CADTH Reimbursement Review
Maralixibat (Livmarli)
Sponsor: Mirum Pharmaceuticals Inc.
Therapeutic area: Alagille syndrome
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Clinical Review
Abbreviations
AE
adverse event
ALGS
Alagille syndrome
ALGSA
Alagille Syndrome Alliance
ALP
alkaline phosphatase
ALT
alanine transaminase
ANCOVA
analysis of covariance
CI
confidence interval
CLF
Canadian Liver Foundation
C-statistic
concordance statistic
CSS
Clinician Scratch Scale
EFS
event-free survival
FSV
fat-soluble vitamin
GALA
Global ALagille Alliance
GGT
gamma-glutamyl transferase
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HR
hazard ratio
HRQoL
health-related quality of life
IBAT
ileal bile acid transporter
IQR
interquartile range
ItchRO
Itch Reported Outcome
ItchRO(Obs)
Itch Reported Outcome (observer)
ItchRO(Pt)
Itch Reported Outcome (patient)
ITT
intent to treat
LOCF
last observation carried forward
LS
least squares
LTE
long-term extension
MID
minimal important difference
mITT
modified intent to treat
PedsQL
Pediatric Quality of Life Inventory
RCT
randomized controlled trial
SAE
serious adverse event
sBA
serum bile acid
SBD
surgical biliary diversion
SD
standard deviation
TFS
transplant-free survival
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Maralixibat (Livmarli), 9.5 mg of maralixibat per mL, oral solution |
Sponsor | Mirum Pharmaceuticals Inc. |
Indication | For the treatment of cholestatic pruritus in patients with Alagille syndrome |
Reimbursement request | As per indication |
Health Canada approval status | Post-NOC |
Health Canada review pathway | Priority review |
NOC date | July 21, 2023 |
Recommended dose | The recommended maintenance dose is 380 mcg/kg once daily in the morning after 1 week of a starting dose of 190 mcg/kg orally once daily. The maximum daily dose in volume for patients above 70 kg is 3 mL. |
NOC = Notice of Compliance.
Introduction
Alagille syndrome (ALGS) is a rare, life-threatening, genetic, complex, multisystem disorder that presents with a range of clinical features, including cholestatic liver disease, failure to thrive, cardiovascular disease, skeletal abnormalities, ocular abnormalities, renal and vascular abnormalities, and distinct facial features.1,2 In most cases, the liver dysfunction associated with ALGS is an early and a serious feature of this genetic condition and typically presents in the first 3 months of life. Elevated levels of serum bile acids (sBAs) and jaundice (elevated bilirubin) are hallmarks of ALGS and indicate the presence of impaired bile flow.3 Clinically important manifestations of cholestasis in ALGS include debilitating and intractable pruritus, disfiguring xanthomas, sleep disturbances, chronic debilitating fatigue, and failure to thrive (i.e., insufficient growth).4,5 ALGS is predominantly caused by mutations in the Jagged-1 gene in greater than 90% of cases6 and in the NOTCH2 gene in approximately 4% of cases, resulting in bile duct paucity, bile flow obstruction, and bile accumulation in the liver. ALGS is inherited in an autosomal-dominant pattern. Cholestasis is defined as a decrease in bile flow due to impaired secretion by hepatocytes or to obstruction of bile flow through intrahepatic or extrahepatic bile ducts.7 Cholestasis leads to hepatic and systemic accumulation of substances normally excreted via the biliary tract, such as bile acids and conjugated bilirubin. Elevated levels of bile acids are hepatotoxic and contribute to disease progression. Bile acids have been shown to induce damage and necrosis in hepatocytes and cholangiocytes and are associated with increased morbidity and mortality in chronic cholestatic diseases.7 The systemic and hepatic accumulation of bile acids and other toxins leads to incapacitating and chronic cholestatic symptoms such as pruritus, and clinical sequelae.
The clinical manifestations of cholestasis associated with ALGS are severe, even in the absence of liver disease, with cholestatic pruritus being the leading cause of liver transplant in patients.8-10 These clinical manifestations present in the first few years of life and as early as 3 months of age. The symptoms include severe and unremitting pruritus (74%),10 xanthomas (disfiguring and sometimes disabling subcutaneous lipid deposits, 40%), chronic fatigue (between 65% and 85%), and growth failure (between 50% and 87%11). In addition, fat-soluble vitamin (FSV) malabsorption and increased risk of bone fractures because of trabeculae malformation can also be present. A second wave of portal hypertension and associated complications occurs later in adolescence.6 Collectively, all cholestasis-related symptoms result in poor health-related quality of life (HRQoL). As cholestasis progresses and symptoms worsen, as described previously, a majority of patients will either receive a liver transplant (50.4%) or die (9.3%) by 18 years of age, with only 40.3% of patients reaching adulthood with their native liver.6,10 The estimated 20-year life expectancy is 75% for patients diagnosed with ALGS, 80% for patients with ALGS who do not require liver transplant, and 60% for patients with ALGS who require liver transplant.12 For patients with ALGS who undergo liver transplant, the estimated 1-year survival rate is 87%.13 The majority of early liver transplantations occur because of complications associated with cholestasis, including pruritus. The reported incidence of ALGS is 1 in 30,000 to 50,000 births.3,11 In the absence of Canadian statistics, the sponsor estimated prevalence (based on 1 in 30,000 live births) to be a total of 1,032 patients with ALGS in 2023.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of maralixibat (Livmarli), 9.5 mg/mL, oral solution, for the treatment of cholestatic pruritus in patients with ALGS aged 2 months and older.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to the CADTH call for input and from clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, the Canadian Liver Foundation (CLF) and the Alagille Syndrome Alliance (ALGSA), provided input. The CLF is the only national health charity committed to supporting people in Canada affected by liver diseases. Based in the US, the ALGSA is a nonprofit organization dedicated to supporting families affected by ALGS globally. The CLF submission included phone and virtual interviews conducted in May 2023 with 8 patients and caregivers in Canada. Of those, 4 respondents had experiences with maralixibat through clinical trials. The ALGSA gathered data online through family surveys (2020), personal conversations, and topic-specific discussions among support or focus groups, including at least 76 members in Canada. Both groups stated that the itchiness (pruritus) is the most bothersome symptom affecting patients’ and caregivers’ lives. For example, the itchiness interrupts patients and families’ sleep, making those affected fatigued, anxious, depressed, irritable, and worried. Patients said they feel isolated at school and that it is challenging to maintain employment. Also, patients and families have difficulty finding a specialist who could recognize and make a proper diagnosis of ALGS and manage disease treatment. Patients and families from both groups want a new therapy that can provide significant relief of itchiness with long-term effects without high risks such as liver transplant and immunosuppression. Patients who have taken maralixibat during clinical trials said that their itchiness has been resolved with minor side effects, such as upset stomach and diarrhea; that they felt more like their true self and were able to engage in normal day-to-day activities; and that their households were also positively affected.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical expert panel stated that cholestatic pruritus remains a very significant management problem for patients with ALGS and their families, due to partial, incomplete, or null response to currently available treatments. Current treatments are used off-label and are supportive in nature. The experts noted that surgical options such as an external or internal biliary diversion can be offered to patients with ALGS with cholestatic pruritus refractory to medical therapies; however, these are not very effective and seldomly used in clinical practice. Finally, the experts stated that between 50% and 75% of patients with cholestatic liver disease will require a liver transplant and that cholestatic pruritus is a leading indication for a transplant. Liver transplant is associated with increased morbidity, mortality, and lifelong immune suppression. As such, the experts noted that there is an unmet need for effective symptomatic and curative treatment for cholestatic pruritus in the indicated patient population.
The clinical experts stated that maralixibat would likely be used in combination with current off-label treatments in patients experiencing ongoing pruritus, and that it is possible some patients could discontinue some of the off-label treatments once they are established on maralixibat and their pruritus is under control. The experts noted that, if easily accessible, maralixibat may be used as an initial therapy for new patients presenting with severe pruritus. The clinical experts stated that the estimated incidence of ALGS in Canada is about 1 in 30,000 to 50,000 live births, with about 200 new cases each year. The experts noted that pediatric patients with ALGS most suited for treatment with maralixibat are those who present with cholestatic pruritus that is persistent with current off-label treatments, which makes up about a third of patients in a clinical expert’s practice. Patients least suited to treatment with maralixibat are those who have minimal liver involvement (i.e., minimal liver enzyme abnormalities and no FSV deficits) and patients who do not experience cholestatic pruritus.
According to the expert panel, a clinically meaningful response to treatment would include a reduction in the frequency and severity of pruritus, a reduction in sleep deprivation among patients and their caregivers, the ability for patients and their caregivers to attend school or work, reduced damage to the patients’ skin, and improved patient weight and growth. The clinical experts consulted on this review noted that response to therapy would likely be evaluated via subjective family reporting of symptoms including itching and sleep disturbances as well as by visual assessments of excoriations on the patient’s skin, which are often indicative of severe cholestatic pruritus. Standard scratch scales are not commonly used in clinical practice, according to the experts. Measurements of sBA could be used to assess response to therapy; however, the experts noted that this is not common in clinical practice due to the high cost and limited availability of such testing in some practice settings. The clinical experts would initially assess patients monthly for approximately 3 months, at which time the frequency of visits would be reduced to every 3 to 6 months if a response to treatment is evident. The clinical experts stated that treatment with maralixibat will likely be lifelong for most patients. The panel noted that treatment discontinuation may be considered if there is no effect on cholestatic pruritus after approximately 6 months of treatment initiation, if a patient’s liver disease progresses and they undergo liver transplant, or due to serious adverse events (SAEs); however, the experts stated that AEs associated with maralixibat are likely self-limited and may be addressed by titrating the dose of maralixibat. The clinical experts noted that a pediatric or adult liver or gastrointestinal specialist would be the preferred specialist to prescribe and monitor treatment with maralixibat.
Clinician Group Input
One clinician from the Canadian Association for the Study of the Liver provided input. The clinician group and 2 clinical experts consulted by CADTH agree on the unmet need, which is a lack of effective therapy specifically indicated for cholestatic pruritus associated with ALGS refractory to current off-label treatments. They also agree that all the existing therapies are not effective at reducing cholestatic pruritus associated with ALGS and that there are no guidelines for treating cholestatic pruritus in patients with ALGS. In alignment with clinical experts, the clinician group stated that treatment goals are mainly improvement in pruritus, improvement in quality of life (i.e., sleep duration), and optimizing nutritional goals (i.e., treating FSV deficiency). Also, both groups agree that patients with ALGS and cholestatic pruritus that is persistent on standard-of-care medical treatment would be an eligible population. The clinician group stated that if a patient’s liver disease progresses and they undergo liver transplant, discontinuation is considered and the clinical experts stated that if there is no effect on itch as measured clinically, then discontinuation is considered after adequate trial — i.e., 6 months. Otherwise, both groups agree that adverse events (AEs) would be an unlikely reason to discontinue since maralixibat is well tolerated. Lastly, all the clinician group and clinical experts agree that maralixibat should be prescribed by a pediatric gastroenterologist or hepatologist. None of the clinician group or clinical experts consulted by CADTH had declared experience with maralixibat.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The drug plans identified implementation issues related to considerations for initiation of therapy and continuation or renewal of therapy. The clinical expert consulted by CADTH weighed evidence from the pivotal trial and other clinical considerations to provide responses to the drug program’s implementation questions. Refer to Table 6 for more details.
Clinical Evidence
Systematic Review
Description of Studies
The pivotal LUM001-304 (ICONIC) trial was an open-label, phase IIb study to evaluate the safety and efficacy of maralixibat in children with ALGS between the ages of 1 and 18 years. A total of 31 patients enrolled into the study, which was conducted at 10 clinical sites in Australia, Europe, and the UK between November 25, 2014, and September 11, 2015. The study comprised an 18-week, open-label run-in period during which all patients received maralixibat, up to 380 mcg/kg/day (0 weeks to 18 weeks); a 4-week, randomized, double-blind, placebo-controlled drug-withdrawal phase (weeks 18 to 22), during which 13 patients continued receiving active treatment while 16 patients shifted to placebo; followed by a 26-week stable-dosing period (weeks 23 to 48), during which all patients received active treatment at doses up to 380 mcg/kg/day; and an optional long-term treatment period. It should be noted that during the long-term extension (LTE) phase (as of week 103) eligible patients could have received a dose of maralixibat of up to 760 mcg/kg/day (given as twice-daily doses of 380 mcg/kg) which is outside of the proposed Health Canada indication of 380 mcg/kg/day. As such, efficacy and safety data after this period is not aligned with the recommended dose. Assessed efficacy outcomes included change in sBA, change in pruritus assessed using the Itch Reported Outcome (ItchRO) observer (Obs) and patient (Pt) tools, change in liver biomarkers and enzymes (alanine transaminase [ALT], alkaline phosphatase [ALP], total and direct bilirubin), change in body height and weight z scores, and HRQoL as measured by the Pediatric Quality of Life Inventory (PedsQL) total score (parent) and the PedsQL Multidimensional Fatigue Scale score (parent). Assessed harms included AEs such as diarrhea, abdominal pain, and FSV deficiency; and SAEs.
In the overall study population (N = 31), there were more males (19 of 31 [61.3%]) than females (12 of 31 [38.7%]) at baseline and in the maralixibat (9 of 13 [69.2%]) and placebo groups (n = 10 of 16 [62.5%]) during the randomized withdrawal (RWD) period. The mean age in the overall study population was 5.4 years (range, 1 to 15 years) and was similar between the maralixibat and placebo groups. Most patients were from Australia and France (9 of 31 [29.0%] each) in the overall study population. ||| |||| |||| ||||| ||| |||||||| ||||||||| || |||| ||| |||| |||||| || ||| ||||||| ||||| ||||||||||| |||| |||| |||||| || ||||||||||| ||||| ||| |||| |||||| || ||| || ||||||| ||||| |||||| ||| |||||||||| |||||||||| |||||| In the overall study population, 8 of 31 (25.8%) of patients had a family history of ALGS (1 of 13 [7.7%] and 7 of 16 [43.8%] in the maralixibat and placebo groups, respectively). All enrolled patients had the Jagged-1 mutation present. Race and ethnicity data were not collected in the ICONIC trial.
Efficacy Results
In the ICONIC trial, the primary efficacy end point was the change in sBA during the 4-week RWD phase in the modified intent-to-treat (mITT) population (patients with sBA reduction ≥ 50% at week 12 or 18). A total of 15 participants were in the mITT population and were analyzed in the primary end point (5 randomized to maralixibat; 10 to placebo). The least squares (LS) mean difference in change from weeks 18 to 22 in sBA between the maralixibat and placebo groups was –117.28 (95% confidence interval [CI], –211.699 to –23.103; P = 0.0464) μmol/L, in favour of maralixibat. A consistent difference was observed in the overall randomized intent-to-treat (ITT) population.
In the ICONIC pivotal trial, the change from weeks 18 to 22 in ItchRO(Obs) weekly average morning severity score was a secondary end point. The LS mean difference between the maralixibat and placebo groups was –1.48 (95% CI, –2.12 to –0.84; P < 0.0001), in favour of maralixibat. In the overall population, there was a decrease (improvement) in ItchRO(Obs) weekly average morning severity score from baseline to week 18 (secondary end point) with a mean change of –1.70 (95% CI, –2.05 to –1.36; P < 0.0001) and from baseline to week 48 (additional end point) with a mean change of –1.62 (95% CI, –2.12 to –1.12; P < 0.0001). The prespecified sensitivity analyses for ItchRO(Obs) weekly average morning severity score was consistent with the results of the ItchRO(Obs). A total of 14 patients met the age cut-off for completion of the ItchRO(Pt) (≥ 9 years of age or ≥ 5 years of age with the assistance of their caregiver) in the pivotal trial. The LS mean difference between the maralixibat and placebo groups from weeks 18 to 22 for the change in ItchRO(Pt) weekly average morning severity score was –1.98 (–3.01 to –0.97; P = 0.0013), in favour of maralixibat. In the overall population, there was a decrease (improvement) in ItchRO(Pt) weekly average morning severity score from baseline to week 18 (secondary end point) with a mean change of –2.07 (95% CI, –2.65 to –1.50; P < 0.0001) and from baseline to week 48 (additional end point) with a mean change of –2.25 (95% CI, –2.84 to –1.67; P < 0.0001).
From weeks 18 to 22, the LS mean difference between the maralixibat and placebo groups for ALP was 10 (95% CI, –52.6 to 72.6; P = 0.7455) U/L compared with placebo. From weeks 18 to 22, the LS mean difference between treatment groups for ALT was 15.1 (95% CI, –25.1 to 55.2; P = 0.4472) U/L. From weeks 18 to 22, the LS mean difference between the maralixibat and placebo groups for total bilirubin was –0.14 (–0.88 to 0.60; P = 0.7000) mg/dL. From weeks 18 to 22, the LS mean difference between the maralixibat and placebo groups for direct bilirubin was –0.02 (95% CI, –0.56 to 0.53; P = 0.9517) mg/dL.
In the overall study population, there was an increase from baseline to week 100 (last observation carried forward [LOCF]) in mean height z scores with a mean change of 0.25 (95% CI, –0.86 to 2.04; P = 0.0216). In the overall study population, there were no major changes from baseline in mean weight z scores at any time point with a mean change from baseline to week 100 (LOCF) of –0.05 (95% CI, –0.12 to 0.23; P = 0.5306).
The pivotal trial assessed HRQoL using the PedsQL as additional efficacy end points, and the LS mean difference from weeks 18 to 22 in the PedsQL total score (parent) between the maralixibat and placebo groups was 2.33 (95% CI, –10.08 to 14.75; P = 0.7018). In the overall population, the mean change in the PedsQL total score (parent) from baseline to week 18 was 10.73 (95% CI, 4.43 to 17.03; P = 0.0016). The LS mean difference for the PedsQL Multidimensional Fatigue Scale score (parent) from weeks 18 to 22 between the maralixibat and placebo groups was 14.03 (95% CI, –2.78 to 30.84; P = 0.0966). In the overall population the mean change in the PedsQL Multidimensional Fatigue Scale score (parent) from baseline to week 18 was 20.30 (95% CI, 8.98 to 31.63; P = 0.0013).
Harms Results
The incidence of AEs was similar during the open-label phase, after RWD phase, and the LTE phase, with at least 25 of 29 patients (86.2%) experiencing any AEs in these treatment periods. During the RWD phase, patients who stayed on maralixibat had a lower incidence of AEs (7 of 13 patients [38%]) compared with patients receiving placebo (12 of 16 patients [75%]). The most frequently reported AEs (> 30% in at least 1 phase) were abdominal pain, pyrexia, diarrhea, nasopharyngitis, vomiting, cough, and pruritus. During the RWD phase, SAEs were reported for 1 of 13 patients (7.7%) receiving maralixibat and 1 of 16 patients (6.3%) receiving placebo. None of the SAEs were considered related to study medication. A total of 6 patients (2 each in the open-label phase, after RWD phase, and the LTE phase) experienced AEs leading to study drug discontinuation. No deaths were noted during the study. During the RWD phase, patients who stayed on maralixibat had a similar incidence of diarrhea and abdominal pain (1 of 13 patients [7.7%]) compared with those on placebo (1 of 16 patients [6.3%]). No patients experienced events associated with FSV deficiency during the RWD phase.
Critical Appraisal
During the open-label phases of the pivotal trial, patients’ and/or caregivers’ knowledge of treatment assignment may have biased subjective outcomes such as ItchRO(Obs), ItchRO(Pt), and PedsQL in favour of maralixibat. Reporting of harms could also have been biased, potentially in favour of maralixibat. Discontinuation was low, with 3 of 31 patients (9.7%) discontinuing due to an AE through to week 48. Regarding differences in baseline characteristics between patients in the maralixibat and placebo groups, the clinical experts noted that patients in the maralixibat group may have had a higher degree of disease severity than those in the placebo group as indicated by the higher sBA, ALT, and bilirubin values, which may have biased results in favour of placebo. Descriptive post hoc data from the ICONIC pivotal trial found that reductions in sBA from baseline to week 48 were associated with reductions in mean ItchRO(Obs) weekly average morning severity scores (Appendix 1). The data may show an association between sBA and ItchRO in some patients, but as the data were descriptive in nature and the assessment was conducted posthoc on a small number of patients (n = 28), it is unclear the extent to which sBA levels may be associated with pruritus in patients with cholestatic liver diseases.
The clinical experts consulted on this review noted that a minimal important difference (MID) of 1 for the ItchRO tool is clinically meaningful; however, the experts noted that such tools are not commonly used in clinical practice. HRQoL was assessed using the PedsQL as an additional efficacy outcome in the pivotal trial and MID estimates of 4 to 5 points align with the clinical experts’ expectations of a clinically meaningful change. It should be noted that the number of patients assessed for the PedsQL Multidimensional Fatigue Scale score was low during the RWD phase, with 9 of 13 patients (69.2%) in the maralixibat group and 12 of 16 patients (75.0%) in the placebo group contributing to the analysis of mean change from weeks 18 to 22. The impact of missing data on this outcome is unclear in the absence of sensitivity analyses.
The clinical experts consulted on this review stated that patients included in the ICONIC trial generally align with the selection criteria for candidates for maralixibat, although patients with mild cholestatic pruritus would not necessarily be excluded from treatment in clinical practice. Nonetheless, the clinical experts did not expect the exclusion of these patients to significantly affect the generalizability of the patient population in this study. The clinical trial only enrolled patients aged 12 months or older with a Jagged-1 mutation; however, the clinical experts note that the trial results would be applicable to patients younger than 12 months as well as patients with a NOTCH2 mutation, respectively. Although race and ethnicity data were not assessed in the pivotal trial, the clinical experts stated that the results would be applicable to the patient population in Canada. The efficacy outcomes measured in the study were of clinical importance to patients and clinicians, including change in sBA. However, the clinical experts noted that the change in sBA is not often assessed in clinical practice due to high costs and logistical limitations, as sBA testing is often sent to specialized laboratories and is not readily available in all gastroenterology practice settings. The clinical experts consulted for this review indicated that although tools such as PedsQL are frequently used in clinical trials, they are not typically used in clinical practice. Furthermore, the double-blind phase in the pivotal ICONIC trial was 4 weeks in length, limiting the ability to assess the long-term efficacy and safety of maralixibat compared with placebo for the indicated dose of 380 mcg/kg/day. While maralixibat has been approved by Health Canada for use in patients for the treatment of cholestatic pruritus in patients with ALGS, aged 2 months and older, the ICONIC trial only enrolled patients aged 12 months or older. As such, there is an absence of comparative efficacy and safety data assessing maralixibat versus placebo among patients aged younger than 12 months in the ICONIC trial due to the challenges of conducting a controlled clinical trial in this age group. However, the trial results are expected to be applicable to patients younger than 12 months based on clinical experts’ feedback. Furthermore, during the LTE phase of the ICONIC pivotal trial (as of week 103) eligible patients could have received a dose of maralixibat of up to 760 mcg/kg/day (given as twice-daily doses of 380 mcg/kg), which is outside of the proposed Health Canada indication of 380 mcg/kg /day. As such, efficacy and safety data after this period is are aligned with the recommended dose.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal study (ICONIC) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluations (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.14,15 Following the GRADE approach, evidence from the pivotal study started as high-certainty evidence and could be rated down for concerns related to study limitations (i.e., internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: change in fasting sBA levels, change in pruritus as measured by ItchRO(Obs) and ItchRO(Pt) weekly average morning severity scores, change in liver biomarkers and enzymes (ALT, ALP, total, and direct bilirubin), change in body height and weight z scores, HRQoL as measured by the PedsQL total score (parent) and the PedsQL Multidimensional Fatigue Scale score (parent), and AEs including SAEs, diarrhea, abdominal pain, and FSV deficiency.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of any (non-null) effect for all outcomes except the ItchRO and PedsQL due to the lack of a formal MID estimate.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for maralixibat versus placebo for the treatment of cholestatic pruritus in pediatric patients with ALGS.
Table 2: Summary of Findings for Maralixibat Versus Placebo for the Treatment of Cholestatic Pruritus in Patients With ALGS
Outcome and follow-upa | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Maralixibat | Difference | |||||
Serum bile acids | |||||||
Change in fasting sBA levels (μmol/L) from weeks 18 to 22 in patients who previously responded to treatment with maralixibat Follow-up: 4 weeks | 15 (1 RCT) | NA | 95.55 | –21.73 (–115.69 to 72.23) | –117.28 (–232.38 to –2.18) | Lowb | Maralixibat may result in a decrease (improvement) in fasting sBA levels compared with placebo. The clinical importance of the decrease is unclear. |
Pruritis | |||||||
Change in pruritus as measured by ItchRO(Obs) weekly average morning severity score from weeks 18 to 22 in patients who previously responded to maralixibat treatment Follow-up: 4 weeks | 31 (1 RCT) | NA | 1.70 | 0.22 (–0.27 to 0.70) | –1.48 (–2.12 to –0.84) | Lowc | Maralixibat may result in a clinically important improvement in ItchRO(Obs) weekly average morning severity score compared with placebo. |
Change in pruritus as measured by ItchRO(Pt) weekly average morning severity score from weeks 18 to 22 in patients who previously responded to maralixibat treatment Follow-up: 4 weeks | 31 (1 RCT) | NA | 1.84 | –0.15 (–0.97 to 0.67) | –1.99 (–3.01 to –0.97) | Lowc | Maralixibat may result in a clinically important improvement in ItchRO(Pt) weekly average morning severity score compared with placebo. |
Biochemical outcomes | |||||||
Change in ALP (U/L) from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NA | –7.2 | 2.8 (–43.6 to 49.1) | 10 (–52.6 to 72.6) | Lowd | Maralixibat may result in little-to-no difference in ALP compared with placebo. There is some uncertainty about the clinical importance of the estimates. |
Change in ALT (U/L) from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NA | 19.4 | 34.5 (5.6 to 63.4) | 15.1 (–25.1 to 55.2) | Lowd | Maralixibat may result in little-to-no difference in ALT compared with placebo. There is some uncertainty about the clinical importance of the estimates. |
Change in total bilirubin (mg/dL) from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NA | 0.46 | 0.32 (–0.23 to 0.86) | –0.14 (–0.88 to 0.60) | Lowd | Maralixibat may result in little-to-no difference in total bilirubin levels compared with placebo. There is some uncertainty about the clinical importance of the estimates. |
Change in direct bilirubin (mg/dL) from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NA | 0.14 | 0.13 (–0.28 to 0.53) | –0.02 (–0.56 to 0.53) | Lowd | Maralixibat may result in little-to-no difference in direct bilirubin levels compared with placebo. There is some uncertainty about the clinical importance of the estimates. |
Height and weight outcomes | |||||||
Change in body height (z scores) from baseline to week 48 Follow-up: 48 weeks | 31 (1 RCT, noncomparative) | NA | NR | NR | 0.18 (–0.02 to 0.23) | Very lowe | The evidence is very uncertain about the effect of maralixibat on body height z scores compared with any comparator. |
Change in body height (z scores) from baseline to week 100 (LOCF) Follow-up: 100 weeks | 31 (1 RCT, noncomparative) | NA | NR | NR | 0.25 (0.04 to 0.46) | Very lowe | The evidence is very uncertain about the effect of maralixibat on body height z scores compared with any comparator. |
Change in body weight (z scores) from baseline to week 48 Follow-up: 48 weeks | 31 (1 RCT, noncomparative) | NA | NR | NR | 0.02 (–0.15 to 0.18) | Very lowe | The evidence is very uncertain about the effect of maralixibat on body weight z scores compared with any comparator. |
Change in body weight (z scores) from baseline to week 100 (LOCF) Follow-up: 100 weeks | 31 (1 RCT, noncomparative) | NA | NR | NR | 0.05 (–0.12 to 0.23) | Very lowe | The evidence is very uncertain about the effect of maralixibat on body weight z scores compared with any comparator. |
HRQoL | |||||||
Change in PedsQL total score (parent) from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NA | –9.03 | –6.69 (–15.97 to 2.59) | 2.33 (–10.08 to 14.75) | Lowf | Maralixibat may result in little-to-no difference in the PedsQL total score (parent) compared with placebo. |
Change in PedsQL Multidimensional Fatigue Scale score (parent) from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NA | –16.99 | –2.96 (–15.67 to 9.74) | 14.03 (–2.78 to 30.84) | Lowg | Maralixibat may result in improvement of the PedsQL Multidimensional Fatigue Scale score (parent) compared with placebo. |
Harms | |||||||
Patients with SAEs from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NR | 63 per 1,000 | 77 per 1,000 (NR) | NR | Very lowh | The evidence is very uncertain about the effect of maralixibat on SAEs compared with placebo. |
Diarrhea, from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NR | 63 per 1,000 | 77 per 1,000 (NR) | NR | Very lowh | The evidence is very uncertain about the effect of maralixibat on the proportion of patients with diarrhea compared with placebo. |
Abdominal pain, from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NR | 63 per 1,000 | 77 per 1,000 (NR) | NR | Very lowh | The evidence is very uncertain about the effect of maralixibat on the proportion of patients with abdominal pain compared with placebo. |
FSV deficiency, from weeks 18 to 22 Follow-up: 4 weeks | 31 (1 RCT) | NR | 0 per 1,000 | 0 per 1,000 (NR) | NR | Very lowh | The evidence is very uncertain about the effect of maralixibat on the proportion of patients with FSV deficiency compared with placebo. |
ALGS = Alagille syndrome; ALP = alkaline phosphatase; ALT = alanine transaminase; CI = confidence interval; FSV = fat-soluble vitamin; HRQoL = health-related quality of life; ItchRO(Obs) = Itch Reported Outcome (observer); ItchRO(Pt) = Itch Reported Outcome (patient); LOCF = last observation carried forward; MID = minimal important difference; NA = not applicable; NR = not reported; PedsQL = Pediatric Quality of Life Inventory; RCT = randomized controlled trial; SAE = serious adverse event; sBA = serum bile acid.
Note: Study limitations (i.e., internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aStatistical testing for all outcomes was not adjusted for multiplicity. The potential for type I error (false-positive results) is increased.
bRated down 2 levels for very serious imprecision; evidence from 1 trial with small sample size. The small sample size raises concerns about the potential for prognostic imbalance and potential overestimation of the true effect. No known MID so target of certainty appraisal was any effect; 95% CI did not cross the null.
cRated down 2 levels for very serious imprecision; evidence from 1 trial with small sample size. The small sample size raises concerns about the potential for prognostic imbalance and potential overestimation of the true effect. The 95% CI did not considerably cross the threshold of importance (based on an MID of 1).
dRated down 2 levels for very serious imprecision; evidence from 1 trial with small sample size. There is no known MID and the clinical experts consulted by CADTH could not provide a threshold of important difference; however, the CADTH review team judged that the effect estimate was likely to correspond with no important difference, and CI was unlikely to include both important benefit and harm.
eIn the absence of a comparator group at the assessed time point, conclusions about efficacy relative to any comparator cannot be drawn and certainty of evidence started at very low. Rated down 2 levels for very serious imprecision, evidence from 1 arm of 1 trial with small sample size.
fRated down 2 levels for very serious imprecision. The 95% CI for difference between groups included possible important benefit and important harm (based on MID of 4 to 5 points).
gRated down 1 level for serious study limitations. Risk of bias due to missing outcome data, results of analysis available for 9 of 13 patients (69.2%) in the maralixibat group and 12 of 16 patients (75.0%) in the placebo group. Rated down 1 level for serious imprecision; the 95% CI for difference between groups included the potential for little-to-no difference (based on MID of 4 to 5 points).
hRated down 1 level for serious indirectness. The clinical experts noted that the 4-week randomized withdrawal period was not sufficient to fully assess the comparative safety of maralixibat compared with placebo for this outcome. Rated down 2 levels for serious imprecision; the sample size is small and the results are based on very few or no events in each group.
Source: ICONIC Clinical Study Report.16 (Note: details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)17
LTE Studies
The pivotal ICONIC trial included an LTE phase described in the Systematic Review section of this report. No other LTE studies were submitted.
Indirect Comparisons
No indirect comparisons were conducted comparing maralixibat with other comparators for this submission.
Studies Addressing Gaps in the Evidence From the Systematic Review
Description of Studies
The sponsor submitted a natural-history comparison study, which is presented in this report, comparing disease outcomes among patients with ALGS treated with maralixibat (N = 84) with an external controls cohort from the Global ALagille Alliance (GALA) clinical research database (n = 469), with follow-up data up to 6 years. Outcomes assessed included event-free survival (EFS), a composite end point of first event of liver decompensation (ascites, variceal bleeding, surgical biliary diversion [SBD], liver transplant, and death), and transplant-free survival (TFS). Of note, the natural-history comparisons were conducted independent of the sponsor (Mirum Pharma).
Results from patient-level data from 3 long-term studies of patients with ALGS treated with maralixibat, including the LUM001-303 (IMAGINE) trial, the ICONIC pivotal trial (LUM001-304), and the IMAGINE-II (LUM001-305) trial to identify predictors of EFS and TFS were submitted by the sponsor and presented in this report.
Efficacy Results
Results from the natural-history comparison study reported a 70% improvement in EFS with maralixibat treatment compared with the GALA control group (hazard ratio [HR] = 0.305; 95% CI, 0.189 to 0.491; P < 0.0001) and a 67% improvement in TFS with maralixibat treatment compared with the GALA control group (HR = 0.332; 95% CI, 0.197 to 0.559; P < 0.0001). Additional relevant evidence assessing patient-level data (n = 76) from 3 ALGS clinical trials (IMAGINE, IMAGINE-II, and ICONIC) stated that clinical parameters (sBA levels, total serum bilirubin, and change in pruritus from baseline as measured by the ItchRO[Obs]) obtained after 48 weeks of maralixibat treatment were potential predictors of subsequent TFS and EFS.
Critical Appraisal
Concerns regarding the natural-history comparison include the potential residual confounding, incomparability in disease severity, and the lack of sBA data available among patients in the GALA registry. Although the study showed statistically and clinically significant reduction in liver transplant, death and other associated events in patients who received maralixibat treatment compared with patients who received standard of care, there is uncertainty in the results and they should therefore be interpreted with caution. Results from the 3 ALGS clinical trials (IMAGINE, IMAGINE-II, and ICONIC) are subject to uncertainty due to various limitations including the limited sample size, a lack of control group, and uncertainty if the improvements in EFS and TFS observed in this analysis are solely the result of improvements in pruritus.
Conclusions
There is an unmet need for symptomatic and curative treatment options for cholestatic pruritus in pediatric patients with ALGS. Patients and clinicians highlighted the need for treatments that reduce the frequency and severity of pruritus and reduce patient and caregiver fatigue. The pivotal phase II, double-blind, placebo-controlled, randomized drug withdrawal trial (ICONIC) was included in this review, which assessed the treatment of cholestatic pruritus in pediatric patients with ALGS (aged 12 months to 18 years). The trial was an exploratory study.
The study demonstrated that maralixibat may result in a decrease in sBA levels and results in a clinically meaningful improvement in pruritus as assessed by the ItchRO(Obs) and ItchRO(Pt) weekly average morning severity scores compared with placebo. It is important to note that the “low” certainty of evidence as assessed by GRADE for sBA and pruritus outcomes is due to the imprecision observed, and not from bias due to study limitations. This imprecision due to small sample size is clearly connected to the nature of the rarity of the disease. Improvements in the PedsQL total score (parent) and the PedsQL Multidimensional Fatigue Scale score (parent) were uncertain. It also remained uncertain whether maralixibat may have increased ALT and ALP levels or resulted in any difference in total and direct bilirubin compared with placebo. Moreover, although the sponsor provided some evidence to support sBA as a predictor of long-term outcomes such as EFS and TFS, the relationship between sBA and severity of cholestatic pruritus still remains uncertain. Despite certain limitations inherent with observational study design, it is likely that there is a significant treatment effect of maralixibat on long-term outcome such as EFS and TFS.
Maralixibat was generally well-tolerated in the ICONIC trial, with limited grade 3 AEs or SAEs. Of note, however, due to the rare nature of the disease, the severity of the condition, and the lack of approved or effective treatments, the study sample size was small and the study was short, in a 4-week randomized duration. Longer-term evidence to support the overall benefit and safety of maralixibat include LTE of the ICONIC trial and the natural-history comparison study. Real-world data currently being collected through the established GALA registry provide the opportunity to continuously monitor the efficacy and safety of maralixibat.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of maralixibat (Livmarli), 9.5 mg/mL, oral solution, for the treatment of cholestatic pruritus in patients with ALGS, 2 months of age and older.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and by clinical expert input. The following has been summarized and validated by the CADTH review team.
ALGS is a rare, life-threatening, genetic, complex, multisystem disorder that presents with a range of clinical features including cholestatic liver disease, failure to thrive, cardiovascular disease, skeletal abnormalities, ocular abnormalities, renal and vascular abnormalities, and distinct facial features.1,2 In most cases, the liver dysfunction associated with ALGS is an early and serious feature of this genetic condition and typically presents in the first 3 months of life. Elevated levels of sBAs and jaundice (elevated bilirubin) are hallmarks of ALGS and indicate the presence of impaired bile flow.3 Clinically important manifestations of cholestasis in ALGS include debilitating and intractable pruritus, disfiguring xanthomas, sleep disturbances, chronic debilitating fatigue, and failure to thrive (i.e., insufficient growth).4,5
ALGS is predominantly caused by mutations in the Jagged-1 gene in greater than 90% of cases6 and in the NOTCH2 gene in approximately 4% of cases, resulting in bile duct paucity, bile flow obstruction, and bile accumulation in the liver. ALGS is inherited in an autosomal-dominant pattern. Children of an individual with ALGS have a 50% chance of inheriting the causative gene mutation in the Jagged-1 or NOTCH2 gene.2 In 50% to 70% of affected individuals, however, the mutation is de novo.2 Due to the diverse role of Notch signalling, disruption of either gene results in a broad range of clinical manifestations, as described previously. Cholestasis is defined as a decrease in bile flow due to impaired secretion by hepatocytes or to obstruction of bile flow through intrahepatic or extrahepatic bile ducts.7 Cholestasis leads to hepatic and systemic accumulation of substances normally excreted via the biliary tract, such as bile acids and conjugated bilirubin. Elevations of sBA up to 100 times the upper limit of normal (ULN) and serum bilirubin up to 30 times the ULN are not uncommon.6 Elevated levels of bile acids are hepatotoxic and contribute to disease progression. Bile acids have been shown to induce damage and necrosis in hepatocytes and cholangiocytes and are associated with increased morbidity and mortality in chronic cholestatic diseases.7 The systemic and hepatic accumulation of bile acids and other toxins leads to incapacitating and chronic cholestatic symptoms such as pruritus, and clinical sequelae.
The clinical manifestations of cholestasis associated with ALGS are severe, even in the absence of liver disease, with cholestatic pruritus being the leading cause of liver transplant in patients.8-10 These clinical manifestations present in the first few years of life and as early as age 3 months. The symptoms include severe and unremitting pruritus (74%10), xanthomas (disfiguring and sometimes disabling subcutaneous lipid deposits, 40%), chronic fatigue (between 65% and 85%), and growth failure (between 50% and 87%11). In addition, FSV malabsorption and increased risk of bone fractures because of trabeculae malformation can also be present. A second wave of portal hypertension and associated complications occurs later in adolescence.6 Collectively, all cholestasis-related symptoms result in poor HRQoL. As cholestasis progresses and symptoms worsen, as described previously, a majority of patients will either receive a liver transplant (50.4%) or die (9.3%) by age 18 years, with only 40.3% of patients reaching adulthood with their native liver.6,10 The estimated 20-year life expectancy is 75% for patients diagnosed with ALGS, 80% for patients with ALGS who do not require liver transplant, and 60% for patients with ALGS who require liver transplant.12 For patients with ALGS who undergo liver transplant, the estimated 1-year survival rate is 87%.13 The majority of early liver transplantations occur because of complications associated with cholestasis, including pruritus.
The reported incidence of ALGS is 1 in 30,000 to 50,000 live births.3,11 Table 3 presents sponsor-submitted estimates of disease incidence based on the patient population in CADTH-participating jurisdictions. In the absence of Canadian statistics, the sponsor estimated prevalence (based on 1 in 30,000 live births) to be a total of 1,032 patients with ALGS in 2023.
Table 3: Estimated Incidence in Each Region
Region | |||| |||||||| || || ||||||||| |
|---|---|
Pan-Canadian (excluding Quebec) | |||||| |
Alberta | |||||| |
British Columbia | |||||| |
Manitoba | |||||| |
New Brunswick | |||||| |
Newfoundland and Labrador | |||||| |
Northwest Territories | |||||| |
Nova Scotia | |||||| |
Nunavut | |||||| |
Ontario | |||||| |
Prince Edward Island | |||||| |
Saskatchewan | |||||| |
Yukon | |||||| |
Non-insured health benefits | |||||| |
Source: Sponsor’s Clinical Summary Report.17
Diagnosis of ALGS can be challenging due to variable presentation of the clinical manifestations. The phenotypic presentation may vary even among individuals from the same family sharing the same genetic mutation.2 Additionally, there is a lack of a strong correlation between mutation type, clinical manifestation, and disease severity.18 The clinical diagnostic criteria for ALGS can be made if 3 of the following 7 major clinical features are present: cholestasis, ophthalmologic abnormalities, characteristic facial features, cardiac defects, skeletal abnormalities, kidney abnormalities, and vasculature. Liver histology showing bile duct paucity and genetic testing is sometimes conducted to diagnose or confirm the diagnosis of ALGS.2,3,6,11 ALGS can be diagnosed through clinical criteria alone; however, molecular genetic testing can also be used for diagnosis or can provide valuable confirmation to clinical diagnosis, especially in milder cases.11 Genetic testing for ALGS is currently available in Canada and covered by the ministries of health.19 Additionally, Prevention (formerly Emory Medical Laboratories, US) provides free genetic panel for patients who have cholestasis. This service is made available to patients in Canada as well.19
In Canada, patients with ALGS are primarily followed in large specialized tertiary care centres covering large catchment areas.19 These centres receive referrals from specialists from smaller community hospitals, general practitioners or pediatricians, and neonatal intensive care units. The main treating team in these tertiary care centres comprises hepatologists or gastroenterologists and other allied professionals. Hepatologists or gastroenterologists are responsible for the primary management of the disease and patient follow-up, as well as coordination with local hepatologists or gastroenterologists for patients with ALGS who reside far from the tertiary care centres. Other allied professionals involved in the care of patients with ALGS are dieticians or nutritionists, social workers, and psychologists. Other care departments are also involved as needed (e.g., cardiology, radiology, genetics, nephrology, neurology, endocrinology).
ALGS pediatric patients who have progressed in their disease and may require liver transplants are referred to 1 of 3 pediatric transplant centres for transplant assessment. These pediatric transplant centres are located at The Hospital for Sick Children (SickKids) in Toronto, Ontario; the University of Alberta Hospital in Edmonton, Alberta; and the Centre hospitalier universitaire Sainte-Justine in Montreal, Quebec. For patients eligible to receive a liver transplant, the centres are also responsible for preparing patients (i.e., administrative, clinical work-up, and so forth) leading up to the procedure. Ultimately, the liver transplant will take place at these centres.
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and by clinical expert input. The following has been summarized and validated by the CADTH review team.
Currently in Canada, there are no medical treatments approved by Health Canada to treat ALGS or the cholestatic pruritus associated with it. According to the clinical experts consulted, there are no clinical practice guidelines for the treatment of ALGS and current treatment approaches focus on improving symptoms. Certain drugs are prescribed off-label based on limited clinical data, many of which have limited or transient efficacy and may have undesirable adverse effects.20 According to the clinical experts consulted, current treatments for cholestatic pruritus include antihistamines, which are considered for mild cases and for their sedative effect. Ursodeoxycholic acid promotes bile excretion and, due to its attractive safety profile, it is typically used early in the management of cholestasis; however, its effectiveness for pruritus is not certain. Rifampicin has been reported to improve but not completely resolve pruritus in approximately 50% of patients with ALGS. Cholestyramine, a bile-acid binding resin, may be considered, but its poor palatability and interference with absorption of other drugs limits its use in clinical practice. Sertraline and naltrexone provide marginal additional benefit; however, according to the clinical experts, their use in pediatric patients is limited and their efficacy to treat pruritus in pediatric patients with ALGS is unclear.
As existing pharmacological therapies often fail in patients with ALGS, surgical alternatives are often required. Surgical interventions to treat cholestasis in ALGS include SBD procedures and liver transplant. SBD procedures (partial internal biliary diversion and ileal exclusion) have been used in ALGS to ameliorate cholestasis with variable results and short-term and long-term surgical and medical complications6,8,10,11,21 and are seldomly used in Canada, according to the clinical experts consulted.
Intractable pruritus or disfiguring xanthomas can be severe enough to warrant liver transplant, even in the absence of liver failure.10,11,22 Most patients with ALGS will undergo a liver transplant in the first 2 decades of life, with only 40.3% of patients reaching adulthood with their original liver. ALGS-associated pruritus is a leading cause of transplant in these patients.6,10 Liver transplant in ALGS has increased the risk of complications, with studies reporting 1-year survival rates below 80%.12 Patients with ALGS who survive liver transplant have the burden of lifelong immunosuppression and other long-term morbidity.11 In particular, children who require transplant at a young age have poorer outcomes. Infants younger than 1 year have higher rates of mortality on the liver transplant waitlist and higher rates of rejection. Children younger than 4 years have lower rates of graft survival.23 Of note, extrahepatic manifestations of ALGS such as significant cardiac disease can often be a contraindication for a major surgery such as liver transplant, leaving these patients with even further limited treatment options.24
In alignment with clinical experts consulted by CADTH, the sponsor stated that the management of cholestasis and related pruritus in patients with ALGS remains largely supportive. Alternatively, surgical treatment may be required for patients, continuing to experience extensive morbidity. The clinical experts consulted by CADTH said that improving symptoms (such as very debilitating pruritus of ALGS that has a significant impact on patient quality of life) is the current treatment focus. Also, the clinical experts noted that there is a lack of curative and symptomatic treatments, and even liver transplant is not necessarily curative for liver disease due to high morbidity.
Drug Under Review
Key characteristics of maralixibat are summarized in Table 5, along with other treatments available for pruritus associated with ALGS.
Maralixibat (Livmarli), 9.5 mg/mL, oral solution, is approved by Health Canada for the treatment of cholestatic pruritus in patients with ALGS. The reimbursement request is the same as the indication to Health Canada. Maralixibat is the first drug approved by the FDA for cholestatic pruritus in patients with ALGS who are aged 3 months and older.25 Maralixibat is indicated for the treatment of cholestatic pruritus in patients with ALGS, aged 2 months and older in the European Union.26 Maralixibat does not have any other indications and has not been previously reviewed by CADTH. The recommended maintenance dose is 380 mcg/kg once daily in the morning after 1 week of a starting dose of 190 mcg/kg orally once daily. The maximum daily dose in volume for patients above 70 kg is 3 mL (Table 4).
Table 4: Individual Dose Volume by Patient Weight
Patient weight (kg) | Days 1 to 7 (190 mcg/kg once daily) | Beginning day 8 (380 mcg/kg once daily) | ||
|---|---|---|---|---|
Volume per day (mL) | Dosing dispenser size (mL) | Volume per day (mL) | Dosing dispenser size (mL) | |
5 to 6 | 0.1 | 0.5 | 0.2 | 0.5 |
7 to 9 | 0.15 | 0.3 | ||
10 to 12 | 0.2 | 0.45 | ||
13 to 15 | 0.3 | 0.6 | 1 | |
16 to 19 | 0.35 | 0.7 | ||
20 to 24 | 0.45 | 0.9 | ||
25 to 29 | 0.5 | 1 | ||
30 to 34 | 0.6 | 1 | 1.25 | 3 |
35 to 39 | 0.7 | 1.5 | ||
40 to 49 | 0.9 | 1.75 | ||
50 to 59 | 1 | 2.25 | ||
60 to 69 | 1.25 | 3 | 2.5 | |
70 or higher | 1.5 | 3 | ||
Source: Sponsor’s Clinical Summary Report.17
As an inhibitor of the ileal bile acid transporter (IBAT), maralixibat interrupts the enterohepatic circulation of bile acids, leading to statistically significant decreases in sBA levels and increased fecal bile acid secretion. Bile acids are synthesized in the liver and are the major lipid components of bile, making up approximately two-thirds of the solute mass of normal human bile.27 The enterohepatic circulation of bile acids acts as a feedback mechanism to maintain bile acid homeostasis and control bile acid production.28 Circulation of abnormal bile acid levels is associated with a variety of illnesses, such as cholestatic liver disease. IBAT is responsible for the active reabsorption of about 90% to 95% of intestinal bile acids in the terminal ileum.27,29,30 Due to its key role in bile acid re-uptake, IBAT is a target for pharmacologic regulation of bile acid reabsorption.31 The bile duct paucity associated with ALGS leads to impaired bile flow, accumulation of bile acids, and cholestatic liver injury. Through the reduction of sBA, maralixibat provides an improvement in cholestatic pruritus in patients with ALGS.
Table 5: Key Characteristics of Maralixibat, UDCA, Rifampin, and Antihistamines
Characteristic | Maralixibat | UDCA | Rifampin | Antihistaminesa |
|---|---|---|---|---|
Mechanism of action | Inhibits the IBAT and interrupts the enterohepatic circulation of bile acids leading to decrease in sBA levels and increased fecal bile acid secretion | Ursodiol, a naturally occurring, exogenous, hydrophilic bile acid, replaces or displaces toxic concentrations of endogenous hydrophobic bile acids that tend to accumulate in cholestatic liver disease32 | Inhibits DNA-dependent RNA polymerase activity in susceptible cells33 According to the clinician group input, it is thought to increase the metabolism of pruritogens through its enzymatic induction in the liver | Hydroxyzine: Antihistamine that blocks H1 receptors, with anticholinergic, antiemetic, and sedative properties34 |
Indicationb | Proposed for the treatment of cholestatic pruritus in patients with ALGS | Not approved for cholestatic pruritus associated with ALGS For the management of cholestatic liver diseases, such as primary biliary cirrhosis32 | Not approved for cholestatic pruritus associated with ALGS | Not approved for cholestatic pruritus associated with ALGS Hydroxyzine: Used in the management of pruritus due to allergic conditions such as chronic urticaria and atopic and contact dermatoses in adults and pediatrics35 |
Route of administration | Oral | Oral | Oral | Oral |
Recommended dose | Initial: 190 mcg/kg/day Maintenance: 380 mcg/kg/day after 1 week of initial treatment | 10 to 20 mg/kg/day | 10 mg/kg/day | Hydroxyzine: In children and adolescents up to 40 kg in weight, the maximum daily dose is 2 mg/kg/day, given in divided doses (maximum daily dose is 80 mg) In children and adolescents more than 40 kg in weight, the maximum daily dose is the same as for adults: 100 mg per day, given in divided doses |
Serious adverse effects or safety issues | FSV deficiency, transaminases increased (AST, ALT), gastrointestinal bleeding, bone fractures | Leukopenia, rash, esophagitis, hypertension32 | Urticaria, thrombocytopenia, intravascular coagulation, ataxia, visual disturbances, liver toxicity, acute interstitial nephritis33 | Hydroxyzine: QTc prolongation, torsade de pointes, cardiac arrest, sudden death (rare)35 |
Other | NA | According to clinician group input, UDCA is used early in the management of cholestasis due to its safety profile | According to clinician group input, rifampin improves pruritus in 50% of patients with ALGS and is well tolerated; preferred drug over cholestyramine | According to the clinical expert, antihistamines are mainly used for their sedative effect, not for antipruritic effect; based on clinician group input, they can be considered for mild cases |
ALGS = Alagille syndrome; ALT = alanine transaminase; AST = aspartate transaminase; FSV = fat-soluble vitamin; IBAT = ileal bile acid transporter; NA = not applicable; QTc = corrected QT interval; RNA = ribonucleic acid; sBA = serum bile acid; UDCA = ursodeoxycholic acid.
aAntihistamines include certrizine hydrochloride, hydroxyzine hydrochloride, diphenhydramine, and so forth.
bHealth Canada–approved indication.
Source: Sponsor’s Clinical Summary Report.17
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient input received by CADTH has been included in the Stakeholder Perspectives section of this report.
Two patient groups, CLF and ALGSA, provided input. CLF is the only national health charity committed to supporting people in Canada affected by the liver diseases. Based in the US, ALGSA is a nonprofit organization dedicated to supporting families affected by ALGS globally. The CLF submission included phone and virtual interviews conducted in May 2023 with 8 patients and caregivers in Canada. Of those, 4 respondents had experiences with maralixibat through clinical trials. The ALGSA gathered data online through family surveys (2020), personal conversations, and topic-specific discussions among support or focus groups, including at least 76 members in Canada. Both groups stated that itchiness (pruritus) is the most bothersome symptom affecting patients’ and caregivers’ lives. For example, itchiness interrupts patients and families’ sleep, making those affected fatigued, anxious, depressed, irritable, and worried. Patients said they feel isolated in school and that it is challenging to maintain employment. Also, patients and families have difficulty finding a specialist who could recognize and make a proper diagnosis of ALGS and manage disease treatment. Patients and families from both groups want a new therapy that can provide significant relief of itchiness with long-term effects without high risks such as liver transplant and immunosuppression. Patients who have taken maralixibat during clinical trials said that their itchiness has been resolved with minor side effects, such as upset stomach and diarrhea, that they could become more of themselves and engage in normal day-to-day activities, and that their households were also positively affected.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the review of maralixibat, a panel of 3 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
The clinical expert panel stated that cholestatic pruritus remains a very significant management problem for patients with ALGS and their families due to partial, incomplete, or null response to currently available treatments. Current treatments are used off-label and are supportive in nature. These treatments include antihistamines, which can be effective for mild cholestatic pruritus and provide a sedative effect to augment sleep; however, the experts noted that antihistamines are rarely effective for treating cholestatic pruritus. Ursodeoxycholic acid promotes bile excretion and, due to its attractive safety profile, it is typically used early in the management of cholestasis; however, its effectiveness for cholestatic pruritus is not certain. Cholestyramine is often unpalatable and rarely used in pediatric patients. Rifampin does provide some symptomatic relief; however, it is usually ineffective in substantially ameliorating or eradicating cholestatic pruritus. Naltrexone, an opioid antagonist, seldomly used in pediatric patients, has been associated with minimal improvement in some children with ALGS but may include symptoms of opioid withdrawal syndrome and gastrointestinal intolerance. Sertraline, a selective serotonin re-uptake inhibitor, has been used in refractory cases in adults; however, there is limited pediatric data available to support its use as an additional therapy for pruritus. The experts noted that surgical options such as an external or internal biliary diversion can be offered to patients with ALGS with cholestatic pruritus who are refractory to medical therapies; however, these are not very effective and seldomly used in clinical practice. Finally, the experts stated that between 50% and 75% of patients with cholestatic liver disease will require a liver transplant and that cholestatic pruritus is a leading indication for a transplant. Liver transplant is associated with increased morbidity, mortality, and lifelong immune suppression. As such, the experts noted that there is an unmet need for effective symptomatic and curative treatment for cholestatic pruritus in the indicated patient population.
Place in Therapy
The clinical expert panel noted that there are currently no established clinical practice guidelines for ALGS and that none of the currently available off-label treatments target the underlying disease mechanism of bile duct paucity. Many patients with ALGS and cholestatic pruritus have inadequately treated pruritus with currently available off-label medical therapy. The clinical experts stated that maralixibat would likely be used in combination with current off-label treatments in patients experiencing ongoing pruritus, and that it is possible some patients could discontinue some of the off-label treatments once they are established on maralixibat and their pruritus is under control. The experts noted that, if easily accessible, maralixibat may be used as an initial therapy for new patients presenting with severe pruritus.
Patient Population
The clinical experts stated that the estimated incidence of ALGS in Canada is about 1 in 30,000 to 50,000 live births with about 200 new cases each year. A clinical diagnosis can be made by the presence of 3 of 7 clinical features (chronic cholestasis, cardiovascular abnormalities, butterfly vertebrae, posterior embryotoxon, renal anomalies, vascular abnormalities, or characteristic facies). ALGS is an autosomal-dominant condition, and a genetic diagnosis can be confirmed in approximately 95% of patients with clinical features. The experts noted that with the advent of accessible molecular testing, most pediatric patients receive genetic testing to confirm their diagnosis. The experts noted that pediatric patients with ALGS most suited for treatment with maralixibat are those who present with cholestatic pruritus that is persistent with current off-label treatments, which is about a third of patients in a clinical expert’s practice. Patients least suited to treatment with maralixibat are those who have minimal liver involvement (i.e., minimal liver enzyme abnormalities and no FSV deficits) and those who do not experience cholestatic pruritus.
Assessing the Response to Treatment
According to the expert panel, a clinically meaningful response to treatment would include a reduction in the frequency and severity of pruritus, a reduction in sleep deprivation among patients and their caregivers, the ability for patients and their caregivers to attend school or work, reduced damage to the patients’ skin, and improved patient weight and growth. The clinical experts consulted on this review noted that response to therapy would likely be evaluated via subjective family reporting of symptoms including itching and sleep disturbances as well as by visual assessments of excoriations on the patient’s skin, which are often indicative of severe cholestatic pruritus. Standard-reporting itch scales such as the Visual Analogue Scale may be used to assess response to treatment, although such scales are not commonly used in clinical practice. according to the experts. Improvements in patient weight and growth are most often assessed using standardized parametrizes including midarm circumference and skin-fold thickness. Measurements of sBA could also be used to assess response to therapy; however, the experts noted that this is not common in clinical practice due to the high cost and the limited availability of such testing in some practice settings. The clinical experts would initially assess patients monthly for approximately 3 months, at which time the frequency of visits would be reduced to every 3 to 6 months if a response to treatment was evident.
Discontinuing Treatment
The clinical experts stated that treatment with maralixibat will likely be lifelong for most patients. The panel noted that treatment discontinuation may be considered if there is no effect on cholestatic pruritus after approximately 6 months of treatment initiation. Treatment may be discontinued if a patient’s liver disease progresses, and they undergo liver transplant. Treatment may be discontinued due to severe AEs; however, the experts stated that AEs associated with maralixibat are likely self-limited and may be addressed by titrating the dose of maralixibat.
Prescribing Considerations
The clinical experts noted that a pediatric or adult liver or GI specialist would be the preferred specialist to prescribe and monitor treatment with maralixibat.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the Stakeholder Perspectives section of this report.
One clinician from the Canadian Association for the Study of the Liver provided input. The clinician group and 2 clinical experts consulted by CADTH agree on the unmet need, which is a lack of effective therapy specifically indicated for cholestatic pruritus associated with ALGS refractory to current off-label treatments. They also agree that all the existing therapies are not effective at reducing cholestatic pruritus associated with ALGS in most patients and that there are no guidelines for treating cholestatic pruritus in patients with ALGS. In alignment with clinical experts, the clinician group stated that treatment goals are mainly improvement in pruritus, improvement in quality of life (i.e., sleep duration), and optimizing nutritional goals (e.g., treating FSV deficiency). Also, both groups agree that patients with ALGS and cholestatic pruritus that is persistent on standard-of-care medical treatment would be an eligible population. The clinician group stated that if a patient’s liver disease progresses and they undergo liver transplant, discontinuation is considered, and the clinical experts stated that if there is no effect on itch as measured clinically then discontinuation is considered after adequate trial — i.e., 6 months. Otherwise, both groups agree that AEs would be an unlikely reason to discontinue since maralixibat is well-tolerated. Lastly, all the clinician group and clinical experts agree that maralixibat should be prescribed by a pediatric gastroenterologist or hepatologist. None of the clinician group or clinical experts consulted by CADTH had declared experience with maralixibat.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 6.
Table 6: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
Most patients in clinical trials had the documented Jagged-1 mutation. Can the study results be extrapolated to patients with other mutations? | According to the clinical experts, the study results can be extrapolated to patients with other mutations (i.e., NOTCH2). |
Are there any therapies, including off-label treatments, that should be trialled before the initiation of maralixibat? | The clinical experts stated that current off-label treatments such as UDCA and rifampicin may be trialled in patients experiencing ongoing pruritus before initiating maralixibat. It is possible that some patients could discontinue some of the off-label treatments once they are established on maralixibat and their pruritus is under control. The experts noted that, if easily accessible, maralixibat may be used as an initial therapy for new patients presenting with severe pruritus before initiating maralixibat. |
Considerations for continuation or renewal of therapy | |
Is a reduction in the ItchRO scale greater than 1 point from baseline clinically significant? Are the ItchRO scales used in clinical practice? | The clinical experts noted that although scales such as the ItchRO are often used in clinical trials, they are not commonly used in clinical practice. The experts noted that changes in pruritus in clinical practice would likely be evaluated via subjective family reporting or patient reporting for older children of symptoms including itching and sleep disturbances as well as by visual assessments of excoriations on the patient’s skin, which are often indicative of severe pruritus. The experts stated that a reduction of 1 point in the ItchRO scale is clinically meaningful. |
ItchRO = Itch Reported Outcome; UDCA = ursodeoxycholic acid.
Clinical Evidence
The objective of CADTH Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of maralixibat, 9.5 mg per mL, oral solution in the treatment of cholestatic pruritus in patients with ALGS. The focus will be placed on comparing maralixibat with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of maralixibat is presented in 2 sections, with CADTH critical appraisal of the evidence included at the end of each section. The first section, the Systematic Review, includes pivotal studies and randomized controlled trials (RCTs) that were selected according to the sponsor’s systematic review protocol. CADTH assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence. There was no indirect evidence submitted for this review.
Included Studies
Clinical evidence from the following are included in the CADTH review and appraised in this document:
One pivotal study identified in systematic review
Two additional studies addressing gaps in evidence.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included pivotal study are summarized in Table 7.
Table 7: Details of the Study Included in the Systematic Review
Detail | ICONIC (LUM001-304) study |
|---|---|
Designs and populations | |
Study design | Multicentre, phase IIb, double-blind, placebo-controlled, randomized drug withdrawal trial with open-label extension |
Locations | 9 clinical sites in 6 countries (Australia, Belgium, France, Poland, Spain, UK) |
Patient enrolment dates | Start date: November 25, 2014 End date: September 11, 2015 |
Randomized (N) | Open-label run-in period (open-label phase: day 1 to week 18): N = 31 Randomized withdrawal phase (weeks 19 to 22):
|
Inclusion criteria | Male and female patients aged between 12 months and 18 years, inclusive, meeting the following key criteria:
|
Exclusion criteria |
|
Drugs | |
Intervention | All patients received maralixibat, up to 380 mcg/kg/day, during the initial open-label treatment period of the study. After completing the 6-week dose-escalation period and the 12-week stable-dosing period, patients were randomized 1:1 in a 4-week, double-blind withdrawal period to placebo or remained on maralixibat. Patients then entered a 26-week, long-term, stable-dosing period, and all patients received maralixibat up to 380 mcg/kg/day. Patients were considered for an initial, 52-week, optional long-term treatment period, if eligible, and received up to 380 mcg/kg/day or the highest tolerated dose below the 380 mcg/kg/day dose |
Comparator(s) | Patients were then considered for the second, optional, long-term follow-up treatment period, if eligible, receiving up to 760 mcg/kg/day (given as twice-daily dose of 380 mcg/kg) |
Study duration | |
Screening phase | Up to 4 weeks |
Run-in phase | 18 weeks (i.e., 6-week dose escalation and 12-week stable dosing) |
Treatment phase |
|
Follow-up phase |
|
Outcomes | |
Primary end point | Mean change from weeks 18 to 22 of fasting sBA levels in patients who previously responded to maralixibat treatment, as defined by a reduction in sBA ≥ 50% from baseline to week 12 or 18 in the mITT population |
Secondary and exploratory end points | Secondary efficacy end points:
Safety end points:
|
Publication status | |
Publications | Gonzales et al. (2021)36 |
AE = adverse event; ALGS = Alagille syndrome; ALP = alkaline phosphatase; ALT = alanine transaminase; b.i.d. = twice a day; CCS = Clinician Scratch Scale; FSV = fat-soluble vitamin; GGT = gamma-glutamyl transferase; GI = gastrointestinal; INR = international normalized ratio; ItchRO = Itch Reported Outcome; ItchRO(Obs) = Itch Reported Outcome (observer); ItchRO(Pt) = Itch Reported Outcome (patient); LDL-C = low-density lipoprotein cholesterol; LOCF = last observation carried forward; mITT = modified intent to treat; sBA = serum bile acid; PedsQL = Pediatric Quality of Life Inventory; ULN = upper limit of normal.
Source: ICONIC Clinical Study Report.16 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)17
The ICONIC pivotal trial was an open-label, phase IIb study with a double-blind, placebo-controlled, randomized drug withdrawal period and a long-term open-label extension (Figure 1). The study was designed to evaluate the safety and efficacy of maralixibat in children with ALGS aged between 12 months and 18 years. A total of 31 patients enrolled into the study which was conducted at 10 clinical sites in Australia, Europe, and the UK. Patients were enrolled between November 25, 2014, and September 11, 2015.
The periods of the study are grouped as follows:
screening period: study eligibility assessments (up to 4 weeks)
open-label phase: 18-week open-label run-in period
RWD phase: 4-week randomized, double-blind, placebo-controlled drug-withdrawal period
after RWD phase: 26-week stable-dosing period
LTE phase: optional long-term treatment periods consisting of a 52-week follow-up treatment period followed by a long-term follow-up treatment period.
Randomization and Masking
All participants were randomly assigned (1:1) in a blinded fashion to continue receiving the same dose of maralixibat or receive placebo for a period of 4 weeks (RWD; Figure 1). Randomization used a permuted block algorithm stratified by predefined response criteria (≥ 50% sBA reduction from baseline to weeks 12 or 18) and with entire blocks (size 4) assigned by study site using SAS software (version 9.4) by an unblinded statistician not involved in the conduct of the trial or analysis of the data. The randomization code was assigned to each participant in sequence in the order of enrolment, and then the participants received the investigational products labelled with the same code. Both maralixibat and placebo were identical in appearance. All participants, investigators, and laboratory staff were masked to treatment allocation.
The Clinical Study Report for the ICONIC pivotal trial presented maralixibat treatment doses as maralixibat chloride. The dosage of maralixibat described in this report are of maralixibat free base as in the proposed Health Canada indication, unless otherwise specified. A dose of 380 mcg/kg maralixibat free base is equivalent to 400 mcg/kg maralixibat chloride.
Upon completion of the 52-week optional follow-up treatment period, patients were considered for the optional long-term follow-up treatment period. If eligible, based on efficacy and safety assessments, patients received up to 760 mcg/kg/day of maralixibat (given as twice-daily doses of 380 mcg/kg), which is not aligned with the approved Health Canada dosage.
Figure 1: Study Design for the ICONIC Pivotal Trial
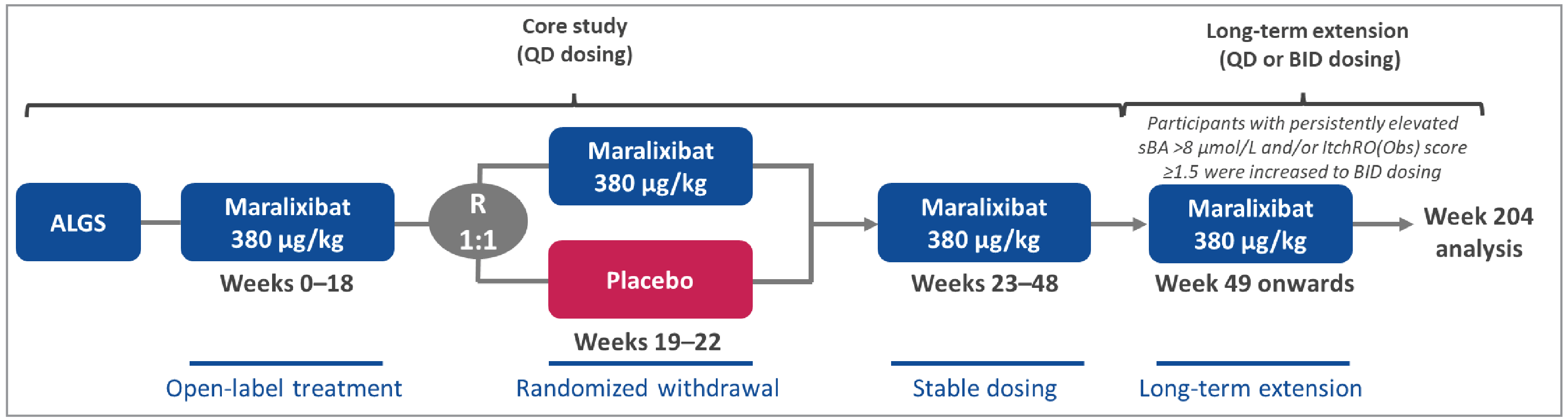
ALGS = Alagille syndrome; BID = twice a day; ItchRO(Obs) = Itch Reported Outcome (observer); QD = every day; R = randomization; sBA = serum bile acid.
Populations
Inclusion and Exclusion Criteria
The ICONIC trial included pediatric patients aged 12 months to 18 years with a diagnosis of ALGS based on diagnostic criteria as shown in Table 8, evidence of cholestasis (described in Table 7), and significant pruritus, defined as an average daily score of greater than 2 on the ItchRO questionnaire for 2 consecutive weeks in the screening period. Key exclusion criteria included chronic diarrhea requiring specific IV fluid or nutritional intervention, surgical disruption of the enterohepatic circulation, liver transplant, decompensated cirrhosis, or history or presence of other concomitant liver disease.
Patients were considered eligible for the initial 52-week optional follow-up treatment period if they had completed the protocol through the week 48 visit with no safety concerns. Patients who had undergone a surgical disruption of the enterohepatic circulation were not eligible to enter into the follow-up treatment period. Patients were considered eligible for the optional long-term follow-up treatment period if they completed the protocol through the week 48 visit with no major safety concerns or discontinued due to safety reasons judged unrelated to maralixibat, and laboratory results had returned to levels acceptable for this patient population or individual and the patient did not meet any of the protocol’s stopping rules at the time of entry into the follow-up period. Patients who were discontinued for other reasons were considered for the follow-up periods on an individual basis. All exclusion criteria mentioned for the core study applied upon entry into the optional long-term follow-up treatment period, with the exception of patients weighing more than 50 kg at screening.
Table 8: ALGS Diagnostic Criteria Used in the ICONIC Pivotal Trial
ALGS family historya | Paucity | Jagged-1 or NOTCH2 mutation | Number of major clinical criteria needed for diagnosisb |
|---|---|---|---|
Present or absent | Present | Identifiedc | Any or no features |
None (proband) | Absent or unknown | Identified | 1 or more features |
None (proband) | Present | Not identifiedd | 3 or more features |
None (proband) | Absent or unknown | Not identified | 4 or more features |
Present | Absent or unknown | Identified | Any or no features |
Present | Present | Not identified | 1 or more features |
Present | Absent or unknown | Not identified | 2 or more features |
ALGS = Alagille syndrome.
aFamily history = ALGS present in a first-degree relative.
bMajor clinical criteria or features for ALGS include: cholestasis consistent cardiac, renal, vascular, ocular, or skeletal involvement; or characteristic Alagille facies.
cIdentified = Jagged-1 or NOTCH2 mutation identified in clinical laboratory.
dNot identified = not identified on screening, or not screened for.
Source: Sponsor-submitted internal package.37
Interventions
Open-Label Phase: Dose-Escalation Period (Day 0 to Week 6)
During the first 6 weeks of the study, the dose-escalation period, the dose of maralixibat was increased at weekly intervals up to 380 mcg/kg/day once daily as follows:
Week 1 dose: 13.3 mcg/kg/day once daily
Week 2 dose: 33.3 mcg/kg/day once daily
Week 3 dose: 66.5 mcg/kg/day once daily
Week 4 dose: 133 mcg/kg/day once daily
Week 5 dose: 266 mcg/kg/day once daily
Week 6 dose: 380 mcg/kg/day once daily
Patients received maralixibat daily for at least 7 days at each dose level during the dose-escalation period.
The dosage and dosing regimen of concomitant drug therapy other than that specified by the protocol was not changed during the first 22 weeks of study, except for weight-based dose adjustments and vitamin supplementation. No new medications used to treat pruritus could be added during the first 22 weeks of the study.
Open-Label Phase: Stable-Dosing Period (Weeks 7 to 18)
Following the 6-week dose-escalation period, patients continued dosing for another 12 weeks at the dose administered at week 6, which may have been 380 mcg/kg/day or the highest tolerated dose below 380 mcg/kg/day.
RWD Phase (Weeks 19 to 22)
At the week 18 visit, patients were randomized 1:1 to continue to receive maralixibat or a corresponding placebo for 4 weeks. Randomization was stratified by whether the patient achieved a greater than 50% reduction in sBA between baseline and weeks 12 or 18.
Long-Term Exposure Period (Weeks 23 to 48)
Following the 4-week study drug RWD period, patients who received placebo returned to the maralixibat dose received during the initial escalation. Patients who were randomized to receive maralixibat during this period continued to receive the same dose of maralixibat and, following week 22, a simulated dose escalation occurred to maintain the blind in the RWD period. Dosing with maralixibat continued in a 26-week, long-term exposure period to complete 48 weeks of study. Dose adjustments were made as needed due to intolerance or change of greater than or equal to 10% in a patient’s body weight since the screening visit.
Fifty-Two–Week Optional Follow-Up Treatment Period
At week 48, the investigator assessed all patients to determine their willingness and eligibility to move into the 52-week optional follow-up treatment period. The following 3 possible scenarios may have occurred:
Patients who were eligible to roll over into the optional follow-up treatment period with no maralixibat dosing interruption or an interruption of less than 7 days were maintained at the same dose level that they had been taking at week 48.
Patients who were eligible to roll over into the optional follow-up treatment period with a maralixibat dosing interruption of 7 days or more were dose escalated beginning at 35 mcg/kg/day up to a maximum of 380 mcg/kg/day or highest tolerated dose.
Patients who did not wish to enter the follow-up treatment period were contacted via telephone by the study site approximately 30 days after the last dose of maralixibat.
Long-Term Optional Follow-Up Treatment Period
Upon completion of the 52-week optional follow-up treatment period, patients eligible for the optional follow-up treatment period either continued receiving the same dose level of maralixibat that they had been taking at the completion of the follow-up period with morning dosing only or started twice-daily dosing. Eligibility for twice-daily dosing was determined based on efficacy as measured by sBA level and ItchRO score. Patients without dosing interruption or interruption of less than 7 continuous days who met sBA and ItchRO eligibility criteria started twice-daily dosing (afternoon dose escalation) up to their maximum tolerated dose or at most a maximum daily dose of 380 mcg/kg twice daily (i.e., 760 mcg/kg/day). Patients with a maralixibat dosing interruption of greater than or equal to 7 days who met sBA and ItchRO eligibility criteria underwent dose escalation starting with 4-times-per-day dosing (at most a maximum daily dose of 380 mcg/kg), followed by an afternoon dose escalation up to their maximum tolerated dose or to a maximum daily dose of 380 mcg/kg twice daily (i.e., 760 mcg/kg/day).
Patients could continue to receive maralixibat until they were eligible to enter another maralixibat study, until maralixibat was available commercially, or until the sponsor stopped the program or development in this indication, whichever occurred first.
Concomitant Medications
Patients were required to discontinue bile acid resins for at least 28 days before screening and for the duration of the study. The dosage and dosing regimen of concomitant drug therapy other than that specified by the protocol should not change during the first 22 weeks of study, with the exception of weight-based dose adjustments and vitamin supplementation. No new medications used to treat pruritus were to be added during the first 22 weeks of the study.
Outcomes
Summarized outcomes are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical expert(s) consulted by CADTH and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected outcomes that were most relevant to inform CADTH expert committee deliberations and finalized this list of outcomes in consultation with members of the expert committee. All summarized efficacy end points identified in Table 9 were assessed using GRADE. Select notable harms outcomes considered important for informing CADTH expert committee deliberations were also assessed using GRADE. A description of outcome measures and their measurement properties is provided in Table 10. Details regarding additional efficacy outcomes not assessed by GRADE can be found in Appendix 1.
Serum Bile Acid
In other pediatric cholestatic diseases (progressive familial intrahepatic cholestasis and biliary atresia), sBA was shown to have prognostic implications, with patients with higher sBA levels being more likely to progress to end-stage liver disease or to undergo liver transplant.38,39 It has not yet been defined what sBA level or reduction can be associated with improved long-term outcome in patients with ALGS. In the pivotal trial, an sBA responder was defined as a patient with a reduction in sBA greater than or equal to 50% from baseline to weeks 12 or 18. An sBA responder at week 48 was defined as a patient with a reduction in sBA greater than or equal to 50% from baseline to week 48. According to the clinical experts consulted by CADTH for this review, there is insufficient evidence on the association between sBA change and pruritis in patients with ALGS.
Itch Reported Outcome
Pruritus was assessed using the ItchRO, which comprises 2 clinical outcome assessment instruments — namely, ItchRO(Obs), the caregiver-reported version, and the ItchRO(Pt), the patient-reported version for patients aged 9 years or older.40 The caregiver-rated ItchRO(Obs) instrument was selected as a tool for assessing pruritus given the expectation, that a large proportion of study patients would be aged younger than age 9 years. The ItchRO(Pt) instrument was included in the ICONIC trial to collect supportive data in children who were old enough to assess their own pruritus (≥ 9 years of age or ≥ 5 years of age with the assistance of their caregiver).
The ItchRO(Obs) consists of a morning and an evening diary entry that asks the caregiver about the severity of the child’s itch-related symptoms using a single item (item 1). The item pertaining to nighttime itching (from bedtime until the time the child woke up) is answered in the morning diary entry, and the item pertaining to daytime itching (from the time the child woke up in the morning until bedtime) is answered in the evening diary entry. Item 1, also called the ItchRO severity score, has a range from 0 to 4, with a higher score indicating increasing itch severity (i.e., 0 = none, 1 = mild, 2 = moderate, 3 = severe, 4 = very severe). The daily score was taken as the highest score of the morning and evening scores, representing the greatest severity of itching over the 24-hour period. The weekly average severity score, defined as the 7-day average of the daily maximum of morning and evening ratings before a study visit, was used in the psychometric validation analysis.
A description of the ItchRO instrument, as well as its psychometric properties and MID estimates, are summarized in Table 10.
Liver Biomarkers and Enzymes
Efficacy end points in the pivotal trial included liver chemistry markers for cholestasis (total and direct bilirubin, ALP) and hepatocellular involvement (ALT). For these parameters, the prespecified secondary end point analyses included 1) change from baseline to weeks 18 and 2) change from weeks 18 to 22. Furthermore, change from baseline to weeks 22, 48, and then every 12 weeks were analyzed as additional efficacy end points.
Health-Related Quality of Life: PedsQL
The PedsQL Generic Core Scales are composed of 23 items to assess pediatric HRQoL measurements across 6 subscales: physical functioning, physical symptoms (only applicable for infants aged 1 to 24 months), emotional functioning, social functioning, cognitive functioning (only applicable for infants aged 1 to 24 months), and school functioning (only applicable for children aged 2 to 18 years). The total scale score encompasses all 23 items. Each item consists of 5-level verbal descriptor response options (0 to 4 points). Items are reverse scored and linearly transformed to a 0 to 100 scale so that higher scores indicate better HRQoL (less negative impact). The PedsQL Multidimensional Fatigue Scale is composed of 18 items across 3 subscales: general fatigue, sleep/rest fatigue, and mental fatigue. Respondents use the scale to indicate how frequently certain fatigue-related symptoms and complaints trouble them. The Multidimensional Fatigue Scale score is computed from all items of the PedsQL Multidimensional Fatigue Scale. Items are reverse scored and linearly transformed to a 0 to 100 scale so that higher scores indicate better HRQoL (fewer symptoms of fatigue). In the ICONIC trial, PedsQL questionnaires were prospectively collected via a caregiver proxy-report during the ICONIC study and analyzed retrospectively. Measurements from baseline and week 48 were included in this analysis.
A description of the PedsQL, its psychometric properties, and MID estimates are summarized in Table 10.
Change in Body Height and Weight
To compare growth parameters to the normal population, z scores were calculated in the ICONIC trial. Changes greater than 0 in height and weight z scores indicate that patients grow faster than their healthy peers (catch-up growth), whereas changes less than 0 indicate a deepening growth deficit. Height and weight z scores are based on a patient’s sex and age at each scheduled visit. For patients younger than 24 months, the WHO growth charts are recommended by the Centers for Disease Control and were used to derive z scores. For patients aged at least 24 months, the Centers for Disease Control growth charts were used to derive z scores.
Harms Outcomes
The harms outcomes assessed in the pivotal trial included the proportion of patients with AEs, SAEs, and notable harms. AEs were any unfavourable and unintended sign, symptom, or disease temporally associated with the study or use of an investigational drug product, which may or may not be related to the study drug. The criteria for SAEs included death; events that were life-threatening, required hospitalization, or prolongation of hospitalization, or resulted in persistent or significant disability or incapacity; was a congenital anomaly or birth defect; or an event that may have required medical or surgical intervention to prevent 1 of the other outcomes listed.
Table 9: Outcomes Summarized From the ICONIC Trial
Outcome measure | Time point | ICONIC trial |
|---|---|---|
Liver-related outcomes | ||
Change in fasting sBA levels (μmol/L) in patients who previously responded to treatment with maralixibat, defined by a reduction in sBA of ≥ 50% from baseline to weeks 12 or 18 | Weeks 18 to 22 | Primary |
Change in pruritus as measured by ItchRO(Obs) and ItchRO(Pt) weekly average morning severity score in patients who previously responded to treatment with maralixibat, defined by a reduction in ItchRO scale of > 1 point from baseline to weeks 12 or 18 | Weeks 18 to 22 | Secondary |
Change in liver biomarkers and enzymes (ALT, ALP, total and direct bilirubin) | Weeks 18 to 22 | Secondary |
Change in body height and weight | Baseline to week 48; baseline to week 100 (LOCF) | Additional efficacy end point |
HRQoL outcomes | ||
Change in PedsQL total score (parent) and PedsQL Multidimensional Fatigue Scale score (parent) | Weeks 18 to 22 | Additional efficacy end point |
Safety outcomes | ||
Patients with SAEs | Weeks 18 to 22 | Safety |
Diarrhea, AE | Weeks 18 to 22 | Safety |
Abdominal pain, AE | Weeks 18 to 22 | Safety |
FSV deficiency, AE | Weeks 18 to 22 | Safety |
AE = adverse event; ALP = alanine phosphatase; ALT = alanine transaminase; FSV = fat-soluble vitamin; HRQoL = health-related quality of life; ItchRO = Itch Reported Outcome; ItchRO(Obs) = Itch Reported Outcome (observer); ItchRO(Pt) = Itch Reported Outcome (patient); LOCF = last observation carried forward; PedsQL = Pediatric Quality of Life Inventory; SAE = serious adverse event; sBA = serum bile acid.
aIdentified as an end point of importance by the clinical experts consulted but not measured in the ICONIC pivotal trial.
Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.17
Source: ICONIC Clinical Study Report.16
Considerations that went into the selection of efficacy outcomes to be summarized and assessed using GRADE include the following:
The primary outcome in the pivotal trial; change in fasting sBA levels (μmol/L) in patients who previously responded to treatment with maralixibat, defined by a reduction in sBA of greater than or equal to 50% from baseline to weeks 12 or 18 was defined as important by the clinical experts consulted and is an outcome that provides the source for key input in the sponsor’s pharmacoeconomic model.
Change in pruritus was defined as a very important outcome by the clinical experts consulted and noted by the patient and clinician group input received, resulting in the selection of the outcome of change in pruritus as measured by ItchRO(Obs) and ItchRO(Pt) weekly average morning severity score in patients who previously responded to maralixibat treatment, defined by a reduction in ItchRO scale of greater than 1 point from baseline to weeks 12 or 18.
Other liver-related outcomes defined as important by the clinical experts consulted included change in liver biomarkers and enzymes (ALT, ALP, total and direct bilirubin).
Change in body height and weight was described as important by the clinical experts consulted and the patient input received.
HRQoL outcomes were defined as important by the clinical experts and patient input received. The PedsQL total score (parent) was selected as it encompasses all 23 items of the PedsQL, and the PedsQL Multidimensional Fatigue Scale score (parent) was selected given the prevalence and severity of fatigue in the indicated patient population as noted by the clinical experts consulted and patient input received.
Safety outcomes defined as important to the clinical experts consulted included SAEs, diarrhea, abdominal pain, and FSV deficiency.
Statistical Analysis
Sample Size and Power Calculation
The planned sample size of 30 evaluable patients with ALGS was based on practical considerations, rather than a desired power for a prespecified difference.
Table 10: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
ItchRO | Comprises 2 clinical outcome assessments: the caregiver- reported version, ItchRO(Obs); and the patient-reported version for patients ≥ 9 years of age, ItchRO(Pt).17 ItchRO(Obs) consists of a morning and evening diary entry that rates severity of itch-related symptoms in a single item. Nighttime itching (from bedtime until waking up) is answered in the morning diary entry, and daytime itching (from waking up until bed time) is answered in the evening diary entry.17 ItchRO severity score ranges from 0 to 4, with the higher score indicating increased itch severity (0 = none, 4 = very severe).17 The daily score is taken from the higher score of the morning and evening scores, representing the greatest severity of itching over the 24-hour period.17 The weekly average severity score is defined as the 7-day average of the daily maximum of morning and evening ratings before a study visit.17 | The 7-day and 28-day average severity scores were used in psychometric validation analyses. Validity: Content validity has been ensured during the development stage through literature review and existing measures of itching; input from clinical experts, patients, and caregivers; and other processes.40 Convergent validity (Pearson r = 0.63 to 0.78) between ItchRO(Obs) and CSS, divergent validity (r = –0.08 to ––0.96) between ItchRO(Obs) and PedsQL scores were noted.41 In known-group analysis, worse ItchRO(Obs) scores were associated with more pruritus-related skin damage, as measured by the CSS.41 Reliability: Test-retest reliability was found to be satisfactory (ICC ≥ 0.70) when measured at screening and repeated at baseline.41 Responsiveness Changes in ItchRO(Obs) scores over time (pretreatment and posttreatment) have not been assessed.40 | Pediatric patients with ALGS: With CIC-itch as an anchor using the change from baseline to week 18, within-person meaningful change is found to be –1.86 to –0.86.17 Based on distribution method (2 SEMs), changes in scores of –0.64 to –0.56 were indicative of meaningful changes.41 |
PedsQL | Self-reported and/or caregiver-reported HRQoL instrument for infants (aged 1 to 12 months), children (aged 13 to 24 months, 2 to 4 years, 5 to 7 years, and 8 to 12 years) and adolescents (aged 13 to 18 years). Pediatric self-report form is used in children aged 5 to 18 years. Parent proxy-report form of child HRQoL is used for children aged 12 months to 18 years.17 There are 23 items in 4 domains: physical functioning (8), emotional functioning (5), social functioning (5), and school functioning (5). Each item is scored in 5-point Likert scale, with options ranging from 0 to 4. Total scale score encompasses all 23 items, with higher scores indicating better HRQoL (less negative impact).17 PedsQL MDFM: Parent-reported PedsQL MDFM addresses 3 domains: general fatigue, sleep/rest fatigue, and cognitive fatigue, with 6 questions per domain.17 | Psychometric properties have not been assessed in pediatric patients with ALGS. | Pediatric patients with chronic health conditions, not including ALGS, for the PedsQL Generic Core Scales: Change of 4 to 5 points is considered to be meaningful.42 |
ALGS = Alagille syndrome; CIC = Caregiver Impression of Change; CSS = Clinician Scratch Scale; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; ItchRO = Itch Reported Outcome; ItchRO(Obs) = Itch Reported Outcome (observer); ItchRO(Pt) = Itch Reported Outcome (patient); MDFM = Multidimensional Fatigue Module; MID = minimal important difference; PedsQL = Pediatric Quality of Life Inventory; SEM = standard error of mean.
Statistical Testing
For the primary efficacy end point, the difference between treatment groups from weeks 18 to 22 in sBA was evaluated using an analysis of covariance (ANCOVA) model with treatment group as a factor, and week 18 sBA as a covariate, assessed in the mITT population. An ANCOVA model that includes the stratification variable sBA responder indicator as an additional covariate was also performed on the ITT population. This model also included the sBA responder covariate by treatment sequence interaction term. The LS mean difference between treatment groups with standard error, 95% CI for the LS mean difference, and P value for testing if the treatment group LS means were equal, was calculated to determine if the change in sBA levels between the treatment groups were statistically significant.
Secondary and other efficacy variables that are continuous measures were analyzed similarly to the primary efficacy analyses in the ITT population using summary statistics, and the majority were assessed by ANCOVA. If there appeared to be a relative significant difference among treatment sequences with respect to baseline characteristics, that baseline variable may have been added to the statistical model as a blocking factor or covariate to determine the effect on treatment. For this analysis, an interaction term for the covariate by treatment sequence was not included. ANCOVA models that only included the baseline value (of the variable of interest) as a single covariate were also included.
Efficacy measures that are categorical binary responder outcomes were analyzed using the chi-square or Fisher’s exact test, as appropriate based on sample sizes. Fisher’s exact test was chosen over the chi-square test if any of the cell counts were less than 5.
Multiple Comparisons
No adjustments were made for multiple comparisons.
As the definition of the pruritus end point used to assess efficacy was not prespecified in the protocol and the type I error was not specifically controlled for, P values for the analyses were treated as nominal.
Handling of Drop-Outs or Missing Data
For ItchRO(Obs) and ItchRO(Pt) weekly average scores, an average of daily scores was computed if at least 4 of the 7 daily ItchRO scores for a 7-day period were reported. If fewer than 4 ItchRO scores were reported, then the weekly average from the previous compliant week was used in an LOCF format. ItchRO(Obs) and ItchRO(Pt) weekly average morning severity scores derived for each week over the 4-week RWD phase were analyzed using ANCOVA and a restricted maximum likelihood (REML)–based repeated-measures approach as the principal sensitivity analysis method. The mixed model for repeated measures (MMRM) analysis model was used for handling missing data, with change from baseline (week 18) as the dependent variable; it included the fixed, categorical effects of treatment group, visit (time point), and treatment-group-by-visit interaction as well as the continuous covariates of baseline and baseline-by-visit interaction. The protocol stated that missing data imputation was not done for other efficacy end points.
In addition to the time points specified in the protocol, efficacy variables analyzed by time point were analyzed (as a sensitivity analysis) at the following LOCF time points: week 18 (LOCF), week 22 (LOCF), week 48 (LOCF), and week 100 (LOCF) time points, where appropriate.
For patients who terminated early from the study before week 100 or were otherwise missing weeks 18, 22, 48, and/or 100, data were imputed in an LOCF approach as follows:
open-label phase: last observation on or before week 18 imputed as week 18 (LOCF)
RWD phase: last observation between week 18 and the end of week 22 imputed as week 22 (LOCF) (only applicable for ItchRO[Obs] weekly average score variables)
after RWD phase: last observation between week 22 and the end of week 48 imputed as week 48 (LOCF) and last observation after week 22 and end of week 100 imputed as week 100 (LOCF).
For PedsQL scale scores, if more than 50% of the items in the scale were missing, the scale score was not computed.
If a patient had a missing sBA, ItchRO, or CSS value at a week required in determining responder status, then the missing change from baseline value would be considered as not meeting the criteria for a responder.
Subgroup Analyses
For ItchRO(Obs) weekly average morning severity scores and sBA levels, summary statistics by analysis visit were conducted for the following subgroups:
Age group (up to 24 months, 2 to 12 years, greater than 12 years)
Baseline sBA (less than 275 μmol/L, greater than or equal to 275 μmol/L)
Baseline total bilirubin (less than 3.8 mg/dL, greater than or equal to 3.8 mg/dL)
Baseline ALT (less than 90 U/L, greater than or equal to 90 U/L)
Baseline ItchRO(Obs) weekly average morning severity score (less than 3 points, greater than or equal to 3 points)
For the subgroup analyses, observed and change from baseline (day 0) values were presented for the open-label and after RWD phases. For the RWD phase, observed and change from week 18 values were presented for the maralixibat and placebo treatment groups.
Detailed statistical analysis of efficacy end points including adjustment factors and sensitivity analyses are presented in Table 11.
Table 11: Statistical Analysis of Efficacy End Points in the ICONIC Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Primary end point: Change in fasting sBA levels (μmol/L) in patients who previously responded to treatment with maralixibat from weeks 18 to 22 (mITT population) | ANCOVA | Treatment group as a fixed effect and baseline value as a covariate were used | LOCF as a sensitivity analysis | An ANCOVA model controlling for sBA responder group, in ItchRO responders, in sBA responders at week 48, and adjusting for a patient hospitalized during the randomized withdrawal phase Ancillary analyses: To account for the drop-outs in the follow-up phase of the study, sensitivity analyses applying 3 different imputation methods (LOCF, MMRM, and multiple imputation) were conducted |
Secondary end point: Change in fasting sBA levels (μmol/L) from baseline to week 18 | Student’s t test | NA | As primary end point | NA |
Secondary end point: Change in pruritus as measured by ItchRO(Obs) and ItchRO(Pt) from weeks 18 to 22 | As primary end point | As primary end point | LOCF Missing visit data were imputed by MMRM as a sensitivity analysis | Assessments controlling for the various baseline characteristics (i.e., sBA responder status) subgroups (i.e., baseline age, sBA, bilirubin, ALT), missing data assumptions, and covariate adjustments Ancillary analyses: To account for the drop-outs in the follow-up phase of the study, sensitivity analyses applying 4 different imputation methods (LOCF, MMRM, REML, and multiple imputation) were conducted |
Secondary end point: Change in pruritus as measured by ItchRO(Obs) and ItchRO(Pt) from baseline to week 18 | Student’s t test | NA | As primary end point | NA |
Secondary end point: Change in liver enzymes (ALT and ALP) and bilirubin (total and direct) from weeks 18 to 22 | As primary end point | As primary end point | As primary end point | An ANCOVA model controlling for sBA responder group, in the mITT population, and in ItchRO responders |
Secondary end point: Change in liver enzymes (ALT and ALP) and bilirubin (total and direct) from baseline to week 18 | Student’s t test | NA | As primary end point | NA |
Additional efficacy end point: Change in body height and body weight z scores from baseline | Student’s t test | NA | As primary end point | Ancillary analyses: To account for the drop-outs in the follow-up phase of the study, sensitivity analyses applying 3 different imputation methods (LOCF, MMRM, and multiple imputation) were conducted |
Additional efficacy end point: Change in PedsQL total score and Multidimensional Fatigue Scale score from weeks 18 to 22 | As primary end point | As primary end point | If more than 50% of the items in the scale are missing, the scale score was not computed | An ANCOVA model controlling for sBA responder group |
ALP = alkaline phosphatase; ALT = alanine transaminase; ANCOVA = analysis of covariance; ItchRO = Itch Reported Outcome; ItchRO(Obs) = Itch Reported Outcome (observer); ItchRO(Pt) = Itch Reported Outcome (patient); LOCF = last observation carried forward; mITT = modified intent to treat; MMRM = mixed model for repeated measures; NA = not applicable; PedsQL = Pediatric Quality of Life Inventory; REML = restricted maximum likelihood; sBA = serum bile acid.
Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.17
Source: ICONIC Clinical Study Report.16
Analysis Populations
Table 12 summarizes the analysis populations in the ICONIC pivotal trial.
Table 12: Analysis Populations of ICONIC (LUM001-304) Study
Population | Definition |
|---|---|
Safety | All patients who were enrolled and received at least 1 dose of the study drug |
ITT | All patients who were enrolled and received at least 1 dose of the study drug |
mITT | All patients who were enrolled, received the study drug through week 18, and had a reduction from baseline in sBA of ≥ 50% at the week 12 or 18 measurement (sBA responder) |
ITT = intent to treat; mITT = modified intent to treat; sBA = serum bile acid.
Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.17
Source: ICONIC Clinical Study Report.16
Results
Patient Disposition
Thirty-six children with ALGS were screened for the ICONIC pivotal trial, 5 (13.9%) of whom were excluded due to screening failure. In total, 31 patients were enrolled into the open-label period of the study, during which 2 (6.5%) discontinued due to AEs. In total, 29 patients entered the RWD period and were randomized to continue maralixibat treatment (n = 13) or administered placebo (n = 16). A total of 28 patients (90.3%) completed the 48-week core study, with 1 (3.4%) patient discontinued due to an AE. After finalization of the core study, 23 patients (79.3%) consented to enter the LTE period at week 48 and 14 (60.9%) remained on study at the time of the interim report. A summary of patient disposition is presented in Table 13.
Baseline Characteristics
In the overall study population (N = 31), there were more males (19 of 31 [61.3%]) than females (12 of 31 [38.7%]) at baseline and in the maralixibat (9 of 13 [69.2%]) and placebo groups (10 of 16 [62.5%]) during the RWD period. The mean age in the overall study population was 5.4 years (range, 1 to 15 years) and was similar between the maralixibat and placebo groups. The majority of the patients were from Australia and France (9 of 31 [29.0%] each in the overall study population). The mean time since the original diagnosis of ALGS was 66.2 months in the overall study population, with 64.5 months in the maralixibat group and 73.2 months in the placebo group during the RWD phase. In the overall study population, 8 of 31 (25.8%) of patients had a family history of ALGS (1 of 13 [7.7%] and 7 of 16 [43.8%] in the maralixibat and placebo group, respectively). All enrolled patients had the Jagged-1 mutation present. During the RWD, 4 of 13 (30.8%) of patients in the maralixibat group had documented bile duct paucity (46.2% unknown), compared with 12 of 16 (75.0%) (18.8% unknown) in the placebo group. The majority of patients in the overall study population (29 of 31 [93.5%]) had used previous antipruritic treatment, with a similar distribution in the maralixibat and placebo groups.
Of note, the mean baseline sBA (μmol/L) was higher in the maralixibat group (317.97) than in the placebo group (249.56). ItchRO(Obs) weekly average morning severity score was similar between the 2 arms (2.88 versus 2.93). All liver biomarkers and enzymes (ALT, ALP, total and direct bilirubin) were consistently higher in the maralixibat group than in the placebo group. Mean baseline 7alpha-hydroxy-4-cholesten-3-one levels were higher in the maralixibat group (14.77 [standard deviation (SD) = 19.874] ng/mL) than in the placebo group (6.53 [SD = 8.728] ng/mL).
The baseline characteristics outlined in Table 14 are limited to those that are most relevant to this review or that were felt to affect the outcomes or interpretation of the study results.
Table 13: Patient Disposition in the ICONIC Trial
Patient disposition | ICONIC (LUM001-304) study | ||||
|---|---|---|---|---|---|
Open-label phase (day 1 to week 18) | Randomized withdrawal phase (weeks 19 to 22) | ARW phase (weeks > 22 to 48) | LTE phase (weeks > 48) | ||
MRX (N = 31) | MRX (N = 13) | Placebo (N = 16) | MRX (N = 29) | MRX (N = 23) | |
Screened, N | 36 | ||||
Reason for screening failure, N (%) | 5 (13.9) | ||||
Failed to meet eligibility criteria, inclusion criteria 12 of study protocola | 4 (11.1) | ||||
Failed to meet eligibility criteria, exclusion criteria 4 of study protocolb | 1 (2.8) | ||||
Enrolled/randomized/continued, N | 31 | 13 | 16 | 29 | 23 |
Safety population,c N | 31 | 13 | 16 | 29 | 23 |
ITT population,d N (%) | 31 (100.0) | 13 (100.0) | 16 (100.0) | 29 (100.0) | 23 (100.0) |
mITT population,e N (%) | 15 (48.4) | 5 (38.5) | 10 (62.5) | 15 (51.7) | 15 (65.2) |
Dose reduced during treatment period, N (%) | 0 | 0 | 0 | 0 | 3 (13.0) |
Completed treatment period,f N (%) | 29 (93.5) | 13 (100.0) | 16 (100.0) | 23 (79.3) | 14 (60.9) |
Discontinued during treatment period,g N (%) | 2 (6.5) | 0 | 0 | 6 (20.7) | 9 (39.1) |
Reason for discontinuation,g N (%) | |||||
Did not consent to PA3 extension | NA | NA | NA | 5 (17.2) | NA |
Did not consent to PA4 extension | NA | NA | NA | NA | 4 (17.4) |
Adverse event | 2 (6.5) | 0 | 0 | 1 (3.4) | 3 (13.0) |
Death | 0 | 0 | 0 | 0 | 0 |
Lost to follow-up | 0 | 0 | 0 | 0 | 0 |
Physician decision | 0 | 0 | 0 | 0 | 1 (4.3) |
Withdrawal by caregiver | 0 | 0 | 0 | 0 | 1 (4.3) |
ALT = alanine transaminase; ARW = after randomized withdrawal; INR = international normalized ratio; ITT = intent to treat; LTE = long-term extension; mITT = modified intent to treat; MRX = maralixibat; NA = not applicable; PA = protocol amendment; sBA = serum bile acid; ULN = upper limit of normal.
Notes: Percentages are based on the number of patients in the safety population within each treatment period and treatment group. Assigned treatment groups are presented.
Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.17
aAverage daily score greater than 2 on the ItchRO questionnaire (maximum possible daily score of 4) for 2 consecutive weeks in the screening period, before dosing. A daily score is the higher of the scores for the morning and evening ItchRO. The average daily score is the sum of all daily scores divided by the number of days the ItchRO was completed.
bDecompensated cirrhosis (ALT 15 times greater than the ULN, INR greater than 1.5 [unresponsive to vitamin K therapy], albumin < 3.0 g/dL, history or presence of clinically significant ascites, variceal hemorrhage, and/or encephalopathy).
cThe safety population includes all patients who were enrolled and received at least 1 dose of study drug.
dThe ITT population includes all patients who were enrolled and received at least 1 dose of study drug.
eThe mITT population includes all patients who were enrolled, received study drug through week 18, and had a reduction from baseline in sBA of greater than or equal to 50% at the weeks 12 or 18 measurement (sBA responder).
fPatients are considered to have completed the treatment period if they were in the study at the time of the data cut-off.
gPatients who did not consent to the optional long-term treatment extensions (PA3 or PA4) are not considered to have completed treatment.
Source: ICONIC Clinical Study Report.16
Table 14: Summary of Baseline Characteristics in the ICONIC Pivotal Trial
Characteristics | All patients (N = 31) | Randomized withdrawal phase (weeks 19 to 22) | |
|---|---|---|---|
Maralixibat (N = 13) | Placebo (N = 16) | ||
Age, in years, mean (SD) | 5.4 (4.25) | 5.5 (5.03) | 5.8 (3.75) |
Sex, male, n (%) | 19 (61.3) | 9 (69.2) | 10 (62.5) |
Sex, female, n (%) | 12 (38.7) | 4 (30.8) | 6 (37.5) |
Country, n (%) | |||
Australia | 9 (29.0) | |||||| | |||||| |
Belgium | 5 (16.1) | |||||| | |||||| |
France | 9 (29.0) | |||||| | |||||| |
Spain | 3 (9.7) | |||||| | |||||| |
Poland | 2 (6.5) | |||||| | |||||| |
UK | 3 (9.7) | |||||| | |||||| |
Family history of ALGS, yes, n (%) | 8 (25.8) | |||||| | |||||| |
Time since original ALGS diagnosis, months, mean (SD) | |||| |||||| | |||| |||||| | |||| |||||| |
Presence of bile duct paucity, n (%) | |||
Yes | 18 (58.1) | 4 (30.8) | 12 (75.0) |
No | 4 (12.9) | 3 (23.1) | 1 (6.3) |
Unknown | 9 (29.0) | 6 (46.2) | 3 (18.8) |
Mutation present, Jagged-1, n (%) | 31 (100.0) | 13 (100.0) | 16 (100.0) |
History of receiving treatment for pruritus, n (%) | |||
Any medication | 29 (93.5) | 12 (92.3) | 15 (93.8) |
UDCA | 25 (80.6) | 10 (76.9) | 13 (81.3) |
Rifampicin | 23 (74.2) | 10 (76.9) | 12 (75.0) |
Naltrexone | 1 (3.2) | 1 (7.7) | 0 (0) |
Sertraline | 1 (3.2) | 0 (0) | 1 (6.3) |
ItchRO(Obs) weekly average morning severity score,b mean (SD) | 2.91 (0.55) | 2.88 (0.54) | 2.93 (0.56) |
Clinician Scratch Scale score,c mean (SD) | 3.3 (0.90) | 3.0 (1.08) | 3.5 (0.73) |
sBA, μmol/L, mean (SD) | 283.43 (210.57) | 317.97 (233.67) | 249.56 (196.80) |
ALT, U/L, mean (SD) | 181.0 (108.56) | 217.8 (149.93) | 147.0 (54.60) |
ALP, U/L, mean (SD) | 601.3 (210.6) | 637.5 (386.0) | 585.1 (169.2) |
Total bilirubin, mg/dL, mean (SD) | 6.09 (5.78) | 6.52 (6.57) | 4.83 (4.27) |
Direct bilirubin, mg/dL, mean (SD) | 4.57 (3.67) | 4.69 (3.80) | 4.04 (3.59) |
Cholesterol, mg/dL, mean (SD) | 512.1 (419.82) | 557.3 (324.0) | 461.0 (353.0) |
7alpha-hydroxy-4-cholesten-3-one, ng/mL, mean (SD) | 10.32 (14.66) | 14.77 (19.87) | 6.52 (8.73) |
ALGS = Alagille syndrome; ALP = alkaline phosphatase; ALT = alanine transaminase; ItchRO(Obs) = Itch Reported Outcome (observer); sBA = serum bile acid; SD = standard deviation; UDCA = ursodeoxycholic acid.
Source: ICONIC Clinical Study Report.16 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)17
Exposure to Study Treatments
Table 15 summarizes study drug exposure and compliance by study phase and by overall study population. Maralixibat doses are shown using maralixibat chloride. In the overall study population, the mean treatment duration was 944 days (SD = 587.07), with a mean average daily dose of 439.8 mcg/kg (SD = 133.47) once daily at the time of the data cut-off. During the RWD phase, the mean duration of study drug exposure was 29.9 days (SD = 4.09) in the maralixibat group and 29.1 days (SD = 2.94) in the placebo group. Study drug compliance was high (≥ 98.0%) throughout all phases of the study.
Concomitant Medications
Table 16 summarizes concomitant medications that treat pruritus by phase for the safety population. In all study phases, most patients (> 82% in all study phases) were taking at least 1 concomitant medication to treat pruritus. At study entry, 74.2% and 80.6% of patients were receiving stable doses of rifampicin and ursodeoxycholic acid, respectively. The most common medications used (> 10% in the open-label phase) included ursodeoxycholic acid, rifampicin, and phenobarbital. No additional concomitant medications with a frequency of greater than 10% were taken in any other study phase.
Efficacy
Efficacy end points in the ICONIC pivotal trial assessing changes from weeks 18 to 22 are shown in Table 17, and changes from baseline to weeks 18 or 48 are shown in Table 18.
Serum Bile Acid
In the ICONIC trial, the primary efficacy end point was the change in sBA during the 4-week RWD phase in the mITT population (patients with sBA reduction ≥ 50% at weeks 12 or 18). A total of 15 patients met this criterion and were analyzed in the primary end point (5 randomized to maralixibat; 10 to placebo by following a prespecified block randomization procedure). The LS mean difference in change from weeks 18 to 22 in sBA between the maralixibat and placebo groups was –117.28 μmol/L (95% CI, –211.699 to –23.103; P = 0.0464), in favour of maralixibat.
Change in sBA during the 4-week RWD phase was also assessed in the ITT population (Figure 2). The LS mean difference in change from weeks 18 to 22 in sBA between the maralixibat (n = 13) and placebo (n = 16) groups was –113.95 μmol/L (95% CI, –212.68 to –15.21; P = 0.0254), in favour of maralixibat.
In the overall ITT population (N = 31 patients), there was a decrease in sBA during the open-label phases from baseline to week 18 (secondary end point) with a mean change of –87.73 μmol/L (95% CI, –133.37 to –42.09; P = 0.0005) and baseline to week 48 (additional end point) with a mean change of –96.44 μmol/L (95% CI, –162.36 to –30.52; P = 0.0058).
The prespecified sensitivity analyses for change in sBA were consistent with the results of the primary efficacy analysis. Ancillary sensitivity analyses that applied 3 different imputation methods (LOCF, MMRM, and multiple imputation) to assesses mean changes in sBA from baseline to week 48 were consistent with the results of the primary efficacy analysis.
Subgroup analyses were generally consistent with the overall results of the pivotal trial.
In a post hoc analysis assessing data from baseline to week 48, sBA reductions of at least 50%, 60%, 70%, 80%, and 90% had associated ItchRO(Obs) reductions of –1.9, –2.1, –2.3, –2.8, and –2.7, respectively (Appendix 1).
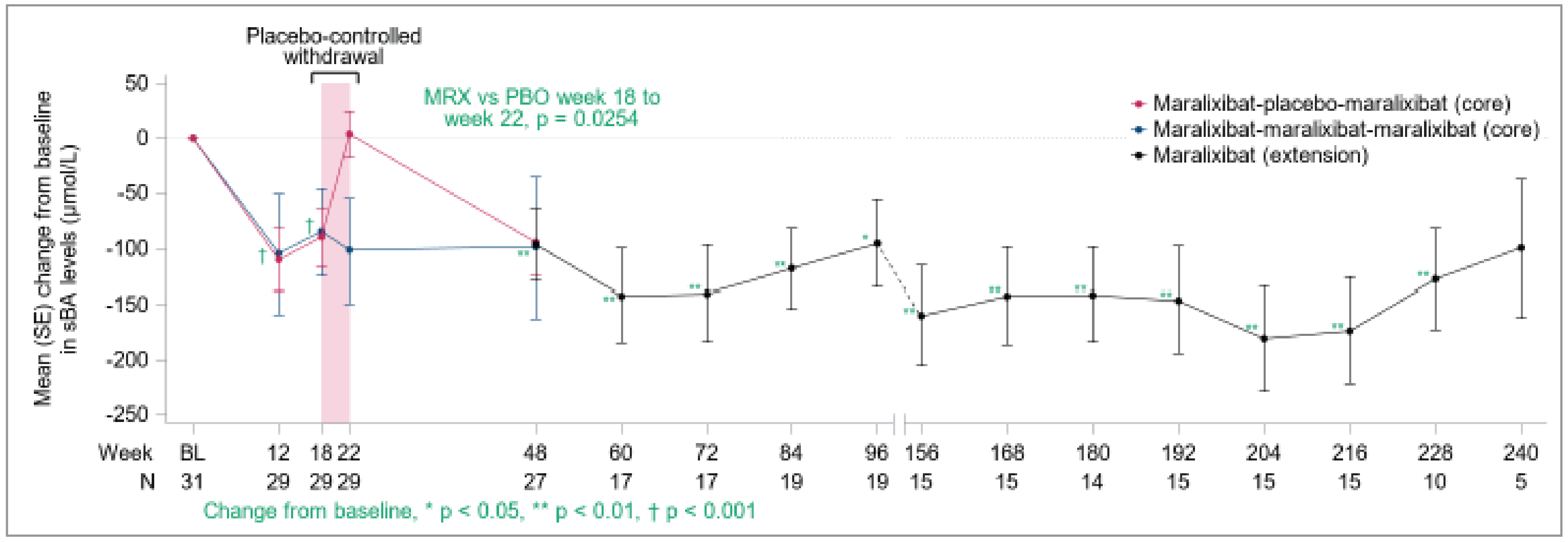
Table 15: Study Drug Exposure and Compliance in the ICONIC Trial — Safety Population
Exposure | ICONIC (LUM001-304) study | ||||||
|---|---|---|---|---|---|---|---|
Open-label phase (day 1 to week 18) | Randomized withdrawal phase (weeks 19 to 22) | ARW phase (weeks > 22 to 48) | LTE phase (weeks > 48) | Overall | |||
Maralixibat q.d. | Maralixibat q.d. | Placebo | Maralixibat q.d. | Maralixibat q.d. | Maralixibat b.i.d. | Maralixibat | |
N = 31 | N = 13 | N = 16 | N = 29 | N = 23 | N = 15 | N = 31 | |
Average daily dose (mcg/kg/day) | |||||||
n | 31 | 13 | 0 | 29 | 23 | 15 | 31 |
Mean (SD) | 302.9 (48.38) | 398.9 (3.96) | NA | 366.4 (29.20) | 385.0 (15.21) | 763.2 (64.29) | 439.8 (133.47) |
Total drug exposure (mcg/kg) | |||||||
n | 31 | 13 | 0 | 29 | 23 | 15 | 31 |
Mean (SD) | 3,7805.1 (907.15) | 11,938.5 (1,656.07) | NA | 66,712.2 (8,042.39) | 153,524.1 (76,034.94) | 546,994.7 (171,012.57) | 483,799.6 (366,040.49) |
Treatment duration (days) | |||||||
n | 31 | 13 | 16 | 29 | 23 | 15 | 31 |
Mean (SD) | 121.4 (22.54) | 29.9 (4.09) | 29.1 (2.94) | 182.1 (15.44) | 404.5 (211.70) | 702.4 (215.76) | 944.3 (587.07) |
Compliance (%) | |||||||
n | 31 | 13 | 16 | 29 | 23 | 15 | 31 |
Mean (SD) | 99.5 (1.10) | 99.7 (0.99) | 99.5 (2.14) | 99.6 (0.89) | 98.7 (2.68) | 98.0 (3.60) | 98.9 (1.89) |
ARW = after randomized withdrawal; b.i.d. = twice daily; LTE = long-term extension; q.d. = every day; SD = standard deviation.
Notes: Compliance (%) equals 100 times (number of days a dose was taken as prescribed divided by treatment duration in days) is calculated for each treatment period, where treatment duration (days) equals date of last dose during the treatment period minus date of first dose during the treatment plus 1 day. A dose was considered “taken as prescribed” if, during q.d. dosing, the patient took any amount of study drug, and during b.i.d. dosing the patient took any amount of both the morning and evening doses. Drug interruptions due to a patient being off-study, between protocol amendments, are not included in the calculation of compliance.
Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.17
Source: ICONIC Clinical Study Report.16
Table 16: Summary of Concomitant Medications That Treat Pruritus in the ICONIC Trial — Safety Population
ATC preferred term | ICONIC (LUM001-304) study | ||||
|---|---|---|---|---|---|
Open-label phase (day 1 to week 18) | Randomized withdrawal phase (weeks 19 to 22) | After randomized withdrawal phase (weeks > 22 to 48) | LTE phase (weeks > 48) | ||
MRX (N = 31) n (%) | MRX (N = 13) n (%) | Placebo (N = 16) n (%) | MRX (N = 29) n (%) | MRX (N = 23) n (%) | |
Number of patients taking any concomitant medications | 28 (90.3) | 12 (92.3) | 14 (87.5) | 25 (86.2) | 19 (82.6) |
Rifampicin | 23 (74.2) | 10 (76.9) | 11 (68.8) | 21 (72.4) | 16 (69.6) |
Phenobarbital | 4 (12.9) | 3 (23.1) | 0 | 3 (10.3) | 1 (4.3) |
Antihistamines for systemic use | 4 (12.9) | 3 (23.1) | 4 (25.0) | 6 (20.7) | 9 (39.1) |
Alimemazine | 1 (3.2) | 1 (7.7) | 0 | 1 (3.4) | 2 (8.7) |
Brompheniramine maleate | 0 | 0 | 0 | 0 | 1 (4.3) |
Cetirizine hydrochloride | 1 (3.2) | 0 | 1 (6.3) | 1 (3.4) | 1 (4.3) |
Chlorphenamine maleate | 0 | 0 | 0 | 0 | 1 (4.3) |
Desloratadine | 0 | 0 | 0 | 0 | 1 (4.3) |
Dexchlorpheniramine maleate | 1 (3.2) | 1 (7.7) | 1 (6.3) | 1 (3.4) | 0 |
Dimetindene maleate | 0 | 0 | 1 (6.3) | 1 (3.4) | 0 |
Ketotifen fumarate | 1 (3.2) | 1 (7.7) | 0 | 1 (3.4) | 0 |
Levocetirizine dihydrochloride | 0 | 0 | 0 | 0 | 1 (4.3) |
Loratadine | 0 | 0 | 0 | 1 (3.4) | 1 (4.3) |
Mequitazine | 0 | 0 | 1 (6.3) | 0 | 0 |
Promethazine hydrochloride | 0 | 0 | 0 | 0 | 1 (4.3) |
Bile and liver therapy | 25 (80.6) | 10 (76.9) | 13 (81.3) | 23 (79.3) | 16 (69.6) |
Ornithine aspartate | 1 (3.2) | 0 | 1 (6.3) | 1 (3.4) | 0 |
Ursodeoxycholic acid | 25 (80.6) | 10 (76.9) | 13 (81.3) | 23 (79.3) | 16 (69.6) |
Other nervous system drugs | 1 (3.2) | 1 (7.7) | 0 | 2 (6.9) | 2 (8.7) |
Naltrexone | 1 (3.2) | 1 (7.7) | 0 | 1 (3.4) | 1 (4.3) |
Naltrexone hydrochloride | 0 | 0 | 0 | 1 (3.4) | 1 (4.3) |
Sertraline | 1 (3.2) | 0 | 1 (6.3) | 1 (3.4) | 1 (4.3) |
ATC = Anatomic Therapeutic Chemical; MRX = maralixibat.
Notes: Patients were counted only once for each ATC or preferred term. Concomitant medications with a start date > 14 days after the patient's last dose of study drug are not included in the presentation. For patients with study drug interruptions, any concomitant medication that starts > 14 days after the last dose before the drug interruption and ended before the study drug was re-initiated will not be considered as a concomitant medication. Included medications are listed from a prespecified dictionary search of medications. Any medication taken during the specified analysis phase(s) will be included in each respective analysis phase and treatment group (where applicable). When the use of concomitant medications continues into subsequent analysis period(s), the medications are counted in each applicable analysis period.
Source: ICONIC Clinical Study Report.16 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)17
Itch Reported Outcome (Observer)
In the ICONIC pivotal trial, the change from weeks 18 to 22 in ItchRO(Obs) weekly average morning severity score was a secondary end point. The LS mean difference between the maralixibat and placebo groups was –1.48 (95% CI, –2.12 to –0.84; P < 0.0001) in favour of maralixibat (Figure 3).
In the overall population, there was a decrease (improvement) in ItchRO(Obs) weekly average morning severity score from baseline to week 18 (secondary end point), with a mean change of –1.70 (95% CI, –2.05 to –1.36; P < 0.0001) and from baseline to week 48 (additional end point) with a mean change of –1.62 (95% CI, –2.12 to –1.12; P < 0.0001).
The prespecified sensitivity analyses for ItchRO(Obs) weekly average morning severity score were consistent with the results of the primary efficacy analysis. Additional sensitivity analyses that applied different imputation methods (LOCF, MMRM, and multiple imputation) to assesses mean changes in ItchRO(Obs) weekly average morning severity score from baseline to week 48 were consistent with the results of the primary efficacy analysis.
Subgroup analyses were generally consistent with the overall results of the pivotal trial, except for where there was a very small number of patients in a given category (n ≤ 4).
Similar results were observed for the ItchRO(Obs) weekly average evening severity score, 4-week average morning severity score, 4-week average evening severity score, and weekly average severity score (daily average of morning and evening scores) in the ITT population, with use of an ANCOVA model.
Itch Reported Outcome (Patient)
A total of 14 patients met the age cut-off for completion of the ItchRO(Pt) (≥ age 9 years or ≥ age 5 years, with the assistance of their caregiver) in the pivotal trial. The LS mean difference between the maralixibat and placebo groups for the change from weeks 18 to 22 in ItchRO(Pt) weekly average morning severity score was –1.98 (–3.01 to –0.97; P = 0.0013) in favour of maralixibat.
In the overall population, there was a decrease (improvement) in ItchRO(Pt) weekly average morning severity score from baseline to week 18 (secondary end point) with a mean change of –2.07 (95% CI, –2.65 to –1.50; P < 0.0001) and from baseline to week 48 (additional end point) with a mean change of –2.25 (95% CI, –2.84 to –1.67; P < 0.0001).
The sensitivity analysis for change in ItchRO(Pt) weekly average morning severity score was consistent with the results of the primary efficacy analysis.
Liver Biomarkers and Enzymes
Alkaline Phosphatase
In the ICONIC pivotal trial, the change from weeks 18 to 22 in ALP was a secondary end point. The LS mean difference between the maralixibat and placebo groups was 10 (95% CI, –52.6 to 72.6; P = 0.7455) U/L.
In the overall population, the mean change in ALP from baseline to week 18 (secondary end point) was –27.8 (95% CI, –72.8 to 17.2; P = 0.2163) U/L and from baseline to week 48 (additional end point) the mean change was –51.5 (95% CI, –95.7 to –7.2; P = 0.0242) U/L.
The sensitivity analyses for change in ALP were consistent with the results of the primary efficacy analysis.
Alanine Transaminase
In the ICONIC pivotal trial, the change from weeks 18 to 22 in ALT was a secondary end point. The LS mean difference between the maralixibat and placebo groups was 15.1 (95% CI, –25.1 to 55.2; P = 0.4472) U/L.
In the overall population, the mean change in ALT from baseline to week 18 (secondary end point) was –1.3 (95% CI, –33.4 to 30.9; P = 0.9358) U/L and the mean change from baseline to week 48 (additional end point) was 17.5 (95% CI, –15.0 to 50.2; P = 0.2787) U/L.
The sensitivity analysis for change in ALT was consistent with the results of the primary efficacy analysis.
Total Bilirubin
In the ICONIC pivotal trial, the change from weeks 18 to 22 in total bilirubin was a secondary end point. The LS mean difference between the maralixibat and placebo groups was –0.14 (–0.88 to 0.60; P = 0.7000) mg/dL.
In the overall population, the mean change in total bilirubin from baseline to week 18 (secondary end point) was –0.47 (95% CI, –1.01 to 0.08; P = 0.0893) mg/dL and the mean change from baseline to week 48 (additional end point) was 0.03 (95% CI, –0.70 to 0.76; P = 0.9285) mg/dL.
The sensitivity analysis for change in total bilirubin was consistent with the results of the primary efficacy analysis.
Direct Bilirubin
In the ICONIC pivotal trial, the change from weeks 18 to 22 in direct bilirubin was a secondary end point. The LS mean difference between the maralixibat and placebo groups was –0.02 (95% CI, –0.56 to 0.53; P = 0.9517) mg/dL.
In the overall population, the mean change in direct bilirubin from baseline to week 18 (secondary end point) was –0.50 (95% CI, –0.90 to –0.11; P = 0.0139) mg/dL and the mean change from baseline to week 48 (additional end point) was –0.24 (95% CI, –0.56 to 0.09; P = 0.1489) mg/dL.
The sensitivity analysis for change in direct bilirubin was consistent with the results of the primary efficacy analysis analyzing direct bilirubin levels change from weeks 18 to 22 and from baseline to week 18.
Change in Body Height and Weight
In the overall study population, there was an increase from baseline in mean height z scores at all time points after weeks 3 to 252. The mean change from baseline to week 100 (LOCF) in height z scores (additional end point) was 0.25 (95% CI, –0.86 to 2.04; P = 0.0216).
In the overall study population, there were no major changes from baseline in mean weight z scores at any time point. The mean change from baseline to week 100/LOCF in weight z scores (additional end point) was –0.05 (95% CI, –0.12 to 0.23; P = 0.5306).
Health-Related Quality of Life
PedsQL Total Score (Parent)
In the ICONIC pivotal trial, the LS mean difference from weeks 18 to 22 in the PedsQL total score (parent) was an additional end point. The LS mean difference between the maralixibat and placebo groups was 2.33 (95% CI, –10.08 to 14.75; P = 0.7018).
In the overall population, the mean change in the PedsQL total score (parent) from baseline to week 18 (additional end point) was 10.73 (95% CI, 4.43 to 17.03; P = 0.0016) and the mean change from baseline to week 48 (additional end point) was 8.94 (95% CI, 1.53 to 16.35; P = 0.0200).
The sensitivity analysis for change in PedsQL total score was consistent with the results of the primary efficacy analysis.
PedsQL Multidimensional Fatigue Scale Score
In the ICONIC pivotal trial, the change from weeks 18 to 22 in the PedsQL Multidimensional Fatigue Scale score (parent) was an additional end point. The LS mean difference between the maralixibat and placebo groups was 14.03 (95% CI, –2.78 to 30.84; P = 0.0966).
In the overall population, the mean change in the PedsQL Multidimensional Fatigue Scale score (parent) from baseline to week 18 (additional end point) was 20.39 (95% CI, 8.91 to 31.87; P = 0.0013) and the mean change from baseline to week 48 (additional end point) was 20.30 (95% CI, 8.98 to 31.63; P = 0.0013).
The sensitivity analysis for change in the PedsQL Multidimensional Fatigue Scale score was consistent with the results of the primary efficacy analysis.
Figure 3: Mean Change From Baseline in ItchRO(Obs) Weekly Average Morning Severity Score Over Time in the ICONIC Trial (ITT Population)
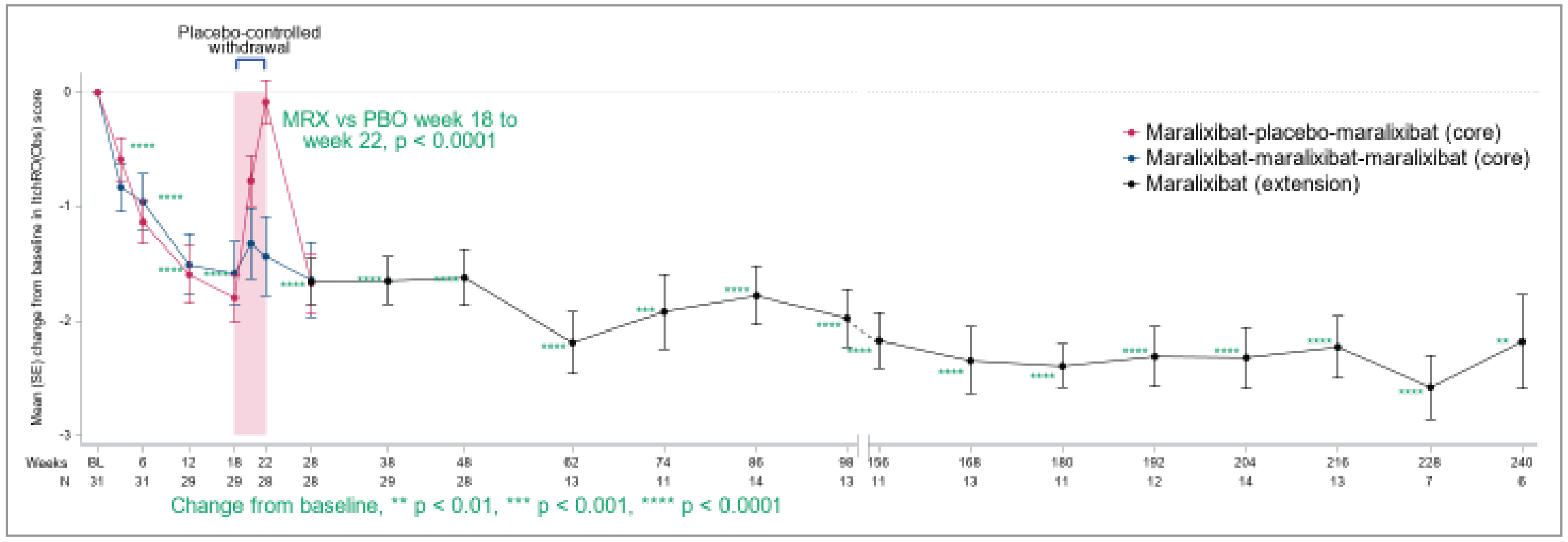
SE = standard error; MRX = maralixibat; PBO = placebo.
Table 17: Summary of Key Efficacy Results in the ICONIC Trial — Randomized Withdrawal Phase, Weeks 18 to 22
Outcomes | Maralixibat N = 13 | Placebo N = 16 |
|---|---|---|
sBA (μmol/L) (mITT)a (primary efficacy end point) | ||
Number of patients contributing to the analysis, n (%) | 5 (38.5) | 10 (62.5) |
Week 18, LS mean (SE) | 100.22 (24.71) | 132.13 (17.40) |
Change from weeks 18 to 22, LS mean (SE) | –21.73 (43.3) | 95.55 (30.49) |
Change from weeks 18 to 22, 95% CI | –115.69 to 72.23 | 29.12 to 161.97 |
LS mean difference (95% CI) | –117.28 (–232.38 to –2.18) | |
P valuebc | 0.0464 | |
sBA (μmol/L) (ITT)d | ||
Number of patients contributing to the analysis, n (%) | 13 (100) | 16 (100) |
Week 18, LS mean (SE) | 209.72 (25.74) | 178.50 (23.17) |
Change from weeks 18 to 22, LS mean (SE) | –18.74 (35.25) | 95.21 (31.69) |
Change from weeks 18 to 22, 95% CI | –91.20 to 53.72 | 30.08 to 160.34 |
LS mean difference (95% CI) | –113.95 (–212.68 to –15.21) | |
P valueb,c | 0.0254 | |
Secondary End points (ITT) | ||
ItchRO(Obs) weekly average morning severity scoree | ||
Number of patients contributing to the analysis, n (%) | 12 (92.3) | 16 (100) |
Week 18, LS mean (SE) | 1.31 (0.24) | 1.12 (0.21) |
Change from weeks 18 to 22, LS mean (SE) | 0.22 (0.23) | 1.70 (0.20) |
Change from weeks 18 to 22, 95% CI | –0.27 to 0.70 | 1.28 to 2.12 |
LS mean difference (95% CI) | –1.48 (–2.12 to –0.84) | |
P valuebc | < 0.0001 | |
ItchRO(Pt) weekly average morning severity scoree | ||
Number of patients contributing to the analysis, n (%)f | 5 (38.5) | 9 (56.3) |
Week 18, LS mean (SE) | 0.78 (0.40) | 0.86 (0.29) |
Change from weeks 18 to 22, LS mean (SE) | –0.15 (0.37) | 1.84 (0.28) |
Change from weeks 18 to 22, 95% CI | –0.97 to 0.67 | 1.23 to 2.45 |
Difference in LS mean (95% CI) | –1.99 (–3.01 to –0.97) | |
P valueb,c | 0.0013 | |
ALP (U/L) | ||
Number of patients contributing to the analysis, n (%) | 13 (100) | 16 (100) |
Week 18, LS mean (SE) | 586.5 (24.01) | 576.1 (21.64) |
Change from weeks 18 to 22, LS mean (SE) | 2.8 (22.55) | –7.2 (20.31) |
Change from weeks 18 to 22, 95% CI | –43.6 to 49.1 | –49.0 to 34.6 |
Difference in LS mean (95% CI) | 10 (–52.6 to 72.6) | |
P valueb,c | 0.7455 | |
ALT (U/L) | ||
Number of patients contributing to the analysis, n (%) | 13 (100) | 16 (100) |
Week 18, LS mean (SE) | 195.3 (19.70) | 162.9 (17.66) |
Change from weeks 18 to 22, LS mean (SE) | 34.5 (14.04) | 19.4 (12.56) |
Change from weeks 18 to 22, 95% CI | 5.6 to 63.4 | –6.4 to 45.2 |
Difference in LS mean (95% CI) | 15.1 (–25.1 to 55.2) | |
P valueb,c | 0.4472 | |
Total bilirubin (mg/dL) | ||
Number of patients contributing to the analysis, n (%) | 13 (100) | 16 (100) |
Week 18, LS mean (SE) | 5.62 (0.39) | 4.72 (0.35) |
Change from weeks 18 to 22, LS mean (SE) | 0.32 (0.27) | 0.46 (0.24) |
Change from weeks 18 to 22, 95% CI | –0.23 to 0.86 | –0.03 to 0.95 |
Difference in LS mean (95% CI) | –0.14 (–0.88 to 0.60) | |
P valueb,c | 0.7000 | |
Direct bilirubin (mg/dL) | ||
Number of patients contributing to the analysis, n (%) | 12 (92.3) | 15 (93.8) |
Week 18, LS mean (SE) | 4.38 (0.26) | 3.67 (0.23) |
Change from weeks 18 to 22, LS mean (SE) | 0.13 (0.20) | 0.14 (0.17) |
Change from weeks 18 to 22, 95% CI | –0.28 to 0.53 | –0.22 to 0.50 |
Difference in LS mean (95% CI) | –0.02 (–0.56 to 0.53) | |
P valueb,c | 0.9517 | |
Additional efficacy end points | ||
PedsQL: total score (parent) | ||
Number of patients contributing to the analysis, n (%) | 12 (92.3) | 16 (100) |
Week 18, LS mean (SE) | 72.64 (3.92) | 69.86 (3.37) |
Change from weeks 18 to 22, LS mean (SE) | –6.69 (4.51) | –9.03 (3.89) |
Change from weeks 18 to 22, 95% CI | –15.97 to 2.59 | –17.03 to –1.02 |
Difference in LS mean (95% CI) | 2.33 (–10.08 to 14.75) | |
P valueb,c | 0.7018 | |
PedsQL: Multidimensional Fatigue Scale score (parent) | ||
Number of patients contributing to the analysis, n (%) | 9 (69.2) | 12 (75.0) |
Week 18, LS mean (SE) | 74.59 (5.31) | 71.07 (3.93) |
Change from weeks 18 to 22, LS mean (SE) | –2.96 (6.05) | –16.99 (5.24) |
Change from weeks 18 to 22, 95% CI | –15.67 to 9.74 | –27.99 to –5.99 |
Difference in LS mean (95% CI) | 14.03 (–2.78 to 30.84) | |
P valueb,c | 0.0966 | |
ALT = alanine transaminase; ALP = alkaline phosphatase; ANCOVA = analysis of covariance; CI = confidence interval; ItchRO(Obs) = Itch Reported Outcome (observer); ItchRO(Pt) = Itch Reported Outcome (patient); ITT = intent to treat; LS = least square; mITT = modified intent to treat; PedsQL = Pediatric Quality of Life Inventory; sBA = serum bile acid; SE = standard error.
Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.17
aThe primary efficacy end point was conducted on the mITT population, including all patients who were enrolled, received study drug through week 18, and had a reduction from baseline in sBA of ≥ 50% at the week 12 or 18 measurement (sBA responder). Estimates are from a mixed model with treatment group as a fixed effect and baseline value as a covariate. Analysis uses a tabulation of fitted summary statistic from ANCOVA in the mITT population.
bThe P value was not adjusted for multiple testing (i.e., the type I error rate was not controlled).
cEstimates are from a mixed model with treatment group as a fixed effect and baseline value as a covariate.
dAnalyses for the primary efficacy outcome variable were also done on the ITT population.
eItchRO scores have a range from 0 to 4, a higher score indicating increasing itch severity.
fPer protocol, only administered to 14 patients at baseline. Children aged at least 9 years complete the ItchRO(Pt). Children aged 5 to 8 years complete the instrument with their caregiver's assistance. There is no ItchRO(Pt) report for patients under age 5 years.
Source: ICONIC Clinical Study Report.16
Table 18: Summary of Efficacy Results From the ICONIC Trial, Overall Maralixibat — Baseline to Week 18 and Baseline to Week 48 (ITT Population)
Time point | ICONIC trial, N = 31 | |||
|---|---|---|---|---|
Number of patients contributing to the analysis, n (%) | Baseline value, mean (SD) | Change from baseline at time point, mean (SE) (95% CI) | P valuea,b | |
sBA (μmol/L) | ||||
Week 18c | 29 (93.5) | 283.43 (210.57) | –87.73 (22.28) (–133.37 to –42.09) | 0.0005 |
Week 48 | 27 (87.1) | –96.44 (32.07) (–162.36 to –30.52) | 0.0058 | |
ItchRO(Obs) weekly average morning severity score | ||||
Week 18c | 29 (93.5) | 2.91 (0.55) | –1.70 (0.17) (–2.05 to –1.36) | < 0.0001 |
Week 48 | 28 (90.3) | –1.62 (0.25) [–2.12 to –1.12] | < 0.0001 | |
ItchRO(Pt) weekly average morning severity score | ||||
Week 18c | 14 (45.2) | 2.90 (0.66) | –2.07 (0.27) (–2.65 to –1.50) | < 0.0001 |
Week 48 | 14 (45.2) | –2.25 (0.27) (–2.84 to –1.67) | < 0.0001 | |
ALP (U/L) | ||||
Week 18c | 29 (93.5) | 601.3 (274.77) | –27.8 (21.97) (–72.8 to 17.2) | 0.2163 |
Week 48 | 28 (90.3) | –51.5 (21.55) (–95.7 to –7.2) | 0.0242 | |
ALT (U/L) | ||||
Week 18c | 29 (93.5) | 181.0 (108.56) | –1.3 (15.70) (–33.4 to 30.9) | 0.9358 |
Week 48 | 28 (90.3) | 17.5 (15.86) (–15.0 to 50.1) | 0.2787 | |
Total bilirubin (mg/dL) | ||||
Week 18c | 29 (93.5) | 6.09 (5.78) | –0.47 (0.26) (–1.01 to 0.08) | 0.0893 |
Week 48 | 28 (90.3) | 0.03 (0.36) (–0.70 to 0.76) | 0.9285 | |
Direct bilirubin (mg/dL) | ||||
Week 18c | 28 (90.3) | 4.57 (3.67) | –0.50 (0.19) (–0.90 to –0.11) | 0.0139 |
Week 48 | 27 (87.1) | –0.24 (0.16) (–0.56 to 0.09) | 0.1489 | |
PedsQL: total score (parent) | ||||
Week 18 | 28 (90.3) | 61.10 (16.99) | 10.73 (3.07) (4.43 to 17.03) | 0.0016 |
Week 48 | 27 (87.1) | 8.94 (3.61) (1.53 to 16.35) | 0.0200 | |
PedsQL – Multidimensional Fatigue Scale score (parent) | ||||
Week 18 | 22 (71.0) | 52.72 (23.30) | 20.39 (5.52) (8.91 to 31.87) | 0.0013 |
Week 48 | 21 (67.7) | 20.30 (5.43) (8.98 to 31.63) | 0.0013 | |
Change in height z score | ||||
Week 48 | 28 (90.3) | –1.67 (1.34) | 0.18 (0.09) (–0.02 to 0.37) | 0.0704 |
Week 100 (LOCF) | 29 (93.5) | 0.25 (0.10) (0.04 to 0.46) | 0.0216 | |
Change in weight z score | ||||
Week 48 | 28 (90.3) | –1.70 (1.18) | 0.02 (0.08) (–0.15 to 0.18) | 0.8225 |
Week 100 (LOCF) | 29 (100.0) | 0.05 (0.08) (–0.12 to 0.23) | 0.5306 | |
ALT = alanine transaminase; ALP = alkaline phosphatase; CI = confidence interval; ITT = intent to treat; ItchRO(Obs) = Itch Reported Outcome (observer); ItchRO(Pt) = Itch Reported Outcome (patient); LOCF = last observation carried forward; PedsQL = Pediatric Quality of Life Inventory; sBA = serum bile acid; SD = standard deviation; SE = standard error.
aThe P value was not adjusted for multiple testing (i.e., the type I error rate was not controlled).
bStudent’s t test used to test if mean change is statistically significant.
cSecondary end point.
Source: ICONIC Clinical Study Report.16 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)17
Harms
Refer to Table 19 for harms data.
Adverse Events
The incidence of AEs was similar during the open-label phase, the after RWD phase, and the LTE phase, with at least 25 of 29 patients (86.2%) experiencing any AEs in these treatment periods (Table 17). During the RWD phase, patients who stayed on maralixibat had a lower incidence of AEs (7 of 13 patients [38%]) compared with patients on placebo (12 of 16 patients [75%]). The most frequently reported AEs (> 30% in at least 1 phase) were abdominal pain, pyrexia, diarrhea, nasopharyngitis, vomiting, cough, and pruritus.
Serious Adverse Events
The incidence of SAEs was similar during the open-label phase (4 of 31 patients [12.9%]) and after RWD phase (5 of 29 patients [17.2%]) and was slightly higher during the LTE phase (6 of 23 patients [26.1%]). During the RWD phase, SAEs were reported for 1 of 13 patients who stayed on maralixibat and 1 of 16 patients on placebo. None of the SAEs were considered related to study medication.
Withdrawals Due to AEs
A total of 6 patients (2 each in the open-label, after RWD, and LTE phases) experienced AEs leading to study drug discontinuation.
Mortality
No deaths were noted during the study.
Notable Harms
Diarrhea
AEs of diarrhea were similar during the open-label phase (13 of 31 patients [41.9%]) and the LTE phase (10 of 23 patients [43.5%]) and were lower during the after RWD phase (7 of 29 patients [24.1%]). During the RWD phase, patients who stayed on maralixibat had a similar incidence of events of diarrhea (1 of 13 patients [7.7%]) compared with those receiving placebo (2 of 16 patients [12.5%]).
Fat-Soluble Vitamin Deficiency
AEs associated with FSV deficiency were similar during the open-label phase (7 of 31 patients [22.6%]) and LTE phase (6 of 23 patients [26.1%]) and were lower during the after RWD phase (1 of 29 patients [3.4%]).
Elevated Transaminases
AEs associated with elevated transaminases were only reported during the LTE phase (4 of 23 patients [17.4%]).
Elevated Bilirubin
AEs associated with elevated bilirubin were only reported during the after RWD phase (1 of 29 patients [3.4%]).
Critical Appraisal
Internal Validity
The ICONIC trial was a phase IIb, placebo-controlled study with open-label extension. Overall, the study, including both the RWD phase and the extension phase, showed considerable evidence with moderate certainty that maralixibat improved sBA, and itch-related outcome. The study is an exploratory trial, with small sample size, Uncertainty remains as to whether maralixibat could improve liver function and patients’ HRQoL. The beneficial effect may only exist in a certain proportion of patients with ALGS in the trial. In particular, after open-label treatment through to week 18, patients remaining in the study were block randomized in a 1:1 ratio to receive maralixibat or placebo in the 4-week RWD phase. The primary efficacy analysis on the changes in mean sBA at week 22 was based on 15 of 29 patients with at least 50% reduction of sBA (responder) at week 12 or 18 (n = 5 in maralixibat, n = 10 in placebo). However, the magnitude of mean change was similar between the selected 15 patients (–117) versus the overall 29 patients (–114), indicating that the treatment effect in the overall randomized study population was primarily driven by those responders representing 50% (15 of 29) of the total study population.
The definition of the pruritus end point used to assess efficacy was not prespecified in the protocol; however, analyses performed using different summaries of the daily pruritus scores (i.e., morning scores, evening scores, weekly average of the 2 daily scores) were in line with the primary analysis.
Subgroup analyses were difficult to interpret due to small sample sizes and a lack of control of multiplicity. During the open-label phases of the pivotal trial, patients’ and/or caregivers’ knowledge of treatment assignment may have biased subjective outcomes such as ItchRO(Obs), ItchRO(Pt), and PedsQL in favour of maralixibat. Reporting of harms could also have been biased, potentially in favour of maralixibat. Discontinuation through to week 48 of the pivotal trial was low, with 3 of 31 patients (9.7%) discontinuing due to an AE through to week 48, with 2 patients discontinuing during the initial open-label phase, and 1 patient discontinuing after the RWD phase.
Differences in baseline characteristics between patients in the maralixibat and placebo group were noted in the ICONIC trial. Those in the maralixibat group were slightly younger, had a smaller proportion of patients with family history of ALGS, had a smaller proportion with known bile duct paucity, and had higher sBA, ALT, and total bilirubin values than those in the placebo group. The clinical experts consulted on this review stated that these baseline differences were not likely to lead to biased results. The clinical experts noted that patients in the maralixibat group may have had a higher degree of disease severity than those in the placebo group as indicated by the higher sBA, ALT, and bilirubin values, which may have biased results in favour of placebo.
Change in sBA was assessed as a primary efficacy end point in the clinical trial. Descriptive posthoc data from the ICONIC pivotal trial found that reductions in sBA from baseline to week 48 were associated with reductions in mean ItchRO(Obs) weekly average morning severity scores (Appendix 1).The data may show an association between sBA and ItchRO in some patients, but as the data were descriptive in nature and the assessment was conducted posthoc on a small number of patients (n = 28), it is unclear the extent to which sBA levels may be associated with pruritus in patients with cholestatic liver diseases.
Table 19: Summary of Harms Results in the ICONIC Trial
Adverse events | ICONIC trial | ||||
|---|---|---|---|---|---|
Open-label phase (day 1 to week 18) | Randomized withdrawal phase (weeks 19 to 22)b | After randomized withdrawal phase (weeks > 22 to 48)b | Long-term extension phase (weeks > 48)b | ||
Maralixibat (N = 31) | Maralixibat (N = 13) | Placebo (N = 16) | Maralixibat (N = 29) | Maralixibat (N = 23) | |
Patients ≥ 1 AE | 30 (96.8) | 7 (53.8) | 12 (75.0) | 25 (86.2) | 23 (100.0) |
Most common AEs,a n (%) | |||||
Ear and labyrinth disorders | 2 (6.5) | 0 | 0 | 3 (10.3) | 3 (13.0) |
Ear pain | 1 (3.2) | 0 | 0 | 3 (10.3) | 1 (4.3) |
Gastrointestinal disorders | 22 (71.0) | 2 (15.4) | 3 (18.8) | 14 (48.3) | 16 (69.6) |
Diarrhea | 13 (41.9) | 1 (7.7) | 1 (6.3) | 5 (17.2) | 7 (30.4) |
Abdominal pain | 12 (38.7) | 1 (7.7) | 1 (6.3) | 6 (20.7) | 12 (52.2) |
Vomiting | 11 (35.5) | 1 (7.7) | 1 (6.3) | 3 (10.3) | 8 (34.8) |
General disorders and administration site conditions | 7 (22.6) | 0 | 3 (18.8) | 10 (34.5) | 12 (52.2) |
Pyrexia | 6 (19.4) | 0 | 2 (12.5) | 7 (24.1) | 10 (43.5) |
Infections and infestations | 21 (67.7) | 6 (46.2) | 4 (25.0) | 15 (51.7) | 17 (73.9) |
Upper respiratory tract infection | 6 (19.4) | 2 (15.4) | 0 | 3 (10.3) | 4 (17.4) |
Nasopharyngitis | 4 (12.9) | 1 (7.7) | 1 (6.3) | 8 (27.6) | 9 (39.1) |
Ear infection | 3 (9.7) | 0 | 0 | 4 (13.8) | 5 (21.7) |
Gastroenteritis | 0 | 0 | 1 (6.3) | 2 (6.9) | 5 (21.7) |
Pharyngitis | 0 | 0 | 1 (6.3) | 0 | 3 (13.0) |
Viral infection | 1 (3.2) | 1 (7.7) | 0 | 1 (3.4) | 4 (17.4) |
Injury, poisoning, and procedural complications | 8 (25.8) | 0 | 1 (6.3) | 6 (20.7) | 11 (47.8) |
Fall | 4 (12.9) | 0 | 0 | 3 (10.3) | 0 |
Contusion | 1 (3.2) | 0 | 0 | 0 | 3 (13.0) |
Investigations | 3 (9.7) | 0 | 0 | 1 (3.4) | 6 (26.1) |
ALT increased | 0 | 0 | 0 | 0 | 4 (17.4) |
Musculoskeletal and connective tissue disorders | 1 (3.2) | 0 | 0 | 1 (3.4) | 8 (34.8) |
Pain in extremity | 0 | 0 | 0 | 0 | 4 (17.4) |
Nervous system disorders | 7 (22.6) | 0 | 1 (6.3) | 2 (6.9) | 5 (21.7) |
Headache | 5 (16.1) | 0 | 0 | 2 (6.9) | 4 (17.4) |
Respiratory, thoracic, and mediastinal disorders | 8 (25.8) | 0 | 0 | 7 (24.1) | 10 (43.5) |
Cough | 3 (9.7) | 0 | 0 | 3 (10.3) | 8 (34.8) |
Oropharyngeal pain | 1 (3.2) | 0 | 0 | 3 (10.3) | 3 (13.0) |
Skin and subcutaneous tissue disorders | 4 (12.9) | 2 (15.4) | 5 (31.3) | 3 (10.3) | 4 (17.4) |
Pruritus | 3 (9.7) | 1 (7.7) | 5 (31.3) | 2 (6.9) | 0 |
SAEs, n (%) | |||||
Patients ≥ 1 SAE | 4 (12.9) | 1 (7.7) | 1 (6.3) | 5 (17.2) | 6 (26.1) |
Most common SAE,a n (%) | |||||
Gastrointestinal disorders | 1 (3.2) | 0 | 0 | 2 (6.9) | 0 |
General disorders and administration site conditions | 0 | 0 | 1 (6.3) | 0 | 2 (8.7) |
Infections and infestations | 2 (6.5) | 1 (7.7) | 0 | 2 (6.9) | 2 (8.7) |
Injury, poisoning, and procedural complications | 1 (3.2) | 0 | 1 (6.3) | 1 (3.4) | 2 (8.7) |
Patients who stopped treatment due to AEs, n (%) | |||||
Patients with ≥ 1 AE leading to study drug discontinuation | 2 (6.5) | 0 | 0 | 2 (6.9) | 2 (8.7) |
Staphylococcal infection | 1 (3.2) | 0 | 0 | 0 | 0 |
Extradural hematoma | 1 (3.2) | 0 | 0 | 0 | 0 |
Subdural hemorrhage | 1 (3.2) | 0 | 0 | 0 | 0 |
Blood bilirubin increased | 0 | 0 | 0 | 1 (3.4) | 0 |
ALT increased | 0 | 0 | 0 | 0 | 2 (8.7) |
Acute kidney injury | 0 | 0 | 0 | 1 (3.4) | 0 |
Deaths, n (%) | |||||
Patients who died | 0 | 0 | 0 | 0 | 0 |
AEs of special interest, n (%) | |||||
Diarrhea events | 13 (41.9) | 1 (7.7) | 2 (12.5) | 7 (24.1) | 10 (43.5) |
Events associated with FSV deficiency | 7 (22.6) | 0 | 0 | 1 (3.4) | 6 (26.1) |
Events associated with elevated transaminases | 0 | 0 | 0 | 0 | 4 (17.4) |
Events associated with elevated bilirubin | 0 | 0 | 0 | 1 (3.4) | 0 |
AE = adverse event; ALT = alanine transaminase; FSV = fat-soluble vitamin; SAE = serious adverse event.
aFrequency ≥ 10%.
bReported in 2 or more patients.
Source: ICONIC Clinical Study Report.16 (Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.)17
Pruritus was assessed using the ItchRO(Obs) and ItchRO(Pt) tool as a secondary outcome measured by patients and/or caregivers using an electronic diary twice per day (morning and evening). Procedures were in place to ensure proper completion of the electronic diary, including training patients and caregivers in the use of the electronic diary during the screening visit, and requiring caregivers and patients 9 years of age or older to have completed at least 10 electronic diary reports (morning and/or evening) during each of 2 consecutive weeks of the screening period; re-training on the use of the diary was offered as appropriate.
There were minimal missing data for the ItchRO(Obs) weekly average morning severity scores during the RWD phase, with only 1 randomized patient (maralixibat group) having a missing ItchRO(Obs) weekly average morning severity score at week 22. Only 9 patients qualified to complete the ItchRO(Pt) tool, with results that were supportive of those attained by the ItchRO(Obs). Predefined sensitivity analyses were conducted to assess the impact of missing data for ItchRO outcomes, and their results were consistent with the primary analysis.
Limited available evidence on the ItchRO tool as assessed among pediatric patients with cholestatic liver disease suggests that it is likely valid and reliable. The clinical experts consulted on this review noted that an MID of 1 for the ItchRO tool is clinically meaningful; however, the experts noted that such tools are not commonly used in clinical practice.
HRQoL was assessed using the PedsQL as an additional efficacy outcome in the pivotal trial. Psychometric properties of the instrument have not been assessed in pediatric patients with ALGS. MID estimates for the PedsQL have not been established in pediatric patients with ALGS; however, MID estimates for the PedsQL Generic Core Scales ranged from 4 to 5 points among a sample of children with and without chronic health conditions, which aligns with the clinical experts’ expectations of a clinically meaningful change. It should be noted that the number of patients assessed for the PedsQL Multidimensional Fatigue Scale score was low during the RWD phase, with 9 of 13 patients (69.2%) in the maralixibat group and 12 of 16 patients (75.0%) in the placebo group contributing to the analysis of mean change from weeks 18 to 22. The impact of missing data on this outcome is unclear in the absence of sensitivity analyses.
Various efficacy outcomes deemed important to the clinical experts were not assessed in the pivotal trial, including liver transplant or TFS, time to liver event, cirrhosis, or renal disease as measured by the gradual loss of kidney function.
External Validity
The clinical experts consulted on this review stated that patients included in the ICONIC trial generally align with the selection criteria for candidates for maralixibat, although patients with mild cholestatic pruritus would not necessarily be excluded from treatment in clinical practice. Nonetheless, the clinical experts did not expect the exclusion of these patients to significantly affect the generalizability of the patient population in this study. The clinical trial only enrolled patients 12 months of age or older with a Jagged-1 mutation; however, the clinical experts note that the trial results would be applicable to patients less than 12 months of age as well as patients with a NOTCH2 mutation, respectively. The experts noted that patients in clinical practice are likely to also receive confirmatory genetic testing, which was not conducted in the pivotal trial. Although race and ethnicity data were not assessed in the pivotal trial, the clinical experts stated that the results would be applicable to the patient population in Canada. The remaining baseline patient characteristics were similar to the indicated patient population in clinical practice in Canada.
The efficacy outcomes measured in the study were of clinical importance to patients and clinicians, including change in sBA. However, the clinical experts noted that the change in sBA is not often assessed in clinical practice due to high costs and logistical limitations, as sBA testing is often sent to specialized laboratories and is not readily available in all gastroenterology practice settings. Change in pruritus and HRQoL outcomes were described as important outcomes by the clinical experts, as well as in the patient and clinician group input received, which noted the need for a treatment that reduces the severity and frequency of pruritus as well as reducing patient and caregiver fatigue. Such outcomes were captured by the ItchRO and the PedsQL tools in the ICONIC trial. The clinical experts consulted for this review indicated that although tools such PedsQL are frequently used in clinical trials they are not typically used in clinical practice. Furthermore, the double-blind phase in the pivotal ICONIC trial was 4 weeks in length, limiting the ability to assess the long-term efficacy and safety of maralixibat compared with placebo for the indicated dose of 380 mcg/kg/day. The durability of treatment effect and long-term safety still remain somewhat uncertain, although the beneficial effect is likely maintained, as reflected by the long-term curve on changes of sBA and ItchRO(Obs).
While maralixibat has been approved by Health Canada for use in patients for the treatment of cholestatic pruritus in patients with ALGS, the ICONIC trial only enrolled patients aged between 1 and 15 years. As such, there is an absence of efficacy and safety data assessing maralixibat versus placebo among patients younger than 12 months due to the challenges of conducting a controlled clinical trial in this age group. However, the trial results are expected to be applicable to patients less than 12 months of age based on clinical experts’ feedback. Furthermore, during the LTE phase of the ICONIC pivotal trial (week 103) eligible patients could have received a dose of maralixibat of up to 760 mcg/kg/day (given as twice-daily doses of 380 mcg/kg) which is outside of the proposed Health Canada indication of 380 mcg/kg /day. As such, efficacy and safety data after this period is not aligned with the recommended dose.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:14,15
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
For RCTs: Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (i.e., internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for maralixibat versus placebo.
LTE Studies
The pivotal ICONIC trial included an LTE phase described in the systematic review section of this report. No other LTE studies were submitted.
Indirect Evidence
No indirect comparisons were conducted comparing maralixibat with other comparators for this submission.
Studies Addressing Gaps in the Pivotal and RCT Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Table 20: Summary of Gaps in the Evidence
Gap in pivotal and RCT evidence | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
There is a lack of evidence to validate the relationship between sBA and pruritus, as well as other events — e.g., transplant or death. | Patient-level data from the 3 long-term studies of patients with ALGS treated with maralixibat (the LUM001-305, LUM001-303, LUM001-304 studies) were included to identify predictors of long-term EFS (with duration of follow-up up to 6 years). | As pruritus is often an indication for liver transplant in patients with ALGS, improvements in pruritus with maralixibat were associated with improved EFS and liver TFS. |
Duration of ICONIC study is not long enough to assess long-term clinical outcomes, such as liver transplant and death. | Natural-history comparison study: A global, open-label, nonrandomized, external-control study to compare time-to-event clinical outcomes, e.g., liver transplant or death, in patients with ALGS treated with maralixibat with selected external controls from the GALA registry. This study followed a prespecified SAP with a duration of follow-up of up to 7 years. |
|
ALGS = Alagille syndrome; CI = confidence interval; EFS = event-free survival; GALA = Global ALagille Alliance; HR = hazard ratio; LTE = long-term extension; RCT = randomized controlled trial; SAP = statistical analysis plan; sBA = serum bile acid; TFS = transplant-free survival.
Source: Sponsor’s Summary of Clinical Evidence,17 GALA SAP.43
Description of Studies
Patient-level data from the 3 long-term studies of patients with ALGS treated with maralixibat (the LUM001-305, LUM001-303, and LUM001-304 studies), and a natural-history comparison study (GALA registry) have been summarized to provide evidence regarding maralixibat for the treatment of cholestatic pruritus in patients with ALGS, aged 2 months and older.
The following gaps in the pivotal and RCT evidence have been identified, which the studies included in this section address.
Population: Although the LUM001-304 (ICONIC) trial did not enrol children aged younger than 12 months with ALGS, there remains an unmet need for the treatment in infants since ALGS is typically diagnosed within the first months of life,1,12 and this population suffers from the clinical burden of cholestasis from a very young age.6,44 In this regard, infants with ALGS may present with the key features of cholestasis: elevations in sBAs, pruritus, hypercholesterolemia, xanthomas, and failure to thrive. The most burdensome clinical feature of cholestasis in ALGS is cholestatic pruritus. Older infants may exhibit typical scratching behaviour and skin mutilations. In younger infants, before development of motor coordination to scratch properly, children may exhibit coarse scratching movement, fussiness, and irritability as signals of pruritus. Therefore, it is clear that pruritus represents a medical need in infancy.40 However, given the rarity of the disease and the differences in presentation of pruritus in this age group, it would be challenging to generate new evidence to demonstrate clinical efficacy in the treatment of cholestatic pruritus in the subgroup of patients with ALGS who are less than age 12 months.
Outcomes: Clinical end points, such as long-term EFS and TFS in patients with ALGS, were not evaluated in the ICONIC study.
Duration: In a life-threatening condition such as ALGS, long-term, randomized, placebo-controlled studies are not possible because the risks of forgoing other treatment options that a patient would otherwise receive outside of a study are so high that it is not feasible or ethical to ask patients to accept those risks.
Patient-Level Data From the LUM001-305 (IMAGINE), LUM001-303 (ICONIC), and LUM001-304 (IMAGINE-II) Studies
To identify predictors of long-term EFS and TFS in patients with ALGS, individual patient data from 3 long-term clinical trials of maralixibat, with up to 6 years of follow-up, were integrated. All trials used ItchRO(Obs) as a primary end point, with a less than 1 point reduction defined as clinically meaningful. Inclusion and exclusion criteria, dosing, and end points were similar for all 3 trials.
Table 21: Details of LTE Studies: LUM001-303 (IMAGINE), LUM001-304 (ICONIC), and LUM001-305 (IMAGINE-II) Studies
Characteristic | LUM001-303 (IMAGINE) studya | LUM001-304 (ICONIC) studyb | LUM001-305 (IMAGINE-II) studyc |
|---|---|---|---|
Designs and populations | |||
Title | An extension study to evaluate the long-term safety and durability of effect of maralixibat in the treatment of cholestatic liver disease in patients with ALGS | Safety and efficacy study of maralixibat with a drug withdrawal period in patients with ALGS | An extension study to evaluate the long-term safety and durability of effect of maralixibat in the treatment of cholestatic liver disease in pediatric patients with ALGS |
Study design | Open-label, single-arm, multicentre, LTE study | Long-term, open-label, single-arm study with a randomized placebo-controlled parallel group period | Single-arm, multicentre, LTE study |
Enrolled (N) | 19 patients | 31 patients | 34 patients |
Key inclusion criteria |
|
|
|
Key exclusion criteria | NR |
| Experienced an AE or SAE related to the study drug during the LUM001-301 protocol that led to the discontinuation of the patient from the core study |
Drugs | |||
Intervention | Dosing of maralixibat with the objective of achieving optimal control of pruritus at a dose level that is tolerated by the patient and up to a maximum daily dose of 280 mcg/kg | Maralixibat orally once a day up to 380 mcg/kg/day up to week 52, followed by an increase in dose orally twice a day during long-term follow-up based on efficacy (sBA level and ItchRO[Obs] score) and safety assessment | Dosing of maralixibat with the objective of achieving optimal control of pruritus at a dose level that is tolerated by the patient and up to a maximum daily dose of 280 mcg/kg orally once daily |
Comparator(s) | NA | NA | NA |
Outcomes | |||
Primary end point | Mean change from maralixibat baseline to week 48 in fasting sBA level | Mean change from weeks 18 to 22 (the RWD period) of fasting sBA levels in patients who had a reduction in sBA ≥ 50% from baseline to weeks 12 or 18 (mITT population) | Mean change from maralixibat baseline to week 48 in fasting sBA levels |
Secondary end points |
|
|
|
Notes | |||
Publications | None | Gonzales, et al. 202145 | None |
AE = adverse event; ALGS = Alagille syndrome; ALP = alkaline phosphatase; ALT = alanine transaminase; AST = aspartate transaminase; FSV = fat-soluble vitamin; GGT = gamma-glutamyl transferase; ItchRO = Itch Reported Outcome; ItchRO(Obs) = Itch Reported Outcome (observer); ItchRO(Pt) = Itch Reported Outcome (patient); LOCF = last observation carried forward; LTE = long-term extension; mITT = modified intent to treat; NA = not applicable; NR = not reported; RWD = randomized withdrawal; SAE = serious adverse event; sBA = serum bile acid; ULN = upper limit of normal.
aThe IMAGO study (NCT01903460) had a similar study design as the ITCH trial but was conducted in the UK and patients were eligible to enrol in an LTE study (IMAGINE; NCT 02047318) for up to 288 weeks.
bThe ICONIC study (NCT02160782) was a double-blind, randomized-withdrawal study of maralixibat in children with ALGS. The ICONIC study was a placebo-controlled, phase IIb study with an open-label extension in children with ALGS (1 to 18 years of age) with more than 3 times the ULN of sBA levels, and intractable pruritus. After 18 weeks of maralixibat, patients were randomized 1:1 to continue maralixibat or receive placebo for 4 weeks, after which all patients received open-label maralixibat up to week 48. Patients could then enrol in an LTE study.
cThe ITCH study (NCT02057692) was a double-blind, RCT conducted in North America of maralixibat for 13 weeks to evaluate the safety and efficacy in the reduction of pruritus and sBA by 3 doses of maralixibat or placebo. Patients enrolled in the ITCH trial were then eligible to enrol in a long-term, open-label extension study (IMAGINE-II) in which all patients received maralixibat (NCT02117713) for up to 220 weeks.
Source: Clinicaltrials.gov — NCT02047318 (LUM001-303),46 NCT02160782 (LUM001-304),47 and NCT02117713 (LUM001-305) trials.48
Populations
Inclusion criteria for this analysis:
Patients who were on maralixibat 48 weeks from the first dose and had laboratory results at 48 weeks
No prior clinical event
Outcomes
EFS was defined as first occurrence of any of the following listed events:
Liver transplant
SBD
Liver decompensation (variceal bleeding, ascites requiring therapy)
Death
TFS was defined as absence of liver transplant or death.
Patients were followed from the start of maralixibat treatment for a minimum of 53 weeks and a maximum of 380 weeks.
Interventions
Maralixibat was administered in the LUM001-303 (IMAGINE) and LUM001-305 (IMAGINE-II) studies, up to 266 mcg/kg/day (doses presented as maralixibat free base).
In the LUM001-304 (ICONIC) study, maralixibat was administered to a final dose of up to 380 mcg/kg/day. Patients were increased to twice-daily dosing starting after week 103.
Statistical Analysis
The statistical analyses conducted are as follows:49
Forty-six predictors were considered, which included, but were not limited to, total bilirubin at week 48, total sBA at week 48, pruritus (as assessed by ItchRO[Obs] 0 to 4 scale) change from baseline to week 48, and age at initiation of maralixibat.
Harrell’s concordance statistic (C-statistic) was computed for each predictor, with the C-statistic defined as the proportion of observations that the variable can order correctly in terms of survival times indicating the goodness of fit.
Once variables and time points with the highest C-statistics were identified, a survival analysis using a Cox regression model for each result was carried out to confirm that each predictor (treated as a continuous variable) was a statistically significant predictor of outcomes of interested assessed at alpha = 0.05 confidence level for 2-sided comparisons.
Variables with a value equal to or greater than 0.7 (indicating a good model) were selected for further analysis.
Cut-offs for each variable were determined via a grid search across the range of values.
Statistical comparisons between the cut-off groups were calculated using a log-rank test.
A total of 84 patients were enrolled across all 3 clinical trials and their LTE studies; however, only 76 patients with complete data, including laboratory values at baseline and week 48 (with a window of less than 12 weeks to more than 6 weeks), were included in integrated analyses.
Results
Baseline Characteristics
This analysis included 76 patients treated with maralixibat, with a median follow-up of 266 weeks (range, 53 to 380). Median duration of maralixibat treatment was 4.7 years (interquartile range [IQR], 1.6 to 4.8). The median age (IQR) of 76 patients was 70 months (33 to 126). The median sBA (IQR) at baseline was 184 µmol/L (78 to 361 µmol/L). The median ItchRO(Obs) score at baseline (IQR) was 2.7 (2.1 to 3.1). The median total bilirubin, ALT, and gamma-glutamyl transferase (GGT) at baseline (IQRs) were 0.03 mmol/L (0.01 to 0.09 mmol/L), 134 U/L (95 to 193 U/L), and 392 U/L (188 to 751 U/L), respectively.
Predictors of EFS
Sixty out of 76 patients remained event-free at the time of analysis. Sixteen patients experienced clinical events: liver transplant (n = 10), decompensation (n = 3), death (n = 2), and SBD (n = 1).
Kaplan-Meier analyses demonstrated that at week 48, lower total bilirubin levels (< 0.07 mmol/L versus ≥ 0.07 mmol/L) and lower sBA levels (< 200 µmol/L versus ≥ 200 µmol/L) were associated with increased EFS (90% versus 43%; P < 0.0001, and 85% versus 49%; P = 0.0010, respectively). A larger reduction in ItchRO(Obs) scores from baseline to week 48 (> 1 point reduction versus ≤ 1 point reduction) and a higher age at initiation of maralixibat were also associated with improved EFS (88% versus 57%; P = 0.0046, and 83% versus 57%; P = 0.0059, respectively) (Figure 4).
Week 48 sBA and change from baseline to week 48 in pruritus (ItchRO[Obs]) were identified as predictors of EFS. Specifically, 6-year EFS improved with a clinically meaningful greater than 1-point ItchRO(Obs) reduction from baseline to W48 (88% versus 57%; P = 0.005) and W48 sBA < 200 µmol/L (85% versus 49%; P = 0.001). These 2 variables had high C-statistics over time, indicating that these cut-offs were stable predictors for 2 to 5 additional years after 48 weeks of maralixibat treatment. These variables were similarly predictive for TFS (Table 22).
Table 22: Predictors of EFS and TFS in Patients With ALGS Treated With Maralixibat
Variable | Better EFS or TFS | Worse EFS or TFS |
|---|---|---|
sBA at week 48 | ||
n | 56 | 18 |
Cut-off | < 200 µmol/L | ≥ 200 µmol/L |
6-year EFS (%) | 85 | 49 |
C-statistic | 0.74 | |
P value | 0.0010 | |
6-year TFS (%) | 90 | 49 |
C-statistic | 0.79 | |
P value | 0.0001 | |
Change from baseline to week 48 ItchRO(Obs) | ||
n | 46 | 30 |
Cut-off | > 1 point reduction | ≤ 1 point reduction |
6-year EFS (%) | 88 | 57 |
C-statistic | 0.70 | |
P value | 0.0046 | |
6-year TFS (%) | 93 | 57 |
C-statistic | 0.77 | |
P value | 0.0007 | |
ALGS = Alagille syndrome; C-statistic = concordance statistic; EFS = event-free survival; ItchRO(Obs) = Itch Reported Outcome (observer); sBA = serum bile acid; TFS = liver transplant-free survival.
Source: Sponsor’s Clinical Summary.17
Figure 4: KM Estimates of 6-Year EFS in Children With ALGS Treated With Maralixibat: sBA Levels at Week 48 and Change in Pruritus (ItchRO[Obs]) From Baseline to Week 48
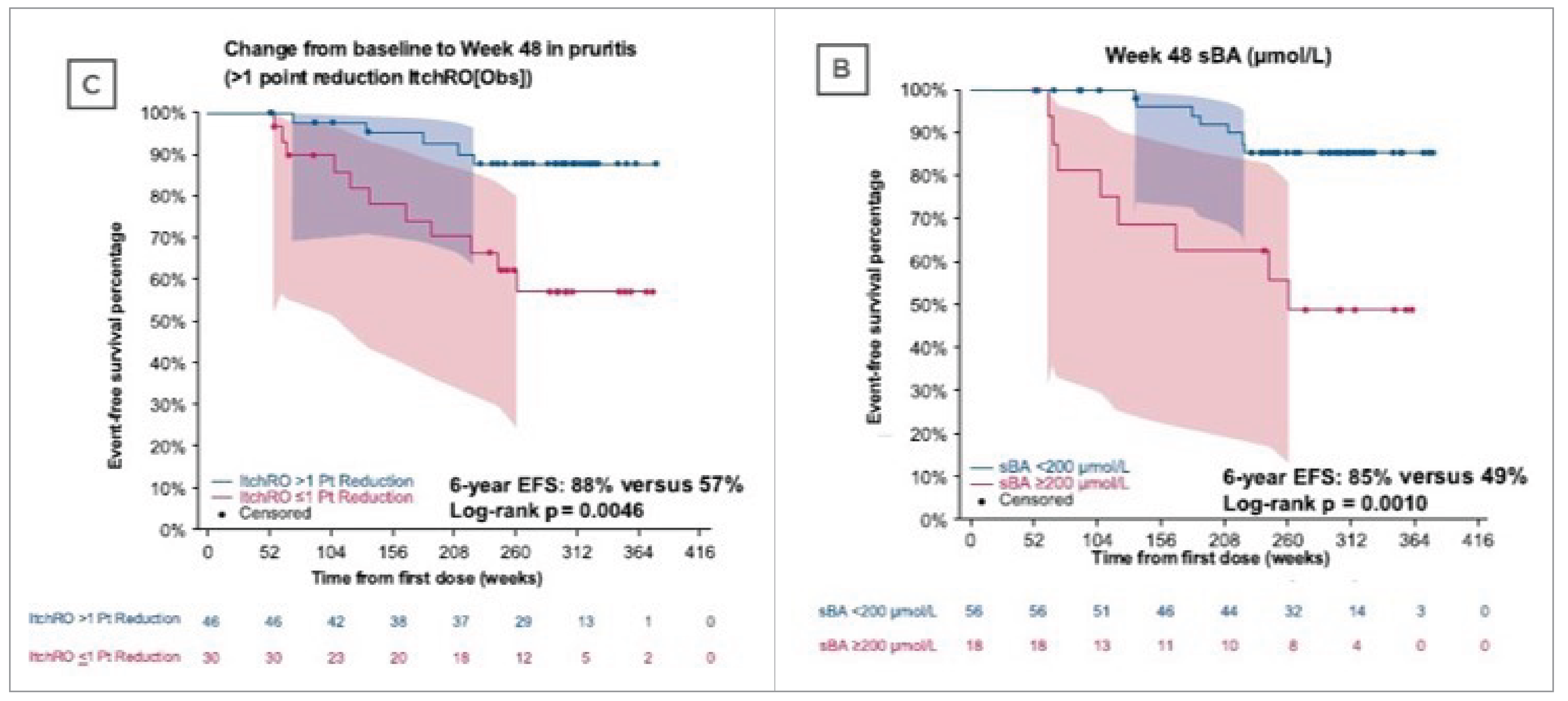
ALGS = Alagille syndrome; EFS = event-free survival; ItchRO = Itch Reported Outcome; ItchRO(Obs) = Itch Reported Outcome (observer); KM = Kaplan-Meier; sBA = serum bile acid.
Discussion
In patients with ALGS, predictors of EFS with maralixibat treatment include sBA (at week 48) and pruritus reduction (from baseline to week 48). These potential markers of disease may help identify disease progression in patients with ALGS treated with maralixibat, and inform long-term outcomes. As pruritus is a leading cause of liver transplant in patients with ALGS, these data also demonstrated that improvements in pruritus with maralixibat are significantly associated with improved EFS and TFS. These data identify potential prognostic markers that may better inform patient and provider discussions of clinical outcomes in patients receiving maralixibat treatment.
One limitation pointed out by the sponsor is the small sample size, although it is considered large for rare diseases. Another limitation the sponsor stated is the absence of liver histology or data from non-invasive techniques to assess hepatic fibrosis. According to the sponsor, it is unclear if improvements in EFS and TFS observed in these analyses were due to improvements in pruritus, which could be the major indication for transplant, or due to improvement or stabilization of hepatic fibrosis and consequent portal hypertension related to the reduction in hepatic retention of toxic bile acids. Lastly, only patients who have shown benefit from maralixibat — i.e., improved pruritus, laboratory parameters, or nutrition — were maintained in the LTE studies with sufficient follow-up that allowed for examination of long-term outcomes. This selection bias may contribute to overestimation of long-term outcomes.
Natural-History Comparison Study: Patients Treated With Maralixibat Versus GALA Natural-History Cohort
A natural-history comparison study was conducted to compare disease outcomes between 2 groups of patients with ALGS: 1) maralixibat-treated study patients and 2) patients who are brown adipose tissue inhibitor–naive from a natural-history global clinical research database (GALA).
The GALA clinical research database, established in 2018, is the only global ALGS database with a robust data collection and appropriate quality-assurance measures in place. GALA is recognized internationally by academic and tertiary referral liver transplant centres and is led by Dr. Binita Kamath at The Hospital for Sick Children in Toronto, with a high level of participation. It is supported by more than 100 physicians, surgeons, scientists, and research coordinators from 35 countries, who contribute more than 1,400 patient records into the clinical database. The GALA clinical research database includes clinical and laboratory data as well as disease outcomes.10
Study Design and Objectives
The study is a global, open-label, nonrandomized, external-control study to compare time-to-event clinical outcomes in patients with ALGS treated with maralixibat with selected external controls from the international registry of the GALA registry. This natural-history comparison followed a prespecified SAP. The statistical team performing the analysis remained blinded to treatment outcomes before the selection of patients from the GALA registry.
The objective is to evaluate the effect of long-term maralixibat treatment on clinical outcomes in patients with ALGS (duration of follow-up is up to 7 years). The prespecified hypothesis is that the time to a clinical event in patients treated with maralixibat is delayed compared with natural-history controls.
There are 2 cohorts in the study: patients with ALGS treated with maralixibat in the LUM001-301, LUM001-302, LUM001-303, LUM001-304, and LUM001-305 studies, and those contained within the natural history or standard of care cohort (GALA registry). The GALA cohort (standard of care) included patients with ALGS who aligned with maralixibat key study entry criteria (listed in the following section) from the registry.
Populations
The GALA registry includes multiple visits per patient from the date of diagnosis of ALGS disease until the patient has an event. Patients are otherwise in follow-up, and the last follow-up will be the date of analysis. Selection of patients for the GALA cohort (standard of care) followed a stepwise procedure of patients’ or visit start times to align with the maralixibat cohort, as follows.
Identification of patients and visits within the GALA registry was selected by the following:
inclusion criteria:
age at inclusion: aged older than 12 months and younger than 18 years
cholestasis, defined by 1 or more of the following:
total sBA greater than 3 times ULN
conjugated or direct bilirubin greater than 0.01 mmol/L
total bilirubin greater than 0.02 mmol/L
GGT greater than 3 times ULN
exclusion criteria:
ALT greater than 15 times ULN
clinical event, defined as liver transplant, SBD, liver decompensation, or death before inclusion
diagnosis of hepatocellular carcinoma by biopsy-proven histopathology
born before 1997
participation in a maralixibat study at any time
patients with only a single visit that do not have any follow-up information available.
Selection of the start of follow-up: A patient may be eligible with multiple visits at different time points. The best visit to represent baseline (i.e., the start of follow-up aligning with maralixibat studies) was selected by maximum likelihood methods. To be specific, the maximum estimated prediction within a patient, based on a stratified logistic regression analysis of all eligible visits, was compared with the maralixibat cohort (yes or no), including covariates of age and total bilirubin.
Balance assessment: The balance between the maralixibat cohort and the GALA cohort at baseline was assessed — i.e., the 2 cohorts should show comparable distributions of the variables such as age, sex, bilirubin (total and direct or conjugated), GGT, and ALT before selection was completed and before describing effects of treatment on the outcome events. If the 2 cohorts were not well balanced, the analysis was repeated with adjustment for confounders. In this case, inverse probability of treatment weights methods were applied using the estimated weights from a propensity score method.
The total number of patients in the GALA clinical research database at the cut-off for the GALA cohort selection and analyses was 1,438, with a total of 12,535 visits. Following the prespecified selection process, a total of 469 patients with 3,906 visits were included in the GALA control group (Figure 5). The maralixibat clinical study data included in this natural-history comparison comprised the aggregated data from all patients treated with maralixibat from the long-term maralixibat ALGS program (N = 84), with follow-up data up to 6 years.
Outcomes
EFS: First occurrence of any of the following listed events (maralixibat versus natural history):
Liver transplant
SBD
Liver decompensation (variceal bleeding, ascites requiring therapy)
Death
A sensitivity analysis included confirmed hepatocellular carcinoma by biopsy-proven histopathological diagnosis as an additional event.
Figure 5: Selection of the GALA Cohort (Standard-of-Care Control Group)
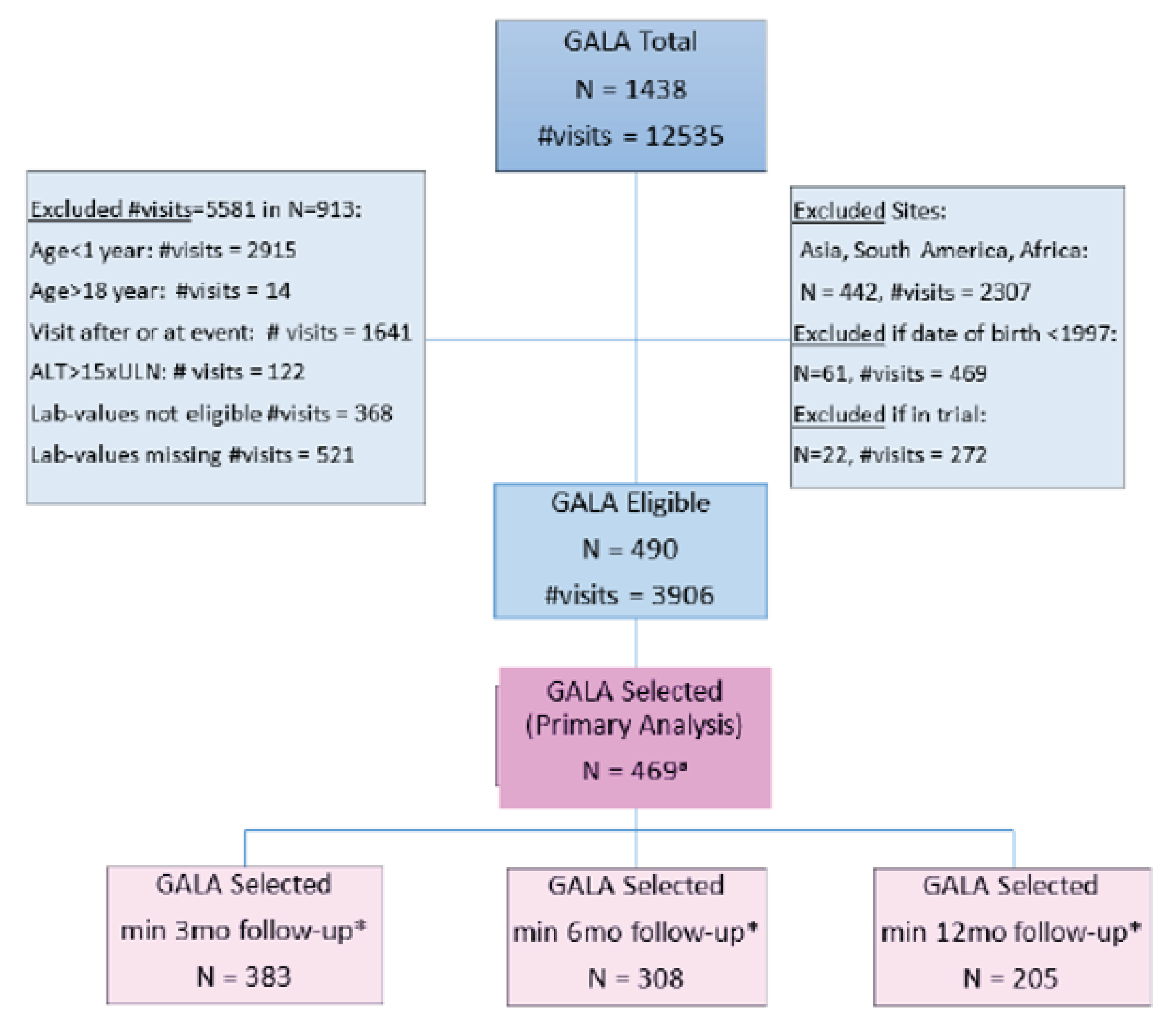
ALT = alanine transaminase; GALA = Global ALagille Alliance; mo = month; ULN = upper limit of normal; yr = year.
Note: Asterisks indicate that, to avoid immortal time bias, all eligible patients must have had 3, 6, or 12 moths of follow-up.
a Among the 490 GALA eligible patients, only 469 patients had all covariates needed to perform the maximum likelihood selection.
Source: Sponsor’s Clinical Summary.17
Statistical Analysis
No formal sample-size calculations were performed. Available patients meeting the selection criteria previously described were analyzed.
The statistical analysis conducted were as follows:
To select the baseline visit for the GALA cohort, logistic regression analysis was used.
Kaplan-Meier analyses were performed, and corresponding figures were generated to compare the time-to-event differences between the maralixibat and GALA cohorts.
Cox proportional hazards models were applied to describe and test differences between the treated and the control cohort, adjusting for potential confounders.
If inverse probability of treatment weights methods were applied (due to imbalance), the analysis was done using the estimated weights.
Sensitivity and subgroup analyses performed included the following:
Inclusion of hepatocellular carcinoma diagnosed by biopsy-proven histopathology as an additional event.
Selection of baseline visit for patients in GALA:
Definition of baseline for GALA cohort: Use first eligible visit.
Definition of baseline for maralixibat and GALA cohorts: Use date of birth instead of selected visit or treatment start.
Subgroup analyses:
Analysis was performed by region: North America versus Europe.
Analysis was performed using overlapping study centres participating in maralixibat studies and GALA registry.
Imputation: For patients treated with maralixibat, the date of the clinical event was used in the time-to-event analysis unless it was unavailable, in which case the date the patient discontinued was used. For patients who discontinued a maralixibat study, follow-up data on outcome events were collected through an appropriate institutional review board/ethics committee approval and consent process.
Missing data: Patients with missing outcome data will be censored at the time of discontinuation.
Results
Baseline Characteristics
Baseline characteristics generally demonstrate balance between the maralixibat cohort and the GALA control group, especially bilirubin, GGT, and ALT (Table 23).
Currently, treatment decisions are based on the management of pruritus and/or disease progression; therefore, measurement of sBA is not typically used for clinical decision-making. For this reason, limited sBA data are available in the GALA cohort (approximately 85% do not have sBA measures), as these are not sampled regularly on a clinical basis, often only at a single time point in some patients (at entry into the registry), and not longitudinally. Compared with patients for whom sBA data were available at baseline in the GALA cohort mean sBA was significantly higher in the maralixibat cohort, suggesting that the patients receiving maralixibat may have had worse disease at baseline; however, that cannot be confirmed given that most patients in the GALA cohort do not have sBA continuously monitored. (Table 23). To address the sBA limitations, a subgroup analysis was performed; it demonstrated results consistent with the primary analysis (Figure 7).
Balance in baseline pruritus levels between the groups could not be assessed as pruritus data are not recorded in the GALA cohort because of the lack of both a validated tool outside of clinical studies and inconsistent data collection across centres, regions, and countries. However, pruritus is a common reason for liver transplant and, as such, is recorded in the GALA cohort as 1 of the options as an indication for liver transplant. In the maralixibat studies, patients were required to have moderate-to-severe pruritus at baseline. The absence of pruritus severity data in the GALA cohort is a limitation of the dataset. Although up to 80% of patients with ALGS have pruritus,11 not all patients with ALGS have moderate-to-severe pruritus. Therefore, the selected GALA control group has likely a larger heterogeneity of pruritus severity, which would include those with a milder pruritus status than those in the maralixibat studies. As such, the presence of pruritus and its severity are unlikely to have influenced results in favour of the maralixibat cohort.
Patients with cardiac disease were not excluded from the maralixibat studies; 82.1% of patients from studies LUM001-301 and LUM001-305 and studies LUM001-302 and LUM001-303 had cardiac disease reported at baseline, and 93.5% had cardiac disease reported at baseline in the LUM001-304 study. Similarly, cardiac disease was not excluded from the GALA cohort selection. It has been previously reported that 91% of patients in the GALA database had cardiac anomalies,10 so similar rates in the selected GALA control group can be expected in the absence of relevant exclusion criteria.
Table 23: Baseline Characteristics of Maralixibat Cohort and GALA Cohort (Control Group)
Variable | Maralixibat cohort N = 84 | GALA cohort N = 469 |
|---|---|---|
Sex, n (%) | ||
Male | 49 (58.3) | 274 (58.4) |
Female | 35 (41.7) | 195 (41.6) |
Age at baseline, years | ||
Median (Q1, Q3) | 5.6 (2.7, 9.9) | 4.3 (2.2, 9.6) |
Region, n (%) | ||
Europe | 41 (48.8) | 229 (48.8) |
North America | 34 (40.5) | 195 (41.6) |
Australia | 9 (10.7) | 45 (9.6) |
Mutation, n (%) | ||
Jagged-1 | 81 (97.6) | 330 (95.1) |
NOTCH2 | 2 (2.4) | 17 (4.9) |
Other/unknown | 1 (0.2) | 37 (9.6) |
Total bilirubin | ||
Median (Q1, Q3), mmol/L | 0.04 (0.01, 0.09) | 0.02 (0.01, 0.13) |
< 0.02, n (%) | 37 (44.0) | 235 (50.1) |
≥ 0.02 | 47 (56.0) | 234 (49.9) |
GGT | ||
Median (Q1, Q3), log10 × ULN | 1.25 (0.93, 1.44) | 1.24 (0.93, 1.52) |
< 3 × ULN, n (%) | 3 (3.6) | 6 (1.3) |
≥ 3 × ULN | 81 (96.4) | 463 (98.7) |
ALT, U/L | ||
Median (Q1, Q3) | 145 (94, 201) | 130 (75, 203) |
sBA,a µmol/L | ||
Median (Q1, Q3) | 200 (81, 371) | 125 (39, 260) |
ALT = alanine transaminase; GALA = Global ALagille Alliance; GGT = gamma-glutamyl transferase; Q = quartile; sBA = serum bile acid; ULN = upper limit of normal.
aBaseline sBA was available for 73 patients in the GALA control group. Approximately 85% values were not captured in the GALA cohort.
Source: Sponsor’s Clinical Summary.17
Efficacy
Event-Free Survival Analysis
The primary analysis was the comparison of the time to the first clinical event between the maralixibat cohort and GALA control group. EFS adjusted for age, sex, bilirubin, and ALT was statistically significantly higher in the maralixibat cohort compared with the GALA control group (HR = 0.305; 95% CI, 0.189 to 0.491; P < 0.0001), indicating a 70% improvement in EFS with maralixibat treatment (Figure 6).
The impact of different clinically meaningful parameters (such as age, sex, serum, levels of total bilirubin, ALT, GGT, region, and year of birth), different baseline definitions, and potential differences in standard of care between regions and centres was also assessed (Figure 7). The immediate events in the GALA control group (Figure 6) may be suggestive of an immortal time bias. Pruning analyses for comparison of EFS that excluded patients who had events in the first 3, 6, or 12 months confirmed significant improvement in EFS with maralixibat treatment (Figure 7).
Liver TFS Analysis
The majority of events in both cohorts were liver transplantations. A separate analysis considered liver TFS, where only events of liver transplant or death were considered. Liver TFS was statistically significantly higher in the maralixibat cohort compared with the GALA control group (HR = 0.332; 95% CI, 0.197 to 0.559, P < 0.0001), indicating a 67% improvement in liver TFS with maralixibat treatment (Figure 8).
For the GALA control group (N = 469), the main reasons for liver transplant were end-stage liver disease and pruritus, followed by growth impairment. In total, there were 24 deaths in the GALA control group. Importantly, there were 2 cardiac-related deaths, with the remaining deaths being most commonly related to liver disease and noncardiac vascular complications. In the maralixibat cohort (N = 84), there were 12 liver transplants reported, and the most common indications were disease progression and pruritus. There were 5 deaths reported in the maralixibat cohort.
Figure 6: Kaplan-Meier Plot for EFS — Maralixibat Cohort Versus GALA Control Group (Primary Analysis)
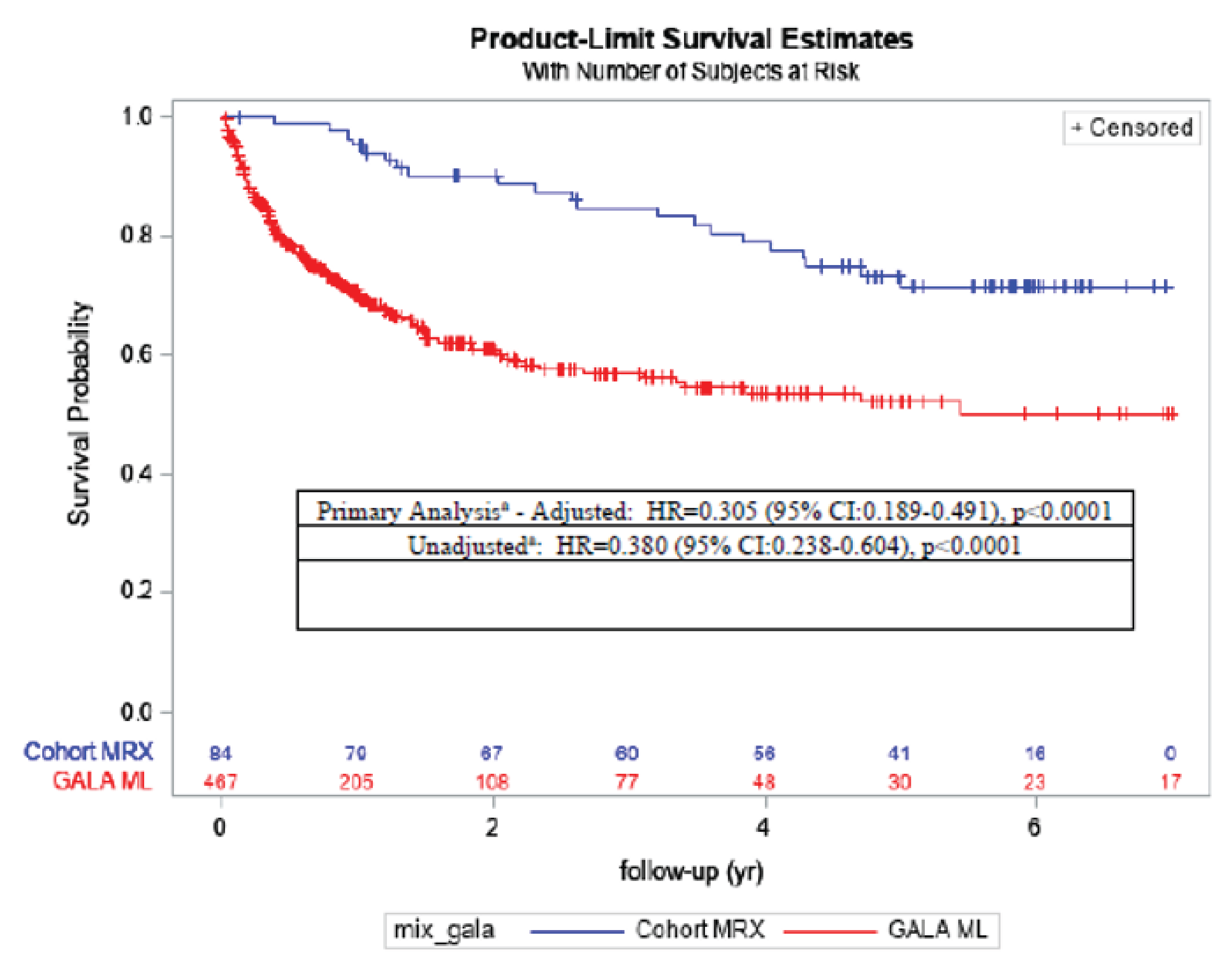
CI = confidence interval; GALA = Global ALagille Alliance; HR = hazard ratio; ML = maximum likelihood; MRX = maralixibat; SAP = statistical analysis plan; vs. = versus; yr = year.
a Cox regression models: Primary: Cox regression – effect of MRX vs. GALA log likelihood test adjusted for age, sex, bilirubin, and ALT (according to the SAP). Unadjusted: Crude model.
Source: Sponsor’s Clinical Summary17
Discussion
The analysis included data from the natural-history clinical research registry and interventional dataset in ALGS. Given the challenge of conducting large, randomized studies in severe, rare pediatric settings, the availability of these datasets has important implications for understanding the effects of maralixibat in ALGS. The breadth of the GALA clinical research database allowed for the selection of a balanced external comparator group to the maralixibat cohort to evaluate EFS and TFS. Important baseline demographic and disease characteristics appeared to be comparable between the 2 groups, except for sBA, which was higher in the maralixibat group than in the control group (200 µmol/L versus 125 µmol/L). Of note, sBA was unavailable in approximately 85% of the patients in the control group. The study showed statistically and clinically significant reduction in liver transplant, death, and other associated events in patients who received maralixibat treatment compared with patients who received standard of care. In conclusion, the natural-history comparison supplements evidence supporting maralixibat as the potential therapeutic option to improve EFS and TFS in patients with ALGS.
Critical Appraisal
Internal Validity
The sponsor attempted to address gaps in evidence by providing long-term outcomes (EFS, TFS) data not available in pivotal trial and a natural-history study to complement the single-arm design of the pivotal trial. Although these attempts partially addressed these gaps, there are still limitations remaining. Primarily, in alignment with the pivotal trial, the relationship between sBA and pruritus has not been fully established.
Moreover, the sponsor applied the external control for maralixibat cohort to address gaps in evidence — i.e., the beneficial effect on long-term clinical outcomes. The sponsor used many methods to minimize selection bias between the comparison groups. First, the statistical team performing the analysis remained blinded to treatment outcomes before the selection of patients from the GALA registry. Second, to balance the baseline characteristics of both cohorts, the sponsor followed a stepwise selection process using a common set of cohort eligibility criteria as the maralixibat trials and a prespecified selection process using logistic regression. The GALA study sites were located in Canada as well as in the US, Europe, and Australia, which share similar health care systems as in Canada. Also, the 2 cohorts were likely recruited from comparable catchment areas to which rare genetic diseases are referred and where genetic testing and specialist care are available. As stated by the sponsor, some of the patients in maralixibat trials are from the same clinical sites as GALA registry clinical sites.
Figure 7: Forest Plot for EFS Analysis
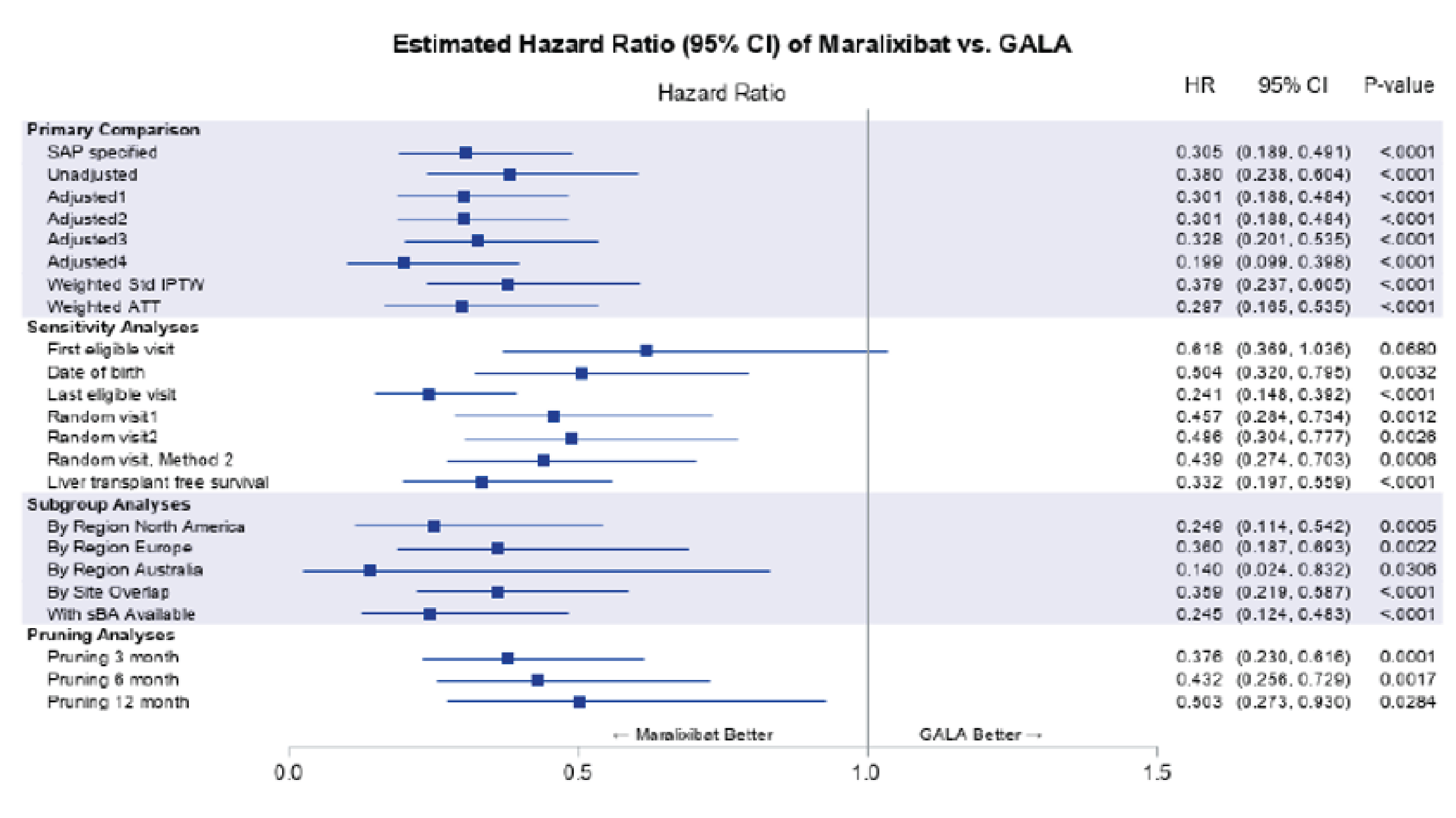
ATT = average treatment effect in the treated; CI = confidence interval; GALA = Global ALagille Alliance; GGT = gamma glutamyltransferase; HR = hazard ratio; IPTW = inverse probability of treatment weights; SAP = statistical analysis plan; sBA = serum bile acid; Std = standardized; vs. = versus.
Notes: SAP specified refers to the Cox regression adjusted for age, sex, total bilirubin, and ALT. Adjusted1 refers to the Cox regression adjusted for age, total bilirubin, and GGT. Adjusted2 refers to the Cox regression adjusted for age, total bilirubin, GGT, ALT, and region. Adjusted3 refers to the Cox regression adjusted for age, total bilirubin, GGT, ALT, sex, and year of birth. Adjusted4 refers to the Cox regression adjusted for age, total bilirubin, GGT, and sBA.
Source: Sponsor’s Clinical Summary.17
Figure 8: Kaplan-Meier Plot for Liver TFS: Maralixibat Cohort Versus GALA Control Group
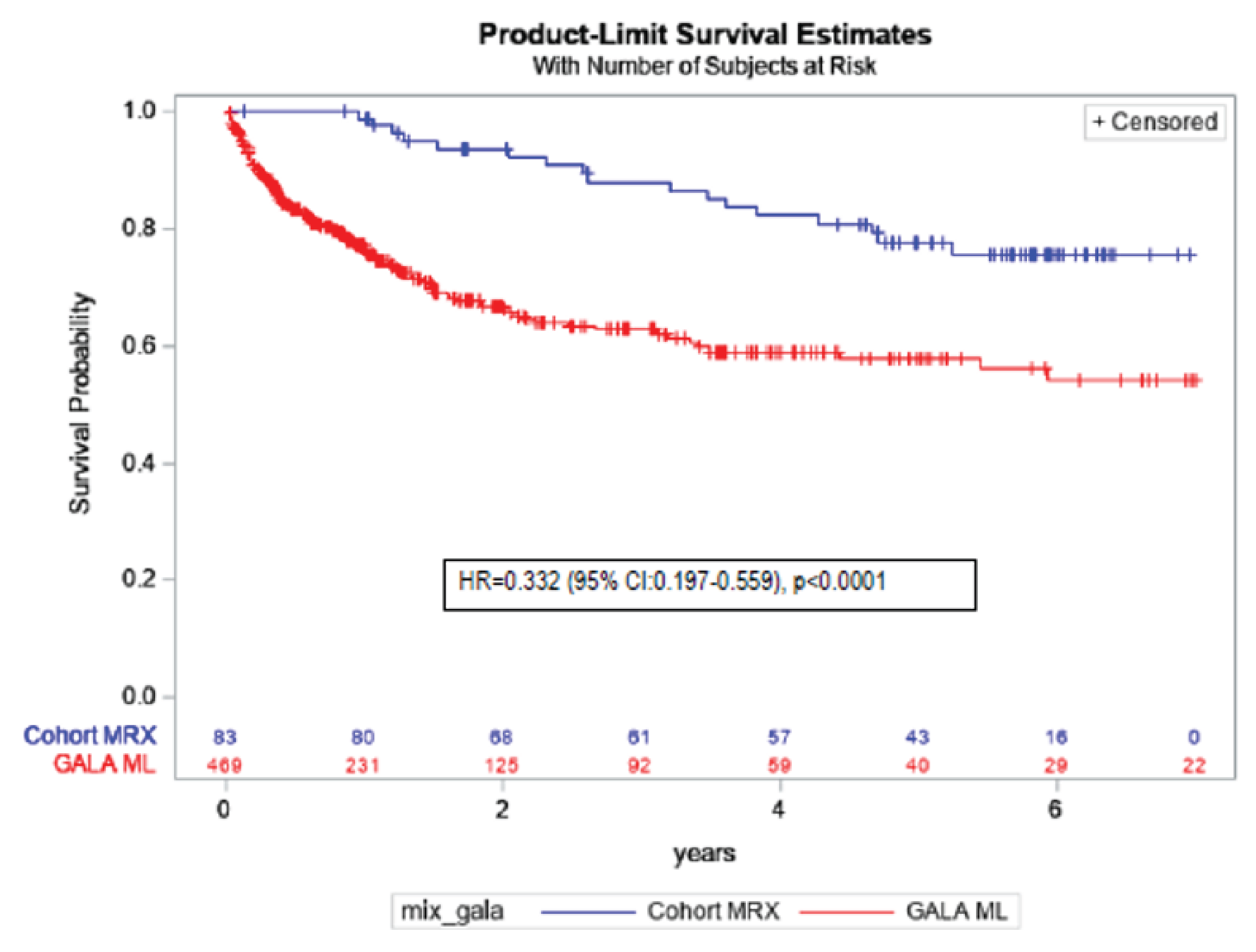
CI = confidence interval; GALA = Global ALagille Alliance; HR = hazard ratio; ML = maximum likelihood; MRX = maralixibat.
Source: Sponsor’s Clinical Summary.17
Although both comparison groups are closely matched with respect to age, sex, genetic mutations, bilirubin (total and direct/conjugated), GGT, and ALT, sBA data were available in only 73 of 469 patients from the GALA registry, with approximately 85% missing information. Moreover, although patients in the GALA registry have confirmed diagnoses of neonatal cholestasis, with the majority having Jagged-1 or NOTCH2 mutations, it is unknown how severe their cholestatic pruritus was in the absence of information on pruritus severity at baseline, which is a significant limitation. Other important prognostic factors besides pruritus severity, such as patients’ history of disease, previous treatments, duration of disease, and comorbidities, are unavailable. Therefore, it is highly likely that many residual confounding factors are still unaccounted for, particularly the disease severity, which could have a significant impact on long-term clinical outcomes such as liver transplant or death. Other factors, such as socioeconomic factors that affect patients’ or physicians’ choice of treatment, as well as differences in clinical settings in different regions, standards of care, and health care systems, could also affect the study results.
Taken together, despite the potential biases due to retrospective cohort design and missing important information on disease characteristics, the cohort study likely showed significant treatment effect of maralixibat on long-term outcome such as EFS and TFS in patients with moderate-to-severe cholestatic pruritus of ALGS.
External Validity
Given the rarity of ALGS and the challenges associated with conducting clinical trials in children, it is extremely difficult to assess the generalizability of long-term effect on clinically outcomes such as liver transplant and death. This study, with external control based on disease registry, may have provided important insights into coping with those evidence gaps. Since most patients treated with maralixibat included in this report have moderate-to-severe cholestatic pruritus (as measured by ItchRO questionnaire), the itch severity in the study may not be representative of that found in the overall ALGS population, which shows a spectrum of itch severity. Missing information on important baseline and disease characteristics, especially information about severity of cholestatic pruritus at baseline, makes it unclear how generalizable the results can be.
Discussion
Summary of Available Evidence
One pivotal trial, the ICONIC trial, met the inclusion criteria for the systematic review conducted by the sponsor. The ICONIC trial (9 sites in 6 countries, N = 31) is a double-blind, placebo-controlled, RWD study with a long-term, open-label extension in children with ALGS (range, 1 to 15 years) designed to evaluate the safety and efficacy of maralixibat. The trial’s primary outcome was the mean change from weeks 18 to 22 of fasting sBA levels in patients who previously responded to maralixibat treatment (n = 15), as defined by a reduction in sBA greater than or equal to 50% from baseline to weeks 12 or 18. Maralixibat was administered according to a dose-escalation schedule over 6 weeks of treatment to a final dose of 380 mcg/kg/day. Patients were eligible to increase to twice-daily dosing for a total of 760 ug/kg/day, starting after week 103, which is outside of the Health Canada recommended dose.
The sponsor submitted a natural-history comparison study, which is presented in this report, comparing disease outcomes among patients with ALGS treated with maralixibat (N = 84) with an external-controls cohort from the GALA disease registry database (n = 469), with follow-up data up to 6 years. Outcomes assessed included EFS (composite end point of first event of liver decompensation [ascites, variceal bleeding], SBD, liver transplant, and death) and TFS (liver transplant and death). Results from patient-level data from 3 long-term studies of patients with ALGS treated with maralixibat, including the LUM001-303 (IMAGINE) trial, the ICONIC pivotal trial (LUM001-304), and the IMAGINE-II (LUM001-305) trial to identify predictors of EFS and TFS was submitted by the sponsor and presented in this report.
Interpretation of Results
Efficacy
The ICONIC pivotal trial included a double-blind phase and appeared to have appropriate methods for blinding, allocation concealment, and randomization during the RWD phase. Although practical constraints associated with sample size are acknowledged, given the rarity of ALGS, small sample size remains a significant limitation, as the level of certainty was rated down across the outcomes. Furthermore, it should be noted that, during open-label phases of the study, patients’ and/or caregivers’ knowledge of treatment assignment may have biased subjective outcomes such as the ItchRO, PedsQL, and report of harms.
Evidence from the pivotal ICONIC trial showed that maralixibat may result in a decrease in sBA from weeks 18 to 22 in the mITT population (n = 15, defined as responders of study patients with an sBA reduction ≥ 50% at weeks 12 or 18), compared with placebo. Subgroup analyses were generally supportive of the findings of the primary analyses. Similar results were demonstrated when change in sBA from weeks 18 to 22 was assessed in the ITT population (N = 31), suggesting that the benefit from maralixibat observed in the trial was primarily from those responders (n = 15), which represented about 50% of the overall study population. Furthermore, trial results found that decreases in sBA levels were maintained over time in the overall study population as demonstrated by change in sBA levels from baseline to week 18 (secondary outcome) and from baseline to week 48 (additional efficacy outcome). The clinical experts consulted for this review indicated that although sBA levels are not measured in clinical practice due to high costs and barriers in access to testing, the results of the primary outcome provide supportive evidence of the mechanism of action of maralixibat. Descriptive posthoc data from the ICONIC pivotal trial suggested that reductions in sBA from baseline to week 48 were associated with reductions in mean ItchRO(Obs) weekly average morning severity scores (Appendix 1). These internal data supported a potential correlation in the improvement between sBA and ItchRO. Yet, according to the clinical expert consulted for this review, there was no substantial and consistent literature on the association between sBA change and itching in patients with ALGS. Moreover, due to the small number of patients (n = 28), it is unclear the extent to which sBA levels may be associated with pruritus in patients with ALGS.
Input from patient and clinician groups and clinical experts highlighted the importance of reduction in pruritus as an important treatment goal for patients. In the ICONC trial, pruritus was assessed using the ItchRO instrument, which has been reported to be generally valid, reliable, and responsive, as assessed either by patients or caregivers (observers). Patients who were dropped off from maralixibat treatment had experienced an increase (worsening) in mean ItchRO(Obs) and ItchRO(Pt) weekly average morning severity scores from weeks 18 to 22, which nearly rebound back to the same levels at baseline, whereas those who continued to receive maralixibat generally maintained lower mean scores during the RWD period. The magnitude of improvement in both measures may be clinically meaningful both in favour of maralixibat according to the MID estimate of 1 point identified in the literature and clinical expert input. Subgroup analyses for the ItchRO(Obs) were generally consistent with the overall results of the pivotal trial, except for where there was a very small number of patients in a given category (n ≤ 4). Similarly, in the overall population, improvements were observed in the ItchRO(Obs) and ItchRO(Pt) weekly average morning severity scores from baseline to week 18 (secondary end point) and baseline to week 48 (additional efficacy end point), providing supportive evidence for maralixibat.
HRQoL was highlighted as an important treatment goal for patients from the input of patient groups and the clinical expert, especially in terms of patient and caregiver fatigue. The pivotal trial assessed HRQoL using the PedsQL, which suggested that maralixibat would likely have contributed to an improvement in the PedsQL total score (parent) and also likely contributed to a considerable reduction of the PedsQL Multidimensional Fatigue Scale score compared with placebo. However, due to a short 4-week randomization phase and, moreover, a small sample size, there was high uncertainty of these findings as result of wide 95% CI in the estimation of the changes, which included null values. Nevertheless, such a trend in improvement of fatigue, in particular, was supported by the LTE phase of the study. In the overall study population, from baseline to week 18 and from baseline to week 48, patients achieved an 8-point improvement in the PedsQL total score and a nearly 20-point improvement in the PedsQL Multidimensional Fatigue Scale score. These changes are larger than the estimated MID of 4 to 5 points for the PedsQL Generic Core Scales in a sample of children with and without chronic health conditions. However, in a single-arm, LTE phase, any improvements over time compared with baseline may not be completely attributable to the treatment itself alone. There might have been other contemporaneous changes that could also contributed to the observed temporal changes overtime. For example, it should be noted that the number of patients assessed for the PedsQL Multidimensional Fatigue Scale score was low (22 out of 31) in the assessment from baseline to week 48. As such, those who completed the questionnaires may be fundamentally different than those who did not (i.e., differences in treatment response). The extent to which the impact of missing data due to selective reporting on the assessment of this outcome is unclear.
Other liver-related outcomes identified as important by the clinical experts consulted included change in liver biomarkers and enzymes. Secondary end points assessed in the RWD phase of the ICONIC trial found that maralixibat may increase ALT and ALP levels and may result in little-to-no difference in total and direct bilirubin compared with placebo. Again, due to the short duration of the RWD phase and the small sample size, the estimation of the differences included a null value across the 95% CIs, indicating the lack of precision, which rendered high uncertainty in these potential improvements. Similarly, no clear and consistent pattern of improvements in ALT and ALP were observed at weeks 18 and 48 in the overall trial population, and no notable changes in total and direct bilirubin during these time periods. The lack of a comparator at these time points and the small sample sizes may have contributed to the high uncertainty of the evidence on improvement of liver biomarkers. The clinical experts noted the importance of change in body height and weight as an important outcome and stated that it is reasonable to assess these outcomes in the indicated patient population in clinical practice. Numerical increases in body height and weight z scores were noted from baseline to week 48 and to week 100; however, the evidence is very uncertain, as conclusions about their efficacy relative to any comparator cannot be drawn.
Results from the natural-history comparison study reported a large improvement in EFS and TFS in the maralixibat cohort compared with standard of care in the control cohort. The study applied an external control based on disease registry, which provided a large sample size with a study design that matched on all known potential confounders including age, sex, genetic mutations, and so forth. However, the disease severity was not well known and therefore, not appropriately controlled, which is a significant concern. Meanwhile, the potential for unknown confounding factors may also have had an impact on the results. Yet it is unlikely that such potential biases would be able to explain all the large differences in EFS and TFS between the comparison groups. In combination with the evidence based on the observed benefit on sBA during the randomized phase of the trial and the potential large improvements over the long-term phase, it could extrapolate that an improvement on EFS and TFS is highly likely. In other words, despite the nonrandomized, retrospective cohort design, this observational study supplements evidence supporting maralixibat as the potential therapeutic option to improve EFS and TFS in patients with ALGS.
Harms
Evidence from the ICONIC trial during the RWD period suggest that maralixibat may result in little-to-no difference in SAEs, diarrhea, abdominal pain, and FSV deficiency compared with placebo. Overall, the most frequently reported AEs (> 30% in at least 1 phase) were abdominal pain, pyrexia, diarrhea, nasopharyngitis, vomiting, cough, and pruritus. A total of 6 patients (2 each in the open-label phase, the after RWD phase, and the LTE phase) experienced AEs leading to study drug discontinuation. According to the clinical experts consulted, the 4-week RWD period was not sufficient to fully assess the comparative safety of maralixibat to placebo. Most AEs were generally of mild-to-moderate severity and no deaths were noted during the study.
Conclusions
There is an unmet need for symptomatic and curative treatment options for cholestatic pruritus in pediatric patients with ALGS. Patients and clinicians highlighted the need for treatments that reduce the frequency and severity of pruritus and reduce patient and caregiver fatigue. The pivotal phase II, double-blind, placebo-controlled, randomized drug withdrawal trial included in this review assessed the treatment of cholestatic pruritus in pediatric patients with ALGS (aged 12 months to 15 years). The trial was an exploratory study.
The study demonstrated that maralixibat may result in a decrease in sBA levels and results in a clinically meaningful improvement in pruritus as assessed by the ItchRO(Obs) and ItchRO(Pt) weekly average morning severity scores compared with placebo. It is important to note that the “low” certainty of evidence as assessed by GRADE for sBA and pruritus outcomes is due to the imprecision observed and not from bias due to study limitations. This imprecision due to small sample size is clearly connected to the nature of the rarity of the disease. Improvements in the PedsQL total score (parent) and the PedsQL Multidimensional Fatigue Scale score (parent) were uncertain. It also remained uncertain whether maralixibat may have increased ALT and ALP levels or resulted in any difference in total and direct bilirubin compared with placebo. Despite certain limitations inherent with observational study design, it is likely that there is a significant treatment effect of maralixibat on long-term outcome such as EFS and TFS.
Maralixibat was generally well-tolerated in the ICONIC trial, with limited grade 3 AEs or SAEs. Of note, however, due to the rare nature of the disease, the study sample size was small and the study was short, in a 4-week randomized duration; therefore, more evidence is needed to support the overall benefit and safety of maralixibat.
References
1.Subramaniam P, Knisely A, Portmann B, et al. Diagnosis of Alagille syndrome-25 years of experience at King's College Hospital. J Pediatr Gastroenterol Nutr. 2011;52(1):84-89. PubMed
2.Saleh M, Kamath B, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet. 2016;9:75-82. PubMed
3.Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251-257. PubMed
4.Osawa Y, Seki E, Adachi M, et al. Role of acid sphingomyelinase of Kupffer cells in cholestatic liver injury in mice. Hepatology. 2010;51(1):237-245. PubMed
5.Mehl A, Bohorquez H, Serrano MS, Galliano G, Reichman TW. Liver transplantation and the management of progressive familial intrahepatic cholestasis in children. World J Transplant. 2016;6(2):278-290. PubMed
6.Kamath BM, Ye W, Goodrich NP, et al. Outcomes of Childhood Cholestasis in Alagille Syndrome: Results of a Multicenter Observational Study. Hepatol Commun. 2020;4(3):387-398. PubMed
7.Monte MJ, Marin JJ, Antelo A, Vazquez-Tato J. Bile acids: chemistry, physiology, and pathophysiology. World J Gastroenterol. 2009;15(7):804-816. PubMed
8.Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut. 2001;49:431-435. PubMed
9.Englert C, Grabhorn E, Burdelski M, Ganschow R. Liver transplantation in children with Alagille syndrome: indications and outcome. Pediatr Transplant. 2006;10(2):154-158. PubMed
10.Vandriel SM, Li LT, She H, et al. Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatology. 2023;77(2):512-529. PubMed
11.Kamath BM, Baker A, Houwen R, Todorova L, Kerkar N. Systematic Review: The Epidemiology, Natural History, and Burden of Alagille Syndrome. J Pediatr Gastroenterol Nutr. 2018;67(2):148-156. PubMed
12.Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology. 1999;29:822-829. PubMed
13.Kamath BM, Yin W, Miller H, et al. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl. 2012;18(8):940-948. PubMed
14.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
15.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
16.Clinical Study Report: LUM001-304. ICONIC: Long-term, open-label study with a double-blind, placebo-controlled, randomized drug withdrawal period of lum001, an apical sodium-dependent bile acid transporter inhibitor (asbti), in patients with Alagille syndrome [internal sponsor's report]. Foster City (CA): Mirum Pharmaceuticals Inc.; 2020 Oct 13.
17.Summary of Clinical Evidence [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Livmarli (Maralixibat), oral solution, 9.5 mg/mL [internal sponsor's package]. Foster City (CA): Mirum Pharmaceuticals Inc.; 2023 May 05.
18.Fischetto R, Palmieri VV, Tripaldi ME, et al. Alagille Syndrome: A Novel Mutation in JAG1 Gene. Front Pediatr. 2019;7:199. PubMed
19.Canadian ALGS and Livmarli Landscape Assessment. Data on file. In: Drug Reimbursement Review sponsor submission: Livmarli (maralixibat), oral solution, 9.5 mg/mL [internal sponsor's package]. Foster City (CA): Mirum Pharmaceuticals Inc.; 2023 May 05.
20.Dull MM, Kremer AE. Newer approaches to the management of pruritus in cholestatic liver disease. Curr Hepatol Rep. 2020;19:86-95.
21.Emerick KM, Whitington PF. Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. Hepatology. 2002;35(6):1501-1506. PubMed
22.Mattei P, von Allmen D, Piccoli D, Rand E. Relief of intractable pruritus in Alagille syndrome by partial external biliary diversion. J Pediatr Surg. 2006;41(1):104-107; discussion 104-107. PubMed
23.Kwong AJ, Ebel NH, Kim WR, et al. OPTN/SRTR 2020 Annual Data Report: Liver. Am J Transplant. 2022;22 Suppl 2:204-309. PubMed
24.Shneider BL. Liver transplantation for Alagille syndrome: the jagged edge. Liver Transpl. 2012;18(8):878-880. PubMed
25.Search Orphan Drug Designations and Approvals: maralixibat Silver Spring (MD): U.S. Food and Drug Administration; 2023: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=401513. Accessed 2023 Jun 1.
26.Product information: Livmarli (maralixibat chloride). (European public assessment report). Amsterdam (NL): European Medicines Agency; 2023: https://www.ema.europa.eu/en/medicines/human/EPAR/livmarli. Accessed 2023 Jun 1.
27.Di Ciaula A, Garruti G, Lunardi Baccetto R, et al. Bile Acid Physiology. Ann Hepatol. 2017;16 Suppl 1:S4-S14. PubMed
28.Reue K, Lee JM, Vergnes L. Regulation of bile acid homeostasis by the intestinal Diet1-FGF15/19 axis. Curr Opin Lipidol. 2014;25(2):140-147. PubMed
29.Al-Dury S, Marschall HU. Ileal Bile Acid Transporter Inhibition for the Treatment of Chronic Constipation, Cholestatic Pruritus, and NASH. Front Pharmacol. 2018;9:931. PubMed
30.Dawson PA. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb Exp Pharmacol. 2011(201):169-203. PubMed
31.Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50(12):2340-2357. PubMed
32.Product monograph: pms-Ursodiol C (Ursodiol). Montreal (QC): Pharmascience; 2017: https://pdf.hres.ca/dpd_pm/00040988.PDF. Accessed 2023 Jun 1.
33.Product monograph: Rifadin (Rifampin). Laval (QC): sanofi-aventis Canada Inc.; 2017: https://pdf.hres.ca/dpd_pm/00040693.PDF. Accessed 2023 Jun 1.
34.Product monograph: Novohydroxyzin (Hydroxyzine Hydrochloride). Scarborough (ON): Teva Canada Limited; 2014: https://www.tevacanada.com/globalassets/canada-scs/product-monographs/novo-hydroxyzin-capsules-10-mg-25-mg--50-mg_pi-feb19_1987-wis-eng.pdf. Accessed 2023 Jun 1.
35.Donkers JM, Roscam Abbing RLP, van de Graaf SFJ. Developments in bile salt based therapies: A critical overview. Biochem Pharmacol. 2019;161:1-13. PubMed
36.Gonzales E, Hardikar W, Stormon M, et al. Maralixibat Treatment Significantly Reduces Pruritus and Serum Bile Acids in Patients with Alagille Syndrome: Results from a Randomized Phase II Study with 4 Years of Follow-Up. SSRN Electronic Journal. 2020.
37.Drug Reimbursement Review sponsor submission: Livmarli (maralixibat), oral solution, 9.5 mg/mL [internal sponsor's package]. Foster City (CA): Mirum Pharmaceuticals Inc.; 2023 May 05.
38.van Wessel DBE, Thompson RJ, Gonzales E, et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J Hepatol. 2020;73(1):84-93. PubMed
39.Harpavat S, Hawthorne K, Setchell KDR, et al. Serum bile acids as a prognostic biomarker in biliary atresia following Kasai portoenterostomy. Hepatology. 2023;77(3):862-873. PubMed
40.Kamath BM, Abetz-Webb L, Kennedy C, et al. Development of a Novel Tool to Assess the Impact of Itching in Pediatric Cholestasis. Patient. 2018;11(1):69-82. PubMed
41.Foster B, Gauthier M, Vig P, Jaecklin T, Kamath B, Andrae DA. Itch reported outcome tool for caregivers of pediatric patients with cholestatic liver disease: an analysis of validation and scoring from the ICONIC maralixibat study. ISPOR; May 18-20, 2020; Virtual.
42.Varni JW, Burwinkle TM, Seid M, Skarr D. The PedsQL 4.0 as a pediatric population health measure: feasibility, reliability, and validity. Ambul Pediatr. 2003;3(6):329-341. PubMed
43.Clinical Study Report: GALA-MRX-ALGS. Long-term outcomes of long-term maralixibat treatment in patients with Alagille Syndrome compared with a natural history cohort from the GALA registry. Statistical Analysis Plan (SAP). In: Drug Reimbursement Review sponsor submission: Livmarli (maralixibat), oral solution, 9.5 mg/mL [internal sponsor's package]. Foster City (CA): Mirum Pharmaceuticals Inc.; 2023 May 05.
44.Ayoub MD, Kamath BM. Alagille Syndrome: Diagnostic Challenges and Advances in Management. Diagnostics (Basel). 2020;10(11). PubMed
45.Gonzales E, Hardikar W, Stormon M, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet. 2021;398(10311):1581-1592. PubMed
46.Shneider BL, Spino CA, Kamath BM, et al. Impact of long-term administration of maralixibat on children with cholestasis secondary to Alagille syndrome. Hepatol Commun. 2022;6(8):1922-1933. PubMed
47.Mirum Pharmaceuticals Inc. NCT02160782: Safety and Efficacy Study of LUM001 (Maralixibat) With a Drug Withdrawal Period in Participants With Alagille Syndrome (ALGS) (ICONIC). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT02160782. Accessed 2023 Jun 5.
48.Mirum Pharmaceuticals Inc. NCT02117713: An Extension Study to Evaluate the Long-Term Safety and Durability of Effect of LUM001 in the Treatment of Cholestatic Liver Disease in Pediatric Subjects With Alagille Syndrome (IMAGINE-II). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT02117713. Accessed 2023 Jun 5.
49.Sokol RJ, Gonzales EM, Kamath BM, et al. Predictors of 6-year event-free survival in alagille syndrome patients treated with maralixibat, an ileal bile acid transporter inhibitor. Hepatology. 2023. PubMed
50.Sokol RJ, Gonzales E, Kamath B, et al. Predictors of 6-year event-free survival in patients with ALGS treated with maralixibat, an IBAT inhibitor. American Association for the Study of Liver Diseases (AASLD) The Liver Meeting Digital Experience; November 12-15, 2021; Virtual.
51.Rovner AJ, Schall JI, Jawad AF, et al. Rethinking growth failure in Alagille syndrome: the role of dietary intake and steatorrhea. J Pediatr Gastroenterol Nutr. 2002;35(4):495-502. PubMed
52.Feranchak AP, Sokol R. Medical and nutritional management of cholestasis in infants and children. Liver disease in children. 2007;3:190-231.
53.Wasserman D, Zemel BS, Mulberg AE, et al. Growth, nutritional status, body composition, and energy expenditure in prepubertal children with Alagille syndrome. J Pediatr. 1999;134(2):172-177. PubMed
54.Kronsten V, Fitzpatrick E, Baker A. Management of cholestatic pruritus in paediatric patients with alagille syndrome: the King's College Hospital experience. J Pediatr Gastroenterol Nutr. 2013;57(2):149-154. PubMed
55.Thebaut A, Habes D, Gottrand F, et al. Sertraline as an Additional Treatment for Cholestatic Pruritus in Children. J Pediatr Gastroenterol Nutr. 2017;64(3):431-435. PubMed
56.Wang KS, Tiao G, Bass LM, et al. Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology. 2017;65(5):1645-1654. PubMed
57.Kriegermeier A, Wehrman A, Kamath BM, Loomes KM. Liver disease in alagille syndrome. Alagille Syndrome. Cham (CH): Springer; 2018:49-65.
58.Sheflin-Findling S, Arnon R, Lee S, et al. Partial internal biliary diversion for Alagille syndrome: case report and review of the literature. J Pediatr Surg. 2012;47(7):1453-1456. PubMed
59.Kamath BM, Ye W, Goodrich NP, et al. Outcomes of Childhood Cholestasis in Alagille Syndrome: Results of a Multicenter Observational Study. Hepatol Commun. 2020;4(3):387-398. PubMed
60.Hori T, Egawa H, Takada Y, et al. Long‐term outcomes after living‐donor liver transplantation for Alagille syndrome: a single center 20‐year experience in Japan. Am J Transplant. 2010;10(8):1951-1952. PubMed
Appendix 1: Detailed Outcome Data
Note this appendix has not been copy-edited.
Table 24: Additional Efficacy Outcomes in the ICONIC Trial
Time point | ICONIC trial, N = 31 | |||
|---|---|---|---|---|
Number of patients contributing to the analysis, n (%) | Baseline value, mean (SD) | Change from baseline at time point, mean (SE) (95% CI) | P value | |
Clinician Scratch Scale | ||||
Week 18 | 29 (93.5) | 3.3 (0.90) | –1.8 (0.28) (–2.3 to –1.2) | < 0.0001 |
Week 48 | 28 (90.3) | –1.8 (0.24) (–2.3 to –1.3) | < 0.0001 | |
Total cholesterol (mg/dL) | ||||
Week 18 | 29 (93.5) | 512.1 (75.40) | –87.1 (25.61) (–139.6 to –34.6) | 0.0020 |
Week 48 | 27 (87.1) | –62.9 (20.61) (–105.3 to –20.6) | 0.0052 | |
Low-density lipoprotein cholesterol (mg/dL) | ||||
Week 18 | 29 (93.5) | 184.9 (10.48) | –27.9 (7.13) (–42.5 to –13.3) | 0.0005 |
Week 48 | 27 (87.1) | –27.8 (10.36) (–49.1 to –6.5) | 0.0126 | |
SD = standard deviation; SE = standard error.
Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.17
Source: ICONIC Clinical Study Report.16
Table 25: ItchRO(Obs) Weekly Morning Average Severity Score by sBA Response in the ICONIC Trial (Safety Population)
sBA responder definition | |||||| || |||||||| || ||| | ||||||||||| |||||| ||||||| | Average change from baseline |
|---|---|---|---|
≥ 50% reduction | || |||||| | |||| | –1.86 |
≥ 60% reduction | || |||||| | |||| | –2.12 |
≥ 70% reduction | |||||| | |||| | –2.31 |
≥ 80% reduction | |||||| | |||| | –2.79 |
≥ 90% reduction | ||||| | |||| | –2.71 |
Normalization (< 8.5 μmol/L) | ||||| | |||| | –3.50 |
ItchRO(OBs) = Itch Reported Outcome (observer); sBA = serum bile acid; SD = standard deviation.
Source: Sponsor submitted internal package.37
Appendix 2: New Data Submitted by Sponsor for Reconsideration
Note this appendix has not been copy-edited.
Itch-Related Symptoms Based on Individual Patient Data for ItchRO(Obs)
As part of the Request for Reconsideration, the sponsor provided a posthoc analysis of individual patient data on ItchRO among the participants who completed 48-week follow-up in ICONIC trial (additional efficacy end point), which is presented in Table 26.
As described in the CADTH initial clinical report, average values of weekly morning severity score indicate a decrease (improvement) in ItchRO(Obs) from baseline to week 18 with a mean change of −1.70 (95% CI, - 2.0 to –1.4;) and from baseline to week 48 with a mean change of −1.6 (95% CI, - 2.1 to −1.1).
When looking at the individual patient values, 12 patients, of which 7 were randomized to placebo and 5 were randomized to maralixibat during the RWD phase, showed decreases in ItchRO score from an average score of about 3 at baseline to an average score of around zero at 48 weeks. The sponsor indicated that change in ItchRO score from a moderate-to-severe itch, indicated by a score of 3 on the ItchRO, to no itch, indicated by a score of less than or equal to 1, represents a binary result and when observed over a period of 48 weeks, does not imply a biased subjective outcome for pruritus measure.
As summarized, the analysis included in the sponsor’s initial submission supports an improvement in itch severity based on the ItchRO(Obs) from baseline to week 48. As a continuous outcome in the trial, change from baseline in ItchRO(Obs) was assessed through an ANCOVA model, using treatment groups as a fixed effect and baseline value as a covariate. It is important to note that changes from baseline to week 48 in ItchRO(Obs) were considered as an additional efficacy end point in the trial. Moreover, caregivers’ knowledge of treatment assignment during the open-label phases of the ICONIC trial might have introduced bias in measuring this subjective outcome.
CADTH acknowledges the observation of patients reporting a score corresponding to no itch at week 48 that was derived from the table of individual patient data, provided in the sponsor’s reconsideration request; however, this is an inappropriate method of analysis that is extremely limited in its interpretability at a population level. Importantly, this analysis has been conducted as posthoc analysis by the sponsor and as such, offers limited evidence regarding durability of the effect of maralixibat.
Table 26: Individual ItchRO(Obs) Data Within Participants Who Completed the Week 48 Study Period (N = 29)
Variable | ItchRO(Obs) Weekly Morning Average | |||
|---|---|---|---|---|
Baseline | Week 18 | Week 22 | Week 48 | |
N | 31 | 29 | 28 | 28 |
Mean (SD) | 2.9 (0.5) | 1.2 (0.8) | 2.2 (1.1) | 1.3 (1.1) |
Mean change from baseline (95% CI) | — | –1.7 (–2 to –1.4) | –0.7 (–1.1 to –0.2) | –1.6 (–2.1 to –1.1) |
Participant A (1) | 2.8 | 2.0 | 2.8 | 2.7 |
Participant B (1) | 2.3 | 2.0 | 2.0 | 1.8 |
Participant C (2) | 4.0 | 1.1 | 3.0 | 0.1 |
Participant D (2) | 3.0 | 1.1 | 2.4 | 1.1 |
Participant E (2) | 2.0 | 0.0 | 0.9 | 0.3 |
Participant F (2) | 3.0 | 1.9 | 3.7 | 2.0 |
Participant G (2) | 2.7 | 1.9 | 3.0 | 1.0 |
Participant H (1) | 3.4 | 0.0 | 0.2 | 0.0 |
Participant I (2) | 2.5 | 0.4 | 3.0 | 0.0 |
Participant J (1) | 2.7 | 0.4 | 1.1 | 0.0 |
Participant K (2) | 3.3 | 1.0 | 3.7 | 0.0 |
Participant L (2) | 2.4 | 0.0 | 2.7 | 1.4 |
Participant M (2) | 2.1 | 1.0 | 2.6 | 0.3 |
Participant N (2) | 2.6 | 0.4 | 2.9 | 1.1 |
Participant O (2) | 3.0 | 2.7 | 3.0 | 2.2 |
Participant P (2) | 2.7 | 2.3 | 3.0 | 3.2 |
Participant Q (1) | 2.4 | 1.9 | 1.9 | 3.7 |
Participant R (1) | 3.1 | 2.0 | 2.7 | NA |
Participant S (1) | 1.9a | 1.1 | 1.6 | 1.9 |
Participant T (1) | 3.3 | 2.0 | 0.1 | 0.7 |
Participant U (2) | 3.0 | 1.3 | 1.6 | 2.0 |
Participant V (1) | 3.1 | 1.0 | 2.1 | 2.0 |
Participant W (2) | 3.4 | 0.9 | 2.0 | 1.7 |
Participant X (1) | 2.4 | 1.1 | 1.0 | 1.4 |
Participant Y (2) | 3.3 | 0.0 | 4.0 | 0.0 |
Participant Z (1) | 3.5 | 0.4 | 0.5 | 0.0 |
Participant AA (1) | 2.7 | 0.1 | 0.6 | 0.0 |
Participant AB (2) | 3.9 | 2.1 | 4.0 | 1.6 |
Participant AC (1) | 3.7 | 2.7 | NA | 3.5 |
ItchRO(Obs) = Itch Reported Outcomes (observer); NA = not available; PBO = placebo; RWD = randomized withdrawal.
Notes:(1) denotes patients randomized to maralixibat during RWD, while (2) denotes patients randomized to PBO during RWD.
This analysis was conducted post hoc.
aThis participant was observed to have an average daily score greater than 2, consistent with study inclusion criteria.
Source: Gonzales, E., et.al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): A randomized phase 2 study. Lancet (2021), issue 10311, 1581 to 92.
Clinician Observed Scratching and/or Physical Evidence of Scratching Based Individual Patient Data on the Clinician Scratch Scale
To support the validation of the changes in ItchRO observed in the ICONIC trial, the sponsor also provided new information based on the Clinician Scratch Scale (CSS) for reconsideration by CDEC. More specifically, individual-level data on clinician-rated severity of cholestatic pruritus measured by the CSS at baseline and week 48 were provided in the reconsideration request (Table 27). CSS contains a 5-point scale, reporting values that range from zero (none) to 4 (cutaneous mutilations, hemorrhage, scarring).
CSS outcome data were predefined as an additional efficacy end point in the ICONIC trial. The individual-level data are considered new information as part of the reconsideration; however, as noted in the CADTH initial Clinical Review report, average CSS baseline values for the ITT population were 3.3, and the average CSS values at week 48 were 1.5.
||| |||||||||| ||||||| |||||||| |||||||| || ||| ||||||||||||||| ||||||| |||||||||||| ||||||| |||| |||||||| || ||| ||| |||||||||| ||| ||||||||| |||| |||||||||||||||||| ||| ||||||||| ||||| |||| || ||||||| ||||| || ||| || ||||||||| ||||| ||||||||||| || |||||||||||||||||| ||||||| |||||||||| || |||| || |||| ||| ||||| ||||||||||| || || |||||||||| || |||||||||| CADTH acknowledges the observed data in certain individuals, but this new information provided by the sponsor is subject to the same limitations noted for the individual-level data for the ItchRO. While the individual-level data may provide some insight or context to the analysis of the ItchRO and CSS reported in the ICONIC trial, this data are very limited in its interpretability. In reference to subjectivity of pruritus measurements, assessed by ItchRO instrument, and its associations with the objective CSS tool, data provided both in sponsor’s initial submission to CADTH and reconsideration request are coming from a published abstract, which offers only limited opportunity to appraise the measurement properties of the 2 instruments (Foster B, Gauthier M, Vig P, Jaecklin T, Kamath B, Andrae DA. Itch reported outcome tool for caregivers of pediatric patients with cholestatic liver disease: an analysis of validation and scoring from the ICONIC maralixibat study. Value Health. 2020;23(5):PIH76).
||||||| | ||||||||||||||||||||||||||||||||||||||||||||||||| | |
|---|---|---|
||||||| | |||||||| | |||| || |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
|||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
||||||||||||||||| | |||||||| | |||||||| |
|||||||||| ||||||| |||||||||||||| ||||||||| ||||||||||||||| |||||||| |||||||||| ||||||| |||||| ||||||| |||||||| |||||| |||||||| |||| ||| |||||||| |||||| ||||||||||||||| ||||||||||
|||||||||| || |||||||| |||| ||||||| || || |||| ||||| || ||| |||||| |||||| ||| |||||||||||| || |||||||||||||| ||| ||||||| ||||||||||||| ||||||||| ||||||| ||||| |||| |||||||||| || |||||||| |||| ||| || ||||||||||| |||||| ||||||| ||||| || || ||||| ||| ||||| |||||||| || || |||||| || |||| || ||| ||||||||||||||| ||||||| |||||| |||| ||| ||||||| ||||||||| |||| |||| ||||||||||| ||| |||||||| || || ||||||| |||||||| ||| ||||||||||| |||||||| |||||||| ||||||||| || |||||||||||| ||||| ||||||| || ||| |||||||||| || |||||||| ||| ||||||||||| ||||||| || || ||||| ||||||| ||||||| |||| || ||| || || |||||||| ||||||| ||||||||||| ||||||| || || ||||| || || |||| ||| |||||| ||| ||||||||||| ||||||||||||||| |||||| ||| |||||||||| || |||||||| |||| ||||||| || || |||| || |||| || ||| ||||| ||| |||| ||| ||||||||||| ||| ||||||| ||||||| ||||||||||||| |||||| ||| ||||||||| |||||| ||||| || ||| || ||| || ||||||||||| ||| |||||||| |||||| ||||||||| |||||| |||||||||| |||||||||| ||| ||||| || ||| || ||| || ||||||||||| ||| |||||||| ||||||| |||||| |||||||||| |||||||||| |||||||| || |||||||| ||||||||||| ||||| || |||||| || |||| ||| || |||| ||| ||| |||||| || |||||||| ||| |||||||||||| ||||| ||||||| ||| ||||||||| |||| ||| || |||||||| ||||||||||| ||||||||||| ||||| ||| ||||||
|||||| | |||| | ||||||||| ||||| | ||||| | || |||||| ||| ||||| ||| |
|---|---|---|---|---|
|||||||||| | ||||||||||| | ||||||||||| | |||| ||||| | |||| |||||| ||| |
|||||||||| | ||||||||||| | ||||||||||||||| | |||| |||||| | |||| |||||| ||||| |
|||||||||| | ||||||||||| | ||||||||||| | || || |||||| | |||| |||||| ||||| |
|||||||||| | ||||||||||| | ||||||||||||||| | || || |||||| | |||| |||||| ||||| |
|||||||||| | ||||||||||| | ||||||||||| | || || |||||| | |||| |||||| ||||| |
|||||||||| | ||||||||||| | ||||||||||||||| | || || |||| | |||| ||||||| ||||| |
|||||||||| | ||||||||||| | ||||||||||| | || || |||||| | |||| ||||||| ||||| |
|||||||||| | ||||||||||| | ||||||||||||||| | |||| |||| | |||| ||||||| ||||| |
|||||||||| |||||||||| | ||||||||||| | ||||||||||| | || || |||||| | |||| ||||||| ||||| |
|||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | |||| |||||| | |||| ||||||| ||||| |
|||||||||| |||||||||| | ||||||||||| | ||||||||||| | || || |||||| | |||| ||||||| ||||| |
|||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | |||| ||||| | |||| |||||| ||||| |
|||||||||| |||||||||| | ||||||||||| | ||||||||||| | || || |||||| | |||| ||||||| ||||| |
|||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | || || ||||| | |||| |||||| ||||| |
|||||||||| |||||||||| | ||||||||||| | ||||||||||| | || || |||||| | |||| |||||| ||||| |
|||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | || || ||||| | |||| |||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||| | || || |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | |||| |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||| | |||| |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | || || |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||| | || || |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | || || |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||| | |||||| | |||| ||||||| |||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | |||| |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||| | ||| |||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | || || |||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||| | |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | |||| |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||| | |||||| | |||| ||||||| ||||| |
||||| |||||||||| |||||||||| | ||||||||||| | ||||||||||||||| | |||||| | |||| ||||||| |||| |
||| |||||||||| ||||||||| ||| |||||||| |||||| |||||| ||| || ||| |||||| || |||||||||||| ||||| ||| ||| || ||| ||||||||| ||||| |||| ||| ||| |||||||| || ||||||||||| |||||||||||||||||||| ||||||||| ||||||||||||||| |||||||| ||||| |||| || ||||| ||||| |||||||||| ||||||||||| |||||||| ||||||
Overall, the sponsor-provided new information included long-term data on subjective (ItchRO(Obs)) and objective (CSS) pruritus measurements among patients in the ICONIC trial. Despite benefits observed across individual patient data, certain considerations were brought up by CADTH reviewers. For instance, changes from baseline to week 48 in pruritus measurements were considered additional efficacy outcomes in the ICONIC trial. Moreover, there was a possibility of biased estimates for subjective measures (ItchRO) during open-label phases of the trial. Importantly, although individual patient improvements may provide supportive evidence to aggregate level data, they remain of limited interpretability at the population level. Due to the outlined limitations, no definitive conclusions can be drawn from the new information provided by the sponsor in the reconsideration request. Similar to the initial CADTH Clinical Review report, improvements in pruritus measurements provided by the sponsor in the reconsideration request provide supportive information regarding the durability of maralixibat treatment effect for cholestatic pruritus in pediatric patients with ALGS.
Pharmacoeconomic Review
Abbreviations
ALGS
Alagille syndrome
BIA
budget impact analysis
BSC
best supportive care
GALA
Global ALagille Alliance
ICER
incremental cost-effectiveness ratio
ItchRO
Itch Reported Outcome
PC
progressive cholestasis with uncontrolled pruritus
QALY
quality-adjusted life-year
sBA
serum bile acid
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Maralixibat (Livmarli), 9.5 mg/mL (oral solution, 30 mL PET bottle) |
Submitted price | Maralixibat, 9.5 mg/mL: $1,787.00 per mL ($53,610.00 per bottle) |
Indication | For the treatment of cholestatic pruritus in patients with ALGS |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | July 21, 2023 |
Reimbursement request | As per indication |
Sponsor | As per Application Overview |
Submission history | Previously reviewed: No |
ALGS = Alagille syndrome; NOC = Notice of Compliance; PET = polyethylene terephthalate.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Cholestatic pruritus in patients aged 12 months and older with ALGS |
Treatment | Maralixibat plus BSC |
Comparator | BSC, comprising UDCAs, rifampin, antihistamines (cetirizine hydrochloride, hydroxyzine hydrochloride), alimemazine tartrate (trimeprazine tartrate), naltrexone, and sertraline |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (94.65 years) |
Key data source | The ICONIC trial and GALA clinical database |
Submitted results | ICER vs. BSC = $2,775,887 per QALY gained (incremental QALYs: 4.11; incremental costs: $11,406,596) |
Key limitations |
|
CADTH reanalysis results |
|
ALGS = Alagille syndrome; BSC = best supportive care; GALA = Global ALagille Alliance; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; sBA = serum bile acid; UDCA = ursodeoxycholic acid; vs. = versus.
Conclusions
The CADTH Clinical Review concluded that evidence from the ICONIC trial suggested that in pediatric patients with Alagille syndrome (ALGS), maralixibat plus best supportive care (BSC) was associated with reduced serum bile acid (sBA) levels and a clinically meaningful improvement in pruritis severity as assessed by the Itch Reported Outcome (ItchRO) tool, compared with BSC alone. The small sample size of the ICONIC trial meant that the comparative safety of maralixibat plus BSC versus BSC alone was uncertain. The sponsor conducted a natural-history comparison study to estimate the long-term clinical efficacy of maralixibat. This study did not control for important baseline covariates, and comparative efficacy is therefore highly uncertain.
The sponsor’s pharmacoeconomic model was premised on a relationship between sBA score and pruritis severity. The evidence to support a proxy relationship between sBA and pruritis severity was highly uncertain. Clinical expert feedback received by CADTH for this review suggested that sBA is not used in clinical assessment, and that clinical decision-making is made based on pruritis severity. The ability of the pharmacoeconomic model to reflect the impact of maralixibat treatment on clinically important outcomes and clinical management is therefore highly uncertain. This uncertainty carries through to the model’s ability to estimate the impact of maralixibat treatment on quality-adjusted survival in ALGS patients. CADTH could not address issues with the model structure and the choice of clinical outcome through reanalysis. The cost-effectiveness of maralixibat plus BSC compared with BSC alone is therefore highly uncertain.
CADTH identified additional limitations with the sponsor’s pharmacoeconomic model. The method for incorporating weight-based dosing lacked face validity, as it assumed that the patient cohort never reached a weight that would result in receiving the maximum dose of maralixibat over the course of their lifetime, even as adults. Due to the limitations in the model, CADTH was not able to estimate a base-case incremental cost-effectiveness ratio (ICER). Given the issues with the model structure, as well as other limitations described (e.g., modelling of patient’s weight over time), CADTH was unable to address these issues.
Using the sponsor’s submitted results, a price reduction of at least 96.5% would be required for maralixibat to be considered cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY) gained. At this price reduction, the per-mL (9.5 mg/mL) cost of maralixibat would need to be reduced from $1,787.00 to $62.55. Due to the identified limitations within the model, the price reduction needed to achieve cost-effectiveness at $50,000 per QALY gained is likely higher.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process — specifically, information that pertains to the economic submission.
Patient input was received from the Canadian Liver Foundation and the Alagille Syndrome Alliance, which collected perspectives of caregivers and patients with ALGS through surveys, focus groups, and interviews. Patients with ALGS reported severe itch (pruritus), jaundice, fatigue, and xanthomas, which affected sleep, vision, and the ability to carry out daily activities. Patients with ALGS and heart or kidney problems were also required to limit their physical activities and undergo dialysis. Current pharmacological treatments being used by patients included medications such as ursodeoxycholic acid, antihistamines (such as diphenhydramine and hydroxyzine), rifampin, cholestyramine, colesevelam, and naltrexone. Other therapeutic interventions included partial external biliary diversion or ileal exclusion for patients with severe ALGS. Treatment goals were identified as consistent relief from pruritus and improvement in patient quality of life. Patients who had experience with maralixibat reported relief from itching as well as mild to severe abdominal cramping and diarrhea from the use of the medication.
Clinician input was received from the Canadian Association for the Study of the Liver, with the participation of a group of experts in liver disease. The clinician input noted that current treatments for patients with ALGS included symptomatic treatments such as cholestyramine, rifampin, naltrexone, and sertraline, as well as antihistamines and ursodeoxycholic acid (usually considered early in treatment). Surgical interventions such as biliary diversion procedures and liver transplant were also considered for patients with drug-refractory pruritus or severe cholestasis with or without complications. Clinically meaningful treatment goals included reduced pruritus, improving nutritional goals, and treating fat-soluble vitamin deficiencies. The clinicians noted that the tool used to assess pruritus in clinical trials is not feasible in clinical practice. Clinically meaningful treatment response was described as a reduction in pruritus and sleep duration, measured through patient inquiry or assessing for excoriation of the skin. The input noted that all patients with ALGS and persistent cholestatic pruritus on standard of care would be eligible for maralixibat. However, treatment with maralixibat should be discontinued in cases of progression of liver disease or liver transplant.
The drug plans noted that there are no therapies approved by Health Canada for the treatment of cholestatic pruritus in ALGS; however, pharmacological treatments are used off-label. The plans also highlighted surgical procedures such as liver transplant, partial external biliary diversion, ileal exclusion, nasobiliary drainage, plasmapheresis, and albumin dialysis as therapeutic options. The drug input highlighted concerns with generalizability of trial results to patients with mutations other than Jagged-1 as well as assessment and monitoring of therapeutic response. The plans noted that the daily weight-based dosing of maralixibat makes it difficult to estimate treatment costs and, therefore, the incremental budget impact.
Several of these concerns were addressed in the sponsor’s model:
The comparator in the model was BSC, to reflect that no other indicated options are available.
Quality of life was incorporated in the sponsor’s model through the use of health utilities captured in the ICONIC trial using the preference-based time trade-off method.
In addition, CADTH addressed some of these concerns as follows:
The sponsor assumed long-term differences in the relative efficacy and safety of maralixibat; however, there is limited evidence to support this assumption. CADTH explored the impact of assuming no differences in comparative efficacy and safety of maralixibat plus BSC compared with BSC alone in a scenario analysis.
CADTH conducted a scenario analysis to explore the impact of starting treatment in adults with ALGS on the estimated budget impact of maralixibat.
CADTH was unable to address the following concerns raised from stakeholder input:
The sponsor used changes in sBA levels as a surrogate marker for pruritis severity and treatment response; however, the relationship between sBA and pruritis severity is highly uncertain.
Economic Review
The current review is for maralixibat (Livmarli) for the treatment of cholestatic pruritus in patients with ALGS.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of maralixibat plus BSC compared with BSC alone for the treatment of cholestatic pruritus in patients with ALGS.1 The base case model population was aligned with the ICONIC trial. The modelled population was aligned with the Health Canada indication and reimbursement request. BSC consisted of a basket of pharmacological treatments such as ursodeoxycholic acid (UDCA), rifampicin, cholestyramine, alimemazine tartrate (trimeprazine tartrate), hydroxyzine hydrochloride (cetirizine hydrochloride, antihistamines), naltrexone, and sertraline.
Maralixibat is available as a solution for oral administration (9.5 mg per mL in a 30 mL polyethylene terephthalate bottle) and administered using 0.5 mL, 1 mL, and 3 mL oral syringes.2 The recommended dose for maralixibat is weight-based and consists of a starting dose of 198 mcg per kg once daily followed by a maintenance dose of 380 mcg per kg after 1 week. The maximum daily dose for patients above 70 kg is 28.5 mg or 3 mL. At the submitted price of $1,787.00 per mL or $188.11 per mg, the cost per maintenance dose was $1,251, $1,608, $1,787, $2,234, $3,127, and $4,021 for body weights of greater than or equal to 17 kg to less than 20 kg, greater than or equal to 20 kg to less than 25 kg, greater than or equal to 25 kg to less than 32 kg, greater than or equal to 32 kg to less than 46 kg, greater than or equal to 46 kg to less than 51 kg, and greater than or equal to 51 kg, respectively. Assuming patients received 84.29 administrations per 12 weeks beyond the first week, the estimated annual costs of maintenance treatment ranged between $456,891and $1,468,579, depending on patient weight. In the pharmacoeconomic model, maralixibat treatment costs were based on the baseline age of 5.35 years, which reflected the average starting age in the ICONIC trial.3 In comparison, BSC was associated with a cost of $327 per patient per 12 weeks. The sponsor assumed no drug wastage and 100% adherence to oral treatments.
The clinical outcomes of interest were QALYs and life-years. The economic analysis was undertaken over a lifetime time horizon (94.65 years) from the perspective of the Canadian public health care payer. Both costs and health outcomes were discounted at an annual rate of 1.5%.
Model Structure
A Markov model with 6 health states and 12-week cycle lengths was submitted by the sponsor (Figure 1). All patients entered the model in the health state characterized by progressive cholestasis with uncontrolled pruritus (PC). Patients who responded to treatment moved to a health state characterized by nonprogressive cholestasis and controlled pruritus. Only patients in the PC health state could have a liver transplant (LTx) and move to the health state characterized by post–liver transplant (Post-LTx). Patients with cardiac disease were assumed to be ineligible for liver transplant and moved to another health state characterized by progressive cholestasis with cardiac complications and uncontrolled pruritus (PC-cardiac). Patients who discontinued treatment moved back to the PC health state characterized. Death was the absorbing health state.
Model Inputs
Baseline patient characteristics were derived from the ICONIC trial,3 a randomized, placebo-controlled, drug-withdrawal study that included participants between the ages of 1 to 18 years. In the base-case pharmacoeconomic model, the average patient starting age was 5.35 years, patients weighed 16.62 kg at baseline, and were more likely to be male (61.3%).
Efficacy data were obtained for maralixibat plus BSC and BSC alone from 2 different sources; the ICONIC trial and the Global ALagille Alliance (GALA) study clinical database.3,4 The sponsor assumed that a less than 50% reduction in sBA levels was reflective of a patient having nonprogressive cholestasis and controlled pruritus. As such, treatment response was defined as a less than 50% reduction in sBA levels and aligned with the primary end point considered in the ICONIC trial. In the absence of head-to-head trial data comparing maralixibat with BSC, the sponsor did not perform any covariate-adjusted indirect comparisons to assess the relative efficacy of maralixibat plus BSC with BSC alone. Instead, the sponsor estimated that 39% of patients on maralixibat plus BSC and || of patients on BSC alone responded to treatment every 12 weeks using the ICONIC trial and the GALA study registry, respectively. Nonresponders and patients who lost response to maralixibat treatment were assumed to receive BSC alone. The sponsor estimated that the probability of treatment discontinuation was ||||| per cycle based on the data observed in the ICONIC trial. The sponsor assumed that treatment discontinuation and response rates were constant over the time horizon
The sponsor assumed that patients in the nonprogressive cholestasis and controlled pruritus and PC-cardiac health states could not have a liver transplant. For patients with uncontrolled pruritus and progressive cholestasis, the sponsor modelled the probability of having a liver transplant using time-to-event data from the GALA clinical database. The sponsor selected a log-normal distribution to extrapolate the liver transplant rates. Model selection was based on statistical fit (Akaike Information Criterion, Bayesian Information Criterion) and visual inspection. The sponsor excluded adverse events from its analysis.
The sponsor estimated that the annual rate of mortality due to a liver transplant was 13% and that the surviving recipients of liver transplant were at an increased mortality risk, using published literature.5-7 The mortality rates for patients with progressive cholestasis and uncontrolled pruritus, with and without cardiac complications, was also extrapolated using the Gompertz distribution and based on time-to-event data observed in the GALA study registry. Apart from this, patients were assumed to have the same mortality as their age- and gender-matched general population.
Utility weights for patients with ALGS were obtained from a sponsor-commissioned study on 200 individuals in the UK aged 18 years using time trade-off methods.1 The sponsor developed vignettes for health states: progressive cholestasis and nonprogressive cholestasis (each defined using sBA level), successful liver transplant, and chronic liver transplant rejection. The vignette descriptions included signs and symptoms that patients may experience due to pruritus. Patient preference between time spent in the target health state against time spent in full health was elicited until point of indifference to estimate health state utility. The sponsor assumed that health utility was the same for patients with progressive cholestasis and uncontrolled pruritus, irrespective of cardiac complications. In the model, the health utility associated with a successful liver transplant was assumed for health state characterized by liver transplant. The utility for health state characterized by post–liver transplant was adjusted for chronic liver transplant rejection.
Costs in the model included drug acquisition, disease management, and liver transplant. Treatment cost associated with BSC was weighted by the proportion of patients receiving UDCA (93.1%), cholestyramine (9.8%), rifampin (33.5%), hydroxyzine hydrochloride (19.4%), naltrexone (13.8%), sertraline (13.8%), and alimemazine tartrate (0.4%).8,9 Dosing was obtained from respective product monograph and published literature.8,10,11 Costs were sourced from the Ontario Drug Benefit Formulary and the Alberta Drug Benefit List.12,13 Disease management costs included costs of health care practitioners, laboratory tests, and hospitalizations, as relevant to each health state. The sponsor quantified health care resource use for each health state using clinical expert opinion obtained by the sponsor.9 Disease management unit costs and liver transplant costs were obtained using the Ontario Ministry of Health and Long-Term Care’s Schedule of Benefits for Professional Services, as well as its Schedule of Benefits for Laboratory Services, home care costs in Ontario, the Canadian Institute for Health Information Patient cost estimator, and published literature.14-18
The sponsor also assumed that ||||| of patients in the PC health state had biliary diversion as a treatment option until the age of 9 years.19 Biliary diversion was associated with a one-time disutility of stoma care and cost of procedure, postsurgical care, stoma care, and ostomy care.16,20,21
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (8,000 iterations), with the deterministic and probabilistic results being similar. The probabilistic findings are presented below.
Base-Case Results
In the base case, the sponsor reported that maralixibat plus BSC was associated with an additional cost of $11,406,596 and 4.11 additional QALYs compared with BSC alone, leading to an ICER of $2,775,887 per QALY gained (Table 3). Maralixibat plus BSC was associated with an additional 3.10 life-years compared with BSC alone. At a WTP threshold of $50,000 per QALY gained, there was a 0% probability of maralixibat being cost-effective compared with BSC.
Of the 4.11 incremental QALYs gained for maralixibat in combination with BSC, only 0.42 (10%) were accrued during the trial period. Although the sponsor assumed no difference in mortality risk due to treatment with maralixibat plus BSC and BSC alone, the sponsor’s pharmacoeconomic model estimated a survival advantage for patients on maralixibat plus BSC relative to BSC alone because patients who had a less than 50% reduction in sBA levels could not have a liver transplant in the sponsor’s model.
Drug acquisition costs accounted for the majority of incremental costs for maralixibat ($11,603,593; 102%), which was partly offset by reduced costs of health care resource use for patients using maralixibat compared with BSC (incremental savings = $196,998).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. reference ($/QALY) |
|---|---|---|---|---|---|---|---|
BSC | 1,019,061 | Reference | 32.08 | Reference | 21.32 | Reference | Reference |
Maralixibat plus BSC | 12,425,656 | 11,406,596 | 35.17 | 3.09 | 25.43 | 4.11 | 2,775,887 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted sensitivity and scenario analyses. Pairwise 1-way sensitivity analyses were conducted using the deterministic model to assess the impact of specific parameters on the ICER, incremental QALYs, and incremental costs. The parameters that had the largest impact on the model’s findings were patient age, treatment discontinuation rate, response rate to maralixibat treatment, utility weight for patients with progressive cholestasis, uncontrolled pruritus and cardiac complications, proportion of patients who were males, response rate to BSC, proportion of patients who did not receive liver transplant due to cardiac complications, utility weight for patients with progressive cholestasis with uncontrolled pruritus and who were transplant recipients, and mortality risk due to liver transplant.
The sponsor provided scenario analyses exploring the impact of adopting alternative time horizon, discount rates, parametric curves for liver transplant rate, and posttransplant mortality, assuming constant liver transplant rate, adopting general mortality rate for liver transplant recipients, and applying utility weights derived from the EQ-5D questionnaire. In all scenarios, maralixibat plus BSC was associated with increased QALYs and costs, with ICERs ranging from $1,764,181 per QALY gained to $5,113,040 per QALY gained compared with BSC alone.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The relative efficacy and safety of maralixibat plus BSC versus BSC alone is uncertain: In the absence of direct and indirect comparative evidence, the effectiveness of maralixibat plus BSC compared with BSC alone was estimated using a naive comparison between the ICONIC trial and data from the GALA clinical research database. The sponsor used patient-level data on 31 patients in the ICONIC trial and on 73 patients in the GALA study registry to calculate the proportion of patients who achieved a greater than or equal to 50% reduction in sBA from their baseline measurement at 12 weeks. Using this approach, the sponsor estimated a response rate of 39% (12/31) with maralixibat plus BSC and || |||||| with BSC alone. The sponsor did not perform a matching-adjusted analysis between the ICONIC trial and the GALA database to correct for imbalances in patient demographics at baseline, which may have confounded treatment outcomes through the influence of unmeasured and unadjusted confounders. Given that sBA plays a central role in the pharmacoeconomic model, the failure to control for baseline sBA as a covariate contributes a high degree of uncertainty to the comparative effectiveness estimates produced by the model. As such, the results of the sponsor’s approach are highly susceptible to bias and introduce substantial uncertainty in the sponsor’s assertion that treatment with maralixibat plus BSC is associated with a higher response rate compared to treatment with BSC alone.
Although the ICONIC trial included a long-term extension phase and the curation of the GALA database is a multiyear effort,4 the sponsor did not assess the long-term comparative efficacy and safety of maralixibat plus BSC compared with BSC alone. Instead, the sponsor assumed no attenuation in response rates estimated at 12 weeks of treatment over the duration of treatment beyond 12 weeks. The unknown long-term comparative efficacy contributes notable uncertainty to the overall estimates of cost-effectiveness, as the sponsor used a lifetime time horizon (94.65 years) and the near entirety of the modelled population survives beyond the 12-week period in the sponsor’s base case.
CADTH was not able to address the lack of direct and indirect comparative evidence. CADTH conducted a scenario analysis exploring the impact of assuming similar long-term efficacy and safety between maralixibat plus BSC and BSC alone (by assuming treatment response to be the same for maralixibat plus BSC and BSC alone and no discontinuation with maralixibat plus BSC).
The model lacks face validity due to the uncertain relationship between sBA level and pruritus severity: The sponsor’s model considers changes in sBA levels as the primary metric of treatment effectiveness and health state occupancy. Clinical expert feedback solicited by CADTH for this review suggested that sBA level is not a patient and clinically important outcome, and that decisions about whether patients are improving are made based on assessment of pruritis severity. The sponsor justified its choice of sBA as a surrogate marker for pruritis severity in part by noting the similarity of change in sBA and ItchRO scores collected during the 4-week randomized withdrawal period of the ICONIC trial (Figure 2). The sponsor also submitted the results of a post hoc analysis of the ICONIC trial, finding a moderate and statistically significant correlation between ItchRO score and a prespecified end point of a greater than or equal to 50% reduction in sBA at week 48 (r = 0.47; P = 0.012).22 This correlation was used to justify the use of greater than or equal to 50% sBA reduction as a proxy for stable and controlled pruritis. Clinical expert feedback received by CADTH for this review did not support the sponsor’s choice of proxy measure, noting that most clinicians did not measure sBA in ALGS patients and that considerable heterogeneity exists in the scientific literature about the relationship between sBA and pruritis severity. The relationship between changes in sBA and pruritis severity is unclear. Given this lack of a clear relationship, and the fact that sBA is not used in clinical decision-making, the sponsor’s model lacks face validity in terms of its ability to reflect health state occupancy either in the short term or long term. Any estimated QALYs produced by this model are therefore highly uncertain.
CADTH was not able to address this issue in reanalysis.
Treatment costs are underestimated: Maralixibat is dosed based on patient weight. The sponsor’s model used the mean age of ICONIC trial participants (5.35 years) as the mean age within the model, and estimated weight based on age. As patients age within the model, the dose of maralixibat changed. Patient weight reached 56.62 kg at age 17 years, beyond which point weight was assumed to remain constant until age 25 years, with further changes to weight at every 10-year interval. The maximum weight achieved by patients in the sponsor’s model was 60.79 kg, occurring when the modelled cohort reached 45 years of age and patient weight dropped back below 60 kg at age 55 years in the model. This weight corresponded with a maximum dose of 2.5 mL (23.75 mg) per day. According to the product monograph,2 the maximum dose of maralixibat is 3 mL (28.5 mg) per day. CADTH notes that 60.79 kg is considerably lower than the average weight of an adult in Canada (approximately 75.6 kg).23 The sponsor’s model assumes that patients will never receive the maximum dose even into adulthood, which lacks face validity and underestimates the cost of maralixibat.
Further, the sponsor’s model does not reflect the dosing used in the ICONIC trial.3 While the product monograph identifies 28.5 mg per day as the maximum dose, ICONIC trial participants received up to 57.0 mg per day (760 mcg/kg/day) in the long-term treatment period of the trial. The impact of long-term dosing was not considered in the calculation of treatment costs, although it is unclear whether this dose would be given outside the context of the ICONIC trial. If the dose increase observed in the ICONIC trial became part of clinical practice, the sponsor’s model would have underestimated the cost of treatment. Given that drug cost represents virtually 100% of the incremental costs associated with maralixibat treatment, this underestimation has serious implications for any estimate of cost-effectiveness.
A scenario analysis was conducted in which CADTH assumed that all patients aged 25 years or older received the maximum dose of 3 mL or 28.5 mg of maralixibat daily.
A scenario analysis was conducted in which the dose of maralixibat was doubled beyond 100 weeks, to a maximum of 57.0 mg per day, for ||| of patients (reflecting the proportion of ICONIC trial patients who received a dose increase in the long-term phase).
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The sponsor obtained mean weight at baseline from the ICONIC trial and the proportion of patients on hydroxyzine hydrochloride from sponsor’s sought clinical expert. | CADTH noted minor differences in the baseline weight and proportion of patients using hydroxyzine hydrochloride reported in the pharmacoeconomic evaluation and the model. These differences had minimal impact on the cost-effectiveness of maralixibat. |
Treatment begins at a mean age of 5.35 years in patients who weigh 18.30 kg. | Acceptable as a simplifying assumption. Although the mean age and weight are aligned with those observed in the ICONIC trial, patient age and weight are key drivers of the cost-effectiveness of maralixibat because the drug dose is dependent on patients’ age and weight. CADTH noted that the ICER associated with maralixibat increased as the patient age and weight at which treatment began increased. |
The sponsor assumed that baseline model characteristics are reflective of patients with moderate to severe pruritis. | Uncertain. Although the sponsor aligned mean age, proportion of males, and mean weight of the modelled population with the ICONIC trial, the sBA levels or pruritis severity at baseline were not specified. Further, the clinical experts noted that changes in sBA levels are not typically used in clinical practice to determine disease severity. It is unclear whether the model reflects moderate to severe pruritis, or all levels of pruritis severity. Given the model’s overall limited ability to reflect the relationship between sBA and pruritis, this limitation likely had a negligible impact. |
The proportion of patients on components of BSC was estimated based on clinical expert opinion obtained by the sponsor. | Acceptable. The proportion of patients on components of BSC is not aligned with the distribution of patients observed on these treatments in the ICONIC trial. CADTH found that changing the distribution of patients had a minimal impact on the cost-effectiveness results because maralixibat is an add-on treatment. CADTH found the assumption of no difference in the distribution of patients when maralixibat is added to BSC to be acceptable as well. |
Health utility weights are uncertain. | Uncertain. The sponsor estimated utility associated with health states, progressive cholestasis, and nonprogressive cholestasis that were defined using sBA levels. The clinical expert noted that the sponsor’s estimates of health utilities seemed reasonable; however, it is unclear if patient preference elicited on signs and symptoms experienced by ALGS patients reflected changes in sBA levels. As such, the estimated QALYs are uncertain. |
The sponsor assumed no adverse events with maralixibat treatment or BSC. | Uncertain. Although the incidence of adverse events with maralixibat plus BSC or BSC alone was observed to be similar in the ICONIC trial, the long-term safety of maralixibat treatment is unknown. |
The sponsor assumed that ALGS patients with uncontrolled pruritus and cardiac complications are not eligible for liver transplant. | This assumption failed to meet face validity as clinical experts did not agree with the sponsor’s assumption that cardiac complications made all ALGS patients ineligible to receive liver transplant. The overall impact of this limitation on the estimated cost-effectiveness was negligible. |
The sponsor estimated a discontinuation rate of ||||| per cycle. | Uncertain. In the CUA, the sponsor estimated a discontinuation rate of ||||| ||| ||||| || |||| ||| ||||. However, in the BIA, the sponsor estimated an annual discontinuation rate of 6.5% in the first year of treatment and 8.7% in subsequent years of treatment with maralixibat. The clinical experts noted that treatment discontinuation is anticipated to be low and maralixibat dose may be titrated, rather than completely stopped, in the case of an adverse event. As such, the discontinuation rate is uncertain. |
The sponsor assumed no difference in mortality risk between maralixibat plus BSC and BSC alone. | Uncertain. As the ICONIC trial did not measure patient survival, there is insufficient evidence to support a survival advantage associated with maralixibat. The CADTH’s Clinical Review found that the results of the natural-history comparison study evaluating relative event-free survival, a composite end point of death, first event of liver decompensation (ascites, variceal bleeding), surgical biliary diversion, and liver transplant, was uncertain due to potential residual confounding, incomparability in disease severity, and the lack of sBA data available among patients in the GALA registry. However, CADTH noted that the sponsor’s pharmacoeconomic model estimated a survival advantage. This was attributed to an increased rate of downstream liver transplants for patients with BSC relative to patients on maralixibat plus BSC. |
ALGS = Alagille syndrome; BIA = budget impact analysis; BSC = best supportive care; CUA = cost-utility analysis; GALA = Global ALagille Alliance; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; sBA = serum bile acid.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Given the limitations CADTH identified with the sponsor’s economic submission, CADTH was unable to use the model to derive robust estimates of the cost-effectiveness of maralixibat or to help quantify the impact of uncertainty. The model’s estimates of state occupancy are based on underlying trial data that have insufficient statistical power to detect difference in trial outcomes and was not controlled for observed or unobserved confounders, which was extrapolated over the near entirety of the cohort’s lifetime. The model was based on a proxy measure for treatment response that has an unclear relationship with the primary outcome used in assessing patients with ALGS. Finally, the model underestimated drug costs, which represent 100% of the incremental cost, by an unknown amount.
When reviewing the sponsor’s base-case results, the probability that maralixibat plus BSC is cost-effective at a WTP threshold of $50,000 per QALY gained was 0%. As such, it is highly unlikely, based on the sponsor’s analysis, that maralixibat would be cost-effective at this threshold.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s base case (Table 5 and Table 10). A 96.5% reduction in the price of maralixibat would be required for maralixibat plus BSC to be considered cost-effective at a WTP threshold of $50,000 per QALY gained compared with BSC alone. However, given both the high degree of uncertainty in the underlying clinical evidence and model structure, and the underestimation of treatment costs, the price reduction needed to achieve this threshold is likely much higher.
CADTH conducted a scenario analysis to explore the impact of assumptions about patient weight and indicated dose. Patients treated with maralixibat were assumed to weigh 74 kg starting at age 25. This change resulted in $13,017,039 of added incremental costs (versus the sponsor’s base case estimate of $11,168,909 per patient), and the ICER increased to $3,194,793 per QALY gained (versus the sponsor’s base case estimate of $2,741,204).
An additional scenario analysis was performed to explore the impact of maralixibat dose increases beyond 100 weeks. In this scenario, 45% of patients received twice the indicated dose of maralixibat until death or discontinuation. This change resulted in $16,087,733 of added incremental costs, and the ICER increased to $3,948,438 per QALY gained.
Table 5: CADTH Price Reduction Analyses
Analysis | ICERs for maralixibat plus BSC vs. BSC ($/QALY) |
|---|---|
Price reduction | Sponsor base case |
No price reduction | $2,741,204 |
10% | $2,462,085 |
20% | $2,183,333 |
30% | $1,904,582 |
40% | $1,625,830 |
50% | $1,347,078 |
60% | $1,068,327 |
70% | $789,575 |
80% | $510,823 |
90% | $232,072 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Issues for Consideration
According to the clinical experts consulted for this review, maralixibat may be used in all cases of pruritus regardless of severity because there is no validated tool used in clinical practice to determine disease severity of ALGS. Should maralixibat be used in all ALGS patients (including those diagnosed using the presence of Jagged-1 gene mutations rather than phenotypic criteria) with cholestatic pruritus, the estimated budget impact may have been underestimated.
The comparative effectiveness and cost-effectiveness of off-label use of maralixibat in patients with conditions other than ALGS but requiring treatment with a similar mechanism as maralixibat is beyond the scope of this review. Should this happen, the incremental budget impact estimated in this review is underestimated.
Overall Conclusions
The CADTH Clinical Review of the ICONIC trial found that maralixibat may result in a decrease in sBA levels and results in a clinically meaningful improvement in pruritus and parent-reported health-related quality of life measures compared with placebo. Maralixibat was generally well tolerated in terms of adverse events. The Clinical Review noted that the small sample size of the ICONIC trial limited the ability to control for type I error, and that strong conclusions about comparative effectiveness could therefore not be drawn. The pharmacoeconomic model derived all estimates of treatment efficacy from the ICONIC trial, and the uncertainty in the trial results translates into uncertainty in the estimates of cost-effectiveness.
CADTH identified additional limitations in the sponsor’s pharmacoeconomic submission. First, the estimates of treatment efficacy compared with BSC were further complicated by the sponsor’s use of a naive comparison to patients from a research database, where sBA levels at baseline were among many other unmeasured and unadjusted confounders. The trial results were also extrapolated over a 94-year time horizon, with 90% of observed incremental QALYs estimated beyond the observation period of the ICONIC trial. The sponsor’s model used a 50% reduction in sBA as a proxy measure for clinical response (i.e., pruritis severity), which was not adequately supported by trial evidence, had unclear support within the literature, and was not supported by clinical expert input solicited by CADTH for this review. The sponsor’s model appeared to underestimate the costs of weight-based treatment by adopting weight estimates that lacked face validity and generalizability to the adult population in Canada. Moreover, the long-term dosing of maralixibat was not aligned with dosing used in the ICONIC trial.
For these reasons, CADTH was not able to generate a robust estimate of cost-effectiveness for maralixibat plus BSC compared with BSC alone. Based on the sponsor’s submitted results, a 96.5% reduction in the price of maralixibat would be required for maralixibat plus BSC to be considered cost-effective compared with BSC alone at a WTP threshold of $50,000 per QALY gained. At this price reduction, the per-mL (9.5 mg/mL) cost of maralixibat would be reduced from $1,787.00 to $62.55. Given the limitations of the trial evidence and the lack of indirect treatment comparison, the uncertain relationship between sBA and pruritis severity, and the methodological concerns identified within the economic model, these estimates remain highly uncertain. CADTH scenario analyses suggest that the incremental cost of maralixibat treatment is likely much higher than the sponsor’s estimate, and that the price reduction needed to ensure cost-effectiveness may be greater than 97.5%.
Maralixibat treatment is associated with a substantial increase in costs per patient over the course of their lifetime. The comparative effectiveness of maralixibat versus BSC is highly uncertain, and no robust estimate of incremental QALYs could be generated in this review. The cost-effectiveness of maralixibat treatment is therefore highly uncertain.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Livmarli (maralixibat), oral solution, 9.5 mg/mL [internal sponsor's package]. Foster City (CA): Mirum Pharmaceuticals Inc.; 2023 May 05.
2.Livmarli (Maralixibat): 9.5 mg/mL, oral solution [product monograph]. Foster City (CA): Mirum Pharmaceuticals, Inc.; 2022 Dec.
3.Clinical Study Report: LUM001-304. ICONIC: Long-term, open-label study with a double-blind, placebo-controlled, randomized drug withdrawal period of lum001, an apical sodium-dependent bile acid transporter inhibitor (asbti), in patients with Alagille syndrome [internal sponsor's report]. Foster City (CA): Mirum Pharmaceuticals Inc.; 2020 Oct 13.
4.Vandriel SM, Li LT, She H, et al. Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatology. 2023;77(2):512-529. PubMed
5.Table: 13-10-0710-01 Mortality rates, by age group. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701.
6.Aberg F GM, Karlsen TH, et al. Differences in long-term survival among liver transplant recipients and the general population: a population-based Nordic study. Hepatology. 2015;61(2):668-677. PubMed
7.Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut. 2001;49:431-435. PubMed
8.Menon J, Shanmugam N, Vij M, Rammohan A, Rela M. Multidisciplinary Management of Alagille Syndrome. J Multidiscip Healthc. 2022;15:353-364. PubMed
9.Canadian ALGS and Livmarli Landscape Assessment. Data on file. In: Drug Reimbursement Review sponsor submission: Livmarli (maralixibat), oral solution, 9.5 mg/mL [internal sponsor's package]. Foster City (CA): Mirum Pharmaceuticals Inc.; 2023 May 05.
10.Kamath BM, Loomes KM, Piccoli DA. Medical management of Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;50(6):580-586. PubMed
11.Thebaut A, Habes D, Gottrand F, et al. Sertraline as an Additional Treatment for Cholestatic Pruritus in Children. J Pediatr Gastroenterol Nutr. 2017;64(3):431-435. PubMed
12.Government of Alberta. Interactive drug benefit list. 2022; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2023 Apr 2.
13.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2024 Apr 2.
14.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2023 Mar 30.
15.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 Mar 30.
16.Patient cost estimator. 2022; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2023 Apr 3.
17.Closing the Gap. Home Care Costs in Ontario—A Complete Breakdown. 2019; https://www.closingthegap.ca/home-care-costs-in-ontario-a-complete-breakdown/ Accessed 2023 Mar 31.
18.Levy AR SB, James D, et al. The costs of change: direct medical costs of solid organ transplantation in British Columbia, Canada, 1995-2003. Value Health. 2009;12(2):282-292. PubMed
19.Gonzales E, Hardikar W, Stormon M, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet. 2021;398(10311):1581-1592. PubMed
20.Inner Good. re Your Ostomy Supplies Covered? FAQs on Insurance Coverage in Canada. https://innergood.ca/are-ostomy-supplies-covered-in-canada/#:~:text=The%20financial%20cost%20of%20living%20with%20an%20ostomy,the%20basics%2C%20including%20stoma%20appliances%20and%20supporting%20systems. Accessed 2023 Apr 3.
21.National Insitute for Health and Care Excellence. Odevixibat for treating progressive familial intrahepatic cholestasis (Highly specialised technologies guidance) 2021: https://www.nice.org.uk/guidance/hst17.
22.Gonzales E VP, Tucker E, et al. Pruritus intensity is associated with cholestasis biomarkers and quality of life measures after maralixibat treatment in children with Alagille syndrome. Hepatology. 2020.
23.Table 23: Measured weight, by age and sex, household population, Canada, 2009 to 2011. Ottawa (ON): Statistics Canada; 2015.
24.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Livmarli (maralixibat), oral solution, 9.5 mg/mL [internal sponsor's package]. Foster City (CA): Mirum Pharmaceuticals Inc.; 2023 May 05.
25.Statistics Canada. Table 17-10-0009-01 Population estimates, quarterly. 2024; https://doi.org/10.25318/1710000901-eng.
26.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. Ottawa (ON): The Conference Board of Canada; 2017: https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326.
27.Alberta Health Interactive Health Data Team. Hospital Inpatient Care Case Costs - CMG/Plex. 2015; https://open.alberta.ca/opendata/hospital-inpatient-care-case-costs-cmgplex.
28.Panectyl (trimeprazine tartrate): 2.5mg and 5mg tablets [product monograph]. Montreal (QC): ERFA Canada: https://pdf.hres.ca/dpd_pm/00033061.PDF.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 6: CADTH Cost Comparison Table
Treatment | Strength/ concentration | Form | Price ($)a | Recommended dosage | Average daily cost ($)b | Average annual cost ($)b |
|---|---|---|---|---|---|---|
Maralixibat (Livmarli) | 9.5 mg/mL | 30 mL vial Solution for oral administration | $1,787.0000 per mL or $188.1052 per mg | Week 1: 190 mcg/kg daily Week 2 and onwards: 380 mcg/kg daily up to 28.5 mg per (or 3 mL) daily for patients above 70 kg | Year 1: 352.99 (5 kg patient) to 5,294.91 (75 kg patient) Beyond year 1: 356.42 (5 kg patient) to 5,346.31 (75 kg patient) | Year 1: 128,843 (5 kg patient) to 1,932,641 (75 kg patient) Beyond year 1: 130,094 (5 kg patient) to 1,951,404 (75 kg patient) |
aSponsor-submitted price.1
bThe cost range is estimated for a patent with a minimum weight of 5 kg and a maximum weight of 75 kg. Annual period assumes 52 weeks or 365 days.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The sponsor’s analysis did not consider patient and clinically important outcomes such patient growth. Moreover, the sponsor’s modelled key efficacy outcome lacked face validity (refer to limitation “The model lacks face validity due to the uncertain relationship between sBA level and pruritus severity”). |
Model has been adequately programmed and has sufficient face validity | No | Model structure lacks face validity. Refer to CADTH critical appraisal section for limitations “The model lacks face validity due to the uncertain relationship between sBA level and pruritus severity” and “Treatment costs are underestimated.” |
Model structure is adequate for decision problem | No | The sponsor’s assumption that patients with uncontrolled pruritus and cardiac complications are ineligible to receive liver transplant does not meet face validity. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | There are instances when the code refers to cells that are empty. This was observed in cells estimating treatment cost on the sheet “Treatment cost engine.” |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Additional information was requested for clarifications on how the proportion of response was estimated and reasons for treatment discontinuation. Some references were missing in the sponsor’s submission and were requested during the review process. |
sBA = serum bile acid.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
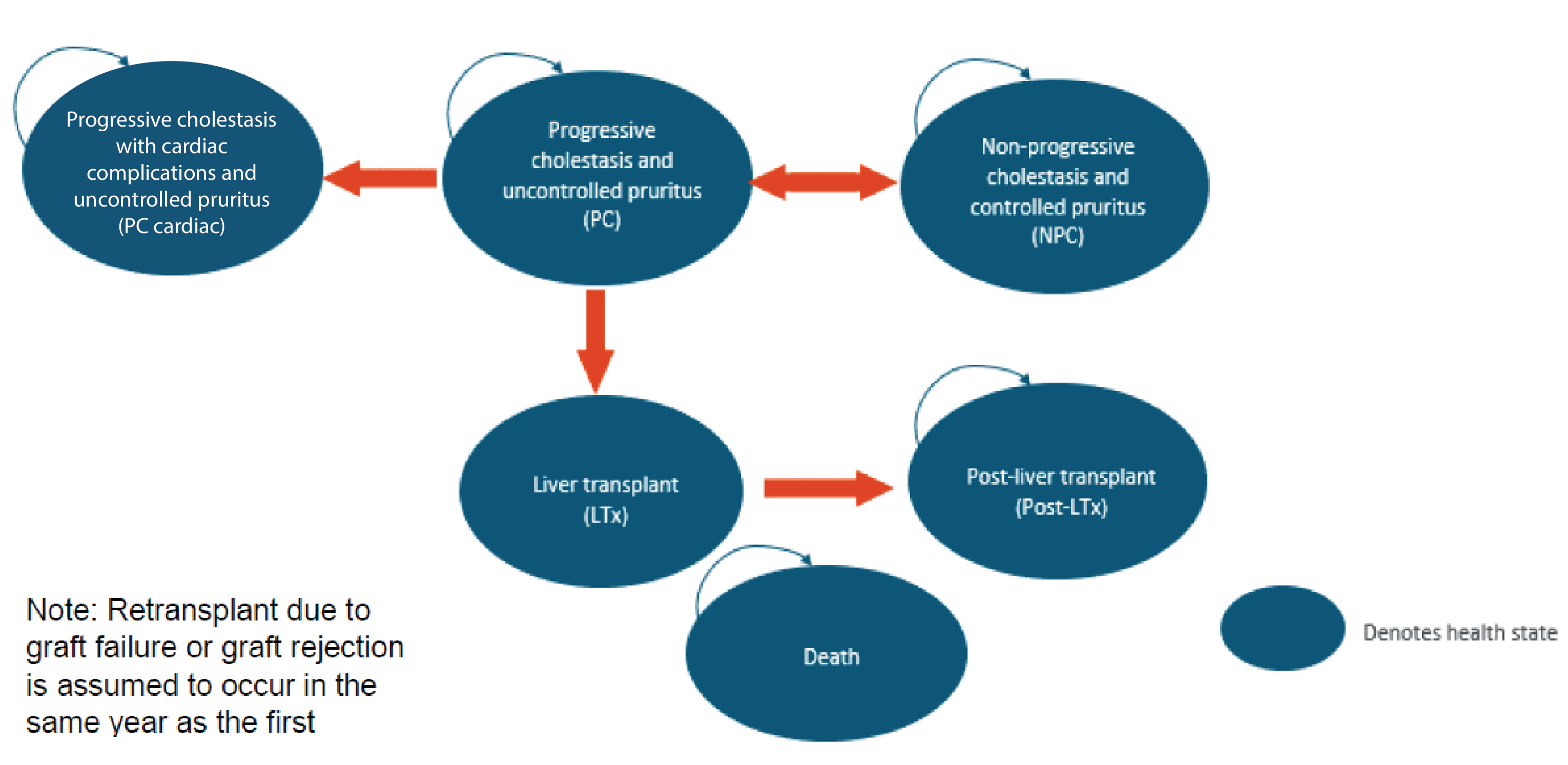
LTx = liver transplant; NPC = non-progressive cholestasis; PC = progressive cholestasis.
Sponsor’s pharmacoeconomic submission.1
Figure 2: Temporal Association Between sBA and ItchRO in the ICONIC Trial
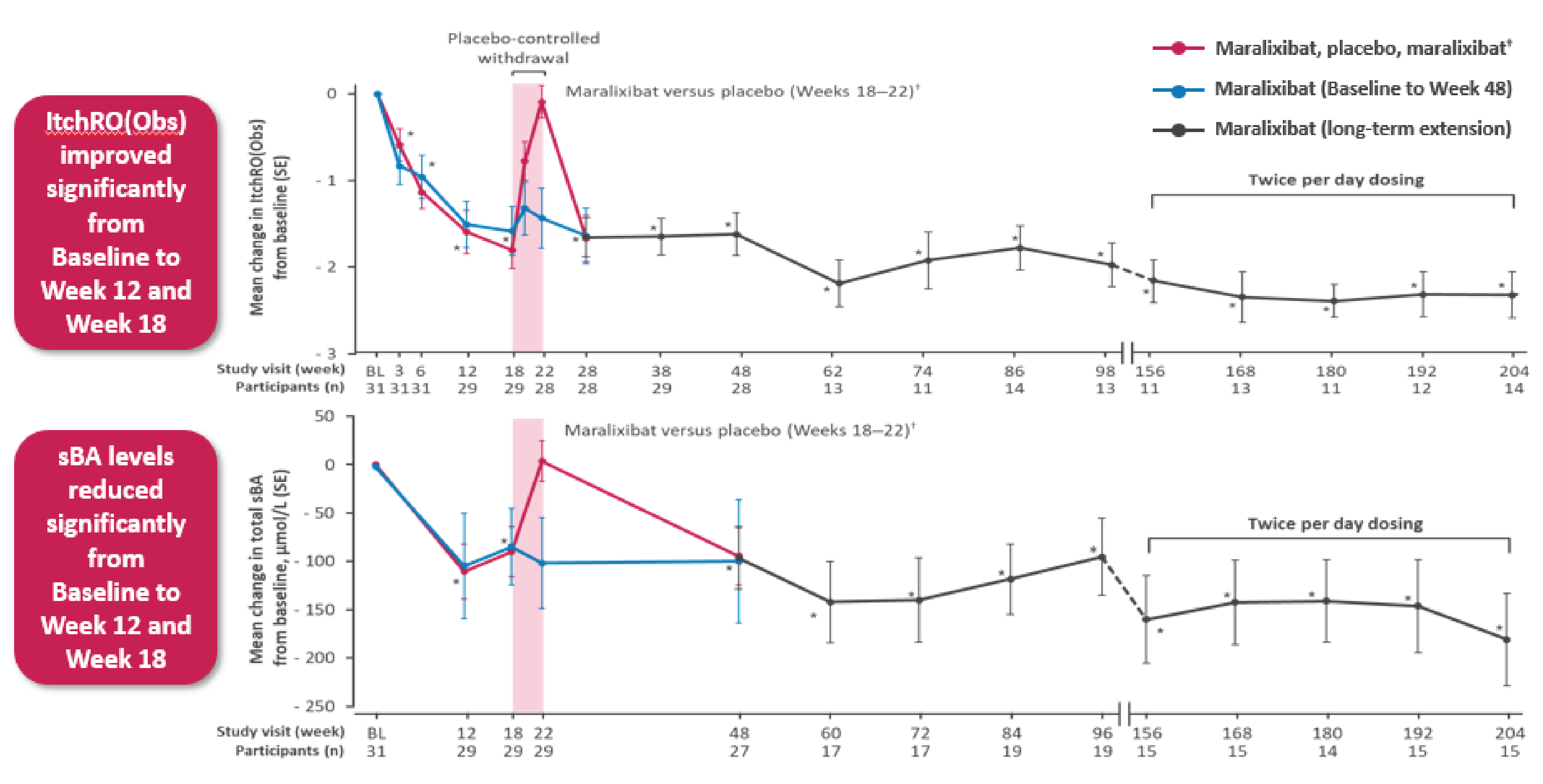
ItchRO = Itch Reported Outcome; sBA = serum bile acid.
Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 8: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Maralixibat + BSC | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 35.161 | 32.085 | 3.075 |
Nonprogressive cholestasis and controlled pruritus | 9.357 | 0.619 | 8.737 |
Progressive cholestasis and uncontrolled pruritus | 15.748 | 17.423 | –1.676 |
Liver transplant | 0.376 | 0.458 | –0.083 |
Post liver transplant | 6.148 | 7.934 | –1.785 |
Progressive cholestasis (cardiac) and uncontrolled pruritus | 3.477 | 5.596 | –2.119 |
Discounted QALYs | |||
Total | 25.405 | 21.330 | 4.074 |
Nonprogressive cholestasis and controlled pruritus | 8.468 | 0.560 | 7.907 |
Progressive cholestasis and uncontrolled pruritus | 9.161 | 10.111 | –0.950 |
Liver transplant | 0.221 | 0.269 | –0.049 |
Post liver transplant | 5.462 | 7.056 | –1.593 |
Progressive cholestasis (cardiac) and uncontrolled pruritus | 2.038 | 3.279 | –1.241 |
Discounted costs ($) | |||
Total | 12,187,825 | 1,018,916 | 11,168,909 |
Treatment cost ($) | 11,384,966 | 20,794 | 11,364,172 |
Health state (resource use) cost ($) | 802,859 | 998,122 | –195,262 |
Adverse event cost ($) | 0 | 0 | 0 |
Societal cost ($) | 0 | 0 | 0 |
ICER ($/QALY) | 2,741,204 | ||
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Scenario Analyses
A series of scenario analyses were performed on the sponsor’s base case to investigate the impact of critical assumptions on the cost-effectiveness of maralixibat plus BSC compared to BSC. These scenario analyses explored the impact of the following model parameters and assumptions on the ICER: assuming equal efficacy and safety between maralixibat plus BSC and BSC alone, assuming patients weigh 74 kg starting at the age of 25 years; and assuming 45% of patients received twice the indicated dose of maralixibat beyond 100 weeks of treatment (Table 9). The cost-effectiveness of maralixibat plus BSC was most notably affected when the treatment efficacy and safety was assumed to be equivalent between maralixibat plus BSC and BSC.
Table 9: Additional Scenario Analysis Results
Analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | BSC | 1,018,916 | 21.330 | Reference |
Maralixibat + BSC | 12,187,825 | 25.405 | 2,741,204 | |
Scenario 1: Assumption of equal efficacy and safety between maralixibat plus BSC and BSC alone | BSC | 1,018,916 | 21.330 | Reference |
Maralixibat + BSC | 1,939,666 | 21.330 | Dominated | |
Scenario 2: Patients assumed to weigh 75.6 kg starting at age 25 years | BSC | 1,022,199 | 21.330 | Reference |
Maralixibat + BSC | 14,039,336 | 25.405 | 3,194,817 | |
Scenario 3: 45% of patients received twice the indicated dose of maralixibat beyond 100 weeks of treatment | BSC | 1,018,916 | 21.330 | Reference |
Maralixibat + BSC | 17,106,649 | 25.405 | 3,948,438 |
BSC = best support care; ICER = incremental cost-effectiveness ratio.
Price Reduction Analyses
A price-reduction analysis was performed on scenario analyses using the sponsor’s base case (Table 10). In CADTH scenario assuming equal efficacy and safety between maralixibat plus BSC and BSC alone, maralixibat was more costly than BSC. Since 100% of incremental cost was driven by drug costs, there was no price reduction at which maralixibat would achieve cost-effectiveness at a $50,000 per QALY gained threshold within this scenario. CADTH notes that the uncertain relationship between sBA and pruritis, as well as other methodological limitations noted above, means that the estimated price reductions are highly uncertain and are likely underestimated.
In scenario analyses assuming patient weigh 74 kg starting at the age 25 years and the indicated dose was increased for 45% of patients beyond 100 weeks of treatment, a price reduction of 97% would be required for maralixibat to be considered optimal at a WTP of $50,000.
Table 10: CADTH Price Reduction Analyses for Scenario Analysis
Analysis | ICERs for maralixibat plus BSC vs. BSC | ||
|---|---|---|---|
Price reduction | Sponsor base case | Scenario 2: Patients assumed to weigh 75.6 kg starting at age 25 years | Scenario 3: 45% of patients received twice the indicated dose of maralixibat beyond 100 weeks of treatment |
No price reduction | $2,741,204 | $3,194,817 | $3,948,438 |
10% | $2,462,085 | $2,870,307 | $3,548,451 |
20% | $2,183,333 | $2,546,226 | $3,148,992 |
30% | $1,904,582 | $2,222,144 | $2,749,533 |
40% | $1,625,830 | $1,898,062 | $2,350,073 |
50% | $1,347,078 | $1,573,980 | $1,950,614 |
60% | $1,068,327 | $1,249,898 | $1,551,155 |
70% | $789,575 | $925,816 | $1,151,696 |
80% | $510,823 | $601,734 | $752,237 |
90% | $232,072 | $277,652 | $352,778 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; vs. = versus.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 11: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor stated that submitted budget impact analysis (BIA)24 assessed the expected budgetary impact of reimbursing maralixibat for the treatment of cholestatic pruritus in patients with ALGS 2 months of age and older. The BIA was undertaken from the perspective of the Canadian public drug plans at base year (2023) and over a 3-year time horizon (2024 to 2026). However, the sponsor’s pan-Canadian estimates did not reflect the aggregated results from provincial budgets (excluding Quebec). Key inputs to the BIA are documented in Table 13.
The sponsor estimated the number of eligible patients for maralixibat treatment using an epidemiologic approach with data obtained from various sources including: Statistics Canada population estimates, published literature and clinical expert opinion.4,24-26 The sponsor estimated that the incidence of ALGS based on presence of JAGGED1 mutations and assumed not all patients with ALGS will present phenotypes that meet the criteria for clinical diagnosis. The sponsor narrowed the population eligible for treatment with maralixibat to patients with a presence of cholestasis and cholestatic pruritis.4 The sponsor removed patients who would receive liver transplant because these patients would not be eligible for treatment with maralixibat. The sponsor also adopted a treatment discontinuation rate of 6.5% in year 1 and 8.7% in year 2 and 3 for patients on maralixibat using data on adverse events that led to discontinuation in the ICONIC trial.3 The comparator included BSC which comprised of a basket of therapeutic options. The sponsor estimated the cost of BSC weighted by the proportion of patients receiving UCDA (93.1%), cholestyramine (9.8%), rifampin (33.5%), hydroxyzine hydrochloride (19.4%), naltrexone (13.8%), sertraline (13.8%) and alimemazine tartrate (0.4%). The sponsor obtained drug acquisition costs from Ontario Drug Benefit formulary and Alberta Drug Benefit List.13,27 Dosing was obtained from respective product monograph and published literature.2,8,10,11,28 The sponsor assumed patients have a mean body weight of 13.96 kg, 16.62 kg, 18.40 kg and 20.05 kg at base year, year 1, year 2, and year 3, respectively. No drug wastage was assumed in estimating treatment costs. Markups and dispensing fees were not included.
Table 12: Summary of Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Annual growth rate | 2.73% |
Projected population (year 1 / year 2 / year 3) | 31,794,295 / 32,661,139 / 33,551,618 |
ALGS incidence | 1 in 30,000 live births |
ALGS adjusted clinical prevalence (percentage of patients who meet clinical criteria for diagnosis) | 1 in 56,000 |
Percentage of patients with presence of cholestasis | 85% |
Percentage of patients with presence of cholestatic pruritis | 74% |
Percentage of patients who reach 18 years of age without a liver transplant and are eligible for treatment with maralixibat | 40% |
Percentage of patients covered by public payer | 67% |
Number of patients eligible for drug under review | 96 / 99 / 101 |
Market uptake (3 years) | |
Uptake (reference scenario) BSC | 100% / 100% / 100% |
Uptake (new drug scenario) Maralixibat BSC | |||||||||||||||||||||||||||| |||||||||||||||||||||||||||| |
Cost of treatment (per patient)a | |
Cost of treatment over year Maralixibat BSC | $498,945 $338 |
ALGS = Alagille syndrome; BSC = best supportive care.
aThe treatment cost over the base year and the adopted time horizon is presented here.
Summary of the Sponsor’s BIA Results
The sponsor estimated the net three-year budget impact of introducing maralixibat for the treatment of cholestatic pruritus in patients with ALGS 2 months of age and older to be $45,035,065 (year 1: $8,969,456; year 2: $15,130,113; year 3: $20,935,496).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The proportion of patients who keep their native liver does not align with the mean age of population considered in the BIA: The sponsor assumed that 40% of patients with ALGS would reach adulthood without a liver transplant for a population with mean ages of 5.35 to 7.35 years using the study by Vandriel et al. (2023). However, Vandriel et al. reported a much higher proportion of patients surviving with their native liver (72%) for patients aged 5 years that decreased to 68% by the age of 7 years.4 The proportion of patients that kept their native liver was 40.3% at the age of 18 years. As such, the proportion of patients with their native liver does not align with the mean age of the population. Given the population in the BIA is between the ages of 5 to 7 years, extrapolating survival beyond this age group is beyond the time horizon adopted by the sponsor. As such, the sponsor has underestimated the proportion of children aged between 5 to 7 years who would survive with their native liver.
The sponsor also assumed that the proportion of patients with a native liver transplant would not be impacted by the availability of maralixibat. However, the clinical experts consulted by CADTH noted treatment with maralixibat may reduce or delay the incidence of liver transplant if it is found to be effective in clinical practice. In this case, the number of eligible patients in the scenario in which maralixibat is covered by the public drug plans would increase.
In CADTH’s reanalysis, CADTH estimated that the average proportion of patients with a native liver (i.e., those who have not had a liver transplant) for a population aged 5.35 to 7.35 years would be 70%. CADTH explored the impact of reducing the proportion of patients with a liver transplant by half in a scenario analysis.
The proportion of patients covered by public drug plans is uncertain: The sponsor estimated that 67.3% of the eligible population in Canada is covered by public drug plans, using data reported in the Sutherland and Ding (2017) report.26 The sponsor considered the proportion of individuals that meet the eligibility criteria for the public coverage in a population considering all ages (including those over the age of 25 years). For example, the sponsor estimated that out of 13,976,300 individuals in Ontario, 5,622,100 or 40.2% of Ontario residents are eligible for public coverage. However, given the sponsor’s submitted BIA was estimated for a patient population with an average age of 5.35 years, it is inappropriate to adopt a coverage rate estimated for all ages. Further, the sponsor’s estimate was based on the assumption that not all pediatric patients eligible for maralixibat treatment would meet the eligibility criteria for coverage by the public health care payer in New Brunswick, Newfoundland and Labrador, Ontario, and Prince Edward Island. However, there are publicly funded programs to cover the cost of high-cost drugs and treatments for rare diseases in these jurisdictions. It is not clear whether patients would be covered under public or private insurance for maralixibat across different jurisdictions, or which private plans would cover maralixibat. No evidence was presented by the sponsor to quantify the proportion of patients that are eligible for treatment in jurisdictions with a mix of public and private coverage. In the absence of a rigorous estimate, CADTH makes no assumption about the proportion of patients eligible for public coverage and makes the conservative assumption that the public system will cover all pediatric patients with severe disease requiring treatment with a high-cost drug.
In reanalysis, CADTH assumed 100% of eligible population is covered by public drug plans.
Treatment discontinuation rate is uncertain: The sponsor adopted a treatment discontinuation rate of 6.5% in year 1 and 8.7% in years 2 and 3 using data from the open-label phase (day 1 to week 18) and long-term extension phase of the ICONIC trial. The reasons for treatment discontinuation included staphylococcal infection, extradural hematoma, subdural hemorrhage, and alanine aminotransferase. The clinical expert noted that these reasons are not indicative of disease progression or related to the effect of drug use. The clinical expert also noted that in the event of adverse events, treatment with maralixibat may be titrated to a lower dose, rather than completely discontinued. As such, the rate of treatment discontinuation is uncertain and may not be aligned with the reasons patients will discontinue treatment in Canadian clinical practice. The sponsor also adopted a lower discontinuation rate (||||| ||| || ||||| || ||||| ||| ||||) in the cost-utility analysis, and this misalignment between the cost-utility analysis and BIA adds to the uncertainty in the treatment discontinuation rate.
In CADTH’s reanalysis, CADTH assumed no treatment discontinuation (i.e., 100% of patients remain on treatment). CADTH explored the impact an annual discontinuation rate of ||||| in a scenario analysis.
Treatment cost of maralixibat did not include drug wastage: The sponsor also assumed no drug wastage in estimating treatment cost of maralixibat. Given the adopted perspective in this BIA, the cost of drug wastage would be incurred by the public health care payer. Excluding cost of drug wastage underestimates the cost of maralixibat. To account for drug wastage, the treatment cost should be estimated based on the number of units dispensed.
In reanalysis, CADTH included the cost of drug wastage using the method built into the sponsor’s BIA model.
Treatment cost of maralixibat is uncertain: Maralixibat is dosed based on patient weight. The sponsor’s model used the mean age of ICONIC trial participants (5.35 years) and estimated weight based on age. In the sponsor’s model, the dosing of maralixibat increased to 8.55 mg by year 3, however, maralixibat may be dosed to a maximum of 28.50 mg. The sponsor has estimated treatment cost of maralixibat based on age and weight of children, however, maralixibat may also be used in adult population. As the age distribution of patients with ALGS (i.e., the proportion of patients in various age categories) is not known, the treatment cost of maralixibat and the estimated budget impact is uncertain. However, the sponsor’s budget impact is underestimated because the sponsor’s model assumes that patients will never receive the maximum dose of maralixibat.
In scenario analysis, CADTH explored the budget impact for patients with the maximum starting age of 25 years and a weight of 75.6 kg.23 In this scenario, the proportion of patient with native liver was assumed to be 40%.
Dose escalation is not considered: The recommended maralixibat dosage in the product monograph is 380 mcg/kg once daily after 1 week. However, maralixibat dose was titrated to doses higher than 380 mcg/kg in the ICONIC trial. After almost 2 years (beyond week 100), approximately 45% of patients (14 of 31) received a maralixibat dose of at least 760 mcg/kg/day. The sponsor assumed no dose escalation for patents who remained on treatment for the first 2 years in the submitted BIA.
In scenario analysis, CADTH assumed a maralixibat dose of 760 mcg/kg/day for 45% of patients in year 3.
The sponsor’s submitted BIA model had programmatic errors: There were programmatic errors noted in the model that were found during model scrutinizing. The user input options for altering treatment discontinuation rates did not impact the values that were used in the calculations. The sponsor also used a macro to update jurisdiction costs, however, changing options to include or exclude markup and dispensing fees did not lead to any changes in the estimated budget impact. The pan-Canadian coverage rate was also hard coded, rather than calculated as a weighted-average of jurisdiction-specific coverage rates. It was that the sum of disaggregated results was similar to the pan-Canadian estimated budget impact but not the same on the sheet presenting disaggregated results. These errors prevented CADTH from ensuring that changes to default values occurred throughout the calculations.
CADTH corrected the programmatic error on inputting alternative discontinuation rates in the “User Inputs” sheet. CADTH could not address other programmatic errors. The estimated budget impact is therefore uncertain.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by assuming the proportion of patients receiving a liver transplant decrease over time, adopting a coverage rate of 100%, assuming no treatment discontinuation and including drug wastage in estimating maralixibat treatment cost (Table 13).
Table 13: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Proportion of patients with a native liver | 40% | 70% |
2. Coverage rate | 67% | 100% |
3. Treatment discontinuation | Year 1: 6.5% Years 2 and 3: 8.7% | Years 1, 2 and 3: 0% |
4. Drug wastage | Excluded | Included |
CADTH base case | CADTH base case (Reanalysis 1 + 2 + 3 + 4) | |
BIA = budget impact analysis.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 14 and a more detailed breakdown is presented in Table 15.
In the CADTH reanalysis, the three-year budget impact of reimbursing maralixibat plus BSC from the public drug plan perspective for the treatment of cholestatic pruritus in patients with ALGS 2 months of age and older increased to $130,727,100 (year 1: $26,649,978; year 2: $44,315,818; year 3: $59,761,303).
Table 14: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $45,035,065 |
CADTH reanalysis 1 | $78,811,364 |
CADTH reanalysis 2 | $66,916,091 |
CADTH reanalysis 3 | $48,780,369 |
CADTH reanalysis 4 | $46,775,811 |
CADTH base case | $131,744,508 |
BIA = budget impact analysis.
CADTH also conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 15. The scenario analyses included:
assuming the proportion of patients with a liver transplant reduced by half (i.e., the proportion of patients with their liver transplant increased to 85%)
adopting an annual discontinuation rate of 2.40%
assuming a treatment starting age of 25 years, a weight of 75.6 kg, and native liver in 40% of the population
assuming a maralixibat dose of 760 mcg/kg/day for 45% of patients in year 3
assuming a price reduction of 96%.
Table 15: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $31,627 | $32,489 | $33,375 | $34,285 | $100,149 |
New drug | $31,627 | $9,001,946 | $15,163,488 | $20,969,781 | $45,135,214 | |
Budget impact | $0 | $8,969,456 | $15,130,113 | $20,935,496 | $45,035,065 | |
CADTH base case | Reference | $82,238 | $84,481 | $86,784 | $89,150 | $260,414 |
New drug | $82,238 | $25,994,182 | $44,402,602 | $61,608,139 | $132,004,923 | |
Budget impact | $0 | $25,909,701 | $44,315,818 | $61,518,989 | $131,744,508 | |
CADTH scenario analysis 1: assuming proportion of patients with a liver transplant decrease by half | Reference | $99,861 | $102,584 | $105,380 | $108,254 | $316,217 |
New drug | $99,861 | $31,564,363 | $53,917,446 | $74,809,883 | $160,291,692 | |
Budget impact | $0 | $31,461,780 | $53,812,065 | $74,701,629 | $159,975,474 | |
CADTH scenario analysis 2: adopting an annual treatment discontinuation rate of 2.40% | Reference | $82,238 | $84,481 | $86,784 | $89,150 | $260,414 |
New drug | $82,238 | $25,371,943 | $43,338,329 | $60,130,720 | $128,840,992 | |
Budget impact | $0 | $25,287,463 | $43,251,545 | $60,041,570 | $128,580,578 | |
CADTH scenario analysis 3: assuming a treatment starting age of 25 years, a weight of 75.6 kg and native liver in 40% of the population | Reference | $181,881 | $186,840 | $191,934 | $197,166 | $575,940 |
New drug | $181,881 | $56,856,333 | $97,119,142 | $134,751,014 | $288,726,490 | |
Budget impact | $0 | $56,669,494 | $96,927,209 | $134,553,848 | $288,150,550 | |
CADTH scenario analysis 4: assuming a maralixibat dose of 760 mcg/kg/day for 45% of patients in year 3 | Reference | $82,238 | $84,481 | $86,784 | $89,150 | $260,414 |
New drug | $82,238 | $25,994,182 | $44,402,602 | $89,291,684 | $159,688,468 | |
Budget impact | $0 | $25,909,701 | $44,315,818 | $89,202,534 | $159,428,053 | |
CADTH scenario analysis 5: assuming 96% price reduction for maralixibat | Reference | $82,238 | $84,481 | $86,784 | $89,150 | $260,414 |
New drug | $82,238 | $1,120,869 | $1,859,417 | $2,549,909 | $5,530,195 | |
Budget impact | $0 | $1,036,388 | $1,772,633 | $2,460,760 | $5,269,780 |
BIA = budget impact analysis.
Ethics Review
Abbreviations
ALGS
Alagille syndrome
GALA
Global ALagille Alliance
HRQoL
health-related quality of life
sBA
serum bile acid
Summary
Alagille syndrome (ALGS) is a rare, life-threatening, genetic disease associated with chronic cholestasis (i.e., impaired bile flow) that can cause chronic, severe, and unremitting cholestatic pruritis (i.e., itch). Patient group, clinician group, clinical expert, and drug program input gathered during this CADTH review, as well as relevant literature, were reviewed to identify ethical considerations relevant to the use of maralixibat to treat cholestatic pruritis in people with ALGS.
Ethical considerations identified in this review included those related to the following:
Diagnosis, treatment, and experiences of people with ALGS: Ethical considerations arising in the context of ALGS highlighted the significant physical, psychosocial, and financial impact of the condition and its associated cholestatic pruritis on patients and their families, and the difficulties and harms associated with delays in accessing a timely diagnosis and routine treatment and care. Families with limited income, with multiple members who have ALGS, or those living far from specialized treatment centres may experience a disproportionate burden in managing the condition and difficulties accessing timely care. There is a significant unmet need for an effective treatment for cholestatic pruritis in ALGS due to its devastating impacts on patients and their families; the limited efficacy of and adverse effects associated with currently available off-label therapies; and the invasive, life-altering nature of surgical treatment alternatives such as liver transplant.
Clinical and economic evidence used in the evaluation of maralixibat: Clinical trial evidence indicated that maralixibat may result in a clinically meaningful decrease in pruritis and may result in little to no difference in serious adverse events compared with placebo; however, there is evidentiary uncertainty concerning its safety and efficacy (particularly concerning its effect on long-term treatment outcomes and health-related quality of life [HRQoL]), which limits the assessment of clinical benefits and harms associated with its use as well as the accuracy of the pharmacoeconomic assessment of cost-effectiveness.
Clinical use and implementation of maralixibat: Clinical experts voiced that they would prescribe maralixibat based on the currently available evidence, given its potential to address a substantial unmet need for the treatment of ALGS-associated cholestatic pruritis with a favourable safety profile. However, given the uncertainty of evidence and the likelihood that maralixibat may not halt the progression of the underlying liver disease causing pruritis (for which there is no curative, nonsurgical treatment), robust, informed consent processes are required in both pediatric and adult contexts. As an orally administered medication, maralixibat is relatively accessible for patients, but equitable access requires attending to potential diagnostic, geographic, and monitoring-related barriers to access.
Health systems: Ethical considerations for health systems related to the implementation of maralixibat highlight the challenges of funding decisions for high-cost drugs for rare diseases, assessments of opportunity costs, and the fair allocation of scarce resources, as well as issues related to pan-Canadian approaches to providing equitable reimbursement and access.
Objectives
To identify and describe ethical considerations associated with the use of maralixibat for the treatment of cholestatic pruritis in people with ALGS, including considerations related to the context of ALGS, evidentiary basis, the use of maralixibat, and health systems.
Research Questions
This report addresses the following research questions:
What ethical considerations arise in the context of ALGS, including those related to its diagnosis, treatment, and outcomes?
What ethical considerations arise related to the evidence (e.g., clinical and economic data) used to evaluate maralixibat?
What ethical considerations arise in the use of maralixibat for patients with ALGS, their caregivers, and clinicians?
What ethical considerations for health systems are involved in the context of maralixibat?
Methods
Overview
To identify ethical considerations relevant to the use of maralixibat in the treatment of cholestatic pruritis in people with ALGS, this Ethics Review report was driven by relevant questions identified in the European Network for Health Technology Assessment (EUnetHTA) Core Model 3.0, Ethics Analysis Domain,1 and supplemented by relevant questions from the Equity Checklist for Health Technology Assessment.2 These guiding questions were organized to respond to the research questions posed, and investigated ethical considerations related to:
Patients living with ALGS and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges related to diagnosis or clinical care; and factors that might prevent patients from gaining access to therapies).
The evidence used to demonstrate the benefits, harms, and value of maralixibat (i.e., ethical considerations in relevant clinical trials, including their representativeness, choice of outcome measures, and appropriateness of analytical methods and models to all population groups; and ethical considerations related to the data or assumptions in the economic evaluation).
The use of maralixibat, including considerations related to benefits and harms to patients, relatives, caregivers, clinicians, or society; and considerations related to access to these therapies.
The uptake of maralixibat in health systems, including considerations related to the distribution of health care resources.
Data Collection: Review of Project Inputs and Literature
Data to inform this Ethics Review report drew from an identification of ethical considerations (e.g., values, norms, or implications for equity, justice, resource allocation, and ethical considerations in the evidentiary basis) in the patient and clinician group, clinical expert, and drug program input collected by CADTH to inform this review, as well as a complementary search of the published literature. Ongoing collaboration and communication with CADTH reviewers working on the clinical and economic reviews for this submission also assisted in the clarification and identification of ethical considerations raised.
Review of Project Inputs
During this CADTH review, a single reviewer collected and considered input from 6 main sources for content related to ethical considerations relevant to addressing the research questions. In addition to the published literature, this report considered the following sources:
the sponsor submission, including information and external references or sources relevant to each of the research questions driving this report
clinician group input received by CADTH from 1 clinician from the Canadian Association for the Study of the Liver
patient input received by CADTH from the Canadian Liver Foundation and the Alagille Syndrome Alliance
drug program input received by CADTH from drug programs participating in the CADTH Reimbursement Review process
discussions with clinical experts (n = 3) directly engaged by CADTH throughout this Reimbursement Review, including through 1 teleconference discussion involving 2 experts and 1 panel discussion involving 3 experts. During these 2 discussions, the ethics reviewer asked the clinical experts targeted questions related to ethical considerations corresponding to the research questions driving this report. All clinical experts were practising gastroenterologists or hepatologists with experience treating people with cholestatic liver disease in Canada. One of the experts had experience treating children with ALGS
engagement with CADTH clinical and economic reviewers to identify domains of ethical interest arising from their respective reviews and relevant questions and sources to further pursue in this report.
Literature Search Methods
An information specialist conducted a literature search on key resources including MEDLINE via Ovid, Philosopher’s Index via Ovid, PsycInfo via Ovid, the Cumulative Index to Nursing and Allied Health Literature via EBSCO, and Scopus. Google Scholar was searched to find additional materials not captured in the major bibliographic databases. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were maralixibat, Alagille syndrome, and pediatric cholestatic pruritus.
CADTH-developed search filters were applied to limit retrieval to citations related to ethical concepts or considerations and qualitative studies. Search terms for equity were also applied to the main concepts to capture additional articles. Duplicates were removed by manual deduplication in EndNote. Retrieval was limited to the English language. The search was completed on May 30, 2023.
Literature Screening and Selection
Literature retrieved according to the search and selection methods detailed previously was screened in 2 stages. First, titles and abstracts of citations retrieved were screened for relevance by a single reviewer. Articles were identified and retrieved for full-text review by a single reviewer if their titles or abstracts identified ethical considerations, provided normative analysis (i.e., focusing on “what ought to be” through argumentation), or presented empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to the experiences, incidence, diagnosis, treatment, or outcomes of ALGS in Canada or similar contexts; or the evidence on, use of, or implications of maralixibat for patients with ALGS. In the second stage, full-text publications categorized as “retrieve” were reviewed by the same reviewer. Texts that included substantive information meeting the aforementioned criteria were included in the review, and reports that did not meet these criteria were excluded. As a parallel process, other sources drawn from relevant bibliographies, relevant key concepts, in consultation with experts, or other CADTH reviewers were retrieved and reviewed using the previously-listed selection criteria.
Data Analysis
Data analysis was driven by the 4 research questions guiding this report and included the collection, coding, and thematic analysis of data drawn from the literature and project inputs. The reviewer conducted 2 iterative cycles of coding and analysis to abstract, identify, and synthesize relevant ethical considerations in the literature and from relevant project inputs.
In the initial coding phase, publications and input sources were reviewed for ethical content (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis). Once identified, claims related to ethical content were coded using methods of qualitative description.3 In the second coding phase, major themes and subcodes were identified through repeated readings of the data,3 and summarized into thematic categories within each guiding domain or research question. Where ethical content did not fit into these categories or domains outlined in the research questions, this was noted, as were discrepancies or conflicts between ethical considerations or values identified between project sources or within thematic categories. Data analysis was iterative, and themes identified in the literature, in project inputs, and during consultations with clinical experts were used to further refine and re-interpret ethical considerations identified.
Data collected and analyzed from these sources were thematically organized and described according to the 4 research questions and domains driving this report. The results of this analysis and its limitations and conclusions are described subsequently.
Results
Description of Included Sources
The literature search identified 191 results. Following title and abstract screening, 147 citations were excluded and 44 potentially relevant publications from the electronic searches were retrieved for full-text review. Of the potentially relevant publications, 33 publications were excluded as they did not discuss ethical considerations of maralixibat or ALGS (n = 28), were captured in an included systematic review (n = 3), focused on ethical considerations relevant to low or middle-income countries (n = 1), or were abstracts reporting the same findings as an included citation (n = 1). Eleven publications met the inclusion criteria and were included in this report. Six additional publications were retrieved from backward searching of included publications’ reference lists or through a manual search.
A total of 17 publications were used to inform this review. Of these publications, 6 discussed ethical considerations in the context of ALGS, including related to diagnosis and treatment; 3 discussed patient and/or family and caregiver experiences in the context of AGLS; 2 provided insight into ethical considerations related to the evidence used to evaluate maralixibat; 1 was selected to provide a broader understanding of diversity in clinical trials; 1 provided insight into considerations for the uptake of maralixibat in health systems specifically; and 4 were selected to provide a broader understanding of the context of ethical considerations for drugs for rare diseases or the pediatric population.
Key Ethical Considerations
Diagnosis, Treatment, and Experiences of People With ALGS
Diagnosis
ALGS is a rare, life-threatening, genetic disease with an estimated incidence of 1 in 30,000 to 50,000 births.4-7 Mutations in the Jagged-1 gene and NOTCH2 gene cause the condition in more than 90% and 2% to 4% of cases, respectively.6,7 The condition is heritable, although 60% of affected individuals have a de novo mutation.4,6,7 Although epidemiological information on ALGS is scarce,4 clinical experts in the published literature and consulted by CADTH reported a suspected equal incidence of the condition across different sexes and races.5
ALGS affects multiple organs, including the liver, wherein a paucity of bile ducts causes chronic cholestasis (i.e., impaired bile flow) that manifests clinically as early as 3 months of age.4,6,7 Hallmark serological findings of cholestasis include elevated serum bile acids (sBAs) and bilirubin, the latter of which causes jaundice. Clinical manifestations of cholestasis can be severe and include unremitting, severe, and debilitating cholestatic pruritis (i.e., itch) in 74% of cases; disfiguring xanthomas; growth failure, fat-soluble vitamin malabsorption and associated bone fractures; and liver damage and portal hypertension.4,7,8 As cholestasis and its associated manifestations progress, most people living with ALGS will either undergo liver transplant or die by the age of 18 years, with only 40.3% reaching adulthood with their native liver.7 Notably, unremitting pruritis is a leading cause of liver transplant in people living with ALGS.4,7-9
Despite its rarity, clinical experts did not anticipate barriers to or inequities in accessing a timely diagnosis of ALGS in Canada. They noted that, in their experience, children with ALGS present with obvious signs and symptoms of cholestasis (e.g., jaundice) that trigger general providers to consult a pediatric hepatologist or gastroenterologist with the knowledge required to diagnose the condition. Clinicians can diagnose ALGS based on the presence of 3 of the 7 major clinical features alone; however, it is common practice to confirm the diagnosis either by liver histology or, more commonly, genetic testing.4-6 These sources reported that genetic testing reduces the risk of misdiagnosis, is helpful in milder cases, and, when positive, can prompt providers to inform family members that they, too, may have the condition.5 The clinical experts emphasized that this genetic testing is easily accessible in Canada, although health care providers need to send samples to the US for processing.
However, despite genetic testing being easily accessible in Canada once ordered, the patient group input and published literature detailed challenges in providing or obtaining an ALGS diagnosis that raise ethical considerations regarding exposure to preventable harm and informed decision-making related to heritable diseases. Diagnosing ALGS can be difficult due to its variable penetrance and clinical manifestations, clinical features similar to those of other conditions (e.g., biliary atresia), as well as little correlation existing between mutation type, clinical manifestations, and disease severity.4-6 Furthermore, in 4% of cases of ALGS, patients meet clinical criteria for the condition but do not have an identified mutation, which may further complicate diagnosis.6 For these reasons, and since not all nonspecialist clinicians are aware of the clinical features of ALGS, patient group input and the published literature reported delayed diagnoses, with adults sometimes unaware they had the condition until their child received a diagnosis.5 Patient group input and the published literature described how misdiagnosis has resulted in some people with the condition receiving invasive surgical procedures associated with poor outcomes or experiencing delayed access to timely and appropriate monitoring for, recognition of, and interventions to mitigate the multisystem implications of ALGS.5 Additionally, without a diagnosis, people with ALGS may miss opportunities for accessing genetic counselling, which was highlighted as an important component of holistic care that can support a person with ALGS in making informed decisions regarding family planning or disclosure of their diagnosis to at-risk family members who may also have the condition.5,8
Difficulties and Disparities in Accessing Care and Treatment
As detailed in the Clinical Review report and the published literature, the monitoring and management of ALGS are lifelong and multidisciplinary, and are typically provided through tertiary care centres capable of providing specialized care.8 As detailed in the Clinical Review report, this care involves multiple health care professionals, including hepatologists or gastroenterologists, other physicians consulted as needed, and allied health care professionals.8 The patient group input and clinical experts noted that while it is possible and common practice for these providers to virtually monitor and treat patients living in areas that are far from specialized care centres, these patients may still experience disproportionate difficulty accessing timely and quality care for ALGS. As noted by clinical experts, the concentration of ALGS cases in Canadian provinces with specialized care centres suggests that there are equity concerns related to the accessibility of care and treatment for those without resources to relocate or travel to such locations.
The patient group input, clinical experts, and published literature emphasized that the emotional, cognitive, financial, and time-related burdens of coordinating and advocating for care fall primarily on the families of people living with ALGS.10 The clinical experts described how parents often continue this coordinating role after their child reaches adulthood, as ALGS is associated with intellectual and learning disabilities that make navigating the health care system difficult, even though pediatric providers often continue treating their patients with ALGS into adulthood. Moreover, caregivers coordinating their child’s care may also be living with the physical features and intellectual disabilities of ALGS, which further challenge their ability to navigate the health care system effectively. Clinical experts also described how families with lower incomes might have greater difficulty accessing important therapies not covered by provincial health plans, including equipment, formula, and vitamins required for optimal nutrition and formal psychosocial support. As a result, families with multiple members with ALGS, living with low income, or living in provinces with less public coverage for necessary treatments may disproportionately experience burdens related to navigating and accessing care.
Difficulties navigating or accessing timely specialist care for ALGS may negatively affect health outcomes. For example, it was reported that, in California, children living with ALGS who were Black made up a disproportionate number of hospital admissions for the condition from 2005 to 2015;11 the clinical experts attributed these findings to the US’s lack of universal health care rather than a known higher incidence of ALGS in children who are Black, as patients in equity-deserving groups may experience greater obstacles in accessing regular specialist and preventive care. The literature search did not capture studies detailing whether there are racial disparities in ALGS-related hospitalizations or outcomes in Canada, so further research would be required to determine whether people with ALGS in equity-deserving groups in Canada may be similarly affected by barriers to accessing care.
Patient and Caregiver Experiences of Cholestatic Pruritis and ALGS
The patient and clinician group input, clinical experts, and published literature reported that living with ALGS is physically, psychosocially, and financially burdensome for patients and their caregivers.4,10,12,13 According to these sources, unremitting pruritis is the most frequent, disturbing, and physically burdensome physical manifestation of the disease.4,12 The sources described this symptom as an intense, “agonizing,” and constant discomfort that is often worse at night, which affects a patient and family’s ability to sleep and causes debilitating fatigue and thus also affects their ability to function and engage in school, social activities, and work.4,12-14 They also noted that pruritis is either incompletely relieved or unrelieved by scratching, available off-label medications, or over-the-counter lotions or remedies, and that constant scratching leads to skin excoriations that can be so severe that parents often report finding blood on their child’s bedsheets.12,13 The clinical experts emphasized that, when it is severe and unremitting, pruritis may even pose a threat to life for older children and adults, who may consider suicide in the absence of alternatives for relief.
Furthermore, the patient group input and the published literature noted that, for parents, the psychological burden of watching and supporting a child suffering from constant itch is compounded with that related to the isolation, worry, uncertainty, and chronic sorrow of having a child with a rare and life-threatening disease.10 Patient group input reported that older children and adults with ALGS similarly experience psychological distress related to their prognosis. According to patient group input and the clinical experts, alongside managing the burden of cholestatic pruritis, people with ALGS and their families must also devote cognitive energy, time, and financial resources toward accessing, coordinating, and managing care and treatment for other health complications associated with the multisystem condition involving frequent medications, appointments, and hospitalizations. Patient group input and the published literature reported that parents of children with ALGS perceived that accessing and advocating for quality care depended on them independently seeking information on the condition and its treatment, often through online resources and connections, as they perceived some health care providers as either lacking knowledge or sharing unhelpful or insufficient information regarding the rare disease and its treatment.10 Online sources could offer encouragement and informational support at times, but also could trigger negative emotions and chronic sorrow (e.g., when including information emphasizing ALGS’s poor prognosis and unmet treatment needs).10
The clinical experts noted that the demands of caring for a child with ALGS are so great that some parents must reduce or cease their employment, making affording noninsured treatments even more difficult. They also noted that some families experience reduced functioning or breakdown because of caregiver burden and ALGS-related stress. As previously detailed, these burdens may disproportionately affect families who have multiple people living with the condition, who live in regions with fewer provincially funded services, or who have limited financial means.
Unmet Needs in the Treatment of Cholestatic Pruritis in ALGS
As described in the clinician group input and by the clinical experts, there are no other Health Canada–approved treatments for ALGS or associated pruritis, and many off-label therapies have limited efficacy and undesirable adverse effects.6 Additionally, patient and clinician group input and the clinical experts noted that many patients with ALGS require multiple off-label pharmacotherapies for cholestatic pruritis, which increases the burden associated with medication management for patients and their families. When these pharmacological options fail to manage debilitating pruritis, surgical alternatives such as biliary diversion procedures or liver transplant become necessary, even without liver failure.4,6,7,9 However, these procedures have variable results, and short- and long-term complications, and may be contraindicated in children experiencing certain extrahepatic features of ALGS.4,7,9 Younger children may be especially at risk of complications following liver transplant, and the procedure causes lifelong immunosuppression and other morbidities, which have devastating impacts on physical and psychosocial well-being.4,9
The clinician and patient group input and clinical experts emphasized a significant unmet need for safe and effective symptomatic and curative treatment for cholestatic pruritis in patients with ALGS. Specifically, patients, families, and clinicians hope for effective and safe relief of itch that will improve HRQoL by improving sleep, functioning in school or the workplace, and psychosocial well-being; reduce skin excoriations from scratching; and prevent or delay the need for invasive surgeries, including liver transplant.
Ethics of Evidence and Evaluation of Maralixibat
As described in detail in the Clinical Review report, the ICONIC (LUM001-304) pivotal trial (n = 31) sought to evaluate the safety and efficacy of maralixibat in children with ALGS aged between 12 months and 18 years. The phase IIb study comprised an 18-week, open-label phase; a 4-week, randomized, double-blind, placebo-controlled drug-withdrawal phase; a 26-week stable-dosing period; and an optional, long-term, open-label extension. The Clinical Review report discusses in detail additional clinical evidence submitted by the sponsor, including a natural-history comparison study using information from the international GALA database; and results from patient-level data from 3 long-term ALGS clinical trials (the IMAGINE, IMAGINE-11, and ICONIC trials) seeking to identify predictors of event-free survival and transplant-free survival. This section focuses on the ICONIC trial.
Although the ICONIC trial did not include participants with the NOTCH2 mutation, adolescents aged 15 years and older, or people treated in North America, and did not report the race and ethnicity of participants, clinical experts did not consider these study population characteristics to be a significant generalizability concern and believed that the pivotal trial results could be extrapolated to people with ALGS in Canada. While it was not possible to appraise the representation of different racial or ethnic groups in the pivotal trial, the representation of racial and other types of diversity in clinical trials has been recognized as being important to building trust in medical research and institutions (which can affect patients’ willingness to pursue treatment), promoting fairness for potential participants and their communities, and producing higher-quality biomedical knowledge.15
Overall, as detailed in the Clinical Review report, the ICONIC trial found that maralixibat was well-tolerated and may result in little to no difference in serious adverse events, diarrhea, abdominal pain, and fat-soluble vitamin deficiency compared with placebo. It was, however, uncertain whether maralixibat may have increased alanine transaminase and alkaline phosphatase levels (i.e., liver enzymes that indicate inflammation or damage to the liver cells at higher levels) compared with placebo.
As detailed in the Clinical Review report, the ICONIC trial demonstrated that maralixibat may result in a decrease in sBA levels, the primary efficacy end point evaluated in the trial, compared with placebo. However, it is not possible to determine whether sBA is associated with itch, the symptom of interest in this review, based on the sponsor-submitted evidence or the published literature identified in the database search informing this report.14 While clinical experts noted that the decrease in sBA was supportive of the drug’s biological mechanism of action, they emphasized that the biomarker is likely a co-factor associated with itch, rather than 1 directly correlated with itch, and thus, that assessing efficacy in relieving pruritis for patients with ALGS in clinical practice would require clinical assessment, as discussed further in the context of considerations related to the use of maralixibat.
The ICONIC trial also showed that maralixibat may result in a clinically meaningful decrease in pruritis, as assessed by the Itch Reported Outcome (observer) and Itch Reported Outcome (patient) weekly average morning severity scores, and reported improvements in the Pediatric Quality of Life Inventory total score (parent) and Multidimensional Fatigue Scale score (parent), although the HRQoL evidence was uncertain. A sponsor-funded and sponsor-conducted subanalysis of the ICONIC trial data also found that participants who had a clinically meaningful treatment response to maralixibat experienced meaningful improvements in fatigue compared with nonresponders.16 However, as detailed in the Clinical Review report, the ICONIC trial was an exploratory study with limitations that hinder certainty regarding maralixibat’s short and long-term safety and efficacy. These limitations included a small sample, the lack of control for type I error, and the potential for patients’ or caregivers’ knowledge of treatment assignment to have biased subjective outcomes in favour of maralixibat during the open-label phases. Additionally, the HRQoL measure used in the ICONIC trial (i.e., the Pediatric Quality of Life Inventory) was not optimized for a pediatric population with cholestatic liver disease.16
While evidentiary uncertainty concerning efficacy and safety is not uncommon in the context of drugs for rare diseases, it hinders the assessment of the balance of harms and benefits of using or forgoing maralixibat, which affects clinical decision-making and informed consent.17 The clinical experts emphasized, however, that despite its uncertainty, they considered the available efficacy and safety evidence sufficient to prescribe maralixibat, especially in the absence of effective and safe alternatives to alleviate suffering associated with unremitting cholestatic pruritis. They noted that real-world data collected through registries or databases could add to the current evidence base, especially concerning maralixibat’s long-term safety and effectiveness (including for event-free and transplant-free survival). Of note, although the GALA database includes data from 4 sites in Canada, it currently focuses on collecting clinical, genetic, and laboratory data, rather than information specifically intended to monitor the safety and effectiveness of maralixibat.18 Database or registry data may help address gaps in the uncertain clinical evidence to better inform shared decision-making regarding the use of maralixibat.
As discussed in greater detail in the Pharmacoeconomic Review report in this review, the choice of primary metric of clinical effectiveness in the pharmacoeconomic model (i.e., sBA, for which the sponsor-submitted evidence did not confirm a direct association with pruritis), the uncertainty concerning short-term and long-term efficacy, and limitations of the comparative effectiveness data limits the ability to accurately model and assess the cost-effectiveness of maralixibat. These limitations affect cost-effectiveness analyses for drugs for rare diseases more generally and present challenges to assessing the opportunity costs — or forgone benefits — associated with reimbursing and resourcing maralixibat over other drugs, which complicates resource-allocation decisions at a health-systems level.17
Ethical Considerations in the Use of Maralixibat
There are several important ethical considerations pertaining to the use of maralixibat, including those related to balancing benefits and harms, informed consent, and access.
Balancing Benefits and Harms
As detailed previously, cholestatic pruritis results in a tremendous physical, psychosocial, and financial burden experienced by patients with ALGS and their families,4,10,12,13 including because currently available off-label treatments have adverse events and limited effectiveness6 and because patients must often resort to invasive, life-altering surgical procedures associated with morbidity and mortality, and psychosocial consequences.4,7,9 According to the clinical experts, some patients with unremitting pruritis may also consider suicide.
Given the suffering and burden caused by unremitting pruritis, and the unmet need for therapies that provide effective and consistent relief, clinical experts noted that they would prescribe maralixibat in both pediatric and adult patients with ALGS based on the available efficacy and safety evidence, despite its limitations and uncertainty, because of its potential to reduce pruritis. Similarly, patient group input emphasized that maralixibat was “life-changing” for patients and their families when effectively alleviating itch, as it lessened physical discomfort; improved sleep and fatigue; alleviated the burden of living with ALGS; and improved feelings of anxiety, depression, guilt, hopelessness, and helplessness. As 1 parent noted about their child, “Before the maralixibat, he just cried, and scratched, and we were up every 2 hours, he was being tortured by his body all the time. [After maralixibat] he began to eat, he slept, he could play, he could think.”
Clinical experts noted that, while the evidence was uncertain, the safety profile of maralixibat was favourable, especially considering its potential to address an unmet need. However, they did note the need to engage in shared decision-making with patients centred on their preferences regarding avoiding commonly reported adverse effects such as diarrhea relative to the possibility of experiencing improvement in pruritis. Regarding the uncertain evidence indicating that maralixibat may increase alkaline phosphatase and alanine transaminase levels, the clinical experts noted that elevations in liver enzymes over time are an expected finding in people living with chronic cholestasis and that a 4-week randomization period was insufficient to assess the impact of the drug on these biomarkers. Instead, they noted that slight elevation or a lack of change in liver function tests indicated that maralixibat was affecting a driver of itch, but not the underlying liver disease, which had implications for informed consent, as discussed in the following section. Drug plan input recommended discontinuing maralixibat in patients with elevated liver enzymes until these biomarkers stabilize.
Informed Consent
Patient and clinician group input and clinical experts emphasized that patients, families, and clinicians living with or treating unremitting cholestatic pruritis are often desperate to try anything to relieve this debilitating symptom. The clinical experts acknowledged that this desperation could cause patients, families, and providers to ascribe less value to the evidentiary limitations regarding or possible risks of using maralixibat. The informed consent process should transparently acknowledge possible benefits, adverse effects, and the current state of evidence (including uncertainty in safety and efficacy data, especially in the long term), and that these conversations should continue in a process of shared decision-making as the evidence base grows. When discussing the potential benefits of maralixibat, clinical experts noted that it would be pertinent to emphasize to patients and their families that maralixibat offers symptomatic treatment of cholestatic pruritis, but that it does not cure its underlying cause (i.e., bile duct paucity) nor necessarily slow the progression of liver disease. Patient group input and the published literature emphasized that patients with ALGS and their families benefit from accurate, open, and realistic information about their prognosis and available treatment options, which reduces feelings of uncertainty and empowers them to make informed decisions.10 However, conversations about a lack of curative options for ALGS may also deepen a patient’s and family’s understanding of the chronic and life-threatening nature of the condition and may exacerbate negative emotional experiences and chronic sorrow, necessitating access to appropriate psychosocial supports.10 The clinical experts also emphasized the importance of involving parents in obtaining informed consent and assent in younger children, who cannot consent themselves.
Access
The clinical experts and drug plan input described how, once prescribed, maralixibat was relatively accessible for patients, as it is administered orally and can be delivered successfully by specialist pharmacies to patients across Canada, including in rural and remote communities. However, patient and clinician group input and the clinical experts described potential barriers to accessing maralixibat related to the diagnosis of ALGS, access to specialist care, and criteria used to assess need and efficacy that may disproportionately affect some groups.
The clinician group input and clinical experts emphasized that hepatologists or gastroenterologists should prescribe, monitor, and manage the titration or discontinuation of maralixibat to ensure safe use, given their specialist knowledge regarding ALGS and cholestatic pruritis. For this reason, the previously detailed challenges to accessing a diagnosis, treatment, and care through these specialists, which disproportionately affect people without a known genetic mutation, who live far from specialist tertiary centres or lack the resources to travel or relocate, may also result in barriers to safely using maralixibat. Furthermore, clinical experts noted that a lack of awareness about maralixibat among physicians could create disparities in access, even for people who are able to access specialty care.
The clinical experts also voiced concern that explicit requirements to use standard itch scales or sBA to assess itch severity of treatment efficacy for the purpose of making prescribing or discontinuation decisions could prevent people who may benefit from maralixibat from receiving it. They noted that children may be unable to describe the severity of their itch,13 some patients in their practice report mild or moderate itch despite clinical indicators they are experiencing severe pruritis (e.g., notably excoriated skin, constant scratching behaviours, and significantly impaired sleep and fatigue), and patients and families experiencing meaningful improvements in itch with maralixibat use may not necessarily grade their itch differently on standard itch scales. Of note, Kamath et al.14 reported no association between clinician-observed and caregiver-observed itch severity, as measured by the Clinician Scratch Scale and Itch Reported Outcome (observer) tool, respectively, highlighting the challenge of assessing itch using these scale-based measures. The clinical experts voiced similar concerns about using serological markers to evaluate eligibility for or the effectiveness of maralixibat. As detailed in the Clinical and Pharmacoeconomic Review reports, it is not possible to determine whether a true correlation between sBA and pruritis exists based on the sponsor-submitted evidence, and the clinical experts did not believe that sBA was an accurate predictor of itch severity. They noted that while sBA levels could, in theory, confirm whether patients not responding to treatment were using the medication as prescribed, making sBA monitoring a requirement for use would create inequities in accessing maralixibat, as the test is expensive and not accessible in some regions in Canada.
To ensure equitable access and the avoidance of potentially harmful premature discontinuation, the clinical experts believed that prescribing clinicians with expertise in assessing cholestatic pruritis should determine eligibility and effectiveness based on their expert, subjective assessment of information collected during a physical examination (e.g., the extent and pattern of skin excoriations and scratching behaviours) and conversations with a patient and their family (i.e., to obtain information about the severity, duration, and frequency of itch; duration and quality of sleep and fatigue; and the extent to which itching impairs function in school, work, and social engagements). They noted that these assessments could be augmented, but not replaced, by data collected through standard itch scales. As 1 clinical expert summarized, “We’re treating suffering…it’s not as though the degree of itch numerically somehow correlates to some other biologic process that we’re also trying to treat…[if] my patient thinks they’re suffering less [while taking maralixibat], then I need to respect that.” The clinical experts confirmed that they could conduct these assessments virtually for clients living far from and unable to travel to in-person visits.
Health Systems Considerations
The reimbursement of maralixibat for cholestatic pruritis in ALGS raises several ethical considerations relating to health systems and resource considerations, including opportunity costs, distributive justice, the ongoing need for specialist care and monitoring, and equitable access and funding across Canada.
While acknowledging that maralixibat is an expensive medication, the clinical experts and the published literature noted that safe and effective pharmacological treatments for unremitting cholestatic pruritis in ALGS could decrease health care resource utilization associated with more invasive methods of managing the symptom, including liver transplant.19 As detailed in the Clinical Review report, despite its limitations, the sponsor-submitted natural-history comparison study may supplement evidence supporting maralixibat as a potential therapeutic option to improve event-free and transplant-free survival in patients with ALGS. Considering further evidence regarding whether maralixibat decreases the long-term demand for liver transplant for patients with ALGS would have implications for understanding the opportunity costs of implementing, or not implementing, the medication within the health care system, as well as for distributive justice and resource-allocation decisions for liver transplant.19
Additionally, the clinical experts did not believe that using maralixibat would significantly increase the frequency of visits to a specialist, monitoring, blood draws, or other testing beyond that already needed for a person with chronic cholestasis. Given the availability of genetic testing to confirm diagnosis reported in the literature and by the clinical experts, it is likely that children with an ALGS diagnosis who are eligible for maralixibat would already have undergone this testing; therefore, making it a requirement for treatment likely would not increase health care spending, although it could pose a barrier to access for the 4% of people with ALGS who do not have a known mutation.5,6
As detailed in the Pharmacoeconomic Review report for this review, the use of sBA as the primary metric of clinical effectiveness in the economic model, the uncertainty of evidence, and limitations in the comparative effectiveness data limit the ability to model and assess the maralixibat’s cost-effectiveness accurately. In turn, this limits the ability to consider opportunity costs or forgone benefits associated with its reimbursement in the context of limited health care budgets. The clinical experts emphasized, however, that while maralixibat is an expensive medication, the rarity of ALGS and the limited number of eligible patients in Canada may limit its overall budget impact.
Justifications grounded in cost-effectiveness reflect utilitarian reasons, which aim to maximize the overall benefit for the greatest number of people.20,21 However, drugs for rare diseases, including maralixibat, are often not deemed to be cost-effective due to their high costs or uncertain magnitude of clinical benefit.20,21 In cases where drugs for rare diseases are not cost-effective or when cost-effectiveness cannot be determined, other considerations of distributive justice may inform and justify reimbursement decisions. In the context of drugs for pediatric rare diseases, these may include justifications based on the prioritization of principles of beneficence or nonabandonment and values such as equity (i.e., recognizing unequal circumstances of patients with significant unmet needs or prioritizing the worse-off) and fair innings (i.e., providing each person with an opportunity to live in good health for a normal life span).17,20-22
Finally, the patient group input and clinical experts emphasized the need for an effective pan-Canadian approach to reimbursing and providing access to maralixibat for patients across the country to ensure equitable access to the medication, including in locations far from tertiary care centres. They noted that funding decisions regarding maralixibat could have implications for equitable access within Canada due to inconsistencies in drug coverage across different provinces and territories.
Limitations
There is very little published literature discussing ethical considerations related to using maralixibat for treating cholestatic pruritis in ALGS, given both the rarity of the disease and the novelty of the drug under review. Nonetheless, this does not imply that ethical considerations in the context of maralixibat for cholestatic pruritis in ALGS are absent. This review of ethical considerations was augmented by drawing from additional resources collected in the course of this Reimbursement Review, including patient group, clinician group, and drug program input, and discussion with clinical experts, as well as engagement with CADTH Clinical and Pharmacoeconomic Review teams, to provide a more comprehensive understanding of the ethical considerations related to the use of maralixibat for the treatment of cholestatic pruritis in ALGS.
Although this Ethics Review report drew on and considered patient group, clinician group, drug program, and clinical expert input, it is possible that more direct engagement with key stakeholders (e.g., direct interviews with patients, caregivers, family members, or decision-makers) on their specific experiences with ALGS and/or maralixibat could have offered additional relevant ethical considerations or domains of analysis.
Conclusion
Input from patient groups, clinician groups, and provincial drug programs, as well as direct engagement with clinical experts and published literature, were reviewed for ethical considerations relevant to the use of maralixibat for the treatment of cholestatic pruritis in patients with ALGS. Ethical considerations in the context of ALGS highlighted the significant physical, psychosocial, and financial impact of the life-threatening condition and its associated cholestatic pruritis on patients, families, and caregivers, as well as harms associated with delays and difficulties in accessing a timely diagnosis and routine treatment and care. Additionally, there is a significant unmet need for an effective treatment for cholestatic pruritis in ALGS due to the limited efficacy of and adverse effects associated with currently available off-label pharmacotherapies and the invasive, life-altering nature of surgical treatment alternatives such as liver transplant. Clinical trial evidence indicated that maralixibat may result in a clinically meaningful decrease in pruritis and in little to no difference in serious adverse events compared with placebo; however, there is evidentiary uncertainty concerning its safety and efficacy (particularly concerning its effect on long-term treatment outcomes and HRQoL), which limits the assessment of clinical benefits and harms associated with its use as well as the pharmacoeconomic assessment of cost-effectiveness.
The clinical experts noted that they would prescribe maralixibat based on the currently available evidence, given the drug’s potential to address a great unmet need for relieving unremitting pruritis with what they considered a favourable safety profile. Patient input similarly emphasized that families were desperate for new treatments providing relief of pruritis and the life-changing improvements maralixibat had on HRQoL when effectively alleviating itch. Robust informed-consent processes are required in both pediatric and adult contexts, including to discuss the limitations of the clinical evidence and the likelihood that the medication does not halt the progression of liver disease. Equitable access requires attending to potential diagnostic, geographic, and monitoring-related barriers to access. Ethical considerations for health systems related to the implementation of maralixibat highlight the challenges of funding decisions for high-cost drugs for rare diseases, assessments of opportunity costs, and the fair allocation of scarce resources, as well as issues related to pan-Canadian approaches to providing equitable reimbursement and access.
Table 1: Details of Included Publications
First author, year | Publication type | Objective | Key ethical considerations | Funding source |
|---|---|---|---|---|
Ayoub, 20206 | Review | To provide a broad clinical overview of ALGS, with a specific focus on diagnostic challenges to gastroenterologists and pathologists, as well as current and future approaches to its management |
| Authors received funding from, or acted as contractors for, Albireo Pharma, Inc., Audentes, and Mirum Pharmaceuticals, Inc. |
Bilhartz, 20169 | Review | To discuss diagnoses, uniqueness, or importance to pediatric liver transplant; the evaluation of a pediatric patient for liver transplant; the system for allocating them a new liver; and postoperative concerns that are unique to the pediatric population |
| None reported |
Chan, 20238 | Review | To describe the epidemiology, clinical features, diagnostic testing, treatment, prognosis, and transplant outcomes of 4 childhood cholestatic liver diseases, including ALGS |
| National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (Award Number T32DK007180) |
Denburg, 202222 | Interview study | To elicit and understand the social values that influence decision-making for public funding of pediatric drugs in Canada |
| The Canadian Institutes of Health Research, the Canadian Child Health Clinician Scientist Program, and the Pierre Elliott Trudeau Foundation |
Glenn, 201410 | Interview study | To explore the lived experiences of mothers of children with ALGS in using online health communications to manage their chronic sorrow |
| None reported |
Gwaltney, 202212 | Interview study | To identify salient concepts in pediatric cholestatic liver disease, develop novel patient-reported and observer-reported outcome instruments, and establish the instruments’ content validity |
| Authors declared employment by Albireo Pharma, Inc. and/or Patient-Centered Solutions, Inc. |
Hobbins, 20225 | Panel discussion summary | To recap a peer exchange discussion among clinicians on the management of ALGS, with topics that included the characteristics of the condition, diagnostic criteria, and genetic testing |
| None reported |
Kacetl, 202220 | Systematic review | To identify ethical questions related to rare diseases and orphan drugs and ethical principles or approaches applied to address them |
| University of Hradec Králové Long Term Development Plan |
Kamath, 20184 | Systematic review | To perform a systematic review of the epidemiology, natural history, and burden of ALGS, with a focus on the liver component |
| Authors received funding or writing assistance from, or reported employment with Alexion Pharmaceuticals, Inc., Jazz Pharmaceuticals Inc., Oxford PharmaGenesis (with funding from Shire International GmbH), Retrophin Inc., and Shire International GmbH |
Kamath, 201813 | Qualitative interview study | To develop a clinical outcome assessment for itching in children with cholestatic pruritis |
| Authors received funding from, declared past or present employment with, or owned stocks in Adelphi Values Ltd., Amplyx Pharmaceuticals, Inc., Bristol-Myers Squibb Company, Endpoint Outcomes, Hyperion Therapeutics Inc. (now Horizon Pharmaceuticals, Inc.), Lumena Pharmaceuticals Inc./ Shire International GmbH, National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases-sponsored ChiLDReN Network, Oxford PharmaGenesis (funded by Shire International GmbH), Patient-Centred Outcomes Research Institute, and Premier Research |
Kamath, 202014 | Subanalysis of clinical trial data | To characterize pruritis, as assessed by the ItchRO instrument, relative to current scales, HRQoL assessments, and associated biomarkers in participants with ALGS at baseline in the ITCH trial |
| Authors declared receiving grants or funding from, or acting as consultants to AbbVie Corporation, Albirea, Audentes, Gilead Sciences, Inc., Intercept Pharmaceuticals, Inc., LogicBio Therapeutics, Inc., Metacrine, Inc., Merck and Co., Inc., Mirum Pharmaceuticals, Inc., the National Center for Advancing Translational Sciences, the National Institute of Diabetes and Digestive and Kidney Diseases, Retrophin, Inc., Shine Pharmaceuticals Limited, and Spruce Biosciences, Inc.; 1 author declared owning stocks in Askelpion Pharmaceuticals, LLC |
Kamath, 202316 | Subanalysis of clinical trial data | To assess the impact of treatment response to maralixibat on HRQoL in children with ALGS |
| Authors received funding or writing support from, or were employed by or contractors for Albireo Pharma, Inc., Audentes, Analysis Group, Inc. (which receives funding from Mirum), CRTS Health Interactions, Laboratories, Mirum Pharmaceuticals, Inc., Third Rock Ventures, Vivet Therapeutics |
Martin, 202111 | Database study | To characterize the demographic, clinical, and socioeconomic factors of liver disease–associated admissions among children between 2005 and 2015 using the Office of Statewide Health Planning and Development hospital claims (California) |
| Transplant and Tissue Engineering Center of Excellence at Stanford University |
Miloh, 202319 | Database study | To assess the economic burden of liver transplant on patients with rare cholestatic liver diseases, including ALGS, in the US |
| Authors received funding from, declared past or current employment with, or owned stocks in AbbVie Corporation, Albireo Pharma, Inc., Analysis Group, Inc., Audentes, Arrowhead Pharmaceuticals, Inc., BioMarin Pharmaceuticals, Inc., Dicerna Pharmaceuticals, Inc., Encoded Therapeutics, Inc., Gilead Sciences, Inc., MedinCell S.A., Merck and Co., Inc., Mirum Pharmaceuticals, Inc., Takeda Pharmaceutical Company, Travere Therapeutics, Inc., and Vertex Pharmaceuticals Incorporated |
Postma, 202221 | Review | To identify ethical challenges associated with assessing the value of orphan drugs using conventional cost-effectiveness analysis as well as alternative and supplemental approaches |
| Pfizer Inc. |
Schwartz, 202315 | Perspective | To identify the ethical considerations related to diverse clinical trial participation |
| Authors received support from Abbott Laboratories, Acorai, Boehringer Ingelheim International GmbH, Cytokinetics Incorporated, Edwards Lifesciences Corporation, Ionis Pharmaceuticals, Inc., Merck and Co., Inc., MyoKardia, Inc., Novartis AG, Aetna Inc., Lown Institute, Tufts Medical Center, VBID Health |
Wagner, 202217 | Review and expert opinion | To identify ethical challenges for appraising interventions for rare diseases, including key ethical tensions as well as approaches and principles for addressing these challenges |
| Institut national d’excellence en santé et en services sociaux (INESSS) |
ALGS = Alagille syndrome; HRQoL = health-related quality of life; HTA = health technology assessment; ItchRO = Itch Reported Outcome; ItchRO(Obs) = Itch Reported Outcome (observer); PedsQL = Pediatric Quality of Life Inventory.
References
1.EUnetHTA. EUnetHTA Joint Action 2 Work Package 8, HTA Core Model version 3.0. 2016; http://www.htacoremodel.info/BrowseModel.aspx. Accessed 2023 Jul 14.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37:e17. PubMed
3.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
4.Kamath BM, Baker A, Houwen R, et al. Systematic Review: The Epidemiology, Natural History, and Burden of Alagille Syndrome. J Pediatr Gastroenterol Nutr. 2018;67(2):148-156. PubMed
5.Hobbins K. Clinicians discuss different paths to diagnosing Alagille syndrome. Contemp Pediatr. 2022;39(7):10-11.
6.Ayoub MD, Kamath BM. Alagille Syndrome: Diagnostic Challenges and Advances in Management. Diagnostics (Basel). 2020;10(11). PubMed
7.Vandriel S, Li LT, She H, et al. Natural History of Liver Disease in a Large International Cohort of Children with Alagille syndrome: Results from The GALA Study. Hepatology. 2022;77. PubMed
8.Chan AP, Venick RS. Childhood Cholestatic Liver Diseases that Persist Into Adulthood: Lessons for the Adult Gastroenterologist. J Clin Gastroenterol. 2023;06:06.
9.Bilhartz JL, Shieck VL. Pediatric Liver Transplantation. Crit Care Nurs Q. 2016;39(3):281-295. PubMed
10.Glenn AD. Using Online Health Communication to Manage Chronic Sorrow: Mothers of Children with Rare Disease Speak, George Mason University; 2014.
11.Martin M, Zou B, Hoang J, Jeong D, Bensen R, Nguyen MH. Racial and Socioeconomic Disparities in Hospitalization of Pediatrics with Liver Disease from 2005 to 2015. Dig Dis Sci. 2021;66(7):2240-2249. PubMed
12.Gwaltney C, Bean S, Venerus M, et al. Development of the Patient- and Observer-Reported PRUCISION Instruments to Assess Pruritus and Sleep Disturbance in Pediatric Patients with Cholestatic Liver Diseases. Adv Ther. 2022;39(11):5126-5143. PubMed
13.Kamath BM, Abetz-Webb L, Kennedy C, et al. Development of a Novel Tool to Assess the Impact of Itching in Pediatric Cholestasis. Patient. 2018;11(1):69-82. PubMed
14.Kamath BM, Spino C, McLain R, et al. Unraveling the Relationship Between Itching, Scratch Scales, and Biomarkers in Children With Alagille Syndrome. Hepatol. 2020;4(7):1012-1018. PubMed
15.Schwartz AL, Alsan M, Morris AA, Halpern SD. Why Diverse Clinical Trial Participation Matters. N Engl J Med. 2023;388(14):1252-1254. PubMed
16.Kamath BM, Goldstein A, Howard R, et al. Maralixibat Treatment Response in Alagille Syndrome is Associated with Improved Health-Related Quality of Life. J Pediatr. 2023;252:68-75.e65. PubMed
17.Wagner M, Goetghebeur M, Ganache I, et al. HTA challenges for appraising rare disease interventions viewed through the lens of an institutional multidimensional value framework. Expert Rev Pharmacoecon Outcomes Res. 2022;23:143 - 152. PubMed
18.The Global ALagille Alliance Study. The GALA Study 2022; https://www.galastudy.com/. Accessed 2023 Jul 14.
19.Miloh T, Goldstein A, Howard R, et al. Costs of pediatric liver transplantation among commercially insured and Medicaid-insured patients with cholestasis in the US. Liver Transpl. 2023;08:08.
20.Kacetl J, Marešová P, Maskuriy R, Selamat A. Ethical Questions Linked to Rare Diseases and Orphan Drugs – A Systematic Review. Risk Manag Healthc Policy. 2020;13:2125-2148. PubMed
21.Postma M, Noone D, Rozenbaum M, et al. Assessing the value of orphan drugs using conventional cost-effectiveness analysis: Is it fit for purpose? Orphanet J Rare Dis. 2022;17. PubMed
22.Denburg AE, Giacomini M, Ungar W, Abelson J. Ethical and Social Values for Paediatric Health Technology Assessment and Drug Policy. Int J Health Policy Manag. 2022;11(3):374-382. PubMed
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Patient to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be patient to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.