CADTH Reimbursement Review
Elexacaftor-Tezacaftor-Ivacaftor and Ivacaftor (Trikafta)
Sponsor: Vertex Pharmaceuticals (Canada) Incorporated
Therapeutic area: Cystic fibrosis, F508del CFTR mutation, 2 to 5 years
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ALT
alanine transaminase
AST
aspartate transaminase
BMI
body mass index
CF
cystic fibrosis
CF CanACT
Cystic Fibrosis Canada’s Accelerating Clinical Trials Network
CF Canada
Cystic Fibrosis Canada
CFQ-R
Cystic Fibrosis Questionnaire–Revised
CI
confidence interval
ELX
elexacaftor
ELX-TEZ-IVA
elexacaftor-tezacaftor-ivacaftor and ivacaftor
FAS
full analysis set
FEV1
forced expiratory volume in the first second
F/F
homozygous for F508del mutation in the CFTR gene
F/G
1F508del mutation and 1 gating mutation in the CFTR gene
F/MF
1 F508del mutation and 1 minimal function mutation in the CFTR gene
F/R117H
1 F508del mutation and 1 R117H mutation in the CFTR gene
F/RF
1 F508del mutation and 1 residual function mutation in the CFTR gene
ITC
indirect treatment comparison
IVA
ivacaftor
MMRM
mixed-effects model for repeated measures
LCI
lung clearance index
LS
least squares
LUM-IVA
lumacaftor-ivacaftor
ppFEV1
percent predicted forced expiratory volume in the first second
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SwCl
sweat chloride
TEZ
tezacaftor
TEZ-IVA
tezacaftor-ivacaftor and ivacaftor
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Elexacaftor-tezacaftor-ivacaftor and ivacaftor (Trikafta):
Patients aged 6 years and older:
|
Sponsor | Vertex Pharmaceuticals (Canada) Incorporated |
Indication | For the treatment of cystic fibrosis (CF) in patients aged 2 years and oldera who have at least 1 F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene |
Reimbursement request | Initiation criteria:
Initial renewal criteria:
Timing for initial renewal assessment: Sponsor requests that the initial renewal criteria be extended from 6 months to 12 months Subsequent renewals annually: The physician must provide evidence of continued treatment benefit with elexacaftor-tezacaftor-ivacaftor and ivacaftor for subsequent renewal of reimbursement |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | October 16, 2023 |
Recommended dose | Patients aged 2 to < 6 years:
|
NOC = Notice of Compliance.
aThis review focuses on patients aged 2 to 5 years.
Introduction
Trikafta is a fixed-dose combination product containing elexacaftor (ELX), tezacaftor (TEZ), and ivacaftor (IVA) co-packaged with IVA (ELX-TEZ-IVA). ELX-TEZ-IVA is available as both oral tablets and oral granules in the following dosing strengths:
Tablets for patients aged 6 years and older:
ELX 50 mg, TEZ 25 mg, and IVA 37.5 mg, co-packaged with a tablet containing IVA 75 mg
ELX 100 mg, TEZ 50 mg, and IVA 75 mg, co-packaged with a tablet containing IVA 150 mg
Granules for patients aged 2 to younger than 6 years:
ELX 100 mg, TEZ 50 mg, and IVA 75 mg (granules) and IVA 75 mg (granules)
ELX 80 mg, TEZ 40 mg, and IVA 60 mg (granules) and IVA 59.5 mg (granules), orally
ELX-TEZ-IVA is indicated for the treatment of cystic fibrosis (CF) in patients aged 2 years and older who have at least 1 F508del mutation in the CFTR gene. A deletion of phenylalanine 508 in the first nucleotide-binding domain (F508del) is the most common mutation in the CFTR gene that results in CF.1 The Canadian Cystic Fibrosis Registry reported 4,344 patients in Canada living with CF in 2019. Of these patients, 87.8% carried at least 1 F508del mutation (47.1% were homozygous, and 40.7% were heterozygous).
This is the third submission to CADTH for ELX-TEZ-IVA. CADTH has previously reviewed ELX-TEZ-IVA for the treatment of CF in patients who have at least 1 F508del mutation in the CFTR gene for those aged 12 years and older (final recommendation issued in August 2021) and those aged 6 years and older (final recommendation issued in June 2022). For both of the previous reviews, the CADTH Canadian Drug Expert Committee recommended that ELX-TEZ-IVA be reimbursed with conditions.2,3 All the indications for ELX-TEZ-IVA were accepted as priority reviews by Health Canada.
The sponsor has requested that the current submission for ELX-TEZ-IVA focus only on those patients aged 2 to 5 years using the new dosage format (i.e., orally administered granules).
The objective of this review is to evaluate the beneficial and harmful effects of ELX-TEZ-IVA at recommended dosages for the treatment of patients aged 2 to 5 years with CF who have at least 1 F508del mutation in the CFTR gene.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from clinical experts consulted by CADTH for the purpose of this review. Complete patient and clinician input received for the current review of ELX-TEZ-IVA is reported in the appendix of this report. The complete input received for the previous CADTH reviews of ELX-TEZ-IVA is available on the CADTH website (refer to reviews for ELX-TEZ-IVA for patients aged 6 to 11 years and 12 years and older).
Patient Input
One patient group, Cystic Fibrosis Canada (CF Canada), responded to CADTH’s call for patient input for the current review of ELX-TEZ-IVA, which is focused on patients aged 2 to 5 years who have at least 1 F508del mutation in the CFTR gene.
The patient group emphasized that CF tremendously impacts those living with the condition, their loved ones, health systems, and society. The most significant clinical impact is in the lungs, where patients experience progressive scarring of their airways and a progressive decline in lung function. Young children who grow older with CF may experience pulmonary exacerbations requiring weeks to months of hospitalization and IV antibiotics. Malnutrition and low body mass index (BMI) are also common consequences of CF among children aged 2 to 5 years. Patients may also experience CF-related comorbidities, such as CF-related diabetes and CF-related liver disease. In addition, CF has a significant impact on socialization, mental health, and isolation among patients and caregivers.
The patient input stated that managing CF requires a demanding treatment routine. As the disease progresses, more time and effort and frequent clinic visits and hospital stays are needed to manage the progressive and debilitating symptoms. This condition has a significant impact on patients’ and caregivers’ day-to-day activities and quality of life, in addition to creating a huge financial burden for families.
According to the patient group input, an ideal treatment for CF would fully address the basic molecular defect in CF and restore normal chloride transport on the cell surface. Patients with CF and their loved ones are seeking treatments that can change the trajectory of the disease, reduce disease symptoms, improve sleep quality and energy levels, and improve both life expectancy and quality of life.
In the patient group input, CF Canada’s Accelerating Clinical Trials Network (CF CanACT) emphasized the importance of early treatment of CF to prevent disease progression and irreversible damage. Extending access to ELX-TEZ-IVA for patients with CF aged 2 to 5 years would be congruent with the secondary prevention paradigm of CF care and would decrease the long-term burden of the disease.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Unmet Needs
Similar to the input from the patient group, the clinical experts consulted by CADTH indicated that there are significant unmet therapeutic needs for patients living with CF. There are no treatments currently available that can meet the most important goals of therapy, which include prolonging survival, preventing the need for lung transplant, slowing the decline in lung function over time, and reversing the course of the disease. In addition, the clinical experts noted that the current standard treatments for CF are burdensome for patients and their caregivers.
Place in Therapy
The clinical experts anticipate that ELX-TEZ-IVA would be used as a preventive therapy, with the goal of initiating treatment before the patient develops significant lung disease. The current treatment paradigm would be significantly altered if ELX-TEZ-IVA can successfully prevent or delay progression to end organ disease (e.g., lung transplant). The clinical experts consulted by CADTH and those who responded to the call for clinician input noted that children aged between 2 and 5 years will often have structural lung disease (e.g., bronchial wall thickening, mucus plugging, or bronchiectasis) but that detection is challenging using the tools available to evaluate lung function in clinical practice (i.e., spirometry) or as part of a research protocol (e.g., lung clearance index [LCI]). These early stages of lung abnormalities can be visualized using CT; therefore, despite younger patients with CF often demonstrating normal lung function, the underlying disease will continue to progress.
All the clinicians who provided input for this review recommended initiating treatment with ELX-TEZ-IVA as soon as possible. This recommendation is aligned with the previously published Canadian Clinical Consensus Guideline for Initiation, Monitoring and Discontinuation of CFTR Modulator Therapies for Patients With Cystic Fibrosis, which also recommends that CFTR modulators be initiated at the youngest age possible, with the goal of attenuating disease progression and improving clinical status. All stakeholders agreed that there are no data to support withholding the initiation of CFTR modulator treatment until clinical symptoms of CF have developed.
Patient Population
For the expanded indication (i.e., patients aged 2 to 5 years), the clinical experts consulted by CADTH noted that nearly all patients would initiate therapy with ELX-TEZ-IVA as soon as possible, provided it is safe to start to treatment. The clinical experts emphasized that ELX-TEZ-IVA has been a transformative and disease-modifying therapy for CF and that it would not be appropriate to wait until the patient shows worsening symptoms, more frequent exacerbations, or a decline in lung function to initiate treatment with ELX-TEZ-IVA.
Applicability of Existing Reimbursement Criteria to Pediatric Patients
In discussions with CADTH, the sponsor noted that nearly all patients aged 12 years and older living in Canada who are eligible for treatment have begun therapy with ELX-TEZ-IVA (some may have elected to discontinue, but all who are interested have been given the opportunity to access the drug). The sponsor similarly stated that all patients aged 6 to 11 years living in Canada who wish to initiate treatment will have begun treatment with ELX-TEZ-IVA by the end of 2023. For those who have initiated treatment with ELX-TEZ-IVA, the sponsor noted that initial renewal criteria had been met for all patients living in Canada who had started the therapy and wanted to continue (i.e., 100% of patients met the renewal criteria recommended by CADTH and/or applied by the public drug programs). The clinical experts consulted expressed general agreement with the sponsor’s position, noting that rates of initial access and renewal are very high within their individual clinics. With nearly all patients aged 6 years or older having now met the initiation and renewal criteria, newly issued CADTH reimbursement criteria focusing exclusively on patients aged 2 to 5 years would effectively replace the previous criteria (i.e., although limited to patients aged 2 to 5 years, all older patients would have already qualified for initiation and renewal).
The clinical experts consulted by CADTH reviewed the existing criteria recommended for patients aged 6 years and older and noted the following:
Baseline measurements: Regarding the baseline measurements that must be completed before initiating treatment with ELX-TEZ-IVA, the clinical experts noted that the following baseline measurements that are currently recommended by CADTH would be problematic to implement, uninformative, and/or not relevant for patients aged 2 to 5 years: baseline forced expiratory volume in the first second (FEV1) (spirometry is not performed in patients younger than 6 years); baseline frequency of pulmonary exacerbations (exacerbations can be infrequent, and it would be challenging establish a reliable baseline); and Cystic Fibrosis Questionnaire–Revised (CFQ-R) respiratory domain scores, which are not routinely obtained for patients in pediatric clinics (typically only when conducting research studies). The clinical experts noted that patients aged 2 to 5 years would have growth parameters monitored in routine clinical practice. However, it was noted that a majority of patients with CF aged 2 to 5 years do not show reductions in age-standardized BMI and that BMI percentile can fluctuate in younger patients, especially following periods of acute illness.
Renewal criteria: Each of the end points are discussed subsequently, with reflection on the applicability of the existing CADTH criteria to the expanded patient population aged 2 to 5 years:
BMI and BMI z scores: The existing criterion is “no decline in BMI (BMI z score in children) at 6 months compared with the baseline BMI assessment.” The clinical experts noted that 6 months is not sufficient to accurately assess the response to treatment and that an assessment of BMI at 12 months would be more appropriate. The longer time was suggested to account for events that could temporarily reduce BMI (e.g., increased physical activity in summer months and growth spurts). It was strongly noted that discontinuation of ELX-TEZ-IVA in such patients would not be clinically appropriate.
Pulmonary exacerbations: The existing criterion is “a decrease in the total number of days for which the patient received treatment with oral and/or IV antibiotics for pulmonary exacerbations compared with the 6-month period prior to initiating treatment OR a decrease in the total number of pulmonary exacerbations requiring oral and/or IV antibiotics compared with the 6-month period prior to initiating treatment.” The clinical experts indicated that pulmonary exacerbations are less frequent in patients aged 2 to 5 years compared with adults and adolescents. The clinical experts suggested that the above renewal criterion would be problematic for patients aged 2 to 5 years. However, it was emphasized that patients who have not experienced a pulmonary exacerbation or those with a very low annual rate of pulmonary exacerbations would still benefit from the treatment. Similar to the criterion for BMI, it was noted that 12 months would be a more appropriate time frame for evaluating changes in pulmonary exacerbations.
CF-related hospitalizations: The existing criterion is “decreased number of CF-related hospitalizations at 6 months compared with the 6-month period prior to initiating ELX-TEZ-IVA treatment.” The clinical experts consulted by CADTH noted that CF-related hospitalization is infrequent and highly variable in patients aged 2 to 5 years. As such, this would be very challenging to implement as a criterion for evaluate response to ELX-TEZ-IVA for the purposes of reimbursement.
Sweat chloride (SwCl) testing: The previous CADTH recommendation did not include SwCl testing as one of the initiation or renewal conditions for ELX-TEZ-IVA. The sponsor has requested that “reduction in sweat chloride” be included as a reimbursement condition for ELX-TEZ-IVA in the current review. The pediatric clinical experts agreed with the prior input from the reviews of ELX-TEZ-IVA in patients aged 6 to 11 years and 12 years and older, noting that SwCl testing should not be used to evaluate the response to ELX-TEZ-IVA for the purposes of drug reimbursement because it is not clearly predictive of clinically important outcomes and only reflects the mechanism of action of CFTR modulators like ELX-TEZ-IVA. The clinical experts also noted that access to SwCl testing can be challenging in some jurisdictions; the timelines to receive the test results can fluctuate; and raised important concerns about the capacity of the health system to accommodate repeated SwCl testing for all patients with at least 1 F508del mutation.
Clinician Group Input
Three groups of clinicians responded to CADTH’s call for input: CF CanACT, the CF Canada Healthcare Advisory Council, and the Canadian Cystic Fibrosis Clinician groups. The input from the clinician groups identified the same unmet medical needs for patients with CF and potential place in therapy for the drug under review as the clinical experts consulted by CADTH.
According to the clinician groups’ input, the treatment paradigm for CF in children aged 2 to 5 years is lifelong. All clinician groups noted that available treatments address the symptoms and complications of CF and attempt to slow down the eventual fatal progression of the disease without effectively addressing the root cause or reversing the course of the disease. The treatments also have significant side effects and numerous drug interactions. The clinician groups emphasized that ELX-TEZ-IVA is the most effective improvement of the existing CFTR modulators as it addresses the underlying disease process, which helps in delaying disease progression and the need for other therapies, including lung transplant. Therefore, any patient with CF who has at least 1 copy of F508del could potentially benefit from ELX-TEZ-IVA.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review processes. The following were identified as key factors that could impact the implementation of a CADTH recommendation for ELX-TEZ-IVA for patients aged 2 to 5 years:
Lack of availability of multiple breath washout testing in most Canadian CF clinics
Potential challenges with identifying objective reimbursement criteria for patients aged 2 to 5 years
The clinical experts consulted by CADTH provided advice on the potential implementation issues raised by the drug programs (refer to the Drug Program Input section).
Clinical Evidence
Systematic Review
Description of Study
The evidence identified in the current review of ELX-TEZ-IVA that addressed the expanded patient population (i.e., those aged 2 to 5 years) included Study 111, a 24-week, open-label, phase III, nonrandomized, single-arm, 2-part (A and B) study. Study 111 was conducted in 2 parts:
Part A (N = 18) consisted of a 15-day treatment period conducted to evaluate the pharmacokinetics and the safety and tolerability of ELX-TEZ-IVA.
Part B (N = 75) consisted of a 24-week treatment period conducted to assess safety and tolerability (primary objective) and pharmacokinetics, pharmacodynamics, and efficacy (secondary objective).
Patients were eligible to be included in Study 111 if they had received a diagnosis of CF and were aged 2 to 5 years (inclusive). All patients had an F508del-CFTR mutation and 1 of the following genotypes: 1 F508del mutation and 1 minimal function mutation in the CFTR gene (F/MF) (69.3%) or homozygous for F508del mutation in the CFTR gene (F/F) (30.7%). Patients were excluded from the study if they had any comorbidities that could impact treatment outcomes or if they had received a prior hematological or solid organ transplant. The trial excluded patients with a history of colonization with Burkholderia cenocepacia, Burkholderia dolosa, and/or Mycobacterium abscessus. Patients were also considered to be ineligible if they reported an acute upper or lower respiratory infection, a pulmonary exacerbation, or changes in therapy (including antibiotics) for pulmonary disease within 4 weeks before the first dose of the study drug. Patients with a history of solid organ or hematological transplant were excluded, as were patients with abnormal laboratory values (e.g., hemoglobin < 10 g/dL), abnormal liver function, or abnormal renal function.
Safety and tolerability were the primary end points in Study 111. The secondary end points were absolute change from baseline in SwCl through 24 weeks and absolute change from baseline in LCI2.5. Changes from baseline in growth parameters (BMI, BMI z score, weight, weight z score, height, and height z score) were evaluated as additional efficacy end points, but no statistical analyses were conducted. Descriptive statistics were provided for pulmonary exacerbations and CF-related hospitalizations in Study 111. LCI2.5 was only evaluated in patients aged at least 3 years at the time of screening (n = 50).
Efficacy Results
Treatment with ELX-TEZ-IVA resulted in a within-group improvement (reduction) in SwCl from baseline through 24 weeks. The least squares (LS) mean absolute change was –57.9 mmol/L (95% confidence interval [CI], –61.3 mmol/L to –54.6 mmol/L; nominal P < 0.0001). The reduction from baseline was observed at all postbaseline assessments (i.e., weeks 4, 12, and 24). The results for the subgroup analyses based on CFTR genotype were –70.0 mmol/L (95% CI, –75.4 mmol/L to –64.5 mmol/L) and –52.6 mmol/L (95% CI, –56.9 mmol/L to –48.4 mmol/L) in the F/F and F/MF groups, respectively.
Among those patients who were assessed, treatment with ELX-TEZ-IVA resulted in an improvement (reduction) in LCI2.5 through 24 weeks. The within-group LS mean absolute change from baseline was –0.83 (95% CI, –1.01 to –0.66; nominal P < 0.0001). The reduction from baseline was observed at all postbaseline assessments (i.e., weeks 4, 12, and 24). The results were similar in the F/F and F/MF genotype subgroups: the LS mean change was –0.89 (95% CI, –1.15 to –0.63) and –0.82 (95% CI, –1.06 to –0.57), respectively.
Sixteen percent of patients experienced a pulmonary exacerbation event through 24 weeks (1 event each), with an annualized event rate of 0.32 per year. One patient experienced a pulmonary exacerbation that required hospitalization. There were no CF-related hospitalizations in Study 111.
The absolute change from baseline in growth end points at 24 weeks was 0.10 (95% CI, 0.00 to 0.20) for BMI z score, 0.02 (95% CI, –0.04 to 0.09) for body weight z score, and –0.06 (95% CI, –0.11 to 0.00) for height z score.
Harms Results
The overall percentage of patients who experienced at least 1 adverse event (AE) was 98.7% (nearly all were mild [62.7%] or moderate [36.0%] in severity), including cough (61.3%); increased alanine aminotransferase (ALT) (10.7%); rhinorrhea (33.3%); increased aspartate aminotransferase (AST) (5.3%); rash (16.0%); pyrexia (34.7%); vomiting (28.0%); COVID-19 (18.7%); nasal congestion (17.3%); upper respiratory tract infection (14.7%); decreased appetite (12.0%); and infective pulmonary exacerbation of CF (10.7%). Two patients (2.7%) experienced serious AEs (SAEs): 1 patient with anal incontinence, urinary incontinence, and abnormal behaviour, and 1 patient with an SAE of infective pulmonary exacerbation of CF. One patient (1.3%) discontinued treatment due to an SAE, and 5 patients (6.7%) had AEs leading to treatment interruption. For AEs of special interest were that 8 patients (10.7%) experienced elevated transaminase events and 15 patients (20.0%) experienced rash events (all events were mild or moderate in severity). Two patients experienced rash events leading to treatment interruption. There were no study discontinuations due to rash events or elevated transaminase events.
Critical Appraisal
Internal Validity
Study 111 was conducted in a manner similar to all other pivotal studies for the use of CFTR modulators in patients aged between 2 and 5 years (i.e., expansion of approval indications for Orkambi4,5 and Kalydeco6,7). Each of these studies was conducted in 2 parts, with Part A involving a small number of patients (n = 18 for Study 111), with a primary objective of evaluating pharmacokinetics, and Part B enrolling more patients (n = 75 for Study 111), with the primary objective of evaluating safety and tolerability. As with the other trials for CFTR modulators in patients aged 2 to 5 years, ELX-TEZ-IVA was administered in an open-label manner in Study 111, and there was no comparator group in either Part A or Part B. The limited number of secondary efficacy end points evaluated in the study were objective and unlikely to be influenced by the open-label administration of a CFTR modulator (i.e., change from baseline in SwCl concentration and change from baseline in LCI2.5).
Pulmonary exacerbations were only evaluated with descriptive statistics, and there were no prebaseline or postbaseline comparisons of event rates. In response to an inquiry from CADTH regarding why pulmonary exacerbations were not included as an efficacy end point, the sponsor reported that, as had been noted in relation to the pediatric trial for patients aged 6 to 11 years, exacerbations occur less frequently in younger patients than in older patients. As Study 111 was a single-arm trial without a defined pretreatment evaluation period, and due to the low pulmonary exacerbation rates in the study population, comparison to a pretreatment event rate was not possible.
External Validity
The eligibility and diagnostic criteria used to screen patients for Study 111 were similar to those used in the other phases of the ELX-TEZ-IVA clinical development program (i.e., Studies 106 and 116 for patients aged 6 to 11 years and Studies 102, 103, 104, and 109 for patients aged 12 years and older). As noted in the previous CADTH review of ELX-TEZ-IVA, these criteria are generally consistent with Canadian clinical practice for diagnosing patients with CF. As all Canadian provinces and territories have instituted newborn screening, diagnosis of CF and confirmation of genotyping would typically occur early in the child’s life (an average of 1 month after birth). As such, no changes would be needed in diagnostic testing requirements to establish patient eligibility based on CF diagnosis and genotype for the revised age range for ELX-TEZ-IVA.
The clinical experts consulted by CADTH noted that the baseline growth parameters for the patients in Study 111 were a reasonable reflection of the typical patient in Canadian practice.
Changes from baseline in lung function were evaluated as a secondary efficacy end point in Study 111 using LCI2.5. This is reflective of regulatory guidance, which has noted that spirometry may not be sensitive enough to detect treatment differences in children with CF. In addition, spirometry is not typically performed in patients younger than 6 years in Canada, and FEV1 has not been used as a clinical trial end point in any CFTR modulator studies for those younger than 6 years. LCI is used in CF clinical trials as it may be more sensitive in identifying early underlying structural deficiencies within the lungs of patients with CF that cannot be detected using spirometry.8,9 Similar to spirometry assessments, the LCI test can be challenging to accurately perform with young children. In Study 111, the sponsor noted the LCI test was only performed on patients who were at least aged 3 years at the time of screening. Although LCI is used as an end point in clinical studies, as noted previously it is not routinely used in Canadian clinical practice, and the clinical relevance of differences in this end point have not been characterized.9,10 The clinical experts consulted by CADTH indicated that LCI is not reliably correlated with FEV1. A literature review conducted by CADTH found that variable correlation was observed between FEV1 and LCI in children.
ELX-TEZ-IVA was added to the existing therapeutic regimens used by the patients, which is reflective of how ELX-TEZ-IVA would be administered in clinical practice. The clinical experts consulted by CADTH indicated that the background therapies used in Study 111 were similar to what would be anticipated in Canadian clinical practice, with the following exceptions: all patients in Canadian practice would be supplementing with vitamins, and the use of mucolytics (i.e., dornase alfa and inhaled hypertonic saline) could be slightly lower for patients aged 2 to 5 years in Canada.
The 24-week study treatment periods were sufficient for observing change from baseline in SwCl and LCI2.5 in Study 111; however, the clinical experts consulted by CADTH suggested that 24 weeks is unlikely to be enough time to observe meaningful changes in BMI for a younger patient population that is relatively healthy. In addition, the absence of a control group in Study 111 limits the ability to interpret the results of change from baseline in the growth parameters.
Table 2: Summary of Key Results From Study 111 Part B
End points | ELX-TEZ-IVA (N = 75) |
|---|---|
Change from baseline in SwCl (mmol/L) (n = 69) | |
Baseline, mean (SD) | 100.7 (11.2) |
Change from baseline, mean (95% CI) | –57.9 (–61.3 to –54.6) |
P value | < 0.0001 |
Change from baseline in LCI2.5 (n = 50) | |
Baseline, mean (SD) | 8.41 (1.48) |
Change from baseline, mean (95% CI) | –0.83 (–1.01 to –0.66) |
P value | < 0.0001 |
Pulmonary exacerbations (n = 75) | |
Patients with events, n (%) | 12 (16.0) |
Number of events | 12 |
Observed event rate per year | 0.32 |
P value | NR |
CF-related hospitalizations (n = 75) | |
Patients with events, n (%) | || || |||| |
P value | NR |
Change from baseline in BMI z score (n = 75) | |
Baseline, mean (SD) | 0.09 (0.85) |
Change from baseline, mean (95% CI) | 0.10 (0.00 to 0.20) |
P value | NR |
Summary of AEs, n (%) | |
≥ 1 AE | 74 (98.7) |
≥ 1 SAE | 2 (2.7) |
AE leading to treatment discontinuation | 1 (1.3) |
AE leading to interruption of treatment | 5 (6.7) |
AEs of special interest, n (%) | |
Elevated transaminase levels | 8 (10.7) |
Rash | 15 (20.0) |
AE = adverse event; BMI = body mass index; CF = cystic fibrosis; CI = confidence interval; LCI2.5 = lung clearance index 2.5; NR = not reported; SAE = serious adverse event; SD = standard deviation; SwCl = sweat chloride.
Source: Sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
Patients who completed Study 111 were eligible to enrol in an open-label extension study. However, the sponsor reported that the interim results of the extension study were not available at the time of filing the application with CADTH.
Indirect Comparisons
Feasibility of Indirect Treatment Comparison in Patients Aged 2 to 5 Years
The sponsor conducted an indirect treatment comparison (ITC) to compare the clinical efficacy of ELX-TEZ-IVA in Study 111 with other CFTR modulators in patients with F/F and F/MF mutations to generate the inputs needed for the cost-effectiveness analysis. A meta-analysis approach via mixed-effects model for repeated measures (MMRM) was used with individual patient-level data from relevant trials; data from all comparators were included in 1 model for each genotype. The sponsor concluded that the ITC was not feasible due to the small number of patients in this age group, which reduced the power to detect differences between ELX-TEZ-IVA, lumacaftor-ivacaftor (LUM-IVA), and/or placebo. As such, the sponsor did not include the ITC comparison in its submission to CADTH and used estimates from the previous CADTH submission for patients aged 6 to 11 years to use as assumptions within its economic model.
ITCs in Patients Aged 6 to 11 Years and 12 Years and Older
To inform the pharmacoeconomic model, the sponsor submitted estimates of clinical efficacy of ELX-TEZ-IVA compared to placebo derived from ITCs previously conducted for patients aged 6 to 11 years and 12 years and older using individual patient data from relevant phase III randomized controlled clinical trials.
The sponsor conducted a single indirect comparison for patients aged 6 to 11 years with an F/F genotype to derive relative estimates of clinical efficacy for ELX-TEZ-IVA versus LUM-IVA, ELX-TEZ-IVA versus placebo, and ELX-TEZ-IVA versus tezacaftor-ivacaftor and ivacaftor (TEZ-IVA). TEZ-IVA is not currently approved by Health Canada or reimbursed by the Canadian public drug programs for use in patients aged 6 to 11 years. To conduct the primary indirect comparisons, the sponsor extracted 24-week individual patient data for those with an F/F genotype from the following studies: Study 106B for ELX-TEZ-IVA (N = 29); pooled data from Study 809-109 and Study 809-011B for LUM-IVA (N = 160); and Study 661-113B for TEZ-IVA (N = 61). Additional sensitivity analyses were performed using 8-week data. The sponsor reported the following indirect estimates of effect for ELX-TEZ-IVA compared with placebo for absolute change from baseline through 24 weeks: || || |||||| || |||||| || |||| for percent predicted FEV1 (ppFEV1). The primary limitations of the ITC were the differences in study design across the included studies (Studies 106B, 809-011B, and 661-113B were single-arm, open-label trials, and Studies 809-109 and 661-115 were double-blind placebo-controlled trials) and the differences in the baseline characteristics.
Studies Addressing Gaps in the Evidence From the Systematic Review
The sponsor did not include any additional studies to address gaps in the pivotal trial evidence.
Conclusions
For patients aged 2 to 5 years, a 24-week, open-label, uncontrolled trial (Study 111 Part B; N = 75) suggested that treatment with ELX-TEZ-IVA resulted in improvements from baseline in lung function (decrease in LCI2.5 from baseline) and CF biomarkers (reduction in SwCl). Study 111 was primarily designed to evaluate the safety, tolerability, and pharmacokinetics of ELX-TEZ-IVA, as the regulatory submission is based on the extrapolation of efficacy data from the studies conducted in older patients with CF (i.e., those showing some measurable level of disease manifestations at baseline). The clinical experts consulted by CADTH noted that, given the mechanism of action and compelling efficacy data in patients aged 6 years and older, ELX-TEZ-IVA would be expected to benefit patients aged 2 to 5 years who have at least 1 508del mutation in the CFTR gene. There is consensus across clinicians and patients that treatment with ELX-TEZ-IVA should be initiated as soon as possible given the clinically meaningful benefits observed in patients who can currently access the treatment. Uncertainty remains regarding the magnitude of the beneficial effect of ELX-TEZ-IVA in very young patients with CF, and future real-world evidence may help address this uncertainty.
Study 111 was limited to patients with an F/F or F/MF genotype. There were no clinical studies conducted with ELX-TEZ-IVA in pediatric patients with 1 F508del mutation and 1 residual function mutation in the CFTR gene (F/RF) or 1 F508del mutation and 1 gating mutation in the CFTR gene (F/G) genotypes; however, the clinical experts noted that ELX-TEZ-IVA would result in clinically meaningful improvements for these patients, based on the evidence reported for ELX-TEZ-IVA in adult patients with F/RF and F/G genotypes and the results in F/F and F/MF pediatric studies. This is consistent with the input from patient and clinician groups, who have indicated that all patients with at least 1 F508del mutation are likely to benefit from treatment with ELX-TEZ-IVA.
ELX-TEZ-IVA was well tolerated in the target patient population (i.e., patients aged 2 to 5 years with at least 1 F508del mutation). SAEs and withdrawals due to AEs were rare in Study 111. The product monograph notes that elevated transaminases have been observed in patients treated with ELX-TEZ-IVA and recommends that ALT and AST be assessed prior to initiating treatment with ELX-TEZ-IVA, every 3 months during the first year of treatment, and annually thereafter. The clinical experts consulted by CADTH noted that the recommendations for monitoring with ELX-TEZ-IVA were not anticipated to result in a substantial increase in the number of clinic visits for patients with CF (particularly after the first year of treatment).
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of ELX-TEZ-IVA (Trikafta; oral granules) at recommended dosages for the treatment of patients aged 2 to 5 years with CF who have at least 1 F508del mutation in the CFTR gene.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
CF, an autosomal recessive condition, is the most common fatal genetic disease affecting children and young adults in Canada. It is caused by mutations in the CFTR gene, which is located on chromosome 7. The CFTR gene encodes a chloride channel that regulates ion and fluid transport across cell membranes. When CFTR is dysfunctional, secretions become tenacious and sticky, resulting in pathology in multiple organs, including the lungs, the large and small intestines, the pancreatic and bile ducts, and the vas deferens. A deletion of phenylalanine 508 in the first nucleotide-binding domain (F508del) is the most common mutation that results in CF.1 The Canadian Cystic Fibrosis Registry reported 4,344 patients in Canada living with CF in 2019. Of these patients, 87.8% carried at least 1 F508del mutation (47.1% were homozygous, and 40.7% were heterozygous).1
More than 2,090 CFTR variants have been identified among patients with CF.1 The CFTR variants have been classified as impaired biosynthesis (class I), defective protein maturation and accelerated degradation (class II), defective regulation of CFTR at the plasma membrane (class III), defective chloride conductance (class IV), diminished CFTR transcription (class V), and accelerated turnover at the cell surface (class VI).11 CFTR variants within classes I to III are associated with severe CF as they are considered nonfunctional, while CFTR variants in classes IV to VI may retain CFTR function.11,12 The F508del mutation is typically considered a class II CFTR mutation and is a severe mutation resulting in significant loss of function of the CFTR protein. A F508del defect causes CFTR to misfold, and thus most of the protein is removed before it can reach the cell membrane. In addition, the F508del mutation in the CFTR gene presents a defect in channel gating as well as being unstable and having more rapid turnover at the cell membrane.13,14 Genotyping for mutations in the CFTR gene is performed routinely on almost all patients with CF in Canada and is also part of the newborn screening process.1
CF results in airway obstruction, chronic endobronchial infection, and inflammation, which ultimately lead to destruction of lung tissue through the development of bronchiectasis and to loss of lung function.15 Although chronic pulmonary therapies instituted early in the disease have reduced the decline in lung function over time, patients who are homozygous for the F508del mutation will develop chronic infection with Pseudomonas and progressive bronchiectasis and airway obstruction. In a cohort of approximately 1,000 healthy young children with CF who did not have Pseudomonas infection at enrolment, there was a greater annual decline in FEV1 over the following 4 years in those who were homozygous for the F508del mutation.16 Chronic endobronchial infection of the airways with bacterial pathogens, such as Pseudomonas aeruginosa (reported in 38% of patients with CF living in Canada in 2019),1 is associated with a more rapid loss of lung function.17 Acute or chronic endobronchial infections result in further destruction of lung tissue and are associated with respiratory morbidity. Lung disease accounts for the vast majority of deaths in patients with CF (over 80%).1,18
Pulmonary exacerbations are associated with lung function decline and mortality and may require treatment with IV antibiotics and hospitalization. The Cystic Fibrosis Foundation has reported that approximately a third of patients with CF will have at least 1 pulmonary exacerbation per year requiring IV antibiotics.19
Maintenance of pulmonary function (FEV1) and fewer respiratory exacerbations are associated with increased survival.20 Pulmonary management of CF therefore aims to clear the airways of secretions and treat lung pathogens to minimize inflammation.
Patients who are homozygous or heterozygous for the F508del mutation typically have pancreatic, gastrointestinal, and nutritional disease as well as progressive pulmonary damage. Gastrointestinal and pancreatic involvement results in pancreatic exocrine insufficiency in most individuals with CF, causing malabsorption of fats and fat-soluble vitamins, which leads to malnutrition. Maintaining adequate nutrition is associated with improved clinical outcomes and longevity for patients with CF.21 Virtually all these patients will have insufficient pancreatic function and will need to take lifelong pancreatic enzyme replacement with every meal as well as fat-soluble vitamin therapy. With increasing age, these patients will develop CF-related diabetes and require therapy with insulin. In 2019, CF-related diabetes was reported in 22.0% of patients with CF living in Canada (33.5% of adults and 3.3% of children).1
The median age of survival in Canada for a child born with CF in 2019 is estimated to be 53.4 years.1 The Canadian Cystic Fibrosis Registry has reported an increase in the median age of death for patients with CF in Canada since the year 2000.1 In 2019 the median age of death was 42.1 years, compared with 27.7 years in 2000, 35.1 years in 2013, and 38.9 years in 2016.1,22,23 There is a clear unmet need for better CF therapies (refer to Patient Group Input and Clinician Input).
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
The goals of CF therapy include preservation of lung function by minimizing pulmonary infection and inflammation; restoration of baseline pulmonary function, symptoms, and level of inflammation after acute respiratory exacerbations; and maintenance of adequate nutrition. The choice of a therapeutic regimen for CF depends on organ involvement. The severity of lung function impairment and the presence of bacterial pathogens are deterministic factors when selecting chronic pulmonary therapy.
Treatments that are approved and/or available can be broadly classified either as therapies used to manage symptoms, complications, and comorbidities of CF or as therapies that aim to correct the underlying defects of the CFTR protein, known as CFTR modulators.
Management of Symptoms, Complications, and Comorbidities
Respiratory treatments consist of physiotherapy and pharmacologic agents, such as inhaled antibiotics (e.g., tobramycin, aztreonam, or colistin), anti-inflammatory agents, or mucolytics (e.g., hypertonic saline and/or dornase alfa).24 Nutritional treatments consist of high-calorie and high-fat diets and pancreatic enzyme replacement for patients with pancreatic insufficiency.18,24 Pulmonary exacerbations are treated with oral or IV antibiotics.25 These treatments do not halt, but only slow, the decline in lung function and the progression of disease.
CFTR Modulators
CFTR modulators are a class of medications that aim to correct the underlying defects of the CFTR protein. The CFTR modulators currently marketed in Canada or other jurisdictions are classified as follows:
Potentiators, which function by increasing the channel-open probability of the CFTR protein at the cell surface. IVA is a CFTR potentiator.
Correctors, which function by improving the conformational stability of the F508del-CFTR protein, resulting in an increased expression of the F508del-CFTR protein at the cell surface. Lumacaftor, TEZ, and ELX are CFTR correctors.
Table 6 provides a summary of the CFTR modulators currently marketed or under review in Canada, the CFTR mutations and age ranges for which they have been approved by Health Canada, and their reimbursement status within the public drug programs. The currently available CFTR modulators are not approved for use in all patients with at least 1 F508del mutation. The approved indications currently cover patients who are homozygous for F508del mutations (Orkambi and Symdeko), patients who are heterozygous for the 508del mutation and have a residual function mutation (Symdeko), or patients who have a non-F508del gating mutation (Kalydeco). Hence, there is a subset of individuals who are heterozygous for the F508del mutation who will not be covered by the existing indications. In addition, the clinical benefit of some of the existing treatments (e.g., Orkambi) has been described as modest; therefore, there remains an unmet medical need for treatments with the potential to offer greater treatment effects and benefits.26 In 2019, CF Canada reported that 658 individuals (216 children and 442 adults) were receiving treatment with CFTR modulators. The number of patients receiving each treatment were 146 receiving Kalydeco, 368 receiving Orkambi, and 186 receiving Symdeko.1
Drug Under Review
Trikafta is a fixed-dose combination product containing ELX, TEZ, and IVA co-packaged with IVA. ELX-TEZ-IVA is available as both oral tablets and oral granules in the following dosing strengths:
Tablets for patients aged 6 years and older:
ELX 50 mg–TEZ 25 mg–IVA 37.5 mg co-packaged with IVA 75 mg
ELX 100 mg–TEZ 50 mg–IVA 75 mg co-packaged with IVA 150 mg
Granules for patients aged 2 to younger than 6 years:
ELX 100 mg–TEZ 50 mg–IVA 75 mg co-packaged with IVA 75 mg
ELX 80 mg–TEZ 40 mg–IVA 60 mg, co-packaged with IVA 59.5 mg
ELX-TEZ-IVA is indicated for the treatment of CF in patients aged 2 years and older who have at least 1 F508del mutation in the CFTR gene. The sponsor has requested that the current submission for ELX-TEZ-IVA focus only on those patients aged 2 to 5 years using the new dosage format (i.e., orally administered granules). The sponsor has requested the reimbursement criteria presented in Table 3.
Table 3: Sponsor Requested Reimbursement Criteria
Category | Requested reimbursement criteria |
|---|---|
Initiation criteria |
|
Renewal criteria |
|
BMI = body mass index; CF = cystic fibrosis; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; INESSS = Institut national d’excellence en santé et services sociaux.
Source: Sponsor Summary of Clinical Evidence.
Mechanism of Action
ELX-TEZ-IVA is the third treatment specifically indicated for the treatment of patients with CF who have F508del mutation(s) in the CFTR gene. This mutation is believed to be associated with misfolding of the CFTR protein, which results in a lower quantity of CFTR expression at the cell surface. In addition to the reduced quantity of the protein, the mutation results in CFTR that is less stable and has defective channel gating compared with wild-type CFTR. Treatment with ELX-TEZ-IVA results in an increased quantity and improved function of the F508del-CFTR protein at the cell surface, through the following mechanisms:26,29,30
ELX and TEZ improve the conformational stability of the F508del-CFTR protein, resulting in an increased expression of the F508del-CFTR protein at the cell surface.
IVA increases the channel-open probability of the CFTR protein at the cell surface.
Recommended Dosage
The recommended dosing of ELX-TEZ-IVA is summarized in Table 4. For patients aged 2 to younger than 6 years who weigh less than 14 kg, the recommended dosage regimen is 1 packet of ELX 80 mg–TEZ 40 mg–IVA 60 mg granules in the morning and 1 packet of IVA 59.5 mg granules in the evening. For those who weigh at least 14 kg, the recommended dosage is 1 packet of ELX 100 mg–TEZ 50 mg–IVA 75 mg granules in the morning and 1 packet of IVA 75 mg granules in the evening.
Both tablets and granules are administered orally and should be taken approximately 12 hours apart with fat-containing food. Table 5 provides a summary of the recommended dosage adjustments for patients with hepatic insufficiency or those receiving concomitant treatment with moderate CYP3A inhibitors (e.g., fluconazole or erythromycin) or strong CYP3A inhibitors (e.g., ketoconazole, itraconazole, posaconazole, voriconazole, telithromycin, or clarithromycin).
Age (weight) | Morning dose | Evening dose |
|---|---|---|
2 to < 6 years (< 14 kg) | 1 packet granules: ELX 80 mg–TEZ 40 mg–IVA 60 mg | 1 packet granules: IVA 59.5 mg |
2 to < 6 years (≥ 14 kg) | 1 packet granules: ELX 100 mg–TEZ 50 mg–IVA 75 mg | 1 packet granules: IVA 75 mg |
6 to < 12 years (< 30 kg) | 2 tablets: ELX 50 mg–TEZ 25 mg–IVA 37.5 mg | 1 tablet: IVA 75 mg |
6 to < 12 years (≥ 30 kg) | 2 tablets: ELX 100 mg–TEZ 50 mg–IVA 75 mg | 1 tablet: IVA 150 mg |
≥ 12 years | 2 tablets: ELX 100 mg–TEZ 50 mg–IVA 75 mg | 1 tablet: IVA 150 mg |
ELX = elexacaftor; IVA = ivacaftor; TEZ = tezacaftor.
Source: Product monograph.29
Table 5: Recommended Dosage Adjustments
Age | Condition | Dosage adjustment |
|---|---|---|
Hepatic insufficiency | ||
2 to < 6 years | Mild (Child-Pugh Class A) | No dose adjustments |
Moderate (Child-Pugh Class B) | Use not recommended: Treatment should only be considered when there is a clear medical need and the benefits are expected to outweigh the risks. If used, ELX-TEZ-IVA should be used with caution at a reduced dose, as follows:
Repeat dosing schedule each week. The evening dose of IVA should not be taken. | |
Severe (Child-Pugh Class C) | Should not be used | |
6 years and older | Mild (Child-Pugh Class A) | No dose adjustments |
Moderate (Child-Pugh Class B) | Use not recommended: Treatment should only be considered when there is a clear medical need and the benefits are expected to outweigh the risks. If used, ELX-TEZ-IVA should be used with caution at a reduced dose, as follows: 2 ELX-TEZ-IVA tablets alternating with 1 ELX-TEZ-IVA tablet, taken in the morning on alternate days. The evening dose of IVA should not be taken. | |
Severe (Child-Pugh Class C) | Should not be used | |
CYP3A inhibitors | ||
2 to < 6 years | Moderate CYP3A inhibitors | Morning: 1 packet ELX-TEZ-IVA granules (day 1); 1 packet IVA granules (day 2) Evening: No dose |
Strong CYP3A inhibitors | Morning: 1 packet ELX-TEZ-IVA granules (twice weekly; 3 to 4 days apart) Evening: No dose | |
6 years and older | Moderate CYP3A inhibitors | Morning: 2 ELX-TEZ-IVA tablets (day 1); 1 IVA tablet (day 2) Evening: No dose |
Strong CYP3A inhibitors | Morning: 2 ELX-TEZ-IVA tablets (twice weekly; 3 to 4 days apart) Evening: No dose | |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; IVA = ivacaftor.
Source: Product monograph.29
Previous CADTH Reviews
This is the third submission to CADTH for ELX-TEZ-IVA. CADTH has previously reviewed ELX-TEZ-IVA for the treatment of CF in patients who have at least 1 F508del mutation in the CFTR gene for patients aged 12 years and older (final recommendation issued in August 2021) and patients aged 6 years and older (final recommendation issued in June 2022). For both of the previous reviews, the CADTH Canadian Drug Expert Committee recommended that ELX-TEZ-IVA be reimbursed with conditions.2,3 All the indications for ELX-TEZ-IVA were accepted as priority reviews by Health Canada.
CADTH has previously reviewed IVA alone for the following indications: patients aged 6 years and older who have a G551D mutation in the CFTR gene; patients aged 6 years and older who have 1 of the following mutations in the CFTR gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R, or G970R; and patients aged 18 years and older who have an R117H mutation in the CFTR gene.31-33 For each of these indications, the CADTH Canadian Drug Expert Committee recommended that IVA be reimbursed with conditions. LUM-IVA was previously reviewed for the treatment of CF in patients who are homozygous for the F508del mutation in the CFTR gene, and it received do not reimburse recommendations in 2016 and 2018.34,35 CADTH was unable to recommend reimbursement for TEZ-IVA as a submission was not filed by the sponsor.36
Key characteristics of ELX-TEZ-IVA and other CFTR modulators approved in Canada are summarized in Table 6.
Table 6: Key Characteristics of CFTR Modulators
Characteristic | Trikafta (ELX-TEZ-IVA) | Orkambi (LUM-IVA) | Symdeko (TEZ-IVA) | Kalydeco (IVA) |
|---|---|---|---|---|
Mechanism of action | CFTR potentiator (IVA) and correctors (ELX and TEZ) | CFTR potentiator (IVA) and corrector (LUM) | CFTR potentiator (IVA) and corrector (TEZ) | CFTR potentiator |
Indicationa | Patients aged ≥ 2 years who have at least 1 F508del mutation in the CFTR gene | Patients aged ≥ 1 year who are homozygous for the F508del mutation in the CFTR gene | Patients aged ≥ 12 years who are homozygous for the F508del mutation or who are heterozygous for the F508del mutation and have 1 of the following CFTR mutations: P67L, D110H, R117C, L206W, R352Q, A455E, D579G, 711 + 3A → G, S945L, S977F, R1070W, D1152H, 2789 + 5G → A, 3272-26A → G, and 3849 + 10kbC → T | Granules (25 mg, 50 mg, and 75 mg) are indicated for the treatment of patients with CF aged 4 months and older and weighing 5 kg to < 25 kg who have one of the following mutations in the CFTR gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R, or R117H Tablets (150 mg) are indicated for the treatment of patients with CF aged 6 years and older and weighing 25 kg or more who have one of the following mutations in the CFTR gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R, or R117H |
Route of administration | Oral tablets and granules | Oral tablets and granules | Oral tablets | Oral tablets and granules |
Recommended dose | Granules:
Tablets:
| Tablets:
Granules
| Morning: TEZ 100 mg–IVA 150 mg Evening: IVA 150 mg | Tablets: IVA 150 mg q.12.h. Granules:
|
Serious adverse effects or safety issues | Product monographs of each of the products include a warning about the risk of elevated transaminases (ALT and AST), and monitoring of liver function is recommended prior to initiating treatment, every 3 months during the first year of treatment, and annually thereafter.29,37-39 Product monograph recommends that ELX-TEZ-IVA not be used in patients with severe hepatic impairment; dosage reduction scenarios are provided in the product monographs for IVA, TEZ-IVA, and LUM-IVA.29,37-39 | |||
CADTH reviews | Aged ≥ 6 years: Reimburse with conditions3 Aged ≥ 12 years: Reimburse with conditions2 | Nonsubmission36 | ||
ALT = alanine transaminase; AST = aspartate transaminase; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ELX = elexacaftor; IVA = ivacaftor; LUM = lumacaftor; LUM-IVA = lumacaftor-ivacaftor; q.12.h. = every 12 hours; TEZ = tezacaftor; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
aHealth Canada–approved indications.
Source: Product monographs for Trikafta, Orkambi, Symdeko, and Kalydeco.29,37-39
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient input received by CADTH have been included in the Stakeholder section of this report.
One patient group, CF Canada, responded to CADTH’s call for patient input for the current review of ELX-TEZ-IVA, which is focused on patients aged 2 to 5 years who have at least 1 F508del mutation in the CFTR gene. CF Canada is a national not-for-profit corporation committed to improving and lengthening the lives of people living with CF through treatments, research, information, and support.
Information from CF Canada was based on focus groups with 6 parents of children younger than 5 years who have at least 1 copy of the F508del mutation; a survey of patients and caregivers with access to ELX-TEZ-IVA, which was conducted in 2021; medical and scientific publications; and the Canadian Cystic Fibrosis Registry. In addition, CF Canada used preliminary findings from phase I of the Global Burden of Disease study, which measures the burden of CF at the individual, family, health system, and societal levels and is considered one of the most comprehensive studies of the burden of CF in the world.
The patient group emphasized that CF has a tremendous impact on those living with the condition, their loved ones, health systems, and society. The most significant clinical impact is on the digestive system and the lungs. Patients experience progressive scarring of their airways and a progressive decline in lung function. Young children who grow older with CF may experience pulmonary exacerbations requiring weeks to months of hospitalization and IV antibiotics. Malnutrition and low BMI are also common consequences of CF among children aged 2 to 5 years. Patients may also experience CF-related comorbidities, such as CF-related diabetes and CF-related liver disease. In addition, CF has a significant impact on socialization, mental health, and isolation among patients and caregivers.
The patient input stated that managing CF requires a demanding treatment routine. As the disease progresses, more time and effort and frequent clinic visits and hospital stays are needed to manage the progressive and debilitating symptoms. This condition has a significant impact on patients’ and caregivers’ day-to-day activities and quality of life, affecting sleep quality, education, career, travel, relationships, and family dynamics and planning, and creates a huge financial burden for families.
According to the patient group input, an ideal treatment in CF would fully address the basic molecular defect in CF and restore normal chloride transport on the cell surface. Patients with CF and their loved ones are seeking treatments that can change the trajectory of the disease, reduce disease symptoms, improve sleep quality and energy levels, and improve both life expectancy and quality of life. Caregivers want their children to have a normal life with better lung function, fewer hospital stays and invasive medical procedures, and a reduction in the treatment burden of daily therapies.
In the patient group input, CF Canada’s clinical trials network (CF CanACT) emphasized the importance of early treatment of CF to prevent disease progression and irreversible damage. Extending access to ELX-TEZ-IVA for patients with CF aged 2 to 5 years would be congruent with the secondary prevention paradigm of CF care and would decrease the long-term burden of the disease.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the review of ELX-TEZ-IVA, a panel of 4 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with CF, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
There are significant unmet therapeutic needs for patients living with CF. There are no treatments currently available that can effectively achieve the most important goals of therapy: prolong survival, prevent the need for lung transplant, slowing the decline in lung function over time, and reverse the course of the disease. In addition, the current standard treatments are burdensome for patients and their caregivers. Patients may not respond or may stop responding over time to the currently available treatments.
Place in Therapy
ELX-TEZ-IVA is a CFTR modulator that functions by increasing the amount of CFTR protein at the cell surface (ELX and TEZ) and by improving the transport of chloride through the CFTR protein (IVA). The mechanism of action for ELX-TEZ-IVA is attractive because it acts directly on the CFTR protein to address the defects that are responsible for the CF phenotype. ELX-TEZ-IVA would be added to existing treatments such as physiotherapy, mucolytics, anti-infectives, and anti-inflammatory treatments (such as azithromycin). The clinical experts noted that ELX-TEZ-IVA would replace earlier CFTR modulators that are significantly less effective (e.g., Orkambi or Kalydeco) and patients currently receiving those drugs would likely be switched to ELX-TEZ-IVA if they meet eligibility and age criteria.
It is anticipated that ELX-TEZ-IVA would be used as a preventive therapy, with the goal of initiating treatment before the patient develops significant lung disease. The current treatment paradigm would be significantly altered if ELX-TEZ-IVA can successfully prevent or delay progression to end organ disease (e.g., lung transplant). The clinical experts consulted by CADTH and those who responded to the call for clinician input noted that children aged between 2 and 5 years will often have structural lung disease (e.g., bronchial wall thickening, mucus plugging, or bronchiectasis) but that detection is challenging using the tools available to evaluate lung function in clinical practice (i.e., spirometry) or as part of a research protocol (e.g., LCI). These early stages of lung abnormalities can be visualized using CT; therefore, although younger patients with CF may demonstrate normal lung function, they have been shown through CT to often have underlying lung abnormalities, which will continue to progress.
All the clinicians who provided input into this review recommended initiating treatment with ELX-TEZ-IVA as soon as possible. This recommendation is aligned with the previously published Canadian Clinical Consensus Guideline for Initiation, Monitoring and Discontinuation of CFTR Modulator Therapies for Patients With Cystic Fibrosis, which also recommends that CFTR modulators be initiated at the youngest age possible, with the goal of attenuating disease progression and improving clinical status. All stakeholders agreed that there are no data to support withholding the initiation of CFTR modulator treatment until clinical symptoms of CF have developed.
Patient Population
The diagnosis of CF is not challenging in routine clinical practice. All provinces and territories have instituted newborn screening for CF, so most people with CF are now identified via newborn screening and have a confirmed diagnosis by 1 month of age (on average). SwCl testing is available and reliably used to confirm the screening test. The provinces and territories have slightly different testing algorithms and CFTR mutation screening panels; however, all provinces and territories have effective processes. Almost 100% of newly diagnosed infants would have both CFTR mutations identified. Infants who are not identified via newborn screening (i.e., false negatives) are usually diagnosed before 1 year of age, after the development of clinical symptoms of CF. There are clear diagnostic guidelines and very little variability in expert opinion. Misdiagnosis and underdiagnosis of CF is exceedingly rare in Canadian clinical practice.
ELX-TEZ-IVA could be used in every patient who meets the Health Canada–approved indication, regardless of their current or past treatment regimens. From a medical perspective, there is no rationale for a patient having to demonstrate an inadequate response or loss of response to prior therapies before initiating treatment with ELX-TEZ-IVA. It would be reasonable to require patients to complete important standard CF therapies at the same time as receiving treatment with ELX-TEZ-IVA. In clinical practice, eligible patients would be identified based on their CFTR genotype, and all patients would be expected to respond to the treatment.
For the expanded indication (i.e., patients aged 2 to 5 years), the clinical experts consulted by CADTH noted that nearly all patients would begin therapy with ELX-TEZ-IVA as soon as possible, provided it was safe to start treatment. The clinical experts emphasized that ELX-TEZ-IVA has been a transformative and disease-modifying therapy for CF and that it would not be appropriate to wait until the patient shows worsening symptoms, more frequent exacerbations, or a decline in lung function to initiate treatment with ELX-TEZ-IVA.
Applicability of Existing Reimbursement Criteria to Pediatric Patients
In discussions with CADTH, the sponsor noted that nearly all patients aged 12 years and older living in Canada who are eligible for treatment have begun therapy with ELX-TEZ-IVA (some may have elected to discontinue, but all who are interested have been given the opportunity to access the drug). The sponsor similarly stated that all patients aged 6 to 11 years living in Canada who wish to will have begun treatment with ELX-TEZ-IVA by the end of 2023. For those who have initiated treatment with ELX-TEZ-IVA, the sponsor noted that initial renewal criteria were met for all patients living in Canada who had started the therapy and wanted to continue (i.e., 100% of patients met the renewal criteria recommended by CADTH and/or applied by the public drug programs). The clinical experts consulted expressed general agreement with the sponsor’s position, noting that rates of initial access and renewal are very high within their individual clinics. With nearly all patients who are at least 6 years or older having met the initiation and renewal criteria, newly issued CADTH reimbursement criteria focusing exclusively on patients aged 2 to 5 years would effectively replace the previous criteria (i.e., although limited to patients aged 2 to 5 years, all older patients would have already qualified for initiation and renewal).
Table 7: CADTH-Recommended Reimbursement Conditions for Patients Aged 6 Years or Older
Category | Reimbursement conditions |
|---|---|
Initiation |
|
Renewal |
|
Discontinuation | Patient has undergone lung transplant |
Prescribing |
|
Feasibility of adoption | The feasibility of adoption of ELX-TEZ-IVA must be addressed |
BMI = body mass index; CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire–Revised; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; FEV1 = forced expiratory volume in the first second.
Baseline Measurements
Regarding the baseline measurements that must be completed prior to initiating treatment with ELX-TEZ-IVA, the clinical experts consulted by CADTH noted that the following baseline measurements currently recommended by CADTH would be problematic to implement, uninformative, and/or not relevant for patients aged 2 to 5 years:
Baseline FEV1: Spirometry is not performed in routine practice in Canada for patients younger than 6 years, and younger patients are unlikely to demonstrate any loss of lung function that can be measured using conventional pulmonary function testing.
Baseline frequency of pulmonary exacerbations: The frequency of pulmonary exacerbations can be quite low in patients aged 2 to 5 years, and it would be challenging to establish a reliable baseline. This would be particularly challenging in situations where a young patient may reach important milestones, such as entering daycare, preschool, or school. In these cases, it could be possible to see an increase in the rate of pulmonary exacerbations, even after initiating treatment with a CFTR modulator, due to increased exposure to bacterial and/or viral infections in these settings.
Baseline nutritional end points (i.e., weight, height, and BMI): As with the older patients, the clinical experts noted that patients aged 2 to 5 years would have growth parameters monitored in routine clinical practice. The clinical experts consulted by CADTH noted that a majority of patients with CF aged 2 to 5 years do not show reductions in age-standardized BMI. According to the CF Canada data registry online, 10.2% of all children with CF aged 2 to 17 years were underweight based on BMI percentile. The experts further noted that BMI percentile can fluctuate in younger patients, especially following periods of acute illness.
CFQ-R: In the previous CADTH reviews of ELX-TEZ-IVA, it was noted that CFQ-R respiratory domain scores are not routinely obtained for patients in pediatric clinics (typically only when conducting research studies). The clinical experts consulted by CADTH confirmed that this remains the case in most Canadian clinics. In addition, the pediatric and caregiver versions of the CFQ-R were designed for use in those aged 6 to 13 years. Although some studies have been conducted to examine the utility of the caregiver version of the CFQ-R for evaluating health-related quality of life in children younger than 6 years, the CFQ-R remains largely a research tool for young patients and has not been applied in Canada or other international jurisdictions for evaluating response to treatment for the purposes of publicly funded drug reimbursement for CFTR modulators. Overall, the clinical experts suggested that the CFQ-R would not be appropriate as a renewal criterion for patients aged 2 to 5 and would be challenging to implement in pediatric clinical practice. No alternative health-related quality of life measure that could be readily implemented in Canadian practice was identified. As noted in the previous review of ELZ-TEZ-IVA for patients aged 6 to 11 years, CF clinics would require additional resources to administer the CFQ-R, document the responses, and track changes in scores over time. Differences in record keeping across Canada (e.g., paper and/or electronic health record systems) were noted as an additional challenge with including CFQ-R assessment in the reimbursement criteria for ELX-TEZ-IVA in pediatric patients.
Assessing Response to Treatment
Each of the end points are discussed below, with reflection on the applicability of the existing CADTH criteria to the expanded patient population (i.e., patients aged 2 to 5 years).
BMI and BMI Z Scores
The CADTH recommendation for patients aged 6 years and older included the following as one of the potential renewal criteria for ELX-TEZ-IVA: no decline in BMI (BMI z score in children) at 6 months compared with the baseline BMI assessment.
As with the previous review of ELX-TEZ-IVA, the clinical experts noted that 6 months would not be sufficient to accurately assess the response to treatment and that an assessment of BMI at 12 months would be more appropriate. The longer time was suggested to account for events that could temporarily reduce BMI (e.g., increased physical activity in summer months and growth spurts). It was strongly noted that discontinuation of ELX-TEZ-IVA in children with temporarily reduced BMI would not be clinically appropriate.
Pulmonary Exacerbations
The CADTH recommendation for patients aged 6 years and older included the following as one of the potential renewal criteria for ELX-TEZ-IVA: A decrease in the total number of days for which the patient received treatment with oral and/or IV antibiotics for pulmonary exacerbations compared with the 6-month period prior to initiating treatment, or a decrease in the total number of pulmonary exacerbations requiring oral and/or IV antibiotics compared with the 6-month period prior to initiating treatment.
Pulmonary exacerbations are less frequent in patients aged 2 to 5 years than in adults and adolescents. The clinical experts consulted by CADTH indicated that this is reflective of clinical practice, where these events are less common in children with relatively normal lung function. The clinical experts suggested that the above-noted renewal criterion would be problematic for the use of ELX-TEZ-IVA in patients aged 2 to 5 years. However, it was emphasized that patients who have not experienced a pulmonary exacerbation or those with a very low annual rate of pulmonary exacerbations would still benefit from the treatment. As with the criterion for BMI, it was noted that 12 months would be a more appropriate time frame for evaluating changes in pulmonary exacerbations.
CF-Related Hospitalizations
The CADTH recommendation for patients aged 6 years and older included the following as one of the potential renewal criteria for ELX-TEZ-IVA: Decreased number of CF-related hospitalizations at 6 months compared with the 6-month period prior to initiating ELX-TEZ-IVA treatment.
The clinical experts consulted by CADTH noted that CF-related hospitalization is infrequent and highly variable in patients aged 2 to 5 years. As such, this criterion would be very challenging to implement to evaluate response to ELX-TEZ-IVA for the purposes of reimbursement.
Sweat Chloride
The previous CADTH recommendation did not include SwCl testing as one of the initiation or renewal conditions for ELX-TEZ-IVA. The sponsor has requested that “reduction in sweat chloride” be included as a reimbursement condition for ELX-TEZ-IVA in the current review. In its comments on the draft report, the sponsor reported that a pooled analysis of phase III and open-label studies suggests that a reduction in SwCl is correlated with improvements in lung function, respiratory symptoms, BMI, and pulmonary exacerbations. However, the pediatric clinical experts agreed with the prior input from the reviews of ELX-TEZ-IVA in patients aged 6 to 11 years and 12 years and older, noting that SwCl testing should be not used to evaluate the response to ELX-TEZ-IVA for the purposes of drug reimbursement because it is not clearly predictive of clinically important outcomes and only reflects the mechanism of action of CFTR modulators like ELX-TEZ-IVA. It was noted that poor adherence with the treatment over a short period of time could increase SwCl (or, conversely, that a patient could only be adherent for a short period of time and still demonstrate considerable reductions in SwCl). The clinical experts also noted that access to SwCl testing can be challenging in some jurisdictions and that the time taken to receive the test results can fluctuate.
The clinical experts consulted by CADTH considered the SwCl renewal criterion proposed by the sponsor (i.e., a reduction in SwCl as defined by a SwCl concentration less than 60 mmol/L or by a reduction in SwCl of at least 30% from baseline) and did not believe this to be a clinically relevant measure for evaluating a patient’s response to treatment. However, they stated that the criterion could be acceptable if some form of objective criteria were also required for patients aged to 2 to 5 years.
CADTH noted that the previously published Canadian Clinical Consensus Guideline for Initiation, Monitoring and Discontinuation of CFTR Modulator Therapies for Patients With Cystic Fibrosis recommended a decrease in SwCl by 20% or 20 mmol/L from baseline at a follow-up visit 3 months after treatment initiation as a renewal criterion.
Discontinuing Treatment
The CADTH recommendation for patients aged 6 years and older stated that reimbursement should be discontinued in patients who have undergone lung transplant. ELX-TEZ-IVA is generally a well-tolerated treatment, and patients who began treatment before age 6 would be expected to remain on the therapy for many years if they continued to benefit. Some of these patients may eventually require a lung transplant; therefore, the discontinuation criterion remains relevant for a recommendation issued for the younger patient population. As was noted in previous reviews, given the expected benefit of ELX-TEZ-IVA on nutrition and growth end points, it is anticipated that clinicians would consider ELX-TEZ-IVA (balancing patient need with risk of possible drug interactions) in patients post–lung transplant. The sponsor expressed that ELX-TEZ-IVA has been shown to be beneficial in patients who have received a lung transplant through improvements in extrapulmonary manifestations of CF.
Prescribing Conditions
As with the previous review of ELX-TEZ-IVA, the only appropriate setting for initiation and monitoring of treatment with ELX-TEZ-IVA remains an adult or pediatric CF clinic. This treatment will typically be initiated and monitored in the outpatient clinic setting by a CF physician and the associated multidisciplinary team (e.g., specialists in respirology, infectious diseases, and gastroenterology). The experts noted that the drug may also be initiated in hospital. It would not be appropriate that a nonspecialty setting or physician would prescribe and monitor treatment with ELX-TEZ-IVA.
Continuation and Subsequent Renewals
The clinical experts emphasized that ELX-TEZ-IVA has the potential to modify the course of disease for patients with CF. When used in older patients, nearly all patients demonstrated sufficient clinical benefit to have reimbursement renewed by the public drug programs. The clinical experts noted that, although objective measures are challenging to implement in clinical practice for patients aged 2 to 5 years, these patients would likely benefit from initiating treatment. The clinical experts noted that subsequent renewals for ELX-TEZ-IVA can be achieved through communication that the patient is continuing to benefit from the treatment and that such an approach could be applied for younger patients, where obtaining meaningful baseline and follow-up measurements of objective criteria would be challenging.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group inputs received by CADTH have been included in the Stakeholder section of this report.
Three groups of clinicians responded to CADTH’s call for input: CF CanACT, the CF Canada Healthcare Advisory Council, and the Canadian Cystic Fibrosis Clinician groups. CF CanACT and the CF Canada Healthcare Advisory Council. CF CanACT operates under the auspices of CF Canada with the purpose of conducting clinical trials in CF and attracting research for new therapies to Canada. CF Canada is a national not-for-profit corporation committed to improving and lengthening the lives of people living with CF through treatments, research, information, and support. Information for this input was gathered from the Canadian Cystic Fibrosis Registry, outcomes of patients who have participated in clinical trials, scientific publications, and experience from treating individuals with CF who received CFTR modulators. In addition, CF Canada used information from the Canadian Clinical Consensus Guideline for Initiation, Monitoring and Discontinuation of CFTR Modulator Therapies for Patients With Cystic Fibrosis.
The input from the clinician groups identified the same unmet medical needs for patients with CF and potential place in therapy for the drug under review as the clinical experts consulted by CADTH.
According to clinician groups’ input, the treatment paradigm for CF in children aged 2 to 5 years is lifelong. It consists of nonmodulator treatments, which include high-calorie, high-fat, high-protein diets; digestive medications; and airway clearance treatments. Consequently, many of these treatments start at the time of diagnosis (including in infancy) and continue every day throughout life. Medications commonly used in CF include antibiotics, mucolytics, bronchodilators, pancreatic enzymes, fat-soluble vitamins, insulin for people with CF-related diabetes, ursodiol for liver disease, and chest physiotherapy. There are also CFTR modulator therapies, which are the first commercially available therapies targeted at correcting the basic defect in CF by improving the production and function of the abnormal CFTR protein. The first-generation (IVA) and second-generation (LUM-IVA and IVA-TEZ-IVA) modulators had a modest but important clinical effect, but the response to the third-generation modulator (ELX-TEZ-IVA) is substantially greater.
Clinician groups noted that there are significant unmet therapeutic needs for patients living with CF. Available treatments address the symptoms and complications of CF and attempt to slow down the eventual fatal progression of the disease without effectively addressing the root cause or reversing the course of the disease. The treatments also have significant side effects and numerous drug interactions. In addition, the current standard treatments are burdensome for patients and their caregivers, which affects medication adherence and the mental health and quality of life of patients and caregivers.
The clinician groups noted that ELX-TEZ-IVA is an improvement on the existing CFTR modulators and the most effective. It addresses the underlying disease process and is complementary to the existing standard of care for CF, which would potentially delay disease progression and thus delay the need for other therapies, including lung transplant. According to the clinician groups, any patient with CF who has at least 1 copy of the F508del mutation could potentially benefit from ELX-TEZ-IVA.
The 2 clinician groups indicated that the outcomes of interest are those that can be assessed during routine visits, which include BMI, frequency of pulmonary exacerbations, number of courses of antibiotics, SwCl levels, and sinopulmonary symptoms. CF Canada added laboratory tests to follow parameters associated with potential side effects (liver enzymes, creatine kinase), sputum microbiology, quality of life questionnaires and mental health screening, fecal elastase testing, and ophthalmological examination.
The clinician groups noted that discontinuation of therapy should be considered in patients with severe side effects, allergies, or the development of signs of worsening liver disease. CF Canada suggested the treatment should also be discontinued for patients aged 2 to 5 years if the patient is not responding to medication. The clinician groups stated that the treatment of patients with CF with ELX-TEZ-IVA should be limited to CF specialists practising at CF clinics.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 8.
Table 8: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Are there any physiological reasons that would caution the extrapolation of data from patients with CF aged 6 years and older treated with ELX-TEZ-IVA to patients aged 2 to 5 years? | The clinical experts consulted by CADTH and those who provided input to CADTH through the call for clinician input all support starting ELX-TEZ-IVA as soon as possible. The clinical experts supported the extrapolation of efficacy data and noted that the data in patients aged 2 to 5 years did not raise additional concerns regarding the safety of ELX-TEZ-IVA. |
Can patients being treated with LUM-IVA (Orkambi) be switched to ELX-TEZ-IVA? If so, are there any special considerations (e.g., additional monitoring)? | The clinical experts consulted by CADTH noted that ELX-TEZ-IVA would replace earlier CFTR modulators that are significantly less effective (e.g., Orkambi) and that patients currently receiving those drugs would likely be switched to ELX-TEZ-IVA if they met eligibility and age criteria. The clinical experts noted that patients with CF are monitored in specialized clinics and that switching from LUM-IVA to ELX-TEZ-IVA would not be anticipated to pose challenges for patients or health care providers. |
Are there specific patient populations in which switching to ELX-TEZ-IVA would be inappropriate? | The clinical experts consulted by CADTH noted that switching would be appropriate for all patients receiving alternative CFTR modulators, provided they met the eligibility and age criteria. |
Considerations for initiation of therapy | |
Can the clinical experts confirm that multiple breath washout tests (e.g., LCI2.5) are only available at specialty clinics at children’s hospitals and not available at all pulmonary function testing clinics? | This measurement is not currently used in routine Canadian clinical practice and would not be practical for the purposes of determining eligibility for ELX-TEZ-IVA reimbursement. |
If children aged 2 to 5 years cannot complete an accurate spirometry (to obtain ppFEV1), and the CFQ-R is not validated in this age group, are there other parameters or biomarkers that could be measured at the time of treatment initiation for the purposes of evaluating response to treatment? | The clinical experts noted that clinically meaningful objective measures of response to ELX-TEZ-IVA are challenging to implement in clinical practice, as patients aged 2 to 5 years often do not show CF symptoms that can be objectively measured in practice using the tools and instruments recommended for those aged 6 years and older. In addition, they are too young for spirometry measurements to be taken, and obtaining baseline measurements of pulmonary exacerbation or CF-related hospitalization is problematic due to low frequency and interpatient variability. Among the criteria currently recommended by CADTH, the BMI z score is the only baseline measurement that would be captured as part of routine practice for patients aged 2 to 5 years. With respect to biomarkers, the clinical experts emphasized the following important considerations regarding sweat chloride:
|
Considerations for continuation or renewal of therapy | |
Are there any clinical benefits that have not been described in the sponsor’s renewal criteria or in the previous CDEC-recommended renewal criteria that should be considered for use as renewal criteria? | The clinical experts emphasized that ELX-TEZ-IVA has the potential to modify the course of disease for patients with CF. When used in older patients, nearly all patients demonstrated sufficient clinical benefit to have reimbursement renewed by the public drug programs. The clinical experts noted that, although objective measures are challenging to implement in clinical practice for patients aged 2 to 5 years, these patients would likely benefit from initiating treatment. The clinical experts noted that subsequent renewals for ELX-TEZ-IVA can be achieved through communication that the patient is continuing to benefit from the treatment and that such an approach could be applied for younger patients, where obtaining meaningful baseline and follow-up measurements of objective criteria would be challenging. |
Can the renewal criteria for patients aged 6 years and older be used for patients aged 2 to 5 years (except for FEV1 and/or CFQ-R)? | The clinical experts consulted by CADTH noted the following regarding the application of the existing reimbursement criteria to patients aged 2 to 5 years:
|
If a patient starts ELX-TEZ-IVA between the ages of 2 and 5 years, when they turn 6 years, can they just follow renewal criteria for the 6 years and older population? | The clinical experts consulted by CADTH noted that the application of the criteria for older patients may be challenging as those aged 2 to 5 years. |
Considerations for discontinuation of therapy | |
The previous CDEC recommendation for ELX-TEZ-IVA included a criterion that reimbursement should be discontinued in patients who have undergone lung transplant. Is this discontinuation criterion appropriate for patients aged 2 to 5 years? | ELX-TEZ-IVA is generally a well-tolerated treatment, and patients who began treatment before age 6 years would be expected to remain on the therapy for many years if they continued to benefit. Some of these patients may eventually require a lung transplant; therefore, the discontinuation criterion remains relevant for a recommendation issued for the younger patient population. |
Are there other discontinuation criteria that public drug plans should consider? | The clinical experts consulted by CADTH did not identify additional objective discontinuation criteria for ELX-TEZ-IVA reimbursement. |
Considerations for prescribing of therapy | |
Currently, there are CDEC-recommended prescribing criteria for the treatment of CF in patients aged 6 years and older who have at least 1 F508del mutation in the CFTR gene:
Are the above prescribing criteria appropriate for patients aged 2 to 5 years? | The only appropriate setting for initiation and monitoring of treatment with ELX-TEZ-IVA remains an adult or pediatric CF clinic. This treatment will typically be initiated and monitored in the outpatient clinic setting by a CF physician and the associated multidisciplinary team (e.g., specialists in respirology, infectious diseases, and gastroenterology). The experts noted that the drug may also be initiated in hospital. It would not be appropriate that a nonspecialty setting or physician would prescribe and monitor treatment with ELX-TEZ-IVA. ELX-TEZ-IVA would not be prescribed in combination with another CFTR modulator. |
Generalizability | |
Is there a clinical desire to use ELX-TEZ-IVA in patients younger than 2 years? | The clinical experts consulted by CADTH and those who provided input to CADTH through the call for clinician input all supported starting ELX-TEZ-IVA as soon as possible. |
Care provision issues | |
No questions. | — |
System and economic issues | |
No questions. | — |
BMI = body mass index; CDEC = CADTH Canadian Drug Expert Committee; CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire–Revised; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; FEV1 = forced expiratory volume in the first second; LCI2.5 = lung clearance index; LUM-IVA = lumacaftor-ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of ELX-TEZ-IVA in the treatment of CF in patients aged 2 to 5 years who have at least 1 F508del mutation in the CFTR gene. The focus will be placed on comparing ELX-TEZ-IVA to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of ELX-TEZ-IVA is presented in 4 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and randomized controlled trials (RCTs) that were selected according to the sponsor’s systematic review protocol. The second section includes sponsor-submitted long-term extension studies (not submitted). The third section includes indirect evidence from the sponsor (derived from a previous submission for ELX-TEZ-IVA). The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence (not submitted).
Included Studies
Clinical evidence from the following are included in the CADTH review and appraised in this document:
1 pivotal study or RCT identified in the systematic review
1 ITC.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Description of Study
Characteristics of the included study are summarized in Table 9.
Table 9: Details of the Study Included in the Systematic Review
Study characteristic | Study 111 (NCT04537793) |
|---|---|
Design and population | |
Study design | Phase III, 2-part (Parts A and B), open-label, multicentre trial |
Locations | Part A: 7 sites: US Part B: 22 sites: US, Australia, Canada, Germany, UK |
Patient enrolment dates | Start date: November 2020 (Part A ended March 2021; Part B began July 2021) End date: June 2022 |
Randomized | All patients received ELX-TEZ-IVA Part A: N = 18 Part B: N = 75 |
Inclusion criteria | Part A: Male and female patients with CF aged 2 to 5 years (inclusive) with F/MF or F/F genotypes. Patients must have weighed ≥ 14 kg on day 1. Part B: Male and female patients with CF aged 2 to 5 years (inclusive) who have at least 1 F508del mutation in the CFTR gene or another ELX-TEZ-IVA–responsive CFTR mutation. Patients must have weighed ≥ 10 kg at the screening visit. |
Exclusion criteria | Patients with clinically significant cirrhosis or portal hypertension Patients with lung infections caused by organisms associated with a rapid decline in pulmonary status Patients who have undergone solid organ or hematological transplant |
Drugs | |
Intervention | Part A: ≥ 14 kg: ELX 100 mg q.d., TEZ 50 mg q.d., IVA 75 mg q.12.h. Part B:
|
Comparator(s) | None |
Study duration | |
Screening phase | Part A: 28 days Part B: 28 days |
Treatment phase | Part A: 15 days Part B: 24 weeks |
Follow-up phase | Part A: 28 days Part B: 28 days |
Outcomes | |
Primary end point | Part A:
Part B: Safety and tolerability assessments as determined by AEs, clinical laboratory values, standard 12-lead electrocardiograms, vital signs, and pulse oximetry |
Secondary and exploratory end points | Secondary Part B:
Exploratory Part B:
|
Publication status | |
Publications | Goralski et al. (2023)40 Clinicaltrials.gov (NCT04537793)41 |
AE = adverse event; BMI = body mass index; CF = cystic fibrosis; ELX = elexacaftor; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; IVA = ivacaftor; LCI2.5 = lung clearance index; PK = pharmacokinetic; q.d. = every day; q.12.h = every 12 hours; TEZ = tezacaftor.
Source: Sponsor’s Summary of Clinical Evidence.
Study Design
The clinical development program for ELX-TEZ-IVA in patients aged 2 to 5 years was designed to demonstrate similar pharmacokinetic exposures and safety as for older patients, with the efficacy demonstrated in patients aged 6 years and older extrapolated to that in patients aged 2 to 5 years. Study 111 (NCT04537793) was an interventional, phase III, nonrandomized, 2-part (A and B), open-label study. In total, 18 patients in Part A and 75 patients in Part B were treated with ELX-TEZ-IVA.40
Part A
The primary objectives of Part A were to evaluate the pharmacokinetics of ELX, TEZ, and IVA when dosed in triple combination and to evaluate the safety and tolerability of ELX-TEZ-IVA.40 This part of Study 111 was initiated on November 19, 2020 (when the first patient gave informed consent) and concluded on March 5, 2021 (when the last patient completed their final visit). This part of the study involved a 4-week (28-day) screening period to assess patients for study eligibility and characteristics at baseline, a 15-day treatment period, and a 4-week (28-day) posttreatment follow-up visit to assess safety and tolerability (Figure 1).40 Part A was conducted at 7 study sites in the US.
Figure 1: Design of Part A of Study 111
ELX/TEZ/IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor.
Source: Clinical Study Report.42
Part B
The primary objective of Part B was to assess safety and tolerability in 75 patients treated with ELX-TEZ-IVA. Secondary objectives included assessing pharmacokinetics, pharmacodynamics, and efficacy.40 The study design of Part B involved a 4-week (28-day) screening period to assess patients for study eligibility and characteristics at baseline, a 24-week treatment period, and a 4-week (28-day) posttreatment follow-up visit to assess safety and tolerability (provided patients had not enrolled in the associated open-label extension study, NCT0515331743) (Figure 2).40 A total of 22 sites were used to conduct Part B, 2 of which were in Canada (The Hospital for Sick Children, Toronto, and British Columbia Children’s Hospital, Vancouver).
Figure 2: Design of Part B of Study 111
ELX/TEZ/IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor.
a The safety follow-up visit was not required for patients who enrolled in the optional open-label extension safety study within 28 days of the last scheduled visit in the treatment period.
Source: Clinical Study Report.42
Populations
Inclusion and Exclusion Criteria
Patients were eligible to be included in Study 111 if they had received a diagnosis of CF and were aged 2 to 5 years (inclusive). In Part A, eligible patients were required to have a F508del-CFTR mutation that was either F/F or F/MF. In Part B, patients were eligible if they had an F508del-CFTR mutation or another ELX-TEZ-IVA–responsive CFTR mutation. Patients were required to be at least 14 kg in Part A and at least 10 kg in Part B. Patients were excluded from the study if they had any comorbidities that could impact treatment outcomes or if they had received a prior hematological or solid organ transplant.
Interventions
Study 111 was a single-arm, sequential assignment trial. Patients in Part A and Part B received ELX-TEZ-IVA for 15 days and 24 weeks, respectively, with a safety follow-up visit conducted 4 weeks after treatment cessation, provided patients had not enrolled in the associated open-label extension study (NCT05153317) within 28 days of their final visit during the treatment period of Part B.
Part A: Patients received ELX 100 mg, TEZ 50 mg, and IVA 75 mg in combination, and IVA 75 mg.
Part B: Patients weighing least 10 kg and less than 14 kg received ELX 80 mg, TEZ 40 mg, and IVA 60 mg in combination, and IVA 59.5 mg; patients weighing at least 14 kg received the same dose as patients in Part A.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 10; the table is followed by descriptions of the outcome measures. The summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as on any outcomes identified as important to this review according to the clinical experts consulted by CADTH and on stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected the end points considered most relevant to inform CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using Grading of Recommendations, Assessment, Development, and Evaluations (GRADE). Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Sweat Chloride
SwCl samples were obtained from patients using an approved collection device. At each time point, 2 samples were collected, 1 from each of the patient’s arms, and sent to a central laboratory for analysis.42 All the included studies evaluated absolute change from baseline in SwCl. Absolute change from baseline in SwCl through 24 weeks was a secondary end point in Study 111.42
Lung Clearance Index
The LCI is a multiple breath washout test that estimates the number of lung volume turnovers required to clear the lung of an inert gas.44 The test is sensitive to changes in the small airways and may be able to detect pulmonary disease in patients with normal FEV1.8,45 LCI2.5 represents the number of lung turnovers required to reduce the end tidal nitrogen concentration to 2.5% of the starting value. The LCI assessments were derived from multiple breath washout testing using nitrogen. Absolute change from baseline in LCI2.5 was a secondary end point of Study 111.42
Pulmonary Exacerbations
Pulmonary exacerbations were evaluated as an exploratory end point in Study 111.42 Pulmonary exacerbations were defined as newly initiated or changed treatment with oral, inhaled, or IV antibiotics and fulfillment of 1 criterion from List A or of 2 criteria from List B (Table 11), within the period from 3 days before the antibiotic start date through to the antibiotic stop date.42
Table 10: Outcomes Summarized From Study 111
Outcome measure | Time point | Type of outcome | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|---|---|
Safety and tolerability | 28 weeksa | Primary | Descriptive statistics | Not applicable | Not applicable | Not applicable |
Sweat chloride, LCI2.5 | 24 weeks | Secondary | MMRM |
|
| No sensitivity analyses |
Pulmonary exacerbations | 24 weeks | Exploratory | Descriptive statistics (annualized event rate) | Not applicable | Not applicable | Not applicable |
CF-related hospitalizations | 24 weeks | Exploratory | ||||
Weight | 24 weeks | Exploratory | MMRM |
|
| No sensitivity analyses |
Weight z score | ||||||
Height | ||||||
Height z score | ||||||
BMI | ||||||
BMI z score |
BMI = body mass index; CF = cystic fibrosis; F/F = homozygous for F508del mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; LCI2.5 = lung clearance index; MMRM = mixed-effects model for repeated measures.
aSafety was assessed at a separate visit occurring 4 weeks after the end of the treatment period.
Source: Clinical Study Report.42
Table 11: Pulmonary Exacerbation Criteria
List | Number of criteria | Criteria |
|---|---|---|
List A | Must meet 1 of these criteria |
|
List B | Must meet 2 of these criteria |
|
FEV1 = forced expiratory volume in the first second.
Source: Clinical Study Report.42
CF-Related Hospitalizations
Complications related to CF that led to hospitalization were recorded in Study 111, in terms of both the number of patients with a hospitalization event and the total number of events. The yearly event rate was also calculated.
Growth Parameters (BMI, Weight, Height, Z Scores)
Absolute change from baseline in BMI, BMI z score, weight, weight z score, height, and height z score at 24 weeks were exploratory end points in Study 111.
Adverse Events
AEs were defined as any untoward medical occurrence in a patient during the study, including newly occurring events or worsening of pre-existing conditions (e.g., increase in severity or frequency). An AE was considered serious if it resulted in any of the following outcomes: death; life-threatening condition; inpatient hospitalization or prolongation of hospitalization; persistent or significant disability, incapacity, congenital anomaly, or birth defect; or an important medical event that jeopardized the patient or required medical or surgical intervention to prevent one of the aforementioned outcomes.
Statistical Analysis
Sample Size and Power Calculation
For Part B, no formal power calculation was performed. The sample size of approximately 70 patients was deemed adequate to meet the primary outcome of safety and tolerability. Assuming a dropout rate of 10% to 20%, approximately 56 to 63 patients were expected to complete the study.
Statistical Testing
As discussed above, an MMRM approach was used to assess the mean absolute change in SwCl from baseline through week 24.46 SwCl values obtained from all available visits up to week 24 were included in the model, with visit as a fixed effect and baseline SwCl and genotype group (F/F or F/MF) as covariates.46 The model was estimated using restricted maximum likelihood.46 Denominator degrees of freedom for the F test of fixed effects were estimated using the Kenward-Roger approximation. An unstructured covariance structure was used to model the within-patient errors.46 If the model estimation did not converge, a compound symmetry covariance structure was used instead.46 Conditional on the observed data and covariates, missing data were assumed to be missing at random; consequently, no imputation of missing data was performed. The results obtained from the model were the average treatment effect through week 24, estimated using all postbaseline visits up to and including week 24.46 The estimated mean change from baseline in SwCl through week 24, along with the corresponding 2-sided 95% CI and P value, was provided.46
The same MMRM structure was then applied to assess mean absolute change in LCI2.5 from baseline through week 24 and to analyze the absolute change from baseline in weight, height, BMI, and associated z scores at week 24 (full analysis set [FAS], Part B).46 Given that efficacy was a secondary objective of Study 111, no adjustments were made for multiplicity and all P values were considered nominal.46
Descriptive analyses were performed for all other end points in Part B.46
Multiple Testing Procedure
There was no adjustment for multiplicity for any of the end points in Study 111.
Subgroup Analyses
The absolute change in SwCl and LCI2.5 from baseline through week 24 was analyzed in each genotype subgroup (i.e., F/F and F/MF). Similar MMRMs as specified in the Statistical Testing section were conducted for each subgroup, with genotype removed from the covariates.46
Analysis Populations
A summary of analysis sets defined in Study 111 is presented in Table 12.
Table 12: Analysis Populations of Study 111
Population | Definition | Application |
|---|---|---|
All patients set | All patients who were enrolled or who received at least 1 dose of ELX-TEZ-IVA | Individual patient data listings and disposition summary tables |
Safety set | All patients who received at least 1 dose of ELX-TEZ-IVA | All safety analyses |
Full analysis set | All enrolled patients who carried the intended CFTR allele mutation and received at least 1 dose of ELX-TEZ-IVA | Baseline characteristics; all efficacy analyses (unless specified) |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor.
Source: Sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
A summary of patient dispositions for Study 111, Part B, is presented in Table 13. A total of 75 patients were enrolled and received at least 1 dose of ELX-TEZ-IVA; 74 patients (98.7%) completed the study. One (1.3%) patient discontinued due to an AE.
Table 13: Summary of Patient Disposition From Study 111 Part B
Patient disposition | ELX-TEZ-IVA (N = 75) |
|---|---|
Screened, n | NR |
Reason for screening failure, n (%) | NR |
Enrolled, n | 75 |
Discontinued from study, n (%) | 1 (1.3) |
Rolled over into extension study, n (%) | 71 (94.7) |
Reason for discontinuation, n (%) | |
Adverse events | 1 (1.3) |
Lost to follow-up | 0 (0) |
Consent withdrawal | 0 (0) |
FAS, N | || || |||| |
Safety, N | 75 |
FAS = full analysis set; NR = not reported.
Source: Clinical Study Report.42
Baseline Characteristics
The baseline characteristics outlined in Table 14 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. The mean age of the patients was 4.1 years, and over half (54.7%) of patients were female. Twenty-three (30.7%) patients had an F/F genotype, and 52 (69.3%) had an F/MF genotype. The baseline values for mean SwCl and mean LCI2.5 were 100.7 mmol/L and 8.41, respectively.
Table 14: Summary of Baseline Characteristics From Study 111 Part B
Characteristic | ELX-TEZ-IVA (N = 75) |
|---|---|
Sex, n (%) | |
Male | 34 (45.3) |
Female | 41 (54.7) |
Age (years) | |
Mean (SD) | 4.1 (1.1) |
Median (range) | 4.0 (2.1 to 6.0) |
≥ 2 to < 3 years, n (%) | 11 (14.7) |
≥ 3 to < 4 years, n (%) | 27 (36.0) |
≥ 4 to < 5 years, n (%) | 22 (29.3) |
≥ 5 to < 6 years, n (%) | 15 (20.0) |
Geographic region, n (%) | |
North America | || || |||||| || |||| |
Europe and Australia | | || || || ||| || |||| |
CFTR genotype, n (%) | |
F/F | 23 (30.7) |
F/MF | 52 (69.3) |
Other | 0 (0.0) |
Baseline values, mean (SD) | |
Weight (kg) | 16.5 (3.2) |
Weight z score | –0.07 (0.89) |
Height (cm) | 101.8 (9.2) |
Height z score | –0.09 (1.10) |
BMI (kg/m2) | 15.79 (1.06) |
BMI z score | 0.09 (0.85) |
Sweat chloride (mmol/L) | 100.7 (11.2) |
LCI2.5 (mmol/L) | 8.41 (1.48) |
Prior CF medications, n (%) | |
Prior use of CFTR modulator | 10 (13.3) |
Prior use of dornase alfa | 33 (44.0) |
Prior use of azithromycin | 8 (10.7) |
Prior use of inhaled antibiotic | 8 (10.7) |
Prior use of any bronchodilator | 51 (68.0) |
Prior use of any inhaled bronchodilator | 51 (68.0) |
Prior use of any inhaled hypertonic saline | 37 (49.3) |
Pseudomonas aeruginosa infection, n (%) | |
Infection in 2 years prior to screening | 14 (18.7) |
BMI = body mass index; CF = cystic fibrosis; F/F = homozygous for F508del mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; LCI2.5= lung clearance index; SD = standard deviation.
Source: Clinical Study Report.42
Exposure to Study Treatments
Table 15 summarizes the exposure to ELX-TEZ-IVA in Study 111.
Table 15: Summary of Patient Exposure From Study 111 Part B
Exposure | ELX-TEZ-IVA (N = 75) |
|---|---|
Total (patient-weeks) | || || |||| |
Duration, mean (SD) | | || || || || || | |
Duration, median (range) | | || || || || || || | | |
Adherence (%) | | || | |
SD = standard deviation.
Source: Clinical Study Report.42
Concomitant Medications and Co-Interventions
A summary of concomitant medications used among patients in Part B of Study 111 is presented in Table 16. The most common concomitant medications were typically used for the management of CF. Pancreatin, sodium chloride, and salbutamol were the most commonly used concomitant medications in Part B.
Table 16: Summary of Concomitant Medications Used in Study 111 Part B
Concomitant medication, n (%) | ELX-TEZ-IVA (N = 75) |
|---|---|
| || || || || || || || || || || || | | || || |||| || || |
|| || || || || || || || | || || |||| || || |
|| || |||| || || | || || |||| || || |
|| || |||| || || | || || |||| || || |
|| || |||| || || | || || |||| || || |
|| || |||| || || | || || |||| || || |
|| || |||| || || | || || |||| || || |
|| || |||| || || | || || |||| || || |
|| || |||| || || | || || |||| || || |
|| || |||| || || | || || |||| || || |
|| || |||| || |||| || |||| || |||| || |||| || |||| || |||| | || || |||| || || |
|| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || |||| || || | || || |||| || || |
|| || |||| || || | || || |||| || || |
Source: Clinical Study Report.42
Efficacy
Key efficacy outcomes assessed in Study 111 included SwCl concentration, LCI2.5, number of pulmonary exacerbations, number of CF-related hospitalizations, as well as body weight, height, BMI, and their respective z scores (Table 17). Specific outcomes are discussed in further detail in the following subsections.
Table 17: Summary of Key Efficacy Results From Study 111 Part B
End point | ELX-TEZ-IVA (N = 75) |
|---|---|
Sweat chloride | |
Patients contributing to the analysis, n | 69 |
Baseline (mmol/L), mean (SD) | 100.7 (11.2) |
Change from baseline (mmol/L), mean (95% CI) | –57.9 (–61.3 to –54.6) |
Treatment group difference versus control (95% CI) | NR |
P value | < 0.0001 |
LCI2.5 | |
Patients contributing to the analysis, n | 50 |
Baseline, mean (SD) | 8.41 (1.48) |
Change from baseline, mean (95% CI) | –0.83 (–1.01 to –0.66) |
Treatment group difference versus control (95% CI) | NR |
P value | < 0.0001 |
Pulmonary exacerbations | |
Patients contributing to the analysis, n | 75 |
Patients with events, n (%) | 12 (16.0) |
Number of events | 12 |
Observed event rate per year | 0.32 |
P value | NR |
CF-related hospitalizations | |
Patients contributing to the analysis, n | || || |||| |
Patients with events, n (%) | || || |||| |
Number of events | || || |||| |
Observed event rate per year | || || |||| |
P value | || || |||| |
Body weight | |
Patients contributing to the analysis, n | || || |||| |
Baseline (kg), mean (SD) | || || |||| |
Change from baseline (kg), mean (95% CI) | || || |||| |
Treatment group difference versus control (95% CI) | || || |||| |
P value | || || |||| |
Body weight z score | |
Patients contributing to the analysis, n | || || |||| |
Baseline, mean (SD) | || || |||| |
Change from baseline, mean (95% CI) | || || |||| |
Treatment group difference versus control (95% CI) | || || |||| |
P value | || || |||| |
Height | |
Patients contributing to the analysis, n | || || |||| |
Baseline (cm), mean (SD) | || || |||| |
Change from baseline (cm), mean (95% CI) | || || |||| |
Treatment group difference versus control (95% CI) | || || |||| |
P value | || || |||| |
Height z score | |
Patients contributing to the analysis, n | 75 |
Baseline, mean (SD) | || || |||| |
Change from baseline, mean (95% CI) | –0.06 (–0.11 to 0.00) |
Treatment group difference versus control (95% CI) | NR |
P value | NR |
BMI | |
Patients contributing to the analysis, n | 75 |
Baseline (kg/m2), mean (SD) | 15.79 (1.06) |
Change from baseline (kg/m2), mean (95% CI) | 0.03 (–0.10 to 0.17) |
Treatment group difference versus control (95% CI) | NR |
P value | NR |
BMI z score | |
Patients contributing to the analysis, n | 75 |
Baseline, mean (SD) | 0.09 (0.85) |
Change from baseline, mean (95% CI) | 0.10 (0.00 to 0.20) |
Treatment group difference versus control (95% CI) | NR |
P value | NR |
BMI = body mass index; CF = cystic fibrosis; CI = confidence interval; LCI2.5 = lung clearance index; NR = not reported; SD = standard deviation.
Source: Sponsor’s Summary of Clinical Evidence.
Sweat Chloride Concentration
In Study 111, SwCl concentration was reported for the FAS as well as for the F/F and F/MF subgroups. The mean baseline concentration was calculated across 71 patients in the FAS (n = 75) and was applied to the F/F (n = 23) and F/MF (n = 52) subgroups (100.7 mmol/L; standard deviation [SD] = 11.2). In the FAS, treatment with ELX-TEZ-IVA resulted in an improvement (reduction) in SwCl by week 4 that was sustained throughout the treatment period (Figure 3). The within-group LS mean absolute change from baseline through week 24 was –57.9 mmol/L (95% CI, –61.3 to –54.6; nominal P < 0.0001). A numerically larger reduction in SwCl was observed in the F/F group (–70.0 mmol/L; 95% CI, –75.4 to –64.5) than in the F/MF group (–52.6 mmol/L; 95% CI, –56.9 to –48.4) (Table 18).
Table 18: Summary of Sweat Chloride Concentration in Study 111 Part B
SwCl analysis | ELX-TEZ-IVA | ||
|---|---|---|---|
All patients N = 75 | Patients with F/F n = 23 | Patients with F/MF n = 52 | |
Patients contributing to the analysis, n | 69 | 22 | 47 |
Baseline (mmol/L), mean (SD) | 100.7 (11.2)a | 100.7 (11.2)a | 100.7 (11.2)a |
End of treatment time point, mean | 24 weeks | 24 weeks | 24 weeks |
Change from baseline (mmol/L), mean (95% CI) | –57.9 (–61.3 to –54.6) | –70.0 (–75.4 to –64.5) | –52.6 (–56.9 to –48.4) |
P value | < 0.0001 | NR | NR |
CI = confidence interval; F/F = homozygous for F508del mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; NR = not reported; SD = standard deviation; SwCl = sweat chloride.
aBaseline was taken across 71 patients.
Source: Clinical Study Report.42
Table 19: Summary of Lung Clearance Index in Study 111 Part B
LCI2.5 analysis | ELX-TEZ-IVA | ||
|---|---|---|---|
All patients N = 63 | Patients with F/F n = 23 | Patients with F/MF n = 52 | |
Patients contributing to the analysis, n | 50 | 17 | 33 |
Baseline LCI2.5, mean (SD) | 8.41 (1.48)a | 8.41 (1.48)a | 8.41 (1.48)a |
End of treatment time point, mean | 24 weeks | 24 weeks | 24 weeks |
Change in LCI2.5 from baseline, mean (95% CI) | –0.83 (–1.01 to –0.66) | –0.89 (–1.15 to –0.63) | –0.82 (–1.06 to –0.57) |
P value | < 0.0001 | NR | NR |
CI = confidence interval; F/F = homozygous for F508del mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; LCI2.5 = lung clearance index; NR = not reported; SD = standard deviation.
aBaseline was taken across 51 patients.
Source: Clinical Study Report.42
Figure 3: Absolute Change in Sweat Chloride by Visit in Study 111 Part B
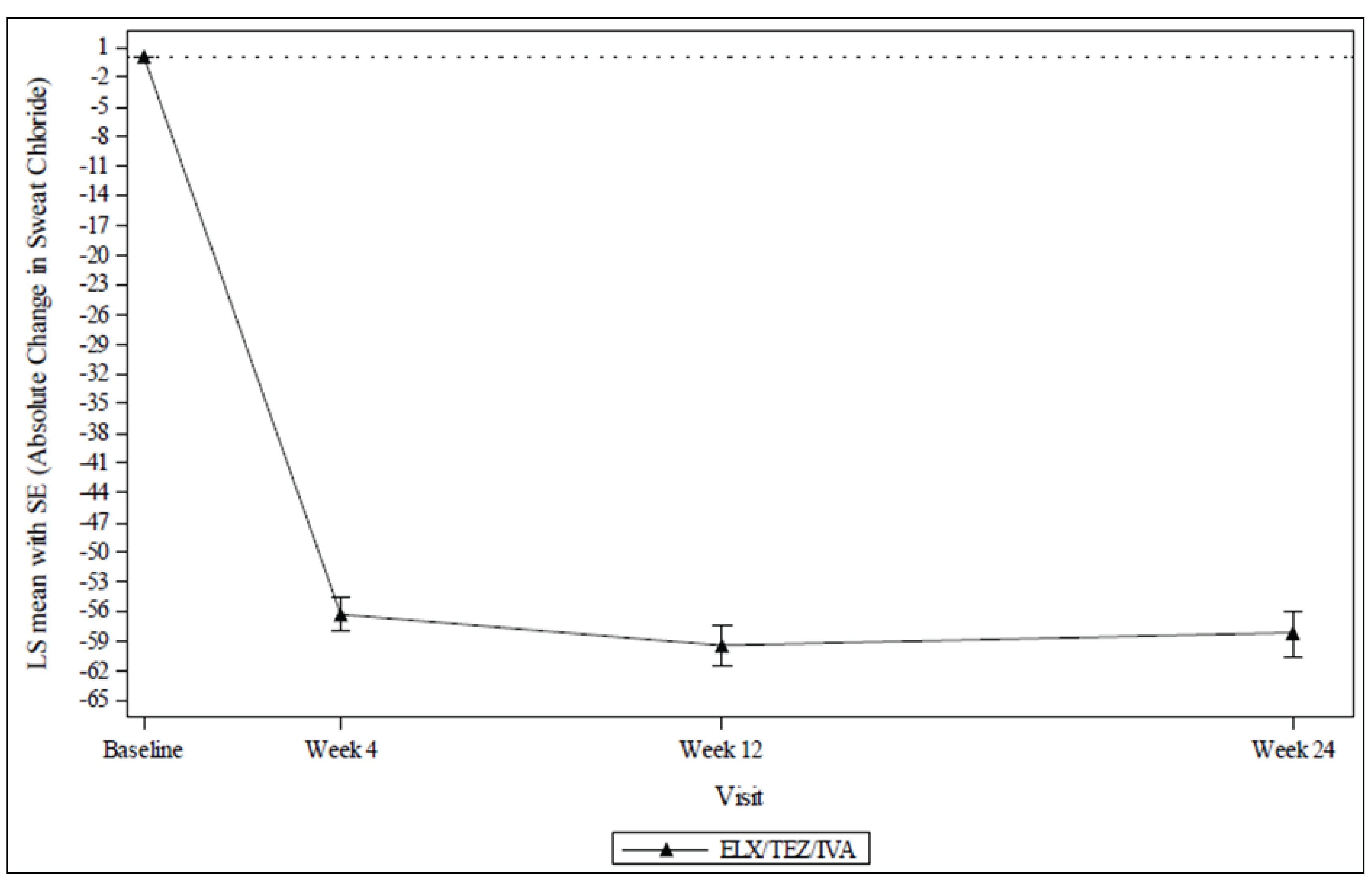
ELX/TEZ/IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; LS = least squares; SE = standard error.
Source: Clinical Study Report.42
Lung Clearance Index
Results for within-group change from baseline in LCI2.5 are summarized in Table 19. LCI2.5 was only evaluated in patients aged 3 years or older at the time of screening (n = 50). Among those patients, treatment with ELX-TEZ-IVA resulted in an improvement (reduction) in LCI2.5 by 24 weeks: the within-group LS mean absolute change from baseline was –0.83 (95% CI, –1.01 to –0.66; nominal P < 0.0001). As shown in Figure 4, the reduction from baseline was observed at all postbaseline assessments (i.e., weeks 4, 12, and 24). The results were similar in the F/F and F/MF genotype subgroups: LS mean change of –0.89 (95% CI, –1.15 to –0.63) and LS mean change of –0.82 (95% CI, –1.06 to –0.57), respectively.
Pulmonary Exacerbations
In Study 111, 16.0% of patients experienced a pulmonary exacerbation event through week 24 (1 event each). This corresponded to a yearly pulmonary exacerbation event rate of 0.32 (Table 20).
Table 20: Summary of Pulmonary Exacerbations in Study 111 Part B
Analysis | ELX-TEZ-IVA (N = 75) |
|---|---|
PEx | |
Patients contributing to the analysis, n | 75 |
End of treatment time point, mean | 24 weeks |
Patients with events, n (%) | 12 (16.0) |
Number of events | 12 |
Observed event rate per year | 0.32a |
P value | NR |
PEx requiring hospitalization | |
Patients with events, n (%) | || || |||| |
Number of events | || || |||| |
Observed event rate per year | || || |||| |
PEx requiring IV antibiotic therapy | |
Patients with events, n (%) | || || |||| |
Number of events | || || |||| |
Observed event rate per year | || || |||| |
PEx requiring hospitalization or IV antibiotic therapy | |
Patients with events, n (%) | || || |||| |
Number of events | || || |||| |
Observed event rate per year | || || |||| |
NR = not reported; PEx = pulmonary exacerbation.
aOne year was counted as 48 weeks (336 days).
Source: Clinical Study Report.42
Body Weight
In Study 111, the mean baseline weight in the FAS was || || |||||| || ||||. The absolute increase in body weight at week 24 was || || |||||| || |||||| || ||||. The mean baseline body weight z score was || || |||||| || ||||. The absolute increase in body weight z score at week 24 was || || |||||| || |||||| || ||||.
Height
In Study 111, the mean baseline height among all 75 patients in the FAS was || || |||||| || ||||. The absolute increase in height at week 24 was || || |||||| || |||||| || ||||. At baseline, the mean height z score was || || |||| || || ||||. The absolute decrease in height z score at week 24 was –0.06 (95% CI, –0.11 to 0.00).
Body Mass Index
In Study 111, the mean baseline BMI for the FAS was 15.79 kg/m2 (SD = 1.06 kg/m2). The absolute increase in BMI at week 24 was || || |||||| || |||||| || |||||| || ||||. The mean baseline BMI z score was 0.09 (SD = 0.85). The absolute increase in BMI z score at 24 weeks was 0.10 (95% CI, 0.00 to 0.20).
Figure 4: Absolute Change in Lung Clearing Index by Visit in Study 111 Part B
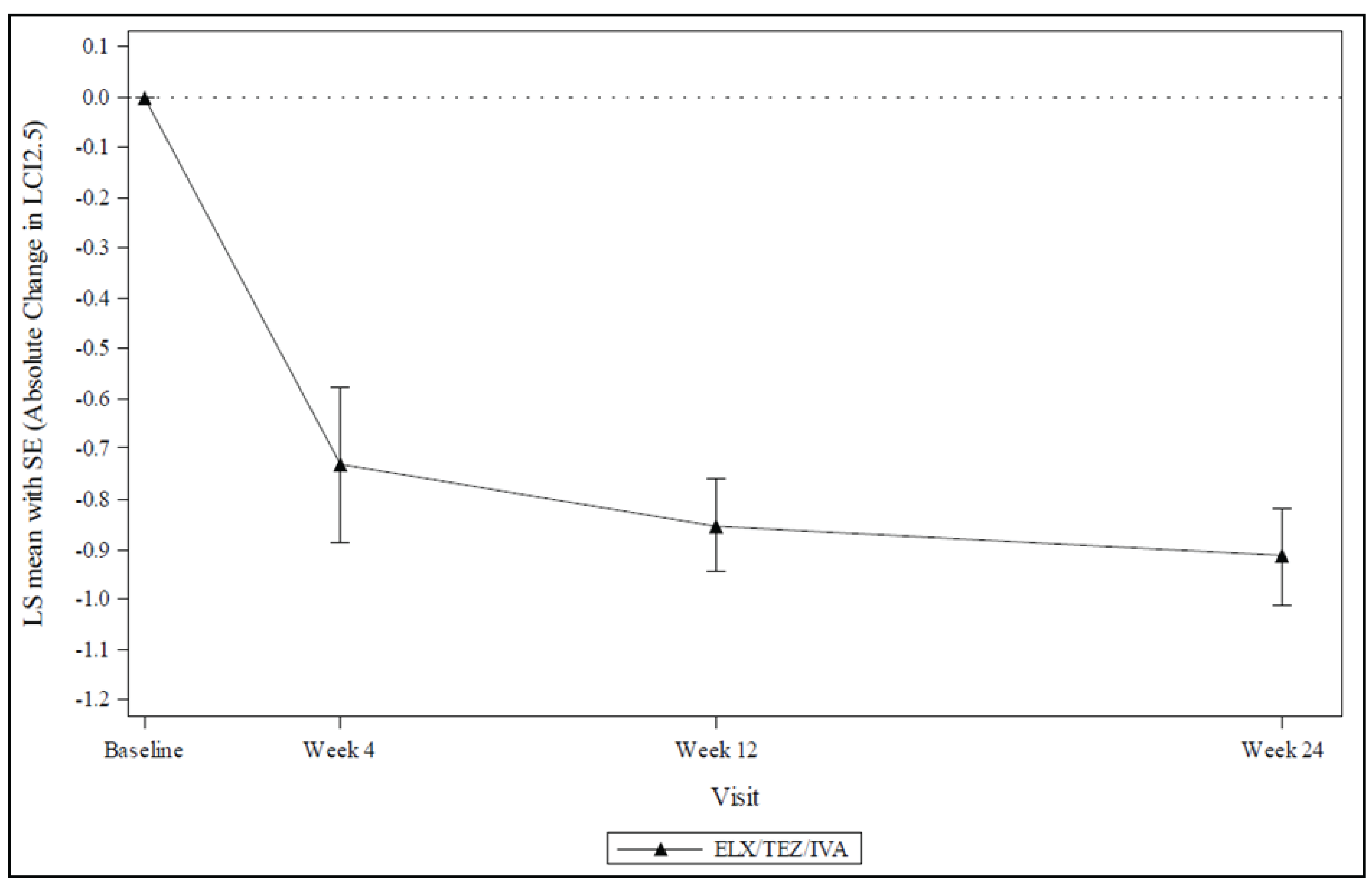
ELX/TEZ/IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; LCI2.5 = lung clearance index; LS = least squares; SE = standard error.
Source: Clinical Study Report.42
CF-Related Hospitalizations
There were no CF-related hospitalizations in Study 111.
Harms
Refer to Table 21 for harms data. Safety and tolerability were evaluated as the primary outcomes in Part B. The overall safety profile of ELX-TEZ-IVA was assessed based on the following safety and tolerability end points: treatment-emergent AEs, clinical laboratory values, standard 12-lead electrocardiograms, vital signs, and pulse oximetry. Rash and elevated transaminase levels were investigated as AEs of special interest.
Adverse Events
In total, 98.7% of patients experienced 1 or more AEs, all of which were either mild (62.7%) or moderate (36.0%) in severity. Overall, common AEs (i.e., in ≥ 10% of patients) were generally consistent with common manifestations and complications of CF in patients aged 2 through 5 years, as well as with the established safety profile of ELX-TEZ-IVA. The AEs most commonly reported among the patients were cough (61.3%), pyrexia (34.7%), and rhinorrhea (33.3%).
Serious Adverse Events
Two patients (2.7%) experienced SAEs. One patient had concurrent SAEs of anal incontinence (mild in severity), urinary incontinence (moderate in severity), and abnormal behaviour (moderate in severity). The other patient had an SAE of infective pulmonary exacerbation of CF (moderate in severity) that was not related to the treatment.
Withdrawals Due to Adverse Events
One patient (1.3%) discontinued treatment due to an SAE of abnormal behaviour. Five patients (6.7%) experienced AEs leading to treatment interruption. Of these 5 patients, 2 experienced rash leading to treatment interruption, 1 experienced anal and urinary incontinence, 1 experienced aggression, and 1 experienced increased levels of ALT, AST, and gamma-glutamyl transferase.
Adverse Events of Special Interest
Eight patients (10.7%) experienced elevated transaminase events and 15 (20.0%) experienced rash events. All events were deemed mild or moderate in severity, and none were serious. Two of the patients experienced rash events leading to treatment interruption. There were no study discontinuations due to rash events or elevated transaminase events.
Table 21: Summary of Harms Results From Study 111 Part B
Adverse events | ELX-TEZ-IVA (N = 75) |
|---|---|
Most common adverse events, n (%)a | |
≥ 1 adverse event | 74 (98.7) |
Cough | 46 (61.3) |
ALT increased | 8 (10.7) |
Rhinorrhea | 25 (33.3) |
AST increased | 4 (5.3) |
Rash | 12 (16.0) |
Pyrexia | 26 (34.7) |
Vomiting | 21 (28.0) |
COVID-19 | 14 (18.7) |
Nasal congestion | 13 (17.3) |
Upper respiratory tract infection | 11 (14.7) |
Decreased appetite | 9 (12.0) |
Infective pulmonary exacerbation of CF | 8 (10.7) |
Headache | || || |||| |
SARS-CoV-2 test positive | || || |||| |
Constipation | || || |||| |
Nasopharyngitis | || || |||| |
Abdominal discomfort | || || |||| |
Diarrhea | || || |||| |
Abdominal pain | || || |||| |
GGT increased | || || |||| |
Irritability | || || |||| |
Serious adverse events, n (%) | |
Patients with ≥ 1 serious adverse event | 2 (2.7) |
Patients who stopped treatment due to adverse events, n (%) | |
Patients who had adverse events leading to treatment discontinuation | 1 (1.3) |
Patients who had adverse events leading to interruption of treatment | 5 (6.7) |
Deaths, n (%) | |
Patients who died | 0 (0) |
Adverse events of special interest, n (%) | |
Elevated transaminase levels | 8 (10.7) |
Rash | 15 (20.0) |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; CF = cystic fibrosis; GGT = gamma-glutamyl transferase.
aAdverse events occurring in at least 5% of patients by preferred term.
Source: Sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
Study 111 was conducted in a manner similar to all other pivotal studies for the use of CFTR modulators in patients aged between 2 and 5 years (i.e., expansion of approval indications for Orkambi4,5 and Kalydeco6,7). Each of these studies was conducted in 2 parts, with Part A involving a small number of patients (n = 18 for Study 111), with a primary objective of evaluating pharmacokinetics, and Part B enrolling more patients (n = 75 for Study 111), with the primary objective of evaluating safety and tolerability.
As with the previously reviewed trials in adults, adolescents, and children aged 6 to 11 years, few patients were discontinued from Study 111 (100% completion for Part A and 98.7% completion for Part B). Adherence to the study treatments was evaluated by counting the number of study drugs at each visit and was reported to be || || |||| for Part A and || || |||| for Part B. In accordance with the study protocols, the use of concomitant medications remained stable throughout the treatment period for all treatment groups.
As with the other trials for CFTR modulators in patients aged 2 to 5 years, ELX-TEZ-IVA was administered in an open-label manner in Study 111, and there was no comparator group for either Part A or Part B. The limited number of secondary efficacy end points evaluated in the study were objective and unlikely to be influenced by the open-label administration of a CFTR modulator (i.e., change from baseline in SwCl concentration and change from baseline in LCI2.5).
There are no globally accepted definitions of pulmonary exacerbations in patients with CF. The definitions used in Study 111 were considered appropriate by regulatory authorities and the clinical experts consulted by CADTH. There was no independent adjudication of pulmonary exacerbation events. Pulmonary exacerbations were only evaluated with descriptive statistics, and there were no prebaseline or postbaseline comparisons of event rates. In response to an inquiry from CADTH regarding why pulmonary exacerbations were not included as an efficacy end point, the sponsor reported that, as had been noted in relation to the pediatric trial for patients aged 6 to 11 years, exacerbations occur less frequently in younger patients than in older patients. As Study 111 was a single-arm trial without a defined pretreatment evaluation period, and due to the low pulmonary exacerbation rates in the study population, comparison to a pretreatment event rate was not possible.
Statistical power calculations were performed for Part A of Study 111 (pharmacokinetic assessment) but were not performed for Part B, as the primary objective was the evaluation of safety and tolerability. The sponsor planned for a sample size of approximately 70 patients; the assumed dropout rate of 10% to 20% was far greater than the 1 patient (1.3%) who did drop out. Enough patients were enrolled and completed both parts of the study for the sponsor to evaluate the primary objectives. The MMRM analyses for the primary evaluations assumed data were missing at random, which may not be a valid assumption. However, the amount of missing data in the trials was low.
The secondary end points were analyzed without statistical testing procedures to control the type I error rate (the sponsor noted that all P values were considered nominal); therefore, the results should be interpreted with caution due to the risk of inflated type I error.
External Validity
The eligibility and diagnostic criteria used to screen patients for Study 111 were similar to those used in the other phases of the ELX-TEZ-IVA clinical development program (i.e., Studies 106 and 116 for patients aged 6 to 11 years and Studies 102, 103, 104, and 109 for patients aged 12 years and older). As noted in the previous CADTH review of ELX-TEZ-IVA, these criteria are generally consistent with Canadian clinical practice for diagnosing patients with CF. As all Canadian provinces and territories have instituted newborn screening, diagnosis of CF and confirmation of genotyping would typically occur early in the child’s life (an average of 1 month after birth). As such, no changes would be needed in diagnostic testing requirements to establish patient eligibility based on CF diagnosis and genotype for the revised age range for ELX-TEZ-IVA.
The clinical experts consulted by CADTH noted that the baseline growth parameters for the patients in Study 111 were a reasonable reflection of the typical patient in Canadian practice.
Changes from baseline in lung function were evaluated as a secondary efficacy end point in Study 111 using LCI2.5. This is reflective of regulatory guidance, which has noted that spirometry may not be sensitive enough to detect treatment differences in children with CF. In addition, spirometry is not typically performed in patients younger than 6 years in Canada, and FEV1 has not been used as a clinical trial end point in any CFTR modulator studies for patients younger than 6 years. LCI is used in CF clinical trials as it may be more sensitive in identifying early underlying structural deficiencies within the lungs of patients with CF that cannot be detected using spirometry.8,9 Similar to spirometry assessments, the LCI test can be challenging to accurately perform with young children. In Study 111, the sponsor noted that the LCI test was only performed on patients aged 3 years or older at the time of screening. Although LCI is used as an end point in clinical studies, it is not routinely used in Canadian clinical practice, and the clinical relevance of differences in this end point have not been characterized.9,10 The clinical experts consulted by CADTH indicated that LCI is not reliably correlated with FEV1. A literature review conducted by CADTH found that variable correlation was observed between FEV1 and LCI in children.
As with all other CFTR modulator phase III trials, Study 111 excluded patients with a history of colonization with B. cenocepacia, B. dolosa, and/or M. abscessus. CF Canada reports that a small minority of patients in Canada have colonization with Burkholderia species (3.2% in 2020), with only 12.9% of those cases reported in children.47 As with previous CADTH reviews of ELX-TEZ-IVA, the clinical experts consulted by CADTH noted that the exclusion of such patients does not reduce the generalizability of the study results.
The proportion of patients in Study 111 who were positive for P. aeruginosa was 18.7%, which is close to the rate reported for children aged 2 to 5 years in the 2019 CF Canada data registry (approximately 14%). The clinical experts consulted by CADTH noted that the rate of P. aeruginosa infection in Study 111 is similar to what would be anticipated in routine Canadian clinical practice.
Similar to other phase III studies for CFTR modulators, including those for ELX-TEZ-IVA for patients aged 6 to 11 years and 12 years and older, Study 111 excluded patients who had a respiratory infection, pulmonary exacerbation, or changes in their therapy for pulmonary disease in the 4 weeks prior to the first dose of the study drug. As in the previous CADTH reviews of ELX-TEZ-IVA,48,49 the clinical experts consulted by CADTH noted that the exclusion of these patients is unlikely to limit the generalizability of the results to the broader patient population with CF.
ELX-TEZ-IVA was added to the existing therapeutic regimens used by the patients, which is reflective of how ELX-TEZ-IVA would be administered in clinical practice. The clinical experts consulted by CADTH indicated that the background therapies used in Study 111 were similar to what would be anticipated in Canadian clinical practice, with the following exceptions: all patients in Canadian practice would be supplementing with vitamins, and the use of mucolytics (i.e., dornase alfa and inhaled hypertonic saline) could be slightly lower for patients aged 2 to 5 years in Canada.
The 24-week study treatment periods were sufficient for observing change from baseline in SwCl and LCI2.5 in Study 111; however, the clinical experts consulted by CADTH suggested that 24 weeks is unlikely to be enough time to observe meaningful changes in BMI for a younger patient population that is relatively healthy. In addition, the absence of a control group in Study 111 limits the ability to interpret the results of change from baseline in the growth parameters.
Patients and caregivers in Study 111 received extensive contact with health professionals over the study period (i.e., 8 clinic visits and 2 phone contacts). This level of contact is not reflective of routine care for patients with CF with relatively stable disease.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Patients who completed Study 111 were eligible to enrol in an open-label extension study. However, the sponsor reported that the interim results of the extension study were not available at the time of filing the application with CADTH.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Feasibility of ITC in patients aged 2 to 5 years: The sponsor conducted an ITC to compare the clinical efficacy of ELX-TEZ-IVA in Study 111 with other CFTR modulators in patients with F/F and F/MF mutations to generate the inputs needed for the cost-effectiveness analysis. A meta-analysis approach via MMRM was used with individual patient-level data from relevant trials; data from all comparators were included in 1 model for each genotype. The sponsor concluded that the ITC was not feasible due to the small number of patients in this age group, which reduced the power to detect differences between ELX-TEZ-IVA, LUM-IVA, and/or placebo. As such, the sponsor did not include the ITC in its submission to CADTH and used estimates from the previous CADTH submission for patients aged 6 to 11 years as assumptions within its economic model.
ITCs in patients aged 6 to 11 years and 12 years and older: To inform the pharmacoeconomic model, the sponsor submitted estimates of clinical efficacy of ELX-TEZ-IVA compared to placebo derived from ITCs previously conducted for patients aged 6 to 11 years and 12 years and older using individual patient-level data from relevant phase III randomized controlled clinical trials.
Table 22: Summary of Methods Used to Inform Acute ppFEV1 Increase in the Cost-Effectiveness Model
Model input | F/F | F/G | F/RF | F/MF |
|---|---|---|---|---|
Acute ppFEV1 increase when simulated patients aged 2 to 5 years turn age 6 years in the cost-effectiveness model. | ITC conducted for patients aged 6 to 11 years was used to inform efficacy for ELX-TEZ-IVA vs. placebo and LUM-IVA vs. placebo. | ITC conducted for patients aged 12 years and older was used to inform efficacy for ELX-TEZ-IVA vs. placebo and relative efficacy observed between patient populations aged 6 to 11 years and 12 years and older. | ITC conducted for patients aged 12 and older was used to inform efficacy for ELX-TEZ-IVA vs. placebo and relative efficacy observed between patient populations aged 6 to 11 years and 12 years and older. | Separate ITC was not conducted. Placebo-adjusted data from Study 116 in patients aged 6 to 11 years were used to inform efficacy. |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ITC = indirect treatment comparison; LUM-IVA = lumacaftor-ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second; vs. = versus.
Description of Indirect Comparison
As shown in Table 23, for patients aged 6 to 11 years, the sponsor conducted an indirect comparison investigating the comparative efficacy of ELX-TEZ-IVA versus other CFTR modulators and placebo for patients with an F/F genotype.50
Table 23: Study Selection Criteria and Methods for Indirect Treatment Comparison
Characteristic | ITC study selection criteria and methods |
|---|---|
Population | Patients with CF aged 6 to 11 years with F/F genotype |
Intervention | ELX 200 mg–TEZ 100 mg–IVA 150 mg (every morning) + IVA 150 (every evening) |
Comparator |
|
Outcome | ppFEV1 |
Study design |
|
Publication characteristics | Not reported |
Exclusion criteria | Not reported |
Databases searched | Not reported |
Selection process | Not reported |
Data extraction process | Not reported |
Quality assessment | Not reported |
CF = cystic fibrosis; ELX = elexacaftor; F/F = homozygous for F508del mutation in the CFTR gene; IVA = ivacaftor; LUM-IVA = lumacaftor-ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second; TEZ = tezacaftor; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
Source: Sponsor’s indirect treatment comparison.50,51
ITC for Patients Aged 6 to 11 Years With F/F Genotype
Study Selection Methods
The criteria used by the sponsor to select studies for inclusion in the ITC for patients aged 6 to 11 years with F/F genotype were as follows:
study design is a phase III trial
population includes patients aged 6 to 11 years with F/F genotype
interventions include ELX-TEZ-IVA, TEZ-IVA, or LUM-IVA
comparators include TEZ-IVA, LUM-IVA, or placebo
study duration of 24 weeks.
A systematic literature search and review was not undertaken by the sponsor to identify studies for inclusion. The sponsor reported that since Vertex Pharmaceuticals is the only manufacturer with relevant CFTR modulators and conducted all the relevant phase III trials, a systematic literature review was unlikely to retrieve any additional relevant evidence.50 CADTH did not identify any additional studies that would have met the inclusion criteria but were not included in the ITC.
ITC Analysis Methods
The ITC analyses for ELX-TEZ-IVA versus LUM-IVA, TEZ-IVA, and placebo, as well as for LUM-IVA and TEZ-IVA versus placebo, were conducted using an MMRM meta-analysis approach and individual patient-level data from patients with an F/F genotype in the relevant treatment groups from each of the included trials. The sponsor reported that an MMRM meta-analysis approach was the most appropriate methodology for the following reasons:
Study 106 was a single-arm trial, which precluded anchored comparisons using Bucher or network meta-analysis methods.
The sponsor (Vertex Pharmaceuticals) has access to the individual patient-level data for each of the relevant comparator groups.
The baseline characteristics were similar across all included studies.
MMRM allows flexibility to account for missing data and aligns more closely with the original study analyses for each of the end points.
The MMRM meta-analysis approach is consistent with the methodology used in the cross-trial comparison requested by the European Medicines Agency Paediatric Committee as part of the European Paediatric Investigation Plan (MAA Module 2.5, Section 4.7.2).
The relevant comparisons for the CADTH review are ELX-TEZ-IVA versus LUM-IVA or IVA (as TEZ-IVA is not approved for use in patients aged 6 to 11 years in Canada). For these comparisons, the sponsor calculated the estimated treatment difference for ppFEV1 (defined as the average of weeks 4, 8, 16, and 24). The MMRMs included treatment group, visit, and treatment-by-visit interaction as fixed effects. The covariates for adjustment were sex and the corresponding baseline variable. A 2-sided 95% CI and a 2-sided P value for the estimated indirect treatment difference were calculated based on normal approximation. The sponsor conducted additional analyses for ELX-TEZ versus TEZ-IVA using 8-week end points, as 24-week data were unavailable. These analyses are reported in this section of the report but are not appraised in detail as TEZ-IVA is not approved for use in patients aged 6 to 11 years in Canada.50
Table 24: Indirect Comparisons for Patients Aged 6 to 11 Years With F/F Genotype
Within-group estimates (study) | End point |
|---|---|
| Absolute change in ppFEV1 (through 24 weeks)a |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; LUM-IVA = lumacaftor-ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
aAnalyses for TEZ-IVA were conducted at 8 weeks.
Source: Sponsor’s indirect treatment comparison.50
Results for ITC Analysis
Included Studies
The evidence network for the studies with patients aged 6 to 11 years who have an F/F genotype is shown in Figure 5. Indirect comparisons were performed for ELX-TEZ-IVA versus placebo, ELX-TEZ-IVA versus LUM-IVA, and ELX-TEZ-IVA versus TEZ-IVA.50
Study Characteristics
Table 25 provides a summary of the characteristics of the studies that were included in the indirect comparison for patients aged 6 to 11 years with an F/F genotype. There were 4 studies included in the primary analysis (Studies 106B, 809-109, 809-011B, and 661-113B), and an additional study involving TEZ-IVA was used in sensitivity analyses (Study 661-115). The single-arm Study 109B provided the estimated treatment effect for ELX-TEZ-IVA. Pooled data from Study 809-109 (a double-blind, placebo-controlled RCT) and the single-arm Study 809-011B provided the estimated treatment effect for LUM-IVA. The estimated effect for the placebo was also derived from Study 809-109. The estimated treatment effects for TEZ-IVA were from the single-arm Study 661-113B in the primary analyses and from Study 661-115 for sensitivity analyses.50
The inclusion criteria were generally similar across the studies. The key exceptions were the inclusion of patients with genotypes other than F/F in Study 106B (patients with an F/MF genotype were also enrolled) and Studies 661-113B and 661-115 (patients with an F/RF genotype were also enrolled) and the lower threshold for ppFEV1 in the inclusion criteria (≥ 40% in Studies 106B, 809-011B, and 661-113B versus ≥ 70% in Studies 809-109 and 661-115). Despite this difference, the baseline ppFEV1 was similar across the studies (summarized in Table 26). All the studies enrolled patients aged between 6 and 11 years. The ppFEV1 was calculated using the Global Lung Function Initiative approach in the trials for ELX-TEZ-IVA (Study 106B) and TEZ-IVA (Studies 661-113B and 661-115) and using the Wang-Hankinson reference equations in the studies for LUM-IVA (Studies 809-109 and 809-011B). The sponsor recalculated the ppFEV1 values from the LUM-IVA studies using the Global Lung Function Initiative normalization approach to align with the approach used in the ELX-TEZ-IVA and TEZ-IVA trials. The end points were generally similar across the studies; however, only Studies 809-109 and 661-115 included a prespecified primary efficacy end point.50
Figure 5: Indirect Comparison Network for Patients Aged 6 to 11 Years With F/F Genotype
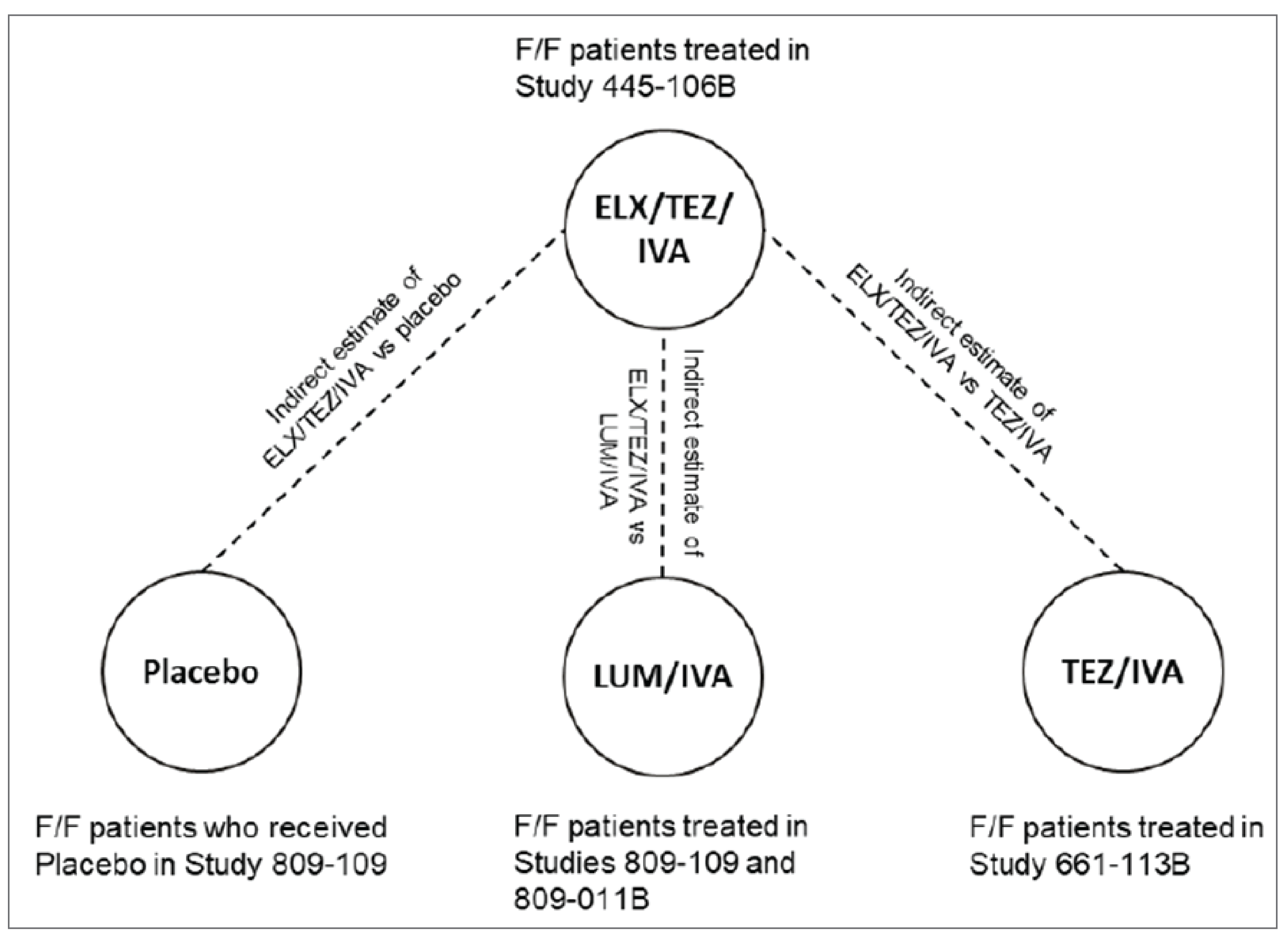
ELX/TEZ/IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; LUM/IVA = lumacaftor-ivacaftor; TEZ/IVA = tezacaftor-ivacaftor and ivacaftor; vs = versus.
Source: Sponsor’s indirect treatment comparison.50
Table 25: Study Characteristics in Patients Aged 6 to 11 Years With F/F Genotype
Characteristic | ELX-TEZ-IVA | LUM-IVA | TEZ-IVA | ||
|---|---|---|---|---|---|
Study 106B | Study 809-109 | Study 809-011B | Study 661-113B | Study 661-115a | |
Study design | Single arm, OL | DB RCT | Single arm, OL | Single arm, OL | DB RCT |
Study population |
|
|
|
|
|
Treatment groups | ELX-TEZ-IVA |
| LUM-IVA | TEZ-IVA |
|
CFTR modulator washout requirements | ≥ 28 days prior to day 1 visit | ≥ 30 days prior to screening visit | ≥ 30 days prior to screening visit | ≥ 30 days prior to day 1 visit (≥ 28 days for LUM-IVA) | ≥ 28 days prior to day 1 visit |
Treatment duration | 24 weeks | 24 weeks | 24 weeks | 24 weeks | 8 weeks |
Baseline ppFEV1 inclusion criteria | ≥ 40% (GLI) | ≥ 70% (Wang equation) | ≥ 40% (Wang equation) | ≥ 40% (GLI) | ≥ 70% (GLI) |
Sample size | ELX-TEZ-IVA: 66 |
| LUM-IVA: 58 | TEZ-IVA: 70 |
|
Primary efficacy end point | NA | Absolute change from baseline in LCI2.5 through week 24 | NA | NA | Absolute change from baseline in LCI2.5 through week 8 |
DB = double blind; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; GLI = Global Lung Function Initiative; ITC = indirect treatment comparison; IVA = ivacaftor; LCI2.5 = lung clearance index; LUM-IVA = lumacaftor-ivacaftor; NA = not applicable; OL = open label; ppFEV1 = percent predicted forced expiratory volume in the first second; RCT = randomized controlled trial; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
aSensitivity analysis only.
Source: Sponsor’s indirect treatment comparison.50
Baseline Characteristics
As shown in Table 26, the baseline and demographic characteristics were generally similar across the studies included in the indirect comparisons for patients aged 6 to 11 years with an F/F genotype. For the comparisons of interest for this review (i.e., ELX-TEZ-IVA versus LUM-IVA or placebo), the baseline ppFEV1, LCI2.5, weight-for-age z score, and BMI-for-age z score were similar across the treatment groups, with the exception of the BMI-for-age z score, which was greater in Study 661-113B (TEZ-IVA), at 0.39 (SD = 0.90) than in the other trials (range, –0.09 [0.86] in the pooled LUM-IVA studies to 0.09 [0.96] in the pooled TEZ-IVA studies). The baseline CFQ-R respiratory domain scores were lower for the placebo (77.1) and LUM-IVA groups (78.5) than for the ELX-TEZ-IVA and TEZ-IVA groups (81.8 and 83.2, respectively).
Table 26: Baseline Characteristics in Patients Aged 6 to 11 Years With F/F Genotype
Characteristic | ELX-TEZ-IVA Study 106B (N = 29) | Placebo Study 809-109 (N = 101) | LUM-IVA Studies 809-109 and 809-011, Part B, pooled (N = 160) | TEZ-IVA | ||
|---|---|---|---|---|---|---|
Study 661-113, Part B (N = 61) | Study 661-115 (N = 42) | Studies 661-113, Part B, and 661-115, pooled (N = 103) | ||||
Sex, n (%) | ||||||
Male | 12 (41.4) | 43 (42.6) | 66 (41.3) | 31 (50.8) | 20 (47.6) | 51 (49.5) |
Female | 17 (58.6) | 58 (57.4) | 94 (58.8) | 30 (49.2) | 22 (52.4) | 52 (50.5) |
Age at screening (years) | ||||||
Mean (SD) | 8.3 (1.9) | 8.9 (1.6) | 8.8 (1.6) | 8.0 (1.8) | 8.5 (1.6) | 8.2 (1.7) |
Median | 8.0 | 9.0 | 9.0 | 8.0 | 9.0 | 8.0 |
ppFEV1 (%), mean (SD) | 87.3 (18.3) | 88.6 (11.1) | 87.5 (13.6) | 91.2 (12.4) | NA | 88.7 (12.9) |
LCI2.5 (lung turnovers), mean (SD) | 10.26 (3.36) | 10.26 (2.24) | 10.25 (2.42) | NA | 9.84 (2.17) | NA |
Weight z score, mean (SD) | –0.23 (0.59) | –0.21 (0.76) | –0.14 (0.90) | 0.18 (0.94) | NA | –0.04 (0.90) |
BMI z score, mean (SD) | –0.10 (0.61) | –0.14 (0.88) | –0.09 (0.86) | 0.39 (0.90) | NA | 0.09 (0.96) |
CFQ-R respiratory domain, mean (SD) | 81.8 (12.0) | 77.1 (15.5) | 78.5 (14.4) | 81.7 (13.9) | NA | 83.2 (12.5) |
BMI = body mass index; CFQ-R = Cystic Fibrosis Questionnaire–Revised; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; LCI2.5 = lung clearance index; LUM-IVA = lumacaftor-ivacaftor; NA = not applicable; ppFEV1 = percent predicted forced expiratory volume in the first second; SD = standard deviation; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
Source: Sponsor’s indirect treatment comparison.50
Indirect Comparison Results
Table 27 provides a summary of the results of the indirect comparisons for studies conducted in patients aged 6 to 11 years with an F/F genotype. The sponsor reported the following indirect estimates of effect for ELX-TEZ-IVA compared with LUM-IVA: || || |||||| || |||||| || |||||| || |||||| || |||||| || |||| for absolute change in ppFEV1 from baseline through 24 weeks.50
Table 27: Results of Indirect Comparison for Patients Aged 6 to 11 Years With F/F Genotype
Absolute change in ppFEV1 from baseline through 24 weeks | ELX-TEZ-IVA Study 445-106B (N = 29) | LUM-IVA Studies 809-109 and 809-011B (N = 160) | TEZ-IVA Study 661-113B (N = 61) | Placebo Study 809-109 (N = 101) |
|---|---|---|---|---|
LS mean within-group (95% CI) P value | || || |||||| || |||||| | | || || |||||| || |||||| | | || || |||||| || |||||| | | || || |||||| || |||||| | |
LS mean between-group difference (95% CI) (ELX-TEZ-IVA versus comparator) P value | || || |||||| || |||||| | | || || |||||| || |||||| | | || || |||||| || |||||| | | || || |||||| || |||||| | |
CI = confidence interval; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; LS = least squares; LUM-IVA = lumacaftor-ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
Source: Sponsor’s indirect treatment comparison.50
Critical Appraisal
The screening phase (4 weeks), treatment phase (24 weeks), and follow-up phase were similar across the studies. The key baseline characteristics were generally similar across the studies, with the exception that the baseline CFQ-R respiratory domain scores were lower for the placebo (77.1) and LUM-IVA groups (78.5) than for the ELX-TEZ-IVA and TEZ-IVA groups (81.8 and 83.2, respectively).
Table 28: CADTH Assessment of Homogeneity for the ITC for Patients Aged 6 to 11 Years With F/F Genotype
Characteristic | Description and handling of potential effect modifiers |
|---|---|
Disease severity |
|
Treatment history | All the trials required patients with prior exposure to a CFTR modulator to undergo a washout period of at least 4 weeks. |
Clinical trial eligibility criteria | Eligibility criteria were generally similar across the included trials, with the following exceptions:
|
Dosing of comparators | All the study drugs were used in accordance with the recommendations in the Canadian product monographs.29,37,39 |
Definitions of end points | The end points included in the ITC were similarly defined and evaluated for each of the included studies. |
Timing of end point evaluation or trial duration | All the end points were calculated using the same approach for the MMRM (e.g., through 24 weeks for ppFEV1). |
Withdrawal frequency | There were few withdrawals from any of the trials included in the ITC. |
Clinical trial setting | All of the studies included in the ITC were phase III studies conducted at specialized CF clinics. |
Study design | The screening phase (4 weeks), the treatment phase (24 weeks), and the follow-up phase were similar across the included studies. Studies 106B, 809-011B, and 661-113B were single-arm, open-label trials. Studies 809-109 and 661-115 were double-blind, placebo-controlled trials. |
BMI = body mass index; CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire–Revised; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ITC = indirect treatment comparison; LCI2.5 = lung clearance index; LUM-IVA = lumacaftor-ivacaftor; MMRM = mixed-effects model for repeated measures; ppFEV1 = percent predicted forced expiratory volume in the first second; RCT = randomized controlled trial; SwCl = sweat chloride; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
ITC for Patients Aged 12 Years and Older With F/G Genotype
Study Selection Methods
The criteria used by the sponsor to select studies for inclusion in the ITC for patients aged 12 years and older with an F/G genotype were as follows:
Study design is a phase III RCT.
Population includes patients aged 12 years and older with an F/G (including 1 F508del mutation and 1 R117H mutation in the CFTR gene [F/R117H]) genotype.
Interventions include ELX-TEZ-IVA or IVA.
Comparators include IVA or placebo.
Study duration is 8 weeks.
It was not reported if a systematic literature search and review was undertaken by the sponsor to identify studies for inclusion; however, CADTH did not identify any additional studies that would have met the inclusion criteria but were not included in the ITC.
ITC Analysis Methods
The indirect comparison for ELX-TEZ-IVA versus placebo in patients aged 12 years and older with an F/G genotype was estimated using the Bucher method for continuous end points, with IVA as the common comparator. The sponsor stated that the Bucher method was considered the most appropriate approach for this indirect comparison because of the 4-week IVA run-in period included in Study 104 (but not in the STRIVE, KONNECTION, or KONDUCT trials). As all the patients in the STRIVE, KONNECTION, and KONDUCT trials were naive to CFTR modulator treatment at baseline, the baselines were not considered to be sufficiently comparable to conduct an individual patient-level data meta-analysis.
The sponsor used MMRMs to estimate the direct treatment effects from each of the studies, which were subsequently used in the Bucher indirect comparison. For ppFEV1, CFQ-R domain scores, and SwCl, the sponsor calculated the estimated treatment difference through 8 weeks. This was calculated using the average of weeks 4 and 8 for Study 104, KONNECTION, and KONDUCT, but only the week 8 measurement for STRIVE (as the trial did not include a week 4 assessment). The sponsor conducted an additional supportive analysis, in which the week 8 assessments were used for all the studies. For BMI-for-age z score and weight-for-age z score, the sponsor calculated the estimated treatment difference at week 8.
Each MMRM included treatment group, visit, and treatment-by-visit interaction as fixed effects and patient as the random effect. Additionally, the MMRM that used data from KONNECTION included treatment period as a fixed effect (as KONNECTION was a crossover trial and the others were parallel group trials). The covariates included for adjustment were based on those included in the MMRM for each of the trials used in the ITC and included age at screening (≥ 12 years to < 18 years, versus ≥ 18 years) and continuous baseline ppFEV1. Age at screening was not included as a covariate for the MMRM that used data from the KONDUCT trial, because only 1 patient was aged at least 12 years but younger than 18 years at the time of screening. Additionally, continuous baseline SwCl was included as a covariate in the MMRM used to estimate the ELX-TEZ-IVA versus IVA treatment effect, as it was included as a covariate in the MMRM of Study 104.
The direct estimates for IVA versus placebo were derived from a meta-analysis of subgroup analyses from 3 studies: STRIVE, KONNECTION, and KONDUCT. A fixed-effect, meta-analysis approach was used to combine the individual treatment effects and estimate the overall treatment effect of IVA versus placebo. The individual effects were weighted by the inverse variance of each effect-size estimate.
A 2-sided 95% CI and a 2-sided P value for each estimated indirect treatment difference were calculated based on normal approximation.
Table 29: Indirect Comparisons for Patients Aged 12 Years and Older With F/G Genotype
Indirect estimate | Direct estimates (study) | End point |
|---|---|---|
ELX-TEZ-IVA versus placebo | ELX-TEZ-IVA versus TEZ-IVA (subgroup data from Study 104) + IVA versus placebo (meta-analysis of STRIVE, KONNECTION, and KONDUCT) | ppFEV1 (through 8 weeks) |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; IVA = ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
Source: Sponsor’s indirect treatment comparison.51
Results for ITC Analysis
Included Studies
The evidence network for the studies with patients aged 12 years and older who have an F/G genotype is shown in Figure 6. Indirect comparison was performed for ELX-TEZ-IVA versus placebo. The direct evidence for ELX-TEZ-IVA versus IVA was derived from a subgroup analysis of Study 104. The direct estimates for IVA versus placebo were derived from a meta-analysis of subgroup analyses from 3 studies: STRIVE, KONNECTION, and KONDUCT.
Figure 6: Indirect Comparison Network for Patients Aged 12 Years and Older With F/G Genotype
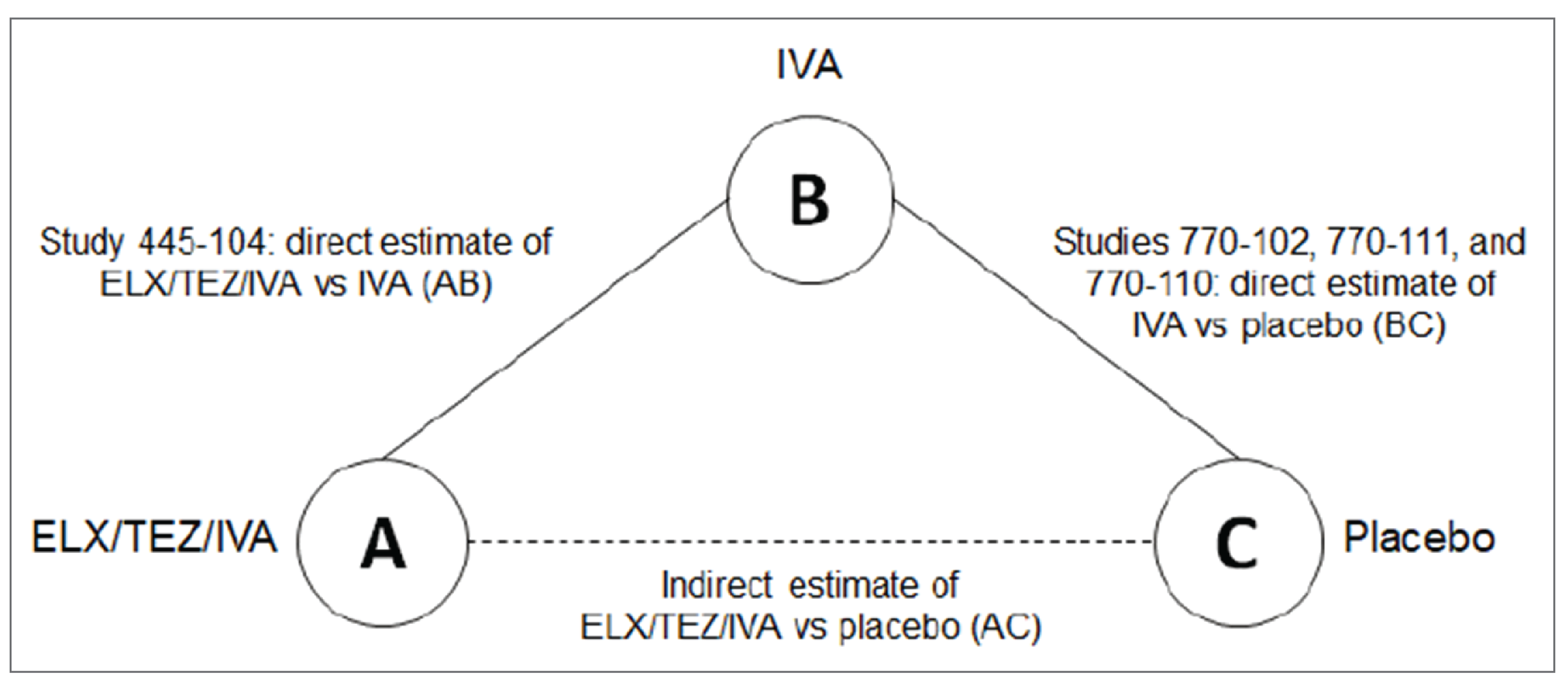
ELX/TEZ/IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; IVA = ivacaftor; vs = versus.
Source: Sponsor’s indirect treatment comparison.51
Study Characteristics
Table 30 provides a summary of the characteristics of the studies included in the indirect comparison for patients aged 12 years and older with an F/G genotype. A key difference across the studies was the use of a 4-week, open-label run-in period in Study 104, in which all patients received treatment with IVA prior to randomization. The studies had different durations for the treatment periods, ranging from 8 weeks in Study 104 and KONNECTION to 48 weeks in STRIVE. The sponsor performed the indirect comparisons using the 8-week time point for all assessments.
The inclusion criteria differed across the studies with respect to age and CFTR genotypes. Patients were required to be aged 12 years or older to be eligible for Study 104 and STRIVE, but patients aged 6 years and older were eligible for KONNECTION and KONDUCT. Study 104 enrolled patients with an F/G (including F/R117H) or an F/RF genotype. In contrast, the other studies were conducted in patients who did not necessarily have an F508del mutation: STRIVE enrolled patients with at least 1 G551D gating mutation; KONNECTION enrolled patients with at least 1 non-G551D gating mutation; and KONDUCT enrolled patients with at least 1 R117H mutation. Given the heterogenous study populations, the sponsor extracted subgroup data for patients who had an F/G genotype: 95 of 258 (37%) for Study 104, 122 of 161 (76%) for STRIVE, 34 of 78 (44%) for KONNECTION, and 39 of 69 (57%) for KONDUCT.
All the studies specified that patients had to have a ppFEV1 of at least 40% at screening to be eligible. Study 104, STRIVE, and KONDUCT all specified an upper threshold for ppFEV1 of 90% at screening to determine eligibility for enrolment; KONNECTION did not specify an upper threshold for ppFEV1.
All the studies specified absolute change in ppFEV1 from baseline as the primary end point. The primary end point was assessed through 8 weeks in Study 104 and KONNECTION and through 24 weeks in STRIVE and KONDUCT. All the studies included changes in BMI, CFQ-R, SwCl, and body weight as additional end points. STRIVE, KONNECTION, and KONDUCT included pulmonary exacerbations as an efficacy end point, and Study 104 did not; therefore, no indirect comparison can be conducted for this end point.
Table 30: Study Characteristics for Patients Aged 12 Years and Older With F/G Genotype
Characteristic | Study 104 (F/G subset) | STRIVE (F/G551D subset) | KONNECTION (F/non-G551D subset) | KONDUCT (F/R117H subset) |
|---|---|---|---|---|
Study population | Patients with F/G (including F/R117H) or F/RF genotypes and aged ≥ 12 years | Patients with ≥ 1 G551D gating mutation and aged ≥ 12 years | Patients with ≥ 1 non-G551D gating mutation and aged ≥ 6 years | Patients with ≥ 1 R117H mutation and aged ≥ 6 years |
Design | DB, active-controlled, parallel group RCT | DB, placebo-controlled parallel group RCT | DB, placebo-controlled, crossover RCT | DB, placebo-controlled, parallel group RCT |
Active run-in period | 4 weeks with TEZ-IVA or IVA | None | None | None |
Treatment period | 8 weeks | 48 weeks | 8 weeks | 24 weeks |
Treatment groups |
|
|
|
|
ppFEV1 inclusion criteria at screening | 40% to 90% | 40% to 90% | ≥ 40% | 40% to 90% for patients aged ≥ 12 years |
Schedule of assessments | Day 1, day 15, week 4, week 8 | Day 1, day 15, week 8, every 4 weeks thereafter | Day 1, week 2, week 4, week 8 of each treatment period | Day 1, week 2, week 4, week 8, week 16, week 24 |
Sample size |
|
|
|
|
Subset of patients included in ITC |
|
|
|
|
Primary efficacy end point | Absolute change in ppFEV1 from baseline through 8 weeks | Absolute change in ppFEV1 from baseline through 24 weeks | Absolute change in ppFEV1 from baseline through 8 weeks | Absolute change in ppFEV1 from baseline through 24 weeks |
Other end points | BMI, CFQ-R, SwCl, weight | BMI, CFQ-R, PEx, SwCl, weight | BMI, CFQ-R, PEx, SwCl, weight | BMI, CFQ-R, PEx, SwCl, weight |
BMI = body mass index; CFQ-R = Cystic Fibrosis Questionnaire–Revised; DB = double blind; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; patients with at least 1 non-G551D gating mutation; F/G551D = 1 F508del mutation and 1 G551D gating mutation; F/non-G551D = 1 F508del mutation and at least 1 non-G551D gating mutation; F/R117H = 1 F508del mutation and 1 R117H mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ITC = indirect treatment comparison; IVA = ivacaftor; PEx = pulmonary exacerbation; ppFEV1 = percent predicted forced expiratory volume in the first second; RCT = randomized controlled trial; SwCl = sweat chloride; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
Source: Sponsor’s indirect treatment comparison.51
Baseline Characteristics
Baseline and demographic characteristics for the F/G studies are summarized in Table 31. The median age of patients differed across the 4 studies, ranging from 22.0 and 25.0 years in the placebo and IVA groups, respectively, of the STRIVE trial to 43.0 and 39.5 years in the placebo and IVA groups, respectively, of the KONDUCT trial. Only 1 patient was younger than 18 years in the relevant subgroup of patients from the KONDUCT trial. The other trials included a subset of patients younger than 18 years at screening, ranging from 13.3% and 16.0% in the IVA and ELX-TEZ-IVA groups, respectively, of Study 104 to 29.4% and 35.3% in the placebo and IVA groups, respectively, of the KONNECTION trial.
Patients in KONNECTION had a higher mean ppFEV1 (79.9% and 78.8% in the placebo and IVA groups, respectively) than those in Study 104 (68.1% and 66.0% in the IVA and ELX-TEZ-IVA groups, respectively), STRIVE (63.9% and 59.6% in the placebo and IVA groups, respectively) and KONDUCT (61.3% and 68.1% in the placebo and IVA groups, respectively). The proportion of male and female patients differed across the studies, with the proportion of female patients ranging from 37.8% and 44.0% in the IVA and ELX-TEZ-IVA groups of Study 104, respectively, to 63.2% and 55.0% in the placebo and IVA groups of the KONDUCT trial, respectively. SwCl levels were substantially lower in Study 104 (47.6 mmol/L and 50.9 mmol/L in the IVA and ELX-TEZ-IVA groups, respectively) than in the other studies, where baseline SwCl ranged from 73.4 mmol/L and 66.1 mmol/L in the placebo and IVA groups of KONDUCT, respectively, to 101.0 mmol/L and 101.1 mmol/L in the placebo and IVA groups, respectively, of the STRIVE trial. Mean BMI at baseline was greater in the KONDUCT trial (24.51 kg/m2 and 27.56 kg/m2 in the placebo and IVA groups, respectively) than in the other trials. Baseline CFQ-R respiratory domain scores differed across the trials, ranging from 59.1 and 70.2 in the placebo and IVA groups, respectively, of the KONDUCT trial to 80.7 and 77.3 in the placebo and IVA groups of the KONNECTION trial, respectively.
Table 31: Baseline Characteristics for Patients Aged 12 Years and Older With F/G Genotype
Characteristic | Study 104 (F/G subset) | STRIVE (F/G551D subset) | KONNECTION (F/non-G551D subset) | KONDUCT (F/R117H subset) | ||||
|---|---|---|---|---|---|---|---|---|
IVA (N = 45) | ELX-TEZ-IVA (N = 50) | Placebo (N = 58) | IVA (N = 64) | Placebo (N = 17) | IVA (N = 17) | Placebo (N = 19) | IVA (N = 20) | |
Sex, n (%) | ||||||||
Male | 28 (62.2) | 28 (56.0) | 28 (48.3) | 30 (46.9) | 9 (52.9) | 9 (52.9) | 7 (36.8) | 9 (45.0) |
Female | 17 (37.8) | 22 (44.0) | 30 (51.7) | 34 (53.1) | 8 (47.1) | 8 (47.1) | 12 (63.2) | 11 (55.0) |
Age at screening (years) | ||||||||
Mean (SD) | 30.7 (11.2) | 33.4 (13.8) | 24.0 (9.5) | 26.0 (10.1) | 28.8 (12.7) | 28.1 (13.2) | 41.2 (12.3) | 37.7 (12.6) |
Median | 29.0 | 32.7 | 22.0 | 25.0 | 26.0 | 26.0 | 43.0 | 39.5 |
Age group at screening (years), n (%) | ||||||||
≥ 12 to < 18 | 6 (13.3) | 8 (16.0) | 15 (25.9) | 16 (25.0) | 5 (29.4) | 6 (35.3) | — | 1 (5.0) |
≥ 18 | 39 (86.7) | 42 (84.0) | 43 (74.1) | 48 (75.0) | 12 (70.6) | 11 (64.7) | 19 (100.0) | 19 (95.0) |
ppFEV1, mean (SD) | 68.1 (16.6) | 66.0 (14.8) | 63.9 (14.4) | 59.6 (15.3) | 79.9 (15.1) | 78.8 (18.9) | 61.3 (14.6) | 68.1 (16.8) |
BMI (kg/m2), mean (SD) | 22.91 (3.39) | 23.71 (3.76) | 22.02 (3.10) | 21.40 (3.04) | 23.49 (5.17) | 23.12 (5.36) | 24.51 (5.30) | 27.56 (5.42) |
Sweat chloride (mmol/L), mean (SD) | 47.6 (19.1) | 50.9 (23.3) | 101.0 (10.3) | 101.1 (9.8) | 91.0 (24.0) | 88.5 (22.3) | 73.4 (16.0) | 66.1 (22.9) |
CFQ-R respiratory domain score, mean (SD) | 75.8 (17.6) | 76.3 (16.4) | 71.9 (16.4) | 70.6 (16.6) | 80.7 (16.7) | 77.3 (14.4) | 59.1 (21.2) | 70.2 (20.7) |
BMI = body mass index; CFQ-R = Cystic Fibrosis Questionnaire–Revised; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/G551D = 1 F508del mutation and 1 G551D gating mutation; F/non-G551D = 1 F508del mutation and at least 1 non-G551D gating mutation; F/R117H = 1 F508del mutation and 1 R117H mutation in the CFTR gene; IVA = ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second; SD = standard deviation.
Source: Sponsor’s indirect treatment comparison.51
Indirect Comparison Results
Table 32 provides a summary of the results of the direct and indirect comparisons for studies conducted in patients aged 12 years and older with an F/G genotype. The indirect comparison results are shown side by side with the direct comparison results for ELX-TEZ-IVA versus IVA (derived from the subgroup analysis from Study 104) and with direct comparison results for IVA versus placebo (derived from the meta-analysis of STRIVE, KONNECTION, and KONDUCT). The sponsor reported the following indirect estimates of effect for ELX-TEZ-IVA compared with placebo: || || |||||| || |||||| ||| || |||||| || |||||| | for absolute change in ppFEV1 from baseline through 8 weeks; || || |||||| || |||||| ||| || |||||| || |||||| | for absolute change in SwCl from baseline through 8 weeks; || || |||||| || |||||| ||| || |||||| || |||||| | for absolute change in BMI from baseline at 8 weeks; and || || |||||| || |||||| ||| || |||||| || |||||| | for absolute change in weight-for-age z score from baseline at 8 weeks.
Indirect estimates of effect for ELX-TEZ-IVA compared with placebo are provided for each of the CFQ-R domains, with favourable effects reported for respiratory symptoms || || |||||| || |||||| ||| || |||||| || |||||| |; physical functioning || || |||||| || |||||| ||| || |||||| || |||||| |; vitality || || |||||| || |||||| ||| || |||||| || |||||| |; health perceptions || || |||||| || |||||| ||| || |||||| || |||||| |; and social functioning || || |||||| || |||||| ||| || |||||| || |||||| |.
Table 32: Results of Direct and Indirect Comparison for Patients Aged 12 Years and Older With F/G Genotype
End point | Direct estimate ELX-TEZ-IVA vs. IVA LSMD (95% CI), P value | Indirect estimates Bucher mean between-group difference (95% CI), P value | |
|---|---|---|---|
Pooled IVA vs. placebo | ELX-TEZ-IVA vs. placebo | ||
Absolute change in ppFEV1 from baseline through 8 weeks | || || |||||| || |||||| | | || || |||||| || |||||| | | || || |||||| || |||||| | |
CI = confidence interval; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; IVA = ivacaftor; LSMD = least squares mean difference; ppFEV1 = percent predicted forced expiratory volume in the first second; vs. = versus.
Source: Sponsor’s indirect treatment comparison.51
Critical Appraisal
Table 33 summarizes the assessment of homogeneity for the ITC of the studies of patients aged 12 years and older with an F/G genotype. The primary difference across the studies included in the ITC was the use of an open-label, 4-week active treatment period with TEZ-IVA in Study 104. This trial was essentially investigating switching to ELX-TEZ-IVA from IVA compared with remaining on IVA for patients with an F/G genotype. This is a different design than the placebo-controlled trials included in the ITC (i.e., KONNECTION, KONDUCT, and STRIVE). The baseline ppFEV1 for patients in Study 104 was assessed after 4 weeks of treatment with a CFTR modulator (IVA). In contrast, the baseline assessments in KONNECTION, KONDUCT, and STRIVE reflected patients who were naive to CFTR modulator therapy. As both the ELX-TEZ-IVA and IVA groups of Study 104 received 4 weeks of treatment with TEZ-IVA, it is unclear if the run-in period would introduce bias into the ITC analysis. (For example, would the mean difference between IVA and ELX-TEZ-IVA be reduced because of the run-in period, or would it be relatively unchanged?)
Randomization was stratified according to F/G or F/RF genotype in Study 104;52 however, randomization was not stratified according to whether the patient had an F508del mutation in STRIVE, KONDUCT, or KONNECTION.53-55 Hence, the selection of the F508del subgroup of patients in the placebo-controlled IVA trials would not have maintained randomization.
The sponsor pooled the patient-level subgroup data from the 3 placebo-controlled IVA studies to derive a single estimate of effect for IVA versus placebo. However, the results for these individual studies demonstrated different effect sizes for ppFEV1 response to IVA. Specifically, the results in the KONDUCT trial (LS mean difference = 4.1%; 95% CI, –1.0 to 9.1) were considerably smaller than the responses in the STRIVE trial (LS mean difference = 10.4%; 95% CI, 8.0 to 12.9) and the KONNECTION trial (LS mean difference = 10.3%; 95% CI, 5.4 to 15.3). This resulted in a pooled estimate of effect for IVA versus placebo of || || |||||| || |||||| ||| ||, which may overestimate the effect of IVA in patients with a F/R117H genotype (i.e., those patients from the KONDUCT trial) and underestimate the effect in patients with an F/G genotype (i.e., those from the STRIVE and KONNECTION trials).
Table 33: CADTH Assessment of Homogeneity for the ITC for Patients Aged 12 Years and Older With F/G Genotype
Characteristic | Description and handling of potential effect modifiers |
|---|---|
Disease severity | ppFEV1: Patients in KONNECTION had a higher mean ppFEV1 (79.9% and 78.8% in the placebo and IVA groups, respectively) than those in Study 104 (68.1% and 66.0% in the IVA and ELX-TEZ-IVA groups, respectively), STRIVE (63.9% and 59.6% in the placebo and IVA groups, respectively), and KONDUCT (61.3% and 68.1% in the placebo and IVA groups, respectively). CFQ-R: Baseline CFQ-R respiratory domain scores differed across the trials, ranging from 59.1 and 70.2 in the placebo and IVA groups of the KONDUCT trial, respectively, to 80.7 and 77.3 in the placebo and IVA groups of the KONNECTION trial, respectively. Age: The median age of patients ranged from 22.0 and 25.0 years in the placebo and IVA groups of the STRIVE trial, respectively, to 43.0 and 39.5 years in the placebo and IVA groups of the KONDUCT trial, respectively. |
Treatment history | Patients in Study 104 underwent open-label treatment with IVA or TEZ-IVA (for those with F/G or F/RF genotypes, respectively) for 4 weeks prior to initiating treatment with the randomized study drugs (i.e., none of the patients were I to CFTR modulator therapy at the time of baseline measurements). |
Clinical trial eligibility criteria | The trial eligibility criteria differed with respect to the following: CFTR genotypes: Study 104 (F/G [including F/R117H] or F/RF); STRIVE (≥ 1 G551D gating mutation); KONNECTION (≥ 1 non-G551D gating mutation), KONDUCT (≥ 1 R117H mutation). Given the heterogenous populations, the subgroup data were used for patients with an F/G genotype. Patient ages: ≥ 12 years in Study 104 and STRIVE; ≥ 6 years in KONNECTION and KONDUCT. ppFEV1: All the studies specified that patients had to have a ppFEV1 of at least 40% at screening to be eligible. Study 104, STRIVE, and KONDUCT all specified an upper threshold for ppFEV1 of 90% at screening to determine eligibility for enrolment; KONNECTION did not specify an upper threshold for ppFEV1. |
Dosing of comparators | The study drugs were used in accordance with recommendations in the Canadian product monographs for ELX-TEZ-IVA and IVA. |
Response in the common comparator (i.e., placebo) | There were differences in the treatment effects in the placebo groups across the 3 placebo-controlled trials: ppFEV1: The change from baseline in the placebo group was greater in the KONNECTION study (–2.3%) than in the STRIVE and KONDUCT trials (–0.5% and –0.1%, respectively). CFQ-R: The CFQ-R scores decreased from baseline in the placebo groups of the STRIVE trial (–2.0) and the KONNECTION trial (–2.8) but increased in the placebo group of KONDUCT (5.1). BMI: There were differences across the 3 trials with respect to BMI: 0.11 kg/m2 (STRIVE) 0.03 kg/m2 (KONNECTION), and 0.28 kg/m2 (KONDUCT). |
Definitions of end points | The end points included in the ITC were similarly defined and evaluated for each of the included studies. |
Timing of end point evaluation or trial duration | There were differences across the individual clinical trials (e.g., evaluation of ppFEV1 through 24 weeks in STRIVE and KONDUCT and through 8 weeks in Study 104 and KONNECTION); however, the sponsor recalculated the results using an MMRM to ensure that all analyses reflected the same approach. |
Withdrawal frequency | There were few withdrawals from any of the trials included in the ITC. |
Clinical trial setting | All 4 studies included in the ITC were phase III RCTs conducted at specialized CF clinics. |
Study design | As shown in Table 30, there were differences in the following aspects of the studies: Design: Study 104, STRIVE, and KONDUCT were all parallel group RCTs, and KONNECTION was a crossover RCT. Run-in period: A key difference across the studies was the use of a 4-week, open-label run-in period in Study 104, where all patients received treatment with IVA prior to randomization. Duration: The studies had different durations for the treatment periods, ranging from 8 weeks in Study 104 and KONNECTION to 48 weeks in STRIVE. |
BMI = body mass index; CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire–Revised; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/R117H = 1 F508del mutation and 1 R117H mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ITC = indirect treatment comparison; IVA = ivacaftor; MMRM = mixed-effects model for repeated measures; ppFEV1 = percent predicted forced expiratory volume in the first second; RCT = randomized controlled trial; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
ITC for Patients Aged 12 Years and Older With F/RF Genotype
Study Selection Methods
The criteria used by the sponsor to select studies for inclusion in the ITC for patients aged 12 years and older with F/RF genotype were as follows:
Study design is a phase III RCT.
Population includes patients aged 12 years and older with an F/RF genotype.
Interventions include ELX-TEZ-IVA or TEZ-IVA.
Comparators include TEZ-IVA or placebo.
Study duration is at least 8 weeks.
It was not reported if a systematic literature search and review was undertaken by the sponsor to identify studies for inclusion; however, CADTH did not identify any additional studies that would have met the inclusion criteria but were not included in the ITC.
ITC Analysis Methods
The indirect comparison for ELX-TEZ-IVA versus placebo in patients aged 12 years and older with an F/RF genotype was estimated using the Bucher method for continuous end points, with TEZ-IVA as the common comparator. The sponsor stated that the Bucher method was considered the most appropriate approach for this indirect comparison because of the 4-week TEZ-IVA run-in period included in Study 104 (but not in the EXPAND trial).
The sponsor used MMRMs to estimate the direct treatment effects from each of the studies, which were subsequently used in the Bucher indirect comparison. For ppFEV1, CFQ-R domain scores, and SwCl, the sponsor calculated the estimated treatment difference through 8 weeks (defined as the average of weeks 4 and 8). For BMI and weight-for-age z score, the sponsor calculated the estimated treatment difference at week 8. The model estimating the treatment difference between ELX-TEZ-IVA and TEZ-IVA included treatment group, visit, and treatment-by-visit interaction as fixed effects, patient as the random effect, and age group at screening (≥ 12 to < 18 years versus ≥ 18 years), continuous baseline ppFEV1, and continuous baseline SwCl as covariates. The model estimating the treatment difference between TEZ-IVA and placebo included treatment group, treatment period, visit within treatment period, and treatment-by-visit interaction as fixed effects, patient as the random effect, and age group at screening (≥ 12 to < 18 years versus ≥ 18 years) and continuous baseline ppFEV1 as covariates. A 2-sided 95% CI and 2-sided P values for the estimated indirect treatment differences were calculated based on normal approximation.
Table 34: Indirect Comparisons for Patients Aged 12 Years and Older With F/RF Genotype
Indirect estimate | Direct estimates (study) | End point |
|---|---|---|
ELX-TEZ-IVA versus placebo | ELX-TEZ-IVA versus TEZ-IVA (subgroup data from Study 104) + TEZ-IVA versus placebo (EXPAND) | ppFEV1 (through 8 weeks) |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ppFEV1 = percent predicted forced expiratory volume in the first second; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
Source: Sponsor’s indirect treatment comparison.51
Results for ITC Analysis
Included Studies
The evidence network for the studies with patients aged 12 years and older who have an F/RF genotype is shown in Figure 7. Indirect comparison was performed for ELX-TEZ-IVA versus placebo. The direct evidence for ELX-TEZ-IVA versus TEZ-IVA was derived from a subgroup analysis of Study 104. The direct estimate for TEZ-IVA versus placebo was from the EXPAND trial.
Figure 7: Indirect Comparison Network for Patients Aged 12 Years and Older With F/RF Genotype
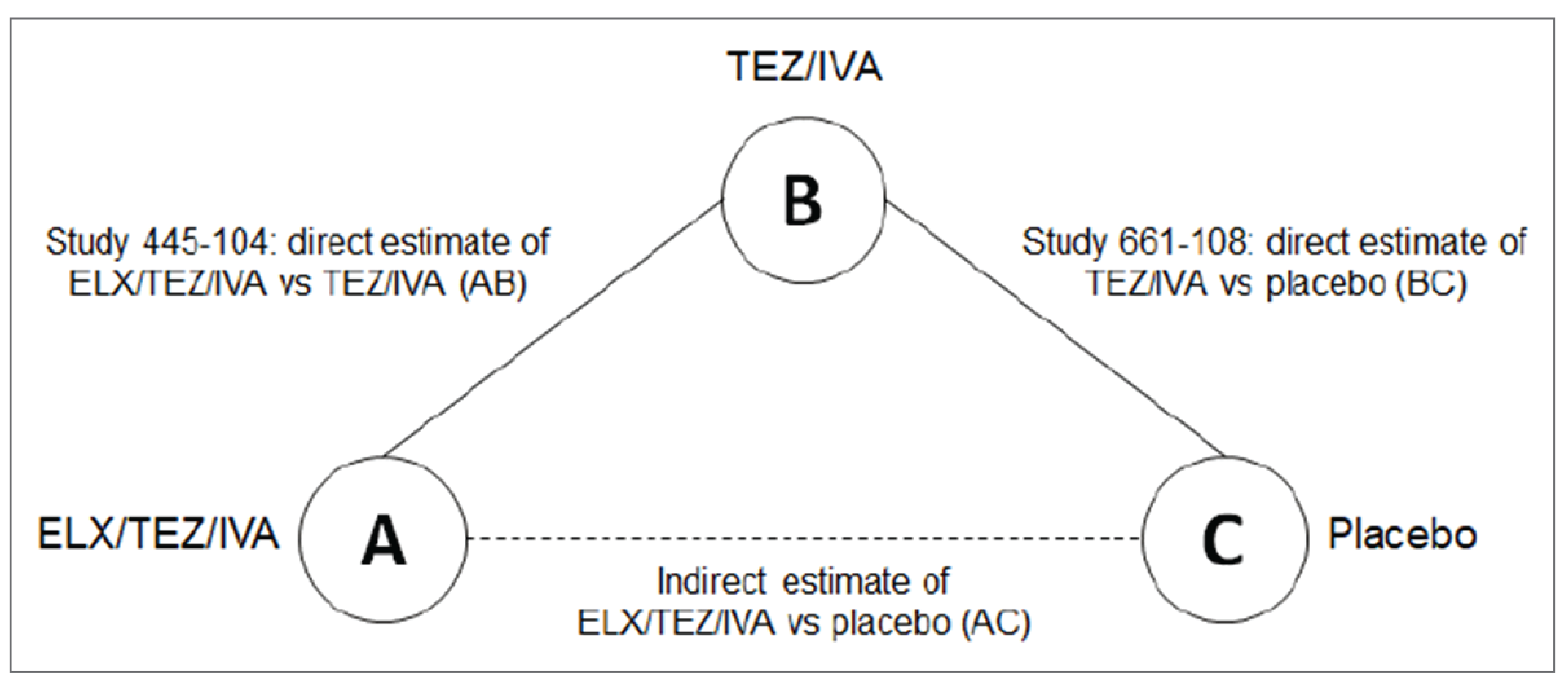
ELX/TEZ/IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; TEZ/IVA = tezacaftor-ivacaftor and ivacaftor; vs = versus.
Source: Sponsor’s indirect treatment comparison.51
Study Characteristics
Table 35 provides a summary of the characteristics of the trials included in the indirect comparison for patients aged 12 years and older with an F/RF genotype (Study 104 and the EXPAND trial). A key difference between the 2 studies was the use of a 4-week, open-label run-in period in Study 104, in which all patients received treatment with TEZ-IVA prior to randomization. The double-blind treatment period was the same in the 2 studies (i.e., 8 weeks). Both trials enrolled patients aged 12 years or older and specified that patients had to have a ppFEV1 between 40% and 90% to be eligible. The inclusion criteria differed across the 2 studies with respect to CFTR genotypes. Study 104 enrolled patients with an F/G (including F/R117H) or an F/RF genotype. The EXPAND trial was conducted only in patients with an F/RF genotype. Given the heterogenous study population of Study 104, the sponsor extracted subgroup data for patients who had an F/RF genotype (163 of 258 [63%]) for use in the indirect comparison.
Both Study 104 and the EXPAND trial specified absolute change in ppFEV1 from baseline as the primary end point but differed in how the assessments were performed (through 8 weeks in Study 104 and using an average of weeks 4 and 8 in EXPAND). Both studies included changes from baseline in BMI, CFQ-R, SwCl, and body weight as additional end points. The EXPAND trial included pulmonary exacerbations as an efficacy end point, and Study 104 did not; therefore, no indirect comparison can be conducted for this end point.
Table 35: Study Characteristics for Patients Aged 12 Years and Older With F/RF Genotype
Characteristic | Study 104 | EXPAND |
|---|---|---|
Study population | Patients with F/G (including F/R117H) or F/RF genotypes and aged ≥ 12 years | Patients with F/RF genotypes and aged ≥ 12 years |
Design | DB, active-controlled, parallel group RCT | DB, placebo-controlled, crossover RCT |
Active run-in period | 4 weeks with TEZ-IVA or IVA | None |
Treatment period | 8 weeks | 8 weeks |
Treatment groups |
|
|
ppFEV1 inclusion criteria at screening | 40% to 90% | 40% to 90% |
Schedule of assessments | Day 1, day 15, week 4, week 8 | Day 1, day 15, week 4, week 8, week 12 of each treatment period |
Sample size |
|
|
Subset of patients included in ITC |
|
|
Primary efficacy end point | Absolute change in ppFEV1 from baseline through 8 weeks | Absolute change in ppFEV1 from baseline through average of week 4 and week 8 measurements |
Other end points | BMI, CFQ-R, SwCl, body weight (Did not include PEx as an efficacy end point) | BMI, CFQ-R, PEx, SwCl, body weight |
BMI = body mass index; CFQ-R = Cystic Fibrosis Questionnaire–Revised; DB = double blind; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/R117H = 1 F508del mutation and 1 R117H mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ITC = indirect treatment comparison; IVA = ivacaftor; PEx = pulmonary exacerbation; ppFEV1 = percent predicted forced expiratory volume in the first second; RCT = randomized controlled trial; SwCl = sweat chloride; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
Source: Sponsor’s indirect treatment comparison.51
Baseline Characteristics
Baseline and demographic characteristics for the F/RF studies are summarized in Table 36. The median age at screening differed between Study 104 (42.0 and 40.3 years in the TEZ-IVA and ELX-TEZ-IVA groups, respectively) and EXPAND (33.0 and 35.0 years in the placebo and TEZ-IVA groups, respectively). The percentage of male and female patients in the 2 studies was similar. The percentage of patients younger than 18 years at screening was greater in the EXPAND trial (14.9% and 13.0% in the placebo and TEZ-IVA groups, respectively) than in Study 104 (3.7% and 8.5% in the TEZ-IVA and ELX-TEZ-IVA groups, respectively). Patients in Study 104 had a higher mean ppFEV1 at baseline (68.1% and 67.8% in the TEZ-IVA and ELX-TEZ-IVA groups, respectively) than those in the EXPAND trial (62.1% and 62.0% in the placebo and TEZ-IVA groups, respectively). Mean BMI at baseline was similar in Study 104 and the EXPAND trial. Baseline SwCl levels were lower in Study 104 (61.4 mmol/L and 64.7 mmol/L in the TEZ-IVA and ELX-TEZ-IVA groups, respectively) than in the EXPAND trial (70.2 mmol/L and 67.0 mmol/L in the placebo and TEZ-IVA groups, respectively). Baseline CFQ-R respiratory domain scores were higher in Study 104 (78.1 and 76.7 in the TEZ-IVA and ELX-TEZ-IVA groups, respectively) than in the EXPAND trial (68.7 and 68.2 in the placebo and TEZ-IVA groups, respectively).
Table 36: Baseline Characteristics for Patients Aged 12 Years and Older With F/RF Genotype
Characteristic | Study 104 | EXPAND | ||
|---|---|---|---|---|
TEZ-IVA (N = 81) | ELX-TEZ-IVA (N = 82) | Placebo (N = 161) | TEZ-IVA (N = 161) | |
Sex, n (%) | ||||
Male | 37 (45.7) | 37 (45.1) | 71 (44.1) | 72 (44.7) |
Female | 44 (54.3) | 45 (54.9) | 90 (55.9) | 89 (55.3) |
Age at screening (years) | ||||
Mean (SD) | 41.3 (14.4) | 40.1 (14.7) | 34.6 (14.4) | 35.6 (14.5) |
Median | 42.0 | 40.3 | 33.0 | 35.0 |
Age group at screening (years), n (%) | ||||
12 to < 18 | 3 (3.7) | 7 (8.5) | 24 (14.9) | 21 (13.0) |
≥ 18 | 78 (96.3) | 75 (91.5) | 137 (85.1) | 140 (87.0) |
ppFEV1, mean (SD) | 68.1 (16.4) | 67.8 (16.3) | 62.1 (14.2) | 62.0 (14.5) |
BMI (kg/m2), mean (SD) | 24.68 (5.22) | 24.29 (5.23) | 24.63 (5.41) | 24.06 (4.74) |
Sweat chloride (mmol/L), mean (SD) | 61.4 (27.3) | 64.7 (27.9) | 70.2 (25.7) | 67.0 (26.8) |
CFQ-R respiratory domain score, mean (SD) | 78.1 (14.7) | 76.7 (16.9) | 68.7 (18.3) | 68.2 (17.5) |
BMI = body mass index; CFQ-R = Cystic Fibrosis Questionnaire–Revised; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ppFEV1 = percent predicted forced expiratory volume in the first second; SD = standard deviation; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
Source: Sponsor’s indirect treatment comparison.51
Indirect Comparison Results
Table 37 provides a summary of the results for the direct and indirect comparisons for studies conducted in patients aged 12 years and older with an F/RF genotype. The indirect comparison results are shown side by side with the direct comparison results for ELX-TEZ-IVA versus TEZ-IVA (derived from the subgroup analysis from Study 104) and direct comparison results for TEZ-IVA versus placebo from the EXPAND trial.
The sponsor reported the following indirect estimates of effect for ELX-TEZ-IVA compared with placebo: || || |||||| || |||||| ||| || ||| for absolute change in ppFEV1 from baseline through 8 weeks; –33.6 mmol/L || || |||||| || |||||| ||| || ||| for absolute change in SwCl from baseline through 8 weeks; 0.31 kg/m2 (95% CI, 0.04 to 0.57; P = 0.0219) for absolute change in BMI from baseline at 8 weeks; and || || |||||| || |||||| ||| || ||| for absolute change in weight-for-age z score from baseline at 8 weeks.
Indirect estimates of effect for ELX-TEZ-IVA compared with placebo are provided for each of the CFQ-R domains, with favourable effects reported for respiratory symptoms || || |||||| || |||||| ||| || |||; physical functioning || || |||||| || |||||| ||| || |||; vitality || || |||||| || |||||| ||| || |||; eating problems || || |||||| || |||||| ||| || |||; health perceptions || || |||||| || |||||| ||| || |||; digestive symptoms || || |||||| || |||||| ||| || |||; and social functioning || || |||||| || |||||| ||| || |||.
Table 37: Results of Direct and Indirect Comparison for Patients Aged 12 Years and Older With F/RF Genotype
End point | Direct estimate LSMD (95% CI), P value | Indirect estimates Bucher mean between-group difference (95% CI), P value ELX-TEZ-IVA vs. placebo | |
|---|---|---|---|
ELX-TEZ-IVA vs. TEZ-IVA | TEZ-IVA vs. placebo | ||
Absolute change in ppFEV1 from baseline through 8 weeks | || || |||||| || |||||| ||| || ||| | || || |||||| || |||||| ||| || ||| | || || |||||| || |||||| ||| || ||| |
CI = confidence interval; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; LSMD = least squares mean difference; ppFEV1 = percent predicted forced expiratory volume in the first second; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor; vs. = versus.
Source: Sponsor’s indirect treatment comparison.51
Critical Appraisal
Table 38 summarizes the assessment of homogeneity for the ITC of the studies of patients aged 12 years and older with an F/RF genotype. The primary difference between Study 104 and the EXPAND trial was the use of an open-label, 4-week active treatment period with TEZ-IVA in Study 104. This trial was essentially investigating switching to ELX-TEZ-IVA from TEZ-IVA compared with remaining on TEZ-IVA for patients with an F/RF genotype. This is a different design than the placebo-controlled EXPAND trial, in which all patients were naive to CFTR modulators at baseline. In contrast, the baseline parameters for patients in Study 104 were assessed after 4 weeks of treatment with a CFTR modulator (TEZ-IVA). As both the ELX-TEZ-IVA and TEZ-IVA groups of Study 104 received 4 weeks of treatment with TEZ-IVA, it is unclear if the run-in period would introduce bias into the ITC analysis (e.g., would the mean difference between TEZ-IVA and ELX-TEZ-IVA be reduced because of the run-in period, or would it be relatively unchanged?).
As noted in Table 38, there are important differences in the baseline and end point values between Study 104 and the EXPAND trial due to the 4 weeks of active treatment that patients in Study 104 received prior to randomization.
Table 38: CADTH Assessment of Homogeneity for the ITC for Patients Aged 12 Years and Older With F/RF Genotype
Characteristic | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Age: Median age at screening was greater in Study 104 (42.0 and 40.3 years in the TEZ-IVA and ELX-TEZ-IVA groups, respectively) than in the EXPAND trial (33.0 and 35.0 years in the placebo and TEZ-IVA groups, respectively). ppFEV1: Study 104 had a higher mean ppFEV1 at baseline (68.1% and 67.8% in the TEZ-IVA and ELX-TEZ-IVA groups, respectively) than the EXPAND trial (62.1% and 62.0% in the placebo and TEZ-IVA groups, respectively). SwCl: Baseline SwCl levels were lower in Study 104 (61.4 mmol/L and 64.7 mmol/L in the TEZ-IVA and ELX-TEZ-IVA groups, respectively) than in the EXPAND trial (70.2 mmol/L and 67.0 mmol/L in the placebo and TEZ-IVA groups, respectively). CFQ-R: Baseline CFQ-R respiratory domain scores were greater in Study 104 (78.1 and 76.7 in the TEZ-IVA and ELX-TEZ-IVA groups, respectively) than in the EXPAND trial (68.7 and 68.2 in the placebo and TEZ-IVA groups, respectively). |
Treatment history | Patients in Study 104 underwent open-label treatment with IVA or TEZ-IVA (for those with F/G or F/RF genotypes, respectively) for 4 weeks prior to initiating treatment with the randomized study drugs (i.e., none of the patients were naive to CFTR modulator therapy at the time of baseline measurements). |
Clinical trial eligibility criteria | The inclusion criteria differed across the 2 studies with respect to CFTR genotypes. Study 104 enrolled patients with an F/G (including F/R117H) or an F/RF genotype. The EXPAND trial was conducted only in patients with an F/RF genotype. Given the heterogenous study population of Study 104, subgroup data were used for the ITC. |
Response in the common comparator (i.e., TEZ-IVA) | Due to the different designs of Study 104 and the EXPAND trial, there are important differences in the change from baseline within the TEZ-IVA groups included in the indirect comparisons:
|
Dosing of comparators | Both ELX-TEZ-IVA and TEZ-IVA were administered in accordance with recommendations in the Canadian product monographs.29,39 However, patients in the TEZ-IVA group of Study 104 would have received this drug for a total of 12 weeks (i.e., 4 weeks in the run-in period and then 8 weeks in the double-blind phase), compared with only 8 weeks of treatment for those in the EXPAND trial. |
Definitions of end points | The end points included in the ITC were similarly defined and evaluated for each of the included studies. |
Timing of end point evaluation or trial duration | Both Study 104 and the EXPAND trial specified absolute change in ppFEV1 from baseline as the primary end point but differed in how the assessments were performed (through 8 weeks in Study 104 and using an average of weeks 4 and 8 in EXPAND); however, the sponsor recalculated the results using an MMRM to ensure that all analysis reflected the same approach. |
Withdrawal frequency | There were few withdrawals from either of the trials included in the ITC. |
Clinical trial setting | Both Study 104 and EXPAND were phase III RCTs conducted at specialized CF clinics. |
Study design | Study 104 and EXPAND were similarly designed, except for the key difference between the studies: the use of a 4-week, open-label run-in period in Study 104, in which all patients received treatment with TEZ-IVA prior to randomization. |
BMI = body mass index; CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire–Revised; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/R117H = 1 F508del mutation and 1 R117H mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ITC = indirect treatment comparison; IVA = ivacaftor; MMRM = mixed-effects model for repeated measures; ppFEV1 = percent predicted forced expiratory volume in the first second; RCT = randomized controlled trial; SwCl = sweat chloride; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor.
Summary of ITCs
Patients Aged 6 to 11 Years
The sponsor conducted a single indirect comparison for patients aged 6 to 11 years with an F/F genotype to derive relative estimates of clinical efficacy for ELX-TEZ-IVA versus LUM-IVA, ELX-TEZ-IVA versus placebo, and ELX-TEZ-IVA versus TEZ-IVA. TEZ-IVA is not currently approved by Health Canada or reimbursed by the Canadian public drug programs for use in patients aged 6 to 11 years. To conduct the primary indirect comparisons, the sponsor extracted 24-week individual patient-level data for patients with an F/F genotype from the following studies: Study 106B for ELX-TEZ-IVA (N = 29); pooled data from Study 809-109 and Study 809-011B for LUM-IVA (N = 160); and Study 661-113B (N = 61) for TEZ-IVA. Additional sensitivity analyses were performed using 8-week data. The sponsor reported the following indirect estimates of effect for ELX-TEZ-IVA compared with placebo for absolute change from baseline through 24 weeks: || || |||||| || |||||| ||| || ||| for ppFEV1.
The primary limitations of the ITC were the difference in study design across the included studies (Studies 106B, 809-011B, and 661-113B were single-arm, open-label trials, and Studies 809-109 and 661-115 were double-blind, placebo-controlled trials) and the differences in baseline characteristics.
Patients Aged 12 Years and Older
For patients with an F/G genotype, indirect comparisons were performed for ELX-TEZ-IVA versus placebo. The direct evidence for ELX-TEZ-IVA versus IVA was derived from a subgroup analysis of Study 104, and the estimates for IVA versus placebo were derived from a meta-analysis of subgroup analyses from 3 studies: STRIVE, KONNECTION, and KONDUCT. The sponsor reported the following indirect estimates of effect for ELX-TEZ-IVA compared with placebo for absolute change from baseline through 8 weeks: || || |||||| || |||||| ||| || ||| for ppFEV1.
For patients with an F/RF genotype, indirect comparisons were performed for ELX-TEZ-IVA versus placebo. The direct evidence for ELX-TEZ-IVA versus TEZ-IVA was derived from a subgroup analysis of Study 104, and the estimates for TEZ-IVA versus placebo were from the EXPAND trial. The sponsor reported the following indirect estimates of effect for ELX-TEZ-IVA compared with placebo for absolute change from baseline through 8 weeks: || || |||||| || |||||| ||| || ||| for ppFEV1.
The primary limitation of the ITC was the difference in study design across the included studies. The ELX-TEZ-IVA study (i.e., Study 104) included an open-label, 4-week active treatment period with TEZ-IVA or IVA prior to randomization. None of the other trials used in the ITC had a similar run-in period; therefore, the study designs, baseline values, and end point values for the common comparator were different. As both the ELX-TEZ-IVA and comparator groups in Study 104 received 4 weeks of treatment with a CFTR modulator, it is unclear if the run-in period would introduce bias into the ITC analysis.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
The sponsor did not include any additional studies to address gaps in the pivotal trial evidence.
Discussion
Summary of Available Evidence
The evidence for the review of ELX-TEZ-IVA in patients aged 2 to 5 years was derived from a review of pivotal and phase III studies. CADTH has previously reviewed ELX-TEZ-IVA for the treatment of CF in patients who have at least 1 F508del mutation in the CFTR gene for patients aged 12 years and older (final recommendation issued in August 2021) and for patients aged 6 years and older (final recommendation issued in June 2022). As reflected in the Canadian product monograph, patients aged 2 to 5 years would receive ELX-TEZ-IVA as oral granules as opposed to tablets.
The current CADTH review is focused only on patients aged 2 to 5 years, and the new evidence consisted of a pivotal, open-label, single-arm trial (Study 111). The regulatory submission for ELX-TEZ-IVA is based on the extrapolation of efficacy data from older age groups to a younger population based on comparable pharmacokinetic exposures and safety. The sponsor noted that the extrapolation of efficacy was appropriate and aligned with the principles described in the International Council for Harmonisation (ICH) of Technical Requirements of Pharmaceuticals for Human Use Guidance: E11(R1) Addendum: Clinical Investigation of Medicinal Products in the Pediatric Population. Specifically, the sponsor noted that the extrapolation was appropriate for the following reasons:
The disease process in patients with CF of all age groups stems from a common etiology of dysfunctional CFTR protein that is targeted by ELX-TEZ-IVA.
The indication is the same.
The outcome of therapy in younger age groups is likely to be comparable to that in older age groups.
The clinical experts consulted by CADTH concurred with the sponsor and concluded that extrapolation of the efficacy data to patients aged 2 to 5 years was appropriate for ELX-TEZ-IVA. The experts noted that all patients with at least 1 F508del mutation would benefit from ELX-TEZ-IVA, given the mechanism of action for the drug.
The sponsor reported that an ITC was attempted to compare the clinical efficacy of ELX-TEZ-IVA in Study 111 with other relevant CFTR modulators in patients with F/F and F/MF mutations to generate the inputs needed for the cost-effectiveness analysis. The sponsor concluded that the ITC was not feasible due to the small number of patients in this age group, which reduced the power to detect differences between ELX-TEZ-IVA, LUM-IVA, and/or placebo. As such, the sponsor did not include the ITC in its submission to CADTH and used estimates from the previous CADTH submissions for patients aged 6 to 11 years (patients with F/F genotype) and patients aged 12 years and older (patients with F/G and F/RF genotypes) as assumptions within its economic model. No indirect comparison was submitted for patients with an F/MF genotype, and the sponsor used the placebo-adjusted data from Study 116 in patients aged 6 to 11 years to inform the economic model.
Interpretation of Results
Efficacy
As previously reported in CADTH reviews of ELX-TEZ-IVA, LCI is not currently used in Canadian clinical practice to evaluate lung function in patients with CF, but it has been recommended as an end point in clinical trials conducted in younger patients.8 This is because spirometry may not be sensitive enough to detect treatment differences in patients who have relatively normal lung function but may still have underlying structural abnormalities in the lungs. In Study 111, ELX-TEZ-IVA demonstrated an improvement from baseline in LCI2.5 through 24 weeks of treatment (absolute reduction = –0.83; 95% CI, –1.01 to –0.66).56 The mean improvements from baseline were observed at all postbaseline assessments (i.e., weeks 4, 12, and 24). In previous CADTH reviews of CFTR modulators in pediatric patients (including ELX-TEZ-IVA for patients aged 6 to 11 years), the sponsor indicated that LCI is correlated with FEV1 (which has been validated as an end point) in its ability to measure airway disease. CADTH’s review of the literature found that the correlation between LCI and FEV1 was variable across studies, an observation supported by the clinical experts consulted by CADTH. Overall, the clinical experts consulted by CADTH indicated that LCI is not currently used in Canadian practice and that there is no consensus in the Canadian clinical community regarding the magnitude of improvement in LCI2.5 that would be considered clinically relevant.
Within-group change from baseline in body weight, height, and BMI is challenging to interpret for patients aged 2 to 5 years, who would be expected to experience growth over the 6-month study period. Study 111 was not powered to evaluate changes in these end points, and no statistical analyses were conducted by the sponsor. The clinical experts consulted by CADTH noted that the results from the previously reviewed studies, where ELX-TEZ-IVA demonstrated improvements from baseline in patients aged 6 to 11 years, are sufficient to conclude that ELX-TEZ-IVA would also improve or maintain growth parameters for patients aged 2 to 5 years. The clinical experts noted that patients with CF living in Canada in the target age range (i.e., 2 to 5 years) are generally well nourished in Canada as a result of early diagnosis through newborn screening and aggressive disease management through specialized clinics. As such, it was noted that the magnitude of improvement in growth parameters following initiation of ELX-TEZ-IVA would be lower in patients aged 2 to 5 years than in older patients.
Pulmonary exacerbations were included as an exploratory end point in Study 111, with no statistical analyses performed to examine change from baseline in the annual exacerbation rate. After 24 weeks of ELX-TEZ-IVA treatment, 12 patients (16.0%) each experienced 1 pulmonary exacerbation (annualized event rate = 0.32 events per year). In response to an inquiry from CADTH regarding why pulmonary exacerbations were not included as an efficacy end point with prebaseline and postbaseline evaluation of exacerbation rates, the sponsor reported that a treatment effect may be difficult to detect in Study 111 given the relative rareness of these events in younger patients relative to older patients. The clinical experts consulted by CADTH noted that the proportion of patients who reported at least 1 pulmonary exacerbation over the 24-week study period was low compared with what would be anticipated for patients aged 2 to 5 years who are not receiving a CFTR modulator. Patients enrolled in Study 111 Part B were enrolled and evaluated between July 2021 and June 2022. The clinical experts noted that exacerbation rates reported in CF clinical trials and those observed in Canadian clinical practice were generally reduced during the COVID-19 pandemic. Physical distancing and the suspension of in-person classes and daycare have been cited as contributing factors to the reduction in exacerbations. Similar observations have been reported in US, where the rate of exacerbations decreased during the COVID-19 pandemic for patients receiving ELX-TEZ-IVA and for those not receiving ELX-TEZ-IVA.57 The clinical experts emphasized that 6 months is generally insufficient to evaluate changes in exacerbation frequency as there is expected to be seasonal variation.
Overall, the clinical experts noted that ELX-TEZ-IVA would be expected to reduce exacerbation frequency and severity in the small subset of patients aged 2 to 5 years who experience frequent exacerbations. For patients who are not experiencing frequent exacerbations, ELX-TEZ-IVA would be expected to help prevent severe exacerbations as the drug will help maintain healthier airways and promote clearance of secretions. However, the clinical experts strongly emphasized that exacerbation frequency will be variable in these young patients and can be influenced by seasonality and exposure to viruses from siblings, daycare, and/or school. There is consensus that ELX-TEZ-IVA will result in meaningful improvements for patients aged 2 to 5 years but that objectively measuring changes in pulmonary exacerbations for the purposes of initiation or renewal of drug reimbursement would be challenging in clinical practice.
Study 111 was limited to patients with an F/F or F/MF genotype. No clinical studies have been conducted with ELX-TEZ-IVA in pediatric patients with F/RF or F/G genotypes; however, the clinical experts noted that ELX-TEZ-IVA would result in clinically meaningful improvements for these patients, based on the evidence reported for ELX-TEZ-IVA in adult patients with F/RF and F/G genotypes and the results in pediatric studies of patients with F/F and F/MF genotypes (both in those aged 6 to 11 years and in those aged 2 to 5 years). This is consistent with the input from patient and clinician groups, who have indicated that all patients with at least 1 F508del mutation would be likely to benefit from treatment with ELX-TEZ-IVA.
Harms
As in the previous reviews of ELX-TEZ-IVA in patients aged 6 years and older, the granular formulation of ELX-TEZ-IVA was well tolerated in the target patient population (i.e., patients aged 2 to 5 years with at least 1 F508del mutation). There have been no updates to the warnings and precautions section of the ELX-TEZ-IVA product monograph since the previous CADTH review.
Serious AEs and withdrawals due to AEs were rare in Study 111. The clinical experts consulted by CADTH noted that intolerance to ELX-TEZ-IVA has been rare in clinical practice with adolescents and adults. The clinical experts consulted by CADTH noted that patients who experience significant AEs following initial treatment with ELX-TEZ-IVA would not likely be completely discontinued from treatment; rather, treatment with ELX-TEZ-IVA would likely be interrupted and the patient would be rechallenged with the drug following resolution of the event(s). This is consistent with the input received from the clinician groups, who noted that discontinuation of therapy should be considered in patients who have clinically significant adverse effects that persist and recur after stopping and reinitiating therapy.
Similar to the development programs for the other CFTR modulators (IVA, LUM-IVA, and TEZ-IVA), patients with abnormal liver function were excluded from the phase III ELX-TEZ-IVA trials. The clinical experts consulted by CADTH noted that most patients who could be eligible for ELX-TEZ-IVA would not have hepatic impairment. The product monograph recommends that the dosage of ELX-TEZ-IVA should be adjusted in patients with moderate hepatic impairment and that the drug should not be used in patients with severe hepatic impairment.29 These recommendations are more restrictive than those in the product monographs for IVA, LUM-IVA, and TEZ-IVA,29,37,38 all of which provide dosage reduction scenarios for patients with CF who have severe hepatic impairment. The clinical experts consulted by CADTH suggested that clinicians may attempt to treat patients with severe hepatic impairment using ELX-TEZ-IVA at a reduced dosage, as opposed to using the reduced dosages of the alternative CFTR modulators, which are unlikely to provide the same level of clinical benefit.
The product monograph notes that elevated transaminases have been observed in patients treated with ELX-TEZ-IVA and recommends that ALT and AST be assessed prior to initiating treatment with ELX-TEZ-IVA, every 3 months during the first year of treatment, and annually thereafter.29 The clinical experts consulted by CADTH noted that the recommendations for monitoring would likely be followed by the clinical community. The clinical experts consulted by CADTH and the clinician groups who provided input noted that patients with CF are typically seen once every 3 months (though this has been less frequent in some cases due to the COVID-19 pandemic). As such, the recommended monitoring regimen for ELX-TEZ-IVA was not anticipated to result in a substantial increase in the number of clinic visits for patients with CF (particularly after the first year of treatment).
Similar to IVA, LUM-IVA, and TEZ-IVA, the product monograph for ELX-TEZ-IVA notes that cases of noncongenital cataracts without impact on vision have been reported in pediatric patients treated with IVA-containing regimens.29,37-39 The product monograph also notes that the patients who demonstrated these events had other risk factors (e.g., corticosteroid use or exposure to radiation); however, a possible risk attributable to treatment with IVA cannot be excluded. As such, it is recommended that pediatric patients beginning treatment with ELX-TEZ-IVA receive baseline and follow-up ophthalmological examinations.29 The clinical experts consulted by CADTH noted that children with CF currently have an ophthalmological examination prior to starting treatment with a CFTR modulator and are monitored on an ongoing basis thereafter.
The sponsor’s indirect comparisons did not investigate the comparative safety of ELX-TEZ-IVA versus IVA, LUM-IVA, or TEZ-IVA.50,51 The clinical trials included in this review demonstrated that ELX-TEZ-IVA does not appear to be associated with the respiratory AEs (e.g., dyspnea and abnormal respiration) reported in the pivotal trials with LUM-IVA.10,34,35,37,58
Other Considerations
The FDA extended the approval of ELX-TEZ-IVA to include an additional 177 mutations in the CFTR gene that have shown to be responsive to ELX-TEZ-IVA based on data from in vitro assays.30,59
Conclusion
For patients aged 2 to 5 years, a 24-week, open-label, uncontrolled trial (Study 111 Part B; N = 75) suggested that treatment with ELX-TEZ-IVA resulted in improvements from baseline in lung function (decrease in LCI2.5 from baseline) and in CF biomarkers (reduction in SwCl). Study 111 was primarily designed to evaluate the safety, tolerability, and pharmacokinetics of ELX-TEZ-IVA, as the regulatory submission is based on the extrapolation of efficacy data from the studies conducted in older patients with CF (i.e., those showing some measurable level of disease manifestations at baseline). The clinical experts consulted by CADTH noted that, given its mechanism of action and the compelling efficacy data in patients aged 6 years and older, ELX-TEZ-IVA would be expected to benefit patients aged 2 to 5 years who have at least 1 508del mutation in the CFTR gene. There is consensus across clinicians and patients that treatment with ELX-TEZ-IVA should be initiated as soon as possible, given the clinically meaningful benefits observed in patients who can currently access the treatment. Uncertainty remains regarding the magnitude of the beneficial effect of ELX-TEZ-IVA in very young patients with CF, and future real-world evidence may help address this uncertainty.
Study 111 was limited to patients with an F/F or F/MF genotype. No clinical studies have been conducted with ELX-TEZ-IVA in pediatric patients with F/RF or F/G genotypes; however, the clinical experts noted that ELX-TEZ-IVA would result in clinically meaningful improvements for these patients, based on the evidence reported for ELX-TEZ-IVA in adult patients with F/RF and F/G genotypes and the results in pediatric studies of patients with F/F and F/MF genotypes. This is consistent with the input from patient and clinician groups, who have indicated that all patients with at least 1 F508del mutation would be likely to benefit from treatment with ELX-TEZ-IVA.
ELX-TEZ-IVA was well tolerated in the target patient population (i.e., patients aged 2 to 5 years with at least 1 F508del mutation). Serious AEs and withdrawals due to AEs were rare in Study 111. The product monograph notes that elevated transaminases have been observed in patients treated with ELX-TEZ-IVA and recommends that ALT and AST be assessed prior to initiating treatment with ELX-TEZ-IVA, every 3 months during the first year of treatment, and annually thereafter. The clinical experts consulted by CADTH noted that the recommendations for monitoring with ELX-TEZ-IVA were not anticipated to result in a substantial increase in the number of clinic visits for patients with CF (particularly after the first year of treatment).
References
1.The Canadian Cystic Fibrosis registry: 2019 annual data report. Toronto (ON): Cystic Fibrosis Canada; 2020: https://www.cysticfibrosis.ca/registry/2019AnnualDataReport.pdf. Accessed 2023 Nov 15.
2.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: elexacaftor / tezacaftor / ivacaftor and ivacaftor (Trikafta - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2021 Sept 16: https://cadth.ca/sites/default/files/DRR/2021/SR0673%20Trikafta%20-%20CADTH%20Final%20Rec%20Revised.pdf. Accessed 2023 Nov 15.
3.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: elexacaftor / tezacaftor / ivacaftor and ivacaftor (Trikafta - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2022 July 6: https://www.cadth.ca/sites/default/files/DRR/2022/SR0710%20Trikafta%20-%20CADTH%20Final%20Rec-meta.pdf. Accessed 2023 May 16.
4.Vertex Pharmaceuticals Incorporated. NCT02797132: Safety and Pharmacokinetic Study of Lumacaftor/Ivacaftor in Subjects Aged 2 Through 5 Years With Cystic Fibrosis, Homozygous for F508del. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2018: https://clinicaltrials.gov/ct2/show/NCT02797132. Accessed 2023 May 19.
5.McNamara JJ, McColley SA, Marigowda G, et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study. The Lancet Respiratory Medicine. 2019;7(4):325-335. PubMed
6.Vertex Pharmaceuticals Incorporated. NCT01705145: Study of Ivacaftor in Cystic Fibrosis Subjects 2 Through 5 Years of Age With a CFTR Gating Mutation. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2016: https://clinicaltrials.gov/ct2/show/study/NCT01705145. Accessed 2023 May 19.
7.Davies JC, Cunningham S, Harris WT, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4(2):107-115. PubMed
8.Report of the workshop on endpoints for cystic fibrosis clinical trials. Amsterdam (NL): European Medicines Agency; 2012: https://www.ema.europa.eu/docs/en_GB/document_library/Report/2012/12/WC500136159.pdf. Accessed 2023 Nov 15.
9.Committee for Medicinal Products for Human Use. Assessment report variation: Orkambi (ivacaftor/lumacaftor). (European public assessment report). Amsterdam (NL): European Medicines Agency; 2017: https://www.ema.europa.eu/en/documents/variation-report/orkambi-epar-assessment-report-variation_en.pdf. Accessed 2023 Nov 15.
10.CADTH Common Drug Review Clinical Review Report: lumacaftor/ivacaftor (Orkambi - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2018 Oct: https://www.cadth.ca/sites/default/files/cdr/clinical/SR0559_Orkambi_CL_Report.pdf. Accessed 2023 Nov 15.
11.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352(19):1992-2001. PubMed
12.Veit G, Avramescu RG, Chiang AN, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27(3):424-433. PubMed
13.Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol Med. 2012;18(2):81-91. PubMed
14.Okiyoneda T, Barriere H, Bagdany M, et al. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329(5993):805-810. PubMed
15.Flume PA, O'Sullivan BP, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176(10):957-969. PubMed
16.Cogen J, Emerson J, Sanders DB, et al. Risk factors for lung function decline in a large cohort of young cystic fibrosis patients. Pediatr Pulmonol. 2015;50(8):763-770. PubMed
17.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34(2):91-100. PubMed
18.Canadian consensus statement on aerosolized antibiotic use in cystic fibrosis. Toronto (ON): Cystic Fibrosis Canada; 2020: https://www.cysticfibrosis.ca/uploads/About%20Us/CFC005%20AEROSOLIZED%20ANTIBIOTIC%20_englishFinal.pdf. Accessed 2023 Nov 15.
19.Cystic Fibrosis Foundation patient registry 2019 annual data report. Bethesda (MD): Cystic Fibrosis Foundation; 2020.
20.Liou TG, Adler FR, Fitzsimmons SC, Cahill BC, Hibbs JR, Marshall BC. Predictive 5-year survivorship model of cystic fibrosis. Am J Epidemiol. 2001;153(4):345-352. PubMed
21.Corey M, McLaughlin FJ, Williams M, Levison H. A comparison of survival, growth, and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J Clin Epidemiol. 1988;41(6):583-591. PubMed
22.The Canadian Cystic Fibrosis registry: 2013 annual report. Toronto (ON): Cystic Fibrosis Canada; 2015: https://www.cysticfibrosis.ca/uploads/cf%20care/Canadian-CF-Registry-2013-FINAL.pdf. Accessed 2023 Nov 15.
23.The Canadian Cystic Fibrosis registry: 2016 annual data report. Toronto (ON): Cystic Fibrosis Canada; 2017: https://www.cysticfibrosis.ca/registry/2016AnnualDataReport.pdf. Accessed 2023 Nov 15.
24.Mogayzel PJ, Jr., Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2013;187(7):680-689. PubMed
25.Flume PA, Mogayzel PJ, Jr., Robinson KA, et al. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. 2009;180(9):802-808. PubMed
26.Center for Drug Evaluation and Research. Multi-Disciplinary Review. Trikafta (elexacaftor, tezacaftor and ivacaftor tablets; ivacaftor tablets). Company: Vertex Pharmaceuticals Incorporated. Application No.: 212273. Approval date: 10/21/2019 (FDA approval package). Rockville (MD): U.S. Food and Drug Administration (FDA); 2019: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212273Orig1s000MultidisciplineR.pdf. Accessed 2023 Nov 15.
27.INESSS. Trikafta - Reimbursement Recommendation. 2022 Aug 5. https://www.inesss.qc.ca/en/themes/medicaments/drug-products-undergoing-evaluation-and-evaluated/extract-notice-to-the-minister/trikafta-fibrose-kystique-6104.html Accessed 2023 Nov 15.
28.Vertex Pharmaceuticals. Cystic Fibrosis Expert Opinion [internal sponsor's report]. 2022.
29.Trikafta®: elexacaftor tezacaftor/ivacaftor tablets and ivacaftor tablets, oral 100 mg/50 mg/75 mg and 150 mg [product monograph]. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2020 Nov 11.
30.Center for Drug Evaluation and Research. Label. Trikafta (elexacaftor, tezacaftor and ivacaftor tablets; ivacaftor tablets). Company: Vertex Pharmaceuticals Incorporated. Application No.: 212273. Approval date: 10/21/2019 (FDA approval package). Rockville (MD): U.S. Food and Drug Administration (FDA); 2020: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212273s000lbl.pdf. Accessed 2023 Nov 15.
31.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: ivacaftor (Kalydeco - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2013 Mar 22: https://www.cadth.ca/sites/default/files/cdr/complete/cdr_complete_Kalydeco_March-25-13_e.pdf. Accessed 2023 Nov 15.
32.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: ivacaftor (Kalydeco - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2014 Dec 19: https://www.cadth.ca/sites/default/files/cdr/complete/cdr_complete_SR0379_Kalydeco_Dec-23-14.pdf. Accessed 2023 Nov 15.
33.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: ivacaftor (Kalydeco - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2015 Nov 19: https://www.cadth.ca/sites/default/files/cdr/complete/SR0430_complete_Kalydeco_R117H_Nov-23-15_e.pdf. Accessed 2023 Nov 15.
34.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: lumacaftor/ivacaftor (Orkambi - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2016 Oct 26: https://www.cadth.ca/sites/default/files/cdr/complete/SR0471_complete_Orkambi-Oct-28-16.pdf. Accessed 2023 Nov 15.
35.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: lumacaftor/ivacaftor (Orkambi - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2018 Sep 26: https://www.cadth.ca/sites/default/files/cdr/complete/SR0559%20Orkambi%20-%20CDEC%20Final%20Recommendation%20October%204%2C%202018.pdf. Accessed 2023 Nov 15.
36.CADTH reimbursement review status: tezacaftor/ivacaftor. 2019; https://www.cadth.ca/tezacaftorivacaftor. Accessed 2023 Nov 15.
37.Orkambi®, lumacaftor/ivacaftor tablets 100 mg/125 mg and 200 mg/125 mg, lumacaftor/ivacaftor granules 100 mg/125 mg and 150 mg/188 mg [product monograph]. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2018 Dec 11: https://pdf.hres.ca/dpd_pm/00048664.PDF. Accessed 2023 Nov 15.
38.Kalydeco®: ivacaftor tablets 150 mg, ivacaftor granules 50 mg per packet, 75 mg per packet [product monograph]. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2019 Jan 25: https://pdf.hres.ca/dpd_pm/00049400.PDF. Accessed 2023 Nov 15.
39.Clinical Study Report: VX18-445-106. A phase 3 study evaluating the pharmacokinetics, safety, and tolerability of VX-445/TEZ/IVA triple combination therapy in cystic fibrosis subjects 6 through 11 years of age [internal sponsor's report]. Boston (MA): Vertex Pharmaceuticals Incorporated; 2020 Nov 11.
40.Goralski JL, Hoppe, J.E., Mall, M.A., McColley, S.A., McKone, E., Ramsey, B., Rayment, J.H., Robinson, P., Stehling, F., Taylor-Cousar, J.L., Tullis, E., Ahluwalia, N., Chin, A., Chu, C., Lu, M., Niu, T., Weinstock, T., Ratjen, F., Rosenfeld, M.; VX20-445-111 Study Group. Phase 3 Open-Label Clinical Trial of Elexacaftor/Tezacaftor/Ivacaftor in Children Aged 2 Through 5 Years with Cystic Fibrosis and at Least One F508del Allele. Am J Respir Crit Care Med. 2023 Jul 1;208(1):59-67. PubMed
41.Vertex Pharmaceuticals Incorporated. NCT04537793: Evaluation of ELX/TEZ/IVA in Cystic Fibrosis (CF) Subjects 2 Through 5 Years. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2020: https://clinicaltrials.gov/ct2/show/NCT04537793. Accessed 2023 Nov 15.
42.Clinical Study Report: A Phase 3 Study Evaluating the Safety, Tolerability, and Pharmacokinetics of Elexacaftor/Tezacaftor/Ivacaftor Triple Combination Therapy in Cystic Fibrosis Subjects 2 Through 5 Years of Age [internal sponsor's report]. Toronto (ON): Vertex Pharmaceuticals Incorporated; 2022.
43.Vertex Pharmaceuticals Incorporated. NCT04545515: A study evaluating the long-term safety and efficacy of elexacaftor/tezacaftor/ivacaftor in cystic fibrosis (CF) subjects 6 years and older and F/MF genotypes. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2023: https://clinicaltrials.gov/ct2/show/NCT05153317. Accessed 2023 May 18.
44.Subbarao P, Milla C, Aurora P, et al. Multiple-Breath Washout as a Lung Function Test in Cystic Fibrosis. A Cystic Fibrosis Foundation Workshop Report. Ann Am Thorac Soc. 2015;12(6):932-939. PubMed
45.Kent L, Reix P, Innes JA, et al. Lung clearance index: evidence for use in clinical trials in cystic fibrosis. J Cyst Fibros. 2014;13(2):123-138. PubMed
46.Vertex Pharmaceuticals. Protocol Number VX20-445-111, Version 3.0, Statistical Analysis Plan [internal sponsor's report]. 2022.
47.The Canadian Cystic Fibrosis registry: 2020 annual data report. Toronto (ON): Cystic Fibrosis Canada; 2022: https://www.cysticfibrosis.ca/registry/2020AnnualDataReport.pdf. Accessed 2023 May 19.
48.CADTH Reimbursement Review Clinical and Pharmacoeconomic Review Report: elexacaftor-tezacaftor ivacaftor and ivacaftor (Trikafta - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/DRR/2021/SR0673-combined-report.pdf. Accessed 2023 Nov 15.
49.CADTH Reimbursement Review Clinical and Pharmacoeconomic Review Report: elexacaftor-tezacaftor ivacaftor and ivacaftor (Trikafta - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/DRR/2022/SR0710-Trikafta_combined.pdf. Accessed 2023 May 19.
50.Drug Reimbursement Review sponsor submission: Trikafta, oral elexacaftor 50 mg / tezacaftor 25 mg / ivacaftor 37.5 mg and ivacaftor 75 mg; elexacaftor 100 mg / tezacaftor 50 mg / ivacaftor 75 mg and ivacaftor 150 mg; [internal sponsor's package]. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2021 Nov 5.
51.Drug Reimbursement Review sponsor submission: Trikafta, elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg tablets and ivacaftor 150 mg tablets; elexacaftor 50 mg/tezacaftor 25 mg/ivacaftor 37.5 mg tablets and ivacaftor 75 mg tablets oral [internal sponsor's package]. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2021 Jan 21.
52.Vertex Pharmaceuticals Incorporated. NCT04105972: A study evaluating the efficacy and safety of VX-445/tezacaftor/ivacaftor in cystic fibrosis subjects, homozygous for F508del. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2020: https://clinicaltrials.gov/ct2/show/NCT04105972. Accessed 2023 Nov 15.
53.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N Engl J Med. 2011;365(18):1663-1672. PubMed
54.Moss RB, Flume PA, Elborn JS, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3(7):524-533. PubMed
55.De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674-680. PubMed
56.Clinical Study Report: VX19-445-116. A phase 3b, randomized, placebo-controlled study evaluating the efficacy and safety of elexacaftor/tezacaftor/ivacaftor in cystic fibrosis subjects 6 through 11 years of age who are heterozygous for the F508del mutation and a minimal function mutation (F/MF) [internal sponsor's report]. Boston (MA): Vertex Pharmaceuticals Incorporated; 2021 Aug 27.
57.Flume PA, Saiman L, Marshall B. The Impact of COVID-19 in Cystic Fibrosis. Arch Bronconeumol. 2022;58(6):466-468. PubMed
58.CADTH Common Drug Review Clinical Review Report: lumacaftor/ivacaftor (Orkambi - Vertex Pharmaceuticals [Canada] Incorporated). Ottawa (ON): CADTH; 2016 Jan 26: https://www.cadth.ca/sites/default/files/cdr/clinical/SR0471_Orkambi_CL_Report.pdf. Accessed 2023 Nov 15.
59.Vertex Pharmaceuticals Incorporated. Vertex announces FDA approvals of Trikafta® (elexacaftor/tezacaftor/ivacaftor and ivacaftor), Symdeko® (tezacaftor/ivacaftor and ivacaftor) and Kalydeco® (ivacaftor) for use in people with CF with certain rare mutations. 2020; https://news.vrtx.com/press-release/vertex-announces-fda-approvals-trikaftar-elexacaftortezacaftorivacaftor-and-ivacaftor. Accessed 2023 Nov 15.
60.Clinical Study Report: study 102. A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of VX-445 combination therapy in subjects with cystic fibrosis who are heterozygous for the F508del mutation and a minimal function mutation (F/MF) [internal sponsor's report]. Toronto (ON): Vertex Pharmaceuticals Incorporated; 2019 Jul 2.
61.Clinical Study Report: study 103. A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of VX-445 combination therapy in subjects with cystic fibrosis who are homozygous for the F508del mutation (F/F) [internal sponsor's report]. Toronto (ON): Vertex Pharmaceuticals Incorporated; 2019 Jun 18.
62.Clinical Study Report: study 104. A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of elexacaftor combination therapy in subjects with cystic fibrosis who are heterozygous for the F508del mutation and a gating or residual function mutation (F/G and F/RF genotypes) [internal sponsor's report]. Toronto (ON): Vertex Pharmaceuticals Incorporated; 2020 Aug 28.
63.Clinical Study Report: study 109. A phase 3b, randomized, double-blind, controlled study evaluating the efficacy and safety of elexacaftor/tezacaftor/ivacaftor in cystic fibrosis subjects, homozygous for F508del [internal sponsor's report]. Toronto (ON): Vertex Pharmaceuticals Incorporated; 2020 Oct 16.
64.Vertex response to March 10, 2021 request for additional information regarding Trikafta CADTH review [internal additional sponsor's information]. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2021 Mar 22.
Appendix 1: Key Results From Studies of Patients Aged 6 to 11 Years
Note that this appendix has not been copy-edited.
Table 39: Summary of Key Results From Pediatric Studies
Analysis | Study 116 | Study 106B (N = 66) | |
|---|---|---|---|
Placebo (N = 61) | ELX-TEZ-IVA (N = 60) | ||
Absolute change in ppFEV1 (%) through week 24 | |||
Baseline mean (SD) | 87.2 (15.8) | 91.4 (13.8) | 88.8 (17.7) |
Patients in analysis | 59 | 59 | 59 |
LS mean change (SE) | –1.5 (1.5) | 9.5 (1.5) | 10.2 (1.2) |
P value within treatment | 0.2977 | < 0.0001 | < 0.0001 |
LSMD (95% CI) | Reference | 11.0 (6.9, 15.1) | NA |
P value versus placebo | Reference | < 0.0001 | NA |
Absolute change from baseline in LCI2.5 through week 24 | |||
Baseline mean (SD) | 9.75 (1.95) | 10.26 (2.22) | 9.77 (2.68) |
Patients in analysis | 61 | 60 | 50 |
LS mean change (SE) | –0.02 (0.16) | –2.29 (0.16) | –1.71 (0.20) |
P value within treatment | 0.8859 | < 0.0001 | < 0.0001 |
LSMD (95% CI) | Reference | –2.26 (–2.71, –1.81) | NA |
P value versus placebo | Reference | < 0.0001 | NA |
Absolute change from baseline CFQ-R respiratory domain through week 24a, b | |||
Baseline mean (SD) | 82.7 (14.1) | 85.7 (11.7) | 80.3 (15.2) |
Patients in analysis | 61 | 60 | 65 |
LS mean change (SE) | 0.5 (1.6) | 5.9 (1.6) | 7.0 (1.1) |
P value within treatment | 0.7693 | 0.0003 | < 0.0001 |
LSMD (95% CI) | Reference | 5.5 (1.0, 10.0) | NA |
P value versus placebo | Reference | 0.0174 | NA |
Absolute change from baseline in sweat chloride through week 24a,b | |||
Baseline mean (SD) | 102.6 (8.6) | 102.8 (10.0) | 102.2 (9.1) |
Patients in analysis | 61 | 60 | 60 |
LS mean change (SE) | –0.9 (1.5) | –52.1 (1.5) | –60.9 (1.4) |
P value within treatment | 0.5241 | < 0.0001 | < 0.0001 |
LSMD (95% CI) | Reference | –51.2 (–55.3, –47.1) | NA |
P value versus placebo | Reference | < 0.0001 | NA |
Pulmonary exacerbations | |||
Patients with event, n (%) | 16 (26.2) (AE only) | 1 (1.7) (AE only) | 4 (6.1) |
Number of events | NA | NA | 4 |
Event rate per year | NA | NA | 0.12 |
Pulmonary exacerbations requiring hospitalization | |||
Patients with event, n (%) | NA | NA | 1 (1.5) |
Number of events | NA | NA | 1 |
Event rate per year | NA | NA | 0.03 |
Pulmonary exacerbations requiring IV antibiotics | |||
Patients with event, n (%) | NA | NA | 1 (1.5) |
Number of events | NA | NA | 1 |
Event rate per year | NA | NA | 0.03 |
Absolute change in BMI z score at week 24 | |||
Baseline mean (SD) | NA | NA | –0.16 (0.74) |
Patients in analysis | NA | NA | 33 |
LS mean (SE) | NA | NA | 0.37 (0.05) |
95% CI of LS mean | NA | NA | (0.26, 0.48) |
P value | NA | NA | < 0.0001 |
Absolute change in body weight z score at week 24 | |||
Baseline mean (SD) | NA | NA | –0.22 (0.76) |
Patients in analysis | NA | NA | 33 |
LS mean (SE) | NA | NA | 0.25 (0.04) |
95% CI of LS mean | NA | NA | (0.16, 0.33) |
P value | NA | NA | < 0.0001 |
Summary of AEs | |||
At least 1 AE | 57 (93.4) | 48 (80.0) | 65 (98.5) |
WDAEs | 0 | 1 (1.7) | 1 (1.5) |
AEs leading to interruption | 0 | 7 (11.7) | 1 (1.5) |
Grade 3/4 AEs | 2 (3.3) | 2 (3.3) | 1 (1.5) |
SAEs | 9 (14.8) | 4 (6.7) | 1 (1.5) |
AEs of special interest | |||
Elevated transaminases | 3 (4.9) | 6 (10.0) | 7 (10.6) |
Discontinuation | 0 | 0 | 0 |
Interruption | 0 | 0 | 0 |
Serious events | 0 | 4 (6.7) | 0 |
Any rash events | 3 (4.9) | 8 (13.3) | 16 (24.2) |
Discontinuation | 0 | 1 (1.7) | 1 (1.5) |
Interruption | 0 | 2 (3.3) | 0 |
Serious events | 0 | 0 | 0 |
AE = adverse event; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; SAE = serious adverse events; WDAE = withdrawal due to adverse events.
Appendix 2: Key Results From Studies of Patients Aged 12 Years and Older
Table 40: Summary of Key Results From Pivotal and Protocol-Selected Studies
Analysis | Study 102 (F/MF) 24 weeks | Study 103 (F/F) 4 Weeks | Study 109 (F/F) 24 weeks | Study 104 (F/G and F/RF) 8 weeks | |||||
|---|---|---|---|---|---|---|---|---|---|
Placebo (N = 203) | ELX-TEZ-IVA (N = 200) | TEZ-IVA (N = 52) | ELX-TEZ-IVA (N = 55) | TEZ-IVA (N = 88) | ELX-TEZ-IVA (N = 87) | Control (N = 126) | ELX-TEZ-IVA (N = 132) | ||
Absolute change in ppFEV1 (%) | |||||||||
BL; mean (SD) | 61.3 (15.5) | 61.6 (15.0) | 60.2 (14.4) | 61.6 (15.4) | 64.2 (15.1) | 63.0 (16.7) | 68.1 (16.4) | 67.1 (15.7) | |
LSM change (SE) | –0.4 (0.5) | 13.9 (0.6) | 0.4 (0.9) | 10.4 (0.9) | 1.0 (0.7) | 11.2 (0.7) | 0.2 (0.5) | 3.7 (0.5) | |
LSMD (95% CI) | 14.3 (12.7 to 15.8) | 10.0 (7.4 to 12.6) | 10.2 (8.2 to 12.1) | 3.5 (2.2 to 4.7) | |||||
P value | < 0.0001 a | < 0.0001 a | < 0.0001 b | < 0.0001 b | |||||
Absolute change in CFQ-R (respiratory domain) | |||||||||
BL; mean (SD) | 70.0 (17.8) | 68.3 (16.9) | 72.6 (17.9) | 70.6 (16.2) | 73.1 (17.6) | 71.2 (19.6) | 77.3 (15.8) | 76.5 (16.6) | |
LSM change (SE) | –2.7 (1.0) | 17.5 (1.0) | –1.4 (2.0) | 16.0 (2.0) | 1.2 (1.5) | 17.1 (1.5) | 1.6 (1.2) | 10.3 (1.2) | |
LSMD (95% CI) | 20.2 (17.5 to 23.0) | 17.4 (11.8 to 23.0) | 15.9 (11.7 to 20.1) | 8.7 (5.3 to 12.1) | |||||
P value | < 0.0001 b | < 0.0001 | < 0.0001 a | < 0.0001 | |||||
Absolute change in BMI (kg/m2) | |||||||||
BL; mean (SD) | 21.31 (3.14) | 21.49 (3.07) | 21.88 (4.12) | 21.75 (3.19) | 21.92 (3.89) | 21.17 (3.43) | 24.05 (4.71) | 24.07 (4.72) | |
LSM change (SE) | 0.09 (0.07) | 1.13 (0.07) | –0.07 (0.07) | 0.53 (0.07) | 0.15 (0.13) | 1.59 (0.13) | 0.16 (0.06) | 0.28 (0.06) | |
LSMD (95% CI) | 1.04 (0.85 to 1.23) | 0.60 (0.41 to 0.79) | 1.44 (1.07 to 1.82) | 0.13 (–0.03 to 0.29) | |||||
P value | < 0.0001 b | < 0.0001 | < 0.0001 | NA | |||||
Absolute change in SwCl (mmol/L) | |||||||||
BL; mean (SD) | 102.9 (9.8) | 102.3 (11.9) | 90.0 (12.3) | 91.4 (11.0) | 89.8 (11.7) | 89.0 (12.2) | 56.4 (25.5) | 59.5 (27.0) | |
LSM change (SE) | –0.4 (0.9) | –42.2 (0.9) | 1.7 (1.8) | –43.4 (1.7) | –3.4 (1.2) | –46.2 (1.3) | 0.7 (1.1) | –22.3 (1.1) | |
LSMD (95% CI) | –41.8 (–44.4 to –39.3) | –45.1 (–50.1 to –40.1) | –42.8 (–46.2 to –39.3) | –23.1 (–26.1 to –20.1) | |||||
P value | < 0.0001 b | < 0.0001 b | < 0.0001 | < 0.0001 | |||||
Pulmonary exacerbations | |||||||||
Patients with evt, n (%) | 76 (37.4) | 31 (15.5) | NA | NA | NA | ||||
Event rate per year | 0.98 | 0.37 | |||||||
Rate ratio (95% CI) | 0.37 (0.25 to 0.55) | ||||||||
P value | < 0.0001 | ||||||||
Pulmonary exacerbations requiring hospitalization | |||||||||
Patients with evt, n (%) | 27 (13.3) | 7 (3.5) | NA | NA | NA | ||||
Event rate per year | 0.24 | 0.07 | |||||||
Rate ratio (95% CI) | 0.29 (0.14 to 0.61) | ||||||||
P value | < 0.0001 | ||||||||
Pulmonary exacerbations requiring IV antibiotics | |||||||||
Patients with evt, n (%) | 42 (20.7) | 9 (4.5) | NA | NA | NA | ||||
Event rate per year | 0.36 | 0.08 | |||||||
Rate ratio (95% CI) | 0.22 (0.11 to 0.43) | ||||||||
P value | < 0.0001 | ||||||||
Time-to-first pulmonary exacerbation | |||||||||
Hazard ratio (95% CI) | 0.34 (0.22 to 0.52) | NA | NA | NA | |||||
P value | < 0.0001 | ||||||||
Time-to-first pulmonary exacerbation requiring hospitalization | |||||||||
Hazard ratio (95% CI) | 0.25 (0.11 to 0.58) | NA | NA | NA | |||||
P value | 0.0011 | ||||||||
Time-to-first pulmonary exacerbation requiring IV antibiotics | |||||||||
Hazard ratio (95% CI) | 0.19 (0.09 to 0.39) | NA | NA | NA | |||||
P value | < 0.0001 | ||||||||
Summary of AEs | |||||||||
At least 1 AE | 193 (96.0) | 188 (93.1) | 33 (63.5) | 32 (58.2) | 81 (92.0) | 77 (88.5) | 83 (65.9) | 88 (66.7) | |
WDAEs | 0 | 2 (1.0) | 0 | 0 | 2 (2.3) | 1 (1.1) | 2 (1.6) | 1 (0.8) | |
Interruption due to AEs | 10 (5.0) | 19 (9.4) | 0 | 0 | 1 (1.1) | 2 (2.3) | 3 (2.4) | 5 (3.8) | |
Grade 3/4 AEs | 15 (7.5) | 19 (9.4) | 1 (1.9) | 0 | 7 (8.0) | 7 (8.0) | 4 (3.2) | 5 (3.8) | |
SAEs | 42 (20.9) | 28 (13.9) | 1 (1.9) | 2 (3.6) | 14 (15.9) | 5 (5.7) | 11 (8.7) | 5 (3.8) | |
Most common AEs | |||||||||
Infective pulmonary exacerbation of CF | 95 (47.3) | 44 (21.8) | 6 (11.5) | 1 (1.8) | 36 (40.9) | 10 (11.5) | 13 (10.3) | 3 (2.3) | |
Sputum increased | 39 (19.4) | 40 (19.8) | 3 (5.8) | 3 (5.5) | 16 (18.2) | 10 (11.5) | 8 (6.3) | 6 (4.5) | |
Headache | 30 (14.9) | 35 (17.3) | 4 (7.7) | 3 (5.5) | 18 (20.5) | 25 (28.7) | 19 (15.1) | 11 (8.3) | |
Cough | 77 (38.3) | 34 (16.8) | 4 (7.7) | 8 (14.5) | 23 (26.1) | 11 (12.6) | 18 (14.3) | 3 (2.3) | |
AEs of special interest | |||||||||
Elevated transaminases | 8 (4.0) | 22 (10.9) | 1 (1.9) | 2 (3.6) | 1 (1.1) | 6 (6.9) | 1 (0.8) | 8 (6.1) | |
Discontinuation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.8) | |
Interruption | 3 (1.5) | 2 (1.0) | 0 | 0 | 0 | 2 (2.3) | 1 (0.8) | 0 | |
Serious events | 1 (0.5) | 0 | 0 | 0 | 0 | 1 (1.1) | 0 | 0 | |
Any rash events | 13 (6.5) | 22 (10.9) | 2 (3.8) | 2 (3.6) | 2 (2.3) | 11 (12.6) | 5 (4.0) | 4 (3.0) | |
Discontinuation | 0 | 1 (0.5) | 0 | 0 | 0 | 0 | 0 | 0 | |
Interruption | 1 (0.5) | 4 (2.0) | 0 | 0 | 0 | 1 (1.1) | 1 (0.8) | 1 (0.8) | |
Serious events | 1 (0.5) | 3 (1.5) | 0 | 1 (1.8) | 0 | 0 | 0 | 0 | |
AE = adverse event; BL = baseline; CF = cystic fibrosis; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene SAE = serious adverse events; TEZ-IVA = tezacaftor-ivacaftor and ivacaftor; URTI = upper respiratory tract infection; WDAE = withdrawal due to adverse events.
Note: This table has not been copy-edited.
aPrespecified primary end point.
bPrespecified key secondary end point.
cPost hoc analysis only reported for the indirect comparison.
Source: Clinical Study Reports60-63 and additional information provided by sponsor.64
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
BSC
best supportive care
CF
cystic fibrosis
CF Canada
Cystic Fibrosis Canada
ELX
elexacaftor
ELX-TEZ-IVA
elexacaftor-tezacaftor-ivacaftor and ivacaftor
F/F
homozygous for F508del mutation in the CFTR gene
F/G
1 F508del mutation and 1 gating mutation in the CFTR gene
F/MF
1 F508del mutation and 1 minimal function mutation in the CFTR gene
F/RF
1 F508del mutation and 1 residual function mutation in the CFTR gene
ICER
incremental cost-effectiveness ratio
IVA
ivacaftor
LUM-IVA
lumacaftor-ivacaftor
PEx
pulmonary exacerbation
ppFEV1
percent predicted forced expiratory volume in the first second
QALY
quality-adjusted life-year
TEZ
tezacaftor
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Elexacaftor-tezacaftor-ivacaftor and ivacaftor (Trikafta) taken orally
|
Submitted price | Elexacaftor 100 mg–tezacaftor 50 mg–ivacaftor 75 mg and ivacaftor 150 mg tablets OR elexacaftor 80 mg–tezacaftor 40 mg–ivacaftor 60 mg and ivacaftor 59.5 mg granules: $840 per daily dose |
Indication | For the treatment of cystic fibrosis (CF) in patients aged 2 years and oldera who have at least 1 F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene |
Health Canada approval status | Pre-NOC |
Health Canada review pathway | Priority review |
NOC date | October 11, 2023 |
Reimbursement request | Confirmed diagnosis with CF with at least 1 F508del mutation in the CFTR gene
|
Sponsor | Vertex Pharmaceuticals (Canada) Incorporated |
Submission history | Previously reviewed: Yes Elexacaftor-tezacaftor-ivacaftor (combination tablet) and ivacaftor
|
NOC = Notice of Compliance.
aThis review is focused on patients aged 2 to 5 years, as per the sponsor’s reimbursement request.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Microsimulation |
Target population | Patients with CF aged 2 to 5 years who have at least 1 F508del-CFTR mutation in the CFTR gene, represented by the following 4 genotypes:
|
Treatment | ELX-TEZ-IVAa with background BSC |
Comparators |
BSC for all genotypes consisted of recommended medications (such as mucolytics, inhaled and oral antibiotics, inhaled hypertonic saline, nutritional supplements, enteral tube feeding, pancreatic enzymes, antifungal agents, and corticosteroids) and physiotherapy |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (97 years) |
Key data sources |
|
Submitted results |
|
Key limitations |
|
CADTH reanalysis results | CADTH conducted a reanalysis that included the removal of the additional benefit of CFTR modulators on the long-term rate of decline in ppFEV1 and PExs; the removal of dynamic pricing; the inclusion of health care costs across the entire model time horizon; the removal of an adjustment to drug acquisition costs based on patient compliance; an assumption of equal hospital and pharmacotherapy costs between treatments; and the removal of a treatment-specific utility increment for patients on ELX-TEZ-IVA. Results of the CADTH reanalysis are as follows:
ELX-TEZ-IVA was not cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained in any scenario conducted by CADTH. A price reduction in excess of 94% for ELX-TEZ-IVA (for both granules and tablets) is required for ELX-TEZ-IVA to be considered cost-effective at a willingness-to-pay threshold of $50,000 for any of the genotypes when compared with BSC. |
BSC = best supportive care; CF = cystic fibrosis; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LUM-IVA = lumacaftor-ivacaftor; LY = life-year; PEx = pulmonary exacerbation; ppFEV1 = percent predicted forced expiratory volume in the first second; QALY = quality-adjusted life-year; vs. = versus.
aThe model assumes that patients aged 2 to 5 years receive ELX-TEZ-IVA granules, whereas patients aged 6 years and older receive ELX-TEZ-IVA tablets.
Conclusions
Results from the 24-week, open-label, uncontrolled Study 111 Part B suggest that treatment with elexacaftor-tezacaftor-ivacaftor and ivacaftor (ELX-TEZ-IVA) resulted in improvements from baseline in lung function (decrease in lung clearance index 2.5 from baseline) and cystic fibrosis (CF) biomarkers (reduction in sweat chloride) in patients aged 2 to 5 years with CF. However, as Study 111 was primarily designed to evaluate the safety, tolerability, and pharmacokinetics of ELX-TEZ-IVA in this younger population, the pharmacoeconomic review is based on the extrapolation of efficacy data from studies conducted in older patients with CF. Patients who completed Study 111 were eligible to enrol in an open-label extension study; however, interim results were not available at the time of filing the application with CADTH.
The clinical experts consulted by CADTH for the current review noted that, given the mechanism of action and compelling efficacy data in patients aged 6 years and older, ELX-TEZ-IVA is expected to benefit patients aged 2 to 5 years who have at least 1 508del mutation in the CFTR gene. However, these conclusions are based on studies with a maximum follow-up period of 192 weeks, and there remains no evidence on the long-term impact of ELX-TEZ-IVA on the rate of decline in percent predicted forced expiratory volume in the first second (ppFEV1) or pulmonary exacerbations (PExs) beyond the trial periods for any genotype or age group.
Beyond uncertainty in the long-term clinical efficacy of ELX-TEZ-IVA, CADTH identified several major limitations with the submitted economic evaluation. The following were addressed in the CADTH reanalysis: the removal of an additional benefit of CFTR modulators on the long-term rate of decline in ppFEV1 and PExs; the removal of dynamic pricing for CFTR modulators; the inclusion of costs for ELX-TEZ-IVA for the period in which a survival benefit was achieved in comparison to best supportive care (BSC); the removal of an adjustment to drug acquisition costs based on patient compliance; and the removal of a treatment-specific utility increment for patients on ELX-TEZ-IVA. The results of the CADTH reanalysis were aligned with the sponsor’s, in that ELX-TEZ-IVA was not cost-effective in any of the genotype subgroups at conventionally acceptable incremental cost-effectiveness ratio (ICER) thresholds. In the CADTH base-case analyses, when compared to BSC, ELX-TEZ-IVA was associated with an ICER of $1,284,953 per quality-adjusted life-year (QALY) gained in patients who are homozygous for F508del mutation in the CFTR gene (F/F genotype); $1,451,526 per QALY gained in patients with 1 F508del mutation and 1 minimal function mutation in the CFTR gene (F/MF genotype); $1,284,853 per QALY gained in patients with 1 F508del mutation and 1 gating mutation in the CFTR gene (F/G genotype); and $1,644,869 per QALY gained in patients with 1 F508del mutation and 1 residual function mutation in the CFTR gene (F/RF genotype). Additionally, ELX-TEZ-IVA was associated with an ICER of $838,687 per QALY gained when compared to lumacaftor-ivacaftor (LUM-IVA) in the F/F genotype population.
The key drivers in the analyses were drug acquisition costs and assumptions in the long-term benefits of ELX-TEZ-IVA, which were uncertain. Treatment with ELX-TEZ-IVA was not cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained in any scenario conducted by CADTH. A price reduction in excess of 94% for ELX-TEZ-IVA (for both granules and tablets) is required for all 4 genotypes for ELX-TEZ-IVA to be considered cost-effective at this threshold in comparison with BSC. Based on a price reduction of this magnitude, the daily cost of ELX-TEZ-IVA would need to be approximately $50.40 per patient. As the majority of patients with CF aged 6 years and older living in Canada are currently being treated with ELX-TEZ-IVA, the sponsor’s submitted economic evaluation assessing the cost-effectiveness of ELX-TEZ-IVA versus BSC or LUM-IVA (for the F/F genotype only) over the entire lifetime of a patient aged 2 to 5 years does not accurately reflect the current landscape of CF treatment in Canada. The cost-effectiveness of starting ELX-TEZ-IVA for patients aged 2 to 5 years versus waiting to initiate treatment at age 6 years and older is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient group input was submitted by Cystic Fibrosis Canada (CF Canada), informed by focus groups with 6 parents of children younger than 5 years who have at least 1 copy of the F508del mutation. Data from the CF Canada 2021 patient and caregiver survey (1,200 responses) on Trikafta access, along with the Canadian Cystic Fibrosis Registry, were also used to inform the patient group input. Patients and caregivers reported that living with CF has a tremendous impact on their lives, affecting physical, psychological, social, and financial components. When asked about disease management, patients noted that managing CF requires a demanding treatment routine involving much time and effort, while frequent clinic visits and hospital stays are needed to manage the progressive and debilitating symptoms. Parents and caregivers of children with CF noted that an ideal treatment in CF would fully address the basic molecular defect in CF and restore normal chloride transport on the cell surface. No input was provided from families that had a child aged 2 to 5 years receiving ELX-TEZ-IVA. Extending access to ELX-TEZ-IVA for patients with CF aged 2 to 5 years would be congruent with the secondary prevention paradigm of CF care and may decrease the long-term burden of the disease.
Clinician input was received from CF Canada’s Accelerating Clinical Trials Network and the CF Canada Healthcare Advisory Council. Clinician input noted that the treatment paradigm for CF in children aged 2 to 5 years is lifelong. It consists of nonmodulator treatments and medications, many of which start at the time of diagnosis (including in infancy) and continue every day throughout life. The clinician input noted that, although not a cure, CFTR modulators are the first available therapies for CF targeted at correcting the basic defect in CF. However, significant unmet therapeutic needs remain for patients living with CF, as available treatments address the symptoms and complications of CF and attempt to slow down the eventual fatal progression of the disease but do not effectively address the root cause or reverse the course of the disease. Currently available therapies, such as LUM-IVA, have significant side effects and numerous drug interactions. In addition, the current standard treatments are burdensome for patients and their caregivers, which affects medication adherence as well as the mental health and quality of life of patients and caregivers. Lastly, the clinician input noted that while ELX-TEZ-IVA may not be the first therapy to address the underlying defect in CF, it still represents an improvement on existing CFTR modulator therapies, and there remains an importance of early treatment of CF to prevent disease progression and irreversible damage.
Feedback from the drug plans indicated interest on if there were any concerns with extrapolation of data from CF patients aged 6 years and older treated with ELX-TEZ-IVA to patients aged 2 to 5 years. They further requested information about the relevance of LUM-IVA in current treatment practices and how the reimbursement of ELX-TEZ-IVA may impact patients currently receiving LUM-IVA (i.e., would patients be eligible to switch, were there any special considerations, and so on). Drug plans also asked, given that patients aged 2 to 5 years would not be able to accurately complete a spirometry, what other parameters or biomarkers could be used for disease diagnosis.
Two of these concerns were addressed in the sponsor’s model:
Relevant comparators for patients with CF aged 2 to 5 years were included.
All patients were assumed to receive standard of care, consisting of antibiotics, mucolytics, and pancreatic enzymes.
In addition, CADTH addressed some of these concerns as follows:
CADTH included disease management costs while on CFTR modulators for the entire time horizon.
CADTH was unable to address the following concerns raised from stakeholder input:
Treatment switching from LUM-IVA to ELX-TEZ-IVA was not included in the cost-utility analysis.
Economic Review
The current review is for ELX-TEZ-IVA for the treatment of CF in patients aged 2 to 5 years who have at least 1 F508del mutation in the CFTR gene.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing ELX-TEZ-IVA in combination with BSC for the treatment of CF in patients aged 2 to 5 years who have at least 1 F508del mutation in the CFTR gene, represented by the F/F, F/M, F/RF, and F/G genotypes. The sponsor compared the submitted drug regimen with BSC alone in all 4 subgroups, as well as with LUM-IVA in the F/F subgroup.1 The modelled population is aligned with a subset of the Health Canada indication, which had not previously been submitted to CADTH: patients aged 2 to 5 years. CADTH has previously reviewed ELX-TEZ-IVA for the treatment of CF in patients aged 6 years and older who have at least 1 F508del mutation in the CFTR gene.2 CADTH focused its review for this submission on the population aged 2 to 5 years.
The recommended dose of ELX-TEZ-IVA is age and weight dependent. Patients aged 2 to 5 years weighing less than 14 kg are to take 1 packet of elexacaftor 80 mg–tezacaftor 40 mg–ivacaftor 60 mg granules in the morning and 1 packet of ivacaftor 59.5 mg granules in the evening.3 Patients aged 2 to 5 years weighing more than 14 kg are to take 1 packet of elexacaftor 100 mg–tezacaftor 50 mg–ivacaftor 75 mg granules in the morning and 1 packet of ivacaftor 75 mg granules in the evening.3 Patients aged 6 to 11 years weighing less than 30 kg are to take 2 tablets, each containing elexacaftor 50 mg–tezacaftor 25 mg–ivacaftor 37.5 mg in the morning and 1 tablet of ivacaftor 75 mg in the evening.3 All other patients have a recommended dose of 2 tablets, each containing elexacaftor 100 mg–tezacaftor 50 mg–ivacaftor 75 mg in the morning and 1 tablet of ivacaftor 150 mg in the evening.3 All morning and evening doses are to be taken approximately 12 hours apart with fat-containing food.3 The daily cost of treatment is $840.00 per day, or an annual cost of $306,810, based on the treatment’s list price, regardless of the strength or form. BSC was selected as the comparator treatment for all genotypes and was defined as recommended medications (such as mucolytics, inhaled and oral antibiotics, inhaled hypertonic saline, nutritional supplements, enteral tube feeding, pancreatic enzymes, antifungal agents, and corticosteroids) and physiotherapy. All patients on CFTR modulator therapies also received BSC. LUM-IVA was also included as a comparator for patients homozygous for F508del-CFTR.
The outcomes of the model included QALYs and life-years over a lifetime horizon of approximately 97 years. The analysis was undertaken from the perspective of the Canadian public health care payer.1 Discounting (1.5% per annum) was applied for both costs and outcomes, and the cycle length was 4 weeks for the first 2 years and annual for the remainder of the model time horizon.1
Model Structure
The sponsor submitted a patient-level microsimulation model (i.e., a microsimulation) used to track CF disease progression and treatment benefits for a typical patient profile of each genotype, informed by various CFTR modulator trials (Figure 1). In the sponsor’s probabilistic base case, 250 average patients per genotype were simulated and the expected costs and clinical effects of ELX-TEZ-IVA, BSC, and LUM-IVA (for the F/F genotype only) were calculated.1 This process was repeated 80 times.
At the beginning of each cycle, the model calculated a patient’s mortality risk based on a Cox proportional hazard model that linked the survival of a patient with CF to 9 risk factors: age, gender, ppFEV1, annual number of PEx, Staphylococcus aureus infection, Burkholderia cepacian infection, CF-related diabetes, weight-for-age z score, and pancreatic sufficiency status.1 For each cycle a patient remained alive, certain patient characteristics were updated, including age and treatment discontinuation for patients aged 2 to 5 years, and age, ppFEV1, weight-for age z score, PEx rate, eligibility for and occurrence of lung transplant, development of CF-related diabetes, and treatment discontinuation for patients aged 6 years and older. Long-term health impacts due to treatment were predicted using clinical outcomes such as median predicted survival, mean time spent in ppFEV1 states, cumulative change in ppFEV1, annual and lifetime PEx rates, and proportion of patients receiving a lung transplant.1 During each cycle, patients would accrue life-years and QALYs; costs were applied at the end of each run of 250 patients for efficiency gains.
Model Inputs
Baseline age-specific risk of death in the model was derived from a cohort-based survival analysis of the Canadian Cystic Fibrosis Registry reported by Stephenson et al.4 Kaplan-Meier curves from this analysis were digitized and fitted using parametric survival analysis to generate a mortality risk for the lifetime time horizon. In the sponsor’s base case, the Gompertz curve was selected as the best-fitting curve, and it was assumed that patients aged 2 to 5 years had 100% survival.1 Mortality was recalculated for each cycle using the Cox proportional hazard model developed by Liou et al. to account for the factors indicted in the Model Structure section.5 The hazard of mortality in the model was assumed to be no lower than for the general population of Canada.1
The patient characteristics used to inform the mortality risk in the model were primarily from the pooled mean baseline characteristics of CFTR modulator trials and were specific to each genotype. Baseline characteristics of age, gender, ppFEV1, and weight-for-age z score for the homozygous F/F genotype were informed by Study 809-011B and Study 809-109 (both LUM-IVA trials), the subset of patients with an F/F genotype from Study 661-113B and Study 661-115 (both tezacaftor-ivacaftor and ivacaftor [TEZ-IVA] trials), and Study 106B (ELX-TEZ-IVA trial). Patients in Study 106B, Study 661-113B, and Study 661-115 may have had a prior history of CFTR modulator use but were required to undergo a 28-day washout period before screening. Their baseline characteristics were therefore considered by the sponsor to be reflective of a CFTR modulator–naive population for the homozygous F/F genotype.6-10 Study 106B and Study 116 (both ELX-TEZ-IVA trials) were further used to inform baseline patient characteristics for the F/MF population.9,11 ENVISION, KONNECTION, and KONDUCT (all trials of IVA monotherapy) were used to inform the characteristics of the F/G population, where patients were not required to have an F508del-CFTR mutation on the second allele.12-14 Lastly, Study 661-113B and Study 661-115 were used to inform the baseline characteristics of the F/RF population.6,8 The baseline rate of PExs requiring IV antibiotics and/or hospitalization was informed by Whiting et al.15 CF-related diabetes status at baseline was based on data from the UK Cystic Fibrosis Registry.16 Patients were assumed not to be at risk of developing CF-related diabetes before age 6, and the annual incidence of CF-related diabetes was applied across all genotypes. It was assumed that the risk of developing CF-related diabetes was equal for patients receiving a CFTR modulator and for those receiving BSC alone.
As noted previously, age, ppFEV1, PExs, and weight-for-age z score were updated at the beginning of each model cycle. Clinical efficacy inputs were derived from the relevant studies. Since ppFEV1 was not available for patients aged 2 to 5 years, the sponsor assumed that lung function, according to ppFEV1, is not tracked until patients turn 6 years in the model, at which point ppFEV1 data from patients aged 6 to 11 years was used. As the assignment of baseline mortality hazards in the model was based on a CFTR modulator–naive population, the analysis required placebo-adjusted estimates of clinical efficacy for CFTR modulators where there were gaps in the available direct evidence. Placebo-adjusted estimates were derived, where necessary, using an indirect treatment comparison with individual patient-level data from relevant phase III randomized controlled trials.1,17
||||| || ||| ||||||| || ||| ||||||||| ||||||| |||||| ||| |||| |||||||| ||||||||| |||| |||||||||| |||| ||||||| || |||| || ||||| |||||||| || ||||||| ||| |||||| |||| |||||||| ||| |||| ||||||||| ||||||||| || |||||| |||| ||| ||| ||||||||| ||| |||||||| |||| | || | |||||| ||| |||||||| ||| ||||||||| ||||||||||| || |||| | || | |||| ||||||| || |||||||||| || ||||| |||||||| || |||||| ||||||||||| |||| ||||||| | ||||| ||||| ||| ||||| |||||||| || |||||| ||| ||||| || |||| ||| ||||||||| |||||||| |||| | || || || |||| ||||| || |||||||| ||| || |||| ||||||||||||| ||| ||| ||| ||||||||||| |||| |||| |||||||| |||| | || || || ||||| ||| |||| |||| || ||||||||| ||| |||||||| ||| ||||||||||| ||| |||| |||||||| ||||| ||||||||||| |||| ||||||| ||||||| ||| ||||| ||||||| ||| ||||||| ||| ||| ||| |||||||||| |||| |||| ||||||| |||| ||| |||||||| ||| ||| |||| ||||||||||| ||| |||||||| || |||||| ||| |||||||||| ||||| |||| |||| ||| |||| | || || |||||||| || ||||| |||||| || |||| ||| ||||||||||| |||||||| || ||| |||||||| |||| | || || |||| ||| ||||||||| ||| ||| |||||||| ||| |||| |||||||||| ||| ||||| ||||||||| |||| |||||||||||| |||| |||| || ||| |||| || ||| ||||| |||||||| |||| ||||| ||| ||||| || ||| |||||||| |||||||||| || ||| ||||| ||||||||| || ||||| |||||||||| ||||||| ||| | || || ||| || ||| ||||| ||| |||||| || ||| ||| ||| |||| ||||||||||||||||
Patients on BSC alone were expected to not have any acute increases in ppFEV1 and were assumed to have a long-term decline in ppFEV1 in line with a study by Leung et al.19 The same rate of decline was applied to all genotypes, except the heterozygous F/RF genotype as they are typically thought to have a milder form of disease and thus a slower rate of decline. In the absence of data on the reduction in the rate of ppFEV1 decline in patients treated with CFTR modulators from ages 2 to 5 years versus patients receiving BSC, the reduction in the rate of ppFEV1 decline was assumed be the rate observed in the registry-matched analysis conducted in patients aged 12 years or older. Patients were assumed to avoid a proportion of lung function decline before the age of 6 years.1 Patients receiving ELX-TEZ-IVA during the maintenance period were assumed to sustain their acute improvements for 192 weeks, based on the results of Study 105. There was no maintenance period for TEZ-IVA. During the post-acute maintenance period, patients on ELX-TEZ-IVA were modelled to have an 89.7% reduction in the rate of ppFEV1 decline compared with patients receiving BSC, with the exception of patients with an F/F genotype, who had an 86.4% reduction in the rate of ppFEV1 decline.1 Patients on LUM-IVA were assumed to experience a 42% reduction in the rate of ppFEV1 decline compared to patients receiving BSC, based on retrospective observational studies.20-22
The baseline rate of occurrence of PExs each cycle after a patient turned 6 years was based on the patient’s ppFEV1 and age, according to a formula derived by Goss and Burns, and was not genotype specific.23 Due to data availability and as a simplifying assumption, PExs were not estimated for patients aged 2 to 5 years. For patients aged 6 to 11 years, CFTR modulators were also assumed to have no treatment effect on PExs requiring IV antibiotics and/or hospitalizations as the observed PEx event rate of ELX-TEZ-IVA remained low over 120 weeks (24 weeks in Study 106B plus an additional 96 weeks in Study 107, an open-label extension study). Once patients on CFTR modulators turned age 12 years, their rate of PExs was adjusted by a rate ratio derived by the sponsor. This rate was based on an assumed additional treatment impact on PExs beyond those impacts explained by improvements in ppFEV1 in patients treated with CFTR modulators, as captured in the Goss and Burns. formula. To account for the potential double counting of the benefit due to the better ppFEV1 observed with CFTR modulators, the sponsor attempted to calibrate the PEx rate ratio for patients receiving a CFTR modulator. Patients on CFTR modulators had their rate of PExs adjusted such that the resulting relative rate of PExs between patients receiving a CFTR modulator and those receiving BSC alone matched the treatment effect on PExs requiring IV antibiotics and/or hospitalizations observed in the pivotal trials.
Patients treated with a CFTR modulator were assumed to experience an acute change in weight-for-age z score from baseline. The magnitude and duration of the acute change was informed by age-specific and genotype-specific clinical trial data. After the initial change was applied upon model entry, a patient’s weight-for-age z score was assumed to remain consistent for the remainder of the simulation.1 Patients with an F/F genotype receiving TEZ-IVA were assumed to have an increase in their weight-for-age z score, applied at age 6 years based on Study 115.24 Patients on BSC alone were expected to not have any acute increases in their weight-for-age z score.
The sponsor’s model also accounted for treatment discontinuation and compliance. Discontinuation rates for the model period corresponding to the trial duration period were obtained from the relevant phase III trials; open-label extension studies were used to inform a “post-acute” phase of the model, in which no patients discontinued treatment.1 Upon CFTR discontinuation, patients were modelled to no longer receive benefits; however, they retained the acute increase in ppFEV1 and weight-for-age z score they had achieved up until the point of discontinuation. In the post-acute period, patients who had discontinued treatment had the same ppFEV1 decline as the age-dependent values of their BSC counterparts. The sponsor further considered treatment compliance to inform treatment costs over the acute period (first 24 weeks).1 Compliance rates from Study 111 in the combined F/F and F/MF population were applied for all genotypes. Patients on TEZ-IVA had compliance rates aligned with Study 115. Treatment compliance in the post-acute period was set to 93% across all CFTR modulators regardless of genotype.1 Compliance was assumed to have no impact on treatment efficacy and to only affect the costs associated with CFTR modulators.
The sponsor-submitted economic model also considered lung transplant and adverse events. The sponsor aligned the rate of lung transplant and mortality risk following transplant with the previous CADTH submission.25 In the base-case analysis, patients with a ppFEV1 threshold of 30% were eligible for lung transplant and the probability of receiving a transplant was 4.6%.1 Adverse events in the model were based on the relevant phase III trials for the respective ages, genotypes, and CFTR modulators.
Costs considered in the model included drug acquisition, monitoring, disease management, pharmacotherapy, diagnostic tests, PExs, adverse events, and lung transplants. The cost of ELX-TEZ-IVA was submitted by the sponsor, whereas the prices of LUM-IVA were obtained from the Ontario Exceptional Access Program formulary.26 For CFTR modulators, including ELX-TEZ-IVA, the sponsor employed a dynamic pricing approach, in which the introduction of a first generic into the market after loss of patent exclusivity would lead to a 25% reduction in the prices of all drugs, and the entry of a second generic would further reduce prices by 50%. These assumptions were based on a pan-Canadian Pharmaceutical Alliance office framework for pricing expectations upon generic entry. Additional costs associated with CFTR modulator use included monitoring costs, consisting of liver function tests and ophthalmologist visits, as per the product monographs, with the costs obtained from the Ontario Schedule of Benefits.27
Annual CFTR modulator monitoring costs were applied as indicated in each CFTR modulator’s associated product monograph.3,28,29 Disease management costs were also captured to include clinician visits, hospitalizations, infection prevention, and management of comorbidities. Such costs were applied in the model by disease severity (based on ppFEV1 thresholds) and were further divided into PEx and non-PEx event costs. Health care resource use associated with routine disease management costs was informed by a sponsor-commissioned burden of illness study and supplemental 2014 data from the Canadian CF Registry.1 Physician and laboratory unit costs were informed by the Ontario Schedule of Benefits; hospitalization costs were derived from a study by Skolnik et al.27,30,31 Informed by published literature suggesting patients on CFTR modulators have reduced CF-related inpatient admission and outpatient IV and antibiotic use, the sponsor adjusted disease management costs for patients on CFTR modulators.32,33 As a result, differential annual inpatient costs and annual pharmacotherapy costs were estimated for patients on BSC alone and for patients on CFTR modulators. The sponsor also excluded disease management costs for patients on CFTR modulators after the similar patient on BSC had died in a given simulation: the patients on CFTR modulators only incurred CFTR modulator therapy costs for the remainder of the time horizon. Lung transplant costs were obtained from Alberta Health Services, with follow-up costs obtained from the literature.34,35 The cost of each adverse event was assumed to be equal to the cost of a single general practitioner assessment.27 All costs were reported in 2023 Canadian dollars.
In the absence of utilities based on a generic instrument (e.g., EQ-5D), the sponsor used an equation developed by Solem et al.,36 which included ppFEV1 and PExs as predictors of an EQ-5D index utility score. For this calculation, each PEx was assumed to last 21.7 days, based on the TRAFFIC and TRANSPORT trials.36 For patients beginning treatment aged 2 to 5 years, the model assumes a utility value regardless of CFTR modulator treatment until the age of 6 years.1 The sponsor also included a treatment-specific utility increment only for patients receiving ELX-TEZ-IVA, as it was felt that the equation by Solem et al. did not capture the impact of treatment on nonrespiratory outcomes. Post–lung transplant utility values were obtained from a study by Whiting et al.15 No disutilities related to adverse events were included in the model, as they were assumed to have minimal impact on patient quality of life.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically, with 250 average patients individually simulated for 80 iterations for the base-case and scenario analyses. The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following section. The sponsor’s base case is based on publicly available list prices for comparators.
Base-Case Results
The sponsor presented results by genotype. For the F/F genotype, ELX-TEZ-IVA was associated with an incremental cost of $3,689,917 and incremental QALYs of 11.2 when compared with LUM-IVA, for an ICER of $329,703 per QALY gained. Compared to BSC, ELX-TEZ-IVA was associated with an incremental cost of $6,714,588 and incremental QALYs of 16.8, for an ICER of $399,484 per QALY gained. For the F/MF genotype, when compared to BSC, ELX-TEZ-IVA was associated with an incremental cost of $6,820,057 and incremental QALYs of 15.9, for an ICER of $429,821 per QALY gained. For the F/G genotype, ELX-TEZ-IVA was associated with an incremental cost of $6,614,327 and incremental QALYs of 17.0, for an ICER of $389,709 per QALY gained versus BSC. For the F/RF genotype, ELX-TEZ-IVA was associated with an incremental cost of $6,791,634 and incremental QALYs of 12.7, for an ICER of $534,587 per QALY gained compared to BSC.
The sponsor also combined all genotypes and presented a weighted ICER by their prevalence and by comparator market share. The overall weighted ICER was $419,571 per QALY gained (incremental costs = $6,716,950; incremental QALYs = 16.0) in comparison with the relevant standard of care.
Table 3: Summary of the Sponsor’s Economic Evaluation Results by Genotype
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
Homozygous for F508del-CFTR (F/F) | |||||
BSC | 973,938 | Reference | 25.0 | Reference | Reference |
ELX-TEZ-IVA | 7,688,526 | 6,714,588 | 41.8 | 16.8 | 399,484 |
LUM-IVA | 3,998,609 | Reference | 30.6 | Reference | Reference |
ELX-TEZ-IVA | 7,688,526 | 3,689,917 | 41.8 | 11.2 | 329,703 |
Heterozygous for F508del-CFTR (F/MF) | |||||
BSC | 788,521 | Reference | 25.3 | Reference | Reference |
ELX-TEZ-IVA | 7,608,578 | 6,820,057 | 41.2 | 15.9 | 429,821 |
Heterozygous for F508del-CFTR (F/G) | |||||
BSC | 1,080,162 | Reference | 24.8 | Reference | Reference |
ELX-TEZ-IVA | 7,694,489 | 6,614,327 | 41.8 | 17.0 | 389,709 |
Heterozygous for F508del-CFTR (F/RF) | |||||
BSC | 737,607 | Reference | 27.9 | Reference | Reference |
ELX-TEZ-IVA | 7,529,241 | 6,791,634 | 40.6 | 12.7 | 534,587 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; LUM-IVA = lumacaftor-ivacaftor; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses pertaining to different discounting values, pricing scenarios for CFTR modulators, and efficacy estimates by using the lung clearance index and adopting a societal perspective (i.e., considering caregiver utility increments and indirect costs resulting from productivity loss due to PExs). The scenario that assumed static pricing for LUM-IVA and ELX-TEZ-IVA had the largest impact, where price reductions for CFTR modulator therapies occurred at the end of patent exclusivity in the base case. This scenario analysis resulted in a weighted ICER of $697,562.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Long-term impact of treatment with ELX-TEZ-IVA or LUM-IVA on ppFEV1 rate of decline is uncertain. In addition to the acute increase in ppFEV1 from treatment with a CFTR modulator, the sponsor assumed that treatment with ELX-TEZ-IVA would result in a 192-week maintenance period of ppFEV1 following this acute increase, based on data from Study 105.1 The sponsor further assumed that treatment with a CFTR modulator would slow the long-term rate of decline in ppFEV1 following this 192-week maintenance period, compared with the rate of decline in patients on BSC. This assumption was informed by the estimated annualized rate of change in ppFEV1 among patients aged 12 years and older with an F/MF or F/F genotype treated with ELX-TEZ-IVA for up to 120 weeks in Study 105, matched via propensity score to untreated registry control patients from the (US) Cystic Fibrosis Foundation Patient Registry.37 While clinical expert feedback received by CADTH noted that there may be a long-term benefit of treatment with a CFTR modulator beyond the maintenance period, in the absence of data to support this assumption, the long-term benefit remains uncertain. This is further compounded by the uncertainty of using long-term data from patients aged 12 years and older for patients initiating treatment between the ages of 2 and 5 years for the entirety of the model time horizon after 192 weeks. The sponsor’s model was not flexible enough to change this relative rate reduction over time, meaning this assumption was applied for nearly 91 years. Overall, these issues resulted in uncertainty surrounding the benefit of ELX-TEZ-IVA on long-term ppFEV1, which may overestimate the total QALYs and may underestimate the costs associated with ELX-TEZ-IVA in the sponsor’s base case.
CADTH removed the long-term relative reduction in the rate of ppFEV1 decline for all CFTR modulators after 192 weeks in the CADTH base case. Inclusion of the long-term relative reduction in the rate of ppFEV1 decline for all CFTR modulators, as included by the sponsor, was explored in a scenario analysis.
Assumption of an additional impact of CFTR modulator therapy on PEx rates beyond its impact explained by improvements in ppFEV1 is highly uncertain. The sponsor used a relationship identified in the literature to determine the PEx rate according to ppFEV1 and age. The sponsor calibrated the first 2 years of PEx rates in the model with the values from Study 102 to determine an additional relative reduction in PExs with ELX-TEZ-IVA beyond its impact explained by changes in ppFEV1. This rate ratio was only applied to the population aged 12 years and older due to a lack of data in the subgroup of patients aged 2 to 11 years. While the clinical experts consulted by CADTH acknowledged that this assumption was plausible, it is uncertain how long this additional benefit would be observed. The sponsor assumed this additional impact on PExs would be applicable for the entire modelled time horizon, starting from when a patient reached 12 years of age, despite having data for only up to week 144 from the open-label extension study. This potentially underestimates the number of PExs patients may experience when on ELX-TEZ-IVA, overestimates the total QALYs, and underestimates the costs of ELX-TEZ-IVA.
CADTH removed the additional reduction in PExs beyond the impact explained by improvements in ppFEV1 in the CADTH base case for all CFTR modulators for the population aged 12 years and older in the period for which there were no observed data. An additional reduction in PExs beyond the impact explained by impact on ppFEV1, included as submitted by the sponsor, was explored in a scenario analysis.
Dynamic pricing for CFTR modulator therapies is uncertain and underestimates drug acquisition costs with ELX-TEZ-IVA and LUM-IVA. In the sponsor’s submitted base-case analysis, dynamic pricing was employed for CFTR modulators including ELX-TEZ-IVA and LUM-IVA. It was assumed that following the loss of patent exclusivity, generics would be introduced in the market, resulting in a 25% price reduction for the CFTR modulators after the first introduction and a 50% price reduction in subsequent years after the second generic launch. While price reductions arising from the availability of generic entrants is possible, there is uncertainty as to if and when price reductions for patent-protected therapies would occur. Patents are frequently extended (i.e., evergreened), leading to uncertainty with the exact timing of entry of a generic, and there is no guaranteed number of generic entries in the market. CADTH guidance states that full costs for ELX-TEZ-IVA at its submitted price for the entire time horizon should be accounted for. In the sponsor’s base case, dynamic pricing reduces the total drug acquisition costs associated with the CFTR modulators, biasing results in their favour.
In the CADTH base-case analysis, dynamic pricing was excluded.
Compliance-adjusted drug costs underestimate the total costs associated with ELX-TEZ-IVA and other CFTR modulators. In the submitted economic evaluation, the sponsor adjusted the price of the CFTR modulators by the assumed compliance rate (93%) in the post-acute period of the model (i.e., the period for which there was no observed data), with the assumption that savings would be incurred by the health care system based on a lack of compliance. There is limited evidence to support the real-world compliance rate, and the sponsor did not adjust treatment efficacy in the model to align accordingly. Additionally, because the drugs would be dispensed regardless of whether the patients were compliant, the public health care payer would bear the full costs of drug acquisition. This adjustment resulted in an underestimate of the drug acquisition costs associated with CFTR modulators, biasing results in favour of ELX-TEZ-IVA when compared to BSC.
CADTH’s reanalysis assumed patients were 100% compliant to ensure all drug acquisition costs were accounted for.
Exclusion of health care costs in the period over which there is a gain in survival leads to an underestimation of the costs associated with ELX-TEZ-IVA. The sponsor failed to consider costs associated with CF care for patients on CFTR modulators after a similar patient on BSC had died and only considered CFTR modulator therapy costs for the remainder of the time horizon. This assumption was made based on the sponsor asserting that accounting for the costs borne by the health care system for the additional period of survival associated with ELX-TEZ-IVA does not align with how society values treatment. This exclusion of costs incurred by the health care system does not reflect the perspective of the public health care payer. This assumption led to an underestimate of the total costs associated with ELX-TEZ-IVA.
CADTH included all costs relevant to the public health care payer in the additional survival period for patients on ELX-TEZ-IVA in the CADTH base case.
Impact of ELX-TEZ-IVA on health care resource use beyond its impact explained by improving lung function is uncertain. Health-state costs in the sponsor’s submitted model were primarily based on ppFEV1, with greater costs for patients with lower ppFEV1. The sponsor included costs associated with inpatient and outpatient hospitalizations, routine antibiotics, and diagnostics. The sponsor adjusted the disease management costs specific to inpatient hospital visits and pharmacotherapy for patients on CFTR modulators, based on studies in the literature that indicated a reduction in CF-related inpatient admissions and outpatient IV and antibiotic use.32,33 As a result, annual inpatient costs and annual pharmacotherapy costs were different for patients on BSC alone and those on CFTR modulators. Upon review of the sponsor’s sources for the reductions in costs associated with inpatient hospital visits and pharmacotherapy, CADTH noted that the studies cited by the sponsor were observational before-and-after studies assessing the impact of CFTR modulator use on relevant costs. These studies did not indicate whether they controlled for patient ppFEV1 or any other factors. As a result, it is difficult to determine whether the magnitude of difference in costs before and after CFTR modulator use observed in these studies was due to ppFEV1 (which was already factored into the sponsor’s submitted model via treatment efficacy) or another factor as asserted by the sponsor. The sponsor’s approach likely underestimated the inpatient hospital visit and outpatient antibiotic use costs associated with CFTR modulator use, biasing results in favour of ELX-TEZ-IVA.
CADTH assumed that inpatient hospital costs and annual pharmacotherapy costs were the same for all patients in the model with a similar ppFEV1, regardless of whether they were receiving a CFTR modulator.
Treatment-specific utility increment for patients on ELX-TEZ-IVA leads to potential overestimation of total benefit. The sponsor based the utility values in the submitted model on an equation by Solem et al. that determines a utility based on the EQ-5D according to a patient’s ppFEV1 and whether they experienced a PEx.36 The sponsor included an additional utility increment for patients on ELX-TEZ-IVA based on an analysis comparing the utility scores (according to the 8-dimension Cystic Fibrosis Questionnaire–Revised) of patients on ELX-TEZ-IVA versus those on placebo in Study 102, adjusting for ppFEV1. This analysis revealed a difference in utility score for patients on ELX-TEZ-IVA in comparison with patients on BSC that was not explained by ppFEV1. The sponsor’s analysis comparing Cystic Fibrosis Questionnaire–Revised scores from the trials did not account for PEx rates, which were already included in the sponsor’s utility estimate, meaning the difference observed in the sponsor’s analysis may be explained by PExs. It is difficult to know what proportion of the difference in utility scores from the trial is attributable to PExs, although PExs were accounted for in the Solem et al. equation. The sponsor should have explicitly modelled other events that it assumed contribute to quality of life, and that are not captured by Solem et al., to allow for greater transparency in what contributes to the quality of life estimates and to what extent. The inclusion of a treatment-specific utility increment for patients on ELX-TEZ-IVA potentially leads to double counting of utility gains, likely biasing results in favour of ELX-TEZ-IVA.
CADTH removed the treatment-specific utility increment with ELX-TEZ-IVA in the CADTH base-case analysis. Treatment-specific utility increments with ELX-TEZ-IVA were explored in a scenario analysis.
Survival benefit with ELX-TEZ-IVA is potentially overestimated, and the model estimate of median predicted survival does not meet face validity. The outputs of the sponsor’s model indicate that the median predicted survival for patients on ELX-TEZ-IVA is between 79.7 and 81.7 years, depending on genotype. In addition, the comparison to BSC resulted in nearly 35 undiscounted, incremental life-years gained for ELX-TEZ-IVA in some genotypes. This result is highly uncertain given the uncertainty of the long-term efficacy of ELX-TEZ-IVA and other CFTR modulators. Furthermore, the survival results for all genotypes suggested a similar survival outcome for patients with CF on ELX-TEZ-IVA compared with the general population. While the clinical experts consulted by CADTH for this review indicated that ELX-TEZ-IVA treatment would likely significantly improve a patient’s survival versus BSC, they noted that it would likely still not be equal to that of the general population. As such, the sponsor’s base-case results overestimated the survival benefit of ELX-TEZ-IVA based on the evidence currently available and did not meet face validity.
While CADTH was unable to directly modify the survival assumptions for ELX-TEZ-IVA, the base-case changes in treatment efficacy corresponded to survival results in the CADTH base case that more accurately reflected clinical expert opinion.
Model lacked transparency, and its programming prevented CADTH from fully exploring the associated uncertainties. The sponsor’s submitted model was programmed with limited transparency, with many inputs and outputs being the result of Visual Basic for Applications coding rather than formula-based operations. CADTH was unable to fully explore the uncertainty with the parameters in the model, although results of the deterministic stepwise analysis met face validity.
CADTH was unable to address this limitation in reanalysis.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Survival for patients between the ages of 2 and 5 years is 100%. | Reasonable. Clinical expert feedback received by CADTH noted that while survival for this age group may not be 100%, mortality would not be due to CF; thus, it is likely a reasonable simplifying assumption. |
An acute increase in ppFEV1 due to ELX-TEZ-IVA for patients aged 12 years and older with an F/G or F/RF genotype is reflective of the pediatric population. | Reasonable. Based on feedback received by clinical experts, the mechanism of action is expected to be identical in younger children. |
Patients aged 2 to 5 years who begin treatment with a CFTR modulator experience an acute increase in ppFEV1 immediately upon turning age 6 years. | Reasonable, according to clinical experts consulted by CADTH. |
The risk of developing CF-related diabetes is the same for patients on CFTR modulators and patients on BSC. | Reasonable, according to clinical experts consulted by CADTH. |
After an initial change, a patient’s weight-for-age z score is assumed to remain constant for the rest of the model time horizon. | Uncertain. Clinical expert feedback received by CADTH noted that while likely reasonable, the value of a weight-for-age z score increase would likely be uncertain once a younger patient transitions to adulthood. |
Rate of lung transplant for patients with ppFEV1 < 40% is 4.6%. | Reasonable. According to clinical experts consulted by CADTH, lung transplant rates have decreased in recent years. |
BSC = best supportive care; CF = cystic fibrosis; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ppFEV1 = percent predicted forced expiratory volume in the first second.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CADTH undertook a stepped analysis, incorporating each change detailed in Table 5 into the sponsor’s model to highlight the impact of each change. Each genotype is presented separately. The summary results of the CADTH reanalyses for the F/F genotype are presented in Table 6. The results for the F/MF, F/RF, and F/G genotypes are presented in Appendix 4.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Reduction in rate of ppFEV1 decline compared with BSC (after 192 weeks) | ELX-TEZ-IVA (F/F): 86.4% ELX-TEZ-IVA (other genotypes): 89.7% LUM-IVA: 42% | No reduction in rate of ppFEV1 decline |
2. Pulmonary exacerbation rate ratio with CFTR modulators compared to BSC | ELX-TEZ-IVA: 0.31 LUM-IVA: 0.46 | 1 for all CFTR modulators |
3. Dynamic pricing of CFTR modulators | 25% price reduction after 16 years for ELX-TEZ-IVA, 6 years for IVA, and 8 years for LUM-IVA 50% price reduction after 17 years for ELX-TEZ-IVA, 7 years for IVA, and 9 years for LUM-IVA | No price reduction over model time horizon |
4. Patient compliance rate in post-acute period | 93% | 100% |
5. Disease management costs during period of survival benefit while on ELX-TEZ-IVA | Not included | Included |
6. ELX-TEZ-IVA impact on inpatient and pharmacotherapy costs (beyond impact on ppFEV1) | Annual inpatient costs
Annual pharmacotherapy costs
| Annual inpatient costs
Annual pharmacotherapy costs
|
7. Treatment-specific utility increment for ELX-TEZ-IVA | Increment of 0.09 included | No utility increment |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 + 7 | |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; IVA = ivacaftor; LUM-IVA = lumacaftor-ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second.
Results from the CADTH base-case analysis showed that for the F/F genotype, ELX-TEZ-IVA was associated with incremental costs of $5,307,519 and incremental QALYs of 6.3 when compared with LUM-IVA, for an ICER of $838,687 per QALY gained. Compared to BSC, ELX-TEZ-IVA was associated with an incremental cost of $10,408,124 and incremental QALYs of 8.1, for an ICER of $1,284,953 per QALY gained. In the heterozygous F/MF genotype, ELX-TEZ-IVA was associated with incremental costs and incremental QALYs of $10,278,140 and 7.1, respectively, for an ICER of $1,451,526 per QALY gained, compared to BSC. In the heterozygous F/G genotype, ELX-TEZ-IVA was associated with $10,312,005 of incremental costs and 8.0 incremental QALYs compared to BSC, resulting in an ICER of $1,284,853 per QALY gained. Lastly, in the heterozygous F/RF genotype, $10,931,906 in incremental costs and 6.6 in incremental QALYs were observed for ELX-TEZ-IVA compared to BSC, for an ICER of $1,644,869 per QALY gained. All analyses were based on publicly available prices. Full results of the CADTH base case can be found in Table 7, with disaggregated results for all genotypes available in Appendix 4.
The sponsor’s model further produced an overall ICER, weighted for each genotype and the relative market shares of the available comparators. The weighted ICER was $1,301,071 per QALY gained. The change to the sponsor’s base case that had the greatest impact on the results was the removal of dynamic pricing due to the introduction of generic options, emphasizing the impact of drug acquisition costs as a key driver of the model.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results (F/F Genotype)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | BSC | 981,477 | 25.1 | Reference |
ELX-TEZ-IVA | 7,753,763 | 42.8 | 381,905 | |
LUM-IVA | 3,999,638 | 30.5 | Reference | |
ELX-TEZ-IVA | 7,753,763 | 42.8 | 306,571 | |
CADTH reanalysis 1: ppFEV1 decline | BSC | 981,477 | 25.1 | Reference |
ELX-TEZ-IVA | 6,752,116 | 35.6 | 546,295 | |
LUM-IVA | 3,720,571 | 27.3 | Reference | |
ELX-TEZ-IVA | 6,752,116 | 35.6 | 363,389 | |
CADTH reanalysis 2: pulmonary exacerbation rate ratio | BSC | 981,477 | 25.1 | Reference |
ELX-TEZ-IVA | 7,779,104 | 42.7 | 384,842 | |
LUM-IVA | 4,025,042 | 30.3 | Reference | |
ELX-TEZ-IVA | 7,779,104 | 42.7 | 302,161 | |
CADTH reanalysis 3: dynamic pricing | BSC | 981,477 | 25.1 | Reference |
ELX-TEZ-IVA | 12,332,427 | 42.8 | 640,106 | |
LUM-IVA | 6,170,089 | 30.5 | Reference | |
ELX-TEZ-IVA | 12,332,427 | 42.8 | 503,232 | |
CADTH reanalysis 4: compliance in the post-acute phase | BSC | 981,477 | 25.1 | Reference |
ELX-TEZ-IVA | 8,307,701 | 42.8 | 413,142 | |
LUM-IVA | 4,258,058 | 30.5 | Reference | |
ELX-TEZ-IVA | 8,307,701 | 42.8 | 330,704 | |
CADTH reanalysis 5: disease management costs during survival benefit period | BSC | 981,477 | 25.1 | Reference |
ELX-TEZ-IVA | 7,905,129 | 42.8 | 390,441 | |
LUM-IVA | 4,147,587 | 30.5 | Reference | |
ELX-TEZ-IVA | 7,905,129 | 42.8 | 306,850 | |
CADTH reanalysis 6: inpatient and pharmacotherapy costs | BSC | 981,477 | 25.1 | Reference |
ELX-TEZ-IVA | 7,888,312 | 42.8 | 389,492 | |
LUM-IVA | 4,103,321 | 30.5 | Reference | |
ELX-TEZ-IVA | 7,888,312 | 42.8 | 309,092 | |
CADTH reanalysis 7: treatment-specific utility for ELX-TEZ-IVA | BSC | 981,477 | 25.1 | Reference |
ELX-TEZ-IVA | 7,753,763 | 40.8 | 429,241 | |
LUM-IVA | 3,999,638 | 30.5 | Reference | |
ELX-TEZ-IVA | 7,753,763 | 40.8 | 364,833 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7): deterministic | BSC | 981,477 | 25.1 | Reference |
ELX-TEZ-IVA | 11,390,106 | 33.1 | 1,290,470 | |
LUM-IVA | 6,116,136 | 26.8 | Reference | |
ELX-TEZ-IVA | 11,390,106 | 33.1 | 829,744 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7): probabilistic | BSC | 973,938 | 25.0 | Reference |
ELX-TEZ-IVA | 11,382,063 | 33.1 | 1,284,953 | |
LUM-IVA | 6,074,544 | 26.7 | Reference | |
ELX-TEZ-IVA | 11,382,063 | 33.1 | 838,687 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; LUM-IVA = lumacaftor-ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second; QALY = quality-adjusted life-year.
Table 7: Summary of the CADTH Base-Case Results by Genotype
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
Homozygous for F508del-CFTR (F/F) | |||||
BSC | 973,938 | Reference | 25.0 | Reference | Reference |
ELX-TEZ-IVA | 11,382,063 | 10,408,124 | 33.1 | 8.1 | 1,284,953 |
LUM-IVA | 6,074,544 | Reference | 26.7 | Reference | Reference |
ELX-TEZ-IVA | 11,382,063 | 5,307,519 | 33.1 | 6.3 | 838,687 |
Heterozygous for F508del-CFTR (F/MF) | |||||
BSC | 788,521 | Reference | 25.3 | Reference | Reference |
ELX-TEZ-IVA | 11,066,661 | 10,278,140 | 32.4 | 7.1 | 1,451,526 |
Heterozygous for F508del-CFTR (F/G) | |||||
BSC | 1,080,162 | Reference | 24.8 | Reference | Reference |
ELX-TEZ-IVA | 11,392,167 | 10,312,005 | 32.8 | 8.0 | 1,284,853 |
Heterozygous for F508del-CFTR (F/RF) | |||||
BSC | 737,607 | Reference | 27.9 | Reference | Reference |
ELX-TEZ-IVA | 11,669,512 | 10,931,906 | 34.5 | 6.6 | 1,644,869 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; LUM-IVA = lumacaftor-ivacaftor; QALY = quality-adjusted life-year.
Scenario Analysis Results
Price reduction analyses were conducted using the sponsor and CADTH base cases, assuming proportional price reductions for ELX-TEZ-IVA (Table 8) for the summary of price reductions. Appendix 4 provides full price reduction analyses for individual genotypes (i.e., F/F, F/MF, F/G, and F/RF) and all genotypes combined (weighted by prevalence and market shares). Note for the F/F genotype comparison with LUM-IVA are also presented. Using the weighted population CADTH base-case analysis, a price reduction in excess of 94% is required for ELX-TEZ-IVA to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY in comparison with BSC for all genotypes. The price reduction required varies by genotype but is smallest for the F/F genotype and is greatest for the F/RF genotype.
Table 8: CADTH Price Reduction Analyses
Analysis | ICER for ELX-TEZ-IVA vs. BSC ($/QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
Homozygous for F508del-CFTR (F/F) | ||
No price reduction | 381,905 | 1,290,470 |
90% | 1,355 | 107,112 |
95% | Dominant | 41,370 |
99% | Dominant | Dominant |
Heterozygous for F508del-CFTR (F/MF) | ||
No price reduction | 406,979 | 1,429,803 |
90% | 11,996 | 130,410 |
95% | Dominant | 58,222 |
99% | Dominant | 471 |
Homozygous for F508del-CFTR (F/G) | ||
No price reduction | 368,537 | 1,295,357 |
90% | Dominant | 100,086 |
95% | Dominant | 33,682 |
99% | Dominant | Dominant |
Homozygous for F508del-CFTR (F/RF) | ||
No price reduction | 502,800 | 1,578,493 |
90% | 20,812 | 146,693 |
95% | Dominant | 67,148 |
99% | Dominant | 3,513 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: All price reduction analysis was conducted deterministically, unless otherwise stated.
CADTH also undertook a series of scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of ELX-TEZ-IVA. These scenarios included the following:
Long-term relative reduction of rates of ppFEV1 with ELX-TEZ-IVA (86.4% and 89.7% for the F/F and other genotypes, respectively) and LUM-IVA (42%), in comparison with BSC were included, according to the sponsor’s base-case assumption.
A reduction in the rate of PExs observed with ELX-TEZ-IVA and LUM-IVA based on observed trial data was applied in the post-maintenance phase for the entire time horizon, according to the sponsor’s base-case assumption.
Treatment-specific utility increment due to benefits beyond improvements explained in lung function and PExs for patients on ELX-TEZ-IVA were included, according to the sponsor’s base-case assumption.
The results of the CADTH scenario analyses are available in Table 9 for the F/F genotype and in Appendix 4 for the F/MF, F/RF, and F/G genotypes. These scenarios highlighted the impact of assuming additional benefit with ELX-TEZ-IVA despite a lack of supporting evidence: none of the scenarios for the F/F genotype population produced an ICER below $836,939 per QALY gained when compared to BSC. These scenario analyses are driven by the high drug acquisition costs with ELX-TEZ-IVA, which offset the estimated QALY gains.
Table 9: CADTH Scenario Analysis Summary — F/F Genotype
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
1. Slower rate of decline in ppFEV1 | BSC | 973,938 | 25.0 | Reference |
ELX-TEZ-IVA | 13,472,245 | 40.0 | 832,726 | |
LUM-IVA | 6,899,343 | 30.2 | Reference | |
ELX-TEZ-IVA | 13,472,245 | 40.0 | 676,763 | |
2. Long-term reduction in pulmonary exacerbations included for CFTR modulators | BSC | 973,938 | 25.0 | Reference |
ELX-TEZ-IVA | 11,425,272 | 33.7 | 1,198,260 | |
LUM-IVA | 6,067,537 | 27.3 | Reference | |
ELX-TEZ-IVA | 11,425,272 | 33.7 | 836,939 | |
3. Inclusion of treatment-specific utility increment for patients on ELX-TEZ-IVA | BSC | 973,938 | 25.0 | Reference |
ELX-TEZ-IVA | 11,382,063 | 34.6 | 1,078,743 | |
LUM-IVA | 6,074,544 | 26.7 | Reference | |
ELX-TEZ-IVA | 11,382,063 | 34.6 | 673,822 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; LUM-IVA = lumacaftor-ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second; QALY = quality-adjusted life-year.
Issues for Consideration
The sponsor-submitted economic evaluation assessed the cost-effectiveness of ELX-TEZ-IVA compared to BSC or LUM-IVA (for the F/F genotype only) over the entire lifetime of a patient aged 2 to 5 years. However, this decision problem does not accurately reflect the current landscape of CF treatment, as the majority of patients aged 6 years and older are currently being treated with ELX-TEZ-IVA. The cost-effectiveness of starting ELX-TEZ-IVA for patients aged 2 to 5 years versus waiting to initiate treatment at age 6 years and older is unknown.
CADTH previously reviewed ELX-TEZ-IVA for the treatment of CF in patients aged 6 years and older who have at least 1 F508del-CFTR mutation.38 The submitted price in that review was the same, with a daily cost of $840, or $280 per tablet. The committee recommended reimbursement of ELX-TEZ-IVA with conditions, including a price reduction of 90%. Results from the current review of ELX-TEZ-IVA were similar, indicating that this drug is not cost-effective at the submitted price and would require a significant price reduction.
Overall Conclusions
Results from the 24-week, open-label, uncontrolled Study 111 Part B suggest that treatment with ELX-TEZ-IVA resulted in improvements from baseline in lung function (decrease in lung clearance index 2.5 from baseline) and CF biomarkers (reduction in sweat chloride) in patients aged 2 to 5 years with CF. However, as Study 111 was primarily designed to evaluate the safety, tolerability, and pharmacokinetics of ELX-TEZ-IVA in this younger population, the pharmacoeconomic review is based on the extrapolation of efficacy data from studies conducted in older patients with CF. Patients who completed Study 111 were eligible to enrol in an open-label extension study; however, interim results were not available at the time of filing the application with CADTH.
As noted in CADTH’s previous review of ELX-TEZ-IVA for patients with CF aged 6 to 11 years,25 the clinical evidence submitted by the sponsor demonstrated that ELX-TEZ-IVA was associated with statistically and clinically significant improvements in acute ppFEV1 and weight-for-age z scores compared with relevant comparators in patients aged 6 to 11 years with F/F or F/MF genotypes. Although no clinical studies were conducted with ELX-TEZ-IVA in pediatric patients with F/RF or F/G genotypes, the clinical expert feedback received during the review noted that ELX-TEZ-IVA would result in clinically meaningful improvements for these patients, based on the evidence reported for ELX-TEZ-IVA in adolescent and adult patients with the F/RF and F/G genotypes and the results of studies of pediatric patients with the F/F and F/MF genotypes. Similar results were reported for patients aged 12 years and older regarding the acute change in ppFEV1 in all genotypes.39
The clinical experts consulted by CADTH for the current review noted that, given the mechanism of action and compelling efficacy data in patients aged 6 years and older, ELX-TEZ-IVA is expected to benefit patients aged 2 to 5 years who have at least 1 508del mutation in the CFTR gene. However, these conclusions are based on studies with a maximum follow-up of 192 weeks, and there remains no evidence on the long-term impact of ELX-TEZ-IVA on the rate of decline in ppFEV1 and PExs rates beyond the trial period for any genotype or age group.
Beyond uncertainty in the long term clinical efficacy of ELX-TEZ-IVA, CADTH identified several major limitations with the submitted economic evaluation. The following were addressed in the CADTH reanalysis: the removal of an additional benefit of CFTR modulators on the long-term rate of decline in ppFEV1 and PExs; the removal of dynamic pricing for CFTR modulators; the inclusion of costs for ELX-TEZ-IVA for the period in which a survival benefit was achieved in comparison to BSC; the removal of an adjustment to drug acquisition costs based on patient compliance; and the removal of a treatment-specific utility increment for patients on ELX-TEZ-IVA. The results of the CADTH reanalysis were aligned with the sponsor’s, in that ELX-TEZ-IVA was not cost-effective in any of the genotype subgroups at conventionally acceptable ICER thresholds. In the CADTH base-case analyses, when compared to BSC, ELX-TEZ-IVA was associated with an ICER of $1,284,953 per QALY gained in the F/F genotype, $1,451,526 per QALY gained in the F/MF genotype, $1,284,853 per QALY gained in the F/G genotype, and $1,644,869 per QALY gained in the F/RF genotype. Additionally, ELX-TEZ-IVA was associated with an ICER of $838,687 per QALY gained when compared to LUM-IVA in the F/F genotype population.
The key drivers in the analyses were drug acquisition costs and assumptions in the long-term benefits of ELX-TEZ-IVA, which were uncertain. Treatment with ELX-TEZ-IVA was not cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained in any scenario conducted by CADTH. A price reduction in excess of 94% for ELX-TEZ-IVA (for both granules and tablets) is required for all 4 genotypes for ELX-TEZ-IVA to be considered cost-effective at this threshold in comparison with BSC. Based on a price reduction of this magnitude, the daily cost of ELX-TEZ-IVA would be approximately $50.40 per patient. As the majority of patients with CF aged 6 years and older are currently being treated with ELX-TEZ-IVA, the sponsor’s submitted economic evaluation assessing the cost-effectiveness of ELX-TEZ-IVA compared to BSC or LUM-IVA (for the F/F genotype only) over the entire lifetime of a patient aged 2 to 5 years does not accurately reflect the current landscape of CF treatment. The cost-effectiveness of starting ELX-TEZ-IVA for patients aged 2 to 5 years versus waiting to initiate treatment at age 6 years or older is unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Epkinly (epcoritamab), 4 mg in 0.8 mL [5mg/mL] and 48 mg in 0.8 mL [60 mg/mL], for injection. Pointe-Claire (QC): AbbVie Corporation; 2023 Nov 14.
2.pan-Canadian Oncology Drug Review Committee (pERC) final recommendation: Elexacaftor-TezacaftorIvacaftor and Ivacaftor (Trikafta). Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/DRR/2021/SR0673%20Trikafta%20-%20CADTH%20Final%20Rec%20Revised.pdf. Accessed 2023 Jun 28.
3.Clinical Study Report: GCT3013-01, update #1. A phase 1/2, open-label, dose-escalation trial of GEN3013 in patients with relapsed, progressive or refractory b-cell lymphoma [internal sponsor's report]. Plainsboro (NJ): Genmab US, Inc.; 2023 Oct 03.
4.Stephenson AL, Tom M, Berthiaume Y, et al. A contemporary survival analysis of individuals with cystic fibrosis: a cohort study. Eur Respir J. 2015;45(3):670-679. PubMed
5.Liou TG, Adler FR, Fitzsimmons SC, Cahill BC, Hibbs JR, Marshall BC. Predictive 5-year survivorship model of cystic fibrosis. Am J Epidemiol. 2001;153(4):345-352. PubMed
6.Walker S, Flume P, McNamara J, et al. A phase 3 study of tezacaftor in combination with ivacaftor in children aged 6 through 11 years with cystic fibrosis. J Cyst Fibros. 2019;18(5):708-713. PubMed
7.Milla CE, Ratjen F, Marigowda G, Liu F, Waltz D, Rosenfeld M. Lumacaftor/Ivacaftor in Patients Aged 6-11 Years with Cystic Fibrosis and Homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017;195(7):912-920. PubMed
8.Davies JC, Sermet-Gaudelus I, Naehrlich L, et al. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in participants 6 through 11 years of age with cystic fibrosis homozygous for F508del or heterozygous for the F508del-CFTR mutation and a residual function mutation. J Cyst Fibros. 2021;20(1):68-77. PubMed
9.Zemanick ET, Taylor-Cousar JL, Davies J, et al. A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am J Respir Crit Care Med. 2021;203(12):1522-1532. PubMed
10.Ratjen F, Hug C, Marigowda G, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017;5(7):557-567. PubMed
11.Barry PJ, Mall MA, Álvarez A, et al. Triple Therapy for Cystic Fibrosis Phe508del-Gating and -Residual Function Genotypes. N Engl J Med. 2021;385(9):815-825. PubMed
12.Moss RB, Flume PA, Elborn JS, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3(7):524-533. PubMed
13.De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674-680. PubMed
14.Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187(11):1219-1225. PubMed
15.Whiting P, Al M, Burgers L, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technol Assess. 2014;18(18):1-106. PubMed
16.Adler AI, Shine BS, Chamnan P, Haworth CS, Bilton D. Genetic determinants and epidemiology of cystic fibrosis-related diabetes: results from a British cohort of children and adults. Diabetes Care. 2008;31(9):1789-1794. PubMed
17.Indirect Treatment Comparison for Epcoritamab in Patients with Relapsed or Refractory Large B-cell Lymphoma based on EPCORE NHL-1 trial [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Epkinly (epcoritamab), 4 mg in 0.8 mL [5mg/mL] and 48 mg in 0.8 mL [60 mg/mL], for injection. New York (NY): OPEN Health; 2023 Mar 10.
18.Mall MA, Brugha R, Gartner S, et al. Efficacy and Safety of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 Through 11 Years of Age with Cystic Fibrosis Heterozygous for F508del and a Minimal Function Mutation: A Phase 3b, Randomized, Placebo-controlled Study. Am J Respir Crit Care Med. 2022;206(11):1361-1369. PubMed
19.Leung GJ, Cho TJ, Kovesi T, Hamid JS, Radhakrishnan D. Variation in lung function and nutritional decline in cystic fibrosis by genotype: An analysis of the Canadian cystic fibrosis registry. J Cyst Fibros. 2020;19(2):255-261. PubMed
20.Loukou I, Moustaki M, Plyta M, Douros K. Long-term clinical outcome of cystic fibrosis paediatric patients presenting with meconium ileus. Acta Paediatr. 2020;109(12):2738-2739. PubMed
21.Ejiofor LCK, Mathiesen IHM, Jensen-Fangel S, et al. Patients with cystic fibrosis and advanced lung disease benefit from lumacaftor/ivacaftor treatment. Pediatr Pulmonol. 2020;55(12):3364-3370. PubMed
22.Muilwijk D, Zomer DD, Gulmans V, Ent CK. P055 Real-world trends in long-term clinical outcomes of lumacaftor/ivacaftor. Journal of Cystic Fibrosis. 2020;19:S70-S71.
23.Goss CH, Burns JL. Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax. 2007;62(4):360-367. PubMed
24.McNamara JJ, McColley SA, Marigowda G, et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2-5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study. Lancet Respir Med. 2019;7(4):325-335. PubMed
25.Drug Reimbursement Review pharmacoeconomic report: Elexacaftor-TezacaftorIvacaftor and Ivacaftor (Trikafta) for Cystic fibrosis, F508del-CFTR mutation, 6 years and older. Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/DRR/2022/SR0710-Trikafta_combined.pdf. Accessed 2023 Jul 10.
26.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 Jun 24.
27.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf.
28.Kalydeco: Ivacaftor tablets 150 mg, Oral Ivacaftor granules 25 mg per packet, 50 mg per packet, 75 mg per packet, Oral [product monograph]. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2022.
29.Orkambi: Lumacaftor / Ivacaftor Tablets 100 mg / 125 mg, Oral, Lumacaftor / Ivacaftor Tablets 200 mg / 125 mg, Oral Lumacaftor / Ivacaftor Granules 75 mg / 94 mg, Oral, Lumacaftor / Ivacaftor Granules 100 mg / 125 mg, Oral Lumacaftor / Ivacaftor Granules 150 mg / 188 mg, Oral [product monograph]. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2023.
30.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 1800 Jan 1.
31.Skolnik K, Ronksley P, Pendharkar S, Wick J, Quon BS, Williamson T. Hospitalization-related costs for canadian cystic fibrosis patients [conference abstract]. Pediatr Pulmonol. 2018;53.
32.Feng LB, Grosse SD, Green RF, Fink AK, Sawicki GS. Precision Medicine In Action: The Impact Of Ivacaftor On Cystic Fibrosis-Related Hospitalizations. Health Aff (Millwood). 2018;37(5):773-779. PubMed
33.Hassan M, Bonafede MM, Limone BL, Hodgkins P, Suthoff ED, Sawicki G. Reduction in pulmonary exacerbations (PEx) after initiation of ivacaftor: a retrospective cohort study among patients with cystic fibrosis (CF) treated in real-world settings [conference abstract]. Journal of Cystic Fibrosis. 2016;15:S58.
34.Vasiliadis HM, Collet JP, Penrod JR, Ferraro P, Poirier C. A cost-effectiveness and cost-utility study of lung transplantation. J Heart Lung Transplant. 2005;24(9):1275-1283. PubMed
35.Government of Alberta. Alberta Interactive Health Data Application. Average cost of lung transplant procedure in 2017/2018. 2019; http://www.ahw.gov.ab.ca/IHDA_Retrieval/redirectToURL.do?cat=201&subCat=486. Accessed 2023 Jun 8.
36.Solem CT, Vera-Llonch M, Tai M, O’Callaghan L. Pulmonary Exacerbations, Lung Dysfunction, And Eq-5d Measures In Adolescents And Adults With Cystic Fibrosis And Homozygous For The F508del-Cftr Mutation. Value Health. 2016;19(3):A116-A117.
37.Lee T, Sawicki GS, Altenburg J, et al. Effect of elexacaftor/tezacaftor/ivacaftor on annual rate of lung function decline in people with cystic fibrosis. J Cyst Fibros. 2023;22(3):402-406. PubMed
38.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: Elexacaftor-TezacaftorIvacaftor and Ivacaftor (Trikafta): Treatment of cystic fibrosis in patients aged 6 years and older who have at least 1 F508del mutation in the cystic fibrosis transmembrane conductance regulator gene. Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/DRR/2022/SR0710%20Trikafta%20-%20CADTH%20Final%20Rec-meta.pdf. Accessed 2023 Jun 20.
39.Drug Reimbursement Review pharmacoeconomic report: Elexacaftor-TezacaftorIvacaftor and Ivacaftor (Trikafta) for Cystic fibrosis, F508del CFTR mutation. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/DRR/2021/SR0673-combined-report.pdf. Accessed 2023 Jul 10.
40.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Epkinly (epcoritamab), 4 mg in 0.8 mL [5mg/mL] and 48 mg in 0.8 mL [60 mg/mL], for injection. Pointe-Claire (QC): AbbVie Corporation; 2023 Nov 14.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 10: CADTH Cost Comparison Table for CFTR Modulator Therapies for Cystic Fibrosis for Patients Aged 2 to 5 Years
Treatment | Strength / concentration | Form | Price ($) | Recommended dosagea | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Elexacaftor- tezacaftor- ivacaftor and ivacaftor (Trikafta) | 100 mg / 50 mg/ 75 mg and 75 mg 80 mg/ 40 mg/ 60 mg and 59.5 mg | Granules Packet | 420.0000b | One granule packet (containing elexacaftor 100 mg - tezacaftor 50 mg - ivacaftor 75 mg or elexacaftor 80 mg - tezacaftor 40 mg - ivacaftor 60 mg) taken in the morning and 1 granule packet (ivacaftor 75 mg or ivacaftor 59.5 mg) taken in the evening approximately 12 hours apart, with fat-containing food | 840.00 | 306,810 |
100 mg/ 50 mg/ 75 mg and 150 mg 50 mg/ 25 mg/ 37.5 and 75 mg | Tablets | Two tablets (each containing elexacaftor 100 mg - tezacaftor 50 mg - ivacaftor 75 mg or elexacaftor 50 mg - tezacaftor 25 mg - ivacaftor 37.5 mg) taken in the morning and 1 tablet (ivacaftor 150 mg or ivacaftor 75 mg) taken in the evening approximately 12 hours apart, with fat-containing food | ||||
CFTR modulator therapies | ||||||
Lumacaftor-ivacaftor (Orkambi) | 100 mg/125 mg | Granules | 170.5357 | One packet twice daily | 682.14 | 249,152 |
Note: All prices are from the Ontario Exceptional Access Program formulary (accessed June 2023)26 unless otherwise indicated, and do not include dispensing fees. Annual costs are based on 365.25 days per year.
aRecommended dosages are from the respective product monographs.3,29
bSponsor-submitted price.1
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | No | Model lacks transparency with regards to programming |
Model structure is adequate for decision problem | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
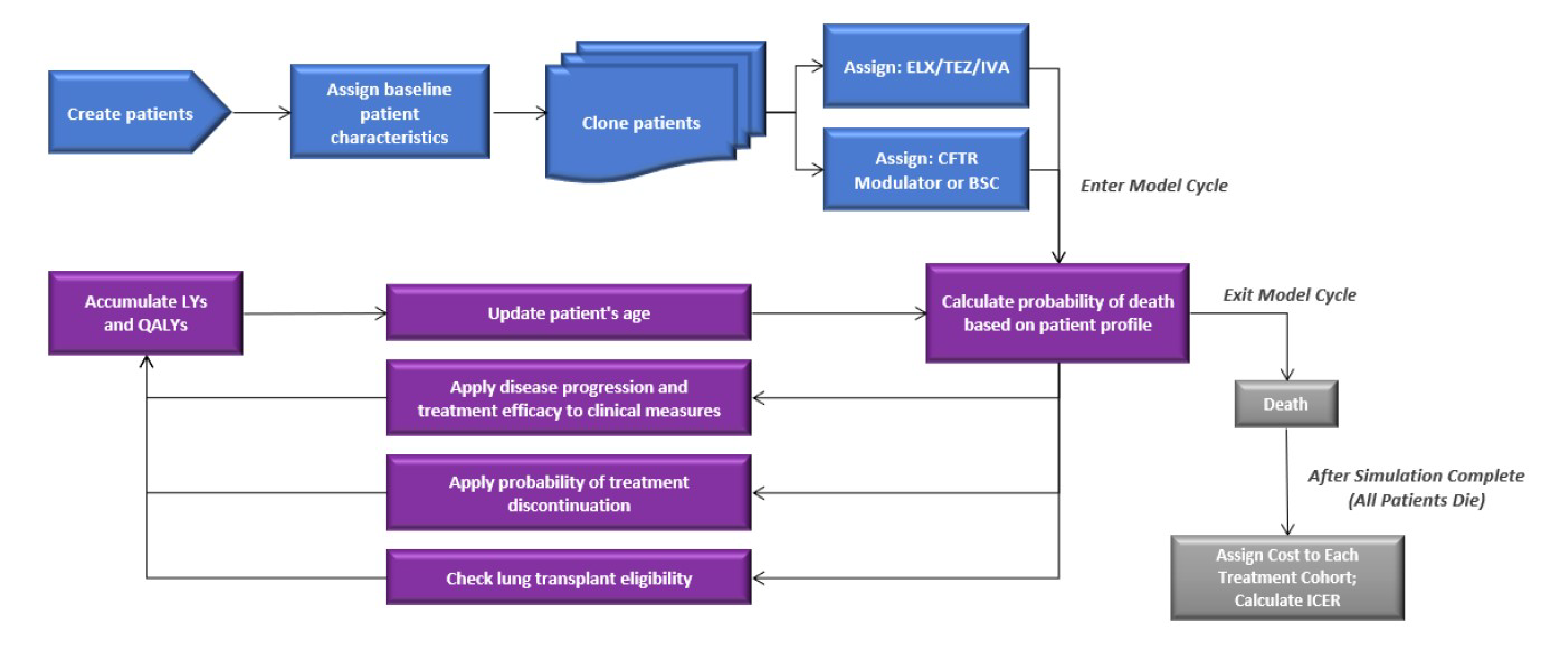
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 12: Disaggregated Summary of CADTH’s Economic Evaluation Results (Deterministic) — F/F Genotype
Parameter | ELX-TEZ-IVA | LUM-IVA | Incremental | ELX-TEZ-IVA | BSC | Incremental |
|---|---|---|---|---|---|---|
Discounted LYs | ||||||
Total | 34.6 | 28.8 | 5.7 | 34.6 | 26.9 | 7.6 |
Discounted QALYs | ||||||
Total | 34.2 | 28.2 | 6.0 | 34.2 | 26.2 | 8.0 |
Discounted costs ($) | ||||||
Total | 11,203,814 | 6,061,356 | 5,142,458 | 11,203,814 | 916,157 | 10,287,657 |
Drug acquisition cost | 10,437,275 | 5,156,352 | 5,280,923 | 10,437,275 | 0 | 10,437,275 |
Non–PEx-related disease management costs | 504,690 | 485,342 | 19,347 | 504,690 | 492,515 | 12,175 |
PEx-related costs | 258,165 | 417,205 | –159,040 | 258,165 | 421,210 | –163,046 |
Lung transplant costs | 0 | 0 | 0 | 0 | 0 | 0 |
Adverse event cost | 3,527 | 2,277 | 1,250 | 3,527 | 2,432 | 1,095 |
Monitoring cost | 157 | 180 | –23 | 157 | 0 | 157 |
Indirect costs | 0 | 0 | 0 | 0 | 0 | 0 |
ICER ($/QALY) | 850,053 | 1,283,744 | ||||
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; F/F = homozygous for F508del mutation in the CFTR gene; LY = life-year; PEx = pulmonary exacerbations; QALY = quality-adjusted life-year.
Table 13: Disaggregated Summary of CADTH’s Economic Evaluation Results (Deterministic) — F/MF Genotype
Parameter | ELX-TEZ-IVA | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 34.9 | 27.2 | 7.7 |
Discounted QALYs | |||
Total | 34.7 | 26.7 | 7.9 |
Discounted costs ($) | |||
Total | 11,129,360 | 742,088 | 10,387,273 |
Drug acquisition cost | 10,532,547 | 0 | 10,532,547 |
Non–PEx-related disease management costs | 495,432 | 442,792 | 52,640 |
PEx-related costs | 96,961 | 296,833 | –199,872 |
Lung transplant costs | 0 | 0 | 0 |
Adverse event cost | 4,263 | 2,463 | 1,800 |
Monitoring cost | 158 | 0 | 158 |
Indirect costs | 0 | 0 | 0 |
ICER ($/QALY) | 1,311,755 | ||
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; LY = life-year; PEx = pulmonary exacerbations; QALY = quality-adjusted life-year.
Table 14: Disaggregated Summary of CADTH’s Economic Evaluation Results (Deterministic) — F/G Genotype
Parameter | ELX-TEZ-IVA | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 35.2 | 27.1 | 8.1 |
Discounted QALYs | |||
Total | 34.9 | 26.2 | 8.6 |
Discounted costs ($) | |||
Total | 11,395,429 | 1,008,352 | 10,387,077 |
Drug acquisition cost | 10,641,712 | 0 | 10,641,712 |
Non–PEx-related disease management costs | 521,095 | 511,913 | 9,182 |
PEx-related costs | 228,337 | 493,520 | –265,182 |
Lung transplant costs | 0 | 470 | –470 |
Adverse event cost | 4,126 | 2,450 | 1,676 |
Monitoring cost | 159 | 0 | 159 |
Indirect costs | 0 | 0 | 0 |
ICER ($/QALY) | 1,204,386 | ||
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; F/G = 1 F508del mutation and 1 gating mutation in the CFTR gene; LY = life-year; PEx = pulmonary exacerbations; QALY = quality-adjusted life-year.
Table 15: Disaggregated Summary of CADTH’s Economic Evaluation Results (Deterministic) — F/RF Genotype
Parameter | ELX-TEZ-IVA | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 36.9 | 29.6 | 7.3 |
Discounted QALYs | |||
Total | 36.8 | 29.2 | 7.6 |
Discounted costs ($) | |||
Total | 11,718,395 | 747,296 | 10,971,100 |
Drug acquisition cost | 11,143,534 | 0 | 11,143,534 |
Non–PEx-related disease management costs | 496,796 | 466,103 | 30,692 |
PEx-related costs | 73,585 | 278,350 | –204,764 |
Lung transplant costs | 0 | 0 | 0 |
Adverse event cost | 4,318 | 2,842 | 1,476 |
Monitoring cost | 161 | 0 | 161 |
Indirect costs | 0 | 0 | 0 |
ICER ($/QALY) | 1,437,829 | ||
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; F/RF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; LY = life-year; PEx = pulmonary exacerbations; QALY = quality-adjusted life-year.
Table 16: Summary of the Stepped Analysis of the CADTH Reanalysis Results (Deterministic) — F/MF Genotype
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | BSC | 793,354 | 25.3 | Reference |
ELX-TEZ-IVA | 7,663,343 | 42.2 | 406,979 | |
CADTH reanalysis 1 – ppFEV1 decline | BSC | 793,354 | 25.3 | Reference |
ELX-TEZ-IVA | 6,639,136 | 34.9 | 608,529 | |
CADTH reanalysis 2 – pulmonary exacerbation rate ratio | BSC | 793,354 | 25.3 | Reference |
ELX-TEZ-IVA | 7,663,082 | 42.0 | 412,396 | |
CADTH reanalysis 3 – dynamic pricing | BSC | 793,354 | 25.3 | Reference |
ELX-TEZ-IVA | 12,156,915 | 42.2 | 673,180 | |
CADTH reanalysis 4 – compliance in the post-acute phase | BSC | 793,354 | 25.3 | Reference |
ELX-TEZ-IVA | 8,210,525 | 42.2 | 439,395 | |
CADTH reanalysis 5 – disease management costs during survival benefit period | BSC | 793,354 | 25.3 | Reference |
ELX-TEZ-IVA | 7,806,674 | 42.2 | 415,470 | |
CADTH reanalysis 6 – inpatient and pharmacotherapy costs | BSC | 793,354 | 25.3 | Reference |
ELX-TEZ-IVA | 7,798,275 | 42.2 | 414,973 | |
CADTH reanalysis 7 – treatment-specific utility for ELX-TEZ-IVA | BSC | 793,354 | 25.3 | Reference |
ELX-TEZ-IVA | 7,663,343 | 40.3 | 458,604 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7) (deterministic) | BSC | 793,354 | 25.3 | Reference |
ELX-TEZ-IVA | 11,058,871 | 32.5 | 1,429,803 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7) (probabilistic) | BSC | 788,521 | 25.3 | Reference |
ELX-TEZ-IVA | 11,066,661 | 32.4 | 1,451,526 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; F/MF = 1 F508del mutation and 1 minimal function mutation in the CFTR gene; LY = life-year; QALY = quality-adjusted life-year; ppFEV1 = percent predicted forced expiratory volume in the first second.
Table 17: Summary of the Stepped Analysis of the CADTH Reanalysis Results (Deterministic) — F/G Genotype
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | BSC | 1,089,164 | 24.8 | Reference |
ELX-TEZ-IVA | 7,773,682 | 42.9 | 368,537 | |
CADTH reanalysis 1 – ppFEV1 decline | BSC | 1,089,164 | 24.8 | Reference |
ELX-TEZ-IVA | 6,741,011 | 35.5 | 529,192 | |
CADTH reanalysis 2 – pulmonary exacerbation rate ratio | BSC | 1,089,164 | 24.8 | Reference |
ELX-TEZ-IVA | 7,761,414 | 42.6 | 375,063 | |
CADTH reanalysis 3 – dynamic pricing | BSC | 1,089,164 | 24.8 | Reference |
ELX-TEZ-IVA | 12,369,724 | 42.9 | 621,931 | |
CADTH reanalysis 4 – compliance in the post-acute phase | BSC | 1,089,164 | 24.8 | Reference |
ELX-TEZ-IVA | 8,328,927 | 42.9 | 399,150 | |
CADTH reanalysis 5 – disease management costs during survival benefit period | BSC | 1,089,164 | 24.8 | Reference |
ELX-TEZ-IVA | 7,929,684 | 42.9 | 377,138 | |
CADTH reanalysis 6 – inpatient and pharmacotherapy costs | BSC | 1,089,164 | 24.8 | Reference |
ELX-TEZ-IVA | 7,907,190 | 42.9 | 375,898 | |
CADTH reanalysis 7 – treatment-specific utility for ELX-TEZ-IVA | BSC | 1,089,164 | 24.8 | Reference |
ELX-TEZ-IVA | 7,773,682 | 40.9 | 413,940 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7) (deterministic) | BSC | 1,089,164 | 24.8 | Reference |
ELX-TEZ-IVA | 11,313,664 | 32.7 | 1,295,357 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7) (probabilistic) | BSC | 1,080,162 | 24.8 | Reference |
ELX-TEZ-IVA | 11,392,167 | 32.8 | 1,284,853 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; F/MF = 1 F508del mutation and 1 gating mutation in the CFTR gene; LY = life-year; QALY = quality-adjusted life-year; ppFEV1 = percent predicted forced expiratory volume in the first second.
Table 18: Summary of the Stepped Analysis of the CADTH Reanalysis Results (Deterministic) — F/RF Genotype
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | BSC | 725,405 | 27.9 | Reference |
ELX-TEZ-IVA | 7,574,624 | 41.5 | 502,800 | |
CADTH reanalysis 1 – ppFEV1 decline | BSC | 725,405 | 27.9 | Reference |
ELX-TEZ-IVA | 6,918,294 | 36.9 | 689,212 | |
CADTH reanalysis 2 – pulmonary exacerbation rate ratio | BSC | 725,405 | 27.9 | Reference |
ELX-TEZ-IVA | 7,596,250 | 41.4 | 508,813 | |
CADTH reanalysis 3 – dynamic pricing | BSC | 725,405 | 27.9 | Reference |
ELX-TEZ-IVA | 11,966,983 | 41.5 | 825,242 | |
CADTH reanalysis 4 – compliance in the post-acute phase | BSC | 725,405 | 27.9 | Reference |
ELX-TEZ-IVA | 8,113,294 | 41.5 | 542,344 | |
CADTH reanalysis 5 – disease management costs during survival benefit period | BSC | 725,405 | 27.9 | Reference |
ELX-TEZ-IVA | 7,688,854 | 41.5 | 511,186 | |
CADTH reanalysis 6 – inpatient and pharmacotherapy costs | BSC | 725,405 | 27.9 | Reference |
ELX-TEZ-IVA | 7,723,595. | 41.5 | 513,736 | |
CADTH reanalysis 7 – treatment-specific utility for ELX-TEZ-IVA | BSC | 725,405 | 27.9 | Reference |
ELX-TEZ-IVA | 7,574,624 | 39.7 | 582,928 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7) (deterministic) | BSC | 725,405 | 27.9 | Reference |
ELX-TEZ-IVA | 11,726,690 | 34.9 | 1,578,493 | |
CADTH base case (1 + 2 + 3 + 4 + 5 + 6 + 7) (probabilistic) | BSC | 737,607 | 27.9 | Reference |
ELX-TEZ-IVA | 11,669,512 | 34.5 | 1,644,869 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; F/MF = 1 F508del mutation and 1 residual function mutation in the CFTR gene; LY = life-year; QALY = quality-adjusted life-year; ppFEV1 = percent predicted forced expiratory volume in the first second.
Scenario Analyses
Table 19: CADTH Price Reduction Analysis — F/F Genotype
Analysis | ICER ($/QALY) for ELX-TEZ-IVA vs. LUM-IVA | ICER ($/QALY) for ELX-TEZ-IVA vs. BSC | ||
|---|---|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis | Sponsor base case | CADTH reanalysis |
No Price Reduction | 306,571 | 829,744 | 381,905 | 1,290,470 |
10% | 245,340 | 662,894 | 339,621 | 1,158,986 |
20% | 184,109 | 496,044 | 297,338 | 1,027,502 |
40% | 61,647 | 162,345 | 212,771 | 764,533 |
60% | Dominant | Dominant | 128,205 | 501,565 |
70% | Dominant | Dominant | 85,921 | 370,080 |
80% | Dominant | Dominant | 43,638 | 238,596 |
90% | Dominant | Dominant | 1,355 | 107,112 |
95% | Dominant | Dominant | Dominant | 41,370 |
99% | Dominant | Dominant | Dominant | Dominant |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; LUM-IVA = lumacaftor-ivacaftor; QALY = quality-adjusted life-year; vs. = versus.
Table 20: CADTH Price Reduction Analysis — F/MF Genotype
Analysis | ICER ($/QALY) for ELX-TEZ-IVA vs. BSC | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No Price Reduction | 406,979 | 1,429,803 |
10% | 363,092 | 1,285,426 |
20% | 275,318 | 996,672 |
40% | 187,544 | 707,918 |
60% | 143,657 | 563,541 |
70% | 99,770 | 419,164 |
80% | 55,883 | 274,787 |
90% | 11,996 | 130,410 |
95% | Dominant | 58,222 |
99% | Dominant | 471 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/MF = heterozygous for F508del and 1 minimal function mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Table 21: CADTH Price Reduction Analysis — F/G Genotype
Analysis | ICER ($/QALY) for ELX-TEZ-IVA vs. BSC | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No Price Reduction | 368,537 | 1,295,357 |
10% | 327,103 | 1,162,549 |
20% | 244,233 | 896,933 |
40% | 161,363 | 631,317 |
60% | 119,928 | 498,510 |
70% | 78,493 | 365,702 |
80% | 37,058 | 232,894 |
90% | Dominant | 100,086 |
95% | Dominant | 33,682 |
99% | Dominant | Dominant |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/G = heterozygous for F508del and 1 gating mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Table 22: CADTH Price Reduction Analysis — F/RF Genotype
Analysis | ICER ($/QALY) for ELX-TEZ-IVA vs. BSC | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No Price Reduction | 502,800 | 1,578,493 |
10% | 449,246 | 1,419,404 |
20% | 342,137 | 1,101,226 |
40% | 235,029 | 783,048 |
60% | 181,475 | 623,959 |
70% | 127,920 | 464,870 |
80% | 74,366 | 305,782 |
90% | 20,812 | 146,693 |
95% | Dominant | 67,148 |
99% | Dominant | 3,513 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/RF = heterozygous for F508del and 1 residual function mutation in the CFTR gene; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Table 23: CADTH Price Reduction Analysis — Weighted Analysis, All Genotypes Combined
Analysis | ICER ($/QALY) for ELX-TEZ-IVA vs. standard of care | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 399,060 | 1,355,183 |
10% | 355,170 | 1,216,867 |
20% | 267,389 | 940,234 |
40% | 179,609 | 663,601 |
60% | 135,719 | 525,285 |
70% | 91,829 | 386,969 |
80% | 47,938 | 248,652 |
90% | 4,048 | 110,336 |
95% | Dominant | 41,178 |
99% | Dominant | Dominant |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Table 24: CADTH Scenario Analysis Summary — F/MF Genotype
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
1. Slower rate of decline in ppFEV1 | BSC | 788,521 | 25.3 | Reference |
ELX-TEZ-IVA | 13,285,699 | 39.4 | 886,656 | |
2. Long-term reduction in pulmonary exacerbations included for CFTR modulators | BSC | 788,521 | 25.3 | Reference |
ELX-TEZ-IVA | 11,121,006 | 32.9 | 1,355,482 | |
3. Inclusion of treatment-specific utility increment for patients on ELX-TEZ-IVA | BSC | 788,521 | 25.3 | Reference |
ELX-TEZ-IVA | 11,066,661 | 33.8 | 1,202,824 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Table 25: CADTH Scenario Analysis Summary — F/G Genotype
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
1. Slower rate of decline in ppFEV1 | BSC | 1,080,162 | 24.8 | Reference |
ELX-TEZ-IVA | 13,478,005 | 39.9 | 821,473 | |
2. Long-term reduction in pulmonary exacerbations included for CFTR modulators | BSC | 1,080,162 | 24.8 | Reference |
ELX-TEZ-IVA | 11,417,054 | 33.5 | 1,190,243 | |
3. Inclusion of treatment-specific utility increment for patients on ELX-TEZ-IVA | BSC | 1,080,162 | 24.8 | Reference |
ELX-TEZ-IVA | 11,392,167 | 34.4 | 1,071,801 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Table 26: CADTH Scenario Analysis Summary — F/RF Genotype
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
1. Slower rate of decline in ppFEV1 | BSC | 737,607 | 27.9 | Reference |
ELX-TEZ-IVA | 13,082,606 | 38.9 | 1,121,384 | |
2. Long-term reduction in pulmonary exacerbations included for CFTR modulators | BSC | 737,607 | 27.9 | Reference |
ELX-TEZ-IVA | 11,665,696 | 34.8 | 1,580,648 | |
3. Inclusion of treatment-specific utility increment for patients on ELX-TEZ-IVA | BSC | 737,607 | 27.9 | Reference |
ELX-TEZ-IVA | 11,669,512 | 36.0 | 1,338,537 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 27: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted an epidemiology-based budget impact analysis (BIA), assessing the expected budgetary impact of reimbursing ELX-TEZ-IVA plus BSC for the treatment of CF patients who are 2 to 5 years of age with at least 1 F508del-CFTR mutation. The analysis was conducted from the perspective of the Canadian public drug plans over a 3-year time horizon (from 2024 to 2026, with 2023 as the base year). Only drug acquisition costs were included. The BIA considered a reference scenario where only LUM-IVA (only available in Ontario, Alberta, Saskatchewan, and the Non-Insured Health Benefits for F/F patients) was available while the new drug scenario included both ELX-TEZ-IVA and LUM-IVA. All patients were assumed to receive background BSC, therefore costs associated with BSC were not considered. The sponsor’s estimates of expected ELX-TEZ-IVA utilization were based on internal estimates, whereas the market size was primarily based on data generated from the CF Canada patient registry and further reduced based on the proportion of patients covered by provincial formularies. Key inputs to the BIA are documented in Table 28.
Additionally, the sponsor made the following key assumptions:
65% of indicated patients would be covered by provincial drug programs.
The market uptake of ELX-TEZ-IVA would be 77%, 83%, and 85% in Years 1 through 3, respectively.
The compliance rate for CFTR modulators was assumed to be 93%.
Genotype-specific subpopulations grow at the same rate as the general CF population.
Drug mark-up and dispensing fees were excluded in the base-case analysis.
Proportion of patients switching from LUM-IVA to ELX-TEZ-IVA are captured in the ELX-TEZ-IVA market share values.
Figure 2: Sponsor’s Estimation of the Size of the Eligible Population (Ontario Example)
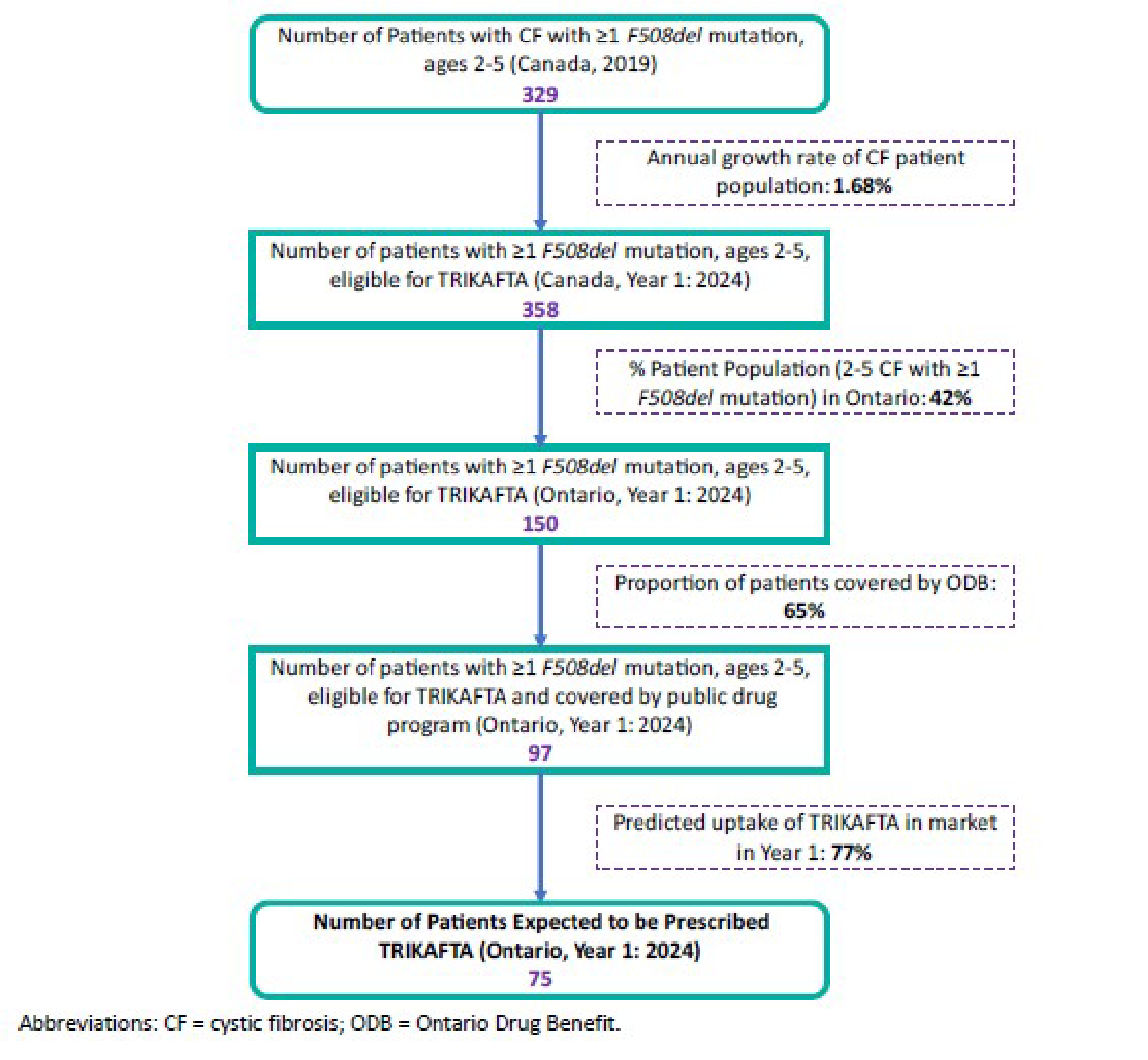
Source: Sponsor’s budget impact submission.40
Table 28: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3) |
|---|---|
Target population | |
Number of patients eligible for drug under review | 179 / 183 / 186 |
Market Uptake (3 years) | |
Uptake (reference scenario) LUM-IVA BSC | 2% / 2% / 2% 98%/ 98%/ 98% |
Uptake (new drug scenario) ELX-TEZ-IVA LUM-IVA BSC | 77%/ 83%/ 85% 2% / 1% / 0% 22%/ 16%/ 15% |
Cost of treatment (per patient) | |
Annual treatment cost, adjusted for 93% compliance ELX-TEZ-IVA LUM-IVA BSC | $285,333 $231,712 $0 |
BSC = best supportive care; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; LUM-IVA = lumacaftor-ivacaftor.
Note: Values may not sum to 100% due to rounding.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s base case BIA estimated that the reimbursement of ELX-TEZ-IVA for the treatment of CF patients who are 2 to 5 years old with at least 1 F508del-CFTR mutation would be $39,435,735 in Year 1, $43,055,265 in Year 2, and $44,667,268 in Year 3. Therefore, the 3-year incremental budget impact would be $127,158,268.
The sponsor conducted several sensitivity analyses to assess the impact of different compliance rates, predicted utilization rates of ELX-TEZ-IVA and IVA, increasing the size of the eligible patient population, and the inclusion of pharmacy upcharges and dispensing fees. All had a large impact on results, with the largest impact associated with the scenario analyses assuming a 10% increase in either eligible population size or market uptake upon ELX-TEZ-IVA availability. Both scenario analyses were associated with a 3-year budget impact of $139,924,982. Another scenario of note is that assuming 100% compliance, where the 3-year incremental budget impact was $136,729,321.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Compliance-adjusted drug costs underestimate the total costs associated with ELX-TEZ-IVA and other CFTR modulators to public drug plans. In alignment with the submitted CEA, the sponsor adjusted the price of CFTR modulators by the assumed compliance rate of 93% claiming that savings would be incurred by public drug plans due to patients not being 100% compliant. As the real-world compliance rate with ELX-TEZ-IVA for patients aged 2 to 5 is not available, the sponsor used compliance data from LUM-IVA which is associated with uncertainty as ELX-TEZ-IVA may have a different compliance rate.40 Additionally, the full complement of ELX-TEZ-IVA would be dispensed, regardless of whether the patient was compliant, thus resulting in the full costs of treatment being incurred by the public drug payer. This underestimated the total costs associated with ELX-TEZ-IVA and its total budget impact.
CADTH assumed patients were 100% compliant in reanalyses, in alignment with the CADTH pharmacoeconomic base case.
Treatment switching from LUM-IVA to ELX-TEZ-IVA may be underestimated. In the sponsor’s base-case analysis, it was assumed that a proportion of patients would gradually switch from LUM-IVA to ELX-TEZ-IVA upon ELX-TEZ-IVA treatment reimbursement (i.e., 33% in Year 1, 67% in Year 2, and 100% in Year 3). Based on clinical expert feedback received by CADTH, it was noted that the treatment switching from LUM-IVA to ELX-TEZ-IVA is expected to occur much more rapidly such that most, if not all, LUM-IVA patients would switch to ELX-TEZ-IVA within the first year of ELX-TEZ-IVA public reimbursement.
CADTH was unable to address this limitation due to the model structure of the submitted BIA. However, an increase in the proportion of patients switching from LUM-IVA to ELX-TEZ-IVA would increase the budget impact of reimbursing ELX-TEZ-IVA by a small amount due to the small proportion of patients currently on LUM-IVA.
Estimated proportion of patients with public coverage for CFTR modulator therapy is uncertain. The sponsor assumed 65% of the population indicated for ELX-TEZ-IVA would have public coverage, thus reducing the total eligible population size by 35%. The evidence cited by the sponsor for this assumption was internal data not available to CADTH, and uncertainty remains as to the proportion of the indicated population who would be covered by public drug plans. There is uncertainty in the proportion of patients with public versus private insurance. If more than 65% of the population would be covered, the anticipated budget impact associated with ELX-TEZ-IVA would be higher. If fewer are covered, the anticipated budget impact would be lower.
CADTH assumed 65% coverage in the base-case analysis, and tested scenarios of 50% coverage and 100% coverage.
CADTH Reanalyses of the BIA
Table 29: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Patient compliance | 93% | 100% |
CADTH base case | Reanalysis 1 | |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; LUM-IVA = lumacaftor-ivacaftor.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 30 and a more detailed breakdown is presented in Table 31.
Based on the CADTH base case, the budget impact of the reimbursement of ELX-TEZ-IVA for the treatment of CF in patients who are 2 to 5 years of age with at least 1 F508del-CFTR mutation is expected to be $42,404,017 in Year 1, $46,295,984 in Year 2, and $48,029,320 in Year 3. Therefore, the 3-year total is $136,729,321. Scenario analyses were conducted around the proportion of patients expected to have public drug coverage. The 3-year budget impact totals for these analyses were $105,176,400 and $210,352,801 when 50% and 100% of patients were assumed to have public coverage, respectively.
Table 30: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted sponsor base case | $127,158,268 |
CADTH base case | $136,729,321 |
BIA = budget impact analysis.
Table 31: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted sponsor base case | Reference | $324,491 | $329,944 | $335,488 | $341,126 | $1,006,559 |
New drug | $324,491 | $39,765,679 | $43,390,753 | $45,008,394 | $128,164,827 | |
Budget impact | $0 | $39,435,735 | $43,055,265 | $44,667,268 | $127,158,268 | |
CADTH base case | Reference | $348,915 | $354,778 | $360,740 | $366,803 | $1,082,321 |
New drug | $348,915 | $42,758,795 | $46,656,724 | $48,396,123 | $137,811,642 | |
Budget impact | $0 | $42,404,017 | $46,295,984 | $48,029,320 | $136,729,321 | |
CADTH scenario analysis: 94% price reduction | Reference | $348,915 | $354,778 | $360,740 | $366,803 | $1,082,321 |
New drug | $348,915 | $2,899,019 | $2,968,951 | $2,903,767 | $8,771,738 | |
Budget impact | $0 | $2,544,241 | $2,608,211 | $2,536,965 | $7,689,417 | |
CADTH scenario: 50% public coverage | Reference | $268,396 | $272,906 | $277,493 | $282,156 | $832,555 |
New drug | $268,396 | $32,891,381 | $35,889,788 | $37,227,787 | $106,008,955 | |
Budget impact | $0.00 | $32,618,474 | $35,612,295 | $36,945,631 | $105,176,400 | |
CADTH scenario: 100% public coverage | Reference | $536,792 | $545,813 | $554,985 | $564,312 | $1,665,109 |
New drug | $536,792 | $65,782,761 | $71,779,575 | $74,455,573 | $212,017,910 | |
Budget impact | $0 | $65,236,949 | $71,224,590 | $73,891,262 | $210,352,801 |
BIA = budget impact analysis.
Ethics Review
Abbreviations
CF
cystic fibrosis
CFTR
cystic fibrosis transmembrane conductance regulator
ELX-TEZ-IVA
elexacaftor-tezacaftor-ivacaftor and ivacaftor
Summary
Cystic fibrosis (CF) is a multiorgan genetic disorder caused by dysfunction in the CFTR gene. This disease primarily impacts the respiratory system, leading to progressive lung damage and loss of lung function sometimes requiring lung transplant, loss of livable years, and diminished quality of life. It additionally affects the gastrointestinal and reproductive systems. Patient group, clinical expert, and drug program input gathered during this CADTH review, as well as relevant literature, were reviewed to identify ethical considerations regarding the use of elexacaftor-tezacaftor-ivacaftor and ivacaftor (ELX-TEZ-IVA) for the treatment of CF in patients aged 2 to 5 years who have at least 1 F508del mutation in the CFTR gene.
Ethical considerations identified in this review included those related to the following:
Diagnosis, treatment, and experiences of CF: Ethical considerations in the context of CF highlighted the physical and psychosocial burden of CF on patients, families, and caregivers.
Evidence and evaluation of ELX-TEZ-IVA in patients aged 2 to 5 years: Clinical trial evidence indicated that ELX-TEZ-IVA was well tolerated in study participants aged 2 to 5 years, with few serious adverse events, although there is a recommendation for ongoing monitoring of liver enzymes. However, because the trial was not primarily designed to assess efficacy, the determination of efficacy in patients aged 2 to 5 years for the purposes of regulatory approval was extrapolated from studies conducted in patients with CF older than 5 years. Extrapolation may offer benefits such as avoiding exposing patient populations deemed vulnerable, such as children, to unnecessary research and extending access to therapy in patient populations that may be difficult to study or cannot be studied in clinical trials. However, extrapolation also presents potential risks if efficacy is not generalizable and thus overestimates or underestimates real-world effectiveness across different populations. Long-term monitoring is required to understand the long-term safety, efficacy, and comparative effectiveness of ELX-TEZ-IVA in patients aged 2 to 5 years. The lack of long-term efficacy and comparative effectiveness data limits the ability to accurately model and assess the cost-effectiveness of ELX-TEZ-IVA for use in patients aged 2 to 5 years.
Use of ELX-TEZ-IVA in patients aged 2 to 5 years: The clinical experts consulted by CADTH noted that, given the efficacy data from patients aged 6 years and older, they expected ELX-TEZ-IVA to benefit patients aged 2 to 5 years who have at least 1 508del mutation in the CFTR gene. As a result, they suggested that they would recommend prescribing ELX-TEZ-IVA for children aged 2 to 5 years, given the expected benefits of preventive treatment in relation to structural lung damage, the lack of effective alternatives, and the generally favourable safety and tolerability profile in this age group. As an orally administered medication, ELX-TEZ-IVA is relatively accessible and easy to administer for patients or their caregivers, including relative to alternate therapies.
Health system considerations: Expensive drugs for rare diseases, such as ELX-TEZ-IVA, raise ethical considerations related to distributive justice and equitable access, the sustainability of health care budgets and consideration of opportunity costs, and fair pricing of pharmaceuticals. As a highly expensive medication, the cost of ELX-TEZ-IVA could present challenges for provincial drug budgets as the reimbursement of ELX-TEZ-IVA may have a disproportionately large budget impact. There is a need to address potential inequities in access due to inconsistent reimbursement and/or insurance coverage across and within jurisdictions in Canada.
Objective
To identify and describe the ethical considerations associated with the use of ELX-TEZ-IVA in the treatment of CF in patients aged 2 to 5 years who have at least 1 F508del mutation in the CFTR gene, including considerations related to the disease context, evidentiary basis, use of the therapy, and considerations relevant to health systems.
Research Questions
This report addresses the following research questions:
What ethical considerations arise in the context of patients with CF aged 2 to 5 years who have at least 1 F508del mutation in the CFTR gene, including considerations related to diagnosis, treatment, and outcomes?
What ethical considerations arise related to the evidence (e.g., clinical and economic data) used to evaluate ELX-TEZ-IVA for patients with CF aged 2 to 5 years?
What ethical considerations arise in the use of ELX-TEZ-IVA for patients aged 2 to 5 years, their caregivers, and their clinicians?
What ethical considerations for health systems are involved in the context of the implementation of ELX-TEZ-IVA for patients aged 2 to 5 years?
Methods
To identify ethical considerations relevant to the use of ELX-TEZ-IVA in the treatment of CF in patients aged 2 to 5 years who have at least 1 F508del mutation in the CFTR gene, this ethics report was driven by relevant questions identified in the EUnetHTA Core Model 3.0,1 Ethics Analysis Domain,1 and supplemented by relevant questions from the Equity Checklist for HTA (ECHTA).2 These guiding questions were organized to respond to the research questions posed and to investigate ethical considerations related to:
patients living with CF and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges related to diagnosis or clinical care; factors that might prevent patients from gaining access to ELX-TEZ-IVA)
the evidence used to demonstrate the benefits, harms, and value of ELX-TEZ-IVA (i.e., ethical considerations in relevant clinical trials, including their representativeness, choice of outcome measures, appropriateness of analytical methods and models to all population groups; ethical considerations related to the data or assumptions in the economic evaluation)
the use of ELX-TEZ-IVA, including considerations related to benefits and harms to patients, relatives, caregivers, clinicians, and society, as well as considerations related to access to these therapies
the uptake of ELX-TEZ-IVA in health systems, including considerations related to the distribution of health care resources.
Data Collection: Review of Project Inputs and Literature
Data to inform this ethics report were drawn from the identification of ethical considerations (e.g., values, norms, or implications related to the harms, benefits, and implications for equity, justice, and resource allocation, and ethical considerations in the evidentiary basis) in the patient and clinician group, clinical expert, and drug program input collected by CADTH to inform this review, as well as from a complementary search of the published literature. Ongoing collaboration and communication with CADTH reviewers working on the clinical and economic reviews for this submission also assisted in the clarification and identification of ethical considerations.
Review of Project Inputs
During this CADTH review, a single reviewer collected and considered input from 6 main sources related to ethical considerations relevant to the research questions guiding this ethics report. In addition to published literature, this report considered the following sources:
The sponsor submission, including noting relevant information and external references or sources relevant to each of the research questions driving this report.
Clinician group input received by CADTH from Cystic Fibrosis Canada’s Accelerating Clinical Trials Network (CF CanACT), Cystic Fibrosis Canada Healthcare Advisory Council, and Canadian Cystic Fibrosis Clinicians.
Patient input received by CADTH from Cystic Fibrosis Canada.
Drug program input received by CADTH from drug programs participating in the CADTH Reimbursement Review process.
Discussions with the clinical experts directly engaged by CADTH over the course of this Reimbursement Review, including consultation meetings with clinical experts and drug program representatives. During discussions, the clinical experts were asked targeted questions related to ethical considerations corresponding to the research questions driving this report. All the clinical experts specialized in the treatment of CF, and all had experience treating pediatric patients with CF in Canada.
Engagement with CADTH economic reviewers to identify domains of ethical interest arising from their respective reviews as well as relevant questions and sources to further pursue in this report.
Literature Search Methods
An information specialist conducted a literature search using key resources, including MEDLINE via Ovid, Philosopher’s Index via Ovid, PsycInfo via Ovid, the Cumulative Index to Nursing and Allied Health Literature via EBSCO, and Scopus. Google Scholar was searched to find additional materials not captured in the major bibliographic databases. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Trikafta along with searches for cystic fibrosis and the F508del mutations of the CFTR gene.
CADTH-developed search filters were applied to limit retrieval to citations related to ethical concepts or considerations and qualitative studies. Search terms for equity were also applied to the main concepts to capture additional articles. Duplicates were removed by manual deduplication in Endnote. Retrieval was limited to the English language. The search was completed on June 7, 2023.
Literature Screening and Selection
Literature retrieved according to the search and selection methods detailed above was screened in 2 stages. First, the titles and abstracts of the citations retrieved were screened for relevance by a single reviewer. Articles were identified and retrieved for full-text review by a single reviewer if their titles or abstracts identified ethical considerations or provided normative analysis (i.e., focusing on “what ought to be” through argumentation) or empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to the experiences, incidence, diagnosis, treatment, or outcomes of CF or related to the evidence on, use of, or implications of ELX-TEZ-IVA for patients with CF. In the second stage, full-text publications categorized as “retrieve” were reviewed by the same reviewer. Texts that included substantive information meeting the aforementioned criteria were included in the review, and reports that did not meet these criteria were excluded. As a parallel process, other sources drawn from relevant bibliographies, relevant key concepts, and consultation with experts or other CADTH reviewers were retrieved and reviewed using the selection criteria listed previously.
Data Analysis
Data analysis was driven by the 4 research questions guiding this report and included the collection, coding, and thematic analysis of data drawn from the literature and project inputs. The reviewer conducted 2 iterative cycles of coding and analysis in NVivo, a qualitative analysis software program, to abstract, identify, and synthesize relevant ethical considerations from the literature and from relevant project inputs.
In the initial coding phase, publications and input sources were reviewed for ethical content (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis). Once identified, claims related to ethical content were coded using methods of qualitative description.3 In the second coding phase, major themes and subcodes were identified through repeated reading of the data3 and were summarized into thematic categories within each guiding domain or research question. If the ethical content did not fit into these categories or into the domains outlined in the research questions, this was noted, as were discrepancies or conflicts in ethical considerations or values identified between project sources or within thematic categories. Data analysis was iterative, and the themes identified in the literature, in project inputs, and during consultations with clinical experts were used to further refine and reinterpret the ethical considerations identified.
The data collected and analyzed from these sources were thematically organized and described according to the 4 research questions driving this report. The results of this analysis and its limitations and conclusions are described in the following sections.
Results
Description of Included Sources
Data to inform this ethics report were drawn from a review of clinician group input, drug program input, and consultation with clinical experts engaged by CADTH for this review. All the clinical experts were active in relevant clinical roles in Canada, and all had experience treating patients with CF. A description and summary of these sources are included in the CADTH Clinical Review Report.
The literature search identified 223 results, and the grey literature search identified 19 additional results, for a total of 242 results. Following title and abstract screening, 209 citations were excluded and 33 potentially relevant publications from the electronic searches were retrieved for full-text review. Of the potentially relevant publications, 12 were excluded because they did not discuss ethical considerations of CF or ELX-TEZ-IVA. A total of 21 publications met the inclusion criteria and were included in this report. An additional 4 articles were identified in a manual search of the references of eligible articles.
A total of 25 publications were used to inform this report. Of these publications, 10 publications discussed ethical considerations in the context of CF, including in relation to diagnosis and treatment; 8 publications discussed patient and/or family and caregiver experiences in the context of CF; 5 publications were selected to provide a broader understanding of diversity in clinical trials; and 2 publications were selected to provide a broader understanding of the context of ethical considerations relating to drugs for rare diseases. Details regarding the characteristics of included publications are reported in Table 1.
Key Ethical Considerations
Diagnosis, Current Treatment Landscape, and Experiences of CF
As noted in the clinical report, the incidence of CF in Canada is approximately 1 per 3,600 live births.4 Approximately 4,300 people with CF live in Canada, of which 8% are children aged 2 to 5 years.4 Of these children, approximately 87% have at least 1 copy of the F508del mutation of the CFTR gene, making them eligible for ELX-TEZ-IVA pending Health Canada approval.5
CF is a multiorgan genetic disorder caused by dysfunction in the CFTR gene.6 This disease primarily impacts the respiratory system, leading to progressive lung damage. It additionally affects the gastrointestinal and reproductive systems.7,8 CF is currently incurable, but substantial improvements have been seen in survival prognosis, with the median age of survival estimated to be 57.3 years in people with CF in Canada.9 Key factors influencing prognosis include duration of time to diagnosis and treatment initiation, lung disease severity, nutritional and socioeconomic conditions, and mental well-being.10
Diagnosis
The introduction of newborn screening for CF across Canada has significantly altered the diagnostic approach for this disease, as newborns with positive screening results are referred for further diagnostic testing. Newborn screening in Canada involves drawing a small sample of blood from the infant’s heel. If there is a positive or inconclusive result, the patient is referred to a CF clinic for further testing. A sweat chloride test is administered, which measures the amount of chloride in a person’s sweat. If the results indicate 60 mmol/L or greater, CF is confirmed.
The establishment of a newborn screening program for CF in Canada and the US has led to improved health outcomes and long-term growth outcomes for patients with CF.11-13 Screening results, however, are not always clear because there have been instances when individuals present with symptoms indicative of CF but they were either were not screened due to the absence of newborn screening or they received a false-negative result during newborn screening.14,15 Such circumstances can result in delayed diagnosis, irreversible progression of the disease, and delayed access to early treatment.16,17 The clinical experts consulted by CADTH noted that misdiagnosis and underdiagnosis of CF are rare in Canada, with most cases confirmed by 1 month of age on average, and most false-negatives resolved and accurately diagnosed by 1 year of age following the development of symptomatic CF.
In addition, CF has historically been perceived as a condition primarily impacting white individuals of European ancestry,18 despite also impacting individuals in other regions, such as the Middle East, Asia, and Latin America.17 This perception is ethically concerning because it can lead to tacit health care practitioner racial bias that can result in delayed diagnosis for patients of non-Caucasian or non-European ancestry. Canadian practitioners need to be aware of this when treating patients born outside of Canada who may not have undergone newborn screening for CF in their country of birth and thus may be undiagnosed. Increasing awareness of CF is also important for patients of non-European descent and their caregivers; US research indicates that these populations have significantly less knowledge and awareness of CF than non-Hispanic white patients.19
Current Treatment Landscape
Clinician input described the treatment approach for CF in children (aged 2 to 5 years) as lifelong. As noted in the clinical report, nonmodulator therapies include high-calorie, high-fat, and high-protein diets; digestive medications; and airway clearance treatments, most of which are initiated at the time of diagnosis. Frequently prescribed medications in CF include antibiotics, mucolytics, bronchodilators, pancreatic enzymes, fat-soluble vitamins, insulin for individuals with CF-related diabetes, and ursodiol for liver disease.
As previously stated, CF is caused by mutations in the CFTR gene. There are more than 2,000 CFTR mutations currently documented.20 Since the introduction of CFTR modulator therapy nearly a decade ago, the treatment of CF has undergone significant transformation, leading to health outcome improvements for individuals with CF. CFTR modulators are mutation-specific therapies. For some mutations, early initiation of CFTR modulators has shown interruption of disease progression in multiple systems.21 The clinical benefits of CFTR modulators include improved lung function, reduced frequency of pulmonary exacerbations, improved weight or body mass index, and enhanced quality of life.21-23
ELX-TEZ-IVA has demonstrated improvements in health outcomes and quality of life in children aged 6 years and older and adults with CF.22,24-26 Additionally, clinician group input noted that ELX-TEZ-IVA is a more efficacious therapy for patients with CF who have at least 1 copy of the F508del mutation than other previously approved CFTR modulators. Furthermore, ELX-TEZ-IVA has the potential to delay disease progression and the need for additional procedures.
ELX-TEZ-IVA is used to treat up to 90% of patients with CF in Canada5 and is already approved for use with, and publicly reimbursed across Canada for, patients with CF who have at least 1 F508del mutation and are aged 6 years and older. Patients with other rare genetic mutations resulting in CF do not currently have access to most approved modulator treatments in Canada. However, there are some exceptions, such as patients with a gating mutation (G551D) who can access ivacaftor once they are aged 6 years or older. The research and development of CFTR modulators for children with rare genotypes who are currently ineligible for ELX-TEZ-IVA due to gene mutation requirements may help expand access to CFTR therapy in a more diverse patient population.27
Because CF is a multiorgan disease, it requires multiple health care professionals for surveillance and management. The clinical experts consulted by CADTH and caregiver perspectives in the literature highlight a need for ELX-TEZ-IVA among children with CF aged 2 to 5 years. This is due to the role ELX-TEZ-IVA can play in the prevention of disease progression, loss of livable years, suffering, while also improving quality of life. The clinical experts noted that there were no data to support withholding the initiation of CFTR modulator treatment until clinical symptoms of CF have developed in children, especially as children younger than 6 years could still develop structural lung disease while presenting as asymptomatic.28,29
Patient, Family, and Caregiver Experiences of CF
Upon learning of a CF diagnosis, caregivers reportedly experience a traumatic response, which can include extended periods of shock and distress and ongoing feelings of loss and grief, followed by a process of adjustment.30-32 Caregivers’ lack of knowledge about CF can further exacerbate this traumatic response.31
CF presents with a wide range of clinical manifestations, varying in severity among individuals, and includes chronic lung infections, progressive deterioration of lungs, progressive loss of lung function, and early mortality. Common complications from CF include digestion difficulties, malnutrition, the development of vitamin deficiencies due to the inability to effectively absorb nutrients, chronic lung infections, CF-related diabetes, and sinus infections.33
Prior to the introduction of CFTR modulators, pediatric treatments for CF included airway clearance techniques and nebulized mucolytics, which need to be maintained for life.34 As per the patient group input, the management of CF necessitates a demanding treatment routine. As the disease advances, it requires an increasing amount of time and effort, and frequent clinic visits and hospital stays to cope with the progressive and debilitating symptoms. Some children have described alternate treatments to be tedious and time-consuming. Caregivers echoed these sentiments, with some reporting that their entire life was dictated by managing their child’s various treatments. Upon initiating ELX-TEZ-IVA, both caregivers and adolescents (aged 12 to 18 years) alike expressed astonishment and disbelief at the significant reduction in treatment burden in addition to the life-changing benefits.34
Ethics of Evidence and Evaluation of ELX-TEZ-IVA
As discussed in detail in the CADTH Clinical Report for this review, ELX-TEZ-IVA was evaluated in Study 111, a 24-week, open-label, phase III, nonrandomized, single-arm, 2-part study. Study 111 was primarily designed to evaluate the safety, tolerability, and pharmacokinetics of ELX-TEZ-IVA in patients aged 2 to 5 years with at least 1 F508del mutation in the CFTR gene. However, as discussed in the following section, the regulatory submission is based on the extrapolation of efficacy data from studies conducted in older patients with CF, as the assessment of efficacy was not a primary objective in Study 111 and assessments of efficacy outcome measures in patients aged 2 to 5 years can be challenging. As discussed in the clinical review, ELX-TEZ-IVA was well tolerated in study participants, with few serious adverse events. The product monograph notes that elevated transaminases (alanine transaminase and aspartate transaminase) have been observed in patients treated with ELX-TEZ-IVA, requiring periodic and ongoing monitoring to understand any potential risks to the liver.
Considerations Related to the Extrapolation of Efficacy
Extrapolation is the practice of making predictions or inferences about outcomes beyond available data. In the context of this CADTH review, and as discussed further in the CADTH Clinical Review Report, this involves extending the efficacy findings from 1 age group (e.g., patients aged 12 years and older) to estimate potential efficacy outcomes in another age group (e.g., patients aged 2 to 5 years). Extrapolation is frequently required in the approval of new medications where preapproval trials cannot encompass all patient subpopulations, age groups, and comorbidities due to study limitations and to the need to avoid exposing vulnerable patient populations, such as children, to unnecessary research.35 Extrapolation is thus commonly used for pediatric therapies to increase availability of information for pediatric use and regulatory approval.36 However, using limited clinical data to generalize a treatment’s efficacy across a wide range of patients may not always be clinically appropriate because this approach may overestimate or underestimate a drug’s efficacy and safety during routine use.35 Concerns about generalizability arise when factors such as patient age, sex, and race or ethnicity may not have been adequately represented during preapproval trials.35 Careful consideration of these differences is particularly important when applying extrapolated data to the pediatric population.35 In the context of this report, developmental stages, physiological changes, and genetic variations should be carefully considered.
The clinical experts consulted by CADTH noted that the assessment of outcome measures in evaluating the efficacy of ELX-TEZ-IVA for children with CF aged 2 to 5 years is challenging. The clinical experts and published literature noted that this population is unable to consciously perform coordinated efforts, such as spirometry, which is used to assess lung function in clinical practice.21 Sedation may be required to perform lung function assessments, which presents ethical concerns regarding risks and recovery (e.g., respiratory depression, allergic reactions, aspiration, inadequate sedation, and, in rare cases, death).21 Moreover, establishing the effectiveness of interventions may require several years in children with minimal disease.21
As noted in the CADTH Clinical Review Report, Study 111 suggested that treatment with ELX-TEZ-IVA resulted in improvements from baseline in lung function and CF biomarkers; however, uncertainty remains about the magnitude of the treatment effect and its comparative effectiveness based on currently available evidence. Nonetheless, the clinical experts noted that, given the efficacy data in patients aged 6 years and older, ELX-TEZ-IVA would be expected to benefit patients aged 2 to 5 years who have at least 1 508del mutation. They noted that there are no physiological differences between children aged 2 to 5 years and adolescents or adults that would affect the underlying disease pathology or mechanism of action of CFTR modulators. Additionally, they noted they would raise these considerations when discussing the use of extrapolated data with families or caregivers concerning the use of ELX-TEZ-IVA. For example, the clinical experts stated that CFTR modulators are associated with a significant decrease in pulmonary exacerbations, reducing hospital admissions, and that, over time, each pulmonary exacerbation results in irreversible lung damage. Consequently, the primary objective in discussing extrapolation with caregivers is to highlight the importance of preventing pulmonary exacerbations. Furthermore, the experts noted that there is evidence suggesting that structural changes can occur early in the disease course, including before the age of 6 years. The goal is therefore to initiate medication early, even before symptoms manifest, recognizing that there might be underlying processes occurring that are not visible or measurable in routine clinical practice.
The clinical experts noted that CF registries may be 1 option for collecting long-term data on the safety and efficacy of ELX-TEZ-IVA in children aged 2 to 5 years but that use of these data to inform reimbursement decisions may be limited because important indicators, such as liver function tests, are not submitted. Rather, such information is known by treating clinicians. Other registry data, such as number of exacerbations, hospital stays, and days of antibiotics, can provide some information, but not a complete picture. The clinical experts stated that, in the absence of traditional outcome measures evaluating response to ELX-TEZ-IVA, they would look at improvement or stabilization in the frequency and severity of pulmonary exacerbations and body mass index to evaluate efficacy, which are not considered reliable indicators of the medication’s benefits in the target patient population. This fact has important implications for renewal criteria for reimbursement by the public drug programs. For example, the CADTH clinical experts have recommended that sweat chloride testing not be used to determine ELX-TEZ-IVA response for the purposes of drug reimbursement, which is consistent with the clinical expert input from previous reviews of ELX-TEZ-IVA in older patients (aged 6 to 11 years and 12 years and older). This decision is because sweat chloride testing does not provide a reliable prediction of clinically significant outcomes; rather, it reflects the mechanism of action of CFTR modulators like ELX-TEZ-IVA.
Clinical experts also highlighted the challenges in accessing sweat chloride testing in certain jurisdictions due to the variability in receiving test results within different time frames and significant concerns regarding the health care system’s ability to accommodate repeated sweat chloride testing for all patients with at least 1 F508del mutation. Because children aged 2 to 5 years will likely not be able to demonstrate such improvements in overt measurable ways, the clinical experts noted that prescribing decisions (e.g., whether to prescribe, renew, or discontinue the medication) would be based on an assessment by the treating specialist clinician. The clinical experts recommended relying on writing clinical letters to support renewals, especially if children have complex care needs (e.g., developmental delays) and cannot perform pulmonary function tests.
Representativeness in Research Participation
Although the randomized controlled trial that informed the Health Canada decision to grant children (aged 6 to 11 years) access to ELX-TEZ-IVA was broadly representative of the patient population most commonly affected by CF,37 there was underrepresentation of racialized groups because participants in Study 111 were primarily white. The primary reason for this is because CF disproportionately affects non-Hispanic white people. Nonetheless, the inclusion of racialized populations in clinical trials is a crucial step toward addressing health inequities and facilitating better access to advancements in CF therapies,38 especially because trial participation is often seen as a quicker path to accessing innovative therapies such as ELX-TEZ-IVA.39 Additionally, diverse clinical trial participation can produce higher-quality biomedical knowledge and ensure fairness for potential participants.38 Lack of interest in trial participation among racialized populations is not the root of this disparity, and research participation can be improved by employing research staff with cultural competence who use community involvement in the research process, such as in developing consent forms, translating documents, and using community liaisons for recruitment.40
Considerations for Pharmacoeconomic Assessments
The lack of long-term efficacy and comparative effectiveness data has implications for the pharmacoeconomic assessment of ELX-TEZ-IVA because it limits the ability to accurately model and assess its cost-effectiveness. This limitation, which may impact cost-effectiveness analyses for drugs for rare diseases more generally, presents challenges for assessing the opportunity costs — or forgone benefits — associated with reimbursing and resourcing a particular intervention over others.41 Understanding opportunity costs is important for informing resource allocation decisions at a health system level.
Ethical Considerations in the Use of ELX-TEZ-IVA
Balancing Benefits and Harms
As previously stated, there is evidence supporting the benefits of using ELX-TEZ-IVA for children and youth aged 6 years and older and for adults.37,42-44 Despite evidentiary limitations, such as the absence of direct evidence of efficacy in patients aged 2 to 5 years, as well as the absence of long-term safety and comparative effectiveness data for this age group, the clinical experts noted that they would recommend prescribing ELX-TEZ-IVA for children aged 2 to 5 years, given the expected benefits of preventive treatment in relation to structural lung damage, the lack of effective alternatives, and the generally favourable safety and tolerability profile in this age group. As an orally administered medication, ELX-TEZ-IVA is accessible and easy to administer for patients or their caregivers, including relative to alternate therapies. Moreover, as noted by the clinical experts and in the literature, there are additional anticipated benefits for patients and their caregivers, including lesser treatment-associated burden on patients and caregivers, decreases in hospitalizations, and equity-associated benefits for patients of lower socioeconomic status.
The clinical experts noted that the side effects of ELX-TEZ-IVA in a pediatric population are reportedly minimal (e.g., elevated liver enzymes and creatine kinase). Long-term evidence on the benefits of this drug is limited for all age groups. The lack of long-term efficacy data on ELX-TEZ-IVA is reportedly raising questions among practitioners and patients alike regarding the sustainability of its benefits in terms of health outcomes and quality of life.34,45 This knowledge gap has implications for the assessment of ELX-TEZ-IVA as it may limit the ability to accurately model and assess its cost-effectiveness. The clinical experts stated that they did not anticipate any physiological differences between children aged 2 to 5 years and older children that would differentially affect the use or efficacy of ELX-TEZ-IVA. However, they noted that younger children with CF face specific vulnerabilities, such as being more susceptible to the impacts of viral illnesses before vaccination and potential consequences arising from the re-emergence of vaccine hesitancy, which could heighten their susceptibility to preventable diseases and result in pulmonary exacerbations.
Additionally, while clinical experts acknowledged the expected benefits associated with the use of ELX-TEZ-IVA, they noted that patients or caregivers may be inclined to reduce or discontinue other treatments due to improvements experienced following the use of ELX-TEZ-IVA, which could lead them to believe that they no longer require additional therapy. This observation aligns with recent research findings.34 As patients with CF may be required to undertake more than 10 complex daily treatments, which can take more than 2 hours a day,27 treatment adherence may become challenging to children who reportedly do not feel affected by CF after taking ELX-TEZ-IVA.34
Sex-related disparities in the outcomes of individuals with CF, including in response to CFTR modulator treatment, have also been reported in other age groups, with female patients exhibiting a higher rate of pulmonary exacerbations and requiring lung transplant at an earlier stage than male patients.46 If ELX-TEZ-IVA is reimbursed for children aged 2 to 5 years, postmarket monitoring of its efficacy across sexes may be warranted to better inform decisions around treatment and overall care for both female and male patients in this age group.
Socioeconomic Considerations
It is reported that low socioeconomic status negatively impacts the social determinants of health of people living with CF.47-50 The clinical experts underscored the positive influence that ELX-TEZ-IVA could have on children with CF from low socioeconomic backgrounds. Notably, the experts emphasized the enhanced equity and improved health outcomes resulting from a decreased treatment burden after transitioning to the orally administered medication, taken twice a day. For instance, children with CF who are living in poverty often face psychosocial challenges, which can hinder their ability to adhere to numerous treatment obligations. However, following the introduction of ELX-TEZ-IVA, these children have shown significant improvements in their overall well-being and treatment management.
Health System Considerations
The reimbursement of ELX-TEZ-IVA for CF raises ethical considerations related to health systems as well as resource considerations, including opportunity costs, the sustainability of Canadian health care budgets, and the shifting use of health care resources as a result of implementation. Clinical experts consulted by CADTH highlighted that initiation of ELX-TEZ-IVA would be expected to increase the use of health care resources (e.g., frequent clinic visits; additional blood work, extended time for treatment education; and more examinations, follow-up appointments, and associated treatment costs) for all age groups. However, they expected that, following the initial treatment period, the burden on health systems would gradually diminish as there would be a reduction in required treatments beyond ELX-TEZ-IVA and a decrease in pulmonary exacerbations and associated hospitalization costs.34,45
Expensive drugs for rare diseases, such as ELX-TEZ-IVA, raise ethical considerations related to distributive justice and equitable access, the sustainability of health care budgets, and the fair pricing of pharmaceuticals.41,51 As a highly expensive medication, the clinical experts noted that the cost of ELX-TEZ-IVA, and CFTR modulators in general, could present a challenge to provincial drug budgets because, although the population of patients with CF may be small within a jurisdiction, the cost of reimbursing ELX-TEZ-IVA could have a disproportionate budget impact on provincial formularies. Reimbursing high-cost drugs for rare diseases, such as ELX-TEZ-IVA for CF, requires consideration of the opportunity costs associated with reimbursing, or not, a therapy over other therapies or services (e.g., including primary and preventive care) and raises questions about the fair and equitable allocation of scarce health care resources.50 The assessment of the opportunity costs of implementing ELX-TEZ-IVA for children aged 2 to 5 years is further complicated by the evidentiary uncertainty about the magnitude and durability of its therapeutic effect.
The clinical experts also noted that inconsistencies in pharmaceutical insurance coverage or reimbursement of ELX-TEZ-IVA within Canada could present access-related delays or challenges for patients (and their families). For example, they noted that families with private drug coverage that did not reimburse very-high-cost therapies such as ELX-TEZ-IVA could be faced with the decision of removing a dependent child from their parents’ insurance plan to access ELX-TEZ-IVA if it was otherwise reimbursed through a provincial drug plan.
Limitations
There is very little published literature that discusses ethical considerations related to the use of ELX-TEZ-IVA for the treatment of CF in a pediatric population, given both the rarity of the disease and the novelty of the drug under review for children aged 2 to 5 years. Nonetheless, this does not imply that ethical considerations in the context of ELX-TEZ-IVA for the treatment of CF are absent. This review of ethical considerations draws from additional resources collected in the course of this Reimbursement Review, including clinician and drug program input, discussion with clinical experts, and engagement with CADTH clinical and pharmacoeconomic review teams, to provide a more comprehensive understanding of the ethical considerations related to the use of ELX-TEZ-IVA for the treatment of CF.
Although this ethics report drew on expert input, it is possible that more direct engagement with key stakeholders (e.g., direct interviews with patients, caregivers, family members, and decision-makers) on their specific experiences with CF and/or ELX-TEZ-IVA could have offered additional relevant ethical considerations or domains of analysis.
Conclusion
Input from patient groups, clinician groups, and provincial drug programs, as well as direct engagement with clinical experts and published literature, were reviewed for ethical considerations relevant to the use of ELX-TEZ-IVA in patients aged 2 to 5 years with CF. Ethical considerations in the context of CF emphasized the physical and psychosocial burden of CF on patients, families, and caregivers. Clinical trial evidence indicated that ELX-TEZ-IVA was well tolerated in study participants aged 2 to 5 years, with few serious adverse events, although there is a recommendation for ongoing monitoring of liver enzymes. However, as the trial was not primarily designed to assess efficacy, the determination of efficacy in patients aged 2 to 5 years for the purposes of regulatory approval was extrapolated from studies conducted in older patients with CF. Extrapolation may offer benefits such as avoiding exposing vulnerable patients, such as children, to unnecessary research and extending access to therapy in patient populations that may be difficult to study or cannot be studied in clinical trials. However, extrapolation also presents potential risks if efficacy is not generalizable and thus overestimates or underestimates real-world effectiveness across different populations. Long-term monitoring is required to understand the long-term safety, efficacy, and comparative effectiveness of ELX-TEZ-IVA in patients aged 2 to 5 years. However, the clinical experts noted that, given the efficacy data in patients aged 6 years and older, they expected ELX-TEZ-IVA to benefit patients aged 2 to 5 years who have at least 1 508del mutation in the CFTR gene. As a result, the clinical experts suggested they would recommend prescribing ELX-TEZ-IVA for children aged 2 to 5 years, given the expected benefits of preventive treatment in relation to structural lung damage, the lack of effective alternatives, and the generally favourable safety and tolerability profile in this age group. Ethical considerations for health systems related to the use of ELX-TEZ-IVA for patients aged 2 to 5 years highlight challenges in funding decisions and in assessments of opportunity costs for expensive drugs for rare diseases as well as the need to address potential inequities in access due to inconsistent reimbursement and/or insurance coverage across and within jurisdictions in Canada.
References
1.EUnetHTA Joint Action 2 Work Package 8. HTA Core Model® version 3.0. 2016; http://www.htacoremodel.info/BrowseModel.aspx. Accessed 2023 Nov 15.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37(1). PubMed
3.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
4.Canadian Cystic Fibrosis Registry. 2020 Annual Data Report: The Canadian Cystic Fibrosis Registry. 2020; https://www.cysticfibrosis.ca/registry/2020AnnualDataReport.pdf. Accessed 2023 Nov 15.
5.Cystic Fibrosis Canada. Trikafta. 2023; https://www.cysticfibrosis.ca/our-programs/advocacy/access-to-medicines/trikafta. Accessed 2023 Nov 15.
6.Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic fibrosis. Lancet. 2021;397(10290):2195-2211. PubMed
7.Milo F, Ciciriello F, Alghisi F, Tabarini P. Lived experiences of people with cystic fibrosis that were not eligible for elexacaftor-tezacaftor-ivacaftor (ETI): A qualitative study. Journal of Cystic Fibrosis. 2023;22(3):414-419. PubMed
8.Quon BS, Rowe SM. New and emerging targeted therapies for cystic fibrosis. BMJ. 2016;352. PubMed
9.Cystic Fibrosis Foundation. Cystic fibrosis foundation patient registry 2021 annual data report. 2022; https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf. Accessed 2023 Nov 15.
10.Chen Q, Shen Y, Zheng J. A review of cystic fibrosis: Basic and clinical aspects. Animal Models and Experimental Medicine. 2021;4(3):220-232. PubMed
11.McKay K, Wilcken B. Newborn screening for cystic fibrosis offers an advantage over symptomatic diagnosis for the long term benefit of patients: the motion for. Paediatr Respir Rev. 2008;9(4):290-294. PubMed
12.Farrell PM, Lai HJ, Li Z, et al. Evidence on improved outcomes with early diagnosis of cystic fibrosis through neonatal screening: enough is enough! The Journal of pediatrics. 2005;147(3):S30-S36. PubMed
13.Mak D, Sykes J, Stephenson A, Lands L. The benefits of newborn screening for cystic fibrosis: The Canadian experience. J Cyst Fibros. 2016;15(3):302-308. PubMed
14.Sosnay PR, White TB, Farrell PM, et al. Diagnosis of cystic fibrosis in nonscreened populations. The Journal of pediatrics. 2017;181:S52-S57. e52.
15.Vaidyanathan S, Trumbull AM, Bar L, Rao M, Yu Y, Sellers ZM. CFTR genotype analysis of Asians in international registries highlights disparities in the diagnosis and treatment of Asian patients with cystic fibrosis. Genetics in Medicine. 2022;24(10):2180-2186. PubMed
16.Wright M, Wright T. Health disparity in CF: Perspectives from a lived experience. Pediatric Pulmonology. 2022;57:S13-S16. PubMed
17.Owusu SK, Morrow BM, White D, et al. Cystic fibrosis in black African children in South Africa: a case control study. J Cyst Fibros. 2020;19(4):540-545. PubMed
18.Silva Filho LVRF, Castaños C, Ruíz HH. Cystic fibrosis in Latin America—improving the awareness. J Cyst Fibros. 2016;15(6):791-793. PubMed
19.Hutchins K, Barr E, Bellcross C, Ali N, Hunt WR. Evaluating Differences in the Disease Experiences of Minority Adults With Cystic Fibrosis. Journal of Patient Experience. 2022;9:1-8. PubMed
20.Bierlaagh MC, Muilwijk D, Beekman JM, van der Ent CK. A new era for people with cystic fibrosis. Eur J Pediatr. 2021;180(9):2731-2739. PubMed
21.Dobra R, Bentley S, Edmondson C, et al. Going the Extra Mile: Why Clinical Research in Cystic Fibrosis Must Include Children. Children. 2022;9(7):1-14. PubMed
22.Heijerman HG, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. The Lancet. 2019;394(10212):1940-1948. PubMed
23.McNamara JJ, McColley SA, Marigowda G, et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study. The Lancet Respiratory Medicine. 2019;7(4):325-335. PubMed
24.Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381(19):1809-1819. PubMed
25.Nichols DP, Paynter AC, Heltshe SL, et al. Clinical effectiveness of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis: a clinical trial. Am J Respir Crit Care Med. 2022;205(5):529-539. PubMed
26.Sutharsan S, McKone EF, Downey DG, et al. Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: a 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. The Lancet Respiratory Medicine. 2022;10(3):267-277. PubMed
27.King JA, Nichols A-L, Bentley S, Carr SB, Davies JC. An update on CFTR modulators as new therapies for cystic fibrosis. Pediatric Drugs. 2022;24(4):321-333. PubMed
28.Guo J, Wang J, Zhang J, Fortunak J, Hill A. Current prices versus minimum costs of production for CFTR modulators. Journal of Cystic Fibrosis. 2022;21(5):866-872. PubMed
29.Stanojevic S, Vukovojac K, Sykes J, Ratjen F, Tullis E, Stephenson AL. Projecting the impact of delayed access to elexacaftor/tezacaftor/ivacaftor for people with Cystic Fibrosis. Journal of Cystic Fibrosis. 2021;20(2):243-249. PubMed
30.Priddis L, Dougall A, Balding E, Barrett A. Cystic fibrosis diagnosis: impact on mothers of affected Australian children. Neonatal Paediatr Child Health Nurs. 2009;12:20-25.
31.Souza TCF, Correa Júnior AJS, Santana MEd, Pimentel IMdS, Carvalho JN. Experiences of family members of children with cystic fibrosis under the light of Callista Roy. Revista Brasileira de Enfermagem. 2020;73.
32.Dingus Keuhlen K. An exploration of the experiences of caregivers raising children with a cystic fibrosis diagnosis and the relationship between anxiety, depression, and quality of life: An online exploratory mixed-methods study [Dissertation]2021.
33.Cystic Fibrosis Canada. What is cystic fibrosis? 2023; https://www.cysticfibrosis.ca/about-cf/what-is-cystic-fibrosis. Accessed 2023 Nov 15.
34.Almulhem M, Harnett N, Graham S, et al. Exploring the impact of elexacaftor-tezacaftor-ivacaftor treatment on opinions regarding airway clearance techniques and nebulisers: TEMPO a qualitative study in children with cystic fibrosis, their families and healthcare professionals. BMJ Open Respiratory Research. 2022;9(1):1-10. PubMed
35.Feldman D, Avorn J, Kesselheim AS. Use of extrapolation in new drug approvals by the US Food and Drug Administration. JAMA Network Open. 2022;5(4):e227958-e227958. PubMed
36.Samuels S, Park K, Bhatt‐Mehta V, et al. Pediatric Efficacy Extrapolation in Drug Development Submitted to the US Food and Drug Administration 2015–2020. The Journal of Clinical Pharmacology. 2023;63(3):307-313. PubMed
37.Zemanick ET, Taylor-Cousar JL, Davies J, et al. A phase 3 open-label study of elexacaftor/tezacaftor/ivacaftor in children 6 through 11 years of age with cystic fibrosis and at least one F508del allele. Am J Respir Crit Care Med. 2021;203(12):1522-1532. PubMed
38.Shemie G, Nguyen MT, Wallenburg J, Ratjen F, Knoppers BM. The equitable implementation of cystic fibrosis personalized medicines in Canada. Journal of Personalized Medicine. 2021;11(5):382. PubMed
39.Dobra R, Davies G, Pike K, et al. Optimising equity of access: how should we allocate slots to the most competitive trials in Cystic Fibrosis (CF)? Journal of Cystic Fibrosis. 2021;20(6):978-985. PubMed
40.McGarry ME, McColley SA. Minorities are underrepresented in clinical trials of pharmaceutical agents for cystic fibrosis. Annals of the American Thoracic Society. 2016;13(10):1721-1725. PubMed
41.Kacetl J, Marešová P, Maskuriy R, Selamat A. Ethical questions linked to rare diseases and orphan drugs - a systematic review. Risk Manag Healthc Policy. 2020;13:2125-2148. PubMed
42.Walker S, Flume P, McNamara J, et al. A phase 3 study of tezacaftor in combination with ivacaftor in children aged 6 through 11 years with cystic fibrosis. J Cyst Fibros. 2019;18(5):708-713. PubMed
43.Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187(11):1219-1225. PubMed
44.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663-1672. PubMed
45.Aspinall SA, Mackintosh KA, Hill DM, Cope B, McNarry MA. Evaluating the effect of kaftrio on perspectives of health and wellbeing in individuals with cystic fibrosis. International Journal of Environmental Research and Public Health. 2022;19(10):1-13. PubMed
46.Wang A, Lee M, Keller A, et al. Sex differences in outcomes of people with cystic fibrosis treated with elexacaftor/tezacaftor/ivacaftor. Journal of Cystic Fibrosis. 2023. PubMed
47.Bailey J, Ladores S. Ethical Issues When Conducting Research in People with Cystic Fibrosis. The Nurse’s Role in Addressing Barriers to Cancer Screening of African Americans: An Integrative. 2018;8(1):48-52.
48.McGarry ME, Neuhaus JM, Nielson DW, Burchard E, Ly NP. Pulmonary function disparities exist and persist in Hispanic patients with cystic fibrosis: a longitudinal analysis. Pediatric Pulmonology. 2017;52(12):1550-1557. PubMed
49.Seear M, Amed S, Dionne J, et al. In support of point-of-care social needs screening: The effects of five social determinants on the health of children with chronic diseases in British Columbia. Paediatrics & Child Health. 2019;24(3):200-208. PubMed
50.Zampoli M, Morrow B, Paul G. Real-world disparities and ethical considerations with access to CFTR modulator drugs: Mind the gap! Frontiers in Pharmacology. 2023;14:1163391. PubMed
51.Wagner M, Goetghebeur MM, Ganache I, et al. HTA challenges for appraising rare disease interventions viewed through the lens of an institutional multidimensional value framework. Expert Rev Pharmacoecon Outcomes Res. 2022:1-10. PubMed
Appendix 1: Details of Included Publications
Note that this appendix has not been copy-edited.
Table 1: Details of Included Publications
First author (year) | Publication type | Objective | Key ethical considerations | Funding source |
|---|---|---|---|---|
Almulhem (2022)34 | Qualitative interview study | To explore the opinions of children with CF, their parents or caregivers, and health care professionals on the impact of ELX-TEZ-IVA, airway clearance techniques, and nebulized treatments. |
| Funded as part of a PhD studentship |
Aspinall (2022)45 | Qualitative interview study | To explore the effects Kaftrio has on an individual’s perception of reality and to what extent the modulator has changed their life beyond just the physical aspects. |
| Knowledge Economy Skills Scholarships (KESS) |
Bailey (2018)47 | Review | To explore the ethical issues associated with conducting research in the population of people with CF who experience health disparities, and to propose recommendations to address ethical issues. |
| None reported |
Dingus Keuhlen (2021)32 | Mixed methods study | To explore, analyze, and expand upon the lived experiences of caregivers raising children with CF and the connection to the systemic influences contributing to their elevated rates of psychological symptoms. | Caregivers experience elevated levels of anxiety and depression while raising their children (aged 0 to 6 years) with CF. | None reported |
Dobra (2021)39 | Survey study | To understand how the CF community thinks slots in competitive trials should be allocated across the UK and whether this should be driven by clinical need or patients’ engagement or adherence or if it should be random. | Inclusion of racialized populations in clinical trials is a crucial step toward addressing health inequities and facilitating better access to advancements in CF therapies because participation is often seen as a quicker path to accessing ELX-TEZ-IVA. | Cystic Fibrosis Trust through the Clinical Trials Accelerator Platform |
Dobra (2022)21 | Review | To explore why it is important to involve children in research. |
| National Institutes of Health Research through a predoctoral fellowship (NIHR300407) |
Guo (2022)28 | Economic analysis | To estimate the minimum costs of production of CFTR modulators, assuming robust generic competition, and to compare them with current list prices to evaluate the feasibility of increased global access to treatment. | Delays in accessing ELX-TEZ-IVA can cause tangible impacts on health outcomes. | None reported |
Hutchins (2022)19 | Survey study | To comparatively explore disease-specific experiences and cultural contributions for persons with CF from racialized populations. | Some racialized populations have significantly less knowledge about CF compared to their non-Hispanic Caucasian counterparts. | Georgia Association of Genetic Counselors student grant |
King (2022)27 | Review | To review the evidence from clinical trials and mounting real-world observational and registry data that demonstrates the impact highly effective modulators have on both pulmonary and extrapulmonary manifestations of CF. | Although drug development for patients who are ineligible for ELX-TEZ-IVA due to gene mutation requirements are reportedly ongoing, there are currently no modulator treatments clinically available for this population. | None reported |
McGarry (2016)40 | Review | To evaluate the representation of racialized populations in pharmacology clinical trials for CF. | Racialized populations are underrepresented in clinical trials of pharmaceutical agents for CF. | National Institute of General Medical Sciences grants 5T32GM007546 to 35 andUL1TR001422 |
McGarry (2017)48 | Review | To compare longitudinal pulmonary function between Hispanic and non-Hispanic white patients with CF. | Low socioeconomic status negatively impacts the social determinants of health of people living with CF. | Cystic Fibrosis Research Institute, National Institutes of Health, Cystic Fibrosis Foundation, and National Science Foundation |
Owusu (2020)17 | Retrospective case-controlled study | To describe and compare the presentation and outcomes of Black African children with CF to those with p.Phe508del genotype. | In the absence of newborn screening, Black African children with CF tend to receive a later diagnosis.
| African Paediatric Fellowship Programme, University of Cape Town, and National Research Foundation of South Africa |
Silva-Filho (2016)18 | Commentary | To assess the current CF situation in some Latin American countries and make suggestions for possible directions for future focus. | CF has historically been perceived as a condition primarily impacting individuals of Caucasian or European ancestry, but the disease is also found in other regions, such as the Middle East, Asia, and Latin America. | Novartis Pharma AG |
Stanojevic (2021)29 | Pharmacoeconomic study | To estimate the potential impact of ELX-TEZ-IVA on morbidity and mortality, and the impact of delayed access. | Delays in accessing ELX-TEZ-IVA can cause tangible impacts on health outcomes. | Cystic Fibrosis Canada |
Vaidyanathan (2022)15 | Database study | To characterize CF in Asian subgroups to address disparities such as delayed diagnosis and poor prognosis. | Newborn screening of CF is less effective among racialized populations. | Stanford University School of Medicine |
Wang (2023)46 | Observational longitudinal study | To examine the differences between sexes in pulmonary exacerbations, ppFEV1, BMI, and Pseudomonas aeruginosa before and after treatment with ELX-TEZ-IVA at a large CF centre in the US. | Males experienced a more significant decrease in pulmonary exacerbations than females following use of ELX-TEZ-IVA. | None reported |
Wright (2022)16 | Commentary | To discuss health disparities, reflecting on authors’ experiences in delayed diagnosis of CF based on race and bias in health care practitioners. | An African American man experienced a lifetime of distress, physical suffering, frequent hospital visits, multiple diagnoses, and inappropriate surgeries because of delayed diagnosis due to the belief that CF only affects non-Hispanic white people. | None reported |
Zampoli (2023)50 | Review | To highlight the current global inequity in access to CFTR drugs and its impact on widening disparities between high-income countries and low- and middle-income countries in CF outcomes and survival. |
| None reported |
BMI = body mass index; CF = cystic fibrosis; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ppFEV1 = percent predicted forced expiratory volume in the first second.
Stakeholder Input
Patient Input
Cystic Fibrosis Canada
About Cystic Fibrosis Canada
Since being founded by parents in 1960, Cystic Fibrosis Canada has grown into a leading organization with a central role engaging people living with cystic fibrosis, parents and caregivers, volunteers, researchers and healthcare professionals, government and donors. We have advanced research and care that has quadrupled life expectancy. We work together to change lives through treatment, research, information and support. Despite our progress we are not yet done. According to the Canadian Cystic Fibrosis Registry, now 50 years old, half of the people with cystic fibrosis who died over the past three years were younger than 37.1 years of age. A child born with cystic fibrosis in 2021 has only a 50% chance of living to 57.3 years. We will keep pushing, keep going further until all people with cystic fibrosis experience — and enjoy everything life has to offer.
Cystic Fibrosis Canada funds basic, discovery science and clinical research, and has helped establish core facilities across the country. We provide financial support to the forty multi-disciplinary cystic fibrosis clinics that see nearly all Canadians living with cystic fibrosis and maintain close relationships with the clinical and research communities. We have invested over $270M in research and clinical care support. The close relationships with the research, clinical and patient communities give us an excellent understanding the disease and how it impacts Canadians with CF, as well as those who serve them. We are the most respected and trusted source for information on cystic fibrosis in Canada and provide an information and resource service to the community that includes publishing a comprehensive resource compendium for the community. In addition, we maintain close relationships with our sister organizations around the world, which allow for the rapid sharing of information and adoption of best practices. In 2018, we launched the Cystic Fibrosis Canada Accelerating Clinical Trials (CF CanACT) network that now includes 10 of the 40 cystic fibrosis clinics serving over 60% of Canadians with cystic fibrosis. CF CanACT also works closely with our international partners to conduct protocol reviews, share Data Safety Monitoring Boards, and help speed clinical trial progress.
Cystic Fibrosis Canada manages the Canadian Cystic Fibrosis Registry (the Registry). The Registry contains the clinical information on nearly all Canadians with cystic fibrosis, living or deceased, with data going back to the 1970’s. The Registry publishes an annual report that describes the current status of the cystic fibrosis population in Canada and national trends over time. The data in the Registry is also used by investigators in Canada and around the world to better understand the disease and the impact of therapeutic efforts as well as to propose improvements to care. When the registry was established 50 years ago, the median age of survival for Canadians with CF was in the mid-teens. Largely due to multi-disciplinary care and access to standard of care drugs, half of Canadians with CF live to be almost 60 years old, but this could change if clinical care and access to CF medicines diminishes.
We work closely with our patient community to advocate to improve their health and well-being. Cystic Fibrosis Canada’s National Advocacy Network has over a hundred well-trained advocates and a basket of tools to help them in their efforts. We’ve been able to help the cystic fibrosis community by amplifying their voices through coordinated efforts that have addressed both national and regional priorities.
Cystic Fibrosis Canada’s contributions have led to significant improvements care and quality of life for people living with cystic fibrosis. As a result, Canada has one of the highest median ages of survival in the world.
Cystic Fibrosis Canada is pleased to provide patient group input to CADTH’s consideration of Trikafta for the treatment of cystic fibrosis (CF) in patients aged 2-5 years who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. We appreciate the consideration CADTH gave to our submission on the 12+ population and to our response and our clinicians and researchers' responses to the draft criteria. We are very pleased with the thoughtful approach the Common Drug Review (CDR) Committee took in reviewing the 6+ file, including accepting the outcomes of the Promise Study (Nichols, D. P. et al. Clinical Effectiveness of Elexacaftor/Tezacftor/Ivacaftor in People with Cystic Fibrosis. Am. J. Resp. Crit. Care (2021) doi:10.1164/rccm.202108- 1986oc.), which provided a compelling set of real-world evidence (RWE) that ultimately informed the CDR’s 2022 recommendation to eliminate the 90% or less lung function initiation criterion it recommended in 2021 for 12+. CF clinicians and researchers share this sentiment. Collectively, we look forward to providing CADTH with a suite of submissions for the 2-5 year old population in Canada to help guide CDEC’s deliberations to ensure the broadest access possible for this life-changing therapy.
Information Gathering
In completing this submission, Cystic Fibrosis Canada conducted focus groups with six parents of children with cystic fibrosis under five years of age who have at least one copy of the F508del mutation. We also utilized findings from our 2021 patient and caregiver survey on access to Trikafta, which received over 1200 responses from our community. We used data from the now 50 year old Canadian Cystic Fibrosis Registry, as well as preliminary findings from Phase I of The Social and Economic Impact of Cystic Fibrosis in Canada: A Burden of Disease Study (herein called “Burden of Disease Study”) conducted by Dalhousie University and Cystic Fibrosis Canada, in partnership with the Conference Board of Canada and funded in part by the Canadian Institutes in Health Research (CIHR). Through this study we conducted a community survey just before Trikafta became widely available in Canada.
Where permission was granted, survey findings were linked to individual registry records, which allows us to stratify data across disease severity, geographic location, age, sex, and other informative data points. Yet to be published, the study measures the burden of CF at the individual, family, health systems and societal levels. It is said to be one of the most comprehensive studies of the burden of CF in the world. Phase II and Phase III follow up studies are in the planning stages and will provide data that demonstrate the impact Trikafta has had on the Canadian CF population.
We also provide insights from relevant medical and scientific publications and perspectives that speak to the health and health related challenges that the 2-5 year old cohort face.
Disease Experience
Cystic fibrosis (CF) is the most common fatal genetic disease affecting children and young adults in Canada. There is no cure. The incidence of CF in Canada is approximately 1/3600 live births. According to the Canadian Cystic Fibrosis Registry, there are approximately 4300 Canadians who live with cystic fibrosis, among which are about 359 children aged 2- to 5- year olds, as of 2021. Approximately 314 of these children have at least one copy of the F508del mutation, making them eligible for Trikafta pending Health Canada approval.
Cystic fibrosis is a complex, progressive, degenerative disease caused by mutations in the gene for the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). There are over 2,090 known mutations. Cystic fibrosis has a tremendous impact on the people who live with it, their loved ones, on our health systems and on society. Every week in Canada, two people are diagnosed with cystic fibrosis, one of them through newborn screening. Every week in Canada, one person with cystic fibrosis will die.
Cystic fibrosis causes various effects on the body, but mainly it affects the digestive system and lungs. The clinical progression of cystic fibrosis can vary greatly from person to person, even with the same mutations. Ultimately, the most significant clinical impact is in the lungs, where patients have difficulty in clearing secretions, which in combination, with aberrant inflammation leads to persistent infections with cycles of inflammation that are ineffective in clearing infections. This leads to progressive scarring of the airways and a progressive and sometimes rapid decline in lung function. Pulmonary/ infection/ cardiovascular complications cause eighty percent of cystic fibrosis fatalities. (The 2021 Annual Data Report of the Canadian Cystic Fibrosis Registry. www.cysticfibrosis.ca/registry/2021AnnualDataReport.pdf (2022).
Thanks to significant progress in treatment and care, most children with cystic fibrosis will reach adulthood. The estimated median survival of Canadians with cystic fibrosis in 2021 was 57.3 years of age. (The 2021 Annual Data Report of the Canadian Cystic Fibrosis Registry. www.cysticfibrosis.ca/registry/2021AnnualDataReport.pdf (2022). There were no deaths amongst 2-5 year old in 2021. In their submission to CADTH for 2-5 year old with CF, the Canadian Cystic Fibrosis Clinicians group noted that:
“CF clinical care has changed enormously in the time since highly effective CFTR modulator treatments (HEMT) have been available for people with CF who have Cystic Fibrosis Transmembrane Regulator (CFTR) mutations that are amenable to this class of medications and w. People with CF (pwCF) are reporting to clinicians that they feel better, and we are noticing in our clinics that most patients show an improvement or stabilization in lung function, hospitalizations are decreasing, the number of lung transplants for CF has plummeted, and projected life expectancy is increasing. However, CF is far from cured. Treatment of CF continues to include a multitude of treatments, including dietary modifications, medications, and airway clearance treatments.”
“However, without the promise that being on HEMTs brings, as young children grow older with cystic fibrosis, they may suffer from pulmonary exacerbations (PEx, flares of lung disease) requiring weeks-to-months of treatment with antibiotics and often requiring hospitalization and I.V. antibiotics. PEx cause rapid decline of lung function and more rapid disease progression and are associated with a greater risk of death. Other consequences of having cystic fibrosis include malnutrition and very low BMI, and cystic fibrosis-related comorbidities like cystic fibrosis-related diabetes (CFRD) and cystic fibrosis-related liver disease.”
Malnutrition and low BMI – or failure to thrive – is common among children with CF who are 2-5 years old. In fact, these symptoms often appear before birth (in utero), at or shortly after birth. Children who are affected by these symptoms largely fall into smaller percentiles in terms of weight and height when compared to their peers who don’t have CF. According to a 2023 study by Mariotti Zani (Mariotti Zani E, Grandinetti R, Cunico D, Torelli L, Fainardi V, Pisi G, Esposito S. Nutritional Care in Children with Cystic Fibrosis. Nutrients. 2023 Jan 17;15(3):479. doi: 10.3390/nu15030479. PMID: 36771186; PMCID: PMC9921127.) et al:
There is a strong association between nutritional status and clinical outcomes in CF patients. In children with CF, malnutrition and poor lung function are the main sources of morbidity. Previous studies found a link between fat absorption and pulmonary outcomes: children who had steatorrhea had more severe symptoms, a higher sweat chloride and an overall worse prognosis than children who had a normal fat absorption [15,16]. The respiratory status and survival in children and adults inversely correlate with the degree of malnutrition, including short stature, lower weight and body mass index (BMI) [17,18]. Lung function at older ages is also associated with the nutritional status in toddlerhood and childhood [19,20,21,22]. Ashkenazi et al. found that a BMI z-score < −0.75 at 10 years of age was associated with a higher rate of lung transplantation in adulthood [22].
One of the first systems that shows symptoms of CF-related disease is often the gastrointestinal tract (GI). The GI symptoms, in fact, can be present even in utero or precede the pulmonary symptoms. In the association between malnutrition and CF, several factors are involved: a lower energy intake, greater energy expenditure, higher essential fatty acid (EFA) turnover, endocrine and exocrine PI (pancreatic insufficiency), enteric inflammation, bacterial overgrowth and impaired bicarbonate secretion… In patients with CF, the primary mechanism of malabsorption is the PI. Approximately 85% of patients with CF develop PI by the age of one [29]. The ductal obstruction is predisposed by the thickened secretions caused by impaired chloride and bicarbonate secretion. The obstructions in the pancreatic ducts damage the transport of the digestive enzymes and pancreatic bicarbonate to the intestinal lumen.
All of the parents we spoke to in our focus groups said that their children had GI issues, and some noted PI and lung function concerns as well:
“(We had) issues with weight and then her liver enzymes have always kind of been elevated in blood work. So obviously there's always a concern of if she's going to have liver disease later on and then, you know, the whole component of the lungs and our ability to control (progression of the disease).” — Parent of a 2-5 year old with CF
“(I worry about) further lung deterioration for them…to me it's all about prevention. She had a clear chess X-ray a year ago but who knows what is gonna show this year, right? It's CF, it can just pop up at any time you know.” — Parent of 2-5 year old child with CF
CF also has a significant impact on socialization, mental health, and isolation. Research has shown that patients with chronic diseases (defined as a condition that persists for longer than three months) can often have anxiety and depression. It is estimated that up to one third of individuals with a serious medical condition will experience depression. Depression is one of the most common complications of chronic illness like cystic fibrosis, and it also affects caregivers. (Duff AJ. Depression in cystic fibrosis; Implications of The International Depression/Anxiety Epidemiological Study (TIDES) in cystic fibrosis. Paediatr Respir Rev. 2015 Oct;16 Suppl 1:2-5. doi: 10.1016/j.prrv.2015.07.006. Epub 2015 Sep 26. PMID: 26410281.) This was heard loud and clear in our focus groups with parents of children 2-5 years of age with CF:
“And then just to touch on the mental health piece… I've been diagnosed with PTSD and anxiety. Like it's just it's definitely been a toll on all of us.” — Mom of a 2-5 year old child with CF
“Our four-year-old grandson has missed out in so much of his life that he deserves more childhood instead of all the time the medications and therapies take away.” — Grandparent of a child with CF
Experiences With Currently Available Treatments
CFTR modulators have been developed to tackle the underlying defect of CF. Although not a cure, these medications partially restore the function of the CFTR protein, a chloride and bicarbonate channel, at the cell surface. CFTR modulators are tailored to correct specific mutations and are an example of precision (personalized) medicine. Correction of CFTR protein function at an early age is congruent with the overall preventative paradigm of CF treatment – early correction will hopefully prevent disease progression and irreversible damage.
All jurisdictions now screen for CF as part of their newborn screening panels, to ensure that infants with cystic fibrosis can start treatment as soon as possible, including starting CFTR modulators such as Kalydeco and Orkambi at the age of two, which many jurisdictions now fund. If a newborn screens positive or inconclusive, the family is referred to a CF clinic, where a sweat chloride test is administered. If the results of the sweat chloride test are above or equal to 60 mmol/L a diagnosis of CF is confirmed.
“I got a phone call from my midwife who just said the newborn screen came back and the results are concerning. I need to talk to you immediately and you need to tell your partner to come home. Luckily my parents happened to be there, so I had people around me. I called my partner and then (our midwife) came over an hour later and told us and (it was) the worst (news): it's very likely that he has cystic fibrosis. I remember that weekend Googling: What are the odds of a mistake on the newborn screen? But they're not very high… then we found out on a Friday and then Monday we had an appointment with the CF clinic here where they did the sweat test and gave us enzymes. I kind of new once they told me certain things like you can taste the salt on your little baby, (which) was true and he wasn't gaining weight and thriving. And they thought it was something to do with (breast milk) supply, right. But my supply was actually pretty good. And so once I started giving him enzymes, like, it was an immediate change from to his poops and also to his comfort… he did have a couple of, like really screamy two first weeks where like he was in stomach pain, I assume. And so, yeah, it changed, I would say like immediately (once he started taking enzymes). So even before the sweat test came back, I was very confident that this was the thing.” — Parent of a child with CF
A typical day in the life of a family with a young child with CF is challenging. Even at the young age of 2 to 5 years, the treatment regime is relentless. There are no days off when it comes to managing CF.
“She wakes up, usually around 7:00 am. (We) get her up, she's still on a bottle, she just won't cut it, so (we give her) Creon and MVW vitamins and vitamin D with that first bottle breakfast. And then we do her 30 minutes of percussions at around 10:00. And then it's snack time. She gets another dose of Creon. Lunch, more Creon. Then it's nap time. She wakes up, then another snack with enzymes, supper with enzymes, and then Ventolin at night. Sorry I forgot to mention Ventolin. I do that at the same time as physio in the morning. And then another dose of Creon with her night-time bottle. That's our routine. And obviously throughout the day she's running around, she's bouncing on the trampoline and she's blowing bubbles. Like I just do anything to keep her active. But she doesn't sit still anyway.” — Parent of a child with CF
She's allowed four (doses of Creon) per day and it's good for two hours, so we are just constantly doing that. So, you give her milk in the morning, you've got two hours to fit in breakfast. If she doesn't want to eat, we're carrying fruit around then until she's allowed to have her next Creon dose. So, then she has that again with lunch. That's usually when we give her supplements and whatnot, and then we do our 25 minutes of physio before her nap, and then she wakes up, we do another Creon dose for a snack and keep a close eye on the clock again for two hours, just in case supper starts early or something like that. And try to make sure that we don't forget…that's our biggest problem right now is sometimes supper is 15 to 30 minutes earlier than her Creon is expired. And so, we have to keep it there and keep a timer on our phone to make sure that we're giving it to her if she's still eating, if that makes sense. — Parent of a child with CF
Our issues have mostly been digestive. Not mostly, they've been entirely digestive. Yeah, fortunately she's like a 95-percentile kid. The enzymes are absolutely working for her. Her pancreatic test is still coming in above 200, so we're very thankful about that. But her liver enzymes are very elevated, and the fear of liver disease is really clear… but like the others said it's all about prevention. — Parent of a 2-5 year old child with CF
We are doing twice a day physio at this point. Thankfully physio goes really, really well. We've always maintained an extreme routine though since she was born it is that the exact time of day at the exact. It's before naps and she associates it with cuddle time and asks for it. Thank God. And otherwise, she is on Creon before every meal. She's on extra vitamins. We have dealt with other medications in the past such as reflux meds. — Parent of a 2-5 year old child with CF
I honestly think that it does play a pretty significant role in in her and the entire families day-to-day life. She has not had the healthiest year, year and a half or so the last the last little while she's been admitted to hospital twice in the last year…she's been on oral antibiotics, I want to say four or five times in in the last year. We've had a number of medication changes. We've had some pretty big changes to her, her daily maintenance treatments and things like that, and she's starting to notice that she's in (junior kindergarten) this year and she had a pretty significant illness. That ran from about mid-October through until January until we found a different combination of medications and it actually involved us having to go in [inpatient care in the hospital] for about a three-week period, and (she does) treatments in school throughout her school day. So even like her first year of school has been impacted by…CF, like what she deals with on a day-to-day basis. — Parent of a child with CF
It’s pretty much like a full time job taking care of baby or toddler. With CF, you know it's enzymes before every meal. — Parent of a 2-5 year old with CF
Physiotherapy, like chest percussions, were a huge struggle probably till about age one and a half when she just started taking our phones and we were able to actually finish it all within that 30-minute block. Otherwise, she does take Creon as well, which she takes good with her applesauce. But again, we're always tweaking the doses because she does end up with oily stools or constipation of one kind or the other. — Parent of a 0–5-year-old with CF
We just started Ventolin, which she has a really hard time with the mask on. She is not a fan. Same thing. We do MVW vitamins and vitamin D drops and that's about it for treatment for now. I know that we'll start nebulizers when she's about 3 or if she were to culture something, but she's always cultured a small amount of. Staph. and they've left it untreated since she's asymptomatic. — Parent of 2-5 year old who lives with CF
A huge one was Creon when she was a newborn. She had the nastiest diaper rash I've ever seen on a baby, and it wasn't even a diaper rash anymore. It was down to the…the skin was torn, she was bleeding. And it was months of this until we met with a wound care specialist who was able to put in like a treatment plan for us, but we couldn't stop the Creon obviously, you know? So, I would say that was the hardest one for her for sure. — Parent of a 2-5 year old with CF
Just constant, constant discontent, constant crying. We knew that it was Creon that was doing it, but we couldn't stop, so we had to continue to push through. And thankfully things have settled. We're still pretty sure she gets frequent stomach aches, but honestly, she's lived with that pain since she was 14 days old, and so I think she's just gotten really used to that. — Parent of 2-5 year old child with CF
Moreover, from a socialization perspective, it can be challenging for both the child and the family as the child starts to understand that they live with CF and that they are “different” than other children their age:
“She has a twin sister, and a younger sister (who) are both very active and she's starting to notice that you know, she has to sit still and have these treatments done a few times a day. And her sisters are around doing whatever they want to do. So, she's probably not at the point where most five-year-olds would be. With the realization she is (different), she (also has) an autism diagnosis, so she's…a little less aware of what everybody else goes through and that she is a bit different, but she's starting to make some of those connections. So, I think it's having a pretty significant impact.” — Parent of 2-5 year old with CF
“I guess like in the sense of like he already sort of has to spend time taking care of his health in a way that's unlike other kids his age. Not all lots of kids his age have to take care of their health in other ways too. But for the sort of the typical kid, they don't have to spend an hour a day doing like a sort of treatment like that. So, time, I guess, is the biggest thing. And then he does not really know that (there are dangers) when the weather changes and stuff but like in the winter and when illness is just like rampant at daycare and in my house, it's there's like a baseline cough that is ever present, and that scares me.” — Parent of 2-5 year old with CF
“When we didn't know or didn't understand as much, like I, I had a cold and I had to be masked up pre COVID, isolated in the house for, you know, till symptoms were clear because I that information wasn't communicated well, whether it was from me, from our team or from, you know, even CF Canada, sometimes that information's not clear on what you need to do sort things out, like, is how is a cold going to kill him or is the cold just going to be a minor inconvenience sort of thing and that that information's not always available sort of thing or easy to easy to handle, and we had to isolate ourselves for long periods at a time.” — Parent of a 2-5 year old with CF
“Having a child with cystic fibrosis, (there are) just (so many) sleepless nights. Absolutely. Worrying about (your child’s health), you're worrying if he's going be able to cough, if he got coughs through the whole night, or what's going to happen and those sorts of things. Absolutely…the bigger impact is on the parent, I think.” — Parent of a 2-5 year old with CF
“I mean, it helps that he hasn't had any major health, adverse health things. You feel like you can be a bit imperfect. But yeah, so that that that piece of advice I think was pretty powerful that sometimes the experience is more important. And so, yeah, like it's important to have a sleepover or to try anyways. And so, it won't mean he'll get like his lung physio later on in It can really have a negative impact on your mental health. It's hard. I'm constantly. Worrying about what I'm hearing like, oh, is that a wheeze? Is that a cough? What's going on with this? Uh, am I doing enough? Like is the physio I'm doing enough? Is, um, like? Did I hear that dosage change correctly like things like that? If you're always doubting yourself because it can have such a huge, like such huge repercussions on everything for her. If I make one little mistake like it's, it's a lot of pressure. And it's, uh, I don't know, it's just a lot.” — Parent of a 2-5 year old with CF
“Disregarding some of the old advice, like spending time with other people who have CF, like to me like the thought that he'll never get to like, play with another kid with CF like that, I don't know why that just makes me really emotional because it's just like our experiences, shared experiences, are so important. And so yeah, if it could like, mean that that piece of medical advice changed that, like, oh yeah, you can like in an outdoor setting, you can spend time with someone else with CF or like, you can do that. I think that that would be really powerful of change in his life.”
“If it meant that, like, it reduced the number of hours he needs to spend in a week doing like, because he'll transition away from us doing lung physio to like the nebulizers and stuff like that in the next year or two. And so, if it meant that it reduced the amount of time, he's doing those things like that would feel like a real gift. If it meant that, you know, he could only like.” — Parent of a 2-5 year old with CF
As the disease advances more time and effort are needed to manage the progressive and debilitating symptoms. Children with cystic fibrosis may need to quit school or go part-time, and their parents may need to leave the work force or undertake part-time work. Our Burden of Disease Study revealed that:
Almost 80% of caregivers were employed; of these 75% were employed full-time.
Many caregivers reduce their hours of work, could not work, or took less demanding jobs to balance caregiving responsibilities.
Inability to work resulted in the loss of health insurance benefits, much needed to afford medications. Due to the nature and volume of treatments and medicines, CF is an expensive disease.
These findings align with what we heard in our focus groups:
When two of my children were first diagnosed, the doctor told me I’d never go back to work again. It is a full-time job keeping my children healthy. From helping with their physio to clear mucus, frequent CF clinic visits, hospital stays, and on top of that ensuring our third child does not feel left out as a healthy child. — Parent of a child with CF
Yeah, he's definitely been passed up for work, passed up for promotions. I myself have had to go down to three days a week because of childcare, finding childcare that can properly take care of (our daughter) has been a major struggle. So yeah, we can't afford five days a week of one-on-one care, nanny care, which is really our only option right now. — Parent of a 2-5 year old with CF
So, I would say the biggest impact is work. You know, like we both have to take time off of work. And my husband owns his own business. So, he's actually needed to kind of like take a bit of a step back from that and like work for an employer so that he can actually, you know, get some benefits out of that and get some vacation time, whatnot, because obviously that's not a reality with self-employment. So that's been like one of the bigger impacts I would say. — Parent of a 2-5 year old with CF
Our Burden of Disease Study also found that CF is an expensive disease and that travel to clinic puts a significant strain on families, noting that the average out of pocket expenses paid per clinic visit is $238. Most CF patients visit clinic four times a year, meaning that it costs almost $1000 out of pocket a year to go to clinic. There are few resources available to offset these costs. Moreover:
People with CF typically attend 4 CF clinic visits a year. On average, people with CF and their caregivers travel 42 Km (range: 13-110 Km) to a CF clinic; 9% (32) of participants live more than 300 Km from a CF clinic; a small but important proportion (5.4%, 19) spend more than a day travelling to and from the CF clinic and require accommodations in addition to travel costs.
In addition to travel costs and time required to attend CF clinic there are several out-of-pocket costs that are paid by people with CF and/or their caregivers, including meals (86.5%, 83), childcare (37.8%, 28), and parking (94% (327).
Again, what we heard in our focus groups supported these findings.
“I think the most assistance that we really need is just financial assistance is definitely needed, because a lot of things are not covered. The CF drug program does help, but there are a lot of things that come out of pocket and until I was able to start working full time and it was pretty tight. So that that assistance is needed, but I think there's also a lot needed for the parents.” — Parent of a 2-5 year old with CF
“We live about 200km from clinic…it takes us takes us almost two hours to get to SickKids and (then) it's just the parking fee, which is $20, which is fine, but that's about 400 kilometers both ways like round trip well that's because of traffic, timing is (key). So, if (we) go at 9:30, it's literally like two plus hours. So, we're leaving at like 6:00 AM. To get down there and park…I've requested a 10:30 appointment at the earliest which they've accommodated now. So at least it's only been an hour and a half. We make lunch to save money, so the trip costs about $50 each way… (up to $125 per visit), which was every six weeks in the beginning.” — Parent of a child with CF
“(Getting a diagnosis) through the genetic screening…normally takes six weeks (or so). Actually, it was a log logging back and forth to the hospital and we were like over 200 kilometers away from (clinic). It was a full trek…leaving at 8:00 AM and back at like 7:00 PM so, so that would be 400 kilometers both ways round trip. So, we do all the tax applications for that and those sorts of things. But yeah, it's a long ordeal.” — Parent of a child with CF
“…and round trip we're talking at least 13 hours of travel (to clinic), close to 1200 kilometers. So, it's a three-day adventure every single time. Fortunately, our clinic is very understanding of how difficult that is, so we do as many zoom calls as possible, but they insist on us seeing us even when she doesn't require any extra testing or anything. They require us to be there for conversations with the whole team. So, we do go there often. And so, I mean all in, that's three days of meals, which is what like a for a family, I don't know, close to $100 a day and then plus fuel, I mean you're talking I don't know, ($700-$800 per trip) probably (each way), four times a year. And then I mean, obviously we have to take off work for that, both of us.” — Parent of a child with CF
CF takes a significant toll on caregivers mental and physical health.
“I had really bad bronchitis recently and even just that feeling of struggling for breath, like imagining him experiencing that or like having to go through a lung transplant, Like, I think that's the scariest part for me for sure. Like and it's the part that you can't really see or control quite so easily. Like there's no way for him at this young age he doesn't get li We did do one called like a multiple breath washout. I'm sure you know what that is but we did it once in Vancouver when we were there because the people running that were can be trial. We're doing this other trial, and he has really good lung function, but we don't get to check that often. Whereas like with the digestive stuff it's like an immediate like you know if he needs more enzymes because his bowel movements are crampy, and the poops look different and. Or he stops gaining weight or something. You can kind of assess that quite easily. Or like, yeah, so it's the lung stuff that you can't see. And it's like that scares me. And I guess the other stuff that scares me is like the fact that there's like stuff that they don't always, they don't really know. Well, maybe they do know, but I don't fully know like why some people develop like CF related diabetes or CF related liver disease, like those kinds of unknowns that are just gonna show up later on for him in life and or may not.ke regular lung function testing done because he he's too young.” — Parent of a 2-5 year old who lives with CF
“I was actually formally diagnosed with PTSD and anxiety. So, I am taking medication for that. But for a while, like, it was a lot of, like, nightmares. And I would actually have dreams about Ella being in, like Hospice and palliative care or her needing a lung transplant at this young age, you know, like. And then there was just like flashbacks of back to like when she was initially diagnosed. It was just a really, really hard time. You know, lots of tears and anger really like why us and stuff like that. So, I do seek support from like a social worker every week we're doing like cognitive behavioral therapy. And then I see a psychiatrist on top of that just for the medication piece. So definitely a huge emotional impact on my side. Andrea, Tanya, mine kind of goes through waves sometimes I'm just. Perfectly content with our life. Like, OK, we've got this covered.”
“Everything's going really well. And then it's like, oh, no, am I actually doing enough? Should I have not done this with him? Our CF teams, mottos kind of like just live life like normal and you know, don't hide him from things. So, then I've kind of been living that way, but then I'll just get smacked in the face with all of this guilt and it's like, was that right? Like I. Just casually watching the good doctor and the CF kids are on there. I'm like, oh my God, why, why am I watching this show?”
“So of course, it's mentally difficult, but I'm just the type of person that kind of rolls with the punches. So, we'll see how it goes day, day by day.” — Parent of a 2-5 year old with CF
“I too have guilt (and see a counsellor), but if you look at the statistics, Canada has the greatest life expectancy rate. And so, I think we're doing things right up here and I'm really grateful that we've got a team that just says, you know, live life and if something happens, we will. We will tackle it together and but believe me, the guilt, the fear, the fear, the unknowing absolutely gets to me sometimes and I'm grateful for my husband. You always bring me back down to earth and helps me, helps to remind me that she's just a normal kid.” — Parent of a 2-5 year old with CF
“Yeah, like we just have sad moments sometimes with when it's a clinic day or like when he's had just like a really harsh sounding cough for a long time. And the thing about Sasha is like when he gets sick, he just like does not really want to stop or slow down and so he's actually really hard sometimes tell are you sick or is this just like a lingering cough or is what's going on. But he will like sometimes cough. So just it sounds rough. And I know he's kind of used to it. But like, I think about when I have those coughs, it's actually like kind of exhausting on your body to be coughing like that. So, hearing that can be something a little bit, like can kind of make my body tense up or recoil a bit sometimes. And then yeah, just like, you know, you have all the typical worries of a parent. Like, I'm a high school teacher, so I work with teenagers, so, you know, I'm just like, oh my God, what happens when all his friends are vaping and, like, he better not freaking ever. You know, like just this stuff where I'm just like, you actually have to be a smarter teenager. And like, that's a really big responsibility for a teenager because they're often make very bad decisions. And so, yeah, he, like, has less room for error in terms of making bad decisions. – Parent of a 2-5 year old with CF
As alluded to in the previous section, cystic fibrosis also takes a toll on the mental health of children who live with it and their caregivers:
“(Yes, CF can be isolating)… we absolutely relate to just not being as close to certain family members or friends who just don't understand what we're dealing with…it makes me think of when I flew up to see my brother and sister-in-law and their kids last summer. And it was the first and last time I'll ever do that, because…no one asked about how she was doing. No one cared that we had to space out our meals in a certain way. They ate without us because I couldn't feed her yet. We had to eat all by herself 45 minutes later, and then when she finally went to bed after a tummy ache and dealing with that, my brother said, so what happens if you just don't give her Creon? Like, who cares? And so, I cried, and we came home early. We got on an early flight and so it absolutely impacts our family. It was months before we spoke. Again, just you flip flops between one family (member) that treats us extremely differently because we have a child with this disease, and they tiptoe around everything and absolutely every conversation has to be about our child's health and then another family member pretends that it doesn't exist and it's not happening.” — Parent of a 2-5 year old with CF
“She's become a lot more resilient than I ever really noticed. But I find that that's really unfair because she shouldn't have to. It's not the forefront of her mind that you know what I'm different and this is being done because of me. But I think we're getting there and I'm getting worried about the impact that it's going to have on her mental health and her self-image.” — Parent of a 2-5 year old with cystic fibrosis
Cystic fibrosis also affects family dynamics and planning:
“How does it impact my social life? I mean, we don't have babysitters at all. We wouldn't. Other than our nanny, who is trained for this, for treating children with special circumstances other than her, we don't really trust very many people to give her any medication. We have taught the grandparents, but they're very, very nervous. So, it doesn't instill a lot of confidence in us, but we'll do it if we have to. But it just doesn't happen. We just don't leave. We just don't leave her. And (when it comes to) family planning and how it's so heavily impacted that even just in my own marriage, you know, one of us wanting another child, one of us saying like, no, we've got our hands full with this one.” — Parent of a 2-5 year old with CF
“We would have to do IVF in order to conceive another child due to our genetic circumstances, and obviously that's pretty tough. That's a pretty tough, huge decision between two people and really hard on our marriage. But we have come to a conclusion (to not have anymore children) that I think is right for our family and right for our marriage, but that doesn't make it easy.” — Parent of a 2-5 year old with CF
“I needed to go through a divorce because of it basically, right. Mmhmm. Just because of conflicting views on (care), (we had) like completely polar views on this. Right. So that's the biggest one and I think it's probably more common than not… that's probably the biggest challenge obviously.” — Parent of a child with CF
“Having a child with cystic fibrosis. Oh for sure. Yeah. Like just sleepless nights on it. Absolutely. Worrying about and stuff like that. You're worrying if he's gonna you know is he gonna be able to cough if he got coughs through the whole night or what's gonna happen and those sorts of things. Absolutely. That's probably the bigger impact is on the parent I think if he. Yeah. No absolutely. What's the day like for him does he understand he has to stick fibrosis or like how to not he. No, he's no he's no clue.”
“I feel like we're pretty similar in the sense of like my relationship with my in-laws and my brother and sister-in-law. It's pretty much like inexistent at this point because they refuse to acknowledge CF, you know, and it's and it's strange like looking at their dynamics with my 4-year-old versus (my daughter with CF). Like they kind of like act like (my daughter with CF) doesn't exist and it's really strange because she's a human being, you know she's not her CF. So, like our social circle has a very has gotten very small as a result of this. We're pretty much just around my parents now because they take the time to, you know, do extra cleaning around the house, do the things that, like, would make me feel comfortable. And then, you know, touching on family planning, Same for us. You know, my husband is actually scheduled to go for a vasectomy next week.”
“Sorry, there's too much information. But, yeah, we're we don't want to take the risk. Like I love my (daughter with CF) and I always want her here. I just I would have so much guilt knowing we have we're carrying the gene and to pass it on knowingly, I just I don't know I couldn't (do that) in terms of quality of life. I feel like it's definitely (made) an impact because we try to isolate ourselves a lot. Like I don't want to unnecessarily put her in a situation where she is going to catch anything. Like you know our cold and flu season was so bad we didn’t do much to be honest. And as a result of that, she is so shy, like painfully shy. She cries around everybody. And it's just that I think that's had an impact on all of us and even my older daughter. Not being able to socialize like her being around children is just like this magical new thing because she doesn't know what that's like.” — Parent of a 2-5 year old with CF
“Prioritizing the experience for everybody and like being less than perfect and how you do everything. And so I'm also like a little bit more relaxed over like if someone gives him enzymes and they, you know, spill a bunch and he doesn't get the full dose, like it's not gonna kill him, right? And so like, I, as I've sort of grown in in this experience like just being okay with like things being imperfect sometimes.” — Parent of a 2-5 year old with CF
Improved Outcomes
Parents want their children to live long, healthy and full lives. More than anything they just want them be regular children. Cystic fibrosis steals many things from people and families. Restful sleep is broken up by coughing fits. Time with friends and loved ones is second to daily physiotherapy and treatment routines. School and work are interrupted by frequent infections and subsequent hospital stays. Family dynamics may be strained by the stress and anxiety of managing a chronic illness. Dreams about the future are clouded by heavy realities of a fatal, rare disease.
But there is hope. There are highly effective medicines that treat the basic defect of cystic fibrosis (CF) rather than just the symptoms and that significantly improve the health outcomes and quality of life for many people with cystic fibrosis. These therapies offer a changed reality for people living with the disease including children 6 years of age and older. Based experience in the 6+ population, Trikafta offers parents a significant amount of hope for their children’s future. The U.S. Food and Drug Agency (FDA), recently approved Trikafta – the single greatest innovation in the history of cystic fibrosis - for 2-5 year old who have one copy of the F508del mutation or certain mutations that are responsive to Trikafta based on lab data. Canada does not yet accept predictive precision medicine data and should ensure that all who can benefit from Trikafta can.
In its submission to CADTH for this cohort, our clinical trials network (CF CanACT) noted:
Elexacaftor/tezacaftor/ivacaftor (ELX/TEZ/IVA; Trikafta™). In vitro, this medication effectively recovers CFTR function in cells with one or two copies of the F508del mutation. It is important to note that ELX/TEZ/IVA is indicated for people with one copy of F508del in the targeted population, whereas two existing therapies – Orkambi and Symdeko – were only indicated for people with two copies of this mutation. This group (one or two copies of F508del) includes ~90% of Canadians with CF, so this represents a significant expansion of the population that is genetically eligible for this class of medications.
In clinical trials, this medication is also extremely effective clinically in people with CF with one or two copies of F508del and has been approved by Health Canada for those age 6 and up. Further, previous CADTH reviews have recommended funding of ELX/TEZ/IVA for those age 6 years and up based on the published efficacy data in both randomized and open label studies – effectiveness/efficacy outcomes assessed in these studies included lung function indices, nutritional indices, quality of life indices, rates of pulmonary exacerbations and sweat chloride concentration.
The next step in the progression of CFTR-modulator-based treatment for Canadian children with CF is the data shown in the recently published study looking at the safety and effectiveness of ELX/TEZ/IVA in children aged 2-5 years2. This study further extends the good safety profile and effectiveness of this medication into this younger age range. Results from this study, showed a decrease in sweat chloride concentration of −57.9 mmol/ with 90% of study participants normalizing their sweat chloride levels to below the range for a diagnosis of CF, absolute change in BMI-for-age z-score was 0.10 (95% CI, 0.00 to 0.20). Lung function as measured by LCI2.5 is a sensitive measure of ventilation inhomogeneity, where increases in Lung clearance Index (LCI2.5) indicate worsening of lung function, in this study the LCI had a positive fall by −0.83 units (95% CI, −1.01 to −0.66). Lastly mean (SD) absolute change in fecal elastase-1 concentration which is a marker of pancreatic insufficiency was 39.5(89.2) μg/g. All these outcome measures demonstrated that ELX/TEZ/IVA led to improvements in CFTR function and lung function and was associated with stable nutritional status over a 24-week treatment period.
Extending access to this medication to this age range would be congruent with the secondary prevention paradigm of CF care and would hopefully result in decreasing the long-term burden of disease for children (and eventually adults) with CF if treatment is initiated early. An ideal treatment in CF would fully address the basic molecular defect in CF and restore normal chloride transport on the cell surface. If applied at an early enough date, complete and early correction of CFTR function would prevent the multisystem downstream effects that are ultimately fatal for this group. Work continues to develop tools to completely correct CFTR function (this may be gene therapy, or perfected small molecule interventions). ELX/TEZ/IVA is the latest “third generation” CFTR modulator. In vitro, this medication provides the greatest restoration of CFTR function observed to date.
Among the parents who participated in our focus groups, whose children have at least one copy of the F508del, like any other parents, they want their children to have hopes and dreams and that their children can live long enough and well enough to pursue them.
“I think it will just affect obviously his day-to-day life and confidence as a person as he gets older. For us it just represents so much hope for the future. There are so many things that people with cystic fibrosis were unable to do, were unable to do easily. In the past and I just really, we just really hope that this just offers her the opportunity to do anything she wants to do in terms of careers, dreams, aspirations, all of the things as well as again, yeah, what Andrea said. Peace of Mind for us so that when she does get a symptom, we're not watching her. Like a hawk to make sure that it doesn't progress from there, you know in in that we don't end up in the hospital or something like that. We think it's just that sense of normalcy that we crave so badly and her digestive issues, we really hope that it helps with that.” — Parent of a 2-5 year old with CF
“I've heard that people like, you know, their sweat tests are even less elevated. Like Alice was like 99. And like if we could get that down to like a moderate range, like I would be so happy and like if I just feel like if we could just like they said it prevent illness. Like, you know, I'm. I'm trying to bubble wrap her kind of right now. If I could just let her live freely and not risk, you know, there's Pseudomonas all over the place. Like, I I don't want to have her live like that. And at the same time, like, I dread having that conversation about like, you know, your life is limited because of this illness. I don't want to tell her, you know, the median is 57 years old. I want her to know that she can Live her life to the fullest. That's the biggest thing.” — Parent of a 2-5 year old with CF
“You know I feel like some people you end up sort of like with pill fatigue when you're swallowing so many pills a day. And so, for like the number of enzymes he has to take every day. If it could just like stay where it is or even like somehow get to be a little bit lower, like that would be so awesome for him. Like yeah, pie in the sky. It's like he takes a pill and then that's all he does to maintain his health. Like that would be the ultimate like dream, but even if it just reduces all of the things by some percentage and if it.”
“But even if it didn't do that, I mean, ultimately the biggest goal is just that it like, allows him to like, you know, perform in the world in the like, in the ways and in the amounts that he wants to and that he doesn't need to. Like he won't have lots of time off away from school for like long hospital stays. He won't need a lung transplant. All that kind of stuff. Like, I don't know, just like the closest to normalcy, I guess, that it could get. But yeah, that's the dream.” — Parent of a 2-5 year old with CF
Experience With Drug Under Review
We were unable to speak to a family that had a 2-5 year old child in clinical trials or that had access to Trikafta through some other mechanism. We did, however, speak to a mom of a child aged 2-5 years old who is taking Orkambi. Despite receiving two “do not list” recommendations from the Common Drug Review, to our knowledge Alberta, Saskatchewan, Manitoba, Ontario, Quebec, and Prince Edward Island provide Orkambi to children aged 2-5 years old with two copies of the F508del mutation under highly restrictive criteria. It would therefore seem illogical and perhaps even harmful to not provide Trikafta to children in this age group, given the tremendous efficacy Trikafta has compared to Orkambi. Orkambi is an earlier generation, less effective drug that is otherwise similar to Trikafta in how it addresses the folding and functional defect of CFTR in the F508del mutation.
The parent of the child who was in Orkambi trials and who continues to be on therapy shared this:
“…during the trial, obviously we had no access to data like around whether anything was shifting for him. But since he's been off the trial and just under the care of our CF team here and just still being given the drug. But our CF team here has done like sweat tests and fecal elastase tests and everything's improved. So, I know that the drug is working on a level that I can't see like he's gone from being.”
“Like there's three levels of pancreatic insufficiency. I can't remember exactly what they were, but he's gone from being in like the most extreme end to being bordering on the like just a little bit insufficient. And if that makes sense, like he's really, he's changed quite a bit. The numbers change quite a bit. And same with the sweat chloride, it's changed quite a bit. It's dropped fairly significantly.”
“…when he was in between being in the trial and then just being compassionately covered, we had to do like a two-week washout. Where he wasn't on the drugs and his poops changed in those two weeks. So again, that's just like what I observed. It might have been a coincidence, but that's what I observed. And then when we did the fecal elastase testing here, it sort of confirmed that that might have actually been true.”
“I guess it would just it's kind of like it's kind of like when you started or can be started (on therapy). It's like it's not like you notice this immediate change, but it's kind of like just a bit of an emotional weight off like just knowing that.”
“There's something on this cellular level that I can't see that's doing even more hard work to like, yeah, I think it would be that initially it's just like this like excitement and emotional weight and stuff of like okay. He's getting the absolute best treatment that he can be getting like this is, this is good. And then yeah, the fantasy would be them that like okay now like real big life stuff can actually change.”
“I (would be open to him starting Trikafta). I trust like there's so much research done around this stuff. Like and I trust the people who are doing that and having participated in the drug trial and just knowing how like rigorous that stuff is, having gone (through it with Orkambi). Like I definitely have a lot of faith in sort of the way that it's done and also just knowing that...this is like such an expensive drug that like no one has been wanting to pay for it but (with Trikafta) they can't refute the evidence. The evidence is so good, so strong. So…if all of these countries around the world are finally paying for this drug then like yeah, it's (our turn). I have no qualms or fears about it.” — Parent of a 2-5 year old with CF
[Trikafta is] clinically shown to work better than Orkambi- which my child is on. — Parent of a child with CF
An adult with CF who was on Orkambi and has transitioned to Trikafta noted:
“Being on Orkambi increased my energy and overall improved my symptoms and it was great. I am thankful that I got to take Orkambi and stabilize my health. It was able to stabilize my health and I felt great. But it did not alleviate as many symptoms as Trikafta. When I started Trikafta it was life changing. It not only alleviated 99% of all mucus in my lungs. It increased my lung function significantly. Being on Trikafta gave me a chance at living a life without an imminent need for a lung transplant. It has allowed me to put my cystic fibrosis on the back burner and it not be the only focus in my life. My CF is more of an inconvenience than a death sentence now that j am taking Trikafta. For me the obvious choice is that Trikafta works significantly better than Orkambi for my body.” — Person living with CF
Companion Diagnostic Test
In its submission, our clinical trials network (CF CanAct) noted that:
Patients would be identified based solely on their genetic mutation, either homozygous or heterozygous for F508del. Genetic mutation is determined in conjunction with NBS across Canada at the time of diagnosis. No other diagnostic test is required.
From a parent’s perspective:
“Well, we have to do the regular throat swabs when we go to clinic and we've got those are those are hard when the nurses do them it's fine like it's fast, it's over fast but we've because I have two young kids in daycare we get sick all the time and so we've taken some throat swabs home just to swab them quickly so that. When he's sick but not super sick, we can do the throat swabs and just get them to the lab. And that is very hard to administer, especially because for a while it'd be like, OK, throat swab and then no swab. Like, yeah, he's gotten swabbed a lot. So those are difficult for me to go administer, but they're not like, you know, all the time either. And then it's just it's difficult for him to tolerate blood work.” — Parent of a 2-5 year old with CF
“In this age group it is important to consider measurements of BMI and overall health including frequency and duration of infections to monitor the health of the child. Sweat chloride is not something that can easily be monitored frequently and there are no lung function tests that are widely available for clinical use in this young age group. It is therefore important to observe growth to ensure it is normal, that the child is thriving and that they are generally healthy.”
Conflict of Interest Declaration — Cystic Fibrosis Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
We worked with CF clinicians and researchers to inform our submission, where required.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Cystic Fibrosis Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca Canada Inc. | X | — | — | — |
Horizon Therapeutics | — | — | — | X |
Organon | — | X | — | — |
Trudell Medical International | — | X | — | — |
Vertex Pharmaceuticals | X | — | — | — |
Vertex Pharmaceuticals (Canada) | — | — | — | X |
Viatris | — | — | X | — |
Clinician Input
Cystic Fibrosis Canada’s Accelerating Clinical Trials Network Executive Committee
About Cystic Fibrosis Canada’s Accelerating Clinical Trials Network Executive Committee
The Physicians who are submitting this proposal are Executive and Steering Committee members of Cystic Fibrosis Canada’s Accelerating Clinical Trials Network (CF CanACT). CF CanACT operates under the auspices of Cystic Fibrosis Canada and its purpose is to conduct world class clinical trials in Cystic Fibrosis (CF) in Canada. This is integral to bringing new therapeutics and better care to CF patients in Canada.
The Physicians represent 14 Cystic Fibrosis Clinics and 10 Clinical trial sites across Canada comprising 60% of the CF population. In addition, these Physicians represent the leading clinical researchers in CF in Canada. A few of these physicians were involved in the clinical trials on the use of Elexacaftor/tezacaftor/ivacaftor in the 2–5-year age group, and while this may be a conflict of Interest, they are in the unique situation of having seen first-hand knowledge of the impact of this drug in those patients.
https://cysticfibrosis.ca/our-programs/clinical-trials-network
Information Gathering
Information supporting this submission was gathered by the following means:
Cystic Fibrosis Canada’s Data Registry which contains individual patient information on people living with CF in Canada.
Outcomes of patients who have participated in clinical trials within the network, especially CFTR modulator trials.
Publications from the scientific literature.
Personal experience of the CF physicians treating patients with CF.
Current Treatments and Treatment Goals
CF in the Canadian Context
Cystic Fibrosis (CF) is the most commonly inherited genetic condition in Canada effecting over 4,300 Canadians, with an incidence of approximately 1 in 3,600 live births. CF is a progressive, degenerative multi-system disease that is caused by a loss of function mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which was first identified at The Hospital for Sick Children in 1989. CF is a multisystem disease, most known for its effects on the lungs and digestive systems.
Since the original description of CF in the 1940s, research into the underlying mechanisms of disease has led directly to the development of novel therapeutics that markedly improved survival and quality of life for people living with CF. Median expected survival has increased from less than one year when the condition was originally described, to 25 years when the Canadian CF Patient Data Registry started reporting this metric in 1984 and crossed the 50-year threshold for the first time in 2012.
Currently in Canada, there are 4185 people living with CF. Of those, 359 are children aged 2-5 years. These children attend outpatient clinics 3 - 4 times per year. Overall, owing to the quality of care received and the fact that pulmonary manifestations of CF build up over time, children in this age range with CF are clinically quite well with relatively few admissions to hospital1.
CF Standards of Care – Symptom-Directed Therapies
Prior to the development of disease modifying interventions (i.e., medications that help recover the function of the CFTR protein), interventions were “symptom-directed” – in other words they focused on the downstream effects of CFTR dysfunction. Specifically, therapies were focused on the maintenance of adequate nutrition, optimization of airway secretion clearance and treatment with antibiotics of the aggressive infections contracted by people with CF.
Despite the “symptomatic” nomenclature, research in CF has clearly demonstrated that aggressive management to PREVENT the development of these downstream complications, aims to slow disease progression. This paradigm of secondary prevention has been the cornerstone of modern CF care for decades.
Newborn screening is the epitome of preventative care in CF. Since 2018, all babies born in Canada are screened for CF at birth. This accounts for 2/3rd of the 160 annual CF diagnoses. The goal of newborn screening is early identification to allow timely implementation of the treatments outlined above to slow the onset of the later complications of CF.
Historically, children with CF died in early childhood of malnutrition. The introduction of aggressive nutritional rehabilitation and pancreatic enzyme replacement therapy was the first great advance in the care of people with CF and allowed survival into later childhood and early adulthood. Modern CF treatment focuses on optimising growth and maintaining adequate nutrition. Due to pancreatic insufficiency, most patients require pancreatic enzyme supplementation in addition to fat soluble vitamin supplementation. This promotes good nutrition and is critically linked to overall health and survival.
As survival improves, the main cause of morbidity and mortality in people with CF is lung damage due to a vicious cycle of retained mucus, infection, inflammation and lung destruction. Breaking this cycle of retained mucus and lung destruction is the mainstay of modern pulmonary CF care. This is achieved by regular daily chest physiotherapy, inhaled mucolytics (e.g., hypertonic saline, Pulmozyme™) and either acute or chronic suppressive inhaled antibiotic therapy (e.g., TOBI™, Cayston™). Often this therapy takes 1-2 hours per day to perform. This strategy aims to slow the evolving lung damage and the resultant decline in lung function that ultimately led to respiratory failure and death. While these treatments are effective, they can only slow this decline.
Given the multisystem impact of CF, complications arise as the patient ages and so all patients have regular screening for complications in various organ systems. Specifically, CF-related diabetes is very prevalent with up to 33% adult patients needing to use insulin. Liver disease is common and, if medical management is unsuccessful, may lead to liver transplantation. Additionally, patients are at risk of early development of osteoporosis. Issues with fertility are common with most men being infertile and women sub-fertile. There is an increased risk of cancer particularly bowel cancer as well as inflammatory bowel disease and celiac disease. All of these conditions are routinely screened in clinic and are part of the preventative paradigm in CF care.
CF care is holistic and emotional wellness is now a significant problem within this patient group. A significant number of patients and/or caregivers are currently suffering from either anxiety or depression. This has become a high priority in this patient group and is the focus of a national incentive. Proactive screening for mental health complications and timely referral to mental health practitioners is now integrated into CF care.
The aim of newborn screening and the time-consuming CF treatment regime is to alter the natural history, control symptoms and reduce morbidity associated with recurrent pulmonary exacerbations and hospitalisations. However, all these therapies address downstream consequences of the genetic defect in CF. Currently, there is no cure for this rare fatal genetic disease, and approved medications aim at altering and slowing the trajectory of lung function decline. Ultimately, when respiratory failure occurs, lung transplantation is the only option to try to extend life expectancy and improve quality of life.
CF Standards of Care - CFTR Modulators
CFTR modulators have been developed to tackle the underlying defect of CF. Although not a cure, these medications restore the function of the CFTR protein, a chloride and bicarbonate channel, at the cell surface. CFTR modulators are tailored to work to correct specific mutations and are an example of precision (personalized) medicine. Correction of CFTR protein function at an early age is congruent with the overall preventative paradigm of CF treatment – early correction with hopefully prevent disease progression and irreversible damage.
The first CFTR modulator commercially available was ivacaftor (IVA; Kalydeco™), which was approved in by Health Canada in 2012 and is now available for those aged 12 months and up. It is effective in patients who have “gating” mutations, only 4% of Canadians with CF. In the 12-to-24-month age group, Ivacaftor has been shown to normalize sweat chloride levels, increase pancreatic function and improve BMI Z scores3. It is an extremely effective medication, which provides substantial clinical benefits of increasing lung function, reducing hospitalizations and improving nutritional status, and real-world evidence of improving survival and decreasing need for lung transplant. It is currently funded by some private plans and most public drug programs.
For patients with 2 copies of the most common CF mutation, F508del (~50% of Canadians with CF), lumacaftor/ivacaftor (LUM/IVA; Orkambi™) and tezacaftor/ivacaftor (TEZ/IVA; Symdeko™) have been developed. Despite Health Canada approval, these medications are not generally reimbursed provincially. (except for Quebec’s ‘patient d’exception’ programme) and consequently only 12% of Canadian CF patients receive these through participation in clinical trials or 3rd party payers.
Elexacaftor/tezacaftor/ivacaftor (ELX/TEZ/IVA; Trikafta™). In vitro, this medication effectively recovers CFTR function in cells with one or two copies of the F508del mutation. It is important to note that ELX/TEZ/IVA is indicated for people with one copy of F508del in the targeted population, whereas the two existing therapies - Orkambi and Smydeko - were only indicated for people with two copies of this mutation. This group (one or two copies of F508del) includes ~90% of Canadians with CF, so this represents a significant expansion of the population that is genetically eligible for this class of medications.
In clinical trials, this medication is also extremely effective clinically in people with CF with one or two copies of F508del and has been approved by Health Canada for those age 6 and up. Further, previous CADTH reviews have recommended funding of ELX/TEZ/IVA for those age 6 years and up based on the published efficacy data in both randomized and open label studies – effectiveness/efficacy outcomes assessed in these studies included lung function indices, nutritional indices, quality of life indices, rates of pulmonary exacerbations and sweat chloride concentration.
The next step in the progression of CFTR-modulator-based treatment for Canadian children with CF is the data shown in the recently published study looking at the safety and effectiveness of ELX/TEZ/IVA in children aged 2-5 years.2 This study further extends the good safety profile and effectiveness of this medication into this younger age range. Results from this study, showed a decrease in sweat chloride concentration of −57.9 mmol/ with 90% of study participants normalizing their sweat chloride levels to below the range for a diagnosis of CF, absolute change in BMI-for-age z-score was 0.10 (95% CI, 0.00 to 0.20). Lung function as measured by LCI 2.5 is a sensitive measure of ventilation inhomogeneity, where increases in Lung clearance Index (LCI2.5) indicate worsening of lung function, in this study the LCI had a decrease (i.e., lung function improved) fall by −0.83 units (95% CI, −1.01 to −0.66). Lastly mean (SD) absolute change in fecal elastase-1 concentration which is a marker of pancreatic insufficiency was 39.5(89.2) μg/g. All these outcome measures demonstrated that ELX/TEZ/IVA led to improvements in CFTR function and lung function and was associated with stable nutritional status over a 24-week treatment period.
Extending access to this medication to this age range would be congruent with the secondary prevention paradigm of CF care and anticipated to result in decreasing the long-term burden of disease for children (and eventually adults) with CF if treatment is initiated early. An ideal treatment in CF would fully address the basic molecular defect in CF and restore normal chloride transport on the cell surface. If applied at an early enough date, complete and early correction of CFTR function would prevent the multisystem downstream effects that are ultimately fatal for this group. Work continues to develop tools to completely correct CFTR function (this may be gene therapy, or perfected small molecule interventions). ELX/TEZ/IVA is the latest “third generation” CFTR modulator. In vitro, this medication provides the greatest restoration of CFTR function observed to date.
Treatment Goals
The goals of treatment with Elexacaftor/tezacaftor/ivacaftor (ELX/TEZ/IVA; Trikafta™) for the 2–5-year-old age group would be to:
Improve and/or stabilizes lung function.
Prevent and/or reduce pulmonary exacerbations.
Improve and/or stabilize nutrition and growth.
Minimize and/or reverse other multisystem complications of CF disease.
Improve emotional wellness.
Improve quality of life
Allow attendance at school with minimal disruption.
Reduce burden of care and number of therapies needed to maintain health.
Prolong life.
References
Cystic Fibrosis Canada. (2023). The Canadian Cystic Fibrosis Registry 2021 Annual Data Report. Toronto, Canada: Cystic Fibrosis Canada.
J Goralski et al. Phase 3 Open-Label Clinical Trial of Elexacaftor/Tezacaftor/Ivacaftor in Children Aged 2 Through 5 Years with Cystic Fibrosis and at Least One F508del Allele. Am J Resp Crit Care Med. 2023; Mar 15, online ahead of print. doi: 10.1164/rccm.202301-0084OC
Rosenfeld M, Wainwright CE, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med. 2018 Jul;6(7):545-553. doi: 10.1016/S2213-2600(18)30202-9. Epub 2018 Jun 7.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Current treatments such as inhaled antibiotics and mucus thinning agents target downstream consequences of CF lung disease (e.g., infection, thick, dehydrated mucus) and therefore do not treat the root cause or reverse the course of disease. These therapies require nebulization and therefore are extremely time-consuming to administer (2-3 hours per day) and thus adversely impact quality of life and school productivity at a critical time of childhood development. This demanding treatment regimen also influences medication adherence and mental health. They do not halt the disease progression rather they are aimed at slowing down the progression of the disease.
Furthermore, there are significant side effects related to the use of LUM/IVA (e.g., chest tightness, blood pressure elevation) and numerous drug-drug interactions. They also provide only a marginal increase to lung function.
Current treatments do not reverse extra-pulmonary manifestations of CF including sinusitis, exocrine pancreatic insufficiency, diabetes, liver disease, and bowel manifestations.
The drug under review would be considered first-line to alter the trajectory of the disease and when started in this young age group and prescribed continuously, may reduce or eliminate the need for symptomatic therapies later in life.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Triple combination modulator therapy (ELX/TEZ/IVA) addresses the underlying disease process in cystic fibrosis (CF) and is added on to current standard of care for CF as a first-line therapy for those patients with the appropriate CF mutations (genotype). This CFTR modulator therapy is specifically targeted to the folding/trafficking/functional defect of the CFTR protein that results from the indicated population’s genetic defect.
The current treatment paradigm for CF divides therapies into those that address the basic defect (CFTR modulators) and those that treat the downstream consequences of the defect (for example: inhaled antibiotics, inhaled mucolytics, bronchodilators, anti-inflammatory drugs, physiotherapy).
The Phase 3 trial in patients 2-5 year older have demonstrated that the addition of ELX/TEZ/IVA to standard of care results in significant improvements in clinically important outcomes of lung function as measured by LCI, pulmonary exacerbations, weight and sweat chloride (1). Consensus guidelines and Standard of Care guidelines already include CFTR modulator therapies (2, 3), but they have not been recommended to replace prior therapies such as inhaled antibiotics that treat consequences of the defect because end-organ damage has already occurred and therefore these treatments remain necessary. Future research will determine if some of these other standard of care therapies can be safely removed for patients on ELX/TEZ/IVA and whether introduction of ELX/TEZ/IVA earlier in life will prevent the need for inhaled antibiotics, inhaled mucolytics, and other standard of care treatments.
ELX/TEZ/IVA is not the first therapy that addresses the underlying defect in CF, but rather it is an improvement on existing CFTR modulator therapies. This therapy would replace other CFTR modulators that are currently available, except for a few mutations where Ivacaftor is still the only modulator approved for. When compared to the Health Canada-approved CFTR modulator (TEZ/IVA), Phase 3 trials in the 6 – 11year old group have demonstrated greater efficacy of ELX/TEZ/IVA (4). ELX/TEZ/IVA is also indicated for a broader CF population than TEZ/IVA, as it has been shown to be effective in patients who have at least one F508del mutation (1). In this sense it is, for F508del heterozygous CF patients, a first-line therapy that addresses the underlying defect in CF.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review.
Patients starting CFTR modulator therapy should already be receiving standard of care treatments as indicated for the extent and characteristics of their disease (e.g., chest physiotherapy for airway clearance, mucolytics, inhaled antibiotics for chronic airway infection, anti-inflammatory therapies, bronchodilators, pancreatic enzymes, fat soluble vitamins, insulin). ELX/TEZ/IVA should be added to this therapy regardless of treatment response to standard of care as it is the only therapy that targets the defect in CFTR. For patients homozygous for F508del currently on TEZ/IVA or LUM/IVA, or those heterozygous with F508del and a mutation indicated for IVA treatment, it is beneficial to switch to ELX/TEZ/IVA.
References
J Goralski et al. Phase 3 Open-Label Clinical Trial of Elexacaftor/Tezacaftor/Ivacaftor in Children Aged 2 Through 5 Years with Cystic Fibrosis and at Least One F508del Allele. Am J Resp Crit Care Med. 2023; Mar 15, online ahead of print. doi: 10.1164/rccm.202301-0084OC.
Ren CL, Morgan RL, Oermann C, Resnick HE, Brady C, et al. Cystic fibrosis pulmonary guidelines: use of cystic fibrosis transmembrane conductance regulator modulator therapy in patients with cystic fibrosis. Ann Am Thorac Soc 2018;15:271–280.
Canadian clinical consensus guideline for initiation of monitoring and discontinuation of CFTE modulator therapies for patients with cystic fibrosis. Cystic Fibrosis Standard of Care documents 2023, https://www.cysticfibrosis.ca/uploads/CFC%20Modulator%20Guidelines_RevisedOct62021%20(003)
Zemanick, Taylor-Cousar, Davies, et al. A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am J Respir Crit Care Med Vol 203, Iss 12, pp 1522–1532, Jun 15, 2021
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients with a single copy of F508del paired with another CF mutation (i.e., F508del heterozygous) that is not a gating or residual function mutation have the greatest unmet need as there are currently no approved CFTR modulator therapies available to them. In patients with a single copy of F508del and a “minimal function” mutation, clinical manifestations can be severe and the drug under review is considered a breakthrough as it leads to clinically significant improvements in lung function.
Patients with two copies of F508del (i.e., F508del homozygous) also have substantial unmet need as only a minority have been able to access LUM/IVA or TEZ/IVA due to the lack of public reimbursement in most provinces. Furthermore, in the minority of patients who have been able to access these therapies, response is variable and side effects can be considerable. The drug under review leads to tremendous improvements beyond the effects related to LUM/IVA and TEZ/IVA and has fewer side effects and drug-drug interactions than LUM/IVA.
Children between 2-5 years old often have clinically significant structural lung disease (bronchial wall thickening, mucus plugging, bronchiectasis) but detection can be challenging as this age group has difficulty performing spirometry which is used in older children and adults to monitor lung disease. Lung clearance index (LCI), measured by the multiple breath washout test, is a sensitive indicator of ventilation inhomogeneity and a higher value indicates greater lung disease (i.e., worse lung function). The majority of children (58%) have elevated LCI at their first preschool visit and this is predictive of lower lung function in their school age years. Imaging studies involving CT and MRI demonstrate structural and functional abnormalities, respectively, within the first few years of life despite “normal” lung function and therefore lung disease progresses in a silent manner. Currently available treatments do not reverse the course of disease or prevent end-organ damage.
As demonstrated in studies of CF infants diagnosed by newborn screening, structural lung damage (airway wall thickening, air trapping due to narrowed airways and mucus obstruction, early bronchiectasis) begins early in life (Sly NEJM 2013). As such, there is no “silent” or pre-symptomatic disease. All cystic fibrosis patients with at least one F508del mutation should be eligible for the drug. A recent MRI imaging study on the effect of ELX/TEZ/IVA in CF patients over the age of 12 years (Graeber AJRCCM 2023) demonstrated improvements in airway wall thickening/bronchiectasis and mucus plugging, the earliest changes noted in infant imaging. Drug-drug interactions may require that certain other medications be changed, or the dosage be changed, but these are few and used infrequently in the CF population.
With Newborn Screening, we have the ability to detect those with CF at birth. The earlier we commence treatment before there are permanent changes in the lung the better. Hence, while ELX/TEZ/IVA is currently approved for 6 years and older, if we commence treatment at two years old, the greater the chance of preventing lung disease.
Which patients would be least suitable for treatment with the drug under review.
All patients homozygous or heterozygous for F508 del would benefit from this drug. We are unsure yet, if other patients who are no homozygous or heterozygous for F508del would benefit from this drug. At least one clinical trial is examining the benefit of ELX/TEZ/IVA in other mutations. The label has been expanded in the US for 170+ additional mutations for which laboratory data indicates a likely benefit however this type of data is not accepted in Canada at this time. There needs to be a mechanism in Canada to ensure access to this life-changing drug for all those who will benefit.
How would patients best suited for treatment with drug under review be identified.
Patients would be identified based solely on their genetic mutation, either homozygous or heterozygous for F508del. Genetic mutation is determined in conjunction with NBS across Canada at the time of diagnosis. No other diagnostic test is required.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
In this age group tests of lung function are not established as a clinical tool and symptoms scores are often in the normal range despite the existence of structural lung changes, as described above The only lung function measure available for this age group is the LCI, however, this test is only available in three CF centres in Canada and is only approved by Health Canada for use in clinical trials. It would not be available for use by all patients as a clinical monitoring tool. As the treatment goal of this progressive disease is to slow decline in lung function and reduce mortality, the most important outcomes are linked to avoiding progression of disease:
The outcomes of interest are those that can be assessed during routine visits. Assessment of response should be done following 12 months of treatment. As discussed, lung function by spirometry cannot be conducted in young children and LCI is not a feasible clinical assessment test to use.
Potential outcomes that can be used include:
Nutritional status has an important long-term impact on outcomes, such that the body mass index percentile (BMI) should be at least maintained.
Pulmonary exacerbations have an important impact on lung function, and so an important outcome is a decrease in the number of exacerbations, or the number of days of antibiotics or the number of courses of antibiotics.
Sweat chloride testing, with a 12-month value less than 60 mmol/L or a reduction of at least 30% from the value obtained at diagnosis.
In addition, there may be other factors that the treating CF physician should consider as evidence of response to treatment including overall well-being and/or improvement in sinopulmonary symptoms.
Improvement or maintenance of any of the above outcomes should be considered as a positive response to treatment.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Clinical trial data in the 2–5-year age group suggested that the majority of adverse events were mild and manageable. Discontinuation of therapy should be considered in patients who have clinically significant adverse effects that persist and recur after stopping and re-initiating therapy.
Examples of these reactions include (but are not limited to):
Elevation of liver function tests beyond the higher range of fluctuations observed in CF patients.
Allergic reactions to treatment
However, the risk-benefit of discontinuing treatment should be considered on a case-by-case basis depending on the severity of the adverse event and risk of stopping treatment on overall health of the CF patient.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Treatment should be limited to CF patients attending the cystic fibrosis clinics accredited by CF Canada. All or nearly all patients indicated for this treatment in the 2-5 year age range would already attend these clinics regularly (typically quarterly).
Additional Information
No additional information.
Conflict of Interest Declarations — Cystic Fibrosis Canada’s Accelerating Clinical Trials Network Executive Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes, we required data from the CF Canada Registry specific to 2 - 5 year old.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Yes, we required data from the CF Canada Registry specific to 2 - 5 year olds.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Jonathan Rayment
Position: Respirologist CF Clinic BC Children’s Hospital, Medical Lead CF CanACT
Date: 23-May-2023
Table 2: COI Declaration for CF CanACT — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex | — | — | — | X* |
Sanofi | X | — | — | — |
*Unrestricted research grant.
Declaration for Clinician 2
Name: Brad Quon
Position: Associate Professor of Medicine, University of British Columbia, Director CF Clinic St. Paul’s Hospital.
Date: 23-May-2023
Table 3: COI Declaration for CF CanACT — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex | X | — | — | — |
AbbVie | X | — | — | — |
Horizon Therapeutics | X | — | — | — |
Declaration for Clinician 3
Name: Felix Ratjen
Position: Professor University of Toronto, Respirologist, Hospital for Sick Children, Toronto.
Date: 23-May-2023
Table 4: COI Declaration for CF CanACT — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex | — | — | X | — |
Declaration for Clinician 4
Name: Larry Lands
Position: Director, Pediatric CF Clinic, Montreal Children’s Hospital, Montreal.
Date: 22-May-2023
Table 5: COI Declaration for CF CanACT — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex | — | X | — | — |
CF Canada Health Care Advisory Council
About CF Canada Health Care Advisory Council
Cystic Fibrosis (CF) Canada is a national not-for-profit corporation committed to improving and lengthening the lives of people living with cystic fibrosis. It is a leading organization with a central role engaging people living with cystic fibrosis, parents and caregivers, volunteers, researchers and healthcare professionals, government and donors to work together to change lives through treatments, research, information and support.
As part of this is the CF Canada Healthcare advisory council, a group composed of 10 Interprofessional healthcare providers and 2 lay members. These members are involved in the provision of care to children and adults with CF in Canada. The council supports CF Canada in developing CF care policy and guidelines to support the CF clinic directors and community.
Information Gathering
The information included in this submission was gathered in several ways:
Experience gained by working with and delivering medical services to people with cystic fibrosis.
Experience treating people with cystic fibrosis who received elexacaftor/tezacaftor/ivacaftor (elexa/teza/iva), ivacaftor, ivacaftor-lumacaftor, and ivacaftor-tezacaftor either during participation in clinical trials, through the Health Canada Special Access Program, and/or since the Health Canada marketing authorization for persons ages 12 years and older.
Review of the medical and scientific literature, including clinical trial results.
The Cystic Fibrosis Canada Canadian Cystic Fibrosis Registry, a collection of patient data and other information regarding CF care and outcomes.
The Canadian Clinical Consensus Guideline for Initiation, Monitoring and Discontinuation of CFTR Modulator Therapies for Patients with Cystic Fibrosis
Current Treatments and Treatment Goals
Cystic fibrosis (CF) is a fatal, progressive genetic disease that affects approximately 4,300 Canadians, with an incidence of approximately 1/3,600 live births. In 2021 there were 98 new cases diagnosed in Canada, with 67 of those diagnosed through provincial newborn screening programs.1 It is a lifelong, chronic, degenerative disease that affects multiple organ systems, most importantly the lungs and the digestive system. According to the Canadian Cystic Fibrosis Registry, 4,388 individuals live with CF, of whom 359 (8%) children are aged between 2-5 years.2
In order to treat the underlying symptoms, people with CF (pwCF) are prescribed a multitude of treatments, including high-calorie high fat high protein diets, digestive medications, and airway clearance treatments. Physiological manifestations of CF start very early in life, before many tests will show significant changes. Consequently, many of these treatments start at the time of diagnosis (including in infancy) and continue every day throughout life. Medications commonly used in CF include acute and chronic antibiotic therapies, mucolytics, bronchodilators, pancreatic enzymes, fat soluble vitamins, insulin for people with cystic fibrosis related diabetes, and ursodiol for liver disease. Chest physiotherapy treatment is prescribed several times a day. The average time for pwCF for to undertake therapy is >2 hours/day. As disease severity increases so does time commitment.
One transformational change was the introduction of newborn screening for CF. This is now standard practice across all of Canada. Children are now diagnosed within 1 month of life instead of a median 2-3 years.
Consequently, therapy is commenced within weeks of life and significantly alters the natural history of CF disease progression. One of the principles of screening is that there must be an accepted treatment for patients with recognized disease. With the introduction of elexa/teza/iva there is now not just an accepted treatment but a highly effective one.
A second transformational change was the introduction of Cystic fibrosis transmembrane regulator (CFTR) modulators, new revolutionary treatments in CF care. Non-modulator treatments are aimed at targeting symptoms, treating exacerbations, and slowing the progression of what is a life-long, degenerative, and fatal disease. CFTR modulators are the first commercially available therapies that are targeted at correcting the basic defect in CF by improving the production and function of the abnormal CFTR protein. Although none of the modulators are a “cure” for CF, the highly effective treatment results in improvement in CFTR production and function, minimizes symptoms, improves clinical parameters such as lung function, body mass index, pulmonary exacerbations, and sweat chloride measurements, and have been shown to have a positive effect on quality of life in pwCF. The second-generation modulators had a modest but important clinical effect, but the response to the third-generation modulator, elexa/teza/iva is substantially greater and more comparable to the response of pwCF with eligible mutations to ivacaftor.
The first- (ivacaftor) and second-generation (ivacaftor-lumacaftor, and ivacaftor-tezacaftor) CFTR modulator medications, were the first commercially available treatments to treat the underlying disease mechanism: a poorly produced and/or malfunctioning CFTR protein. Ivacaftor is currently approved for pwCF ages 4 months and up who have one or two of a small number of CFTR mutations, representing only about 4% of Canadians with CF. Alone, ivacaftor is not effective in pwCF who have two copies of the most common F508del mutation3 or who carry one F508del and another mutation not responsive to ivacaftor. Lumacaftor-ivacaftor (Orkambi) and Tezacaftor-ivacaftor (Symdeko) is currently indicated in Canada for pwCF ages 4 months and older or >12 years, respectively. However, the response to both these modulators in clinical trials was more modest than observed with elexacaftor-tezacaftor-ivacaftor.4,5 Due to cost and reimbursement issues, many pwCF who are eligible have not been able to access this medication and off label use in the 6-11 year age group has been negligible.2 Although the true cost to payers of these medications has been confidential, based on the list price there is no cost advantage to prescribing lumacaftor-ivacaftor or tezacaftor-ivacaftor to pwCF who are eligible for elexa/teza/iva. Due to cost and reimbursement policies, many pwCF eligible for lumacaftor-ivacaftor have not been able to access the medication.
Since elexa/teza/iva received market authorization, access to the medication was initially limited by type of insurance and province of residence. However, it is now fully available for all patients with CF who have 1 copy of F508del aged 6 years and older. From the CF Canada registry (2021) 75% of CF patients are now eligible and 33% were already receiving a highly effective CFTR modulator. The Canadian Clinical Consensus Guideline for use of CFTR modulator therapies in pwCF, provides comprehensive recommendations for criteria for initiation, monitoring and response for all patients with CF. Within this guideline, the recommendation is that all patients with eligible mutations and a diagnosis of CF should have the opportunity to be treated with a highly effective CFTR modulator.
In addition to these treatments, the recommendations for CF care include routine medical visits to the cystic fibrosis clinic every three months. Additional visits may be required due to illness or for closer follow up of progressing symptoms, severe disease, or pre- and post-transplant care. Hospitalizations and home intravenous treatments may be required for acute respiratory infections or other complications of cystic fibrosis, such as distal intestinal obstructive syndrome. According to the 2021 Cystic Fibrosis Registry report, Canadians with CF had over 17,000 clinic visits that year and logged 16,000 hospital days and 10,000 home IV treatment days.1 This is trending down and likely reflects, in part, the impact of highly effective CFTR modulator therapy in those CF patients having access.
Lung transplant is a treatment for end-stage CF pulmonary disease. It comes with risk factors and additional treatment burden and does not address CF disease in other organ systems. The median length of survival after lung transplant reported in the 2021 Registry report was 10.6 years, so it is not a cure, and the direct cost of medical care involved in lung transplant is around $1,000,000. Lung transplantation is only offered at four centres in Canada, and relocating to one of these centres (Toronto, Montreal, Edmonton, or Vancouver) is required during parts of the transplant process.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Most of the currently available treatments only treat the symptoms and complications of CF and attempt to slow down the eventual fatal progression of the disease. Most patients die from end stage respiratory failure.
While ivacaftor is a highly effective CFTR modulator, it is effective for only about 4% of Canadians with CF and is not effective for CF patients who are homozygous for F508del or who carry one F508del mutation and a mutation that is not treatable by ivacaftor. These patients make up approximately 88% of people with CF.
The only currently available modulator for pwCF ages 2-5 years is lumacaftor-ivacaftor. While it has shown positive effects on respiratory, body mass index, and quality of life measures, the effects are modest4, cost- effectiveness concerns have led to barriers to access for many and is only available through 3rd party payers in many provinces. Tezacaftor-ivacaftor is not approved by Health Canada for children under 12 years and the effect size for this drug has not been as substantial as that shown by highly effective modulators.5,6
For pwCF ages 6 years and older, clinical studies and real-life clinical experience of elexa/teza/iva in Canadian provincial CF Clinics has shown a positive therapeutic response. In the PROMISE study, subjects with mild CF lung disease showed significant improvements in ppFEV1, sweat chloride measurement, CFQ-R Respiratory domain score, and BMI. This is despite these patients being perceived to have milder disease.7
Although, newborn screening has been shown to alter the natural history CF, it has been shown that these newly diagnosed patients still have evidence of lung disease and other manifestations of CF early in life. These changes are progressive, cumulative, and ultimately lead to death despite early initiation of standard treatment. Access to a highly effective CFTR modulator has been shown to significantly alter these early changes. As illustrated by the improvement in sweat chloride and lung clearance index in an open label study of children aged 2-5 receiving elexa/teza/iva.8 Similar findings were seen in children aged 6-11 years who are now receiving commercial drug in Canada and so it is likely these findings are transferable.
Most patients with CF are pancreatic insufficient requiring the need to tale pancreatic enzymes to absorb food and grow. Data have reported the recovery of pancreatic function in some patients on CFTR modulators. Early introduction has been key. Similarly, preservation or stabilisation of pancreatic function has helped reduce the frequency of CF Related diabetes.
Development of progressive, non-reversible lung disease and antibiotic resistance in bacteria involved in acute and chronic lung infections due to repeated antibiotic exposure make treatment of infection more challenging with time.
Adherence to treatment and simplifying treatment burden is a major concern for people with CF and for CF clinicians. This is paramount in the 2-5 year old group where treatment is solely dependent on the parent or caregiver. Burden of care is high from the time of diagnosis and increases with age and the severity of the disease. A recent study by the James Lind Alliance Priority Setting Partnership (JLA PSP) in cystic fibrosis surveyed people with CF, parents of children with CF, and health care workers to determine perceived priorities for CF research. Important themes than emerged were that the lived experience of treatment burden in CF is high, that it extends beyond just the time taken to perform routine daily treatments, and that the impact on daily life varies. Adherence to the more burdensome treatments, such as nebulized antibiotics and airway clearance are often the first to be missed. Of the subset of people with CF who answered questions regarding work and education, “87% felt that their treatments get in the way of their job or career and 77% (168/217) in the way of their education. Two thirds (67%) reported that their treatments get in the way of family relationships, relationship with a partner (69%), and relationships with friends (75%). An impact of treatments on socializing and on sports and hobbies was reported by 81% and 80%, respectively.9 Treatments need to be able to be easily integrated into daily life, to not form a barrier that limits participation in important activities during childhood, such as school, family life, peer relationships, sports, hobbies, and play.
Other treatments to treat the molecular basis of the disease, such as gene therapy, are currently under development and still within the pre-clinical stage. This has potential but will likely target single organ CF dysfunction rather than systemically as seen in CFTR modulators.
The development of a highly effective CFTR modifier such as elexacaftor/tezacaftor/ivacaftor fills a niche in CF care that is not currently occupied by another equally effective treatment. The fact that it is an oral medication will have a huge impact on adherence and started earlier in life will modify disease progression and prevent other therapies being required.
There will remain approximately 10% of people with CF in Canada who have CFTR mutations that do not respond to any of the current CFTR modulator therapies. Research continues to find a treatment for this group.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Currently, elexa/teza/iva is a unique therapy that restores function CFTR to the cell surface and is complimentary to additional existing CF therapies. In future CF treatment this will likely reduce the need for current CF therapies. The Canadian guidelines recommend that elexa/teza/iva is added on to existing therapies for a duration of one year and response to therapy evaluated and recorded as part of the national CANimpact study. In addition, this study will evaluate the impact of elexa/teza/iva in decreasing the number and intensity of other routine CF treatments.10
The impact on lung transplantation in CF has been impressive. Since the introduction elexa/teza/iva the need for lung transplantation within the community has fallen by 60%.1
While elexa/teza/iva is not the first CFTR modulator developed, it is so far the most effective for the majority of CF. Of the 3721 patients aged 6 years and older 87% will be eligible for the medication. Expanding access to 2- 5-year-old will allow access to another 314 patients who represent 8% of the Canadian CF population. This will have lifelong benefit, as mentioned previously, because the medication addresses the underlying disease process i.e., decreased production and lack of function of the defective CFTR protein.
A Canadian group used a microsimulation transition model to estimate the effect of the introduction of elexa/teza/iva on the Canadian CF population. In this model, the number of persons with severe lung disease decreased by 60%, the number of pulmonary exacerbations decreased by 19%, and the number of lung transplants decreased by 146 during the period 2021-2030 if the medication is introduced by 2021. Decreasing the need for acute treatments and lung transplant would be a shift in the role of these treatments in the disease.9
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The recent Canadian guidelines clearly define patients who should be treated with a CFTR modulator and recommend all patients with CF who have at least one copy of F508del. The Canadian Cystic Fibrosis clinic system is well established and covers almost all persons with CF in Canada. Patients likely to respond to this medication have been well described in the clinical studies and will be identified by their CF clinic based on genotype of their CFTR mutations. The criteria for diagnosing CF are well established and standardized, and the appropriate tests are available at CF clinics. Most patients are now being diagnosed through CF newborn screening programs. Early initiation of therapy is key to this patient group.
Because CF is a genetic, progressive chronic disease, manifestations of the disease start in early life. Treatment with this potentially disease altering medication should not be held until persons become more symptomatic or until lung function deteriorates below an arbitrary threshold.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Spearheaded by the Health Advisory Group at Cystic Fibrosis Canada, a group of Canadian CF clinicians have developed standardized guidelines for all patients started on CFTR modulator treatment and for assessing response.10 In addition to the regular clinic visits every three months, an additional visit has been recommended 1 month after starting therapy with elexa/teza/iva to assess the initial response to therapy, to screen for side effects, and to address patient concerns. At follow up visits, outcomes measured include a history and physical exam, measurement of height/weight and calculation of BMI, laboratory tests to follow parameters associated with potential side effects (liver enzymes, creatine kinase), sputum microbiology, quality of life questionnaires and mental health screening, and a review of prescribed therapies. Fecal elastase and sweat chloride levels will be monitored at intervals. Regular follow up with a yearly ophthalmological examination is also recommended. These outcomes align with those identified in the clinical trials and with normal standard care of patients with CF.
For children aged 2-5 years standard lung function is not reliable and is not performed and consequently is not valid as a response to therapy. The measurement of lung clearance index, as reported in the study, is not approved for clinical use in Canada and is only available in 3 Canadian centers as a research tool. CT scans require sedation and are therefore not recommended for routine surveillance. An outcome of particularly interest is recovery of pancreatic function. 95% of CF patients are pancreatic insufficient. Early treatment of patients receiving ivacaftor showed there was recovery of pancreatic function. Similarly, this has been described in real world experience.
The improvements that have been measured in most clinical trials include pulmonary function testing, pulmonary exacerbations and antibiotic use, weight and nutritional status, and quality of life. In clinical practice, patients have reported feeling better, having fewer symptoms such as cough or shortness of breath, having less difficulty maintaining a healthy weight, missing less work or school due to hospitalization for pulmonary exacerbations, and stabilization of the disease. Increased attention to quality-of-life measures and screening measures to detect mental health issues have led to these aspects also being included in response to treatment clinically.
These treatment responses include some quantifiable measures (pulmonary function, BMI) that should not vary across physicians. Whether a pwCF is admitted to hospital or treated as an outpatient for a pulmonary exacerbation may be physician, centre, or location dependent and may also be affected by other factors.
However, the effect of the medication on disease stability should not vary greatly by physician. Quality of life measures and patient reported symptoms should also not be practitioner dependent.
Criteria for determining response to therapy has been clearly identified by the Canadian Guideline for children aged 6 years or less, including recommendations for dose interruption and discontinuation.
What factors should be considered when deciding to discontinue treatment with the drug under review?
As with any treatment, discontinuation should be considered if a severe side effect, allergy, or other adverse event occurs. With this medication, the development of signs of worsening liver disease or other significant side effects may require stopping treatment. The Canadian guidelines provide recommendation for dose reduction or discontinuation and potential side effects. For 2-5 year old patients’ discontinuation should be considered if the child is not responding to medication as per the parent and CF team opinion.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Almost all patients with cystic fibrosis in Canada are followed at accredited hospital based Cystic Fibrosis clinics, which are staffed by professionals who have the training and experience in diagnosing, treating, and monitoring people with CF. The pediatric CF clinics would be treating 2-5 year old with this medication.
Experience is already gained with the medication in the population who have already receive it over the last 2 years.
References
Cystic Fibrosis Canada. (2022). The Canadian Cystic Fibrosis Registry 2021 Annual Data Report. Toronto, Canada: Cystic Fibrosis Canada.
Unpublished data obtained from the Canadian Cystic Fibrosis Registry. Data inquiry December 2021.
Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 2012;142(3):718-724. doi:10.1378/chest.11-2672.
Milla CE, Ratjen F, Marigowda G, Liu F, Waltz D, Rosenfeld M; VX13-809-011 Part B Investigator Group *. Lumacaftor/Ivacaftor in Patients Aged 6-11 Years with Cystic Fibrosis and Homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017 Apr 1;195(7):912-920. doi: 10.1164/rccm.201608- 1754OC.
Zemanick ET, Taylor-Cousar JL, Davies J, et al. A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am J Respir Crit Care Med. 2021 Jun 15;203(12):1522-1532. doi: 10.1164/rccm.202102-0509OC
Flume PA, Biner RF, Downey DG, et al.; VX14-661-110 study group. Long-term safety and efficacy of tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years or older who are homozygous or heterozygous for Phe508del CFTR (EXTEND): an open-label extension study. Lancet Respir Med. 2021 Jul;9(7):733-746. doi: 10.1016/S2213-2600(20)30510-5.
Nichols DP, Paynter AC, Heltshe SL, et al for the PROMISE Study Group. Clinical effectiveness of Elexacaftor/Tezacaftor/Ivacaftor in people with cystic fibrosis. Am J Respir Crit Care Med. 2021, article in press, doi: 10.1164/rccm.202108-1986OC
Goralski JL, et al. Phase 3 Open-Label Clinical Trial of Elexacaftor/Tezacaftor/Ivacaftor in Children Aged 2 Through 5 Years with Cystic Fibrosis and at Least One F508del Allele. Am J Respir Crit Care Med. 2023 Mar 15. doi: 10.1164/rccm.202301-0084OC. Epub ahead of print. PMID: 36921081
Davies G, Rowbotham NJ, Smith S, Elliot ZC, Gathercole K, Rayner O, et al. Characterising burden of treatment in cystic fibrosis to identify priority areas for clinical trials. J Cyst Fibros 2020;19(3):499–502.
Canadian Observational Study Evaluating the Long-term IMPACT of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators on People With CF (Can-IMPACT CF) NCT05200429
Conflict of Interest Declarations — CF Canada Health Care Advisory Council
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation.
Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Unpublished Data form the CF Canada Registry.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr Mark Chilvers
Position: Chair, CF Canada Health Care Advisory Council
Date: 23-05-2023
Table 6: COI Declaration for CF Canada Health Care Advisory Council — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex | — | X | — | — |
Canadian Cystic Fibrosis Clinicians
About Canadian Cystic Fibrosis Clinicians
The Canadian Cystic Fibrosis Clinicians group is made up of the clinic directors and physicians associated with the accredited cystic fibrosis clinics in Canada. Clinic directors and cystic fibrosis clinic staff physicians have special training, expertise, and experience in delivering multi-system medical care and support to people with cystic fibrosis.
They collaborate to help improve and advance clinical care for persons with cystic fibrosis, both at individual clinic, provincial, and national levels.
Information Gathering
The information included in this submission was gathered in several ways:
Personal experience gained by working with and delivering medical services to people with cystic fibrosis.
Personal experience treating people with cystic fibrosis who received elexacaftor/tezacaftor/ivacaftor and ivacaftor, either during participation in clinical trials, through the Health Canada Special Access Program, and/or since the Health Canada marketing authorization for persons ages 6 years and older
Review of the medical and scientific literature, including clinical trial results.
The Cystic Fibrosis Canada Canadian Cystic Fibrosis Registry, a collection of patient data and other information regarding CF care and outcomes.
The Canadian Clinical Consensus Guideline for Initiation, Monitoring and Discontinuation of CFTR Modulator Therapies for Patients with Cystic Fibrosis.
Current Treatments and Treatment Goals
Cystic Fibrosis (CF) is a life shortening, progressive, multi-system genetic disease that affects approximately 4,300 Canadians, with an incidence of approximately 1/3,600 live births. There are just over 300 children with CF in the age 2-5 year age group in Canada.
CF clinical care has changed enormously in the time since highly effective CFTR modulator treatments (HEMT) have been available for people with CF who have Cystic Fibrosis Transmembrane Regulator (CFTR) mutations that are amenable to this class of medications. People with CF (pwCF) are reporting to clinicians that they feel better, and we are noticing in our clinics that most patients show an improvement or stabilization in lung function, hospitalizations are decreasing, the number of lung transplants for CF has plummeted, and projected life expectancy is increasing. However, CF is far from cured. Treatment of CF continues to include a multitude of treatments, including dietary modifications, medications, and airway clearance treatments.
HEMT medications have already been reviewed by CADTH and are now commonly available for pwCF in Canada. Elexacaftor- tezacaftor-ivacaftor (EThad already been reviewed for pwCF with at least one F508del CFTR mutation ages 6 years and older. Since this application extends the review of elexacaftor-tezacaftor-ivacaftor (ETI) down to the age of 2 years and higher, we will mainly address the 2-5 year age group not covered in the previous reviews.
The pre-school years are a time of wonder and challenge for both children and their caregivers. Toddlers are discovering the world and their place as individuals in it. However, they are still figuring out how to manage actions, feelings, and impulses, and a lack of understanding and sometimes inability to express their feelings can lead to the famous terrible 2s (which last though most of the period under review). They are also mastering new skills and growing quickly. This is an important time of development and sets the stage for developing the skills needed to function later in the family setting, with others, and in school. However, when a child this age has a chronic illness, it puts an extra strain on both the child’s physical development as well as their social and emotional development. Medical procedures can promote fear, insecurity, and a feeling of lack of control. Parents have to add on an extra role as provider or supervisor of both daily care and treatments as well as episodic clinic visits and medical interventions. This adds an extra stress on top of the normal challenges, joys, and rewards of parenting toddlers.
We know that the pathologic changes of CF begin to manifest in pre-natal life and infancy. Structural lung changes are detectable in infants and young children with CF. Early attention on nutrition is associated with better long-term outcomes. Early treatment of CF associated infections is also associated with less rapid progression of CF disease. Treatments start at diagnosis, even in infants, and continue throughout life. In the 2–5-year age group not yet eligible for elexacaftor-tezacaftor-ivacaftor (ETI) treatment, common daily treatments include a high fat, high protein diet, supplemental pancreatic enzymes, inhaled mucolytics, bronchodilators, fat soluble vitamin supplementation, chest physiotherapy, and sometimes inhaled antibiotics, ursodiol for liver disease, and inhaled antibiotics. Oral antibiotics are prescribed for some respiratory exacerbations, and while hospitalizations are less common in this age group than in older pwCF, some pre-school children with CF (pcCF) require hospitalization and treatment for respiratory and gastrointestinal manifestations of CF disease.
Regular treatments incur financial costs for families and the healthcare system, but there are also indirect costs to pcCF and their families, including the time spent on CF care and treatments, clinic appointments (recommended at least every 3 months), lost time at work due to appointments, and illnesses, lost daycare/pre-school time, the effects of illness of childhood development, and the stress of knowing that the child has a life-limiting illness. And in recent years, there was the added stressor of having a child who may show respiratory symptoms even when not acutely ill during a respiratory illness pandemic and the effect of this on the child and family. Currently, there is also the added concern of knowing that there exists a treatment (ETI) that can help treat the disease but that is not yet available for their child.
The most important goals for treatment of CF are decreasing the burden and progression of the disease. As noted, this starts at the very beginning. Even in the era of newborn screening, when CF can be detected and diagnosed shortly after birth, young children still have significant manifestations of the disease that are progressive. An ideal treatment would be one that did not significantly increase the burden of treatment of the disease, would be easy for caregivers to administer and children to take, and would be easy to include in daily life and would not make them stick out excessively from other children. It would significantly decrease symptoms, have a positive effect on the multiple organ systems affected by CF, and slow disease progression. Decreasing or slowing the onset of pancreatic insufficiency would be a major impact, since early nutritional status is associated with later disease burden and feeding children with CF can be a stressful and difficult challenge for caregivers. Toddlers can be notoriously challenging when it comes to feeding, so adding on the burden of maintaining a high-fat, high-protein diet and the effect of poor weight gain on future health encountered in a child with CF further magnifies the common challenges of feeding a toddler.
The cost of HEMTs is a barrier that cannot be ignored. The $50,000 QALY standard frequently used in calculations of cost- effectiveness was not developed for medications and diseases that are present and progressive throughout the lifespan, starting in infancy for most pwCF. We are aware that this is a very expensive medication, but it is also difficult to place a cost on the significant burden of the disease on young, developing children and the effect that early disease and disease progression will have on their health and other aspects of their lives. We call on all parties concerned to help make these medications available to Canadians with CF in a equitable manner at a reasonable and bearable cost.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Currently available treatments for most children under 6 years with CF help to slow disease progression and treat the manifestations and sequelae of the disease, but they do not address the underlying cause of the disease, which is lack of adequate production or function of the cystic fibrosis transmembrane regulator protein. They mainly focus on the symptoms and complications of CF and do not treat the biochemical basis of the disease.
Ivacaftor is a HEMT that has been shown to be effective in pwCF ages 2-5 years with a CFTR gating mutation. A clinical trial showed improvement in sweat chloride measurements, body mass index (BMI) z-score, and fecal elastase (a measure of pancreatic exocrine function). An open label extension study showed that improvements in these measures were maintained. In clinical trials, the response to ETI in participants with at least one copy of F508del is of a similar magnitude and significance to the response seen to ivacaftor in the CFTR gating mutation group.
However, the effects of lumacaftor-ivacaftor, the only CFTR modulator currently approved in the 2 years and older age group and only for pwCF who have two copies of the F508del CFTR mutation, showed more modest improvements in study outcomes.
Lumacaftor-ivacaftor is not universally covered by payers in Canada and is not available for pwCF who only have one F508del mutation.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Elexacaftor-tezacaftor-ivacaftor addresses the biochemical mechanism at the root of CF – abnormal transmembrane chloride transport due to a lack of and/or malfunctioning of the CFTR protein. Currently, CFTR modulators are added on to current CF therapies. Because they are taken orally twice a day, the additional burden of treatment is limited. Studies are ongoing to determine if the use of HEMT will allow pwCF to discontinue some other treatments, but only short-term study results are currently available and this is a topic of study and discussion in the CF community.
Because we know that the disease manifestations in CF occur from infancy, ETI would be a first-line, early treatment. Children and other pwCF stating ETI would already be treated with other standard CF therapies prior to starting ETI.
Because CF is a genetic, progressive chronic disease, manifestations of the disease start in early life. Treatment with this potentially disease altering medication should not be withheld until persons become more symptomatic or until lung function deteriorates below an arbitrary threshold.
Because the response to ETI in clinical trials has been shown to be superior to that of lumacaftor-ivacaftor, side effect profiles are generally comparable, and the cost in Canada is also comparable, there are no advantages in prescribing lumacaftor-ivacaftor instead of ETI in pwCF ages 2 and older with eligible CFTR mutations.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Because the response to ETI is different depending on the type of mutation in CFTR, it is currently approved for persons who carry at least one F508del mutation. Some other mutations that show a response to ETI have been added to the indication for therapy in the United States. Almost all pwCF in Canada are diagnosed and followed by accredited CF clinics. The identification of those eligible and likely to benefit from treatment will be identified based on genotype and evaluation by an experienced CF clinician. Issues with diagnosis are extremely unlikely. Publication of the Canadian Clinical Consensus Guideline for Initiation, Monitoring and Discontinuation of CFTR Modulator Therapies for Patients with Cystic Fibrosis provides a standardized approach for determining whether pwCF are eligible for treatment with CFTR modulators.
The Consensus Guideline states: “CFTR modulators should be initiated at the YOUNGEST age possible with the goal of attenuating disease progression and improving clinical status.”
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Monitoring outcomes in the 2-5 year age group is not as simple as in older pwCF. General outcomes that are commonly monitored in all CF clinics at this age include general health (energy, development, well-being), laboratory tests, growth (height, weight, BMI), pulmonary exacerbation, other organ system complications (liver disease, gastrointestinal manifestations, nasal and sinus disease), antibiotic use, and hospitalizations/home IV antibiotic courses.
Some CF clinics can monitor sweat chloride, quality of life measures, and advanced radiologic testing, but these measures are not universally available for monitoring therapy at all centres. In some centres, the sweat chloride labs can only manage the number of tests needed for diagnosis of new CF patients. Advanced radiologic testing, such as chest CT or MRI, is often difficult to obtain for children in this age group in smaller centres and adds the extra burden and risk of required sedation to obtain usable images.
In young children, routine measurement of lung function is complicated and often difficult. Techniques used in clinical trials, such as lung clearance index, are not available in all centres and the devices commonly used in the trials do not have Health Canada approval for clinical use (research use only). Lung function testing in this age group either requires complicated involuntary techniques or is unreliable due to the child’s inability to perform voluntary maneuvers such as spirometry. Most children can reliably perform spirometry before the age of 8 years, but that is out of the age range currently under discussion.
Because of the effect of the medication on general health and the common measures performed in clinic, these should be sufficient to monitor for response to therapy.
What factors should be considered when deciding to discontinue treatment with the drug under review?
As with any treatment, discontinuation should be considered if a severe side effect, allergy, or other adverse event occurs. With this medication, the development of signs of worsening liver disease or other significant side effects may require stopping treatment. The Canadian Clinical Consensus guidelines provide recommendation for dose reduction or discontinuation.
Most adverse effects in the clinical trial of ETI in the 2-5 year age group did not require discontinuing the medication.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Because regular follow up in the CF clinic is recommended at least every three months, pwCF on ETI can be followed closely for clinical effect and monitoring for adverse events.
Additional Information
When families receive the news that their infant has been diagnosed with CF, they commonly run the gamut of emotions: joy at having a new infant, mourning the perceived impossibility of fulfilling their dreams for their new child once the diagnosis is confirmed, having to add CF treatments and therapies to the normal tasks related to raising children, fear that each illness, even common childhood illnesses, could lead to weight loss, lung damage, and faster progression of their child’s CF. The stakes of periods of picky eating during the toddler years are higher and add to the stress already present. Leaving a child with a caregiver for a night out or a weekend away involves finding someone they can trust with daily therapies and medications. We see young children who already require hospitalizations and IV antibiotic treatments, gastrostomy or nasogastric feeding tubes, or other investigations and procedures. While difficult enough for older children, teens, and adults with CF, they are much more challenging in young children who cannot understand why.
We have seen how HEMTs have improved the health and lives of pwCF ages 6 and up. We know that health in early childhood, especially in those with CF, is crucial to early development and future health. Extending availability of HEMT to all pwCF ages 2 and older who can benefit from this treatment will help get that earlier start on improving the symptoms and slowing the progression of CF.
HEMT provides pwCF and those close to them home and an improved ability to handle the burden of the disease and allows those dreams for the future to seem more possible.
Conflict of Interest Declarations — Canadian Cystic Fibrosis Clinicians
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation.
Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Cystic Fibrosis Canada provided an updated list of the email addresses of CF clinic directors.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No help was received for analyzing or collection information from outside the clinician group. We referenced the Cystic Fibrosis Canada annual registry report for demographic data.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Martha L. McKinney, MD MPH
Position: Pediatric Respirologist and CF Clinic Physician, Stollery Children’s Hospital, Edmonton, AB
Date: 15-05-2023
Table 7: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex Pharmaceuticals | X | — | — | — |
Declaration for Clinician 2
Name: Nancy Porhownik
Position: Respirologist, CF Clinic Director, Winnipeg, MB
Date: May 22, 2023
Table 8: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Name: Elizabeth Anne Hicks
Position: Associate professor, University of Alberta, Pediatric Respiratory Medicine and Pediatric Respirologist, Alberta Health Services (Stollery Children’s Hospital), Edmonton, AB
Date: 22/05/2023
Table 9: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex Pharmaceuticals | X | — | — | — |
Declaration for Clinician 4
Name: Tamizan Kherani MD MSc
Position: Pediatric respirologist and pediatric cystic fibrosis medical director, Stollery Children’s Hospital
Date: May 23, 2023
Table 10: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex Pharmaceuticals | — | — | — | X |
Declaration for Clinician 5
Name: Abid Lodhi
Position: Pediatric CF Clinic Director, Regina, SK
Date: 21 May 2023
Table 11: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Steven Kent
Position: CF Clinic Director, VGH Pediatrics, Victoria, BC
Date: 22-05-2023
Table 12: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex | X | — | — | — |
Declaration for Clinician 7
Name: Larry Lands
Position: Director, Pediatric Respiratory Medicine and Pediatric Cystic Fibrosis Clinic, Montreal Children’s Hospital- McGill University Health Centre
Date: 22-05-2023
Table 13: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 7
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex Pharmaceuticals | — | X | — | — |
Declaration for Clinician 8
Name: Mary Jane Smith, MD FRCPC
Position: Cystic Fibrosis Clinic Director, Janeway Children’s Health and Rehab Centre, St. John’s NL
Date: 05-23-2023
Table 14: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 8
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex Pharmaceuticals | X | — | — | — |
Declaration for Clinician 9
Name: Winnie Leung, MD FRCPC
Position: Adult respirologist, Medical Director of Edmonton Adult Cystic Fibrosis Clinic
Date: 23 May 2023
Table 15: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 9
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex Pharmaceuticals | X | — | — | — |
Declaration for Clinician 10
Name: Melinda Solomon
Position: Pediatric Respirologist and CF Clinic Director, Hospital for Sick Children, Toronto, ON
Date: 22-05-2023
Table 16: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 10
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex | — | X | — | — |
Declaration for Clinician 11
Name: Dr. Mark Chilvers
Position: BC
Date: 22-05-2023
Table 17: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 11
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex Pharmaceuticals | — | X | — | — |
Declaration for Clinician 12
Name: Grace Lam
Position: Assistant Professor and Adult CF Respirologist at the University of Alberta Hospital
Date: 24-05-2023
Table 18: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 12
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche Diagnostics | — | — | — | X* |
Boehringer Ingelheim | X | — | — | — |
Respiplus | X | — | — | — |
*Research funding.
Declaration for Clinician 13
Name: Nita Chauhan, MD FRCPC
Position: Pediatric Respirologist and Pediatric CF Clinic Director, Jim Pattison Children’s Hospital, Saskatoon, SK
Date: 24-05-2023
Table 19: COI Declaration for Canadian Cystic Fibrosis Clinicians — Clinician 13
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Vertex Pharmaceuticals | X | — | — | — |
Other Clinicians who did not provide conflict of interest information but who endorsed the submission:
Dr. Linda Pedder, MD, Pediatric CF Clinic director, McMaster University
Dr. Joe Reisman, MD, Pediatric CF Clinic director, Children’s Hospital of Eastern Ontario
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.