholds suggested by the clinical experts (for all other outcomes).
Results of GRADE Assessment
Table 2: Summary of Findings for Efgartigimod Alfa Versus Placebo for Patients With AChR-Ab+
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Efgartigimod alfa | Difference | |||||
Activities of daily living | |||||||
MG-ADL score (0 [best] to 24 [worst]) | |||||||
Responders (≥ 2-point reduction for 4 consecutive weeks) during cycle 1 Follow-up: 8 weeks | 129 (1 RCT) | OR = 4.95 (2.21 to 11.53) | 30 per 100 | 68 per 100 (NR) | 38 more per 100 (22 to 56 more per 100) | Moderatea | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important increase in the proportion of MG-ADL responders during the first treatment cycle when compared with placebo. |
Early responders (≥ 2-point reduction during first 2 weeks) during cycle 1 Follow-up: 2 weeks | 129 (1 RCT) | OR = 3.94 (1.77 to 9.15) | 25 per 100 | 57 per 100 (NR) | 32 more per 100 (NR)b | Moderatec | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important increase in the proportion of early MG-ADL responders during the first treatment cycle when compared with placebo. |
LSM change from cycle baseline at week 4 of cycle 1 (points) Follow-up: 4 weeks | 129 (1 RCT) | NA | –1.3 | –4.1 (–5.0 to –3.2) | –2.8 (–3.8 to –1.9) | Moderated | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important improvement in activities of daily living during the first treatment cycle when compared with placebo. |
Responders (≥ 2-point reduction for 4 consecutive weeks) during cycle 2e Follow-up: 8 weeks of cycle 2 | 94 (1 RCT) | OR = 8.19 (2.88 to 25.73) | 26 per 100 | 71 per 100 (NR) | 45 more per 100 (NR)b | Lowf | As an add-on to conventional therapy, efgartigimod alfa may result in a clinically important increase in the proportion of MG-ADL responders during the second treatment cycle when compared with placebo. |
LSM change from cycle baseline at week 4 of cycle 2 (points)e Follow-up: 8 weeks of cycle 2 | 98 (1 RCT) | NA | –0.4 | –4.4 (–5.4 to –3.3) | –3.9 (–5.1 to –2.8) | Moderateg | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important improvement in activities of daily living during the second treatment cycle when compared with placebo. |
Mean % time with a clinically meaningful (≥ 2-point) improvement Follow-up: 126 days | 129 (1 RCT) | NA | 26.7 | 48.7 (36.5 to 60.9) | 22.1 (10.9 to 33.2) | Moderateh | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important increase in the percentage of time with a CMI in MG-ADL total score when compared with placebo. |
Disease severity | |||||||
QMG responders (≥ 3 points reduction for 4 consecutive weeks) during cycle 1 Follow-up: 8 weeks | 129 (1 RCT) | OR = 10.84 (4.18 to 31.20) | 14 per 100 | 63 per 100 | 49 more per 100 (35 to 65 more per 100) | Moderatei | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important increase in the proportion of QMG responders during the first treatment cycle when compared with placebo. |
Time to re-treatment | |||||||
Qualified for re-treatment Follow-up: 168 days | 129 (1 RCT) | NA | 89 per 100 | 88 per 100 | 1 less per 100 (NR)b | Moderatej | As an add-on to conventional therapy, efgartigimod alfa likely results in little to no difference in the proportion of patients who qualify for re-treatment when compared with placebo. |
HRQoL | |||||||
LSM change from baseline in MG-QoL15r score (0 [best] to 30 [worst]) at week 4 of cycle 1 (points)e Follow-up: 4 weeks | 123 (1 RCT) | NA | –1.76 | –7.21 (–8.80 to –5.63) | –5.45 (–7.22 to –3.69) | Moderatek | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important improvement in HRQoL as measured by the MG-QoL15r during the first treatment cycle when compared with placebo. |
Mean change from baseline in EQ-5D VAS (0 [worst] to 100 [best]) at week 4 of cycle 1 (points)e Follow-up: 4 weeks | 123 (1 RCT) | NA | 2.76 | 16.04 (NR) | 13.28 (8.32 to 18.24) | Moderatel | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important improvement in HRQoL as measured by the EQ-5D VAS during the first treatment cycle when compared with placebo. |
LSM change from baseline in MG-QoL15r score (0 [best] to 30 [worst]) at week 4 of cycle 2 (points)e Follow-up: 4 weeks of cycle 2 | 89 (1 RCT) | NA | 0.35 | –5.09 (–6.73 to –3.47) | –5.45 (–7.28 to –3.62) | Lowm | As an add-on to conventional therapy, efgartigimod alfa may result in a clinically important improvement in HRQoL as measured using the MG-QoL15r during the second treatment cycle when compared with placebo. |
Mean change from baseline in EQ-5D-5L VAS (0 [worst] to 100 [best]) at week 4 of cycle 2 (points)e Follow-up: 4 weeks of cycle 2 | 89 (1 RCT) | NA | 1.92 | 14.16 (9.69 to 18.63) | 12.24 (7.33 to 17.16) | Lown | As an add-on to conventional therapy, efgartigimod alfa may result in a clinically important improvement in HRQoL as measured using the EQ-5D VAS during the second treatment cycle when compared with placebo. |
Other clinical outcomes | |||||||
MG-related hospitalizationse Follow-up: 26 weeks | 129 (1 RCT) | NR | 5 per 100 | 0 (NR) | 5 less per 100 (NR)b | Very lowo | As an add-on to conventional therapy, the evidence is very uncertain about the effect of efgartigimod alfa on the number of hospitalizations when compared with placebo. |
MG exacerbationse Follow-up: 26 weeks | 129 (1 RCT) | NR | 44 per 100 | 26 per 100 (NR) | 18 less per 100 (NR)b | Lowp | As an add-on to conventional therapy, efgartigimod alfa may result in a clinically important reduction in MG exacerbations when compared with placebo. |
MG crisise Follow-up: 26 weeks | 129 (1 RCT) | NR | 1 per 100 | 0 (NR) | 1 less per 100 (NR)b | Very lowo | As an add-on to conventional therapy, the evidence is very uncertain about the effect of efgartigimod alfa on MG crises when compared with placebo. |
Adverse events of special interest | |||||||
Infections Follow-up: 26 weeks | 129 (1 RCT) | NR | 34 per 100 | 45 per 100 (NR) | 10 more per 100 (7 less to 27 more per 100) | Moderateq | As an add-on to conventional therapy efgartigimod alfa likely results in a clinically important increase in the proportion of patients experiencing 1 or more infection when compared with placebo. |
AChR-Ab+ = acetylcholine receptor antibody positive; CI = confidence interval; CMI = clinically meaningful improvement; HRQoL = health-related quality of life; LSM = least squares mean; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item; NA = not applicable; NR = not reported; OR = odds ratio; QMG = Quantitative Myasthenia Gravis; RCT = randomized controlled trial; VAS = visual analogue scale.
a–1 level for serious imprecision. The 95% CI excludes the threshold of a 20% difference between groups, as informed by the clinical experts; however, the sample size and number of events does not meet the optimal information size.
bUpon request, the sponsor did not provide the 95% CI for the between-group difference (indicated that it was not calculable).
c–1 level for serious imprecision. No CI was available for judging the precision of the comparative effect estimate. The number of events does not meet the optimal information size.
d–1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a suggested minimally important difference of 2 points, as defined in the trial and agreed upon by the clinical experts.
eIn the trial, statistical testing for these efficacy outcomes was not adjusted for multiplicity. The results are considered as supportive evidence.
f–1 level for serious risk of bias. Not all patients completed the second treatment cycle, so prognostic balance between the groups cannot be ensured. –1 level for serious imprecision. No CI was available for judging precision of the comparative effect estimate. The number of events does not meet the optimal information size.
g–1 level for serious risk of bias. Not all patients completed the second treatment cycle, so prognostic balance between the groups cannot be ensured.
h–1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a 10% to 15% difference between groups, as informed by the clinical experts.
i–1 level for serious imprecision. The 95% CI excludes the threshold of a 20% difference between groups, as informed by the clinical experts; however, the sample size and number of events does not meet the optimal information size.
j–1 level for serious imprecision. No CI was available for judging the precision of the comparative effect estimate. The number of events does not meet the optimal information size.
k–1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a difference of 5 points between groups, as informed by the clinical experts.
l–1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a difference of 10 points between groups, as informed by the clinical experts.
m–1 level for serious risk of bias. Not all patients completed the second treatment cycle, so prognostic balance between the groups cannot be ensured. –1 level for serious imprecision. The 95% CI includes the possibility of a clinically important effect favouring efgartigimod alfa, based on a threshold of a difference of 5 points between groups, as informed by the clinical experts.
n–1 level for serious risk of bias. Not all patients completed the second treatment cycle, so prognostic balance between the groups cannot be ensured. –1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a difference of 10 points between groups, as informed by the clinical experts.
o–1 level for serious risk of bias. The analyses of these outcomes were undertaken post hoc, so there is risk of bias in the selection of the reported result. –2 levels for very serious imprecision. No CI was available for judging the precision of the comparative effect estimate. The number of events does not meet the optimal information size; there were very few or no events in either group.
p–1 level for serious risk of bias. The analyses of these outcomes were undertaken post hoc, so there is risk of bias in the selection of the reported result. –1 level for serious imprecision. No CI was available for judging the precision of the comparative effect estimate. The number of events does not meet the optimal information size.
q–1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a 10% difference between groups, as informed by the clinical experts.
Sources: Clinical Study Report;30 the sponsor’s submission1; and sponsor-provided additional information.36,37 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
Description of Studies
The ADAPT+ study (ARGX-113-1705) is a long-term, single-arm, open-label, multicentre, phase III follow-on study of patients who enrolled in the ADAPT study (ARGX-113-1704; NCT03669588). The primary objective was to evaluate the long-term safety and tolerability of efgartigimod alfa in the AChR-Ab+ subgroup, and the secondary objective was to evaluate safety and tolerability in the overall population (AChR-Ab+ and AChR-Ab–). Efficacy data were collected as exploratory end points. The ADAPT+ study was conducted at 51 sites, including 41 sites in 14 countries or regions that had 1 or more patient roll over from the ADAPT study. Data were collected over a 3-year period in 2 sequential parts (part A: 1 year, part B: 2 years maximum). Part B was added as a protocol amendment to ensure accessibility to efgartigimod alfa until it became commercially available or available through an expanded access program. Results of the long-term extension phase up to 14 cycles (ADAPT+) are also presented in this report. The ADAPT+ trial was still ongoing at the time of this review; therefore, the long-term efficacy and safety outcomes of ADAPT+ were based on interim analysis (IA4 and IA5). MG-ADL, QMG, and safety outcomes were assessed in the long-term extension study. At the data cut-off (June 30, 2022), 151 patients had rolled over from the ADAPT trial, regardless of treatment or placebo group, into ADAPT+ and 145 patients had received at least 1 partial or complete dose of efgartigimod alfa in ADAPT+.
Efficacy Results
In terms of MG-ADL response (up to 14 cycles) and QMG response (up to 7 cycles), evidence from the long-term, open-label, extension (ADAPT+) trial appeared consistent with those from the randomized controlled period. Patients who switched from placebo to efgartigimod alfa experienced numeric improvements from baseline in MG-ADL and QMG in each cycle. However, interpretation of these data was limited by the open-label and descriptive nature of the extension study.
Harms Results
Safety data from the long-term extension phase appeared consistent with that observed in the double-blind phase with no new safety signals reported.
Critical Appraisal
Internal Validity
The ADAPT+ study was limited by its open-label and noncomparative design because it was uncertain whether the results observed were attributable to the effects of the drugs including other treatments or the natural history of disease. Furthermore, the missing outcome data and the small sample size toward the end of ADAPT+ made it difficult to draw any firm conclusions on the efficacy and safety of efgartigimod alfa. Due to its open-label nature of ADAPT+, the subjective outcomes (e.g., rates of self-reported AEs) were at risk of bias and potentially in favour of the intervention (i.e., efgartigimod alfa). It is noteworthy that ADAPT+ had fewer scheduled visits for outcome assessments compared with the ADAPT study. ADAPT+ only collected MG-ADL data at week 3 of each study; however, the maximum clinical effect in ADAPT was observed at weeks 4 to 5 of a cycle. Furthermore, the longer-term safety and tolerability profile of efgartigimod alfa treatment was hard to determined due to the rates of AEs and SAEs may be underestimated with the less frequent assessment schedule. In ADAPT+, efficacy was assessed as an exploratory outcome using the patient-reported MG-ADL and physician-reported QMG scales. Patients were to remain on their stable dose and regimen of concomitant gMG treatment during part A of ADAPT+. Given the presence of rollover effects, efficacy results related to part B may be difficult to interpret due to that changes were permitted in part B, including changes in the type, dose, or regimen of the concomitant gMG treatment as well as the additional use of other treatments. Therefore, the confounding effects of other therapies cannot be eliminated in part B. In terms of outcome measures, some important long-term outcomes reported by patients and clinicians were not measured (e.g., HRQoL, exacerbations) in the ADAPT+.
External Validity
Because the patients who took part in the open-label, long-term safety extension phase were originally from the pivotal ADAPT trial, it is reasonable to expect that the same limitations to generalizability are relevant to the extension phase. Given the nature of noncomparative study design, it is not possible to compare the effectiveness and tolerability of efgartigimod alfa as an add-on treatment of gMG against other add-on treatments (e.g., IVIg).
Indirect Comparisons
Description of Studies
To date, there have been no clinical trials directly comparing the efficacy of efgartigimod alfa with other treatments in patients diagnosed with gMG. Due to this gap in evidence, the sponsor submitted an indirect treatment comparison (ITC) that included a systematic literature review (SLR)38 and a network meta-analysis (NMA)1 that provide comparative evidence of the efficacy of efgartigimod alfa relative to ravulizumab and IVIg. The eligible interventions for the ITC were limited to those used in Canada for the treatment of gMG to ensure that the comparators were relevant to the Canadian settings. After feasibility assessment, the following 5 studies were considered eligible to be included in the NMA: 2 studies comparing efgartigimod alfa with placebo,28,39 2 studies comparing IVIg with placebo,40,41 and 1 study comparing ravulizumab with placebo.42 All NMAs were performed using a Bayesian framework. Placebo was chosen as the reference treatment for all analyses, given its presence as an anchor treatment in all studies and the outcomes assessed in the network. The clinical end points used for ITC estimates included change from baseline in MG-ADL and QMG as these were the most consistently reported outcomes in all studies included in the NMA. Primary analyses were performed at the primary assessment time points for all included studies, ranging from 4 to 26 weeks, and sensitivity analyses were performed at or plus or minus 2 weeks of week 4, which was the primary assessment time point in the ADAPT trial.
As the sponsor’s reimbursement request is limited to patients in the AChR-Ab+ subpopulation, comparators relevant to that group were used in the primary ITC analysis.
Efficacy
Primary Analyses
The mean differences for change from baseline in MG-ADL were –2.64 (95% credible interval [CrI], –4.16 to –1.12) for efgartigimod alfa versus IVIg, and –0.91 (95% CrI, –2.25 to 0.39) for efgartigimod alfa versus ravulizumab. The mean differences for change from baseline in QMG were –4.39 (95% CrI, –6.95 to –1.81) for efgartigimod alfa versus IVIg, and –2.89 (95% CrI, –4.72 to –1.12) for efgartigimod alfa versus ravulizumab. A change of 2 points in the MG-ADL score and 3 points in the QMG score was estimated to be the threshold of clinical significance in patients with MG.
Sensitivity analyses results for change from baseline in MG-ADL and QMG at or plus or minus 2 weeks of week 4 were consistent with results of the primary analyses.
Additional Analyses
The mean differences for change from baseline in MG-ADL were –2.64 (95% CrI, –4.18 to –1.12) for efgartigimod alfa versus IVIg, –0.92 (95% CrI, –2.25 to 0.43) for efgartigimod alfa versus ravulizumab, and –1.93 (95% CrI, –3.87 to 0.07) for efgartigimod alfa versus rituximab. The mean differences for change from baseline in QMG were –4.39 (95% CrI, –7.01 to –1.83) for efgartigimod alfa versus IVIg, –2.89 (95% CrI, –4.72 to –1.06) for efgartigimod alfa versus ravulizumab, and –2.71 (95% CrI, – 5.56 to –0.2) for efgartigimod alfa versus rituximab.
Harms
No analysis of harms was reported in the sponsor-submitted ITC report.
Critical Appraisal
The SLR used to identify relevant studies was methodologically sound in terms of the sponsor using a comprehensive literature search strategy as well as performing study selection, data extraction, risk-of-bias assessments in duplicate, and providing a list of excluded studies and justifying the exclusions. However, it was unclear in the ITC report whether the feasibility assessment was carried out by a single or multiple assessors. By conducting a feasibility assessment, the sponsor excluded all head-to-head trials, including those comparing efficacy of IVIg treatment versus PE, which may have reduced the information to inform the NMA. The risk of bias of included studies in the SLR was assessed per individual study; however, this may differ according to the outcomes of each study. Analyses were run using a Bayesian framework with placebo as the reference treatment, which was deemed appropriate. Change from baseline in MG-ADL and QMG scores were considered the best source of comparative efficacy data for this NMA, although these outcomes were not primary or secondary end points of the ADAPT trial. The studies that did not report on MG-MDL or QMG were excluded even if they reported other relevant outcomes, which may have biased the results, although the extent of bias is uncertain. All trials included in the ITC had sufficiently similar study designs and a common comparison group (placebo). However, there were some important differences between the trials included in the NMA that increase the uncertainty of the analyses. All included studies employed a dosing schedule involving spaced infusions, but only ADAPT used individual patient response to determine subsequent cycles of treatment. The studies included in the ITC analyses ranged in follow-up time from 4 to 26 weeks. All studies allowed the use of concomitant standard-of-care treatments (e.g., corticosteroids, NSISTs), but detailed information on the breakdown of actual concomitant medications used was not available. In many studies, baseline data were not reported consistently, such as for MGFA at baseline, use of steroids or NSISTs at baseline, disease duration, and history of thymectomy. The primary analyses conducted at the primary assessment time point for all trials could be biased against ADAPT, as they could exclude the best responders to efgartigimod alfa, whereas ITCs conducted at week 4 only could be biased against any treatments that demonstrated improved responses over time. Therefore, sensitivity analyses were performed at or plus or minus 2 weeks of week 4 to improve the robustness of the ITCs and align with the primary assessment time point of the ADAPT trial.
The results were reported as mean differences and 95% Crls. The evidence is imprecise in the effect estimates from the NMA due to the sparseness of data, with wide Crls. In addition, heterogeneity between the included studies can potentially introduce bias into the study estimates observed between the comparators. Because all comparator studies were performed with patients who were exclusively AChR-Ab+, all ITC analyses included only patients from the AChR-Ab+ subpopulation, which aligns with the reimbursement request submitted by the sponsor and the approved Health Canada indication. Another important limitation of the presented ITC is the lack of safety and HRQoL data. The results of this ITC are highly uncertain given the inconsistency between trials with respect to dosing regimen (individualized dosing for efgartigimod alfa versus continuous dosing for the comparators), variability in eligibility criteria, and study follow-up times. The ITC estimates were too imprecise to draw a conclusion about the comparative effect of efgartigimod alfa relative to alternative treatments on change from baseline in MG-ADL and QMG.
Conclusions
One double-blind, RCT of patients with gMG was included in this review.
Evidence from the ADAPT trial showed that, compared with placebo, treatment with efgartigimod alfa as an add-on to standard conventional therapy likely results in a clinically meaningful benefit in terms of the proportion of MG-ADL and QMG responders, and HRQoL (as assessed using the MG-QoL15r and EQ-5D VAS) after cycle 1 of treatment relative to placebo among adult patients with AChR-Ab+ gMG whose symptoms persist despite a stable dose of standard-of-care (concomitant gMG treatment) treatment with AChEIs, corticosteroids, and/or NSISTs (moderate certainty). Similar benefit was observed in cycle 2, although there is less certainty in these results because not all randomized patients participated in this cycle. Efgartigimod alfa likely results in a clinically important increase in the percentage of time with a meaningful MG-ADL improvement compared with placebo (moderate certainty). Maximum benefit appeared to occur at approximately 4 weeks of each cycle. Efgartigimod alfa may result in a clinically important reduction in MG exacerbations relative to placebo (low certainty); the results were inconclusive for MG-related hospitalizations and MG crises due to low numbers of events reported for these outcomes. The safety profile of efgartigimod alfa reported in the ADAPT trial was considered as expected and commonly seen in existing gMG therapies. In terms of MG-ADL and QMG scores and safety profile, evidence from the long-term, open-label, extension (ADAPT+) trial appeared consistent with those from the randomized controlled period in the patients who were AChR-Ab+. However, interpretation of the long-term data was limited by the open-label and descriptive nature of the extension study. The results of the sponsor-submitted NMA suggest that relative to IVIg and ravulizumab, efgartigimod alfa may provide a benefit with respect to change in MG-ADL and QMG scores; however, the 95% CrIs for the effect estimates included the possibility of trivial effects (i.e., only small, nonclinically important differences between groups) and no difference (in the case of change in MG-ADL score relative to ravulizumab). No difference in efficacy in terms of change from baseline in MG-ADL and QMG scores could be concluded for efgartigimod alfa relative to rituximab due to wide 95% CrIs (which included the possibility of clinically important benefit favouring efgartigimod alfa), and methodological limitations. There is no evidence for the effect of efgartigimod alfa on HRQoL or harms outcomes relative to any other active treatment.
Introduction
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
MG is a rare, chronic, neuromuscular autoimmune disease mediated by pathogenic autoantibodies that target structural components of the neuromuscular junction, impairing neuromuscular transmission, and leading to muscle weakness and fatigue,2-4 extensively disrupting the ability to perform normal daily activities and profoundly impairing HRQoL. Many patients initially present with symptoms affecting only the eye muscles (i.e., ocular MG). Approximately 85% of patients go on to develop gMG, with generalized weakness affecting the neck, trunk, limbs, and bulbar and respiratory muscles. The characteristic feature of gMG is fluctuating fatigable muscle weakness, although there is heterogeneity in the specific muscles that are affected. Patients with gMG experience symptoms that negatively impact HRQoL.5 The disease has a fluctuating natural history, with MG exacerbation (an increase in symptoms in patients who were previously asymptomatic or minimally symptomatic, defined as a ≥ 3-point worsening in QMG score versus baseline) and myasthenic crisis (muscle weakness causing life-threatening difficulties with breathing and swallowing that requires ventilator support) can occur gradually or without warning.6 The serological profile of gMG can be defined by the presence of autoantibodies to particular receptors, which can affect treatment decisions and disease prognosis.43 Approximately 85% of patients with gMG are AChR-Ab+ and as many as 15% are AChR-Ab–.7,8 An estimated 1% to 10% of patients do not have AChR antibodies but do have autoantibodies against MuSK-Ab+ or autoantibodies against LRP4-Ab+, which also lead to a decrease in AChRs.3 The MGFA classification system is a tool used to categorize gMG based on clinical features and/or disease severity.44 The classification ranges from Class I (i.e., ocular weakness only) to Class V (i.e., patients require intubation, with or without mechanical ventilation, except when employed during routine postoperative management or myasthenic crisis); MGFA Class II to V are used to characterize gMG. Class II, Class III, and Class IV represent patients with mild, moderate, and severe muscle weakness, respectively.9 The incidence of MG in Canada is estimated at 23 cases per 1 million person-years, with a prevalence of 32 cases per 100,000 adults (0.032%) in Canada.10-12 Thus, there are approximately 8,121 patients with MG across the CADTH-participating drug programs (0.032% × 25,376,703 adult patients in CADTH-participating drug programs in 2023). Among adults with MG, approximately 85% are anticipated to progress to gMG, which corresponds to approximately 6,903 adult patients with gMG in Canada.
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
The clinical experts CADTH consulted for this review indicated that the goal of treatment in most patients with gMG is to reduce disease symptoms as well as adverse effects of MG therapy to allow the patient to function as they would normally with good HRQoL. Other goals of treatment include avoiding MG exacerbations and myasthenic crises, minimizing hospitalizations and ICU admissions, and reducing the numbers and doses of therapies (especially corticosteroid use) required for symptom control. The available main therapies of gMG include AChEIs, corticosteroids, NSIST, rituximab, IVIg, PE or PP, and terminal complement inhibitors (i.e., ravulizumab and eculizumab). According to the clinical experts consulted by CADTH for this review, the first-line standard of care (i.e., the conventional therapy) for MG are AChEIs, corticosteroids, and NSISTs (e.g., azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine, tacrolimus, and methotrexate). Mild to moderate gMG (MGFA Class II or IIIa) is initially treated symptomatically with AChEIs (usually pyridostigmine);13 the onset of benefit occurs in hours to days. If this provides insufficient symptom relief, IST with corticosteroids (usually prednisone) is administered;14 maximal responses typically occur 2 to 6 months later, after which slow tapering of corticosteroids is begun. In patients who do not respond to corticosteroids, who have significant comorbidities such that long-term corticosteroid treatment is contraindicated, or whose doses of corticosteroids cannot be tapered, treatment with NSISTs15 and/or immunomodulatory drugs, including rituximab, may be initiated.16 The clinical experts stated that the onset of benefit from NSISTs occurs in months to years (approximately 9 to 18 months for azathioprine and mycophenolate mofetil). Rituximab is a monoclonal antibody directed against the CD20 receptor on B-lymphocytes. Rituximab has not been approved as a gMG treatment by Health Canada. International guidelines have identified evidence for the use of rituximab in patients with MuSK-Ab+ gMG who have an insufficient response to other immunotherapies; and it is considered a treatment option in Canada for patients with refractory gMG who are AChR-Ab+, according to surveys conducted with clinical experts.16 According to the clinical experts, for patients with moderate to severe gMG, especially those who have respiratory or bulbar weakness, IVIg, PE, or PP may be administered17,18 in addition to rituximab, either at the time of IST initiation or to treat MG exacerbation or myasthenic crisis. Critical care, including ICU admission and ventilator support, may be required for patients experiencing myasthenic crisis. Thymectomy may also be considered in a small group of patients.16 As MG symptoms improve, doses of AChEIs, corticosteroids, and then other ISTs are reduced and the frequency of IVIg, PE, or PP is also reduced until the minimal maintenance therapy required for remission is identified. Patients with refractory gMG who are AChR-Ab+ may be candidates for the complement inhibitor eculizumab. While eculizumab received a recommendation for reimbursement with conditions in 2020,19 price negotiations concluded without an agreement in December 2022.20 A survey of 7 expert clinicians from across 6 provinces indicated that eculizumab would be another treatment option if it were to be approved and funded, while ravulizumab would be a treatment option for patients who have an inadequate response to conventional therapy.18 In April 2023, CADTH issued a draft “do not reimburse” recommendation for ravulizumab in this indication.21 The clinical experts consulted by CADTH for this review emphasized that most patients with gMG (more than 80%) will respond well to currently available treatments: although these cannot cure the disease, excellent symptom control is achieved in most patients and prognosis is generally good in terms of muscle strength and function as well as HRQoL. Despite treatment with conventional therapy (AChEIs, corticosteroids, and/or NSISTs), many patients continue to experience disease burden and symptoms that impact their HRQoL,22-27 and treatment-related side effects may be severe. Approximately 25% of patients with gMG are able to achieve pharmacological remission (i.e., symptom control with conventional therapy), but only 8% achieve clinical remission (i.e., no symptoms off-treatment for more than a year),45 which highlights the need for additional options to improve disease control. A survey of 7 clinicians across 6 provinces18 indicated that the characteristics of patients enrolled in the ADAPT trial were generally aligned with the characteristics of patients with gMG in their practice. Furthermore, the clinicians indicated that they saw the greatest benefit of efgartigimod alfa treatment for patients with AChR-Ab+ who require chronic IVIg or PE add-on treatment. Therefore, based on these opinions and the clinical evidence from ADAPT and ADAPT+, the sponsor proposes that efgartigimod alfa be used as an add-on treatment for adult patients with AChR-Ab+ gMG whose symptoms persist despite adequate treatment with conventional therapy (any combination of AChEIs, corticosteroids, and/or NSISTs, at the treating physician’s discretion).1
Drug Under Review
Key characteristics of efgartigimod alfa and other treatments available for adult patients with gMG are summarized in Table 3. Efgartigimod alfa is a first-in-class human IgG1 antibody Fc-fragment that blocks FcRn.28,29 Efgartigimod alfa is supplied as a 20 mg/mL solution, and 10 mg/kg is administered as an IV infusion over 1 hour once weekly for 4 doses (i.e., weeks 0 to 3). In patients weighing 120 kg or more, the recommended dose of efgartigimod alfa is 1,200 mg (3 vials) per infusion. Efgartigimod alfa binds to FcRn, resulting in the reduction of circulating IgG including autoantibodies. Efgartigimod alfa does not affect the levels of other immunoglobulins (IgA, IgD, IgE, or IgM) or of albumin. IgG autoantibodies are the underlying cause of the pathogenesis of MG. They impair neuromuscular transmission by binding to AChRs, MuSK, and LRP4. Efgartigimod alfa reduces the levels of pathogenic IgG autoantibodies.29
Efgartigimod alfa received a Health Canada Notice of Compliance for the treatment of adult patients with gMG who are AChR-Ab+ on September 19, 2023. The sponsor’s reimbursement request is that efgartigimod alfa be reimbursed as an add-on therapy for adult patients with gMG who are anti-acetylcholine receptor (AChR) antibody positive, whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs, which is a subgroup of the approved Health Canada indication.1
Table 3: Key Characteristics of Efgartigimod Alfa and Other Drugs Used for the Treatment of gMG
Characteristics | Efgartigimod alfa | Ravulizumab | Eculizumab | AChEIs (e.g., pyridostigmine) | Immunosuppressive therapy (e.g., corticosteroids, steroid-sparing agents, rituximab) | IVIg | PE or PP |
|---|---|---|---|---|---|---|---|
Mechanism of action | Human IgG1 antibody crystallizable fragment engineered for increased affinity to FcRn | Terminal complement inhibitor | Terminal complement inhibitor | Cholinesterase inhibitor | Suppression of production of AChR antibodies | Unknown | Removal of AChR antibodies |
Relevant indicationa | For the treatment of adult patients with gMG who are AChR-Ab+ | For the treatment of adult patients with gMG who are AChR-Ab+ | Adult patients with gMG | For the symptomatic treatment of myasthenia gravis | NA | NA | NA |
Route of administration | IV | IV | IV | Orally | Orally, IV | IV | IV |
Recommended dose | The recommended dosage of efgartigimod alfa is 10 mg/kg administered as an IV infusion over 1 hour once weekly for 4 weeks. In patients weighing ≥ 120 kg, the recommended dose of efgartigimod alfa is 1,200 mg (3 vials) per infusion. Administer subsequent treatment cycles based on clinical evaluation. The frequency of efgartigimod alfa treatment cycles may vary by patient | 2,400 mg to 3,000 mg (loading); 3,000 mg to 3,600 mg every 8 weeks starting 2 weeks after loading dose (maintenance)b | 900 mg weekly for 4 weeks followed by 1,200 mg 1 week later (loading); 1,200 mg every 2 weeks (maintenance)c | 60 mg per day to 1,500 mg per day | Various | 1 g/kg to 2 g/kg administered over 2 to 5 days | 1 plasma volume to 1.5 plasma volumes daily, usually 5 to 6 exchanges |
Serious adverse effects or safety issues | Infections: As efgartigimod alfa causes transient reduction in IgG levels, the risk of infections may increase. The most common infections observed in clinical trials were upper respiratory tract infections | Infections, including serious meningococcal infections | Infections, including serious meningococcal infections | Increased salivation and fasciculation, diarrhea, nausea, vomiting | Infections, infusion reactions | Infusion reactions | Infection, bleeding, thrombosis, transfusion reactions |
Ab+ = antibody positive; AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; gMG = generalized myasthenia gravis; FcRn = neonatal crystallizable fragment receptor; IgG1 = immunoglobulin G subclass 1; IVIg = intravenous immunoglobulin; NA = not applicable; PE = plasma exchange; PP = plasmapheresis.
aRelevant Health Canada–approved indications.
bSupplemental ravulizumab doses of 1,200 mg to 1,800 mg are given after PE or PP, and supplemental doses of 600 mg are given after IVIg.
cSupplemental eculizumab doses of 300 mg to 600 mg are given after PE or PP.
Sources: Product monographs for efgartigimod,29 ravulizumab,46 eculizumab,47 and pyridostigmine;48 and the sponsor’s submission.1
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient input(s) received by CADTH have been included in the Stakeholder section of this report.
CADTH received 1 patient group submission from MDC. MDC is a health charity that supports people affected by muscular dystrophies and related muscle diseases in Canada. MDC’s mission is to enhance the lives of those impacted by neuromuscular disorders by continually working to provide ongoing support and resources while searching for a cure through well-funded research.
MDC identified and contacted adults living with MG to participate in a survey and semistructured interviews. Surveys were shared with members via e-blasts, personalized invites, and online patient groups. MDC also conducted a journey mapping project for adults living with MG via virtual interviews, roundtable sessions, surveys, and HRQoL measures (EQ VAS, EQ-5D, MG-ADL, MG-QoL). MDC collected information from 108 individuals impacted by MG, including 39 males and 69 females between ages 21 to 76 from all provinces in Canada. MDC used a qualitative descriptive approach, employing a constant comparison technique, to produce a thematic analysis.
Respondents indicated that MG has a significant impact on productivity, level of fatigue, energy levels, quality of sleep, respiratory health, mobility, strength, independence, relationships and social participation, eyes and vision, speech, and swallowing. They also explained that the impact of MG extends beyond physical symptoms and affects their mental health, quality of life, and the well-being of their families.
Some of the respondents explained the impact of MG using the following phrases:
I had to retire early
I am fearful of the next MG crisis
I am on disability leave
MG has financial impacts
I feel useless at home
feeling tired
need a full day to recover if I do too much
after 10 minutes of housework then have to rest
feel my lungs are weaker
had to go on a ventilator in ICU
choking on food or saliva interferes with breathing
can’t even walk inside my house
always keep a walker or cane nearby because you never know when the MG will flare up
can't sleep at night because it aches
unable to drive
on bad days I feel like a prisoner in my own house
I miss out on socializing
quite disabled and dependent on others
depressing knowing that there is no cure and that this is my way of life now
scared to be alone
have to puree my foods
my voice sounds very strained after a short conversation
I had slurred speech as though I was intoxicated
I frequently go cross-eyed
double vision interferes with reading
multiple acute hospitalizations.
When the respondents were asked how their MG was being managed with the available treatments, 3 main themes emerged from the analysis: negative experiences with steroids (e.g., adverse effects; costs); the slow onset of medication effects (e.g., example, 1 patient respondent indicated, “My doctor told me it could take 6, maybe even 9, months for the treatment to take effect”; another patient respondent stated, “Imagine living half the year waiting for a drug to show benefit and then to find out you need to be switched to something else”); and a feeling of going through a process of trial and error with medications. Although the treatment had a positive impact on health outcomes, the participants had concerns about the long-term and sustained benefits of supportive treatments.
Regarding the improved outcomes, the patient group identified 3 aspects of MG that they wanted better controlled: decreased intensity of disease exacerbations and medication side effects, maintenance of independence, and fewer serious hospital admissions. The method, duration, and frequency of medication administration and the convenience and cost of treatment were considered very important to the patients and caregivers. They preferred less travel, fewer hospital visits, and less invasive methods of treatment. HRQoL was noted to be a key priority over convenience of the drug. Respondents stated that they would be willing to deal with side effects of medications if these aspects of MG were better controlled. They also stated that although their current medications decreased the number of exacerbations, they do not have an impact on overall quality of life. They expected new treatments to help them become independent, stop the myasthenic crises, address the respiratory and general weakness, be easier to swallow (for pills), reduce pain, not lead to diabetes, be target treatment for MG instead of generally immunosuppressive, be less expensive, work quickly, be a single daily dose in the morning.
One respondent had received Vyvgart as a participant in a clinical trial and explained that this medication replaced the need for IVIg, the effects appeared to be quicker compared with other therapies, the infusion time was shorter than expected, treatment was received less frequently compared with other therapies, and they experienced fewer side effects compared with other therapies. This respondent highlighted that although diarrhea was a problem and not unique to Vyvgart, it was manageable after the first cycle of treatment.
All the respondents had undergone diagnostic blood testing, and many had undergone single-fibre electromyography to confirm diagnosis. A total of 80% of the respondents reported difficulty receiving a diagnosis. Based on early findings of the MG journey mapping project, MDC reported 7 years to be the average time from first bothersome symptom to diagnosis, with the range up to 23 years. According to MDC, the majority of respondents found the process of testing and diagnosis cost-effective but lengthy, with many missed opportunities, delays in diagnosis, and misdiagnoses (such as stroke and Bell palsy), resulting in incurred costs. Those who were diagnosed during a MG crisis or hospitalization (25% of respondents) reported a smooth diagnosis.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise on the diagnosis and management of the condition for which the drug is indicated. Clinical experts are an essential part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of gMG.
Unmet Needs
The clinical experts indicated that approximately 90% of patients respond to current treatments, but response is often partial, meaning that there are still symptoms that affect quality of life and function. Besides prednisone and rescue treatments, immunosuppressive therapies take a very long time to act (e.g., azathioprine takes at least a year). That means that patients may be exposed to higher doses of steroids for longer periods, and experience persistent symptoms for long periods, before even knowing whether a medication will be effective. Current treatments are nontargeted, causing overall more diffuse immunosuppression, and there is an increased risk of cancer with long-term use. Also, there is an increased risk of steroid-related AEs with prolonged doses, which requires periodic monitoring. The clinical experts also indicated that the fraction of patients considered to have refractory gMG varies according to the definition, but that 10% to 20% of the total population is a reasonable estimate. The risk of generalized immunosuppression is principally related to infection (whether azathioprine affects risk of malignancy is debated, and the long-term data are limited).
Place in Therapy
According to the clinical experts consulted by CADTH for this review, patients with gMG usually start with pyridostigmine (symptomatic treatment) but most patients will need disease-modifying treatment with immunosuppression, most commonly prednisone. Depending on severity, age, and comorbidities, an NSIST (e.g., azathioprine, mycophenolate mofetil, tacrolimus) may be started soon after diagnosis or later (e.g., inability to reduce dose of steroids). Some patients with severe disease at onset (e.g., crisis or severe symptoms) may receive a rescue treatment such as IVIg or PE early for fast improvement while immunosuppression kicks in. A few of patients receive chronic IVIg or PE, but some are dependent on these. The treatment goals are to achieve minimal symptoms or remission, with the fewest AEs from treatments. Patients express the need to improve their ability to perform their daily life activities, reduce fatigue, and be able to care for their family, work, or carry out home obligations.
The clinical experts stated that efgartigimod alfa has a specific mechanism of action related to the pathophysiology of MG (reduction of IgG levels, including AChR antibodies), but that the same could be said about pyridostigmine. Efgartigimod alfa reduces levels of IgG but does not affect the process of producing AChR antibodies. The clinical experts indicated that they do not foresee efgartigimod alfa being used as first-line treatment. Rather it would be suitable for individuals without response to available treatments, for those dependent on IVIg or PE, or for those with very severe disease, to bridge the delayed action of standard immunosuppressive therapies. Patients should receive standard treatments first, as these will be satisfactory in a large number of patients. The clinical experts also stated that most treatments for patients with gMG (for example, prednisone, azathioprine, mycophenolate mofetil, tacrolimus, rituximab, and others) are given off label given the lack of RCT evidence. IVIg or PE used for rescue therapy is different from IVIg or PE used chronically (i.e., maintenance therapies, once a month); only a small number of patients use IVIg or PE chronically. Eculizumab or ravulizumab are used too rarely at present to include them as comparators. One clinical expert indicated that it could be argued that efgartigimod alfa be tried as an initial therapy by patients for whom pyridostigmine alone was ineffective; however, the cost is likely to be a major barrier.
Patient Population
The clinical experts indicated that efgartigimod alfa will provide a new treatment for patients with gMG who are AChR-Ab+ and probably for patients who are MuSK-Ab+. However, whether patients with gMG who are seronegative (e.g., AChR-Ab–, MuSK-Ab–) respond to efgartigimod alfa is unknown because few seronegative patients were included in the ADAPT trial. One clinical expert indicated that, based on efgartigimod alfa’s mechanism of action, AChR-Ab+, MuSK-Ab+, and likely “seronegative” patients would be expected to respond (“seronegative” patients are generally assumed to have as-yet-unidentified autoantibodies).
Assessing the Response Treatment
The clinical experts indicated that in most clinics (not academic) patients are not given standardized assessments. In academic settings, clinicians use validated measures. The MG-ADL used in trials is easy to use, and can be easily incorporated into routine clinical practice and all settings. The clinical experts recommended using the MG-ADL for patients receiving active treatment at all visits, as this enables following the clinical course. An improvement (reduction) of 2 points is considered significant. Apart from using symptom scores to assess improvements, overall function, ability to return to work, and so on, are also assessed. The ability to reduce or stop chronic use of corticosteroids, IVIg, or PE is an important outcome when considering the use of efgartigimod alfa. Some patients are dependent on chronic IVIg or PE, so it is important to be able to wean off these. Some patients have multiple hospitalizations due to MG, so reducing or avoiding these is also important. The frequency of assessments depends on patients’ symptoms or stability. Patients whose symptoms are well-controlled are typically seen by clinicians every 6 months but can be seen more frequently (e.g., every 2 to 3 months) in case of worsening health, new medications, and so ono. For efgartigimod alfa, most responders were fast, but a small proportion of responders lagged, and response could be seen at the second cycle. Therefore, 2 to 3 months to assess response to cycle 1 or the need for a new cycle is reasonable. Patients would then need to be seen at 6 months to determine nonresponders. For responders, subsequent assessments every 3 to 6 months could be done to determine if new cycles are needed.
Discontinuing Treatment
The clinical experts indicated that efgartigimod alfa should be discontinued if there is no response to treatment (no improvement in symptoms/function); if a severe AE (e.g., severe infusion reactions) occurs; if rescue treatment (IVIg or PE, or increased dose of steroids) is needed; or if chronic use of corticosteroids, IVIg, or PE cannot be reduced.
Prescribing Considerations
The clinical experts indicated that patients should be under the care of a neurologist with experience diagnosing and treating MG, usually a neuromuscular specialist. The infusion itself can be arranged at infusion clinics.
Additional Considerations
None.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group input(s) received by CADTH have been included in the Stakeholder section of this report.
CADTH received 1 clinician group submission from NMD4C, which was launched in January 2020 with funding from the Canadian Institutes of Health Research and MDC. The mission of NMD4C is to improve the care, research, and treatment of neuromuscular diseases for all people living in Canada. Its vision is to be a comprehensive, inclusive, open, and enduring network through which stakeholders can share expertise and data and collaborate on joint activities and research for the benefit of all patients in Canada.
Clinicians with experience in treating gMG were asked to contribute to this submission. The information presented in this submission was gathered from one-to-one discussions with the lead author and through group discussions.
NMD4C stated that conventional treatment options for gMG have been based on symptomatic therapy, short-term rescue immunotherapy, and long-term immunosuppressive therapy. Moreover, nonspecific immunosuppressants have been only partially effective and many patients do not attain stable remission, with 10% to 20% not responding or intolerant to these drugs.
According to NMD4C some of the unmet needs of the standard treatments are side effects, lack of effectiveness for all patients, long periods of treatment, and transient effectiveness. Another unmet need in this field is the lack of therapeutic options for seronegative patients.
NMD4C noted that patients with AChR antibodies in their system will most likely respond to the drug under review. Patients with MuSK antibodies and those who are double seronegative might respond. Patients who get worse quickly, particularly patients with MG crisis, are most in need of an intervention that works quickly, but patients who have symptoms restricted to their ocular muscles are unlikely to require such rapid intervention with the drug under review. The patients best suited for treatment with the drug under review are identified by clinician examination and judgment supplemented by assessment of activities of daily living using scales that reflect severity of disease, such as the QMG, MGII, and SSQ. If these are not available, then antibody testing needs to be done, although this can be delayed, according to the clinician group. Currently, there is no clear way at this point to predict which patients are more likely to respond except the presence of AChR antibodies.
NMD4C indicates that diagnosis of double seronegative patients is an issue because cluster antibodies to both acetylcholine receptor and MuSK may be present but need to be tested specifically, which can take weeks.
The clinician group indicated that to reduce the risk of underdiagnosis, serological testing as well as single-fibre electromyography and repetitive nerve stimulation studies are necessary. To determine patients’ response to therapy, scales such as the MG-ADL, QMG, MGII, and SSQ at 2 and 4 weeks are required; after that, the assessment should be based on the patient’s status. The clinician group noted that a clinically meaningful response to treatment used in the clinical trials is 2 or more points on the ADL and 3 or more points on the QMG scale. For the SSQ, the clinician group suggested that levels above 72% indicate general satisfaction. In case of lack of response, discontinuation of treatment should be considered.
The clinician group mentioned that usual Ig treatment for MG can be effective but place a significant burden on the Canadian health care system and that supplies can be at risk in situations such as a pandemic. They think the drug under review is likely to replace Ig therapies.
In summary, the clinician group’s input is aligned with the input provided by the clinical experts consulted by CADTH.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Issues with the choice of comparator in the submitted trial(s):
| The complement inhibitors (eculizumab and ravulizumab) are mechanistically different from efgartigimod alfa and would likely have a different role in therapy. Therefore, the lack of direct comparisons of efgartigimod alfa with these 2 is not really a problem. |
Considerations for initiation of therapy | |
Prior therapies required for eligibility: The requested indication for efgartigimod alfa is as an add-on therapy to conventional therapy, which may include AChEIs, CSs, and/or NSISTs in patients who are AChR-Ab+. Although the sponsor included rituximab in the list of comparators, it was not included in the studied indication because it is only used in patients who are not AChR-Ab+. Patients treated with either rituximab or eculizumab within 6 months of screening were also excluded from the study. The sponsor envisions the place in therapy of efgartigimod alfa to be considered as an add-on as an alternative to immunoglobulins after use of NSISTs and/or CSs as depicted in Figure 1. The indication is for addition of efgartigimod alfa to 1 or a combination of the 3 conventional therapy classes. The ADAPT trial allowed inclusion of patients on any combination of conventional gMG treatment, which was limited to AChEIs, steroids, and NSISTs and did not require the patients to have received or discontinued use of any specific treatment. It is unclear if a patient’s eligibility for addition of efgartigimod alfa would require trials of medications from all 3 classes, or from 1 or 2 classes? | Figure 1 is a reasonable depiction of efgartigimod alfa’s place in therapy, although the clinical experts said that they suspected that cost would drive clinicians to use IVIg first. Unless efgartigimod alfa ends up being priced similarly to current conventional therapies, it is likely that unsuccessful trials of all 3 classes (AChEIs, corticosteroids, and NSISTs) will be prerequisites to the use of efgartigimod alfa. In addition, in ADAPT not all included patients had refractory gMG; that was, not all patients needed to have multiple therapies that failed. Technically, from a data and mechanistical perspective, there was no need to have a failed trial of all conventional meds. Realistically, and mostly driven by price, efgartigimod alfa should not be offered as first-line treatment, but rather after patients have tried conventional treatments. So, the diagram looks realistic. |
Eligibility for re-treatment: In the ADAPT trial, if a patient was an MG-ADL responder during a previous cycle and lost response, that patient could qualify for re-treatment. Loss of response was defined as a < 2-point reduction in the MG-ADL total score during the cycle, compared to the baseline value for that cycle. Re-treatment in subsequent cycles was permitted if they met all of the following criteria:
This allowed for a maximum of 3 treatment cycles during the 26-week study. According to the sponsor and the product monograph, following cycle 1, treatment with efgartigimod alfa can be given on an as-needed basis according to clinical assessment, and thus would vary by patient. This poses a unique challenge for drug plans when instating an approval if there is no certainty on whether a patient will be re-treated and at what frequency. | The other clinical expert indicated that from an economic perspective, re-treatment with efgartigimod alfa on an as-need basis would likely result in savings, as based on data some patients had relatively long stretches between cycles. However, it will make implementation difficult for clinicians based on the need for more frequent monitoring to decide appropriate time for re-treatment. Therefore, as a prescriber, approval for 3 cycles initially would be reasonable to assess response to treatment; further approvals would be conditional on demonstrating benefit. The clinician can then tailor cycles (e.g., some patients may take longer to use all cycles). |
Special subtypes (not explicitly mentioned in the indication) to consider separately for eligibility: Considering patients who had used eculizumab within 6 months of screening were excluded from the study and there were no comparisons to ravulizumab, would patients who experienced failure with either 1 or both drugs be considered for treatment with efgartigimod alfa? | Yes. Failure to respond to rituximab or eculizumab (or ravulizumab) would absolutely not preclude consideration of efgartigimod alfa. These patients should be considered for efgartigimod alfa. |
Considerations for continuation or renewal of therapy | |
Challenges related to assessment and monitoring of therapeutic response:
Can the clinical expert(s) confirm if use of the MG-ADL reflects best practices when treating patients with gMG in Canada? If not, is there another tool or outcome that would better align with how patients are monitored in the Canadian practice setting? | One clinical expert indicated that they prefer a clinician-driven assessment (like the QMG). The other clinical expert indicated that most neurologists in Canada do not use standardized outcome measures for MG. The standardized outcome measures are mostly used in academic centres. This clinical expert uses MGII, which combines patient-reported outcomes and physician examination, but considered the MG-ADL or equivalent acceptable. Both clinical experts agreed that MG-ADL is extremely easy to use and implement. And both clinical experts agreed that regardless of outcome measurement tools, it is more important that patients be assessed by neurologists with experience and expertise in the management of MG. |
Considerations for discontinuation of therapy | |
Definition of loss of response, absence of clinical benefit, or disease progression Efgartigimod alfa is administered as needed, based on clinical response (physician assessment and patient-reported outcomes). How many times would a patient require re-treatment due to loss of response before being considered for discontinuation? Likewise, if a patient has a need for increased frequency of dosing, would consideration be given to discontinuation of efgartigimod alfa? It would be helpful to have a clear definition of loss of response and disease progression that would indicate the need for discontinuation, defined according to MG-ADL parameters and/or frequency of dosing. | One clinical expert stated that how many unsuccessful re-treatments would be needed before concluding that efgartigimod alfa does not work would vary from clinician to clinician, but they would probably stop it after 2 unsuccessful re-treatments. There would not be a good rationale to give efgartigimod alfa at a frequency greater than every 1 week to 2 weeks. The other clinical expert indicated that, based on ADAPT, patients had no response after 2 cycles (no significant improvement or worsening) should discontinue the treatment. |
Considerations for prescribing of therapy | |
Dosing, schedule/frequency, dose intensity The medication is given as a 1-hour infusion once weekly for 4 weeks (this being cycle 1). Following the initial dose, subsequent doses and frequency are dependent on clinical response, and thus may vary by patient. There was no further clarity provided in the product monograph regarding frequency of dosing. The sponsor estimates that patients with AChR-Ab+ required a mean number of 4.72 cycles per year, with approximately 24% of patients requiring < 3.5 cycles per year. Bearing in mind that each cycle consists of up to 4 weekly infusions, to a maximum of 3 vials per infusion, this would mean up to 48 vials annually if at maximum dose and at an average of 4 treatments per year. It is unclear if there is a minimum amount of time that should exist between cycles. Is there a minimum frequency before administering a subsequent dose? In the ADAPT trial, the median time between the last infusion in cycle 1 and the start of cycle 2 was 7 weeks (mean of 10 weeks). In a real-world study of utilization patterns, the sponsor noted an average gap of 50 to 58 days between the last infusion of cycle 1 and the start of cycle 2. In the long-term extension study, ADAPT+, subsequent cycles were only started if the patient completed the fourth infusion of the previous cycle at least 4 weeks prior. If consistent with this information, would re-treatments with efgartigimod alfa require a minimum of 4 weeks after the last infusion before initiating the next cycle? | Both clinical experts agreed that waiting at least 4 weeks before initiating a re-treatment cycle seems rational. |
Drug administration Administration is by IV infusion only and requires a trained health care professional. The sponsor expects the infusion to be most commonly administered in a patient’s home and less commonly at an infusion clinic. Given this information, a trained health care professional would be required to make home visits to complete the administration. | For CDEC consideration. No clinical expert response required. |
Concerns related to accessing clinical specialists and/or special settings Administration will require in-home services or infusion clinics. Although the sponsor states that it is committed to providing standardized access to all patients, including those in remote areas, how this accessibility will be provided is a potential concern. | For CDEC consideration. No clinical expert response required. |
Concerns related to combination usage Would there be any potential combination usage of efgartigimod alfa with eculizumab or ravulizumab, specifically considering that Health Canada issued a NOC for ravulizumab plus conventional therapy in the treatment of patients with AChR-Ab+ gMG? | One clinical expert indicated that efgartigimod alfa might be combined with either eculizumab or ravulizumab (as the mechanisms are different), but the cost would make this difficult to justify. The other expert stated that theoretically, efgartigimod alfa might be combined with either eculizumab or ravulizumab as they have different mechanisms. But it is hard to know if the combination would be clinically superior to either alone because there are no data. They would not support concurrent use. Rather, they suspect eculizumab or ravulizumab would be used sequentially if no response to one. |
Care provision issues | |
Drug preparation, storage, and administration or dispensing Administration is by IV infusion only. It requires reconstitution and administration by a trained health care professional, and up to 3 vials may be needed per dose, depending on the patient’s weight (10 mg/kg). | For CDEC consideration. No clinical expert response required. |
System and economic issues | |
Concerns regarding the anticipated budget impact and sustainability At the submitted price, efgartigimod alfa is significantly more expensive than conventional therapy and immunoglobulin or PE therapies but comparable to the cost of ravulizumab. | For CDEC consideration. No clinical expert response required. |
Presence of confidential negotiated prices for comparators At this time, ravulizumab has not received a positive funding recommendation for gMG or gone through pricing negotiations so it is difficult to make a comparison with these unknowns. It is awaiting CDEC reconsideration. Although not mentioned as a comparator by the sponsor, eculizumab is another treatment for gMG and its pricing negotiations ended without agreement. The reimbursement status of eculizumab for gMG across the jurisdictions is not publicly known. | For CDEC consideration. No clinical expert response required. |
AChEI = acetylcholinesterase inhibitor; AChR-Ab+ = AChR antibody positive; CDEC = Canadian Drug Expert Committee; CMI = clinically meaningful improvement; CS = corticosteroid; Fc = fragment crystallizable; gMG = generalized myasthenia gravis; IgG = immunoglobulin G; IMP = investigational medicinal product; IVIg = intravenous immunoglobulin; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGII = Myasthenia Gravis Impairment Index; NOC = notice of compliance; NSIST = nonsteroidal immunosuppressive therapy; PE = plasma exchange; QMG = Quantitative Myasthenia Gravis.
Figure 1: Anticipated Place in Therapy of Efgartigimod Alfa for Anti-AChR Antibody-Positive gMG
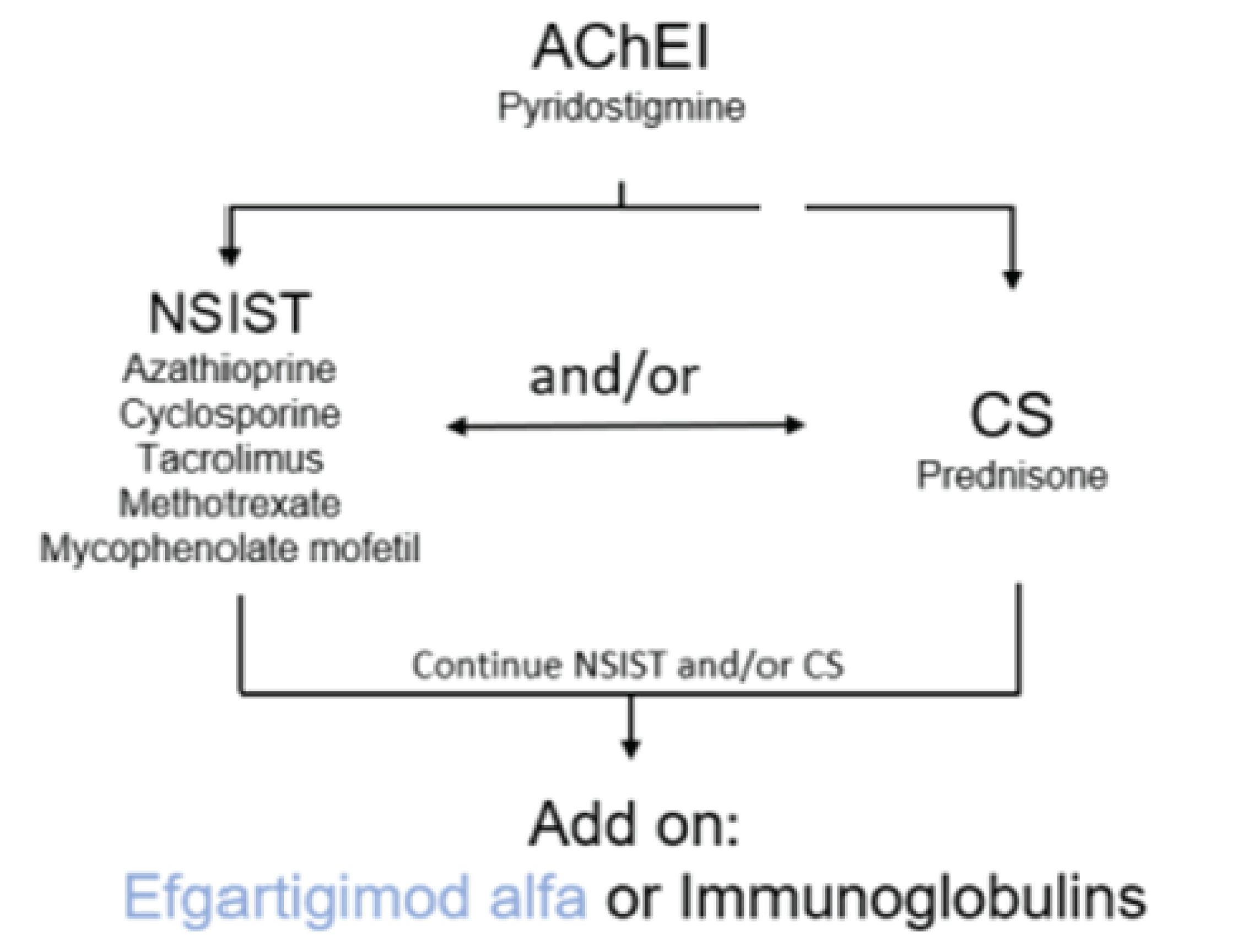
AChEI = acetylcholinesterase inhibitor; CS = corticosteroid; NSIST = nonsteroidal immunosuppressive therapy.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of efgartigimod alfa (VYVGART), 20 mg/mL solution, IV infusion in the treatment of adult patients with gMG. The focus is on comparing efgartigimod alfa to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence provided by the sponsor is presented in 3 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence provided by the sponsor.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this report:
1 pivotal phase III double-blind RCT (ADAPT) identified in the systematic review
1 long-term extension study (ADAPT+)
1 ITC.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Description of Studies
Key characteristics of the ADAPT trial are shown in Table 5, and a summary of its design is shown in Figure 1. The ADAPT trial (ARGX-113-1704; NCT03669588; N = 167)30 was a randomized, double-blind, placebo-controlled, multicentre, phase III trial designed to evaluate the efficacy, safety, and tolerability of efgartigimod alfa and its impact on HRQoL and normal daily activities in patients with gMG. The study has been completed and the data cut-off (last patient completed) was April 6, 2020. Patients were recruited from 56 centres in the US, Europe, Japan, and Canada, including 4 patients were enrolled at 3 sites in Canada. The primary objective was to evaluate the efficacy of efgartigimod alfa as assessed by the percentage of MG-ADL responders in the AChR-Ab+ population after the first treatment cycle. The secondary objectives were to evaluate the safety and tolerability of efgartigimod alfa in the overall population and in subgroups and to evaluate the efficacy of efgartigimod alfa based on:
the percentage of QMG responders after the first treatment cycle in the AChR-Ab+ population
the percentage of MG-ADL responders after the first treatment cycle in the overall population (patients with AChR-Ab+ and AChR-Ab–)
the percentage of time that patients show a CMI in the MG-ADL total score during the study (up to and including day 126) in the AChR-Ab+ population
the time to qualify for re-treatment in the AChR-Ab+ population (up to 168 days)
the percentage of early MG-ADL responders after the first treatment cycle in the AChR-Ab+ population.
After a 2-week screening period, patients were randomized in a 1:1 ratio to receive 4 IV infusions of efgartigimod alfa 10 mg/kg or a matching placebo administered at weekly intervals in addition to their ongoing treatment for gMG. Randomization was based on a permuted block strategy with interactive response technology, and stratification was based on Japanese or non-Japanese status, AChR-Ab+ or AChR-Ab– status, and use of NSISTs or non-NSISTs as concomitant therapy for gMG. The patients, investigators, study staff, and sponsor were blinded to patient treatment assignment. Unblinding of treatment was permitted in the event of a medical emergency, in which case the patient was discontinued from the study. The total study duration was up to 28 weeks divided into the 2-week screening period and a 26-week treatment period. Patients started the 26-week treatment period with an initial 8-week treatment cycle that comprised a 3-week treatment period (4 infusions on weeks 0 to 3) and a 5-week follow-up period. At the end of the first treatment cycle, patients started an intertreatment cycle of variable length depending on the patient’s clinical response to efgartigimod alfa or placebo. If a patient was an MG-ADL responder during a previous treatment cycle and lost response, that patient could qualify for re-treatment. Loss of response was defined as a less than 2-point reduction in the MG-ADL total score during the cycle compared to the baseline value for that cycle). Re-treatment in subsequent cycles was permitted if patients met all the following criteria:
completed the prior treatment cycle (i.e., the 3-week treatment period and 5-week follow-up period).
had an MG-ADL total score greater than or equal to 5 points with more than 50% of the total score attributed to non-ocular symptoms.
the subsequent cycle did not start after day 127 and could be completed within the 26-week treatment period.
Based on these criteria, a maximum of 3 treatment cycles was possible during the 26-week study; patients who completed the study were eligible to enrol in a long-term extension study, ADAPT+ (refer to Section 3).
Table 5: Details of the ADAPT Trial
Detail | ADAPT |
|---|---|
Design and population | |
Study design | Randomized, double-blind, placebo-controlled, multicentre, phase III trial |
Locations | 56 clinical sites in the US, Europe, Japan, and Canada (4 patients at 3 Canadian sites) |
Patient enrolment dates | First patient consented: August 22, 2018 Last patient completed: April 6, 2020 |
Randomized (N) | 167 patients enrolled and randomized (84 patients to the efgartigimod alfa group and 83 patients to the placebo group) |
Inclusion criteria |
Patients were required to be on a stable dose of standard of care (concomitant gMG treatment) before screening. Concomitant gMG treatment was limited to AChEIs, steroids, and NSISTs. |
Exclusion criteria |
|
Drugs | |
Intervention | Efgartigimod alfa 10 mg/kg administered as a 1-hour IV infusion weekly for 4 infusions (days 1, 8, 15, and 22) in each cycle. For each administration, 125 mL of IMP was infused over 1 hour. The maximum permitted efgartigimod alfa dose per infusion is 1,200 mg. Subsequent cycles of treatment (within the 26-week treatment period) could be initiated based on clinical response, which was measured using the MG-ADL scale. |
Comparator(s) | Matched placebo was administered as a 1-hour infusion weekly for 4 infusions (days 1, 8, 15, and 22) in each cycle. For each administration, 125 mL of IMP was infused over 1 hour. Subsequent cycles of treatment (within the 26-week treatment period) could be initiated based on clinical response, which was measured using the MG-ADL scale. |
Study duration | |
Screening phase | 2 weeks before treatment initiation |
Treatment phase | A 26-week treatment period divided into:
|
Follow-up phase | Patients could potentially enter the extension study ADAPT+ (ARGX-113-1705; NCT03770403) to receive open-label efgartigimod alfa IV 10 mg/kg for up to 3 years. Patients could enter ADAPT+ if they:
|
Outcomes | |
Primary end point | Percentage of patients in the AChR-Ab+ population who, after the first cycle (C1), had a reduction of ≥ 2 points on the MG-ADL total score (compared to baseline of the first cycle [C1B]) for ≥ 4 consecutive weeks with the first of these decreases occurring ≤ 1 week after the last infusion of IMP. |
Secondary and exploratory end points | Secondary:
Tertiary end points:
Exploratory:
|
Publication status | |
Publications | Saccà et al. (2023)50 US National Library of Medicine (2023)51 |
Ab+ = antibody positive; Ab– = antibody negative; AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; C1 = cycle 1; C1B = baseline of cycle 1; CMI = clinically meaningful improvement; CnB = baseline of cycle n; gMG = generalized myasthenia gravis; IgG = immunoglobulin G; IMP = investigational medicinal product; IVIg = intravenous immunoglobulin; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MGFA = Myasthenia Gravis Foundation of America; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item; NSIST = nonsteroidal immunosuppressive therapy; PE = plasma exchange; q.7.d. = every 7 days; QMG = Quantitative Myasthenia Gravis; SEB = study entry baseline; VAS = visual analogue scale.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sources: ADAPT Clinical Study Report (ADAPT 2020),30 and the sponsor’s submission.1
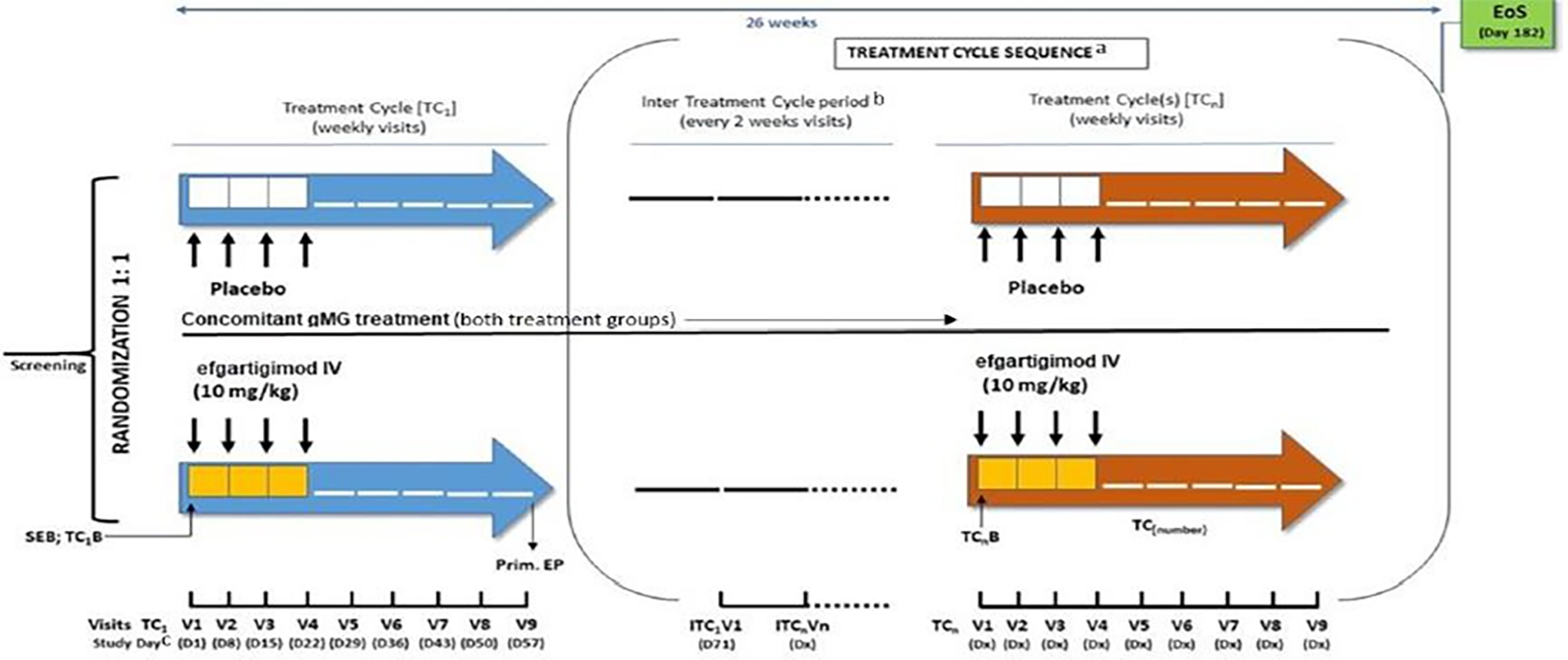
D = study day; EoS = end of study; gMG = generalized myasthenia gravis; ITC1V1 = intertreatment cycle 1 visit 1; ITCnVn = intertreatment cycle n visit n; Prim. EP = primary end point; SEB = study entry baseline; TC = treatment cycle; TCB = treatment cycle baseline; V = visit.
Note: A study cycle is defined as a treatment cycle and an intertreatment cycle.
a The cycle sequence could have been repeated as many times as necessary as long as the last treatment cycle did not begin after day 127.
b The intertreatment cycle period varied from patient to patient.
c Time windows for clinic visits were ± 1 day for treatment cycle visits and ± 2 days for intertreatment cycle visits.
Source: ADAPT Clinical Study Report.30
Populations
Inclusion and Exclusion Criteria
The eligibility criteria for the ADAPT trial are shown in Table 5. Patients were eligible for inclusion in the ADAPT study if they were adults (aged ≥ 18 years), had a diagnosis of Class II to IV gMG based on the MGFA system, had any serotype of gMG (AChR-Ab+, AChR-Ab–, MuSK, or LRP4), and had an MG-ADL score of greater than or equal to 5 (with > 50% of the score caused by non-ocular symptoms) despite being on a stable dose of concomitant conventional therapy before screening (AChEIs, steroids, and/or NSISTs), which was continued throughout the trial. There was no requirement to have received or discontinued the use of any specific treatment. A maximum of 20% of patients with AChR-Ab–were to be enrolled. Patients were required to be on a stable dose of standard of care before screening. For AChEIs, this meant no dose change for 2 weeks before screening; for steroids, 3 months or longer of treatment and no dose change for 1 month before screening; and for NSISTs, 6 months or longer of treatment and no dose change for 3 months before screening. The standard of care was allowed to be used alone or in combination. Patients were excluded if they received treatment with IVIg or PE within 1 month of screening, treatment with rituximab or eculizumab in the 6 months before screening or underwent thymectomy in the 3 months before screening. Patients were also excluded if they had MGFA Class I or V gMG, worsening muscle weakness secondary to concurrent infection/medication, seropositivity or tested positive for active viral infection (e.g., hepatitis B, hepatitis C, HIV), a known severe infection or major episode in last 8 weeks, a history of non-MG autoimmune disease that would interfere with an accurate assessment of clinical symptoms, and documented lack of clinical response to PE.
Interventions
Efgartigimod alfa IV 10 mg/kg or matching placebo was administered as a 1-hour infusion every 7 days for 4 infusions (days 1, 8, 15, and 22) in each cycle. The maximum permitted efgartigimod alfa dose per infusion was 1,200 mg. Patients were to maintain their stable concomitant conventional therapy during treatment with efgartigimod alfa or placebo. Efgartigimod alfa and the matched placebo were identical in physical appearance and supplied in identical containers. If a significant change (> 10%) in body weight was observed, the dose was recalculated to ensure that efgartigimod alfa dose was 10 mg/kg. When delayed for more than 3 days, a dose was not administered to ensure that 2 consecutive doses were administered 3 or more days apart. Treatment adherence per cycle was defined according to the following formula: (number of doses received ÷ 4) × 100%.
Subsequent treatment cycles were initiated as needed to permit personalized treatment based on the patient’s clinical response, which was measured by the validated MG-ADL scale. Loss of response was defined as a less than 2-point reduction in the MG-ADL total score in a specific cycle compared to the baseline for that cycle. After treatment cycle 1, the frequency of re-treatment (i.e., initiation of subsequent cycles) was based on clinical response as measured using the MG-ADL. To be re-treated, a patient must have met all of the following re-treatment criteria: completed the prior treatment cycle (i.e., the 3-week treatment period and the 5-week follow-up period), have an MG-ADL total score greater than or equal to 5 points with more than 50% of the total score attributed to non-ocular symptoms, and the subsequent cycle must have started no later than day 127 and have been completed within the 26-week study duration. If the patient was an MG-ADL responder in a previous cycle and lost the response, then the patient could qualify for re-treatment if the re-treatment criteria were met. Loss of response is defined as a less than 2-point reduction in the MG-ADL total score in a particular cycle compared to that cycle’s baseline.
Rescue therapy was permitted for patients with protocol-defined MG clinical deterioration (new or worsening of respiratory and/or bulbar symptoms or a ≥ 2-point increase in individual non-ocular items on the MG-ADL) and whose health the investigator considered to be in jeopardy if rescue therapy was not provided. Patients who were provided rescue therapy were discontinued from study treatment but were still followed according to the assessment schedule. Rescue therapy options included PE, IVIg, immunoadsorption, any new type of corticosteroid, or any increased dose of current corticosteroid used as a standalone therapy or combined with another treatment.
Prohibited medications during the study, which would result in study discontinuation, included any other IgG therapy; change in the type, dose, or regimen of concomitant treatment (replacing, adding, and/or removing treatment or adjusting the dose and/or frequency), even if used for non-gMG indications; any monoclonal antibody for immunomodulation; and vaccines.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by CADTH and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered to be most relevant to inform CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE. For the purpose of this review, only information on the patients with AChR-Ab+ is presented in this report.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | ADAPT AChR-Ab+ population |
|---|---|---|
MG-ADL responder | Cycle 1 and cycle 2 | Primary outcome in cycle 1, exploratory outcome in cycle 2 |
Early MG-ADL responders | Cycle 1 | Key secondary outcome in cycle 1 |
Percentage of time with a CMI of MG-ADL | Up to 126 days | Key secondary outcome in cycle 1 |
Time to qualify for re-treatment | Up to 168 days | Key secondary outcome |
Change from cycle baseline in MG-ADL total score | Cycle 1 and cycle 2 | Exploratory |
QMG responder | Cycle 1 | Key secondary outcome |
MG-QoL15r | Cycle 1 | Exploratory |
EQ-5D-5L | Cycle 1 | Exploratory |
Hospitalizations | Week 26 | Post hoc analysis |
Exacerbations | Week 26 | Post hoc analysis |
Myasthenic crisis | Week 26 | Post hoc analysis |
Safety | Week 26 | Exploratory |
Notable harms (AESIs) (i.e., infections and infestations) | Week 26 | Exploratory |
AChR-Ab+ = acetylcholine receptor antibody positive; AESI = adverse event of special interest; CMI = clinically meaningful improvement; EQ-5D-5L = 5-Level EQ-5D; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item; QMG = Quantitative Myasthenia Gravis.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sources: ADAPT Clinical Study Report;30 Howard et al. (2021).28
The MG-ADL is an 8-item patient-reported outcome tool used to assess MG symptoms and their effects on daily activities. The scale includes 2 items on daily life activities (ability to brush teeth or comb hair and limitations in the ability to rise from a chair) and 6 MG symptom categories (diplopia, chewing, voice and speech, ptosis, swallowing, and respiratory). Each item was graded on a 4-point symptom severity scale (0 = normal to 3 = most severe), with the total score from 0 to 24; higher scores indicate worse symptoms (Table 7). Scoring of the MG-ADL was performed by a trained and certified evaluator. A CMI on the MG-ADL has been defined as a 2-point reduction in total score,52,53 which was used to define CMIs for responders and nonresponders. Minimal symptom expression is a score of 0 or 1.52 End points in the ADAPT trial related to the MG-ADL that were relevant to this review included:
The primary efficacy end point: Percentage of patients with AChR-Ab+ who were MG-ADL responders in the first treatment cycle (an MG-ADL responder was defined as a patient with a ≥ 2-point improvement [reduction] in MG-ADL score, sustained for ≥ 4 consecutive weeks, with the first improvement occurring by week 4 of the cycle [≤ 1 week after the fourth infusion]).
Secondary end point: Percentage of MG-ADL responders in cycle 1 in the overall population (i.e., AChR-Ab+ and AChR-Ab–).
Secondary end point: Percentage of time patients with AChR-Ab+ showed a CMI in their MG-ADL score (≥ 2-point reduction) up to day 126.
Secondary end point: Time from week 4 to qualify for re-treatment in the AChR-Ab+ population (i.e., a < 2-point reduction in the MG-ADL total score and MG-ADL total score of ≥ 5 points with > 50% of the total score attributed to non-ocular symptoms).
Secondary end point: Percentage of early MG-ADL responders in cycle 1 in the AChR-Ab+ population (MG-ADL responders with first MG-ADL improvement of ≥ 2 points occurring by week 2).
Various tertiary and exploratory end points related to characterizing the time to onset of effect and magnitude of effect in MG-ADL scores, change over time (total score and individual items), duration and magnitude of MG-ADL response, and repeatability of the effect from the second cycle onward.
The QMG quantifies disease severity based on impairments of body functions and structures, as defined by the International Classification of Functioning, Disability and Health.54 The QMG includes 13 investigator-rated items that measure endurance or fatigability, accounting for disease state fluctuations. The QMG score is composed of the following items: ocular (2 items), facial (1 item), bulbar (2 items), gross motor (6 items), axial (1 item), and respiratory (1 item).55 A trained evaluator performed scoring on the QMG scale. The items were scored on a 4-point severity scale (0 = no symptoms to 3 = severe symptoms), and permissible scores range from 0 to 39, with higher scores indicative of greater disease severity (Table 7). A CMI on the QMG has been defined as a 3-point reduction in the QMG score.56 End points in the ADAPT trial related to the QMG scale that were relevant to this review included:
Secondary end point, the percentage of QMG responders (defined as a ≥ 3-point improvement in the total QMG score for ≥ 4 consecutive weeks with the first improvement occurring by week 4 of cycle 1) in the AChR-Ab+ population.
The MG-QoL15r is a 15-item survey of a patient’s perceived HRQoL that addresses attributes known to be meaningful to a patient with gMG, such as psychological well-being and social functioning. The MG-QoL15r helps inform the clinician about the patient’s perception of the extent of and dissatisfaction with MG-related dysfunction. The patient uses a 3-point (0 = not at all, to 2 = very much) Likert scale to assess statements regarding the domains of mobility (9 items), symptoms (3 items), general contentment (1 item), and emotional well-being (2 items). The maximum possible score is 30 points, and higher scores reflect greater MG-related dysfunction (Table 7). An MID or CMI on the MG-QoL15r has not been determined.57 In the ADAPT trial, the only tertiary end point involving the MG-QoL15r was the change from baseline in the total MG-QoL15r score.
The EQ-5D-5L is a standardized instrument developed by the EuroQol Group to provide a simple, generic measure of health status for clinical and economic appraisal.58 A VAS is included in the questionnaire, and respondents were asked to mark their health status from 0 to 100 on the day the interview was conducted, with a score of 0 corresponding to “the worst health you can imagine” and of 100 corresponding to “the best health you can imagine.” The sponsor is not aware of a reported MID or CMI value for EQ-5D VAS in patients with gMG.
gMG exacerbation was defined as a greater than or equal to 3-point worsening in QMG score versus baseline. In the overall population, a post hoc analysis was also conducted to estimate the proportions of patients with exacerbation (a ≥ 3-point worsening in QMG score versus baseline), which was compared between the treatment arms using the chi-square test.59
Safety Outcomes
Safety was assessed through the incidence of TEAEs and changes in clinical laboratory values, vital signs, and electrocardiogram results.28 The TEAEs were coded using MedDRA central coding dictionary and were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0. Events within the MedDRA system organ class “infections and infestations” were defined as AESIs. All AEs were assessed, documented, and reported in accordance with International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Guideline for Good Clinical Practice. The safety and disease severity follow-up visits continued until day 182.
Table 7: Summary of Outcome Measures and Their Measurement Properties in the ADAPT Trial
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
MG-ADL questionnaire | An 8-item patient-reported outcome measure assessing MG symptoms and functional activities related to activities of daily living and producing a total score ranging from 0 to 24, where higher scores indicate greater severity of symptoms. The MG-ADL is composed of items related to patients’ assessment of functional disability secondary to ocular (2 items), bulbar (3 items), respiratory (1 item), and gross motor or limb impairment (2 items).52 | Validity The MG-ADL highly correlated with the MGC (r = 0.85; 1) and MG-QoL15 (r = 0.76; P < 0.0001) (n = 87).53 Correlation of the MG-ADL score and physician impression of change between the visits was strong (r = 0.70; P < 0.0001) (n = 76).53 Reliability Test-retest reliability coefficient of 93.7% among 20 patients, with lower bound of the 95% CI at 87.3%, tested twice within 1 week.53 Responsiveness The MG-ADL was assessed at 2 visits, where the mean improvement in score in patients who improved, based on the gold standard, was 3.88 (SD = 2.72) (n = 76).53 Note the measurement properties of the subcomponents of the scale have not been investigated. | A 2-point improvement in MG-ADL score was a threshold that provided the best balance of sensitivity (n = 26) and specificity (n = 50) when referenced to MG-QoL15 and physician impression of change for predicting clinical improvement at the level of the individual for patients with MG.53 |
QMG scale | A 13-item direct physician assessment scoring system that quantifies disease severity, based on impairments of body functions and structures. Total QMG scores range from 0 to 39, where higher scores indicate greater disease severity. The QMG score is composed of the following items: ocular (2 items), facial (1 item), bulbar (2 items), gross motor (6 items), axial (1 item), and respiratory (1 item).55 | Validity Construct validity was assessed through correlations with the MMT (r = 0.69 in 303 patients60 and r = 0.73 in 53 patients).61 Reliability Internal consistency assessed via Cronbach alpha value was 0.74 for the QMG, demonstrating an acceptable threshold (n = 251).62,63 Test-retest reliability was studied in 209 stable patients assessed 2 weeks apart. The intraclass correlation coefficient for the total scores was 0.88 (95% CI, 0.85 to 0.91).62,63 Responsiveness The index of responsiveness (signal-to-noise ratio) was 1.45 (n = 53).61 Note that the measurement properties of the subcomponents of the scale have not been investigated. | Based on an interrater reliability of 1.342 SD, any change in the QMG score of up to 2.6 points was expected to occur due to variability of repeated observations. Therefore, a change of 2.6 points was estimated to be the threshold of clinical significance in patients with MG (n = 5 with MG and n = 4 otherwise healthy).55 Using the anchor-based method with the patients’ perception of overall improvement as assessed using a VAS, there was some evidence that the MID should be higher in patients with higher baseline QMG scores, where the MID with mild to moderate MG (QMG ≤ 16) was estimated to be 2 points (n = 38), compared to patients with higher baseline values (QMG > 16), for whom the estimated MID was 3 points (n = 12).56,57 |
MG-QoL15r | A total of 3 items in the original MG-QoL15 scale were reworded to improve its clinimetric properties and face and content validity. The wording, “e.g., double vision,” was added to the ocular item; “work at home” was added to the work item; and other limitations of personal independence were added to the driving item. The revised version uses a 3-response option scale (0 = not at all; 1 = somewhat; 2 = very much), with higher scores indicating worse quality of life over the past few weeks. The maximum possible score is 30 points. | The psychometric properties of MG-QoL15r, QMG, MG-ADL, and MGC were evaluated and compared to response to disease change in patients with autoimmune MG (N = 872).64 Validity Construct validity was demonstrated for MG-QoL15r with QMG (Pearson correlation coefficient [r] = 0.550), MG-ADL (r = 0.701), and MGC (r = 0.635). For discriminant validity, the MG-QoL15r scores were different between patients based on their MGFA classification and MGC scores.64 Reliability Internal consistency reliability was demonstrated by the Cronbach alpha of 0.93 for MG-QoL15r.64 Responsiveness For responsiveness to change, the Pearson correlation coefficients between changes in MG-QoL15r and QMG after treatment were 0.423.64 | An MID for patients with MG has not been estimated. |
EQ-5D-5L | A generic, self-reported measure of health status comprising 2 parts.58 The descriptive system consists of 5 dimensions (mobility, self-care, usual activities, pain or discomfort and anxiety or depression). Each dimension has 5 increasing levels of severity or response. The responses are used to generate a health state profile (5-digit code) which can be converted to a summary index score based on societal preference weights. Index scores range from < 0 to 1, with higher scores representing higher health utility.58 Patient’s perceived health status on that day is also rated using the VAS, ranging from 0 (worst imaginable health) to 100 (best imaginable health).58 | The validity, reliability, and responsiveness to change have not been investigated in patients with MG. | An MID for patients with MG has not been estimated. |
CI = confidence interval; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15 = Myasthenia Gravis Quality of Life 15-item; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item; MGFA = Myasthenia Gravis Foundation of America; MID = minimally important difference; MMT = Manual Muscle Test; QMG = Quantitative Myasthenia Gravis; SD = standard deviation; VAS = visual analogue scale.
Statistical Analysis
Clinical Trial End Points
The data cut-off date (last patient completed the study) was April 6, 2020. A summary of the statistical analyses of efficacy end points is presented in Table 8. The total MG-ADL, QMG, and MG-QoL15r changes from the baseline of cycle 1 between treatment group differences were analyzed using mixed models for repeated measures. The model included available data up to week 8 on treatment, visit and treatment-by-visit interaction terms as fixed effects, with baseline value and stratification factors as covariates. The within-subject correlation was modelled by assuming an unstructured covariance matrix for the error terms.
Sample Size and Power Calculation
The proportion of MG-ADL responders in the placebo group was hypothesized to be 30%. The treatment difference was assumed to be 29% in favour of the efgartigimod alfa group, representing a weighted average treatment difference of 35% for patients with AChR-Ab+ (80% of enrolled patients) and 5% for patients with AChR-Ab– (20% of enrolled patients). A 10% dropout rate was assumed.
Based on the assumptions above, a sample size of 150 patients would provide 96% power to detect a treatment difference of 35% (2-sided significance level of 5%) in the rate of responders among 120 patients in the primary population (patients with AChR-Ab+).28
Statistical Testing
The statistical analysis methods are presented in Table 8. The results summarized in this submission are from the final analyses of the ADAPT trial.
The primary end point was tested using a 2-sided exact test using a logistic regression model with baseline MG-ADL total score as a covariate and the following 3 stratification factors: AChR-Ab status (seropositive versus seronegative); NSISTs (taking versus not taking); and Japanese nationality (yes versus no). The treatment effect was presented as an OR with a 95% CI and 2-sided P value (5% 2-sided alpha level).
If the primary end point met significance, the 5 secondary end points were tested in a strict hierarchical order (refer to Table 15) to control the type I error rate using a 5% 2-sided significance level for each analysis. If an end point did not meet significance at the 5% significance level, the subsequent end points in the hierarchical order were not evaluated.
The results of tertiary and exploratory end points were summarized descriptively.
A similar logistic regression model was used to analyze parameters related to the MG-ADL and QMG scales as for the primary efficacy end point.
The percentage of time that patients had a CMI in their MG-ADL total score was analyzed using an analysis of covariance (ANCOVA) model with treatment (as randomized) and baseline total score as covariates; the model was stratified for the stratification variables (race and concomitant gMG treatment).
The time to qualify for re-treatment, as monitored by the MG-ADL total score, was analyzed using the Kaplan-Meier time-to-event analysis (stratified log-rank test), stratified for the stratification variables.
Analysis Populations
The analysis sets are presented in Table 9. The ITT analysis set included all patients who were randomized. Efficacy analyses were performed in the modified intention-to-treat (mITT) population, which included all randomized patients who had a valid baseline MG-ADL assessment and at least 1 postbaseline MG-ADL assessment. The per-protocol analysis set was a subset of the mITT population that included patients with 3 or more of the 4 infusions (in any order) without any major protocol deviations. The per-protocol analysis set was used for sensitivity analyses of the primary and secondary end points. In both the mITT and per-protocol analysis sets, patients were analyzed in the group to which they were randomized. The AChR-Ab+ population was defined based on the stratification factor as randomized. The safety analysis set included all patients who received at least a partial dose of investigative product. Patients were analyzed based on the treatment received.
Data Imputation Methods
In general, the main reasons for missing data (i.e., due to missing visit or missing value(s) on specific questions) on an efficacy scale were classified as:
Missing data due to MG disease worsening or missing data due to an AE
Missing data not due to MG disease worsening (e.g., vacation) or an AE
Missing data in an analysis window as visit was performed out of window
Missing data due to a missing value(s) on an efficacy scale (due to technical reasons, patient not able to perform the test with reason not linked to MG disease status, missing answer to a question, and so on).
Reason a resulted in data missing not at random, whereas reasons b, c, and d can reasonably be considered as data missing at random. The following rules were therefore intended for the handling of missing visit(s) or missing value(s):
A patient who dropped out or was lost-to-follow-up was treated as a nonresponder if they had not qualified as an MG-ADL responders before.
Intermittent missing data for only 1 of 4 postonset (O) consecutive analysis windows:
If the missing data followed 1 of the following score patterns, then the patient was considered as having achieved an MG-ADL response if that the missing data “m” was not due to reason a):
OXmXX
OmXXX
OXXXmX
OXXmX
If no such patterns were present, then the patient was considered as not having achieved an MG-ADL response.
Intermittent missing data following the onset of response at greater than or equal to 2 of 4 consecutive postonset analysis windows. Regardless of the reason for missing data, the patient was considered as not having a sustained response.
Using a missing-is-failure imputation method as a sensitivity analysis to assess the imputation impact for missing values.
Subgroup Analyses
For MG-ADL and QMG response, the following subgroups were prespecified (but not adjusted for multiplicity), with response rates and differences in response rate (efgartigimod alfa – placebo) shown together with the 95% Wald confidence limits:
age category at baseline (18 to < 65 years versus ≥ 65 years)
sex (male versus female)
race (Black or African American versus Asian versus white)
region (US versus Japan versus rest of the world)
baseline MG-ADL score (5 to 7 versus 8 to 9 versus ≥ 10)
number of cycles (1 versus 2 versus 3).
Additional post hoc analyses were conducted to evaluate MG-ADL and QMG response rates in additional clinically meaningful subgroups:
duration of disease (< 3 years versus 3 to < 6 years versus ≥ 6 years)
concomitant therapies (AChEI only versus any steroid versus any NSIST)
prior thymectomy (yes versus no)
prior NSIST use (≥ 1 versus none)
with and without prior treatment failure (prior exposure to ≥ 2 immunosuppressive therapies or treatment with ≥ 1 immunosuppressive therapy and requiring IVIg or PE multiple times within 1 year before study inclusion).
Sensitivity Analyses
Sensitivity analyses were conducted for the primary and secondary end points on the per-protocol analysis set. A second sensitivity analysis was conducted for the primary end point on the mITT population by using missing-is-failure imputation.
Table 8: Statistical Analysis of Efficacy End Points in the ADAPT Trial
End point | ADAPT | |||
|---|---|---|---|---|
Statistical model | Stratification and adjustment factors | Handling of missing data | Sensitivity analyses | |
Primary end point: Percentage of patients who, after the first cycle, have a decrease of ≥ 2 points in total MG-ADL score (compared to cycle 1 baseline) for ≥ 4 consecutive weeks with the first of these decreases occurring ≤ 1 week after the last infusion of the IMP in the AChR-Ab+ population | 2-sided exact test (using logistic regression) | Stratification: AChR- Ab status (positive vs. negative); NSISTs (taking vs. not taking), and Japanese nationality (yes vs. no) | Considered responders: Data “m” missing NOT due to a worsening of the disease or an AE for only 1 of 4 postonset (O) analysis windows with the following patterns: OXmXX, OmXXX, OXXXmX, or OXXmX. Considered nonresponders:
|
|
Secondary end point 1: Proportion of QMG responders (defined as a ≥ 3-point improvement in the total QMG score for ≥ 4 consecutive weeks with the first improvement occurring by week 4 of cycle 1) in the AChR-Ab+ population | 2-sided exact test (using logistic regression) | Stratification: NSISTs (taking vs. not taking) and Japanese nationality (yes vs. no) |
| |
Secondary end point 2: Percentage of MG-ADL responders in cycle 1 in the overall population (i.e., AChR-Ab+ and AChR-Ab–) | 2-sided exact test (using logistic regression) | Stratification: AChR- Ab status (positive vs. negative); NSISTs (taking vs. not taking); and Japanese nationality (yes vs. no) |
| |
Secondary end point 3: Proportion of time patients showed a CMI in MG-ADL score, in the AChR-Ab+ population, up to day 126 | ANCOVA | Randomized treatment group and stratification variables included as factors, and baseline total MG-ADL score included as a covariate |
| |
Secondary end point 4: Time from day 28 (1 week after the fourth infusion in cycle 1) to not having a CMI in the AChR-Ab+ population | Kaplan-Meier time-to-event analysis:
| Stratification: NSISTs (taking vs. not taking) and Japanese nationality (yes vs. no) |
| |
Secondary end point 5: Proportion of early MG-ADL responders in cycle 1 (MG- ADL responders with first MG-ADL improvement of ≥ 2 points occurring by week 2) in the AChR-Ab+ population | 2-sided exact test (using logistic regression) | Stratification: NSISTs (taking vs. not taking) and Japanese nationality (yes vs. no) |
| |
Ab = antibody; Ab+ = antibody positive; Ab– = antibody negative; AChR = acetylcholine receptor; AE = adverse event; ANCOVA = analysis of covariance; C1B = baseline of cycle 1; CMI = clinically meaningful improvement; IMP = investigational medicinal product; m = missing data; MG-ADL = Myasthenia Gravis Activities of Daily Living; mITT = modified intention to treat; NSIST = nonsteroidal immunosuppressive therapy; QMG = Quantitative Myasthenia Gravis; vs. = versus.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sources: ADAPT Clinical Study Report;30 Howard et al. (2021).28
Table 9: Analysis Populations in the ADAPT Trial
Population | Definition | Application |
|---|---|---|
All randomized patients (ITT) | All patients who were randomized into the study (randomized defined by a complete randomization date in the database or any information that confirmed randomization). | Hospitalizations, MG exacerbation and MG crisis |
mITT | All randomized patients with a value for the MG-ADL total score at baseline and at least 1 postbaseline time point. | Efficacy |
Per protocol | A subset of the mITT set of patients with ≥ 3 out of 4 infusions (in any order) without a major protocol deviation reported. | Sensitivity analysis of primary and secondary end points |
Safety analysis set | All patients who received at least a partial dose of IMP. | General characteristics, safety, and immunogenicity |
IMP = investigational medicinal product; ITT = intention to treat; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; mITT = modified intention to treat.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sources: ADAPT Clinical Study Report;30 Howard et al. (2021).28
Results
Patient Disposition
Patient disposition in the AChR-Ab+ population is summarized in Table 10.
Of the 216 patients screened for inclusion, 129 patients were enrolled and randomized to the efgartigimod alfa group (65 patients) or the placebo group (64 patients), with 117 patients (90.7%) completing treatment and 121 patients (93.8%) completing the study. The primary reason for discontinuation from treatment was the occurrence of a TEAE; 2 patients (3.1%) in both efgartigimod alfa group and the placebo group discontinued treatment for this reason. Administration of rescue therapy resulted in protocol-mandated treatment discontinuation for 1 patient (1.5%) in the efgartigimod alfa group and 2 patients (3.1%) in the placebo group.
Table 10: Summary of Patient Disposition From the ADAPT Study Included in the Systematic Review (AChR-Ab+ Population, Safety Analysis Set)
Patient disposition | Efgartigimod alfa (N = 65) | Placebo (N = 64) |
|---|---|---|
Screened, na | 216 | |
Screening failure, n | 49 | |
Did not meet inclusion criteria | 32 | |
Sponsor decision | 1 | |
Withdrew consent | 2 | |
Seronegative enrolment cap reached | 11 | |
Othera | 3 | |
Randomized, n (%) | 65 | 64 |
Completed treatment, n (%) | 62 (95.4) | 55 (85.9) |
Discontinued treatment, n (%) | 3 (4.6) | 9 (14.1) |
Adverse events | 2 (3.1) | 2 (3.1) |
Prohibited medications | 0 (0) | 1 (1.6) |
Rescue therapy | 1 (1.5) | 2 (3.1) |
Sponsor decision | 0 (0) | 1 (1.6) |
Withdrawal by patient | 0 (0) | 3 (4.7) |
Completed study, n (%) | 63 (96.9) | 58 (90.6) |
Discontinued from study, n (%) | 2 (3.1) | 6 (9.4) |
Adverse events | 1 (1.5) | 0 (0) |
Physician decision | 0 (0) | 1 (1.6) |
Sponsor decision | 1 (1.5) | 0 (0) |
Withdrawal by patient | 0 (0) | 4 (6.3) |
Other (%) | 0 (0) | 1 (1.6) |
Cycle 1 | 65 (100) | 64 (100) |
Completed treatment, n (%) | 63 (96.9) | 56 (87.5) |
Discontinued treatment, n (%) | 2 (3.1) | 8 (12.5) |
Reason for treatment discontinuation, n (%) | ||
Adverse events | 1 (1.5) | 2 (3.1) |
Prohibited medications | 0 | 1(1.6) |
Rescue therapy | 1 (1.5) | 2 (3.1) |
Sponsor decision | 0 | 1 (1.6) |
Withdrawal by patient | 0 | 2 (3.1) |
Cycle 2 | 51 (100) | 43 (100) |
Completed treatment, n (%) | 50 (98) | 42 (97.7) |
Discontinued treatment, n (%) | 1 (2) | 1 (2.3) |
Reason for discontinuation, n (%) | ||
Adverse events | 1 (2.0) | 0 (0) |
Withdrawal by patient | 0 (0) | 1 (2.3) |
Cycle 3 | ||
Completed treatment, n (%) | 7 (100) | 1 (100) |
mITT analysis set, n | 65 | 64 |
Per-protocol analysis set, n | 55 | 53 |
Safety analysis set, n | 65 | 64 |
AChR-Ab+ = acetylcholine receptor antibody positive; mITT = modified intention to treat.
aData are only available for the full population as randomization happens after screening; available in Figure 1 of the Howard et al. (2021) pivotal clinical trial publication provided to CADTH as part of the original submission.
Sources: Sponsor’s submission;1 sponsor-provided additional information.36
Baseline Characteristics
In the AChR-Ab+ population, 129 patients were enrolled and randomized (65 patients in the efgartigimod alfa group and 64 patients in the placebo group). The mean age was 44.7 years (standard deviation [SD] = 14.9 years) and 49.2 years (SD = 15.5 years) in the efgartigimod alfa and placebo groups, respectively. Most patients were female (66.7%), and white (85.3%). Demographic characteristics were similar in both the efgartigimod alfa and placebo groups (Table 11). The baseline clinical characteristics were generally similar in both the efgartigimod alfa and placebo groups, although some baseline imbalances were noted (Table 12). There were baseline imbalances between the efgartigimod alfa and placebo groups by age category (≥ 65 years: 12.3% and 20.3%, respectively) and by sex (female: 70.8% and 62.5%, respectively), across the treatment groups.
The mean time since diagnosis was 9.31 years. The most frequently reported MGFA class at screening was Class III to IIIb (n = 37; 28.69%), which reflects a patient population with moderate weakness affecting muscles other than the ocular muscle and ocular muscle weakness of any severity. Most patients had previously undergone thymectomy for MG (n = 75; 58.14%), with mean time since that procedure 10.93 years. The proportion of patients who underwent thymectomy was higher in the efgartigimod alfa group than in the placebo group (69.2% versus 46.9%). The mean total MG-ADL score at baseline was 8.8 points (range, 5 to 16), and 54 patients (42%) had scores of 8 to 9 points, which reflects a moderate symptom burden. A total of 7 (11%) patients in the efgartigimod alfa group and 4 (6.0%) in the placebo group had MG-ADL total scores of 5, and 24 (37%) patients in the efgartigimod alfa group and 17 (27%) in the placebo group had MG-ADL total scores greater than or equal to 10.
Similar results were observed for the mean QMG score at baseline, which was 15.6 points in the overall population, which reflects a overall moderate severity of patients with gMG (based on MGFA QMG severity classification).65 Patients also had impaired HRQoL based on median baseline MG-QoL15r scores of 16 to 17 points (out of a possible 30, higher scores indicate more severe impairment) across the efgartigimod alfa and placebo groups.
All patients had previously received 1 or more classes of conventional therapy for gMG, and large proportions had previously received 2 or more (96.1%) or 3 or more classes of conventional therapy (79.1%); no notable differences were observed between treatment arms. Based on the disease characteristics at baseline, these patients had persistent symptoms and impaired HRQoL despite previously receiving up to 3 classes of conventional therapy (AChEIs, corticosteroids, and/or NSISTs). Of note, despite their disease burden, these patients were still receiving stable doses of conventional therapies at baseline (i.e., they did not discontinue or progress beyond conventional therapy). These concomitant conventional therapies are summarized in the Exposure to Study Treatments section.
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Table 11: Summary of Baseline Demographic Characteristics From the ADAPT Study (Safety Analysis Set)
Characteristics | AChR-Ab+ population | |
|---|---|---|
Efgartigimod alfa (N = 65) | Placebo (N = 64) | |
Age (years) | ||
Mean (SD) | 44.7 (14.97) | 49.2 (15.54) |
Median (range) | 43.0 (19 to 78) | 46.5 (19 to 81) |
Age category, n (%) | ||
18 to < 65 years | 57 (87.7) | 51 (79.7) |
≥ 65 years | 8 (12.3) | 13 (20.3) |
Sex at birth, n (%) | ||
Female | 46 (70.8) | 40 (62.5) |
Male | 19 (29.2) | 24 (37.5) |
Race, n (%) | ||
American Indian or Alaska Native | 2 (3.1) | 0 |
Asian | 7 (10.8) | 4 (6.3) |
Black or African American | 1 (1.5) | 3 (4.7) |
White | 54 (83.1) | 56 (87.5) |
Multiple | 1 (1.5) | 0 |
Not reported | 0 | 1 (1.6) |
Ethnicity, n (%) | ||
Hispanic or Latino | 5 (7.7) | 2 (3.1) |
Japanese | 6 (9.2) | 4 (6.3) |
Not Hispanic or Latino | 54 (83.1) | 57 (89.1) |
Not reported | 0 | 1 (1.6) |
Weight (kg) | ||
Mean (SD) | 81.61 (29.823) | 79.51 (19.521) |
Median (range) | 74.00 (49.0 to 228.7) | 74.20 (41.2 to 118.1) |
AChR = acetylcholine receptor antibody positive; SD = standard deviation.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: ADAPT Clinical Study Report.30
Table 12: Summary of Baseline Clinical Characteristics From the ADAPT Study (Safety Analysis Set)
Characteristics | AChR-Ab+ population | |
|---|---|---|
Efgartigimod alfa (N = 65) | Placebo (N = 64) | |
Time since gMG diagnosis (years) | ||
n | 65 | 64 |
Mean (SD) | 9.68 (8.251) | 8.93 (8.214) |
Median (range) | 7.36 (1.0 to 45.3) | 6.15 (0.2 to 36.1) |
MG-ADL total score | ||
n | 65 | 64 |
Mean (SD) | 9.0 (2.48) | 8.6 (2.14) |
Median (range) | 9.0 (5 to 15) | 8.0 (5 to 16) |
MG-ADL total score category, n (%) | ||
n | 65 | 64 |
< 5 | ||||||| | |||||| |
5 to 7 | || |||||| | || |||||| |
8 to 9 | || |||||| | || |||||| |
≥ 10 | || |||||| | || |||||| |
Total QMG score | ||
n | 65 | 62 |
Mean (SD) | 16.0 (5.14) | 15.2 (4.39) |
Median (range) | 16.0 (4 to 28) | 15.5 (6 to 24) |
Baseline MG-QoL15r | ||
n | 65 | 64 |
Mean (SD) | 15.7 (0.78) | 16.6 (0.68) |
Median (range) | 16.0 (11.0 to 21.0) | 17.0 (12.5 to 21.0) |
Total MGC | ||
n | 65 | 64 |
Mean (SD) | 18.6 (6.08) | 18.1 (5.18) |
Median (range) | 19.0 (3 to 33) | 18.0 (8 to 29) |
AChR-Ab status, n (%) | ||
n | 65 | 64 |
Positive | 65 (100) | 64 (100) |
Negative | 0 | 0 |
MuSK-Ab status, n (%) | ||
n | 65 | 64 |
Positive | 0 | 0 |
Negative | 65 (100) | 64 (100) |
MGFA class at screening, n (%) | ||
n | 65 | 64 |
II to IIa | 14 (21.5) | 14 (21.9) |
II to IIb | 14 (21.5) | 11 (17.2) |
III to IIIa | 15 (23.1) | 19 (29.7) |
III to IIIb | 20 (30.8) | 17 (26.6) |
IV to IVa | 1 (1.5) | 3 (4.7) |
IV to IVb | 1 (1.5) | 0 |
Thymectomy for MG, n (%) | ||
n | 65 | 64 |
No | 20 (30.8) | 34 (53.1) |
Yes | 45 (69.2) | 30 (46.9) |
Time since thymectomy (years), mean (SD) | 10.31 (8.27) | 11.56 (8.82) |
Prior gMG treatment, n (%) | ||
n | 65 | 64 |
≥ 1 prior therapy | || ||||| | || ||||| |
≥ 2 prior therapies | || |||||| | || |||||| |
≥ 3 prior therapies | || |||||| | || |||||| |
≥ 1 prior NSIST | || |||||| | || |||||| |
≥ 2 prior NSISTs | || |||||| | || |||||| |
≥ 3 prior NSISTs | | ||||| | | ||||| |
IVIg | || |||||| | || |||||| |
Plasma exchange | || |||||| | || |||||| |
Plasmapheresis | || |||||| | || |||||| |
Any prior AChEI | 62 (95.4) | 61 (95.3) |
Any prior steroid | 49 (75.4) | 57 (89.1) |
Any prior NSIST | 47 (72.3) | 43 (67.2) |
Prior AChEI only | || |||||| | | ||||| |
Prior steroid only | | ||||| | | ||||| |
Prior NSIST only | | ||||| | | ||| |
Prior steroid plus NSIST plus AChEI | || |||||| | || |||||| |
Prior steroid plus AChEI only | | ||||| | || |||||| |
Prior AChEI plus NSIST only | | ||||| | | ||||| |
Prior steroid plus NSIST only | | ||||| | | ||||| |
Prior gMG treatment failure, n (%)a | 40 (61.5) | 41 (64.1) |
Ab = antibody; Ab+ = antibody positive; AChR = acetylcholine receptor; AChEI = acetylcholinesterase inhibitor; gMG = generalized myasthenia gravis; IVIg = intravenous immunoglobulin; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MGFA = Myasthenia Gravis Foundation of America; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item; min = minimum; MuSK = muscle-specific kinase; NSIST = nonsteroidal immunosuppressive therapy; QMG = Quantitative Myasthenia Gravis; SD = standard deviation.
Notes: Scores range from 0 points to 24 points for the MG-ADL, 0 points to 39 points for the QMG, 0 points to 50 points for the MGC, and 0 points to 30 points for the MG-QoL15r; higher scores are indicative of worse quality of life.
Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aA post hoc analysis was conducted where “prior treatment failure” was defined as prior exposure to ≥ 2 immunosuppressive therapies or treatment with ≥ 1 immunosuppressive therapy and requiring PE or IVIg multiple times within 1 year before study inclusion.32
Sources: ADAPT Clinical Study Report;30 data on file.32
Exposure to Study Treatments
Study Treatments
All patients received at least 1 dose of efgartigimod alfa or placebo during the study and the mean duration in the study (i.e., from the first dose until end of the study) was not reported in the sponsor’s evidence summary for the patients who were AChR-Ab+. The cumulative duration of treatment exposure was not reported for the patients who were AChR-Ab+ in the sponsor’s evidence summary either.
Patients in either group received a maximum of 3 cycles in the 26-week treatment period. Therefore, data regarding cycle duration after the first cycle would be cut-off at the end of the 26-week treatment period and thus not represent the true duration of those treatment cycles.
The number of patients per cycle was balanced between the efgartigimod alfa group (only 1 cycle: n = 14 [21.54%] patients; 2 cycles: n = 44 [67.7%] patients; 3 cycles: n = 7 [10.77%] patients) and the placebo group (only 1 cycle: n = 21 [32.82%] patients; 2 cycles: n = 42 [65.6%] patients; 3 cycles: n = 1 [1.59%] patients). An overview of the number of patients in each treatment group who received the treatment, and the cycle durations are summarized by cycle in Table 13.
Table 13: Summary of Patient Exposure From Studies Included in the Systematic Review
Exposure | AChR-Ab+ population | |
|---|---|---|
Efgartigimod alfa (N = 65) | Placebo (N = 64) | |
Total, patient-years | NR | NR |
Days of treatment, mean (SD) | NR | NR |
Cycle 1 | ||
Cycle duration (days), mean (SD) | 91.2 (38.32) | 99.6 (48.49) |
Cycle duration (days), median (min, max) | 71.0 (58 to 185) | 71.5 (16 to 190) |
Number of patients with infusions in cycle 1 | 65 | 64 |
4 infusions, n (%) | || |||||| | || |||||| |
3 infusions, n (%) | | ||||| | | ||||| |
2 infusions, n (%) | ||| | | ||||| |
1 infusion, n (%) | ||| | | ||||| |
Cycle 2 | ||
Cycle duration (days), mean (SD) | |||| |||||| | |||| ||||||| |
Cycle duration (days), median (min, max) | |||| |||| |||| | |||| |||| |||| |
Number of patients with infusions in cycle 2 | 51 | 43 |
4 infusions, n (%) | || |||||| | || |||||| |
3 infusions, n (%) | | ||||| | ||| |
2 infusions, n (%) | ||| | | ||||| |
1 infusion, n (%) | ||| | ||| |
Cycle 3 | ||
Cycle duration (days), mean (SD) | |||| ||||||| | |||| |||| |
Cycle duration (days), median (min, max) | |||| |||| ||| | |||| |||| |
Number of patients with infusions in cycle 3 | 7 | 1 |
4 infusions, n (%) | | |||||| | | ||||| |
3 infusions, n (%) | ||| | ||| |
2 infusions, n (%) | | |||||| | ||| |
1 infusion, n (%) | | |||||| | ||| |
Number of patients received only 1 cycle, n (%) | 14 (21.54) | 21 (32.82) |
Number of patients received only 2 cycles, n (%) | 44 (67.7) | 42 (65.6) |
Number of patients received only 3 cycles, n (%) | 7 (10.77) | 1 (1.59) |
Ab+ = antibody seropositive; AChR = acetylcholine receptor; max = maximum; min = minimum; NC = not calculable; SD = standard deviation.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: ADAPT Clinical Study Report.30
Concomitant Medications and Cointerventions
Concomitant gMG treatment was defined as any MG treatment received at a stable dosage during screening and maintained throughout the study (Table 14).
In the AChR-Ab+ population, the categories of concomitant gMG treatment were:
Any AChEI: 57 patients (87.7%) in the efgartigimod alfa group and 57 patients (89.1%) in the placebo group.
Any steroid: 46 patients (70.8%) in the efgartigimod alfa group and 51 patients (79.7%) in the placebo group.
Any NSIST: 40 patients (61.5%) in the efgartigimod alfa group and 37 (57.8%) patients in the placebo group. Detailed information on the most common NSIST used for the AChR-Ab+ population was not presented in the sponsor’s evidence summary.
In the AChR-Ab+ subgroup, concomitant gMG treatment in the efgartigimod alfa and placebo groups often involved 2 classes of therapy (21.5% versus 34.4%, respectively) or 3 classes of therapy (49.2% versus 46.9%, respectively).
Table 14: Summary of Concomitant Medications in the ADAPT Study (Safety Analysis Set)
Exposure | AChR-Ab+ population | |
|---|---|---|
Efgartigimod alfa (N = 65) | Placebo (N = 64) | |
Any steroid, n (%) | 46 (70.8) | 51 (79.7) |
Any NSIST, n (%) | 40 (61.5) | 37 (57.8) |
Any AChEI, n (%) | 57 (87.7) | 57 (89.1) |
One class, n (%) | || |||||| | || |||||| |
Steroid | | ||||| | | ||||| |
NSIST | | ||||| | | ||||| |
AChEI | || |||||| | | ||||| |
Two classes, n (%) | || |||||| | || |||||| |
Steroid plus NSIST | | ||||| | | ||||| |
Steroid plus AChEI | | |||||| | || |||||| |
NSIST plus AChEI | | ||||| | | ||||| |
All 3 classes, n (%) | || |||||| | || |||||| |
Ab+ = antibody positive; AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; NSIST = nonsteroidal immunosuppressive therapy.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: ADAPT Clinical Study Report.30
Rescue Medication Use
Treatment discontinuation due to rescue therapy was reported for 3 patients (2.3%) in the AChR-Ab+ population, 1 (1.5%) patient in the efgartigimod alfa group, and 2 (3.1%) patients in the placebo group.
Efficacy
Overall results from the primary and key secondary end points for AChR-Ab+ population are presented in Table 15.
Table 15: Summary of MG-ADL and QMG Efficacy Results in the AChR-Ab+ Population (mITT Population)
Variables | Efgartigimod alfa (N = 65) | Placebo (N = 64) |
|---|---|---|
MG-ADL responders during cycle 1 | ||
N | 65 | 64 |
Responders, n (%) | 44 (67.7) | 19 (29.7) |
Risk difference (efgartigimod alfa – placebo), % (95% CI) | 38 (22.05 to 55.96) | |
OR vs. control (95% CI) | 4.951 (2.213 to 11.528) | |
P value | < 0.0001 | |
Early MG-ADL responders during cycle 1 | ||
N | 65 | 64 |
Responders, n (%) | 37 (56.9) | 16 (25.0) |
Risk difference (efgartigimod alfa – placebo), % (95% CI) | 31.9 (NR) | |
P value | Not testeda | |
MG-ADL total score CMI percentage of time | ||
N | 65 | 64 |
Percentage of time of CMI (%), LSM (95% CI) | 48.71 (36.517 to 60.912) | 26.65 (14.12 to 39.15) |
Absolute between-group LSM difference (efgartigimod alfa – placebo), % (95% CI) | 22.07 (10.94 to 33.18) | |
P value | 0.0001 | |
Time to qualify for re-treatment (since week 4 visit) | ||
N | 65 | 64 |
Patients who qualified for re-treatment, n (%) | || |||||| | || |||||| |
Patients censored, n (%) | | |||||| | | |||||| |
Risk difference (efgartigimod alfa – placebo), % (95% CI) | |||| ||||||||| | |
Time to qualify for re-treatment since week 4 visit (days) | ||
Median (95% CI) | |||| |||||| ||||| | ||| ||||| ||||| |
P value | |||||| |||||| | |
Percentage of MG-ADL responders during cycle 2 | ||
N | 51 | 43 |
Responders, n (%) | 36 (70.6) | 11 (25.6) |
Risk difference (efgartigimod alfa – placebo), % (95% CI) | 45.0 (NR) | |
OR vs. control (95% CI) | 8.19 (2.88 to 25.73) | |
P value | 0.001 | |
QMG responders during cycle 1 | ||
N | 65 | 64 |
Responders, n (%) | 41 (63.1) | 9 (14.1) |
Risk difference (efgartigimod alfa – placebo), % (95% CI) | 49 (34.5 to 63.5) | |
OR vs. control (95% CI) | 10.84 (4.18 to 31.20) | |
P value | < 0.0001 | |
AChR-Ab+ = acetylcholine receptor antibody positive; CI = confidence interval; CMI = clinically meaningful improvement; gMG = generalized myasthenia gravis; LSM = least squares mean; MG-ADL = Myasthenia Gravis Activities of Daily Living; mITT = modified intention to treat; NR = not reported; OR = odds ratio; QMG = Quantitative Myasthenia Gravis; vs. = versus.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aThis outcome was not tested statistically due to earlier failure of the hierarchy.
bThe hierarchy test failed at this end point.
Sources: ADAPT Clinical Study Report;1,30 sponsor provided additional information.66
Activities of Daily Living (MG-ADL)
Percentage of MG-ADL Responders During Cycle 1
Primary Analysis
Percentage of MG-ADL responders in the AChR-Ab+ population during cycle 1 was assessed as the primary outcome. In the AChR-Ab+ population, the MG-ADL responder criterion was met by 44 patients (67.7%) in the efgartigimod alfa group and 19 patients (29.7%) in the placebo group. The between-group difference was 38% (95% CI, 22.05% to 53.96%). The OR was 4.95 (95% CI, 2.21 to 11.53; P < 0.0001) (Table 15).
Subgroup Analysis
Various post hoc subgroup analyses were conducted to evaluate the primary end point in clinically relevant subgroups within the AChR-Ab+ population (Table 16).1,33,67 Results among the subgroups analyzed were generally consistent with the main analysis.
Table 16: Subgroup Analyses of Cycle 1 MG-ADL Response in the AChR-Ab+ Population (Post Hoc Analyses, ADAPT)
Prior MG therapy | Efgartigimod alfa AChR-Ab+, % (n of N) | Placebo AChR-Ab+, % (n of N) | Treatment difference, % (95% CI) |
|---|---|---|---|
According to prior therapiesa | |||
Any prior MG therapy | 67.7 (44 of 65) | 29.7 (19 of 64) | 38.0 (22.05 to 53.96) |
Prior AChEI only | |||| |||||| | ||| ||||| | |||| ||||||| |||||| |
Any prior steroid | |||| ||||||| | |||| ||||||| | |||| ||||||| |||||| |
Any prior NSIST | 63.8 (30 of 47) | 30.2 (13 of 43) | 33.6 (14.18 to 53.02) |
Steroids plus NSIST plus AChEI | |||| ||||||| | |||| ||||||| | |||| ||||||| |||||| |
According to concomitant therapiesa | |||
Any concomitant MG therapy | 67.7 (44 of 65) | 30.2 (19 of 63) | 37.5 (21.48 to 53.59) |
AChEI only | 84.6 (11 of 13) | 16.7 (1 of 6) | 67.9 (32.26 to 100.0) |
Any steroid | 63.0 (29 of 46) | 29.4 (15 of 51) | 33.6 (14.90 to 52.37) |
No steroid | |||| ||||||| | |||| |||||| | |||| ||||||| |||||| |
Any NSIST | 65.0 (26 of 40) | 29.7 (11 of 37) | 35.3 (14.40 to 56.14) |
No NSIST | |||| ||||||| | |||| |||||| | |||| ||||||| |||||| |
Steroids plus NSIST plus AChEI | |||| ||||||| | |||| |||||| | |||| ||||||| |||||| |
Published subgroup analysesb | |||
Disease duration | |||
Disease duration < 3 years | 78.6 (11 of 14) | 23.5 (4 of 17) | 55.0 (25.6 to 84.5) |
Disease duration 3 to < 6 years | 85.7 (12 of 14) | 53.3 (8 of 15) | 32.4 (1.2 to 63.6) |
Disease duration ≥ 6 years | 56.8 (21 of 37) | 21.9 (7 of 32) | 34.9 (13.4 to 56.3) |
Prior treatment failure | |||
Prior treatment failurec | 67.5 (27 of 40) | 31.7 (13 of 41) | 35.8 (15.5 to 56.1) |
No prior treatment failurec | 68.0 (17 of 25) | 26.1 (6 of 23) | 41.9 (16.3 to 67.5) |
Thymectomy | |||
Prior thymectomy | 60.0 (27 of 45) | 26.7 (8 of 30) | 33.3 (12.0 to 54.7) |
No prior thymectomy | 85.0 (17 of 20) | 32.4 (11 of 34) | 52.6 (30.5 to 74.8) |
Ab+ = antibody positive; AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; CI = confidence interval; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; mITT = modified intention to treat; NSIST = nonsteroidal immunosuppressive therapy.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aPost hoc analyses of Argenx data on file (April 21, 2023).1 Selected data presented by Karam et al. (2021).67
bAdapted from Bril et al. (2022)33 and Sacca et al. (2023).32
cPrior treatment failure is defined as prior exposure to ≥ 2 immunosuppressive therapies or treatment with ≥ 1 immunosuppressive therapy and requiring plasma exchange or IV immunoglobulin multiple times within 1 year before study inclusion.
Sources: ADAPT Clinical Study Report;30 sponsor’s submission.1
Sensitivity Analysis
Per-protocol analysis was done as a sensitivity analysis of MG-ADL responders in the AChR-Ab+ population during cycle 1. The results of this per-protocol analysis were consistent with the results in the mITT set. In the AChR-Ab+ population, the MG-ADL responder criterion was met in || ||||||| patients in the efgartigimod alfa group compared to 18 (34.0%) patients in the placebo group (|| |||| |||||||| ||||||| |||||).30
Percentage of MG-ADL Responders During Cycle 2
Percentage of MG-ADL responders during cycle 2 was assessed as exploratory outcome (Table 15 and Figure 2). In the AChR-Ab+ population, the proportion of MG-ADL responders in each group during cycle 2 appeared to be similar to that in cycle 1 in the AChR-Ab seropositive population. MG-ADL responders were reported in 36 of 51 patients (70.6%) in the efgartigimod alfa group and 11 of 43 patients (25.6%) in the placebo group in cycle 2. The between-group difference was 45.0% (95% CI, NR). The OR was 8.19 (95% CI, 2.88 to 25.73; P < 0.001).66
Early MG-ADL Responders
Early MG-ADL responders (i.e., responded at week 2 of the cycle) in the AChR-Ab+ population was assessed as a key secondary outcome. Early MG-ADL response occurred in 37 patients (56.9%) in the efgartigimod alfa group and 16 patients (25.0%) in the placebo group. The between-group difference was 31.9% (95% CI, NR). The || |||| ||| was |||| ||||| || |||||| | ||||| | ||||||.66
Percentage of Time with a CMI of MG-ADL Score
In the AChR-Ab+ population, the percentage of time with a CMI (reduction of ≥ 2 points versus study entry baseline) in the MG-ADL total score was assessed as a key secondary outcome. It was observed that the least squares mean (LSM) percentage of time with a CMI up to day 126 was 48.7% (standard error [SE] = 6.2) in the efgartigimod alfa group compared to 26.6% (SE = 6.3) in the placebo group. The between-group difference was 22.1% (95% CI, 10.9% to 33.2%; in favour of efgartigimod alfa; P = 0.0001) (Table 15).
Time to Qualify for Re-Treatment
Time to qualify for re-treatment in the AChR-Ab+ Population was assessed as a key secondary outcome. Since the week 4 visit, || ||||||| patients in the efgartigimod alfa group and || ||||||| patients in the placebo group qualified for re-treatment. The between-group difference was ||||| |||| ||| ||| ||||||||. The median time to qualify for re-treatment || |||| ||||||| ||||| |||| || |||| ||||| in the efgartigimod alfa group and 8 days (95% CI, 1.0 day to 30.0 days) in the placebo group, respectively (||| ||||| | ||||| | ||||||). The statistical testing hierarchy was broken at this end point (Table 15 and Table 17). The Kaplan-Meier plot for time to qualification for re-treatment in the AChR-Ab+ population is presented in Figure 2. The hazard ratio was not provided.1
Table 17: Time to Qualify for Re-Treatment in the AChR-Ab+ Population (mITT Analysis Set)
Variables | ADAPT (AChR-Ab+ population) | |
|---|---|---|
Efgartigimod alfa (N = 65) | Placebo (N = 64) | |
Day 14 | ||
N | || | || |
Patients who qualified for re-treatment, n (%) | || | || |
Patients censored in each group, n (%) | | ||| | | ||||| |
Risk difference (efgartigimod alfa vs. placebo), % (95% CI) | ||||| |||||| || |||||| | |
Event rate, % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| |
Day 28 | ||
N | || | || |
Patients who qualified for re-treatment, n (%) | || | || |
Patients censored in each group, n (%) | | ||| | | ||||| |
Risk difference (efgartigimod alfa vs. placebo), % (95% CI) | ||||| |||||| || |||||| | |
Event rate, % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| |
Day 42 | ||
N | || | || |
Patients who qualified for re-treatment, n (%) | || | || |
Patients censored in each group, n (%) | | ||| | | ||||| |
Risk difference (efgartigimod alfa vs. placebo), % (95% CI) | |||| |||||| || |||| | |
Event rate, % (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| |
All time (up to 168 days) | ||
N | || | || |
Patients who qualified for re-treatment, n (%) | || |||||| | || |||||| |
Patients censored, n (%) | | |||||| | | |||||| |
Risk difference (efgartigimod alfa vs. placebo), % (95% CI) | |||| |||| | |
AChR+ = acetylcholine receptor antibody positive; CI = confidence interval; mITT = modified intention to treat; NR = not reported; vs. = versus.
Sources: Sponsor’s submission;1 and sponsor-provided additional information.36
Figure 3: Time to Qualify for Re-Treatment in the AChR-Ab+ Population (mITT Analysis Set) [Redacted]

|||| || ||||||| ||| ||||||||||| || ||| |||||||| |||||||||| ||||| |||||||| |||||||| ||||| |||||| | |||||| |||| |||||||||| || |||||||| ||||||||| ||| ||||||||||| || || ||| ||||| || |||||| ||||| |||| | |||||| || ||||||| |||||||| || |||||||||||| |||| ||||| ||| || ||||||| ||||||||| ||| ||||||||||||||
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sources: ADAPT Clinical Study Report;30 sponsor’s submission.1
MG-ADL Total Score Mean Change From Cycle Baseline During Cycle 1 and Cycle 2
The MG-ADL total score changes from baseline in cycle 1 and cycle 2 are presented in Table 18 and Figure 3. Figure 3 shows that the maximum improvement from study entry baseline in MG-ADL total score occurred at week 4 during cycle 1 and cycle 2 in the efgartigimod alfa group of the AChR-Ab+ population.
At week 4 of cycle 1, the LSM change from study entry baseline in the MG-ADL total score was –4.104 points (95% CI, –5.007 points to –3.201 points) in the efgartigimod alfa group and –1.269 (95% CI, –2.199 to –0.339) in the placebo group. The between-group difference in the LSM change from baseline at week 4 of cycle 1 was –2.84 (95% CI, –3.8 to –1.9; P < 0.0001) (Table 18). At the end of cycle 1, the LSM change from study entry baseline in the MG-ADL total score was –1.56 (95% CI, –2.52 to –0.60) in the efgartigimod alfa group and –1.19 (95% CI, –2.18 to –0.18) in the placebo group. The between-group difference in the LSM change from baseline at week 4 of cycle 1 was –0.37 (95% CI, –1.45 to 0.71; P = 0.4978) (Table 18).
Table 18: MG-ADL Total Score LSM Change From Cycle Baseline During Cycle 1 and Cycle 2 in the AChR-Ab+ Population (mITT Population)
Outcomes | ADAPT (AChR-Ab+ population) | |
|---|---|---|
Efgartigimod alfa (N = 65) | Placebo (N = 64) | |
Change from baseline in MG-ADL score at week 4 of cycle 1 | ||
n | 65 | 63 |
LSM change (95% CI) | ||||| | |||||| |||||| | |||||| | |||||| |||||| |
Difference in LSM change, (EFG – PBO) (95% CI) | ||||| ||||||| |||||| | |
P value | < 0.0001a | |
Change from baseline in MG-ADL score at the end of cycle 1b | ||
n | 65 | 63 |
LSM change (95% CI) | ||||| | |||||| |||||| | ||||| | |||||| |||||| |
Difference in LSM change, (EFG – PBO) (95% CI) | ||||| ||||||| ||||| | |
P value | | | ||||||| | |
Change from cycle 2 baseline in MG-ADL score at week 4 of cycle 2 | ||
n | 46 | 52 |
LSM change (95% CI) | ||||| | |||||| |||||| | ||||| | |||||| ||||| |
Difference in LSM change, (EFG – PBO) (95% CI) | ||||| ||||||| |||||| | |
P value | < 0.0001a | |
Change from cycle 2 baseline in MG-ADL score at the end of cycle 2b | ||
n | 46 | 52 |
LSM change (95% CI) | ||||| | |||||| |||||| | |||| | |||||| ||||| |
Difference in LSM change, (EFG – PBO) (95% CI) | ||||||||||||| |||||| | |
P value | | | ||||||| | |
AChR-Ab+ = acetylcholine receptor antibody positive; CI = confidence interval; LSM = least squares mean; MG-ADL = Myasthenia Gravis Activities of Daily Living; mITT = modified intention to treat.
aP values were not adjusted for multiplicity.
bThe treatment period is defined as a 26-week period divided into 8-week treatment cycles; therefore, the end of cycle n was defined as week 8
Sources: Sponsor’s submission1 and the sponsor-provided additional information.66
Figure 4: MG-ADL Total Score Mean Change From Cycle Baseline During Cycle 1 and Cycle 2 in the AChR-Ab+ Population (mITT Analysis Set)
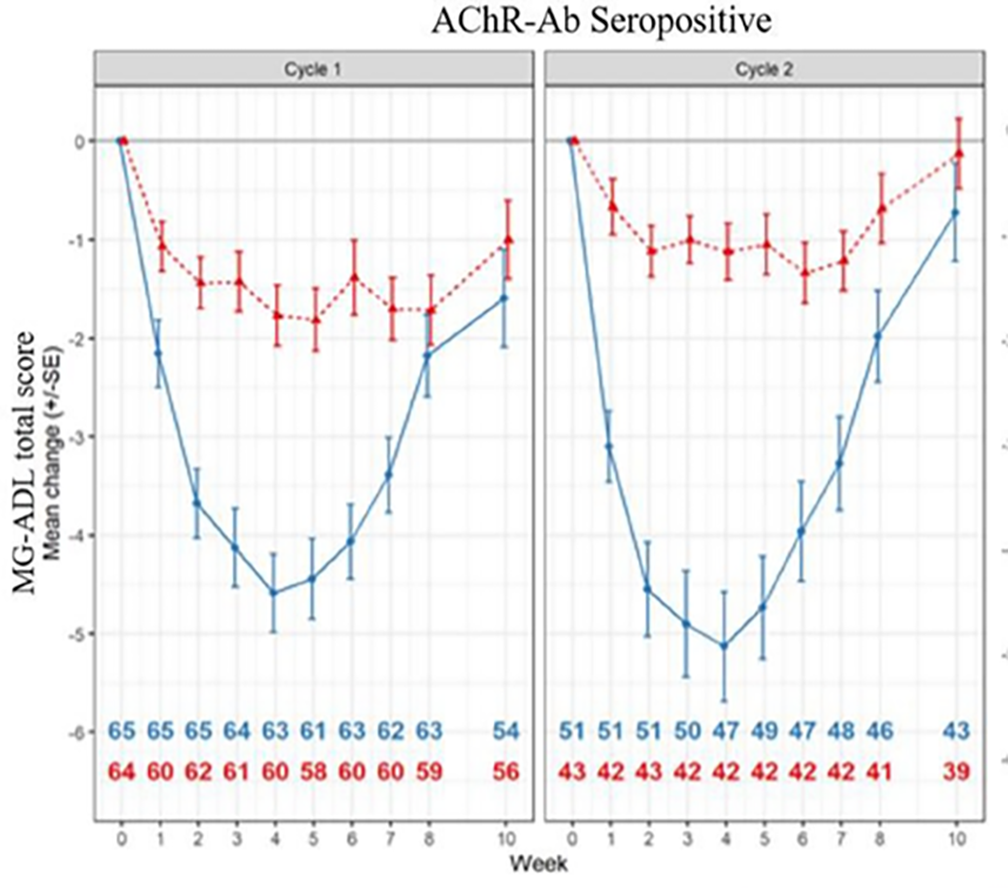
Ab = antibody; AChR = acetylcholine receptor; MG-ADL = Myasthenia Gravis Activities of Daily Living; mITT = modified intention to treat; SE = standard error.
Notes: The MG-ADL was scored from 0 to 24, with higher scores indicating more severe disease.
The red dotted line represents changes over time in the efgartigimod alfa group; the blue solid line represents changes over time in the placebo group.
Sources: ADAPT Clinical Study Report;30 sponsor’s submission.1
Figure 5: MG-ADL Total Score Mean Change From Cycle Baseline During Cycle 1 and Cycle 2 in the Overall Population (mITT Analysis Set) [Redacted]

Ab = antibody; AChR = acetylcholine receptor; MG-ADL = Myasthenia Gravis Activities of Daily Living; mITT = modified intention to treat; SE = standard error.
Notes: The MG-ADL was scored from 0 to 24, with higher scores indicating more severe disease.
The red curve represents changes over time in the efgartigimod alfa group; the blue represents changes over time in the placebo group.
Sources: ADAPT Clinical Study Report;30 sponsor’s submission.1
Disease Severity (Assessed With QMG)
Quantitative Myasthenia Gravis
The percentage of QMG responders during cycle 1 within the AChR-Ab+ population was assessed as the first key secondary outcome. The QMG responder criterion was met by 41 patients (63.1%) in the efgartigimod alfa group and 9 patients (14.1%) in the placebo group. The between-group difference was 49% (95% CI, 34.5% to 63.5%). The OR was 10.84 (95% CI, 4.18 to 31.20; P < 0.0001) (Table 15, Figure 4).
Figure 6: Percentage of QMG Responders During Cycle 1 in the AChR-Ab+ Population (mITT Analysis Set)
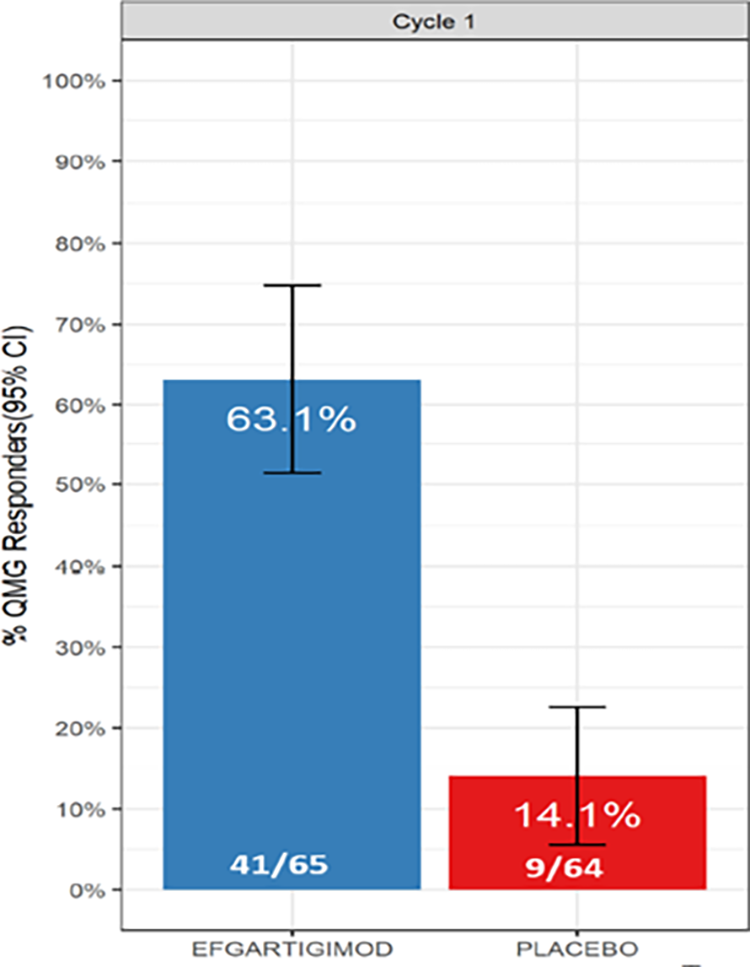
AChR-Ab+ = acetylcholine receptor antibody positive; CI = confidence interval; mITT = modified intention to treat; QMG = Quantitative Myasthenia Gravis.
Sources: ADAPT Clinical Study Report30 and the sponsor’s submission.1
Figure 7: Percentage of QMG Responders During [Redacted] in the AChR-Ab+ Population (mITT Analysis Set) [Redacted]

AChR-Ab+ = acetylcholine receptor antibody positive; CI = confidence interval; mITT = modified intention to treat; QMG = Quantitative Myasthenia Gravis.
Sources: ADAPT Clinical Study Report30 and the sponsor’s submission.1
Health-Related Quality of Life
Revised Myasthenia Gravis Quality of Life 15-Item Scale
Mean Change From Baseline During Cycle 1 and Cycle 2 (mITT Analysis Set)
MG-QoL15r mean change from baseline during cycle 1 and cycle 2 was assessed as an exploratory outcome (Table 19).
The LSM change in MG-QoL15r score from study baseline to week 4 of cycle 1 was –7.21 (95% CI, –8.79 to –5.63) in the efgartigimod alfa group and –1.76 (95% CI, –3.38 to –0.14) in the placebo group. The between-group difference in LSM change from study baseline at week 4 of cycle 1 was –5.45 (95% CI, –7.22 to –3.69; P < 0.0001). At the end of cycle 1, the LSM change from study baseline was –4.07 (95% CI, –5.69 to –2.45) in the efgartigimod alfa group and –1.70 (95% CI, –3.37 to –0.04) in the placebo group. The between-group difference in LSM change from study baseline at week 4 of cycle 1 was –2.37 (95% CI, –4.21 to –0.53; P = 0.0123).
At cycle 2, at week 4, the LSM change from study baseline was –5.09 (95% CI, –6.73 to –3.47) in the efgartigimod alfa group and 0.35 (95% CI, –1.44 to 2.14) in the placebo group. The between-group difference in LSM change from study baseline at week 4 of cycle 2 was –5.45 (95% CI, –7.28 to –3.62; P < 0.0001). At the end of cycle 2, the LSM change from study baseline was –1.71 (95% CI, –3.34 to –0.08) in the efgartigimod alfa group and 0.35 (95% CI, –1.41 to 2.11) in the placebo group. The between-group difference in LSM change from study baseline at the end of cycle 2 was –2.06 (95% CI, –3.85 to –0.28; P = 0.0239).
Table 19: Summary of MG-QoL15r and EQ-5D VAS Results from the ADAPT Trial (AChR-Ab+)
Outcomes | ADAPT (AChR-Ab+) | |
|---|---|---|
Efgartigimod alfa (N = 65) | Placebo (N = 64) | |
Change from baseline in MG-QoL15r score at week 4 of cycle 1 | ||
n | 63 | 60 |
LSM change (95% CI) | ||||| ||||||| |||||| | ||||| ||||||| |||||| |
Difference in LSM change (95% CI) | –5.45 ||||||| |||||| | |
P value | < 0.0001 | |
Change from baseline in MG-QoL15r score at the end of cycle 1b | ||
n | 63 | 59 |
LSM change (95% CI) | ||||| ||||||| |||||| | ||||| ||||||| |||||| |
Difference in LSM change (95% CI) | –2.37 ||||||| |||||| | |
P value | 0.0123a | |
Change from cycle 2 baseline in MG-QoL15r score at week 4 of cycle 2 | ||
n | 47 | 42 |
LSM change (95% CI) | ||||| ||||||| |||||| | |||| ||||||| ||||| |
Difference in LSM change (95% CI) | –5.45 |||||||| ||||||| | |
P value | < 0.0001a | |
Change from cycle 2 baseline in MG-QoL15r score at the end of cycle 2b | ||
n | 46 | 41 |
LSM change (95% CI) | ||||| ||||||| |||||| | |||| ||||||| ||||| |
Difference in LSM change (95% CI) | –2.06 ||||||| |||||| | |
P value | 0.0239a | |
Change from baseline in EQ-5D VAS at week 4 of cycle 1 | ||
n | 63 | 60 |
LSM change (95% CI) | ||||| ||||||| |||||| | |||| ||||||| ||||| |
Difference in LSM change (EFG – PBO) (95% CI) | 13.28 |||||| |||||| | |
P value | < 0.0001a | |
Change from baseline in EQ-5D VAS at the end of cycle 1b | ||
n | 63 | 59 |
LSM change (95% CI) | |||| ||||||| ||||| | |||| |||||||| ||||| |
Difference in LSM change, (EFG – PBO) (95% CI) | 0.69 ||||||| ||||| | |
P value | 0.7784a | |
Change from cycle 2 baseline in EQ-5D-5L VAS at week 4 of cycle 2 | ||
n | 47 | 42 |
LSM change (95% CI) | ||||| |||||| |||||| | |||| ||||||| ||||| |
Between-group absolute mean difference in LSM change, (EFG – PBO) (95% CI) | 12.24 |||||| |||||| | |
P value | < 0.0001a | |
Change from cycle 2 baseline in EQ-5D-5L VAS at the end of cycle 2b | ||
n | 46 | 41 |
LSM change (95% CI) | |||| ||||||| ||||| | |||| ||||||| ||||| |
Between-group absolute mean difference in LSM change, (EFG – PBO) (95% CI) | –0.10 ||||||| ||||| | |
P value | 0.9693a | |
AChR-Ab+ = AChR antibody positive; CI = confidence interval; EFG = efgartigimod alfa group; LSM = least squares mean; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item; PBO = placebo group; VAS = visual analogue scale.
aP values were not adjusted for multiplicity.
bThe treatment period is defined as a 26-week period divided into 8-week treatment cycles; therefore, the end of cycle n was defined as week 8.
Sources: The sponsor’s submission1 and the sponsor-provided additional information.36
Figure 8: MG-QoL15r Least Squares Mean Change From Baseline During Cycle 1 and Cycle 2 in the AChR-Ab+ Population (mITT Analysis Set)
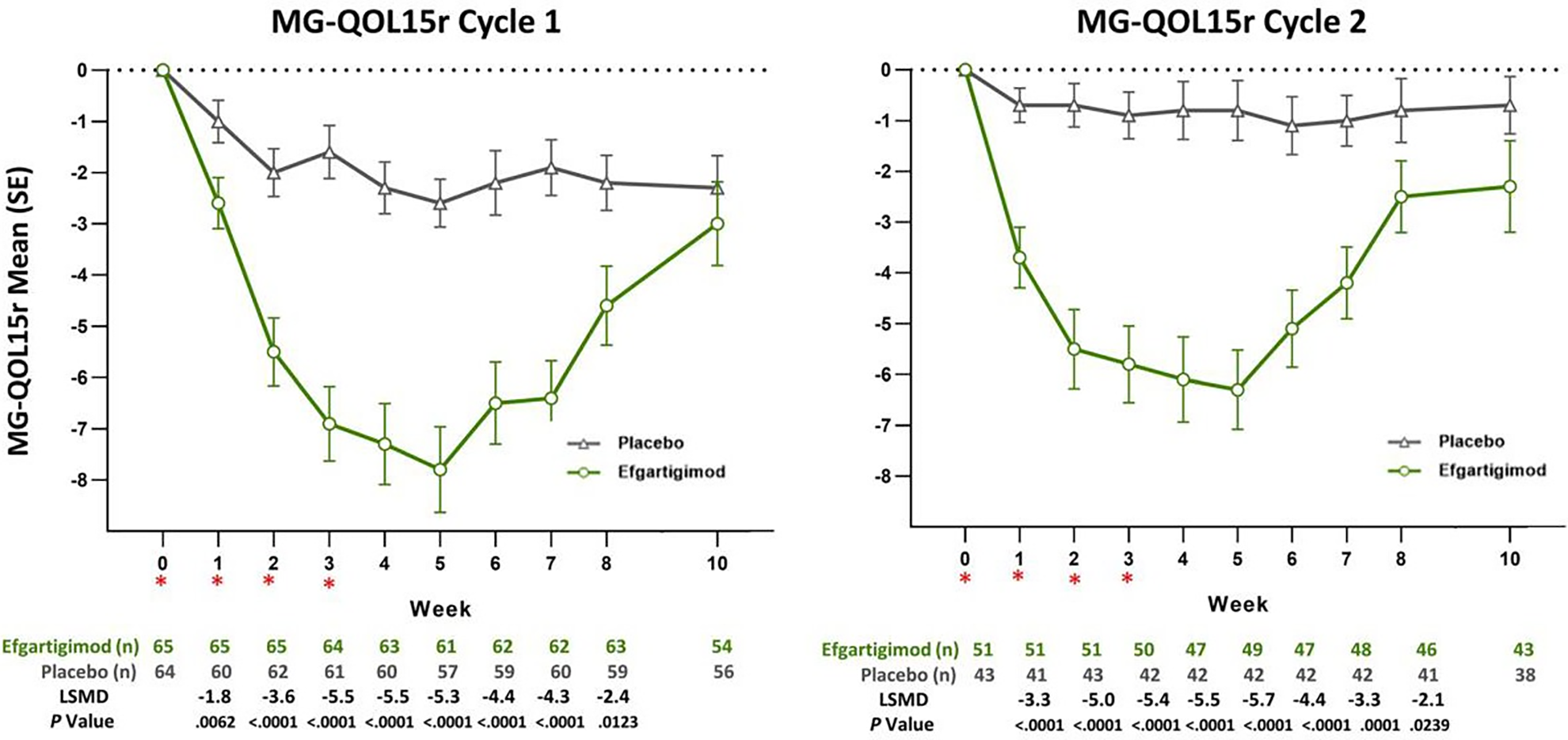
AChR-Ab+ = acetylcholine receptor antibody positive; LSM = least squares mean; LSMD = least squares mean difference; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item; mITT = modified intention to treat; SE = standard error.
Note: The MG-QoL15r was scored from 0 to 30, with higher scores indicating more impaired quality of life and decreases indicating improved quality of life.
* Indicates treatment administration (efgartigimod alfa or placebo).
Source: Sacca F et al. J Neurol. 2023;270:2096-2105. Reprinted with accordance with Creative Commons Attribution 4.0 International License (CC BY 4.0): https://creativecommons.org/licenses/by/4.0/50
EQ-5D
EQ-5D VAS Mean Change From Baseline
EQ-5D VAS mean change from baseline during cycle 1 and cycle 2 was assessed as an exploratory outcome (Table 19).
At week 4 of cycle 1, the LSM change from baseline was 16.04 (95% CI, 11.37 to 20.71) in the efgartigimod alfa group and 2.76 (95% CI, –2.05 to 7.56) in the placebo group. The between-group difference in LSM change from baseline at week 4 of cycle 1 was 13.28 (95% CI, 8.32 to 18.24; P < 0.0001). At the end of cycle 1, the LSM change from baseline was 4.61 (95% CI, –0.01 to 9.24) in the efgartigimod alfa group and 3.92 (95% CI, –0.09 to 8.69) in the placebo group. The between-group difference in LSM change from baseline to the end of cycle 1 was 0.69 (95% CI, –4.19 to 5.59; P = 0.7784).
At week 4 of cycle 2, the LSM change from baseline was 14.16 (95% CI, 9.69 to 18.63) in the efgartigimod alfa group and 1.92 (95% CI, –3.03 to 6.87) in the placebo group. The between-group difference in LSM change from baseline at week 4 of cycle 2 was 12.24 (95% CI, 7.33 to 17.16; P < 0.0001). At the end of cycle 2, the LSM change from baseline was 0.16 (95% CI, –4.53 to 4.86) in the efgartigimod alfa group and 0.27 (95% CI, –4.87 to 5.41) in the placebo group. The between-group difference in LSM change from baseline to the end of cycle 2 was –0.10 (95% CI, –5.40 to 5.19; P = 0.9693).
Figure 9: EQ-5D-5L VAS Mean Change From Baseline in the AChR-Ab+ Population (mITT Analysis Set)
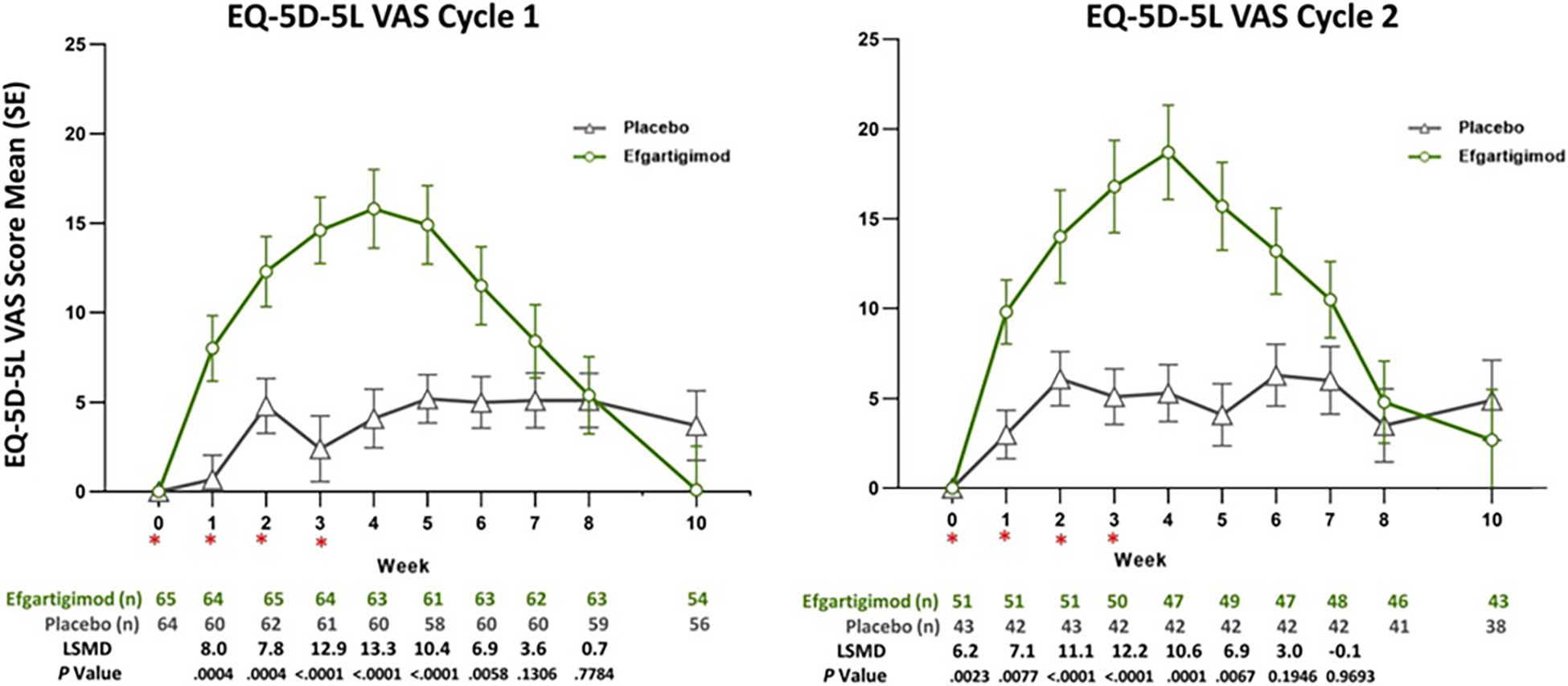
Ab+ = antibody seropositive; AChR = acetylcholine receptor; EQ-5D-5L = 5-Level EQ-5D; LSM = least squares mean; LSMD = least squares mean difference; mITT = modified intention to treat; SE = standard error; VAS = visual analogue scale.
Note: The cycle 2 panel contains a transposition error, where the values shown in the black line with triangles start under week 0 instead of week 1.
* Indicates treatment administration (efgartigimod alfa or placebo).
Source: Sacca F et al. J Neurol. 2023;270:2096-2105. Reprinted with accordance with Creative Commons Attribution 4.0 International License (CC BY 4.0): https://creativecommons.org/licenses/by/4.0/50
Other Clinical Outcomes (Post Hoc Analysis)
MG Hospitalizations
Post hoc analysis of hospitalizations was conducted using data from the ITT population of ADAPT.59 In that analysis, the numbers of all-cause and gMG-related hospitalizations were combined with follow-up times to calculate the incidence rates of hospitalizations, which were compared between the efgartigimod alfa and placebo arms for the AChR-Ab+ population. By week 26, no patients in the efgartigimod alfa group and 3 (4.69%) patients in placebo group had MG hospitalizations. The between-group difference (efgartigimod alfa – placebo) was –4.69% (95% CI, NR) (Table 20). The incidence of gMG-related hospitalizations was 0 per 100 patient-years and 10.98 per 100 patient-years in patients in the efgartigimod alfa and placebo groups, respectively.36,59 The between-group difference was 10.98 per 100 patient-years (95% CI, –23.42 per 100 patient-years to 1.47 per 100 patient-years).
Table 20: Results of Other Clinical Outcomes in the ADAPT Trial (AChR-Ab+ Population, Week 26) (mITT)
Outcomes | ADAPT (AChR-Ab+ population) | |
|---|---|---|
Efgartigimod alfa (N = 65) | Placebo (N = 64) | |
Proportion of patients with MG hospitalizations by week 26 | ||
Patients with events, n | 0 | 3 |
Percentage of patients with events, % (95% CI) | 0 (0) | 4.69 (NR) |
Risk difference (EFG – PBO), % (95% CI) | –4.69 (NR) | |
Incidence rate per 100 follow-up years, % (95% CI) | 0 (0) | 10.98 (NR) |
Between-group difference (EFG – PBO), % (95% CI) | –10.98 (–23.42 to 1.47) | |
Proportion of patients with MG exacerbations by week 26 | ||
Patients with events, n | 17 | 27 |
Percentage of patients with events, % (95% CI) | 26.1 (NR) | 44.3 (NR) |
Risk difference (EFG – PBO), % (95% CI) | 18.2 (NR) | |
Proportion of patients with MG crisis by week 26 | ||
Patients with events, n (%) | ||| | ||| |
Percentage of patients with events, % (95% CI) | | ||| | ||| |||| |
Risk difference (EFG – PBO), % (95% CI) | |||| |||| | |
Incidence rate per 100 FU years, % (95% CI) | |||| | |||||||| |
Between-group difference (EFG – PBO), % (95% CI) | ||||| |||||||| ||||| | |
P value | |||||| | |
AChR-Ab+ = acetylcholine receptor antibody positive; CI = confidence interval; EFG = efgartigimod alfa group; mITT = modified intention to treat; MG = myasthenia gravis; NR = not reported; PBO = placebo group.
Notes: MG hospitalization, MG exacerbation, and MG crisis were assessed as post hoc analyses. P values were not adjusted for multiplicity for type I error control.
Sources: Sponsor’s submission1 and the sponsor-provided additional information.36
MG Exacerbation
gMG exacerbation was defined as a greater than or equal to 3-point worsening in QMG score versus baseline. Among patients who were AChR-Ab+, gMG exacerbations were identified in 17 (26.1%) in the efgartigimod alfa group and 27 (44.3%) in the placebo group. The between-group absolute risk difference was –18.2% (95% CI, NR; nominal P = 0.033).36,59
MG Crisis
At week 26, In patients with AChR-Ab+, use of rescue therapy was reported | |||| in the efgartigimod alfa group | ||||||| ||||||| in the placebo group. The between-group difference was |||||| ||| ||| ||| ||||||||. The incidence rate per 100 follow-up years, was ||| in the efgartigimod alfa group and |||| in the placebo group. The between-group incidence rate difference was |||||| |||||| |||||| || ||||| ||||||||.
Harms
Only those harms identified in the sponsor’s evidence summary review protocol are reported below.
AEs in the population of patients who were AChR-Ab+ are presented in Table 21.
Adverse Events
During the randomized controlled period, among patients in the AChR-Ab+ group, TEAEs were reported in || ||||||| patients in the efgartigimod alfa group and || ||||||| patients in the placebo group (Table 21). The most common TEAEs (occurring in ≥ 10% patients in any arm) were headache (EFG versus PBO, ||||| ||| |||||); nasopharyngitis (EFG versus PBO, ||||| ||||||||), upper respiratory tract infection |||||||| ||| ||||||, and diarrhea (EFG versus PBO | |||| ||| |||||).
Serious Adverse Events
During the randomized controlled period, treatment-emergent SAEs in patients in the AChR-Ab+ population were reported in ||| |||||| patients in the efgartigimod alfa group and | |||||| patients in the placebo group (Table 21).
Withdrawal of Treatment Due to Adverse Events
During the randomized controlled period, TEAEs that led to discontinuation of treatment among patients in the AChR-Ab+ population were reported for | |||||| patients in the efgartigimod alfa group and | |||||| patients in the placebo group. No specific TEAE that led to discontinuation of the investigational medicine product was reported for more than 1 patient in either treatment group.
Mortality
During the randomized controlled period, no death was reported in either group (Table 21).
Notable Harms (Adverse Events of Special Interest)
In the ADAPT trial, AESIs were defined as infections and infestations. During the randomized controlled period, as shown in Table 21, AESIs were reported for || ||||||| patients in the efgartigimod alfa group and || ||||||| patients in the placebo group. The most frequently reported treatment-emergent AESIs (≥ 3% of patients in either group) among patients in the AChR-Ab+ population were: nasopharyngitis (EFG versus PBO: ||||| ||||||||); upper respiratory tract infection |||||| ||| ||||); urinary tract infection ||||| ||| |||||| bronchitis (|||| ||| |||||; and influenza (|||| ||| ||||| (Table 21).
Table 21: Adverse Events in the AChR-Ab+ Population (Safety Analysis Set)
Adverse events | ADAPT (AChR-Ab+ population) | |
|---|---|---|
Efgartigimod alfa (N = 65) | Placebo (N = 64) | |
≥ 1 TEAE, n (%) | || |||||| | || |||||| |
Common TEAEs (≥ 3% patients with TEAE in any arm), n (%) | ||
Abdominal pain | | ||||| | | ||||| |
Diarrhea | | ||||| | | |||||| |
Nausea | | ||||| | | ||||| |
Fatigue | | ||||| | | ||||| |
Bronchitis | | ||||| | | ||||| |
Influenza | | ||||| | | ||||| |
Nasopharyngitis | | |||||| | || |||||| |
Upper respiratory tract infection | | |||||| | | ||||| |
Urinary tract infection | | ||||| | | ||||| |
Contusion | | ||||| | | ||||| |
Procedural headache | | ||||| | |||| |
Back pain | | ||||| | | ||||| |
Myalgia | | ||||| | |||| |
Pain in extremity | |||| | | ||||| |
Dizziness | | ||||| | | ||||| |
Headache | || |||||| | || |||||| |
Myasthenia gravis | | ||||| | | ||||| |
Paresthesia | | ||||| | | ||||| |
Cough | | ||||| | | ||||| |
Oropharyngeal pain | | ||||| | | ||||| |
Hypertension | | ||||| | | ||||| |
Serious adverse events, n (%) | ||
Patients with 1 SAE, n (%) | | ||||| | | ||||| |
Most common SAEs (≥ 2% patients with SAE in any arm, n (%) | ||
Blood and lymphatic system disorders | | ||||| | |||| |
Thrombocytosis | | ||||| | |||| |
Cardiac disorders | |||| | | ||||| |
Myasthenia gravis | |||| | | ||||| |
Myasthenia gravis crisis | |||| | | ||||| |
Patients who stopped treatment due to adverse events, n (%) | | ||||| | | ||||| |
Deaths, n (%) | | ||| | | ||| |
Adverse events of special interest, n (%) | ||
Infections and infestations, n (%) | || |||||| | || |||||| |
Most common AESI (≥ 3% of patients with an AESI in any arm), n (%) | ||
Bronchitis | | ||||| | | ||||| |
Influenza | | ||||| | | ||||| |
Nasopharyngitis | | |||||| | || |||||| |
Upper respiratory tract infection | | |||||| | | ||||| |
Urinary tract infection | | ||||| | | ||||| |
AChR-Ab+ = acetylcholine receptor antibody positive; AESI = adverse event of special interest; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Sources: Sponsor’s submission;1 sponsor-provided additional information.36
Critical Appraisal
Internal Validity
Appropriate methods of randomization, blinding, and allocation concealment were reported. Outcomes were assessed using validated scales incorporating physician and patient assessments, and end points requiring a combination of CMI and sustained effect. However, minimally important between-group differences, that is, the thresholds used for the GRADE for all outcomes, were not available. Therefore, clinical expert opinion was used to inform the thresholds to determine whether the between-group difference observed for each outcome are clinically meaningful. Appropriate statistical methods were used. Multiplicity adjustments were made for the primary and 5 key secondary outcomes to control for family-wise type I error (probability of making more than 1 type I error); the hierarchy failed at time to re-treatment (result was not statistically significant). Other outcomes (e.g., tertiary, exploratory, and outcomes assessed in cycle 2) were not adjusted for multiplicity, so there is an increased risk of false-positive conclusions for statistically significant results.
Not all patients participated in the second cycle; therefore, there is a risk of selection bias. The extent and direction of the bias on the outcomes assessed during the second treatment cycle cannot be determined.
In addition, although the overall baseline demographic and clinical disease characteristics were mostly balanced, there was some notable imbalance between the efgartigimod alfa and placebo groups in AChR-Ab+ population. For example, more patients in the efgartigimod alfa group had an MG-ADL score greater than or equal to 10 than in placebo group (EFG versus PBO: ||||| ||| ||||| in AChR-Ab+ population), fewer patients in the efgartigimod alfa group had prior combination use of steroid plus AChEI than in placebo group (EFG versus PBO | |||| ||| ||||| in AChR-Ab+ population); and the proportion of patients who underwent thymectomy was higher in the efgartigimod alfa group than in the placebo group (EFG versus PBO: 69.2% versus 46.9% in AChR-Ab+ population). Furthermore, more patients in the efgartigimod alfa group used concomitant AChEIs than in placebo group (EFG versus PBO: ||||| ||| |||| in AChR-Ab+ population); fewer patients in the efgartigimod alfa group used concomitant steroid plus AChEI than that in placebo group (EFG versus PBO: ||||| ||| ||||| in in AChR-Ab+ population). Whether these baseline imbalances introduced bias is uncertain. The clinical experts consulted by CADTH for this review indicated that these observed imbalances were unlikely to have significantly affected the study results.
The treatment discontinuations were relatively infrequent in the efgartigimod alfa group compared with the placebo group (4.6% versus 14.1%, respectively). Protocol violations, prohibited drug use, and rescue therapy were all infrequent in both arms, all of which are unlikely to impact the study results. There was no indication from the efficacy or harms data that unblinding of patients or study personnel had occurred.
Furthermore, the changes from the study baseline to the end of the study (i.e., by week 26) were not assessed. The clinical experts CADTH consulted for this review indicated that the assessment at cycle 1 is acceptable. The results of other clinical outcomes (MG hospitalization, MG exacerbations, and MG crisis) were based on post hoc analyses. The study was not powered to assess the between-group difference of these outcomes. Therefore, the results should be interpreted with consideration of limitations of the statistics.
External Validity
Of the patients who were AChR-Ab+, 63% had experienced prior gMG treatment failure (also known as refractory gMG).32 Thirty-seven percent of patients were classified as having responded inadequately to the existing standard-of-care gMG therapy. According to the clinical experts CADTH consulted for his review, the population included in the ADAPT trial is adequately reflective of the population of patients in Canadian clinical settings who experience unmet needs in the treatment of gMG.
According to the clinical experts consulted by CADTH for this review, the demographic and disease characteristics and prior treatment histories of the patients enrolled in the ADAPT trial were reflective of the population of adult patients with gMG in Canada. The eligibility criteria for patients with MGFA Class II to IV gMG and an MG-ADL total score of 5 or more would select appropriately for patients with symptomatic gMG most in need of intervention. Patients with MGFA Class V gMG were excluded from the ADAPT trial, and very few patients with MGFA Class IV gMG were included (3.9% in the AChR-Ab+ population). Whether the findings of the ADAPT trial can be generalized to patients with MGFA Class IV or MGFA Class V is uncertain. The clinical experts consulted by CADTH for this review indicated that a subset of patients with MGFA Class I (ocular MG) or V and MG-ADL scores less than 5, who were excluded from the trial, would be suitable for treatment. Specifically, the clinical experts indicated that patients with ocular MG or mild symptoms can still be refractory to other therapies, and patients with MGFA Class V (on a ventilator) who have no contraindications would potentially benefit from efgartigimod alfa. However, the results of the trial cannot be directly generalized to these groups of patients.
For patients with gMG, 1 of the treatment goals is to reduce or discontinue the use of steroids. However, changes to concomitant medications, including steroids, were not allowed per the study protocol. Therefore, there is no information available about whether efgartigimod alfa would have an impact on steroid use among patients with gMG who are eligible for this treatment.
Furthermore, It is uncertain whether the findings derived from the ADAPT trial can be generalized to patients who are MuSK-Ab+. The clinical experts CADTH consulted for this review indicated that efgartigimod alfa will provide a new treatment for patients with gMG who are AChR-Ab+ and probably for those who are MuSK-Ab+. The clinician group input for this review also indicated that patients with MuSK antibodies might respond to efgartigimod alfa; however, whether these patients would benefit could not be confirmed based on ADAPT trial evidence.
All patients in the AChR-Ab+ population had previously received 1 or more prior therapies, with a large proportion of patients having received 2 or more prior therapies (96.1% in the AChR-Ab+ only populations) or 3 prior or more therapies (79.1% in the AChR-Ab+ only populations). In addition, during the trial, in patients who were AChR-Ab+, use of concomitant medications was 23.3% for 1 class of concomitant medications, 27.9% for 2 class of concomitant medications, and 48.1% for 3 class of concomitant medications. The clinical experts CADTH consulted for this review indicated that both the prior therapies and the concomitant medications used in the trial are aligned with clinical practice.
One of the inclusion criteria for recruiting patients into the ADAPT trial was an MG-ADL total score of 5 or more points. Of the patients who were AChR-Ab+, a total of | ||||||| patients in the efgartigimod alfa group, and | |||||| patients in the placebo group had an MG-ADL total score of 5 at the baseline of the study. The sponsor’s rationale for using the cut-off of 5 or more points was to align with the expected real-world usage of efgartigimod alfa and the unmet medical need for an additional treatment alternative to IVIg. The sponsor indicated that at the time of designing the trial, it was understood that patients who maintained an MG-ADL score of less than 5 would not need efgartigimod alfa. However, CADTH noticed that an MG-ADL total score cut-off of 6 or more points was an inclusion criterion in previous gMG clinical trials (i.e., in the REGAIN trial for eculizumab for gMG and in the CHAMPION trial for ravulizumab for gMG). Whether lowering the cut-off from 6 to 5 points for the MG-ADL total score potential has an impact on the patient’s response to efgartigimod alfa is uncertain. The clinical experts CADTH consulted for this review indicated that severity status based on the MG-ADL was subjective and a difference of 1 point may not be clinically important in terms of disease severity. In addition, the proportion of patients with an MG-ADL total score of 5 points was relatively low. Therefore, it is unlikely to have a meaningful impact on patients’ response to the treatment. Furthermore, the ADAPT trial was a placebo-controlled trial, so it does not provide any comparative efficacy or harms information on efgartigimod alfa relative to existing gMG therapies (e.g., AChEIs, steroids, NSIST, IVIg, PE, complement C5 inhibitors). Finally, the change from baseline of MG-ADL total score, MG-QoL15r score, and EQ-5D VAS score were assessed at cycle 1 and cycle 2. However, the overall efficacy of these outcomes at the end of the 26-week trial is unknown. The long-term comparative efficacy of efgartigimod alfa versus placebo still needs to be further established.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:34,35
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs is initially considered to be of high certainty and can be rated down if there are concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For this review, the target of the certainty of evidence assessment was based on the presence of absence of a clinically important effect, as informed by MIDs suggested by the sponsor and agreed upon by the clinical experts consulted by CADTH for this review (for change from baseline in MG-ADL score and QMG score), or by thresholds suggested by the clinical experts (for all other outcomes).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for efgartigimod alfa versus placebo for the AChR-Ab+ population.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Description of Studies
The ADAPT+ study (ARGX-113-1705) is a long-term, single-arm, open-label, multicentre, phase III follow-on study of patients who enrolled in the ADAPT study (ARGX-113-1704; NCT03669588). The primary objective was to evaluate the long-term safety and tolerability of efgartigimod alfa in the AChR-Ab+ subgroup and the secondary objective was to evaluate safety and tolerability in the overall population (AChR-Ab+ and AChR-Ab–). Efficacy data were collected as exploratory end points. A final Clinical Study Report is available for ADAPT+ (data cut-off: June 30, 2022).68
The ADAPT+ study was conducted at 51 sites, including 41 sites in 14 countries or regions that had 1 or more patients who could roll over from the ADAPT study. Data were collected over a 3-year period in 2 sequential parts (part A: 1 year, part B: 2 years maximum) (Figure 7). Part B was added as a protocol amendment to ensure accessibility to efgartigimod alfa until it became commercially available or available through an expanded access program. At the data cut-off (June 30, 2022), 151 patients had rolled over from the ADAPT trial, regardless of whether they were in the treatment or placebo group, into ADAPT+, and 145 patients had received at least 1 partial or complete dose of efgartigimod alfa in ADAPT+.
Figure 10: Study Design for the ADAPT+ Trial
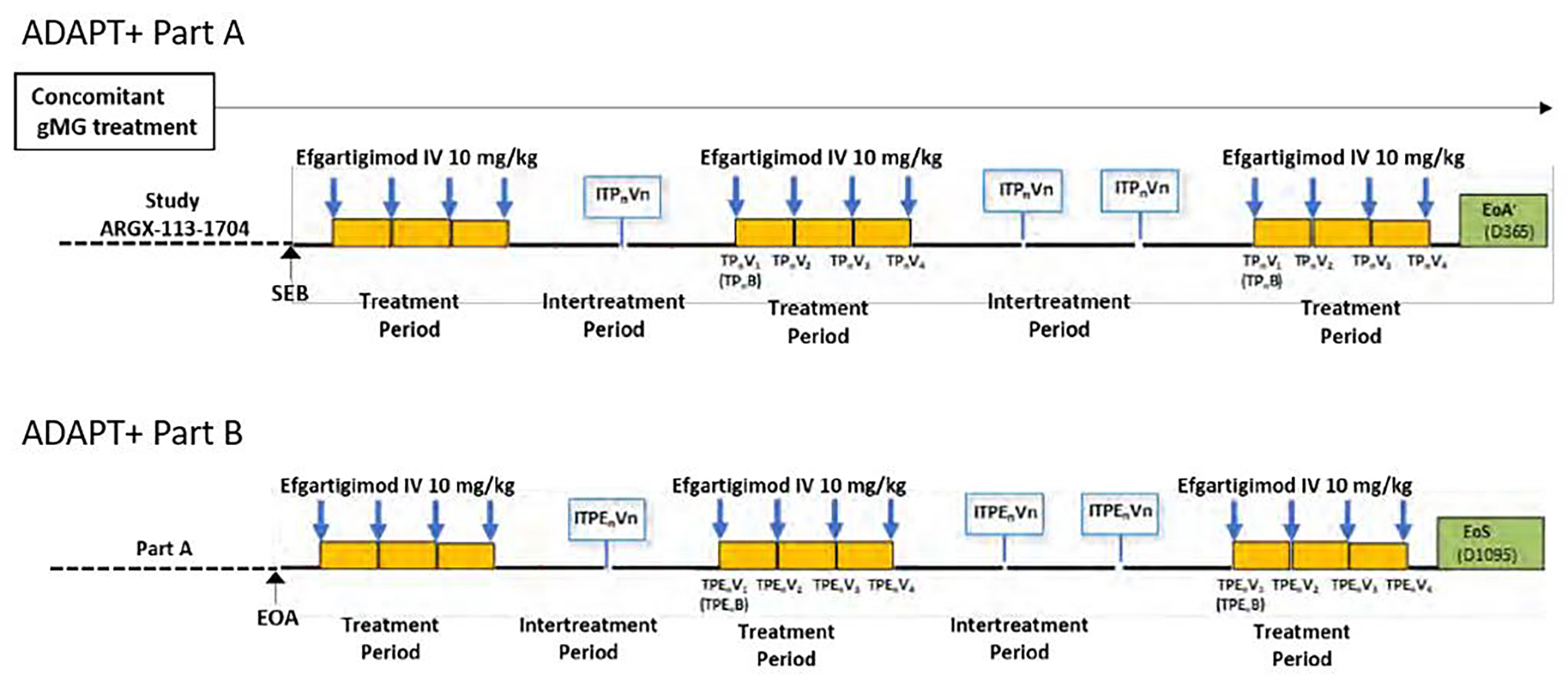
D = day; EoA = end of part A; gMG = generalized myasthenia gravis; ITPnVn = visit n of intertreatment period n; SEB = study entry baseline; TPnB = baseline of treatment period n; TPnVn = visit n of treatment period n.
Note: As with the ADAPT study, a study cycle was defined as a treatment period plus the subsequent intertreatment period.
Source: ADAPT+ final Clinical Study Report (data cut-off: June 30, 2022).68
Populations
As in the ADAPT trial, patients were required to be receiving a stable dose of concomitant gMG treatment before ADAPT+ entry. Consenting patients who could comply with the study procedures were eligible to roll over into ADAPT+ if they had participated in the ADAPT trial and:
reached the end of study at day 182 in the ADAPT trial
needed (re-)treatment while in the ADAPT trial, but could not complete a treatment cycle within the ADAPT study period (these patients could immediately roll over to ADAPT+ and receive efgartigimod alfa)
discontinued early from randomized treatment in the ADAPT trial for reasons other than pregnancy, rescue therapy, or an SAE
had a temporary interruption from randomized treatment in the ADAPT trial (these patients could be offered the option to roll over into ADAPT+).
The exclusion criteria for ADAPT+ were generally consistent with those of the ADAPT trial. Patients were also excluded from rolling over to ADAPT+ if they had discontinued from randomized treatment or the ADAPT study because of pregnancy, need for rescue therapy, or an SAE.
Interventions
Efgartigimod alfa treatment was administered in ADAPT+ using the dosing calculation and schedule from the ADAPT trial: efgartigimod alfa 10 mg/kg administered as a 1-hour IV infusion (total volume: 125 mL) every 7 days for 4 infusions (days 1, 8, 15, and 22) in each cycle. The maximum efgartigimod alfa dose per infusion was 1,200 mg (for patients weighing ≥ 120 kg).
Consistent with the ADAPT study criteria, subsequent treatment and re-treatment cycles in part A of ADAPT+ were started when the following criteria were met:
the patient had completed the previous cycle’s treatment period (i.e., after visit 4)
the patient had an MG-ADL total score of greater than or equal to 5 points (> 50% of score due to non-ocular symptoms)
the patient had a less than 2-point reduction in MG-ADL total score compared to:
the last cycle’s baseline in ADAPT for patients receiving their first treatment period in ADAPT+
the previous cycle’s baseline for patients in any cycle of ADAPT+.
Subsequent re-treatment cycles in Part B of ADAPT+ were started when the following criteria were met:
the patient had completed the previous cycle’s treatment period (i.e., after visit 4)
the investigator judged that efgartigimod alfa treatment was in the best interest of the patient
4 weeks or longer (1 calendar month) had elapsed since the last efgartigimod alfa infusion.
Prior therapy, concomitant therapy, and prohibited medications were defined as in the ADAPT trial. Use of prohibited medications would result in discontinuation of efgartigimod alfa treatment only in part A; part B did not restrict concomitant gMG treatment if it was based on standard clinical practice. Patients who required rescue therapy for protocol-defined MG clinical deterioration (as in the ADAPT trial) were discontinued from the study.
Outcomes
Efficacy outcomes in ADAPT+ were assessed using the MG-ADL tool (part A and part B) and the QMG tool (part A only) in the AChR-Ab+ population, in the AChR-Ab– population, and in the overall population (seropositive and seronegative). For the purpose of this review, only information on the patients with AChR-Ab+ are presented in this report. Efficacy outcome definitions, as well as the cut-offs for CMIs, were consistent between the ADAPT and ADAPT+ studies. A CMI on the MG-ADL has been defined as a 2-point reduction in the total score,52,53 and minimal symptom expression is a score of 0 or 1.52 A CMI on the QMG has been defined as a 3-point reduction in the QMG score.56
Due to the slightly different visit schedules in ADAPT+ (no visits were scheduled at weeks 4 to 6 of a cycle) and the ADAPT study (every week for the first 8 weeks of a cycle), ADAPT+ only collected MG-ADL data at week 3 of each study cycle.
Safety outcomes in ADAPT+ were assessed in the same manner as in the ADAPT study, with coding according to MedDRA (version 23.0) and severity grading according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0.
Statistical Analysis
All analyses were descriptive and performed using the safety analysis set from ADAPT+ (all patients who received ≥ 1 dose or partial dose), with subgrouping according to AChR-Ab status (Ab+ versus AB–) and the treatments received in the ADAPT and ADAPT+ trials (efgartigimod alfa in ADAPT and efgartigimod alfa in ADAPT+ [EFG-EFG] versus placebo in ADAPT and efgartigimod alfa in ADAPT+ [PBO-EFG]) (Table 22). Summary statistics were calculated for absolute values and changes from ADAPT+ baseline (overall) or changes from baseline of each cycle (i.e., the change for each cycle).
The rollover patients set, which included all patients who rolled over from ADAPT, was not used for any analyses.
Table 22: Patients in the Safety Analysis Set (ADAPT+)
Population | Efgartigimod alfa in both ADAPT and ADAPT+, N | Placebo in ADAPT and efgartigimod alfa in ADAPT+, N | Total efgartigimod alfa, N |
|---|---|---|---|
Rollover patients set | Not applicable | Not applicable | 151 |
Safety analysis set | 77 | 68 | 145 |
AChR-Ab– population | 16 | 18 | 34 |
AChR-Ab+ population | 61 | 50 | 111 |
Ab+ = antibody positive; Ab– = antibody negative; AChR = acetylcholine receptor antibody.
Notes: The rollover set includes all patients who rolled over from the ADAPT study, while the safety analysis set includes all patients who rolled over and received ≥ 1 dose or partial dose. AChR-Ab serostatus was determined based on the stratification factor at the time of randomization in the ADAPT study.
Source: ADAPT+ Clinical Study Report.68
Patient Disposition
Baseline Characteristics
Demographic characteristics of patients who were AChR-Ab+ were enrolled in ADAPT+ are shown in Table 23, stratified by the treatments received in the ADAPT and ADAPT+ trials. Patient characteristics were generally similar across the reported groups. Mean age was 47.0 years (SD = 14.8 years), and most patients were white (86.9%) and female (71.0%).
Table 23: Baseline Demographic Characteristics in ADAPT+ (Safety Analysis Set)
Characteristics | AChR-Ab+ population | ||
|---|---|---|---|
Efgartigimod alfa in both ADAPT and ADAPT+ (N = 61) | Placebo in ADAPT and efgartigimod alfa in ADAPT+ (N = 50) | Total patients who received efgartigimod alfa (N = 111) | |
Age (years) | |||
n | ||| | ||| | 111 |
Mean (SD) | |||| ||||||| | |||| ||||||| | 47.1 (15.52) |
Median (range) | ||||||||| ||| | ||||||||| ||| | 45.0 (19 to 81) |
Age category, n (%) | |||
18 to < 65 years | || |||||| | || |||||| | 93 (83.8) |
≥ 65 years | | |||||| | || |||||| | 18 (16.2) |
Weight (kg) | |||
N | ||| | ||| | 111 |
Mean (SD) | |||||||||||||| | |||||||||||||| | 81.37 (25.583) |
Median (range) | ||||||||||| |||||| | |||||||||||| |||||| | 74.00 (46.0 to 227.5) |
Sex, n (%) | |||
Female | || |||||| | || |||||| | 75 (67.6) |
Male | || |||||| | || |||||| | 36 (32.4) |
Race, n (%) | |||
American Indian or Alaska Native | | ||||| | ||| | 2 (1.8) |
Asian | | |||||| | | ||||| | 8 (7.2) |
Black or African American | | ||||| | | ||||| | 3 (2.7) |
White | || |||||| | || |||||| | 97 (87.4) |
Multiple | | ||||| | ||| | 1 (0.9) |
Ethnicity, n (%) | |||
Hispanic or Latino | | ||||| | | ||||| | 7 (6.3) |
Japanese | | ||||| | | ||||| | 7 (6.3) |
Not Hispanic or Latino | || |||||| | || |||||| | 97 (87.4) |
AChR-Ab+ = acetylcholine receptor antibody positive; SD = standard deviation.
Source: ADAPT+ Clinical Study Report.68
Baseline disease characteristics of patients with AChR-Ab+ who enrolled in ADAPT+, stratified by the treatments received in the ADAPT trial and ADAPT+, are shown in Table 24. Patient characteristics were generally similar across the reported groups. As in the ADAPT trial, patients in the AChR-Ab+ population who rolled over to ADAPT+ had a mean time since diagnosis of 9.7 years (SD = 7.9 years) with mean scores of 9.5 points (SD = 3.1 points) for MG-ADL total score and 15.3 points (SD = 5.7 points) for QMG total score.
Table 24: Baseline Disease Characteristics in ADAPT+ (Safety Analysis Set)
Characteristics | AChR-Ab+ population | ||
|---|---|---|---|
Efgartigimod alfa in both ADAPT and ADAPT+ (N = 61) | Placebo in ADAPT and efgartigimod alfa in ADAPT+, (N = 50) | Total efgartigimod alfa (N = 111) | |
Time since diagnosis (years) | |||
n | ||| | ||| | 111 |
Mean (SD) | ||| ||||| | ||| ||||| | 9.7 (7.9) |
Median (range) | |||||||||| ||||| | |||||||||| ||||| | 6.92 (1.4 to 45.7) |
Total MG-ADL | |||
n | ||| | ||| | 111 |
Mean (SD) | ||| |||||| | ||| |||||| | 9.5 (3.09) |
Median (range) | |||| ||| ||| | ||| ||| ||| | 9.0 (3 to 18) |
Total MG-ADL category, n (%) | |||
< 5 | || |||||| | || |||||| | 54 (48.6) |
5 to 7 | | ||||| | ||| | 1 (0.9) |
8 to 9 | || |||||| | || |||||| | 32 (28.8) |
≥ 10 | || |||||| | || |||||| | 24 (21.6) |
Total QMG score | |||
n | ||| | ||| | 111 |
Mean (SD) | |||| |||||| | |||| |||||| | 15.3 (5.65) |
Median (range) | |||| ||| ||| | |||| ||| ||| | 16.0 (3 to 34) |
Concomitant gMG treatment, n (%) | |||
NSISTs | || |||||| | || |||||| | 67 (60.4) |
No NSISTs | || |||||| | || |||||| | 44 (39.6) |
AChR-Ab status, n (%) | |||
Seropositive | || ||||| | || ||||| | 111 (100) |
Seronegative | ||| | ||| | 0 |
MuSK-Ab status, n (%) | |||
Seropositive | ||| | ||| | 0 |
Seronegative | || ||||| | || ||||| | 111 (100) |
Ab = antibody; AChR-Ab+ = acetylcholine receptor antibody positive; gMG = generalized myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MuSK = muscle-specific kinase; NSIST = nonsteroidal immunosuppressive therapy; QMG = Quantitative Myasthenia Gravis; SD = standard deviation.
Source: ADAPT+ Clinical Study Report.68
Patient Disposition
No disposition information was reported for the seropositive population.
Exposure to Study Treatments
Study Treatments
Data regarding extent of exposure for patients who were AChR-Ab+ were not reported in the sponsor’s evidence summary.
Concomitant Medications and Cointerventions
Patients were to remain on their stable dose and regimen of concomitant gMG treatment (without replacing, adding, removing, or adjusting the dose and/or frequency of treatments) during part A of ADAPT+. During part B, changes in the type, dose, or regimen of the concomitant gMG treatment were permitted, as was the additional use of other treatments (e.g., PE, IVIg, immunoadsorption, any new corticosteroid type, or an increase in the dose of current corticosteroid).
As shown in Table 24, concomitant gMG treatments at baseline were classified as NSISTs (60.4% of patients in the AChR-Ab+ population) or non-NSISTs (39.6% of patients in the AChR-Ab+ population).
Subsequent Treatment
Results
Efficacy
Efficacy results were descriptively reported as exploratory outcomes based on validated clinical scales: the patient-reported MG-ADL (part A and part B) and the physician-reported QMG (only part A). Clinically meaningful mean improvements in symptoms were observed during each cycle based on both scales, with improvements that were repeatable across multiple cycles and occurred regardless of previous efgartigimod alfa treatment (i.e., in the both the group that received efgartigimod alfa in both ADAPT and ADAPT+ and the group that received placebo in ADAPT and efgartigimod alfa in ADAPT+). These clinically meaningful improvements (e.g., 2 or more points on MG-ADL and 3 or more points on QMG) were also observed in 88.7% of patients with 2 or more points on MG-ADL and 64.4% of patients with 3 or more points on QMG of the AChR-Ab+ subgroups. Furthermore, in AChR-Ab+ subgroups, the mean improvement in MG-ADL score was greater than or equal to 5 points across all cycles, which is greater than the suggested MID of 2 points.
Exploratory: MG-ADL Scores (Part A and Part B)
Data at all assessment points up to cycle 14 (due to limited patient data at later cycles) are shown graphically for the AChR-Ab+ population (Figure 8) These results show that, across all the groups, the largest mean MG-ADL response (reduction from baseline) appeared to be reached around week 3 of each cycle (data were not recorded at weeks 4 or 5 of ADAPT+, which is where the maximum response was observed in the ADAPT trial; there were also no data for week 6) and a return to approximately cycle baseline sometime during week 7 to week 11.
Within the AChR-Ab+ population, the mean change from the baseline of the first cycle to week 3 in the MG-ADL score (5.0 points; SE = 0.33 points) was greater than the suggested MID of 2 points. During the first 10 cycles of ADAPT+, an improvement in MG-ADL score of 2 points or more was observed for 88.7% of the patients who received efgartigimod alfa (EFG-EFG or PBO-EFG) and an improvement of 5 points or more was observed for 62.0%. At the final ADAPT+ analysis (data cut-off: June 30, 2022), mean change in MG-ADL score was greater than or equal to 5 points for all cycles except cycle 16 (SE = 4.9 points) in the total efgartigimod alfa group of the AChR-Ab+ population.
Figure 11: MG-ADL Total Score Mean Change From Cycle Baseline in the AChR-Ab+ Population (Safety Analysis Set) [Redacted]

Source: ADAPT+ final Clinical Study Report (Figure 3, data cut-off: June 30, 2022).68
Exploratory: QMG Scores (Part A Only)
As with the MG-ADL results, QMG data are presented for the cycle baseline and the week 3 time point within each cycle because the ADAPT+ visit schedule was less intensive than the ADAPT trial schedule. Data at all assessment points up to cycle 7 (only collected during the first 1 year of ADAPT+) are shown graphically for the AChR-Ab+ population (Figure 9).
Consistent with the MG-ADL results, in the AChR-Ab+ population, the mean change from cycle baseline in the QMG score was greater than the MID of 3 points across all time points.
During the first 7 cycles of ADAPT+, an improvement of greater than or equal to 3 points in QMG score was observed for 64.4% of patients with AChR-Ab+ gMG who received efgartigimod alfa (EFG-EFG or PBO-EFG) and an improvement of greater than or equal to 6 points was observed for 36.0%.
Figure 12: QMG Total Score Mean Change From Cycle Baseline in the AChR-Ab+ Population (Safety Analysis Set) [Redacted]

||| ||||| ||||| |||| |||||| |||| ||||| |||||||| || ||| |||||||| |||||||||| ||||||| |||||||| |||| |||| ||||| |||||| | ||||| ||| ||| ||||| ||||| |||| |||||| |||| ||||| |||||||| |||| ||||| | || ||||| | || ||| ||||| || |||||||||||| ||||||||||| ||| ||||||| |||| |||||| ||||||||||| |||| ||||| |||||||| |||||||| || |||| | || |||| ||||| || |||| |||||||||||| |||| | |||||||||||| |||| ||| ||||||| | |||||||||||| |||| ||||||| ||| ||||||||| || ||| |||||| |||| |||||||| |||||||| ||||||| || ||| ||| || |||| |||||| ||| ||||||| ||||||||| |||||||| ||||||| |||| ||||| | || ||||| || |||||||||| | |||||||||||||||||| |||||||| ||||||||| ||| | |||||||||||| |||||||||| ||||||| || | |||||||| |||||| ||||||| |||||| ||||| |||||||| ||||| |||||| | |||| |||||||| |||| ||| ||||||68
Harms
Some safety outcomes were reported for the AChR-Ab+ population in the sponsor’s Clinical Study Report. A summary of harms data collected in ADAPT+ up to the data cut-off (December 15, 2022) is shown in Table 25.
Adverse Events
The most commonly reported TEAEs were headache (|| ||||||||| |||||||) nasopharyngitis (|| ||||||||| |||||||), COVID-19 (|| ||||||||| |||||), and diarrhea (|| ||||||||| |||||||). These results are consistent with the safety profile of efgartigimod alfa in ADAPT.
Serious Adverse Events
SAEs occurred in || |||||||| |||||). The most common SAE was MG (| ||||||||| ||||||).
Adverse Events of Special Interest
TEAEs in the system organ class of infections and infestations were classified as AESIs and were reported by 35 patients in the group that received efgartigimod alfa in both ADAPT and ADAPT+ (57.4%) and 26 patients in the group that received placebo in ADAPT and efgartigimod alfa in ADAPT+ (52.0%).
Table 25: Summary of Harms in ADAPT+ in the AChR-Ab+ Population (Safety Analysis Set)
Adverse events | Efgartigimod alfa in both ADAPT and ADAPT+ (N = 61) | Placebo in ADAPT and efgartigimod alfa in ADAPT+, (N = 50) | Total efgartigimod alfa (N = 111) |
|---|---|---|---|
Most common treatment-emergent adverse events, n (%) | |||
Patients with ≥ 1 treatment-emergent adverse event | || |||||| | || |||||| | || |||||| |
Occurring in ≥ 5% of total patients receiving efgartigimod alfa | |||
Headache | || |||||| | || |||||| | || |||||| |
Nasopharyngitis | | |||||| | | |||||| | || |||||| |
COVID-19 | || |||||| | | ||||| | || |||||| |
Diarrhea | | ||||| | | |||||| | || |||||| |
Urinary tract infection | | ||||| | | ||||| | || ||||| |
Arthralgia | | ||||| | | ||||| | | ||||| |
Pyrexia | | ||||| | | ||||| | | ||||| |
Nausea | | ||||| | | ||||| | | ||||| |
Myasthenia gravis | | ||||| | | ||||| | | ||||| |
Hypertension | | ||||| | | ||||| | | ||||| |
Back pain | | ||||| | | ||||| | | ||||| |
Serious adverse events, n (%) | |||
Patients with ≥ 1 serious treatment-emergent adverse event | || |||||| | || |||||| | || |||||| |
Occurring in ≥ 2 patients from total patients receiving efgartigimod alfa | |||
Myasthenia gravis | | ||||| | | ||||| | | ||||| |
COVID-19 | | ||||| | | ||||| | | ||||| |
COVID-19 pneumonia | | ||||| | | ||| | | ||||| |
Pneumonia | | ||| | | ||||| | | ||||| |
Myasthenia gravis crisis | | ||||| | | ||||| | | ||||| |
Adverse events of special interest, n (%) | |||
Infections and infestations, n (%) | || |||||| | || |||||| | || |||||| |
Occurring in ≥ 5% of total patients receiving efgartigimod alfa | |||
Nasopharyngitis | | |||||| | | |||||| | || |||||| |
COVID-19 | || |||||| | | ||||| | || |||||| |
Urinary tract infection | | ||||| | | ||||| | || ||||| |
AChR-Ab+ = acetylcholine receptor antibody positive.
aDeaths during ADAPT+ were caused by acute myocardial infarction, septic shock, myasthenia gravis crisis, lung neoplasm malignant, and death (1 case each).
bThis event resulted in death.
Source: ADAPT+ final Clinical Study Report (data cut-off: December 15, 2022).68
Critical Appraisal
Internal Validity
The ADAPT+ study was limited by its open-label and noncomparative design. Because there was no comparator, it cannot be confirmed whether the observed results are attributable to the effects of the drug or to natural history. Furthermore, the missing outcome data and small sample size toward the end of ADAPT+ made it difficult to draw any firm conclusions on the efficacy and safety of efgartigimod alfa. Because the study was open label and nonblinded, the subjective outcomes (e.g., rates of self-reported AEs and MG-ADL and QMG scores) are at risk of bias, most likely in favour of the intervention (i.e., efgartigimod alfa), at least for efficacy outcomes. The population enrolled in ADAPT+ had demographic and clinical characteristics that were consistent with their characteristics at entry into the ADAPT study. The design, eligibility criteria, and efgartigimod alfa dosing regimen were consistent in both studies, although it is noteworthy that ADAPT+ had fewer scheduled visits for outcome assessments (no visits were scheduled at weeks 4 to 6 of a cycle) compared with the ADAPT trial (every week for the first 8 weeks of a cycle); ADAPT+ only collected MG-ADL data at week 3 of each study. Furthermore, the longer-term safety and tolerability profile of efgartigimod alfa treatment was hard to determine because the rates of AEs and SAEs may be underestimated, since ADAPT+ had fewer scheduled visits for outcome assessments and MG-ADL data were collected only at week 3 of each study cycle, instead of 3 times (week 3, 4, and 5 of each cycle) in ADAPT trial.
In ADAPT+, efficacy was assessed as an exploratory outcome using the patient-reported MG-ADL and physician-reported QMG scores. Patients were to remain on their stable dose and regimen of concomitant gMG treatment during part A of ADAPT+. Given the presence of rollover effects from part A, efficacy results related to part B may be difficult to interpret because changes were permitted in part B in the type, dose, or regimen of the concomitant gMG treatment and in the use of other treatments. Therefore, the effects of other therapies cannot be eliminated as contributors to treatment response in part B. Although no detailed safety outcomes for the AChR-Ab+ population were reported in sponsor’s evidence summary and Clinical Study Report (i.e., ADAPT+), some safety outcomes were reported for the AChR-Ab+ population in the Clinical Study Report.68 These appear to be consistent with the safety profile of efgartigimod alfa observed in the ADAPT trial.
External Validity
Because the patients who took part in the open-label long-term safety extension phase were originally from the pivotal ADAPT trial, it is reasonable to expect that the same limitations to generalizability are relevant to the open-label long-term safety extension phase. Given the nature of noncomparative study design, it is not possible to compare the effectiveness and tolerability of efgartigimod alfa as add-on treatment of gMG against others add-on treatment (e.g., ravulizumab, eculizumab, and IVIg). In terms of outcome measures, some important long-term outcomes reported by patients and clinicians (e.g., HRQoL, MG exacerbations) were not measured in the ADAPT+ trial.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
The objective of this section is to summarize and critically appraise available indirect evidence comparing efgartigimod alfa to other therapies for the treatment of patients with gMG in Canadian settings.
Description of the Sponsor-Submitted ITC
The efficacy and safety of efgartigimod alfa as add-on therapy for adult patients with AChR-Ab+ gMG whose symptoms persist despite adequate doses of conventional therapy (AChEIs, corticosteroids, and/or NSISTs) have been previously assessed in the ADAPT trial.28 However, no head-to-head evidence of efgartigimod alfa compared against other treatments for gMG was available for this review. Due to this gap in evidence, the sponsor submitted an ITC, including an SLR38 and an NMA,1 that provides comparative evidence of the efficacy of efgartigimod alfa relative to ravulizumab and IVIg. Data from this ITC were used to inform the pharmacoeconomic model.
Indirect Treatment Comparison Design
Objectives of the Sponsor’s ITC
The objective of the sponsor’s ITC was to assess the comparative effectiveness of efgartigimod alfa compared with other therapies used in Canada for the treatment of patients with gMG whose symptoms are not well-controlled by the conventional treatment options.
Study Selection Methods
The sponsor conducted a systematic literature search to identify studies assessing any chronic or acute pharmaceutical interventions for adult patients with gMG, and to evaluate the comparative efficacy and safety of efgartigimod alfa and alternative treatments in this patient setting. Methods and results were described as outlined by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement and the corresponding extension statement for the NMA.69 Table 26 shows the study selection criteria and key aspects of the methods for the systematic review.
An electronic search of the literature was conducted on February 14, 2020, and updated on April 7, 2022. The search was conducted using the following databases: MEDLINE, 1946 to present (OVID); MEDLINE In-Process and Other Non-Indexed Citations (OVID); MEDLINE Epub Ahead of Print (OVID); Embase, 1980 to present (OVID); Cochrane Central Register of Controlled Trials (CENTRAL) (Wiley); Cochrane Database of Systematic Reviews (CDSR) (Wiley); PubMed (NLM) — e-publications only; Database of Abstracts of Reviews of Effects (DARE); Health Technology Assessment (HTA) database; International HTA (INAHTA) database; Conference Proceedings Citation Index-Science (CPCI-S), 1990 to present (Web of Science, Clarivate Analytics); International Society for Pharmacoeconomics and Outcomes Research (ISPOR) Presentations Database. Supplementary searches were conducted using ClinicalTrials.gov, EU Clinical Trials Register, and the WHO International Clinical Trials Registry Platform (WHO ICTRP), and the proceedings of recent conferences since March 1, 2020: European Academy of Neurology Congress, American Academy of Neurology annual meeting, and Peripheral Nerve Society annual meeting. A search for other published NMAs was not explicitly carried out for the SLR. The study screening, selection, data extraction, and risk-of-bias appraisal processes were conducted by 2 independent reviewers. Disagreements between researchers were resolved through discussion or, if necessary, through consultation with a third researcher.
Table 26: Study Selection Criteria and Methods for Systematic Review
Characteristics | Indirect comparison |
|---|---|
Population | Adult patients with gMG |
Intervention | Efgartigimod alfa Acetylcholinesterase inhibitors Corticosteroids Azathioprine, mycophenolate mofetil, cyclosporine, tacrolimus, methotrexate Immunoglobulin therapy (IV and subcutaneous) Plasmapheresis Cyclophosphamide Rituximab Eculizumab Zilucoplan Rozanolixizumab Ravulizumab |
Comparator | Placebo Standard of care with or without placebo and/or background medication Active intervention (i.e., head-to-head trials) |
Outcome | Efficacy or HRQoL
Safety
|
Study designs | RCTs (phases I to IV) Randomized crossover trials Long-term follow-up studies (e.g., open-label follow-up studies with continuation of treatment) |
Publication characteristics | Only published studies were included |
Exclusion criteria | Conference presentations published on or after March 1, 2020; English languagea |
Databases searched | MEDLINE, 1946 to present (OVID); MEDLINE In-Process and Other Non-Indexed Citations (OVID); MEDLINE Epub Ahead of Print (OVID); Embase, 1980 to present (OVID); Cochrane Central Register of Controlled Trials (CENTRAL) (Wiley); Cochrane Database of Systematic Reviews (CDSR) (Wiley); PubMed (NLM) — e-publications only; Database of Abstracts of Reviews of Effects (DARE) (CRD); Health Technology Assessment (HTA) database (CRD); International HTA (INAHTA) database; Conference Proceedings Citation Index-Science (CPCI-S), 1990 to present (Web of Science, Clarivate Analytics);b International Society for Pharmacoeconomics and Outcomes Research (ISPOR) Presentations Database |
Selection process | Two reviewers independently screened the abstracts identified by the literature search after duplicate citations were removed, with disagreements settled by discussion or involvement of a third reviewer if needed. The same process was followed for review of full-text articles to establish final study selection. |
Data extraction process | A comprehensive data extraction form, created in Microsoft Excel (Microsoft Corporation, Seattle, Washington, US), was used to compile the data. Data extracted from clinical studies included study design, interventions, primary and secondary end points, inclusion/exclusion criteria, patient baseline characteristics, duration of follow-up, key efficacy outcomes (MG-ADL, QMG, MGC, and MG-QoL) and safety outcomes (incidence of any AEs, TEAEs, SAEs, mortality, and treatment discontinuation). For this systematic literature review, the only subgroup of interest was the AChR-Ab+ population. |
Quality assessment | Risk-of-bias assessments were performed using the CRD Guidance for Undertaking Reviews in Health Care.70 Included studies were evaluated using a fixed set of domains of bias focused on different aspects of trial design, conduct, and reporting. Disagreements between researchers were resolved through discussion or, if necessary, by consulting a third researcher. |
AChR-Ab+ = acetylcholine receptor antibody positive; AE = adverse event; CRD = Centre for Reviews and Dissemination; gMG = generalized myasthenia gravis; HRQoL = health-related quality of life; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15 = Myasthenia Gravis Quality of Life 15-item; QMG = Quantitative Myasthenia Gravis; RCT = randomized controlled trial; SAE = serious adverse event; SLR = systematic literature review; TEAE = treatment-emergent adverse event.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aPublications in languages other than English were tagged during screening for record-keeping purposes but were not included.
bData were not extracted; reference lists were cross-checked for additional relevant records.
Sources: Sponsor-submitted Indirect Treatment Comparison and Systematic Literature Review reports.1,38
From the evidence base created from the SLR, the sponsor further identified studies that were potentially eligible for an ITC based on a set of additional criteria listed in Table 27. Studies were required to be placebo-controlled randomized trials of treatments for adults with AChR-Ab+ gMG, reporting on at least 1 of the following outcomes: MG-ADL responders, or change from baseline in MG-ADL score; and QMG responders, or change from baseline in QMG score. The eligible interventions for the ITC were limited to those used in Canada for the treatment of gMG to ensure that the comparators were relevant to Canadian settings. Eligible interventions to be included in the NMA were efgartigimod alfa, ravulizumab, maintenance IVIg or subcutaneous immunoglobulin, and PE. Ravulizumab has been approved by Health Canada for the treatment of adult patients with AChR-Ab+ gMG and is currently under review by CADTH. Eculizumab was not considered a relevant comparator for this ITC by the sponsor because as it has only been studied in patients with AChR-Ab+ and refractory gMG. Treatment guidelines71 indicate that eculizumab is expected to continue to be considered as a last resort for patients who have not experienced treatment success on other immunotherapies, which is different from the expected place in therapy for efgartigimod alfa. In addition, eculizumab is not currently publicly funded in Canada. Rituximab was not considered by the sponsor to be a key comparator for efgartigimod alfa in patients with AChR-Ab+ gMG and thus was not included in the primary ITC analysis.
Table 27: Feasibility Assessment Study Selection Criteria
Inclusion criteria | Exclusion criteria |
|---|---|
Population | |
Adults (aged 18 years or older) who have been diagnosed with AChR-Ab+ gMG |
|
Interventions | |
Interventions relevant to the Canadian market, selected from the following:
|
|
Comparators | |
Placebo | Any active intervention; no comparator (single-arm trials) |
Outcomes | |
Studies reporting on at least 1 of the following outcomes:
| Studies not reporting any of the listed outcomes (e.g., safety only, steroid-sparing effect only) |
Study design and document type | |
Randomized controlled trials |
|
AChR-Ab+ = acetylcholine receptor antibody positive; gMG = generalized myasthenia gravis; IVIg = intravenous immunoglobulin; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living scale; MGC = Myasthenia Gravis Composite; QMG = Quantitative Myasthenia Gravis; SCIg = subcutaneous immunoglobulin.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
aTreatments commonly included in the conventional therapy for gMG were considered as a part of placebo treatment and thus not eligible for inclusion as independent treatment arms.
Sources: Sponsor-submitted Indirect Treatment Comparison report and Systematic Literature Review report.1,38
Risk-of-Bias Assessment
Risk-of-bias assessments were performed by 2 independent reviewers using the CRD Guidance for Undertaking Reviews in Health Care.70 Included studies were evaluated using a fixed set of domains of bias focused on different aspects of trial design, conduct, and reporting. Eight specific domains were examined: random sequence generation, allocation concealment, blinding of participants, blinding of investigators, blinding of outcome assessors, incomplete outcome data, selective reporting, and other potential sources of bias that may affect internal or external validity and generalizability of the study findings to the general population. Disagreements between researchers were resolved through discussion or, if necessary, through consultation with a third researcher.
Feasibility Assessment
Prior to conducting analyses, groups of studies informing distinct potential NMAs were compared in detail to validate the exchangeability assumption underlying the methodology. The following elements of study design were compared for similarity in methods across studies: blinding, study phase, geographical distribution of study centres, and the time-periods of enrolment and follow-up. Decisions to conduct distinct NMAs were made based on the degree and potential impact of cross-trial heterogeneity among the trials that would be indirectly compared.
ITC Analysis Methods
Analytical Framework
All NMAs were performed using a Bayesian framework.72-74 Placebo was chosen as the reference treatment for all analyses, given its presence as an anchor treatment in all studies and the outcomes assessed in the network. All analyses only included patients with AChR-Ab+ gMG as all comparator studies were performed exclusively in the AChR-Ab+ subpopulation. Primary analyses were conducted based on efficacy estimates observed at each trial’s primary assessment time point or, if unreported, at the end of the randomized controlled period. A sensitivity analysis was conducted at or plus or minus 2 weeks of week 4, which was the primary time point of assessment in ADAPT.
Evidence Network
Evidence network diagrams were created to visualize the evidence base for each analysis, where similar interventions identified among the studies (based on dosage and frequency of administration) were grouped as treatment nodes. Studies included in the network were meant to inform the NMAs. In the network diagram, treatment nodes were sized to reflect the proportionate numbers of patients randomized to each treatment, with larger nodes representing more patients.
Model Effects, Iterations, and Convergence
Given the paucity of links between multiple studies in the evidence network, a random effects NMA was not considered feasible, and fixed effects NMAs were performed for all outcomes. All models were built using 4 unique sets of initial values and were based on burn-in and sampling durations of 20,000 iterations or more. Convergence was monitored quantitatively using the latest implementation of the Gelman-Rubin diagnostic (Rhat) based on 4 chains.75 This new implementation captures nonconvergence from stationary but nonoverlapping chains, overlapping nonstationary chains, chains with heavy tails, and chains with different variance. Samples were considered to have converged if Rhat was equal to or less than 1.05. After convergence was reached, there was concern about whether there were sufficient independent samples for stable estimates. The newest version of effective sample size and Monte Carlo standard error (MCSE) estimation were used to ensure that there were enough samples after convergence to support the inference.75 If the rank-normalized effective sample size was greater than 400 (i.e., 100 per chain), then samples were taken to ensure that the MCSE was small enough to allow for stable estimates to at least 1 decimal place.75 All assessments of effective sample size and MCSE were produced for each reported parameter.72,76
Model Priors
By default, vague prior distributions that assume no pre-existing information, according to the National Institute for Health and Clinical Excellence (NICE),77 were assigned for the treatment effects and trial baselines (Table 28).
Table 28: Vague Prior Distributions Used in Network Meta-Analyses
Parameter | Outcome | Prior distribution |
|---|---|---|
Baselines, unadjusted models (mu) | MG-ADL | dnorm (0,100) |
QMG | ||
Basic parameters (d) | MG-ADL | dnorm (0,100) |
QMG |
MG-ADL = Myasthenia Gravis Activities of Daily Living; QMG = Quantitative Myasthenia Gravis.
Note: Details included in the table are from the Sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted Indirect Treatment Comparison report.1
Assessment of Consistency
While the use of an unrelated mean effects model (i.e., an inconsistency model) was planned to test for inconsistency, there were no independent closed loops in the evidence network; therefore, no analyses evaluating consistency of direct and indirect evidence were performed.74 Decisions to conduct distinct NMAs were made based on the degree and potential impact of cross-trial heterogeneity of studies that would be compared indirectly.
Outcome Measures
The efficacy outcomes included change from baseline in MG-ADL and QMG at the primary assessment time points, ranging from 4 to 26 weeks. The primary analysis was conducted in patients with AChR-Ab+ gMG based on efficacy estimates observed at each trial’s primary assessment time point or, if unreported, at the end of the randomized controlled period. For ADAPT, the primary assessment time point was week 4. An NMA for continuous, arm-based outcomes was used to compare MG-ADL and QMG. Data inputs from the individual trials were the mean change from baseline and standard errors. Pairwise comparisons of interventions estimated from NMAs were expressed using mean differences with 95% CrIs.
Table 29: ITC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | An NMA for continuous, arm-based outcomes was used to compare MG-ADL and QMG. Data inputs were the mean change from baseline and standard errors. |
Priors | By default, vague prior distributions that assume no pre-existing information according to NICE Decision Support Unit Technical Support Document 377 were assigned for the treatment effects and trial baselines. |
Assessment of consistency | The exchangeability and transitivity assumptions of NMA indicate that the analyzed network is consistent, meaning that there is no evidence of disagreement between the direct and indirect evidence being combined. Inconsistency can be thought of as the statistical realization of the violation of the transitivity assumption. While the use of an unrelated mean effects model (i.e., an inconsistency model) was planned to test for inconsistency), there were no independent closed loops in the evidence network and therefore no analyses evaluating consistency of direct and indirect evidence were performed.74 |
Assessment of convergence | Convergence was monitored quantitatively using the latest implementation of the Gelman-Rubin diagnostic (Rhat) based on 4 chains.75 This new implementation captures nonconvergence from stationary but nonoverlapping chains, overlapping nonstationary chains, chains with heavy tails, and chains with different variance. Samples were considered to have converged if Rhat was ≤ 1.05. |
Outcomes | Pairwise comparisons of interventions estimated from NMAs are presented with mean differences with 95% credible intervals for all outcomes. All estimates from NMAs are conditional on the assumptions underpinning them. These assumptions include all those that are relied on in the analysis of the original trial data in addition to those described below. Violation of these assumptions can influence the direction and magnitude of the point estimate as well as the precision with which it is estimated (i.e., credible and confidence intervals). |
Follow-up time points | The studies included in this feasibility assessment ranged in follow-up time from 4 to 26 weeks. The ADAPT study was unique in that it measured patient response during the first cycle, after 4 weeks of treatment, and then during subsequent cycles wherein only patients requiring additional treatments for symptom management, as determined by clinicians, were given additional doses. Primary analyses in this report were conducted on the primary time points for all included studies. |
Sensitivity analyses | Analyses conducted at the primary time point for all trials could be biased against ADAPT, as they could exclude the best responders to efgartigimod alfa, whereas ITCs conducted at 4 weeks only could be biased against any treatments that demonstrated improved responses over time. Therefore, sensitivity analyses were performed at or within ± 2 weeks of week 4, which was the primary time point of assessment in ADAPT. |
Subgroup analysis | All analyses, including additional analyses, were conducted in patients with AChR-Ab+ gMG. |
Methods for pairwise meta-analysis | All NMAs were performed using a Bayesian framework. The chosen reference treatment for all analyses was placebo, given its presence as the anchor treatment across all studies and outcomes assessed in the network. Network diagrams were drawn to visualize the evidence base for each analysis. In these figures, treatment nodes are sized to reflect the proportionate numbers of patients randomized to each treatment, with larger nodes signifying more patients. Lines that connect nodes signify the presence of ≥ 1 RCTs that directly compare treatments, with the thickness of each line reflecting the number of RCTs informing the comparison; thicker lines signify more RCTs comparing treatments. Given the paucity of multistudy connections in the evidence network, a random effects NMA was deemed infeasible and fixed effects NMA was performed for all outcomes. |
AChR-Ab+ = acetylcholine receptor antibody positive; ITC = indirect treatment comparison; gMG = generalized myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; NICE = National Institute for Health and Clinical Excellence; NMA = network meta-analysis; QMG = Quantitative Myasthenia Gravis; RCT = randomized controlled trial.
Note: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted Indirect Treatment Comparison report.1
Results of ITC
Summary of Included Studies
The electronic search for clinical evidence identified 2,171 clinical and 1,503 economic citations across the databases searched. An additional 1,575 citations were collected from supplementary searching, including recent conference abstracts, registers, websites, and hand-searches. Out of these 5,429 citations, 2,113 were identified as duplicates and were removed. Of the 3,136 unique citations, a total of 2,811 were excluded during title and abstract screening because they did not meet the prespecified inclusion criteria. Out of the remaining 325 citations, 239 were excluded at the full-text screening phase. Overall, 80 records were eligible for inclusion in the SLR, representing 40 clinical studies. After feasibility assessment, the following 5 studies conducted with patients with AChR-Ab+ gMG were included in the NMA: 2 trials comparing efgartigimod alfa with placebo;28,39 2 trials comparing IVIg with placebo;40,41 and 1 trial comparing ravulizumab with placebo42 (Table 30). The authors performed a risk-of-bias assessment that showed a low or unclear risk of bias for most of the included studies.
Eligibility Criteria
Inclusion and exclusion criteria were generally similar across studies. All trials other than ADAPT included only the AChR-Ab+ subpopulation. Patients with a recent thymectomy were generally excluded, although the exclusion period ranged from 3 to 12 months before across studies. All trials excluded patients who had recently received either IVIg, subcutaneous immunoglobulin, or PE treatment and patients with very mild disease (e.g., MGFA Class I). All studies excluded patients under the age of 18, except for CHAMPION MG (ravulizumab versus placebo) which was restricted to patients aged 15 or older, although the mean age for patients in that study was not meaningfully different from the age of patients in the other trials. Most of the studies excluded patients with a history of malignancy (3 of 5) or treatment with rituximab within the past 6 to 12 months (4 of 5). Less common exclusion criteria included pregnancy (2 of 5), MG crisis (2 of 5), and renal impairment (2 of 5).
Treatment Definitions and Concomitant Therapies
Treatment definitions were generally similar between studies. All studies allowed for the use of concomitant standard-of-care treatments (i.e., steroids or NSISTs), but detailed information on the breakdown of the actual concomitant medications used was not available. The sponsor assumed that these treatments did not interact with the therapies under consideration and that any effect was additive. As such, the sponsor considered the placebo arm of the evidence network to be inclusive of standard-of-care therapies available in Canada.
Efficacy Outcomes
In all eligible studies, results for MG-ADL and QMG scores were reported in different formats (e.g., proportion of responders, or mean change from baseline). The mean change from baseline in MG-ADL and QMG scores were the most consistently reported outcomes; thus, trials that did not report these outcomes were excluded after a feasibility assessment. Change from baseline in MG-ADL and QMG scores were neither primary nor secondary end points for the ADAPT trial but given that those outcomes were reported in all other eligible studies and that these end points were required for the cost-effectiveness model, they were calculated for use in the NMA.
Table 30: Summary of Included Trials in the Sponsor-Submitted ITC
Detail | ADAPT28 | Howard et al. (2019)39 (NCT02965573) | Champion-MG42 | NCT0247395240 | Wolfe et al. (2002)41 |
|---|---|---|---|---|---|
Intervention | Efgartigimod alfa vs. placebo | Efgartigimod alfa vs. placebo | Ravulizumab vs. placebo | IVIg vs. placebo | IVIg vs. placebo |
Study design | Randomized, phase III, double-blind | Randomized, phase II, double-blind | Randomized, phase III, double-blind | Randomized, phase II, double-blind | Randomized, double-blind |
Population | Patients with gMG (including patients with AChR-Ab+ gMG) | Patients with AChR-Ab+ gMG | Patients with AChR-Ab+ gMG | Patients with AChR-Ab+ gMG | Patients with AChR-Ab+ gMG |
Number of patients at randomization | 167 patients, including 84 in the efgartigimod alfa group, and 83 in the placebo group | 24 patients, including 12 in the efgartigimod alfa group, and 12 in the placebo group | 175 patients, including 86 in the ravulizumab group and 89 in the placebo group | 62 patients, including 30 in the IVIg group, and 32 in the placebo group | 15 patients, including 6 in the IVIg group, and 9 in the placebo group |
Total duration | 26 weeks | 11 weeks | 26 weeks | 24 weeks | 42 days |
Inclusion criteria |
|
|
|
|
|
Exclusion criteria |
|
|
|
|
|
Concomitant therapies | Stable dose of at least 1 treatment (AChEI, CS, or NSIST) |
| Stable conventional therapy permitted | Conventional therapy |
|
End points |
|
|
|
|
|
Ab+ = antibody positive; Ab– = antibody negative; AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; CS = corticosteroid; gMG = generalized myasthenia gravis; Ig = immunoglobulin; IMIg = intramuscular immunoglobulin; ITC = indirect treatment comparison; LRP4 = lipoprotein receptor-related protein 4; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MGFA = Myasthenia Gravis Foundation of America; MG-QoL15 = Myasthenia Gravis Quality of Life 15-item; MuSK = muscle-specific kinase; NSIST = nonsteroidal immunosuppressive therapy; PE = plasma exchange; PP = plasmapheresis; QMG = Quantitative Myasthenia Gravis; SCIg = subcutaneous immunoglobulin; vs. = versus.
Source: Sponsor-submitted Indirect Treatment Comparison report.1
Availability of Data for IVIg
Very few studies reported on the efficacy of IVIg versus placebo for gMG. Most of the IVIg studies identified compared IVIg with PE or considered it as an acute treatment. Only Wolfe et al. (2002)41 and NCT0247395240 reported IVIg maintenance therapy compared to placebo for the treatment of gMG. The Wolfe et al. (2002)41 study was the only study to report both MG-ADL and QMG outcomes, and it was terminated early due to insufficient IVIg inventories and had a very small sample size (n = 15). The NCT02473952 study40 had a larger sample size (n = 62) and ran to completion, but MG-ADL had to be imputed to increase the sample size to inform efficacy of IVIg in the NMA. The MG-ADL data from the NCT02473952 study were imputed based on the QMG data using the Bayesian bivariate meta-analysis model.
Baseline Characteristics
Baseline characteristics reported by the included studies are presented in Table 31. In many studies, data were not reported consistently, such as for MGFA at baseline, the use of steroids or NSISTs at baseline, disease duration, and history of thymectomy. Race, sex, MG-ADL at baseline, and QMG at baseline were generally comparable across those studies with available data. The main exceptions to this were the Wolfe et al. (2002) study, which enrolled younger patients with lower baseline MG-ADL and QMG scores, and CHAMPION MG, which enrolled higher proportions of Asian, Black, African American, or other ethnicity patients.
The included studies were assessed for homogeneity. Important differences across trials for key characteristics are summarized in Table 32.
Table 31: Baseline Characteristics of Patients With AChR-Ab+ in the ITC
Characteristic | ADAPT (N = 129) | Howard et al. (2019) (N = 24) | NCT02473952 (N = 62) | Wolfe et al. (2002) (N = 15) | CHAMPION MG (N = 175) |
|---|---|---|---|---|---|
Any history of thymectomy, % | 58.1 | 50.0 | NR | NR | NR |
Female, % | 66.7 | 62.5 | 53.2 | NR | 51.0 |
Race, % | |||||
Asian | 8.5 | 4.2 | 1.6 | NR | 18.0 |
Black or African American | 3.1 | 4.2 | 1.6 | NR | 30.0 |
Other or not reported | 3.2 | 0.0 | 1.6 | NR | 6.0 |
White | 85.3 | 91.7 | 95.2 | NR | 73.0 |
Age (years), mean (SD) | 46.9 (15.4) | 49.4 (17.4) | 51.2 (15.6) | 41.9 (NR) | 55.6 (15.1) |
MG duration (years), mean (SD) | 9.3 (8.3) | 10.8 (10.3) | NR | NR | 9.9 (9.3) |
Baseline MG-ADL score, mean (SD) | 8.8 (2.3) | 8.0 (2.6) | NR | 5.7 (3.8) | 9.0 (2.5) |
Baseline QMG score, mean (SD) | 15.6 (4.8) | 13.2 (5.9) | NR | 9.9 (3.7) | 14.7 (5.2) |
Baseline MGFA class, % | |||||
II | 41.1 | 54.2 | NR | NR | 44.0 |
III | 55.8 | 41.7 | NR | NR | 49.0 |
IV | 3.9 | 4.2 | NR | NR | 6.0 |
Use of steroids or NSISTs at baseline, % | 86.0 | NR | NR | NR | 90.0 |
AChR-Ab+ = acetylcholine receptor antibody positive; ITC = Indirect Treatment Comparison; MG = myasthenia gravis; MGFA = Myasthenia Gravis Foundation of America; MG-ADL = Myasthenia Gravis Activities of Daily Living; NR = not reported; NSIST = nonsteroidal immunosuppressive therapy; QMG = Quantitative Myasthenia Gravis; SD = standard deviation.
Source: Sponsor-submitted Indirect Treatment Comparison Report.1 Details included in the table are from the Sponsor’s Summary of Clinical Evidence.
Table 32: Assessment of Homogeneity
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Patients with MGFA Class I were excluded from all studies. Baseline MG-ADL and QMG scores were generally similar between studies, except for the Wolfe et al. (2002) study, which enrolled somewhat healthier patients. |
Trial eligibility criteria | Inclusion and exclusion criteria were generally similar across studies. All trials other than ADAPT only included patients with AChR-Ab+ gMG. Patients with a recent thymectomy were generally excluded, although the exclusion period ranged from 3 to 12 months across studies. All trials excluded patients with very mild disease (e.g., MGFA Class I). All studies excluded patients younger than 18 years, with the exception of CHAMPION MG, which was limited to patients 15 years or older, although the mean age of patients in this study was not meaningfully different from the age of patients in the other studies. Most of the studies excluded patients with a history of malignancy (3 of 5). Less common exclusion criteria included pregnancy (2 of 5), myasthenic crisis (2 of 5), and renal impairment (2 of 5). |
Treatment history | All trials excluded patients who had recently received IVIg, SCIg, or PE treatment. Most of the studies excluded patients with a history of treatment with rituximab within the past 6 to 12 months (4 of 5). |
Dosing of comparators | All studies employed a dosing schedule involving spaced infusions, but only ADAPT used individual patient response to determine subsequent cycles of treatment. |
Placebo response | Placebo response was generally comparable between studies. |
Definitions of end points | There was considerable variation in primary and secondary end points across the included studies. The MG-ADL responder definition in ADAPT was different from any of the other comparator trials included in the feasibility assessment. Responders per MCID for MG-ADL and QMG were only reported in ADAPT and CHAMPION MG, while changes from baseline MG-ADL and QMG scores were the best reported, appearing in 3 and 4 studies, respectively. Although these continuous outcomes were not primary or secondary end points for ADAPT, those end points could be calculated using the Clinical Study Report68 and were the best source of comparative efficacy data for this NMA. |
Timing of end point evaluation | The included studies varied in follow-up time, and primary assessment time points ranged from 4 to 26 weeks. The ADAPT study was unique in that it measured patient response during the first cycle, after 4 weeks of treatment, and then during subsequent cycles, when only patients requiring additional treatments for symptom management, as determined by clinicians, received additional doses. Analyses conducted at the primary time point for all trials could be biased against ADAPT, as they could exclude the best responders to efgartigimod alfa, whereas ITCs conducted at week 4 only could be biased against any treatments that demonstrated improved responses over time. Primary analyses in this report were conducted on the primary time points for all included studies, and a sensitivity analysis was used to compare all trials at ± 2 weeks of week 4 to allow more studies to be included. |
Study design | All studies were placebo-controlled, randomized, double-blind trials. |
AChR-Ab+ = acetylcholine receptor antibody positive; ITC = indirect treatment comparison; IVIg = intravenous immunoglobulin; MCID = minimum clinically important difference; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGFA = Myasthenia Gravis Foundation of America; NMA = network meta-analysis; PE = plasma exchange; QMG = Quantitative Myasthenia Gravis; SCIg = subcutaneous immunoglobulin.
Note: Details included in the table are from the Sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted Indirect Treatment Comparison report.1
Evidence Network
The evidence network was constructed as part of a feasibility assessment, which was built for each outcome of interest based on data availability. The evidence networks for change from baseline in MG-ADL and QMG included 5 RCTs (Figure 10). Two connections were informed by 2 studies (efgartigimod alfa versus placebo,28,39 and IVIg versus placebo40,41), and another connection (ravulizumab versus placebo42) was informed by 1 study.
Figure 13: Network Diagram for Change From Baseline in MG-ADL and QMG at Weeks 4 to 26
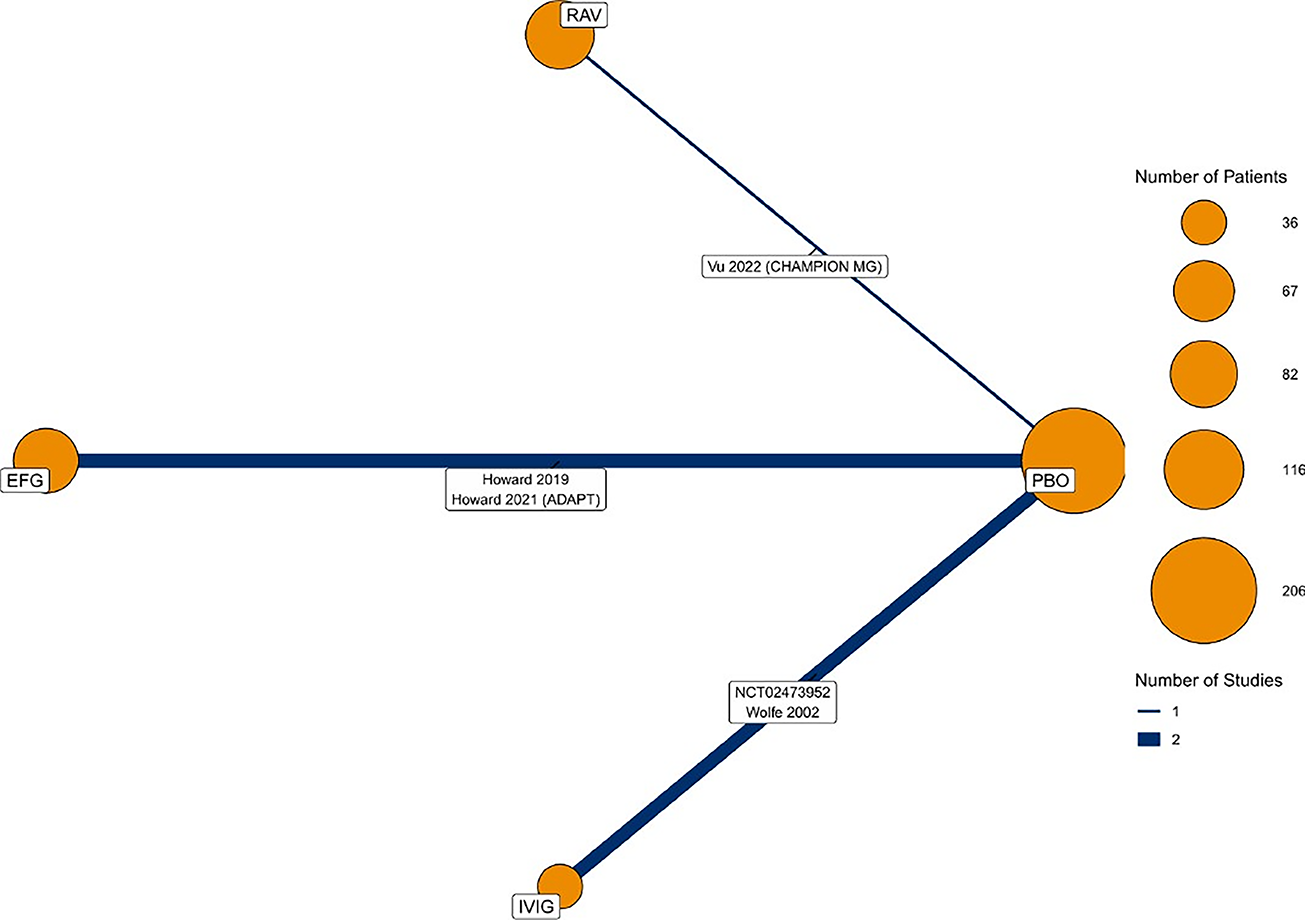
EFG = efgartigimod alfa; IVIg = intravenous immunoglobulin; MG-ADL = Myasthenia Gravis Activities of Daily Living; PBO = placebo; QMG = Quantitative Myasthenia Gravis; RAV = ravulizumab.
Source: Sponsor-submitted Indirect Treatment Comparison report.1
Efficacy
Changes from baseline in MG-MDL and QMG scores from the individual studies included in the ITC analysis are presented in Table 33.
Table 33: Change From Baseline in MG-MDL and QMG Scores From Individual Trials: AChR-Ab+ Population
Study | Treatment | N | Change from baseline in MG-ADL score (points) | Change from baseline in QMG score (points) | ||
|---|---|---|---|---|---|---|
Mean (SE) | Primary time point (weeks) | Mean (SE) | Primary assessment time point (weeks) | |||
ADAPT | Efgartigimod alfa | 65 | –4.60 (0.40) | 4 | –6.20 (0.66) | 4 |
Placebo | 64 | –1.80 (0.31) | 4 | –1.00 (0.37) | 4 | |
Howard et al. (2019) | Efgartigimod alfa | 12 | –3.50 (1.10) | 11 | –4.80 (2.40) | 11 |
Placebo | 12 | –0.80 (1.20) | 11 | –2.10 (1.60) | 11 | |
NCT02473952 | IVIg | 30 | –3.31a (0.58a) | 24b | –4.60 (0.93) | 24 |
Placebo | 32 | –2.22a (0.58a) | 24b | –2.70 (1.10) | 24 | |
Wolfe et al. (2002) | IVIg | 6 | –0.30 (0.82) | 6 | 0.00 (1.55) | 6 |
Placebo | 9 | –2.60 (0.80) | 6 | –1.60 (0.90) | 6 | |
CHAMPION MG | Ravulizumab | 86 | –3.12 (0.38) | 26 | –2.85 (0.45) | 26 |
Placebo | 89 | –1.42 (0.35) | 26 | –0.80 (0.45) | 26 | |
AChR+ = acetylcholine receptor antibody positive; IVIg = intravenous immunoglobulin; MG-ADL = Myasthenia Gravis Activities of Daily Living; QMG = Quantitative Myasthenia Gravis; SE = standard error.
aMG-ADL results for NCT02473952 were imputed.
bThe MG-ADL results for NCT02473952 were imputed based on the study’s QMG results at the same time point.
Note: Details included in the table are from the Sponsor’s Summary of Clinical Evidence.
Source: Sponsor-submitted Indirect Treatment Comparison report.1
This section summarizes the results of the ITC comparing efgartigimod alfa versus IVIg and versus ravulizumab for MG-ADL and QMG.
Primary analyses were conducted on the primary assessment time points for all included studies, from 4 to 26 weeks. The mean differences for change from baseline in MG-ADL were –2.64 (95% CrI, –4.16 to –1.12) for efgartigimod alfa versus IVIg, and –0.91 (95% CrI, –2.25 to 0.39) for efgartigimod alfa versus ravulizumab. The mean differences for change from baseline in QMG were –4.39 (95% CrI, –6.95 to –1.81) for efgartigimod alfa versus IVIg, and –2.89 (95% CrI, –4.72 to –1.12) for efgartigimod alfa versus ravulizumab. A change of 2 points in the MG-ADL score and 3 points in the QMG score was estimated to be the threshold of clinical significance in patients with MG.
Sensitivity Analyses
As the primary assessment time points varied across studies from 4 to 26 weeks, sensitivity analyses for change from baseline in MG-ADL and QMG scores were performed at or plus or minus 2 weeks of week 4 (the timing of the largest mean treatment response in ADAPT). For comparator studies that did not report data at week 4, assessments conducted at plus or minus 2 weeks of week 4 were included in the analysis. A total of 4 studies reported change from baseline in MG-ADL score in this time frame (at 4 ± 2 weeks). Wolfe et al. (2002)41 was the only study to report change from baseline in MG-ADL score for IVIg within this time frame (at week 6). Imputed MG-ADL data for IVIg estimated for NCT0247395240 were not included as imputations leveraged efficacy data across all reported time points included in the primary analysis.
The mean differences for change from baseline in MG-ADL were –4.91 (95% CrI, –7.37 to –2.53) for efgartigimod alfa versus IVIg, and –1.54 (95% CrI, –2.84 to –0.23) for efgartigimod alfa versus ravulizumab. The mean differences for change from baseline in QMG score were –6.53 (95% CrI, –10.3 to –2.86) for efgartigimod alfa versus IVIg, and –3.20 (95% CrI, –5.01 to –1.38) for efgartigimod alfa versus ravulizumab.
Additional Analyses
As the sponsor’s reimbursement request is limited to patients who are AChR-Ab+, comparators that the sponsor considered relevant to that group were used in the primary ITC analysis. Because the ADAPT trial had a mixed population, it was feasible to perform an ITC using the available comparator studies and efficacy data from the ADAPT trial for the AChR-Ab+ subgroup. Within the AChR-Ab– subpopulation, patients with gMG who tested positive for antibodies against MuSK receptors are often treated with rituximab; therefore, the sponsor included rituximab as a comparator for the additional analyses to provide some evidence on the AChR-Ab– subpopulation. The sponsor assumed that the ITCs derived from the AChR-Ab+ subpopulation would apply equally to the AChR-Ab– population.
Only the BeatMG78 trial, which compared the efficacy of rituximab with placebo, was found to be eligible for inclusion in the additional analyses after the feasibility assessment; however, only patients with AChR-Ab+ gMG were included in this study. The patient population in the BeatMG78 trial was different from that in the ADAPT trial, representing a population with milder disease severity (i.e., baseline MG-ADL of 4.9 in BeatMG versus 8.8 in ADAPT), and shorter disease duration (i.e., average disease duration of 5.5 years in BeatMG versus 9.3 years in ADAPT). BeatMG78 was a US-based, phase II, multicentre, randomized, double-blind study with a planned study duration of 52 weeks. In total, 52 patients were enrolled from across the US, all of whom tested positive for antibodies to AChR. Patients were randomized 1:1 to receive placebo or 2 cycles of IV rituximab over 4 weekly infusions per cycle of 375 mg/m2, separated by 6 months. A total of 25 patients were randomized to receive rituximab, and 27 to receive placebo. The primary end points of BeatMG were steroid-sparing effect (proportion achieving at least 75% reduction in mean daily prednisone dose in the 4 weeks before week 52 with clinical improvement, or no significant worsening compared to the 4-week period before randomization) and safety outcomes. Additional secondary end points included Myasthenia Gravis Composite (MGC) and QMG scores from baseline to week 52 with exploratory clinical outcomes measuring change in MG-ADL, MG-QoL15, MG exacerbation rate, and biomarkers. The risk-of-bias assessment showed that the BeatMG trial had a low or unclear risk of bias.
Overall, 6 RCTs were included in the additional analyses, including 2 for efgartigimod alfa, 2 for IVIg, 1 for ravulizumab, and 1 for rituximab (Figure 11).
Figure 14: Network Diagram for Additional Analyses for Change From Baseline in MG-ADL and QMG Scores
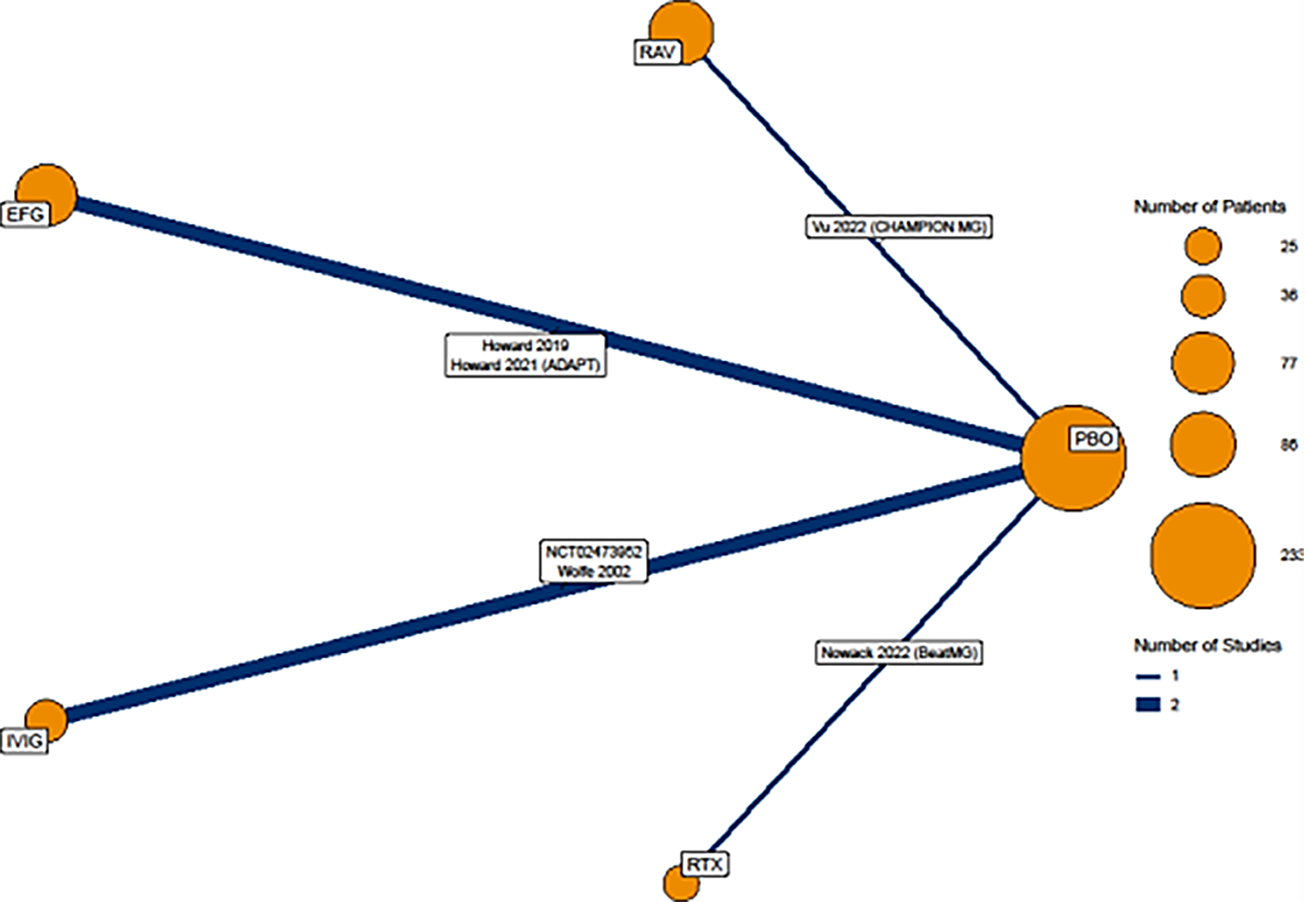
EFG = efgartigimod alfa; IVIG = intravenous immunoglobulin; PBO = placebo; RAV = ravulizumab; RTX = rituximab.
Source: Sponsor-submitted Indirect Treatment Comparison report.1
Efficacy
The mean differences from baseline in MG-ADL were –2.64 (95% CrI, –4.18 to –1.12) for efgartigimod alfa versus IVIg, –0.92 (95% CrI, –2.25 to 0.43) for efgartigimod alfa versus ravulizumab, and –1.93 (95% CrI, –3.87 to 0.07) for efgartigimod alfa versus rituximab. The mean differences from baseline in QMG scores were –4.39 (95% CrI, –7.01 to –1.83) for efgartigimod alfa versus IVIg, –2.89 (95% CrI, –4.72 to –1.06) for efgartigimod alfa versus ravulizumab, and –2.71 (95% CrI, – 5.56 to 0.2) for efgartigimod alfa versus rituximab.
Harms
According to the sponsor, safety analyses were not conducted in the ITC given the heterogeneity in the methodology associated with safety end points across studies and the unreliability of safety data derived in the clinical trial context.
Critical Appraisal of Sponsor-Submitted ITC
The SLR used to identify relevant studies was methodologically sound in terms of the sponsor using a comprehensive literature search strategy as well as performing study selection, data extraction, risk-of-bias assessment in duplicate, and providing a list of excluded studies and justifying the exclusions. However, it was unclear in the ITC report whether the feasibility assessment was carried out by a single or multiple assessors. By conducting a feasibility assessment, the sponsor excluded all head-to-head trials, including those comparing efficacy of IVIg treatment versus PE, which may have reduced the information in NMA. In addition, a search for other published network meta-analyses was not explicitly carried out for the SLR; as such, it is not clear whether there are other NMAs in this disease area, and if so, whether their conclusions are similar. The risk of bias of included studies in the SLR was assessed per individual study; however, it may be different depending on the study outcomes. The literature search was conducted on February 14, 2020, and updated on April 7, 2022, so it is considered relatively up-to-date at the time of this review. The eligible interventions for the ITC were limited to those used in Canada for the treatment of gMG to ensure that the comparators were relevant to Canadian settings. After feasibility assessment, 5 studies were considered eligible to be included in the NMA, including 2 studies for efgartigimod alfa, 2 studies for IVIg, and 1 study for ravulizumab. Studies were required to report at least 1 of the following outcomes: MG-ADL responders or change from baseline in MG-ADL score; and QMG responders or change from baseline in QMG score. Thus, trials that did not report on those outcomes were excluded, even if they reported other relevant outcomes, such that comparisons for outcomes relevant to patients, clinicians, and drug plans have been excluded (e.g., HRQoL, exacerbations, hospitalizations).
All NMA analyses were performed using a Bayesian framework. Given the paucity of multistudy connections in the evidence network, a random effects NMA was deemed infeasible, and fixed effects NMA was performed for all outcomes. Other limitations of the NMA relate to data sparseness, and network structure. The networks for analyses were sparse (i.e., 3 comparisons with few trials), and the assessment of statistical consistency was not possible as no closed loops were included. All trials included in the ITC had sufficiently similar study designs and a common comparison group (placebo). However, there were some important differences between the trials included in the NMA that increase the uncertainty of the analyses. All included studies employed a dosing schedule involving spaced infusions, but only ADAPT used individual patient response to determine subsequent cycles of treatment. The studies included in the ITC analyses ranged in follow-up time from 4 to 26 weeks. Patients with a recent thymectomy were generally excluded from all studies, although the exclusion period ranged from 3 to 12 months across the studies. All studies allowed for the use of concomitant standard-of-care treatments, but details on the breakdown of the actual concomitant medications used were not available; as a result, it is not clear whether concomitant medication use was similar across the trials. In many studies, baseline data were not reported consistently, such as for MGFA at baseline, use of steroids or NSISTs at baseline, disease duration, and history of thymectomy. As such, it was not possible to ascertain whether the patient populations were similar enough across the trials to combine in NMA. Primary analyses in this report were conducted on the primary assessment time points for all included studies, which could be biased against ADAPT as they could exclude the best responders to efgartigimod alfa. The ITC results were reported as mean differences and 95% Crls. The evidence is imprecise in the effect estimates from the NMA due to the sparseness of data, with wide CrIs, and the upper and lower boundaries of the CrIs suggest the potential for different conclusions regarding the efficacy of efgartigimod alfa relative to the comparator drugs (i.e., important differences and little to no difference [or trivial effects]). In addition, heterogeneity between the included studies, both in terms of patient characteristics (which are uncertain because many baseline characteristics of patients across trials were not reported) and methods, would be expected to introduce bias into the study estimates observed between the comparators. Another important limitation of the presented ITC is the lack of safety data. Without this, it is not possible to evaluate the combined relative efficacy and safety of efgartigimod alfa relative to the comparator drugs. Given the absence of a clear conclusion with respect to efficacy, the influence of safety may become of higher priority to physicians and patients considering treatment with efgartigimod alfa. Outcomes that are important to patients, such as HRQoL, were also not analyzed in this ITC.
The sponsor stated that patients with AChR-Ab– gMG who tested positive for antibodies to MuSK are often treated with rituximab; therefore, rituximab was found to be relevant only to the AChR-Ab– subpopulation. Thus, rituximab was not included in the primary ITC analysis, but was included in the additional analyses to provide some evidence for the AChR-Ab– subpopulation. However, additional analyses included only patients with AChR-Ab+ gMG from 6 studies, including BeatMG for rituximab. The clinical experts consulted by CADTH for this review indicated that a study published in 2022,79 which was not identified in the sponsor’s search and therefore was not included in the ITC, provides evidence for the efficacy and safety of rituximab for early onset gMG in patients tested positive for antibodies to AChR. The sponsor assumed that the ITCs derived from the AChR-Ab+ subpopulation would be equally applicable to the AChR-Ab– subpopulation. However, the clinical experts consulted for this review indicated that there is no clinical evidence available to support that assumption, as no RCTs have been conducted in patients with AChR-Ab– gMG given the small number of patients. Because all comparator studies were performed exclusively with patients with AChR-Ab+ gMG, all analyses included only patients from the AChR-Ab+ subpopulation, which aligns with the sponsor-submitted reimbursement request criteria and revised proposed Health Canada indication.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Studies addressing gaps in the systematic review evidence are not available.
Discussion
Summary of Available Evidence
One phase III, double-blind, placebo-controlled RCT (ADAPT N = 169),30 with an open-label extension period of up to 14 cycles (ADAPT+)68,80 and a sponsor-submitted ITC were included in this review. The objective of the ADAPT trial was to evaluate the efficacy and safety of efgartigimod alfa added on to the conventional therapy versus placebo added to the conventional therapy in adult patients with gMG whose symptoms persist despite a stable dose of standard-of-care (concomitant gMG treatment) treatment with AChEIs, corticosteroids, and/or NSISTs. All patients had MGFA Class II to IV gMG and MG-ADL total scores of 5 or more. In the AChR-Ab+ population, the mean age was 44.7 to 49.2 years, and most patients were white (83.1% to 87.5%). All (N = 129, 100%) patients had received 1 prior therapy, 124 (96.2%) patients had received 2 or more prior therapies and 102 (79.1%) patients had received 3 or more prior therapies. The majority had previously received 2 or more (96.2%) or 3 or more (79.1%) different classes of conventional therapy medication (any combination of AChEIs, corticosteroids, and/or NSISTs, at the physician’s discretion). In the patients who were AChR-Ab+, 41 (63%) in the efgartigimod alfa group and 37 (58%) in the placebo group had received 3 classes of prior therapy (steroid plus NSIST plus AChEI). Eleven (16.9%) patients in the efgartigimod alfa group and 23 (36%) in the placebo group had received any 2 of the 3 (steroid, NSIST, and AChEI) classes of prior therapy. In addition, of the patients who were AChR-Ab+, 63% had experienced prior gMG treatment failure (also known as patients with refractory gMG)32 and 37% had responded inadequately to the existing standard-of-care gMG therapy.1,81
According to the clinical experts consulted by CADTH for the review, the baseline characteristics of the ADAPT trial population were broadly representative of the population of patients in Canada who have gMG that is refractory to the standard conventional therapy or whose symptoms are inadequately controlled with the existing standard conventional therapy for gMG. Patients were randomized 1:1 to receive efgartigimod alfa or a matching placebo in cycle 1 (i.e., for the first 8 weeks), followed by an individualized treat-as-needed regimen based on each patient’s MG-ADL response. All patients received a stable concomitant treatment during the trial. The primary outcome of the study was the percentage of patients with AChR-Ab+ who were MG-ADL responders in the first treatment cycle. Key secondary outcomes included percentage of time of CMI in MG-ADL score (≥ 2-point reduction) up to day 126; time from week 4 to qualify for re-treatment; percentage of early MG-ADL responders in cycle 1 (i.e., MG-ADL ≥ 2 points occurred by week 2); and change from cycle baseline in MG-ADL total score during cycle 1 and cycle 2. Change from cycle baseline in HRQoL (MG-QoL15r score, EQ-5D VAS) during cycle 1 and cycle 2 were assessed as tertiary or exploratory outcomes. Post hoc analyses were performed for MG hospitalization, MG exacerbation, MG crisis, and use of rescue therapy. It should also be noted that, although the duration of the ADAPT trial was designed to be 26 weeks, the primary, key secondary, and the HRQoL outcomes at the end of the study (i.e., week 26) were not assessed. Instead, the outcomes were assessed at the ends of cycle 1 and cycle 2.
Results of the long-term extension phase up to 14 cycles (ADAPT+) are also presented in this report. The ADAPT+ trial is ongoing at the time of this review. Therefore, the long-term efficacy and safety outcomes of ADAPT+ were based on interim analysis (IA4 and IA5). MG-ADL, QMG, and safety outcomes were assessed in the long-term extension study.
The sponsor-submitted SLR with NMA, which evaluated the relative efficacy of efgartigimod alfa against IVIg and ravulizumab in patients with AChR-Ab+ gMG, is also included in this report. The primary analysis included 5 studies, 2 studies comparing efgartigimod alfa with placebo,28,39 2 studies comparing IVIg with placebo,40,41 and 1 study comparing ravulizumab with placebo.42 The ITC was performed using the Bayesian framework with placebo as the reference treatment. The clinical end points used for ITC estimates included change from baseline in MG-MDL and QMG scores, although these outcomes were not primary or secondary end points for ADAPT.
Interpretation of Results
Efficacy
Activities of Daily Living (MG-ADL)
MG-ADL Responders During Cycle 1 and Cycle 2
MG-ADL responders during cycle 1 in the population of patients who were AChR-Ab+ was the primary outcome in the ADAPT trial. Thirty-eight percent (95% CI, 22% to 56%) more patients in the efgartigimod alfa group than in the placebo group achieved an improvement in MG-ADL score of 2 points or more during cycle 1. According to clinical experts, this indicated a clinically meaningful benefit compared to the placebo group in terms of MG-ADL response during cycle 1. Various post hoc subgroup analyses were conducted for MG-ADL responders during cycle 1. These results suggest that efgartigimod alfa produces improvements in MG-ADL response compared to placebo, regardless of prior therapies, concomitant therapies, disease duration, thymectomy, and prior treatment failure; however, it should be noted that the trial was not powered to detect subgroup differences.1,30,32,33 In terms of the MG-ADL responders, similar benefit was observed in in cycle 2.
Early MG-ADL responders: Early responders (those who responded at week 2 of the cycle 1) in the population of patients who were AChR-Ab+ was assessed as a fifth key secondary outcome. Because the statistical testing hierarchy was broken at the fourth secondary end point (i.e., time to qualify for re-treatment), the percentage of patients in the AChR-Ab+ population who were early MG-ADL responders was not statistically tested based on statistical plan in the protocol. Nevertheless, within the AChR-Ab+ population, a higher proportion of patients in the efgartigimod alfa group achieved MG-ADL improvement of 2 points or more at week 2 than in placebo group, that is 31.9% (95% CI, NR) more patients were MG-ADL responders at week 2 than in the placebo group. The clinical experts consulted by CADTH for this review considered the between-group difference to be clinically important. The percentage of early MG-ADL responders during cycle 2 was not assessed and not reported in the sponsor’s evidence summary.
Percentage time of the MG-ADL CMI up to day 126: Among those in the AChR-Ab+ population, the percentage of time with a CMI in the MG-ADL total score up to day 126 was assessed as a third key secondary outcome and was included in the hierarchy test to control for type I error. According to the clinical experts, the percentage of time with a CMI in the MG-ADL total score in the efgartigimod alfa group was clinically meaningfully longer (efgartigimod alfa group – placebo group = 22.07%; 95% CI, 10.94 to 33.18; P = 0.0001) compared with that in the placebo group.
MG-ADL change from cycle baseline: During cycle 1 and cycle 2, the changes from cycle baseline in MG-ADL score were assessed as exploratory outcomes in patients who were AChR-Ab+. At week 4 of cycle 1, the reduction (improvement) in MG-ADL total score in the efgartigimod alfa group was greater than that in the placebo group (efgartigimod alfa group – placebo group = –2.84; 95% CI, –3.8 to –1.9; P < 0.0001). However, since this was assessed as an exploratory outcome and with no multiplicity adjustment (i.e., it was not included in the hierarchy test), there is an increased risk of type I error. Nevertheless, the results are supportive of, and aligned with, the responders analysis so the concern about a type I error is reduced. It should be noted that the maximum MG-ADL change from cycle baseline improvement of treatment with efgartigimod alfa occurred at approximately week 4 of the cycle. The magnitude of the improvement and the comparative benefit of efgartigimod alfa compared with placebo tended to smaller toward the end of the cycle. Similar results were observed in cycle 2.
Time to Re-Treatment
Time to qualify for re-treatment in the patients who were AChR-Ab+ was assessed as the fourth key secondary outcome. The median time to qualify for re-treatment in the efgartigimod alfa group was numerically but not significantly greater than the time in the placebo group (median = || |||| ||| | ||||| ||||||||||||| |||||||| ||||||||). As such, the statistical hierarchy test was broken at this point. Since the week 4 visit, the proportion of patients who qualified for re-treatment appeared to be similar in both groups (efgartigimod alfa group – placebo group = 1.4%, 95% CI, NR). The clinical experts that CADTH consulted for this review indicated that the results show that approximately half of the patients usually need re-treatment around week 6 of the treatment cycle.
Disease Severity (Assessed With QMG)
QMG Responder During Cycle 1
The percentage of QMG responders among patients who were AChR-Ab+ was assessed as the first key secondary outcome. It was reported that 49.0% more patients (95% CI, 34.5% to 63.5%) in the efgartigimod alfa group achieved QMG response compared with in the placebo group. According to the clinical experts, the benefit of treatment with efgartigimod alfa compared with placebo was considered clinically meaningful.
Health-Related Quality of Life
HRQoL (i.e., MG-QoL15r and EQ-5D VAS) was assessed as an exploratory outcome in patients who were AChR-Ab+. The changes from cycle baseline in MG-QoL15r and EQ-5D VAS scores were assessed for cycle 1 and cycle 2. At week 4 of cycle 1, the reduction (improvement) in MG-QoL15r score in the efgartigimod alfa group appeared greater than that in placebo group (efgartigimod alfa group – placebo group = –5.45; 95% CI, | |||||| || ||||||; P < 0.0001). The increase (improvement) in EQ-5D VAS score in the efgartigimod alfa group also appeared to be greater than that in placebo group (efgartigimod alfa group – placebo group = 13.28; 95% CI, |||| || |||||; nominal P < 0.0001). Because HRQoL was assessed as an exploratory outcome with no multiplicity adjustment (i.e., it was not included in the statistical hierarchy test), there is an increased risk of a type I error; however, the results provide supportive evidence. Although there is no established MID for either the MG-QoL15r or the EQ-5D VAS among patients with gMG, the clinical experts consulted by CADTH for this review considered the between-group differences to be clinically meaningful. It should be noted that both the maximum MG-QoL15r and EQ-5D VAS improvements with efgartigimod alfa appeared to occur at approximately week 4 of the cycle. The magnitude of the improvement and the comparative benefit of efgartigimod alfa compared with placebo tended to smaller toward the end of the cycle. Similar results were observed in cycle 2.
Other Clinical Outcomes (MG Hospitalization, MG Exacerbations, MG Crisis, Rescue Therapy Use, and Survival)
In patients who were AChR-Ab+, during the 26 weeks double-blind period, MG hospitalization events, MG crisis events, and rescue therapy use were very low in both groups. These outcomes (as well as MG exacerbations) were analyzed in post hoc analyses. and the study were not powered to assess between-group difference of these outcomes. No death was reported during the 26-week study period; however, follow-up may not have been long enough to adequately assess this outcome.
Long-term, open-label, extension (ADAPT+): In terms of MG-ADL response (up to 14 cycles) and QMG response (up to 7 cycles), evidence from the long-term, open-label, extension (ADAPT+) trial appeared consistent with that from the randomized controlled period. Patients who switched from placebo to efgartigimod alfa experienced numeric improvements from baseline in MG-ADL and QMG scores in each cycle. However, interpretation of these data was limited by the open-label and descriptive nature of the extension study.
ITC: In the sponsor’s submitted ITC, the primary analysis used data from the primary time point of assessment for each study, and a sensitivity analysis was conducted using data at or plus or minus 2 weeks of week 4, to align with the primary assessment time point of the ADAPT trial. The results of the sponsor-submitted NMA suggest that relative to IVIg and ravulizumab, efgartigimod alfa may provide a benefit with respect to change in MG-ADL and QMG scores; however, the 95% CrIs for the effect estimates included the possibility of trivial effects (i.e., only small, nonclinically important differences between groups) and no difference (in the case of change in MG-ADL score relative to ravulizumab). No difference in efficacy in terms of change from baseline in MG-ADL and QMG scores could be concluded for efgartigimod alfa relative to rituximab due to wide 95% CrIs (which included the possibility of clinically important benefit favouring efgartigimod alfa), and methodological limitations. There is additional uncertainty in these results due to the heterogeneity between trials with respect to differences in dosing regimens of drugs, variability in patient characteristics (and lack of reporting of many patient characteristics across the trials), and variability in study follow-up times. The network was sparse (included few trials) and consistency in direct and indirect effects could not be tested due to a lack of closed loops. Another important limitation presented by the ITC is the lack of safety and HRQoL data. Additional analyses were conducted with the inclusion of rituximab as a comparator drug to provide some evidence on the AChR-Ab– subpopulation. However, only patients with AChR-Ab+ gMG from 6 studies, including the BeatMG trial for rituximab, were included in the additional analyses. Thus, all ITC analyses were performed only in patients with AChR-Ab+ gMG, which aligns with the sponsor-submitted reimbursement request and may not be fully generalized to the overall gMG population (including patients who are AChR-Ab–). A search for other published network meta-analyses was not explicitly carried out for the SLR. However, the sponsor identified a recently published exploratory NMA comparing efgartigimod alfa, ravulizumab, rituximab, eculizumab, zilucoplan, and rosanoliximab in patients with gMG, with efficacy results of efgartigimod alfa versus ravulizumab consistent with those presented in the sponsor-submitted ITC.
Harms
Reduction of side effects was identified in the patient input for this review as of interest for patients with gMG. The ADAPT trial, including its randomized controlled period, provided some relevant information regarding the safety profile of efgartigimod alfa in the treatment of gMG. However, it did not provide direct comparative evidence regarding the side effects of efgartigimod alfa versus other MG therapies. In the AChR-Ab+ population, during the randomized controlled period, proportions of patients with TEAEs in the efgartigimod alfa group appeared similar to that in the placebo group (efgartigimod alfa group versus placebo group: ||||| ||| |||||) in the ADAPT trial. The proportions of patients with SAEs were low in both groups and appeared numerically lower in the efgartigimod alfa group than in the placebo group (|||| ||| |||||) in the ADAPT trial. Withdrawals due to AEs occurred in similar proportions in both the efgartigimod alfa and placebo groups (|||| ||| |||||) in the ADAPT trial. No deaths were reported during the double-blind period; however, the follow-up time was likely insufficient to measure differences in this outcome. The main notable harms (i.e., the AESIs for this review) were in the MedDRA system organ class “infections and infestations,” and were reported in a numerically higher proportion of patients in the efgartigimod alfa group than in the placebo group (efgartigimod alfa group versus placebo group: ||||| ||| |||||); however, there is some uncertainty in this conclusion because the 95% CI includes the possibility of clinically important harm (i.e., a decrease in infections and infestations with the use of efgartigimod alfa relative to placebo). The proportions of patients in the AChR-Ab+ population with infusion reactions were not reported in the Clinical Study Report. No meningococcal infections were reported. According to the clinical experts CADTH consulted for this review, the TEAEs reported in the ADAPT trial were as expected and are commonly seen with other existing immunosuppressive treatments, for example, complement C5-inhibitor treatment of gMG.
However, some limited safety data from the long-term extension phase appeared consistent with that observed in the double-blind phase with no new safety signals reported.
Safety analyses were not conducted in the sponsor-submitted ITC. As such, the risk for harms with efgartigimod alfa treatment relative to any other active comparator is not known.
Conclusion
One double-blind, RCT of patients with gMG was included in this review.
Evidence from the ADAPT trial showed that, compared with placebo, treatment with efgartigimod alfa as an add-on to standard conventional therapy likely results in a clinically meaningful benefit in terms of the proportion of MG-ADL and QMG responders, and HRQoL (as assessed using the MG-QoL15r and EQ-5D VAS) after cycle 1 of treatment relative to placebo among adult patients with AChR-Ab+ gMG whose symptoms persist despite a stable dose of standard-of-care (concomitant gMG treatment) treatment with AChEIs, corticosteroids, and/or NSISTs (moderate certainty). Similar benefit was observed in cycle 2, although there is less certainty in these results because not all randomized patients participated in this cycle. Efgartigimod alfa likely results in a clinically important increase in the percentage of time with a meaningful MG-ADL improvement compared with placebo (moderate certainty). Maximum benefit appeared to occur at approximately 4 weeks of each cycle. Efgartigimod alfa may result in a clinically important reduction in MG exacerbations relative to placebo (low certainty); the results were inconclusive for MG-related hospitalizations and MG crises due to low numbers of events reported for these outcomes. The safety profile of efgartigimod alfa reported in the ADAPT trial was considered as expected and commonly seen in existing gMG therapies. In terms of MG-ADL and QMG scores and safety profile, evidence from the long-term, open-label, extension (ADAPT+) trial appeared consistent with those from the randomized controlled period in the patients who were AChR-Ab+. However, interpretation of the long-term data was limited by the open-label and descriptive nature of the extension study. The results of the sponsor-submitted NMA suggest that relative to IVIg and ravulizumab, efgartigimod alfa may provide a benefit with respect to change in MG-ADL and QMG scores; however, the 95% CrIs for the effect estimates included the possibility of trivial effects (i.e., only small, nonclinically important differences between groups) and no difference (in the case of change in MG-ADL score relative to ravulizumab). No difference in efficacy in terms of change from baseline in MG-ADL and QMG scores could be concluded for efgartigimod alfa relative to rituximab due to wide 95% CrIs (which included the possibility of clinically important benefit favouring efgartigimod alfa), and methodological limitations. There is no evidence for the effect of efgartigimod alfa on HRQoL or harms outcomes relative to any other active treatment.
References
1.Efgartigimod alfa (Vyvgart) for generalized myasthenia gravis: systematic review and network meta analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg/mL solution for intravenous use. Vaughan (ON): argenx Canada Inc.; 2023 May 9.
2.Dresser L, Wlodarski R, Rezania K, Soliven B. Myasthenia gravis: epidemiology, pathophysiology and clinical manifestations. J Clin Med. 2021;10(11):2235. PubMed
3.Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S. Myasthenia gravis - autoantibody characteristics and their implications for therapy. Nat Rev Neurol. 2016;12(5):259-268. PubMed
4.Juel VC, Massey JM. Myasthenia gravis. Orphanet J Rare Dis. 2007;2:44. PubMed
5.Paul RH, Nash JM, Cohen RA, Gilchrist JM, Goldstein JM. Quality of life and well-being of patients with myasthenia gravis. Muscle Nerve. 2001;24(4):512-516. PubMed
6.Wendell LC, Levine JM. Myasthenic crisis. Neurohospitalist. 2011;1(1):16-22. PubMed
7.Lazaridis K, Tzartos SJ. Autoantibody specificities in myasthenia gravis; implications for improved diagnostics and therapeutics. Front Immunol. 2020;11:212. PubMed
8.Meriggioli MN, Sanders DB. Muscle autoantibodies in myasthenia gravis: beyond diagnosis? Expert Rev Clin Immunol. 2012;8(5):427-438. PubMed
9.Jaretzki A, 3rd, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology. 2000;55(1):16-23. PubMed
10.Breiner A, Widdifield J, Katzberg HD, Barnett C, Bril V, Tu K. Epidemiology of myasthenia gravis in Ontario, Canada. Neuromuscul Disord. 2016;26(1):41-46. PubMed
11.Ndegwa S, Mierzwinski-Urban M. CADTH horizon scan: emerging drugs for generalized myasthenia gravis. Can J Health Technol. 2022;2(2). https://www.cadth.ca/sites/default/files/hs-eh/EH0099%20Drugs%20for%20myasthenia%20gravis%20Final.pdf. Accessed 2023 Jul 25. PubMed
12.Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol. 2010;10(1):46. PubMed
13.Farmakidis C, Pasnoor M, Dimachkie MM, Barohn RJ. Treatment of myasthenia gravis. Neurol Clin. 2018;36(2):311-337. PubMed
14.Schneider-Gold C, Gajdos P, Toyka KV, Hohlfeld RR. Corticosteroids for myasthenia gravis. Cochrane Database Syst Rev. 2005;2(CD002828). PubMed
15.Sanders DB, Wolfe GI, Benatar M, al e. International consensus guidance for the management of myasthenia gravis: executive summary. Neurology. 2016;87(4):419-425. PubMed
16.Narayanaswami P, Sanders DB, Wolfe G. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021;96(3):114-122. PubMed
17.Off-label use of intravenous immunoglobulin for neurological conditions: a review of clinical efectiveness (CADTH Rapid response report: summary with critical appraisal). Ottawa (ON): CADTH; 2018: https://www.cadth.ca/sites/default/files/pdf/htis/2018/RC0962%20Neurology%20Off-Label%20IVIG%20Final.pdf. Accessed 2023 Jul 25.
18.Canadian clinician survey: current and future treatment landscape of gMG [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg/mL solution for intravenous use. Vaughan (ON): argenx Canada Inc.; 2023 May 9.
19.CADTH Drug Reimbursement Expert Review Committee final recommendation: eculizumab (Soliris) in adult patients with refractory generalized myasthenia gravis (gMG). Ottawa (ON): CADTH; 2020 Oct: https://www.cadth.ca/sites/default/files/cdr/complete/SR0605%20Soliris%20MG%20-%20CDEC%20Final%20Recommendation%20October%2021%2C%202020_for%20posting.pdf. Accessed 2023 Jul 25.
20.pan-Canadian Pharmaceutical Alliance. Negotiations: Soliris (eculizumab). 2022; https://www.pcpacanada.ca/negotiation/21306. Accessed 2023 Jul 25.
21.CADTH Drug Reimbursement Expert Review Committee draft recommendation: ravulizumab (Ultomiris) for adult patients with AChR-Ab+ gMG. Ottawa (ON): CADTH; 2023 Apr: https://www.cadth.ca/sites/default/files/DRR/2023/SR0765%20Ultomiris%20-%20Draft%20CADTH%20Recommendation%20April%2013%2C%202023%20for%20posting.pdf. Accessed 2023 Jul 25.
22.Twork S, Wiesmeth S, Klewer J, Pohlau D, Kugler J. Quality of life and life circumstances in German myasthenia gravis patients. Health Qual Life Outcomes. 2010;8:129. PubMed
23.Cutter G, Xin H, Aban I, et al. Cross-sectional analysis of the myasthenia gravis patient registry: disability and treatment. Muscle Nerve. 2019;60(6):707-715. PubMed
24.Petersson M, Feresiadou A, Jons D, et al. Patient-reported symptom severity in a nationwide myasthenia gravis cohort: cross-sectional analysis of the Swedish GEMG study. Neurology. 2021;97:e1382-e1391. PubMed
25.Ruiter AM, Verschuuren J, Tannemaat MR. Prevalence and associated factors of fatigue in autoimmune myasthenia gravis. Neuromuscul Disord. 2021;31(7):612-621. PubMed
26.Andersen LK, Jakobsson AS, Revsbech KL, Vissing J. Causes of symptom dissatisfaction in patients with generalized myasthenia gravis. J Neurol. 2022;269(6):3086-3093. PubMed
27.Dewilde S, Philips G, Paci S. Patient-reported burden of myasthenia gravis: baseline results of the international prospective, observational, longitudinal real-world digital study MyRealWorld-MG. BMJ Open Print. 2023;13(1):e066445. PubMed
28.Howard JF, Jr., Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526-536. PubMed
29.Vyvgart (efgartigimod alfa): 400 mg/20mL (20 mg/mL) solution for intravenous use [product monograph]. Mississauga (ON): Quality & Compliance Service Inc; 2023 Sep 19.
30.Clinical Study Report: ARGX-113-1704. A randomized, double-blind, placebo- controlled, multicenter phase 3 trial to evaluate the efficacy, safety and tolerability of ARGX-113 in patients with myasthenia gravis having generalized muscle weakness (ADAPT) [internal sponosor's report]. Zwijnaarde (Ghent) (BE): argenx BV; 2020 Aug 18.
31.Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg/mL solution for intravenous use [internal sponsor's package]. Vaughan (ON): argenx Canada Inc; 2023 May 9.
32.Sacca F, De Bleecker JL, Verschuuren J, et al. Efgartigimod demonstrates consistent improvements in patients with generalised myasthenia gravis regardless of prior treatment failures [abstract] [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg/mL solution for intravenous use. Vaughan (ON): argenx Canada Inc.; 2023 May 9.
33.Bril V, Vu T, Brauer E, Kerstens R, Howard JF. Efgartigimod demonstrates consistent improvements in generalized myasthenia gravis across patient subgroups, including early in diagnosis [abstract] [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg/mL solution for intravenous use. Vaughan (ON): argenx Canada Inc.; 2023 May 9.
34.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
35.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
36.argenx Canada Inc. response to June 1 & 29, 2023 CADTH request for additional information regarding Vyvgart (efgartigimod alfa) for generalized myasthenia gravis [internal additianal sponsor's information]. Vaughan (ON): argenx Canada Inc.; 2023 Jul 7.
37.argenx Canada Inc. response to June 19, 2023 CADTH request for additional information regarding Vyvgart (efgartigimod alfa) for generalized myasthenia gravis [internal additianal sponsor's information]. Vaughan (ON): argenx Canada Inc.; 2023 Jun 26.
38.Generalized myasthenia Gravis: systematic review, version: 2.0 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg/mL solution for intravenous use. Vaughan (ON): argenx Canada Inc.; 2023 May 9.
39.Howard JF, Jr., Bril V, Burns TM, et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology. 2019;92(23):e2661-e2673. PubMed
40.Grifols Therapeutics LLC. NCT02473952: A study to evaluate the efficacy and safety of IGIV-C in symptomatic subjects with generalized myasthenia gravis. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2019 Mar 5: https://clinicaltrials.gov/study/NCT02473952. Accessed 2023 Jun 2.
41.Wolfe GI, Barohn RJ, Foster BM, et al. Randomized, controlled trial of intravenous immunoglobulin in myasthenia gravis. Muscle Nerve. 2002;26(4):549-552. PubMed
42.Vu T, Meisel A, Mantegazza R, et al. Terminal complement inhibitor ravulizumab in generalized myasthenia gravis. NEJM Evid. 2022;1(5). PubMed
43.Gilhus NE. Myasthenia gravis. N Engl J Med. 2016;375(26):2570-2581. PubMed
44.Jaretzki A, 3rd, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Ann Thorac Surg. 2000;70(1):327-334. PubMed
45.Lehnerer S, Jacobi J, Schilling R, et al. Burden of disease in myasthenia gravis: taking the patient's perspective. J Neurol. 2022;269:3050-3063. PubMed
46.Alexion Pharma GmbH. Ultomiris (ravulizumab): 10 mg/mL & 100 mg/mL concentrate for solution for intravenous infusion [product monograph]. Mississauga (ON): Alexion Pharma Canada Corp; 2023 Jan 6: https://pdf.hres.ca/dpd_pm/00069134.PDF. Accessed 2023 Jul 25.
47.Soliris (eculizumab for injection): 30 mL parenteral solution (10 mg/mL) [product monograph]. Zurich (CZ): Alexion Pharma GmbH; 2021 Mar 25: https://pdf.hres.ca/dpd_pm/00060546.PDF. Accessed 2023 Jul 25.
48.Mestinon (pyridostigmine bromide): 60 mg tablets and 180 mg slow release tablets [product monograph]. Laval (QC): Valeant Canada LP; 2019 Jul 3: https://pdf.hres.ca/dpd_pm/00052033.PDF. Accessed 2023 Jul 25.
49.Correction to Lancet Neurol 2021; 20: 526-36. Lancet Neurol. 2021 Aug;20(8):e5. doi: 10.1016/S1474-4422(21)00216-7. Erratum for: Lancet Neurol. 2021 Jul;20(7):526-536.
50.Saccà F, Barnett C, Vu T, et al. Efgartigimod improved health-related quality of life in generalized myasthenia gravis: results from a randomized, double-blind, placebo-controlled, phase 3 study (ADAPT). J Neurol. 2023;270:2096-2105. PubMed
51.argenx. NCT03669588: An efficacy and safety study of ARGX-113 in patients with myasthenia gravis who have generalized muscle weakness (ADAPT). ClinicalTrial.gov. Bethesda (MD): U.S. National Library of Medicine; 2022 Feb 8: https://clinicaltrials.gov/ct2/show/NCT03669588. Accessed 2023 Jul 25.
52.Wolfe GI, Herbelin L, Nations SP, Foster B, Bryan WW, Barohn RJ. Myasthenia gravis activities of daily living profile. Neurology. 1999;52(7):1487-1489. PubMed
53.Muppidi S, Wolfe GI, Conaway M, Burns TM. MG-ADL: still a relevant outcome measure. Muscle Nerve. 2011;44(5):727-731. PubMed
54.World Health Organization. International Classification of Functioning, Disability and Health (ICF). 2023; https://icd.who.int/dev11/l-icf/en. Accessed 2023 Jul 25.
55.Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan WW. Reliability testing of the quantitative myasthenia gravis score. Ann N Y Acad Sci. 1998;841:769-772. PubMed
56.Katzberg HD, Barnett C, Merkies IS, Bril V. Minimal clinically important difference in myasthenia gravis: outcomes from a randomized trial. Muscle Nerve. 2014;49(5):661-665. PubMed
57.Barnett C, Herbelin L, Dimachkie MM, Barohn RJ. Measuring clinical treatment response in myasthenia gravis. Neurol Clin. 2018;36(2):339-353. PubMed
58.van Reenen M JB, Stolk E, Boye K, Herdman M, Kennedy-Martin M, Kennedy-Martin T, Slaap B. EQ-5D-5L user guide. 2019: https://euroqol.org/publications/user-guides. Accessed 2022 Nov 4.
59.Qi CZ, Dewilde S, Gelinas D, Brauer E, Philips G. Hospitalization and exacerbation estimates of efgartigimod vs. conventional therapy in generalized myasthenia gravis patients: a post-hoc analysis of the phase 3 ADAPT study [poster abstract] [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg/mL solution for intravenous use. Vaughan (ON): argenx Canada Inc.; 2023 May 9.
60.Sanders DB, Tucker-Lipscomb B, Massey JM. A simple manual muscle test for myasthenia gravis: validation and comparison with the QMG score. Ann N Y Acad Sci. 2003;998:440-444. PubMed
61.Bedlack RS, Simel DL, Bosworth H, Samsa G, Tucker-Lipscomb B, Sanders DB. Quantitative myasthenia gravis score: assessment of responsiveness and longitudinal validity. Neurology. 2005;64(11):1968-1970. PubMed
62.Barnett C, Merkies IS, Katzberg H, Bril V. Psychometric properties of the quantitative myasthenia gravis score and the myasthenia gravis composite scale. J Neuromuscul Dis. 2015;2(3):301-311. PubMed
63.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
64.Luo Y, Dong X, Peng Y, et al. Evaluation of outcome measures for myasthenia gravis subgroups. J Clin Neurosci. 2021;91:270-275. PubMed
65.Myasthenia Gravis Foundation of America. The Quantitative Myasthenia Gravis (QMG) test. 2023; https://myasthenia.org/Professionals/Resources-for-Professionals. Accessed 2023 Jul 25.
66.argenx Canada Inc. response to July 4, 2023 CADTH request for additional information regarding Vyvgart (efgartigimod alfa) for generalized myasthenia gravis [internal additianal sponsor's information]. Vaughan (ON): argenx Canada Inc.; 2023 Jul 14.
67.Karam C, Vu T, Bril V. Efgartigimod treatment of patients with generalized myasthenia gravis demonstrated consistent improvements across all muscle subgroups, and regardless of background immunosuppressive therapy [abstract] [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg/mL solution for intravenous use. Vaughan (ON): argenx Canada Inc.; 2023 May 9.
68.Clinical Study Report: ARGX-113-1705. A long-term, single-arm, open-label, multicenter, phase 3 follow-on study of ARGX-113-1704 to evaluate the safety and tolerability of ARGX-113 in patients with myasthenia gravis having generalized muscle weakness (ADAPT+, IA5) [internal sponsor's report]. Zwijnaarde (Ghent) (BE): argenx BV; 2023 Mar 21.
69.Hutton B, Salanti G, Caldwell DM, et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: checklist and explanations. Ann Intern Med. 2015;162(11):777-784. PubMed
70.Systematic Reviews. CRD's guidance for undertaking reviews in health care. York (GB): Centre for Reviews and Dissemination; 2009: https://www.york.ac.uk/media/crd/Systematic_Reviews.pdf. Accessed 2023 May 28.
71.Narayanaswami P, Sanders DB, Wolfe G. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021;93(3):114-122. PubMed
72.Dias S, Sutton AJ, Ades AE, Welton NJ. Evidence synthesis for decision making 2: a generalized linear modeling framework for pairwise and network meta-analysis of randomized controlled trials. Med Decis Making. 2013;33(5):607-617. PubMed
73.Dias S, Sutton AJ, Welton NJ, Ades AE. Evidence synthesis for decision making 3: heterogeneity--subgroups, meta-regression, bias, and bias-adjustment. Med Decis Making. 2013;33(5):618-640. PubMed
74.Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G. Evidence synthesis for decision making 4: inconsistency in networks of evidence based on randomized controlled trials. Med Decis Making. 2013;33(5):641-656. PubMed
75.Vehtari A, Gelman A, Simpson D, Carpenter B, Bürkner PC. Rank-normalization, folding, and localization: an improved R-hat for assessing convergence of MCMC. Bayesian Anal. 2019;16(2):667-718.
76.Spiegelhalter D, Best N, Carlin B, Van Der Linde A. Bayesian measures of model complexity and fit. J R Stat Soc. 2002;64(4):583-639.
77.Dias S, Sutton AJ, Welton NJ, Ades AE. NICE DSU Technical Support Document 3: Heterogeneity: subgroups, meta-regression, bias and bias-adjustment. Sheffield (GB): Decision Support Unit, ScHARR, University of Sheffield; 2012: https://www.ncbi.nlm.nih.gov/books/NBK395886/pdf/Bookshelf_NBK395886.pdf. Accessed 2023 Jul 25.
78.Nowak RJ, Coffey CS, Goldstein JM, et al. Phase 2 trial of rituximab in acetylcholine receptor antibody-positive generalized myasthenia gravis. Neurology. 2022;98(4):e376-e389. PubMed
79.Piehl F, Eriksson-Dufva A, Budzianowska A, et al. Efficacy and safety of rituximab for new-onset generalized myasthenia gravis: the RINOMAX randomized clinical trial. JAMA Neurol. 2022;79(11):1105-1112. PubMed
80.Clinical Study Report: ARGX-113-1705. A long-term, single-arm, open-label, multicenter, phase 3 follow-on study of ARGX-113-1704 to evaluate the safety and tolerability of ARGX-113 in patients with myasthenia gravis having generalized muscle weakness (ADAPT+, IA4) [internal sponsor's report]. Zwijnaarde (Ghent) (BE): argenx BV; 2022 Aug 10.
81.argenx Canada Inc. response to June 1, 2023 CADTH request for additional information regarding Vyvgart (efgartigimod alfa) for generalized myasthenia gravis [internal additianal sponsor's information]. Vaughan (ON): argenx Canada Inc.; 2023 Jun 5.
Pharmacoeconomic Review
Abbreviations
Ab+
antibody positive
Ab–
antibody negative
AChEI
acetylcholinesterase inhibitor
AChR
acetylcholine receptor
AE
adverse event
BIA
budget impact analysis
gMG
generalized myasthenia gravis
HR
hazard ratio
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
IVIg
intravenous immunoglobulin
MDC
Muscular Dystrophy Canada
MG
myasthenia gravis
MG-ADL
Myasthenia Gravis Activities of Daily Living
MGFA
Myasthenia Gravis Foundation of America
MuSK
muscle-specific kinase
NIHB
Non-Insured Health Benefits
NMA
network meta-analysis
NSIST
nonsteroidal immunosuppressive therapy
ODB
Ontario Drug Benefit
PE
plasma exchange
QALY
quality-adjusted life-year
QMG
Quantitative Myasthenia Gravis
SCIg
subcutaneous immunoglobulin
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Efgartigimod alfa (Vyvgart), 20 mg/mL solution for IV use |
Submitted price | Efgartigimod alfa: $7,900.00, 400 mg single-dose vial |
Indication | For the treatment of adult patients with generalized myasthenia gravis who are anti-acetylcholine receptor (AChR) antibody positive |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 19, 2023 |
Reimbursement request | As an add-on therapy for adult patients with AChR-Ab+ gMG whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or nonsteroidal immunosuppressive therapies |
Sponsor | argenx Canada Inc. |
Submission history | Previously reviewed: No |
AChR-Ab+ = acetylcholine receptor antibody positive; AChEI = acetylcholinesterase inhibitor; gMG generalized myasthenia gravis; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with gMG who are AChR-Ab+ |
Treatment | Efgartigimod alfa plus conventional therapy consisting of AChEIs, CSs, and/or NSISTs) |
Comparators | Blood products (chronic immunoglobulin or PE) plus conventional therapy; conventional therapy alone. |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (53 years) |
Key data source | ADAPT, a randomized, multicentre, double-blind, placebo-controlled trial; ADAPT+ extension study; sponsor-submitted NMA |
Submitted results | Efgartigimod alfa plus conventional therapy was associated with an ICER of $283,913 per QALY gained (incremental costs = $1,084,669; incremental QALYs = 3.82) compared to conventional therapy alone. |
Key limitations |
|
CADTH reanalysis results |
|
AChR-Ab+ = acetylcholine receptor antibody positive; AChEI = acetylcholinesterase inhibitor; CS = corticosteroid; gMG = generalized myasthenia gravis; ICER = incremental cost-effectiveness ratio; LY = life-year; MGFA = Myasthenia Gravis Foundation of America; MG-ADL = Myasthenia Gravis Activities of Daily Living; NMA = network meta-analysis; NSIST = nonsteroidal immunosuppressant therapy; PE = plasma exchange; QALY = quality-adjusted life-year; WTP = willingness to pay.
Conclusions
The CADTH Clinical Review concluded that evidence from the ADAPT trial suggests that in adult patients with Myasthenia Gravis Foundation of America (MGFA) Class II to IV myasthenia gravis (MG) at screening whose symptoms persist despite treatment with acetylcholinesterase inhibitors (AChEIs), corticosteroids, and/or nonsteroidal immunosuppressive therapies (NSISTs) (i.e., Myasthenia Gravis Activities of Daily Living [MG-ADL] score remains at ≥ 5), efgartigimod alfa as an add-on to conventional therapy likely results in a clinically meaningful benefit in terms of the proportion of “MG-ADL responders,” proportion of “quantitative myasthenia gravis (QMG) responders,” and health-related quality of life (HRQoL), after cycle 1 of treatment, as well as after cycle 2 (albeit with less certainty), compared with placebo, in the population of patients who are acetylcholine receptor (AChR) antibody positive (Ab+). The comparative efficacy of efgartigimod alfa versus active comparators is uncertain in the absence of direct comparative evidence. Overall, the CADTH Clinical Review concluded that the network meta-analyses (NMAs) for patients with AChR-Ab+ gMG suggests that efgartigimod alfa may provide benefit in terms of change from baseline MG-ADL relative to intravenous immunoglobulin (IVIg). However, the results were uncertain due to wide 95% credible intervals and methodological limitations. No difference in efficacy in terms of change from baseline MG-ADL could be concluded for efgartigimod alfa relative to rituximab or ravulizumab due to methodological limitations and wide 95% credible intervals, which included both the possibility of clinical benefits favouring efgartigimod alfa as well as no difference between treatments.
CADTH undertook reanalyses to address some of the key limitations, resulting in a CADTH base case in which efgartigimod alfa plus conventional therapy was associated with an incremental cost-effectiveness ratio (ICER) of $1,764,628 per quality-adjusted life-year (QALY) gained compared with rituximab plus conventional therapy. The findings of the CADTH reanalysis were generally aligned with those submitted by the sponsor: efgartigimod alfa is not a cost-effective treatment option for adult patients with gMG who are AChR-Ab+ at a willingness to pay (WTP) of $50,000 per QALY gained. An 84% price reduction to reduce the unit price of efgartigimod alfa from $7,900 to $1,264 per 400 mg vial would be required for efgartigimod alfa to be considered cost-effective at this threshold of $50,000 per QALY gained. At this price reduction, the drug acquisition cost per 4-week treatment cycle with efgartigimod alfa would be $10,112 to $15,168 per patient, depending on body weight, or $47,729 to $71,593 per year, assuming 4.72 treatment cycles annually.
CADTH identified several additional limitations with the sponsor’s economic evaluation that could not be addressed. The cost-effectiveness of efgartigimod alfa plus conventional therapy in the full Health Canada indication is unknown as patients in MGFA Class I and V, and in patients with MG-ADL total score of less than 5 were excluded from the ADAPT trial and ADAPT+. CADTH was unable to validate all model inputs, most critically the individual patient data informing the transition probabilities between MG-ADL-based health states for patients receiving efgartigimod alfa or conventional therapy alone, and subsequently the transition probabilities of comparators derived from the sponsor’s submitted NMAs. In addition, results from the sponsor’s NMA were associated with wide credible intervals indicating substantial clinical uncertainty which was not reflected within the sponsor’s economic model. CADTH was also unable to alter the assumption that all patients receiving rituximab and blood products will cease transitioning through health states after 6 and 1 model cycles, respectively, an assumption that likely biases results in favour of efgartigimod alfa. Taken together, there remains considerable uncertainty in the cost-effectiveness results.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process specifically, information that pertains to the economic submission.
Patient input was received from Muscular Dystrophy Canada (MDC), which collected patient perspectives from 108 respondents aged between 21 and 76 years and from all provinces in Canada, and the majority of whom had a confirmed diagnosis of gMG. An online survey and semistructured virtual interviews were used to inform a qualitative descriptive approach and produce a thematic analysis. MDC identified the following themes in terms of the impact of MG symptoms: productivity; fatigue, energy levels, and quality of sleep; respiratory health; mobility and strength; independence; relationships and social participation; and vision, speech, and swallowing. Patients also identified nonphysical impacts affecting their mental health, quality of life, and the well-being of their families. Themes identified by MDC around current treatments included negative experiences with steroids, the slow onset of medication effects, and a feeling of trial and error with medications. Patients reported experience with prednisone, pyridostigmine, azathioprine, mycophenolate mofetil, IVIg, and thymectomy, and reported difficulty accessing rituximab. IVIg was reported by several patients as effective or helpful but time consuming and wearing off too quickly. In terms of gaps, MDC reported themes of patients wanting decreased intensity of exacerbations and side effects, maintenance of independence, and less serious hospital admissions. Patients were reported as stating their current medications seemed to decrease the number of exacerbations but not impact overall quality of life. Only 1 respondent indicated they had received efgartigimod alfa as part of a clinical trial, sharing that it had replaced their need for IVIg. As well, compared to other MG medications the patient had received, efgartigimod alfa had a quicker effect and shorter infusion time, and had less severe side effects, with only manageable diarrhea being noted.
Clinician input was received from the Neuromuscular Disease Network for Canada. The clinician group noted current treatments for patients with MG included symptomatic treatments such as AChEIs, short-term rescue immunotherapy such as plasma exchange (PE) or IVIg, and long-term immunosuppressive therapy such as corticosteroids and nonsteroidal immunosuppressives (azathioprine, cyclosporine, mycophenolate mofetil, and tacrolimus). The group emphasized that standard therapies are often transiently effective, require long periods to observe benefits, may have side effects, and may not be effective for all patients. Although IVIg and subcutaneous Ig (SCIg) were accepted as effective by the clinician group, both place significant burden on the health system and supplies can be periodically at risk. Clinically meaningful treatment goals included an improvement of 2 or more points on the MG-ADL or 3 or more on the QMGS with levels greater than 72% on the SSQ indicating general satisfaction. Patients identified as most likely to respond to efgartigimod alfa were those who have AChR-Ab+ MG, although those with muscle-specific kinase (MuSK) antibodies or who are double seronegative were also thought to potentially respond.
The drug plans noted that efgartigimod alfa has individualized frequency of re-treatment after the first cycle, raising challenges for plans instating approval when there is no certainty regarding if or when a patient will be retreated. The plans also noted considerations for initiation, continuation, prescribing, administration, and discontinuation of therapy, as well as uncertainty in the future reimbursement status of ravulizumab and the lack of the inclusion of eculizumab as a comparator.
Several of these concerns were addressed in the sponsor’s model:
Model health states were based on the MG-ADL scale, which considers many of the symptoms mentioned by patients including double vision and difficulty swallowing.
IVIg was considered a relevant comparator to efgartigimod.
Inclusion of quality of life and mortality impacts for chronic corticosteroid use.
In addition, CADTH addressed some of these concerns as follows:
inclusion of rituximab as a comparator.
CADTH was unable to address the following concerns raised from stakeholder input:
A lack of robust comparative efficacy and safety data between efgartigimod alfa and other add-on treatments to conventional therapy.
Economic Review
The current review is for efgartigimod alfa (Vyvgart) for adults with gMG.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted an economic analysis to examine the cost-effectiveness of efgartigimod alfa as an add-on to conventional therapy compared to conventional therapy alone, blood products (chronic immunoglobulin or PE) plus conventional therapy in adults with gMG who are AChR-Ab+.1 Conventional therapy was defined as AChEIs, corticosteroids, and/or NSISTs. The base-case model population was aligned with a subgroup of the ADAPT trial, comprised of adults with AChR-Ab+ gMG classified as MGFA Class II to IV, with an MG-ADL score of at least 5 (with 50% of the MG-ADL score due to non-ocular symptoms).2 The Health Canada indication does not specify MGFA class or MG-ADL score.3 The modelled population, therefore, was not aligned with the Health Canada indication. The sponsor’s reimbursement request is for efgartigimod alfa as an add-on therapy in adults with AChR-Ab+ gMG whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs. Ravulizumab was considered as a comparator in a scenario analysis. Rituximab was considered only in an analysis considering the overall population of patients with gMG (i.e., patients with AChR-Ab+ and AChR-Ab– gMG, consistent with the full population of the ADAPT trial, which is broader than the Health Canada indication because it included patients with AChR-Ab– gMG).
Efgartigimod alfa is available as a solution for IV infusion (400 mg in 20 mL single-dose vials).3 The recommended dose of efgartigimod alfa is 10 mg/kg of body weight administered once weekly for 4 weeks, with a dose of 1,200 mg in patients weighing 120 kg or more. Subsequent cycles of treatment should be based on clinical evaluation and may vary by patient. At the submitted price of $7,900.00 per 400 mg vial,1 the average cost per 4-week course is $63,200 for a patient weighing between 41 kg to 80 kg, and up to $94,800 for patients weighing more than 80 kg, assuming wastage of excess medication in vials. Assuming a mean of 4.72 annual treatment cycles of efgartigimod alfa, as reportedly administered to patients who were AChR-Ab+ in the pooled ADAPT and ADAPT+ data,1 the annual cost of treatment is $298,304 to $447,456, depending on patient weight.
The clinical outcome of interest was QALYs. The sponsor adopted a lifetime time horizon (53 years), with the analyses conducted from the perspective of a publicly funded health care payer. Future costs and benefits were discounted at a rate of 1.5% per year, and the model cycle length was 28 days.
Model Structure
The sponsor’s model consisted of a Markov model with 6 health states, 4 of which were based on MG-ADL score, along with MG crisis, and death (Figure 1). Patients entered the model distributed across the MG-ADL 5 to 7, MG-ADL 8 to 9, and MG-ADL 10 or greater health states based on the proportion of patients with gMG in each of these categories at baseline of the ADAPT trial (Table 11). Patients receiving efgartigimod alfa plus conventional therapy or conventional therapy alone could then transition into other health states every 28 days, based on individual patient data from the ADAPT trial (data cut-off: April 6, 2020) and ADAPT+ extension study (data cut-off: January 31, 2022) for the first 20 weeks. Thereafter, the cohort using conventional therapy was assumed to remain in the same health state unless they developed myasthenic crisis or died, while patients using efgartigimod alfa recycled the transition probabilities from week 16 to 20, unless they discontinued efgartigimod alfa, developed a crisis, or died. Patients using blood products were assumed to remain in the same MG-ADL state after cycle 1, barring crisis or death. In the model, patients receiving efgartigimod alfa plus conventional therapy received 4 weekly efgartigimod alfa infusions (1 treatment cycle) and were then off treatment for an additional 4 weeks (1 model cycle) before starting another treatment cycle, with the exception of patients in the MG-ADL < 5 health state, who remained off treatment with efgartigimod alfa unless they transitioned into an MG-ADL health state of 5 or higher. Conventional therapy was assumed to continue for these patients, regardless of when their next cycle of efgartigimod alfa took place. Similarly, patients receiving blood products plus conventional therapy were not treated with blood products when in the MG-ADL less than 5 health state.
Patients could permanently discontinue their add-on therapy for 2 reasons: first, a nonresponse stopping rule in which patients receiving efgartigimod alfa or rituximab would switch to conventional therapy alone due to nonresponse or, second, discontinuation due to unplanned reasons, in which patients could discontinue their add-on therapy over time (efgartigimod alfa, rituximab) over time or at a single time point (blood products). Patients in the MG-ADL 5 to 7, MG-ADL 8 to 9, or MG-ADL 10 or greater health states had a flat risk of myasthenic crisis (Table 11), which was associated with an increased risk of death. All patients in the myasthenic crisis health state transitioned to the MG-ADL 10 or greater health state the following cycle, assuming they survived the crisis.
Model Inputs
The baseline population characteristics used to inform the model were based on the AChR-Ab+ subgroup of the ADAPT trial, a randomized, double-blind, placebo-controlled, multicentre trial that included patients aged at least 18 years (mean = 47 years), who had AChR-Ab+ (77%) or AChR-Ab– (23%) gMG, and who had MGFA Class II to IV disease and an MG-ADL score of at least 5.2 Trial patients with AChR-Ab+ gMG had a mean MG-ADL score of 9.0 at baseline (distribution of baseline MG-ADL scores can be found in Table 11), and 67% were female.2
Treatment effects, characterized by changes in MG-ADL score between clinical trial assessment time points, were informed by pooled individual patient data from the AChR-Ab+ subgroup of the ADAPT trial and ADAPT+ extension study (modified to account for different time points of measurement) for patients receiving efgartigimod alfa or conventional therapy alone for the first 20 weeks. After 20 weeks, transition probabilities from weeks 16 to 20 were recycled unless the patient had a crisis or died. According to the sponsor, for patients receiving blood products, the inverse of the MG-ADL change for efgartigimod alfa versus chronic immunoglobulin obtained from the sponsor-conducted NMA was applied to the efgartigimod alfa MG-ADL scores at the individual patient level at week 4, and the effect was assumed to be retained until crisis or death, or during a 1-time discontinuation stopping point in the second cycle (33% of patients) based on 2 of 6 patients receiving IVIg who discontinued due to severe headaches by day 22 in a 2002 randomized trial.4
In each cycle, patients could experience clinical events (i.e., myasthenic exacerbations requiring hospitalization or crises). The rates per cycle of experiencing these events were determined by MG-ADL health state (Table 11). Patients could also experience adverse events (AEs), based on the rates of grade 3 or higher treatment-emergent AEs reported in the efgartigimod alfa and placebo groups of the ADAPT trial, for efgartigimod alfa and conventional therapy, respectively. AE rates for blood products were assumed to be equal to conventional therapy.1
Mortality within the MG-ADL model health states was assumed to be equal to that of the general population. Risk of mortality was assumed to increase from that of the age- and sex-matched general population due to the use of low-dose (hazard ratio [HR] = 1.60) or high-dose corticosteroids (HR = 3.48), based on a sponsor-conducted systematic literature review.1 Patients in the MG crisis health state also had a 5% risk of death during their cycle in that state.5 Because the use of high-dose corticosteroids and crisis risk was directly associated with whether a patient had an MG-ADL score of 5 or higher (Table 11), MG-ADL health state indirectly affected mortality rate.
Health state utility values were determined primarily by MG-ADL state (Table 11), and were based on a mixed model regression of MyRealWorld MG study longitudinal EQ-5D data.6 A disutility of –0.725 was applied to the MG-ADL score of less than 5 utility weight for patients in the crisis health state, leading to a utility in the crisis state of 0.118 for the 28 days patients were assumed to be in that state. A disutility of 0.20 was applied for patients having an exacerbation lasting an average of 14 days.5 High-dose corticosteroid use was associated with an additional disutility of –0.18, while the disutility associated with low-dose corticosteroid use was –0.07, based on an average of disutilities reported in the literature for other conditions.7,8 Disutilities for AEs ranged from –0.01 to –0.14 and lasted a mean of 14 days.1,9
Costs in the model included drug acquisition, treatment administration, routine monitoring, complications associated with chronic corticosteroid use, exacerbation and crisis treatment, AEs, and end of life care, and were inflated to 2023 Canadian dollars if applicable. The proportion of patients using each drug within conventional therapy, whether used alone or with an add-on therapy, was based on clinical expert feedback elicited by the sponsor.1 Acquisition costs of drugs used in conventional therapy ($133 per model cycle) were from the Ontario Drug Benefit (ODB) formulary,10,11 while the cost of blood products (IVIg, SCIg, or PE; weighted average of $13,963 per induction, $7,832 per treatment cycle) was derived from the literature.12,13 Administration costs were derived from the literature, and amounted to $846 per treatment cycle for efgartigimod alfa and blood products.12,14 Monitoring costs included general practitioner, nurse, hospital outpatient, physiotherapist, and neurology specialist visits, with MG-ADL health state–specific visit frequencies6,15 (Table 11).1 The cost of treating an MG crisis or exacerbation was assumed to be $114,903 and $25,548, respectively, derived from the CADTH review of eculizumab for gMG.5 AE management costs were based on Canadian Institute for Health Information (CIHI) patient cost estimator codes, and ranged from $1,828 to $8,276 per event.16 Costs associated with conditions related to chronic corticosteroid use (e.g., increased health care utilization) were $935 and $407 per cycle for high-dose and low-dose corticosteroids, respectively, derived from a sponsor-conducted systematic literature review.1
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations) for the base-case and scenario analyses. The deterministic and probabilistic results were similar. The probabilistic findings are presented below. All results are based on publicly available list prices. During the review period, CADTH noted an error in the sponsor’s model such that not all traces were summing to zero. The sponsor submitted a corrected model file. The results presented subsequently are reflective of the sponsor’s corrected model file.
Base-Case Results
In the sponsor’s base case, the sponsor reported that efgartigimod alfa plus conventional therapy was associated with an additional cost of $1,084,669 and 3.82 additional QALYs when compared to conventional therapy alone, resulting in an ICER of $283,913 per QALY gained (Table 3). Efgartigimod alfa plus conventional therapy dominated blood products plus conventional therapy (i.e., was associated with higher costs and fewer QALYs). In addition, the analysis report that efgartigimod alfa plus conventional therapy was associated with an additional 0.96 and 0.88 life-years compared to conventional therapy and blood products plus conventional therapy, respectively. Efgartigimod alfa plus conventional therapy had a 5.2% chance of being cost-effective at a WTP threshold of $50,000 per QALY. At the end of the time horizon, less than 1% of patients were alive.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Conventional therapy | 828,519 | 12.98 | Reference |
Efgartigimod alfa + conventional therapy | 1,913,189 | 16.80 | 283,913 |
Dominated treatments | |||
Blood products + conventional therapy | 2,169,667 | 13.35 | Dominated by efgartigimod alfa |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Analysis results are based on publicly available list prices.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted a variety of probabilistic scenario analyses, including adding ravulizumab as a comparator, altering the discount rate, adding a societal perspective, adjusting the threshold for high-dose corticosteroids to 10 mg per day, disassociating efgartigimod alfa drug costs from MG-ADL health states in favour of average trial dosing, utilizing the utility values from the ADAPT trial, and assuming vial sharing. The associated ICERs for efgartigimod alfa plus conventional therapy resulting from these scenarios ranged from $215,215 to $417,052 per QALY gained compared to conventional therapy alone.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The full Health Canada population was not modelled: The sponsor submitted an analysis estimating the cost-effectiveness of efgartigimod alfa plus conventional therapy among patients with AChR-Ab+, MGFA Class II to IV gMG with an MG-ADL score of at least 5, with effectiveness informed by the AChR-Ab+ subgroup of the ADAPT trial and ADAPT+ extension study. Because the ADAPT study excluded patients with MGFA Class I and V gMG as well as patients with an MG-ADL score of less than 5, and the Health Canada indication specifies adults with gMG without restriction based on severity, the modelled population is narrower than the Health Canada–indicated population. In addition, the Health Canada product monograph does not specify in either the indication or the recommended dose section that efgartigimod must be given as add-on to conventional therapy. As such, the sponsor’s analyses reflect the cost-effectiveness of efgartigimod alfa plus conventional therapy in only a subset of the indicated population, and only in combination with conventional therapy. Clinical expert opinion elicited by CADTH indicated that a subset of patients with MGFA Class I of V disease or having MG-ADL scores of less than 5 might potentially benefit from treatment with efgartigimod alfa, but that trial results cannot be directly generalized to them.
Finally, the sponsor’s reimbursement request is that efgartigimod alfa be reimbursed as an add-on therapy for adult patients with AChR-Ab+ gMG whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs, which is a subgroup of the proposed Health Canada indication. According to the CADTH Clinical Review report, the proportion of people enrolled in the ADAPT trial whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs is uncertain. Because the model was populated based on the ADAPT trial, the sponsor and CADTH base case are reflective of the ADAPT trial population (a subset of the Health Canada indication) rather than the Health Canada indication or the sponsor’s reimbursement request.
CADTH was unable to address this limitation owing to a lack of clinical data. As noted in the CADTH Clinical Review, the efficacy of efgartigimod alfa in patients with MGFA Class I and V gMG or in patients with an MG-ADL total score of less than 5 cannot be directly generalized from trial data that excluded such patients. As such, the clinical effectiveness and cost-effectiveness of efgartigimod alfa in patients with MGFA Class I and V gMG as well as patients with an MG-ADL total score of less than 5 is unknown, as is the cost-effectiveness of efgartigimod alfa plus conventional therapy in the full Health Canada–indicated population. In addition, because the proportion of people enrolled in the ADAPT trial whose symptoms persisted despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs is uncertain, the clinical efficacy and cost-effectiveness of efgartigimod alfa plus conventional therapy in the reimbursement request population is also unknown.
Rituximab was excluded as a comparator: The sponsor excluded rituximab as a relevant comparator because rituximab is not indicated for gMG and is of uncertain efficacy in refractory AChR-Ab+ gMG.1 However, rituximab is considered a treatment option for patients with refractory AChR-Ab+ according to clinical experts from Canada, both those consulted by the sponsor1 and those consulted by CADTH, as well as within international guidelines.17-19 Although rituximab was included as a comparator in additional NMAs provided by the sponsor that were intended to establish relative efficacy in the population of patients who are AChR-Ab–, patients included in these NMAs were exclusively AChR-Ab+, including those in the BeatMG trial used to inform rituximab efficacy.20 As such, the population of these additional NMAs that include rituximab aligns with AChR-Ab+ gMG population of the Health Canada indication and the sponsor’s reimbursement request. Furthermore, rituximab is explicitly funded by some provincial drug plans for the treatment of refractory gMG under exceptional access programs (e.g., Saskatchewan21) and is accessible in other jurisdictions to some patients for gMG, according to clinical expert feedback elicited by CADTH without limitation by serotype.
In reanalysis, rituximab was included as a comparator using transition probabilities provided within the sponsor’s submitted model. These transition probabilities were derived from NMA results applied to individual patient data from the ADAPT trial in a manner similar to how the transition probabilities for blood products were derived. As rituximab is not funded in all jurisdictions for the treatment of gMG and is used less frequently than blood products or conventional therapy alone (refer to Appendix 5), a scenario analysis was also conducted excluding rituximab.
Relative efficacy of comparators is highly uncertain and could not be validated: The ADAPT trial and ADAPT+ extension study compared efgartigimod alfa with placebo, which was assumed to be representative of conventional therapy in the sponsor’s economic evaluation due to the use of concomitant AChEIs, corticosteroids, and/or NSISTs at stable doses in both treatment groups. There have been no head-to-head trials of efgartigimod alfa versus other active comparators (i.e., rituximab, IVIg, and ravulizumab). The sponsor therefore conducted NMAs to inform the comparative efficacy of efgartigimod alfa versus other active comparators. According to the CADTH Clinical Review report, the sponsor-submitted NMAs were highly uncertain given the sparse network, inability to test for consistency due to an absence of closed loops, and inconsistency between included trials with respect to dosing regimens, variability in patient characteristics, and study follow-up times. Overall, the CADTH Clinical Review concluded that the NMA for patients with AChR-Ab+ gMG suggests that efgartigimod alfa may provide benefit in terms of change from baseline MG-ADL relative to IVIg. However, the results were uncertain due to wide 95% credible intervals and methodological limitations. No difference in efficacy in terms of change from baseline MG-ADL could be concluded for efgartigimod alfa relative to rituximab or ravulizumab due to the wide 95% credible intervals, which included no difference between treatments, and methodological limitations.
In the sponsor’s model, transition probabilities for efgartigimod alfa plus conventional therapy and conventional therapy alone were based on individual patient data from the AChR-Ab+ subgroup of the ADAPT trial and the ADAPT+ extension study. According to the sponsor, transition probabilities for non-ADAPT trial comparators (e.g., blood products such as IVIg) were derived by applying the difference in change from baseline MG-ADL score between efgartigimod alfa and the comparator from the NMA to individual patient data for efgartigimod alfa. This was used to estimate what each patient’s score would have been if they had instead received the comparator. For example, a patient whose MG-ADL score changed from 12 to 8 at week 4 with efgartigimod alfa would be assumed to have a week 4 MG-ADL score of 11 if they instead received IVIg, based on the 2.64-point difference in change from baseline score reported in the sponsor’s NMA for efgartigimod alfa and IVIg (refer to the CADTH Clinical Report). However, except for 3 examples described by the sponsor in response to an additional information request made by CADTH, neither the original nor adjusted individual patient data were provided to CADTH, and therefore the transition matrices driving both absolute and relative efficacy could not be validated (i.e., transition matrices for all treatments were hard-coded). In addition, the sponsor did not consider the wide credible intervals reported for each comparison (e.g., the credible interval for difference in MG-ADL score reported for efgartigimod alfa compared to IVIg was –4.16 to –1.12). As such, the uncertainty resulting from imprecision in the comparative efficacy of efgartigimod alfa relative to the comparator treatments has not been reflected within the model.
CADTH was unable to address this limitation in reanalysis. The CADTH Clinical Review concluded that in terms of MG-ADL score, efgartigimod alfa likely results in a clinically meaningful benefit compared to placebo (based on the AChR-Ab+ subgroup of the ADAPT trial) and may provide benefit relative to IVIg (based on indirect evidence for the AChR-Ab+ population). However, the CADTH Clinical Review concluded that no difference was found in terms of change from baseline in MG-ADL for efgartigimod alfa relative to rituximab due to wide credible intervals and methodological limitations. Due to the lack of direct evidence and limitations with the indirect comparative evidence utilized by the sponsor in the pharmacoeconomic analysis, the cost-effectiveness of efgartigimod alfa compared to IVIg, rituximab, and ravulizumab is highly uncertain and this uncertainty has not been adequately captured in the sponsor’s analysis. In addition, the sponsor’s parameterization of comparator transition probabilities could not be fully validated by CADTH.
Assumptions leading to reductions of crises, corticosteroid use, and mortality were inappropriate: The sponsor’s model estimated that patients receiving efgartigimod alfa experienced reductions in the average number of crises of 42% and 44%, compared to blood products plus conventional therapy and conventional therapy alone, respectively. These reductions occurred due to the assumption that patients in the MG-ADL less than 5 health state had no risk of crisis, whereas those in health states associated with MG-ADL scores of 5 or more would. There is insufficient evidence from the ADAPT trial, in which only 1 patient experienced a myasthenic crisis, to support such reduced rates. According to clinical expert opinion elicited by CADTH, while risk of crisis may be correlated to MG-ADL score and to the particular symptoms being experienced, MG-ADL score is not used as a predictor of crisis in clinical practice and a risk of crisis remains at any MG-ADL score. This association between reduction in risk of crisis with use of efgartigimod alfa was not supported by the clinical evidence and was not considered plausible according to clinical expert feedback.
The sponsor also assumed that patients in the MG-ADL less than 5 state would never receive high-dose corticosteroids.1 Clinical expert opinion elicited by CADTH did not agree with this assumption, noting that some patients may achieve low MG-ADL scores due to their use of high-dose steroids. Change in corticosteroid use was not an outcome of the ADAPT trial; doses of concomitant medications, including corticosteroids, were held stable during the trial except in the case of rescue therapy. As such, the ADAPT trial could not demonstrate an association between efgartigimod alfa and reduction in corticosteroid use.
Because both myasthenic crisis and corticosteroid dosage were associated with increased mortality in the sponsor’s model, these 2 assumptions led to the sponsor’s model reporting that patients receiving efgartigimod alfa would live, on average, 11.8 months longer than those receiving blood products or conventional therapy alone. The association between efgartigimod alfa and reduced mortality was not demonstrated in either of the clinical trials (mortality was not an outcome in the ADAPT trial or ADAPT+ extension study and no deaths occurred in the clinical trials) or by real-world evidence, and clinical expert opinion elicited by CADTH did not consider such an extrapolation to be plausible without data to support it.
To negate the undemonstrated survival advantage conferred by the sponsor’s model to patients using efgartigimod alfa and due to structural limitations precluding such a modification, CADTH assumed that patients in the MG-ADL less than 5 health state had the same risk of crisis and the same probability of using high-dose corticosteroids as patients in health states associated with higher MG-ADL scores in the CADTH base-case reanalyses.
Utility values of MG-ADL health states are likely underestimated: The sponsor’s base-case utility value set was derived from a sponsor-funded real-world study in which patients could download a mobile app and complete questionnaires regarding their diagnosis, condition status in terms of MGFA classification, symptom status including MG-ADL score, and quality of life including the EQ-5D-5L.6 Limitations of the study included possible selection bias due to the proactive and digital nature of data collection as well as the inability to validate user-entered data. Furthermore, this study used a UK value set to map EQ-5D scores to utilities,22 and thus the utilities used in the sponsor’s base case do not reflect preferences in Canada. Clinical expert opinion elicited by CADTH indicated that the utility values from the MyRealWorld MG dataset appeared too low for most health states, and also indicated that the MG-ADL 10 or higher health state was too broad to lump together into a single patient experience. An alternate utility value set derived from EQ-5D-5L data collected during the ADAPT trial and mapped with a Canadian value set23 was provided within the sponsor’s model. Finally, utility values should be specific to a clinically homogenous group of patients.24 According to clinical expert opinion elicited by CADTH, the broad range of patient symptoms and experiences that are included in a health state ranging from MG-ADL scores of 10 to 24 represents a highly varied set of patients who are unlikely to experience similar levels of quality of life.
In the CADTH base-case reanalyses, in order to align with the source of the clinical efficacy data, clinical expert opinion elicited by CADTH, and the use of a Canadian valuation, CADTH assumed the utility value set derived from pooled EQ-5D data from the ADAPT trial. Due to the structure of the model, CADTH was unable to further distinguish patients with MG-ADL scores at or near 10 from those with scores substantially above 10. A scenario analysis was instead conducted in which the lower MyRealWorld MG utility was assumed for the MG-ADL 10 or greater health state to explore the uncertainty inherent in grouping patients with such a broad range of MG-ADL scores into a single health state.
The model structure does not adequately reflect gMG in clinical practice: The sponsor’s model assumed that all patients receiving blood products remained in the same health state after the first cycle. The only transitions patients receiving blood products could make after this time point was if they experienced a myasthenic crisis or died. In contrast, patients receiving efgartigimod alfa and conventional therapy could continue transitioning through health states until week 20. Further, beyond week 20, patients receiving efgartigimod alfa continued to cycle through the week 16 to 20 transition probabilities due to the on-and-off nature of treatment with efgartigimod alfa. This issue also applied in the analysis including rituximab as a comparator, although these patients remained in the same health state after the sixth model cycle.
CADTH was unable to address this limitation owing to the sponsor’s model structure not incorporating long-term transition probabilities for comparator treatments. As patients receiving active comparator treatments either remained in the same health state achieved at the first or sixth model cycle or worsened upon experiencing crisis, whereas patients receiving efgartigimod alfa could potentially improve over time, the efficacy of efgartigimod alfa relative to comparators is likely overestimated in CADTH’s base case.
The model also assumed that all patients who experienced a myasthenic crisis entered the MG-ADL 10 or greater health state following their cycle in crisis. As such, patients receiving blood products who experienced a myasthenic crisis after the first model cycle (or after the sixth for those using rituximab when included), remained in the MG-ADL 10 or greater health state for their remaining time in the model regardless of the initial state from which they experienced the crisis, with the exception of patients receiving efgartigimod alfa (because these patients may continue cycling through week 16 to 20 transition probabilities). Clinical expert opinion elicited by CADTH did not find this plausible for several reasons. First, the clinical experts indicated that patients would most likely return to their previous health state after a crisis, with some patients potentially improving due to receiving subsequent therapies and/or more robust follow-up post-crisis. Second, in assuming that patients receiving efgartigimod alfa could improve following a crisis while those receiving a comparator treatment could not throughout most of the model’s time horizon, the sponsor’s model inappropriately biases results in favour of efgartigimod alfa.
Due to the structure of the model, CADTH was unable to return patients to their previous health state after a crisis and was therefore unable to fully adjust for this limitation. To partially adjust for the issue, CADTH reanalyses assumed that following a crisis, patients would exit crisis in a distribution more reflective of Canadian clinical practice, informed by clinical expert estimates elicited by CADTH.
Discontinuation assumptions were highly uncertain: The sponsor’s model included an efgartigimod alfa stopping rule for patients who do not respond to treatment such that 18% of patients receiving efgartigimod alfa were assumed to discontinue therapy, based on the proportion of patients in the ADAPT trial who did not respond to either of the first 2 treatment cycles with efgartigimod alfa. In the analysis including rituximab, 35% of patients receiving rituximab were assumed to discontinue due to nonresponse at 1 year, derived from the BeatMG trial.20 As these nonresponse rates are naively derived from differing sources and do not account for differences in trial population or time point of assessment, it is unclear what the relative nonresponse rates and time points would be if applied to the same population. Neither discontinuation nor nonresponse rates were outcomes considered in the sponsor’s NMA; as such, there is no direct or indirect evidence available to inform these estimates. Long-term discontinuation rates were also extrapolated from individual trial data for efgartigimod alfa and rituximab (when relevant), while for blood products, a 1-time discontinuation probability of 33% was applied at week 4, based on 2 of 6 patients discontinuing IVIg due to headaches as reported by Wolfe et al. (2002).4 In addition, the model assumes that patients who discontinue their add-on therapy remain on conventional therapy alone for the remainder of their lives. According to clinical expert opinion elicited by CADTH, patients who do not adequately respond or otherwise need to stop 1 add-on therapy are likely to try another.
CADTH was unable to adjust for this limitation in reanalysis.
Model lacked transparency: The sponsor’s submitted model was complex and transition probabilities were difficult to trace or validate and were poorly labelled and inadequately described in the submitted report. When conflated with the lack of transparency in the derivation of the transition probabilities described in this section, CADTH could neither conduct a full validation nor adequately test alternate assumptions.
CADTH could not address this limitation and notes that a thorough validation of the sponsor’s model was not possible.
In addition, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Background therapies used in the ADAPT trial represent conventional therapy. | Uncertain. The ADAPT trial compared efgartigimod alfa to placebo, rather than to conventional therapy. Instead, patients in both groups received a stable dose of conventional therapies including AChEIs, CSs, and/or NSISTs. As this concomitant conventional therapy was required to remain stable except in the case of rescue therapy, efgartigimod alfa was not compared to any individual or combination conventional therapy as it would typically be used in clinical practice, i.e., altering doses or adding additional medications to suit patients’ current symptoms or other needs. As such, the cost-effectiveness of efgartigimod alfa compared to customizable conventional therapies is uncertain. |
Exclusion of eculizumab as a comparator and restriction of ravulizumab to scenario analysis. | Acceptable. Despite the overlap in Health Canada indications between eculizumab25 and efgartigimod alfa3 as well as a conditional positive recommendation from CADTH,26 eculizumab is not funded by jurisdictional drug plan payers27 and will thus not be displaced by efgartigimod alfa, should it be funded. Ravulizumab recently received a Do Not List recommendation for the treatment of AChR-Ab+ gMG from CDEC,28 and thus is unlikely to be funded for the treatment of gMG. Ravulizumab was dominated by efgartigimod alfa when included in both the sponsor’s analysis and CADTH reanalyses. |
AEs sourced from individual trials and assumption. | Inappropriate. The sponsor’s combining of AE rates from multiple sources (ADAPT2 for efgartigimod alfa and conventional therapy, BeatMG20 for rituximab when relevant, blood products assumed the same as conventional therapy) constitutes a naive comparison that does not account for between-trial differences. Ideally, an NMA would have been conducted to estimate the relative safety of comparators. In addition, AE rates sourced from the ADAPT trial include those of all trial patients rather than those of the AChR-Ab+ subgroup of interest. However, AE rates are not a key driver of model results, and thus higher value information regarding relative AE rates is not expected to substantially impact reported ICERs. |
Incidence of AEs in the blood product plus conventional therapy group was assumed to be equivalent to the placebo group (i.e., receiving conventional therapy only) of the ADAPT trial. | Uncertain; however, this assumption is likely conservative. |
Pricing of blood products. | Unknown. In the absence of publicly available list prices for immunoglobulins, the sponsor has used an estimate for the cost per unit of IVIg from the literature. It is unknown whether this cost is reflective of costs currently paid in Canada. As such, the true cost-effectiveness of efgartigimod alfa compared to blood products is unknown. |
High dose of CSs was defined as ≥ 15 mg per day. | Acceptable. |
Mortality multipliers and HRQoL decrements for CS use. | Uncertain. The sponsor’s model applies mortality multipliers for patients using low-dose and high-dose CSs, based on studies that reported a large range of hazard ratios associated with chronic CS use. It is unclear how generalizable such multipliers are to the gMG population, nor the extent of confounding included due to the association of higher steroid use with more severe disease in these conditions. Similarly, utility decrements applied in the sponsor’s model for CS use are derived from studies exploring HRQoL and CS use in patients with lupus or any condition requiring chronic CS use, and thus generalizability and confounding may similarly be at issue. While it is likely that the application of such mortality multipliers and utility decrements partially double counts impacts already captured in crisis and exacerbation rates and health state utility values associated with MG-ADL scores, CADTH did not revise these estimates as other reanalyses were conducted which equalized crisis rates and CS usage across health states, negating the issue. |
Disutility in myasthenic crisis of 0.72. | Uncertain. The sponsor assumed a disutility for myasthenic crisis based on the one used for the CADTH review of eculizumab for gMG.26 In that review, the crisis disutility was based on the change in EQ-5D score for a single patient measured the week before and after experiencing a crisis. While the application of this disutility in the current submission was not associated with the lack of face validity outlined in the eculizumab review, it remains a highly uncertain value. However, given other changes made during reanalyses, CADTH did not further explore alternate assumptions for the disutility of a crisis. |
Chronic IVIg administered every 3 weeks. | Uncertain. According to clinical expert feedback received for this review, chronic IVIg is administered every 4 weeks in Canadian clinical practice, while patient input indicates some patients may receive it more frequently than every 4 weeks. Because IVIg is dominated by rituximab, this change has little effect on the ICER for efgartigimod alfa but was included as a scenario analysis. |
Cost per exacerbation for the included population is expected to be equal to that of exacerbations in patients with refractory gMG. | Inappropriate. According to clinical expert input elicited by CADTH, patients with refractory gMG would be expected to have more severe exacerbations on average than patients meeting the Health Canada indication for efgartigimod alfa. As these patients will be less severe, they are less likely to require hospital admission or postexacerbation nursing care than assumed during the eculizumab review. CADTH reviewers found that this assumption had a limited effect on overall cost-effectiveness; as such, this value was left unadjusted for in the CADTH base case. |
AChR-Ab+ = acetylcholine receptor antibody positive; AChEI = acetylcholinesterase inhibitor; AE = adverse event; CDEC = Canadian Drug Expert Committee; CS = corticosteroid; gMG = generalized myasthenia gravis; HRQoL = health-related quality of life; ICER = incremental cost-effectiveness ratio; IVIg = intravenous immunoglobulin; MG-ADL = Myasthenia Gravis Activities of Daily Living; NMA = network meta-analysis; NSIST = nonsteroidal immunosuppressive therapy; QALY = quality-adjusted life-year.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH reanalyses addressed a number of key limitations of the submitted model, as summarized in Table 5. The CADTH base-case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CADTH also corrected the sponsor’s analysis to run without randomization seeding and to include probabilistic variation for disease severity at baseline. All probabilistic reanalyses were run for 5,000 iterations.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Randomization method for probabilistic analyses | Seeded | Unseeded |
2. Disease severity at baseline for probabilistic analyses | Deterministic | Probabilistic |
Changes to derive the CADTH base case | ||
1. Rituximab included as a comparator | Excluded | Included |
2. Risk of crisis equalized between health states | Crisis probability per 28-day cycle: MG-ADL < 5: 0% | Crisis probability per 28-day cycle: All MG-ADL health states: 0.09% |
3. Proportion using high-dose CSs equalized between health states | Percentage of patients using CSs who are on high dose: MG-ADL < 5: 0% | Percentage of patients using CSs who are on high dose: All MG-ADL health states: 55.91% |
4. Utility source | MyRealWorld MG dataa | ADAPT trial, treatments pooleda |
5. Altered post-crisis recovery distributionb | MG-ADL < 5: 0% MG-ADL 5 to 7: 0% MG-ADL 8 to 9: 0% MG-ADL ≥ 10: 100% | MG-ADL < 5: 5% MG-ADL 5 to 7: 10% MG-ADL 8 to 9: 25% MG-ADL ≥ 10: 60% |
CADTH base case | 1 + 2 + 3 + 4 + 5 | |
CS = corticosteroid; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living.
aHealth utilities by health state can be found in Table 11.
bBased on clinical expert opinion elicited by CADTH. When run probabilistically as part of the CADTH base case, this reanalysis also included an alteration of the Dirichlet distribution mean and uncertainty such that a total of 100 patients were assumed of whom 5, 10, 25, and 60 were in the MG-ADL < 5, 5 to 7, 8 to 9, and ≥ 10 health states, respectively, following a crisis.
CADTH’s base-case results are presented in Table 6. CADTH undertook a stepped analysis, incorporating each change proposed in Table 5 to the sponsor’s base case to highlight the impact of each change in Table 12. Step-wise results for individual reanalyses, as well as disaggregated results for the CADTH base case (Table 13), are presented in Appendix 4. When considering the population of adult patients with AChR-Ab+ gMG, treatment with efgartigimod alfa plus conventional therapy was associated with higher costs (incremental: $1,195,367) and higher QALYs (incremental: 0.68) compared to rituximab plus conventional therapy, leading to an ICER of $1,764,628 per QALY gained (Table 6). Efgartigimod alfa had a 0.1% chance of being cost-effective at a WTP threshold of $50,000 per QALY gained. Total life-years gained were equal across all comparators in the CADTH reanalysis. As such, all incremental QALYs associated with efgartigimod alfa are due to quality-of-life assumptions derived from more time spent in less severe MG-ADL health states versus comparator treatments. Almost all QALYs (98%) associated with efgartigimod alfa were accrued in the post-trial period, and less than 1% of patients were alive at the end of the time horizon. Nearly all of the incremental costs for efgartigimod alfa were drug costs and all incremental QALYs for efgartigimod alfa were accrued in the MG-ADL less than 5 health state (Table 13).
Table 6: Summary of the CADTH Reanalysis Results
Drug | Total costs | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor-corrected base-case analysis | |||
Conventional therapy | 832,506 | 12.96 | Reference |
Efgartigimod alfa + conventional therapy | 1,914,156 | 16.80 | 281,939 |
Dominated treatments | |||
Blood products + conventional therapy | 2,175,502 | 13.33 | Dominated by efgartigimod alfa |
CADTH base-case reanalysis | |||
Rituximab + conventional therapy | 774,526 | 15.70 | Reference |
Efgartigimod alfa + conventional therapy | 1,969,893 | 16.38 | 1,764,628 |
Dominated treatments | |||
Conventional therapy | 818,381 | 15.36 | Dominated by rituximab |
Blood products + conventional therapy | 2,210,045 | 15.47 | Dominated by rituximab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor-submitted base case and CADTH’s base case (Table 7). The results indicate that a price reduction of 84% would be required for efgartigimod alfa plus conventional therapy to be considered cost-effective compared to rituximab plus conventional therapy at a WTP of $50,000 per QALY gained.
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for efgartigimod alfa vs. comparators ($/QALY) | |
|---|---|---|
Price reduction (price per 400 mg vial) | Sponsor base case, as submitted | CADTH reanalysis |
No price reduction ($7,900) | WTP < 283,913: conventional therapy 283,913 ≤ WTP: efgartigimod alfa | WTP < 1,764,628: rituximab 1,764,628 ≤ WTP: efgartigimod alfa |
10% ($7,110) | WTP < 247,580: conventional therapy 247,580 ≤ WTP: efgartigimod alfa | WTP < 1,560,339: rituximab 1,560,339 ≤ WTP: efgartigimod alfa |
20% ($6,320) | WTP < 211,214: conventional therapy 211,214 ≤ WTP: efgartigimod alfa | WTP < 1,356,050: rituximab 1,355,050 ≤ WTP: efgartigimod alfa |
30% ($5,530) | WTP < 174,849: conventional therapy 174,849 ≤ WTP: efgartigimod alfa | WTP < 1,151,762: rituximab 1,151,762 ≤ WTP: efgartigimod alfa |
40% ($4,740) | WTP < 138,483: conventional therapy 138,483 ≤ WTP: efgartigimod alfa | WTP < 947,473: rituximab 947,473 ≤ WTP: efgartigimod alfa |
50% ($3,950) | WTP < 102,118: conventional therapy 102,118 ≤ WTP: efgartigimod alfa | WTP < 743,185: rituximab 743,185 ≤ WTP: efgartigimod alfa |
60% ($3,160) | WTP < 65,752: conventional therapy 65,752 ≤ WTP: efgartigimod alfa | WTP < 538,896: rituximab 538,896 ≤ WTP: efgartigimod alfa |
70% ($2,370) | WTP < 29,387: conventional therapy 29,387 ≤ WTP: efgartigimod alfa | WTP < 334,607: rituximab 334,607 ≤ WTP: efgartigimod alfa |
80% ($1,580) | WTP ≤ 0: efgartigimod alfa (dominant) | WTP < 130,319: rituximab 130,319 ≤ WTP: efgartigimod alfa |
90% ($790) | WTP ≤ 0: efgartigimod alfa (dominant) | WTP ≤ 0: efgartigimod alfa (dominant) |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus; WTP = willingness to pay.
Scenario analyses were conducted using CADTH reanalyses for both populations to investigate the impact of:
including ravulizumab as a comparator treatment
excluding rituximab as a comparator treatment
utilizing the sponsor’s original utility value from the MyRealWorld MG study for the MG-ADL score of 10 or greater health state (i.e., 0.458).
Results of these scenarios are presented in Appendix 4 (Table 14). Ravulizumab plus conventional therapy was dominated by efgartigimod alfa because it was more expensive and yielded fewer QALYs. When rituximab was excluded, efgartigimod alfa plus conventional therapy was associated with an ICER of $1,113,373 compared to conventional therapy alone. When the lower utility value derived from the MyRealWorld MG study was reapplied for the MG-ADL 10 or greater health state, the ICER for efgartigimod alfa compared to rituximab decreased to $975,385 per QALY gained. The sensitivity of the results to the MG-ADL 10 or greater utility value highlights the impact and uncertainty inherent in both the inclusion of patients with disparate range of symptoms and experiences within a single health state, as well as the assumption that a proportion of people will remain in the MG-ADL 10 or greater health state for the remainder of the time horizon after crisis. The utility value of the MG-ADL 10 or greater health state would likely be less influential if health state transitions could continue to occur for all comparators for the duration of the time horizon, particularly after crisis.
Issues for Consideration
Differing budget holders: While the use of efgartigimod alfa would be associated with additional costs from both the Canadian drug plan payer perspective and the overall health care payer perspective, the reimbursement of efgartigimod alfa for gMG would likely reduce the use of blood products (IVIg, SCIg), which may result in some savings to alternate budget holders. A potential reduction in the use of blood products is also relevant given ongoing reports of blood and plasma shortages in Canada.29
Overall Conclusions
The CADTH Clinical Review concluded that evidence from the ADAPT trial suggests that in adult patients with MGFA Class II to IV MG at screening, whose symptoms persist despite treatment with AChEIs, corticosteroids, and/or NSISTs (i.e., MG-ADL score remains at ≥ 5), efgartigimod alfa as an add-on to conventional therapy likely results in clinically meaningful benefit in terms of the proportion of MG-ADL responders, proportion of QMG responders, and HRQoL, after cycle 1 of treatment, as well as after cycle 2 albeit with less certainty, compared with placebo, in the AChR-Ab+ population. The comparative efficacy of efgartigimod alfa versus active comparators is uncertain in the absence of direct comparative evidence. Overall, the CADTH Clinical Review concluded that the NMAs for patients with AChR-Ab+ gMG suggests that efgartigimod alfa may provide benefit in terms of change from baseline MG-ADL relative to IVIg. However, the results were uncertain due to wide 95% credible intervals and methodological limitations. No difference in efficacy in terms of change from baseline MG-ADL could be concluded for efgartigimod alfa relative to rituximab or ravulizumab due to methodological limitations and wide 95% credible intervals (which included within their range both the possibility of clinical benefit favouring efgartigimod alfa and no difference between treatments).
CADTH undertook reanalyses to address some of the key limitations in the sponsor’s pharmacoeconomic base case by including rituximab as a comparator, equalizing risk of crisis as well as corticosteroid dose used across health states, using health state utilities as measured in the ADAPT trial, and adjusting the distribution of health states for patients exiting a crisis. CADTH was unable to address uncertainty in the comparative clinical data, the grouping of disparate patient experiences into a single high-symptom health state, the uncertain and disparate discontinuation assumptions, or the lack of transparency in the model. Because several key limitations remained unresolved, the reanalysis performed by CADTH is associated with uncertainty.
In adult patients with AChR-Ab+ gMG, efgartigimod alfa plus conventional therapy was associated with an ICER of $1,764,628 per QALY gained (incremental costs: $1,195,367; incremental QALYs: 0.68) compared to rituximab plus conventional therapy. Both the sponsor’s submitted base-case analysis and CADTH’s reanalysis indicated that at the submitted price, efgartigimod alfa is not a cost-effective treatment option for adults with AChR-Ab+ gMG. An 84% price reduction to the unit price of efgartigimod alfa from $7,900, to $1,264 per 400 mg vial would be required for efgartigimod alfa to be considered cost-effective at a WTP threshold of $50,000 per QALY gained. At this price reduction, the drug acquisition cost per 4-week treatment cycle with efgartigimod alfa would be $10,112 to 15,168 per patient depending on body weight, or $47,729 to $71,593 per year, when 4.72 treatment cycles are assumed annually.
CADTH identified several additional limitations with the economic analyses submitted by the sponsor that could not be addressed. The cost-effectiveness of efgartigimod alfa plus conventional therapy in the full Health Canada indication (adults with AChR-Ab+ gMG) is unknown as patients in MGFA Class I and V, and in patients with MG-ADL total score of less than 5 were excluded from the ADAPT trial and ADAPT+. CADTH was unable to validate all model inputs, most critically the individual patient data informing the transition probabilities between MG-ADL-based health states for patients receiving efgartigimod alfa or conventional therapy alone, and subsequently the transition probabilities of comparators derived from the sponsor’s submitted NMAs. In addition, results from the sponsor’s NMA were associated with wide credible intervals indicating substantial clinical uncertainty, which was not reflected within the sponsor’s economic model. CADTH was also unable to alter the assumption that all patients receiving rituximab and blood products will cease transitioning through health states after 6 and 1 model cycles, respectively, an assumption that likely biases results in favour of efgartigimod alfa. Taken together, there remains considerable uncertainty in the cost-effectiveness results.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg / mL solution for intravenous use. Vaughan (ON): argenx Canada Inc.; 2023 May 9.
2.Howard JF, Jr., Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526-536. PubMed
3.Vyvgart (efgartigimod alfa): 400 mg/20 mL (20 mg/mL) solution, for intravenous use [product monograph]. Mississauga (ON): Quality & Compliance Services Inc; 2023 Sep 19.
4.Wolfe GI, Barohn RJ, Foster BM, et al. Randomized, controlled trial of intravenous immunoglobulin in myasthenia gravis. Muscle Nerve. 2002;26(4):549-552. PubMed
5.CADTH Drug Reimbursement Review pharmacoeconomic report: eculizumab (Soliris) for adult patients with generalized myasthenia gravis. Ottawa (ON): CADTH; 2020 Dec: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0605-soliris-mg-pharmacoeconomic-review-report.pdf. Accessed 2023 Jun 14.
6.Dewilde S, Philips G, Paci S, et al. Patient-reported burden of myasthenia gravis: baseline results of the international prospective, observational, longitudinal real-world digital study MyRealWorld-MG. BMJ Open. 2023;13(1):e066445. PubMed
7.Sullivan PW, Ghushchyan VH, Globe G, Sucher B. Health-related quality of life associated with systemic corticosteroids. Qual Life Res. 2017;26(4):1037-1058. PubMed
8.Bexelius C, Wachtmeister K, Skare P, Jonsson L, Vollenhoven R. Drivers of cost and health-related quality of life in patients with systemic lupus erythematosus (SLE): a Swedish nationwide study based on patient reports. Lupus. 2013;22(8):793-801. PubMed
9.Sullivan PW, Slejko JF, Sculpher MJ, Ghushchyan V. Catalogue of EQ-5D scores for the United Kingdom. Med Decis Making. 2011;31(6):800-804. PubMed
10.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2023: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 Jun 05.
11.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Jun 5.
12.Furlan JC, Barth D, Barnett C, Bril V. Cost-minimization analysis comparing intravenous immunoglobulin with plasma exchange in the management of patients with myasthenia gravis. Muscle Nerve. 2016;53(6):872-876. PubMed
13.Blackhouse G, Gaebel K, Xie F, et al. Cost-utility of Intravenous Immunoglobulin (IVIG) compared with corticosteroids for the treatment of Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) in Canada. Cost Eff Resour Alloc. 2010;8:14. PubMed
14.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
15.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 Mar 1.
16.Patient cost estimator. 2022; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2023 May 5.
17.Sussman J, Farrugia ME, Maddison P, Hill M, Leite MI, Hilton-Jones D. Myasthenia gravis: Association of British Neurologists' management guidelines. Pract Neurol. 2015;15(3):199-206. PubMed
18.Narayanaswami P, Sanders DB, Wolfe G, et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 update. Neurology. 2021;96(3):114-122. PubMed
19.Fowler S, Herrington J, Koopman W, Ricci M. Care of the patient with myasthenia gravis. In: Blissitt P, ed. Chicago (IL): American Association of Neuroscience Nurses; 2013: https://aann.org/uploads/Publications/CPGs/AANN14_CPGMysGravis.pdf. Accessed 2023 Jul 20.
20.Nowak RJ, Coffey CS, Goldstein JM, et al. Phase 2 trial of rituximab in acetylcholine receptor antibody-positive generalized myasthenia gravis: the BeatMG study. Neurology. 2021;98(4):e376-389. PubMed
21.Saskatchewan Drug Plan: search formulary. 2022; http://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2023 Jun 05.
22.van Hout B, Janssen MF, Feng YS, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health. 2012;15(5):708-715. PubMed
23.Xie F, Pullenayegum E, Gaebel K, et al. A time trade-off-derived value set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. PubMed
24.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2023 Jul 25.
25.Soliris (eculizumab): 30 mL parenteral solution (10 mg/mL) for injection [product monograph]. Zurich (CH): Alexion Pharma GmbH; 2021 Mar 25: https://pdf.hres.ca/dpd_pm/00060546.PDF. Accessed 2023 Jun 5.
26.CADTH Drug Reimbursement Expert Review Committee final recommendation: eculizumab (Soliris) for adult patients with refractory generalized myasthenia gravis. Ottawa (ON): CADTH; 2020 Oct: https://www.cadth.ca/sites/default/files/cdr/complete/SR0605%20Soliris%20MG%20-%20CDEC%20Final%20Recommendation%20October%2021%2C%202020_for%20posting.pdf. Accessed 2023 Jul 22.
27.pan-Canadian Pharmaceutical Alliance. Soliris (eculizumab) for myasthenia gravis. 2022; https://www.pcpacanada.ca/negotiation/21306. Accessed 2023 Aug 1.
28.CADTH Drug Reimbursement Expert Review Committee final recommendation: ravulizumab (Ultomiris) for AChR antibody-positive generalized myasthenia gravis. 2023 Aug; https://www.cadth.ca/sites/default/files/DRR/2023/SR0765Ultomiris%20-%20Confidential%20Final%20CADTH%20Recommendation%20August%2024%2C%202023%20revised.pdf. Accessed 2023 Aug 30.
29.Pasieka C. Canadian Blood Services needs thousands more donors to roll up their sleeves. Toronto (ON): CBC News; 2023 Jun 17: https://www.cbc.ca/news/canada/toronto/canadian-blood-services-needs-thousands-more-canadians-to-roll-up-their-sleeves-1.6879312.
30.Ultomiris (ravulizumab): 10 mg/mL & 100 mg/mL concentrate for solution for intravenous use [product monograph]. Zurich (CH): Alexion Pharma GmbH; 2023 Jan 6: https://pdf.hres.ca/dpd_pm/00069134.PDF. Accessed 2023 Jun 05.
31.CADTH Reimbursement Review: ravulizumab (Ultomiris) for paroxysmal nocturnal hemoglobinuria. Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/DRR/2022/SR0700CL-Ultomiri_combined.pdf. Accessed 2023 Jun 14.
32.Alberta Health. Interactive Drug Benefit List. 2023; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2023 Jun 05.
33.Farmakidis C, Pasnoor M, Dimachkie MM, Barohn RJ. Treatment of myasthenia gravis. Neurol Clin. 2018;36(2):311-337. PubMed
34.DeltaPA. Ottawa (ON): IQVIA; 2023: https://www.iqvia.com/. Accessed 2023 Jun 05.
35.Heckmann JM, Rawoot A, Bateman K, Renison R, Badri M. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol. 2011;11:97. PubMed
36.Mestinon (pyridostigmine bromide): 60 mg tablets, 180 mg slow release tablets [product monograph]. Laval (QC): Bausch Health, Canada Inc; 2019 Jul 3: https://pdf.hres.ca/dpd_pm/00052033.PDF. Accessed 2023 Jun 5.
37.Melzer N, Ruck T, Fuhr P, et al. Clinical features, pathogenesis, and treatment of myasthenia gravis: a supplement to the Guidelines of the German Neurological Society. J Neurol. 2016;263(8):1473-1494. PubMed
38.Table: 17-10-0005-01. Population estimates on July 1st, by age and sex Ottawa (ON): Statistics Canada; 2023: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2023 June 15.
39.About the Non-Insured Health Benefits program. 2023; https://www.sac-isc.gc.ca/eng/1576790320164/1576790364553#a4. Accessed 2023 Jun 1.
40.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. Ottawa (ON): The Conference Board of Canada; 2017: https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326. Accessed 2023 Jul 25.
41.Breiner A, Widdifield J, Katzberg HD, Barnett C, Bril V, Tu K. Epidemiology of myasthenia gravis in Ontario, Canada. Neuromuscul Disord. 2016;26(1):41-46. PubMed
42.Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S. Myasthenia gravis - autoantibody characteristics and their implications for therapy. Nat Rev Neurol. 2016;12(5):259-268. PubMed
43.Lazaridis K, Tzartos SJ. Autoantibody specificities in myasthenia gravis; implications for improved diagnostics and therapeutics. Front Immunol. 2020;11:212. PubMed
44.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Vyvgart (efgartigimod alfa), 20 mg / mL solution for intravenous use. Vaughan (ON): argenx Canada Inc.; 2023 May 9.
45.Procedures for CADTH reimbursement reviews. Ottawa (ON): CADTH; 2023 Nov: https://www.cadth.ca/sites/default/files/Drug_Review_Process/CADTH%20Drug%20Reimbursement%20Review%20Procedures.pdf. Accessed 2023 Jul 25.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Complement Inhibitors Indicated for the Treatment of Generalized Myasthenia Gravis
Treatment | Strength or concentration | Form | Price ($) | Recommended dosage | Average daily cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Efgartigimod alfa (Vyvgart) | 20 mg/mL | 400 mg vial solution for IV use | 7,900.0000a | Loading: 10 mg/kg (max 1,200 mg) weekly for 4 weeks. Subsequent treatment cycles are based on clinical evaluation and may vary by patient.3 | Patient weight 41 to 80 kg: 816.71b Patient weight above 80 kg: 1,225.07b | Patient weight 41 to 80 kg: 298,304b Patient weight above 80 kg: 447,456b |
Ravulizumab (Ultomiris) | 10 mg/mL | 30 mL single-dose vial of concentrate for solution for IV infusion | 7,296.6700c | Loading dose at weeks 0, then maintenance dose at week 2 and every 8 weeks thereafter based on weight as followsd: ≥ 40 kg to < 60 kg Loading: 2,400 mg; Maintenance: 3,000 mg ≥ 60 kg to < 100 kg Loading: 2,700 mg; Maintenance: 3,300 mg ≥ 100 kg Loading: 3,000 mg; Maintenance: 3,600 mg30 | First year: 1,558.13 Subsequent years: 1,433.27 | First year: 569,108 Subsequent years: 523,503 |
Eculizumab (Soliris) | 10 mg/mL | 300 mg single-use vial | 6,675.3000e | Loading: 900 mg weekly for 4 weeks, then 1,200 mg for the fifth dose 1 week later Maintenance: 1,200 mg every 2 weeks thereafter25 | First year: 1,943.78 Subsequent years: 1,907.23 | First year: 709,966 Subsequent years: 696,615 |
Note: Costs assume a 365.25-day year and wastage of excess medication in vials. Weight-based dosing assumes an 80 kg patient unless otherwise specified, consistent with the mean body weight reported for patients in the ADAPT trial.2
aSponsor-submitted price.1
bAssumes an average of 4.72 4-week courses per year, as reportedly administered to patients with AChR-Ab+ gMG in pooled ADAPT and ADAPT+ data.1 Cost per 4-week course is $63,200 for patients weighing 41 kg to 80 kg and $94,800 for patients weighing more than 80 kg.
cPrice submitted for CADTH’s review of Ultomiris for paroxysmal nocturnal hemoglobinuria.31
dPatients weighting less than 40 kg have different dosing quantities and schedules. Daily and annual costs assume an 80 kg patient. For patients switching from eculizumab, the loading dose of ravulizumab is given 2 weeks after the last eculizumab infusion. Maintenance doses are, then, given every 8 weeks, starting 2 weeks after the loading dose.
eAlberta formulary, accessed June 20, 2023. Note that Alberta does not fund eculizumab for generalized myasthenia gravis.32
Table 9: CADTH Cost Comparison Table for Other Treatments for Generalized Myasthenia Gravis
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Average daily cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Other biologics | ||||||
Rituximab (biosimilars) | 10 mg/mL | 10 mL 50 mL Vial for IV infusion | 297.0000 1,485.0000 | 375 mg/m2 weekly for 4 doses | NA | Cost per course: 8,316 |
Alternate dosing: 1 g, followed by 1 g 2 weeks later, and then every 6 months | First year: 31.90 Subsequent years: 16.26 | First year: 11,652 Subsequent years: 5,940 | ||||
Glucocorticoids | ||||||
Prednisone (Winpred, generics) | 1 mg 5 mg 50 mg | Tablet | 0.1214 0.0220 0.1735 | Initiate at 10 to 20 mg/day, increase by 5 mg/day per week until stable remission (target 1 mg/kg/day) | 0.04 to 0.31 | 16 to 112 |
Alternate dosing: Initiate at 60 to 80 mg/day, then taper after improvement | 0.21 to 0.31 | 77 to 112 | ||||
Immunosuppressive agents | ||||||
Azathioprine (generics) | 50 mg | Tablet | 0.2405 | Initiate at 50 mg/day for 5 days, and then, escalate to 2.5 to 3 mg/kg/dayd | 0.96 to 1.20 | 351 to 439 |
Cyclophosphamide (Procytox, generics) | 25 mg 50 mg | Tablet | 0.3545 0.4773 | 500 mg/m2 to 1,000 mg/m2 every month for 6 months | NA | Cost per course: 52 to 103 |
500 mg 1,000 mg 2,000 mg | IV vial, powder for injection | 97.8000b 177.2700b 326.0000b | NA | Cost per course: 1,063 to 1,956 | ||
Cyclosporine (generics) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.7115 0.7870 1.5350 3.0720 | Starting dose: 100 mg twice daily Target dose: 5 to 6 mg/kg/day in 2 divided doses, adjust for serum trough level of 75 to 150 ng/mL | 12.29 to 15.34 | 4,489 to 5,606 |
Methotrexate (generic, Metoject SC) | 2.5 mg 10 mg | Tablet | 0.2513 2.7983c | 10 mg/week to 20 mg/week, orally or SC | 0.14 to 0.29 | 52 to 105 |
10 mg/mL 25 mg/mL | Vial for injection | 8.9200 12.5000 | 1.27 to 2.55 | 465 to 930 | ||
10 mg/0.2 mL 12.5 mg/0.25 mL 15 mg/0.3 mL 17.5 mg/0.35 mL 20 mg/0.4 mL 22.5 mg/0.45 mL 25 mg/0.5 mL | Prefilled syringe for SC use | 29.6400 31.2000 24.5700 24.0000 26.2500 26.2500 29.2500 | 3.75 to 4.23 | 1,370 to 1,547 | ||
Mycophenolate mofetil (generics) | 250 mg | Capsule | 0.3712 | 1,000 mg twice daily | 2.97 | 1,084 |
500 mg | Tablet | 0.7423 | 2.98 | 1,087 | ||
Mycophenolate Sodium (generics) | 180 mg 360 mg | Enteric tablet | 0.9989 1.9977 | 720 mg twice dailye | 7.99 | 2,917 |
Tacrolimus (generics) | 0.5 mg 1 mg 5 mg | Capsule | 1.4775 1.8900 9.4650 | 3 to 5 mg per dayf | 2.67 to 9.47 | 2,071 to 3,458 |
Cholinesterase inhibitors | ||||||
Pyridostigmine (Mestinon, generics) | 60 mg | Tablet | 0.2673 | 60 mg to 120 mg every 3 to 8 hours while awake | 0.53 to 3.20 | 195 to 1,172 |
180 mg | SR tablet | 1.3384 | 2.68 | 978 | ||
Blood products | ||||||
IV immunoglobulin | 10,340 per exacerbationg | |||||
Plasma exchange | 7,600 per exacerbationg | |||||
Note: All prices are from the Ontario Drug Benefit Formulary (accessed June 2023), unless otherwise indicated, and do not include dispensing fees. All cost calculations for agents with weight or body surface area-based dosing was calculated using the mean body surface area of 1.8 m2 and mass of 80 kg. Drug wastage was included. Dosing is from a study by Farmakidis et al., unless otherwise indicated.33
aOntario Drug Benefit Formulary Exceptional Access Program (accessed June 2023).10
bDelta PA database wholesale prices (accessed June 20, 2023).34
cSaskatchewan Drug Plan formulary (accessed June 20, 2023).21
dAzathioprine dosing was obtained from published literature.35
eMyfortic product monograph, dose indicated for the prophylaxis of organ rejection in patients receiving allogeneic renal transplants, confirmed with clinical experts as also use for gMG.36
fTacrolimus dose reported for patients with therapy-refractory myasthenia gravis in clinical features, pathogenesis, and treatment of myasthenia gravis: a supplement to the Guidelines of the German Neurological Society.37
gThe cost of IV immunoglobulin and plasma exchange, totalling $8,277 and $6,084, respectively, in 2014 dollars,12 was for rescue therapy and included cost of blood products and hospital costs and was inflated to 2023 dollars by CADTH. Due to confidential prices of IV immunoglobulin products and plasma exchange, chronic treatment cost is unknown.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Rituximab was inappropriately excluded from the reimbursement request population. Refer to limitation “Rituximab was excluded as a comparator in the reimbursement request population.” |
Model has been adequately programmed and has sufficient face validity | No | Data underlying the transition probabilities and conversion of NMA results was not provided, and therefore could not be validated. The MG-ADL 10+ health state is inclusive of patients with a wide range of symptoms and quality of life impacts that are inadequately summarized within that single state. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Uncertainty in the NMA results (i.e., the wide credible intervals) was not adequately reflected in the derivation of transition probabilities for rituximab, blood products, and ravulizumab. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Some parameters required complete reprogramming of their probabilistic distributions when changed and did not switch to probabilistic parameters when the PSA was run (e.g., disease severity at baseline). Uncertainty in the relative clinical efficacy of NMA-based comparators was not considered and could not be validated. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Explanations for the sponsor’s methodology was often vague, split into multiple sections, or confusingly worded. The model sometimes lacked adequate labelling to clearly understand the trace and tunnel states. |
MG-ADL = Myasthenia Gravis Activities of Daily Living. NMA = network meta-analysis; PSA = probabilistic sensitivity analysis.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
MG-ADL = Myasthenia Gravis Activities of Daily Living.
Note: While the model figure shows that patients can transition from MG-ADL < 5 to crisis, and from crisis back to any of the MG-ADL states, the submitted model sets the probability of transitioning from MG-ADL < 5 to crisis at 0%, and the probability of transitioning from crisis to MG-ADL ≥ 10% to 100%.
Source: Sponsor’s pharmacoeconomic submission.1
Table 11: Model Input Parameters by MG-ADL Health State
MG-ADL health state | Disease severity at baseline | Per-cycle probability of MG crisis | Per-cycle probability of exacerbation requiring hospitalization | Health state utilitya | Proportion patients on high-dose CS | Disease monitoring costs per cycle |
|---|---|---|---|---|---|---|
MG-ADL < 5 | 0% | 0% | 0.9% | ||||| | 0% | $61 |
MG-ADL 5 to 7 | 26% | 0.09% | 2.0% | ||||| | 56.7% | $78 |
MG-ADL 8 to 9 | 42% | 0.09% | 3.9% | ||||| | 56.7% | $109 |
MG-ADL ≥ 10 | 32% | 0.09% | 15.5% | ||||| | 56.7% | $126 |
CS = corticosteroid; MG-ADL = Myasthenia Gravis Activities of Daily Living.
aHealth state utility for patients in myasthenic crisis was 0.118. Risk of death per myasthenic crisis was 5%. Additional disutilities were applied for corticosteroid use, exacerbations, and adverse events. Alternate utilities were provided based on EQ-5D-5L data from the ADAPT trial utilities for the health states as follows: MG-ADL < 5 = |||||, MG-ADL 5 to 7 = |||||, MG-ADL 8 to 9 = |||||, MG-ADL 10+ = |||||, with crisis associated with a utility of |||||.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 12: Summary of the Step-Wise CADTH Base-Case Reanalysis Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor’s base case, as submitted (deterministic) | |||
Conventional therapy | 828,320 | 12.93 | Reference |
Efgartigimod alfa + conventional therapy | 1,924,657 | 16.72 | 288,794 |
Blood products + conventional therapy | 2,166,857 | 13.30 | Dominated by efgartigimod alfa |
Sponsor’s base case, as submitted (probabilistic) | |||
Conventional therapy | 828,519 | 12.98 | Reference |
Efgartigimod alfa + conventional therapy | 1,913,189 | 16.80 | 283,913 |
Blood products + conventional therapy | 2,169,667 | 13.35 | Dominated by efgartigimod alfa |
Sponsor’s base case, corrected (probabilistic)a | |||
Conventional therapy | 832,506 | 12.96 | Reference |
Efgartigimod alfa + conventional therapy | 1,914,156 | 16.80 | 281,939 |
Blood products + conventional therapy | 2,175,502 | 13.33 | Dominated by efgartigimod alfa |
CADTH reanalysis 1 – rituximab included (deterministic) | |||
Rituximab + conventional therapy | 753,802 | 14.36 | Reference |
Conventional therapy | 828,320 | 12.93 | Dominated by rituximab |
Efgartigimod alfa + conventional therapy | 1,924,657 | 16.72 | 495,981 |
Blood products + conventional therapy | 2,166,857 | 13.30 | Dominated by rituximab, efgartigimod alfa |
CADTH reanalysis 2 – risk of crisis equal across MG-ADL health states (deterministic) | |||
Conventional therapy | 867,779 | 12.52 | Reference |
Efgartigimod alfa + conventional therapy | 2,014,069 | 15.79 | 350,840 |
Blood products + conventional therapy | 2,273,730 | 12.83 | Dominated by efgartigimod alfa |
CADTH reanalysis 3 – high-dose CS equalized across MG-ADL health states (deterministic) | |||
Conventional therapy | 836,672 | 12.36 | Reference |
Efgartigimod alfa + conventional therapy | 1,938,876 | 15.09 | 403,910 |
Blood products + conventional therapy | 2,176,918 | 12.64 | Dominated by efgartigimod alfa |
CADTH reanalysis 4 – health state utilities from ADAPT trial, treatments pooled (deterministic) | |||
Conventional therapy | 828,320 | 15.89 | Reference |
Efgartigimod alfa + conventional therapy | 1,924,657 | 18.21 | 472,931 |
Blood products + conventional therapy | 2,166,857 | 16.11 | Dominated by efgartigimod alfa |
CADTH reanalysis 5 – altered post-crisis health state distribution (deterministic) | |||
Conventional therapy | 782,158 | 13.21 | Reference |
Efgartigimod alfa + conventional therapy | 1,903,554 | 16.85 | 307,602 |
Blood products + conventional therapy | 2,110,876 | 13.57 | Dominated by efgartigimod alfa |
CADTH base case (1 + 2 + 3 + 4 + 5, deterministic) | |||
Rituximab + conventional therapy | 774,379 | 15.65 | Reference |
Conventional therapy | 817,419 | 15.32 | Dominated by rituximab |
Efgartigimod alfa + conventional therapy | 1,977,115 | 16.33 | 1,769,633 |
Blood products + conventional therapy | 2,208,650 | 15.43 | Dominated by rituximab |
CADTH base case (1 + 2 + 3 + 4 + 5, probabilistic) | |||
Rituximab + conventional therapy | 774,526 | 15.70 | Reference |
Conventional therapy | 818,381 | 15.36 | Dominated by rituximab |
Efgartigimod alfa + conventional therapy | 1,969,893 | 16.38 | 1,764,628 |
Blood products + conventional therapy | 2,210,045 | 15.47 | Dominated by rituximab |
CS = corticosteroid; ICER = incremental cost-effectiveness ratio; IVIg = intravenous immunoglobulin; MG-ADL = Myasthenia Gravis Activities of Daily Living; QALY = quality-adjusted life-years.
aThe sponsor’s corrected key scenario is presented probabilistically as the corrections only apply to probabilistic analyses. When run deterministically, the sponsor’s corrected key scenario is the same as the submitted base case.
Table 13: Disaggregated Summary of CADTH’s Economic Evaluation Results
Component | Efgartigimod alfa + conventional therapy | Rituximab + conventional therapy | Blood products + conventional therapy | Conventional therapy alone |
|---|---|---|---|---|
Life-years | ||||
MG-ADL < 5 | 11.83 | 6.84 | 4.63 | 4.06 |
MG-ADL 5 to 7 | 5.19 | 5.65 | 7.50 | 7.35 |
MG-ADL 8 to 9 | 3.38 | 5.40 | 4.41 | 4.50 |
MG-ADL ≥ 10 | 3.19 | 5.69 | 7.05 | 7.67 |
Crises | 0.02 | 0.02 | 0.02 | 0.02 |
Total | 23.61 | 23.61 | 23.61 | 23.61 |
Quality-adjusted life-years | ||||
MG-ADL < 5 | 8.79 | 5.09 | 3.44 | 3.02 |
MG-ADL 5 to 7 | 3.62 | 3.94 | 5.23 | 5.13 |
MG-ADL 8 to 9 | 2.17 | 3.47 | 2.83 | 2.89 |
MG-ADL ≥ 10 | 1.80 | 3.20 | 3.97 | 4.32 |
Crises | –0.00 | –0.00 | –0.00 | –0.00 |
Total | 16.38 | 15.70 | 15.47 | 15.36 |
Costs ($) | ||||
Drug acquisition | 1,421,957 | 83,578 | 1,330,314 | 41,245 |
Administration | 15.220 | 10,885 | 137,639 | 0 |
Disease monitoring | 24,723 | 28,168 | 29,080 | 29,652 |
Exacerbations | 281,966 | 424,923 | 487,924 | 518,744 |
CS-related chronic complications | 162,971 | 162,989 | 163,003 | 163,007 |
Crises | 30,584 | 30,526 | 30,524 | 30,530 |
AEs | 23,974 | 24,960 | 23,065 | 26,704 |
End of life | 8,498 | 8,498 | 8,498 | 8,498 |
Total | 1,969,893 | 774,526 | 2,210,045 | 818,381 |
AE = adverse event; CS = corticosteroid; MG-ADL = Myasthenia Gravis Activities of Daily Living.
Scenario Analyses
Table 14: Summary of Scenario Analyses Conducted on CADTH Base-Case Reanalysis
Drug | Total costs | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
CADTH base-case results | |||
Rituximab + conventional therapy | 774,526 | 15.70 | Reference |
Conventional therapy | 818,381 | 15.36 | Dominated by rituximab |
Efgartigimod alfa + conventional therapy | 1,969,893 | 16.38 | 1,762,628 |
Blood products + conventional therapy | 2,210,045 | 15.47 | Dominated by rituximab, efgartigimod alfa |
Scenario 1 – Ravulizumab included as a comparator | |||
Rituximab + conventional therapy | 773,054 | 15.72 | Reference |
Conventional therapy | 820,485 | 15.36 | Dominated by rituximab |
Efgartigimod alfa + conventional therapy | 1,963,912 | 16.39 | 1,775,980 |
Blood products + conventional therapy | 2,213,030 | 15.48 | Dominated by rituximab, efgartigimod alfa |
Ravulizumab + conventional therapy | 2,694,216 | 16.09 | Dominated by efgartigimod alfa |
Scenario 2 – Rituximab excluded as a comparator | |||
Conventional therapy | 819,708 | 15.37 | Reference |
Efgartigimod alfa + conventional therapy | 1,961,601 | 16.39 | 1,113,373 |
Blood products + conventional therapy | 2,217,140 | 15.48 | Dominated by efgartigimod alfa |
Scenario 3 – Lower utility associated with MG-ADL 10+ health state | |||
Rituximab + conventional therapy | 775,428 | 14.47 | Reference |
Conventional therapy | 819,808 | 13.68 | Dominated by rituximab |
Efgartigimod alfa + conventional therapy | 1,968,553 | 15.69 | 975,385 |
Blood products + conventional therapy | 2,213,105 | 13.94 | Dominated by rituximab, efgartigimod alfa |
gMG = generalized myasthenia gravis; ICER = incremental cost-effectiveness ratio; MG-ADL = Myasthenia Gravis Activities of Daily Living; QALY = quality-adjusted life-years.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 15: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
NIHB = Non-Insured Health Benefits; PE = plasma exchange.
Summary of Sponsor’s BIA
In the submitted base-case budget impact analysis (BIA), the sponsor assessed the introduction of efgartigimod alfa for adult patients with AChR-Ab+ gMG, the Health Canada–indicated population. A key scenario analysis assessing the introduction of efgartigimod alfa for adult patients with AChR-Ab+ gMG whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs was also conducted.
The BIA was undertaken from the perspective of a Canadian public payer over a 3-year time horizon (2025 to 2027) using an epidemiological approach. The sponsor compared a reference scenario in which patients received treatments currently used for the treatment of gMG in Canada (i.e., conventional therapy alone and blood products plus conventional therapy) to a new drug scenario in which efgartigimod alfa was reimbursed. The sponsor’s analysis included drug acquisition costs including those for conventional therapy; dispensing fees and markups were not included in the base case. Data for the model were obtained from various sources including Statistics Canada,38 Non-Insured Health Benefits (NIHB) annual reports,39 the published literature,12,13,40-43 ODB formulary list prices,11 clinical expert opinion elicited by the sponsor, and the sponsor’s internal data.1,44 Key inputs to the BIA are documented in Table 16. Key assumptions include:
The prevalence of MG can be represented by estimates from 2013.
The population of the NIHB can be added to the population of adults in the Canadian provinces.
Efgartigimod alfa will be used by the same number of patients regardless of whether it is reimbursed for all patients with AChR-Ab+ gMG or limited to those whose symptoms are inadequately controlled on conventional therapy.
Rituximab will not be used to treat patients with AChR-Ab+ gMG, nor will it be displaced by efgartigimod alfa.
75% of market uptake for efgartigimod alfa will displace blood products while 25% will displace conventional therapy.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimated Health Canada–indicated population (year 1 / year 2 / year 3) | Sponsor’s estimated reimbursement request population (year 1 / year 2 / year 3) |
|---|---|---|
Target population | ||
Adult population across Canada (base year) | 25,737,572a | |
MG prevalence | 32 per 100,00041 | |
Proportion with gMG | 85%42 | |
Proportion who are AChR-Ab+ | 85%43 | |
Proportion who are inadequately controlled with conventional therapy | NA | 23.57%b |
Proportion eligible for public coverage | 72.11%40 | |
Number of patients eligible for drug under review | 4,351 / 4,411 / 4,471 | 1,021 / 1,035 / 1,049 |
Proportion of patients assumed to be in first year of their assigned treatment | 50%c | |
Market uptake (reference scenario, 3 years) d | ||
Efgartigimod alfa plus conventional therapy | 0% | 0% |
Conventional therapy alone | 80.5% | 25.0% |
Blood products plus conventional therapy | 19.5% | 75.0% |
Rituximab plus conventional therapy | 0% / 0% / 0% | 0% / 0% / 0% |
Market uptake (new drug scenario, 3 years)d | ||
Efgartigimod alfa plus conventional therapy | 4.02% / 6.21% / 7.36% | 17.04% / 26.36% / 31.24% |
Conventional therapy alone | 79.50% / 78.95% / 78.66% | 20.74% / 18.41% / 17.19% |
Blood products plus conventional therapy | 16.49% / 14.84% / 13.98% | 62.22% / 55.23% / 51.57% |
Rituximab plus conventional therapy | 0% / 0% / 0% | 0% / 0% / 0% |
Cost of treatment (per patient per year) | ||
Efgartigimod alfa plus conventional therapy | $360,2191 | |
Conventional therapy alone | $1,79211 | |
Blood products plus conventional therapy | ||
Rituximab plus conventional therapy | NAe | |
AChR-Ab+ = acetylcholine receptor antibody positive; gMG = generalized myasthenia gravis; NA = not applicable.
aSum of the adult populations (18+ years) of all provinces except for Quebec, plus the client population of NIHB who are 20+ years of age. The base year and years 1 through 3 population estimates for all provinces were linearly forecast from 2017 to 2022 Statistics Canada population estimates, while that of the NIHB was linearly forecast from 2017 through 2022 NIHB annual reports.38,39
bA weighted average estimate of the proportion of patients who are still symptomatic despite treatment with CT, based on a survey of Canadian clinicians conducted by the sponsor.1 Only applied in the reimbursement request population.
cAssumption, the average cost of induction and maintenance years is assumed for all comparators where it is relevant.
dCited as based on information gathered from interviews with clinicians in Canada and internal market research.44
eRituximab was not included within the updated BIA submitted by the sponsor in September 2023. The annual cost of rituximab used in previous versions of the BIA was $13,672.11
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analysis suggest that the reimbursement of efgartigimod alfa for adults with AChR-Ab+ gMG whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs (the reimbursement request population) would be associated with an incremental cost of $49,087,097 in Year 1, $76,971,159 in Year 2, and $92,481,720 in Year 3, for a 3-year incremental budget impact of $218,539,976. The sponsor’s results for the Health Canada–indicated population (adults with AChR-Ab+ gMG regardless of prior treatment) were identical to those for the reimbursement population.
A scenario analysis including ravulizumab as a comparator for the reimbursement request population was also provided, where ravulizumab was assumed to be expanding into the market taking approximately 17% / 26% / 31% of the reference scenario market share in years 1, 2, and 3, with efgartigimod taking 19% / 32% /39% of the new drug scenario market proportionally from each comparator. The estimated incremental cost of reimbursing efgartigimod alfa under this scenario was $38,053,823 in Year 1, $47,566,693 in Year 2, and $48,627,615 in Year 3, for a 3-year total budgetary impact of $134,248,131.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Market uptake and comparator displacement do not reflect the Health Canada indication: For the Health Canada–indicated population, the estimated market displacement appears to reflect the expected reimbursement criteria (i.e., patients with persistent symptoms despite adequate conventional therapy) in terms of total displacement and comparators displaced, leading to a budget impact assessment which is identical for both populations. As such, the sponsor has assumed that efgartigimod alfa will only be used in the reimbursement request population, regardless of whether the full Health Canada indication is reimbursed. According to clinical expert opinion elicited by CADTH, if efgartigimod alfa were reimbursed for its full indicated population (i.e., without restriction by persistent symptoms despite conventional therapy) it would be used by more patients than if it is restricted to those with persistent symptoms. That is, if reimbursed for the full Health Canada–indicated population, it is expected that some patients without persistent symptoms would use efgartigimod alfa. How many more patients might receive efgartigimod alfa in this case was deemed to be uncertain based on clinical expert opinion elicited by CADTH, with estimates ranging from up to 4 times more patients receiving efgartigimod alfa if funded for its full indication than if it was limited to patients with persistent symptoms despite adequate conventional therapy, to up to 50% of all patients with AChR-Ab+ gMG receiving it. Experts additionally noted that, if reimbursed for the full Health Canada–indicated population, efgartigimod alfa could be used as an initial therapy to bridge patients through the delay in efficacy of nonsteroidal immunosuppressive therapies, in which case it could be used by greater than 50% of patients with AChR-Ab+ gMG.
To explore this limitation, CADTH conducted a scenario analysis assuming that uptake of efgartigimod alfa could be up to 50% of all patients with AChR-Ab+ gMG by Year 3 (15% and 30% assumed in years 1 and 2, respectively), to represent patients who would receive it who have AChR-Ab+ gMG but do not meet the reimbursement request criterion of being symptomatic despite adequate conventional therapy.
NIHB population was inappropriately calculated: The sponsor calculated the total population of CADTH-participating drug plans by adding the population of the provinces as reported by Statistics Canada, excluding Quebec, to the population of NIHB clients. NIHB clients living within the borders of a province are counted within provincial population data, thus the NIHB population was double counted in the sponsor’s analysis. Additionally, NIHB clients residing within Ontario are covered primarily by ODB if they are younger than 25 years or older than 65 years of age. The sponsor considered NIHB clients who were at least 20 years as adults for the purpose of the model due to the structure of NIHB population data; however, this leaves out clients aged 18 or 19 years. Finally, the sponsor forecast future NIHB client numbers using 2017 to 2021 data, however the NIHB 2021 to 2022 annual report is now available.
In CADTH reanalyses, the number of NIHB clients who are adults was calculated by summing all age groups from 20 to 65+, as well as 2/5 of the population within the 15 to 19 age bracket, for each available year (2017 to 2022), and then linearly forecast future years. Adult NIHB clients living within each provincial jurisdiction were subtracted from that province’s population for each available year, with the exception of adults under 25 years and those 65 years and older residing in Ontario. The NIHB client populations of each Atlantic province were assumed to be proportional to the overall populations of those provinces.
Rituximab was excluded as a comparator: As discussed in the appraisal of the pharmacoeconomic analysis, the sponsor excluded rituximab as a relevant comparator despite its inclusion as a treatment option for patients with refractory AChR-Ab+ gMG in Canadian clinical practice and international guidelines,1,17-19 and its funding for this population in some jurisdictions. According to clinical expert opinion elicited by CADTH, rituximab is used more frequently for the treatment of AChR-Ab+ gMG than AChR-Ab– gMG and would be displaced by efgartigimod alfa for in the AChR-Ab+ population, should it be funded.
In CADTH reanalyses, rituximab was included as a comparator for both populations, with a reference case market share of 2% when considering the Health Canada–indicated population, and 5% when considering the reimbursement request population. In both populations, rituximab was assumed to be displaced by efgartigimod alfa in proportion to its reference case market share.
Proportion of eligible patients who will be publicly funded is uncertain and differs by comparator: The sponsor assumed a public coverage rate of 72%, based on the population-based public drug coverage proportions reported by the Conference Board of Canada in 2017.40 However, due to the rarity of gMG, the expense and IV administration of efgartigimod alfa as well as rituximab and IVIg, and the full public funding of blood products (albeit under non-drug plan budgets), the proportion of patients who would be eligible for public reimbursement of efgartigimod alfa and its add-on comparators is expected to be higher than the average for general prescription coverage, including conventional therapy. This was consistent with clinical expert feedback elicited by CADTH indicating that a very high proportion of patients accessing rituximab for gMG were doing so through public reimbursement. The model was not structured to accept differing public reimbursement proportion assumptions for different treatment groups.
In CADTH reanalyses, 100% of eligible patients were assumed to be publicly funded. This proportion was reverted back to the sponsor’s assumption in scenario analyses.
Drug plan payer perspective: According to the Procedures for CADTH Reimbursement Reviews, the BIA base case should be undertaken from the perspective of a pan-Canadian drug plan program.45 As such, costs relating to the use of blood products (i.e., IVIg, SCIg, PE) are not funded by jurisdictional drug plan budgets and should thus be excluded from the drug plan perspective. Additionally, the sponsor assumed that patients receiving IVIg would have it administered every 3 weeks, and that no patients would receiving PE. Clinical expert opinion elicited by CADTH indicated that when used chronically, IVIg is typically limited to once every 4 weeks in current Canadian clinical practice, although patient input submitted for this review indicated some patients do receive IVIg more frequently. Additionally, clinical experts estimated that approximately 5% to 10% of patients with gMG receiving blood products receive PE rather than immunoglobulins.
Costs associated with the use of blood products were excluded from the drug plan perspective. Blood product costs were included within a health care system perspective, which also included administration costs. For this perspective, CADTH assumed that 5% of patients using blood products would receive PE. A scenario analysis was conducted where IVIg was limited to once every 4 weeks.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s submitted analyses by correcting the double-counting of NIHB clients, including rituximab as a comparator in both populations and adjusting its assumed market share, increasing the proportion of eligible patients who would be publicly reimbursed, removing the cost of blood products from the drug plan perspective, and including the cost of administration and revising the usage assumptions for blood products for the health care system perspective. The changes applied to derive the CADTH base case and key scenario analysis for both perspectives are described in Table 17.
Table 17: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case and key scenario reanalyses | ||
1. NIHB adjustments (base year 2024) | NIHB-funded adult population: 653,147 Total included adult population: 25,737,572 | NIHB-funded adult population: 650,698 Total included adult population: 25,203,237 |
2. Rituximab as a comparator | Not included | HC population: 2%a RR population: 5% Rituximab is included and displaced proportionally to reference case market share |
3. Proportion of eligible patients publicly reimbursed | 72% | 100% |
4a. Blood product costs (drug plan perspective) | Included | Set to $0 |
4b. Administration and blood product costs (health care system perspective) | Administration costs excluded 0% receive PE | Administration costs included 5% receive PE |
CADTH base case (drug plan perspective) | 1 through 4a | |
CADTH combined reanalysis (health care system perspective) | 1 through 3 and 4b | |
Ab+ = antibody seropositive; AChR = acetylcholine receptor; BIA = budget impact analysis; HC = Health Canada (indicated); IVIg = intravenous immunoglobulin; NIHB = Non-Insured Health Benefits; PE = plasma exchange; RR = reimbursement request.
aThe sponsor submitted a new BIA model in September 2023 which considered the Health Canada–indicated population of patients with gMG who are AChR-Ab+, but removed the flexibility of including rituximab as a comparator. This issue could be worked around for the reimbursement request population by substituting rituximab inputs into the scenario intended to represent the inclusion of ravulizumab. However, this workaround was not feasible for the Health Canada–indicated population without a complete reprogramming of the submitted model. Therefore, all reanalyses considering the Health Canada–indicated population were conducted on an older version of the BIA model, updated with the sponsor’s new market share inputs for the overall population of patients who are AChR-Ab+.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19. For the full Health Canada–indicated population (i.e., adults with AChR-Ab+ gMG), CADTH reanalyses suggest that the reimbursement of efgartigimod alfa in combination with conventional therapy will be associated with a 3-year budgetary incremental cost of $378,513,999 when considering the drug plan perspective, or $294,458,021 when considering the health care system perspective. Of note, these analyses assume that the same number of patients would be eligible and receive efgartigimod alfa as if it was reimbursed according to the reimbursement request criteria. Results for the reimbursement request population (i.e., adults with AChR-Ab+ gMG whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs) were similar and only differed due to different market share assumptions for rituximab in the 2 populations.
Table 18: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total | |
|---|---|---|
Health Canada–indicated populationa,b | Reimbursement request populationc | |
Submitted base case | $218,539,976 | $218,539,976 |
CADTH reanalysis 1 — NIHB population revisions | $210,137,173 | $210,137,173 |
CADTH reanalysis 2 — Rituximab as a comparator | $219, 561,332 | $221,093,367 |
CADTH reanalysis 3 — 100% public reimbursement | $303,079,043 | $303,079,043 |
CADTH reanalysis 4a — no blood product costs | $278,850,243 | $278,850,243 |
CADTH reanalysis 4b — administration and revised blood product costs | $215,691,536 | $215,691,536 |
CADTH base case (drug plan perspective) | $378,513,999 | $378,137,376 |
CADTH combined reanalysis (health care system perspective) | $294,458,021 | $296,681,086 |
Ab+ = antibody seropositive; AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; BIA = budget impact analysis; gMG = generalized myasthenia gravis; NIHB = Non-Insured Health Benefits; NSIST = nonsteroidal immunosuppressive therapy.
aHealth Canada–indicated population: adult patients with AChR-Ab+ gMG.
bThe sponsor submitted a new BIA model in September 2023 which considered the Health Canada–indicated population of patients with gMG who are AChR-Ab+, but removed the flexibility of including rituximab as a comparator. This issue could be worked around for the reimbursement request population by substituting rituximab inputs into the scenario intended to represent the inclusion of ravulizumab. However, this workaround was not feasible for the Health Canada–indicated population without a complete reprogramming of the submitted model. Therefore, all reanalyses considering the Health Canada–indicated population were conducted on an older version of the BIA model, updated with the sponsor’s new market share inputs for the overall AChR-Ab+ population.
cReimbursement request population: adult patients with AChR-Ab+ gMG whose symptoms persist despite adequate treatment with AChEIs, corticosteroids, and/or NSISTs.
CADTH conducted additional scenario analyses (Table 19) to highlight the uncertainty associated with the potential budget impact. For both populations, and for the health care payer perspective, a scenario was conducted assuming IVIg would be used every 4 weeks. From the drug plan perspective, scenarios were conducted assuming the price reduction resulting from the CADTH economic evaluation reanalysis and reverting the proportion of patients who will be publicly reimbursed to 72% and, for the Health Canada–indicated population, assuming that the uptake of efgartigimod alfa could be up to 50% of all patients with AChR-Ab+ gMG by year 3 (15% and 30% assumed in years 1 and 2, respectively) if funded for its full indication.
Table 19: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Health Canada–Indicated population | ||||||
Submitted base case | Reference | $94,170,121 | $95,490,490 | $96,810,860 | $98,131,229 | $290,432,579 |
New drug | $94,170,121 | $144,577,587 | $173,782,019 | $190,612,949 | $508,972,555 | |
Budget impact | $0 | $49,087,097 | $76,971,159 | $92,481,720 | $218,539,976 | |
CADTH base case (drug plan perspective) | Reference | $11,825,523 | $11,992,422 | $12,159,320 | $12,326,218 | $36,477,959 |
New drug | $11,825,523 | $97,002,961 | $145,472,131 | $172,516,866 | $414,991,958 | |
Budget impact | $0 | $85,010,539 | $133,312,812 | $160,190,648 | $378,513,999 | |
CADTH combined reanalysis (health care system perspective) | Reference | $138,657,011 | $140,613,931 | $142,570,851 | $144,527,770 | $427,712,552 |
New drug | $138,657,011 | $206,746,320 | $246,279,106 | $269,145,146 | $722,170,572 | |
Budget impact | $0 | $66,132,389 | $103,708,256 | $124,617,376 | $294,458,021 | |
CADTH scenario 1: IVIg every 4 weeks (health care system perspective) | Reference | $116,979,928 | $118,630,911 | $120,281,894 | $121,932,876 | $360,845,681 |
New drug | $116,979,928 | $188,159,108 | $229,315,427 | $252,949,185 | $670,423,721 | |
Budget impact | $0 | $69,528,198 | $109,033,534 | $131,016,309 | $309,578,040 | |
CADTH scenario analysis 2: 84% price reduction (drug plan perspective) | Reference | $11,825,523 | $11,992,422 | $12,159,320 | $12,326,218 | $36,477,959 |
New drug | $11,825,523 | $25,546,740 | $33,415,087 | $37,867,463 | $96,829,290 | |
Budget impact | $0 | $13,554,318 | $21,255,768 | $25,541,245 | $60,351,331 | |
CADTH scenario 3: 72% public reimbursement (drug plan perspective) | Reference | $8,371,311 | $8,489,458 | $8,607,606 | $8,725,753 | $25,822,817 |
New drug | $8,371,311 | $68,668,582 | $102,980,000 | $122,125,020 | $293,773,602 | |
Budget impact | $0 | $60,179,123 | $94,372,395 | $113,399,267 | $267,950,785 | |
CADTH scenario 4: higher uptake of efgartigimod alfa | Reference | $11,825,523 | $11,992,422 | $12,159,320 | $12,326,218 | $36,477,959 |
New drug | $11,825,523 | $329,485,935 | $655,983,441 | $1,100,094,912 | $2,085,564,288 | |
Budget impact | $0 | $317,493,513 | $643,824,121 | $1,087,768,694 | $2,049,086,329 | |
Reimbursement request population | ||||||
Submitted key scenario | Reference | $80,249,366 | $81,374,551 | $82,499,736 | $83,624,921 | $247,499,208 |
New drug | $80,249,366 | $130,461,648 | $159,470,895 | $176,106,641 | $466,039,184 | |
Budget impact | $0 | $49,087,097 | $76,971,159 | $92,481,720 | $218,539,976 | |
CADTH key scenario (drug plan perspective) | Reference | $3,328,544 | $3,375,521 | $3,422,498 | $3,469,475 | $10,267,493 |
New drug | $3,328,544 | $88,301,474 | $136,602,662 | $163,500,733 | $388,404,869 | |
Budget impact | $0 | $84,925,953 | $133,180,165 | $160,031,258 | $378,137,376 | |
CADTH combined reanalysis (health care system perspective) | Reference | $114,679,395 | $116,297,910 | $117,916,424 | $119,534,938 | $353,749,272 |
New drug | $114,679,395 | $182,929,577 | $222,407,644 | $245,093,136 | $650,430,358 | |
Budget impact | $0 | $66,631,668 | $104,491,220 | $125,558,198 | $296,681,086 | |
CADTH Scenario 1: IVIg every 4 weeks (health care system perspective) | Reference | $95,629,850 | $96,979,511 | $98,329,172 | $99,678,833 | $294,987,516 |
New drug | $95,629,850 | $166,903,034 | $207,982,652 | $231,440,078 | $606,325,764 | |
Budget impact | $0 | $69,923,523 | $109,653,480 | $131,761,245 | $311,338,248 | |
CADTH scenario analysis 1: 84% price reduction (drug plan perspective) | Reference | $3,328,544 | $3,375,521 | $3,422,498 | $3,469,475 | $10,267,493 |
New drug | $3,328,544 | $16,845,253 | $24,545,619 | $28,851,329 | $70,242,201 | |
Budget impact | $0 | $13,469,732 | $21,123,121 | $25,381,855 | $59,974,708 | |
CADTH scenario 2: 72% public reimbursement (drug plan perspective) | Reference | $2,356,282 | $2,389,537 | $2,422,793 | $2,456,048 | $7,268,378 |
New drug | $2,356,282 | $62,508,782 | $96,701,286 | $115,742,482 | $274,952,551 | |
Budget impact | $0 | $60,119,245 | $94,278,494 | $113,286,434 | $267,684,173 | |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Muscular Dystrophy Canada
About Muscular Dystrophy Canada
Muscular Dystrophy Canada is registered with CADTH.
Muscular Dystrophy Canada (MDC) supports people affected by muscular dystrophies and related muscle diseases. Together, these rare conditions are referred to as “neuromuscular disorders.” Neuromuscular disorders are a group of diseases that weaken the body’s muscles. The causes, symptoms, age of onset, severity and progression vary depending on the exact diagnosis and the individual.
Since 1954, Muscular Dystrophy Canada has been the leading health charity and voice of the neuromuscular community in Canada. MDC is a sophisticated network of informed professionals, service specialists, and volunteers who deeply understand neuromuscular disorders. MDC represents 30,896 Canadians impacted by neuromuscular disorders including 12,047 persons with neuromuscular disorders, and 19,155 family members/caregivers.
MDC’s mission is to enhance the lives of those impacted by neuromuscular disorders by continually working to provide ongoing support and resources while relentlessly searching for a cure through well-funded research.
MDC has a full spectrum of programs, services, and supports for the thousands of Canadians of all ages living with a neuromuscular disorder that include systems navigation, education and knowledge translation, access to financial supports for critical life-changing equipment and services to improve quality of life, peer-to-peer networking, emotional support, evidence- based information for new treatments, medical advances, and clinical trials and advocacy. Plus, MDC invests in transformative research to work towards more answers, therapies, and hopefully, potential cures.
Funded by Canadians from coast to coast, our investment in the research community is advancing the development of important new treatments. Our programs and services play a critical role in informing and supporting members of the neuromuscular community by funding equipment to improve daily life; hosting family and caregiver retreats; providing emotional and educational support; and with providing access to vital resources and support systems. Our advocacy efforts focus on enhancing public policy at all levels of government to bring about positive change. We are currently working to bring new treatments and trials to Canada. Advances in medicine have resulted in individuals with neuromuscular disorders living longer but not necessarily living better. As their disorder progresses and changes, so do their needs and financial strains.
Our desire is to provide support through all stages of disease progression by providing the tools, resources and support individuals need to live a full and rich life.
At the MDC, we follow the principle Nothing About Us Without Us closely. Individuals with Myasthenia Gravis and their circle of support are actively involved in every aspect of our organization - from leadership and decision-making roles to serving on committees and participating in collaborative research efforts. By integrating the perspectives and experiences of those affected by Myasthenia Gravis, we strive to ensure that our efforts are aligned with the needs and priorities of the patient community.
Myasthenia gravis (MG) is one of the neuromuscular disorders that falls under MDC’s umbrella. There is expected to be approximately 10, 000 patients affected by MG in Canada.
MG is a rare and chronic autoimmune disease in which autoantibodies attack specific proteins in the neuromuscular junction, resulting in muscle weakness. Many patients develop generalized MG resulting in severe fatigable muscle weakness with difficulties in facial expression, speech, swallowing, and mobility.
Information Gathering
CADTH is interested in hearing from a wide range of patients and caregivers in this patient input submission. Describe how you gathered the perspectives: for example, by interviews, focus groups, or survey; personal experience; or a combination of these. Where possible, include when the data were gathered; if data were gathered in Canada or elsewhere; demographics of the respondents; and how many patients, caregivers, and individuals with experience with the drug in review contributed insights. We will use this background to better understand the context of the perspectives shared.
Muscular Dystrophy Canada has Neuromuscular Service Support Staff in all provinces across Canada. As part of the System Navigation Program, the Neuromuscular Service Support Staff provide front-line support to thousands of Canadians affected by neuromuscular disorders. The program operates on collaboration and patient engagement principles. Neuromuscular Service Support Staff work directly with patients and family members to identify non-medical needs (e.g., housing, transportation, access to equipment, information on clinical trials) and provide them access to the right resources in a personalized customized manner. Neuromuscular Service Support Staff work in partnership with patients and their families to address barriers, network and make connections with others in the community, share education materials and resources, enhance life skills and self-coping strategies, embrace inclusion and ultimately provide supports to help positively improve the overall well-being and quality of life of the patient and their family members.
The Neuromuscular Service Support Staff identified and contacted adults living with Myasthenia gravis to participate in a healthcare experience survey (available in English and French) and semi- structured virtual (phone, Zoom) interviews. We shared the survey with members by e-blasts, personalized invites and Canadian patient online groups (i.e., Canadian Snowflakes -Myasthenia Gravis Support Group).
MDC also conducted a Myasthenia Gravis Canadian Journey Mapping project during this period, where 1-hour interviews, roundtable sessions, surveys, health-related quality of life measures (i.e., EQ-VAS, EEQ-5D, MG-ADL, MG QOL) were completed.
The following submission reflects data from a total of 108 individuals impacted by MG, the majority of which have a confirmed diagnosis of generalized Myasthenia Gravis through clinical reports. The respondents included 39 males and 69 females between ages 21 to 76 from all provinces in Canada.
We sought the opinion on the value of having efgartigimod alfa (Vyvgart) approved for use in Canada for those affected by generalized Myasthenia Gravis. A qualitative descriptive approach, employing the technique of constant comparison, was used to produce a thematic analysis. We have included patients’ quotes to ensure their voices are captured in this report and to provide context for quantitative elements. A report capturing all patient comments is also available for review. We are also open to sharing the results of the Canadian Myasthenia Gravis Journey Mapping project.
Disease Experience
CADTH involves clinical experts in every review to explain disease progression and treatment goals. Here we are interested in understanding the illness from a patient’s perspective. Describe how the disease impacts patients’ and caregivers’ day-to-day life and quality of life. Are there any aspects of the illness that are more important to control than others?
We asked participants to describe how Myasthenia gravis affects their daily life and quality of life, as well as which aspects of the condition are more important to manage. Based on the responses, the survey identified 7 key themes that were frequently reported, listed in order of frequency: 1) significant impact on productivity, 2) significant impact on fatigue, energy levels and quality of sleep, 3) significant impact on respiratory health, 4) significant impact on mobility and strength, 5) significant impact on independence, 6) significant impact on relationships and social participation, 7) significant impact on eyes/vision and speech and swallowing.
Individuals affected by Myasthenia gravis conveyed through their quotes that the impact of MG extends beyond physical symptoms, and that it affects their mental health, quality of life, and the wellbeing of their families.
Significant Impact on Productivity (at Work and Home)
“I am unable to work, need to rest frequently, need help with activities like washing my hair, etc.”
“I had to retire from my job because of MG.”
“I retired because the stress of my job plus MG did not mix well.”
“I have worked while experiencing a MG crisis – it was horrible. I am fearful for the next crisis.”
“I am on disability leave because of my MG.”
“I am no longer able to work and rely fully on my husband for my meals, clean home and being moved from one place to another.”
“I wasn’t able to work today because I was very tired. It gets in the way of my ability to work. And this impacts my finances in a huge way.”
“I am not able to do the work I was once able to because I can’t strain my eyes and read for more than 20 minutes.”
“I feel useless at home. Everything now falls on my husband. Taking care of the children, cleaning, cooking and taking care of everything that revolves around IVIG treatment. MG is unreliable and my ability to support is unreliable.”
“I had to move to part time and modified work.”
“I can walk into the office but might need to be carried out by my partner. I no longer feel productive or competent at work.”
“MG forced me into retiring earlier than I would have otherwise.”
“I can no longer work. This impacts my finances and how I spend money.”
Significant Impact on Fatigue/Energy Levels, and Quality of Sleep
“I am unable to do anything without feeling tired.”
“I can do one task and then need a break.”
“Think of the spoon theory: In the theory, each spoon represents a finite unit of energy. Healthy people may have an unlimited supply of spoons, but people with MG have to think carefully and plan ahead on where to spend their energy to just to get through the day.”
“I experience fatigue daily at some point, usually in the evening.”
“I get very tired from even a conversation or reading an article.”
“If I overdo things I will need to rest. It could hit me the same day or the next day. I may be fatigued a good part of the day.”
“If I do too much, I know I will pay for it the next day. I will need a full day to recover. Imagine how you feel after travelling on different time zones, this is me every time I do something as simple as going to get groceries or cleaning my home.”
“I tire very easily. Do 10 minutes of housework then have to rest. Some days l can do this and some days I can’t do anything.”
“I typically require a 15- or 20-minute rest after having a shower!”
“Most days I have to sleep for a couple of hours in the afternoon due to fatigue.”
From our MG Canadian Journey Mapping project, we used photo voice methodology. One of the participants shared:
“I use this metaphor of “I feel like Cinderella” or I have to do X, Y, Z before I turn into a pumpkin. Daily, I guess this is what I feel. All of us share this experience that you only have so many things you can do in a particular day and particularly any task that exacerbates MG symptoms. Pretty much every day, if I wake up feeling quite good, I realize, like Cinderella at midnight the coach turns into the pumpkin and that was it. She has to go home and resume a sort of oppressed life. I’m not suggesting mine is oppression, but at a certain point in the day when I run out of energy and the MG starts to flare up, I have to shut down whatever plans I have or whatever I’m doing and just be able to rest and not make things worse so that’s new.”
Significant Impact on Respiratory Health
“The most bothersome aspect of MG is definitely the impact on breathing.”
“I can feel my lungs are weaker because of MG.”
“I am immunosuppressed because of the MG drugs, and I feel weak respiratory wise. The combination of the two is awful.”
“It is scary how difficult it is to breathe sometimes and that’s why I have a ventilator nearby.”
“I had to go on a ventilator in ICU three times now because of MG crises.”
“Choking on food or saliva interferes with breathing as diaphragm muscles become weak.” “Breathing is most affected, limiting my ability to walk, climb stairs, or bend over to tie my shoes.”
“I have terrible shortness of breath.”
Significant Impact on Mobility & Strength
“My legs are weak. By the time I get to the top of the stairs, I have to drag my legs up to the top of the stairs.”
“I have lost the ability to walk without support.”
“Sometimes I can walk into the grocery store but will need to carry out by a family member or use a wheelchair on the way out.”
“I used to walk around the neighbourhood after dinner, now I can’t even walk inside my house.”
“I can walk short distances but always keep a walker or cane nearby because you never know when the MG will flare up or when I will turn into a rag doll.”
“I can't walk without a walker, I can't stand for any length of time, can't sleep at night because it aches.”
“I can’t do stairs anymore. We had to remodel our entryway because it was becoming increasingly difficult to get inside the house on my own.”
“Some days it is difficult to just walk. Muscles seem be tense and not allowing me to do things.”
“I cannot walk down the street without falling. I cannot hold up a blow dryer to dry my hair.”
Significant Impact on Independence, and Social Participation
“I am unable to drive because of the weakness in my eyes and generalized weakness.”
“MG has taken a huge toll on my relationships and on my ability to carry activities out on my own. I rely on my partner for many reasons and there is guilt.”
“Standing to cook or do dishes takes 3x longer. On bad/weak days I feel like a prisoner in my own house.”
“We are not able to travel because heat bothers me, stress is a trigger for MG.”
“I have symptoms every day. Difficulty completing activities of daily living. No longer able to work. Can only drive short distances. I miss out on socializing due to mobility and fatigued.”
“I need to take Mestinon daily and have to try hard to avoid a Myasthenia flare up. Prior to diagnosis, I have spent long weeks and even months quite disabled and dependent on others for care. It affects social life, professional life, and all areas of my life.”
“I feel like I can’t be left alone – I feel I am on the verge of the next myasthenic crisis.”
“Not able to do dishes and laundry and everyday normal tasks. some days it's not bad and then it will go for a couple of days and then it will flare up.”
“Not being able to do anything with others. I can't get in a vehicle and can't lift my legs. I can't go out to see other people. I did have a scooter, but it got burned up and I don't have a scooter anymore.”
“Visiting with friends and family tire me out. Can’t get to church. Can’t go to play darts. It is very depressing knowing that there is no cure and that this is my way of life now.”
“I am very restricted in my abilities and require assistance. Loss of independence, social interaction and employment.”
“Loss of independence is awful. I can’t do activities on spur of moment, have to be carefully planned and at times have to decline, have had to drop out of some activities.”
“It has tremendously affected my independence. I cannot drive. They took my license away. I don't have a scooter. Not being able to be with my friends or anything. The only way to see people is for them to come to me.”
“I am a very independent person and now I am scared to be alone for long periods of time.”
“I can't drive at night or for long periods, I can't clean my own house, I can't cook for long periods of time, etc.”
“I have to have someone drive me to any appointments out of time. I also have to have help with some activities for daily living.”
“I have to ask my husband to puree my foods and brush my hair. I have lost complete independence and that is the most bothersome aspect of MG.”
Significant Impact on Eyes, Speech, and Swallowing
“I have to puree my foods and chug it down.”
“I tend to choke on my own saliva and food.”
“I had slurred speech as though I was intoxicated.”
“My voice gives out on me and sounds very strained after a short conversation sometimes.”
“Not being able to speak is very hard.”
“The ability to swallow and have my facial muscles work properly is very important as it affects my daily life at work. When they don’t, it’s very frustrating because you cannot take too much Mestinon to correct it. It’s time released and dosage is every 4 hours.”
“Choking on food or saliva interferes with breathing as diaphragm muscles become weak.”
“I think people not understanding or even knowing about it as it’s one of those invisible illnesses. I’m not in a wheelchair and outwardly appear to be “fine”, but it’s what’s going on inside is something only I know unless I start slurring my speech. When that happens, people who don’t know me or anything about me having MG, might think I’m intoxicated.
“Breathing is often affected, limiting my ability to walk, climb stairs, or bend over to tie my shoes.”
“One of the first symptoms of MG is a defect in eyesight and then a weakness in muscle, if one is lucky and MG is diagnosed at an early age (I was 24 and I think that I was fortunate that I had knowledgeable physicians) that the shock of the diagnosis is easier to accept.”
“It affects my eyes the most.”
“I was diagnosed 34 years ago with MG and have been on Mestinon for the whole time. I do get fatigued when I am in a situation that requires a lot of speaking (work, meetings) in my mouth, face, throat and eyes. I have to get lots of rest and prepare ahead of time with my Mestinon so it will be controlled.”
“I frequently go cross eyed.”
“Ptosis, difficulty chewing and swallowing. Multiple acute hospitalizations.”
“Double vision interferes with reading.”
“I have double vision and just could not do ordinary everyday things that others take for granted, like drive myself for coffee!”
“Double vision is the most bothersome as it affects my ability to drive, read, etc.”
Experiences With Currently Available Treatments
CADTH examines the clinical benefit and cost-effectiveness of new drugs compared with currently available treatments. We can use this information to evaluate how well the drug under review might address gaps if current therapies fall short for patients and caregivers.
Describe how well patients and caregivers are managing their illnesses with currently available treatments (please specify treatments). Consider benefits seen, and side effects experienced and their management. Also consider any difficulties accessing treatment (cost, travel to clinic, time off work) and receiving treatment (swallowing pills, infusion lines).
When MDC asked how MG is being managed with available treatments or therapies, three main themes emerged in response: negative experiences with steroids/prednisone, the slow onset of medication effects, and a feeling of trial and error with medications. People affected by MG reported that while supportive treatments have had positive health outcomes, there are concerns about the long-term and sustained benefits of these treatments, as highlighted by quotes from individuals.
Negative Experience With Prednisone
“The first thing the neurologist put me on was prednisone. But it was also the first thing I keep asking to be tapered off of. The side effects are awful.”
“I was placed on prednisone straight away, but the moodiness and weight gain killed me.”
“If I don't take my medication, I'd be dead. No side effects except I was on prednisone and getting depressed and putting on weight. It's a bad pill to be on so the doctor cut it back.”
“Prednisone helped MG a bit at first, but I don’t know how helpful it is now.”
“I am on prednisone, but I don't like it. I can't afford it and have to choose between food and medication, and it causes diabetes which is my main concern.”
“I have been on prednisone for four years. It took several rounds of IVIGs waiting for Mycophenolate to work.”
“I was put on prednisone increased to 50 mg. Not helping mg. Put my blood sugars out of wak. So had to go on insulin. That gave me neuropathy with nerve pain and numbness. Put on cellcept 500 mg 2 times a day while slowly decreasing prednisone. And increased cellcept to 750. So now I am taking mestinon 30 mg 3 times a day, cellcept 750 2times a day, and prednisone 2.5 mg every other day. My swallowing is somewhat better as it doesn’t happen as often. I still get cross eyed and still get tired easily.”
Conventional Treatments Take a Long Time to Take Effect
“My doctor told me it could take 6 maybe even 9 months for the treatment to take effect.”
“Imagine living half the year waiting for a drug to show benefit and then to find out you need to be switched to something else.”
“I was told it would take a while for the benefits to kick in.”
“My whole life revolves around MG. I feel the effects of lack of IVIG close to the end of the month. Then I am knocked for a day or two after IVIG. It takes effect but loses its effect by the third week.”
Experience with Trial and Error
“Treatments have been many and honestly too overwhelming. It’s a guessing game i.e., trial and error. Seronegative patients not eligible for any of the advanced treatments. Not fair. I get IVIG which means I am stuck at the hospital for up to 7 hours 2 days every 3 weeks. Immune suppressants caused frequent infections and pneumonia; prednisone caused a vascular necrosis in both hips resulting in fractures.”
“It feels like I am put on a drug only to see if I will fail it or it will work enough to stop me from complaining.”
“I have been tried on so many drugs and so many different dosages. It feels like a big game of trial and error – which is not how you want to feel about your treatment plan.”
“I want to be given Rituxan, but my doctor says I am not worse enough for that… I want to be given a chance to try a different treatment.”
“It feels like the treatments I have tried only half address or control MG. And so, then I am tried on something else. Another line of treatment.”
Experience With IVIG
“IVIG is really the one thing that worked for me.”
“Prednisone- huge negative psychological symptoms with psychosis Azathioprine- not effective IVIG- my savior. Every two weeks.”
“Standard treatments such as IVIG has helped. Equally important are the dietary, relaxation, exercise and physio routines I practice daily.”
“Mestinon — does not seem to have an effect on my symptoms. Azathioprine — started taking in January of 2022. I don’t think it’s made any improvement for me. IVIG — used last Christmas at the time of my diagnosis because my symptoms were mostly bulbar, and neurologist was concerned that I could be headed towards crisis. IVIG worked for me, and I felt so much better… for a couple of weeks Also used before my thymectomy to make sure that I was as strong as possible before surgery.”
Improved Outcomes
CADTH is interested in patients’ views on what outcomes we should consider when evaluating new therapies. What improvements would patients and caregivers like to see in a new treatment that is not achieved in currently available treatments? How might daily life and quality of life for patients, caregivers, and families be different if the new treatment provided those desired improvements? What trade-offs do patients, families, and caregivers consider when choosing therapy?
Patients identified three aspects of MG that they want better controlled, these included: decreased intensity of exacerbations and side effects, maintenance of independence, and less serious hospital admissions. Patients stated that they would be willing to deal with side effects of medications if these aspects of MG were better controlled. Patients stated that current medications seem to be decreasing the number of exacerbations but not the impact on overall quality of life.
Desires for treatment include:
“A treatment that helps me to work, to care for my children, to help around the house.”
“Target treatment for MG would be something that would stop the myasthenic crises.”
“A treatment that address the respiratory and general weakness would be important.”
“Sometimes I have trouble swallowing the pills and they get stuck in my throat. I would love them to be more of a capsule that floats when you swallow rather than a chalky pill. If it gets stuck it’s awful because it starts to dissolve, and the taste is just awful!”
“Something that could take away the aches and the pain all over especially in my legs. I can't sleep in a bed. I sleep in a remote-controlled chair.”
“I would like a treatment that does not lead to diabetes.”
“Target treatment for MG instead of general immunosuppression.”
“I would like to see more options available. I would also like to see costs of infusions to be lowered.”
“I would like a drug that is convenient to take, takes effect quickly and so benefits are observed – like Mestinon and IVIG but doesn’t have that low period that requires recovery.”
“I would love to have a drug that I could take once a day in the morning and that could be time released over 24 hours. Right now, I have to make sure I take my Mestinon 30 minutes prior to eating and that can be tricky sometimes to schedule when I’m not in control of that or at work.”
“A treatment that lasts long and doesn’t take so long to work.”
“I would like a treatment that addresses all symptoms without creating side effects that are sometimes worse than the symptoms would certainly be nice, though. Remission for all.”
“More muscle strength and stamina. Would love to be able to go for a walk.”
“I would like for there to be treatments that don't cause other serious problems like compromised immunity, cancer, etc.”
“Less side effects, something that would improve quality of life and regain our independence.”
When patients, families, and caregivers evaluate different therapies, they take into account factors such as how the treatment is delivered, potential side effects, duration and frequency of treatments, convenience (e.g., travel time and parking for clinic visits), and financial impact (costs). It was consistently found that therapies with low invasiveness, minimal hospital visits, low risk of side effects, and low cost were highly valued. Patients appreciated treatments that could be administered outside of the hospital (i.e., at home or with community health resources), allowing them to have more control and flexibility. Patients not only valued but which symptoms a drug addressed/managed and how few side effects there were. Health related quality of life was noted as a key priority vs. convenience of a drug. Participants mentioned they travelled 13 hours at times for specialist care or IVIG or plasma exchange and so they will move mountains as long as the treatment is right and will help with minimizing the negative experiences that come with MG. If families were considering switching to a different therapy, they would weigh the potential side effects of the new therapy against those of the current therapy. They would also consider the ease of access to the treatment and whether it was covered by private or provincial insurance.
Experience With Drug Under Review
CADTH will carefully review the relevant scientific literature and clinical studies. We would like to hear from patients about their individual experiences with the new drug. This can help reviewers better understand how the drug under review meets the needs and preferences of patients, caregivers, and families.
How did patients have access to the drug under review (for example, clinical trials, private insurance)? Compared to any previous therapies’ patients have used, what were the benefits experienced? What were the disadvantages? How did the benefits and disadvantages impact the lives of patients, caregivers, and families? Consider side effects and if they were tolerated or how they were managed. Was the drug easier to use than previous therapies? If so, how? Are there subgroups of patients within this disease state for whom this drug is particularly helpful? In what ways? If applicable, please provide the sequencing of therapies that patients would have used prior to and after in relation to the new drug under review. Please also include a summary statement of the key values that are important to patients and caregivers with respect to the drug under review.
Only one adult indicated they received the drug under review as part of the clinical trial. In short, the individuals shared:
“Vyvgart completely replaced the need for IVIG for me. It took much quicker to see the impact of the drug, and infusion time was less than I expected. I also didn’t feel as if I needed to go in frequently to get the next dose. Side effects were not as bad as some of the other drugs I have taken for MG, but diarrhea was a problem – this happened with a few other drugs as well where I was not able to leave my house because of fear of needing the bathroom constantly. But the diarrhea was manageable after the first cycle and my doctor mentioned I was tolerating the drug very well.”
Companion Diagnostic Test
If the drug in review has a companion diagnostic, please comment. Companion diagnostics are laboratory tests that provide information essential for the safe and effective use of particular therapeutic drugs. They work by detecting specific biomarkers that predict more favourable responses to certain drugs. In practice, companion diagnostics can identify patients who are likely to benefit or experience harms from particular therapies or monitor clinical responses to optimally guide treatment adjustments.
What are patient and caregiver experiences with the biomarker testing (companion diagnostic) associated with regarding the drug under review?
Consider:
Access to testing: for example, proximity to testing facility, availability of appointment.
Testing: for example, how was the test done? Did testing delay the treatment from beginning? Were there any adverse effects associated with testing?
Cost of testing: Who paid for testing? If the cost was out of pocket, what was the impact of having to pay? Were there travel costs involved?
How patients and caregivers feel about testing: for example, understanding why the test happened, coping with anxiety while waiting for the test result, uncertainty about making a decision given the test result.
100% reported that they did have diagnostic testing completed with at least a blood test; but many also had single fiber electromyography to confirm diagnosis. 80% of respondents reported significant difficulty getting diagnosed. Early findings of the Canadian MG Journey Mapping project indicate 7 years from time of first bothersome symptom to diagnosis, with the range up to 23 years. The vast majority found it to be a cost-effective but lengthy process with many missed opportunities. They noted significant diagnostic odyssey - delays, misdiagnoses and costs incurred. For those who received a diagnosis as part of a crisis or medical event/hospitalization, the diagnosis was reported as smooth (25%). Below are quotes that further highlight the experiences of patients and caregivers with the testing:
Dismissive
“I was worked up for a stroke and Bell’s palsy... and when it wasn’t either of those, I was told to go home. That was the end of testing.”
“I went to the ER five times before I was seen by neurology.”
“I was told my results didn’t show anything. Then they called me and said maybe there is something, it might be artifact. Turns out I have a rare form of MG.”
“I have sero negative MG and was dismissed many times because the results did not match up with what the doctor expected for typical MG.”
Easy/Smooth Experience with Testing
“There were many tests.”
“Easy access to testing. I had headaches from the testing, and it started with a twitch with my left eye. My doctor sent me to the hospital and the doctors confirmed I had MG. It was covered by the province (Ontario). I had to pay for gas to go to the hospital.”
“OHIP covered the cost of the testing. But getting to the right test was a mission.”
“It took a bit of time and a few visits to my GP, walk-in clinics and ER departments before we came up with the possibility of MG. Eventually, my GP ordered one simple blood test that showed that I am ACHR+. He then sent out a referral request for a neurologist.”
“I was rushed to hospital because I couldn't breathe. I had just had a triple bypass and valve replaced two weeks prior was only five minutes away from hospital and diagnosed within the hour of arrival I believe they did blood tests. OHIP payed then but now living in BC. treatment started right away.”
Delayed Diagnosis
“I visited 3 doctors in two different countries before getting properly diagnosed. It took 4 years.”
“I had to pay for the blood test which gets sent to the University of British Columbia. I was tested for the generalized form of MG which I do not have. It was ruled out and I was told I do not have MG. Because I was not tested for MuSK MG, I was hospitalized for 3 months. The blood test that was $50.00 was missed and so the hospital stay was very costly to the system.”
“I went to the doctor and then was sent on a huge runaround of doctors. I was sent to a dentist as they thought it was TMJ! After a few months I finally was sent to a neurologist, but he wouldn’t even consider me because I was only 24. My family doctor was amazing and even sat with me in his office and had me describe my symptoms while he flipped through his medical book. He was the one who thought I had MG. Finally, after a few months of my above symptoms happening to me daily, I woke up one morning and I could not swallow my own saliva! I sounded drunk, couldn’t speak, move my tongue, etc. that I headed to Emergency, and they dealt with it asap. There they tested me with the tension test and came to a diagnosis.”
“I have had a very hard time getting diagnosed. There has not been agreement among the physicians who have assessed me. Some say I have MuSK MG based on clinical assessment and also positive MuSK antibodies. The physician who I was sent to did not believe I have MG because I did not have a positive SFEMG. She did not believe my symptoms were caused by MG and she did not consider my antibodies for MuSK relevant at all. It has been in reliably frustrating dealing with physicians like her. I am very relieved to have a neurologist now who understands there can be quite a diversity in MG presentations.”
“It took almost 2 years after that to finally get positive blood results. All genetic testing and muscle biopsy done in that time.”
Costs Related to Diagnosis
“Testing done through academic centre so no cost to me.”
“Pretty much right after that, I was scheduled for a thymectomy within a month and put on Mestinon. In Canada, there was no payment for the surgery, but the drugs were expensive. Fortunately, I had good benefits coverage at work. I haven’t had coverage for the past 8 years so that’s out of pocket for me and costly. Mestinon monthly is about $125 which is not a lot I realize but it is on top of everything else. It’s another expense for sure but a vital one.”
“Was tested at one neurologist who sent me for bloodwork at a cost of $145.00. He then sent me to a neuromuscular specialist who was 60 miles away. He tested me and had more blood work done. The bloodwork was sent from Toronto to Vancouver. It took 4 months for the results. Then Covid came along and had difficulty getting a follow up appointment. So, after 18 months I was diagnosed with generalized mg. Yes, I had to pay for travelling and parking. It was an extra cost out of my budget.”
“I saw one neurologist who sent me for a blood test because he thought I may have myasthenia gravis, but he said it came back negative. He thought I may have had a stroke. That was negative. I had double vision, weak muscles, I couldn’t without falling down, so I was covered in bruises. The doctor more or less told me it was all in my head. I was falling at work, I was falling downstairs, and I had to lift my leg from the gas pedal in my car to the break with my hand because it would not move by itself. I went back to see this doctor and showed him how I was covered in bruises, and he sent me to see someone at the University Hospital. There the Neurologist give me a test, and the doctor said you have MG. All in all, it took two years. Most frustrating.”
Anything Else?
Is there anything else specifically related to this drug review that CADTH reviewers or the expert committee should know?
“CDEC should know that just because there are drugs approved for generalized MG, we are not good. We are not cured. We need better treatment options and paths for improvement. This drug has potential to cut back on time for infusion and to show positive effects, especially on mobility and energy. We desperately need you to vote positively in favour of this drug. We sit on online forums and groups with those in the US and are envious that they have access and are doing very well with Vyvgart – but it is out of reach for us. It is time to do the right thing.”
Conflict of Interest Declaration — Muscular Dystrophy Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Muscular Dystrophy Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
argenx | — | — | — | X* |
*$58, 720 – all for educational initiatives that did not involve the company at all.
Clinician Input
Neuromuscular Disease Network for Canada
About Neuromuscular Disease Network for Canada
The Neuromuscular Disease Network for Canada (NMD4C) is a new pan-Canadian network that brings together the country’s leading clinical, scientific, technical, and patient expertise to improve care, research, and collaboration in neuromuscular disease. https://neuromuscularnetwork.ca/.
Launched in January 2020 with funding from the Canadian Institutes of Health Research (CIHR) and Muscular Dystrophy Canada (MDC), NMD4C builds on existing national initiatives such as the Canadian Neuromuscular Disease Registry (CNDR), the Canadian Pediatric Neuromuscular Group (CPNG), and the former neuromuscular network CAN-NMD. The mission of NMD4C is to improve the care, research, and treatment of NMDs for all Canadians. Its vision is to be a comprehensive, inclusive, open and enduring network through which Canadian stakeholders can share expertise and data and collaborate on joint activities and research for the benefit of Canadian patients.
The network’s goals are to:
Formalize and sustain a network of NMD stakeholders united around a cohesive three-year work plan;
Train and educate the next generation of NMD stakeholders (clinicians, scientists, and patient advocates);
Raise the standard of care for NMD and access to therapies across Canada;
Strengthen biomedical and clinical infrastructure to build research capacity in Canada.
Since its inception, NMD4C has grown to more than 500 members with the majority having expertise in neurology and physical medicine and rehabilitation. NMD4C provides leadership and evidence-based support to improve access to approved novel treatments. We published a Canadian guidance on gene replacement therapy in spinal muscular atrophy (SMA), provided guidance on NMD respiratory care and vaccination during the COVID pandemic, and developed a variety of knowledge translation products.
As NMD4C members and neuromuscular clinicians across Canada with significant clinical expertise in the management of patients with generalized myasthenia gravis, we are writing to offer our strong support for favorable benefit access for efgartigimod alfa as a treatment option in Canada.
Information Gathering
Clinicians with experience treating generalized Myasthenia Gravis, including clinicians with experience with ravulizumab, eculizumab and efgartigimod alfa and standards of care for gMG were asked to contribute to this submission. These expert clinicians contribute to the knowledge of gMG and its treatments and are involved in clinical and observational research, clinical guidelines development and health technology assessment. The clinicians contributing herein are familiar with the data from clinical trials on treatments for gMG, and, specifically, for efgartigimod alfa. The information presented in this submission was gathered from 1:1 discussion with lead author, Dr. Vera Bril, and group discussions.
Current Treatments and Treatment Goals
Fundamentally, the gMG treatment goal is to achieve a complete remission, pharmacological remission or minimal manifestation status (i.e., asymptomatic or no disease-related functional limitation) with minimal adverse events (AEs) (Lascano et al., 2021, Alhaidar et al., 2022).
It is noted that conventional treatment options for gMG have been based on symptomatic therapy (e.g., acetylcholinesterase inhibitors), short-term rescue immunotherapy (e.g., plasma exchange and intravenous immunoglobulins) and long-term immunosuppressive therapy (e.g., corticosteroids and nonsteroidal immunosuppressants) (Menon and Bril 2022, Lascano et al., 2021 and Habib et al., 2020).
Further, non-specific immunosuppressants, such as corticosteroids, azathioprine, cyclosporine, mycophenolate and tacrolimus, have been only partially effective in controlling disease symptoms; however, many patients fail to attain a complete or stable remission, with 10–20% of patients not responding or intolerant to these agents (Alhaidar et al., 2022, Vanoli et al. 2022).
Moreover, these agents may take several weeks to many months to be effective and are frequently associated with burdensome and intolerable side effects (Alhaidar et al., 2022, Vanoli et al., 2022).
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
We would like to emphasize that standard treatments for gMG are often transiently effective, may require long treatment periods for benefits to be observed, may have side-effects and may not be effective for all patients.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The pivotal trial evidence indicates that efgartigimod alfa addresses the underlying cause of myasthenia gravis by decreasing circulating pathogenic antibody and will do so with minimal side effects and without significant immunosuppression.
Specifically, treatment with efgartigimod alfa showed significant decreases in disease symptoms that were consistent across 2 MG-specific scales. Clinically and statistically significant improvements in function (MG-ADL responders, 67.7%) and strength (QMG responders, 63.1%) were observed in efgartigimod alfa treated AChR-Ab+ patients compared to placebo (29.7% and 14.1%, respectively) (Howard et al 2021). The clinical response was also established quickly with over 50% patients of MG-ADL responders having an onset of effect by week 1with nearly 85% achieving an MG-ADL response by week 2 (Howard et al., 2021). Treatment response was repeatable with additional cycles of treatment. This positive response was seen in patients who were poorly controlled although 70-80% were on steroids and about ½ were on steroids plus non-steroidal immunosuppressants at baseline indicating failure of standard therapies. At the completion of only two treatment cycles, 78.5% of patients achieved MG-ADL responder status (Howard et al., 2021). These were MG patients with baseline burdensome disease (ADL of 9 and QMGS of 16) despite other therapies. A not insignificant proportion of patients (40%) achieved minimum symptom expression (equivalent to MG-ADL of 0 or 1) which clearly is an impressive outcome for gMG patients and has the potential to improve the standard of care for Canadian patients affected with gMG. In terms of safety, efgartigimod alfa was well tolerated, and there were no infusion-related reactions.
Another unmet need in our practices is related to seronegative patients. Although efgartigimod alfa improved MG-ADL, QMG and MSE in seronegative patients if evaluated using a combined function and strength scale, a larger study is needed to validate the findings. Given the lack of therapeutic alternatives, efgartigimod alfa may be considered as treatment in this population.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review? Which patients are most likely to respond to treatment with drug under review?
Those most likely to respond are those with acetylcholine receptor antibodies in their system. Those with MuSK antibodies might respond as might those who are double seronegative.
Which patients are most in need of an intervention?
Those needing intervention most are those getting worse quickly and therefore need a therapy that works quickly such as efgartigimod alfa (within 1-2 weeks in most patients). Of particular concern are those who may develop MG crisis rapidly.
Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
MG is so variable that prediction of rapid worsening is difficult. Those patients who have symptoms restricted to only ocular muscles are unlikely to require such rapid intervention with this therapy.
How would patients best suited for treatment with drug under review be identified (e.g., clinician examination/judgement, laboratory tests (specify), diagnostic tools (specify))?
The patients best suited for treatment would be identified by clinician examination/judgement supplemented by assessment of MG activities of daily living and other scales that reflect severity of disease such as the quantitative myasthenia gravis score, the MG Impairment Index, and the single simple question. If not available, then antibody testing needs to be done, but can be delayed.
Are there any issues related to diagnosis?
The diagnosis in those who are double-seronegative is less understood. Cluster antibodies to both acetylcholine and MuSK may be present but need to be tested for specifically and this can take weeks.
Is a companion diagnostic test required?
The clinical scales as indicated above are used to assess disease severity.
Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)?
It is absolutely likely that under diagnosis occurs in clinical practice. Appropriate investigations including serological testing as well as single fibre EMG and repetitive nerve stimulation studies are necessary in evaluating the patients.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with drug under review?
There is nothing clear at this point that predicts those likely to respond other than the presence of acetylcholine receptor antibodies.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed? Are outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Many people use MG ADL today to assess their patients and this should be used if efgartigimod alfa is administered. Also, other scales are used such as the QMGS, MGII, SSQ. So, the MG ADL would be the minimum required to assess the patients. The response has to be assessed depending on the severity of MG in the patient; so, at 2 weeks would be required in most and then at 4 weeks, and after that directed by the patient’s status. Mahy of the scales are reliably tested using virtual means so in person clinic visits are not necessarily required and facilitate frequent and rapid assessment.
What would be considered a clinically meaningful response to treatment? Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
The clinically meaningful response to treatment should follow those used in the clinical trials such as 2 or more points on the ADL or 3 or more points on the QMGS. Of course, how much this matters in an individual patient will be determined by their starting severity and many will improve more than this. The ADL measures those activities of the patient on a daily basis. The QMGS is an assessment of impairments. The MGII measures impairments including the element of fatigue and is mostly patient reported. The SSQ is an overall gestalt of how the patient feels with respect to this MG and levels above 72% indicate general satisfaction with their state. Different physicians may use different scales, but one would expect some uniformity in the changes in the scale needed to assess response.
What factors should be considered when deciding to discontinue treatment with the drug under review?
The lack of response to treatment should lead the clinician to discontinue treatment and the way to measure response are listed above. If treatment is started, then the interval to measure response should be a minimum of 2 weeks if the patient is not doing well. So, for example, there needs to be time for the response to work similar to if intravenous immunoglobulin is administered. It should be noted that some patients need 2 cycles of therapy to show a response. (Howard et al., 2021).
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
It is generally accepted that that IVIg and SCIg treatments are effective in MG patients are utilized in certain clinical settings, but Ig treatment places significant burden on the Canadian health care system and that supplies can be at risk periodically (such as in the pandemic). In consideration of the trial evidence, efgartigimod alfa is an excellent treatment option for patients who are candidates for or are intolerant to IVIg or SCIg therapy. We think that FcR inhibitors, such as efgartigimod alfa, are likely to replace Ig therapies.
Additional Information
In closing we strongly endorse access to efgartigimod alfa as a treatment option in Canada. We thank CADTH for the opportunity to provide clinician input on the efgartigimod alfa submission. My colleagues and I would be pleased to provide additional information and/or clarification.
Conflict of Interest Declarations — Neuromuscular Disease Network for Canada
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Vera Bril
Position: Professor of Neurology
Date: May 3, 2023
Table 2: COI Declaration for Neuromuscular Disease Network for Canada — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Grifols | — | X | — | — |
CSL | — | — | — | X |
UCB | — | — | — | X |
argenx | — | — | — | X |
Takeda | — | — | — | X |
Alnylam | — | — | X | — |
Octapharma | — | — | — | X |
Akcea | — | — | — | X |
Ionis | — | — | — | X |
Sanofi | X | — | — | — |
Momenta (J&J) | — | — | — | X |
Roche | X | — | — | — |
Janssen | — | — | X | — |
AZ-Alexion | — | — | — | X |
Novo-Nordisk | X | — | — | — |
Immunovant | — | — | — | X |
Japan Tobacco | X | — | — | — |
Declaration for Clinician 2
Name: Catherine Elizabeth Pringle
Position: Associate Professor (Neurology), University of Ottawa
Date: May 10, 2023
Table 3: COI Declaration for Neuromuscular Disease Network for Canada — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
argenx | — | — | X | — |
Declaration for Clinician 3
Name: Hans Katzberg
Position: Associate Professor of Medicine, University of Toronto
Date: May 11, 2023
Table 4: COI Declaration for Neuromuscular Disease Network for Canada — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Akcea | X | — | — | — |
Alnylam | X | — | — | — |
UCB | — | — | X | — |
CSL Behring | — | X | — | — |
Alexion | — | X | — | — |
argenx | X | — | — | — |
Octapharma | X | — | — | — |
Roche | X | — | — | — |
Merz | — | X | — | — |
Dyne | X | — | — | — |
Terumo | X | — | — | — |
Declaration for Clinician 4
Name: Dubravka Dodig, MD, FRCP C
Position: Neuromuscular Neurologist
Date: 11-05-2023
Table 5: COI Declaration for Neuromuscular Disease Network for Canada — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
argenx | — | X | — | — |
Alnylam | X | — | — | — |
Akcea | X | — | — | — |
Sanofi | — | — | — | X |
AZ-Alexion | — | — | X | — |
Declaration for Clinician 5
Name: Angela Genge
Position: Neurologist
Date: 15-05-2023
Table 6: COI Declaration for Neuromuscular Disease Network for Canada — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Quralis | — | — | — | X |
AL-S Pharma | — | — | — | X |
AZ Alexion | — | X | — | — |
Amylyx | — | — | X | — |
MTPA | — | X | — | — |
Sanofi | X | — | — | — |
UCB | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.