CADTH Reimbursement Review
Upadacitinib (Rinvoq)
Sponsor: AbbVie Corporation
Therapeutic area: Crohn disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
5-ASA
5-aminosalicylate
AE
adverse event
AESI
adverse event of special interest
AO
as observed
AP
abdominal pain
bio-RR
biologic therapy–intolerant or inadequate responder
CCC
Crohn’s and Colitis Canada
CD
Crohn Disease
CDAI
Crohn's Disease Activity Index
CI
confidence interval
CMH
Cochran-Mantel-Haenszel
CPK
creatine phosphokinase
CR-70
decrease of at least 70 points from baseline in the Crohn’s Disease Activity Index
CR-100
decrease of at least 100 points from baseline in the Crohn’s Disease Activity Index
CrI
credible interval
CRP
C-reactive protein
EIM
extra-intestinal manifestation
EMA
European Medicines Agency
GI
gastrointestinal
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HBI
Harvey-Bradshaw Index
HRQoL
health-related quality of life
IBD
inflammatory bowel disease
IBDQ
Inflammatory Bowel Disease Questionnaire
IL
interleukin
ITC
indirect treatment comparison
ITT1
intention-to-treat (part 1 or cohort 1 of study)
JAK
Janus kinase
MID
minimally important difference
MMRM
mixed model for repeated measures
NRI-NC
nonresponder imputation with no special data-handling to account for COVID-19
PRO
patient-reported outcome
RCT
randomized controlled trial
SAE
serious adverse event
SES-CD
Simple Endoscopic Score for Crohn’s Disease
SF
stool frequency
TNF
tumour necrosis factor
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Upadacitinib (Rinvoq), 15 mg, 30 mg, and 45 mg once daily, extended-release tablets, oral |
Sponsor | AbbVie Corporation |
Indication | For the treatment of adult patients with moderately to severely active Crohn disease who have demonstrated prior treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy |
Reimbursement request | Consistent with indication |
Health Canada approval status | Approved |
Health Canada review pathway | Standard |
NOC date | October 12, 2023 |
Recommended dose | Adults aged 18 to 64 years:
Adults aged ≥ 65 years:
|
NOC = Notice of Compliance.
Introduction
Crohn disease (CD) is a chronic and progressive form of inflammatory bowel disease (IBD) that leads to significant disability and negatively affects a patient’s health-related quality of life (HRQoL).1-3 It is characterized by recurrent, uncontrolled inflammation that can affect any part of the gastrointestinal (GI) tract from mouth to anus, and primarily affects the ileum, colon, and rectum.4,5 Common symptoms include diarrhea, abdominal pain, fatigue, fever, rectal bleeding, loss of appetite, weight loss, and malnutrition.6,7 Complications associated with CD include bowel obstructions, fistulas, anal fissures, intra-abdominal and other abscesses, and ulcers.8,9
Canada has the highest incidence and prevalence of IBD in the world.10,11 According to the 2018 Impact of Inflammatory Bowel Disease in Canada report, approximately 270,000 Canadians were living with IBD, of whom 135,000 had CD. For every 100,000 Canadians, 16.3 new cases of CD are diagnosed each year.12
Diagnosis of CD requires a combination of a medical history and physical examinations.6 Key biomarkers used in laboratory tests include C-reactive protein (CRP), erythrocyte sedimentation rate, and fecal calprotectin.6,13-15 In active CD, CRP levels can range from 5 mg/L to 20 mg/L, depending on disease severity, compared to typical baseline levels of 1 mg/L.15 Diagnostic imaging, including endoscopy, radiology, and ultrasound, are also used to examine findings of typical CD cases, including bowel obstruction, stenosis, fistulae, abscesses, and atrophy.14,16-19
Upadacitinib is a selective and reversible Janus kinase (JAK) inhibitor engineered to have greater inhibitory potency against JAK1 proteins versus JAK2, JAK3, and TYK2 in human cellular assays. Upadacitinib preferentially inhibits signalling by JAK1 or JAK1/JAK3 with functional selectivity over cytokine receptors that signal via pairs of JAK2 proteins.20,21
The indication for upadacitinib is for the treatment of adult patients with moderately to severely active CD who have demonstrated prior treatment failure (i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy). The sponsor’s reimbursement request is consistent with the indication.
Upadacitinib is administered orally. The recommended dosing schedule involves 2 phases; a 12-week induction phase consisting of 45 mg upadacitinib once daily, followed by an ongoing maintenance phase. The recommended dosage of upadacitinib for maintenance treatment of patients aged between 18 and 64 years is 15 mg or 30 mg once daily, based on patient presentation; a dosage of 30 mg once daily may be appropriate for patients with high disease burden (such as refractory or severe disease) or those who do not show adequate therapeutic benefit with 15 mg once daily. The lowest effective dose for maintenance should be used. For patients who are aged at least 65 years, the recommended maintenance dosage is 15 mg once daily. In patients responding to induction or maintenance treatment with upadacitinib, corticosteroids may be reduced and/or discontinued in accordance with standard of care.22,23
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from 1 clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, Crohn and Colitis Canada (CCC) and the GI Society, provided input for this review. CCC gathered information from a report published in 2018 (Impact of Inflammatory Bowel Disease in Canada), a survey, and interviews with patients who participated in the upadacitinib clinical trials. The patient input provided by the GI Society was based on surveys, interviews, a patient roundtable, and media interactions.
The 2 patient groups emphasized that CD has a tremendous impact on every aspect of a person’s life. The most frequent symptoms associated with CD reported by the patients are diarrhea, rectal bleeding, abdominal pain, and weight loss. Other symptoms included inflammation of the eyes or joints, ulcers of the mouth or skin, tender and inflamed nodules on the shins, anemia, anxiety, and stress. Both CCC and the GI Society stated that being unable to predict when the next urgent need for a bowel movement would occur, and the inability to control the flare, had a significant negative impact on the personal and social lives of patients with CD.
The GI Society described the treatment of CD as multifaceted as it involves managing symptoms and consequences of the disease and reducing inflammation. Patients also rely on medications to reduce the need for surgery. First-line treatments include 5-aminosalicylate (5-ASA) and corticosteroids to reduce inflammation in moderate to severe cases of CD. When 1 medication fails, patients must try another. According to the patient input, these treatments are inconvenient therapies that make it difficult for patients to keep a normal routine. As described by the GI Society, JAK inhibitors (such as the drug under review) are a newer class of medication. Unlike other biologics delivered by infusion therapy, JAK inhibitors are easier and more convenient to take as they are in pill form. Patients have difficulty achieving remission or adequate symptom relief despite available treatment options. Even after surgery to repair fistulas and fissures or removal of diseased bowel tissue, CD symptoms tend to recur in most patients.
Improved outcomes described as important by the patient groups included symptom mitigation and a reduction in preventable patient suffering. Managing unpredictable and frequent bowel movements, pain, and fatigue were also noted as important by CCC respondents. Unmet patient needs were noted by CCC to vary among individuals depending on their specific symptoms and life circumstances. Both patient groups emphasized the importance of a treatment option that is easy to administer and can provide symptom relief, achieve remission, and improve subsequent HRQoL.
Three patients from CCC and 2 patients from the GI Society group who had experience with upadacitinib reported near-immediate improvements in their health, alleviation of disease symptoms, and either no side effects or few mild side effects, such as weight gain. Patients noted the convenience of pill-based administration and the lack of need to refrigerate the medication or attend a clinic for infusions.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The clinical expert consulted by CADTH for this review indicated that there are profound treatment gaps in the management of IBD, including CD. Over time, transmural damage results in complications (e.g., stenosis and penetrating complications that often require surgery). Early treatment initiation is key to limiting disease activity and stopping progression. Although several effective drugs are available for the treatment of moderate to severe CD, there are significant limitations in efficacy, in addition to the frequency of loss of response over time and reduced efficacy with the introduction of each subsequent biologic after failure. This was described as the greatest treatment challenge in the management of CD.
The clinical expert noted that both primary nonresponse and secondary loss of response are common in the treatment of CD with advanced therapies. Therapies that remain efficacious in patients who have experience with biologics are therefore needed.
Clinical trial design has historically focused on clinical symptoms of response and remission, which may not always correlate with objective measures such as endoscopic remission and mucosal healing. The clinical expert pointed out that long-term longitudinal studies to evaluate the modification of bowel damage are lacking as most clinical trials are up to 2 years in duration.
According to the clinical expert, some treatments are particularly inconvenient and can affect a patient’s lifestyle due to the need to visit an infusion clinic for a few hours every 4 to 8 weeks. Therapies given by subcutaneous injection are more convenient, but patients may find them painful. No orally administered advanced therapies for CD are currently available.
The clinical expert noted that the current treatment paradigm for adults with moderately to severely active CD is complex and dictated by disease phenotype. Conventional therapies (e.g., steroids and immune suppressants) are not typically used in isolation over the long-term, and most patients with moderately to severely active disease go on to receive advanced therapy, such as a biologic. The clinical expert noted that the first therapy to be prescribed has the best chance of resulting in improvement and healing due to the aforementioned pattern of a lower likelihood of a robust response with subsequent advanced therapies. Selecting the optimal therapy from the start is a challenge and is based on disease phenotype, disease severity, and the risks and expected onset of action of each available therapy. Particularly severe disease would warrant selection of a therapy with rapid onset, high efficacy, and steroid-sparing effects (e.g., anti–tumour necrosis factors [TNFs] or anti–interleukin [IL]-23 and IL-12/IL-23).
Nearly half of patients have extra-intestinal manifestations (EIMs) of CD, which can be disabling, and only a select few of the currently available medications address them, with a preference for the anti-TNF category. Approximately a quarter of patients with CD have fistulizing perianal disease, which is a marker of severe disease, and again anti-TNFs are the preferred therapeutic option for this subpopulation. Other options for patients with these disease phenotypes are needed.
The clinical expert indicated that upadacitinib would be used as a first-line drug for patients receiving advanced therapies for CD, and that no mechanism-, efficacy-, or sequencing-based argument requires the failure of other advanced drugs before initiation of upadacitinib.
The clinical expert acknowledged that there is increasing off-label use of combination therapies with complementary mechanisms of effect in particularly severe, high-risk patients with prior drug failures, surgeries, or other markers of disabilities. The expert stated that combinations would typically include a low-risk, safe drug, such as an anti-integrin, with other more systemically active drugs. This may become relevant in the case of upadacitinib. However, the expert emphasized that this use is limited to dire situations in which there is a risk of extensive surgery or disability.
According to the clinical expert, patients with confirmed pathologic or histologic diagnosis of moderate to severe CD are typically diagnosed by a gastroenterologist. Misdiagnosis is rare, but diagnosis may be delayed, as previously noted. Patients with EIMs (e.g., inflammatory arthropathy, peripheral or axial) are a priority for treatment. Although there are no clear “stages” of CD, objective measures such as endoscopic activity and the requirement for, or dependence on, corticosteroids are important, while the presence of clinical symptoms is not as critical. There are no established predictors of disease response.
The clinical expert noted that assessment of response in clinical practice differs from clinical trials due to logistics and patient preference. The most easily accessed marker of response is improvement in clinical symptoms (abdominal pain [AP] and frequency of soft or liquid stools in particular), but this is weakly correlated with objective markers of disease activity and may be heterogeneous according to disease phenotype. For example, patients with bowel stricture may experience constipation instead of diarrhea, and patients with prior surgeries may have differing symptoms caused by anatomic alteration rather than inflammation. Objective measures of disease activity are important, particularly endoscopy (i.e., ileocolonoscopy). The clinical expert noted that, although the clinical trials assessed endoscopic outcomes at 12 weeks, endoscopy is rarely performed at 12 weeks in clinical practice, and instead is typically performed at 6 to 9 months, and can be challenging to repeat. Other objective measures may include biomarkers (CRP and fecal calprotectin) as well as noninvasive intestinal ultrasound scans.
The clinical expert indicated treatment discontinuation should be considered in a manner similar to that of other advanced therapies for adults with CD, and based on a combination of clinical symptoms and objective data that support primary nonresponse or loss of response, including:
persistence or worsening of clinical symptoms, most importantly diarrhea and AP
persistence or worsening of endoscopic activity
worsening or persistent elevation of biomarkers, including CRP and fecal calprotectin
worsening or development of complications (including strictures and penetrating disease) on cross-sectional imaging
dependence on or need for recurrent courses of corticosteroids (e.g., 2 or more full courses of oral prednisone within 1 year, but details may be debated)
development of adverse events (AEs), weighed on a case-by-case basis depending on treatability and severity of the AE; all patients should be vaccinated appropriately (e.g., for varicella) to avoid any preventable AEs potentially associated with treatment
circumstances when patients with severe disease may require a single course of corticosteroids, which may not preclude ongoing maintenance and therefore the need for discontinuation would be judged by the treating physician.
Prescription of upadacitinib should not be limited to IBD specialists, as general gastroenterologists would have the expertise required to initiate therapy. General internists with a special interest in IBD and/or GI may have sufficient experience and training to prescribe upadacitinib, and this may be important for accessibility in rural regions of Canada.
The clinical expert noted that initiation criteria should be similar to those for other biologics. However, the expert emphasized that the current requirements for previously failed therapies are not up to date with clinical practice. In particular, the current requirements for prior drug failures in prescribing advanced therapies includes 5-ASA, which was considered by the clinical expert to be out of date due to its known lack of efficacy in this population. The clinical expert noted that, in clinical practice, this results in short-term prescriptions of 5-ASA to meet the requirements when it is not expected to have patient benefit, and the expert recommended that this not be a requirement for prior treatment failures when prescribing upadacitinib. The expert noted that, if treatment is interrupted for at least 2 weeks, the patient may need to undergo another course of induction therapy.
Clinician Group Input
One clinician group, the Canadian IBD Specialist Group, responded to CADTH’s call for input. The input was based on a discussion held by the group in March 2023.
The clinician group emphasized that CD has a tremendous impact on the physical, emotional, and social aspects of those living with the disease, affecting HRQoL and causing a significant economic burden. The current treatment paradigm for CD includes 5-ASA, corticosteroids, immune modifiers, and biologics that include anti-TNFs, anti-integrin, and anti–IL-12/IL-23 and anti–IL-23 agents.
The input from the clinician groups identified the same unmet medical needs for patients with CD and potential place in therapy for the drug under review as the clinical experts consulted by CADTH.
The clinician group identified significant unmet therapeutic needs for patients living with moderate to severe CD. There is a lack of safe and effective treatments that could rapidly improve endoscopic appearance, maintain long-term improvement and remission, and reduce the risk of complications and need for surgery.
In addition to relieving clinical symptoms, the clinician group emphasized that the goal of treatment should focus on changing the course of disease for patients with CD, preventing further intestinal damage, avoiding disability, and reducing the overall cost of care.
The clinician group noted that upadacitinib has a novel mechanism of action, and it is the first oral therapy for CD to be evaluated to meet the treatment goals. According to the clinician group, any patient with an inadequate response or intolerance to corticosteroids or multiple advanced therapies and those with 1 or more extraintestinal IBD manifestations could benefit from upadacitinib.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that may affect the implementation of a CADTH recommendation for upadacitinib:
relevant comparators
consideration for continuation or renewal of therapy
considerations for discontinuation of therapy
consideration for prescribing of therapy
care provision issues
system and economic issues.
The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug program. Table 5 provides more details.
Clinical Evidence
Systematic Review
Description of Studies
Three phase III, double-blind, placebo-controlled, multicentre, international randomized controlled trials (RCTs) are included in this review. Two of the RCTs were induction studies in adult patients with moderately to severely active CD and a history of either biologic failure (U-EXCEED) or biologic and/or conventional therapy failure (U-EXCEL). Patients in both induction studies were randomized in a 2:1 ratio to receive upadacitinib 45 mg or placebo once daily. The primary results for randomized cohorts were evaluated at 12 weeks (“part 1”), although patients who did not achieve an adequate response could carry on to part 2 or 3 for extended induction. An adequate response was defined as a decrease of 30% or more in average daily very soft or liquid stool frequency (SF) and/or a decrease of 30% or more in the average daily AP score (neither worse than baseline). The third RCT (U-ENDURE) was a maintenance study of upadacitinib 15 mg or 30 mg once daily versus placebo in patients who had achieved an adequate response in either the U-EXCEED or U-EXCEL trial, and the primary results for re-randomized patients from part 1 of the induction studies were evaluated at 52 weeks. Patients who had carried on to extended induction therapy in either of the induction studies and thereafter achieved a response could also enrol in cohort 2 or 3 of the U-ENDURE trial, which were not randomized. The coprimary outcomes in all trials included clinical remission (based on patient-reported outcomes [PROs] or the Crohn Disease Activity Index [CDAI]), and endoscopic response (based on the Simple Endoscopic Score for Crohn Disease [SES-CD]). Other important outcomes included endoscopic remission, the proportion of patients who discontinued corticosteroid use for CD and achieved clinical remission (among patients taking corticosteroids at induction baseline), the proportion of patients who achieved both clinical remission and endoscopic remission, change in HRQoL (using the Irritable Bowel Disease Questionnaire [IBDQ]), a decrease of at least 100 points from baseline in the CDAI (CR-100), resolution of EIMs in patients who had EIMs at induction baseline, the proportion who experienced CD-related hospitalizations or surgeries, and the proportion who experienced harms, including serious adverse events (SAEs) or adverse events of special interest (AESIs).
The U-EXCEED trial enrolled 624 patients across 229 sites in 39 countries; the U-EXCEL trial enrolled 526 patients across 209 sites in 42 countries; and the U-ENDURE trial enrolled 901 patients across 277 sites in 43 countries. In the double-blind cohorts of the U-EXCEED, U-EXCEL, and U-ENDURE trials, there were slightly more male than female patients (53.5%, 53.8%, and 55.4%, respectively), and the mean ages were 38.1, 39.6, and 37.0 years, respectively. The majority of enrolled patients were white (approximately 70% in each trial, followed by Asian, Black, multiple races (unspecified in the study), and American Indian or Alaska Native). The mean durations of CD were 9.4 years in the U-EXCEED trial, 6.1 years in the U-EXCEL trial, and 7.2 years in the U-ENDURE trial; the differences were expected given that the U-EXCEL trial included patients who may not have experienced failure with biologics, indicating an earlier point in treatment history on average. Overall, most enrolled patients in the randomized cohorts had a history of biologic failure (100% in the U-EXCEED trial by design, 45.4% in the U-EXCEL trial, and 75.6% in the U-ENDURE trial).
Efficacy Results: Induction
Induction Outcomes (12 Weeks)
Clinical Remission as Measured by PROs at 12 Weeks: In the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved clinical remission as measured by PROs among those treated with upadacitinib 45 mg compared with those who received placebo. In the U-EXCEED trial, the response-rate difference compared to placebo was 25.9% (95% confidence interval [CI], 18.7% to 33.1%), and in the U-EXCEL trial it was 28.7% (95% CI, 20.9% to 36.4%).
Results were consistent across subgroups based on the number of prior biologics failed and the analysis for clinical remission as measured by PROs at 12 weeks.
Clinical Remission as Measured by CDAI at 12 Weeks: In the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved clinical remission as measured by CDAI among those treated with upadacitinib 45 mg compared to those who received placebo. In the U-EXCEED trial, the response-rate difference compared to placebo was 17.9% (95% CI, 10.0% to 25.8%) and in the U-EXCEL trial it was 20.8% (95% CI, 12.7% to 28.8%).
Results were consistent across subgroups based on the number of prior biologics failed and the analysis for clinical remission as measured by CDAI at 12 weeks.
Endoscopic Response at 12 Weeks: In the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved endoscopic response among patients treated with upadacitinib 45 mg compared to those who received placebo. In the U-EXCEED trial, the response-rate difference compared to placebo was 31.2% (95% CI, 25.5% to 37.0%) and in the U-EXCEL trial it was 33.0% (95% CI, 26.2% to 39.9%).
Results were consistent across subgroups based on the number of prior biologics failed and the analysis for endoscopic response at 12 weeks.
Endoscopic Remission at 12 Weeks: In the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved endoscopic remission among those treated with upadacitinib 45 mg compared with those who received placebo. In the U-EXCEED trial, the difference compared to placebo was 16.8% (95% CI, 12.0% to 21.6%) and in the U-EXCEL trial it was 21.8% (95% CI, 15.8% to 27.8%).
Discontinuation of Corticosteroid Use and Clinical Remission as Measured by CDAI at 12 Weeks Among Patients Taking Corticosteroids at Baseline: In the U-EXCEED and U-EXCEL trials, a higher proportion of patients treated with upadacitinib 45 mg discontinued corticosteroid use and had CDAI clinical remission at week 12 compared to the placebo group. In the U-EXCEED trial, the difference compared to placebo was 22.5% (95% CI, 11.1% to 34.0%) and in the U-EXCEL it was 27.7% (95% CI, 15.7% to 39.8%).
Results were similar for discontinuation of corticosteroid use and clinical remission as measured by PROs at 12 weeks.
Clinical Remission as Measured by CDAI and Endoscopic Remission at 12 Weeks: | |||||||| | |||||||| |||||| |||||||||| || ||||||||||||||||||| |||||||||||| || || || |||||||| ||||||||| || |||| || |||||||||| ||||||||| || || |||||| || |||||||| |||||||||| |||||||| || ||||||| || ||||| |||| || |||| || |||||| || ||||| |||| ||||| || ||||||| |||||||||| |||||||| |||||||||||||
Change From Baseline in IBDQ Total Score at 12 Weeks: In the U-EXCEED and U-EXCEL trials, there was a larger within-group change from baseline in IBDQ total score in patients treated with upadacitinib 45 mg than in patients treated with placebo. The between-group difference compared to placebo (least squares mean) was 24.3 (95% CI, 17.2 to 31.5) in the U-EXCEED trial and 21.8 (95% CI, 15.6 to 28.1) in the U-EXCEL trial.
CR-100 at 12 Weeks
In the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved CR-100 among patients treated with upadacitinib 45 mg than in those who received placebo. In the U-EXCEED trial, the difference compared to placebo was 22.8% (95% CI, 14.4% to 31.2%), and in the U-EXCEL it was 19.8% (95% CI, 11.3% to 28.4%).
Resolution of EIMs at 12 Weeks in Patients With EIMs at Baseline: In the U-EXCEED trial, resolution of EIMs at week 12 in patients with any EIMs at baseline was 32.8% for upadacitinib 45 mg versus 21.7% for PBO (between-group difference = 11.5%; 95% CI, −1.5% to 24.4%). In the U-EXCEL trial, resolution of EIMs at week 12 in patients with any EIMs at baseline was 28.5% for upadacitinib 45 mg versus 20.9% for placebo (between-group difference = 9.0%; 95% CI, −1.9% to 19.9%). In both cases, the 95% CI crossed the threshold between potential benefit and potential harm (i.e., null).
Proportion With CD-Related Hospitalizations Through 12 Weeks: No substantial differences were observed in the proportion of patients with CD-related hospitalizations between patients treated with upadacitinib 45 mg in the U-EXCEED trial (20 of 324) and the U-EXCEL trial (13 of 350) compared to patients treated with placebo (15 of 171 and 9 of 176, respectively). The differences compared to placebo were −2.6% (95% CI, −7.6% to 2.4%) in the U-EXCEED trial and −1.4% (95% CI, −5.2% to 2.4%) in the U-EXCEL trial. In both cases, the 95% CI crossed the threshold between potential benefit and potential harm (i.e., null).
Proportion With CD-Related Surgeries Through 12 Weeks: |||||| ||||||||| ||||||| | ||||||||||| ||||||||||| |||||||||| ||||||||| |||| |||||||| || |||||||||| || ||||||||||||| || || ||||| |||||||| || | ||||||| ||||| || |||| |||| || ||||| || ||||| || |||||||| || |||| |||||| || ||||| || |||||||| || |||| |||||| || || || ||||||| || ||||||||| ||||||| ||||||||| |||||||| ||||||||| |||| |||||| ||||||
Maintenance Outcomes (52 Weeks)
Clinical Remission as Measured by PROs at 52 Weeks: In the U-ENDURE trial, the upadacitinib 15 mg and 30 mg groups had higher percentages of patients who achieved response rates in clinical remission as measured by PROs compared to the placebo group. The differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups, were 21.9% (95% CI, 13.7% to 30.0%) and 31.8% (95% CI, 23.2% to 40.3%), respectively.
The results were similar for the subgroups of patients for whom treatment had failed using 1 or more prior biologics, 1 prior biologic, and more than 1 prior biologic, as well as for those in the 30 mg group for whom treatment with 0 prior biologics had failed. However, for the subgroup of 0 prior biologics failed in the 15 mg group, the difference (versus placebo) was smaller, and the 95% CI crossed the null value (11.7%; −9.1% to 32.5%).
Clinical Remission as Measured by CDAI at 52 Week: The upadacitinib 15 mg and 30 mg groups had higher percentages of patients who achieved clinical remission as measured by CDAI compared to the placebo group. The differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups, were 23.7% (95% CI, 15.2% to 32.1%) and 32.8% (95% CI, 23.9% to 41.6%), respectively.
Results in the subgroups based on the number of prior biologics failed were consistent with the analysis for both dosage groups.
Endoscopic Response at 52 Weeks: The upadacitinib 15 mg and 30 mg groups had higher percentages of patients who achieved an endoscopic response compared to the placebo group. The differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups were 21.0% (95% CI, 13.6% to 28.4%) and 33.7% (95% CI, 26.0% to 41.3%), respectively.
Results in the subgroups based on number of prior biologics failed were consistent with the analysis for both dosage groups.
Endoscopic Remission at 52 Weeks: The upadacitinib 15 mg and 30 mg groups had higher percentages of patients who achieved endoscopic remission compared to the placebo group. The differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups, were 14.4% (95% CI, 7.7% to 21.0%) and 23.6% (16.1% to 31.0%), respectively.
Discontinuation of Corticosteroid Use at Least 90 Days Prior to Week 52 and Clinical Remission as Measured by CDAI at 52 Weeks Among Patients Taking Corticosteroids for CD at Induction Baseline.
Among patients taking corticosteroids for CD at induction baseline, a higher proportion of patients in the upadacitinib 15 mg and 30 mg groups discontinued corticosteroid use and had CDAI clinical remission at week 52 compared to the placebo group. The differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups, were 35.4% (95% CI, 23.3% to 47.5%) and 32.3% (95% CI, 20.1% to 44.5%), respectively. Among all patients (i.e., not limited to those taking corticosteroids at induction baseline), the differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups, were 23.8% (95% CI, 15.5% to 32.1%) and 32.2% (95% CI, 23.4% to 40.9%), respectively.
Clinical Remission as Measured by CDAI and Endoscopic Remission at 52 Weeks: A higher proportion of patients in the upadacitinib 15 mg and 30 mg groups had both CDAI clinical remission and endoscopic remission at week 52 compared to the placebo group. The differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups were 12.2% (95% CI 6.3% to 18.1%) and 19.8% (95% CI 13.0% to 26.6%), respectively.
Change From Baseline in IBDQ Total Score at 52 Weeks: The within-group change from baseline in IBDQ total score was larger in the patients treated with upadacitinib (15 mg or 30 mg) than in those treated with placebo. The between-group difference (least squares mean) was 12.9 (95% CI, 4.3 to 21.4) when the upadacitinib 15 mg group was compared to the placebo group and 18.1 (95% CI, 9.8, 26.4) when the upadacitinib 30 mg group was compared to the placebo group. Only the between-group difference in the latter comparison (i.e., 30 mg upadacitinib versus placebo) was greater than the reported minimally important difference (MID) of 16 points in the IBDQ total score for patients with CD; the 95% CIs of both comparisons include values both greater than and less than this MID.
CR-100 at 52 Weeks
There was a higher percentage of patients who achieved CR-100 among patients treated with upadacitinib 30 mg or 15 mg compared with placebo. The differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups, were 27.1% (95% CI, 18.3% to 35.8%) and 36.4% (95% CI, 27.5% to 45.2%), respectively.
Resolution of EIMs at 52 Weeks in Patients With EIMs at Induction Baseline: The proportion of patients who achieved resolution of EIMs at week 52 in those with any EIMs at induction baseline was 24.6% (upadacitinib 15 mg), 35.6% (upadacitinib 30 mg), and 15.2% (placebo). The difference versus placebo was 9.6% (95% CI, −3.4% to 22.6%) for upadacitinib 15 mg and 22.0% (95% CI, 9.3% to 34.8%) for upadacitinib 30 mg. For the 15 mg dose but not the 30 mg dose, the 95% CI crossed the threshold between potential benefit and potential harm (i.e., null).
Proportion With CD-Related Hospitalizations Through 52 Weeks: No substantial differences were observed in the percentages of patients who experienced CD-related hospitalizations across the upadacitinib 30 mg, upadacitinib 15 mg, and placebo groups. The differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups were −0.78% (95% CI, −10.4% to 8.8%) and −4.17 (95% CI, −13.1% to 4.7%), respectively. In both cases, the 95% CI was wide and crossed the threshold between potential benefit and potential harm (i.e., null).
Incidence of CD-Related Surgeries Through 52 Weeks: |||||| || ||||||||| ||||||| ||||| || |||| ||||||||| |||| || |||||||||| ||||||||| || || ||||||||||||| |||| |||||||| || |||| |||||||||| |||||| |||||||||||||| || | |||||||||||| || || || || |||||| |||||||| || || ||||||| ||||| |||| |||| |||| || ||||| || ||||| || |||| |||||| || |||||| ||||||||||||| || |||| |||||| | | ||| |||| | ||||||| || ||||||||| ||||||| ||||||||| ||||||| || ||||||||| |||| |||||| ||||||
Harms Results
Across the trials, AEs were common and were experienced by approximately 58% to 76% of patients. In the placebo-controlled parts of the trials, the rates of AEs and withdrawals due to AEs were generally similar between treatment groups. SAEs were reported for approximately 7% to 15% of patients across the different treatment groups and cohorts of the included trials, and they were approximately similar between patients treated with upadacitinib and those who received placebo in the comparative cohorts. Some of the most frequently reported SAEs among all trials were GI disorders, infections, and infestations.
An evaluation of SAEs using Grading of Recommendations Assessment, Development and Evaluation (GRADE) found that upadacitinib induction or maintenance may result in little to no difference in the incidence of SAEs compared to placebo in a 12-week or 52-week time period.
Selection of AESIs was based on safety concerns reported for other JAK inhibitors, upadacitinib data obtained from preclinical studies, and the upadacitinib development program, as well as customary regulatory concerns for novel small-molecule drugs. Across the trials, AESIs of serious infection, opportunistic infection, herpes zoster, adjudicated GI perforation, anemia, neutropenia, lymphopenia, elevated creatine phosphokinase (CPK), hepatic disorder, renal dysfunction, and adjudicated venous thromboembolic events were observed. The most commonly reported AESIs (≥ 4% in any part or cohort of any included trial) included anemia, lymphopenia, serious infections, infections and infestations, herpes zoster, hepatic disorder, and elevated CPK. One adjudicated cardiovascular event was observed in a patient who was treated with placebo in part 1 of the U-EXCEL trial. Malignancies (all types), malignancies (excluding nonmelanoma skin cancer, and nonmelanoma skin cancer were rare in the U-ENDURE trial and not observed in the induction trials (U-EXCEED and U-EXCEL). No cases of lymphoma or active tuberculosis were observed in any included trial.
Critical Appraisal
All 3 trials were phase III, double-blind, placebo-controlled, multicentre assessments of several important clinical, endoscopic, and HRQoL-related outcomes. A review of internal validity raised no concerns related to study design (e.g., method of randomization, concealment of allocation, maintenance of blinding, or balance of patient characteristics between treatment groups). The U-ENDURE maintenance study included an enriched population as only patients with a response to and adequate tolerance of the study drug during induction could enrol, but this is representative of the reality of clinical practice. All trials included nonrandomized cohorts to accommodate patients who needed more than 12 weeks of induction to reach an adequate response; although not represented in the primary analysis, these patients reflect a minority of patients in real-world practice. Only the randomized data are discussed in detail here. In the U-ENDURE trial, patients who enrolled after achieving a response at 12 weeks of induction were re-randomized, which preserved the strength of randomized study design. Additionally, the use of separate induction and maintenance studies was consistent with European Medicines Agency (EMA) guidance for the development of drugs for the treatment of CD. Discontinuation rates were potentially imbalanced, with higher placebo-group withdrawals due to lack of efficacy in the U-EXCEL trial, and they were generally high during the U-ENDURE trial (20% to 28% across cohorts and treatment groups).
The clinical expert consulted by CADTH indicated that the study populations were wholly representative of the target population of adults with moderate to severe CD and a history of treatment failure. The dosage of the intervention, upadacitinib, was 45 mg once daily during the induction studies and either 15 mg or 30 mg once daily during the maintenance study. The clinical expert noted that treatment of moderate to severe CD in clinical practice would lean more often toward a maintenance dosage of 30 mg once daily due to evidence of greater efficacy and a reluctance to potentially undertreat, given the irreversible nature of the bowel damage that can occur. However, the clinical expert and the product monograph also noted that patients should be treated with the lowest effective dose in the interest of safety, and that the approach to dosing may vary by treating physician and disease severity. All 3 RCTs were placebo-controlled, and there is a lack of direct, head-to-head comparisons of active therapies. The 3 RCTs were unique among CD trials in that there was a mandatory tapering of corticosteroids, which was considered to be reasonably similar to clinical practice. Overall, the selected primary and key secondary outcomes were relevant to decision-making and/or clinical practice, and adequately reflected measures of both efficacies and harms. The duration of follow-up was appropriate for the induction and maintenance phase of treatment. However, when measuring the proportion of patients who experienced events such as hospitalizations or surgeries related to CD, both a 12-week and 52-week time frame were considered to be inadequate to experience a difference between arms, which contributed to uncertainty in interpreting these outcomes. Additionally, the clinical expert noted that, in clinical practice, endoscopy is not typically conducted at 12 weeks but rather after 6 to 9 months of initiating treatment due to practical limitations and the invasiveness of the procedure. This logistical limitation was also considered by the expert to be a factor in decision-making around dosing, as patients without symptoms may be experiencing endoscopic activity that would not be seen until the procedure could be completed.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to informing CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.24,25 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down in response to concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: clinical remission as measured by PROs, clinical remission as measured by CDAI, endoscopic response, endoscopic remission, discontinuation of corticosteroid use for CD and CDAI clinical remission in patients taking corticosteroids at induction baseline, endoscopic remission and CDAI clinical remission, change in IBDQ, CR-100, resolution of EIMs among patients who had EIMs at induction baseline, CD-related hospitalization, CD-related surgery, and SAEs.
The induction studies (U-EXCEED and U-EXCEL) were assessed together due to their similarities in population and study design and are reported in Table 2. The maintenance study (U-ENDURE) is reported separately in Table 3, and GRADE assessments were conducted independently for the 2 dosages of upadacitinib maintenance therapy (15 mg or 30 mg once daily).
Indirect Comparisons
One indirect treatment comparison (ITC) was submitted by the sponsor to estimate the relative efficacy and safety of upadacitinib versus advanced therapies for the treatment of adult patients with moderately to severely active CD.27,28
Description of Studies
Studies included in the ITC enrolled adult and adolescent patients with moderately to severely active CD receiving advanced treatments for CD, namely upadacitinib, vedolizumab, ustekinumab, risankizumab, adalimumab, and infliximab. Efficacy outcomes included clinical outcomes (remission and response), endoscopic outcomes (remission and response), and safety outcomes (any AE, SAE, serious infection, or AE leading to discontinuation), which generally aligned with the outcomes that were important to patients and clinicians.
Efficacy Results
The results of the ITC suggest that upadacitinib was ||| |||||||||||| ||||||||| |||| other advanced therapies for clinical remission and response induction. The endoscopic outcomes networks were sparse with only | treatments, making it challenging to |||| |||||| ||||||||||| from the data. Moreover, the credible intervals (CrIs) for risk-difference point estimates, including those |||| |||||||| || ||||| |||| |||||||||| |||| indicating imprecise treatment-effect estimates. ||||| |||| |||||||||| ||||||||||||| |||||| |||||||||| || |||, particularly for the maintenance phase, |||||||||| meaningful conclusions whether upadacitinib is |||| || |||| ||||||||||| than the comparators |||| ||||| ||||.
Table 2: Summary of Findings for Upadacitinib Induction Versus Placebo for Patients With Moderately to Severely Active CD and a History of Treatment Failure
Outcome and follow-up | Patients, N (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|
Clinical remission | ||||
Proportion of patients with clinical remission as measured by PROs Follow-up: 12 weeks | 1,021 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Higha | UPA 45 mg induction results in a clinically important increase in the proportion of patients with clinical remission as measured by PROs at 12 weeks compared to placebo |
Proportion of patients with clinical remission as measured by CDAIs Follow-up: 12 weeks | 1,021 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Moderateb | UPA 45 mg induction likely results in a clinically important increase in the proportion of patients with clinical remission as measured by CDAI at 12 weeks compared to placebo |
Endoscopic response | ||||
Proportion of patients with endoscopic response Follow-up: 12 weeks | 1,021 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Highc | UPA 45 mg induction results in a clinically important increase in the proportion with endoscopic response at 12 weeks compared to placebo |
Endoscopic remission | ||||
Proportion of patients with endoscopic remission Follow-up: 12 weeks | 1,021 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Highc | UPA 45 mg induction results in a clinically important increase in the proportion with endoscopic remission at 12 weeks compared to placebo |
Discontinuation of corticosteroid use and CDAI clinical remission | ||||
Proportion of patients who discontinued corticosteroid use for CD and had clinical remission as measured by CDAI among patients receiving corticosteroids at baseline Follow-up: 12 weeks | 358 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Moderated | UPA 45 mg induction likely results in a clinically important increase in the proportion of patients who discontinue corticosteroids for CD and have clinical remission as measured by CDAI (among patients who were receiving corticosteroids at baseline) at 12 weeks compared to placebo |
Endoscopic remission and CDAI clinical remission | ||||
Proportion of patients with endoscopic remission and clinical remission as measured by CDAI Follow-up: 12 weeks | 1,021 (2 RCTs) | U-EXCEED: ||| || | || || |||| |||| || || |||||||||||||| || | |||| |||||||||||| | |||| || |||| ||||| |||||||||| | |||||||| |||| U-EXCEL: ||| || | | | |||| |||| || || || ||||||||||| || | |||| |||||||||||| | |||| | |||| || |||||||||||||| | |||||||| || | |||| | | UPA 45 mg induction results in a |||||||||| ||||||||| |||||||| in the proportion of patients with both endoscopic and clinical remission (as measured by CDAI) at 12 weeks compared to placebo |
HRQoL (IBDQ) | ||||
Change from baseline in IBDQ total score (range of score: 32 [worst HRQoL] to 224 [best HRQoL]; least squares mean change [95% CI]) Follow-up: 12 weeks | 848 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Highe | UPA 45 mg induction results in a clinically important improvement in IBDQ at 12 weeks compared to placebo |
CR-100 | ||||
Proportion with CR-100 Follow-up: 12 weeks | 1,021 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Highf | UPA 45 mg induction results in a clinically important increase in the proportion with CR-100 at 12 weeks compared to placebo |
Resolution of EIMs | ||||
Proportion with resolution of EIMs among patients who had EIMs at baseline Follow-up: 12 weeks | 420 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Moderateg | UPA 45 mg induction likely results in little to no clinically important difference in the proportion with resolution of EIMs at 12 weeks compared to placebo, among patients who had EIMs at induction baseline |
CD-related hospitalization | ||||
Proportion with CD-related hospitalization Follow-up: 12 weeks | 1,021 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Lowh | UPA 45 mg induction may result in little to no difference in the proportion with CD-related hospitalization at 12 weeks compared to placebo. There is some uncertainty about the clinical importance of the estimates |
CD-related surgery | ||||
Proportion with CD-related surgery Follow-up: 12 weeks | 1,021 (2 RCTs) | U-EXCEED: ||| || || || || |||| |||| || ||||||||||||| || || |||| |||||||||||| ||||| || |||| | |||||||||| ||||||| |||| | || ||||| || || |||||| U-EXCEL: ||| || || || || |||| |||| || ||||||||||||| || || |||| |||||||||||| |||| || |||| |||||||||||| ||||||| |||| || || ||||| || || ||||| | ||| | | UPA 45 mg induction ||||||||| || |||||| || || |||||||||| in the proportion with CD-related surgery at 12 weeks compared to placebo; there is some uncertainty about the clinical importance of the estimates |
SAEs | ||||
Proportion of patients who experienced any SAE Follow-up: 12 weeks | 1,021 (2 RCTs) | U-EXCEED:
U-EXCEL:
| Moderatei | UPA 45 mg induction likely results in little to no difference in the proportion with SAEs at 12 weeks compared to placebo; there is some uncertainty about the clinical importance of the estimates |
CD = Crohn disease; CDAI = Crohn Disease Activity Index; CI = confidence interval; CR-100 = a decrease of at least 100 points from baseline in the CDAI; EIM = extra-intestinal manifestation; HRQoL = health-related quality of life; IBDQ = Inflammatory Bowel Disease Questionnaire; PRO = patient-reported outcome; RCT = randomized controlled trial; SAE = serious adverse event; UPA = upadacitinib.
Note: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aA difference of 15% between groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
bRated down 1 level for serious imprecision as the 95% CI for the between-group difference for each trial crossed the MID of 15% identified by the clinical expert consulted by CADTH for this outcome.
cA difference of 5% between groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
dRated down 1 level for serious imprecision as the 95% CI for the between-group difference in the U-EXCEED trial crossed the threshold of 15% that was identified by the clinical expert consulted by CADTH for this outcome.
eA MID of ≥ 16 points on the IBDQ was identified from the literature as clinically important. Although the lower boundary of the 95% CI in the U-EXCEL trial was 15.6, this was not considered a serious imprecision due to its proximity to 16.
fA difference of 15% between groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome. Although the lower boundaries of the 95% CIs were below this threshold, given the sample size and the proximity of the lower bound of the confidence intervals to the estimated threshold across both trials, the imprecision was not considered serious.
gRated down 1 level for serious concern regarding imprecision because the point estimates are below the difference of 15% between groups identified by the clinical expert as clinically important, and the upper bound of the 95% CIs include the possibility of an important benefit.
hRated down 2 levels for serious concerns regarding indirectness and imprecision. Longer-term outcome assessment would be required to compare the effect of treatment more meaningfully on these outcomes. The point estimates are close to null and the 95% CIs cross the null threshold. The clinical expert consulted by CADTH could not provide a threshold of important difference; however, the CADTH review team judged that the effect estimate and CI were unlikely to include any important effect.
iRated down 1 level for serious concerns regarding imprecision. As no 95% CI of the difference was available, the optimal information size approach was used to judge imprecision. The clinical expert consulted by CADTH could not provide a threshold of important difference; however, the CADTH review team judged that the effect estimate and CI were unlikely to include any important effect.
Source: Clinical Study Reports of U-EXCEED26 and U-EXCEL.22 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 3: Summary of Findings for Upadacitinib Maintenance Versus Placebo for Patients With Moderately to Severely Active CD and History of Treatment Failure
Outcome and follow-up | Dose of UPA | Patients, N (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|---|
Clinical remission | |||||
Proportion of patients with clinical remission as measured by PROs Follow-up: 52 weeks | 15 mg | 334 (1 RCT) |
| Moderatea | UPA 15 mg maintenance likely results in a clinically important increase in the proportion of patients with clinical remission as measured by PROs at 52 weeks compared to placebo |
30 mg | 333 (1 RCT) |
| Highb | UPA 30 mg maintenance results in a clinically important increase in the proportion of patients with clinical remission as measured by PROs at 12 weeks compared to placebo | |
Proportion of patients with clinical remission as measured by CDAI Follow-up: 52 weeks | 15 mg | 334 (1 RCT) |
| Highb | UPA 15 mg maintenance results in a clinically important increase in the proportion of patients with clinical remission as measured by CDAI at 52 weeks compared to placebo |
30 mg | 333 (1 RCT) |
| Highb | UPA 30 mg maintenance results in a clinically important increase in the proportion of patients with clinical remission as measured by CDAI at 52 weeks compared to placebo | |
Endoscopic response | |||||
Proportion of patients with endoscopic response Follow-up: 52 weeks | 15 mg | 334 (1 RCT) |
| Highc | UPA 15 mg maintenance results in a clinically important increase in the proportion of patients with endoscopic response at 52 weeks compared to placebo |
30 mg | 333 (1 RCT) |
| Highc | UPA 30 mg maintenance results in a clinically important increase in the proportion of patients with endoscopic response at 52 weeks compared to placebo | |
Endoscopic remission | |||||
Proportion of patients with endoscopic remission Follow-up: 52 weeks | 15 mg | 334 (1 RCT) |
| Highc | UPA 15 mg maintenance results in a clinically important increase in the proportion of patients with endoscopic remission at 52 weeks compared to placebo |
30 mg | 333 (1 RCT) |
| Highc | UPA 30 mg maintenance results in a clinically important increase in the proportion of patients with endoscopic remission at 52 weeks compared to placebo | |
Discontinuation of corticosteroid use and CDAI clinical remission | |||||
Proportion of patients who discontinued corticosteroid use for CD for at least 90 days at week 52 and had clinical remission as measured by CDAI among patients who were receiving corticosteroids at induction baseline Follow-up: 52 weeks | 15 mg | 124 (1 RCT) |
| Moderated | UPA 15 mg maintenance likely results in a clinically important increase in the proportion of patients who discontinue corticosteroids for CD and have clinical remission as measured by CDAI (among patients who were receiving corticosteroids at induction baseline) at 52 weeks compared to placebo |
30 mg | 124 (1 RCT) |
| Moderated | UPA 30 mg maintenance likely results in a clinically important increase in the proportion of patients who discontinue corticosteroids for CD and have clinical remission as measured by CDAI (among patients who were receiving corticosteroids at induction baseline) at 52 weeks compared to placebo | |
Endoscopic remission and CDAI clinical remission | |||||
Proportion of patients with endoscopic remission and clinical remission as measured by CDAI Follow-up: 52 weeks | 15 mg | 334 (1 RCT) |
| Highc | UPA 15 mg maintenance results in a clinically important increase in the proportion of patients with both endoscopic and clinical remission (as measured by CDAI) at 52 weeks when compared to placebo |
30 mg | 333 (1 RCT) |
| Highc | UPA 30 mg maintenance results in a clinically important increase in the proportion of patients with both endoscopic and clinical remission (as measured by CDAI) at 52 weeks when compared to placebo | |
HRQoL (IBDQ) | |||||
Change from baseline in IBDQ total score (range of score: 32 [worst HRQoL] to 224 [best HRQoL]; least squares mean change) Follow-up: 52 weeks | 15 mg | 119 (1 RCT) |
| Moderatee | UPA 15 mg maintenance likely results in little to no difference in IBDQ at 52 weeks when compared with placebo |
30 mg | 135 (1 RCT) |
| Moderatef | UPA 30 mg maintenance likely results in a clinically important improvement in IBDQ at 52 weeks when compared with placebo | |
CR-100 | |||||
Proportion with CR-100 Follow-up: 52 weeks | 15 mg | 334 (1 RCT) |
| Highc | UPA 15 mg maintenance results in a clinically important increase in the proportion of patients with CR-100 at 52 weeks compared with placebo |
30 mg | 333 (1 RCT) |
| Highc | UPA 30 mg maintenance results in a clinically important increase in the proportion of patients with CR-100 at 52 weeks compared with placebo | |
Resolution of EIMs | |||||
Proportion with resolution of EIMs among patients who had EIMs at baseline Follow-up: 52 weeks | 15 mg | 127 (1 RCT) |
| Lowg | UPA 15 mg maintenance may result in little to no clinically important increase in the proportion of patients with resolution of EIMs at 52 weeks when compared with placebo, among patients who had EIMs at induction baseline |
30 mg | 139 (1 RCT) |
| Moderateh | UPA 30 mg maintenance likely results in a clinically important increase in the proportion of patients with resolution of EIMs at 52 weeks compared with placebo, among patients who had EIMs at induction baseline | |
CD-related hospitalization — maintenance | |||||
Proportion with of CD-related hospitalization Follow-up: 52 weeks | 15 mg | 334 (1 RCT) |
| Lowi | UPA 15 mg maintenance may result in little to no difference in CD-related hospitalizations at 52 weeks compared to placebo; there is some uncertainty about the clinical importance of the estimates |
30 mg | 333 (1 RCT) |
| Lowi | UPA 30 mg maintenance may result in little to no difference in CD-related hospitalizations at 52 weeks compared to placebo; there is some uncertainty about the clinical importance of the estimates | |
CD-related surgery — maintenance | |||||
Incidence rate (n of 100 patient-years) of CD-related surgery Follow-up: 52 weeks | 15 mg | 334 (1 RCT) | |||| |||||| |||| | |||||| || ||||||| || ||||||||||||| |||||| || || |||||||||||||| |||| |||| |||||||||| ||||||||| || || || ||||| |||||| ||||| || |||||| ||||| | ||| | | UPA 15 mg maintenance ||| |||||| || |||||| || || |||||||||| in the incidence of CD-related hospitalizations at 52 weeks compared to placebo; ||||| || |||| |||||||||||||||| | |||||||| |||||||||| || || |||||||||| |
30 mg | 333 (1 RCT) | |||| |||||| |||| || |||||| || ||||||| | ||||||||||||| |||||| || || |||||||||||||| |||| |||| |||||||||| ||||||||| || || || ||||| |||||| ||||| || |||||| ||||| | ||| | | UPA 30 mg maintenance ||| |||||| || |||||| || || |||||||||| in the incidence of CD-related hospitalizations at 52 weeks compared to placebo; ||||| || |||| |||||||||||||||| |||||||| |||||||||| || || |||||||||| | |
SAEs — maintenance | |||||
Proportion of patients who experienced any SAE Follow-up: 52 weeks | 15 mg | 452 (1 RCT) |
| Moderatej | UPA 15 mg maintenance likely results in little to no difference in SAEs at 52 weeks compared to placebo; there is some uncertainty about the clinical importance of the estimates |
30 mg | 450 (1 RCT) |
| Moderatej | UPA 30 mg maintenance likely results in little to no difference in SAEs at 52 weeks compared to placebo; there is some uncertainty about the clinical importance of the estimates | |
CD = Crohn disease; CDAI = Crohn Disease Activity Index; CI = confidence interval; CR-100 = decrease of at least 100 points from baseline in the CDAI; EIM = extra-intestinal manifestation; HRQoL = health-related quality of life; IBDQ = Inflammatory Bowel Disease Questionnaire; MID = minimally important difference; PRO = patient-reported outcome; RCT = randomized controlled trial; SAE = serious adverse event; UPA = upadacitinib.
Note: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision as the 95% CI of each trial crossed the difference of 15% between groups identified by the clinical expert consulted by CADTH as clinically important for this outcome.
bA difference of 15% between groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
cA difference of 5% between groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
dRated down 1 level for serious concerns regarding imprecision. As no 95% CI of the difference was available, the optimal information size approach was used to judge imprecision. There is no established MID.
eRated down 1 level for serious concern regarding imprecision because the point estimate was lower than the literature-reported MID of ≥ 16 points on the IBDQ, and the 95% CI crossed the MID.
fRated down 1 level for serious concern regarding imprecision because the 95% CI crossed the literature-reported MID of 16 points or higher on the IBDQ.
gRated down 2 levels for very serious concerns regarding imprecision because the point estimate was lower than the difference of 15% between groups identified by the clinical expert consulted by CADTH as clinically important for this outcome, and the 95% CI crossed the clinical importance threshold.
hRated down 1 level for serious concerns regarding imprecision because and the 95% CI crossed the difference of 15% between groups identified by the clinical expert consulted by CADTH as clinically important for this outcome.
iRated down 2 levels for serious concerns regarding indirectness and imprecision. Longer-term outcome assessment would be required to compare the effect of treatment more meaningfully on these outcomes. The point estimates are close to null and the 95% CIs crossed the null threshold. These outcomes may not have been tested for multiplicity in the trial and should be considered as supportive evidence. The clinical experts consulted by CADTH could not provide a threshold of important difference; however, the CADTH review team judged that the effect estimate and CI were unlikely to include any important effect.
jRated down 1 level for serious concern regarding imprecision. No 95% CI of the difference was available. As there is no established MID, the optimal information size approach was used. The clinical experts consulted by CADTH could not provide a threshold of important difference; however, the CADTH review team judged that the effect estimate and CI were unlikely to include any important effect.
Source: Clinical Study Reports of U-ENDURE.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms Results
Induction-phase safety results had |||| |||| || || ||||||| || |||| for comparisons of upadacitinib versus other advanced treatments. Likewise, ||||||||||||| || |||||||||| |||| |||| ||||||||| comparative safety conclusions |||| ||||| |||| |||| ||||||||||.
Critical Appraisal
The networks included relatively few direct comparisons between active treatments or were small (e.g., endoscopic outcomes). In general, based on the baseline patient characteristics, the clinical expert was of the opinion that the patients in the studies were similar and generalizable to those who could receive upadacitinib in Canadian clinical practice. However, patient characteristics (and potential treatment-effect modifiers) varied across the studies for disease duration, CRP and fecal calprotectin values, and concomitant medication use, and it is likely that differences in patients’ experience with previous treatments (number and type) introduced further bias to the analysis. There was heterogeneity in the duration of induction and maintenance phases and in the definition of induction response (which dictated entry to the maintenance phase); re-randomization into the maintenance phase invalidated the assumption of a common placebo comparator; and studies used different definitions for safety outcomes, creating issues when analyzing harms results. ||| ||||||||||||| ||||| ||||||||| ||||||||| ||||||||||||| |||||||||||||| || |||| |||| |||| || ||||||||||| || |||| |||||||||| |||||||| | |||||| ||||||||||| ||||||| |||||||||||| | |||||| || ||||| |||| ||||| |||||||| || |||||||||| || |||| ||||||||||.
Conclusions
Three phase III, multicentre, double-blind RCTs evaluated the efficacy and safety of upadacitinib compared to placebo in adult patients with moderately to severely active CD and a history of treatment failure. Two of the RCTs evaluated induction therapy of upadacitinib 45 mg once daily while a third RCT evaluated maintenance therapy of upadacitinib 15 mg once daily and 30 mg once daily.
Compared to placebo at 12 weeks, upadacitinib 45 mg induction results in an increase in the proportion of patients who have clinical remission as measured by PROs; endoscopic response, endoscopic remission, both endoscopic remission and clinical remission as measured by CDAI, and CR-100; and an improvement in HRQoL based on IBDQ. It also likely results in an increase in the proportion who discontinued corticosteroid use for CD with concurrent clinical remission as measured by CDAI (in patients who were taking corticosteroids at baseline), and in the proportion with clinical remission as measured by CDAI. It likely results in an increase in the proportion of patients who have resolution of EIMs among those who had EIMs at baseline, and it may result in little to no difference in CD-related hospitalization, or |||||||||| ||||||||| at 12 weeks. Compared to placebo at 52 weeks, outcomes with upadacitinib 15 mg and 30 mg maintenance were generally consistent with the induction-related outcomes, albeit with slightly more imprecision in some outcomes for the 15 mg dose, and for the outcome of IBDQ for either dose. The time frame was considered insufficient to detect differences in the outcomes of CD-related hospitalization and |||||||||| |||||||||. The evidence shows that upadacitinib likely results in little to no difference in SAEs compared to placebo, and no new safety signals were identified. Longer-term data are currently being collected in substudy 2 of the U-ENDURE trial, but results are not yet available.
There is a data gap in head-to-head, direct evidence between upadacitinib and other advanced therapies for CD. Indirect evidence provided by the sponsor demonstrated | |||||||||| |||||||||| || |||||||| || |||||| |||||||| between upadacitinib and other advanced therapies during induction or maintenance, but the ITCs |||||||| |||| ||||||||||| ||||||||||| || |||||||||| |||||||||||||| |||||||||| |||||||||| ||||||||||| for both efficacy and safety outcomes.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of an oral induction dose of upadacitinib 45 mg once daily and maintenance doses of 15 mg or 30 mg once daily in the treatment of CD in adults with moderately to severely active disease who have demonstrated prior treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
Crohn disease is a chronic progressive form of IBD that leads to significant disability and negatively affects HRQoL.1-3 It is characterized by recurrent, uncontrolled inflammation that can affect any part of the GI tract from mouth to anus,8 primarily affecting the ileum (i.e., small intestine), colon (i.e., beginning of the large intestine), and rectum.4,5 As CD is a fluctuating disease, patients experience periods of flare-ups.29
It is thought that CD is triggered by a disturbance of the intestinal epithelial barrier due to environmental factors and/or genetic susceptibility.30,31 The disruption of the epithelial barrier and the normal homeostasis of gut microbiota and mucosal immune system stimulates aberrant activation of the gut immune response.21 Common symptoms of CD include diarrhea, AP, fatigue, fever, and rectal bleeding. Other symptoms include loss of appetite, weight loss, and malnutrition.6,7 Complications associated with CD can include bowel obstructions, fistulas, anal fissures, intra-abdominal and other abscesses, and ulcers that often require hospitalization or surgery.8,9
Canada has the highest incidence and prevalence of IBD in the world.10,11 The incidence of CD varies across the Canadian provinces, with the highest rate reported in Nova Scotia at 22.6 per 100,000 persons, whereas in Alberta, British Columbia, Manitoba, Ontario, Quebec, and Saskatchewan rates range from 8.8 to 16.6 per 100,000 persons.10,32 According to the 2018 Impact of Inflammatory Bowel Disease in Canada report, approximately 270,000 Canadians were living with IBD, of whom 135,000 had CD. For every 100,000 Canadians, 16.3 new cases of CD are diagnosed each year.12 This translates to more than 6,000 new cases annually. The number of people living with IBD is expected to exceed 400,000, or approximately 1% of the population, in 2030.12
Diagnosis of CD requires a combination of a medical history and physical examinations.6 Laboratory testing measures key biomarkers, including CRP, erythrocyte sedimentation rate, and fecal calprotectin.6,13-15 In active CD, depending on disease severity, CRP levels can range from 5 to 20 mg/L compared to typical baseline levels of 1 mg/L.15 Diagnostic imaging, including endoscopy, radiology, and ultrasound, are also used to examine findings of typical CD cases, including bowel obstruction, stenosis, fistulae, abscesses, and, atrophy.14,16-19
For many patients with CD, symptoms are chronic and intermittent, and disease activity and severity can vary widely. Disease severity is measured using the CDAI and Harvey-Bradshaw Index (HBI), which are designed to evaluate bowel-related symptoms, including SF, AP, arthritis and/or arthralgia, uveitis, skin and mouth lesions, and perianal disease. While the HBI is commonly used in routine gastroenterology practice, the CDAI remains the most common comparable end point across biologics in CD.33 Less precision is expected with the HBI as it is a subset of the CDAI (e.g., the HBI uses single-day readings, only 5 of the 8 CDAI variables, and sums variables instead of applying weighted coefficients).33 The correlation coefficient between HBI and CDAI is reportedly between 0.80 and 0.93.33,34 The IBDQ incorporates social and emotional symptoms to give a meaningful indication of the impact of the disease on quality of life.35,36 In addition, the Crohn Disease Endoscopic Index of Severity and SES-CD are scoring systems based on endoscopic observations of the size and location of ulceration and lesions.37
Standards of Therapy
Contents within this section were informed by materials submitted by the sponsor and clinical expert input. The following summary was validated by the CADTH review team.
Selection of therapy is based on the location, extent, phenotype, and severity of disease.38 As there is no cure for CD, the therapeutic goal is to induce and maintain clinical and endoscopic remission and reduce the need for long-term corticosteroid use.38 Treatment of CD with pharmacotherapies is typically divided into induction, in which the goal is to achieve control of inflammation within a short period of time (e.g., 3 months or less), followed by maintenance, in which that control is sustained beyond 3 months.38,39 The clinician group input, patient group input, and the clinical expert consulted by CADTH agreed that treatment goals for patients include short-term improvement of symptoms, and intermediate- to long-term maintenance of overall wellness and ability to return to work, school, and regular functions of daily life, with reductions in pain, fatigue, bowel movements, and improvement in HRQoL. While symptomatic control and clinical remission are important treatment goals, the clinical expert consulted by CADTH and published treatment guidelines for CD38,39 emphasize that the modern treatment paradigm increasingly values the objective outcomes of endoscopic remission and mucosal healing, which may not always be correlated with clinical measures or symptom burden but are important in the prevention of downstream complications of CD.
Two major categories of pharmacotherapies are used to treat CD: conventional and biologic.38 The conventional therapies include corticosteroids (e.g., prednisone), 5-ASA, and immunomodulators (e.g., azathioprine, cyclosporine, methotrexate, and 6-mercaptopurine).38 Conventional therapies, particularly 5-ASA, are typically used in first-line treatment of patients with mild to moderate disease despite poor efficacy due to the risks associated with more effective therapies (i.e., biologics).38 Corticosteroids are primarily used as rescue medication to treat CD flares, and systemic formulations are effective in short-term induction of remission in patients with moderate to severe CD, but are not appropriate for long-term use due to the associated side effects and poor efficacy in achieving mucosal healing.38 Reducing dependence on corticosteroid use is a common goal in treatment of CD.38
For moderate to severe CD, more effective therapies are required.38 Biologic therapies for CD include TNF-alpha antagonists (e.g., infliximab and adalimumab), integrin inhibitors (e.g., vedolizumab), and IL-12/IL-23 inhibitors (e.g., ustekinumab). Selection of biologic therapy depends on patient phenotype and treatment experience, risks and benefits of each therapy, and patient preference. If patients develop an inadequate response, loss of response, or intolerance to the biologic agents, treatment will be escalated to higher doses or to newer biologic classes. Patients with serious complications or who have medically refractory disease may be candidates for surgery (e.g., total colectomy or ileostomy), which is associated with important morbidities.
Drug Under Review
Upadacitinib is a selective and reversible JAK inhibitor engineered to have greater inhibitory potency for JAK1 versus JAK2, JAK3, and TYK2 in human cellular assays.40 Upadacitinib preferentially inhibits signalling by JAK1 or JAK1/JAK3 with functional selectivity over cytokine receptors that signal via pairs of JAK2 proteins. JAK inhibitors such as upadacitinib can inhibit intracellular downstream signalling elicited by multiple cytokines known to have a role in the inflammatory environment in CD.41 For example, pro-inflammatory cytokines such as the IL-23 pathway have a substantial role in the pathogenesis of CD.42,43 IL-23 interacts with the IL-23 receptor to activate JAK2 and TYK2, stimulating the activation of key immune cells through the P38 MAPK, PI3K-Akt and nuclear factor kappa–beta signalling pathways.20,21
The approved indication for upadacitinib is for the treatment of adult patients with moderately to severely active CD who have demonstrated prior treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy. The sponsor’s reimbursement request is consistent with the approved indication. Upadacitinib is administered orally. The recommended dosing schedule is in 2 phases: a 12-week induction phase consisting of 45 mg upadacitinib once daily, followed by an ongoing maintenance phase. The recommended dosage of upadacitinib for maintenance treatment for patients who are aged 18 to 64 years is 15 mg or 30 mg upadacitinib once daily, based on patient presentation; 30 mg once daily may be appropriate for patients with high disease burden (such as in cases of refractory or severe disease) or those who do not show adequate therapeutic benefit with 15 mg once daily, and the lowest effective dose for maintenance should be used. For patients who are aged 65 years or older, the recommended maintenance dose is 15 mg once daily. In patients who are responding to induction or maintenance treatment with upadacitinib, corticosteroids may be reduced and/or discontinued in accordance with standard of care.22,23 Key characteristics of upadacitinib and other treatments available for CD are summarized in Table 4.
Table 4: Key Characteristics of Upadacitinib, Adalimumab, Infliximab, Ustekinumab, Vedolizumab, and Risankizumab
Characteristic | Upadacitinib | Adalimumab | Infliximab | Ustekinumab | Vedolizumab | Risankizumab |
|---|---|---|---|---|---|---|
Mechanism of action | A selective JAK inhibitor that demonstrates activity against JAK1 JAK2, JAK3, and TYK2 | Anti-TNF; human IgG1 monoclonal antibody; binds and blocks TNF-alpha and its interaction with p55 and p75 cell-surface TNF receptors | Anti-TNF; IgG1-kappa monoclonal antibody that neutralizes the biological activity of TNF-alpha by specifically binding to its receptors | Human IgG1 monoclonal antibody; neutralizes cellular responses mediated by IL-12 and IL-23 | IgG1 monoclonal antibody; binds to human alpha 4 beta 7 integrin, acting as a gut-selective anti-inflammatory biologic | Humanized IgG1 monoclonal antibody that binds to the p19 subunit of human IL-23 cytokine and inhibits IL-23 signalling in cell-based assays, including the release of the pro-inflammatory cytokine IL-17 |
Indicationa | Patients with moderately to severely active CD who have demonstrated prior treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy | Reducing signs and symptoms and inducing and maintaining clinical remission in adults with moderately to severely active CD who have responded inadequately to conventional therapy Reducing signs and symptoms and inducing clinical remission in adults with moderately to severely active CD who have lost response or are intolerant to infliximab | Reduction of signs and symptoms, induction and maintenance of clinical remission and mucosal healing and reduction of CS use in adults with moderately to severely active CD who have had an inadequate response to a CS and/or aminosalicylate Adults with fistulizing CD who have not responded despite conventional treatment | Patients with moderately to severely active CD who have had an inadequate response, loss of response, or were intolerant to either CS or immunomodulators or 1 or more TNF antagonists, or who were CS-dependent | Patients with moderately to severely active CD who have had an inadequate response, lost response, or were intolerant to immunomodulators or a TNF antagonist; or have had an inadequate response or are intolerance to, or demonstrated dependence on, a CS | Patients with moderately to severely active CD who have had an inadequate response, intolerance, or demonstrated dependence to corticosteroids; or an inadequate response, intolerance, or loss of response to immunomodulators or biologic therapies |
Route of administration | Oral | SC | IV | IV (induction) and SC (maintenance) | IV (induction and maintenance) and SC (maintenance) | IV (induction) and SC (maintenance) |
Recommended dose | Adults (moderate to severe CD) Induction: 45 mg daily (q.d.) for 12 weeks Maintenance: 15 mg or 30 mg upadacitinib q.d. | Adult CD Induction: 160 mg at week 0, 80 mg at week 2 Maintenance: 40 mg q.2.w. beginning at week 4; dose escalation for patients with a disease flare or nonresponse | Adults (moderate to severe CD) Induction: 5 mg/kg at weeks 0, 2, 6 Maintenance: 5 mg/kg q.8.w. 10 mg/kg for incomplete responders Adults (fistulizing CD) Induction: 5 mg/kg at weeks 0, 2, 6 Maintenance: 5 mg/kg q.8.w. or 10 mg/kg q.8.w. for those with relapse following an initial response | Adult CD Induction: tiered weight-based dose approximating 6 mg/kg IV at week 0 Maintenance: 90 mg SC at week 8 and q.8.w. thereafter Alternative maintenance: 90 mg SC at week 12 and q.12.w. thereafter; may switch to q.8.w. for inadequate response | Adults (moderate to severe CD) IV formulation: Induction: 300 mg at weeks 0, 2, 6 Maintenance: 300 mg q.8.w following induction treatment SC formulation: Maintenance: 108 mg q.8.w following the induction treatment with IV infusion | Adults (moderate to severe CD) Induction: 600 mg administered by IV infusion at week 0, week 4, and week 8 Maintenance: 360 mg administered by SC injection at week 12, and q.8.w |
Serious adverse effects or safety issues |
|
|
|
|
|
|
CD = Crohn disease; CS = corticosteroid; IgG1 = immunoglobin G1; IL = interleukin; JAK = Janus kinase; q.d. = once daily; q.2.w. = every 2 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; SC = subcutaneous; TNF = tumour necrosis factor.
aHealth Canada indication.
Source: Product monographs of upadacitinib (Rinvoq)4,4 risankizumab (Skyrizi),45 vedolizumab (Entyvio),46 infliximab (Remicade),47 adalimumab (Humira),48 and ustekinumab (Stelara).49
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient inputs received by CADTH are included in the Stakeholder section of this report.
Two patient groups, CCC and the GI Society, provided input for this review. The CCC is a national, volunteer-based charity dedicated to finding cures for patients with CD and ulcerative colitis and improving the lives of affected children and adults. The GI Society is committed to improving the lives of people with GI and liver conditions, supporting research, advocating for appropriate patient access to health care, and promoting GI and liver health.
Information was gathered by CCC from a report published in 2018 (Impact of Inflammatory Bowel Disease in Canada), a 2022 survey involving 1,700 respondents (687 with moderate to severe CD), and interviews with 3 patients who participated in the upadacitinib clinical trials. The patient input provided by the GI Society was based on surveys and interviews, including a 2015 survey on biologics and biosimilars involving 423 Canadian patients with IBD, a 2018 survey on the unmet needs involving 432 Canadians with IBD, a 2020 survey on the unmet needs of IBD involving 579 respondents, a 2020 survey on biosimilars completed by 145 respondents (most of whom had IBD), a 2022 survey on the IBD patient journey completed by 54 Canadians with IBD, a 2022 focus group with patients living with CD, interviews with 2 patients with CD who participated in the upadacitinib trial, one-on-one conversations with patients with IBD, a patient roundtable, and media interactions.
The 2 patient groups emphasized that CD has a tremendous impact on every aspect of a person’s life. Common symptoms experienced by patient respondents included diarrhea, nausea, unpredictable urgency to use the washroom, bloating, vomiting, rectal bleeding, AP, weight loss, stress and mental health disorders, anemia resulting from blood loss, skin conditions, adhesions or scar tissue, bowel obstruction, eye inflammation, fistulas and abscesses (perianal, anal, internal, or intra-abdominal), stricture, arthritis of the spine, liver conditions, and cancer. EIMs can include fever, inflammation of the eyes or joints (arthritis), ulcers of the mouth or skin, tender and inflamed nodules on the shins, and numerous other conditions. Anxiety, stress, and mental health are major factors, and the symptoms were described by respondents as relentless, unpredictable, embarrassing, and scary. Respondents to both patient groups stated that the inability to predict when the next urgency of bowel movements or flare would occur had a significant negative impact on the personal and social lives of patients with CD.
The GI Society described the treatment of CD as multifaceted as it involves managing symptoms and consequences of the disease and reducing inflammation. Patients also rely on medications to reduce the need for surgery. When 1 medication fails, patients must try another. First-line treatments include 5-ASA and corticosteroids to reduce inflammation in moderate to severe cases of CD. According to the patient input, these treatments are inconvenient therapies that make it difficult for patients to keep a normal routine. Also, if a patient has significant diarrhea, then the rectal medications may be difficult to hold in place for sufficient time to be effective. Immunosuppressive drugs reduce dependence on steroids and help patients who have a steroid-resistant disease; however, these treatments take a long time (e.g., as much as 6 months or more) for patients to experience results. A variety of biologic therapies are used to treat CD when older medications fail to relieve symptoms. As described by the GI input society, JAK inhibitors (such as the drug under review) are a newer class of medication. Unlike other biologics delivered by infusion therapy, JAK inhibitors are easier and more convenient to take as they are in pill form. Despite available treatment options, patients have difficulty achieving remission or adequate symptom relief. Even after surgery to repair fistulas and fissures or removal of diseased bowel tissue, CD symptoms tend to recur in most patients.
Improved outcomes noted as important by the patient groups included symptom mitigation such that patients can work productively; attend school and social events; and perform basic activities of daily living such as errands, maintaining a home, and raising children. Other improved outcomes of interest identified by respondents included reducing preventable patient suffering, such as debilitating symptoms, secondary illnesses (e.g., depression and anxiety), loss of family and social interactions, and reliance on health care resources such as hospital stays, surgeries, and other medications. Managing unpredictable and frequent bowel movements, pain, and fatigue were also considered important by CCC respondents. Unmet patient needs were noted by the CCC to vary between individuals depending on their specific symptoms and life circumstances. Both patient groups emphasized the importance of a treatment option that is easy to administer and can provide symptom relief, achieve remission, and improve subsequent HRQoL.
Three patients from the CCC and 2 patients from the GI Society group with experience with upadacitinib reported near-immediate improvements in their health, and alleviation of symptoms of their CD, with no side effects or few mild side effects such as weight gain. Patients noted the convenience of pill-based administration and the lack of a need to refrigerate the medication or attend a clinic for infusions.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of CD.
Unmet Needs
The clinical expert indicated there are profound treatment gaps in the management of IBD, including CD. A significant portion of patients do not exhibit symptoms associated with disease activity. Subclinical disease progression is common and patients often present at diagnosis with significant established, longstanding disease characterized by deep ulcers in the bowel. Over time, transmural damage leads to complications (e.g., stenosis and penetrating complications that often require surgery). Early treatment initiation is key to limiting disease activity and stopping progression. Although a number of effective drugs are available for the treatment of moderate to severe CD, there are significant limitations in efficacy. For example, the expert noted that the average likelihood of endoscopic remission across drugs ranges from approximately 20% to 40%, and emphasized the frequency of loss of response over time and the reduced efficacy with the introduction of each subsequent biologic after failure. This was described as the greatest treatment challenge in the management of CD.
The clinical expert added that primary nonresponse and secondary loss of response are both common in the treatment of CD with advanced therapies, creating a need for therapies that remain efficacious in patients with experienced with biologics.
Clinical trial design has historically focused on clinical symptoms of response and remission, which may not always correlate with objective measures, such as endoscopic remission and mucosal healing. The clinical expert noted that long-term longitudinal studies to evaluate the modification of bowel damage are lacking as most clinical trials are up to 2 years in duration.
The clinical expert acknowledged that some treatments are particularly inconvenient and affect a patient’s lifestyle due to the need to visit an infusion clinic for a few hours every 4 to 8 weeks. Therapies given by subcutaneous injection are more convenient, but patients may find them painful. No available orally administered advanced therapies are currently available to treat CD.
Place in Therapy
The clinical expert noted that the current treatment paradigm for adults with moderately to severely active CD is complex and dictated by disease phenotype. Conventional therapies (e.g., steroids and immune suppressants) are not typically used in isolation over the long-term, and most patients with moderately to severely active disease would go on to receive advanced therapy, such as a biologic drug. The clinical expert noted that the first therapy to be prescribed has the best chance for improvement and healing due to the aforementioned pattern of a lower likelihood of a robust response with subsequent advanced therapies. Selecting the optimal therapy from the start is a challenge and is based on disease phenotype, disease severity, and the risks and expected onset of action of each available therapy. For example, particularly severe disease would warrant selection of a therapy with rapid onset, high efficacy, and steroid-sparing effects (e.g., anti-TNFs or anti–IL-23 and anti–IL-12/IL-23).
Nearly half of patients have EIMs of CD, which can be disabling, and only a select few medications that are currently available address them, with a preference for the anti-TNF category. Approximately a quarter of patients with CD have fistulizing perianal disease, which is a marker of severe disease, and again anti-TNFs are the preferred therapeutic option for this subpopulation. Other options are needed for patients with these disease phenotypes.
The clinical expert indicated that upadacitinib would be used as a first-line option for patients receiving advanced therapies for CD, and that there is no mechanistic, efficacy, or sequencing-based argument to require the failure of other advanced drugs before initiation of upadacitinib.
The clinical expert also acknowledged increased off-label use of combination therapies with complementary mechanisms of effect in particularly severe, high-risk patients with prior drug failures, surgeries, or other markers of disabilities. According to the expert, combinations would typically include a low-risk, safe drug, such as an anti-integrin, with other more systemically active drugs. This may become relevant in the case of upadacitinib. However, the expert emphasized that this use is limited to dire situations in which there is a risk of extensive surgery or disability.
Patient Population
The clinical expert stated that patients with confirmed pathologic or histologic diagnosis of moderate to severe CD are typically diagnosed by a gastroenterologist. Misdiagnosis is rare, but a diagnosis may be delayed as previously described. Patients with EIMs (e.g., inflammatory arthropathy, peripheral or axial) are a priority for treatment. Although there are no clear “stages” of CD, objective measures such as endoscopic activity and the requirement or dependence on corticosteroids are important, while the presence of clinical symptoms is not as critical. There are no established predictors of disease response.
Assessing the Response Treatment
The clinical expert noted that assessment of response in clinical practice differs from clinical trials due to logistics and patient preference. The most easily accessed marker of response is improvement in clinical symptoms (particularly AP and frequency of soft or liquid stools), but this is only weakly correlated with objective markers of disease activity and may be heterogeneous according to disease phenotype. For example, patients with bowel stricture may experience constipation instead of diarrhea, and patients with prior surgeries may have differing symptoms caused by anatomic alteration rather than inflammation. Objective measures of disease activity, particularly endoscopy (i.e., ileocolonoscopy), are important. The clinical expert added that, although the clinical trials assessed endoscopic outcomes at 12 weeks, in clinical practice endoscopy is rarely performed at 12 weeks, and instead is typically performed at 6 to 9 months and can be challenging to repeat. Other objective measures may include biomarkers (CRP and fecal calprotectin) as well as noninvasive cross-sectional imaging, including intestinal ultrasound.
Discontinuing Treatment
The clinical expert indicated that treatment discontinuation should be considered in a manner similar to that of other advanced therapies for adults with CD, and include a combination of clinical symptoms and objective data to support primary nonresponse or loss of response:
persistence or worsening of clinical symptoms, most importantly diarrhea and AP
persistence or worsening of endoscopic activity
worsening or persistent elevation of biomarkers including CRP and fecal calprotectin
worsening or development of complications (including strictures and penetrating disease) on cross-sectional imaging
dependence on or need for recurrent courses of corticosteroids (e.g., 2 or more full courses of oral prednisone within 1 year, but details may be debated)
development of AEs should be weighed on case-by-case basis depending on treatability and severity of the AE; all patients should be vaccinated appropriately (e.g., varicella) to avoid any preventable AEs potentially associated with treatment
patients with severe disease may require a single course of corticosteroids, which again may not preclude ongoing maintenance, and the need for discontinuation would therefore be judged by the treating physician.
Prescribing Considerations
Prescription of upadacitinib should not be limited to IBD specialists, as general gastroenterologists would have the expertise required to initiate therapy. General internists with a special interest in IBD and/or GI may have sufficient experience and training to prescribe upadacitinib, and this may be important for accessibility in rural regions of Canada.
The clinical expert noted that initiation criteria should be similar to other biologics. However, the expert emphasized that the current requirements for previously failed therapies are not up to date with clinical practice. In particular, the current requirements for prior drug failures in prescribing advanced therapies includes 5-ASA, which the clinical expert considered to be out of date due to its known lack of efficacy in this population. The clinical expert noted that, in clinical practice, this results in short-term prescriptions of 5-ASA to meet the requirements when it is not expected to have patient benefit, and the expert recommended that this not be included as a requirement for prior treatment failures when prescribing upadacitinib.
The expert noted that, if treatment is interrupted for at least 2 weeks, the patient may need to undergo induction therapy again.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by 1 clinician group. The full original clinician group input received by CADTH is included in the Stakeholder section of this report.
One clinician group, the Canadian IBD Specialist Group, responded to CADTH’s call for input. This group consists of specialists in gastroenterology from across Canada caring for patients with CD. The clinician group input was based on a discussion held by the Canadian IBD Specialist Group in March 2023. The group reviewed the safety and efficacy data from the upadacitinib phase III trials, described the CD burden in Canada, and discussed the unmet treatment needs of CD, treatment goals, and how access to upadacitinib could benefit patients and society in the short and long-term.
The clinician group emphasized that CD has a tremendous impact on the physical, emotional, and social aspects, including HRQoL, of those living with the disease, and causes a significant economic burden. The current treatment paradigm for CD includes 5-ASA, corticosteroids, immune modifiers, and biologics that include anti-TNF, anti-integrin, and anti–IL-12/IL-23 and anti–IL-23 agents.
The input from the clinician groups identified the same unmet medical needs for patients with CD and potential place in therapy for the drug under review as the clinical experts consulted by CADTH.
The clinician group noted that there are significant unmet therapeutic needs for patients living with moderate to severe CD. There is a lack of safe and effective treatments that could rapidly improve the endoscopic appearance, maintain long-term improvement and remission, and reduce the risk of complications and the need for surgery. The group claimed that there is a high rate of surgery and postoperative recurrence in patients with CD despite currently available treatment options.
In addition to relieving clinical symptoms, the clinician group emphasized that the goal of treatment should focus on changing the course of disease for patients with CD, preventing further intestinal damage, avoiding disability, and reducing the overall cost of care.
The clinician groups noted that upadacitinib, with its selective blockade of the JAK1 signalling pathway, has a novel mechanism of action, and it is the first oral therapy for CD that has ever been evaluated to meet the treatment goals, treat symptoms of CD, improve the underlying inflammation, and provide sustained clinical and endoscopic outcomes.
According to the clinician group, any patient with inadequate response or intolerance to corticosteroids or multiple advanced therapies and those with 1 or more extraintestinal IBD manifestations could benefit from upadacitinib. It could also be used by patients with moderately to severely active CD who prefer an orally administered drug or those in whom there may be advantages to having both rapid onset and offset of action without concerns about immunogenicity or the altered pharmacokinetics that may occur with the use of biologics. However, for patients with ocular complications of CD, or where perianal disease is the dominant disease manifestation, the use of anti-TNF drugs would be preferred.
The clinician group indicated that the outcomes of interest include a meaningful improvement in symptoms as measured by SF and AP, symptomatic remission and off corticosteroids for 3 to 6 months, a decrease in biomarkers (CRP and fecal calprotectin) of inflammatory activity in the first 3 months, and a robust endoscopic response by 6 to 12 months. The group added that HRQoL should be taken into consideration.
The clinician group noted that discontinuation of therapy should be considered if there is a worsening of symptoms, inadequate clinical response to therapy by the end of the 12-week induction period, or severe AEs. The clinician group stated that treatment with upadacitinib should be prescribed by a CD specialist. Assessment of short-term clinical response should be made within 8 to 12 weeks of initiating therapy, biomarker response should be assessed by 3 to 6 months, and endoscopic response by 6 to 12 months.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s Reimbursement Review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Pivotal trials were placebo-controlled. There are no direct head-to-head trials with other therapies used for the treatment of CD. Would other active therapies have been a more informative comparator? | Several other advanced therapies are available for adults with moderately to severely active CD for which there is a lack of head-to-head data with upadacitinib, and a head-to-head comparison between upadacitinib and other advanced therapies would have been more informative. |
Considerations for initiation of therapy | |
Should eligibility criteria for upadacitinib be based on the initiation criteria used by each of the public drug plans for other biologic treatments currently reimbursed for the treatment of adult patients with moderately to severely active CD who have had an inadequate response, a loss of response, or intolerance to conventional and/or biologic therapies? | Initiation criteria should be similar to existing criteria for other biologic treatments currently reimbursed for the treatment of adult patients with moderately to severely active CD who have had an inadequate response, a loss of response, or intolerance to conventional and/or biologic therapies, but with the caveat that current initiation criteria for the biologic treatments require previous treatment with 5-ASA, which according to the clinical expert is known to be an ineffective therapy in this population. Initiation criteria for upadacitinib and other advanced or biologic therapies for this population should not require prior experience with 5-ASA. |
Considerations for continuation or renewal of therapy | |
Should renewal criteria for upadacitinib be based on the renewal criteria used by each of the public drug plans for other biologic treatments currently reimbursed for the treatment of adult patients with moderately to severely active CD? | Renewal criteria should be similar to the renewal criteria used by each of the public drug plans for other biologic treatments currently reimbursed for the treatment of adult patients with moderately to severely active CD. |
Considerations for discontinuation of therapy | |
Does induction need to be repeated if there is an interruption in treatment? | Induction needs to be repeated if there is an interruption of 2 weeks or more. |
Should discontinuation criteria for upadacitinib be based on the discontinuation criteria used by each of the public drug plans for other biologic treatments currently reimbursed for the treatment of adult patients with moderately to severely active CD? | Discontinuation criteria should be similar to the discontinuation criteria used by each of the public drug plans for other biologic treatments currently reimbursed for the treatment of adult patients with moderately to severely active CD. |
Should patients achieve clinical response to induction therapy after 12 weeks of treatment with upadacitinib to continue to maintenance therapy? | Induction patients should achieve clinical response to induction therapy after 12 weeks of treatment with upadacitinib to continue maintenance therapy with upadacitinib. |
Considerations for prescribing of therapy | |
Is it appropriate to use upadacitinib in combination with other JAK inhibitors or biologics? Are there any concerns for clinical practice? | There is increasing off-label use of combination therapies with complementary mechanisms of effect in particularly severe, high-risk patients with prior drug failures, surgeries, or other markers of disabilities. Combinations would typically include a low-risk, safe drug such as an anti-integrin with other more systemically active drugs. Such combination therapies would be limited to dire situations in which there is a risk of extensive surgery or disability. Upadacitinib should not be combined with other therapies with higher risks and more systematically active drugs, such as other JAK inhibitors. |
Should upadacitinib only be prescribed by a physician experienced in the diagnosis and management of CD? | Prescribing of upadacitinib should not be limited to IBD specialists, as general gastroenterologists would have the expertise required to initiate therapy. The clinical expert also noted that general internists with a special interest in IBD and/or GI may have sufficient experience and training to prescribe upadacitinib, which may be important for accessibility in rural regions of Canada. |
Care provision issues | |
Upadacitinib has a black-box warning for increased risk of infections, malignancy and thromboses as these events have been reported. Canadian labelling for all JAK inhibitors was updated in November 2022 to include risks of serious heart-related problems, thrombosis, and malignancies. This was a precautionary measure based on a Health Canada review of tofacitinib and whether these risks would apply to baricitinib and upadacitinib. | Comment to inform CDEC deliberations. |
System and economic issues | |
There are negotiated confidential prices for the biosimilars of adalimumab and infliximab. There is also a negotiated price for vedolizumab. Is there any reason a public plan should pay a significant price premium for upadacitinib? | Comment to inform CDEC deliberations. |
The submission for upadacitinib includes the marketed price for a 45 mg tablet; however, there is no 45 mg tablet marketed or approved by Health Canada. Currently only the 15 mg and 30 mg tablets are marketed. The included price is 101.8100 per 45 mg tablet. Will the 45 mg strength be marketed in Canada? | The sponsor confirmed that the 45 mg tablet is currently approved and marketed in Canada. The DIN is 02539721. |
5-ASA = 5-aminosalicylates; CD = Crohn disease; CDEC = CADTH Canadian Drug Expert Committee; IBD = inflammatory bowel disease; JAK = Janus kinase.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of upadacitinib (Rinvoq) 15 mg, 30 mg, and 45 mg once daily extended-release oral tablets for the treatment of adult patients with moderately to severely active CD who have demonstrated prior treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy. The focus is on comparing upadacitinib to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of upadacitinib is presented in 3 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from 3 RCTs and a single ITC are included in the CADTH review and appraised in this document.
Systematic Review
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Description of Included Studies
Based on a systematic literature review, characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Detail | U-EXCEED | U-EXCEL | U-ENDURE |
|---|---|---|---|
Designs and populations | |||
Study design | Multicentre, double-blind, randomized, placebo-controlled phase III study | Multicentre, double-blind, randomized, placebo-controlled phase III study | Multicentre, double-blind, randomized, placebo-controlled, maintenance and long-term extension phase III study |
Locations | 229 sites in 39 countries including Africa, Europe, North America (including Canada), South America, Oceania, and Asia | 209 sites in 42 countries including Africa, Europe, North America (including Canada), South America, Oceania, and Asia | 277 sites in 43 countries including Africa, Europe, North America (including Canada), South America, Oceania, and Asia |
Patient enrolment dates | Start date: November 2017 End date: August 2021 | Start date: December 2017 End date: January 2022 | Start date: March 2018 End date: March 2022 |
Randomized (N) | N = 624 Part 1 (randomized double-blind 2:1 to UPA or matching placebo for a 12-week induction period):
Part 2 (not randomized):
Part 3 (not randomized):
| N = 526 Part 1 (randomized double-blind 2:1 to UPA or matching placebo for a 12-week induction period):
Part 2 (not randomized):
| N = 901 Cohort 1 (randomized in a 1:1:1 ratio to placebo, UPA 30 mg, or UPA 15 mg):
Cohort 2:
Cohort 3 (not randomized):
|
Inclusion criteria |
|
|
|
Exclusion criteria |
|
|
|
Drugs | |||
Intervention | Part 1: Oral UPA (45 mg once daily) for 12 weeks for double-blind induction period Part 2: Oral UPA (45 mg once daily) for 12 weeks during the open-label induction period Part 3 (Extended treatment): Patients who did not achieve clinical response at week 12 in Parts 1 and 2 were able to participate in Part 3, a 12-week extended treatment period consisting of 3 cohorts):
| Part 1: Oral UPA (45 mg once daily) for 12 weeks Part 2 (Extended treatment): Patients who did not achieve clinical response at week 12 in part 1 were able to participate in part 2, a 12-week extended treatment period consisting of 2 cohorts:
| Substudy 1 Cohort 1: Patients who received the 12-week induction treatment with UPA 45 mg (including those who did not achieve clinical response with placebo and then received UPA 45 mg for 12 weeks) and achieved clinical response in the U-EXCEED and U-EXCEL trials were re-randomized to either UPA 30 mg daily, UPA 15 mg daily, or matching placebo in a 1:1:1 ratio Cohort 2: Patients who received the 12-week induction treatment with placebo and achieved clinical response in the U-EXCEED and U-EXCEL trials continued to receive placebo Cohort 3: Patients who did not achieve clinical response after the 12-week induction treatment with UPA 45 mg and received the 12-week extended treatment with UPA 30 mg and achieved clinical response in the U-EXCEED and U-EXCEL trials continued to receive UPA 30 mg daily |
Comparator(s) | Placebo, orally (once daily) for 12 weeks | Placebo, orally (once daily) for 12 weeks | Placebo, orally (once daily) for 52 weeks |
Study duration | |||
Screening phase | NR | NR | NR |
Run-in phase | NR | NR | NR |
Treatment phase | Part 1: 12 weeks (oral UPA 45 mg once daily) Part 2: 12 weeks (oral UPA 45 mg once daily) Part 3:
| Part 1: 12 weeks (oral UPA 45 mg once daily) Part 2:
| From randomization of maintenance group to long-term extension group Cohort 1: 52 weeks (oral UPA 15 mg or 30 mg once daily or placebo once daily) Cohort 2: 52 weeks (placebo once daily) Cohort 3: 52 weeks (oral UPA 30 mg once daily) |
Follow-up phase | Follow-up visit 30 days from the last dose of the study drug | Follow-up visit 30 days from the last dose of the study drug | Follow-up visit 30 days from the last dose of the study drug |
Outcomes | |||
Primary end point | Coprimary end points for FDA regulatory purposes:
Coprimary end points for EMA regulatory purposes:
| Coprimary end points for FDA regulatory purposes:
Coprimary end points for EMA regulatory purposes:
| Coprimary end points for FDA regulatory purposes:
Coprimary end points for EMA regulatory purposes:
|
Secondary and exploratory end points | FDA:
EMA:
| FDA:
EMA:
| FDA:
EMA:
|
Publication status | |||
Publications | NCT03345836 Loftus et al. (2023) | NCT03345849 Loftus et al. (2023) | NCT03345823 M14 to 430 Loftus et al. (2023) |
AE = adverse event; AP = abdominal pain; CD = Crohn disease; CDAI = Crohn Disease Activity Index; CR-100 = decrease of at least 100 points from baseline in the Crohn Disease Activity Index; EIM = extra-intestinal manifestation; EMA = European Medicines Agency; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; IBDQ = inflammatory Bowel Disease Questionnaire; NR = not reported; PRO = patient-reported outcome: SES-CD = Simple Endoscopic Score for Crohn Disease; SF = stool frequency; UPA = upadacitinib.
Source: Sponsor’s Summary of Clinical Evidence.
The objective of the randomized study of the U-EXCEED trial was to evaluate the efficacy and safety of upadacitinib 45 mg as an induction treatment compared to placebo in patients with moderately to severely active CD. This study was a multicentre, randomized, double-blind, placebo-controlled trial. Eligible patients were adults with a confirmed diagnosis of moderately to severely active CD for at least 3 months who had responded inadequately or were intolerant to biologic therapy. A total of 624 patients were enrolled and randomized between November 2017 and August 2021. Of the 229 study sites across 39 countries, 15 were in Canada. No screening phase was reported. More than 30% of enrolled patients had demonstrated an inadequate response or intolerance to 3 or more biologics. Treatment was split into 3 parts. In part 1, 495 patients were randomized double-blind 2:1 to upadacitinib or matching placebo for a 12-week induction period. Patients were stratified by baseline corticosteroid use (yes or no), endoscopic disease severity (SES-CD < 15 or ≥ 15), and number of prior biologics (> 1 or ≤ 1). In part 2, a total of 129 patients from part 1 were enrolled for a 12-week open-label induction period of an oral daily dose of upadacitinib 45 mg. Patients who achieved a clinical response in part 1 and 2 were eligible to enter a double-blind maintenance portion of the U-ENDURE trial and patients who did not achieve a clinical response in part 1 or 2 were enrolled in a 12-week part 3 extended treatment period. The extended treatment period was divided into 3 cohorts. Patients in cohort 1 who received placebo in part 1 received double-blind induction treatment with upadacitinib 45 mg daily for 12 weeks until week 24. Patients in cohort 2 who received upadacitinib 45 mg in part 1 were given double-blind induction treatment with upadacitinib 30 mg daily for 12 weeks until week 24. Patients in cohort 3 who received open-label upadacitinib 45 mg in part 2 were given open-label upadacitinib 30 mg daily for 12 weeks until week 24.
Figure 1: Study Design Schematic for U-EXCEED
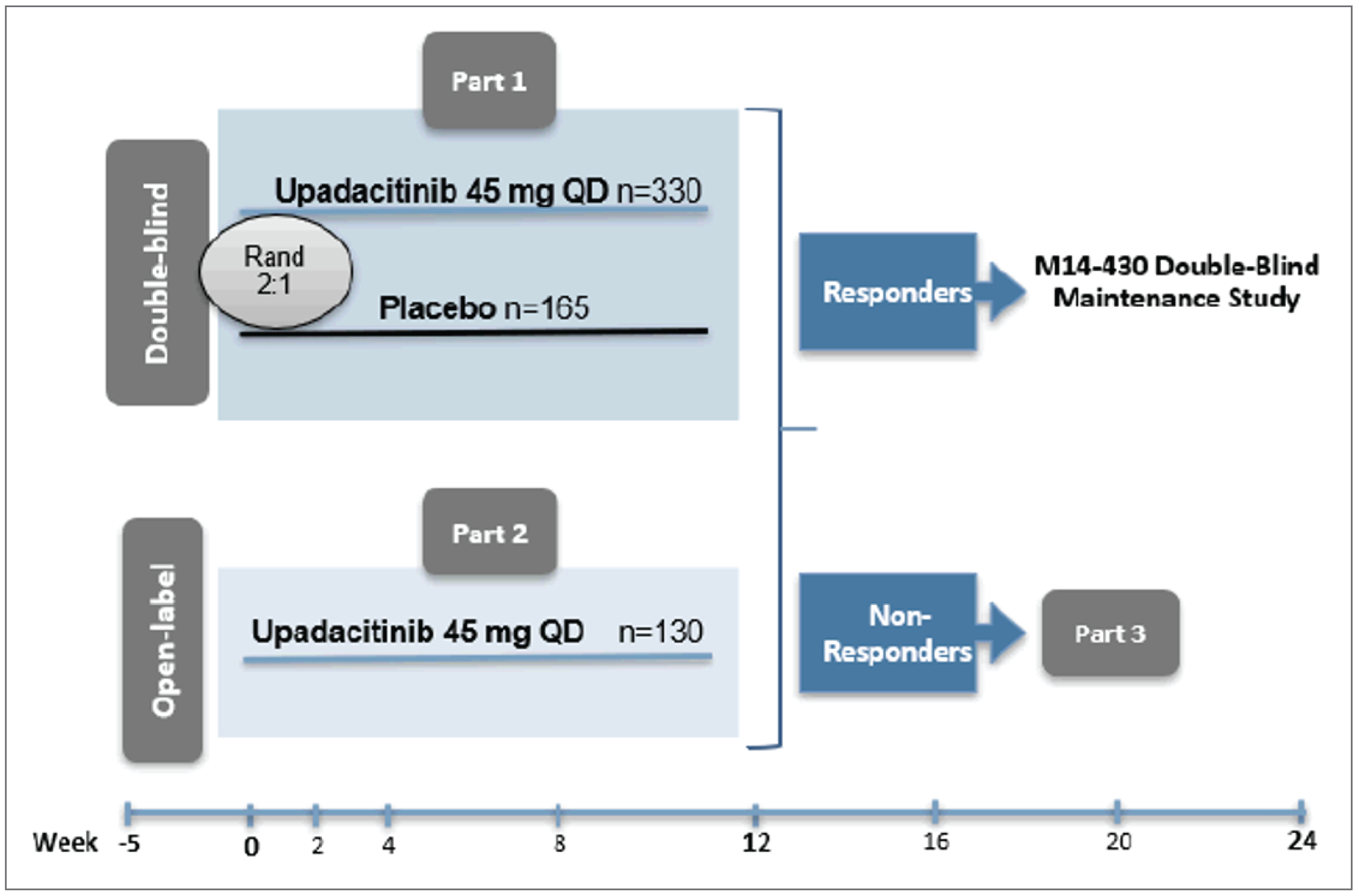
QD = once daily; Rand = randomized.
Source: U-EXCEED Clinical Study Report.26
The U-EXCEL study shared the same objective and study design as the U-EXCEED study but had different patient eligibility criteria. Eligible patients were adults with a confirmed diagnosis of moderately to severely active CD for at least 3 months who were biologic therapy–intolerant or inadequate responders to conventional therapy but had not failed biologic therapy (non-bio-IR population) and patients who were biologic therapy–intolerant or inadequate responders to 1 or more biologic agents for CD (bio-IR population). A total of 526 patients were enrolled and randomized between December 2017 and January 2022. There were 209 study sites across 42 countries, including 17 sites in Canada. No screening phase was reported. Treatment was split into 2 parts. In part 1, a total of 526 patients were randomized double-blind 2:1 to upadacitinib 45 mg or matching placebo for a 12-week induction period. Patients were stratified based on baseline steroid use (yes or no), endoscopic disease severity (SES-CD < 15 or ≥ 15) and number of prior biologics (0, 1, or > 1). Patients who achieved a clinical response in part 1 were eligible to enter a double-blind maintenance portion of the U-ENDURE trial, and patients who did not achieve a clinical response in part 1 were enrolled in a 12-week part 2 extended treatment period, which was categorized into 2 cohorts. Patients in cohort 1 who received placebo in part 1 were given double-blind induction treatment with upadacitinib 45 mg daily for 12 weeks until week 24. Patients in cohort 2 who received upadacitinib 45 mg in part 1 were given double-blind induction treatment with upadacitinib 30 mg daily for 12 weeks until week 24.
Figure 2: Study Design Schematic for U-EXCEL
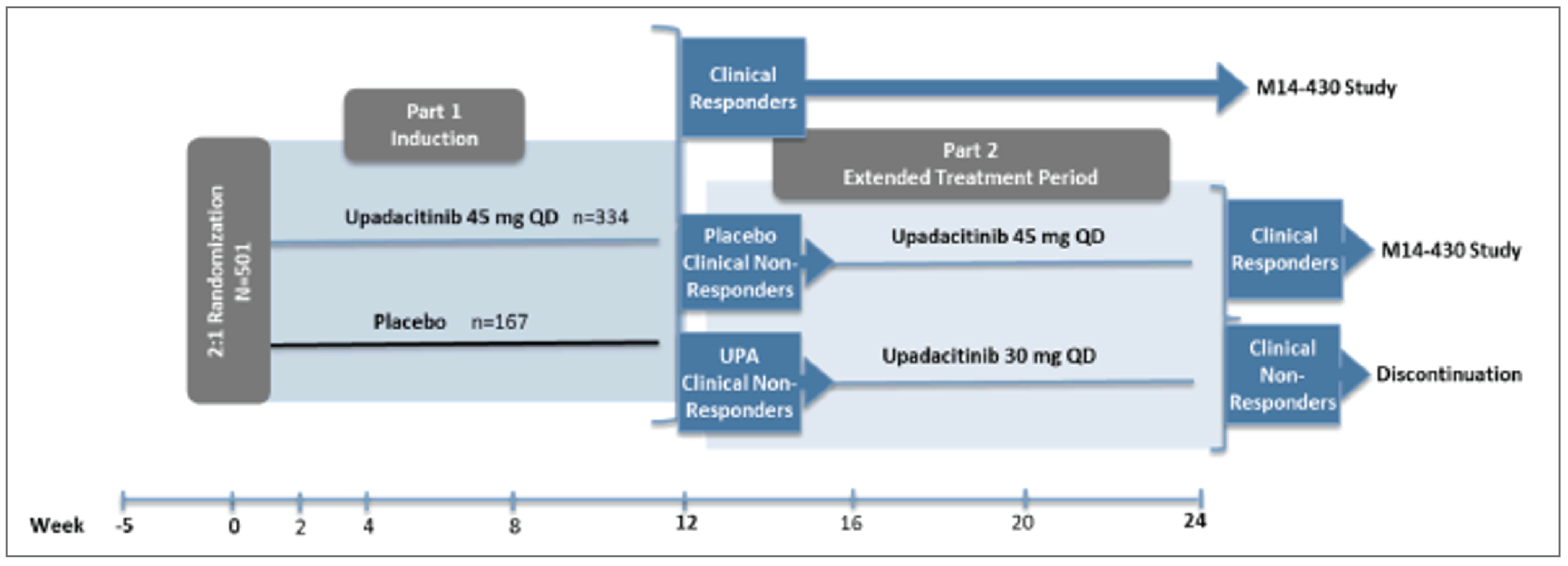
QD = once daily; UPA = upadacitinib.
Source: U-EXCEL Clinical Study Report.22
The U-ENDURE trial was designed to evaluate the efficacy and safety of upadacitinib 15 mg and 30 mg daily as maintenance treatments versus placebo in patients with moderately to severely active CD who responded to upadacitinib induction treatment in the U-EXCEED and U-EXCEL trials. This study was a multicentre, randomized, double-blind, placebo-controlled, long-term extension trial. A total of 901 patients were enrolled between March 2018 and March 2022. The 277 study sites across 43 countries included 17 sites in Canada. No screening phase was reported. Treatment was split into substudy 1 (maintenance) and substudy 2 (long-term extension). In cohort 1 of substudy 1, patients from the U-EXCEED or U-ENDURE trial who received 45 mg of upadacitinib for the 12-week induction (including patients who did not initially achieve a clinical response with placebo and received a second 12-week induction of upadacitinib 45 mg) and achieved a clinical response were re-randomized to either upadacitinib 30 mg daily, upadacitinib 15 mg daily, or matching placebo in a 1:1:1 ratio. Patients were stratified by prior population (blinded bio-IR, open-label bio-IR, and non–bio-IR status) in the induction studies, as well as by clinical remission (as measured by PROs) and endoscopic response. In cohort 2, patients who received placebo and achieved a clinical response during the induction studies continued to receive placebo. In cohort 3, patients who achieved a clinical response during the extended treatment period with 30 mg upadacitinib after not achieving a clinical response in the induction period continued to receive 30 mg upadacitinib daily in the U-ENDURE trial. Reported results in this section are presented for substudy 1, which included a 52-week maintenance treatment for patients previously enrolled in the U-EXCEL or U-EXCEED studies. Substudy 2 of the U-ENDURE trial includes a 240-week long-term extension study, which is ongoing.
Figure 3: Study Design Schematic for U-ENDURE
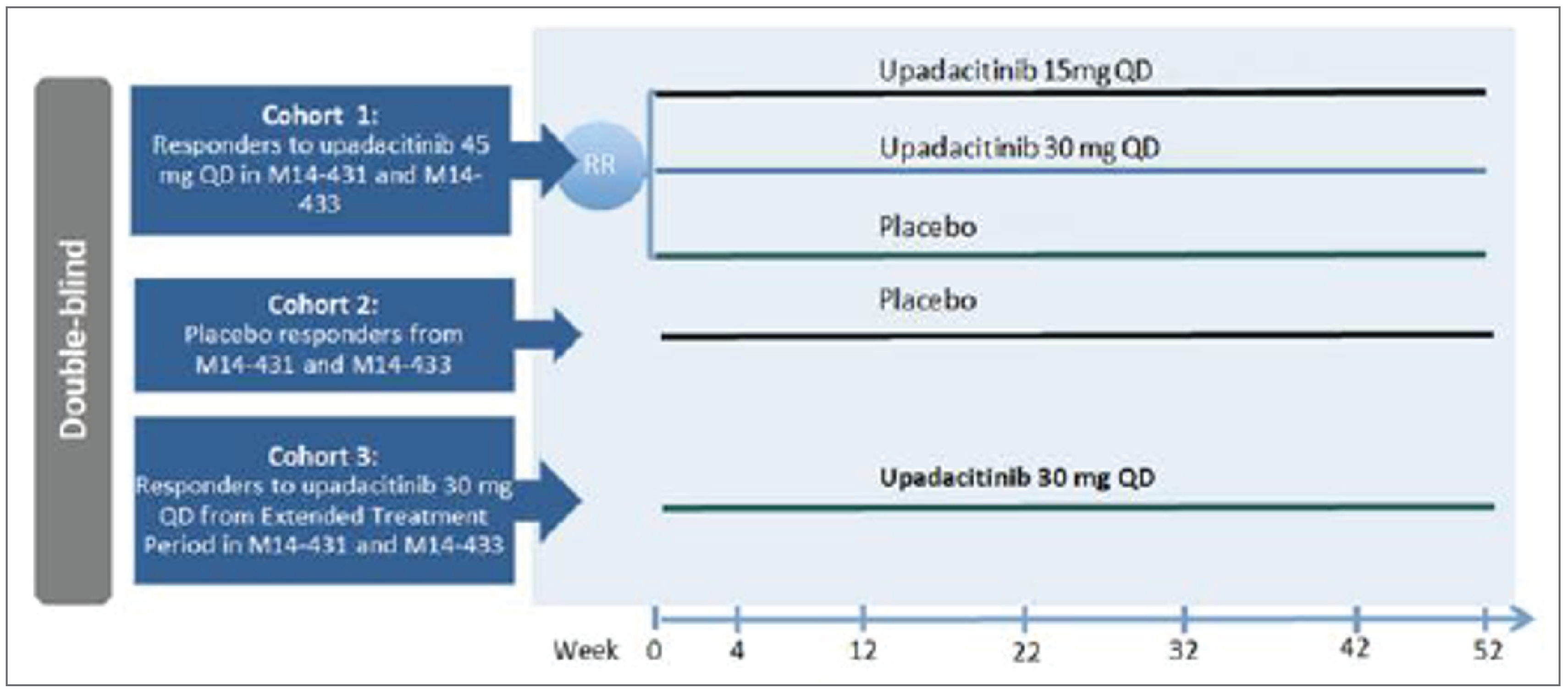
QD = once daily.
Source: U-ENDURE Clinical Study Report.23
Populations
Inclusion and Exclusion Criteria
The U-EXCEED trial enrolled patients who had an inadequate response or were intolerant to 1 or more biologics for CD (bio-IR population).
The U-EXCEL trial enrolled patients who had an inadequate response or intolerance to conventional therapies but had not failed biologic therapy (non-bio-IR population), and patients who had an inadequate response or were intolerant to 1 or more biologics for CD (bio-IR population).
Patients in the U-EXCEED and U-EXCEL trials had a confirmed diagnosis of CD (an average daily soft or liquid SF of at least 4 and/or average daily AP score of at least 2). Patients were not included if they had a current diagnosis of ulcerative or indeterminate colitis, fulminant colitis, ongoing abscess, symptomatic bowel strictures, or ileoanal pouch, or were planning bowel surgery.
Patients in the U-ENDURE trial had a confirmed diagnosis of CD, achieved a clinical response, and completed the induction studies. Patients were not included if they had hypersensitivity or a history of an AE with recurring infections.
Interventions
All 3 trials compared upadacitinib against placebo. Upadacitinib was administered for 12 weeks at 45 mg once daily for parts 1 and 2, and 30 or 45 mg once daily for part 3 of the U-EXCEED trial. For the U-EXCEL trial, upadacitinib was administered for 12 weeks at 45 mg once daily for part 1, and at 30 mg or 45 mg once daily for part 2. Patients enrolled in the U-ENDURE trial received upadacitinib for 52 weeks at 15 or 30 mg once daily for cohort 1, and 30 mg once daily for cohort 3. In all 3 trials, the matching placebo was identical in appearance to the upadacitinib tablets and packaging. The investigators, study-site personnel, data-monitoring committee, and enrolled patients were to remain blinded to treatment assignment throughout the study, except in the event of a medical situation that required unblinding.
Prior and Concomitant Medications
For the induction trials (U-EXCEL and U-EXCEED), patients must have discontinued biological therapy before the first dose of the study drug as specified in the washout procedures. Biologics and medications containing strong CYP3A inhibitors and inducers were prohibited during the study periods. Patients were allowed to enrol with concomitant use of a systemic oral corticosteroid, methotrexate, aminosalicylate, or CD-related antibiotics on stable doses.
The mandatory tapering schedule for corticosteroids began at week 4. All patients continuing in the study were to have corticosteroids discontinued no later than week 11. Patients who did not achieve a clinical response at week 12 and enter part 2 of either study without having completed the steroid taper were to resume tapering at week 16. For patients who entered the study on a corticosteroid dose that was between 2 specific levels in the tapering schedule, the dose beginning at baseline was to be rounded up to the closest higher dose. Patients were to reduce their corticosteroid dose weekly by a set amount dictated by their baseline dose of prednisone, prednisolone, budesonide, methylprednisolone, or hydrocortisone. Patients were not allowed to be on both budesonide (for CD) and prednisone (or equivalent) simultaneously.
Initiation of locally acting (rectal or suppository) or systemic corticosteroids for any reason was prohibited during the study and considered a protocol deviation. The use of inhaled or topical (except rectal or suppository) corticosteroids was not restricted.
Patients receiving a stable dose of CD-related antibiotics, aminosalicylates, or methotrexate were to maintain their concomitant treatments without dose changes through the end of the study; initiating and/or changing doses of these medications was prohibited during the study, except for decreases in the event of moderate to severe treatment-related toxicities. Use of setons as concomitant therapy was authorized for patients with perianal fistulas.
In the maintenance trial (U-ENDURE substudy 1), patients on concomitant CD-related antibiotics were allowed to discontinue them at study entry. Patients on concomitant methotrexate or aminosalicylate were to continue them on stable doses through the end of the study. Doses of CD-related antibiotics, aminosalicylates, or methotrexate could be decreased in the event of treatment-related toxicities or demonstrated inadequate response and need for rescue therapy. Patients who were on a corticosteroid were encouraged to continue the mandatory gradual taper, in which doses were reduced on a weekly basis according to baseline dose.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures and a summary of the interpretation and validity of PROs in Table 8. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review by the clinical expert consulted by CADTH and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points that were considered to be most relevant to inform CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing CADTH’s expert committee deliberations were also assessed using GRADE.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | U-EXCEED | U-EXCEL | U-ENDURE substudy 1 |
|---|---|---|---|---|
Clinical remission via PROs |
| FDA: Ranked secondary (12 weeks)a EMA: Coprimary (12 weeks)a | FDA: Ranked secondary (12 weeks) a EMA: Coprimary (12 weeks)a | FDA: Ranked secondary (52 weeks)a EMA: Coprimary (52 weeks)a |
Clinical remission via CDAI |
| FDA: Coprimary (12 weeks)a EMA: Ranked secondary (12 weeks)a | FDA: Coprimary (12 weeks)a EMA: Ranked secondary (12 weeks)a | FDA: Coprimary (52 weeks)a EMA: Ranked secondary (52 weeks)a |
Endoscopic response |
| FDA and EMA: Coprimary (12 weeks)a | FDA and EMA: Coprimary (12 weeks)a | FDA and EMA: Coprimary (52 weeks)a |
Endoscopic remission |
| FDA and EMA: Ranked secondary (12 weeks)a | FDA and EMA: Ranked secondary (12 weeks)a | FDA and EMA: Ranked secondary (52 weeks)a |
Proportion of patients who discontinue corticosteroid use for CD and achieve clinical remission as measured by CDAI at week 12, in patients taking corticosteroids for CD at baseline | At 12 weeks (induction) | FDA: Ranked secondary (12 weeks)a EMA: Additional nonranked outcome (12 weeks) | FDA: Ranked secondary (12 weeks)a EMA: Additional nonranked outcome (12 weeks) | NA |
Proportion of patients who discontinued corticosteroid use for CD at least 90 days before week 52 and achieved clinical remission as measured by CDAI at week 52, in patients taking corticosteroids for CD at induction baseline | At 52 weeks (maintenance) | NA | NA | FDA: Ranked secondary (52 weeks)a EMA: Additional nonranked outcome (52 weeks) |
Clinical remission as measured by CDAI and endoscopic remission |
| Additional nonranked outcome (12 weeks) | Additional nonranked outcome (12 weeks) | FDA: Ranked secondary outcome (52 weeks)a EMA: Additional nonranked outcome (52 weeks) |
IBDQ, change in total score from baseline |
| FDA and EMA: Ranked secondary (12 weeks)a | FDA and EMA: Ranked secondary (12 weeks)a | FDA and EMA: Ranked secondary outcome (52 weeks)a |
CR-100 |
| FDA and EMA: Ranked secondary (12 weeks)a | FDA and EMA: Ranked secondary (12 weeks)a | FDA and EMA: Ranked secondary outcome (52 weeks)a |
Resolution of EIMs in patients who had EIMs at induction baseline |
| FDA and EMA: Ranked secondary (12 weeks)a | FDA and EMA: Ranked secondary (12 weeks)a | FDA and EMA: Ranked secondary outcome (52 weeks)a |
Proportion with serious AEs |
| Safety | Safety | Safety |
Proportion of patients with hospitalizations related to CD |
| FDA and EMA: Ranked secondary (12 weeks)a | FDA and EMA: Ranked secondary (12 weeks) a | FDA and EMA: Ranked secondary outcome (52 weeks)a |
Occurrence of CD-related surgeries |
| Additional nonranked outcome (12 weeks) | Additional non-ranked outcome (12 weeks) | Additional nonranked outcome (52 weeks) |
AE = adverse event; CD = Crohn disease; CDAI = Crohn Disease Activity Index; CR-100 = decrease of at least 100 points from baseline in the Crohn Disease Activity Index; EIM = extra-intestinal manifestation; EMA = European Medicines Agency; IBDQ = Inflammatory Bowel Disease Questionnaire; PRO = patient-reported outcome.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Source: Clinical Study Reports of U-EXCEED,22 U-EXCEL,23 and U-ENDURE.26 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Clinical Remission via PROs or via CDAI
The primary outcomes were the same across all 3 trials, assessed at 12 weeks in the U-EXCEL and U-EXCEED trials, and at 52 weeks in the U-ENDURE trial. The primary outcomes were clinical remission as measured by PROs for EMA regulatory purposes, and clinical remission as measured by CDAI for FDA regulatory purposes.
Both the PRO-based and CDAI-based metrics of clinical remission were included in CADTH’s assessment because, depending on jurisdiction of regulatory body (i.e., FDA versus EMA), both were considered end points coprimary with endoscopic response. Among the clinical expert consulted by CADTH, the clinician group input submitted to the review, the design of the included trials, and previous publications of trials in CD, there is a diversity of opinion on preference between PRO- and CDAI-based clinical remission for decision-making. As a result, CADTH concluded it was prudent to assess both.
Clinical remission as measured by PROs was defined as an average daily very soft or liquid SF of less than equal to 2.8 and average daily AP score of less than equal to 1.0 and both not greater than baseline, based on patient diary entries in the prior 7 days. Very soft and liquid stools were assessed using the Bristol Stool Chart, which patients were trained to recognize and calculate with during the screening period.
The National Cooperative Crohn Disease Study Group developed the CDAI using prospective data from 187 visits of 112 patients with CD.50 The CDAI is a disease-specific index that is considered the standard for assessing CD activity in clinical trials, but is also considered impractical for real-world practice. The overall score is based on the sum of the weighted value of 8 domains and ranges between 0 to 600; a score of 150 is the threshold between remission and active disease.51,52 Scores ranging between 150 to 219 indicate mild to moderate CD, scores between 250 and 450 indicate moderate to severe CD, and scores above 450 indicate very severe CD.50,53 Scores for each item are derived using entries from patient diaries from the 7 days preceding each visit. The items are assessed on the daily sum per week and include the following: number of liquid or very soft stools; AP score (from 0 to 3), general well-being (from 0 to 4); sum of events per week of arthritis and/or arthralgia, mucocutaneous lesions, iris and/or uveitis, anal disease, external fistula, or fever above 37.8°C; antidiarrheal use; abdominal mass (none = 0, equivocal = 2, present = 5); a hematocrit level of 47 (males) or 42 (females); and 100 × (1 – [body weight + standard weight]).50
Endoscopic Response (SES-CD)
Endoscopic response was a coprimary outcome in all 3 trials for both the EMA and FDA. It was assessed at 12 weeks in the U-EXCEL and U-EXCEED trials, and at 52 weeks in the U-ENDURE trial. Endoscopic response was defined as a decrease of greater than 50% from baseline in SES-CD (or for patients with an SES-CD of 4 at baseline of the induction study, at least a 2-point reduction from baseline), as scored by the central reviewer.
The SES-CD was designed to assess 4 endoscopic items: size of ulcers, ulcerated surface, affected surface, and presence of narrowing.54 Each item is scored from 0 to 3 with a total score ranging from 0 to 56. Higher scores indicate more severe disease.
Endoscopic Remission (SES-CD)
Endoscopic remission was defined as an SES-CD of 4 or lower and at least 2-point reduction from baseline and no subscore greater than 1 in any individual variable, as scored by the central reviewer. It was assessed at 12 weeks in the U-EXCEED and U-EXCEL trials, and at 52 weeks in the U-ENDURE trial.
Corticosteroid Discontinuation (for CD) and Clinical Remission as Measured by CDAI
The 12-week outcome evaluated in the U-EXCEED and U-EXCEL trials was the proportion of patients who discontinued corticosteroids and had clinical remission as measured by CDAI among those who had been receiving corticosteroids at baseline.
The 52-week outcome evaluated in the U-ENDURE trial was the proportion of patients who had not used a corticosteroid to treat CD for at least 90 days before week 52 and who had clinical remission as measured by CDAI among those who had used a corticosteroid at induction baseline.
The trials also reported corticosteroid discontinuation and clinical remission as measured by PROs. Although not formally assessed in this review because clinical remission as measured by PROs does not have validated thresholds for disease severity or MIDs, brief comparisons are made in the results section between the CDAI-based and PRO-based results. Results were also generated for the subset of patients who were taking corticosteroids at baseline; again, although not formally assessed, the results are discussed in comparison with the outcome of interest.
Clinical Remission as Measured by CDAI and Endoscopic Remission
The proportion of patients who achieved both clinical remission as measured by CDAI and endoscopic remission as measured by SES-CD as previously described. It was assessed at 12 weeks in the U-EXCEED and U-EXCEL trials, and at 52 weeks in the U-ENDURE trial.
The trials also reported clinical remission as measured by PROs and endoscopic remission. Although not formally assessed in this review, brief comparisons between the CDAI-based end point and the PRO-based end point are made in the results section.
IBDQ, Change in Total Score From Baseline
The IBDQ is a disease-specific instrument composed of 32 Likert-scaled items in the form of a physician-administered questionnaire.55,56 The questionnaire is divided into 4 dimensions: bowel symptoms (10 items), systemic symptoms (5 items), emotional function (12 items), and social function (5 items). Patients are asked to recall symptoms and quality of life from the last 2 weeks, and each item is graded on a 7-point Likert scale, where 1 represents the worst situation and 7 represents the best situation. The total score ranges from 32 to 224, where higher scores represent better quality of life. Scores of patients in remission typically range from 170 to 190. It was assessed at 12 weeks in the U-EXCEED and U-EXCEL trials, and at 52 weeks in the U-ENDURE trial.
CR-100
A CR-100 was defined as a decrease of at least 100 points in CDAI from baseline, which has been suggested by the FDA and EMA as a meaningful response threshold.57 It was assessed as a proportion of patients achieving CR-100 at 12 weeks in the U-EXCEED and U-EXCEL trials, and 52 weeks in the U-ENDURE.
Resolution of EIMs in Patients With EIMs at Induction Baseline
Extra-intestinal manifestations were defined as manifestations of CD in areas of the body other than the digestive tract, including the eyes, skin, joints, mouth, and liver. It is not clear how the resolution of EIMs was defined.
Proportion With SAE
All AEs reported from the time of study drug administration until 30 days following discontinuation of study drug administration were collected, whether solicited or spontaneously reported by the patient. Patients who discontinued study drug treatment but continued to participate in the study had SAEs and nonserious AEs collected for the remainder of their study participation. In addition, SAEs and protocol-related nonserious AEs were collected from the time the patient signed the study-specific informed consent.
An AE was defined as any untoward medical occurrence in a patient given a pharmaceutical product, which does not necessarily have a causal relationship with this treatment. If an AE met any of the following criteria, it was considered to be serious:
death of patient
life-threatening
hospitalization or prolonging of hospitalization
congenital anomaly
persistent or significant disability or incapacity
important medical event requiring medical or surgical intervention to prevent serious outcome.
Proportion of Patients With Hospitalizations Due to Crohn Disease
The proportion of patients who had any hospitalizations due to CD at 12 weeks or at 24 weeks. A hospitalization was defined as any event that resulted in an admission to the hospital for any length of time and does not include visits to an emergency department visits or admission to an outpatient facility.
Other Outcomes
Additional AESIs, which were reported but not assessed using GRADE, include:
serious infections
opportunistic infections
herpes zoster
active tuberculosis
malignancy (all types)
adjudicated gastrointestinal perforations
adjudicated cardiovascular events (e.g., major adverse cardiac event)
anemia
neutropenia
lymphopenia
renal dysfunction
hepatic disorder
elevated CPK
adjudicated embolic and thrombotic events (noncardiac, non–central nervous system).
Additional outcomes reported only to inform the economic evaluation are summarized in Appendix 1:
5-Level EQ-5D at 12 and 52 weeks.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Clinical remission via PROs | Defined as average daily very soft or liquid SF ≤ 2.8 and average daily AP score ≤ 1.0 and both not greater than baseline. This consists of the SF (i.e., number of liquid or very soft stools) and AP (i.e., abdominal pain rating) components of CDAI and was assessed by patient diary entries. | Construct validity: Based on data from a trial investigating methotrexate vs. placebo in 141 patients with chronically active CD who had received prednisone therapy for a minimum of 3 months,58 Khanna et al. (2015)59 conducted regression analyses against CDAI and found that SF/AP scores of 8, 14, and 34 points correlated with CDAI scores of 150, 220, and 450 points (R2 = 0.76), respectively, and that SF/AP-change scores of 4, 5, and 8 points correlated with CDAI change scores of 50, 70, and 100 points (R2 = 0.71), respectively. Responsiveness: The Guyatt responsiveness statistic was 0.48 (95% CI, 0.30 to 0.75) for SF/AP score (conventional thresholds of 0.2, 0.5 and 0.8 indicate small, moderate, and large degrees of responsiveness, respectively).59 | No information regarding MID in patients with CD was identified. |
CDAI | The CDAI is a disease-specific index used to assess the severity of CD. The CDAI consists of 8 items, each of which is independently weighted, including SF (weight: 2), abdominal pain (weight: 5), general well-being (weight: 7), sum of 6 findings (weight: 20), antidiarrheal use (weight: 30), hematocrit level (weight: 6), and body weight (weight: 1). The overall CDAI score is based on the sum of the weighted value of each item and ranges from 0 to 600, where a score of 150 is defined as the threshold between remission and active disease. Scores ranging between 150 to 219 indicate mild to moderate CD, scores between 250 to 450 indicate moderate to severe CD, and scores above 450 indicate very severe CD.50,53 | Construct validity: The items included in the CDAI were selected by gastroenterologists and are based on accepted features of CD.53 Criterion validity: Generally, the CDAI does not demonstrate any significant correlation between the overall score and objective measurements such as mucosal healing. However, the lack of correlation may not be indicative of a lack of criterion validity due to the multifaceted nature of CD.53 Predictability is another component of criterion validity. One study demonstrated that CDAI scores increased 2 months preceding exacerbations of CD and decreased 1 month following exacerbations of CD, therefore demonstrating criterion validity.53 Test-retest reliability: the index provided good to very good test-retest reliability evaluated based on 2 successive visits for 32 patients.50,53 The CDAI was subsequently re-evaluated and re-derived using data collected from 1,058 patients and demonstrated little difference compared to the original formulation; therefore, the original version was recommended.60 | The FDA and EMA have suggested that a change of 100 points in CDAI is a meaningful response (i.e., enhanced clinical response).57 |
Endoscopic outcomes (SES-CD score) | The SES-CD was designed for the assessment of 4 endoscopic items, including size of ulcers, ulcerated surface, affected surface, and presence of narrowing.54 Each item is to be scored 0 to 3 with a total score ranging from 0 to 56. Higher scores indicate more severe disease. | Construct validity: Daperno et al. (2004)61 validated SES-CD against CDEIS in 70 patients with CD and found a strong correlation between the 2 instruments (multiple correlation coefficient = 0.920; 95% CI, 0.8740 to 0.9497). After construction of SES-CD, Daperno et al. (2004)61 validated SES-CD against CDEIS in a sample of 121 patients with CD. The Pearson and the Spearman rank correlation coefficients between SES-CD and CDEIS were 0.887 (95% CI, 0.8418 to 0.9199) and 0.910 (95% CI, 0.8734 to 0.9364) (P < 0.001), respectively. In a review, estimates of correlation between SES-CD and the CDAI ranged from 0.15 to 0.92.62 Intra- and inter-rater reliability: In a study of 50 patients with CD Khanna et al. (2016)63 found that the ICC for intra-rater agreement for SES-CD was 0.91 (95% CI, 0.89 to 0.95). The corresponding ICC for inter-rater agreement was 0.83 (95% CI, 0.75 to 0.88). | No information regarding the MID of SES-CD in patients with CD was identified. |
IBDQ | The IBDQ is a physician-administered questionnaire developed by Guyatt et al. to assess HRQoL in patients with IBD.55,56,64 It is a 32-item Likert-based questionnaire, divided into 4 dimensions (bowel symptoms [10 items], systemic symptoms [5 items], emotional function [12 items], and social function [5 items]). Patients are asked to recall symptoms and quality of life from the last 2 weeks, with responses graded on a 7-point Likert scale (1 being the worst situation, 7 being the best) with the total IBDQ score ranging between 32 and 224 (i.e., higher scores represent better quality of life). Scores of patients in remission typically range from 170 to 190. | This questionnaire has been validated in a variety of settings, countries, and languages.64,65 Discriminant validity: A review64 of 9 validation studies on the IBDQ in patients with IBD reported that the IBDQ was able to differentiate clinically important differences between patients with disease remission and patients with disease relapse. Responsiveness: All 6 studies that evaluated the IBDQ for sensitivity to change found that changes in HRQoL correlated to changes in clinical activity in patients with CD.64 | A study conducted by Gregor et al.66 noted that a clinically meaningful improvement in quality of life would be an increase of at least 16 points in the IBDQ total score or 0.5 points or more per question in patients with CD. |
EQ-5D-5L | The EQ-5D-5L is a generic self-reported HRQoL outcome measure that may be applied to a variety of health conditions and treatments.67 The first 2 components of the EQ-5D-5L assess 5 domains: mobility, self-care, usual activities, pain/discomfort and anxiety/depression.67 Each domain has 5 levels: no problem; slight problems; moderate problems; severe problems; and extreme problems. A descriptive system that classifies respondents (aged ≥ 12 years) based on the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. The EQ-5D-5L has 5 possible levels for each domain and respondents are asked to choose the level that reflects their health state for each of the 5 domains resulting in 3,125 possible health states.67 The EQ-5D-5L tool has been applied to a wide range of health conditions and treatments, including IBD.68,69 The EQ-5D-5L index score is generated by applying a multi-attribute utility function to the descriptive system.70 Different utility functions are available that reflect the preferences of specific populations (e.g., US or UK). Scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively. The second component of the EQ-5D-5L is a 10 cm VAS that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” Respondents are asked to rate their health by drawing a line from an anchor box to the point on the VAS that best represents their health on that day. The EQ-5D-5L produces 3 types of data for each respondent:
| Discriminant validity: In a study validating EQ-5D-5L in 206 patients with CD,71 the EQ-5D-5L had an average Shannon index, H′, of 1.18. and an average Shannon evenness index, J′, of 0.51. Convergent validity: In a single study,71 the correlation coefficients between the EQ-5D-5L and its VAS ranged from −0.091 for self-care to −0.525 for pain/discomfort. Between EQ-5D-5L and CDAI scores across all dimensions, the correlation coefficients ranged from −0.028 for mobility to 0.182 for pain and/or discomfort. The correlation coefficients between EQ-5D-5L and CDAI ranged from 0.044 for anxiety and/or depression to 0.285 for pain and/or discomfort. | No information regarding MID of EQ-5D-5L in patients with CD was identified. |
AP = abdominal pain; CD = Crohn disease; CDAI = Crohn Disease Activity Index; CDEIS = Crohn Disease Endoscopic Index of Severity; CI = confidence interval; EMA = European Medicines Agency; EQ-5D-5-Level = 5-Level EQ-5D questionnaire; HRQoL = health-related quality of life; IBD = inflammatory bowel disease; IBDQ = Inflammatory Bowel Disease Questionnaire; ICC = intraclass correlation coefficient; MID = minimal important difference; PRO = patient-reported outcome; SES-CD = Simple Endoscopic Score for Crohn Disease; SF = stool frequency; VAS = visual analogue scale.
Statistical Analysis
A summary of statistical analyses by end point is presented in Table 9.
Primary End Points
Endoscopic response and clinical remission as measured by PROs or CDAI were the coprimary end points in all trials. All primary and secondary end points were analyzed based on the intention-to-treat population for part 1 in each study (ITT1). The comparison between the treatment groups for the coprimary end points was performed using the Cochran-Mantel-Haenszel (CMH) test and was adjusted for stratification factors including baseline corticosteroid use (yes or no), endoscopic disease severity (SES-CD < 15 or ≥ 15), and number of prior biologics used (> 1 or ≤ 1) in the U-EXCEED trial. In the U-EXCEL trial, stratification was based on baseline corticosteroid use (yes or no), endoscopic disease severity (SES-CD < 15 or ≥ 15) and number of prior biologics with prior inadequate response or intolerance (0, 1, or > 1). A CMH-based 2-sided 95% CI for the difference between the treatment groups compared to placebo was constructed for all end points. Clinical remission according to both PROs and CDAI used 3 sensitivity analyses in both the U-EXCEED and U-EXCEL trials: a “9-2 approach” (using a 7-day window before a visit, excluding 2 days around the endoscopic procedure; i.e., a total of 9 days before the visit excluding the day before and the day of endoscopy), a nonresponder imputation with no special data-handling to account for COVID-19 (NRI-NC), and “as observed” (AO). Endoscopic remission was analyzed using the NRI-NC and AO approaches in all trials.
For all primary and secondary end points, the primary approach for handling missing data was nonresponder imputation while incorporating multiple imputation to handle data missing due to COVID-19. Moreover, if the average daily very soft or liquid SF or AP score or CDAI at week 12 were missing, the nonresponder-imputation approach was applied for the clinical remission as measured by PROs at week 12 and clinical remission as measured by CDAI at the week 12 end points, respectively. Patients who discontinued the study drug before week 12 were considered “not achieved” for clinical remission or endoscopic response end points in both the U-EXCEED and U-EXCEL trials. The method used to treat missing patients was also applied to the U-ENDURE trial at week 52.
In the U-ENDURE trial, the coprimary end points between the upadacitinib and placebo group were analyzed using the CMH test and stratified by randomization factors (prior induction population), clinical remission (as measured by PROs), status (yes or no), and endoscopic response status (yes or no). A multiple-testing procedure was used to control of the type I error rate at a 0.05 significance level comparing the upadacitinib dose group to the placebo group followed by a test using a 0.025 significance level.
Secondary End Points
Categorical secondary efficacy variables were analyzed using a CMH test for stratification variables. A CMH-based 2-sided 95% CI for the difference between treatment groups versus placebo was constructed for all end points. The same stratification factors were used for each trial. Covariates including continuous secondary efficacy variables with repeated measurements, such as the change in baseline for IBDQ total scores for all trials, were analyzed using a mixed model for repeated measures (MMRM). Hospitalizations due to CD during the induction period at week 12 (U-EXCEED and U-EXCEL trials) were analyzed based on the observed data using the AO approach only. For the U-EXCEED and U-EXCEL trials, all other secondary efficacy end points were presented using both the NRI-NC and AO approaches. For the ENDURE trial, the continuous secondary efficacy variables with repeated measurements also used multiple imputation incorporating a return to baseline. All other efficacy end points except for the exposure-adjusted rate for CD-related hospitalizations (which used only the AO approach) were presented using primary-analysis NRI-NC and AO approaches. For all primary and secondary end points with a small sample size or small number of responders (e.g., occurrence of CD-related hospitalizations during part 1), upadacitinib and placebo groups were compared using a chi-square test (or Fisher's exact test if more than 20% of the cells had expected counts of less than 5).
Table 9: Statistical Analysis of Efficacy End Points in Each Trial
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
U-EXCEED and U-EXCEL | ||||
| CMH test Chi-square test | Stratified by the randomization stratification factors (baseline steroid use, endoscopic disease severity, and number of prior biologics based on ITT1 population) | Nonresponder imputation incorporating MI to handle missing data due to COVID-19a | 1. Sensitivity analysis calculated based on the 9 to 2 approachb 2. As observedc 3. NRI-NC |
| 1. As observedc 2. NRI-NC | |||
Change in baseline for IBDQ total score at week 12 | MMRMd ANCOVAe | Stratified by the randomization stratification factors (baseline steroid use, endoscopic disease severity, and number of prior biologics based on ITT1 population) | MMRMd | NA |
| CMH test Chi-square test | As observedc | ||
U-ENDURE | ||||
| CMH test Chi-square test | The randomization was stratified by prior population (blinded bio-IR, open-label bio-IR, and non–bio-IR status) in the induction studies, as well as the clinical remission (as measured by PROs) and endoscopic response | Nonresponder imputation incorporating MI to handle missing data due to COVID-19a | 1. As observedc 2. NRI-NC |
Change from induction baseline in IBDQ total score at week 52 | MMRMd ANCOVAe | Nonresponder imputation incorporating MI to handle missing data due to COVID-19a | MMRMd RTB-MIf | NA |
| CMH test Chi-Square test | As observedc | ||
ANCOVA = analysis of covariance; bio-IR = inadequate response or intolerant to 1 or more biologics; CD = Crohn disease; CDAI = Crohn Disease Activity Index; CMH = Cochran-Mantel-Haenszel; CR-100 = decrease of at least 100 points from baseline in the Crohn Disease Activity Index; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; IBDQ = Inflammatory Bowel Disease Questionnaire; ITT1 = intention-to-treat (part 1 or cohort 1 of study); MI = multiple imputation; MMRM = mixed model for repeated measures; NA = not applicable; NRI-NC = nonresponder imputation with no special data-handling to account for COVID-19; PRO = patient-reported outcome; RTB-MI = return-to-baseline multiple imputation.
aNRI-NC categorized any patients who did not have an evaluation during a prespecified window (either due to missing assessment or due to early withdrawal from the study) as a nonresponder for the visit. The only exception is that patients with missing data due to COVID-19 infection or logistical restriction were handled by MI and the patients will be characterized as responders or nonresponders based on MI imputed values. In addition, at and after the CD-related corticosteroids intercurrent event and on after the date of initiation of CD-related confounding medications after premature discontinuation of study drug, patients were considered nonresponders.
bUsing a 7-day window before a visit, excluding 2 days around the endoscopic procedure (i.e., a total of 9 days before the visit excluding the day before and the day of endoscopy).
cAll available measurements are used for analysis regardless of intercurrent events and do not include imputed values for missing data.
dThe repeat measurement analysis will be conducted using a mixed model including observed measurements at all postbaseline visits up to the end of the analysis period. The mixed model includes the categorical fixed effects of treatment, visit and treatment-by-visit interaction, stratification factors at randomization, and the continuous fixed covariates of induction baseline and week 0 measurement.
eFor continuous efficacy variables that are collected at only 1 postbaseline visit (such as the Simplified Endoscopic Score of Crohn Disease), ANCOVA will be used. The model includes the categorical fixed effects of treatment, stratification factors at randomization, and the continuous fixed covariates of induction baseline and week 0 measurement.
fRTB-MI is multiple imputation incorporating return-to-baseline to handle visits after occurrence of intercurrent events. The least squares mean, 95% CI and standard error are synthetic result based on ANCOVA with baseline FACIT-F, week 0 FACIT-F, stratification factors (prior induction), population (non–bio-IR, double-blind bio-IR, and open-label bio-IR), clinical remission as measured by PRO status (yes or no), and endoscopic response status (yes or no).
Source: Sponsor’s Summary of Clinical Evidence.
Sample Size and Power Calculation
The U-EXCEED trial enrolled 624 patients, with 495 assigned to the randomized, double-blind, part 1 phase. Assuming a rate of 12% for clinical remission as measured by PROs in the placebo group and 29% in the upadacitinib group at week 12, a total sample size of 495 patients randomized in a 2:1 ratio (330 patients in the upadacitinib group and 165 patients in the placebo group) would be sufficient to detect at least a 17% difference between the treatment groups in clinical remission rates at week 12 using Fisher’s exact test with at least 95% power at a 2-sided significance level of 0.05. Assuming a rate of 20% for clinical remission as measured by CDAI in the placebo group and 40% in the upadacitinib group at week 12, a same sample size of 495 patients would be sufficient to detect at least a 20% difference between the treatment groups in clinical remission rates at week 12 using Fisher’s exact test with at least 95% power at a 2-sided significance level of 0.05.
The U-EXCEL trial had a sample size of 526, with all 526 assigned to the randomized, double-blind, part 1 phase. Assuming a clinical remission rate of 15% in the placebo group and 33% in the upadacitinib group at week 12, a total sample size of 501 patients randomized in a 2:1 ratio (334 patients in the upadacitinib group and 167 patients in the placebo group) would be sufficient to detect at least an 18% difference between the treatment groups in clinical remission rates at week 12 using Fisher’s exact test with at least 95% power at a 2-sided significance level of 0.05. Assuming a clinical remission rate of 21.5% as measured by CDAI in the placebo group and 45% in the upadacitinib group at week 12, this sample size would be sufficient to detect at least a 23.5% difference between the treatment groups in clinical remission rates at week 12 using Fisher’s exact test with at least 95% power at a 2-sided significance level of 0.05. Assuming an endoscopic response rate of 11.5% in the placebo group and 28.5% in the upadacitinib group at week 12, this sample size would be sufficient to detect at least a 17% difference between the treatment groups in endoscopic response rates at week 12 using Fisher’s exact test with at least 95% power at a 2-sided significance level of 0.05.
The U-ENDURE trial enrolled 901 patients, with 674 assigned to cohort 1. Assuming a week 52 clinical remission (as measured by PROs) rate of 42% for 1 of the upadacitinib dose groups and 17% for the placebo group, a total sample size of 501 patients randomized in a 1:1:1 ratio (167 patients each in the upadacitinib 30 mg daily, upadacitinib 15 mg daily, and placebo groups) would have approximately 99% power to detect at least a 25% difference between the treatment and placebo groups in clinical remission rates at week 52 using Fisher’s exact test at a 2-sided significance level of 0.025. Assuming a week-52 clinical remission (as measured by CDAI) rate of 50% for 1 of the upadacitinib dose groups and 22% for the placebo group, this would have approximately 99% power to detect at least a 28% difference between the treatment and placebo groups in clinical remission rates at week 52 using Fisher’s exact test at a 2-sided significance level of 0.025. Assuming an endoscopic response rate of 35% for 1 of the upadacitinib dose groups and 17% for the placebo group, this sample size would have approximately 94% power to detect at least a difference between the treatment and placebo groups of 18% in endoscopic response rates at week 52 using Fisher’s exact test at a 2-sided significance level of 0.025.
Statistical Testing
In both the U-EXCEED and U-EXCEL trial, the overall type I error rates of the coprimary and the secondary end points were controlled using a fixed-sequence, multiple-testing procedure as well as the Holm procedure. Multiplicity adjustment was applied to the additional efficacy end points. The analyses for additional efficacy end points were performed at the nominal alpha level of 0.05 (2-sided). Covariates such as categorical variables used a CMH test with a 95% CI. Continuous end points with repeated measurements collected longitudinally were analyzed using an MMRM and those collected at only 1 postbaseline visit were analyzed using analysis of covariance with a 95% CI.
In the U-ENDURE trial, the overall type I error rate of the coprimary and the secondary end points were controlled using a graphical multiple-testing procedure. Coprimary end points were tested using a 2-sided alpha level of 0.025 for each upadacitinib dose compared to placebo based on ITT1 for cohort 1. No statistical comparisons were performed for cohort 2 and cohort 3. The analysis of additional efficacy end points was performed at a nominal alpha level of 0.05 (2-sided) for each dose versus placebo. No multiplicity adjustment was applied to the additional efficacy end points. Similar to the other trials, covariates such as categorical end points used a CMH test with a 95% CI. Continuous secondary end points with repeated measurements were analyzed using an MMRM, and those collected at only 1 postbaseline visit were analyzed using analysis of covariance with a 95% CI.
Subgroup Analyses
Each trial conducted predefined subgroup analyses for several baseline demographic and disease-related variables, including sex, age, race, biomarkers, and clinical metrics of disease severity, treatment history, disease duration, and others. In this report, subgroup results for a number of prior biologics failed are presented for the coprimary outcomes. The clinical expert consulted by CADTH also noted that potentially important subgroups include patients with EIMs at baseline and patients with perianal disease; however, these subgroups were not prespecified in the clinical trials.
For the subgroup analysis, 95% CIs and point estimates for each treatment group as well as for treatment differences between the upadacitinib and placebo groups were presented. No P value was provided. If a subgroup population was less than 10% of the planned study size, subgroup analyses were not presented for that category. Comparability between the upadacitinib and placebo groups was presented for each dose, and no multiplicity adjustment was applied to the end points.
Analysis Populations
The analysis sets for each study are summarized in Table 10.
Table 10: Analysis Populations of U-EXCEED, U-EXCEL, and U-ENDURE
Study | Population | Definition | Application |
|---|---|---|---|
U-EXCEED | ITT1 | All randomized patients who received at least 1 dose of a double-blinded study drug in part 1; patients in this population will be analyzed according to the treatment to which they are randomized to (induction period) | Efficacy and baseline analyses in part 1 |
ITT2 | Includes all patients who have received at least 1 dose of the study drug in part 2 (induction period) | Efficacy and baseline analyses in part 2 | |
ITT3 | Includes all patients who have received at least 1 dose of the study drug in part 3 (extended treatment) | Efficacy and baseline analyses in part 3 | |
Safety | All patients who received at least 1 dose of the study drug; the “as treated” was determined by the most frequent dose regimen received in the analysis period The safety population for part 1 (SA1) includes all patients who received at least 1 dose of the study drug in part 1 The safety population for part 2 (SA2) includes all patients who received at least 1 dose of the study drug (UPA 45 mg) in part 2 The safety population for part 3 (SA3) includes all patients who received at least 1 dose of the study drug (UPA 30 mg or 45 mg) in part 3 The all-UPA safety population (SA-UPA) includes all patients who received at least 1 dose of upadacinitib in part 1 or part 2 or part 3 | Safety data and analysis of adverse events | |
PP | Per-protocol patients include a subset of ITT patients who did not have major protocol deviations; patients were included in the analysis according to the treatment group to which they were randomized | Efficacy analyses | |
U-EXCEL | ITT1 | All randomized patients who received at least 1 dose of a double-blinded study drug during part 1; patients in this population will be analyzed according to the treatment to which they are randomized (induction period) | Efficacy and baseline analyses for part 1 |
ITT2 | All patients who received at least 1 dose of the study drug in part 2 (extended treatment period) | Efficacy and baseline analyses for part 2 | |
Safety | All patients who received at least 1 dose of the study drug in part 1; the “as treated” was determined by the most frequent dose regimen received in the analysis period The safety population for part 1 (SA1) includes all patients who received at least 1 dose of the study drug in part 1 The safety population for part 2 (SA2) includes all patients who received at least 1 dose of the study drug (UPA 45 mg or 30 mg) in part 2 The all-UPA safety population (SA-UPA) includes all patients who received at least 1 dose of upadacinitib in part 1 or part 2 | Safety data and analysis of adverse events | |
PP | Per-protocol patients include a subset of ITT patients who did not have major protocol deviations | Efficacy analyses | |
U-ENDURE | ITT1 | All randomized patients who received at least 1 dose of the study drug in substudy 1; patients in this population will be analyzed according to the treatment to which they are randomized | Efficacy and baseline analyses for cohort 1 |
ITT2 | The subset of ITT population who have been enrolled in cohort 2, including patients who a) completed 52 weeks of study treatment or b) were enrolled at least 52 weeks prior but prematurely withdrew from the study | Efficacy and baseline analyses for cohort 2 | |
ITT3 | The subset of ITT population who were enrolled in cohort 3, including patients who a) completed 52 weeks of study treatment or b) were enrolled at least 52 weeks prior but prematurely withdrew from the study | Efficacy and baseline analyses for cohort 3 | |
Safety | All patients who received at least 1 dose of the study drug; the “as treated” was determined by the most frequent dose regimen received in the analysis period. SA population: all patients who received at least 1 dose of the study drug in substudy 1 SA1 population: the subset of SA population who were in cohort 1 SA2 population: the subset of SA population who were enrolled in cohort 2. SA3 population: the subset of SA population who were enrolled in cohort 3. The all-UPA safety population (SA-UPA) included all patients who received at least 1 dose of upadacinitib in substudy 1 | Safety data and analysis of adverse events | |
Per protocol | Per-protocol patients include a subset of ITT patients who did not have major protocol deviations; patients were included in the analysis according to the treatment group to which they were randomized | Efficacy analyses |
ITT = intention-to-treat population; ITT1 = intention-to-treat population in part 1, ITT2 = intention-to-treat population in part 2, ITT3 = intention-to-treat population in part 3, SA = safety population, UPA = upadacitinib.
Source: Sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
In the part 1 double-blind period of the U-EXCEED and U-EXCEL trials, which informs the 12-week outcomes, 11.1% and 8.0% of patients discontinued from the studies, respectively. Discontinuation was typically due to AEs, withdrawal by patients, or lack of efficacy. Comparing the placebo and upadacitinib groups, the rate of discontinuation was similar but slightly higher in the placebo group.
In substudy 1 of the U-ENDURE trial, which informs the 52-week outcomes, discontinuation occurred in almost a quarter of randomized patients; ||||||| ||||||||||||||| |||||||| | || |||||||||| |||||||| ||||||| || || ||||||||| ||||||| || |||||||||||||||| |||||||||||| || || || || |||||||||| || ||||||||| || |||||||||||||||| || || ||||| || ||||| || || ||||||||| |||||| || |||| || ||||||| |||||||| |||||||| || |||||||||||| || || || || |||||||| ||||||||| ||||||| ||||||||||||||| |||||||| || ||||| || ||||| || ||||||||| |||||||||||||
Table 11: Summary of Patient Disposition in U-EXCEED
Patient disposition | Part 1 (double-blind) | |||| || | |||| || |||||||| ||||||||| | |||| ||| | ||||
|---|---|---|---|---|---|---|---|---|
PBO (N = 171) | UPA 45 mg (N = 324) | Total (N = 495) | ||| || |||| | |||||||| || | | ||| || ||||||| | |||||||| || | ||| || ||||||| | |
Randomized, N (%) | 171 (100.0) | 324 (100.0) | 495 (100.0) | || | || | || | || | || |
Treated, N (%) | 171 (100.0) | 324 (100.0) | 495 (100.0) | ||| ||||||| | | ||||||| | | ||||||| | ||| ||||||| | | ||||||| |
Discontinued study drug N (%) | 22 (12.9) | 33 (10.2) | 55 (11.1) | | ||||| | | |||||| | | |||||| | | |||||| | | |||||| |
Discontinued from study, N (%) | 22 (12.9) | 33 (10.2) | 55 (11.1) | | ||||| | | |||||| | | |||||| | | |||||| | | |||||| |
Adverse events | 5 (2.9) | 17 (5.2) | 22 (4.4) | | ||||| | | |||||| | | ||||| | | ||||| | | ||||| |
Withdrawal by patients | 8 (4.7) | 8 (2.5) | 16 (3.2) | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Loss to follow-up | 0 (0.0) | 1 (0.3) | 1 (0.2) | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Lack of efficacy | 8 (4.7) | 4 (1.2) | 12 (2.4) | | ||||| | | ||||| | | ||||| | | ||||| | | |||||| |
COVID-19 infection | 0 (0.0) | 0 (0.0) | 0 (0.0) | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
COVID-19 logistical restrictions | 0 (0.0) | 0 (0.0) | 0 (0.0) | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Other | 1 (0.6) | 3 (0.9) | 4 (0.8) | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
ITT, N | 171 | 324 | 495 | ||| | || | || | ||| | || |
PP, N | 150 | 299 | 449 | || | || | || | || | || |
Safety, N | 171 | 324 | 495 | ||| | || | || | ||| | || |
ITT = intention-to-treat; NA = not applicable; NR = not reported; OL = open label; PBO = placebo; PP = per protocol; UPA = upadacitinib.
Sources: U-EXCEED Clinical Study Report.26 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 12: Summary of Patient Disposition in U-EXCEL
Patient disposition | Part 1 (double-blind) | |||| ||||||||| ||||||||| | ||||
|---|---|---|---|---|---|---|
PBO (N = 176) | UPA 45 mg (N = 350) | Total (n = 526) | ||||||| ||||||| | | ||| || |||||| || | | |||||||| ||||| | |
Randomized, N (%) | 176 (100.0) | 350 (100.0) | 526 (100.0) | || | || | || |
Treated, N (%) | 176 (100.0) | 350 (100.0) | 526 (100.0) | | ||||||| | | ||||||| | ||| ||||||| |
Discontinued study drug N (%) | 21 (11.9) | 22 (6.3) | 43 (8.2) | | |||||| | | |||||| | | |||||| |
Discontinued from study, N (%) | 22 (12.5) | 20 (5.7) | 42 (8.0) | | |||||| | | |||||| | | |||||| |
Adverse events | 8 (4.5) | 12 (3.4) | 20 (3.8) | | ||||| | | ||||| | | ||||| |
Withdrawal by patients | 6 (3.4) | 3 (0.9) | 9 (1.7) | | ||||| | | ||||| | | ||||| |
Loss to follow-up | 0 (0.0) | 1 (0.3) | 1 (0.2) | | ||||| | | ||||| | | ||||| |
Lack of efficacy | 8 (4.5) | 3 (0.9) | 11 (2.1) | | ||||| | | ||||| | | ||||| |
Covid-19 infection | 0 (0.0) | 0 (0.0) | 0 (0.0) | | ||||| | | ||||| | | ||||| |
Covid-19 logistical restrictions | 0 (0.0) | 0 (0.0) | 0 (0.0) | | ||||| | | ||||| | | ||||| |
Other | 0 (0.0) | 1 (0.3) | 1 (0.2) | | ||||| | | ||||| | | ||||| |
ITT, N | 176 | 350 | 526 | || | || | ||| |
PP, N | 142 | 301 | 443 | || | || | || |
Safety, N | 176 | 350 | 526 | || | || | ||| |
ITT = intention-to-treat; NR = not reported; PBO = placebo; PP = per protocol; UPA = upadacitinib.
Sources: U-EXCEL Clinical Study Report.22 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 13: Summary of Patient Disposition in U-ENDURE
Patient disposition | Cohort 1 | |||||| | | |||||| | | |||
|---|---|---|---|---|---|---|
PBO (N = 165) | UPA 15 mg (N = 169) | UPA 30 mg (N = 168) | Total (N = 502) | ||||| ||| | ||| || ||| | |
Randomized, N (%) | 165 (100.0) | 169 (100.0) | 168 (100.0) | 502 (100.0) | ||| ||||||| | | ||||||| |
Treated, N (%) | 165 (100.0) | 169 (100.0) | 168 (100.0) | 502 (100.0) | ||| ||||||| | ||| ||||||| |
Discontinued study drug, N (%) | 43 (26.1) | 44 (26.0) | 34 (20.2) | 121 (24.1) | ||| ||||||| | ||| ||||||| |
Without receiving OL rescue UPA 30 mg q.d. | 18 (10.9) | 23 (13.6) | 20 (11.9) | 61 (12.2) | ||| ||||||| | ||| ||||||| |
Completed study drug, N (%) | 122 (73.9) | 125 (74.0) | 134 (79.8) | 381 (75.9) | ||| ||||||| | ||| ||||||| |
Without receiving OL UPA 30 mg q.d. | 45 (27.3) | 88 (52.1) | 105 (62.5) | 238 (47.4) | ||| ||||||| | ||| ||||||| |
After receiving OL UPA 30 mg q.d. | 77 (46.7) | 37 (21.9) | 29 (17.3) | 143 (28.5) | ||| ||||||| | ||| ||||||| |
Discontinued from substudy 1, N (%) | 43 (26.1) | 43 (25.4) | 34 (20.2) | 120 (23.9) | ||| ||||||| | ||| ||||||| |
Discontinued without receiving OL UPA 30 mg q.d. | 18 (10.9) | 23 (13.6) | 20 (11.9) | 61 (12.2) | ||| ||||||| | ||| ||||||| |
Adverse events | 6 (3.6) | 10 (5.9) | 9 (5.4) | 25 (5.0) | ||| ||||||| | ||| ||||||| |
Withdrawal by patients | 4 (2.4) | 4 (2.4) | 8 (4.8) | 16 (3.2) | ||| ||||||| | ||| ||||||| |
Loss to follow-up | 1 (0.6) | 2 (1.2) | 1 (0.6) | 4 (0.8) | ||| ||||||| | ||| ||||||| |
Lack of efficacy | 6 (3.6) | 5 (3.0) | 1 (0.6) | 12 (2.4) | ||| ||||||| | ||| ||||||| |
COVID-19 infection | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||| ||||||| | ||| ||||||| |
COVID-19 logistical restrictions | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||| ||||||| | ||| ||||||| |
Other | 1 (0.6) | 2 (1.2) | 1 (0.6) | 4 (0.8) | ||| ||||||| | ||| ||||||| |
Discontinued after receiving OL UPA 30 mg q.d. | 25 (15.2) | 20 (11.8) | 14 (8.3) | 59 (11.8) | ||| ||||||| | ||| ||||||| |
Adverse events | 4 (2.4) | 6 (3.6) | 3 (1.8) | 13 (2.6) | ||| ||||||| | ||| ||||||| |
Withdrawal by patients | 1 (0.6) | 3 (1.8) | 2 (1.2) | 6 (1.2) | ||| ||||||| | ||| ||||||| |
Loss to follow-up | 1 (0.6) | 1 (0.6) | 0 (0.0) | 2 (0.4) | ||| ||||||| | ||| ||||||| |
Lack of efficacy | 16 (9.7) | 8 (4.7) | 9 (5.4) | 33 (6.6) | ||| ||||||| | ||| ||||||| |
Covid-19 infection | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||| ||||||| | ||| ||||||| |
Covid-19 logistical restrictions | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||| ||||||| | ||| ||||||| |
Other | 3 (1.8) | 2 (1.2) | 0 (0.0) | 5 (1.0) | ||| ||||||| | ||| ||||||| |
ITT, N | 165 | 169 | 168 | 502 | ||| ||||||| | ||| ||||||| |
PP, N | 156 | 150 | 152 | NR | ||| ||||||| | ||| ||||||| |
Safety, N | 223 | 221 | 229 | 673 | ||| ||||||| | ||| ||||||| |
ITT = intention-to-treat; OL = open-label; PBO = placebo, PP = per protocol, q.d. = once daily; UPA = upadacitinib.
Sources: U-ENDURE Clinical Study Report.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
In all 3 trials, baseline characteristics appeared to be relatively balanced between treatment arms. In each trial, there were slightly more male patients than female patients. The mean age was approximately 37 to 40 years in each trial. The majority of patients (approximately 70% to 75%) were white across all trials.
The median CD duration was 9.4 years in the U-EXCEED trial, 5.7 years in the U-EXCEL trial, and 7.6 years in the U-ENDURE trial. The longer median CD duration in the U-EXCEED trial may be reflective of the inclusion criterion requirement of inadequate response or intolerance to biologic therapy only, in contrast to the U-EXCEL trial, in which patients may have had an inadequate response or intolerance to conventional and/or biologic therapy; conventional therapy tends to occur earlier in the treatment pathway.
In the U-EXCEED trial, patients in part 1 had failed no more than 1 biologic (39.2%), 2 biologics (29.7%), or 3 or more biologics (31.1%). More than 95% of patients had prior failure to anti-TNF agents, and vedolizumab or natalizumab, whereas approximately 36% had prior failure to ustekinumab. In the U-EXCEL trial, patients were divided into subgroups based on biologic exposure: bio-IR patients had prior failure or intolerance to biologics, while non–bio-IR patients did not, but may not have necessarily been biologic-naive. Among the non–bio-IR patients in part 1 of the U-EXCEL trial, 8.7% had prior exposure to biologic therapy. Among bio-IR patients in part 1, the distribution of the number of biologics was roughly similar to that in the U-EXCEED trial (36.0%, 31.8%, and 32.2%, respectively). As the U-ENDURE cohorts comprised patients from the U-EXCEED and U-EXCEL trial, both bio-IR patients (75.6% in cohort 1) and non–bio-IR patients (24.4% in cohort 1) were included in the study, with similar proportions of patients having experienced 1, 2, or at least 3 prior biologic failures as in the parent studies.
The median baseline CDAI was 299.80 in part 1 of the U-EXCEED trial, 285.00 in part 1 of the U-EXCEL trial, and 300.50 in cohort 1 of the U-ENDURE trial, all of which fall into the “moderate to severe” category of CD activity. The median baseline SES-CD in part 1 of the U-EXCEED trial was 13.0, in part 1 of the U-EXCEL trial it was 12.0, and in cohort 1 of U-ENDURE trial it was 14.0, suggesting that patients in the U-ENDURE had more severe disease in terms of endoscopic outcomes, and patients in the U-EXCEL had less severe disease than the other trials. As the total score ranged from 0 to 56 and there was no reported MID for SES-CD, it is unknown whether these differences are substantial.
The studies were similar or different in the CD location as measured by SES-CD. The majority of patients in part 1 or cohort 1 of each trial (approximately 49% to 50%) had ileal-colonic CD, followed by colonic-only CD (approximately 35% to 41%), and finally ileal-only CD (approximately 12% to 17%). The U-EXCEL trial had the highest proportion of ileal-only CD out of the 3 included trials. The average daily frequency of very soft or liquid stools ranged from 5.5 to 5.9, and the average daily AP score was approximately 1.9 in all 3 trials. Baseline corticosteroid use ranged from approximately 36% to 38%. Baseline immunosuppressant use was less common (3.7% to 7.5%).
Exposure to Study Treatments
| | |||||||| |||||| | |||| |||||||| || ||||||||| |||||||| |||||| |||| |||| |||| || |||| |||| ||||||||| || | |||| || ||||||||||||| |||||| |||| || ||||| || |||||||| |||||||| || || || |||||||||||| | || |||||| |||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| |||||||||| ||||||||
Table 14: Summary of Baseline Characteristics of Patients in U-EXCEED
Characteristic | Part 1 (double-blind) | |||| ||| | |||| | |||||||| ||||||||| | ||| ||||||| | ||||
|---|---|---|---|---|---|---|---|---|
PBO (N = 171) | UPA 45 mg (N = 324) | Total (N = 495) | ||| ||||||| | ||||||| || | ||| || || ||| | |||||||| ||| | || || |||| | | |
Female, N (%) | 75 (43.9) | 155 (47.8) | 230 (46.5) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Male, N (%) | 96 (56.1) | 169 (52.2) | 265 (53.5) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Age, N (%) | ||||||||
Mean, years (SD) | 37.5 (12.12) | 38.4 (13.71) | 38.1 (13.18) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
18 to < 40 years | 96 (56.1) | 187 (57.7) | 283 (57.2) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
40 to < 65 years | 71 (41.5) | 122 (37.7) | 193 (39.0) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
≥ 65 years | 4 (2.3) | 15 (4.6) | 19 (3.8) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Race, N (%) | ||||||||
White | 126 (73.7) | 230 (71.0) | 356 (71.9) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Black or African American | 6 (3.5) | 19 (5.9) | 25 (5.1) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Asian | 38 (22.2) | 69 (21.3) | 107 (21.6) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
American Indian or Alaska Native | 1 (0.6) | 1 (0.3) | 2 (0.4) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Multiple | 0 | 5 (1.5) | 5 (1.0) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Body mass index (kg/m2), N (%) | ||||||||
Mean (SD) | 23.90 (6.19) | 24.16 (5.98) | 24.07 (6.04) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
< 18.5 | 34 (19.9) | 48 (14.8) | 82 (16.6) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
≥ 18.5 to < 25 | 81 (47.4) | 160 (49.4) | 241 (48.7) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
≥ 25 to < 30 | 28 (16.4) | 68 (21.0) | 96 (19.4) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
≥ 30 | 28 (16.4) | 48 (14.8) | 76 (15.4) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
CD duration (years) | ||||||||
Median (minimum to maximum) | 9.8261 (0.6352 to 46.0726) | 9.2512 (0.4901 to 55.1677) | 9.3908 (0.4901 to 55.1677) | |||||||||||||| | |||||||||||||||| | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Biological failure history, N (%) | ||||||||
≤ 1 | 68 (39.8) | 126 (38.9) | 194 (39.2) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
2 | 55 (32.2) | 92 (28.4) | 147 (29.7) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
≥ 3 | 48 (28.1) | 106 (32.7) | 154 (31.1) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Prior failure to anti-TNF drug, N (%) | ||||||||
Yes | 164 (95.9) | 308 (95.1) | 472 (95.4) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Prior vedolizumab or natalizumab failure, N (%) | ||||||||
Yes | 47 (27.5) | 99 (30.6) | 146 (29.5) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Prior ustekinumab failure, N (%) | ||||||||
Yes | 57 (33.3) | 118 (36.4) | 175 (35.4) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Baseline CDAI | ||||||||
Median (minimum to maximum) | 303.00 (112.0 to 545.0) | 298.80 (102.0 to 627.0)a | 299.80 (102.0 to 627.0)b | ||||||||||||| |||||| | |||||||||||||| |||||| | |||||||||||||| |||||| | |||||||||||||| |||||| | ||||||||||||| |||||| |
Baseline SES-CD | ||||||||
Median (minimum to maximum) | 13.0 (4 to 41) | 13.0 (4 to 40) | 13.0 (4 to 41) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
CD location per SES-CD, N (%) | ||||||||
Ileal only | 23 (13.5) | 48 (14.8) | 71 (14.3) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Colonic only | 68 (39.8) | 112 (34.6) | 180 (36.4) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Ileal-colonic | 80 (46.8) | 164 (50.6) | 224 (49.3) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Average daily very soft or liquid stool frequency | ||||||||
Mean (SD) | 6.0929 (3.3355) | 5.7299 (3.3603)c | 5.8555 (3.3528)d | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Average daily abdominal pain score | ||||||||
Mean (SD) | 1.7955 (0.6849) | 1.8508 (0.6913)c | 1.8317 (0.6889)d | |||||| |||||||| | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Baseline corticosteroid use (yes or no), N (%) | ||||||||
Yes | 60 (35.1) | 108 (33.3) | 168 (33.9) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Baseline immunosuppressant use (yes or no), N (%) | ||||||||
Yes | 13 (7.6) | 24 (7.4) | 37 (7.5) | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
CD = Crohn disease; CDAI = Crohn Disease Activity Index; OL = open label; PBO = placebo; SD = standard deviation; SES-CD = Simplified Endoscopic Score of Crohn Disease; TNF = tumour necrosis factor; UPA = upadacitinib.
an = 322.
bn = 493.
cn = 323.
dn = 494.
Sources: Sponsor’s Summary of Clinical Evidence.
Table 15: Summary of Baseline Characteristics of Patients in U-EXCEL
Characteristic | Part 1 (double-blind) | |||| ||||||| ||||||||| | |||
|---|---|---|---|---|---|
Placebo (N = 176) | UPA 45 mg (N = 350) | Total (N = 526) | ||||||| |||| ||| | ||| |||||||| |||| | |
Female, N (%) | 82 (46.6) | 161 (46.0) | 243 (46.2) | ||| ||||||| | ||| ||||||| |
Male, N (%) | 94 (53.4) | 189 (54.0) | 283 (53.8) | ||| ||||||| | ||| ||||||| |
Age, N (%) | |||||
Mean, years (SD) | 39.3 (13.63) | 39.7 (13.71) | 39.6 (13.67) | ||| ||||||| | ||| ||||||| |
18 to < 40 years | 91 (51.7) | 193 (55.1) | 284 (54.0) | ||| ||||||| | ||| ||||||| |
40 to < 65 years | 80 (45.5) | 142 (40.6) | 222 (42.2) | ||| ||||||| | ||| ||||||| |
≥ 65 years | 5 (2.8) | 15 (4.3) | 20 (3.8) | ||| ||||||| | ||| ||||||| |
Race, N (%) | |||||
White | 130 (73.9) | 258 (73.7) | 388 (73.8) | ||| ||||||| | ||| ||||||| |
Black or African American | 4 (2.3) | 17 (4.9) | 21 (4.0) | ||| ||||||| | ||| ||||||| |
Asian | 36 (20.5) | 73 (20.9) | 109 (20.7) | ||| ||||||| | ||| ||||||| |
Multiple | 6 (3.4) | 2 (0.6) | 8 (1.5) | ||| ||||||| | ||| ||||||| |
Body mass index (kg/m2), N (%) | |||||
Mean (SD) | 25.61 (6.97) | 24.47 (5.96) | 24.85 (6.3) | ||| ||||||| | ||| ||||||| |
< 18.5 | 22 (12.5) | 53 (15.1) | 75 (14.3) | ||| ||||||| | ||| ||||||| |
≥ 18.5 to < 25 | 69 (39.2) | 158 (45.1) | 227 (43.2) | ||| ||||||| | ||| ||||||| |
≥ 25 to < 30 | 53 (30.1) | 76 (21.7) | 129 (24.5) | ||| ||||||| | ||| ||||||| |
≥ 30 | 32 (18.2) | 63 (18.0) | 95 (18.1) | ||| ||||||| | ||| ||||||| |
CD duration (years) | |||||
Median (minimum to maximum) | 5.6810 (0.2765 to 46.2752) | 6.6516 (0.0575 to 52.1123) | 6.0794 (0.0575 to 52.1123) | ||||||||||||||| || | ||||||||||||||| | |
Biological use or failure history, N (%) | |||||
Bio-IR | 78 (44.3) | 161 (46.0) | 239 (45.4) | ||| ||||||| | ||| ||||||| |
Non–bio-IR | 98 (55.7) | 189 (54.0) | 287 (54.6) | ||| ||||||| | ||| ||||||| |
Prior exposure to biologic therapy among non–bio-IR patients, N (%) | |||||
N (non–bio-IR) | 98 | 189 | 287 | ||| ||||||| | ||| ||||||| |
Yes | 9 (9.2) | 16 (8.5) | 25 (8.7) | ||| ||||||| | ||| ||||||| |
Biological failure history, N (%) | |||||
N (bio-IR) | 78 | 161 | 239 | ||| ||||||| | ||| ||||||| |
1 | 28 (35.9) | 58 (36.0) | 86 (36.0) | ||| ||||||| | ||| ||||||| |
2 | 24 (30.8) | 52 (32.3) | 76 (31.8) | ||| ||||||| | ||| ||||||| |
≥ 3 | 26 (33.3) | 51 (31.7) | 77 (32.2) | ||| ||||||| | ||| ||||||| |
Prior failure to anti-TNF drug, N (%) | |||||
N (bio-IR) | 78 | 161 | 239 | ||| ||||||| | ||| ||||||| |
Yes | 75 (96.2) | 157 (97.5) | 232 (97.1) | ||| ||||||| | ||| ||||||| |
Prior vedolizumab or natalizumab failure, N (%) | |||||
N (bio-IR) | 78 | 161 | 239 | ||| ||||||| | ||| ||||||| |
Yes | 25 (32.1) | 49 (30.4) | 74 (31.0) | ||| ||||||| | ||| ||||||| |
Prior ustekinumab failure, N (%) | |||||
N (bio-IR) | 78 | 161 | 239 | ||| ||||||| | ||| ||||||| |
Yes | 33 (42.3) | 64 (39.8) | 97 (40.6) | ||| ||||||| | ||| ||||||| |
Baseline CDAI | |||||
Median (minimum to maximum) | 290.50 (89.5 to 530.0) | 284.00 (62.0 to 543.8) | 285.00 (62.0 to 543.8) | |||||| |||||| |||||| | |||||| |||||| |||||| |
Baseline SES-CD | |||||
Median (minimum to maximum) | 12.0 (4 to 35) | 12.0 (4 to 38) | 12.0 (4 to 38) | ||| ||||||| | ||| ||||||| |
CD location per SES-CD, N (%) | |||||
Ileal only | 27 (15.3) | 58 (16.6) | 85 (16.2) | ||| ||||||| | ||| ||||||| |
Colonic only | 57 (32.4) | 121 (34.6) | 178 (33.8) | ||| ||||||| | ||| ||||||| |
Ileal-colonic | 92 (52.3) | 171 (48.9) | 263 (50.0) | ||| ||||||| | ||| ||||||| |
Average daily very soft or liquid stool frequency | |||||
Mean (SD) | 5.0857 (2.8366) | 5.1864 (2.6130) | 5.1527 (2.6876) | |||||| |||||||| | |||||| |||||||| |
Average daily abdominal pain score | |||||
Mean (SD) | 1.9064 (0.6942) | 1.8917 (0.6795) | 1.8966 (0.6839) | |||||| |||||||| | |||||| |||||||| |
Baseline corticosteroid use (yes, no), N (%) | |||||
Yes | 64 (36.4) | 126 (36.0) | 190 (36.1) | ||| ||||||| | ||| ||||||| |
Baseline immunosuppressant use (yes, no), N (%) | |||||
Yes | 3 (1.7) | 13 (3.7) | 16 (3.0) | | ||||| | | ||||| |
bio-IR = biologic therapy–intolerant or inadequate responder; CD = Crohn disease; CDAI = Crohn Disease Activity Index; OL = open label; SD = standard deviation; SES-CD = Simplified Endoscopic Score of Crohn Disease; TNF = tumour necrosis factor; UPA = upadacitinib.
an = 349.
bn = 525.
Sources: Sponsor’s Summary of Clinical Evidence.
Concomitant Medications and Cointerventions
The list of concomitant medications shown in the tables in the following section were deemed appropriate by clinical data repository experts. Treatments may be recommended practice, rather than actual practice. ||||| || || |||||||| ||||||||||| ||||||||||| || || |||||||||||||| || |||| |||||| |||||| |||| |||| |||| |||||| || |||||||||| |||||||| || |||||||| | |||| |||||| ||||||||||| |||||||||| |||||||||| || ||||||||||| | |||| |||||| |||||||||| |||||||||| || |||| | |||||||||||| ||||||||||| |||||||||| || || |||||||| ||||| |||||| || |||||| |||| ||||| || |||||| |||
In the U-EXCEED and U-EXCEL trials, all patients were to undergo mandatory tapering beginning at week 4, with the goal of discontinuing corticosteroid treatment by week 11 (i.e., before the end of the induction period). In the U-ENDURE trial, at week 0, all patients who were taking corticosteroids at baseline in the U-EXCEED or U-EXCEL trials and had not completed the taper were to continue the mandatory tapering beginning at week 4, with the goal of discontinuing corticosteroid treatment by week 11.
Table 16: Summary of Baseline Characteristics of Patients in U-ENDURE
Characteristic | Cohort 1 | |||||| | | |||||| | | |||
|---|---|---|---|---|---|---|
Placebo (N = 165) | UPA 15 mg (N = 169) | UPA 30 mg (N = 168) | Total (N = 502) | |||||| |||| | ||| || ||||| || | |
Female, N (%) | 77 (46.7) | 67 (39.6) | 75 (44.6) | 219 (43.6) | ||| ||||||| | ||| ||||||| |
Male, N (%) | 88 (53.3) | 102 (60.4) | 93 (55.4) | 283 (56.4) | ||| ||||||| | ||| ||||||| |
Age, N (%) | ||||||
Mean, years (SD) | 38.1 (13.03) | 38.1 (13.46) | 37.0 (13.27) | 37.7 (13.24) | ||| ||||||| | ||| ||||||| |
18 to < 40 years | 97 (58.8) | 102 (60.4) | 101 (60.1) | 300 (59.8) | ||| ||||||| | ||| ||||||| |
40 to < 65 years | 62 (37.6) | 62 (36.7) | 60 (35.7) | 184 (36.7) | ||| ||||||| | ||| ||||||| |
≥ 65 years | 6 (3.6) | 5 (3.0) | 7 (4.2) | 18 (3.6) | ||| ||||||| | ||| ||||||| |
Race, N (%) | ||||||
White | 119 (72.1) | 118 (69.8) | 114 (67.9) | 351 (69.9) | ||| ||||||| | ||| ||||||| |
Black or African American | 11 (6.7) | 6 (3.6) | 7 (4.2) | 24 (4.8) | ||| ||||||| | ||| ||||||| |
Asian | 35 (21.2) | 43 (25.4) | 45 (26.8) | 123 (24.5) | ||| ||||||| | ||| ||||||| |
American Indian or Alaska Native | 0 | 0 | 0 | 0 | ||| ||||||| | ||| ||||||| |
Multiple | 0 | 2 (1.2) | 2 (1.2) | 4 (0.8) | ||| ||||||| | ||| ||||||| |
Body mass index (kg/m2), N (%) | ||||||
Mean (SD) | 24.64 (6.65) | 24.10 (6.035) | 24.17 (6.56) | 24.30 (6.40) | ||| ||||||| | ||| ||||||| |
< 18.5 | 26 (15.8) | 32 (18.9) | 28 (16.7) | 86 (17.1) | ||| ||||||| | ||| ||||||| |
≥ 18.5 to < 25 | 76 (46.1) | 74 (43.8) | 85 (50.6) | 235 (46.8) | ||| ||||||| | ||| ||||||| |
≥ 25 to < 30 | 31 (18.8) | 26 (15.4) | 24 (14.3) | 81 (16.1) | ||| ||||||| | ||| ||||||| |
≥ 30 | 32 (19.4) | 37 (21.9) | 31 (18.5) | 100 (19.9) | ||| ||||||| | ||| ||||||| |
CD duration (years) | ||||||
Median (minimum to maximum) | 7.6030 (0.3258 to 48.7283) | 7.8713 (0.2765 to 40.0767) | 7.2197 (0.3422 to 44.9281) | 7.4606 (0.2765 to 48.7283) | ||||||||||||| | |||||||||||||| |
Biological failure history, N (%) | ||||||
Bio-IR | 126 (76.4) | 124 (73.4) | 127 (75.6) | 377 (75.1) | ||| ||||||| | ||| ||||||| |
Non–bio-IR | 39 (23.6) | 45 (26.6) | 41 (24.4) | 125 (24.9) | ||| ||||||| | ||| ||||||| |
Biological failure history, among bio-IR patients, N (%) | ||||||
N (bio-IR) | 126 | 124 | 127 | 377 | ||| ||||||| | ||| ||||||| |
1 | 52 (41.3) | 52 (41.9) | 43 (33.9) | 147 (39.0) | ||| ||||||| | ||| ||||||| |
2 | 32 (25.4) | 31 (25.0) | 35 (27.6) | 98 (26.0) | ||| ||||||| | ||| ||||||| |
≥ 3 | 42 (33.3) | 41 (33.1) | 49 (38.6) | 132 (35.0) | ||| ||||||| | ||| ||||||| |
Prior failure to anti-TNF drug among bio-IR patients, N (%) | ||||||
N (bio-IR) | 126 | 124 | 127 | 377 | ||| ||||||| | ||| ||||||| |
Yes | 118 (93.7) | 117 (94.4) | 123 (96.9) | 358 (95.0) | ||| ||||||| | ||| ||||||| |
Prior vedolizumab or natalizumab failure among bio-IR patients, N (%) | ||||||
N (bio-IR) | 126 | 124 | 127 | 377 | ||| ||||||| | ||| ||||||| |
Yes | 38 (30.2) | 39 (31.5) | 43 (33.9) | 120 (31.8) | ||| ||||||| | ||| ||||||| |
Prior ustekinumab failure among bio-IR patients, N (%) | ||||||
N (bio-IR) | 126 | 124 | 127 | 377 | ||| ||||||| | ||| ||||||| |
Yes | 48 (38.1) | 41 (33.1) | 49 (38.6) | 138 (36.6) | ||| ||||||| | ||| ||||||| |
Baseline CDAI | ||||||
N | 164 | 168 | 168 | 500 | ||| ||||||| | ||| ||||||| |
Median (mininum to maximum) | 305.90 (114.4 to 509.0) | 283.50 (102.0 to 657.0) | 300.50 (153.8 to 543.8) | 299.50 (102.0 to 657.0) | ||||||||||||| | |||||||||||||| |
Average daily very soft or liquid stool frequency | ||||||
N | 165 | 168 | 168 | 501 | ||| | || |
Mean (SD) | 5.6003 (2.8025) | 5.3755 (3.2652) | 5.5355 (2.7927) | 5.5032 (2.9582) | |||||| |||||||| | |||||| |||||||| |
Average daily abdominal pain score | ||||||
N | 165 | 168 | 168 | 501 | ||| | || |
Mean (SD) | 1.9492 (0.6586) | 1.8485 (0.7005) | 1.9419 (0.6026) | 1.9130 (0.6554) | |||||| |||||||| | |||||| |||||||| |
Baseline SES-CD | ||||||
N | 165 | 169 | 168 | 502 | ||| | || |
Median (minimum to maximum) | 13.0 (4 to 35) | 14.0 (4 to 40) | 14.0 (4 to 41) | 14.0 (4 to 41) | ||| ||||||| | ||| ||||||| |
CD location per SES-CD, N (%) | ||||||
Ileal only | 24 (14.5) | 22 (13.0) | 20 (11.9) | 66 (13.1) | ||| ||||||| | ||| ||||||| |
Colonic only | 67 (40.6) | 62 (36.7) | 70 (41.7) | 199 (39.6) | ||| ||||||| | ||| ||||||| |
Ileal-colonic | 74 (44.8) | 85 (50.3) | 78 (46.4) | 237 (47.2) | ||| ||||||| | ||| ||||||| |
Baseline corticosteroid use (yes or no), N (%) | ||||||
Yes | 61 (37.0) | 63 (37.3) | 63 (37.5) | 187 (37.3) | ||| ||||||| | ||| ||||||| |
Baseline immunosuppressant use (yes, no), N (%) | ||||||
Yes | 11 (6.7) | 5 (3.0) | 9 (5.4) | 25 (5.0) | | ||||| | | ||||| |
bio-IR = biologic therapy–intolerant or inadequate responder; CD = Crohn disease; CDAI = Crohn Disease Activity Index; SD = standard deviation; SES-CD = Simplified Endoscopic Score of Crohn Disease; TNF = tumour necrosis factor, UPA = upadacitinib.
Sources: Sponsor’s Summary of Clinical Evidence.
Table 17: Summary of Patient Exposure in U-EXCEED [Redacted]
|||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | ||
|---|---|---|---|---|---|---|
|||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |
||||| ||||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
||||||||| |||| |||| || |||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
||||||||| |||||| ||||| || | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
||||||||| || || |||||| | | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||||||| || |||||||||||||| || |||||||||||| || ||| |||||| || ||||||||| || |||||| || |||||||| |||||||||| || |||||||||||||||||||||| ||||||| |||||||| || || ||||| | |||| ||
Note: This table has been redacted at the request of the sponsor.
Table 18: Summary of Patient Exposure in U-EXCEL [Redacted]
|||||||| | |||| ||| | |||| ||| | ||
|---|---|---|---|---|
|||| ||| | |||| ||| | |||| ||| | ||| || || ||||| ||| | |
|||||| ||||||||||||| | |||| ||| | |||| ||| | |||| ||| | || |
||||||||| |||| |||| || |||| | |||| ||| | |||| ||| | |||| ||| | |||| ||||||| |
||||||||| |||||| ||||| |||| || |||| | |||| ||| | |||| ||| | |||| ||| | |||| | ||| |
||||||||| || || |||||| | | |||| ||| | |||| ||| | |||| ||| | |||| |
|||||||||||||| || |||||||||||||| || |||||||||||| || ||| |||||| || ||||||||| || |||||| || |||||||| |||||||||| || |||||||||||||||||||||| ||||||| |||||||| || || ||||| | |||| ||
Note: This table has been redacted at the request of the sponsor.
Table 19: Summary of Patient Exposure in U-ENDURE [Redacted]
|||||||| | |||| ||| | |||| ||| | |||| ||| | |||
|---|---|---|---|---|---|---|
|||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | ||| || || |||||||| | |
|||||| ||||||||||||| || ||||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | || |
||||||||| |||| |||| || |||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
||||||||| |||||| ||||| |||| || |||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
||||||||| || || |||||| | | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||||||| || |||||||||||||| || |||||||||||| || ||| |||||| || ||||||||| || |||||| || |||||||| |||||||||| || |||||||||||||||||||||| ||||||| |||||||| || || ||||| | |||| ||
Note: This table has been redacted at the request of the sponsor.
Table 20: Summary of Concomitant Medication in U-EXCEED [Redacted]
|||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | ||||
|---|---|---|---|---|---|---|---|---|
|||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |
|||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
||||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
||||||||||||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
|||||||||||||| || |||||||||||||| || |||||||||||| || ||| |||||| || ||||||||| || |||||| || |||||||| |||||||||| || |||||||||||||||||||||| ||||||| |||||||| || || ||||| | |||| || ||||||||| ||||||| || |||||||| |||||||||
Note: This table has been redacted at the request of the sponsor.
Table 21: Summary of Concomitant Medication in U-EXCEL
|||||||| | |||||| |||| | |||||| |||| | ||||
|---|---|---|---|---|---|---|
|||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |
|||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||||| || |||||||||||||| || |||||||||||| || ||| |||||| || ||||||||| || |||||| || |||||||| |||||||||| || |||||||||||||||||||||| ||||||| |||||||| || || ||||| | |||| || ||||||||| ||||||| || |||||||| |||||||||
Note: This table has been redacted at the request of the sponsor.
Table 22: Summary of Concomitant Medication in U-ENDURE [Redacted]
|||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||
|---|---|---|---|---|---|---|
|||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |
|||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
|||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
||||||||||||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
||||||||||||| || |||||||||||||| || |||||||||||| || ||| |||||| || ||||||||| || |||||| || |||||||| |||||||||| || |||||||||||||||||||||| ||||||| |||||||| || || ||||| | |||| || ||||||||| ||||||| || |||||||| |||||||||
Note: This table has been redacted at the request of the sponsor.
Efficacy
Induction Outcomes (12 Weeks)
Subgroup results for the primary outcomes (clinical remission by PROs, clinical remission by CDAI, and endoscopic response) by number of prior biologics failed are reported in Appendix 1 (Table 36 for the U-EXCEED trial and Table 37 for the U-EXCEL trial). Subgroups in the U-EXCEED trial included 1 or fewer biologics failed, and more than 1 biologic failed. Subgroups in the U-EXCEL trial included 0 prior biologics failed, 1 or more prior biologic failed, and exactly 1 prior biologic failed.
Clinical Remission as Measured by PROs at 12 Weeks
In the ITT1 population of the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved clinical remission as measured by PROs among those treated with upadacitinib 45 mg compared with placebo. In the U-EXCEED trial, the response-rate difference compared to placebo was 25.9% (95% CI, 18.7% to 33.1%), and in the U-EXCEL it was 28.7% (95% CI, 20.9% to 36.4%).
Results were consistent across subgroups based on number of prior biologics failed and the ITT1 analysis for clinical remission as measured by PROs at 12 weeks.
Clinical Remission as Measured by CDAI at 12 Weeks
In the ITT1 population of the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved clinical remission as measured by CDAI among patients treated with upadacitinib 45 mg compared with placebo. In the U-EXCEED trial, the response-rate difference compared to placebo was 17.9% (95% CI, 10.0% to 25.8%) and in the U-EXCEL it was 20.8% (95% CI, 12.7% to 28.8%).
Results were consistent across subgroups based on number of prior biologics failed and the ITT1 analysis for clinical remission as measured by CDAI at 12 weeks.
Endoscopic Response at 12 Weeks
In the ITT1 population of the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved an endoscopic response among patients treated with upadacitinib 45 mg compared to placebo. In the U-EXCEED trial, the response-rate difference compared to placebo was 31.2% (95% CI, 25.5% to 37.0%) and in the U-EXCEL it was 33.0% (95% CI, 26.2% to 39.9%).
Results were consistent across subgroups based on number of prior biologics failed and the ITT1 analysis for endoscopic response at 12 weeks.
Endoscopic Remission at 12 Weeks
In the ITT1 population of the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved endoscopic remission among patients treated with upadacitinib 45 mg compared with placebo. In the U-EXCEED trial, the rate difference compared to placebo was 16.8% (95% CI, 12.0% to 21.6%) and in the U-EXCEL it was 21.8% (95% CI, 15.8% to 27.8%).
Discontinuation of Corticosteroid Use and Clinical Remission as Measured by CDAI at 12 Weeks Among Patients Taking Corticosteroids at Baseline
In the ITT1 population of the U-EXCEED and U-EXCEL trials, a higher proportion of patients treated with upadacitinib discontinued corticosteroid use and had CDAI clinical remission at week 12 compared to the placebo group. In the U-EXCEED trial, the rate difference compared to placebo was 22.5% (95% CI, 11.1% to 34.0%) and in the U-EXCEL it was 27.7% (95% CI, 15.7% to 39.8%).
Results were similar for discontinuation of corticosteroid use and clinical remission as measured by PROs at 12 weeks.
Clinical Remission as Measured by CDAI and Endoscopic Remission at 12 Weeks
||||| || || |||||||| ||||||||||| ||||||||||| || || |||||||||||||| || |||| |||||| |||||| |||| |||| |||| |||||| || |||||||||| |||||||| || |||||||| | |||| |||||| ||||||||||| |||||||||| |||||||||| || ||||||||||| | |||| |||||| |||||||||| |||||||||| || |||| | |||||||||||| ||||||||||| |||||||||| || || |||||||| ||||| |||||| || |||||| |||| ||||| || |||||| |||||||| || || |||||||| ||||||||||| ||||||||||| || || |||||||||||||| || |||| |||||| |||||| |||| |||| |||| |||||| || |||||||||| |||||||| || |||||||| | |||| |||||| ||||||||||| |||||||||| |||||||||| || ||||||||||| | |||| |||||| |||||||||| |||||||||| |
Change From Baseline in IBDQ Total Score at 12 Weeks
In the ITT1 population of U-EXCEED and U-EXCEL, there was a larger within-group change from baseline in IBDQ total score in patients treated with upadacitinib compared with those who received placebo. The between-group difference compared to placebo (least squares mean) was 24.3 (95% CI, 17.2 to 31.5) in the U-EXCEED trial and 21.8 (95% CI, 15.6 to 28.1) in the U-EXCEL trial.
CR-100 at 12 Weeks
In the ITT1 population of the U-EXCEED and U-EXCEL trials, a higher percentage of patients achieved CR-100 among patients treated with upadacitinib 45 mg compared with those who received placebo. In the U-EXCEED trial, the difference compared to placebo was 22.8% (95% CI, 14.4% to 31.2%), and in the U-EXCEL it was 19.8% (95% CI, 11.3% to 28.4%).
Resolution of EIMs at 12 Weeks in Patients With EIMs at Baseline
In the U-EXCEED trial, resolution of EIMs at week 12 in patients with any EIMs at baseline was 32.8% for upadacitinib 45 mg versus 21.7% for placebo (difference = 11.5%; 95% CI, −1.5% to 24.4%). In the U-EXCEL trial, resolution of EIMs at week 12 in patients with any EIMs at baseline was 28.5% for upadacitinib 45 mg versus 20.9% for placebo (difference = 9.0%; 95% CI, −1.9% to 19.9%). In both cases, the 95% CI crossed the threshold between potential benefit and potential harm (i.e., null).
Proportion With CD-Related Hospitalizations Through 12 Weeks
No substantial differences were observed in the proportion of patients with CD-related hospitalizations between the upadacitinib-treated groups of the U-EXCEED trial (20 of 324) and the U-EXCEL trial (13 of 350) compared to the placebo-treated groups (15 of 171 and 9 of 176, respectively). The differences compared to placebo were −2.6% (95% CI, −7.6% to 2.4%) in the U-EXCEED trial and −1.4% (95% CI, −5.2% to 2.4%) in the U-EXCEL trial. In both cases, the 95% CI crossed the threshold between potential benefit and potential harm (i.e., null).
Proportion With CD-Related Surgeries Through 12 Weeks
||||| || || |||||||| ||||||||||| ||||||||||| || || |||||||||||||| || |||| |||||| |||||| |||| |||| |||| |||||| || |||||||||| |||||||| || |||||||| | |||| |||||| ||||||||||| |||||||||| |||||||||| || ||||||||||| | |||| |||||| |||||||||| |||||||||| || |||| | |||||||||||| ||||||||||| |||||||||| || || |||||||| ||||| |||||| || |||||| |||| ||||| || |||||| |||
Maintenance Outcomes (52 Weeks)
Subgroup results for the primary outcomes (clinical remission by PROs, clinical remission by CDAI, and endoscopic response) by number of prior biologics failed are reported in Appendix 1 (Table 38). Subgroups in the U-ENDURE included 0 prior biologics failed, at least 1 prior biologic failed, exactly 1 prior biologic failed, and greater than 1 biologic failed.
Clinical Remission as Measured by PROs at 52 Weeks
In the ITT1 population of the U-ENDURE trial, the upadacitinib 15 mg and 30 mg groups had higher percentages of patients who achieved response rates in clinical remission as measured by PROs compared to the placebo group. The differences compared to placebo were 21.9% (95% CI, 13.7% to 30.0%) and 31.8% (95% CI, 23.2% to 40.3%), respectively.
The results were similar for the subgroups of 1 or more prior biologic failed, 1 prior biologic failed, and more than 1 prior biologic failed, as well as 0 prior biologics failed in the 30 mg group. However, for the subgroup of 0 prior biologics failed in the 15 mg group, the difference (versus placebo) was smaller, and the 95% CI crossed the null value (11.7%; 95% CI, −9.1% to 32.5%).
Clinical Remission as Measured by CDAI at 52 Week
In the ITT1 population of the U-ENDURE trial, the upadacitinib 15 mg group and 30 mg group had higher percentages of patients who achieved clinical remission according to CDAI compared to the placebo group. The differences compared to placebo were 23.7% (95% CI, 15.2% to 32.1%) and 32.8% (95% CI, 23.9% to 41.6%), respectively.
Results in the subgroups based on number of prior biologics failed were consistent with the ITT1 analysis for both dosage groups.
Endoscopic Response at 52 Weeks
In the ITT1 population of the U-ENDURE trial, the upadacitinib 15 mg group and 30 mg group had higher percentages of patients who achieved an endoscopic response compared to the placebo group. The differences compared to placebo were 21.0% (95% CI, 13.6% to 28.4%) and 33.7% (95% CI, 26.0% to 41.3%), respectively.
Results in the subgroups based on the number of prior biologics failed were consistent with the ITT1 analysis for both dosage groups.
Endoscopic Remission at 52 Weeks
In the ITT1 population of the U-ENDURE trial, the upadacitinib 15 mg group and 30 mg group had higher percentages of patients who achieved endoscopic remission compared to the placebo group. The differences compared to placebo were 14.4% (95% CI, 7.7% to 21.0%) and 23.6% (16.1% to 31.0%), respectively.
Discontinuation of Corticosteroid Use at Least 90 Days Prior to Week 52 and Clinical Remission as Measured by CDAI at 52 Weeks Among Patients Taking Corticosteroids for CD at Induction Baseline
In the ITT1 population of the U-ENDURE trial among patients taking corticosteroids for CD at induction baseline, a higher proportion of patients in the upadacitinib 15 mg and 30 mg groups discontinued corticosteroid use and had CDAI clinical remission at week 52 compared to the placebo group. The differences compared to placebo were 35.4% (95% CI, 23.3% to 47.5%) and 32.3% (95% CI, 20.1% to 44.5%), respectively. Among all patients (i.e., not limited to those taking corticosteroids at induction baseline), the differences compared to placebo were 23.8% (95%: 15.5% to 32.1%) and 32.2% (95% CI, 23.4% to 40.9%), respectively.
Clinical Remission as Measured by CDAI and Endoscopic Remission at 52 Weeks
In the ITT1 population of the U-ENDURE trial, a higher proportion of patients in the upadacitinib 15 mg group and 30 mg group had both CDAI clinical remission and endoscopic remission at week 52 compared to the placebo group. The differences compared to placebo were 12.2% (95% CI 6.3% to 18.1%) and 19.8% (95% CI 13.0% to 26.6%), respectively.
Change From Baseline in IBDQ Total Score at 52 Weeks
In the ITT1 population of the U-ENDURE trial, there was a larger within-group change from baseline in IBDQ total score in the patients treated with upadacitinib (15 mg or 30 mg) compared with those receiving placebo. The between-group differences (least squares mean) compared to placebo were 12.9 (95% CI, 4.3 to 21.4) and 18.1 (95% CI, 9.8, 26.4), respectively. Only the between-group difference in the latter comparison (i.e., 30 mg upadacitinib versus placebo) was greater than the reported MID of 16 points in the IBDQ total score for patients with CD; the 95% CIs of both comparisons included values both greater than and less than this MID.
CR-100 at 52 Weeks
In the ITT1 population of the U-ENDURE trial, a higher percentage of patients achieved CR-100 among patients treated with upadacitinib 30 mg or 15 mg compared with placebo. The differences between the upadacitinib 15 mg and placebo groups, and between the upadacitinib 30 mg and placebo groups were 27.1% (95% CI, 18.3% to 35.8%), and 36.4% (95% CI, 27.5% to 45.2%), respectively.
Resolution of EIMs at 52 Weeks in Patients With EIMs at Induction Baseline
In the U-ENDURE trial, the proportions of patients who achieved resolution of EIMs at week 52 with any EIMs at induction baseline were 24.6% (upadacitinib 15 mg), 35.6% (upadacitinib 30 mg), and 15.2% (placebo). The differences versus placebo were 9.6% (95% CI, −3.4% to 22.6%) for upadacitinib 15 mg and 22.0% (95% CI, 9.3% to 34.8%) for upadacitinib 30 mg. For the 15 mg dose but not the 30 mg dose, the 95% CI crossed the threshold between potential benefit and potential harm (i.e., null).
Proportion With CD-Related Hospitalizations Through 52 Weeks
In the ITT1 population of U-ENDURE, no substantial differences were observed in the percentages of patients who experienced CD-related hospitalizations across the upadacitinib 30 mg, upadacitinib 15 mg, and placebo groups. The differences compared to placebo were −0.78% (95% CI, −10.4% to 8.8%) and −4.17 (95% CI, −13.1% to 4.7%), respectively. In both cases, the 95% CI was wide and crossed the threshold between potential benefit and potential harm (i.e., null).
Incidence of CD-Related Surgeries Through 52 Weeks
||||| || || |||||||| ||||||||||| ||||||||||| || || |||||||||||||| || |||| |||||| |||||| |||| |||| |||| |||||| || |||||||||| |||||||| || |||||||| | |||| |||||| ||||||||||| |||||||||| |||||||||| || ||||||||||| | |||| |||||| |||||||||| |||||||||| || |||| | |||||||||||| ||||||||||| |||||||||| || || |||||||| ||||| |||||| || |||||| |||| ||||| || |||||| |||||||| || || |||||||| ||||||||||| ||||||||||| || || ||||||||||||| |||||| |||||| |||| |||| |||| |||||| || |||||||||| |||||||| || |||||||| | |||| |||||| ||||||||||| |||||||||| |||||||||| || ||||||||||| | |||| |||||| |||||||||| |||||||||| || |||| | |||||||||||| ||||||||||| ||
Harms
Adverse Events
Crohn disease, nasopharyngitis, acne, anemia, and influenza were some of the most commonly reported AEs across all trials. Across the 3 trials, treatment groups, and cohorts, AEs were common and experienced by approximately 58% to 76% of patients. In the comparative parts and cohorts, the rate of AEs between the placebo and active-treatment groups were generally similar. The rate of any AE was slightly higher in patients treated with upadacitinib relative to those who received placebo in part 1 of the U-EXCEL trial but slightly higher in the those who received placebo in cohort 1 of the U-ENDURE trial.
Serious Adverse Events
SAEs occurred in approximately 7% to 15% of patients across the different treatment groups and cohorts of the included trials. Some of the most frequently reported SAEs among all trials were GI disorders and infections and infestations. In placebo-controlled parts and cohorts, SAEs among patients receiving upadacitinib were less common (U-EXCEED), similar (U-EXCEL), or more common (U-ENDURE) compared to patients receiving placebo.
Withdrawal Due to Adverse Events
Withdrawals due to AEs varied across parts and cohorts between the included trials. In the U-EXCEED trial, patients in part 1 discontinued due to AEs at a rate of 4.1% (placebo group) and 5.6% (upadacitinib group), ||||| || || |||||||| ||||||||||| ||||||||||| || || |||||||||||||| || |||| |||||| |||||| |||| |||| |||| |||||| || |||||||||| |||||||| || |||||||| | |||| |||||| ||||||||||| ||.The most common reason was GI disorders.
In part 1 of the U-EXCEL trial, 5.7% and 4.3% of patients receiving placebo or upadacitinib discontinued due to AEs. ||||| || || |||||||| ||||||||||| ||||||||||| || || |||||||||||||| || |||| |||||| |||||| |||| |||| |||| |||||| || |||||||||| |||||||| || |||||||| | |||| |||||| ||||||||||| |||||| The most common reasons for discontinuation due to AE were GI disorders.
Table 23: Summary of Key Efficacy Results From Studies Included in the Systematic Review
Variable | U-EXCEED week 12; ITT1 population; NRI-NC | U-EXCEL week 12; ITT1 population; NRI-NC | U-ENDURE week 52; ITT1 population; NRI-NC | ||||
|---|---|---|---|---|---|---|---|
UPA 45 mg N = 324 | Placebo N = 171 | UPA 45 mg N = 350 | Placebo N = 176 | UPA 15 mg N = 169 | UPA 30 mg N = 168 | Placebo N = 165 | |
Clinical remission as measured by PROs | |||||||
n responders of total N (%; 95% CI) | 129 of 324 (39.8; 34.5 to 45.1) | 24 of 171 (14.0; 8.8 to 19.2) | 178 of 350 (50.7; 45.5 to 56.0) | 39 of 176 (22.2; 16.0 to 28.3) | 60 of 169 (35.5; 28.3 to 42.7) | 78 of 168 (46.4; 38.9 to 54.0) | 24 of 165 (14.4; 9.0 to 19.8) |
Difference compared to placebo percentage (95% CI); FDA P value (EMA P value) | 25.9 (18.7 to 33.1); < 0.0001 (< 0.0001) | 28.7 (20.9 to 36.4); < 0.0001 (< 0.0001) | 21.9 (13.7 to 30.0); < 0.0001 (< 0.0001) | 31.8 (23.2 to 40.3); < 0.0001 (< 0.0001) | NA | ||
Clinical remission as measured by CDAI | |||||||
n responders of total N (%; 95% CI) | 126 of 324 (38.9; 33.6 to 44.2) | 36 of 171 (21.1; 4.9 to 27.2) | 173 of 350 (49.5; 44.2 to 54.8) | 51 of 176 (29.1; 22.4 to 35.8) | 63 of 169 (37.3; 30.0 to 44.6) | 80 of 168 (47.6; 40.1 to 55.2) | 25 of 165 (15.1; 9.6 to 20.6) |
Difference compared to placebo percentage (95% CI); FDA P value (EMA P value) | 17.9 (10.0 to 25.8); < 0.0001 (< 0.0001) | 20.8 (12.7 to 28.8); < 0.0001 (< 0.0001) | 23.7 (15.2 to 32.1); < 0.0001 (< 0.0001) | 32.8 (23.9 to 41.6); < 0.0001 (< 0.0001) | NA | ||
Endoscopic response | |||||||
n responders of total N (%; 95% CI) | 112 of 324 (34.6; 29.4 to 39.8) | 6 of 171 (3.5; 0.8 to 6.3) | 159 of 350 (45.5; 40.3 to 50.8) | 23 of 176 (13.1; 8.1 to 18.0) | 47 of 169 (27.6; 20.8 to 34.4) | 67 of 168 (40.1; 32.7 to 47.6) | 12of 165 (7.3; 3.3 to 11.2) |
Difference compared to placebo percentage (95% CI); FDA P value (EMA P value) | 31.2 (25.5 to 37.0); < 0.0001 | 33.0 (26.2 to 39.9); < 0.0001 (< 0.0001) | 21.0 (13.6 to 28.4); < 0.0001 | 33.7 (26.0 to 41.3); < 0.0001 | NA | ||
Endoscopic remission | |||||||
n responders of total N (%; 95% CI) | 62 of 324 (19.1; 14.9 to 23.4) | 4 of 171 (2.3; 0.1 to 4.6) | 101 of 350 (28.9; 24.2 to 33.7) | 13 of 176 (7.4; 3.5 to 11.3) | 32 of 169 (19.1; 13.1 to 25.0) | 48 of 168 (28.6; 21.8 to 35.5) | 9 of 165 (5.5; 2.0 to 9.0) |
Difference compared to placebo percentage (95% CI); FDA P value (EMA P value) | 16.8 (12.0 to 21.6); < 0.0001 (< 0.0001) | 21.8 (15.8 to 27.8); < 0.0001 (< 0.0001) | 14.4 (7.7 to 21.0); < 0.0001 (< 0.0001) | 23.6 (16.1 to 31.0); < 0.0001 (< 0.0001) | NA | ||
Discontinuation of corticosteroid use and CDAI clinical remissiona | |||||||
n responders of total N (%; 95% CI) | 37 of 108 (34.3; 25.3 to 43.2) | 7 of 60 (11.7; 3.5 to 19.8) | 54 of 126 (42.9; 34.2 to 51.5) | 10 of 64 (15.7; 6.8 to 24.7) | 25 of 63 (39.7; 27.6 to 51.8) | 25 of 63 (39.7; 27.6 to 51.8) | 3 of 61 (4.9; 0.0 to 10.3) |
Difference compared to placebo percentage (95% CI); P value a | 22.5 (11.1 to 34.0); < 0.0001 | 27.7 (15.7 to 39.8); < 0.0001 | 35.4 (23.3 to 47.5); < 0.0001 | 32.3 (20.1 to 44.5); < 0.0001 | NA | ||
Clinical remission as measured by CDAI and endoscopic remission | |||||||
n responders of total N (%; 95% CI) | |||||| ||||| |||| || |||||| | ||||| |||| |||| || ||||| | |||||| ||||| ||||| || |||||| | |||||| |||| |||| | 25 of 169 (14.8; 9.5 to 20.2) | 39 of 168 (23.2; 16.8 to 29.6) | 6 of 165 (3.7; 0.8 to 6.5) |
Difference compared to placebo percentage (95% CI); P valueb | |||| |||| || |||||| ||||||| | |||| |||| || |||||| ||||||| | 12.2 (6.3 to 18.1); < 0.0001 | 19.8 (13.0 to 26.6); < 0.0001 | NA | ||
Change from induction baseline in IBDQ total score | |||||||
N | 280 | 130 | 304 | 134 | 78 | 94 | 41 |
Within-group change from baseline, least squares mean (95% CI) | 46.0 (41.7 to 50.2) | 21.6 (15.7 to 27.6) | 46.3 (42.5 to 50.0) | 24.4 (19.0 to 29.8) | 59.3 (52.9 to 65.6) | 64.5 (58.3 to 70.7) | 46.4 (38.5 to 54.3) |
Between-group difference compared to placebo, least squares mean (95% CI); FDA P value (EMA P value) | 24.3 (17.2 to 31.5); < 0.0001 (< 0.0001) | 21.8 (15.6 to 28.1); < 0.0001 (< 0.0001) | 12.9 (4.3 to 21.4); 0.0033 (0.0033) | 18.1 (9.8 to 26.4); < 0.0001 (< 0.0001) | NA | ||
CR-100 | |||||||
n responders of total N (%; 95% CI) | 164 of 324 (50.5; 45.1 to 56.0) | 47 of 171 (27.5; 20.8 to 34.2) | 198 of 350 (56.6; 51.4 to 61.8) | 66 of 176 (37.3; 30.1 to 44.5) | 70 of 169 (41.4; 34.0 to 48.8) | 86 of 168 (51.2; 43.6 to 58.7) | 25 of 165 (15.2; 9.7 to 20.6) |
Difference compared to placebo percentage (95% CI); FDA P value (EMA P value) | 22.8 (14.4 to 31.2); < 0.0001 (< 0.0001) | 19.8 (11.3 to 28.4); < 0.0001 (< 0.0001) | 27.1 (18.3 to 35.8); < 0.0001 | 36.4 (27.5 to 45.2); < 0.0001 | NA | ||
Resolution of EIMs in patients with EIMs at induction baseline | |||||||
n responders of total N (%; 95% CI) | 43 of 131 (32.8; 24.8 to 40.9) | 13 of 60 (21.7; 11.2 to 32.1) | 43 of 151 (28.5; 21.3 to 35.7) | 16 of 78 (20.9; 11.8 to 30.1) | 15 of 61 (24.6; 13.8 to 35.4) | 26 of 73 (35.6; 24.6 to 46.6) | 10 of 66 (15.2; 6.5 to 23.8) |
Difference compared to placebo percentage (95% CI); FDA P value (EMA P value) | 11.5 (−1.5 to 24.4); 0.0833 (0.0833) | 9.0 (−1.9 to 19.9); 0.1044 (0.1044) | 9.6 (−3.4 to 22.6); 0.1476 (0.1476) | 22.0 (9.3 to 34.8); 0.0007 (0.0007) | NA | ||
CD-related hospitalization | |||||||
n patients with CD-related hospitalization of total N (%; 95% CI) | 20 of 324 (6.2; 3.6 to 8.8) | 15 of 171 (8.8; 4.5 to 13.0) | 13 of 350 (3.7; 1.7 to 5.7) | 9 of 176 (5.1; 1.9 to 8.4) | 13 of 169 (11.2249; 5.1231 to 17.3267) | 10 of 168 (7.8282 [2.9763 to 12.6801]) | 10 of 165 (12.0022 [4.5633 to 19.4411]) |
Difference compared to placebo (95% CI); FDA P value (EMA P value) | −2.6 (−7.6 to 2.4); 0.2834 (0.2834) | −1.4 (−5.2 to 2.4); 0.4494 (0.4494) | −0.78 (−10.3985 to 8.8440); 0.8742 (0.8742) | −4.17 (−13.0553 to 4.7074); 0.3570 (0.3570) | NA | ||
CD-related surgery | |||||||
n patients with CD-Related surgeries of total N (%; 95% CI) | ||||| |||| |||| || | ||||| |||| |||| || | ||||| |||| |||| || | ||||| |||| |||| || | ||||| |||| | ||||| |||| | ||||| |||| |
Incidence rate (n of 100 patient-years); 95% CI) | || | || | || | || | |||||| ||||||| || | | |||||| ||||||| || | | |||||| ||||||| || || |
Rate difference compared to placebo (95% CI); P valueb | ||| ||||| || ||||| |||||| | ||| ||||| || ||||| |||||| | || | || | || | ||
Incidence rate (n of 100 patient-years) difference compared to placebo (95% CI); P value | || | || | |||| |||||||| || ||| | |||| |||||||| || | | || | ||
D = Crohn disease; CDAI = Crohn Disease Activity Index; CI = confidence interval; CR-100 decrease of at least 100 points from baseline in the Crohn Disease Activity Index; EIM = extra-intestinal manifestation; EMA = European Medicines Agency; IBDQ = Inflammatory Bowel Disease Questionnaire; ITT1 = intention-to-treat (part 1 or cohort 1 of study); NA = not applicable; NRI-NC = nonresponder imputation with no special data-handling to account for COVID-19; PRO = patient-reported outcome; UPA = upadacinitib.
aThe 12-week outcome evaluated in the U-EXCEED and U-EXCEL trials was defined as the proportion of patients who discontinued corticosteroids and had clinical remission as measured by CDAI among patients who had been receiving corticosteroids at baseline. The 52-week outcome evaluated in the U-ENDURE trial was defined as the proportion of patients without corticosteroid use for CD for at least 90 days before week 52 and who had clinical remission as measured by CDAI among patients taking corticosteroids for CD at induction baseline.
bIndicates P values that have not been adjusted for multiple testing.
Source: Clinical Study Reports for U-EXCEED,22 U-EXCEL,23 and U-ENDURE.26 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
In cohort 1 of the U-ENDURE trial the proportion of patients who stopped treatment due to an AE was 3.6% in the placebo group, 7.2% in the upadacitinib 15 mg group, and 5.7% in the upadacitinib in the 30 mg group. | |||||| |||| || | |||| || |||||||| ||||||||| || || ||| |||| || ||||| ||||||||||||| Again, the most common reason was GI disorders; however, infections and infestations were also a relatively common reason compared to the other trials.
Of the parts and cohorts investigated in the included trials, the greatest proportion of patients who discontinued study treatment due to AEs was 10.3% in the part 3, double-blind, extended treatment arm of the U-EXCEED trial.
Mortality
One patient died due to an AE in the upadacitinib 45 mg group of the U-EXCEED trial, ||| || ||||||| |||| || || |||||||||||| || ||||||||||||||| || || || || || ||||||| |||||. Causes of death included infectious shock and COVID-19 infection.
Notable Harms
The AESIs were selected based on safety concerns reported for other JAK inhibitors, upadacitinib data obtained from preclinical studies, and the upadacitinib development program, as well as customary regulatory concerns for novel small-molecule drugs.
Across the trials, AESIs of serious infection, opportunistic infection, herpes zoster, adjudicated gastrointestinal perforation, anemia, neutropenia, lymphopenia, elevated CPK, hepatic disorder, renal dysfunction, and adjudicated venous thromboembolic events were observed. The most commonly reported AESIs (≥ 4% in any part or cohort of any included trial) were anemia, lymphopenia, serious infections, infections and infestations, herpes zoster, hepatic disorder, and elevated CPK. One adjudicated cardiovascular event was observed in the U-EXCEL in a patient from part 1 treated with placebo. Malignancies (all types), malignancies (excluding nonmelanoma skin cancer), and nonmelanoma skin cancer occurred rarely in the U-ENDURE trial and were not observed in the induction trials (i.e., U-EXCEED and U-EXCEL). No events of lymphoma or active tuberculosis were observed in any included trial.
Table 24: Summary of Key Harms Data in U-EXCEED
Adverse event | Part 1 (double-blind) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | ||
|---|---|---|---|---|---|---|---|---|
PBO (N = 171) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |
Most common treatment-emergent adverse events (reported at ≥ 2% incidence rate), n (%) | ||||||||
Any adverse event | 112 (65.5) | 221 (68.2) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Nasopharyngitis | 5 (2.9) | 23 (7.1) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Headache | 9 (5.3) | 20 (6.2) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Crohn disease | 23 (13.5) | 19 (5.9) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Upper respiratory tract infection | 5 (2.9) | 17 (5.2) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Anemia | 10 (5.8) | 16 (4.9) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Acne | 4 (2.3) | 15 (4.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Nausea | 8 (4.7) | 15 (4.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Pyrexia | 8 (4.7) | 13 (4.0) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Abdominal pain | 12 (7.0) | 10 (3.1) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Increased blood CPK | 4 (2.3) | 9 (2.8) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Cough | 5 (2.9) | 9 (2.8) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Influenza | 2 (1.2) | 9 (2.8) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Urinary tract infection | 2 (1.2) | 8 (2.5) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Vomiting | 4 (2.3) | 8 (2.5) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Arthralgia | 11 (6.4) | 7 (2.2) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Constipation | 2 (1.2) | 7 (2.2) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Serious adverse events, n (%) | ||||||||
Any serious adverse event | 17 (9.9) | 30 (9.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Blood and lymphatic system disorders | 1 (0.6) | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Ear and labyrinth disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
GI disorders | 14 (8.2) | 14 (4.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Hepatobiliary disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Infections and infestations | 3 (1.8) | 9 (2.8) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Metabolism and nutrition disorders | 1 (0.6) | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Musculoskeletal and connective tissue | 1 (0.6) | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Psychiatric disorders | 1 (0.6) | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Renal and urinary disorders | 0 | 2 (0.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Reproductive system and breast disorders | 0 | 2 (0.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Respiratory, thoracic, and mediastinal disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
General disorders and administration site | NR | NR | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Injury, poisoning, and procedural complications | NR | NR | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Skin and subcutaneous tissue disorders | NR | NR | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Patients who stopped treatment due to adverse events, n (%) | ||||||||
Any adverse event | 7 (4.1) | 18 (5.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Blood and lymphatic system disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Cardiac disorders | 1 (0.6) | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Eye disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
GI disorders | 6 (3.5) | 9 (2.8) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
General disorders and administration-site conditions | 1 (0.6) | 3 (0.9) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Infections and infestations | 0 | 4 (1.2) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Investigationsa | 0 | 2 (0.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Metabolism and nutrition disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Musculoskeletal and connective tissue | 0 | 2 (0.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Nervous system disorders | 0 | 2 (0.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Psychiatric disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Respiratory, thoracic, and mediastinal | 0 | 2 (0.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Skin and subcutaneous tissue disorders | 0 | 3 (0.9) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Deaths, n (%) | ||||||||
All deaths | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Deaths occurring > 30 days after last dose of study drug | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Adverse events of special interest, n (%) | ||||||||
Serious infections | 3 (1.8) | 9 (2.8) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Opportunistic infection excluding tuberculosis and herpes zoster | 0 | 2 (0.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Active tuberculosis | 0 | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Herpes zoster | 0 | 5 (1.5) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Adjudicated GI perforation | 0 | 1 (0.3) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Anemiab | 11 (6.4) | 22 (6.8) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Neutropenia | 0 | 4 (1.2) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Lymphopenia | 2 (1.2) | 6 (1.9) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Elevated CPK | 4 (2.3) | 9 (2.8) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Hepatic disorder | 6 (3.5) | 8 (2.5) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Renal dysfunction | 0 | 2 (0.6) | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Malignancies (all types) | 0 | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Malignancies excluding NMSC | 0 | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
NMSC | 0 | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Lymphoma | 0 | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Adjudicated cardiovascular events | 0 | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
Adjudicated thrombotic events | 0 | 0 | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| | |||| ||| |
CPK = creatine phosphokinase; GI = gastrointestinal; NMSC = nonmelanoma skin cancer; NR = not reported; OL = open-label; PBO = placebo; q.d. = once daily; SAE = serious adverse event; UPA = upadacitinib.
aInvestigations include an increase in either alanine transaminase, aspartate transaminase, blood creatine, hepatitis B DNA, decreased lymphocyte count, or Mycobacterium tuberculosis complex test-negative.
bRates of adverse events of special interest per 100 patient-years of exposure.
Sources: Sponsor’s Summary of Clinical Evidence.
Table 25: Summary of Key Harms Data in U-EXCEL
Adverse event | Part 1 (double-blind) | |||| |||||||| ||||||||| | ||
|---|---|---|---|---|
Placebo (N = 176) | UPA 45 mg q.d. (N = 350) | ||||||| |||| ||||| ||| | ||| || |||||| || || || || | | |
Most common treatment-emergent adverse events (reported at ≥ 2% incidence rate), n (%) | ||||
Any adverse event | 103 (58.5) | 219 (62.6) | |||| ||| | |||| ||| |
Acne | 1 (0.6) | 24 (6.9) | |||| ||| | |||| ||| |
Anemia | 8 (4.5) | 22 (6.3) | |||| ||| | |||| ||| |
Nasopharyngitis | 6 (3.4) | 16 (4.6) | |||| ||| | |||| ||| |
Headache | 7 (4.0) | 15 (4.3) | |||| ||| | |||| ||| |
Nausea | 8 (4.5) | 15 (4.3) | |||| ||| | |||| ||| |
Pyrexia | 2 (1.1) | 15 (4.3) | |||| ||| | |||| ||| |
Abdominal pain | 5 (2.8) | 13 (3.7) | |||| ||| | |||| ||| |
Crohn disease | 18 (10.2) | 13 (3.7) | |||| ||| | |||| ||| |
Rash | 4 (2.3) | 13 (3.7) | |||| ||| | |||| ||| |
Increased blood creatine phosphokinase | 0 | 12 (3.4) | |||| ||| | |||| ||| |
Upper respiratory tract infection | 4 (2.3) | 12 (3.4) | |||| ||| | |||| ||| |
Fatigue | 7 (4.0) | 11 (3.1) | |||| ||| | |||| ||| |
Influenza | 0 | 11 (3.1) | |||| ||| | |||| ||| |
Constipation | 3 (1.7) | 10 (2.9) | |||| ||| | |||| ||| |
Herpes zoster | 0 | 10 (2.9) | |||| ||| | |||| ||| |
Arthralgia | 8 (4.5) | 9 (2.6) | |||| ||| | |||| ||| |
Back pain | 3 (1.7) | 7 (2.0) | |||| ||| | |||| ||| |
Diarrhea | 7 (4.0) | 7 (2.0) | |||| ||| | |||| ||| |
Serious adverse events, n (%) | ||||
Any serious adverse event | 12 (6.8) | 24 (6.9) | |||| ||| | |||| ||| |
Blood and lymphatic system disorders | 0 | 2 (0.6) | |||| ||| | |||| ||| |
Gastrointestinal disorders | 8 (4.5) | 13 (3.7) | |||| ||| | |||| ||| |
General disorders and administration site | 0 | 1 (0.3) | |||| ||| | |||| ||| |
Hepatobiliary disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| |
Infections and infestations | 3 (1.7) | 4 (1.1) | |||| ||| | |||| ||| |
Injury, poisoning, and procedural complications | 0 | 0 | |||| ||| | |||| ||| |
Metabolism and nutrition disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| |
Musculoskeletal and connective tissue | 1 (0.6) | 0 | |||| ||| | |||| ||| |
Nervous system disorders | 1 (0.6) | 2 (0.6) | |||| ||| | |||| ||| |
Renal and urinary disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| |
Respiratory, thoracic, and mediastinal disorders | 0 | 0 | |||| ||| | |||| ||| |
Skin and subcutaneous tissue disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| |
Patients who stopped treatment due to adverse events, n (%) | ||||
Any adverse event | 10 (5.7) | 15 (4.3) | |||| ||| | |||| ||| |
Blood and lymphatic system disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| |
Gastrointestinal disorders | 7 (4.0) | 8 (2.3) | |||| ||| | |||| ||| |
Hepatobiliary disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| |
Immune system disorders | 1 (0.6) | 0 | |||| ||| | |||| ||| |
Infections and infestations | 1 (0.6) | 0 | |||| ||| | |||| ||| |
Investigationsa | 1 (0.6) | 2 (0.6) | |||| ||| | |||| ||| |
Nervous system disorders | 0 | 3 (0.9) | |||| ||| | |||| ||| |
Psychiatric disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| |
Renal and urinary disorders | 0 | 0 | |||| ||| | |||| ||| |
Skin and subcutaneous tissue disorders | 0 | 1 (0.3) | |||| ||| | |||| ||| |
Deaths, n (%) | ||||
All deaths | 0 | 0 | |||| ||| | |||| ||| |
Deaths occurring ≤ 30 days after last dose of study drug | 0 | 0 | |||| ||| | |||| ||| |
Deaths related to COVID-19 | 0 | 0 | |||| ||| | |||| ||| |
Adverse events of special interest, n (%) | ||||
Serious infections | 3 (1.7) | 4 (1.1) | |||| ||| | |||| ||| |
Opportunistic infection excluding tuberculosis and herpes zoster | 0 | 0 | |||| ||| | |||| ||| |
Active tuberculosis | 0 | 0 | |||| ||| | |||| ||| |
Herpes zoster | 0 | 10 (2.9) | |||| ||| | |||| ||| |
Adjudicated gastrointestinal perforation | 0 | 0 | |||| ||| | |||| ||| |
Anemiab | 8 (4.5) | 28 (8.0) | |||| ||| | |||| ||| |
Neutropenia | 1 (0.6) | 9 (2.6) | |||| ||| | |||| ||| |
Lymphopenia | 6 (3.4) | 5 (1.4) | |||| ||| | |||| ||| |
Elevated creatine phosphokinase | 0 | 12 (3.4) | |||| ||| | |||| ||| |
Hepatic disorder | 4 (2.3) | 10 (2.9) | |||| ||| | |||| ||| |
Renal dysfunction | 0 | 0 | |||| ||| | |||| ||| |
Malignancies (all types) | 0 | 0 | |||| ||| | |||| ||| |
Malignancies excluding NMSC | 0 | 0 | |||| ||| | |||| ||| |
NMSC | 0 | 0 | |||| ||| | |||| ||| |
Lymphoma | 0 | 0 | |||| ||| | |||| ||| |
Adjudicated cardiovascular events | 1 (0.6) | 0 | |||| ||| | |||| ||| |
Adjudicated thrombotic events | 0 | 0 | |||| ||| | |||| ||| |
NMSC = nonmelanoma skin cancer; q.d. = once daily; UPA = upadacitinib.
aInvestigations include an increase in either alanine transaminase, aspartate transaminase, blood creatine, hepatitis B DNA, decreased lymphocyte count, or Mycobacterium tuberculosis complex test-negative.
bRates of adverse events of special interest per 100 patient-years of exposure.
Sources: Sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
Three RCTs were included in this review, 2 of which (U-EXCEED and U-EXCEL) evaluated upadacitinib 45 mg once daily versus placebo during induction therapy and 1 of which (U-ENDURE) evaluated upadacitinib 30 or 15 mg once daily versus placebo during maintenance therapy. All 3 trials were phase III, double-blind, placebo-controlled, multicentre studies that included several hundred patients and numerous important clinical, endoscopic, and HRQoL-related outcomes. The U-EXCEED trial included patients who had prior failure of biologic therapies, while the U-EXCEL trial included patients with either prior failure of biologics or prior failure of conventional treatments. The U-ENDURE trial, as a maintenance study, included patients from both the U-EXCEED and U-EXCEL studies. There were no concerns with regard to internal validity related to study design (e.g., method of randomization, concealment of allocation, maintenance of blinding, or balance of patient characteristics between treatment arms).
Although the studies included some nonrandomized cohorts, such as those who did not adequately respond during the initial 12-week induction period, these cohorts were not included in the primary efficacy analyses discussed in this report, and so do not impart any increased risk of bias to interpretation of the study results. However, patients who required extended induction (i.e., those involved in part 2 or 3 of the U-EXCEED or U-EXCEL trial) are representative of a proportion of patients in clinical practice, and the focus of this report on part 1 (i.e., 12-week induction) of each study chosen due to the RCT design of this period. In the U-ENDURE trial, cohort 1 (which was the focus of the primary analyses) only included patients who had achieved a response during 12 weeks of induction; cohorts 2 and 3 included patients who had achieved a response during extended induction periods. The clinical expert consulted by CADTH agreed with this approach for the purpose of decision-making regarding the efficacy of upadacitinib. Safety-related results were reported here for all cohorts.
Table 26: Summary of Key Harms Data in U-ENDURE
Adverse event | Cohort 1 | |||||| | | |||||| | | ||
|---|---|---|---|---|---|
Placebo (N = 223) | UPA 15 mg q.d. (N = 221) | UPA 30 mg q.d. (N = 229) | |||||| |||| | ||| || || ||||| | |
Most common adverse events (reported in ≥ 5% of patients), n (%) | |||||
Any adverse event | 169 (75.8) | 165 (74.7) | 176 (76.9) | |||| ||| | |||| ||| |
Crohn disease | 57 (25.6) | 40 (18.1) | 19 (8.3) | |||| ||| | |||| ||| |
COVID-19 | 8 (3.6) | 11 (5.0) | 14 (6.1) | |||| ||| | |||| ||| |
Arthralgia | 14 (6.3) | 7 (3.2) | 15 (6.6) | |||| ||| | |||| ||| |
Pyrexia | 5 (2.2) | 7 (3.2) | 15 (6.6) | |||| ||| | |||| ||| |
Upper respiratory tract infection | 8 (3.6) | 10 (4.5) | 12 (5.2) | |||| ||| | |||| ||| |
Nasopharyngitis | 10 (4.5) | 13 (5.9) | 8 (3.5) | |||| ||| | |||| ||| |
Herpes zoster | 4 (1.8) | 5 (2.3) | 12 (5.2) | |||| ||| | |||| ||| |
Acne | 6 (2.7) | 3 (1.4) | 12 (5.2) | |||| ||| | |||| ||| |
Anemia | 12 (5.4) | 7 (3.2) | 8 (3.5) | |||| ||| | |||| ||| |
Nausea | 13 (5.8) | 7 (3.2) | 8 (3.5) | |||| ||| | |||| ||| |
Rash | 12 (5.4) | 5 (2.3) | 9 (3.9) | |||| ||| | |||| ||| |
Serious adverse events, n (%) | |||||
Any serious adverse event | 31 (13.9) | 26 (11.8) | 24 (10.5) | |||| ||| | |||| ||| |
Blood and lymphatic system disorders | 1 (0.4) | 0 | 1 (0.4) | |||| ||| | |||| ||| |
Cardiac disorders | 0 | 0 | 1 (0.4) | |||| ||| | |||| ||| |
Gastrointestinal disorders | 18 (8.1) | 15 (6.8) | 11 (4.8) | |||| ||| | |||| ||| |
General disorders and administration-site conditions | 1 (0.4) | 0 | 0 | |||| ||| | |||| ||| |
Hepatobiliary disorders | 0 | 1 (0.5) | 1 (0.4) | |||| ||| | |||| ||| |
Infections and infestations | 9 (4.0) | 7 (3.2) | 11 (4.8) | |||| ||| | |||| ||| |
Injury, poisoning and procedural complications | 0 | 2 (0.9) | 2 (0.9) | |||| ||| | |||| ||| |
Investigationsa | 0 | 1 (0.5) | 0 | |||| ||| | |||| ||| |
Neoplasms benign, malignant, and unspecified (including cysts and polyps) | 0 | 1 (0.5) | 2 (0.9) | |||| ||| | |||| ||| |
Nervous system disorders | 1 (0.4) | 1 (0.5) | 0 | |||| ||| | |||| ||| |
Renal and urinary disorders | 4 (1.8) | 1 (0.5) | 0 | |||| ||| | |||| ||| |
Respiratory, thoracic, and mediastinal disorders | 1 (0.4) | 2 (0.9) | 0 | |||| ||| | |||| ||| |
Vascular disorders | 0 | 0 | 1 (0.4) | |||| ||| | |||| ||| |
Immune system disorders | NR | NR | NR | |||| ||| | |||| ||| |
Patients who stopped treatment due to adverse events, n (%) | |||||
Any adverse event | 8 (3.6) | 16 (7.2) | 13 (5.7) | |||| ||| | |||| ||| |
Blood and lymphatic system disorders | 0 | 1 (0.5) | 2 (0.9) | |||| ||| | |||| ||| |
Gastrointestinal disorders | 4 (1.8) | 8 (3.6) | 3 (1.3) | |||| ||| | |||| ||| |
Hepatobiliary disorders | 0 | 0 | 1 (0.4) | |||| ||| | |||| ||| |
Infections and infestations | 2 (0.9) | 4 (1.8) | 0 | |||| ||| | |||| ||| |
Investigations | 1 (0.4) | 2 (0.9) | 2 (0.9) | |||| ||| | |||| ||| |
Musculoskeletal and connective tissue disorders | 1 (0.4) | 0 | 0 | |||| ||| | |||| ||| |
Neoplasms benign, malignant, and unspecified (including cysts and polyps) | 0 | 1 (0.5) | 3 (1.3) | |||| ||| | |||| ||| |
Nervous system disorders | 0 | 1 (0.5) | 0 | |||| ||| | |||| ||| |
Respiratory, thoracic, and mediastinal disorders | 0 | 1 (0.5) | 0 | |||| ||| | |||| ||| |
Skin and subcutaneous tissue disorders | 0 | 0 | 2 (0.9) | |||| ||| | |||| ||| |
General disorders and administration site | NR | NR | NR | |||| ||| | |||| ||| |
Renal and urinary disorders | NR | NR | NR | |||| ||| | |||| ||| |
Vascular disorders | NR | NR | NR | |||| ||| | |||| ||| |
Deaths, n (%) | |||||
Patients who died | 0 | 0 | 0 | |||| ||| | |||| ||| |
Adverse events of special interest, n (%) | |||||
Serious infections | 9 (4.0) | 7 (3.2) | 11 (4.8) | |||| ||| | |||| ||| |
Opportunistic infections, excluding tuberculosis and herpes zoster | 0 | 1 (0.5) | 1 (0.4) | |||| ||| | |||| ||| |
Active tuberculosis | 0 | 0 | 0 | |||| ||| | |||| ||| |
Herpes zoster | 5 (2.2) | 6 (2.7) | 12 (5.2) | |||| ||| | |||| ||| |
Adjudicated gastrointestinal perforation | 1 (0.4) | 1 (0.5) | 1 (0.4) | |||| ||| | |||| ||| |
Anemiab | 13 (5.8) | 12 (5.4) | 10 (4.4) | |||| ||| | |||| ||| |
Neutropenia | 1 (0.4) | 2 (0.9) | 5 (2.2) | |||| ||| | |||| ||| |
Lymphopenia | 10 (4.5) | 4 (1.8) | 9 (3.9) | |||| ||| | |||| ||| |
Elevated creatine phosphokinase | 3 (1.3) | 5 (2.3) | 8 (3.5) | |||| ||| | |||| ||| |
Hepatic disorder | 2 (0.9) | 7 (3.2) | 11 (4.8) | |||| ||| | |||| ||| |
Renal dysfunction | 2 (0.9) | 0 | 0 | |||| ||| | |||| ||| |
Malignancies (all types) | 0 | 1 (0.5) | 2 (0.9) | |||| ||| | |||| ||| |
Malignancies excluding nonmelanoma skin cancer | 0 | 1 (0.5) | 2 (0.9) | |||| ||| | |||| ||| |
Nonmelanoma skin cancer | 0 | 0 | 0 | |||| ||| | |||| ||| |
Lymphoma | 0 | 0 | 0 | |||| ||| | |||| ||| |
Adjudicated major adverse cardiovascular event | 0 | 0 | 0 | |||| ||| | |||| ||| |
Adjudicated venous thromboembolic event | 0 | 0 | 1 (0.4)c | |||| ||| | |||| ||| |
NR = not reported; q.d. = once daily; UPA = upadacitinib.
aInvestigations include an increase in either alanine transaminase, aspartate transaminase, blood creatine, hepatitis B DNA, decreased lymphocyte count, or Mycobacterium tuberculosis complex test-negative.
bRates of adverse events of special interest per 100 patient-years of exposure.
cOne patient referring to other venous thrombosis.
dOne patient referring to arterial thromboembolic events (noncardiac, nonneurologic).
Sources: Sponsor’s Summary of Clinical Evidence.
Patients who responded during the 12-week induction period of the U-EXCEED or U-EXCEL trial could proceed to the U-ENDURE trial and were re-randomized upon entry. This design allows for an evaluation of whether the response was maintained in the absence or presence of continued upadacitinib therapy and preserves the strengths of the randomized study design. Additionally, the use of separate induction and maintenance studies is consistent with EMA guidance for the development of drugs for the treatment of CD. This design also reflects clinical practice in that only patients who responded to induction therapy would continue onto maintenance; however, from a research perspective, this is an enriched population (i.e., responders who could tolerate treatment) that does pose some limitations for interpretability.
The coprimary end points of clinical remission (based on CDAI or PROs) and endoscopic response were considered appropriate by the clinical expert consulted by CADTH. Endoscopic remission and endoscopic response were described by the clinical expert as objective measures that are of particular importance in managing patients with moderately to severely active CD. Analyses of the coprimary outcomes were appropriately adjusted for multiplicity, as were most of the outcomes evaluated in this report, except for the proportion of patients experiencing SAEs, CD-related hospitalizations, or CD-related surgeries.
Discontinuation rates exceeded 10% in part 1 of the U-EXCEED trial in all treatment groups but were similar across groups. However, in part 1 of the U-EXCEL trial, substantially more patients withdrew from the study in the placebo group (12.5%) than from the upadacitinib 45 mg group (5.7%), and the primary reason for discontinuation among patients treated with placebo was a lack of efficacy. |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| |. As the primary and secondary efficacy results did not include the extended induction periods of either trial, they are not expected to affect interpretation of the results.
In the maintenance substudy 1 of the U-ENDURE trial, discontinuation rates were high, ranging from 20% to 28% across cohorts and treatment groups. ||| ||||||| |||| || ||||||||||||||| || || |||||| |||||||| |||||||| ||||||||| |||||||| ||||||| |||||| |||||||| || || |||||||||| ||||||| | || |||||| ||||||. Although higher discontinuation rates are not surprising in a longer-term study, they do raise concerns about data integrity due to the high amount of imputation required and given the nature of the disease. Because a lack of efficacy was such a common reason for discontinuation among patients receiving placebo, the nonresponder imputation used in the trial primary analyses may be an accurate approach.
External Validity
The U-EXCEED, U-EXCEL, and U-ENDURE clinical trials were large, multicentre, international studies that recruited patients aged 18 to 75 years inclusive with a confirmed diagnosis of moderate to severe CD at least 3 months before baseline, and a history of an inadequate response or intolerance to biologic therapy (U-EXCEED) or conventional and/or biologic therapy (U-EXCEL). Patients in the U-ENDURE trial were those from the induction trials who achieved a response to therapy.
The clinical expert consulted by CADTH indicated that the study populations were wholly representative of the target population of adults with moderate to severe CD and a history of treatment failure. The expert described this as a strength of the study design, as the enrolled patients appeared to have more severe disease and would not be expected to simply get better over time without intervention. The clinical expert noted that patients suitable for potential treatment with upadacitinib who may have been excluded by the U-EXCEED or U-ENDURE eligibility criteria would be rare and represent complex, severe cases, such as those with extensive prior surgery in whom further resections would be potentially inappropriate. The expert noted that it was reasonable to exclude such cases from the clinical trials, and that this did not present a concern for generalizability given that these would be uncommon, medically fragile, highly complex cases treated on a patient-by-patient basis. There were no substantial concerns with respect to generalizability regarding the eligibility criteria and baseline characteristics of the included patient populations, with the exception that the U-ENDURE study included an enriched population of responders who tolerated induction therapy; however, this reflects clinical practice in that only those same patients would receive maintenance therapy in practice.
The dosages of the intervention, upadacitinib, were 45 mg once daily during the induction studies and either 15 mg or 30 mg once daily during the maintenance study. The clinical expert consulted by CADTH pointed out that clinical practice in moderate to severe CD would lean more commonly toward a maintenance dosage of 30 mg once daily due to evidence of higher efficacy and reluctance to potentially undertreat, and due to the irreversible nature of bowel damage that can occur. However, the clinical expert and the product monograph also noted that patients should be treated with the lowest effective dose in the interest of safety, and the expert indicated that the approach to dosing may vary by practice.
All 3 RCTs were placebo-controlled. Although suitable from a research perspective, there is a lack of direct evidence comparing active therapies head-to-head.
The 3 RCTs required mandatory tapering of corticosteroids. The clinical expert consulted by CADTH indicated that attempts at tapering are common in clinical practice and the time frame and intervals of tapering the dose are somewhat similar to that of the included clinical trials.
Several subgroups were defined a priori, including by number of prior biologics failed. The clinical expert indicated that 2 other subgroups of interest would be patients with perianal disease and patients with EIMs at baseline; however, these were not reported in the clinical trials, with the exception that the “resolution of EIMs” outcome was only evaluated in patients who did have EIMs at induction baseline.
The clinical expert noted that clinical remission as measured by CDAI is an outcome used only in clinical trials and is not relevant to clinical practice, but clinical remission as measured by PROs was also reported and was generally consistent with CDAI-related results. Overall, the selected primary and key secondary outcomes were relevant to decision-making and/or clinical practice, and adequately reflected measures of both efficacies and harms. The duration of follow-up was appropriate for the induction and maintenance phases of treatment. However, when measuring the proportion of patients who experienced events such as hospitalizations or surgeries related to CD, both 12-week and 52-week time frames were considered insufficient to experience a difference between groups, which contributed to uncertainty in interpreting these outcomes. The clinical expert also noted that endoscopy is not typically conducted at 12 weeks in clinical practice, but after 6 to 9 months of initiating treatment due to practical limitations and the invasiveness of the procedure. The expert considered this logistical limitation to be a factor in decisions regarding dosing, as patients without symptoms may be experiencing endoscopic activity that would not be seen until the procedure could be completed.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to informing CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.24,25
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimated effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimated effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. For the majority of outcomes, thresholds for clinically important effect were estimated by the clinical expert consulted by CADTH, including for metrics of clinical and endoscopic response or remission and resolution of EIMs. The threshold for IBDQ was based on literature. No thresholds were identified for the proportion of patients with SAEs, CD-related hospitalizations, or CD-related surgeries.
The induction studies (U-EXCEED and U-EXCEL) were assessed together for 12-week induction-related outcomes and summarized narratively according to outcome because they were similar in population, design, and outcome measures. The U-ENDURE trial was assessed for 52-week maintenance-related outcomes.
Results of GRADE Assessments
Induction
Table 2 presents the GRADE summary of findings for upadacitinib induction (45 mg once daily) versus placebo in adults with moderately to severely active CD and a history of treatment failure.
Maintenance
Table 3 presents the GRADE summary of findings for upadacitinib maintenance (15 mg or 30 mg once daily) versus placebo in adults with moderately to severely active CD and a history of treatment failure.
Long-Term Extension Studies
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Description of Studies
Substudy 2 of the U-ENDURE trial is an ongoing long-term extension study of substudy 1, and results are not yet available.
Substudy 2 aims to evaluate the safety and efficacy of long-term administration of upadacitinib in patients with moderately to severely active CD. Substudy 2 will enrol patients who completed substudy 1 and patients from the U-EXCEED trial (part 3 or cohort 3) who received open-label upadacitinib 30 mg once daily in the extended treatment period and achieved a clinical response at week 24. The duration of substudy 2 will be up to 244 weeks, including a 240-week period and a 30-day follow-up visit from the last dose of the study drug. During substudy 1, patients received upadacitinib, either 15 mg once daily or 30 mg once daily, for 52 weeks; patients will receive either placebo or upadacitinib during substudy 2.
Adverse events will be reported as a primary outcome for the long-term extension study.
Indirect Evidence
Contents within this section were informed by materials submitted by the sponsor. The following summary was validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
The clinical trials included in the pivotal and RCT evidence did not provide direct evidence of the comparative efficacy and safety of upadacitinib to other advanced therapies for the proposed Health Canada indication. The objective of this section is to summarize and critically appraise an ITC submitted by the sponsor that assessed the relative efficacy and safety of upadacitinib versus other advanced therapies in the treatment of adult patients with moderately to severely active CD.27,28 This summary also informs the pharmacoeconomic evaluation.
Description of Indirect Comparison
The sponsor-submitted ITC was based on a systematic literature review to assess the clinical efficacy and safety of upadacitinib compared to other advanced therapies in patients with CD, namely |||| |||| |||| |||| | |||.27,28 The selection criteria and methods for the ITC are presented in Table 27.
Table 27: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor [Redacted]
||||||||||||||| | |||||||| |||||||||| |
|---|---|
|||||||||| | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| || |
|||||||||||| | |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| || |
|||||||||| | ||||||| |
||||||| | |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| || |
||||| ||||||| | |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| || |
||||||||||| ||||||||||||||| | |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| || |
||||||||| |||||||| | |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| || |
||||||||| |||||||| ||||||| | |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| || |
||||||||| ||||||| | |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| |
|||| |||||||||| ||||||| | |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| ||| |
||||||| |||||||||| | |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||||||||||| |||||||||||| || | |||| |||||| | ||||| |
| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || |||||
Note: This table has been redacted at the request of the sponsor.
ITC Design
Objectives
The objective was to conduct a systematic review and ITC to estimate the relative efficacy and safety of upadacitinib versus advanced therapies relevant to Canadian drug plans for the treatment of adult patients with moderately to severely active CD.
Study Selection Methods
|||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||
ITC Analysis Methods
The analysis methods for the ITC are presented in Table 28.
|||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| ||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | ||||
Table 28: Analysis Methods for ITC Submitted by the Sponsor [Redacted]
Methods | Description |
|---|---|
Analysis methods | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| |
Data imputation and assumptions | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || |||||||| |
Priors | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| |
Assessment of model fit | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || |||||||| |
Assessment of consistency | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| |
Assessment of convergence | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | |
Baseline risk adjustment | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| |
Risk-difference link | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || |||||| |
Outcomes | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || |||||| |
Follow-up time points | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || | |
Construction of nodes | ||| |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || |||||||||||||| |
Sensitivity analyses | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || |||| |
Subgroup analysis | |||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| ||||||||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || || |
Methods for pairwise meta-analysis | ||| ||||||||| |
| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || |||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |
Note: This table has been redacted at the request of the sponsor.
Source: Sponsor-submitted ITC technical report and sponsor’s Summary of Clinical Evidence.27,28
Results of ITC
Summary of Included Studies
|||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| |
Characteristics of the included studies are presented in Appendix 1, Table 39. |||||| |||||||| ||||||||| || |||| || ||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| |
Baseline patient characteristics at induction are presented in Appendix 1, Table 40. Data were organized by study subpopulation |||||| || || |||| where available, rather than for the entire study population. The mean proportion of males in each study population varied from ||||| || ||||| and the mean age of patients in the study subpopulations varied from |||| ||||| || |||| years. Patients in most studies had a mean disease duration of between | ||||| | || |||||| |||||| |||||||| || ||||| |||| ||||||| ||||||||| || || ||||||| |||| |||| |||||||||||| |||| || || |||| |||| |||| || |||||| |||| ||||||| || |||| || || |||| ||||||| |||| ||||||| || ||||| || ||||| || |||||| || ||||| |||||||| || |||| |||| ||||| |||||||||||| |||||| |||| ||||||| |||| ||||| || |||| |||||||||||| |||| ||||||| | ||||| |||| |||||| || |||||| ||||| |||||| |||||||| || |||||||||| || |||||||| || |||| ||||| |||||||||| || |||| |||||| |||||||| |||||| |||| ||||| || ||||| || | |||||||||| |||||||||||||||||||||| |||||| |||| || || |||||| Baseline patient characteristics at maintenance are presented in Appendix 1, Table 41.
||||||||| || || ||||||| |||||||||| |||| || |||||||||| | |||||||| ||||||| || ||| |||| || ||||| | |||||| |||| || || ||||| || |||||||| |||| || |||| |||| || ||||| ||||||| ||||||||| |||| || ||||| ||||||| || ||||||||| ||||||||| |||||| |||||||||| |||| || ||||||| ||||||||| |||||||| ||||| || |||||||| ||||||||||||||| |||| | ||||| | |||||||| || ||||||||||| ||||||||||The assessment of homogeneity is presented in Table 29.
Table 29: Assessment of Homogeneity for ITC Submitted by the Sponsor [Redacted]
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | ||||||||| ||||||||| |||||| ||||||||| |||| |||| |||||||||| |||||||| ||||||| ||||||||| |||||||| |||| |||||| || ||||||||||||||||||||||||||||||||||||| ||||||| |||||||| ||||||| |||||| || || |||||| |||| ||||||| || ||||| ||||| || ||||| || ||||||||||| ||||||| || ||||| || ||||| ||||||| || |||| | || ||||| ||| ||||||| || ||||| || ||||| || |||||| | ||||| ||||||||||| ||||||| || ||||| ||||| |||||||||||| |||||||||||| |||| ||||| |||| |||||| |||| ||||||| | ||||| |
Treatment history | ||||||||| ||||||||| |||||| ||||||||| |||| |||| |||||||||| |||||||| ||||||| ||||||||| |||||||| |||| |||||| || ||||||| |||||||||||||||||||||||||||||| ||||||| | ||||| ||||||| |||||||| ||||||| |||||| || || |||||| |||| ||||||| || ||||| ||||| || ||||| || ||||||||||| ||||||| || ||||| || ||||| ||||||| || |||| | || ||||| ||| ||||||| || ||||| || ||||| || |||||| | ||||| ||||||||||| ||||||| || ||||| ||||| |||||||||||| |||||||||||| |||| ||||| |||| |||||| |||| ||||||| | ||||| |||| |||||| || |||||| ||||| |||||| |||||| |
Trial eligibility criteria | ||||||||| ||||||||| |||||| ||||||||| |||| |||| |||||||||| |||||||| ||||||| ||||||||| |||||||| |||| |||||| || ||||||| ||||||||||||||||||||||||||||| ||||||| | ||||| ||||||| |||||||| ||||||| |||||| || || |||||| |||| ||||||| || ||||| ||||| || ||||| || ||||||||||| ||||||| || ||||| || ||||| ||||||| || |||| | || ||||| ||| ||||||| || ||||| || ||||| || |||||| | ||||| ||||||||||| ||||||| || ||||| ||||| |||||||||||| |||||||||||| |||| ||||| |||| |||||| |||| ||||||| | ||||| |||| |||||| || |||||| ||||| |||||| ||||||||||||||| ||||||||| |||||| ||||||||| |||| |||| |||||||||| || |
Dosing of comparators | |||||||||| |||||| |||||||||||| ||||||||| ||||||||||||||||||| ||||||| |||||||||||| ||||||||||| |||||||||||| ||||||||||||||||||||| ||||| |||||||||||| |||||||||| ||||||||||||||||||||| ||||||| |||||||||||| ||||||||||||||||||||| |||||||||| ||||||||| |||||||||||| ||||||||| |||||||||||||||||||| ||||| |||||||||||| |||||||||||| |||||||||||||||| |
Placebo response | ||||||||| ||||||||| |||||| ||||||||| |||| |||| |||||||||| |||||||| ||||||| ||||||||| |||||||| |||| |||||| || ||||||| |||||||||||||||||||||||||||||||| ||||||| | ||||| ||||||| |||||||| ||||||| |||||| || || |||||| |||| ||||||| || ||||| ||||| || ||||| |
Definitions of end points | ||||||||| ||||||||| |||||| ||||||||| |||| |||| |||||||||| |||||||| ||||||| ||||||||| |||||||| |||| |||||| || ||||||| |||||||||||||||||||||||||||| ||||||| | ||||| ||||||| |||||||| ||||||| |||||| || || |||||| |||| ||||||| || ||||| ||||| || ||||| || ||||||||||| ||||||| || ||||| || ||||| ||||||| || |||| | || ||||| ||| ||||||| || ||||| || ||||| || |||||| | ||||| ||||||||||| ||||||| || ||||| ||||| |||||||||||| |||||||||||| |||| ||||| |||| |||||| |||| ||||||| | ||||| |||| |||||| || |||||| ||||| |||||| ||||||||||||||| ||||||||| |||||| ||||||||| |||| |||| |||||||||| |||||||| ||||||| ||||||||| |||||||| |||| |||||| || ||||||| ||||||||||||||||||||||||||||| ||||||| | ||||| ||||||| |||||||| ||||||| |||||| || || |||||| |||| ||||||| || ||||| ||||| || ||||| || ||||||||||| ||||||| || ||||| || ||||| ||||||| || |||| | || ||||| ||| ||||||| || ||||| || ||||| || |||||| | ||||| |
Timing of end point evaluation | ||||||||| ||||||||| |||||| ||||||||| |||| |||| |||||||||| |||||||| ||||||| ||||||||| |||||||| |||| |||||| || ||||||| ||||||||||||||||||||||||||||| ||||||| | ||||| ||||||| |||||||| ||||||| |||||| || || |||||| |||| ||||||| || ||||| ||||| || ||||| || ||||||||||| ||||||| || ||||| || ||||| ||||||| || |||| | || ||||| ||| ||||||| || ||||| || ||||| || |||||| | ||||| ||||||||||| ||||||| || ||||| ||||| |||||||||||| |||||||||||| |||| ||||| |||| |||||| |||| ||||||| | ||||| |||| |||||| || |||||| ||||| |||||| |||||| |
Withdrawal frequency | ||||||||| ||||||||| |||||| ||||||||| |||| |||| |||||||||| ||||||| ||||||||| |||||||| |||| |||||| || ||||||| ||||||||||||||||||||||||||||||| |
Clinical trial setting | ||||||||| ||||||||| |||||| ||||||||| |||| |||| ||| |
Study design | ||||||||| ||||||||| |||||| ||||||| |
| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || |||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| ||||
Note: This table has been redacted at the request of the sponsor.
Source: Sponsor-submitted ITC technical report and sponsor’s Summary of Clinical Evidence.27,28
Results
|||||| |||||||| ||||||||| || |||| || ||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| ||||||| |||||||| ||||||||| || |||| || ||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||| ||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| | |||||||||||| |||| || || ||||||||| |||||| |||||||||| |||||||| ||||||||| || |||| || ||||||||| | |||| || ||||||||||||||| | ||||| |||||| |||||||||| |||||||||||| || | |||| |||||| |
Induction-Phase Efficacy Outcomes: CDAI Remission, CR-100, Endoscopic Remission, and Endoscopic Response for Upadacitinib 45 mg
The efficacy results for the induction phase are summarized in Table 30. Many of the results comparing upadacitinib to advanced therapies had CrIs that |||||||| | ||||. The treatment-effect-estimated CrIs for upadacitinib versus the comparators were |||||||||||||, including those that |||||||| | ||||, indicating ||||||||||| || || |||||||| || || |||| || |||||||||| ||||||||||||| |||||| |||||||| || | ||||||||||| |||| ||||| |||| |||||||||| |||||||| || |||||||| ||||||||| |||| ||||||||||| ||||||| |||||||||||| || |||||| || ||||| |||| |||||||| |||||||||| | || |||||| || |||||
|||| |||||| ||||| ||||| |||||||||| |||||| |||| |||||||||| || |||||||| ||||||||| || ||||||||| ||||||| || ||||| || || | |||| |||||||| | |||| |||||| |||| |||||| |||||||||| |||| | || ||||||| ||||| |||| |||||| || || || |||| |||||| ||||| ||||| |||||||||| |||||| |||| |||||||||| || |||||||| ||||||||| || ||||||||| ||||||| || ||||| || || | |||| |||||||| | |||| |||||| |||| |||||| |||||||||| |||| | || ||||||| ||||| |||| |||||| || || |||||| |||||| ||||| ||||| |||||||||| |||||| |||| |||||||||| || |||||||| ||||||||| || ||||||||| ||||||| || ||||| || || | |||| |||||||| | |||| |||||| |||| |||||| |||||||||| |||| | || ||||||| ||||| |||| |||||| || || |||||| |||||| ||||| ||||| |||||||||| |||||| |||| |||||||||| || |||||||| ||||||||| || ||||||||| ||
Maintenance-Phase Efficacy Outcomes: CDAI Remission for Upadacitinib 15 mg and Upadacitinib 30 mg
The efficacy results for the maintenance phase are summarized in Table 31. Similar to the induction efficacy outcomes, many of the CrIs for the maintenance outcomes |||||||| || |||| || | |||| |||| |||||||||| ||||| |||||||||| |||||||||||. Another ||||| that |||||| || |||||||||||||| of the results is that only patients ||| |||||||| ||||||||| |||||||| ||||| ||||||| ||||||||||| |||||| |||||||| were eligible for and were re-randomized into the maintenance phase. Maintenance results therefore reflect only |||||||||| || ||||||| || |||||||||||||||| |||||||| ||||| |||| |||| ||||||| |||| ||||||||| |||||||||| |||| |||| |||| ||||||||||||||| || || |||||| || ||||||| |||| ||||| || ||||||| ||||||| |||||||||| ||||| |||||||. The same limitations of the induction phase apply to the maintenance phase ||||||||| |||||||||| ||||||||||| ||||| |||||||||||| |||||| ||||| |||||||| ||||||||| || || |||| ||||| ||||.
Table 30: Risk Difference for Induction-Phase Efficacy Outcomes for ITC Submitted by the Sponsor [Redacted]
|||||||||| | ||||| |||||||| || |||||||||| | |
|---|---|---|
||| |||||||||| || |||||| | |||||||||| | |
|||| |||||||| |||||||||| || |||| |||| | ||
||||||| | |||| |||||| ||||| | |||| |||||| ||||| |
|||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||| || |||| |||| | ||
||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| |||||||||| || |||| |||| | ||
||||||| | |||| |||||| ||||| | |||| ||||||| ||||| |
|||| | |||| |||||| ||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| ||||||||| || |||| |||| | ||
||||||| | |||| |||||| ||||| | |||| ||||||| ||||| |
|||| | |||| |||||| ||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||
Note: This table has been redacted at the request of the sponsor.
Table 31: Risk Difference for Maintenance-Phase Efficacy Outcomes for ITC Submitted by the Sponsor [Redacted]
|||||||||| | ||| |||||||||| | |||||| | | |||||||||| | |||||| |
|---|---|---|
||||| |||||||| || |||||||||| ||||| |||||||| |||||||||| || |||| |||| | ||
||||||| | |||| |||||| |||| | |||| |||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | |||| ||||||| ||||| | || |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||||| | |||| ||||||| ||||| | || |
||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||| | ||||| ||||||| ||||| | ||||| ||||||| |||||| |
||||| |||||||| || |||||||||| ||||| |||||||| |||||||||| || |||| |||| | ||
||||||| | |||| |||||| ||||| | |||| |||||| ||||| |
|||||||||| | |||| |||||| ||||| | |||| |||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| |||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| |||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| |||||| ||||| |
|||||||| | |||| ||||||| ||||| | || |
|||||||||| | |||| ||||||| ||||| | |||| |||||| ||||| |
||||||||| | |||| ||||||| ||||| | |||| |||||| ||||| |
||||||||||| | |||| ||||||| ||||| | || |
||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | |||| ||||||| ||||| | |||| |||||| ||||| |
|||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| |||||||||||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||
Note: This table has been redacted at the request of the sponsor.
Induction-Phase Safety Outcomes: Any AEs, SAEs, Serious Infections, and AEs Leading to Discontinuation for Upadacitinib 45 mg
The efficacy results for the induction phase are summarized in Table 32. All of the comparisons of upadacitinib versus other advanced therapies |||||||| |||| for any AEs, SAEs, serious infections, or AEs leading to discontinuation and the |||||| with analyzing the efficacy outcomes similarly apply to the safety outcomes.
Maintenance-Phase Safety Outcomes: Any AEs, SAEs, Serious Infections, and AEs Leading to Discontinuation for Upadacitinib 15 mg and Upadacitinib 30 mg
The efficacy results for the maintenance phase are summarized in Table 33. All the results for upadacitinib versus other comparators |||||||| | |||| for any AEs, SAEs, and serious infections, as did most of the results for the AEs leading to discontinuation outcomes. Definitions for safety outcomes |||||| |||||| ||||||| |||||||||||| || | ||||||||||||| ||||||||||||| || |||||||||| |||||| |||||||||| || ||||||||||. The safety results were similarly affected by the limitations listed for the maintenance-phase efficacy outcomes and should be considered when interpreting the results.
Table 32: Risk Difference for Induction-Phase Safety Outcomes for ITC Submitted by the Sponsor [Redacted]
|||||||||| | ||||| |||||||| || |||||||||| |
|---|---|
||||||| |||||||||| | |
||| || | ||||||| || |||| |||| | |
||||||| | |||| ||||||| ||||| |
|||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| |
|||||||| | |||| ||||||| ||||| |
||||||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| |
|||| || ||||||| || |||| |||| | |
||||||| | ||||| ||||||| ||||| |
|||||| | ||||| ||||||| ||||| |
|||| | ||||| ||||||| ||||| |
||||||||| | |||| ||||||| ||||| |
|||||||| | |||| ||||||| ||||| |
|||||| | |||| |||||||| ||||| |
||||||| |||||||||| || ||||||| || |||| |||| | |
||||||| | |||| ||||||| ||||| |
|||| | |||| |||||| ||||| |
|||||| | |||| ||||||| ||||| |
||||||||| | |||| ||||||| ||||| |
|||||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| |
||| ||||||| || ||||||||||||||| || ||||||| || |||| |||| | |
||||||| | ||||| ||||||| ||||| |
|||||||| | ||||| ||||||| ||||| |
|||| | |||| ||||||| ||||| |
||||||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| |
|||||| | |||| ||||||| ||||| |
|||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||
Note: This table has been redacted at the request of the sponsor.
Table 33: Risk Difference for Maintenance-Phase Safety Outcomes for ITC Submitted by the Sponsor [Redacted]
|||||||||| | |||||||| |||||||| || |||||||||| | |||||||| |||||||| || |||||||||| |
|---|---|---|
||| | | ||||||| || |||| |||| | ||
||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||| | ||||| ||||||| ||||| | || |
||||||||| | ||||| ||||||| ||||| | | ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | || | |||| ||||||| ||||| |
|||| | ||||||| || |||| |||| | ||
||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||| | || | ||||| ||||||| ||||| |
|||||||| | |||| ||||||| ||||| | || |
||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||| |||||||||| || ||||||| || |||| |||| | ||
||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | ||||| ||||||| ||||| | || |
||||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||||| | ||||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | || | |||| ||||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||| ||||||| || ||||||||||||||| || ||||||| || |||| |||| | ||
||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||| | ||||| ||||||| |||||| | ||||| ||||||| |||||| |
||||||||||| | ||||| ||||||| ||||| | ||||| ||||||| ||||| |
|||||||| | || | ||||| ||||||| ||||| |
|||||||| | |||| ||||||| ||||| | || |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
|||||||||| | |||| ||||||| ||||| | |||| ||||||| ||||| |
||||||||| | |||| |||||| ||||| | |||| |||||| ||||| |
|||||||| | |||| |||||| ||||| | |||| |||||| ||||| |
|||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| |||||||||||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||
Note: This table has been redacted at the request of the sponsor.
|||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| |||||||||||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| |||||||||||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||| | ||||||| ||||||| |||||||| |||||||| || ||||| || || |||||| | ||||| ||||||| |||||||| |||||||| || ||||| || || || |||||||||| |||||||| |||||||| || || |||| || |||| |||||||| |||||||| || ||||| || | || |||||| || |||||||| ||||||||
Critical Appraisal of ITC
Overall, the systematic literature review methods appeared to be acceptable, with literature screening and data extraction performed in duplicate and quality assessment conducted using ||| |||||||| |||| || |||| ||||. All studies were found to have a low risk of bias, and none were excluded from the ITC due to risk of bias.
For the most part, studies eligible to be included in the ITC met the indication for the current drug under review, with the exception of | studies that included adolescent patients as young as 15 years. The clinical expert consulted by CADTH stated that the inclusion of adolescent patients would not meaningfully change the interpretation or generalizability of the results to an adult patient population, the latter of which is stated in the Health Canada indication for upadacitinib. It was unclear what proportion of the study populations were adolescents and how much this could have biased the results because ||||||||||| ||||||||||||||| || ||||| ||||||| |||| |||||||||. Based on the ITC feasibility assessment, | study was not included in the base-case analysis because it was considerably different in terms of baseline characteristics for body mass, disease duration, CDAI, and rate of immunomodulator use, which were considered to be potential treatment-effect modifiers. Unlike the other studies in the ITC, the upadacitinib studies’ inclusion criteria did not use CDAI scores and instead used 2 key CD symptom criteria to determine patient eligibility. CDAI was still measured in the upadacitinib studies allowing for posthoc analyses of patients who met the CDAI 220-to-450-point range from all studies. Although the sponsor stated that the patient characteristics and efficacy results of the primary and posthoc analyses in the upadacitinib studies were similar, the selection of patients with a CDAI score of 220 to 450 from the overall populations breaks randomization and its benefits of balancing known and unknown (or unmeasured) effect modifiers. As a result, it is unclear what bias this may introduce to the upadacitinib analysis.
The networks were sparsely populated, with relatively few nodes centred around a single connection (placebo) in a star geometry. Furthermore, most closed loops were between different doses of individual drugs and, consequently, the evidence was essentially all indirect, increasing uncertainty in the estimates for each outcome, and the consistency assumption could not be assessed. This was particularly notable for the endoscopic outcomes, which contained only | studies for the non–bio-IR population and | studies for the bio-IR population. Among the networks for CDAI outcomes and safety outcomes, there were few closed loops, making it challenging to assess the consistency assumption. The sponsor’s analysis of the ITC concluded that there was a lack of evidence of consistency; nonetheless, this is inconclusive of evidence for consistency between direct and indirect evidence.
The clinical expert confirmed that the list of potential treatment-effect modifiers identified in the ITC technical report were clinically relevant, but to be comprehensive the expert would have also included EIMs and previous surgical interventions. The clinical expert was of the opinion that, based on the baseline patient characteristics, the patients in the ITC reflect those who may be eligible for treatment with upadacitinib in Canada. However, complete information on all of the potentially important effect modifiers was not available due to the limited reporting of baseline patient demographics and medical histories from the individual trials. Using the information that was available, the notable differences in patient characteristics between studies suggest the similarity assumption was violated. For example, for the induction phase, ||||| |||| ||||||||||| || |||||||| || || ||||||| |||||| || || ||||||| | ||||||| |||| || || ||||| | ||||| |||||||||||| |||||| ||||| || |||| ||||||| || ||||||||||| |||||||||| || || |||||||| |||||| || |||||| | |||||||||||||||| | || ||||||| ||||||||| ||||||||||| |||||||||| | |||||||||||||| |||||||| | ||||||||| || |||||||| |||| |||||||| |||||||||||||| || ||||| |||||| || ||||||||| ||||||| |||||||| | || |||| |||| |||||||| || ||||| || || || |||||| ||||||| ||||| It is also likely that there were differences in patients’ experience with previous treatments (number and type) used for CD given the ongoing development and increasing availability of new drugs; however, these data were not clearly reported. It was unclear if patients received an adequate trial to determine failure or intolerance, and how these outcomes were defined.
In addition to patient characteristics, heterogeneity related to study design and outcomes was an issue in the ITC. For example, the duration of the induction trials ranged from 4 weeks to 12 weeks. The duration of the maintenance phases ranged from 44 weeks to 60 weeks, although the clinical expert did not expect the variability to introduce bias in the interpretation. The sponsor justified the variable durations as consistent with the primary end points of the studies, the length of time during which the treatments are expected to be fully effective, and the times that most likely align with treatment labels. While the clinical expert did not expect there to be bias due to the variable induction durations, outcomes were assessed based on a specific time point rather than on the patient’s response to treatment and the induction period. There was considerable variability in the definition of induction response, which dictated transition from the induction phase into the maintenance phase of the studies. In the upadacitinib ||| ||| studies, induction response was defined as a decrease of 30% or more in average daily SF and/or a decrease of 30% or more in average daily AP score (neither worse than baseline). The |||| ||| studies defined induction simply as a decrease of at least 70 points from baseline in the CDAI (CR-70), while the ||| study used CR-100. In the ||| study, induction was defined using CR-70 and a 25% reduction in total CDAI score from baseline. Because the CR-100 outcome was used in the ITC analyses and most of the studies reported this value, missing data were imputed for 2 studies |||| || |||| that only reported CR-70 data. Imputation was based on the studies that provided CR-100 data and used the average relative difference between CR-70 and CR-100 for the treatment groups |||||| |||| |||| ||||| ||||. No sensitivity analyses were reported that explored the potential impact of this assumption on the results, and it is therefore unclear if this approach was reasonable. |||||||||||||||| |||||||||| ||||||| |||| |||| ||||||||| ||||||||||||| || | ||||||| || ||||| ||||||||| || ||||||| || | ||||| ||||||||| ||||||||. Still, only ||| | drugs contributed to the indirect comparison for induction-phase endoscopic outcomes and no analysis could be performed for ||| ||||||||||| |||||, which is a limitation as this outcome was identified as important for this reimbursement review. Definitions of safety outcomes varied across studies and, due to inconsistent data reporting, results could not be separated by ||| || | population and it is unclear how harms compared between the populations.
The re-randomization of patients upon entering the maintenance phase negates the assumption of a common placebo comparator across studies. ||| || ||||||||| |||||| ||||| ||||||||| ||||||| |||||||||| |||||||| |||| |||||||||| |||||| ||||||||||||||| | ||||||| |||||| |||| |||| || | || ||||||| || |||| || ||||||||||| || ||||||||||||| || || ||||||| ||||||||| || ||||||||||| ||||||| || || ||||||| |||||||||| || || |||||||| |||||| |||| || || |||| || ||||||| || |||||||| ||||||||||| || ||||||| ||||||||||||||| || ||||| ||||| ||||||| However, it is a proxy and may not account for all sources of heterogeneity. As well, it does not adjust for differences in study design, conduct, or analysis. ||| || ||||||||| |||||| |||||||||||| |||| || |||||| |||||||| ||||||||| ||||| |||||||| |||| |||||||||||. The |||||| networks also meant that fixed-effect models were generally reported over random-effects models, which is important given the differences between the 2 models when heterogeneity between comparisons is suspected. Use of a risk-difference network meta-analysis framework as the base case due in part to the lack of ability to calculate |||||||| |||| |||||||| |||||| is not considered by CADTH reviewers to be sufficient to address heterogeneity between studies. In particular, the risk-difference link would not likely account for differences arising from the temporal issues related to the apparent heterogeneity between study characteristics and designs. Relative treatment effects (in the form of |||) were reported and generally agreed with the || results; however, only the ||| ||| ||| between the various treatments versus placebo were reported, and analyses showed ||||||||||| between treatment comparisons without the |||||| ||||| ||||||||| || ||||||| ||||||||||||||||||| |||||||| |||| ||||||||| ||||||||| ||||||||| || ||||||| || || |||||||||| |||| ||||||| |||||||||| |||||||||||| | ||||||| || |||||||||| |||| || |||| || |||||||| ||||||| |||||||||| ||||||||| |||||||||||||| | |||| ||||| ||||| |||| |||| || |||||||||||||| |||||||| |||| || |||| ||||||||| | ||||||||||||| || || |||| |||| |||||||| |||||||||| |||||||| ||||| || |||||||||| || |||||| || ||||||| || ||||||||| ||||| || |||||||||| ||||||||||| || |||||||||| |||||||||||||||| |||||| || ||||| ||||||||| | || ||||||||||| ||||||||||| ||||||||||| ||||| || |||| |||||| | ||||| |||||||||
Discussion
Summary of Available Evidence
Three phase III, double-blind, placebo-controlled, multicentre, international RCTs were included in this review. Two of the RCTs were induction studies in adult patients with moderately to severely active CD, and a history of biologic failure (U-EXCEED) or a biologic and/or conventional therapy failure (U-EXCEL). Patients in both induction studies were randomized in a 2:1 ratio to receive upadacitinib 45 mg once daily or placebo. The primary results for randomized cohorts were evaluated at 12 weeks, although patients who did not achieve an adequate response could carry on to part 2 or 3 for extended induction. The third RCT was a maintenance study of upadacitinib 15 or 30 mg once daily versus placebo in patients who had achieved an adequate response in either the U-EXCEED or U-EXCEL trial, and the primary results were evaluated at 52 weeks among re-randomized patients from part 1 of the induction studies. Patients who had carried on to extended induction therapy in either of the induction studies, and thereafter achieved a response, could also enrol in U-ENDURE cohort 2 or 3, which were not randomized. The coprimary outcomes in all trials included clinical remission (as measured by PROs or CDAI), and endoscopic remission. Other important outcomes included endoscopic response, proportion of patients who discontinued corticosteroid use for CD and achieved clinical remission (among patients taking corticosteroids at induction baseline), proportion of patients who achieved both clinical remission and endoscopic remission, change in HRQoL (using IBDQ), CR-100, resolution of EIMs in patients who had EIMs at induction baseline, proportion who experienced CD-related hospitalizations |||||||||, and proportion who experienced harms including SAEs or AESIs.
The U-EXCEED trial enrolled 624 patients across 229 sites in 39 countries, the U-EXCEL trial enrolled 526 patients across 209 sites in 42 countries, and the U-ENDURE trial enrolled 901 patients across 277 sites in 43 countries. In the double-blind cohorts of the U-EXCEED, U-EXCEL, and U-ENDURE trials, there were slightly more male than female patients (53.5%, 53.8%, and 55.4%, respectively), and the mean ages were 38.1 years, 39.6 years, and 37.0 years, respectively. The majority of enrolled patients were white (approximately 70% in each trial, followed by Asian, Black, multiple races, and American Indian or Alaska Native). The mean duration of CD was 9.4 years in the U-EXCEED trial, 6.1 years in the U-EXCEL trial, and 7.2 years in the U-ENDURE trial; the differences were expected given that the U-EXCEL trial included patients who may not have experienced failure with biologics, indicating an earlier point in treatment history on average. Overall, most enrolled patients had a history of biologic failure (100% in the U-EXCEED trial by design, 45.4% in the U-EXCEL trial, and 75.6% in the U-ENDURE trial).
One long-term extension, substudy 2 of U-ENDURE, is ongoing, with no results currently available.
One ITC was submitted by the sponsor to estimate the relative efficacy and safety of upadacitinib versus advanced therapies for the treatment of adult patients with moderately to severely active CD.27,28 Studies included ||||| |||||||||| patients with moderately to severely active CD and advanced treatments for CD (||||||||||||| |||| |||| |||| |||| || |||). |||||||| outcomes included |||||||| |||||||||| || |||||||||||||||||||| |||||||||| || |||||||||| || |||||| |||||||| |||| || |||| ||||||| |||||||||| || || ||||||| || ||||||||||||||||| which generally aligned with the outcomes deemed most important to patients and clinicians.
Interpretation of Results
Efficacy
Induction
Evidence from the U-EXCEED and U-ENDURE trials showed that upadacitinib 45 mg once daily induction results in an increase in clinical remission as measured by PROs, and likely results in an increase in clinical remission as measured by CDAI at 12 weeks when compared to placebo. Induction of upadacitinib 45 mg once daily also results in an increase in CR-100 at 12 weeks compared to placebo. The thresholds for CD activity using CDAI are relatively well established. For the between-group comparison in percentage of responders, the clinical expert consulted by CADTH provided estimates of MIDs to support analyses of these and other outcomes that were reported as response rates; a difference of 15% between groups was considered clinically meaningful for the outcomes of clinical remission, CR-100, endoscopic response, remission, and resolution of EIMs. Although there was slightly more imprecision in the result for clinical remission as measured by CDAI (compared remission as measured by PROs), this analysis is based on the MIDs that may be somewhat subjective, and opinions may vary between clinicians, given that no published literature was identified to provide estimates for the MIDs for these outcomes. For clinical remission as measured by PROs, the thresholds used in the study were considered appropriate by the clinical expert, but opinions may vary among clinicians in that regard. Although clinical remission (and therefore symptomatic improvement) are important outcomes in CD and are expected to improve patient HRQoL, the clinical expert emphasized that these may not always correlate with objective measures of disease activity as assessed through endoscopy.
The induction trials also showed that, compared to placebo at 12 weeks, induction of upadacitinib 45 mg once daily results in an increase in the proportion of patients who achieve endoscopic remission, an endoscopic response, and the combination outcome of endoscopic response with clinical remission (as measured by CDAI). Endoscopic remission and response are both particularly important outcomes as they are objective measures of inflammatory activity in the bowels, which is believed by the clinical expert to be the driver of downstream outcomes, such as the need for surgery or hospitalizations related to CD. The clinical expert noted that the evaluation of endoscopic outcomes is a relatively new approach to measuring treatment outcomes in CD, whereas historical treatment goals and trials focused primarily on resolution of clinical symptoms. The expert added that endoscopic improvement reflects an arguably more important treatment outcome compared with clinical response due to the expected relationship between endoscopic disease activity and irreversible bowel damage. Accordingly, the outcome of a patient achieving both clinical (symptomatic) and endoscopic remission is particularly important. The between-group MIDs used to evaluate the certainty in these outcomes were also subjective and only supplied by the expert due to a lack of published literature evaluating MIDs; as the expert described endoscopic outcomes as generally harder to achieve than clinical outcomes in the treatment of CD, a 5% difference between groups was considered clinically meaningful. As expected by the clinical expert consulted for this review, numerically fewer patients achieved endoscopic remission compared to the proportion who achieved clinical remission. However, the proportions who achieved clinical remission were somewhat similar to the proportions who achieved endoscopic response. Also as expected, numerically fewer patients achieved the combination outcome of both endoscopic remission and clinical remission. However, all of the endoscopy-related outcomes exceeded the estimated between-group threshold of 5% for clinical importance.
Unlike the clinical trial design, endoscopic measures in real-world clinical practice may not be commonly evaluated at 12 weeks and may be difficult to repeat due to scheduling, resource constraints, and the nature of the test. The clinical expert anticipated that evaluation of these endoscopic outcomes would more likely be performed at approximately 6 months from treatment initiation. Additionally, beneficial endoscopic outcomes are expected to be associated with benefits in downstream modification of bowel damage, hospitalizations, and surgeries over a patient’s lifetime, but there is a lack of direct data for these longer-term outcomes given the typical 2-year duration of clinical trials. Although hospitalizations and surgeries were measured, the short duration of the induction trials was considered insufficient to experience a difference in these outcomes; as expected, the trial data showed that induction with upadacitinib may result in little to no difference in these outcomes at 12 weeks.
The trials assessed other secondary outcomes that were considered important in the treatment of CD. Results from the induction trials demonstrated that upadacitinib 45 mg once daily results in an improvement in IBDQ at 12 weeks, and likely an increase in the proportion of patients who discontinued corticosteroids for CD and had clinical remission as measured by CDAI, as well as the proportion of patients with resolution of EIMs among those with EIMs at baseline. The clinical expert pointed out that resolution of EIMs is of particular importance in subgroups of patients who have consequential EIMs that reduce their HRQoL, and who may have few options for therapies that have been shown to improve resolution of EIMs.
Head-to-head trial data are not available to compare induction with upadacitinib 45 mg once daily to other active therapies used in this population. Based on indirect evidence submitted by the sponsor, the ITCs ||| || |||| | ||||||||||| |||||||||| in efficacy between upadacitinib 45 mg once daily and the other evaluated therapies for ||| ||||||| during induction, but this ||| || |||||||||| || ||||||| |||||||||| efficacy due to |||| |||||||||||| |||||||||||||| || ||||||||||||| in the ITCs.
Maintenance
Overall, maintenance outcomes were consistent with induction outcomes, with some differences in the level of certainty. The major considerations were discussed previously, and will only be briefly reiterated here with respect to the outcomes of 52-week maintenance therapy. Based on 52-week results from the U-ENDURE trial, maintenance therapy of upadacitinib 30 mg once daily (compared to placebo) results in an increase in the proportion of patients who achieve clinical remission as measured by PROs, clinical remission as measured by CDAI, endoscopic response, endoscopic remission, discontinuation of corticosteroids with accompanying clinical remission as measured by CDAI, both endoscopic remission and concurrent clinical remission as measured by CDAI, and CR-100. It also likely results in an improvement in IBDQ at 52 weeks compared to placebo, and resolution of EIMs among patients who had EIMs at induction baseline. However, maintenance therapy of upadacitinib 30 mg once daily may result in little to no difference in CD-related hospitalizations or |||||||||| |||||||||; the 52-week time frame of this outcome was considered insufficient to detect any meaningful difference for these specific outcomes, and longer-term data would be more valuable to draw any conclusions.
The results of maintenance therapy of upadacitinib 15 mg once daily compared to placebo were generally numerically less substantial than for the higher dose. Based on GRADE, the direction and clinical significance of the results was the same as previously described for 30 mg once daily for most outcomes, with exceptions that will be detailed here. For the outcomes of clinical remission as measured by PROs, the change in IBDQ score from baseline, and the proportion with resolution of EIMs among patients who had EIMs at induction baseline, there was more imprecision resulting from 95% CIs that crossed the expert-provided MID thresholds. As such, maintenance therapy of upadacitinib 15 mg once daily likely results in more clinical remission as measured by PROs, may result in an improvement of IBDQ, and may result in more resolution of EIMs in patients who had EIMs at induction baseline, when compared to placebo at 52 weeks. Analysis of CD-related hospitalizations and CD-related surgeries had the same issues as previously described.
Head-to-head trial data are not available to compare upadacitinib maintenance therapy (15 mg or 30 mg once daily) to other active therapies used in this population. Based on indirect evidence submitted by the sponsor, the ITCs ||| || |||| || ||||||||||| |||||||||| |||||||||| between either dose of upadacitinib and the other evaluated therapies for ||| ||||||| during maintenance, but this ||| | |||||||||| ||||||||| |||||||||| efficacy due to |||| |||||||||||| |||||||||||||| || ||||||||||||| || | ||||.
Harms
Across all the cohorts of the 3 included trials, AEs were common but the frequency of AEs, SAEs, and withdrawals due to AEs were generally similar between patients treated with upadacitinib and those treated with placebo at 12 weeks and at 52 weeks. No new safety signals were identified. Upadacitinib, like other advanced therapies for moderate to severe CD, is associated with substantial AESIs that occur in a notable number of patients. However, some AESIs related to opportunistic infections can be fully mitigated by appropriate vaccination before starting treatment. According to the clinical expert, patients are already routinely counselled on the risks of infection, malignancy, and other AESIs associated with advanced therapies for CD, and that there are no new concerns related to upadacitinib compared with AEs already identified and noted in the product monograph.
Head-to-head trial data are not available to compare the safety of upadacitinib to other active therapies. Based on indirect evidence submitted by the sponsor, the ITCs ||| | |||| | ||||||||||| |||||||||| || || |||||||||| || |||||||| |||| || |||| ||||| ||||||| ||||||||||| || || ||||||| || ||||||||||||||| when comparing upadacitinib (induction with 45 mg once daily, maintenance with 15 mg once daily, or maintenance with 30 mg once daily) and the other evaluated therapies, ||| |||| | | |||||||||| || ||||||| |||||||||| |||||| || |||| |||||||||||| |||||||||||||| | ||||||||||||| || | ||||.
Conclusion
Three phase III, multicentre, double-blind RCTs evaluated the efficacy and safety of upadacitinib compared to placebo in adult patients with moderately to severely active CD and a history of treatment failure. Two of the RCTs evaluated induction therapy of upadacitinib 45 mg once daily while a single RCT evaluated maintenance therapies of upadacitinib 15 mg once daily and 30 mg once daily.
Compared to placebo at 12 weeks, upadacitinib 45 mg induction results in an increase in the proportion of patients who have clinical remission as measured by PROs, endoscopic response, endoscopic remission, both endoscopic remission and clinical remission as measured by CDAI, CR-100, and an improvement in HRQoL based on IBDQ. It also likely results in an increase in the proportion of patients who discontinued corticosteroid use for CD with concurrent clinical remission as measured by CDAI (in patients who were taking corticosteroids at baseline), and in the proportion with clinical remission as measured by CDAI. It likely results in an increase in the proportion of patients who have resolution of EIMs among those with EIMs at baseline, but little to no difference in CD-related hospitalization, or |||||||||| ||||||||| at 12 weeks. Compared to placebo at 52 weeks, upadacitinib 15 mg and 30 mg maintenance therapies were generally consistent with the induction-related outcomes, albeit with slightly more imprecision in some outcomes for the 15 mg dose, and for the outcome of IBDQ for either dose. The time frame was considered insufficient to detect differences in the outcomes of CD-related hospitalization and |||||||||| |||||||||. The evidence shows that upadacitinib likely results in little to no difference in SAEs compared to placebo, and no new safety signals were identified. Longer-term data are currently being collected in substudy 2 of the U-ENDURE trial, but results are not yet available.
There is a data gap with respect to head-to-head, direct evidence between upadacitinib and other advanced therapies for CD. Indirect evidence provided by the sponsor demonstrated |||||||||| |||||||||| || |||||||| || |||||| |||||||| between upadacitinib and other advanced therapies during induction or maintenance, but the ITCs |||||||| |||| ||||||||||| ||||||||||| || |||||||||| |||||||||||||| |||||||||| |||||||||| ||||||||||| for both efficacy and safety outcomes.
References
1.Colombel JF. Measuring disability in IBD: the IBD disability index. Gastroenterol Hepatol (N Y). 2013;9(5):300-302. PubMed
2.Høivik ML, Moum B, Solberg IC, Henriksen M, Cvancarova M, Bernklev T. Work disability in inflammatory bowel disease patients 10 years after disease onset: results from the IBSEN Study. Gut. 2013;62(3):368-375. PubMed
3.Le Berre C, Ananthakrishnan AN, Danese S, Singh S, Peyrin-Biroulet L. Ulcerative colitis and Crohn’s disease have similar burden and goals for treatment. Clin Gastroenterol Hepatol. 2020;18(1):14-23. PubMed
4.Cámara RJ, Ziegler R, Begré S, Schoepfer AM, von Känel R. The role of psychological stress in inflammatory bowel disease: quality assessment of methods of 18 prospective studies and suggestions for future research. Digestion. 2009;80(2):129-139. PubMed
5.Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19:5a-36a. PubMed
6.Baumgart DC, Sandborn WJ. Crohn's disease. Lancet. 2012;380(9853):1590-1605. PubMed
7.Wilburn J, Twiss J, Kemp K, McKenna SP. A qualitative study of the impact of Crohn's disease from a patient's perspective. Frontline Gastroenterol. 2017;8(1):68-73. PubMed
8.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785-1794. PubMed
9.Peyrin-Biroulet L, Loftus EV, Jr., Colombel JF, Sandborn WJ. The natural history of adult Crohn's disease in population-based cohorts. Am J Gastroenterol. 2010;105(2):289-297. PubMed
10.Benchimol EI, Bernstein CN, Bitton A, et al. The impact of inflammatory bowel disease in canada 2018: a scientific report from the Canadian Gastro-Intestinal Epidemiology Consortium to Crohn's and Colitis Canada. J Can Assoc Gastroenterol. 2019;2:S1-S5. PubMed
11.Kaplan GG, Bernstein CN, Coward S, et al. The impact of inflammatory bowel disease in Canada 2018: epidemiology. J Can Assoc Gastroenterol. 2019;2:S6-S16. PubMed
12.Coward S, Clement F, Benchimol EI, et al. A29 The rising prevalence of inflammatory bowel disease in Canada: analyzing the past to predict the future. J Can Assoc Gastroenterol. 2018;1(Suppl 2):47-48.
13.Cappello M, Morreale GC. The role of laboratory tests in Crohn's Disease. Clin Med Insights Gastroenterol. 2016;9:51-62. PubMed
14.Feuerstein JD, Cheifetz AS. Crohn Disease: epidemiology, diagnosis, and management. Mayo Clin Proc. 2017;92(7):1088-1103. PubMed
15.Iskandar HN, Ciorba MA. Biomarkers in inflammatory bowel disease: current practices and recent advances. Transl Res. 2012;159(4):313-325. PubMed
16.Kilcoyne A, Kaplan JL, Gee MS. Inflammatory bowel disease imaging: current practice and future directions. World J Gastroenterol. 2016;22(3):917-932. PubMed
17.Magro F, Langner C, Driessen A, et al. European consensus on the histopathology of inflammatory bowel disease. J Crohns Colitis. 2013;7(10):827-851. PubMed
18.Spiceland CM, Lodhia N. Endoscopy in inflammatory bowel disease: role in diagnosis, management, and treatment. World J Gastroenterol. 2018;24(35):4014-4020. PubMed
19.Stenczel ND, Purcarea MR, Tribus LC, Oniga GH. The role of the intestinal ultrasound in Crohn's disease diagnosis and monitoring. J Med Life. 2021;14(3):310-315. PubMed
20.Bell GM, Reynolds G, Isaacs JD. Biologic therapies in non-rheumatic diseases: lessons for rheumatologists? Nat Rev Rheumatol. 2011;7(9):507-516. PubMed
21.Schmitt H, Neurath MF, Atreya R. Role of the IL23/IL17 pathway in Crohn's disease. Front Immunol. 2021;12:622934. PubMed
22.Clinical Study Report M14-431: a multicenter, randomized, double-blind, placebo-controlled induction study of the efficacy and safety of upadacitinib (ABT-494) in subjects with moderately to severely active Crohn's Disease who have inadequately responded to or are intolerant to biologic therapy [internal sponsor's report]. St. Laurent (QC): AbbVie; 2022.
23.Clinical Study Report M14-430: a multicenter, randomized, double-blind, placebo-controlled maintenance and long-term extension study of the efficacy and safety of upadacitinib (ABT-494) in subjects with Crohn's Disease who completed the studies M14-431 or M14-433 [internal sponsor's report]. St. Laurent (AC): Abbvie; 2022.
24.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406. PubMed
25.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. PubMed
26.Clinical Study Report M14-433: a multicenter, randomized, double-blind, placebo controlled induction study of the efficacy and safety of upadacitinib (ABT-494) in subjects with moderately to severely Active Crohn's Disease who have inadequately responded to or are intolerant to conventional and/or biologic therapies [internal sponsor's report]. St. Laurent (QC): AbbVie; 2022.
27.Sponsor clinical summary of evidence: Rinvoq for the treatment of patients with moderate to severe active Crohn's Disease [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib): 15 mg, 30 mg, 45 mg extended release tablets. Saint-Laurent (QC): AbbVie Corporation; 2023 Jun 5.
28.The relative efficacy and safety of upadacitinib (upa) versus other advanced treatments for patients with active moderate-to-severe Crohn’s Disease (CD): a Bayesian Network Meta-analysis (NMA) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib): 15 mg, 30 mg, 45 mg extended release tablets. Saint-Laurent (QC): AbbVie Corporation; 2023 Jun 5.
29.Torres J, Mehandru S, Colombel JF, Peyrin-Biroulet L. Crohn's disease. Lancet. 2017;389(10080):1741-1755. PubMed
30.Boyapati R, Satsangi J, Ho GT. Pathogenesis of Crohn's disease. F1,000Prime Rep. 2015;7:44.
31.Liu Z, Yadav PK, Xu X, et al. The increased expression of IL-23 in inflammatory bowel disease promotes intraepithelial and lamina propria lymphocyte inflammatory responses and cytotoxicity. J Leukoc Biol. 2011;89(4):597-606. PubMed
32.Impact of inflammatory bowel disease in Canada. Toronto (ON): Crohn's and Colitis Canada; 2018: https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2018-Impact-Report-LR.pdf. Accessed 2023 Jun 20.
33.Best WR. Predicting the Crohn's Disease Activity Index from the Harvey-Bradshaw Index. Inflamm Bowel Dis. 2006;12(4):304-310. PubMed
34.Vermeire S, Schreiber S, Sandborn WJ, Dubois C, Rutgeerts P. Correlation between the Crohn's disease activity and Harvey-Bradshaw indices in assessing Crohn's disease severity. Clin Gastroenterol Hepatol. 2010;8(4):357-363. PubMed
35.Papay P, Ignjatovic A, Karmiris K, et al. Optimising monitoring in the management of Crohn's disease: a physician's perspective. J Crohns Colitis. 2013;7(8):653-669. PubMed
36.Williet N, Sandborn WJ, Peyrin-Biroulet L. Patient-reported outcomes as primary end points in clinical trials of inflammatory bowel disease. Clin Gastroenterol Hepatol. 2014;12(8):1246-1256.e1246. PubMed
37.Turner D, Ricciuto A, Lewis A, et al. STRIDE-II: an update on the selecting therapeutic targets in inflammatory bowel disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): determining therapeutic goals for treat-to-target strategies in IBD. Gastroenterology. 2021;160(5):1570-1583. PubMed
38.Lichtenstein GR, Loftus EV, Isaacs KL, Regueiro MD, Gerson LB, Sands BE. ACG clinical guideline: management of Crohn's Disease in adults. Am J Gastroenterol. 2018;113(4):481-517. PubMed
39.Torres J, Bonovas S, Doherty G, et al. ECCO guidelines on therapeutics in Crohn's disease: medical treatment. J Crohns Colitis. 2019;14(1):4-22. PubMed
40.Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O'Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;16(12):843-862. PubMed
41.Parmentier JM, Voss J, Graff C, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018;2:23. PubMed
42.Singh S, Kroe-Barrett RR, Canada KA, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. MAbs. 2015;7(4):778-791. PubMed
43.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585-600. PubMed
44.PrRinvoq® (upadacitinib): 15 mg, 30 mg, 45 mg extended-release oral tablets [product monograph]. St-Laurent (QC): AbbVie Corporation; 2023 Oct 11.
45.PrSkyrizi® (risankizumab): 150 mg in 1 mL (150 mg/mL), 360 mg in 2.4 mL (150 mg/mL), 75 mg in 0.83 mL (90 mg/mL) sterilie solution for subcutaneous injection; 600 mg in 10 mL (60 mg/mL) sterile solution for intravenous infusion [product monograph]. St-Laurent (QC): AbbVie; 2023 May 15: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/SKYRIZI_PM_EN.pdf. Accessed 2023 Dec 6.
46.PrEntyvio® (vedolizumab): 300 mg / vial powder for concentrate for solution for intravenous infusion, 108 mg / 0.68 mL solution for subcutaneous injection in a pre-filled syringe or pen [product monograph]. Toronto (ON): Takeda Canada; 2020 Sep 10: https://pdf.hres.ca/dpd_pm/00058003.PDF. Accessed 2023 Dec 6.
47.PrRemicade® (infliximab): 100 mg/vial powder for solution for intravenous infusion [product monograph]. Toronto (ON): Janssen; 2021.
48.PrHumira® (adalimumab): 40 mg in 0.8 mL (50 mg/mL), 10 mg in 0.1 mL (100 mg/mL), 20 mg in 0.2 mL (100 mg/mL), 40 mg in 0.4 mL (100 mg/mL), 80 mg in 0.8 mL (100 mg/mL) sterile solution for subcutaneous injection [product monograph]. St. Laurent (QC): AbbVie; 2022 Sep 16.
49.PrStelara® (ustekinumab): 45 mg/0.5 mL, 90 mg/1.0 mL solution for subcutaneous injection, 130 mg/26 mL (5 mg/mL) solution for intravenous infusion [product monograph]. Toronto (ON): Janssen; 2018 Nov 9: https://pdf.hres.ca/dpd_pm/00048246.PDF. Accessed 2023 Dec 6.
50.Best WR, Becktel JM, Singleton JW, Kern F, Jr. Development of a Crohn's Disease activity index. National cooperative Crohn's Disease study. Gastroenterology. 1976;70(3):439-444. PubMed
51.Peyrin-Biroulet L, Panés J, Sandborn WJ, et al. Defining disease severity in inflammatory bowel diseases: current and future directions. Clin Gastroenterol Hepatol. 2016;14(3):348-354.e317. PubMed
52.Peyrin-Biroulet L, Reinisch W, Colombel JF, et al. Clinical disease activity, C-reactive protein normalisation and mucosal healing in Crohn's disease in the SONIC trial. Gut. 2014;63(1):88-95. PubMed
53.Yoshida EM. The Crohn's disease activity index, its derivatives and the inflammatory bowel disease questionnaire: a review of instruments to assess Crohn's disease. Can J Gastroenterol. 1999;13(1):65-73. PubMed
54.Daperno M, D'Haens G, Van Assche G, et al. Development and validation of a new, simplified endoscopic activity score for Crohn's Disease: the SES-CD. Gastrointest Endosc. 2004;60(4):505-512. PubMed
55.Irvine EJ. Development and subsequent refinement of the inflammatory bowel disease questionnaire: a quality-of-life instrument for adult patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1999;28(4):S23-27. PubMed
56.Guyatt G, Mitchell A, Irvine EJ, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96(3):804-810. PubMed
57.Sandborn WJ, Feagan BG, Hanauer SB, et al. A review of activity indices and efficacy end points for clinical trials of medical therapy in adults with Crohn's disease. Gastroenterology. 2002;122(2):512-530. PubMed
58.Feagan BG, Rochon J, Fedorak RN, et al. Methotrexate for the treatment of Crohn's disease. The North American Crohn's Study Group Investigators. N Engl J Med. 1995;332(5):292-297. PubMed
59.Khanna R, Zou G, D'Haens G, et al. A retrospective analysis: the development of patient reported outcome measures for the assessment of Crohn's disease activity. Aliment Pharmacol Ther. 2015;41(1):77-86. PubMed
60.Best WR, Becktel JM, Singleton JW. Rederived values of the eight coefficients of the Crohn's disease activity index (CDAI). Gastroenterology. 1979;77(4 Pt 2):843-846. PubMed
61.Zheng X, Li M, Wu Y, et al. Assessment of pediatric Crohn's disease activity: validation of the magnetic resonance enterography global score (MEGS) against endoscopic activity score (SES-CD). Abdom Radiol. 2020;45(11):3653-3661. PubMed
62.Khanna R, Nelson SA, Feagan BG, et al. Endoscopic scoring indices for evaluation of disease activity in Crohn's disease. Cochrane Database Syst Rev. 2016(8):CD010642. PubMed
63.Khanna R, Zou G, D'Haens G, et al. Reliability among central readers in the evaluation of endoscopic findings from patients with Crohn's disease. Gut. 2016;65(7):1119-1125. PubMed
64.Pallis AG, Mouzas IA, Vlachonikolis IG. The inflammatory bowel disease questionnaire: a review of its national validation studies. Inflamm Bowel Dis. 2004;10(3):261-269. PubMed
65.Chen XL, Zhong LH, Wen Y, et al. Inflammatory bowel disease-specific health-related quality of life instruments: a systematic review of measurement properties. Health Qual Life Outcomes. 2017;15(1):177. PubMed
66.Gregor JC, McDonald JW, Klar N, et al. An evaluation of utility measurement in Crohn's disease. Inflamm Bowel Dis. 1997;3(4):265-276. PubMed
67.Brooks R. The EuroQol Group after 25 years. Dordrecht (NL): Springer; 2012.
68.Feng YS, Kohlmann T, Janssen MF, Buchholz I. Psychometric properties of the EQ-5D-5L: a systematic review of the literature. Qual Life Res. 2021;30(3):647-673. PubMed
69.Buchholz I, Janssen MF, Kohlmann T, Feng YS. A systematic review of studies comparing the measurement properties of the three-level and five-level versions of the EQ-5D. Pharmacoeconomics. 2018;36(6):645-661. PubMed
70.Sinnott PL, Joyce VR, Barnett P. Guidebook: preference measurement in economic analysis. Menlo Park (CA): Health Economics Resource Center; 2007: https://www.herc.research.va.gov/files/BOOK_419.pdf. Accessed 2023 Dec 6.
71.Rencz F, Lakatos PL, Gulácsi L, et al. Validity of the EQ-5D-5L and EQ-5D-3L in patients with Crohn's disease. Qual Life Res. 2019;28(1):141-152. PubMed
72.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: a generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. Sheffield (UK): University of Sheffield; 2011: https://www.sheffield.ac.uk/nice-dsu/tsds/full-list. Accessed 2023 Jun 20.
73.Higgins J, Thomas J, Chandler J, et al. Cochrane handbook for systematic reviews of interventions. 2nd edition. Chichester (UK): John Wiley & Sons; 2019.
74.Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE DSU Technical Support Document 4: inconsistency in networks of evidence based on randomized controlled trials. Sheffield (UK): University of Sheffield; 2011: https://www.sheffield.ac.uk/nice-dsu/tsds/full-list. Accessed 2023 Jun 20.
75.Chen B, Gao X, Zhong J, et al. Efficacy and safety of adalimumab in Chinese patients with moderately to severely active Crohn's Disease: results from a randomized trial. Therap Adv Gastroenterol. 2020;13:1756284820938960. PubMed
76.Ustekinumab for moderately to severely active Crohn’s disease after previous treatment. NICE technology appraisal guidance [TA456]. London (UK): National Institute for Health and Care Excellence; 2017: https://www.nice.org.uk/guidance/ta456/resources/ustekinumab-for-moderately-to-severely-active-crohns-disease-after-previous-treatment-pdf-82604848449733. Accessed 2023 Jun 20.
77.Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet. 2002;359(9317):1541-1549. PubMed
78.Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology. 2007;132(1):52-65. PubMed
79.Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006;130(2):323-333; quiz 591. PubMed
80.Sandborn WJ, Rutgeerts P, Enns R, et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann Intern Med. 2007;146(12):829-838. PubMed
81.Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. 2013;369(8):711-721. PubMed
82.Sands BE, Sandborn WJ, Van Assche G, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease in patients naïve to or who have failed tumor necrosis factor antagonist therapy. Inflamm Bowel Dis. 2017;23(1):97-106. PubMed
83.Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn's disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147(3):618-627.e613. PubMed
84.Feagan BG, Sandborn WJ, Gasink C, et al. Ustekinumab as induction and maintenance therapy for Crohn's Disease. N Engl J Med. 2016;375(20):1946-1960. PubMed
85.Rutgeerts P, Gasink C, Chan D, et al. Efficacy of ustekinumab for inducing endoscopic healing in patients with Crohn's disease. Gastroenterology. 2018;155(4):1045-1058. PubMed
86.Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med. 1997;337(15):1029-1035. PubMed
87.Vermeire S, D'Haens GR, Baert FJ, et al. 950 The visible 2 phase 3 study of efficacy and safety of vedolizumab SC for moderate-to-severe Crohn's disease [abstract]. Gastroenterology. 2020;158(6 Suppl 1):S-194.
88.Vermeire S, D’Haens G, Baert F, et al. Efficacy and safety of subcutaneous vedolizumab in patients with moderately to severely active Crohn’s disease: results from the VISIBLE 2 randomised trial. J Crohns Colitis. 2022;16(1):27-38. PubMed
89.Watanabe M, Hibi T, Lomax KG, et al. Adalimumab for the induction and maintenance of clinical remission in Japanese patients with Crohn's disease. J Crohns Colitis. 2012;6(2):160-173. PubMed
90.Watanabe K, Motoya S, Ogata H, et al. Effects of vedolizumab in Japanese patients with Crohn's disease: a prospective, multicenter, randomized, placebo-controlled Phase 3 trial with exploratory analyses. J Gastroenterol. 2020;55(3):291-306. PubMed
Appendix 1: Additional Outcome Data
Note the tables in this appendix have not been copy-edited.
Table 34: EQ-5D-5L Index Value Results at 12 Weeks [Redacted]
|||||||| | ||||||||| |||| ||||||||||| |||| | | |||||||||||| ||||||||||| |||| | | ||
|---|---|---|---|---|
|||||||||||| || | | ||||||| | |||||||||||| || | ||||||| | |
|||||||| |||| | ||| | ||| | ||| | ||| |
|||||||| |||| | ||||| | ||||| | ||||| | ||||| |
||||| |||| | ||||| | ||||| | ||||| | ||||| |
|||||| |||| |||||||| |||||||||||||| | ||||
||||| ||||||| |||| | ||||| | ||||| | ||||| | ||||| |
||| | | ||||| || ||||| | ||||| || ||||| | ||||| || ||||| | ||||| || ||||| |
|||||||| ||||| | |||||| | |||||| | |||||| | |||||| |
|||||| |||| |||||||| |||||||||||||| |||||||||||| || || || || |||||||| | ||||
||||| ||||||| |||| | ||||| | ||||| | ||
||| | | ||||| || ||||| | ||||| || ||||| | ||
|||||||| ||||| | |||||| | |||||| | ||
||||||| | ||||||| | ||||||| | ||
||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||| |||||||| |||||| | || |||| |||||||||| |||||| |||||||||||||||||| ||||||||||| |||||||| || | |||||| || |||||||||||| |||||||||| |||||| | ||||||||||||| |||||||| ||||| ||||||| || |||||||| | ||||||
Note: This table has been redacted at the request of the sponsor.
Table 35: EQ-5D-5L Index Value Results at 52 Weeks [Redacted]
|||||||| | ||||||||||||||| || |||| ||||||||||| |||| | | ||
|---|---|---|---|
|||||||||||| || | | |||||||||||| || | | ||||||| | |
|||||| |||| ||||||||| |||||||| |||||||||||||| | |||
|||||||| |||| | || | || | || |
||||||||| |||||||| |||| | ||||| | ||||| | ||||| |
||||| ||||||| |||| | ||||| | ||||| | ||||| |
||| | | |||||| ||||| | |||||| ||||| | |||||| ||||| |
|||||||| ||||| | |||||| | |||||| | |||||| |
|||||| |||| ||||||||| |||||||| |||||||||||||| || |||||||| | |||
||||| ||||||| |||| | ||||| | ||||| | || |
||| | | |||||| ||||| | ||||||| ||||| | || |
|||||||| ||||| | |||||| | |||||| | || |
||||||| | |||||| | |||||| | || |
|||||| |||| |||||||| |||| ||||||||||||||| | |||
|||||||| |||| | || | || | || |
|||| ||||||||| |||| | ||||| | ||||| | ||||| |
||||| ||||||| |||| | ||||| | |||||| | |||||| |
||| | | |||||| ||||| | ||||||| ||||| | ||||||| ||||| |
|||||||| ||||| | |||||| | |||||| | |||||| |
|||||| |||| |||||||| |||| |||||||||||||| | |||||||| | |||
||||| ||||||| |||| | ||||| | ||||| | || |
||| | | |||||| ||||| | ||||||| ||||| | || |
|||||||| ||||| | |||||| | |||||| | || |
||||||| | |||||| | |||||| | || |
||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||| |||||||| |||||| | || |||| |||||||||| |||||| |||||||||||||||||| ||||||||||| |||||||| || | |||||| || |||||||||||| |||||||||| |||||| | ||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||| ||||||||
Note: This table has been redacted at the request of the sponsor.
Subgroup Results
Table 36: Subgroup Results (Number of Prior Biologics Failed) for Primary Efficacy Outcomes in U-EXCEED
Outcome | UPA 45 mg q.d. | Placebo |
|---|---|---|
Clinical Remission as measured by PROs at week 12, ITT1 population; NRI-NC | ||
Subgroup: ≤ 1 Prior Biologics Failed | ||
N responders/total N (% [95% CI]) | 59/126 (46.8 [38.1 to 55.5]) | 14/68 (20.6 [11.0 to 30.2]) |
Response-rate difference compared to placebo percentage (95% CI) | 26.2 (13.3 to 39.2) | |
Subgroup: > 1 Prior Biologics Failed | ||
N responders/total N (% [95% CI]) | 10/103 (9.7 [4.0 to 15.4]) | 70/198 (35.4 [28.7 to 42.0]) |
Response-rate difference compared to placebo percentage (95% CI) | 28.0 (17.2 to 38.7) | |
Clinical Remission as measured by CDAI at week 12, ITT1 population; NRI-NC | ||
Subgroup: ≤ 1 Prior Biologics Failed | ||
N responders/total N (% [95% CI]) | 58/126 (46.0 [37.3 to 54.7]) | 20/68 (29.4 [18.6 to 40.2]) |
Response-rate difference compared to placebo percentage (95% CI) | 16.6 (2.7, 30.5) | |
Subgroup: > 1 Prior Biologics Failed | ||
N responders/total N (% [95% CI]) | 68/198 (34.3 [27.7 to 40.9]) | 16/103 (15.5 [8.5, 22.5]) |
Response-rate difference compared to placebo percentage (95% CI) | 18.8 (9.1 to 28.4) | |
Endoscopic response at week 12, ITT1 population; NRI-C | ||
Subgroup: ≤ 1 Prior Biologics Failed | ||
N responders/total N (% [95% CI]) | 60/126 (47.6 [38.9 to 56.4]) | 3/68 (4.4 [0.0 to 9.3]) |
Response-rate difference compared to placebo percentage (95% CI) | 43.2 (33.2 to 53.2) | |
Subgroup: > 1 Prior Biologics Failed | ||
N responders/total N (% [95% CI]) | 52/198 (26.3 [20.1 to 32.4]) | 3/103 (2.9 [0.0 to 6.2]) |
Response-rate difference compared to placebo percentage (95% CI) | 36.3 (28.1 to 44.4) | |
CDAI = Crohn Disease Activity Index; CI = confidence interval; ITT1 = intention-to-treat part one; LS = least squares; NRI-C = nonresponder imputation while incorporating multiple imputation to handle missing data due to COVID-19; q.d. = once daily; PRO = patient-reported outcome; UPA = upadacitinib.
Sources: U-EXCEED Clinical Study Report.26 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 37: Subgroup Results (Number of Prior Biologics Failed) for Primary Efficacy Outcomes in U-EXCEL
Outcome | UPA 45 mg q.d. | Placebo |
|---|---|---|
Clinical remission as measured by PROs at week 12, ITT1 population; NRI-NC | ||
Subgroup: 0 Prior Biologics Failed (Non-Bio-IR Population) | ||
N responders/total N (% [95% CI]) | 102/189 (54.1 [47.0 to 61.2]) | 28/98 (28.6 [19.6, 37.5]) |
Response-rate difference compared to placebo percentage (95% CI) | 25.5 (14.1 to 36.9) | |
Subgroup: ≥ 1 Prior Biologics Failed (Bio-IR Population) | ||
N responders/total N (% [95% CI]) | 75/161 (46.8 [39.1 to 54.6]) | 11/78 (14.1 [6.4 to 21.8]) |
Response-rate difference compared to placebo percentage (95% CI) | 32.7 (21.8 to 43.7) | |
Subgroup: 1 Prior Biologic Failed (Within Bio-IR Population) | ||
N responders/total N (% [95% CI]) | 32/58 (55.9 [43.0 to 68.8]) | 6/28 (21.4 [6.2 to 36.6]) |
Response-rate difference compared to placebo percentage (95% CI) | 34.4 (14.5 to 54.4) | |
Clinical Remission as measured by CDAI at week 12, ITT1 population; NRI-NC | ||
Subgroup: 0 Prior Biologics Failed (Non-Bio-IR Population) | ||
N responders/total N (% [95% CI]) | 103/189 (54.3 [47.1 to 61.4]) | 39/98 (39.8 [30.1 to 49.5]) |
Response-rate difference compared to placebo percentage (95% CI) | 14.5 (2.4 to 26.5) | |
Subgroup: ≥ 1 Prior Biologics Failed (Bio-IR Population) | ||
N responders/total N (% [95% CI]) | 71/161 | 12/78 (15.6 [7.5 to 23.8]) |
Response-rate difference compared to placebo percentage (95% CI) | 28.3 (17.0 to 39.5) | |
Subgroup: 1 Prior Biologic Failed (Within Bio-IR Population) | ||
N responders/total N (% [95% CI]) | 30/58 (51.5 [38.6 to 64.4]) | 8/28 (28.6 [11.8 to 45.3]) |
Response-rate difference compared to placebo percentage (95% CI) | 22.9 (1.8 to 44.1) | |
Endoscopic response at week 12, ITT1 population; NRI-C | ||
Subgroup: 0 Prior Biologics Failed (Non-Bio-IR Population) | ||
N responders/total N (% [95% CI]) | 98/189 (52.0 [44.9, 59.2]) | 16/98 (16.3 [9.0 to 23.6]) |
Response-rate difference compared to placebo percentage (95% CI) | 14.5 (−8.8 to 37.7) | |
Subgroup: ≥ 1 Prior Biologics Failed (Bio-IR Population) | ||
N responders/total N (% [95% CI]) | 61/171 (37.9 [30.4 to 45.4]) | 7/78 (9.0 [2.6, 15.3]) |
Response-rate difference compared to placebo percentage (95% CI) | 29.0 (19.1 to 38.8) | |
Subgroup: 1 Prior Biologic Failed (Within Bio-IR Population) | ||
N responders/total N (% [95% CI]) | 27/58 (46.6 [33.7 to 59.4]) | 4/28 (14.3 [1.3 to 27.2]) |
Response-rate difference compared to placebo percentage (95% CI) | 32.3 (14. to, 50.5) | |
CDAI = Crohn Disease Activity Index; CI = confidence interval; ITT1 = intention-to-treat part one; LS = least square; mg = milligrams; N = number; NRI-C = nonresponder imputation while incorporating multiple imputation to handle missing data due to COVID-19; q.d. = once daily; PRO = patient-reported outcome; UPA = upadacitinib.
Sources: U-EXCEL Clinical Study Report.22 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 38: Subgroup Results for Primary Efficacy Outcomes in U-ENDURE
Outcome | UPA 15 mg q.d. | UPA 30 mg q.d. | Placebo |
|---|---|---|---|
Clinical Remission as measured by PROs at week 52, ITT1 Population; NRI-C | |||
Subgroup: 0 Prior Biologics Failed (Non-Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 20/45 (44.4 [29.9 to 59.0]) | 13/39 (32.7 [17.8 to 47.6]) | 13/39 (32.7 [17.8 to 47.6]) |
Response-rate difference compared to placebo percentage (95% CI) | 11.7 (−9.1 to 32.5) | 25.8 (4.6 to 47.0) | NA |
Subgroup: ≥ 1 Prior Biologics Failed (Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 40/124 (32.3 [24.0 to 40.5]) | 54/127 (42.5 [33.9 to 51.1]) | 11/126 (8.7 [3.8 to 13.7]) |
Response-rate difference compared to placebo percentage (95% CI) | 23.5 (13.9 to 33.1) | 33.8 (23.8 to 43.7) | NA |
Subgroup: 1 Prior Biologic Failed (Within Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 18/52 (34.6 [21.7 to 47.5]) | 24/43 (55.8 [41.0 to 70.7]) | 8/52 (15.4 [5.6 to 25.2]) |
Response-rate difference compared to placebo percentage (95% CI) | 19.2 (3.0 to 35.5) | 40.4 (22.6 to 58.2) | NA |
Subgroup: > 1 Prior Biologic Failed (Within Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 22/72 (30.6 [19.9 to 41.2]) | 30/84 (35/7 [25.5 to 46.0]) | 3/74 (4.1 [0.0 to 8.5]) |
Response-rate difference compared to placebo percentage (95% CI) | 26.5 (20.5 to 42.8) | 26.5 (15.0 to 38.1) | NA |
Clinical Remission as measured by CDAI at Week 52, ITT1 Population; NRI-C | |||
Subgroup: 0 Prior Biologics Failed (Non-Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 21/45 (46.7 [32.1 to 61.2]) | 23/41 (56.1 [40.9 to 71.3]) | 10/39 (25.5 [11.7 to 39.2]) |
Response-rate difference compared to placebo percentage (95% CI) | 21.2 (1.2 to 41.2) | 30.6 (10.1 to 51.1) | NA |
Subgroup: ≥ 1 Prior Biologics Failed (Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 42/124 (33.9 [25.5 to 42.2]) | 57/127 (44.9 [36.2 to 53.5]) | 15/126 (11.9 [6.3 to 17.6]) |
Response-rate difference compared to placebo percentage (95% CI) | 22.0 (11.9 to 32.0) | 33.0 (22.6 to 43.3) | NA |
Subgroup: 1 Prior Biologic Failed (Within Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 20/52 (38.5 [25.2 to 51.7]) | 25/43 (58.1 [43.4 to 72.9]) | 7/52 (13.5 [4.2 to 22.7]) |
Response-rate difference compared to placebo percentage (95% CI) | 25.0 (8.8 to 41.2) | 44.7 (27.3 to 62.1) | NA |
Subgroup: > 1 Prior Biologic Failed (Within Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 22/72 (30.6 [19.9 to 41.2]) | 32/84 (38.1 [27.7 to 48.5]) | 8/74 (10.8 [3.7 to 17.9]) |
Response-rate difference compared to placebo percentage (95% CI) | 19.7 (7.0 to 32.5) | 27.3 (14.7 to 39.9) | NA |
Endoscopic response at week 52, ITT1 population; NRI-NC | |||
Subgroup: 0 Prior Biologics Failed (Non-Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 18/45 (39.8 [25.0 to 54.5]) | 18/41 (43.9 [28.7 to 59.1]) | 7/39 (17.9 [5.9 to 30.0]) |
Response-rate difference compared to placebo percentage (95% CI) | 21.8 (2.8 to 40.9) | 26.0 (6.6 to 45.3) | NA |
Subgroup: ≥ 1 Prior Biologics Failed (Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 29/129 (23.2 [15.7 to 30.6]) | 49/127 (38.9 [30.4 to 47.4]) | 5/126 (4.0 [0.6 to 7.4]) |
Response-rate difference compared to placebo percentage (95% CI) | 19.2 (11.0 to 27.4) | 34.9 (25.9 to 44.1) | NA |
Subgroup: 1 Prior Biologic Failed (Within Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 13/52 (25.0 [13.2 to 36.8]) | 23/43 (54.4 [39.4 to 69.5]) | 4/52 (7.7 [0.4 to 14.9]) |
Response-rate difference compared to placebo percentage (95% CI) | 17.3 (3.5 to 31.1) | 46.7 (30.0 to 63.4) | NA |
Subgroup: > 1 Prior Biologic Failed (Within Bio-IR Population) | |||
N responders/total N (% [95% CI]) | 16/72 (21.9 [12.2 to 31.5]) | 26/84 (31.0 [21.1 to 40.8]) | 1/74 (1.4 [0.0 to 4.0]) |
Response-rate difference compared to placebo percentage (95% CI) | 20.5 (10.5 to 30.5) | 29.6 (19.4 to 39.8) | NA |
CDAI = Crohn Disease Activity Index; CI = confidence interval; ITT1 = intention-to-treat part one; LS = least square; mg = milligrams; N = number; NA = not applicable; NRI-NC = nonresponder imputation while incorporating multiple imputation to handle missing data due to COVID-19; q.d. = once daily; PRO = patient-reported outcome; UPA = upadacitinib.
Note: Study M14 to 431 = U-EXCEED; Study M14 to 433 = U-EXCEL.
Sources: U-ENDURE Clinical Study Report.23 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Additional Data Related to the ITC
Table 39: RCTs Meeting the Inclusion Criteria for the ITC [Redacted]
||||| | |||||| || | |||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||| | |||||| || |
|---|---|---|---|---|---|---|---|---|---|---|---|
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
|||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||||||| | |||||| || | ||||| | |||||| || | |||||| || | |||||| || | |||||| || | |||||| || | |||||| | |||||| | |||||| | |||||| || |
||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||| |||||||| |||||| | || |||| |||||||||| |||||||||||||||||||||| ||||||||||| |||||||| || | |||||| || |||||||||||| |||||||||| |||||| | ||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| |
Note: This table has been redacted at the request of the sponsor.
Table 40: Summary of Baseline Characteristics at Induction for Studies in the ITC [Redacted]
||||| | |||||||||| | ||||||||| | |||| | ||||| | | |||| ||||||| | |||| ||||| | |||||| | ||||||| | |||||| | ||| | ||||| | | |||||| | |||| |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
||||||| ||||||||| ||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||| ||||||||| ||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||| |||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||| |||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||| |||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||| |||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||| |||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| |||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| |||||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| |||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||| ||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||||| |||| ||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||||| |||| ||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||||| |||| ||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||||| |||| ||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||||| |||| ||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| |||||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| |||||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| |||||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||| ||||||||||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||| ||||||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||| ||||||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||| ||||| ||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||| ||||| ||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||| ||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||| ||||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||||| ||||||||| |||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
|||||||| ||||||||| || | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||| ||||| ||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||| ||||| ||| | ||||||||| | |||| | |||| | ||||||||| | ||||||||| | ||||||||| | |||| | |||| | |||| | ||||||||| | |||| | |||| |
||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||| |||||||| |||||| | || |||| |||||||||| |||||| |||||||||||||||||| ||||||||||| ||||||||
Note: This table has been redacted at the request of the sponsor.
Table 41: Summary of Baseline Characteristics at Maintenance for Studies in the ITC [Redacted]
||||| ||||||| | ||||||||| | |||| | ||||| | | ||||| | |||| |||| | |||||| | ||||| | ||||||| | |||| || | ||||| ||||| | |||||| | |||| |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|||||| ||||| |||| | ||||||||||||||| | ||| | ||| | | |||||| | || | ||| ||||| | ||| | ||| | ||| | ||| | ||| | || |
||||| ||||||||| | ||||||||||||||| | ||| | ||| | |||| |||| | |||| ||||| | || | ||| | ||| | ||| | ||| | ||| | ||| |
||||||| ||||| | | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
||||||| ||||| | | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||| | | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||| | | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||| | | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| ||||||||| | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| ||||||||| | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| ||||||||| | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| ||||||||| | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| ||||||||| | ||||||||||||||| | ||| | ||| | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| ||||||||| | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
||||||| | | ||||||||||||||| | ||| | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
||||||| | | ||||||||||||||| | ||| | ||| | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| |||| | ||||||||| ||| | || | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||| |||| | ||||||||||||| | || | || | | |||||| | | |||||| | | |||||| | ||| | ||| | ||| | ||| | || | || |
|||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||| |||||||| |||||| | || |||| |||||||||| |||||| |||||||||||||||||| ||||||||||| ||||||||| |||| ||||||||||| ||||| |||||| |||||||||||| || |||||| || |||||||||||| ||||| |||||| ||||||||||| |||| |||||||| |||||| |||||||||||||| ||||||| ||||||||| |||||||||||||| |||| || |||| |||||||||| ||||||| |||||||| ||||||| || || |||| | |||||| || ||||||||||||||| ||||||||| |||| |||||||||||| ||||| |||||| ||
Note: This table has been redacted at the request of the sponsor.
Figure 4: Network of Included RCTs With Induction-Phase CDAI Outcomes for CCF Patients [Redacted]

Note: This figure has been redacted at the request of the sponsor.
Figure 5: Network of Included RCTs With Induction-Phase CDAI Outcomes for BF Patients [Redacted]

Note: This figure has been redacted at the request of the sponsor.
Figure 6: Network of Included RCTs With Induction-Phase Endoscopic Outcomes for CCF Patients [Redacted]

Note: This figure has been redacted at the request of the sponsor.
Figure 7: Network of Included RCTs With Induction-Phase Endoscopic Outcomes for BF Patients [Redacted]

Note: This figure has been redacted at the request of the sponsor.
Figure 8: Network of Included RCTs With Induction-Phase Safety Outcomes for All Patients [Redacted]

Note: This figure has been redacted at the request of the sponsor.
Figure 9: Network of Included RCTs With Maintenance-Phase CDAI Outcomes for CCF Patients [Redacted]

Note: This figure has been redacted at the request of the sponsor.


Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BF
biologic failure
CCF
conventional care failure
CD
Crohn disease
CDAI
Crohn’s Disease Activity Index
CR-100
decrease of at least 100 points from baseline in the Crohn’s Disease Activity Index
ICER
incremental cost-effectiveness ratio
IL
interleukin
NMA
network meta-analysis
pCPA
pan-Canadian Pharmaceutical Alliance
QALY
quality-adjusted life-year
UPA15
upadacitinib 45 mg induction dose and 15 mg maintenance dose
UPA30
upadacitinib 45 mg induction dose and 30 mg maintenance dose
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Upadacitinib (Rinvoq), 15 mg, 30 mg, 45 mg extended-release oral tablets |
Submitted price | Upadacitinib 15 mg: $51.68 per tablet Upadacitinib 30 mg: $76.96 per tablet Upadacitinib 45 mg: $101.81 per tablet |
Indication | For the treatment of adult patients with moderately to severely active Crohn disease who have demonstrated prior treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 12, 2023 |
Reimbursement request | As per indication |
Sponsor | AbbVie Corporation |
Submission history | Previously reviewed: Yes Ankylosing spondylitis:
Atopic dermatitis:
Psoriatic arthritis:
Rheumatoid arthritis:
|
NOC = Notice of Compliance; DMARD = disease-modifying antirheumatic drug; NSAID = nonsteroidal anti-inflammatory drug.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree followed by a Markov model |
Target population | Patients with moderately to severely active Crohn disease with an inadequate response, loss of response, or intolerance to conventional care (CCF subgroup) or biologic therapy (BF subgroup). |
Treatments | Upadacitinib 45 mg for induction plus 15 mg for maintenance. Upadacitinib 45 mg for induction plus 30 mg for maintenance. |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (60 years) |
Key data sources | Network meta-analyses; effectiveness of upadacitinib informed by the U-EXCEED, U-EXCEL, and U-ENDURE trials. |
Submitted results | CCF subgroup:
BF subgroup:
|
Key limitations |
|
CADTH reanalysis results |
|
BF = biologic failure; CCF = conventional care failure; CD = Crohn disease; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose.
Conclusions
Based on the CADTH clinical review, upadacitinib results in an increase in the proportion of adult patients who experience a clinical response and clinical remission among those with moderately to severely active Crohn disease (CD) and a history of treatment failure compared with placebo. There are no direct head-to-head trials comparing upadacitinib with biologic treatments for CD in either the conventional care failure (CCF) or biologic failure (BF) subgroup, and indirect evidence submitted by the sponsor suggests that there may be no meaningful difference in efficacy or safety between upadacitinib and other advanced treatments during either the induction or maintenance phases. However, substantial imprecision and unresolved heterogeneity were noted in the sponsor’s network meta-analyses (NMAs), precluding meaningful conclusions for both efficacy and safety outcomes among treatments.
Based on the sponsor’s base case, upadacitinib is not a cost-effective treatment for moderately to severely active CD in either the CCF or BF subgroup. Given that the sponsor-submitted NMAs suggest that there may be no difference between upadacitinib and biologic drugs in terms of clinical response and clinical remission, there is insufficient evidence to suggest that upadacitinib should be priced higher than currently available biologic treatments for moderately to severely active CD. To ensure cost-effectiveness, upadacitinib should be priced no more than the lowest-cost biologic that is funded in the population to be reimbursed.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient group input was received from the Gastrointestinal Society and Crohn and Colitis Canada. Patient input described the symptoms of CD, including diarrhea, rectal bleeding, abdominal pain, unpredictable and urgent bowel movements, bloating, pain, and fatigue, as well as inflammation of the eyes or joints, ulcers of the mouth or skin, tender and inflamed nodules on the shins, anemia, anxiety, and stress. Respondents noted that current treatments were not convenient due to storage requirements for at-home treatments and travel time and expenses required for treatments administered at a clinic. A desire was expressed for a treatment that alleviates symptoms and reduces flares, is easy to administer and noninvasive, has minimal side effects, and can improve their ability to work. Respondents with upadacitinib experience reported improved quality of life and health due to symptom improvement and the ability to taper off corticosteroids. Reported side effects included weight gain and infected hair follicles.
Clinician input was received from the Canadian Inflammatory Bowel Disease Specialist Group. The group noted that current treatments for CD include corticosteroids (prednisone, budesonide, and methylprednisolone), immunomodulators (azathioprine, 6-mercaptopurine, and methotrexate), anti–tumour necrosis factors (infliximab and adalimumab), anti–interleukin (IL)-12/IL-23 (ustekinumab), anti-integrin (vedolizumab), anti–IL-23 (risankizumab), and surgery. The group noted that endoscopy is the gold standard in detecting active disease, and noted the importance of treatments that achieve improvement in endoscopic appearance (endoscopic response), endoscopic remission, and mucosal healing, as achieving these metrics reduces flares, hospitalizations, and surgeries, and improves patients’ quality of life and reduces future disability. The group noted that the Crohn Disease Activity Index (CDAI) is used in clinical trials but does not fully capture disease severity as it does not necessarily reflect disease prognosis or course and correlates poorly with endoscopic disease severity. The clinician group noted that upadacitinib could be used as a first-line advanced therapy for patients with moderately to severely active CD or as later-line therapy for those who had an inadequate response to 1 or more previous biologic therapies. Clinicians pointed out that herpes zoster infections can occur with upadacitinib but that this can be mitigated by vaccination.
CADTH participating drug plans noted the absence of head-to-head trials comparing upadacitinib with other therapies used for the treatment of CD and that the upadacitinib monograph contains serious warnings and precautions pertaining to an increased risk of infections, malignancy, and thromboses. Drug plans commented on the issues involved in discontinuing therapy as it was unclear whether a patient would need to repeat induction dosing if there is an interruption in upadacitinib treatment. Drug plans noted the presence of confidential prices for an adalimumab biosimilar, infliximab, an infliximab biosimilar, and vedolizumab, and noted that risankizumab was omitted from the sponsor’s base case.
Several of these concerns were addressed in the sponsor’s model:
Treatment response was modelled in the sponsor’s submission based on CDAI score. Endoscopic response was incorporated in scenario analyses.
Quality of life was incorporated in the sponsor’s model by use of the EQ-5D questionnaire data captured in the upadacitinib trials. However, the EQ-5D is unlikely to capture all symptoms of CD that were noted by patients to affect quality of life.
The cost-effectiveness of upadacitinib compared to biologic drugs for CD (excluding risankizumab) was considered, although uncertainty in the indirect clinical data limits conclusions that can be drawn.
Loss of productivity was included in a scenario analysis.
In addition, CADTH addressed some of these concerns by including risankizumab in a scenario analysis.
CADTH was unable to address the following concerns raised from stakeholder input:
Risankizumab was included in a scenario analysis.
Herpes zoster infections in the economic model; costs related to vaccination were not included.
Economic Review
The current review is for upadacitinib (Rinvoq) for adult patients with moderately to severely active CD who have demonstrated prior treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis that compared upadacitinib with an adalimumab biosimilar, an infliximab biosimilar, vedolizumab, vedolizumab subcutaneous, ustekinumab, and conventional care (assumed to comprise corticosteroids [prednisone and budesonide], aminosalicylates [mesalazine and sulfasalazine], and immunomodulators [azathioprine, methotrexate, and 6-mercaptopurine]).1 The sponsor submitted 2 cost-utility analyses to assess the cost-effectiveness of upadacitinib among subgroups of patients with moderately to severely active CD who have demonstrated prior treatment failure (i.e., inadequate response, loss of response, or intolerance) to either conventional care (hereafter referred to as the conventional care failure [CCF] subgroup) and those who have demonstrated the same treatment failure to biologic therapy (the biologic failure [BF] subgroup). The modelled CCF and BF subgroups are in line with the reimbursement request and Health Canada–indicated population and were based on patients enrolled in the U-EXCEL, U-EXCEED, and U-ENDURE trials.
Upadacitinib is available as 15 mg, 30 mg, and 45 mg oral extended-release tablets at submitted prices of $51.68, $76.96, and $101.81, respectively.2 The recommended dosage is 45 mg of upadacitinib once daily for 12 weeks (induction), followed by a maintenance dosage of 15 mg once daily or 30 mg once daily, with the choice between the 2 maintenance dosages based on “patient presentation.”2 Hereafter, the regimens that include 45 mg of upadacitinib for induction and either 15 mg or 30 mg of upadacinitib for maintenance are referred to as UPA15 and UPA30, respectively, unless otherwise specified. The annual per-patient costs of upadacitinib in the first year are expected to be $23,074 for patients who receive UPA15 and $30,178 for patients who receive UPA30. In the second year and beyond, the annual per-patient costs are $18,876 for UPA15 and $28,110 for UPA30.1 The sponsor’s analysis included an additional arm, in which 60% of patients were assumed to receive UPA15 and 40% to receive UPA30 as the maintenance dose (annual per-patient costs: $25,916 in year 1 and $22,570 in subsequent years).
The analysis was conducted from the perspective of the Canadian public health care payer. Costs, life years, and quality-adjusted life-years (QALYs) were estimated over a lifetime horizon (60 years; 2-week cycle length), discounted at a rate of 1.5% per annum.
Model Structure
The sponsor submitted a model consisting of a decision tree (reflecting the induction period; Figure 1) and a Markov model (16 alive health states plus death, reflecting the maintenance period; Figure 2). The decision tree included a primary response period (from treatment initiation to the response assessment) with the length of the induction period ranging from 4 to 12 weeks (based on the respective product monographs). Patients entered the decision tree on treatment, and treatment response was evaluated at the end of the induction period by use of the CDAI score. Treatment response was defined as a decrease of at least 100 points from baseline in CDAI (CR-100). Patients deemed to be treatment responders entered the Markov model on their current treatment and remained on treatment unless they discontinued due to a lack of CR-100 response. The sponsor assumed there was no maximum duration of biologic therapy. Patients deemed to be nonresponders after the induction period were assumed to receive conventional care for the remainder of the model horizon. Patients who initiated the treatment on conventional care remained on conventional care irrespective of their response status.
The Markov model included 16 alive health states: moderate to severe CD (220 ≤ CDAI < 600), mild CD (150 ≤ CDAI < 220), remission (CDAI < 150), and surgery; each repeated for 4 matrices: “On low-dose biologics after response,” “On high dose biologics after response,” “Conventional care after no response,” and “Conventional care after response” (Figure 2). Treatment responders from the induction period entered the Markov model in the moderate to severe health state within the “On low-dose after response” matrix, while nonresponders entered the Markov model in the “Conventional care after no response” matrix. In each subsequent cycle, patients could transition between CDAI-based health states, remain in their current state, discontinue treatment, or die. The sponsor assumed that patients who lose response to the low dose of advanced treatment and subsequently escalate to the high dose of the same treatment retain the low-dose efficacy (i.e., they maintain the same probability of a treatment response but incur higher costs). Patients who discontinued advanced treatment in the Markov model transitioned to the “Conventional care after no response” matrix. In each cycle, a proportion of patients in the moderate to severe health state of each matrix were assumed to undergo surgery. After surgery, patients stayed in a postsurgery tunnel state for 8 weeks and then transitioned to a CDAI-based health state (i.e., remission, mild, or moderate to severe disease) unless they died.
Model Inputs
The baseline characteristics in the model were based on the U-EXCEL and U-EXCEED trials for the CCF subgroup (54.5% male, 71.95 kg, mean age of 40.18 years) and BF subgroup (53.6%, 69.75 kg, mean age of 38.32 years), respectively.1 The sponsor assumed that 47.6% of patients in each subgroup had moderate disease (220 ≤ CDAI < 300) and that 52.4% had severe disease (CDAI ≥ 300) based on posthoc analysis of the U-EXCEL and U-EXCEED trials.
Clinical efficacy inputs in the model were derived from sponsor-conducted NMAs for the CCF and BF subgroups and included CR-100 response rates (for the induction phase) and CR-100 remission rates (for both induction and maintenance phases). The sponsor assumed that UPA30 and the weighted upadacitinib groups had the same CR-100 response rate as the UPA15 group. All biosimilars were assumed to have the same efficacy as the branded drug. Both forms of vedolizumab were assumed to have the same efficacy. In the BF subgroup, the sponsor assumed that, due to data limitations, the CR-100 response rate for infliximab would be equal to that of adalimumab. Incidence rates for grade 3 or 4 adverse events (AEs) (i.e., serious infections, tuberculosis, lymphoma, hypersensitivity, and skin reactions) were estimated using clinical trial data for upadacitinib and risankizumab, and sourced from the literature for all other comparators.1,3 The 7% annual probability of surgery for patients with moderate to severe CD assumed by the sponsor was taken from the literature.3 Dose-escalation data were obtained from the U-ENDURE trial for upadacitinib and from the literature for ustekinumab and adalimumab,1,3 while vedolizumab and infliximab were assumed to have the same rate of dose escalation as ustekinumab and adalimumab, respectively.
Mortality was based on all-cause mortality data from Statistics Canada and weighted by the proportion of male patients in the CCF and BF subgroups based on the U-EXCEED and U-EXCEL trials. The risk of death was assumed by the sponsor to be independent of the treatment received.
Health-state utility values for the remission, mild, and moderate to severe health states were derived from 5-Level EQ-5D questionnaire data collected for the U-EXCEED, U-EXCEL, and U-ENDURE trials, with observations pooled across the CCF and BF subgroups and mapped to the 3-Level EQ-5D.4 The utility value for the surgery health state was obtained from the literature.5 Patients in a given health state were assumed to have the same health-state utility value irrespective of treatment received. The model incorporated utility decrements for the impact of AEs, which were sourced from the literature.6-10 No disutility was applied for surgical complications.
The economic model included costs related to drug acquisition and administration, health care resource use, AEs, surgery, and surgical complications. Drug acquisition costs for upadacitinib were based on prices submitted by the sponsor.1 All other drug acquisition costs were sourced from IQVIA DeltaPA,11 with the exception of the price of risankizumab, which was obtained from AbbVie Canada.1 The reference case included wastage for infliximab. Acquisition costs for treatments included in conventional care were obtained from the Ontario Drug Benefit Formulary and Alberta Drug Benefit List, with the average daily cost for conventional care based on each treatment’s relative usage informed by expert opinion.12,13 Administration costs were included for subcutaneous injections (first administration only)1,14 and IV infusions.15,16 Health care resource use by CDAI health state (patient monitoring, laboratory tests and procedures, physician visits, hospitalizations for surgery, surgical complications, and AEs) was estimated by the sponsor based on expert input, and costs were obtained from the Ontario Schedule of Benefits for Physician Services, the Ontario Schedule of Benefits for Laboratory Services, and the Ontario Case Costing Initiative. Costs were inflated to 2023 dollars using the health care consumer price index from Statistics Canada.16-20
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,500 iterations). The deterministic and probabilistic results were similar, and the probabilistic findings are presented below. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3. The sponsor submitted 2 subgroup analyses (CCF and BF) to reflect the overall indicated population.
Base-Case Results
Among patients in the CCF subgroup, UPA15 was more costly (incremental costs: $210,986) and more effective (incremental QALYs: 1.90) than conventional care, resulting in an incremental cost-effectiveness ratio (ICER) of $110,911 per QALY gained over a 60-year time horizon, while UPA30 was extendedly dominated (Table 3). In the sponsor’s sequential analysis, UPA15 was the optimal treatment in 0.67% of iterations at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained, while UPA30 had a 0% probability of being considered optimal.
Results were driven by predicted differences in QALYs between upadacitinib and comparators (incremental QALYs for UPA15 versus conventional care: 1.90) and drug acquisition costs (Appendix 3). Drug acquisition costs represent 54% and 59% of total costs for UPA15 and UPA30, respectively. The sponsor’s model estimates that upadacitinib will generate 21.88 to 22.02 QALYs over a lifetime horizon; of these, 98% of the incremental benefits accrue after the first year of treatment (i.e., beyond the treatment duration of the U-ENDURE trial).
Table 3: Summary of Sponsor’s Economic Evaluation Results — Conventional Care Failure Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Conventional care | 339,106 | 19.98 | Reference |
UPA15 | 550,092 | 21.88 | 110,911 vs. conventional care |
Infliximab biosimilar | 789,476 | 22.56 | 354,155 vs. UPA15 |
Dominated therapies | |||
Vedolizumab-SC | 550,052 | 21.36 | Extendedly dominated by UPA15, UPA-weighted, UPA30 |
UPA-weighteda | 573,948 | 21.92 | Extendedly dominated by UPA30, infliximab biosimilar |
UPA30 | 611,824 | 22.02 | Extendedly dominated by infliximab biosimilar, UPA15, UPA-weighted |
Ustekinumab | 719,871 | 21.84 | Dominated by UPA15, UPA-weighteda UPA30 |
Vedolizumab | 730,318 | 21.51 | Dominated by UPA15, UPA-weighteda UPA30, ustekinumab |
Adalimumab biosimilar | 1,041,885 | 20.99 | Dominated by vedolizumab subcutaneous, UPA15, UPA-weighteda UPA30, ustekinumab, vedolizumab, infliximab biosimilar |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; UPA = upadacitinib; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose.
aIn the “UPA-weighted” group, the sponsor assumed that 60% of patients would receive UPA15 as maintenance and 40% would receive UPA30 as maintenance.
Source: Sponsor’s pharmacoeconomic submission.1
Among patients in the BF subgroup, UPA15 was more costly and less effective compared to an infliximab biosimilar (i.e., UPA15 was dominated by infliximab biosimilar), such that UPA15 would not be chosen as the optimal treatment regardless of a decision-maker’s WTP threshold. In the sponsor’s sequential analysis, UPA30 was associated with an ICER of $399,240 (incremental costs: $124,549; incremental QALYs: 0.31) per QALY gained compared to the infliximab biosimilar. At a WTP of $50,000 per QALY, the probability of UPA15 being cost-effective was 0.07% in this subgroup; UPA30 had a 0% probability of being cost-effective.
Results were driven by predicted differences in QALYs between upadacitinib and comparators (1.96 incremental QALYs for UPA15 versus conventional care) and drug acquisition costs (Appendix 3). In total, drug acquisition costs reflect 62% and 67% of total costs for UPA15 and UPA30, respectively. The sponsor’s model estimates that upadacitinib will generate 22.52 to 22.97 QALYs over a lifetime horizon; of these, 98% of the incremental benefits accrue after the first year of treatment (i.e., beyond the treatment duration of the U-ENDURE trial).
Table 4: Summary of Sponsor’s Economic Evaluation Results — Biologic Failure Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Conventional care | 349,375 | 20.56 | Reference |
Infliximab biosimilar | 567,351 | 22.66 | 103,861 vs. conventional care |
UPA30 | 691,900 | 22.97 | 399,240 vs. infliximab biosimilar |
Dominated therapies | |||
Vedolizumab-SC | 508,761 | 21.63 | Extendedly dominated by infliximab biosimilar, UPA30 |
Ustekinumab | 604,007 | 21.69 | Dominated by infliximab biosimilar |
UPA15 | 624,997 | 22.52 | Dominated by infliximab biosimilar |
UPA-weighteda | 649,344 | 22.65 | Dominated by infliximab biosimilar |
Vedolizumab | 654,178 | 21.61 | Dominated by vedolizumab subcutaneous, infliximab biosimilar, ustekinumab, UPA15, UPA-weighteda |
Adalimumab biosimilar | 896,121 | 21.01 | Dominated by vedolizumab subcutaenous, infliximab biosimilar, ustekinumab, UPA15, UPA-weighteda vedolizumab, UPA30 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; UPA = upadacitinib; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose.
aIn the “UPA-weighted” group, the sponsor assumed that 60% and 40% of patients would receive UPA15 and UPA30 as maintenance. respectively.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several probabilistic scenario analyses, including adopting alternative modelling assumptions (i.e., 10- or 20-year horizon; 0% or 3% discount rate; separate states for moderate and severe CD; excluding AEs), alternative cost perspectives (i.e., societal perspective, including patient and caregiver productivity loss, and travel and parking costs), and alternative assumptions related to time to treatment discontinuation, discontinuation rates, starting doses, concomitant care, vial sharing, utility values, and comparators (i.e., excluding the infliximab biosimilar in the BF subgroup and including risankizumab). Deterministic scenario analyses included adopting high-dose efficacy for patients whose doses escalate and using endoscopic response to measure treatment response. Base-case results remained robust across most analyses, with the following exceptions: when maximum treatment duration was 3 years, upadacitinib was not cost-effective at any cost threshold (i.e., both maintenance doses were dominated or extendedly dominated among both subgroups); when AEs were excluded; when high-dose efficacy for patients that dose-escalate was adopted; and when treatment response was assessed via endoscopic response.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative efficacy and safety of upadacitinib is uncertain. There is a lack of direct head-to-head evidence comparing upadacitinib to relevant biologic comparators, including vedolizumab, ustekinumab, adalimumab, infliximab, and risankizumab. To inform efficacy in the pharmacoeconomic model (i.e., CR-100 and remission), the sponsor conducted NMAs to estimate the relative efficacy of upadacitinib in the CCF and BF subgroups in the induction and maintenance phases. As noted in the CADTH Clinical Review Report, indirect evidence provided by the sponsor suggests that there may be no meaningful difference in efficacy between upadacitinib and relevant comparators during either phase, although the presence of substantial imprecision and unresolved heterogeneity precludes meaningful conclusions. Health-related quality of life was not assessed in the sponsor-submitted NMAs.
The rate of grade 3 and 4 AEs (i.e., serious infections, tuberculosis, lymphoma, hypersensitivity, and skin reactions) in the pharmacoeconomic model for upadacitinib and comparators was based on naive comparisons, without adjustment or accounting for differences in patient characteristics. Due to the direct use of clinical trial data, it is not possible to determine if any observed differences between the therapies were due solely to the treatment or, rather, to bias or confounding factors. Although serious infections were included as an outcome in the sponsor’s NMA, these data were not utilized in the pharmacoeconomic model. In addition, the AEs included in the model do not capture the range of AEs of special interest to clinicians (e.g., herpes zoster infections). Although the pivotal upadacitinib CD trials showed few serious AEs, the upadacitinib product monograph includes serious warnings and precautions about serious infections, malignancies, thrombosis, and major cardiovascular events.2
Given the lack of direct evidence and limitations with the sponsor’s NMAs, the comparative efficacy and safety of upadacitinib to other advanced therapies available for the treatment of CD in the CCF and BF subgroups are uncertain. As the sponsor-submitted NMAs suggest that there may be no differences in efficacy or safety between upadacitinib and other advanced treatments, it is uncertain whether upadacitinib provides a net benefit relative to currently funded biologic treatments. CADTH was unable to address this limitation in a reanalysis.
The long-term treatment effectiveness of upadacitinib is uncertain. Evidence of the long-term effectiveness of upadacitinib beyond 52 weeks is not available. In the pharmacoeconomic model, the sponsor assumed that patients who remain on upadacitinib maintain the efficacy of upadacitinib estimated from the NMAs for the duration of treatment, without consideration of potential waning of treatment effect. Clinical expert input received by CADTH for this review indicated that treatment effectiveness has been observed to wane with other advanced treatments used to treat CD and that waning of effectiveness may be possible with upadacitinib over time. Given that the majority (98%) of the incremental QALYs predicted by the sponsor’s model in both subgroups were derived from extrapolated findings rather than observed benefit, the lack of long-term data and the lack of consideration of potential waning of effectiveness introduce considerable uncertainty into the analysis.
This limitation could not be addressed by CADTH due to a lack of clinical data. The direction and magnitude of the impact of this limitation is unknown given that the comparative rate of potential effectiveness waning with upadacitinib versus other treatments for CD is unknown.
The downstream effects of surgery and surgical complications were inadequately modelled. The sponsor’s model includes costs associated with surgery, but does not differentiate between the causes of surgery (e.g., stricture) or the types of surgeries for CD (e.g., resection and anastomosis and permanent ileostomy), and does not account for the impact of surgery and surgical complications on quality of life, risk of recurrence, and risk of future complications. For example, clinical input received by CADTH suggests that a permanent ileostomy would be expected to significantly reduce a patient’s quality of life; however, in the sponsor’s model all patients who undergo surgery return 8 weeks later to 1 of the CDAI health states with utility and transition probabilities similar to those of patients who never underwent surgery.
This limitation could not be addressed by CADTH due to the structural limitations of the sponsor’s model. The magnitude of the impact on the cost-effectiveness of upadacitinib is unknown.
The model lacks transparency: The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is over-written with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical, as it remains unclear whether the model is running inappropriately by overriding errors.
CADTH was unable to address this limitation and notes that a thorough validation of the sponsor’s model was not possible.
Additionally, the following key assumptions (Table 5) made by the sponsor were appraised by CADTH.
Table 5: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The sponsor submitted separate subgroup analyses for patients with prior CCF and for patients with prior BF. | Reasonable. Clinical expert input received by CADTH for this review supports considering CCF and BF subgroups separately as the natural history of the disease (i.e., duration and surgical history) typically differs between these groups. |
The sponsor’s pharmacoeconomic analysis included a weighted arm consisting of 60% of patients receiving UPA15 and 40% receiving UPA15, with efficacy for this arm based on a weighted average of the efficacy of UPA15 and UPA30. | Not appropriate. The assumption that an individual patient in the blended group would have efficacy equal to the weighted efficacy of UPA30 and UPA15 does not reflect the efficacy of either dose. It would have been more appropriate to provide an overall weighted ICER derived from the ICERs for UPA30 and UPA15. However, the proportion of patients expected to receive UPA30 vs. UPA15 is highly uncertain. Clinical expert input received by CADTH indicated that, in clinical practice, patients with moderately to severely active CD are expected to receive 30 mg as the maintenance dose, given evidence suggesting higher efficacy than UPA15 and a reluctance among clinicians to potentially undertreat CD due to the irreversible nature of bowel damage that can occur, although the approach to determining the optimal dosage may vary by practice. The draft Health Canada monograph for upadacitinib indicates that patients should receive the lowest effective dose. |
Patients who discontinued advanced treatment were assumed to receive only conventional care for the remainder of their lifetime. | Not appropriate. Clinical expert input received by CADTH for this review indicated that patients who do not exhibit a positive response to biologic treatment are likely to switch to an alternative therapy. |
CDAI response was used to define patient response and disease severity. | Not appropriate. Clinical expert input received by CADTH noted that CDAI is used primarily in clinical trials and not in clinical practice. Objective measures such as endoscopic outcomes are preferred in practice, given that patient-reported symptoms may not correlate well with the extent of bowel disease. Treatment decisions in practice would be based on endoscopy after 6 to 9 months treatment, not on patient-reported symptoms. Fecal (calprotectin) levels and serum biomarkers of inflammation (C-reactive protein) may also be used to guide treatment decisions along with endoscopic response. |
The sponsor adopted a higher probability of having a skin reaction with adalimumab than other anti-TNF agents (i.e., infliximab) from a previous NICE submission (adalimumab: 10.37%; infliximab: 0.72%). | Uncertain. The probability of AEs in the pharmacoeconomic model was based on a naive comparison, which does not account for differences in patient populations, outcome definitions, or treatment durations. Should the relative rate of skin reactions with adalimumab be lower in clinical practice than modelled, the incremental QALYs between upadacitinib and adalimumab may be lower than predicted by the sponsor’s analysis. |
EQ-5D-5L scores were mapped to EQ-5D-3L index values using an algorithm by Hernández Alava et al.4 | Uncertain. Mapping from EQ-5D-5L to EQ-5D-3L is unnecessary and adds uncertainty to both the precision and validity of the utility estimates. |
Discontinuation from treatment was assumed equal across all advanced treatments and was based on upadacitinib data from U-ENDURE. | Reasonable. Clinical expert input received by CADTH suggests that it is likely reasonable to assume equal discontinuation across therapies. |
The sponsor assumed that patients who lose response to a low-dose advanced treatment and subsequently receive the high dose of the same biologic maintain the same probability of a treatment response while incurring the cost of the higher does. | Reasonable. Clinical expert input received by CADTH noted that escalating to a higher dose with the aim of re-establishing a patient's response is reasonable. |
There is no maximum duration of treatment. | Reasonable. Clinical expert input received by CADTH indicated that if patients are responding to treatment, they are likely to remain on treatment indefinitely. |
The efficacy and safety of biosimilar drugs was assumed to be equal to that of the brand name original drug. The branded biologic treatments adalimumab (Humira) and infliximab (Remicade) were omitted from the sponsor’s base case. | Reasonable. The clinical experts consulted by CADTH found this assumption acceptable. CADTH notes that the exclusion of branded treatments was a conservative assumption as biosimilar treatments were assumed to have equivalent efficacy and safety at a lower acquisition cost. |
CD is not expected to be associated with excess mortality. | Reasonable. The clinical experts consulted by CADTH found this assumption acceptable. |
AE = adverse event; BF = biologic failure; CCF = conventional care failure; CD = Crohn disease; CDAI = Crohn Disease Activity Index; EQ-5D-5L = 5-Level EQ-5D questionnaire; 3-Level EQ-5D questionnaire; QALY = quality-adjusted life-year; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH was unable to address the lack of head-to-head comparative clinical data, uncertainty in the long term efficacy of upadacinitib, and the downstream effects of surgery and surgical complications. Given these limitations, CADTH was unable to provide a more reliable estimate of the cost-effectiveness of UPA.
When reviewing the sponsor’s corrected base-case results (Appendix 4, Table 10, Table 11), in which the price of vedolizumab was updated to reflect the current public list price, the probabilities of UPA15 being cost-effective at a WTP threshold of $50,000 per QALY gained were 0.87% in the CCF subgroup and 0% in the BF subgroup, while the probability of UPA30 being cost-effective was 0% in both subgroups. In both subgroups, results were driven by predicted differences in QALYs between UPA15, with approximately 97% of the predicted benefits with upadacitinib derived from extrapolated findings rather than observed benefit. Drug acquisition costs account for approximately 54% to 67% of total costs in each subgroup.
Scenario Analysis Results
CADTH undertook price-reduction analyses based on the sponsor’s base-case results. For the CCF subgroup, the sponsor’s base case suggests that 39% and 88% price reductions for UPA15 and UPA30, respectively, would be required to achieve cost-effectiveness relative to conventional care at a threshold of $50,000 per QALY (Appendix 4, Table 14). For the BF subgroup, the sponsor’s base case suggests that 45% and 62% price reductions for UPA15 and UPA30, respectively, would be required to make upadacitinib cost-effective relative to conventional care at a threshold of $50,000 per QALY (Appendix 4, Table 15).
CADTH undertook a scenario analysis to explore the impact of including risankizumab on the cost-effectiveness of upadacitinib (Appendix 4, Table 17), using the sponsor-provided option to do so. The results of this scenario analysis are in line with the sponsor’s base case: upadacitinib is not a cost-effective treatment for moderately to severely active CD in either the CCF or BF subgroup. Among patients in the CCF subgroup, UPA15 was associated with an ICER of $111,167 compared to conventional care, while UPA30 was extendedly dominated by infliximab biosimilar and UPA15. In the BF subgroup, UPA30 was associated with an ICER of $394,382 and UPA15 was dominated by infliximab biosimilar.
Issues for Consideration
An adalimumab biosimilar, an infliximab biosimilar, and vedolizumab have successfully completed negotiations with the pan-Canadian Pharmaceutical Alliance (pCPA) and are listed on public formularies. It is likely that the treatments are reimbursed by jurisdictional drug plans at confidential prices that are less than publicly available list prices. Additionally, risankizumab is currently undergoing negotiations with the pCPA;21 should negotiations conclude with a letter of intent, the price of risankizumab paid by the drug plans may be lower than what is incorporated in the sponsor’s pharmacoeconomic model.
Risankizumab was omitted from the sponsor’s base case. Risankizumab recently received a positive recommendation from CADTH and is currently under consideration for negotiation with the pCPA for the treatment of moderately to severely active CD.21,22 The sponsor-submitted NMAs suggest no meaningful difference in efficacy between upadacitinib and risankizumab.
Upadacitinib may be self-administered and is the only oral advanced therapy drug currently available for the indicated population. Ease of administration was noted as an important aspect of treatment for patients.
CADTH has previously reviewed adalimumab, risankizumab, vedolizumab, ustekinumab, and infliximab for CD.22-27 The cost-effectiveness reported in these evaluations may not be directly comparable to those in the current review, due to differences in model structure, clinical effectiveness parameters, health-state utility values, and cost inputs.
Overall Conclusions
Based on the CADTH clinical review, upadacitinib results in an increase in the proportion of adult patients who experience a clinical response and clinical remission among those with moderately to severely active CD and a history of treatment failure compared with placebo. There are no direct head-to-head trials comparing upadacitinib with biologic treatments for CD in either the CCF or BF subgroup, and indirect evidence submitted by the sponsor suggests that there may be no meaningful differences in efficacy or safety between upadacitinib and other advanced treatments during either the induction or maintenance phases. However, substantial imprecision and unresolved heterogeneity were noted in the sponsor’s NMAs, precluding meaningful conclusions for both efficacy and safety outcomes among treatments.
CADTH was unable to address the lack of head-to-head comparative clinical data, uncertainty in the long term efficacy of upadacitinib, and the downstream effects of surgery and surgical complications. Given these limitations, CADTH was unable to provide a more reliable estimate of the cost-effectiveness of upadacitinib. Based on the sponsor’s submitted base-case results, upadacitinib is not a cost-effective treatment for moderately to severely active CD in either the CCF or BBF subgroup. In the CCF subgroup, relative to conventional care, UPA15 had an ICER of $110,911 per QALY gained, while UPA30 was extendedly dominated. In the BF subgroup, UPA30 had an ICER of $399,240 compared with infliximab biosimilar, while UPA15 was dominated by infliximab biosimilar. In both subgroups, the probability of upadacitinib being the optimal treatment strategy at a WTP threshold of $50,000 per QALY gained was less than 1%, and a price reduction would be required for upadacitinib to be considered cost-effective relative to currently available advanced treatments for this indication.
The cost-effectiveness of upadacitinib relative to other advanced treatments for CD, in both the subgroup of patients with prior failure of conventional treatments or with prior failure of biologics, is uncertain due to a lack of robust comparative data and limitations in the sponsor’s analysis that could not be addressed in CADTH’s reanalyses. Notably, the sponsor-submitted NMAs suggest that there may be no difference in clinical outcomes (e.g., clinical response and clinical remission) between upadacitinib and biologic drugs, and health-related quality of life was not assessed in the sponsor’s NMAs. As such, there is insufficient evidence to suggest that upadacitinib should be priced higher than advanced treatments for moderately to severely active CD. To ensure cost-effectiveness, upadacitinib should be priced no more than the lowest-cost biologic that is funded in the population to be reimbursed.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib), 15 mg, 30 mg, 45 mg, extended-release oral tablets. Pointe-Claire (QC): AbbVie Corporation; 2023 Jun 5.
2.PrRinvoq® (upadacitinib): 15 mg, 30 mg, 45 mg extended-release oral tablets [product monograph]. St-Laurent (QC): AbbVie; 2023 Oct 11.
3.Ustekinumab for moderately to severely active Crohn’s disease after previous treatment. NICE Technology appraisal guidance [TA456]. London (UK): National Institute for Health and Care Excellence; 2017: https://www.nice.org.uk/guidance/ta456/resources/ustekinumab-for-moderately-to-severely-active-crohns-disease-after-previous-treatment-pdf-82604848449733. Accessed 2023 Mar 13.
4.Hernández Alava M, Pudney S, Wailoo A, Chrysanthou G. Estimating the relationship between EQ-5D-5L and EQ-5D-3L: results from an English population study. Sheffield (UK): Policy Research Unit in Economic Evaluation of Health and Care Interventions. Universities of Sheffield and York.
5.Blackhouse G, Assasi N, Xie F, et al. Canadian cost-utility analysis of initiation and maintenance treatment with anti-TNF-α drugs for refractory Crohn's disease. J Crohns Colitis. 2012;6(1):77-85. PubMed
6.Brown RE, Hutton J, Burrell A. Cost effectiveness of treatment options in advanced breast cancer in the UK. Pharmacoeconomics. 2001;19(11):1091-1102. PubMed
7.Porco TC, Lewis B, Marseille E, Grinsdale J, Flood JM, Royce SE. Cost-effectiveness of tuberculosis evaluation and treatment of newly-arrived immigrants. BMC Public Health. 2006;6:157. PubMed
8.Hornberger J, Reyes C, Lubeck D, Valente N. Economic evaluation of rituximab plus cyclophosphamide, vincristine and prednisolone for advanced follicular lymphoma. Leuk Lymphoma. 2008;49(2):227-236. PubMed
9.Beusterien KM, Davies J, Leach M, et al. Population preference values for treatment outcomes in chronic lymphocytic leukaemia: a cross-sectional utility study. Health Qual Life Outcomes. 2010;8:50. PubMed
10.Beusterien KM, Szabo SM, Kotapati S, et al. Societal preference values for advanced melanoma health states in the United Kingdom and Australia. Br J Cancer. 2009;101(3):387-389. PubMed
11.IMS Health Quintiles via DeltaPA Formulary Prices 2022. [Ottawa (ON)]: IQVIA.
12.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario Drug Benefit formulary/Comparative Drug Index. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Mar 13.
13.Government of Alberta. Interactive drug benefit list. 2022; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2023 Mar 13.
14.Government of Canada. Job Bank; wages for registered nurse (NOC 3012). All regions across Canada. 2023; https://www.jobbank.gc.ca/wagereport/occupation/993. Accessed 2023 Mar 13.
15.Hughes A, Marshall JK, Moretti ME, Ungar WJ. A cost-utility analysis of switching from reference to biosimilar infliximab compared to maintaining reference infliximab in adult patients with Crohn's disease. J Can Assoc Gastroenterol. 2021;4(1):48. PubMed
16.Table: 18-10-0004-08. Consumer Price Index, monthly, percentage change, not seasonally adjusted, Canada, provinces, Whitehorse and Yellowknife — health and personal care. Ottawa (ON): Statistics Canada; 2023: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1810000408. Accessed 2023 Mar 13.
17.Schedule of benefits for laboratory services. Toronto (ON): Ontario Ministry of Health; 2020.
18.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health,; 2022.
19.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Ministry of Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2023 Jun 8.
20.Physician survey of Crohn’s Disease management in Canada. [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib), 15 mg, 30 mg, 45 mg, extended-release oral tablets. Pointe-Claire (QC): AbbVie Corporation; 2023 Jun 5.
21.Skyrizi (risankizumab). In: pCPA brand name drug negotiations status. Toronto (ON): pan-Canadian Pharmaceutical Alliance (pCPA); 2023: https://www.pcpacanada.ca/negotiation/22367. Accessed 2023 Aug 9.
22.CADTH reimbrusement review: risankizumab (Skyrizi) for Crohn's disease. Can J Health Technol. 2023;3(8).
23.Humira (adalimumab) for Crohn's disease. CADTH drug reimbursement review. Ottawa (ON): CADTH; 2007: https://www.cadth.ca/adalimumab-2. Accessed 2023 Aug 9.
24.Entyvio (vedolizumab) for Crohn's disease. CADTH drug reimbursement review. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/vedolizumab-0. Accessed 2023 Aug 9.
25.Ustekinumab (Stelara) for Crohn's disease. CADTH drug reimbursement review. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/ustekinumab-1. Accessed 2023 Aug 9.
26.Inflectra (infliximab) for Crohn's disease and ulcerative colitis. CADTH drug reimbursement review. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/infliximab-2. Accessed 2023 Aug 9.
27.Inflectra (infliximab) for rheumatoid arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, psoriatic arthritis, plaque psoriasis. CADTH drug reimbursement review. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/infliximab-3. Accessed 2023 Aug 9.
28.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario Drug Benefit formulary/Comparative Drug Index. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Aug 1.
29.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 Aug 23.
30.Saskatchewan Drug Plan: search formulary. 2023; http://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2023 Aug 1.
31.pan-Canadian Budget Impact Analysis report: Rinvoq® (upadacitinib) for the treatment of adult patients with moderate-to-severe active Crohn's disease [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib): 15 mg, 30 mg, 45 mg extended release tablets. Saint-Laurent (QC): AbbVie Corporation; 2023 Jun 5.
32.GPM data - CD Market from March 2020 to January 2023. [Ottawa (ON)]: IQVIA; 2023: https://www.iqvia.com/solutions/real-world-evidence/real-world-data-and-insights. Accessed 2023 Mar 13.
33.Ustekinumab (Stelara) for Crohn's disease. CADTH Common Drug Review: pharmacoeconomic review report. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0501_Stelara_PE_Report.pdf. Accessed 2023 Jun 5.
Appendix 1: Cost-Comparison Table
Note this appendix has not been copy-edited.
The comparators presented in Table 6 have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing product listing agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 6: CADTH Cost-Comparison Table for Crohn Disease
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Upadacitinib (Rinvoq) | 15 mg 30 mg 45 mg | Tablet | 51.6810a 76.9600a 101.8100a | Induction: 45 mg once daily for 12 weeks Maintenance: 15 mg or 30 mg once daily | Year 1: 63.22 to 82.68 Year 2+: 51.68 to 76.96 | Year 1: 23,074 to 30,178 Year 2+: 18,864 to 28,090 |
Relevant active comparators | ||||||
Adalimumab (Humira) | 20 mg in 0.2 mL 40 mg in 0.8 mL | Pen or prefilled syringe | 397.0500b 794.1000 | Induction: 160 mg at week 0, then 80 mg at week 2 Maintenance: 40 mg every 2 weeks beginning at week 4 | Year 1: 67.44 Year 2+: 56.57 | Year 1: 24,617 Year 2+: 20,647 |
Adalimumab Biosimilarc | 20 mg in 0.2 mL 40 mg in 0.8 mL | Prefilled syringe or prefilled pen | 235.6350 471.2700 | Induction: 160 mg at week 0, followed by 80 mg at week 2 Maintenance: 40 mg every 2 weeks beginning at week 4 | Year 1: 40.03 Year 2+: 33.57 | Year 1: 14,609 Year 2+: 12,253 |
Infliximab (Remicade) | 100 mg/vial | Vial for IV infusion | 987.5600b | Induction: 5 mg/kg at weeks 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Year 1: 86.58 Year 2+: 70.35 | Year 1: 31,602 Year 2+: 25,677 |
Infliximab Biosimilar (Avsola, Renflexis) | 100 mg/vial | Vial for IV infusion | 493.0000 | Induction: 5 mg/kg at weeks 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Year 1: 43.22 Year 2+: 35.12 | Year 1: 15,776 Year 2+: 12,818 |
Infliximab Biosimilar (Inflectra) | 100 mg/vial | Vial for IV infusion | 525.0000 | Induction: 5 mg/kg at weeks 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Year 1: 46.03 Thereafter: 37.40 | Year 1: 16,800 Thereafter: 13,650 |
Risankizumab (Skyrizi) | 360 mg in 2.4 mL | Cartridge | 4,593.1400d | Induction: 600 mg by IV infusion at weeks 0, 4, and 8 Maintenance: 360 mg by SC injection at week 12, and every 8 weeks thereafter | Year 1: 113.26 Year 2+: 81.80 | Year 1: 41,338 Year 2+: 29,855 |
600 mg in 10 mL | Vial for IV infusion | |||||
Vedolizumab (Entyvio) | 300 mg/vial | Vial for IV infusion | 3,401.8600b | Induction: 300 mg at weeks 0, 2, and 6 Maintenance: 300 mg every 8 weeks thereafter | Year 1: 74.56 Year 2+: 60.58 | Year 1: 27,215 Year 2+: 22,112 |
108 mg injection | Prefilled syringe or prefilled pen | 850.4600b | Induction: 300 mg by IV infusion at least twice Maintenance: 108 mg by SC injection every 2 weeks thereafter | Year 1: 76.89 Year 2+: 60.58 | Year 1: 28,065 Year 2+: 22,112 | |
Ustekinumab (Stelara) | 45 mg/0.5 mL 90 mg/1.0 mL 130 mg/vial | Prefilled syringe Vial for IV infusion | 4,593.1400 2,080.0000e | Induction: At week 0, 260 mg for patients weighing < 55 kg, 390 mg for patients weighing 55 to 85 kg, and 520 mg for patients3 85 kg Maintenance: 90 mg by SC injection every 8 weeks thereafter | Year 1: 92.60 Year 2+: 81.80 | Year 1: 33,799 Year 2+: 29,855 |
Note: All prices are from the Ontario Drug Benefit Formulary (accessed August 2023), unless otherwise indicated and assume wastage.28 Mark-ups and dispensing fees are excluded. Annual period assumes 52 weeks or 365 days. For dosing that depends on weight, CADTH assumed a patient weight of 70 kg, unless otherwise indicated. Recommended doses are based on the respective product monographs.
aSponsor’s submitted price.
bOntario Drug Benefit Formulary Exceptional Access Program (accessed August 2023).29
cConsisting of either Abrilada, Amgevita, Hadlima, Hulio, Hyrimoz, Idacio, Simlandi, or Yuflyma.
dRisankizumab CADTH Reimbursement Review.22
eSaskatchewan Formulary list price (accessed August 2023).30
Appendix 2: Submission Quality
Note this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | Risankizumab, which is currently undergoing negotiations with the pCPA for the treatment of moderately to severely active CD, was omitted as a comparator from the sponsor's base-case analysis. The sponsor-submitted NMAs suggest no meaningful difference in efficacy between UPA and risankizumab. |
Model has been adequately programmed and has sufficient face validity | No | The model includes numerous IFERROR statements. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical, as it remains unclear whether the model is running inappropriately by overriding errors. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | CADTH notes that the sponsor did not transparently provide the comparator market shares in the new drug scenario in the budget impact analysis. |
NMA = network meta-analysis; pCPA = pan-Canadian Pharmaceutical Alliance; UPA = upadacitinib.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
Figure 1: Decision-Tree Model Structure
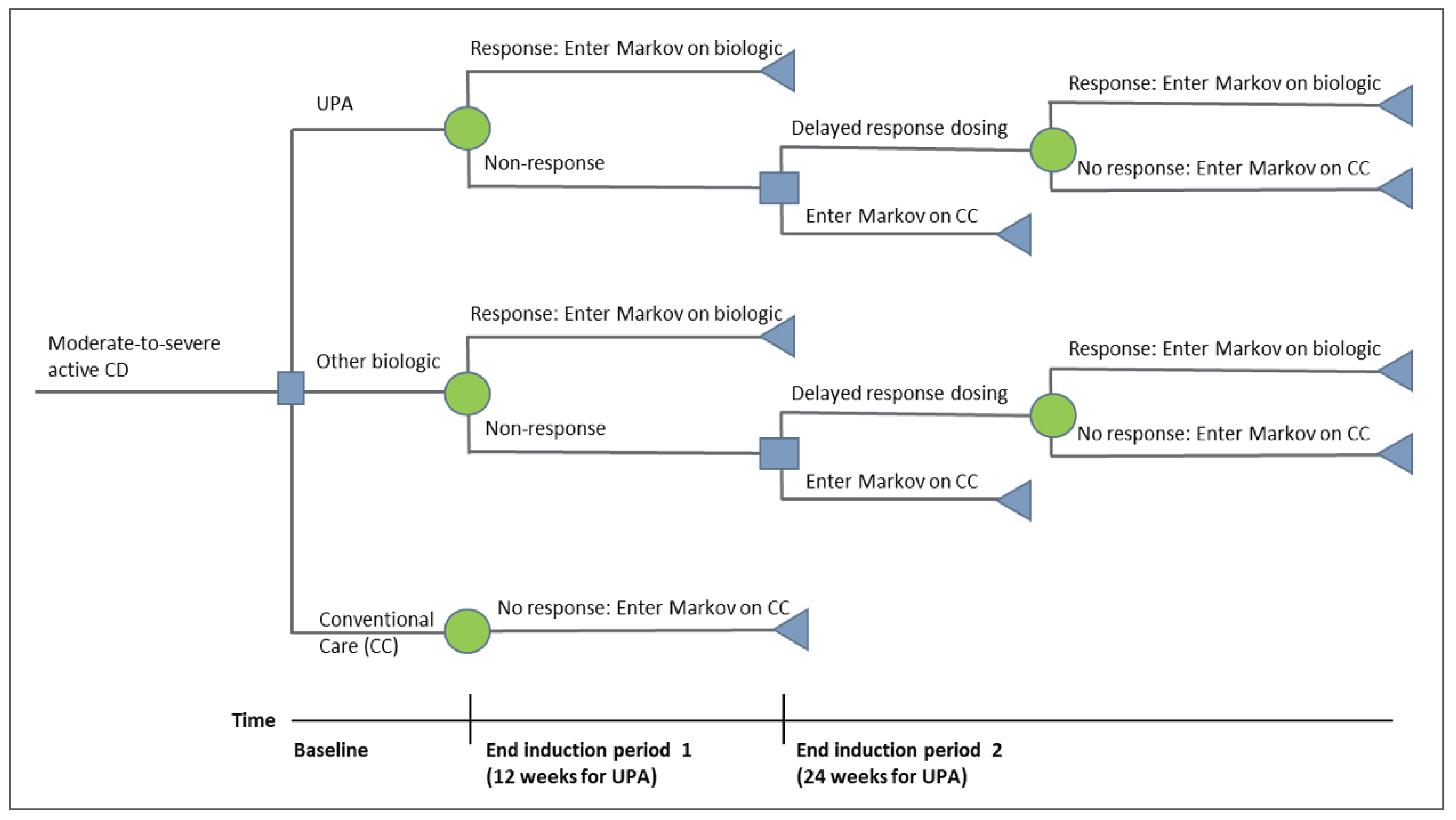
CC = conventional care; CD = Crohn disease; UPA = upadacitinib.
Source: Sponsor’s submission.1
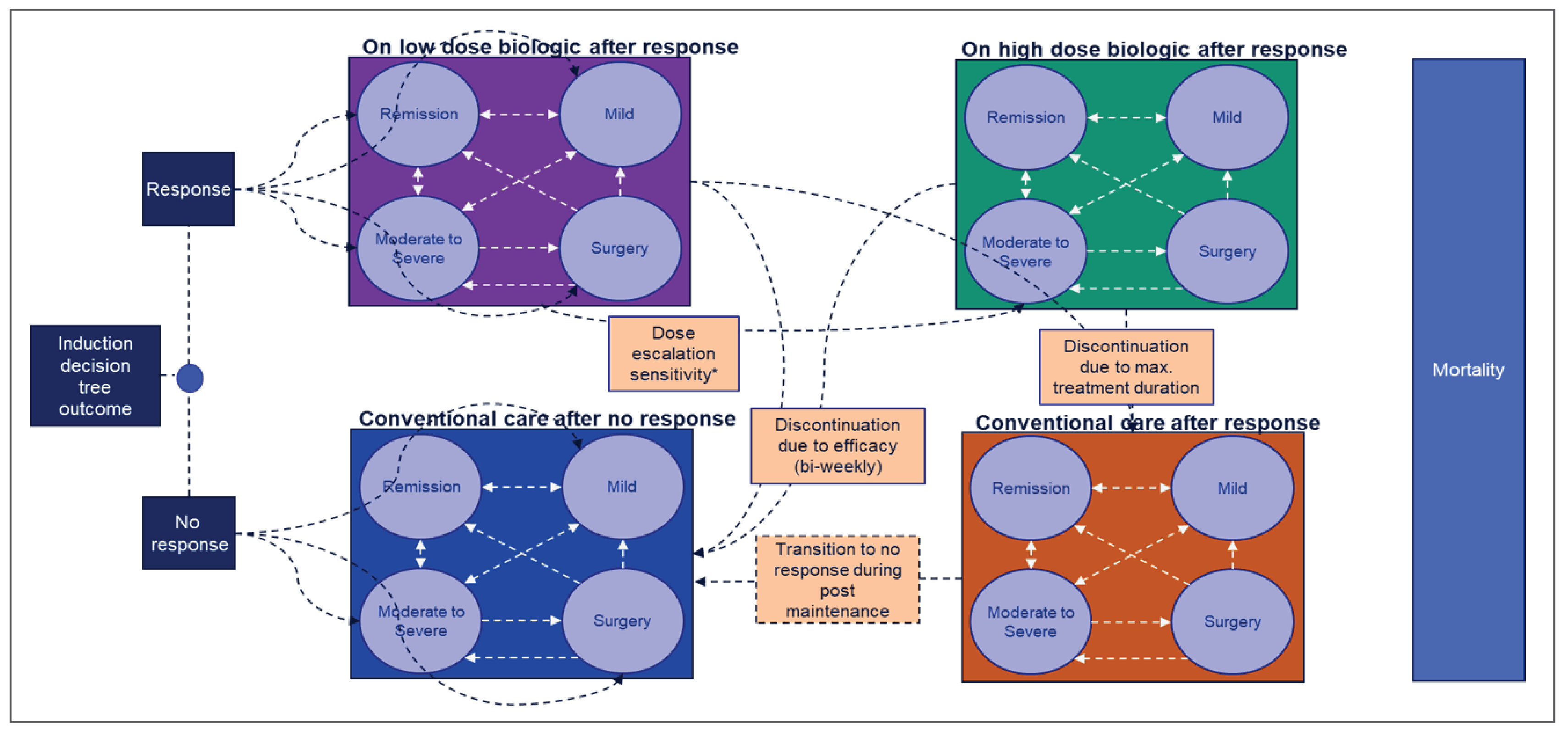
Detailed Results of the Sponsor’s Base Case
Table 8: Disaggregated Results of the Sponsor’s CCF Subgroup Base Case
Parameter | CC | UPA15a | UPA30b | UPA-weightedc | UST | ADA-Bio | IFX-Bio | VDZ | VDZ-SC |
|---|---|---|---|---|---|---|---|---|---|
Discounted LYs | |||||||||
Total | 42.52 | 42.52 | 42.52 | 42.52 | 42.52 | 42.52 | 42.52 | 42.52 | 42.52 |
Discounted QALYs | |||||||||
Remission (CDAI < 150) | 0.86 | 6.92 | 7.62 | 7.12 | 6.03 | 8.09 | 8.34 | 5.28 | 4.49 |
Mild (150 ≤ CDAI ≤ 220) | 6.89 | 6.7 | 6.29 | 6.58 | 8.28 | 6.99 | 8.71 | 7.67 | 8.13 |
Moderate-severe (CDAI 220+) | 12.37 | 8.4 | 8.24 | 8.35 | 7.74 | 7.36 | 5.83 | 8.74 | 8.92 |
Surgery | 0.04 | 0.03 | 0.03 | 0.03 | 0.03 | 0.03 | 0.02 | 0.03 | 0.03 |
Postsurgery | 0.22 | 0 15 | 0.14 | 0 15 | 0.14 | 0 13 | 0.1 | 0 15 | 0.16 |
AE Utility Decrements (Biologic Related) | 0 | −0.02 | −0.03 | −0.03 | −0.13 | −1.31 | −0.15 | −0.08 | −0.08 |
AE Utility Decrements (CC Related) | −0.4 | −0.29 | −0.28 | −0.29 | −0.26 | −0.3 | −0.3 | −0.29 | −0.29 |
Total | 19.98 | 21.88 | 22.02 | 21.92 | 21.84 | 20.99 | 22.56 | 21.51 | 21.36 |
Discounted costs ($) | |||||||||
Biologic Acquisition | 0 | 299,633 | 361,661 | 323,329 | 434,168 | 326,394 | 489,196 | 432,712 | 267,772 |
Biologic Administration | 0 | 0 | 0 | 0 | 14,596 | 4,661 | 22,528 | 20,386 | 2,579 |
CC | 29,019 | 20,542 | 20,447 | 20,516 | 18,742 | 21,586 | 21,814 | 20,719 | 20,822 |
CDAI HS (On Biologic) | 0 | 10,165 | 9,689 | 10,027 | 14,746 | 12,788 | 20,144 | 11,230 | 11,748 |
CDAI HS (Off Biologic) | 59,951 | 35,371 | 35,099 | 35,294 | 29,067 | 29,125 | 16,928 | 35,931 | 36,223 |
CDAI HS (Postsurgery - Off Biologic) | 317 | 214 | 210 | 213 | 198 | 189 | 149 | 223 | 228 |
Surgical Procedures | 92,830 | 62,574 | 61,381 | 62,230 | 57,788 | 55,231 | 43,662 | 65,337 | 66,698 |
Surgical complications | 961 | 648 | 636 | 644 | 598 | 572 | 451 | 676 | 690 |
AEs (Biologic) | 0 | 10,574 | 12,852 | 11,463 | 49,231 | 475,296 | 57,228 | 31,780 | 31,402 |
Adverse events (CC) | 156,028 | 110,372 | 109,851 | 110,233 | 100,737 | 116,043 | 117,376 | 111,324 | 111,890 |
Total | 339,106 | 550,092 | 611,824 | 573,948 | 719,871 | 1,041,885 | 789,476 | 730,318 | 550,052 |
ADA = adalimumab; AE = adverse event; BF = biologic failure; CC = conventional care; CCF = conventional care failure; CDAI = Crohn Disease Activity Index; HS = Health State; IFX = infliximab; SC = subcutaneous injection; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose; UPA-weighted = upadacitinib weighted (60% receive 45 mg induction dose + 15 mg maintenance dose and 40% receive 45 mg induction dose + 30 mg maintenance dose); UST = ustekinumab; VDZ = vedolizumab.
aUPA45 as induction dose and UPA15 as maintenance dose.
bUPA45 as induction dose and UPA30 as maintenance dose.
cIn the “UPA-weighted” group, the sponsor assumed that 60% of patients would receive UPA15 as maintenance and 40% would receive UPA30 as maintenance.
Table 9: Disaggregated Results of the Sponsor’s BF Subgroup Base Case
Parameter | CC | UPA15a | UPA30b | UPA-weightedc | UST | ADA-Bio | IFX-Bio | VDZ | VDZ-SC |
|---|---|---|---|---|---|---|---|---|---|
Discounted LYs | |||||||||
Total | 44.26 | 44.26 | 44.26 | 44.26 | 44.26 | 44.26 | 44.26 | 44.26 | 44.26 |
Discounted QALYs | |||||||||
Remission (CDAI < 150) | 0.88 | 5.98 | 8.28 | 6.64 | 3.53 | 4.66 | 8.55 | 3.55 | 3.64 |
Mild (150 ≤ CDAI ≤ 220) | 7.07 | 8.49 | 7.17 | 8.11 | 8.55 | 8.21 | 5.95 | 8.08 | 8.02 |
Moderate-severe (CDAI 220+) | 12.76 | 8.16 | 7.64 | 8.01 | 9.78 | 9.28 | 8.43 | 10.14 | 10.12 |
Surgery | 0.05 | 0.03 | 0.03 | 0.03 | 0.04 | 0.03 | 0 03 | 0.04 | 0.04 |
Postsurgery | 0.22 | 0 14 | 0.13 | 0.14 | 0 17 | 0.16 | 0 15 | 0 18 | 0 18 |
AE utility decrements (biologic related) | 0 | −0.03 | −0.04 | −0.03 | −0.09 | −0.99 | −0.09 | −0.06 | −0 06 |
AE utility decrements (CC related) | −0.42 | −0.25 | −0.24 | −0.25 | −0.3 | −0.34 | −0.36 | −0.32 | −0.32 |
Total | 20.56 | 22.52 | 22.97 | 22.65 | 21.69 | 21.01 | 22.66 | 21.61 | 21.63 |
Discounted costs ($) | |||||||||
Biologic acquisition | 0 | 390,102 | 462,489 | 415,726 | 298,394 | 256,523 | 248,420 | 337,774 | 207,858 |
Biologic administration | 0 | 0 | 0 | 0 | 10,014 | 3,695 | 13,119 | 15,910 | 2,088 |
CC | 29,879 | 17,881 | 17,532 | 17,782 | 21,356 | 24,683 | 25,575 | 23,005 | 22,772 |
CDAI HS (On Biologic) | 0 | 14,967 | 13,458 | 14,532 | 10,831 | 11,183 | 8,746 | 9,211 | 9,157 |
CDAI HS (off biologic) | 61,803 | 30,793 | 29,892 | 30,538 | 40,802 | 38,490 | 36,943 | 43,526 | 43,493 |
CDAI HS (Postsurgery - off biologic) | 327 | 208 | 195 | 204 | 250 | 237 | 215 | 259 | 259 |
Surgical procedures | 95,727 | 60,970 | 57,016 | 59,837 | 73,296 | 69,538 | 63,034 | 75,886 | 75,720 |
Surgical complications | 991 | 631 | 590 | 619 | 759 | 720 | 652 | 785 | 784 |
AEs (biologic) | 0 | 13,291 | 16,449 | 14,481 | 33,372 | 358,358 | 33,137 | 24,168 | 24,235 |
Adverse events (CC) | 160,649 | 96,152 | 94,279 | 95,624 | 114,934 | 132,694 | 137,509 | 123,654 | 122,396 |
Total | 349,375 | 624,997 | 691,900 | 649,344 | 604,007 | 896,121 | 567,351 | 654,178 | 508,761 |
ADA = adalimumab; AE = adverse event; BF = biologic failure; CC = conventional care; CCF = conventional care failure; CDAI = Crohn Disease Activity Index; HS = Health State; IFX = infliximab; SC = subcutaneous injection; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose; UPA-weighted = upadacitinib weighted (60% receive 45 mg induction dose + 15 mg maintenance dose and 40% receive 45 mg induction dose + 30 mg maintenance dose); UST = ustekinumab; VDZ = vedolizumab.
aUPA45 as induction dose and UPA15 as maintenance dose.
bUPA45 as induction dose and UPA30 as maintenance dose.
cIn the “UPA-weighted” group, the sponsor assumed that 60% of patients would receive UPA15 as maintenance and 40% would receive UPA30 as maintenance.
Table 10: Summary of the Sponsor’s Economic Evaluation Results — CCF Subgroup
Analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case | Conventional care | 339,106 | 19.98 | Reference |
UPA15a | 550,092 | 21.88 | 110,911 | |
Infliximab biosimilar | 789,476 | 22.56 | 354,155 | |
Vedolizumab-SC | 550,052 | 21.36 | Extendedly dominated by UPA15, UPA-weighted, UPA30 | |
UPA-weightedb | 573,948 | 21.92 | Extendedly dominated by UPA30, infliximab biosimilar | |
UPA30c | 611,824 | 22.02 | Extendedly dominated by infliximab biosimilar, UPA, UPA-weighted | |
Ustekinumab | 719,871 | 21.84 | Dominated by UPA15, UPA-weighted, UPA30 | |
Vedolizumab | 730,318 | 21.51 | Dominated by UPA15, UPA-weighted, UPA30, ustekinumab | |
Adalimumab biosimilar | 1,041,885 | 20.99 | Dominated by vedolizumab-SC, UPA15, UPA-weighted, UPA30, ustekinumab, vedolizumab, infliximab biosimilar | |
Sponsor’s corrected base cased | Conventional care | 341,739 | 19.97 | Reference |
UPA15a | 551,526 | 21.87 | 110,267 | |
Infliximab biosimilar | 789,833 | 22.54 | 354,281 | |
Vedolizumab-SC | 560,779 | 21.35 | Dominated by UPA15 | |
UPA-weightedb | 575,674 | 21.91 | Extendedly dominated by UPA30, infliximab biosimilar | |
UPA30c | 613,617 | 22.01 | Extendedly dominated by infliximab biosimilar, UPA, UPA-weighted | |
Ustekinumab | 721,569 | 21.82 | Dominated by UPA15, UPA-weighted, UPA30 | |
Vedolizumab | 745,990 | 21.50 | Dominated by UPA15, UPA-weighted, UPA30, ustekinumab | |
Adalimumab biosimilar | 1,044,019 | 20.98 | Dominated by UPA15, vedolizumab-SC, UPA-weighted, UPA30, ustekinumab, vedolizumab, infliximab biosimilar |
CCF = conventional care failure; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; SC = subcutaneous injection; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose; UPA-weighted = upadacitinib weighted (60% receive 45 mg induction dose + 15 mg maintenance dose and 40% receive 45 mg induction dose + 30 mg maintenance dose).
aUPA45 as induction dose and UPA15 as maintenance dose.
bIn the “UPA-weighted” group, the sponsor assumed that 60% of patients would receive UPA15 as maintenance and 40% would receive UPA30 as maintenance.
cUPA45 as induction dose and UPA30 as maintenance dose.
dPrice of vedolizumab corrected. Original: $3,290.00 per 300 mg vial; $822.50 per 108 mg syringe for SC injection. Corrected: $3,401.86 per 300 mg vial; $850.46 per 108 mg syringe per SC injection, as per the publicly available list price in Ontario.29
Table 11: Summary of Sponsor’s Economic Evaluation Results — BF Subgroup
Analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case | Conventional care | 349,375 | 20.56 | Reference |
Infliximab biosimilar | 567,351 | 22.66 | 103,861 | |
UPA30a | 691,900 | 22.97 | 399,240 | |
Vedolizumab-SC | 508,761 | 21.63 | Extendedly dominated by infliximab biosimilar, UPA30 | |
Ustekinumab | 604,007 | 21.69 | Dominated by infliximab biosimilar | |
UPA15b | 624,997 | 22.52 | Dominated by infliximab biosimilar | |
UPA-weightedc | 649,344 | 22.65 | Dominated by infliximab biosimilar | |
Vedolizumab | 654,178 | 21.61 | Dominated by vedolizumab-SC, infliximab biosimilar, ustekinumab, UPA15, UPA-weighted | |
Adalimumab biosimilar | 896,121 | 21.01 | Dominated by vedolizumab-SC, infliximab biosimilar, ustekinumab, UPA15, UPA-weighted, vedolizumab, UPA30 | |
Sponsor’s corrected base cased | Conventional care | 350,229 | 20.57 | Reference |
Infliximab biosimilar | 567,881 | 22.67 | 103,736 | |
UPA30a | 692,481 | 22.98 | 394,382 | |
Vedolizumab-SC | 516,532 | 21.63 | Extendedly dominated by infliximab biosimilar, UPA30 | |
Ustekinumab | 604,756 | 21.69 | Dominated by infliximab biosimilar | |
UPA15b | 625,272 | 22.53 | Dominated by infliximab biosimilar | |
UPA-weightedc | 649,830 | 22.66 | Dominated by infliximab biosimilar | |
Vedolizumab | 665,805 | 21.61 | Dominated by vedolizumab-SC, infliximab biosimilar, ustekinumab, UPA15, UPA-weighted | |
Adalimumab biosimilar | 895,270 | 21.02 | Dominated by vedolizumab-SC, infliximab biosimilar, ustekinumab, UPA15, UPA-weighted, vedolizumab, UPA30 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; SC = subcutaneous injection; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose; UPA-weighted = upadacitinib weighted (60% receive 45 mg induction dose + 15 mg maintenance dose and 40% receive 45 mg induction dose + 30 mg maintenance dose).
aUPA45 as induction dose and UPA30 as maintenance dose.
bUPA45 as induction dose and UPA15 as maintenance dose.
cIn the “UPA-weighted” group, the sponsor assumed that 60% of patients would receive UPA15 as maintenance and 40% would receive UPA30 as maintenance.
dPrice of vedolizumab corrected. Original: $3,290.00 per 300 mg vial; $822.50 per 108 mg syringe for SC injection. Corrected: $3,401.86 per 300 mg vial; $850.46 per 108 mg syringe per SC injection, as per the publicly available list price in Ontario.29
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note this appendix has not been copy-edited.
Table 12: CADTH Price-Reduction Analyses
Analysis | ICERs for UPA vs. comparators ($ per QALY)a | |
|---|---|---|
Price reduction | CCF subgroup | BF subgroup |
No price reduction | WTP < 111,702 conventional care 111,702 < WTP < 320,562: UPA15 320,562 < WTP: infliximab biosimilar | WTP < 138,440: conventional care 138,440 < WTP < 153,263: UPA-weighted 153,263 < WTP: UPA30 |
10% | WTP < 95,788: conventional care 95,788 < WTP < 352,823: UPA15 352,823 < WTP < 358,083: UPA-weighted 358,083 < WTP: infliximab biosimilar | WTP < 118,939: conventional care 118,939 < WTP < 121,924: UPA15 121,924 < WTP < 136,693: UPA-weighted 136,693 < WTP: UPA30 |
20% | WTP < 79,875: conventional care 79,875 < WTP < 313,194: UPA15 313,194 < WTP < 349,570: UPA-weighted 349,570 < WTP < 407,684: UPA30 407,684 < WTP: infliximab biosimilar | WTP < 99,096: conventional care 99,096 < WTP < 107,007: UPA15 107,007 < WTP < 120,122: UPA-weighted 120,122 < WTP: UPA30 |
30% | WTP < 63,961: conventional care 63,961 < WTP < 273,565: UPA15 273,565 < WTP < 305,475: UPA-weighted 305,475 < WTP < 461,782: UPA30 461,782 < WTP: infliximab biosimilar | WTP < 79,354: conventional care 79,354 < WTP < 92,090: UPA15 92,090 < WTP < 103,552: UPA-weighted 103,552 < WTP: UPA30 |
38.8% | WTP < 49,958: conventional care 49,958 < WTP < 238,961: UPA15 238,961 < WTP < 266,672: UPA-weighted 266,672 < WTP < 509,378: UPA30 509,378 < WTP: infliximab biosimilar | NA |
40% | WTP < 48,048: conventional care 48,048 < WTP < 233,936: UPA15 233,936 < WTP < 261,380: UPA-weighted 261,380 < WTP < 515,879: UPA30 515,879 < WTP: infliximab biosimilar | WTP < 59,612: conventional care 59,612 < WTP < 77,173: UPA15 77,173 < WTP < 86,982: UPA-weighted 86,982 < WTP: UPA30 |
44.9% | NA | WTP < 49,938: conventional care 49,938 < WTP < 69,864: UPA15 69,864 < WTP < 78,862: UPA-weighted 78,862 < WTP: UPA30 |
50% | WTP < 32,135: conventional care 32,135 < WTP < 194,307: UPA15 194,307 < WTP < 217,285: UPA-weighted 217,285 < WTP < 569,976: UPA30 569,976 < WTP: infliximab biosimilar | WTP < 39,869: conventional care 39,869 < WTP < 62,256: UPA15 62,256 < WTP < 70,411: UPA-weighted 70,411 < WTP: UPA30 |
60% | WTP < 16,211: conventional care 16,211 < WTP < 154,678: UPA15 154,678 < WTP < 173,191: UPA-weighted 173,191 < WTP < 624,073: UPA30 624,073 < WTP: infliximab biosimilar | WTP < 20,127: conventional care 20,127 < WTP < 47,339: UPA15 47,339 < WTP < 53,841: UPA-weighted 53,841 < WTP: UPA30 |
62.4% | NA | WTP < 15,389: conventional care 15,389 < WTP < 43,759: UPA15 43,759 < WTP < 49,864: UPA-weighted 49,864 < WTP: UPA30 |
70% | WTP < 308: conventional care 308 < WTP < 115,049: UPA15 115,049 < WTP < 129,096: UPA-weighted 129,096 < WTP < 678,170: UPA30 678,170 < WTP: infliximab biosimilar | NA |
80% | WTP < 75,420: UPA15 75,420 < WTP < 85,001: UPA-weighted 85,001 < WTP < 732,268: UPA30 732,268 < WTP: infliximab biosimilar | NA |
88% | WTP < 43,716: UPA15 43,716 < WTP < 49,725: UPA-weighted 49,725 < WTP < 775,545: UPA30 775,545 < WTP: infliximab biosimilar | NA |
BF = biologic failure; ICER = incremental cost-effectiveness ratio; NA = not applicable; vs. = versus; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose; UPA-weighted = upadacitinib weighted (60% receive 45 mg induction dose + 15 mg maintenance dose and 40% receive 45 mg induction dose + 30 mg maintenance dose).
Note: Only nondominated comparators are presented. Reported points were chosen based on the price reduction at which UPA entered the cost-effectiveness frontier and the price reduction at which it became cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
aDeterministic analyses based on the sponsor’s base case.
Table 13: Summary of Scenario Analyses
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
CCF subgroup | ||||
Sponsor’s corrected base case | Conventional care | 341,739 | 19.97 | Reference |
UPA15a | 551,526 | 21.87 | 110,267 | |
Infliximab biosimilar | 789,833 | 22.54 | 354,281 | |
Vedolizumab-SC | 560,779 | 21.35 | Dominated by UPA15 | |
UPA-weightedb | 575,674 | 21.91 | Extendedly dominated by UPA30, infliximab biosimilar | |
UPA30c | 613,617 | 22.01 | Extendedly dominated by infliximab biosimilar, UPA, UPA-weighted | |
Ustekinumab | 721,569 | 21.82 | Dominated by UPA15, UPA-weighted, UPA30 | |
Vedolizumab | 745,990 | 21.50 | Dominated by UPA15, UPA-weighted, UPA30, ustekinumab | |
Adalimumab biosimilar | 1,044,019 | 20.98 | Dominated by UPA15, vedolizumab-SC, UPA-weighted, UPA30, ustekinumab, vedolizumab, infliximab biosimilar | |
CADTH scenario analysis: Risankizumab includedd | Conventional care | 336,676 | 19.99 | Reference |
UPA15a | 548,871 | 21.90 | 111,167 | |
Infliximab biosimilar | 788,688 | 22.58 | 355,996 | |
Vedolizumab-SC | 557,156 | 21.38 | Dominated by UPA15 | |
UPA30c | 610,438 | 22.04 | Extendedly dominated by infliximab biosimilar, UPA15 | |
Risankizumab | 709,209 | 21.56 | Dominated by UPA15, UPA30 | |
Ustekinumab | 718,257 | 21.86 | Dominated by UPA15, UPA30 | |
Vedolizumab | 742,995 | 21.53 | Dominated by UPA15, UPA30, risankizumab, ustekinumab | |
Adalimumab biosimilar | 1,035,878 | 21.01 | Dominated by UPA15, vedolizumab-SC, UPA30, risankizumab, ustekinumab, vedolizumab, infliximab biosimilar | |
BF subgroup | ||||
Sponsor’s corrected base case | Conventional care | 350,229 | 20.57 | Reference |
Infliximab biosimilar | 567,881 | 22.67 | 103,736 | |
UPA30c | 692,481 | 22.98 | 394,382 | |
Vedolizumab-SC | 516,532 | 21.63 | Extendedly dominated by infliximab biosimilar, UPA30 | |
Ustekinumab | 604,756 | 21.69 | Dominated by infliximab biosimilar | |
UPA15a | 625,272 | 22.53 | Dominated by infliximab biosimilar | |
UPA-weightedb | 649,830 | 22.66 | Dominated by infliximab biosimilar | |
Vedolizumab | 665,805 | 21.61 | Dominated by vedolizumab-SC, infliximab biosimilar, ustekinumab, UPA15, UPA-weighted | |
Adalimumab biosimilar | 895,270 | 21.02 | Dominated by vedolizumab-SC, infliximab biosimilar, ustekinumab, UPA15, UPA-weighted, vedolizumab, UPA30 | |
CADTH scenario analysis: Risankizumab includedd | Conventional care | 350,293 | 20.57 | Reference |
Infliximab biosimilar | 567,849 | 22.67 | 103,569 | |
UPA30c | 692,578 | 22.98 | 399,241 | |
Vedolizumab-SC | 516,551 | 21.63 | Extendedly dominated by infliximab biosimilar, UPA30 | |
Ustekinumab | 604,631 | 21.69 | Dominated by infliximab biosimilar | |
UPA15a | 625,325 | 22.53 | Dominated by infliximab biosimilar | |
Vedolizumab | 666,058 | 21.61 | Dominated by vedolizumab-SC, infliximab biosimilar, ustekinumab, UPA15 | |
Risankizumab | 704,356 | 22.11 | Dominated by infliximab biosimilar, UPA15, UPA30 | |
Adalimumab biosimilar | 898,008 | 21.02 | Dominated by vedolizumab-SC, infliximab biosimilar, ustekinumab, UPA15, vedolizumab, UPA30, risankizumab | |
BF = biologic failure; CCF = conventional care failure; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; SC = subcutaneous injection; UPA15 = upadacitinib 45 mg induction dose and 15 mg maintenance dose; UPA30 = upadacitinib 45 mg induction dose and 30 mg maintenance dose; UPA-weighted = upadacitinib weighted (60% receive 45 mg induction dose + 15 mg maintenance dose and 40% receive 45 mg induction dose + 30 mg maintenance dose).
aUPA45 as induction dose and UPA15 as maintenance dose.
bIn the “UPA-weighted” group, the sponsor assumed that 60% of patients would receive UPA15 as maintenance and 40% would receive UPA30 as maintenance.
cUPA45 as induction dose and UPA30 as maintenance dose.
dRisankizumab included using the sponsor-provided option to do so. UPA-weighted arm excluded.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 14: Summary of Key Takeaways
Key Takeaways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the budget impact of reimbursing upadacitinib for use by adult patients with moderately to severely active CD and a prior treatment failure (i.e., an inadequate response to, loss of response to, or intolerance to at least 1 of conventional and/or biologic therapy).31 The BIA was undertaken from the perspective of a Canadian public payer (excluding Quebec) over a 3-year time horizon (January 2024 to December 2026), using a claims-based approach. The number of patients with CD receiving biologic treatment from February 2021 and January 2023 was estimated using the IQVIA GPM database.32 The sponsor estimated the proportion of patients covered by a public drug plan (57.3%) based on internal data for the number of patients receiving branded adalimumab from a public drug plan as of December 2020.
The sponsor’s analysis included drug acquisition costs and considered wastage, mark-ups, and dispensing fees. Data were obtained from IQVIA Delta PA,11 internal projections, U-EXCEL and U-EXCEED trials,1 previous CADTH reimbursement reviews,33 and published literature. Key inputs to the BIA are documented in Table 19.
Key assumptions included:
Branded adalimumab and infliximab will no longer be available in year 2 and 3 of the BIA based on the implementation of a nonmedical switch (NMS) policy (i.e., only biosimilar adalimumab and infliximab will be used).
Upadacitinib market shares will come proportionally from all currently reimbursed therapies (through new patients or switches).
All patients will receive UPA45 in the induction phase. In the maintenance phase, 60% of patients were assumed to receive UPA15 and 40% assumed to receive UPA30.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Number of CD patients treated with biologic therapy | 27,008 / 32,433 / 39,973 |
Market uptake (3 years) | |
Uptake (reference scenario) Upadacitinib Adalimumab Adalimumab biosimilar Infliximab Infliximab biosimilar Vedolizumab Ustekinumab | 0.00% / 0.00% / 0.00% 0.84% / 0.00% / 0.00% 25.65% / 23.96% / 21.07% 1.08% / 0.00% / 0.00% 52.95% / 57.30% / 61.68% 16.67% / 16.05% / 14.78% 2.81% / 2.69% / 2.47% |
Uptake (new drug scenario) Upadacitiniba | 4.06% / 9.15% / 14.42% |
Cost of treatment (per patient per year)b | |
Upadacitinibc Adalimumab Adalimumab biosimilar Infliximab Infliximab biosimilar Vedolizumab Ustekinumab | $22,570 to $25,916 $20,718 to $23,823 $12,295 to $14,138 $25,765 to $31,602 $12,862 to $15,776 $21,458 to $26,320 $29,958 to $35,878 |
CD = Crohn disease.
aThe sponsor assumed that upadacitinib market shares would come proportionally from all comparators. Disaggregated uptake in the new drug scenario was not provided for comparators.
bEstimated based on an annual cost including the induction dose in the first year of treatment and an annual cost with the maintenance dose only in subsequent years.
cAll patients were assumed to receive UPA45 in the induction phase, with 60% of patients assumed to receive UPA15 and 40% to receive UPA30 in the maintenance phase.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the 3-year budget impact of reimbursing upadacitinib for adult patients with moderately to severely active CD who have demonstrated prior treatment failure would be $67,140,041 (year 1: $7,325,987; Year 2: $20,226,831; year 3: $39,587,222).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Use of a claims-based approach to estimate market size: The sponsor estimated the market size based on the IQVIA GPM database, which consists of claims data. Using that database, the sponsor assumed that the number of CD patients treated with biologic therapy would be 27,008 in year 1, 32,433 in year 2 and, 39,973 in year 3. The sponsor did not specify how the claims data were filtered to consider only claims for CD (e.g., compared to other indications for biologic drugs, such as rheumatoid arthritis, ankylosing spondylitis, ulcerative colitis, plaque psoriasis), or how the number of claims aligns with the Health Canada–indicated population (i.e., moderately to severely active CD and prior failure of at least 1 conventional or biologic treatment).2,31 Further, the sponsor assumed that all claims for comparators were for the indication of interest but did not provide supporting evidence for this assumption. Given that the claims database does not specify the indication and the proportion of claims pertaining to use for other indications is unknown, using a claims-based approach to estimate market size introduces significant uncertainty in the estimated market size as it potentially overestimates the size of the eligible patient population.
CADTH was unable to address the limitations of a claims-based approach to estimate budget impact.
Additional limitations were identified but were not considered to be key limitations. Given the limitations associated with the use of a claims-based approach by the sponsor, CADTH was unable to address these limitations.
The estimated proportion of patients that would be eligible for public coverage is uncertain: The sponsor estimated that 57% of patients would be eligible for upadacitinib via public drug plans, based on the number of CD patients covered by a public drug plan and receiving branded adalimumab as of December 2020 (data from the AbbVie Care Support Program). No further details were provided about the type of data in that source or how it was gathered and analyzed. Patients receiving public drug coverage of adalimumab may not be fully representative of the indicated population’s eligibility for public drug plan coverage.
The average annual cost of upadacitinib may be underestimated: The sponsor has assumed that 60% of eligible patients receive the 45 mg induction dose and 15 mg maintenance dose while 40% of eligible receive the 45 mg induction dose and 30 mg maintenance dose. Clinical expert input received by CADTH for this review indicated that most patients would be expected to receive the 30 mg maintenance dose of upadacitinib. The expected annual cost of upadacitinib is expected to be $23,074 to $30,178 in year 1 and $18,864 to $28,090 in subsequent years, depending on maintenance dose (Refer to Appendix 1, Table 11).
The market uptake of upadacitinib is uncertain: The sponsor’s submitted base case assumed that 4.06%, 9.15%, and 14.42% of eligible patients would receive upadacitinib in year 1, year 2, and year 3, respectively, based on the sponsor’s internal estimates and expert opinion.31 Clinician input received by CADTH for this review indicated that uptake of upadacitinib may be up to double that estimated by the sponsor. CADTH notes that the sponsor attempted to address this uncertainty by performing a series of one-way sensitivity analyses on their base case.
The price of drugs paid by public drug plans is uncertain: The sponsor’s analysis was based on publicly available list prices for all comparators. Adalimumab biosimilar, infliximab biosimilar, and vedolizumab have gone through negotiations at pCPA, and the prices paid by public drug plans are not known.
Omission of potentially relevant comparators: Risankizumab was excluded from the sponsor’s base case. Risankizumab received a positive recommendation from CADTH and is currently under considerations for negotiations with the pCPA.21,22 Should risankizumab become reimbursed on public formularies during the BIA analysis horizon, it will be considered a relevant comparator to upadacitinib for this indication.
CADTH Reanalyses of the BIA
In the absence of more reliable estimates to inform the key parameters of the BIA, the sponsor’s submitted base case was maintained. CADTH expects that the budget impact of reimbursing upadacitinib for the treatment of moderate to severe CD will be sensitive to more reliable inputs which may affect the market size calculation.
Table 16: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $407,430,989 | $441,484,720 | $504,301,914 | $612,792,703 | $1,558,579,337 |
New drug | $407,430,989 | $448,810,707 | $524,528,746 | $652,379,925 | $1,625,719,378 | |
Budget impact | $0 | $7,325,987 | $20,226,831 | $39,587,222 | $67,140,041 |
Stakeholder Input
Patient Input
Gastrointestinal Society
About Gastrointestinal Society
The GI (Gastrointestinal) Society is committed to improving the lives of people with GI and liver conditions, supporting research, advocating for appropriate patient access to healthcare, and promoting gastrointestinal and liver health.
We are a national charity formed in 2008 on the groundwork of its partner organization, the Canadian Society of Intestinal Research (CSIR), which was founded in Vancouver in 1976. We receive national and international attention, simply because we have earned the respect of both the gastrointestinal medical community and Canadians who battle GI and liver issues daily. Our English and French websites received 6,903,208 pageviews by 5,174,016 unique visitors in 2022.
All our programs and services focus on providing Canadians with trusted, commercial-free, medically-sound information on gut (including obesity) and liver diseases and disorders in both official languages. Our BadGut® lectures, quarterly Inside Tract® newsletter, pamphlets, support groups, and educational videos arm Canadians with the information they require to better understand and manage their specific needs. We also work closely with healthcare professionals and governments at all levels toward system-wide improvements in care and treatment.
Information Gathering
The information we used to complete this submission was obtained primarily through questionnaires and interviews:
2015 survey on biologics and biosimilars (then called subsequent entry biologics) completed by 423 Canadians (English: 317 and French: 106) with inflammatory bowel disease (IBD), including Crohn’s disease and ulcerative colitis
2018 survey on the unmet need in IBD completed by 432 Canadians with IBD
2020 survey completed by 579 respondents regarding the unmet needs of IBD
2020 survey on biosimilars with 145 respondents, most of whom had IBD (some had other inflammatory conditions)
2022 survey about the IBD patient journey with 54 Canadian respondents with IBD
2022 focus group with several persons living with Crohn’s disease so we could map the patient journey and animate it, which is available on our website at http://badgut.org/patient-journeys and we encourage your reviewers to watch this short video
one-to-one interviews with 2 individuals living with Crohn’s disease who participated in a clinical trial for upadacitinib and received the trial drug
We also had contact with patients affected by IBD through one-to-one conversations at our BadGut® Lectures; a patient roundtable; recent phone/email/social media interactions with individuals who have IBD; and stories submitted over time from patients.
Disease Experience
Crohn’s disease is an inflammatory bowel disease (IBD) that can arise at any age, commonly occurring in young people. It is a systemic disease, and inflammation can involve any area throughout the entire digestive tract. There is an increased risk for those who have a family member with the condition. Currently, Canada has among the highest prevalence and incidence yet reported in the world, with approximately 135,000 diagnosed individuals.
Diarrhea, rectal bleeding, abdominal pain, and weight loss are some of the common recurring symptoms of Crohn’s disease. Inflammation decreases the intestine’s absorptive surfaces, triggering watery stools that can lead to fecal urgency and poor control of bowel function. Low red blood cell count (anemia) can result from blood loss due to ulcerations in the intestine and from general malnutrition due to decreased nutrient absorption and the debilitating effects of the disease.
Some patients have extra-intestinal manifestations, including fever, inflammation of the eyes or joints (arthritis), ulcers of the mouth or skin, tender and inflamed nodules on the shins, and numerous other conditions. Anxiety, stress, and mental health are major factors.
Crohn’s disease often has a profound effect on an individual’s life – physically, emotionally, and socially, both at home and at school or in the workplace. Symptoms can be relentless, embarrassing, and scary. The severity of the disease can fluctuate, making it necessary to go through routine testing, reassessments, and medication changes. It is particularly difficult for children and young adults, since it often affects a person’s sense of self.
More than anything, patients have told us that sustained remission/treatment response is more important than relieving any one symptom of Crohn’s disease. As a chronic disease, it is never just one flare that dominates the impact of the disease, but the constant concern that there will be future flares, possibly worse than the last, at unpredictable times, which can disastrously disrupt their lives.
The following quotes are from individuals describing what it feels like during a Crohn’s disease flare, and what their biggest concern is, in their own words:
“Your gut aches and burns and there is often blood in the toilet. You lose your appetite and weight, unhealthily! My biggest concern is I’m going to run out of meds to help!”
“It’s like I can’t control anything, I feel weak and can barely get up. My biggest concern is usually when I see blood and determining at what point to go to the ER.”
“The pain is worse than childbirth... and I have 3 kids...1 labour without drugs.”
“Worst flu symptoms, fatigue, lethargy, like swallowing glass and chili and then having constipation and diarrhea at the same time. Gut cramps and hunger cramps at the same time. Want to die. Biggest concern is needing a toilet at all times with zero minutes waiting time.”
“It feels like my guts are in a vise. The nausea can be so bad I can’t move or even vomit and the diarrhea is so painful I’ll be literally screaming in the bathroom.”
“The worst part is fear of irreversible permanent damage that will affect your day-to-day life forever.”
“It is so exhausting and feels like it will never end. You start to question if you can still live the life you planned. And no-one gives you a break.”
“A flare can come out of nowhere and completely disrupt your life. Pain can sometimes be so bad that it keeps you in bed. You mostly spend life either asleep or on the toilet. My biggest concern during a flare is being able to keep up with my responsibilities (work, school, social, etc.).”
“It feels like your body is betraying you. You can’t plan anything in advance because you don’t know how your body will feel on a day-to-day basis.”
“There’s a huge element of fear and worry and being faced with mortality at such a young age.”
It’s one thing to read a list of common symptoms or data on how Crohn’s disease affects patients, but it is the individual stories of these patients, as summarized above, which astound us and motivate us to support patients’ need for more diversity in effective treatments. In addition, treatments should improve quality of life, not cause more symptoms, pain, frustration, or hardship.
Experiences With Currently Available Treatments
The treatment of Crohn’s disease is multi-faceted; it includes managing the symptoms and consequences of the disease along with therapies targeted to reduce the underlying inflammation. Typically, a patient starts on one type of treatment and, if there is inadequate response, then switches to another type.
5-ASA helps to settle acute inflammation and, for some patients, keeps the inflammation inactive when taken on a long-term basis (maintenance). To reduce inflammation in moderate to severe cases of Crohn’s disease, corticosteroids can help. For topical relief in the colon, corticosteroids are available in rectal formulations. These are inconvenient therapies that make it difficult for patients to keep a normal routine. Also, if a patient has significant diarrhea, then the rectal medications may be difficult to hold in place for sufficient time to be effective. Immunosuppressive agents reduce dependence on steroids and help patients who have steroid-resistant disease, but it could take up to six months or more of therapy to see results.
Biologics treat Crohn’s disease when older medications fail to relieve symptoms. However, there are a variety of mechanisms through which they work. A newer class of medication for IBD, Janus kinase (JAK) inhibitors, typically work faster than other immunosuppressive medications, pose no risk for immunogenicity, unlike biologics, and are easier and more convenient to take since they are in pill form. However, upadacitinib (Rinvoq®) is currently the only medication of this class available for Crohn’s disease.
While there are a few options available, patients still have a lot of difficulty obtaining remission or adequate symptom relief. In one of our surveys, we asked patients if the currently available medications are adequate to control their disease. Only 24% of those with IBD thought that the available medications are adequate, 56% found them to be only somewhat adequate, and 20% not at all adequate. Patients are still suffering, and they need new and effective options to achieve mucosal healing and reduce the debilitating symptoms of Crohn’s disease.
Patients also rely on medications to reduce the need for surgery. This can include surgical treatment to repair fistulas and fissures or more complicated procedures such as the removal of all visible and microscopic disease in the bowel, and they might then need a stoma to collect stool. With the loss of colon function, bowel movements can occur frequently and have high liquid content. Sometimes multiple surgeries are necessary. Even after surgery, Crohn’s disease symptoms tend to recur in as many as 75% of patients.
Improved Outcomes
Patients affected by Crohn’s disease need access to medications that work. Inadequate access to medication results in preventable patient suffering (e.g., continual, debilitating disease symptoms; secondary illnesses such as depression and anxiety disorders; and loss of family/social interactions). It also leads to unnecessary usage of healthcare resources (e.g., hospital stays, surgeries, diagnostic procedures, other medications) and a ripple effect of financial burden on the government and taxpayers (e.g., through inability to work, long-term disability claims, biologic-related debt, and even bankruptcy).
We know that biologics are effective at treating Crohn’s disease; these medications have revolutionized treatment for inflammatory conditions. In one of our surveys, 63% of respondents reported symptom reduction on a biologic and 23% reported confirmed remission. Many of these individuals had been suffering for years trying to find a treatment that works. Unfortunately, due to several factors, including non-medical switching policies adding stresses and burdens on patients’ continuity of care as well as loss of control on treatment choice, some patients are no longer taking biologics at all. Many patients already feel powerless and are fearful of the future impacts of the disease on their health.
When the Crohn’s disease patient receives the right medication at the right time and for the right duration – as determined between physician and patient – these individuals can live full, rewarding lives as productive, valuable citizens who participate in the workforce and community. However, since patients are unique, they respond differently to various medications, and in some cases stop responding to medications after using them for some time, it is important to have a variety of options available.
Experience With Drug Under Review
We interviewed two patients who participated in clinical trials for upadacitinib in Canada. Both continue to take the medication.
Patient #1
She received a diagnosis in her 40s, but her gastroenterologist had suspected Crohn’s disease for several years and had been unable to detect it in her bowel. She knew she had a sensitive stomach since eating garlic and greasy foods, and undergoing a lot of stress, were some of the triggers for her symptoms. She experienced flares with symptoms of nausea, diarrhea, and cramping, that usually lasted for a few days to a couple weeks. However, a few years ago she experienced the worst flare she’s ever had. It started with watery, bloody stools and continued for two months. This gave her gastroenterologist the opportunity to find the inflammation and give her a diagnosis. “I was only working [at] about 10% capacity but I had good management that understood what was happening. If I wasn’t an office nurse and had an operational job, I never would’ve been able to do that. Grocery shopping was horrible, and I was going to the bathroom every 10 minutes.”
Her gastroenterologist recommended corticosteroids for treatment, which she rejected. She did not want the side effects, especially since they would only induce remission and reduce inflammation but not maintain it in the long-term. He also offered biologics and she rejected this as well. “I didn’t want to plan my life going to the hospital and spending a few hours there getting an infusion done, especially with the COVID pandemic.” Her gastroenterologist then discussed with her the clinical trial for upadacitinib, which she was eligible for.
It has now been a year and a half since she started the clinical trial. “Within a week, it was a night and day difference,” she said. When asked what side effects she experienced, she described having a bit of weight gain and infected hair follicles, but they were minor compared to the medication’s impact on her quality of life. “The side effects are so mild that I can’t complain.” She also had swollen joints before, but this has now improved. She goes in every 10 weeks to meet with the study nurses and her gastroenterologist. The pill is easy to take as it is only once a day, which she takes in the morning. “Oral medication makes travelling easy. I don’t have to schedule my life around I.V. infusions. It’s very discreet. Nobody has to know you’re taking it.”
She contracted COVID-19 in August, and at that time, her healthcare team instructed her to stop taking the medication. Within a day or two, her symptoms were back and felt worse than COVID. She also knows that stressful situations can trigger her bowel. She was laid off and had to look for a new job but being on the medication kept her symptoms under control and helped her cope.
She is now in remission for the first time since her flare years ago. “I’m very grateful for this medication and I just love how it lets you be normal again and be able to live and not have to worry. Your bowels are not your first thought all the time. Now it allows you to be more spontaneous and go out and have fun.”
Patient #2
He started his path to diagnosis during the height of the pandemic in 2020. He was preparing for his finals for his teaching practicum when he began to experience severe symptoms. He had briefly experienced similar symptoms a few years ago, so he didn’t think anything of them at first. Although they started to subside, he went in to see his doctor who immediately referred him to a gastroenterologist that same day and booked him an appointment for a colonoscopy. He said, “I would’ve lost my colon if I hadn’t done anything about it.” His gastroenterologist then gave him a prescription for Mezavant®.
Everything was fine for him until a few months after his son was born when he began experiencing more flare ups and joint pain. He went to the emergency room and the doctor diagnosed it as gout. After repeated episodes of joint pain, they diagnosed it as rheumatoid arthritis, which his gastroenterologist said was a common comorbidity of Crohn’s disease. He was then put on a rolling prescription for a low dose of prednisone that he took daily. However, this did not stop his flare ups and joint pain. “It was like clockwork. Every two months, I found myself in the ER since I couldn’t walk, couldn’t move, and then they’d pump me with a larger dose of prednisone.”
Seeing that this was not a sustainable way of managing his Crohn’s and arthritis, his gastroenterologist discussed biologic therapy as a treatment option. He rejected this option. “It was a biologic injection every six weeks meaning I had to take time off work since it takes 3-4 hours a day. At the time, where I was at financially, I had to prepare for that, and it was a little difficult for me. The cost was significant, especially not knowing if it would be covered by my providers. It discouraged me because it’s difficult to go through a medical episode and have medical care and you may not be able to afford it and may not be able to move. Something I can do at home and take a tablet is a better option. It just made more sense for me since it allows me to not have to take time off work and lose wages and time. I already have to take time off for colonoscopies every year.”
His gastroenterologist then shared with him the clinical trial for upadacitinib. Since then, he has had no issues or side effects for two years and counting. He had a colonoscopy last week where his specialist said there were no signs of Crohn’s disease in his bowel. He is still taking a low dose of Mezavant® once a day but tapered off the prednisone for his arthritis within a few weeks of taking the study drug. “I feel great. I don’t even have to think about Crohn’s disease. I consider myself to be back to normal.”
Companion Diagnostic Test
Not applicable.
Anything Else?
Despite the pandemic and ongoing strains on the healthcare system, we thank the enduring efforts of healthcare professionals in caring for patients. Both patients we interviewed spoke about the significant impact that a physician can have on a patient’s life. Here is what they said:
“It’s a game changer for Crohn’s when you have a healthcare team that understands the disease and having somebody that listens and is very supportive of patients.”
“I’m very, very thankful for being referred to my gastroenterologist. I don’t want to think about what it could’ve been if it was somebody different.”
Conflict of Interest Declaration — Gastrointestinal Society
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Yes. We are incredibly grateful for the time and input that we received from the patients who had direct experience with upadacitinib. We also have a wide range of individuals from across the country who respond to our surveys and requests for real information on what it is like to live with Crohn’s disease.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Gastrointestinal Society
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | — | X |
Crohn’s and Colitis Canada
About Crohn’s and Colitis Canada
Crohn’s and Colitis Canada website (https://crohnsandcolitis.ca/)
Crohn’s and Colitis Canada is the only national, volunteer-based health charity focused on finding the cures for Crohn’s disease and ulcerative colitis, the two main forms of inflammatory bowel disease (UC) and improving the lives of children and adults affected by these diseases.
Crohn’s and Colitis Canada is one of the top health charity funders of Crohn’s and colitis research in the world, investing over $140 million in research since our founding in 1974. The organization also delivers on its promise through patient programs, advocacy and awareness. We help improve the quality of lives today by:
Sharing accurate and reliable information on treatments, research and issues related to life with Crohn’s and colitis through website, print materials, webinars and live events;
Increasing public washroom access through the GoHere program;
Raising awareness about these Canadian diseases with bilingual public communication;
Offering kids with Crohn’s or colitis camp experience;
Providing a peer support program to newly diagnosed people; and
Advocating on behalf of the patients and caregivers on priority concerns and needs.
Crohn’s and Colitis Canada is comprised of approximately 65,000 supporters including volunteers, donors or individuals interested in engaging with the organization. There is no paid membership. Crohn’s and Colitis Canada is governed by a national volunteer Board of Directors. The organization has a network of volunteer-led Chapters in 46 communities across the country, offering information, events, fundraising opportunities and encouragement. There are thousands of volunteers from coast-to-coast supporting Crohn’s and Colitis Canada’s mission.
Information Gathering
Information summarized in this submission was compiled from a variety of sources, including Crohn’s and Colitis Canada’s 2018 report “Impact of Inflammatory Bowel Disease (IBD) in Canada”, a survey conducted in early 2022 to better understand the priority needs and concerns of IBD patients and their caregivers (1,700 respondents; 687 with moderate to severe Crohn’s disease) and live interviews with three patients who participated in Rinvoq / upadacitinib clinical trial. The results from the patient survey provide a window into how patients with moderate to severe CD live and manage their symptoms. Of those surveyed, 63% had a diagnosis of CD, 34% had ulcerative colitis, while the rest had other forms of IBD. Patients came from all over Canada and varied in age. The vast majority of respondents, some 76%, were female 76%, 23% were male and 1% non-binary.
Disease Experience
CD is a life-long, episodic, autoimmune disease that primarily affects the large intestine. CD can be diagnosed in all age groups, but most diagnoses are amongst youth, young adults (16 – 30 years) and seniors. The majority of Canadians living with CD are working-age Canadians. CD can affect anywhere in the gastrointestinal system, from mouth to bum. Symptoms include unpredictable urgent bowel movements, bloody diarrhea, bloating, unbearable pain and often debilitating fatigue. CD unfortunately affects every aspect of a person’s life from family, friends and work activities.
When asked what symptoms they have experienced, most reported stress and mental health disorders (69%), followed by anal fissures and hemorrhoids (61%), joint inflammation & arthritis (60%), malnutrition and weight loss (60%), anemia (58%), skin conditions (40%), adhesions/scar tissue (32%), bowel obstruction (32%) and surgery/bowel resection (30%), eye inflammation (29%), perianal or anal fistulas and abscesses (27%), internal (or intra-abdominal) fistulas or abscesses (26%), stricture (23%), ankylosing spondylitis/arthritis of the spine (12%), liver conditions (8%), and cancer (5%).
Nearly nine-in-ten (87%) agree that most people don’t understand what IBD is and 56% of patients agree that their family and friends don’t understand what they are going through. Nearly all patients surveyed reported they continue to experience symptoms of IBD. Asked what symptoms they’d experienced in the previous two weeks, 100% reported fatigue, 99% abdominal pain/cramps, 98% pain, 97% constipation, 95% gas, 79% bloating, 78% urgency to use bathroom, 73% diarrhea, 39% nausea and vomiting, 38% diminished appetite and weight loss. Nearly half (48%) believed that different treatment options could make them feel better. Thinking back to when they were first diagnosed, 35% of patients noted that they hid aspects of their diagnosis from friends, coworkers and classmates. Nearly half (48%) felt they could not be open about their condition as they’d like. Nearly as many (47%) felt isolated as a result of their IBD, with 43% reporting that their condition has had a negative impact on their romantic relationships with spouses/partners.
A significant proportion of patients have adjusted their lifestyle and expectations with 72% agreeing that they’d changed the expectations they had of themselves or that they are always adapting their lifestyle to account for their IBD and 45% agreeing that they’d changed their career goals.
Due to unpredictable urgency of bowel movements, accidents are not uncommon, especially when a patient is experiencing a flare. Patients often hide their disease from work colleagues, friends and even relatives because of the perceived stigma of the condition being a “poop” disease. Unable to predict when their next flare will occur and how to control their flare, isolation, stress and anxiety are companions to the patient’s disease journey. In extreme cases, patients have thought of suicide because of their inability to control/cope with the impacts of CD on their personal and social lives, as well as consequences in their career or school. 6 in 10 respondents felt that their CD has impacted their romantic relationships. Chronic fatigue and anemia are also consequences of CD. Crohn’s disease is perceived as a lonely disease in that 9 in 10 felt that most people do not understand what CD is and almost 6 in 10 feeling isolated because of their CD.
All three patients we interviewed experienced severe and debilitating symptoms. The first, interviewee 1, a nurse, was going to the washroom about fifty times a day at the time of her diagnosis, blood in her stool, cramps, bloating.
Reports Interviewee 1:
“I couldn't sleep. I was having to get up all night to go to the washroom and really it was awful. I worked 5 minutes from my home, and I was scared to drive from work from home to work, but still had to continue working. So spent most of my workday in the bathroom and yeah, even to go grocery shopping was it was terrifying and embarrassing because you didn't know if someone else was in the washroom when it hits you… It was very embarrassing. It was very constricting. No opportunity to be spontaneous, like the thought of going to travel anywhere… There’s no way I could have gone anywhere. We love going to Disneyland. The thought of waiting in the line for a ride. There’s no way I would have been able to. We have two dogs at home. I couldn’t even take them for a walk because I was too afraid that something would happen five minutes into the walk. So super constricting and like I say, embarrassing. Like, you don’t want to tell everybody that you know, you need to go to the bathroom 800 times a day. And just you know… zero quality of life during that time…[I] didn’t want to eat anything… I didn’t want to drink water because as soon as anything came in, it came right out… It really impacted sleep, food, social life. Just every aspect of quality of life…The other piece I forgot to mention this…I get almost like rheumatoid arthritis symptoms where all my joints are very stiff and sore, and it impacts literally every joint. And so that was debilitating too. That’s one that always worries me about coming back too because…I’m only 53…the thought of having to deal with that for another 20-30 years is not the greatest thing to look forward to.”
The second, Interviewee 2, a substitute teacher, was told at the time of diagnosis that he was probably “two to three weeks from losing [his] colon”. He was started to a medication to “help with the inflammation” and later when he developed severe joint pain Prednisone.
Reports Interviewee 2:
“The medication that I was on was working, but it wasn't working to its full potential…to the point where I was struggling to go to the bathroom…I kept having these flare ups where I developed arthritis which is a common side effect that comes with Crohn's. Plus, I was also dealing with joint pain where there were days where I couldn't move at all.”
The third, Interviewee 3, a shift worker, reports needing to “run into the bathroom between seven and eleven times a day” while dealing with constant fatigue, a result of all the blood he was losing with each visit to the bowel movement:
Reports Interviewee 3:
“I work shift work, which requires me to work 12-hour shifts on a desk. I don’t really have the freedom to kind of leave and walk around all the time. So, you know, it impacted work. I love playing sports, so that impacts sports when you know, every time you get a chance, you have to run off the field or off the rink to go to the bathroom and then come back. My fatigue levels were insane to keep up with, and I don't drink coffee, so it was very hard for me lots of days to wake up and actually be productive. On my days off, I spent most of the time just sitting around doing nothing. It also affected, you know, my relationship with my wife because she always wanted to do stuff and I just didn't have the energy. I wasn't in a good mood…The biggest worry was the amount of blood I was losing every time I went to the washroom. That was the big problem… In that last six months there, I started to have so much blood in my stool that…my energy levels went down and that’s when it became concerning.”
Experiences With Currently Available Treatments
Canadians have one of the highest rates of prevalence of Crohn’s disease, however, when compared with other Western countries, there are fewer treatment options available for people with moderate to severe forms of CD. 6 out of 10 of the respondents feared running out of treatment options. That currently available treatments are suboptimal to treat CD is apparent where 8 out of 10 of the respondents hoped for better treatments to better manage their disease.
In spite of being on treatments, over 7 in 10 respondents indicated that they experienced diarrhea, bloating and unpredictable urgency to use the washroom at least some of the days. 5 in 10 experienced rectal bleeding and nausea and vomiting.
Only one of the three patients we interviewed had experience with other therapies. This patient had been given a drug to control the symptoms of their CD (they didn’t recall the name of the drug) alongside Prednisone. According to the patient:
“They gave me a bunch of Prednisone steroid and sent me on my way, and it would clear things up… Everything would be OK and then it would come back on again. The joint pain would come back on again… So, I would go through those episodes where the joint pain would come up and I’d still have issues internally where things weren’t working. And then I would end up going to the hospital. I would end up getting steroid treatments and more Prednisone… They would just pump me full of higher doses of Prednisone to pump everything through and then send me on my way.”
Improved Outcomes
Patients seek any treatments that can mitigate their symptoms to protect a patient’s ability to work productively, attend school and social events, and even basic daily necessities like leaving the house to run errands or have the energy to maintain a household or raise children. Quality of life could be greatly improved in CD patients if their flares are brought into remission and current treatment options do not appear to be addressing the symptoms of most concern for CD patients.
When asked which factors are the most important in managing their CD, 8 in 10 indicated (along with medication) unpredictable and frequent bowel movements, pain and fatigue. In spite of their treatments, over 7 in 10 respondents indicated that they experienced diarrhea, bloating and unpredictable urgency to use the washroom at least some of the days. 5 in 10 experienced rectal bleeding and nausea and vomiting.
When asked about other important aspects of treatment options, taking fewer medications and minimizing chronic steroid use were scored highly; where at least 7 in 10 respondents scored 7 and above for fewer medications and 9 in 10 for minimizing chronic steroid use (on a sliding scale of 0 (not important at all) to 10 (extremely important), with an additional option of “I don’t know”).
The three patients interviewed had very different ideas about improved outcomes and trade-offs based on their circumstances. For example, one had experienced extreme fatigue resulting from blood loss in stool over a six-month period, so for him the most important improvement he would be looking for was alleviation of this particular symptom. Another patient who’d experienced frequent bowel movements accompanied by arthritis and extreme joint pains said that alleviation of these symptoms was most important, but that he’d “put up with some aches and pains” in order to “manage needing to go to the washroom”. The third, a nurse, said that her ideal drug would be “something that was easy [to administer]” that was “non-invasive with minimal side effects” that “could get rid of the rheumatoid arthritis symptoms too”.
All three cited convenience as being an important attribute to them. Though none had prior experience of infusions, they were aware of the other treatment options.
Interviewee 1:
“Being able to be spontaneous and not having to schedule your life around your IV infusions, that that's a huge thing as well. And I also just feel like there's the opportunity for more even dosing… you're [not] getting a big dose of IV… then it's wearing off and then you have to have another high dose. So those ups and downs, so really quality of life would be impacted greatly in in just that that work life balance and being able to not have to schedule your life around IV infusions… Anything that you can do at home without having to expose yourself to everything in the hospital and having to sit there and infuse to me would be much, much preferable.”
Interviewee 2:
“Now going for an infusion, to my understanding has to be every however many weeks and you have to go to a clinic to do it, which means not only do you have to take a day off work, you gotta facilitate all that stuff so that you can be looked after… make sure you get a ride home… It was rumored at the time when we talked about it that... each treatment can be up to $10,000… we don't know if they would have been covered or if they would have been covered fully or partially or what.”
Interviewee 3:
“As far as I understand, most of the treatments revolve around some sort of injection right now. And to me, I don't care about needles. That doesn't bug me. But I know some treatments require going in for a shot. When it comes to a home remedy that requires whatever solution you're using has to be frozen or kept in a freezer. So now if you want to travel, if you want to go and do things for a weekend, you have to make sure that you know you have a freezer pack or you have to have some sort of way to maintain that product so that you can use it. And to me, that's…There's no logic in that really. For everyday life right now, you're hindering people just to take care of themselves… If I had a solution that requires a freezer, let’s say I go, you know, visit family somewhere, I have to make sure they have a freezer available. If I go out to the lake, I got to make sure the freezer is available. It becomes a hindrance to other people and to myself where I can go when I can go places whereas a pill, I mean, you just throw that in your bag and you’re good to go. You just take it when you need to take it.”
Experience With Drug Under Review
Key values of Rinvoq: All three patients reported near immediate improvements in their health, including alleviation of the symptoms of their CD. None of the interviewees reported significant side effects from using the drug. Two said they weren’t aware of any side effects and the third said they experienced some acne and some weight gain since going on the drug. All patients we interviewed spoke to the convenience of being able to take a pill, rather than having to report to a clinic for infusions, the fact that it needn’t be refrigerated being less of a hindrance to travel or visits with friends or family.
All three patients interviewed accessed the drug through participation in a clinical trial program. Experience with other drugs and therapies was fairly limited as most had only recently been diagnosed with CD.
Interviewee 1:
“It’s been a complete game changer!”
Prior to getting into the Rinvoq clinicial trial, the interviewee had been struggling with frequent bowel movements, up to 50 in a single day, many of them bloody, fatigue, abdominal pain, rheumatoid arthritis and other symptoms of Crohn’s disease. She describes herself as having “zero quality of life during that time”, adding she “didn't want to eat anything…didn't even want to drink water…because as soon as anything came in, it came right out…It really impacted sleep, food, social life. Just every aspect of quality of life.” She reports that within a week of starting treatment on Rinvoq that her frequency of bowel movements went from 50 times a day to “probably three to four times a day”, that her rheumatoid arthritis had improved, as had her quality of life:
“My joints feel better. My guts feel better. And just my overall quality of life is greatly, greatly improved. I don’t hesitate to do things like drive for three hours or go on a plane. I’ve gone to Disneyland twice now with being on the medication and it hasn’t been an issue whatsoever. So yeah, I can take my dogs for walks again. I can go to the gym. I can do all those things that I wanted to do before. I can go swimming, where I couldn’t do that before.”
“I think I'm much more pleasant. I'm able to spend time with family. I'm able to go and do the task that I need to do so things like grocery shopping, things like going, going to work, doing those sorts of things. I'm much more pleasant because I'm not in pain and I'm not in the washroom. You get very dangerously close to being depressed and some people may cross that line when you can't do anything, and you just feel quite helpless. So, it's really given control back around… what you can and can't do. It's allowed me to travel with my family again. It allows me to go out to eat at a restaurant again. So really, it's just allowed me to have that freedom to spend time with them again.”
Interviewee 2:
“Night and day. Right now, everything is to the point where I can live that normal life. The only thing I have to do is take a tablet each day…a five second task that allows me to continue living the life I want to live.”
The second interviewee had been diagnosed with CD nearly three years previous following what he’d initially thought was an episode of food poisoning. His symptoms included abdominal pain, fatigue, frequent bowel movements, constipation, arthritis and severe joint pain which can only be described as debilitating, leaving him in bed or on the couch and unable to work or spend time with his wife and son. Initially treated using conventional therapy, the level of Prednisone needed to control his symptoms and manage his joint pain rapidly increased to the point where he and his doctor both agreed it was unsustainable. He is currently on Rinvoq due to his having participated in the clinical trial. He reports he “very rarely” has to worry these days “about where the bathroom is” or that he’s “not going to be able to walk today” or if he’s “going to be able to work”.
Getting on Rinvoq has had significant positive impacts on the second interviewee’s day-to-day life, family life and working life:
“I can live my life as close to what it was before everything happened… I can do everything that my wife does… everything that I could before, even before we had our son. Just this past summer, we spent a week, and we went up into the mountains. We could climb mountains. There aren’t a lot of bathrooms up in the mountain and the physical ability to go up a mountain is challenging, so you don’t have to worry about, you know, can I make it to the top?... So, it allows me to contribute to my family where my wife doesn’t have to worry about what’s going to happen or should we have another baby, because we’re trying to have another baby, and it’s like we don’t let what I have get in the way because of this. It allows me to live my life as a family man and do many other things along with it.”
The interviewee had been working during this period as a substitute teacher and his condition had a significant adverse impact on his ability to work. He figures that he likely had to turn down at least 20% of the opportunities he was offered. More than this, he figures “There was missed opportunities that could have turned into full time work or a longer period of work. I’m at the point now where I’m able to take long period work because I’m making those connections because I’m consistently working. I don’t have to say ‘Sorry, I can’t come in today because I can’t move right now.’ Schools want people that are reliable and that they don’t have to say, ‘You’re not going to be here’ or ‘We can’t rely on you, so we’re going to go with the next person.’”
Interviewee 3:
“When [my doctor] told me it was a pill a day, I was like, sign me up. Let’s go. It doesn’t get easier than that.”
The third interviewee went almost directly into the clinical trial after diagnosis with CD. He had been experiencing quite a lot of blood in his stools over a six-month period which resulted in extreme fatigue. According to this patient:
“Overall, I'd say the last six months up until my diagnosis and then my entry into the trial, it was pretty hard to really have motivation to do anything other than work. My health sprang back within a couple weeks. I was back to myself, back to energetic. Stools were back down to one to two a day and normal. I could go back to eating whatever I wanted. So, I guess that impacted my family, my relationship because now we could go back to the way things used to be. We didn't have to worry about certain situations and certain vacations and stuff like that. We could just enjoy what we previously enjoyed.”
Companion Diagnostic Test
None of the three patients had any issues resulting from the companion diagnostics associated with their clinical trial. All three lived within a short distance from the clinic and hospital. The costs of the diagnostics were covered so none were out of pocket. One stated that he experienced a little anxiety around tests where the results were not as positive as were hoped but was grateful for the information and insights that came from the testing. Another said she found it embarrassing having to carry around a stool sample and a little inconvenient having to take time off work to get to the lab to drop off samples or to the hospital for endoscopies or colonoscopies, but other than that everything was fine.
Anything Else?
Interviewee 1:
“Once you start the medication and the results were so fast, which is also another big bonus because I know with the IV biologics they do take some time to get up in your system, and this is so quick and the results were so dramatic for me that I wish everybody who had Crohn's disease or colitis was able to try this medication.”
Interviewee 2:
“I think with something that's little, that comes in a little tablet, that you can take orally each day, I think there's a lot of promise there that, you know, people can get on the track to being better and… it can be more readily accessible than having to tie up clinics and infusion labs and whatever it is that people need to go through to do that… And they don't have to have any lost time because of it. So, take it how you will, I believe that we're on to something here.”
Interviewee 3:
“As far as I know, basically you could take this drug and within I think 24 hours, it's starting to work and then if you stop taking the drug within 24 hours, it clears your system. I don't see why you couldn't go get a prescription for this. Take a pill within a day. You're feeling better and then you could continue to take pills per day until you see. Or until I guess you, you're feeling better and then you can drop the pill and it clears your system. I mean, to me, it's almost like a like an Advil if you think about it. I’d say that everything with this drug’s been pretty straightforward and pretty simple, so hopefully everything goes well, and we'll start to see it on the shelf.”
Conflict of Interest Declaration — Crohn’s and Colitis Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
The first survey was conducted in collaboration with Leger who performed the initial analysis of the data.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Programs, research and medical conferences, educational brochures, kid’s camps, post-secondary scholarships as well as outreach and advocacy activities on behalf of Canadians living with Crohn’s and colitis. The vast majority of Crohn’s and Colitis Canada’s funding comes from individual donors contributing to fundraising events such as the Gutsy Walk. Crohn’s and Colitis Canada is participating in this review as part of our advocacy for Canadians living with inflammatory bowel disease and does not endorse or recommend the use of specific products or treatment or attribute of any product. No sponsor was involved in developing the content of this submission.
Table 2: Financial Disclosures for Crohn’s and Colitis Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | — | X |
Clinician Input
Canadian IBD Specialist Group
About Canadian IBD Specialist Group
Our clinician group is an ad hoc group of 24 gastroenterologists from across Canada, the Canadian IBD Specialist Group. We include physicians who are national and international experts in the diagnosis and management of inflammatory bowel diseases (IBD). Collectively, we have over 200 years of clinical experience in caring for patients with Crohn’s disease and our group members have published over 2000 scientific peer-reviewed publications. Members of our group have figured prominently in the development of the Canadian Association of Gastroenterology (CAG) Canadian Consensus Guidelines on the treatment of Crohn’s disease (2019) with 2 members of our group having been co-primary authors. Furthermore, several of our group members figured prominently in the 2018 Crohn’s Colitis Canada impact of IBD in Canada report. We have come together at this time in an effort to help our patients with Crohn’s disease (CD) access new, highly effective and safe therapies as they become available following Health Canada approval with the ultimate goal of addressing gaps and unmet needs in the treatment landscape.
Information Gathering
On March 7, 2023, the Canadian IBD Specialist Group met virtually to discuss the current CD treatment landscape. The meeting consisted of individual presentations of data supporting the topics included in this submission, including currently available treatments, treatment goals, and unmet needs, were reviewed and discussed. In addition, a review of the safety and efficacy data from the upadacitinib phase III development program was presented. These presentations were followed by workshops in which participants reflected on the data presented and their own clinical experiences and offered their personal expert opinions on the above topics. The final discussion focused on the CD burden in Canada and how upadacitinib would best fit into the management paradigms for Crohn’s disease given the current burden of disease, the existing unmet needs and the available evidence regarding the effectiveness and safety of upadacitinib. The group considered how access to upadacitinib could fulfill these unmet needs in the treatment landscape and how this might benefit patients and society in the short and long term.
The “consensus” captured during the meeting serves to guide the input to the Canadian Agency for Drugs and Technology in Health (CADTH) for upadacitinib reported here. This report was reviewed by the group present as well as additional experts who did not attend the March 7th meeting but had expressed interest in providing their expertise and opinion into this submission. The summary of the discussion is captured herein.
Current Treatments and Treatment Goals
Disease Overview and Background
CD is an inflammatory bowel disease (IBD) characterized by recurrent, chronic, uncontrolled, idiopathic immune-mediated inflammation that can affect any part of the gastrointestinal (GI) tract from mouth to anus.1 Although inflammation in CD can occur anywhere in the GI tract, it most commonly affects the small intestine and/or colon.2,3 As CD is a chronic but fluctuating disease, patients will go through periods in which the disease flares up, is active and causes disabling symptoms.4 Symptoms of CD include persistent diarrhea (loose, watery, or frequent bowel movements), abdominal cramping, abdominal pain, fever and, at times, rectal bleeding.5 These may also be accompanied by loss of appetite, weight loss, malnutrition and fatigue.5 The hallmark of disease activity is the inflammation which can be seen on endoscopic, radiographic or histologic evaluation. Endoscopy remains the gold standard in diagnosing and detecting active disease, but other modalities can provide additional and sometimes complementary information.
CD can be classified according to the Montreal classification, which considers age of onset (≤16, 17-40, >40 years), disease location (terminal ileum, colon, ileocolon, upper GI tract), and disease behaviour (non-stricturing/non-penetrating, stricturing, penetrating).3
Historically, disease activity in CD is generally assessed in clinical trials using tools that measure signs and symptoms of the disease and in clinical practice by subjective assessment of signs and symptoms.6 Overall, disease activity can be assessed using patient-reported outcomes (PROs) as well as the Crohn’s Disease Activity Index (CDAI). A CDAI score of ≤150 indicates clinical remission, which has been used in many of the phase III clinical trials for CD therapies.7-17 The CDAI incorporates both subjective and objective components, including hematocrit, bodyweight, the presence of an abdominal mass on physical examination, the use of medications, and the presence of a number of extraintestinal manifestations (EIM).18
However, the CDAI is only done at a given point in time in the patient’s disease course and it does not truly capture the overall severity of the disease since the score is not necessarily reflective of disease prognosis or disease course. In addition to the CDAI, complete CD assessment should consider factors such as overall risk profile (for disease relapse or recurrence and the occurrence of irreversible complications) and the disease impact on the patient. Risk factors that have been associated with a higher incidence of relapse or a more aggressive/complicated disease course include clinical factors (younger age, smoking, longer disease duration, need for corticosteroids soon after diagnosis, and fistulizing perianal CD), laboratory markers (low hemoglobin, low albumin, high C-reactive protein (CRP) and high fecal calprotectin levels), disease location, disease burden, and most notably the endoscopic appearance (the presence of deep ulcers).19-26
For patient-reported symptoms, those most frequently attributable to CD activity are stool frequency and abdominal pain.27,28 Two PRO measures that can be used to gauge symptom remission are PRO2 (stool frequency and pain) and PRO3 (stool frequency, pain, and general well-being). These measures have been validated and shown to be responsive to treatment-associated changes in disease activity.29 A PRO2 score of <8 corresponds to a CDAI score of <150 (clinical remission).27,28 The PRO2 has been employed as a treatment endpoint in clinical trials outside of the United States.
The CDAI and PRO2 correlate poorly with scores of endoscopic disease severity and with fecal (calprotectin) and serum biomarkers of inflammation CRP.29-31 Thus, to detect and assess disease activity and align with current treatment target recommendations, patients should also be periodically evaluated for disease activity using objective measures such as fecal calprotectin testing, other non-invasive testing and periodic endoscopic evaluation according to the Canadian IBD Specialist Group to ensure that the therapy is controlling disease activity, not only symtpoms.32
Disease Burden
Canada has the highest prevalence of IBD in the world.33 According to the 2018 Impact of IBD in Canada Report, approximately 270,000 Canadians were living with IBD, of which 135,000 had CD. For every 100,000 Canadians, 16.3 new cases of CD are diagnosed each year.34 This translates to over 6000 new cases each year. By 2030, the number of people living with IBD is expected to rise to over 400,000, or approximately 1% of the population.34 Studies have shown that up to one third of patients require hospitalization within the first year after diagnosis and more than half within five years.35
IBD can be diagnosed at any age but has a typical age of onset in adolescence or early adulthood, at a time when patients are pursuing employment, building families, and reaching key milestones.36-38 In addition to the tremendous impact that IBD has on quality of life (QoL), people living with IBD face myriad challenges, including prolonged symptoms due to late or inappropriate diagnosis, social stigma of having a chronic disease that affects toileting habits, difficulty with excursions due to limited or uncertain access to bathroom facilities, affordability of medications, diminished employment prospects, limited community-based supports, and inequitable access to health care services and specialists.39-41
A 2015 systematic review of publications relating to CD, its economic burden, and impact on health-related quality of life (HRQoL) found that CD in the US and Europe together was associated with annual total costs of nearly €30 billion, more than half due to indirect costs.42 Among CD patients, HRQoL was consistently and statistically significantly lower when compared with normal populations, due to physical, emotional, and social effects.42
The Canadian IBD Specialist Group underscored that the broader psychosocial effects among their CD patient populations are often overlooked. These can emerge or worsen when the disease is uncontrolled despite management with currently available treatment options. The Group noted that after several treatment options fail to control symptoms, patients often experience deteriorating mental health due to stress, despair, and a loss of hope that manifests as anxiety and depression. The Group cited that patients with uncontrolled CD often undergo a loss of productivity, missing days or sometimes weeks of school or work, especial during flare ups. The Group reported that symptoms of CD that persist lead many patients to forego opportunities for advancements at school, or promotions at work for fear of not being able to function adequately on any added responsibilities. According to the Group, social isolation and strained intimacy in relationships are also issues experienced and reported by many patients.
Minimal data is available on caregiver costs, and even less in a Canadian context. In a U.S. study of pediatric IBD patients using health insurance databases, 200 patients with CD and their caregivers were compared to age-matched controls without IBD and their caregivers. Unadjusted annual hours of work loss were 214.4 ± 171.5 and 169.6 ± 157.5 for caregivers of CD patients compared to controls, translating to annual lost productivity costs of USD $5,243 and USD $4,121 per caregiver, respectively.43 The Canadian IBD Specialist Group stated that CD often poses a huge financial burden for families of patients. The Group noted that direct caregivers often stay home as well to care for a CD patient during flares and if they undergo surgery, which contributes to a further loss of income to the family. The burden of caregiving also often has ripple effects on the entire family unit in terms of emotional and psychological impact, according to the Group.
Direct Costs in Canada
Direct healthcare costs of IBD that encompass the costs of medically necessary services and treatments paid for by public and private payers, including hospital-based care, outpatient physician consultations, prescription medications, diagnostic testing, complex continuing care, and home care, was estimated to be at least CAD$1.28 billion in 2018.33 In a population-based study from Manitoba, patients with CD cost the healthcare system CAD$4,232 per person annually.44
About one in five Canadian adults with CD is hospitalized every year.41,45 This hospitalization is often due to incomplete control of the disease with currently available therapy, in particular therapies that fail to have a significant impact on the endoscopic activity of the disease. In a Canadian population-based study, 2.3% of hospitalized IBD patients were re-hospitalized within one month of discharge, while 5.6% were readmitted to hospital within six months and 7.7% within 12 months.46 The average length of CD-related hospitalizations was 8.8 days. The population-based study from Manitoba found that 0.74% of patients with IBD were admitted to an intensive care unit (ICU) every year, which was higher than for matched controls.47 The risk of ICU admission was greater for CD patients than ulcerative colitis patients as compared to matched controls.47
Of patients hospitalized for CD in Canada, 16% undergo an intestinal resection during their first hospitalization.46 In a systematic review and meta-analysis of population-based studies, 16.3%, 33.3% and 46.6% of persons with CD required surgery within one, five, and ten years of diagnosis, respectively.48 The most common operation was a limited intestinal resection. Of persons who undergo surgery, 24.2% and 35.0% undergo repeat surgery within five and ten years, respectively.49
Prior to the introduction of biologic therapies to treat IBD, prescription drugs accounted for less than 25% of costs while hospitalizations accounted for more than 50% of direct costs of IBD care.50,51 In the post-biologic era, prescription drugs account for approximately 30% of IBD-related health care costs internationally.52-56 Mean annual hospitalization costs, however, decreased by 12% in the year following anti-tumour necrosis factor (anti-TNF) initiation, from CAD$6,419 to $5,627 per person. Similar decreases in inpatient care costs have been observed in Alberta (decreasing from CAD$2,715 to $968 in the year before and after infliximab initiation).57 Outpatient costs appear to be similar before and after treatment with infliximab.58-61 Overall, 14.2% of adults with CD are using the anti-TNF biologic agents infliximab and adalimumab.62,63
In summary, despite the rising costs of care associated with increasing biologics, their use has been associated with improvements in health outcomes and QoL among IBD patients. This is evidenced by declining rates of hospitalizations and surgeries. However, current therapies continue to fall short in achieving our endoscopic goals of endoscopic remission and mucosal healing which has been associated with decrease in flares, hospitalization, and surgery (please refer to the section below).
Indirect Costs
Canadian-specific data on indirect health related costs of IBD are sparse across all domains of indirect costs, including costs linked to decreased professional development, caregiver burden, and out-of-pocket purchases among IBD patients as well as costs incurred by Canadian children with IBD and their families. In particular, the rates of absenteeism, presenteeism, and premature retirement among Canadian IBD patients require further study to gauge more accurately the indirect health-related costs of IBD in Canada.
Extrapolating from multiple sources, the total indirect health-related cost of IBD in Canada in 2018 is estimated to be CA$1.29 billion.33 However, this may be a significant underestimate as costs relating to presenteeism, reduced achievement, and caregiver burden could not be estimated and did not factor into this extrapolation.
In a survey study of 744 individuals living with IBD in Manitoba, reduced workplace productivity (i.e. presenteeism) during the previous 14 days was reported by 37% of individuals, including a reduction for 1-2 days by 18% of patients, for 3-9 days by 16% of patients, and on most days by 3% of patients.67 Overall, working persons with IBD may expect to miss an additional 3.5 to 7.5 days from work annually due to illness compared to non-IBD persons. Based on the average Canadian salary in 2016 from Statistics Canada reports (CAD$956.50 per week or CAD$49,738 per year), the estimated mean annual per patient cost related to medical absenteeism is CAD$752 (range CAD$478 to CAD$1,025.34 In 2018, it is estimated that there are 97,809 Canadian working-age adults (age 18-64) with CD.68 Based on an estimated workforce rate of 68% among persons with CD living in Canada, roughly 66,510 persons would be eligible to experience medical absenteeism.69
Extrapolating annual retirement rates from a German study to working age Canadians with IBD, 430 persons with CD may be expected to retire each year in Canada, assuming that all working-age persons with IBD would otherwise be employed.34,70 Using the mean retirement ages from the German study of roughly 43 among CD patients, and the average earnings for Canadians in 2016, the average lifetime lost wages from premature retirement are calculated to be CAD$1,044,498 per person with CD (based on an average retirement age among working Canadians of 64).68,70 Aggregated across all IBD retirees each year, this equates to roughly CAD$449 million among persons with CD in permanent lost wages annually, assuming a similar wage distribution among IBD retirees and non-retirees.
In 2012, the cost of premature death among IBD persons in Canada was estimated to be CAD$9.4 million. In 2016, IBD specific premature deaths would result in 675 lost years of productivity and roughly CAD$33 million in permanent lost wages (CAD$746,070 per decedent) accrued annually across all working-age IBD persons (over and above lost wages due to premature mortality from non-IBD related causes).34 The rise in estimated indirect costs of premature mortality in people with IBD from 2012 to 2016 is the result of (1) higher wages; and (2) a higher number of deaths due to IBD, likely because of the increasing prevalence of IBD in Canada. Population-based studies from Ontario and Manitoba report higher mortality rates among patients with CD compared to the general population, particularly among young and middle-aged individuals.71,72 According to Statistics Canada data from 2010-2014, there are an average of 33 deaths directly resulting from CD per year.73
Based on cost estimates for sick days and short-term disability, premature retirement, premature death, and out-of-pocket expenses, the total indirect health-related cost to the Canadian economy due to IBD is estimated to be close to CAD$1.29 billion in 2018, or roughly CAD$4,781 per person with IBD. The largest component of this cost is related to lost productivity, particularly premature retirement (CAD$629 million). Importantly, this estimate does not consider presenteeism costs, caregiver costs and the costs of reduced professional development, which may be substantial but could not be accurately estimated due to insufficient data. The estimated annual cost due to medical absenteeism is speculated be as high as CAD$1.57 billion.33
Current Available Treatments for Moderate To Severe CD
CD patients in Canada are treated with variety of therapies depending on the characteristics of their disease. Treatments include 5-aminosalicylates (5-ASA), corticosteroids, immune modifiers, and biologics which include anti-TNF, anti-integrin, and anti-IL 12/23 and anti-IL 23 agents.74 For moderately to severely active disease therapies include corticosteroids, immunomodulators, and biologics. The therapies discussed here are supported by the CAG Consensus Practice guidelines published in 2018. The traditional approach has been to treat patients with corticosteroids during periods of disease flare to reduce symptoms and induce remission.74 However, these drugs are not prescribed on a long-term basis due to side effects and poor effectiveness for maintaining remission.74 The requirement for corticosteroids is usually the indicator for the need for more advanced therapies (biologics). Therefore, for patients who are refractory to corticosteroids, who are dependent on corticosteroids to keep symptoms under some degree of control or who have disease that is severe enough to require initiation of corticosteroids, biologics are typically initiated.74 According to the Canadian IBD Specialist Group, the use of highly effective therapies early in the disease course affords the best chance to change the natural history of the disease. This is supported by Canadian Consensus Guidelines.32 The group reviewed the treatment landscape and the pros and cons of each therapy which is outlined here.
Corticosteroids
Prednisone and budesonide are effective medications in patients with moderately to severely active CD.5 Methylprednisolone can be given intravenously (IV) in patients who are hospitalized. These medications non-specifically suppress the immune system. The limitations of corticosteroids are their inability to maintain clinical remission and the fact that they have not been shown to be associated with mucosal healing.
The Group agreed that they are very effective agents for the treatment of symptoms but are associated with significant short- and long-term side effects. They should not be used as a maintenance medication.75 Moreover, the group agreed that corticosteroids do not meet the modern treatment goals which include endoscopic remission and mucosal healing.
Immunomodulators
Also referred to as immunosuppressants, drugs such as azathioprine, 6-mercaptopurine (6-MP), and methotrexate, may be used to help decrease corticosteroid dependency and may help maintain disease remission.76,77,78,79 In a study of 141 patients with active or steroid dependent CD, methotrexate was more effective than placebo at inducing (19.1% vs. 39.4%, p = 0.025) and maintaining remission (39% vs. 65%, p = 0.04).78,79 The purine analogs, azathioprine and 6-MP, have been shown to be more effective than placebo for maintenance of symptomatic remission in CD, although the quality of evidence is low and there is no information on the ability of these treatments to achieve and maintain endoscopic mucosal improvement or healing.80 In addition, these therapies are associated with tolerability issues in the short term and potential for serious toxicity or complications with long term use.
The group agreed that immunomodulators may be effective agents for the treatment of corticosteroid-dependent disease but once again they do not meet the modern treatment goals which include endoscopic remission and mucosal healing and have never been shown to decrease the rates of hospitalization or surgeries.
Biologics
Anti-TNF (infliximab and adalimumab)
The advent of anti-TNF therapy and its ability to effectively induce and maintain remission while sparing corticosteroids transformed the management of CD when these agents were introduced into the treatment paradigms. They have also been associated with mucosal healing and for those who respond a reduction in hospitalization and surgery rates has been demonstrated.81
Anti-TNF biologics infliximab and adalimumab are currently approved in Canada for moderate to severe CD.32 They work by blocking the action of tumour necrosis factor-alpha (TNF-α), a key pro-inflammatory cytokine in the immune cascade. The ACCENT I study of 573 patients investigated if maintenance infliximab therapy in CD patients can provide better long-term efficacy than no further treatment after a single-dose induction given by intravenous infusion. Results showed that patients who received maintenance therapy were two times more likely to maintain clinical remission compared with those who received placebo (OR: 2.7, 95% CI: 1.6–4.6).82 The median time to loss of response was 46 weeks in the treatment group vs. 19 weeks in the placebo group.
The SONIC trial investigated the efficacy of infliximab, azathioprine, and a combination of the two drugs to induce and maintain corticosteroid-free clinical remission in patients with moderate to severe CD.83 The primary aim of this study was to evaluate the rate of corticosteroid-free clinical remission at week 26. Disease severity was evaluated using the CDAI and IBD-Questionnaire (IBDQ) scores as well as with direct visualization of mucosal healing with ileocolonoscopy at week 26. The greatest percentage of patients in whom corticosteroid-free remission was achieved was observed with combination therapy (43.9%), although results were significantly better with infliximab-based monotherapy (30.1%) as compared to azathioprine alone (16.5%). Therefore, SONIC demonstrated that to achieve the best clinical and endoscopic results therapy consisting of infliximab in combination with a purine anti-metabolite was needed.
The CLASSIC-I trial was a short 4-week dose-ranging study evaluating the efficacy of the anti-TNF drug adalimumab in CD.84 In the study, a relatively small number of patients (299) were randomly assigned to receive placebo or one of three different adalimumab induction dosing regimens at weeks 0 and 2. The adalimumab treated patients received either 40 mg/20 mg, 80 mg/40 mg or 160 mg/80 mg. Compared to placebo, the only induction loading dose regimen that achieved statistical significance for remission rates was 160 mg/80 mg. While the CLASSIC-I study demonstrated that adalimumab was effective in inducing remission by week 4, the CHARM study went on to show that adalimumab was equally effective at maintaining remission in patients with moderate to severe CD, either at a dose of 40 mg weekly or 40 mg every other week.85 At week 56, those on adalimumab were 1.5 to 2 times more likely to have maintained remission compared to placebo.
The EXTEND study was a phase 4 study and was the first study designed to evaluate mucosal healing as the primary endpoint.86 The results demonstrated that adalimumab could provide early and sustained mucosal healing in patients with moderate to severe ileocolonic CD. Higher rates of mucosal healing with adalimumab compared with placebo were observed by week 12 (27% vs. 13%, p = 0.056) but did not achieve statistical significance.
The anti-TNF drug infliximab has also been found to be effective in resulting in clinical closure and maintenance of closure of perianal fistulas in patients with Crohn’s disease.122,123 It is currently the only medical therapy that has this indication on its label in Canada.
Due to the recent expiry of patents for infliximab and adalimumab, several biosimilars have been approved for use in IBD.87,88 Biosimilars are analogous in structure but not identical to the original.87 The use of biosimilars may provide some advantages over the originator biologic drug, with the most relevant one being an easing of the economic burden of anti-TNF treatment.89 However, by definition these biosimilar drugs are clinically indistinguishable from the originator biologic with respect to efficacy and safety.
The group agreed that anti-TNF therapy has been a significant advance in the treatment of CD. However, anti-TNF agents have limitations which include immunogenicity, the need for combination with immunomodulators, and significant rates of loss of response over time. Furthermore, the Group identified that between 30% and 50% of patients in their practices are receiving doses that are higher or more frequently administered than those of the labelled dosing. The group speculated whether this was due to the limited data available regarding optimal dosing required for achieving mucosal healing or possibly due to a lack of a broad dosing range having been evaluated in phase 2/3 with no plateau effect seen.
Anti-IL 12/23 (ustekinumab)
In 2016, ustekinumab was approved in Canada for use in moderate to severe CD.90,91 To investigate its efficacy in CD, two 8-week placebo-controlled induction trials (UNITI-1 and UNITI-2) and one 44-week maintenance trial (IM-UNITI) were undertaken.15 Results of the three trials showed consistent superiority with ustekinumab over placebo in inducing and maintaining remission in patients with moderate to severe CD. At week 44, patients receiving maintenance doses of ustekinumab every 8, or 12 weeks were more likely to be in remission than placebo (53.1% vs. 48.8% vs. 35.9%, respectively; p < 0.05). The study included patient previously exposed to anti-TNF therapy. However, the improvement in endoscopic indices was modest at best and did not achieve statistical significance.
The group agreed that ustekinumab is effective in treating patients with moderately to severely active CD, both those who are naïve to biologic therapy and those exposed to anti-TNF. Its strength is a balance between efficacy and safety. Like anti-TNF therapy, the experience across the group was that in clinical practice 30-40% of patients required dosing outside of the approved dosing intervals. Additionally, the lack of robust mucosal healing in the phase three program was cited by some as a weakness.
Anti-integrin (vedolizumab)
Vedolizumab is a biologic approved for use in moderate to severe CD.32 Vedolizumab blocks α4β7-integrin on the surface of lymphocytes thus interrupting their homing to inflamed tissue in the gut, by blocking this migration it helps reduce inflammation. Vedolizumab is deemed gut-selective as α4β7-expressing lymphocytes only home to the gut.92 The GEMINI-2 study investigated the efficacy of vedolizumab at inducing and maintaining remission in 368 CD patients.13 The primary endpoint was to assess clinical remission (CDAI score ≤150) at week 6. In the study, vedolizumab induction therapy was moderately more likely than placebo to result in remission at week 6 (14.5% vs. 6.8%; p = 0.02). However, by week 52, patients who had an initial response to induction therapy had higher rates of clinical remission and glucocorticoid-free remission than placebo (21.6%) when vedolizumab was given 4- or 8-weekly (36.4% and 39% respectively) (p = 0.004 and p < 0.001, respectively). The effect of vedolizumab induction at week 6 was modest. The GEMINI-3 study focused specifically on the efficacy of vedolizumab with previous anti-TNF failure The results did not show a significant difference between vedolizumab and placebo at week 6 (15.2% vs. 12.1% respectively, p = 0.433) but there was a modest benefit at week 10 (26.6% vs. 12.1% respectively, p = 0.001). The phase III program did not include an assessment of endoscopic endpoints.
The group agreed that vedolizumab is effective in treating bio-naïve patients with moderately to severely active CD. Its biggest strength is its safety profile due to its gut selectivity. Limitations include its modest clinical efficacy, the lack of robust data for endoscopic endpoints and its limited utility in anti-TNF exposed patients and its lack of utility for the management of some extra-intestinal manifestations of Crohn’s disease.
Anti IL23 (Risankizumab)
Risankizumab was approved by Health Canada in late 2022 for use in the treatment of moderate to severe CD. In two Phase III induction studies (ADVANCE and MOTIVATE) a total of 1579 patients were randomized to receive induction dosing with placebo, risankizumab 600 mg iv or risankizumab 1200 mg iv at weeks 0, 4 and 8.121 At week 12 clinical remission (the co-primary endpoint), using stool frequency and abdominal pain criteria, was observed more frequently in the patients receiving Risankizumab 600 mg (43.5% in ADVANCE and 34.6% in MOTIVATE) as compared to the placebo treated patients (21.7% in ADVANCE and 19.3% in MOTIVATE. Endoscopic response (the other co-primary endpoint) was observed in 40.3% and 28.8% of patients receiving risankizumab 600 mg in the ADVANCE and MOTIVATE trials, respectively. This was significantly greater than the frequency of endoscopic response seen in the placebo patients (12% and 11.2% respectively). The long-term efficacy and safety of risankizumab were tested in the subsequent maintenance study (FORTIFY).124 In that trial, patients who had responded to induction dosing with risankizumab in either the ADVANCE or MOTIVATE trials were randomized to receive placebo (n=184), risankizumab 180 mg (n=179) or risankizumab 360 mg (n = 179) by subcutaneous injection given every 8 weeks out to 52 weeks.124 Greater clinical remission and endoscopic response rates were reached with 360 mg risankizumab vs. placebo with CDAI clinical remission reached in 52% of patients vs. 41% of placebo treated (Risankizumab withdrawal) patients (p=0.0054). Endoscopic response was achieved in 47% of patients in the 360 mg group vs. 22% of patients in the placebo group (p<0.0001). Efficacy was observed irrespective of prior intolerance or inadequate response to other advanced therapies. Risankizumab subcutaneous maintenance therapy was deemed safe and well tolerated in patients. Although these studies showed improvements in both clinical and endoscopic outcomes compared with placebo the therapy involves both intravenous and subcutaneous dosing which the group felt are not preferred modes of administration for many patients. In addition, although absolute response and remission rates are generally favorable there was still a significant proportion of patients who did achieve the endpoints or treatment goals.
Surgery
Surgery is necessary in CD when medications prove ineffective or if complications arise, such as fistulae, abscesses, scarring, and narrowing of the bowel.93 Complications are believed to arise due to uncontrolled inflammation at the bowel level (i.e., the lack of mucosal healing). Because damage to the bowel and the associated complications are frequently irreversible and do not respond to existing medical therapies their occurrence frequently requires surgical resection to adequately manage and return the patient to improved health. In most cases, surgery involves resection of the diseased segment of the bowel. Historically the endoscopic post -operative recurrence rate after an ileal resection is 50-70%. This leads to many patients requiring 2nd and 3rd surgeries. Surgery is associated with significant morbidity, impairment in quality of life and even mortality. Some patients will require an ostomy, an ileostomy, or a colostomy.93
The group agreed that surgery should be performed in patients who have developed complications or who have limited small intestinal disease. However, surgery is generally not a preferred option for patients and, although it can produce very good short to medium term results with respect to quality of life the frequent recurrence of CD in areas of the intestine that were previously not involved is problematic and as a result medical therapies that can reduce the need for surgery while maintaining or improving health related quality of life are needed. In 2023, the ongoing need for surgery in patients with CD is likely a reflection that the existing therapies are not able to consistently achieve all of the important treatment goals.
Treatment Goals
Treatment goals in IBD have evolved over recent years recognizing that treating only to symptom control is inadequate and leaves patients at risk of developing progressive and complicated disease. As discussed above, it is these complications that often lead to the need for surgery. Surgery is not the optimal answer due to the high rate of post-operative recurrence and the morbidity and mortality associated with surgery. One of the most important concepts in Crohn’s disease is the disconnect between the presence and absence of symptoms and control of inflammation. Patients may be asymptomatic (i.e., be in symptomatic remission) but continue to have significant underlying endoscopic activity and inflammation.94,95
Recognizing this disconnect between symptoms and inflammatory activity, in 2015, the International Organization for the Study of IBD (IOIBD) initially outlined that treatment targets/goals should NOT rely only on achieving symptomatic relief but should also include mucosal healing.96 In 2021, the updated Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE-II) initiative of the IOIBD reiterated the most important treatment target /goals for patients with CD as clinical remission, normalization of biomarkers and endoscopic healing (EH), restoration of quality of life (QoL), and absence of disability.97 Relief of symptoms is acknowledged as important because this is what impacts patients in their present daily lives. However, it is paramount to have therapies that can achieve improvement in the endoscopic appearance (endoscopic response), endoscopic remission, and mucosal healing. Achieving these endpoints, has been demonstrated to reduce the chance of future flares, hospitalizations, and surgeries.98,99 As such, they have the potential to improve patients’ future QoL and reduce future disability.
In recognition of the importance of having therapies that not only lead to symptomatic remission but can improve the endoscopic appearance of the bowel towards a goal of achieving mucosal healing and changing the natural history of the disease, regulatory authorities now include endoscopic response as a co-primary endpoint in phase III registrational trials of moderate to severe CD.100
The Canadian IBD Specialist Group noted that use of a treatment that induces symptomatic remission but also promotes healing that can be seen on endoscopic imaging would ideally be used for all patients early in the course of their disease before irreversible intestinal damage or complications have occurred. According to the Group, treatment goals now focus on changing the course of disease for CD patients, preventing further intestinal damage, avoiding disability, and reducing the overall cost of care. However, the Group discussed that this expanded target is not achievable in most patients using currently available treatments. As such, the Group underscored the need for more robust therapy options in the long-term care of CD patients.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Important unmet needs remain in the treatment of moderate to severe CD. There is need for a safe and easily administered therapy that rapidly induces symptomatic remission and can be used to maintain symptomatic remission. Importantly there is need for a therapy that can also improve the endoscopic appearance of the bowel and maintain this over the long-term so as to reduce the risk of complications and the need for surgery. Avoidance of chronic symptoms, complications and surgery will ultimately improve quality of life and reduce disability due to the disease or the consequences of its surgical management.
The presently available therapies each have their limitations which will be discussed below. These limitations are highlighted for the currently available biologics as the Canadian IBD Specialist Group flagged the need for dose escalation to levels that are off label to achieve control in CD treatment in a substantial percentage of their patient population (ranging from 30-50% depending on the agent). In real-world clinical practice, dosage levels did not appear to perform as well as those used in clinical trials in specific patient populations. The Group also underscored that this frequent dose escalation in clinical practice has a cumulative negative impact on the patient-physician relationship and serves to sow seeds of doubt and a loss of confidence in patients towards their therapeutic options and towards their care providers who are recommending these therapeutic options to them.
Limitations Associated With Current Treatments
Corticosteroids
Prednisone and budesonide are orally administered for the treatment of CD and can be highly effective for the acute treatment of inflammatory disease flares. However, safety, especially associated with long-term use, is a concern and, as such, corticosteroids are not recommended for maintenance therapy.33,75 The Canadian IBD Specialist Group concurred stating that risk of infection and other potentially irreversible adverse events are higher with corticosteroid use.101,102 There was also a consensus within the Group with respect to the relatively high risks for short-term cosmetic (e.g. skin thinning, acne, hirsutism, cushingoid or ‘moon face’ appearance) and neuropsychological (e.g. hypomanic reactions, insomnia, psychosis) impacts that have been well documented in clinical studies and seen frequently in clinical practice. Although budesonide, a topically active corticosteroid that has high first pass metabolism in the liver to inactive metabolites, has a more favorable side effect profile compared to prednisone, it is limited by virtue of the fact that it is only effective in patients who have mild to moderate disease limited to only the terminal ileum with or without involvement of the right side of the colon. Budesonide has also been shown not to prevent clinical recurrence of disease.125 This has also been the experience of the Group with respect to the long-term use of prednisone, with members of the Group voicing concerns over the limited ability of corticosteroids to treat inflammation and maintain clinical remission in CD over the long-term as well the increasing potential for serious side effects with chronic therapy.
Prolonged corticosteroid therapy has been shown to be associated with an increase in mortality in patients with CD.102 There is also a significant positive correlation between having C. difficile infection and more corticosteroid and antibiotic exposure, and increased disease activity, worse QoL, and increased health care utilization (all p < 0.01).103
Corticosteroids were the mainstay for induction therapy until the late 1990s when evidence began showing that they induce complete clinical remission in 48% and partial clinical remission in 32% of patients with active CD. However, 20% of patients were found to be resistant from the onset, and at their 1-year follow-up, 45% of the patients who responded initially had become steroid-dependent, with only 32% of patients had maintained a prolonged clinical response.105,106 It became apparent that corticosteroids were ineffective at maintaining remission, reducing flares, or preventing disease recurrence.105-109 The French GETAID study provided further proof that corticosteroids were not disease-modifying agents with limited evidence to indicate their ability to achieve endoscopic mucosal healing or preventing endoscopic relapse.110 In that study, patients were given prednisolone 1 mg/kg for 7 weeks, and although over 90% of patients were deemed to have had clinical response only 29% achieved endoscopic and clinical remission, with 71% still showing active endoscopic lesions. In fact, 9% of patients had worsening endoscopic lesions despite symptomatic improvement.110 Corticosteroids are still typically used during periods of disease flare but as there is a risk of patient reliance along with worsening CD, they should not be used long term. In addition, according to the Group, the known and previously experienced side effects frequently result in patient reluctance or refusal to go back on corticosteroid therapy, even when needed.
The Canadian IBD Specialist Group would advise against the use of corticosteroids for maintenance therapy but recognize that treatment failure on prednisone is still required on most formularies prior to prescribing a biologic or advanced therapy. The Group would advise use of corticosteroids in the short term in times of inflammatory disease flare in moderate to severe CD but this therapy typically ends up being a bridge to more advanced therapies that can more effectively and safely maintain remission and more consistently result in improvement of the mucosal appearance of the intestine.
Immunomodulators
The advantages of using thiopurines (azathioprine or 6-mercaptopurine (6-MP)) are their steroid-sparing effects. However, their slow onset of action (8-12 weeks) makes them ineffective for short-term induction therapy in active, symptomatic disease.111 With regard to immune modifiers azathioprine and methotrexate, the Group noted that tolerability issues, inferior efficacy compared to advanced therapies such as biologics, and delayed onset of action are limitations. Added to these are concerns over increased risk of infections, purine associated pancreatitis, myelosuppression, hepatotoxicity, and malignancy, particularly lymphoma and nonmelanoma skin cancer.112-114
The Group agreed that immunomodulators may be effective agents for the treatment of corticosteroid-dependent disease but, once again, they do not meet the modern treatment goals which include endoscopic remission and mucosal healing and have never been shown to decrease the rates of hospitalization or surgeries.
Biologics
Anti-TNF (infliximab and adalimumab)
The Canadian IBD Specialist Group has encountered patient reluctance when prescribing the anti-TNF biologics infliximab and adalimumab due to perceptions of risk and long-term safety concerns, with many patients requesting to be taken off these biologics. Compared to newer biologics used for the management of CD, the Group has found that the mode and frequency of administration of this class of biologics can pose an obstacle to their use as many patients have an aversion to intravenous administration (infliximab) and many others are unhappy with the frequency of subcutaneous dosing required with adalimumab, particularly in the 30 to 50% of patients who require dose escalations which typically involve more frequent dosing. This requirement for dose escalation can compound the treatment obstacles discussed above and complicates the management journey for patients.
Anti-TNF drugs are effective in the management of CD, but treatment failure is not an uncommon occurrence with these medications. Treatment failure can entail either non-response or loss of response or, in some cases, side effects that the patient finds to be unacceptable or serious side effects that have the potential to be life-threatening or lead to disability. The personalized anti-TNF therapy in CD study (PANTS) aimed to identify specific clinical and pharmacokinetic factors that predicted primary non-response.115 The investigators performed a multivariate analysis which demonstrated that the only factor independently associated with primary non-response was low drug concentration at week 14 for both infliximab and adalimumab. For both drugs, suboptimal drug concentrations at week 14 predicted immunogenicity, with the formation of anti-drug antibodies.
Anti-TNF therapies (biosimilars included) have transformed the care of patients with IBD, redefining our standards, however, it has become obvious that they are not universally effective, with 30-50% of patients being primary non-responders and with further attrition from subsequent loss of response (mechanistic escape, immunogenicity, or intolerance).116 There is also the real risk of infectious complications attributable to non-specific inhibition of TNF-mediated immunologic cascades.116,117
The Canadian IBD Specialty Group agreed that anti-TNF therapy has been a significant advance in the treatment of CD. However, this class of therapy has limitations which include immunogenicity, the need for combination with immunomodulators in order to maximize outcomes, and loss of response over time. Immunogenicity, with the formation of anti-drug antibodies, is a limitation of the anti-TNF drugs because of its strong association with inadequate response or loss of response. The risk of developing anti-drug antibodies is increased if treatment is stopped or interrupted and then resumed. The Group felt that this is a limitation for anti-TNF therapy because there are frequently instances in which treatment needs to be delayed or interrupted or when dosing is not administered according to schedule due to patient adherence. Furthermore, the group identified that between 30-50% of patients in their practices are receiving doses that are outside of the labelled dosing. The Group speculated as to whether this was due to the limited data available for achieving mucosal healing at standard doses and a desire to maximize serum drug levels in the absence of any dose or drug level related toxicity. Alternatively, the Group speculated as to whether this might be due to the fact that the Phase 2/3 development anti-TNF programs did not test a sufficiently broad range of doses to allow determination of an optimal dose or drug level for both clinical and endoscopic outcomes.
Anti-IL 12/23 (ustekinumab)
Ustekinumab use in the Group’s experience shows sub-optimal efficacy on endoscopic endpoints and is often used at higher off-label doses to achieve remission. The Group noted that the current on-label dosage of this biologic is often inadequate to achieve the desired symptomatic and endoscopic outcomes without a dose escalation. According to the Group, this need for off-label dose escalation occurs in up to 30-50% of patients and can erode trust between the patient and their specialist as treatment continues to fail to elicit an adequate response after many weeks of therapy. However, the Group did find that ustekinumab performed better than vedolizumab at resolving some extra-intestinal manifestations (EIMs) of CD.
The SEAVUE trial was a randomized controlled trial that directly compared ustekinumab with the anti-TNF drug adalimumab over a period of approximately 1 year.126 Patients in that study had disease that was, on average, of shorter duration than the patients in most of the other clinical development programs. As such, these patients would have been more likely to experience clinical and endoscopic response. The SEAVUE study showed no important differences in clinical or endoscopic outcomes with endoscopic remission seen in 28.5% of patients treated with ustekinumab. Ustekinumab had a slightly better side effect profile and tolerability as compared with adalimumab.
The group agreed that ustekinumab is effective in treating patients with moderately to severely active CD, both in those who are naïve to biologic therapy and those who have been exposed to anti-TNF therapy. Its strength is a balance between efficacy and safety. Like anti-TNF therapy, the experience across the Group was that in clinical practice 30-50% of patients required dosing outside of the approved dosing intervals. Additionally, the lack of robust mucosal healing in the Phase III program was cited by some as a weakness and not meeting the current needs of therapy.
Anti IL23 (risankizumab)
Given the relatively short time since Health Canada approval of the risankizumab CD indication, in October 2022, the Canadian IBD Specialist Group’s experience with the drug outside of the clinical development program has been somewhat limited. However, it was felt that similar to ustekinumab, risankizumab provides a very good balance between efficacy and safety. Furthermore, in the ADVANCE, MOTIVATE and FORTIFY studies patients who had previously received ustekinumab therapy were allowed to enrol in the studies. It was found that a proportion of those patients still responded to risankizumab even though they had inadequate response to ustekinumab and that this rate of response was numerically greater than the placebo rate of response.
The Group felt that the results reported in the Phase 2/3 risankizumab studies provide more robust evidence for improvement in endoscopic outcomes than other therapeutic classes. However, as discussed above, after 52 weeks of risankizumab therapy, 52% of patients had CDAI clinical remission and 47% had endoscopic response. This means that there are still approximately half of patients who did not meet the desired clinical and endoscopic outcomes. In clinical practice, these patients could potentially benefit from the availability of other treatment options.
Another potential limitation of risankizumab therapy is the route of administration with the requirement for both intravenous (induction) and subcutaneous (maintenance) dosing.
Anti-integrin (vedolizumab)
The Canadian IBD Specialist Group would not advise use of vedolizumab in patients with large ulcers or high-risk endoscopic profiles as the absolute rates of ulcer healing are inadequate. The Group also deemed that this agent has limited efficacy as a second line option following the most commonly used class of biologic therapy – the anti-TNF drugs. In addition, several ‘real-world’ studies have demonstrated that vedolizumab may be more beneficial in patients who are biologic-naïve and those with an inflammatory phenotype. The Group concurred that vedolizumab would not be the first choice for severely active CD due to the comparatively lower efficacy. This diminished efficacy typically prompts a dose escalation every from every 8 to every 4 weeks in 30-50% of patients. The slower onset of action seen with vedolizumab – significant increase in symptomatic remission compared with placebo was only starting to be seen by 10 weeks of treatment in the clinical development program - may necessitate the co-administration of a corticosteroid to achieve sufficiently rapid and robust initial disease control. This then increases patient risk of infection and other adverse events, according to the Group, and this negates the potential advantage that the “gut selectivity” of vedolizumab confers as compared to other advanced therapy classes with respect to infectious complications. This concern over vedolizumab’s inability to induce remission by 6 weeks and the lack of mucosal healing data do not provide compelling evidence for its routine use in CD.
The Group agreed that vedolizumab is modestly effective in treating bio-naïve patients with moderately to severely active CD. Its greatest strength is its safety profile, likely due to its gut selectivity and lack of effect on systemic immune response. Limitations include its modest clinical efficacy, the lack of robust data for endoscopic endpoints and its limited utility in anti-TNF exposed patients.
In summary, none of the available therapies meet the current needs of CD patients in the short or long-term. This is underscored by the fact that, despite the availability of biologics, there is still a significant proportion of patients who suffer with medically refractory disease. Up to 40% of CD patients are primary non-responders to an anti-TNF agent prescribed as a first line advanced therapy and up to 46% experience a loss of response during maintenance treatment with an anti-TNF agent within 1 year of starting therapy. Furthermore, 35-50% of non-responders to an anti-TNF agent fail to respond adequately to a second anti-TNF agent.119 Loss of response can also occur with other classes of biologics. Among CD patients on vedolizumab, the pooled incidence of loss of response is 47.9 per 100 patient-years. Among patient on ustekinumab, 34% experienced a loss of response to maintenance therapy at a median of 47.4 weeks while 16% required a dose escalation.118,119
Given that, despite recent advances in CD therapy, there are still patients who have medically refractory disease or who develop disease complications while on medical therapy, there is still a significant requirement for surgical management for patients with CD. Furthermore, post-operative recurrence of CD has continued to be a frequent finding despite the use of biologics (primarily anti-TNFs) for post-operative prophylaxis. Over the last decade, the probability of surgery has been reported to be between 30% and 66% within 15 years of diagnosis, with clinical relapse and reoperation rates of 50-60% and 28-45%, respectively.118
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Despite a number of available advanced therapies for the treatment of moderate to severely active CD there is still a need for novel targeted therapies with new mechanisms of action that provide sustained clinical and endoscopic outcomes as well as having acceptable and comparable safety profiles across different patient populations when used as long-term maintenance therapy. The Janus kinase (JAK) -STAT signalling pathway plays a pivotal role in the activity of several key pro-inflammatory cytokines. The JAK-STAT intracellular signalling that occurs in response to the binding of the cytokines to their extracellular receptor binding sit ultimately leads to nuclear transcription changes that result in increased inflammatory activity.127 The JAK-STAT pathway has been shown to be important in the pathogenesis of Crohn’s disease and several other immune mediated diseases including psoriatic arthritis, rheumatoid arthritis and atopic dermatitis.120 Therefore, specific blockade of the JAK-STAT pathway has the potential to have an ameliorating effect on the inflammation that occurs in these disorders. Blockade of these pathways has been demonstrated with the use of upadacitinib in patients with CD.128
JAK inhibitors block the ATP binding site of JAKs and interfere with the phosphorylation that is necessary for JAK activity. Upadacitinib is an orally administered small molecule inhibitor of the JAK complex. It has high affinity and selectivity for the JAK1 subtype and, as a result, avoids some of the side effects and toxicity that can be observed with other less selective JAK inhibitors. Upadacitinib has a rapid onset of action and, when treatment is stopped, it has a fast offset of action. It is the first oral therapy for CD that has ever been evaluated to meet the treatment goals that are highly valued in the treatment of CD and discussed above. Specifically, both clinical and endoscopic endpoints (co-primary endpoints) were shown to be improved during both induction and maintenance phases of therapy. As such, upadacitinib is the first oral therapy to demonstrate in a Phase III program that it not only is able to treat symptoms of CD but also improve the underlying inflammation that defines CD and which, if left unchecked, can lead to disease complications, need for hospitalization and surgery.
The Phase III upadacitinib program included CD patients with moderately to severely active disease. Eligible patients were enrolled in of 2 parallel 12-week induction studies (U-EXCEL129 and U-EXCEED130) followed by a 1 year maintenance study (U-ENDURE131) in patients who experienced clinical response in one of the two induction studies. All of the patients in U-EXCEED had failed 1 or more prior biologic therapies and about 45% of those in U-EXCEL had failed 1 or more prior biologic therapies with 55% not having failed prior biologics. In the U-EXCEL study 50.5% of the 350 patients who received upadacitinib 45 mg daily achieved clinical remission at week 12 compared with 22.2% of the 176 patients randomized to placebo. In the U-EXCEED study clinical remission was achieved in 39.8% of the 324 patients on upadacitinib 45 mg daily compared with 14% of the 171 placebo treated patients. Endoscopic response (the co-primary outcome measure) was observed in 45.5% of upadacitinib patients in U-EXCEL and 13.1% of placebo patients and in the U-EXCEED study in 24.6% of upadacitinib patients and 3.5% of placebo patients. All of these differences were highly statistically significant. Furthermore, for the approximately 35% of patients who were receiving corticosteroid therapy at study baseline (i.e. those who were steroid refractory or dependent) there was a forced steroid taper after week 4 so that patients were to have discontinued steroids by week 12. In the patients taking corticosteroids at baseline 44.4% of those receiving upadacitinib in U-EXCEL and 37% of those in U-EXCEED were in steroid-free clinical remission at week 12 as compared with placebo treated patient rates of 12.5% and 6.7% (p ≤ .0001 for both studies).
All co-primary endpoints, which entailed clinical remission PRO criteria (average daily stool frequency and abdominal pain score) and endoscopic response at week 12, were met in both trials with upadacitinib (p values ≤0.0001). In addition, upadacitinib therapy resulted in significant rates of steroid free clinical remission and is the first orally administered agent to demonstrate the ability to induce remission while allowing steroid tapering. Efficacy of upadacitinib was observed in both biologic naïve and biologic failure patient populations.
The U-ENDURE maintenance study enrolled 502 patients who had experienced a clinical response to upadacitinib 45 mg daily in one of the two induction studies.131 Eligible patients were randomized to receive, placebo, 15 or 30 mg of upadacitinib daily and were followed out to 1 year. Clinical remission, based on stool frequency and abdominal pain, was achieved in 46.4% of the 168 patients in the upadacitinib 30 mg daily group, 35.5% of the 169 patients in the upadacitinib 15 mg daily group and in 14.4% of the 165 patients who received placebo (i.e. those who received only upadacitinib induction therapy). Of the patients who had achieved clinical remission at the end of induction therapy 60 to 65% of them were still in remission at the end of 52 weeks of treatment as compared with approximately 20% of patients receiving placebo during the maintenance phase of the trial (i.e. upadacitinib withdrawal patients). Endoscopic response was observed in 40.1%, 27.6% and 7.3% of the upadacitinib 30 mg daily, upadacitinib 15 mg daily and placebo/upadacitinib withdrawal groups, respectively.
The safety of upadacitinib was observed to be acceptable and comparable to that of the biologics and immunomodulators commonly used to treat CD. In the induction studies, numerically more herpes zoster infections, serum transaminase elevations and creatine phosphokinase (CPK) elevations were observed in patients receiving upadacitinib as compared with those receiving placebo. The liver enzyme and CPK elevations were not associated with any clinical symptoms or clinical disease manifestations. In the maintenance study, treatment emergent adverse events were no more frequent in the upadacitinib arms as compared with placebo and, specifically, there was no signal suggesting any increase in cardiovascular events in patients receiving upadacitinib maintenance therapy.
Thus, selective blockade of the JAK1 signalling pathway is a new mechanism of action that shows efficacy in patients with moderate to severe CD refractory to conventional therapy or one or more categories of biologic therapies, including anti-TNF drugs, vedolizumab and ustekinumab. The Canadian IBD Specialist Group felt that the results reported are the strongest of a non-corticosteroid orally administered agent ever studied in CD, especially in light of the refractory patient population studied and the fact that upadacitinib demonstrated rapid responses with the ability to improve endoscopic appearance in a period as short as 12 weeks. Importantly, upadacitinib was able to induce remission while also allowing tapering of corticosteroids. Over half of patients - both biologic naïve patients and patients who had inadequate response to biologics - responded to induction therapy thus making them eligible for maintenance therapy. As a result, the impact of upadacitinib across the CD patient population is potentially quite meaningful and could encompass a broad segment of the CD patient population. This impressive efficacy was not at the expense of safety over 1 year of therapy.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Overall, the clinical trial outcomes indicate that a broad range of use of upadacitinib is possible in clinical practice – from first line advanced therapy to treatment of patients with inadequate response or intolerance to corticosteroids or multiple advanced therapies. Importantly, the robust endoscopic data could translate into changing the course of disease with upadacitinib.
The Canadian IBD Specialist Group felt that based upon the very good clinical and endoscopic response data, the durability of those clinical and endoscopic responses, the steroid sparing effect, the acceptable safety profile and the ease of administration, that upadacitinib would be an excellent choice to be used as first line advanced therapy in those patients with moderate to severely active CD who prefer an orally administered drug or for those in whom there may be advantages to having both rapid onset and offset of action without concerns about immunogenicity or the altered pharmacokinetics that may occur with the use of biologics in patients with more severe CD. Given its demonstrated efficacy in other immune mediated inflammatory disorders (e.g. rheumatoid arthritis, psoriatic arthritis, atopic dermatitis) the use of upadacitinib could be preferred over some of the other advanced therapies in patients who have one or more extraintestinal manifestations of IBD.
In addition, the Group also felt that upadacitinib could also be used in patients who were inadequate responders to 1 or more previous biologic therapies given the robust data for clinical and endoscopic outcomes in that patient subpopulation. In particular, an orally administered small molecule such as upadacitinib may have particular advantages over large molecule biologics in patients with severe or extensive intestinal mucosal inflammation and low serum albumin where increased loss of biologic drugs into the intestinal lumen results in reduced serum drug levels and reduced efficacy. Such alteration in pharmacokinetics is not observed with small molecule drugs such as upadacitinib.
Upadacitinib has not been specifically studied in patients with perianal fistulizing CD. As such, in patients where perianal disease is the dominant disease manifestation the use of another class of advanced therapy, such as the anti-TNF drugs, where more robust data on efficacy in the treatment of perianal fistulizing disease is available would be preferred. Similarly, patients with ocular complications of CD such as uveitis would be best served being treated with an anti-TNF drug.
There was consensus among the Group that durability, good endoscopic mucosal healing combined with the high efficacy and the overall safety of upadacitinib positions this new therapy in CD as a prime candidate to help resolve many of the unmet needs in the treatment landscape. The Group noted that these properties will also reduce the need for very frequent monitoring of patients as is done with many conventional therapies that have higher adverse event risk profiles. The high efficacy, rapid onset of treatment response and the lack of requirement for dose adjustment with upadacitinib will also help re-establish patient trust in their treatment choice and in their care providers. The Group also felt confident that upadacitinib efficacy and durability as a first line biologic agent will help drive down the risk of hospitalization and need for surgical interventions in the long-term.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The upadacitinib development program was one of the first clinical trial programs that aligns with what is done in clinical practice and also aligns with disease management strategies outlined in STRIDE-II. The Canadian IBD Specialist Group recommends that management strategies strive for complete remission, which is defined as both symptomatic and endoscopic remission. This aligns with the recommendations of the Canadian Consensus Guidelines.
In the first three months of therapy, a meaningful improvement in symptoms as measured by elements of the PRO2 (stool frequency and abdominal pain) should be demonstrated. Patients would be expected to be in symptomatic remission and off corticosteroids by 3 to 6 months. Symptomatic improvement should be accompanied be a decrease in biomarkers (C-reactive protein and fecal calprotectin) of inflammatory activity in the first three months. Although, the program demonstrated robust endoscopic response in the first three months, the group would not assess endoscopic activity until 6-12 months into therapy as long as clinical or biomarker targets are being met or approached. In addition, the consensus group recognized that because of the substantial impact of CD on a patient’s daily life activities and health related quality of life, it is imperative to consider the patient’s perspective when making treatment decisions and the timing of those decisions. In many instances, factors that influence patient decisions relating to therapy choice and goals of therapy are not the same as those of the treating clinician.
What factors should be considered when deciding to discontinue treatment with the drug under review?
The Canadian IBD Specialist Group would recommend discontinuing treatment with upadacitinib if there is worsening of symptoms or if there is an inadequate clinical response to therapy by the end of the 12-week induction period. However, based on available clinical data demonstrating that more than half of patients will respond to therapy over the first 3 months, it is not anticipated that this would be a common occurrence. The Group recognizes that there is often a disconnect between symptoms and objective evidence of inflammation in Crohn’s disease. As such, the Group would caution against using only clinical symptoms or only biomarker evidence of inflammation to make decisions about discontinuing therapy and would suggest that these decisions include an assessment of both clinical response and biomarker response. In instances where there is an inadequate response to upadacitinib as first line advanced therapy in moderate to severe CD, then a switch to another class of therapy is warranted, according to the Group.
Adverse events that are severe enough to require discontinuation of therapy during the induction phase of therapy are uncommon (less than 5%) and in the U-EXCEL and U-EXCEED studies were no more common in patients treated with upadacitinib as compared to patients treated with placebo. Herpes zoster infection is one adverse event that can occur with upadacitinib and with the JAK inhibitor class of drugs in general. The Group anticipates that this risk will be mitigated in clinical pracctice by the use of vaccination but, should an infection occur, it would only require temporary interruption of upadacitinib therapy until healing of zoster lesions.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
In the clinical experience of the Canadian IBD Specialist Group, upadacitinib should be prescribed by a provider with significant experience and expertise in treating patients with CD working in a specialty clinic setting. Since upadacitinib is a once daily orally administered drug with no immediate post-administration serious adverse effects it can be taken by the patient at home or other setting and does not require any immediate post-dosing monitoring. Specialist monitoring of response to therapy (clinical/symptomatic; biomarker; endoscopic) is necessary to ensure that the patient is meeting the pre-determined goals of therapy and to monitor for any possible adverse effects of therapy. The frequency of these assessments may vary according to individual patient factors such as the severity of the disease at treatment initiation and patient age and comorbidities, but, in general, an assessment of short-term clinical response should be made within 8 to 12 weeks of initiating therapy, biomarker response should be assessed by 3 to 6 months and endoscopic response by 6 to 12 months. At any point, if a patient is experiencing worsening CD symptoms or is experiencing a possible adverse effect of treatment they should be assessed at that time.
Additional Information
Summary From the Canadian IBD Specialist Group
Despite the availability of increased numbers of advanced (biologic) therapies for the treatment of Crohn’s disease over the past 10 years there are still significant gaps and unmet needs that need to be addressed in the management of moderate to severe Crohn’s disease. Upadacitinib meets many of these needs with its convenience being orally administered, its high rates of short and long term clinical symptomatic response, its impact on extra-intestinal manifestations of CD and its ability to improve the intestinal mucosal appearance while maintaining a favorable therapeutic index. The observed improvement in mucosal appearance is extremely important since mucosal healing has been shown to be associated with reduced hospitalizations and need for surgery which result in improved quality of life and reduced disability.
Conflict of Interest Declarations — Canadian IBD Specialist Group
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
Not applicable.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
Not applicable.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: A. Hillary Steinhart, MD FRCPC
Position: Gastroenterologist, Professor of Medicine, University of Toronto
Date: 18-04-2022
Table 3: COI Declaration for Canadian IBD Specialist Group — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Amgen | X | — | — | — |
BioJAMP | X | — | — | — |
BMS | X | — | — | — |
Celgene | X | — | — | — |
Celltrion | X | — | — | — |
Fresenius Kabi | — | X | — | — |
Janssen | — | — | X | — |
McKesson | X | — | — | — |
Mylan Pharmaceuticals | X | — | — | — |
NKS Pharmacy | X | — | — | — |
Organon | X | — | — | — |
Pendopharm | X | — | — | — |
Pfizer | — | X | — | — |
Sandoz | X | — | — | — |
Takeda | — | — | X | — |
Declaration for Clinician 2
Name: Vipul Jairath
Position: Professor of Medicine at the Schulich School of Medicine
Date: 27-04-2022
Table 4: COI Declaration for Canadian IBD Specialist Group — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Alimentiv | — | — | — | X |
Arena Pharmaceuticals | X | — | — | — |
Asahi Kasei Pharma | X | — | — | — |
Asieris | X | — | — | — |
AstraZeneca | X | — | — | — |
Avoro Capital | X | — | — | — |
Bristol Myers Squibb | — | X | — | — |
Celltrion | X | — | — | — |
Eli Lilly | — | X | — | — |
Endpoint Health | X | — | — | — |
Ferring | X | — | — | — |
Flagship Pioneering | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Galapagos | X | — | — | — |
Gilde Healthcare | X | — | — | — |
Glaxo-Smith-Klein | X | — | — | — |
Genentech | X | — | — | — |
Gilead | X | — | — | — |
Janssen | — | — | X | — |
Merck | X | — | — | — |
Mylan | X | — | — | — |
Metacrine | X | — | — | — |
Pandion Pharma | X | — | — | — |
Pendopharm | X | — | — | — |
Pfizer | — | — | X | — |
ProtagonistTherapeutics | X | — | — | — |
Reistone Biopharma | X | — | — | — |
Roche | X | — | — | — |
Sandoz | X | — | — | — |
Second Genome | X | — | — | — |
Sorriso | X | — | — | — |
Takeda Pharmaceuticals | — | — | X | — |
Teva | X | — | — | — |
Topivert | X | — | — | — |
Ventyx | X | — | — | — |
Vividion | X | — | — | — |
Declaration for Clinician 3
Name: Charles N. Bernstein
Position: Distinguished Professor of Medicine, University of Manitoba
Date: 02-03-2023
Table 5: COI Declaration for Canadian IBD Specialist Group — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
Janssen | X | — | — | — |
Pfizer | X | — | — | — |
Takeda | X | — | — | — |
Bristol Myers Squibb | X | — | — | — |
Sandoz | X | — | — | — |
Amgen | X | — | — | — |
Eli Lilly | X | — | — | — |
JAMP Pharmaceuticals | X | — | — | — |
Roche | X | — | — | — |
Declaration for Clinician 4
Name: Sanchit Bhasin, MD, FRCPC
Position: Gastroenterologist – Saskatchewan Health Authority; Regina General Hospital; Assistant Professor- University of Saskatchewan
Date: 30-03-2023
Table 6: COI Declaration for Canadian IBD Specialist Group — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Pfizer | — | — | X | — |
Takeda | X | — | — | — |
AbbVie | — | X | — | — |
Janssen | — | X | — | — |
Declaration for Clinician 5
Name: Mark Borgaonkar
Position: Staff Physician, Eastern Health
Date: 30-03-2023
Table 7: COI Declaration for Canadian IBD Specialist Group — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Amgen | — | X | — | — |
AstraZeneca | X | — | — | — |
BIOJAMP | X | — | — | — |
BMS | X | — | — | — |
Celltrion | X | — | — | — |
Janssen | — | — | X | — |
Pendopharm | X | — | — | — |
Pfizer | — | — | X | — |
Sandoz | X | — | — | — |
Takeda | — | — | X | — |
Declaration for Clinician 6
Name: Brian Bressler
Position: Staff Gastroenterologist
Date: 04-05-2023
Table 8: COI Declaration for Canadian IBD Specialist Group — Clinician 6
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | — | X |
Alimentiv | — | — | — | X |
BMS | — | X | — | — |
Celltrion | X | — | — | — |
Eli Lilly | X | — | — | — |
Gilead | — | X | — | — |
Janssen | — | — | — | X |
Organon | — | X | — | — |
Pfizer | — | — | — | X |
Sandoz | X | — | — | — |
Takeda | — | — | — | X |
Viatris | — | X | — | — |
Declaration for Clinician 7
Name: Sharyle Fowler
Position: Associate Professor, University of Saskatchewan, Saskatoon, SK
Date: 24-04-2023
Table 9: COI Declaration for Canadian IBD Specialist Group — Clinician 7
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Janssen | — | — | X | — |
Takeda | — | — | X | — |
Pendopharm | X | — | — | — |
Pfizer | X | — | — | — |
Bristol Myers Squibb | X | — | — | — |
Roche | X | — | — | — |
Amgen | X | — | — | — |
Sandoz | X | — | — | — |
Declaration for Clinician 8
Name: Daniel Green
Position: MD
Date: 14-03-2023
Table 10: COI Declaration for Canadian IBD Specialist Group — Clinician 8
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Takeda | X | — | — | — |
Janssen | — | X | — | — |
AbbVie | — | X | — | — |
Amgen | X | — | — | — |
BMS | X | — | — | — |
Celltrion | — | X | — | — |
Fresenius Kabi | — | X | — | — |
JAMP | X | — | — | — |
Merck | X | — | — | — |
Declaration for Clinician 9
Name: John Igoe
Position: Gastroenterologist, NB
Date: 02-04-2023
Table 11: COI Declaration for Canadian IBD Specialist Group — Clinician 9
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Janssen | X | — | — | — |
Takeda | — | X | — | — |
Pfizer | X | — | — | — |
GSK | — | X | — | — |
BIOJAMP | X | — | — | — |
Lupin | — | X | — | — |
Knight Pharma | X | — | — | — |
Bausch | X | — | — | — |
Intercept | X | — | — | — |
Declaration for Clinician 10
Name: Peter L Lakatos
Position: Professor of Medicine
Date: 31-03-2023
Table 12: COI Declaration for Canadian IBD Specialist Group — Clinician 10
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | X | — | — |
Amgen | — | X | — | — |
BIOJAMP | X | — | — | — |
BMS | X | — | — | — |
Fresenius Kabi | — | X | — | — |
Genetech | X | — | — | — |
Janssen | — | X | — | — |
Merck | X | — | — | — |
Mylan | X | — | — | — |
Organon | X | — | — | — |
Pendopharm | X | — | — | — |
Pfizer | — | — | X | — |
Roche | X | — | — | — |
Sandoz | — | X | — | — |
Takeda | — | — | X | — |
Tillots | X | — | — | — |
Viatris | X | — | — | — |
Declaration for Clinician 11
Name: Yvette Leung
Position: Associate Clinical Professor
Date: 21-03-2023
Table 13: COI Declaration for Canadian IBD Specialist Group — Clinician 11
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
BMS | — | X | — | — |
Jannsen | — | X | — | — |
AbbVie | — | — | X | — |
Frenius Kabus | X | — | — | — |
Amgen | X | — | — | — |
Takeda | — | — | X | — |
Pfizer | — | — | X | — |
Lilly | — | X | — | — |
Pendopharm | X | — | — | — |
BIOJAMP | X | — | — | — |
Declaration for Clinician 12
Name: Christopher Ma
Position: Assistant Professor, University of Calgary
Date: 14-03-2023
Table 14: COI Declaration for Canadian IBD Specialist Group — Clinician 12
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | — | X |
Alimentiv Inc. | — | — | — | X |
American College of Gastroenterology | — | — | X | — |
Amgen | — | — | X | — |
AVIR Pharma Inc. | — | X | — | — |
BIOJAMP | — | X | — | — |
Bristol Myers Squibb | — | — | X | — |
Celltrion | — | X | — | — |
Ferring | — | — | — | X |
Fresenius Kabi | — | — | X | — |
Janssen | — | — | X | — |
McKesson | — | X | — | — |
Mylan | — | X | — | — |
Takeda | — | — | — | X |
Pendopharm | — | X | — | — |
Pfizer | — | — | — | X |
Prometheus Biosciences Inc. | — | X | — | — |
Roche | — | X | — | — |
Sanofi | — | X | — | — |
Tillotts Pharma | — | X | — | — |
Springer Publishing | — | — | X | — |
Declaration for Clinician 13
Name: Mark MacMillan
Position: Gastroenterology, MD, FRCPC, CAGF
Date: 14-03-2023
Table 15: COI Declaration for Canadian IBD Specialist Group — Clinician 13
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
BIOJAMP | X | — | — | — |
Janssen | X | — | — | — |
Organon | X | — | — | — |
PendoPharm | X | — | — | — |
Pfizer | X | — | — | — |
Takeda | X | — | — | — |
Vantage | X | — | — | — |
Declaration for Clinician 14
Name: John Marshall
Position: Professor of Medicine
Date: 20-03-2023
Table 16: COI Declaration for Canadian IBD Specialist Group — Clinician 14
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | — | X |
AstraZeneca | X | — | — | — |
Amgen | — | — | X | — |
BMS | — | X | — | — |
Celltrion | X | — | — | — |
Ferring | — | X | — | — |
Fresenius Kabi | — | X | — | — |
Janssen | — | — | X | — |
Lilly | — | X | — | — |
Organon | X | — | — | — |
Pfizer | — | — | X | — |
Sandoz | — | X | — | — |
Takeda | — | — | X | — |
Viatris | — | X | — | — |
Declaration for Clinician 15
Name: Jeffrey McCurdy
Position: Gastroenterologist, University of Ottawa
Date: 19-03-2023
Table 17: COI Declaration for Canadian IBD Specialist Group — Clinician 15
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | X | — | — |
Fresenius Kabi | X | — | — | — |
BMS | X | — | — | — |
Ferring | X | — | — | — |
Janssen | — | X | — | — |
Pfizer | X | — | — | — |
Takeda | — | — | X | — |
Declaration for Clinician 16
Name: Neeraj Narula
Position: Director of IBD Clinic at Hamilton Health Sciences
Date: 20-03-2023
Table 18: COI Declaration for Canadian IBD Specialist Group — Clinician 16
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Janssen | — | X | — | — |
Takeda | — | X | — | — |
Eli Lilly | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Pfizer | — | X | — | — |
Viatris | X | — | — | — |
Sandoz | X | — | — | — |
Iterative Health | — | — | X | — |
Innomar Strategies | — | X | — | — |
Declaration for Clinician 17
Name: Kerri Novak, MD
Position: Clinical Associate Professor of Medicine, University of Calgary
Date: 22-03-2023
Table 19: COI Declaration for Canadian IBD Specialist Group — Clinician 17
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | X | — | — |
Amgen | X | — | — | — |
Bristol Myers Squibb | X | — | — | — |
Celltrion | X | — | — | — |
Eli Lilly | X | — | — | — |
Fresnius Kabi | X | — | — | — |
Janssen | X | — | — | — |
Organon | X | — | — | — |
Pendopharm | X | — | — | — |
Pfizer | — | X | — | — |
Takeda | — | X | — | — |
Declaration for Clinician 18
Name: Remo Panaccione
Position: Gastroenterologist, Professor of Medicine, University of Calgary
Date: 13-03-2023
Table 20: COI Declaration for Canadian IBD Specialist Group — Clinician 18
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Alimentiv | — | — | X | — |
Amgen | X | — | — | — |
Arena Pharmaceuticals | X | — | — | — |
AstraZeneca | X | — | — | — |
Bristol Myers Squibb | — | X | — | — |
Boehringer Ingelheim | X | — | — | — |
Eli Lilly | — | X | — | — |
Ferring | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Galapagos | X | — | — | — |
Gilead Sciences | X | — | — | — |
Glaxo-Smith-Klein | X | — | — | — |
JAMP Biomed | X | — | — | — |
Janssen | — | — | X | — |
Merck | X | — | — | — |
Mylan | X | — | — | — |
Oppilan | X | — | — | — |
Organon | — | X | — | — |
Pandion Pharma | X | — | — | — |
Pfizer | — | — | X | — |
Progenity | X | — | — | — |
Protagonist Therapeutics | X | — | — | — |
Roche | X | — | — | — |
Satisfai Health | X | — | — | — |
Sandoz | X | — | — | — |
Sublimity | X | — | — | — |
Takeda Pharmaceuticals | — | — | X | — |
Viatris | X | — | — | — |
Declaration for Clinician 19
Name: Greg Rosenfeld
Position: Clinical Associate Professor of Medicine
Date: 22-03-2023
Table 21: COI Declaration for Canadian IBD Specialist Group — Clinician 19
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | — | X |
Amgen | X | — | — | — |
BIOJAMP | X | — | — | — |
BMS | X | — | — | — |
Ferring | — | — | — | X |
Fresenius-Kabi | X | — | — | — |
Organon | X | — | — | — |
Pfizer | — | — | — | X |
Takeda | — | X | — | — |
Viatris | X | — | — | — |
Declaration for Clinician 20
Name: Seth Shaffer
Position: Assistant Professor of Medicine, at University of Manitoba
Date: 31-03-2023
Table 22: COI Declaration for Canadian IBD Specialist Group — Clinician 20
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Pfizer Canada | X | — | — | — |
AbbVie Canada | X | — | — | — |
Takeda Canada | X | — | — | — |
Janssen Canada | X | — | — | — |
Declaration for Clinician 21
Name: Chris Sheasgreen
Position: Clinical Assistant Professor, Dalhousie University
Date: 07-04-2023
Table 23: COI Declaration for Canadian IBD Specialist Group — Clinician 21
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | X | — | — |
Janssen | — | — | X | — |
BIOJAMP | X | — | — | — |
Merck | X | — | — | — |
Pfizer | X | — | — | — |
Takeda | — | X | — | — |
Declaration for Clinician 22
Name: Jesse Siffledeen, MD, FRCPC, MSc
Position: Gastroenterologist, Covenant Health, Edmonton, AB
Date: 24-04-2023
Table 24: COI Declaration for Canadian IBD Specialist Group — Clinician 22
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Janssen | — | — | X | — |
Takeda | — | — | X | — |
Fresenius Kabi | — | X | — | — |
BMS | X | — | — | — |
JAMP | — | — | X | — |
Lupin | X | — | — | — |
Celltrion | X | — | — | — |
Pendopharm | X | — | — | — |
Amgen | X | — | — | — |
Declaration for Clinician 23
Name: Michael Stewart
Position: Gastroenterologist, Halifax, Nova Scotia
Date: 02-04-2022
Table 25: COI Declaration for Canadian IBD Specialist Group — Clinician 23
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Amgen | X | — | — | — |
Janssen | — | X | — | — |
Pfizer | X | — | — | — |
Takeda | X | — | — | — |
Sandoz | X | — | — | — |
Bristol Myers Squibb | X | — | — | — |
Declaration for Clinician 24
Name: Brian G. Feagan, MD
Position: Professor of Medicine, Epidemiology and Biostatistics
Date: 27-04-2023
Table 26: COI Declaration for Canadian IBD Specialist Group — Clinician 24
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbolerIS | X | — | — | — |
AgomAB | X | — | — | — |
Allianthera | X | — | — | — |
Avoro Capital | X | — | — | — |
Boxer | X | — | — | — |
Cytoki | X | — | — | — |
Disc Medicine | X | — | — | — |
Duality | X | — | — | — |
Ecor1 | X | — | — | — |
Equillium | X | — | — | — |
Eermium | X | — | — | — |
First Wave | X | — | — | — |
First Word Group | X | — | — | — |
Galen Atlantica | X | — | — | — |
Hinge Bio | X | — | — | — |
Hot Spot Therapeutics | X | — | — | — |
JAKAcademy | X | — | — | — |
LEK Consulting | X | — | — | — |
LifeSci Capital | X | — | — | — |
Lument AB | X | — | — | — |
MiroBio | X | — | — | — |
Orphagen | X | — | — | — |
Pandion | X | — | — | — |
Play to Know AG | X | — | — | — |
Progenity | X | — | — | — |
Q32 Bio | X | — | — | — |
Rebiotix | X | — | — | — |
Silverback Therapeutics | X | — | — | — |
Ysios | X | — | — | — |
Ysopia | X | — | — | — |
Zealand Pharma | X | — | — | — |
Amgen | — | X | — | — |
AnaptysBio | — | X | — | — |
AMT | — | X | — | — |
Arena | — | X | — | — |
Atomwise | — | X | — | — |
BIOJAMP | — | X | — | — |
Biora Therapeutics | — | X | — | — |
Galapagos | — | X | — | — |
Galen Atlantica | — | X | — | — |
Genentech/Roche | — | X | — | — |
Index Pharma | — | X | — | — |
Imhotex | — | X | — | — |
Immunic Therapeutics | — | X | — | — |
Kaleido Biosciences | — | X | — | — |
Landos Biopharma | — | X | — | — |
Mylan | — | X | — | — |
Origo BioPharma | — | X | — | — |
Pendopharm | — | X | — | — |
Tigenix | — | X | — | — |
Thelium | — | X | — | — |
Ventyx Biosciences | — | X | — | — |
AbbVie | — | — | X | — |
Janssen | — | — | X | — |
Takeda | — | — | X | — |
Pfizer | — | — | X | — |
Eli Lilly | — | — | X | — |
Boehringer Engleheim | — | — | X | — |
Celsius Therapeutics | — | — | X | — |
Celgene/BMS | — | — | X | — |
Connect Biopharm | — | — | X | — |
Gilead | — | — | X | — |
Gossamer | — | — | X | — |
GSK | — | — | X | — |
Morphic Therapeutics | — | — | X | — |
Prometheus | — | — | X | — |
Sanofi | — | — | X | — |
Seres Therapeutics | — | — | X | — |
Surrozen | — | — | X | — |
Teva | — | — | X | — |
Tillotts | — | — | X | — |
References
Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011; 140:1785–1794.
Camara RJ, Ziegler R, Begre S, et al. The role of psychological stress in inflammatory bowel disease: Quality assessment of methods of 18 prospective studies and suggestions for future research. Digestion. 2009;80(2):129-139.
Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular, and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19: Suppl A:5-36.
Torres J, Mehandru S, Colombel JF, et al. Crohn’s disease. Lancet. 2017;389(10080):1741-1755.
Wilburn J, Twiss J, Kemp K, et al. A qualitative study of the impact of Crohn’s disease from a patient’s perspective. Frontline Gastroenterol. 2017;8(1):68-73.
Levesque BG, Sandborn WJ, Ruel J, et al. Converging goals of treatment of inflammatory bowel disease from clinical trials and practice. Gastroenterology 2015; 148:37–51 e1.
Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 2006; 130:323–333, quiz 591.
Sandborn WJ, Rutgeerts P, Enns R, et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann Intern Med 2007; 146:829–838.
Sandborn WJ, Feagan BG, Stoinov S, et al. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007; 357: 228–238.
Schreiber S, Khaliq-Kareemi M, Lawrance IC, et al. Maintenance therapy with certolizumab pegol for Crohn’s disease. N Engl J Med 2007; 357: 239–250.
Sandborn WJ, Colombel JF, Enns R, Feagan BG, Hanauer SB, Lawrance IC, Panaccione R, Sanders M, Schreiber S, Targan S, van Deventer S, Goldblum R, Despain D, Hogge GS, Rutgeerts P; International Efficacy of Natalizumab as Active Crohn's Therapy (ENACT-1) Trial Group; Evaluation of Natalizumab as Continuous Therapy (ENACT-2) Trial Group. Natalizumab induction and maintenance therapy for Crohn's disease. N Engl J Med. 2005 Nov 3;353(18):1912-25.
Targan SR, Feagan BG, Fedorak RN, Lashner BA, Panaccione R, Present DH, Spehlmann ME, Rutgeerts PJ, Tulassay Z, Volfova M, Wolf DC, Hernandez C, Bornstein J, Sandborn WJ; International Efficacy of Natalizumab in Crohn's Disease Response and Remission (ENCORE) Trial Group. Natalizumab for the treatment of active Crohn's disease: results of the ENCORE Trial. Gastroenterology. 2007 May;132(5):1672-83.
Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med 2013; 369: 711–721.
Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology 2014; 147: 618–627.
Feagan BG, Sandborn WJ, Gasink C, et al. Ustekinumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med 2016; 375: 1946–1960.
Rutgeerts P, Gasink C, Chan D, et al. Efficacy of ustekinumab for inducing endoscopic healing in patients with Crohn’s disease. Gastroenterology 2018; 155: 1045–1058.
Schreiber S, Ben-Horin S, Leszczyszyn J, et al. Randomized controlled trial: subcutaneous vs intravenous infliximab CT-P13 maintenance in inflammatory bowel disease. Gastroenterology 2021; 160: 2340–2353.
Ferrante M, Panaccione R, Baert F, et al. Risankizumab as maintenance therapy for moderately to severely active Crohn’s disease: results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase III FORTIFY maintenance trial. Lancet 2022; 399: 2031–46
Beaugerie L, Seksik P, Nion-Larmurier I, et al. Predictors of Crohn’s disease. Gastroenterology 2006; 130:650–656.
Loly C, Belaiche J, Louis E. Predictors of severe Crohn’s disease. Scand J Gastroenterol 2008; 43:948–954.
Beaugerie L, Sokol H. Clinical, serological, and genetic predictors of inflammatory bowel disease course. World J Gastroenterol 2012; 18:3806–3813.
Gisbert JP, Marin AC, McNicholl AG, et al. Systematic review with metaanalysis: the efficacy of a second anti-TNF in patients with inflammatory bowel disease whose previous anti-TNF treatment has failed. Aliment Pharmacol Ther 2015; 41:613–623.
Kiss LS, Papp M, Lovasz BD, et al. High-sensitivity C-reactive protein for identification of disease phenotype, active disease, and clinical relapses in Crohn’s disease: a marker for patient classification? Inflamm Bowel Dis 2012; 18:1647–1654.
Mao R, Xiao YL, Gao X, et al. Fecal calprotectin in predicting relapse of inflammatory bowel diseases: a meta-analysis of prospective studies. Inflamm Bowel Dis 2012; 18:1894–1899.
Ghaly S, Murray K, Baird A, et al. High vitamin D-binding protein concentration, low albumin, and mode of remission predict relapse in Crohn’s disease. Inflamm Bowel Dis 2016; 22:2456–2464.
Qin G, Tu J, Liu L, et al. Serum albumin and C-reactive protein/albumin ratio are useful biomarkers of Crohn’s disease activity. Med Sci Monit 2016; 22:4393–4400.
Khanna R, Zou G, D’Haens G, et al. A retrospective analysis: the development of patient reported outcome measures for the assessment of Crohn’s disease activity. Aliment Pharmacol Ther 2015; 41:77–86.
Khanna R, D’Haens G, Feagan BG, et al. Patient reported outcome measures derived from the Crohn’s Disease Activity Index: correlation between PRO2 and PRO3 scores and CDAI-defined clinical thresholds [abstract P176]. J Crohns Colitis 2014;8: S135.
Cellier C, Sahmoud T, Froguel E, et al. Correlations between clinical activity, endoscopic severity, and biological parameters in colonic or ileocolonic Crohn’s disease: a prospective multicentre study of 121 cases—The Groupe d’Etudes Therapeutiques des Affections Inflammatoires Digestives. Gut 1994 ;35 :231–235.
Sipponen T, Savilahti E, Kolho KL, et al. Crohn’s disease activity assessed by fecal calprotectin and lactoferrin: correlation with Crohn’s disease activity index and endoscopic findings. Inflamm Bowel Dis 2008; 14:40–46.
Jones J, Loftus EV Jr, Panaccione R, et al. Relationships between disease activity and serum and fecal biomarkers in patients with Crohn’s disease. Clin Gastroenterol Hepatol 2008; 6:1218–1224.
Panaccione R, Steinhart AH, Bressler B, Khanna R, Marshall JK, Targownik L, Afif W, Bitton A, Borgaonkar M, Chauhan U, Halloran B, Jones J, Kennedy E, Leontiadis GI, Loftus EV Jr, Meddings J, Moayyedi P, Murthy S, Plamondon S, Rosenfeld G, Schwartz D, Seow CH, Williams C, Bernstein CN. Canadian Association of Gastroenterology Clinical Practice Guideline for the Management of Luminal Crohn's Disease. Clin Gastroenterol Hepatol. 2019 Aug;17(9):1680-1713.
Crohn’s and Collitis Canada. 2018 Impact of Inflammatory Bowel Disease in Canada. https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2018-Impact-Report-LR.pdf
Coward S, Clement F, Benchimol E, et al. The rising prevalence of inflammatory bowel disease in Canada: Analyzing the past to predict the future. Journal of the Canadian Association of Gastroenterology. 2018; 1(Supp 2): A-29.
Golovics PA, Lakatos L, Mandel MD, et al. Prevalence and predictors of hospitalization in Crohn’s disease in a prospective population-based inception cohort from 2000–2012. World J Gastroenterol 2015; 21:7272–7280.
Becker HM, Grigat D, Ghosh S, et al. Living with inflammatory bowel disease: A Crohn’s and Colitis Canada survey. Can J Gastroenterol Hepatol. 2015;29(2):77-84.
Bernstein CN. Review article: Changes in the epidemiology of inflammatory bowel disease clues for aetiology. Aliment Pharmacol Ther. 2017;46(10):911-919.
Lix LM, Graff LA, Walker JR, et al. Longitudinal study of quality of life and psychological functioning for active, fluctuating, and inactive disease patterns in inflammatory bowel disease. Inflamm Bowel Dis. 2008;14(11):1575-1584.
Vavricka SR, Spigaglia SM, Rogler G, et al. Systematic evaluation of risk factors for diagnostic delay in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18(3):496-505.
Schoepfer AM, Dehlavi MA, Fournier N, et al. Diagnostic delay in Crohn’s disease is associated with a complicated disease course and increased operation rate. Am J Gastroenterol. 2013;108(11):1744-1753.
Benchimol EI, Manuel DG, Mojaverian N, et al. Health services utilization, specialist care, and time to diagnosis with inflammatory bowel disease in immigrants to Ontario, Canada: A population-based cohort study. Inflamm Bowel Dis. 2016;22(10):2482-2490.
Floyd DN, Langham S, Severac HC, et al. The economic and quality-of-life burden of Crohn’s disease in Europe and the United States, 2000 to 2013: a systematic review. Dig Dis Sci 2015; 60:299–312.
Kahn SA, Lin CW, Ozbay B, et al. Indirect costs and family burden of pediatric Crohn’s disease in the United States. Inflamm Bowel Dis. 2017;23(12):2089-2096.
Bernstein CN, Longobardi T, Finlayson G, et al. Direct medical cost of managing IBD patients: A Canadian population-based study. Inflamm Bowel Dis. 2012;18(8):1498-1508.
Longobardi T, Walker JR, Graff LA, et al. Health service utilization in IBD: Comparison of self-report and administrative data. BMC Health Serv Res. 2011; 11:137.
Nguyen GC, Bollegala N, Chong CA. Factors associated with readmissions and outcomes of patients hospitalized for inflammatory bowel disease. Clin Gastroenterol Hepatol. 2014;12(11):1897-1904.
Marrie RA, Garland A, Peschken CA, et al. Increased incidence of critical illness among patients with inflammatory bowel disease: A population-based study. Clin Gastroenterol Hepatol. 2014;12(12):2063-2070.
Frolkis AD, Dykeman J, Negron ME, et al. Risk of surgery for inflammatory bowel diseases has decreased over time: A systematic review and meta-analysis of population-based studies. Gastroenterology. 2013;145(5):996-1006.
Frolkis AD, Lipton DS, Fiest KM, et al. Cumulative incidence of second intestinal resection in Crohn’s disease: A systematic review and meta-analysis of population-based studies. Am J Gastroenterol. 2014;109(11):1739-1748.
Bassi A, Dodd S, Williamson P, et al. Cost of illness of inflammatory bowel disease in the UK: A single centre retrospective study. Gut. 2004;53(10):1471-1478.
Blomqvist P, Ekbom A. Inflammatory bowel diseases: Health care and costs in Sweden in 1994. Scand J Gastroenterol. 1997;32(11):1134-1139.
Niewiadomski O, Studd C, Hair C, et al. Health care cost analysis in a population-based inception cohort of inflammatory bowel disease patients in the first year of diagnosis. J Crohns Colitis. 2015;9(11):988-996.
Burisch J, Vardi H, Pedersen N, et al. Costs and resource utilization for diagnosis and treatment during the initial year in a European inflammatory bowel disease inception cohort: An ECCO-EpiCom Study. Inflamm Bowel Dis. 2015;21(1):121-131.
van der Valk ME, Mangen MJ, Leenders M, et al. Healthcare costs of inflammatory bowel disease have shifted from hospitalisation and surgery towards anti-TNFalpha therapy: Results from the COIN study. Gut. 2014;63(1):72-79.
Park KT, Colletti RB, Rubin DT, et al. Health insurance paid costs and drivers of costs for patients with Crohn’s disease in the United States. Am J Gastroenterol. 2016;111(1):15-23.
Aldeguer X, Sicras-Mainar A. Costs of ulcerative colitis from a societal perspective in a regional health care area in Spain: A database study. Gastroenterol Hepatol. 2016;39(1):9-19.
Loomes DE, Teshima C, Jacobs P, et al. Health care resource use and costs for Crohn’s disease before and after infliximab therapy. Can J Gastroenterol. 2011;25(9):497-502.
Wan GJ, Kozma CM, Slaton TL, et al. Inflammatory bowel disease: Healthcare costs for patients who are adherent or non-adherent with infliximab therapy. J Med Econ. 2014;17(6):384-393.
Sprakes MB, Ford AC, Suares NC, et al. Costs of care for Crohn’s disease following the introduction of infliximab: A single-centre UK experience. Aliment Pharmacol Ther. 2010;32(11-12):1357-1363.
Lindsay JO, Chipperfield R, Giles A, et al. A UK retrospective observational study of clinical outcomes and healthcare resource utilisation of infliximab treatment in Crohn’s disease. Aliment Pharmacol Ther. 2013;38(1):52-61.
Waters HC, Vanderpoel JE, Nejadnik B, et al. Resource utilization before and during infliximab therapy in patients with inflammatory bowel disease. J Med Econ. 2012;15(1):45-52.
Church P, Walters T, Benchimol E, et al. Steroid-free remission among Canadian pediatric inflammatory bowel disease patients. Can J Gastroenterol Hepatol. 2016;2016(4792898):7-8.
Targownik LE, Tennakoon A, Leung S, et al. Temporal trends in initiation of therapy with tumor necrosis factor antagonists for patients with inflammatory bowel disease: A population-based analysis. Clin Gastroenterol Hepatol. 2017;15(7):1061-1070.
Rocchi A, Benchimol EI, Bernstein CN, et al. Inflammatory bowel disease: A Canadian burden of illness review. Can J Gastroenterol 2012;26(11):811-817.
Nguyen GC, Nugent Z, Shaw S, et al. Outcomes of patients with Crohn’s disease improved from 1988 to 2008 and were associated with increased specialist care. Gastroenterology. 2011;141(1):90-97.
Nugent Z, Singh H, Targownik LE, et al. Predictors of emergency department use by persons with inflammatory bowel diseases: A population-based study. Inflamm Bowel Dis. 2016;22(12):2907-2916.
Shafer LA, Walker JR, Chhibba T, et al. Association between IBD, disability, and reduced work productivity (presenteeism): A population-based study in Manitoba, Canada. Gastroenterology. 2017;152(5):152.
Statistics Canada. Average weekly earnings (including overtime), by province and territory. 2017.
Rogala L, Miller N, Graff LA, et al. Population-based controlled study of social support, self-perceived stress, activity and work issues, and access to health care in inflammatory bowel disease. Inflamm Bowel Dis. 2008;14(4):526-535.
Stallmach A, Dennler U, Marschall U, et al. Patient-relevant endpoints in inflammatory bowel diseases – have changes occurred in Germany over the past twelve years? J Crohns Colitis. 2015;9(5):390-397.
Bernstein CN, Nugent Z, Targownik LE, et al. Predictors and risks for death in a population-based study of persons with IBD in Manitoba. Gut. 2015;64(9):1403-1411.
Nguyen GC, Bernstein CN, Benchimol EI. Risk of surgery and mortality in elderly-onset inflammatory bowel disease: A population-based cohort study. Inflamm Bowel Dis. 2017;23(2):218-223.
Statistics Canada. Table 102-0531 – Deaths, by cause, Chapter XI: Diseases of the digestive system (K00 to K93), age group and sex, Canada, Annual. 2017.
Sadowski DC, Bernstein CN, Bitton A, et al. Canadian Association of Gastroenterology Clinical Practice Guidelines: The use of tumour necrosis factor-alpha antagonist therapy in Crohn’s disease. Can J Gastroenterol. 2009;23(3):185-202.
Vavricka SR, Schoepfer AM, Scharl M, et al. Steroid use in Crohn’s disease. Drugs. 2014;74(3):313-324.
Hazlewood GS, Rezaie A, Borman M, et al. Comparative effectiveness of immunosuppressants and biologics for inducing and maintaining remission in Crohn’s disease: A network meta-analysis. Gastroenterology. 2015;148(2):344-354.
Present DH, Korelitz BI, Wisch N, et al. Treatment of Crohn’s disease with 6-mercaptopurine. A long-term, randomized, double-blind study. N Engl J Med 1980; 302: 981–987.
Feagan BG, Rochon J, Fedorak RN, et al. Methotrexate for the treatment of Crohn’s disease. The North American Crohn’s Study Group Investigators. N Engl J Med 1995; 332: 292–297.
Feagan BG, Fedorak RN, Irvine EJ, et al. A comparison of methotrexate with placebo for the maintenance of remission in Crohn’s disease. North American Crohn’s Study Group Investigators. N Engl J Med 2000; 342: 1627–1632.
Kumar A, Cole A, Segal J, Smith P, Limdi JK. A review of the therapeutic management of Crohn's disease. Therap Adv Gastroenterol. 2022 Feb 17;15: 1-19.
Ben-Horin S, Kopylov U, Chowers Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun Rev. 2014;13(1):24-30.
Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 2002; 359: 1541–1549.
Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med 2010; 362: 1383–1395.
Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 2006; 130: 323–333.
Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 2007; 132: 52–65.
Rutgeerts P, Van Assche G, Sandborn WJ, et al. Adalimumab induces and maintains mucosal healing in patients with Crohn’s disease: data from the EXTEND trial. Gastroenterology 2012; 142: 1102–1111.
Papamichael K, Van Stappen T, Jairath V, et al. Review article: pharmacological aspects of anti-TNF biosimilars in inflammatory bowel diseases. Aliment Pharmacol Ther 2015; 42: 1158–1169.
Peyrin-Biroulet L, Danese S, Cummings F, et al. Anti-TNF biosimilars in Crohn’s Disease: a patient-centric interdisciplinary approach. Expert Rev Gastroenterol Hepatol 2019; 13: 731–738.
Bhat S, Limdi JK, Cross RK, et al. Does similarity breed contempt? A review of the use of biosimilars in inflammatory bowel disease. Dig Dis Sci 2021; 66: 2513–2532.
Hansen T, Targownik LE. Ustekinumab for the treatment of Crohn’s disease. Expert Rev Gastroenterol Hepatol. 2016;10(9):989-994.
Kavanaugh A, Ritchlin C, Rahman P, et al. Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: results of an integrated analysis of radiographic data from the phase III, multicentre, randomised, doubleblind, placebo-controlled PSUMMIT-1 and PSUMMIT-2 trials. Ann Rheum Dis 2014; 73: 1000–1006.
Haddley K. Vedolizumab for the treatment of inflammatory bowel disease. Drugs Today (Barc). 2014;50(4):309-319.
Vilchez V, Lightner AL. Surgical Management of Crohn's Disease. Gastroenterol Clin North Am. 2022 Jun;51(2):353-367.
Cellier C. et al. Correlations between clinical activity, endoscopic severity, and biological parameters in colonic or ileocolonic Crohn's disease. A prospective multicentre study of 121 cases. The Groupe d'Etudes Thérapeutiques des Affections Inflammatoires Digestives Gut. 1994; 35:231-235; Lewis JD, et al. Inflamm Bowel Dis 2020;26(2):304-313.
Panaccione R, Colombel JF, Louis E, et al. Evolving definitions of remission in Crohn’s disease. Inflamm Bowel Dis 2013; 19:1645–1653.
Peyrin-Biroulet L, et al. Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE): Determining Therapeutic Goals for Treat-to-Target. Am J Gastroenterol. 2015 Sep;110(9):1324-38.
Turner D, Ricciuto A, Lewis A, et al. STRIDE-II: an update on the selecting therapeutic targets in inflammatory bowel disease (STRIDE) initiative of the International Organization for the Study of IBD (IOIBD): determining therapeutic goals for treat-to-target strategies in IBD. Gastroenterology 2021; 160: 1570–83.
Shah SC, Colombel JF, Sands BE, et al. Narula N. Systematic review with meta-analysis: mucosal healing is associated with improved long-term outcomes in Crohn's disease. Aliment Pharmacol Ther. 2016 Feb;43(3):317-33.
Yzet C, Diouf M, Le Mouel et al, Complete Endoscopic Healing Associated with Better Outcomes Than Partial Endoscopic Healing in Patients with Crohn's Disease. Clin Gastroenterol Hepatol. 2020 Sep;18(10):2256-2261.
https://clinicaltrials.gov/ct2/show/NCT03105128 Accessed October 16, 2022
van der Goes MC, Jacobs JW, Boers M, Andrews T, Blom-Bakkers MA, Buttgereit F, Caeyers N, Cutolo M, Da Silva JA, Guillevin L, Kirwan JR, Rovensky J, Severijns G, Webber S, Westhovens R, Bijlsma JW. Monitoring adverse events of low-dose glucocorticoid therapy: EULAR recommendations for clinical trials and daily practice. Ann Rheum Dis. 2010 Nov;69(11):1913-9.
Lewis JD, Scott FI, Brensinger CM, et al. Increased mortality rates with prolonged corticosteroid therapy when compared with antitumor necrosis factor-alpha-directed therapy for inflammatory bowel disease. Am J Gastroenterol 2018; 113: 405–417.
Anderson A, Click B, Ramos-Rivers C, et al. Lasting impact of clostridium difficile infection in inflammatory bowel disease: A propensity score matched analysis. Inflamm Bowel Dis. 2017;23(12):2180-2188.
XXXXXXXXXXXX
Munkholm P, Langholz E, Davidsen M, et al. Frequency of glucocorticoid resistance and dependency in Crohn’s disease. Gut 1994; 35: 360–362.
Faubion WA Jr, Loftus EV Jr, Harmsen WS, et al. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology 2001; 121: 255–260.
Summers RW, Switz DM, Sessions JT Jr, et al. National Cooperative Crohn’s Disease Study: results of drug treatment. Gastroenterology 1979; 77: 847–869.
Steinhart AH, Ewe K, Griffiths AM, et al. Corticosteroids for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev 2003; 4: CD000301.
Kuenzig ME, Rezaie A, Kaplan GG, et al. Budesonide for the induction and maintenance of remission in Crohn’s disease: systematic review and meta-analysis for the cochrane collaboration. J Can Assoc Gastroenterol 2018; 1: 159–173.
Modigliani R, Mary JY, Simon JF, et al. Clinical, biological, and endoscopic picture of attacks of Crohn’s disease. Evolution on prednisolone. Groupe d’Etude Therapeutique des Affections Inflammatoires Digestives. Gastroenterology 1990 ; 98 : 811–818.
Chande N, Patton PH, Tsoulis DJ, et al. Azathioprine or 6-mercaptopurine for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev 2015 ; 10 : CD000067.
Kappelman MD, Farkas DK, Long MD, et al. Risk of cancer in patients with inflammatory bowel diseases: a nationwide population-based cohort study with 30 years of follow-up evaluation. Clin Gastroenterol Hepatol 2014; 12: 265–273.
Kotlyar DS, Lewis JD, Beaugerie L, et al. Risk of lymphoma in patients with inflammatory bowel disease treated with azathioprine and 6-mercaptopurine: a meta-analysis. Clin Gastroenterol Hepatol 2015; 13: 847–858.
Cuffari C, Theoret Y, Latour S, et al. 6-mercaptopurine metabolism in Crohn’s disease: correlation with efficacy and toxicity. Gut 1996; 39: 401–406.
Kennedy NA, Heap GA, Green HD, et al. Predictors of anti-TNF treatment failure in anti-TNF-naive patients with active luminal Crohn’s disease: a prospective, multicentre, cohort study. Lancet Gastroenterol Hepatol 2019; 4: 341–353.
Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med 2003; 348: 601–608.
Bonovas S, Fiorino G, Allocca M, et al. Biologic therapies and risk of infection and malignancy in patients with inflammatory bowel disease: a systematic review and network meta-analysis. Clin Gastroenterol Hepatol 2016; 14: 1385–1397.
Golovics PA, Mandel MD, Lovasz BD, et al. Inflammatory bowel disease course in Crohn’s disease: is the natural history changing? World J Gastroenterol 2014; 20: 3198–3207.
Roda G, Jharap B, Neeraj N, Colombel JF. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin Transl Gastroenterol. 2016 Jan 7;7(1):e135.
Schett G, McInnes IB, Neurath MF. Reframing Immune-Mediated Inflammatory Diseases through Signature Cytokine Hubs. N Engl J Med. 2021 Aug 12;385(7):628-639.
D'Haens G, Panaccione R, Baert F, et al. Risankizumab as induction therapy for Crohn's disease: results from the phase III ADVANCE and MOTIVATE induction trials. Lancet. 2022 May 28;399(10340):2015-2030
Present DH, Rutgeerts P, Targan S, et al. Infliximab for the treatment of fistulas in patients with Crohn's disease. NEnglJMed. 1999;340(18):1398-405.
Sands BE, Anderson F, Bernstein CB, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. NEnglJMed 2004; 2004;350:876-85.
Ferrante M, Panaccione R, Baert F, et al. Risankizumab as maintenance therapy for moderately to severely active Crohn’s disease: results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase III FORTIFY maintenance trial. Lancet 2022;399:2031-2046
Benchimol EI, Seow CH, Otley AR, Steinhart AH. Cochrane Database Syst Rev. 2009 Jan 21;(1):CD002913. doi: 10.1002/14651858.CD002913.pub2
Sands B, Irving PM, Hoop T, et al. Ustekinumab versus adalimumab for induction and maintenance therapy in biologic-naive patients with moderately to severely active Crohn’s disease: a multicentre, randomised, double-blind, parallel-group, phase IIIb trial. Lancet 2022;399:2200-2211.
Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus Kinase (JAK) inhibitors for inflammatory diseases. J Med Chem 2014;57:5023-37
Aguilar D, et al. Randomized Controlled Trial Substudy of Cell-specific Mechanisms of Janus Kinase Inhibition With Upadacitinib in the Crohn's Disease Intestinal Mucosa: Analysis from the CELEST Study. Inflamm Bowel Dis. 2021 Nov 15;27(12):1999-2009. doi: 10.1093/ibd/izab116
Loftus EV Jr, et al. Efficacy and Safety of Upadacitinib Induction Therapy in Patients With Moderately to Severely Active Crohn’s Disease: Results From a Randomized Phase III U-EXCEL Study. Am J Gastroenterol 2022;117:S736
Colombel JF, et al. Efficacy and safety of upadacitinib induction therapy in patients with moderately to severely active Crohn's disease who failed prior biologics: Results from a randomized phase III U-EXCEED study. Gastroenterology. 2022;162(7): S-1394.
Panes J, Loftus EV Jr, Lacerda AP, et al. Efficacy and Safety of Upadacitinib Maintenance Therapy in Patients with Moderately to Severely Active Crohn’s Disease: Results from a Randomized Phase III U-ENDURE Maintenance Study. Oral presentation at: the American College of Gastroenterology Annual Scientific Meeting; October 21-26, 2022; Charlotte, NC
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.