CADTH Reimbursement Recommendation
Efgartigimod Alfa (Vyvgart)
Indication: For the treatment of adult patients with generalized myasthenia gravis who are anti-acetylcholine receptor antibody positive
Sponsor: argenx Canada Inc.
Final recommendation: Reimburse with conditions
Recommendation
Summary
What Is the CADTH Reimbursement Recommendation for Vyvgart?
CADTH recommends that Vyvgart be reimbursed by public drug plans for the treatment of adult patients with generalized myasthenia gravis (gMG) if certain conditions are met.
Which Patients Are Eligible for Coverage?
Vyvgart should only be covered to treat patients who have a diagnosis of class II to IV gMG based on the Myasthenia Gravis Foundation of America (MGFA) system, tested positive for anti–acetylcholine receptor (AChR) antibodies, and have a Myasthenia Gravis Activities of Daily Living (MG-ADL) scale score of at least 5. Vyvgart should only be covered to treat patients if their symptoms persist despite a stable dose of conventional therapy with acetylcholinesterase inhibitors (AChEIs), corticosteroids (CSs), and/or nonsteroidal immunosuppressants (NSISTs).
What Are the Conditions for Reimbursement?
Vyvgart should not be reimbursed when given during a gMG exacerbation (i.e., moment when patient experience weakness in some or all muscles, without needing assistance to breath) or crisis (i.e., moment when respiratory muscles are too weak, limiting air flow in and out of lungs, and as a result, patient is unable to breathe), or within 3 months of thymectomy (i.e., surgical removal of thymus gland). Vyvgart should only be reimbursed if prescribed by or in consultation with a neurologist with expertise in managing patients with gMG, and the cost of Vyvgart is reduced. Vyvgart should not be used concomitantly with rituximab or complement inhibitors.
Why Did CADTH Make This Recommendation?
Evidence from a clinical trial (ADAPT) demonstrated that treatment with Vyvgart was associated with a meaningful improvement in gMG daily activity, reduction in disease activity, and improvement in health-related quality of life for patients whom symptoms persisted despite a stable dose of conventional therapy.
Vyvgart met some of the identified patient needs, including sustained efficacy benefit (based on the long-term open-label extension trial ADAPT+), with manageable side effects.
Based on CADTH’s assessment of the health economic evidence, Vyvgart does not represent good value to the health care system at the public list price. A price reduction is therefore required.
Based on public list prices, Vyvgart is estimated to cost the public drug plans approximately $379 million over the next 3 years.
Additional Information
What Is gMG?
Myasthenia gravis (MG) is a condition that causes muscle weakness. In some patients, symptoms remain exclusive to the eyes (ocular MG); however, most patients either are diagnosed with or progress within a few years to gMG, which affects the head, neck, and other muscles. Symptoms of gMG include eyelid drooping and double vision, altered facial expression, difficulty chewing and swallowing food, difficulty speaking, and, in patients with more severe disease, problems with limb movement and breathing.
Unmet Needs in gMG
Symptom control can be achieved with standard treatment for most patients with gMG; however, in some patients, symptom control cannot be achieved with any standard treatment. For these patients, fewer treatment options exist.
How Much Does Vyvgart Cost?
Treatment with Vyvgart is expected to cost approximately $63,200 to $94,800 per patient per course, or $298,304 to $447,456 per patient per year, depending on patient weight and assuming 4.72 courses per year.
Recommendation
The CADTH Canadian Drug Expert Committee (CDEC) recommends that efgartigimod alfa be reimbursed for the treatment of adult patients with gMG who are anti-AChR antibody positive only if the conditions listed in Table 1 are met.
Rationale for the Recommendation
Evidence from 1 phase III, multicentre, double-blind (DB), randomized, placebo-controlled trial (ADAPT) demonstrated that compared with placebo, treatment with efgartigimod alfa results in added clinical benefit in adult patients with gMG who are anti-AChR antibody positive. ADAPT demonstrated that, after cycle 1 of treatment, efgartigimod alfa compared with placebo was associated with a statistically significant and clinically meaningful improvement in terms of the proportion of MG-ADL responders (the between-group mean difference [efgartigimod alfa minus placebo]: 38%; 95% confidence interval [CI], 22% to 56%; the odds ratio versus placebo (95% CI) was 4.951; 95% CI, 2.213 to 11.528; P < 0.0001); the proportion of qualitative myasthenia gravis (QMG) responders (the between-group mean difference [efgartigimod alfa minus placebo]: 49.0%; 95%CI, 34.5% to 63.5%; the odds ratio versus placebo was 10.84; 95% CI, 4.18 to 31.20; P = 0.0001); and the percentage of time with a meaningful MG-ADL score improvement during 126 days of follow-up (48.7% versus 26.6%, P = 0.0001). There were largely no notable differences in adverse events (AEs). In terms of MG-ADL score, QMG, and safety profile, evidence from the long-term open-label extension (ADAPT+) trial appeared consistent.
Despite the available treatment options, there remains an unmet therapeutic need for effective treatment options for patients with this rare and chronic condition, specifically for patients with refractory gMG and those with inadequately controlled gMG despite treatment with conventional therapies (e.g., AChEIs, CSs, and/or NSISTs). Patients expressed a need for treatments with sustained efficacy and reduced side effects that enhance independence (e.g., method, frequency, setting of delivery). Based on the evidence reviewed, CDEC concluded that efgartigimod alfa met some of the needs identified. The efficacy results from the ADAPT trial demonstrated meaningful benefit (improvement in gMG daily activity, reduction in disease activity, and improvement in health-related quality of life), suggesting sustained benefit for up to 14 cycles in the long-term open-label extension (ADAPT+) trial. Efgartigimod alfa may offer more convenience in terms of fast onset (proportion of early MG-ADL responders) and longer period between infusions in a subpopulation (e.g., potentially compared to some IV immunoglobulin [IVIg] regimens).
Using the sponsor-submitted price for efgartigimod alfa and publicly listed prices for all other drug costs, the incremental cost-effectiveness ratio for efgartigimod alfa plus conventional therapy was $1,764,628 per quality-adjusted life-year (QALY) compared with rituximab plus conventional therapy. At this incremental cost-effectiveness ratio, efgartigimod alfa plus conventional therapy is not cost-effective at a $50,000 per QALY willingness to pay threshold for adults with anti-AChR antibody-positive gMG. A price reduction is required for efgartigimod alfa to be considered cost-effective at a $50,000 per QALY threshold.
Table 1: Reimbursement Conditions and Reasons
Reimbursement condition | Reason | Implementation guidance |
|---|---|---|
Initiation | ||
1. Treatment with efgartigimod alfa should be reimbursed for adult patients with gMG who have all of the following: 1.1. positive serologic test for anti-AChR antibodies 1.2. an MG-ADL score at baseline of ≥ 5 1.3. MGFA class II to IV disease 1.4. symptoms persist, despite a stable dose of conventional therapy with AChEIs, CSs, and/or NSISTs. | The results from one phase III, multicentre, DB, randomized, placebo-controlled trial (ADAPT) demonstrated that compared with placebo, treatment with efgartigimod alfa results in added clinical benefit in adult patients with gMG who are anti-AChR antibody positive. ADAPT enrolled adult patients (aged ≥ 18 years) with gMG who tested positive for anti-AChR antibodies, had an MG-ADL score ≥ 5, MGFA class of II to IV, and symptoms persist despite a stable dose of conventional therapy with AChEIs, CSs, and/or NSISTs at baseline. | Stable dose may be defined as adequate trial (as determined by the treating physician) of at least 1 of AChEIs, CSs, and/or NSISTs in the previous 12 months. CDEC noted that rituximab may be available in some jurisdictions; however, CDEC heard from the clinical experts that access to rituximab remains a barrier for some patients. |
2. Efgartigimod alfa should not be initiated: 2.1. during a gMG exacerbation or crisis, or 2.2. within 3 months of thymectomy. | Patients with class V MGFA who had thymectomy less than 3 months before screening were excluded from the ADAPT trial. The efficacy and harms of efgartigimod alfa in such patients are unknown. | |
3. MG-ADL score must be measured and provided by the physician at baseline. | Baseline MG-ADL score was measured in the ADAPT trial and was used to determine response to treatment. | |
4. The maximum duration of initial authorization is 3 cycles. | According to the clinical experts, approval for 3 cycles initially would be reasonable to assess response to treatment. | Considerations for maximum duration of initial authorization for approximately 6 months would be reasonable given that the treatment phase in the ADAPT trial was a 26-week treatment period. |
Renewal | ||
5. Reimbursement of treatment with efgartigimod alfa should be continued if, after the initial 3 cycles of treatment, there is documented improvement in MG-ADL score of 2 points or greater. Reassessment should occur every 12 months thereafter. | According to the clinical experts, clinically meaningful responses would be reflected by improvements in disease symptoms (approximately 2 points on the MG-ADL scale). | Based on clinical expert opinion, after first initial 3 cycles of efgartigimod alfa, if a patient has responded, treatment would be given as long as the patient continues to have a clinically meaningful response (i.e., continuous cycles without defined duration as long as the patient continues to have a clinically meaningful response). In terms of maximum duration of treatment, treatment with efgartigimod alfa would probably be given as long as efgartigimod alfa continued to be effective, or disease spontaneously remitted. |
6. For subsequent renewal, the physician must provide proof of no worsening of MG-ADL score. | This will ensure patients are maintaining their response to treatment with efgartigimod alfa. | Based on clinical expert opinion, there is the possibility of efgartigimod alfa being used for 1 year or more years. If a patient had responded to efgartigimod alfa after the 3 initial cycles and was stable for a year, but worsens afterward, this patient who continued to receive treatment cycles after the initial 3 cycles but was no longer receiving efgartigimod alfa (i.e., was an initial responder but was no longer receiving treatment) can reinitiate therapy, as long as they met the initiation criteria. The patient would not be expected to try standard care (AChEIs, CSs, and/or NSISTs) again. |
Prescribing | ||
7. Efgartigimod alfa should be prescribed by or in consultation with a neurologist with expertise in managing patients with gMG. | Accurate diagnosis and follow-up of patients with gMG is important to ensure that efgartigimod alfa is prescribed to appropriate patients. | |
8. Efgartigimod alfa should not be used concomitantly with rituximab or complement inhibitors. | The efficacy and safety of efgartigimod alfa in combination with rituximab, eculizumab, and/or ravulizumab is unknown. | |
Pricing | ||
9. A reduction in price | The ICER for efgartigimod alfa plus conventional therapy is $1,764,628 per QALY when compared with rituximab plus conventional therapy. A price reduction of 84% would be required for efgartigimod alfa plus conventional therapy to achieve an ICER of $50,000 per QALY compared to rituximab plus conventional therapy. | |
Feasibility of adoption | ||
10. The feasibility of adoption of efgartigimod alfa must be addressed. | At the submitted price, the incremental budget impact of efgartigimod alfa is expected to be greater than $40 million per year. | |
AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; DB = double blind; CDEC = CADTH Canadian Drug Expert Committee; CS = corticosteroid; gMG = generalized myasthenia gravis; ICER = incremental cost-effectiveness ratio; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGFA = Myasthenia Gravis Foundation of America; NSIST = nonsteroidal immunosuppressant; QALY = quality-adjusted life-year.
Discussion Points
CDEC discussed the rarity of this condition and noted that despite its low incidence, there are treatment options available for patients (e.g., azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine, tacrolimus, methotrexate, prednisone). However, CDEC acknowledged that not all treatment options may be available to every patient in every jurisdiction. CDEC acknowledged that there is an unmet need for effective therapy for patients with refractory gMG and patients with inadequately controlled gMG despite trial of conventional therapies (e.g., AChEIs, CSs, and/or NSISTs). CDEC noted that among the patients who were anti-AChR antibody positive in the ADAPT trial, 63% had disease that had failed to respond to prior gMG treatments and were considered refractory (i.e., prior exposure to ≥ 2 immunosuppressive therapies or treatment with ≥ 1 immunosuppressive therapy and requiring plasma exchange (PE) or IV immunoglobulin multiple times within 1 year before study inclusion).
CDEC noted that according to the clinical experts, approximately 90% of patients respond to current treatments and that response is often partial, resulting in the continuation of symptoms that affect quality of life and function. CDEC noted that the overall treatment goal of gMG is to improve quality of life, followed by reduce treatment burden and treatment-related morbidities, and to maintain adequate disease control. CDEC reviewed evidence from the ADAPT trial and noted that while improvements in health-related quality of life (HRQoL) were exploratory, the results were considered clinically meaningful.
CDEC discussed needs identified by patients, including for decreased intensity of exacerbations and less serious hospital admissions. CDEC acknowledged the ad hoc analysis, which reported low incidences of MG exacerbations and MG-related hospitalizations.
CDEC also discussed patients’ desire for fewer treatment-emergent adverse events (TEAEs). While the ADAPT trial did not provide direct comparative evidence regarding the adverse effects of efgartigimod alfa versus other MG therapies, CDEC noted that TEAEs appeared similar in both the efgartigimod alfa and placebo groups and that there were no treatment-related deaths in either group. CDEC did, however, acknowledge that in terms of AEs of special interest, infections and infestations events were higher in the efgartigimod alfa group, which was also acknowledged in the Health Canada product monograph.
CDEC acknowledged the possibility of treatment burden being higher initially compared to conventional therapy given that efgartigimod alfa is an add-on therapy. While treatment burden may be impacted initially, CDEC noted that the improvement in quality of life observed in the ADAPT trial was clinically meaningful.
While the proportion of early MG-ADL responders (i.e., responder within 2 weeks) during cycle 1 were descriptive in nature, the results were considered clinically meaningful. Moreover, while the meaningful benefits (improvement in gMG daily activity, reduction in disease activity, and improvement in HRQoL) observed in the ADAPT trial involved short 8-week cycles, and that longer comparative evidence were not available; nonetheless, evidence from the long-term open-label extension (ADAPT+) trial suggests sustained benefit for up to 14 cycles, although interpretation of the long-term results was limited by the open-label and descriptive nature of the extension study.
CDEC discussed the results of the sponsor-submitted network meta-analysis (NMA), which suggested that relative to IVIg and ravulizumab, efgartigimod alfa may provide a benefit with respect to change in MG-ADL and QMG scores; however, the 95% credible intervals (CrIs) for the effect estimates included the possibility of trivial effects (i.e., only small, non–clinically important differences between groups) and no difference (in the case of change in MG-ADL score relative to ravulizumab). No difference in efficacy in terms of change from baseline in MG-ADL and QMG scores could be concluded for efgartigimod alfa relative to rituximab due to wide 95% CrIs (which included the possibility of clinically important benefit favouring efgartigimod alfa), and methodological limitations.
Background
MG is a rare, chronic, neuromuscular autoimmune disease mediated by pathogenic autoantibodies that target structural components of the neuromuscular junction, impairing neuromuscular transmission and leading to muscle weakness and fatigue. Many patients initially present with symptoms affecting only the eye muscles (i.e., ocular MG). Approximately 85% of patients go on to develop generalized weakness affecting the neck, trunk, limbs, and bulbar and respiratory muscles (gMG). Patients with gMG experience symptoms that negatively impact HRQoL. The disease has a fluctuating natural history, and MG exacerbation (an increase in symptoms in patients who were previously asymptomatic or minimally symptomatic, defined as a ≥ 3-point worsening in QMG score versus baseline) and myasthenic crisis (muscle weakness causing life-threatening difficulties with breathing and swallowing and requiring ventilator support) can occur gradually or without warning. Approximately 85% of patients with gMG are anti-AChR antibody positive and as many as 15% of patients with gMG are negative for anti-AChR antibodies. An estimated 1% to 10% of patients do not have anti-AChR antibodies but do have autoantibodies against muscle-specific kinase (MuSK) antibody positive or autoantibodies against lipoprotein receptor-related protein-4 positive, which also lead to a decrease in anti-AChR antibodies. The MGFA classification system is a tool used to categorize gMG based on clinical features and/or disease severity. The classification ranges from class I (i.e., ocular weakness only), to class II, class III, and class IV (which represent mild, moderate, and severe muscle weakness respectively), to class V (i.e., defined by intubation, with or without mechanical ventilation, except when employed during routine postoperative management, and myasthenic crisis). The incidence of MG in Canada is estimated at 23 cases per 1 million person-years, with a prevalence of 32 cases per 100,000 adults (0.032%). Thus, there are approximately 8,121 patients with MG across the CADTH-participating drug programs (0.032% × 25,376,703 adults in CADTH-participating drug programs in 2023). Approximately 85% of adults with MG are anticipated to progress to gMG, which corresponds to approximately 6,903 adults with gMG in Canada.
The clinical experts that CADTH consulted for this review indicated that the goal of treatment in patients with gMG is to reduce disease symptoms as well as the adverse effects of MG therapy and to allow the patient to function and work normally with good HRQoL. Other goals of treatment include avoiding MG exacerbations and myasthenic crisis, minimizing hospitalizations and intensive care unit admissions, and reducing the numbers and doses of therapies (especially corticosteroid use) required for symptom control. The available main therapies for gMG include AChEIs, CSs, NSISTs, rituximab, IVIgs, PEs and/or plasmapheresis (PPs), and terminal complement inhibitors (i.e., ravulizumab and eculizumab). According to the clinical experts consulted by CADTH for this review, the first-line standards of care (i.e., the conventional therapy) for MG are AChEIs, CSs, and NSISTs (e.g., azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine, tacrolimus, methotrexate).
Mild to moderate gMG (MGFA class II or IIIa) is initially treated symptomatically with AChEIs (usually pyridostigmine), and the onset of benefit occurs in hours to days. If this provides insufficient symptom relief, immunosuppressive therapy (IST) with corticosteroids (usually prednisone) is administered, and maximum responses typically occur 2 to 6 months later, after which a slow tapering of CSs is begun. In patients who do not respond to CSs, who have significant comorbidities such that long-term CS treatment is contraindicated, or in whom doses of CSs cannot be tapered, treatment with a NSISTs and/or immunomodulatory drugs (including rituximab) may be initiated. The clinical experts stated that the onset of benefit from NSISTs occurs in months to years (approximately 9 to 18 months for azathioprine and mycophenolate). While rituximab has not been approved as a gMG treatment by Health Canada, it is considered a treatment option in Canada for patients with refractory anti-AChR antibody-positive disease according to surveys conducted with clinical experts. According to the clinical experts, in patients with moderate to severe gMG, especially those who have respiratory or bulbar weakness, IVIg, PE, or PP may be administered in addition to rituximab, either at the time of IST initiation or to treat MG exacerbation or myasthenic crisis. As MG symptoms improve, doses of AChEIs, steroids, and NSISTs are reduced and the frequency of IVIg, PE, or PP is reduced until the minimal maintenance therapy required for remission is identified. Patients with refractory gMG who are anti-AChR antibody positive may be candidates for the complement inhibitor eculizumab. While eculizumab received a recommendation for reimbursement with conditions in 2020, price negotiations concluded without an agreement in December 2022. A survey of 7 expert clinicians across 6 provinces in Canada indicated that ravulizumab would be another option for patients who have an inadequate response to conventional therapy if it were to be approved and funded. In April 2023, CADTH issued a draft “do not reimburse” recommendation for ravulizumab in this indication. The clinical experts consulted by CADTH for this review emphasized that most patients with gMG (more than 80%) respond well to currently available treatments; although these cannot cure the disease, excellent symptom control is achieved in most patients and prognosis is generally good in terms of muscle strength and function as well as HRQoL. Despite treatment with conventional therapy (AChEIs, CSs, and/or NSISTs), many patients continue to experience disease burden and symptoms that impact their HRQoL, as well as treatment-related side effects that may be severe.
Efgartigimod alfa is a first-in-class human immunoglobulin (Ig) G1 antibody crystallizable fragment that blocks the neonatal crystallizable fragment receptor. Efgartigimod alfa is supplied as a 20 mg/ mL solution, 10 mg/kg administered as an IV (IV) infusion over 1 hour once weekly for 4 doses (i.e., Weeks 0 to 3). In patients weighing 120 kg or more, the recommended dose of efgartigimod alfa is 1,200 mg (3 vials) per infusion. Efgartigimod alfa reduces the levels of pathogenic IgG autoantibodies. Efgartigimod alfa received a Health Canada Notice of Compliance for the treatment of adult patients with gMG who are anti-AChR antibody positive on September 19, 2023. The sponsor’s reimbursement request is that efgartigimod alfa be reimbursed as an add-on therapy for adult patients with anti-AChR antibody-positive gMG whose symptoms persist despite adequate treatment with AChEIs, CSs, and/or NSISTs, which is a subgroup of the approved Health Canada indication.
Sources of Information Used by the Committee
To make its recommendation, the committee considered the following information:
a review of 1 phase III, DB, placebo-controlled randomized controlled trial (RCT) (ADAPT) in adults with gMG whose symptoms persisted despite a stable dose of conventional therapy (concomitant gMG treatment) treatment with AChEIs, CSs, and/or NSISTs
patient perspectives gathered by 1 patient group, Muscular Dystrophy Canada (MDC)
input from the public drug plans that participate in the CADTH review process
2 clinical specialists with expertise diagnosing and treating patients with gMG
input from 1 clinician group, the Neuromuscular Disease Network for Canada (NMD4C)
a review of the pharmacoeconomic model and report submitted by the sponsor.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
CADTH received 1 patient group submission from the MDC, which identified and contacted adults living with MG to participate in a survey and semistructured interviews.
Respondents indicated that MG has a significant impact on productivity, fatigue, energy levels, quality of sleep, respiratory health, mobility, strength, independence, relationships and social participation, eyes and vision, speech, and swallowing. They also explained that the impact of MG extends beyond physical symptoms, and affects their mental health, quality of life, and the wellbeing of their families.
Some of the respondents indicated that they feel that their lungs are weaker; that they had to go on a ventilator in the intensive care unit; that they choke on their food or that their saliva interferes with breathing; that they cannot walk, even inside their house; that they always keep a walker or cane nearby because MG can flare up at any time; that they cannot sleep at night because of MG-related aches; and that they are unable to drive. Some indicated that they had slurred speech, frequently go cross-eyed, have double vision that interferes with reading, and have experienced multiple acute hospitalizations.
When asked about how MG is being managed with available treatments, 3 main themes emerged from the analysis: negative experiences with steroids (e.g., adverse effects, costs); the slow onset of medication effects; and a feeling of trial and error with medications. Regarding the improved outcomes, the patient group identified 3 aspects of MG that they want better controlled, including decreased intensity of exacerbations and side effects, maintenance of independence, and less serious hospital admissions. The administering method, duration, frequency, and convenience of treatment, as well as the cost, are very important to patients and caregivers. They prefer less travel, fewer hospital visits, and less invasive methods of treatment. HRQoL was noted as a key priority over convenience of a drug.
The respondents stated that they would be willing to deal with side effects of medications if the previously noted aspects of MG were better controlled. They also stated that although current medications decrease the number of exacerbations, they do not have an impact on overall quality of life. They are looking for new treatments to help them become independent, stop myasthenic crises, address respiratory and general weakness, be easier to swallow (for pills), reduce pain, not lead to diabetes, be target treatment for MG instead of general immunosuppression, be less expensive, work quickly, and be a 1 daily dose in the morning.
One participant had received Vyvgart as a participant of a clinical trial and explained that this medication replaced the need for IVIg, the effects appeared quicker compared with other therapies, the infusion time was less than expected, treatment was received less frequently compared with other therapies, and they experienced fewer side effects compared with other therapies. This patient respondent highlighted that while diarrhea was a problem and not unique to Vyvgart, it was manageable after the first cycle.
All of the respondents had experienced diagnostic blood testing and many had single fibre electromyography to confirm diagnosis. A total of 80% of the respondents reported difficulty getting diagnosed. MDC, based on early findings of the MG Journey Mapping Project, reported 7 years from time of first bothersome symptom to diagnosis, with the range up to 23 years. According to MDC, the majority of respondents found the process of testing and diagnosis cost-effective but lengthy, with many missed opportunities, delayed diagnosis, misdiagnosis (such as stroke and Bell palsy), and costs incurred. Those who were diagnosed during a crisis or hospitalization reported a smooth diagnosis (25%).
Clinician Input
Input From Clinical Experts Consulted by CADTH
The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of gMG.
The clinical experts indicated that approximately 90% of patients respond to current treatments; however, response is often partial, meaning that there are still symptoms that affect quality of life and function. According to the clinical experts, besides prednisone and rescue treatments, ISTs take very long to act (e.g., azathioprine takes at least a year). The clinical experts explained that this means that patients may be exposed to higher doses of steroids for longer, and with persistent symptoms for long, before even knowing whether this medication will be effective or not. Current treatments are nontargeted, so overall cause more diffuse immunosuppression, and there is an increased risk of cancer with long-term use. Also, the risk of all steroid-related AEs increases with prolonged doses. The clinical experts also indicated that the fraction of patients whose disease is “refractory” varies according to definition, however 10% to 20% of the total population is a reasonable estimate. According to the clinical experts consulted by CADTH for this review, patients with gMG usually start by receiving pyridostigmine, but most patients will need disease-modifying treatment with an immunosuppressant, most commonly prednisone. Depending on severity, age, and comorbidities, an NSIST (e.g., azathioprine, mycophenolate, tacrolimus) may be started early after diagnosis, or later (for example, when it is not possible to reduce steroid dose). Some patients with severe disease at onset (e.g., crisis or severe symptoms) may receive a rescue treatment such as IVIg or PE early, to have fast improvement while immunosuppression begins. Few patients receive chronic IVIg or PE, and some of these patients are dependent on these treatments. According to the clinical experts, the treatment goals is to achieve minimal symptoms or remission, with the least amount of AEs form treatments. Patients expressed a need to improve the ability to perform their daily life activities, reduce fatigue, and improve their ability to care for their families and work or home obligations.
The clinical experts stated that efgartigimod alfa has a specific mechanism of action related to the pathophysiology of MG (reduction of IgG levels, including anti-AChR antibodies). Efgartigimod alfa reduces levels of IgG, but efgartigimod alfa does not affect the process of producing anti-AChR antibodies. The clinical experts indicated that they do not foresee that efgartigimod alfa will be used as a first-line treatment, rather that it would be suitable for individuals whose disease does not show adequate response to available treatments, those dependent on IVIg or PE, or those with very severe disease to bridge gap of delayed action of standard ISTs. The clinical experts noted that patients should have received standard conventional treatments first, as standard conventional treatments will be satisfactory for a large number of patients. The clinical experts also stated that most treatments for patients with gMG are given off label given a lack of RCT evidence (i.e., prednisone, azathioprine, mycophenolate, tacrolimus, rituximab). The clinical experts noted that IVIg or PE used as a rescue treatment is different from IVIg or PE used chronically (i.e., maintenance therapies, once a month); and only a small number of patients use IVIg or PE chronically. According to the clinical experts, eculizumab or ravulizumab are used too rarely at present to include them as comparators. One clinical expert indicated that if cost was not an issue, one could argue that efgartigimod alfa could be tried as an initial therapy in patients in whom pyridostigmine alone was ineffective, however, they noted that the cost is likely to be a major barrier. The clinical experts indicated that efgartigimod alfa will provide a new treatment for patients with gMG who are anti-AChR antibody positive and probably for patients with antibody-positive MuSK. However, whether patients with gMG who are seronegative would respond to efgartigimod alfa is unknown because few of these patients were included in ADAPT trial. The clinical experts indicated that in most clinics patients do not have standardized assessments, though in academic settings, clinician use validated measures. The clinical experts noted that the MG-ADL scale used in trials is easy to use and can be easily incorporated into routine clinical practice and all settings (i.e., community, hospital, and academic). The clinical experts recommended using the MG-ADL scale for all visits in patients with active treatment, as this allows for following clinical course. The clinical experts stated that an improvement (reduction) of 2 points is considered significant. Apart from symptom scores, overall function and ability to return to work were also assessed. The clinical experts expressed that the ability to reduce or stop chronic use of CSs, IVIg, or PE is an important outcome when considering the use of efgartigimod alfa. They explained that some patients are dependent on chronic IVIg or PE, and weaning of these treatments is important. The clinical experts also highlighted that reduction or avoidance of hospitalization due to MG is an important outcome. N. Frequency of assessments depends on symptoms and patient stability. Patients whose symptoms are well controlled typically seen by clinicians every 6 months; however, they can be seen more frequently (e.g., every 2 to 3 months) in case of worsening, new medications, and so forth. For efgartigimod alfa, most responses were fast, however a small proportion of responders lagged, and response could be seen at the second cycle. Therefore, the clinical experts suggested that 2 to 3 months may be needed to assess response to cycle 1 and/or to assess for need of new cycle. An assessment at 6 months may be needed to determine those whose disease does not respond. For these patients, assessments could then be done every 3 to 6 months to determine if new cycles will be needed.
The clinical experts indicated that efgartigimod alfa should be discontinued if a patient has no response to treatment (i.e., no improvement in symptoms and/or function), if a patient experienced a severe AEs (e.g., severe infusion reaction), if patients need rescue treatment (IVIg or PE, or increased dose of steroids), or there is an inability to reduce chronic use of corticosteroids, IVIg, or PE. The clinical experts indicated that patients should be under the care of a neurologist with experience in diagnosing and treating MG, usually a neuromuscular specialist. The infusion itself can be arranged at infusion clinics.
Clinician Group Input
CADTH received 1 clinician group submission from the NMD4C.
NMD4C stated that conventional treatment options for gMG have been based on symptomatic therapy, short-term rescue immunotherapy, and long-term IST. Moreover, nonspecific immunosuppressants have been only partially effective and many patients do not attain stable remission, with 10% to 20% being intolerant to these drugs or having disease that does not respond.
According to the NMD4C, some of the unmet needs of the standard treatments are side effects, not being effective for all patients, long period of treatment, and transient effectiveness. Another unmet need in this field is the lack of therapeutic options for patients who are seronegative.
The NMD4C noted that patients who are AChR antibody positive will most likely respond to efgartigimod alfa. Patients with MuSK antibodies and those who are double seronegative may respond. Patients who get worse quickly, particularly those with MG crisis, are most in need of an intervention that works quickly, but patients who have symptoms restricted to only ocular muscles are unlikely to require such rapid intervention with efgartigimod alfa. To identify the patients best suited for treatment with efgartigimod alfa, clinician examination and judgment supplemented by assessment of MG activities of daily living, using scales that reflect severity of disease, such as the quantitative MG score, the MG Impairment Index, and the single simple question (SSQ). If not available, then antibody testing needs to be done, but according to the clinician group, this can be delayed. There is nothing clear at this point to predict which patients are more likely to have disease that responds, except the presence of AChR antibodies.
The NMD4C indicates that diagnosis of patients who are double seronegative is an issue as cluster antibodies to both AChR and MuSK may be present but need to be tested specifically, which can take weeks.
The clinician group indicated that to determine patients’ response to therapy, scales such as the MG-ADL, QMG, Myasthenia Gravis Impairment Index, and SSQ at 2 and 4 weeks is required and after that the assessment should be based on the patient’s status. The clinician noted that a clinically meaningful response to treatment used in the clinical trials is 2 or more points on the MG-ADL and 3 or more points on the QMG. For the SSQ, the clinician group suggested that levels above 72% indicate general satisfaction. In case of lack of response, discontinuation of treatment should be considered.
The clinician group mentioned that usual Ig treatment for MG can be effective but place a significant burden on the Canadian health care system and that supplies can be at risk in situations such as the pandemic. They indicated that efgartigimod alfa is likely to replace Ig therapies.
In summary, the clinician group’s input is aligned with the input provided by the clinical experts consulted by CADTH.
Drug Program Input
The drug programs provided input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 2.
Table 2: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Issues with the choice of comparator in the submitted trial(s)
| According to the clinical experts, the complements inhibitors (i.e., eculizumab, ravulizumab) are mechanistically different from efgartigimod alfa and would likely have a different role in therapy so the lack of direct comparison with these 2 is not a problem. CDEC agreed with the clinical experts. |
Considerations for initiation of therapy | |
Prior therapies required for eligibility The requested indication for efgartigimod alfa is for add-on therapy to conventional therapy, which may include AChEIs, CSs, and/or NSISTs in patients with anti-AChR antibody-positive disease. Although the sponsor included rituximab in the list of comparators, it was not included in the studied indication because it is only used in patients whose disease is not anti-AChR antibody-positive. Patients treated with either rituximab or eculizumab within 6 months of screening were also excluded from the study. The sponsor envisions the place in therapy of efgartigimod alfa to be considered as an add-on alternative to immunoglobulins after use of NSISTs and/or CSs as depicted in Figure 1. The indication is for addition of efgartigimod alfa to 1 or a combination of the 3 conventional therapy classes. The ADAPT trial allowed the inclusion of patients on any combination of conventional gMG treatment, which was limited to AChEIs, steroids, and NSISTs, and did not require the patients to have received or discontinued use of any specific treatment. It is unclear if a patient’s eligibility for addition of efgartigimod alfa would require trials of medications from all 3 classes or from 1 or 2 classes. | According to the clinical experts, Figure 1 is a reasonable depiction of efgartigimod alfa’s place in therapy, although the clinical expert suspect that cost will drive clinicians to use IVIg first. Unless efgartigimod alfa ends up being priced similar to current conventional therapies, it is likely that unsuccessful trials of all 3 classes (AChEIs, CSs, and NSISTs) will be a prerequisite to the use of efgartigimod alfa. The clinical experts stated that in addition, in the ADAPT trial, not all included patients were refractory (i.e., not all patients needed to have disease that failed to respond to multiple therapies). So, technically from a data and mechanistical perspective, there is no need to have a failed trial of all conventional medications. Realistically, and mostly driven by price, efgartigimod alfa should not be offered in the first line but rather after a patient has tried conventional treatments, which makes the diagram realistic. CDEC acknowledged the clinical expert’s response and agreed that failure of all 3 conventional therapies would not be required, rather considering trial of at least 1 of AChEIs, CSs, and/or NSISTs in the previous 12 months. |
Eligibility to re-treatment In the ADAPT trial, if a patient was an MG-ADL responder during a previous cycle and then their disease lost response, that patient could qualify for re-treatment. Loss of response was defined as a < 2-point reduction in the MG-ADL total score during the cycle compared to the baseline value for that cycle. Re-treatment in subsequent cycles was permitted if the patient met all of the following criteria:
This allowed for a maximum of 3 treatment cycles during the 26-week study. According to the sponsor and the product monograph, following cycle 1, treatment with efgartigimod alfa can be given on an as-needed basis, according to clinical assessment, and thus would vary by patient. This poses a unique challenge for drug plans when instating an approval if there is no certainty on whether a patient will be re-treated and at what frequency. | The other clinical expert indicated that from an economic perspective, re-treatment with efgartigimod alfa on an as-need basis likely result in savings, as based on data some patients had relatively long stretches between cycles. However, it will pose an implementation difficulty for clinicians based on the need for more frequent monitoring to decided appropriate time for re-treatment. Therefore, as a prescriber, approval for 3 cycles initially would be reasonable to assess response to treatment; further approvals would be conditional on demonstrating benefit. Clinician can then tailor cycles (e.g., some patients may take longer to use all cycles). CDEC agreed with the clinical expert that this approach would be reasonable. |
Special subtypes (not explicitly mentioned in the indication) to consider separately for eligibility Considering that patients who had received eculizumab within 6 months of screening were excluded from the study, and there were no comparisons to ravulizumab, would patients who failed either 1 or both drugs be considered for treatment with efgartigimod alfa? | Yes, failure to respond to rituximab or eculizumab (or ravulizumab) would not preclude consideration of efgartigimod alfa. These patients should be considered for efgartigimod alfa. CDEC agreed with the clinical expert that this approach would be reasonable. |
Considerations for continuation or renewal of therapy | |
Challenges related to assessment and monitoring of therapeutic response
Can the clinical experts confirm if the MG-ADL scale is reflective of best practices when treating patients with gMG in Canada? If not, is there another tool or outcome that would better align with how patients are monitored in the Canadian practice setting? | One clinical expert indicated that they prefer a clinician-driven assessment (like the QMG). The other clinical expert indicated that most neurologists in Canada do not use standardized outcomes measurement in MG. The standard outcome measurements are mostly used in academic centres. The clinical expert uses MGII, which combines PRO and examination. But the MG-ADL scale or equivalent is certainly acceptable. Both clinical experts agree that the MG-ADL scale is extremely easy to use and to implement. And both clinical experts agreed that regardless of outcome measurement tools, more important than the tools, patients should be assessed by neurologists with experience and/or expertise in the management of MG. CDEC noted the clinical expert response and suggested that the MG-ADL scale would be reasonable given its ease of use and that it was used in the ADAPT trial. |
Considerations for discontinuation of therapy | |
Definition of loss of response, absence of clinical benefit, or disease progression Efgartigimod alfa is administered on an as-needed based for clinical response (physician assessment and PROs). How many times would a patient require re-treatment due to loss of response before being considered for discontinuation? Likewise, if a patient has a need for increased frequency of dosing, would consideration be given to discontinuation of efgartigimod alfa? It would be helpful to have a clear definition of loss of response and disease progression that would indicate the need for discontinuation, defined according to MG-ADL scale parameters and/or frequency of dosing. | One clinical expert stated that how many unsuccessful re-treatments would be needed before concluding that efgartigimod alfa does not work would vary from clinician to clinician, but that they would probably stop it after 2 unsuccessful re-treatments. There would not be a good rationale to give efgartigimod alfa at a frequency greater than every 1 to 2 weeks. The other clinical expert indicated that based on the ADAPT trial, patients who disease does not respond after 2 to 3 cycles (no significant improvement or worsening) should discontinue the treatment. CDEC agreed with the clinical expert, it would be reasonable based on the ADAPT trial, to discontinue the treatment if a patient had no response after 3 cycles (no significant improvement or worsening). |
Considerations for prescribing of therapy | |
Dosing, schedule and/or frequency, dose intensity The medication is given as a 1-hour infusion once weekly for 4 weeks (this being cycle 1). Following the initial dose, subsequent doses and frequency are dependent on clinical response, and thus may vary by patient. There is no further clarity provided in the product monograph regarding frequency of dosing. The sponsor estimates that patients with anti-AChR antibody-positive disease required a mean (SD) number of 4.72 cycles per year, with approximately 24% of patients requiring < 3.5 cycles per year. Bearing in mind that each cycle consists of up to 4 weekly infusions, to a maximum of 3 vials per infusion, this would mean up to 48 vials annually if at maximum dose and with an average of 4 treatments per year. It is unclear if there is a minimum amount of time that should exist between cycles. Is there a minimum frequency before administering a subsequent dose? In the ADAPT trial, the median time between the last infusion of cycle 1 and the start of cycle 2 was 7 weeks (mean of 10 weeks). In a real-world study of use patterns, the sponsor noted an average gap of 50 days to 58 days between the last infusion of cycle 1 and the start of cycle 2. In the long-term extension study, ADAPT+, subsequent cycles were only started if the patient completed the fourth infusion of the previous cycle at least 4 weeks prior. If consistent with this information, would re-treatments with efgartigimod alfa require a minimum of 4 weeks following last infusion before initiating next cycle? | Both experts and CDEC agreed that waiting at least 4 weeks before initiating a re-treatment cycle seems rational. The clinical experts stated that they would plan 3 cycles before concluding there was no response, with 4 weeks between each treatment. The clinical experts stated that based on the RCT (the ADAPT trial), a number of patients whose disease does not respond to cycle 1 see a response with cycle 2, and an even smaller number of patients may yet see a response by cycle 3. Therefore, after cycle 1 if no response is achieved, the experts stated that they would still prescribe 1 to 2 cycles to get these late responders; and if there is a clinical response, they would maintain cycles given efficacy. The clinical experts highlighted that the minimum length between each cycle treatments is 4 weeks based on the ADAPT+ trial. The experts stated that they would not wait for the patient to deteriorate before the next cycle of treatment, and instead would treat the patient before clinical deterioration or at minimal deterioration. |
Drug administration Administration is by IV infusion only and requires a trained health care professional. The sponsor expects the infusion to be most commonly administered in a patient’s home and less commonly at an infusion clinic. Given this information, a trained health care professional would be required to make home visits to complete the administration. | Comment from the drug programs to inform CDEC deliberations. |
Concerns related to accessing clinical specialists and/or special settings Administration will require in-home services or infusion clinics. Although the sponsor states a commitment to providing standardized access to all patients, including remote areas, how this accessibility will be provided is a potential concern. | Comment from the drug programs to inform CDEC deliberations. |
Concerns related to combination usage Would there be any potential combination usage of efgartigimod alfa with eculizumab or ravulizumab, specifically considering that Health Canada issued a NOC for ravulizumab plus conventional therapy in the treatment of patients with anti-AChR antibody-positive gMG? | One clinical expert indicated that efgartigimod alfa might be combined with either eculizumab or ravulizumab (as the mechanisms are different), but the cost would make this difficult to justify. The other expert stated that theoretically, efgartigimod alfa might be combined with either eculizumab or ravulizumab as they have different mechanisms. But indicated that it would be hard to know if the combination would be clinically superior to either alone as there are no data related to these combinations. As such, the expert stated that they would not support concurrent use. Rather, they anticipated that they would be used sequentially if there was no response to 1 of the drugs. CDEC acknowledged the clinical experts’ response and noted that there was no evidence reviewed for the combination of efgartigimod alfa with eculizumab or ravulizumab. |
Care provision issues | |
Drug preparation, storage, administration or dispensing Administration is by IV infusion only. It requires reconstitution and administration by a trained health care professional, and up to 3 vials may be needed per dose, depending on weight (10 mg/kg). | Comment from the drug programs to inform CDEC deliberations. |
System and economic issues | |
Concerns regarding the anticipated budget impact and sustainability At the submitted price, efgartigimod alfa is significantly more expensive than conventional therapy and immunoglobulin and/or PE therapies but comparable to the cost of ravulizumab. | Comment from the drug programs to inform CDEC deliberations. |
Presence of confidential negotiated prices for comparators At this time, ravulizumab has not received a positive funding recommendation for gMG or gone through pricing negotiations so it is difficult to make a comparison with these unknowns. It is awaiting CDEC reconsideration. Although not mentioned as a comparator by the sponsor, eculizumab is another treatment for gMG and its pricing negotiations ended without agreement. The reimbursement status of eculizumab for gMG across the jurisdictions is not currently known. | Comment from the drug programs to inform CDEC deliberations. |
AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; CDEC = CADTH Canadian Drug Expert Committee; IgG1 = immunoglobulin G1; IMP = investigational medicinal product IVIg = IV immunoglobulin; CS = corticosteroid; gMG = generalized myasthenia gravis; MGII = Myasthenia Gravis Impairment Index; NOC = Notice of Compliance; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; NSIST = nonsteroidal immunosuppressive therapy; PE = plasma exchange; PRO = patient-reported outcome; QMG = qualitative myasthenia gravis; RCT = randomized controlled trial; SD = standard deviation.
Figure 1: Anticipated Place in Therapy of Efgartigimod Alfa for Anti-AChR Antibody-Positive gMG
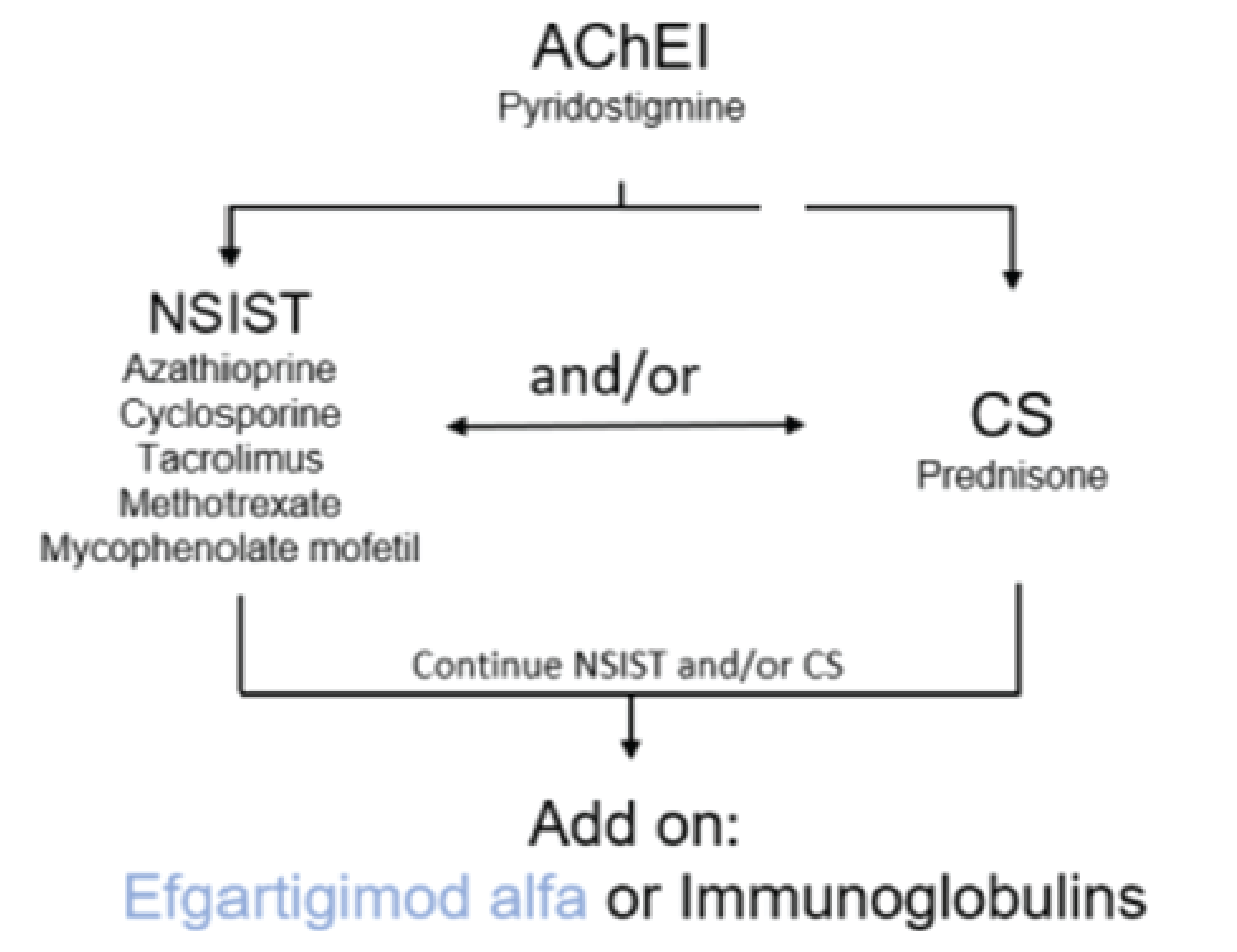
AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; CS = corticosteroids; gMG = generalized myasthenia gravis; NSIST = nonsteroidal immunosuppressant.
Clinical Evidence
Systematic Review
Description of Studies
One phase III, DB, placebo-controlled RCT (ADAPT; N = 169) is included in the systematic review. The objective of the ADAPT trial was to evaluate the efficacy and safety of efgartigimod alfa added on to conventional therapy versus placebo added to conventional therapy in adult patients with gMG whose symptoms persist despite a stable dose of conventional therapy (concomitant gMG treatment) with AChEIs, CSs, and/or NSISTs. Patients were required to be on a stable dose of conventional therapy before screening (i.e., for AChEIs, no dose change for 2 weeks before screening; for CSs, ≥ 3 months of treatment and no dose change for 1 month before screening; and for NSISTs, ≥ 6 months of treatment and no dose change for 3 months before screening). All patients were MGFA class II to IV with an MG-ADL scale total score 5 or more. The mean age was 44.7 to 49.2 years, and most patients were white (83.1% to 87.5%). In the AChR antibody-positive population, 129 (100%) patients received 1 prior | || || || || || || || patients receive 2 prior therapies, and || || || patients received 3 or more prior therapies. The majority had been pretreated with 2 or more || || || or 3 or more || || || different classes of conventional therapy medication (any combination of AChEIs, CSs, and/or NSISTs at the physician’s discretion). For patients who were AChR antibody positive, those who received 3 classes of prior therapy (CS plus NSIST plus AChEI) were || || || in the efgartigimod alfa group and || || || in the placebo group. || || || || || || patients in the efgartigimod alfa and || || || in the placebo group had received any 2 of the 3 prior therapies (i.e., CS, NSIST, and/or AChEI). In addition, among the patients who were AChR antibody positive, 63% patients had not responded to prior gMG treatments (also known as patients with refractory gMG) and 37% had disease that had not failed on a prior treatment, but had inadequately responded to the existing standard of gMG therapy.
Patients were randomized 1:1 to receive efgartigimod alfa or a matching placebo in cycle 1 (i.e., for the first 8 weeks), followed by an individualized treat-as-needed regimen based on the patient’s MG-ADL scale response. All patients received a stable concomitant treatment during the trial. The primary outcome of the study was percentage of anti-AChR antibody-positive patients who were MG-ADL responders in the first treatment cycle (an MG-ADL responder was defined as a patient with a ≥ 2-point improvement [reduction] in MG-ADL score). Key secondary outcomes included the percentage of MG-ADL responders in cycle 1 in the overall population (i.e., anti-AChR antibody positive and anti-AChR antibody negative); the percentage of time patients who were anti-AChR antibody-positive showed a clinical meaningful improvement in MG-ADL score (≥ 2-point reduction) up to day 126; time from week 4 to qualify for re-treatment in the anti-AChR antibody-positive population; percentage of early MG-ADL responders in cycle 1 in the anti-AChR antibody-positive population (i.e., MG-ADL score change of ≥ 2 points occurred by week 2); and change from cycle baseline in MG-ADL total score at cycle 1 and cycle 2. Change from cycle baseline in HRQoL (Component Myasthenia Gravis Quality of Life 15 – Revised [MG-QoL15r] score, EQ visual analogue scale [VAS]) at cycle 1 and cycle 2 were assessed as tertiary or exploratory outcomes. Post hoc analysis was performed for gMG hospitalization, gMG exacerbation, and gMG crisis. It should also be noted that, although the ADAPT trial duration was designed for 26 weeks, the primary, key secondary, and HRQoL outcomes at the end of the study (i.e., week 26) were not assessed. Instead, the outcomes were assessed at the end of cycle 1 and cycle 2.
Efficacy Results
Patients With Anti-AChR Antibody-Positive Disease
Myasthenia Gravis Activities of Daily Living
MG-ADL responder during cycle 1 and cycle 2: MG-ADL responders during cycle 1 in patients who were anti-AChR antibody positive was the primary outcomes in the ADAPT trial. Thirty-eight percent (95% CI, 22% to 56%) more patients in the efgartigimod alfa group (those with anti-AChR antibody-positive randomized to receive efgartigimod alfa) than in the placebo group achieved a 2 or greater point MG-ADL scale improvement during cycle 1. The between-group difference was considered clinically meaningful by the clinical experts consulted by CADTH.
Various post hoc subgroup analyses were conducted for MG-ADL responders during cycle 1. Consistent with the primary analysis, these results demonstrate that efgartigimod alfa produces improvements in MG-ADL response compared to placebo, regardless of prior therapies, concomitant therapies, disease duration, thymectomy, and prior treatment failure; however, the trial was not powered to detect subgroup differences. In terms of the MG-ADL responders, similar benefit was observed in cycle 2.
Early MG-ADL responders: Early MG-ADL responders (responded at week 2 of the cycle 1) in the patients who were anti-AChR antibody positive was assessed as a fifth key secondary outcome. Because the statistical testing hierarchy was broken at the fourth secondary end point (i.e., time to quality re-treatment), the percentage of patients in the anti-AChR antibody-positive population who were early MG-ADL responders was not statistically tested based on statistical plan in the protocol. Nevertheless, within the anti-AChR antibody-positive population, a higher proportion of patients in the efgartigimod alfa group achieved a 2 point or greater MG-ADL scale improvement at week 2 than in placebo group (between-group difference = 31.9%; 95% CI, not reported). The between-group difference was considered clinically meaningful by the clinical experts consulted by CADTH. The percentage of MG-ADL early responders during cycle 2 was not assessed and not reported in the sponsor’s evidence summary.
Percentage time of the MG-ADL clinical meaningful improvement up to day 126: Among patients who were anti-AChR antibody positive, the percentage of time with a clinical meaningful improvement in the MG-ADL total score up to day 126 was assessed as a third key secondary outcome and was included in the hierarchy test to control the type I error. According to the clinical experts, the percentage of time with a clinical meaningful improvement in the MG-ADL total score in the efgartigimod alfa group was a clinically meaningfully longer (efgartigimod alfa minus placebo = 22.07%, || || |||| || |||| || ||; P = 0.0001) than that in placebo group.
MG-ADL change from cycle baseline: During cycle 1 and cycle 2, the change from cycle baseline of MG-ADL score was assessed as the exploratory outcome in patients who were anti-AChR antibody positive. At week 4 of cycle 1, the reduction (improvement) of MG-ADL total score in the efgartigimod alfa group was larger than that in the placebo group (efgartigimod alfa minus placebo = −2.84, || || || || || || || | P < 0.0001). This was assessed as an exploratory outcome and with no multiplicity adjustment (not included in the hierarchy test); therefore, there is an increased risk of type I error. However, the findings were aligned with the responder analysis and were considered clinically meaningful by the clinical experts consulted by CADTH. It should be noted that the maximum MG-ADL score change from cycle baseline with efgartigimod alfa appeared to occur at approximately week 4 of the cycle. The magnitude of the improvement and the comparative benefit of efgartigimod alfa compared with placebo tended to smaller at the end of the cycle. Similar results were observed in cycle 2.
Time to re-treatment: Time to qualify for re-treatment in the patients who were anti-AChR antibody positive was assessed as a fourth key secondary outcome. The median time to qualify for re-treatment in the efgartigimod alfa group was numerically but not significantly greater than the time in the placebo group (median = 35 days; 95%CI, 29 days to 43 days; versus 8 days; 95% CI, 1 day to 30 days; respectively; log-rank P = 0.2604). The statistically hierarchy test was broken at this point. || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || || The clinical expert CADTH consulted for this review indicated that the results likely showed that about half of the patients would need to receive re-treatment around week 6 of the treatment cycle.
Disease Severity (Assessed With QMG)
QMG responder during cycle 1: The percentage of QMG responders among patients who were anti-AChR antibody positive was assessed as the first key secondary outcome. It was reported that the 49.0% more patients (95% CI, 34.5% to 63.5%) in the efgartigimod alfa group achieved a QMG response compared with the placebo group (odds ratio = 7.1; 95% CI, 3.24 to 16.49; P < 0.0001). According to the clinical experts, this benefit of treatment with efgartigimod alfa compared with placebo was considered clinically meaningful.
HRQoL
HRQoL (i.e., MG-QOL15 r and EQ VAS) was assessed as an exploratory outcome among patients who were anti-AChR antibody positive. The change from cycle baseline of MG-QOL15r and EQ VAS were assessed for cycle 1 and cycle 2. At week 4 of cycle 1, the reduction (improvement) of MG-QOL15r in the efgartigimod alfa group was greater than that in placebo group (efgartigimod alfa minus placebo = −5.45; 95%CI −7.221 to −3.685; P < 0.0001). The increase (improvement) in EQ VAS score in efgartigimod alfa group was greater than in placebo group (efgartigimod alfa minus placebo = 13.28; 95% CI, 8.32 to 18.24; P < 0.0001). As HRQoL was assessed as an exploratory outcome with no multiplicity adjustment (not included in the hierarchy test), there is an increased risk of type I error; however, the results provide supportive evidence. Although there is no known minimal important difference for either the MG-QoL15r or EQ VAS among patients with gMG, the clinical experts considered the results to be clinically meaningful. It should be noted that the maximum MG-QOL15r and EQ VAS improvement with efgartigimod alfa occurred at approximately week 4 of the cycle. The magnitude of the improvement and the comparative benefit of efgartigimod alfa versus placebo tended to be smaller at the end of the cycle. Similar results were observed in cycle 2.
Other Clinical Outcomes (MG Hospitalization, MG Exacerbations, and MG Crisis)
In patients who were anti-AChR antibody positive, during the 26 weeks DB period, the event rates for MG hospitalization and MG crisis were low in both groups. MG exacerbations were identified in 17 patients (26.2%) in the efgartigimod alfa group and 27 patients (44.3%) in the placebo group. The between-group absolute risk difference (efgartigimod alfa minus placebo) was −18.2 (95% CI, not reported). The results of MG hospitalization, MG exacerbations, and MG crisis were based on post hoc analyses. Therefore, the results for these outcomes were inconclusive.
Harms Results
Reduction of side effects was identified in the patient input for this review as of interest for patients with gMG. The ADAPT trial, including its randomized controlled period and open-label extension, provided relevant information regarding the safety profile of efgartigimod alfa in the treatment of gMG. However, it did not provide direct comparative evidence regarding the adverse effects of efgartigimod alfa versus other active gMG therapies. In the anti-AChR antibody-positive population, during the randomized controlled period, the proportion of patients with TEAEs in the efgartigimod alfa group appeared similar to that in the placebo group (efgartigimod alfa versus placebo = 75.4% versus 84.4%). The proportion of patients with serious AEs was low in both groups and appeared lower in the efgartigimod alfa group than in placebo group (4.6% versus 9.4%) in the ADAPT trial. Withdrawals due to AEs occurred in similar proportions in both the efgartigimod alfa and placebo groups (3.1% versus 4.7%) in the ADAPT trial. No deaths were reported during the DB period; however, the length of follow-up in the trial may not have been long enough to assess this outcome with certainty. The main notable harm (i.e., AEs of special interest for this review) in the system organ class was infections and infestations, which was reported in higher proportion of patients in the efgartigimod alfa group than the placebo group (44.6% versus 34.4%). No meningococcal infections were reported. According to the clinical expert CADTH consulted for this review, the TEAEs reported in the ADAPT trial were expected and commonly seen in existing immunosuppressive treatments and complement C5 inhibitor treatments in gMG.
Critical Appraisal
Appropriate methods of randomization, blinding, and allocation concealment were reported. Outcomes were assessed using validated scales that incorporate physician and patient assessment, as well as end points requiring a combination of clinically meaningful improvement and sustained effect. However, minimal important between-group differences, that is the thresholds used for the GRADE for all outcomes, are not available. Therefore, clinical expert opinion informed the thresholds to determine whether the between-group differences observed for each outcome are clinically meaningful or not. Appropriate statistic method was used in the ADAPT trial. Multiplicity adjustment was used for the primary and 5 key secondary outcomes to control the family-wise type I error (probability of making more than 1 type I error). Overall, the ADAPT trial was relatively well designed; however, there are several potential key limitations of the ADAPT trial, including some notable imbalance of baseline disease characteristics between groups. For example, the proportion of patients who had an MG-ADL total score of 10 or greater, prior combination use of a CS and AChEI, and underwent thymectomy were imbalanced between groups. Furthermore, the proportion of patients who used concomitant AChEIs and concomitant CSs plus AChEIs were also imbalanced between the 2 treatment groups. Whether these baseline imbalances may have introduced bias is uncertain. However, the clinical experts consulted by CADTH for this review indicated that these observed imbalances were unlikely to significantly affect the study results. The efficacy outcome assessment for patients with anti-AChR antibody-negative disease were assessed as sensitivity or subgroup analysis. In addition, the ADAPT trial was a placebo-controlled trial. The comparative efficacy information comparing efgartigimod alfa with existing gMG therapies (e.g., AChEIs, CSs, NSISTs, IVIgs, PEs, C5 complement inhibitors) are unknown. Furthermore, MG-ADL total score change from cycle baseline, HRQoL, and all outcomes examined in cycle 2 were assessed as either tertiary or exploratory outcomes, which were not included in the hierarchy test and were not controlled for type I error. Therefore, the results of all of those tertiary and exploratory analysis should be interpreted with consideration of this limitation. Finally, reduction of steroid use and reduction of high dose of steroid use is 1 treatment goal with efgartigimod alfa; however, these outcomes could not be assessed as, because of the study design, the concomitant treatments were not allowed to change unless used for rescue. The impact of efgartigimod alfa on changes in MG medications could not be evaluated because this was not allowed per the study protocol.
According to clinical experts CADTH consulted for this review, the population included in the ADAPT trial reflects the patients who experience unmet needs in the treatment of gMG in Canadian clinical settings. However, patients with MGFA I (ocular MG) and MGFA V were excluded from the ADAPT trial, so whether the findings of the ADAPT trial can be generalized to patients with MGFA I (ocular M) or MGFA V are uncertain. The clinical experts CADTH consulted for this review indicated that efgartigimod alfa will provide a new treatment for patients with gMG who have anti-AChR antibody-positive disease and likely also for patients have MuSK antibody-positive disease. The clinician group input for this review also indicated that patient with MuSK antibodies might respond to efgartigimod alfa. It is uncertain whether the findings derived from the ADAPT trial can be generalized to patients who were anti-AChR antibody negative, MuSK or double negative anti-AChR antibody negative and MuSK antibody negative. The number of patients with anti-AChR antibody-negative disease were relatively small and the ADAPT trial was not powered for testing the statistically significant between-group difference. Therefore, the comparative efficacy results of efgartigimod alfa versus placebo for patients with anti-AChR antibody-negative disease were inconclusive. In addition, 6 patients (3.6%) were MuSK antibody positive and there was no sensitivity or subgroup analysis for patients who were MuSK antibody positive. The comparative efficacy of efgartigimod alfa versus placebo for patients who were MuSK antibody positive was unknown. Therefore, the findings for overall population were mainly driven by the patients with anti-AChR antibody-positive disease.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group. Following the GRADE approach, evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and the input received from the patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: activities of daily living (proportion of MG-ADL responders in cycle 1 and cycle 2); proportion of early MG-ADL responders in cycle 1; mean proportion of time with a clinically meaningful (≥ 2 point) improvement in MG-ADL score (follow-up = 126 days); time to qualify for re-treatment (up to 168 days); MG-ADL total score change from cycle baseline at week 4 of cycle 1 and cycle 2; disease severity (measured with QMG); HRQoL (MG-QoL15r and EQ VAS change from cycle baseline at week 4 in cycle 1 and cycle 2); and other clinical outcomes (MG hospitalization, MG exacerbation, and MG crisis by week 26) and notable harms (i.e., infections and infestations).
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For this review, the target of the certainty of evidence assessment was based on the presence of absence of a clinically important effect, as informed by the minimal important differences suggested by the sponsor and agreed upon by the clinical experts consulted by CADTH for this review (for change from baseline in MG-ADL score and QMG score), or by thresholds suggested by the clinical experts (for all other outcomes).
Table 3 presents the summary of findings for efgartigimod alfa versus placebo for patients with AChR antibody-positive disease.
Long-Term Extension Studies
Description of Studies
The ADAPT+ study (ARGX-113-1705) is a long-term, single-arm, open-label, multicentre, phase III follow-on study of patients who enrolled in the ADAPT study (ARGX-113-1704, NCT03669588). The primary objective was to evaluate the long-term safety and tolerability of efgartigimod alfa in the anti-AChR antibody-positive subgroup and the secondary objective was to evaluate safety and tolerability in the overall population (anti-AChR antibody positive and negative). Efficacy data were collected as exploratory end points. The ADAPT+ study was conducted at 51 sites, including 41 sites in 14 countries and/or regions that had 1 or more patient roll over from the ADAPT study. Data were collected over a 3-year period in 2 sequential parts (part A = 1 year; part B = 2 years maximum). Part B was added as a protocol amendment to ensure accessibility to efgartigimod alfa until it became commercially available or available through an expanded access program. The results of the long-term extension phase up to 14 cycles (ADAPT+) are also presented in this report. The ADAPT+ trial was still ongoing at the time of this review; therefore, the long-term efficacy and safety outcome of the ADAPT+ trial was based on interim analyses 4 and 5. MG-ADL score, QMG score, and safety outcomes were assessed in the long-term extension study. At the data cut-off (June 30, 2022), 151 patients had rolled over from the ADAPT trial into the ADAPT+ trial, regardless of treatment or placebo group, and 145 patients had received 1 or more partial or complete dose of efgartigimod alfa in the ADAPT+ trial.
Efficacy Results
In terms of MG-ADL (up to 14 cycles) and QMG response (up to 7 cycles), evidence from the ADAPT+ trial appeared consistent with that from the randomized controlled period. Patients who switched from placebo to efgartigimod alfa experienced numeric improvements from baseline in MG-ADL and QMG score in each cycle. However, interpretation of these data was limited by the open-label and descriptive nature of the extension study.
Harms Results
Safety data from the long-term extension phase appeared consistent with those observed in the DB phase with no new safety signals reported.
Table 3: Summary of Findings for Efgartigimod Alfa Versus Placebo For patients With Anti-AChR Antibody-Positive gMG
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Efgartigimod alfa | Difference | |||||
Activities of daily living: MG-ADL score (0 [best] to 24 [worst]) | |||||||
Responders (≥ 2-point reduction for 4 consecutive weeks) during cycle 1 Follow-up: 8 weeks | 129 (1 RCT) | OR = 4.95 (2.21 to 11.53) | 30 per 100 | 68 per 100 (NR) | 38 more per 100 (22 to 56 more per 100) | Moderatea | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important increase in the proportion of MG-ADL responders during the first treatment cycle when compared with placebo. |
Early responders (≥ 2-point reduction during first 2 weeks) during cycle 1 Follow-up: 2 weeks | 129 (1 RCT) | || || |||| || |||| || || | 25 per 100 | 57 per 100 (NR) | 32 more per 100 (NR)b | Moderatec | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important increase in the proportion of early MG-ADL responders during the first treatment cycle when compared with placebo. |
LSM change from cycle baseline at week 4 of cycle 1, points Follow-up: 4 weeks | 129 (1 RCT) | NA | || || || | || || |||| || || | || || |||| || || | Moderated | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important improvement in activities of daily living during the first treatment cycle when compared with placebo. |
Responders (≥ 2-point reduction for 4 consecutive weeks) during cycle 2e Follow-up: 8 weeks of cycle 2 | 94 (1 RCT) | OR = || || |||| || |||| || || | 26 per 100 | 71 per 100 (NR) | 45 more per 100 (NR)b | Lowf | As an add-on to conventional therapy, efgartigimod alfa may result in a clinically important increase in the proportion of MG-ADL responders during the second treatment cycle when compared with placebo. |
LSM change from cycle baseline at week 4 of cycle 2, pointse Follow-up: 8 weeks of cycle 2 | 98 (1 RCT) | NA | || || || | || || |||| || || | || || |||| || || | Moderateg | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important improvement in activities of daily living during the second treatment cycle when compared with placebo. |
Mean % time with a clinically meaningful (≥ 2-point) improvement Follow-up: 126 days | 129 (1 RCT) | NA | 26.7 | 48.7 (|| || |||| ) | 22.1 (|| || ||||) | Moderateh | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important increase in the percentage of time with a clinically meaningful improvement in MG-ADL total score when compared with placebo. |
Disease severity | |||||||
QMG responders (≥ 3 points reduction for 4 consecutive weeks) during cycle 1 Follow-up: 8 weeks | 129 (1 RCT) | OR = 10.84 (4.18 to 31.20) | 14 per 100 | 63 per 100 | 49 more per 100 (35 to 65 more per 100) | Moderatei | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important increase in the proportion of QMG responders during the first treatment cycle when compared with placebo. |
Time to re-treatment | |||||||
Qualified for re-treatment Follow-up: 168 days | 129 (1 RCT) | NA | 89 per 100 | 88 per 100 | 1 less per 100 (NR)b | Moderatej | As an add-on to conventional therapy, efgartigimod alfa likely results in little to no difference in the proportion of patients who qualify for re-treatment when compared with placebo. |
HRQOL | |||||||
LSM change from baseline in MG-QoL15r score (0 [best] to 30 [worst]) at week 4 of cycle 1, pointse Follow-up: 4 weeks | 123 (1 RCT) | NA | || || || | || || |||| || || | −5.45 || || || || | Moderatek | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important improvement in HRQoL as measured by the MG-QoL15r during the first treatment cycle when compared with placebo. |
Mean change from baseline in EQ VAS (0 [worst] to 100 [best]) at week 4 of cycle 1, pointse Follow-up: 4 weeks | 123 (1 RCT) | NA | || || || | || || || | 13.28 || || |||| || || | Moderatel | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important improvement in HRQoL as measured by the EQ VAS during the first treatment cycle when compared with placebo. |
LSM change from baseline in MG-QoL15r score (0 [best] to 30 [worst]) at week 4 of cycle 2, pointse Follow-up: 4 weeks of cycle 2 | 89 (1 RCT) | NA | || || || | || || || | −5.45 || || |||| || || | Lowm | As an add-on to conventional therapy, efgartigimod alfa may result in a clinically important improvement in HRQoL as measured by the MG-QoL15r during the second treatment cycle when compared with placebo. |
Mean change from baseline in EQ VAS (0 [worst] to 100 [best]) at week 4 of cycle 2, pointse Follow-up: 4 weeks of cycle 2 | 89 (1 RCT) | NA | || || || | || || || | 12.24 || || |||| || || | Lown | As an add-on to conventional therapy, efgartigimod alfa may result in a clinically important improvement in HRQoL as measured by the EQ VAS during the second treatment cycle when compared with placebo. |
Other clinical outcomes | |||||||
MG-related hospitalizationse Follow-up: 26 weeks | 129 (1 RCT) | NR | 5 per 100 | 0 (NR) | 5 less per 100 (NR)b | Very lowo | As an add-on to conventional therapy, the evidence is very uncertain about the effect of efgartigimod alfa on the number of hospitalizations when compared with placebo. |
MG exacerbationse Follow-up: 26 weeks | 129 (1 RCT) | NR | 44 per 100 | 26 per 100 (NR) | 18 less per 100 (NR)b | Lowp | As an add-on to conventional therapy, efgartigimod alfa may result in a clinically important reduction in MG exacerbations when compared with placebo. |
|| || |||| || |||| || |||| || |||| || || | || || || | || || || | || || || | || || || | || || || | || || || | || || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |
Adverse events of special interest | |||||||
Infections Follow-up: 26 weeks | 129 (1 RCT) | NR | 34 per 100 | 45 per 100 (NR) | 10 more per 100 (7 less to 27 more per 100) | Moderateq | As an add-on to conventional therapy, efgartigimod alfa likely results in a clinically important increase in the proportion of patients experiencing 1 or more infection when compared with placebo. |
AChR = acetylcholine receptor; CI = confidence interval; gMG = generalized myasthenia gravis; HRQoL = health-related quality of life; LSM = least squares means; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MG-QoL15r = Component Myasthenia Gravis Quality of Life 15 – Revised; NA = not available; NR = not reported; OR = odds ratio; QMG = Quantitative Myasthenia Gravis; RCT = randomized controlled trial; VAS = visual analogue scale.
aRated down 1 level for serious imprecision. The 95% CI excludes the threshold of a 20% difference between groups, as informed by the clinical experts; however, the sample size and number of events do not meet the optimal information size.
bUpon request, the sponsor did not provide the 95% CI for the between-group difference (they indicated that it was not calculable).
cRated down 1 level for serious imprecision. No CI was available for judging the precision of the comparative effect estimate. The number of events does not meet the optimal information size.
dRated down 1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a suggested minimally important difference of 2 points, as defined in the trial and agreed upon by the clinical experts.
eIn the trial, statistical testing for these efficacy outcomes was not adjusted for multiplicity. The results are considered as supportive evidence.
fRated down 1 level serious risk of bias. Not all patients completed the second treatment cycle, so prognostic balance between the groups cannot be ensured. This also meets a −1 level of certainty for serious imprecision. No CI was available for judging precision of the comparative effect estimate. The number of events does not meet the optimal information size.
gRated down 1 level for serious risk of bias. Not all patients completed the second treatment cycle, so prognostic balance between the groups cannot be ensured.
hRated down 1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a 10% to 15% difference between groups, as informed by the clinical experts.
iRated down 1 level for serious imprecision. The 95% CI excludes the threshold of a 20% difference between groups, as informed by the clinical experts; however, the sample size and number of events do not meet the optimal information size.
jRate down 1 level for serious imprecision. No CI was available for judging the precision of the comparative effect estimate. The number of events does not meet the optimal information size.
kRated down 1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a difference of 5 points between groups, as informed by the clinical experts.
lRated down 1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a difference of 10 points between groups, as informed by the clinical experts.
mRated down 1 level for serious risk of bias. Not all patients completed the second treatment cycle, so prognostic balance between the groups cannot be ensured. Rated down 1 level for serious imprecision. The 95% CI includes the possibility of a clinically important effect favouring efgartigimod alfa, based on a threshold of a difference of 5 points between groups, as informed by the clinical experts.
nRated down 1 level for serious risk of bias. Not all patients completed the second treatment cycle, so prognostic balance between the groups cannot be ensured. Rated down 1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a difference of 10 points between groups, as informed by the clinical experts.
oRated down 1 level for serious risk of bias. The analyses of these outcomes were undertaken post hoc, so there is risk of bias in the selection of the reported result. Rated down 2 levels for very serious imprecision. No CI was available for judging the precision of the comparative effect estimate. The number of events does not meet the optimal information size; there were very few or no events in either group.
pRated down 1 level for serious risk of bias. The analyses of these outcomes were undertaken post hoc, so there is risk of bias in the selection of the reported result. Rated down 1 level for serious imprecision. No CI was available for judging the precision of the comparative effect estimate. The number of events does not meet the optimal information size.
qRated down 1 level for serious imprecision. The 95% CI includes the possibility of a trivial effect, based on a threshold of a 10% difference between groups, as informed by the clinical experts.
Source: Clinical Study Report, the sponsor’s submission, additional information provided by the sponsor. The details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
The ADAPT+ study was limited by its open-label and noncomparative design, as it was uncertain whether the results observed may be attributable to the effects of the drugs, including other treatments or natural history of disease. Furthermore, the missing outcomes data and small sample size toward the end of the ADAPT+ trial led to difficulties drawing any firm conclusion on the efficacy and safety of efgartigimod alfa. Due to its open-label nature, the subjective outcomes, (e.g., rates of self-reported AEs), were at risk of bias, potentially in favour of the intervention (i.e., efgartigimod alfa). It is noteworthy that the ADAPT+ trial had fewer scheduled visits for outcome assessments compared with the ADAPT trial. The former only collected MG-ADL score data at week 3 of each study; however, the maximum clinical effect in the ADAPT trial was observed at weeks 4 to 5 of a cycle. Furthermore, the longer-term safety and tolerability profile of efgartigimod alfa was hard to determine as the rates of AEs and serious AEs may be underestimated with less frequent assessment schedule. In the ADAPT+ trial, efficacy was assessed as an exploratory outcome using the patient-reported MG-ADL and physician-reported QMG scales, and patients were to remain on their stable dose and regimen of concomitant gMG treatment during part A of the ADAPT+ trial. Given the presence of rollover effects, efficacy results related to part B may be difficult to interpret as changes were permitted in part B, including changes in the type, dose, or regimen of the concomitant gMG treatment, as well as the additional use of other treatments. Therefore, the confounding effects of other therapies cannot be eliminated in part B. In terms of outcome measures, some important long-term outcomes reported by patients and clinicians were not measured (e.g., HRQoL and exacerbations in the ADAPT+ trial).
External Validity
Because the patients who took part in the open-label long-term safety extension phase were originally from the pivotal ADAPT trial, it is reasonable to expect that the same limitations to generalizability are relevant to the open-label long-term safety extension phase. Given the nature of noncomparative study design, it is not possible to compare the effectiveness and tolerability of efgartigimod alfa as add-on treatment for gMG against others add-on treatments (e.g., IVIg).
Indirect Comparisons
Description of Studies
To date, there have been no clinical trials directly comparing the efficacy of efgartigimod alfa with other treatments in patients diagnosed with gMG. Due to this gap in evidence, the sponsor submitted an indirect treatment comparison (ITC), including a systematic literature review (SLR) and an NMA that provided comparative evidence of the efficacy of efgartigimod alfa relative to ravulizumab and IVIg. The eligible interventions for the ITC were limited to those used in Canada for the treatment of gMG to ensure that the comparators were relevant to the Canadian settings. After feasibility assessment, the following 5 studies were considered eligible for inclusion in the NMA: 2 studies comparing efgartigimod alfa with placebo, 2 studies comparing IVIg with placebo, and 1 study comparing ravulizumab with placebo. All NMAs were performed using a Bayesian framework. Placebo was chosen as the reference treatment for all analyses, given its presence as an anchor treatment in all studies and the outcomes assessed in the network. The clinical end points used for the ITC estimates included change from baseline in MG-ADL and QMG score as these were the most consistently reported outcomes in all studies included in the NMA. Primary analyses were performed at the primary assessment time points for all included studies, ranging from 4 to 26 weeks, and sensitivity analyses were performed at or within ± 2 weeks of week 4, which was the primary assessment time point in ADAPT.
As the sponsor’s reimbursement request is limited to the anti-AChR antibody-positive subpopulation, comparators relevant to that group were used in the primary ITC analysis.
Efficacy
Primary Analyses
The mean differences for change from baseline in MG-ADL score were –2.64 (95% CrI, –4.16 to –1.12) for efgartigimod alfa versus IVIg, and –0.91 (95% CrI, –2.25 to 0.39) for efgartigimod alfa versus ravulizumab. The mean differences for change from baseline in QMG score were –4.39 (95% CrI, –6.95 to –1.81) for efgartigimod alfa versus IVIg, and –2.89 (95% CrI, –4.72 to –1.12) for efgartigimod alfa versus ravulizumab. A change of 2 points in the MG-ADL score and 3 points in the QMG score was estimated to be the threshold of clinical significance in patients with MG.
The sensitivity analyses results for change from baseline in MG-ADL and QMG score at or within of week 4 were consistent with results of the primary analyses.
Additional Analyses
The mean differences for change from baseline in MG-ADL score were –2.64 (95% CrI, –4.18 to –1.12) for efgartigimod alfa versus IVIg, –0.92 (95% CrI, –2.25 to 0.43) for efgartigimod alfa versus ravulizumab, and –1.93 (95% CrI, –3.87 to 0.07) for efgartigimod alfa versus rituximab. The mean differences for change from baseline in QMG score were –4.39 (95% CrI, –7.01 to –1.83) for efgartigimod alfa versus IVIg, –2.89 (95% CrI, –4.72 to –1.06) for efgartigimod alfa versus ravulizumab, and –2.71 (95% CrI, – 5.56 to –0.2) for efgartigimod alfa versus rituximab.
Harms
No analysis of harms was reported in the sponsor-submitted ITC report.
Critical Appraisal
The SLR used to identify relevant studies was methodologically sound in terms of the sponsor using a comprehensive literature search strategy as well as performing study selection, data extraction, and risk of bias assessment in duplicate, and providing a list of excluded studies and justifying the exclusions. However, it was unclear in the ITC report whether the feasibility assessment was carried out by a single or multiple assessors. By conducting a feasibility assessment, the sponsor excluded all head-to-head trials, including those comparing the efficacy of IVIg treatment versus PE, which may have reduced the data informing the NMA. The risk of bias of including studies in the SLR was assessed per individual study; however, it may be different depending on the study outcomes. Analyses were run using a Bayesian framework with placebo as the reference treatment, which was deemed appropriate. Changes from baseline in MG-ADL and QMG scores were considered the best source of comparative efficacy data for this NMA, although these outcomes were not primary or secondary end points for the ADAPT trial. The studies that did not report on MG-MDL or QMG score were excluded even if they reported other relevant outcomes, which may have biased the results, although the extent of bias is uncertain. All trials included in the ITC had sufficiently similar study designs and a common comparison group (placebo). However, there were some important differences between the trials included in the NMA that increase the uncertainty of the analyses. All included studies employed a dosing schedule involving spaced infusions, but only the ADAPT trial used individual patient response to determine subsequent cycles of treatment. The studies included in the ITC analyses ranged in follow-up time from 4 to 26 weeks. All studies allowed the use of concomitant conventional therapy treatments (e.g., CSs, NSISTs), but detailed information on the breakdown of the actual concomitant medications used was not available. In many studies, baseline data were not reported consistently, such as for MGFA at baseline, use of steroids or NSISTs at baseline, disease duration, and history of thymectomy. The primary analyses conducted at the primary assessment time point for all trials could be biased against the ADAPT trial, as they could exclude the best responders to efgartigimod alfa, whereas ITCs conducted at week 4 only could be biased against any treatments that demonstrated improved responses over time. Therefore, sensitivity analyses were performed at or within ± 2 weeks of week 4 to improve the robustness of the ITCs and align with the primary assessment time point of the ADAPT trial.
The results were reported as mean differences and 95% Crls. The evidence is imprecise in the effect estimates from the NMA due to the sparseness of data, with wide Crls. Additionally, heterogeneity between the included studies would be expected to introduce bias into the study estimates observed between the comparators. As all comparator studies were performed exclusively in patients who are anti-AChR antibody positive, all ITC analyses included only patients from the anti-AChR antibody-positive subpopulation, which aligns with the reimbursement request submitted by the sponsor and the approved Health Canada indication. Another important limitation of the presented ITC is the lack of safety and HRQoL data. The results of this ITC are highly uncertain given the inconsistency between trials with respect to dosing regimen (individualized dosing for efgartigimod alfa versus continuous dosing for the comparators), variability in eligibility criteria, and study follow-up times. The ITC estimates were too imprecise to draw a conclusion about the comparative effect of efgartigimod alfa relative to alternative treatments on change from baseline in MG-ADL and QMG scores.
Economic Evidence
Table 4: Cost and Cost-Effectiveness
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with anti-AChR antibody-positive gMG |
Treatment | Efgartigimod alfa plus CT (consisting of AChEIs, CSs, and/or NSISTs) |
Dose regimen | 10mg/kg of body weight administered once weekly for 4 weeks, with subsequent cycles based on clinical evaluation, varying by patient |
Submitted price | $7,900 per 400 mg vial |
Treatment cost | $63,200 to $94,800 per patient per course, or 298,304 to $447,456 per patient per year, depending on patient weight and assuming 4.72 courses per year |
Comparators | Blood products (chronic immunoglobulin and/or PE) plus CT; CT alone. |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (53 years) |
Key data source | ADAPT, a randomized multicentre, double-blind, placebo-controlled trial; the ADAPT+ extension study; a sponsor-submitted NMA |
Key limitations |
|
CADTH reanalysis results | CADTH undertook reanalyses to address several key limitations, including adding rituximab plus CT as a comparator; equalizing the risk of crisis as well as the dose of CS used across health states; using health state utilities derived from the ADAPT trial; and adjusting the distribution of health states for patients exiting a crisis. In the CADTH base case, compared with rituximab plus CT, efgartigimod alfa was associated with an ICER of $1,764,628 per QALY gained (incremental costs = $1,195,367; incremental QALYs = 0.68). A price reduction of 84% (from $7,900 to $1,264 per 400 mg vial) would be needed for efgartigimod alfa to be cost-effective at a WTP threshold of $50,000 per QALY gained compared to rituximab. |
AChR = acetylcholine receptor; AChEI = acetylcholinesterase inhibitor; CS = corticosteroid; CT = conventional therapy; gMG = generalized myasthenia gravis; ICER = incremental cost-effectiveness ratio; LY = life-year; MFGA = Myasthenia Gravis Foundation of America; MG-ADL = Myasthenia Gravis Activities of Daily Living; NMA = network meta-analysis; NSIST = nonsteroidal immunosuppressants; PE = plasma exchange; QALY = quality-adjusted life-year; WTP = willingness to pay.
Budget Impact
CADTH identified the following key limitations with the sponsor’s analysis:
The market uptake and comparator displacement do not reflect the Health Canada indication.
The sponsor’s derivation of the Non-Insured Health Benefits population was inappropriately calculated.
Rituximab was excluded as a comparator.
The proportion of patients eligible for public reimbursement is uncertain and likely differs by comparator.
The analyses were not conducted from a drug plan payer perspective as blood products are not funded by drug plan programs.
The proportion of patients who receive PE was underestimated.
CADTH reanalyses corrected the double-counting of the Non-Insured Health Benefits population, included rituximab as a comparator in both populations, assumed 100% of patients would be publicly reimbursed, and considered both a drug plan payer perspective excluding the cost of blood products, as well as a health care system perspective, where administration costs were included and the cost of blood products was adjusted to reflect PE usage.
CADTH reanalyses suggest that:
For the Health Canada–indicated population, reimbursement of efgartigimod alfa plus conventional therapy for adults with gMG may be associated with a budgetary increase of $378,513,999 over 3 years (year 1 = $85,010,539; year 2 = $133,312,812; year 3 = $160,190,648). This estimate does not consider the likelihood that patients with gMG, beyond those meeting the reimbursement request criteria, would access efgartigimod alfa, and thus may be an underestimation of the cost of reimbursing efgartigimod alfa for the full indicated population.
For the reimbursement request population, reimbursement of efgartigimod alfa plus conventional therapy for adults with anti-AChR antibody-positive gMG whose symptoms persist despite adequate treatment with conventional therapy, efgartigimod alfa may be associated with a budgetary increase of $378,137,376 over 3 years (year 1 = $84,925,953; year 2 = $133,180,165; year 3 = $160,031,258).
The estimated budget impact of reimbursing efgartigimod alfa, in combination with conventional therapy, is sensitive to the perspective taken (drug plan versus health care system), the price of efgartigimod alfa, the proportion of patients who are publicly reimbursed, and the number of additional patients who might receive efgartigimod alfa if funding is not limited to the reimbursement request.
CDEC Information
Members of the Committee
Dr. James Silvius (Chair), Dr. Sally Bean, Mr. Dan Dunsky, Dr. Edward Xie, Mr. Bob Gagne, Dr. Ran Goldman, Dr. Peter Jamieson, Mr. Morris Joseph, Dr. Christine Leong, Dr. Kerry Mansell, Dr. Alicia McCallum, Dr. Srinivas Murthy, Dr. Trudy Huyghebaert, Dr. Danyaal Raza, Dr. Emily Reynen, and Dr. Peter Zed
Meeting date: October 25, 2023
Regrets: Three expert committee members did not attend.
Conflicts of interest: None
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third-party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for noncommercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.