Drugs, Health Technologies, Health Systems
Reimbursement Review
Secukinumab (Cosentyx)
Sponsor: Novartis Pharmaceuticals Canada Inc.
Therapeutic area: Hidradenitis suppurativa
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AN
abscesses and inflammatory nodules
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CrI
credible interval
DIC
deviance information criterion
DLQI
Dermatology Life Quality Index
EQ VAS
EQ-5D visual analogue scale
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HiSCR
Hidradenitis Suppurativa Clinical Response
HLT
high-level term
HRQoL
health-related quality of life
HS
hidradenitis suppurativa
IL
interleukin
ITC
indirect treatment comparison
LS
least squares
NICE
National Institute for Health and Care Excellence
NMA
network meta-analysis
NRS
numerical rating scale
NRS30
at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale
OR
odds ratio
OTC
over the counter
RCT
randomized controlled trial
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
SMQ
Standardised MedDRA (Medical Dictionary for Regulatory Activities) Query
SOC
system organ class
VAS
visual analogue scale
Executive Summary
Submission Update Provided by the Sponsor Dated April 24, 2024
The review of secukinumab was accepted as a pre–Notice of Compliance submission and the Clinical Report was initially drafted based on the draft product monograph. In consideration of the revisions included in the final product monograph, specifically the indication and dosage and administration sections for hidradenitis suppurativa (HS) (summarized in Table 1), additional information relevant to the updated product monograph was extracted from the SUNSHINE and SUNRISE studies (collectively referred to as the SUNNY trials) and the indirect treatment comparison (ITC) submitted by the sponsor. This included results on the comparison between the secukinumab 300 mg every 4 weeks dosage group versus placebo from the SUNNY trials and versus adalimumab from the ITC for the outcomes of interest to this review.
Table 1: Summary of the Revisions to the Product Monograph of Secukinumab
Draft product monograph | Revised (final) product monograph |
|---|---|
Indication | |
Proposed for the treatment of adult patients with moderate to severe hidradenitis suppurativa. | For the treatment of adult patients with moderate to severe hidradenitis suppurativa (acne inversa) who have responded inadequately to conventional systemic hidradenitis suppurativa therapy. |
Recommended dose | |
A 300 mg dose of secukinumab by subcutaneous injection with initial dosing at weeks 0, 1, 2, 3, and 4, followed by a maintenance dose of 300 mg every 2 weeks. Each 300 mg dose is given as 1 subcutaneous injection of 300 mg or as 2 subcutaneous injections of 150 mg. | 300 mg of secukinumab by subcutaneous injection with initial dosing at weeks 0, 1, 2, 3, and 4, followed by monthly maintenance dosing (every 4 weeks). Based on clinical response, the maintenance dose can be increased to 300 mg every 2 weeks. Each 300 mg dose is given as 1 subcutaneous injection of 300 mg or as 2 subcutaneous injections of 150 mg. |
Note: Notice of Compliance was issued on May 17, 2024.
Source: Product monograph for secukinumab.1
Systematic Review Evidence on the Monthly Maintenance Dose of Secukinumab
Detailed outcome data to address the revisions made to the product monograph are presented in Appendix 2.
Results
Sixteen-Week Placebo-Controlled Treatment Period 1
Overall, the direction of treatment effect based on the key efficacy results was consistent between the biweekly and monthly maintenance dosing of secukinumab versus placebo. Statistical significance cannot be claimed for the primary analysis results at week 16 for the abscesses and inflammatory nodules (AN) count and at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale (NRS30 skin pain response) at week 16, despite the P value being less than 0.005, from the SUNSHINE trial for the secukinumab monthly maintenance dosage group versus placebo because the result for the primary end point (at least a 50% decrease in AN count with no increase in the number of abscesses and/or in the number of draining fistulas [HiSCR50 response]), which was a prior end point in the testing hierarchy, was not statistically significant. Results for these end points should be considered as supportive evidence. Overall, no notable differences in the frequency of adverse events (AEs) between the study drug groups were identified in each study.
Entire Study Period
The entire study period consisted of a 16-week placebo-controlled treatment period (treatment period 1), a 36-week treatment period (treatment period 2), and an 8-week follow-up. The results at week 52 were noncomparative and presented descriptively. Overall, the direction of treatment effect based on the key efficacy results was consistent between the biweekly and monthly maintenance dosing of secukinumab. Additionally, no notable differences in the frequency of AEs between the study drug groups were identified in each study.
Critical Appraisal
In general, no notable differences in the study population between the 3 study drug groups (i.e., secukinumab 300 mg every 2 weeks, secukinumab 300 mg every 4 weeks, and placebo) was identified in each study. As such, the limitations discussed for the primary and exploratory efficacy analyses at week 16 and week 52 of the biweekly maintenance dosing are applicable to the corresponding analyses of the monthly maintenance dosing. Overall, no concerns of serious risk of bias and no major issues with the generalizability of the results to the target population and Canadian practice were identified in the appraisal of the SUNNY trials. Notably, there was no active or placebo comparator group for the assessments made at week 52. As such, the ability to draw causal conclusions about the 52-week results is limited because the noncomparative design does not facilitate distinguishing between the effect of treatment, placebo effects, and natural history.
Network Meta-Analyses on the Monthly Maintenance Dose of Secukinumab
Detailed outcome data to address the revisions made to the product monograph are presented in Appendix 2.
The primary evidence network was informed by 4 studies (PIONEER 1, PIONEER 2, SUNSHINE, and SUNRISE) and was limited to patients who were biologic-naive. All results were based on the induction phase of the trials (12 to 16 weeks). Overall, the results for the secukinumab every 4 weeks dosage group were similar to the secukinumab every 2 weeks dosage group. The findings for the secukinumab every 4 weeks dosage group were inconclusive, showing 95% credible intervals (CrIs) that were wide and included the null for secukinumab versus adalimumab in biologic-naive patients. The results of the sensitivity analyses that included biologic-naive and biologic-experienced patients were also inconclusive. These analyses shared the same limitations as discussed for the secukinumab every 2 weeks comparison in the indirect evidence section of this report.
Pharmacoeconomic Review on the Monthly Maintenance Dose of Secukinumab
Economic Impact
The economic review compared secukinumab 300 mg every 2 weeks with standard of care and adalimumab. At the committee meeting, it was noted that the comparison with adalimumab was more relevant, and this informed the pricing condition. As no robust evidence was provided that indicated secukinumab produced better health outcomes than adalimumab, the pricing condition was: “Secukinumab should be negotiated so that it does not exceed the drug program cost of treatment with the least costly form of adalimumab reimbursed for the treatment of HS.” This statement is not unique to 2-week dosing. The same pricing condition would apply to the new draft monograph, which also allows for 4-week dosing, unless the committee felt the 4-week dose would result in better or worse health outcomes for patients relative to adalimumab.
Budget Impact
The budget impact analysis (BIA) was conducted assuming twice-weekly dosing. If dosing every 4 weeks were implemented, this would reduce the drug costs associated with secukinumab and therefore lower the BIA. However, it is unclear how many patients would be placed on this dosing schedule and how many would remain on this schedule. It is also uncertain whether a less frequent dose schedule would increase the size of the market of patients willing to try a biologic; if so, this would increase the budget impact. Overall, there was considerable uncertainty around the size of the original BIA, with the estimate by Canada’s Drug Agency (CDA-AMC) being substantially lower than the sponsor’s submitted BIA (CDA-AMC 3-year BIA: $9,547,349; sponsor-submitted 3-year BIA: $76,542,993). As such, a reimbursement condition was added to the recommendation text stating that uncertainty associated with the BIA must be addressed (refer to reimbursement condition 10). The presence of a different dosing schedule would further increase the uncertainty associated with the BIA.
Executive Summary Before Submission Update
The following report reflects the draft product monograph before the aforementioned revisions were made, hence the focus on the maintenance dose of 300 mg of secukinumab administered every 2 weeks, unless otherwise specified.
An overview of the submission details for the drug under review is provided in Table 2.
Table 2: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Secukinumab (Cosentyx) 75 mg/0.5 mL,a 150 mg/1 mL, and 300 mg/2 mL solution for injection and 150 mg powder for solution for injection.b |
Sponsor | Novartis Pharmaceuticals Canada Inc. |
Indication | For the treatment of adult patients with moderate to severe hidradenitis suppurativa (acne inversa) who have responded inadequately to conventional systemic hidradenitis suppurativa therapy. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | May 17, 2024 |
Recommended dose | 300 mg of secukinumab by subcutaneous injection with initial dosing at weeks 0, 1, 2, 3, and 4, followed by monthly maintenance dosing (every 4 weeks). Based on clinical response, the maintenance dose can be increased to 300 mg every 2 weeks. Each 300 mg dose is given as 1 subcutaneous injection of 300 mg or as 2 subcutaneous injections of 150 mg. |
NOC = Notice of Compliance.
aSecukinumab (Cosentyx) 75 mg/0.5 mL solution for injection is not applicable to the indication for hidradenitis suppurativa.
bThe single-use vial is not available in Canada.
Introduction
HS is a chronic, debilitating skin condition characterized by abscesses that lead to tissue destruction and scarring on the skin, particularly in the skin folds such as the axillae, groin, and perineum.2,3 HS is thought to involve a combination of factors, including immune and endocrine dysregulation, genetics, and bacterial infection.4-10 Key symptoms of HS are pain, itch, malodorous discharge, burning sensations, and local warmth.2,11 The onset of HS typically occurs after puberty, mostly occurring in the second or third decade of life.12 The estimated prevalence of HS in North America and Europe is approximately 1% of the population.13-17 A study of patients with HS living in Canada suggested that approximately 44% of patients have stage 2 disease and 12% of patients have stage 3 disease.18
The clinical experts consulted by CDA-AMC for this review indicated that systemic antibiotics are the first-line systemic therapies in the treatment of HS. The experts indicated that tetracyclines are the most commonly utilized antibiotic class, with prescriptions for doxycycline and tetracycline exceeding those for minocycline. The experts further indicated that clindamycin combined with rifampin and IV ertapenem are used much less frequently than tetracyclines. In general, the North American clinical management guidelines for HS19 (published in 2019) indicate that systemic antibiotics are used as adjunctive therapy in advanced disease because they result in lower response rates and increased recurrence. The experts indicated that patients with moderate to severe HS that has not responded to systemic antibiotic therapy would be eligible for adalimumab, the only biologic therapy currently approved by Health Canada for use in HS. This is aligned with the guidelines,19 which advise on treatment with adalimumab in patients with moderate to severe disease. Other biologics that are not approved for use in HS but are discussed in the guidelines19 for the treatment of moderate to severe HS include infliximab, anakinra, and ustekinumab. The experts indicated that topical therapy that has resulted in a partial response in a patient with moderate to severe HS may be continued as adjunct therapy before starting systemic therapy. More specifically, the guidelines19 reference treatment with topical clindamycin and resorcinol.
When considering the unmet need in HS, the clinician group, Canadian Hidradenitis Suppurativa Foundation, which provided input for this review, and the experts indicated that not all patients experience a response to the currently available treatment options, including adalimumab. The clinician group and the experts also agreed that HS becomes refractory to systemic therapies, including adalimumab, and up-dosing of adalimumab is common to maintain response in clinical practice. The clinician group also indicated that the current management options for HS are not effective in inducing remission.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of secukinumab 150 mg/1 mL and 300 mg/2 mL solution by subcutaneous (SC) injection in the treatment of HS in adult patients with moderate to severe HS.
Note that secukinumab was previously reviewed by CADTH for psoriatic arthritis, ankylosing spondylitis, and plaque psoriasis.
Patient Group and Clinician Group Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from the clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
The Canadian Skin Patient Alliance, HS Heroes, and Hidradenitis and Me Support Group collaboratively provided input for this review. Patient input was gathered from the results of a survey (N = 547) published in the 2020 National Report of Patients’ Experiences Living with Hidradenitis Suppurativa and a patient survey hosted by the patient groups from March 28 to May 23, 2023 (N = 15). Of note, 4 patients from the 2023 patient survey reported prior experience with secukinumab. All respondents indicated their HS lesions are chronic, with the majority being active lesions.
More than 80% of respondents to the 2020 survey reported HS negatively impacted their work performance, social interactions, and intimacy with their partner. Respondents to the 2020 survey reported being worried about odour, staining of clothes, and the unpredictable onset of painful disease flares. Nearly all respondents to the 2020 survey reported experiencing some degree of moderate pain daily; only 11% of survey respondents considered their pain to be well controlled and 46% considered their pain to be poorly controlled. Similarly, respondents to the 2023 survey reported that HS has a severe impact (drainage, severe pain, and lesions) on their day-to-day life. Respondents to the 2023 survey highlighted the high costs associated with wound care and treatment for HS and the high level of anxiety and irritation due to living with HS. When considering unmet needs, 1 respondent to the 2020 survey described their experience with HS as “so painful, so disgusting, and so life-altering.”
In the 2020 survey, respondents reported trying an average of 15 different medications, surgical procedures, home treatments, and lifestyle modifications to manage symptoms, with only a few reporting any significant improvement. Eighty-two percent of survey respondents reported receiving a long course of antibiotics, with 11% reporting improvement in symptoms. Twenty-seven percent of survey respondents reported using biologics, with 38% reporting symptomatic improvement. Other treatments reported by the survey respondents were corticosteroid injections, carbon dioxide lasers, radiotherapy, incision and drainage, and surgical intervention. Overall, 13% of survey respondents reported satisfaction with their current treatments. Respondents reported the following side effects with currently available treatments: back pain, headache, intestinal problems, and fatigue.
The main treatment goals described by the 2020 survey respondents were to achieve symptom control, cure HS, and be able to enjoy personal relationships. Moreover, based on input from the patient groups, patients expressed that they would derive emotional, physical, and daily life benefits with effective therapy. While describing their experience with the current drug under review, 2 of 4 respondents indicated secukinumab to be effective in reducing HS lesions, pain, and the need for wound care. One respondent reported achieving complete resolution of HS lesions and disease remission, while 1 reported treatment discontinuation due to ineffectiveness.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The clinical experts indicated that not all patients respond to currently available treatment options, including adalimumab. The experts estimated that 40% to 60% of patients would experience a partial response to adalimumab and 20% of patients experience a good response to adalimumab. The experts also indicated that HS becomes refractory to systemic therapies, including adalimumab. The experts anticipated secukinumab to be an alternative to adalimumab as a second-line systemic drug used after the failure of systemic antibiotics. The experts anticipated secukinumab would be offered to patients with HS that has not responded to, developed AEs to, or have contraindications to adalimumab. The experts indicated that secukinumab can be offered as the patient’s first biologic therapy. As such, the experts concluded that it may cause a slight shift in the current treatment paradigm. According to the experts, the patient population best suited for treatment with secukinumab are patients with moderate to severe HS that has not responded to systemic antibiotic therapy or antibiotic therapy and adalimumab and who are eligible for adalimumab (i.e., as an alternative to adalimumab).
The experts identified the following as outcomes used in clinical practice to assess response to treatment: lesion count (abscess, nodule, and fistula), pain scale, number of sites involved, extent of disease, and patient-reported outcomes such as the Dermatology Life Quality Index (DLQI), activities of daily living, and health-related quality of life (HRQoL). The experts highlighted the importance of the number of sites involved — a reduction in lesion count with new sites of involvement would likely be interpreted by the patient as treatment failure. The experts indicated that outcomes are typically assessed every 3 to 6 months. When deciding to discontinue treatment with secukinumab, the experts indicated they would consider the following: disease progression, less than 50% improvement after 6 months of treatment, and severe AEs related to secukinumab, such as severe inflammatory bowel disease.
Clinician Group Input
One clinician group, Canadian Hidradenitis Suppurativa Foundation, provided input for this review, with 2 clinicians contributing to this input. When considering unmet needs, the clinician group indicated that current management options are not able to completely control the disease and are not effective in inducing remission; furthermore, some patients may lose benefit with treatment. The clinician group further indicated that a higher dose of the medication (i.e., adalimumab) may be required in patients with severe disease to maintain efficacy. The clinician group noted that adalimumab is the only biologic option approved in Canada for the treatment of HS. According to the clinician group, off-label alternative biologics include infliximab, ustekinumab, interleukin (IL) 17 (IL-17) inhibitors, and IL-1 inhibitors; however, these alternative treatments are offered to patients depending on coverage and compassionate programs.
The clinician group suggested that secukinumab may be an alternative treatment option for patients who would have not demonstrated efficacy with the current standard of care (i.e., secukinumab should be offered as a biologic alternative when treatment with systemic antibiotics after 12 weeks have failed). When considering patients who would be best suited for treatment with the drug under review, the clinician group identified patients with moderate to severe HS (i.e., Hurley stage 2 and 3).
To determine response to treatment, the clinician group suggested achievement of a 50% reduction in abscesses and sinuses with no new lesions after initiation of therapy with secukinumab. The clinician group further suggested patient-reported outcomes, such as pain, odour, and drainage management, as alternative outcome measures.
Drug Program Input
Input was obtained from the drug programs that participate in the Reimbursement Review process. The following items were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for secukinumab:
relevant comparators
consideration for initiation of therapy
consideration for continuation or renewal of therapy
consideration for prescribing of therapy
generalizability
care provision issues
system and economic issues.
The clinical experts consulted by CDA-AMC provided advice on the potential implementation issues raised by the drug programs (Table 5).
Clinical Evidence
Systematic Review
Description of Studies
Two phase III, randomized, double-blind, placebo-controlled, parallel-group trials, SUNSHINE (N = 541) and SUNRISE (N = 543), assessed whether 2 SC secukinumab dose regimens improved HiSCR50 response from baseline compared with placebo after 16 weeks of treatment in adult patients (≥ 18 years) with moderate to severe HS. The outcomes measured in the trials and selected for a Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment were response to treatment and disease severity (HiSCR50 and AN count), disease worsening (patients experiencing flares), symptoms (NRS30 skin pain), HRQoL (DLQI and EQ-5D health state assessment), and notable harms (infections and infestations; candida infections; malignant or unspecified tumours; neoplasms benign, malignant, or unspecified, including cysts and polyps; squamous cell carcinoma of an HS-affected area; and inflammatory bowel disease). Baseline characteristics were generally similar between groups and across trials. Across trials, the mean age of patients ranged from 35.5 years (SD = 10.75) in the placebo group in the SUNSHINE trial to 37.3 years (standard deviation [SD] = 11.48) in the secukinumab group in the SUNRISE trial. At baseline, most patients were categorized with Hurley stage 2 disease severity, ranging from 51.1% of patients (92 of 180) in the secukinumab group in the SUNRISE trial to 67.2% of patients (121 of 180) in the placebo group in the SUNSHINE trial, across trials. At baseline, patients with Hurley stage 3 disease severity ranged from 28.3% of patients (51 of 180) in the placebo group in the SUNSHINE trial to 45.6% (82 of 180 patients) in the secukinumab group in the SUNRISE trial, across trials. The proportions of patients with 1 to 11 anatomic regions with at least 1 fistula, inflammatory nodule, or abscess were generally well balanced between groups and across trials. The mean baseline AN count across trials ranged from 12.8 (SD = 8.15) in the placebo group in the SUNSHINE trial to 13.9 (SD = 9.93) in the secukinumab group in the SUNRISE trial.
Note that 2 different dose regimens were assessed in both trials; however, only the maintenance dose of 300 mg of secukinumab administered every 2 weeks is included in the Health Canada indication. Therefore, only the results of the dose regimen of every 2 weeks are summarized in this report.
Efficacy Results
Response to Treatment and Disease Severity
Hidradenitis Suppurativa Clinical Response
Both the SUNSHINE and SUNRISE studies met the primary end point, achievement of HiSCR50 response at week 16, for the secukinumab 300 mg every 2 weeks dose regimen. In the SUNSHINE trial, the marginal risk difference in HiSCR50 response at week 16 between secukinumab and placebo was █████ (96% confidence interval [CI], ████ ██ █████) (odds ratio [OR] = 1.75; 96% CI, ████ ██ ████; P value = 0.0070), in favour of secukinumab. In the SUNRISE trial, the marginal risk difference in HiSCR50 response at week 16 between secukinumab and placebo was █████ (96% CI, ████ ██ █████) (OR = 1.64; 96% CI, ████ ██ ████; P value = 0.0149), also in favour of secukinumab. The sensitivity analysis, supplementary analysis, and tipping point analysis results for HiSCR50 response at week 16 were generally consistent with and supportive of the primary analysis results for the secukinumab 300 mg every 2 weeks dose regimen in both studies. The results of the subgroup analysis by the key subgroups (concomitant antibiotic use, body weight stratum, previous use of systemic biologics, Hurley stage, and baseline AN count) are generally consistent with the primary analysis, with the exception of the results experienced by patients with Hurley stage 1 in the SUNRISE trial.
The proportion of patients achieving an HiSCR50 response observed at week 52 was an exploratory end point in both studies. In the SUNSHINE trial, █████ (██ of 117 patients; 95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of 58 patients; 95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved HiSCR50 response at week 52. In the SUNRISE trial, █████ (██ of 137 patients; 95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of 64 patients; 95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved HiSCR50 response at week 52.
Abscesses and Inflammatory Nodules Count
Both the SUNSHINE and SUNRISE studies met the secondary end point, percentage change from baseline in AN count at week 16, for the secukinumab 300 mg every 2 weeks dose regimen; this secondary end point was tested in a hierarchical manner to control for type I error rate. In the SUNSHINE trial, the least squares (LS) mean difference in percentage change from baseline in AN count at week 16 between secukinumab and placebo was −23.05 (96% CI, ██████ ██ ██████; P < 0.0001), in favour of secukinumab. In the SUNRISE trial, the LS mean difference in percentage change from baseline in AN count at week 16 between secukinumab and placebo was −16.33 (96% CI, ██████ ██ █████; P = 0.0051), also in favour of secukinumab. The sensitivity analysis and tipping point analysis results for AN count at week 16 were generally consistent with and supportive of the primary analysis results for the secukinumab 300 mg every 2 weeks dose regimen in both studies.
The percentage change from baseline in AN count observed at week 52 was an exploratory end point in both studies. In the SUNSHINE trial, the mean percentage change from baseline in AN count at week 52 was █████ (95% CI, █████ ██ █████) in the secukinumab group and █████ (95% CI, █████ ██ █████) in the placebo-to-secukinumab group. In the SUNRISE trial, the mean percentage change from baseline in AN count at week 52 was █████ (95% CI, █████ ██ █████) in the secukinumab group and █████ (95% CI, ████ ██ █████) in the placebo-to-secukinumab group.
Remission
Disease remission was not measured in the SUNSHINE and SUNRISE trials.
Disease Worsening
Flare
Only the SUNSHINE study met the secondary end point, experience of any flares at week 16, for the secukinumab 300 mg every 2 weeks dose regimen; this secondary end point was tested in a hierarchical manner to control for type I error. In the SUNSHINE trial, the marginal risk difference in flares at week 16 between secukinumab and placebo was ██████ (96% CI, ██████ ██ █████) (OR = 0.42; 96% CI, ████ ██ ████; P value = 0.0010), in favour of secukinumab. In the SUNRISE trial, the marginal risk difference in flares at week 16 between secukinumab and placebo was █████ (96% CI, ██████ ██ ████) (OR = 0.68; 96% CI, ████ ██ ████; P value = 0.0732). The sensitivity analysis and tipping point analysis results of flares at week 16 were generally consistent with and supportive of the primary analysis results for secukinumab 300 mg every 2 weeks dose regimen in both studies.
The proportion of patients experiencing any flares observed at week 52 was an exploratory end point in both studies. In the SUNSHINE trial, █████ (██ of 138 patients; 95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of 65 patients; 95% CI, █████ ██ █████) in the placebo-to-secukinumab group experienced any flares at week 52. In the SUNRISE trial, █████ (██ of 151 patients; 95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of 67 patients; 95% CI, █████ ██ █████) in the placebo-to-secukinumab group experienced any flares at week 52.
Symptoms
Skin Pain
The secondary end point, achievement of NRS30 (reduction in skin pain at its worst) at week 16, for the secukinumab 300 mg every 2 weeks dose regimen was met based on pooled data from the SUNSHINE and SUNRISE studies in patients with a baseline numerical rating scale (NRS) score of 3 or more; this secondary end point was tested in a hierarchical manner to control for type I error. The marginal risk difference in NRS30 at week 16 between secukinumab and placebo was █████ (96% CI, ████ ██ █████) (OR = ████; 96% CI, ████ ██ ████; P value = 0.0003), in favour of secukinumab. The tipping point analysis results of NRS30 at week 16 were supportive of the primary analysis results for secukinumab 300 mg every 2 weeks dose regimen in both studies.
The proportion of patients achieving NRS30 observed at week 52 was an exploratory end point based on pooled data from both trials in patients with baseline NRS of 3 or more. Based on the pooled data, █████ (███ of ███ patients) (95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of ██ patients) (95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved NRS30 at week 52.
Health-Related Quality of Life
Dermatology Life Quality Index
The proportion of patients achieving DLQI response observed at week 16 was an exploratory end point in both studies. In the SUNSHINE trial, the risk difference in DLQI response at week 16 between secukinumab and placebo was ██████ (95% CI, █████ ██ ██████) (OR = ████; 95% CI, ████ ██ ████; P value = ██████). In the SUNRISE trial, the risk difference in DLQI response at week 16 between secukinumab and placebo was █████ (95% CI, ██████ ██ ██████) (OR = ████; 95% CI, ████ ██ ████; P value = ██████).
The proportion of patients with a DLQI response observed at week 52 was an exploratory end point in both studies. In the SUNSHINE trial, 51.0% (49 of 96 patients; 95% CI, █████ ██ █████) in the secukinumab group and 50.0% (25 of 50 patients; 95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved DLQI response at week 52. In the SUNRISE trial, 55.2% (64 of 116 patients; 95% CI, █████ ██ █████) in the secukinumab group and 47.5% of patients (29 of 61; 95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved DLQI response at week 52.
The change from baseline in DLQI total score observed at week 16 was an exploratory end point in both studies. In the SUNSHINE trial, the mean difference in absolute change from baseline in DLQI total score at week 16 between secukinumab and placebo was ████ (95% CI, ████ ██ ████). In the SUNRISE trial, the mean difference in absolute change from baseline in DLQI total score at week 16 between secukinumab and placebo was ████ (95% CI, ████ ██ ████).
The change from baseline in DLQI total score observed at week 52 was an exploratory end point in both studies. In the SUNSHINE trial, the mean absolute change from baseline in DLQI total score at week 52 was ████ (95% CI, ████ ██ ████) in the secukinumab group and ████ (95% CI, ████ ██ ████) in the placebo-to-secukinumab group. In the SUNRISE trial, the mean absolute change from baseline in DLQI total score at week 52 was ████ (95% CI, ████ ██ ████) in the secukinumab group and ████ (95% CI, ████ ██ ████) in the placebo-to-secukinumab group.
EQ-5D Health State Assessment (Visual Analogue Scale)
The change from baseline in the EQ-5D health state assessment visual analogue scale (EQ VAS) observed at week 16 was an exploratory end point in both studies. In the SUNSHINE trial, the mean difference in absolute change from baseline in the EQ VAS score at week 16 between secukinumab and placebo was ███ (95% CI, ████ ██ ███). In the SUNRISE trial, the mean difference in absolute change from baseline in the EQ VAS score at week 16 between secukinumab and placebo was ███ (95% CI, ███ ██ ████).
The change from baseline in the EQ VAS score observed at week 52 was an exploratory end point in both studies. In the SUNSHINE trial, the mean absolute change from baseline in the EQ VAS score at week 52 was ███ (95% CI, ███ ██ ████) in the secukinumab group and ███ (95% CI, ███ ██ ████) in the placebo-to-secukinumab group. In the SUNRISE trial, the mean absolute change from baseline in the EQ VAS score at week 52 was ████ (95% CI, ███ ██ ████) in the secukinumab group and ███ (95% CI, ████ ██ ███) in the placebo-to-secukinumab group.
Harms Results
Adverse Events
In treatment period 1, the proportion of patients with any AE was generally similar between groups and across trials, ranging from 62.8% of patients (113 of 180) in the secukinumab group in the SUNRISE trial to 67.4% of patients (122 of 181) in the secukinumab group in the SUNSHINE trial.
The most common AEs (frequency ≥ 5% in any group) reported in the SUNSHINE trial were nasopharyngitis (11.0% [20 of 181 patients] in the secukinumab group compared with 7.2% [13 of 180 patients] in the placebo group), headache (9.4% [17 patients] compared with 7.8% [14 patients], respectively), hidradenitis (6.1% [11 patients] compared with 13.3% [24 patients], respectively), and diarrhea (2.8% [5 patients] compared with 5.0% [9 patients], respectively).
The most common AEs (frequency ≥ 5% in any group) reported in the SUNRISE trial were headache (11.7% [21 of 180 patients] in secukinumab group compared with 8.2% [15 of 183 patients] in placebo group), nasopharyngitis (7.2% [13 patients] compared with 8.7% [16 patients], respectively), hidradenitis (5.6% [10 patients] compared with 7.7% [14 patients], respectively), upper respiratory tract infection (5.0% [9 patients] compared with 3.8% [7 patients], respectively), and diarrhea (4.4% [8 patients] compared with 7.1% [13 patients], respectively).
In the entire study period, the proportion of patients with any AE continued to be generally similar across trials, ranging from 80.1% (209 of 261 patients) in the any-secukinumab group in the SUNRISE trial to 85.1% (154 of 181 patients) in the secukinumab group in the SUNSHINE trial. The most common AEs (frequency ≥ 10% in any group) reported in both trials were headache, nasopharyngitis, and hidradenitis.
Serious Adverse Events
In treatment period 1, the proportion of patients with any serious adverse event (SAE) was generally similar between groups and across trials, ranging from 1.7% (3 of 181 patients) in the secukinumab group to 3.3% (6 of 180 patients) in the placebo group in the SUNSHINE trial. The most common SAE (frequency ≥ 1% in any group in both trials) reported was hidradenitis in 0.6% (1 of 181 patients) in the secukinumab group and 1.1% (2 of 180 patients) in the placebo group in the SUNSHINE trial, and 0.6% (1 of 180 patients) in the secukinumab group and no patients in the placebo group in the SUNRISE trial.
In the entire study period, the proportion of patients with any SAE was generally similar across trials, ranging from 6.8% (18 of 266 patients) in the any-secukinumab group in the SUNSHINE trial to 10.6% (19 of 180 patients) in the secukinumab group in the SUNRISE trial. In both trials, the most common SAE (frequency ≥ 1% in any group) reported was hidradenitis, reported in 1.7% (3 of 181 patients) in the secukinumab group and 1.5% (4 of 266 patients) in the any-secukinumab group in the SUNSHINE trial, and 2.2% (4 of 180 patients) in the secukinumab group and 1.9% (5 of 261 patients) in the any-secukinumab group in the SUNRISE trial. In the SUNRISE trial, 2 SAEs, acute kidney injury and pyrexia, were reported in 1.1% (2 of 180 patients) in the secukinumab group and 0.8% (2 of 261 patients) in the any-secukinumab group.
Withdrawals Due to Adverse Events
In treatment period 1, the proportion of patients who stopped treatment due to any AE was generally similar between groups and across trials, ranging from 0.6% (1 of 180 patients) in the placebo group to 2.8% (5 of 181 patients) in the secukinumab group in the SUNSHINE trial. No AE that led to treatment discontinuation was reported in 1% or more of patients in any group in either the SUNSHINE or SUNRISE trial.
In the entire study period, the proportion of patients who stopped treatment due to any AE was generally similar across trials, ranging from 3.4% (9 of 261 patients) in the any-secukinumab group in the SUNRISE trial to 5.5% (10 of 181 patients) in the secukinumab group in the SUNSHINE trial. Similar to treatment period 1, no AE that led to treatment discontinuation was reported in 1% or more of patients in any group in either trial.
Mortality
In treatment period 1 and the entire study period, no deaths were reported in either trial.
Notable Harms
In general, AEs of special interest (notable harms) were similar between the secukinumab and placebo groups and across trials in treatment period 1. For infections and infestations (system organ class [SOC]), the risk difference was ████ (95% CI, █████ ██ ████) in the SUNSHINE trial and █████ (95% CI, █████ ██ ████) in the SUNRISE trial. For candida infections (high-level term [HLT]), the risk difference was █████ (95% CI, █████ ██ ████) in the SUNSHINE trial and ████ (95% CI, █████ ██ ████) in the SUNRISE trial. For malignant and unspecified tumour (Standardised MedDRA [Medical Dictionary for Regulatory Activities] Query [SMQ]), the risk difference was █████ (95% CI, █████ ██ ████) in the SUNSHINE trial and █████ (95% CI, █████ ██ ████) in the SUNRISE trial. For neoplasms benign, malignant, and unspecified (including cysts and polyps), the risk difference was █████ (95% CI, █████ ██ ████) in the SUNSHINE trial and █████ (95% CI, █████ ██ ████) in the SUNRISE trial. In treatment period 1, no patients were reported with squamous cell carcinoma of an HS-affected area or inflammatory bowel disease.
In the entire study period, patients with any notable harms continued to be generally similar across trials. Patients reported with infections and infestations (SOC) ranged from 51.7% (93 of 180 patients) in the secukinumab group in the SUNRISE trial to 58.6% of patients (106 of 181) in the secukinumab group in the SUNSHINE trial. Patients reported with candida infections (HLT) ranged from 5.4% of patients (14 of 261) in the any-secukinumab group to 6.7% (12 of 180 patients) in the secukinumab group in the SUNRISE trial. The proportion of patients reported with a malignant or unspecified tumour (SMQ) or neoplasm (benign, malignant, or unspecified, including cysts and polyps) was less than 5% of patients in each group for both trials. Similar to treatment period 1, no patients were reported with squamous cell carcinoma of an HS-affected area or inflammatory bowel disease in the entire study period.
Critical Appraisal
The SUNSHINE and SUNRISE trials were randomized, double-blind, and placebo-controlled. Randomization was stratified by region, concomitant antibiotic use, and body weight. The proportions of patients with the relevant medical history and disease characteristics (effect modifiers) at baseline were generally well balanced between the secukinumab and placebo groups in both trials. There were slightly more patients with Hurley stage 3 disease in the secukinumab group versus placebo. The experts indicated that Hurley stage 3 disease is more severe and difficult to treat and, as such, potential bias against secukinumab may have been introduced in analyses that were unadjusted for this characteristic; however, the magnitude is unclear and could be small. Of note, there was no active or placebo comparator group for the assessments made at week 52 and, as such, the ability to draw definitive conclusions about the 52-week results is limited due to the potential for confounding.
A statistical testing strategy was implemented in both trials to control for type I error rate at the level of the individual studies and at the level of the pooled dataset of both studies. Exploratory end point analyses, including DLQI, EQ-5D health state assessment, and efficacy outcomes at week 52 were not adjusted for multiple comparisons and are therefore at an increased risk of false-positive results. Subgroup analyses were not adjusted for multiple testing; moreover, the ability to draw definitive conclusions about the results is limited because of the relatively small sample size of most subgroups.
There is evidence in the literature to support the measurement properties of HiSCR20,21 as a measure of response to treatment and the clinical importance of HiSCR50 in patients with HS.20,22 There is also evidence in the literature to support the validity of the patient-reported outcomes, NRS30,23 DLQI,24 and EQ-5D health state assessment,25 as a measure of skin pain and HRQoL in patients with HS. Furthermore, there is evidence to support the clinical importance of NRS30 skin pain (albeit only a 30% threshold was suggested and not in patients with HS)26-28 and DLQI response (estimated minimal important difference [MID] of 5 points in patients with HS)22 as defined in the trials. Note that an MID in the EQ-5D health state assessment has not been estimated in patients with HS.
According to the experts, the inclusion and exclusion criteria used in the trials were considered standard in HS. Although some potential candidates for treatment (identified by the experts) were excluded from the trials, the experts indicated the results would likely be applicable in those patients (e.g., patients with fewer than 5 inflammatory lesions). The experts agreed that the criteria for the use of rescue therapy and the options for rescue therapy used in the trials generally reflected clinical practice. According to the feedback from the experts, aside from minocycline, which is used less commonly in practice in Canada, the concomitant use of antibiotics in the antibiotic strata and nonopioid analgesics in the trials was consistent with clinical practice and aligned with the guidelines.19,29 Although topical antibiotic therapy was prohibited in the trials, the experts anticipated that patients would continue topical antibiotic therapy while on treatment with secukinumab if they had previously experienced a partial response to the topical antibiotic therapy.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform the expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.30,31
For RCTs: Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
For single arms of trials (not presented in the summary of findings table): Although GRADE guidance is not available for noncomparative studies, the CDA-AMC review team assessed the noncomparative (52 weeks) outcomes for study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty with no opportunity for rating up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for HiSCR50 response, AN count, flares, NRS30 skin pain, DLQI response, and EQ-5D health state assessment was set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review. The reference point for the certainty of evidence assessment for DLQI total score was set according to the presence or absence of an important effect based on the threshold identified in the literature. The reference points for the certainty of evidence assessment for notable harms was set according to the presence or absence of an important effect based on thresholds informed by the clinical experts.
For the GRADE assessments, findings from the SUNSHINE and SUNRISE studies were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and the input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
Response to treatment and disease severity: HiSCR50 and AN count
Disease worsening: Flares
Symptoms: NRS30 skin pain
HRQoL: DLQI and EQ-5D health state assessment
Notable harms: Infections and infestations; candida infections; malignant or unspecified tumours; neoplasms (benign, malignant, or unspecified, including cysts and polyps); squamous cell carcinoma of an HS-affected area; and inflammatory bowel disease.
Results of GRADE Assessments
Secukinumab Versus Placebo
Table 3 presents the GRADE summary of findings for secukinumab 300 mg every 2 weeks versus placebo as well as secukinumab 300 mg every 4 weeks versus placebo. Note that the data presented in the table on GRADE summary of findings are based on data provided by the sponsor following the submission update dated April 24, 2024 (details in Appendix 2).
Table 3: Summary of Findings for Secukinumab Versus Placebo for Patients With Hidradenitis Suppurativa
Outcome and follow-up | Dose and N (studies) | Relative and absolute effects | Certainty | What happens |
|---|---|---|---|---|
Response to treatment and disease severity | ||||
HiSCR50 response: Proportion of patients with ≥ 50% decrease in AN count with no increase in the number of abscesses and/or in the number of draining fistulas (96% CI for the q.2.w. dosing and 99% CI for the q.4.w. dosing) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE Odds ratio: 1.75 █████ ██ █████
| Moderatea | Secukinumab 300 mg every 2 weeks likely results in a clinically meaningful increase in the proportion of patients with HiSCR50 response when compared with placebo. |
SUNRISE Odds ratio: 1.64 █████ ██ █████
| ||||
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE Odds ratio: 1.48 █████ ██ █████
| Lowb | Secukinumab 300 mg every 4 weeks may result in a clinically meaningful increase in the proportion of patients with HiSCR50 response when compared with placebo. | |
SUNRISE Odds ratio: 1.90 █████ ██ █████
| ||||
AN count: LS mean percentage change from baseline (96% CI for the q.2.w. dosing and 99% CI for the q.4.w. dosing) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE
| Moderatec | Secukinumab 300 mg every 2 weeks likely results in a clinically meaningful reduction in AN count when compared with placebo. |
SUNRISE
| ||||
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE
| Moderatec | Secukinumab 300 mg every 4 weeks likely results in a clinically meaningful reduction in AN count when compared with placebo. | |
SUNRISE
| ||||
Disease worsening | ||||
Flares: Proportion of patients with ≥ 25% increase in AN count with a minimum increase of 2 AN relative to baseline (96% CI for the q.2.w. dosing and 99% CI for the q.4.w. dosing) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE Odds ratio: 0.42 █████ ██ █████
| Lowd | Secukinumab 300 mg every 2 weeks may result in a clinically meaningful decrease in the proportion of patients experiencing flares when compared with placebo. |
SUNRISE Odds ratio: 0.68 █████ ██ █████
| ||||
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE Odds ratio: 0.71 █████ ██ █████
| Lowe | Secukinumab 300 mg every 4 weeks may result in a clinically meaningful decrease in the proportion of patients experiencing flares when compared with placebo. | |
SUNRISE Odds ratio: 0.49 █████ ██ █████
| ||||
Symptoms | ||||
NRS30 skin pain (scored from 0 = no skin pain to 10 = skin pain as bad as you can imagine): Proportion of patients with ≥ 30% reduction and ≥ 2-unit reduction in the patient’s global assessment of skin pain (96% CI for the q.2.w. dosing and 99% CI for the q.4.w. dosing) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE and SUNRISE (pooled data) Odds ratio: ████ █████ ██ █████
| Moderatef | Secukinumab 300 mg every 2 weeks likely results in a clinically meaningful increase in the proportion of patients with NRS30 skin pain response when compared with placebo. |
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE and SUNRISE (pooled data) Odds ratio: ████ █████ ██ █████
| Moderatef | Secukinumab 300 mg every 4 weeks likely results in a clinically meaningful increase in the proportion of patients with NRS30 skin pain response when compared with placebo. | |
Health-related quality of life | ||||
DLQI response: Proportion of patients with ≥ 5-point reduction in DLQI total score (95% CI) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 551 (2 RCTs) | SUNSHINE Odds ratio: ████ █████ ██ █████
| Moderateg | Secukinumab 300 mg every 2 weeks likely results in a clinically meaningful increase in the proportion of patients with DLQI response when compared with placebo. |
SUNRISE Odds ratio: ████ █████ ██ █████
| ||||
Secukinumab 300 mg every 4 weeks: 607 (2 RCTs) | SUNSHINE Odds ratio: ████ █████ ██ █████
| Highh | Secukinumab 300 mg every 4 weeks results in a clinically meaningful increase in the proportion of patients with DLQI response when compared with placebo. | |
SUNRISE Odds ratio: ████ █████ ██ █████
| ||||
DLQI total score (0 [no effect at all on patient’s life] to 30 [extremely large effect on patient’s life]), mean absolute change from baseline (95% CI) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 590 (2 RCTs) | SUNSHINE
| Highi | Secukinumab 300 mg every 2 weeks results in little-to-no clinically meaningful difference in the DLQI total score when compared with placebo. |
SUNRISE
| ||||
Secukinumab 300 mg every 4 weeks: 588 (2 RCTs) | SUNSHINE
| Highi | Secukinumab 300 mg every 4 weeks results in little-to-no clinically meaningful difference in the DLQI total score when compared with placebo. | |
SUNRISE
| ||||
EQ-5D health state assessment (VAS score) (0 [worst imaginable health state] to 100 [best imaginable health state]), mean absolute change from baseline (95% CI) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 585 (2 RCTs) | SUNSHINE
| Lowj | Secukinumab 300 mg every 2 weeks may result in a clinically meaningful improvement in the EQ-5D health state assessment when compared with placebo. |
SUNRISE
| ||||
Secukinumab 300 mg every 4 weeks: 586 (2 RCTs) | SUNSHINE
| Moderatek | Secukinumab 300 mg every 4 weeks likely results in little-to-no clinically meaningful difference in the EQ-5D health state assessment when compared with placebo. | |
SUNRISE
| ||||
Notable harms | ||||
Infections and infestations (SOC), n (95% CI) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE Relative risk: ████ █████ ██ █████
| Lowl | Secukinumab 300 mg every 2 weeks may result in little-to-no difference in infections and infestations when compared with placebo. |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE Relative risk: ████ █████ ██ █████
| Lowl | Secukinumab 300 mg every 4 weeks may result in little-to-no difference in infections and infestations when compared with placebo. | |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
Candida infections (HLT), n (95% CI) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE Relative risk: ████ █████ ██ █████
| Lowm | Secukinumab 300 mg every 2 weeks may result in little-to-no difference in candida infections when compared with placebo. |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE Relative risk: ████ █████ ██ █████
| Lowm | Secukinumab 300 mg every 4 weeks may result in little-to-no difference in candida infections when compared with placebo. | |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
Malignant or unspecified tumours (SMQ), n (95% CI) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE Relative risk: ████ █████ ██ █████
| Very lown | The evidence is very uncertain about the effect of secukinumab 300 mg every 2 weeks on malignant or unspecified tumours when compared with placebo. |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE Relative risk: ████ █████ ██ █████
| Very lowo | The evidence is very uncertain about the effect of secukinumab 300 mg every 4 weeks on malignant or unspecified tumours when compared with placebo. | |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
Neoplasms benign, malignant, or unspecified, including cysts and polyps (SOC), n (95% CI) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE Relative risk: ████ █████ ██ █████
| Very lown | The evidence is very uncertain about the effect of secukinumab 300 mg every 2 weeks on neoplasms benign, malignant, and unspecified (including cysts and polyps) when compared with placebo. |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE Relative risk: ████ █████ ██ █████
| Very lowo | The evidence is very uncertain about the effect of secukinumab 300 mg every 4 weeks on neoplasms (benign, malignant, or unspecified, including cysts and polyps) when compared with placebo. | |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
Squamous cell carcinoma of HS-affected area (PT), n (95% CI) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE Relative risk: ███ █████████
| Very lown | The evidence is very uncertain about the effect of secukinumab 300 mg every 2 weeks on squamous cell carcinoma of HS-affected area when compared with placebo. |
SUNRISE Relative risk: ███ █████████
| ||||
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE Relative risk: ███ █████████
| Very lown | The evidence is very uncertain about the effect of secukinumab 300 mg every 4 on squamous cell carcinoma of HS-affected area when compared with placebo. | |
SUNRISE Relative risk: ███ █████████
| ||||
Inflammatory bowel disease (NMQ), n (95% CI) Follow-up: 16 weeks | Secukinumab 300 mg every 2 weeks: 724 (2 RCTs) | SUNSHINE Relative risk: ███ █████████
| Very lown | The evidence is very uncertain about the effect of secukinumab 300 mg every 2 weeks on inflammatory bowel disease when compared with placebo. |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
Secukinumab 300 mg every 4 weeks: 723 (2 RCTs) | SUNSHINE Relative risk: ███ █████████
| Very lown | The evidence is very uncertain about the effect of secukinumab 300 mg every 4 weeks on inflammatory bowel disease when compared with placebo. | |
SUNRISE Relative risk: ████ █████ ██ █████
| ||||
AN = abscesses and inflammatory nodules; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; DLQI = Dermatology Life Quality Index; HiSCR = Hidradenitis Suppurativa Clinical Response; HRQoL = health-related quality of life; HS = hidradenitis suppurativa; LS = least squares; NA = not applicable; NMQ = SMQ, narrow; NRS = numeric rating scale; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; OR = odds ratio; PT = preferred term; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RCT = randomized controlled trial; RR = risk ratio; SMQ = Standardised MedDRA Query; SOC = system organ class.
Notes: Data presented in this table are based on data provided by the sponsor following the April 24, 2024, submission update (details in Appendix 2).
Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
Applicable to all outcomes of importance in this table: Although some potential candidates for treatment with secukinumab were excluded from the SUNNY trials, in consultation with the 2 clinical experts consulted by CDA-AMC for the purpose of this review, it was concluded that the results are likely generalizable to those patients and, as such, the review team did not rate down for indirectness.
Applicable to the primary and secondary end points in the SUNNY trials: The analysis of the secondary end point (flares at week 16) on the secukinumab q.2.w. dosing group failed to meet statistical significance in the statistical hierarchy in the SUNRISE trial. The analysis of the primary end point, HiSCR50 response at week 16, on the secukinumab q.4.w. dosing group failed to meet statistical significance in the statistical hierarchy in the SUNSHINE trial and, as such, all subsequent tests of the secondary end points were considered not statistically significant. These can be considered as supportive evidence only.
Applicable to the patient-reported outcomes (NRS30 skin pain and HRQoL measures) — analysis of these outcomes was not adjusted for multiplicity and as such, results are considered supportive evidence. Although the outcome measures were subjective, in consideration of the low rates of discontinuation and the double-blind trial design, did not rate down for risk of bias.
Applicable to outcomes for which the analysis did not adjust for Hurley stage (DLQI total score and EQ-5D health state assessment [VAS score]) — in consideration of the small baseline imbalance in Hurley stage 3 (effect modifier identified by the clinical experts) between groups, did not rate down for risk of bias.
aRated down 1 level for serious imprecision; data from both trials show secukinumab may provide benefit or little-to-no benefit, based on a conservative threshold of 100 more per 1,000 patients (50 to 100 per 1,000 was suggested by clinical experts).
bRated down 1 level for serious inconsistency; although the 99% CIs are largely overlapping, there is large variability in the point estimates where SUNSHINE suggests little-to-no important difference while SUNRISE suggests a clinically important benefit. Rated down 1 level for serious imprecision; data from both trials show secukinumab may provide benefit or little-to-no benefit based on a conservative threshold of 100 more per 1,000 patients (50 to 100 per 1,000 was suggested by clinical experts). Although the boundaries of the 99% CIs least favourable to the intervention include the possibility of harm, it was concluded that it did not considerably cross the null (i.e., not a substantial harm); therefore, imprecision was rated down by 1 level only.
cRated down 1 level for serious imprecision; data from both trials show secukinumab may provide benefit or little-to-no benefit based on a conservative threshold of 10% difference (5% to 10% difference was suggested by clinical experts).
dRated down 1 level for serious inconsistency; although the 96% CIs are largely overlapping, there is large variability in the point estimates; where SUNSHINE suggest a clinically important benefit while SUNRISE suggest little-to-no difference. Rated down 1 level for serious imprecision. Data from both trials show secukinumab may provide benefit or little-to-no benefit, based on a conservative threshold of 100 less per 1,000 patients (50 to 100 per 1,000 was suggested by clinical experts).
eDid not rate down for inconsistency; although there is some variability in the point estimates, the 99% CIs are largely overlapping and the following concerns in imprecision that led to the rating down of the level of certainty in the evidence was felt to sufficiently reflect the level of certainty in the evidence. Rated down 2 levels for very serious imprecision; based on a conservative threshold of 100 less per 1,000 patients (50 to 100 per 1,000 was suggested by clinical experts), data from the trials show secukinumab may provide benefit or little-to-no benefit and includes the possibility of harm. The boundary of the 99% CI least favourable to the intervention includes the possibility of harm and it was concluded that it did considerably cross the null (i.e., a substantial harm); therefore, imprecision was rated down by 2 levels.
fRated down 1 level for serious imprecision. Data from the pooled results show secukinumab may provide benefit or little-to-no benefit, based on a conservative threshold of 100 more per 1,000 patients (50 to 100 per 1,000 was suggested by clinical experts).
gRated down 1 level for serious imprecision; data from the trials show secukinumab may provide benefit or little-to-no benefit based on a conservative threshold of 50 more per 1,000 patients (as suggested by clinical experts).
hData from the trials show secukinumab may provide benefit based on a conservative threshold of 50 more per 1,000 patients (as suggested by clinical experts).
iA treatment difference of at least 5 points is considered clinically meaningful (based on literature findings and aligned with clinical expert input); data from both trials show secukinumab may provide a trivial (or no) effect.
jRated down 1 level for serious inconsistency. Minimal overlap of the 95% CIs was considered. Rated down 1 level for serious imprecision. Based on a conservative threshold of 5 points (as suggested by clinical experts), data from both trials show secukinumab may provide benefit or little-to-no benefit.
kRated down 1 level for serious imprecision. Based on a conservative threshold of 5 points (as suggested by clinical experts), data from both trials show secukinumab may provide benefit or little-to-no-benefit.
lIn the absence of a threshold for clinical importance, the null was used. Rated down 2 levels for very serious imprecision. Based on the null, data from both trials show secukinumab may provide benefit and harm.
mIn the absence of a threshold for clinical importance, the null was used. Rated down 2 levels for very serious imprecision. There are very few events; ratio of the upper to the lower bound of the 95% CIs associated with the relative risk from both trials are greater than 3.0; therefore, the number of events is likely far from meeting the optimal information size.32
nIn the absence of a threshold for clinical importance, the null was used. Rated down 1 level for serious indirectness. Follow-up was not sufficiently long to observe events. Rated down 2 levels for very serious imprecision. Few or no events were observed because of insufficient follow-up.
oIn the absence of a threshold for clinical importance, the null was used. Rated down 1 level for serious indirectness. Follow-up was not sufficiently long to observe events. Rated down 2 levels for very serious imprecision. Few or no events were observed because of insufficient follow-up. The ratio of the upper to the lower bound of the 95% CI associated with the relative risk from the trial is greater than 3.0; therefore, the number of events is likely far from meeting the optimal information size.32
Sources: SUNSHINE Clinical Study Report,33 SUNRISE Clinical Study Report,34 and sponsor response to requests on June 19, 2023, July 5, 2023, and May 22, 2024, from CDA-AMC for additional information regarding the secukinumab review.35-37
Long-Term Extension Study
The extension study, NCT04179175, assessing the effects of noninterrupted versus interrupted and long-term treatment of 2 dose regimes of secukinumab in patients with HS was ongoing and no results were available at the time of this report.38
Indirect Comparisons
Description of Studies
The sponsor submitted a network meta-analysis (NMA) that assessed the short-term efficacy (12 to 16 weeks) of secukinumab versus adalimumab for the treatment of adults with moderate to severe HS. The base-case Bayesian NMA was informed by 4 RCTs and limited to patients who were biologic-naive (N = 1,462).
Efficacy Results
For secukinumab 300 mg every 2 weeks versus adalimumab 40 mg weekly, the results of the NMA were inconclusive, showing 95% CrIs that were wide and included the null for HiSCR50, skin pain NRS30 response, the proportion of patients with flares or who achieved a DLQI score of 0 or 1. The change from baseline in AN count and DLQI total score and the multinomial model that examined HiSCR25, HiSCR50, and HiSCR75 response thresholds (i.e., a decrease of 25%, 50%, or 75%, respectively, in the AN count with no increase in the number of abscesses or draining fistulae compared with baseline) also showed a 95% CrI that included the null. The sensitivity analyses that included biologic-naive and biologic-experienced patients showed similar findings.
Harms Results
No safety end points were analyzed in the NMA.
Critical Appraisal
No major issues were identified by CDA-AMC on the methods used to conduct the systematic review or the statistical methods used in the NMA. The evidence networks were sparse, and the analyses were limited to short-term efficacy outcomes at the end of the induction period. There was heterogeneity present for some patient characteristics (e.g., the distribution of males, smokers, and Hurley stage), as well as study characteristics (treatment duration, definition of NRS30 response, and imputation methods for missing study data). Most effect estimates lacked precision, showing 95% CrIs that included the null. Thus, it is unclear whether secukinumab is superior, inferior, or had comparable efficacy to adalimumab 40 mg once daily. The comparative safety is unknown, as there were no safety end points analyzed in the NMA.
Studies Addressing Gaps in the Evidence From the Systematic Review
No additional studies were submitted by the sponsor for this review.
Conclusion
Collectively, the evidence from the SUNSHINE study (N = 541) and the SUNRISE study (N = 543) (referred to as the SUNNY trials) demonstrated that 16 weeks of treatment with secukinumab 300 mg every 2 weeks (biweekly maintenance dosing) likely results in a clinically meaningful improvement in HS (measured by HiSCR50 response, AN lesion count, and NRS30 skin pain response), and may result in a clinically meaningful decrease in the proportion of patients experiencing flares when compared with placebo in patients aged 18 years and older with moderate to severe HS. Uncertainty in the evidence is primarily because, in all cases, the CIs included the potential that the difference compared with placebo is small and unimportant. Findings for secukinumab 300 mg every 4 weeks (monthly maintenance dosing) were similar, but the certainty of evidence was lower for HiSCR50 response because there was some inconsistency across trials in the magnitude of the effect and statistical significance was not reached in the SUNSHINE trial. Impacts on exploratory HRQoL end points for both dosing regimens were difficult to interpret, as the findings differed based on how the change in DLQI and EQ-5D scores were analyzed. Overall, no serious risk of bias concern and no serious concern about the generalizability of results to the population of interest was identified in the appraisal of the placebo-controlled phase of the trials. The results from the sponsor-conducted NMA were inconclusive for the assessment of short-term efficacy (12 to 16 weeks) of secukinumab (both dose regimens) versus adalimumab, showing 95% CrIs that were wide and included the null for all outcomes tested (HiSCR50, AN count, skin pain, flares, HRQoL). Overall, no new concerns with the harms of secukinumab were identified based on the harms data observed with follow-up at 16 and 52 weeks across the SUNNY trials. In the absence of a comparator group for the 52-week follow-up time point, it is not possible to draw definitive conclusions about the harms (and efficacy) of secukinumab versus any comparator, including placebo.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of secukinumab 150 mg/1 mL and 300 mg/2 mL solution by SC injection in the treatment of HS in adult patients with moderate to severe HS.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical experts input. The following has been summarized and validated by the CDA-AMC review team.
HS is a chronic, debilitating skin condition characterized by abscesses that lead to tissue destruction and scarring on the skin, particularly in the skin folds, such as the axillae, groin, and perineum.2,3 HS is thought to involve a combination of factors, including immune and endocrine dysregulation, genetics, and bacterial infection.4-10 Key symptoms of HS are pain, itch, malodorous discharge, burning sensations, and local warmth.2,11 The pain caused by HS impacts physical functioning and activities of daily living,39 and HS flares impact work and productivity.40-43 Additionally, HS has a negative impact on emotional and social functioning,40,44 sexual health,45 and the quality of life of patients living in Canada.44 HS is associated with comorbidities such as metabolic syndrome, diabetes mellitus, inflammatory bowel disease,46-48 and anxiety and depression.49
Smoking is a risk factor for the development of HS; approximately 90% of patients with HS are smokers,50 and smoking is associated with more severe HS presentations compared with nonsmokers.8 A meta-analysis investigating the association between current smoking status and HS found there was a 4.2 times higher risk of developing HS in active smokers.51
The onset of HS typically occurs after puberty, mostly occurring in the second or third decade of life.12 A survey of patients with HS, including 30% of patients living in Canada, reported an average age of diagnosis of 32 years.52 The estimated prevalence of HS in North America and Europe is approximately 1% of the population.13-17 A study of patients with HS living in Canada suggested that approximately 44% of patients have stage 2 disease and 12% of patients have stage 3 disease.18
Diagnosis is based on a visual examination of patients and the reporting of symptoms by patients.53,54 The Dessau criteria are used to diagnose HS by characterizing recurrent, painful, or suppurating nodules that are present on 2 or more occasions within 6 months.55 In general, the diagnosis of HS is based on 3 diagnostic criteria: lesion morphology (e.g., nodules, abscesses, or fistulas), distribution of lesions (e.g., axillary, inframammary, or perineal), and chronicity and recurrence (e.g., whether more than 2 lesions are present during a time period of 6 months or more).56 Imaging may also be used to confirm the diagnosis and staging of HS; skin ultrasonography and MRI scans can detect specific features of HS.57-59
The severity of HS is evaluated using the Hurley staging system, which is based on the extent of inflammatory lesions, skin tunnels, and scarring.60-62 Disease severity is classified into 3 groups: Hurley stage 1 (mild disease typically presenting as inflammatory nodules or abscess formation without sinus tracts and scarring), Hurley stage 2 (moderate disease typically presenting as recurrent abscesses and nodules with sinus tract formation or scarring either as single or multiple widely separated lesions), and Hurley stage 3 (severe and refractory disease typically presenting as diffuse or near-diffuse involvement with multiple interconnected sinus tracts, scarring, and abscesses across an entire area).
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical experts input. The following has been summarized and validated by the CDA-AMC review team.
The clinical experts consulted by CDA-AMC for the purpose of this review identified the following as goals of therapy in HS: reduce active inflammatory lesion count, achieve pain relief (i.e., discontinue opioids, if possible), prevent progression (i.e., absence of disease occurrence in new body areas), reduce need for surgery and improve surgical outcomes (i.e., reduce visits to primary care provider and emergency department), improve quality of life, and improve productivity, if applicable.
The experts indicated that systemic antibiotics are the first-line systemic therapies in the treatment of HS; a 3-month trial is typically required to assess efficacy. The experts indicated that tetracyclines are the most commonly utilized antibiotic class, with prescriptions for doxycycline and tetracycline exceeding those for minocycline. The experts further indicated that clindamycin combined with rifampin and IV ertapenem are used much less frequently than tetracyclines. In general, the North American clinical management guidelines for HS19 (published in 2019) indicated that systemic antibiotics are used as adjunctive therapy in advanced disease due to lower response rates and increased recurrence. Specific to moderate to severe disease, the guidelines19 reference treatment with moxifloxacin, metronidazole, and rifampin in combination as second- or third-line therapy. The guidelines19 note that the duration and frequency of antibiotic use should balance treatment benefit with the risk of antibiotic resistance.
The experts indicated that patients with moderate to severe HS that has not responded to systemic antibiotic therapy would be eligible for adalimumab, the only biologic therapy currently approved by Health Canada for use in HS. This is aligned with the guidelines,19 which advise on treatment with adalimumab in patients with moderate to severe disease. Note that some adalimumab biosimilars are listed for the indication under review in most jurisdictions in Canada. Other biologics that are not approved for use in HS but are discussed in the guidelines19 for moderate to severe HS include infliximab, anakinra, and ustekinumab.
The experts further indicated that topical therapy may be continued as adjunct therapy in a patient with moderate to severe HS who has experienced a partial response to the topical therapy before starting systemic therapy. More specifically, the North American clinical management guidelines for HS19 reference treatment with topical clindamycin and resorcinol; however, they are associated with risk of bacterial resistance and contact dermatitis, respectively.
Additionally, lifestyle modifications and alternative treatments, antibacterial washes, wound care, laser treatment, surgical intervention, immunosuppressants, hormonal therapy, and retinoids in the management of HS; steroid therapy in the management of HS flares; and analgesics in the management of pain due to HS are discussed in the guidelines.19,29 Of note, the experts indicated that systemic immunosuppressants (i.e., methotrexate, azathioprine, and cyclosporine) and retinoids (i.e., isotretinoin, acitretin, and alitretinoin) are no longer routinely used in practice for HS due to their poor efficacy, and systemic steroids are not suitable for long-term management of HS due to their potential side effects.
Drug Under Review
Key characteristics of secukinumab are summarized in Table 4 with the other treatments available for moderate to severe HS.
Secukinumab is indicated for the treatment of adult patients with moderate to severe HS (acne inversa) who have responded inadequately to conventional systemic HS therapy. The reimbursement request is as per the Health Canada indication. The recommended dose is 300 mg of secukinumab by SC injection with initial dosing at weeks 0, 1, 2, 3, and 4, followed by monthly maintenance dosing (every 4 weeks). Based on clinical response, the maintenance dose can be increased to 300 mg every 2 weeks. Each 300 mg dose is given as 1 SC injection of 300 mg or as 2 SC injections of 150 mg.1
Secukinumab is a human immunoglobulin G1 kappa monoclonal antibody that selectively binds to IL-17A, a naturally occurring cytokine involved in inflammatory and immune responses. IL-17A is upregulated in HS lesions compared with skin lesions in patients with psoriasis and healthy controls. Secukinumab targets IL-17A, inhibiting the interaction it has with IL-17 receptors, thus inhibiting the release of cytokines and chemokines, which promote inflammation.1
Note that secukinumab has been previously reviewed by CDA-AMC for psoriatic arthritis, ankylosing spondylitis, and plaque psoriasis.
Table 4: Key Characteristics of Secukinumab and Adalimumab
Characteristic | Secukinumab | Adalimumab |
|---|---|---|
Mechanism of action | Secukinumab is a human IgG1 kappa monoclonal antibody that targets IL-17A, inhibiting the interaction it has with IL-17 receptors, thus inhibiting the release of cytokines and chemokines which promote inflammation. | Adalimumab is a recombinant human IgG1 monoclonal antibody that binds with high affinity and specificity to soluble TNF alpha but not lymphotoxin (TNF beta). |
Indicationa | For the treatment of adult patients with moderate to severe HS (acne inversa) who have responded inadequately to conventional systemic HS therapy. | Treatment of active moderate to severe HS in adult and adolescent patients (12 years to 17 years weighing ≥ 30 kg) who have not responded to conventional therapy (including systemic antibiotics). |
Route of administration | Subcutaneous | Subcutaneous |
Recommended dose | 300 mg of secukinumab by subcutaneous injection with initial dosing at weeks 0, 1, 2, 3, and 4, followed by monthly maintenance dosing (every 4 weeks). Based on clinical response, the maintenance dose can be increased to 300 mg every 2 weeks. | An initial dose of 160 mg, followed by an 80 mg dose 2 weeks later. The maintenance dose is 40 mg every week beginning 4 weeks after the initial dose. |
Serious adverse effects or safety issues | Patients should be monitored for signs and symptoms of active tuberculosis and inflammatory bowel disease. | Contraindicated in patients with severe infections such as sepsis, tuberculosis, and opportunistic infections, and with moderate to severe heart failure (NYHA class III or IV). |
HS = hidradenitis suppurativa; IgG1 = immunoglobulin G1; IL = interleukin; NYHA = New York Heart Association; TNF = tumour necrosis factor.
aHealth Canada–approved indication.
Sources: Product monographs for secukinumab and adalimumab.1,63
Patient Group and Clinician Group Perspectives
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website publicly accessible here.
Patient Group Input
This section was prepared by the CDA-AMC review team based on the input provided by patient groups.
The Canadian Skin Patient Alliance, HS Heroes, and Hidradenitis and Me Support Group collaboratively provided input for this review. Patient input was gathered from the survey results from the 2020 National Report of Patients’ Experiences Living With Hidradenitis Suppurativa and a patient survey hosted by the patient groups from March 28 to May 23, 2023. The 2020 survey gathered responses from 537 individuals with HS, of which 73 (14%) were from Canada, 267 (50%) were from the US, and 67 (12%) were from the UK. The greatest proportion of participants living in Canada were from Ontario (41%), followed by Alberta (18%). In total, the 2023 survey received 15 responses; all respondents were from Canada, with the majority being from Ontario (47%), followed by Alberta (27%). Four patients who responded to the 2023 survey mentioned using secukinumab previously; 1 of these patients mentioned stopping it during treatment because of ineffectiveness.
More than 80% of respondents from the 2020 survey reported that HS negatively impacted their work performance (81%), social interactions, and intimacy with their partner. Patients reported being worried about the odour, staining of clothes, and the unpredictable onset of very painful disease flares. Intimacy and family life were affected by living with HS, as reported by 87% and 68% of survey respondents, respectively. Nearly 70% of respondents reported feelings of depression because of a variety of stressors related to HS. Per the 2020 national report, nearly all patients reported experiencing some degree of moderate pain daily (5.3 on a scale of 10). Only 11% of all respondents considered their pain well controlled, while 46% reported poorly controlled pain. Moreover, 51% of patients reported self-managing with difficulty the process of accessing prescriptions. Respondents from the 2023 survey identified the severe impact of HS on their day-to-day life, including drainage, severe pain, lesions that make it challenging to walk, and challenges to find clothes. The costs of wound care and treatments were reported to be high, as was the high anxiety and irritation from living with HS. All patients reported their HS lesions to be chronic, with the majority of patients constantly having active HS lesions.
Misdiagnosis and delayed diagnosis of HS have been identified as an issue for patients in Canada with HS, often translating into worse symptoms and more advanced disease by the time patients reached the point of being offered a treatment plan. According to a 2020 CPSA report, people living in Canada who responded to a survey reported a median of 7 years from symptom onset to HS diagnosis, and the average age of diagnosis was 30 years. The survey respondents reported facing many unmet needs, and 1 respondent described the condition as “so painful, so disgusting, and so life-altering.”
In the 2020 survey, respondents reported trying an average of 15 different medications, surgical procedures, home treatments, or lifestyle modifications to help manage symptoms, with only a few finding any significant improvement. Fifty-three percent of respondents in Canada reported receiving at least 1 corticosteroid injection, while 18% reported receiving injections more than 10 times. Additionally, 74% reported having at least 1 boil or cyst incised or drained, and 19% reported undergoing this procedure more than 10 times. Moreover, 82% of survey participants reported getting a long course of antibiotics, with 11% reporting improvement in symptoms. Other treatments reported by the respondents were carbon dioxide lasers (26% effective), radiotherapy (33%), incision and drainage (23%), and surgical therapies other than incision and drainage (39%). Only 27% of respondents mentioned using biologics, with 38% reporting symptomatic improvement. Overall, only 13% of respondents reported being satisfied with the current treatments. In addition, less than 35% of respondents reported using available treatments; based on this information, the patient groups suggested that access to and affordability of treatments for HS are challenges that patients face. Back pain, headache, intestinal problems, and fatigue were some of the side effects reported by the respondents regarding currently available treatments.
The main treatment goals described by the 2020 survey participants were to control HS symptoms (90%), cure HS completely (71%), and be able to enjoy personal relationships (69%). Moreover, according to the input from the patient groups, patients expressed that they would derive emotional, physical, and daily life benefits with effective therapy. While describing the experience with the current drug under review, 2 of the 4 patients indicated secukinumab was effective in reducing their HS lesions and pain and decreasing the need for wound care, whereas 1 patient reported having complete resolution of lesions and remission of HS. All patients taking secukinumab reported concerns with the cost of treatment and an inability to afford it.
Clinician Input
Input From the Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of HS.
Unmet Needs
The experts indicated that not all patients respond to currently available treatment options, including adalimumab; the experts estimated that 40% to 60% of patients would have a partial response to adalimumab and 20% of patients would have a good response to adalimumab. The experts also indicated that HS becomes refractory to systemic therapies, including adalimumab; up-dosing of adalimumab is commonly used to maintain response in clinical practice.
Place in Therapy
The experts suggested it would be reasonable to recommend at least 1 adequate trial of systemic antibiotic therapy before initiating treatment with secukinumab, given the relative cost of antibiotics versus biologics.
The experts anticipated secukinumab being an alternative treatment option to adalimumab as a second-line systemic drug used after the failure of systemic antibiotics. The experts anticipated secukinumab being offered to patients with HS that has not responded to, developed AEs to, or have contraindications to adalimumab. The experts indicated that secukinumab can be offered as the patient’s first biologic therapy. As such, the experts concluded that it may cause a slight shift in the current treatment paradigm.
The experts expected that secukinumab will be used in combination with topical therapies, antiandrogen therapies, intralesional corticosteroids, and deroofing surgery. The experts also expected that patients treated with secukinumab may also receive treatment with systemic antibiotic therapy for flares.
Patient Population
According to the experts, the patient population best suited for treatment with secukinumab are patients with moderate to severe HS that has not responded to systemic antibiotic therapy or antibiotic therapy and adalimumab and who are eligible for adalimumab (i.e., as an alternative to adalimumab).
According to the experts, the patient population least suitable for treatment with secukinumab are patients with mild HS (which would extend beyond the Health Canada indication), no prior experience with systemic therapy, and relative contraindication(s) to IL-17 inhibitors, such as active inflammatory bowel disease, active infection, malignancy, and severe candidiasis.
The experts indicated that HS is a clinical diagnosis made by dermatologists (i.e., diagnostic tests are not required) and misdiagnosis is common before the patient presents to a dermatologist.
Assessing the Response to Treatment
The experts identified the following as outcomes used in clinical practice to assess response to treatment: lesion count (abscess, nodule, and fistula), pain scale, number of sites involved, extent of disease, and patient-reported outcomes such as DLQI, activities of daily living, and HRQoL. The experts indicated that outcomes are typically assessed every 3 to 6 months.
The experts also noted the importance of the number of sites involved, as a reduction in lesion count with new sites of involvement would likely be interpreted as treatment failure by the patient.
Discontinuing Treatment
When deciding to discontinue treatment with secukinumab, the experts indicated they would consider the following: disease progression, less than 50% improvement after 6 months of treatment, and severe AEs related to secukinumab, such as severe inflammatory bowel disease.
Prescribing Considerations
The experts suggested community or home treatment, under the direction of a dermatologist, would be an appropriate setting for treatment with secukinumab.
Clinician Group Input
This section was prepared by the CDA-AMC review team based on the input provided by 1 clinician group.
Clinician group input on the review of secukinumab was received from a Canadian Hidradenitis Suppurativa Foundation group. Two clinicians provided input for this review.
The clinician group mentioned that the management of the disease is currently based on staging. The group also mentioned that adalimumab is the only approved biologic option in Canada. They also added that there are no additional Health Canada–approved options for patients with HS that has not responded to adalimumab. The clinician group pointed out that some off-label alternative biologics, such as infliximab, ustekinumab, IL-17 inhibitors, and IL-1 inhibitors are offered to patients, depending on the availability of extended benefits coverage or compassionate programs. The clinician group further added that current management options are not effective in inducing remission in patients with HS.
While describing treatment gaps or unmet needs, the clinician group noted that current management options are not fully able to control the disease. They also mentioned that for patients with severe disease, higher doses are sometimes required to maintain efficacy despite the initiation of adalimumab, whereas some patients also lose benefits. The clinician group described the role of secukinumab in addressing a different mechanism of action in the pathogenesis of HS, thus providing the clinicians with an alternative treatment option. More specifically, the clinician group suggested that secukinumab may be an alternative treatment option for patients who have not experienced efficacy with the current standard of care (i.e., secukinumab should be offered as a biologic alternative to patients with HS that has not responded to treatment with systemic antibiotics after 12 weeks).
While describing patients who would be best suited for treatment with the drug under review, the clinician group mentioned those with moderate to severe HS (Hurley stage 2 and 3). While mentioning the outcomes that would indicate a response, the clinician group referred to the currently available RCT evidence, suggesting the achievement of a 50% reduction in abscesses and sinuses with no new lesions after initiation of therapy with secukinumab. The group also suggested alternative measures, particularly in the forms of PATIENT-REPORTED outcomes such as pain, odour, and drainage management. The group also suggested that the decision to initiate secukinumab in the management of HS should be decided by a dermatologist.
Drug Program Input
The drug programs provide input on each drug being reviewed through our Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and the corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
In the context of currently available treatment options for moderate to severe HS in Canada, is placebo an appropriate comparator? | The clinical experts indicated that placebo is an appropriate comparator. However, the experts noted that the ideal comparator would be other biologics, in particular, adalimumab. |
Patients in the antibiotic strata were allowed to enter the SUNSHINE and SUNRISE trials on a stable dose of permitted antibiotics. Could this have affected the results? Should antibiotics be considered a relevant comparator? | The clinical experts agreed that patients in the antibiotic strata entering the trials on a stable dose of a permitted antibiotic would have an impact on the results. The clinical experts agreed that antibiotics should be considered a relevant comparator. The CDA-AMC team notes that the proportion of patients in the secukinumab every 2 weeks and placebo groups that enrolled in the antibiotic strata were similar between groups in both the SUNSHINE and SUNRISE trials. |
Adalimumab received a positive recommendation for the indication under review and has established criteria in its recommendation. Adalimumab is listed for this indication (as well as some biosimilars) in most jurisdictions. | Comment from the drug programs to inform CDEC deliberations. |
Considerations for initiation of therapy | |
The proposed indication is pre-NOC; should the considerations for initiation of therapy be more restrictive or aligned with adalimumab? | The clinical experts agreed it would be reasonable for the considerations for the initiation of secukinumab to be aligned with the reimbursement recommendation and conditions for adalimumab. |
Should patients with HS need to fail a conventional treatment, such as oral antibiotics, as was included in the adalimumab recommendation, before starting secukinumab? | The clinical experts indicated it would be reasonable to recommend at least 1 adequate trial of systemic antibiotic therapy before initiating treatment with secukinumab. The clinical experts agreed that secukinumab should be prescribed only after a failed trial of antibiotics, similar to the conditions for adalimumab. |
Should 1 biologic be preferred over other biologics in the treatment of HS? | In the absence of a direct treatment comparison with relevant comparators, the clinical experts suggested that whether 1 biologic should be preferred over other biologics should be based on clinician judgment. |
Considerations for continuation or renewal of therapy | |
Consider alignment with criteria for adalimumab. | Comment from the drug programs to inform CDEC deliberations. |
Considerations for prescribing of therapy | |
How do both secukinumab and adalimumab fit into therapy? | In terms of its place in therapy, the clinical experts anticipated that secukinumab will, like adalimumab, be a second-line systemic drug used after the failure of systemic antibiotics. The clinical experts indicated that secukinumab may be offered to patients with HS that has not responded to, have contraindications to, or experienced adverse events related to adalimumab. Additionally, the clinical experts suggested secukinumab may be offered before adalimumab as the patient’s first biologic therapy. |
There may be interest in combining secukinumab with other biologics because of different mechanisms of action. Would this be a concern? | The clinical experts indicated that in practice, it is highly unlikely that 2 biologics would be combined in the treatment of HS. |
Should prescribing of secukinumab be consistent with adalimumab or managed separately? | The clinical experts suggested that secukinumab should be prescribed by a dermatologist. |
There may be limited access to specialists in some regions. | Comment from the drug programs to inform CDEC deliberations. |
Generalizability | |
Patients were excluded if they had 20 or more fistulae at baseline, had ongoing active conditions requiring treatment with a prohibited medication (e.g., systemic biological immunomodulating treatment, live vaccines, or other investigational treatments), and patients with fewer than 5 lesions, or lesions in only 1 area, or had been diagnosed for less than 1 year. Should patients with these characteristics be considered for treatment with secukinumab as well? | The clinical experts noted that patients with fewer than 5 inflammatory lesions and patients who have a history of numerous lesions may be candidates for treatment in clinical practice, as HS fluctuates in disease severity independent of treatment. Regarding the use of prohibited medications in the SUNNY trials, the clinical experts anticipated that patients would remain on topical antibiotic therapy while on treatment with secukinumab if the patient experienced a partial response to the topical antibiotic therapy before receiving secukinumab. The clinical experts also noted that opioid analgesics can occasionally be prescribed for patients with HS. Additionally, the clinical experts noted that patients with previous exposure to an IL-17 inhibitor would be candidates for treatment in clinical practice. |
Care provision issues | |
Patients were allowed to continue antibiotic and topical therapy in the studies. Is this a required or recommended practice? Are antibiotic and topical therapy considered adjunctive therapy? | The CDA-AMC team notes that oral antibiotics (minocycline or doxycycline up to 100 mg twice daily) and unplanned surgery or intervention (e.g., intralesion steroid administration) were permitted as rescue therapies in the SUNSHINE and SUNRISE trials. The CDA-AMC team also notes that oral antibiotics in the antibiotic strata only (tetracycline up to 500 mg twice daily, minocycline up to 100 mg twice daily, and doxycycline up to 100 mg twice daily), and systemic antibiotics for the treatment of acute systemic infectious disease, both related to and unrelated to HS, were permitted as medically warranted. Additionally, daily topical OTC antiseptics and wound care dressings were permitted concomitant therapies in both trials. The clinical experts indicated that the drugs considered as concomitant and/or adjunctive therapy in the treatment of HS would depend on the clinician. |
System and economic issues | |
If secukinumab is recommended as a first-line option, this will have significant budget impact. | Comment from the drug programs to inform CDEC deliberations. |
Adalimumab and its biosimilars have achieved confidential negotiated prices. | Comment from the drug programs to inform CDEC deliberations. |
CDA-AMC = Canada’s Drug Agency; CDEC = Canadian Drug Expert Committee; HS = hidradenitis suppurativa; IL = interleukin; NA = not applicable; NOC = Notice of Compliance; OTC = over the counter.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of secukinumab 150 mg/1 mL and 300 mg/2 mL solution by SC injection in the treatment of adult patients with moderate to severe HS. The focus will be placed on comparing secukinumab with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of secukinumab is presented in 2 sections, with our critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor. No long-term extension studies and no additional studies were submitted by the sponsor.
Included Studies
Clinical evidence from the following is included in the CDA-AMC review and appraised in this document:
2 pivotal studies identified in systematic review
1 ITC.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Detail | SUNSHINE | SUNRISE |
|---|---|---|
Design and population | ||
Study design | Phase III, multicentre, randomized, double-blind, placebo-controlled, parallel-group study | |
Locations | 111 sites in North America (4 sites in Canada), South America, Europe, Asia, Oceana | 108 sites in North America (3 sites in Canada), South America, Europe, Asia, Oceana, and Africa |
Patient enrolment dates | FPFV: January 31, 2019 | FPFV: February 25, 2019 |
Randomized (N) | N = 541:
| N = 543:
|
Inclusion criteria |
| |
Exclusion criteria (key) |
| |
Drug | ||
Intervention | Loading dose of secukinumab 300 mg SC at baseline and then once weekly for 4 weeks, followed by secukinumab 300 mg SC every 2 weeks or every 4 weeks | |
Comparator | Placebo SC once weekly for 5 weeks, followed by placebo every 2 weeks or every 4 weeks | |
Study duration | ||
Screening phase | Up to 4 weeks | |
Treatment phase |
| |
Follow-up phase | 8-week posttreatment follow-up or enrolment into a 4-year long-term extension phase | |
Outcomes | ||
Primary end point | Proportion of patients achieving HiSCR50 at week 16 | |
Secondary and exploratory end points | Secondary:
Exploratory:
| |
Publication status | ||
Publications |
|
|
AN = abscesses and inflammatory nodules; CRP = C-reactive protein; DLQI = Dermatology Life Quality Index; ESR = erythrocyte sedimentation rate; FPFV = first patient first visit; HiSCR = Hidradenitis Suppurativa Clinical Response; HS = hidradenitis suppurativa; HS-PGA = Hidradenitis Suppurativa Physician’s Global Assessment; IL = interleukin; mHSS = modified Hidradenitis Suppurativa Score; NRS = numerical rating scale; OTC = over the counter; PGI-S = Patient Global Impression–Severity; PGIC = Patient Global Impression of Change; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SC = subcutaneous; VAS = visual analogue scale; WPAI-SHP = Work Productivity and Activity Impairment–Specific Health Problem.
Sources: SUNSHINE Clinical Study Report,33 SUNRISE Clinical Study Report,34 SUNSHINE Clinical Study Report Week 52,65 and SUNRISE Clinical Study Report Week 52.66 Details included in the table are from the sponsor’s summary of clinical evidence.67
The SUNSHINE (N = 541) and SUNRISE (N = 543) trials were identically designed multicentre, randomized, double-blind, placebo-controlled, parallel-group studies assessing the short- and long-term efficacy, safety, pharmacokinetics, and tolerability of 2 SC secukinumab dose regimens in adult patients (≥ 18 years) with moderate to severe HS. The SUNSHINE trial was conducted at 111 sites, including 4 sites in Canada, and the SUNRISE trial was conducted at 108 sites, including 3 sites in Canada; all patients in Canada were enrolled from Ontario and Quebec. The primary objective of both studies was to demonstrate the efficacy of 2 secukinumab dose regimens compared with placebo with respect to HiSCR after 16 weeks of treatment.
The screening period (to assess patient eligibility and to wash out and/or taper any prohibited medications) was up to 4 weeks from baseline, followed by the 16-week placebo-controlled treatment period 1 and 36-week treatment period 2 (Figure 1). Patients who discontinued from the study, or who completed the study but did not enter the extension study (NCT04179175), were required to complete an 8-week posttreatment follow-up. For patients rolling over to the extension study, week 52 was the end-of-study visit. For patients continuing to the posttreatment follow-up, week 60 was the end-of-study visit.
At baseline, patients were randomized through interactive response technology in a 1:1:0.5:0.5 ratio to receive secukinumab 300 mg every 2 weeks, secukinumab 300 mg every 4 weeks (with placebo between doses to be indistinguishable from the every 2 weeks dosing regimen), placebo to secukinumab 300 mg every 2 weeks, or placebo to secukinumab 300 mg every 4 weeks, respectively. Randomization was stratified by region, concomitant antibiotic use (yes or no), and body weight (< 90 kg or ≥ 90 kg). Note that up to 40% of patients were permitted to enter the study on treatment with a stable dose of antibiotics.
The data cut-off date was September 23, 2021, in the SUNRISE trial and October 1, 2021, in the SUNSHINE trial for the primary efficacy analysis at week 16. The database lock date was August 9, 2022, in the SUNRISE trial and August 17, 2022, in the SUNSHINE trial.
Note that 2 different dose regimens were assessed in both trials; however, only the maintenance dose of 300 mg of secukinumab administered every 2 weeks is included in the Health Canada indication. Therefore, only the results of the dose regimen of every 2 weeks are summarized in this report.
Figure 1: Study Design of SUNSHINE and SUNRISE Trials
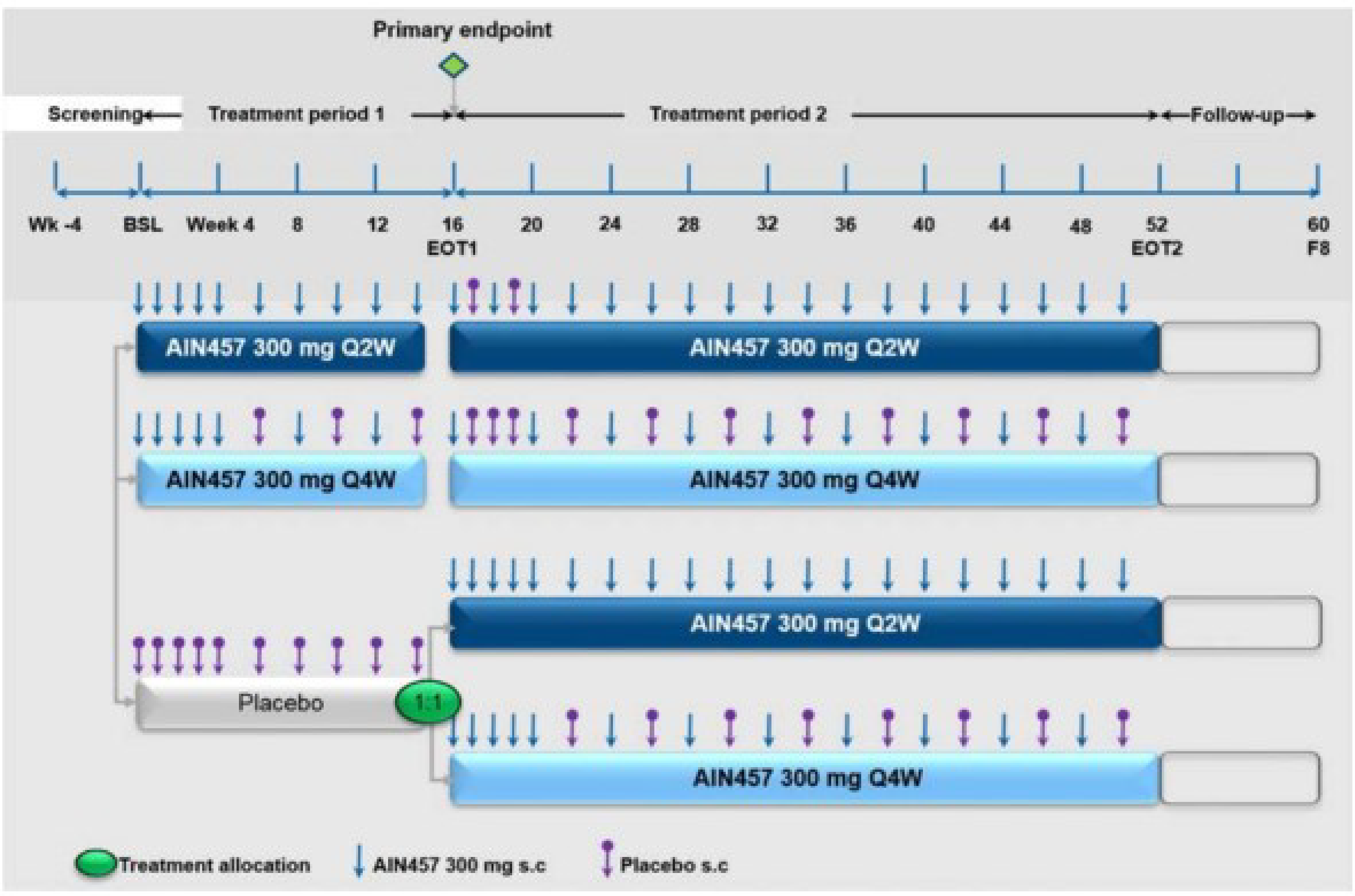
AIN457 = secukinumab; BSL = baseline; EOT1 = end of treatment 1; EOT2 = end of treatment 2; F8 = end of follow-up visit at week 60; Q2W = every 2 weeks; Q4W = every 4 weeks; s.c = subcutaneous.
Notes: Treatment allocation for the patients in the placebo arm switching to the secukinumab arms at week 16 was performed at the randomization visit in a 1:1 ratio and did not account for potential discontinuations during treatment period 1.
The only patients who entered follow-up were those who discontinued treatment prematurely during treatment period 1 or 2 or who did not enrol in the extension study.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34
Populations
Inclusion and Exclusion Criteria
The eligibility criteria were the same across both trials (Table 6). Briefly, adult patients (≥ 18 years) with moderate to severe HS who had a diagnosis of HS at least 1 year before baseline were included. Moderate to severe HS was defined as a total of at least 5 inflammatory lesions (abscesses and/or inflammatory nodules) affecting at least 2 distinct anatomic areas. Patients were required to agree to daily use of topical over-the-counter (OTC) antiseptics on areas affected by HS lesions while on study treatment. Patients were excluded if they had a total fistula count of 20 or more at baseline, or other active skin disease or condition that may interfere with the assessment of HS. Patients were excluded if they had prior exposure to secukinumab or to any other drug targeting the IL-17 receptor. Additionally, patients with a history of lymphoproliferative disease or any known malignancy or history of malignancy of any organ system treated or untreated within the past 5 years were excluded from the trials.
Interventions
Study Drugs
The interventions administered were the same in both studies. Secukinumab 300 mg and placebo solutions for SC injection were supplied in a 2 mL prefilled syringe. The study treatments were identical in appearance, packaging, labelling, and administration schedule. At baseline, patients were instructed by site staff on how to self-inject using the prefilled syringe. All doses of the study treatment were self-administered (injected in a nonaffected area of the skin) by the patient or a trained caregiver either at the study site or at home at predefined visits.
In treatment period 1 (defined as baseline to before week 16 dosing), all patients received a single SC injection of the blinded study drug once a week (induction) at baseline and weeks 1, 2, 3, and 4. Thereafter, the frequency of the study drug injections was every 2 weeks for all patients to maintain the treatment blind: secukinumab every 2 weeks (secukinumab 300 mg every 2 weeks group), secukinumab alternating with placebo every 2 weeks (secukinumab 300 mg every 4 weeks group), or placebo (placebo-to-secukinumab groups) (Figure 1). Patients who completed treatment period 1 entered treatment period 2.
In treatment period 2 (defined as post week 16 dosing through week 52), all patients received secukinumab. At week 16, all patients previously assigned to the placebo group received a reinduction consisting of a single SC injection of the blinded study drug once a week at weeks 16, 17, 18, 19, and 20. Note that patients who were randomized to receive secukinumab in treatment period 1 received placebo at weeks 17, 18 (only in the secukinumab every 4 weeks group), and 19 to maintain the blind. Thereafter, all patients received study drug injections every 2 weeks: secukinumab every 2 weeks (secukinumab 300 mg every 2 weeks group and placebo to secukinumab 300 mg every 2 weeks group) or secukinumab alternating with placebo every 2 weeks (secukinumab 300 mg every 4 weeks group and placebo to secukinumab 300 mg every 4 weeks group) until week 50 (Figure 1).
Dose adjustments and/or interruptions of the study drug were not permitted; however, a dose could be temporarily interrupted if, in the opinion of the investigator, a patient experienced a significant safety risk. Study treatment could be restarted at the next scheduled visit after the safety risk had been resolved.
Rescue Therapies
During treatment period 1 (specifically at weeks 4, 8, and 12), if a patient experienced an increase in AN count (e.g., total AN count ≥ 150% of the weighted average of the AN count at screening and baseline) with a minimum increase of 3 lesions, then an oral antibiotic (minocycline or doxycycline up to 100 mg twice daily) could be provided as rescue medication; the dosing regimen must have remained stable until week 16. Note that there was no requirement regarding the duration of increased AN count in the criteria for use of oral antibiotic rescue medication.36
At any time, if a patient experienced an acutely painful single lesion that required an immediate intervention, then the investigator could perform an unplanned surgery or intervention for the lesion such as excision, drainage, or intralesion steroid administration. Note that other HS-related surgeries could not be performed until after week 16. Evaluations at study visits occurred before any interventions were performed.
Concomitant Therapies
Patients were instructed to use daily topical OTC antiseptics on areas affected by HS lesions. Additionally, use of wound care dressings on HS wounds was permitted.
For patients in the antibiotic strata, treatment with tetracycline up to 500 mg twice daily, minocycline up to 100 mg twice daily, or doxycycline up to 100 mg twice daily on a stable dose was permitted, defined as a dose regimen that had not changed in the previous 28 days before baseline and was anticipated to remain stable for at least 16 weeks of treatment. Systemic antibiotics for the treatment of acute systemic infectious disease both related and unrelated to HS were permitted as required.
Patients on a stable dose of a nonopioid analgesic could continue the analgesic, provided the dose was stable in the 14 days before baseline and was anticipated to remain stable for at least 16 weeks of treatment. Note that “as needed” was not considered a stable dose of a nonopioid analgesic. If a patient presented with HS-related uncontrolled pain during the study, ibuprofen and acetaminophen were permitted. If the HS-related pain was uncontrolled with ibuprofen or acetaminophen at the maximal dose as per the local label during the study, patients could be prescribed tramadol at a dose of up to 100 mg oral every 4 hours, not to exceed 400 mg in 24 hours.
Prohibited Treatments
The following concomitant medications for the treatment of HS were prohibited: topical antibiotic therapies, systemic antibiotics (nonantibiotic strata), systemic corticosteroids, and surgeries other than those permitted as rescue therapy. Additionally, systemic biologic and nonbiologic immunomodulating treatments, opioid analgesics, live vaccines, and any other investigational treatment were prohibited. Note that patients could have received these prohibited therapies before beginning study treatment if they had completed the prespecified washout period (Table 37 in Appendix 1).
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted by CDA-AMC and input from patient groups, clinician groups, and public drug plans. Using the same considerations, the CDA-AMC review team selected end points that were considered to be most relevant to inform our expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select harms outcomes considered important for informing our expert committee deliberations were also assessed using GRADE.
The following were considerations that went into the selection of the efficacy outcomes summarized in this report and assessed using GRADE:
According to the input from the patient groups, the main treatment goals described by patients were to control HS symptoms, cure HS completely, and be able to enjoy personal relationships.
The clinical experts consulted by CDA-AMC for this review identified the following as outcomes used in clinical practice to assess response to treatment: lesion count (abscess, nodule, and fistula), pain scale, number of sites involved, extent of disease, and patient-reported outcomes such as DLQI, activities of daily living, and HRQoL.
The clinician group identified the following outcomes that are used to determine response to treatment: HiSCR50 and patient-reported outcomes such as pain, odour, and drainage management.
The experts identified remission, defined as free (clearance) of active inflammatory lesions (i.e., abscess or nodules) while on treatment over a 6-month period, as an important outcome in making clinical decisions in practice. Note that remission was not measured in the SUNSHINE and SUNRISE trials.
All notable harms of interest identified by the experts and input from patient and clinician groups and public drug plans are summarized in this report; however, only the notable harms included in Table 7 were assessed using GRADE. As advised by the experts, infections and infestations, malignant or unspecified tumours, and squamous cell carcinoma of an HS-affected area were included because secukinumab is an immune modulatory drug. Candidiasis was included because IL-17 inhibitors, such as secukinumab, are associated with a risk of candidiasis.68 Inflammatory bowel disease was included because it is identified in the warnings and precautions of the product monograph for secukinumab.1
The experts indicated that long-term assessment (e.g., 2 years) would be meaningful considering HS is a life-long disease.
The following were considerations that went into the exclusion of efficacy outcomes in the report:
There is evidence in the literature to support the clinical importance of HiSCR50 response in patients with HS;20,22 moreover, the experts agreed on the clinical importance of this threshold. Additionally, the experts considered other HiSCR thresholds to be important but not essential in making clinical decisions (e.g., achieving HiSCR25 can be used to identify patients with HS that responds early to treatment). As such, results for HiSCR25, HiSCR50, HiSCR75, HiSCR90, and HiSCR100, based on observed data, are included in Appendix 1, which also inform the pharmacoeconomic model submitted to CDA-AMC.
The experts indicated that the modified Hidradenitis Suppurativa Score (mHSS) is not commonly used in clinical practice to assess patients with HS in Canada.
When considering the outcome measures, mHSS, HS Physician’s Global Assessment, Patient Global Impression–Severity, and Patient Global Impression of Change, the experts suggested disease severity would be adequately measured by the included outcome measures (HiSCR50, AN count, and pain), which align with the outcomes described previously and are used in clinical practice to assess response to treatment.
The experts identified disease recurrence, defined as the return of active inflammatory lesions (i.e., abscess or nodules) or, more specifically, a patient presenting with multiple inflammatory abscesses and nodules after being free of inflammatory lesions in the preceding 6 months, as an important outcome in making clinical decisions in practice. However, the experts suggested that disease recurrence could be adequately captured in HiSCR (e.g., loss of HiSCR50, HiSCR75, or HiSCR90 response).
Table 7: Outcomes Summarized From the SUNSHINE and SUNRISE Trials
Outcome measure | Time point | SUNSHINE and SUNRISE |
|---|---|---|
Response to treatment and disease severity | ||
Proportion of patients achieving an HiSCR50 responsea | At week 16 | Primaryb |
At week 52 | Exploratory | |
Percentage change from baseline in AN count | At week 16 | Secondaryb |
At week 52 | Exploratory | |
Remissionc | Not evaluated in the SUNSHINE and SUNRISE trials | |
Disease worsening | ||
Proportion of patients experiencing any flaresd | At week 16 | Secondaryb |
At week 52 | Exploratory | |
Symptoms | ||
Proportion of patients achieving NRS30 (skin pain)e | At week 16 | Secondaryb |
At week 52 | Exploratory | |
Health-related quality of life | ||
Proportion of patients achieving DLQI responsef | At week 16 | Exploratory |
At week 52 | Exploratory | |
Change from baseline in DLQI total score | At week 16 | Exploratory |
At week 52 | Exploratory | |
Change from baseline in EQ-5D health state assessment (VAS score) | At week 16 | Exploratory |
At week 52 | Exploratory | |
Notable harmsg | ||
Infections and infestations | At week 16 | Exploratory |
At week 52 | Exploratory | |
Candidiasis | At week 16 | Exploratory |
At week 52 | Exploratory | |
Malignant or unspecified tumours | At week 16 | Exploratory |
At week 52 | Exploratory | |
Squamous cell carcinoma of HS-affected area | At week 16 | Exploratory |
At week 52 | Exploratory | |
Inflammatory bowel disease | At week 16 | Exploratory |
At week 52 | Exploratory | |
AN = abscesses and inflammatory nodules; CDA-AMC = Canada’s Drug Agency; DLQI = Dermatology Life Quality Index; GRADE = Grading of Recommendations Assessment, Development and Evaluation; HiSCR = Hidradenitis Suppurativa Clinical Response; HS = hidradenitis suppurativa; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; VAS = visual analogue scale.
aHiSCR50 was defined as at least a 50% decrease in AN count compared with baseline with no increase in the number of abscesses and/or in the number of draining fistulas from baseline in the SUNSHINE and SUNRISE trials.
bStatistical testing for these end points was adjusted for multiple comparisons (i.e., hierarchal testing).
cThe clinical experts consulted by CDA-AMC for this review identified remission, defined as free (clearance) of active inflammatory lesions (i.e., abscess or nodules) while on treatment over a 6-month period, as an important outcome in making clinical decisions in practice. Note that remission was not measured in the SUNSHINE and SUNRISE trials.
dFlare was defined as at least a 25% increase in AN count with a minimum increase of 2 AN relative to baseline in the SUNSHINE and SUNRISE trials.
eNRS30 (skin pain) was defined as at least a 30% reduction and at least 2-unit reduction from baseline in the patient's global assessment of skin pain on the NRS for pain at its worst averaged over the last 7 days in the SUNSHINE and SUNRISE trials.
fDLQI response was defined as a decrease of ≥ 5 points on DLQI total score from baseline in the SUNSHINE and SUNRISE trials.
gAll notable harms of interest identified by the clinical experts consulted by CDA-AMC and input from patient and clinician groups and public drug plans are summarized in this report; however, only the notable harms included in this table were assessed using GRADE. As advised by the clinical experts, infections and infestations, malignant or unspecified tumours, and squamous cell carcinoma of an HS-affected area were included because secukinumab is an immune modulatory drug. Candidiasis was included because IL-17 inhibitors, such as secukinumab, are associated with a risk of candidiasis.68 Inflammatory bowel disease was included because it is identified in the warnings and precautions of the product monograph for secukinumab.1
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34 Details included in the table are from the sponsor’s summary of clinical evidence.67
Efficacy Outcomes
HS Clinical Response and Individual Lesion Count Assessment
In both the SUNSHINE and SUNRISE trials, achievement of HiSCR50 response at week 16 was the primary end point, and HiSCR50 response at week 52 was an exploratory end point. In both trials, percentage change from baseline in AN count at week 16 and week 52 were secondary and exploratory end points, respectively. Response to treatment (HiSCR50 achievement) was defined as at least a 50% reduction in AN count with no increase in the number of abscesses and/or in the number of draining fistulas from baseline to week 16, where increase was defined as at least 1 abscess or 1 draining fistula more than the baseline value. Additionally, HiSCR25, HiSCR50, HiSCR75, HiSCR90, and HiSCR100, which describe a decrease of at least 25%, 50%, 75%, 90%, 100% in AN count with no increase in the number of abscesses and/or in the number of draining fistulae compared with baseline, at week 16 and week 52 based on observed data were exploratory end points in both trials. The HiSCR was derived from the individual lesion counts of abscesses, nodules, and fistulae at scheduled visits. Any existing and newly observed lesions were counted by area by the physician at screening, in treatment period 1 (at baseline, week 2, week 4, and every 4 weeks thereafter to week 16), in treatment period 2 (at week 18, week 20, every 4 weeks thereafter to week 52), and at posttreatment follow-up (at week 60).
The HiSCR is used to measure response to treatment based on lesion counts. Areas affected by HS, including right and left axillary, right and left gluteal, right and left inguinal–femoral, perineal, pubic, sternal, and right and left submammary, were assessed by the physician for the presence of inflammatory nodules, abscesses, draining fistulae, total fistulae, and other lesions (Table 8).
Table 8: Definitions of HS Lesions
HS lesions | Definition |
|---|---|
Inflammatory nodules | Raised, deep-seated, 3-dimensional, round, tender, erythematous, infiltrated, and possibly pyogenic granuloma lesions with a diameter of > 10 mm |
Abscesses | Inflammatory, painful, tender but fluctuating mass, with a diameter of > 10 mm and surrounded by an erythematous area, with the middle area containing purulent material |
Draining fistulae | Sinus tracts, raised, tender but fluctuating longitudinal tunnels of variable length and depth, with communications to skin surface, draining purulent fluid |
Fistulae | Sinus tracts, raised, tender but fluctuating longitudinal tunnels of variable length and depth, with communications to skin surface, with or without purulent discharge |
HS = hidradenitis suppurativa.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34 Details included in the table are from the sponsor’s summary of clinical evidence.67
Flares
In both trials, experience of flares at week 16 and week 52 were secondary and exploratory end points, respectively. A flare was defined as at least a 25% increase in AN count with a minimum increase of 2 AN relative to baseline. The occurrence of a flare was calculated from the lesion count assessment.
Skin Pain
In both trials, achievement of NRS30 at week 16 and week 52 were secondary and exploratory end points, respectively. Achievement of NRS30 was defined as at least a 30% reduction and at least 2-unit reduction from baseline in the patient's global assessment of skin pain at its worst on the NRS. Patients completed a daily diary of their skin pain at screening and in treatment period 1 (from baseline through week 16). Patients also completed skin pain assessments on a weekly basis in treatment period 2 (from week 16 through week 52) and at the posttreatment follow-up (at week 60).
The NRS is a patient-reported, segmented numeric version of the VAS that is used to measure the intensity of skin pain at its worst as well as the average intensity of skin pain due to HS in the last 24 hours. The range of the NRS includes whole numbers from 0 (no skin pain) to 10 (skin pain as bad as you can imagine).
Dermatology Life Quality Index
In both trials, achievement of DLQI response at week 16 and week 52 as well as change from baseline in DLQI total score at week 16 and week 52 were exploratory end points. Response according to DLQI was defined as a decrease of 5 points or more on the DLQI total score from baseline. The DLQI was completed in treatment period 1 (at baseline and weeks 2, 4, 12, and 16) and treatment period 2 (at weeks 28 and 52).
The DLQI is a 10-item, self-administered, general dermatology disability index used to assess HRQoL in adults with skin diseases (e.g., eczema, psoriasis, and acne). The questionnaire consists of 6 domains, including symptoms and feelings, daily activities, leisure, work and school, personal relationships, and treatment. The recall period is the last week. Each item is scored on a 4-point scale from 0 (not relevant or not at all) to 3 (very much). The total score is the sum of the scores of each item, ranging from 0 (no effect at all on patient’s life) to 30 (extremely large effect on patient’s life), with a higher score indicating greater impairment in HRQoL.
EQ-5D Health State Assessment
In both trials, change from baseline in EQ-5D health state assessment at week 16 and week 52 were exploratory end points. The EQ-5D was completed in treatment period 1 (at baseline and weeks 2, 4, 12, and 16) and treatment period 2 (at weeks 28 and 52).
The EQ-5D-3L is a self-administered, generic instrument consisting of the EQ-5D descriptive system and the EQ VAS that is used to assess patient health status. The descriptive system consists of the following 5 dimensions: mobility, self-care, usual activities, pain and discomfort, severe problems, and anxiety and depression. Each dimension has 3 response levels: no problem, moderate problem, and severe problems. The VAS is used to assess the patient’s self-rated health on a vertical 20 cm VAS where the end points are labelled 0 (worst imaginable health state) and 100 (best possible health state). The recall period is the day of the assessment.
Harms Outcomes
In both the SUNSHINE and SUNRISE trials, AEs were assessed at screening and throughout the study.
Notable harms for secukinumab included hypersensitivity, injection site reaction, infections and infestations, candidiasis, malignant or unspecified tumours, squamous cell carcinoma of an HS-affected area, inflammatory bowel disease, and suicidal ideation or behaviour or suicide attempt.
Table 9: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
HiSCR | A physician-assessed measure of response to treatment20 based on the status of 3 types of lesions: abscesses, inflammatory nodules, and draining fistulae. HiSCR50 is defined as:
| Validity: In 1 study of patients with HS, there was weak to moderate correlation (Spearman rho −0.61 to −0.27) with physician-rated assessments (Hurley stage, MSS, and HS-PGA) and PRO measures (pain VAS, DLQI, and WPAI-SHP).20 Test–retest reliability: Strong correlation of AN count between screening and baseline visit (ICC = 0.91).20 Responsiveness: HiSCR50 achievers showed meaningful improvements in all PROs.20,21 | Several studies suggest that a threshold of 50% (i.e., HiSCR50) is clinically meaningful to assess response.20,22 No MID was found in the literature. |
NRS30 | Patient-reported measure of pain intensity in the past 24 hours ranging from 0 (no skin pain) to 10 (skin pain as bad as you can imagine). NRS30 was defined as a ≥ 30% reduction and at least 2 units of reduction from baseline in the patient’s global assessment of skin pain at its worst.23 | Construct-related and predictive validity: NRS30 converged well with criteria measures, correlating significantly with all physician and PRO assessments (DLQI, EQ-5D-3L [pain/discomfort], EQ-5D-3L VAS score, PGI-S, PGIC, AN count, mHSS, HiSCR). Absolute values of Somers D were 0.2256 to 0.5622 at week 16.23 NRS30 response at week 16 was a strong predictor for EQ-5D-3L response (pain/discomfort) (OR = 3.14; 95% CI, 1.36 to 7.22; P = 0.0073) and for a greater DLQI score (OR = 4.05; 95% CI, 1.85 to 8.90; P = 0.0005) at week 52.23 Reliability: No evidence identified. Responsiveness: A large to moderate effect was observed by week 16 in most clinical and PRO measures among patients with NRS30 response. At week 52, the majority of measures assessed had a greater extent of responsiveness among patients with NRS30 response compared with patients who did not experience NRS30 response.23 | Several studies suggest that a threshold of about 30% (i.e., NRS30) is clinically meaningful for within-patient change in pain. These studies were not specific to HS.26-28 |
DLQI | Self-administered HRQoL assessment tool for adults with skin diseases. Includes 6 domains, with the sum corresponding to a total score. The range of scores is 0 to 30, with a higher score indicating lower HRQoL.22 | Validity: In 1 study of patients with HS, DLQI was moderately to strongly correlated with EQ-5D-5L, EQ VAS, DLQI-relevant, and Skindex-16 total scores (range, 0.49 to 0.99).24 There were weak or moderate correlations with HS-PGA (0.35 to 0.43) and MSS (0.32 to 0.39).24 Reliability and responsiveness: No evidence identified. | Among adult patients with moderate to severe HS and baseline DLQI ≥ 5, the MID for improvement was defined as a decrease of at least 5 units.22 |
EQ VAS | Generic self-reported HRQoL assessment tool that uses a vertical 20 cm VAS where the end points are labelled “Best imaginable health state” and “Worst imaginable health state.”69 | Validity: In 1 study of patients with HS, those with more severe disease (Hurley stage 3) had lower VAS scores than those with Hurley stage 1 or 2. There were moderate correlations between VAS scores and the number of lesions (r = 0.280).25 Reliability: No evidence identified. Responsiveness: No evidence identified. | In a reanalysis of data from 2 RCTs conducted by Jensen et al., the findings suggested that 100 mm VAS ratings of 0 mm to 4 mm could be considered no pain; 5 mm to 44 mm, mild pain; 45 mm to 74 mm, moderate pain; and 75 mm to 100 mm, severe pain.28 No MID was found from the literature. |
AN = abscesses and inflammatory nodules; CI = confidence interval; DLQI = Dermatology Life Quality Index; EQ VAS = EQ-5D visual analogue scale; HiSCR = Hidradenitis Suppurativa Clinical Response; HRQoL = health-related quality of life; HS = hidradenitis suppurativa; HS-PGA = Hidradenitis Suppurativa Physician’s Global Assessment; HSS = Hidradenitis Suppurativa Score; ICC = intraclass correlation coefficient; mHSS = modified Hidradenitis Suppurativa Score; MID = minimal important difference; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; OR = odds ratio; PGI-S = Patient Global Impression–Severity; PGIC = Patient Global Impression of Change; PRO = patient-reported outcome; RCT = randomized controlled trial; VAS = visual analogue scale; Work Productivity and Activity Impairment–Specific Health Problem.
Statistical Analysis
Analysis of clinical end points in the SUNSHINE and SUNRISE trials is summarized in Table 10.
Sample Size and Power Calculation
The sample size for both trials was driven by the primary end point, HiSCR50 achievement at week 16. Both studies were independently powered to address the primary end point and secondary end points of AN count and flares. The initial plan was to randomize 471 patients in a 1:1:0.5:0.5 ratio to the study drug. To account for the impact of COVID-19, a protocol amendment was made to increase the number of randomized patients by 15% to 541 patients to ensure that power was maintained throughout the trial.
For the testing of the primary end point, a response rate of 30% for placebo was assumed, based on the PIONEER I and II trials comparing adalimumab with placebo.70 The initial sample size of 471 patients was sufficient to achieve 93% power for demonstration of a 20% difference of secukinumab 300 mg every 2 weeks over placebo, assuming a secukinumab response rate of 50%, based on the primary end point.
Expected responses for the primary and secondary end points were based on the results from the PIONEER I and II trials:70
A difference of at least 18% in favour of secukinumab versus placebo was assumed for the mean percentage change from baseline in AN count. The approximate sample size for analysis of this end point was based on a simple t test. The initial sample size of 471 patients was expected to achieve 92% power in testing secukinumab 300 mg every 2 weeks versus placebo, assuming an SD of 46%.
A placebo rate of 35% was assumed for flares. The initial sample size of 471 patients was expected to achieve 98% power in demonstrating a 20% difference of secukinumab 300 mg every 2 weeks over placebo, assuming a rate of 15% for secukinumab 300 mg every 2 weeks.
A placebo rate of 23% was assumed for NRS30. Assuming that 80% of patients would qualify for the NRS30 analysis, a total sample size of 942 patients across both studies was expected to achieve 85% power to demonstrate a difference of 13% of secukinumab 300 mg every 2 weeks group over placebo, assuming a rate of 36% for secukinumab 300 mg every 2 weeks.
Primary End Point
The primary analysis method for HiSCR50 response at week 16 was logistic regression with treatment group, Hurley stage, baseline AN count, geographical region, use of antibiotics, and baseline body weight as explanatory variables. ORs were computed for comparisons of secukinumab dose regimens versus placebo based on the logistic regression model fit.
Secondary End Points
Both studies included the following secondary end points:
percentage change from baseline in AN count at week 16
proportion of patients who experience at least 1 HS flare over 16 weeks; a flare was defined as at least a 25% increase in AN count, with a minimum increase of 2 AN relative to baseline
proportion of patients achieving NRS30 at week 16 in patients with baseline NRS ≥ 3; NRS30 was defined as at least 30% reduction and at least 2-unit reduction from baseline in the patient’s global assessment of skin pain.
The analysis method for percentage change from baseline in AN count at week 16 was an analysis of covariance (ANCOVA) model with treatment group, Hurley stage, baseline AN count, geographical region, use of antibiotics, and baseline body weight as explanatory variables.
The analysis method for flares over 16 weeks and NRS30 at week 16 was logistic regression with treatment group, Hurley stage, geographical region, use of antibiotics, baseline body weight, baseline value of the variable being assessed, and study (NRS30 only) as explanatory variables. ORs were computed for the comparisons of the secukinumab dose regimens versus placebo based on the logistic regression model fit.
Exploratory End Points
For exploratory end point analyses at week 16, the corresponding P values for pairwise comparisons were not adjusted for multiple comparisons and no formal confirmatory hypothesis testing was planned.
The analysis method for DLQI response at week 16 was logistic regression based on observed data, with treatment group, Hurley stage, geographical region, use of antibiotics, baseline body weight, and baseline DLQI score as explanatory variables. ORs were computed for the comparisons of the secukinumab dose regimens versus placebo based on the logistic regression model fit.
The analyses of the DLQI total score and EQ-5D health state assessment were descriptive and based on observed data only.
The analyses of HiSCR25, HiSCR50, HiSCR75, HiSCR90, and HiSCR100 were descriptive and based on observed data only.
Exploratory end point analyses at week 52 were descriptive and used only observed data. A Kaplan-Meier analysis based on observed values for the cumulative incidence of the flare-free period up to week 52 was presented for each treatment group; patients without any flares were censored at the last known visit.
Testing Strategy
There was initially an equal alpha split for the testing of each dose versus placebo, but this was later amended to allocate four-fifths of the alpha to the biweekly maintenance dosing following results of another study (CAIN457A2324), suggesting it may be reasonable to believe there would be a better treatment effect with biweekly maintenance dosing compared with monthly maintenance dosing.
The primary and secondary end points (percentage change from baseline in AN count at week 16 and proportion of patients experiencing any flares at week 16) were tested in each study separately. The secondary end point, NRS30 at week 16, was tested using the pooled data from both studies. To control for type I error at the level of the individual studies and at the level of the pooled dataset of both studies, a 5% 2-sided type I error rate was used and split, and the following testing strategy was implemented (Figure 6 in Appendix 1).
The primary end point in both dose regimens was first tested, with 4% alpha allocated to test secukinumab every 2 weeks against placebo, and 1% to test secukinumab every 4 weeks against placebo. For each dose regimen, if the primary hypothesis was rejected, then the allocated alpha was passed to the first secondary end point (percentage change from baseline in AN count at week 16) in the testing strategy for the same dose regimen and, subsequently, to the next secondary end point (flares over 16 weeks). If statistical significance was achieved for the primary end point and the 2 secondary end points for a secukinumab dose regimen, then the respective allocated alpha was passed on to test the end points of the other secukinumab dose regimen if they did not already achieve statistical significance at the initial allocated alpha.
Statistical testing for the secondary end point, NRS30 at week 16, was dependent on the statistical significance having been achieved in both studies for the primary end point on the same secukinumab dose regimen. Additionally, the allocated alpha for this secondary end point could be passed from 1 dose regimen for which statistical significance was achieved to the other dose regimen. The secondary end point, NRS30 was based on pooled data from the SUNNY trials; the initial significance level for the skin pain hypothesis was set to alpha squared minus alpha (1-sided alpha is 0.025) and could be increased, dependent on which hypotheses were rejected; the subtraction of alpha squared was to account for the maximum possible type I error for HiSCR50 response and percentage change in AN count and flares in both studies.
Note that, based on an interim futility and efficacy analysis that was performed for the SUNNY trials when 40% of patients in both studies had completed week 16, a Haybittle-Peto–type adjustment of the 1-sided levels of significance was applied in the analysis of the primary and secondary end points. However, the adjustment was deemed to be very small (0.00001) by the investigator and they concluded that it did not have any impact on the results; thus, the original significance levels are referenced in the results. Therefore, the primary and secondary end points (AN count and flares) were tested in a hierarchical order with 1-sided significance level of 0.02 for the biweekly maintenance dosing and 0.005 for the monthly maintenance dosing.
Data Imputation Methods
Missing data for the primary and secondary end points were addressed using multiple imputation. To account for different postrandomization events, missing data were multiply imputed using a reference-based approach or based on a missing at random assumption, depending on the intercurrent event or unrelated to an intercurrent event. Note that for the intake of any prohibited medications, such events were ignored and all observed values were considered. Any observations after a patient had permanently discontinued study treatment for reasons other than AEs or lack of efficacy were discarded. For 2 intercurrent events, intake of rescue medication and permanent discontinuation of study treatment because of AEs or lack of efficacy, a patient was either considered a nonresponder, or a score of 0 (no change) would be imputed in the analysis of the percentage change from baseline in AN count at week 16. In the analysis of NRS30, a patient was also considered a nonresponder in the intercurrent event of prohibited medication intake.
Imputations were performed on the continuous variables from which the imputed binary outcomes were then constructed. More specifically, for the primary end point analysis, the number of abscesses, inflammatory nodules, and draining fistulae were imputed, from which the response variable was then constructed. Since these lesion count variables were used to derive HiSCR, AN count, and flares, 1 set of multiple imputations of the lesion count variables was generated for consistency between the analyses of these end points. For the analysis of NRS30, the values of the patient’s global assessment of skin pain were imputed, from which the imputed binary outcome was then constructed.
Sensitivity and Supplementary Analyses
A tipping point analysis was implemented for both the primary and secondary end points to assess the robustness of the multiple imputation approach.
A sensitivity analysis using a weighted average of the screening and baseline visits (compared with the value at baseline that was used in the primary analysis) was performed for the primary end point and the 2 secondary end points (AN count and flares). For inflammatory nodules, abscesses, and draining fistulae, a weighted average based on screening visit 1 (with a weight of 1/6), screening visit 2 (with a weight of 2/6), and randomization visit (with a weight of 3/6) was taken. Note that this weighted average was used during the study to identify the need for rescue medication.
A supplementary analysis was implemented for only the primary end point and all attributes of this end point remained the same as defined for the primary end point; however, the intercurrent events were considered differently, per protocol.
Subgroup Analysis
The primary and secondary end points of both trials were evaluated using the following subgroup variables:
concomitant antibiotics use
body weight stratum (< 90 kg, ≥ 90 kg)
geographical region
age
gender
race
previous use of systemic biologics
C-reactive protein levels (< 5, 5 to 10, ≥ 10)
erythrocyte sedimentation rate levels (< 20, ≥ 20)
Hurley stage (1, 2, 3)
baseline AN count (≤ 10, > 10)
baseline disease duration (< 2 years, 2 to 5 years, 5 to 10 years, ≥ 10 years).
To assess for any potential difference in treatment effect between subgroups, logistic regression models were used to test for an interaction between treatment and the subgroups. A separate model was used to assess each subgroup with treatment group weight, baseline AN count, and the interaction between treatment and the assessed subgroup as covariates. For the primary end point, the P values of the interaction terms did not support any assumption of a difference in the treatment effect between the subgroups. Note that the subgroup analyses were not adjusted for multiple testing.
Of the subgroups listed previously, the clinical experts consulted by CDA-AMC for this review identified concomitant antibiotic use, body weight stratum, previous use of systemic biologics, Hurley stage, and baseline AN count as patient demographic or disease characteristics that would be expected to influence the efficacy of treatment (i.e., effect modifiers).
Table 10: Statistical Analysis of the Efficacy End Points in the SUNSHINE and SUNRISE Trials
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
HiSCR50 response at week 16 | Logistic regression | Explanatory variables:
| Missing data were multiply imputed based on the end point strategy related to intercurrent events or missing at random assumption for all missing values not related to intercurrent events. |
|
Percentage change from baseline in AN count at week 16 | ANCOVA | Same as row 1 | Same as row 1 | Same as row 1 |
Flare(s) over 16 weeks | Logistic regression | Same as row 1 | Same as row 1 | Same as row 1 |
NRS30 (skin pain) at week 16 (pooled analysis of both studies) | Logistic regression | Explanatory variables:
| — | Tipping point analysis |
DLQI response at week 16 | Logistic regression | Explanatory variables:
| Missing data were not imputed; analyses were based only on observed data. | None |
Change from baseline in DLQI total score at week 16 | Summary statistics | None | Missing data were not imputed; analyses were based only on observed data. | None |
Change from baseline in EQ-5D health state assessment (VAS score) at week 16 | Summary statistics | None | Missing data were not imputed; analyses were based only on observed data. | None |
AN = abscesses and inflammatory nodules; ANCOVA = analysis of covariance; DLQI = Dermatology Life Quality Index; HiSCR50 = Hidradenitis Suppurativa Clinical Response 50, i.e., a decrease of 50% or greater in the AN count with no increase in the number of abscesses or draining fistulae compared with baseline; NRS = numerical rating scale; VAS = visual analogue scale.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34 Details included in the table are from the sponsor’s Summary of Clinical Evidence.67
Analysis Populations
The analysis sets used in both trials are summarized in Table 11. The definitions of each analysis population were the same across both trials. Additionally, the full analysis set was the same as the randomized set. The analysis of efficacy end points was based on the full analysis set.
For all analyses in treatment period 1, placebo to secukinumab 300 mg every 2 weeks group and placebo to secukinumab 300 mg every 4 weeks group were pooled together (placebo group). The any-secukinumab group included the patients who received secukinumab at initial randomization and the patients who switched from placebo to secukinumab.
Table 11: Analysis Populations of the SUNSHINE and SUNRISE Trials
Population | Definition | Application |
|---|---|---|
Randomized analysis set | Included all randomized patients. Patients were analyzed according to their treatment assignment at randomization. | Analysis of patient disposition, protocol deviations, demographics, and baseline characteristics. |
Full analysis set | Included all patients who were assigned a study treatment. Patients were analyzed according to their treatment assignment at randomization. If the actual stratum of patients was different to the assigned stratum, the actual stratum was used in the analyses. | Analysis of efficacy end points. Note the full analysis set was the same as the randomized set. |
Safety analysis set | Included all patients who received at least 1 dose of study treatment. Patients were analyzed according to the study treatment they actually received. | Analysis of study treatment data, number of treatment injections, duration of exposure to study treatment, prior and concomitant treatments, and safety evaluations. |
Notes: Patients who were misrandomized were excluded and patients who had experienced a serious violation of good clinical practice at their site were excluded. Patients who are misrandomized are those who did not advance past screening but had been randomized by the investigator before eligibility was finally assessed; however, they were not treated.
Treatment received is defined as the randomized or assigned treatment if the patient took at least 1 dose of that treatment, or the first treatment received if the randomized or assigned treatment was never received (patient received the wrong treatment during the entire study).
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34 Details included in the table are from the sponsor’s Summary of Clinical Evidence.67
Results
Although 2 different dosing schedules were assessed in the SUNSHINE and SUNRISE trials, only the maintenance dose of 300 mg of secukinumab administered every 2 weeks is included in the Health Canada indication. Therefore, only the results of the dosing schedule of every 2 weeks were summarized in this report.
Patient Disposition
A summary of patient disposition during treatment period 1 in both trials is presented in Table 12. Patient disposition was generally similar between groups and across trials. A total of 676 and 687 patients were screened for eligibility in the SUNSHINE and SUNRISE trials, respectively. The most common reason for screen failure was due to screen failure (15.5% [105 patients] in the SUNSHINE trial and 13.5% [93 patients] in the SUNRISE trial). In the SUNSHINE trial, 181 patients were randomized to the secukinumab group and 180 patients to the placebo group. In the SUNRISE trial, 180 patients were randomized to the secukinumab group and 183 patients to the placebo group.
In the SUNSHINE trial, 7.2% (13 patients) in the secukinumab group and 4.4% (8 patients) in the placebo group discontinued from the study. In the SUNRISE trial, 5.6% (10 patients) and 8.7% (16 patients) randomized to secukinumab and placebo, respectively, discontinued from the study. Of the patients who were randomized to secukinumab and placebo, the most common reason for study discontinuation was patient decision (3.3% [6 patients] and 2.8% [5 patients], respectively, in the SUNSHINE trial, and 3.3% [6 patients] and 3.8% [7 patients], respectively, in the SUNRISE trial).
After completing treatment period 1, patients initially randomized to the placebo group were switched to a secukinumab regimen, while patients initially randomized to secukinumab continued to receive their regimen. A summary of patient disposition during the entire study period in both trials is presented in Table 13. In the SUNSHINE trial, ██ of ███ randomized patients (█████) in the secukinumab group discontinued from the study. In the SUNRISE trial, ██ of ███ randomized patients (█████) in the secukinumab group discontinued from the study. Similar to treatment period 1, the most common reason for study discontinuation was patient decision in the secukinumab group (██|█████| patients in the SUNSHINE trial and ██|████| patients in the SUNRISE trial).
Table 12: Summary of Patient Disposition From the SUNSHINE and SUNRISE Trials in Treatment Period 1
Disposition | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. | Placebo | Secukinumab q.2.w. | Placebo | |
Screened, N | 676 | 687 | ||
Reason for screening failure, n (%) | ||||
Screen failure | 105 (15.5) | 93 (13.5) | ||
Patient decision | 17 (2.5) | 37 (5.4) | ||
Physician decision | | █████ | | █████ | ||
Lost to follow-up | | █████ | | █████ | ||
Technical problems | | █████ | | █████ | ||
Protocol deviation | | █████ | | █████ | ||
Adverse event | | █████ | | █████ | ||
Randomized, N | 181 | 180 | 180 | 183 |
Discontinued from study, n (%) | 13 (7.2) | 8 (4.4) | 10 (5.6) | 16 (8.7) |
Reason for discontinuation, n (%) | ||||
Patient decision | 6 (3.3) | 5 (2.8) | 6 (3.3) | 7 (3.8) |
Lost to follow-up | 3 (1.7) | 2 (1.1) | 1 (0.6) | 2 (1.1) |
Adverse events | 2 (1.1) | 0 | 1 (0.6) | 4 (2.2) |
Technical problems | 1 (0.6) | 0 | 1 (0.6) | 1 (0.5) |
Physician decision | 1 (0.6) | 1 (0.6) | 0 | 0 |
Lack of efficacy | 0 | 0 | 1 (0.6) | 1 (0.5) |
Pregnancy | 0 | 0 | 0 | 1 (0.5) |
Randomized analysis set, N | 181 | 180 | 180 | 183 |
Full analysis set, N | 181 | 180 | 180 | 183 |
Safety analysis set, N | 181 | 180 | 180 | 183 |
q.2.w. = every 2 weeks.
Note: In the SUNSHINE trial, 3 patients were excluded from the randomized set per the statistical analysis plan: 1 patient was misrandomized in interactive response technology, 1 patient experienced a serious violation of good clinical practice, and 1 patient experienced a serious breach. In the SUNRISE trial, 1 patient was misrandomized in the interactive response technology and excluded from the randomized set, per the analysis plan.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34 Details included in the table are from the sponsor’s Summary of Clinical Evidence.67
Table 13: Summary of Patient Disposition From the SUNSHINE and SUNRISE Trials in Entire Study Period
Patient disposition | SUNSHINE | SUNRISE | ||||
|---|---|---|---|---|---|---|
Secukinumab q.2.w. | Placebo to secukinumab q.2.w. | Any secukinumab q.2.w. | Secukinumab q.2.w. | Placebo to secukinumab q.2.w. | Any secukinumab q.2.w. | |
Randomized set, N | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Discontinued from study, n (%) | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Reason for study discontinuation, n (%) | ||||||
Patient decision | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Lost to follow-up | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Adverse events | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Lack of efficacy | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Physician decision | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Pregnancy | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Technical problems | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Full analysis set, N | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Safety analysis set, N | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
q.2.w. = every 2 weeks.
Notes: The entire study period included treatment period 1, treatment period 2, and the follow-up period.
In the SUNSHINE trial, 3 patients were excluded from the randomized set per the statistical analysis plan: 1 patient was misrandomized in interactive response technology, 1 patient experienced a serious violation of good clinical practice, and 1 patient experienced a serious breach. In the SUNRISE trial, 1 patient was misrandomized in the interactive response technology and excluded from the randomized set, per the analysis plan.
Patients in the placebo group who discontinued in treatment period 1 and did not receive secukinumab are not included in this table. The safety analysis set included only patients who received the active treatment (secukinumab).
Sources: SUNSHINE65,66Clinical Study Report Week 52 and SUNRISE Clinical Study Report Week 52. Details included in the table are from the sponsor’s summary of clinical evidence.67
Protocol Deviations
A summary of the protocol deviations that occurred during treatment period 1 in both trials is presented in Table 14. The number of patients with at least 1 protocol deviation was similar between groups and across trials, with approximately one-third of patients reporting at least 1 protocol deviation. Most protocol deviations were related to treatment deviation; in the SUNSHINE trial, this was reported in ██ of 181 (██████) patients in the secukinumab group and ██ of 180 (█████) patients in the placebo group, and in the SUNRISE trial, treatment deviation was reported in ██ of 180 (█████) patients in the secukinumab group and ██ of 183 (█████) patients in the placebo group. Note that the “treatment deviation” category was related mainly to drug administration at home versus at the site.
Table 14: Summary of Protocol Deviations From the SUNSHINE and SUNRISE Trials in Treatment Period 1 (Randomized Set)
Protocol deviation | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
Patients with at least 1 protocol deviation | | █████ | | ████ | | █████ | █████ |
Treatment deviationa | | █████ | | ████ | | █████ | █████ |
Prohibited concomitant medication | | █████ | | ████ | | █████ | █████ |
Selection criteria not met | | █████ | █████ | | █████ | █████ |
Otherb | | █████ | █████ | | █████ | |█████ |
q.2.w. = every 2 weeks.
Note: A patient with multiple protocol deviations in the same category was counted only once in that category. Patients could have protocol deviations in more than 1 protocol deviation category.
aThe “treatment deviation” category was related mainly to drug administration at home vs. at the site.
bThe “other” category covers protocol deviations that do not fall into any of the previous categories (selection criteria not met, patient withdrawal as per protocol, treatment deviation, or prohibited concomitant medication) but that impact the completeness, accuracy, and/or reliability of the study data or the patient’s rights, safety, or well-being. For example, a procedure was performed after the patient withdrew consent, the study site was not compliant with good clinical practice, or primary end point assessment was missed.
Sources: Source: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34 Details included in the table are from the sponsor’s Summary of Clinical Evidence.67
A summary of the protocol deviations that occurred during the entire study period in both trials is presented in Table 15. A similar proportion of patients in the secukinumab group across trials reported at least 1 protocol deviation (███ of 181 |█████| patients in the SUNSHINE trial and ███ of 180 |█████| patients in the SUNRISE trial). Similar to treatment period 1, most protocol deviations in the secukinumab group were related to treatment deviation (██ of 181 |█████| patients in the SUNSHINE trial and ██ of 180 |█████| patients in the SUNRISE trial).
Table 15: Summary of Protocol Deviations From the SUNSHINE and SUNRISE Trials in Entire Study Period (Randomized Set)
Protocol deviation | SUNSHINE | SUNRISE | ||||
|---|---|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo to secukinumab q.2.w. (N = 90) | Any secukinumab q.2.w. (N = 271) | Secukinumab q.2.w. (N = 180) | Placebo to secukinumab q.2.w. (N = 90) | Any secukinumab q.2.w. (N = 270) | |
Patients with at least 1 protocol deviation | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Treatment deviation | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Prohibited concomitant medication | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Selection criteria not met | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
Othera | | █████ | | █████ | | █████ | | █████ | | █████ | | █████ |
q.2.w. = every 2 weeks.
Notes: The entire study period comprised treatment period 1, treatment period 2, and the follow-up period.
A patient with multiple occurrences of a protocol deviation in the same category was counted only once in that category. Patients could have protocol deviations in more than 1 protocol deviation category.
aThe “other” category covers protocol deviations that do not fall into the previous categories (selection criteria not met, patient withdrawal as per protocol, treatment deviation, or prohibited concomitant medication) but that impact the completeness, accuracy, and/or reliability of the study data or the patient’s rights, safety, or well-being (e.g., a procedure was performed after the patient withdrew consent, the study site was not compliant with good clinical practice, or the assessment of the primary end point was missed).
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week 52.66 Details included in the table are from the sponsor’s summary of clinical evidence.67
Baseline Characteristics
A summary of baseline characteristics from both the SUNSHINE and SUNRISE trials in treatment period 1 is presented in Table 16. Baseline characteristics were generally similar between groups and across trials. Across trials, the mean age of patients ranged from 35.5 years (SD = 10.75) in the placebo group in the SUNSHINE trial to 37.3 years (SD = 11.48) in the secukinumab group in the SUNRISE trial. Across trials, the proportion of patients who were male ranged from 42.6% (78 of 183 patients) in the placebo group to 45.6% (82 of 180 patients) in the secukinumab group in the SUNRISE trial, and patients who were female ranged from 54.4% (98 of 180 patients) in the secukinumab group to 57.4% (105 to 183 patients) in the placebo group in the SUNRISE trial. The majority of patients across trials were white, ranging from 73.9% (133 of 180 patients) in the secukinumab group in the SUNRISE trial to 80.1% (145 of 181 patients) in the SUNSHINE trial. Across trials, the mean body mass index ranged from 31.42 kg/m2 (SD = 7.382) in the placebo group in the SUNRISE trial to 32.64 kg/m2 (SD = 7.904) in the secukinumab group in the SUNSHINE trial.
In terms of smoking status, more than half of the patients were current smokers; across trials, the proportion of patients who were current smokers ranged from 52.5% (95 of 181 patients) in the secukinumab group in the SUNSHINE trial to 57.9% (106 of 183 patients) in the placebo group in the SUNRISE trial. Across trials, the proportion of patients with obesity ranged from ████ (██ of 180 patients) in the placebo group in the SUNSHINE trial to █████ (██ of 180 patients) in the secukinumab group in the SUNRISE trial. Across trials, the proportion of patients with diabetes mellitus ranged from ████ (||| of 180 patients) in the secukinumab group to ████ (||| of 183 patients) in the placebo group in the SUNRISE trial. One patient in each group in the SUNRISE trial was reported to have hyperandrogenism; no patients in the SUNSHINE trial were reported to have hyperandrogenism.
In the SUNSHINE trial, the mean time since HS symptom onset across trials ranged from ████ years (SD = ████) in the placebo group to ████ years (SD = ████) in the secukinumab group. At baseline, most patients were categorized with Hurley stage 2 disease severity, ranging from 51.1% (92 of 180 patients) in the secukinumab group in the SUNRISE trial to 67.2% (121 of 180 patients) in the placebo group in the SUNSHINE trial. At baseline, patients with Hurley stage 3 disease severity ranged from 28.3% (51 of 180 patients) in the placebo group in the SUNSHINE trial to 45.6% (82 of 180 patients) in secukinumab group in the SUNRISE trial. The proportions of patients with 1 to 11 anatomic regions with at least 1 total fistula, inflammatory nodule, or abscess were generally well balanced between groups and across trials.
Across trials, the mean baseline AN count ranged from 12.8 (SD = 8.15) in the placebo group in the SUNSHINE trial to 13.9 (SD = 9.93) in the secukinumab group in the SUNRISE trial. The mean baseline NRS score ranged from 5.0 (SD = 2.61) in the placebo group in the SUNSHINE trial to 5.4 (SD = 2.42) in the secukinumab group in the SUNRISE trial. The mean baseline DLQI total score ranged from ████ (SD = ████) in the placebo group in the SUNSHINE trial to ████ (SD = ████) in the secukinumab group in the SUNRISE trial.
In the SUNSHINE trial, 13.8% (25 of 181 patients) in the secukinumab group and 10.6% (19 of 180 patients) in the placebo group enrolled in the antibiotic strata. Similarly, in the SUNRISE trial, 9.4% (17 of 180 patients) in the secukinumab group and 10.4% (19 of 183 patients) in the placebo group enrolled in the antibiotic strata.
The baseline characteristics outlined in Table 16 are limited to those that are most relevant to this review or that were felt by the review team to affect the outcomes or interpretation of the study results.
A summary of the baseline characteristics from both the SUNSHINE and SUNRISE trials in the entire study period is presented in Table 17. The baseline characteristics in the secukinumab group were generally similar between trials and generally similar to the baseline characteristics in treatment period 1.
Table 16: Summary of Baseline Characteristics From the SUNSHINE and SUNRISE Trials in Treatment Period 1 (Randomized Analysis Set)
Characteristic | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
Demographics and other baseline characteristics | ||||
Age (years), mean (SD) | 37.1 (12.53) | 35.5 (10.75) | 37.3 (11.48) | 36.2 (11.25) |
Age (years), median (range) | ██ ██ ██ ██ | ██ ██ ██ ██ | ██ █ ██ ███ | ██ ██ ██ ██ |
Age group (years), n (%) | ||||
< 30 | 58 (32.0) | 51 (28.3) | 52 (28.9) | 57 (31.1) |
30 to < 40 | 56 (30.9) | 70 (38.9) | 48 (26.7) | 65 (35.5) |
40 to < 65 | 64 (35.4) | 58 (32.2) | 77 (42.8) | 59 (32.2) |
≥ 65 | 3 (1.7) | 1 (0.6) | 3 (1.7) | 2 (1.1) |
Sex, n (%) | ||||
Male | 79 (43.6) | 78 (43.3) | 82 (45.6) | 78 (42.6) |
Female | 102 (56.4) | 102 (56.7) | 98 (54.4) | 105 (57.4) |
Race, n (%) | ||||
American Indian or Alaska Native | 1 (0.6) | 2 (1.1) | 7 (3.9) | 8 (4.4) |
Asian | 19 (10.5) | 24 (13.3) | 16 (8.9) | 19 (10.4) |
Black of African American | 15 (8.3) | 12 (6.7) | 18 (10.0) | 12 (6.6) |
Native Hawaiian or other Pacific Islander | 0 | 0 | 1 (0.6) | 0 |
White | 145 (80.1) | 139 (77.2) | 133 (73.9) | 143 (78.1) |
Multiplea | 1 (0.6) | 3 (1.7) | 4 (2.2) | 1 (0.5) |
Not reported | 0 | 0 | 1 (0.6) | 0 |
Weight (kg), mean (SD) | ██ ██ ██ ██ | ██ █ ██ ███ | ██ █ ██ ██ | ███ ██ ██ █ |
Weight groups in kg, n (%) | ||||
< 90 | 82 (45.3) | 83 (46.1) | 86 (47.8) | 92 (50.3) |
≥ 90 | 99 (54.7) | 97 (53.9) | 94 (52.2) | 91 (49.7) |
BMI (kg/m2), mean (SD) | 32.64 (7.904) | 31.97 (7.053) | 31.90 (7.788) | 31.42 (7.382) |
Smoking status, n (%) | ||||
Never | ███ █ █ ███ | ██ █ ██ ███ | ██ █ ██ ███ | ██ █ ██ ███ |
Current | 95 (52.5) | 101 (56.1) | 97 (53.9) | 106 (57.9) |
Former | 26 (14.4) | 30 (16.7) | 32 (17.8) | 24 (13.1) |
Relevant medical history | ||||
Obesity, n (%) | ██ ██ ██ ██ | ██ ██ ██ ██ | ██ █ ██ ███ | ██ ██ ██ ██ |
Diabetes mellitus, n (%) | ██ ██ ██ ██ | ██ ██ ██ ██ | █ █ ██ ███ | ██ ██ ██ ██ |
Type 1 diabetes mellitus, n (%) | ███ █ ██ ██ | ███ █ ██ ██ | ███ █ █ ███ | ██ ██ ██ ██ |
Type 2 diabetes mellitus, n (%) | ██ █ ██ ██ | ███ █ █ ███ | ███ █ ██ █ | ██ ██ ██ ██ |
Hyperandrogenism, n (%) | ███ █ ██ ██ | ██ █ ██ ███ | ███ █ ██ ██ | ██ ██ ██ ██ |
Disease history and baseline disease characteristics | ||||
Baseline Hurley stage, n (%) | ||||
1 | 7 (3.9) | 8 (4.4) | 6 (3.3) | 3 (1.6) |
2 | 104 (57.5) | 121 (67.2) | 92 (51.1) | 110 (60.1) |
3 | 70 (38.7) | 51 (28.3) | 82 (45.6) | 70 (38.3) |
Family history of HS, n (%) | ██ ██ ██ ██ | ███ █ ██ ██ | ███ █ ██ ██ | ███ █ ██ ██ |
Time since HS symptom onset (years), mean (SD) | ██ ██ ██ ██ | ███ █ ██ ██ | ██ █ ██ ███ | █ ███ ██ ██ |
Time since diagnosis of HS (years), n | 181 | 180 | 180 | 182 |
Mean (SD) | 7.4 (7.98) | 7.5 (7.00) | 7.1 (7.04) | 7.0 (6.65) |
Number of anatomic regionsb with at least 1 total fistula, inflammatory nodule, or abscess,c n (%) | ||||
1 | ██ █ ██ ███ | ██ █ ██ ███ | ██ ██ ██ ██ | ███ █ ██ ██ |
2 | ██ █ ██ ███ | ██ ██ ██ ██ | ████ ██ ██ | ██ █ ██ ███ |
3 | ██ █ ██ ███ | ██ █ ██ ███ | ████ ██ ██ | ███ █ ██ ██ |
4 | ██ █ ██ ███ | ███ █ ██ ██ | ████ ██ ██ | ██ ███ ███ |
5 | ██ █ ██ ███ | ██ ██ ██ ██ | ████ ██ ██ | ███ █ ██ ██ |
6 | ██ █ ██ ███ | ██ ██ ██ ██ | ████ ██ ██ | ██ ██ ██ ██ |
7 | ██ █ ██ ███ | ██ ██ ██ ██ | ████ ██ ██ | ███ █ ██ ██ |
8 | ██ █ ██ ███ | ███ █ ██ █ | ████ ██ ██ | ██ █ ██ ███ |
9 | ██ █ ██ ███ | ██ █ ██ ███ | ████ ██ ██ | ██ █ ██ ███ |
10 | ██ █ ██ ███ | ███ █ ██ ██ | ██ ██ ██ ██ | ███ █ ██ ██ |
11 | ██ █ ██ ███ | ███ █ ██ ██ | ███ ██ ███ | ██ █ ██ ███ |
Patients with at least 1 lesion in perineum,c,d n (%) | ██ █ ██ ███ | ███ █ ██ ██ | ███ ██ ███ | ██ █ ██ ███ |
Baseline AN count, n | 181 | 180 | 180 | 183 |
Mean (SD) | 12.9 (9.60) | 12.8 (8.15) | 13.9 (9.93) | 12.8 (8.45) |
Baseline inflammatory nodule count, n | 181 | 180 | 180 | 183 |
Mean (SD) | 10.1 (7.80) | 10.1 (6.99) | 10.0 (7.71) | 9.6 (6.77) |
Baseline abscess count, n | 181 | 180 | 180 | 183 |
Mean (SD) | 2.9 (4.26) | 2.7 (3.76) | 3.9 (5.41) | 3.2 (4.96) |
Baseline draining fistulae count, n | 181 | 180 | 180 | 183 |
Mean (SD) | 2.9 (3.41) | 2.4 (3.16) | 3.0 (3.63) | 2.6 (3.24) |
Baseline total fistulae count, n | 181 | 180 | 180 | 183 |
Mean (SD) | ██ ██ ██ ██ | ██ █ ██ ███ | ██ ██ ██ ██ | ██ ██ ██ ██ |
Baseline NRS, n | 163 | 162 | 166 | 166 |
Mean (SD) | 5.2 (2.51) | 5.0 (2.61) | 5.4 (2.42) | 5.3 (2.48) |
Patients with NRS ≥ 3, n (%) | ██ █ ██ ███ | ██ █ ██ ███ | ███ █ ██ ██ | █████ ██ ██ |
Baseline DLQI total score, n | 164 | 163 | 161 | 175 |
Mean (SD) | ██ █ ██ ███ | ██ █ ██ ███ | ███ ██ ██ | ██ █ ██ ███ |
Patients with DLQI total score ≥ 5,c n/m (%) | ██ █ ██ ███ | ██ █ ██ ███ | ███ █ ██ ██ | ██ █ ██ ███ |
Patients enrolled in the antibiotic strata, n (%) | 25 (13.8) | 19 (10.6) | 17 (9.4) | 19 (10.4) |
AN = abscesses and inflammatory nodules; BMI = body mass index; DLQI = Dermatology Life Quality Index; HS = hidradenitis suppurativa; m = number of patients with nonmissing DLQI total score at the baseline; mHSS = modified Hidradenitis Suppurativa Score; NA = not applicable; NR = not reported; NRS = numerical rating scale; q.2.w. = every 2 weeks; SD = standard deviation.
aRace – multiple means multiple entries were selected in the electronic case report form.
bAnatomic regions were defined as the following: intermammary area; submammary or inframammary area, left; submammary or inframammary area, right; buttock, left; buttock, right; inguinocrural fold, left; inguinocrural fold, right; perianal; perineal; axilla, left; axilla, right; neck; abdomen; chest; back; pubic region; genital region; upper extremity, left; upper extremity, right; lower extremity, left; lower extremity, right.
cBased on the full analysis set.
dPerineum included the genital, perianal, perineal, and pubic anatomic regions. Lesions included total fistula, inflammatory nodule, or abscess.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34-36 Details included in the table are from the sponsor’s summary of clinical evidence.67
Table 17: Summary of Baseline Characteristics From the SUNSHINE and SUNRISE Trials in Entire Study Period (Randomized Analysis Set)
Characteristic | SUNSHINE | SUNRISE | ||||
|---|---|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo to secukinumab q.2.w. (N = 90) | Any secukinumab q.2.w. (N = 271) | Secukinumab q.2.w. (N = 180) | Placebo to secukinumab q.2.w. (N = 90) | Any secukinumab q.2.w. (N = 270) | |
Demographics and other baseline characteristics | ||||||
Age (years), mean (SD) | ██ █ █ ██ | ███ █ ██ | ██ ██ ██ | ██ █ █ ██ | ███ ███ | ██ █ █ ██ |
Age (years), median (range) | ██ █ █ ██ | ██ █ █ ██ | ██ █ █ ██ | ██ █ █ ██ | ███ █ ██ | ██ █ █ ██ |
Age group (years), n (%) | ██ █ █ ██ | ██ █ █ ██ | ██ ██ ██ | ██ █ █ ██ | █ █ ██ ██ | ██ █ ██ █ |
< 30 | ██ █ █ ██ | ██ █ █ ██ | ██ █ █ ██ | ██ █ █ ██ | ███ ███ | ██ █ █ ██ |
30 to < 40 | ██ █ █ ██ | ██ █ █ ██ | ██ ██ ██ | ██ █ █ ██ | █ █ ██ ██ | ██ █ ██ █ |
40 to < 65 | ██ █ █ █ | ██ █ █ ██ | █ █ ██ ██ | ██ █ ███ | ██ █ ███ | ██ █ ███ |
≥ 65 | ██ █ █ ██ | ██ █ █ ██ | ██ █ █ ██ | ██ █ ███ | ██ ██ █ | ██ █ █ ██ |
Sex, n (%) | █ ██ █ ██ | █ ██ █ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | ██ █ █ ██ |
Male | ██ ██ ██ | ██ █ ███ | ██ ██ ██ | ██ █ █ ██ | ██ █ █ ██ | ██ █ ██ █ |
Female | █ ██ █ ██ | ██ █ ███ | █ █ ██ ██ | ██ █ █ ██ | ███ █ ██ | ██ █ █ ██ |
Race, n (%) | █ █ ██ ██ | ██ ██ ██ | ████ ███ | ██████ | ██ ██ ██ | ██ ██ ██ |
American Indian or Alaska Native | ██ ██ ██ | ████ ██ | ███ ████ | ███ ███ | ███ ███ | ███ ███ |
Asian | ███ █ ██ | █ ██ █ ██ | ██ ██ ██ | ██ █ █ ██ | ██ ██ ██ | ██ █ █ ██ |
Black of African American | █ █ ██ ██ | █ █ ██ ██ | ██ █ ██ █ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Native Hawaiian or other Pacific Islander | █ ███ ██ | ███ █ ██ | ██ ██ ██ | ███ █ ██ | ██ ██ ██ | ██ ██ ██ |
White | █ █ ██ ██ | ███ █ ██ | ██ █ █ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Multiplea | █ ███ ██ | ███ █ ██ | ██ █ ███ | ██ █ █ ██ | ███ ███ | ██ ██ ██ |
Not reported | ██ █ ███ | ███ █ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Weight (kg), mean (SD) | ██ ██ ██ | ███ █ ██ | ██ ██ ██ | █ █ ██ ██ | ██ █ ██ █ | ██ █ ██ █ |
Weight groups in kg, n (%) | ██ █ █ ██ | ███ █ ██ | ██ █ █ ██ | █ █ ██ ██ | █ █ ██ ██ | █ █ ██ ██ |
< 90 | ██ █ █ ██ | ███ █ ██ | ██ ██ ██ | ██ ██ █ █ | ██ ██ ██ | ██ █ █ ██ |
≥ 90 | ██ █ █ ██ | ██ █ ███ | ██ ██ ██ | ██ ██ ██ | █ ██ ███ | ███ ██ █ |
BMI (kg/m2), mean (SD) | ██ █ █ ██ | ███ █ ██ | ██ █ █ ██ | ██ █ ██ | ██ █ ██ █ | ██ ██ ██ |
Smoking status, n (%) | ██ █ ███ | ███ █ ██ | ██ █ ███ | ███ █ ██ | ██ █ ███ | █ ██ ███ |
Never | ██ ██ ██ | ██ █ ███ | ██ █ ███ | ██ ██ ██ | ██ ██ ██ | ██ █ ███ |
Current | ███ ███ | ██ ██ ██ | █ █ ██ ██ | ██ █ ██ █ | ██ ██ ██ | ██ ██ ██ |
Former | ██ ████ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | ██ █ ██ █ |
Disease history and baseline disease characteristics | ||||||
Baseline Hurley stage, n (%) | ||||||
1 | ██ ██ ██ | ██ ██ ██ | ███ ████ | ██ █ █ ██ | ██ ██ ██ | ██ ██ ██ |
2 | ██ ██ ██ | ██ █ ███ | ██ █ ███ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
3 | ██ ██ ██ | ███ ███ | ███ ████ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Family history of HS, n (%) | ██ █ █ ██ | ███ ███ | ██ █ ███ | ██ ██ ██ | ██ █ ███ | ██ █ █ ██ |
Time since HS symptom onset (years), mean (SD) | ██ ██ ██ | ███ ███ | ████ ███ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Time since diagnosis of HS (years), n | █ █ ██ ██ | ███ ███ | ██ █ █ ██ | ███ ██ | ██ ██ ██ | ██ █ █ ██ |
Mean (SD) | █ █ █ ██ | ███ ███ | ██ █ █ ██ | ███ ███ | ██ ██ ██ | ██ ██ ██ |
Baseline AN count, n | ██ █ █ ██ | ███ ███ | ██ █ ███ | ██ ██ ██ | ███ ███ | ██ ██ ██ |
Mean (SD) | ██ █ ██ █ | ███ ███ | ████ ███ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Baseline inflammatory nodule count, n | ███ ███ | ██ █ ██ | ██ █ █ █ | ██ ██ ██ | ██ █ ██ | ██ ██ ██ |
Mean (SD) | ███ ██ | ██ ██ ██ | ██ █ ███ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Baseline abscess count, n | ██ █ ███ | ███ █ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | █ █ ██ ██ |
Mean (SD) | ███ ███ | ███ ███ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Baseline draining fistulae count, n | ██ ██ ██ | ██ ██ ██ | ████ ███ | ██ █ █ ██ | ██ ██ ██ | ██ ██ ██ |
Mean (SD) | ███ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | ██ █ █ ██ |
Baseline total fistulae count, n | ███ ███ | ██ ██ ██ | ██ █ █ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Mean (SD) | ███ ███ | ██ █ ███ | ██ ██ ██ | █ █ █ ██ | ██ ██ ██ | ██ ██ ██ |
Baseline NRS, n | ██ █ ███ | ██ █ ███ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Mean (SD) | ██ █ ███ | ███ ███ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ | ██ ██ ██ |
Baseline DLQI total score, n | ██ ██ ██ | ███ ███ | ██ █ ███ | ██ █ ██ █ | ██ ██ ██ | ██ ██ ██ |
Mean (SD) | ███ ███ | ██ ██ ██ | ██ ██ ██ | ██ █ █ ██ | ██ ██ ██ | ██ ██ ██ |
AN = abscesses and inflammatory nodules; BMI = body mass index; DLQI = Dermatology Life Quality Index; HS = hidradenitis suppurativa; NRS = numerical rating scale; q.2.w. = every 2 weeks; SD = standard deviation.
Note: The entire study period included treatment period 1, treatment period 2, and the follow-up period.
aRace – multiple means multiple entries were selected in the electronic case report form.
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week 52.66 Details included in the table are from the sponsor’s Summary of Clinical Evidence.67
A summary of prior HS therapies in the SUNSHINE and SUNRISE trials is presented in Table 18. The proportion of patients with reported prior exposure to any HS medications was generally similar between groups and across trials. Overall, █████ (███ of 180 patients) in the placebo group in the SUNSHINE trial to █████ (███ of 180 patients) in the secukinumab group in the SUNRISE trial reported prior exposure to any HS medications. In both trials, the majority (approximately █████) of patients in each group reported prior systemic antibiotics use, with the most common reason (approximately █████) for discontinuation of systemic antibiotics being lack of efficacy; these proportions were generally similar between groups and across trials. In particular, ||████ (██ of 181 patients) in the secukinumab group and █████ (██ of 180 patients) in the placebo group reported prior exposure to adalimumab and the reason for discontinuation in the SUNSHINE trial. Similarly, █████ (██ of 180 patients) in the secukinumab group and █████ (██ of 183 patients) in the placebo group reported exposure to adalimumab and the reason for discontinuation in the SUNRISE trial. In both trials, the majority (| █████ of patients with prior adalimumab use) in each group discontinued because of lack of efficacy; the proportion of patients who discontinued adalimumab because of lack of efficacy was generally similar between groups and across trials.
Table 18: Summary of Prior HS Therapies From the SUNSHINE and SUNRISE Trials (Safety Set)
Therapy | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
Patients with ≥ 1 HS therapy, n (%) | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Topical therapy | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Reason for discontinuation of topical therapy | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Lack of efficacy | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Lack of tolerability | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Other | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Systemic antibiotics | 146 (80.7) | 150 (83.3) | 151 (83.9) | 151 (82.5) |
Reason for discontinuation of systemic antibiotics | ||||
Lack of efficacy | ███ ██████ | ██ █████ | ███ ██████ | ███ ████ |
Lack of tolerability | ███ ██████ | ██ █████ | ███ ██████ | ███████ |
Other | ███ ██████ | ██ █████ | ███ ██████ | ███ ████ |
Missing | ███ ██████ | ███████ | ███ ██████ | ██ ████ |
Systemic biologic therapy | 44 (24.3) | 46 (25.6) | 36 (20.0) | 48 (26.2) |
Reason for discontinuation of systemic biologic therapy | ||||
Lack of efficacy | ███ ██████ | ██ █████ | ███ ██████ | ███████ |
Lack of tolerability | ███ ██████ | ███ ███ | ███ ██████ | ███ ████ |
Other | ███ ██████ | ██ ████ | ███ ██████ | ███████ |
Adalimumaba | ███ ██████ | ███████ | ███ ██████ | ██ █████ |
Reason for discontinuation of adalimumabb | ███ ██████ | ███████ | ███ ██████ | ██ █████ |
Lack of efficacy | ███ ██████ | ███████ | ███ ██████ | ███████ |
Lack of tolerability | ███ ██████ | ███████ | ███ ██████ | ██ █████ |
Otherc | ███ ██████ | ███████ | ███ ██████ | ███████ |
Other systemic therapy (excluding biologics and antibiotics) | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Other | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Prior surgery for HS, n (%)a | 71 (39.2) | 72 (40.0) | 78 (43.3) | 78 (42.6) |
CDA-AMC = Canada’s Drug Agency; HS = hidradenitis suppurativa; q.2.w. = every 2 weeks.
Note: A patient with multiple types of therapy is counted only once in the total row. A patient can have multiple medications discontinued for different reasons within a type of therapy.
aBased on the randomized analysis set. In the SUNSHINE trial, 41 patients in the secukinumab group and 44 patients in the placebo group reported prior adalimumab discontinuation; 41 patients in the secukinumab group and 43 patients in the placebo group had nonmissing reason of discontinuation. In the SUNRISE trial, 35 patients in the secukinumab group and 46 patients in the placebo group reported prior adalimumab discontinuation; 34 patients in the secukinumab group and 44 patients in the placebo group had nonmissing reason of discontinuation.
bBased on the full analysis set. The N was the number of patients with prior use of adalimumab for HS.
cIf treatment discontinuation was not due to lack of efficacy or lack of tolerability, then the other category was selected in the case report form.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34,36 Details included in the table are from the sponsor’s Summary of Clinical Evidence.67
Exposure to Study Treatments
A summary of patient exposure to treatment in the SUNSHINE and SUNRISE trials during treatment period 1 is presented in Table 19. Exposure to treatment was generally similar between groups and across trials. At week 16, the proportion of patients with exposure to the study treatment ranged from █████ (███ of 181 patients) in the secukinumab group to █████ (███ of 180 patients) in the placebo group in the SUNSHINE trial, across trials. Note that not all patients had undergone 16 weeks of treatment at the time of data cut-off. The mean duration of treatment ranged from █████ (SD = █████) days in the placebo group in the SUNSHINE trial to █████ (SD = █████) days in the placebo group in the SUNRISE trial. In the secukinumab group, the mean total number of secukinumab injections was ███ (SD = ████) in the SUNSHINE trial and ███ (SD = ████) in the SUNRISE trial. The cumulative exposure to secukinumab was ████ patient-years in the SUNSHINE trial and ████ patient-years in the SUNRISE trial.
Table 19: Summary of Patient Exposure From the SUNSHINE and SUNRISE Trials in Treatment Period 1 (Safety Set)
Exposure | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
Any exposure, n (%)a | 181 (100.0) | 180 (100.0) | 180 (100.0) | 183 (100.0) |
≥ 1 week | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
≥ 2 weeks | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
≥ 3 weeks | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
≥ 4 weeks | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
≥ 8 weeks | ███ ██████ | ███████ | ███ ██████ | ███████ |
≥ 12 weeks | ███ ██████ | ███████ | ███ ██████ | ███████ |
≥ 16 weeks | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Duration (days), mean (SD)b | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Duration (days), median (range) | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Total number of injections, mean (SD) | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Total number of secukinumab injections | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Total number of placebo injections | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Patient time (patient-years)c | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
q.2.w. = every 2 weeks; SD = standard deviation.
aFor those patients who came earlier than day 112 for the week 16 visit, the treatment period was shorter than 16 weeks, and therefore these patients were not included in the “≥ 16 weeks” row but were counted in that category for the entire study period if they continued treatment after day 112.
bDuration of exposure to study treatment is defined as min (end date of treatment period 1, last dose date plus 84 days) minus start date of study treatment plus 1.
cPatient time in patient-years was calculated as a sum of individual patient durations in days divided by 365.25.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34 Details included in the table are from the sponsor’s Summary of Clinical Evidence.67
A summary of patient exposure to treatment in the SUNSHINE and SUNRISE trials during the entire study period and at the time of database lock is presented in Table 20. At week 52, █████ (███ of 181 patients) in the secukinumab group in the SUNSHINE trial and █████ (███ of 180 patients) in the secukinumab group in the SUNRISE trial had exposure to the study drug. In the secukinumab group, the mean duration of exposure to treatment was █████ days (SD = █████) the SUNSHINE trial and █████ days (SD = █████) in the SUNRISE trial. In the secukinumab group, the mean total number of secukinumab injections was ████ (SD = ████) in the SUNSHINE trial and ████ (SD = ████) in the SUNRISE trial. The cumulative exposure to secukinumab in the secukinumab group was █████ patient-years in the SUNSHINE trial and █████ patient-years in the SUNRISE trial.
Table 20: Summary of Patient Exposure From the SUNSHINE and SUNRISE Trials in Entire Study Period (Safety Set)
Exposure | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Any secukinumab q.2.w. (N = 266) | Secukinumab q.2.w. (N = 180) | Any secukinumab q.2.w. (N = 261) | |
Any exposure, n (%) | ||||
≥ 24 weeks | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
≥ 32 weeks | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
≥ 40 weeks | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
≥ 52 weeks | ███ ██████ | ███████ | ███ ██████ | ██ ████ |
Duration (days), mean (SD)a,b | ███ ██████ | ███████ | ███ ██████ | ███████ |
Duration (days), median (range) | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Total number of injections, mean (SD) | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Total number of secukinumab injections | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Total number of placebo injections | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Patient time (patient-years)c | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
q.2.w. = every 2 weeks; SD = standard deviation.
Notes: The entire study period included treatment period 1, treatment period 2, and the follow-up period.
The database lock was August 17, 2022, in the SUNSHINE trial and August 9, 2022, in the SUNRISE trial.
aDuration of exposure to study treatment is defined as end date of treatment period 1 last dose date plus 84 days minus the start date of study treatment plus 1.
bFor placebo-to-secukinumab switchers, exposure after the first intake of secukinumab is counted in the any-secukinumab groups accordingly.
cPatient time in patient-years was calculated as the sum of individual patient durations in days divided by 365.25.
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week 52.66 Details included in the table are from the sponsor’s Summary of Clinical Evidence.67
Concomitant Therapy
A summary of concomitant therapy from the SUNSHINE and SUNRISE trials in treatment period 1 is presented in Table 21. In treatment period 1, the use of any concomitant therapy was similar between groups and across trials, with the majority of patients (approximately █████) in each group using concomitant therapies. In the SUNSHINE trial, ibuprofen was used by █████ (██ of 181 patients) in the secukinumab group and █████ (██ of 180 patients) in the placebo group, and paracetamol was used by █████ (██ of 181 patients) in the secukinumab group and █████ (██ of 180 patients) in the placebo group. Similarly, in the SUNRISE trial, ibuprofen was used by █████ (██ of 180 patients) in the secukinumab group and █████ (██ of 183 patients) in the placebo group, and paracetamol was used by █████ (██ of 180 patients) in the secukinumab group and █████ (██ of 183 patients) in the placebo group. Tramadol, ibuprofen with paracetamol, doxycycline, minocycline, tetracycline, spironolactone, ethinyl estradiol, and cyproterone acetate were used by less than ███ of patients in each group and were generally similar between groups and across trials.
In the SUNSHINE trial, a total of ████ (██ of 181 patients) in the secukinumab group and █████ (██ of 180 patients) in the placebo group underwent a concomitant medical procedure and/or significant nondrug therapy (Table 21). In the SUNRISE trial, a total of █████ (██ of 180 patients) in the secukinumab group and █████ (██ of 183 patients) in the placebo group underwent a concomitant medical procedure and/or significant nondrug therapy. Note that no specific concomitant medical procedure and/or significant nondrug therapy was reported in more than ██ of patients in each group, across trials.
A summary of concomitant therapy from the SUNSHINE and SUNRISE trials in the entire study period is presented in Table 22. In the entire study period, the use of any concomitant therapy continued to be similar across trials, with the majority of patients (approximately ███ ██████) in each group using concomitant therapies. In both trials in the entire study period, the proportion of patients in the secukinumab group using any of the permitted concomitant therapies or any of the concomitant therapies anticipated to be used (according to the clinical experts consulted by CDA-AMC for this review), were generally similar to the proportions in treatment period 1, with a slightly higher proportion of patients using ibuprofen or paracetamol in the SUNSHINE trial (approximately one-half of patients in the secukinumab group).
In both trials, approximately one-third of patients in each group underwent a concomitant medical procedure and/or significant nondrug therapy (Table 22). Note that no concomitant medical procedure and/or significant nondrug therapy was reported in more than ██ of patients in each group, across trials.
Table 21: Summary of Concomitant Therapy From the SUNSHINE and SUNRISE Trials in Treatment Period 1 (Safety Set)
Therapy | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
Patients with ≥ 1 concomitant therapy, n (%) | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Permitted concomitant therapy | ||||
Ibuprofen | ███ ██████ | ████████ | ███ ██████ | ███ ████ |
Paracetamol | ███ ██████ | ████████ | ███ ██████ | ███ ████ |
Doxycycline | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Tramadol | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Minocycline | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Tetracycline | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Ibuprofen with paracetamol | ███ ██████ | ████████ | ███ ██████ | ███ ████ |
Paracetamol with tramadol | ███ ██████ | ████████ | ███ ██████ | ███ ████ |
Anticipated by the clinical experts to be possible concomitant therapy | ||||
Spironolactone | ███ ██████ | ████████ | ███ ██████ | ███ ████ |
Ethinyl estradiol | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Cyproterone acetate | ███ ██████ | ████████ | ███ ██████ | ███ ████ |
Isotretinoin | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Patients with ≥ 1 concomitant medical procedure and/or significant nondrug therapy, n (%) | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
NR = not reported; q.2.w. = every 2 weeks.
Note: A patient with multiple occurrences within an ATC class is counted only once in the total row.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34
Table 22: Summary of Concomitant Therapy From the SUNSHINE and SUNRISE Trials in Entire Study Period (Safety Set)
Therapy | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Any secukinumab q.2.w. (N = 266) | Secukinumab q.2.w. (N = 180) | Any secukinumab q.2.w. (N = 261) | |
Patients with ≥ 1 concomitant therapy, n (%) | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Permitted concomitant therapy | ||||
Ibuprofen | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Paracetamol | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Doxycycline | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Tramadol | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Minocycline | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Tetracycline | ███ ██████ | ███ ████ | ███ ██████ | ██ █████ |
Ibuprofen with paracetamol | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
Paracetamol with tramadol | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Advised by the clinical experts as anticipated possible concomitant therapy | ||||
Spironolactone | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
Ethinyl estradiol | ███ ██████ | ███████ | ███ ██████ | ███████ |
Cyproterone acetate | ███ ██████ | ███████ | ███ ██████ | ███████ |
Isotretinoin | ███ ██████ | ███████ | ███ ██████ | ███ ████ |
Patients with ≥ 1 concomitant medical procedure and/or significant nondrug therapy, n (%) | ███ ██████ | ███████ | ███ ██████ | ███ ████ |
NR = not reported; q.2.w. = every 2 weeks.
Note: A patient with multiple occurrences within an ATC class is counted only once in the total row.
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week 52.66
Rescue Therapy
A summary of rescue therapy used in the SUNSHINE and SUNRISE trials is presented in Table 23 and Table 24. In both studies, relatively few patients required any rescue medication or any rescue therapy procedure during either treatment period 1 or the entire study period.
Table 23: Summary of Rescue Therapy From the SUNSHINE and SUNRISE Trials (Safety Set)
Therapy | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
Treatment period 1 | ||||
Patients requiring any rescue HS medication, n (%) | ███ ██████ | ███████ | ███ ██████ | ███ ████ |
Patients requiring rescue intervention with systemic antibiotics, n (%) | ███ ██████ | ███████ | ███ ██████ | ███ ████ |
Patients requiring rescue intervention with intralesion corticosteroid injections, n (%) | ███ ██████ | ███████ | ███ ██████ | ███████ |
Patients with ≥ 1 rescue therapy procedure, n (%) | ███ ██████ | ███ ████ | ███ ██████ | ███ ████ |
HS = hidradenitis suppurativa; q.2.w. = every 2 weeks.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34
Table 24: Summary of Rescue Therapy From the SUNSHINE and SUNRISE Trials (Safety Set)
Therapy | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Any secukinumab q.2.w. (N = 266) | Secukinumab q.2.w. (N = 180) | Any secukinumab q.2.w. (N = 261) | |
Entire study period | ||||
Patients with ≥ 1 rescue medication, n (%) | ███ ██████ | ███ █████ | ███ ██████ | ███████ |
Patients with ≥ 1 rescue therapy procedure, n (%) | ███ ██████ | ███ ████ | ███ ██████ | ███████ |
HS = hidradenitis suppurativa; q.2.w. = every 2 weeks.
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week 52.66
Efficacy
Response to Treatment and Disease Severity
Hidradenitis Suppurativa Clinical Response
Both the SUNSHINE and SUNRISE studies met the primary end point, achievement of HiSCR50 response at week 16, for the secukinumab 300 mg every 2 weeks dose regimen (Table 25). In the SUNSHINE trial, the marginal risk difference in HiSCR50 response at week 16 between secukinumab and placebo was █████ (96% CI, ████ ██ █████) (OR = 1.75; 96% CI, ████ ██ ████; P value = 0.0070) in favour of secukinumab. In the SUNRISE trial, the marginal risk difference in HiSCR50 response at week 16 between secukinumab and placebo was █████ (96% CI, ████ ██ █████) (OR = 1.64; 96% CI, ████ ██ ████; P value = 0.0149), also in favour of secukinumab. The sensitivity analysis, supplementary analysis, and tipping point analysis results for HiSCR50 response at week 16 were generally consistent with and supportive of the primary analysis results for the secukinumab 300 mg every 2 weeks dose regimen in both studies.
A summary of the results from the key subgroups analysis of the primary end point in both trials (concomitant antibiotic use, body weight stratum, previous use of systemic biologics, Hurley stage, and baseline AN count) are presented in Table 26. The results of the subgroups analysis by the key subgroups listed previously are generally consistent with the primary analysis, with the exception of the results by patients with Hurley stage 1 in the SUNRISE trial (█████| of ||| patients] in the secukinumab group compared with █████ of ||| patients] in the placebo group achieved HiSCR50 response at week 16).
The proportion of patients achieving HiSCR50 response observed at week 52 (exploratory end point) in both studies is presented in Table 27. In the SUNSHINE trial, █████ (██ of 117 patients) (95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of 58 patients) (95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved HiSCR50 response at week 52. In the SUNRISE trial, █████ (██ of 137 patients) (95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of 64 patients) (95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved HiSCR50 response at week 52.
The proportion of patients in each group with a decrease of at least 25%, 50%, 75%, 90%, or 100% in AN count with no increase in the number of abscesses and/or in the number of draining fistulae compared with baseline (i.e., an HiSCR25, HiSCR50, HiSCR75, HiSCR90, or HiSCR100 response, respectively), observed at week 16 and week 52 (exploratory end points) in both studies, is presented in Table 38 and Table 39 in Appendix 1.
Abscesses and Inflammatory Nodules Count
Both the SUNSHINE and SUNRISE studies met the secondary end point, AN count at week 16, for the secukinumab 300 mg every 2 weeks dose regimen; this secondary end point was tested in a hierarchical manner to control for type I error (Table 25). In the SUNSHINE trial, the LS mean difference in percentage change from baseline in AN count at week 16 between secukinumab and placebo was −23.05 (96% CI, ██████ ██ ██████; P < 0.0001) in favour of secukinumab. In the SUNRISE trial, the LS mean difference in percentage change from baseline in AN count at week 16 between secukinumab and placebo was −16.33 (96% CI, ██████ ██ █████; P = 0.0051), also in favour of secukinumab. The sensitivity analysis and tipping point analysis results of AN count at week 16 were generally consistent with and supportive of the primary analysis results for the secukinumab 300 mg every 2 weeks dose regimen in both studies.
The percentage change from baseline in AN count observed at week 52 (exploratory end point) in both studies is presented in Table 27. In the SUNSHINE trial, the mean percentage change from baseline in AN count at week 52 was █████ (95% CI, █████ ██ █████) in the secukinumab group and █████ (95% CI, █████ ██ █████) in the placebo-to-secukinumab group. In the SUNRISE trial, the mean percentage change from baseline in AN count at week 52 was █████ (95% CI, █████ ██ █████) in the secukinumab group and █████ (95% CI, █████ ██ █████) in the placebo-to-secukinumab group.
Remission
Disease remission was not measured in the SUNSHINE and SUNRISE trials.
Disease Worsening
Flare
Only the SUNSHINE study met the secondary end point, experience of any flares at week 16, for the secukinumab 300 mg every 2 weeks dose regimen; this secondary end point was tested in a hierarchical manner to control for type I error (Table 25). In the SUNSHINE trial, the marginal risk difference in flares at week 16 between secukinumab and placebo was ██████ (96% CI, ██████ ██ █████) (OR = 0.42; 96% CI, ████ ██ ████; P value = 0.0010), in favour of secukinumab. In the SUNRISE trial, the marginal risk difference in flares at week 16 between secukinumab and placebo was █████ (96% CI, ██████ ██ ████) (OR = 0.68; 96% CI, ████ ██ ████; P value = 0.0732). The sensitivity analysis and tipping point analysis results of flares at week 16 were generally consistent with and supportive of the primary analysis results for secukinumab 300 mg every 2 weeks dose regimen in both studies.
The proportion of patients experiencing any flares observed at week 52 (exploratory end point) in both studies is presented in Table 27. In the SUNSHINE trial, █████ (██ of 138 patients) (95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of 65 patients) (95% CI, █████ ██ █████) in the placebo-to-secukinumab group experienced any flares at week 52. In the SUNRISE trial, █████ (██ of 151 patients) (95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of 67 patients) (95% CI, █████ ██ █████) in the placebo-to-secukinumab group experienced any flares at week 52. The Kaplan-Meier estimates of time to disease flare up observed up to week 52 is presented in Figure 2 for SUNSHINE and Figure 3 for SUNRISE; patients without any flares were censored at the last known visit.
Symptoms
Skin Pain
The secondary end point, achievement of NRS30 (skin pain at its worst) at week 16, for the secukinumab 300 mg every 2 weeks dose regimen was met based on pooled data from the SUNSHINE and SUNRISE studies in patients with baseline NRS of 3 or more; this secondary end point was tested in a hierarchical manner to control for type I error (Table 25). The marginal risk difference in NRS30 at week 16 between secukinumab and placebo was █████ (96% CI, ████ ██ █████) (OR = ████; 96% CI, ████ ██ ████; P value = 0.0003), in favour of secukinumab. The tipping point analysis results of NRS30 at week 16 were supportive of the primary analysis results for secukinumab 300 mg every 2 weeks dose regimen in both studies.
The proportion of patients achieving NRS30 observed at week 52 (exploratory end point) based on pooled data from both trials in patients with baseline NRS of 3 or more is presented in Table 28. Based on the pooled data, █████ (███ of ███ patients) (95% CI, █████ ██ █████) in the secukinumab group and █████ (██ of ██ patients) (95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved NRS30 at week 52.
Health-Related Quality of Life
Dermatology Life Quality Index
The proportion of patients achieving DLQI response observed at week 16 (exploratory end point) in both studies is presented in Table 25. In the SUNSHINE trial, the risk difference in DLQI response at week 16 between secukinumab and placebo was ██████ (95% CI, █████ ██ ██████) (OR = ████; 95% CI, ████ ██ ████; P value = ██████). In the SUNRISE trial, the risk difference in DLQI response at week 16 between secukinumab and placebo was █████ (95% CI, ██████ ██ ██████) (OR = ████; 95% CI, ████ ██ ████; P value = ██████).
The proportion of patients achieving DLQI response observed at week 52 (exploratory end point) in both studies is presented in Table 27. In the SUNSHINE trial, 51.0% (49 of 96 patients) (95% CI, █████ ██ █████) in the secukinumab group and 50.0% (25 of 50 patients) (95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved DLQI response at week 52. In the SUNRISE trial, 55.2% (64 of 116 patients) (95% CI, █████ ██ █████) in the secukinumab group and 47.5% (29 of 61 patients) (95% CI, █████ ██ █████) in the placebo-to-secukinumab group achieved DLQI response at week 52.
The change from baseline in DLQI total score observed at week 16 (exploratory end point) in both studies is presented in Table 25. In the SUNSHINE trial, the mean difference in absolute change from baseline in DLQI total score at week 16 between secukinumab and placebo was ████ (95% CI, ████ ██ ████). In the SUNRISE trial, the mean difference in absolute change from baseline in DLQI total score at week 16 between secukinumab and placebo was ████ (95% CI, ████ ██ ████).
The change from baseline in DLQI total score observed at week 52 (exploratory end point) in both studies is presented in Table 27. In the SUNSHINE trial, the mean absolute change from baseline in DLQI total score at week 52 was ████ (95% CI, ████ ██ ████) in the secukinumab group and ████ (95% CI, ████ ██ ████) in the placebo-to-secukinumab group. In the SUNRISE trial, the mean absolute change from baseline in DLQI total score at week 52 was ████ (95% CI, ████ ██ ████) in the secukinumab group and ████ (95% CI, ████ ██ ████) in the placebo-to-secukinumab group.
EQ-5D Health State Assessment (VAS)
The change from baseline in EQ-5D health state assessment (EQ VAS) observed at week 16 (exploratory end point) in both studies is presented in Table 25. In the SUNSHINE trial, the mean difference in absolute change from baseline in EQ VAS score at week 16 between secukinumab and placebo was ███ (95% CI, ████ ██ ███). In the SUNRISE trial, the mean difference in absolute change from baseline in EQ VAS score at week 16 between secukinumab and placebo was ███ (95% CI, ███ ██ ████).
Table 25: Summary of Key Efficacy Results From the SUNSHINE and SUNRISE Trials at Week 16 (Full Analysis Set)
Outcome | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
Response to treatment and disease severity | ||||
HiSCR50 response at week 16a | ||||
n in 100 imputations/N evaluable (%) | 81.5/181 (45.0) | 60.7/180 (33.7) | 76.2/180 (42.3) | 57.1/183 (31.2) |
95% CI | ███ ██ ███ | ███ ██ ███ | ███ █ ████ | ███ ██ ███ |
OR (96% CI) | 1.75 ████ ██ ████ | 1.64 ████ ██ ████ | ||
P valueb | 0.0070 | 0.0149 | ||
Risk, percent (96% CI) | ███ ██ ███ | ███ █ ████ | █████ ███ | ███ ██ ███ |
Marginal risk difference (96% CI) | ████ ██ ████ | ████ ██ ████ | ||
AN count at week 16c | ||||
N evaluabled | ███ ██ ███ | ███ ██ ███ | ███ ██ ███ | ███ ██ ███ |
Mean baseline AN count (SE) | ███ ██ ███ | ███ ██ ███ | ███ █ ████ | ███ ██ ███ |
Mean week 16 AN count (SE) | ███ ██ ███ | ███ ██ ███ | ███ ██ ███ | ███ ██ ███ |
Mean percentage change from baseline (SE)e | −46.8 (3.33) | −24.3 (4.33) | −39.3 (4.43) | −22.4 (4.84) |
LS mean difference estimate (96% CI) | −23.05 ████ ██ ████ | −16.33 ████ ██ ████ | ||
P valueb | < 0.0001 | 0.0051 | ||
Disease worsening | ||||
Flares at week 16a | ||||
n in 100 imputations/N evaluable (%) | 27.8/181 (15.4) | 52.2/180 (29.0) | 36.1/180 (20.1) | 49.5/183 (27.0) |
95% CI | ███ ██ ███ | ███ ██ ███ | ███ ██ ██ | ███ ██ ███ |
OR (96% CI) | 0.42 ████ ██ ████ | 0.68 ████ ██ ████ | ||
P valueb | 0.0010 | 0.0732 | ||
Risk, percent (96% CI) | ███ ██ ███ | ███ ██ ███ | ███ █████ | ███ ██ ███ |
Marginal risk difference (96% CI) | ████ ██ ████ | ████ ██ ████ | ||
Symptom | ||||
NRS30 (skin pain)f,g at week 16 | SUNSHINE and SUNRISEh | NA | ||
n in 100 imputations/N evaluable (%) | 97.2/266 (36.6) | 57.8/251 (23.0) | NA | NA |
95% CI | ███ ██ ███ | ███ ██ ███ | NA | NA |
OR (96% CI) | 2.08 ████ ██ ████ | NA | ||
P valueb | 0.0003 | NA | ||
Risk, percent (96% CI) | ███ █ ████ | ████ █ ███ | NA | NA |
Marginal risk difference (96% CI) | ████ ██ ████ | NA | ||
Health-related quality of life | ||||
DLQI responsei,j at week 16 | ||||
N | ███ ██ ███ | ███ ██ ███ | ███ ██ ███ | ███ ██ ███ |
n/N evaluable (%) | 64/134 (47.8) | 37/128 (28.9) | 54/144 (37.5) | 46/145 (31.7) |
95% CI | ███ ██ ███ | ███ ██ ███ | ███ ██ ███ | ████ ██ ██ |
Risk difference (95% CI) | ████ ██ ████ | ████ ██ ████ | ||
OR (95% CI) | ████ ██ ████ | ████ ██ ████ | ||
P valuek | ████ ██ ████ | ████ ██ ████ | ||
DLQI total score at week 16 | ||||
N evaluable | ███ ██ ███ | ███ ██ ███ | ███ ██ ███ | ███ ██ ███ |
Mean baseline DLQI score (SD) | ███ ██ ███ | ███ ██ ███ | ████ █ ███ | ███ █ ████ |
Mean week 16 DLQI score (SD)l | ███ ██ ███ | ███ ██ ███ | ███ █ ████ | ███ █ ████ |
Mean absolute change from baseline (SD) | ███ ██ ███ | ███ ██ ███ | ████ █ ███ | ███ ██████ |
Difference in mean change (SD) | ████ ██ ████ | ████ ██ ████ | ||
95% CIm | ████ ██ ████ | ████ ██ ████ | ||
Mean percentage change from baseline (SD) | █████ ████ | ███ █ ████n | ███ ██ ███ | ███ █ ████o |
EQ-5D health state assessment (EQ VAS score) at week 16p | ||||
N evaluable | 143 | 143 | 150 | 149 |
Mean baseline EQ VAS score (SD) | ███ █ ████ | ██ ██ ████ | ███ █ ████ | ███ █ ████ |
Mean week 16 EQ VAS score (SD)l | 67.7 ████ ██ ████ | 63.4 ████ ██ ████ | 69.6 ████ ██ ████ | 63.2 ████ ██ ████ |
Mean absolute change from baseline (SD) | █████ ████ | ███ █ ████ | ███ █ ████ | ███ █ ████ |
Difference in mean change (SD) | ████ ██ ████ | ████ ██ ████ | ||
95% CIm | ████ ██ ████ | ████ ██ ████ | ||
Mean percentage change from baseline (SD) | ██ ██ ████ | ██ ██ ████ | ████ ████ | ███ ██████ |
AN = abscesses and inflammatory nodules; ANCOVA = analysis of covariance; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; DLQI = Dermatology Life Quality Index; EQ VAS = EQ-5D visual analogue scale; HiSCR = Hidradenitis Suppurativa Clinical Response; LS = least squares; NA = not applicable; NRS = numerical rating scale; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; q.2.w. = every 2 weeks; SD = standard deviation; SE = standard error.
Note: The placebo group included patients randomized to placebo to secukinumab 300 mg every 2 weeks group and placebo to secukinumab 300 mg every 4 weeks group before their first intake of active secukinumab.
aCovariates included in the logistic regression model: treatment group, Hurley stage, baseline AN count, geographical region, use of antibiotic, and baseline body weight.
bOne-sided nominal P value is presented. P value was adjusted for multiple testing using a predefined testing hierarchy. The primary and secondary end points (AN count and flares) were tested in a hierarchical order (1-sided significance level of 0.02). The secondary end point, NRS30 (skin pain) was based on pooled data from the SUNSHINE and SUNRISE trials.
cCovariates included in the ANCOVA model: treatment group, Hurley stage, baseline AN count, geographical region, use of antibiotic, and baseline body weight.
dAN count at baseline and week 16 was based on the number of patients evaluable (observed data).
eMean is the pooled mean over 100 imputations. Standard error is the pooled standard error over 100 imputations.
fCovariates included in the logistic regression model: treatment group, Hurley stage, baseline NRS, geographical region, use of antibiotic, baseline body weight, study.
gNRS is the numeric rating scale of the patient's global assessment of skin pain at its worst (averaged over the last 7 days). Only patients with a baseline NRS ≥ 3 are included. NRS30 is defined as at least a 30% reduction and at least 2-unit reduction from baseline NRS.
hAt week 16, the NRS30 skin pain was analyzed based on pooled data from the SUNSHINE and SUNRISE trials.
iDLQI response was defined as a decrease of ≥ 5.0 points on the DLQI total score. Only patients with a baseline DLQI score ≥ 5.0 points were included.
jCovariates included in the logistic regression model: treatment group, Hurley stage, baseline DLQI, geographical region, use of antibiotic, baseline body weight.
kTwo-sided P value is presented. The P value for pairwise comparisons was not adjusted for multiple comparisons and no formal confirmatory hypothesis testing was planned.
lFor each postbaseline visit, only patients with a value from both the baseline and respective postbaseline visit were included.
mThe 95% CI was based on t statistics.
nA total of 142 patients were evaluable.
oA total of 149 patients were evaluable.
pHigher score values indicate a better quality of life status in the EQ-5D health state assessment.
Sources: SUNSHINE Clinical Study Report,33 SUNRISE Clinical Study Report,34 and sponsor response to June 19, 2023 and July 5, 2023, CDA-AMC requests for additional information regarding the secukinumab review.35,36 Details included in the table are from the sponsor’s summary of clinical evidence.67
Table 26: Summary of Subgroup Analysis Results of Primary End Point From the SUNSHINE and SUNRISE Trials at Week 16 (Full Analysis Set)
Outcome | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
Current antibiotic use | ||||
HiSCR50 response in patients with concomitant antibiotic use at week 16 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | █████ ██████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
HiSCR50 response in patients without concomitant antibiotic use at week 16 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
Body weight stratum | ||||
HiSCR50 response in patients with a body weight of < 90 kg at week 16 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
HiSCR50 response in patients with a body weight of ≥ 90 kg at week 16 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
Previous exposure to biologics | ||||
HiSCR50 response in patients with previous exposure to biologics at week 16 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (96% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
HiSCR50 response in patients without previous exposure to biologics at week 16 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
Hurley stage | ||||
HiSCR50 response in patients with Hurley stage 1 at week 16 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
HiSCR50 response in patients with Hurley stage 2 at week 16 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
HiSCR50 response in patients with Hurley stage 3 at week 16 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
Baseline AN count | ||||
HiSCR50 response in patients with baseline AN count ≤ 10 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
HiSCR50 response in patients with baseline AN count > 10 | ||||
n in 100 imputations/N evaluable (%) | ██████ █████ | ██████ █████ | ██████ █████ | ██████ █████ |
OR (95% CI) | ███████ ██████ | ███████ ██████ | ||
P valuea | ███████ ██████ | ███████ ██████ | ||
AN = abscesses and inflammatory nodules; CI = confidence interval; HiSCR50 = Hidradenitis Suppurativa Clinical Response 50, i.e., a decrease of 50% or greater in the AN count with no increase in the number of abscesses or draining fistulae compared with baseline; OR = odds ratio; q.2.w. = every 2 weeks.
Notes: The placebo group included patients randomized to placebo to secukinumab 300 mg every 2 weeks group and placebo to secukinumab 300 mg every 4 weeks group before their first intake of active secukinumab.
Covariates included in the logistic regression model: treatment group, weight, and baseline AN count.
aTwo-sided P value is presented. The P value was not adjusted for multiple testing.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34
The change from baseline in EQ VAS score observed at week 52 (exploratory end point) in both studies is presented in Table 27. In the SUNSHINE trial, the mean absolute change from baseline in EQ VAS score at week 52 was ███ (95% CI, ███ ██ ████) in the secukinumab group and ███ (95% CI, ███ ██ ████) in the placebo-to-secukinumab group. In the SUNRISE trial, the mean absolute change from baseline in EQ VAS score at week 52 was ████ (95% CI, ███ ██ ████) in the secukinumab group and ███ (95% CI, ████ ██ ███) in the placebo-to-secukinumab group.
Table 27: Summary of Efficacy Results From the SUNSHINE and SUNRISE Trials at Week 52 (Full Analysis Set)
Outcome | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo to secukinumab q.2.w. (N = 90) | Secukinumab q.2.w. (N = 180) | Placebo to secukinumab q.2.w. (N = 90) | |
Response to treatment and disease severity | ||||
HiSCR50 response at week 52 | ||||
n/N evaluable (%) | ████ ████ | ████ ████ | ████ ████ | ███ █████ |
95% CI | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
AN count at week 52 | ||||
N evaluable | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Mean baseline AN count (SE) | ████ ████ | ████ ████ | ████ ████ | ████ ███ |
Mean week 52 AN count (SE) | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Mean percentage change from baseline (SE) | ████ ████ | ███ █████ | ████ ████ | ████ ████ |
95% CI | ████ ████ | ███ █████ | ████ ████ | ███ █████ |
Disease worsening | ||||
Flares at week 52 | ||||
n/N evaluable (%) | ████ ████ | ███ █████ | ████ ████ | ████ ████ |
95% CI | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Health-related quality of life | ||||
DLQI response at week 52a | ||||
N | ████ ████ | ███ █████ | ███ █████ | ████ ████ |
n/N evaluable (%) | ████ ████ (51.0) | ████ ████ (50.0) | ████ ███ (55.2) | ████ ████ (47.5) |
95% CI | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
DLQI total score at week 52 | ||||
N evaluable | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Mean baseline DLQI score (SE) | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Mean week 52 DLQI score (SE)b | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Mean absolute change from baseline (SE) | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
95% CI | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Mean percentage change from baseline (SE) | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
EQ-5D health state assessment (EQ VAS) at week 52 | ||||
N evaluable | 103 | 54 | 122 | 60 |
Mean baseline EQ VAS score (SE) | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Mean week 52 EQ VAS score (SE)b | 70.7 ████ ██████ | 68.3 ██████ ██████ | 72.0 ███████ ██████ | 69.2 ████ ██████ |
Mean absolute change from baseline (SE) | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
95% CI | ████ ████ | ████ ████ | ████ ████ | ████ ████ |
Mean percentage change from baseline (SE) | ████ ████ | ████ ████ | ████ ███ | ████ ████ |
AN = abscesses and inflammatory nodules; CI = confidence interval; DLQI = Dermatology Life Quality Index; EQ VAS = EQ-5D visual analogue scale; HiSCR = Hidradenitis Suppurativa Clinical Response; LS = least squares; q.2.w. = every 2 weeks; SE = standard error.
aDLQI response was defined as a decrease of ≥ 5.0 points on the DLQI total score. Only patients with a baseline DLQI score ≥ 5.0 points were included.
bFor each postbaseline visit, only patients with a value from both the baseline and respective postbaseline visit were included.
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week 52,66 and sponsor responses to July 5, 2023, request for additional information regarding our review of secukinumab.36 Details included in the table are from the sponsor’s summary of clinical evidence.67
Table 28: Summary of Efficacy Results From the SUNSHINE and SUNRISE Trials at Week 52 (Full Analysis Set)
Outcome | SUNSHINE and SUNRISEb | |||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo to secukinumab q.2.w. (N = 90) | Secukinumab q.2.w. (N = 180) | Placebo to secukinumab q.2.w. (N = 90) | |
Symptoms | ||||
NRS30 (skin pain) at week 52a | ||||
N | ████ ████ | ████ ████ | ||
n/N evaluable (%) | ████ ████ | ████ ████ | NA | NA |
95% CI | ████ ████ | ████ ████ | NA | NA |
CI = confidence interval; NA = not applicable; NRS = numerical rating scale; q.2.w. = every 2 weeks.
aNRS is the numeric rating scale of the patient’s global assessment of skin pain at its worst (averaged over the last 7 days). Only patients with a baseline NRS ≥ 3 are included. NRS30 is defined as at least 30% reduction and at least 2-unit reduction from baseline NRS.
bAt week 52, the NRS30 skin pain was analyzed based on pooled data from the SUNSHINE and SUNRISE trials.
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week 52,66 and sponsor responses to July 5, 2023, request for additional information regarding our review of secukinumab.36 Details included in the table are from the sponsor’s summary of clinical evidence.67
Figure 2: Kaplan-Meier Estimates of Time to Disease Flare up to Week 52 From the SUNSHINE Trial (Full Analysis Set) — Redacted

Figure 3: Kaplan-Meier Estimates of Time to Disease Flare up to Week 52 From the SUNRISE Trial (Full Analysis Set) — Redacted

Harms
A summary of harms data from the SUNSHINE and SUNRISE trials in treatment period 1 and the entire study period is presented in Table 29 and Table 30, respectively.
Adverse Events
In treatment period 1, the proportion of patients with any AE was generally similar between groups and across trials, ranging from 62.8% (113 of 180 patients) in the secukinumab group in the SUNRISE trial to 67.4% (122 of 181 patients) in the secukinumab group in the SUNSHINE trial.
The most common AEs (frequency ≥ 5% in any group) reported in the SUNSHINE trial were nasopharyngitis (11.0% [20 of 181 patients] in secukinumab group compared with 7.2% [13 of 180 patients] in placebo group), headache (9.4% [17 patients] compared with 7.8% [14 patients], respectively), hidradenitis (6.1% [11 patients] compared with 13.3% [24 patients], respectively), and diarrhea (2.8% [5 patients] compared with 5.0% [9 patients], respectively). The most common AEs (frequency ≥ 5% in any group) reported in the SUNRISE trial were headache (11.7% [21 of 180 patients] in secukinumab group compared with 8.2% [15 of 183 patients] in placebo group), nasopharyngitis (7.2% [13 patients] compared with 8.7% [16 patients], respectively), hidradenitis (5.6% [10 patients] compared with 7.7% [14 patients], respectively), upper respiratory tract infection (5.0% [9 patients] compared with 3.8% [7 patients], respectively), and diarrhea (4.4% [8 patients] compared with 7.1% [13 patients], respectively).
In the entire study period, the proportion of patients with any AE continued to be generally similar across trials, ranging from 80.1% (209 of 261 patients) in the any-secukinumab group in the SUNRISE trial to 85.1% (154 of 181 patients) in the secukinumab group in the SUNSHINE trial. The most common AEs (frequency ≥ 10% in any group) reported in both trials were headache, nasopharyngitis, and hidradenitis. AEs reported in 5% or more of patients in any group in both trials included diarrhea, upper respiratory tract infection, pyrexia, arthralgia, pruritus, intertrigo, urinary tract infection, oropharyngeal pain, eczema, COVID-19, hypertension, and folliculitis (Table 30).
Serious Adverse Events
In treatment period 1, the proportion of patients with any SAE was generally similar between groups and across trials, ranging from 1.7% (3 of 181 patients) in the secukinumab group to 3.3% (6 of 180 patients) in the placebo group in the SUNSHINE trial. The most common SAE (frequency ≥ 1% in any group in both trials) reported was hidradenitis in 0.6% (1 of 181 patients) in the secukinumab group and 1.1% (2 of 180 patients) in the placebo group in the SUNSHINE trial, and 0.6% (1 of 180 patients) in the secukinumab group and no patients in the placebo group in the SUNRISE trial.
In the entire study period, the proportion of patients with any SAE was generally similar across trials, ranging from 6.8% (18 of 266 patients) in the any-secukinumab group in the SUNSHINE trial to 10.6% of patients (19 of 180 patients) in the secukinumab group in the SUNRISE trial. The most common SAE (frequency ≥ 1% in any group) in both trials reported was hidradenitis in 1.7% of patients (3 of 181 patients) in the secukinumab group and 1.5% of patients (4 of 266 patients) in the any-secukinumab group in the SUNSHINE trial, and 2.2% (4 of 180 patients) in the secukinumab group and 1.9% (5 of 261 patients) in the any-secukinumab group in the SUNRISE trial. In the SUNRISE trial, each SAE acute kidney injury and pyrexia, was reported in 1.1% (2 of 180 patients) in the secukinumab group and 0.8% (2 of 261 patients) in the any-secukinumab group.
Withdrawals Due to Adverse Events
In treatment period 1, the proportion of patients who stopped treatment because of any AE was generally similar between groups and across trials, ranging from 0.6% (1 of 180 patients) in the placebo group to 2.8% (5 of 181 patients) in the secukinumab group in the SUNSHINE trial. No AE that led to treatment discontinuation was reported in 1% of patients or more in any group in both trials.
In the entire study period, the proportion of patients who stopped treatment because of any AE was generally similar across trials, ranging from 3.4% (9 of 261 patients) in the any-secukinumab group in the SUNRISE trial to 5.5% (10 of 181 patients) in the secukinumab group in the SUNSHINE trial. Similar to treatment period 1, no AE that led to treatment discontinuation was reported in 1% of patients or more in any group in both trials.
Mortality
In treatment period 1 and the entire study period, no deaths were reported in either trial.
Notable Harms
In general, AEs of special interest (notable harms) were similar between the secukinumab and placebo groups and across trials in treatment period 1. For infections and infestations (SOC), the risk difference was ████ (95% CI, █████ ██ ████) in the SUNSHINE trial and █████ (95% CI, █████ ██ ████) in the SUNRISE trial. For candida infections (HLT), the risk difference was █████ (95% CI, █████ ██ ████) in the SUNSHINE trial and ████ (95% CI, █████ ██ ████) in the SUNRISE trial. For malignant and unspecified tumour (SMQ), the risk difference was █████ (95% CI, █████ ██ ████) in the SUNSHINE trial and █████ (95% CI, █████ ██ ████) in the SUNRISE trial. For neoplasms (benign, malignant, and unspecified, including cysts and polyps), the risk difference was █████ (95% CI, █████ ██ ████) in the SUNSHINE trial and █████ (95% CI, █████ ██ ████) in the SUNRISE trial. No patients were reported with squamous cell carcinoma of an HS-affected area or inflammatory bowel disease in treatment period 1.
Table 29: Summary of Harms Results From the SUNSHINE and SUNRISE Trials in Treatment Period 1 (Safety Set)
Adverse events | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
Treatment-emergent adverse events, n (%)a | ||||
Patients with any adverse event | 122 (67.4) | 120 (66.7) | 113 (62.8) | 116 (63.4) |
Nasopharyngitis | 20 (11.0) | 13 (7.2) | 13 (7.2) | 16 (8.7) |
Headache | 17 (9.4) | 14 (7.8) | 21 (11.7) | 15 (8.2) |
Hidradenitis | 11 (6.1) | 24 (13.3) | 10 (5.6) | 14 (7.7) |
Diarrhea | 5 (2.8) | 9 (5.0) | 8 (4.4) | 13 (7.1) |
Upper respiratory tract infection | 5 (2.8) | 4 (2.2) | 9 (5.0) | 7 (3.8) |
Serious adverse events, n (%)b | ||||
Patients with any SAE | 3 (1.7) | 6 (3.3) | 6 (3.3) | 5 (2.7) |
Hidradenitis | 1 (0.6) | 2 (1.1) | 1 (0.6) | 0 (0.0) |
Patients who stopped treatment because of adverse events, n (%)b | ||||
Patients with any WDAE | 5 (2.8) | 1 (0.6) | 1 (0.6) | 4 (2.2) |
Mortality, n (%) | ||||
Deaths | 0 | 0 | 0 | 0 |
Adverse events of special interest, n (%) | ||||
Hypersensitivity (SMQ) | 12 (6.6) | 9 (5.0) | 7 (3.9) | 7 (3.8) |
Hypersensitivity | NR | NR | NR | NR |
Drug hypersensitivity | NR | NR | 1 (0.6) | 0 |
Injection site reaction | 0 | 0 | 0 | 0 |
Infections and infestations (SOC) | 59 (32.6) | 53 (29.4) | 52 (28.9) | 62 (33.9) |
Infections and infestations | 59 (32.6) | 53 (29.4) | 52 (28.9) | 62 (33.9) |
RR estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
RD estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
Candida infections (HLT) | 2 (1.1) | 4 (2.2) | 5 (2.8) | 2 (1.1) |
RR estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
RD estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
Malignant or unspecified tumour (SMQ) | 0 | 1 (0.6) | 0 | 1 (0.5) |
RR estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
RD estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
Neoplasms benign, malignant, or unspecified (including cysts and polyps) | 0 | 1 (0.6) | 2 (1.1) | 4 (2.2) |
RR estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
RD estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
Squamous cell carcinoma of HS-affected area | 0 | 0 | 0 | 0 |
RR estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
RD estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
Inflammatory bowel disease | 0 | 0 | 0 | 0 |
RR estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
RD estimate (95% CI) | ████ █████ ██ █████ | ████ █████ ██ █████ | ||
Suicidal ideation and behaviour (SMQ) | ██ █ ██ ███ | ██ █ █████ | ███ ██ ███ | ██ ██ █ ██ |
Suicidal ideation | ██ █ ██ ███ | ██ █ █████ | ███ ██ ███ | ██ █ ██ ██ |
Suicide attempt | 1 (0.6) | 0 | ████ ██ ███ | █ ████ ███ |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; HLT = high-level term; HS = hidradenitis suppurativa; NA = not applicable; NR = not reported; q.2.w. = every 2 weeks; RD = risk difference; RR = risk ratio; SAE = serious adverse event; SMQ = Standardised MedDRA Query; SOC = system organ class; WDAE = withdrawal due to adverse event.
Notes: The placebo group included patients randomized to placebo to secukinumab 300 mg every 2 weeks group and placebo to secukinumab 300 mg every 4 weeks group before their first intake of active secukinumab.
A patient with multiple AEs with the same preferred term is counted only once for that preferred term.
SMQs are validated, standard sets of MedDRA terms used to support signal detection and monitoring and represent a variety of safety topics of regulatory interest. SMQs include narrow and/or broad terms; narrow terms are highly likely to represent the condition of interest.71
aFrequency ≥ 5% in any group.
bFrequency ≥ 1% in any group.
Sources: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report, and sponsor response to July 5, 2023, CDA-AMC requests for additional information regarding the secukinumab review.34,36 Details included in the table are from the sponsor’s summary of clinical evidence.67
In the entire study period, patients with any notable harms continued to be generally similar across trials. Patients reported with infections and infestations (SOC) ranged from 51.7% (93 of 180 patients) in the secukinumab group in the SUNRISE trial to 58.6% (106 of 181 patients) in the secukinumab group in the SUNSHINE trial. Patients reported with candida infections (HLT) ranged from 5.4% (14 of 261 patients in the any-secukinumab group to 6.7% (12 of 180 patients) in the secukinumab group in the SUNRISE trial. The proportion of patients reported with a malignant or unspecified tumour (SMQ) or neoplasm (benign, malignant, or unspecified, including cysts and polyps) was less than 5% of patients in each group for both trials. Similar to treatment period 1, no patients were reported with squamous cell carcinoma of an HS-affected area or inflammatory bowel disease in the entire study period.
Table 30: Summary of Harms Results From the SUNSHINE and SUNRISE Trials in Entire Study Period (Safety Set)
Adverse events | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Any secukinumab q.2.w. (N = 266) | Secukinumab q.2.w. (N = 180) | Any secukinumab q.2.w. (N = 261) | |
Treatment-emergent adverse events, n (%)a | ||||
Patients with any adverse event | 154 (85.1) | 220 (82.7) | 147 (81.7) | 209 (80.1) |
Headache | 33 (18.2) | 39 (14.7) | 31 (17.2) | 39 (14.9) |
Nasopharyngitis | 32 (17.7) | 40 (15.0) | 21 (11.7) | 28 (10.7) |
Hidradenitis | 19 (10.5) | 31 (11.7) | 22 (12.2) | 28 (10.7) |
Pyrexia | 13 (7.2) | 16 (6.0) | 9 (5.0) | 11 (4.2) |
Diarrhea | 11 (6.1) | 12 (4.5) | 13 (7.2) | 19 (7.3) |
Arthralgia | 11 (6.1) | 14 (5.3) | 7 (3.9) | 11 (4.2) |
Pruritus | 11 (6.1) | 15 (5.6) | 8 (4.4) | 11 (4.2) |
Intertrigo | 10 (5.5) | 11 (4.1) | 4 (2.2) | 6 (2.3) |
Upper respiratory tract infection | 9 (5.0) | 12 (4.5) | 13 (7.2) | 16 (6.1) |
Urinary tract infection | 9 (5.0) | 10 (3.8) | 7 (3.9) | 8 (3.1) |
Oropharyngeal pain | 9 (5.0) | 11 (4.1) | 8 (4.4) | 8 (3.1) |
Eczema | 8 (4.4) | 9 (3.4) | 10 (5.6) | 11 (4.2) |
COVID-19 | 6 (3.3) | 7 (2.6) | 10 (5.6) | 13 (5.0) |
Hypertension | 6 (3.3) | 8 (3.0) | 11 (6.1) | 14 (5.4) |
Folliculitis | 4 (2.2) | 6 (2.3) | 9 (5.0) | 9 (3.4) |
Serious adverse events, n (%)b | ||||
Patients with any SAE | 13 (7.2) | 18 (6.8) | 19 (10.6) | 22 (8.4) |
Hidradenitis | 3 (1.7) | 4 (1.5) | 4 (2.2) | 5 (1.9) |
Acute kidney injury | 0 | 0 | 2 (1.1) | 2 (0.8) |
Pyrexia | 0 | 0 | 2 (1.1) | 2 (0.8) |
Patients who stopped treatment because of any adverse event, n (%)b | ||||
Patients with any WDAE | 10 (5.5) | 11 (4.1) | 7 (3.9) | 9 (3.4) |
Mortality, n (%) | ||||
Deaths | 0 | 0 | 0 | 0 |
Adverse events of special interest, n (%) | ||||
Hypersensitivity (SMQ) | 29 (16.0) | 35 (13.2) | 23 (12.8) | 28 (10.7) |
Hypersensitivity | 0 | 0 | 0 | 0 |
Drug hypersensitivity | NR | NR | 1 (0.6) | 1 (0.4) |
Injection site reaction | 0 | 0 | 0 | 1 (0.4) |
Infections and infestations (SOC) | 106 (58.6) | 140 (52.6) | 93 (51.7) | 136 (52.1) |
Infections and infestations | 106 (58.6) | 139 (52.3) | 91 (50.6) | 134 (51.3) |
Candida infections (HLT) | 11 (6.1) | 15 (5.6) | 12 (6.7) | 14 (5.4) |
Malignant or unspecified tumour (SMQ) | 1 (0.6) | 1 (0.4) | 1 (0.6) | 1 (0.4) |
Neoplasms benign, malignant, or unspecified (including cysts and polyps) | 2 (1.1) | 2 (0.8) | 7 (3.9) | 7 (2.7) |
Squamous cell carcinoma of HS-affected area | 0 | 0 | 0 | 0 |
Inflammatory bowel disease | 0 | 0 | 0 | 0 |
Suicidal ideation and behaviour (SMQ) | ██ ██████ | ██ ██ █ ███ | ██ ███ ███ | ████ ████ |
Suicidal ideation | 1 (0.6) | 1 (0.4) | 0 | 0 |
Suicidal attempt | 1 (0.6) | 1 (0.4) | 0 | 0 |
HLT = high-level term; HS = hidradenitis suppurativa; NR = not reported; q.2.w. = every 2 weeks; SAE = serious adverse event; SMQ = Standardised MedDRA Query; SOC = system organ class; WDAE = withdrawal due to adverse event.
Notes: The entire study period included treatment period 1, treatment period 2, and the follow-up period.
The any-secukinumab 300 mg every 2 weeks group included patients randomized to secukinumab 300 mg every 2 weeks plus patients randomized to placebo to secukinumab 300 mg every 2 weeks group after their first intake of secukinumab.
A patient with multiple adverse events with the same preferred term is counted only once for that preferred term.
SMQs are validated, standard sets of MedDRA terms used to support signal detection and monitoring and represent a variety of safety topics of regulatory interest. SMQs include narrow and/or broad terms; narrow terms are highly likely to represent the condition of interest.71
aFrequency ≥ 5% in any treatment group.
bFrequency ≥ 1% in any treatment group.
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week 52.66 Details included in the table are from the sponsor’s summary of clinical evidence.67
Critical Appraisal
Internal Validity
The SUNSHINE and SUNRISE trials were randomized, double-blind, and placebo-controlled. Randomization was stratified by region, concomitant antibiotic use, and body weight. Based on input from the clinical experts consulted by CDA-AMC for the purpose of this review, the following patient demographic and disease characteristics were identified as effect modifiers: smoking status, obesity, diabetes, androgen excess in female patients, inadequate response to a prior biologic, baseline lesion count, number of anatomic sites involved, involvement of perineum, and baseline disease severity. There were slightly more patients with Hurley stage 3 disease in the secukinumab group versus placebo. The experts indicated that Hurley stage 3 disease is more severe and difficult to treat and, as such, potential bias against secukinumab may have been introduced in analyses that were unadjusted for this characteristic; however, the magnitude is unclear and could be small. Although randomization was not stratified by the aforementioned variables identified by the experts, any potential impact on the efficacy results was not expected because the proportions of patients with the relevant medical history and disease characteristics (effect modifiers) at baseline were generally well balanced between the secukinumab and placebo groups in both trials (other than Hurley stage, as discussed previously). Of note, there was no active or placebo comparator group for the assessments made at week 52 and, as such, the ability to draw definitive conclusions about the 52-week results is limited because of the potential for confounding.
The experts indicated that the list of prohibited medications in the trials and durations of the washout periods can be considered reasonable, such that no continued effect would be expected after the prespecified washout period and therefore the medications would be unlikely to have an impact on the efficacy assessment of secukinumab. Concomitant use of permitted therapies, medical procedures, significant nondrug therapies, and those therapies that the 2 clinical experts anticipate would be concomitant with secukinumab in practice, was generally balanced between groups and across trials in treatment period 1 and in the entire study period. Additionally, relatively few patients (≤ 11%) in both trials required any rescue medication and/or any rescue therapy procedure. Therefore, any potential impact on the efficacy results due to concomitant therapies and rescue therapy use was not expected. The experts indicated that the daily use of topical OTC antiseptics on areas affected by HS while on study treatment was not reflective of clinical practice; however, the experts did not expect this inclusion criterion to impact the efficacy results.
A statistical testing strategy was implemented in both trials to control for type I error at the level of the individual studies and at the level of the pooled dataset of both studies. Exploratory end point analyses were not adjusted for multiple comparisons and are therefore at an increased risk of false-positive results. The subgroup analyses were not adjusted for multiple testing; moreover, the ability to draw definitive conclusions about the results is limited because of the relatively small sample size of most subgroups.
Missing data for the primary and secondary end points were addressed using multiple imputation, while missing data for the exploratory outcomes at week 16 were not imputed and the respective analyses were based only on observed data. For the primary and secondary end points, the sensitivity analysis, supplementary analysis, and tipping point analysis were generally consistent with and supportive of the primary analyses. For the exploratory outcomes at week 16, the amount of missing data observed was relatively low (< 20%), with the exception of DLQI total score and EQ-5D health state assessment in the SUNSHINE trial (approximately 20%); however, the amount of missing data observed was relatively balanced between groups in both trials. As such, any potential impact on the efficacy results at week 16 because missing data was not expected. Missing data for the exploratory outcomes at week 52 were not imputed and based only on observed data. The amount of missing data observed was relatively high (> 20%) in both the secukinumab and placebo-to-secukinumab groups in both trials. Therefore, there was potential for bias because of the relatively high amount of missing data in the efficacy results at week 52; however, the direction and magnitude of this potential bias is unknown.
Although approximately 30% of patients in each group from both trials had at least 1 protocol deviation in treatment period 1, the proportions of patients were generally well balanced between groups and across trials. Most patients had a protocol deviation related to treatment deviation, which was mainly related to drug administration at home versus at the site. As such, any potential impact on the efficacy results because of protocol deviations during treatment period 1 was not expected. In the entire study period of both trials, more than 60% of patients in each group had at least 1 protocol deviation, with treatment deviation continuing to be the most common category. As such, there was a potential impact on the efficacy results at week 52 because of the relatively high proportions of patients with a protocol deviation; however, the direction and magnitude of this potential impact is unknown.
Treatment response according to HiSCR and the occurrence of flares was based on lesion counts. Areas affected by HS were assessed by the physician for the presence of inflammatory nodules, abscesses, draining fistulae, total fistulae, and other lesions. Therefore, the primary and secondary outcomes, HiSCR, AN count, and flares, are potentially subjective measures of lesion count. Moreover, central adjudication was not performed and the experts suggested there could potentially be interobserver variation in lesion counts (e.g., resolving nodules may be counted by some clinicians but not others). However, considering the double-blind design, the concern for the potentially subjective measures of lesion count to have an impact on the efficacy results is low. Note that there is evidence in the literature to support the measurement properties of HiSCR20,21 and the clinical importance of HiSCR50 in patients with HS.20,22
The patient-reported outcomes, NRS for skin pain due to HS, DLQI score, and EQ-5D health state assessment are also potentially subjective measures of skin pain and HRQoL. However, considering the low rates (< 10%) of discontinuation in treatment period 1 and the double-blind design, the concern for the potentially subjective measures of skin pain and HRQoL to have an impact on the efficacy results is low. Note that there is evidence in the literature to support the validity of the NRS30,23 DLQI,24 and EQ VAS25 in patients with HS. Furthermore, there is evidence to support the clinical importance of NRS30 skin pain (albeit a threshold of only 30% was suggested, and not in patients with HS)26-28 and DLQI response (estimated MID of 5 points in patients with HS)22 as defined in the trials. Note that an MID in the EQ-5D health state assessment has not been estimated in patients with HS.
External Validity
According to the clinical experts consulted by CDA-AMC for this review, the inclusion and exclusion criteria used in the SUNSHINE and SUNRISE trials were considered standard for trials in patients with HS. The trials excluded patients with fewer than 5 inflammatory lesions, a history of multiple lesions, previous exposure to any IL-17 inhibitors, and other active skin disease or condition that may interfere with the assessment of HS; however, based on feedback from the experts, these criteria would not preclude a patient from treatment with secukinumab. The experts further noted that patients with any known malignancy or history of malignancy of any organ system treated or untreated within the past 5 years would be considered for treatment in practice on a case-by-case basis in consultation with the patient’s oncologist. Based on the baseline characteristics, patients in both trials were reflective of the patient population that would be considered as candidates for treatment with secukinumab in practice in Canada. This was supported by feedback from the clinical experts consulted by CDA-AMC.
At baseline, patients were instructed by site staff on how to self-inject using the prefilled syringe. All doses of the study treatment were self-administered by the patient or a trained caregiver either at the study site or at home; this aligns with the product monograph for secukinumab.1 The experts indicated that patients would continue to receive treatment from week 16 to week 52 despite no response or incomplete response. Note that completing treatment period 1 (defined as baseline to before week 16 dosing) was the only criterion used in the trials to enter treatment period 2 (defined as post week 16 dosing through week 52).
The experts agreed that the criteria for the use of rescue therapy and the options for rescue therapy used in both trials generally reflected clinical practice. The experts acknowledged that some clinicians prefer antibiotics, while others prefer surgical intervention as rescue therapy. There was no requirement in either trial regarding the duration of increased AN count in the criteria for use of an oral antibiotic rescue medication, but the experts consulted by CDA-AMC indicated that patients who experience an increase in AN count that occurs for a longer period of time (months) would likely receive rescue therapy in practice.
Based on feedback from the experts, aside from minocycline, which is used less commonly in practice in Canada, the concomitant use of antibiotics in the antibiotic strata and nonopioid analgesics in the trials were consistent with clinical practice and aligned with the North American clinical management guidelines for HS (published in 2019).19,29 Of note, although topical antibiotic therapy was prohibited in the trials, the experts anticipated that patients would continue topical antibiotic therapy while on treatment with secukinumab if they previously experienced a partial response to the topical antibiotic therapy.
The comparator in both trials was placebo, which can be considered appropriate in evaluating the efficacy of secukinumab. The investigator noted that the use of placebo would minimize the number of patients who would need to be exposed to a treatment that, at the time of conducting the trials, was without a confirmed benefit-risk assessment in HS. The investigator further noted that demonstration of efficacy using either a superiority or noninferiority design versus an active drug would require a large, potentially unfeasible sample size in an indication with a relatively low prevalence; the estimated prevalence of HS in North America and Europe is approximately 1% of the population.13-17
The experts identified the following as outcomes used in clinical practice to assess response to treatment: lesion count (abscess, nodule, and fistula), pain scale, number of sites involved, extent of disease, and patient-reported outcomes such as DLQI, activities of daily living, and HRQoL; therefore, the assessments made in the trials generally aligned with clinical practice. The experts indicated that outcomes are typically assessed every 3 to 6 months. The experts considered the assessment of efficacy of secukinumab at the 16-week time point to be reasonable and agreed that response to treatment would be expected to occur in 16 weeks. Additionally, the experts indicated that long-term assessment (e.g., 2 years) would be meaningful, considering HS is a life-long disease. Nevertheless, the experts considered the long-term assessment of the efficacy and safety of secukinumab at the 52-week time point to be reasonable.
In the entire study period, approximately 20% or more of patients in each group of each study discontinued, mostly because of patient decision; study discontinuation because of AEs or lack of efficacy was relatively low (< 5%). The experts indicated that patients with HS are often not adherent to treatment and suggested that possible reasons for discontinuation could include improvement in HS, lack of response to treatment, and inconvenience of travelling to clinical trial visits. Regardless, the experts considered a 20% rate of study discontinuation to be reasonable.
Monthly Maintenance Dosing
In general, no notable differences in the study populations between study drug groups (secukinumab 300 mg every 2 weeks, secukinumab 300 mg every 4 weeks, and placebo groups) were identified in either study. As such, the limitations discussed for the primary and exploratory efficacy analyses at week 16 and week 52 of the biweekly maintenance dosing are applicable to the corresponding analyses of the monthly maintenance dosing.
Overall, no serious risk of bias concerns and no major issues with the generalizability of the results to the target population and Canadian practice were identified in the appraisal of the placebo-controlled phase of the SUNNY trials. Notably, there was no active or placebo comparator group for the assessments made at week 52 and there is a concern for potential bias (albeit of unknown direction and magnitude) because of the relatively high amount (> 20%) of missing data observed at week 52 in both trials. As such, the ability to draw causal conclusions about the 52-week results is limited because the noncomparative design does not facilitate distinguishing between the effect of treatment, placebo effects, and natural history.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform the CDA-AMC expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:30,31
High certainty: We are very confident that the true effect lies close to the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
For RCTs: Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
For single arms of trials (not presented in the summary of findings table): Although GRADE guidance is not available for noncomparative studies, the CDA-AMC review team assessed the noncomparative (52 weeks of) outcomes for study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials starts at very low certainty, with no opportunity for rating up.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for HiSCR50 response, AN count, flares, NRS30 skin pain, DLQI response, and EQ-5D health state assessment was set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review. The reference point for the certainty of evidence assessment for DLQI total score was set according to the presence or absence of an important effect based on the threshold identified in the literature. The reference points for the certainty of evidence assessment for notable harms (infections and infestations; candida infections; malignant or unspecified tumours; neoplasms benign, malignant, or unspecified, including cysts and polyps; squamous cell carcinoma of an HS-affected area; and inflammatory bowel disease) was set according to the presence or absence of an important effect based on thresholds informed by the clinical experts.
For the GRADE assessments, findings from the SUNSHINE and SUNRISE studies were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures.
Results of GRADE Assessments
Secukinumab Versus Placebo
Table 3 presents the GRADE summary of findings for secukinumab 300 mg every 2 weeks versus placebo as well as secukinumab 300 mg every 4 weeks versus placebo. Note that the data presented in the table on GRADE summary of findings is based on data provided by the sponsor following the submission update dated April 24, 2024 (details in Appendix 2).
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
There was no direct evidence from the pivotal trials that compared secukinumab with biologics or other systemic therapies used to treat HS. This section appraises the indirect evidence submitted by the sponsor.
Description of Indirect Treatment Comparison
The sponsor submitted an NMA that compared the short-term efficacy of secukinumab with adalimumab in patients with moderate to severe HS.72
Indirect Treatment Comparison Design
Objectives
The objective of the ITC was to assess the relative efficacy of secukinumab compared with other relevant approved therapy for moderate to severe HS.
Study Selection Methods
The sponsor conducted a systematic review of the literature to identify relevant evidence informing the efficacy and safety of biologics and other systemic therapies, surgery, or light-based therapies, in adolescents (≥ 12 years of age) and adults with moderate to severe HS. A focused literature search was conducted in April 2021 and updated in August 2022, which was screened using the criteria listed in Table 31. Additional criteria were applied to select studies to inform the ITC, which focused on biologic comparators. Of note, only the results from the 2021 search were used to inform the ITC.
Table 31: Study Selection Criteria for SLR and ITC and Methods for Indirect Comparisons
Characteristics | SLR | Indirect comparison |
|---|---|---|
Population | Adolescents and adults (≥ 12 years of age) with moderate to severe HS; studies with a mixed population were included if they presented the subgroup data for the population of interest | Adult patients (≥ 18 years of age) with moderate to severe HS |
Interventions |
| Secukinumab |
Comparatorsa |
|
|
Outcomes | None specified |
|
Study designsb |
|
|
Publication characteristics | Publications were limited to the English language and those published between database inception and the date of when the SLR was conducted | Publications were limited to the English language |
Exclusion criteria | None specified | None specified |
Sources searched |
| |
Selection process | Screening was conducted independently by 2 reviewers. Any discrepancies at either title and abstract or full-text review stage were resolved by discussion with a third reviewer. | |
Data extraction process | Data extraction was performed by 2 independent reviewers in a prespecified data extraction grid. A third independent reviewer undertook a quality check of the data extraction for accuracy and completeness by reviewing 100% of the extracted articles. | |
Quality assessment | Risk of bias was assessed using the questions listed in the NICE quality assessment for clinical trials. The quality of trials was considered during the feasibility assessment to determine the trials included in the ITC, and to address the uncertainty within the NMAs. It is unclear if assessment was done by a single reviewer or in duplicate with consensus, as the methods were not reported. | |
AN = abscesses and inflammatory nodules; DLQI = Dermatology Life Quality Index; HiSCR = Hidradenitis Suppurativa Clinical Response; HS = hidradenitis suppurativa; HTA = health technology assessment; ITC = indirect treatment comparison; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; RCT = randomized controlled trial; SLR = systematic literature review.
aNot applicable for single-group studies.
bStudies with English abstracts where the full-text articles are in a non–English language were excluded from the review but listed and shared with Novartis.
Sources: Sponsor-submitted ITC report.72 Details included in the table are from the sponsor’s summary of clinical evidence.67
ITC Analysis Methods
A feasibility assessment was conducted based on the results of the systematic review to determine the best approach for conducting the ITC. The criteria and methods used to conduct the feasibility assessment are listed in Table 31.
The feasibility assessment also reviewed the following:
Connectivity of networks for each outcome of interest based on the available data across studies and comparability of outcome definitions.
Designs and characteristics of studies of interest (including treatment regimens, study designs, and eligibility criteria) were reviewed and compared to assess the level of heterogeneity.
Potential treatment-effect modifiers and prognostic factors in patients with HS were identified to assess whether imbalances between studies could lead to heterogeneity in the network or biased estimated effects.
Baseline characteristics across studies of interest for the ITC were compared.
Methods to analyze the different outcomes of interest, assumptions of the ITC, and sensitivity analyses were determined based on the previously mentioned assessments.
The ITC was conducted using fixed- and random-effects Bayesian NMA models that compared secukinumab with adalimumab. The sponsor stated that a number of biologics were considered for inclusion in the ITC; however, as adalimumab was the only comparator with Health Canada approval, only trials assessing adalimumab were included in the construction of networks for the ITC. The base-case analyses were limited to patients who were biologic-naive, and the sensitivity analyses included a larger evidence base in biologic-naive and biologic-experienced patient populations.
For this review, the efficacy outcomes of interest were the proportion of patients with HS clinical response (3 thresholds: HiSCR25, HiSCR50, HiSCR75), HS flares, skin pain NRS30 response, and DLQI response (score of 0 or 1). Also of interest was the percent change from baseline in AN count and change from baseline in DLQI. The analyses were based on outcomes assessed at the end of the induction period for each clinical trial.
The authors of the ITC state the NMAs were conducted in accordance with National Institute for Health and Care Excellence (NICE) Decision Support Unit guidelines.73,74 All analyses were run using noninformative priors, with model fit assessed based on the deviance information criterion (DIC). Statistical heterogeneity in pairwise comparisons was examined based on the I2 value and Cochran Q test. The selection of the preferred model (i.e., fixed versus random effects) was based on the DIC with the lower value, the presence of statistical heterogeneity, and the assessment of convergence. The analysis of binary end points used a binomial likelihood and a logit link function to derive estimated ORs and 95% CrIs. Mean differences and 95% CrIs were estimated for continuous outcomes. The proportion of patients who achieved different thresholds of response were analyzed using a multinomial model, where patients were categorized as follows: less than HiSCR25, HiSCR25 to less than HiSCR50, HiSCR50 to less than HiSCR75, HiSCR75 to less than HiSCR100. Details related to the ITC methods are summarized Table 32.
The sponsor stated that it was necessary to impute study data to complete some of the analyses. HiSCR25 and HiSCR75 data were imputed for the PIONEER studies from the estimated transition probabilities issued from an ordered categorical NMA, with treatment effects measured on the probit scale based on data reported by NICE in its adalimumab submission papers.75 The number of patients who achieved DLQI response was also imputed for PIONEER, based on the percentage of responders, and the estimated proportion of patients who had a baseline DLQI total score of at least 5 at baseline (i.e., the approximated sample size for that end point).
Table 32: Indirect Comparison Analysis Methods
Methods | Description |
|---|---|
Analysis methods | A Bayesian NMA was conducted using both fixed- and random-effect models. A 3-chain MCMC analysis was conducted using a minimum of 20,000 burn-in iterations followed by 20,000 iterations to assess the posterior distribution of parameters for the fixed-effects models. A minimum of 100,000 burn-in iterations followed by 100,000 iterations were conducted to assess the posterior distribution of parameters for the random-effect models.
An adjustment was required to account for trials with 3 or more treatment groups. The multigroup trials were considered to estimate a vector of random effects vs. only 1, which was computed using a conditional distribution of the multivariate normal distribution to account for correlation of treatment effects within the study. All models for all end points were constructed following the NICE Decision Support Unit Technical Support Document 2 and Document 4 guidelines and were conducted using WinBUGS version 1.4 software.73,74 |
Priors | Noninformative prior distributions for unknown parameters were used to ensure that the posterior distributions were driven by the data distributions. |
Assessment of model fit | Models with the lower DIC value with considerations for heterogeneity were used to determine the use of random or fixed effects for each model. |
Assessment of consistency | The assessment of consistency was made for loops of evidence within the networks. The loops identified in the networks included placebo, adalimumab, and bimekizumab. The direct and indirect evidence were compared using the Bucher method.76 |
Assessment of convergence | Convergence was assessed using Gelman-Rubin plots. |
Assessment of heterogeneity | An assessment of statistical heterogeneity was conducted using the Cochran Q test (10% significance level) and the I2 (> 50%) from the pairwise meta-analyses, with forest plots to illustrate the heterogeneity. |
Outcomes | Continuous:
Binary:
Ordered categorical outcomes:
|
Missing data imputations | Missing data imputation methods could differ across trials for the different end points of interest (e.g., nonresponder imputation, multiple imputation, LOCF, or observed case). |
Follow-up time points | Outcomes analyzed at the end of each study’s induction period (12 to 16 weeks). |
Construction of nodes | Each dosage regimen was analyzed as a separate node. |
Subgroup analysis | None. |
Sensitivity analyses | Included a larger evidence base in a mixed population (biologic-naive and biologic-experienced). |
Methods for pairwise meta-analysis | Methods for the meta-analysis were not reported, aside from using both random- and fixed-effects models. |
AN = abscesses and inflammatory nodules; CI = confidence interval; CrI = credible interval; CRP = C-reactive protein; DIC = deviance information criterion; DLQI = Dermatology Life Quality Index; HiSCR = Hidradenitis Suppurativa Clinical Response; LOCF = last observation carried forward; MCMC = Markov chain Monte Carlo; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; OR = odds ratio; RD = risk difference; RR = relative risk.
Sources: Sponsor-submitted ITC report72 and sponsor’s summary of clinical evidence.67
Results of ITC
Summary of Included Studies
The evidence to inform the ITC was based on the April 2021 literature search and did not include studies identified in the 2022 update. A total of 30 studies met the inclusion criteria of the systematic review, of which 17 were RCTs, 13 were single-arm trials, and 2 were open-label extension studies. Five additional studies were identified in the 2022 update to the literature search, but these trials were not used to inform the ITC.
After applying the more restrictive inclusion criteria used for the ITC, a total of 13 studies for 7 different biologics were included in the feasibility assessment. The sponsor stated that since adalimumab was the only biologic approved for use in HS by the European Medicines Agency (EMA) and the FDA, it was deemed the comparator of interest for the ITC. During the feasibility assessment, preliminary NMAs were conducted for biologic-naive and biologic-experienced patients for HiSCR50, flares, and skin pain NRS30 end points. Two adalimumab trials were excluded; 1 because of heterogeneity across baseline characteristics (HS0001 study), and the other because it was deemed outside the scope, as it assessed the efficacy of adalimumab in combination with surgery (SHARPS study). Thus, the primary evidence network was informed by 4 studies (PIONEER 1, PIONEER 2, SUNSHINE, and SUNRISE) and limited to patients who were biologic-naive. The sensitivity analyses included all comparative trials assessing adalimumab and secukinumab in biologic-experienced and biologic-naive patients and were informed by 5 or 6 RCTs, depending on the outcome.
Base-Case Analyses
The trials included in the base-case analyses were double-blind, phase III, placebo-controlled RCTs. The sponsor’s ITC report states the inclusion and exclusion criteria for the 2 trials were similar, except for the inclusion of biologic-experienced patients in the SUNNY trials. In the base-case analysis, only data for the subgroup of patients with no prior exposure to biologics were included in the analysis. In the PIONEER studies, patients received placebo or adalimumab 40 mg SC every week to the end of the induction phase. From the SUNNY trials, both the secukinumab every 2 weeks and every 4 weeks treatment groups were included in the model, but only data from the every 2 weeks group have been summarized in this report. The PIONEER trials included 308 and 326 patients, and the SUNNY trials included 541 and 543 patients in total for the 3 treatment groups.
The proportion of males ranged from 43% to 46% versus 31% to 41%, and the proportion of smokers was 50% to 58% versus 53% to 67% in the SUNNY and PIONEER trials, respectively. The proportion of patients with Hurley stage 2 or stage 3 was 51% to 67% and 28% to 46% in the SUNNY trials, compared with 52% to 55% and 45% to 48% in the PIONEER studies. The duration of disease was 6.6 years to 8.2 years in the SUNNY trials and was not reported in the PIONEER studies.
The ITC was based on outcomes assessed at the end of the induction phase, which was 12 weeks in duration in the PIONEER studies and 16 weeks in the SUNNY trials. The same definition of HiSCR50 was used in both studies. The PIONEER studies did not assess HiSCR25 and HiSCR75 thresholds; thus, these data were imputed. There were differences for the skin pain NRS30 end point. The PIONEER trial required at least a 30% reduction and at least a 1-unit decrease in the pain score. In the SUNNY trials, patients were required to have at least a 2-unit decrease in skin pain score from its worst. In both the PIONEER and SUNNY trials, the end point was measured in patients with a baseline score of 3 or higher. No information was provided on how DLQI response was defined, except that patients should achieve a score of 0 or 1 at the end of treatment. Some variation across studies was noted in the placebo response rate for the binary outcomes (Table 33). The change from baseline in the placebo groups was not reported for AN count or DLQI score.
The missing data imputation methods differed between the studies, with binary outcomes from the SUNNY trials based on multiple imputation (HiSCR, skin pain NRS30, flares) or nonresponder imputation (DLQI response), whereas, in the PIONEER studies, the data were based on nonresponder imputation (HiSCR, skin pain NRS30, flares) or were calculated from the percentage of responders and estimated sample size (DLQI response). Continuous end points were based on observed data for the change in DLQI score, or multiple imputation for AN in the SUNNY trials, and on last observation carried forward in the PIONEER studies.
The authors of the ITC rated the PIONEER trials as having an unclear risk of bias related to allocation concealment and a low risk of bias for other domains. No risk of bias assessment was reported for the SUNNY trials.
Table 33: Placebo Rate for Studies Included in the ITC — Base-Case Analysis
Trial | HiSCR50 (%) | Flares (%) | Skin pain NRS30 (%) | DLQI response (%) |
|---|---|---|---|---|
PIONEER I | 26.0 | ████ | 24.8 | ████ |
PIONEER II | 27.6 | ████ | 20.7 | ████ |
SUNSHINE | ████ | ████ | ████ | ████ |
SUNRISE | ████ | ████ | ████ | ████ |
DLQI = Dermatology Life Quality Index; HiSCR50 = Hidradenitis Suppurativa Clinical Response; ITC = indirect treatment comparison; NRS30 = at least a 30% reduction and either a minimum 2-unit reduction (SUNNY trials) or 1-unit reduction (PIONEER trials) from baseline in skin pain at its worst as measured by a numerical rating scale.
Source: Sponsor-submitted ITC report.72
Sensitivity Analyses
A sensitivity analysis was conducted that included both biologic-naive and biologic-experienced patients with adalimumab as the comparator. Three outcomes were analyzed (HiSCR50, NRS30, and flares) and the evidence network included 5 or 6 RCTs, depending on the outcome.
No specific assessment of clinical and methodological heterogeneity was conducted by the sponsor for the 6 trials. The PIONEER and SUNNY trials were included in all sensitivity analyses. The 2 additional trials had a sample size of 88 (study HS0001) and 206 patients (SHARPS study) who were treated with adalimumab, bimekizumab, or placebo for 12 weeks. Study HS0001 restricted inclusion to patients who were biologic-naive, whereas the SHARPS trial did not specifically exclude biologic-experienced patients. Of note, the objective of the SHARPS study was to evaluate adalimumab in combination with surgery, and treatment effects may be confounded by this co-intervention. According to the sponsor’s ITC report, the populations of the trials had similar exclusion criteria related to fistulae count, other skin conditions or comorbidities, prohibited medications, history of hypersensitivity to the study drug constituents, and pregnancy.
Skin pain in the SUNNY and PIONEER trials assessed NRS30 for patients with a baseline score of 3 or higher, while study HS0001 did not restrict assessment to patients with certain baseline values. In addition, the definition NRS30 in the SUNRISE and SUNSHINE trials was defined as a 30% reduction and at least a 2-unit reduction from baseline, while it was defined as a 30% reduction and at least a 1-unit reduction from baseline in other trials. According to the sponsor’s ITC report, other end points were consistently measured across the studies. The PIONEER, HS0001, and SHARPS trials were rated in the sponsor’s ITC report as having a low risk of bias (PIONEER and HS0001 were at unclear risk of bias for allocation concealment). The SUNNY trials were not appraised in the sponsor’s report; however, the CDA-AMC review of these trials did not identify any serious risk of bias concerns.
Results
For the base-case analysis, a total of 4 studies (PIONEER I, PIONEER II, SUNSHINE, SUNRISE) and 4 treatments (2 secukinumab regimens, adalimumab, and placebo) were included in the network of biologic-naive patients treated for HS. The network used for each end point (HiSCR50, flares, skin pain NRS30, AN count, DLQI response, DLQI total score) is depicted in Figure 4.
Figure 4: ITC Network — Base-Case Analysis
ITC = indirect treatment comparison; Q1W = every week; Q2W = every 2 weeks; Q4W = every 4 weeks.
Source: Sponsor-submitted ITC report.72
For the base-case analyses, random-effects models were preferred for all end points except for DLQI response, DLQI change from baseline, and the multinomial model, where fixed-effects models were selected. In most cases, the DIC values were similar between the 2 models, and random effects were chosen due to the presence of some statistical heterogeneity (HiSCR50, NRS30) or based on the convergence assessment and the shape of the network. Fixed-effects models were selected for the DLQI end points based on the convergence diagnostics and the lack of statistical heterogeneity detected, even though for DLQI response, the DIC value was lower for the random-effects model ██████ compared with the fixed-effects model ██████. For the multinomial model, the random-effects model did not converge; thus, the results were based on the fixed-effects model.
The results of the base-case NMAs are summarized in Table 34 and Table 35. For HiSCR50 response, skin pain NRS30 response, and proportion of patients with flares at 12 to 16 weeks, the results showed 95% CrIs that included the null for secukinumab every 2 weeks versus placebo and compared with adalimumab 40 mg every week.
The binomial model for HiSCR50 response reported an OR of ████ ████ ████ ████ ██ ████ for secukinumab versus placebo, and an OR of ████ ████ ████ ████ ██ █████ for secukinumab versus adalimumab. The multinomial model showed relative risks and 95% CrIs that favoured secukinumab versus placebo for the HiSCR25, HiSCR50, and HiSCR70 response thresholds, but for the comparison with adalimumab, the 95% CrIs included the null.
The NMA estimates for DLQI response favoured secukinumab versus placebo, but not versus adalimumab. The NMA for the percent change in AN count and change from baseline in DLQI score also favoured secukinumab versus placebo but not versus adalimumab.
Table 34: ITC Results of the Base-Case Analyses — Biologic-Naive Population
Outcome | N studies (patients), | Adalimumab q.1.w. vs. placebo | Secukinumab q.2.w. vs. placebo | Secukinumab q.2.w. vs. adalimumab q.1.w. |
|---|---|---|---|---|
OR (95% CrI) | ||||
HiSCR50b | | ██████ ██ | | ██████ ██ | | ████████ ██ | | ████████ ██ |
Skin pain NRS30b | | ██████ ██ | | ██████ ██ | | ████████ ██ | | ████████ ██ |
Flaresb | | ██████ ██ | | ██████ ██ | | ████████ ██ | | ████████ ██ |
DLQI responsec | | ██████ ██ | | ██████ ██ | | ████████ ██ | | ████████ ██ |
Mean difference in % change from baseline (95% CrI) | ||||
AN countd | | ██████ ██ | | ██████ ██ | | ████████ ██ | | ████████ ██ |
Mean difference in change from baseline (95% CrI) | ||||
DLQI total scoree | | ██████ ██ | | ██████ ██ | | ████████ ██ | | ████████ ██ |
AN = abscesses and inflammatory nodules; CrI = credible interval; FE = fixed effects; DLQI = Dermatology Life Quality Index; HiSCR = Hidradenitis Suppurativa Clinical Response; HRQoL = health-related quality of life; ITC = indirect treatment comparison; LOCF = last observation carried forward; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; OR = odds ratio; q.1.w. = every week; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
Note: Values in bold indicate the 95% CrI excludes the null value. An OR greater than 1 and a negative mean difference favour the treatment vs. the comparator.
aPatient totals include those in the secukinumab q.4.w. groups of the SUNNY trials.
bResults were obtained using multiple imputation for the SUNNY trials and nonresponder imputation for the PIONEER trials.
cResults were obtained using nonresponder imputation for the SUNNY trials. For the PIONEER trials, the sample size was derived using 90% of the arms’ sample size, as well as the number of events based on the reported percentages of responders in publications.
dResults were obtained using multiple imputation for the SUNNY trials and LOCF for the PIONEER trials. Mean difference in percentage change from baseline was adjusted on treatment group, weight, and baseline AN count for the SUNNY trials. A negative value indicates symptomatic improvement in treatment vs. control group.
eResults were obtained using the observed data for the SUNNY trials and the LOCF for the PIONEER trials. DLQI is scored from 0 to 30, with a higher score indicating greater impairment in HRQoL.
Source: Sponsor-submitted ITC report.72
Table 35: ITC Results for HiSCR25, HiSCR50, and HiSCR75 at the End of the Induction Phase in the Biologic-Naive Population — Multinomial Model
Outcome | N studies (patients), | Adalimumab q.1.w. vs. placebo | Secukinumab q.2.w. vs. placebo | Secukinumab q.2.w. vs. adalimumab q.1.w. |
|---|---|---|---|---|
RR (95% CrI) | ||||
HiSCR25 | ████████ ██ | ████████ ██ | ████████ ██ | ████████ ██ |
HiSCR50 | ████████ ██ | ████████ ██ | ████████ ██ | |
HiSCR75 | ████████ ██ | ████████ ██ | ████████ ██ | |
CrI = credible interval; FE = fixed effect; HiSCR = Hidradenitis Suppurativa Clinical Response; ITC = indirect treatment comparison; NR = not reported; RR = relative risk; q.1.w. = every week; q.2.w. = every 2 weeks.
Note: Values in bold indicate the 95% CrI excludes the null value. RR values greater than 1 favour the treatment vs. the comparator.
Source: Sponsor-submitted ITC report.72
The evidence network for the sensitivity analyses that included biologic-naive and -experienced patients is shown in Figure 5. Relative to the base-case analysis, this network included 1 additional comparator (bimekizumab). For the outcomes of HiSCR50 and flares, 2 other studies were included (HS0001, SHARPS) and, for NRS30, 1 study was added (HS0001). The sensitivity analysis results for DLQI response, change in DLQI, AN count, and the multinomial model were not reported in the sponsor’s ITC report.
Table 36 summarizes the results of the sensitivity analyses. The OR of HiSCR50, skin pain NRS30 response, and flares showed 95% CrIs that included the null for secukinumab versus placebo and versus adalimumab.
Figure 5: ITC Network — Sensitivity Analysis
ITC = indirect treatment comparison; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; Q1W = every week; Q2W = every 2 weeks; Q4W = every 4 weeks.
Note: The network for the skin pain NRS30 did not include the SHARPS trial.
Source: Sponsor-submitted ITC report.72
Table 36: ITC Results of the Sensitivity Analyses — Biologic-Naive and Biologic-Experienced Population
Outcome | N studies (patients), | Adalimumab q.1.w. vs. placebo | Secukinumab q.2.w. vs. placebo | Secukinumab q.2.w. vs. adalimumab q.1.w. |
|---|---|---|---|---|
OR (95% CrI) | ||||
HiSCR50b | | ████████ ██ | | ██████ ██ | | ██████ ██ | | ████████ ██ |
Skin pain NRS30b | | ████████ ██ | | ██████ ██ | | ██████ ██ | | ████████ ██ |
Flaresb | | ████████ ██ | | ██████ ██ | | ██████ ██ | | ████████ ██ |
CrI = credible interval; HiSCR = Hidradenitis Suppurativa Clinical Response; ITC = indirect treatment comparison; NR = not reported; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; OR = odds ratio; q.1.w. = every week; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
aPatient totals include those in the secukinumab q.4.w. groups of the SUNNY trials.
bResults were obtained using multiple imputation for the SUNNY trials and nonresponder imputation for the PIONEER trials.
Note: Values in bold indicate the 95% CrI excludes the null value. OR values greater than 1 favour the treatment vs. the comparator.
Source: Sponsor-submitted ITC report.72
Critical Appraisal of ITC
The systematic review was conducted using standard methods, based on inclusion and exclusion criteria specified a priori, and with screening and data extraction completed independently in duplicate. The additional criteria applied to select trials for the ITC were clearly specified. The risk of bias was not assessed for all studies included in the systematic review; specifically, the SUNSHINE and SUNRISE studies were not evaluated. The other trials included in the ITC were rated as having a low risk of bias by the authors of the ITC. Our appraisal of the SUNNY trials did not identify any serious risk of bias concerns.
The ITC was conducted using Bayesian NMA methods that, according to the NMA’s authors, were consistent with NICE methodologic guidance. No major issues were identified by CDA-AMC with the statistical methods used. Both fixed- and random-effects models were analyzed, and the stated rationale for selecting the preferred model for each outcome appeared to be appropriate.
With regard to clinical heterogeneity across the included studies, the NMA’s authors identified several differences in the design of the trials and the clinical characteristics of the patients enrolled. Some trials included a mixed population of biologic-naive and biologic-experienced patients; to address this, the base-case analysis was limited to patients who were biologic-naive. This required using subgroup data from the SUNNY trials; however, randomization was not stratified by treatment history in these studies. Thus, confounders may not be fully balanced between the active and control groups in the biologic-naive subgroup.
The NMA’s authors stated there were differences across studies in the proportion of males, smoking status, disease duration, and Hurley stage. The clinical experts consulted for this review identified smoking status, obesity, the number of baseline lesions or disease severity, body site affected (e.g., perineum), inadequate response to prior biologics and hyperandrogenism in women, as potential treatment-effect modifiers. There was limited information provided in the ITC report on some of these characteristics; thus, it was difficult for CDA-AMC to evaluate whether there were imbalances between trials. Moreover, there were differences in the follow-up duration, the definition of NRS30 skin pain response, and the imputation of missing data within the trials. The impact of these differences on the findings is unclear; however, they challenge the plausibility of the underlying transitivity assumption. Inconsistency was assessed using appropriate methods; however, there were no closed loops relevant to the comparison of interest (i.e., all evidence was indirect).
Overall, the networks were sparse, with the base-case model including 4 RCTs, and the sensitivity analyses including 5 or 6 studies. All comparisons between secukinumab and adalimumab were affected by wide CrIs, introducing uncertainty about which treatment was favoured. The analyses were limited to short-term outcomes at the end of the induction period (12 to 16 weeks); thus, longer-term comparative efficacy is unknown. No safety end points were evaluated in the ITC.
With respect to external validity, the dose of adalimumab in the ITC was 40 mg per week, but the clinical experts consulted stated that the dose may be increased to 80 mg weekly. Thus, the comparator treatment may not fully reflect clinical practice.
Discussion
Summary of Available Evidence
Two phase III, double-blind, RCTs (SUNSHINE, N = 541; SUNRISE, N = 543) assessed whether secukinumab 300 mg every 2 weeks and secukinumab 300 mg every 4 weeks increased the proportion of patients with HiSCR50 response, defined as at least a 50% reduction in AN count with no increase in the number of abscesses and/or the number of draining fistulas from baseline compared with placebo after 16 weeks of treatment in patients aged 18 years and older with moderate to severe HS (defined as a total of at least 5 inflammatory lesions affecting at least 2 distinct anatomic areas). Other outcomes of interest include change in disease severity measured by AN count, disease worsening measured by patients experiencing flares, HS symptoms measured by NRS30 skin pain response, and HRQoL measured by DLQI and EQ-5D health state assessment. Notable harms include infections and infestations; candida infections; malignant or unspecified tumours; neoplasms benign, malignant, or unspecified (including cysts and polyps); squamous cell carcinoma of an HS-affected area; and inflammatory bowel disease. The mean age of patients randomized to each study drug group was similar — approximately 36 years — and ranged from 18 to 73 years across trials. Most patients presented with Hurley stage 2 HS at baseline, ranging from 56.7% of all patients (308 of 543) randomized in the SUNRISE trial to 61.4% of all patients (332 of 541) randomized in the SUNSHINE trial. Patients presenting with Hurley stage 3 HS at baseline ranged from 34.0% of all patients (184 patients) randomized in the SUNSHINE trial to 40.5% of all patients (220 patients) randomized in the SUNRISE trial. Across trials, approximately 80% of randomized patients had prior experience with systemic antibiotics and approximately 20% of randomized patients had prior experience with adalimumab, with most patients discontinuing therapy because of lack of efficacy.
In the absence of direct comparative evidence of secukinumab versus other relevant comparators, the sponsor submitted 1 NMA that assessed the short-term efficacy (12 to 16 weeks) of secukinumab versus adalimumab for the treatment of adults with moderate to severe HS. The base-case Bayesian NMA was informed by 4 RCTs and limited to patients who were biologic-naive (N = 1,462).
The extension study, NCT04179175, assessing the effects of noninterrupted versus interrupted and long-term treatment of 2 dose regimens of secukinumab in patients with HS was ongoing, and no results were available at the time of this report.
Interpretation of Results
Efficacy
Patient groups identified the following unmet needs in the treatment of patients living with HS: a safe and effective therapy that can control HS symptoms (e.g., reduction in lesions and pain) and a therapy that can induce disease remission. The SUNNY trials demonstrated that 16 weeks of treatment with secukinumab 300 mg every 2 weeks (biweekly maintenance dosing) likely results in a clinically meaningful improvement in HS, measured by HiSCR50 response, AN lesion count, and NRS30 skin pain response, and may result in a clinically meaningful decrease in the proportion of patients experiencing flares when compared with placebo. Uncertainty in the evidence is primarily because, in all cases, the CIs included the potential that the difference compared with placebo is small and unimportant. Findings for secukinumab 300 mg every 4 weeks (monthly maintenance dosing) were similar, but the certainty of evidence was lower for HiSCR50 response due to some inconsistency across trials in the magnitude of effect. Of note, the analysis of the primary end point, HiSCR50 response at week 16 for the secukinumab monthly maintenance dosing group did not meet statistical significance in the statistical hierarchy in the SUNSHINE trial. As such, all subsequent tests of the secondary end points for the monthly maintenance dosing group in the SUNSHINE trial are considered as supportive evidence only. Additionally, the analysis of the secondary end point, flares at week 16, for the secukinumab biweekly maintenance dosing group did not meet statistical significance in the statistical hierarchy in the SUNRISE trial.
The subgroup analysis results for HiSCR50 response at week 16 in the SUNNY trials appear to be directionally aligned with the primary analysis for the biweekly and monthly maintenance dosing of secukinumab versus placebo, regardless of concomitant antibiotic use, body weight cut-off, previous use of systemic biologics, Hurley stage (except Hurley stage 1), and baseline AN count. However, these subgroup analysis results are considered supportive analyses, and the small sample size of some group limits the ability to interpret the results.
Overall, the exploratory analysis results on the HRQoL outcomes in the SUNNY trials were variable, depending on how the outcomes were analyzed. More specifically, the SUNNY trials demonstrated that 16 weeks of treatment with secukinumab likely results in a clinically meaningful, favourable treatment effect in the HRQoL of patients with HS, as measured by DLQI response (i.e., ≥ 5-point reduction) when compared with placebo. Uncertainty in the evidence for the biweekly maintenance dosing group is also primarily because of concerns regarding imprecision (potential for little-to-no difference). When analyzed as a continuous variable, the trials showed that secukinumab results in little-to-no clinically meaningful difference in the DLQI total score when compared with placebo. When HRQoL was measured using the EQ VAS at 16 weeks’ follow-up, the trials showed that the biweekly maintenance dosing may result in a clinically meaningful improvement when compared with placebo, while the monthly maintenance dosing likely results in little-to-no clinically meaningful difference when compared with placebo. Of note, the DLQI and EQ VAS are not disease-specific outcome measures and, as such, may not capture all relevant impacts of HS on HRQoL.
In consultation with the clinical experts, it was concluded that long-term assessment is important in the context of HS being a life-long disease. At 52 weeks of follow-up (exploratory end point), the results of the SUNNY trials are suggestive of a potential for a favourable treatment effect with secukinumab. The findings at this time point are at risk of bias (albeit the direction and magnitude are unknown) because of the relatively high amount of missing data in both trials and, in the absence of a comparator group, it is not possible to draw causal conclusions about the efficacy of secukinumab versus any comparator, including placebo. Additionally, the effect of secukinumab versus any comparator on disease remission and recurrence (important outcomes according to the clinical expert input) in patients with HS is unknown, as they were not assessed in the SUNNY trials.
In consultation with the clinical experts, it was concluded that the ideal comparator is adalimumab (or any other biologic) to assess the efficacy of secukinumab in patients with moderate to severe HS. In the absence of direct evidence versus other biologics, the sponsor provided a, NMA that assessed short-term efficacy (12 to 16 weeks) versus adalimumab. For secukinumab 300 mg every 2 weeks versus adalimumab 40 mg weekly, the results of the NMA were inconclusive, showing 95% CrIs that were wide and included the null for all outcomes tested (HiSCR50, AN count, skin pain, flares, and HRQoL). The sensitivity analyses that included biologic-naive and biologic-experienced patients showed similar findings. Overall, the results for the secukinumab every 4 weeks dosage group were similar to the secukinumab every 2 weeks dosage group. Based on these data, it is unclear whether secukinumab is superior, inferior, or has similar efficacy to adalimumab. Although CDA-AMC did not identify any major issues with the methods used to conduct the ITC, there was some heterogeneity in the patient and study characteristics across the trials used to inform the analysis, which may have challenged the underlying assumption of transitivity. Overall, the network was sparse, with only 4 RCTs included in the base-case analyses. The clinical experts consulted noted that the dose of adalimumab is often increased to 80 mg once weekly; thus, the dose used in the NMA may not reflect clinical practice, which may affect the generalizability of the results.
The clinician groups and the clinical experts consulted by CDA-AMC identified the following challenges in practice in the treatment of patients with HS: not all patients’ HS respond to current management options and patients’ HS can become refractory to systemic therapies, including adalimumab. Of note, adalimumab is currently the only approved biologic option for HS in Canada, while alternative biologics used off-label are offered to patients, depending on coverage and access. Based on the available evidence and input from the clinician groups and the clinical experts, secukinumab likely addresses these concerns, as it provides an alternative biologic treatment option for patients with moderate to severe HS whose HS has not adequately responded to the current standard of care (i.e., systemic antibiotics). Additionally, the clinical experts anticipated secukinumab being offered to patients who have experience with adalimumab (i.e., patients whose HS has not adequately responded to adalimumab, patients who have experienced AEs with adalimumab, and patients who have contraindications to adalimumab), and secukinumab may also be offered to patients as their first biologic therapy.
Harms
Overall, no notable differences in the frequency of AEs between the study drug groups were identified in either of the SUNNY trials during the 16-week placebo-controlled treatment phase and the 52-week noncomparative treatment phase.
An increased risk of infection and new onset or exacerbated inflammatory bowel disease are described in the product monograph for secukinumab.1 Overall, the evidence from the SUNNY trials suggests that 16 weeks of treatment with secukinumab may result in little-to-no difference in the occurrence of infections and infestations or candida infections compared with placebo; however, there was important uncertainty because of a low number of events and/or wide CIs. The effect of secukinumab on the occurrence of malignant or benign neoplasms (including malignant or unspecified tumours; benign, malignant, or unspecified neoplasms; or squamous cell carcinoma of an HS-affected area) or inflammatory bowel disease was very uncertain because the follow-up period was considered too short to assess these harms and, as a result, few or no events were observed. Harms of secukinumab were not assessed in the sponsor-submitted NMA.
In consultation with the clinical experts, no major concern with the harm profile of secukinumab was identified based on the data observed at the 16-week and 52-week follow-up time points across trials that would be expected to impact therapeutic decision-making. In the absence of a comparator group for the 52-week follow-up time point, it is not possible to draw definitive conclusions about the harms of secukinumab versus any comparator, including placebo.
Conclusion
Collectively, the evidence from the SUNSHINE (N = 541) and SUNRISE (N = 543) studies (referred to as the SUNNY trials) demonstrated that 16 weeks of treatment with secukinumab 300 mg every 2 weeks (biweekly maintenance dosing) likely results in a clinically meaningful improvement in HS (measured by HiSCR50 response, AN lesion count, and NRS30 skin pain response), and may result in a clinically meaningful decrease in the proportion of patients experiencing flares, when compared with placebo, in patients aged 18 years and older with moderate to severe HS. Uncertainty in the evidence is primarily because, in all cases, the CIs included the potential that the difference compared with placebo is small and unimportant. The findings for secukinumab 300 mg every 4 weeks (monthly maintenance dosing) were similar, but the certainty of the evidence was lower for HiSCR50 response because there was some inconsistency across trials in the magnitude of the effect, and statistical significance was not reached in the SUNSHINE trial. Impacts on exploratory HRQoL end points for both dosing regimens were difficult to interpret, as the findings differed based on how the change in DLQI and EQ-5D scores were analyzed. Overall, no serious risk of bias concern and no serious concern about the generalizability of results to the population of interest was identified in the appraisal of the placebo-controlled phase of the trials. The results from the sponsor-conducted NMA were inconclusive on the assessment of short-term efficacy (12 to 16 weeks) of secukinumab (both dose regimens) versus adalimumab, showing 95% CrIs that were wide and included the null for all outcomes tested (HiSCR50, AN count, skin pain, flares, and HRQoL). Overall, no new concerns with the harms of secukinumab were identified based on the harms data observed with follow-up at 16 and 52 weeks across the SUNNY trials. In the absence of a comparator group for the 52-week follow-up time point, it is not possible to draw definitive conclusions about the harms (and efficacy) of secukinumab versus any comparator, including placebo.
References
1.Cosentyx (secukinumab): 75 mg/0.5 mL, 150 mg/1 mL, 300 mg/2 mL solution for injection, 150 mg powder for solution for injection [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2024.
2.Sabat R, Jemec GBE, Matusiak L, Kimball AB, Prens E, Wolk K. Hidradenitis suppurativa. Nat Rev Dis Primers. 2020;6(1):18. PubMed
3.Zouboulis CC, Desai N, Emtestam L, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29(4):619-644. PubMed
4.Kelly G, Hughes R, McGarry T, et al. Dysregulated cytokine expression in lesional and nonlesional skin in hidradenitis suppurativa. Br J Dermatol. 2015;173(6):1431-1439. PubMed
5.Ingram JR. The genetics of hidradenitis suppurativa. Dermatol Clin. 2016;34(1):23-28. PubMed
6.Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. PubMed
7.Pink AE, Simpson MA, Desai N, Trembath RC, Barker JNW. gamma-Secretase mutations in hidradenitis suppurativa: new insights into disease pathogenesis. J Invest Dermatol. 2013;133(3):601-607. PubMed
8.Margesson LJ, Danby FW. Hidradenitis suppurativa. Best Pract Res Clin Obstet Gynaecol. 2014;28(7):1013-1027. PubMed
9.Guet-Revillet H, Jais JP, Ungeheuer MN, et al. The microbiological landscape of anaerobic infections in hidradenitis suppurativa: a prospective metagenomic study. Clin Infect Dis. 2017;65(2):282-291. PubMed
10.Nikolakis G, Join-Lambert O, Karagiannidis I, Guet-Revillet H, Zouboulis CC, Nassif A. Bacteriology of hidradenitis suppurativa/acne inversa: a review. J Am Acad Dermatol. 2015;73(5 Suppl 1):S12-18. PubMed
11.Nguyen TV, Damiani G, Orenstein LAV, Hamzavi I, Jemec GB. Hidradenitis suppurativa: an update on epidemiology, phenotypes, diagnosis, pathogenesis, comorbidities and quality of life. J Eur Acad Dermatol Venereol. 2021;35(1):50-61. PubMed
12.Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60(4):539-561; quiz 562-533. PubMed
13.Garg A, Kirby JS, Lavian J, Lin G, Strunk A. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153(8):760-764. PubMed
14.Ingram JR, Jenkins-Jones S, Knipe DW, Morgan CLI, Cannings-John R, Piguet V. Population-based Clinical Practice Research Datalink study using algorithm modelling to identify the true burden of hidradenitis suppurativa. Br J Dermatol. 2018;178(4):917-924. PubMed
15.Jemec GB, Heidenheim M, Nielsen NH. The prevalence of hidradenitis suppurativa and its potential precursor lesions. J Am Acad Dermatol. 1996;35(2 Pt 1):191-194. PubMed
16.Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: results from two case-control studies. J Am Acad Dermatol. 2008;59(4):596-601. PubMed
17.Lachaine J, Miron A, Shear N, Alhusayen R. The prevalence and incidence of hidradenitis suppurativa in Canada: results from a population-based survey. Value Health. 2016;19(3):A123.
18.HS patient journey study: patient phase report. Prepared for Novartis by MD Analytics [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Secukinumab (Cosentyx), 300mg, 150mg solution for subcutaneous injection. 2022.
19.Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81(1):91-101. PubMed
20.Kimball AB, Jemec GBE, Yang M, et al. Assessing the validity, responsiveness and meaningfulness of the Hidradenitis Suppurativa Clinical Response (HiSCR) as the clinical endpoint for hidradenitis suppurativa treatment. Br J Dermatol. 2014;171(6):1434-1442. PubMed
21.Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30(6):989-994. PubMed
22.Kimball AB, Tzellos T, Calimlim BM, Teixeira HD, Geng Z, Okun MM. Achieving hidradenitis suppurativa response score is associated with significant improvement in clinical and patient-reported outcomes: post hoc analysis of pooled data from PIONEER I and II. Acta Derm Venereol. 2018;98(10):932-937. PubMed
23.Statistical Analysis Plan. A randomized, double-blind, multicenter study assessing short (16 weeks) and long-term efficacy (up to 1 year), safety, and tolerability of 2 subcutaneous secukinumab dose regimens in adult patients with moderate to severe hidradenitis suppurativa (SUNSHINE) [internal sponsor's report]. In Drug Reimbursement Review sponsor submission: Secukinumab (Cosentyx), 300mg, 150mg solution for subcutaneous injection. Montreal (QC): Novartis Pharmaceuticals Canada Inc 2021 Oct 21.
24.Gergely LH, Gáspár K, Brodszky V, et al. Validity of EQ-5D-5L, Skindex-16, DLQI and DLQI-R in patients with hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2020;34(11):2584-2592. PubMed
25.Matusiak L, Bieniek A, Szepietowski JC. Psychophysical aspects of hidradenitis suppurativa. Acta Derm Venereol. 2010;90(3):264-268. PubMed
26.Farrar JT, Berlin JA, Strom BL. Clinically important changes in acute pain outcome measures: a validation study. J Pain Symptom Manage. 2003;25(5):406-411. PubMed
27.Farrar JT, Young JP, Jr., LaMoreaux L, Werth JL, Poole MR. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain. 2001;94(2):149-158. PubMed
28.Jensen MP, Chen C, Brugger AM. Interpretation of visual analog scale ratings and change scores: a reanalysis of two clinical trials of postoperative pain. J Pain. 2003;4(7):407-414. PubMed
29.Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: A publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81(1):76-90. PubMed
30.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
31.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
32.Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines 6. Rating the quality of evidence--imprecision. J Clin Epidemiol. 2011;64(12):1283-1293. PubMed
33.Clinical Study Report (Primary efficacy analysis): CAIN457M2301. A randomized, double-blind, multicenter study assessing short (16 weeks) and long-term efficacy (up to 1 year), safety, and tolerability of 2 subcutaneous secukinumab dose regimens in adult patients with moderate to severe hidradenitis suppurativa (SUNSHINE) [internal sponsor's report]. Basel (CH): Novartis; 2023 Apr 5.
34.Clinical Study Report (Primary efficacy analysis): CAIN457M2302. A randomized, double-blind, multicenter study assessing short (16 weeks) and long-term efficacy (up to 1 year), safety, and tolerability of 2 subcutaneous secukinumab dose regimens in adult patients with moderate to severe hidradenitis suppurativa (SUNRISE) [internal sponsor's report]. Basel (CH): Novartis; 2023 Apr 4.
35.Novartis response to June 19, 2023 CADTH request for additional information regarding secukinumab (Cosentyx) CADTH review [internal sponsor's report]. Montreal (QC): Novartis Pharmaceuticals Canada Inc; 2023 Jun 23.
36.Novartis response to July 5, 2023 CADTH request for additional information regarding secukinumab (Cosentyx) CADTH review [internal sponsor's report]. Montreal (QC): Novartis Pharmaceuticals Canada Inc; 2023 Jul 14.
37.Novartis response to May 22, 2024 CADTH request for additional information regarding secukinumab (Cosentyx) CADTH review [internal sponsor's report]. Montreal (QC): Novartis Pharmaceuticals Canada Inc; 2024 May 28.
38.Novartis response to June 5, 2023 CADTH request for additional information regarding secukinumab (Cosentyx) CADTH review [internal sponsor's report]. Montreal (QC): Novartis Pharmaceuticals Canada Inc; 2023 Jun 8.
39.Esmann S, Jemec GB. Psychosocial impact of hidradenitis suppurativa: a qualitative study. Acta Derm Venereol. 2011;91(3):328-332. PubMed
40.Kimball AB, Crowley JJ, Papp K, et al. Baseline patient-reported outcomes from UNITE: an observational, international, multicentre registry to evaluate hidradenitis suppurativa in clinical practice. J Eur Acad Dermatol Venereol. 2020;34(6):1302-1308. PubMed
41.Peris K, Lo Schiavo A, Fabbrocini G, et al. HIDRAdisk: validation of an innovative visual tool to assess the burden of hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2019;33(4):766-773. PubMed
42.Sandhu VK, Shah M, Piguet V, Alavi A. The impact of hidradenitis suppurativa on work productivity and activity impairment. Br J Dermatol. 2020;182(5):1288-1290. PubMed
43.Gáspár K, Hunor Gergely L, Jenei B, et al. Resource utilization, work productivity and costs in patients with hidradenitis suppurativa: a cost-of-illness study. Expert Rev Pharmacoecon Outcomes Res. 2022;22(3):399-408. PubMed
44.Alavi A, Farzanfar D, Lee RK, Almutairi D. The contribution of malodour in quality of life of patients with hidradenitis suppurativa. J Cutan Med Surg. 2018;22(2):166-174. PubMed
45.Alavi A, Farzanfar D, Rogalska T, Lowes MA, Chavoshi S. Quality of life and sexual health in patients with hidradenitis suppurativa. Int J Womens Dermatol. 2018;4(2):74-79. PubMed
46.Cartron A, Driscoll MS. Comorbidities of hidradenitis suppurativa: a review of the literature. Int J Womens Dermatol. 2019;5(5):330-334. PubMed
47.Tzellos T, Yang H, Mu F, Calimlim B, Signorovitch J. Impact of hidradenitis suppurativa on work loss, indirect costs and income. Br J Dermatol. 2019;181(1):147-154. PubMed
48.Garg A, Neuren E, Cha D, et al. Evaluating patients' unmet needs in hidradenitis suppurativa: results from the Global Survey Of Impact and Healthcare Needs (VOICE) Project. J Am Acad Dermatol. 2020;82(2):366-376. PubMed
49.Kouris A, Platsidaki E, Christodoulou C, et al. Quality of life and psychosocial implications in patients with hidradenitis suppurativa. Dermatology. 2016;232(6):687-691. PubMed
50.Hana A, Booken D, Henrich C, et al. Functional significance of non-neuronal acetylcholine in skin epithelia. Life Sci. 2007;80(24-25):2214-2220. PubMed
51.Acharya P, Mathur M. Hidradenitis suppurativa and smoking: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82(4):1006-1011. PubMed
52.Scarred for life: a national report of the patient experience living with hidradenitis suppurativa. Ottawa (ON): Canadian Skin Patient Alliance; 2017.
53.Zouboulis CC, Bechara FG, Dickinson-Blok JL, et al. Hidradenitis suppurativa/acne inversa: a practical framework for treatment optimization - systematic review and recommendations from the HS ALLIANCE working group. J Eur Acad Dermatol Venereol. 2019;33(1):19-31. PubMed
54.Ballard K, Shuman VL. Hidradenitis suppurativa. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022: https://www.ncbi.nlm.nih.gov/books/NBK534867/. Accessed by sponsor, no date provided.
55.Hood C, Shanmugam VK. Hidradenitis suppurativa—diagnosis and management. J Nurse Pract. 2019;15(10):713-716.
56.Johnston LA, Alhusayen R, Bourcier M, et al. Practical guidelines for managing patients with hidradenitis suppurativa: an update. J Cutan Med Surg. 2022;26(2_suppl):2s-24s. PubMed
57.Wortsman X, Jemec GB. Real-time compound imaging ultrasound of hidradenitis suppurativa. Dermatol Surg. 2007;33(11):1340-1342. PubMed
58.Wortsman X, Jemec GBE. A 3D ultrasound study of sinus tract formation in hidradenitis suppurativa. Dermatol Online J. 2013;19 6:18564.
59.Zouboulis CC, Del Marmol V, Mrowietz U, Prens EP, Tzellos T, Jemec GB. Hidradenitis suppurativa/acne inversa: criteria for diagnosis, severity assessment, classification and disease evaluation. Dermatology. 2015;231(2):184-190. PubMed
60.Jemec GB. Clinical practice. Hidradenitis suppurativa. N Engl J Med. 2012;366(2):158-164. PubMed
61.Micheletti RG. Natural history, presentation, and diagnosis of hidradenitis suppurativa. Semin Cutan Med Surg. 2014;33(3 Suppl):S51-53. PubMed
62.Prens LM, Rondags A, Volkering RJ, et al. The refined Hurley classification: the inter-rater and intrarater reliability and face validity. Br J Dermatol. 2019;181(6):1335-1337. PubMed
63.Humira (adalimumab injection): 40 mg in 0.8 mL, sterile solution (50 mg/mL), 10 mg in 0.1 mL sterile solution (100 mg/mL), 20 mg in 0.2 mL sterile solution (100 mg/mL), 40 mg in 0.4 mL sterile solution (100 mg/mL), 80 mg in 0.8 mL sterile solution (100 mg/mL) for subcutanous injection [product monograph]. St-Laurent (QC): AbbviVie Corporation; 2022.
64.Kimball AB, Jemec GBE, Alavi A, et al. Secukinumab in moderate-to-severe hidradenitis suppurativa (SUNSHINE and SUNRISE): week 16 and week 52 results of two identical, multicentre, randomised, placebo-controlled, double-blind phase 3 trials. Lancet. 2023. PubMed
65.Clinical Study Report (Full report): CAIN457M2301. A randomized, double-blind, multicenter study assessing short (16 weeks) and long-term efficacy (up to 1 year), safety, and tolerability of 2 subcutaneous secukinumab dose regimens in adult patients with moderate to severe hidradenitis suppurativa (SUNSHINE) [internal sponsor's report]. Basel (CH): Novartis; 2022 Apr 5.
66.Clinical Study Report (Final report): CAIN457M2302. A randomized, double-blind, multicenter study assessing short (16 weeks) and long-term efficacy (up to 1 year), safety, and tolerability of 2 subcutaneous secukinumab dose regimens in adult patients with moderate to severe hidradenitis suppurativa (SUNRISE) [internal sponsor's report]. Basel (CH): Novartis; 2022 Apr 5.
67.Sponsor summary of clinical evidence for secukinumab (Cosentyx) for hidradenitis suppurativa [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Secukinumab (Cosentyx), 300mg, 150mg solution for subcutaneous injection. Montreal (QC): Novartis Pharmaceuticals Canada Inc; 2023 May 10.
68.Davidson L, van den Reek J, Bruno M, et al. Risk of candidiasis associated with interleukin-17 inhibitors: a real-world observational study of multiple independent sources. Lancet Reg Health Eur. 2022;13:100266. PubMed
69.Yang Y, Brazier J, Longworth L. EQ-5D in skin conditions: an assessment of validity and responsiveness. Eur J Health Econ. 2015;16(9):927-939. PubMed
70.Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375(5):422-434. PubMed
71.MedDRA. Standardised MedDRA Queries (SMQs). https://www.meddra.org/how-to-use/tools/smqs. Accessed 2023 Jun 21.
72.Indirect treatment comparison in hidradenitis suppurative: technical report version 2.0 [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Secukinumab (Cosentyx), 300mg, 150mg solution for subcutaneous injection. Montreal (QC): Novartis Pharmaceuticals Canada Inc; 2022.
73.Dias S, Welton J, Sutton A, Ades AE. NICE DSU technical support document 2: a generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials Sheffield (GB): University of Sheffield; 2016: https://www.sheffield.ac.uk/nice-dsu/tsds/full-list. Accessed by sponsor, no date provided.
74.Dias S, Welton J, Sutton A, Caldwell D, Lu G, Ades AE. NICE DSU technical support document 4: inconsistency in networks of evidence based on randomised controlled trials Sheffield (GB): University of Sheffield; 2014: https://www.sheffield.ac.uk/nice-dsu/tsds/full-list. Accessed by sponsor, no date provided.
75.Single technology appraisal: adalimumab for treating moderate to severe hidradenitis suppurativa. (Committee Papers ID812). London (GB): National Institute for Health and Care Excellence; 2016: https://www.nice.org.uk/guidance/ta392/documents/committee-papers. Accessed 2023 Jun 6.
76.Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol. 1997;50(6):683-691. PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 37: Summary of Prohibited Medications From the SUNSHINE and SUNRISE Trials
Prohibited medication | Before randomization | Use between randomization and last dose of study drug | Action take |
|---|---|---|---|
Prior treatment with secukinumab or other drugs blocking IL-17 A/F or IL-17R | Not permitted | Not permitted | Patient not eligible |
Systemic biological immunomodulating treatment (e.g., adalimumab, infliximab, ustekinumab, anakinra, natalizumab) | 12 weeks or 5 half-lives, whichever is longer, washout period required | Not permitted | Discontinue use or discontinue study treatment |
Systemic nonbiologic immunomodulating treatment (e.g., methotrexate, cyclosporine A, retinoids, apremilast) | 28 days washout period required | Not permitted | Discontinue use or discontinue study treatment |
Topical antibiotic therapies for the treatment of HS | 14 days washout period required | Not permitted until week 16 | Discontinue use or discontinue study treatment if not medically warranted |
Antibiotics for the treatment of HS (nonantibiotic strata) | 28 days washout period required | Not permitted until week 16, except for rescue treatment | Discontinue use or discontinue study treatment if not medically warranted |
Systemic corticosteroids for the treatment of HS | 28 days washout period required | Not permitted | Discontinue use or discontinue study treatment |
Opioid analgesics (including tramadol) for both HS-related and non–HS-related pain | 14 days washout period required | Not permitted | Discontinue use or discontinue study treatment |
Surgeries for the treatment of HS other than permitted as rescue therapy | Not permitted within 6 weeks before randomization | Not permitted until week 16 | Discontinue study treatment |
Live vaccines | Not permitted within 6 weeks before randomization | Not permitted | Discontinue study treatment |
Any investigational treatment or participation in any interventional trial | 28 days or 5 half-lives, whichever is longer, washout period required | Not permitted | Discontinue study treatment |
HS = hidradenitis suppurativa; IL = interleukin.
Source: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34
Figure 6: Statistical Testing Strategy From the SUNSHINE and SUNRISE Trials
q2w = once every 2 weeks; q4w = once every 4 weeks.
Notes: M2301 refers to the SUNSHINE trial and M2302 refers to the SUNRISE trial.
The hypotheses included in the testing strategy were the following:
H1 and H’1: secukinumab 300 mg every 2 weeks SC was not different to placebo regimen with respect to HiSCR after 16 weeks of treatment.
H2 and H’2: secukinumab 300 mg every 4 weeks SC was not different to placebo regimen with respect to HiSCR after 16 weeks of treatment.
H3 and H’3: secukinumab 300 mg every 2 weeks SC was not different to placebo regimen with respect to percentage change from baseline in AN count at week 16.
H4 and H’4: secukinumab 300 mg every 4 weeks SC was not different to placebo regimen with respect to percentage change from baseline in AN count at week 16.
H5 and H’5: secukinumab 300 mg every 2 weeks SC was not different to placebo regimen with respect to flare over 16 weeks of treatment.
H6 and H’6: secukinumab 300 mg every 4 weeks SC was not different to placebo regimen with respect to flare over 16 weeks of treatment.
H7 and H’7: secukinumab 300 mg every 2 weeks SC was not different to placebo regimen with respect to NRS30 at week 16.
H8 and H’8: secukinumab 300 mg every 4 weeks SC was not different to placebo regimen with respect to NRS30 at week 16.
Source: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34
Table 38: Summary of HiSCR Response at Various Thresholds From the SUNSHINE and SUNRISE Trials at Week 16 (Full Analysis Set)
Outcome | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Placebo (N = 183) | |
HiSCR response criterion, n/m (%) | ||||
HiSCR25 | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
95% CI | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
HiSCR50 | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
95% CI | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
HiSCR75 | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
95% CI | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
HiSCR90 | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
95% CI | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
HiSCR100 | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
95% CI | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
AN = abscesses and inflammatory nodules; CI = confidence interval; n = number of patients with observed response; m = number of patients evaluable; q.2.w. = every 2 weeks.
Note: HiSCR25, HiSCR50, HiSCR75, HiSCR90, HiSCR100 describes a decrease of at least 25%, 50%, 75%, 90%, 100% in AN count with no increase in the number of abscesses and/or in the number of draining fistulae compared with baseline.
Source: SUNSHINE Clinical Study Report33 and SUNRISE Clinical Study Report.34 Details included in the table are from the sponsor’s summary of clinical evidence.67
Table 39: Summary of HiSCR Response at Various Thresholds From the SUNSHINE and SUNRISE Trials at Week 52 (Full Analysis Set)
Outcome | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Placebo to secukinumab q.2.w. (N = 90) | Secukinumab q.2.w. (N = 180) | Placebo to secukinumab q.2.w. (N = 90) | |
HiSCR response criterion, n/m (%) | ||||
HiSCR25 | | ████████ ██ | | ████ ██ | | ████████ ██ | |█████ ██ |
95% CI | | ████████ ██ | | ████ ██ | | ████████ ██ | |█████ ██ |
HiSCR50 | | ████████ ██ | | ████ ██ | | ████████ ██ | █████ ██ |
95% CI | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
HiSCR75 | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
95% CI | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
HiSCR90 | | ████████ ██ | | ████ ██ | | ████████ ██ | | ████ ██ |
95% CI | | ████████ ██ | ████ ██ | | ████████ ██ | █████ ██ |
HiSCR100 | | ████████ ██ | ████ ██ | | ████████ ██ | █████ ██ |
95% CI | | ████████ ██ | ████ ██ | | ████████ ██ | █████ ██ |
n = number of patients with observed response; m = number of patients evaluable.
Note: HiSCR25, HiSCR50, HiSCR75, HiSCR90, HiSCR100 describes a decrease of at least 25%, 50%, 75%, 90%, 100% in AN count with no increase in the number of abscesses and/or in the number of draining fistulae compared with baseline.
Source: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week 52.66 Details included in the table are from the sponsor’s Summary of Clinical Evidence.67
Appendix 2: Additional Outcome Data to Address Revisions to the Product Monograph
Please note that this appendix has not been copy-edited.
Additional relevant data were extracted from the SUNNY trials (the SUNSHINE and SUNRISE studies) and ITC submitted by the sponsor to address the revisions to the recommended dosing regimen in the product monograph (revisions are summarized in Table 1 in the executive summary section). This included results of the comparison between secukinumab 300 mg every 4 weeks dosing (hereafter referred to as the secukinumab monthly maintenance group) versus placebo from the SUNNY trials and versus adalimumab from the ITC for the outcomes of interest to this review.
Clinical Evidence
Systematic Review
Results (Placebo-Controlled Treatment Period 1; Secukinumab Monthly Maintenance Dosing)
The data summarized subsequently reflect the data cut-off date of October 1, 2021, for the primary efficacy analysis at week 16 from the SUNSHINE study and September 23, 2021, for the primary efficacy analysis at week 16 from the SUNRISE study.
Patient Disposition
SUNSHINE study — A total of 93.9% (169 of 180 patients) of patients in the secukinumab monthly maintenance group and 95.6% (172 of 180 patients) in the placebo group completed treatment period 1. A total of 6.1% (11 patients) of patients in the secukinumab monthly maintenance group and 4.4% (8 patients) of patients in the placebo group discontinued the study. The most frequently reported primary reason for study discontinuation in either study drug group was patient decision — 5.0% (9 patients) of patients in the secukinumab monthly maintenance group and 2.8% (5 patients) of patients in the placebo group. No notable differences in other reasons for study discontinuation between study drug groups (lost to follow-up and physician decision) were identified.
SUNRISE study — A total of 93.9% (169 of 180 patients) of patients in the secukinumab monthly maintenance group and 91.3% (167 of 183 patients) in the placebo group completed treatment period 1. A total of 6.1% (11 patients) of patients in the secukinumab monthly maintenance group and 8.7% (16 patients) of patients in the placebo group discontinued the study. The most frequently reported primary reason for study discontinuation in either study drug group was patient decision — 3.9% (7 patients) of patients in the secukinumab monthly maintenance group and 3.8% (7 patients) of patients in the placebo group. No notable differences in other reasons between study drug groups (AE, lack of efficacy, lost to follow-up, pregnancy, and technical problems) were identified.
Protocol Deviations
SUNSHINE study — A total of █████ (██ of 180 patients) of patients in the secukinumab monthly maintenance group and █████ (██ of 180 patients) of patients in the placebo group had at least 1 protocol deviation. The most frequently reported protocol deviation in either study drug group was treatment deviation primarily related to home versus site drug administration —█████ (██ patients) of patients in the secukinumab monthly maintenance group and █████ (██ patients) in the placebo group. No notable differences in other reported protocol deviations (prohibited concomitant medication, selection criteria not met, and other) between study drug groups were identified.
SUNRISE study — A total of █████ (██ of 180 patients) of patients in the secukinumab monthly maintenance group and █████ (██ of 183 patients) of patients in the placebo group had at least 1 protocol deviation. The most frequently reported protocol deviation in either study drug group was treatment deviation primarily related to home versus site drug administration —█████ (██ patients) of patients in the secukinumab monthly maintenance group and █████ (██ patients) in the placebo group. No notable differences in other reported protocol deviations (prohibited concomitant medication, selection criteria not met, and other) between study drug groups were identified.
Baseline Characteristics
SUNSHINE study — The mean age of patients in the secukinumab monthly maintenance group was 35.7 years (SD = 11.71 years) and in the placebo group was 35.5 years (SD = 10.75 years). A total of 55.6% (100 of 180 patients) of patients in the secukinumab monthly maintenance group and 56.7% (102 of 180 patients) of patients in the placebo group were female; a total of 44.4% (80 patients) of patients and 43.3% (78 patients) of patients were male, respectively. Most patients were white — 81.1% (146 patients) of patients in the secukinumab monthly maintenance group and 77.2% (139 patients) of patients in the placebo group. No notable differences in other races (Asian, Black or African American, American Indian or Alaska Native, and multiple races) between study drug groups were identified. The mean BMI was 32.78 kg/m2 (SD = 7.897 kg/m2) in the secukinumab monthly maintenance group and 31.97 kg/m2 (SD = 7.053 kg/m2) in the placebo group. Most patients’ smoking status was current 53.3% (96 patients) of patients in the secukinumab monthly maintenance group and 56.1% (101 patients) of patients in the placebo group, followed by smoking statuses of never (approximately ███ of patients in each group) then former (approximately 15% of patients in each group).
Most patients had Hurley stage 2 HS: 59.4% (107 patients) of patients in the secukinumab monthly maintenance group and 67.2% (121 patients) of patients in the placebo group — and Hurley stage 3 HS — 35.0% (63 patients) of patients in the secukinumab monthly maintenance group and 28.3% (51 patients) of patients in the placebo group — at baseline (i.e., somewhat more severe disease in the secukinumab group). The mean time since diagnosis of HS was similar between study groups — 6.6 years (SD = 6.73 years) in the secukinumab monthly maintenance group and 7.5 years (SD = 7.00 years) in the placebo group. No notable differences in other baseline disease characteristics (AN count, draining fistulae count, NRS, and DLQI total score) between study drug groups were identified (Table 40). A total of █████ (██ patients) of patients in the secukinumab monthly maintenance group and ████ (██ patients) of patients in the placebo group have a medical history of obesity. A total of ████ (██ patients) in the secukinumab monthly maintenance group and ████ (|||| patients) of patients in the placebo group have a medical history of type 2 diabetes mellitus.
SUNRISE study — The mean age of patients in the secukinumab monthly maintenance group was 35.5 years (SD = 11.41 years) and in the placebo group was 36.2 years (SD = 11.25 years). A total of 57.2% (103 of 180 patients) of patients in the secukinumab monthly maintenance group and 57.4% (105 of 183 patients) of patients in the placebo group were female; a total of 42.8% (77 patients) of patients and 42.6% (78 patients) of patients were male, respectively. Most patients were white — 77.2% (139 patients) of patients in the secukinumab monthly maintenance group and 78.1% (143 patients) of patients in the placebo group. No notable differences in other races (Asian, Black or African American, American Indian or Alaska Native, and multiple races) between study drug groups were identified. The mean BMI was 31.98 kg/m2 (SD = 7.478 kg/m2) in the secukinumab monthly maintenance group and 31.42 kg/m2 (SD = 7.382 kg/m2) in the placebo group. Most patients’ smoking status was current 50.0% (90 patients) of patients in the secukinumab monthly maintenance group and 57.9% (106 patients) of patients in the placebo group, followed by smoking statuses of never (approximately ███ of patients in each group) then former (approximately 15% of patients in each group).
Most patients had Hurley stage 2 HS: 58.9% (106 patients) of patients in the secukinumab monthly maintenance group and 60.1% (110 patients) of patients in the placebo group — and Hurley stage 3 HS — 37.8% (68 patients) of patients in the secukinumab monthly maintenance group and 38.3% (70 patients) of patients in the placebo group — at baseline. The mean time since diagnosis of HS was similar between study groups — 8.2 years (SD = 8.42 years) in the secukinumab monthly maintenance group and 7.0 years (SD = 6.65 years) in the placebo group. No notable differences in other baseline disease characteristics (AN count, draining fistulae count, NRS, and DLQI total score) between study drug groups were identified (Table 40). A total of █████ (██ patients) of patients in the secukinumab monthly maintenance group and ████ (██ patients) of patients in the placebo group have a medical history of obesity. A total of ████ (||| patients) in the secukinumab monthly maintenance group and ████ (██ patients) of patients in the placebo group have a medical history of type 2 diabetes mellitus.
Prior Therapies
SUNSHINE study — Approximately █████ of patients in the study reported prior experience with treatment for HS. No notable differences in HS treatment history (topical therapy, systemic antibiotics, systemic biologic therapy, prior surgery for HS, and others not specified) between study drug groups were identified. Specifically, a total of 82.8% (149 of 180 patients) of patients in the secukinumab monthly maintenance group and 83.3% (150 of 180 patients) of patients in the placebo group reported previous exposure to systemic antibiotics. A total of 21.7% (39 patients) of patients in the secukinumab monthly maintenance group and 25.6% (46 patients) of patients in the placebo group reported previous exposure to systemic biologic therapy. More specifically, a total of █████ (██ patients) of patients in the secukinumab monthly maintenance group and █████ (██ patients) of patients in the placebo group reported previous exposure to adalimumab. The most frequently reported reason for discontinuation of prior HS therapies was lack of efficacy.
SUNRISE study — Approximately █████ of patients in the study reported prior experience with treatment for HS. No notable differences in HS treatment history (topical therapy, systemic antibiotics, systemic biologic therapy, prior surgery for HS, and others not specified) between study drug groups were identified. Specifically, a total of 84.4% (152 of 180 patients) of patients in the secukinumab monthly maintenance group and 82.5% (151 of 183 patients) of patients in the placebo group reported previous exposure to systemic antibiotics. A total of 23.3% (42 patients) of patients in the secukinumab monthly maintenance group and 26.2% (48 patients) of patients in the placebo group reported previous exposure to systemic biologic therapy. More specifically, a total of █████ (██ patients) of patients in the secukinumab monthly maintenance group and █████ (██ patients) of patients in the placebo group reported previous exposure to adalimumab. The most frequently reported reason for discontinuation of prior HS therapies was lack of efficacy.
Concomitant and Rescue Therapies
SUNSHINE study — A total 13.9% (25 patients) of patients in the secukinumab monthly maintenance group and 10.6% (19 patients) of patients in the placebo group enrolled in the antibiotic strata. Approximately █████ of patients in the study used concomitant medications. The most commonly used concomitant medications were analgesics, specifically ibuprofen and paracetamol (no notable differences between study drug groups were identified). A total of ████ (||| of 180 patients) of patients in the secukinumab monthly maintenance group and ████ (||| of 180 patients) of patients in the placebo group required at least 1 rescue medication; and ████ (||| patients) and ████ (██ patients), respectively, required surgical intervention (removal or drainage).
SUNRISE study — A total 11.7% (21 patients) of patients in the secukinumab monthly maintenance group and 10.4% (19 patients) of patients in the placebo group enrolled in the antibiotic strata. Approximately █████ of patients in the study used concomitant medications. The most commonly used concomitant medications were analgesics, specifically ibuprofen and paracetamol (no notable differences between study drug groups were identified). A total of ████ (||| of 180 patients) of patients in the secukinumab monthly maintenance group and ████ (||| of 183 patients) of patients in the placebo group required at least 1 rescue medication; and ████ (||| patients) and ████ (||| patients), respectively, required surgical intervention (removal or drainage).
Exposure to Study Treatment
SUNSHINE study — The mean duration of exposure to study treatment was █████ days (SD = █████ days; range = ██ ██ ███ days) in the secukinumab monthly maintenance group and █████ days (SD = █████ days; range = | ██ ███ days) in the placebo group. The cumulative exposure was ████ patient-years in the secukinumab monthly maintenance group and ████ patient-years in the placebo group.
SUNRISE study — The mean duration of exposure to study treatment was █████ days (SD = █████ days; range = ██ ██ ███ days) in the secukinumab monthly maintenance group and █████ days (SD = █████ days; range = ██ ██ ███ days) in the placebo group. The cumulative exposure was ████ patient-years in the secukinumab monthly maintenance group and ████ patient-years in the placebo group.
Efficacy
SUNNY trials — A summary of the key efficacy results on the secukinumab 300 mg (every 2 weeks and every 4 weeks dosage groups) versus placebo from the SUNNY trials at week 16 is presented in Table 40. Overall, the direction of treatment effect based on the key efficacy results was consistent between the biweekly and monthly maintenance dosing of secukinumab versus placebo. It should be noted that statistical significance cannot be claimed for the primary analysis results of AN count and NRS30 skin pain response at week 16, despite the P value being less than 0.005, from the SUNSHINE trial for the secukinumab monthly maintenance dosage group versus placebo because the result for the primary end point (HiSCR50 response), a prior end point in the testing hierarchy, was not statistically significant. These should be considered as supportive evidence.
Similar to the secukinumab 300 mg every 2 weeks dosing, the sensitivity analysis, supplementary analysis (only applicable to the primary end point), and tipping point analysis results of HiSCR50 response, AN count, flares, and skin pain at week 16 were generally consistent with and supportive of the primary analysis results for the secukinumab 300 mg every 4 weeks dosing from both trials.
Similar to the secukinumab 300 mg every 2 weeks dosing, the subgroup analysis results of the primary end point by concomitant antibiotic use, body weight stratum, previous use of systemic biologics, Hurley stage, and baseline AN count for the monthly maintenance dosage group from the SUNNY trials are generally consistent in direction with the primary analysis; however, some subgroups were small. Also similar to the biweekly maintenance dosing, the exception is the subgroup analysis by patients with Hurley stage 1 in the SUNRISE trial — █████ (||| of ||| patients) of patients in the secukinumab 300 mg every 2 weeks dosage group and █████ (||| of ||| patients) of patients in the secukinumab 300 mg every 4 weeks dosage group compared with █████ (||| of ||| patients) of patients in the placebo group had HiSCR50 response at week 16.
Harms
SUNNY trials — A summary of harms with secukinumab 300 mg (every 2 weeks and every 4 weeks dosage groups) in comparison with placebo from the SUNNY trials at week 16 is presented in Table 41. Overall, no notable differences in the frequency of AEs between study drug groups were identified in each study.
Table 40: Summary of Key Efficacy Results From the SUNNY Trials at Week 16 (Full Analysis Set)
Outcome | SUNSHINE | SUNRISE | ||||
|---|---|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Secukinumab q.4.w. (N = 180) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Secukinumab q.4.w. (N = 180) | Placebo (N = 183) | |
Response to treatment and disease severity | ||||||
HiSCR50 response at week 16a,b | ||||||
OR (96% or 99% CI) | 1.75 ██ ██ █████ | 1.48 ██ ██ █████ | Reference | 1.64 ██ ██ █████ | 1.90 ██ ██ █████ | Reference |
1-sided P valuec | 0.0070 | 0.0418 | Reference | 0.0149 | 0.0022 | Reference |
Risk, percent (96% or 99% CI) | ██ █████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
Marginal risk difference (96% or 99% CI) | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
AN count at week 16d | ||||||
m | ██ █████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
Baseline AN count, mean (SE) | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
Week 16 AN count, mean (SE) | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
Percentage change from baseline, mean (SE) | −46.8 (3.33) | −42.4 (4.01) | −24.3 (4.33) | −39.3 (4.43) | −45.5 (4.08) | −22.4 (4.84) |
LS difference estimate, mean (96% or 99% CI) | −23.05 ██ █ ████ | −18.46 ██ █ ████ | Reference | −16.33 ██ █ ████ | −22.94 ██ █ ████ | Reference |
1-sided P valuec | < 0.0001 | ██ █ ███e | Reference | 0.0051 | ██ █ ████ | Reference |
Disease worsening | ||||||
Flares at week 16b | ||||||
OR (96% or 99% CI) | 0.42 ██ ██ █████ | 0.71 ██ ██ █████ | Reference | 0.68 ██ ██ █████ | 0.49 ██ ██ █████ | Reference |
1-sided P valuec | 0.0010 | 0.0926 | Reference | 0.0732 | 0.0049 | Reference |
Risk, percent (96% or 99% CI) | 15.3 (NR) | 23.1 (NR) | 29.3 (NR) | 20.3 (NR) | 15.5 (NR) | 26.8 (NR) |
Marginal risk difference (96% or 99% CI) | −14.1 ██ ██ █████ | −6.3 ██ ██ █████ | Reference | −6.6 ██ ██ █████ | −11.3 ██ ██ █████ | Reference |
Symptom | ||||||
NRS30 (skin pain) at week 16f | ||||||
SUNSHINE and SUNRISE (pooled) | ||||||
OR (96% or 99% CI) | 2.08 ██ ██ █████ | 1.77 ██ ██ █████ | Reference | NA | ||
1-sided P value | 0.0003c | 0.0044e | Reference | |||
Risk, percent (96% or 99% CI) | ███████ | ██ █████ | ██ █████ | |||
Marginal risk difference (96% or 99% CI) | ██ █████ | ██ ████ | ██ █████ | |||
Health-related quality of life | ||||||
DLQI response at week 16g | ||||||
N | ██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
n/m (%) | 64/134 (47.8) | 62/128 (48.4) | 37/128 (28.9) | 54/144 (37.5) | 67/142 (47.2) | 46/145 (31.7) |
95% CI | ██ █████ | ██ █████ | ██ █████ | ██ ████ | ██ █████ | ██ █████ |
Risk difference (95% CI) | ██ █████ | ██ █████ | ██ ████ | ██ ████ | ███████ | ██ ████ |
OR (95% CI) | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ███ ████ | ██ █████ |
2-sided P valueh | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
DLQI total score at week 16 | ||||||
m | ███ ████ | ███ █ ███ | ███████ | ███ ███ | ███ ████ | ██ █████ |
Baseline DLQI score, mean (SD) | ██ █████ | ██ █████ | █ ██████ | ██ █████ | ███ ████ | ██ █████ |
Week 16 DLQI score, mean (SD) | ███ ████ | ███ ████ | ██ █████ | ██ █████ | ███ ████ | ███ ████ |
Absolute change from baseline, mean (SD) | ███ ████ | ███ ████ | ██ █████ | ██ █████ | ███ ████ | ██ █████ |
Difference in change from baseline, mean (SD) | ███ ████ | ██ █████ | ██ █████ | ██ █████ | ███ ████ | ███ ████ |
95% CIi | ██ █████ | ███ ████ | ██ ████ | ██ ████ | ███ ███ | ██ ████ |
m | ███ ████ | ███ ████ | ██ █████ | ███ ███ | ███ ████ | ███ █ ███ |
Percentage change from baseline, mean (SD) | ██ █ ████ | ██ █ ████ | ███████ | ██ ██ ██ | ██ ██ ███ | ███ ████ |
EQ-5D health state assessment (VAS score) at week 16 | ||||||
m | 143 | 139 | 143 | 150 | 155 | 149 |
Baseline VAS score, mean (SD) | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
Week 16 VAS score, mean (SD) | 67.7 ██ ██ █████ | 67.6 ██ ██ █████ | 63.4 ██ ██ █████ | 69.6 ██ ██ █████ | 67.6 ██ ██ █████ | 63.2 ██ ██ █████ |
Absolute change from baseline, mean (SD) | ██ █ ████ | ██ █ ████ | ███ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
Difference in change from baseline, mean (SD) | █ █████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
95% CIi | ███████ | ██ █ ███ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
Percentage change from baseline, mean (SD) | ██ █ ███ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ | ██ █ ████ |
AN = abscesses and inflammatory nodules; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; DLQI = Dermatology Life Quality Index; HiSCR = Hidradenitis Suppurativa Clinical Response; = LS = least squares; m = number of patients evaluable; n = number of patients with observed response; NA = not applicable; NR = not reported; NRS = numerical rating scale; OR = odds ratio; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SD = standard deviation; SE = standard error.
Notes: Values in bold indicate statistically significant based on the predefined testing hierarchy. The primary and secondary end points (AN count and flares) were tested in a hierarchical order (1-sided significance level of 0.02 for the q.2.w. dosing and 0.005 for the q.4.w. dosing). Note that based on an interim futility and efficacy analysis performed for the SUNNY trials when 40% of patients in both studies completed week 16, a Haybittle-Peto-type adjustment of the 1-sided levels of significance was applied in the analysis of the primary and secondary end points; however, the adjustment was deemed to be very small (0.00001) by the investigator and they concluded that it did not have any impact on the results, thus, the original significant levels are referenced in the results. The secondary end point, NRS30 (skin pain) was based on pooled data from the SUNNY trials; the initial significance level for the skin pain hypothesis was set to alpha squared minus alpha (1-sided alpha is 0.025) and can be increased dependent on which hypothesis were rejected — the subtraction of alpha squared was to account for the maximum possible type I error for HiSCR50 response, percentage change in AN count and flare in both studies.
For secukinumab q.2.w. vs. placebo, 96% CIs are presented; for secukinumab q.4.w. vs. placebo, 99% CIs are presented.
The placebo group included patients randomized to placebo to secukinumab 300 mg every 2 weeks dosage group and placebo to secukinumab 300 mg every 4 weeks dosage group before their first intake of active secukinumab.
aHiSCR50 response was defined as at least a 50% reduction in AN count with no increase in the number of abscesses and/or in the number of draining fistulas from baseline to week 16, where increase was defined as at least 1 abscess or 1 draining fistula more than the baseline value.
bCovariates included in the logistic regression model: treatment group, Hurley stage, baseline AN count, geographical region, use of antibiotic, and baseline body weight.
cP value was adjusted for multiple testing using a predefined testing hierarchy.
dCovariates included in the ANCOVA model: treatment group, Hurley stage, baseline AN count, geographical region, use of antibiotic, and baseline body weight.
eAlthough the P value is < 0.005, statistical significance cannot be claimed because the result for the primary end point (HiSCR50 response), a prior end point in the testing hierarchy, was not statistically significant.
fNRS is the numeric rating scale of the patient's global assessment of skin pain at its worst (averaged over the last 7 days). NRS30 is defined as at least 30% reduction and at least 2-unit reduction from baseline NRS. Only patients with a baseline NRS ≥ 3 are included. Covariates included in the logistic regression model: treatment group, Hurley stage, baseline NRS, geographical region, use of antibiotic, baseline body weight, study.
gDLQI is a general dermatology disability index used to assess HRQoL in adults with skin diseases. DLQI response was defined as a decrease of ≥ 5.0 points on the DLQI total score. Only patients with a baseline DLQI score ≥ 5.0 points were included. Covariates included in the logistic regression model: treatment group, Hurley stage, baseline DLQI, geographical region, use of antibiotic, baseline body weight.
hThe P value for pairwise comparisons was not adjusted for multiple comparisons and no formal confirmatory hypothesis testing was planned.
iThe 95% CI was based on t-statistics.
Sources: SUNSHINE Clinical Study Report,33 SUNRISE Clinical Study Report,34 and sponsor response to June 19, 2023, July 5, 2023, and May 22, 2024, CDA-AMC requests for additional information regarding the secukinumab review.35-37
Table 41: Summary of Harms Results From the SUNNY Trials in Treatment Period 1 (Safety Set)
Adverse events | SUNSHINE | SUNRISE | ||||
|---|---|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Secukinumab q.4.w. (N = 180) | Placebo (N = 180) | Secukinumab q.2.w. (N = 180) | Secukinumab q.4.w. (N = 180) | Placebo (N = 183) | |
Treatment-emergent adverse events, n (%)a | ||||||
Patients with any AE | 122 (67.4) | 118 (65.6) | 120 (66.7) | 113 (62.8) | 114 (63.3) | 116 (63.4) |
Nasopharyngitis | 20 (11.0) | 16 (8.9) | 13 (7.2) | 13 (7.2) | 9 (5.0) | 16 (8.7) |
Headache | 17 (9.4) | 20 (11.1) | 14 (7.8) | 21 (11.7) | 17 (9.4) | 15 (8.2) |
Hidradenitis | 11 (6.1) | 5 (2.8) | 24 (13.3) | 10 (5.6) | 11 (6.1) | 14 (7.7) |
Diarrhea | 5 (2.8) | 13 (7.2) | 9 (5.0) | 8 (4.4) | 7 (3.9) | 13 (7.1) |
Upper respiratory tract infection | 5 (2.8) | 6 (3.3) | 4 (2.2) | 9 (5.0) | 3 (1.7) | 7 (3.8) |
Serious adverse events, n (%)b | ||||||
Patients with any SAE | 3 (1.7) | 3 (1.7) | 6 (3.3) | 6 (3.3) | 6 (3.3) | 5 (2.7) |
Hidradenitis | 1 (0.6) | 0 | 2 (1.1) | 1 (0.6) | 0 | 0 |
Patients who stopped treatment because of adverse events, n (%)b | ||||||
Patients with any WDAE | 5 (2.8) | 1 (0.6) | 1 (0.6) | 1 (0.6) | 4 (2.2) | 4 (2.2) |
Mortality, n (%) | ||||||
Deaths | 0 | 0 | 0 | 0 | 0 | 0 |
Adverse events of special interest, n (%) | ||||||
Hypersensitivity (SMQ) | 12 (6.6) | 9 (5.0) | 9 (5.0) | 7 (3.9) | 5 (2.8) | 7 (3.8) |
Hypersensitivity | NR | NR | NR | NR | NR | NR |
Drug hypersensitivity | NR | NR | NR | 1 (0.6) | 0 | 0 |
Injection site reaction | 0 | 1 (0.6) | 0 | 0 | 1 (0.6) | 0 |
Infections and infestations | 59 (32.6) | 51 (28.3) | 53 (29.4) | 52 (28.9) | 59 (32.8) | 62 (33.9) |
Infections and infestations (SOC) | 59 (32.60) | 52 (28.89) | 53 (29.44) | 52 (28.89) | 59 (32.78) | 62 (33.88) |
RR estimate (95% CI) | ██ █ ████ | ██ ██ ███ | █ ███ | █ █ ████ | █ ██ ███ | █ ███ |
RD estimate, % (95% CI) | ██ ██ ███ | ███ █ ███ | █████ | ██ ██ ███ | ██████ | █████ |
Candida infections (HLT) | 2 (1.10) | 1 (0.56) | 4 (2.22) | 5 (2.78) | 5 (2.78) | 2 (1.09) |
RR estimate (95% CI) | ██ █ █ ███ | █ ██ ████ | ██ ███ | ████ ████ | ██ ██ ██ | █████ |
RD estimate, % (95% CI) | ███ ███ | ██ █ ███ | █████ | ███ █ ███ | ██ █ ███ | █ ████ |
Malignant and unspecified tumour (SMQ) | 0 | 0 | 1 (0.56) | 0 | 2 (1.11) | 1 (0.55) |
RR estimate (95% CI) | ███ █ ███ | ██ █████ | ██ ███ | ██ ██ ███ | ██ ██ ███ | ██ ███ |
RD estimate, % (95% CI) | ██ █ ████ | ███ ████ | █████ | ████ ████ | ███ ████ | █ ███ |
Neoplasms benign, malignant, and unspecified (including cysts and polyps) (SOC) | 0 | 1 (0.56) | 1 (0.56) | 0 | 2 (1.11) | 2 (1.09) |
RR estimate (95% CI) | ███ █ ███ | ██ █ ████ | █████ | ██ █ ████ | ██ ██ ██ | █████ |
RD estimate, % (95% CI) | ██ ██ ███ | ██ █ ████ | █████ | ████ ███ | ██ █ ████ | █████ |
Squamous cell carcinoma of HS-affected area (PT) | 0 | 0 | 0 | 0 | 0 | 0 |
RR estimate (95% CI) | ███ █ ███ | ███ █ ███ | █████ | ██ █ ████ | ██ ██ ███ | █████ |
RD estimate, % (95% CI) | ███ ████ | ██ █████ | █████ | █ ██ ████ | ██ ██ ██ | █████ |
Inflammatory bowel disease (NMQ) | ██ ██ ██ | ██ █████ | ██ ███ | ██ ██ ██ | ██ ████ | ███ ██ |
RR estimate (95% CI) | ██ ██ ███ | ██ ██ ███ | █████ | ██ ███ ██ | █ ██████ | █████ |
RD estimate, % (95% CI) | ██ ██ ███ | ██ █████ | █████ | ██ ████ | ███ ████ | ██ ███ |
Suicidal ideation and behaviour (SMQ) | ███ ████ | ███ ████ | █████ | ███ █████ | ██ ███ | ██ ███ |
Suicidal ideation | ███ ████ | ████ ███ | █████ | ██ ██ ██ | ██ █ ███ | █████ |
Suicide attempt | 1 (0.6) | 0 | 0 | ███ █ ██ | ██ █ ██ | ██ █ █ |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; HLT = high-level term; HS = hidradenitis suppurativa; NA = not applicable; NMQ = SMQ, narrow; NR = not reported; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RD = risk difference; RR = risk ratio; SAE = serious adverse event; SMQ = Standardised MedDRA Query; SOC = system organ class; WDAE = withdrawal due to adverse event.
Notes: A patient with multiple AEs with the same preferred term is counted only once for that preferred term.
SMQs are validated, standard sets of MedDRA terms used to support signal detection and monitoring and represent a variety of safety topics of regulatory interest. SMQs include narrow and/or broad terms; narrow terms are highly likely to represent the condition of interest.71
The placebo group included patients randomized to placebo to secukinumab 300 mg every 2 weeks dosage group and placebo to secukinumab 300 mg every 4 weeks dosage group before their first intake of active secukinumab.
aFrequency ≥ 5% in any group.
bFrequency ≥ 1% in any group.
Sources: SUNSHINE Clinical Study Report,33 SUNRISE Clinical Study Report,34 and sponsor response to July 5, 2023 and May 22, 2024, CDA-AMC requests for additional information regarding secukinumab CDA-AMC review.36,37
Results (Entire Study Period; Secukinumab Monthly Maintenance Dosing)
Only data on the secukinumab monthly maintenance dosing, which included patients who were randomized to receive secukinumab 300 mg every 4 weeks at the study entry, are summarized subsequently, as it was considered to be most relevant to inform the expert committee deliberations.
The data summarized subsequently reflect the final database lock date of August 17, 2022, for the entire study period from the SUNSHINE study and August 9, 2022, for the entire study period from the SUNRISE study. The entire study period included treatment period 1 (16 weeks), treatment period 2 (36 weeks), and a follow-up period (8 weeks).
Patient Disposition
SUNSHINE study: A total of █████ (███ of 180 patients) of patients in the secukinumab monthly maintenance group completed the study. A total of █████ (██ patients) of patients in the secukinumab monthly maintenance group discontinued the study. The most frequently reported primary reason for study discontinuation was patient decision (████████ patients). All other reasons were reported in less than ██ of patients in the secukinumab monthly maintenance group, including physician decision, lost to follow-up, lack of efficacy, and AE.
SUNRISE study: A total of █████ (███ of 180 patients) of patients in the secukinumab monthly maintenance group completed the study. A total of █████ (██ patients) of patients in the secukinumab monthly maintenance group discontinued the study. The most frequently reported primary reason for study discontinuation was patient decision (████████ patients). All other reasons were reported in less than ██ of patients in the secukinumab monthly maintenance group, including lost to follow-up, AE, lack of efficacy, physician decision, technical problems, death, and pregnancy.
Protocol Deviations
SUNSHINE study: A total of █████ (███ of 180 patients) of patients in the secukinumab monthly maintenance group had at least 1 protocol deviation. The most frequently reported protocol deviation in the secukinumab monthly maintenance group was treatment deviation (█████|██ patients). Other protocol deviations reported in the secukinumab monthly maintenance group included the use of prohibited concomitant medication (█████|██ patients), selection criteria not met (████||| patients), and all other reasons (█████ |██ patients).
SUNRISE study: A total of █████ (███ of 180 patients) of patients in the secukinumab monthly maintenance group had at least 1 protocol deviation. The most frequently reported protocol deviation in the secukinumab monthly maintenance group was treatment deviation (█████|██ patients]). Other protocol deviations reported in the secukinumab monthly maintenance group included the use of prohibited concomitant medication (█████|██ patients), selection criteria not met (████|██ patients), and all other reasons (█████|██ patients).
Concomitant and Rescue Therapies
SUNSHINE study — A total of █████ (███ of 180 patients) of patients in the secukinumab monthly maintenance group used concomitant medications. The most commonly used concomitant medications continued to be analgesics, specifically ibuprofen (█████|███ patients) and paracetamol (█████|██ patients). A total of ████ (██ patients) of patients in the secukinumab monthly maintenance group required at least 1 rescue medication and █████ (██ patients) required surgical intervention.
SUNRISE study — A total of █████ (███ of 180 patients) of patients in the secukinumab monthly maintenance group used concomitant medications. The most commonly used concomitant medications continued to be analgesics, specifically ibuprofen (█████|███ patients) and paracetamol (█████|██ patients). A total of ████ (██ patients) of patients in the secukinumab monthly maintenance group required at least 1 rescue medication and ████ (██ patients) required surgical intervention.
Exposure to Study Treatment
SUNSHINE study — The mean duration of exposure to study treatment was █████ days (SD = █████ days; range = ██ ██ ███ days) in the secukinumab monthly maintenance group. The cumulative exposure was █████ patient-years in the secukinumab monthly maintenance group.
SUNRISE study — The mean duration of exposure to study treatment was █████ days (SD = █████ days; range = ██ ██ ███ days) in the secukinumab monthly maintenance group. The cumulative exposure was █████ patient-years in the secukinumab monthly maintenance group.
Efficacy
SUNNY trials — A summary of the key efficacy results on the secukinumab 300 mg (every 2 weeks and every 4 weeks dosage groups) from the SUNNY trials at week 52 is presented in Table 42. Overall, the direction of estimates for each outcome based on the key efficacy results was consistent between the biweekly and monthly maintenance dosing of secukinumab.
Harms
SUNNY trials — A summary of harms with secukinumab 300 mg (every 2 weeks and every 4 weeks dosage groups) from the SUNNY trials at week 52 is presented in Table 43. Overall, no notable differences in the frequency of AEs between study drug groups were identified in each study.
Approximately ███ of patients in each study drug group across trials experienced an AE at the 16-week follow-up, with the most common AE being nasopharyngitis or headache in the secukinumab groups. At the follow-up time point of 52 weeks, approximately ███ of patients in each group across trials experienced an AE, with the most common AE still being headache and nasophyngitis.
No greater than ██ of patients in each study drug group across trials experienced an SAE at the 16-week follow-up. At the follow-up time point of 52 weeks, no greater than ███ of patients in each group across trials experienced an SAE.
No greater than ██ of patients in each study drug group across trials experienced a withdrawal due to an adverse event (WDAE) at the 16-week follow-up. At the follow-up time point of 52 weeks, no greater than ██ of patients in each group across trials experienced a WDAE.
At the follow-up time point of 52 weeks, 1 patient in the secukinumab monthly maintenance dosing group in the SUNRISE trial died; the cause was myocardial infarction.
Table 42: Summary of Efficacy Results From the SUNNY Trials at Week 52 (Full Analysis Set)
Outcome | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Secukinumab q.4.w. (N = 180) | Secukinumab q.2.w. (N = 180) | Secukinumab q.4.w. (N = 180) | |
Response to treatment and disease severity | ||||
HiSCR50 response at week 52 | ||||
n/m (%) | ████ ███ ████ | ██ ██ ██ ████ | ███ ██ ████ | ██ ███ ████ |
95% CI | ████ ███ ████ | ██ ███ ██ ███ | ██ ███ █ ███ | ███ ███ ███ |
AN count at week 52 | ||||
m | ████ ███ ███ | ██ ████ ███ | ██ ███ ████ | █ ███ █ ████ |
Mean baseline AN count (SE) | ██ ███ ██ ███ | ██ ███ ██ ███ | █ ███ █ ████ | ██ ██ █████ |
Mean week 52 AN count (SE) | ██ ███ ██ ███ | ██ ███ ██ ███ | ███ ███ ███ | ██ ██ █ ███ |
Mean percentage change from baseline (SE) | ███ ██ ██ ████ | ██ ████ ████ | ██ ██ ██ ███ | ██ ███ ████ |
95% CI | ███ ██ ██ ████ | ███ █ ██ ████ | ███ ██ ████ | ███ ██ ████ |
Disease worsening | ||||
Flares at week 52 | ||||
n/m (%) | ███ ██ ██ ████ | ███ █ ██ ████ | ███ ██ ████ | ███ ███ ███ |
95% CI | ███ ██ ██ ████ | ███ █ ██ ████ | ███ █ ██ ███ | ███ █ ██ ███ |
Symptoms | ||||
NRS30 (skin pain) at week 52 | SUNSHINE and SUNRISE pooled | NA | ||
N | ███ ██ ██ ████ | ███ ███ ████ | ||
n/m (%) | ███ ██ ██ ████ | ███ █ ██ ████ | ||
95% CI | ███ ██ ██ ████ | ███ █ ██ ████ | ||
Health-related quality of life | ||||
DLQI response at week 52 | ||||
N | ███ ██ ██ ████ | ███ █ ██ ████ | ███ ██ ████ | ███ ██ ██ ██ |
n/m (%) | 49/96 (51.0) | 45/97 (46.4) | 64/116 (55.2) | 48/101 (47.5) |
95% CI | ███ ██ ██ ████ | ███ █ ██ ████ | ███ ██ ████ | ███ ██ ████ |
DLQI total score at week 52 | ||||
m | ███ ██ ██ ████ | ████ █ ██ ███ | ████ ██ ██ | ███ ██████ |
Mean baseline DLQI score (SE) | ███ █ ██ ████ | ██████ ███ | ████ ██ ██ | ████ ████ |
Mean week 52 DLQI score (SE) | ███ ██ ██ ███ | ███ ███ ████ | ██████ ████ | ████ ██ ███ |
Mean absolute change from baseline (SE) | ████ ██ ██ ███ | ███ █ ██ ████ | ██ █ ██ ████ | ██ ██ ██ ███ |
95% CI | ████ ██ ██ ███ | ███ ██ █ ████ | ███ ██ ████ | ███ █ █ ████ |
Mean percentage change from baseline (SE) | ███ ██ ██ ████ | ███ █ ██ ████ | ████ ██ ███ | ██ ███ ████a |
EQ-5D health state assessment (VAS) at week 52 | ||||
m | 103 | 103 | 122 | 112 |
Mean baseline EQ-5D VAS score (SE) | ████ ██ ██ ███ | ███ ███ ████ | ███ ██ ████ | ███ ██ ████ |
Mean week 52 EQ-5D VAS score (SE) | 70.7 ██████ | 72.7 ██████ | 72.0 ██████ | 72.3 ██████ |
Mean absolute change from baseline (SE) | ████ ██ ████ | ████ ██ ████ | ███ ██ ████ | ███ ██ ████ |
95% CI | ██████ █████ | ███ ██ █████ | ██████ ████ | ███ ██ ████ |
Mean percentage change from baseline (SE) | █████ ██████ | ████ ██ ████ | ████ ██████ | ███ ██ ████ |
AN = abscesses and inflammatory nodules; CI = confidence interval; DLQI = Dermatology Life Quality Index; HiSCR = Hidradenitis Suppurativa Clinical Response; LS = least squares; m = number of patients evaluable; n = number of patients with observed response; NA = not applicable; NR = not reported; NRS = numerical rating scale; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SE = standard error.
Note: The entire study period included treatment period 1, treatment period 2, and the follow-up period.
aThe number of patients evaluated was 112.
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week52,66 and sponsor response to July 5, 2023 request for additional information regarding secukinumab CDA-AMC review.36
Table 43: Summary of Harms Results From the SUNNY Trials in the Entire Study Period (Safety Set)
Adverse events | SUNSHINE | SUNRISE | ||
|---|---|---|---|---|
Secukinumab q.2.w. (N = 181) | Secukinumab q.4.w. (N = 180) | Secukinumab q.2.w. (N = 180) | Secukinumab q.4.w. (N = 180) | |
Treatment-emergent adverse events, n (%)a | ||||
Patients with any AE | 154 (85.1) | 154 (85.6) | 147 (81.7) | 153 (85.0) |
Headache | 33 (18.2) | 32 (17.8) | 31 (17.2) | 27 (15.0) |
Nasopharyngitis | 32 (17.7) | 24 (13.3) | 21 (11.7) | 18 (10.0) |
Hidradenitis | 19 (10.5) | 20 (11.1) | 22 (12.2) | 23 (12.8) |
Serious adverse events, n (%)b | ||||
Patients with any SAE | 13 (7.2) | 9 (5.0) | 19 (10.6) | 15 (8.3) |
Hidradenitis | 3 (1.7) | 3 (1.7) | 4 (2.2) | 0 |
Sweat gland infection | ██████ | ██████ | ██████ | ██████ |
Patients who stopped treatment because of any adverse event, n (%)b | ||||
Patients with any WDAE | 10 (5.5) | 5 (2.8) | 7 (3.9) | 9 (5.0) |
Mortality, n (%) | ||||
Deaths | 0 | 0 | 0 | 1 (0.6) |
Myocardial infarction | ██████ | ██████ | ██████ | ██████c |
Adverse events of special interest, n (%) | ||||
Hypersensitivity (SMQ) | 29 (16.0) | 24 (13.3) | 23 (12.8) | 19 (10.6) |
Hypersensitivity | 0 | 1 (0.6) | 0 | 1 (0.6) |
Drug hypersensitivity | NR | NR | 1 (0.6) | 0 |
Injection site reaction | 0 | 1 (0.6) | 0 | 2 (1.1) |
Infections and infestations (SOC) | 106 (58.6) | 95 (52.8) | 93 (51.7) | 95 (52.8) |
Infections and infestations | 106 (58.6) | 94 (52.2) | 91 (50.6) | 95 (52.8) |
Candida infections (HLT) | 11 (6.1) | 8 (4.4) | 12 (6.7) | 8 (4.4) |
Malignant and unspecified tumour (SMQ) | 1 (0.6) | 0 | 1 (0.6) | 2 (1.1) |
Neoplasms benign, malignant and unspecified (including cysts and polyps) | 2 (1.1) | 4 (2.2) | 7 (3.9) | 3 (1.7) |
Squamous cell carcinoma of an HS-affected area | 0 | NR | 0 | NR |
Inflammatory bowel disease | 0 | NR | 0 | 1 (0.6) |
Suicidal ideation and behaviour (SMQ) | ██████ | ██████ | ██████ | ██████ |
Suicidal ideation | 1 (0.6) | 0 | 0 | 1 (0.6) |
Suicide attempt | 1 (0.6) | 0 | 0 | 1 (0.6)d |
AE = adverse event; HLT = high-level term; HS = hidradenitis suppurativa; NA = not applicable; NR = not reported; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SAE = serious adverse event; SMQ = Standardised MedDRA Query; SOC = system organ class; WDAE = withdrawal due to adverse event.
Notes: A patient with multiple AEs with the same preferred term is counted only once for that preferred term.
SMQs are validated, standard sets of MedDRA terms used to support signal detection and monitoring and represent a variety of safety topics of regulatory interest. SMQs include narrow and/or broad terms; narrow terms are highly likely to represent the condition of interest.71
The entire study period included treatment period 1, treatment period 2, and the follow-up period.
aFrequency ≥ 10% in any treatment group.
bFrequency ≥ 1% in any treatment group.
cThis patient had pre-existing aortic valve stenosis, experienced a myocardial infarction, and died on day 219.
dPreferred term is intentional overdose.
Sources: SUNSHINE Clinical Study Report Week 5265 and SUNRISE Clinical Study Report Week.52.66
Indirect Comparisons
Results
The primary evidence network was informed by 4 studies (PIONEER 1, PIONEER 2, SUNSHINE, and SUNRISE), and limited to patients who were biologic-naive. The sensitivity analyses included all comparative trials assessing adalimumab and secukinumab in biologic-experienced and biologic-naive patients, and were informed by 5 or 6 RCTs, depending on the outcome. All results were based on the induction phase of the trials (12 to 16 weeks).
Overall, the results for the secukinumab every 4 weeks dosage group were similar to the secukinumab every 2 weeks dosage group and shared the same limitations. The results for both secukinumab dosage groups are shown in Table 44 and Source: Sponsor-submitted ITC report.72
Table 45 for the biologic-naive base-case and multinomial analyses, and in Source: Sponsor-submitted ITC report.72
Table 46 for the sensitivity analyses that included biologic-naive and biologic-experienced patients. Refer to the ITC section of this report for a description of the methods and critical appraisal of the ITC.
Table 44: NMA Results for the Secukinumab q.4.w. Comparison — Base-Case Analyses in the Biologic-Naive Population
Outcome | N studies (patients), | Secukinumab q.2.w. vs. adalimumab q.1.w. | Secukinumab q.4.w. vs. placebo | Secukinumab q.4.w. vs. adalimumab q.1.w. |
|---|---|---|---|---|
OR (95% CrI) | ||||
HiSCR50b | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
Skin pain NRS30b | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
Flaresb | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
DLQI responsec | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
Mean difference in % change from baseline (95% CrI) | ||||
AN countd | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
Mean difference in change from baseline (95% CrI) | ||||
DLQI total scoree | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
AN = abscesses and inflammatory nodules; CrI = credible interval; DLQI = Dermatology Life Quality Index; FE = fixed effect; HiSCR = Hidradenitis Suppurativa Clinical Response; LOCF = last observation carried forward; NMA = network meta-analysis; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; OR = odds ratio; q.1.w. = every week; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RE = random effects.
Note: Values in bold indicate 95% CrI exclude the null value. OR greater than 1, and negative mean difference favour the treatment vs. the comparator.
aPatient totals include those in the secukinumab q.2.w. and q.4.w. groups of the SUNNY trials.
bResults were obtained using the multiple imputation for SUNNY trials and nonresponder imputation for PIONEER trials.
cResults were obtained using nonresponder imputation for SUNNY trials. For the PIONEER trials the sample size was derived using 90% of the arms sample size, as well as the number of events based on the reported percentages of responders in publications.
dResults were obtained using the multiple imputation for SUNNY trials and LOCF for PIONEER trials. Mean difference in % change from baseline were adjusted on treatment group, weight, and baseline AN count for SUNNY trials. Negative value indicates symptomatic improvement in treatment vs. control group.
eResults were obtained using the observed data for SUNNY trials and LOCF for PIONEER trials. DLQI is scored from 0 to 30, with a higher score indicating greater impairment in HRQoL.
Source: Sponsor-submitted ITC report.72
Table 45: NMA Results for HiSCR 25, 50, and 75 at End of Induction Phase for the Secukinumab q.4.w. Comparison — Multinomial Model in the Biologic-Naive Population
Outcome | N Studies (patients), | Secukinumab q.2.w. vs. adalimumab q.1.w. | Secukinumab q.4.w. vs. placebo | Secukinumab q.4.w. vs. adalimumab q.1.w. |
|---|---|---|---|---|
RR (95% CrI) | ||||
HiSCR25 | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
HiSCR50 | | ████████ ██ | | ████████ ██ | | ████████ ██ | |
HiSCR75 | | ████████ ██ | | ████████ ██ | | ████████ ██ | |
CrI = credible interval; FE = fixed effect; HiSCR = Hidradenitis Suppurativa Clinical Response; NMA = network meta-analysis; RR = relative risk; q.1.w. = every week; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
Note: Values in bold indicate 95% CrI exclude the null value. RR values greater than 1 favour the treatment vs. the comparator.
Source: Sponsor-submitted ITC report.72
Table 46: NMA Results of the Secukinumab 300 mg q.4.w. Comparison — Sensitivity Analyses in Biologic-Naive and Biologic-Experienced Population
Outcome | N studies (patients), | Secukinumab q.2.w. vs. adalimumab q.1.w. | Secukinumab q.4.w. vs. placebo | Secukinumab q.4.w. vs. adalimumab q.1.w. |
|---|---|---|---|---|
OR (95% CrI) | ||||
HiSCR50b | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
Skin pain NRS30b | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
Flaresb | | ██████ ██ | | ████████ ██ | | ████████ ██ | | ████████ ██ |
AN = abscesses and inflammatory nodules; CrI = credible interval; DLQI = Dermatology Life Quality Index; HiSCR = Hidradenitis Suppurativa Clinical Response; ITC = indirect treatment comparison; NMA = network meta-analysis; NR = not reported; NRS30 = at least a 30% reduction and at least a 2-unit reduction from baseline in skin pain at its worst as measured by a numerical rating scale; OR = odds ratio; q.1.w. = every week; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RE = random effects.
aPatient totals include those in the secukinumab q.2.w. and q.4.w. groups of the SUNNY trials.
bResults were obtained using the multiple imputation for SUNNY trials and nonresponder imputation for PIONEER trials.
Note: values in bold indicate 95% CrI exclude the null value. OR values greater than 1 favour the treatment vs. the comparator.
Source: Sponsor-submitted ITC report.72
Pharmacoeconomic Review
Abbreviations
AE
adverse event
AN
abscesses and inflammatory nodules
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
HiSCR
Hidradenitis Suppurativa Clinical Response
HRQoL
health-related quality of life
HS
hidradenitis suppurativa
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
NICE
National Institute for Health and Care Excellence
SOC
standard of care
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Secukinumab (Cosentyx) solution for subcutaneous injection |
Submitted pricea |
|
Indication | Proposed: Adult patients with moderate to severe hidradenitis suppurativa (HS) |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Standard review |
NOC date | Anticipated: October 19, 2023 |
Reimbursement request | As per indication |
Sponsor | Novartis Pharmaceuticals Canada Inc. |
Submission history | Previously reviewed: Yes Indication: Psoriatic arthritis and ankylosing spondylitis:
Indication: Plaque psoriasis:
|
NOC = Notice of Compliance.
aReflects submitted prices for Ontario, British Columbia, Nova Scotia, and Newfoundland and Labrador (and the highest prices submitted). Other prices were submitted for the other provinces and incorporated into the budget impact analysis. Only the highest price was used in the cost-utility analysis.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation |
|
Target population | Adults with moderate to severe HS who have not responded to conventional therapy |
Treatment | Secukinumab |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | LYs, QALYs |
Time horizon | Lifetime (44 years) |
Key data sources |
|
Submitted results | Sequential results:
|
Key limitations |
|
CDA-AMC reanalysis results | CDA-AMC incorporated the following changes to address the identified limitations for the base case:
|
CDA-AMC = Canada’s Drug Agency; HS = hidradenitis suppurativa; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
Conclusions
Two sponsor-submitted, phase III, randomized, double-blind, placebo-controlled trials, SUNSHINE and SUNRISE, comparing secukinumab with placebo in adult patients (≥ 18 years) with moderate to severe hidradenitis suppurativa (HS) demonstrated that 16 weeks of treatment with secukinumab likely results in a clinically important improvement in response to treatment and severity of HS as measured by Hidradenitis Suppurativa Clinical Response (HiSCR) 50 (HiSCR50) response. For secukinumab 300 mg every 2 weeks versus adalimumab 40 mg weekly, the results of a network meta-analysis were inconclusive, showing 95% credible intervals that were wide and included the null for all outcomes tested (HiSCR50, abscesses and inflammatory nodules (AN) count, skin pain, flares, and health-related quality of life [HRQoL]). The economic evaluation was based on secukinumab impact on the HiSCR score.
CDA-AMC incorporated the following changes to address the identified limitations for the base case: assuming equivalent response and discontinuation rates between adalimumab and secukinumab, increasing rates of treatment discontinuation after 1 year to account for potential treatment waning (4.61% per 4-week cycle).
Adalimumab is the only biologic for HS approved in Canada; therefore, it is the most relevant comparator in patients who are being considered for a biologic. In the CDA-AMC base-case analysis, relative to adalimumab, secukinumab was associated with a near-zero gain (< 0.01) in quality-adjusted life-years (QALYs) and higher costs ($25,558), resulting in an incremental cost-effectiveness ratio (ICER) of $2,884,183 per QALY gained. Based on this analysis, a 51% price reduction would be required for secukinumab to achieve cost-effectiveness at a threshold of $50,000 per QALY relative to adalimumab. However, no robust evidence was provided in this submission to indicate that secukinumab has a superior treatment effect relative to adalimumab and the assumptions informing the cost-effectiveness analysis are uncertain. Therefore, to ensure cost-effectiveness relative to adalimumab, the total drug costs associated with the secukinumab regimen should not exceed the total drug costs associated with the biosimilar adalimumab regimen.
In patients whose HS fails to respond on a biologic, standard of care (SOC) is the only treatment option available, as there are no other biologics for HS approved in Canada. Relative to SOC, secukinumab was associated with a higher QALY gain (0.15) and higher costs ($47,026), resulting in an ICER of $321,446 per QALY gained. At a threshold of $50,000 per QALY gained, a price reduction of 81% would be required to ensure cost-effectiveness relative to SOC; however, this assumes that the response rate to secukinumab treatment is the same regardless of prior exposure to a biologic.
Outstanding uncertainty remains due to limitations with the sponsor-submitted model, for example, the impact on health gains and because the costs of titrating the adalimumab dose upward to achieve a better response was not explored.
Patient and Clinician Group Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participate in the CDA-AMC review process.
CDA-AMC received patient input from a collaboration between the Canadian Skin Patient Alliance (CSPA), HS Heroes, and Hidradenitis and Me Support Group (2 of which received some financial support from the sponsor). This collaboration combined the results from the 2020 National Report of Patients’ Experiences Living with Hidradenitis Suppurativa (N = 537; 73 [14%] lived in Canada), and a patient survey hosted on the communications channels of CSPA, HS Heroes, and the HS and Me Support Group (March to May 2023; N = 15; all lived in Canada). Patient input indicated that the treatment goals were to control symptoms, cure HS, and be able to enjoy personal relationships. Most respondents (61%) were dissatisfied with the currently available treatments. They reported anxiety, high costs with wound care and drugs, and difficulty accessing prescriptions (51%); a minority reported well-controlled pain (11%). Most had tried an average of 15 different medications (although 35% had not used any), surgical procedures, home treatments, and/or lifestyle modifications, with only a few finding any significant improvement. The most used treatment was a long course of antibiotics (received by 82% of survey participants; only 11% of whom responded to treatment). Only 27% of respondents had used biologics (of whom 38% reporting symptomatic improvement). HS-related expenses ranged from $65 to $262 per month without biologics (costs for prescription drugs were included but expenses were mostly for nonprescription items such as soaps, bath products, creams, wound care, and other nonprescription treatments or therapies) to $150 to $1,200 per month with biologics (adalimumab being the only biologic available). Costs were highly dependent on whether the patient had insurance or received coverage from the sponsor. Only 4 patients had used secukinumab previously (3 under a randomized controlled trial; 1 under compassionate care). None of the patients noted side effects. One patient discontinued because of ineffectiveness; 2 found it effective in reducing lesions, pain, and the need for wound care; and 1 reported complete resolution of lesions and remission. All reported concerns with the cost of treatment and inability to afford it.
Clinician input was received from the Canadian Hidradenitis Suppurativa Foundation, a not-for-profit organization that helps dermatologists in Canada better manage HS. For all patients, disease management involves lifestyle changes and managing pain, odour, and drainage. Currently, the guidelines for managing HS are based on staging:
Hurley stage 1: Includes gentle cleaning with cleansers, application of topical clindamycin, and injection of targeted therapy with intralesional triamcinolone. May include additional adjunctive therapy with topical resorcinol, oral zinc supplementation, laser hair removal, or antiandrogens for female patients.
Hurley stage 2: Addition of an oral antibiotic (usually tetracycline for up to 12 to 16 weeks).
Recurrent episodes and/or Hurley stage 3: Multiple courses of antibiotics are often necessary. Adalimumab is the only approved biologic option (about half of patients on adalimumab experience some response, specifically, a reduction in inflammatory nodules and abscesses). When a patient’s HS does not respond to adalimumab, off-label alternative biologics are offered (e.g., infliximab, ustekinumab, interleukin [IL]-17, and IL-1 inhibitors), depending on the availability of extended benefits coverage or compassionate programs.
Current management methods are not effective in inducing remission in patients. Higher doses of adalimumab are sometimes required to maintain efficacy and some patients lose benefit. Secukinumab addresses a different mechanism of action, providing clinicians with an alternative first-line treatment when systemic antibiotics for 12 weeks is not effective. Secukinumab may have fewer potential side effects and a lower frequency of administration, which should allow for better adherence compared to the currently approved biologic.
The drug plan input noted uncertainty regarding the positioning of secukinumab in therapy (first line, second line, or both) and whether the Notice of Compliance should be more restrictive, similar to adalimumab (i.e., conditional on a lack of response to systemic conventional therapy). The drug plans commented on the need for clarity on whether to expect secukinumab to be used in combination with other biologics (because of their different mechanisms of action) or SOC drugs (as adjunctive therapy), which could have significant implications for budget impact.
Several of these concerns were addressed in the sponsor’s model:
Clinical effectiveness was based on treatment response measured by the HiSCR, which used the percentage decrease in abscesses and inflammatory nodules count compared with baseline with no increase in the number of abscesses and/or draining fistulas.
HRQoL was included in the model through health state utility values applied to the response-based health states that capture pain and depression and anxiety outcomes.
Health care utilization to manage pain and wound care was incorporated into the model as the frequency of visits (outpatient,, emergency department, and wound care visits and hospitalizations) and applied to the response-based health states.
CDA-AMC was unable to address the following concerns raised by the patient and clinician group input:
The efficacy of secukinumab or adalimumab following treatment failure could not be evaluated directly because the structure of the model does not allow sequential treatment with another biologic.
The efficacy of secukinumab given in combination with adalimumab could also not be determined, given the lack of data for this scenario.
Economic Review
The current review is for secukinumab (Cosentyx) for adults with moderate to severe HS, with or without prior exposure to anti–tumour necrosis factor (TNF) therapies, that has not responded to conventional therapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of secukinumab compared with adalimumab or SOC in adults with moderate to severe HS with or without prior exposure to anti-TNF therapies, who have not responded to conventional therapy.1 The indication in the monograph is not explicitly conditional on nonresponse to conventional therapy, leaving it open to interpretation. The SOC comprised a basket of other drugs (antibiotics, retinoids, and immunosuppressants). The modelled population is consistent with the reimbursement request.
Secukinumab is available in single-use prefilled syringes or pens for self-administered subcutaneous injection of 75 mg, 150 mg, or 300 mg of the drug; however, the 300 mg form was not priced or considered within the submission. The recommended dosage for secukinumab is 300 mg per week for 5 weeks followed by 300 mg every 2 weeks.2 Based on the recommended dosing and a unit cost of $882.59 per 150 mg syringe or pen, the annual cost of treatment is $51,190 in the first year (plus a 1-time self-injection training fee of $56.74) and $45,895 every year thereafter, as calculated by the sponsor.1
The recommended dosage for adalimumab is 160 mg at week 0, 80 mg at week 2, 40 mg at week 4, and 40 mg weekly thereafter. The annual cost of treatment with adalimumab is $25,449 in the first year (plus a 1-time self-injection training fee of $56.74) and $24,506 thereafter, based on a unit cost for biosimilars of $471.27 per 40 mg syringe or pen.1
For SOC, it was assumed a proportion of patients would be receiving certain drugs from a basket of other drugs (antibiotics, retinoids, and immunosuppressants), while some patients would receive none (approximately 35%). The annual cost of SOC treatment was estimated to be $227.85. An overview of the 4-week cycle costs for SOC adopted in the sponsor’s model is provided in Table 10.
The outcomes modelled included QALYs and life-years over a lifetime time horizon of 44 years and a cycle length of 4 weeks. The base-case analysis was conducted from the perspective of the Canadian public health care system, with costs and outcomes discounted at 1.5% annually.
Model Structure
A diagrammatic representation of the sponsor’s model can be found in Figure 1. The sponsor submitted a 6-state Markov model with 4 “response to treatment” health states, an “off-treatment” health state (not displayed in Figure 1), and a “death” state. Response was based on HiSCR, defined by the sponsor as “high response” (HiSCR ≥ 75), “response” (HiSCR 50 to 74), “partial response” (HiSCR 25 to 49), and “nonresponse” (HiSCR < 25). Response was used to determine discontinuation at the end of the induction phase (16 weeks for secukinumab and 12 weeks for adalimumab) and at the end of the maintenance phase (52 weeks for any treatment).
Patients starting on biologics entered the model in the induction phase in the nonresponse health state and could either transition to another response state, remain in the nonresponse health state (but continue on biologics plus SOC drugs), or die. Patients could discontinue biologics and move to the off-treatment state, where they continued to receive only SOC drugs indefinitely. Discontinuation of biologics was assumed to occur depending on a combination of length of time on biologic treatment and the response to treatment using 3 different assumptions:
Induction phase (16 weeks for secukinumab and 12 weeks for adalimumab): Patients with no response or partial response (HiSCR < 50) moved to the off-treatment state and biologics were immediately discontinued; patients with a response or high response (HiSCR ≥ 50) could move to the response, partial response, or high response health states.
Maintenance phase (up to week 52): Patients could continue to move between different levels of response states. Patients who moved to the no or partial response health states continued therapy during the maintenance phase. During the maintenance phase, a flat rate of patients discontinued for reasons unrelated to treatment efficacy; these patients moved to the off-treatment health state. At the end of week 52, all patients in the partial response and no response states discontinued biologics and moved to the off-treatment state.
Week 53 onward: Patients who remained on biologics (in the response and high response states) no longer transitioned between response states (sustained efficacy) and could move to the death or off-treatment state, assuming a flat discontinuation rate applied over time.
Patients starting on SOC entered the model in the induction phase in the no response state and could either transition to another response state, remain in the no response state, or move to the death state. Patients on SOC who moved to the off-treatment state continued to receive SOC drugs indefinitely.
No elevated risk of death was attributed to the condition, treatment, or any other events. Response to treatment, discontinuation, and adverse events (AEs) were assumed to be treatment-dependent. Utilities and resource use were assumed to be health state–dependent. Patients who move to the off-treatment state experience utility values equal to the average of the partial response and no response states.
Model Inputs
The baseline patient characteristics in the sponsor’s model were aligned with the SUNSHINE and SUNRISE trials3 (mean age 36.2 years; 56.3% female).
Clinical efficacy, defined as the proportion of patients in each HiSCR category every 4 weeks, was extracted directly from different trial arms (i.e., no comparative measures of effect were used, such as relative risk or odds ratio from the indirect treatment comparison [ITC]).
For the induction phase, HiSCR data for secukinumab and SOC were pooled from their respective arms from the SUNSHINE and SUNRISE trials.3 For adalimumab, the HiSCR data were taken from the PIONEER trials (weeks 4 to 12, fixed-effects meta-analysis of PIONEER trials from the National Institute for Health and Care Excellence [NICE] adalimumab submission4) and then lowered by 23.5% based on data from Montero-Vilchez et al.,5 assuming the use of biosimilars would lead to worse results. The comparison of adalimumab versus secukinumab was therefore informed by a naive comparison of the PIONEER and the SUNSHINE and SUNRISE trials.
For the maintenance phase, HiSCR data were pooled from the secukinumab arms from the SUNSHINE and SUNRISE trials3 (weeks 16 to 52). For adalimumab, the same data were complemented with data from Jemec et al.6 and, again, efficacy was lowered by 23.5% to assume a lower efficacy of biosimilar adalimumab. In the SOC arm, all patients were moved into the off-treatment state at week 16, where patients continued to receive SOC drugs indefinitely and utility values were assumed to be equal to the average of the partial response and no response states. After week 52, it was assumed patients on biologics who remained in the partial response and no response states would discontinue treatment and transition into the off-treatment state where they would continue to receive SOC drugs indefinitely. It was assumed patients in the high response and response states remain in the same state indefinitely unless they discontinued or died. This was based on an assumption due to a lack of long-term data.
The main reason for discontinuing biologics in the model was due to a lack of clinical response. It was assumed all patients would complete the induction phase of treatment, after which 100% of patients with an HiSCR of less than 50 would discontinue treatment. After the induction phase, it was assumed at 52 weeks that 100% of patients would discontinue if their HiSCR decreased below 50 at this time point. An additional discontinuation rate was considered, separate from the clinical response, which was applied equally across all clinical response states during the maintenance phase. This discontinuation rate would reflect patients discontinuing because of AEs or tolerability of treatment for example. For secukinumab, this rate was sourced from the SUNSHINE and SUNRISE trials3 and was found to be 1.8% per 4-week cycle. For adalimumab, this rate was sourced from the NICE submission4 and inflated by 43% based on data from Roccuzzo et al.7 resulting in a discontinuation rate of 2.63% per 4-week cycle. After week 52, a discontinuation rate for biologics of 0.47% per 4-week cycle (data sourced from Corbett et al.) was applied equally for secukinumab and adalimumab.8 Discontinuation from SOC never occurred, as patients in the off-treatment state in the model continued to receive SOC.
AE data were sourced from the individual arms of the SUNSHINE and SUNRISE trials3 at week 16 for both secukinumab and SOC, and from week 52 of the PIONEER trials from the NICE submission for adalimumab.4
Data on general population mortality in Canada were used to estimate mortality risk in all health states.9
Utilities were dependent on health state (HiSCR ≥ 75 = 0.78; HiSCR 50 to 74 = 0.72; HiSCR 25 to 49 = 0.58; < HiSCR 25 = 0.47) and sourced from the PIONEER trials from the NICE adalimumab submission.4 CDA-AMC notes that EQ-5D data were also collected in the SUNSHINE and SUNRISE trials3 but were not considered in this submission. Patients in the off-treatment state had utility values equal to the average of the partial response and no response states. The disutility of AEs (sourced from a community-based EQ-5D index score for a wide variety of chronic conditions in the UK10) and surgeries (no source provided) were also included.
Costs included drug acquisition costs for secukinumab, adalimumab, and the basket of drugs included in the SOC, self-injection training for biologics, AE costs, and health care resource use costs (concerning nonsurgery- and surgery-related outpatient visits, hospitalizations, wound care visits, and emergency department visits). No administration costs were included for SOC, as care was assumed to be self-administered. SOC costs were also assumed to incur while patients were on any biologic therapy. Relevant costs were inflated to 2023 Canadian dollars. Drug acquisition costs for all drugs were sourced from the Ontario Drug Benefit Formulary.11 AE costs were sourced from the Ontario Case Costing Initiative and calculated as weighted averages based on the estimated relative percentage in each treatment arm.12 Health care resource use costs were dependent on health state occupancy (and independent of treatments received) and their frequency by health state was based on a survey of physicians (n = 40) who actively treated patients in the UK with moderate to severe HS and validated with clinicians in Canada (the same clinicians used in past submissions to NICE4 and CDA-AMC13). Physicians were surveyed regarding the frequency of each type of resource use, stratified by health state and severity, and weighted based on the proportions of patients in each disease severity category, as observed in the PIONEER clinical trials. The cost of these resources was sourced from the Ontario Schedule of Benefits and fees,14 Ontario Case Costing Initiative,12 and the Ontario nurses collective agreement (with benefits and vacation).
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (2,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
Based on a sequential analysis, secukinumab was associated with incremental costs of $116,119 and 0.46 incremental QALYs in comparison with adalimumab, resulting in an ICER of $254,840 per QALY gained (Table 3).
Results were driven by the incremental QALYs gained in higher response states (i.e., there is no difference in mortality across treatments) and drug acquisition costs. Most incremental costs associated with secukinumab are attributed to drug acquisition costs ($162,164), slightly offset by a decrease in health care resource use costs (−$5,520 versus adalimumab; −$9,233 versus SOC). Disaggregated results are presented in Appendix 3 (Table 11, Table 12, Table 13).
Table 3: Summary of the Sponsor’s Economic Evaluation Results (Probabilistic)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
SOC | 159,299 | 16.40 | Reference |
Adalimumab | 195,333 | 16.69 | 124,425 versus SOC |
Secukinumab | 311,452 | 17.15 | 254,840 versus adalimumab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The submitted analysis is based on the publicly available prices for all treatments, including comparator treatments.
Source: Sponsor’s pharmacoeconomic submission, Table 23.1
Sensitivity and Scenario Analysis Results
The sponsor conducted scenario analyses that included a societal perspective, a shorter time horizon (30 years versus lifetime), different discount rates (0%, 3%), excluding costs of SOC drugs while on biologics, assuming a dose escalation of adalimumab, excluding disutilities of AEs or surgeries, and varying the health care resource use (± 25%).
Most of the scenarios did not substantially change the ICERs or the rank of the treatments on the sequential analysis (i.e., adalimumab was still the next best alternative to SOC as first line). The only scenario analysis in which the results changed was the 1 in which an escalation of the adalimumab dose (to 80 mg weekly from week 1) was assumed, resulting in adalimumab being less effective and more costly (dominated) by secukinumab.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Comparative clinical efficacy versus adalimumab is highly uncertain: There is an absence of head-to-head clinical evidence comparing secukinumab with adalimumab for the treatment of HS. The sponsor conducted an ITC to provide comparative clinical effectiveness data. The CDA-AMC Clinical Review Report appraised the submitted ITC and identified the results as inconclusive, showing wide 95% confidence intervals that included the null for all outcomes tested (HiSCR50, AN count, skin pain, flares, and HRQoL) for patients who have and have not been exposed to a biologic. The CDA-AMC Clinical Review concluded it is unclear whether secukinumab is superior, inferior, or has similar efficacy to adalimumab.
Despite the availability of an ITC, results from this analysis were not considered in the sponsor-submitted economic evaluation. The efficacy parameters (% patients in each HiSCR state) were drawn directly from separate studies and arms. More critically, the comparison of adalimumab versus secukinumab was informed by a naive comparison of the PIONEER and SUNSHINE and SUNRISE trials. A naive comparison breaks randomization and does not account for potential differences in patient populations between trials. For example, in the PIONEER trials, placebo response was 26% to 28%. In the SUNSHINE trials, placebo response was 33% to 35%. Therefore, it would be biased and inaccurate to compare absolute response rates, as the difference in response may be attributable to confounding factors. Instead, a comparison of relative differences, as per an ITC, is more appropriate. The sponsor also did not use measures of relative effect, such as relative risk ratios or odds ratios, to inform differences between treatment arms. This makes the probabilistic analysis inaccurate, as it assumes no correlation between response rates among treatments.
Finally, the sponsor assumed biosimilar adalimumab would be worse than originator adalimumab. However, feedback from the clinical experts consulted by CDA-AMC noted the retrospective study used to justify the adjustment of the efficacy parameters for adalimumab is insufficient to suggest that biosimilars are worse than originators. The study that was used to inform this difference was based on a retrospective observational analysis of 17 patients;5 therefore, given the small sample size and inability to adjust for confounding, the evidence was not considered sufficient to confirm a worsening of biosimilar effectiveness. Likewise, the study explored the impact of switching patients from originator adalimumab to biosimilar adalimumab, but did not address any potential perception bias that might have been introduced, depending on how the clinicians presented the switch to the patients (i.e., clinician’s positive or negative attitude toward biosimilars). Given there is no direct head-to-head evidence comparing adalimumab biosimilar with secukinumab, conclusions about comparative efficacy cannot be drawn.
In the CDA-AMC reanalyses, secukinumab and adalimumab were assumed to produce equivalent results, as no evidence was presented that suggests secukinumab is superior to adalimumab. Reanalyses were conducted by assuming adalimumab and secukinumab have equivalent response rates and discontinuation rates. CDA-AMC notes that due to the model structure (adalimumab and secukinumab having different induction periods), it was not possible to ensure QALYs were exactly equivalent between arms.
Structural limitations with the model: In the model, when a patient discontinued therapy they moved into the off-treatment state. Based on the sponsor’s discontinuation rules, patients who do not respond (HiSCR < 25) or partially respond (HiSCR 25 to HiSCR 49) both move to the off-treatment state. In the off-treatment state, patients are assumed to have a utility equal to a weighted average of those in the no response and partial response states. This means when a patient in the partial response state comes off therapy their utility decreases and when a patient in the nonresponse state comes off therapy their utility increases; no justification is provided to support this. The model assumes that an HiSCR score measured before coming off treatment has no impact on future quality of life, i.e., a patient with an HiSCR score of 90 will have the same outcomes as a patient with an HiSCR score of less than 25, once they come off treatment; this assumption is uncertain. The clinical experts consulted by CDA-AMC also noted that patients will come off SOC treatments and then return over time, as these are not meant to be continuously used. Overall, the model structure is not granular enough to fully consider the impact of what will happen to patients in the long-term once they discontinue a biologic.
Second, the sponsor’s model does not allow patients who discontinue secukinumab to move to adalimumab. Given these drugs have different mechanisms of action, a patient who does not respond to secukinumab could be treated with adalimumab instead, given the lack of other approved treatment options. The ability to explore this scenario was not programmed into the sponsor’s model.
Finally, according to feedback from the clinical experts, in provinces that allow for dose escalation, patients with suboptimal response from 52 weeks onward (HiSCR < 50 and/or loss of response), might not discontinue biologics but instead have their dose increased. This would require a model structure that allows the patients to continue moving between response states (with different doses for those with HiSCR < 50) instead of moving them to the off-treatment state.
CDA-AMC could not address the limitations associated with the sponsor’s model structure. The impact of discontinuation on long-term health and cost outcomes is uncertain. Given the lack of comparative head-to-head data comparing adalimumab and secukinumab, addressing this limitation would unlikely influence the conclusions that can be drawn around the cost-effectiveness of secukinumab relative to adalimumab.
Uncertainty regarding the long-term clinical effectiveness of biologics: In the sponsor’s pharmacoeconomic submission, the effects of biologics were assumed to be sustained over the lifetime model horizon (approximately 44 years) after the response assessment at 52 weeks (i.e., patients in high response and response states no longer transition between response states and remain in the response state until discontinuation or death). The potential waning of treatment effect over time in terms of HiSCR was not explored in the sponsor’s model, besides applying a flat annual discontinuation rate to those patients still on treatment (0.47% per 4-week cycle = 6% annually). Feedback from the clinical experts consulted by CDA-AMC noted it may be feasible for treatment benefits to be sustained, but there is limited clinical evidence to support the assumption that it would persist for such a lengthy time horizon for so many patients. One study shows that, for patients with HS on adalimumab, the proportion of patients on treatment at 12 and 24 months was 56.3% and 30.5%, respectively, which was predominantly determined by effectiveness.15
Due to the structure of the submitted model, the only mechanism that can address the sustained response assumption is to assume higher discontinuation rates after 52 weeks. Given there is no evidence to suggest secukinumab has a long-term sustained treatment effect, but there is evidence that shows biologics in HS have a waning effect over time, CDA-AMC conducted a reanalysis where long-term discontinuation rates are higher. CDA-AMC notes this may overestimate cost-effectiveness, as it assumes patients discontinue therapy when efficacy wanes, which the clinical experts noted may not be the case in clinical practice because of the lack of alternatives.
A scenario analysis was conducted that assumed long-term treatment effectiveness and patients remaining on therapy with a HiSCR between 25 and 49.
Cost-effectiveness by treatment line is uncertain: The model assessed a pooled population that included patients who were biologic-naive and those who were biologic-exposed. In the SUNSHINE and SUNRISE trials, approximately one-quarter of patients were biologic-exposed. There is insufficient evidence to conclude whether prior biologic exposure influences the efficacy of secukinumab. In the first-line setting in Canada, as noted by the sponsor and the clinical experts, patients would receive only adalimumab. In the second-line setting, SOC is the only alternative treatment available. Therefore, comparing secukinumab with SOC is more appropriate when considering cost-effectiveness in the second-line setting, and a comparison with adalimumab is more appropriate when considering cost-effectiveness in the first-line setting. Therefore, to be considered cost-effective in both settings, secukinumab would have to be cost-effective against both adalimumab and SOC.
CDA-AMC was unable to address this limitation.
Additionally, the following key assumptions were made by the sponsor and appraised by CDA-AMC (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
HiSCR response rates are assumed to be independent of the patient’s baseline characteristics. | Uncertain. This issue affected both biologic arms equally and, despite the uncertainty, it is unlikely to change the comparative results between secukinumab versus adalimumab. |
Resource use parameters (hospital admissions, surgeries, wound care) are not based on utilization data but on a survey of clinicians. | Uncertain. The source of data and lack of face validity for some cost components (e.g., the number of “visits for wound care NOT due to HS surgery” are more frequent for patients with HiSCR 75 than those with HiSCR 25). This is unlikely to substantively impact the cost-effectiveness results, especially when assuming similar efficacy between biologics, given the current clinical evidence. |
Mortality is assumed to be the same as the general population and the same across treatments—irrespective of health state or response rate. | Likely appropriate and unlikely to substantively impact the cost-effectiveness results, as there is no evidence of a mortality impact associated with treatment. |
Disutility due to surgery was considered in the base case. | Uncertain. The disutility of AEs (sourced from a community-based EQ-5D index score for a wide variety of chronic conditions in the UK10) and surgeries (no source) were both included but could arguably be considered as double-counting, given the assumption on health state–dependent utilities. However, this is unlikely to substantively impact the cost-effectiveness results, as they were also applied in the off-treatment state and the total disutility because of surgeries in total represents only < 1% of the net QALYs across all arms. |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; HiSCR = Hidradenitis Suppurativa Clinical Response; QALY = quality-adjusted life-year.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
Several limitations with the sponsor’s submission could not be adequately addressed (i.e., lack of head-to-head comparative clinical data, uncertainty regarding long-term clinical effectiveness, and lack of sequential treatment with biologics within the model structure design). The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with the clinical experts. CDA-AMC undertook a reanalysis that assumed equal efficacy between secukinumab and adalimumab. Details on the changes made to derive the CDA-AMC reanalysis are presented in Table 5. The summary results of the CDA-AMC reanalyses are presented in Table 6 (disaggregated results are presented in Appendix 4).
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CDA-AMC base case | ||
1. Response to treatment and consequent discontinuation (CDA-AMC base case) | Distribution among HiSCR response states informed by naive comparison (no comparative measures of effect were used); the proportion of patients in each response state were drawn directly from separate arms of the PIONEER and the SUNSHINE and SUNRISE trials. Adjustments to adalimumab efficacy (lowered by 23.5%) and discontinuation (increased by 43%) were made assuming biosimilars have a worse performance than the originator. | Distribution among HiSCR states after induction and at week 52 were assumed to be equivalent between adalimumab and secukinumab. Discontinuation rates for secukinumab and adalimumab in year 1 were assumed the same (based on the secukinumab data). For the SOC arm, the same input values that were submitted by the sponsor for treatment response and discontinuation were maintained. |
2. Long-term treatment efficacy | Assumed outcomes at 52 weeks were permanent until treatment discontinuation. Probability of treatment discontinuation was assumed to be 6% annually (0.47% per 4-week cycle). | Implemented a treatment waning effect by assuming the probability of treatment discontinuation would be 45.8% annually (4.61% per 4-week cycle). |
CDA-AMC = Canada’s Drug Agency; HiSCR = Hidradenitis Suppurativa Clinical Response; SOC = standard of care.
In the CDA-AMC base case, secukinumab was associated with a total cost of $205,374 and 16.58 QALYs compared with $179,816 and 16.57 QALYs for patients receiving adalimumab, and $158,348 and 16.43 QALYs for patients receiving SOC. Based on a sequential analysis, the ICER for secukinumab compared with adalimumab was $2,884,183 per QALY gained. The ICER for secukinumab versus SOC was $321,446 per QALY gained, which is relevant to consider in patients who have tried and discontinued adalimumab. At the current public prices, there is a 0% probability of secukinumab being cost-effective at any willingness-to-pay threshold. Detailed information and disaggregated results are presented in Table 14 in Appendix 4.
Table 6: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results (Probabilistic)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | SOC | 159,612 | 16.49 | Reference |
Adalimumab | 195,648 | 16.78 | 125,206 | |
Secukinumab | 311,512 | 17.24 | 253,438 | |
Reanalysis 1 Adalimumab and secukinumab have the same response and discontinuation rates in year 1 (deterministic) | SOC | 159,612 | 16.49 | Reference |
Adalimumab | 233,832 | 17.21 | 102,941 | |
Secukinumab | 311,512 | 17.24 | 3,237,764 | |
Reanalysis 2 Higher long-term discontinuation rates (deterministic) | SOC | 159,612 | 16.49 | Reference |
Adalimumab | 175,002 | 16.55 | 260,303 | |
Secukinumab | 206,490 | 16.64 | 369,103 | |
CDA-AMC base case (reanalysis 1 + 2; probabilistic) | SOC | 158,348 | 16.43 | Reference |
Adalimumab | 179,816 | 16.57 | 156,349 | |
Secukinumab | 205,374 | 16.58 | 2,884,183 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments.
Scenario Analysis Results
CDA-AMC performed price reduction analyses based on the sponsor base case and the CDA-AMC base-case reanalysis (Table 7). Based on the CDA-AMC base case, a price reduction of approximately 51% would be required for secukinumab to be cost-effective relative to adalimumab. In the model, patients discontinue adalimumab sooner than secukinumab, as adalimumab has a shorter induction period (4 weeks versus 5 weeks); this means patients on secukinumab spend longer on therapy than adalimumab, even if equivalent efficacy is assumed, resulting in higher costs. Relative to SOC, an 81% price reduction would be required for secukinumab to be cost-effective at a threshold of $50,000 per QALY.
Table 7: CDA-AMC Price Reduction Analyses
Analysis | ICERs for SEC versus SOC $/QALY | ICERs for SEC versus ADA (biologic-naive) $/QALY | ||
|---|---|---|---|---|
Price reduction | Sponsor base case | CDA-AMC reanalysis | Sponsor base case | CDA-AMC reanalysis |
No price reduction | 204,162 | 321,446 | 254,840 | 2,844,183 |
10% | 182,402 | 287,929 | 219,251 | 2,298,519 |
20% | 160,643 | 254,413 | 183,662 | 1,752,854 |
30% | 138,883 | 220,896 | 148,072 | 1,207,190 |
40% | 117,124 | 187,379 | 112,483 | 661,526 |
50% | 95,365 | 153,863 | 76,894 | 115,861 |
60% | 73,605 | 120,346 | 41,305 | Dominant (SEC is as effective but less costly than ADA) |
70% | 51,846 | 86,829 | 5,716 | Dominant |
80% | 30,086 | 53,312 | Dominant (SEC is as effective but less costly than ADA) | Dominant |
90% | 8,327 | 19,796 | Dominant | Dominant |
ADA = adalimumab; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; SEC = secukinumab; SOC = standard of care.
CDA-AMC undertook 2 scenario analyses to explore the impact of alternate assumptions on the cost-effectiveness of secukinumab versus adalimumab and SOC for HS to assess uncertainty surrounding some other key assumptions.
SOC costs: CDA-AMC updated the list of drugs included in the SOC (overall regimen daily costs) and the proportion of patients assumed to receive each drug from the SOC basket (Table 8), based on feedback from the clinical experts after reviewing current guidelines.
Continuation of biologic therapy with a partial response over time: The clinical experts noted that in clinical practice, patients may not discontinue biologics upon loss of effect over time because of the lack of treatment alternatives. To explore this scenario, CDA-AMC changed the rules of discontinuation at the end of the induction phase (week 12 for adalimumab and week 16 for secukinumab) and at week 52, so that patients in both arms (adalimumab and secukinumab) stayed on biologic therapy if they had an HiSCR of 25 to 49. Patients with an HiSCR of less than 25 were assumed to discontinue therapy, and the flat annual discontinuation rate for year 2 was reverted to the estimate proposed by the sponsor (6% annually).
The results of these scenario analyses are presented in Table 15 (Appendix 4) and were largely consistent with the CDA-AMC base-case reanalysis and price reduction estimates. CDA-AMC notes that scenario analysis 2 estimates large QALY gains for biologics relative to SOC. This is primarily driven by large utility reductions in patients with HiSCR scores of less than 25 versus those with scores from 26 to 50. There is a considerable amount of uncertainty in the relative differences in utility estimates across HiSCR scores; therefore, the QALY gains in this scenario may be overestimated.
Issues for Consideration
CDA-AMC notes that price negotiations on secukinumab with the pan-Canadian Pharmaceutical Alliance have concluded with a letter of intent arrangement for ankylosing spondylitis, psoriatic arthritis and psoriasis, and moderate to severe plaque.
The product monograph for secukinumab notes a 300 mg form is available; this dose was not included as part of the sponsor’s submission (no price information provided). In terms of environmental impact (e.g., disposal of syringes and pens, footprint with distribution) and convenience for the patients, it would be beneficial to have only 1 injection instead of 2 to achieve the same recommended dose.
The sponsor notes its analysis is for patients with moderate to severe HS that did not respond to conventional therapy. This is narrower than the proposed Health Canada indication, which is for moderate to severe HS with no mention of failure of conventional therapy. However, the sponsor’s analysis utilizes data from the SUNSHINE and SUNRISE (SUNNY) trials and allows a comparison to be made with SOC. Likewise, the clinical experts consulted by CDA-AMC anticipate secukinumab to be used as a second-line systemic drug used after the failure of systemic antibiotics.
Overall Conclusions
Two sponsor-submitted, phase III, randomized, double-blind, placebo-controlled trials, SUNSHINE and SUNRISE, comparing secukinumab with placebo in adult patients (≥ 18 years) with moderate to severe HS demonstrated that 16 weeks of treatment with secukinumab likely results in a clinically important improvement in response to treatment and a reduction in the severity of HS as measured by HiSCR50 response. For secukinumab 300 mg every 2 weeks versus adalimumab 40 mg weekly, the results of a network meta-analysis were inconclusive, showing 95% credible intervals that were wide and included the null for all outcomes tested (HiSCR50, AN count, skin pain, flares, and HRQoL). The economic evaluation was based on secukinumab’s impact on the HiSCR score.
CDA-AMC incorporated the following changes to address the identified limitations for the base case: assuming equivalent response and discontinuation rates between adalimumab and SEC, increasing rates of treatment discontinuation after 1 year to account for potential treatment waning (4.61% per 4-week cycle).
Adalimumab is the only biologic approved in Canada for HS and, therefore, is the most relevant comparator in patients who are being considered for a biologic. In the CDA-AMC base-case analysis, relative to adalimumab, secukinumab was associated with a near-zero QALY gain (< 0.01) but higher costs ($25,558), resulting in an ICER of $2,884,183 per QALY gained. Based on this analysis, a 51% price reduction would be required for secukinumab to achieve cost-effectiveness at a threshold of $50,000 per QALY relative to adalimumab. However, no robust evidence was provided in this submission to indicate that secukinumab has a superior treatment effect relative to adalimumab, and the assumptions informing the cost-effectiveness analysis are uncertain. Therefore, to ensure cost-effectiveness relative to adalimumab, the total drug costs associated with the secukinumab regimen should not exceed the total drug costs associated with the biosimilar adalimumab regimen.
In patients who have already received and discontinued a biologic, SOC is the only treatment option available, as there are no other biologics for HS approved in Canada. Relative to SOC, secukinumab was associated with a higher QALY gain (0.15) and higher costs ($47,026), resulting in an ICER of $321,446 per QALY gained. At a threshold of $50,000 per QALY gained, a price reduction of 81% would be required to ensure cost-effectiveness relative to SOC; however, this assumes that treatment response rate to secukinumab is the same regardless of prior exposure to a biologic.
Outstanding uncertainty remains due to limitations with the sponsor-submitted model, for example, the impact on health gains and costs of titrating the dose of adalimumab upward to achieve a better response were not explored. Likewise, the model did not allow consideration of the patients who would switch to adalimumab after treatment failure with secukinumab. These limitations likely result in the underestimation of both the long-term costs and health impacts of patients who initially start treatment with secukinumab.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Secukinumab (Cosentyx), 300mg, 150mg solution for subcutaneous injection. Montreal (QC): Novartis Pharmaceuticals Canada Inc; 2023 May 10.
2.Cosentyx (secukinumab): 75 mg/0.5 mL, 150 mg/1 mL, 300 mg/2 mL solution for injection, 150 mg powder for solution for injection [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc; 2024.
3.Kimball AB, Jemec GBE, Alavi A, et al. Secukinumab in moderate-to-severe hidradenitis suppurativa (SUNSHINE and SUNRISE): week 16 and week 52 results of two identical, multicentre, randomised, placebo-controlled, double-blind phase 3 trials. Lancet. 2023;401(10378):747-761. PubMed
4.National Institute for Health and Care Excellence. Adalimumab for treating moderate to severe hidradenitis suppurativa. (Technology appraisal guidance TA392) 2016 Jun 22; https://www.nice.org.uk/guidance/ta392. Accessed 2023 May 12.
5.Montero-Vilchez T, Cuenca-Barrales C, Rodriguez-Tejero A, Martinez-Lopez A, Arias-Santiago S, Molina-Leyva A. Switching from adalimumab originator to biosimilar: clinical experience in patients with hidradenitis suppurativa. J Clin Med. 2022;11(4). PubMed
6.Jemec GBE, Okun MM, Forman SB, et al. Adalimumab medium-term dosing strategy in moderate-to-severe hidradenitis suppurativa: integrated results from the phase III randomized placebo-controlled PIONEER trials. Br J Dermatol. 2019;181(5):967-975. PubMed
7.Roccuzzo G, Rozzo G, Burzi L, et al. Switching from adalimumab originator to biosimilars in hidradenitis suppurativa: what's beyond cost-effectiveness? Dermatol Ther. 2022;35(11):e15803. PubMed
8.Corbett M, Soares M, Jhuti G, et al. Tumour necrosis factor-α inhibitors for ankylosing spondylitis and non-radiographic axial spondyloarthritis: a systematic review and economic evaluation. Health Technol Assess. 2016;20(9):1-334, v-vi. PubMed
9.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed by sponsor, no date provided.
10.Sullivan PW, Slejko JF, Sculpher MJ, Ghushchyan V. Catalogue of EQ-5D scores for the United Kingdom. Med Decis Making. 2011;31(6):800-804. PubMed
11.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Jan 30.
12.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2022: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 May 12.
13.CADTH common drug review: pharmacoeconomic review report. adalimumab (Humira -AbbVie Corporation). Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0455_HumiraHS_PE_Report.pdf. Accessed 2024 Sep 5.
14.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 May 12.
15.Prens LM, Bouwman K, Aarts P, et al. Adalimumab and infliximab survival in patients with hidradenitis suppurativa: a daily practice cohort study. Br J Dermatol. 2021;185(1):177-184. PubMed
16.Johnston LA, Alhusayen R, Bourcier M, et al. Practical guidelines for managing patients with hidradenitis suppurativa: an update. J Cutan Med Surg. 2022;26(2_suppl):2s-24s. PubMed
17.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Secukinumab (Cosentyx), 300mg, 150mg solution for subcutaneous injection. Montreal (QC): Novartis Pharmaceuticals Canada Inc; 2023 May 10.
18.Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed by sponsor, no date provided.
19.First Nations and Inuit Health Branch. Non-insured health benefits program: annual report 2020/ 2021. Toronto (ON): Indigenous Services Canada; 2022.
20.PDCI Market Access. Canadian Health Reimbursement and Insurance(CHRIS). Model Version 1.25. Model year 2022. 2022.
21.Cosentyx cumulative patients served. Data on file [internal sponsor's document]. In: Drug Reimbursement Review sponsor submission: Secukinumab (Cosentyx), 300mg, 150mg solution for subcutaneous injection. Montreal (QC): Novartis Pharmaceuticals Canada Inc; 2022.
22.Gill N, Gniadecki R. Incidence and prevalence of hidradenitis suppurativa: a systematic review and meta-analysis [non peer-reviewed preprint]. medRxiv. 2019.
23.Jfri A, Nassim D, O’Brien E, Gulliver W, Nikolakis G, Zouboulis CC. Prevalence of hidradenitis suppurativa: a systematic review and meta-regression analysis. JAMA Dermatol. 2021;157(8):924-931. PubMed
24.HS patient journey study: patient phase report. Prepared for Novartis by MD Analytics [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Secukinumab (Cosentyx), 300mg, 150mg solution for subcutaneous injection. Montreal (QC): Novartis Pharmaceutical Canada Inc; 2022.
25.Choi ECE, Phan PHC, Oon HH. Hidradenitis suppurativa: racial and socioeconomic considerations in management. Int J Dermatol. 2022;61(12):1452-1457. PubMed
26.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage (Appendix C). Ottawa (ON): The Conference Board of Canada; 2017: https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326. Accessed 2023 Jul 10.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on clinical expert feedback. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Moderate to Severe HS
Treatment | Strength | Form | Price ($)a | Recommended dosageb | Daily cost ($)c | Annual cost ($) |
|---|---|---|---|---|---|---|
Secukinumab (Cosentyx) | 75 mg/0.5 mL × 1 | Single-use prefilled syringe or autoinjector (pen) for SC injection | 772.5000d | 300 mg for 5 weeks, 300 mg every 2 weeks thereafter starting at week 6 | Year 1: 138.17 Year 2 and onward: 126.08 | Year 1: 50,465 Year 2 and onward: 46,052 |
150 mg/mL × 1 | 882.5900 | |||||
150 mg/mL × 2 | 1,765.1800d | |||||
Biologics | ||||||
Adalimumab (biosimilars) | 40 mg/0.4 mL × 1 | Single-use prefilled syringe or autoinjector (pen) for SC injection | 471.2700 | 160 mg at week 0, 80 mg at week 2, 40 mg at week 4 and 40 mg weekly thereafter | Year 1: 71.20 Year 2 and onward: 67.32 | Year 1: 26,004 Year 2 and onward: 24,590 |
40 mg/0.8 mL × 1 | 471.2700 | |||||
80 mg/0.8 mL × 1 | 942.5400 | |||||
Systemic antibiotics | ||||||
Clindamycin (generics) | 150 mg | Capsule | 0.2217 | 300 mg twice daily for 12 weeks | 0.89 | 324 |
300 mg | 0.4434 | 0.89 | ||||
Dapsone (generic) | 100 mg | Tablet | 0.7031 | 50 mg to 150 mg twice daily for 12 weeks (assumed 100 mg b.i.d.) | 1.41 | 514 |
Tetracycline (generics) | 250 mg | Capsule | 0.0670 | 500 mg twice daily for 12 weeks | 0.27 | 98 |
Doxycycline (generics) | 100 mg | Tablet | 0.5860 | 100 mg to 200 mg twice daily for 12 weeks (assumed 100 mg b.i.d.) | 1.17 | 428 |
Minocycline (generics) | 50 mg | Capsule | 0.1101 | 100 mg daily for 12 weeks | 0.22 | 78 |
Minocycline (generics) | 100 mg | Capsule | 0.2125 | 100 mg daily for 12 weeks | 0.21 | |
Rifampicin (generics) | 150 mg | Capsule | 0.8460 | 300 mg twice daily for 12 weeks | 3.38 | 973 |
Rifampicin (generics) | 300 mg | Capsule | 1.3317 | 300 mg twice daily for 12 weeks | 2.66 | |
Immunosuppressants | ||||||
Prednisone (generics) | 5 mg | Tablet | 0.0220 | 40 mg/d, taper off 10 mg weekly over 2 to 4 weeks | 0.18 | 64 |
Oral retinoids | ||||||
Isotretinoin (generics) | 10 mg | Capsule | 0.9313 | 40 mg/day to 60 mg/day (assumed 50 mg/day) | 3.77 | 1,379 |
Isotretinoin (generics) | 20 mg | Capsule | 1.89 | |||
Isotretinoin (generics) | 30 mg | Capsule | 2.3734 | |||
Isotretinoin (generics) | 40 mg | Capsule | 1.9003 | |||
Hormone therapies | ||||||
Oral contraceptives (various) | 21 or 28 tabs | Tablet | 6.21 | Assumed Yasmin 21 price | 6.21 | 2,268 |
Pain management | ||||||
Tramadol HCL and acetaminophen | 37.5 mg and 325 mg | Tablet | 0.6264 | 1 or 2 tablets every 4 to 6 hours as needed, to a maximum of 8 tablets daily | 2.5056 | 915 |
Acetaminophen and codeine | 300 mg and 30 mg | Tablet | 0.1300 | 1 or 2 tablets every 4 hours as needed | 0.78 | 285 |
Triamcinolone acetonide (intralesional) | 50 mg/5 mL | Injection suspension | 19.45 | 10 mg/mL is used, with a maximum total dose of 40 mg per treatment session (assumed 10 mg) | 3.89 | 1,421 |
CDA-AMC = Canada’s Drug Agency; SC = subcutaneous; SOC = standard of care.
aAll prices are from the Ontario Drug Benefit Formulary (accessed between June 28 and July 10, 2023, unless otherwise indicated) and do not include dispensing fees; actual prices reimbursed by plans may be lower than those publicly listed or submitted to CDA-AMC.
bDosing for secukinumab and adalimumab is from product monographs. Dosing for other drugs included in the SOC basket are from clinical experts’ feedback after considering the 2022 CDA-AMC guidelines.16
CDaily costs are calculated based on the lowest price of the highest concentration to reach the recommended dose (fewer pills for the patient to ingest).
dHighest sponsor’s submitted price used in the economic evaluation representing the price for Ontario, British Columbia, Nova Scotia, and Newfoundland and Labrador.1 Province-specific prices were provided with the submission and appropriately used in the budget impact analysis.
eMost drugs are not given continuously throughout the year, but this represents the average daily cost over the period of time the patient remains on the drug.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | No | Poor modelling practices were employed (refer to CDA-AMC critical appraisal section). |
Model structure is adequate for decision problem | No | Refer to CDA-AMC appraisal regarding structural limitations |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CDA-AMC appraisal regarding structural limitations, uncertainty around efficacy and treatment lines |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to CDA-AMC appraisal regarding structural limitations, uncertainty around efficacy and treatment lines |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Technical documentation was discrepant with model implementation (refer to CDA-AMC critical appraisal section) for several issues (i.e., undisclosed health state, off-treatment state utilities, double-counting of SOC drug costs in induction period). Poor modelling practices (e.g., key model inputs were sampled for multiple spreadsheets requiring navigating multiple sheets to trace the input value). |
CDA-AMC = Canada’s Drug Agency; SOC = standard of care.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
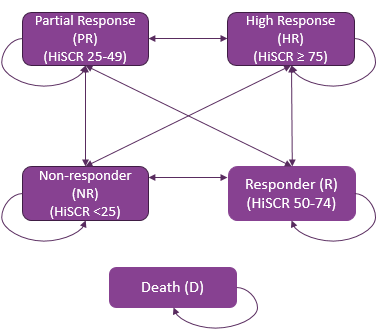
D = death; HiSCR = Hidradenitis Suppurativa Clinical Response; HR = High response; NR = non-responder; PR = partial response; R = responder
Source: Sponsor’s pharmacoeconomic submission.1
Table 10: Four-Week Model Cycle Costs for SOC Treatment for HS
Drug options in SOC | Total costs per 4-week cycle ($) | |
|---|---|---|
% patients using each drug | Total cost per daily dose | |
Oral tetracyclines | 8.6% | 0.07 |
Clindamycin | 8.6% | 0.22 |
Rifampin | 8.6% | 1.22 |
Acitretin | 8.6% | 2.28 |
Isotretinoin | 8.6% | 1.90 |
Dapsone | 8.6% | 0.70 |
Ciclosporin | 8.6% | 0.79 |
Oral prednisone | 5.0% | 0.17 |
Total costs per 4-week cycle | — | $17.53 |
HS = hidradenitis suppurativa; SOC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Mean Probabilistic Discounted Base-Case Costs — Disaggregated
Intervention | Total costs | Drug costs | SOC treatment costs | Administration costs | Resource use costs | AE costs |
|---|---|---|---|---|---|---|
SEC | $311,467 | $162,164 | $6,701 | $57 | $133,334 | $9,210 |
ADA | $195,330 | $39,974 | $7,116 | $57 | $138,854 | $9,329 |
SOC | $159,294 | $0 | $7,549 | $0 | $142,567 | $9,178 |
ADA = adalimumab; AE = adverse events; SEC = secukinumab; SOC = standard of care
Table 12: Mean Probabilistic Discounted Base-Case Life-Years — Disaggregated
Intervention | Total LYs | LYs (HiSCR ≥ 75) | LYs (HiSCR 50 to 74) | LYs (HiSCR 25 to 49) | LYs (HiSCR < 25) | Off-treatment |
|---|---|---|---|---|---|---|
SEC | 32.81 | 2.39 | 0.73 | 0.07 | 0.21 | 29.41 |
ADA | 32.81 | 0.98 | 0.30 | 0.11 | 0.20 | 31.23 |
SOC | 32.81 | 0.03 | 0.05 | 0.03 | 0.21 | 32.49 |
ADA = adalimumab; HiSCR = Hidradenitis Suppurativa Clinical Response; LYs = life-years; SEC = secukinumab; SOC = standard of care.
Table 13: Mean Probabilistic Discounted Base-Case Quality-Adjusted Life-Years — Disaggregated
Intervention | Total QALYs | QALYs | QALYs (HiSCR 50 to 74) | QALYs | QALYs (HiSCR < 25) | Off-treatment | AE disutility |
|---|---|---|---|---|---|---|---|
SEC | 17.15 | 1.87 | 0.52 | 0.04 | 0.10 | 15.39 | 0.77 |
ADA | 16.70 | 0.76 | 0.22 | 0.06 | 0.09 | 16.34 | 0.78 |
SOC | 16.41 | 0.03 | 0.03 | 0.02 | 0.10 | 17.01 | 0.78 |
AE = adverse events; SEC = secukinumab; ADA = adalimumab; AE = adverse events; HiSCR = Hidradenitis Suppurativa Clinical Response; QALYs = quality-adjusted life-years; SEC = secukinumab; SOC = standard of care.
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Scenario Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 14: Disaggregated Summary of CDA-AMC’s Economic Evaluation Results (Probabilistic)
Parameter | SEC | ADA | SOC | Incremental (SEC versus ADA) | Incremental (SEC versus SOC) |
|---|---|---|---|---|---|
Discounted LYs | |||||
Total | 32.82 | 32.82 | 32.82 | 0 | 0 |
Discounted QALYs | |||||
Total | 16.58 | 16.57 | 16.43 | < 0.01 | 0.15 |
By health state | |||||
HiSCR ≥ 75 | 0.38 | 0.37 | 0.03 | < 0.01 | 0.35 |
HiSCR 50 to 74 | 0.13 | 0.12 | 0.03 | < 0.01 | 0.10 |
HiSCR 25 to 49 | 0.04 | 0.04 | 0.02 | < 0.01 | 0.02 |
HiSCR < 25 | 0.10 | 0.09 | 0.10 | < 0.01 | < 0.01 |
Off treatment | 16.70 | 16.72 | 17.03 | −0.02 | −0.33 |
AE disutility | 0.77 | 0.78 | 0.77 | < −0.01 | < 0.01 |
Discounted costs ($) | |||||
Total costs | 205,374 | 179,816 | 158,348 | 25,558 | 47,026 |
Biologic drug costs | 49,033 | 23,350 | 0 | 25,684 | 49,033 |
SOC treatment | 7,261 | 7,271 | 7,550 | −10 | −289 |
Administration | 57 | 57 | 0 | 0 | 57 |
Resource use costs | 139,846 | 139,883 | 141,631 | −37 | −1,786 |
AE costs | 9,185 | 9,264 | 9,176 | −79 | 9 |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SEC = secukinumab, ADA = adalimumab; SOC = standard of care; HiSCR = Hidradenitis Suppurativa Clinical Response; AE = adverse events.
Scenario Analyses
Table 15: Summary of CDA-AMC’s Scenario Analyses Results (Probabilistic)
Drug | Total Costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
CDA-AMC base case | |||
SOC | 158,348 | 16.43 | Reference for SOC comparison |
ADA | 179,816 | 16.57 | Reference for ADA comparison |
SEC | 205,374 | 16.58 | 2,884,183 versus ADA; 321,446 versus SOC |
Scenario 1: SOC costs from update “basket” of drugs | |||
SOC | 162,380 | 16.44 | Reference for SOC comparison |
ADA | 183,885 | 16.57 | Reference for ADA comparison |
SEC | 209,305 | 16.58 | 2,938,622 versus ADA; 326,801 versus SOC |
Scenario 2: HiSCR25 to HiSCR49 remain on biologics at end of induction and week 52 | |||
SOC | 158,380 | 16.41 | Reference for SOC comparison |
ADA | 269,850 | 17.37 | Reference for ADA comparison |
SEC | 382,157 | 17.40 | 3,401,325 versus ADA; 226,069 versus SOC |
ADA = adalimumab; AE = adverse event; CDA-AMC = Canada’s Drug Agency; HiSCR = Hidradenitis Suppurativa Clinical Response; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SEC = secukinumab; SOC = standard of care.
Appendix 5: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 16: Summary of Key Takeaways
Key takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted budget impact analysis assessed the introduction of secukinumab for the treatment of adult patients with moderate to severe HS who have not responded to conventional therapy.17 The analysis took the perspective of CDA-AMC-participating Canadian public drug plans using a top-down epidemiological approach, per province, and incorporated drug acquisition costs. A time horizon of 3 years was taken. The target population size was estimated using the number of adults 18 years and older, percentage covered by public plans, the prevalence of HS, percentage of patients diagnosed with HS, percentage of patients with moderate and severe HS, percentage treated with biologics, percentage of market share of biosimilars and yearly HS population growth. The reference scenario only included adalimumab, assumed 100% biosimilars. The new drug scenario considered the reimbursement of adalimumab and secukinumab. It was assumed that all patients on treatment remain on treatment throughout the time horizon of the model. Treatment switching and discontinuation were not included. Costs for background therapies, including antibiotics and topical treatments, were not included in this analysis. Dispensing fees, mark-ups, and co-pays were included in the base-case analysis. Key inputs to the BIA are documented in Table 18.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
BC: 4,478,159; AB: 3,513,321; SK: 820,591; MB: 999,029 ON: 12,331,423; NB: 665,738; NS: 841,558; NL: 436,613 PE: 138,517; NIHB: 669,990 | |
% covered by public plans20 | BC: 41%; AB: 39%; SK: 34%; MB: 35%; ON: 48%; NB: 35% NS: 39%; NL: 39%; PE: 40%; NIHB: 100% |
HS Prevalence3 | 1% |
Percentage of patients diagnosed with HS4 | 19.0% |
Percentage with moderate and severe HS (Hurley stage 2 or 3)21 | 56% |
Proportion treated with a biologic21 | 14% |
Adalimumab biosimilar | 100% |
Yearly Patient Population Growth (incidence)22 | 2.85% |
Number of patients eligible for Cosentyx Existing patients New patients Total patients | Base-year / year 1 / year 2 / year 3 1,676 / 1,723 / 1,771 / 1,820 48 / 48 / 49 / 50 1,723 / 1,771 / 1,820 / 1,871 |
Market share (4 years) | |
Market share (reference scenario) Existing patients Adalimumab New patients Adalimumab | Base-year / year 1 / year 2 / year 3 100% / 100% / 100% / 100% 100% / 100% / 100% / 100% |
Market share (new drug scenario) Existing patients Adalimumab Secukinumab New patients Adalimumab Secukinumab | Base-year / year 1 / year 2 / year 3 100% / 46% / 41% / 39% 0% / 54% / 59% / 61% 100% / 69% / 63% / 59% 0% / 31% / 37% / 41% |
Annual cost of treatment excluding mark-up and dispensing fees (per patient)a | |
Adalimumab (First-year/ Maintenance) Secukinumab (First-year/ Maintenance) | $25,919 / $24,506 $50,307 / $45,895 |
AB = Alberta; BC = British Columbia; MB = Manitoba; NB = New Brunswick; NL = Newfoundland and Labrador; NS = Nova Scotia; ON = Ontario; PE = Prince Edward Island; QC = Quebec; SK = Saskatchewan; NIHB = Non-Insured Health Benefits.
aOntario price. Provinces have specific prices, mark-ups, dispensing fees, and co-payments. Provincial-specific estimates were used in the model. Annual costs were calculated based on the monograph doses for each biologic, including loading doses.
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget impact of funding secukinumab for adult patients with moderate to severe HS (existing and new patients; first or second-line treatment) is $26,122,088 in year 1, $24,572,772 in year 2, and $25,848,133 in year 3, for a 3-year total of $76,542,993.
In the sponsor’s scenario analyses, varying (lowering or increasing) several parameters and scenario assumptions (i.e., percentage covered by the public plan, percentage of patients diagnosed with HS, percentage of patients with moderate and severe HS, percentage treated with biologics, prevalence, excluding dispensing fees and co-pays) resulted in the increased costs to the drug plans over 3 years to be between $29,095,917 and $145,417,781. The only scenario analysis in which funding secukinumab was cost-saving (saved $3,938,896 over 3 years) is when assumed that all patients receiving adalimumab received higher doses (80mg weekly, doubled the recommended in the monograph).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the results of the BIA:
Prevalence and diagnosis rate of HS is uncertain. The proportion of patients with diagnosed HS who receive a biologic is uncertain. In the sponsor’s BIA prevalence of HS is estimated to be 1% and of those 19% are expected to receive a diagnosis resulting in a prevalence of diagnosed HS of 0.19%. Finally, it was assumed 56% have moderate/severe HS and 14% will receive a biologic. Estimates from the literature show the prevalence of HS is highly variable with some estimates being as low as 0.03% and others being as high as 4%.23 The sponsor’s prevalence estimate was taken from a study that notes uncertainty in the true prevalence estimate. The proportion of diagnosed cases was taken from a 2016 NICE technology appraisal report based on market research conducted by Abbvie consisting of an online survey of 60 respondents (30 dermatologists, 15 general surgeons, and 15 plastic surgeons). The data for the 14% of biologic use were based on private market research conducted by Novartis on 39 patients (performed in Canada24).
Clinical experts consulted for this review noted that there is a large degree of uncertainty regarding HS prevalence and diagnosis rates in Canada. However, the number of patients on biologic treatment for HS was seen as less uncertain given the small number of patients treated in clinical practice. In British Columbia, it was expected around 100 patients are currently on a publicly funded biologic and an additional 20 patients may have tried adalimumab in the past and discontinued. This would indicate that either the proportion of patients with HS who receive a biologic is very low or the prevalence and/or diagnosis rate is substantially lower than 1% and 19%, respectively. It was noted that low biologic uptake may be due to the low availability of dermatologists and aversion by some to prescribe a biologic. However, this may be different from province to province.
In the CDA-AMC base case, biologic uptake was assumed to be 3.5% while a prevalence rate of 1% and a diagnosis rate of 19% were maintained. The 3.5% estimate was back-calculated by assuming approximately 120 patients in British Columbia received adalimumab for HS and public coverage rates in British Columbia are 73%.
It was noted that uptake in other provinces may be higher depending on access to dermatologists. Therefore, a scenario analysis was conducted that increased the size of the population on a biologic by 50%.
Public coverage rates are uncertain. To estimate the proportion of patients who would be eligible for public coverage, the sponsor estimated average public coverage rates for patients older than 18 years. For patients older than 18 years, the sponsor looked at the number of patients older than 65 years, on social assistance, or those without a primary source of private insurance. According to clinical experts, it was noted that HS patients are more likely to be from a lower socioeconomic status.25 Therefore, patients may be more likely to be on social assistance or not have private insurance relative to the Canadian average. Second, patients older than 65 years make up a very small proportion of patients with HS. This was evidenced from the SUNSHINE and SUNRISE trials that showed patients older than 65 years made up less than 2% of the trial population. The sponsor’s calculation of public coverage rates was driven primarily by patients older than 65 years.
CDA-AMC used the “Understanding the gap” report26 to estimate public coverage rates. The report highlights the proportion of patients in each jurisdiction who are enrolled in a public program, eligible for a public program, or have private insurance, stratified by age. CDA-AMC assumed all patients enrolled in a public plan would have this treatment publicly covered. CDA-AMC also assumed all without private insurance would also receive public coverage. Based on the SUNRISE/SUNSHINE trials, CDA-AMC assumed 30% of patients would be under the age of 24 and 69% would be between 25 to 64 and 1% would be 65 and older.
CDA-AMC acknowledges this may still be an underestimate as it assumes private coverage in the HS population matches the general population in Canada. This also assumes that private insurance will always be the first payer. Some programs, such as trillium in Ontario, still provide public coverage if private insurance does not cover 100% of prescription drug costs. A scenario analysis was conducted that assumed 50% public coverage for plans that had an estimated public coverage rate below this.
Growth in the population receiving biologics was underestimated. At the start of the analysis, it is assumed all prevalent patients who will ever receive a biologic will have already received one. Only patients who have yet to receive a diagnosis of HS will have no experience with a biologic. This may underestimate the size of the population base of patients who have a current diagnosis of HS and have yet to receive a biologic. Clinical experts consulted by CDA-AMC noted that there is expected to be a year-to-year growth of 10% for treatment with biologics based on uptake of adalimumab in HS since 2018.
CDA-AMC assumed a 10% growth rate in the size of the population receiving biologics each year.
Switching from patients on adalimumab is overestimated. The sponsor assumes after 3 years 61% of patients currently on adalimumab will have switched over to secukinumab. According to clinical experts for this review given there is no head-to-head evidence comparing the 2 therapies it is unlikely that such a large number of clinicians would recommend patients switching over to a new therapy. Clinical experts noted that in patients who have no response to adalimumab who also do not respond to a higher dose then a switch to secukinumab may be warranted.
CDA-AMC assumed 30% of patients will have switched to secukinumab from adalimumab after 3 years starting with 10% in year 1 with a linear uptake.
Costs associated with biologic use are overestimated. The sponsor assumes that patients will remain on biologics indefinitely with no patients discontinuing therapy for any reason. In the submitted economic analysis it is assumed that after 12 weeks, individuals with an HiSCR score of less than 50 will discontinue therapy. Likewise, at 52 weeks it was assumed any patients whose HiSCR score has dropped below 50 will discontinue. Clinical experts consulted by CDA-AMC noted that due to the lack of alternatives patients are unlikely to discontinue once they start on adalimumab. If strict adherence to response rates from the trial are enforced, then discontinuation rates will likely be around 50% after 1 year.
CDA-AMC was unable to explore the impact of this as a scenario analysis due to uncertain programming of the sponsors BIA which relied on macros that overrode user inputs. If patients discontinue therapy, then this will decrease the costs associated with secukinumab. However, patients may switch to adalimumab after treatment failure with secukinumab which would increase the budget impact of having an additional biologic.
The sponsor’s BIA does not reflect current practice. First, patients who do not respond to adalimumab may have their dose titrated up to 80 mg every week to achieve a better response. The sponsor explored this but only by allowing increasing the adalimumab dose for all patients rather than just those who do not respond. Second, if funded in the proposed indication then patients who fail on secukinumab could switch to adalimumab as it is the only alternative treatment option available. The submitted BIA does not allow consideration of this. Finally, the sponsor’s BIA does not include the SOC. Evidence shows that some patients do not respond to biologic therapy, and some discontinue due to AEs. In the sponsor’s base case, they assume 100% of patients who received adalimumab would remain on it and not switch to the SOC. In existing patients, the SOC is a relevant comparator for some patients who tried and failed to respond on adalimumab and these patients would likely switch to secukinumab given there are no other indicated therapies for HS.
CDA-AMC could not address these limitations. These factors will not influence the model’s ability to estimate the costs associated with secukinumab acquisition costs but do prohibit an accurate estimation of the budget impact relative to current practice.
The sponsor’s BIA is narrower than the proposed Health Canada population. The proposed Health Canada population is for the treatment of adult patients with moderate to severe HS. However, the sponsors BIA notes the analysis is focused on patients for the treatment of adult patients with moderate to severe HS who have not responded to conventional therapy. This is technically a narrower population than the full Health Canada indication. However, CDA-AMC notes the sponsor applies no limiting criteria to only consider patients who fail conventional therapy. It is assumed of those with moderate to severe HS only 14% receive a biologic but this was considered an overestimate by the experts. As the sponsor does not include SOC as a comparator in the analysis if the approved indication was broader than that of adalimumab the BIA could not accurately address this.
CDA-AMC could not address this limitation. Clinical experts consulted by CDA-AMC anticipate secukinumab to be used as a second-line systemic drug used after failure of systemic antibiotics, so the BIA aligns with anticipated uptake of secukinumab.
CDA-AMC Reanalyses of the BIA
Table 18: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
Excluded dispensing fees, mark-ups, and co-pays | ||
Changes to derive the CDA-AMC base case | ||
Reanalysis 1: Population size | Assumed 14% of patients receive a biologic | Assumed 120 in BC receive or have received a biologic. From this number, back calculated a biologic uptake rate of 3.5% which was then applied to all provinces |
Reanalysis 2: Coverage rates | Assumed average public coverage rates apply to HS patients inclusive of patients older than 65 years. British Columbia: 41% Alberta: 39% Saskatchewan: 34% Manitoba: 35% Ontario: 48% New Brunswick: 35% Nova Scotia: 38% Prince Edward Island: 40% Newfoundland and Labrador: 39% NIHB: 100% | Assuming the greater of the proportion of patients enrolled in a public program and the proportion of patients without private insurance, calculated using data from the “Understanding the gap” report26 AND Assuming 30% of patients would be younger than 24 years, 69% would be between 25 and 64 years, and 1% would be older than 65 years, as per the SUNNY trials. British Columbia: 73% Alberta: 35% Saskatchewan: 46% Manitoba: 57% Ontario: 46% New Brunswick: 27% Nova Scotia: 27% Prince Edward Island: 28% Newfoundland and Labrador: 29% NIHB: 100% |
Reanalysis 3: Biologic growth | Assumed the population receiving a biologic grows by 2.5% every year | Assumed the population receiving a biologic grows by 10% every year |
Reanalysis 4: Switching from adalimumab to secukinumab | Proportion of patients switching from adalimumab to secukinumab:
| Proportion of patients switching from adalimumab to secukinumab:
|
CDA-AMC base case | Correction + reanalysis 1 + 2 + 3 + 4 | |
BC = British Columbia; HS = hidradenitis suppurativa; NIHB = Non-Insured Health Benefit.
Note: All reanalyses are performed after including all corrections.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 19 and a more detailed breakdown is presented in Table 20. In the CDA-AMC base case, the 3-year budget impact of reimbursing secukinumab for adult patients with moderate to severe HS is expected to be $9,547,349 (year 1: $1,717,030; year 2: $3,091,377; year 3: $4,738,942).
Table 19: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total budget impact |
|---|---|
Submitted base case | $76,542,993 |
CDA-AMC correction (removing dispensing fees, mark-ups, and co-pays) | $73,199,136 |
CDA-AMC reanalysis 1 — population size | $18,299,784 |
CDA-AMC reanalysis 2 — public coverage rates | $80,769,500 |
CDA-AMC reanalysis 3 — growth in biologic use | $87,011,386 |
CDA-AMC reanalysis 4 — proportion of patients switching from adalimumab | $27,056,308 |
CDA-AMC base case (correction + reanalysis 1 + 2 + 3 + 4) | $9,547,349 |
BIA = budget impact analysis
Note: Analysis is based on the publicly available prices of the comparator treatments.
CDA-AMC also conducted additional scenario analyses to address remaining uncertainty, using the CDA-AMC base case. Results are provided in Table 20.
Assuming 50% public coverage for plans that had an estimated public coverage rate below this (AB, SK, ON, NB, NS, NFL, PEI)
Assuming a 50% increase in the size of the population on a biologic therapy if broader access to dermatologists (from 3.5% to 5.25%)
Table 20: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $44,767,147 | $46,004,702 | $47,280,566 | $48,591,700 | $141,876,968 |
New drug | $44,765,516 | $72,126,790 | $71,853,338 | $74,439,833 | $218,419,961 | |
Budget impact | $0 | $26,122,088 | $24,572,772 | $25,848,133 | $76,542,993 | |
CDA-AMC base case | Reference | NR | $13,475,113 | $14,710,970 | $16,058,481 | $44,244,563 |
New drug | NR | $15,192,143 | $17,802,346 | $20,797,423 | $53,791,913 | |
Budget impact | NR | $1,717,030 | $3,091,377 | $4,738,942 | $9,547,349 | |
CDA-AMC scenario analysis 1 — minimum 50% public coverage | Reference | NR | $14,936,829 | $16,306,735 | $17,800,418 | $49,043,982 |
New drug | NR | $16,835,550 | $19,724,584 | $23,039,464 | $59,599,598 | |
Budget impact | NR | $1,898,721 | $3,417,849 | $5,239,046 | $10,555,616 | |
CDA-AMC scenario analysis 2 — larger population accessing biologics | Reference | NR | $20,212,669 | $22,066,454 | $24,087,721 | $66,366,845 |
New drug | NR | $22,788,215 | $26,703,519 | $31,196,134 | $80,687,869 | |
Budget impact | NR | $2,575,546 | $4,637,065 | $7,108,413 | $14,321,024 |
BIA = budget impact analysis; NR = not reported using the sponsor-provided model.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.