CADTH Reimbursement Review
Somapacitan (Sogroya)
Sponsor: Novo Nordisk Canada Inc.
Therapeutic area: Growth hormone deficiency (GHD)
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
AHV
annualized height velocity
ANCOVA
analysis of covariance
BMI
body mass index
CI
confidence interval
CORD
Canadian Organization for Rare Disorders
CrI
credible interval
DIC
deviance information criterion
GeNeSIS
Genetic and Neuroendocrinology of Short-Stature International Study
GHD-CIM
Growth Hormone Deficiency–Child Impact Measure
EMA
European Medicines Agency
ETD
estimated treatment difference
FE
fixed effects
FAS
full analysis set
G-DAT
Growth Hormone Device Assessment Tool
GH
growth hormone
GHD
growth hormone deficiency
GHD-CTB
Growth Hormone Deficiency–Child Treatment Burden
GHD-PPQ
Growth Hormone Patient Preference Questionnaire
GHD-PTB
Growth Hormone Deficiency–Parent Treatment Burden
GRADE
Grading of Recommendations Assessment, Development, and Evaluation
hGH
human growth hormone
IGF-1
insulin-like growth factor 1
IGFBP-3
insulin-like growth factor binding protein 3
ITC
indirect treatment comparison
LAR
legally authorized representative
MID
minimal important difference
MMRM
mixed-model for repeated measures
NMA
network meta-analysis
PY
patient-year
RCT
randomized controlled trial
RE
random effects
SD
standard deviation
SDS
standard deviation score
SGA
small for gestational age
SLR
systematic literature review
TB-CGHD-O
Treatment Burden Measure–Child Growth Hormone Deficiency–Observer
TB-CGHD-P
Treatment Burden Measure–Child Growth Hormone Deficiency–Parent
TRIM-CGHD-O
Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Somapacitan (Sogroya), 5 mg/1.5 mL (3.3 mg/mL), 10 mg/1.5 mL (6.7 mg/mL), 15 mg/1.5 mL (10 mg/mL) as a prefilled pen for subcutaneous injection |
Sponsor | Novo Nordisk Canada Inc. |
Indication | For the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone (growth hormone deficiency).a |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | July 26, 2023 |
Recommended dose | Recommended initiation dose: 0.16 mg/kg body weight once weekly. The maintenance dose can be adjusted based on the patient and their response. |
NOC = Notice of Compliance.
aSogroya was approved by Health Canada on July 26, 2023. According to the indication specified in the product monograph (point 1.1), “Pediatrics (2.5 years old to epiphyseal fusion): The efficacy and safety of Sogroya in pediatric patients aged 2.5 years to 11 years experiencing growth failure due to growth hormone deficiency have been established in clinical trials. The efficacy and safety of Sogroya have not been established in patients younger than 2.5 years of age. Data on the efficacy and safety of Sogroya in patients aged from 12 to less than 18 years are limited. Pediatric patients with a history or presence of malignancy, including intracranial tumours, were not studied in clinical trials.”1
Introduction
Growth hormone deficiency (GHD) is a rare disease caused by impaired secretion of growth hormone (GH) by the pituitary gland, which affects patients’ growth, body composition, metabolic profile, bone mineral density, and quality of life.2,3 In a cohort of 850 children in Canada treated with GH, among whom 526 had GHD, 72% (379 out of 526) were diagnosed with predominantly organic rather than idiopathic GHD, particularly intracranial tumours and congenital pituitary abnormalities.4 Congenital causes of GHD can be linked to a number of gene mutations; some of these mutations may be located on GH1, HESX1, X-linked recessive genes such as SOX3 and BTK, and many others.2 The physical manifestation of GHD can vary depending on the types of cells affected, the age of onset, and the combination of genetic mutations.5 Estimates for the prevalence of pediatric GHD are sparse, with no specific Canadian data, although studies across Europe, the US, and China suggest an estimated prevalence ranging from 1 in 30,000 to 1 in 5,600.3,6-8 Estimates of pediatric GHD suggest a prevalence range of 1 in 4,000 to 10,000 children worldwide, suggesting a prevalence of approximately 1,600 children in Canada.9,10
GH stimulation testing is considered the standard diagnostic tool for GHD. The treatment of GHD is injections of synthetic GH.11 Canadian and US guidelines recommend treatment with GH in cases of extreme shortness in children and adolescents with GHD to attain normal adult height.12 Treatment with GH should begin as soon as possible for patients to optimize their growth velocity and final adult height,9,13 and be continued until a patient reaches their full adult height and bone maturity, or when their height velocity (HV) is less than 2 cm/year.14-16 The clinical expert consulted for this review indicated that somatropin and somatrogon are administered as daily or weekly subcutaneous injections for the treatment of pediatric GHD. Somatropin, administered as a daily subcutaneous injection, has traditionally been the primary GH used for the treatment of GHD. In March 2022, somatrogon (Ngenla), a Health Canada–approved GH, received a reimbursement recommendation from CADTH for the treatment of pediatric GHD as a weekly subcutaneous injection,14 but received a negative funding recommendation from the Institut national d’excellence en santé et en services sociaux (INESSS) due to the potential for greater pain at the injection site and the need for higher doses.17 According to input from the sponsor and the clinical expert consulted by CADTH, treatment goals for GHD include optimizing final adult height, restoring metabolic functions associated with GHD, reducing injection burden, improving treatment adherence, and optimizing quality of life. According to clinical expert input, 1 of the limitations associated with current treatments is that they require injections and daily (or near daily) injections, which can lead to suboptimal adherence, resulting in suboptimal clinical outcomes, e.g., the treatment goal of improvement in height is affected.3,18,19
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of somapacitan 5 mg/1.5 mL (3.3 mg/mL), 10 mg/1.5 mL (6.7 mg/mL), and 15 mg/1.5 mL (10 mg/mL) as a prefilled pen for subcutaneous use in the long term treatment of pediatric patients experiencing growth failure due to an inadequate secretion of endogenous GH.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, the Canadian Organization for Rare Disorders (CORD) and the MAGIC Foundation, provided input for the treatment of GHD. CORD recruited participants through a patient membership list and parents attending a European summit. It then conducted interviews, whereas the MAGIC Foundation gathered data from surveys. A total of 12 parents (6 from the list and 6 from the summit) participated in the interviews conducted by CORD. Among these 12 participants, 4 were from Canada (Ontario), 2 were from the US, and 6 were from Europe. All of the participants who responded to the MAGIC Foundation’s surveys were from the US, and none of their current members in Canada had experience with a long-acting GH (LAGH). The children represented in the CORD input ranged in age from 4 to 15 years old, and those from the MAGIC Foundation input were between 3 and 18 years of age.
When parents were asked by CORD about the impact of the disease on patients’ and caregivers’ day-to-day life and quality of life, they expressed going through a variety of emotions, such as denial, blame, sadness, acceptance, and compassion. Parents from both groups reported a variety of psychological and social impacts on the child and the family, particularly due to short stature. In the MAGIC Foundation’s input, parents mentioned that their children were shorter than their peers, were fatigued, lacked concentration and appetite, had poor stamina, and were often very sick before beginning GH treatment.
Patients from both groups were reported to have been on GH therapy. All patients in the CORD group were reported to have experience with daily subcutaneous injections of somatropin, and 4 patients were reported to have current experience with LAGH therapy (somatrogon, lonapegsomatropin, and somapacitan-beco) through clinical trials, compassionate access, or reimbursement or insurance. On the other hand, some patients from the MAGIC Foundation group were reported to have experience with both daily and weekly injections, whereas others were reported to be strictly on daily injections.
Parents from the CORD group described the daily administration of an injection every night as the most consistent challenge. These parents also recognized the importance of GH therapy, despite the challenges and worries about the future. Parents from the MAGIC Foundation group mentioned the high cost of GH treatment and dependence on insurance companies in getting the treatment for children in the US. When parents were asked by the CORD group about the outcomes to consider when evaluating new therapies, an injection that lasted longer and was easier to administer was a desired change. While describing the experience with the current drug under review, all 4 respondents having experience with LAGH therapy shared positive feedback, such as positive impact on the child’s and the family’s quality of life; some described this impact as “transformational” and “life-changing.”
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The expert indicated that the current treatment paradigm for children with GHD is to offer recombinant GH. The treatment goals are: to restore height by improving HV (with the target of achieving a near-final adult height that is close to the patient’s midparental heights), to restore metabolic health, to address hypoglycemia (especially in the neonatal or infantile periods), and to restore well-being. The expert pointed out that several brands of somatropin are available to treat GHD. They noted that somapacitan can be used in the treatment of pediatric GHD by those who are willing to try it or who are using somatropin and want to switch to once-weekly injections. The expert noted that the patients most in need of somapacitan would be those who experience significant pain or anxiety from the injections to the extent that there is a threat to optimal adherence to daily somatropin. The expert indicated that treatment responses include change in absolute height and height standard deviation score (SDS), change in HV and HV SDS, and change in insulin-like growth factor 1 [IGF-1] level and IGF-1 SDS). According to the clinical expert, patients prescribed somapacitan should be under the care of pediatric endocrinologists and pediatric endocrine nurses in either community settings or academic referral centres.
Clinician Group Input
Clinician group input on the review of somapacitan was received from Canadian Pediatric Endocrinology Nurses. A total of 5 nurses provided input for this review.
Canadian Pediatric Endocrinology Nurses mentioned that daily somatropin injections are used as the current treatment paradigm for GHD. These injections are administered to increase growth, stabilize blood sugar levels, increase bone density, and increase muscle development. The Pediatric Endocrinology Nurses group described the treatment gaps or unmet needs of currently available treatments. Issues with current treatments include: poor compliance in patients with daily injections, anxiety with daily injections, lack of availability of GH, and the need for a treatment with improved compliance, better tolerance, and formulations with improved convenience. The group indicated that somapacitan could be used as a first-line treatment for GHD if approved or funded.
While describing which patients would be best suited for treatment with the drug under review, the clinician group mentioned those who experience needle anxiety, have compliance issues, are in a complex social situation, or have remote living conditions. The group added that patients with GHD can be identified by, for example, clinician examination, GH stimulation testing, bone age, and IGF-1 level. The group emphasized that without GH treatment (either daily or weekly), patients with GHD will not grow and could have hypoglycemia, decreased bone density, poor muscle development, and altered body composition. The clinician group pointed out that improved growth velocity and normalized glucose in infants would be considered a clinically meaningful response to treatment. The group also added that factors such as achieving final adult height, closed epiphyses, and a growth rate of less than 2 cm/year should be considered when deciding to discontinue treatment with the drug under review. The clinician group noted that the patients must be diagnosed, treated, monitored, and prescribed by a pediatric endocrinologist.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for somapacitan:
relevant comparators
considerations for initiation of therapy
consideration for continuation or renewal of therapy
considerations for discontinuation of therapy
consideration for prescribing of therapy
generalizability
system and economic issues.
The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug program. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
One pivotal phase III, open-label, randomized controlled trial (RCT) comparing somapacitan (0.16 mg/kg once weekly) with Norditropin (somatropin) (0.034 mg/kg once daily) via subcutaneous infusion in prepubertal children with GHD who had no prior exposure to GH therapy or IGF-1 treatment (N = 200) was identified. Patients were randomized to either somapacitan or Norditropin (somatropin) in a 2:1 ratio for a 52-week randomized controlled period. The primary end point was estimated mean at week 52 in HV (cm/year) at week 52. The key secondary end points were change from baseline in height SDS and HV SDS at week 52. The key exploratory end points were Growth Hormone Deficiency–Child Impact Measure (GHD-CIM) as well as estimated mean scores in Growth Hormone Deficiency–Child Treatment Burden (GHD-CTB) and Growth Hormone Deficiency–Parent Treatment Burden (GHD-PTB) at week 52. HV reflects the change rate in height over time and was converted to be expressed in cm/year in the current review. Height SDS and HV SDS are end points that reflect whether the auxologic parameters are within a normal range (usually −2 to 2) considering age and gender, and standards of these measurements have been developed and used in clinical settings. The GHD-CIM, GHD-CTB, and GHD-PTB are disease-specific questionnaires that measure the impact or burden of GH treatment on children with GHD and their caregivers in terms of symptoms, physical functioning, social well-being, emotional well-being, and interference in daily life activities. Each of these 3 questionnaires has subdomains and total scores that are presented on a normalized range of 0 to 100, with a lower score indicating a better health state.
At baseline, the mean age was 6.38 years (standard deviation [SD] = 2.23) in the somapacitan arm and 6.43 years (SD = 2.42) in the Norditropin (somatropin) arm. More male patients (74.5%) were enrolled than female patients (25.5%). The majority of patients were white (57%) followed by Asian (37%), and 88% had GHD that was idiopathic (cause unknown). The median peak level of GH was higher in the somapacitan arm (5.2 mcg/L) than in the Norditropin (somatropin) arm (3.9 mcg/L). In terms of concomitant medications at baseline, a lower proportion of patients used thyroid hormones in the somapacitan arm (8.3%) than in the Norditropin (somatropin) arm (14.7%). After initiation of randomization, overall, concomitant medications were used by a higher proportion of patients in the somapacitan arm (66.7%) than in the Norditropin (somatropin) arm (58.8%), based on the detailed documentation of patient records; the difference did not have a potential impact on study results.
Efficacy Results
The key efficacy results from the REAL 4 trial are summarized in Table 2. The full analysis set was used for the auxologic response outcomes, and the observation datasets were used for the patient-reported outcomes.
Auxologic Response
The mean HV at baseline was 4.3 cm/year (SD = 1.4) in the somapacitan arm and 4.1 cm/year (SD = 1.4) in the Norditropin (somatropin) arm. The estimated mean at week 52 in HV was 11.2 cm/year and 11.7 cm/year in the somapacitan and Norditropin (somatropin) arms, respectively, with an estimated treatment difference (ETD) of −0.5 cm/year (95% confidence interval [CI], −1.1 to 0.2), which demonstrated noninferiority of somapacitan to Norditropin (somatropin) based on a prespecified noninferiority margin of −1.8 cm/year.
The improvements in height SDS and HV SDS (supportive secondary end points that were measured as change from baseline scores at week 52) were comparable between the 2 treatment arms. The baseline mean height SDS was −2.99 (SD = 1.02) in the somapacitan arm and −3.47 (SD = 1.52) in the Norditropin (somatropin) arm. The ETD for height SDS was −0.05 (95% CI, −0.18 to 0.08). The estimated mean change from baseline scores was 1.25 and 1.30 in the somapacitan and Norditropin (somatropin) arms, respectively. The baseline mean HV SDS was −2.35 (SD = 1.51) in the somapacitan arm and −2.52 (SD = 1.55) in the Norditropin (somatropin) arm. The ETD for HV SDS was −0.78 (95% CI, −1.63 to 0.08). The estimated mean change from baseline scores was 8.05 and 8.82 in the somapacitan and Norditropin (somatropin) arms, respectively.
Patient-Reported Outcomes
The improvements were comparable between the treatment arms at week 52 in total GHD-CIM scores (ETD = 1.8 points; 95% CI, −2.9 to 6.6) and GHD-CTB scores (ETD = −2.4 points; 95% CI, −5.7 to 0.9). The result of the GHD-PTB total score was in favour of somapacitan compared with Norditropin (somatropin) at week 52, with an ETD of −6.0 points (95% CI, −10.0 to −2.1).
Harms Results
The key harm results from the randomized controlled period of the REAL 4 trial are summarized in Table 2. The full analysis set was used for all of the safety outcomes.
During the 52-week treatment period, adverse events (AEs) were reported by 71.2% of patients who received somapacitan compared with 60.3% of patients who received Norditropin (somatropin). The most frequently reported AEs were headache (12.1%), nasopharyngitis (11.4%), and pain in extremity (9.1%) in the somapacitan arm, and nasopharyngitis (10.3%), pyrexia (10.3%), and headache (8.8%) in the Norditropin (somatropin) arm.
No withdrawal from treatment due to treatment-emergent AEs or deaths were reported among patients in either arm.
The occurrences of the 2 notable harms were comparable between the treatment arms at week 52, with a relative risk of |||| |||| ||| |||| || ||||| for injection-site reactions (5.3% versus 5.9% in the somapacitan and Norditropin [somatropin] arms, respectively) and |||| |||| ||| |||| || |||||| for injection-site pain (1.5% in both arms). Of note, the relative risk and 95% CI for injection-site reactions and injection-site pain were not part of the statistical analysis plan and were requested by CADTH to allow for the imprecision of the findings to be assessed.
Other Results
The other outcome identified as relevant and important, treatment discontinuation (or adherence to therapy), is summarized in Table 2. One patient was discontinued from treatment with somapacitan due to a violation of the inclusion and/or exclusion criteria. No patients discontinued treatment in the Norditropin (somatropin) arm. The mean therapy adherence rate, measured with a patient e-diary device for electronic data recording, was higher in the somapacitan arm than in the Norditropin (somatropin) arm (96% versus 88%).
Critical Appraisal
Appropriate methods of randomization were used. The proportion of patients who used thyroid hormones was lower in the somapacitan arm than in the Norditropin (somatropin) arm (8.3% versus 14.7%); whether this difference might impact the study results was uncertain, according to the clinical expert consulted by CADTH. The efficacy and harms outcomes were analyzed using the full analysis set (FAS); all the patient-reported outcomes were analyzed for the observation datasets in countries where these outcomes were available; the impact of the missing data in 4 countries was unknown. The open-label study design could potentially increase the risk of bias for the subjective assessment of patient-reported outcomes, such as health-related quality of life, and AEs such as injection-site reactions (including pain, bruising, hematoma, and swelling) and injection-site pain. Ethnicity and race are significant predictors of HV; however, the REAL 4 trial did not stratify this factor (only Japan was separated from the rest-of-the-world region, possibly because of different peak GH levels used in GHD diagnosis). Whether the lack of adjustment by different regions in the analysis might affect the results was uncertain. The study sample size calculation in the REAL 4 trial was based on an assumption of an SD of 3.5 cm/year and a noninferiority margin of −1.8 cm/year for HV. Most of the patients enrolled in the REAL 4 trial were from the US (|||), Japan (|||), Russia (|||||), India (|||), Korea (||), and Ukraine (||), among others. ||| ||||||| (|||| of overall study population) ||| ||| allocated to the somapacitan group was from Canada and whether |||| ||||||| belonged to an Indigenous population was unknown. A total of 57.0% of the patients were white and 37.0% were Asian. At baseline, the patients were aged between 2.5 years and 11 years for boys, and 2.5 years and 10 years for girls. Overall, there was a greater difference in the proportion of boys (74.5%) than girls (25.5%) in the REAL 4 trial compared with the Genetic and Neuroendocrinology of Short-Stature International Study (GeNeSIS) study (63% male versus 37% female in the GHD cohort of patients in Canada).4 For context, the GeNeSIS study is a phase IV prospective observational study that evaluated the outcomes of GH treatment in pediatric patients in Canada that compared the findings for the American pediatric patients in the study versus the overall global population. The REAL 4 trial enrolled only patients who had no prior exposure to GH or IGF-1 treatment. The clinical expert commented that these differences between patients in the REAL 4 trial and those encountered in Canadian clinical practice are less likely to impact the trial results. The majority of the patients in the REAL 4 trial had idiopathic GHD (88.0%) versus organic (12%),20 which was different from the GHD patient cohort in the GeNeSIS study (28% had idiopathic and 72% had organic GHD).4 The clinical expert indicated that idiopathic GHD may be accompanied by other conditions that might influence GHD; thus, it was uncertain whether the results would be impacted by the fact that this patient characteristic in the GeNeSIS trial differs from that in the REAL 4 trial. The clinical expert noted there were patients excluded from the trial that clinicians would want to treat with GH therapy, if needed, such as patients with cancerous brain tumours and patients with a condition that precludes them from a height evaluation (e.g., patients with cerebral palsy who cannot stand). According to the clinical expert consulted by CADTH, the concomitant medications, the comparator used, and the administration of the study treatments in the REAL 4 trial were reflective of those encountered in Canadian clinical practice.
Summary of Findings and Certainty of the Evidence Using the Grading of Recommendations Assessment, Development, and Evaluation Framework
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) framework was used to assess the certainty of the evidence for outcomes considered most relevant to inform deliberations by CADTH’s expert committee; a final certainty rating was determined as outlined by the GRADE Working Group.21,22 Following the GRADE approach, the evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and the input received from patient and clinician groups and the public drug plans. The following list of outcomes was finalized with the input of the members of the CADTH expert committee: HV (cm/year, measured as estimated mean at follow-up), height SDS, HV SDS and GHD-CIM (measured as change from baseline in these values), GHD-CTB and GHD-PTB (measured as estimated mean at follow-up), injection-site reactions and injection-site pain (measured as occurrence of these AEs), and treatment discontinuation (measured as the proportion of patients who discontinued the treatment).
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty-of-evidence assessment was the presence or absence of an important effect based on the thresholds for HV, height SDS, and HV SDS informed by the clinical expert consulted for this review. The target of the certainty-of-evidence assessment was the presence or absence of any (non-null) effect for GHD-CIM, GHD-CTB, GHD-PTB, injection-site reactions, injection-site pain, and treatment discontinuation due to the lack of a formal minimal important difference (MID) estimate.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for somapacitan versus Norditropin (somatropin) in prepubertal children with GHD.
Long-Term Extension Studies
REAL 4 Long-Term Safety Extension Phase
Description of Studies
The REAL 4 study design is described in the main body of this report. A summary of the long-term safety extension phase (week 52 to week 104) is presented in this section.23 The results presented in the sponsor’s interim analysis reflect the data available as of December 22, 2022.
Interventions
Patients were randomized (2:1) to receive either once-weekly somapacitan 0.16 mg/kg or daily Norditropin (somatropin) 0.034 mg/kg for the 52-week main trial period. For the subsequent safety extension trial period, which was planned for an additional 3 years following the main trial period and up to 208 weeks, all patients were allocated to once-weekly somapacitan 0.16 mg/kg.
Table 2: Summary of Findings for Somapacitan Versus Norditropin (Somatropin) for Pediatric Patients With GHD
Outcome and follow-up (follow-up was at 52 weeks) | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Norditropin (somatropin) | Somapacitan | Difference | |||||
Auxologic response | |||||||
Height velocity, estimated mean at follow-up, cm/year | 200 (1 RCT) | NA | 11.7 | 11.2 (NR) | −0.5 (−1.1 to 0.2) | Higha,b,c | Somapacitan results in little to no difference (i.e., a noninferior effect) on height velocity when compared with Norditropin (somatropin) |
Height SDS (range −10 to 10 with 0 representing a height equivalent to the population mean), change from baseline | 200 (1 RCT) | NA | 1.30 | 1.25 (NR) | −0.05 (−0.18 to 0.08) | Higha,b,c | Somapacitan results in little to no difference in the change from baseline in height SDS when compared with Norditropin (somatropin) |
Height velocity SDS (range −10 to 10, with 0 representing a height velocity equivalent to the population mean); change from baseline | 200 (1 RCT) | NA | 8.82 | 8.05 (NR) | −0.78 (−1.63 to 0.08) | Moderatea,b,d | Somapacitan likely results in a smaller increase from baseline in height velocity SDS when compared with Norditropin (somatropin) |
Patient-reported outcomes assessed with questionnaires for functioning or disease burden | |||||||
Disease-specific functioning as measured with GHD-CIM (TRIM-CGHD-O, 0 [best] to 100 [worst]); change from baseline, points | 138 (1 RCT) | NA | −10.9 | −9.0 (NR) | 1.8 (−2.9 to 6.6) | Lowb,e,f | Somapacitan may result in little to no difference in disease-specific functioning when compared with Norditropin (somatropin) |
Child treatment burden as measured with GHD-CTB (TB-CGHD-O, 0 [best] to 100 [worst]), estimated mean at follow-up, points | 169 (1 RCT) | NA | 13.1 | 10.7 (NR) | −2.4 (−5.7 to 0.9) | Lowb,g,h | Somapacitan may reduce child treatment burden when compared with Norditropin (somatropin); the clinical importance of the reduction is uncertain |
Caregiver treatment burden as measured with GHD-PTB (TB-CGHD-P, 0 [best] to 100 [worst]), estimated mean at follow-up, points | 176 (1 RCT) | NA | 14.7 | 8.7 (NR) | −6.0 (−10.0 to −2.1) | Moderateb,i,j | Somapacitan probably reduces caregiver treatment burden when compared with Norditropin (somatropin); the clinical importance of the reduction is uncertain |
Notable harms | |||||||
Injection-site reactions, including pain, bruising, hematoma, and swelling | 200 (1 RCT) | RR = 0.90 (0.27 to 2.97)k | 59 per 1,000 | 53 per 1,000 (NR) | 6 fewer per 1,000 (from 74 fewer to 62 more per 1,000) | Moderateb,l,m | Somapacitan likely results in little to no difference in injection-site reactions when compared with Norditropin (somatropin); there is some uncertainty about the clinical importance of the estimates |
Injection-site pain | 200 (1 RCT) | RR = 1.03 (0.10 to 11.16)k | 15 per 1,000 | 15 per 1,000 (NR) | No difference per 1,000 (from 35 fewer to 36 more per 1,000) | Moderateb,l,m | Somapacitan likely results in little to no difference in injection-site pain when compared with Norditropin (somatropin); there is some uncertainty about the clinical importance of the estimates |
Treatment discontinuation | |||||||
Treatment discontinuation | 200 (1 RCT) | RR = 1.56 (0.06 to 37.70) | 0 per 1,000n | 8 per 1,000 (NR)n | 8 more per 1,000 (from 7 fewer to 22 more per 1,000) | Highb,l,o | Somapacitan results in little to no difference in treatment discontinuation when compared with Norditropin (somatropin); there is some uncertainty about the clinical importance of the estimates |
CI = confidence interval; GH = growth hormone; GHD = growth hormone deficiency; GHD-CIM = Growth Hormone Deficiency–Child Impact Measure; GHD-CTB = Growth Hormone Deficiency–Child Treatment Burden; GHD-PTB = Growth Hormone Deficiency–Parent Treatment Burden; MID = minimal important difference; NA = not applicable; NR = not reported; RCT = randomized controlled trial; RR = relative risk; SDS = standard deviation score; TB-CGHD-O = Treatment Burden Measure–Child Growth Hormone Deficiency–Observer; TB-CGHD-P = Treatment Burden Measure–Child Growth Hormone Deficiency–Parent; TRIM-CGHD-O = Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. For all the outcomes in this table, statistical testing was not adjusted for multiplicity; there is an increased risk of type I error (false-positive results).
aThe CADTH review team identified a potential risk of bias due to several factors, including a baseline imbalance in GH peak level at baseline that was higher in the somapacitan group (median 5.2 mcg/L) than in the Norditropin (somatropin) group (3.9 mcg/L), an imbalance in the thyroid hormone use at baseline that was lower in the somapacitan group (8.3%) than in the Norditropin (somatropin) group (14.7%), and a higher treatment adherence rate in the somapacitan group (96%) than in the Norditropin (somatropin) group (88%), as measured with a patient e-diary device to record dosing; however, the certainty of evidence was not rated down for risk of bias because it was uncertain whether these imbalances might raise the potential for bias in favour of somapacitan.
bIndirectness did not result in the level of certainty being rated down. Differences between the patients in clinical practice and those included (e.g., GH therapy–naive, no intracranial tumour) or excluded (due to prepubertal age and potential exclusion of patients who could not physically stand up due to, for example, significant spinal abnormalities and congenital abnormalities) in the single RCT that informed the evidence were noted; however, these differences were not considered serious enough to result in important differences in the observed effect, according to the clinical expert consulted by CADTH.
cImprecision did not result in the level of certainty being rated down. The point estimate and both the lower and upper boundaries of the 95% CI of the between-group comparison indicate trivial or no clinically meaningful difference, according to the clinical expert consulted by CADTH; for HV, the 95% CI excludes the noninferiority margin (−1.8 cm/year).
dThe level of certainty was rated down by 1 level for serious imprecision. No specific threshold was established but, according to the clinical expert consulted by CADTH, the point estimate (−0.78) and the lower boundary of the 95% CI (−1.63) could be considered a clinical meaningful worse efficacy for somapacitan vs. Norditropin (somatropin), while the upper boundary of the 95% CI (0.08) suggests no clinically meaningful difference between the 2 groups.
eThe level of certainty was rated down by 1 level for serious risk of bias due to the potential for bias in favour of somapacitan (once-weekly injections) compared with Norditropin (somatropin) (once-daily injections) arising from the open-label nature of the study and the subjective nature of the outcome. The impact of missing outcome data (31% of the patients) is unclear.
fThe level of certainty was rated down by 1 level for serious imprecision. Based on the MID identified in the literature (5 points based on within-group data), the point estimate suggested little to no difference, while the upper bound of the 95% CI suggested an increase in the GHD-CIM score compared with Norditropin (somatropin).
gThe level of certainty was rated down by 1 level for serious risk of bias due to potential for bias in favour of somapacitan (once-weekly injections) compared with Norditropin (somatropin) (once-daily injections) arising from the open-label nature of the study and the subjective nature of the outcome. The impact of missing outcome data (15.5% of the patients) is unclear.
hThe level of certainty was rated down by 1 level for serious imprecision. The CADTH review team was unable to confirm with the clinical expert consulted by CADTH whether the MID identified in the literature (6 points based on the within-group data provided by the sponsor) would be suitable to assess a between-group difference; therefore, the null was used to assess certainty. The point estimate suggested a decrease in the GHD-CTB score, while the upper bound of the 95% CI suggested an increase in the GHD-CTB score compared with Norditropin (somatropin).
iThe level of certainty was rated down by 1 level for serious risk of bias due to potential for bias in favour of somapacitan (once-weekly injections) compared with Norditropin (somatropin) (once-daily injections) arising from the open-label nature of the study and the subjective nature of the outcome. The impact of missing outcome data (12% of the patients) is unclear.
jImprecision did not result in the level of certainty being rated down. The CADTH review team was unable to confirm with the clinical expert consulted by CADTH about whether the MID identified in the literature (7 points based on within-group data, provided by the sponsor) would be suitable to assess a between-group difference; therefore, the null was used to assess certainty.
kThe RR and 95% CI were not part of the statistical analysis plan; they were requested by CADTH to allow for an assessment of the imprecision of the findings.
lThe level of certainty was rated down by 1 level for serious risk of bias due to potential for bias in favour of somapacitan (once-weekly injections) compared with Norditropin (somatropin) (once-daily injections) arising from the open-label nature of the study and the subjective nature of the outcome.
mImprecision did not result in the level of certainty being rated down. The clinical expert consulted by CADTH could not provide a threshold of important difference; however, the CADTH review team judged that the effect estimate and 95% CI were unlikely to include any important effect.
nTreatment discontinuation is for any reason and not limited to adverse events. Treatment discontinuation due to adverse events in both treatment arms in the REAL 4 trial was 0. One patient in the main study discontinued somapacitan due to a violation of inclusion and/or exclusion criteria. A total of 132 patients in the somapacitan arm and 131 patients in the Norditropin (somatropin) arm completed the main treatment period.
oRisk of bias did not result in the level of certainty being rated down. One patient from the somapacitan arm discontinued the treatment due to a violation of the study protocol; no patients in the Norditropin (somatropin) arm discontinued the treatment.
Source: REAL 4 Clinical Study Report (week 52).20
Results
Patient Disposition: A total of 199 patients were exposed in the safety extension (following week 52). Four patients in the somapacitan to somapacitan group and 1 patient in the Norditropin (somatropin) to somapacitan group discontinued the trial product in the safety extension period (from week 52 to week 104). None of the discontinuations were due to AEs. All patients in the extension period who discontinued were withdrawn from the trial.
Exposure to Study Treatments: In the safety extension period (week 52 to 104), the mean duration of exposure to somapacitan and mean adherence were similar between the somapacitan to somapacitan group and the Norditropin (somatropin) to somapacitan group. The mean adherence in the somapacitan to somapacitan group was 94.2% from week 0 to 104, suggesting consistently high adherence throughout the entire study.
Efficacy
The sponsor’s interim analysis includes data for up to week 104 in the safety extension phase. The key efficacy outcomes evaluated in the REAL 4 main trial phase were evaluated descriptively for the long-term safety extension phase.
After 104 weeks of treatment, the HV observed in the patients who had been treated with somapacitan during the full trial period was similar to the HV observed in the patients who switched from Norditropin (somatropin) to somapacitan after week 52. After 104 weeks of treatment, the increase in change from baseline in height SDS and HV SDS observed in the patients treated with somapacitan during the full trial period was similar to the change observed in the patients who were switched from Norditropin (somatropin) to somapacitan at week 52. The mean observed change from baseline after 104 weeks in the IGF-1 SDS and the insulin-like growth factor binding protein 3 (IGFBP-3) SDS was similar in both groups.
The Growth Hormone Patient Preference Questionnaire (GH-PPQ) assessing GH treatment preference was used to evaluate the switch from Norditropin (somatropin) to somapacitan after the final visit in the main phase of the trial at week 52. The questionnaire was completed for patients by the parent or legally authorized representative (LAR) 4 weeks after the patient had switched from Norditropin (somatropin) to somapacitan (week 56). Parents for 50 of the 68 patients who were switched from Norditropin (somatropin) to somapacitan at week 52 responded to the questionnaire. Of these 50 respondents, 45 preferred somapacitan to somatropin, while 5 respondents answered that they had no preference. None of the respondents preferred Norditropin (somatropin). The majority of the respondents (38 out of 45) who preferred somapacitan had a strong or very strong preference for its once-weekly treatment regimen over daily Norditropin (somatropin) injections. The main reasons selected in the GH-PPQ for the preference included: “number of times needing to do injections,” “less worried about remembering to give the injections,” and “child [is] less worried or annoyed by getting injections.” Thirty-five of the 45 parents preferring somapacitan stated they expected higher adherence to the current once-weekly regimen versus the Norditropin (somatropin) regimen; 1 parent expected the patient to be more adherent to Norditropin (somatropin), while the remaining 9 parents had no preference with regard to expected adherence to either drug.
Harms
The safety profile of once-weekly somapacitan administered to children with GHD for up to 104 weeks was similar to the well-known safety profile for daily Norditropin (somatropin). No new safety or local tolerability issues were identified.
In patients with an IGF-1 SDS above 2, no trend was seen in the amount or type of AEs reported at 2 or more consecutive visits compared with the remaining patients in the relevant treatment groups. There were no dose reductions due to AEs for these patients. The majority of AEs were of mild severity.
Critical Appraisal
Internal Validity
REAL 4 is an open-label trial, which may influence the perception of improvement by patients and clinicians, particularly for outcomes that are subjective in measurement and interpretation (e.g., patient-reported outcomes, subjective AEs). In the REAL 4 trial, only prepubertal children were enrolled into the main trial to avoid the pubertal growth spurt interfering with the treatment effect.
The efficacy outcomes were presented using descriptive statistics, so no statistical inferences were possible. The safety profile with somapacitan from the main phase to the long-term extension phase was consistent in the REAL 4 trial.
External Validity
Because the patients who took part in the open-label long-term safety extension phase were originally from the REAL 4 trial and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to the open-label long-term safety extension phase.
Indirect Comparisons
Direct comparative evidence exists between somapacitan and Norditropin (somatropin) 0.034 mg/kg once daily from the REAL 3 and REAL 4 trials, but there is a gap in evidence comparing somapacitan with somatrogon. Indirect comparisons were required to address this gap. One indirect treatment comparison (ITC) was submitted by the sponsor, which has been summarized and appraised in this report.
Description of Studies
Based on a systematic literature review (SLR) conducted by the sponsor, network meta-analyses (NMAs) were conducted to indirectly compare somapacitan with somatrogon based on phase II and phase III RCTs. Data for somapacitan 0.16 mg/kg/week versus somatropin (Norditropin 0.034 mg/kg/day) was informed by the REAL 3 and REAL 4 trials, while data for somatrogon 0.66 mg/kg/week versus somatropin (Genotropin 0.034 mg/kg/day) was informed by the Opko II and Opko III trials in the base-case analyses and, additionally, by the Opko JPN trial (Genotropin 0.025 mg/kg/day) in a scenario analysis because it differed in study design, patient population, dose escalation of somatrogon, and dose of somatropin relative to all other included trials. In all included studies, patients were randomized to treatment with an LAGH or short-acting GH (SAGH) for at least 26 weeks. The trials enrolled treatment-naive prepubescent children with GHD and had mostly similar patient eligibility criteria. Patient baseline characteristics differed across trials with regard to age, percent male, racial distribution, peak GH, and height SDS; other characteristics such as IGF-1 SDS, HV SDS, GHD cause, body mass index (BMI) and height could not be compared due to lack of reporting.
Efficacy Results
The sponsor conducted NMAs assessing annualized HV (AHV), HV SDS, and height SDS. Data sufficient to form a network was available at week 26 and week 52 for AHV and height SDS, but only at week 26 for HV SDS. No statistically significant differences were identified between somapacitan and somatrogon for any outcome in the base-case analyses. Although some differences were identified for the lower dose of somatropin assessed in the Opko JPN trial, the lower dose is not considered relevant.
Quantitative comparisons were not conducted for longer-term outcomes (week 104 and week 156) because the Opko II and Opko III extensions were single-arm trials, so there was no direct comparative data available to inform the somatrogon node. The results were presented narratively but could not be compared.
Harms Results
Short-Term Outcomes (52 Weeks)
Quantitative comparisons were not performed for any harm outcomes, though justification was not provided by the sponsor for this decision. The proportion of patients experiencing at least 1 AE across trials varied from approximately 60% to 100% through 52 weeks of treatment. Serious AEs were observed in 0 to 3 patients per treatment arm in all cases except the somapacitan arm of the REAL 4 trial, in which 6 patients (4.5%) experienced serious AEs. Severe AEs did not occur in the REAL 3 trial, occurred in few patients in the REAL 4 trial (from 1.5% in the Norditropin [somatropin] arm to 3.0% in the somapacitan arm), and occurred in from 5.2% (somatropin arm) to 8.3% (somatrogon arm) in the OPKO III trial. Although very few AEs were observed in the REAL 3, Opko II, and Opko JPN trials, the sample sizes of each of the treatment arms were very small.
In the REAL trials, the number of injection-site pain events ranged from zero to very few in both the somapacitan and Norditropin (somatropin) arms. In the Opko III and Opko JPN trials, there were high rates of injection-site pain reported in the somatrogon arms (39.4% and 72.7%, respectively). However, in the Opko III and JPN trials, there were also more frequent events of injection-site pain in the somatropin arms (25.2% and 13.6%, respectively), which contrasts with the 0% and 1.5% event rates in somatropin-treated patients from the REAL 3 and REAL 4 trials, respectively. Severe injection-site pain did not occur in any patients in either the REAL 3 or 4 trials, regardless of treatment assignment, whereas 1 somatrogon-treated patient in the Opko II trial, and 4.6% of somatrogon-treated patients and 2.6% of somatropin-treated patients in the Opko III trial experienced severe injection-site pain.
Overall, rates of antibodies varied considerably across the trials. Analyses showed patients who were positive for antibodies did not experience reduced efficacy or safety issues compared with those without antibodies.
Long-Term Outcomes (Beyond 52 Weeks)
Overall, continued treatment with somapacitan and somatrogon was well generally tolerated, with no new safety signals identified. The proportion of patients experiencing at least 1 AE ranged from approximately 70% to 81% across the extensions of the REAL 3 (week 52 to 156), Opko II (week 52 to 208), and Opko III (week 52 to 104) trials. Most reported AEs were mild to moderate in severity.
In the REAL 3 trial extension (week 52 to 156), 3 patients (6.7%) treated with somapacitan and 1 patient (7.1%) treated with Norditropin (somatropin) experienced injection site–related AEs. All injection site–related AEs were considered to be mild in severity. No injection-site AE data were available from the extension trials of somatrogon.
Similar to results from the 52-week outcomes, neutralizing antibodies were observed with somatrogon but not with somapacitan. Where reported, analyses indicated the presence of antibodies did not have any impact with respect to efficacy outcomes.
Critical Appraisal
In the base-case NMAs, there were some between-trial imbalances between studies in race, gender, and mean age at baseline, and differences in eligibility criteria related to age range, peak GH, and bone age. The sponsor stated these were not expected to have modified any effects; however, it did not provide any support for this statement. Notably, height SDS and HV SDS are standardized for age and gender by definition; therefore, the potential effect of imbalances in age and gender (but not race) may be partially mitigated inherently in these outcomes, however, that was not the case for AHV. Ultimately, the potential magnitude and direction of bias is unknown, which obscures the interpretation of the results. The clinical expert consulted by CADTH identified that other potential prognostic factors and effect modifiers include the cause of GH deficiency (idiopathic versus organic), and isolated GHD versus multiple pituitary hormone deficiencies; however, these characteristics could not be compared between trials due to a lack of reporting.
The networks were sparse. To account for potentially high heterogeneity, the sponsor used informative priors, though acknowledged that it was potentially problematic because data informing the priors were not sourced externally from the included trials. The heterogeneity of the trials may therefore still bias the results. Additionally, the network had no closed loops, so assessing for consistency was not possible.
The sparse network, small sample sizes, between-trial heterogeneity, and wide credible intervals (CrIs) indicate inconclusive results. Ultimately, there is no evidence that there is a clinically meaningful difference between somatropin and somatrogon; however, whether somatropin and somatrogon can be said to be clinically equivalent is likewise inconclusive. Furthermore, there were no quantitative comparisons of safety, and concerning differences between trials in the rate of injection-site pain in the common comparator arm (somatropin 0.034 mg/kg once daily) that were not explained. As a result, no conclusions can be drawn regarding the comparative safety of somapacitan versus somatrogon.
Studies Addressing Gaps in the Evidence From the Systematic Review
One other relevant study (REAL 3) was a phase II, multicentre, open-label, dose-finding trial that compared somapacitan versus Norditropin (somatropin) after 156 weeks of treatment, with a long-term (additional 4 years in plan and 1 year of data available) single-arm extension phase. The CADTH review focused on data for somapacitan with a dosage of 0.16 mg/kg/week, as per the proposed product monograph.
Description of Studies
Fourteen patients were enrolled in the 0.16 mg/kg/week somapacitan arm and 14 patients were enrolled in the Norditropin (somatropin) arm. The mean age of these patients was 6 years; ||||| of them were males and ||||| were females (cohort 1). The majority of patients were Asian (|||) followed by white (|||), and had idiopathic GHD (|||). The median GH peak level was higher in the somapacitan arm (5.28 mcg/L) than in the Norditropin (somatropin) arm (4.55 mcg/L). Pediatric patients of other ages were also analyzed in the long term safety extension phase: cohort 2 enrolled patients younger than 2.5 years (N = 1) and cohort 3 enrolled patients of pubertal age and up to 17 years (N = 9 for patients exposed in the long term extension and N = 1 for those who completed). Similar outcomes were reported at week 26 (primary time point for the dose-finding trial phase) and week 52 (extension phase). Harms outcomes were reported at weeks 104 and 208 in the single-arm safety extension phase. For cohorts 2 and 3, only harms outcomes were reported.
Efficacy Results
The results showed comparable effects in AHV at week 26 (primary end point), with an ETD of 1.67 cm/year (95% CI, −0.22 to 3.56); and the estimated mean HVs for somapacitan and Norditropin (somatropin) were 13.08 cm/year and 11.41 cm/year, respectively. The difference was similar at week 52 (1.8 cm/year; 95% CI, 0.5 to 3.1), but was smaller at week 156 (0.8 cm/year; 95% CI, −0.4 to 2.1). The point estimates were in favour of somapacitan compared with Norditropin (somatropin) for other auxologic response outcomes, including height SDS, HV SDS, and height (cm) at both week 26 and 52.
As of the pharmacodynamic end points, there was an increase in IGF-1 SDS with somapacitan compared with Norditropin (somatropin) at week 26 (ETD = 1.17; 95% CI, 0.38 to 1.95) and at week 52 (ETD = 1.56; 95% CI, 0.66 to 2.46). There was an increase in IGFBP-3 SDS at week 52 with an ETD of 0.93 (95% CI, 0.13 to 1.73).
The point estimates were in favour of somapacitan compared with Norditropin (somatropin) for the patient-reported outcomes of GHD-CIM, GHD-CTB, and GHD-PTB at week 52, but the 95% CI of the differences included the null.
Harms Results
During the 156-week treatment period, AEs were reported by 100% of patients in both the somapacitan and Norditropin (somatropin) arms. The most frequently reported AEs were pyrexia (29%), nasopharyngitis (21%), and vomiting (21%) in the somapacitan arm, and nasopharyngitis (21%), influenza (21%), and rhinitis (21%) in the Norditropin (somatropin) arm.
No patients in the somapacitan arm compared with 14% of patients in the Norditropin (somatropin) arm withdrew from treatment due to treatment-emergent AEs (2 patients in the Norditropin [somatropin] treatment arm discontinued treatment due to AEs that included nephrotic syndrome and drug hypersensitivity). No deaths were reported among patients in either arm.
The occurrence of 2 notable harms was comparable between the treatment arms at week 156: injection-site reactions were 7% in both arms, and injection-site pain was 7% in the somapacitan arm and 0 in the Norditropin (somatropin) arm.
No new safety issues were identified in the long term safety extension phase for any of the cohorts.
Other Outcomes
A comparable proportion of patients discontinued treatment with somapacitan (|||||) and Norditropin (somatropin) (|||||) at week 156, with reasons that included AEs (|||||| |||||), study protocol violation (|||| ||||||), and withdrawn by parent or guardian (|||| ||||||) in the somapacitan and Norditropin (somatropin) arms. Adherence to therapy was 92.2% in the somapacitan arm and 87.2% in the Norditropin (somatropin) arm at week 156.
Critical Appraisal
In general, internal validity and external validity for the REAL 3 trial were similar to those for the REAL 4 trial. Although the treatment and follow-up duration was up to 3 years for both efficacy and harms outcomes, with an additional 4 years of follow-up for safety, and patients of a wider range of age were enrolled (cohorts 2 and 3) in the REAL 3 trial, the generalizability of the study was limited by its small sample size. The use of an open-label design may bias the reporting of patient-reported outcomes (e.g., health-related quality of life) and AEs such as injection-site reactions (including pain, bruising, hematoma, and swelling) and injection-site pain. Furthermore, because of the lack of a control arm in the extension phases, the interpretation of the results is limited.
Conclusions
One phase III, multicentre, open-label study (REAL 4) compared the subcutaneous injection of somapacitan 0.16 mg/kg once weekly with Norditropin (somatropin) 0.034 mg/kg once a day in prepubertal children (aged 2.5 years to 10 years for girls and 2.5 years to 11 years for boys) with GHD at week 52. Somapacitan results in little to no difference in HV and height SDS (high certainty), and likely results in a smaller improvement from baseline in HV SDS (moderate certainty) when compared with Norditropin (somatropin). Somapacitan may result in little to no difference in disease-specific functioning when compared with Norditropin (somatropin) (low certainty). Somapacitan was associated with a reduction in child treatment burden (low certainty) and caregiver treatment burden (moderate certainty), compared with Norditropin (somatropin). Somapacitan results in little to no difference in treatment discontinuation when compared with Norditropin (somatropin) (moderate certainty). The evidence shows that somapacitan likely results in little to no difference in the notable harms, injection-site reactions and injection-site pain, when compared with Norditropin (somatropin) (moderate certainty). No new safety signals were identified from the longer-term single-arm studies; however, long-term comparative data were not available.
In the NMAs, there were no statistically significant differences identified between somapacitan and somatrogon (0.66 mg/kg/week) for the outcomes of AHV, height SDS, or HV SDS at week 26, nor in AHV or height SDS at week 52; however, the interpretation of the NMA is limited by the sparse network, small sample sizes, between-trial heterogeneity, and wide CrIs across all assessed outcomes. Disease-specific functioning, child treatment burden, and safety-related outcomes were not assessed in the NMAs.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of somapacitan 5 mg/1.5 mL (3.3 mg/mL), 10 mg/1.5 mL (6.7 mg/mL), 15 mg/1.5 mL (10 mg/mL) as a prefilled pen for subcutaneous use in the long term treatment of pediatric patients experiencing growth failure due to an inadequate secretion of endogenous GH (i.e., GHD).
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
Overview of the Condition
GHD is a rare disease caused by impaired secretion of GH by the pituitary gland, which affects patients’ growth, body composition, metabolic profile, bone mineral density, and quality of life.2,3 GHD can be congenital (genetic and/or associated with brain malformations) or organic, also referred to as acquired (due to damage caused by tumours, physical trauma, inflammation, brain infections, or radiotherapy), and may occur in isolation (isolated GHD) or in conjunction with other pituitary hormone deficits (multiple pituitary hormone deficiency).2 The majority of patients with GHD (up to 70%) have isolated GHD with no known cause (idiopathic).24 This statistic seems different from the Canadian context; in a cohort of 850 children in Canada treated with GH, 526 of which had GHD, 72% (379 out of 526) were diagnosed with predominantly organic (particularly intracranial tumours and congenital pituitary abnormalities) rather than idiopathic GHD.4 Congenital causes of GHD can be linked to a number of gene mutations; some of these mutations may be located on GH1, HESX1, or X-linked recessive genes such as SOX3 and BTK, and many others.2
The clinical manifestation of GHD can vary, depending on the types of hypothalamic-pituitary lesions, the age of onset, and the combination of genetic mutations.5 For example, infants with GHD may not initially display obvious signs of the condition, but they may experience poor weight gain and lethargy, whereas GHD in adolescents may be associated with growth retardation and delayed puberty.2 The key clinical features of GHD include delayed dentition, an immature face, a prominent forehead and depressed midfacial development, a high-pitched voice, increased fat mass that is predominantly centrally distributed, male hypogenitalism, decreased muscle mass, and delayed puberty.2,25,26 In children and adolescents, GH is responsible for increasing bone length and density and is involved in regulating body composition.2,27 GH also has antagonizing effects on insulin and glucose metabolism, affecting whole-body lipid metabolism. GH has the ability to induce insulin resistance, which is important for preventing hypoglycemia.28,29 These regulatory processes mediated by GH contribute to the overall growth and maturation of children and adolescents into adults.
Estimated Disease Prevalence
Estimates for prevalence of pediatric GHD are sparse, with no specific Canadian data, although studies across Europe, the US, and China suggest an estimated prevalence ranging from 1 in 30,000 to 1 in 5,600.3,6-8 Estimates of pediatric GHD suggest a prevalence of 1 in 4,000 to 10,000 children worldwide, suggesting a prevalence of approximately 1,600 children in Canada.9,10
Diagnosis of the Condition
Diagnostic Testing Requirements
While there is no gold-standard diagnostic test for GHD, a multifaceted approach, including comprehensive clinical and auxological assessments, is used to diagnose children with GHD.30 Typically, other potential causes of short stature, such as genetic, hormonal, metabolic, and/or psychogenic causes, will need to be explored and excluded before the evaluation of GHD in children with short stature.31 The Growth Hormone Research Society consensus guidelines recommend that assessments of GHD include a detailed medical history (gestational age, birth history, family history), anthropometric measures, static biochemical tests (IGF-1, IGFBP-3), imaging studies (bone age, MRI examination of the sella), and molecular genetics.12,25,32 The Canadian Pediatric Endocrine Group consensus criteria for GH testing in Canada specifically includes the following measurements of auxologic and bone age:
growth velocity of less than 25% of bone age documented over a 12-month period
short stature, defined as being less than 2 SDs from midparental height (defined as a child’s estimated adult height based on the parent’s height), or bone age delayed by 2 SDs compared with chronological age, or the presence of other significant features associated with GH deficiency.33
GH stimulation tests are performed to support the diagnosis of GHD and to measure the ability of the body to produce GH. A GH stimulation test involves administering a pharmacologic drug (e.g., arginine, glucagon, clonidine) to stimulate the pituitary gland to release GH and subsequently measuring GH levels.34 Patients undergo 2 GH stimulation tests, which may be conducted consecutively on the same day or across multiple days. Specifically, the Canadian Pediatric Endocrine Group Consensus Criteria for the Diagnosis of Growth Hormone Deficiency in Canada indicates the criteria for a diagnosis of GHD involves 2 pharmacologic stimulation tests with peak GH concentrations of less than 10 mcg/L, or 1 pharmacologic test with peak GH concentrations of less than 3 mcg/L with abnormality present on MRI.33 Thresholds for confirmation of GHD may vary based on the type of assay used for testing; however, the assays used in Canada for GH stimulation tests may typically range from 5.6 mcg/L to 8 mcg/L.
Availability of Diagnostic Testing
GH stimulation testing is a key diagnostic tool for GHD and usually occurs in hospital settings in Canada. According to the clinical expert consulted by CADTH, patients living in rural or geographically remote areas have a greater challenge in going to the specialty centres to have the GH stimulation testing done. The testing is done by pediatric endocrinologists who have access to or privileges at children’s hospitals (or, less commonly, in community hospitals) that have the qualified nurses, protocols, and facilities to conduct them properly. Two GH stimulation tests are usually required and are conducted consecutively on the same day. A single pharmacologic test may be sufficient for someone known to have multiple pituitary hormone deficiencies or a hypoplastic anterior pituitary or ectopic pituitary. GH stimulation testing is considered the standard of care for the diagnosis of GHD.
Health Care Resources
Treatment Phase
Patients receiving somapacitan (Sogroya) will be initiated with a dose of 0.16 mg/kg body weight once weekly. Maintenance dosing of somapacitan can be adjusted based on the patient and their response.1
Patients who switch from another daily GH treatment to somapacitan should choose the preferred day to begin the weekly dose and stop daily GH treatment 8 to 24 hours before their first dose of somapacitan. Patients who switch from another GH therapy administered weekly are recommended to continue their weekly schedule. According to the product monograph, patients should be prescribed somapacitan by a health care provider but will self-administer or receive treatment with the help of a caregiver or guardian with proper training.1
A key indicator of GHD is HV. The determination of HV should be made through serial measurements occurring at approximately 6-month intervals; the frequency of clinical visits may depend on the age of the child, with infants being seen more frequently at every 3 months, while adolescents may be seen every 4 to 6 months.2,25 During these routine clinical visits, in addition to the measurement of height, additional assessments may occur, including the assessment of puberty, side effects from treatment, need for potential dose adjustments, adherence, and treatment satisfaction. Additional laboratory measurements will also need to take place approximately every 3 to 6 months to assess for proper physiological development and any hormone deficiencies (i.e., IGF-1 levels, thyroid function, pituitary hormone deficiencies, and glucose tolerance). Assessments of bone age may also occur every year.35
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
The primary treatment of GHD is injections of synthetic GH, which has been available since 1985.11 Canadian and US guidelines recommend treatment with GH to normalize extreme shortness in children and adolescents with GHD to attain normal adult height.12 Treatment with GH should begin as soon as possible for patients to achieve their optimal growth velocity and final adult height,9,13 and be continued until a patient reaches their full adult height and bone maturity, or when their HV is less than 2 cm/year.14-16 Somatropin, administered as a daily subcutaneous injection, has traditionally been the primary GH used for treatment of GHD. In March 2022, somatrogon (Ngenla), a Health Canada–approved GH, received CADTH’s reimbursement recommendation for treatment of pediatric GHD as a weekly subcutaneous injection,14 but received a negative funding recommendation by INESSS due to the potential for greater pain at the injection site and higher doses.17
If left untreated, GHD may become permanent upon reaching adulthood. Other complications from untreated GHD may also include insulin resistance, increase in adipose tissue and decreased lean body mass, decreased bone strength, and increased blood pressure.36 According to the sponsor and clinical expert input, treatment goals for GHD include optimizing final adult height, restoring metabolic functions associated with GHD, reducing injection burden, improving treatment adherence, and optimizing quality of life.
The clinical expert consulted for this review indicated that all products in Table 3 may be used as treatments for pediatric GHD administered as daily or weekly subcutaneous injections. The choice of synthetic GH for treatment of GHD may be based on several factors, such as consideration of patient preference, cost, and financial and device support offered by the sponsor of the GH therapy.
According to the clinical expert input, 1 of the limitations associated with current treatments is that they require injections and daily (or near daily) injections, which can lead to suboptimal adherence, resulting in suboptimal clinical outcomes, e.g., the treatment goal of improvement in height is affected.3,18,19 The daily dosing frequency can be burdensome to both patients and caregivers, and can cause anxiety around injection (e.g., needle phobia) and associated pain for patients.37 Caregivers of pediatric patients may also feel uneasy about administering therapy, as parents often worry about causing their child pain.37 Storage and administration of treatment can influence treatment adherence as can the patient’s and caregiver’s lives. Devices with less burdensome storage requirements that are easy to use and have advanced design in size, weight, and strength required to administer the injection (which is of particular importance for pediatric patients who self-administer treatment) may improve patient adherence to the treatment.19,38
Drug Under Review
Indication and Reimbursement Request
The dossiers for somapacitan (Sogroya) were submitted to CADTH as a pre–Notice of Compliance submission, with the Notice of Compliance issued on July 26, 2023. Somapacitan underwent a standard review at Health Canada for the long-term treatment of pediatric patients experiencing growth failure due to an inadequate secretion of endogenous GH (GHD).
Dosing and Administration
Somapacitan is recommended to be initiated at a dose of 0.16 mg/kg body weight once weekly for treatment-naive patients and patients switching from daily GH (somatropin), under the supervision of an experienced health care provider. The dose of somapacitan is to be individualized and adjusted based on the patient’s response to treatment. Treatment with somapacitan may be administered at any time of day and is to be injected subcutaneously in the abdomen, thighs, buttocks, or upper arms. It is recommended that the injection site be rotated every week.1
Patients switching from daily human GH to once-weekly somapacitan should choose the preferred day for the weekly dose and end daily treatment the day prior, or at least 8 hours before taking the first dose of once-weekly somapacitan. Patients switching from a weekly human GH to once-weekly somapacitan are recommended to continue their once-weekly dosing schedule. When GHD persists after growth completion, it is recommended that GH treatment be continued to achieve full somatic adult development, including lean body mass and bone mineral accrual.1
Mechanism of Action
Somapacitan is a 23.3 kDa human GH derivative (99% similarity to endogenous GH) linked to a small noncovalent albumin-binding moiety that facilitates reversible endogenous albumin binding to delay somapacitan elimination. The mechanism of action of somapacitan is either directly via the GH receptor and/or indirectly through the IGF-1 produced in tissues throughout the body. When GH deficiency is treated with somapacitan, a normalization of body composition (i.e., decreased body fat mass, increased lean body mass) and of metabolic action is achieved. Somapacitan stimulates skeletal growth in pediatric patients with GHD as a result of its effects on the growth plates (epiphyses) of bones.1
Prescribing
According to the Health Canada product monograph, somapacitan treatment should be initiated and monitored by physicians who are appropriately qualified and experienced in the diagnosis and management of patients with the condition for which somapacitan is indicated. Switching a patient from another type or brand of GH should be done by a physician who has experience in the diagnosis and management in GHD.1
Key characteristics of somapacitan are summarized in Table 3, along with other treatments available for GHD.
Table 3: Key Characteristics of Somapacitan (Sogroya), Somatropin (Norditropin, Nutropin AQ, Humatrope, Genotropin, Omnitrope, Saizen), and Somatrogon (Ngenla)
Detail | Somapacitan (Sogroya) | Somatropin (Norditropin, Nutropin AQ, Humatrope, Genotropin, Omnitrope, Saizen) | Somatrogon (Ngenla) |
|---|---|---|---|
Mechanism of action | Either directly via the GH receptor and/or indirectly through the IGF-1 produced in tissues throughout the body, predominantly by the liver. When GHD is treated with somapacitan, a normalization of body composition (i.e., decreased body fat mass, increased lean body mass) and of metabolic action is achieved. Somapacitan distributes to the hypertrophic zone and primary spongiosa in the epiphysis of the proximal tibia of GH-deficient hypophysectomized rats. The distribution of somapacitan to peripheral tissues is comparable to human GH. Somapacitan stimulates skeletal growth in pediatric patients with GHD as a result of its effects on the growth plates (epiphyses) of bones. | Norditropin: Somatropin (as well as endogenous GH) binds to a dimeric GH receptor in the cell membrane of target cells resulting in intracellular signal transduction and a host of pharmacodynamic effects. Some of these pharmacodynamic effects are primarily mediated by IGF-1 produced in the liver and also locally (e.g., skeletal growth, protein synthesis), while others are primarily a consequence of the direct effects of somatropin (e.g., lipolysis). Nutropin AQ: This is a human GH produced by recombinant DNA technology. The amino acid sequence of the somatropin protein is identical to that of pituitary-derived human GH. The treatment of children experiencing a lack of adequate secretion of endogenous GH results in an increase in growth rate and an increase in IGF-1. Humatrope: This stimulates linear growth in pediatric patients whose bodies do not produce an adequate (normal amount) of endogenous GH and in children who are short in stature in association with Turner syndrome, idiopathic short stature, SHOX deficiency, and whose height did not catch up (after being identified as small for gestational age at birth). Treating pediatric patients with GH deficiency and patients with Turner syndrome with Humatrope produces an increased growth rate and IGF-1 concentrations similar to those seen in therapy with human GH of pituitary origin. Genotropin: Polypeptide hormone of recombinant DNA origin. The amino acid sequence of the product is identical to that of human GH of pituitary origin. Stimulates linear growth in children with GH deficiency. Omnitrope: Human GH produced by recombinant DNA technology. The amino acid sequence of the somatropin protein is identical to that of pituitary-derived human GH. The treatment of pediatric patients who have GHD results in linear growth and normalizes concentrations of IGF-1. Saizen: Polypeptide hormone consisting of 191 amino acid residues; its structure is identical to that of GH extracted from human pituitary glands. It is produced by recombinant DNA technology in a mammalian cell expression system. It provides an exogenous supply of human GH for those patients lacking the ability to produce adequate endogenous supplies. | Binds to the GH receptor initiating a signal transduction cascade, resulting in changes in growth and metabolism. Somatrogon binding leads to the activation of the STAT5B signalling pathway and increases the serum concentration of IGF-1. |
Indicationa | Long-term treatment of pediatric patients who have growth failure due to inadequate secretion of endogenous GHD. | Norditropin: Long-term treatment of children with growth failure due to an inadequate secretion of endogenous GH (GHD). Nutropin AQ: Long-term treatment of children who have growth failure due to hormone inadequacy. Humatrope: Long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of normal endogenous GH and whose epiphyses are not closed. Genotropin: Long-term treatment of children who have growth failure due to an inadequate secretion of endogenous GH (GHD). Omnitrope: Long-term treatment of children, who have growth failure due to an inadequate secretion of endogenous GH (GHD). Saizen: Long-term treatment of children with growth failure due to inadequate secretion of normal endogenous GH. | Long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous GH (GHD). |
Route of administration | SC | Norditropin: SC Nutropin AQ: SC or intramuscular injection Humatrope: SC Genotropin: SC Omnitrope: SC Saizen: SC | SC |
Recommended dose | Once weekly at 0.16 mg/kg. | Norditropin: Up to 0.043 mg/kg/day. Nutropin AQ: At a dose of 0.3 mg/kg/week (approximately 0.90 IU/kg/week) administered in divided daily doses. Humatrope: At 0.18 mg/kg/week (daily equivalent dose of 0.026 mg/kg/day) to be administered on 3 alternate days, or 6 to 7 times per week, or daily. Genotropin: At a dose of 0.16 mg/kg to 0.24 mg/kg body weight/week divided into 6 to 7 doses. Omnitrope: At a dose of 0.16 to 0.24 mg/kg body weight per week divided into 6 to 7 doses. Saizen: At 0.2 mg/kg body weight per week. The dose can be increased to 0.27 mg/kg/week if there is insufficient response to treatment. | 0.66 mg/kg per week. |
Serious adverse effects or safety issues | Must not be used when there is any evidence of neoplastic activity. Contraindicated in adult patients with acute critical illness experiencing complications following open heart surgery, abdominal surgery, multiple accidental trauma, acute respiratory failure, or similar conditions. | Norditropin: Somatropin is contraindicated in patients with PWS who are severely obese, have a history of upper airway obstruction or sleep apnea, or have severe respiratory impairment. There have been reports of sudden death when somatropin was used in such patients. Norditropin is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed PWS. Treatment with Norditropin should be discontinued at the time of renal transplant. Nutropin AQ: Not to be used for growth promotion in pediatric patients with closed epiphyses. Humatrope: Not to be used for growth promotion in pediatric patients with closed epiphyses. Genotropin: Contraindicated in patients with PWS who have uncontrolled diabetes, or active psychosis, or active cancer. Omnitrope: Not to be used when there is any evidence of neoplastic activity or in pediatric patients with closed epiphyses. Saizen: Not to be used for growth promotion in pediatric patients with closed epiphyses or patients with active neoplasia. | Contraindicated in patients with active malignancy and in patients with closed or fused epiphyses. |
Other | NA | NA | NA |
GH = growth hormone; GHD = growth hormone deficiency; IGF-1 = insulin-like growth factor 1; NA = not applicable; PWS = Prader-Willi syndrome; SC = subcutaneous.
aHealth Canada–approved indication.
Source: Product monographs.1,39-45
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient inputs received by CADTH have been included in the stakeholder section of this report.
Two patient groups, CORD and the MAGIC Foundation, provided input for the treatment of GHD. CORD recruited participants through patient membership list and parents attending a European summit and then conducted interviews, whereas the MAGIC Foundation gathered data from surveys. A total of 12 parents (6 from the list and 6 from the summit) participated in the interviews conducted by CORD. Among these 12 participants, 4 were from Canada (Ontario), 2 were from the US, and 6 were from Europe. All participants responding to the MAGIC Foundation’s surveys were from the US, and none of their current members in Canada had experience with an LAGH. The children represented in the CORD input ranged in age from 4 to 15 years, and those from the MAGIC Foundation input were between 3 and 18 years of age.
When parents were asked by CORD about the impact of the disease on patients’ and caregivers’ day-to-day life and quality of life, they expressed going through a variety of emotions, such as denial, blame, sadness, acceptance, and compassion. Parents from both groups reported a variety of psychological and social impacts on the child and the family, particularly due to short stature. In the MAGIC Foundation’s input, parents mentioned that their children were shorter than their peers, were fatigued, lacked concentration and appetite, had poor stamina, and were often very sick before beginning GH treatment.
Patients from both groups were reported to have been on GH therapy. All patients in the CORD group were reported to have experience with daily subcutaneous injections of somatropin, and 4 patients were reported to have current experience with an LAGH (somatrogon, lonapegsomatropin, and somapacitan-beco) obtained through clinical trials, compassionate access, or reimbursement or insurance. On the other hand, some patients from the MAGIC Foundation group were reported to have experience with both daily and weekly injections, whereas others were reported to be strictly on daily injections.
Parents from the CORD group described the daily administration of an injection every night as the most consistent challenge. These parents also recognized the importance of GH therapy despite the challenges and worries about the future. Parents from the MAGIC Foundation group mentioned the high cost of GH treatment and dependence on insurance companies in getting the treatment for children in the US. When parents were asked about the outcomes to consider when evaluating new therapies by the CORD group, an injection that lasted longer and was easier to administer was a desired change. While describing the experience with the current drug under review, all 4 respondents who had experience with LAGH therapy shared positive feedback, such as having a positive impact on the child’s and the family’s quality of life; some described this impact as “transformational” and “life-changing.”
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of pediatric GHD.
Unmet Needs
The clinical expert indicated that the current treatment paradigm for patients with GHD is to offer recombinant GH (i.e., somatropin), generally with a starting dose of 0.18 mg/kg/week by daily injection (if hypoglycemia is a concern) or, commonly, 6 out of 7 days per week (to provide 1 day per week as a break from injections). According to the clinical expert, the treatment goals are: to restore height by improving HV (with the target of achieving a near-final adult height that is close to the patient’s midparental target heights), to restore metabolic health, to address hypoglycemia (especially in the neonatal or infantile periods), and to restore well-being.
Place in Therapy
According to the clinical expert, several brands of somatropin currently on the market are available to treat GHD. They noted that somapacitan can be used in the treatment of pediatric GHD by those who are willing to try it or who are already using somatropin and want to switch to once-weekly injections because they can no longer tolerate daily injections.
The clinical expert noted that using the ethical principle of justice, it could be appropriate to recommend that patients try somatropin first, especially if somapacitan is significantly more expensive than somatropin. The clinical expert did not believe that all patients should be required to use somatropin before somapacitan. According to the clinical expert, the ethical principle of beneficence requires that physicians prevent harm; if there is an available option that reduces the pain and anxiety of daily injections and is similar in treatment and safety outcomes to the conventional version, then physicians should prescribe the option that reduces harm. Nonetheless, the clinical expert highlighted that somapacitan can be prescribed for treatment-naive patients.
Patient Population
The clinical expert noted that patients who are similar to the ones enrolled in the REAL 4 trial (prepubertal, younger children) are most likely to respond to treatment with somapacitan, although uncertainties remain as to whether patients with other conditions that can affect growth would respond as well, since they were excluded from the trial. According to the clinical expert, an active control, Norditropin (somatropin), was used as the comparison in the REAL 4 trial; hence somapacitan probably confers greater benefit to those with more severe GHD, those with initial lower pretreatment levels of IGF-1, and those with a lower HV SDS.
The clinical expert noted that patients with GHD who were most in need of intervention would be those who experience significant pain or anxiety from the injections to the extent that there is a threat to optimal adherence to daily somatropin.
The clinical expert pointed out that although there are limitations in the diagnosis of GHD using examinations of IGF-1 level and peak GH, the likelihood of overdiagnosis in Canada is probably lower than in other countries, based on findings of the GeNeSIS study,4 which means that pediatric endocrinologists in Canada tend to be more stringent in diagnosing GHD. The clinical expert noted that underdiagnosis of GHD may happen more within the subgroup of children who acquire GHD after treatment for cancer.
The clinical expert noted that although no confirmed predictors have been identified, children who have more severe GHD, initiate GH treatment at a younger age, stay on therapy for a longer duration, and are adherent to therapy are more likely to respond to somapacitan, based on extrapolation from the results of the somatropin studies.
Assessing the Treatment Response
According to the clinical expert, the main outcomes that are used in clinical practice to determine whether a patient is responding to GH therapy include: change in absolute height and height SDS, change in HV and HV SDS, and change in IGF-1 level (and IGF-1 SDS).
Discontinuing Treatment
The clinical expert pointed out that GH therapy is necessary for children with GHD and, therefore, a concerted effort should be made to understand the cause of suboptimal growth before discontinuing somapacitan (or any other GH therapy), including excluding causes that are not related to the GH therapy itself, such as the lack of adherence to therapy for multiple reasons, other comorbid causes of suboptimal growth (nutrition, celiac disease, additional pituitary hormone deficiencies), and injection-technique issues.
According to the clinical expert, a contraindication that precludes ongoing use of somapacitan (or any other GH therapy) is when the epiphyses are closed. Adverse effects such as glucose intolerance, IGF-1 levels that exceed 2 SDS, raised intracranial pressure, slipped caput femoral epiphyses, and avascular necrosis would pause treatment but not necessarily discontinue it. When a patient develops a cancer (primary or secondary), GH therapy would be discontinued (but potentially restarted in the future).
Prescribing Considerations
The clinical expert indicated that somapacitan should be prescribed by board-certified pediatric endocrinologists who can diagnose, treat, and monitor the patients. A pediatric endocrine nurse should be involved in initiating recombinant GH, providing ongoing support, and double-checking that the patient is adhering to prescribed therapy. According to the clinical expert, treatment with somapacitan could happen in community settings or academic referral centres.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the stakeholder section of this report.
Clinician group input on the review of somapacitan was received from the Canadian Pediatric Endocrinology Nurses group. A total of 5 nurses provided input for this review.
Canadian Pediatric Endocrinology Nurses mentioned that daily somatropin injections are being used as the current treatment paradigm for GHD. These injections are administered to increase growth, stabilize blood sugar levels, and increase bone density and muscle development. The group described the treatment gaps or unmet needs of currently available treatments. Issues with current treatments include: poor compliance in patients with daily injections, anxiety with daily injections, lack of availability of GH, and the need for a treatment with improved compliance, better tolerance, and formulations with improved convenience. The group indicated that the drug under review could be used as a first-line treatment for GHD if approved and funded.
While describing which patients would be best suited for treatment with the drug under review, the clinician group mentioned those who experience needle anxiety, have compliance issues, are in a complex social situation, or have remote living conditions. The group also added that patients with GHD can be identified by, for example, clinician examination, GH stimulation testing, bone age, IGF-1 level, and so forth. The group emphasized that without GH (either daily or weekly), patients will not grow and could have hypoglycemia, decreased bone density, poor muscle development, and altered body composition. The clinician group pointed out that improved growth velocity and normalized glucose in infants would be considered a clinically meaningful response to treatment. They also added that factors such as achieving final adult height, closed epiphyses, and a growth rate of less than 2 cm/year should be considered when deciding to discontinue treatment with the drug under review. The clinician group noted the patients must be diagnosed, treated, monitored, and prescribed by a pediatric endocrinologist.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The sponsor submitted a pivotal trial that compared somapacitan and Norditropin (somatropin), which is a once-daily GH. There are no comparative head-to-head trials between somapacitan and Ngenla (Somatrogon, reviewed in October 2021) which is also a once-weekly GH. The sponsor submitted an indirect comparison comparing somapacitan, somatropin, and somatrogon. | For consideration by CDEC; no clinical expert response required. |
Some provinces and territories (e.g., Ontario) fund GH through alternative pathways, e.g., the special drug program at the Hospital for Sick Children (SickKids) in Toronto. Typically, NIHB would not cover pediatric patients experiencing growth failure due to an inadequate secretion of endogenous GH (GH deficiency), as they would apply for funding through the available provincial or territorial routes. | For consideration by CDEC; no clinical expert response required. |
Considerations for initiation of therapy | |
It is possible for patients who are taking daily injections to want to transition to subcutaneous weekly injections if the latter shows efficacy in growth. Often, requests are still received to continue funding somapacitan therapy when the patients in the trial (the study population included girls who were aged 2.5 years to 10 years and boys who were 2.5 years to 11 years of age) have reached adulthood. | For consideration by CDEC; no clinical expert response required. |
If possible, consider aligning with Ngenla (somatrogon). | For consideration by CDEC; no clinical expert response required. |
Considerations for continuation or renewal of therapy | |
Consider aligning with Ngenla (somatrogon). | For consideration by CDEC; no clinical expert response required. |
Considerations for discontinuation of therapy | |
Consider aligning with Ngenla (somatrogon). Question for the clinical expert: Study populations included girls aged 2.5 to 10 years and boys aged 2.5 to 11 years. Often, requests are received to continue funding therapy once the patient has reached adulthood. Should the somapacitan therapy be discontinued when the patient has reached adulthood? | The clinical expert noted that the indication changes when the patient becomes 18 years and older as it changes to adult GH deficiency, and an adult endocrinologist is involved. The individual together with the adult endocrinologist would decide whether to continue GH therapy. |
Considerations for prescribing of therapy | |
Consider aligning with Ngenla (somatrogon). | For consideration by CDEC; no clinical expert response required. |
Generalizability | |
Populations outside the indication or reimbursement request but of interest to jurisdictions comprise patients who are postpuberty or adults with GH deficiency. | For consideration by CDEC; no clinical expert response required. |
System and economic issues | |
Ngenla completed confidential drug pricing negotiations. | For consideration by CDEC; no clinical expert response required. |
CDEC = CADTH Canadian Drug Expert Committee; GH = growth hormone; NIHB = Non-Insured Health Benefits.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of somapacitan 5 mg/1.5 mL (3.3 mg/mL), 10 mg/1.5 mL (6.7 mg/mL), 15 mg/1.5 mL (10 mg/mL) as a prefilled pen for subcutaneous use in the long term treatment of pediatric patients experiencing growth failure due to an inadequate secretion of endogenous GH (i.e., GHD). The focus will be placed on comparing somapacitan with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of somapacitan is presented in 4 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
one pivotal study (RCT) identified and included in systematic review
one long-term extension study
one ITC
one additional study addressing gaps in evidence.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included study are summarized in Table 5. Refer to Appendix 1 for the detailed patient inclusion and exclusion criteria.
One pivotal multicentre, multinational, randomized, open-label, active-controlled, phase III trial (REAL 4) met the inclusion criteria for the sponsor’s systemic review, in which a total of 86 sites randomized 200 patients (Table 5). The REAL 4 trial investigated the efficacy and safety of a once-weekly dosing of somapacitan compared with a once-daily dosing of Norditropin (somatropin) in patients with GHD who were at least 2.5 years of age and younger than 11 years for boys and younger than 10 years for girls. The trial consisted of a 52-week main treatment period that ended on November 10, 2021,20 plus a 156-week (3-year) safety extension period. The total trial duration was 4 years (the 3-year extension period was still ongoing as of the March 18, 2022, cut-off) and the follow-up period was a minimum of 30 days (Figure 1). The main body of this report presents data from the main trial phase for the REAL 4 trial up to week 52. Data from the safety extension phase (week 52 to 104) are presented in the clinical evidence section under long-term extension studies.
Table 5: Details of Study Included in the Systematic Review
Detail | REAL 4 |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, randomized, open-label, active-controlled study |
Locations |
|
Patient enrolment dates |
|
Randomized (N) | Total N = 200:
|
Key inclusion criteria |
|
Key exclusion criteria |
|
Drugs | |
Intervention | Somapacitan 0.16 mg/kg once a week through a prefilled pen for subcutaneous injection. |
Comparator | Norditropin (somatropin) 0.034 mg/kg once a day through a prefilled pen for subcutaneous injection. |
Study duration | |
Screening phase | 2 weeks |
Main trial | 52 weeks |
Long-term safety extension (single-arm phase) | 156 weeks |
Follow-up phase | 30 days |
Outcomes | |
Primary end point | Height velocity at week 52 |
Secondary and exploratory end points | Secondary Efficacy:
Safety:
Exploratory Patient-reported outcomes:
|
Publication status | |
Publications | Miller et al., 202246 Clinicaltrials.gov Identifier: NCT03811535 |
CDC = Centers for Disease Control and Prevention; FPFV = first patient first visit; G-DAT = Growth Hormone Device Assessment Tool; GH = growth hormone; GHD = growth hormone deficiency; GHD-CIM = Growth Hormone Deficiency–Child Impact Measure; GHD-CTB = Growth Hormone Deficiency–Child Treatment Burden; GHD-PTB = Growth Hormone Deficiency–Parent Treatment Burden; IGF-1 = insulin-like growth factor 1; IGFBP-3 = insulin-like growth factor binding protein 3; LPLV = last patient last visit; SD = standard deviation; SDS = standard deviation score; TB-CGHD-O = Treatment Burden Measure–Child Growth Hormone Deficiency–Observer; TB-CGHD-P = Treatment Burden Measure–Child Growth Hormone Deficiency–Parent; TRIM-CGHD-O = Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer; WHO = WHO.
Note: 1 additional report was included (Miller et al., 202246).
aActual key study date.
bPlanned key study date.
Source: REAL 4 trial week 5220 and week 10423 Clinical Study Reports.
Patients were randomized 2:1 to receive either somapacitan or Norditropin (somatropin) during the 52-week main trial period. Randomization was stratified by region (Japan versus the rest of the world, possibly because of different peak GH levels used in GHD diagnosis), age group (< 6 years versus ≥ 6 years at randomization), gender (boys versus girls), and GH peak level (< 7.0 ng/mL versus ≥ 7.0 ng/mL). During the subsequent 3-year safety extension trial period, all patients were treated with once-weekly somapacitan. Both trial products were administered as subcutaneous injections. The trial was open-label; no blinding was conducted.
Figure 1: Study Design for the REAL 4 Trial
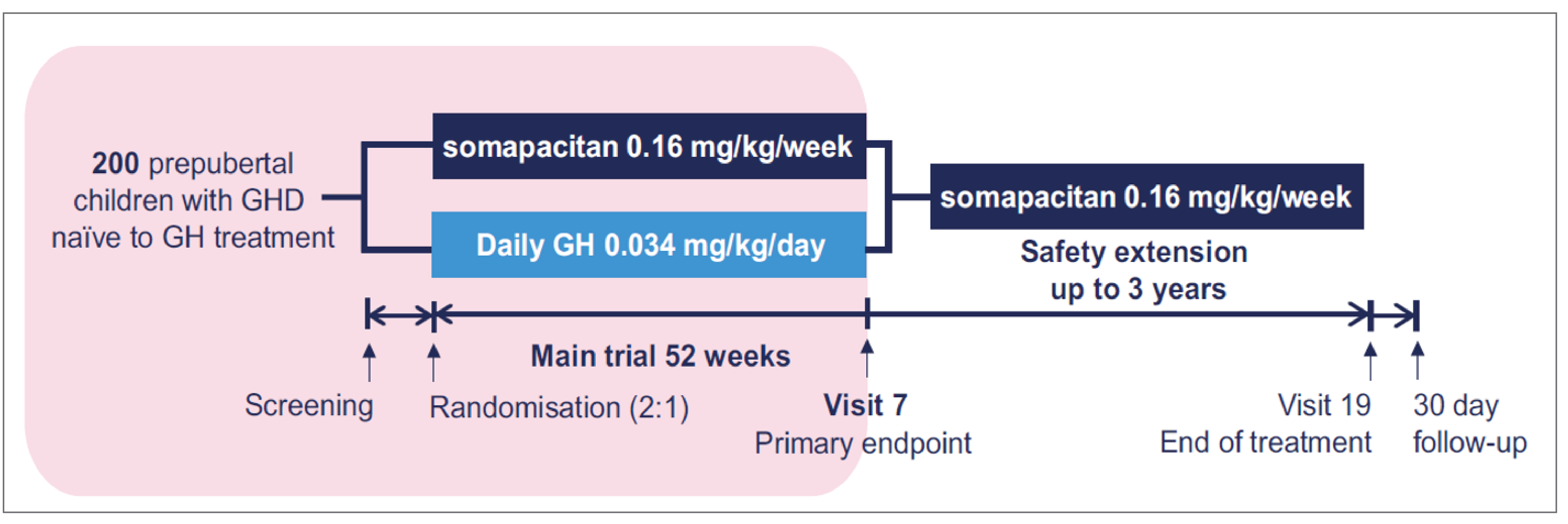
GH = growth hormone; GHD growth hormone deficiency.
Note: Reprinted from Miller et al., 2022. Creative Commons Attribution License 4.0.46
Source: Miller et al., 2022,46 and the week 5220 and week 10423 Clinical Study Reports for the REAL 4 trial.
Populations
Inclusion and Exclusion Criteria
REAL 4 included prepubertal children who were at least 2.5 years old and younger than 11 years for boys and younger than 10 years for girls who had a diagnosis of GHD confirmed by 2 different GH stimulation tests performed in the last 12 months before screening, defined as a peak GH level of 10.0 ng/mL or lower, who had impaired height (at least 2 SD below the mean) and impaired HV (below the 25th percentile) for chronological age and gender, and who had a mean IGF-1 level at least 1 SD below the standardized mean for age and gender. Patients were naive to other GH therapies or IGF-1 treatments. Key exclusion criteria included children with any clinically significant abnormality likely to affect growth or the ability to evaluate growth using standing or length measurements, concomitant administration of other treatments that may have an effect on growth, and a prior history or presence of malignancy and/or intracranial tumour. Additional exclusion criteria are shown in Table 5. Of note, for study sites in Japan, the confirmed diagnosis of GHD was determined by 1 GH stimulation test for patients with intracranial organic disease or symptomatic hypoglycemia and 2 different GH stimulation tests for patients in other countries, defined as peak GH level of 6 ng/mL or lower by assay using recombinant GH standard within 12 months before screening.
Interventions
Somapacitan was provided in a liquid formulation in the PDS290 pen-injector device and administered subcutaneously. In the REAL 4 trial, somapacitan was administered at a dose of 0.16 mg/kg/week throughout the main 52-week trial period and the 3-year extension period. Injections were given subcutaneously and 3 PDS290 pen-injector strengths (5 mg/1.5 mL, 10 mg/1.5 mL, and 15 mg/1.5 mL) were used in the trial based on the patient’s body weight.
The active control, Norditropin (somatropin), was provided in a liquid formulation in the FlexPro pen-injector device at a dose of 0.034 mg/kg/day for subcutaneous administration and was administered for the 52-week main trial period.
In the REAL 4 trial, when patients self-administered the trial product at home, compliance with trial-product administration was assessed and documented at each visit.
If AEs with a probable relationship to the trial product were persistent but allowed continuation in the trial, as judged by the investigator, dose reduction in consecutive steps of 25% of the current dose was considered at the investigator’s discretion. If after consecutive dose-reduction steps the AEs persisted, the patient’s treatment was discontinued according to the treatment discontinuation or withdrawal criteria. When the AE was resolved, the dose could be resumed to the original planned dose at the investigator’s discretion.
If IGF-1 SDS exceeds 2.5 SDS at 2 consecutive visits, Novo Nordisk would inform the investigator. The current dose would then have to be reduced by 25%.
No prior exposure to GH therapy and/or IGF-1 treatment was permitted in either of the pivotal trials.
At visit 2, the patients were provided with an e-diary device for the electronic recording of data. Information about the injection of the trial product was recorded in the e-diary device to provide data on adherence to therapy.
Outcomes
A list of the efficacy end points assessed is provided in Table 6, followed by descriptions of the outcome measures and their measurement properties in Table 7. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review, according to the clinical expert consulted by CADTH and the stakeholder input from the patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected outcome measures considered to be most relevant to inform CADTH’s expert committee deliberations and finalized this list of outcome measures in consultation with members of the expert committee. All summarized efficacy outcome measures were assessed using GRADE and included HV, height SDS and HV SDS. Patient-reported outcomes that reflect patient quality of life or burden from disease and treatment, including GHD-CIM, GHD-CTB, and GHD-PTB, were considered important by the patient groups and were assessed using GRADE. Select notable harms outcomes and treatment discontinuations (or adherence to therapy) that were considered important for informing the deliberations of CADTH’s expert committee were also to be assessed using GRADE. Two notable harms were selected for GRADE assessment: injection-site reactions and injection-site pain, which were recognized as important by the clinical expert consulted by CADTH and could be associated with treatment adherence, which plays a role in GH therapy efficacy in terms of growth. Also, these harms provide the source for a key input in the sponsor’s pharmacoeconomic model.
Table 6: Outcomes Summarized From the REAL 4 Trial
Outcome measure | Time point | REAL 4 |
|---|---|---|
Height velocity | Week 52 | Primary |
Height SDS | Week 52 | Secondary |
Height velocity SDS | Week 52 | Secondary |
GHD-CIM (TB-CGHD-O) | Week 26 and 52 | Exploratory |
GHD-CTB (TB-CGHD-P) | Week 26 and 52 | Exploratory |
GHD-PTB (TRIM-CGHD-O) | Week 26 and 52 | Exploratory |
GHD-CIM = Growth Hormone Deficiency–Child Impact Measure; GHD-CTB = Growth Hormone Deficiency–Child Treatment Burden; GHD-PTB = Growth Hormone Deficiency–Parent Treatment Burden; SDS = standard deviation score; TB-CGHD-O = Treatment Burden Measure–Child Growth Hormone Deficiency–Observer; TB-CGHD-P = Treatment Burden Measure–Child Growth Hormone Deficiency–Parent; TRIM-CGHD-O = Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer.
Source: REAL 4 trial Clinical Study Report (week 52).20
Height Velocity
The primary efficacy end point was change in HV at week 52 measured in cm/year using the following equation:
HV is derived from the height measurement that reflects the rate of change in human stature over time, and an AHV (over 1 year) is commonly used as a measure of growth.47,48 In the REAL 4 trial, the calculation of HV was based on the difference between the heights at week 52 and baseline and the value was converted to be expressed in cm/year using the preceding formula. Standing height was measured by a trained person blinded to treatment allocation using a calibrated wall-mounted stadiometer, preferably using the same stadiometer at the same time of day (± 2 hours, compared with baseline). The average of the standing height, measured 3 consecutive times in centimetres or inches and rounded to 1 decimal, was used to determine the patient’s height during the visit.
Other Auxologic Response
Other auxologic responses were secondary end points in the REAL 4 trial, evaluated as change from baseline to week 52 in height SDS and HV SDS. SDS-related measurements are typically used as end points for outcomes such as height and HV because they are influenced by age and gender. Standards of these measurements have been developed and used by clinicians treating GHD.12
Height SDS was derived using the Centers for Disease Control and Prevention’s 25 standards, and HV SDS was derived using Prader standards as reference data.
Patient-Report Outcomes
The following patient-reported outcome questionnaires were completed:
the GHD-CIM, previously called the Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer (TRIM-CGHD-O)
the GHD-CTB, previously called the Treatment Burden Measure–Child Growth Hormone Deficiency–Observer (TB-CGHD-O)
the GHD-PTB, previously called the Treatment Burden Measure–Child Growth Hormone Deficiency–Parent (TB-CGHD-P)
the Growth Hormone Device Assessment Tool (G-DAT).
These 4 questionnaires were completed only in countries where they were available.
The TRIM-CGHD-O is a disease-specific questionnaire that measures the impact of GH treatment on symptoms, physical health, and social and emotional well-being of children with GHD. Based on the responses, an overall score as well as domain-specific scores can be derived. The updated name of the TRIM-CGHD-O questionnaire is GHD-CIM. A lower score indicates a better health state. The scores of the GHD-CIM range from 0 to 100. Anchor-based patient and clinician ratings of severity of disease suggest a preliminary MID of 5 points for the overall score, 5 points for the physical functioning domain score, 7 points for the emotional well-being domain score, and 5 points for the social well-being domain score.49
The TB-CGHD-O is a disease-specific questionnaire that measures the physical burden of GH treatment as well as its burden on emotional well-being and interference in the activities of daily life of children with GHD. Based on the responses, an overall score as well as domain-specific scores can be derived. The updated name of the TB-CGHD-O questionnaire is the GHD-CTB. The scores of the GHD-CTB range from 0 to 100, with a lower score indicating a better health state. Anchor-based patient and clinician ratings of severity of disease suggest a preliminary MID of 6 points for the physical domain score, 9 points for the emotional domain score, and 6 points for the interference domain score.50
The TB-CGHD-P is a disease-specific questionnaire that measures the burden of GH treatment on the emotional well-being of the parent or guardian as well as the interference in the activities of daily life of the parent or guardian. Based on the responses, an overall score and domain-specific scores can be derived. The updated name of the TB-CGHD-O questionnaire is the GHD-PTB. The scores of the GHD-PTB range from 0 to 100, with a lower score indicating a better health state. Anchor-based patient and clinician ratings of severity of disease suggest a preliminary MID of 10 points for the emotional domain score, and 6 points for the interference domain score.50
The G-DAT is a questionnaire that includes 6 questions completed by the parent or LAR to gather information on how the parents feel about their child’s GH device. The final question was “Overall, how difficult or easy is it to use the device?” Answers were scored on a 5-level scale from “very difficult” to “very easy.”
Safety
AE recording and laboratory measurements for safety were conducted when patients fulfilled the prespecified study visits at weeks 4, 13, 26, 39, and 52.46,51 An AE was defined as any untoward medical event that occurred during the period when a patient was receiving a product but that did not necessarily have a causal relationship with the treatment. An injection-site reaction is defined as “an injection site reaction considered clinically significant by the investigator.” Digital photos were required to be taken of the injection-site reaction at the time of identification as frequently as judged necessary by the investigator, and then evaluated by an external dermatologist. Pain (general or at the injection site) was assessed as part of the injection-site reactions noted during the visual and manual inspection performed by the investigator at each visit.51
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
GHD-CIM (TRIM-CGHD-O) | TRIM-CGHD-O is a disease-specific questionnaire that measures the impact of GH treatment on symptoms, physical health, and social and emotional well-being of children with GHD. Based on the responses, an overall score as well as domain-specific scores can be derived. The updated name of the TRIM-CGHD-O questionnaire is GHD-CIM. A lower score indicates a better health state. GHD-CIM scores range from 0 to 100.49 | Validity: Known-groups validity hypotheses for the social well-being and emotional well-being domains were significant (P < 0.05) for children with GHD aged 9 to < 13 years.49 Reliability: All coefficients exceeded the threshold of 0.70, indicating internal consistency reliability for all domain scores and for the overall score in children with GHD aged 9 to < 13 years.49 Responsiveness: Associated effect sizes ranged from –0.40 to –0.58, indicating that the GHD-CIM is sensitive to change.49 | Anchor-based patient and clinician ratings of severity of disease suggest a preliminary MID of 5 points for overall score, 5 points for physical functioning domain score, 7 points for emotional well-being domain score, and 5 points for social well-being domain score (within-patient change).49 |
GHD-CTB (TB-CGHD-O) | TB-CGHD-O is a disease-specific questionnaire that measures the physical burden of GH treatment as well as the burden of GH treatment on emotional well-being and interference in daily life activities of children with GHD. Based on the responses, an overall score as well as domain-specific scores can be derived. The updated name of the TB-CGHD-O questionnaire is GHD-CTB. GHD-CTB scores range from 0 to 100, with a lower score indicating a better health state.50 | Validity: All but 1 of the convergent validity hypotheses were proven (r > 0.40). Known-groups validity hypotheses were significant for length of time to administer the injections (P < 0.001 for the physical, emotional, and overall domains, and P < 0.01 for the interference domain).50 Reliability: Internal consistency was acceptable for all measures (Cronbach alpha > 0.70). Test–retest reliability was acceptable for the physical, emotional, and overall domains.50 Responsiveness: Associated effect sizes ranged from −0.27 to −0.57, indicating that the measures are sensitive to change.50 | Anchor-based patient and clinician ratings of severity of disease suggest a preliminary MID of 6 points for the physical domain score, 9 points for the emotional domain score, and 6 points for the interference domain score.50 |
GHD-PTB (TB-CGHD-P) | TB-CGHD-P is a disease-specific questionnaire that measures the burden of GH treatment on the emotional well-being of the parent or guardian as well as the interference in the daily life activities of the parent or guardian. Based on the responses, an overall score as well as domain-specific scores can be derived. The updated name of the TB-CGHD-O questionnaire is GHD-PTB. GHD-PTB scores range from 0 to 100, with a lower score indicating a better health state.50 | Validity: All convergent validity hypotheses were proven (r > 0.40). Known-groups validity was supported by the GHD-PTB scores that were able to discriminate between whether the parent or guardian gave the injections more often than the child for the emotional domain (P < 0.05).50 Reliability: Internal consistency was acceptable for all measures (Cronbach alpha > 0.70). Test–retest reliability was acceptable for the emotional and overall domains.50 Responsiveness: Associated effect sizes ranged from −0.74 to −0.69, indicating that the measures are sensitive to change.50 | Anchor-based patient and clinician ratings of severity of disease suggest a preliminary MID of 10 points for the emotional domain score, and 6 points for the interference domain score.50 |
G-DAT | G-DAT is a questionnaire that includes 6 questions that were completed by the patient’s parent or LAR to gather information on how the parents feel about their child’s growth hormone device. The final question was “Overall, how difficult or easy is it to use the device?” Answers were scored on a 5-level scale from “Very difficult” to “Very easy.” | Validity, reliability, and responsiveness: No evidence found. | No evidence found. |
G-DAT = Growth Hormone Device Assessment Tool; GHD-CIM = Growth Hormone Deficiency–Child Impact Measure; GHD-CTB = Growth Hormone Deficiency–Child Treatment Burden; GHD-PTB = Growth Hormone Deficiency–Parent Treatment Burden; LAR = legally authorized representative; MID = minimal important difference; TB-CGHD-O = Treatment Burden Measure–Child Growth Hormone Deficiency–Observer; TB-CGHD-P = Treatment Burden Measure–Child Growth Hormone Deficiency–Parent; TRIM-CGHD-O = Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer.
Statistical Analysis
Clinical Trial End Points
Different estimand strategies for the FDA and European Medicines Agency (EMA) were used based on recommendations from the health authorities (Table 8). For the FDA, the treatment policy strategy was to use the calculated treatment difference in mean AHV at week 52 between somapacitan and Norditropin (somatropin) for all randomized patients with GHD, regardless of treatment adherence or initiation of ancillary therapy. For the EMA, the hypothetical strategy (no ancillary therapy available) used the treatment difference between somapacitan and Norditropin (somatropin) in mean AHV at week 52 when ancillary therapy had not been available before week 52, i.e., assuming no initiation of ancillary therapy in children with GHD. For the FDA strategy, the primary analysis of the primary end point was based on the FAS. For the EMA strategy, the primary analysis of the primary end point was based on the FAS, but the data assessed after the discontinuation of randomized treatment were disregarded. A tipping-point analysis was conducted for the EMA strategy, since 1 patient discontinued treatment. As there were no missing values at week 52 for the FDA strategy, no imputations were done.
The following 2 observation periods were used in the trial:
“On-treatment” period: Including first administration and up until the last trial contact, visit 7, or 14 days after last administration, whichever came first.
“In-trial” period: Including first administration and up until visit 7 or the last trial contact, whichever came first.
The secondary efficacy end points (bone age, height SDS, HV SDS) were analyzed based on the in-trial observation period during the main trial period. Data were analyzed as the change from screening (week 0) to visit 7 (week 52). The time interval used for the derivation of the baseline HV was defined as the period from the date of the pretrial height assessment (a minimum of 6 months and a maximum of 18 months before screening) to the date of the randomization visit.
Change in height SDS and HV SDS were analyzed using the same analysis model that was used for analyzing the primary end point for the primary estimand (treatment policy strategy), except that baseline height SDS and baseline HV SDS, respectively, were used as covariates in the model instead of baseline height. The estimate for the treatment difference at week 52 was reported with the corresponding 95% CI and P value.
Change in bone age was analyzed using an analysis of covariance (ANCOVA) model on the change in bone age or chronological age assessed at week 52. The model included treatment, gender, age group, region, GH peak group, and gender by age group by region interaction term as factors and bone age or chronological age at screening as a covariate. The treatment difference estimate was reported with the corresponding 95% CI and P value. Patients for whom no postrandomization data were available for the analyzed end point were not included in the analysis.
The pharmacodynamic end points (IGF-1 SDS and IGFBP-3 SDS) were analyzed based on the on-treatment observation period and the in-trial observational period using the mixed-model for repeated measures (MMRM). The patient-reported outcomes were analyzed based on the “on-treatment” observational period using an MMRM.
The supportive secondary safety end points (fasting plasma glucose, homeostatic model assessment, hemoglobin A1C levels) were analyzed using descriptive statistics based on the on-treatment observation period and the in-trial observation period. Data were measured as a change from screening (visit 1) to visit 7 (week 52). Homeostasis model assessment (HOMA) was reported as steady-state beta cell function (HOMA-B) and insulin resistance (HOMA-IR).
According to the protocol, the missing week 52 values were planned to be imputed. The missing week 52 values were imputed using a Markov chain Monte Carlo method. This imputation was carried out for each treatment group separately and 100 copies of the dataset were generated (seed = 5,297). The number of copies was increased if the estimation process did not result in robust estimates.
Sample Size and Power Calculation
The sample size was determined based on the primary end point of HV using a noninferiority margin of −1.8 cm/year and a 1-sided 2-group t test with a significance level of 2.5% for a 2:1 randomization ratio between somapacitan and Norditropin (somatropin). Previous trials that have allowed ancillary therapy or the retrieval of landmark data included in the analysis are limited; therefore, a conservative SD of 3.5 cm/year for HV at week 52 was chosen. With an SD of 3.5 cm/year and 90% power for the primary analysis, a sample size of 192 patients was calculated under the assumption of a true difference in AHV of 0 cm/year between the 2 treatment groups and no true difference in AHV between retrieved patients (i.e., subjects discontinuing randomized treatment but have landmark visit data) and patients staying on randomized treatment at the landmark visit. The sample size for the EMA was determined using the same assumptions as for the estimand for the FDA; for a total of 192 patients, the power was 88%.
Statistical Testing
The noninferiority of somapacitan was confirmed when the lower boundary of the 2-sided 95% CI was above −1.8 cm/year or equivalent when the P value for the 1-sided test of H0 (D ≤ −1.8 cm/year) versus HA (D > −1.8 cm/year) was less than 2.5%, where D was the mean treatment difference (somapacitan minus Norditropin [somatropin]). If noninferiority was confirmed, superiority was considered confirmed if the lower boundary of the 2-sided 95% CI was above 0 cm/year. If superiority was confirmed, the P value from the 2-sided superiority test was also reported. A tipping-point analysis was conducted for the primary outcome (and estimands) and based on a penalty for imputed values in the somapacitan arm of 1.8 cm/year, giving a power of 87% (adjusted treatment effect = 0.9 × 0 minus 0.05 × 1.8 = −0.09), under the assumption that landmark visit data (corresponding to an analysis that performs an imputation under the noninferiority null method) is not available for 5% of patients.
Subgroup Analyses
No subgroup analyses were planned or conducted.
Table 8: Statistical Analysis of Efficacy End Points for the REAL 4 Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
REAL 4 | ||||
Mean height velocity at week 52 |
|
| Missing week 52 values were imputed using a Markov chain Monte Carlo method. This imputation was carried out for each treatment group separately and 100 copies of the dataset were generated (seed = 5,297). The number of copies was increased if the estimation process did not result in robust estimates. | Tipping-point analysis |
Mean change from baseline in height SDS at week 52 | ANCOVA | None | ||
Mean change from baseline in height velocity SDS at week 52 | ANCOVA | None | ||
Mean change from baseline in GHD-CIM (TB-CGHD-O) at weeks 26 and 52 | MMRM |
| Missing week 52 values were imputed using a Markov chain Monte Carlo method. This imputation was carried out for each treatment group separately and 100 copies of the dataset were generated (seed = 5,297). The number of copies was increased if the estimation process did not result in robust estimates. | None |
Mean change from baseline in GHD-CTB (TB-CGHD-P) at weeks 26 and 52 | MMRM | None | ||
Mean change from baseline in GHD-PTB (TRIM-CGHD-O) at weeks 26 and 52 | MMRM | None | ||
G-DAT | Descriptive statistics | NA | ||
GH-PPQ | ||||
ANCOVA = analysis of covariance; G-DAT = Growth Hormone Device Assessment Tool; GH = growth hormone; GHD-CIM = Growth Hormone Deficiency–Child Impact Measure; GHD-CTB = Growth Hormone Deficiency–Child Treatment Burden; GHD-PPQ = Growth Hormone Patient Preference Questionnaire; GHD-PTB = Growth Hormone Deficiency–Parent Treatment Burden; MMRM = mixed-model for repeated measures; NA = not applicable; SDS = standard deviation score; TB-CGHD-O = Treatment Burden Measure–Child Growth Hormone Deficiency–Observer; TB-CGHD-P = Treatment Burden Measure–Child Growth Hormone Deficiency–Parent; TRIM-CGHD-O = Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer.
Source: REAL 4 trial Clinical Study Report (week 52).20
Analysis Populations
The efficacy end points in the REAL 4 trial were analyzed based on the FAS, which included all randomized patients as randomized (Table 9). The safety population included all randomized patients who received at least 1 dose of treatment, analyzed as treated.
Results
Patient Disposition
In the REAL 4 trial, 200 patients were randomized. One patient discontinued treatment with somapacitan due to a violation of the inclusion and/or exclusion criteria. Refer to Table 10 for the data.
Table 9: Analysis Populations of REAL 4
Population | Definition | Application |
|---|---|---|
FAS | All randomized patients, analyzed as randomized | Efficacy end points |
Per-protocol set | Patients from the FAS who did not violate any inclusion or exclusion criteria and had used the randomized treatment for at least 47 weeks (for patients receiving somapacitan) or 329 days (for patients receiving Norditropin [somatropin]) corresponding to 90% of the planned exposure, during the main trial period, analyzed as treated | Used to confirm the robustness of the primary statistical analysis |
Safety analysis set | All randomized patients who received at least 1 dose of treatment, analyzed as treated | Evaluation of safety end points |
FAS = full analysis set.
Source: REAL 4 trial Clinical Study Report (week 52).20
Table 10: Summary of Patient Disposition From REAL 4 (Week 52)
Patient disposition | Somapacitan | Norditropin (somatropin) |
|---|---|---|
Screened, N | 348 | |
Reason for screening failure, n (%) | ||
Did not meet the ninth inclusion criterion of IGF-1 < −1.0 | ||| |||||| | |
Did not meet the fifth inclusion criterion of impaired height velocity | || ||||| | |
Did not meet the fourth inclusion criterion of impaired height | ||||| | |
Did not meet the seventh inclusion criterion of bone age less than chronological age | ||||| | |
Did not meet the eighth inclusion criterion of body mass index > the fifth and < 95th percentile | ||||| | |
Did not meet the 11th inclusion criterion of no intracranial tumour | ||||| | |
Did not meet the third inclusion criterion of confirmed diagnosis of growth hormone deficiency | ||||| | |
Did not meet the 10th inclusion criterion of adequate and stable hormone replacement therapies for any other hormone deficiency for ≥ 90 days before randomization | ||||| | |
Met any of the exclusion criteria | || ||||| | |
Randomized, N (%) | 132 (100) | 68 (100) |
Discontinued from study, n (%) | 0 | 0 |
Reason for discontinuation from study, n (%) | ||
Withdrawn by parent or guardian | 0 | 0 |
Discontinued from treatment, n (%) | 1 (0.8) | 0 |
Reason for discontinuation from treatment, n (%) | ||
Adverse events | 0 | 0 |
Protocol violation | 1 (0.8) | 0 |
Withdrawn by parent or guardian | 0 | 0 |
FAS, N (%) | 132 (100) | 68 (100) |
PP, N (%) | 122 (92.4) | 64 (94.1) |
Safety, N (%) | 132 (100) | 68 (100) |
FAS = full analysis set; IGF-1 = insulin-like growth factor 1; PP = per protocol.
Source: REAL 4 trial Clinical Study Report (week 52).20
Baseline Characteristics
The baseline characteristics outlined in Table 11 are limited to those considered to be most relevant to this review or which were believed to affect the outcomes or interpretation of the study results.
In general, the patient demographics and disease characteristics were similar between groups. The mean age of patients enrolled in the REAL 4 trial was 6.38 years (SD = 2.23) and 6.43 years (SD = 2.42) in the somapacitan and Norditropin (somatropin) groups, respectively. Overall, the proportion of boys (74.5%) was higher than that of girls (25.5%); most patients were white (57%), with fewer patients who were Asian (37%) or Black (1 patient, 0.5% of total), or who were either of another race or their race was unknown (0.8% to 5.3%).
The mean BMI values of patients were 15.7 kg/m2 (SD = 1.59) and 15.59 kg/m2 (SD = 1.38) in the somapacitan and Norditropin (somatropin) groups, respectively. The proportion of idiopathic GHD in the somapacitan group (87.1%) was slightly lower than in the Norditropin (somatropin) group (89.7%). Most of the growth-related parameters in the somapacitan group (higher GH peak level, greater HV, less divergence from the normal range of −2 to 2 in HV SDS and height SDS) were slightly superior to those in the Norditropin (somatropin) group.
Exposure to Study Treatments
In the REAL 4 trial, 1 patient from the somapacitan group discontinued treatment during the 52-week duration (Table 10). The majority of patients received the planned treatment, with a mean adherence of 95.8% among patients receiving somapacitan and 88.3% among patients receiving Norditropin (somatropin), indicating possibly more missing doses among patients on Norditropin (somatropin). The mean and median duration of on-treatment days were similar between treatment groups (Table 12).
Table 11: Summary of Baseline Characteristics From the REAL 4 Trial (Full Analysis Set)
Characteristic | Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) |
|---|---|---|
Age, years | ||
Mean (SD) | 6.38 (2.23) | 6.43 (2.42) |
Median (range) | 6.15 (2.5 to 10.8) | 6.15 (2.7 to 11.0) |
< 6 years, n (%) | 64 (48.5) | 33 (48.5) |
≥ 6 years, n (%) | 68 (51.5) | 35 (51.5) |
Sex, n (%) | ||
Male | 99 (75.0) | 50 (73.5) |
Female | 33 (25.0) | 18 (26.5) |
Race, n (%) | ||
Asian | 46 (34.8) | 28 (41.2) |
Black or African American | 0 | 1 (1.5) |
White | 78 (59.1) | 36 (52.9) |
Other | 1 (0.8) | 0 |
Not reported | 7 (5.3) | 3 (4.4) |
Height, cm | ||
Mean (SD) | 102.3 (12.5) | 100.2 (15.0) |
Median (range) | 100.4 (74.9 to 128.2) | 97.9 (58.2 to 128.6) |
Weight, kg | ||
Mean (SD) | 16.69 (4.60) | 16.01 (4.95) |
Median (range) | 15.15 (9.2 to 31.2) | 14.80 (5.2 to 27.4) |
BMI, kg/m2 | ||
Mean (SD) | 15.70 (1.59) | 15.59 (1.38) |
Median (range) | 15.32 (13.2 to 22.8) | 15.38 (12.6 to 18.8) |
GHD cause, n (%) | ||
Idiopathic | 115 (87.1) | 61 (89.7) |
Organic | 17 (12.9) | 7 (10.3) |
GH peak, mcg/L | ||
Mean (SD) | 4.93 (2.50) | 4.10 (2.77) |
Median (range) | 5.20 (0.30 to 9.80) | 3.90 (0 to 9.50) |
Mother’s height, cm | ||
Mean (SD) | 159.3 (7.6) | 158.1 (7.1) |
Median (range) | 160.0 (124.5 to 180.0) | 157.5 (146.0 to 173.8) |
Father’s height, cm | ||
Mean (SD) | 171.5 (8.7) | 170.3 (8.0) |
Median (range) | 171.5 (135.0 to 191.0) | 170.1 (152.0 to 192.0) |
Baseline mean (SD) | ||
Height velocity (cm/year) | 4.3 (1.4) | 4.1 (1.4) |
Height velocity SDS | −2.35 (1.51) | −2.52 (1.55) |
Height SDS | −2.99 (1.02) | −3.47 (1.52) |
IGF-1 SDS | −2.03 (0.97) | −2.33 (1.03) |
IGFBP-3 SDS | −1.89 (1.12) | −2.18 (1.27) |
BMI = body mass index; GH = growth hormone; GHD = growth hormone deficiency; IGF-1 = insulin-like growth factor 1; IGFBP-3 = insulin-like growth factor binding protein 3; SD = standard deviation; SDS = standard deviation score.
Source: REAL 4 trial Clinical Study Report (week 52).20
Table 12: Summary of Patient Exposure From the REAL 4 Trial (Full Analysis Set)
Exposure | Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) |
|---|---|---|
Main trial period (week 52) | ||
Total, days | 48,734 | 25,166 |
Duration, days, mean (standard deviation) | 369.2 (28.8) | 370.1 (3.7) |
Duration, days, median (range) | 370 (56 to 458) | 369 (364 to 387) |
Adherence, mean (%)a | 95.8 | 88.3 |
aRepresents adherence according to diary: Number of administered dosings reported in diary divided by the number of planned dosings multiplied by 100.
Source: REAL 4 trial Clinical Study Report (week 52).20
Concomitant Medications and Co-Interventions
Fewer patients in the somapacitan group (38.6%) than in the Norditropin (somatropin) group (41.2%) received concomitant medication at baseline, whereas more patients in the somapacitan group (66.7%) initiated concomitant medication than in the Norditropin (somatropin) group (58.8%) after randomization. The most frequently used medications at baseline included thyroid hormones (8.3% versus 14.7% in the somapacitan and Norditropin (somatropin) arms, respectively), and vitamin D and analogues (6.8% versus 5.9%). After randomization, the most frequently used medications were anilides (18.9% versus 16.2%) and propionic acid derivatives (13.6% versus 13.2%). Refer to Table 13 for the most frequently reported concomitant medications.
Table 13: Summary of Concomitant Medications From the REAL 4 Trial by ATC (Full Analysis Set)
Variable | Concomitant medication at baseline | Concomitant medication initiated after randomization | ||
|---|---|---|---|---|
Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | |
All medications, n (%) | ||||
All medications | 51 (38.6) | 28 (41.2) | 88 (66.7) | 40 (58.8) |
Medications by ATC, n (%) | ||||
Thyroid hormones | 11 (8.3) | 10 (14.7) | 9 (6.8) | 3 (4.4) |
Vitamin D and analogues | 9 (6.8) | 4 (5.9) | 8 (6.1) | 3 (4.4) |
Piperazine derivatives | 4 (3.0) | 4 (5.9) | 5 (3.8) | 2 (2.9) |
Calcium, combinations with vitamin D and/or other drugs | 5 (3.8) | 0 | 1 (1.5) | |
Leukotriene receptor antagonists | 2 (1.5) | 3 (4.4) | 5 (3.8) | 4 (5.9) |
Other antihistamines for systemic use | 1 (0.8) | 3 (4.4) | 7 (5.3) | 7 (10.3) |
Selective beta2 adrenoreceptor agonists | 2 (1.5) | 2 (2.9) | 7 (5.3) | 4 (5.9) |
Melatonin receptor agonists | 2 (1.5) | 1 (1.5) | 0 | 2 (2.9) |
Multivitamins with minerals | 3 (2.3) | 0 | 0 | 1 (1.5) |
Anilides | 1 (0.8) | 1 (1.5) | 25 (18.9) | 11 (16.2) |
Mucolytics | NR | NR | 9 (6.8) | 4 (5.9) |
Calcium | 2 (1.5) | 0 | NR | NR |
Iron preparations | 1 (0.8) | 1 (1.5) | NR | NR |
Multivitamins, other combinations | 1 (0.8) | 1 (1.5) | NR | NR |
Osmotically acting laxatives | 0 | 2 (2.9) | 0 | 1 (1.5) |
Other lipid-modifying drugs | 2 (1.5) | 0 | 2 (1.5) | 0 |
Various alimentary tract and metabolism products | 1 (0.8) | 1 (1.5) | 1 (0.8) | 0 |
Corticosteroids | 1 (0.8) | 0 | 2 (1.5) | 1 (1.5) |
Corticosteroids, potent (group III) | 0 | 1 (1.5) | 2 (1.5) | 0 |
Enzyme preparations | 1 (0.8) | 0 | NR | NR |
Epinephrine | 1 (0.8) | 0 | 1 (0.8) | 0 |
Heparin group | 0 | 1 (1.5) | 1 (0.8) | 0 |
Heparins or heparinoids for topical use | 1 (0.8) | 0 | 1 (0.8) | 0 |
Iron trivalent, oral preparations | 1 (0.8) | 0 | 1 (0.8) | 0 |
Multivitamins, plain | 1 (0.8) | 0 | 0 | 1 (1.5) |
Other antiepileptics | 1 (0.8) | 0 | NR | NR |
Other emollients and protectives | 1 (0.8) | 0 | 1 (0.8) | 0 |
Propionic acid derivatives | 0 | 1 (1.5) | 18 (13.6) | 9 (13.2) |
Tonics (dietary supplement) | 1 (0.8) | 0 | NR | NR |
Vitamins, other combinations | 1 (0.8) | 0 | 0 | 1 (1.5) |
Multiple ATC | 25 (18.9) | 14 (20.6) | 54 (40.9) | 25 (36.8) |
ACT = anatomic therapeutic chemical classification; NR = not reported.
Source: REAL 4 trial Clinical Study Report (week 52).20
Efficacy
Height Velocity
There was a −0.5 cm/year difference (95% CI, −1.1 to 0.2) in the estimated mean at follow-up between the somapacitan and Norditropin (somatropin) groups from baseline to week 52 (Figure 2), confirming that the noninferiority as the lower boundary of the 95% CI was above the prespecified noninferiority margin of −1.8 cm/year.
Height SDS
The change in the height SDS values from baseline to week 52 for somapacitan and Norditropin (somatropin) were nearly identical, with an ETD of −0.05 (95% CI, −0.18 to 0.08). The observed mean “height SDS” values in both treatment groups increased consistently from week 0 to week 52. The estimated versus the observed mean changes in height SDS from baseline to week 52 were similar: the estimated mean changes were 1.25 for somapacitan and 1.30 for Norditropin (somatropin), and the observed mean changes were 1.21 (SD = 0.54) for somapacitan and 1.37 (SD = 0.69) for Norditropin (somatropin).
Figure 2: HV at Week 52 in the REAL 4 Trial (FAS)
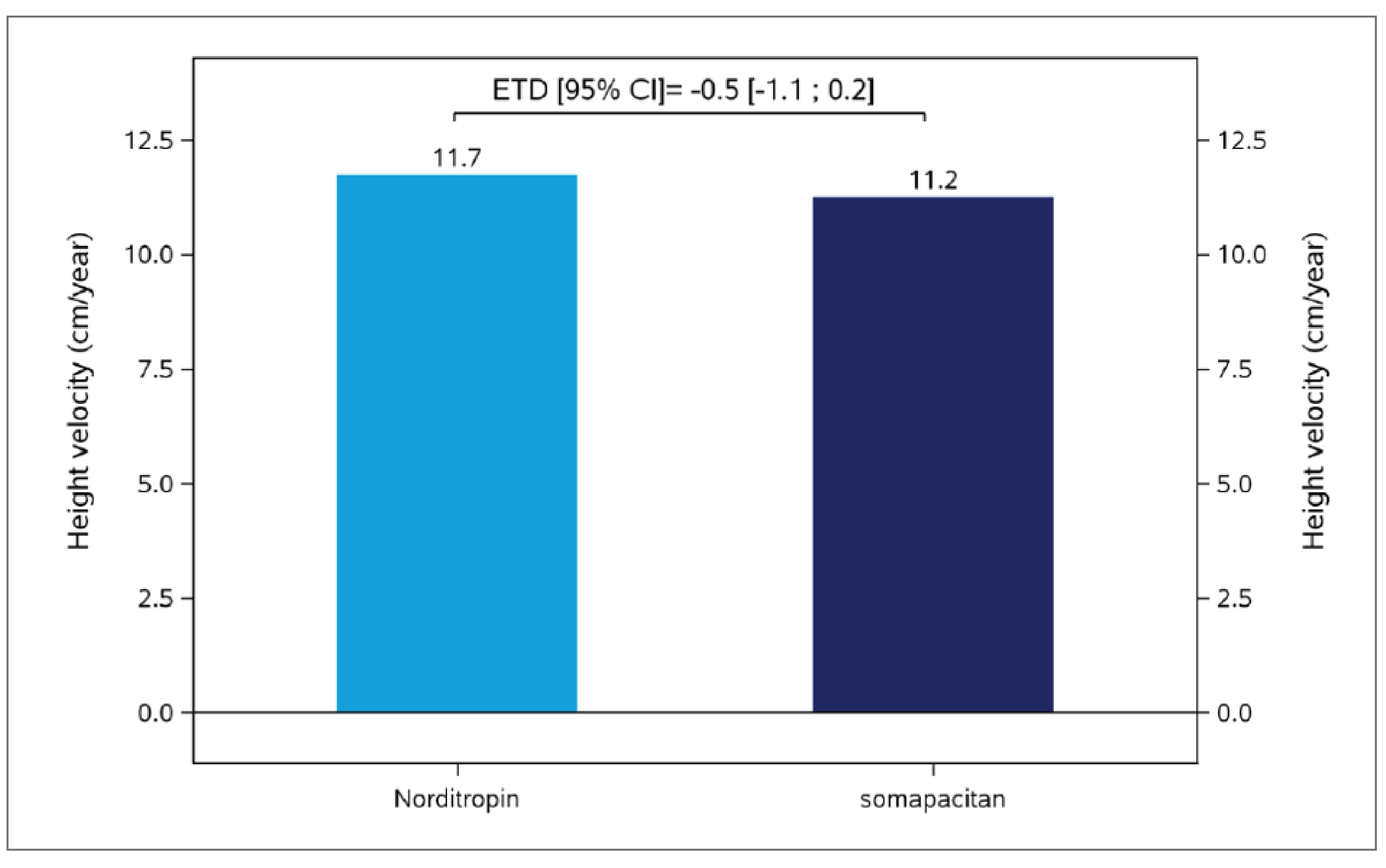
CI = confidence interval; ETD = estimated treatment difference; HV = height velocity; FAS = full analysis set.
Note: Figure was based on the estimated mean. HV at week 52 was analyzed using an analysis of covariance model with treatment, gender, age group, region, growth hormone peak group, and gender by age group by region interaction term as factors, and baseline height as covariate. There were no missing values at week 52, so no multiple imputation was done.
Source: REAL 4 trial Clinical Study Report (week 52).20
Height Velocity SDS
There was a −0.78 difference in the increase in HV SDS between somapacitan and Norditropin (somatropin) at week 52 (95% CI, −1.63 to 0.08). The observed mean (i.e., HV SDS values for the 2 treatment groups using in-trial data) increased from week 0 to week 26, changing from −2.35 (SD = 1.51) to 8.95 (SD = 4.39) and from −2.52 (SD = 1.55) to 9.73 (SD = 4.62); overall, these increases were maintained to week 52 with values of 7.97 (SD = 3.36) and 8.97 (SD = 4.38) for the somapacitan and Norditropin (somatropin) groups, respectively.
A summary of the key efficacy results is shown in Table 14.
Table 14: Summary of Key Efficacy Results From the REAL 4 Trial (FAS)
Variable | Week 26 | Week 52 | ||
|---|---|---|---|---|
Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | |
Auxologic response | ||||
Height velocity (cm/year) | ||||
Baseline (cm/year), mean (SD) | 4.3 (1.4) | 4.1 (1.4) | 4.3 (1.4) | 4.1 (1.4) |
End point (cm/year), observed mean (SD) | 12.2 (3.5) | 12.9 (3.7) | 11.2 (2.5) | 11.8 (2.9) |
End point (cm/year), estimated meana | NR | NR | 11.2 | 11.7 |
Estimated treatment difference (95% CI)a,b | NR | −0.5 (−1.1 to 0.2) | ||
P value | NR | NA | ||
Height SDS | ||||
Baseline, mean (SD) | −2.99 (1.02) | −3.47 (1.52) | −2.99 (1.02) | −3.47 (1.52) |
Observed mean (SD) change from baseline | 0.73 (0.38) | 0.82 (0.45) | 1.21 (0.54) | 1.37 (0.69) |
Estimated mean change from baselinea | NR | NR | 1.25 | 1.30 |
Estimated treatment difference (95% CI)a | NR | −0.05 (−0.18 to 0.08) | ||
P value | NR | 0.4247 | ||
Height velocity SDS | ||||
Baseline, mean (SD) | −2.35 (1.51) | −2.52 (1.55) | −2.35 (1.51) | −2.52 (1.55) |
Observed mean (SD) change from baseline | 8.95 (4.39) | 9.73 (4.62) | 7.97 (3.36) | 8.97 (4.38) |
Estimated mean change from baselinea | NR | NR | 8.05 | 8.82 |
Estimated treatment difference (95% CI)a | NR | −0.78 (−1.63 to 0.08) | ||
P value | NR | 0.0743 | ||
ANCOVA = analysis of covariance; CI = confidence interval; GH = growth hormone; NA = not applicable; NR = not reported; SD = standard deviation; SDS = standard deviation score.
Note: In-trial period results are reported.
aAnalysis conducted using an ANCOVA model, with treatment, gender, age group, region, GH peak group, and gender by age group by region interaction term as factors, and baseline assessment as a covariate.
bSuperiority was not confirmed. Noninferiority was confirmed; the noninferiority margin is −1.8 cm/year.
Source: REAL 4 trial Clinical Study Report (week 52).20
Patient-Reported Outcomes
GHD-CIM (Previously TRIM-CGHD-O)
Improvements were observed for the 3 domain scores and the total score for both treatment groups at the end of the main trial period of 52 weeks, and all of these changes from the baseline scores were similar between somapacitan and Norditropin (somatropin) (Table 15, Figure 3).
Table 15: Summary of GHD-CIM (TRIM-CGHD-O) Scores From the REAL 4 Trial (Full Analysis Set)
Variable | REAL 4 week 26 | REAL 4 week 52 | ||
|---|---|---|---|---|
Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | |
Physical functioning score | ||||
Baseline mean (SD) | 34.3 (15.0) | 34.7 (15.7) | 34.3 (15.0) | 34.7 (15.7) |
Number of patients | 88 | 44 | 91 | 46 |
Estimated change from baselinea | −3.8 | −3.0 | −4.3 | −5.1 |
Estimated treatment difference (95% CI)a | −0.8 (−6.1 to 4.5) | 0.8 (−4.6 to 6.2) | ||
P value | 0.7655 | 0.7686 | ||
Emotional well-being score | ||||
Baseline mean (SD) | 19.2 (18.5) | 22.7 (23.6) | 19.2 (18.5) | 22.7 (23.6) |
Number of patients | 78 | 37 | 81 | 44 |
Estimated change from baselinea | −3.6 | −7.9 | −9.2 | −10.0 |
Estimated treatment difference (95% CI)a | 4.3 (−2.9 to 11.5) | 0.8 (−5.8 to 7.4) | ||
P value | 0.2362 | 0.8064 | ||
Social well-being score | ||||
Baseline mean (SD) | 37.5 (21.1) | 42.8 (20.7) | 37.5 (21.1) | 42.8 (20.7) |
Number of patients | 84 | 39 | 85 | 46 |
Estimated change from baselinea | −4.1 | −11.6 | −14.1 | −13.2 |
Estimated treatment difference (95% CI)a | 7.5 (−0.5 to 15.6) | −0.9 (−9.1 to 7.2) | ||
P value | 0.0675 | 0.8217 | ||
Total score | ||||
Baseline mean (SD) | 30.6 (14.1) | 34.2 (15.9) | 30.6 (14.1) | 34.2 (15.9) |
Number of patients | 87 | 43 | 90 | 48 |
Estimated change from baselinea | −3.2 | −8.7 | −9.0 | −10.9 |
Estimated treatment difference (95% CI)a | 5.6 (0.5 to 10.6) | 1.8 (−2.9 to 6.6) | ||
P value | 0.0307 | 0.4451 | ||
CI = confidence internal; GH = growth hormone; GHD-CIM = Growth Hormone Deficiency–Child Impact Measure; MMRM = mixed-model for repeated measures; SD = standard deviation; TRIM-CGHD-O = Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer.
aAnalysis conducted using an MMRM model, with treatment, gender, age group, region, GH peak group, and gender by age group by region interaction term as factors and baseline value as a covariate, all nested within week as a factor.
Source: REAL 4 trial Clinical Study Report (week 52).20
Figure 3: Forest Plot of Change From Baseline in GHD-CIM (TRIM-CGHD-O) From the REAL 4 Trial at Week 52 (FAS)
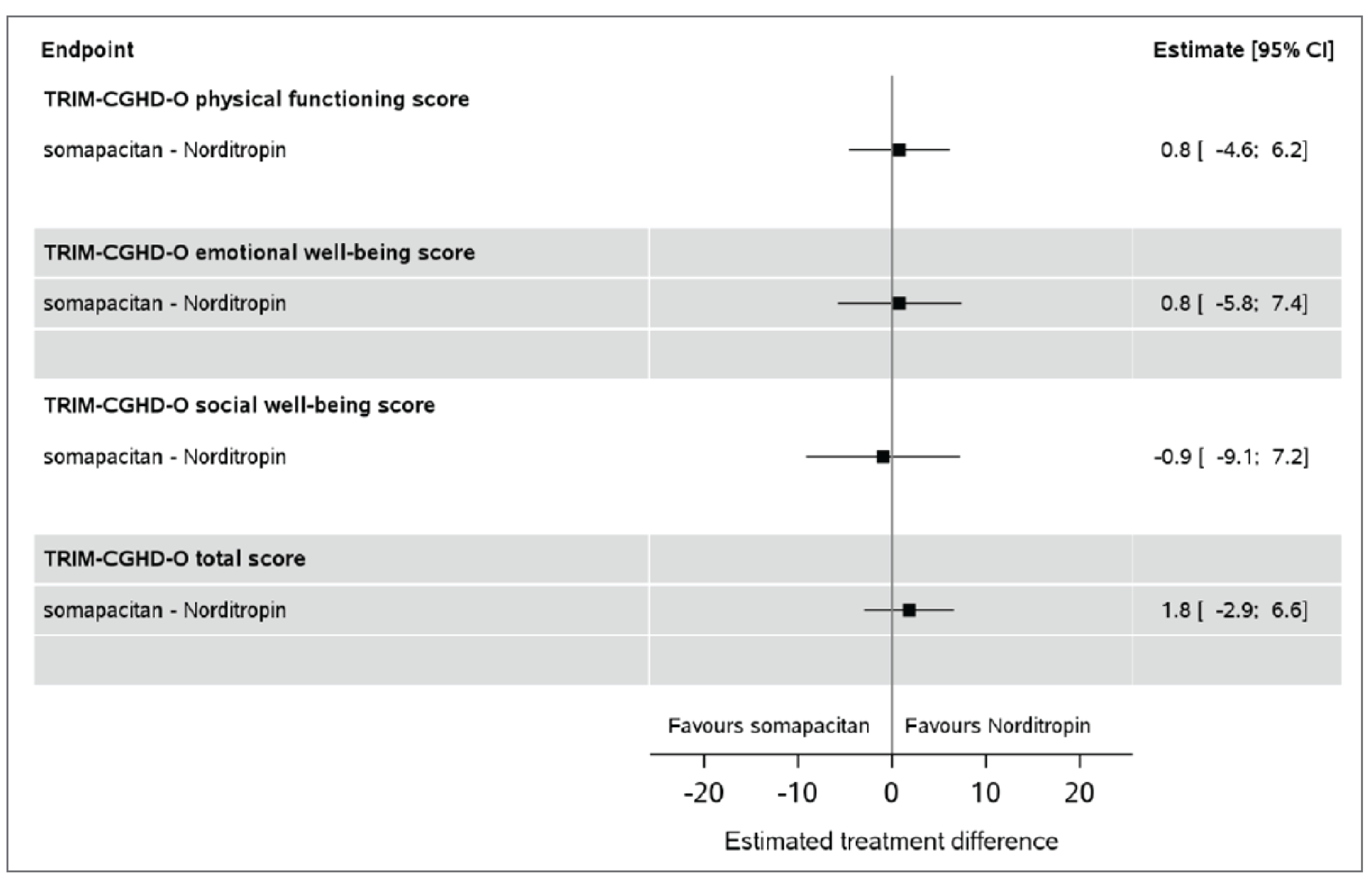
CI = confidence internal; GHD-CIM = Growth Hormone Deficiency–Child Impact Measure; TRIM-CGHD-O = Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer.
Note: The figure depicts REAL 4 trial results for the on-treatment period.
Source: REAL 4 trial Clinical Study Report (week 52).20
GHD-CTB (Previously TB-CGHD-O)
Improvements were observed for the 3 domains (physical, emotional, interference) and the total score for both treatment groups at the end of the main trial period of 52 weeks, and all of the changes from baseline scores were similar between somapacitan and Norditropin (somatropin) (Table 16, Figure 4).
Table 16: Summary of GHD-CTB (TB-CGHD-O) Scores From the REAL 4 Trial (Full Analysis Set)
Variable | REAL 4 week 26 | REAL 4 week 52 | ||
|---|---|---|---|---|
Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | |
Physical functioning score | ||||
Number of patients | 107 | 53 | 116 | 57 |
Estimated meana | 13.5 | 16.3 | 11.6 | 14.5 |
Estimated treatment difference (95% CI)a | −2.7 (−7.1 to 1.6) | −2.9 (−6.8 to 1.0) | ||
P value | 0.2184 | 0.1466 | ||
Emotional well-being score | ||||
Number of patients | 103 | 50 | 112 | 58 |
Estimated meana | 18.1 | 16.9 | 15.5 | 19.1 |
Estimated treatment difference (95% CI)a | 1.2 (−5.0 to 7.3) | −3.5 (−9.5 to 2.4) | ||
P value | 0.7117 | 0.2435 | ||
Interference score | ||||
Number of patients | 105 | 53 | 117 | 59 |
Estimated meana | 5.8 | 9.6 | 5.2 | 6.4 |
Estimated treatment difference (95% CI)a | −3.8 (−7.2 to −0.3) | −1.3 (−3.9 to 1.3) | ||
P value | 0.0336 | 0.3345 | ||
Total score | ||||
Number of patients | 99 | 50 | 112 | 57 |
Estimated meana | 12.3 | 14.0 | 10.7 | 13.1 |
Estimated treatment difference (95% CI)a | −1.7 (−5.4 to 2.0) | −2.4 (−5.7 to 0.9) | ||
P value | 0.3631 | 0.1552 | ||
CI = confidence interval; GH = growth hormone; GHD-CTB = Growth Hormone Deficiency–Child Treatment Burden; MMRM = mixed-model for repeated measures; SD = standard deviation; TB-CGHD-O = Treatment Burden Measure–Child Growth Hormone Deficiency–Observer.
Note: The figure depicts REAL 4 trial results for the on-treatment period.
aAnalysis conducted using an MMRM model, with treatment, gender, age group, region, GH peak group, and gender by age group by region interaction term as factors and baseline value as a covariate, all nested within week as a factor.
Source: REAL 4 trial Clinical Study Report (week 52).20
GHD-PTB (Previously TB-CGHD-P)
The difference in the decrease (improved condition) in domain scores between somapacitan and Norditropin (somatropin) from baseline to week 52 was −5.3 (95% CI, −10.0 to −0.70) in the emotional domain, −6.7 (95% CI, −11.6 to −1.9) in the interference domain, and −6.0 (95% CI, −10.0 to −2.1) in the total score (Table 17 and Figure 4).
Table 17: Summary of GHD-PTB (TB-CGHD-P) Scores From REAL 4 (FAS)
Variable | REAL 4 (week 26) | REAL 4 (week 52) | ||
|---|---|---|---|---|
Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | |
Emotional well-being score | ||||
Number of patients | 109 | 54 | 117 | 59 |
Estimated meana | 14.9 | 21.4 | 12.4 | 17.7 |
Estimated treatment difference (95% CI)a | −6.6 (−11.9 to −1.2) | −5.3 (−10.0 to −0.7) | ||
P value | 0.0167 | 0.0242 | ||
Interference score | ||||
Number of patients | 109 | 54 | 117 | 59 |
Estimated meana | 5.2 | 11.0 | 4.9 | 11.6 |
Estimated treatment difference (95% CI)a | −5.8 (−10.3 to −1.2) | −6.7 (−11.6 to −1.9) | ||
P value | 0.013 | 0.0067 | ||
Total score | ||||
Number of patients | 109 | 54 | 117 | 59 |
Estimated meana | 10.0 | 16.3 | 8.7 | 14.7 |
Estimated treatment difference (95% CI)a | −6.3 (−10.5 to −2.0) | −6.0 (−10.0 to −2.1) | ||
P value | 0.0039 | 0.0031 | ||
CI = confidence interval; GH = growth hormone; GHD-PTB = Growth Hormone Deficiency–Parent Treatment Burden; MMRM = mixed-model for repeated measures; TB-CGHD-P = Treatment Burden Measure–Child Growth Hormone Deficiency–Parent.
Note: The figure depicts REAL 4 trial results for the on-treatment period.
aAnalysis conducted using an MMRM model, with treatment, gender, age group, region, GH peak group, and gender by age group by region interaction term as factors and baseline value as a covariate, all nested within week as a factor.
Source: REAL 4 trial Clinical Study Report (week 52).20
Growth Hormone Device Assessment Tool
The G-DAT questionnaire was used to evaluate and compare the ease of use for the somapacitan and Norditropin (somatropin) devices in the study at week 26. The somapacitan and Norditropin (somatropin) devices were evaluated as “easy” or “very easy” to use (96% and 96%, respectively) and store (95% and 94%, respectively).
Figure 4: Forest Plot of Change From Baseline in GHD-CTB (TB-CGHD-O) and GHD-PTB (TB-CGHD-P) From REAL 4 at Week 52 (FAS)
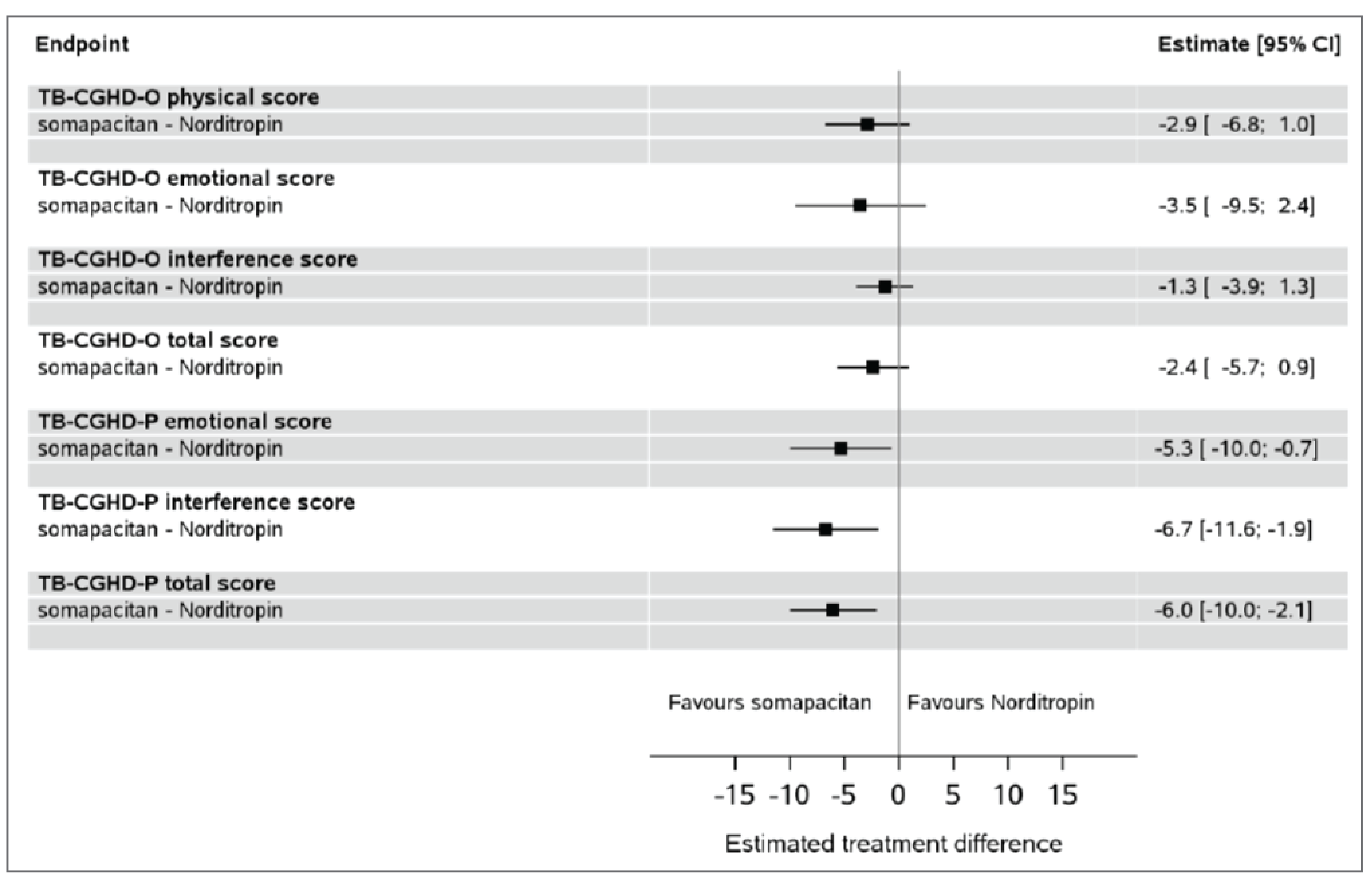
CI = confidence interval; GHD-CTB = Growth Hormone Deficiency–Child Treatment Burden; GHD-PTB = Growth Hormone Deficiency–Parent Treatment Burden; TB-CGHD-O = Treatment Burden Measure–Child Growth Hormone Deficiency–Observer; TB-CGHD-P = Treatment Burden Measure–Child Growth Hormone Deficiency–Parent.
Note: The figure depicts REAL 4 trial results for the on-treatment period.
Source: REAL 4 trial Clinical Study Report (week 52).20
Harms
The REAL 4 trial reported the harms at the end of the main trial period at week 52. Overall, the safety profile of somapacitan was similar to the safety profile of once-daily Norditropin (somatropin) (Table 18). Ninety-four patients (71.2%) reported a total of 310 AEs in the somapacitan group compared with 41 patients (60.3%) reporting a total of 147 AEs in the Norditropin (somatropin) group. The AE reporting rates observed were 232.3 AEs per 100 patient-years (PYs) for somapacitan and 212.8 AEs per 100 PYs for Norditropin (somatropin). The most frequently reported AEs were headache (12%), nasopharyngitis (11%), and pain in extremity (9%) in the somapacitan group, and nasopharyngitis (10%), pyrexia (10%), and headache (9%) in the Norditropin (somatropin) group.
Serious Adverse Events
In the REAL 4 trial, 6 patients (4.5%) reported a total of 8 serious AEs (SAEs) in the somapacitan arm (6.0 SAEs per 100 PYs), and 2 patients (2.9%) reported a total of 3 SAEs in the Norditropin (somatropin) arm (4.3 SAEs per 100 PYs).
Withdrawal Due to Adverse Events
In the REAL 4 trial, no patients discontinued treatment or withdrew from the study due to AEs.
Mortality
No deaths were reported during the main REAL 4 trial period (up to week 52).
Notable Harms
Injection-Site Reactions
Comparably low proportions of children experienced injection-site reactions in the somapacitan group (5.3%) and the Norditropin (somatropin) group (5.9%) in the REAL 4 trial at week 52.
Antibodies
All antibody-positive samples were negative for in vitro neutralizing antibodies. Low-titre, non-neutralizing antibodies were detected in 20 somapacitan-treated patients (15.2%) and 7 patients (10.3%) treated with Norditropin (somatropin). Of these, 2 patients (1.5%) in the somapacitan group and 1 child (1.5%) in the Norditropin (somatropin) group had at least 2 consecutive antibody-positive samples.
IGF-1 SDS Greater Than 2
The mean IGF-1 SDS up to week 52 remained below 2.0 for both treatment groups. In the patients treated with somapacitan, 5 patients (3.8%) had an IGF-1 SDS above 2 at 2 or more consecutive visits during the 52-week treatment period. In the patients treated with Norditropin (somatropin), 2 patients (2.9%) had an IGF-1 SDS above 2.
No trend was seen in the amount or type of AEs reported in patients with an IGF-1 SDS above 2 at 2 or more consecutive visits compared with the remaining children in the relevant groups. None of the patients with an IGF-1 SDS above 2 at 2 or more consecutive visits had any treatment interruptions or dose reductions.
Injection-Site Infections
The incidence of injection-site infections was zero in the REAL 4 trial.
Injection-Site Pain
Two patients (1.5%) in the somapacitan group versus 1 patient (1.5%) in the Norditropin (somatropin) group experienced injection-site pain.
Dysglycemia and Glucose Metabolism
In the REAL 4 trial, 3 patients in each treatment arm (2.3% in the somapacitan group versus 4.4% in the Norditropin [somatropin] group) experienced dysglycemia.
There were no clinically relevant changes from baseline in mean fasting blood glucose or hemoglobin A1C levels and changes were similar for somapacitan and Norditropin (somatropin). Hemoglobin A1C mean values were similar between somapacitan (SD = 5.31%; SD = 0.28%) and Norditropin (somatropin) (5.31%; SD = 0.33%) at week 52. The increase in insulin from baseline was in line with what is expected following GH treatment in children with GHD and was similar between the somapacitan and Norditropin (somatropin) treatment groups. There were no reports of type 2 diabetes during the trial.
Critical Appraisal
Internal Validity
Randomization in the REAL 4 trial was performed using an appropriate methodology with adequate allocation concealment (i.e., web-based, interactive response system), and stratification was based on relevant prognostic factors, i.e., geographical region (Japan versus the rest of the world, with different diagnosis criteria for the 2 regions), sex (boys, girls), age (< 6 years, ≥ 6 years), and GH peak level (< 7.0 ng/mL, ≥ 7.0 ng/mL). Overall, baseline characteristics were generally similar and balanced between groups.
Table 18: Summary of Harms Results From the REAL 4 Trial at Week 52 (Full Analysis Set)
Adverse events | Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) |
|---|---|---|
Most common adverse events, n (%) | ||
≥ 1 adverse event | 94 (71.2) | 41 (60.3) |
Headache | 16 (12.1) | 6 (8.8) |
Nasopharyngitis | 15 (11.4) | 7 (10.3) |
Pyrexia | 10 (7.6) | 7 (10.3) |
Pain in extremity | 12 (9.1) | 2 (2.9) |
Bronchitis | 4 (3.0) | 5 (7.4) |
Vomiting | 6 (4.5) | 4 (5.9) |
Influenza | 3 (2.3) | 2 (2.9) |
Rhinitis | 5 (3.8) | 3 (4.4) |
SAEs, n (%) | ||
Patients with ≥ 1 SAEa | 6 (4.5) | 2 (2.9) |
Patients who stopped treatment due to adverse events, n (%) | ||
Patients who stopped | 0 | 0 |
Deaths, n (%) | ||
Patients who died | 0 | 0 |
Adverse events of special interest, n (%) | ||
Injection-site reactionb | 7 (5.3) | 4 (5.9) |
Relative risk (95% CI)c | |||| |||||| ||||| | Reference |
Risk difference (95% CI)c | ||||| ||||||| ||||| | Reference |
Two consecutive positive GH antibody samples | 2 (1.5) | 1 (1.5) |
IGF-1 SDS > 2 at 2 (or more) consecutive visits | 5 (3.8) | 2 (2.9) |
Relative risk (95% CI) | |||| |||||| ||||| | Reference |
Risk difference (95% CI) | |||| ||||||| ||||| | Reference |
Injection-site infection | 0 | 0 |
Injection-site pain | 2 (1.5) | 1 (1.5) |
Relative risk (95% CI)c | |||| |||||| |||||| | Reference |
Risk difference (95% CI)c | |||| ||||||| ||||| | Reference |
Dysglycemiad,e | 3 (2.3) | 3 (4.4) |
Relative risk (95% CI) | |||| |||||| ||||| | Reference |
Risk difference (95% CI) | ||||| |||||||| ||||| | Reference |
CI = confidence interval; GH = growth hormone; IGF-1 = insulin-like growth factor 1; NR = not reported; SAE = serious adverse event; SDS = standard deviation score.
aIn the REAL 4 trial, SAEs occurred in 6 patients in the somapacitan treatment arm (1 patient had vomiting and dehydration; 1 patient had otitis media and adenoidal hypertrophy; asthma, headache, umbilical hernia, and cryptorchism occurred in 1 patient each) and 2 patients in the Norditropin (somatropin) treatment arm (1 patient had gastroenteritis and acute adrenocortical insufficiency; 1 patient had COVID-19).
bInjection-site reaction (including bruising, pain, hematoma, hypersensitivity, and swelling).
cThese analyses were not part of the statistical analysis plan and were requested by CADTH to allow for an assessment of the imprecision of the findings.
dDysglycemia is defined as either fasting plasma glucose outside normal range (3.61 mmol/L to 5.83 mmol/L) or hemoglobin A1C in blood outside normal range (4% to 6%).
eIn the REAL 4 trial, a number of events of dysglycemia were reported in the somapacitan treatment arm (1 hyperglycemia, 1 impaired fasting glucose, and 1 increased blood glucose) and in the Norditropin (somatropin) treatment arm (2 hyperglycemia and 1 increased hemoglobin A1C).
Source: REAL 4 trial Clinical Study Report (week 52).20
The proportion of idiopathic GHD was slightly lower in the somapacitan arm (87.1%) than in the Norditropin (somatropin) arm (89.7%). The baseline growth-related parameters, such as height SDS, were closer to the normal range of −2 to 2 (−2.99 versus −3.47) in the somapacitan arm versus the Norditropin (somatropin) arm, respectively. The baseline median GH peak was higher in the somapacitan arm than in the Norditropin (somatropin) arm (5.2 mcg/L versus 3.9 mcg/L, respectively). ||||| |||||||||||||| |||||||||| ||||| |||| |||||| ||| ||||||| || |||||| || ||||||||||||
Most patients were included in the FAS for efficacy and harms outcomes. One patient (0.8%) in the somapacitan group discontinued from treatment in the REAL 4 trial due to a protocol violation versus no patients in the Norditropin (somatropin) group. The analysis set of patients was adequate.
All of the patient-reported outcomes were analyzed for the observation datasets in countries where these data were available. Patients in 4 participating countries (Latvia, Poland, Serbia, and Spain) did not complete the patient-reported outcome questionnaires; the impact of these missing data was unknown.
REAL 4 was an open-label study design, which could potentially increase the risk of bias, particularly for the subjective assessment of patient-reported outcomes (i.e., GHD-CIM, GHD-CTB, GHD-PTB, G-DAT) whereas the use of an open-label study design would not have affected the evaluation of treatment effect on height-related outcome measures. Height (cm) assessments were performed in an assessor-blinded manner. IGF-1 and IGFBP-3 were objective measures with standardized measurement or as measured by a central laboratory.
According to the sponsor’s Clinical Study Report for the REAL 4 trial, there were a limited number of previous trials that had allowed ancillary therapy or the retrieval of the landmark data included in the analysis, and no relevant data were available for children with GHD; thus, the noninferiority margin of −1.8 cm/year for the primary end point was chosen based on placebo-controlled trial results for children who were born small for gestational age (SGA) from the GHLIQUID-1424 trial.52 According to the sponsor, a noninferiority margin of −1.8 cm/year to −2.0 cm/year has been applied in previous trials on different brands of somatropin in children with GHD.20 According to the clinical expert consulted by CADTH, the growth-related outcomes assessment at 1 year (52 weeks) was adequate because the best auxologic responses happen in the first year of a GH therapy.
Among the most frequently used medications at baseline, the proportion of patients who used thyroid hormones (8.3%) was lower in the somapacitan arm than in the Norditropin (somatropin) arm (14.7%). Whether this difference might impact the study results was uncertain, according to the clinical expert consulted by CADTH. After initiation of randomization, overall, a higher proportion of patients used a concomitant medication in the somapacitan arm (66.7%) than in the Norditropin (somatropin) arm (58.8%), based on the detailed documentation of patient records, but this difference would not have had an impact on the study results, according to the clinical expert.
The clinical expert noted that pediatric patients and their caregivers may have needle phobia, and caregivers may be under pressure when they administer the treatment. As noted in the patient and clinician group inputs for this review, patients and caregivers often prefer less frequent subcutaneous injections.
No significant protocol deviations were noted in either group in the REAL 4 trial. The sensitivity analyses and per-protocol analysis (these data are not included in this report) were completed and supported the conclusion of noninferiority for the primary end point.
Overall, the statistical methods used in both trials are appropriate. However, it is unknown what the impact of missing data would be on patient-reported outcome measures. The study stratified region into 2 categories (Japan versus the rest of the world) and included region in the various multivariate analyses. However, REAL 4 included Asian countries, such as Korea and India; moreover, the Asian population represented at least one-third of the total patients who received somapacitan. Ethnicity and race are significant predictors of HV; however, the REAL 4 trial did not stratify this factor (only Japan was separated from the rest-of-the-world region possibly because of different peak GH levels used in GHD diagnose). Whether the lack of adjustment by different regions in the analysis might affect the results was uncertain.
External Validity
The REAL 4 trial was conducted globally, and most patients were enrolled from the US (|||), Japan (|||), Russia (|||||), India (|||), Korea (||), and Ukraine (||), among others. ||| ||||||| (|||| of overall study population) ||| ||| allocated to the somapacitan group was from Canada; whether |||| ||||||| belonged to an Indigenous population was unknown. A total of 57.0% of the patients were white and 37.0% were Asian. At baseline, patient age ranged from 2.5 years to 11 years for boys and 2.5 years to 10 years for girls. Overall, there was a greater difference in the higher proportion of boys (74.5%) versus girls (25.5%) in the REAL 4 trial compared with the difference in proportions in the GeNeSIS study, which reported data on children treated with somatropin (63% male versus 37% female in the GHD cohort in Canada).4 The REAL 4 trial only enrolled patients who had no prior exposure to GH or IGF-1 treatment. The clinical expert commented that these differences between patients in the REAL 4 trial versus what is encountered in Canadian clinical practice are not likely to impact the trial results. About 58% of the patients who were screened entered the trial; the main reason for screening failure was that the patient did not meet the inclusion criterion of having an IGF-1 SDS of less than −1.0. This was not considered too significantly different from what would be seen in clinical practice, according to the clinical expert.
The majority of the patients in the REAL 4 trial (88.0%) had idiopathic GHD (and 12% had organic),20 which was different from the GHD patient cohort in the GeNeSIS study (28% had idiopathic and 72% had organic).4 The clinical expert indicated that idiopathic GHD may be accompanied by other conditions that might influence GHD; thus, it was uncertain whether the results would be impacted when this patient characteristic in clinical practice is different from that in the REAL 4 trial. For example, it is uncertain whether the results would remain similar if, in clinical practice, there are more instances of organic GHD versus idiopathic. The clinical expert noted there were patients excluded from the trial that clinicians would want to treat with GH therapy, if needed, such as patients with cancerous brain tumours and patients with a condition that precludes them from a height evaluation (e.g., patients with cerebral palsy who cannot stand).
According to the clinical expert consulted by CADTH, the concomitant medications used in the REAL 4 trial were reflective of those encountered in Canadian clinical practice; the comparator used was appropriate in a Canadian clinical context, and the administration of the study treatments (dosages and settings) is in line with routine clinical practice in Canada. The clinical expert commented that the efficacy and safety outcomes used in the REAL 4 trial were generally clinically relevant and important to clinicians and patients in Canada.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined, as defined by the GRADE Working Group:21,22
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, the evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or relative to the null. The target of the certainty-of-evidence assessment was the presence or absence of an important effect based on thresholds informed by the clinical expert consulted for this review for HV, height SDS, and HV SDS. The target of the certainty-of-evidence assessment was the presence or absence of any (non-null) effect for GHD-CIM, GHD-CTB, GHD-PTB, injection-site reactions, injection-site pain, and treatment discontinuation due to the lack of a formal MID estimate.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for somapacitan versus Norditropin (somatropin) in prepubertal children with GHD.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following were summarized and validated by the CADTH review team.
REAL 4 Long-Term Safety Extension Phase
Description of Studies
The REAL 4 study design is described in the main body of the sponsor’s Clinical Study Report.23 A summary of the long-term safety extension phase (week 52 to week 104) is presented in this section of the CADTH Clinical Review Report. The results presented in the sponsor’s interim analysis of the REAL 4 trial reflect the data available as of December 22, 2022.
Populations
The inclusion and exclusion criteria for patients enrolled in the REAL 4 study were described in the main body of the sponsor’s report.
Interventions
Patients were randomized (2:1) to receive either once-weekly somapacitan 0.16 mg/kg or daily Norditropin (somatropin) 0.034 mg/kg for the 52-week main trial period. For the subsequent safety extension trial period, which was planned for an additional 3 years following the main trial period and up to 208 weeks, all patients have been allocated to once-weekly somapacitan 0.16 mg/kg.
Outcomes and Statistical Analysis
There were no primary or secondary efficacy growth-related end points (i.e., HV, HV SDS, height SDS, or bone age) defined after 52 weeks.
The following secondary efficacy outcomes were defined at week 104 and presented using descriptive statistics:
change in IGF-1 SDS
change in IGFBP-3 SDS.
The GH-PPQ at week 56 is also presented using descriptive statistics.
The 2 data observation periods (in-trial and on-treatment) that were utilized for the primary estimand, HV, and the 2 pharmacodynamic end points, IGF-1 SDS and IGFBP-3 SDS in the main Clinical Trial Report, were not defined past week 52 (visit 7). For completeness, observation periods similar to the main 52-week period were included for data in the safety extension period (weeks 52 to 104). As all patients discontinuing the trial product in the safety extension period were withdrawn from the trial, the results for the 2 observation periods from week 52 to 104 are nearly identical.
Results
Patient Disposition
Patient disposition for the long-term safety extension phase of the REAL 4 trial is presented in Table 19. A total of 199 patients were exposed in the safety extension (following week 52). Four patients in the somapacitan to somapacitan group and 1 patient in the Norditropin (somatropin) to somapacitan group discontinued the trial product in the safety extension period (from week 52 to week 104). None of the discontinuations were due to AEs. All patients who discontinued during the extension period were withdrawn from the trial.
Table 19: Patient Disposition in the REAL 4 Safety Extension Phase
Patient disposition | REAL 4a | |
|---|---|---|
Somapacitan group | Norditropin (somatropin) to somapacitan crossover group | |
Randomized, N (%) | 132 (100) | 68 (100) |
Exposed to somapacitan in safety extension period, N (%) | 131 (99.2) | 68 (100) |
Discontinued from study, N (%) | 4 (3.0) | 1 (1.5) |
Reason for discontinuation, n(%) | ||
Lost to follow-up | 1 (0.8) | 0 |
Physician decision | 0 | 1 (1.5) |
Withdrawn by parent or guardian | 1 (0.8) | 0 |
Other | 2 (1.5) | 0 |
Discontinued from treatment, N (%) | 4 (3.0) | 1 (1.5) |
Reason for discontinuation, n(%) | ||
At the discretion of the investigator | 0 | 1 (1.5) |
Lost to follow-up | 1 (0.8) | 0 |
Other | 3 (2.3) | 0 |
aPatient disposition presented for the safety extension period (week 52 to week 104).
Source: REAL 4 trial Clinical Study Report (week 104).23
Exposure to Study Treatments
In the safety extension period (week 52 to 104), the mean duration of exposure to somapacitan and mean adherence were similar between the somapacitan to somapacitan group and the Norditropin (somatropin) to somapacitan group (Table 20). The mean adherence in the somapacitan to somapacitan group was 94.2% from week 0 to 104, suggesting consistently high adherence throughout the entire study.
Table 20: Exposure to Somapacitan in the Safety Extension Phase of the REAL 4 Trial From Week 52 to Week 104
Exposure | Somapacitan group (N = 131) | Norditropin (somatropin) to somapacitan crossover group (N = 68) |
|---|---|---|
Total, days | 46,039 | 24,498 |
Duration, mean (SD) | 351.4 (41.9) | 360.3 (8.4) |
Duration, median (range) | 359 (44 to 386) | 359 (344 to 405) |
Adherence, mean (%) | 90.3 | 88.8 |
SD = standard deviation.
Source: Week 104 Clinical Study Report for the REAL 4 trial.23
Efficacy
The sponsor’s interim analysis includes data up to week 104 in the safety extension phase. The key efficacy outcomes evaluated in the REAL 4 main trial phase were evaluated descriptively for the long-term safety extension phase and are summarized in Table 21.
After 104 weeks of treatment, the HV observed in the patients treated with somapacitan for the full period is similar to the HV observed in the patients who switched from Norditropin (somatropin) to somapacitan after week 52. After 104 weeks of treatment, a similar increase in change from baseline in height SDS and HV SDS was observed in the patients treated with somapacitan for the full period and the patients who switched from Norditropin (somatropin) to somapacitan at week 52. The mean observed IGF-1 SDS and IGFBP-3 SDS change from baseline after 104 weeks was also similar in both groups.
Table 21: Summary of Efficacy End Points in the Safety Extension Phase of the REAL 4 Trial at Week 104
Efficacy end points | Somapacitan group (N = 132) | Norditropin (somatropin) to somapacitan crossover group (N = 68) |
|---|---|---|
Height velocity, cm/year | ||
Baseline mean (SD) | 4.3 (1.4) | 4.1 (1.4) |
N at week 104 | 127 | 67 |
Observed mean (SD) at week 104 | 8.4 (1.5) | 8.7 (1.8) |
Height SDS | ||
Baseline mean (SD) | −2.99 (1.02) | −3.47 (1.52) |
N at week 104 | 127 | 67 |
Observed mean (SD) change from baseline at week 104 | 1.75 (0.72) | 1.97 (0.98) |
Height velocity SDS | ||
Baseline mean (SD) | −2.35 (1.51) | −2.52 (1.55) |
N at week 104 | 127 | 67 |
Observed mean (SD) change from baseline at week 104 | 5.21 (2.58) | 5.62 (3.15) |
Pharmacodynamic end points | ||
IGF-1 SDS | ||
Baseline mean (SD) | −2.03 (0.97) | −2.33 (1.03) |
N at week 104 | 127 | 66 |
Observed mean (SD) change from baseline at week 104 | 1.78 (0.98) | 2.05 (1.33) |
IGFBP-3 SDS | ||
Baseline mean (SD) | −1.89 (1.12) | −2.18 (1.27) |
N at week 104 | 127 | 66 |
Observed mean (SD) change from baseline at week 104 | 1.43 (0.84) | 1.49 (0.95) |
CI = confidence interval; IGF-1 = insulin-like growth factor 1; IGFBP-3 = insulin-like growth factor binding protein 3; SD = standard deviation; SDS = standard deviation score.
Note: In-trial period results are reported.
Source: REAL 4 trial Clinical Study Report (week 104).23
The GH-PPQ, which assesses GH treatment preference, was used to evaluate the switch from Norditropin (somatropin) to somapacitan after the final visit in the main phase of the trial at week 52. The questionnaire was completed by the patient’s parent or LAR 4 weeks after the patient had switched from Norditropin (somatropin) to somapacitan (week 56). Parents of 50 of the 68 patients who were switched from Norditropin (somatropin) to somapacitan at week 52 responded to the questionnaire. Of these 50 respondents, 45 preferred somapacitan to Norditropin (somatropin), while 5 respondents answered that they had no preference. None of the respondents preferred Norditropin (somatropin). The majority (38 out of 45) of the respondents preferring somapacitan had a strong or very strong preference for the once-weekly (somapacitan) over the once-daily (Norditropin [somatropin]) treatment regimen. The main reasons selected in the GH-PPQ for the preference included: “number of times needing to do injections,” “less worried about remembering to give the injections,” and “child [is] less worried or annoyed by getting injections.” Thirty-five of the 45 parents who preferred somapacitan stated they expected higher adherence to the current once-weekly regimen than to Norditropin (somatropin); 1 parent expected the patient would be more adherent to Norditropin (somatropin), while the remaining 9 parents had no preference with regard to expected adherence to either drug.
Harms
A summary of harms for the REAL 4 safety extension phase is presented in Table 22.
Table 22: Summary of Harms Results From the REAL 4 Trial Safety Extension Phase Up to Week 104
Adverse events | Somapacitan group, week 52 to 104 (N = 31) | Norditropin (somatropin) to somapacitan crossover group, week 52 to 104 (N = 68) | Somapacitan group, week 0 to 104 (N = 132) |
|---|---|---|---|
Most common adverse events, n (%) | |||
≥ 1 adverse event | 82 (62.6) | 39 (57.4) | 114 (86.4) |
Headache | 3 (2.3) | 3 (4.4) | 19 (14.4) |
Nasopharyngitis | 11 (8.4) | 5 (7.4) | 21 (15.9) |
Pyrexia | 5 (3.8) | 2 (2.9) | 14 (10.6) |
Pain in extremity | 5 (3.8) | 0 | 15 (11.4) |
COVID-19 | 6 (4.6) | 5 (7.4) | 9 (6.8) |
Bronchitis | 5 (3.8) | 2 (2.9) | 8 (6.1) |
Vomiting | 5 (3.8) | 1 (1.5) | 8 (6.1) |
Cough | 5 (3.8) | 0 | 9 (6.8) |
Rhinitis | 3 (2.3) | 0 | 8 (6.1) |
Influenza | 5 (3.8) | 0 | 8 (6.1) |
Serious adverse events, n (%) | |||
Patients with ≥ 1 SAE | 3 (2.3) | 0 | 9 (6.8) |
Patients who stopped treatment due to adverse events, n (%) | |||
Patients who stopped | 0 | 0 | 0 |
Deaths, n (%) | |||
Patients who died | 0 | 0 | 0 |
Adverse events of special interest, n (%) | |||
Injection-site reaction | 5 (3.8) | 4 (5.9) | 7 (5.3) |
Two consecutive positive GH antibody samples | 3 (2.3) | 3 (4.4) | 4 (3.0) |
IGF-1 SDS > 2 at 2 consecutive visits | 10 (7.6) | 4 (5.9) | 10 (7.6) |
GH = growth hormone; IGF-1 = insulin-like growth factor 1; SAE = serious adverse event; SDS = standard deviation score.
Source: REAL 4 trial Clinical Study Report (week 104).23
The safety profile of once-weekly somapacitan administered to children with GHD for up to 104 weeks was similar to the well-known safety profile for daily Norditropin (somatropin). No new safety or local tolerability issues were identified.
In patients with an IGF-1 SDS above 2, no trend was seen in the amount or type of AEs reported at 2 or more consecutive visits compared with the remaining patients in the relevant treatment groups. There were no dose reductions due to AEs for these patients. The majority of AEs were of mild severity.
Critical Appraisal
Internal Validity
REAL 4 is an open-label trial, which may influence the perception of improvement by patients and clinicians, particularly for outcomes that are subjective in measurement and interpretation (e.g., patient-reported outcomes, subjective AEs). In the REAL 4 trial, only prepubertal children were enrolled into the main trial to avoid the pubertal growth spurt interfering with the treatment effect.
The efficacy outcomes were presented using descriptive statistics, so no statistical inferences were possible. The safety profile for somapacitan from the main trial phase to the long-term extension phase was consistent in the REAL 4 trial.
External Validity
The patients who took part in the open-label long-term safety extension phase were originally from the REAL 4 trial and the eligibility criteria remained the same; therefore, it is reasonable to expect that the same limitations to generalizability are relevant to the safety extension phase. In addition, the results in week 104 may be more representative of what might be observed in a long-term clinical setting. Of note, for example, the mean observed change in HV in both arms was not as high at week 104 as it was at week 52. It should also be noted that according to the clinical expert consulted by CADTH, the assessment of growth-related outcomes at 52 weeks was adequate because the best auxologic responses happen in the first year of a GH therapy.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
Direct comparative evidence exists between somapacitan and somatropin (Norditropin 0.034 mg/kg once daily) from the REAL 3 and REAL 4 trials, but there is a gap in evidence comparing somapacitan with somatrogon. Indirect comparisons were required to address this gap. One ITC was submitted by the sponsor, which has been summarized and appraised in this report.
Description of the ITC
An SLR was conducted by the sponsor to identify evidence for inclusion in the ITC, as described in Table 23. One ITC was submitted by the sponsor comparing somapacitan with other LAGH treatments for GHD on height-related outcomes within the first year of treatment in prepubescent boys and girls. The sponsor also assessed the feasibility of conducting similar ITCs at time points beyond the first year of treatment, but concluded that it was infeasible.
Table 23: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Eligibility criteria |
|---|---|
Population | Treatment-naive prepubescent boys and girls aged ≥ 2 to ≤ 12 years with GHD and impaired HV |
Intervention | Somapacitan 0.16 mg/kg/week |
Comparator | Comparator LAGH of interest: somatrogon 0.66 mg/kg/week Other LAGH comparators included in the SLR but not relevant to the Canadian context:
Common comparator:
|
Outcome | Efficacy outcomes assessed in ITCs:
Safety outcomes included for descriptive comparisons only:
Other outcomes included in SLR but not ITCs or descriptive comparisons:
Time points of ITCs and comparisons:
|
Study designs | Studies with English-language abstracts:
|
Publication characteristics | Published and unpublished sources included |
Exclusion criteria | Population: patients diagnosed with 1 of the other indications for growth hormones (e.g., small for gestational age) Intervention/comparator:
Outcomes: studies not reporting efficacy or safety evidence Study design:
Language: non-English articles without English abstract Date: no restriction Countries: no restriction |
Databases searched | Databases (May 19, 2022, no date limit):
Conference proceedings (2017 to 2022):
Clinical trial registries:
HTA submissions:
Government and international bodies:
|
Selection process |
|
Data-extraction process |
|
Quality assessment |
|
AE = adverse event; AHV = annualized height velocity; EPHPP = Effective Public Health Practice Project; GHD = growth hormone deficiency; HV = height velocity; IGF-1 = insulin-like growth factor 1; IGFBP-3 = insulin-like growth factor binding protein 3; ITC = indirect treatment comparison; LAGH = long-acting growth hormone; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; SAGH = short-acting growth hormone; RCT = randomized controlled trial; SDS = standard deviation score; SLR = systematic literature review; STA = single technology appraisal.
aThe reference lists of relevant SLRs and clinical guidance information were screened to ensure no relevant study was missed.
Source: Sponsor-submitted NMA report53 and accompanying SLR report.54
ITC Design
Objectives
The objective of the ITC was to generate evidence on the comparative effectiveness of somapacitan versus other LAGHs for GHD.
Study Selection Methods
The eligibility criteria for the SLR that informed the ITCs are detailed in Table 23. In summary, English-language clinical studies were included that evaluated LAGHs in pediatric patients with GHD from 2 to 12 years of age against any comparator and reported efficacy or safety results. Patients with indications for GHs other than height deficiency were not considered relevant. The databases searched were Embase, MEDLINE, and the Cochrane Library, in addition to several conferences and grey literature sources. Study screening was conducted in duplicate. Data extraction was conducted by 1 analyst and quality-checked by a different analyst. Quality assessments were performed using the tools described in Table 23 by 1 statistician.
The sponsor evaluated the feasibility of assessing outcomes at weeks 26, 52, 104, and 156. The sponsor-conducted ITC included Bayesian NMAs evaluating 3 efficacy outcomes (AHV, HV SDS, and height SDS) at weeks 26 and 52. However, longer-term outcomes were primarily informed by single-arm trials, so anchored analyses such as NMAs were not feasible. Longer-term efficacy outcomes and 2 safety outcomes (injection-site reactions and antibodies) were compared qualitatively only. Although the SLR included additional outcomes, the sponsor selected these as the most clinically relevant to compare for the purpose of addressing this data gap.
ITC Analysis Methods
Feasibility Assessment
The similarity of patient characteristics across 5 trials was assessed qualitatively. The assessment of patient characteristics focused on the following characteristics identified by the sponsor as potential prognostic factors:
age
gender
race
height SDS or AHV SDS
peak GH
Network Structure
In trials with multiple doses of the same LAGH treatment, only the recommended dose (or intended dose if market authorization was pending) was used for analysis.
Within this network, multiple arms of somatropin were combined and considered as a single somatropin node, which was a common comparator for other treatments. A key assumption of this network was that the somatropin treatment arms (Genotropin and Norditropin) were considered similar enough to be combined into a single treatment node.
In the base-case network, somatropins of the same dose were pooled (0.034 mg/kg/day). An alternative network was conducted as a scenario analysis in which the trial where the somatropin dose differs (Opko JPN trial, 0.025 mg/kg/day) was connected, as a separate node, to the network through the common comparator somatrogon node.
Analysis Model and Implementation
All outcomes in the analysis were continuous. Height SDS was modelled in terms of the mean change in the outcome of interest from baseline, whereby it was assumed that the mean changes from baseline in trials follow a normal distribution. AHV and HV SDS were modelled in terms of the mean at the time point of interest, again assuming mean values at the time point of interest in trials follow a normal distribution. All analyses of continuous outcomes were performed using a normal likelihood, identity link model.
The programming language R (version 4.1.3) was used for implementation. Analysis was conducted utilizing function “nma” in version 0.4.2 of the publicly available package multinma.55 Four chains of 10,000 iterations were run: 5,000 for burn-in and 5,000 for sampling.
Consistency of Evidence
As the network contains no loops, a formal assessment of consistency was not performed.
Prior Distributions
The prior represents the prior probability distribution; a vague prior contains no information about the parameters of interest. Vague priors are used for the study-specific treatment effect μi and treatment effect sizes relative to reference treatment 1 (d1k) in the form of a normal distribution, with a mean of 0 and variance of 10,000, as recommended in NICE Technical Support Document 2.56
||| || ||| |||| || ||| ||||||| ||| ||||||| ||||| |||||||||||| ||||| |||||| |||| ||| |||| ||| ||||||| ||||| ||||||||||||| || ||| |||||| ||||||| |||||| |||||||| ||||||||||| |||| ||| ||| ||||| ||| ||||||||||||| ||| ||| ||||| ||||||||| || |||||| ||| ||||| |||| |||||||| |||||||| ||| |||||||||| ||||||||||||||||| ||||||||||||| |||| ||||| |||||||| ||| |||| ||||||||| ||| ||||||||| |||||||| ||||||||| ||| ||| ||||||||| |||||||||| ||||||| ||||| ||| ||||| ||| |||| |||| ||||||| ||| ||||||| ||||| || ||| || ||| ||||||||||||||| ||||||||||||| |||||| ||| ||| |||||||||
Measures of Model Complexity and Fit
Model comparisons (fixed effects [FE] versus random effects [RE]) were based on comparing the average residual deviance (i.e., residual deviance divided by the number of data points), in addition to the deviance information criterion (DIC), and the convergence of the models.56 In general, a model is favoured if it has a lower DIC (a difference of 3 to 5 points or more between the DIC from the FE and RE models indicates a strong preference for the model with the lower DIC) and a posterior residual deviance closer to the number of data points. Model fit was assessed in the base-case network and the same model was applied to the alternative-scenario analysis network.
Data Selection and Transformation
A number of trials reported both arm- and trial-level data (e.g., treatment differences with measures of uncertainty); in such cases, arm-level data were used in the analysis. All trials reported mean change from baseline for height SDS, so no additional data transformation was needed. For HV SDS, posthoc analyses on Novo Nordisk trials were conducted to align with reported outcomes in other trials (having originally been modelled as mean change from baseline). The process of identifying and obtaining data has been described in detail in the statistical analysis plan.
Estimates of Treatment Effect and Credible Intervals
The results are presented as median treatment differences and an associated 95% CrI. The 95% CrI can be interpreted as an interval such that there is a 95% probability that the true value lies within the calculated interval.
For each end point in the base-case network, a plot of relative treatment estimates with the associated 95% CrI was presented, including the results of both the FE and RE models (Table 24).
Table 24: Indirect Treatment Comparison 1 Analysis Methods
Methods | Description |
|---|---|
Analysis methods | All outcomes in the analysis were continuous. Height SDS was modelled in terms of the mean change in the outcome of interest from baseline, whereby it was assumed that the mean changes from baseline in trials follow a normal distribution. Annualized HV and HV SDS were modelled in terms of the mean at the time point of interest, again assuming mean values at the time point of interest in trials follow a normal distribution. All analyses of continuous outcomes were performed using a normal likelihood, identity link model. |
Priors | ||| || ||| |||| || ||| ||||||| ||| ||||||| ||||| |||||||||||| ||||| |||||| |||| ||| |||| ||| ||||||| ||||| ||||||||||||| || ||| |||||| ||||||| |||||||||||||| ||||||||||| |||| ||| ||| ||||| ||| ||||||||||||| ||| ||| ||||| ||||||||| || |||||| ||| ||||| |||| |||||||| |||||||| ||| |||||||||||||||||||||||||||| ||||||||||||| |||| ||||| |||||||| ||| |||| ||||||||| ||| ||||||||| |||||||| ||||||||| ||| ||| ||||||||| |||||||||| |||||||||||| ||| ||||| ||| |||| |||| |||||| ||| ||||||| ||||| || ||| || ||| ||||||||||||| |
Assessment of model fit | Model comparisons (FE vs. RE) were based on comparing the average residual deviance (i.e., residual deviance divided by the number of data points), in addition to the DIC, and the convergence of the models. In general, a model is favoured if it has a lower DIC (a difference of 3 to 5 points or more between the DIC from the FE and RE models indicates a strong preference for the model with the lower DIC) and a posterior residual deviance closer to the number of data points. Model fit was assessed in the base-case network; then, the same model was applied to the alternative network. |
Assessment of consistency | As the network contains no loops, a formal assessment of consistency was not performed. |
Assessment of convergence | Four chains of 10,000 iterations were run: 5,000 for burn-in and 5,000 for sampling. |
Outcomes | A number of trials reported both arm- and trial-level data (e.g., treatment differences with measures of uncertainty) and, in such cases, arm-level data are used in the analysis. All trials reported mean change from baseline for height SDS, so no additional data transformation was needed. For HV SDS, posthoc analyses on Novo Nordisk trials were conducted to align with reported outcomes in other trials (having originally been modelled as mean change from baseline). Different measures of uncertainty were reported across the different trials. SDs and CIs were transformed to SEs, respecting the normal distribution of all the variables. Where measures of uncertainty for each treatment arm are missing, this quantity was imputed as the mean of the uncertainties of the same treatment class (LAGH vs. SAGH) in other trials of similar size (grouped as having treatment arms with < 40 patients or ≥ 40 patients). If outcomes were reported in the same way, data were extracted only for the preferred source and following consistent rules (primary manuscript preferred over conference abstracts, then clinicaltrials.gov, then internal documents). When reported, least squares means were preferred because they would have been adjusted to control for potential imbalances in patient characteristics, most commonly: age, sex, peak GH, region, and 1 height variable (e.g., height at baseline, height SDS at baseline). If 1 of the sources presented results only in plots while another reported exact results in tables or text, then the latter was used, for precision. When uncertainty was missing in the preferred source, but available in another source, the latter was used. For the Novo Nordisk trials, where data had not been published, additional analyses were conducted and the results added to the data-extraction table. |
Follow-up time points | 26 weeks and 52 weeks (short-term efficacy end points) |
Construction of nodes | In trials with multiple doses of the same LAGH treatment, only the recommended dose (or intended dose if market authorization is pending) was used for analysis. Within this network, there were multiple arms of somatropin which have been combined and considered as a single somatropin node, which was a common comparator for other treatments. A key assumption of this network was that the somatropin treatment arms were considered similar enough to be combined into a single treatment node, which was seen as feasible because Genotropin and Norditropin are both somatropin products. In the base-case network, somatropins of the same dose were pooled (0.034 mg/kg/day). In the alternative network, instead of pooling all arms where the somatropin dose is in the labelling range for pediatric GHD (0.024 to 0.034 mg/kg/day), the only trial where the somatropin dose differs (Opko JPN trial, 0.025 mg/kg/day) was connected to the network through the somatrogon node. The ~30% reduction in somatropin dose, going from 0.034 mg/kg/day to 0.025 mg/kg/day, has been evaluated to be too great for the treatments to be considered similar, according to the sponsor. In the alternate network, both the relative treatment difference for the somatropin 0.034 mg/kg/day dose and 0.025 mg/kg/day dose are presented. |
Sensitivity analyses | None |
Subgroup analysis | None |
Methods for pairwise meta-analysis | None |
CI = confidence interval; DIC = deviation information criterion; FE = fixed effects; GH = growth hormone; GHD = growth hormone deficiency; HV = height velocity; LAGH = long-acting growth hormone; RE = random effects; SAGH = short-acting growth hormone; SDS = standard deviation score; SE = standard error.
Source: Sponsor-submitted network meta-analysis53 and accompanying SLR report.54
Results of ITC
Summary of Included Studies
Overview
In total, the SLR identified 22 records comprising 10 unique studies. For the Canadian context, irrelevant comparators were excluded, so the final number of studies was smaller. Ultimately, the ITCs included 7 records informing 5 studies, 3 of which assessed somatrogon versus somatropin (the Opko II, Opko III, and Opko JPN trials), and 2 of which assessed somapacitan versus somatropin (REAL 3 and REAL 4 trials). The studies are summarized briefly in Table 25, and the evidence networks are displayed in Figure 5 (base case) and Figure 6 (alternative scenario, including somatotropin 0.025 mg/kg/day from the Opko JPN study).
The primary end point was AHV in all trials: 4 assessed the primary end point at 52 weeks, while 1 (REAL 3) assessed the primary end point at 26 weeks. Generally, when reported, the analysis of continuous end points was similar across trials, with trials considering treatment, age, sex, peak GH, region, and height in their analysis. However, 1 trial (Opko II) did not report on the use of methods such as ANCOVA.
Table 25: Trials Eligible to Inform Short-Term Efficacy Outcomes
Trial | Intervention | Comparator | Primary outcome | Model used and variables considered for primary outcome |
|---|---|---|---|---|
REAL 3 | Somapacitan: 0.04 mg/kg/week, 0.08mg/kg/week, or 0.16 mg/kg/week | Somatropin: 0.034 mg/kg/day | AHV at 26 weeks | MMRM: Treatment, age group, sex, region, and sex by age group interaction term as factors; baseline height as a covariate; all nested within week as a factor |
REAL 4 | Somapacitan: 0.16 mg/kg/week | Somatropin: 0.034 mg/kg/day | AHV at 52 weeks | ANCOVA: Treatment, sex, age group, region, GH peak group, and sex by age group by region interaction term as factors; baseline height as covariate |
Opko II | Somatrogon: 0.25 mg/kg/week, 0.48 mg/kg/week, and 0.66 mg/kg/week | Somatropin: 0.034 mg/kg/day | AHV at 52 weeks | — |
Opko III | Somatrogon: 0.66 mg/kg/week | Somatropin: 0.034 mg/kg/day | AHV at 52 weeks | ANCOVA: Stratification classes for treatment, age group, peak GH value during stimulation test, region, and sex; baseline height SDS as a covariate |
Opko JPN | Somatrogon: Escalating doses of 0.25 mg/kg/week, 0.48 mg/kg/week, and 0.66 mg/kg/week for 2 weeks at each dose, then 0.66 mg/kg/week | Somatotropin: 0.025 mg/kg/day | AHV at 52 weeks | ANCOVA: Treatment and sex as factors; peak GH value during stimulation test and baseline height SDS as covariates |
AHV = annualized height velocity; ANCOVA = analysis of covariance; GH = growth hormone; MMRM = mixed-model for repeated measures; SDS = standard deviation score.
Source: Sponsor-submitted network meta-analysis53 and accompanying SLR report.54
Trial Design
In all trials, patients were randomized to treatment with an LAGH or an SAGH (somatropin) for at least 26 weeks. All randomized phases were open-label between LAGHs and somatropin; however, the REAL 3 trial was double-blinded within LAGH doses, and the REAL 3 and REAL 4 trials applied observer blinding to the primary end point. Four of the 5 trials evaluated somatropin at the same dose used in the Novo Nordisk–sponsored trials (0.034 mg/kg/day), while 1 trial (Opko JPN) evaluated somatropin at a lower dose and was therefore not included in the base-case analyses. Additionally, dose escalation for somatrogon-treated patients occurred in the first 6 weeks of the Opko JPN trial (0.25 mg/kg/week, 0.48 mg/kg/week, and 0.66 mg/kg/week; 2 weeks each) with the remaining 46 weeks at a dose of 0.66 mg/kg/week.
Across the trials, dose adjustments were made as needed based on IGF-1 assessments. There were some differences with respect to how much the dose was reduced, the criteria to justify a dose adjustment, and the timing of IGF-1 measurements.
The patient eligibility criteria are summarized in Table 26. The trials enrolled treatment-naive prepubescent children with GHD. Across the trials, there were slight differences in inclusion criteria for disease severity as defined by GH peak levels: the REAL 3 trial required 7 ng/mL or less; the REAL 4, Opko II, and Opko III trials all required 10 ng/mL or less; and the Opko JPN trial required 6 ng/mL or less. There were also slight differences in the age ranges of the patients enrolled, but the trials generally enrolled patients aged from 2 to 3 years to 10 to 11 years and all trials had different age caps for boys (1 year younger) than girls. Tanner development stages for testes and breasts were also described in the eligibility criteria for the REAL 3 and 4 trials, as previously described; this was not described in any of the included Opko trials. All trials required impaired height, but the Opko III trial uniquely did not require an SDS for height of less than −2 for patient eligibility. All trials required an AHV below the 25th percentile and excluded children born SGA.
Table 26: Patient Eligibility Criteria for the Studies Included in the ITCs
Key eligibility criterion | REAL 3 | REAL 4 | Opko II | Opko III | Opko JPN |
|---|---|---|---|---|---|
Age | Prepubertal children:
| Prepubertal children:
| Prepubertal children:
| Prepubertal children:
| Prepubertal children:
|
GHD and multiple pituitary hormone deficiency | Allowed | NR | Isolated GHD or GHD as part of multiple pituitary hormone deficiency | Patients with congenital causes of multiple pituitary hormone deficits were eligible, but hydrocortisone and/or L-thyroxin replacement doses had to be stable for a minimum of 3 months before enrolment | NR |
Diagnosis | Confirmed by 2 different unprimed GH provocation tests defined as peak plasma GH level of ≤ 7 ng/mL with no prior exposure to GH therapy and/or IGF-1 treatment For children with 3 or more pituitary hormone deficiencies, only 1 GH stimulation test was required | Confirmed diagnosis of GHD by 2 different GH tests performed in the last 12 months before randomization, defined as a peak GH level of ≤ 10 ng/mL using the WHO International Standard for Somatropin (98/574) | Confirmed by 2 different GH provocation tests defined as a peak plasma GH level of ≤ 10 ng/mL using a validated assay | Confirmed by 2 different GH provocation tests defined as a peak plasma GH level of ≤ 10 ng/mL using a validated assay | Confirmed by 2 different GH provocation tests, defined as a peak serum GH level of ≤ 6.0 ng/mL, or ≤ 16 ng/mL for a GH-releasing peptide-2 provocation test |
Bone age | Bone age less than chronological age | NR | No older than chronological age, and should not be > 9 years for girls and > 10 years for boys | Not older than chronological age, < 10 years for females and < 11 years for males | NR |
Treatment history | No prior exposure to any GH therapy and/or IGF-1 treatment | No prior exposure to any GH therapy and/or IGF-1 treatment | No prior exposure to any recombinant GH therapy | No prior exposure to any recombinant GH therapy | No prior exposure to any recombinant GH therapy |
Height | Impaired height (at least 2.0 SD below mean) for chronological age and sex | Impaired height (at least 2.0 SD below mean) for chronological age and sex | Impaired, defined as at least 2 SDs below the mean for chronological age and sex (height SDS ≤ −2.0)a | Height had to be impaired but was not required to be < −2 SDS for inclusion | Height SDS ≤ −2 |
HV | Annualized HV below the 25th percentile, or below −0.7 SD, for chronological age and sex,a calculated over a minimum time span of 6 months and maximum 18 months before screening | Annualized HV below the 25th percentile for chronological and sex | Annualized HV below the 25th percentile for chronological age (HV SDS < −0.7)a | Annualized HV below the 25th percentile for chronological age (HV < −0.7 SDS) | Impaired, below the 25th percentile for chronological age |
BMI | BMI > fifth and < 95th percentile according to CDC BMI-for-age growth charts | NR | BMI within ± 2 SD of mean BMI for the chronological age and sexb | Not malnourished (defined as BMI < −2 SDS, age- and sex-standardized) | Not malnourished (defined as BMI < −2 SDS, age- and sex-standardized) |
IGF-1 | NR | IGF-1 < −1.0 SDS | At least 1 SD below the mean IGF-1 level standardized for age and sex (IGF-1 ≤ −1.0 SDS) | At least 1 SD below the age- and sex-standardized mean IGF-1 level (SDS ≤ −1) | At least 1 SD below the age- and sex-standardized mean IGF-1 levels (SDS ≤ −1) |
Notable exclusion criteriac | Children born small for gestational age, i.e., birth weight and/or birth length < 2 SD for gestational age | Children born small for gestational age, i.e., birth weight and/or birth length < 2 SD for gestational age | Children born small for gestational age, i.e., birth weight and/or birth length < 2 SD for gestational age | Children born small for gestational age, i.e., birth weight and/or birth length < 2 SD for gestational age | Children born small for gestational age (definition NR) |
BMI = body mass index; GH = growth hormone; GHD = growth hormone deficiency; HV = height velocity; IGF-1 = insulin-like growth factor 1; ITC = indirect treatment comparison; NR = not reported; SD = standard deviation; SDS = standard deviation score.
aAccording to standard growth charts by Prader et al.57
bAccording to the 2000 Centers for Disease Control and Prevention standards.58
cNot exhaustive.
Sources: Publications of the Opko II,59 Opko III,60 and Opko JPN61 trials.
Patient Baseline Characteristics
The baseline characteristics for the Opko II, Opko III, and Opko JPN trials are summarized in Table 27. For any studies that evaluated multiple doses of somatrogon (i.e., Opko II), only the 0.66 mg/kg/week dose is of interest, and so the cohorts randomized to other doses of somatrogon are not described here.
There was a large spread in the proportions of male patients across trials, from approximately 50% male in the Opko JPN trial to approximately 70% in the REAL 4 trial. There was also considerable variability with respect to race, with the Opko JPN trial exclusively enrolling Asian patients, while more than 90% of the patients enrolled in the Opko II trial were white. Additionally, the mean age in the Opko III trial was higher than in other trials, including REAL 4 (7.7 years versus 6.4 years, respectively). Height SDS and AHV SDS were slightly lower in the Opko II trial compared with other trials.
Table 27: Summary of Patient Baseline Characteristics in the Opko II, Opko III, and Opko JPN Trials
Characteristic | Opko II (NCT01592500) | Opko III (NCT02968004) | Opko JPN (NCT03874013) | |||
|---|---|---|---|---|---|---|
Somatrogon | Genotropin | Somatrogon | Genotropin | Somatrogon | Genotropin | |
Sample size | ||||||
n | 14 | 11 | 109 | 115 | 22 | 22 |
Age, years | ||||||
Mean (SD) | 6.1 (2.2) | 5.7 (1.9) | 7.83 (NR) | 7.61 (NR) | 5.28 (1.84) | 6.78 (2.34) |
Median (range) | NR (3 to 10) | NR (4 to 9) | NR (3.01 to 11.96) | NR (3.05 to 11.85) | NR | NR |
< 6 years, n (%) | NR | NR | NR | NR | NR | NR |
≥ 6 years, n (%) | NR | NR | NR | NR | NR | NR |
Sex, n (%) | ||||||
Male | 9 (64.3) | 8 (72.7) | 82 (75.2) | 79 (68.7) | 9 (40.9) | 12 (54.5) |
Female | 5 (35.7) | 3 (37.3) | 27 (24.3) | 36 (31.3) | 13 (59.1) | 10 (45.5) |
Race, n (%) | ||||||
White | 14 (100) | 10 (90.0) | 81 (74.3) | 86 (74.8) | NR | NR |
Asian | NR | NR | 24 (22.0) | 21 (18.3) | 22 (100%) | 22 (100%) |
Black or African American | NR | NR | 0 | 2 (1.7) | NR | NR |
American Indian or Alaska Native | NR | NR | 1 (0.9) | 0 | NR | NR |
Native Hawaiian or Other Pacific Islander | NR | NR | 0 | 1 (0.9) | NR | NR |
Other | 0 | 1 (9.1) | 3 (2.8) | 5 (4.3) | NR | NR |
Not reported | NR | NR | NR | NR | NR | NR |
Height, cm | ||||||
Mean (SD) | NR | NR | NR | NR | NR | NR |
Median (range) | NR | NR | NR | NR | NR | NR |
Weight, kg | ||||||
Mean (SD) | NR | NR | NR | NR | NR | NR |
Median (range) | NR | NR | NR | NR | NR | NR |
BMI, kg/m2 | ||||||
Mean (SD) | NR | NR | NR | NR | NR | NR |
Median (range) | NR | NR | NR | NR | NR | NR |
GHD cause, n (%) | ||||||
Idiopathic | NR | NR | NR | NR | NR | NR |
Organic | NR | NR | NR | NR | NR | NR |
GH peak, mcg/L | ||||||
Mean (SD) | 3.97 (2.97) | 3.82 (2.78) | 5.45 (2.81) | 5.76 (2.59) | NR | NR |
Median (range) | NR | NR | NR (0.10 to 9.93) | NR (0.10 to 9.90) | NR | NR |
Mother’s height, cm | ||||||
Mean (SD) | NR | NR | NR | NR | NR | NR |
Median (range) | NR | NR | NR | NR | NR | NR |
Father’s height, cm | ||||||
Mean (SD) | NR | NR | NR | NR | NR | NR |
Median (range) | NR | NR | NR | NR | NR | NR |
Baseline mean (SD) | ||||||
Height velocity, cm/year | NR | NR | NR | NR | NR | NR |
Height velocity SDS | −3.01 (1.42) | −3.29 (1.91) | NR | NR | NR | NR |
Height SDS | −4.21 (1.45) | −4.22 (1.58) | −2.94 (1.29) | −2.78 (1.27) | −2.61 (0.44) | −2.53 (0.40) |
IGF-1 SDS | NR | NR | NR | NR | −1.39 (0.90) | −1.62 (0.84) |
IGFBP-3 SDS | NR | NR | NR | NR | NR | NR |
BMI = body mass index; GH = growth hormone; GHD = growth hormone deficiency; HV = height velocity; IGF-1 = insulin-like growth factor 1; IGFBP-3 = insulin-like growth factor binding protein 3; NR = not reported; SD = standard deviation; SDS = standard deviation score.
Sources: Publications of the Opko II,59 Opko III,60 and Opko JPN61 trials.
Summary of the Assessment of Homogeneity for the Sponsor-Submitted ITC
A summary of the assessment of homogeneity was not provided by the sponsor in its summary of clinical evidence. CADTH has summarized the details in Table 28.
Table 28: Assessment of Homogeneity for the Sponsor-Submitted ITC
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Some differences were observed in eligibility criteria with respect to disease severity (examine the trial eligibility criteria row). There was between-trial heterogeneity in baseline age, sex, and race, which could impact disease severity, prognosis, and/or treatment effect. |
Treatment history | All included trials only permitted treatment-naive patients. |
Trial eligibility criteria | Trial eligibility criteria were generally similar across trials, except that the Opko III trial did not require a < −2 SDS in height for eligibility, and GH peak level requirements varied: the REAL 3 trial required ≤ 7 ng/mL; the REAL 4, Opko II, and Opko III trials all required ≤ 10 ng/mL; and the Opko JPN trial required ≤ 6 ng/mL. The trials also differed in whether there were criteria pertaining to bone age and IGF-1. All trials required annualized HV below the 25th percentile and excluded children born small for gestational age. |
Dosing of comparators | The common comparator (somatropin 0.034 mg/kg/day) was the same dose for all included trials except Opko JPN (0.025 mg/kg/day), which was included only in a scenario analysis. In the REAL trials, the somatropin brand was Norditropin, whereas in the Opko trials, it was Genotropin. |
Comparator response | There were substantial differences between the AEs experienced by somatropin-treated patients in the REAL trials compared with the Opko trials in terms of injection-site pain. In the REAL trials, 0 to 1 (1.5%) of somatropin-treated patients had experienced injection-site pain in the REAL 3 and REAL 4 trials at 52 weeks. In the Opko III trial, 29 (25.2%) somatropin-treated patients had experienced injection-site pain at 52 weeks. |
Definitions of end points | The definition and measurement of height-related end points were similar between all included trials. Height was generally measured 3 consecutive times. For the mean height for chronological age and sex, all trials except Opko JPN referred to the CDC and the standards by Prader.57 The Opko JPN trial referred instead to Japanese national data. |
Timing of end point evaluation | Overall, most trials reported the 3 outcomes of interest at week 26 and 52, with some exceptions. Longer-term outcomes were available from fewer trials, and comparative data after week 52 were only available from the REAL 3 trial. |
Withdrawal frequency | There were no concerning imbalances in withdrawal frequency:
|
Clinical trial setting | All trials included outpatient (clinic) and at-home procedures. |
Study design | All studies were open-label randomized controlled trials in which patients were randomized to either a LAGH or SAGH for at least 26 weeks, except REAL 3 was double-blinded within LAGH doses, and both REAL 3 and REAL 4 applied observer blinding of the primary end point. Opko JPN (not included in the base-case NMA) differed from all other trials in the study design with regard to dose escalation. |
AE = adverse event; CDC = Centers for Disease Control and Prevention; FAS full analysis set; GH = growth hormone; GHDS = growth hormone deficiency syndrome; HV = height velocity; IGF-1 = insulin-like growth factor 1; ITC = indirect treatment comparison; LAGH = long-acting growth hormone; NMA = network meta-analysis; SAGH = short-acting growth hormone; SAS = safety analysis set; SD = standard deviation; SDS = standard deviation score.
Source: Sponsor-submitted NMA report53 and accompanying SLR report.54
Results
Evidence Networks
The evidence networks are presented in Figure 5 for the base case and in Figure 6 for the alternative scenario that includes the Opko JPN trial. The availability of data by outcome and time point from each trial is summarized in Table 29.
Figure 5: Base-Case Scenario Evidence Network

d = day; QD = once daily; wk = week.
Source: Sponsor-submitted network meta-analysis.53
Figure 6: Alternative Scenario Evidence Network

d = day; QD = once daily; wk = week.
Note: Trials not included in the base-case evidence network are in light grey.
Source: Sponsor-submitted network meta-analysis.53
Table 29: Overview of Data Availability Across Included Trials for Each Outcome and Time Point
Outcome | REAL 3 | REAL 4 | Opko II | Opko III | Opko JPN (alt. only) | Number of studies in network |
|---|---|---|---|---|---|---|
AHV at 52 weeks | Yes | Yes | Yes | Yes | Yes | 4 (5 alt.) |
AHV at 26 weeks | Yes | Yes | Yes | Yes | Yes | 4 (5 alt.) |
HV SDS at 52 weeks | Yes | Yes | Yes | No | No | 4 (no alt.) |
HV SDS at 26 weeks | Yes | Yes | No | No | No | Not applicable |
Height SDS at 52 weeks | Yes | Yes | Yes | Yes | Yes | 4 (5 alt.) |
Height SDS at 26 weeks | Yes | Yes | Yes | Yes | Yes | 4 (5 alt.) |
AHV = annualized height velocity; alt. = alternative scenario; HV = height velocity; SDS = standard deviation score.
Source: Sponsor-submitted network meta-analysis.53
Annualized HV
Four of the 5 trials were included in the base-case network (all trials except Opko JPN), and all 5 trials were included in the alternative network (Table 30).
Results from the base-case NMA are presented in Table 31. While the difference in DIC and residual deviation between the FE and RE models did not indicate a strong model preference, there was a slight decrease with the RE model, suggesting a possible better model fit. Results from the RE model showed no differences between somapacitan and somatrogon or somatropin at week 26 or week 52. Results were generally similar with the FE model.
Results from the alternative network demonstrated that somapacitan improved AHV more than low-dose somatropin (0.025 mg/kg/day) at 52 weeks (difference of −1.61; 95% CrI, −3.23 to −0.16). No other differences between somatrogon and somapacitan were observed.
Table 30: Results From the Included Trials — AHV
Trial | Arm | Week 26 | Week 52 | ||
|---|---|---|---|---|---|
Mean | Difference (95% CI) | Mean | Difference (95% CI) | ||
REAL 3 | Somapacitan 0.16 mg/kg/week | 12.9 (SE, 0.67) | 1.7 (−0.2 to 3.6) | 11.7 (SE, 0.45) | 1.8 (0.5 to 3.1) |
Somatropin 0.034 mg/kg/day | 11.4 (SE, 0.66) | 9.9 (SE, 0.46) | |||
REAL 4 | Somapacitan 0.16 mg/kg/week | 12.25 (SE, 0.27) | −0.51 (−1.41 to 0.39) | 11.2 (SE, 0.19) | −0.5 (−1.1 to 0.2) |
Somatropin 0.034 mg/kg/day | 12.76 (SE, 0.37) | 11.7 (SE, 0.27) | |||
Opko II | Somatrogon 0.66 mg/kg/week | 13.5 (SD, 5) | NR | 11.93 (SD, 3.5) | NR |
Somatropin 0.034 mg/kg/day | 15 (SD, 2.9) | 12.5 (SD, 2.9) | |||
Opko III | Somatrogon 0.66 mg/kg/week | 10.59 (95% CI, 9.96 to 11.22) | 0.55 (−0.13 to 1.23) | 10.1 (95% CI, 9.58 to 10.63) | 0.33 (−0.24 to 0.89) |
Somatropin 0.034 mg/kg/day | 10.04 (95% CI, 9.47 to 10.62) | 9.78 (95% CI, 9.29 to 10.26) | |||
Opko JPN (alternative scenario only) | Somatrogon 0.66 mg/kg/week | 10.35 | 1.88 (0.74 to 3.03) | 9.65 | 1.79 (0.91 to 2.61) |
Somatropin 0.034 mg/kg/day | 8.47 | 8.47 | |||
AHV = annualized height velocity; CI = confidence interval; HV = height velocity; NR = not reported; SDS = standard deviation score; SE = standard error.
Source: Sponsor-submitted network meta-analysis.53
Table 31: Results From the Base-Case NMA for AHV at 26 and 52 Weeks
Parameter | Difference (95% CrI) | |
|---|---|---|
Fixed-effects model | Random-effects model | |
Week 26 | ||
Somatropin vs. somapacitan | 0.12 (−0.69 to 0.93) | 0.04 (−0.96 to 0.98) |
Somatrogon vs. somapacitan | 0.54 (−0.63 to 1.70) | 0.42 (−1.09 to 1.73) |
DIC | 17.14 | 16.62 |
Residual deviance | 11.13 | 10.33 |
Week 52 | ||
Somatropin vs. somapacitan | 0.09 (−0.50 to 0.66) | −0.02 (−0.86 to 0.68) |
Somatrogon vs. somapacitan | 0.32 (−0.53 to 1.21) | 0.19 (−1.08 to 1.25) |
DIC | 20.13 | 18.34 |
Residual deviance | 14.17 | 11.84 |
AHV = annualized height velocity; CrI = credible interval; DIC = deviance information criteria; NMA = network meta-analysis.
Note: Results are presented as median treatment differences and an associated 95% CrI for comparators vs. somapacitan. Positive differences favour comparator treatments.
Source: Sponsor-submitted NMA report.53
Table 32: Results From the Included Trials — HV SDS
Trial | Arm | Week 26 | Week 52 | ||
|---|---|---|---|---|---|
Mean | Difference (95% CI) | Mean | Difference (95% CI) | ||
REAL 3 | Somapacitan 0.16 mg/kg/week | 7.19 (SE, 0.9) | 1.61 (−0.97 to 4.19) | 5.72 (SE, 0.58) | 1.64 (−0.02 to 3.31) |
Somatropin 0.034 mg/kg/day | 5.58 (SE, 0.92) | 4.07 (SE, 0.59) | |||
REAL 4 | Somapacitan 0.16 mg/kg/week | 6.62 (SE, 0.33) | −0.62 (−1.74 to 0.49) | 5.62 (SE, 0.25) | −0.82 (−1.68 to 0.04) |
Somatropin 0.034 mg/kg/day | 7.24 (SE, 0.46) | 6.44 (SE, 0.35) | |||
Opko II | Somatrogon 0.66 mg/kg/week | NR | NR | 6.57 (SD, 1.2) | NR |
Somatropin 0.034 mg/kg/day | NR | 7.38 (SD, 0.88) | |||
Opko III | Somatrogon 0.66 mg/kg/week | NR | NR | NR | NR |
Somatropin 0.034 mg/kg/day | NR | NR | |||
Opko JPN (alternative scenario only) | Somatrogon 0.66 mg/kg/week | NR | NR | NR | NR |
Somatropin 0.034 mg/kg/day | NR | NR | |||
AHV = annualized height velocity; CI = confidence interval; HV = height velocity; NR = not reported; SD = standard deviation; SDS = standard deviation score; SE = standard error.
Source: Sponsor-submitted network meta-analysis.53
Results from the base case are presented in Table 33. While the difference in DIC and residual deviation between the FE and RE models did not indicate a strong model preference, there was a slight decrease with the RE model, suggesting a possible better model fit. Overall, no differences between somatrogon and somapacitan were observed in the RE analysis. Results were generally similar in the FE analysis.
Of note, results for the somapacitan versus somatrogon comparison differ in that the results numerically favoured somatrogon in the AHV analysis but favoured somapacitan in the HV SDS analysis. In either case, results did not indicate that somapacitan and somatrogon differed.
Height SDS
Four of the 5 trials were included in the base-case network (all trials except Opko JPN), and all 5 trials were included in the alternative network (Table 34).
Results from the base case are presented in Table 35. While the difference in DIC and residual deviation between the FE and RE models did not indicate a strong model preference, there was a slight decrease with the RE model, suggesting a possible better model fit. Overall, no differences between somatrogon and somapacitan were observed in the RE analysis. Results were generally similar in the FE analysis.
Results from the alternative network demonstrated that somapacitan improved height SDS more than low-dose somatropin (0.025 mg/kg/day) at 52 weeks (difference of −0.4; 95% CI, −0.72 to −0.11). No other differences between somatrogon and somapacitan were observed.
HV SDS
Data on HV SDS was available from 3 trials at week 52 (REAL 3, REAL 4, Opko II) and 2 trials at week 26 (REAL 3, REAL 4). A network for HV SDS at 26 weeks was determined to be not feasible. An alternative network was not constructed for HV SDS, as no data were available from the Opko JPN trial (Table 32).
Table 33: Results From the Base-Case NMA for Height Velocity SDS at 52 Weeks
Parameter | Difference (95% CrI) at 52 weeks | |
|---|---|---|
Fixed-effects model | Random-effects model | |
Somatropin vs. somapacitan | 0.26 (−0.49 to 1.01) | 0.15 (−0.90 to 1.07) |
Somatrogon vs. somapacitan | −0.55 (−1.65 to 0.57) | −0.66 (−2.21 to 0.77) |
Deviance information criteria | 16.73 | 15.20 |
Residual deviance | 11.76 | 9.84 |
CrI = credible interval; NMA = network meta-analysis; SDS = standardized deviation scores.
Results are presented as median treatment differences and an associated 95% CrI for comparators vs. somapacitan. Positive differences favour comparator treatments.
Source: Sponsor-submitted NMA report.53
Table 34: Results From the Included Trials — Height SDS
Trial | Arm | Week 26 | Week 52 | ||
|---|---|---|---|---|---|
Change | Difference (95% CI) | Change | Difference (95% CI) | ||
REAL 3 | Somapacitan 0.16 mg/kg/week | 0.87 (SE, 0.08) | 0.16 (−0.06 to 0.38) | 1.42 (SE, 0.1) | 0.35 (0.05 to 0.65) |
Somatropin 0.034 mg/kg/day | 0.71 (SE, 0.08) | 0.71 (SE, 0.08) | |||
REAL 4 | Somapacitan 0.16 mg/kg/week | 0.73 (SE, 0.03) | −0.09 (−0.20 to 0.02) | 1.24 (SE, 0.03) | −0.05 (−0.18 to 0.08) |
Somatropin 0.034 mg/kg/day | 0.82 (SE, 0.04) | 0.82 (SE, 0.04) | |||
Opko II | Somatrogon 0.66 mg/kg/week | 0.90 (SD, 0.39) | NR | 1.45 (SD, 0.61) | NR |
Somatropin 0.034 mg/kg/day | 1.00 (SD, 0.35) | 1.00 (SD, 0.35) | |||
Opko III | Somatrogon 0.66 mg/kg/week | 0.54 (95% CI, 0.48 to 0.61) | 0.06 (−0.01 to 0.13) | 0.92 (95% CI, 0.82 to 1.02) | 0.05 (−0.06 to 0.16) |
Somatropin 0.034 mg/kg/day | 0.48 (95% CI, 0.42 to 0.54) | 0.48 (95% CI, 0.42 to 0.54) | |||
Opko JPN (alternative scenario only) | Somatrogon 0.66 mg/kg/week | 0.58 | 0.26 (0.12 to 0.41) | 0.94 | 0.42 (0.23 to 0.61) |
Somatropin 0.034 mg/kg/day | 0.31 | 0.31 | |||
AHV = annualized height velocity; CI = confidence interval; HV = height velocity; NR = not reported; SDS = standard deviation score; SE = standard error.
Source: Sponsor-submitted network meta-analysis.53
Table 35: Results From Base Case for Height SDS at 26 and 52 Weeks
Parameter | Difference (95% CrI) | |
|---|---|---|
Fixed-effects model | Random-effects model | |
Week 26 | ||
Somatropin vs. somapacitan | 0.05 (−0.04 to 0.14) | 0.04 (−0.07 to 0.14) |
Somatrogon vs. somapacitan | 0.1 (−0.02 to 0.21) | 0.09 (−0.08 to 0.22) |
Deviance information criteria | 17.10 | 16.57 |
Residual deviance | 11.11 | 10.35 |
Week 52 | ||
Somatropin vs. somapacitan | 0 (−0.10 to 0.11) | −0.02 (−0.17 to 0.11) |
Somatrogon vs. somapacitan | 0.04 (−0.12 to 0.20) | 0.02 (−0.20 to 0.21) |
Deviance information criteria | 19.46 | 18.00 |
Residual deviance | 13.44 | 11.70 |
CrI = credible interval; SDS = standardized deviation score.
Results are presented as median treatment differences and an associated 95% CrI for comparators vs. somapacitan. Positive differences favour comparator treatments.
Source: Sponsor-submitted network meta-analysis.53
Long-Term Efficacy Outcomes
Long-term efficacy data beyond 52 weeks was informed by the trials summarized in Table 36, and the results from each extension trial are summarized in Table 37. Quantitative ITCs were not feasible due to differences in study design and the lack of common comparator arms, given the single-arm nature of most of the long-term data.
Only the REAL 3 trial reported comparative data at week 104 and 156. The difference in AHV between the somapacitan and somatropin treatment arms at week 156 was 0.8 cm/year (95% CI, −0.4 to 2.1). Differences in the other 2 outcomes were not formally evaluated but were numerically similar for HV SDS and height SDS. Results were not provided by the sponsor for any additional efficacy outcomes.
Table 36: Trials Informing Long-Term Efficacy Outcomes at 104 or 156 Weeks
Trial | Details of switching from main trial period to long-term period | AHV | HV SDS | Height SDS |
|---|---|---|---|---|
REAL 3 | After 26 weeks in the main trial period, patients receiving somapacitan and somatropin continued at the same dose for an additional 26 weeks. Then, all patients receiving somapacitan transitioned to 0.16 mg/kg/week, while patients receiving somatropin continued for an additional 104 weeks. Thereafter, patients entered a further 208-week extension using somapacitan 0.16 mg/kg/week only. |
|
|
|
REAL 4 | Patients from the RCT entered a 3-year extension period with somapacitan 0.16 mg/kg/week. | NR | NR | NR |
OPKO II | After 52 weeks in the main trial period, patients receiving somatrogon continued at the same dose for an additional 52 weeks, and patients receiving somatropin were randomized to 1 of the 3 somatrogon doses (0.25 mg/kg/week, 0.48 mg/kg/week, or 0.66 mg/kg/week) for 52 weeks. Thereafter, all patients transitioned to somatrogon 0.66 mg/kg/week. |
| NR |
|
OPKO III | After 52 weeks in the main trial period, patients from receiving somatrogon continued at the same dose, and patients receiving somatropin switched to somatrogon 0.66 mg/kg/week. |
| NR |
|
AHV = annualized height velocity; HV SDS = height velocity standard deviation score; NR = not reported; RCT = randomized controlled trial; SDS = standard deviation score.
Source: Sponsor-submitted network meta-analysis.53
Table 37: Long-Term Efficacy Results From Included Trials
Arm | AHV | Height SDS | HV SDS | |||
|---|---|---|---|---|---|---|
N | Mean | N | Change | N | Change | |
REAL 3 extension | ||||||
Week 52 (baseline) | ||||||
Somapacitan 0.16 mg/kg/week | 14 | 11.7 (SE, 0.45) | 14 | 1.5 (SD, 0.9) | 14 | 8.6 (SD, 3.2) |
Somatropin 0.034 mg/kg/day | 14 | 9.9 (SE, 0.46) | 14 | 1.0 (SD, 0.5) | 14 | 7.4 (SD, 4.1) |
Week 104 | ||||||
Somapacitan 0.16 mg/kg/week | 14 | 9.2 (SD, 1.7) | 14 | 2.2 (SD, 1.2) | 14 | 6.4 (SD, 3.0) |
Somatropin 0.034 mg/kg/day | 14 | 9 (SD, 2.3) | 14 | 1.7 (SD, 0.7) | 14 | 6.6 (SD, 3.2) |
Week 156 | ||||||
Somapacitan 0.16 mg/kg/week | 14 | 8.4 (SD, 1.7) | 14 | 2.7 (SD, 1.4) | 14 | 5.3 (SD, 3.0) |
Somatropin 0.034 mg/kg/day | 14 | 7.6 (SD, 2.0) | 14 | 2.1 (SD, 0.9) | 14 | 5.3 (SD, 3.9) |
Opko II extensiona | ||||||
Week 52 (baseline) | ||||||
Somatrogon 0.66 mg/kg/week | 13 | 11.93 | 41 | −2.61 (mean) | NR | NR |
Week 104 | ||||||
Somatrogon 0.66 mg/kg/week | 14 | 8.81 | 48 | −2.06 (mean) | NR | NR |
Week 156 | ||||||
Somatrogon 0.66 mg/kg/week | 44 | 7.18 | 43 | −1.6 (mean) | NR | NR |
Week 208 | ||||||
Somatrogon 0.66 mg/kg/week | 38 | 7.12 | 38 | −1.27 (mean) | NR | NR |
Opko III extensionb,c | ||||||
Week 52 (baseline) | ||||||
Somatrogon to somatrogon 0.66 mg/kg/week | NR | NR | NR | −2.01 (SD, 1.07) (mean) | NR | NR |
Somatropin to somatrogon 0.034 mg/kg/day | NR | NR | NR | −1.94 (SD, 1.13) (mean) | NR | NR |
Week 104 | ||||||
Somatrogon to somatrogon 0.66 mg/kg/week | 84 | 7.98 (SD, 1.81) | 84 | 0.42 (SD, 0.33) (change); −1.46 (SD, 0.87) (mean) | NR | NR |
Somatropin to somatrogon 0.034 mg/kg/day | 78 | 8.23 (SD, 1.88) | 78 | 0.49 (SD, 0.33) (change); −1.28 (SD, 0.78) (mean) | NR | NR |
AHV = annualized height velocity; CI = confidence interval; HV = height velocity; NR = not reported; SD = standard deviation; SDS = standard deviation score; SE = standard error.
Note: Weeks indicate time on treatment (including time from randomized phase).
aNo assessment of uncertainty in the Opko II trial for these outcomes was performed.
bFor the Opko III trial at weeks 52 and 104, AHV is reported among those who continued on somatrogon 0.66 mg/kg/week from the main trial, while height SDS is reported among all patients entering the extension, regardless of the dose received in the main trial.
cUnits were not provided by the sponsor but are assumed to represent mean (95% CI).
Source: Sponsor-submitted network meta-analysis.53
Harms
Harms outcomes were not evaluated quantitatively in the indirect analysis. The sponsor summarized injection AEs and antibodies qualitatively.
Harms Outcomes at 52 Weeks
Overall AEs
The qualitative comparison of overall AEs (e.g., any AEs, serious AEs, severe AEs) at 52 weeks is summarized in Table 38. The proportion of patients experiencing at least 1 AE across trials varied from approximately 60% to 100% through 52 weeks of treatment. Serious AEs were observed in 0 to 3 patients per treatment arm in all cases except the somapacitan arm of the REAL 4 trial, in which 6 (4.5%) of 132 patients experienced serious AEs. Severe AEs did not occur in the REAL 3 trial, occurred in few patients in the REAL 4 trial (1.5% in the somatropin arm compared with 3.0% in the somapacitan arm), and 5.2% in the somatropin arm compared with 8.3% in somatrogon arm in the OPKO III trial. Although very few AEs were observed in the REAL 3, Opko II, and Opko JPN trials, the sample sizes of each of the treatment arms were very small.
Table 38: Overall AEs Through 52 Weeks of Treatment in the Included Trials
Trial | Arm | N | Any AE, n (%) | Serious AE, n (%) | Severe AE, n (%) |
|---|---|---|---|---|---|
REAL 3 | Somapacitan 0.16 mg/kg/week | 14 | 13 (92.9) | 1 (7.1) | 0 |
Somatropin 0.034 mg/kg/day | 14 | 14 (100) | 1 (7.1) | 0 | |
REAL 4 | Somapacitan 0.16 mg/kg/week | 132 | 94 (71.2) | 6 (4.5) | 4 (3.0) |
Somatropin 0.034 mg/kg/day | 68 | 41 (60.3) | 2 (2.9) | 1 (1.5) | |
Opko II | Somatrogon 0.66 mg/kg/week | 14 | 10 (71.4) | 0 | 0 |
Somatropin 0.034 mg/kg/day | 11 | 8 (72.7) | 0 | 0 | |
Opko III | Somatrogon 0.66 mg/kg/week | 109 | 95 (87.2) | 3 (2.8) | 9 (8.3) |
Somatropin 0.034 mg/kg/day | 115 | 97 (84.3) | 2 (1.7) | 6 (5.2) | |
Opko JPN | Somatrogon 0.66 mg/kg/week | 22 | 22 (100) | 2 (9.1) | 2 (9.1) |
Somatropin 0.034 mg/kg/day | 22 | 19 (86.4) | 2 (9.1) | 2 (9.1) |
AE = adverse event.
Source: Sponsor-submitted network meta-analysis.53
Injection-Site AEs
Injection-related AEs are summarized in Table 39. The studies varied in what kinds of AEs were reported. Injection-site AEs overall were not reported in the Opko trials but occurred in a similar number of patients in the REAL 4 trial between the somapacitan and somatropin arms. Injection-site pain was reported in the REAL 3 and 4 trials and in the Opko III and Opko JPN trials. In the REAL trials, the number of injection-site pain events was zero to very few in both the somapacitan and somatropin arms. In the Opko III and Opko JPN trials, there were high rates of injection-site pain reported in the somatrogon arms (39.4% and 72.7%, respectively). However, in the Opko III and JPN trials, there were also much more frequent events of injection-site pain in the somatropin arms (25.2% and 13.6%, respectively) which contrasts with the 0% and 1.5% event rates in the somatropin-treated patients from the REAL 3 and REAL 4 trials, respectively.
Severe injection-site pain did not occur in any patients in the REAL 3 or 4 trials, regardless of treatment assignment, whereas 1 somatrogon-treated patient in the Opko II trial, and 4.6% of somatrogon-treated patients and 2.6% of somatropin-treated patients in the Opko III trial, experienced severe injection-site pain.
Table 39: Injection AEs Through 52 Weeks of Treatment in the Included Trials
Trial | Arm | N | Injection-site AE n (%) | Injection-site pain n (%) | Severe injection-site pain n (%) |
|---|---|---|---|---|---|
REAL 3 | Somapacitan 0.16 mg/kg/week | 14 | 0 | 0 | 0 |
Somatropin 0.034 mg/kg/day | 14 | 0 | 0 | 0 | |
REAL 4 | Somapacitan 0.16 mg/kg/week | 132 | 7 (5.3) | 2 (1.5) | 0 |
Somatropin 0.034 mg/kg/day | 68 | 4 (5.9) | 1 (1.5) | 0 | |
Opko II | Somatrogon 0.66 mg/kg/week | 14 | NR | NR | 1 (7.1) |
Somatropin 0.034 mg/kg/day | 11 | NR | NR | 0 | |
Opko III | Somatrogon 0.66 mg/kg/week | 109 | NR | 43 (39.4) | 5 (4.6) |
Somatropin 0.034 mg/kg/day | 115 | NR | 29 (25.2) | 3 (2.6) | |
Opko JPN | Somatrogon 0.66 mg/kg/week | 22 | NR | 16 (72.7) | 0 |
Somatropin 0.034 mg/kg/day | 22 | NR | 3 (13.6) | 0 |
AE = adverse event; NR = not reported.
Source: Sponsor-submitted network meta-analysis.53
Antibodies at 52 Weeks
In the REAL 3 trial, 1 patient (7.1%) treated with somatropin had persistent non-neutralizing anti–human GH (hGH) antibodies of low titre, and 2 patients (14.2%) treated with somapacitan 0.16 mg/kg/week had 1 single transient measurement of low-titre, non-neutralizing, anti-somapacitan antibodies during the 52-week treatment period. No neutralizing anti-hGH or anti-somapacitan antibodies were observed. In the REAL 4 trial, 2 patients (1.5%) treated with somapacitan, and 1 patient treated with somatropin (1.5%) had 2 or more consecutive positive non-neutralizing antibody samples during the 52-week treatment period. No neutralizing anti-hGH or anti-somapacitan antibodies were observed.
In the Opko II trial, 2 patients (14.3%) treated with somatrogon 0.66 mg/kg/week and 1 patient (9.1%) treated with somatropin had antidrug antibodies. There was no incidence of anti-CTP (cytidine triphosphate) antibodies. In the Opko III trial, 84 patients (77.1%) treated with somatrogon 0.66 mg/kg/week tested positive for antidrug antibodies compared with 18 patients (15.6%) treated with somatropin. In another trial of somatrogon (Opko JPN), 18 patients (81.8%) treated with somatrogon 0.66 mg/kg/week, and 4 patients (18.2%) treated with somatropin tested positive for antidrug antibodies; 2 patients (9.1%) treated with somatrogon tested positive for neutralizing antibodies at 1 visit.
Overall, the rates of antibodies varied considerably across the trials. Analyses showed patients who were positive for antibodies did not experience reduced efficacy or safety issues compared with those without antibodies.
Harms Outcomes in Extension Studies
Overall AEs
Overall, continued treatment with somapacitan and somatrogon was generally well tolerated, with no new safety signals identified. The proportion of patients experiencing at least 1 AE ranged from approximately 70% to 81% across the extensions of the REAL 3 (week 52 to 156), Opko II (week 52 to 208), and Opko III (week 52 to 104) trials. Most of the reported AEs were mild to moderate in severity.
In the extension of the REAL 3 (week 52 to 156) trial, serious AEs occurred in 4 (8.9%) of the pooled somapacitan-treated patients and 2 (14.3%) of the somatropin-treated patients. In the extension of the Opko II trial (week 52 to 208), serious AEs occurred in 3 (6.3%) of the somatrogon-treated patients. Serious AEs were not reported in the Opko III trial.
Severe AEs occurred in 0 of the somapacitan-treated patients and 1 (7.1%) of the somatropin-treated patients in the REAL 3 trial extension (week 52 to 156). Severe AEs were not reported in the Opko II and Opko III extension studies.
Injection-Site AEs
In the REAL 3 trial extension (week 52 to 156), 3 patients (6.7%) treated with somapacitan and 1 patient (7.1%) treated with somatropin experienced injection site–related AEs. All injection site–related AEs were considered to be mild in severity.
No data were available from the extension trials of somatrogon.
Antibodies
In the REAL 3 extension, over the entire 3 years of treatment (i.e., baseline to week 156), low-titre non-neutralizing antibodies were observed in 10 patients (22.2%) treated with somapacitan and 1 patient (7.1%) treated with somatropin. Seven patients (15.6%) treated with somapacitan experienced transient antibodies at a single time point, and 3 (6.7%) had more than 1 positive sample. The patient treated with somatropin experienced persistent anti-hGH antibodies of low titre from visit 4 to 16. All antibody samples were negative for neutralizing antibodies.
In the Opko II trial extension, 17 patients (35.4%) had low titres of anti-somatrogon antibodies, of which 3 had transient antibodies. All tests were negative for neutralizing antibodies. In the Opko III trial extension, among the patients in the somatrogon 0.66 mg/kg/week to somatrogon 0.66 mg/kg/week group, 84 of 109 patients (77%) had antidrug antibodies; 2 of these patients tested positive for neutralizing antibodies.
Similar to results from the 52-week outcomes, neutralizing antibodies were observed with somatrogon but not with somapacitan. Where reported, the analyses indicated that the presence of antibodies did not have any impact with respect to efficacy outcomes.
Critical Appraisal of Sponsor-Submitted ITC
An ITC was required to estimate the comparative effect estimates of somapacitan versus somatrogon; the choice of studies was appropriate to address the current gap in direct evidence. All studies had a common comparator (somatropin), albeit the somatropin arms in the REAL 3 and REAL 4 trials used a different brand (Norditropin) than that used in the Opko II, Opko III, and Opko JPN trials (Genotropin). The 2 brands were assumed to be equivalent and were pooled into 1 node in the NMA, given that they contain the same active ingredient. NICE previously noted there is no evidence of any difference between these brands,62 and this assumption was considered acceptable by the clinical expert consulted by CADTH. The Opko JPN trial differed from all other included trials in the study design, dose of the intervention and comparator, patient eligibility criteria, and patient characteristics at baseline, so it was appropriate that it was excluded from the base-case network and included in a separate, alternative-scenario network, where it informed an additional treatment node. Given the heterogeneity between the networks when the Opko JPN trial is included and given that the study population and dosages of treatments informed by the Opko JPN trial are not as relevant for the Canadian context, the results from this scenario analysis will not be discussed at length.
The ITC was conducted using NMA methodology, which was reasonable, given there were multiple studies informing multiple comparisons when the Opko JPN trial was included. However, since the target comparison is somapacitan versus somatrogon with a common comparator, simple Bucher analyses may have been preferable in the base case where the Opko JPN trial is excluded. A simple Bucher would be expected to generate results similar to the Bayesian FE model of the NMAs.
The sponsor quantitatively assessed the outcomes of AHV, height SDS, and HV SDS at 26 and 52 weeks in the NMAs. These outcomes were identified as the most important efficacy outcomes in this review, so the selection of efficacy outcomes was considered to be appropriate. However, the sponsor did not quantitatively assess any safety-related outcomes, and justification for this decision was not provided. It was not feasible to conduct quantitative ITCs for outcomes beyond 52 weeks due to a lack of comparative data and differences in the study designs of the relevant extension studies; therefore, a qualitative comparison was reported instead.
In the base-case NMAs, there were some between-trial imbalances between studies in race, gender, and mean age at baseline, and differences in eligibility criteria related to age range, peak GH, and bone age. The sponsor stated these were not expected to have any effect modification, but did not provide any support for this statement. Ultimately, the potential magnitude and direction of bias is unknown, which complicates the interpretation of the results. The clinical expert consulted by CADTH indicated that other potential prognostic factors or effect modifiers include the cause of GH deficiency (idiopathic versus organic), and isolated GHD versus multiple pituitary hormone deficiencies, but these characteristics could not be compared between trials due to a lack of reporting.
The networks were sparse. To account for potentially high heterogeneity, the sponsor utilized informative priors, but acknowledged that it was potentially problematic because the data informing the priors were not sourced externally from the included trials. The heterogeneity of the trials may therefore still bias the results. Additionally, the network had no closed loops, so assessing for consistency was not possible. The modelling of AHV and HV SDS was done in terms of the mean at the time point of interest, with an assumption by the sponsor that the mean values followed a normal distribution; however, it was not reported whether the sponsor tested whether this assumption holds. If the assumption does not hold, there is a concern that the results in mean difference were biased.
An important flag regarding the potential heterogeneity between trials is the differences in the trial outcomes for the arm used as a common comparator which, in this case, was somatropin 0.034 mg/kg/day. Among only somatropin-treated patients, there were substantial differences in the incidence of harms outcomes when comparing the REAL trials with the Opko trials. In the REAL 4 trial, 1 (1.5%) of the 68 somatropin-treated patients experienced injection-site pain while, in the Opko III trial, 29 (25.2%) of the 115 somatropin-treated patients experienced injection-site pain. Although the source of this difference is unknown, it may be related to differences in the treatment practices occurring at the treatment centres, different excipients in the different brands of somatropin used in the REAL versus Opko trials, and/or differences in how these events were recorded in the trials. Because the method of assessing injection-site pain was not reported in the REAL 3 and REAL 4 trials, it is unknown whether the cause of this difference is isolated to this outcome.
The sponsor’s conclusion that failure to detect a difference means that the efficacy is equivalent is somewhat uncertain. The sparse network, small sample sizes, and wide CrIs indicate inconclusive results, especially when considered together with the uncontrolled between-trial heterogeneity and inconsistency in AE results for the somatropin arms. Ultimately, there is no evidence that there is a clinically meaningful difference between somatropin and somatrogon, but whether the 2 drugs can be said to be clinically equivalent is likewise inconclusive.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by the materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
One open-label, randomized, phase II trial (REAL 3) was identified as a study addressing gaps in the evidence. The characteristics of this study are summarized in Table 40. Refer to Appendix 1 for the detailed patient inclusion and exclusion criteria.
REAL 3 was a randomized, multinational, multiple-dose, dose-finding, open-label, parallel-group trial investigating the efficacy and safety of once-weekly somapacitan compared with once-daily Norditropin (somatropin) in GH treatment-naive children (aged ≥ 2.5 and ≤ 10.0 years) with GHD. A total of 29 study sites randomized 59 patients. The main trial period ended on September 13, 2018. The trial consisted of a 26-week main trial period followed by a 26-week extension trial period, a 104-week safety extension trial period, a 208-week long-term safety extension trial period, and a 30-day follow-up period (Figure 7). Patients received once-weekly somapacitan at 1 of 3 different dose levels or once-daily Norditropin (somatropin) for the 26-week main and extension trial periods in a 4-arm parallel-group design.
Figure 7: Study Design for the REAL 3 Trial

Note: Reprinted (and modified) from Sävendahl et al., 2020.64 Creative Commons Attribution License 4.0.
Source: Sävendahl et al., 2020,63,64 and the week 26,67 week 156,68 and week 20869 Clinical Study Reports from the REAL 3 trial.
Table 40: Details of Studies Addressing Gaps in the Systematic Review Evidence
Detail | REAL 3 Study |
|---|---|
Study design | Phase II, multicentre, randomized, open-label, active-controlled study |
Locations |
|
Patient enrolment dates |
|
Randomized (N) | Total N = 59:
|
Key inclusion criteria |
|
Key exclusion criteria |
|
Intervention | Somapacitan 0.16 mg/kg, 0.08 mg/kg, or 0.04 mg/kg administered once a week through a prefilled pen for subcutaneous injection |
Comparator | Norditropin (somatropin) 0.034 mg/kg once a day through a prefilled pen for subcutaneous injection |
Screening phase | 3 weeks |
Main trial | 26 weeks |
Extension | 26 weeks |
Safety extension | 104 weeks |
Long-term safety extension | 208 weeks |
Follow-up phase | 30 days |
Primary end point | Height velocity at week 26 |
Secondary and exploratory end points | Secondary Efficacy:
Exploratory Patient-reported outcomes:
|
Publications |
BMI = body mass index; CDC = Centers for Disease Control and Prevention; FPFV = first patient first visit; G-DAT = Growth Hormone Device Assessment Tool; GH = growth hormone; GHD = growth hormone deficiency; GHD-CIM = Growth Hormone Deficiency–Child Impact Measure; GHD-CTB = Growth Hormone Deficiency–Child Treatment Burden; GHD-PTB = Growth Hormone Deficiency–Parent Treatment Burden; IGF-1 = insulin-like growth factor 1; IGFBP-3 = insulin-like growth factor binding protein 3; LPLV = last patient last visit; NA = not applicable; RCT = randomized controlled trial; SD = standard deviation; SDS = standard deviation score; TB-CGHD-O = Treatment Burden Measure–Child Growth Hormone Deficiency–Observer; TB-CGHD-P = Treatment Burden Measure–Child Growth Hormone Deficiency–Parent; TRIM-CGHD-O = Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer.
Note: 3 additional reports were included (Sävendahl et al., 2020;63,64 Sävendahl et al., 2022;65 and Sävendahl et al., 202366).
aActual key study date.
bPlanned key study date.
Source: Week 26,67 week 156,68 and week 20869 REAL 3 trial Clinical Study Reports.
Patients were randomized in a 1:1:1:1 ratio by a web-based randomization system (interactive web response system [IWRS]) to receive either once-weekly somapacitan (0.04 mg/kg, 0.08 mg/kg, or 0.16 mg/kg) or once-daily Norditropin (somatropin) during the main trial period (26 weeks) and the extension trial period (26 weeks). The randomization was stratified by region (Japan versus the rest of the world), age (< 6 years versus > 6 years), and gender in the rest-of-the-world region to minimize bias. After completing the main and extension trial periods (week 52), all patients initially randomized to double-blinded somapacitan were allocated to open-label somapacitan (0.16 mg/kg/week) treatment for the 104-week safety extension trial period. Patients randomized to Norditropin (somatropin) continued treatment with Norditropin (somatropin) (0.034 mg/kg/day). After completing the safety extension trial period (week 156), patients randomized to Norditropin (somatropin) were allocated to open-label somapacitan (0.16 mg/kg/week) for the 208-week long-term safety extension period. Cohorts 2 and 3, enrolled in the 208-week long-term safety extension trial period, were all allocated to open-label somapacitan (0.16 mg/kg/week).
The main trial period was double-blinded regarding different dose levels of once-weekly somapacitan, but was open-label with regard to daily Norditropin (somatropin) as the active control arm. The open-label design was chosen to enable comparison of local tolerability (daily versus weekly injections) and allowed for the comparison of patient satisfaction for dosing frequency. The 104-week safety extension and 208-week long-term safety extension trial periods were open-label, as only 1 dose level for somapacitan was used.
Populations
REAL 3 included prepubertal children aged 2 years and 26 weeks and older and less than 10 years old for boys and less than 9 years old for girls with a confirmed diagnosis of GHD by 2 different GH tests performed in the last 12 months before screening, defined as a peak GH level of 7 ng/mL or less. The patients included in the study were to have impaired height (≥ 2 SDs below the mean height for chronological age and gender at screening) and impaired HV, defined as an AHV below the 25th percentile for chronological age and gender or an SD of −0.7 or less according to the Prader standard, calculated over a time span of a minimum 6 months and a maximum of 18 months before screening. At screening, the mean IGF-1 level had to be at least 1 SD below the mean IGF-1 level standardized for age and gender. Patients were naive to treatment with other GH therapies or IGF-1 treatments. Key exclusion criteria included children with any clinically significant abnormality likely to affect growth or the ability to evaluate growth with standing or length measurements, patients born SGA, concomitant administration of other treatments that may have an effect on growth, and a prior history or presence of malignancy and/or intracranial tumour.
Interventions
Somapacitan was provided in a liquid formulation in the PDS290 pen-injector device and administered subcutaneously. In the REAL 3 trial, somapacitan was administered at 1 of 3 different doses (0.04 mg/kg, 0.08 mg/kg, or 0.16 mg/kg) throughout the 26-week main and extension trial periods, and at a dose of 0.16 mg/kg/week for the 104-week safety extension trial period and the 208-week long-term safety extension. The active control, Norditropin (somatropin), was provided in a liquid formulation in the FlexPro pen-injector device at a dose of 0.034 mg/kg/day for subcutaneous administration. In the REAL 3 trial, Norditropin (somatropin) was administered for 156 weeks.
Patients in the somapacitan and Norditropin (somatropin) treatment arms were trained by the site staff in the use of the pen injector to administer the trial drugs. The first dose was administered by the patient (at visit 2) under the supervision of the site staff. The subsequent doses were administered by the patient at home. When patients self-administered a trial product at home, compliance with trial-product administration was assessed and documented at each visit.
Outcomes
The outcomes measured in the REAL 367,68 trial were similar to those in the REAL 4 trial.20,51 An exemption was that the G-DAT was not measured in the REAL 3 trial.
Pharmacodynamic Parameters
The change from baseline in IGF-1 SDS and IGFBP-3 SDS was assessed at weeks 26 and 52. Blood samples were drawn for assessment for IGF-1 and IGFBP-3 before trial drug administration, if planned on a sampling day. The following equation was used to calculate IGF-1 SDS (median, skewness, and SD were based on reference tables published by Bidlingmaier et al.70,71). GF-1 SDS equals begin fraction open parenthesis “IGF-1 value over median” close parenthesis raised to the skewness power minus 1 over the product of skewness and standard deviation.
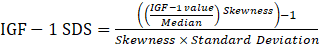
Statistical Analysis
Clinical Trial End Points
Annualized HV was analyzed using an MMRM, with treatment, age group, sex, region, and sex by age group interaction term as factors and height at baseline as a covariate, all nested within week as a factor. From the MMRM, the treatment differences at week 26 between the 3 somapacitan treatment arms and Norditropin (somatropin) control were estimated with the corresponding 95% CI.
The secondary efficacy end points, including change in height SDS, HV SDS, IGF-1 SDS, and IGFBP-3 SDS from baseline, were analyzed using an MMRM similar to the MMRM used for the primary analysis of the primary end point, with baseline assessment as covariate and treatment, age group, sex, region, and sex by age group interaction term as factors, all nested within week as a factor. The analyses were based on assessments from week 13, 26, 39, and 52. Changes from baseline to week 156 in height SDS, HV SDS, IGF-1 SDS, IGFBP-3 SDS, GHD-CIM, and the TB-CGHD-O and TB-CGHD-P questionnaires were analyzed using descriptive statistics.
The patient-reported outcome-based end points were analyzed using an ANCOVA, with treatment, age group, sex, region, and sex by age group interaction as factors. For change from baseline in GHD-CIM, the baseline score value was added to the model as a covariate. The GHD-CIM patient-reported outcome analyses were based on assessments from week 26 and week 52.
When an entire visit was missed and it was not possible to reschedule the visit in the allowed time window, attempts were made to ensure information was collected by telephone. Patients were invited for the next scheduled visit according to scheduling. To ensure patients had sufficient trial product until the next scheduled dispensing, the patient was required to collect their additional doses as soon as possible.
Sample Size and Power Calculation
In the REAL 3 trial, sample size calculations (cohort 1) were based on an assumption of an SD of 3.1 cm/year for HV after 26 weeks of treatment and the use of a delta value of −3.8 cm/year (where delta corresponds to the noninferiority margin in a noninferiority trial) and a 1-sided significance level of 2.5%. This resulted in 15 patients per treatment arm (somapacitan 0.04 mg/kg, somapacitan 0.08 mg/kg, somapacitan 0.16 mg/kg, and Norditropin [somatropin]). The maximum expected dropout rate during the trial was 7%, which resulted in a randomization of 15 patients per treatment arm. This ensured an 87% power and achieved a 95% CI for the ETD that was completely above the chosen delta value when the somapacitan and Norditropin (somatropin) treatment arms were compared, given that the 2 treatments were equal.
Statistical Testing
In the REAL 3 trial, the 1-sided test used for the primary end point was based on an alpha level of 2.5%. All other statistical tests conducted were 2-sided at the 5% significance level. Age group was defined as a factor with 2 levels: younger than 6 years old or 6 years of age or older. No adjustment for multiple testing was applied. Region was defined as a factor with 2 levels: Japan and all other countries.
Analysis Populations
The efficacy and safety end points in the REAL 3 trial were analyzed in all randomized patients who received at least 1 dose of treatment, analyzed as treated (Table 41).
Table 41: REAL 3 Trial Analysis Populations
Population | Definition | Application |
|---|---|---|
FAS | All randomized patients who received at least 1 dose of randomized treatment, analyzed as treated | Primary analysis of the primary end point |
Per-protocol set | Patients from the FAS who have not violated any inclusion or exclusion criteria and have used the randomized treatment for at least 22 weeks (for patients receiving somapacitan) or 154 days (for patients receiving Norditropin [somatropin)]) during the main trial period; analyzed as treated | Analysis of the primary end point included in the main trial period |
Safety analysis set | All randomized patients who received at least 1 dose of randomized treatment, analyzed as treated | Evaluation of safety end points |
FAS = full analysis set.
Source: REAL 3 trial Clinical Study Reports for week 26,67 week 156,68 and week 208.69
Results
Patient Disposition
In the REAL 3 trial, 59 patients were randomized and exposed to treatment. Of these, 53 patients (89.9%) completed the 26-week main trial period, the 26-week extension period, and the 104-week safety extension trial period (156 weeks). One patient in the somapacitan 0.04 mg/kg/week to 0.16 mg/kg/week arm withdrew from the study during the main trial period. Eight patients discontinued the trial product before the end of the 104-week safety extension trial period: 6 in the somapacitan treatment arms (4 patients in the 0.04 mg/kg/week to 0.16 mg/kg/week group, 1 patient in the 0.08 mg/kg/week to 0.16 mg/kg/week group, and 1 patient in the 0.16 mg/kg/week to 0.16 mg/kg/week group) and 2 in the Norditropin (somatropin) treatment arm. Two children who were randomized in error and discontinued treatment early were excluded from the FAS. Refer to Table 42 for the data.
Table 42: Summary of Patient Disposition From the REAL 3 Trial at Week 156
Patient disposition | REAL 3 | |
|---|---|---|
Somapacitan pooled | Norditropin (somatropin) | |
Screened, N | 86 | |
Reason for screening failure, n (%) | ||
Did not meet the eighth inclusion criterion of IGF-1 < −1.0 | || |||||| | |
Did not meet the seventh inclusion criterion of BMI > fifth and < 95th percentile according to CDC BMI-for-age growth charts | ||||| | |
Did not meet the sixth inclusion criterion of impaired HV, defined as annualized height velocity below the 25th percentile for chronological age and gender or ≤ −0.7 SD | ||||| | |
Did not meet the fifth inclusion criterion of impaired height, defined as ≥ 2 SDs below the mean height for chronological age and gender at screening according to the standards of the CDC | ||||| | |
Did not meet the ninth inclusion criterion of bone age less than chronological age at screening | ||||| | |
Did not meet the third inclusion criterion of IGF-1 level | ||||| | |
Did not meet the second inclusion criterion of prepubertal children | ||||| | |
Met any of the exclusion criteria | ||||| | |
Randomized, N (%) | 45 (100) | 14 (100) |
Discontinued from study, n (%) | 1 (2.2) | 0 |
Reason for discontinuation from study, n (%) | ||
Withdrawn by parent or guardian | 1 (2.2) | 0 |
Discontinued from treatment, n (%) | 6 (13.3) | 2 (14.3) |
Reason for discontinuation from treatment, n (%) | ||
Adverse events | 0 | 2 (14.3) |
Protocol violation | 4 (8.9) | 0 |
Withdrawn by parent or guardian | 2 (4.4) | 0 |
FAS, N | 43 (95.6) | 14 (100) |
PP, N | 41 (91.1) | 12 (85.7) |
Safety, N | 45 (100) | 14 (100) |
BMI = body mass index; CDC = Centers for Disease Control and Prevention; FAS = full analysis set; HV = height velocity; IGF-1 = insulin-like growth factor 1; NR = not reported; PP = per protocol; SD = standard deviation.
Note: Patient disposition presented for the main trial period, extension period and safety extension trial period (156 weeks).
Source: REAL 3 trial Clinical Study Report for week 26,67 week 156,68 and week 208.69
Baseline Characteristics
In general, the patient demographics were similar between groups. In the REAL 3 trial, most patients were Asian (|||) and white (|||||), while 1 patient was Black (||||). The proportion of idiopathic GHD in the somapacitan 0.16 mg/kg/week group (92.9%) was higher than in the Norditropin (somatropin) group (85.7%) (Table 43).
Table 43: Summary of Baseline Characteristics From the REAL 3 Trial (Full Analysis Set)
Characteristic | Somapacitan 0.16 mg/kg/week (N = 14) | Norditropin (somatropin) (N = 14) |
|---|---|---|
Age, years | ||
Mean (SD) | 6.11 (2.33) | 5.95 (1.99) |
Median (range) | 5.90 (2.8 to 9.8) | 5.77 (3.5 to 9.6) |
< 6 years, n (%) | 8 (57.1) | 7 (50.0) |
≥ 6 years, n (%) | 6 (42.9) | 7 (50.0) |
Sex, n (%) | ||
Male | 8 (57.1) | 9 (64.3) |
Female | 6 (42.9) | 5 (35.7) |
Race, n (%) | ||
Asian | 8 (57.1) | 6 (42.9) |
Black or African American | 0 | 1 (7.1) |
White | 6 (42.9) | 7 (50.0) |
Other | 0 | 0 |
Not reported | 0 | 0 |
Height, cm | ||
Mean (SD) | 97.74 (16.41) | 98.35 (13.81) |
Median (range) | 95.17 (64.3 to 123.0) | 98.55 (78.5 to 120.5) |
Weight, kg | ||
Mean (SD) | 14.88 (5.23) | 15.46 (5.03) |
Median (range) | 14.05 (6.2 to 25.4) | 14.60 (9.1 to 27.0) |
BMI, kg/m2 | ||
Mean (SD) | 15.14 (1.15) | 15.59 (1.42) |
Median (range) | 14.75 (13.8 to 17.2) | 15.45 (13.6 to 18.6) |
GHD cause, n (%) | ||
Idiopathic | 13 (92.9) | 12 (85.7) |
Organic | 1 (7.1) | 2 (14.3) |
GH peak, mcg/L | ||
Mean (SD) | 4.13 (2.44) | 3.97 (1.96) |
Median (range) | 5.28 (0.05 to 6.88) | 4.55 (0.80, 6.56) |
Mother’s height, cm | ||
Mean (SD) | NR | NR |
Median (range) | NR | NR |
Father’s height, cm | ||
Mean (SD) | NR | NR |
Median (range) | NR | NR |
Baseline mean (SD) | ||
Height velocity (cm/year) | 3.76 (1.46) | 3.50 (1.59) |
Height velocity SDS | −2.85 (1.84) | −3.14 (2.14) |
Height SDS | −3.83 (2.03) | −3.38 (1.05) |
IGF-1 SDS | −2.04 (1.02) | −2.07 (0.74) |
IGFBP-3 SDS | −1.60 (1.45) | −1.75 (0.94) |
BMI = body mass index; GH = growth hormone; GHD = growth hormone deficiency; IGF-1 = insulin-like growth factor 1; IGFBP-3 = insulin-like growth factor binding protein 3; NR = not reported; SD = standard deviation; SDS = standard deviation score.
Source: REAL 3 trial Clinical Study Report (week 26).67
Exposure to Study Treatments
A summary of patient exposure from the REAL 3 trial at week 52 and up to week 156 is shown in Table 44.
Table 44: Summary of Patient Exposure From the REAL 3 Trial (Full Analysis Set)
Exposure | Somapacitan 0.16 mg/kg/week (N = 14) | Norditropin (somatropin) (N = 14) |
|---|---|---|
Week 52 | ||
Total, days | 2,614 | 2,434 |
Duration, days, mean (SD) | 186.7 (4.7) | 173.9 (37.1) |
Duration, days, median (range) | 185 (182 to 200) | 183 (45 to 189) |
Adherence, mean (%)a | 95.8 | 89.9 |
Up to week 156 | ||
Total, days | 14,665 | 12,901 |
Duration, days, mean (SD) | 1,047.5 (194.7) | 921.5 (367.6) |
Duration, days, median (range) | 1,100 (371 to 1,103) | 1,093 (45 to 1,188) |
Adherence, mean (%)a | 92.2 | 87.2 |
NA = not applicable; SD = standard deviation.
aRepresents adherence according to diary: Number of reported dosings from diary in adherence divided by number of planned dosings multiplied by 100.
Source: Week 2667 and week 15668 REAL 3 trial Clinical Study Reports.
In the REAL 3 trial, 14 patients (32.6%) in the somapacitan groups and 5 patients (35.7%) in the Norditropin (somatropin) group received concomitant medication at baseline.
Efficacy
Height Velocity
In the REAL 3 trial, there was a 1.7 cm/year (95% CI, −0.2 to 3.6) difference in the increase in HV between somapacitan and Norditropin (somatropin) at week 26. Similarly, at week 52, this difference was 1.8 cm/year (95% CI, 0.5 to 3.1) (Table 45). The difference at week 156 was 0.8 cm/year (95% CI, −0.4 to 2.1 cm/year).
Height SDS
There was an increase of 0.16 (95% CI, −0.06 to 0.38) in height SDS with somapacitan compared with Norditropin (somatropin) at week 26 and an increase of 0.35 (95% CI, 0.05 to 0.65) at week 52.
HV SDS
There was a difference of 1.61 (95% CI, −0.97 to 4.19) in the increase in the HV SDS when comparing somapacitan with Norditropin (somatropin) at 26 weeks, and a similar difference at 52 weeks (1.64; 95% CI, −0.02 to 3.31); of note, however, is that the results included the null.
Pharmacodynamic End Points
In the REAL 3 trial, there was a statistically significant increase in IGF-1 SDS with somapacitan 0.16 mg/kg/week compared with Norditropin (somatropin) after 26 weeks (ETD = 1.17; 95% CI, 0.38 to 1.95) and 52 weeks (ETD = 1.56; 95% CI, 0.66 to 2.46) of treatment. There was also a statistically significant increase in the IGFBP-3 SDS at week 52 with somapacitan 0.16 mg/kg/week compared with Norditropin (somatropin) (ETD = 0.93; 95% CI, 0.13 to 1.73).
Table 45: Summary of Key Efficacy Results From the REAL 3 Trial (Full Analysis Set)
Variable | Week 26 | Week 52 | ||
|---|---|---|---|---|
Somapacitan 0.16 mg/kg/week (N = 14) | Norditropin (somatropin) (N = 14) | Somapacitan 0.16 mg/kg/week (N = 14) | Norditropin (somatropin) (N = 14) | |
Auxologic response | ||||
Height velocity (cm/year) | ||||
Baseline (cm/year), mean (SD) | 3.8 (1.5) | 3.5 (1.6) | 3.8 (1.5) | 3.5 (1.6) |
End point (cm/year), observed mean (SD) | 12.9 (3.5) | 11.3 (3.3) | 11.5 (2.6) | 9.8 (2.3) |
End point (cm/year), estimated meana | 13.08 | 11.41 | 11.7 | 9.9 |
Estimated treatment difference (95% CI)a | 1.67 (−0.22 to 3.56) | 1.8 (0.5 to 3.1) | ||
P value | NR | NR | ||
Height SDS | ||||
Baseline, mean (SD) | −3.83 (2.03) | −3.38 (1.05) | −3.83 (2.03) | −3.38 (1.05) |
Observed mean (SD) change from baseline | 0.88 (0.52) | 0.66 (0.37) | 1.45 (0.86) | 0.98 (0.50) |
Estimated mean change from baselinea | 0.87 | 0.71 | 1.42 | 1.07 |
Estimated treatment difference (95% CI)a | 0.16 (−0.06 to 0.38) | 0.35 (0.05 to 0.65) | ||
P value | 0.1549 | 0.0216 | ||
Height velocity SDS | ||||
Baseline, mean (SD) | −2.85 (1.84) | −3.14 (2.14) | −2.85 (1.84) | −3.14 (2.14) |
Observed mean (SD) change from baseline | 10.01 (4.67) | 9.02 (5.03) | 8.60 (3.15) | 7.41 (4.08) |
Estimated mean change from baselinea | 9.85 | 8.23 | 8.38 | 6.73 |
Estimated treatment difference (95% CI)a | 1.61 (−0.97 to 4.19) | 1.64 (−0.02 to 3.31) | ||
P value | 0.2147 | 0.0531 | ||
Height (cm) | ||||
Baseline (cm), mean (SD) | 97.7 (16.4) | 98.3 (13.8) | 97.7 (16.4) | 98.3 (13.8) |
Change from baseline (cm), mean (SD) | 6.6 (1.9) | 5.7 (1.6) | 11.7 (3.0) | 9.8 (2.3) |
End point (cm), observed mean (SD) | 104.3 (15.5) | 104.1 (13.6) | 109.5 (14.8) | 108.1 (12.8) |
Estimated treatment difference (95% CI) | |||| ||||||| ||||| | |||| |||||| ||||| | ||
P value | NR | NR | ||
Body weight (kg) | ||||
Baseline (cm), mean (SD) | 14.88 (5.23) | 15.46 (5.03) | 14.88 (5.23) | 15.46 (5.03) |
Change from baseline (cm), mean (SD) | 2.71 (1.07) | 1.43 (1.01) | 4.74 (1.67) | 3.86 (2.14) |
End point (cm), observed mean (SD) | 17.59 (5.94) | 16.89 (5.59) | 19.62 (5.88) | 19.31 (6.47) |
Estimated treatment difference (95% CI) | |||| |||||| ||||| | |||| ||||||| ||||| | ||
P value | NR | NR | ||
Bone age versus chronological age ratio | ||||
Baseline, mean (SD) | NR | NR | 0.601 (0.199) | 0.556 (0.148) |
End point, mean (SD) | NR | NR | 0.092 (0.138) | 0.021 (0.110) |
Estimated treatment difference (95% CI)a | NR | NR | ||
P value | NR | NR | ||
Pharmacodynamic end points | ||||
IGF-1 SDS | ||||
Baseline, mean (SD) | −2.04 (1.02) | −2.07 (0.74) | −2.04 (1.02) | −2.07 (0.74) |
Observed mean (SD) change from baseline | 3.00 (1.43) | 1.86 (0.81) | 3.29 (1.73) | 1.67 (1.78) |
Estimated mean change from baselinea | 3.28 | 2.11 | 3.37 | 1.81 |
Estimated treatment difference (95% CI)a | |||| |||||| ||||| | |||| |||||| ||||| | ||
P value | 0.0046 | 0.0011 | ||
IGF-1 (ng/mL) | ||||
Baseline, geometric mean [coefficient of variation (%)]) | 54.68 (35.75) | 48.52 (18.82) | 54.68 (35.75) | 48.52 (18.82) |
Change from baseline (ng/mL), mean (SD) | 156.08 (92.71) | 80.17 (37.77) | 179.83 (89.75) | 83.84 (67.33) |
End point (ng/mL), geometric mean [coefficient of variation (%)]) | 210.76 (103.12) | 128.69 (39.59) | 234.51 (92.93) | 132.36 (56.75) |
Estimated treatment difference (95% CI) | ||||| ||||||| |||||| | ||||| ||||||| |||||| | ||
P value | NR | NR | ||
IGFBP-3 SDS | ||||
Baseline, mean (SD) | −1.60 (1.45) | −1.75 (0.94) | −1.60 (1.45) | −1.75 (0.94) |
End point, mean (SD) | −0.14 (1.06) | −0.18 (0.81) | 0.21 (0.94) | −0.8 (1.53) |
Observed mean (SD) change from baseline | 1.45 (1.33) | 1.58 (0.99) | 1.81 (1.39) | 0.95 (1.88) |
Estimated mean change from baselinea | 1.68 | 1.76 | 2.04 | 1.11 |
Estimated treatment difference (95% CI)a | −0.09 (−0.72 to 0.54) | 0.93 (0.13 to 1.73) | ||
P value | 0.7842 | 0.0234 | ||
CI = confidence interval; IGF-1 = insulin-like growth factor 1; IGFBP-3 = insulin-like growth factor binding protein 3; MMRM = mixed-model for repeated measures; NA = not applicable; NR = not reported; SD = standard deviation; SDS = standard deviation score.
aMMRM with treatment, age group, sex, region, and sex by age group interaction as factors and baseline assessment as a covariate, all nested within week as a factor.
Source: Week 2667 and week 15668 REAL 3 trial Clinical Study Reports.
Patient-Reported Outcomes
GHD-CIM (Previously TRIM-CGHD-O)
In the REAL 3 trial, a greater reduction in GHD-CIM scores from baseline to week 52 was observed in patients in the somapacitan 0.16 mg/kg/week group compared with those in the Norditropin (somatropin) group for all 3 domains (emotional well-being, physical health, social well-being) and the total score (Table 46). No ETDs achieved statistical significance.
GHD-CTB (Previously TB-CGHD-O)
In the REAL 3 trial, a lower estimated mean value for all 3 domains (physical functioning, emotional well-being, and interference) and the overall score at 52 weeks was observed in patients in the somapacitan group compared with those in the Norditropin (somatropin) group. The physical functioning and interference scores for somapacitan 0.16 mg/kg/week were significantly improved compared with Norditropin (somatropin), with an ETD in the physical functioning score of −9.59 (95% CI, −18.21 to −0.96), versus an ETD in the interference score of −10.45 (95% CI, −17.10 to −3.80) (Table 47).
Table 46: Summary of GHD-CIM (TRIM-CGHD-O) Scores From the REAL 3 Trial at Week 52 (Full Analysis Set)
Variable | Somapacitan 0.16 mg/kg/week (N = 14) | Norditropin (somatropin) (N = 14) |
|---|---|---|
Physical functioning score | ||
Baseline mean (SD) | 32.6 (19.1) | 28.8 (17.8) |
Number of patients | 14 | 13 |
Estimated change from baselinea | −5.54 | −2.80 |
Estimated treatment difference (95% CI)a | −2.74 (−11.29 to 5.81) | |
P value | 0.522 | |
Emotional well-being score | ||
Baseline mean (SD) | 16.5 (24.6) | 22.0 (27.1) |
Number of patients | 14 | 12 |
Estimated change from baselinea | −15.45 | −6.11 |
Estimated treatment difference (95% CI)a | −9.34 (−23.76 to 5.07) | |
P value | 0.1979 | |
Social well-being score | ||
Baseline mean (SD) | 34.2 (21.7) | 37.8 (19.0) |
Number of patients | 14 | 14 |
Estimated change from baselinea | −20.04 | −9.92 |
Estimated treatment difference (95% CI)a | −10.12 (−25.60 to 5.37) | |
P value | 0.1952 | |
Total score | ||
Baseline mean (SD) | 27.8 (11.7) | 30.0 (16.7) |
Number of patients | 14 | 13 |
Estimated change from baselinea | −13.85 | −6.41 |
Estimated treatment difference (95% CI)a | −7.43 (−16.90 to 2.03) | |
P value | 0.1207 | |
CI = confidence interval; GHD-CIM = Growth Hormone Deficiency–Child Impact Measure; MMRM = mixed-model for repeated measures; SD = standard deviation; TRIM-CGHD-O = Treatment-Related Impact Measure–Child Growth Hormone Deficiency–Observer.
aMMRM with treatment, age group, sex, region, and sex by age group interaction as factors and baseline assessment as a covariate, all nested within week as a factor.
Source: REAL 3 trial Clinical Study Report (week 156).68
Table 47: Summary of GHD-CTB (TB-CGHD-O) Scores From the REAL 3 Trial at Week 52 (Full Analysis Set)
Variable | Somapacitan 0.16 mg/kg/week (N = 14) | Norditropin (somatropin) (N = 14) |
|---|---|---|
Physical functioning score | ||
Number of patients | 14 | 13 |
Estimated meana | 4.71 | 14.29 |
Estimated treatment difference (95% CI)a | −9.59 (−18.21 to −0.96) | |
P value | 0.0301 | |
Emotional well-being score | ||
Number of patients | 14 | 12 |
Estimated meana | 6.15 | 15.77 |
Estimated treatment difference (95% CI)a | −9.62 (−22.30 to 3.05) | |
P value | 0.1334 | |
Interference score | ||
Number of patients | 14 | 13 |
Estimated meana | −0.06 | 10.39 |
Estimated treatment difference (95% CI)a | −10.45 (−17.10 to −3.80) | |
P value | 0.0027 | |
Total score | ||
Number of patients | 14 | 12 |
Estimated meana | 3.54 | 10.43 |
Estimated treatment difference (95% CI)a | −6.88 (−14.07 to 0.31) | |
P value | 0.0602 | |
CI = confidence interval; GH = growth hormone; GHD-CTB = Growth Hormone Deficiency–Child Treatment Burden; MMRM = mixed-model for repeated measures; SD = standard deviation; TB-CGHD-O = Treatment Burden Measure–Child Growth Hormone Deficiency–Observer.
aIn the REAL 3 trial, the analysis was conducted using an MMRM model, with treatment, age group, sex, region, and sex by age group interaction as factors and baseline assessment as a covariate, all nested within week as a factor. In the REAL 4 trial, the MMRM model had treatment, gender, age group, region, GH peak group, and gender by age group by region interaction term as factors and baseline value as a covariate, all nested within week as a factor.
Source: REAL 3 trial Clinical Study Report (week 156).68
GHD-PTB (Previously TB-CGHD-P)
In the REAL 3 trial, a lower estimated mean value for 2 domains and the overall score was observed in patients in the somapacitan 0.16 mg/kg/week group compared with those in the Norditropin (somatropin) group at week 52 (Table 48).
Table 48: Summary of GHD-PTB (TB-CGHD-P) Scores From the REAL 3 Trial at Week 52 (Full Analysis Set)
Variable | Somapacitan 0.16 mg/kg/week (N = 14) | Norditropin (somatropin) (N = 14) |
|---|---|---|
Emotional well-being score | ||
Number of patients | 14 | 13 |
Estimated meana | 13.06 | 21.05 |
Estimated treatment difference (95% CI)a | −7.99 (−21.35 to 5.36) | |
P value | 0.2348 | |
Interference score | ||
Number of patients | 14 | 13 |
Estimated meana | 3.63 | 7.52 |
Estimated treatment difference (95% CI)a | −3.89 (−12.71 to 4.92) | |
P value | 0.379 | |
Total score | ||
Number of patients | 14 | 13 |
Estimated meana | 8.34 | 14.28 |
Estimated treatment difference (95% CI)a | −5.94 (−15.58 to 3.69) | |
P value | 0.2209 | |
CI = confidence interval; GHD-PTB = Growth Hormone Deficiency–Parent Treatment Burden; MMRM = mixed-model for repeated measures; SD = standard deviation; TB-CGHD-P = Treatment Burden Measure–Child Growth Hormone Deficiency–Parent.
aMMRM with treatment, age group, sex, region, and sex by age group interaction as factors and baseline assessment as a covariate, all nested within week as a factor.
Source: REAL 3 trial Clinical Study Report (week 156).68
Harms
Overall, the safety profile of once-weekly somapacitan was similar to the safety profile of once-daily Norditropin (somatropin) (Table 49).
Adverse Events
In the REAL 3 trial, AE rates were similar between the somapacitan treatment arms (258.1 AEs per 100 PYs for the pooled somapacitan group) and the Norditropin (somatropin) arm (267.7 AEs per 100 PYs). The most common AEs observed in at least 10% of patients (AEs observed in at least 2 patients in 1 or more treatment arms) included pyrexia, influenza, nasopharyngitis, constipation, vomiting, rhinitis allergic, and gastroenteritis. The majority of AEs were of mild (87.2%) or moderate (12.1%) severity, and 3 AEs were classified as severe. The majority of AEs (93.9%) were considered unlikely to be related to the trial product by the investigator.
Serious Adverse Events
Up to week 156 in the REAL 3 trial, the event rates for SAEs were similar for the somapacitan and Norditropin (somatropin) groups (6.5 SAEs per 100 PYs and 8.5 SAEs per 100 PYs, respectively).
Withdrawal Due to Adverse Events
Up to week 156, 2 patients discontinued treatment due to AEs in the Norditropin (somatropin) group (1 due to nephrotic syndrome; 1 due to drug hypersensitivity).
Mortality
No deaths were reported up to week 156 in the REAL 3 trial.
Notable Harms
Injection-Site Reactions
A low proportion of children experienced injection-site reactions in both the somapacitan group (1.7%) and the Norditropin (somatropin) group (1.7%) in the REAL 3 trial at week 156.
Antibodies
During the 156 weeks of the REAL 3 trial, low-titre non-neutralizing antibodies were observed in 10 somapacitan-treated patients. In 7 patients in the somapacitan groups, somapacitan antibodies were observed at single time points: 4 patients in the 0.04 mg/kg/week to 0.16 mg/kg/week group, 2 patients in the 0.08 mg/kg/week to 0.16 mg/kg/week group, and 1 patient in the 0.16 mg/kg/week to 0.16 mg/kg/week group.
In 3 patients, positive samples were observed in 2 consecutive samples in the somapacitan 0.16 mg/kg/week to 0.16 mg/kg/week group, 3 consecutive samples in the somapacitan 0.04 mg/kg/week to 0.16 mg/kg/week group, and 4 consecutive samples in the somapacitan 0.08 mg/kg/week to 0.16 mg/kg/week group. One child in the Norditropin (somatropin) treatment arm had persistent anti-hGH antibodies of low titre from visit 4 (week 13) to visit 16 (follow-up visit for the present trial period). All antibody-positive samples were negative for in vitro neutralizing antibodies.
IGF-1 SDS Greater Than 2
In the REAL 3 trial, 3 patients in the somapacitan 0.16 mg/kg/week group and 2 in the Norditropin (somatropin) group had IGF-1 SDS values above 2 at 2 or more consecutive visits. No trend was seen in the amount or type of AEs reported in patients with IGF-1 SDS above 2 at 2 or more consecutive visits, compared with the remaining children in the relevant groups. None of the patients with an s SDS above 2 at 2 or more consecutive visits had any treatment interruptions or dose reductions.
Injection-Site Infection
No patients had an injection-site infection up to week 156 in the REAL 3 trial.
Injection-Site Pain
Up to week 156 in the REAL 3 trial, 1 patient (7.1%) versus no patients experienced injection-site pain in the somapacitan and Norditropin (somatropin) groups, respectively.
Table 49: Summary of Harms Results From the REAL 3 Trial at Week 156 (Full Analysis Set)
Adverse events | Somapacitan 0.16 mg/kg/week (N = 14) | Norditropin (somatropin) (N = 14) |
|---|---|---|
Most common adverse events, n (%) | ||
≥ 1 adverse event | 14 (100) | 14 (100) |
Headache | 1 (7.1) | 2 (14.3) |
Nasopharyngitis | 3 (21.4) | 3 (21.4) |
Pyrexia | 4 (28.6) | 2 (14.3) |
Pain in extremity | 1 (7.1) | 1 (7.1) |
Bronchitis | 1 (7.1) | 2 (14.3) |
Vomiting | 3 (21.4) | 1 (7.1) |
Influenza | 2 (14.3) | 3 (21.4) |
Rhinitis | 1 (7.1) | 3 (21.4) |
Serious adverse events, n (%) | ||
Patients with ≥ 1 SAEa | 2 (14.3) | 2 (14.3) |
Patients who stopped treatment due to adverse events, n (%), n (%) | ||
Patients who stoppedb | 0 | 2 (14.3) |
Deaths, n (%) | ||
Patients who died | 0 | 0 |
Adverse events of special interest, n (%) | ||
Injection-site reactionc | 1 (7.1) | 1 (7.1) |
Two consecutive positive GH antibody samples | 1 (7.1) | 1 (7.1) |
IGF-1 SDS > 2 at 2 (or more) consecutive visits | 3 (21.4) | 2 (14.3) |
Injection-site infection | 0 | 0 |
Injection-site pain | 1 (7.1) | 0 |
Dysglycemiad | 0 | 1 (7.1) |
GH = growth hormone; IGF-1 = insulin-like growth factor 1; SAE = serious adverse event; SDS = standard deviation score.
aIn the REAL 3 trial, SAEs occurred in 2 patients in the somapacitan 0.16 mg/kg/week treatment arm (1 patient had tonsillitis, generalized edema, and vomiting; 1 patient had anaphylactic shock) and in 2 patients in the Norditropin (somatropin) treatment arm (1 patient had adenoidectomy; 1 patient had respiratory syncytial virus bronchitis and nephrotic syndrome).
bIn the REAL 3 trial, 2 patients in the Norditropin (somatropin) treatment arm discontinued treatment due to adverse events (nephrotic syndrome and drug hypersensitivity).
cInjection-site reactions included pain, bruising, hematoma, swelling, hypersensitivity, skin atrophy, lipoatrophy, subcutaneous hemorrhage, and hip deformity.
dIn the REAL 3 trial, 1 patient in the Norditropin (somatropin) treatment arm had 2 events of abnormal glucose metabolism (impaired fasting glucose and abnormal blood glucose).
Source: REAL 3 trial Clinical Study Report (week 156).68
Dysglycemia and Glucose Metabolism
Up to week 156 in the REAL 3 trial, no patients versus 1 patient (1.7%) experienced dysglycemia in the somapacitan and Norditropin (somatropin) groups, respectively. There were no apparent clinically relevant changes in mean fasting plasma glucose after 156 weeks of treatment in either of the treatment groups. For all treatment groups, the mean values for fasting plasma glucose at week 156 were similar to the mean values at baseline and in reference ranges. There were no apparent clinically relevant changes in mean hemoglobin A1C from baseline to week 156 in either of the treatment groups. All mean values of hemoglobin A1C were within the reference ranges throughout the trial.
REAL 3 Long-Term Safety Extension Phase
Description of Studies
A summary of the long-term safety extension phase for the REAL 3 trial is presented subsequently.69 The results presented in this interim analysis reflect the data available as of November 19, 2021.
After completing the safety extension trial period (week 156), patients randomized to Norditropin (somatropin) were allocated to open-label somapacitan (0.16 mg/kg/week) for a 208-week long-term safety extension period. The patients in cohorts 2 and 3, who were enrolled in the 208-week long-term safety extension trial period were all allocated to open-label somapacitan (0.16 mg/kg/week).
Populations
The inclusion criteria for patients who completed the main trial, extension, and safety extension period were described in the main body of the report. The key inclusion and exclusion criteria for cohort 2 and 3 are presented in Table 50. Refer to Appendix 1 for the detailed patient inclusion and exclusion criteria.
Interventions
After completing the safety extension trial period (week 156), patients randomized to Norditropin (somatropin) were allocated to open-label somapacitan (0.16 mg/kg/week) for the 208-week long-term safety extension period. The patients in cohorts 2 and 3, enrolled in the 208-week long-term safety extension trial period, were all allocated to open-label somapacitan (0.16 mg/kg/week).
Table 50: Inclusion and Exclusion Criteria for Cohort 2 and 3 in the REAL 3 Safety Extension Phase
Details | Cohort 2 | Cohort 3 |
|---|---|---|
Key inclusion criteria |
|
|
Key exclusion criteria |
|
|
BMI = body mass index; GH = growth hormone; GHD = growth hormone deficiency; IGF-1 = insulin-like growth factor 1; SD = standard deviation; SDS = standard deviation score.
Source: REAL 3 trial Clinical Study Report (week 208).69
Outcomes
The primary end point for cohorts 2 and 3 is the incidence of AEs, including injection-site reactions, during at least 13 weeks and up to 208 weeks of treatment with somapacitan in children with GHD.
The supportive secondary safety end point for cohort 1 is the incidence of AEs, including injection-site reactions, and the occurrence of anti-somapacitan and anti-hGH antibodies for up to 364 weeks of treatment.
Statistical Analysis
The addition of cohorts 2 and 3 is based on a regulatory request from the FDA and no formal sample size calculation was performed for these cohorts. The analyses of cohorts 2 and 3 will include all patients who received at least 1 dose of somapacitan 0.16 mg/kg/week.
AEs were analyzed using descriptive statistics and summarized by cohort.
Results
Baseline Characteristics
For cohort 1, the demographics were proportionally similar between the 14 patients in the Norditropin (somatropin) to somapacitan group and the 43 patients in the somapacitan 0.04 mg/kg/week, 0.08 mg/kg/week, and 0.16 mg/kg/week pooled group. No clinically relevant differences in characteristics were evident at baseline.
In cohorts 1 and 2, all children were prepubertal (Tanner stage I) at screening. In cohort 3, the 2 previously treated children had entered puberty (Tanner stage > 1) and, among the 7 treatment-naive children, 5 were prepubertal and 2 had entered puberty at screening.
Patient Disposition
Patient disposition for the long-term safety extension phase of REAL 3 is presented in Table 51. In cohort 1, a total of 50 children were exposed in the long term safety extension trial period following week 156. No children in cohort 1 discontinued treatment from week 156 to 208. Among the 51 children who completed the long-term safety extension trial period (208 weeks), 1 patient was following protocol version 1, where visit 7 (week 52) was the last visit. This patient was counted as a completer of the subsequent treatment periods but was not exposed to treatment in the long term safety extension trial period.
In cohorts 2 and 3, a total of 15 children were screened for inclusion (recruitment still ongoing). In cohorts 2 and 3, no children withdrew from the trial or discontinued treatment before the cut-off date of the present (week 208) report. That analysis includes data for patients in cohorts 2 and 3 up to visit 20 (52 weeks of treatment) or the cut-off date of August 26, 2021, whichever came first.
Table 51: Patient Disposition for the REAL 3 Trial, Safety Extension Phase
Patient disposition | Cohort 1 | Cohorts 2 and 3 | ||
|---|---|---|---|---|
Somapacitan pooled | Norditropin (somatropin) | Cohort 2 | Cohort 3 | |
Screened, N | 86 | 15 | ||
Randomized, N (%) | 45 (100) | 14 (100) | — | — |
Exposed in long-term safety extension, n (%) | 39 (86.7) | 11 (78.6) | 1 (100) | 9 (100) |
Discontinued from study, n (%) | 2 (4.4) | 0 | 0 | 0 |
Completed long-term safety extension, n (%) | 39 (86.7) | 12 (85.7) | 1 (100) | 1 (11.1) |
Source: REAL 3 trial Clinical Study Report (week 208).69
Exposure to Study Treatments
In cohort 1, a total of 45 children were exposed to somapacitan for up to 208 weeks and 14 children were exposed to Norditropin (somatropin) for up to 156 weeks. Among the 14 children who started on Norditropin (somatropin) at week 0, 11 were switched to somapacitan for up to 52 weeks. The median exposure was similar across treatment groups. In cohort 2, 1 child was exposed to somapacitan for 366 days. In cohort 3, 2 treatment-naive children were exposed to somapacitan for up to 52 weeks, with a median exposure of 338 days, and 7 previously treated children were exposed to somapacitan for up to 52 weeks, with a median exposure of 161 days (Table 52).
Table 52: Summary of Patient Exposure for the REAL 3 Trial, Safety Extension Phase
Exposure | Cohort 1 | Cohorts 2 and 3 | |||
|---|---|---|---|---|---|
Somapacitan pooled (N = 45) | Norditropin (somatropin)/ somapacitan (N = 11) | Cohort 2 previously treated (N = 1) | Cohort 3 treatment-naive (N = 2) | Cohort 3 previously treated (N = 7) | |
Total, days | 59,585 | 3,941 | 366 | 676 | 1,022 |
Duration, mean (SD) | 1,324.1 (403.8) | 358.3 (27.8) | 366 | 338.0 (59.4) | 146.0 (75.7) |
Duration, median (range) | 1,471 (14 to 1,512) | 366 (275 to 374) | 366 | 338 (296 to 380) | 161 (7 to 242) |
Adherence, mean (%) | 96.8 | 96.2 | 100 | 76.1 | 67.4 |
SD = standard deviation.
Source: REAL 3 trial Clinical Study Report (week 208).69
Efficacy
The sponsor’s interim analysis includes data for cohort 1 up to week 208, and data for cohorts 2 and 3 obtained up to visit 20 or the cut-off date of August 26, 2021, whichever came first.
Efficacy outcomes were evaluated descriptively for the long-term safety extension phase and are briefly summarized here. In cohort 1, between 156 and 208 weeks, the HV showed overall parallel development for the 2 treatment groups, ending up at mean HV values after 208 weeks of 7.4 cm/year (SD = 1.6) and 6.6 cm/year (SD = 1.6) for the somapacitan pooled and Norditropin (somatropin) to somapacitan group, respectively. Overall, HV, HV SDS and height SDS, IGF-1 SDS, IGFBP-3 SDS, bone age progression, and patient-reported outcome questionnaires showed similar development from 156 to 208 weeks for the 2 treatment groups.
The single previously treated patient in cohort 2 showed an increase in HV from 7.0 cm/year at week 13 to 9.2 cm/year at week 52. No baseline assessment was available for this patient. For cohort 3, there were 2 treatment-naive patients with measurements from baseline to week 26, with mean HV values increasing from 5.3 cm/year (SD = 3.5) to 8.0 cm/year (SD = 0.1). For 1 of these patients, an HV measurement at 52 weeks was also available (7.7 cm/year).
The mean HV values (n previously treated patients with measures) for cohort 3 at baseline (n = 7), week 13 (n = 6), and week 26 (n = 3) were 7.4 cm/year (SD = 2.0), 5.0 cm/year (SD = 2.7), and 6.5 cm/year (SD = 0.8), respectively, demonstrating maintenance of achieved HV in the previously treated patients. The effect on other height-based measures (HV SDS, height SDS), pharmacodynamic parameters (IGF-1 SDS, IGFBP-3 SDS), and patient-reported outcome questionnaires was similar to what was observed with HV.
Harms
Overall, the safety profile of once-weekly somapacitan administered subcutaneously to children with GHD for up to 208 weeks was similar to the well-known safety profile of daily GH (e.g., Norditropin [somatropin]). No new safety issues were identified. No local tolerability issues were identified. Similar AE reporting rates were observed for the somapacitan 0.04 mg/kg/week, 0.08 mg/kg/week, and 0.16 mg/kg/week pool and the somapacitan 0.16 mg/kg/week pool (248.2 and 232.9 AEs per 100 PYs) compared with the Norditropin (somatropin) group (270.8 AEs per 100 PYs). In comparison, a lower AE rate was reported in the Norditropin (somatropin) to somapacitan 0.16 mg/kg/week group (111.2 AEs per 100 PYs) but the low exposure in this treatment group should be considered. The majority of the AEs were nonserious and of mild or moderate severity.
Critical Appraisal
Internal Validity
The major limitation related to the REAL 3 trial is the small sample size, which resulted in high uncertainty in the results due to the lack of precision. Furthermore, REAL 3 is an open-label trial, which may influence outcomes that are subjective in measurement and interpretation (e.g., patient-reported outcomes, subjective AEs). In the REAL 3 trial, only prepubertal children were enrolled into the main trial to avoid the pubertal growth spurt interfering with the treatment effect. For the 208-week long-term safety extension study in the REAL 3 trial, 2 additional cohorts with different age groups were enrolled at the request of the FDA. The reason for adding these additional cohorts was to enrol children with GHD in additional age groups for whom treatment may be relevant. However, at the time of the last data cut-off, only 15 children were screened for both cohorts 2 and 3, and only 2 children were included in the current analysis. The low number of participants made it challenging to interpret the results and draw any conclusions.
The efficacy outcomes were presented using descriptive statistics, so no statistical inferences were possible, which limited the interpretation of the results. The safety profile for somapacitan from the main phase to the long-term extension phase was consistent in the REAL 3 trial.
External Validity
The major limitations in external validity for the randomized controlled period in the REAL 3 trial (up to week 156) were similar to those of the REAL 4 trial and were related to the characteristics of enrolled patients in terms of age (prepubertal children), race (50% were Asian and 46% were white), being GH treatment-naive, and the majority (89%) having idiopathic GHD, and the exclusion of patients with an intracranial tumour and potential exclusion of patients who could not stand up (e.g., patients with significant spinal abnormalities and congenital abnormalities were excluded in the REAL 4 trial).
Because the patients who took part in the open-label long-term safety extension phase were originally from the REAL 3 trial and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to the open-label long-term safety extension phase. The 2 additional cohorts in the REAL 3 long-term safety extension phase may have different demographics and baseline characteristics than those in cohort 1 from the main trial. Since the recruitment of these additional cohorts is still ongoing and the current number of patients is very low, it is not possible to compare the generalizability.
Discussion
Summary of Available Evidence
This systematic review included evidence of the benefits and harms of somapacitan from 1 pivotal trial with a single-arm extension period, 1 ITC, and 1 other relevant study.
The pivotal trial (REAL 4) was a phase III, multicentre, open-label trial that compared once-weekly somapacitan with once-daily Norditropin (somatropin) after 52 weeks of treatment, with a long-term (additional 3 years in plan and 1 year of data available), single-arm extension phase. Two hundred GH therapy–naive children with GHD (mean age, 6.4 years; 74.5% were male and 25.5% were female) were randomized in a 2:1 ratio to either somapacitan or Norditropin (somatropin) groups and measures of growth (HV, height SDS, HV SDS), and patient-reported outcomes (GHD-CIM, GHD-CTB, GHD-PTB) at week 52 (time point for primary end point) were compared. Harms and adherence to therapy were also recorded.
The sponsor conducted NMAs assessing AHV, HV SDS, and height SDS to indirectly compare somapacitan with somatrogon. Data sufficient to form a network was available at week 26 and 52 for AHV and height SDS, but only at week 26 for HV SDS. No NMAs were conducted for any safety-related outcomes and were not feasible for any longer-term efficacy outcomes. No published NMAs were included.
One other relevant sponsor-submitted study (REAL 3) was a phase II, multicentre, open-label, dose-finding trial that compared somapacitan with Norditropin (somatropin) after 156 weeks of treatment, with a long-term (additional 4 years in plan and 1 year of data available), single-arm extension phase. Fourteen patients were enrolled in the 0.16 mg/kg/week somapacitan group and 14 patients were enrolled in the Norditropin (somatropin) group. The mean age of these patients was 6 years; 60.7% of them were male and 39.3% were female (cohort 1). Pediatric patients of other ages (cohort 2 enrolled patients younger than 2.5 years, and cohort 3 enrolled patients of pubertal age and up to 17 years of age) were also analyzed in the long term safety extension phase. The outcomes were similar to those in the REAL 4 trial.
Interpretation of Results
Efficacy
Auxologic Response
Evidence from the pivotal, phase III REAL 4 trial showed that somapacitan is noninferior to Norditropin (somatropin) in HV achievement at week 52 in prepubertal children with GHD, addressing a relevant and important treatment outcome noted by both patients and clinicians and at an adequate time point of measurement for a GH therapy, according to the clinical expert consulted by CADTH. Adult or near-adult height is considered the clinical end point for assessing the efficacy of GH therapy in pediatric patients, and HV can be a meaningful surrogate end point when there is a need to assess shorter-term efficacy outcomes before patients reach adulthood.72 Though final or near-adult height was mentioned as an important outcome by the clinical expert, it was not included in the list of outcomes for GRADE assessment. During the consultation with the clinical expert, the clinical expert noted that the 52-week follow-up time point is ideal to assess response to a GH therapy and the most feasible; however, final height would not be expected to be achieved at this time point for all patients (this would require longer follow-up, potentially many years); the inclusion of only prepubertal patients with GHD was considered adequate, as it is ideal to initiate a GH therapy at a young age. SDS-related measures are typically used for end points that are influenced by age, sex, race, and geographic location, such as height, HV, and IGF-1. Change in height SDS, which can reflect the growth of pediatric patients with GHD relative to the population of children without GHD who have the same or similar demographic characteristics, is often used as a surrogate outcome to assess treatment efficacy in both clinical practice and patient research.72 In the REAL 4 trial after 52 weeks of treatment, the somapacitan and Norditropin (somatropin) treatment arms showed similar efficacy in change from baseline for height SDS and HV SDS. Monitoring HV and change in height SDS at an interval of every 6 to 12 months is also valuable to assess whether the dose or the type of GH therapy is adequate. According to Bang et al. and the perspective of the Growth Hormone Research Society 2019 Workshop experts, when 1 or more of the 3 criteria (change in HV of < 2 cm/year, change in height SDS of < 0.3 per year, or HV SDS < 0) are met during the first 6 to 12 months after initiation of the GH therapy, it may indicate an inadequate response with that therapy in patients with GHD.73,74 These parameters were not observed in patients who received somapacitan in either the REAL 4 or REAL 3 trials at week 52, which suggests an adequate response to the therapy in the surrogate, growth-related end points.
In the NMAs, the sparse network, small sample sizes, between-trial heterogeneity, and wide CrIs indicate inconclusive results across all of the assessed outcomes. Ultimately, there is no evidence that there is a clinically meaningful difference between somatropin and somatrogon, as there were no statistically significant differences identified in AHV, height SDS, or HV SDS at week 26, nor in AHV and height SDS at week 52. However, whether somatropin and somatrogon can be said to be clinically equivalent is inconclusive. Additionally, longer-term outcomes could not be quantitatively compared through an NMA due to a lack of data. The REAL 3 and REAL 4 trials evaluated similar outcomes, each with up to 3 years of efficacy outcomes available. As stated by the clinical expert, greater auxologic response and efficacy (larger ETD) were observed in both arms at week 26 and week 52, which was in the first year after initiation of GH therapy. The ability to interpret the results of the REAL 3 trial is limited, mainly due to its small sample size.
Pharmacodynamic Outcomes
Although data on IGF-1 SDS, IGF-1 level, and IGFBP-3 SDS in the REAL 4 trial are reported in Appendix 2, because they were not identified as the most important end points by any groups that provided input, these biochemical biomarkers may play a role in aiding GH dose adjustment. Although no consensus research results have found a direct correlation between IGF-1 level and the clinical end points, IGF-1 is the most optional measure among those currently available and, ideally, IGF-1 SDS should be close to 0 during the treatment.72 The IGF-1 SDS can also serve as a long-term safety marker during GH treatment because a too-high level of IGF-1 would be harmful, e.g., an IGF-1 SDS greater than 2 at 2 (or more) consecutive visits was prespecified as a safety outcome in both the REAL 4 and REAL 3 trials.20,68,72 In both trials, there was a small or no increase in IGF-1 SDS and IGFBP-3 SDS with somapacitan compared with Norditropin (somatropin), and the mean IGF-1 SDS levels were within the normal range (−2 to 2) in both arms at week 26 and/or week 52. No pharmacodynamic outcomes were assessed in the ITCs.
Patient-Reported Outcomes
In the REAL 4 trial, statistically significant differences were found in the measurement of parent treatment burden (in both total score and the subdomain scores of emotional well-being and interference for GHD-PTB) that were in favour of somapacitan compared with Norditropin (somatropin). The efficacy in GHD-CIM and GHD-CTB was similar between the 2 arms. In the REAL 3 trial, 2 subdomains of GHD-CTB (physical functioning and interference scores) demonstrated a greater improvement with somapacitan than with Norditropin (somatropin); however, the efficacy in the third domain (emotional well-being) and total score for the GHD-CTB, GHD-CIM, and GHD-PTB was similar between the 2 arms. Furthermore, it was uncertain whether the detected differences were clinically meaningful because the point estimates and/or boundaries of the 95% CIs did not exceed their MIDs. Other types of studies prefer using qualitative methods to explore the perspectives and attitudes of the participating patients and their caregivers, which might be valuable to understanding the reasons why there were no significant differences in the questionnaire data—despite evident differences in subcutaneous injection frequencies between the 2 treatments. The result of the G-DAT that evaluated the ease of device use showed similarly high proportions of patients in both arms who rated the device as “easy” or “very easy” to use. No patient-reported outcomes were assessed in the ITCs.
Harms
It appears there were similar results in safety outcomes, including the notable harms of injection-site reactions and injection-site pain. No new safety signals were identified from the longer-term single-arm studies; however, long-term comparative data were not available. As per the information provided by the sponsor, both injection-site reactions and injection-site pain (which was assessed as part of the injection-site reactions) were assessed and recorded during the visual and manual inspections performed by the investigator at each visit. There is the potential for the under-reporting of these AEs (i.e., the occurrence of the injection-site reactions and, particularly, injection-site pain), as they could be more frequent during treatment administration at home than was reported in the REAL 4 and REAL 3 trials; therefore, the results of the comparisons of injection-site pain between the 2 groups or with other therapies, if measured using the patients’ own records, become uncertain.
In the ITCs comparing somapacitan with somatrogon, there were no quantitative comparisons of safety, and there were concerning differences between trials in the rate of injection-site pain in the common comparator arm (somatropin 0.034 mg/kg once daily) that was not explained. As a result, no conclusions can be drawn regarding the comparative safety of somapacitan versus somatrogon.
Other Considerations
According to the clinical expert consulted by CADTH, treatment discontinuation (or adherence to therapy) is important and can impact the efficacy of a GH therapy. The expert noted that the adherence rates (which were high in both the somapacitan and Norditropin (somatropin) groups, with a higher rate in the somapacitan group) reported in the REAL 4 (96% for somapacitan versus 88% for Norditropin [somatropin]) and REAL 3 (92% for somapacitan versus 87% for Norditropin [somatropin]) trials were higher than in clinical practice. The proportion of patients who discontinued treatment for any reason was similar between the somapacitan and Norditropin (somatropin) groups in the REAL 4 trial. A possible reason for this could be that, compared with usual practice, the treatment arms in a clinical trial have more stringent conditions (regular follow-up visits, stricter patient screening criteria, and more examinations), which could make it more likely that patients will adhere to the treatment. Also, the expert indicated it is possible that patients who inject weekly are more likely associated with a higher adherence rate and a lower treatment discontinuous rate than those who inject daily for 6 days a week, especially among the patients or caregivers who have needle phobia, and a better GH treatment adherence would benefit the growth achievement. More studies with a long follow-up duration are necessary to evaluate the association between the magnitude of missing doses and the effects of a GH therapy and to investigate the factors that may influence treatment adherence.
Conclusion
One phase III, multicentre, open-label study (REAL 4) compared subcutaneous injection of somapacitan 0.16 mg/kg once weekly with Norditropin (somatropin) 0.034 mg/kg once a day in prepubertal children (aged 2.5 years to 10 years for girls and 2.5 years to 11 years for boys) with GHD at week 52. Somapacitan results in little to no difference in HV and height SDS (high certainty), and likely results in a smaller improvement from baseline in HV SDS (moderate certainty) when compared with Norditropin (somatropin). Somapacitan may result in little to no difference in disease-specific functioning when compared with Norditropin (somatropin) (low certainty). Somapacitan was associated with a reduction in child treatment burden (low certainty) and caregiver treatment burden (moderate certainty), compared with Norditropin (somatropin). Somapacitan results in little to no difference in treatment discontinuation when compared with Norditropin (somatropin) (moderate certainty). The evidence shows that somapacitan likely results in little to no difference in the notable harms, injection-site reactions and injection-site pain, when compared with Norditropin (somatropin) (moderate certainty). No new safety signals were identified from the longer-term single-arm studies; however, long-term comparative data were not available.
In the NMAs, there were no statistically significant differences identified between somapacitan and somatrogon (0.66 mg/kg/week) for the outcomes of AHV, height SDS, or HV SDS at week 26, nor in AHV or height SDS at week 52; however, the interpretation of the NMA is limited by the sparse network, small sample sizes, between-trial heterogeneity, and wide CrIs across all assessed outcomes. Disease-specific functioning, child treatment burden, and safety-related outcomes were not assessed in the NMAs.
References
1.SOGROYA® (somapacitan injection) pre-filled pen for subcutaneous use 5 mg/1.5 mL (3.3 mg/mL), 10 mg/1.5 mL (6.7 mg/mL), 15 mg/1.5 mL (10 mg/mL) [product monograph]. Mississauga (ON): Novo Nordisk Canada Inc.; 2023 Jul 26.
2.Hage C, Gan H-W, Ibba A, et al. Advances in differential diagnosis and management of growth hormone deficiency in children. Nature Reviews Endocrinology. 2021;17(10):608-624. PubMed
3.Ranke MB. Short and Long-Term Effects of Growth Hormone in Children and Adolescents With GH Deficiency. Frontiers in Endocrinology. 2021;12(950). PubMed
4.Deal C, Kirsch S, Chanoine JP, et al. Growth hormone treatment of Canadian children: results from the GeNeSIS phase IV prospective observational study. CMAJ Open. 2018;6(3):E372-e383. PubMed
5.Taylor M, Couto-Silva AC, Adan L, et al. Hypothalamic-pituitary lesions in pediatric patients: endocrine symptoms often precede neuro-ophthalmic presenting symptoms. J Pediatr. 2012;161(5):855-863. PubMed
6.Brod M, Alolga SL, Beck JF, Wilkinson L, Højbjerre L, Rasmussen MH. Understanding burden of illness for child growth hormone deficiency. Qual Life Res. 2017;26(7):1673-1686. PubMed
7.Thomas M, Massa G, Craen M, et al. Prevalence and demographic features of childhood growth hormone deficiency in Belgium during the period 1986-2001. Eur J Endocrinol. 2004;151(1):67-72. PubMed
8.Vimpani GV, Vimpani AF, Lidgard GP, Cameron EH, Farquhar JW. Prevalence of severe growth hormone deficiency. Br Med J. 1977;2(6084):427-430. PubMed
9.Stanley T. Diagnosis of growth hormone deficiency in childhood. Curr Opin Endocrinol Diabetes Obes. 2012;19(1):47-52. PubMed
10.Pfizer Inc. Health canada approves ngenla (somatrogon) injection for pediatric growth hormone deficiency. 2021; https://www.pfizer.ca/en/media-centre/health-canada-approves-ngenla-somatrogon-injection-pediatric-growth-hormone-deficiency. Accessed 2023 Jun 2.
11.Richmond E, Rogol AD. Treatment of growth hormone deficiency in children, adolescents and at the transitional age. Best Practice & Research Clinical Endocrinology & Metabolism. 2016;30(6):749-755. PubMed
12.Grimberg A, DiVall SA, Polychronakos C, et al. Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin-Like Growth Factor-I Deficiency. Horm Res Paediatr. 2016;86(6):361-397. PubMed
13.Hardin DS. Treatment of short stature and growth hormone deficiency in children with somatotropin (rDNA origin). Biologics. 2008;2(4):655-661. PubMed
14.CADTH Reimbursement Recommendation: Somatrogon (Ngenla). Ottawa (ON): CADTH; 2022 March: https://www.cadth.ca/sites/default/files/DRR/2022/SR0683%20Ngenla%20-%20CADTH%20Final%20Rec.pdf. Accessed 2023 May 15.
15.Rose S, Cook D, Fine M. Growth Hormone Therapy Guidelines: Clinical and Managed Care Perspectives. AJPB® Translating Evidence-Based Research Into Value-Based Decisions®. 2014;6(5).
16.Nwosu BU, Jasmin G, Parajuli S, Rogol AD, Wallace EC, Lee AF. Long-term GH Therapy Does Not Advance Skeletal Maturation in Children and Adolescents. Journal of the Endocrine Society. 2021;5(5). PubMed
17.NGENLA (RETARD DE CROISSANCE). Quebeq (QC): INESSS; 2022: https://www.inesss.qc.ca/en/themes/medicaments/drug-products-undergoing-evaluation-and-evaluated/extract-notice-to-the-minister/ngenla-retard-de-croissance-5853.html. Accessed 2023 Jun 2.
18.Cutfield WS, Derraik JG, Gunn AJ, et al. Non-compliance with growth hormone treatment in children is common and impairs linear growth. PLoS One. 2011;6(1):e16223. PubMed
19.Fisher BG, Acerini CL. Understanding the growth hormone therapy adherence paradigm: a systematic review. Horm Res Paediatr. 2013;79(4):189-196. PubMed
20.Clinical Study Report: NN8640-4263. REAL 4 main period (up to week 52). A trial comparing the effect and safety of once weekly dosing of somapacitan with daily Norditropin® in children with growth hormone deficiency [internal sponsor's report]. Bagsvaerd, Denmark: Novo Nordisk. 2022 Mar 18.
21.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of clinical epidemiology. 2011;64(4):401-406. PubMed
22.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of clinical epidemiology. 2020;119:126-135. PubMed
23.Clinical Study Report: NN8640-4263. REAL 4 main period (up to week 104). A trial comparing the effect and safety of once weekly dosing of somapacitan with daily Norditropin® in children with growth hormone deficiency [internal sponsor's report]. Bagsvaerd, Denmark: Novo Nordisk. 2023 Feb 9.
24.Song K, Jin SL, Kwon AR, et al. Etiologies and characteristics of children with chief complaint of short stature. Annals of Pediatric Endocrinology & Metabolism. 2015;20:34 - 39. PubMed
25.GHR Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab. 2000;85(11):3990-3993. PubMed
26.Webb EA, Dattani MT. Diagnosis of growth hormone deficiency. Endocr Dev. 2010;18:55-66. PubMed
27.Boot AM, Engels MA, Boerma GJ, Krenning EP, De Muinck Keizer-Schrama SM. Changes in bone mineral density, body composition, and lipid metabolism during growth hormone (GH) treatment in children with GH deficiency. J Clin Endocrinol Metab. 1997;82(8):2423-2428. PubMed
28.Møller N, Jørgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. 2009;30(2):152-177. PubMed
29.Kopchick JJ, Berryman DE, Puri V, Lee KY, Jorgensen JOL. The effects of growth hormone on adipose tissue: old observations, new mechanisms. Nature Reviews Endocrinology. 2020;16(3):135-146. PubMed
30.Overview, Human growth hormone in children (review of NICE technology appraisal guidance 42). Human growth hormone (somatropin) for the treatment of growth failure in children. London (UK): National Institute for Health and Care Excellence; 2010: https://www.nice.org.uk/guidance/ta188/resources/human-growth-hormone-somatropin-for-the-treatment-of-growth-failure-in-children-pdf-82598502860485. Accessed 2023 Jun 2.
31.Ranke MB, Wit JM. Growth hormone — past, present and future. Nature Reviews Endocrinology. 2018;14(5):285-300. PubMed
32.Chinoy A, Murray PG. Diagnosis of growth hormone deficiency in the paediatric and transitional age. Best Practice & Research Clinical Endocrinology & Metabolism. 2016;30(6):737-747. PubMed
33.CPEG. CPEG Consensus Criteria for the Diagnosis of Growth Hormone Deficiency in Canada - October 2010. Ottawa (ON): Canadian Pediatric Endocrine Group (CPEG); 2010: https://cpeg-gcep.net/sites/default/files/upload/bookfiles/CPEG%20Consensus%20GH%20Guidelines.pdf. Accessed 2023 Jun 2.
34.Growth Hormone Stimulation Test. New York City (NY): Icahn School of Medicine at Mount Sinai; 2022: https://www.mountsinai.org/health-library/tests/growth-hormone-stimulation-test#:~:text=The%20growth%20hormone%20(GH)%20stimulation,vein%20to%20raise%20hGH%20levels. Accessed 2023 Jun 2.
35.Drug Reimbursement Review clinical guidance report: somatrogon (Ngenla) for the treatment of growth hormone deficiency. Vol 2. Ottawa (ON): CADTH; May 2022: https://www.cadth.ca/somatrogon. Accessed 2023 Jun 2.
36.Dunger D, Darendeliler F, Kandemir N, Harris M, Rabbani A, Kappelgaard AM. What is the evidence for beneficial effects of growth hormone treatment beyond height in short children born small for gestational age? A review of published literature. J Pediatr Endocrinol Metab. 2020;33(1):53-70. PubMed
37.Brod M, Højbjerre L, Alolga SL, Beck JF, Wilkinson L, Rasmussen MH. Understanding Treatment Burden for Children Treated for Growth Hormone Deficiency. Patient. 2017;10(5):653-666. PubMed
38.Dumas H, Panayiotopoulos P, Parker D, Pongpairochana V. Understanding and meeting the needs of those using growth hormone injection devices. BMC Endocrine Disorders. 2006;6(1):5. PubMed
39.PrNorditropin NordiFlex® (Somatropin injection): Pre-filled disposable pen 5 mg/1.5 mL, 10 mg/1.5 mL and 15 mg/1.5 mL. Solution for subcutaneous administration. Growth Hormone [Product Monograph]. Mississauga (ON) Novo Nordisk Canada Inc.; 2020 Mar 23.
40.PrNUTROPIN AQ® NuSpin® somatropin injection solution; NuSpin® injection device prefilled with cartridge: NUTROPIN AQ® NuSpin® 5 (5 mg/2 mL) NUTROPIN AQ® NuSpin® 10 (10 mg/2 mL) NUTROPIN AQ® NuSpin® 20 (20 mg/2 mL). Growth Hormone [Product Monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2018 Jun 5.
41.PrHUMATROPE® (somatropin for injection): Biosynthetic Human Growth Hormone of Recombinant DNA Origin: 6, 12, 24mg cartridges Sterile Lyophilized Powder and Diluent. Lilly Standard. Growth Stimulant [Product Monograph]. Toronto (ON): ELI LILLY CANADA INC.; 2021 Sep 16.
42.PrGENOTROPIN® GoQuick TM Somatropin [rDNA origin] for injection Lyophilized Powder for reconstitution 5 mg, 5.3 mg, 12 mg pre-filled pen, GoQuick and PrGENOTROPIN®MiniQuickTM Somatropin [rDNA origin] for injection Lyophilized Powder for reconstitution 0.2 mg, 0.4 mg, 0.6 mg, 0.8 mg, 1.0 mg, 1.2 mg, 1.4 mg, 1.6 mg, 1.8 mg, and 2.0 mg prefilled syringe, MiniQuick. Human Growth Hormone [Product Monograph]. Kirkland (QC): Pfizer Canada ULC; 2020 Jul 28.
43.Pr OMNITROPE® somatropin rDNA origin for injection Lyophilized Powder for solution: 5.8 mg/vial Solution for Injection: 5 mg/1.5 mL, 10 mg/1.5 mL, 15 mg/1.5 mL. Pharmaceutical Standard: Ph.Eur./USP. Human Growth Hormone [Product Monograph]. Boucherville (QC): Sandoz Canada Inc.; 2016 Dec 12.
44.PrSaizen® Somatropin for Injection Lyophilized Powder for reconstitution 3.33 mg/vial, 5 mg/vial PrSaizen® click.easy Somatropin for Injection Lyophilized Powder for reconstitution 8.8 mg (5.83 mg/mL) PrSaizen® Somatropin Solution for Injection in a Cartridge 6 mg (5.83 mg/mL), 12 mg (8 mg/mL), 20 mg (8 mg/mL). Pharmaceutical Standard: Professed Therapeutic Classification: Human Growth Hormone [Producat Monograph]. Mississauga (ON): EMD Serono; 2016 Dec 9.
45.PrNGENLA® Somatrogoninjection 24 mg/1.2 mL (20 mg/mL), pre-filled penfor subcutaneous use 60 mg/1.2 mL (50 mg/mL), pre-filled penfor subcutaneous use recombinant DNA technology Human Growth Hormoneanalogue [Product Monograph]. Kirkland (QC): Pfizer Canada ULC; 2021 Oct 26.
46.Miller BS, Blair JC, Rasmussen MH, et al. Weekly Somapacitan is Effective and Well Tolerated in Children With GH Deficiency: The Randomized Phase 3 REAL4 Trial. J Clin Endocrinol Metab. 2022;107(12):3378-3388. PubMed
47.Brook CG, Hindmarsh PC, Healy MJ. A better way to detect growth failure. Br Med J (Clin Res Ed). 1986;293(6556):1186. PubMed
48.Voss LD, Wilkin TJ, Bailey BJ, Betts PR. The reliability of height and height velocity in the assessment of growth (the Wessex Growth Study). Arch Dis Child. 1991;66(7):833-837. PubMed
49.Brod M, Højby Rasmussen M, Vad K, et al. Psychometric Validation of the Growth Hormone Deficiency-Child Impact Measure (GHD-CIM). Pharmacoecon Open. 2021;5(3):505-518. PubMed
50.Brod M, Rasmussen MH, Alolga S, et al. Psychometric Validation of the Growth Hormone Deficiency-Child Treatment Burden Measure (GHD-CTB) and the Growth Hormone Deficiency-Parent Treatment Burden Measure (GHD-PTB). Pharmacoecon Open. 2023;7(1):121-138. PubMed
51.Protocol: NN8640-4263. REAL 4 Protocol: A trial comparing the effect and safety of once weekly dosing of somapacitan with daily Norditropin® in children with growth hormone deficiency [internal sponsor's report]. Bagsvaerd, Denmark: Novo Nordisk. 2023 Feb 8.
52.Ranke MB, Traunecker R, Martin DD, et al. IGF-I and IGF binding protein-3 levels during initial GH dosage step-up are indicators of GH sensitivity in GH-deficient children and short children born small for gestational age. Horm Res. 2005;64(2):68-76. PubMed
53.Comparative efficacy and safety of somapacitan and somatrogon in paediatric growth hormone deficiency [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: somapacitan once weekly subcutaneous injection. Bagsvaerd, Denmark: Novo Nordisk. 2022 Oct 28.
54.Systematic literature review to assess the effectiveness and safety of long-acting growth hormones in pediatric growth hormone deficiency (GHD) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: somapacitan once weekly subcutaneous injection. Bagsvaerd, Denmark: Novo Nordisk. 2022 Jul 11.
55.Philippo D. R package multinma: Bayesian Network Meta-Analysis of Individual and Aggregate Data. https://cran.r-project.org/web/packages/multinma/multinma.pdf. Accessed 2023 Jun 2.
56.Dias S W, N.J., Sutton, A.J. & Ades, A.E. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials. London (UK): National Institute for Health and Care Excellence. 2011. www.nicedsu.org.uk. Accessed 2023 Jun 2.
57.Prader A, Largo RH, Molinari L, Issler C. Physical growth of Swiss children from birth to 20 years of age. First Zurich longitudinal study of growth and development. Helv Paediatr Acta Suppl. 1989;52:1-125. PubMed
58.Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, et al. CDC growth charts: United States. Adv Data. 2000(314):1-27. PubMed
59.Zelinska N, Iotova V, Skorodok J, et al. Long-Acting C-Terminal Peptide-Modified hGH (MOD-4023): Results of a Safety and Dose-Finding Study in GHD Children. J Clin Endocrinol Metab. 2017;102(5):1578-1587. PubMed
60.Deal CL, Steelman J, Vlachopapadopoulou E, et al. Efficacy and Safety of Weekly Somatrogon vs Daily Somatropin in Children With Growth Hormone Deficiency: A Phase 3 Study. J Clin Endocrinol Metab. 2022;107(7):e2717-e2728. PubMed
61.Horikawa R, Tanaka T, Hasegawa Y, et al. Efficacy and Safety of Once-Weekly Somatrogon Compared with Once-Daily Somatropin (Genotropin®) in Japanese Children with Pediatric Growth Hormone Deficiency: Results from a Randomized Phase 3 Study. Horm Res Paediatr. 2022;95(3):275-285. PubMed
62.Final appraisal determination–Human growth hormone (somatropin) for the treatment of growth failure in children (review of NICE technology appraisal guidance 42). London (UK): National Institute for Health and Care Excellence. 2010. https://www.nice.org.uk/guidance/ta188/resources/human-growth-hormone-somatropin-for-the-treatment-of-growth-failure-in-children-pdf-82598502860485. Accessed 2023 Jun 2.
63.Corrigendum to: “Once-Weekly Somapacitan vs Daily GH in Children With GH Deficiency: Results From a Randomized Phase 2 Trial”. J Clin Endocrinol Metab. 2020;105(12):e4982. PubMed
64.Sävendahl L, Battelino T, Brod M, et al. Once-Weekly Somapacitan vs Daily GH in Children With GH Deficiency: Results From a Randomized Phase 2 Trial. J Clin Endocrinol Metab. 2020;105(4):e1847-1861. PubMed
65.Sävendahl L, Battelino T, Højby Rasmussen M, Brod M, Saenger P, Horikawa R. Effective GH Replacement With Once-weekly Somapacitan vs Daily GH in Children with GHD: 3-year Results From REAL 3. J Clin Endocrinol Metab. 2022;107(5):1357-1367. PubMed
66.Sävendahl L, Battelino T, Højby Rasmussen M, et al. Weekly Somapacitan in GH Deficiency: 4-Year Efficacy, Safety, and Treatment/Disease Burden Results From REAL 3. The Journal of Clinical Endocrinology & Metabolism. 2023. PubMed
67.Clinical Study Report: NN8640-4172. REAL 3 analysis of main trial period (26 weeks): A Randomised, Multinational, Active-controlled, (Open-labelled), Dose Finding, (Double-blinded), Parallel Group Trial Investigating Efficacy and Safety of Once-weekly Somapacitan (NNC0195-0092) Treatment Compared to Daily Growth Hormone Treatment (Norditropin® Flexpro®) in Growth Hormone Treatment Naïve Pre-pubertal Children with Growth Hormone Deficiency [internal sponsor's report]. Bagsvaerd, Denmark: Novo Nordisk. 2018 Sep 13.
68.Clinical Study Report: NN8640-4172. REAL 3 extension and safety extension trial periods (up to 156 weeks): A Randomised, Multinational, Active-controlled, (Open-labelled), Dose Finding, (Double-blinded), Parallel Group Trial Investigating Efficacy and Safety of Once-weekly Somapacitan (NNC0195-0092) Treatment Compared to Daily Growth Hormone Treatment (Norditropin® Flexpro®) in Growth Hormone Treatment Naïve Pre-pubertal Children with Growth Hormone Deficiency [internal sponsor's report]. Bagsvaerd, Denmark: Novo Nordisk. 2021 May 12.
69.Clinical Study Report: NN8640-4172. REAL 3 long-term safety extension period (up to 208 weeks): A Randomised, Multinational, Active-controlled, (Openlabelled), Dose Finding, (Double-blinded), Parallel Group Trial Investigating Efficacy and Safety of Once-weekly Somapacitan (NNC0195-0092) Treatment Compared to Daily Growth Hormone Treatment (Norditropin® Flexpro®) in Growth Hormone Treatment Naïve Pre-pubertal Children with Growth Hormone Deficiency [internal sponsor's report]. Bagsvaerd, Denmark: Novo Nordisk. 2022 Feb 2.
70.Corrigendum to: “Reference Intervals for Insulin-Like Growth Factor-1 (IGF-I) From Birth to Senescence: Results From a Multicenter Study Using a New Automated Chemiluminescence IGF-I Immunoassay Conforming to Recent International Recommendations”. J Clin Endocrinol Metab. 2020;105(12). PubMed
71.Bidlingmaier M, Friedrich N, Emeny RT, et al. Reference intervals for insulin-like growth factor-1 (igf-i) from birth to senescence: results from a multicenter study using a new automated chemiluminescence IGF-I immunoassay conforming to recent international recommendations. J Clin Endocrinol Metab. 2014;99(5):1712-1721. PubMed
72.Johannsson G, Bidlingmaier M, Biller BMK, et al. Growth Hormone Research Society perspective on biomarkers of GH action in children and adults. Endocr Connect. 2018;7(3):R126-r134. PubMed
73.Bang P, Ahmed SF, Argente J, et al. Identification and management of poor response to growth-promoting therapy in children with short stature. Clin Endocrinol (Oxf). 2012;77(2):169-181. PubMed
74.Collett-Solberg PF, Ambler G, Backeljauw PF, et al. Diagnosis, Genetics, and Therapy of Short Stature in Children: A Growth Hormone Research Society International Perspective. Horm Res Paediatr. 2019;92(1):1-14. PubMed
Appendix 1: Detailed Patient Inclusion and Exclusion Criteria
Note this appendix has not been copy-edited.
Detailed Patient Inclusion and Exclusion Criteria for the REAL 4 Study
Inclusion Criteria
Patients are eligible to be included in the trial only if all of the following criteria apply:
Informed consent of the patient’s parent or LAR and child assent, as age-appropriate consent must be obtained before any trial-related activities
The parent or LAR of the child must sign and date the Informed Consent Form (according to local requirements)
The child must sign and date the Child Assent Form or provide oral assent (if required according to local requirements)
Prepubertal children:
Boys:
Age ≥ 2 years and 26 weeks and < 11.0 years at screening
Testis volume < 4 mL
Girls:
Age ≥ 2 years and 26 weeks and < 10.0 years at screening
Tanner stage I for breast development (no palpable glandular breast tissue)
Confirmed diagnosis of GH deficiency determined by 2 different GH stimulation tests performed within 12 months before randomization, defined as a peak GH level of ≤ 10.0 ng/mL using the WHO International Somatropin 98/574 standard
If only 1 GH stimulation test is available before screening, then confirmation of GH deficiency by second and different GH stimulation test must be done
For children with at least 2 additional pituitary hormone deficiencies (other than GH deficiency) only 1 GH stimulation test is needed
For Japan: Confirmed diagnosis of GH deficiency within 12 months before screening as determined by 1 GH stimulation test for patients with intracranial organic disease or symptomatic hypoglycemia and 2 different GH stimulation tests for other patients defined as peak GH level of ≤ 6 ng/mL by assay using recombinant GH standard.
If only 1 GH stimulation test is available before screening, then confirmation of GH deficiency by second and different GH stimulation test must be done.
For children with at least 2 additional pituitary hormone deficiencies only 1 GH stimulation test is needed.
Impaired height defined as at least 2.0 SDs below the mean height for chronological age and gender at screening according to the standards of Centers for Disease Control and Prevention
Impaired HV, defined as AHV below the 25th percentile for chronological age and gender according to the standards of Prader calculated over a time span of a minimum 6 months and a maximum of 18 months before screening
No prior exposure to GH therapy or IGF-1 treatment
Bone age less than chronological age at screening
BMI > fifth and < 95th percentile according to Centers for Disease Control and Prevention, Body Mass Index-for-age growth charts
IGF-1 < −1.0 SDS at screening, compared with age- and gender-normalized range measured by a central laboratory
Hormone replacement therapies for any other hormone deficiency should be adequate and stable for at least 90 days before randomization
No intracranial tumour confirmed by MRI or computer tomography scan. An image or scan taken within 9 months before screening can be used as screening data if the medical evaluation and conclusion is available
Exclusion Criteria
Patients are excluded from the trial if any of the following criteria apply:
Known or suspected hypersensitivity to trial product(s) or related products
Previous participation in this trial. Participation is defined as randomization
Receipt of any investigational medicinal product within 3 months before screening or participation in another clinical trial at time of randomization
Any known or suspected clinically significant abnormality likely to affect growth or the ability to evaluate growth with standing height measurements:
Turner Syndrome (including mosaicisms)
Chromosomal aneuploidy and significant gene mutations causing medical “syndromes” with short stature, including but not limited to Laron syndrome, Noonan syndrome, Prader-Willi syndrome, abnormal SHOX-1 gene analysis, or absence of GH receptors
Significant spinal abnormalities including but not limited to scoliosis, kyphosis and spina bifida variants
Congenital abnormalities (causing skeletal abnormalities), including but not limited to Russell-Silver Syndrome or skeletal dysplasias
Family history of skeletal dysplasia
Children born SGA (birth weight and/or birth length < −2 SDS for gestational age according to national standards)
Children diagnosed with diabetes mellitus or screening values from central laboratory of
fasting plasma glucose ≥ 126 mg/dL (7.0 mmol/L) or
hemoglobin A1C ≥ 6.5%
Current inflammatory diseases requiring systemic corticosteroid treatment for longer than 2 consecutive weeks within the last 3 months before screening
Children requiring inhaled glucocorticoid therapy at a dose of greater than 400 mcg/day of inhaled budesonide or equivalents for longer than 4 consecutive weeks within the last 12 months before screening
Concomitant administration of other treatments that may have an effect on growth, e.g., but not limited to methylphenidate for treatment of attention-deficit/hyperactivity disorder
Diagnosis of attention-deficit/hyperactivity disorder
Prior history or presence of malignancy including intracranial tumours
Prior history or known presence of active hepatitis B or hepatitis C (exceptions to this exclusion criterion is the presence of antibodies due to vaccination against hepatitis B)
Any disorder which, in the opinion of the investigator, might jeopardize the patient’s safety or compliance with the protocol
The patient or the parent or LAR is likely to be noncompliant in respect to trial conduct, as judged by the investigator.
Detailed Patient Inclusion and Exclusion Criteria for the REAL 3 Study
Inclusion Criteria and Exclusion Criteria for Cohort 1
Inclusion Criteria (Cohort 1)
For an eligible patient, all inclusion criteria must be answered “yes.”
Informed consent of patient’s parent or LAR and child assent, as age-appropriate obtained before any trial-related activities. Trial-related activities are any procedures that are carried out as part of the trial, including activities to determine suitability for the trial.
The parent or LAR of the child must sign and date the Informed Consent Form (according to local requirements).
The child must sign and date the Child Assent Form or provide oral assent (if required according to local requirements).
Prepubertal children
Boys: Tanner stage I for pubic hair and testis volume < 4 mL, age ≥ 2 years and 26 weeks and ≤ 10.0 years.
Girls: Tanner stage I for breast development (no palpable glandular breast tissue) and pubic hair, age ≥ 2 years and 26 weeks and ≤ 9.0 years.
Confirmed diagnosis of GHD within 12 months before screening as determined by 2 different GH stimulation tests, defined as a peak GH level of ≤ 7.0 ng/mL. For children with 3 or more pituitary hormone deficiencies only 1 GH stimulation test is needed.
FOR JAPAN ONLY: Confirmed diagnosis of GHD within 12 months before screening as determined by 1 GH stimulation tests for patients with intracranial organic disease or symptomatic hypoglycemia and 2 different GH stimulation test for other patients, defined as a peak GH level of ≤ 6 ng/mL by assay using recombinant GH standard. END OF TEXT ONLY APPLICABLE FOR JAPAN.
No prior exposure to GH therapy and/or IGF-1 treatment.
Height of at least 2.0 SDs below the mean height for chronological age and gender according to the standards of Centers for Disease Control and Prevention for patients aged 2 to 20 years: Girls/boys stature-for-age and weight-for-age percentiles at screening.
Annualized HV (HV) at screening below the 25th percentile for chronological age and gender or below −0.7 SD score for chronological age and sex, according to the standards of Prader calculated over a time span of a minimum of 6 months and a maximum of 18 months before screening.
BMI percentile > fifth and < 95th percentile according to Centers for Disease Control and Prevention BMI-for-age growth charts.
IGF-1 SDS < −1.0 at screening, compared with age- and sex-normalized range according to central laboratory measurements.
Bone age (X-ray of left hand and wrist, central reviewed according to the Greulich and Pyle atlas) less than chronological age at screening. An X-ray taken within 13 weeks before screening can be used as screening data if the image is available and meets requirements for central reading.
Exclusion Criteria (Cohort 1)
For an eligible patient, all exclusion criteria must be answered “no.”
Previous participation in this trial. Participation is defined as randomization.
Receipt of any investigational medicinal product within 3 months before screening.
FOR BRAZIL ONLY: Participation in other trials within 1 year (defined as 365 days) before screening visit (visit 1) unless there is a direct benefit to the participating patient; decision to include made at the investigator’s discretion. END OF TEXT ONLY APPLICABLE FOR BRAZIL
Any clinically significant abnormality likely to affect growth or the ability to evaluate growth with standing measurements:
Chromosomal aneuploidy and significant gene mutations causing medical “syndromes” with short stature, including but not limited to Turner syndrome, Laron syndrome, Noonan syndrome, or absence of GH receptors.
Congenital abnormalities (causing skeletal abnormalities), including but not limited to Russell-Silver Syndrome, skeletal dysplasia.
Significant spinal abnormalities including but not limited to scoliosis, kyphosis, and spina bifida variants.
Poorly controlled or uncontrolled pituitary insufficiencies or primary hormonal deficiencies of other axes (e.g., thyroid-stimulating hormone/T4, adrenocorticotropic hormone/cortisol, and vasopressin deficiency) in children who have been on stable replacement therapy for less than 6 months for thyroid replacement therapy, and less than 3 months for other hormonal deficiencies before screening.
Children born SGA (SGA = birth weight and/or birth length < −2 SD for gestational age).
Children diagnosed with diabetes mellitus or fasting blood glucose ≥ 126 mg/dL (7.0 mmol/L), or hemoglobin A1C ≥ 6.5% at screening, determined by central laboratory.
Current inflammatory diseases (e.g., but not limited to arthritis, inflammatory bowel diseases) requiring systemic corticosteroid treatment for longer than 2 consecutive weeks within the last 3 months before screening.
Children requiring inhaled glucocorticoid therapy (e.g., asthma) at a dose of greater than 400 mcg/day of inhaled budesonide or equivalents for longer than 4 consecutive weeks within the last 12 months before screening.
Concomitant administration of other treatments that may have an effect on growth, e.g., but not limited to methylphenidate for treatment of attention-deficit/hyperactivity disorder.
Prior history or presence of malignancy and/or intracranial tumour.
Prior history or presence of active hepatitis B and/or hepatitis C (exceptions to this exclusion criterion is the presence of antibodies due to vaccination against hepatitis B and hepatitis C).
Clinically significant abnormal electrocardiogram at screening, as evaluated by investigator.
Any clinically significant abnormal laboratory screening tests, as judged by the investigator.
Any disorder which, in the opinion of the investigator, might jeopardize the patient’s safety or compliance with the protocol.
The patient and/or the patient’s parent or LAR are likely to be noncompliant in respect to trial conduct, as judged by the investigator.
Inclusion Criteria and Exclusion Criteria for Cohort 2
Inclusion Criteria (Cohort 2)
For an eligible patient, all inclusion criteria must be answered “yes,” if applicable.
Informed consent of parent or patient’s LAR. Trial-related activities are any procedures that are carried out as part of the trial, including activities to determine suitability for the trial. The parent or LAR of the child must sign and date the Informed Consent Form (according to local requirements).
Age < 2 years and 26 weeks and a minimum body weight of 5 kg at screening.
Confirmed diagnosis of GHD, the GHD diagnosis must be confirmed by investigator according to local practice.
For GH treatment-naive patients, no prior exposure to GH therapy and/or IGF-1 treatment.
For GH treatment-naive patients, IGF-1 SDS < −1.0 at screening, compared with age- and sex-normalized range, according to central laboratory measurements.
Exclusion Criteria (Cohort 2)
For an eligible patient, all exclusion criteria must be answered “no.”
Previous participation in this trial. Participation is defined as randomization.
Receipt of any investigational medicinal product within 3 months before screening.
Any clinically significant abnormality likely to affect growth:
Chromosomal aneuploidy and significant gene mutations causing medical “syndromes” with short stature, including but not limited to Turner syndrome, Laron syndrome, Noonan syndrome, or absence of GH receptors.
Congenital abnormalities (causing skeletal abnormalities), including but not limited to Russell-Silver Syndrome, skeletal dysplasias.
Significant spinal abnormalities including but not limited to scoliosis, kyphosis, and spina bifida variants.
Poorly controlled or uncontrolled pituitary insufficiencies or primary hormonal deficiencies of other axes (e.g., thyroid-stimulating hormone/T4, adrenocorticotropic hormone/cortisol, and vasopressin deficiency) as judged by to investigator.
Children born SGA (SGA = birth weight and/or birth length < −2 SD for gestational age).
Children suspected of or diagnosed with diabetes mellitus.
Current inflammatory diseases (e.g., but not limited to arthritis, inflammatory bowel diseases) requiring systemic corticosteroid treatment as judged by investigator.
Children requiring inhaled glucocorticoid therapy (e.g., asthma).
Concomitant administration of other treatments that may have an effect on growth, e.g., but not limited to methylphenidate for treatment of attention-deficit/hyperactivity disorder.
Prior history or presence of malignancy and/or intracranial tumour.
Prior history or presence of active hepatitis B and/or hepatitis C (exceptions to this exclusion criterion is the presence of antibodies due to vaccination against hepatitis B and hepatitis C).
Any clinically significant abnormal laboratory screening tests, as judged by the investigator.
Any disorder which, in the opinion of the investigator, might jeopardize the patient’s safety or compliance with the protocol.
The patient and/or the patient’s parent or LAR are likely to be noncompliant in respect to trial conduct, as judged by the investigator.
Inclusion Criteria and Exclusion Criteria for Cohort 3
Inclusion Criteria (Cohort 3)
For an eligible patient, all inclusion criteria must be answered “yes,” if applicable.
Informed consent of parent or patient’s LAR and child assent, as age-appropriate obtained before any trial-related activities. Trial-related activities are any procedures that are carried out as part of the trial, including activities to determine suitability for the trial.
The parent or LAR of the child must sign and date the Informed Consent Form (according to local requirements).
The child must sign and date the Child Assent Form or provide oral assent (if required according to local requirements).
Age:
Girls: > 9 years and ≤ 17.0 years at screening.
Boys: > 10 years and ≤ 17.0 years at screening.
Confirmed diagnosis of GHD:
for GH treatment-naive patients, confirmed diagnosis within 12 months before screening as determined by 2 different GH stimulation tests, defined as a peak GH level of ≤ 7.0 ng/mL. For children with 3 or more pituitary hormone deficiencies only 1 GH stimulation test is needed. FOR JAPAN ONLY: Confirmed diagnosis of GHD within 12 months before screening as determined by 1 GH stimulation tests for patients with intracranial organic disease or symptomatic hypoglycemia and 2 different GH stimulation test for other patients, defined as a peak GH level of ≤ 6 ng/mL by assay using recombinant GH standard. END OF TEXT ONLY APPLICABLE FOR JAPAN.
for non-naive, confirmed GHD diagnosis by investigator according to local practice.
For GH treatment-naive patients, no prior exposure to GH therapy and/or IGF-1 treatment.
BMI percentile > fifth and < 95th percentile according to Centers for Disease Control and Prevention BMI-for-age growth charts.
For GH treatment-naive patients, IGF-1 SDS < −1.0 at screening, compared with age- and sex-normalized range according to central laboratory measurements.
Bone age (X-ray of left hand and wrist, central reviewed according to Greulich and Pyle atlas) less than chronological age at screening. An X-ray taken within 13 weeks before screening can be used as screening data if the image is available and meets requirements for central reading.
Open epiphyses; defined as bone age < 14 years for females and bone age < 16 years for males.
Exclusion Criteria (Cohort 3)
For an eligible patient, all exclusion criteria must be answered “no.”
Previous participation in this trial. Participation is defined as randomization.
Receipt of any investigational medicinal product within 3 months before screening.
Any clinically significant abnormality likely to affect growth or the ability to evaluate growth with standing measurements:
Chromosomal aneuploidy and significant gene mutations causing medical “syndromes” with short stature, including but not limited to Turner syndrome, Laron syndrome, Noonan syndrome, or absence of GH receptors.
Congenital abnormalities (causing skeletal abnormalities), including but not limited to Russell-Silver Syndrome, skeletal dysplasias.
Significant spinal abnormalities including but not limited to scoliosis, kyphosis, and spina bifida variants.
Poorly controlled or uncontrolled pituitary insufficiencies or primary hormonal deficiencies of other axes (e.g., thyroid-stimulating hormone/T4, adrenocorticotropic hormone/cortisol, and vasopressin deficiency) as judged by investigator.
Children born SGA (SGA = birth weight and/or birth length < −2 SD for gestational age).
Children diagnosed with diabetes mellitus or fasting blood glucose ≥ 126 mg/dL (7.0 mmol/L), or hemoglobin A1C ≥ 6.5% at screening, determined by central laboratory.
Current inflammatory diseases (e.g., but not limited to arthritis, inflammatory bowel diseases) requiring systemic corticosteroid as judged by the investigator.
Children requiring inhaled glucocorticoid therapy (e.g., asthma) at a dose of greater than 400 mcg/day of inhaled budesonide or equivalents for longer than 4 consecutive weeks within the last 12 months before screening.
Concomitant administration of other treatments that may have an effect on growth, e.g., but not limited to methylphenidate for treatment of attention-deficit/hyperactivity disorder.
Prior history or presence of malignancy and/or intracranial tumour.
Prior history or presence of active hepatitis B and/or hepatitis C (exceptions to this exclusion criterion is the presence of antibodies due to vaccination against hepatitis B and hepatitis C).
Clinically significant abnormal electrocardiogram at screening, as evaluated by investigator.
Any clinically significant abnormal laboratory screening tests, as judged by the investigator.
Any disorder which, in the opinion of the investigator, might jeopardize the patient’s safety or compliance with the protocol.
The patient and/or the patient’s parent or LAR are likely to be noncompliant in respect to trial conduct, as judged by the investigator.
A positive pregnancy test at screening (only applicable for female patients who have had menarche).
Appendix 2: Detailed Outcome Data
Note this appendix has not been copy-edited.
Refer to Table 53 for the supportive outcome data from the REAL 4 trial.
Auxologic Response
Height
The change in height (cm) from baseline to week 52 between somapacitan and Norditropin (somatropin) were nearly identical (treatment difference ||||| ||| ||| ||| ||||| || || |||| ||). The observed mean (SD) height SDS values in both treatment groups increased consistently from week 0 to week 52. The mean increases (SD) from baseline to week 52 in height of 11.3 cm (2.6) and 12.0 cm (3.0) for somapacitan and Norditropin (somatropin), respectively.
Bone Age
The bone age versus chronological age ratio was below 1 for most patients at week 52. Analysis showed that the change in bone age versus chronological age ratio between somapacitan and Norditropin (somatropin) was nearly identical (ETD −0.02; 95% CI, −0.06 to 0.01) from baseline to week 52.
Table 53: Summary of Supportive Data From REAL 4 (FAS)
Variable | Week 26 | Week 52 | ||
|---|---|---|---|---|
Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | Somapacitan (N = 132) | Norditropin (somatropin) (N = 68) | |
Auxologic response | ||||
Height (cm) | ||||
Baseline (cm), mean (SD) | 102.3 (12.5) | 100.2 (15.0) | 102.3 (12.5) | 100.2 (15.0) |
Change from baseline (cm), mean (SD) | 6.2 (1.8) | 6.6 (1.9) | 11.3 (2.6) | 12.0 (3.0) |
End point (cm), observed mean (SD) | 108.7 (11.9) | 107.2 (13.7) | 113.6 (11.6) | 112.2 (13.4) |
Estimated treatment difference (95% CI) | ||||| ||||||| ||||| | ||||| ||||||| ||||| | ||
P value | NR | NR | ||
Body weight (kg) | ||||
Baseline (cm), mean (SD) | 16.69 (4.60) | 16.01 (4.95) | 16.69 (4.60) | 16.01 (4.95) |
Change from baseline (cm), mean (SD) | 2.98 (1.06) | 1.87 (0.88) | 5.30 (1.66) | 3.92 (1.71) |
End point (cm), observed mean (SD) | 19.79 (5.29) | 18.01 (5.16) | 21.99 (5.65) | 19.93 (5.72) |
Estimated treatment difference (95% CI) | |||| |||||| ||||| | |||| |||||| ||||| | ||
P value | NR | NR | ||
Bone age versus chronological age ratio | ||||
Baseline, mean (SD) | NR | NR | 0.06 | 0.08 |
End point, mean (SD) | NR | NR | 0.06 | 0.08 |
Estimated treatment difference (95% CI)a | NR | −0.02 (−0.06 to 0.01) | ||
P value | NR | 0.17 | ||
Pharmacodynamic end points | ||||
IGF-1 SDS | ||||
Baseline, mean (SD) | −2.03 (0.97) | −2.33 (1.03) | −2.03 (0.97) | −2.33 (1.03) |
End point, mean (SD) | 0.80 (1.26) | −0.28 (1.14) | 0.28 (1.28) | 0.10 (1.09) |
Observed mean (SD) change from baseline | 2.81 (1.27) | 2.03 (1.01) | 2.32 (1.27) | 2.41 (1.09) |
Estimated mean change from baselinea | NR | NR | 2.36 | 2.33 |
Estimated treatment difference (95% CI)a | NR | 0.03 (−0.30, 0.36) | ||
P value | NR | 0.8544 | ||
IGF-1 (ng/mL) | ||||
Baseline, geometric mean [coefficient of variation (%)]) | 47.5 (82.8) | 37.31 (115.0) | 47.5 (82.8) | 37.31 (115.0) |
Change from baseline (ng/mL), mean (SD) | 149.86 (80.73) | 92.85 (54.80) | 127.66 (77.93) | 122.18 (63.61) |
End point (ng/mL), geometric mean [coefficient of variation (%)] | 189.04 (50.3) | 126.07 (64.9) | 166.62 (52.1) | 157.04 (52.0) |
Estimated treatment difference (95% CI) | ||||| ||||||| |||||| | |||| ||||||| |||||| | ||
P value | NR | NR | ||
IGFBP-3 SDS | ||||
Baseline, mean (SD) | −1.89 (1.12) | −2.18 (1.27) | −1.89 (1.12) | −2.18 (1.27) |
End point, mean (SD) | −0.39 (1.02) | −0.81 (1.12) | −0.33 (0.96) | −0.46 (0.94) |
Observed mean (SD) change from baseline | 1.48 (0.99) | 1.34 (0.99) | 1.58 (0.92) | 1.68 (1.02) |
Estimated mean change from baselinea | NR | NR | 1.61 | 1.61 |
Estimated treatment difference (95% CI)a | NR | 0.01 (−0.22, 0.23) | ||
P value | NR | 0.9616 | ||
ANCOVA = analysis of covariance; CI = confidence interval; GH = growth hormone; IGF-1 = insulin-like growth factor 1; IGFBP-3 = insulin-like growth factor binding protein 3; NA = not applicable; NR = not reported; SD = standard deviation; SDS = standard deviation score.
Note: In-trial period results are reported.
aANCOVA with treatment, gender, age group, region, GH peak group, and gender by age group by region interaction term as factors, and baseline assessment as a covariate.
bSuperiority was not confirmed. Noninferiority was confirmed; the noninferiority margin is −1.8 cm/year.
Source: REAL 4 trial Clinical Study Report (week 52).20
Pharmacodynamic End Points
The change in IGF-1 SDS, IGF-1 (ng/mL) and IGFBP-3 SDS levels from baseline to week 52 between somapacitan and Norditropin (somatropin) were nearly identical with ETD (95% CI) of 0.03 (−0.30 to 0.36), |||| ||||||| ||||||, and 0.01 (−0.22 to 0.23), respectively. Mean IGF-1 SDS levels and IGFBP-3 SDS levels increased from baseline levels and were within normal range (−2 to 2), in both treatment groups at week 52.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
GHD
growth hormone deficiency
HRQoL
health-related quality of life
HSDS
height standard deviation score
ICER
incremental cost-effectiveness ratio
NMA
network meta-analysis
NIHB
Non-Insured Health Benefits
QALY
quality-adjusted life-year
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Somapacitan (Sogroya), prefilled pen for subcutaneous injection |
Submitted price | Somapacitan prefilled pen:
|
Indication | For the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone (Growth Hormone Deficiency (GHD)) |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | July 26, 2023 |
Reimbursement request | As per indication |
Sponsor | Novo Nordisk Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov cohort model |
Target population | Pediatric patients who have growth failure due to inadequate secretion of endogenous growth hormone. |
Treatment | Somapacitan (once-weekly injection) |
Comparators | Somatrogon (once-weekly injection) Somatropin (once-daily injection), consisting of all branded somatropin products weighted by market share |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | 11.6 years |
Key data sources |
|
Submitted results | Somapacitan was dominant compared with somatrogon and somatropin (i.e., somapacitan was associated with lower total costs and more QALYs). |
Key limitations |
|
CADTH reanalysis results |
|
GH = growth hormone; GHD = growth hormone deficiency; HSDS = height standard deviation score; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Conclusions
Based on the sponsor-submitted clinical evidence from the phase III REAL 4 trial for pediatric patients with growth hormone deficiency (GHD), somapacitan results in little to no difference in the change from baseline in height standard deviation score (HSDS), notable harms, injection-site reactions and injection-site pain, and treatment discontinuation when compared with Norditropin-branded somatropin. Using the Grading of Recommendations Assessment, Development, and Evaluation framework (GRADE), CADTH categorized this evidence as having moderate to high certainty. Data from the REAL 4 extension trial and the phase II REAL 3 trial were considered descriptive in nature; therefore, there is greater uncertainty in the findings from these studies. The sponsor-submitted network meta-analysis (NMA) suggested there were no statistically significant differences identified between somapacitan and somatrogon for HSDS at week 26 and week 52; however, several sources of uncertainty identified with the NMA limit the interpretation of these results, such as the sparse network, small sample sizes, between-trial heterogeneity, and wide credible intervals across all assessed outcomes. No conclusions can be drawn regarding the comparative safety of somapacitan, somatrogon, and somatropin.
The CADTH base-case analysis resulted in an incremental cost-effectiveness ratio (ICER) for somapacitan of $275,250 per quality-adjusted life-year (QALY) gained when compared against somatropin; somatrogon was dominated (associated with greater total costs and no additional QALYs). The cost-effectiveness of somapacitan and somatrogon varied by model run due to the input distributions in a probabilistic model, while the cost-effectiveness of somapacitan relative to somatropin was impacted by the weighted cost of somatropin. CADTH noted that the gain in QALYs associated with somapacitan is due solely to the disutility associated with patients receiving daily somatropin treatments. Based on the assumptions underpinning the CADTH base case, a price reduction of at least 13% would be required for somapacitan to be considered cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained compared with a weighted basket of somatropins. If the cost of somatropin were considered using the lowest-cost branded somatropin only, CADTH noted that a price reduction of at least 41% would be required for somapacitan to be no more costly than the least costly somatropin. Based on these price reductions, the cost of somapacitan would range from $174.16 to $258.27 per prefilled pen (5 mg/1.5 mL).
The cost-effectiveness of somapacitan is highly sensitive to the cost of somatropin (e.g., dosing and the cost of the different brands of somatropin), the incidence of injection-site pain associated with somatrogon, and the disutility associated with receiving daily injections – which was assessed in scenario analysis. If disutility associated with more frequent injections is removed, somapacitan is dominated by somatropin. There is insufficient clinical evidence to justify a price premium for somapacitan over currently available growth hormone treatments for GHD. To ensure cost-effectiveness, somapacitan should be priced no more than the lowest cost growth hormone comparator used to treat GHD that is funded.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient group input was received from the Canadian Organization for Rare Disorders, which was based on interviews with 12 caregivers of children aged 4 to 15 years who had been diagnosed with GHD. Fifty percent of the respondents were situated in Ontario, whereas the remaining respondents were distributed across the US and Europe. Respondents discussed their experience of living with GHD and their experience with currently available treatments. Caregivers emphasized the impact of GHD on their children's height, the delayed onset of puberty, and the psychological and social impacts. Caregivers reported facing difficulties administering daily injections and expressed a desire for a treatment in either a pill form, a powder that could be dissolved in a drink, or an injection that lasted longer and was easier to administer. Four respondents with experience in long-acting growth-hormone therapy highlighted its positive impact on their quality of life, specifically in alleviating the administration challenges associated with daily injections.
Clinician input was received from Canadian Pediatric Endocrinology Nurses based on 3 studies reviewed.1-3 The input noted that the current treatment paradigm for GHD involves daily somatropin injections, which are used to increase growth, stabilize blood sugar levels, and increase bone density and muscle development. The clinician input noted that in clinical practice, improved growth velocity and normalized glucose in infants would be considered a meaningful response to treatment. They described how current daily injections have poor compliance, are often the source of anxiety, and emphasized the need for treatments that are better tolerated, can improve compliance, and have greater convenience. The clinician input noted that patients with needle anxiety, compliance issues, complex social situations, or remote living circumstances would benefit from somapacitan. Furthermore, they recommended that patients receiving somapacitan should be diagnosed, treated, monitored, and prescribed by a pediatric endocrinologist.
The drug plan input noted that the pivotal trials submitted by the sponsor only compared somapacitan with somatropin, specifically Norditropin, and did not compare somapacitan with somatrogon. Additional input noted that some provinces and territories have alternative funding pathways for growth hormone treatment, such as special drug programs at hospitals and, as such, the Non-Insured Health Benefits (NIHB) program would typically not cover pediatric patients with GHD, as patients would apply for reimbursement through provincial or territorial routes. As the clinical trial demonstrated noninferiority of somapacitan to somatropin, the drug plans noted that patients receiving daily injections would likely want to transition to weekly injections. The drug plan input noted that the sponsor for somatrogon completed confidential drug price negotiations.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s base-case analysis compared somapacitan with a mix of somatropin treatments (Genotropin, Humatrope, Norditropin, Nutropin, Omnitrope, Saizen) and somatrogon.
Health-related quality of life (HRQoL) was included in the model for patients with GHD. Caregiver HRQoL was included in a scenario analysis.
The sponsor included disutilities for patients receiving daily injections. Utility decrements were not applied to patients receiving weekly treatments.
The sponsor assumed improved compliance for weekly treatments compared with daily treatments.
In addition, CADTH addressed some of these concerns, as follows:
In the sponsor’s base-case analysis, somapacitan was assumed to only displace somatrogon. In its scenario analyses, CADTH assumed that somapacitan would displace all comparators proportionally.
CADTH was unable to address the following concerns raised from stakeholder input:
Estimate of alternative coverage rates for growth hormone treatments among the NIHB population.
Economic Review
The current review is for somapacitan (Sogroya) for the long-term treatment of pediatric patients who have GHD due to an inadequate secretion of endogenous growth hormone.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing somapacitan against somatrogon and somatropin for the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone. The modelled population was aligned with the Health Canada–indicated population and reimbursement request.4,5
Somapacitan is available in prefilled pens containing 5 mg, 10 mg, or 15 mg in 1.5 mL of solution at a submitted cost of $297.20, $594.40, and $891.60, respectively.4,5 The sponsor calculated the average annual costs for all treatments by assuming a starting patient weight of 16.46 kg5 and weight increases over time, based on a prior appraisal (Technology Appraisal 188)6 by the National Institute for Health and Care Excellence (NICE). The sponsor assumed no wastage of unused product for both single and multiuse treatments resulting from storage wastage, preparation wastage, last-dose wastage, device-setting wastage, and adherence wastage. The recommended pediatric initiation dose for somapacitan is 0.16 mg/kg administered once weekly by subcutaneous injection with the dose adjusted and individualized as necessary. Assuming patients remain on the initiation dose, the average annual per-patient cost is $15,423. Somatrogon was assumed to be administered weekly at a dose of 0.66 mg/kg, resulting in an average annual cost of $15,423. The branded somatropins included by the sponsor (i.e., Genotropin, Humatrope, Norditropin, Nutropin, Omnitrope, and Saizen) were assumed to be administered at a daily dose of 0.034 mg/kg. Somatropin was included as a single comparator that reflected a mix of somatropin products; the total drug acquisition cost of somatropin was therefore calculated as an average cost weighted by the market shares of the various somatropin products (Appendix 3, Table 10), which resulted in an average annual cost of $10,979 to $19,458.5 The branded somatropin products were assumed to have equivalent efficacy and safety to support the single comparator approach, based on the prior CADTH review of somatrogon.5,7
The economic evaluation was conducted over a time horizon of 11.6 years (i.e., as the starting age of the model cohort is 6.4 years and the sponsor assumed that pediatric patients with GHD would stop their growth hormone treatment when they reach 18 years of age) from the perspective of the Canadian publicly funded health care system. Costs and clinical outcomes (life-years and QALYs) were discounted at 1.5% per annum; a half-cycle correction was included.
Model Structure
The sponsor submitted a Markov model consisting of 4 health states: child alive and on treatment, child alive and off treatment, adult alive and off treatment, and an absorbing death state (Appendix 3, Figure 1).5 Each model cycle was 1 year in duration. Patients entered the model in the child alive and on treatment health state and received either somapacitan or a comparator growth hormone treatment. Patients could only discontinue treatment during the first year and transition to the child alive and off treatment health state. Patients who discontinued treatment were not eligible to restart growth hormone treatment. After the first year, patients remained in their existing health states until reaching 18 years of age, at which time they would all transition to the adult alive and off treatment health state. All 3 alive health states allowed for transition to the absorbing death health state.
Model Inputs
Baseline patient characteristics in the model were reflective of the REAL 4 clinical trial (i.e., proportion male = 74.5%; average age = 6.4 years; patient weight = 16.46 kg; baseline HSDS = −3.15).8 The baseline age defines when growth hormone treatment is initiated and thus contributes directly to the duration of treatment and the modelled time horizon. In each model cycle, age- and gender-specific patient weight increases were sourced from NICE Technology Appraisal 188.9
Efficacy data for somapacitan were based on the secondary efficacy end point in the REAL 4 study, the change in HSDS from baseline. This was used to derive the gain in height after 1 year post baseline. The subsequent 2-year and 3-year HSDS estimates were obtained from the REAL 4 extension study and REAL 3 study, respectively. It was assumed that the HSDS rate of change that patients experience at the end of year 3 would be sustained for the remainder of the model’s time horizon. Equal efficacy was assumed across treatments and thus equivalent HSDS scores were used for somapacitan, somatrogon, and somatropin in the sponsor’s base case.
The sponsor derived treatment compliance and 1-year discontinuation rates from the REAL 4 study for somapacitan and somatropin. Equal compliance rates were assumed between somapacitan and somatrogon. Across all treatments, the sponsor applied a 5% proportional reduction of HSDS in patients who were noncompliant with treatment, based on a study by Maggio et al.10 All treatments were assumed to have the same proportion of patients discontinuing treatment within the first year and the sponsor assumed that patients who discontinue treatment would maintain their last observed HSDS for the remainder of the model. Moreover, the sponsor assumed that all patients who remain on treatment at the end of year 1 would remain on treatment until 18 years of age.
The model incorporated adverse events (AEs), specifically, the annual incidence of injection-site pain. Incidence rates for somapacitan and somatropin were obtained from the REAL 4 study. The sponsor assumed that all somatropins have the same annual incidence rate. Incidence rates of injection-site pain for somatrogon were sourced from Deal et al.11 Furthermore, the sponsor assumed that the treatment did not impact the risk of mortality. All-cause mortality data from Statistics Canada were modelled annually and were weighted by the cohort’s proportion of males.
Health state utility values were derived from a study that examined the influence of short stature, via HSDS, on HRQoL among adults from the general adult population in the UK via the 3-Level EQ-5D (EQ-5D-3L) questionnaire.12 Utility values from this study formed the basis for a linear interpolation to derive utility scores by HSDS in the economic model. Additionally, the sponsor adjusted the health state utilities by age and sex based on a regression algorithm by Ara and Brazier.13 Disutilities associated with injection-site pain (−0.03 per year) and frequency of administration were considered in the base case (−0.009 per year). The disutility values were obtained from a time trade-off study conducted by the sponsor.14,15
The costs captured in the model included drug acquisition, monitoring, and AEs. Drug acquisition costs for somapacitan were based on the sponsor’s submitted price,5 and all other drug costs were informed by either the Ontario Exceptional Access Program or a prior CADTH reimbursement report.7,16 In scenario analyses, the sponsor explored the impact of a 10% loss due to mechanical wastage for treatments that require reconstitution; however, the term mechanical wastage was not explicitly defined. Resource utilization costs related to patient monitoring were included in the model. The sponsor assumed that routine monitoring visits and blood tests occurred at a constant rate. Additionally, children undergoing treatment were assumed to undergo a pituitary function test and, except for the first year of treatment, they were also assumed to require an annual hand X-ray. Monitoring costs were obtained from the Ontario Schedule of Benefits for Physician Services, apart from hand X-rays, which were obtained from the Ontario Schedule of Facility Costs.17,18 AE costs, specifically injection-site pain, were obtained from the Ontario Schedule of Benefits for Physician Services and were assumed to require a pediatric consultation to manage.17
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
In the sponsor’s base case, somapacitan was dominant (i.e., less costly, more effective) when compared with somatrogon and somatropin. Somapacitan was associated with an estimated cost of $122,489 and 8.70 QALYs over the modelled time horizon (Table 4), which resulted in an incremental gain of 0.13 and 0.10 QALYs and incremental cost savings of $552 and $966 compared with somatrogon and somatropin, respectively. Approximately 73% of the incremental QALYs were gained in the extrapolated portion of the model (i.e., after the first 3 years as observed in the REAL 3 trial) in comparison with both somatrogon and somatropin. At the end of the 11.6-year time horizon, 99.9% of patients remained alive for all comparators.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Somapacitan | 122,489 | 8.70 | Reference |
Somatrogon | 123,041 | 8.57 | Dominated |
Somatropin | 123,455 | 8.60 | Dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.5
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses. These analyses involved considering alternative time horizons, discount rates, assumptions related to efficacy, the inclusion of drug wastage, and the average age at which patients start treatment. The sponsor conducted a scenario analysis from a societal perspective that included caregiver disutilities for injection-site pain and caregiver disutilities for patients receiving daily injections. The sponsor’s results for all scenario analyses were aligned with the base case, in that somapacitan remained dominant in all scenarios.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Dosing for somatropin does not align with the dose commonly used in Canadian clinical practice: In the economic model, the sponsor assumed that the average administered dose for all somatropin products was 0.24 mg/kg/week. Clinical expert feedback obtained by CADTH for this review indicated that the average dose applied in the sponsor’s base case was higher than the average dose commonly used in Canadian clinical practice, which is typically 0.18 mg/kg/week, and noted that less than 10% of patients are administered a higher dose. Furthermore, the clinical expert feedback obtained by CADTH noted that once patients have reached their final adult height, typically between 16 and 18 years old, the daily dose is reduced to 0.3 mg to 0.5 mg per patient per day. The average administered dose in Canadian clinical practice, per clinical expert feedback obtained by CADTH, is supported by the results from the Canadian cohort of the multinational phase IV prospective observational Genetic and Neuroendocrinology of Short-Stature International Study (GeNeSIS) of children with various causes of short stature, including patients with GHD who were treated with growth hormone.19 Deal et al. reported that the average somatropin dose administered to children with GHD within the Canadian cohort over a 5-year period was 0.18 mg/kg/week and further indicated that mean growth hormone dosages in Canada were lower than those administered in the US or globally. As drug costs are a key driver of model results, the sponsor’s assumption of a higher dose for all somatropin products overestimated the total costs associated with somatropin, thus biasing cost-effectiveness results in favour of somapacitan and somatrogon.
To align with the dosing expected in Canadian clinical practice, the CADTH base case altered the dosing for somatropin treatments to 0.18 mg/kg/week, per Deal et al.19 CADTH notes that this value was held constant in the CADTH reanalyses, as the clinical expert feedback obtained by CADTH noted there was limited variability in this dose. CADTH was unable to explore the impact of dose tapering due to the model structure.
Uncertainty in the incidence of injection-site pain and associated disutility: The sponsor considered the impact of injection-site pain as the main AE and assumed that somatrogon had a much higher rate of injection-site pain (39.4%) than somapacitan and somatropin (1.5% for each treatment). The annual incidence of injection-site pain was obtained from 2 different sources: REAL 4 for somatropin and somapacitan, and Deal et al. for somatrogon.8,11 Deal et al. reported injection-site pain rates for somatropin and somatrogon and, notably, the reported rates of injection-site pain for somatropin are significantly higher than those reported in REAL 4 (25.2% versus 1.5%). CADTH’s evaluation of the clinical evidence highlighted an unexplained and significant difference in injection-site pain reported for somatropin in the 2 clinical trials. This disparity suggests the presence of between-trial heterogeneity, which could undermine the transitivity assumption needed for interpreting NMAs. Moreover, in the economic analysis, the sponsor did not control for the larger rate of injection-site pain reported by Deal et al. for somatropin and utilized the absolute values for the proportion of patients who reported injection-site pain from REAL 4. This was inappropriate. Clinical expert feedback obtained by CADTH for this review noted that somatrogon may be associated with higher rates of injection-site pain but that almost all patients, irrespective of which treatment they are receiving, will report injection-site pain, and that it would be uncommon for a patient not to report injection-site pain. Moreover, the sponsor assumed that each event of injection-site pain would require a pediatric consultation. Clinical expert feedback obtained by CADTH deemed this assumption inappropriate, as patients very rarely require a medical visit for injection-site pain.
Lastly, the sponsor applied a disutility of 0.03 per year for patients who experience injection-site pain based on a time trade-off study conducted by the sponsor.15 The sponsor did not detail its study methodology and, as such, there is uncertainty regarding the magnitude of the disutility associated with injection-site pain.
CADTH addressed this limitation by assuming the annual rate of injection-site pain for somatrogon was equivalent to somapacitan and somatropin, as there was no conclusive evidence supporting a higher rate of injection-site pain for somatrogon. Moreover, CADTH removed the pediatric consultation cost associated with injection-site pain. Uncertainty surrounding the injection-site pain disutility could not be fully addressed; however, as all treatments have an equal rate of injection-site pain in reanalyses, the impact of the disutility on results was minimal.
Reduction in effectiveness due to noncompliance is inappropriate: In the economic model, the sponsor assumed patients receiving weekly treatments have greater compliance than patients receiving daily treatments (95.8% versus 88.3%), based on data from REAL 4. Clinical expert feedback obtained by CADTH deemed this reasonable. However, the sponsor applied an additional 5% reduction in effectiveness for all treatments due to noncompliance based on a retrospective study of patients receiving growth hormone treatment by Maggio et al.10 As treatment efficacy already takes compliance into account, further reducing efficacy based on suboptimal compliance would result in double-counting the impact of noncompliance. Furthermore, Maggio et al. did not find statistically significant results linking height increase and treatment adherence and as such, it was inappropriate to use this study as the basis for further reducing efficacy.10 As the sponsor reported higher rates of compliance for weekly treatments, this assumption favours somapacitan and somatrogon.
CADTH addressed this limitation by assuming no further reduction in effectiveness due to noncompliance.
The magnitude of the disutility associated with daily somatropin injections in comparison with weekly injections is uncertain: The sponsor assumed there was a disutility (−0.009 per year) associated with daily injections of somatropin compared with weekly injections of somapacitan and somatrogon. Clinical expert feedback obtained by CADTH for this review noted that the assumption that a weekly injection compared with a daily injection would be associated with an improvement in a patient’s quality of life was reasonable, since any change or reduction in the number of injections is important to patients. The utility decrement used in the sponsor’s submission was based on a time trade-off study conducted by the sponsor.15 The sponsor did not detail its study methodology and, as such, there is uncertainty regarding the magnitude of the disutility associated with receiving daily injections instead of weekly injections.
CADTH was unable to address this limitation. In a scenario analysis, CADTH explored the impact of no utility decrement for patients receiving daily treatments.
Uncertainty in market share distribution of branded somatropin treatments: The economic model used an average cost weighted by market shares of various somatropin products (Genotropin, Humatrope, Norditropin, Nutropin, Omnitrope, and Saizen). Market shares were estimated based on IQVIA claims data from 2022. However, there is uncertainty regarding the assumed market share distribution for each product, as the sponsor’s estimates are not indication-specific and somatropins are used across multiple indicated populations. The claims database lacks indication details, making it unknown what proportion of claims were for other indications. Moreover, although the sponsor’s data suggested that 60% of the market share of the treatments indicated for GHD was held by the 2 somatropins with the highest publicly available prices (Humatrope and Saizen), CADTH was unable to validate the sponsor’s information supporting this claim. Clinical expert feedback obtained by CADTH noted a recent shortage of Humatrope and Norditropin. Given that drug costs significantly influence model results, the assumed market share distribution could greatly impact somatropin’s cost and the resulting cost-effectiveness of somapacitan.
CADTH was unable to address this limitation. In a scenario analysis, CADTH compared the cost-effectiveness of somapacitan with the least costly somatropin product (Genotropin), and the most costly somatropin product (Humatrope).
There is uncertainty with the use of utility scores by HSDS: In the economic model, the sponsor assumed a linear relationship between HRQoL and height based on a study which assessed EQ-5D-3L utility scores by HSDS.12 This study found that mean EQ-5D scores were lower in adults with a height shorter than the average person’s, with the EQ-5D score decreasing as the HSDS moves further away from average. The sponsor further conducted a linear interpolation analysis using the Christensen et al. study to predict patient utility by HSDS, such that patient utility scores could be measured in 0.1 HSDS increments rather than 0.5. Additionally, the sponsor incorporated an age- and sex-specific utility and disutility adjustment based on a regression algorithm by Ara and Brazier.13 The applicability of these results to the sponsor’s submitted model and patients with GHD or growth hormone insufficiency is associated with some uncertainty, including the generalizability of these values in adults to children with GHD or growth hormone insufficiency, as well as the validity of the linear interpolation. As there were only minor differences in the final HSDS change from baseline predicted by the model, the use of these utility values has minimal impact on model results.
CADTH was unable to address this limitation.
Drug costs are likely underestimated due to the exclusion of wastage: In the base case, the sponsor calculated drug costs by deriving the cost per milligram and did not consider wastage (i.e., storage wastage, preparation wastage, last-dose wastage, device-setting wastage, and adherence wastage). As a result, treatment costs were likely underestimated because the potential drug wastage resulting from unused product was not considered. Growth hormone treatments are typically dispensed based on dosage form or device type rather than by milligram and, frequently, the device can only administer a dose at set increments. Therefore, costs should have been calculated based on the full price of the device (e.g., rounding up to the nearest full pen). By not accounting for these factors, the sponsor’s submitted base case does not accurately capture annual drug acquisition costs.
CADTH addressed this limitation by adjusting drug cost calculations to account for drug wastage and assuming a 10% loss for treatments requiring reconstitution.
Presence of calculation errors in the probabilistic results of the sponsor’s model: The sponsor’s model contained calculation errors related to patient weight. Specifically, the calculation for females was inconsistent with the calculation for males and resulted in an underestimation of treatment costs.
CADTH corrected this by revising the weight-based calculation for females, multiplying their weight in kilograms by the proportion of females.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH performed reanalyses to, where possible, address key limitations in the submitted economic model, as summarized in Table 5. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical expert feedback obtained by CADTH. CADTH was unable to address limitations regarding the uncertainty of disutility associated with daily injections versus weekly injections, the applicability of utility scores measured by HSDS, and the market share distributions of various branded somatropin treatments.
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients start treatment at 6.4 years of age. | Reasonable. Clinical expert feedback obtained by CADTH noted that GHD is typically treated earlier, generally between 3 and 4 years of age if symptoms are present in infancy. |
A time horizon of 11.6 years (i.e., until age 18) was adopted in the sponsor’s base case. | Reasonable. According to the CADTH Guidelines for the Economic Evaluation of Health Technologies: Canada, the time horizon selected in the economic model should be long enough to capture the costs and effects of treatment. This treatment is administered in childhood and is expected to continue until the patient reaches their final adult height, which occurs approximately at a bone age of 14 for females and 16 for males. Clinical expert feedback obtained by CADTH noted that patients may continue receiving treatment with a reduced dose upon reaching their final adult height. |
All treatments have identical HSDS changes from the baseline. | Likely appropriate. CADTH’s appraisal of the clinical evidence found somapacitan to be noninferior with somatropin and found somatropin and somatrogon to have no meaningful difference clinically. However, CADTH cannot state with certainty that somatropin and somatrogon are clinically equivalent. |
The HSDS benefit observed at the end of year 3 will be sustained throughout the model’s time horizon. | Inappropriate. Clinical expert feedback obtained by CADTH noted that the treatment benefits likely cease earlier than 18 years of age, as final adult height is achieved at a bone age of 14 for females and 16 for males. |
The discontinuation rate was assumed to be the same for all patients, based on data from somapacitan from the REAL 4 study. | Uncertain. The CADTH Clinical Review concluded that treatment discontinuation may be increased with somapacitan vs. Norditropin, but the increase was small. |
Patients who discontinue treatment maintain their last observed HSDS. | Inappropriate. Clinical expert feedback obtained by CADTH indicated that patients who discontinue growth hormone treatment would have their HSDS decrease over time. |
Discontinuation was considered only in the first year of treatment. Upon discontinuation, patients are assumed not to reinitiate growth hormone treatment. | Inappropriate. Clinical expert feedback obtained by CADTH indicated that patients who discontinue treatment would likely switch to an alternative treatment due to the metabolic consequences of GHD. |
No mortality difference between model comparators. | Appropriate. |
The various somatropin brands in the model are assumed to have equivalent effectiveness and safety. A similar dose is assumed for all somatropin brands. | Appropriate. |
GHD = growth hormone deficiency; HSDS = height standard deviation score.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
Patient weight | Patient weight for females was calculated by multiplying the percentage year-over-year increase in weight by the proportion of females. | Patient weight for females was calculated by multiplying their age-specific weight in kilograms by the proportion of females. |
Changes to derive the CADTH base case | ||
1. Dosing for somatropin treatments | 0.24 mg/kg/week | 0.18 mg/kg/week |
2. Injection-site pain | Somatrogon annual rate of injection pain = 39.4% Management cost per injection-site pain event = $84.45 | Somatrogon annual rate of injection pain = 1.5% Management cost per injection-site pain event = $0 |
3. Drug wastage | Drug wastage excluded | Drug wastage included |
4. Reduction in effectiveness due to noncompliance | 5% | 0% |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 | |
CADTH undertook a stepped analysis, incorporating each change proposed in Table 6 into the sponsor’s base case to highlight the impact of each change. Full and disaggregated results as well as additional reanalyses are presented in Appendix 4.
Results from the CADTH base case suggest that over an 11.6-year time horizon, somapacitan is more costly than somatropin and associated with a gain of 0.094 QALYs, while it is less costly than somatrogon and associated with a gain of 0.001 QALYs. The results suggest that somapacitan has an ICER of $275,250 per QALY gained when compared with somatropin. Somatrogon is dominated by (more costly and no more effective than) somapacitan. CADTH notes that the cost-effectiveness of somapacitan and somatrogon varied by model run due to the stochastic nature of the probabilistic model. Deterministic results indicate equivalent QALYs for somapacitan and somatrogon.
In the CADTH base case, somapacitan had a 10% probability of being cost-effective at a WTP threshold of $50,000 per QALY when compared with somatropin and a 39% probability when compared with somatrogon. Consistent with the sponsor’s submitted analysis, 73% of the QALYs associated with somapacitan were accrued in the post-trial period and 99.9% of patients were still alive at the end of the modelled time horizon.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s results and CADTH’s base case. The CADTH base case suggested that a price reduction of at least 13% for somapacitan would be required for it to achieve cost-effectiveness relative to somatropin at a $50,000 per-QALY threshold (Table 7). However, CADTH noted that a price reduction of 16% would be required for somapacitan to be no more costly than the mix of somatropin treatments. Furthermore, a price reduction of at least 41% would be required for somapacitan to be no more costly than the least costly somatropin.
Table 6: Summary of the CADTH Reanalysis Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Somatropin | 134,577 | 8.5806 | Reference |
Somapacitan | 160,513 | 8.6748a | $275,250 vs. somatropin |
Somatrogon | 162,263 | 8.6739a | Dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CADTH reanalyses are based on publicly available prices of comparator treatments and do not reflect confidential, negotiated prices.
aProbabilistic results report minor differences in QALYs (0.0009) between somapacitan and somatrogon. These differences are solely attributable to the stochastic nature of the probabilistic model. Deterministic analysis results indicate equivalent QALYs for the modelled time horizon. CADTH notes that the presence of somapacitan and somatrogon on the efficiency frontier varied by model run. The magnitude of the ICER was consistent.
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for somapacitan vs. relevant comparators ($/QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | Somapacitan was dominant | WTP threshold < $273,158: Somatropin WTP threshold > $273,158: Somapacitan |
10% | Somapacitan was dominant | WTP threshold < $102,287: Somatropin WTP threshold > $102,287: Somapacitan |
13.1% | Somapacitan was dominant | WTP threshold < $49,316: Somatropin WTP threshold > $49,316: Somapacitan |
16% | Somapacitan was dominant | Dominant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; WTP = willingness to pay.
Note: Only nondominated comparators are presented. Results reported are based on deterministic analyses. Dominant means a treatment is more effective and less costly.
CADTH undertook scenario analyses to explore the impact of alternative assumptions on the cost-effectiveness of somapacitan, which are outlined as follows:
Assuming no disutility is associated with receiving daily injections.
Assuming the market share for somatropin treatments is captured entirely by the lowest-priced somatropin (Genotropin).
Assuming the market share for somatropin treatments is captured entirely by the highest-priced somatropin (Humatrope).
The results of these analyses are presented in Appendix 4, Table 14. Results were most sensitive to the scenario in which disutilities associated with daily injections were removed, as somapacitan and somatrogon were dominated by somatropin (i.e., somatropin was less costly and associated with more QALYs). Although QALY differences are due to the stochastic nature of probabilistic analysis, deterministic analyses indicate all treatments have equal QALYs. Assuming that the market share for somatropin treatments is captured entirely by the lowest-priced treatment resulted in an ICER of $695,015 per QALY gained for somapacitan when compared with somatropin. Assuming that the market share for somatropin treatments is captured entirely by the highest-priced treatment (somatropin), somapacitan is the lowest-costing treatment. Therefore, somatrogon has an ICER of $617,481 per QALY gained when compared with somapacitan, while somatropin is dominated. CADTH notes that in scenario analyses 2 and 3, the presence of somapacitan and somatrogon on the efficiency frontier varied by model run due to the stochastic nature of the probabilistic model. Deterministic results using the CADTH base case indicate equivalent QALYs for somapacitan and somatrogon.
Issues for Consideration
Potential off-label use of somapacitan: The sponsor is requesting that somapacitan be reimbursed for the long-term treatment of pediatric patients who have GHD. However, it is possible that somapacitan will be used off-label for other conditions that relevant comparator products are also indicated for, such as idiopathic short stature, short stature homeobox–containing gene deficiency, small for gestational age, and growth failure in children due to chronic renal failure. The potential for off-label use of somapacitan may be associated with considerable costs to the drug plans. However, it should be noted that the possibility of off-label use is not unique to somapacitan and applies to the other available somatrogon and somatropin products on the market in Canada.
Stopping rules: In practice, treatment with growth hormone therapies is expected to be continued until the target height has been achieved. In the submitted model, patients on treatment were not assumed to achieve their target height until age 18 and were assumed to be on treatment until then. No other stopping rules were assessed by the sponsor or CADTH.
Availability and pricing comparators: The sponsor of somatrogon successfully completed negotiations with the pan-Canadian Pharmaceutical Alliance (pCPA) and is listed on public formularies. Of note, the CADTH Canadian Drug Expert Committee (CDEC) recommended that somatrogon be reimbursed only if it does not cost more than the least costly somatropin.20 Therefore, it is likely that somatrogon will be reimbursed by jurisdictional drug plans at confidential prices that are lower than the publicly available list prices.21 CADTH noted that the prices of the branded somatropin products differed across jurisdictions, and it is unclear whether there are negotiated prices within CADTH-participating drug plans. Additionally, clinical expert feedback obtained by CADTH for this review noted a recent shortage of multiple branded somatropin treatments (i.e., Humatrope and Norditropin).
Overall Conclusions
Based on the sponsor-submitted clinical evidence from the phase III REAL 4 trial for pediatric patients with GHD, somapacitan results in little to no difference in the change from baseline in HSDS, notable harms, injection-site reactions and injection-site pain, and treatment discontinuation when compared with Norditropin brand somatropin. Using GRADE, CADTH categorized this evidence as having moderate to high certainty. Data from the REAL 4 extension trial and the REAL 3 trial were considered descriptive in nature and, therefore, there is greater uncertainty in the findings from these studies. The sponsor-submitted NMA suggested there were no statistically significant differences identified between somapacitan and somatrogon for HSDS at week 26 and week 52; however, several sources of uncertainty identified with the NMA limit the interpretation of these results, such as the sparse network, small sample sizes, between-trial heterogeneity, and wide credible intervals across all assessed outcomes. No conclusions can be drawn regarding the comparative safety of somapacitan, somatrogon, and somatropin.
CADTH identified several limitations in the sponsor’s pharmacoeconomic analysis that have notable implications on the cost-effectiveness of somapacitan. CADTH revised the dose for all somatropin products to align with the dose commonly used in Canadian clinical practice, aligned the annual rate of injection-site pain for somatrogon with that of somapacitan and somatropin, removed the pediatric consultation visit cost association with patients who experience an event of injection-site pain, included drug wastage, and removed the reduction in effectiveness due to noncompliance. The CADTH base-case analysis resulted in an ICER for somapacitan of $275,250 per QALY gained when compared against somatropin; somatrogon was dominated (associated with greater total costs and no additional QALYs). The cost-effectiveness of somapacitan and somatrogon varied by model run due to the input distributions in a probabilistic model, while the cost-effectiveness of somapacitan relative to somatropin was impacted by the weighted cost of somatropin. CADTH noted that the gain in QALYs associated with somapacitan is solely due to the disutility associated with patients receiving daily somatropin treatments.
Based on the assumptions underpinning the CADTH base case, a price reduction of at least 13% would be required for somapacitan to be considered cost-effective at a WTP threshold of $50,000 per QALY gained compared with a weighted basket of somatropins. If the cost of somatropin was considered using the lowest-cost branded somatropin only, CADTH noted that a price reduction of at least 41% would be required for somapacitan to be no more costly than the least costly somatropin. Based on these price reductions, the cost of somapacitan would range from $174.16 to $258.27 per prefilled pen (5 mg/1.5 mL).
The cost-effectiveness of somapacitan is highly sensitive to the cost of somatropin (e.g., dosing and the cost of the brand of somatropin being considered), the incidence of injection-site pain associated with somatrogon, and the disutility associated with receiving daily injections, which was assessed in a scenario analysis. If the disutility associated with more frequent injections is removed, somapacitan is dominated by somatropin. There is insufficient clinical evidence to justify a price premium for somapacitan over currently available growth hormone treatments for GHD. To ensure cost-effectiveness, somapacitan should be priced no more than the lowest-cost growth hormone comparator used to treat GHD that is funded.
References
1.Sävendahl L, Battelino T, Brod M, et al. Once-Weekly Somapacitan vs Daily GH in Children With GH Deficiency: Results From a Randomized Phase 2 Trial. J Clin Endocrinol Metab. 2020;105(4):e1847-1861. PubMed
2.Sävendahl L, Battelino T, Højby Rasmussen M, Brod M, Saenger P, Horikawa R. Effective GH Replacement With Once-weekly Somapacitan vs Daily GH in Children with GHD: 3-year Results From REAL 3. J Clin Endocrinol Metab. 2022;107(5):1357-1367. PubMed
3.Miller BS, Blair JC, Rasmussen MH, et al. Weekly Somapacitan is Effective and Well Tolerated in Children With GH Deficiency: The Randomized Phase 3 REAL4 Trial. J Clin Endocrinol Metab. 2022;107(12):3378-3388. PubMed
4.SOGROYA® (somapacitan injection) 5mg/1.5mL, 10mg/1.5mL, 15mg/1.5mL pre-filled pen for subcutaneous use [product monograph]. Mississauga (ON): Novo Nordisk Canada, Inc; 2023 Jul 26.
5.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Sogroya (somapacitan), 5mg/1.5mL, 10mg/1.5mL, 15mg/1.5mL injection. Mississauga (ON): Novo Nordisk Canada, Inc.; 2023 Apr 27.
6.TA188. Human growth hormone (somatropin) for the treatment of growth failure in children. London (UK): National Institute for Health and Care Excellence; 2010: https://www.nice.org.uk/guidance/ta188/resources/human-growth-hormone-somatropin-for-the-treatment-of-growth-failure-in-children-pdf-82598502860485. Accessed [sponsor supplied citation].
7.CADTH Reimbursement Review Somatrogon (Ngenla). Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/DRR/2022/SR0683-Ngenla-CombinedReport.pdf. Accessed [sponsor supplied citation].
8.Clinical Study Report: NN8640-4263. REAL 4 main period (up to week 52). A trial comparing the effect and safety of once weekly dosing of somapacitan with daily Norditropin® in children with growth hormone deficiency. Bagsvaerd, Denmark: Novo Nordisk; 2022 Mar 18.
9.Takeda A, Cooper K, Bird A, et al. Recombinant human growth hormone for the treatment of growth disorders in children: a systematic review and economic evaluation. Health Technol Assess. 2010;14:42. PubMed
10.Maggio MC, Vergara B, Porcelli P, Corsello G. Improvement of treatment adherence with growth hormone by easypod device: experience of an Italian centre. Ital J Pediatr. 2018;44(1):113. PubMed
11.Deal CL, Steelman J, Vlachopapadopoulou E, et al. Efficacy and Safety of Weekly Somatrogon vs Daily Somatropin in Children With Growth Hormone Deficiency: A Phase 3 Study. J Clin Endocrinol Metab. 2022;107(7):e2717-e2728. PubMed
12.Christensen TL, Djurhuus CB, Clayton P, Christiansen JS. An evaluation of the relationship between adult height and health-related quality of life in the general UK population. Clinical Endocrinology. 2007;67(3):407-412. PubMed
13.Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health. 2010;13(5):509-518. PubMed
14.Kirsch S, Butler G, de Fries Jensen L, Boegelund M, Håkan-Bloch J. PCR3 Pain-Free Injections for Growth Hormone Deficiency (GHD) Improve Quality of Life: A Time Trade-Off (TTO) Study in the UK and Canada. Value in Health. 2023;26(6, Supplement):S312.
15.Novo Nordisk. Time trade-off study [Data on File]. 2022 Jan.
16.Ontario Ministry of Health. Exceptional Access Program (EAP). 2023; https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed [sponsor supplied citation].
17.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed [sponsor supplied citation].
18.Ontario Ministry of Health. Schedule of Facility Fees For Independent Health Facilities https://www.health.gov.on.ca/en/pro/programs/ohip/sob/facility/indep_health_facilities.pdf. Accessed [sponsor supplied citation].
19.Deal C, Kirsch S, Chanoine JP, et al. Growth hormone treatment of Canadian children: results from the GeNeSIS phase IV prospective observational study. CMAJ Open. 2018;6(3):E372-e383. PubMed
20.CADTH Remimbursement Recommendation: Somatrogon (Ngenla). Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/DRR/2022/SR0683%20Ngenla%20-%20CADTH%20Final%20Rec.pdf. Accessed [sponsor supplied citation].
21.Pan-Canadian Pharmaceutical Alliance. Ngenla (somatrogon). 2022: https://www.pcpacanada.ca/negotiation/21798. Accessed 2023 Jul 18.
22.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Sogroya (somapacitan), 5mg/1.5mL, 10mg/1.5mL, 15mg/1.5mL injection. Mississauga (ON): Novo Nordisk Canada, Inc.; 2023 Apr 27.
23.Saskatchewan Drug Plan: search formulary. 2022; http://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed [sponsor supplied citation].
24.Grimberg A, Huerta-Saenz L, Grundmeier R, et al. Gender Bias in U.S. Pediatric Growth Hormone Treatment. Scientific Reports. 2015;5(1):11099. PubMed
25.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. Ottawa (ON): The Conference Board of Canada; 2017: https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326. Accessed [sponsor supplied citation].
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Human Growth Hormones to Treat Growth Hormone Deficiency
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Somapacitan (Sogroya) | 5 mg / 1.5 mL 10 mg / 1.5 mL 15 mg / 1.5 mL | Prefilled pen | 297.2000a 594.4000a 891.6000a | 0.16 mg/kg weekly | 42.34 | 15,454 |
Somatrogon (Ngenla)b | 24 mg / 1.2 mL | Prefilled pen | 345.8400 | 0.66 mg/kg weekly | 42.64 | 15,563 |
60 mg / 1.2 mL | 864.6000 | |||||
Daily growth hormone treatment | ||||||
Somatropin (Genotropin) | 5.3 mg 12 mg | Prefilled pen | 154.5840 349.9880 | 0.16 to 0.24 mg/kg/week, in 6 to 7 doses | 20.75 to 31.64 | 7,575 to 11,550 |
0.6 mg 0.8 mg 1.0 mg 1.2 mg 1.4 mg 1.6 mg 1.8 mg 2.0 mg | Single-use prefilled syringe | 17.5000 23.3329 29.1657 35.0000 40.8314 46.6657 52.4986 58.3314 | 20.76 to 31.21 | 7,578 to 11,392 | ||
Somatropin (Humatrope) | 6 mg / 3.15 mL 12 mg / 3.15 mL 24 mg / 3.15 mL | Cartridge | 315.2500 630.5000 1,261.0100 | 0.18 to 0.30 mg/kg/week in 3 to 7 doses | 42.32 to 72.55 | 15,447 to 26,481 |
Somatropin (Norditropin) | 5 mg / 1.5 mL 10 mg / 1.5 mL 15 mg / 1.5 mL | Prefilled pen | 194.7000 389.4000 584.1000 | Up to 0.3 mg/kg/week, divided in daily doses | 51.74 | 18,886 |
Somatropin (Nutropin AQ) | 5 mg / 2 mL 10 mg / 2 mL 20 mg / 2mL | Cartridge | 203.7100 407.4200 814.8400 | Up to 0.3 mg/kg/week, divided in daily doses | 54.14 | 19,760 |
Somatropin (Omnitrope) | 5 mg 10 mg | Vial | 155.8000 311.6000 | 0.16 to 0.24 mg/kg/week | 22.2 to 33.29 | 8,102 to 12,152 |
15 mg / 1.5 mL | Cartridge | 467.4000c | 23.05 to 33.29 | 8,413 to 12,152 | ||
Somatropin (Saizen) | 5 mg | Vial | 224.0500 | 0.20 to 0.27 mg/kg/week in 3 to 7 doses | 39.9 to 54.02 | 14,563 to 19,716 |
6 mg (5.83 mg/mL) 12 mg (8 mg/mL) 20 mg (8 mg/mL) | Cartridge | 268.8300 537.6600 896.1000 | 39.77 to 54.50 | 14,517 to 19,893 | ||
Note: All prices are from the Ontario Exceptional Access Program16 (accessed July 2023), unless otherwise indicated, and do not include dispensing fees. Weight-based doses use a weight of 31.08 kg per the sponsor’s average patient weight estimate in the budget impact analysis.22 Annual costs are based on rounding the number of units required in a calendar year to the nearest integer. Daily costs are based on annual costs divided by 365. No other wastage costs have been considered.
aSponsor’s submitted price.
bSomatrogon received a pricing condition from CADTH Canadian Drug Expert Committee (CDEC) in March 2022, for the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone (growth hormone deficiency) in that it should only be reimbursed if it does not cost more than the least costly somatropin.20
cSaskatchewan Formulary list price23 (accessed in July 2023).
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | No | The sponsor incorrectly calculated average patient weight by multiplying the percentage year-over-year weight increase for females by the proportion of females |
Model structure is adequate for decision problem | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
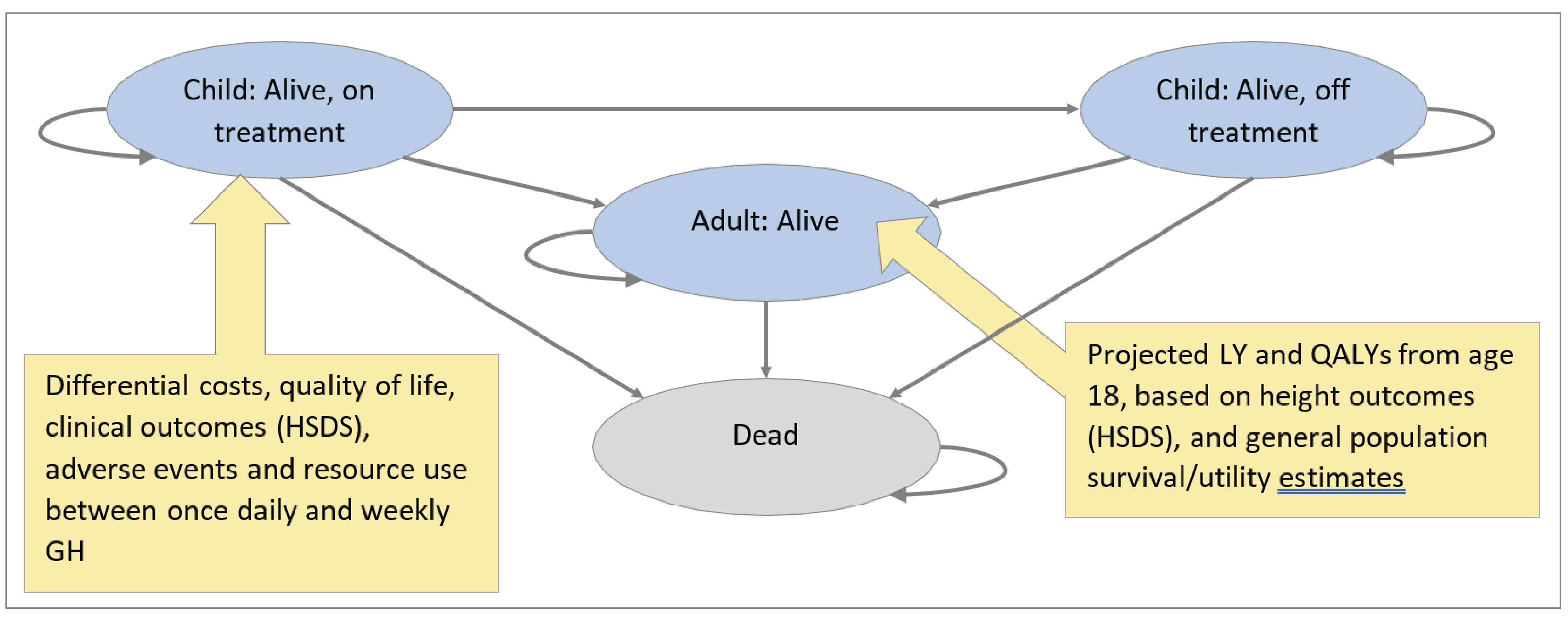
GH = growth hormone; HSDS = height standard deviation score; LY = life-year; = QALY = quality-adjusted life-year.
Source: Sponsor’s submission.5
Table 10: Market Shares of Branded Somatropins
Branded somatropin | Market share (%) |
|---|---|
Genotropin | 7.8 |
Humatrope | 29.0 |
Norditropin | 12.3 |
Nutropin | 4.2 |
Omnitrope | 15.7 |
Saizen | 31.1 |
Source: Sponsor’s submission.5
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 11: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Somapacitan | Somatropin | Somatrogon |
|---|---|---|---|
Discounted LYs | |||
Total | 10.4839 | 10.4839 | 10.4839 |
Discounted QALYs | |||
Total | 8.6748 | 8.5806 | 8.6739 |
Child on tx: patient utility | 8.5562 | 8.5540 | 8.5545 |
Child on tx: patient disutility | −0.0048 | −0.0971 | −0.0048 |
Child off tx: patient utility | 0.0541 | 0.0543 | 0.0548 |
Adult patient utility | 0.0693 | 0.0694 | 0.0694 |
Discounted costs ($) | |||
Total | $160,513 | $134,577 | $162,263 |
Child on tx: treatment | $152,468 | $126,533 | $154,218 |
Child on tx: monitoring | $7,990 | $7,990 | $7,990 |
Child on tx: AEs | $0 | $0 | $0 |
Child off tx: monitoring | $26 | $26 | $26 |
Adult: monitoring. | $29 | $29 | $29 |
AE = adverse event; LY = life-year; QALY = quality-adjusted life-year; tx = treatment.
Table 12: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | Somapacitan | 122,489 | 8.6952 | Reference |
Somatrogon | 123,455 | 8.5699 | Dominated | |
Somatropin | 123,041 | 8.5983 | Dominated | |
Sponsor’s corrected base case | Somapacitan | 160,997 | 8.6949 | Reference |
Somatrogon | 162,183 | 8.5762 | Dominated | |
Somatropin | 162,234 | 8.6013 | Dominated | |
CADTH reanalysis 1: Dosing for somatropin treatments | Somatropin | 123,774 | 8.5725 | Reference |
Somapacitan | 161,254 | 8.6700 | $384,246 | |
Somatrogon | 161,801 | 8.5442 | Dominated | |
CADTH reanalysis 2: Injection-site pain | Somapacitan | 160,191 | 8.6441 | Reference |
Somatrogon | 161,223 | 8.6454 | $789,428 | |
Somatropin | 162,230 | 8.5512 | Dominated | |
CADTH reanalysis 3: Drug wastage | Somapacitan | 161,024 | 8.6796 | Reference |
Somatrogon | 161,803 | 8.5540 | Dominated | |
Somatropin | 175,886 | 8.5845 | Dominated | |
CADTH reanalysis 4: Reduction in effectiveness due to noncompliance | Somapacitan | 161,477 | 8.6825 | Reference |
Somatrogon | 161,761 | 8.5576 | Dominated | |
Somatropin | 162,890 | 8.5893 | Dominated | |
CADTH base case (reanalysis 1 + 2 + 3 + 4) | Somatropin | 134,577 | 8.5806 | Reference |
Somapacitan | 160,513 | 8.6748 | $275,250 | |
Somatrogon | 162,263 | 8.6739 | Dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: CADTH reanalyses are based on publicly available prices of comparator treatments and do not reflect confidential, negotiated prices.
Scenario Analyses
Table 13: CADTH Scenario Analyses
Scenario | CADTH base case | CADTH scenario |
|---|---|---|
1. Disutility associated with daily injections | Included disutility for daily injections (−0.009) | Excluded disutility associated with daily injections |
2a. Lowest-priced somatropin market share | Norditropin = 12.3% Saizen = 31.3% Omnitrope = 15.7% Nutropin = 4.2% Genotropin = 7.8% Humatrope = 29.0% | Norditropin = 0% Saizen = 0% Omnitrope = 0% Nutropin = 0% Genotropin = 100% Humatrope = 0% |
2b. Highest-priced somatropin market share | Norditropin = 12.3% Saizen = 31.3% Omnitrope = 15.7% Nutropin = 4.2% Genotropin = 7.8% Humatrope = 29.0% | Norditropin = 0% Saizen = 0% Omnitrope = 0% Nutropin = 0% Genotropin = 0% Humatrope = 100% |
Table 14: Summary of Scenario Analyses Conducted on CADTH Base Case
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CADTH base case | Somatropin | 134,577 | 8.5806 | Reference |
Somapacitan | 160,513 | 8.6748 | $275,250 | |
Somatrogon | 162,263 | 8.6739 | Dominated | |
CADTH scenario analysis: Disutility associated with daily injections | Somatropin | 134,950 | 8.7046 | Reference |
Somatrogon | 160,499 | 8.7043 | Dominated | |
Somapacitan | 161,518 | 8.7027 | Dominated | |
CADTH scenario analysis: Lowest-priced somatropin market share | Somatropin | 96,197 | 8.6170 | Reference |
Somapacitan | 161,676 | 8.7112 | $695,015 | |
Somatrogon | 162,130 | 8.7086 | Dominated | |
CADTH scenario analysis: Highest-priced somatropin market share | Somapacitan | 161,356 | 8.6403 | Reference |
Somatrogon | 162,364 | 8.6420 | $617,481 | |
Somatropin | 164,273 | 8.5474 | Dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference
Note: CADTH reanalyses are based on publicly available prices of comparator treatments and do not reflect confidential, negotiated prices.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 15: Summary of Key Takeaways
Key takeaways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the sponsor-submitted budget impact analysis (BIA), the sponsor assessed the budget impact of somapacitan for the treatment of pediatric patients with GHD, in line with the Health Canada indication.22 The BIA was undertaken from the perspective of the Canadian public payer over a 3-year time horizon (2024 to 2026) using an epidemiologic approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the (NIHB) program. Data used to inform the model were obtained from various sources including published literature, IQVIA databases, the sponsor’s internal data, and input from clinical expert feedback obtained by the sponsor. Key inputs to the BIA are documented in Table 15.
In the reference scenario, the sponsor assumed that patients aged 3 to 17 years of age would be eligible to receive somatrogon or various branded somatropin (i.e., Genotropin, Humatrope, Norditropin, Nutropin, Omnitrope, Saizen). In the new-drug scenario, somapacitan was assumed to capture market shares from somatrogon only. The sponsor’s analysis included drug acquisition costs, excluding wastage; dispensing fees and markups were not included in the base case. The sponsor utilized a weight table for children with GHD sourced from Takeda et al., which resulted in an average weight of 31.08 kg, based on the premise that children will be equally distributed within the age range of 3 to 17 years.9 This weight was assumed constant throughout the model’s time horizon. Additionally, the sponsor assumed that all somatropins were administered at a dose of 0.034 mg/kg per day. Recommended dosing for somapacitan and somatrogon were retrieved from the respective product monographs.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3, if appropriate) |
|---|---|
Target population | |
Population aged 3 to 17 | 4,163,202 |
Prevalence of GHD | 0.025%24 |
Proportion receiving treatment | 3 to 16 years old: 95.0%a 17 years old: 40.0%a |
Proportion covered by public plan | 52.0%22 |
Number of patients eligible for drug under review | 517 / 519 / 521 |
Market uptake (3 years) | |
Uptake (reference scenario) Somapacitan Somatrogon Genotropin Humatrope Norditropin Nutropin AQ Omnitrope Saizen | 0% / 0% / 0% 35% / 45% / 55% 5.1% / 4.31% / 3.53% 18.81% / 15.92% / 13.03% 7.97% / 6.74% / 5.52% 2.74% / 2.32% / 1.9% 10.19% / 8.63% / 7.06% 20.19% / 17.08% / 13.97% |
Uptake (new-drug scenario) Somapacitan Somatrogon Genotropin Humatrope Norditropin Nutropin AQ Omnitrope Saizen | 8.75% / 22.5% / 41.25% 26.25% / 22.5% / 13.75% 5.1% / 4.31% / 3.53% 18.81% / 15.92% / 13.03% 7.97% / 6.74% / 5.52% 2.74% / 2.32% / 1.9% 10.19% / 8.63% / 7.06% 20.19% / 17.08% / 13.97% |
Cost of treatment (per patient) | |
Cost of treatment over 1 year Somapacitan Somatrogon Genotropin Humatrope Norditropin Nutropin AQ Omnitrope Saizen | $15,423.11 $15,423.44 $10,978.67 $19,457.93 $15,155.85 $15,857.21 $12,127.80 $17,440.52 |
GHD = growth hormone deficiency.
aBased on sponsor’s internal estimates.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the 3-year budget impact of reimbursing somapacitan for the long-term treatment of pediatric patients with GHD to result in cost savings of −$122 (year 1: −$15; year 2: −$38; year 3: −$70).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Dosing for somatropin does not align with the dose commonly used in Canadian clinical practice: In the submitted BIA the sponsor assumed that the average administered dose for all somatropin products was 0.24 mg/kg/week. Clinical expert feedback obtained by CADTH for this review indicated that the average dose applied in the sponsor’s base case was higher than the average dose commonly used in Canadian clinical practice, which is typically 0.18 mg/kg/week, and further noted that less than 10% of patients are administered a higher dose. Furthermore, the clinical expert feedback obtained by CADTH noted that once patients have reached their final adult height, typically between 16 to 18 years old, the daily dose is reduced to 0.3 to 0.5 mg per day. As the sponsor’s BIA includes patients 3 to 17 years old, the average dose for older pediatric patients is likely significantly overestimated. Deal et al. reported that the average somatropin dose administered to children with GHD within the Canadian cohort over a 5-year period was 0.18 mg/kg/week and further indicated that mean growth hormone dosages in Canada were lower than those administered in the US or globally.19 As drug costs are a key driver of model results, the sponsor’s assumption of a higher dose for all somatropin products overestimated the total costs associated with somatropin, and biasing cost-effectiveness results in favour of somapacitan and somatrogon.
To align with the dosing expected in Canadian clinical practice, the CADTH base case altered the dosing for somatropin treatments to 0.18 mg/kg/week, as administered in Deal et al. 2018.19 CADTH was unable to explore the impact of an alternative dose for different age groups due to model structure.
Uncertainty regarding the anticipated market shares and comparator displacement of somapacitan: The sponsor’s base case assumed that 8.75%, 22.5%, and 41.25% of eligible patients would receive somapacitan in year 1, year 2, and year 3, respectively, based on expert opinion.22 Moreover, the sponsor assumed that somapacitan would only capture market shares from somatrogon. Clinical expert feedback obtained by CADTH for this review noted that uptake will likely be lower because physicians may not have as much training in monitoring IGF-1 levels with once-weekly treatments as somatrogon, the alternative once-weekly treatment, is still slowly being implemented into clinical practice. The clinical expert feedback obtained by CADTH noted that it was inappropriate of the sponsor to assume that somapacitan would only displace somatrogon as it was likely to displace all comparators fairly equally as clinicians will be weighing the once-weekly injections against somatropins in general, instead of against specific brands of somatropin.
CADTH addressed this limitation by assuming somapacitan displaced all comparators proportionate to their respective market share and revised the market uptake rates for somapacitan based on clinical expert input for this review. Estimated market uptake rates for somapacitan were assumed to be 5%, 12.5%, and 27.5% in year 1, year 2, and year 3, respectively. CADTH explored uncertainty in the uptake of somapacitan in scenario analyses to reflect the sponsor’s market uptake estimates.
Uncertainty regarding the market shares of treatments included in the reference scenario: In the reference scenario, the sponsor estimated somatrogon’s uptake to be 55% by year 3 (year 1: 35%; year 2 = 45%; year 3 = 55%) based on expert opinion and assumed that uptake was equal across provinces. Clinical expert feedback obtained by CADTH noted that although somatrogon concluded negotiations with the pCPA and has a letter of intent with CADTH jurisdictions,21 the market shares for somatrogon were significantly higher than expected as it is still being slowly implemented into clinical practice. At the time CADTH conducted this review, somatrogon was being funded only by the Saskatchewan, Ontario, Manitoba, and NIHB public plans. As such, the market shares for somatrogon are overestimated for the majority of CADTH-participating drug programs.
Additionally, the sponsor estimated market shares for somatropin treatments based on IQVIA claims data but did not specify how the claims data were filtered to align with the Health Canada–indicated population of pediatric patients with GHD. Further, the sponsor assumed that all claims were for the indication of interest, despite these drugs having other indications. Given the fact that the claims database does not specify the indication and the proportion of claims pertaining to use for other indications is unknown, the sponsor’s methods introduce uncertainty in the estimated market size as it potentially overestimates the size of the eligible patient population. The clinical expert feedback obtained by CADTH noted that there is also a shortage of Humatrope and Norditropin.
CADTH addressed this limitation by assuming somatrogon captured 17.5%, 22.5%, and 27.5% of the market across all provinces in years 1, 2, and 3, respectively. Market shares previously attributed to somatrogon were proportionally distributed across somatropin treatments.
Drug costs are likely underestimated due to the exclusion of wastage: In the sponsor’s submitted pharmacoeconomic and budget impact analyses, the sponsor calculated drug costs by deriving the cost per milligram and did not consider wastage of any kind. As a result, all treatment costs were likely underestimated because the sponsor's calculations did not consider the potential drug wastage resulting from unused product (Refer to the CADTH Appraisal of the Sponsor’s Economic Evaluation). To align the BIA with CADTH’s reanalysis of the submitted pharmacoeconomic review, drug cost calculations were adjusted to consider the drug wastage.
CADTH addressed this limitation by adjusting drug cost calculations to account for drug wastage.
The proportion of patients covered by public drug plans was likely underestimated: The sponsor assumed that the proportion of children with GHD assumed to be enrolled in public drug programs was 52% based on the sponsor’s internal data. It is more appropriate to use the proportion of patients eligible, rather than enrolled, as the market size will be determined by all eligible for public coverage, and the BIA should consider all patients eligible regardless of whether they are presently enrolled. Should somapacitan be reimbursed by public plans, it is assumed that all eligible patients for this treatment would enrol for public coverage. CADTH acknowledges that some who are eligible for public coverage may also have coverage through a private plan. Additionally, as the proportion of patients eligible for public plan coverage varies by province, jurisdiction-specific estimates should be utilized when available.
CADTH additionally notes that the NIHB population was inappropriately calculated by the sponsor. NIHB clients residing within Ontario who are under 25 years of age are eligible for reimbursement by the Ontario Drug Benefit Program (ODB) and thus should be counted as ODB clients rather than NIHB clients for the purposes of modelling the budgetary impact of reimbursing somapacitan.
In the CADTH base case, the jurisdiction-specific proportion of patients eligible for public drug plan coverage was used to estimate the market size for somapacitan,25 and the eligible population of NIHB clients was recalculated by considering NIHB clients residing in Ontario to be part of Ontario’s eligible population.
Additional limitations were identified but were not considered to be key limitations. These limitations include:
Misalignment of model inputs between the sponsor-submitted economic analysis and BIA. CADTH noted that several model inputs and assumptions in the BIA were not aligned with the economic analysis submitted by the sponsor. First, in the economic analysis, the sponsor assumed an annual rate of discontinuation across all treatments of 0.758% per year based on data from REAL 4 but assumes that all patients remain on treatment for the full 3-year horizon in the BIA.8 Second, the sponsor assumed patients start treatment at 6.4 years old and 3 years old in the economic analysis and BIA, respectively. Third, the sponsor assumed that 74.5% of the population was male in the economic analysis and 68% of the population was male in the BIA.8,19 Fourth, the sponsor assumed that patients continue to receive growth hormone therapy until the age of 18 in the economic analysis. However, in the BIA, the sponsor assumed that at the age of 17, only 40% of patients with GHD continue to receive treatment. None of the misalignments are expected to have a notable impact on the estimated budgetary impact of reimbursing somapacitan.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by aligning the daily dose of somatropin products with clinical expert feedback obtained by CADTH for this review, including drug wastage, incorporating the proportion of patients eligible for public drug plan coverage, correcting the NIHB population size, halving the estimated market share of somatrogon, assuming somapacitan displaced all comparators proportionate to their market share, and aligning somapacitan’s market uptake with clinical expert input received for this review (Table 17).
Table 17: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
None. | — | — |
Changes to derive the CADTH base case | ||
1. Drug wastage | Drug wastage excluded | Drug wastage included |
2a. Public drug plan coverage | Based on the proportion of patients covered by public drug programs (52%) | Based on the proportion of patients eligible for public drug programs (jurisdiction-specific; range 7% to 100%) |
2b. NIHB population | NIHB population (base year) = 217,916 | NIHB population (base year) = 175,352 |
3. Somatrogon market shares | Year 1 = 35%; year 2 = 45%; year 3 = 55% | Year 1 = 17.5%; year 2 = 22.5%; year 3 = 27.5% |
4. Comparator displacement | Somapacitan was assumed to only displace somatrogon. | Somapacitan was assumed to displace all comparators proportionate to their market share. |
5. Dosing for somatropin treatments | 0.24 mg/kg/week | 0.18 mg/kg/week |
6. Somapacitan market uptake | Year 1 = 8.75%; year 2 = 22.5%; year 3 = 41.25% | Year 1 = 5%; year 2 = 12.5%; year 3 = 27.5% |
CADTH base case | Reanalyses 1 + 2a + 2b + 3 + 4 + 5 + 6 | |
NIHB = Non-Insured Health Benefits.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19. In the CADTH base case, the 3-year budget impact is expected to be $458,079 (year 1: $52,450; year 2: $128,547; year 3: $277,081) should somapacitan be reimbursed as per the sponsor’s reimbursement request (i.e., for the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone). CADTH notes reanalyses 1 through 3 have a minor impact on the budgetary impact. The budgetary impact can be largely attributed to somapacitan displacing somatropin at a cost and dose reflective of Canadian clinical practice.
Table 18: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | −122 |
CADTH reanalysis 1: Include drug wastage | −22,260 |
CADTH reanalysis 2a: Public drug plan coverage | −125 |
CADTH reanalysis 2b: NIHB and Ontario population | −120 |
CADTH reanalysis 3: Somatrogon market shares | −122 |
CADTH reanalysis 4: Comparator displacement | −184,853 |
CADTH reanalysis 5: Daily dose for somatropin treatments | −122 |
CADTH reanalysis 6: Somapacitan market uptake | −76 |
CADTH reanalysis 7: (1 + 2a + 2b + 3) | −22,379 |
CADTH reanalysis 8: (1 + 2a + 2b + 3 + 4) | −512,818 |
CADTH base case (1 + 2a + 2b + 3 + 4 + 5 + 6) | 458,079 |
BIA = budget impact analysis; NIHB = Non-Insured Health Benefits.
Note: CADTH reanalyses are based on publicly available prices of comparator treatments and do not reflect confidential, negotiated prices.
CADTH conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 19.
Assuming somapacitan market uptake reflects the sponsor’s estimates of 8.75%, 22.5%, and 41.25% in year 1, year 2, and year 3, respectively.
Assuming that the price of somapacitan is reduced by 13.1%, the price reduction at which somapacitan would be considered cost-effective at a WTP threshold of $50,000 per QALY in the CADTH reanalysis of the cost-utility analysis (refer to Table 7).
Table 19: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 8,338,946 | 8,297,051 | 8,279,872 | 8,262,422 | 24,839,345 |
New drug | 8,338,946 | 8,297,036 | 8,279,834 | 8,262,353 | 24,839,223 | |
Budget impact | 0 | −15 | −38 | −70 | −122 | |
CADTH base case | Reference | 6,903,474 | 6,968,175 | 7,020,872 | 7,073,879 | 21,062,926 |
New drug | 6,903,474 | 7,020,626 | 7,149,419 | 7,350,960 | 21,521,005 | |
Budget impact | 0 | 52,450 | 128,547 | 277,081 | 458,079 | |
CADTH scenario analysis: somapacitan market uptake | Reference | 6,903,474 | 6,968,175 | 7,020,872 | 7,073,879 | 21,062,926 |
New drug | 6,903,474 | 7,059,963 | 7,252,257 | 7,489,501 | 21,801,721 | |
Budget impact | 0 | 91,788 | 231,385 | 415,622 | 738,795 | |
CADTH sensitivity analysis 13.1% price reduction | Reference | 6,903,474 | 6,968,175 | 7,020,872 | 7,073,879 | 21,062,926 |
New drug | 6,903,474 | 6,968,113 | 7,017,613 | 7,059,826 | 21,045,552 | |
Budget impact | 0 | −62 | −3,259 | −14,053 | −17,374 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Canadian Organization for Rare Disorders
About Canadian Organization for Rare Disorders
The Canadian Organization for Rare Disorders (CORD) is Canada’s national network for organizations representing all those with rare disorders. CORD provides a strong common voice to advocate for health policy and a healthcare system that works for those with rare disorders. The Canadian Organization for Rare Disorders works with governments, researchers, clinicians and industry to promote research, diagnosis, treatment and services for all rare disorders in Canada.
Website: www.raredisorders.ca
Information Gathering
Recruitment: Participants were recruited through two sources, one local and one international. An outreach was made through the CORD patient membership list requesting parents of children with Growth Hormone Deficiency (GHD) to participate in an interview about their experience of living with GHD and any therapies they have used.
Respondents were asked to indicate: (1) whether their child or children had been diagnosed with GHD; (2) whether they had been prescribed any form of Growth Hormone Therapy (GHT); (3) whether their child or children were currently taking GHT; and (4) whether they would be willing to participate in a brief interview about a new form of GHT. They were also encouraged to send the request to any other parents that might be interested. Ten parents responded to the outreach and six subsequently participated in an interview.
The second outreach was to parents attending a European summit for patients with several different types of rare diseases, including conditions or syndromes affecting growth and stature. Among the volunteers, we recruited six who were parents of children with Growth Hormone Deficiency. Parents were individually contacted by email and provided with the interview questions. Interviews were conducted by telephone, zoom or WhatsApp. All interviews were conducted in English and each lasted between 20 and 30 minutes. The interviews were all conducted by the same interviewer who took notes during the conversation. None of the interviews were recorded.
The children represented ranged in age from four to 15 years old.
Four of the parents and patients resided in Ontario. Two families lived in the United States, and six lived in Europe (Sweden, Germany, France, United Kingdom), Eight of the patients were female and four male. All patients represented were diagnosed with Growth Hormone Deficiency.
Disease Experience
Parents relayed various experiences toward getting a diagnosis and living with GHD. Overall, they reported going through a variety of emotions, including denial, blame, sadness, acceptance, and compassion.
“Up until she was three years old, she was just like a “model” baby, meeting every milestone physically, cognitively, and socially. I am not sure when her growth started to slow down, but we suddenly realized she was measuring in the lower percentile for her age group. Her pediatrician initially said it was nothing to worry about; kids go through slow and rapid growth periods. But she ordered some blood tests just to rule out any underlying causes. That was the first time we learned about the possibility of growth hormone deficiency, which the doctor said was very rare, about 1 in 10,000 children, so nothing to worry about. We went home, looked up everything we could about “GHD”; because it was a genetic condition and we knew no one in our families who had it, we convinced ourselves it was not likely. It was only after she ordered a number of other tests, including an MRI, that we started to get worried. And then we got news of the unlikely. And then we had to decide what to do.”
“Ian was our second child. Our first was five when he was diagnosed with [Growth Hormone Deficiency]. So, when Ian’s growth slowed down at about 18 months, we had him tested right away. We were already prepared and even before we got the results, we asked. “When should we start treatment?”
“My grandmother was very short so we weren’t too concerned by the fact that Ann-Lise was small but when she turned 13 and showed no signs of puberty, we asked our family doctor whether we should be concerned. He recommended some blood tests and then x-rays and MRI. He referred us to a pediatric endocrinologist who gave us a diagnosis of Growth Hormone Deficiency. Frankly, we were still not convinced that “being short” was a medical condition that needed to be treated. That’s when he explained what it really means to be “hormone” deficient and the other problems that can happen when she gets older, like heart disease and bone disease.”
Many parents are not prepared for a diagnosis of GHD as a serious hormonal, or endocrinological, disease. Some said that they initially did not want to stigmatize their child with diagnosis of a medical condition and were also afraid it would impact their future, for example, ability to get employment or insurance. According to one mother, “We thought of being short as just being at one end of a physical spectrum like being tall or heavy or near-sighted.”
When asked, parents did report a variety of psychological and social impacts on the child and the family. “Being a boy, it was harder to be shorter than his sister and the other girls in his class.” “He was teased a lot, not just because of his size but also because he was timid and not very good in sports.”
“Because of her size and the fact that she looked a lot younger, she was always treated as if she were a small child, which was very frustrating for her. She just didn’t feel like she fit in with others, which caused her to become depressed and even more isolated.”
Experiences With Currently Available Treatments
All of the children represented have been taking Growth Hormone Therapy for a while, from two to 10 years. All had experience with daily sub-cutaneous injections of somatropin (several brand names) and four patients have (current) experience with long-acting GHD (somatrogon, lomapegsomatropin, and somapacitan-beco). The families’ experience with the standard daily therapy can be summarized into xx themes.
Why Treat?
According to some parents, treating with GHT was a “no brainer” while others expressed considerable caution, ambivalence, and/or anxiety over the decision to administer hormone therapy.
“His doctor said the hormones were synthetic but worked just like natural protein; it has been used for many years and was very safe. But we still weren’t convinced until we spoke to some other parents who said they had no regrets and were very pleased with the results.”
“We didn’t want to make a decision that would require her to do this [injections] for the rest of her life. We asked whether it was necessary, and her doctor said it wasn’t absolutely essential but highly recommended by experts.”
“We didn’t feel that being short was a disability, but we were also told that hormone therapy wasn’t just about height but also protecting other organs. For example, without treatment, patients are at greater risk for heart disease and bone fractures. That shifted our thinking about the balance of benefits and risks to choosing hormone therapy.”
“We asked if we could wait until he was older, but our pediatrician showed us the research; the younger the child was when started, the more he or she would gain in additional height; some who started very young were close to normal height.”
Challenges of Administration
Perhaps the most consistent challenge, beyond any adverse events, was the challenge of daily administration injection every night.
“It really breaks your heart; in the beginning we tried to catch him off guard, to do it at different times. But now we start to prepare for the injection about one hour in advance. We try to make it a ceremony or a “fun event” but it never gets easy. And you never get over the guilt.”
“It usually takes both of us to do it. Depending on his mood, one of us will try to distract him, bribe him, or even hold him and the other does the injection. We really feel it has an impact on how he feels about us as parents. It’s challenging and sad.”
“When she was nine years old, she decided she wanted to do it herself. We were worried but it has turned out to be the best thing that has happened. She takes the responsibility very seriously and is very proud.”
“We have never felt comfortable letting her stay over at a friend’s house or go to camp. We tried letting her stay with her grandparents, but it turned out to be a dismal failure. Maybe next year when she is older and able to do the injections herself.”
Benefits of Treatment
Overall, despite the challenges and worries about the future, parents recognized the value of growth hormone therapy.
“Within the first year of starting treatment, he seemed to grow more than he had the previous year. He also seemed to have more energy and was more out-going.”
Improved Outcomes
Parents were asked what would make treatment better or more acceptable, other than a permanent cure. Parents expressed changes they would like for their children: a pill instead of an injection, a powder that you could dissolve and just drink; an injection that lasted longer and was easier to administer. All of the parents were aware that there were new therapies that were a “once-a-week” injection, and some were aware that these were already or would soon be available in Canada.
“It would change our lives if we didn’t have to do an injection every night.”
“I think the relationship with our son would be much better, less fights and shorter bedtime routines.”
“It would mean she could do sleepovers,”
“We would feel a lot more confident letting him do his own injections. We worry about compliance as he gets older.”
“if they can make slow-release insulin, heart medication that you can take every two weeks, and a pill for psoriasis, even an HIV-treatment that you can take once a month (we see a lot of drug ads on American TV), surely they can make a growth hormone that lasts longer than one day. We’re talking about children and infants.”
Experience With Drug Under Review
Of the 12 respondents, four (one-third) had experience with a long-acting growth hormone therapy (somatrogon, lomapegsomatropin, and somapacitan-beco) through clinical trials, compassionate access or reimbursement/insurance. Two of the families lived in Canada and two others in the USA.
All parents were overwhelmingly positive about the impact on the child’s and the family’s quality of life.” Some even described the impact as “transformational” and ‘life changing’.”
“For the first time in years, since forever, bedtime was not a battleground or source of tension. We read a book, played a game, and turned out the light with no trauma.”
“When we were offered the new drug, we actually hesitated because we were worried it wouldn’t work as well. We did the research, talked to our doctor and decided it was worth the risk. Of course, we can’t really say whether the growth is or will be exactly the same, and maybe we will never know. What we do know is that our family lives are so much better, not just at bedtime but all the time. I guess we never realized how much stress there was around the injections.”
“Our ten-year old son, who has been on the therapy for five years, had never complained about the daily injection; in fact, he seemed to be more concerned about making us feel bad. In comparison to stories from other parents, we felt very lucky. We were pretty surprised, then, when we were given the chance to try the once-a-week drug, he was so eager to do it. And we were surprised by his reaction: ‘It’s like getting a birthday present every night.’ What he meant was that when he was younger, we used to offer to let him skip his injections on his birthday or Christmas. We hadn’t done that recently, but I guess he remembered.”
“We think every child with GHD should get access to this therapy. I realize it may not work better to improve height but it does make all our lives so much better. And it will allow children to stay on therapy.”
“We know families that have broken up over this condition; I think the daily injection was as much to blame as the condition itself. It really feels sometimes like that saying, “sometimes, the cure is worse than the disease.” Not really, but sometimes you just wish you could stop or take a break and you feel you can’t.”
Companion Diagnostic Test
There are no additional tests for the long-acting therapy except for regular testing to measure hormone levels, other biomarkers, and growth, but these are done regularly as part of routine monitoring regardless of which form of the drug.
Anything Else?
It is tempting to dismiss a change in formulation to allow for less frequent dosing, from daily to once a week, as not that important. But clearly to the patients and families, the change constitutes a very important improvement. We cannot minimalize the impact on the psychological well-being and the mental health of the child and the family. Healthcare should not inflict pain and trauma when there are options to lessen these adverse effects. Moreover, as we have realized with other conditions, a weekly injection will have a significant benefit in improving adherence among children and adolescents, will improve self-reliance and self-respect, and will inevitably lead to better health outcomes.
Conflict of Interest Declaration — Canadian Organization for Rare Disorders
Did you receive help from outside your patient group to complete this submission?
No outside help was received to complete the submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No outside help was received to analyze data.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Canadian Organization for Rare Disorders
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novo Nordisk | X | — | — | — |
The MAGIC Foundation
About The MAGIC Foundation
The MAGIC Foundation is a charitable non-profit organization created to provide support services for the families of children afflicted with a wide variety of chronic and/or critical disorders, syndromes, and diseases that affect a child’s growth. The mission is to reduce the emotional and physical trauma cause by growth disorders, resulting in healthier, happier children and consequently, adults.
Information Gathering
Data was collected using surveys distributed to our growth hormone deficiency population of members. All members who responded were form the United States. None of our current Canadian members have had experience with long-acting growth hormone. All participants had children between the ages of 3-18 years old who are currently on growth hormone. Some have been on both daily and weekly injections; others have been strictly on daily injections. Families surveyed have between 1-4 children in their household diagnosed with growth hormone deficiency.
Disease Experience
Parents have submitted that their children were shorter than their peers, fatigued, lacking concentration, lack of appetite, poor stamina, and were often very sick before beginning growth hormone treatment. Often psychological issues came about from bullying due to short stature.
“Our son was sick nearly every day and since being on GH he is healthy, fights off infection well, and it has improved his balance, coordination and muscle tone. Height is an added bonus which, by the way took our son from the 1% for height and brought him up to 85% in 2.5 hrs. Our son also has pulmonary valve stenosis and has had a balloon valvuplasty and has not needed intervention again since being on GH. GH does so much more than give height, it creates cell growth within all areas of the body effecting all organs and giving such an amazing quality of life. It's liquid gold in our family and we are so incredibly happy that our son has access to GH and has it covered with insurance.”
“He began treatment at 14.75 and his appetite and stamina approved within a month of beginning treatment. As he grew in height and confidence, his smile and laughter returned. He is now 18 and nearly at the end of his treatment. He is a competitive boxer, has a solid friend group, and no longer socially isolates. I attribute growth hormone treatment to both his physical and emotional health now.”
Experiences With Currently Available Treatments
Due to the high cost of growth hormone treatment, families are often at the mercy of their insurance companies as to whether or not treatment is possible for children in the United States. When denied the treatment, symptoms worsen, and the psychological difficulties prevail.
Daily injections are burdensome to children’s social lives, and while treatments have had positive outcomes, most families who have been able to switch to the long-acting growth hormone, have had better adherence to treatment, producing better results. Medications that need to be refrigerated have also cause challenges for certain lifestyles.
"My kids just moved to weekly shots, and it is life changing. Lots less stress."
"As explained above it gives an overall well being and creates and promotes a healthy body overall. Our son hasn't had side effects with the exception of occasional site bruising and swelling."- On daily injections.
"It’s nice to have once shots a week. They feel less like medical kids having weekly shots v daily shots."- On weekly injections.
Conflict of Interest Declaration — The MAGIC Foundation
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures for The MAGIC Foundation
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novo Nordisk | — | — | — | X |
Pfizer | — | — | — | X |
Ascendis | — | — | — | X |
Clinician Input
Pediatric Endocrinology Nurses
About Pediatric Endocrinology Nurses
We are a group of pediatric Endocrinology nurses reviewing this medication.
Information Gathering
We reviewed:
Once-Weekly Somapacitan vs Daily GH in Children With GH Deficiency: Results From a Randomized Phase 2 Trial Lars Sävendahl,1 Tadej Battelino,2 Meryl Brod,3 Michael Højby Rasmussen,4 Reiko Horikawa,5 Rasmus Vestergaard Juul,4 and Paul Saenger6 on behalf of the REAL 3 study group*
Effective GH Replacement With Once-weekly Somapacitan vs Daily GH in Children With GHD: 3-year Results From REAL 3 Lars Sävendahl,1, Tadej Battelino,2, Michael Højby Rasmussen,3 Meryl Brod,4 Paul Saenger,5 and Reiko Horikawa6
Weekly Somapacitan is Effective and Well Tolerated in Children With GH Deficiency: The Randomized Phase 3 REAL4 Trial Bradley S. Miller,1 Joanne C. Blair,2 Michael Højby Rasmussen,3 Aristides Maniatis,4 Rasmus Juul Kildemoes,3 Jun Mori,5 Michel Polak,6 Rikke Beck Bang,7 Volker Böttcher,8 Stefano Stagi,9 and Reiko Horikawa10
Current Treatments and Treatment Goals
Daily Somatropin (growth hormone) injections
Used to increase growth, stabilize blood sugar levels, increase bone density and muscle development
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Poor compliance in patients with daily injections
Availability of growth hormone
Anxiety with daily injections
Treatments are needed that are better tolerated
Treatments are needed to improve compliance
Formulations are needed to improve convenience.
Please describe limitations associated with current treatments (e.g., adverse events, administration, etc., if applicable).
Refer to above.
Place in Therapy
How would the drug under review fit into the current treatment paradigm? Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments?
No.
Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy?
No.
Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment?
As a first line treatment if approved/funded.
Would the drug under review be reserved for patients who are intolerant to other treatments or in whom other treatments are contraindicated?
Refer to above.
Is the drug under review expected to cause a shift in the current treatment paradigm?
Yes, if funded.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with drug under review. Please provide a rationale for your perspective.
No, it would be offered to patients and the patient/family would decide if this were best for them.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Which patients are most likely to respond to treatment with drug under review?
Patients who experience the following:
Needle anxiety
compliance issues
complex social situation
remote living circumstances.
Which patients are most in need of an intervention?
Refer to above.
Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
No.
How would patients best suited for treatment with drug under review be identified (e.g., clinician examination/judgement, laboratory tests (specify), diagnostic tools (specify)).
If found to be growth hormone deficient (clinician examination, growth hormone stimulation testing, bone age, IGF-1 etc.) family would be offered this drug.
Are there any issues related to diagnosis?
Yes, without growth hormone (either daily or weekly) patients will not grow, could have hypoglycemia, decreased bone density, poor muscle development, and/or altered body composition.
Is a companion diagnostic test required?
Refer to above.
Is it likely that misdiagnosis occurs in clinical practice (e.g., under diagnosis)?
Unlikely.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with drug under review?
Yes.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed? Are outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Yes.
What would be considered a clinically meaningful response to treatment? Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Improved growth velocity
Normalized glucose in infants
No difference between physicians.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Until final adult height achieved
Closed epiphyses
Growth rate < 2 cm/year.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Must be diagnosed, treated, monitored and prescribed by a pediatric endocrinologist.
Additional Information
It is beneficial to patients and families to have the option of a once weekly growth hormone injection.
Conflict of Interest Declarations — Pediatric Endocrinology Nurses
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Yes. We requested the studies from Novo Nordisk but analyzed the information ourselves.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Table 3: Financial Disclosures for Pediatric Endocrinology Nurses
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novo Nordisk | X | — | — | — |
Sandoz | X | — | — | — |
EMD Serono | X | — | — | — |
Pfizer | X | — | — | — |
Ipsen | X | — | — | — |
Declaration for Clinician 1
Name: Kristen Langdon
Position: Pediatric Endocrine Nurse
Date: 27/04/2023
Table 4: COI Declaration for Pediatric Endocrinology Nurses — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novo Nordisk | X | — | — | — |
Sandoz | X | — | — | — |
EMD Serono | X | — | — | — |
Pfizer | X | — | — | — |
Ipsen | X | — | — | — |
Declaration for Clinician 2
Name: Brenda Fraser
Position: Pediatric Endocrine Nurse
Date: 27/04/2023
Table 5: COI Declaration for Pediatric Endocrinology Nurses — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novo Nordisk | X | — | — | — |
Sandoz | X | — | — | — |
EMD Serono | X | — | — | — |
Pfizer | X | — | — | — |
Ipsen | X | — | — | — |
Declaration for Clinician 3
Name: Julianne McKernan
Position: Pediatric Endocrine Nurse
Date: 27/04/2023
Table 6: COI Declaration for Pediatric Endocrinology Nurses — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novo Nordisk | X | — | — | — |
Sandoz | X | — | — | — |
EMD Serono | X | — | — | — |
Pfizer | X | — | — | — |
Ipsen | X | — | — | — |
Declaration for Clinician 4
Name: Carmen Lin Carriere
Position: Pediatric Endocrine Nurse
Date: 27/04/2023
Table 7: COI Declaration for Pediatric Endocrinology Nurses — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novo Nordisk | X | — | — | — |
Sandoz | X | — | — | — |
EMD Serono | X | — | — | — |
Pfizer | X | — | — | — |
Ipsen | X | — | — | — |
Declaration for Clinician 5
Name: Janette Rees
Position: Pediatric Endocrine Nurse
Date: 27/04/2023
Table 8: COI Declaration for Pediatric Endocrinology Nurses — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novo Nordisk | X | — | — | — |
Sandoz | X | — | — | — |
EMD Serono | X | — | — | — |
Pfizer | X | — | — | — |
Ipsen | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.